Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к способу получения простого фторметил-1,1,1,3,3,3-гексафторизопропилового эфира (севофлурана), который широко используется в качестве лекарственных средств и, в частности, ингаляционных анестетиков.
Уровень техники
[0002]
Простой фторметил-1,1,1,3,3,3-гексафторизопропиловый эфир (севофлуран) широко используется в качестве безопасного для использования ингаляционного анестетика. Как описано в патенте США № 4250334 (патентная литература 1), севофлуран может синтезироваться посредством добавления концентрированной серной кислоты и фтористого водорода к параформальдегиду, нагрева полученной реакционной смеси и добавления 1,1,1,3,3,3-гексафторизопропилового спирта (HFIP) по каплям к смеси. Представляющее интерес вещество (то есть севофлуран) можно собирать вместе с непрореагировавшим веществом (например, HFIP) посредством сбора газа, генерируемого в реакционной системе.
[0003]
В рассмотренной выше реакции синтеза севофлурана генерируются различные побочные продукты. Среди них, побочный продукт, который является сложным для отделения, представляет собой простой бис(фторметиловый) эфир. Однако сообщается, что простой бис(фторметиловый) эфир можно эффективно удалять посредством приведения реакционной смеси для севофлурана в контакт с "кислотой Бренстеда, такой как концентрированная серная кислота, с кислотой Льюиса или с кислотой, иммобилизованной на смоле или на чем-либо подобном" (патентная литература 2: патент Японии № 2786106). Альтернативно, простой бис(фторметиловый) эфир можно эффективно удалять посредством приведения его в контакт с цеолитом (патентная литература 3: патент Японии № 3240043).
[0004]
В дополнение к этому известно, что непрореагировавший HFIP можно эффективно удалять посредством приведения органического слоя, содержащего севофлуран, в "контакт с основным водным раствором гидроксида натрия или чем-либо подобным" (патентная литература 4: патент Японии № 4087488).
[0005]
Другой побочный продукт, содержащийся в севофлуране, представляет собой простой фторметил-1,1,3,3,3-пентафторизопропениловый эфир (как правило, упоминаемый как "соединение A"). Это соединение рассматривается как соединение, которое генерируется, когда севофлуран подвергается воздействию реакции дегидрофторирования. Когда севофлуран приводится в контакт с сильным основанием, таким как гидроксид натрия, может генерироваться малое количество этого соединения. В дополнение к этому он постепенно генерируется во время дистилляции и очистки севофлурана (смотри следующую формулу).
[0006]
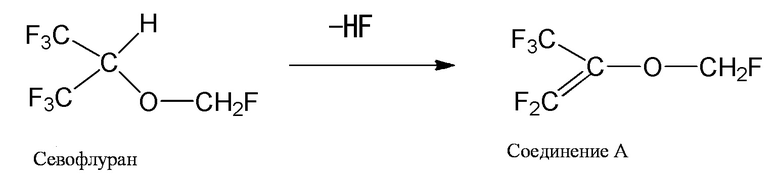
[0007]
Соединение A само по себе представляет собой стабильное соединение, так что оно не деградирует легко даже при нагреве. В дополнение к этому оно демонстрирует псевдоазеотропное поведение вместе с севофлураном. По этой причине после того как соединение A генерируется во время очистки севофлурана, отделение соединения A от севофлурана (очистка севофлурана) часто становится сложным.
[0008]
В качестве технологии для решения этой проблемы, патент Японии № 2786108 (патентная литература 5) описывает попытки, предпринимаемые для осуществления "дистилляции и очистки в присутствии ингибитора деградации, такого как гидрофосфат натрия". Показано, что реакция для деградирования севофлурана до соединения A во время дистилляции может существенно ингибироваться с помощью такой технологии, и малое количество соединения A концентрируется в основном в первой фракции дистилляции, так что севофлуран, полученный в качестве главной фракции дистилляции, по существу не содержит соединения A. В результате, севофлуран, содержащий соединение A, на уровне, не превышающем предела детектирования, успешно получается в качестве главной фракции дистилляции.
Список цитирований
Патентная литература
[0009]
Патентная литература 1: патент США № 4250334
Патентная литература 2: патент Японии № 2786106
Патентная литература 3: патент Японии № 3240043
Патентная литература 4: патент Японии № 4087488
Патентная литература 5: патент Японии № 2786108
Сущность изобретения
Техническая проблема
[0010]
Способ из Патентной литературы 5 представляет собой превосходный способ, с помощью которого ингибируется генерирование соединения A в качестве побочного продукта при дистилляции и очистке севофлурана, так что главная фракция дистилляции (фракция севофлурана), полученная посредством дистилляции, не содержит соединения A. Другими словами, как описано в Примерах 2-№1, №2 из Патентной литературы 5, когда сырой севофлуран, содержащий соединение A при концентрации 10-30 м.д., дистиллируется с добавлением двухосновного фосфата натрия в качестве ингибитора деградации, севофлуран, по существу не содержащий соединения A (это означает, что соединение A содержится при концентрации меньше чем 1 м.д.; это же применимо и в дальнейшем), может быть получен в качестве главной фракции дистилляции, при этом соединение A концентрируется в основном в первой фракции дистилляции.
[0011]
Однако в рассмотренном выше способе недостатком является то, что имеется большая вероятность потерь севофлурана. Другими словами, как указано выше, соединение A демонстрирует псевдоазеотропное поведение вместе с севофлураном. По этой причине, когда "севофлуран, содержащий соединение A", дистиллируется в соответствии с Патентной литературой 5, является неизбежным то, что часть севофлурана дистиллируется в первой фракции дистилляции вместе с соединением A. Более конкретно, в соответствии с Примером 2-№1, №2 из Патентной литературы 5, генерирование соединения A во время дистилляции может значительно ингибироваться. При этом выход дистилляции (доля извлечения севофлурана в качестве главной фракции дистилляции) остается на уровне 71%-72%. С другой стороны, количество "первой фракции дистилляции" достигает 10% от извлекаемого количества "главной фракции дистилляции". Содержание севофлурана в первой фракции дистилляции фактически достигает 99,6%-99,8%. В результате, "первая фракция дистилляции" в основном состоит из севофлурана, что приводит в результате к "потерям относительно большого количества севофлурана для отделения малого количества соединения A".
[0012]
При рассмотренных выше обстоятельствах, авторы настоящего изобретения предприняли попытки подвергнуть снова "первую фракцию дистилляции", полученную с помощью рассмотренного выше способа непосредственно прецизионной дистилляции с тем, чтобы выделить севофлуран. В результате, однако, когда дистилляция осуществляется при таком же количестве теоретических ступеней и при таком же коэффициенте дефлегмации, соединение A постоянно детектируется от начала и почти до конца дистилляции (другими словами, площадь пика не уменьшается до уровня меньше чем 1 м.д. при газохроматографическом анализе с FID (с пламенно-ионизационным детектором)) (смотри "Сравнительный пример 1"). При этом посредством осуществления снова дистилляции с увеличенным количеством теоретических ступеней по сравнению с количеством, используемым для сбора первой фракции дистилляции, может быть извлечена первая фракция дистилляции, в которой соединение A дополнительно концентрируется, делая возможным получение малого количества севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции. Однако такая технология требует использования множества дистилляционных колонн при различных условиях, и всего-навсего вызывает усложнение работы. Таким образом, такой способ не может рассматриваться в качестве способа эффективного получения севофлурана высокой чистоты.
[0013]
При этом соединение A, которое представляет собой вещество, которое вызывает понижение эффективности дистилляции, имеет структуру, до которой севофлуран дегидрофторируется. По этой причине, считается, что, если HF получает возможность для воздействия на соединение A, соединение A может преобразовываться в севофлуран при добавлении HF (смотри формулы, следующие далее).
[0014]
Соединение A+HF (безводный) → севофлуран
Соединение A+HF (безводный)+концентрированная серная кислота → севофлуран
[0015]
Однако даже если безводный HF получает возможность для воздействия на соединение A, рассмотренная выше реакция добавления не происходит. Даже в системе, в которую добавляют концентрированную серную кислоту в качестве ускорителя реакции, реакция не происходит так, как ожидается.
[0016]
Как описано выше, имеется потребность в новом способе, с помощью которого соединение A можно удалять из "севофлурана, содержащего соединение, A" таким образом, что можно извлекать севофлуран высокой чистоты.
Решение проблемы
[0017]
В результате интенсивных исследований для достижения указанной выше цели, авторы настоящего изобретения обнаружили, что посредством приведения соединения A в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30", можно дать возможность соединению A для постепенного взаимодействия, с тем, чтобы оно преобразовывалось в виды химических частиц (в настоящем документе упоминаются как "соединение X"), имеющие неустановленную структуру, которые могут отделяться от севофлурана посредством дистилляции.
[0018]
В настоящее времени, конкретная молекулярная структура "соединения X" не установлена. Однако авторы настоящего изобретения подтвердили то явление, что когда 1,2-дихлорэтан (иногда сокращенно упоминается в настоящем документе как "DCE") получает возможность для совместного существования в качестве вещества внутреннего стандарта и приводится в контакт с указанной выше композицией, площадь газохроматографического пика соединения А по отношению к пику DCE уменьшается со временем (смотри "Пример 1", ниже). Это, несомненно, говорит о том, что во время работы индуцируется химическая реакция, приводящая в результате к уменьшению содержания соединения A. Изначально, соединение A не представляет собой соединение, которое может легко деградировать при нагреве. В дополнение к этому, соединение A не вызывает указанной выше дополнительной реакции, даже когда оно приводится в контакт с безводным HF. То есть, имеется тот факт, что химическая реакция между соединением A и жидкостью, содержащей воду в качестве главного компонента, имеет место при относительно низкой температуре примерно равной комнатной температуре (эта реакция упоминается в настоящем документе как "стадия 1").
[0019]
На основе рассмотренных выше наблюдений, авторы настоящего изобретения привели "севофлуран, содержащий малое количество соединения A", в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30". В результате, со временем происходит значительное уменьшение количества соединения А по отношению к количеству сосуществующего совместно с ним севофлурана, это говорит о том, что севофлуран является стабильным при таких условиях, в то время как соединение A селективно вызывает химическую реакцию (эта стадия реакции в настоящем документе упоминается как "стадия 1a").
[0020]
Затем, после окончания стадии 1a, органический слой отделяется от водного слоя (от композиции HF и воды), обрабатывается посредством промывки водой и подвергается воздействию прецизионной дистилляции в присутствии ингибитора деградации. В результате, "севофлуран, по существу не содержащий соединения A (это означает, что отношение площади газохроматографического пика с FID соответствует концентрации меньше чем 1 м.д.; это же применяется и в дальнейшем)", успешно получается в качестве компонента главной фракции дистилляции в количестве, которое существенно больше, чем в случае неосуществления рассмотренной выше стадии 1a (стадии 2).
[0021]
Далее, анализируется жидкость, остающаяся в форме остатков после дистилляции (или донных остатков в танке), полученная в результате стадии 2. Неожиданно, эта жидкость, как обнаружено, содержит "простой полиэфир", описываемый следующей далее формулой:
[0022]

[0023]
(где R1 и R2, каждый, независимо представляют собой водород, C1-C10 алкильную группу или галогеналкильную группу (галоген: фтор, хлор или бром), n представляет собой целое число от 1 до 10, и при этом R1 и R2 не представляют собой водорода).
