Настоящее изобретение относится к водным дисперсиям сополимеров, и к их получению и применению, особенно в области автомеханической окончательной отделки.
Предыдущий уровень техники раскрывает сополимеры, имеющие структуру ядро/оболочка.
Документ ЕР 1218434 В1 раскрывает привитой сополимер на основе полиуретана и их применение для получения материалов покрытия, клеящих веществ и герметизирующих составов.
Документы ЕР 1185568 В1 и ЕР 1173491 В1 описывают сополимеры, имеющие структуру ядро/оболочка, внешняя часть которого полностью сшита, где ядро состоит из полиакрилата и оболочка может состоять из полиуретана.
Документ ЕР 1330480 В1 описывает сополимеры, где ядро состоит из сульфид-содержащих полиуретанов и оболочка из полиакрилата.
Сополимеры, известные из предыдущего уровня техники, находятся в форме водных дисперсий и могут быть применены для получения водных материалов покрытия, особенно для водоразбавляемых базовых покрытий.
Эти водоразбавляемые базовые покрытия могут быть применены в так называемом методе базовое покрытие/самовосстанавливающееся покрытие, метод окраски по влажному нижнему покрытию без предварительной сушки, для цветного покрытия и/или покрытия для эффекта многослойной красочной системы.
Известные методы нуждаются в улучшении, особенно относительно возникновения крошечных отверстий, видимых как очень маленькие отверстия в самовосстанавливающемся покрытии и основе покрытия. Способность скрывать мелкие отметины и адгезия в случае многослойной окончательной отделки являются не надлежащими в случае покрытий известных из предыдущего уровня техники.
Задача, которая лежит в основе настоящего изобретения, таким образом, состоит в предоставлении водной дисперсии сополимера типа, описанного выше, которая может быть применена для связующего вещества слоя цветного и/или эффекта многослойной красочной системы, улучшенной по сравнению с предыдущим уровнем техники.
Более конкретно, у этого слоя должно быть только очень немного крошечных отверстий, если таковые имеются, и/или более высокий предел возникновения точечных проколов. Предел возникновения точечных проколов относится к толщине сухой базовые покрытия, выше которой появляются крошечные отверстия. В то же время способность скрывать мелкие отметины и адгезия в случае многослойной окончательной отделки, например, в форме повторной окончательной отделки, должны быть улучшены.
Было найдено, что, неожиданно, сложная задача, лежащая в основе этого изобретения, достигнута, обеспечив
водную дисперсию, включающую, по меньшей мере один сополимер, сополимер, который получают следующим путем
(I) первоначально загружают водную дисперсию, по меньшей мере, одного полиуретана, и затем
(II) полимеризуют смесь олефиново ненасыщенных мономеров в присутствии полиуретана из (I),
где
(а) применяют водорастворимый инициатор,
(б) дозированное добавление олефиново ненасыщенных мономеров проводят таким образом, что концентрация 6.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров, в растворе реакции не превышена в ходе всей продолжительности реакции, и
(в) смесь олефиново ненасыщенных мономеров включает, по меньшей мере, один полиолефиново ненасыщенный мономер.
Изобретение также относится к способу получения изобретенных дисперсий, к применению этих водных дисперсий в материалах покрытия, особенно материалах базовые покрытия, к водоразбавляемым материалам базовые покрытия, включающим вышеупомянутые дисперсии, и к способу получения многослойных красочных систем.
Крошечные отверстия представляют собой очень маленькие отверстия в самовосстанавливающемся покрытии и основе покрытия, которые видимы в готовом покрытии. Они представляют собой один из наиболее распространенных поверхностных дефектов при нанесении аэрозольных красок. Дефекты, называемые крошечными отверстиями в описании и доступных примерах, которые следуют, характеризуются воронкообразными отверстиями, которые продолжаются в цилиндрические трубки в порядке размера отпечатков пузырьков, которые имеют средний диаметр открытия воронки приблизительно 300-700 мкм и средний диаметр трубки приблизительно 15-60 мкм, и и проходят через самовосстанавливающееся покрытие и базовое покрытие до достижения покрытия ниже. Они могут быть результатом выходящих газов, таких как заключенный воздух или испаряющиеся жидкости.
Термин "(мет)акрилат" должно относится в этом документе далее и к акрилату, и к метакрилату.
Сополимер в контексте настоящего изобретения относится к частицам полимера, сформированным из разных полимеров. Он явным образом включает оба полимера, соединенных ковалентно друг с другом и те, в которых различные полимеры связаны друг с другом адгезией. Комбинации двух типов связываний также охвачены этим определением.
Изобретенные дисперсии и способ получения этих дисперсий характеризуются стадиями их получения.
На первой стадии получения, первоначально загружают водную дисперсию полиуретановой смолы.
Подходящие насыщенные или ненасыщенные полиуретановые смолы описаны, например, в
- Заявка на патент Германии DE 19948004 А1, стр. 4 линия 19 - стр. 11 линия 29 (полиуретановый преполимер В1)
- Европейская заявка на патент ЕР 0228003 А1, стр. 3 линия 24 - стр. 5 линия 40,
- Европейская заявка на патент ЕР 0634431 А1, стр. 3 линия 38 - стр. 8 линия 9, или
- международная заявка на патент WO 92/15405, стр. 2 линия 35 - стр. 10 линия 32.
Для получения полиуретановой смолы, предпочтение отдают применению алифатических, циклоалифатических, алифатически-циклоалифатических, ароматических, алифатически-ароматических и/или циклоалифатически-ароматических полиизоцианатов, известных специалисту в данной области техники. Особое предпочтение отдают алифатическим и алифатически-циклоалифатическим полиуретановым смолам.
Спиртовые компоненты, применяемые для получения полиуретановых смол, предпочтительно представляют собой насыщенные и ненасыщенные полиолы, известные специалисту в данной области техники, и необязательно, в незначительных количествах, также моноспирты. Особенно диолы и необязательно, в незначительных количествах, триолы применяют для появления отраслей. Примеры подходящих полиолов представляют собой насыщенные или олефиново ненасыщенные полиэстер полиолы и/или полиэтер полиолы. Применяемые полиолы представляют собой особенно полиэстер полиолы, особенно такие, которые имеют среднюю молекулярную массу от 400 до 5000 г/моль (измеренную посредством осмометрии с использованием давления паров).
Предпочтительно, полиуретан, первоначально загруженный в водную дисперсию, представляет собой гидрофильно стабилизированный полиуретан. Для гидрофильной стабилизации или для увеличения способности диспергироваться в водной среде, полиуретановая смола может содержать особые ионные группы и/или группы, которые могут быть преобразованы в ионные группы (потенциально ионные группы). Такие полиуретановые смолы названы в контексте настоящего изобретения как ионно гидрофильно стабилизированные полиуретановые смолы. Аналогично могут присутствовать неионогенные гидрофильньно модифицированные группы. Предпочтение отдают, тем не менее, ионно гидрофильно стабилизированным полиуретанам. Более определенно, модифицируемые группы представляют собой
- функциональные группы, которые могут быть преобразованы в катионы при помощи нейтрализующих агентов и/или кватернизующих агентов, и/или катионные группы (катионная модификация)
или
- функциональные группы, которые могут быть преобразованы в анионы при помощи нейтрализующих агентов, и/или анионные группы (анионная модификация)
или
- неионогенные гидрофильный группы (неионогенная модификация)
или
- комбинации вышеупомянутых групп.
Как известно специалисту в данной области техники, функциональные группы для катионной модификации представляют собой, например, первичные, вторичные и/или третичные аминогруппы, вторичные сульфидные группы и/или третичные фосфиновые группы, особенно третичные аминогруппы и вторичные сульфидные группы (функциональные группы, которые могут быть преобразованы в катионные группы при помощи нейтрализующих агентов и/или кватернизующих агентов). Упоминание дополнительно должно быть сделано к катионным группам, полученным из вышеупомянутых функциональных групп, применяя нейтрализующие агенты и/или кватернизующие агенты, известные специалисту в данной области техники, такие как первичные, вторичные, третичные и/или четвертичные аммониевые группы, третичные сульфониевые группы и/или четвертичные фосфониевые группы, особенно четвертичные аммониевые группы и третичные сульфониевые группы.
Как известно, функциональные группы для анионной модификации представляют собой, например, группы карбоновой кислоты, сульфоновой кислоты и/или фосфоновой кислоты, особенно группы карбоновой кислоты (функциональные группы, которые могут быть преобразованы в анионные группы при помощи нейтрализующих агентов), и анионные группы, полученные из вышеупомянутых функциональных групп, применяя нейтрализующие агенты, известные специалисту в данной области техники, такие как карбоксилатные, сульфонатные и/или фосфонатные группы.
Функциональные группы для неионогенной гидрофильной модификации представляют собой предпочтительно поли(оксиалкилен) группы, особенно поли(оксиэтилен) группы.
Ионные гидрофильньные модификации могут быть введены в полиуретановую смолу при помощи мономеров, содержащих ионные или потенциально ионные группы. Неионогенные модификации вводят как боковые или концевые группы в молекулы полиуретана, например, через включение поли(этиленоксид) полимеров. Гидрофильные модификации введены, например, применяя соединения, содержащие, по меньшей мере, одну группу реакционноспособную к изоцианатным группам, предпочтительно, по меньшей мере, одну гидроксильную группу. Для введения ионной модификации, можно применять мономеры, которые, так же как и модифицируемые группы содержат, по меньшей мере, одну гидроксильную группу. Для введения неионогенных модификаций, предпочтение отдают применению простых полиэфиров диолов и/или алкокси поли(оксиалкилен) спиртов, известных специалисту в данной области техники.
Предпочтение отдают добавлению, по меньшей мере, одного растворителя к первоначально загружаемой полиуретановой дисперсии, растворитель является смешивающимся в любом отношении с водой и в любом отношении со смесью олефиново ненасыщенных мономеров. Особенно подходящими растворителями являются N-метилпирролидон, N-этилпирролидон и простые эфиры спирты, такие как особенно метоксипропанол.
Как следующая стадия получения, проводится полимеризация смеси олефиново ненасыщенных мономеров в присутствии полиуретана методами так называемой свободнорадикальной эмульсионной полимеризации в присутствии, по меньшей мере, одного инициатора полимеризации.
Применяемый инициатор полимеризации должен быть водорастворимым инициатором. Примерами подходящих инициаторов являются пероксодисульфат калия, пероксодисульфат натрия или пероксодисульфат аммония, и также перекись водорода, трет-бутил гидропероксид, 2,2'-азобис(2-амидоизопропан)дигидрохлорид, 2,2'-азобис(N,N'-диметиленизобутирамидин) дигидрохлорид или 2,2'-азобис(4-цианопентановая кислота). Инициаторы применяют или отдельно, или в смеси, например, смеси перекиси водорода и персульфата натрия.
Также возможно применять известные окислительно-восстановительные системы инициатора в качестве инициаторов полимеризации. Такие окислительно-восстановительные системы инициатора содержат, по меньшей мере, одно пероксид-содержащее соединение в комбинации с окислительно-восстановительным соинициатором, например, восстанавливающие соединения серы, например, бисульфиты, сульфиты, тиосульфаты, дитиониты или тетратионаты щелочных металлов и аммониевые соединения, натрий гидроксиметансульфинат дигидрат и/или тиомочевина. Например, возможно применять комбинации пероксодисульфатов с гидросульфитами щелочных металлов или аммония, например, пероксодисульфат аммония и дисульфит аммония. Массовое соотношение пероксид-содержащих соединений к окислительно-восстановительным соинициаторам предпочтительно составляет от 50:1 до 0.05:1. В комбинации с инициаторами или окислительно-восстановительными системами инициатора, дополнительно возможно применять катализаторы на основе переходных металлов, например, соли железа, соли никеля, соли кобальта, соли марганца, соли меди, ванадиевые соли или соли хрома, такие как сульфат железа(II), хлорид кобальта(II), сульфат никеля(II), хлорид меди(I), ацетат марганца(II), ацетат ванадия(III), хлорид марганца(II). В пересчете на мономеры, эти соли переходных металлов применяют обычно в количествах 0.1-1000 млн.д.. Например, возможно применять комбинации перекиси водорода с солями железа(II), например, 0.5-30% перекись водорода и 0.1-500 млн.д. соли Мора.
Инициаторы предпочтительно применяют в количестве 0.05-20 мас. %, предпочтительно 0.05-10 мас. %, более предпочтительно 0.1-5 мас. %, в пересчете на общую массу олефиново ненасыщенных мономеров.
