Изобретение относится к способу получения многослойной красочной системы, в которой базовое покрытие или непосредственно последовательные базовые покрытия получают непосредственно на металлической основе, покрытой отвержденным покрытием, наносимым электрохимическим способом (электрохимическое покрытие), прозрачный слой лакового покрытия (лаковое покрытие) получают непосредственно на одном базовом покрытии или на самом верхнем из множества покрытий, а потом одно или более базовых покрытий и лаковое покрытие совместно отверждают. Настоящее изобретение дополнительно относится к многослойной красочной системе, которая была получена с помощью способа в соответствии с изобретением.
Область техники
Многослойные красочные системы на металлических основах, например, многослойные красочные системы в автомобильной промышленности, являются известными. В общем случае, многослойные красочные системы такого вида включают, если смотреть со стороны основы наружу, электрохимическое покрытие, слой, который наносится непосредственно на электрохимическое покрытие и обычно называется грунтовочным покрытием, по крайней мере, одно покрытие, которое включает цветные пигменты и/или пигменты эффекта и обычно называется базовым покрытием, и лаковое покрытие.
Основной состав и функции этих слоев и композиций для покрытия, которые являются необходимыми для формирования этих слоев, то есть материалы для покрытия, которое наносится электрохимическим путем, так называемые грунтовки, композиции покрытия, которые включают цветные пигменты и/или пигменты эффекта и являются известными как материалы базового покрытия, а также материалы лакового покрытия, являются известными. Например, электрохимическое покрытие, которое наносится путем электрофореза, служит в основном для защиты основы от коррозии. Так называемое грунтовочное покрытие служит, главным образом, для защиты от механических нагрузок, например, для защиты от ударов мелкими камнями, и дополнительно для выравнивания неровностей в основе. Следующий слой, который называется базовым покрытием, главным образом, является ответственным за создание эстетических свойств таких, как цвет и/или эффекты такие, как флоп-эффект, в то время как лаковое покрытие, которое следует за ним, служит, в частности, для придания многослойной красочной системе стойкости к царапинам и глянца.
Такие многослойные красочные системы, в общем случае, получают путем первого нанесения или напыления электрохимического покрытия, в частности, катодного электрохимического покрытия, которое наносится с помощью электрофореза на металлическую основу, например, кузов автомобиля. Перед нанесением покрытия, которое наносится электрохимическим путем, металлическая основа может быть предварительно обработана различными способами; например, является возможным применять известные конверсионные покрытия такие, как фосфатные покрытия, в частности, покрытия при использовании фосфата цинка. Процесс нанесения электрохимического покрытия в общем случае осуществляется в приемлемых ваннах для электрохимического покрытия. После такого нанесения основу с покрытием извлекают из ванны, необязательно, промывают и подвергают испарению и/или промежуточному высушиванию, а нанесенное электрохимическим путем покрытие окончательно отверждают. Целевая толщина пленки составляет примерно от 15 до 25 микрометров. В дальнейшем так называемая грунтовка наносится непосредственно на отвержденное электрохимическое покрытие, необязательно подвергается испарению и/или промежуточному высушиванию, а затем отверждению. Для того чтобы отвержденное грунтовочное покрытие могло выполнять вышеуказанные задачи, целевая толщина пленки составляет, например, от 25 до 45 микрометров. После этого так называемые базовое покрытие, которое включает цветные пигменты и/или пигменты эффекта, непосредственно наносят на отвержденное грунтовочное покрытие и подвергают необязательному испарению и/или промежуточному высушиванию, и лаковое покрытие непосредственно наносят на базовое покрытие полученное, таким образом, без отдельного отверждения. В дальнейшем базовое покрытие и лаковое покрытие, которое при необходимости, также было подвергнуто испарению и/или предварительному промежуточному высушиванию, совместно отверждают (метод окраски по влажному слою). В то время как отвержденное базовое покрытие, в принципе, имеет сравнительно низкую толщину пленки, например, от 10 до 30 микрометров, целевая толщина пленки для отвержденного лакового покрытия, составляет, например, от 30 до 60 микрометров, для того, чтобы достичь описанных эксплуатационных свойств. Грунтовка, базовое покрытие и лаковое покрытие могут быть нанесены, например, с помощью методов нанесения, которые являются известными квалифицированным специалистам в данной области техники, путем пневматического и/или электростатического напыление. В настоящее время грунтовочное покрытие и базовое покрытие все чаще используют в виде водных материалов для покрытия, по крайней мере, из экологических соображений.
Многослойные красочные системы такого вида и способы их получения описываются, например, в DE 199 48 004 А1, от стр. 17 строка 37 до стр. 19 строка 22, или также в DE 100 43 405 С1, колонка 3 абзац [0018] и от колонки 8 абзац [0052] до колонки 9 абзац [0057], в сочетании с колонкой 6 абзац [0039] до колонки 8 абзац [0050].
Даже несмотря на то, что многослойные красочные системы, полученные таким образом, могут в общем случае соответствовать требованиям, предъявляемым в автомобильной промышленности к эксплуатационным свойствам и эстетическому профилю, упрощение сравнительно сложного описанного производственного процесса по экологическим и экономическим причинам в настоящее время является предметом повышенного внимания производителей автомобилей.
Например, существуют подходы, в которых делается попытка обойтись без отдельного этапа отверждения композиции для покрытия, которая наносится непосредственно на отвержденное электрохимическое покрытие (для композиции покрытия, которая называется как грунтовочное покрытие в контексте описанного выше стандартного способа), а также одноверменно уменьшить толщину пленочного покрытия, полученного из этой композиции для покрытия. В специальной области это пленочное покрытия, которое, таким образом, не отверждают отдельно, часто называют базовым покрытием (но уже не грунтовочным покрытием), или первым базовым покрытием, в отличие от второго базового покрытия, которое наносится на него. Кроме того, было сделано несколько попыток полностью избавиться от этого пленочного покрытия (только в этом случае так называемое базовое покрытие получают непосредственно на электрохимическом покрытии, которое покрыто сверху лаковым покрытием, без отдельного этапа отверждения, а это означает, что аналогичным образом можно обойтись без отдельного этапа отверждения). Вместо отдельного этапа отверждения и дополнительного заключительного этапа отверждения, может, таким образом, существовать только заключительный этап отверждения после нанесения всех пленочных покрытий, которые наносятся на электрохимическое покрытие.
В частности, отсутствие отдельного этапа отверждения для композиции покрытия, нанесенной непосредственно на электрохимическое покрытие, является весьма предпочтительным с экологической и экономической точки зрения. Это обусловлено тем, что это приводит к экономии энергии, и общий производственный процесс, конечно, может осуществляться гораздо более точно и быстро.
Вместо отдельного этапа отверждения, является, таким образом, предпочтительным, чтобы пленочное покрытие, полученное непосредственно на электрохимическом покрытии, подвергалось только испарению при комнатной температуре и/или промежуточному высушиванию при повышенных температурах, без проведения операции отверждения, что, как известно, постоянно требуют повышенных температур отверждение и/или длительного времени отверждения.
Однако является проблематичным, что необходимые эксплуатационные эстетические свойства часто не могут быть получены в настоящее время при такой форме производства.
Например, отказ от отдельного этапа отверждения пленочного покрытия, которое наносится непосредственно на электрохимическое покрытие, например, первое базовое покрытие, перед нанесением дополнительных композиций для покрытия, например, второго материала базового покрытия и материала лакового покрытия, может привести к возникновению нежелательных включений воздуха, растворителя и/или влаги, что может проявляться в форме пузырьков ниже поверхности всей красочной системы и может разрушать ее при заключительном отверждении.
Отверстия, образующиеся в красочной системе, которые также называются как микроотверстия, приводят к неблагоприятному внешнему виду. Количество органического растворителя и/или воды, которое возникает в процессе общего наслоения материала первого базового покрытия, второго базового покрытия и лакового покрытия, и количество воздуха, который введен в процессе нанесения, является слишком большим для общего количества, чтобы выйти из красочной системы на заключительной стадии отверждения при отсутствии образования дефектов. В случае традиционного процесса получения, как описывается выше, при котором так называемое грунтовочное покрытие отверждается отдельно перед получением обычно сравнительно тонкого базового покрытия (которое, таким образом, включает в себя только сравнительно немного воздуха, органического растворителя и/или воды), к решению этой проблемы, конечно, предъявляются гораздо меньшие требования.
Однако при получении многослойных красочных систем, в которых применение композиции для покрытия, которая называется грунтовочным покрытием, в стандартном процессе полностью устраняется, то есть, системы, в которых только один материал так называемого базового покрытия непосредственно наносят на отвержденное электрохимическое покрытие, описанные проблемы, связанные с микроотверстиям, также часто встречаются. Это происходит по той причине, что в соответствии с применением и использованием многослойной красочной системы, которую получают, полное отсутствие покрытия, которое называется как грунтовочное покрытие в стандартном процессе, в общем случае требует более толстого базового покрытия по сравнению со стандартными системами для того, чтобы получить желаемые свойства. Таким образом, в этом случае также общая толщина пленочного покрытия, которое должно быть отверждено на заключительном этапе отверждения, является намного больше, чем в стандартном процессе, что может привести к возникновению соответствующих проблем с микроотверстиями.
Дополнительный фактор заключается в том, что замена композиций для покрытия на основе органических растворителей водными композициями для покрытия становится все более важной в наше время для того, чтобы удовлетворить растущие требования в отношении охраны окружающей среды.
Соответственно, было бы выгодно иметь способ получения многослойных красочных систем, в которых является возможным избежать отдельного этапа отверждения, как описывается выше, для композиции покрытия, которая непосредственно наносится на электрохимическое покрытие, и полученная многослойная красочная система, тем не менее, имеет отличную стабильность в отношении микроотверстий.
Проблема, рассматриваемая в настоящем изобретении, соответственно, заключается в том, чтобы отыскать способ получения многослойной красочной системы на металлических основах, в которой композиция для покрытия наносится непосредственно на электрохимическое покрытие, не отверждается отдельно, но в котором эта композиция для покрытия, вместо этого, отверждается на совместном этапе отверждения с дополнительными пленочными покрытиями, которые наносятся на последующих этапах. Несмотря на этот способ упрощения, полученные многослойные красочные системы должны иметь отличную устойчивость к образованию микроотверстий так, чтобы многослойные красочные системы, в частности, соответствовали эстетическим требованиям производителей автомобилей и их потребителей. Кроме того, должно быть возможным, таким образом, в соответствии с требованиями и индивидуальной областью применения обеспечить многослойные красочные системы, в которых одна или более композиция(ий) для покрытия, которая(ые) размещается(ются) между электрохимическим покрытием и лаковым покрытием, может(могут) иметь вариабельную толщину покрытия, и в которых не возникает проблем с микроотверстиями, в частности, при относительно высокой толщине покрытия. В то же время композиция для покрытия, которая наносится на отвержденное электрохимическое покрытие, но перед материалом лакового покрытия, должна быть водной для того, чтобы соответствовать растущим требованиям экологического профиля красочных систем.
Было обнаружено, что упомянутые проблемы решаются с помощью нового способа получения многослойной красочной системы (М) на металлическом субстрате (S), который включает:
(1) получение отвержденного электрохимического покрытия (E.1) на металлической основу (S) путем электрофоретического нанесения электрохимического покрытия (е.1) на основу (S) и последующее отверждение электрохимического покрытия (е.1),
(2) получение (2.1) базового покрытия (В.2.1) или (2.2) непосредственно последовательных базовых покрытий (В.2.2.x) непосредственно на отвержденном электрохимическом покрытии, (Е.1) путем (2.1) нанесения материала водного базового покрытия (b.2.1) непосредственно на электрохимическое покрытие (Е.1) или (2.2) нанесения материалов множества базовых покрытий (b.2.2.х) в непосредственной последовательности на покрытие, которое наносится электрохимическим путем (Е.1),
(3) получение лакового покрытия (K) непосредственно на (3.1) базовом покрытии (В.2.1) или (3.2) самом верхнем базовом покрытии (В.2.2.x) путем нанесения материала лакового покрытия (k) непосредственно на (3.1) базовое покрытие (В.2.1) или (3.2) на самое верхнее базовое покрытие (В.2.2.x),
(4) совместное отверждение (4.1) базового покрытия (В.2.1) и лакового покрытия (K) или (4.2) базовых покрытий (В.2.2.x) и лакового покрытия (K),
где
материал базового покрытия (b.2.1) или, по крайней мере, один из материалов базового покрытия (b.2.2.х) включает,
по крайней мере, одну водную дисперсию, которая включает, по крайней мере, один сополимер (CP), где указанный сополимер (CP) получают путем:
(i) первоначальной загрузки водной дисперсии, по крайней мере, одного полиуретана, и затем
(ii) полимеризации смеси олефиновых ненасыщенных мономеров в присутствии полиуретана из (i), где
(a) используют растворимый в воде активатор,
(b) олефиновые ненасыщенные мономеры дозируют таким образом, что концентрация 6,0% по весу в расчете на общее количество олефиновых ненасыщенных мономеров, которые используются для полимеризации, в реакционном растворе не превышается в течение всего времени реакции, и
(c) смесь олефиновых ненасыщенных мономеров включает, по крайней мере, один полиолефиновый ненасыщенный мономер,
и включает,
по крайней мере, один линейный гидроксифункцональный продукт реакции (R), имеющий кислотное число менее, чем 20 мг KOH/г, получение которого вовлекает применение, по крайней мере, одного соединения (v), содержащего две функциональные группы (v.1) и алифатический или аралифатический радикал гидрокарбила (v.2), который размещается между функциональными группами и содержит от 12 до 70 атомов углерода.
Упомянутый выше способ также называется в данной заявке ниже как способ в соответствии с изобретением и, соответственно, образует часть предмета изобретения в соответствии с настоящим изобретением. Предпочтительные воплощения способа в соответствии с изобретением могут быть найдены в описании, которое представлено ниже, а также в зависимых пунктах формулы.
Настоящее изобретение дополнительно обеспечивает многослойную красочную систему, которая была получена с помощью способа в соответствии с изобретением.
Способ в соответствии с изобретением позволяет осуществлять получение многослойных красочных систем при отсутствии отдельного этапа отверждения для пленочного покрытия, полученного непосредственно на электрохимическом покрытии. С целью большей ясности, это пленочное покрытие называется как базовое покрытие в контексте настоящего изобретения. Вместо отдельного отверждения, это базовое покрытие совместно отверждают вместе с любыми дополнительными базовыми покрытиями, которые располагаются под лаковым покрытием, и лаковым покрытием. Несмотря на это, использование способа в соответствии с изобретением приводит к получению многослойных красочных систем, которые имеют отличную стабильность в отношении микроотверстий, так, что даже относительно высокая толщина покрытия соответствующих пленок базового покрытия может быть получена без какой-либо потери эстетического качества. Является также возможным сформировать соответствующие базовые покрытия при использовании водной композиции покрытия для того, чтобы удовлетворить требования охраны окружающей среды.
Прежде всего, следует разъяснить некоторые термины, которые используются в настоящем изобретении.
Если делается ссылка в контексте настоящего изобретения на официальный стандарт без указания официального срока действия, то это, конечно, означает текущую версию стандарта на дату подачи или, если на эту дату не существовало текущей версии стандарта, то последнюю текущую версию.