[0024]
Наиболее типичные примеры виды химических частиц рассмотренного выше простого полиэфира представляют собой "простой полиэфир 1" и "простой полиэфир 2", описанные ниже. Остатки, остающиеся после дистилляции (донные остатки в танке) после окончания стадии 2 имеют тенденцию к удерживанию больших количеств этих генерируемых простых полиэфиров (смотри Примеры ниже).
[0025]
(CF3)2CHO-CH2-O-CH(CF3)2 <Простой полиэфир 1>
1,1,1,3,3,3-Гексафтор-2-[[2,2,2-трифтор-1-(трифторметил)-этокси]метокси]пропан
(CF3)2CHO-CH2-O-CH2-O-CH(CF3)2 <Простой полиэфир 2>
2,2'-[Оксибис(метиленокси)]бис[1,1,1,3,3,3-гексафторпропан]
[0026]
"Простой полиэфир 3" и "простой полиэфир 4" могут генерироваться, в дополнение к рассмотренным выше простым полиэфирам или вместо них, в зависимости от времени или температуры дистилляции на стадии 2.
[0027]
(CF3)2CHO-(CH2-O)3-CH(CF3)2 <Простой полиэфир 3>
2,2'-[Окситрис(метиленокси)]бис[1,1,1,3,3,3-гексафторпропан]
(CF3)2CHO-(CH2-O)4-CH(CF3)2 <Простой полиэфир 4>
2,2'-[Окситетра(метиленокси)]бис[1,1,1,3,3,3-гексафторпропан]
[0028]
Такой "простой полиэфир" не представляет собой соединения, которое генерируется посредством приведения севофлурана в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30". С учетом этого, предполагается, что "простой полиэфир" получается из "соединения X", генерируемого в результате стадии 1, и, следовательно, соединение X индуцирует некоторый вид химической реакции во время дистилляции, что дает в результате генерирование простого полиэфира.
[0029]
Здесь, известно, что "простой полиэфир" (независимо от целого числа, представленного n) может преобразовываться в севофлуран, когда он приводится в контакт с HF и ускорителем реакции (особенно предпочтительно, с концентрированной серной кислотой) (патент Японии № 3441735). На основе этого открытия, авторы настоящего изобретения подтвердили генерирование севофлурана на значительном уровне посредством предоставления возможности HF и концентрированной серной кислоте для воздействия на остатки (донные остатки в танке), остающиеся после стадии 2 (стадии дистилляции) по настоящему изобретению. Другими словами, по желанию, можно преобразовывать донные остатки в танке, остающиеся после стадии 2, в севофлуран посредством предоставления возможности остаткам для взаимодействия с HF и концентрированной серной кислотой (эта стадия реакция упоминается как "стадия 3"). Можно получать севофлуран в качестве главной фракции дистилляции посредством дистилляции сырого севофлурана, полученного на стадии 3 ("стадия 4").
[0030]
Можно уменьшать количество соединения A в "смешанной композиции севофлурана и соединения A" для того, чтобы собирать "севофлуран, по существу не содержащий соединения A" посредством осуществления "стадии 1a" и "стадии 2" в сочетании. Также можно эффективно использовать остатки (донные остатки в танке или остатки на дне танка), остающиеся после "стадии 2", посредством осуществления в дальнейшем "стадии 3" и "стадии 4" в сочетании. Соответственно, вариант осуществления, в котором все стадии, стадия 1a, стадия 2, стадия 3 и стадия 4, объединяются, представляет собой особенно предпочтительный вариант осуществления настоящего изобретения.
[0031]
Как описано выше, авторы настоящего изобретения обнаружили, что соединение A можно преобразовывать в соединение X при конкретных условиях (стадия 1). На основе этого открытия, авторы настоящего изобретения обнаружили каждую из стадий 1a-4, описанных выше. В результате, авторы настоящего изобретения успешно осуществили заметное подавление "потерь севофлурана при удалении соединения A из севофлурана посредством дистилляции", которое является особенно проблематичным в способе из Патентной литературы 5. В более предпочтительном варианте осуществления, становится возможным преобразование "соединения X", содержащегося в донных остатках в танке, остающихся после стадии 2, которое представляет собой промежуточный продукт соединения A, в севофлуран. В результате, предлагается способ получения севофлурана, который является заметно усовершенствованным по сравнению с обычными способами.
[0032]
Конкретно, настоящее изобретение охватывает следующие далее изобретения.
[0033]
[Изобретение 1]
Способ уменьшения количества соединения A, включающий следующую стадию:
стадия 1 приведения простого фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединения A) в контакт с композицией, содержащей фтористый водород и воду при массовом отношении от 1:1 до 1:30.
[0034]
[Изобретение 2]
Способ получения 2-й органической жидкости, включающий следующую стадию:
стадия 1a приведения жидкости (1-й органической жидкости), содержащей севофлуран и простой фторметил-1,1,3,3,3-пентафторизопропениловый эфир (соединение A), в контакт с композицией, содержащей фтористый водород и воду при массовом отношении от 1:1 до 1:30, с получением при этом следующей жидкости (i) или (ii) (2-й органической жидкости): (i) органической жидкости, содержащей севофлуран и соединение A в количестве, которое меньше, чем количество в 1-й органической жидкости; или (ii) органической жидкости, содержащей севофлуран и по существу не содержащей соединения A.
[Изобретение 3]
Способ в соответствии с Изобретением 2, где температура во время контакта составляет 0°C-60°C.
[0035]
[Изобретение 4]
Способ в соответствии с Изобретением 2 или 3, где контакт осуществляется при совместном существовании с гексафторизопропиловым спиртом (HFIP).
[0036]
[Изобретение 5]
Способ получения севофлурана, по существу не содержащего соединения A, включающий следующую стадию:
стадия 2 дистилляции 2-й органической жидкости, полученной с помощью способа в соответствии с любым из пунктов 2-4, в присутствии ингибитора деградации, с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
[0037]
[Изобретение 6]
Способ в соответствии с Изобретением 5, где ингибитор деградации, используемый на стадии 2, представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из NaHCO3, Na2B4O7, H3BO4, C6H4(COOK)(COOH), Na2SO3, Na2HPO4, CH3COONa и Na3PO4.
[0038]
[Изобретение 7]
Способ в соответствии с Изобретением 5 или 6, дополнительно включающий следующую стадию:
стадия 3 приведения безводного фтористого водорода и ускорителя реакции в контакт с остатками (3-й органической жидкостью), остающимися после дистилляции на стадии 2, с получением при этом жидкости (4-й органической жидкости), в которой, по меньшей мере, часть компонентов в 3-й органической жидкости преобразуется в севофлуран.
[0039]
[Изобретение 8]
Способ в соответствии с Изобретением 7, дополнительно включающий следующую стадию:
стадия 4 дистилляции 4-й органической жидкости с получением севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
[0040]
[Изобретение 9]
Способ в соответствии с любым из Изобретений 2-8, где "1-я органическая жидкость" получается в качестве первой фракции дистилляции на следующей далее стадии:
стадия A дистилляции севофлурана в присутствии ингибитора деградации с тем, чтобы собрать первую фракцию дистилляции.
[0041]
[Изобретение 10]
Способ в соответствии с Изобретением 9, где ингибитор деградации, используемый на стадии A, представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из NaHCO3, Na2B4O7, H3BO4, C6H4(COOK)(COOH), Na2SO3, Na2HPO4, CH3COONa и Na3PO4.
[0042]
[Изобретение 11]
Способ получения севофлурана, по существу не содержащего соединения A, включающий следующие стадии:
стадия 1b приведения жидкости (1-й органической жидкости), содержащей севофлуран и простой фторметил-1,1,3,3,3-пентафторизопропениловый эфир (соединение A)", в контакт с композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30, в присутствии гексафторизопропилового спирта (HFIP) при 0°C-60°C, с получением при этом следующей жидкости (i) или (ii) (2-й органической жидкости): (i) органической жидкости, содержащей севофлуран и соединение A в количестве, которое меньше, чем его количество в 1-й органической жидкости; или (ii) органической жидкости, содержащей севофлуран и по существу не содержащей соединения A; и
стадия 2 дистилляции 2-й органической жидкости в присутствии ингибитора деградации, с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
[0043]
[Изобретение 12]
Способ в соответствии с Изобретением 11, дополнительно включающий следующие стадии:
стадия 3 приведения остатков (3-й органической жидкости), остающихся после дистилляции на стадии 2, в контакт с безводным фтористым водородом и ускорителем реакции, с получением при этом жидкости (4-й органической жидкости), в которой, по меньшей мере, часть 3-й органической жидкости преобразуется в севофлуран; и
стадия 4 дистилляции 4-й органической жидкости в присутствии ингибитора деградации, с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
Преимущественные воздействия изобретения
[0044]
Настоящее изобретение является преимущественным в том, что соединение A может преобразовываться в соединение X, которое может легко отделяться от севофлурана (стадия 1).
[0045]
В дополнение к этому, в другом варианте осуществления, настоящее изобретение является преимущественным в том, что можно селективно предоставлять возможность соединению A в "1-й органической жидкости" для взаимодействия, с получением при этом "2-й органической жидкости", имеющей уменьшенное содержание соединения A или по существу не содержащей соединения A (стадия 1a).
[0046]
Кроме того, еще в одном варианте осуществления, настоящее изобретение является преимущественным в том, что можно получать севофлуран, по существу не содержащий соединения A, в значительно большем количестве по сравнению со случаем неосуществления стадии 1a с использованием в качестве исходных материалов "1-й органической жидкости", которую обычно сложно использовать эффективно (стадия 1a и стадия 2).
[0047]
Кроме того, еще в одном варианте осуществления, настоящее изобретение является дополнительно преимущественным в том, что неожиданно становится возможным получение севофлурана с использованием, в качестве исходных материалов, остатков (донные остатки в танке), остающихся после дистилляции на стадии 2 (стадия 1a и стадии 2-4).
[0048]
В соответствии с настоящим изобретением, становится возможным получение севофлурана с использованием, в качестве исходных материалов, "севофлурана, содержащего соединение A (1-й органической жидкости)", который сложно использовать эффективно, тем самым предлагается улучшенный способ получения севофлурана.
Краткое описание чертежей
[0049]
[Фигура 1] Фигура 1 иллюстрирует блок-схему соотношения между следующими далее техническими терминами, используемыми в настоящем документе: "стадия A", "стадия 1a", "стадия 2", "стадия 3", "стадия 4", "1-я органическая жидкость", "2-я органическая жидкость", "3-я органическая жидкость" и "4-я органическая жидкость".
Описание вариантов осуществления
[0050]
Далее, настоящее изобретение описывается подробно. Рамки настоящего изобретения не ограничиваются этим. Настоящее изобретение может осуществляться с соответствующими изменениями без отклонения от того, что предлагает настоящее изобретение.
Настоящее описание включает часть содержания, как описано в описании и/или на чертежах заявки на патент Японии № 2017-019555, или всю ее, она является приоритетным документом для настоящей заявки. Все публикации, включая документы предыдущего уровня техники, заявки на патенты, публикации патентов и другие патентные документы, цитируемые в настоящем документе, включаются в настоящий документ в качестве ссылок во всей своей полноте.
