Настоящее изобретение относится к группе соединений, которые содержат хроменовое ядро и которые обладают способностью ингибировать пролиферацию лимфоцитов посредством блокирования взаимодействия TCR с Nсk, поэтому такие соединения можно применять для лечения заболеваний или состояний, при которых такое взаимодействие инициирует осложнения, такие как реакции отторжения трансплантата, иммунные или аутоиммунные заболевания или пролиферативные заболевания.
УРОВЕНЬ ТЕХНИКИ
Аутоиммунные и воспалительные заболевания, такие как астма, рассеянный склероз, аллергии, ревматоидный артрит, болезнь Крона или псориаз, представляют собой разнородную группу заболеваний, при которых адаптивная иммунная система, в частности, с помощью Т-лимфоцитов, атакует собственные антигены организма. Принято считать, что Т-клетки находятся в центре всех иммунологических механизмов. Т-клетки могут распознавать как чужеродные, так и собственные антигены и активировать иммунный ответ против них. Т-клетки распознают антигены с помощью Т-клеточного рецептора (TCR), ответственного за передачу сигналов в цитоплазму. Действительно, тот факт, что гаплотип главного комплекса гистосовместимости (ГКГС) является наиболее важным фактором генетического риска для аутоиммунных заболеваний человека, ставит Т-клетки в центр всех иммунопатологических событий.
Т-клетка распознает антигенный пептид, связанный с ГКГС (пГКГС), через Т-клеточный рецептор антигена (TCR) и способна переводить небольшие различия в химическом составе пГКГС в различные количественные и качественные результаты. В то время как существуют различные механизмы контроля предотвращения активации Т-клеток, несущих TCR со значительным сродством к ГКГС, загруженным с аутопептидами, в том числе подавление потенциально аутореактивных Т-клеток в процессе созревания в тимусе, эти механизмы являются в некоторой степени недостаточным у пациентов, у которых развиваются аутоиммунные заболевания, и аутореактивные Т-клетки активируются и расширяются, преодолевая гомеостатические контроли.
При стимуляции TCR активируется и претерпевает конформационные изменения, в результате чего получается набор различных белков, образующих "TCR сигналосому", ответственную за трансдукцию сигнала и активацию клеток. Этот комплекс включает цитозольный белок Nck, который связывается с PRS мотивом (богатой пролином последовательностью), присутствующим в CD3e субъединице рецептора Т-клеток. В результате стабилизируется конформационное изменение TCR, и эффективно передается сигнал активации.
Современные методы лечения иммунных заболеваний представляют собой иммуносупрессивные стратегии, а не толерогенные/иммуномодулирующие подходы. Азатиоприн, метотрексат, микофенолат и кладрибин являются цитостатиками. Другие методы лечения вызывают истощение Т-клеток (алемтузумаб, анти-СD52) или их удержание в лимфатических узлах (финголимод). При другом подходе в качестве мощной стратегии (BG-12) также применяют непрямую модуляцию иммунной системы. Поэтому, несмотря на центральную роль сигнала TCR для активации Т-клеток при аутоиммунных заболеваниях, усилия последнего времени, направленные на модулирование активации Т-клеток, сосредоточены на модулировании дополнительных стимулирующих сигналов, рецепторов цитокинов и т.д. с последующим отсутствием специфичности и большого числа сопутствующих побочных эффектов.
Для того чтобы разработать конкретную иммуномодулирующую терапию, многочисленные усилия множества различных исследовательских групп были сосредоточены на описании роли Nck в активации Т-клеток. Nck приписывают важную роль в функционировании зрелых Т-клеток на основании исследований у нокаут-мышей, лишенных Nck1 во всех тканях и лишенных Nck2 условно только на Т-клетках. В этих моделях число периферических Т-клеток, экспрессирующих TCR с низкой авидностью к собственным антигенам, резко сократилось, и наблюдалось общее снижение активации Т-клеток путем стимуляции слабыми антигенами. Кроме того, важность Nck была также рассмотрена путем создания костномозговых химер, и было показано, что PRS мотив (место связывания Nck в TCR) имеет важное значение для активации зрелых Т-клеток слабыми, но не сильными агонистами. Аналогичным образом мутации в последовательности PRS изменяли способность мышей активировать адаптивный иммунный ответ in vivo. Кроме того, пептидный ингибитор с высоким сродством к домену SH3.1 Nck изменяет набор сигналосом TCR, на основании чего предполагают, что набор Nck является важным первым шагом в передаче сигналов TCR, который представляет собой мишень для модулирования иммунного ответа.
В документе WO 2010/064707 описан ряд соединений, полученных из 2Н-хромена, для профилактики или лечения заболевания, вызванного нежелательной инфильтрацией лимфоцитов, опосредованной сфингозин-1-фосфатным рецептором (S1P1).
В документе WO 2012/042078 также описаны производные хромена, обладающие способностью ингибировать взаимодействие TCR-Nck в Т-клетках, и их применение для лечения аутоиммунных заболеваний, воспалительных заболеваний или отторжения трансплантата.
Поэтому было бы желательно разработать новые соединения, которые способны ингибировать взаимодействие TCR-Nck в Т-лимфоцитах и которые являются хорошими потенциальными лекарственными средствами. Такие соединения должны проявлять хорошую активность в фармакологическом испытании in vivo, хорошую пероральную абсорбцию при пероральном введении, а также быть метаболически стабильными и иметь благоприятный фармакокинетический профиль. Кроме того, такие соединения не должны быть токсичными и должны вызывать минимум побочных эффектов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
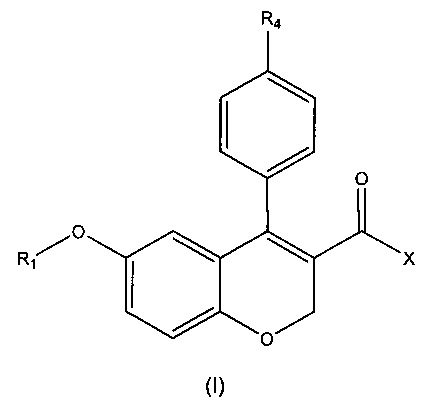
Первый аспект настоящего изобретения относится к соединению формулы (I)
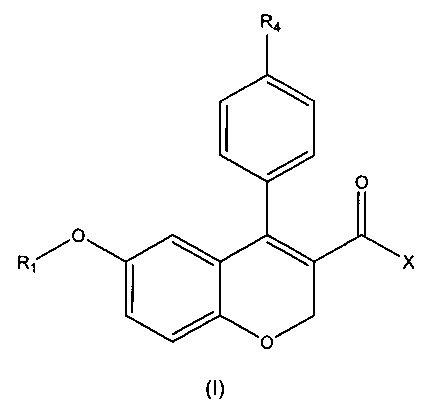
или его фармацевтически приемлемой соли, изомеру или сольвату, где:
R1 выбирают из водорода, замещенного или незамещенного C1-C6 алкила, замещенного или незамещенного C3-C6 циклоалкила, замещенного или незамещенного арила или замещенного или незамещенного гетероарила, -COR5, -C(O)OR5, -C(O)NR5R6, -CNR5;
X выбирают из -ОН или -NR2R3;
R2 и R3 независимо выбирают из водорода, замещенного или незамещенного C1-C8 алкила, замещенного или незамещенного С3-C6 циклоалкила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -COR7, -C(O)OR7, -C(O)NR7R8, -CNR7, -OR7, -NR7R8 и -NR7C(O)R8;
или R2 и R3 вместе с атомом азота, с которым они связаны, образуют замещенный или незамещенный гетероцикл;
R4 представляет собой галоген;
R5, R6, R7 и R8 независимо выбирают из водорода, C1-C4 алкила, C3-C6 циклоалкила, арила, гетероарила и галогена.