Нанесение композиция для покрытия на основу или получение пленочного покрытия на основе понимают следующим образом. Соответствующая композиция для покрытия наносится таким образом, что пленочное покрытие, которое получают, таким образом, размещается на основе, но с необходимостью не требует того, чтобы находиться в непосредственном контакте с основой. Другие слои, например, могут также размещаться между пленочным покрытием и основой. Например, на этапе (1) отвержденное электрохимическое покрытие (Е.1) наносят на металлическую основу (S), но конверсионное покрытие, как описывается ниже, такое, как покрытие фосфатом цинка, может также размещаться между основой и электрохимическим покрытием.
Тот же принцип применяется к нанесению композиции для покрытия (b) на пленочное покрытие (А), полученное при использовании другой композиции для покрытия (а), или к получению пленочного покрытия (В) на другом пленочном покрытии (А), которое размещается, например, на металлической основе (S). Пленочное покрытие (В) с необходимостью не требует того, чтобы находиться в контакте со слоем покрытия (А), а просто должно размещаться сверху него, то есть, на стороне пленочного покрытия (А) верхней частью от металлической основы.
В противовес этому, нанесение композиции для покрытия непосредственно на основу или получение пленочного покрытия непосредственно на основе понимается так, как описано ниже. Соответствующая композиция для покрытия наносится таким путем, что пленочное покрытия, которое получают, таким образом, размещается на основе и находится в непосредственном контакте с основой. Таким образом, более конкретно никакой другой слой не размещается между пленочным покрытием и основой. Конечно, то же самое применяется к нанесению композиция для покрытия (b) непосредственно на пленочное покрытие (А), полученное с помощью другой композиции для покрытия (а), или получению пленочного покрытия (В) непосредственно на другом пленочном покрытии (А), которое размещается, например, на металлической основе (S). В этом случае два пленочных покрытия находятся в непосредственном контакте, то есть, размещаются непосредственно один поверх другого. В частности, не существует дополнительного слоя между пленочными покрытиями (А) и (В).
Конечно, этот же принцип применяется к непосредственному последовательному нанесению композиции для покрытия или получению непосредственно последовательных пленочных покрытий.
В контексте настоящего изобретения "испарение", "промежуточное высушивание" и "отверждение" понимаются в том значении, которое является известным квалифицированному специалисту в данной области техники в связи со способами получения многослойных красочных систем.
Таким образом, термин "испарение" понимается, в принципе, в качестве термина для обозначения испарения или обеспечения возможности испарения органических растворителей и/или воды в композиции для покрытия в производстве красочной системы, как правило, при температуре окружающей среды (то есть, при комнатной температуре), например, при температуре от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин. При осуществлении операции испарения органические растворители и/или вода, присутствующие в применяемой композиции покрытия, испаряются. Поскольку композиция для покрытия все еще остается свободнотекучей, по крайней мере, непосредственно после нанесения и в начале осуществления операции испарения, она может расплываться во время проведения операции испарения. Это происходит, по крайней мере, по той причине, что композиция, которая наносится путем распыления, как правило, применяется в виде капель и не имеет однородной толщины. Тем не менее, она является свободнотекучей благодаря присутствующим органическим растворителям и/или воде, и может, таким образом, формировать однородное, гладкое пленочное покрытие путем растекания. В то же время, органические растворители и/или вода испаряются постепенно, так, что сравнительно гладкое пленочное покрытие формируется после фазы испарения, которое содержит меньшее количество воды и/или растворителя, по сравнению с наносимой композицией покрытия. Однако после осуществления операции испарения пленочное покрытие все еще не находится в состоянии готовности к использованию. Например, оно более не является свободнотекучим, но по-прежнему является мягким и/или липким, а в некоторых случаях - только частично высушенным. В частности, пленочное покрытие все еще не является отвержденным, как описывается ниже.
Промежуточное высушивание, таким образом, также понимается как испарение или обеспечение возможности испарения органических растворителей и/или воды из композиции для покрытия, которая наносится при производстве красочной системы, как правило, при температуре, повышенной по отношению к температуре окружающей среды, например, при температуре от 40 до 90°C, в течение периода, например, от 1 до 60 мин. При осуществлении операции промежуточного высушивания нанесенная композиция покрытия также, таким образом, теряет часть органических растворителей и/или воды. Что касается конкретной композиции для покрытия, то, как правило, бывает так, что промежуточное высушивание, по сравнению с испарением, происходит, например, при более высоких температурах и/или в течение более длительного периода времени, так что, по сравнению с испарением, более высокое содержание органических растворителей и/или воды устраняется из нанесенного пленочного покрытия. Тем не менее, промежуточное высушивание не обеспечивает получения пленочного покрытия в состоянии, готовом к использованию, т.е. отвержденного пленочного покрытия, как описывается ниже. Типичная последовательность операций испарения и промежуточного высушивания будет включать, например, испарение пленочного покрытия, нанесенного при температуре окружающей среды в течение 5 мин., с последующим промежуточным высушиванием при 80°C в течение 10 мин. Тем не менее, никакое убедительное разграничение этих двух терминов не является необходимым или предполагаемым. Из соображений ясности, следует сказать, что эти термины используются для того, чтобы понять, что операция отверждения, описанная ниже, может предшествовать вариабельному и последовательному кондиционированию пленочного покрытия, в котором - в зависимости от состава покрытия, температуры испарения и время испарения - более высокое или более низкое содержание органических растворителей и/или воды, присутствующей в композиции покрытия может испаряться. При определенных обстоятельствах содержание полимеров, присутствующих в композициях покрытий в качестве связующих веществ, даже на этой ранней стадии, может вызывать поперечное сшивание или переплетение, как описано ниже. Тем не менее, ни испарение, ни промежуточная операция высушивания не обеспечивают получения готового к использованию пленочного покрытия, как это достигается путем отверждения, описанного ниже. Соответствующим образом, отверждение четко отграничено от операций испарения и промежуточного высушивания.
Соответственно, отверждение пленочного покрытия понимается в значении превращения такой пленки до готового к применению состояния, то есть, до состояния, в котором основа, обеспеченная соответствующим пленочным покрытием, может транспортироваться, храниться и использоваться по целевому назначению. В частности, отвержденное пленочное покрытие уже не является мягким или липким, но было доведено до твердого состояния пленочного покрытия, которое не подвергается какому-либо дополнительному значимому изменению своих свойств таких, как твердость или адгезия к субстрату, даже при дополнительном воздействии условий отверждения, как описывается ниже.
Как является хорошо известным, композиции покрытия могут в принципе быть отвержденными физически и/или химически в зависимости от присутствующих компонентов таких, как связующие агенты и поперечно сшивающие агенты. В этом случае химическое отверждение, термохимическое отверждение и актинохимическое отверждение представляют собой варианты. Если идет речь о способности к термохимическому отверждению, то композиция для покрытия может представлять собой такую, которая способна к самосшиванию и/или внешнему сшиванию. Указание того, что композиция для покрытия способна к самосшиванию и/или внешнему сшиванию в контексте настоящего изобретения следует понимать для обозначения того, что эта композиция для покрытия включает полимеры такие, как связующие агенты и, необязательно, перекрестно сшивающие агенты, которые могут соответствующим образом связываться друг с другом. Лежащие в основе этого механизмы, а также используемые связующие агенты и перекрестно сшивающие агенты описываются ниже.
В контексте настоящего изобретения термин "способный к физическому отверждению" или термин "физическое отверждение" означает образование отвержденного пленочного покрытия посредством высвобождения растворителя из растворов полимеров или полимерных дисперсий, где отверждение достигается с помощью внутреннего переплетения полимерных цепей.
В контексте настоящего изобретения термин "способный к термохимическому отверждению" или термин "термохимическое отверждение" означает перекрестное сшивание, которое инициируется с помощью химической реакции реактивных функциональных групп красочного покрытия (образование отвержденного пленочного покрытия), при этом является возможным обеспечить энергию активации для этих химических реакций с помощью термальной энергии. Это может вовлекать реакцию различных взаимно комплементарных функциональных групп друг с другом (комплементарные функциональные группы) и/или образование отвержденного слоя на основе реакции аутореактивных групп, то есть, функциональных групп, которые взаимодействуют с группами того же вида. Примеры приемлемых комплементарных реактивных функциональных групп и аутореактивных функциональных групп являются известными, например, из немецкой патентной заявки DE 199 30 665 А1, стр. 7 строка 28 до стр. 9 строка 24.
Такое перекрестное сшивание может представлять собой самосшивание и/или внешнее сшивание. Если, например, комплементарные реактивные функциональные группы уже присутствуют в органическом полимере, который используется как связующий агент, например, полиэстер, полиуретан или поли(мет)акрилат, то присутствует самосшивание. Внешнее сшивание присутствует, например, тогда, когда (первый) органический полимер, содержащий определенные функциональные группы, например, гидроксильные группы, вступает в реакцию с поперечно сшивающим агентом, который является известным сам по себе, например, полиизоцианатом и/или меламиновой смолой. Сшивающий агент, таким образом, содержит реактивные функциональные группы комплементарные реактивным функциональным группам, присутствующим в (первом) органическом полимере, который используется в качестве связующего агента.
В частности, в случае внешнего сшивания используются однокомпонентная и мультикомпонентная системы, в частности, двухкомпонентные системы, которые являются известными сами по себе.
В однокомпонентных системах, которые подвергаются поперечному сшиванию, например, органические полимеры в качестве связующих агентов и сшивающих агентов, присутствуют вместе друг с другом, то есть, в одном компоненте. Предпосылкой этого является то, что компоненты, которые должны быть поперечно сшиты, подвергаются реакции друг с другом, то есть, вступают в реакцию отверждения только при относительно высоких температурах, например, при температурах выше 100°C. В ином случае компоненты, которые подвергаются поперечному сшиванию, должны храниться отдельно друг от друга и быть смешаны друг с другом только незадолго перед нанесением на основу для того, чтобы избежать преждевременного, по крайней мере, частичного термохимического отверждения (см. двухкомпонентные системы). Примером комбинации является такая гидрокси-функциональных полиэстеров и/или полиуретанов с меламиновыми смолами и/или блокированными полиизоцианатами в качестве сшивающих агентов.
В двухкомпонентных системах компоненты, которые подвергаются поперечному сшиванию, например, органические полимеры в качестве связующих агентов и сшивающих агентов, присутствуют отдельно, по крайней мере, в двух компонентах, которые соединяются незадолго до применения. Эта форма выбирается тогда, когда компоненты, которые подвергаются поперечному сшиванию, вступают в реакцию друг с другом даже при температурах окружающей среды или слегка повышенных температурах, например, при температурах от 40 до 90°C. Примером комбинации является такая гидрокси-функциональных полиэстеров и/или полиуретанов и/или поли(мет)акрилатов со свободными полиизоцианатами в качестве сшивающих агентов.
Является также возможным, чтобы органический полимер в качестве связующего агента имел как самосшивающиеся, так и внешне сшивающиеся функциональные группы, и потом соединялся со сшивающими агентами.
В контексте настоящего изобретения термин "способный к актинохимическому отверждению" или термин "актинохимическое отверждение" понимается в значении того факта, что отверждение осуществляется при использовании актиничного излучения, в частности, электромагнитного излучения такого, как ближнее инфракрасное (NIR) и УФ излучение, в частности, УФ излучение, и корпускулярного излучения, такого, как пучки электронов, для отверждения. Отверждение при использовании УФ излучения обычно вызывается фотоинициаторами радикалов или катионов. Типичные способные к актиничному отверждению функциональные группы представляют собой двойные связи углерод-углерод, для которых обычно используются фотоинициаторы свободных радикалов. Актиничное отверждение, таким образом, также основывается на химическом поперечном сшивании.
Безусловно, при отверждении композиции для покрытия, описанной как химически отверждаемая, всегда возможно также возникновение физического отверждения, то есть, взаимного переплетения полимерных цепей. Тем не менее, такая композиция для покрытия описывается как химически отверждаемая в данном случае.
Из изложенного выше следует, что в соответствии с природой композиции для покрытия и компонентами, которые в ней присутствуют, отверждение осуществляется с помощью различных механизмов, которые, конечно, требует также различных условий отверждения, в частности, различных температур отверждения и времени отверждения.
В случае чисто физического отверждения композиции для покрытия отверждение осуществляется предпочтительно при температуре от 15 до 90°C в течение периода времени от 2 до 48 часов. В этом случае отверждение может, таким образом, отличаться от операции испарения и/или промежуточного высушивания просто длительностью кондиционирования пленочного покрытия. Кроме того, различия между испарением и промежуточным высушиванием не является значимым. Можно было бы, например, сначала подвергнуть испарению или промежуточному высушиванию пленочное покрытие, полученное путем нанесения способной к физическому отверждению композиции для покрытия при температуре от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин., а затем поддерживать при температуре 50°C в течение периода времени 5 часов.
Предпочтительно, композиции покрытия для применения в способе в соответствии с изобретением, то есть, материалы электрохимического покрытия, материалы водного базового покрытия и материалы лакового покрытия, однако, являются, по крайней мере, способными к термохимическому отверждению, в частности, предпочтительно способными к термохимическому отверждению и внешнему поперечному сшиванию.
В принципе, в контексте настоящего изобретения отверждение однокомпонентных систем осуществляют предпочтительно при температурах от 100 до 250°C, предпочтительно от 100 до 180°C, в течение периода времени от 5 до 60 мин., предпочтительно от 10 до 45 мин., поскольку эти условия в общем случае являются необходимыми для превращения пленочного покрытия в отвержденное пленочное покрытие с помощью реакций химического поперечного сшивания. Соответствующим образом, любая фаза испарения и/или промежуточного высушивания, которая предшествует отверждению, осуществляется при более низких температурах и/или в течение более коротких периодов времени. В таком случае, например, испарение может осуществляться при температурах от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин., и/или промежуточного высушивания при температуре, например, от 40 до 90°C в течение периода времени, например, от 1 до 60 мин.
В принципе, в контексте настоящего изобретения отверждение двухкомпонентных систем осуществляют при температурах, например, от 15 до 90°C, предпочтительно от 40 до 90°C, в течение периода времени от 5 до 80 мин., предпочтительно от 10 до 50 мин. Соответствующим образом, любая фаза испарения и/или промежуточного высушивания, которая предшествует отверждению, осуществляется при более низких температурах и/или в течение более коротких периодов времени. В таком случае, например, больше не имеет смысла проводить различие между терминами "испарение" и "промежуточное высушивание". Любая фаза испарения и/или промежуточного высушивания, которая предшествует отверждению, может осуществляется, например, при температуре от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин., но, по крайней мере, при более низких температурах и/или в течение более коротких периодов времени, чем отверждение, которое осуществляют после этого.
Это, конечно, не исключает отверждения двухкомпонентной системы при более высоких температурах. Например, на стадии (4) способа в соответствии с изобретением, который описан подробно ниже, базовое покрытие или множество базовых покрытий отверждается/отверждаются вместе с лаковым покрытием. Если обе однокомпонентная и двухкомпонентная системы присутствуют в пленках, например, однокомпонентное базовое покрытие и двухкомпонентное лаковое покрытие, то совместное отверждение, конечно, определяется условиями отверждения, необходимыми для однокомпонентной системы.
Все температуры, примеры которых приведены в контексте настоящего изобретения, понимается как комнатная температура, при которой присутствует основа с покрытием. Это, таким образом, не означает, что сама основа должна иметь определенную температуру.
Способ в соответствии с изобретением
В способе в соответствии с изобретением многослойная красочная система формируется на металлической основе (S).
Полезные металлические основы (S) включают, в принципе, основы, содержащие или состоящие, например, из железа, алюминия, меди, цинка, магния и их сплавов, а также стали в широком разнообразие различных форм и композиций. Предпочтение отдается железным и стальным основам, например, типичным железным и стальным основам, как используется в автомобильной промышленности. Основы могут, в принципе, находиться в любой форме, это означает, например, что они могут, например, представлять собой простые листы или также сложные компоненты такие, как, в частности, автомобильные кузова и их части.