[0051]
Технические термины и выражения, используемые в настоящем документе, определяются так, как приведено ниже.
"Стадия 1":
Стадия 1 представляет собой стадию приведения простого фторметил-1,1,3,3,3-пентафторизопропенилового эфира (упоминаемого как "соединение A") в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30" (тем самым делая возможным преобразование соединения A в соединение X для уменьшения количества соединения A).
[0052]
"Стадия A":
Стадия A представляет собой стадию дистилляции севофлурана в присутствии ингибитора деградации, тем самым собирая первую фракцию дистилляции (первая фракция дистилляции, собранная на стадии A, используется в качестве "1-й органической жидкости" на стадии 1a в особенно предпочтительном варианте осуществления настоящего изобретения).
[0053]
"Стадия 1a":
Стадия 1a представляет собой стадию приведения "жидкости (1-й органической жидкости), содержащей севофлуран и простой фторметил-1,1,3,3,3-пентафторизопропениловый эфир (далее упоминаемый как "соединение A")", в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30", с получением при этом "2-й органической жидкости". Конкретно, стадия 1a может представлять собой стадию, заставляющую соединение A взаимодействовать в результате контакта с тем, чтобы преобразовывать "1-ю органическую жидкость" во "2-ю органическую жидкость" (стадия 1a, в частности, характеризуется тем, что "1-я органическая жидкость" используется в качестве реакционного материала на стадии 1) (отметим, что "2-я органическая жидкость" означает: (i) органическая жидкость, содержащая севофлуран и соединение A в количестве, которое меньше, чем количество в "1-й органической жидкости"; или (ii) органическая жидкость, содержащая севофлуран и по существу не содержащая соединения A; это же применяется и в дальнейшем).
[0054]
"Стадия 1b":
Стадия 1b представляет собой стадию приведения "1-й органической жидкости, содержащей севофлуран и простой фторметил-1,1,3,3,3-пентафторизопропениловый эфир (далее упоминаемый как "соединение A")", в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30", в присутствии гексафторизопропилового спирта (HFIP) при 0°C-60°C, с получением при этом "2-й органической жидкости". Конкретно, стадия 1b может представлять собой стадию, заставляющую соединение A взаимодействовать в результате контакта с тем, чтобы преобразовывать "1-ю органическую жидкость" во "2-ю органическую жидкость".
[0055]
В дополнение к этому, стадия 1b представляет собой особенно предпочтительный вариант осуществления стадии 1a. Когда необходимо отличать стадию 1b от стадии 1с с помощью более широкой концепции, указанная выше стадия называется "стадия 1b"; однако пояснение к стадии 1a может непосредственно применяться к стадии 1b, если не указано иного.
[0056]
"Стадия 2":
Стадия 2 представляет собой стадию дистилляции "2-й органической жидкости", полученной на стадии 1a (или на стадии 1b) в присутствии ингибитора деградации, с получением при этом севофлурана, по существу не содержащего соединения A, в качестве фракции дистилляции.
[0057]
"Стадия 3":
Стадия 3 представляет собой стадию приведения безводного HF и ускорителя реакции в контакт с остатками (или с донными остатками в танке, которые упоминаются как "3-я органическая жидкость"), остающимися после стадии 2, с получением при этом жидкости (4-й органической жидкости), в которой, по меньшей мере, часть компонента в "3-й органической жидкости" преобразуется в севофлуран.
[0058]
"Стадия 4":
Стадия 4 представляет собой стадию дистилляции "4-й органической жидкости" в присутствии ингибитора деградации, с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
[0059]
"1-я органическая жидкость":
1-я органическая жидкость представляет собой жидкую композицию, содержащую севофлуран и соединение A.
[0060]
"2-я органическая жидкость":
2-я органическая жидкость представляет собой композицию, полученную посредством воздействия на 1-ю органическую жидкость на стадии 1a (или на стадии 1b), она представляет собой: (i) органическую жидкость, содержащую севофлуран и соединение A в количестве, которое меньше, чем количество в "1-й органической жидкости"; или (ii) органическую жидкость, содержащую севофлуран и по существу не содержащую соединения A.
[0061]
"3-я органическая жидкость":
3-я органическая жидкость соответствует остаткам (или донные остаткам в танке), остающимся после дистилляции на стадии 2 (отметим, что это 3-я органическая жидкость неожиданно содержит "простой полиэфир").
[0062]
"4-я органическая жидкость":
4-я органическая жидкость представляет собой органическую жидкость, полученную на стадии 3, которая содержит, по меньшей мере, севофлуран.
[0063]
"По существу не содержащий соединения A":
Выражение "по существу не содержащий соединения A" означает, что концентрация соединения A в жидкости представляющей интерес, вычисленная на основе площади пика, составляет меньше чем 1 м.д. при газохроматографическом анализе с FID.
[0064]
Далее, рассмотренные выше стадии, будут описываться подробно в примерном порядке операций.
[0065]
[1] Стадия 1
Стадия 1 представляет собой стадию приведения соединения A в контакт с "композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30". Соответственно, соединение A получает возможность для взаимодействия, так что может быть получено "соединение X", имеющее неустановленную структуру (оно может быть преобразовано в "соединение X").
[0066]
Как описано выше, малое количество соединения A может быть получено посредством приведения севофлурана в контакт с основанием, таким как гидроксид натрия. В дополнение к этому, соединение A постепенно генерируется во время дистилляции и очистки севофлурана. Аутентичное получение соединения A (соединения A, имеющего чистоту 99% или больше) может быть осуществлено посредством предоставления возможности севофлурану для взаимодействия с литием бис(триметилсилил)амидом, служащим в качестве супероснования, в безводном тетрагидрофурановом растворителе при -70°C - -60°C, с последующей дистилляцией и очисткой (Пример 1, описанный ниже, представляет собой экспериментальный пример, использующий этот аутентичный препарат) в соответствии с Journal of Fluorine Chemistry, vol. 45 (2), November 1989, P. 239 to P. 253.
[0067]
Можно осуществлять стадию 1 посредством предоставления возможности фтористому водороду (HF) и воде для воздействия на соединение A (жидкость). Порядок приведения трех химических частиц (соединения A, фтористого водорода и воды) в контакт друг с другом не является как-либо ограниченным. Однако является особенно предпочтительным предварительное смешивание фтористого водорода и воды для приготовления "фтористоводородной кислоты", а затем приведение фтористоводородной кислоты в контакт с соединением A с точки зрения простоты манипуляций.
[0068]
Здесь, выражение "композиция, содержащая фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30", означает, что композиция содержит эти два вида химических частиц при таком массовом отношении. Также можно сделать возможным совместное существование других видов химических частиц (например, севофлурана, HFIP или серной кислоты).
[0069]
Реакция на стадии 1 не осуществляется при безводных условиях (то есть, так сказать, когда используется безводный фтористый водород). По этой причине, вода должна присутствовать в 1-кратном или большем количестве по массе по сравнению с фтористым водородом. В то же время, когда вода присутствует в количестве более чем 30-кратном по массе по сравнению с фтористым водородом, фтористый водород избыточно разбавляется, что предотвращает осуществление реакции до достаточной степени. Другими словами, "массовое отношение фтористого водорода (HF) и воды от 1:1 до 1:30" является требованием для стадии 1.
[0070]
В частности, когда "массовое отношение фтористого водорода (HF) и воды" составляет 1:1-1:10, химическая активность соединения A становится существенно выше, это является особенно предпочтительным.
[0071]
В то же время, "массовое отношение соединения A и фтористого водорода" не является как-либо ограниченным. Когда количество фтористого водорода является слишком малым, это понижает скорость реакции. Когда количество фтористого водорода на 1 г соединения A составляет 1 г или больше, скорость реакции имеет тенденцию к увеличению, что является предпочтительным. В частности, когда количество фтористого водорода на 1 г соединения A составляет 4 г или больше, реакция осуществляется очень быстро, что является особенно предпочтительным.
[0072]
Отметим, что реакция на стадии 1 также зависит от концентрации соединения A в реакционной системе. Как описано на "стадии 1a (которая представляет собой один из вариантов осуществления стадии 1)", ниже, даже если содержание фтористого водорода в 4 раза больше, чем содержание соединения A, например, приготавливается реакционный раствор с "большим количеством севофлурана" и "малым количеством (например, 1000 м.д. (0,1%) или меньше) соединения A", скорость реакции может и не повыситься до достаточной степени, когда концентрация соединения A является слишком низкой. В таком случае, как описано ниже в Примерах, является предпочтительным, чтобы массовое отношение соединения A и HF составляло 1:100 или больше. В случае, в котором количество соединения A является очевидно малым (на уровне м.д.), массовое отношение может быть увеличено до высокого уровня, например, до 1:103-1:106. Как установлено выше, массовое отношение соединения A и HF необязательно должно определяться однозначно. Необходимо отметить, что даже если HF или вода присутствует в количестве, которое существенно больше, чем количество соединения A, это не дает в результате ингибирования реакции или индуцирования новой побочной реакции. Следовательно, желательным является, чтобы специалисты в данной области могли соответствующим образом определять и изменять количества HF и воды, которые должны использоваться, для того, чтобы оптимизировать условия, которые легко делают возможным взаимодействие соединения A при осуществлении этой стадии.
[0073]
Реакционный контейнер, выложенный нержавеющей сталью, железом, фтористой смолой или чем-либо подобным, предпочтительно используется в качестве реактора на стадии 1. Когда используется фтористый водород, стеклянный реакционный контейнер является неподходящим.
[0074]
На стадии 1, предпочтительным является осуществление перемешивания для эффективного приведения соединения A (органического слоя) в контакт с "водным слоем, содержащим воду и HF". Конкретная технология перемешивания не является ограниченной. Моторизованная роторная мешалка может использоваться предпочтительно в масштабе массового производства. Можно также использовать магнитную мешалку, в лабораторном масштабе. В дополнение к способу, использующему "мешалку", можно использовать способ, где сам реактор встряхивают, или способ, где органический слой и водный слой смешиваются посредством протекания реакционного раствора (например, реакционный раствор получает возможность для прохождения через трубу). Эти способы также соответствуют "перемешиванию" на стадии 1 по настоящему изобретению.
[0075]
Температура реакции на стадии 1 (температура во время указанного выше контакта) не является как-либо ограниченной; однако она предпочтительно составляет 0°C-60°C. Когда она ниже 0°C, скорость реакции понижается. Когда она выше 60°C, фтористоводородная кислота имеет тенденцию к улетучиванию, что создает в результате сложности при манипуляциях. Более предпочтительно, температура реакции на стадии 1 составляет 15°C-45°C, а особенно предпочтительно, 25°C-40°C. Когда температура реакции находится в таком диапазоне температур, реакция получает возможность для относительно спокойного осуществления на стадии 1, и улучшаются манипуляции.
[0076]
Является предпочтительным осуществление реакции на стадии 1 в условиях плотной герметизации. Альтернативно, можно осуществлять реакцию в открытых условиях, когда используется система, которая может собирать и удалять улетучивающийся побочный продукт.