Другой аспект настоящего изобретения относится к соединению формулы (II):
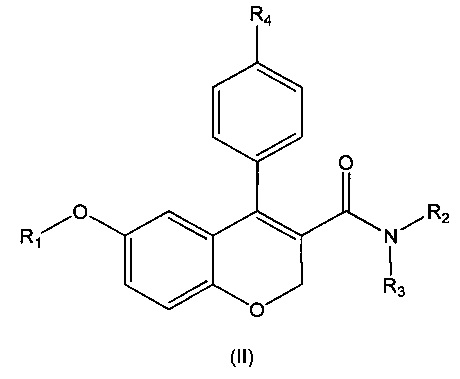
или его фармацевтически приемлемой соли, изомеру или сольвату, где:
R1 выбирают из водорода, замещенного или незамещенного C1-C6 алкила, замещенного или незамещенного С3-С6 циклоалкила, замещенного или незамещенного арила или замещенного или незамещенного гетероарила, -COR5, -C(O)OR5, -C(O)NR5R6, -CNR5;
R2 и R3 независимо выбирают из водорода, замещенного или незамещенного C1-C8 алкила, замещенного или незамещенного C3-C6 циклоалкила, замещенного или незамещенного арила, замещенного или незамещенного гетероарила, -COR7, -C(O)OR7, -C(O)NR7R8, -CNR7,-OR7, -NR7R8 и -NR7C(O)R8;
или R2 и R3 вместе с атомом азота, с которым они связаны, образуют замещенный или незамещенный гетероцикл;
R4 представляет собой галоген;
R5, R6, R7 и R8 независимо выбирают из водорода, C1-C4 алкила, C3-C6 циклоалкила, арила, гетероарила и галогена.
В настоящем изобретении термин "алкил" относится к радикалам углеводородных цепей, линейных или разветвленных, содержащих от 1 до 6 атомов углерода, и предпочтительно от 1 до 4, и связанных с остальной частью молекулы одинарной связью, например, к метилу, этилу, н-пропилу, изо-пропилу, н-бутилу, трет-бутилу, втор-бутилу, н-пентилу, н-гексилу и т.д. Алкильные группы могут быть необязательно замещены одним или несколькими заместителями, такими как галоген, гидроксил, алкоксил, карбоксил, карбонил, циано, ацил, алкоксикарбонил, амино, нитро, меркапто и алкилтио.
В настоящем изобретении термин "циклоалкил" относится к стабильному 3-6-членному моноциклическому радикалу, предпочтительно 3-членному, насыщенному или частично ненасыщенному, и который состоит только из атомов углерода и водорода, таким как циклопропил, циклопентил, циклогексил, и который может быть необязательно замещен одной или более группами, такими как алкил, галоген, гидроксил, алкоксил, карбоксил, циано, карбонил, ацил, алкоксикарбонил, амино, нитро, меркапто и алкилтио.
В настоящем изобретении термин "арил" относится к ароматической карбоциклической цепи, содержащей от 6 до 18 атомов углерода, предпочтительно от 6 до 14 атомов углерода и более предпочтительно от 6 до 8, и может состоять из одного или нескольких колец, и в последнем случае может быть с разделенными и/или конденсированными кольцами. Неограничивающие примеры арильных групп представляют собой фенил, нафтил, инденил и т.д. Предпочтительно арильная группа представляет собой фенил или нафтил. Арильные группы могут быть необязательно замещены одним или более заместителями, такими как алкил, галоген, гидроксил, алкоксил, карбоксил, карбонил, циано, ацил, алкоксикарбонил, амино, нитро, меркапто и алкилтио.
Термин "гетероарил" относится к арильной группе, содержащей по меньшей мере один гетероатом, выбранный из следующей группы: азот, кислород или сера.
В настоящем изобретении термин "гетероцикл" относится к стабильному моноциклическому или бициклическому радикалу, содержащему от 3 до 15 членов, который является ненасыщенным, насыщенным или частично насыщенным и который состоит из атомов углерода и по меньшей мере одного гетероатома, выбранного из следующей группы: азот, кислород или сера. Предпочтительно он содержит от 4 до 8 членов с одним или более гетероатомами, более предпочтительно от 5 до 6 членов с одним или более гетероатомами. Примерами гетероарила могут быть, не ограничиваясь ими: азепины, индолы, имидазолы, изотиазолы, тиадиазолы, фуран, тетрагидрофуран, бензимидазол, бензотиазол, пиперидин, пирролидин, пиперазин, пурин, хинолин. Предпочтительно гетероциклическая группа представляет собой пирролидин или пиперазин. Гетероциклические группы необязательно могут быть замещены в любом из своих положений одним или более заместителями, такими как алкил, галоген, гидроксил, алкоксил, карбоксил, карбонил, циано, ацил, алкоксикарбонил, амино, нитро, меркапто и алкилтио.
Термин "галоген" относится к фтору, хлору, брому или йоду.
В предпочтительном варианте реализации настоящего изобретения R1 представляет собой замещенный или незамещенный C1-C4 алкил.
В более предпочтительном варианте реализации настоящего изобретения R1 представляет собой CH3.
В другом более предпочтительном варианте реализации настоящего изобретения R1 представляет собой C1-C4 алкил, замещенный посредством С3-С6 циклоалкила. В еще более предпочтительном варианте реализации настоящего изобретения R1 представляет собой -CH2-циклопропильную группу.
В другом предпочтительном варианте реализации настоящего изобретения R2 представляет собой Н.
В другом предпочтительном варианте реализации настоящего изобретения R3 представляет собой замещенный или незамещенный C1-C4 алкил.
В более предпочтительном варианте реализации настоящего изобретения R3 представляет собой группу -CH2CH3.
В другом более предпочтительном варианте реализации настоящего изобретения R3 представляет собой C1-C4 алкил, замещенный посредством группы - NR'R'', где R' и R'' независимо выбраны из Н или C1-C4 алкила. В другом еще более предпочтительном варианте реализации настоящего изобретения R3 представляет собой группу -CH2-CH2-N(CH3)2.
В другом предпочтительном варианте реализации настоящего изобретения R2 и R3 образуют замещенный или незамещенный насыщенный 5-членный гетероцикл.
В другом предпочтительном варианте реализации настоящего изобретения R2 и R3 образуют замещенный или незамещенный насыщенный 6-членный гетероцикл.
В другом более предпочтительном варианте реализации настоящего изобретения насыщенный гетероцикл по меньшей мере в одном из его положений замещен посредством C1-C4 алкила.
В другом более предпочтительном варианте реализации настоящего изобретения насыщенный 6-членный гетероцикл содержит вставку из дополнительного атома азота, незамещенного или замещенного посредством C1-C4 алкила.
В другом предпочтительном варианте реализации настоящего изобретения R3 представляет собой насыщенный 6-членный гетероцикл, содержащий вставку из дополнительного атома азота, незамещенного или замещенного посредством C1-C4 алкила.
В другом предпочтительном варианте реализации настоящего изобретения R4 представляет собой фтор.
В другом предпочтительном варианте реализации настоящего изобретения соединение формулы (I) выбирают из следующего списка:
- (4-(4-фторфенил)-6-метокси-2Н-хромен-3-ил)(пирролидин-1-ил)метанон,
- N-этил-4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоксамид,
- (4-(4-фторфенил)-6-метокс-2Н-хромен-3-ил)(4-метилпиперазин-1-ил) метанон,
- N-(2-(диметиламино)этил)-4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоксамид,
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(пирролидин-1-ил)метанон,
- 6-(циклопропилметокси)-N-этил-4-(4-фторфенил)-2Н-хромен-3-карбоксамид,
и
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(4-метилпиперазин-1-ил)метанон,
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(пирролидин-1-ил)метанон,
- 6-(циклопропилметокси)-N-этил-4-(4-фторфенил)-2Н-хромен-3-карбоксамид,
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(4-метилпиперазин-1-ил)метанон,
- 6-(циклопропилметокси)-N-(2-(диметиламино)этил)-4-(4-фторфенил)-2Н-хромен-3-карбоксамид
В другом предпочтительном варианте реализации настоящего изобретения X представляет собой -OH.