Результат применения растворимого в воде инициатора состоит в том, что олефиново ненасыщенные мономеры, которые добавляют к первоначально загружаемой водной дисперсии, могут реагировать сразу же, чтобы предоставить олигомеры. Эти олигомеры обладают более низкой тенденцией проникновения в частицы полиуретана в первоначально заряженной дисперсии, чем более маленькие мономеры.
Полимеризация соответственно проводится при температуре 0-160°С, предпочтительно 60-95°С.
В этом контексте, предпочтение отдают работе с исключением кислорода, предпочтительно в потоке азота. В целом полимеризацию выполняют при стандартном давлении, но также возможно применение более низких давлений или более высоких давлений, особенно когда применяют температуры полимеризации выше точки кипения мономеров и/или растворителей.
Изобретенные полимеры получают свободнорадикальной водной эмульсионной полимеризации, и возможно добавлять сурфактанты или защитные коллоиды к реакционной среде. Список подходящих эмульгаторов и защитных коллоидов может быть найден, например, в Houben Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], volume XIV/1 Makromolekulare Stoffe [Macromolecular substances], Georg Thieme Verlag, Stuttgart 1961, p. 411 ff.
Важным фактором для получения изобретенной водной дисперсии является регулирование условий реакции полимеризации смеси олефиново ненасыщенных мономеров в присутствии полиуретана. Это выполняют способом "точнодозированной полимеризации".
Точнодозированную полимеризацию в контексте настоящего изобретения рассматривают как такую, которая представляет собой эмульсионную полимеризацию, в которой содержание остаточных мономеров в растворе реакции минимизируют на протяжении всей реакции, обозначая, что олефиново ненасыщенные мономеры дозируют таким образом, что концентрация 6.0 мас. %, предпочтительно 5.0 мас. %, более предпочтительно 4.0 мас. %, в растворе реакции, в каждом случае в пересчете на общее количество олефиново ненасыщенных мономеров, не превышена в ходе всей продолжительности реакции.
Концентрация мономеров в растворе реакции может быть определена здесь при помощи газовой хроматографии.
Концентрация мономеров в растворе реакции, также упоминаемые здесь как свободные мономеры, может регулироваться различными путями.
Одно средство поддержания концентрации свободных мономеров на низком уровне состоит в том, чтобы выбрать очень низкую скорость дозирования смеси олефиново ненасыщенных мономеров. Когда скорость дозированного добавления достаточно низкая все мономеры могут реагировать очень быстро, сразу как только они находятся в растворе реакции, это дает возможность гарантировать, что концентрация свободных мономеров минимизируется.
Так же как и скорость дозирования, важно, чтоб в растворе реакции всегда присутствовало достаточно свободных радикалов, таким образом, чтоб дозировано введенные мономеры могли каждый реагировать очень быстро. С этой целью условия реакции предпочтительно должны быть выбраны таким образом, что подача инициатора уже начата перед началом дозированного добавления олефиново ненасыщенных мономеров. Предпочтение отдают началу дозированного добавления, по меньшей мере, на 5 минут ранее, более предпочтительно, по меньшей мере, на 10 минут ранее. Предпочтительно, по меньшей мере, 10 мас. %, более предпочтительно по меньшей мере 20 мас. %, наиболее предпочтительно по меньшей мере 30 мас. %, инициатора, в каждом случае в пересчете на общее количество инициатора, добавляют перед началом дозированного добавления олефиново ненасыщенных мономеров.
Количество инициатора является важным фактором для достаточного наличия свободных радикалов в растворе реакции. Количество инициатора должно быть выбрано таким образом, что достаточно свободных радикалов доступны в любое время, таким образом, что дозировано введенные мономеры могут реагировать. Если количество инициатора увеличивается, также возможно, что реагируют более высокие количества мономеров в то же время.
Дополнительным фактором, который определяет скорость реакции, является структура мономеров.
Концентрация свободных мономеров, таким образом, может регулироваться посредством взаимосвязи количества инициатора, скорость добавления инициатора, скорость добавления мономеров, и через выбор мономеров. И замедление дозированного добавления и увеличение количества инициатора, а также раннее начало добавления инициатора, служите цели поддержания концентрации свободных мономеров ниже упомянутых выше пределов.
В любой момент реакции, может быть определена концентрация свободных мономеров при помощи газовой хроматографии. Типичные параметры для определения следующие: 50 м капиллярная колонка с диоксидом кремния с фазой полиэтиленгликоля или 50 м капиллярная колонка с диоксидом кремния с фазой полидиметилсилоксана, газ-носитель гелий, инжектор для ввода проб с делением потока 150°С, температурный режим термостата 40-220°С, пламенно-ионизационный детектор, температура детектора 275°С, внутренний стандарт изобутилакрилат.
Так как любая концентрация свободных мономеров, которая близко подходит к пределу для точнодозированной полимеризации, должна быть определена этим анализом, например, из-за очень низкого уровня реакционно-способных олефиново ненасыщенные мономеры, вышеупомянутые параметры могут быть использованы для контроля реакции. В этом случае, например, может быть уменьшена скорость дозирования мономеров или увеличено количество инициатора.
Подходящие олефиново ненасыщенные мономеры могут быть моно- или полиолефиново ненасыщенные.
Примеры подходящих моноолефиново ненасыщенных мономеров включают моноолефиново ненасыщенные мономеры на основе (мет)акрилата, виниловые моноолефиново ненасыщенные мономеры, альфа-бета-ненасыщенные карбоновые кислоты, и аллиловые соединения.
Моноолефиново ненасыщенные мономеры на основе (мет)акрилата могут, например, представлять собой (мет)акриловую кислоту и сложные эфиры, нитрилы, или амиды (мет)акриловой кислоты.
Предпочтение отдают сложным эфирам (мет)акриловой кислоты, имеющей олефиново ненасыщенный радикал R.
 или
или 
Радикал R может быть алифатическим или ароматическим. Радикал R предпочтительно является алифатическим.
Радикал R может представлять собой, например, алкильный радикал, или содержать гетероатомы. Примерами радикалов R, содержащих гетероатомы, являются простые эфиры. Радикал R предпочтительно представляет собой алкильный радикал.
Если R представляет собой алкильный радикал, он может быть линейным, разветвленным или циклическим алкильным радикалом. Во всех трех случаях, алкильные радикалы могут представлять собой незамещенные алкильные радикалы или алкильные радикалы, замещенные функциональными группами. Алкильный радикал предпочтительно имеет 1-20, более предпочтительно 1-10, атомов углерода.
Особенно преимущественно подходящими мононенасыщенными сложными эфирами (мет)акриловой кислоты, имеющей незамещенный алкильный радикал, являются метил (мет)акрилат, этил (мет)акрилат, пропил (мет)акрилат, изопропил (мет)акрилат, н-бутил (мет)акрилат, изобутил (мет)акрилат, трет-бутил (мет)акрилат, амил (мет)акрилат, гексил (мет)акрилат, этилгексил (мет)акрилат, 3,3,5-триметилгексил (мет)акрилат, стеарил (мет)акрилат, лаурил (мет)акрилат, циклоалкил (мет)акрилаты, такие как циклопентил (мет)акрилат, изоборнил (мет)акрилат и циклогексил (мет)акрилат, очень особое предпочтение отдают н- и трет-бутил (мет)акрилату и метил метакрилату.
Подходящие мононенасыщенные сложные эфиры (мет)акриловой кислоты, имеющей замещенный алкильный радикал, предпочтительно, могут представлять собой замещенные одной или несколькими гидроксильными группами.
Особенно преимущественно подходящие мононенасыщенные сложные эфиры (мет)акриловой кислоты, имеющей алкильный радикал, замещенный одной или несколькими гидроксильными группами, представляют собой 2-гидроксиэтил (мет)акрилат, 2-гидроксипропил (мет)акрилат, 3-гидроксипропил (мет)акрилат, 3-гидроксибутил (мет)акрилат и 4-гидроксибутил (мет)акрилат.
Виниловые мононенасыщенные мономеры могут представлять собой мономеры, имеющие олефиново ненасыщенный радикал R' на виниловой группе.

Радикал R' может быть алифатическим или ароматическим, предпочтение отдают ароматическим радикалам.
Радикал R' радикал может представлять собой гидрокарбильный радикал, или содержать гетероатомы.
Примеры радикалов R', содержащих гетероатомы, представляют собой простые эфиры, сложные эфиры, амиды, нитрилы и гетероциклы.
Радикал R' предпочтительно представляет собой гидрокарбильный радикал.
Если R' представляет собой гидрокарбильный радикал, он может быть замещен гетероатомами или незамещен, предпочтение отдают незамещенным радикалам.
Радикал R' предпочтительно представляет собой ароматический гидрокарбильный радикал.
Особенно предпочтительными виниловыми олефиново ненасыщенными мономерами являются винилароматические углеводороды, особенно винилтолуол, альфа-метилстирол и особенно стирол.
Когда присутствуют гетероатомы, предпочтение отдают олефиново ненасыщенным мономерам, таким как акрилонитрил, метакрилонитрил, акриламид, метакриламид, N,N-диметилакриламид, винил ацетат, винил пропионат, винил хлорид, N-винилпирролидон, N-винилкапролактам, N-винилформамид, N-винилимидазол и N-винил-2-метилимидазолин.
Примеры подходящих полиолефиново ненасыщенных мономеров включают сложные эфиры (мет)акриловой кислоты, имеющие олефиново ненасыщенный радикал R'', и аллиловые простые эфиры моно- или многоатомных спиртов. Радикал R'' может представлять собой аллильный радикал или (мет)акриловий сложный эфир.
 или
или 
Предпочтительные полиолефиново ненасыщенные мономеры включают этиленгликоль ди(мет)акрилат, 1,2-пропиленгликоль ди(мет)акрилат, 1,3-пропиленгликоль ди(мет)акрилат, бутан-1,4-диол ди(мет)акрилат, неопентилгликоль ди(мет)акрилат, 3-метилпентандиол ди(мет)акрилат, диэтиленгликоль ди(мет)акрилат, триэтиленгликоль ди(мет)акрилат, тетраэтиленгликоль ди(мет)акрилат, дипропиленгликоль ди(мет)акрилат, трипропиленгликоль ди(мет)акрилат, гександиол ди(мет)акрилат и аллил (мет)акрилат.
К тому же, предпочтительные полиолефиново ненасыщенные соединения включают акриловие и метакриловие сложные эфиры спиртов, имеющие более, чем 2 группы ОН, например, триметилолпропан три(мет)акрилат или глицерил три(мет)акрилат, а также триметилолпропан ди(мет)акрилат моноаллиловый простой эфир, триметилолпропан (мет)акрилат диаллиловый простой эфир, пентаэритритол три(мет)акрилат моноаллиловый простой эфир, пентаэритритол ди(мет)акрилат диаллиловый простой эфир, пентаэритритол (мет)акрилат триаллиловый простой эфир, триаллилсахароза и пентааллилсахароза.
Особое предпочтение отдают применению аллил метакрилата.
Смесь олефиново ненасыщенных мономеров включает, по меньшей мере, один полиолефиново ненасыщенный мономер. Смесь олефиново ненасыщенных мономеров предпочтительно также включает один или несколько мононенасыщенных сложных эфиров (мет)акриловой кислоты, имеющих незамещенный алкильный радикал.
Смесь олефиново ненасыщенных мономеров содержит предпочтительно 0.1-6.0 моль %, более предпочтительно 0.1-2.0 моль %, наиболее предпочтительно 0.1-1.0 моль %, полиолефиново ненасыщенных мономеров.
Смесь олефиново ненасыщенных мономеров содержит предпочтительно 0.1-6.0 моль %, более предпочтительно 0.1-2.0 моль %, наиболее предпочтительно 0.1-2.0 моль %, аллил метакрилата. Более предпочтительно, помимо аллил метакрилата, дополнительные полиолефиново ненасыщенные мономеры не присутствуют в смеси.
Смесь олефиново ненасыщенных мономеров содержит предпочтительно <10.0 мас. %, более предпочтительно <5.0 мас. %, винилароматических углеводородов, в пересчете на общее количество олефиново ненасыщенных мономеров. Наиболее предпочтительно винилароматические углеводороды не присутствуют в смеси олефиново ненасыщенных мономеров.
В предпочтительном варианте осуществления, смесь олефиново ненасыщенных мономеров содержит:
- 98.0-99.5 мас. % одного или нескольких мононенасыщенных сложных эфиров (мет)акриловой кислоты, имеющих незамещенные алкильные радикалы, где алкильные радикалы имеют длину 1-10 атомов углерода, и
- 0.5-2.0 мас. % одного или нескольких полиненасыщенных сложных эфиров (мет)акриловой кислоты,
в каждом случае в пересчете на общее количество олефиново ненасыщенных мономеров.