[0077]
Реакционный продукт (соединение X) на стадии 1 не идентифицируется. Он может представлять собой вид химических частиц, которые трудно уловить с помощью газохроматографического анализа. Отметим, что в результате наблюдения авторов настоящего изобретения, на этой стадии не генерируется никаких твердых компонентов (нерастворимых компонентов), и органический слой обычно остается прозрачным до и после реакции. Соответственно, можно предсказать состояние хода реакции посредством предоставления возможности для стандартного вещества, которое является инертным по отношению к взаимодействию (например, 1,2-дихлорэтану (DCE)) с соединением A, существовать совместно с ними, с тем, чтобы определить отношение площадей газохроматографических пиков соединения A и DCE. Это представляет собой особенно предпочтительным способ для понимания состояния хода реакции.
[0078]
Время, необходимое для осуществления стадии 1, изменяется в зависимости от условий; однако оно, как правило, составляет от 10 минут до 12 часов (720 минут). Нет необходимости продолжать реакцию на стадии 1 до достижения доли преобразования 100%. Если можно деградировать, по меньшей мере, часть соединения A, это было бы преимущественным для последующего сбора севофлурана по отношению к количеству деградированного соединения A. По этой причине, доля преобразования в реакции на стадии 1 не является обязательно строго определенной. В предпочтительном примере, стадия 1 завершается после истечения определенного периода времени (например, от 1 до 5 часов). Время реакции для стадии 1 может определяться соответствующим образом из соображений времени, необходимого для осуществления других стадий получения севофлурана (стадии реакции и стадии очистки).
[0079]
В дополнение к непрореагировавшему соединению A, малое количество низкомолекулярного продукта может быть подтверждено в органическом слое после окончания стадии 1. Однако обычно невозможно детектировать "простой полиэфир", который должен присутствовать в остатках (донные остатки в танке) после стадии 2, описанной ниже. В дополнение к этому, севофлуран не генерируется значительно на этой стадии, и даже если севофлуран может детектироваться, его количество обычно остается на уровне следов или меньше.
[0080]
[2] Стадия A (дистилляция на стадии A)
Стадия A представляет собой стадию дистилляции севофлурана в присутствии ингибитора деградации с тем, чтобы собрать "первую фракцию дистилляции" перед стадией 1a, описанной ниже.
[0081]
"Первая фракция дистилляции", собранная на стадии A, представляет собой жидкую композицию, содержащую севофлуран и соединение A. В настоящем изобретении, предпочтительным является использовать первую фракцию дистилляции в качестве "1-й органической жидкости" на стадии 1a с точки зрения цели настоящего изобретения (дистилляция на этой стадии далее иногда упоминается как "дистилляция на стадии A" для того, чтобы отличить ее от других стадий дистилляции по настоящему изобретению).
[0082]
Севофлуран можно предпочтительно синтезировать в соответствии с Патентной литературой 1 и очищать с помощью способов, описанных в Патентной литературе 2-4. "Дистилляция" осуществляемая в соответствии с Патентной литературой 5 после способа очистки (или до или после способа очистки), соответствует "дистилляции на стадии A".
[0083]
Отметим, что главная цель Патентной литературы 5 и "стадии 2" и "стадии 4", описанных ниже, заключается в сборе "главной фракции дистилляции", которая соответствует "севофлурану, по существу, не содержащему соединения A". При этом "дистилляция на стадии A" характеризуется сбором "первой фракции дистилляции", которая соответствует "севофлурану, содержащему соединение A".
[0084]
Ингибиторы деградации, которые можно использовать для "дистилляции на стадии A", представляют собой гидроксид, гидрофосфат, фосфат, бикарбонат, борат или сульфит щелочного металла, соль щелочного металла и уксусной кислоты или фталевой кислоты или борной кислоты, как описано в Патентной литературе 5. Примеры гидроксида щелочного металла включают NaOH и KOH. Гидрофосфат щелочного металла представляет собой гидрофосфат или дигидрофосфат щелочного металла. Их конкретные примеры включают Na2HPO4, NaH2PO4, K2HPO4 и KH2PO4. Примеры фосфата щелочного металла включают метафосфат или полифосфат щелочного металла, а также ортофосфат щелочного металла. Их конкретные примеры включают Na3PO4, K3PO4, (NaPO3)3, (NaPO3)4, (KPO3)3 и (KPO3)4. Примеры бикарбоната щелочного металла включают NaHCO3 и KHCO3. Примеры бората щелочного металла включают диборат, метаборат, тетраборат, пентаборат, гексаборат и октаборат щелочного металла. Их конкретные примеры включают NaBO2, Na2B4O7, NaB5O8, Na2B6O10, Na2B8O18, Na4B2O5, KBO2, K2B4O7, KB5O8, K2B6O10 и K2B8O18. Примеры сульфита щелочного металла включают Na2SO3 и K2SO3. В дополнение к этому, примеры соли щелочного металла и уксусной кислоты включают CH3COONa и CH3COOK.
[0085]
Примеры соли щелочного металла и фталевой кислоты включают соли щелочного металла и o-фталевой кислоты, м-фталевой кислоты и п-фталевой кислоты. Их конкретные примеры включают o-C6H4(COOK)(COOH), m-C6H4(COOK)(COOH), p-C6H4(COOK)(COOH), o-C6H4(COONa)(COOH), m-C6H4(COONa)(COOH) и p-C6H4(COONa)(COOH). Среди добавок, описанных выше, примеры особенно предпочтительных добавок, имеющих воздействие сильного предотвращения деградации севофлурана, включают NaHCO3, Na2B4O7, H3BO4, C6H4(COOK)(COOH), Na2SO3, Na2HPO4, CH3COONa и Na3PO4. Среди них, H3BO4, C6H4 (COOK) (COOH), Na2HPO4, CH3COONa, и тому подобное, имеющие более превосходные воздействия, являются более предпочтительными добавками.
[0086]
Ингибитор деградации может непосредственно добавляться в твердом состоянии. В таком случае, количество, которое должно добавляться, соответствующим образом, составляет от 0,01% масс. до 10% масс., предпочтительно, от 0,05% масс. до5% масс., а более предпочтительно, от 0,1% масс. до 1% масс. по отношению к количеству севофлурана, которое должно обрабатываться.
[0087]
Можно также добавлять ингибитор деградации в форме водного раствора. В таком случае, его концентрация не является как-либо ограниченной; однако она составляет, соответствующим образом, от 0,01% масс. до уровня насыщения (насыщенного раствора), предпочтительно, от 0,1% масс. до 10% масс., а более предпочтительно от 1% масс. до 5% масс. В дополнение к этому, количество ингибитора деградации, добавляемое в форме водного раствора, не является как-либо ограниченным, и таким образом, соответствующее количество, для добавления, может выбираться в зависимости от концентрации водного раствора. Например, когда концентрация водного раствора устанавливается как 1% масс., это количество, соответствующим образом, составляет от 1% масс. до 200% масс., предпочтительно, от 3% масс. до 100% масс., а более предпочтительно, от 5% масс. до 50% масс., по отношению к севофлурану, который должен обрабатываться.
[0088]
Севофлуран, который представляет собой исходные материалы для дистилляции на стадии A, может содержать примеси или соединение A (отметим, что использование очищенного севофлурана, по существу не содержащего соединения A, не является как-либо ограниченным). Также, в случае дистилляции в присутствии какого-либо ингибитора деградации, описанного выше, небольшое количество соединения A может генерироваться в танке. Когда дистилляция осуществляется при таких условиях, соединение A как целое собирается как "первая фракция дистилляции", делая по существу возможным исключение попадания соединения A в "главную фракцию дистилляции". Однако, как установлено выше, невозможно предотвратить попадание севофлурана в первую фракцию дистилляции даже с помощью такой операции дистилляции, в которой предоставляется возможность для его совместного существования с ингибитором деградации.
[0089]
Дистилляционная колонна, используемая для дистилляции на стадии A, не является как-либо ограниченной. Однако предпочтительно может использоваться дистилляционная колонна, заполненная структурированной насадкой или неструктурированной насадкой. Примеры структурированной насадки включают насадки Sulzer, Mellapack, Techno-pack и Flexi-pack. Примеры неструктурированной насадки включают Heli-pack, кольца Рашига и насадку Диксона.
[0090]
Количество теоретических ступеней для дистилляции на стадии A не является как-либо ограниченным; однако оно может составлять от 2 до 50. В частности, предпочтительно, оно составляет от 3 до 30, а более предпочтительно, от 5 до 20.
[0091]
Коэффициент дефлегмации обычно составляет от 0,5 до 50, предпочтительно, от 1 до 30, а более предпочтительно, от 1 до 20.
[0092]
Давление, прикладываемое во время дистилляции на стадии A, не является как-либо ограниченным. Является простым и предпочтительным осуществление дистилляции при обычном давлении. В таком случае, когда главным компонентом, который должен дистиллироваться, является севофлуран, температура дистилляции (температура верхней части колонны) при сборе главной фракции дистилляции равна температуре кипения севофлурана, которая составляет 58°C-59°C. Способ дистилляции компонентов, имеющих более низкие температуры кипения, перед сбором главной фракции дистилляции соответствует "сбору первой фракции дистилляции". Как установлено выше, по существу нет разницы между температурами кипения соединения A и севофлурана. Фактически, первая фракция дистилляции состоит в основном из севофлурана. По этой причине сложно установить граничную точку между температурой для сбора первой фракции дистилляции и температурой для сбора главной фракции дистилляции. Соответственно, предпочтительно непрерывно определять соответствующим образом газохроматографическую композицию каждой фракции во время дистилляции и непрерывно собирать "первую фракцию дистилляции", в то время как детектируется соединение A, а затем собирать "главную фракцию дистилляции", когда подтверждается, что соединение A является по существу недетектируемым (составляет меньше чем 1 м.д.).
[0093]
В предпочтительном варианте осуществления настоящего изобретения, "первая фракция дистилляции" используется в качестве исходных материалов (1-я органическая жидкость) на следующей далее стадии 1a.
[0094]
[3] Стадия 1a
Стадия 1a представляет собой особенно предпочтительный вариант осуществления стадии 1, описанной выше. Другими словами, стадия 1a совпадает со стадией 1 в терминах "смешивания соединения A с HF и водой для осуществления реакции". В то же время, стадия 1a характеризуется тем, что соединение A, служащее в качестве исходных материалов, получают как "жидкую смесь севофлурана и соединения A (1-я органическая жидкость)".
[0095]
Способ получения "1-й органической жидкости", служащей в качестве исходных материалов, не является как-либо ограниченным; однако предпочтительно использовать жидкость, собранную как "первая фракция дистилляции", во время "дистилляции на стадии A", описанной выше.
[0096]
В дополнение к этому, "1-я органическая жидкость" преобразуется во "2-ю органическую жидкость" на стадии 1a. Как установлено выше, "2-я органическая жидкость" означает: (i) органическая жидкость, содержащая севофлуран и соединение A в количестве, которое меньше, чем количество в "1-й органической жидкости"; или (ii) органическая жидкость, содержащая севофлуран и по существу не содержащая соединения A. Различие между (i) и (ii) заключается в том, осуществляется ли полностью преобразование соединения A в соединение X.