Перед этапом (1) способа в соответствии с изобретением металлические основы (S) могут быть предварительно обработаны способом, который является известным сам по себе, то есть, например, очищены и/или обеспечены известными конверсионными покрытиями. Очистка может быть осуществлена механическим способом, например, с помощью протирки, шлифовки и/или полировки, и/или химическим путем с помощью методов травления поверхности в ваннах с кислотой или щелочью, например, при использовании хлористоводородной кислоты или серной кислоты. Конечно, очистка при использовании органических растворителей или водных моющих средств также является возможной. Предварительная обработка путем нанесения конверсионных покрытий, в частности, с помощью фосфатирования и/или хромирования, предпочтительно фосфатирования, также может иметь место. Предпочтительно, чтобы металлические основы имели, по крайней мере, конверсионное покрытие, в частности, были фосфатированы, предпочтительно с помощью цинк фосфатирования.
На этапе (1) способа в соответствии с изобретением отвержденное электрохимическое покрытие (Е.1) получают на металлической основе (S) путем электрофоретического нанесения материала электрохимического покрытия (е.1) на основу (S) и последующего отверждения материала электрохимического покрытия (е.1).
Материал электрохимического покрытия (е.1), используемый на этапе (1) способа в соответствии с изобретением, может представлять собой материал катодного или анодного покрытия, которое наносится электрохимическим путем. Предпочтительным является материал катодного электрохимического покрытия. Материалы электрохимического покрытия давно известны специалистам в данной области техники. Таковые представляют собой водные материалы для покрытия, включающие анионные или катионные полимеры в качестве связующих агентов. Эти полимеры содержат функциональные группы, которые потенциально являются анионными, т.е. могут быть превращены в анионные группы, например, группы карбоновой кислоты, или функциональные группы, которые потенциально являются катионными, т.е. могут быть превращены в катионные группы, например, аминогруппы. Превращение в заряженные группы, как правило, осуществляется за счет использования соответствующих нейтрализующих агентов (органические амины (анионные), органические карбоновые кислоты такие, как муравьиная кислота (катионные)), которые затем приводят к образованию анионных или катионных полимеров. Материалы электрохимического покрытия, как правило, и, таким образом, предпочтительно, включают в себя типичные антикоррозионные пигменты. Материалы катодного электрохимического покрытия, предпочтительные в контексте настоящего изобретения, включают катодные эпоксидные смолы, в частности, в сочетании с блокированными полиизоцианатами, которые являются известными сами по себе. В качестве примера ссылка делается на материалы электрохимического покрытия, описанные в публикациях WO 9833835 Al, WO 9316139 Al, WO 0102498 А1 и WO 2004018580 А1.
Материал электрохимического покрытия (е.1), таким образом, предпочтительно представляет собой, по крайней мере, способный к термохимическому отверждению материал для покрытия, и, в частности, способный к внешнему поперечному сшиванию. Материал электрохимического покрытия (е.1) предпочтительно представляет собой однокомпонентную композицию для покрытия. Предпочтительно, материал электрохимического покрытия (е.1) включает гидрокси-функциональную эпоксидную смолу в качестве связующего агента и полностью блокированный полиизоцианат в качестве поперечно сшивающего агента. Эпоксидная смола предпочтительно является катодной и, в частности, содержит аминогруппы.
Электрофоретическое нанесение такого материала электрохимического покрытия (е.1), которое осуществляется на этапе (1) способа в соответствии с изобретением, также является известным. Нанесение осуществляют с помощью электрофореза. Это означает, что металлическую заготовку, которая подвергается покрытию, сначала погружают в ванну для травления, содержащую материал покрытия, и электрическое поле постоянного тока между металлической деталью и противоэлектродом. Обрабатываемая деталь, таким образом, действует в качестве электрода; нелетучие компоненты материала электрохимического покрытия мигрируют, поскольку описанный заряд полимеров, используемых в качестве связующих агентов, с помощью электрического поля наносится на основу и осаждается на основе, образуя пленку электрохимического покрытия. Например, в случае катодного электрохимического покрытия основа, таким образом, связывается с катодом, а ионы гидроксида, которые образуются там посредством электролиза воды, нейтрализуют катионный связующий агент так, что он откладывается на основе и формирует слой покрытия, наносимого электрохимическим путем. В этом случае таким образом осуществляется нанесение с помощью способа электрофоретического погружения.
После электролитического нанесения материала электрохимического покрытия (е.1), основа с нанесенным покрытием (S) изымается из ванны, необязательно промывается с помощью, например, промывочных растворов на основе воды, потом необязательно испаряется и/или подвергается промежуточному высушиванию, и материал нанесенного электрохимического покрытия, наконец, отверждается.
Материал нанесенного электрохимического покрытия (е.1) (или в виде еще неотвержденного нанесенного электрохимического покрытия) подвергается испарению, например, при температуре от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин., и/или промежуточному высушиванию при температуре предпочтительно от 40 до 90°C в течение периода времени, например, от 1 до 60 мин.
Материал электрохимического покрытия (е.1), нанесенного на основу (или в виде еще неотвержденного нанесенного электрохимического покрытия) предпочтительно отверждается при температурах от 100 до 250°C, предпочтительно от 140 до 220°C, в течение периода времени от 5 до 60 мин., предпочтительно от 10 до 45 мин., что обеспечивает получение отвержденного электрохимического покрытия (Е.1).
Указанные условия испарения, промежуточного высушивания и отверждения применяются, в частности, к предпочтительному случаю, когда материал электрохимического покрытия (е.1) представляет собой однокомпонентную композицию для покрытия, способную к термохимическому отверждению, как описывается выше. Тем не менее, это не исключает возможности того, что материал электрохимического покрытия представляет собой композицию для покрытия, которая отверждается другим способом и/или при этом используется другой способ испарения, промежуточной сушки и условия отверждения.
Толщина слоя отвержденного электрохимического покрытия составляет, например, от 10 до 40 микрометров, предпочтительно от 15 до 25 микрометров. Все величины толщины пленки, указанные в контексте настоящего изобретения, следует понимать в качестве толщины сухой пленки. Толщина пленки, таким образом, является таковой отвержденной пленки, которая рассматривается. Таким образом, если указывается то, что материал покрытия наносится при определенной толщине пленки, то это следует понимать для обозначения того, что материал для покрытия наносится так, что указанная толщина пленки образуется после отверждения.
На этапе (2) способа в соответствии с изобретением (2.1) получают базовое покрытие (В.2.1) или получают (2.2) непосредственно последовательные базовые слои (В.2.2.x). Покрытия получают путем нанесения (2.1) водного материала базового покрытия (b.2.1) непосредственно на отвержденное электрохимическое покрытие (Е.1) или (2.2) путем непосредственного последовательного нанесения множества материалов базовых покрытий (b.2.2.х) на отвержденное электрохимическое покрытие (Е.1).
Непосредственное последовательное нанесение множества материалов базового покрытия (b.2.2.х) на отвержденное электрохимическое покрытие (Е.1), таким образом, следует понимать для обозначение того, что первый материал базового покрытия первым наносится непосредственно на электрохимическое покрытие, а потом второй материал базового покрытия непосредственно наносят на материал первого базового покрытия. Любой третий материал базового покрытия потом непосредственно наносится на материал второго базового покрытия. Эта операция может потом повторяться аналогично для дополнительных материалов базового покрытия (то есть, четвертого, пятого и т.д. базового покрытия).
Базовое покрытие (В.2.1) или первое базовое покрытие (В.2.2.x) после получения, таким образом, размещается непосредственно на отвержденном электрохимическом покрытии (E.1).
Термины "материал базового покрытия" и "базовое покрытие" по отношению к композициям покрытия, которые наносится, и пленочным покрытиям, которые получают на этапе (2) способа в соответствии с изобретением, используются для большей ясности. Базовые покрытия (В.2.1) и (В.2.2.x) не отверждаются отдельно, а предпочтительно отверждаются вместе с материалом лакового покрытия. Отверждение, таким образом, осуществляется аналогично отверждению так называемых материалов базового покрытия, которые используются в стандартном способе, описанном во введении. В частности, композиции для покрытия, используемые на стадии (2) способа в соответствии с изобретением не отверждаютсяя отдельно, как и композиции покрытий, которые называются грунтовками в контексте стандартного способа.
Водный материал базового покрытия (b.2.1), используемый на этапе (2.1), подробно описывается ниже. Однако он предпочтительно является, по крайней мере, способным к термохимическому отверждению, и он, в частности, является способным к внешнему поперечному сшиванию. Предпочтительно, когда материал базового покрытия (b.2.1) представляет собой однокомпонентную композиция для покрытия. Предпочтительно, когда материал базового покрытия (b.2.1) включает комбинацию, по крайней мере, одного гидрокси-функционального полимера в качестве связующего агента, выбранного из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов и сополимеров упомянутых полимеров, например, полиуретан-полиакрилатов, и, по крайней мере, одной меламиновой смолы в качестве поперечно сшивающего агента.
Материал базового покрытия (b.2.1) может наноситься с помощью способов, известных специалисту в данной области техники для нанесения жидкой композиции покрытия, например, путем погружения, нанесения покрытия с удалением излишков с помощью планки, распыления, прокатки или подобных им. Предпочтение отдается использованию способов нанесения путем распыления, например, распыления сжатым воздухом (пневматическое нанесение), безвоздушного распыления, распыления при высокоскоростном вращении, нанесения с помощью электростатического распыления (ESTA), необязательно, в сочетании с применением горячего распыления, например, горячевоздушного распыления. Наиболее предпочтительно, когда материал базового покрытия (b.2.1) наносится путем пневматического распыления или наносится путем электростатического распыления. Нанесение материала базового покрытия (b.2.1), таким образом, формирует базовое покрытие (В.2.1), то есть, покрытие материал базового покрытия (b.2.1) наносится непосредственно на электрохимическое покрытие (Е.1).
После нанесения полученный материал базового покрытия (b.2.1) или соответствующее базовое покрытие (В.2.1) подвергается испарению, например, при температуре от 15 до 35°C в течение периода времени, например, от 0,5 до 30 мин. и/или промежуточному высушиванию при температуре предпочтительно от 40 до 90°C в течение периода времени, например, от 1 до 60 мин. Предпочтение отдается первому испарению при температуре от 15 до 35°C в течение периода времени от 0,5 до 30 мин. и затем промежуточному высушиванию при температуре от 40 до 90°C в течение периода времени, например, от 1 до 60 мин. Описанные условия испарения и промежуточного высушивания применяются, в частности, к предпочтительному случаю, когда материал базового покрытия (b.2.1) представляет собой способную к термохимическому отверждению однокомпонентную композиция для покрытия. Тем не менее, это не исключает возможности того, что материал базового покрытия (В.2.1) представляет собой композицию для покрытия, которая отверждается иным образом, и/или такую, для которой используются другие условия испарения и/или промежуточного высушивания.
Базовое покрытие (В.2.1) не отверждается на этапе (2) способа в соответствии с изобретением, то есть, предпочтительно не подвергается воздействию температур выше 100°C в течение периода времени не более длительного, чем 1 мин. и, в частности, предпочтительно не подвергается воздействию температур выше 100°C вообще. Это ясно и однозначно видно из стадии (4) способа в соответствии с изобретением, описанного ниже. Поскольку базовое покрытие не отверждается до стадии (4), оно не может отверждаться на более ранней стадии (2), поскольку отверждение на этапе (4) не будет возможным на этой стадии.
Водный материал базового покрытия (b.2.2.х), используемый на этапе (2.2) способа в соответствии с изобретением, также описывается более подробно ниже. По крайней мере, один из материалов базового покрытия (b.2.2.х), который используется на этапе (2.2), предпочтительно все те, которые используются на этапе (2.2), однако, предпочтительно являются способными, по крайней мере, к термохимическому отверждению, в частности, предпочтительно внешнему поперечному сшиванию. Предпочтительно, по крайней мере, один материал базового покрытия (b.2.2.х) представляет собой однокомпонентную композиция для покрытия; такая композиция предпочтительно применяется ко всем материалам базового покрытия (b.2.2.х). Предпочтительно, когда, по крайней мере, один из материалов базового покрытия (b.2.2.х) включает комбинацию, по крайней мере, одного гидрокси-функционального полимера в качестве связующего агента, выбранного из группы, состоящей из полиуретанов, полиэстеров, полиакрилатов и сополимеров упомянутых полимеров, например, полиуретан-полиакрилатов, и, по крайней мере, одной из меламиновых смол в качестве поперечно сшивающего агента. Это предпочтительно относится ко всем материалам базового покрытия (b.2.2.х).
Материал базового покрытия (b.2.2.х) может наносится с помощью способов, известных специалисту в данной области техники для нанесения жидкой композиции покрытия, например, путем погружения, нанесения покрытия с удалением излишков с помощью планки, распыления, прокатки или подобных им. Предпочтение отдается использованию способов нанесения путем распыления, например, распыления сжатым воздухом (пневматическое нанесение), безвоздушного распыления, распыления при высокоскоростном вращении, электростатического распыления (ESTA), необязательно, в сочетании с применением горячего распыления, например, горячевоздушного распыления. Наиболее предпочтительно, когда материал базового покрытия (b.2.1) наносится путем нанесения с помощью пневматического распыления или нанесения путем электростатического распыления.
На этапе (2.2) способа в соответствии с изобретением предлагается приведенная ниже номенклатура. Материал базового покрытия и базовые покрытия, в общем случае, обозначаются как (b.2.2.х) и (В.2.2.x), в то время как x может заменяться другими приемлемыми буквами в номенклатуре специфического индивидуального материала базового покрытия и базовых покрытий.
Первый материал базового покрытия и первое базовое покрытие может обозначаться как а, и самый верхний материал базового покрытия и самое верхнее базовое покрытие может обозначаться как z. Эти два материала базового покрытия или базовые покрытия всегда присутствуют на этапе (2.2). Любые слои, расположенные между ними, могут обозначаться последовательно как b, с, d и так далее.
Нанесение первого материала базового покрытия (b.2.2.а), таким образом, формирует базовое покрытие (В.2.2.а) непосредственно на отвержденном электрохимическом покрытии (Е.1). По крайней мере, одно дополнительное базовое покрытие (В.2.2.x) потом получают непосредственно на базовом покрытии (В.2.2.а). Если получают множество дополнительных базовых покрытий (В.2.2.x), то их получают в прямой последовательности. Например, является возможным получать ровно одно дополнительное базовое покрытие (В.2.2.x), и в этом случае оно потом размещается непосредственно под лаковым покрытием (K) в многослойной красочной системе, полученной в конечном счете и, таким образом, оно может называется как базовое покрытие (B.2.2.z) (смотри также Фигуру 2). Является также возможным, например, когда получают два дополнительных базовых покрытия (В.2.2.x), в этом случае покрытие, полученное непосредственно на базовом покрытии (В.2.2.а), может обозначаться как (В.2.2.b), а покрытие, которое, в конечном счете, размещается непосредственно под лаковым покрытием (K), в свою очередь, обозначается как (B.2.2.z) (смотри также Фигуру 3).
Материалы базового покрытия (b.2.2.х) могут быть идентичными или различными. Также является возможным получать множество базовых покрытий (В.2.2.x) при использовании одного и того же материала базового покрытия, и одно или более дополнительных базовых покрытий (В.2.2.x) при использовании одного или более других материалов базового покрытия.