Предпочтительно, к смеси олефиново ненасыщенных мономеров добавляют, по меньшей мере, один растворитель, растворитель является смешивающимся в любом отношении с водой и в любом отношении со смесью олефиново ненасыщенных мономеров. Особенно подходящие растворители представляют собой N-метилпирролидон, N-этилпирролидон и простые эфиры спирты, такие как особенно метоксипропанол.
В результате описанного способа получения сополимеры в изобретенной водной дисперсии имеют структуру ядро/оболочка, которая может быть достигнута посредством определенного процесса получения. Эта структура ядро/оболочка характеризуется ядром, включающим, по меньшей мере, один полиуретан, и оболочкой, включающей, по меньшей мере, полимер, который был получен полимеризацией олефиново ненасыщенных мономеров.
Описанная структура ядро/оболочка достигается через определенные условия реакции точно дозированной полимеризации. В ходе всей продолжительности реакции, нет больших количеств олефиново ненасыщенных мономеров, которые могут проникнуть в частицы полиуретана, при наличии первоначальной загрузки полиуретана. На основании постоянного присутствия свободных радикалов во время добавления мономера в водной фазе, которые обеспечены растворимым в воде инициатором, олигомеры сразу формируются при добавлении, которые больше не могут проникать в полиуретан. Тогда они полимеризируются на поверхности полиуретана.
В предпочтительном варианте осуществления, массовое соотношение ядра к оболочке составляет 80:20-20:80, более предпочтительно 60:40-40:60.
Микроструктура сополимеров может быть исследована при помощи трансмиссионной электронной микроскопии: с этой целью в полимер необходимо включить структуры, которые могут быть сделаны поддающимися противопоставлению, предпочтительно посредством соединений тяжелых металлов. Предпочтительно это достигают во время свободнорадикальной сополимеризации включением последовательностей полидиена в сополимер, предпочтение отдают сополимеризации изопрена или бутадиена с олефиново ненасыщенными соединениями и аллиловыми соединениями.
Для анализа из водной дисперсии должна быть сформирована пленка полимера, которая, после противопоставления в атмосфере обогащенной тетраоксидом осмия или рутения, может быть исследована электронной микроскопией.
Сополимеры предпочтительно имеют размер частиц (z средняя) 60-130 нм, более предпочтительно 70-115 нм, измеренный посредством фотокорреляционной спектроскопии при помощи Malvern Nano S90 (от Malvern Instruments) при 25±1°С. Блок, оборудованный 4 мВт He-Ne лазером при длине волны 633 нм, охватывает диапазон размеров от 1 до 3000 нм.
Сополимеры могут быть сшитыми. Содержание геля изобретенной водной дисперсии предпочтительно составляет 40-97 мас. %, более предпочтительно 75-90 мас. %, в каждом случае в пересчете на твердые частицы в дисперсии.
Содержание геля может быть определено гравиметрически, высушиванием дисперсии замораживанием, определением общей массы полимера, высушенного замораживанием, и затем экстракцией полимера в избытке тетрагидрофурана (соотношение тетрагидрофурана к полимеру, высушенному замораживанием = 300:1) при 25°С в течение 24 часов. Нерастворимую фракцию удаляли и высушивали в сушильном шкафу с циркуляцией воздуха при 50°С в течение 4 часов. После этого, высушенную нерастворимую фракцию взвесили и эту часть сформировали с общей массой полимера, высушенного замораживанием. Полученное значение соответствует содержанию геля.
Средняя молярная масса сополимеров предпочтительно составляет 3*107 г/моль - 8.5*109 г/моль, среднюю молярную массу определяли при помощи рассеяния лазерного излучения под малым углом.
Кислотное число сополимеров составляет 0-220 мг KOH/г твердой смолы, предпочтительно 0-40 мг KOH/г твердой смолы, более предпочтительно 0-25 мг KOH/г твердой смолы. Число ОН составляет меньше, чем 70 и предпочтительно меньше, чем 20 мг KOH/г твердой смолы.
Кислотное число может быть определено на основе DIN EN ISO 2114 в гомогенном растворе THF/вода (9 об. частей THF и 1 об. часть дистиллированной воды) с этаноловым раствором гидроксида калия.
Число ОН может быть определено на основе R.-P.  , R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), при помощи уксусного ангидрида в присутствии 4-диметиламинопиридина как катализатора в растворе тетрагидрофурана (THF)/диметилформамида (DMF) при комнатной температуре, с полным гидролизом оставшегося избытка уксусного ангидрида после ацетилирования и потециометрическим обратным титрованием уксусной кислоты спиртовым раствором гидроксида калия.
, R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), при помощи уксусного ангидрида в присутствии 4-диметиламинопиридина как катализатора в растворе тетрагидрофурана (THF)/диметилформамида (DMF) при комнатной температуре, с полным гидролизом оставшегося избытка уксусного ангидрида после ацетилирования и потециометрическим обратным титрованием уксусной кислоты спиртовым раствором гидроксида калия.
Выгодно применять изобретенную дисперсию для получения материалов покрытия, особенно для получения базовых покрытий.
Настоящая заявка также относится к водоразбавляемому базовому покрытию, включающему изобретенную дисперсию.
Водоразбавляемое базовое покрытие обычно включает красящие пигменты и/или пигменты оптических эффектов.
Такие красящие пигменты и пигменты для эффекта описаны для специалиста в данной области техники, например, в  Lacke und Druckfarben [
Lacke und Druckfarben [ Dictionary of Coating Materials and Printing Inks], Georg Thieme Verlag, Stuttgart, New York, 1998, стр. 176 и 451.
Dictionary of Coating Materials and Printing Inks], Georg Thieme Verlag, Stuttgart, New York, 1998, стр. 176 и 451.
Пигменты для эффекта представляют собой, например, пигменты для металлического эффекта, например, алюминиевые пигменты, золотые бронзовые пигменты, окисленные бронзовые пигменты и/или алюминий оксидные пигменты железа, перламутровые пигменты, например, перламутровая эссенция, основной карбонат свинца, висмут оксид хлорид и/или металлические пигменты окисной слюды и/или другие пигменты для эффекта, например микронизированный диоксид титана, пластинчатый графит, пластинчатый оксид железа, многослойные пигменты для эффекта, составленные из пленок PVD и/или жидкокристаллических полимерных пигментов.
Пропорция пигментов предпочтительно находится в диапазоне 1.0-40.0 мас. %, предпочтительно 2.0-20.0 мас. %, более предпочтительно 5.0-15.0 мас. %, в пересчете на общую массу пигментированного водоразбавляемого базового покрытия.
Изобретенное базовое покрытие может включать связующие вещества отверждаемые физически, термически или термически и с фотохимическим излучением.
В контексте настоящего изобретения, термин "физическое отверждение" означает образование пленки отделением растворителей от растворов полимеров или дисперсий полимеров. Обычно, не нужно никаких сшивающих агентов для этой цели.
В контексте настоящего изобретения, термин "термическое отверждение" означает сшивание слоя материала покрытия инициируемое теплом, в котором или отдельно присутствует сшивающий агент или иначе самосшивающие связующие вещества применяют в основном материале покрытия. Сшивающий агент содержит реакционно-способные функциональные группы, взаимодействующие с реакционно-способными функциональными группами, присутствующими в связующих веществах. Он, как правило, упоминается специалистами как внешний сшиватель. Если дополнительные реакционно-способные функциональные группы или авто-реакционно-способные функциональные группы, то есть группы, которые реагируют с группами того же самого вида, уже присутствуют в молекулах связующего вещества, то самосвязывающие вещества присутствуют. Примеры подходящих дополнительных реакционно-способных функциональных групп и автореакционно-способных функциональных групп известны из Заявка на патент Германии DE 19930665 А1, стр. 7 линия 28 - стр. 9 линия 24.
В контексте настоящего изобретения, фотохимическое излучение следует понимать как электромагнитное излучение, такое как ближнее инфракрасное (БИК), УФ излучение, особенно УФ излучение, и корпускулярное излучение, такое как электронные пучки. Отверждение УФ излучением обычно инициируется свободнорадикальными или катионными фотоинициаторами.
Если применяют термическое отверждение и отверждение с фотохимическим излучением вместе, соответственно также делают "двойное отверждение".
В настоящем изобретении, предпочтение отдают базовым покрытиям способным отверждаться термически или термически и с фотохимическим излучением, то есть при помощи двойного отверждения.
Особенно предпочтительными являются такие базовые покрытия, которые содержат полиуретановую смолу как связующее вещество и аминовую смолу или блокированный или неблокированный полиизоцианат, предпочтительно аминовую смолу, как сшивающий агент. Наряду с аминовыми смолами, предпочтение отдают особенно меламиновым смолам.
Предпочтительно, дополнительно присутствует тиксотропная добавка. Подходящими тиксотропными добавками являются неорганические тиксотропные добавки из группы слоистых силикатов. Особенно подходящими являются силикаты литий-алюминий-магния.
Как и неорганические тиксотропные добавки, тем не менее, также можно применять одну или несколько органических тиксотропных добавок. Их предпочтительно выбирают из группы, включающей тиксотропные добавки сополимера (мет)акриловой кислоты (мет)акрилата, например, коммерческий продукт Viscalex HV30 (Ciba, BASF) и полиуретановые тиксотропные добавки, например, коммерческий продукт DSX® 1550 от Cognis. Тиксотропные добавки сополимера (мет)акриловой кислоты - (мет)акрилата относятся к таким, которые, как и акриловая кислота и/или метакриловая кислота, также содержат один или несколько акриловых сложных эфиров (то есть акрилатов) и/или один или несколько метакриловых сложных эфиров (то есть метакрилатов). Общей чертой тиксотропных добавок сополимера (мет)акриловой кислоты (мет)акрилата является то, что они проявляют существенное повышение вязкости в щелочной среде, то есть при значениях pH>7, особенно >7.5, через образование солей акриловой кислоты и/или метакриловой кислоты, то есть через образование карбоксилатных групп. Если применяют (мет)акриловые сложные эфиры, которые образуются из (мет)акриловой кислоты и C1-С6-алканола, то получают существенно неассоциативные тиксотропные добавки сополимера (мет)акриловой кислоты - (мет)акрилата, например, вышеупомянутый Viscalex HV30. Существенно неассоциативные тиксотропные добавки сополимера (мет)акриловой кислоты - (мет)акрилата также упоминаются в литературе как ASE тиксотропные добавки ("щелочнорастворимые/поддающиеся разбуханию эмульсионные"). Тем не менее, применяемые тиксотропные добавки сополимера (мет)акриловой кислоты - (мет)акрилата также включают те, которые называются HASE тиксотропные добавки ("Гидрофобно Модифицированные Анионные Растворимые Эмульсии"). Их получают, когда алканолы применяемые, вместо или в дополнение к C1-С6-алканолам, являются такими, которые имеют большее количество атомов углерода, например, 7-30, или 8-20, атомов углерода. HASE тиксотропные добавки имеют существенно ассоциированное тиксотропное действие. Применяемые тиксотропные добавки сополимера (мет)акриловой кислоты - (мет)акрилата являются неподходящими в качестве связующих смол, из-за своих тиксотропных свойств; они такжн не влючены среди физически, термически или термически и фотохимически отверждаемых связующих веществ, упомянутых как связующие вещества, и также явным образом отличающихся от связывающих веществ на основе поли(мет)акрилата, которые могут быть применены в композициях изобретенного базового покрытия. Полиуретановые тиксотропные добавки следует понимать, как ассоциативные тиксотропные добавки, упоминаемые в литературе как HEUR ("Гидрофобно Модифицированные Этиленоксид-Уретановые Реологические Модификаторы"). Химическими терминами, они являются неионогенными разветвленными или неразветвленными блок-сополимерами образованными из цепочек полиэтиленоксида (иногда также цепочек полипропиленоксида), которые связаны друг с другом через уретановые связи и которые несут концевые длиннрцепочечные алкильные или алкиленовые группы, имеющие 8-30 атомов углерода. Обычными алкильными группами явялются, например, додециловые или стеариловые группы, обычной алкениловой группой является, например, олеиловая группа, обычной арильной группой является фенильная группа, и обычной алкилированной арильной группой является, например, нонилфенильная группа. Из-за своих тиксотропных свойств и структуры, полиуретановые тиксотропные добавки не подходят в качестве физически, термически или термически и физически отверждаемых связующих смол. Они, также, явным образом отличаются от полиуретанов, которые могут быть применены как связующие вещества в композициях изобретенного базового покрытия.