В другом предпочтительном варианте реализации настоящего изобретения соединение формулы (I) представляет собой 4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоновую кислоту.
Другой аспект настоящего изобретения относится к применению соединения формулы (I), как описано выше, для получения лекарственного средства.
Другой аспект настоящего изобретения относится к применению соединения формулы (I), как описано выше, для получения лекарственного средства для лечения заболеваний или расстройств, опосредованных взаимодействием TCR-Nck в Т-лимфоцитах.
На протяжении всего данного описания термины "лечение" заболевания, "лечить" заболевание или другие грамматически связанные выражения относятся к лечебной терапии, а также паллиативному лечению или профилактическому лечению такого заболевания.
В предпочтительном варианте реализации настоящего изобретения заболевание или расстройство опосредованное взаимодействием TCR-Nck в Т-лимфоцитах, выбирают из отторжения трансплантата, иммунных, аутоиммунных и воспалительных заболеваний, нейродегенеративных заболеваний, гематологических заболеваний и пролиферативных заболеваний.
В более предпочтительном варианте реализации настоящего изобретения заболевание или расстройство, опосредованные взаимодействием TCR-Nck в Т-лимфоцитах, выбирают из отторжения трансплантата, ревматоидного артрита, псориатического артрита, псориаза, диабета I типа, осложнений, связанных с диабетом, рассеянного склероза, системной красной волчанки, атопического дерматита, опосредованных тучными клетками аллергических реакций, лейкозов, лимфом и тромбоэмболических и аллергических осложнений, связанных с лейкозами и лимфомами.
Другой аспект настоящего изобретения относится к соединению формулы (I) для его применения для лечения заболеваний или расстройств, опосредованных взаимодействием TCR-Nck в Т-лимфоцитах.
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение формулы (I), как описано выше, и одно или более фармацевтически приемлемых вспомогательных веществ.
Соединения, описанные в настоящем изобретении, их фармацевтически приемлемые соли и/или сольваты, а также фармацевтические композиции, которые их содержат, можно применять вместе с другими дополнительными лекарственными средствами, чтобы обеспечить комбинированную терапию. Указанные дополнительные лекарственные средства могут быть частью той же самой фармацевтической композиции или, в качестве альтернативы, могут быть обеспечены в виде отдельной композиции для ее одновременного или неодновременного введения в фармацевтическую композицию, содержащую соединение формулы (I) или его изомер, сольват или фармацевтически приемлемую соль.
Если не указано иное, соединения согласно настоящему изобретению также включают соединения, которые отличаются только присутствием одного или более изотопно обогащенных атомов. Например, соединения, имеющие указанную структуру, за исключением замены водорода дейтерием или тритием или замены углерода 13С или 14С-обогащенным углеродом или 15N-обогащенным азотом, находятся в пределах объема настоящего изобретения.
Соединения формулы (I) для терапевтического применения получают в твердой форме или в форме водной суспензии в фармацевтически приемлемом разбавителе. Такие препараты можно вводить любым подходящим способом введения, для которого указанный препарат будет приготовлен фармацевтически пригодным для выбранного способа введения способом. В конкретном варианте реализации настоящего изобретения введение соединения формулы (I), полученного согласно настоящему изобретению, осуществляют пероральным, местным, ректальным или парентеральным (включая подкожный, внутрибрюшинный, внутрикожный, внутримышечный, внутривенный и т.д.) путем. Обзор различных фармацевтических форм введения лекарственных средств и вспомогательных веществ, необходимых для их получения, можно найти, например, в "Treaty of Galenic Pharmacy" С. Fauli i Trillo, 1993  , SA Ediciones, Madrid, или в других распространенных или аналогичных испанской, европейской или американской Фармакопеям.
, SA Ediciones, Madrid, или в других распространенных или аналогичных испанской, европейской или американской Фармакопеям.
Для применения в терапии соединения формулы (I), их изомеры, соли, или сольваты предпочтительно будут находиться в приемлемой или по существу чистой фармацевтической форме, т.е. иметь фармацевтически приемлемый уровень чистоты, исключая фармацевтические добавки, которые являются обычными, такие как разбавители и носители, и не включая материалы, которые считаются токсичными при уровнях обычной дозировки. Уровни чистоты для активного вещества составляют предпочтительно выше 50%, более предпочтительно выше 70%, более предпочтительно выше 90%. В предпочтительном варианте реализации настоящего изобретения они составляют выше 95% соединения формулы (I), или его изомеров, солей или сольватов.
Соединения согласно настоящему изобретению могут быть в кристаллической форме в виде свободных соединений или в виде сольватов, и подразумевается, что обе эти формы находятся в пределах объема настоящего изобретения. В настоящем документе термин "сольват" в контексте настоящего описания включает как фармацевтически приемлемые сольваты, т.е. сольваты соединения формулы (I), которые можно применять при производстве лекарственного средства, так и фармацевтически неприемлемые сольваты, которые могут быть полезны при получении солей или сольватов, которые являются фармацевтически приемлемыми. Природа фармацевтически приемлемого сольвата не имеет решающего значения при условии, что он фармацевтически приемлем. В конкретном варианте реализации настоящего изобретения сольват представляет собой гидрат. Сольваты могут быть получены обычными способами сольватации, хорошо известными специалистам в данной области техники.
Соединения согласно настоящему изобретению, представленные формулой (I), и, в частности, конкретные соединения, относящиеся к этой общей формуле, описанной выше, могут включать изомеры, в зависимости от наличия кратных связей (например, Z, Е), включая оптические изомеры или энантиомеры, в зависимости от наличия хиральных центров. Эти изомеры, энантиомеры или отдельные диастереоизомеры и их смеси входят в объем настоящего изобретения. Индивидуальные энантиомеры или диастереоизомеры, как и их смеси, могут быть разделены обычными способами.
Другим аспектом настоящего изобретения является способ лечения заболеваний или расстройств, опосредованных взаимодействием TCR-Nck в Т-клетках, причем указанный способ включает введение терапевтически эффективного количества соединения формулы (I) нуждающемуся в этом пациенту.
В контексте настоящего описания термин "терапевтически эффективное количество" относится к количеству активного вещества, достаточному для получения желаемого действия, при котором симптомы заболевания ослабляются. Доза не должна быть применена в количествах, которые вызывают нежелательные побочные эффекты, при которых клиническая оценка делает их неблагоприятными и терапевтически непригодными для лечения. Как правило, доза будет варьироваться в зависимости от возраста, состояния, пола и степени заболевания пациента, а также от способа и частоты введения, и будет определяться в каждом конкретном случае.
Другой аспект настоящего изобретения относится к способу получения соединения формулы (I), как описано выше, включающему следующие этапы:
а) обеспечение взаимодействия соединения формулы (II) с соединением формулы (III) и соединением формулы (IV)
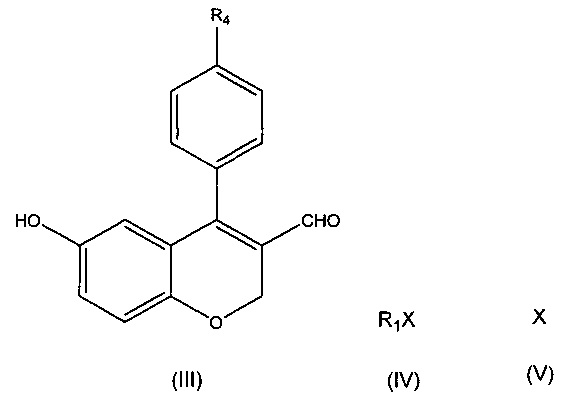
где R1, Х и R4 имеют значения, указанные в п. 1, и
b) обеспечение превращения соединения формулы (III) в соединение формулы (I) за один или более этапов.