[0097]
Содержание соединения A, используемого в качестве исходных материалов в "1-й органической жидкости" на этой стадии, не является как-либо ограниченным. Когда "1-я органическая жидкость" получается с помощью "стадии A", содержание соединения A в "1-й органической жидкости" зависит от содержания соединения A в сыром севофлуране перед стадией A, и оно может изменяться в зависимости от условий осуществления дистилляции. Содержание соединения A в "1-й органической жидкости" обычно составляет от 5 м.д. до 10000 м.д., а, как правило, от 10 до 1000 м.д. Однако даже если содержание соединения A выше или ниже чем указанный выше диапазон, стадию 1a можно осуществлять. В результате уменьшения содержания соединения A, преимущества настоящего изобретения достигаются в достаточной степени.
[0098]
Как объясняется выше, реакция деградации совместно существующего в реакционной смеси севофлурана не индуцируется при условиях стадии 1a. На стадии 1a, севофлуран, который представляет собой компонент, остается как есть. При этом соединение A химически изменяется со временем таким образом, что соединение A преобразуется в "соединение X", которое можно легко отделить от севофлурана. Когда осуществляется стадия 1a, севофлуран действует в качестве вещества внутреннего стандарта. Следовательно, нет необходимости в добавлении стандартного вещества, такого как DCE. После начала реакции, можно понять состояние хода реакции посредством последовательного определения отношения площадей газохроматографических пиков севофлурана и соединения A.
[0099]
Различия между "стадией 1" и "стадией 1a", описанными выше, заключается только в том, содержится ли "севофлуран, который является инертным по отношению к реакции", в качестве главного компонента, или нет. В этом отношении, условия, описанные для "стадии 1" могут опять же применяться как условия для "массового отношения HF и воды", "массового отношения соединения A и HF", "материалов в реакторе", "способа перемешивания", "температуры реакции", и тому подобное.
[0100]
Как объясняется выше, когда главный компонент в "1-й органической жидкости", служащей в качестве исходных материалов, представляет собой севофлуран, в случае стадии 1a, абсолютное количество соединения A обычно является очень малым. По этой причине, в случае стадии 1a, "массовое отношение соединения A:HF", как тенденция, является очень высоким, в то время как соединение A обычно является разбавленным большим количеством севофлурана. В этом случае, как объясняется в разделе стадии 1, выше, является предпочтительным устанавливать массовое отношение таким образом, что количество HF избыточно превышает количество соединения A во многих случаях. Кроме того, скорость реакции на стадии 1a также зависит от "массового отношения HF и воды", "способа перемешивания" и "температуры реакции". Следовательно, чтобы понимать состояние хода реакции, является желательным, чтобы отношение площадей газохроматографических пиков севофлурана и соединения A могло определяться последовательно, это является предпочтительным при установлении первичных условий.
[0101]
Необходимо отметить, что цель "стадии 1a" в соответствии с настоящим изобретением представляет собой "уменьшение" содержания соединения A, содержащегося в "1-й органической жидкости", и это не обязательно означает, что содержание должно быть уменьшено до нуля. Другими словами, в случае, когда определенное количество (содержание) соединения A может быть уменьшено в "1-й органической жидкости" на стадии 1a, даже если дистилляция осуществляется на стадии 2 (описанной ниже) при таком же количестве теоретических ступеней и при таком же коэффициенте дефлегмации как на предыдущей "стадии A", количество "первой фракции дистилляции" уменьшается в ответ на уменьшение количества соединения A, тем самым делая возможным сбор "севофлурана, по существу не содержащего соединения A", в качестве главной фракции дистилляции. Реакция на стадии 1a занимает очень продолжительное время для завершения реакции в зависимости от условий, которые могут быть скорее недостаточными. По этой причине, имеется вполне разумная возможность для предварительного определения времени, необходимого для стадии 1a, чтобы осуществлять реакцию при заданном массовом отношении, способе перемешивания и температуре реакции, а затем, для завершения реакции на этой стадии (даже если доля преобразования не достигает 100%) в пределах заданного времени.
[0102]
Например, в предпочтительном варианте осуществления, стадия 1a предпочтительно осуществляется в течение от 30 минут до 5 часов, а более предпочтительно, от 30 минут до 2 часов, а затем завершается.
[0103]
В дополнение к этому, когда реакция на стадии 1a представляет собой неоднородную реакцию жидкость-жидкость, присутствие поверхностно-активного вещества может ускорить реакцию. В частности, когда гексафторизопропиловый спирт (HFIP), который представляет собой амфипатическое вещество, считающееся поверхностно-активным веществом, получает возможность для совместного существования, доля уменьшения содержания соединения A имеет тенденцию к повышению, даже если все другие условия являются одинаковыми (смотри Пример 3).
[0104]
HFIP представляет собой исходные материалы, используемые для синтеза севофлурана в Патентной литературе 1, и по этой причине, он часто содержится в реакционном растворе для синтеза севофлурана. В дополнение к этому, сам HFIP не вызывает деградации севофлурана. Следовательно, при осуществлении стадии 1a, является особенно предпочтительным предоставление возможности HFIP для совместного существования. Когда используется HFIP, его количество предпочтительно составляет от 0,001 г до 20 г, а более предпочтительно, от 0,01 г до 10 г на 1 г севофлурана в "1-й органической жидкости".
[0105]
В одном из особенно предпочтительных вариантов осуществления, HFIP получает возможность для совместного существования и реакция (контакт, описанный выше) осуществляется при 0°C-60°C на стадии 1a по настоящему изобретению. Когда реакция осуществляется при таких условиях, она имеет тенденцию к получению в результате ускорения деградации, в частности, соединения A до соединения X. Стадия 1a, осуществляемая при таких условиях, иногда упоминается в настоящем документе как "стадия 1b", как объясняется выше.
[0106]
Стадия 1a может осуществляться в присутствии серной кислоты. Поскольку серная кислота также представляет собой исходные материалы, используемые для синтеза севофлурана с помощью способа, описанного в Патентной литературе 1, она часто содержится в реакционном растворе после завершения реакции. Авторы настоящего изобретения обнаружили, что, когда осуществляется стадия 1a по настоящему изобретению, совместное существование серной кислоты не ингибирует как-либо реакцию, и нет таких фактов, что генерируется побочный продукт, который сложно отделять от севофлурана. Однако если количество серной кислоты (H2SO4) является исключительно большим, это может вызвать дезактивацию "воды", необходимой для стадии 1a. Следовательно, не является предпочтительным, чтобы масса серной кислоты была, например, существенно больше, чем масса воды. Обычно серная кислота в таком исключительно большом количестве не вводится в реакционную систему. Однако в случае, в котором количество серной кислоты является большим по некоторым причинам, является предпочтительным предоставить возможность для присутствия воды при массе, которая, по меньшей мере, равна, а предпочтительно в 2 раза или более превышает массу серной кислоты.
[0107]
После завершения стадии 1a, может осуществляться разделение двух слоев в соответствии с обычным способом, таким образом, что можно собрать органический слой. Таким образом, собранный органический слой представляет собой "2-ю органическую жидкость". "2-я органическая жидкость", содержит севофлуран в качестве главного компонента, как и в случае "1-й органической жидкости", служащей в качестве исходных материалов; однако содержание соединения A во 2-й органической жидкости значительно ниже, чем содержание в "1-й органической жидкости". Отметим, что поскольку органический слой обычно содержит HF, используемый на стадии 1a, является предпочтительным осуществление операции очистки, такой как промывка щелочным водным раствором или промывка водой, для собранного органического слоя с целью уменьшения нагрузки для системы, используемой на следующей далее стадии 2 (дистилляции). Конкретно, является желательным осуществление либо промывки щелочным водным раствором, либо промывки водой, по меньшей мере, один раз.
[0108]
[4] Стадия 2 (дистилляция на стадии 2)
Стадия 2 представляет собой стадию дистилляции "2-й органической жидкости", полученной на стадии 1a (или на "стадии 1b", что является особенно предпочтительным вариантом осуществления стадии 1), в присутствии ингибитора деградации для того, чтобы получить в качестве главной фракции дистилляции, севофлуран, по существу не содержащий соединения A. Чтобы отличить дистилляцию на этой стадии от "дистилляции на стадии A", описанной выше, дистилляция на этой стадии иногда упоминается в настоящем документе как "дистилляция на стадии 2". Отметим, что предмет изобретения, полученный посредством "дистилляции на стадии A" представляет собой "первую фракцию дистилляции", в то время как предмет изобретения, полученный посредством "дистилляции на стадии 2", представляет собой "главную фракцию дистилляции".
[0109]
Как описано подробно выше, в результате реакции на стадии 1a, содержание соединения A во "2-й органической жидкости" значительно ниже, чем его содержание в "1-й органической жидкости". По этой причине, можно дополнительно собрать, в качестве главной фракции дистилляции, "севофлуран, по существу не содержащий соединения A", посредством дистилляции "2-й органической жидкости" на стадии 2, например, при таких же условиях как "дистилляция на стадии A" (в терминах количества теоретических ступеней, коэффициента дефлегмации или чего-либо подобного).
[0110]
Поскольку "дистилляция на стадии 2" совпадает с "дистилляцией на стадии A" в терминах "дистилляции севофлурана", условия, описанные подробно в разделе "дистилляция на стадии A", могут применяться снова в качестве условий дистилляции на стадии 2 (тип или количество ингибитора деградации, дистилляционное устройство, количество теоретических ступеней, коэффициент дефлегмации, давление и температура дистилляции). Отметим, что дистилляция на стадии 2 отличается от "дистилляции на стадии A" тем, что предмет изобретения для стадии 2 представляет собой не первую фракцию дистилляции, а главную фракцию дистилляции. Другими словами, "первая фракция дистилляции" может собираться в то время, когда компонент A детектируется как фракция во время дистилляции, а когда соединение A становится собирать невозможно, может собираться "главная фракция дистилляции".
[0111]
При осуществлении "дистилляции на стадии A" перед дистилляцией на стадии 2, как правило, осуществляют "дистилляцию на стадии 2" при таких же условиях как "дистилляция на стадии A", что является особенно рациональным. Это связано с тем, что обе стадии совпадают в том, что дистиллируется севофлуран, так что севофлуран получается в качестве главной фракции дистилляции. Другими словами, задача дистилляции на стадии 2 заключается в извлечении севофлурана до возможной степени в качестве "главной фракции дистилляции" из первой фракции дистилляции при условиях, которые уже оптимизированы на стадии A. Следовательно, нет необходимости в значительном увеличении количества теоретических ступеней или чего-либо подобного вместо оптимизации условий "дистилляции на стадии A" для осуществления дистилляции более строгим образом в течение более продолжительного периода времени. Отметим, что, при желании, "дистилляция на стадии 2" может осуществляться при более строгих условиях.