К тому же, водоразбавляемое базовое покрытие может содержать, по меньшей мере, одну добавку. Примерами таких добавок являются соли, которые термически разлагаются без остатка или существенно без остатка, физически, термически и/или фотохимически радиоактивно-отверждаемые смолы, другие, чем полиуретановые смолы в качестве связующих веществ, дополнительные сшивающие агенты, органические растворители, реакционноспособные разбавители, прозрачные пигменты, наполнители, красители, растворимый в молекулярной дисперсии, наночастицы, светостабилизаторы, антиоксиданты, деаэрирующие агенты, эмульгаторы, смазки, ингибиторы полимеризации, инициаторы свободнорадикальных полимеризаций, усилители адгезии, выравнивающие добавки, помощники пленкообразования, агенты конродирующие оседание (SCAs), добавка, придающая огнеупорные свойства, ингибиторы коррозии, воски, сиккативы, биоциды и матирующие вещества.
Подходящие добавки вышеупомянутого типа известны, например, из
- Заявка на патент Германии DE 19948004 А1, стр. 14 линия 4 - стр. 17 линия 5,
- патент Германии DE 10043405 С1, колонка 5, параграфы [0031]-[0033].
Они применяются в обычных и известных количествах.
Содержание твердых частиц применяемых базовых покрытий в соответствии с изобретением может изменяться согласно требованиям каждого отдельного случая. Прежде всего, содержание твердых частиц диктуется вязкостью, требуемой для нанесения, особенно распылительного нанесения, и таким образом, оно может быть отрегулировано специалистом в данной области техники на основании его общих знаний специалиста, необязательно при помощи нескольких исследовательских тестов.
Содержание твердых частиц базовых покрытий предпочтительно составляет 5-70 мас. %, более предпочтительно 10-65 мас. % и особенно предпочтительно 15-60 мас. %.
Содержание твердых частиц понимают, чтобы означать массовую пропорцию, которая остается как остаток при испарительном концентрировании под фиксированными условиями. Твердые частицы определяли согласно DIN EN ISO 3251 при 130°С, 60 мин, начальная масса 1.0 г.
Применяемые базовые покрытия в соответствии с изобретением могут быть получены, применяя процессы смешивания и установки для смешивания общепринятые и известные для получения базовых покрытий.
Изобретенные базовые покрытия могут быть применены в виде однокомпонентной (1K), двухкомпонентной (2K) или многокомпонентной (3K, 4K) систем. Предпочтение отдают (1K) системам.
В однокомпонентных (1K) системах, связующее вещество и сшивающий агент присутствуют совместно друг с другом, т.е. в одном компоненте. Предпосылка для этого то, что эти два компонента сшиваются друг с другом только при относительно высоких температурах и/или при облучении актиничным излучением.
В двухкомпонентных (2K) системах, например, связующее вещество и сшивающий агент присутствуют отдельно в, по меньшей мере, двух компонентах, которые только быстро соединяют перед нанесением. Такую форму выбирают, когда связующее вещество и сшивающий агент и без того реагируют друг с другом при комнатной температуре. Материал покрытий такого типа особенно применяют для покрытия термически чувствительных основ, особенно в автомеханической повторной отделке.
Пигментированное водоразбавляемое базовое покрытие, применяемое в соответствии с изобретением, может быть нанесено на основу толщинами слоя обычными в пределах автомеханической индустрии, в диапазоне от, например, 5 до 100 микрометров, предпочтительно 5-60 микрометров (толщина слоя после операции сушки). Это включает применение, например, известных способов, таких как распыление, нанесение покрытия с удалением излишков с помощью планки, покраска, наливание, нанесение покрытия погружением, пропитывание, орошение или нанесение покрытия валиком. Предпочтение отдают применению распылительных способов нанесения, таких как распыление сжатым воздухом, безкомпрессорное распыление, высокоскоростное вращение, электростатическое распыление (ESTA), необязательно скомбинированных с нанесением горячим распылением, например, распыление горячим сжатым воздухом.
После того, как пигментированное водоразбавляемое базовое покрытие было нанесено, его можно высушить известными способами. Например, (1K) базовые покрытия могут быть подданы испарению при комнатной температуре на протяжении от 1 до 60 минут и далее предпочтительно высушены при, необязательно, слегка повышенных температурах от 30 до 80°С. Испарение и высушивание, как следует понимать в контексте настоящего изобретения, означают испарение органических растворителей и/или воды, в результате которого материал покрытия становится суше, но не затвердевает, или образуется пленка пока еще не полностью сшитого материала покрытия.
Затем наносят стандартное коммерческое самовосстанавливающееся покрытие аналогично стандартными методами, толщина сухого слоя снова находится в пределах стандартных диапазонов, например, 5-100 микрометров. Такие самовосстанавливающиеся покрытия известны специалисту в данной области техники.
После нанесения самовосстанавливающегося покрытия, оно может быть поддано испарению при комнатной температуре, например, на протяжении от 1 до 60 минут, и необязательно высушено. Затем самовосстанавливающееся покрытие отверждают вместе с нанесенным пигментированным базовым покрытием. В ходе этого могут иметь место, например, реакции сшивания, которые дают изобретенную многослойную цветную и/или для эффекта красочную систему на основе. Отверждение предпочтительно проводят термически или термически с актиничным излучением при температурах 20-200°С.
Дополнительным объектом настоящего изобретения является способ получения многослойной цветной и/или для эффекта красочной системы, в котором
(а) пигментированное водоразбавляемое базовое покрытие наносят на основу,
(б) формируют полимерную пленку из материала покрытия, нанесенного на стадии (а),
(в) наносят самовосстанавливающееся покрытие на слой базового покрытия, полученного таким образом, и затем
(г) слой базового покрытия отверждают вместе со слоем самовосстанавливающегося покрытия,
который включает применение на стадии (а), пигментированного водоразбавляемого базового покрытия, при чем указанное базовое покрытие включает изобретенную дисперсию.
Все детали, поданные выше, в отношении изобретенной дисперсии и водоразбавляемого базового покрытия также применяют к изобретенному применению и к способу согласно изобретение. Это особенно также применяют ко всем предпочтительным, особенно предпочтительным и очень особенно предпочтительным признакам.
Преимущественно, изобретенное водоразбавляемое базовое покрытие могут быть применены для покрытия кузовов автомобилей и/или пластиковых частей для установки в кузовах автомобилей.
С помощью способа согласно изобретение, возможно покрасить металлические и неметаллические основы, особенно пластиковые основы, предпочтительно автомобильных кузовов или их части.
Изобретение иллюстрируют далее при помощи примеров.
Примеры
Если не указано иначе, гидроксильное число в примерах, которые следуют, определяли на основе R.-P.  , R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), с помощью уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофуран (ТНР)/диметилформамид (DMF) при комнатной температуре, с полным гидролизом оставшегося избытка уксусного ангидрида после ацетилирования и потециометрическим обратным титрованием уксусной кислоты спиртовым раствором гидроксида калия. Время ацетилирования 60 мин было достаточным во всех случаях, чтобы обеспечить полное превращение.
, R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), с помощью уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофуран (ТНР)/диметилформамид (DMF) при комнатной температуре, с полным гидролизом оставшегося избытка уксусного ангидрида после ацетилирования и потециометрическим обратным титрованием уксусной кислоты спиртовым раствором гидроксида калия. Время ацетилирования 60 мин было достаточным во всех случаях, чтобы обеспечить полное превращение.
Если не указано иначе, кислотное число в примерах, которые следуют, определяли на основе DIN EN ISO 2114 в гомогенном растворе THF/вода (9 об. частей THF и 1 об. часть дистиллированной воды) этаноловым раствором гидроксида калия.
Если не указано иначе, содержание твердых частиц, также упоминаемое здесь как твердые частицы, определяли в примерах, которые следуют, согласно DIN EN ISO 3251 при 130°; 60 мин, начальная масса 1.0 г.
Среднечисловую молярную массу (Mn) в примерах, которые следуют, если не указано иначе, определяли при помощи 10.00 парофазного осмометра (от Knauer) на концентрационных сериях в толуоле при 50°С согласно Е.  , G.
, G.  , K.-F. Arndt, "Leitfaden der Polymercharakterisierung" [Guidelines for Polymer Characterization], Akademie-Verlag, Berlin, стр. 47-54, 1982.
, K.-F. Arndt, "Leitfaden der Polymercharakterisierung" [Guidelines for Polymer Characterization], Akademie-Verlag, Berlin, стр. 47-54, 1982.
Полиуретановые дисперсии
Пример D-P1
Получение дисперсии ненасыщенного полиэстеруритана, имеющего альфа-метилстириловые группы
Дисперсию альфа-метилстирилсодержащего полиуретана получали на основе описания патента DE 19948004 В4, стр. 27, пример 1, "Herstellung eines  Polyurethans (В)" [Получение изобретенного полиуретана (В)], за исключением того, что твердые частицы в полученной дисперсии составили только 29% амин 35 мас. %. В пересчете на аддукт (В2), упомянутый в описании патента DE 19948004 В4, пример получения 1, аддукт получили из моноэтаноламина и триметилолпропана вместо диэтаноламина:
Polyurethans (В)" [Получение изобретенного полиуретана (В)], за исключением того, что твердые частицы в полученной дисперсии составили только 29% амин 35 мас. %. В пересчете на аддукт (В2), упомянутый в описании патента DE 19948004 В4, пример получения 1, аддукт получили из моноэтаноламина и триметилолпропана вместо диэтаноламина:
Для этой цели, реакционный сосуд, оборудованный мешалкой, внутренним термометром, обратным холодильником и электрическим нагревателем сначала загрузили в атмосфере азота 200.0 мас. частями метилэтилкетона, 800.0 мас. частями N-метилпирролидона и 221.3 мас. частями моноэтаноламина (от BASF SE) при 20°С. По каплям к этой смеси на протяжении полутора часов были добавлены 778.7 мас. частей 1-(1-изоцианато-1-метилэтил)-3-(1-метилэтинил)бензола (TMI® (МЕТА) Ненасыщенный Алифатический Изоцианат, от Cytec), имеющий содержание изоцианата 20.4 мас. % изоцианата, таким образом, что температура реакции не превышала 40°С. Полученную в результате реакционную смесь перемешивали до тех пор, пока не могли больше обнаружить свободных изоцианатных групп. После этого, реакционную смесь стабилизировали 200 млн.д. гидрохинона.
Теоретическое содержание твердых частиц полученного таким образом раствора описанного аддукта составляло 50 мас. %.
Потом, в дополнительном реакционном сосуде, оборудованном мешалкой, внутренним термометром, обратным холодильником и электрическим нагревателем, 431.7 мас. частей линейного полиэстера полиола и 69.7 мас. частей диметилолпропионовой кислоты (от GEO Speciality Chemicals) растворяли в 355.8 мас. частях метилэтилкетона и 61.6 мас. частях N-метилпирролидона в атмосфере азота. Линейный полиэстер полиол был получен заблаговременно из димеризованной жирной кислоты (Pripol® 1012, от Uniqema), изофталовой кислоты (от BP Chemicals) и гексан-1,6-диола (от BASF SE) (массовое соотношение исходных материалов: димерной жирной кислоты к изофталовой кислоте к гексан-1,6-диолу = 54.00:30.02:15.98) и имеет гидроксильное число 73 мг KOH/г твердых частиц и среднечисловую молярную массу 1379 г/моль.
К полученному раствору добавляли при 45°С, 288.6 мас. частей изофорон диизоцианата (Basonat® I, от BASF SE), имеющего содержание изоцианата 37.75 мас. %. После того, как прекратилась экзотермическая реакция, реакционную смесь постепенно нагревали до 80°С, перемешивая при этом. Перемешивание при этой температуре продолжалось до тех пор, пока содержание изоцианата раствора не составляло 3.2 мас. % и не было таким постоянно. После этого, реакционную смесь охлаждали до 65°С, и добавляли 85.2 мас. частей вышеописанного аддукта вместе с 21.8 мас. частями триметилолпропана (от BASF SE). Полученную в результате реакционную смесь перемешивали при 65°С до тех пор, пока содержание изоцианата раствора не падало до 1.0 мас. %. Теперь добавляли 22.2 мас. % диэтаноламина (от BASF SE) и содержание изоцианатных групп мониторили до тех пор, пока не могли больше обнаружить свободные изоцианатные группы. Полученный рабавленный полиуретан перемешивали с 139.7 мас. частями метоксипропанола и 43.3 мас. частями триэтиламина (от BASF SE). Через 30 минут после добавления амина, температуру раствора понижали до 60°С, и затем добавляли 1981 мас. частями деионизированной воды, при этом перемешивая в течение 30 минут. Метилэтилкетон отгоняли из полученной дисперсии при 60°С под сниженным давлением. После этого, любые потери растворителя и воды восполняли.