Нанесенный материал базового покрытия (b.2.2.х) в общем случае подвергается испарению и/или промежуточному высушиванию отдельно и/или вместе. Также на этапе (2.2) предпочтение отдается испарению при температуре от 15 до 35°C в течение периода времени от 0,5 до 30 мин. и промежуточному высушиванию при температуре от 40 до 90°C в течение периода времени, например, от 1 до 60 мин. Последовательность операций испарения и/или промежуточного высушивания н индивидуальных или множественных базовых покрытий (В.2.2.x) может быть скорректирована в соответствии с требованиями в каждом индивидуальном случае. Описанные выше предпочтительные условия испарения и промежуточного высушивания применяются, в частности, к предпочтительному случаю, когда, по крайней мере, один материал базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), включает(ют) способную к термохимическому отверждению однокомпонентную композиции покрытия. Однако это не исключает возможности того, что материалы базового покрытия (b.2.2.х) представляют собой композиции покрытия, которые являются способными к отверждению иным способ и/или что используются другие условия испарения и/или промежуточного высушивания.
Некоторые предпочтительные варианты последовательностей материалов базового покрытия (b.2.2.х) являются следующими.
Вариант а) является возможным получать первое базовое покрытие при нанесении путем электростатического напыления (ESTA) первого материала базового покрытия, и получать дополнительное базовое покрытие непосредственно на первом базовом покрытии путем пневматического распыления того же материала базового покрытия. Несмотря на то, что два базовых покрытия, таким образом, основываются на одном и том же материале базового покрытия, нанесение очевидным образом осуществляется в два этапа так, что материал базового покрытия, о котором идет речь в способе в соответствии с изобретением, соответствует первому материалу базового покрытия (b.2.2.а) и дополнительному материалу базового покрытия (b.2.2.z). Перед пневматическим нанесением первое базовое покрытие предпочтительно подвергается кратковременному испарению, например, при температуре от 15 до 35°C в течение от 0,5 до 3 мин. После пневматического распыления опять осуществляют испарение, например, при температуре от 15 до 35°C в течение от 0,5 до 30 мин., и затем проводят промежуточное высушивание при температуре от 40 до 90°C в течение периода времени от 1 до 60 мин. Описанная структура часто также называется структурой однослойного базового покрытия, полученного при использовании двух нанесений (один раз путем ESTA, один раз путем пневматического распыления). Однако поскольку при реальной OEM отделке технические условия оборудования для окрашивания означают, что определенный временной отрезок всегда проходит между первым нанесением и вторым нанесением, в котором основу, например, кузов автомобиля, подвергают кондиционированию при температуре от 15 до 35°C, например, и, следовательно, выпаривают, при этом характеристика первой структуры в качестве структуры двухслойного базового покрытия является более ясной в формальном смысле. Этот вариант этапа (2.2) предпочтительно выбирают тогда, когда используемый материал базового покрытия (b.2.2.х) (или двух используемых идентичных материалов базового покрытия (b.2.2.а) и (b.2.2.z)) включает пигменты эффекта, как описывается ниже. В то время как нанесение ESTA может гарантировать хорошую передачу материала или только небольшую потерю краски при применении, пневматическое распыление, которое осуществляют после, обеспечивает хорошее выравнивание пигментов эффекта и, следовательно, хорошие свойства всей красочной системы, в частности, высокий флоп-эффект.
Вариант b) является также возможным получать первое базовое покрытие путем электростатического напыления (ESTA) первого материала базового покрытия непосредственно на отвержденное электрохимическое покрытие, испарения и/или промежуточного высушивания указанного первого материала базового покрытия, и потом получать второе базовое покрытие путем непосредственного нанесения второго материала базового покрытия, отличного от первого материала базового покрытия. В этом случае материал второго базового покрытия может так же, как описывается в Варианте а), наносится первым путем электростатического напыления (ESTA), а затем путем нанесения с помощью пневматического распыления, в результате чего два непосредственно последовательных базовых покрытия, которые оба являются такими на основе второго материала базового покрытия, получают непосредственно на первом базовом покрытии. Между нанесениями и/или после нанесений, конечно, является возможным испарение и/или промежуточное высушивание. Вариант (b) этапа (2.2) предпочтительно выбирают тогда, когда базовое покрытие для подготовки окрашивания, как описывается ниже, является первым, которое получают непосредственно на электрохимическом покрытии, и потом, в свою очередь, осуществляют двойное нанесение материала базового покрытия, включающего пигменты эффекта, или нанесение материала базового покрытия, включающего цветные пигменты. В этом случае первое базовое покрытие основывается на материале базового покрытия для подготовки окрашивания, второе и третье базовые покрытия - на материале базового покрытия, включающем пигменты эффекта, или одно дополнительное базовое покрытие - на дополнительном материале базового покрытия, включающем цветные пигменты.
Вариант с) Является также возможным получать три последовательных базовых покрытия непосредственно на отвержденном электрохимическом покрытии, в этом случае базовые покрытия являются такими на основе трех различных материалов базового покрытия. Например, является возможным получать базовое покрытия для подготовки окрашивания, дополнительное покрытие на основе материала базового покрытия, включающего цветные пигменты и/или пигменты эффекта, и дополнительное покрытие на основе второго материал базового покрытия, включающего цветные пигменты и/или пигменты эффекта. Между и/или после индивидуальных нанесений, и/или после всех трех нанесений вновь является возможным осуществлять испарение и/или промежуточное высушивание.
Предпочтительные варианты в контексте настоящего изобретения, таким образом, включают получение двух или трех базовых покрытий на этапе (2.2) способа в соответствии с изобретением, при этом предпочтение в этом контексте отдается получению двух непосредственно последовательных базовых покрытий при использовании того же материала базового покрытия, и особое преимущество отдается получению первого их двух базовых покрытий путем ESTA нанесения и второго из этих двух базовых покрытий - путем пневматического напыления. В этом случае является предпочтительным получение структуры трехслойного базового покрытия, такого, что базовое покрытие, полученное непосредственно на отвержденном электрохимическом покрытии, основывается на материале базового покрытия для подготовки окрашивания. Второе и третье покрытия являются такими на основе одного и того же материала базового покрытия, которое предпочтительно включает пигменты эффекта, или на первом материале базового покрытия, включающем цветные пигменты и/или пигменты эффекта, и отличного второго материала базового покрытия, включающего цветные пигменты и/или пигменты эффекта.
Базовые покрытия (В.2.2.x) не отверждаются на этапе (2) способа в соответствии с изобретением, то есть, предпочтительно не подвергаются воздействию температур более чем 100°C в течение периода времени не более длительного, чем 1 мин., и предпочтительно не подвергаются воздействию температур более чем 100°C вообще. Это является ясным и однозначно очевидным из этапа (4) способа в соответствии с изобретением, описанного ниже. Поскольку базовые покрытия не подвергаются отверждению до этапа (4), они не могут отверждаться на более раннем этапе (2), поскольку отверждение на этапе (4) не будет возможным в этом случае.
Нанесение материалов базового покрытия (b.2.1) и (b.2.2.х) осуществляется таким образом, что базовое покрытие (В.2.1) и индивидуальные базовые покрытия (В.2.2.x) после отверждения, которое осуществляется на этапе (4), имеют индивидуальную толщину покрытия, например, от 5 до 40 микрометров, предпочтительно от 6 до 35 микрометров, в частности, предпочтительно от 7 до 30 микрометров. На этапе (2.1) предпочтительно получают более высокую толщину покрытия от 15 до 40 микрометров, предпочтительно от 20 до 35 микрометров. На этапе (2.2) индивидуальные базовые покрытия имеют, как бы то ни было, сравнительно более низкую толщину покрытия, в этом случае общая структура опять-таки имеет толщина покрытия в пределах порядка величины одного базового покрытия (В.2.1). Например, в случае двух базовых покрытий, первое базовое покрытие (В.2.2.а) предпочтительно имеет толщину покрытия от 5 до 35 и, в частности, от 10 до 30 микрометров, а второе базовое покрытие (B.2.2.z) предпочтительно имеет толщину покрытия от 5 до 30 микрометров, в частности, от 10 до 25 микрометров.
На этапе (3) способа в соответствии с изобретением лаковое покрытие (K) непосредственно наносят на (3.1) базовое покрытие (В.2.1) или (3.2) на самое верхнее базовое покрытие (B.2.2.Z). Это получение осуществляют путем приемлемого нанесения материала лакового покрытия (k).
Материал лакового покрытия (k) может, в принципе, представлять собой любую прозрачную композицию для покрытия, известную специалисту в данной области техники, в этом контексте. Такие включают водные композиции покрытия или композиции покрытия на основе растворителей, которые могут быть рецептированы в виде однокомпонентных или двухкомпонентных композиций покрытия, или многокомпонентных композиций покрытия. Кроме того, материалы порошкообразных смесей лакового покрытия также являются приемлемыми. Предпочтение отдается материалам лакового покрытия на основе растворителя.
Используемые материалы лакового покрытия (k) могут, в частности, быть термохимически и/или актинохимически отверждаемыми. В частности, они являются способными к термохимическому отверждению и внешнему поперечному сшиванию. Предпочтение отдается двухкомпонентным материалам лакового покрытия.
Прозрачные композиции покрытия, таким образом, типично и предпочтительно включают, по крайней мере, один (первый) полимер в качестве связующего агента, имеющего функциональные группы, и, по крайней мере, один поперечно сшивающий агент, который имеет функциональности, комплементарные функциональным группам связующего агента. Предпочтение отдается применению, по крайней мере, одного гидрокси-функционального поли(мет)акрилатного полимера в качестве связующего агента и полиизоцианата в качестве поперечно сшивающего агента.
Приемлемые материалы лакового покрытия описываются, например, в WO 2006042585 A1, WO 2009077182 А1 или также в WO 2008074490 А1.
Материал лакового покрытия (k) наносится с помощью способов, известных специалисту в данной области техники для нанесения жидкой композиции покрытия, например, путем погружения, нанесения покрытия с удалением излишков с помощью планки, распыления, прокатки или подобных им. Предпочтение отдается использованию способов нанесения путем распыления, например, распыления сжатым воздухом (пневматическое нааыление) и электростатического напыления (ESTA).
После нанесения материал лакового покрытия (k) или соответствующее лаковое покрытие (K) подвергается испарению или промежуточному высушиванию при температуре от 15 до 35°C в течение периода времени от 0,5 до 30 мин. Условия испарения и промежуточного высушивания такого вида используются, в частности, для предпочтительного случая, когда материал лакового покрытия (k) является способным к термохимическому отверждению двухкомпонентной композиции для покрытия. Однако это не исключает возможность того, что материал лакового покрытия (k) представляет собой композицию для покрытия, отверждаемую иным образом, и/или того, что могут использоваться другие условия испарения и/или промежуточного высушивания.
Нанесение материала лакового покрытия (k) осуществляется таким образом, что лаковое покрытие после отверждение, которое осуществляется на этапе (4), имеет толщину покрытия, например, от 15 до 80 микрометров, предпочтительно от 20 до 65 микрометров, в частности, предпочтительно от 25 до 60 микрометров.
Является понятным, что объем способа в соответствии с изобретением не исключает нанесения дополнительной композиции покрытия, например, дополнительных материалов лакового покрытия, после нанесения материала лакового покрытия (k) и получения дополнительного пленочного покрытия таким образом, например, дополнительных лаковых покрытий. Такие дополнительные пленочные покрытия потом также отверждаются на этапе (4), описанном ниже. Предпочтительно, однако, когда наносится только один материал лакового покрытия (k), и потом онотверждается так, как описывается на этапе (4).
На этапе (4) способа в соответствии с изобретением осуществляется сочетанное отверждение (4.1) базового покрытия (В.2.1) и лакового покрытия (K) или (4.2) базовых покрытий (В.2.2.x) и лакового покрытия (K).
Сочетанное отверждение предпочтительно осуществляется при температурах от 100 до 250°C, предпочтительно от 100 до 180°C, в течение периода времени от 5 до 60 мин., предпочтительно от 10 до 45 мин. Условия отверждения такого вида применяются, в частности, к предпочтительному случаю, когда базовое покрытие (В.2.1) или, по крайней мере, одно из базовых покрытий (В.2.2.x), предпочтительно все базовые покрытия (В.2.2.x), является/являются такими на основе способной к термохимическому отверждению однокомпонентной композиции для покрытия. Причиной этого, как описывается выше, является то, что такие условия в общем случае необходимы для достижения отверждения, как описывается выше, в такой однокомпонентной композиции для покрытия. Если материал лакового покрытия (k) представляет собой, например, способную к термохимическому отверждению однокомпонентную композицию для покрытия, то лаковое покрытие (K), которое рассматривается, конечно, также отверждается при этих условиях. То же самое очевидным образом применяется к предпочтительному случаю, когда материал лакового покрытия (k) представляет собой способную к термохимическому отверждению двухкомпонентную композицию для покрытия.
Однако приведенные выше утверждения не исключают возможности того, что материалы базовых покрытий (b.2.1) и (b.2.2.х) и материалы лакового покрытия (k) представляют собой композиции покрытия, которые отверждаются иным образом, и/или того, что используются иные условия отверждения.
После этапа (4) способ в соответствии с изобретением завершается, результатом чего является получение многослойной красочной системы в соответствии с изобретением.
Материал базового покрытия для применения в соответствии с изобретением:
Материал базового покрытия (b.2.1) для применения в соответствии с изобретением включает специфическую водную дисперсию, включающую, по крайней мере, один специфический сополимер (CP), предпочтительно точно один сополимер (CP).
Сополимер в контексте настоящего изобретения относится к полимерам, которые формируются из полимеров различных типов, например полиуретана и (мет)акрилатного полимера. Это явным образом включает как полимеры, которые ковалентно связываются друг с другом, так и те, в которых различные полимеры являются связанными друг с другом путем адгезии. Комбинации обоих видов связывания также охватываются этим определением. Термин "(мет)акрилат" охватывает акрилаты, метакрилаты и их смеси.
Сополимер (CP) получают путем
(i) первоначальной загрузки водной дисперсии, по крайней мере, одного полиуретана, и затем
(ii) полимеризации смеси олефиновых ненасыщенных мономеров в присутствии полиуретана из этапа (i),
где
а. используют растворимый в воде активатор,
b. олефиновые ненасыщенные мономеры дозируют таким образом, что концентрация на уровне 6,0% по весу в расчете на общее количество олефиновых ненасыщенных мономеров, которые используются для полимеризации, в реакционном растворе не превышается в течение всего времени проведения реакции, и
c. смесь олефиновых ненасыщенных мономеров включает, по крайней мере, один полиолефиновый ненасыщенный мономер.
На первом этапе получения сначала загружают водную дисперсию полиуретановой смолы.
Приемлемые насыщенные или ненасыщенные полиуретановые смолы описываются, например, в
- немецкой патентной заявке DE 19948004 А1, от стр. 4 строка 19 до стр. 11 строка 29 (полиуретановый преполимер B1),
- европейская патентная заявка ЕР 0228003 А1, от стр. 3 строка 24 до стр. 5 строка 40,
- европейская патентная заявка ЕР 0634431 А1, от стр. 3 строка 38 до стр. 8 строка 9, или
- международная патентная заявка WO 92/15405, от стр. 2 строка 35 до стр. 10 строка 32.
Полиуретановую смолу получают при использовании сначала, предпочтительно алифатических, циклоалифатических, алифатических-циклоалифатических, ароматических, алифатических-ароматических и/или циклоалифатических-ароматических полиизоцианатов, известных специалисту в данной области техники. Особое предпочтение отдается алифатическим и алифатическим-циклоалифатическим полиуретановым смолам.