В описании и формуле изобретения слово "включать" и его варианты не предназначены для исключения других технических характеристик, добавок, компонентов или этапов. Для специалистов в данной области другие задачи, преимущества и признаки настоящего изобретения станут очевидны частично из описания и практического применения настоящего изобретения. Следующие примеры и чертежи приведены в качестве иллюстрации и не предназначены для ограничения настоящего изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
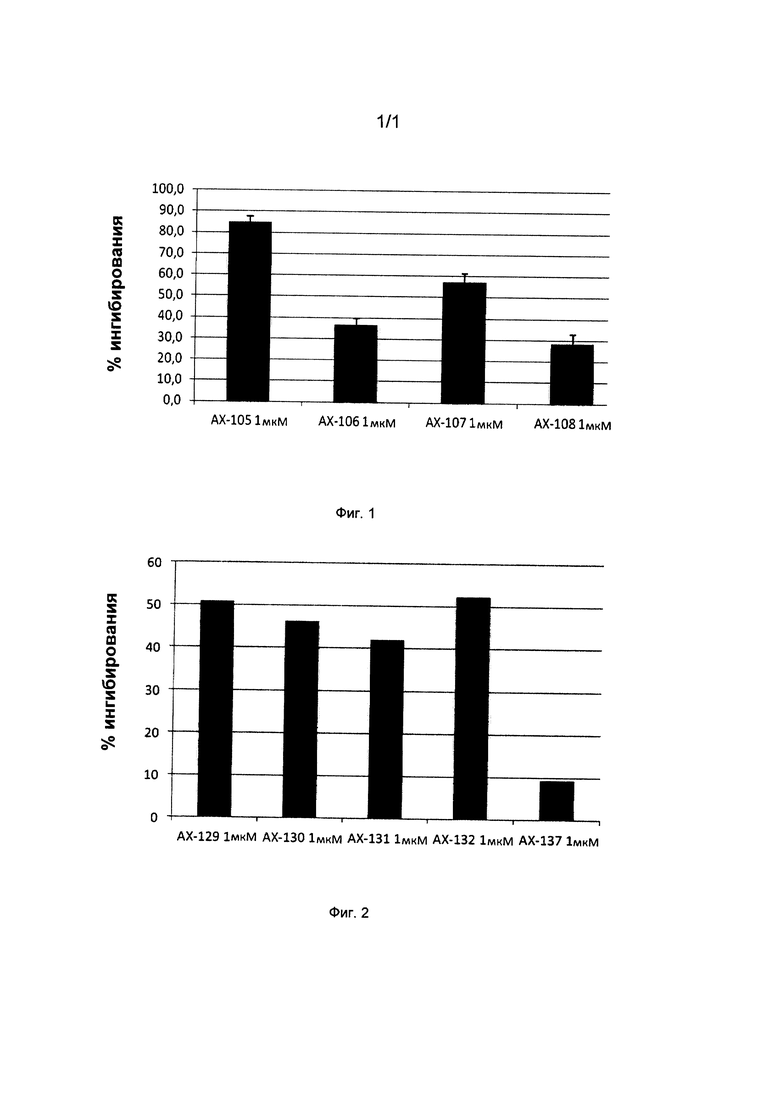
Фиг. 1. Показывает возможность ингибирования пролиферации Т-лимфоцитов для каждого из исследуемых соединений АХ-105, АХ-106, АХ-107 и АХ-108 согласно настоящему изобретению.
Фиг. 2. Показывает возможность ингибирования пролиферации Т-лимфоцитов для каждого из исследуемых соединений АХ-129, АХ-130, АХ-131, АХ-132 и АХ-137 согласно настоящему изобретению.
ПРИМЕРЫ
Настоящее изобретение будет проиллюстрировано с помощью испытаний, проведенных авторами настоящего изобретения, которые показывают эффективность соединений согласно настоящему изобретению.
Пример 1: Синтез соединений согласно настоящему изобретению.
Схема синтеза для АХ-105-АХ-108
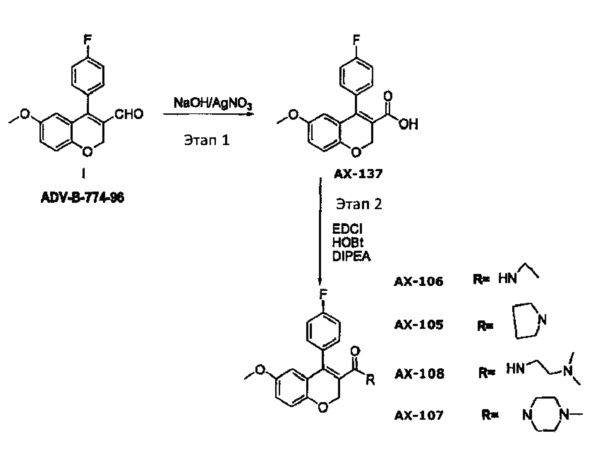
Синтез соединения АХ-137: Смесь Соединения 1 (3 г, 1 экв.), AgNO3 (3,5 г, 2 экв.) в этаноле (30 мл) и NaOH (1,7 г, 4 экв.), растворенную в воде (30 мл), кипятили с обратным холодильником при 85°C и перемешивали при этой температуре в течение 4 часов. Реакцию контролировали посредством ТСХ. После завершения смесь подкисляли 1 М HCl и экстрагировали с помощью дихлорметана (2×30 мл). Объединенный органический слой промывали водой (20 мл), солевым раствором (10 мл) и высушивали над безводным сульфатом натрия. Выпаривание органического слоя при пониженном давлении давало 1,5 г целевого продукта с чистотой 96,2%, определенной посредством ВЭЖХ.
1Н ЯМР (CDCl3) δ 7,17-7,08 (m, 4Н), 6,89-6,87 (d, 1Н), 6,84-6,81 (m, 1Н), 6,21-6,20 (d, 1Н), 4,96 (s, 2Н), 3,61 (s, 3H). Теоретические данные МС для C17H13FO4: 300,28; М++1 экспериментальные: 301,0.
Синтез соединения АХ-105: Смесь АХ-137 (0,2 г, 1 экв.), EDCI (0,14 г, 1,1 экв.), HOBt (0,09 г.1 экв.), пирролидина (0,06 г, 1,2 экв.) и диизопропилэтиламина (DIPEA) (0,17 г, 2 экв.) в ТГФ (5 мл) облучали микроволнами в течение 10 мин. По истечении этого времени ТГФ выпаривали и остаток промывали насыщенным раствором бикарбоната натрия и экстрагировали с помощью дихлорметана (DCM) (2×10 мл). Объединенный органический слой промывали водой (20 мл), солевым раствором (10 мл) и высушивали над безводным сульфатом натрия. Выпаривание органического слоя при пониженном давлении давало 135 мг целевого продукта с чистотой 98%, определенной посредством ВЭЖХ.
1Н ЯМР (CDCl3) δ 7,37-7,33 (m, 2Н), 7,10-7,06 (t, 2Н), 6,89-6.87 (d, 1Н), 6,78-6,75 (dd, 1Н), 6,46-6,45 (d, 1Н), 4,82 (s, 2Н), 3,66 (s, 3H), 3,31-3,27 (t, 2Н), 3,02 (b, 2Н), 1,68-1,63 (m, 2Н), 1,60-1,54 (m, 2Н). Теоретические данные МС для C21H20FNO3: 353,4; М++1 экспериментальные: 354,1.