Дисперсия альфа-метилстирил-содержащего полиуретана, полученная таким образом, имела содержание твердых частиц 29.0 мас. %; кислотное число составляло 34.0 мг KOH/г содержания твердых частиц и pH составляло 7.0 (измеренное при 23°С).
Пример D-P2
Получение дисперсии насыщенного полиэстеруретана, имеющего изотридециловые группы
Дисперсию полиуретана получали как в сравнительном примере DP-1, за исключением того, что он не имел альфа-метилстироловых групп, но имел изотридециловые группы.
В реакционном сосуде, оборудованном мешалкой, внутренним термометром, обратным холодильником и электрическим нагревателем, 440.1 мас. частей линейного полиэстер полиола, полученного из димеризованной жирной кислоты (Pripol® 1012, от Uniqema), изофталевой кислоты (от BP Chemicals) и гексан-1,6-диола (BASF SE) (массовое соотношение исходных материалов: димерная жирная кислота к изофталевой кислоте к гексан-1,6-диолу = 54.00:30.02:15.98), имеющего гидроксильное число 73 мг KOH/г твердых частиц и среднечисловую молярную массу 1379 г/моль, а также 71.1 мас. частей диметилолпропионовой кислоты (от GEO Speciality Chemicals), растворили в 362.7 мас. частей метилэтилкетона и 62.8 мас. частей N-метилпирролидона в атмосфере азота. К раствору, полученному в результате, при 45°С, добавляли 294.3 мас. частей изофорон диизоцианата (Basonat® I, от BASF SE), имеющего содержание изоцианата 37.75 мас. %. После того, как экзотермическая реакция прекратилась, реакционную смесь постепенно нагревали до 80°С, при этом перемешивая. Перемешивание продолжали при этой температуре до тех пор, пока содержание изоцианата раствора не составляло 3.2 мас. % и было постоянным. После этого, реакционную смесь охлаждали до 70°С, и добавляли 33.1 мас. частей изотридецилового спирта (Isotridecanol N, от BASF SE) вместе с 19.3 мас. частями триметилолпропана (от BASF SE) и 25.6 мас. частями метилэтилкетона. Полученную в результате реакционную смесь перемешивали при 70°С до тех пор, пока содержание изоцианата раствора не падало до 0.8 мас. %. В этот момент добавляли 19.6 мас. % диэтаноламина (от BASF SE) и содержание изоцианатных групп измеряли до тех пор, пока не могли больше обнаружить никаких свободных изоцианатных групп. Полученный разбавленный полиуретан перемешивали с 123 мас. частями метоксипропанола и 43.8 мас. частями триэтиламина (от BASF SE). Через 30 минут после добавления амина, температуру раствора понижали до 60°С, и затем, после 30 минут, добавляли 2009 мас. частями деионизированной воды при этом перемешивая. Метилэтилкетон отгоняли из полученной дисперсии при 60°С под сниженным давлением. После этого, любые потери растворителя и воды восполняли.
Дисперсия полиуретана, полученная таким образом, имела содержание твердых частиц 29.0 мас. %; кислотное число составляло 34.1 мг KOH/г содержания твердых частиц и pH составлял 7.0 (измеренное при 23°С).
Пример D-P3
Получение дисперсии насыщенного полиэтеруретана с политетраметиленоксидными сегментами
В реакционный сосуд, оборудованный мешалкой, внутренним термометром, обратным холодильником и электрическим нагревателем, 685.8 мас. частей линейного альфа, с омега-гидрокси концом политетраметиленоксида (PolyTHF® 2000, от BASF SE), имеющего гидроксильное число 56.1 мг KOH/г твердых частиц и среднюю молекулярную массу 2000 г/моль, 138.0 мас. частей диметилолпропионовой кислоты (от GEO Speciality Chemicals), 0.01 мас. части дилаурата дибутилолова и 685.8 мас. частей метилэтилкетона растворяли в атмосфере азота при 70°С. К полученному раствору добавляли 418.8 мас. частей м-тетраметилксилол диизоцианата (TMXDI® (Meta) Алифатический Изоцианат, от Cytec), имеющего содержание изоцианата 34.4 мас. %. Затем, реакционную смесь постепенно нагревали до 80°С, перемешивая при этом, и выдерживали при этой температуре до тех пор, пока содержание изоцианата раствора не составляло 1.8 мас. % и не было таким постоянно. После этого, 87.4 мас. частей триметилолпропана (от BASF SE) и 26.1 мас. частей метилэтилкетона быстро добавляли к реакционной смеси. Полученную в результате реакционную смесь перемешивали при 80°С до тех пор, пока не могли больше обнаружить свободные изоцианатные группы. Полученный разбавленный полиуретан перемешивали с 41.2 мас. частями диметилэтаноламина и перемешивали в течение дополнительных 60 минут. После этого, реакционную смесь полностью перенесли, постоянно перемешивая при этом в течение периода 30 минут, во второй реакционный сосуд, который был уже первоначально загружен 1732 мас. частями деионизированной воды, которая предварительно была нагрета до 70°С. Метилэтилкетон отгоняли из полученной дисперсии при 60°С под сниженным давлением. После этого, любые потери воды восполняли.
Дисперсия полиуретана, полученная таким образом, имела содержание твердых частиц 38.0 мас. %; кислотное число составляло 43.0 мг KOH/г содержания твердых частиц и pH составлял 6.2 (измеренное при 23°С).
Пример D-P4
Получение известной дисперсии серосодержащего насыщенного полиэстеруретана
Согласно ЕР 1330480 В1, полиэстер полиола согласно примеру получения 1, стр. 15, применяли, чтобы получить преполимер полиуретана согласно примеру получения 2, стр. 15, который реагировал с тиодиэтанолом согласно примеру 1, стр. 16 "Die Herstellung eines Polyurethans und einer  Dispersion hiervon" [Получение изобретенного полиуретана и его водной дисперсии], нейтрализовали триэтиламином и затем преобразовали в водную дисперсию полиуретана. Метилэтилкетон отгоняли из полученной дисперсии при 60°С под сниженным давлением. После этого, любые потери воды восполняли.
Dispersion hiervon" [Получение изобретенного полиуретана и его водной дисперсии], нейтрализовали триэтиламином и затем преобразовали в водную дисперсию полиуретана. Метилэтилкетон отгоняли из полученной дисперсии при 60°С под сниженным давлением. После этого, любые потери воды восполняли.
Дисперсия серосодержащего полиуретана, полученная таким способом, имела содержание твердых частиц 27.3 мас. %; кислотное число составляло 34.4 мг KOH/г содержания твердых частиц, уровень нейтрализации составлял 84% и pH составлял 7.1 (измеренное при 23°С).
Дисперсия сополимера полиуретан-полиакрилата
Пример D-A1
Получение исходной дисперсии сополимера полиэстеруретан-полиакрилата, происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
На основе описания патента DE 19948004 В4, стр. 27, пример 2, "Die Herstellung der  eines
eines  Mischpolymerisats 1" [Получение исходной дисперсии изобретенного сополимера 1], и описания патента ЕР 1218434 В1, стр. 24, пример 2, "Die Herstellung der eines Pfropfmischpolymerisats 1" в атмосфере азота, 1937.8 мас. частей дисперсии полиуретана согласно примеру DP-1 разбавили 856.9 мас. частями деионизированной воды и нагревали до 85°С. При этой температуре, при стандартном давлении, смесь 160.7 мас. частей стирола, 160.7 мас. частей метилметакрилата, 120.3 мас. частей н-бутилакрилата и 120.3 мас. частей гидроксиэтилметакрилата равномерно добавляли к дисперсии, перемешивая при этом в течение 3.5 часов. С начала добавления смеси мономеров, в пределах 4 часов добавляли раствор 8.4 мас. частей трет-бутил пероксиэтилгексаноата в 134.9 мас. частях метоксипропанола. Полученную в результате реакционную смесь перемешивали при 85°С до тех пор, пока все мономеры прореагировали (общее содержание мономеров (ГХ) относительно дисперсии <0.1%).
Mischpolymerisats 1" [Получение исходной дисперсии изобретенного сополимера 1], и описания патента ЕР 1218434 В1, стр. 24, пример 2, "Die Herstellung der eines Pfropfmischpolymerisats 1" в атмосфере азота, 1937.8 мас. частей дисперсии полиуретана согласно примеру DP-1 разбавили 856.9 мас. частями деионизированной воды и нагревали до 85°С. При этой температуре, при стандартном давлении, смесь 160.7 мас. частей стирола, 160.7 мас. частей метилметакрилата, 120.3 мас. частей н-бутилакрилата и 120.3 мас. частей гидроксиэтилметакрилата равномерно добавляли к дисперсии, перемешивая при этом в течение 3.5 часов. С начала добавления смеси мономеров, в пределах 4 часов добавляли раствор 8.4 мас. частей трет-бутил пероксиэтилгексаноата в 134.9 мас. частях метоксипропанола. Полученную в результате реакционную смесь перемешивали при 85°С до тех пор, пока все мономеры прореагировали (общее содержание мономеров (ГХ) относительно дисперсии <0.1%).
Во время свободнорадикальной полимеризации, с интервалами 30 минут, содержание свободных мономеров определяли при помощи газовой хроматографии (ГХ) (ГХ: 50 м капиллярная колонка с диоксидом кремния с фазой полиэтиленгликоля или 50 м капиллярная колонка с диоксидом кремния с фазой полидиметилсилоксана, газ-носитель гелий, инжектор для ввода проб с делением потока 150°С, температурный режим термостата 40-220°С, пламенно-ионизационный детектор, температура детектора 275°С, внутренний стандарт изобутилакрилат), и наиболле высокое содержание мономеров относительно дисперсии определяли после 30 мин при 1.8 мас. % (11.1 мас. % на основе винилового мономера).
Полученная исходная дисперсия сополимера имела, как описано в первичном примере, очень хорошую стойкость при хранении. Содержание твердых частиц в ней составляло 32.5 мас. %, кислотное число составляло 18.3 мг KOH/г содержания твердых частиц и ее pH составляло 7.2 (измеренное при 23°С). Размер частиц (z осреднение) при помощи фотокорреляционной спектроскопии составлял 104 нм. При помощи газовой хроматографии (ГХ: 50 м капиллярная колонка с диоксидом кремния с фазой полиэтиленгликоля или 50 м капиллярная колонка с диоксидом кремния с фазой полидиметилсилоксана, газ-носитель гелий, инжектор для ввода проб с делением потока 250°С, температурный режим термостата 40-220°С, пламенно-ионизационный детектор, температура детектора 275°С, внутренний стандарт н-пропилгликоль), определили содержание 6.0 мас. % метоксипропанола и 1.7 мас. % н-метилпирролидона.
После экстракции тетрагидрофураном высушенного замораживанием полимера содержание геля, определенное гравиметрически, составляло 71.5 мас. %. Для этой цели, дисперсию высушивали замораживанием, определяли массу полимера, высушенного замораживанием, потом полимер экстрагировали при 25°С в избытке тетрагидрофурана (соотношение тетрагидрофурана к сополимеру, высушенному замораживанием = 300:1) в течение 24 часов. Нерастворенное содержимое (содержание геля) изолировали, высушивали при 50°С в сушильном шкафу с циркуляцией воздуха в течение 4 часов, и затем взвешивали повторно.
Пример D-A2
Получение исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от насыщенного полиэстеруретана, имеющего изотридециловые группы
Аналогично примеру получения D-A1, первичную дисперсию получали заменой дисперсии полиуретана, имеющей альфа-метилстириловые группы, согласно примеру получения D-P1 для такой же массы дисперсии полиуретана, имеющей изотридециловые группы, полученной согласно примеру получения D-Р2.
Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли путем газовой хроматографии аналогично примеру D-A1, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 2.0 мас. % (12.4 мас. % в пересчете на виниловый мономер).
Полученная первичная дисперсия сополимера имеет очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.2 мас. %, кислотное число составляло 18.1 мг KOH/г содержания твердых частиц и pH составлял 7.3 (измеренное при 23°С). Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 112 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 5.7 мас. % метоксипропанола и 1.2 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 70.2 мас. %. Пример D-A3
Получение исходной дисперсии полиэстеруретан-полиакрилатного сополимера (изопреновый сомономер), происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
Как и в примере получения D-A1, первичную дисперсию сополимера получали, за исключением того, что 15 моль % виниловых мономеров меняли на изопрен: таким образом, 1937.8 мас. частей дисперсии полиуретана согласно примеру получения D-P1 разбавляли 857.0 мас. частями деионизированной воды и нагревали до 85°С. При этой температуре, смесь 145.2 мас. частей стирола, 145.2 мас. частей метил метакрилата, 108.7 мас. частей н-бутил акрилата, 108.7 мас. частей гидроксиэтил метакрилата и 54.3 мас. частей изопрена равномерно добавляли к дисперсии, перемешивая при этом под калибровочным давлением азота 3 бар на протяжении всего курса 3.5 часа. С началом добавления смеси мономера, на протяжении четырех часов добавляли раствор 8.4 мас. частей трет-бутил пероксиэтилгексаноата в 134.9 мас. частях метоксипропанола. Полученную в результате реакционную смесь перемешивали далее при 85°С до тех пор, пока все мономеры не прореагировали.