[0112]
С другой стороны, в случае, когда дистилляция осуществляется при условиях дистилляции, которые являются менее строгими, чем условия "дистилляции на стадии A" (это означает, что количество теоретических ступеней или коэффициент дефлегмации уменьшается), может оказаться невозможным сбор "главной фракции дистилляции" (севофлурана, по существу не содержащего соединения A) даже посредством осуществления "дистилляции на стадии 2", хотя это зависит от степени уменьшения содержания соединения A на стадии 1a. В случае, в котором главная фракция дистилляции не может собираться посредством осуществления дистилляции на стадии 2 при таких менее строгих условиях, уменьшение содержания соединения A посредством реакции на стадии 1a становится нецелесообразным, что не является предпочтительным. В то же время, в случае, когда имеется значительное уменьшение содержания соединения A как результат осуществления стадии 1a, дистилляция может быть возможной, даже делая условия дистилляции для "дистилляции на стадии 2" менее строгими, чем условия "дистилляции на стадии A". В этом случае, преимущественным является то, что дистилляция на стадии 2 может осуществляться проще. Другими словами, оптимальные условия "дистилляции на стадии 2" определяются также в зависимости от условий для предыдущей стадии 1a. По этой причине является предпочтительным регулировать условия на основе знаний специалистов в данной области посредством отслеживания уровня сбора севофлурана в качестве главной фракции дистилляции.
[0113]
Как установлено выше, хотя нет каких-либо конкретных ограничений на условия "дистилляции на стадии 2," можно сэкономить рабочее время на стадии дистилляции и эффективно использовать систему дистилляции посредством осуществления "дистилляции на стадии 2" при таких же условиях (например, используя ту же самую дистилляционную колонну), как условия осуществляемой ранее "дистилляции на стадии A", что может представлять собой предпочтительный вариант осуществления.
[0114]
Севофлуран, извлекаемый в качестве "главной фракции дистилляции" при "дистилляции на стадии 2", может добавляться к продукту севофлурана. Конкретно, извлеченная таким образом "главная фракция дистилляции" представляет собой севофлуран, который не может собираться на стадии A, это означает, что севофлуран может извлекаться посредством стадии 2 как результат осуществления предыдущей стадии 1a.
[0115]
"Первую фракцию дистилляции" также собирают во время "дистилляции на стадии 2". "Первая фракция дистилляции" представляет собой "севофлуран, содержащий соединение A на значительном уровне", как в случае первой фракции дистилляции на стадии A (даже если "2-я органическая жидкость", служащая в качестве исходных материалов для дистилляции на стадии 2, не содержит соединения A, малое количество соединения A может генерироваться во время дистилляции). Эта "первая фракция дистилляции" может удаляться, поскольку ее количество является относительно малым. Альтернативно, она может собираться для повторного использования в качестве исходных материалов на "стадии 1a" (1-я органическая жидкость).
[0116]
В то же время, остатки (донные остатки в танке), остающиеся после сбора главной фракции дистилляции во время дистилляции на "стадии 2", упоминаются в настоящем документе как "3-я органическая жидкость". Эта "3-я органическая жидкость" может удаляться из системы. Авторы настоящего изобретения, однако, обнаружили, что "3-я органическая жидкость" содержит "простой полиэфир", который может представлять собой исходные материалы для получения севофлурана. По этой причине, при желании, "стадия 3", описанная ниже, может осуществляться с использованием "3-й органической жидкости" в качестве исходных материалов.
[0117]
Как установлено выше, типичные примеры "простого полиэфира", используемого в настоящем документе, представляют собой "простой полиэфир 1" и "простой полиэфир 2". Обычно эти простые полиэфиры генерируются в больших количествах. Когда время или температура в течение дистилляции на стадии 2 продлевается или увеличивается, соответственно, имеется тенденция к генерированию "простого полиэфира 3" и "простого полиэфира 4", в дополнение к этим простым полиэфирам или вместо них.
[0118]
[5] Стадия 3
Стадия 3 представляет собой стадию приведения "3-й органической жидкости", полученной в качестве донных остатков в танке после стадии 2, в контакт с безводным HF и ускорителем реакции (особенно предпочтительно, с серным ангидридом), с получением при этом "4-й органической жидкости", в которой, по меньшей мере, часть "простого полиэфира" в "3-й органической жидкости" преобразуется в севофлуран.
[0119]
Как установлено выше, имеются различные типы "простого полиэфира" в 3-й органической жидкости после прекращения дистилляции на стадии 2. Обычно 3-я органическая жидкость находится в форме смешанной композиции различных типов простых полиэфиров. Однако несмотря на обилие конкретных химических видов простых полиэфиров, эти простые полиэфиры могут преобразовываться в севофлуран посредством приведения "3-й органической жидкости" в контакт с безводным HF и ускорителем реакции в соответствии с описанием патента Японии № 3441735. В большинстве случаев, количество собранной 3-й органической жидкости меньше, чем количество "1-й органической жидкости", служащей в качестве исходных материалов на стадии 1a. По этой причине, можно также осуществлять стадию 3 после осуществления стадии 1 и 2 для множества загрузок, для того, чтобы достичь определенного извлекаемого количества 3-й органической жидкости.
[0120]
Примеры ускорителя реакции включают: кислоты Бренстеда, такие как дымящаяся серная кислота, концентрированная серная кислота, серная кислота, фторсерная кислота, безводная фосфорная кислота, фосфорная кислота и трифторметансульфоновая кислота; и кислоты Льюиса, такие как тетрахлорид титана, хлорид алюминия, пентахлорид сурьмы, трифторид алюминия, серный ангидрид и пентафторид сурьмы. Среди них, дымящаяся серная кислота, концентрированная серная кислота, серная кислота (80% масс. или больше), фторсерная кислота, фосфорная кислота или их смесь является предпочтительными. Концентрированная серная кислота является особенно предпочтительной.
[0121]
Температура реакции не является как-либо ограниченной; однако она составляет 10°C-100°C, а предпочтительно, 35°C-80°C. Генерируемый севофлуран может дистиллироваться вместе с непрореагировавшими исходными материалами наружу из реакционной системы в указанном выше диапазоне температур, который является предпочтительным. Когда температура реакции ниже 10°C, реакция замедляется, что является непрактичным, а когда она выше 100°C, реакция избыточно ускоряется, делая сложным контроль реакции, что не является предпочтительным.
[0122]
Поскольку давление реакции не оказывает существенного влияния на реакцию, оно не является как-либо ограниченным. Обычно реакция может осуществляться при 0,1-1 МПа.
[0123]
Реакция на стадии 3 может также осуществляться в присутствии формальдегида или параформальдегида.
[0124]
Отношение смешивания реакционных реагентов, используемых в способе по настоящему изобретению, описывается следующим образом: a: когда используется формальдегид, количество молей формальдегида добавляется к такому же общему количеству молей оксиметиленовой группы "простого полиэфира" в "3-й органической жидкости"; b: когда HFIP используется в сочетании, количество молей HFIP добавляется к такому же количеству молей гексафторизопропенильной группы "простого полиэфира" в "3-й органической жидкости"; c: количество молей HF; d: количество молей ускорителя реакции.
[0125]
Величина b/a обычно составляет от 0,5 до 5, а предпочтительно, от 0,7 до 3.
[0126]
Величина c/a обычно составляет от 1 до 50, а предпочтительно, от 3 до 30 (для HF является предпочтительным, чтобы он присутствовал, по меньшей мере, на эквимолярном уровне по отношению к оксиметиленовым группам для улучшения выхода реакция).
[0127]
Здесь, d представляет собой количество молей произвольного компонента. Когда используется произвольный компонент, величина d/a обычно соответствует молярной кратности от 0,5 до 20, а предпочтительно, молярной кратности от 0,7 до 5,0.
[0128]
Предпочтительно, на стадии 3 воды нет. Нет необходимости строго контролировать концентрацию влажности с использованием устройства для титрования влажности по Карлу Фишеру; однако не является предпочтительным активное добавление воды в реакционную систему, как в случае стадии 1 или стадии 1a. Количество воды, желательно, составляет 0,01 моль или меньше, когда количество молей "a", рассмотренное выше, составляет 1 моль. В дополнение к этому, когда в качестве ускорителя реакции используется дымящаяся серная кислота, серный ангидрид или что-либо подобное, даже если очень малое количество воды имеется в реакционной системе, такой ускоритель реакции захватывает (дезактивирует) воду, давая в результате по существу безводные условия, что является предпочтительным.
В дополнение к этому, как и в случае на стадии 1 или стадии 1a, реакция на стадии 3 эффективно осуществляется при перемешивании, что является предпочтительным.
[0129]
Вариант осуществления реакции на стадии 3 не является как-либо ограниченным. Реакция осуществляется либо в условиях плотной герметизации, либо в открытых условиях так, что "4-я органическая жидкость", в которой, по меньшей мере, часть "простого полиэфира" в 3-й органической жидкости преобразуется в севофлуран, может быть получена со временем.
[0130]
Когда реакция осуществляется при открытых условиях, продукт севофлурана имеет температуру кипения от 58°C до 59°C, при которой реакция на стадии 3 может осуществляться в достаточной степени. Следовательно, технология смешивания 3-й органической жидкости, фтористого водорода, ускорителя реакции, как рассмотрено выше (особенно предпочтительно, концентрированной серной кислоты), и, при необходимости, параформальдегида, в определенных количествах и постепенного повышения температуры для того, чтобы вызвать осуществление реакции примерно при 60°C, является предпочтительной. После генерирования севофлурана, этот севофлуран быстро становится парообразным при такой температуре. Посредством улавливания таких паров с использованием водной охлаждающей ловушки или чего-либо подобного, можно собирать генерируемый севофлуран. Севофлуран, собранный с помощью такого способа, представляет собой сырой севофлуран, и следовательно, он может содержать HF или простой полиэфир в качестве реакционного материала; однако компоненты с высокими температурами кипения могут быть удалены. Следовательно, является предпочтительным собирать сырой севофлуран с помощью такой технологии (сырой севофлуран, полученный посредством сбора паров указанным выше образом, попадает в рамки "4-й органической жидкости").
[0131]
Поскольку "4-я органическая жидкость", полученная выше, обычно содержит HF, используемый в качестве исходных материалов, является предпочтительным осуществление операции очистки, такой как промывка щелочным водным раствором или промывка водой, для собранного органического слоя, чтобы уменьшить нагрузку для системы, используемой на следующей далее стадии 4 (дистилляции). Конкретно, является желательным осуществление промывки щелочным водным раствором или промывки водой, по меньшей мере, один раз.
[0132]
[6] Стадия 4 (дистилляция на стадии 4)
Стадия 4 (дистилляция на стадии 4) представляет собой стадию получения севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции посредством дистилляции "4-й органической жидкости", полученной на стадии 3, в присутствии ингибитора деградации.
[0133]
Эта стадия может осуществляться способом стадии 2 за исключением того, что количество объекта дистилляции меньше, чем его количество на стадии 2 (дистилляции на стадии 2). Тип или количество ингибитора деградации, количество теоретических ступеней дистилляции, способ переключения сбора первой фракции дистилляции на сбор главной фракции дистилляции и другие условия, описанные на стадии 2, могут снова применяться на этой стадии.
[0134]
Количество собранной "4-й органической жидкости" меньше, чем количество "2-й органической жидкости", служащей в качестве исходных материалов на стадии 2. По этой причине, можно также осуществлять стадию 4 после осуществления стадии 1 и 2 для множества загрузок, с целью достижения определенного извлекаемого количества 4-й органической жидкости. Однако на этой стадии, постольку, поскольку можно дистиллировать 4-ю органическую жидкость для сбора севофлурана высокой чистоты (севофлурана, по существу не содержащего соединения A), конкретный способ работы не ограничивается.