Спиртовые компоненты, которые используются для получения полиуретановых смол предпочтительно представляют собой насыщенные и ненасыщенные полиолы, известные специалисту в данной области техники, и необязательно, в минимальных количествах, также моноспирты. В частности, диолы и, необязательно, в минимальных количествах, триолы используются для введения ветвления. Примеры приемлемых полиолов представляют собой насыщенные или олефиновые ненасыщенные полиэстер полиолы и/или полиэтер полиолы. В частности, используемые полиолы представляют собой полиэстер полиолы, в частности, те, которые имеют среднюю молекулярную массы от 400 до 5000 г/моль. Если специфически не указано иное, то среднечисловая молекулярная масса в контексте настоящего изобретения определяется с помощью осмоса при давлении насыщенных паров. Измерение осуществляли при использовании осмометра давления пара (модель 10.00 от Knauer) на сериях концентрации компонента при исследовании в толуоле при 50°C, с бензофеноном в качестве калибровочного вещества для определения константы экспериментальной калибровки используемого прибора (в соответствии с 
 , K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Berlin, стр. 47-54, 1982, где бензил использовали в качестве калибровочного вещества).
, K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Berlin, стр. 47-54, 1982, где бензил использовали в качестве калибровочного вещества).
Полиуретан, который изначально загружают в водную дисперсию, предпочтительно представляет собой гидрофильно стабилизированный полиуретан. Для гидрофильной стабилизации и/или для повышения способности к диспергированию в водной среде, присутствующая полиуретановая смола предпочтительно может содержать определенные ионные группы и/или группы, которые могут быть превращены в ионные группы (потенциально ионные группы). Полиуретановые смолы такого вида в контексте настоящего изобретения называются как ионно гидрофильно стабилизированные полиуретановые смолы. Кроме того, могут присутствовать неоинные гидрофильно модифицирующие группы. Однако предпочтительными являются ионно гидрофильно стабилизированные полиуретановые смолы. Более точно, модифицирующие группы альтернативно представляют собой:
- функциональные группы, которые могут быть превращены в катионы при использовании нейтрализующих агентов и/или кватернизующих агентов, и/или катионные группы (катионная модификация)
или
- функциональные группы, которые могут быть превращены в анионы при использовании нейтрализующих агентов и/или анионные группы (анионная модификация)
или
- неионные гидрофильные группы (неоионная модификация) или
- комбинации упомянутых выше групп.
Как сможет оценить квалифицированный специалист в данной области техники, функциональные группы для катионной модификации представляют собой, например, первичные, вторичные и/или третичные аминогруппы, вторичные сульфидные группы и/или третичные фосфиновые группы, в частности, третичные аминогруппы и вторичные сульфидные группы (функциональные группы, которые могут быть превращены в катионные группы при использовании нейтрализующих агентов и/или кватернизирующих агентов). Следует также упомянуть катионные группы - группы, полученные из упомянутых выше функциональных групп при использовании нейтрализующих агентов и/или кватернизирующих агентов, известных специалисту в данной области техники, таких, как первичные, вторичные, третичные и/или четвертичные группы аммония, третичные группы сульфония и/или четвертичные группы фосфония, в частности, четвертичные группы аммония и третичные группы сульфония.
Как является хорошо известным, функциональные группы для анионной модификации представляют собой, например, группы карбоновой кислоты, сульфоновой кислоты и/или группы фосфоновой кислоты, в частности, группы карбоновой кислоты (функциональные группы, которые могут быть превращены в анионные группы при использовании нейтрализующих агентов), а также анионные группы - группы, полученные из упомянутых выше функциональных групп при использовании нейтрализующих агентов, известных квалифицированному специалисту в данной области техники таких, как группы карбоксилата, сульфоната и/или фосфоната.
Функциональные группы для неионной гидрофильной модификации предпочтительно представляют собой группы поли(оксиалкилена), в частности, группы поли(оксиэтилена).
Ионно гидрофильные модификации могут быть введены в полиуретановую смолу с помощью мономеров, которые содержат ионные или потенциально ионные группы. Неоионные модификации вводят, например, посредством введения полимеров поли(этилен)оксида в качестве боковых или терминальных групп в молекулы полиуретана. Гидрофильные модификации вводят, например, с помощью соединений, которые содержат, по крайней мере, одну группу, реактивную по отношению к группам изоцианата, предпочтительно, по крайней мере, одну гидроксильную группу. Ионные модификация могут быть введены при использовании мономеров, которые так же, как и модифицирующие группы, содержат, по крайней мере, одну гидроксильную группу. Для введения неоионной модификации предпочтение отдается использованию полиэтердиолов и/или алкоксиполи(оксиалкилен)спиртов, известных специалисту в данной области техники.
Предпочтение отдается прибавлению, по крайней мере, одного органического растворителя к изначально загруженной дисперсии полиуретана, при этом указанный органический растворитель предпочтительно является способным смешиваться с водой в любом соотношении, а также со смесью олефиновых ненасыщенных мономеров в любом соотношении. Приемлемые органические растворители представляют собой N-метилпирролидон, N-этилпирролидон и этеры многоатомного спирта, такие, как метоксипропанол, в частности, однако следует отметить, что можно обойтись без растворителей на основе пирролидона только из экологических соображений. Однако количество органического растворителя выбирают таким образом, чтобы сохранялся водный характер дисперсии.
Второй этап получения, полимеризация смеси олефиновых ненасыщенных мономеров в присутствии полиуретана, осуществляется с помощью способов, которые называются свободно-радикальной эмульсионной полимеризацией, в присутствии, по крайней мере, одного инициатора полимеризации.
Используемый инициатор полимеризации должен быть растворимым в воде инициатором. Примеры приемлемых инициаторов представляют собой пероксодисульфат калия, пероксодисульфат натрия или пероксодисульфат аммония, а также перекись водорода, трет-бутил гидропероксид, 2,2'-азобис(2-амидоизопропан) дигидрохлорид, 2,2'-азобис(N,N'-диметиленизобутирамидин) дигидрохлорид или 2,2'-азобис(4-циано)пентановоевую кислоту. Инициаторы используют либо отдельно, либо в смеси, например, в смесях перекиси водорода и персульфата натрия.
Известные окислительно-восстановительные инициирующие системы также могут использоваться в качестве инициаторов полимеризации. Такие окислительно-восстановительные инициирующие системы включают, по крайней мере, одно содержащее пероксид соединение в комбинации с окислительно-восстановительным соинициатором, например, восстанавливающими соединениями серы, например, бисульфитами, сульфитами, тиосульфатами, дитионитами и тетратионатами щелочных металлов и соединений аммония, дигидратом гидроксиметансульфината натрия и/или тиомочевины. Например, является возможным использовать комбинации пероксодисульфатов с щелочными металлами или гидросульфитами аммония, например, пероксодисульфатом аммония и дисульфитом аммония. Весовое соотношение содержащих пероксид соединений к окислительно-восстановительным соинициаторам предпочтительно составляет от 50:1 до 0,05:1. В сочетании с инициаторами или системами окислительно-восстановительного инициатора является дополнительно возможным использовать катализаторы на основе переходных металлов, например, солей железа, солей никеля, солей кобальта, солей марганца, солей меди, солей ванадия или солей хрома, такие, как сульфат железа(II), хлорид кобальта(II), сульфат никеля(II), хлорид меди(I), ацетат марганца(II), ацетат ванадия(III), хлорид марганца(II). Основанные на мономерах, эти соли переходных металлов, как правило, используются в количестве от 0,1 до 1000 частей на миллион. Например, является возможным использовать комбинации перекиси водорода с солями железа(II), например, от 0,5 до 30% перекиси водорода и от 0,1 до 500 частей на миллион соли Мора.
Инициаторы предпочтительно используют в количестве от 0,05 до 20% по весу, предпочтительно от 0,05 до 10%, более предпочтительно от 0,1 до 5% по весу, в расчете на общую массу олефиновых ненасыщенных мономеров, используемых для полимеризации. Термины "общее количество" и "общий вес" являются эквивалентными.
Результат использования растворимого в воде инициатора заключается в том, что олефиновые ненасыщенные мономеры, которые добавляют в первоначально загружаемую водную дисперсию, могут вступать в реакцию немедленно с образованием олигомеров. Эти олигомеры имеют меньшую тенденцию к проникновению в полиуретановые частицы дисперсии, которая первоначально загружается, чем меньшие по размерам мономеры.
Полимеризация приемлемым образом осуществляется, например, при температуре, большей чем от 0 до 160°C, предпочтительно от 60 до 95°C.
Предпочтение отдается работе при исключении доступа кислорода, предпочтительно в токе азота. В общем случае, полимеризацию проводят при нормальном давлении, но также можно использовать более низкое давление или более высокое давление, в частности, когда температура полимеризации является выше температуры кипения используемых мономеров и/или органических растворителей.
Сополимеры (CP) для использования в соответствии с изобретением получают путем свободно-радикальной водной эмульсионной полимеризации, и в этом случае поверхностно-активные вещества или защитные коллоиды могут быть добавлены в реакционную среду. Перечень пригодных эмульгаторов и защитных коллоидов приведен, например, у Houben Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], том XIV/1 Makromolekulare Stoffe [Macromolecular Substances], Georg Thieme Verlag, Stuttgart 1961, стр. 411 ff.
Важным фактором получения водных дисперсий для применения в соответствии с настоящим изобретением, которые включают сополимер (CP), является контроль условий реакции полимеризации смеси олефиновых ненасыщенных мономеров в присутствии полиуретана. Это осуществляется в соответствии со способом, который называется полимеризацией "экономной дозировки", "подачи порциями с опорожнением бункера" или "точной дозировки".
Полимеризация при подаче порциями с опорожнением бункера в контексте настоящего изобретения рассматривается как эмульсионная полимеризация, при которой содержание остаточных мономеров в реакционном растворе сведено к минимуму во время реакции, а это означает, что дозированное добавление олефиновых ненасыщенных мономеров осуществляют таким образом, что концентрация 6,0% по весу, предпочтительно 5,0% по весу, более предпочтительно 4,0% по весу, особенно предпочтительно 3,5% по весу, в каждом случае из расчета на общую сумму олефиновых ненасыщенных мономеров, используемых для полимеризации, не превышается в течение всего времени реакции.
В этом контексте дополнительное предпочтение отдается диапазонам значений концентрации олефиновых ненасыщенных мономеров от 0,01 до 6,0% по весу, предпочтительно от 0,02 до 5,0% по весу, более предпочтительно от 0,03 до 4,0% по весу, в частности, от 0,05 до 3,5% по весу, в каждом случае из расчета на общую сумму ненасыщенных олефиновых мономеров, используемых для полимеризации. Например, самое высокое содержание (или концентрация), способное к определению во время реакции, может составлять 0,5% по весу, 1,0% по весу, 1,5% по весу, 2,0% по весу, 2,5% по весу или 3,0% по весу, в то время как все дополнительные определяемые значения являются ниже значений, указанных здесь. Термин "концентрация" в этом контексте таким образом, является эквивалентным термину "содержание".
Концентрацию мономеров, которые упоминаются в данной заявке в дальнейшем как свободные мономеры, в реакционном растворе можно регулировать различными способами.
Один из способов минимизации концентрации свободных мономеров представляет собой выбор очень низкого дозированного расхода для смеси олефиновых ненасыщенных мономеров. Когда скорость дозированного добавления является настолько низкой, что все мономеры могут вступать в реакцию очень быстро, как только они попадают в реакционный раствор, то можно гарантировать, что концентрация свободных мономеров будет сведена к минимуму. Помимо скорости дозирования важно, чтобы достаточное количество свободных радикалов всегда присутствовало в реакционном растворе, таким образом, чтобы каждый из мономеров, которые вносят в виде доз, мог вступить в реакцию очень быстро. С этой целью условия реакции предпочтительно должны быть выбраны таким образом, чтобы исходный инициатор уже начал свое действие до начала дозированного добавления олефиновых ненасыщенных мономеров.
Предпочтительно дозированное добавление начинают, по крайней мере, за 5 минут, более предпочтительно, по меньшей мере, за 10 минут перед этим. Предпочтительно, по крайней мере, 10% от веса инициатора, более предпочтительно, по крайней мере, 20% от веса, наиболее предпочтительно, по крайней мере, 30% от веса инициатора, в каждом случае из расчета на общую сумму инициатора, добавляют до начала дозированного добавления олефиновых ненасыщенных мономеров.
Количество инициатора является важным фактором для достаточного присутствия свободных радикалов в реакционном растворе. Количество инициатора должен быть выбрано таким образом, чтобы достаточное количество свободных радикалов было доступно в любое время так, чтобы введенные дозированные мономеры могли вступать в реакцию. Если количество инициатора увеличивается, также является возможным подвергать реакции большее количество мономеров в одно и то же время.
Дополнительный фактором, который может определять скорость реакции, является структура мономеров, т.е., в частности, их структурные свойства и реакционная способность, которая вытекает из них.
Концентрация свободных мономеров может, таким образом, контролироваться путем взаимодействия количества инициатора, скорости добавления инициатора, скорости добавления мономера, а также за счет выбора мономеров. Как замедление дозированного добавления, так и увеличение количества инициатора, а также раннее начало добавления инициатора, служат конкретной целью поддержания концентрации свободных мономеров ниже указанных выше границ.
Концентрация мономеров в реакционном растворе может быть определена с помощью газовой хроматографии в любой момент в процессе реакции. Типичные параметры для определения с помощью газовой хроматографии являются следующими: 50 м капиллярная колонка на основе кремнезема с фазой полиэтиленгликоля или 50 м капиллярная колонка с фазой полидиметилсилоксана, гелий в качестве газа-носителя, инжектор для ввода проб с делением потока 150°C, температура печи от 40 до 220°C, пламенно-ионизационный детектор, температура детектора 275°C, внутренний стандарт: изобутилакрилат. В контексте настоящего изобретения концентрацию мономеров предпочтительно определяют с помощью газовой хроматографии, в частности, при соблюдении указанных выше параметров.
Этот анализ позволяет определить концентрацию свободных мономеров, близкую к граничному значению, для полимеризации при точной дозировке, например, благодаря тому, что при высоком содержании олефиновых ненасыщенных мономеров, имеющих низкую реакционную способность, вышеуказанные параметры могут быть использованы для управления реакцией. В этом случае, например, скорость дозирования мономеров можно уменьшить и/или количество инициатора можно увеличить.
Приемлемые ненасыщенные олефиновые мономеры могут быть моно- или полиолифенически ненасыщенными. Предпочтительным является, когда присутствуют, по крайней мере, один ненасыщенный моноолефиновый мономер и, по крайней мере, один ненасыщенный полиолефиновый мономер.
Примеры подходящих моноолефиновых ненасыщенных мономеров включают винильные моноолефиновые ненасыщенные мономеры такие, как, в частности, моноолефиновые ненасыщенные мономеры на основе (мет)акрилата и аллильных соединений. Примерами могут служить также альфа-, бета-ненасыщенные карбоновые кислоты. Предпочтение отдается использованию, по крайней мере, но не обязательно исключительно, моноолефиновых ненасыщенных мономеров на основе (мет)акрилата.
Моноолефиновые ненасыщенные мономере на основе (мет)акрилата могут, например, представлять собой (мет)акриловую кислоту и сложные эфиры, нитрилы или амиды (мет)акриловой кислоты.
Предпочтение отдается эстерам (мет)акриловой кислоты, которые имеют неолефиновый ненасыщенный R радикал.