Полученная первичная дисперсия имеет очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.3 мас. %, кислотное число составляло 18.3 мг KOH/г и pH составлял 7.2 (измеренное при 23°С). Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 115 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 6.0 мас. % метоксипропанола и 1.7 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 70.8 мас. %.
Пример D-A4
Получение исходной дисперсии полиэстеруретан-полиакрилатного сополимера (без стиролового сомономера), происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
Как и в примере получения D-A1, первичную дисперсию сополимера получали, за исключением того, что пропорцию стирола заменяли такой же массой метил метакрилата и содержание твердых частиц понижали до 28%:
Таким образом, в атмосфере азота, 1689.6 мас. частей дисперсии полиуретана разбавляли 1133.5 мас. частями деионизированной воды и 142.4 мас. частями N-метилпирролидона, и нагревали до 85°С. При этой температуре, при стандартном давлении, смесь 280.3 мас. частей метил метакрилата, 104.9 мас. частей н-бутил акрилата и 104.9 мас. частей гидроксиэтил метакрилата равномерно добавляли к дисперсии, перемешивая при этом на протяжении всего курса 3.5 часа. С началом добавления смеси мономера, на протяжении четырех часов добавляли раствор 7.4 мас. частей трет-бутил пероксиэтилгексаноата в 37.0 мас. частях метоксипропанола. Полученную в результате реакционную смесь перемешивали далее при 85°С до тех пор, пока все мономеры не прореагировали.
Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 1.0 мас. % (7.1 мас. % в пересчете на виниловый мономер).
Полученная первичная дисперсия сополимера, как и стирол-содержащая дисперсия, имела очень хорошую стабильность при хранении. Ее содержание твердых частиц регулировали до 28.0 мас. %; кислотное число составляло 18.1 мг KOH/г содержания твердых частиц и pH составлял 7.2 (измеренное при 23°С). Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 102 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 2.8 мас. % метоксипропанола и 5.8 мас. % N-метилпирролидона. Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 72.0 мас. %. Пример D-A5
Получение исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от насыщенного, серасодержащего полиэстеруретана (без растворителей)
Согласно ЕР 1330480 В1, пример 2, стр. 16, "Die Herstellung der  eines
eines  Polymergemischs" [Получение исходной дисперсии изобретенной полимерной смеси], 2285.8 мас. частей дисперсии полиуретана согласно примеру DP-4 первоначально загружали в атмосфере азота и нагревали до 82°С. При этой температуре, при стандартном давлении, смесь 118.8 мас. частей стирола, 207.7 мас. частей гидроксипропил метакрилата, 59.8 мас. частей н-бутил акрилата, 88.9 мас. частей метил метакрилата и 118.8 мас. частей трет-бутилциклогексил акрилата равномерно добавляли к дисперсии, перемешивая при этом на протяжении всего курса 4 часов. С началом добавления смеси мономера, на протяжении 4.5 часов добавляли 29.9 мас. частей трет-бутил пероксиэтилгексаноата. После окончания подачи инициатора, полимеризацию продолжали при 82°С на протяжении одного часа. Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров определяли после 30 мин, которое должно составлять 1.6 мас. % в пересчете на 41.9 мас. % дисперсии (7.6 мас. % в пересчете на виниловый мономер).
Polymergemischs" [Получение исходной дисперсии изобретенной полимерной смеси], 2285.8 мас. частей дисперсии полиуретана согласно примеру DP-4 первоначально загружали в атмосфере азота и нагревали до 82°С. При этой температуре, при стандартном давлении, смесь 118.8 мас. частей стирола, 207.7 мас. частей гидроксипропил метакрилата, 59.8 мас. частей н-бутил акрилата, 88.9 мас. частей метил метакрилата и 118.8 мас. частей трет-бутилциклогексил акрилата равномерно добавляли к дисперсии, перемешивая при этом на протяжении всего курса 4 часов. С началом добавления смеси мономера, на протяжении 4.5 часов добавляли 29.9 мас. частей трет-бутил пероксиэтилгексаноата. После окончания подачи инициатора, полимеризацию продолжали при 82°С на протяжении одного часа. Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров определяли после 30 мин, которое должно составлять 1.6 мас. % в пересчете на 41.9 мас. % дисперсии (7.6 мас. % в пересчете на виниловый мономер).
Полученную первичную дисперсию разбавляли также 590.3 мас. частями воды, так, чтобы получить содержание твердых частиц как в ЕР 1330480 В1, пример 2, 34.8 мас. %.
Кислотное число составляло 19.6 мг KOH/г содержания твердых частиц и pH дисперсии составлял 7.4 (измеренное при 23°С). Уровень нейтрализации составлял 80.2%. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 83 нм. Полученная дисперсия не содержит растворитель.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 0.0 мас. %.
Пример D-A6
Получение исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от насыщенного, серасодержащего полиэстеруретана, и добавлением метоксипропанола и N-метилпирролидона
Аналогично примеру D-A5, получали сополимер.
Полученную первичную дисперсию, имеющую содержание твердых частиц 41.9 мас. %, после конца продолженной полимеризации, разбавляли также 77.0 мас. частями метоксипропанола, 206.5 мас. частей N-метилпирролидона и 306.8 мас. частями воды, так, чтобы получить в результате содержание твердых частиц 34.8 мас. %, аналогично сравнительному примеру D-A5.
Кислотное число составляло 18.5 мг KOH/г содержания твердых частиц и pH составлял 7.2 (измеренное при 23°С). Уровень нейтрализации составлял 80.0%. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 81 нм. Путем газовой хроматографии, аналогично примеру D-A1, определяли содержание 2.2 мас. % метоксипропанола и 5.9 мас. % N-метилпирролидона. Как и в примере D-A5, не было возможно определить никакого содержания геля (содержание геля = 0.0 мас. %).
Пример D-B1
Получение изобретенной исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
Для получения исходной дисперсии изобретенного сополимера, в атмосфере азота, 1961.2 мас. частей дисперсии альфа-метилстирил-содержащего полиуретана согласно примеру получения D-P1 разбавляли 40.0 мас. частями метоксипропанола (0.07% в пересчете на полиуретан) и 686.5 мас. частями деионизированной воды, и нагревали до 80°С. После содержание реактора нагревали до 80°С, 0.6 мас. части пероксодисульфата аммония растворяли в 35.7 мас. частях деионизированной воды и добавляли в реактор при стандартном давлении. Далее, при этом постоянно перемешивая, смесь 301.6 мас. частей метил метакрилата, 261.6 мас. частей н-бутил акрилата, 5.6 мас. частями аллил метакрилата (0.87 моль % в пересчете на общий виниловый мономер) и 134.9 мас. частей N-метилпирролидона равномерно добавляли на протяжении всего курса 5 часов. С началом добавления смеси мономера, раствор 1.1 мас. частей пероксодисульфата аммония в 71.3 мас. частях деионизированной воды аналогично добавляли на протяжении пяти часов.
Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 0.5 мас. % (3.1 мас. % в пересчете на виниловый мономер).
После одновременного окончания дозированного добавления мономера и инициатора, полученную в результате реакционную смесь перемешивали при 80°С на протяжении дополнительного часа и затем охлаждали до комнатной температуры.
Полученная первичная дисперсия сополимера имеет очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.5%, кислотное число составляло 18.8 мг KOH/г содержания твердых частиц и pH составлял 7.0. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 96 нм. Аналогично примеру D-А1, путем газовой хроматографии, определяли содержание 2.7 мас. % метоксипропанола и 5.7 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 80.3 мас. %.
Пример D-B2
Получение изобретенной исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от насыщенного полиэстеруретана, имеющего изотридециловые группы
Аналогично примеру получения D-B1, первичную дисперсию получали заменой дисперсии полиуретана, имеющей альфа-метилстириловые группы согласно примеру получения D-P1 для такой же массы дисперсии полиуретана, имеющей изотридециловые группы согласно примеру получения D-P2. Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 0.5 мас. % (3.1 мас. % в пересчете на виниловый мономер).
Полученная первичная дисперсия сополимера имела очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.4 мас. %, кислотное число составляло 18.4 мг KOH/г содержания твердых частиц и pH составлял 6.9 (измеренное при 23°С). Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 102 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 2.4 мас. % метоксипропанола и 5.1 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 79.1 мас. %.
Пример D-B3
Получение изобретенной исходной дисперсии полиэстеруретан-полиакрилатного сополимера (изопреновый сомономер), происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
Как и в примере получения D-B1, в соответствии с изобретением, первичную дисперсию сополимера получали, за исключением того, что 15 моль % виниловых мономеров меняли на изопрен: таким образом, 1961.2 мас. частей дисперсии альфа-метилстирил-содержащего полиуретана согласно примеру получения D-P1 разбавляли 40.0 мас. частями метоксипропанола и 686.5 мас. частями деионизированной воды, и нагревали до 80°С. При 80°С, 0.6 мас. части пероксодисульфата аммония, растворенные в 35.7 мас. частях деионизированной воды, добавляли в реактор под калибровочным давлением азота 3 бар. Далее, поддерживая давление и постоянно перемешивая, смесь 271.8 мас. частей метил метакрилата, 235.7 мас. частей н-бутил акрилата, 5.9 мас. частями аллил метакрилата (0.87 моль % в пересчете на общий виниловый мономер) 55.3 мас. частей изопрена и 134.9 мас. частей N-метилпирролидона равномерно добавляли на протяжении всего курса шести часов. С началом добавления смеси мономера, раствор 1.1 мас. частей пероксодисульфата аммония в 71.3 мас. частях деионизированной воды также добавляли на протяжении шести часов. После одновременного окончания дозированного добавления мономера и инициатора, полученную в результате реакционную смесь перемешивали при 80°С на протяжении дополнительного часа и затем охлаждали до комнатной температуры.
Полученная первичная дисперсия привитого сополимера имеет очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.2 мас. %, кислотное число 18.8 мг KOH/г содержания твердых частиц и pH составлял 7.0 (измеренное при 23°С). Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 97 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 2.8 мас. % метоксипропанола и 5.6 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 78.0 мас. %.
Пример D-B4
Получение изобретенной исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от ненасыщенного полиэстеруретана, имеющего альфа-метилстириловые группы
Для получения исходной дисперсии изобретенного сополимера, в атмосфере азота, 1689.7 мас. частей дисперсии альфа-метилстирил-содержащего полиуретана согласно примеру получения D-P1 разбавляли 40.3 мас. частями метоксипропанола (0.08% в пересчете на полиуретан) и 993.8 мас. частями деионизированной воды, и нагревали до 80°С. Один раз нагревали содержание реактора до 80°С, 0.7 мас. части пероксодисульфата аммония, растворенные в 46.1 мас. частях деионизированной воды, добавляли в реактор при стандартном давлении. Далее, при этом постоянно перемешивая, согласно примеру получения D-A4, смесь 277.6 мас. частей метил метакрилата, 103.8 мас. частей н-бутил акрилата и 103.8 мас. частей гидроксиэтил метакрилата, и дополнительно 4.8 мас. частей аллил метакрилата (0.86 моль % в пересчете на общий виниловый мономер) вместе с 145.7 мас. частями N-метилпирролидона, равномерно добавляли на протяжении всего курса пяти часов. С началом добавления смеси мономера, раствор 1.5 мас. частей пероксодисульфата аммония в 92.2 мас. частях деионизированной воды аналогично добавляли на протяжении пяти часов.
Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично примеру D-A1 путем газовой хроматографии, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 0.5 мас. % (3.6 мас. % в пересчете на виниловый мономер).
После одновременного окончания дозированного добавления мономера и инициатора, полученную в результате реакционную смесь перемешивали при 80°С на протяжении дополнительного часа и затем охлаждали до комнатной температуры.
Полученная первичная дисперсия сополимера имеет очень хорошую стабильность при храпении. Ее содержание твердых частиц составляло 28.0 мас. %, кислотное число составляло 18.2 мг KOH/г содержания твердых частиц и pH составлял 7.0. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 110 нм. Аналогично примеру D-А1, путем газовой хроматографии, определяли содержание 2.9 мас. % метоксипропанола и 5.9 мас. % N-метилпирролидона. Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 81.2 мас. %.