Синтез соединения АХ-106: Смесь АХ-137 (0,2 г 1 экв.), EDCI (0,14 г, 1,1 экв.), HOBt (0,09 г, 1 экв.), этиламина HCI (0,06 г, 1,2 экв.) и диизопропилэтиламина (0,17 г, 2 экв.) в ТГФ (5 мл) облучали микроволнами в течение 10 мин. По истечении этого времени ТГФ выпаривали и остаток промывали насыщенным раствором бикарбоната натрия и экстрагировали с помощью дихлорметана (2×10 мл). Объединенный органический слой промывали водой (20 мл), солевым раствором (10 мл) и высушивали над безводным сульфатом натрия. Очистка сырого продукта с помощью колоночной хроматографии (9% метанола в дихлорметане) давала 100 мг целевого продукта с чистотой 99,3%, определенной посредством ВЭЖХ. 1Н ЯМР (CDCl3) δ 7,29-7,27 (m, 2Н), 7,20-7,15 (m, 2Н), 6,89-6,86 (d, 1Н), 6,79-6,76 (dd, 1Н), 6,24-6,23 (d, 1Н), 3,63 (s, 3H), 3,11-3,04 (q, 2Н), 0,78-0,74 (t, 3H). Теоретические данные МС для C19H18FNO3: 327,35; М++1 экспериментальные: 328,1.
Синтез соединения АХ-107: Смесь АХ-137 (0,1 г, 1 экв.), EDCI (0,07 г, 1,1 экв), HOBt (0,05 г, 1 экв.), N-метилпиперазина (0,04 г, 1,2 экв.) и диизопропилэтиламина (0,09 г, 2 экв.) в ТГФ (5 мл) облучали микроволнами в течение 10 мин. По истечении этого времени ТГФ выпаривали и остаток промывали насыщенным раствором бикарбоната натрия и экстрагировали с помощью дихлорметана (2×10 мл). Объединенный органический слой промывали водой (20 мл), солевым раствором (10 мл) и высушивали над безводным сульфатом натрия. Выпаривание органического слоя при пониженном давлении давало 68 мг целевого продукта с чистотой 94,9%, определенной посредством ВЭЖХ.
1Н ЯМР (CDCl3) δ 7,31 (b, 2Н), 7,12-7,08 (t, 2Н), 6,89-6,87 (d, 1Н), 6,79-6,76 (dd, 1Н), 6,44 (m, 1Н), 4,95-4,91 (d, 1Н), 4,68-4,64 (d, 1Н), 3,66 (s, 3H), 3,5-3,7 (m, 3H), 3,04 (b, 1Н), 2,3-2,27 (b, 1H), 2,13 (b, 4H), 2,03 (b, 1H), 1,49 (b, 1H). Теоретические данные МС для C22H23FN2O3: 382,4; М1 экспериментальные: 383,0.
Синтез соединения АХ-108: Смесь АХ-137 (0,2 г, 1 экв), EDCI (0,14 г, 1,1 экв), HOBt (0,09 г, 1 экв), N,N-диметилэтилендиамина (0,06 г, 1,2 экв) и диизопропилэтиламина (0,17 г, 132 экв) в ТГФ (5 мл) облучали микроволнами в течение 10 мин. По истечении этого времени ТГФ выпаривали и остаток промывали насыщенным раствором бикарбоната натрия и экстрагировали с помощью дихлорметана (2×10 мл). Объединенный органический слой промывали водой (20 мл), солевым раствором (10 мл) и высушивали над безводным сульфатом натрия. Очистка сырого продукта с помощью колоночной хроматографии (6% метанола в дихлорметане) получали 80 мг целевого продукта с чистотой 97,7%, определенной посредством ВЭЖХ.
1Н ЯМР (CDCl3) δ 7,28-7,25 (m, 2Н), 7,17-7,13 (t, 2Н), 6,88-6,86 (d, 1Н), 6,78-6,75 (dd, 1Н), 6,22-6,21 (d, 1Н), 5,80 (b, 1Н), 3,63 (s, 3H), 3,13-3,09 (q, 2Н), 2,08-2,05 (t, 2Н), 1,97 (s, 6Н). Теоретические данные МС для C21H23ClFN2O3: 370,42; М++1 экспериментальные: 371,0.
Схема синтеза для АХ-129-АХ-132
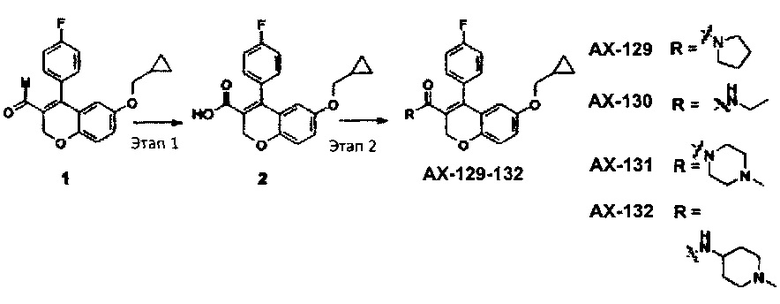
Раствор NaClO2 (1,07 г, 0,003 моль) и NaH2PO4 (1,63 г, 0,01 моль) в воде (2,5 мл) добавляли к раствору соединения 1 (1,2 г, 0,003 моль) и 2-метил-2-бутена (3,92 мл, 0,037 моль) в трет-BuOH (25 мл) при комнатной температуре и перемешивали при той же температуре до тех пор, пока исходные вещества не были израсходованы. После примерно 30 мин трет-BuOH выпаривали и полученный раствор подкисляли с помощью 2N HCl (pH 3-4). Полученное твердое вещество отфильтровывали и высушивали под вакуумом с получением кислоты 2 в виде бледно-желтого твердого вещества (1,1 г, выход 88%), которую брали в дальнейшем для получения амидов АХ-129-АХ-132 без дополнительной очистки.
1Н ЯМР (CDCl3, 400 МГц) δ 7,23 (d, J=6,8 Гц, 1Н), 7,13-7,07 (m, 3H), 6,88-6,80 (m, 2Н), 6,23 (s, 1Н), 4,94 (s, 2Н), 3,58 (d, J=6,8 Гц, 2Н), 1,13 (m, 1Н), 0,58 (d, J=7,6 Гц, 2Н), 0,25 (d, J=4,4 Гц, 2Н). ES-MC [М-1]+: 339,1.
Синтез соединения АХ-129:
EDCI (164 мг, 1,85 ммоль) и HOBt (85 мг, 0,63 ммоль) добавляли к раствору кислоты 2 (194 мг, 0,57 ммоль), пирролидина (32 мг, 0.45 ммоль) и диизопропилэтиламина (220 мг, 1,71 ммоль) в ТГФ (10 мл) и всю смесь облучали микроволновым излучением (900 Вт) в течение 4 мин. ТГФ концентрировали до минимального объема; реакционную массу разбавляли ледяной водой (20 мл) и экстрагировали этилацетатом (3×25 мл). Объединенный органический экстракт высушивали над Na2SO4 и концентрировали на роторном испарителе с получением неочищенного остатка, который очищали с помощью FCC (SiO2, смеси гексан-EtOAc) с получением амида АХ-129 (100 мг, выход 45%) в виде не совсем белого твердого вещества.
1Н ЯМР (ДМСО-d6) δ 7,30-7,28 (m, 4Н), 6,88-6,80 (m, 1Н), 6,29 (s, 1Н), 4,77 (s, 2Н), 3,63 (d, J=6,8 Гц, 2H), 3,11 (m, 2Н), 3,03 (s, 2Н), 1,60-1,51 (m, 4Н), 1,10 (m, 1H), 0,49 (d, J=8,0 Гц, 2H), 0,23 (d, J=4,4 Гц, 2H). ES-MC [M+1]+: 391,4.