 или
или 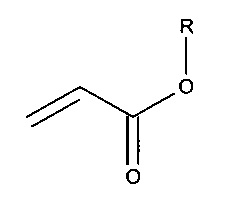
R радикал может быть алифатическим или ароматическим. R радикал предпочтительно является алифатическим.
R радикал может, например, представлять собой алкильный радикал или содержать гетероатомы. Примеры R радикалов, содержащих гетероатомы, представляют собой этеры. Предпочтение отдается использованию, по крайней мере, но не обязательно ограничивается таковыми, как мономеры, в которых R радикал представляет собой алкильный радикал.
Если R представляет собой алкильный радикал, он может, например, быть линейным, разветвленным или циклическим алкильным радикалом. Во всех случаях он может включать незамещенные алкильные радикалы или алкильные радикалы, замещенные функциональными группами. Алкильный радикал имеет предпочтительно от 1 до 20, более предпочтительно от 1 до 10 атомов углерода.
В частности, предпочтительные мононенасыщенные эстеры (мет)акриловой кислоты, содержащие ненасыщенный алкильный радикал, представляют собой метил (мет)акрилат, этил (мет)акрилат, пропил (мет)акрилат, изопропил (мет)акрилат, н-бутил (мет)акрилат, изобутил (мет)акрилат, трет-бутал (мет)акрилат, амил (мет)акрилат, гексил (мет)акрилат, этилгексил (мет)акрилат, 3,3,5-триметилгексил (мет)акрилат, стеарил (мет)акрилат, лаурил (мет)акрилат, циклоалкил (мет)акрилаты такие, как циклопентил (мет)акрилат, изоборнил (мет)акрилат и циклогексил (мет)акрилат, особое предпочтение отдается н- и трет-бутил (мет)акрилату, метил (мет)акрилату и этил (мет)акрилату.
Приемлемые мононенасыщенные эстеры (мет)акриловой кислоты, которые имеют замещенный алкильный радикал, могут предпочтительно быть замещенными одной или более гидроксильными группами.
В частности, предпочтительные мононенасыщенные эстеры (мет)акриловой кислоты, которые имеют алкильный радикал, замещенный одной или более гидроксильными группами, представляют собой 2-гидроксиэтил (мет)акрилат, 2-гидроксипропил (мет)акрилат, 3-гидроксипропил (мет)акрилат, 3-гидроксибутил (мет)акрилат и 4-гидроксибутил (мет)акрилат.
Возможные дополнительные виниловые мононенасыщенные мономеры представляют собой мономеры, которые имеют неолефиновый ненасыщенный R' радикал в группе винила.

R' радикал может быть алифатическим или ароматическим, предпочтение отдается ароматическим радикалам.
R' радикал может представлять собой радикал гидрокарбила или содержать гетероатомы. Примеры R' радикалов, содержащих гетероатомы, представляют собой этеры, эстеры, амид, нитрилы и гетероциклы. Предпочтительно, когда R' радикал представляет собой радикал гидрокарбила. Если R' является радикалом гидрокарбила, то он может быть замещенным или незамещенным гетероатомами, предпочтение отдается незамещенным радикалам. Предпочтительно, R' радикал представляет собой ароматический радикал гидрокарбила.
В частности, предпочтительные дополнительные виниловые олефиновые ненасыщенные мономеры представляют собой винилароматические углеводороды, в частности, винилтолуол, альфа-метилстирен и, в частности, стирен.
Дополнительные предпочтительные мономеры, содержащие гетероатомы, представляют собой ненасыщенные олефиновые мономеры такие, как акрилонитрил, метакрилонитрил, акриламид, метакриламид, N-диметилакриламид, винилацетат, винилпропионат, винилхлорид, N-винилпирролидон, N-винилкапролактам, N-винилформамид, N-винилимидазол и N-винил-2-метилимидазолин.
Примеры приемлемых полиолефиновых ненасыщенных мономеров включают эстеры (мет)акриловой кислоты, содержащие ненасыщенный олефиновый Rʺ радикал, и аллильные этеры многоатомных спиртов.
 или
или 
Rʺ радикалы могут, например, представлять собой аллильный радикал или радикал (мет)акрилового эстера.
Предпочтительные полиолефиновые ненасыщенные мономеры представляют собой этиленгликоль ди(мет)акрилат, пропилен 1,2-гликоль ди(мет)акрилат, пропилен 2,2-гликоль ди(мет)акрилат, бутан-1,4-диол ди(мет)акрилат, неопентил гликоль ди(мет)акрилат, 3-метилпентандиол ди(мет)акрилат, диэтиленгликоль ди(мет)акрилат, триэтиленгликоль ди(мет)акрилат, тетраэтиленгликоль ди(мет)акрилат, дипропиленгликоль ди(мет)акрилат, трипропиленгликоль ди(мет)акрилат, гександиол ди(мет)акрилат и аллил (мет)акрилат.
Предпочтительные полиолефиновые ненасыщенные соединения также представляют собой акриловые и метакриловые эстеры спиртов, которые имеют более двух групп ОН, например, триметилолпропан три(мет)акрилат или глицерил три(мет)акрилат, а также моноаллильный этер триметилолпропан ди(мет)акрилата, диаллильный этер триметилолпропан (мет)акрилата, моноаллильный этер пентаэритритил три(мет)акрилата, диаллильный этер пентаэритритил ди(мет)акрилата, триаллильный этер пентаэритритил (мет)акрилата, триаллилсахарозу и пентааллилсахарозу.
Особое предпочтение отдается использованию аллилметакрилата в качестве полиолефинового ненасыщенного мономера.
Смесь ненасыщенных олефиновых мономеров включает, по крайней мере, один полиолефиновый ненасыщенный мономер. Предпочтительно, когда смесь олефиновых ненасыщенных мономеров также включает один или более мононенасыщенных эстеров (мет)акриловой кислоты, которые содержат незамещенный алкильный радикал.
Предпочтительно, когда смесь олефиновых ненасыщенные мономеров содержит от 0,1 до 6,0 мол. %, более предпочтительно от 0,1 до 2,0 мол. %, наиболее предпочтительно от 0,1 до 1.0 мол. %, полиолефиновых ненасыщенных мономеров.
Предпочтительно, когда смесь ненасыщенных олефиновых мономеров содержит от 0,1 до 6,0 мол. %, более предпочтительно от 0,1 до 2,0 мол. %, наиболее предпочтительно от 0,1 до 2,0 мол. %, аллилметакрилата. Более предпочтительно, когда кроме аллилметакрилата никаких дополнительных полиолефиновых ненасыщенных мономеров не присутствуют в смеси.
Предпочтительно, когда смесь олефиновых ненасыщенных мономеров содержит менее чем 10,0% по весу, более предпочтительно менее чем 5,0% по весу, винилароматических углеводородов в расчете на общее количество олефиновых ненасыщенных мономеров, которые используются в полимеризации. Наиболее предпочтительно, никаких винилароматических углеводородов не содержится в смеси олефиновых ненасыщенных мономеров. В частности, является предпочтительным, когда используются олефиновые ненасыщенных мономеры, которые имеют ароматические группы в количестве менее чем 10,0% по весу, более предпочтительно менее чем 5,0% по весу, в расчете на общее количество олефиновых ненасыщенных мономеров, используемых в полимеризации. В частности, никаких ненасыщенных олефиновых мономеров, имеющих ароматические группы, не присутствует в смеси олефиновых ненасыщенных мономеров.
Из этого следует, что винилароматические углеводороды, указанные выше как предпочтительные, в частности, винилтолуол, альфа-метилстирен и стирен, без сомнения, являются предпочтительными только в пределах группы мономеров, содержащих ароматические группы. Несмотря на это, указанные мономеры предпочтительно не используются в контексте настоящего изобретения. Тем не менее, в случае, если использование таких мономеров является вариантом, то в каждом конкретном случае предпочтение отдается применению мономеров, содержащих ароматические группы, которые обозначаются как предпочтительные.
В предпочтительном варианте осуществления изобретения смесь олефиновых ненасыщенных мономеров, включает:
- от 98,0 до 99,5% по весу одного или более мононенасыщенных эстеров (мет)акриловой кислоты, которые содержат незамещенные алкильные радикалы, где алкильные радикалы предпочтительно имеют от 1 до 10 атомов углерода, и
- от 0,5 до 2,0% по весу одного или более полиненасыщенных эстеров (мет)акриловой кислоты,
в каждом случае из расчета на общее количество олефиновых ненасыщенных мономеров, которые используются в полимеризации.
Предпочтение отдается прибавлению, по крайней мере, одного растворителя к смеси олефиновых ненасыщенных мономеров, при этом указанный растворитель предпочтительно является смешиваемым в любом соотношении с водой и в любом соотношении со смесью ненасыщенных олефиновых мономеров. Приемлемые органические растворители представляют собой N-метилпирролидон, N-этилпирролидон и неполные эфиры многоатомных спиртов, такие, как метоксипропанол, в частности, при этом следует отметить, что можно обойтись без растворителей на основе пирролидона только по экологическим причинам. Однако количество органического растворителя выбирают таким образом, чтобы сохранялся водный характер дисперсии, которую, в конечном счете, получают.
В соответствии с описанным способом получения сополимеры в водной дисперсии в соответствии с изобретением, в частности, имеют структуру ядро-оболочка, которая может быть достигнута при использовании описанного способа получения. Эта структура ядро-оболочка характеризуется наличием ядра, содержащего, по крайней мере, один полиуретан, и оболочки, содержащей, по крайней мере, один полимер, который был получен в результате полимеризации олефиновых ненасыщенных мономеров.
Описанная структура ядро-оболочка достигается за счет специфических условий реакции полимеризации при точной дозировке. В течение всего времени реакции никогда не бывает каких-либо больших количеств олефиновых ненасыщенных мономеров, которые могут проникать в полиуретановые частицы, в присутствии первоначально загруженного полиуретана. Свободные радикалы, которые обеспечиваются водорастворимым инициатором, всегда присутствующие во время добавления мономера в водную фазу, формируют непосредственно при добавлении олигомеры, которые больше не могут проникать в полиуретан. Они затем полимеризуется на поверхности полиуретана.
В предпочтительном варианте осуществления изобретения массовое соотношение ядра к оболочке составляет от 80:20 до 20:80, более предпочтительно от 60:40 до 40:60. В данном случае имеется в виду, что соотношение количества компонентов, используемых для получения ядра (стадия (I), полиуретан) и оболочки (стадия (II), смесь олефиновых ненасыщенных мономеров).
Предпочтительно, когда сополимеры (CP) в водной дисперсии имеют размер частиц (z среднее) от 60 до 130 нм, более предпочтительно от 70 до 115 нм, измеренный с помощью фотонно-корреляционной спектроскопии при использовании Malvern Nano S90 (от Malvern Instruments) при температуре 25±1°C. Устройство, оснащенное 4 мВт He-Ne-лазером с длиной волны 633 нм, охватывает диапазон размеров от 1 до 3000 нм.
Сополимеры (CP) могут быть предпочтительно поперечно сшитыми. Содержание геля водной дисперсии в соответствии с изобретением предпочтительно составляет от 40 до 97% по весу, более предпочтительно от 75 до 90% по весу, в каждом случае из расчета на твердые частицы дисперсии.
Содержание геля может быть определено гравиметрически путем замораживания-высушивания дисперсии, определяя при этом общую массу лиофилизированного полимера (соответствует твердому веществу дисперсии в контексте определения содержания геля), а затем экстрагируя полимер в избытке тетрагидрофурана (отношение тетрагидрофурана к сублимированному полимеру=300:1) при 25°C в течение 24 часов. Нерастворимые фракции удаляют и высушивают в печи с циркуляцией воздуха при 50°C в течение четырех часов. После этого высушенную нерастворимую фракцию взвешивают и определяют показатель при использовании общей массы лиофилизированного полимера. Полученное значение соответствует содержанию геля.
Средневзвешенная молярная масса сополимеров (CP) предпочтительно составляет от 3*107 г/моль до 8,5*109 г/моль, причем можно определить средневзвешенную молекулярную массу методом рассеяния лазерного света под малым углом.
Кислотное число сополимеров (CP) предпочтительно составляет от 0 до 220 мг KOH/г твердой смолы, предпочтительно, от 0 до 40 мг KOH/г твердой смолы, более предпочтительно от 0 до 25 мг KOH/г твердой смолы. Щелочное число предпочтительно составляет менее 70 мг KOH/г твердой смолы, предпочтительно менее 20 мг KOH/г твердой смолы. Термины "твердая смола" и "твердое вещество" по отношению к полимеру или дисперсии полимера, являются эквивалентными. Таким образом, они относятся, в частности, к твердым веществам или содержанию твердых веществ в дисперсии полимера, как описывается ниже.
Кислотное число может быть определено на основе DIN EN ISO 2114 в гомогенном растворе ТГФ/вода (9 частей по объему ТГФ и 1 часть по объему дистиллированной воды) при использовании спиртового раствора гидроксида калия.
Щелочное число может быть определено на основе R.-P.  , R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), при использовании уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофурана (ТГФ)/диметилформамида (ДМФ) при комнатной температура путем полного гидролиза избытка уксусного ангидрида, оставшегося после ацетилирования и проведения потенциометрического обратного титрования уксусного ангидрида при использовании спиртового раствора гидроксида калия.
, R. Gnauck и R. Algeier, Plaste und Kautschuk, 20, 274 (1982), при использовании уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофурана (ТГФ)/диметилформамида (ДМФ) при комнатной температура путем полного гидролиза избытка уксусного ангидрида, оставшегося после ацетилирования и проведения потенциометрического обратного титрования уксусного ангидрида при использовании спиртового раствора гидроксида калия.
Водные дисперсии, по крайней мере, одного сополимера (CP) предпочтительно имеют содержание твердых веществ от 15 до 45% по весу, особенно предпочтительно от 25 до 35% по весу. Содержание твердых веществ такого рода может быть установлено без каких-либо проблем путем использования соответствующих количеств органических растворителей и, в частности, воды, в процессе получения сополимеров и/или путем соответствующего разбавления после получения.
Содержание сополимеров (CP) предпочтительно находится в диапазоне от 2,0 до 30,0% по весу, более предпочтительно от 3,0 до 20,0% по весу, особенно предпочтительно от 4,0 до 15,0% по весу, в каждом случае из расчета на общую массу пигментированного водного материала базового покрытия (В.2.1).
Материал базового покрытия (В.2.1) для использования в соответствии с настоящим изобретением содержит, по крайней мере, один специфический продукт реакции (R), предпочтительно только один продукт реакции (R).
Материал базового покрытия (b.2.1) для применения в соответствии с изобретением включает, по крайней мере, один специфический продукт реакции (R), предпочтительно точно один продукт реакции (R).
Продукты реакции являются линейными. Линейные продукты реакции могут быть, в принципе, получены путем превращения бифункциональных реагентов, в этом случае соединение реагентов посредством реакции функциональных групп приводит к получению линейной, т.е. катенированной структуры. Так, например, если продукт реакции представляет собой полимер, то полимерный скелет имеет линейный характер. Если продукт реакции представляет собой, например, полиэстер, то используемые реагенты, могут быть диолами и двухосновными карбоновыми кислотами, и в этом случае последовательность эфирных связей в продукте реакции имеет линейный характер. В предпочтительном варианте получения продукта реакции (R), таким образом, используются в основном бифункциональные реагенты. Другие реагенты такие, как, в частности, монофункциональные соединения, соответствующим образом используются предпочтительно только в небольших количествах, если вообще используются. В частности, используются, по крайней мере, 80 мол. %, предпочтительно, по крайней мере, 90 мол. % бифункциональных реагентов, а наиболее предпочтительно, используются исключительно бифункциональные реагенты. Если используются дополнительные реагенты, то они предпочтительно являются выбранными исключительно из группы монофункциональных реагентов. Однако предпочтительно, когда используются исключительно бифункциональные реагенты.