Пример D-B5
Получение изобретенной исходной дисперсии полиэстеруретан-полиакрилатного сополимера, происходящего от насыщенного, серасодержащего полиэстеруретана (разбавляемого растворителем)
Для получения исходной дисперсии изобретенного сополимера, в атмосфере азота, 2101.9 мас. частей дисперсии полиуретана согласно сравнительному примеру DP-4 разбавляли 40.1 мас. частями метоксипропанола (0.07% в пересчете на полиуретаны) и 513.3 мас. частями деионизированной воды, и нагревали до 80°С. Один раз нагревали содержание реактора до 80°С, 1.1 мас. частей пероксодисульфата аммония растворяли в 36.4 мас. частях деионизированной воды добавляли в реактор при стандартном давлении. Далее, при этом постоянно перемешивая, согласно сравнительному примеруы D-A5 и D-A6, смесь 108.3 мас. частей стирола, 189.4 мас. частей гидроксипропил метакрилата, 54.6 мас. частей н-бутил акрилата, 81.1 мас. частей метил метакрилата, 108.3 мас. частей трет-бутилциклогексил акрилата и дополнительно 4.5 мас. частями аллил метакрилата (0.87 моль % в пересчете на общий виниловый мономер) вместе с 186.0 мас. частей N-метилпирролидона равномерно добавляли на протяжении всего курса пяти часов. С началом добавления смеси мономера, раствор 2.2 мас. частей пероксодисульфата аммония в 72.8 мас. частях деионизированной воды аналогично добавляли на протяжении пяти часов. Во время свободнорадикальной полимеризации, при интервалах 30 минут, содержание свободных мономеров определяли аналогично предыдущему примеру путем газовой хроматографии, и самое высокое общее содержание мономеров в пересчете на дисперсию определяли после 30 мин, которое должно составлять 0.6 мас. % (3.7 мас. % в пересчете на виниловый мономер).
После одновременного окончания дозированного добавления мономера и инициатора, полученную в результате реакционную смесь перемешивали при 80°С на протяжении дополнительного часа и затем охлаждали до комнатной температуры.
Полученная первичная дисперсия сополимера имела хорошую стабильность при хранении. Ее содержание твердых частиц составляло 33.0 мас. %, кислотное число составляло 20.1 мг KOH/г содержания твердых частиц и pH составлял 7.0. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 72 нм. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 2.1 мас. % метоксипропанола и 6.0 мас. % N-метилпирролидона.
Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 42.1 мас. %.
Пример D-C1
Получение изобретенной исходной дисперсии полиэтеруретан-полиакрилатного сополимера, происходящего от насыщенного полиэтеруретана
Для получения исходной дисперсии изобретенного сополимера, в атмосфере азота, 1496.7 мас. частей политетраметиленоксид-содержащей дисперсии полиуретана согласно примеру получения D-P3 разбавляли 40.0 мас. частями метоксипропанола (0.07% в пересчете на полиуретан) и 1150.9 мас. частями деионизированной воды, и нагревали до 80°С. Один раз нагревали содержание реактора до 80°С, 0.6 мас. части пероксодисульфата аммония, растворенного в 35.7 мас. частях деионизированной воды, добавляли в реактор при стандартном давлении. Далее, при этом постоянно перемешивая, смесь 301.6 мас. частей метил метакрилата, 261.6 мас. частей н-бутил акрилата, 5.6 мас. частями аллил метакрилата (0.87 моль %) и 134.9 мас. частей N-метилпирролидона равномерно добавляли на протяжении всего курса шести часов. С началом добавления смеси мономера, раствор 1.1 мас. частей пероксодисульфата аммония в 71.3 мас. частях деионизированной воды также добавляли на протяжении шести часов. После одновременного окончания дозированного добавления мономера и инициатора, полученную в результате реакционную смесь перемешивали при 80°С на протяжении дополнительного часа и затем охлаждали до комнатной температуры.
Полученная первичная дисперсия сополимера имела очень хорошую стабильность при хранении. Ее содержание твердых частиц составляло 32.8 мас. %, кислотное число составляло 23.2 мг KOH/г содержания твердых частиц и pH составлял 6.2. Аналогично примеру D-A1, путем газовой хроматографии, определяли содержание 1.1 мас. % метоксипропанола и 3.9 мас. % N-метилпирролидона. Размер частиц (z средние), измеренный при помощи фотокорреляционной спектроскопии, составлял 103 нм. Аналогично примеру D-A1, содержание геля, определенное гравиметрически после экстракции тетрагидрофураном, составляло 85.2 мас. %.
Трансмиссионная электронная микроскопия на непигментированных полимерных пленках, изопрен-содержащих полиуретан-полиакрилатных сополимеров
Чтобы исследовать морфологию, получили свободные пленки исходных дисперсий, и их контрастировали при помощи тетраоксида осмия и исследовали с помощью трансмиссионного электронного микроскопа.
Для этой цели, одну каплю исходной дисперсии в каждом случае добавляли к 10 мл деионизированной воды и хорошо перемешивали. Далее, применяли поверхность медной сетки, чтобы захватить одну каплю сильноразбавленной исходной дисперсии, сформированной таким образом, и пленку полимера высушивали под глубоким вакуумом. Это сопровождалось конрастированием пленки в атмосфере, обогащенной тетраоксидом осмия в закрытом сосуде.
Анализ электронной микроскопией выполнили в светопропускании (таб. 1, фиг. 1). Он включал визуализацию спектральных частиц, имеющих размер частиц меньше, чем 150 нм в непрерывной матрице темного или светлого вида, который соответствует морфологии ядро-оболочка:
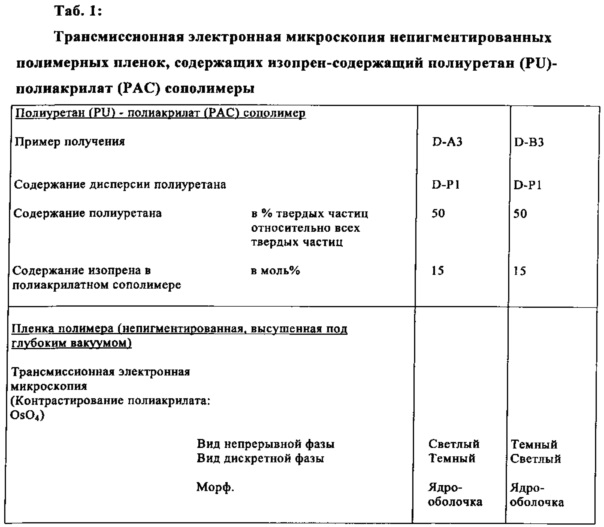
Эксперимент выше, показывает, что структура ядро/оболочка сополимеров изобретенных дисперсий (D-B3) противоположна структуре сополимеров известные из предыдущего уровня техники (D-A3).
Получение серебристых водоразбавляемых базовых покрытий
Компоненты, перечисленные в таблицах 2а, 2б, 3а и 3б под "водной фазой" перемешивают вместе в последовательности установленной, чтобы получить водную смесь. В следующем этапе, компоненты, перечисленные под "органической фазой" применяют, чтобы произвести органическую смесь. Органическую смесь добавляют к водной смеси. Потом смесь перемешивают в течение 10 мин и корректируют с помощью деионизированной воды и N,N-диметилэтаноламина (от BASF SE) до pH 8.0 и вязкости, подходящей для распыления.
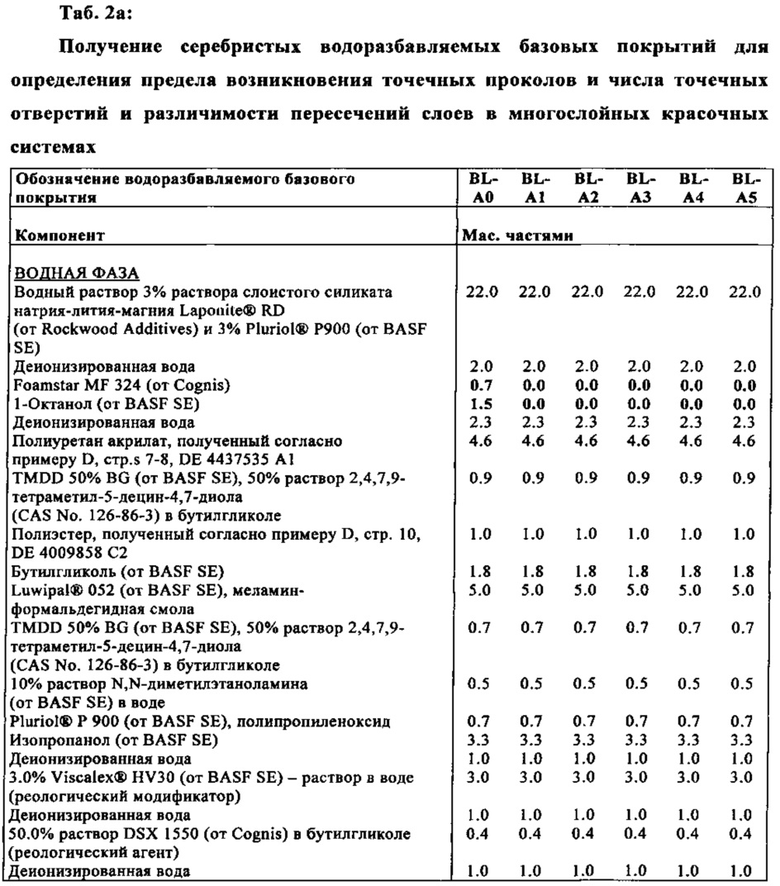









Сравнительные эксперименты между водоразбавляемыми основами покрытия BL-A0 - BL-A5 с изобретенными водоразбавляемыми основами покрытия BL-B1 - BL-B4 относительно предела возникновения точечных проколов, числа точечных отверстий и различимости пересечений слоев в многослойных красочных системах
Чтобы определить предел возникновения точечных проколов, число точечных отверстий и способность скрывать пересечения слоев, исследовали многослойные красочные системы, содержащие водоразбавляемые основы покрытия BL-A0 - BL-B4.
При этом водоразбавляемая основа покрытия BL-A0 содержала Foamstar MF 324 (от Cognis) и 1-октанол (от BASF SE), все другие водоразбавляемые основы покрытия BL-A1 - BL-B4 были свободны от этих двух компонентов.
Чтобы определить предел возникновения точечных проколов и число точечных отверстий, многослойные красочные системы производили с водоразбавляемыми основами покрытий согласно следующему основному способу:
Стальную панель размерами 30 см × 50 см, покрытую грунт-шпатлевочным покрытием, обеспечили тонкой полосной клеящего вещества вдоль одной продольной кромки, для того, чтобы иметь возможность определить различия в толщине слоев после покрытия. Водоразбавляемую основу покрытия наносили электростатически в клинообразной форме. Полученную в результате пленку водоразбавляемой основы покрытия поддали испарению при комнатной температуре в течение одной минуты и затем высушивали при 70°С в сушильном шкафу с циркуляцией воздуха в течение 10 минут. Обычно двухкомпонентное самовосстанавливающиеся покрытие наносят на высушенную пленку водоразбавляемой основы покрытия. Полученную в результате пленку самовосстанавливающегося покрытия поддали испарению при комнатной температуре в течение 20 минут. Затем, пленку водоразбавляемой основы покрытия и пленку самовосстанавливающегося покрытия выдерживали вместе в сушильном шкафу с циркуляцией воздуха при 140°С в течение 20 минут. Толщина слоя пленки выдержанного самовосстанавливающегося покрытия была постоянной (±1 мкм) поверх всего листа, с толщиной пленки самовосстанавливающегося покрытия 38-42 мкм.
После визуальной оценки крошечных отверстий в полученной в результате клиновидной многослойной красочной системе, определяли толщину покрытия для предела возникновения точечных проколов.
В добавление, сравнивали способность скрывать пересечения слоев:
С этой целью, свернутую в спираль панель размерами 30×60 см покрасили грунт-шпатлевкой и грунт-шпатлевку подвергали термической обработке при 160°С в течение периода 20 минут. Кусочек абразивной бумаги, имеющей 800 частиц на см2 поверхности применяли, чтобы отшлифовать пересечение на листе, которое имеет глубину 2-4 мкм и ширину борозды 2 см. Затем, соответствующую основу покрытия наносили толщиной покрытия 8-12 мкм. Затем, полученную в результате панель высушивали при 80°С в течение 10 минут, и наносили обычное и известное двухкомпонентное самовосстанавливающееся покрытие на высушенную пленку водоразбавляемой основы покрытия. Затем, пленку водоразбавляемой основы покрытия и пленку самовосстанавливающегося покрытия выдерживали вместе в сушильном шкафу с циркуляцией воздуха при 140°С в течение 20 минут.