Синтез соединения АХ-130:
АХ-130: АХ-130 (130 мг, выход 76%) получали в виде бледно-желтого клейкого твердого вещества, начиная с кислоты 2 (300 мг, 0,882 ммоль) и замещая пирролидин в описанной выше методике этиламином.
1Н ЯМР (ДMCO-d6, 400 МГц): δ 7,63 (t, J=4,8 Гц, 1Н), 7,27-7,25 (m, 4Н), 6,87-6,79 (m, 2Н), 6,17 (s, 2Н), 4,78 (s, 1Н), 3,61 (d, J=5,1 Гц, 2Н), 2,9 (m, 2Н), 1,08 (m, 1Н), 0,69 (t, J=7,2 Гц, 3H), 0,48 (d, J=8,0 Гц, 2Н), 0,22 (d, J=4,4 Гц, 2Н). ES-MC [М+1]+: 368,1.
Синтез соединения АХ-131:
АХ-131: АХ-131 (120 мг, 23% выход) получали в виде бледно-желтого клейкого твердого вещества, начиная с кислоты 2 (200 МГ, 0,588 ммоль) и замещая пирролидин в описанной выше методике N-метилпиперазином. 1Н ЯМР (ДMCO-d6, 400 МГц): δ 7,32 (m, 4Н), 6,91-6,84 (m, 2Н), 6,27 (s, 1Н), 4,81 (s, 2Н), 4,30 (m, 1Н), 3,89 (m, 1Н), 3,63 (d, J=6,8 Гц, 2Н), 3,18 (m, 1Н), 2,75-2,56 (m, 4Н), 1,09 (m, 1Н), 0,49 (d, J=8,0 Гц, 2Н), 0,23 (d, J=4,4 Гц, 2Н). ES-MC [М+1]+: 423,1.
Синтез соединения АХ-132:
TBTU (514 мг, 1,6 ммоль) добавляли к раствору кислоты 2 (218 мг, 0,64 ммоль) в смеси CH2Cl2 (12 мл), ДМФ (2,5 мл) и ДИЭА (289 мг, 2,244 ммоль) при N2; перемешивали при комнатной температуре. Через 1 час добавляли 4-амино-1-метилпиперидин (221 мг, 1,923 ммоль) и всю реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную массу разбавляли CH2Cl2 (70 мл) и промывали водой (2×25 мл). Органический слой высушивали над Na2SO4 и концентрировали на роторном испарителе с получением неочищенного остатка, который очищали с помощью FCC (SiO2: МеОН-CH2Сl2 смеси) с получением амида АХ-132 (100 мг, выход 39%) в виде бледно-желтого масла.
1Н ЯМР (ДMCO-d6, 400 МГц): δ 7,58 (m, 1Н), 7,28-7,26 (m, 4Н), 6,16 (s, 1Н), 4,78 (s, 2Н), 3,61 (d, J=6,4 Гц, 2Н), 3,45 (m, 1Н), 2,62 (m, 4Н), 2,21-2,00 (m, 6Н), 1,43 (m, 2Н), 1,40-1,00 (m, 6Н), 1,08 (m, 1Н), 0,48 (d, J=7,6 Гц, 2Н), 0,21 (m, 2Н). ES-MC [М+1]+: 437,3.
Синтез соединения АХ-137:
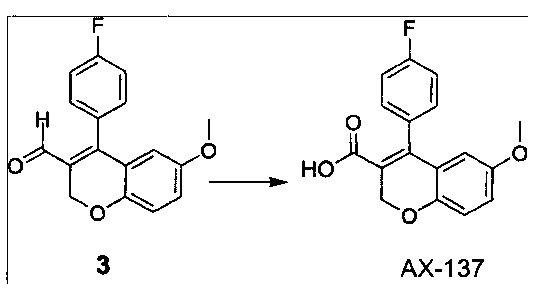
АХ-137: раствор NaOH (1,7 г, 0,04 моль), растворенный в воде (30 мл), добавляли к перемешанному раствору альдегида 3 (3,0 г, 0,01 моль), AgNO3 (3,5 г, 0,02 моль) в этаноле (30 мл) и реакционную массу кипятили с обратным холодильником при температуре 85°C в течение 4 часов. Реакционную среду подкисляли с помощью 1 М HCl (pH=2-3) и экстрагировали с помощью CH2Cl2 (2×70 мл). Объединенный органический экстракт промывали солевым раствором (50 мл) с последующим добавлением воды (100 мл); высушивали над безводным Na2SO4 и концентрировали с получением АХ-137 (1,5 г, выход 47%).
1Н ЯМР (CDCl3, 400 МГц) δ 7,17-7,08 (m, 4Н), 6,89-6,87 (d, J=8,8 Гц, 1Н), 6,83 (dd, J=8,8, 3,2 Гц, 1Н), 6,21 (d, J=3,2 Гц, 1Н), 4,96 (s, 2Н), 3,61 (s, 3H). ES-MC [М+1]+: 301,0.
Пример 2: Ингибирование пролиферации Т-клеток, индуцированной стимуляцией TCR.
Влияние соединений АХ-105, АХ-106, АХ-107 и АХ-108 на способность TCR индуцировать пролиферацию Т-лимфоцитов оценивали для первичных Т-лимфоцитов, полученных из крови здоровых доноров-людей (МКПК, мононуклеарные клетки периферической крови). МКПК добровольцев выделяли центрифугированием венозной крови в градиенте плотности фиколл-пак плюс. Очищенные клетки (НВТ, Т-клетки, разделенные на нейлоновой вате) культивировали в трех повторах в 96-луночных планшетах (0,5×105/лунка) в 200 мкл полной среды и стимулировали с помощью ОКТЗ (10 мкг/мл) или ОКТЗ (1 мкг/мл) плюс CD28 в присутствии или в отсутствие различных соединений в концентрациях от 1 до 10 мкМ. Культуры инкубировали в течение трех дней и анализировали после добавления 0,5 мкКи [3Н] тимидина/лунка в течение последних 12 часов культивирования. Включенную в ДНК радиоактивность определяли с помощью жидкостного сцинтилляционного счетчика. При делении клеток радиоактивными становились дочерние клетки, что позволяло иметь представление о степени пролиферации клеток. Ингибирующая способность испытуемых соединений показана на фигуре 1.
Кроме того, действие соединений АХ-129, АХ-130, АХ-131, АХ-132 и АХ-137 также анализировали в мононуклеарных клетках периферической крови (МКПК) от здоровых добровольцев; при этом клетки выделяли центрифугированием венозной крови в градиенте плотности фиколл-пак плюс. Для очистки Т-клетки МКПК пропускали через колонку с нейлоновой ватой. Для анализа клеточного деления очищенные Т-клетки окрашивали карбоксифлуоресцеин-сукцинимидил эфиром (КФСЭ). Меченые клетки (0,5×105/лунка) культивировали в трех повторах в 96-луночных планшетах в 200 мкл полной среды и стимулировали с помощью иммобилизованного ОКТЗ (1 мкг/мл) в присутствии или в отсутствие испытуемых соединений, указанных ранее, в течение 6 дней, и процент пролиферирующих клеток (определяемых как низкая флуоресценция КФСЭ) определяли с помощью проточной цитометрии. Ингибирующая способность исследуемых соединений показана на фигуре 2.
Эти анализы позволяют давать оценку и подтверждение способностям соединений ингибировать пролиферацию Т клеток и, следовательно, способствуют проверке таких соединений в качестве кандидатов для разработки новых терапевтических способов лечения аутоиммунных заболеваний, опосредованных Т-клетками.
Пример 3: Испытание in vivo на модели диабета 1 типа.