[0135]
Примеры
Настоящее изобретение будет описываться более подробно ниже со ссылками на следующие далее Примеры. Однако настоящее изобретение не ограничивается ими.
[Пример 1]
[0136]
(Стадия 1)
Соединение A (чистота: 99% или больше) (5 г), HF (20 г), воду (100 г) и 1,2-дихлорэтан (DCE), служащий в качестве вещества внутреннего стандарта (15 г), смешивают, с последующим перемешиванием в воздухонепроницаемом контейнере из политетрафторэтиленовой смолы в течение 5 часов при 20°C-25°C, с осуществлением при этом реакции. В ходе реакции, отношение площадей пиков [соединение A]/[DCE] определяют с помощью газохроматографического анализа с FID через 1-часовые интервалы. Отметим, что образец жидкости приводится в контакт с NaF для дегидрофторирования, а затем анализируется с помощью газовой хроматографии (время удерживания для соединения A составляет примерно 5,2 минуты, а время удерживания для DCE составляет примерно 16,5 минуты при условиях газовой хроматографии).
[0137]
В результате, хотя отношение площадей пиков [соединение A]/[DCE] непосредственно перед началом реакции составляет 0,57, оно становится равным 0,48, 0,38, 0,31, 0,25 и 0,19 через 1, 2, 3, 4 и 5 часов, соответственно, после начала реакции. Другими словами, по прохождении 5 часов, отношение площадей пиков уменьшается до одной трети от исходного уровня. Не детектируется конкретного главного пика в качестве пика продукта; однако подтверждается, что соединение A химически изменяется на стадии 1.
[0138]
В дополнение к этому севофлуран не детектируется в значительной степени в реакционной смеси. Обнаружено, что рассмотренная выше операция по существу не преобразует соединение A в севофлуран.
[Пример 2]
[0139]
(Стадия A)
Стандартное вещество соединения A (используемое в Примере 1) используют таким образом, что приготавливают "севофлуран, содержащий соединение A при концентрации 100 м.д.". Полученный севофлуран (1000 г) вводят в стеклянный дистилляционный танк. В дополнение к этому к нему добавляют 1% водный раствор гидрофосфата натрия (70 г), с последующей дистилляцией при обычном давлении с использованием дистилляционной колонны с 10 теоретическими ступенями при коэффициенте дефлегмации от 5 до 20.
[0140]
Дистиллят анализируют с помощью газовой хроматографии с FID и собирают в качестве "первой фракции дистилляции", при этом соединение A детектируется при концентрации 1 м.д. или более. Затем, когда соединение A детектируется на уровне ниже чем 1 м.д., дистиллят собирают в качестве "главной фракции дистилляции".
[0141]
В результате извлеченное количество "первой фракции дистилляции" составляет 267 г, в ней содержание соединения A составляет 341 м.д. В то же время извлеченное количество "главной фракции дистилляции" составляет 720 г, в ней не детектируется соединения A (меньше чем 1 м.д.) (выход сбора главной фракции дистилляции=72%).
[0142]
(Стадия 1a)
"Первая фракция дистилляции (содержание соединения A: 341 м.д.)", полученная выше (на стадии A) (240 г), вводится в автоклав из нержавеющей стали, и к ней добавляют "водный раствор HF (приготовленный посредством растворения 10 г безводного HF в 50 г воды)". Автоклав закрывают, с последующим перемешиванием с использованием мешалки (температура реакции=20°C-25°C).
[0143]
Реакцию прекращают через 5 часов после начала реакции. Органический слой в автоклаве извлекают и промывают водой. Затем органический слой анализируют с помощью газовой хроматографии. В результате, содержание соединения A составляет 123 м.д. Другими словами, содержание соединения A значительно уменьшается по прохождении 5 часов (доля преобразования: 63%). Общее количество остающегося органического слоя промывают "водным раствором гидроксида натрия" таким образом, что кислоты удаляются.
[0144]
(Стадия 2)
Общее количество органического слоя (после промывки), полученного выше (стадия 1a), вводят в дистиллятор из нержавеющей стали, к нему добавляют 17 г 1% водного раствора гидрофосфата натрия, с последующей дистилляцией при обычном давлении с использованием дистилляционной колонны с 10 теоретическими ступенями при коэффициенте дефлегмации от 5 до 20.
[0145]
Дистиллят анализируют с помощью газовой хроматографии с FID и собирают в качестве "первой фракции дистилляции", при этом соединение A детектируется при концентрации 1 м.д. или более. Затем, после того как подтверждается, что содержание соединения A меньше чем 1 м.д., дистиллят собирают в качестве "главной фракции дистилляции".
[0146]
В результате, извлекают 76 г "первой фракции дистилляции", в которой содержание соединения A составляет 330 м.д. При этом извлекают 128 г "главной фракции дистилляции", в которой соединение A не детектируется (его содержание меньше чем 1 м.д.). Как описано в "Сравнительном примере 1", ниже, обнаружено, что невозможно получить главную фракцию дистилляции посредством простой дистилляции первой фракции дистилляции, полученной выше (стадия A). В то же время на этой стадии (стадия 2), главная фракция дистилляции (в количестве, которое хоть и небольшое, но находится на значимом уровне) успешно извлекают. Это вероятно связано с тем, что содержание соединения A может быть уменьшено на рассмотренной выше стадии (стадия 1a).
[0147]
В дополнение к этому получают 31 г "донных остатков в танке" (отметим, что все компоненты, имеющие температуры кипения выше, чем температура кипения главной фракции дистилляции, считаются "донными остатками в танке"). "Донные остатки в танке" анализируют с помощью газовой хроматографии с FID. В результате "простой полиэфир 1", "простой полиэфир 2", "простой полиэфир 3" и севофлуран детектируются при концентрации 42%, 6%, 1% и 20%, соответственно (при этом соединение A не детектируется). В дополнение к этому, детектируемый севофлуран считается севофлураном, который не дистиллируется, но остается в танке во время дистилляции, он иной, чем севофлуран, который генерируется посредством реакции во время дистилляции.
[0148]
(Стадии 3 и 4)
"Донные остатки в танке" (31 г), полученные на стадии 2, вводятся в автоклав из нержавеющей стали, и к ним добавляют 98% серную кислоту (100 г) и фтористый водород (200 г). Смесь постепенно нагревают в течение 4 часов до 65°C.
[0149]
Пары, генерируемые во время реакции, захватываются с использованием водяной ловушки, и полученный органический слой промывают водой. Органическое вещество (26 г) извлекают.
[0150]
Полученное органическое вещество анализируют с помощью газовой хроматографии с FID. Это органическое вещество, как обнаружено, содержит севофлуран при концентрации 96,3%.
[0151]
Органическое вещество промывают водным раствором гидроксида натрия, с последующей дистилляцией при условиях, описанных на стадии 2. Соответственно, получают 14 г севофлурана (чистота: 99,9% или больше).
[0152]
Как описано выше, посредством осуществления стадии 3 и 4 в сочетании, можно дополнительно извлекать севофлуран высокой чистоты, который невозможно собрать посредством обычных технологий.
[Пример 3]
[0153]
(Стадия 1b)
В Примере 3 и в следующих далее примерах, реакция, соответствующая стадии 1b, осуществляется в малом масштабе (в масштабе одна десятая от Примера 2) в качестве модельного эксперимента, сходного со стадией 1a Примера 2 (с использованием малого герметизируемого реактора из нержавеющей стали).
[0154]
Сначала стандартное вещество соединения A и севофлуран смешивают таким образом, чтобы приготовить "севофлуран, содержащий соединение A при концентрации 340 м.д.". Этот севофлуран обозначается как реакционный материал в Примере 3 и в следующих далее примерах.
[0155]
"Водный раствор HF (приготовленный посредством растворения 1,0 г безводного HF в 5,0 г воды)" добавляют к 24 г "севофлурана, содержащего соединение A при концентрации 340 м.д.". Затем добавляют 1,0 г HFIP, и автоклав закрывают. Начинают перемешивание с использованием магнитной мешалки, и внутреннюю температуру поддерживают при 20°C-25°C.
[0156]
По прохождении 5 часов осуществляют газовую хроматографию с FID для определения. Доля преобразования соединения A оценивается как 73%. Когда добавляют HFIP, доля преобразования немного улучшается от 63% до 73%, по сравнению с Примером 2.
[Пример 4]
[0157]
Реакция, соответствующая стадии 1b, осуществляется посредством дополнительного добавления 1,0 г концентрированной серной кислоты при условиях, описанных на стадии 1b Примера 3.
[0158]
По прохождении 5 часов осуществляют газовую хроматографию с FID для определения. Доля преобразования соединения A оценивается как 71%. Другими словами, результаты Примера 4 не очень отличаются от результатов Примера 3. Обнаружено, что реакция не ингибируется даже в присутствии серной кислоты.
[Пример 5]
[0159]
Реакция, соответствующая стадии 1b, осуществляется при условиях, описанных на стадии 1b Примера 4, при температуре реакции 45°C.
[0160]
По прохождении 5 часов, осуществляют газовую хроматографию с FID для определения. Доля преобразования соединения A составляет 81%. Обнаружено, что повышение температуры может вызывать определенное увеличение скорости реакции.
[0161]
[Сравнительный пример 1]
"Стадию A" из Примера 2 повторяют в таком же масштабе при таких же условиях. В результате, извлекают 260 г "первой фракции дистилляции" (содержание соединения A=350 м.д.). Общее количество этой первой фракции дистилляции снова подвергают непосредственному воздействию дистилляции при обычном давлении (количество теоретических ступеней устанавливается равным 10 и коэффициент дефлегмации устанавливается при 5-20, как на "стадии A" Примера 2) без стадии 1с с добавлением 18 г 1% водного раствора гидрофосфата натрия.
[0162]
Однако детектируемый уровень соединения A не уменьшается до уровня "ниже предела детектирования (1 м.д.)" при рассмотренных выше условиях. В результате "главная фракция дистилляции" не извлекается. Другими словами, обнаружено, что невозможно извлечь севофлуран высокой чистоты посредством простого повторения дистилляции "первой фракции дистилляции" без стадии 1a.
[0163]
[Сравнительный пример 2]
Делаются попытки осуществления реакции, как описано выше, с использованием таких же реакционных материалов и такого же реакционного контейнера посредством приведения "соединения A (5 г) и безводного HF (20 г)" в контакт друг с другом без добавления "воды (100 г)", используемой в качестве реакционного материала в "Примере 1 (стадия 1)", выше, (с использованием 1,2-дихлорэтана (DCE) (15 г) в качестве вещества внутреннего стандарта).
[0164]
Отношение площадей пиков [соединение A]/[DCE] составляет 0,57 непосредственно перед началом реакции. Однако в результате определения отношения площадей пиков [соединение A]/[DCE] посредством газохроматографического анализа с FID через 5 часов, отношение площадей пиков [соединение A]/[DCE] остается почти неизменным (0,56). Таким образом, считается, что соединение A практически не взаимодействует при безводных условиях.
Промышленное применение
[0165]
Настоящее изобретение является преимущественным в том, что соединение A может преобразовываться в соединение X, которое может легко отделяться от севофлурана (стадия 1).