Полезные функциональные группы для реагентов включают функциональные группы, известные специалисту в данной области техники в этом контексте. Комбинации реагентов, имеющих приемлемые функциональные группы, которые могут связываться друг с другом, и, таким образом, могут служить для получения продукта реакции, также являются в принципе известными. То же самое относится и к условиям реакции, необходимым для связывания. Предпочтительными функциональными группами для реагентов являются гидроксильные, карбоксильные группы, иминогруппы, группы карбамата, аллофанатные группы, тиогруппы, ангидридные группы, эпоксигруппы, изоцианатные, метилоловые группы, группы метилолакриламидного этера, группы силоксанов и/или аминогруппы, особенно предпочтительно, гидроксильные и карбоксильные группы. Предпочтительные комбинации функциональных групп, которые могут быть связаны друг с другом, представляют собой гидроксильные и карбоксильные группы, группы изоцианата и гидроксила, изоцианатные группы и аминогруппы, эпоксигруппы и карбоксильные группы и/или эпоксигруппы и аминогруппы; при выборе функциональных групп следует обеспечить, чтобы гидроксильная функциональность и кислотное число, описанные ниже, получены в продукте реакции. Особое предпочтение отдается сочетанию гидроксильных и карбоксильных групп. В этом варианте осуществления, по крайней мере, один реагент, таким образом, имеет гидроксильные группы, и, по крайней мере, один дополнительный реагент имеет карбоксильные группы. Предпочтение отдается использованию комбинации реагентов с дигидроксигруппами и дикарбоксильными функциональными группами. Осуществление реакции этих реагентов при использовании способа, который известен сам по себе, обеспечивает получение продуктов реакции, содержащих связи эстера.
Продукт реакции является гидроксифункциональным. Предпочтительно, чтобы реагенты были преобразованы таким образом, чтобы линейные молекулы, которые образуются, имели две терминальные гидроксильные группы. Это означает, что одна гидроксильная группа присутствует в каждом из двух концов каждой из полученных молекул.
Продукт реакции имеет кислотное число менее чем 20, предпочтительно менее 15, особенно предпочтительно менее 10 и наиболее предпочтительно менее чем 5 мг KOH/г. Таким образом, он предпочтительно имеет лишь очень небольшое количество групп карбоновой кислоты. Если явным образом не указано иное, то кислотное число в контексте настоящего изобретения определяется в соответствии с DIN 53402.
Описанная гидроксильная функциональная группа, как и низкое кислотное число, могут быть получены, например, с помощью способа, который является известным сам по себе, путем применения соответствующих соотношений реагентов, имеющих соответствующие функциональные группы. В предпочтительном случае, где при получении используют реагенты с дигидрокси функциональными группами и дикарбокси функциональными группами, таким образом, применяется приемлемый избыток компонента с дигидрокси функциональными группами. В этом контексте дополнительно следует пояснить следующее: исключительно из статистических соображений, одна реальная реакция, конечно, не обеспечивает получения молекул, имеющих, например, желаемую (ди)гидроксильную функциональную группу. Однако выбор соответствующих условий, например, избыток реагентов с дигидрокси-функциональными группами и проведение реакции до достижения желаемого количества кислоты, гарантирует, что продукты конверсии или молекулы, которые составляют продукт реакции, являются функциональными гидроксигруппами, по крайней мере, в среднем. Специалист в данной области техники знает, как выбрать подходящие условия.
При получении продукта реакции, по крайней мере, одно соединение (v), которое используется или преобразовывается в реагент, имеет две функциональные группы (v.1) и алифатический или аралифатический гидрокарбильный радикал (v.2), который расположен между двумя функциональными группами и имеет от 12 до 70, предпочтительно от 22 до 55 и более предпочтительно от 30 до 40 атомов углерода. Соединения (v), таким образом, состоит из двух функциональных групп и радикала гидрокарбила. Полезные функциональные группы, конечно, включают описанные выше функциональные группы, в частности, гидроксильные и карбоксильные группы. Алифатические гидрокарбильные радикалы являются известными как такие, которые являются ациклическими или циклическими, насыщенными или ненасыщенными, неароматическими радикалами гидрокарбила.
Аралифатические гидрокарбильные радикалы являются такими, которые содержат как алифатические, так и ароматические структурные единицы.
Усредненная молекулярная масса продуктов реакции может изменяться в широких пределах и составляет, например, от 600 до 40000 г/моль, в частности, от 800 до 10000 г/моль, наиболее предпочтительно от 1200 до 5000 г/моль. Если явно не указано иное, то усредненная молекулярная масса в контексте настоящего изобретения определяется с помощью осмоса при давлении насыщенных паров. Измерение осуществляется при использовании осмометра при давлении пара (модель 10.00 от Knauer) на сериях концентраций исследуемого компонента в толуоле при 50°C, при использовании бензофенона в качестве калибровочного вещества для определения экспериментальной калибровки константы используемого прибора (в соответствии с Е.  , G.
, G.  , K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Berlin, pp. 47-54, 1982, где бензил используется в качестве калибровочного вещества).
, K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Berlin, pp. 47-54, 1982, где бензил используется в качестве калибровочного вещества).
Предпочтительные соединения (v) представляют собой димерные жирные кислоты или присутствуют в димерных жирных кислотах. При получении продуктов реакции (R), димерные жирные кислоты, таким образом, используются предпочтительно, но не исключительно, в качестве соединения (v). Димерные жирные кислоты (также известные как димеризованные жирные кислоты или димеры кислот), в общем случае и в частности, в контексте настоящего изобретения представляют собой смеси, приготовленные путем олигомеризации ненасыщенных жирных кислот. Их получают, например, путем каталитической димеризации растительных ненасыщенных жирных кислот, используемые исходные материалы, в частности, являются ненасыщенными C12-С22 жирными кислотами. Связи образуются, главным образом, по механизму Дильса-Альдера, и результат, в зависимости от количества и положения двойных связей в жирных кислотах, используемых для получения димерных жирных кислот, представляет собой смеси преимущественно димерных продуктов, имеющих циклоалифатические, линейные алифатические, разветвленные алифатические, а также C6 ароматические углеводородные группы, между карбоксильными группами. В зависимости от механизма и/или какой-либо последующей гидрогенизации алифатические радикалы могут быть насыщенными или ненасыщенными, а фракция ароматических групп может также варьировать. Радикалы между группами карбоновой кислоты содержат, например, от 24 до 44 атомов углерода. Для получения жирные кислоты, которые содержат 18 атомов углерода, используются с предпочтением, и, таким образом, димерный продукт имеет 36 атомов углерода. Радикалы, которые соединяют карбоксильные группы димерных жирных кислот, предпочтительно не имеют никаких ненасыщенных связей и никаких ароматических углеводородных радикалов.
В контексте настоящего изобретения C18 жирные кислоты, таким образом, используются с предпочтением для получения. Особое предпочтение отдается применению линоленовой, линолевой и/или олеиновой кислот.
В зависимости от режима реакции описанная выше олигомеризации приводит к получению смесей, содержащих в основном димерные молекулы, а также тримерные молекулы и мономерные молекул, а также другие побочные продукты. Очистку, как правило, осуществляют с помощью дистилляции. Коммерческие димерные жирные кислоты обычно содержат, по крайней мере, 80% по весу димерных молекул, вплоть до 19% по весу тримерных молекул, и не более чем 1% по весу мономерных молекул и других побочных продуктов.
Предпочтение отдается использованию димерных жирных кислот, которые состоят, по крайней мере, на 90% по весу, предпочтительно, по крайней мере, на 95% по весу, наиболее предпочтительно, по крайней мере, на 98% по весу, из молекул димерной жирной кислоты.
В контексте настоящего изобретения предпочтение отдается использованию димерных жирных кислот, которые состоят, по крайней мере, на 90% по весу из димерных молекул, менее чем на 5% по весу из тримерных молекул, и менее чем на 5% по весу из мономерных молекул и других побочных продуктов. Особое предпочтение отдается использованию димерных жирных кислот, которые состоят на 95-98% по весу из димерных молекул, менее чем на 5% по весу из тримерных молекул и менее чем на 1% по весу из мономерных молекул и других побочных продуктов. Аналогично, с особым предпочтением используются димерные жирные кислоты, состоящие, по крайней мере, на 98% по весу из димерных молекул, менее чем на 1,5% по весу из тримерных молекул, и менее чем на 0,5% по весу из мономерных молекул и других побочных продуктов. Фракции мономерных, димерных и тримерных молекул, а также других побочных продуктов в димерных жирных кислотах могут быть определены, например, с помощью газовой хроматографии (ГХ). В этом случае перед анализом ГХ димерные жирные кислоты превращают в соответствующие метиловые эстеры с помощью способа при использовании трифторида бора (см. DIN EN ISO 5509), а затем анализируют с помощью ГХ.
Фундаментальным идентификатор "димерных жирных кислот" в контексте настоящего изобретения, таким образом, является то, что их получение включает олигомеризацию ненасыщенных жирных кислот. Эта олигомеризация приводит, главным образом, другими словами, к получению предпочтительно, по крайней мере, 80% по весу, более предпочтительно, по крайней мере, 90% по весу, еще более предпочтительно, по крайней мере, 95% по весу и, более предпочтительно, по крайней мере, 98% по весу, димерных продуктов. Тот факт, что олигомеризация, таким образом, приводит к получению преимущественно димерных продуктов, содержащих ровно две молекулы жирных кислот, оправдывает это обозначение, которое является распространенным в любом случае. Альтернативным выражением для соответствующего термина "димерные жирные кислоты", следовательно, является "смесь, содержащая димеризованные жирные кислоты". Использование димерных жирных кислот, таким образом, автоматически предполагает использование бифункциональных соединений (v). Это также оправдывает утверждение в контексте настоящего изобретения, что димерные жирные кислоты предпочтительно используют в качестве соединения (v). Это происходит по той причине, что соединения (v), по-видимому, представляют собой основной компонент смесей, которые называются димерными жирными кислотами. Таким образом, если димерные жирные кислоты используются в качестве соединений (v), то это означает, что эти соединения (v) используются в форме соответствующих смесей с описанными выше мономерными и/или тримерными молекулами и/или другими побочными продуктами.
Димерные жирные кислоты, которые используются, могут быть получены как коммерческие продукты. Примеры включают Radiacid 0970, Radiacid 0971, Radiacid 0972, Radiacid 0975, Radiacid 0976 и Radiacid 0977 от Oleon, Pripol 1006, Pripol 1009, Pripol 1012 и Pripol 1013 от Croda, Empol 1008, Empol 1061 и Empol 1062 от BASF SE и Unidyme 10 и Unidyme TI от Arizona Chemical.
Дополнительные предпочтительные соединения (v) представляют собой димерные диолы или присутствуют в димерных диолах. Димерные диолы уже давно известны и также упоминаются в научной литературе в качестве димерных жирных спиртов. Они представляют собой смеси, которые получают, например, путем олигомеризации ненасыщенных жирных кислот или их эстеров с последующей гидрогенизациецй групп кислоты или эстера, или путем олигомеризации ненасыщенных жирных спиртов. Используемые исходные материалы могут представлять собой ненасыщенные C12-С22 жирные кислоты или их эстеры, или ненасыщенные C12-С22 жирные спирты. Радикалы гидрокарбила, которые связывают гидроксильные группы в димерных диолах, определяются таким же образом, что и радикалы гидрокарбила, которые разделяют карбоксильные группы димерных жирных кислот.
Например, DE-11 98 348 описывает их получение путем димеризации ненасыщенных жирных спиртов с основными соединениями щелочноземельных металлов при температуре выше 280°C.
Они также могут быть получены путем гидрогенизации димерных жирных кислот и/или их эстеров, как описано выше, в соответствии с German Auslegeschrift DE-B-17 68 313. При условиях, описанных в данном источнике, не только карбоксильные группы жирных кислот подвергаются гидрогенизации до гидроксильных групп, но и любые двойные связи, которые все еще присутствует в димерных жирных кислотах или их эстерах, также частично или полностью гидрогенизируются. Кроме того, можно проводить гидрогенизацию таким образом, что двойные связи полностью изменяются в процессе гидрогенизации. В этом случае получают ненасыщенные димерные диолы. Предпочтительно, когда гидрогенизацию проводят таким образом, что двойные связи весьма существенно гидрогенизируются.
Другой способ получения димерных диолов включает димеризацию ненасыщенных спиртов в присутствии катализаторов на основе кремнезема/оксида алюминия и основных соединений щелочных металлов, в соответствии с международной заявкой WO 91/13918.
Независимо от способов, описанных для получения димерных диолов, предпочтение отдается использованию этих димерных диолов, которые были получены из С18-жирных кислот или их эстеров, или С18-жирных спиртов. Таким образом, образуются преимущественно димерные диолы с 36 атомами углерода.
Димерные диолы, которые были получены при использовании указанных выше промышленных процессов, всегда имеют варьирующее количество тримерных триолов и монофункциональных спиртов. В целом, доля димерных молекул составляет более чем 70% по весу, а остаток представляет собой тримерные молекулы и мономерные молекулы. В контексте настоящего изобретения можно использовать либо эти димерные диолы, либо более чистые димерные диолы, содержащие более 90% по весу димерных молекул. Особое предпочтение отдается димерным диолам с более чем 90 до 99% по весу содержанием димерных молекул, а предпочтение среди них отдается, в свою очередь, тем димерным диолам, двойные связи и/или ароматические радикалы которых, были, по крайней мере, частично или полностью гидрогенизированы. Альтернативное выражение, которое соответствует термину "димерные диолы", таким образом, представляет собой выражение "смесь, включающая димеры полученные путем димеризации жирных спиртов". Использование димерных диолов, таким образом, автоматически предполагает использование бифункциональных соединений (v). Это также оправдывает утверждение в контексте настоящего изобретения, что димерные диолы используются в качестве соединений (v). Это происходит благодаря тому, что соединения (v), по-видимому, представляют собой основной компонент смесей, указанных в качестве димерных диолов. Таким образом, если димерные диолы используются в качестве соединений (v), то это означает, что эти соединения (v) используются в форме соответствующих смесей с описанными выше мономерными и/или тримерными молекулами и/или другими побочными продуктами.
Предпочтительно, среднее значение гидроксильных функциональностей димерных диолов должно составлять от 1,8 до 2,2.
В контексте настоящего изобретения особое предпочтение отдается, таким образом, использованию таких димерных диолов, которые могут быть получены путем гидрогенизации из описанных выше димерных жирных кислот. Особое предпочтение отдается тем димерным диолам, которые состоят на ≥ 90% по весу из димерных молекул, на ≤ 5% по весу из тримерных молекул и на ≤ 5% по весу из мономерных молекул и других побочных продуктов и/или имеют гидроксильную функциональность на уровне от 1,8 до 2,2.
Особое предпочтение отдается использованию таких диолов, которые могут быть получены путем гидрогенизации из димерных жирных кислот, которые состоят на 95 до 98% по весу из димерных молекул, менее чем на 5% по весу из тримерных молекул, и менее чем на 1% по весу из мономерных молекул и других побочных продуктов. Особое предпочтение, аналогичным образом, отдается использованию таких диолов, которые могут быть получены путем гидрогенизации из димерных жирных кислот, которые состоят на ≥ 98% по весу из димерных молекул, на ≤ 1,5% по весу из тримерных молекул и на ≤ 0,5% по весу из мономерных молекул и других побочных продуктов.