Различимость пересечения слоев в многослойной красочной системе определяли визуально и оценивали по шкале от 1 до 5. По этой шкале, высокий числовой показатель означает хорошую различимость пересечения слоев и низкий числовой показатель хорошую маскировку пересечений слоев.
Таблицы 4 и 5 дают обзор результатов определения предела возникновения точечных проколов, числа точечных отверстий и способности скрывать пересечения слоев для многослойных красочных систем:


Сравнительные тесты между водоразбавляемыми основами покрытия BL-C1 - BL-C3 и изобретенными водоразбавляемыми основами покрытия BL-C4 - BL-C7 относительно улучшения результатов поперечного сечения
Чтобы определить поперечное сечение, исследовали многослойные красочные системы, содержащие сравнительные водоразбавляемые основы покрытия BL-C1 - BL-C3 и изобретенные водоразбавляемые основы покрытия BL-C4 - BL-C7.
Это сделали, применяя панель размерами 10 см × 20 см, которая имела грунт-шпатлевочное покрытие стандартной коммерческой грунт-шпатлевки, в качестве подложки. Во время производства этой подложки, грунт-шпатлевку высушивали косвенно при 80°С в течение периода 10 мин и затем подвергали термической обработке при 190°С в течение периода 30 мин.
Прежде всего, материал основы покрытия согласно таблице 6, в каждом случае наносили на панель пневматическим способом. После, материал основы покрытия, который поддали испарению при комнатной температуре в течение 1 мин, материал основы покрытия высушивали косвенно в сушильном шкафу с циркуляцией воздуха при 70°С в течение периода 10 мин. Затем, материал двухкомпонентного самовосстанавливающегося покрытия FF99-0345 коммерчески доступного от BASF аналогично наносили пневматически и, после испарения при комнатной температуре в течение 20 мин, два слоя подвергали термической обработке при температуре 160°С в течение периода 30 мин. Результатом была подвергнутая слишком длительной термической обработке многослойная красочная система.
После охлаждения подвергнутой слишком длительной термической обработке многослойной красочной системы, красочные системы очистили. Затем, особый материал основы покрытия согласно таблице 3 нанесли пневматически. После, материал основы покрытия, который поддали испарению при комнатной температуре в течение 1 минуты, материал основы покрытия высушили косвенно в сушильном шкафу с циркуляцией воздуха при 70°С в течение периода 10 мин. Затем, двухкомпонентный заново отделанный материал самовосстанавливающегося покрытия, состоящий из FF23-0500 и SC29-0093 коммерчески доступных от BASF нанесли пневматически и, после испарения при комнатной температуре в течение 20 мин, два слоя подвергли термической обработке при температуре 80°С в течение периода 30 мин. Результатом была заново отделанная красочная система.
Адгезию исследовали на подвергнутом неполной термической обработке, заново отделанном покрытии красочной системы, стертой, подвергнутой слишком длительной термической обработке многослойной красочной системе, тестом поперечного сечения.
Тест поперечного сечения проводили по DIN 2409. Оценку результатов теста поперечного сечения проводили по DIN EN ISO 2409.
Таблица 6 дает обзор результатов теста поперечного сечения со сравнением различных водоразбавляемых материалов основы покрытия:


| название | год | авторы | номер документа |
|---|---|---|---|
| ПИГМЕНТНЫЕ ПАСТЫ, ВКЛЮЧАЮЩИЕ ВОДНУЮ ДИСПЕРСИЮ СОПОЛИМЕРА | 2014 |
|
RU2678197C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2675915C1 |
| ВОДНЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ ПОЛИМЕРЫ, ПОЛУЧЕННЫЕ МНОГОСТАДИЙНЫМ СПОСОБОМ, И КОМПОЗИЦИИ МАТЕРИАЛА ПОКРЫТИЯ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2743013C2 |
| ВОДНЫЕ СВЯЗУЮЩИЕ ВЕЩЕСТВА ДЛЯ ПОКРЫТИЙ С УЛУЧШЕННЫМ БЛЕСКОМ | 2007 |
|
RU2434910C2 |
| ВОДНЫЕ ДИСПЕРСИИ, ВКЛЮЧАЮЩИЕ ПОЛИМЕРЫ, ИЗГОТОВЛЯЕМЫЕ МНОГОСТАДИЙНЫМ СПОСОБОМ, И КОМПОЗИЦИИ ПОКРЫВАЮЩИХ АГЕНТОВ, ВКЛЮЧАЮЩИЕ ИХ | 2016 |
|
RU2675564C1 |
| ВОДНЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ ПО МЕНЬШЕЙ МЕРЕ ОДНУ АЛКИДНУЮ СМОЛУ И ПО КРАЙНЕЙ МЕРЕ ОДИН ПОЛИМЕРИЗАТ ПО МЕНЬШЕЙ МЕРЕ С ОДНИМ (МЕТ)АКРИЛАТНЫМ СЕГМЕНТОМ | 2008 |
|
RU2478677C2 |
| КАРБОКСИФУНКЦИОНАЛЬНЫЕ ПРОДУКТЫ РЕАКЦИИ НА ОСНОВЕ ПРОСТОГО ПОЛИЭФИРА И ВОДНЫЕ ГРУНТОВОЧНЫЕ МАТЕРИАЛЫ, ВКЛЮЧАЮЩИЕ ПРОДУКТЫ РЕАКЦИИ | 2016 |
|
RU2708390C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2667274C1 |
| КАРБОКСИФУНКЦИОНАЛЬНЫЕ ПРОДУКТЫ РЕАКЦИИ НА ОСНОВЕ ПРОСТОГО ПОЛИЭФИРА И ВОДНЫЕ ГРУНТОВОЧНЫЕ МАТЕРИАЛЫ, ВКЛЮЧАЮЩИЕ ПРОДУКТЫ РЕАКЦИИ | 2016 |
|
RU2707886C1 |
| СОСТАВ ДЛЯ ПОКРЫТИЯ НА ВОДНОЙ ОСНОВЕ И ИЗГОТОВЛЕНИЕ МНОГОСЛОЙНЫХ КРАСОЧНЫХ СИСТЕМ С ПРИМЕНЕНИЕМ УКАЗАННОГО СОСТАВА ДЛЯ ПОКРЫТИЯ | 2014 |
|
RU2677334C1 |

Настоящее изобретение относится к водной дисперсии для получения материалов покрытия, к водоразбавляемому базовому покрытию, применимому для покрытия автомобильных кузовов и/или частей для установления на автомобильные кузова, способу получения многослойной цветной и/или для эффекта красочной системы и к способу получения водной дисперсии. Указанная водная дисперсия включает, по меньшей мере, один сополимер, полученный в результате полимеризации олефиново ненасыщенных мономеров в присутствии водной дисперсии полиуретана с применением водорастворимого инициатора. Олефиново ненасыщенные мономеры дозируют так, чтобы их концентрация в растворе реакции не превышала в ходе всей продолжительности реакции 6.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров. Смесь олефиново ненасыщенных мономеров включает, по меньшей мере, один полиолефиново ненасыщенный мономер. Покрытия, полученные с использованием указанных дисперсий, обладают высоким пределом возникновения точечных проколов и, следовательно, наименьшим количеством поверхностных дефектов, а также улучшенной адгезией в случае многослойной окончательной отделки. 6 н. и 11 з.п. ф-лы, 4 ил., 6 табл., 6 пр.
1. Водная дисперсия для получения материалов покрытия, включающая, по меньшей мере, один сополимер, причем сополимер получен путем
(I) первоначально загружают водную дисперсию, по меньшей мере, одного полиуретана, и затем
(II) полимеризуют смесь олефиново ненасыщенных мономеров в присутствии полиуретана из (I),
где
(а) применяют водорастворимый инициатор в количестве от 0.05 до 20 мас. % в пересчете на общее количество олефиново ненасыщенных мономеров,
(б) дозированное добавление олефиново ненасыщенных мономеров проводят таким образом, что концентрация 6.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров, в растворе реакции не превышена в ходе всей продолжительности реакции, и
(в) смесь олефиново ненасыщенных мономеров включает, по меньшей мере, один полиолефиново ненасыщенный мономер.
2. Водная дисперсия по п. 1, где дозированное добавление олефиново ненасыщенных мономеров проводят таким образом, что концентрация 5.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров, в растворе реакции не превышена в ходе всей продолжительности реакции.
3. Водная дисперсия по п. 2, где дозированное добавление олефиново ненасыщенных мономеров проводят таким образом, что концентрация 4.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров, в растворе реакции не превышена в ходе всей продолжительности реакции.
4. Водная дисперсия по любому из пп. 1-3, где смесь олефиново ненасыщенных мономеров содержит 0.1-6.0 моль % полиолефиново ненасыщенных мономеров.
5. Водная дисперсия по п. 4, где смесь олефиново ненасыщенных мономеров содержит 0.1-2.0 моль % полиолефиново ненасыщенных мономеров.
6. Водная дисперсия по п. 4, где смесь олефиново ненасыщенных мономеров содержит 0.1-6.0 моль % аллил метакрилата и не содержит дополнительно каких-либо полиолефиново ненасыщенных мономеров.
7. Водная дисперсия по п. 6, где смесь олефиново ненасыщенных мономеров содержит 0.1-2.0 моль % аллил метакрилата и не содержит дополнительно каких-либо полиолефиново ненасыщенных мономеров.
8. Водная дисперсия по любому из пп. 1-3, 5-7, где смесь олефиново ненасыщенных мономеров содержит <10.0 мас. % винилароматических мономеров, в пересчете на общее количество олефиново ненасыщенных мономеров.
9. Водная дисперсия по п. 8, где смесь олефиново ненасыщенных мономеров содержит <5.0 мас. % винилароматических мономеров, в пересчете на общее количество олефиново ненасыщенных мономеров.
10. Водная дисперсия по любому из пп. 1-3, 5-7, 9, где смесь олефиново ненасыщенных мономеров не содержит каких-либо винилароматических мономеров.
11. Водная дисперсия по любому из пп. 1-3, 5-7, 9, где водная дисперсия имеет содержание геля 40-90 мас. %, в пересчете на твердые частицы в дисперсии.
12. Водная дисперсия по любому из пп. 1-3, 5-7, 9, где сополимер содержит, по меньшей мере,
(A) ядро, включающие, по меньшей мере, один полиуретан, и
(B) оболочку, включающую, по меньшей мере, один полимер, который получали полимеризацией олефиново ненасыщенных мономеров.
13. Применение водной дисперсии по любому из пп. 1-12 для получения материалов покрытия.
14. Водоразбавляемое базовое покрытие, включающее водную дисперсию по любому из пп. 1-12.
15. Способ получения многослойной цветной и/или для эффекта красочной системы, в которой
(I) пигментированное водоразбавляемое базовое покрытие наносят на основу,
(II) формируют полимерную пленку из материала покрытия, нанесенного на стадии (I),
(III) наносят самовосстанавливающееся покрытие на слой базового покрытия, полученного таким образом, и затем
(IV) слой базового покрытия отверждают вместе со слоем самовосстанавливающегося покрытия,
который включает применение на стадии (I) пигментированного водоразбавляемого базового покрытия, причем указанное базовое покрытие включает водную дисперсию по любому из пп. 1-12.
16. Применение водоразбавляемого базового покрытия по п. 14 для покрытия автомобильных кузовов и/или пластиковых частей для установления на автомобильные кузова.
17. Способ получения водной дисперсии, отличающийся стадиями получения:
(I) первоначально загружают водную дисперсию, по меньшей мере, одного полиуретана, и затем
(II) полимеризуют смесь олефиново ненасыщенных мономеров в присутствии полиуретана из (I),
где
(а) применяют водорастворимый инициатор,
(б) дозированное добавление олефиново ненасыщенных мономеров проводят таким образом, что концентрация 6.0 мас. %, в пересчете на общее количество олефиново ненасыщенных мономеров в растворе реакции не превышена в ходе всей продолжительности реакции, и
(в) смесь олефиново ненасыщенных мономеров включает, по меньшей мере, один полиолефиново ненасыщенный мономер.
| US 6632915 B1, 14.10.2003 | |||
| US 6770702 B1, 03.08.2004 | |||
| US 20040234487 A1, 25.11.2004 | |||
| RU 2005129538 A, 10.02.2006 | |||
| WO 1991015528 A, 17.10.1991. |