[0166]
В дополнение к этому, в другом варианте осуществления, настоящее изобретение является преимущественным в том, что можно давать возможность соединению A в "1-й органической жидкости" для селективного взаимодействия, с получением при этом "2-й органической жидкости", содержащей уменьшенное содержание соединения A или по существу не содержащей соединения A (стадия 1a).
[0167]
Кроме того, еще в одном из вариантов осуществления, настоящее изобретение является преимущественным в том, что можно получать севофлуран, по существу не содержащий соединения A, в значительно большем количестве, по сравнению со случаем неосуществления стадии 1a, с использованием в качестве исходных материалов, "1-й органической жидкости", что обычно является сложным для эффективного использования (стадия 1a и стадия 2).
[0168]
Кроме того, еще в одном варианте осуществления, настоящее изобретение является дополнительно преимущественным в том, что неожиданно является возможным получать севофлуран с использованием, в качестве исходных материалов, остатков (донные остатки в танке), остающихся после дистилляции на стадии 2 (стадия 1a и стадии 2-4).
[0169]
В соответствии с настоящим изобретением становится возможным получать севофлуран с использованием в качестве исходных материалов "севофлурана, содержащего соединение A (1-й органической жидкости)", что было сложным эффективно использовать, тем самым предлагается улучшенный способ получения севофлурана.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ДЛЯ ПРОМЫВКИ КОНТЕЙНЕРА ДЛЯ ХРАНЕНИЯ СЕВОФЛУРАНА И СПОСОБ ДЛЯ ХРАНЕНИЯ СЕВОФЛУРАНА | 2016 |
|
RU2685412C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1-ДИБРОМ-1-ФТОРЭТАНА | 2014 |
|
RU2670757C2 |
| АЗЕОТРОПНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ 2,3,3,3-ТЕТРАФТОРПРОПЕН И ФТОРИСТЫЙ ВОДОРОД, И ИХ ИСПОЛЬЗОВАНИЕ В СПОСОБАХ ПОЛУЧЕНИЯ 2,3,3,3-ТЕТРАФТОРПРОПЕНА | 2006 |
|
RU2422427C2 |
| ДВУХСТАДИЙНЫЙ СПОСОБ ПОЛУЧЕНИЯ 2-(ФТОРМЕТОКСИ)-1,1,1,3,3,3-ГЕКСАФТОРИЗОПРОПАНА (СЕВОФЛУРАНА) | 2007 |
|
RU2368597C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(ФТОРМЕТОКСИ)-1,1,1,3,3,3-ГЕКСАФТОРИЗОПРОПАНА (СЕВОФЛУРАНА) | 2024 |
|
RU2830201C1 |
| НОВОЕ АЛИЦИКЛИЧЕСКОЕ ДИОЛЬНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2646220C2 |
| СОЛЬ ПЕРФТОРКАРБОНОВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2453529C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКОКСИЛИРОВАННОГО СПИРТА | 2005 |
|
RU2380348C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ МОНОФТОРМЕТИЛОВЫХ ЭФИРОВ | 1998 |
|
RU2169724C2 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ФТОРИСТОГО ВОДОРОДА | 1998 |
|
RU2194007C2 |
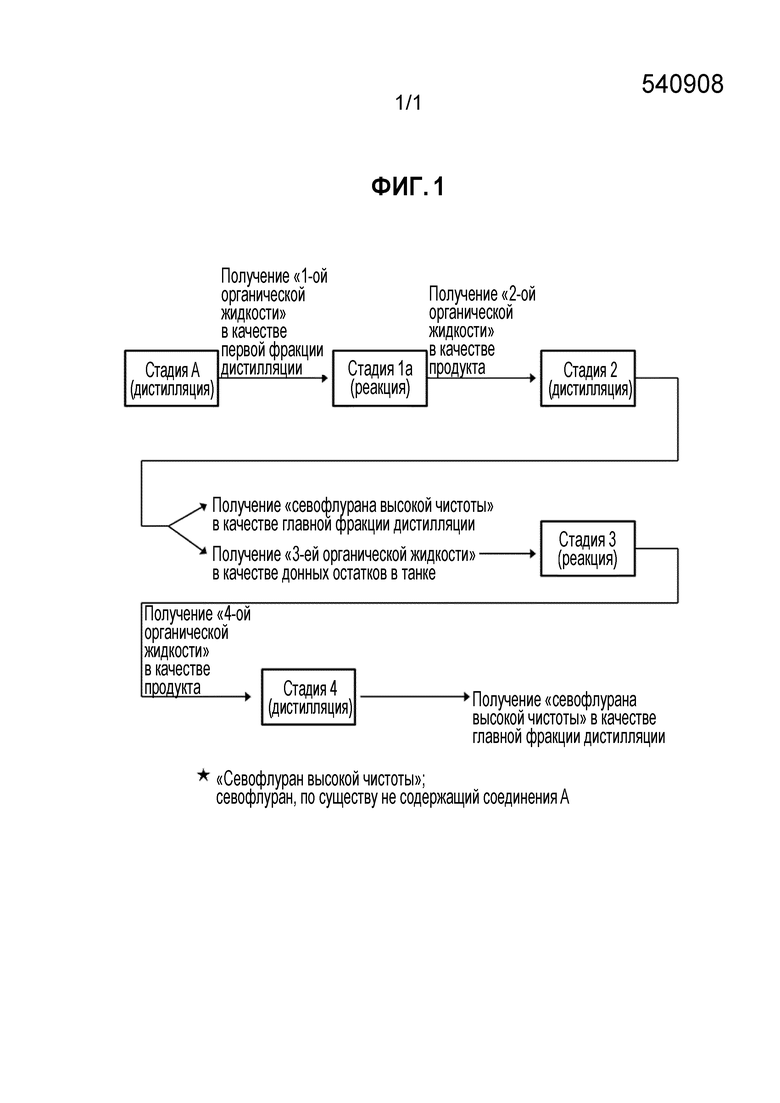
Настоящее изобретение направлено на удаление простого фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединение А) из севофлурана с целью получения севофлурана высокой чистоты. Один из объектов изобретения относится к способу получения севофлурана, по существу не содержащего соединения A, включающему следующие стадии: приведение композиции, содержащей фтористый водород и воду при массовом отношении от 1:1 до 1:30, в контакт с органической жидкостью, содержащей севофлуран и соединение A, с получением органической жидкости, содержащей соединение A в количестве, которое меньше, чем его количество в исходной органической жидкости, и дистилляцию полученной органической жидкости в присутствии ингибитора деградации. 4 н. и 8 з.п. ф-лы, 1 ил., 7 пр.
1. Способ уменьшения количества фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединение A), включающий следующую стадию:
стадия 1 приведения соединения A в композиции в контакт с другой композицией, содержащей фтористый водород и воду при массовом отношении от 1:1 до 1:30.
2. Способ получения 2-й органической жидкости, которая представляет собой (i) органическую жидкость, содержащую севофлуран и фторметил-1,1,3,3,3-пентафторизопропениловый эфир (соединение A) в количестве, которое меньше, чем количество в 1-й органической жидкости, содержащей севофлуран и соединение А; или (ii) органическую жидкость, содержащую севофлуран и по существу не содержащую соединения A, включающий следующую стадию:
стадия 1a приведения жидкости (1-й органической жидкости), содержащей севофлуран, и простого фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединение A) в контакт с композицией, содержащей фтористый водород и воду при массовом отношении от 1:1 до 1:30, с получением 2-й органической жидкости.
3. Способ по п.2, в котором температура во время контакта составляет 0°C-60°C.
4. Способ по п.2 или 3, в котором контакт осуществляется при совместном существовании с гексафторизопропиловым спиртом (HFIP).
5. Способ получения севофлурана, по существу не содержащего фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединение A), включающий следующую стадию:
стадия 1 получения 2-й органической жидкости с помощью способа по любому из пп.2-4; и
стадия 2 дистилляции 2-й органической жидкости (i) в присутствии ингибитора деградации с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
6. Способ по п.5, в котором ингибитор деградации, используемый на стадии 2, представляет собой по меньшей мере одно соединение, выбранное из группы, состоящей из NaHCO3, Na2B4O7, H3BO4, C6H4(COOK)(COOH), Na2SO3, Na2HPO4, CH3COONa и Na3PO4.
7. Способ по п.5 или 6, дополнительно включающий следующую стадию:
стадия 3 приведения безводного фтористого водорода и ускорителя реакции в контакт с остатками (3-й органической жидкостью), остающимися после дистилляции на стадии 2, с получением при этом жидкости (4-й органической жидкости), в которой, по меньшей мере, часть компонента в 3-й органической жидкости преобразуется в севофлуран.
8. Способ по п.7, дополнительно включающий следующую стадию:
стадия 4 дистилляции 4-й органической жидкости с получением севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
9. Способ по любому из пп.2-8, в котором 1-ю органическую жидкость получают в качестве первой фракции дистилляции на следующей стадии:
стадия A дистилляции севофлурана в присутствии ингибитора деградации с тем, чтобы собрать первую фракцию дистилляции.
10. Способ по п.9, в котором ингибитор деградации, используемый на стадии A, представляет собой по меньшей мере одно соединение, выбранное из группы, состоящей из NaHCO3, Na2B4O7, H3BO4, C6H4(COOK)(COOH), Na2SO3, Na2HPO4, CH3COONa и Na3PO4.
11. Способ получения севофлурана, по существу не содержащего фторметил-1,1,3,3,3-пентафторизопропенилового эфира (соединение A), включающий следующие стадии:
стадия 1b приведения жидкости (1-й органической жидкости), содержащей севофлуран и соединение A, в контакт с композицией, содержащей фтористый водород (HF) и воду при массовом отношении от 1:1 до 1:30, в присутствии гексафторизопропилового спирта (HFIP) при 0°C-60°C с получением 2-й органической жидкости, которая представляет собой (i) органическую жидкость, содержащую севофлуран и соединение A в количестве, которое меньше, чем количество в 1-й органической жидкости; или (ii) органическую жидкость, содержащую севофлуран и по существу не содержащей соединения A; и
стадия 2 дистилляции 2-й органической жидкости (i) в присутствии ингибитора деградации с получением севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
12. Способ по п.11, дополнительно включающий следующие стадии:
стадия 3 приведения остатков (3-й органической жидкости), остающихся после дистилляции на стадии 2, в контакт с безводным фтористым водородом и ускорителем реакции с получением жидкости (4-й органической жидкости), в которой по меньшей мере часть 3-й органической жидкости преобразуется в севофлуран; и
стадия 4 дистилляции 4-й органической жидкости в присутствии ингибитора деградации с получением при этом севофлурана, по существу не содержащего соединения A, в качестве главной фракции дистилляции.
| WO 1999044978 A1, 10.09.1999 | |||
| Способ получения алкенбромгидринов с - с | 1977 |
|
SU701985A1 |
| US 4328376 A1, 04.05.1982 | |||
| Преобразователь двоично-десятичногоКОдА B КОд ВОСьМиСЕгМЕНТНОгОиНдиКАТОРА | 1979 |
|
SU822172A1 |
| EA 200701073 A1, 28.12.2007. | |||