Развитие диабета контролировали ежедневно и регистрировали положительно, если уровни глюкозурии обнаруживали выше 250 мг/дл в результате двух последовательных измерений ежедневно для модели RIP-mOVA или каждые две недели для модели мышей с диабетом без ожирения. Влияние лечения на уровень заболеваемости и выживаемость оценивали для модели RIP-mOVA, как это было для инсулита. Гистологическую оценку инсулита секций Н&Е, пропитанных парафином и фиксированных в формалине, давали с помощью слепого испытания, используя следующую систему классификации: этап 0, нет инвазии; этап 1, периинсулит; этап 2, инвазия >25%; этап 3, инвазия >75%; и этап 4, оставшиеся островки. На втором этапе терапевтическое действие соединений оценивали путем лечения мышей RIP-mOVA, когда у них уже развился диабет. В ходе этой второй фазы соединения испытывали на модели мышей с диабетом без ожирения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХРОМЕНА, ЗАМЕЩЁННЫЕ ПОСРЕДСТВОМ АЛКОКСИДА, В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ TCR-NCK | 2014 |
|
RU2658910C2 |
| ПРОИЗВОДНЫЕ ХРОМЕНА | 2011 |
|
RU2574164C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| ХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА, ИНГИБИРУЮЩЕГО ТРАНСПОРТЕР УРАТОВ | 2016 |
|
RU2715229C2 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
| 5-АРОМАТИЧЕСКОЕ АЛКИНИЛЗАМЕЩЕННОЕ БЕНЗАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2695371C2 |
| МОНОМЕРНЫЕ ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДНОГО АНТИБИОТИКА | 2005 |
|
RU2424248C2 |
| ЗАМЕЩЕННЫЕ 2-МЕТИЛИДЕН-5-(ФЕНИЛАМИНО)-2,3-ДИГИДРОТИОФЕН-3-ОНЫ ДЛЯ ЛЕЧЕНИЯ ЛЕЙКОЗОВ С ТРАНСЛОКАЦИЯМИ MLL-ГЕНА И ДРУГИХ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2656603C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| СОЕДИНЕНИЯ-ИНГИБИТОРЫ EGFR | 2017 |
|
RU2751341C2 |
Изобретение относится к группе соединений формулы (I), содержащих хроменовое ядро, или их фармацевтически приемлемым солям, где: R1 представляет собой незамещенный C1-C6 алкил или C1-C6 алкил, замещенный посредством С3-C6 циклоалкила; X выбран из -ОН или -N(R2)(R3); R2 и R3 независимо выбраны из водорода, незамещенного C1-C6 алкила, C1-C6 алкила, замещенного посредством -N(R')(R''), причем R' и R'' независимо выбраны из Н или С1-С4 алкила, или насыщенного 6-членного гетероцикла, который содержит атом N, незамещенный или замещенный посредством С1-С4 алкила; или R2 и R3 вместе с атомом азота, с которым они связаны, образуют 5- или 6-членный насыщенный гетероцикл, где указанный гетероцикл содержит по меньшей мере один гетероатом, выбранный из азота, кислорода или серы, и где гетероцикл необязательно замещен посредством С1-С4 алкила; R4 представляет собой галоген, а также к способу их получения. Эти соединения обладают способностью ингибировать пролиферацию лимфоцитов, опосредованную взаимодействием TCR с Nсk, в связи с чем настоящее изобретение также относится к фармацевтическим композициям на их основе и применению этих соединений для лечения заболеваний или состояний, при которых указанное взаимодействие вызывает осложнения, такие как реакции отторжения трансплантата, иммунные или аутоиммунные заболевания, воспалительные заболевания или пролиферативные заболевания. 6 н. и 30 з.п. ф-лы, 2 ил., 3 пр.
1. Соединение формулы (I)
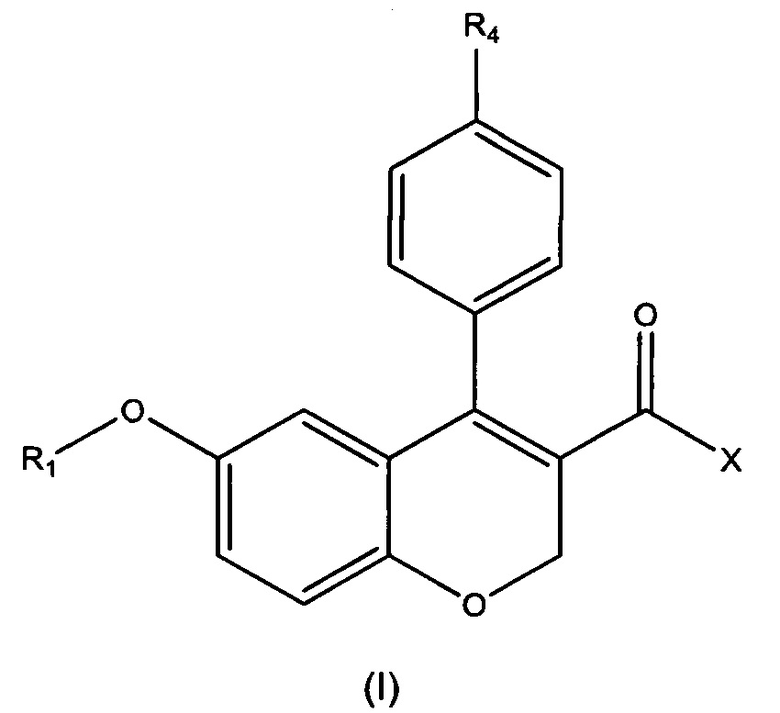
или его фармацевтически приемлемая соль, где:
R1 представляет собой незамещенный C1-C6 алкил или C1-C6 алкил, замещенный посредством С3-C6 циклоалкила;
X выбран из -ОН или -N(R2)(R3);
R2 и R3 независимо выбраны из водорода, незамещенного C1-C6 алкила, C1-C6 алкила, замещенного посредством -N(R')(R''), причем R' и R'' независимо выбраны из Н или С1-С4 алкила, или насыщенного 6-членного гетероцикла, который содержит атом N, незамещенный или замещенный посредством С1-С4 алкила;
или R2 и R3 вместе с атомом азота, с которым они связаны, образуют 5- или 6-членный насыщенный гетероцикл, где указанный гетероцикл содержит по меньшей мере один гетероатом, выбранный из азота, кислорода или серы, и где гетероцикл необязательно замещен посредством С1-С4 алкила;
R4 представляет собой галоген.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающиеся тем, что R1 представляет собой незамещенный С1-С6 алкил.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, отличающиеся тем, что R1 представляет собой -СН3.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающиеся тем, что R1 представляет собой C1-C6 алкил, замещенный посредством С3-С6 циклоалкила.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, отличающиеся тем, что R1 представляет собой -СН2-циклопропил.
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, отличающиеся тем, что X представляет собой -N(R2)(R3).
7. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 представляет собой Н.
8. Соединение по п. 7 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой незамещенный C1-С6 алкил.
9. Соединение по п. 8 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой -СН2-СН3.
10. Соединение по п. 7 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой C1-С6 алкил, замещенный -N(R')(R''), причем R' и R'' независимо выбраны из Н или С1-С4 алкила.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой -СН2-СН2-N(СН3)2.
12. Соединение по п. 7 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой насыщенный 6-членный гетероцикл, который содержит атом N, незамещенный или замещенный посредством С1-С4 алкила.
13. Соединение по п. 12 или его фармацевтически приемлемая соль, отличающиеся тем, что R3 представляет собой 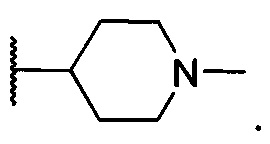
14. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 и R3 вместе с атомом азота, с которым они связаны, образуют незамещенный насыщенный 5-членный гетероцикл.
15. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 и R3 вместе с атомом азота, с которым они связаны, образуют незамещенный насыщенный 6-членный гетероцикл.
16. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 и R3 вместе с атомом азота, с которым они связаны, образуют насыщенный 5-членный гетероцикл, замещенный по меньшей мере в одном из его положений посредством С1-С4 алкила.
17. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 и R3 вместе с атомом азота, с которым они связаны, образуют насыщенный 6-членный гетероцикл, замещенный по меньшей мере в одном из его положений посредством С1-С4 алкила.
18. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R2 и R3 вместе с атомом азота, с которым они связаны, образуют насыщенный 6-членный гетероцикл, содержащий вставку из дополнительного атома азота, незамещенного или замещенного посредством С1-С4 алкила.
19. Соединение по п. 14 или его фармацевтически приемлемая соль, отличающиеся тем, что -N(R2)(R3) представляет собой 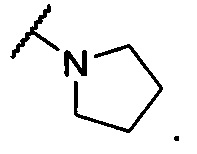
20. Соединение по п. 17 или п. 18 или его фармацевтически приемлемая соль, отличающиеся тем, что -N(R2)(R3) представляет собой 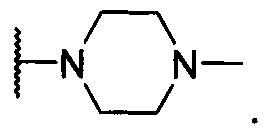
21. Соединение по любому из пп. 1-5 и 7-19 или его фармацевтически приемлемая соль, отличающиеся тем, что R4 представляет собой фтор.
22. Соединение по п. 6 или его фармацевтически приемлемая соль, отличающиеся тем, что R4 представляет собой фтор.
23. Соединение по п. 20 или его фармацевтически приемлемая соль, отличающиеся тем, что R4 представляет собой фтор.
24. Соединение по п. 1 или его фармацевтически приемлемая соль, выбранные из следующего списка:
- (4-(4-фторфенил)-6-метокси-2Н-хромен-3-ил)(пирролидин-1-ил)метанон,
- N-этил-4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоксамид,
- (4-(4-фторфенил)-6-метокси-2Н-хромен-3-ил)(4-метилпиперазин-1-ил)метанон,
- N-(2-(диметиламино)этил)-4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоксамид,
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(пирролидин-1-ил)метанон,
- 6-(циклопропилметокси)-N-этил-4-(4-фторфенил)-2Н-хромен-3-карбоксамид,
- (6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-ил)(4-метилпиперазин-1-ил)метанон,
- 6-(циклопропилметокси)-N-(2-(диметиламино)этил)-4-(4-фторфенил)-2Н-хромен-3-карбоксамид.
25. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающиеся тем, что X представляет собой -ОН.
26. Соединение по п. 25 или его фармацевтически приемлемая соль, которое представляет собой 4-(4-фторфенил)-6-метокси-2Н-хромен-3-карбоновую кислоту или 6-(циклопропилметокси)-4-(4-фторфенил)-2Н-хромен-3-карбоновую кислоту.
27. Применение соединения формулы (I) по любому из пп. 1-26 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболевания или расстройства, выбранного из отторжения трансплантата, иммунных, аутоиммунных и воспалительных заболеваний, нейродегенеративных заболеваний, гематологических заболеваний и пролиферативных заболеваний.
28. Применение соединения формулы (I) по любому из пп. 1-26 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболевания или расстройства, опосредованного взаимодействием TCR-Nck в Т-лимфоцитах.
29. Применение по п. 28, отличающееся тем, что заболевание или расстройство, опосредованное взаимодействием TCR-Nck в Т-лимфоцитах, выбирают из отторжения трансплантата, иммунных, аутоиммунных и воспалительных заболеваний, нейродегенеративных заболеваний, гематологических заболеваний и пролиферативных заболеваний.
30. Применение по любому из пп. 27-29, отличающееся тем, что заболевание или расстройство, опосредованное взаимодействием TCR-Nck в Т-лимфоцитах, выбирают из отторжения трансплантата, ревматоидного артрита, псориатического артрита, псориаза, диабета I типа, осложнений, связанных с диабетом, рассеянного склероза, системной красной волчанки, атопического дерматита, опосредованных тучными клетками аллергических реакций, лейкозов, лимфом и тромбоэмболических и аллергических осложнений, связанных с лейкозами и лимфомами.
31. Фармацевтическая композиция для лечения заболевания или расстройства, опосредованного взаимодействием TCR-Nck в Т-лимфоцитах, содержащая соединение формулы (I) по любому из пп. 1-26 или его фармацевтически приемлемую соль и одно или более фармацевтически приемлемых вспомогательных веществ.
32. Фармацевтическая композиция по п. 31, отличающаяся тем, что заболевание или расстройство, опосредованное взаимодействием TCR-Nck в Т-лимфоцитах, выбрано из отторжения трансплантата, иммунных, аутоиммунных и воспалительных заболеваний, нейродегенеративных заболеваний, гематологических заболеваний и пролиферативных заболеваний.
33. Фармацевтическая композиция для лечения заболевания или расстройства, выбранного из отторжения трансплантата, иммунных, аутоиммунных и воспалительных заболеваний, нейродегенеративных заболеваний, гематологических заболеваний и пролиферативных заболеваний, содержащая соединение формулы (I) по любому из пп. 1-26 или его фармацевтически приемлемую соль и одно или более фармацевтически приемлемых вспомогательных веществ.
34. Фармацевтическая композиция по любому из пп. 31-33, отличающаяся тем, что заболевание или расстройство выбрано из отторжения трансплантата, ревматоидного артрита, псориатического артрита, псориаза, диабета I типа, осложнений, связанных с диабетом, рассеянного склероза, системной красной волчанки, атопического дерматита, опосредованных тучными клетками аллергических реакций, лейкозов, лимфом и тромбоэмболических и аллергических осложнений, связанных с лейкозами и лимфомами.
35. Способ получения соединения формулы (I) по любому из пп. 1-26 или его фармацевтически приемлемой соли, включающий следующий этап:
а) превращение альдегидной группы соединения формулы (III) или его соли в карбоксильную группу
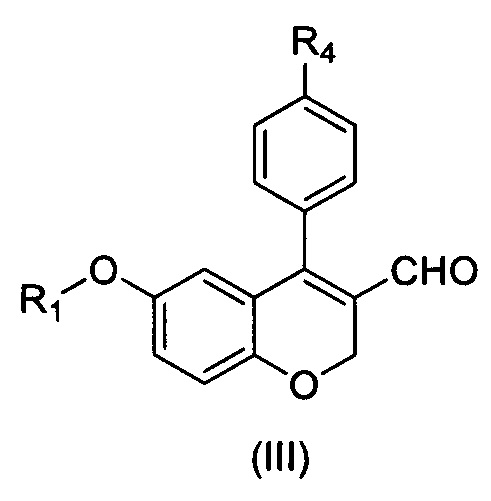 ,
,
где R1 и R4 имеют значения, указанные в п. 1.
36. Способ по п. 35, дополнительно включающий взаимодействие полученного на этапе а) соединения, содержащего карбоксильную группу, или его соли с соединением формулы (IV) или его солью:
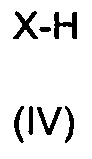 ,
,
где X представляет собой -N(R2)(R3), как определено в п. 1.
| БЛОК ДЛЯ УСТРОЙСТВА ЗАЩИТЫ ОТ ПЕРЕНАПРЯЖЕНИЙ И СООТВЕТСТВУЮЩЕЕ УСТРОЙСТВО ЗАЩИТЫ ОТ ПЕРЕНАПРЯЖЕНИЙ | 2012 |
|
RU2623503C2 |
| WO 2010009069 A1, 21/01.2010 | |||
| ПРОИЗВОДНЫЕ ХРОМАНА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ. | 1998 |
|
RU2223269C2 |
| ПРОИЗВОДНЫЕ ХРОМАНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2233277C2 |