Димерные жирные кислоты, которые могут быть использованы для получения димерных диолов, содержат, как уже было укащано выше, в соответствии с режимом проведения реакции как алифатические, так и, возможно, ароматические молекулярные фрагменты. Алифатические молекулярные фрагменты могут быть затем разделены на линейные и циклические фрагменты, которые, в свою очередь, могут быть насыщенными или ненасыщенными. С помощью гидрогенизации ароматические и ненасыщенные алифатические молекулярные фрагменты могут быть превращены в соответствующие насыщенные алифатические молекулярные фрагменты. Димерные диолы, которые используются в качестве компонента (v), соответственно, могут быть насыщенными или ненасыщенными. Димерные диолы предпочтительно являются алифатическими, в частности, алифатическими и насыщенными.
В контексте настоящего изобретения предпочтение отдается использованию таких димерных диолов, которые могут быть получены путем гидрогенизации групп карбоновой кислоты предпочтительно насыщенных алифатических димерных жирных кислот.
Особое предпочтение отдается использованию таких диолов, которые могут быть получены путем гидрогенизации из димерных жирных кислот, которые состоят на ≥ 98% по весу из димерных молекул, на ≤ 1,5% по весу из тримерных молекул, и на ≤ 0,5% по весу из мономерных молекул и других побочных продуктов.
Более предпочтительно, когда димерные диолы имеют гидроксильное число от 170 до 215 мг KOH/г, еще более предпочтительно от 195 до 212 мг KOH/г и, в частности, от 200 до 210 мг KOH/г, как определяется с помощью DIN ISO 4629. Более предпочтительно димерные диолы имеют вязкость от 1500 до 5000 мПа, еще более предпочтительно 1800 до 2800 мПа (25°C, Brookfield, ISO 2555).
Димерные диолы для применения со значительным преимуществом включают коммерческие продукты Pripol® 2030 и, в частности, Priopol® 2033 от Uniqema, или Sovermol® 908 от BASF SE.
Предпочтительные продукты реакции (R) получают путем реакции димерных жирных кислот с алифатическими, аралифатическими или ароматическими дигидрокси-функциональными соединениями. Алифатические соединения представляют собой неароматические органические соединения. Они могут быть линейными, циклическими или разветвленными. Возможные примеры соединений представляют собой те, которые включают две гидроксильные группы и алифатический радикал гидрокарбила. Возможными также являются соединения, которые содержат как атомы кислорода, присутствующие в двух гидроксильных группах, так и другие гетероатомы такие, как кислород или азот, в частности, кислород, например, в виде соединяющих связей этера и/или эстера. Аралифатические соединения представляют собой те, которые содержат как алифатические, так и ароматические структурные единицы. Предпочтительно, однако, когда продукты реакции (R) получают путем взаимодействия димерных жирных кислот с алифатическими дигидрокси-функциональными соединениями.
Алифатические, аралифатические или ароматические гидроксифункциональные соединения предпочтительно имеют среднюю молекулярную массу от 120 до 6000 г/моль, в частности, предпочтительно от 200 до 4500 г/моль.
Указание среднечислового молекулярного веса, таким образом, означает, что предпочтительные дигидроксифункциональные соединения представляют собой смеси различных больших дигидроксифункциональных молекул. Дигидроксифункциональные соединения предпочтительно представляют собой полиэтердиолы, полиэстердиолы или димерные диолы.
Является предпочтительным в контексте настоящего изобретения, чтобы димерные жирные кислоты и алифатические, аралифатические и/или ароматические, предпочтительно алифатические, дигидроксифункциональные соединения вступали в реакцию друг с другом в молярном соотношении от 0,7/2,3 до 1,6/1,7, предпочтительно от 0,8/2,2 до 1,6/1,8 и наиболее предпочтительно от 0,9/2,1 до 1,5/1,8. В результате избытка гидроксильных групп таким образом получают гидроксифункциональные продукты реакции, которые дополнительно имеют низкое кислотное число. С помощью уровня избытка можно регулировать молекулярную массу продукта реакции. Если используется только небольшой избыток гидроксифункционального реагента, то результат будет, соответственно, представлять собой получение продуктов с длинной цепью, так как только в этом случае гарантируется существенное превращение присутствующих кислотных групп. В случае более значительного избытка гидроксифункционального реагента, результат будет представлять собой, соответственно, продукты реакции с более короткой цепью. Среднечисловая молекулярная масса продуктов реакции, конечно, также зависит от молекулярной массы реагентов, например, предпочтительно алифатических дигидроксифункциональных соединений. Среднечисловая молекулярная масса предпочтительных продуктов реакции может изменяться в широких пределах и составляет, например, от 600 до 40000 г/моль, в частности, от 800 до 10000 г/моль, наиболее предпочтительно от 1200 до 5000 г/моль.
Предпочтительные продукты реакции, таким образом, могут также быть описаны как линейные соединения блочного типа А-(В-А)n. В этом случае, по крайней мере, один тип блоков основывается на соединении (v). Предпочтительно, в основе блоков В лежат димерные жирные кислоты, т.е. соединения (v). В основе блоков А предпочтительно лежат алифатические дигидроксифункциональные соединенияя, в частности, предпочтительно алифатические полиэтердиолы, полиэстердиолы или димерные диолы. В последнем случае соответствующий продукт реакции, таким образом, исключительно основывается на соединениях (v), соединенных друг с другом.
Весьма предпочтительные продукты реакции (R) получают путем реакции димерных жирных кислот, по крайней мере, с одним алифатическим гидроксифункциональным соединением общей структурной формулы (I):

где R представляет собой С3-С6 алкиленовый радикал, а n выбирают таким образом, что соединение формулы (I) имеет среднечисловую молекулярную массу от 120 до 6000 г/моль, димерные жирные кислоты и соединения формулы (I) используют в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловую молекулярную массу от 600 до 40000 г/моль и кислотное число менее чем 10 мг KOH/г.
В очень предпочтительном варианте осуществления n в данной заявке выбирается таким образом, что соединение формулы (I) имеет среднечисловую молекулярную массу от 450 до 2200 г/моль, в частности, от 800 до 1200 г/моль. R предпочтительно представляет собой C3 или C4 алкиленовый радикал. Более предпочтительно, он представляет собой радикал изопропилена или радикал тетраметилена. Наиболее предпочтительно, когда соединение формулы (I) представляет собой полипропиленгликоль или политетрагидрофуран. Димерные жирные кислоты и соединения формулы (I) используются в данном случае предпочтительно в молярном соотношении от 0,7/2,3 до 1,3/1,7. В этом варианте осуществления полученный продукт реакции имеет среднечисловую молекулярную массу от 1500 до 5000 г/моль, предпочтительно от 2000 до 4500 г/моль и наиболее предпочтительно от 2500 до 4000 г/моль.
Кроме того, наиболее предпочтительные продукты реакции (R) получают путем взаимодействия димерных жирных кислот, по крайней мере, с одним гидроксифункциональным соединением общей структурной формулы (II):

где
R представляет собой бивалентный органический радикал, включающий от 2 до 10 атомов углерода,
R1 и R2 каждый независимо представляют собой линейный или разветвленный радикал алкилена, содержащий от 2 до 10 атомов углерода,
X и Y каждый независимо представляет собой О, S или NR3, где R3 представляет собой водород или алкильный радикал, содержащий от 1 до 6 атомов углерода, и
m и n, соответственно, являются выбранными таким образом, что соединение формулы (II) имеет среднечисловой молекулярный вес от 450 до 2200 г/моль,
где компоненты (а) и (b) используются в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловой молекулярный вес от 1200 до 5000 г/моль и кислотное число менее чем 10 мг KOH/г.
В структурной формуле (II) R представляет собой бивалентный органический радикал, включающий от 2 до 10 атомов углерода и, предпочтительно, от 2 до 6 атомов углерода. Радикал R может, например, быть алифатическим, ароматическим или аралифатическим. Радикал R, а также атомы углерода и атомы водорода, может также содержать гетероатомы, например, О или N. Радикал может быть насыщенным или ненасыщенным. R предпочтительно представляет собой алифатический радикал, содержащий от 2 до 10 атомов углерода, более предпочтительно, представляет собой алифатический радикал, содержащий от 2 до 6 атомов углерода, и наиболее предпочтительно представляет собой алифатический радикал, содержащий от 2 до 4 атомов углерода. Например, R радикал представляет собой C2H4, C3H6, C4H8 или С2Н4-О-С2Н4.
R1 и R2 каждый независимо представляют собой линейные или разветвленные радикалы алкилена, содержащие от 2 дол 10 атомов углерода, предпочтительно от 2 до 6 атомов углерода, и более предпочтительно от 3 до 5 атомов углерода. Эти радикалы предпочтительно содержат только углерод и водород.
В соединениях структурной формулы (II), все n R1 радикалов и все m R2 радикалов могут быть идентичными. Однако также является возможным, что присутствуют различные виды радикалов R1 и R2. Предпочтительно, когда все R1 и R2 радикалы являются идентичными.
Весьма предпочтительно, когда R1 и R2 представляют собой C4 или C5 алкилен, в частности, радикал тетраметилена или пентаметилена. В очень предпочтительном варианте осуществления настоящего изобретения оба радикала R1 и R2 представляют собой радикалы пентаметилена.
X и Y каждый независимо представляет собой О, S или NR3, где R3 представляет собой водород или алкильный радикал, содержащий от 1 до 6 атомов углерода. Предпочтительно, X и Y каждый независимо представляет собой О или NR3; более предпочтительно, каждый из них независимо представляет собой О и NH; наиболее предпочтительно, когда X и Y представляют собой О.
Индексы m и n являются соответствующим образом выбраны таким образом, что соединения структурной формулы (II) имеют среднечисловой молекулярный вес от 450 дол 2200 г/моль, предпочтительно от 500 до 1400 г/моль, более предпочтительно от 500 до 1200 г/моль.
Полиолы полиэстера общей структурной формулы (I) могут быть получены с помощью первого способа, где соединения HX-R-YH выступают в качестве исходных соединений, а цепи полиэстера с концевыми гидроксильными группами полимеризуются на исходном соединении путем полимеризации с раскрытием кольца лактонов гидроксикарбоновых кислот HO-R1-COOH и НО-R2-COOH. В соответствии со вторым способом является также возможным сначала получать полиэстеры с альфа-гидрокси-гамма-карбокси-концами, например, путем полимеризации с раскрытием кольца лактонов гидроксикарбоновых кислот HO-R1-COOH и HO-R2-COOH, или путем поликонденсации гидроксикарбоновых кислот HO-R1-COOH и HO-R2-COOH. Полиэстеры с альфа-гидрокси-гамма-карбокси-концами могут потом, в свою, очередь, вступать в реакцию с соединениями HX-R-YH при использовании реакции конденсации с получением диолов полиэстера для применения в соответствии с изобретением.
Соответствующие процессы описываются, например, у German Offenlegungsschrift 2234265 " Polylactone" [Hydroxyl-terminal polylactones] от заявителя Stamicarbon N.V.
Polylactone" [Hydroxyl-terminal polylactones] от заявителя Stamicarbon N.V.
Димерные жирные кислоты и соединения формулы (II) используются предпочтительно в молярном соотношении от 0,7/2,3 до 1,3/1,7. В этом варианте реализации полученный продукт реакции предпочтительно имеет среднечисловой молекулярный вес от 1200 до 5000 г/моль, предпочтительно от 1200 до 4500 г/моль и наиболее предпочтительно от 1300 до 4500 г/моль.
Аналогичным образом наиболее предпочтительные продукты реакции (R) получают путем реакции димерных жирных кислот с димерными диолами, где димерные жирные кислоты и димерные диолы используют в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловую молекулярную массу от 1200 до 5000 г/моль и кислотное число менее чем 10 мг KOH/г.
Предпочтительные димерные диолы уже были описаны выше. В данной заявке является предпочтительным, чтобы димерные жирные кислоты и димерные диолы использовались в молярном соотношении от 0,7/2,3 до 1,3/1,7. Полученный в результате продукт реакции в соответствии с данной заявкой предпочтительно имеет среднечисловую молекулярную массу от 1200 до 5000 г/моль, предпочтительно от 1300 до 4500 г/моль, и весьма предпочтительно от 1500 до 4000 г/моль.
Из приведенных выше утверждений следует, что продукты реакции (R) получают путем исключительного использования соединений (v). Например, можно получить продукты реакции при использовании описанных выше предпочтительных димерных жирных кислот и димерных диолов. Оба класса соединений представляют собой соединения (v) или оба класса соединений представляют собой смеси соединений, содержащие бифункциональные соединения (v). Тем не менее, является в равной степени возможным получать продукты реакции (R) с помощью осуществления реакции соединений (v), предпочтительно димерных жирных кислот, с другими органическими соединениями, в частности, такими, которые имеют структурные формулы (I) и (II).
В контексте настоящего изобретения является предпочтительным, когда от 30 до 100 мол. %, по крайней мере, одного соединения (v) используют при получении продуктов реакции. Если используется только соединение (v), то является очевидным, что используются, по крайней мере, два соединения (v).
Содержание продуктов реакции (R) предпочтительно находится в диапазоне от 0,1 до 15% по весу, предпочтительно от 0,5 до 12% по весу, более предпочтительно от 0,75 до 8% по весу, в каждом случае из расчета на общую массу пигментного водного материала базового покрытия (b.2.1).
Продукт реакции в соответствии с изобретением, как правило, плохо растворим в водных системах. Поэтому предпочтительно использовать его непосредственно в производстве материала пигментного водного базового слоя (b.2.1), а не добавлять к иным образом полученной композиции для покрытия только по завершении получения.
Материал базового покрытия (b.2.1) для применения в соответствии с изобретением предпочтительно включает, по крайней мере, один пигмент. Под пигментами понимают цветные пигменты и/или пигменты визуального эффекта, которые известны сами по себе. Более предпочтительно, если материал базового покрытия содержит пигмент визуального эффекта.
Такие цветные пигменты и пигменты эффекта являются известными квалифицированному специалисту в данной области техники и описываются, например, в  Lacke und Druckfarben, Georg Thieme Verlag, Stuttgart, New York, 1998, стр. 176 и 451. Термины "красящий пигмент" и "цветной пигмент" являются взаимозаменяемыми, подобно терминам "пигмент визуального эффекта" и "пигмент эффекта".
Lacke und Druckfarben, Georg Thieme Verlag, Stuttgart, New York, 1998, стр. 176 и 451. Термины "красящий пигмент" и "цветной пигмент" являются взаимозаменяемыми, подобно терминам "пигмент визуального эффекта" и "пигмент эффекта".
Предпочтительные пигменты эффекта представляют собой, например, пластинчатые металлические пигменты такие, как пластинчатые алюминиевые пигменты, золотистую бронзу, окисленную бронзу и/или пигменты на основе оксида железа-алюминия, перламутровые пигменты такие, как перламутровая эссенция-краситель, основной карбонат свинца, хлорид оксида висмута и/или оксид металла, пигменты слюды и/или другие пигменты такие, как ламеллярный (пластинчатый) графит, пластинчатый оксид железа, пигменты многослойного эффекта, которые состоят из PVD пленок и/или жидкокристаллических полимерных пигментов. Особое предпочтение отдается пластинчатым металлическим пигментам эффекта, в частности, пластинчатым алюминиевым пигментам.
Типичные цветные пигменты, в частности, включают неорганические красящие пигменты такие, как белые пигменты такие, как диоксид титана, пигменты на основе окиси цинка, сульфида цинка или литопона; черные пигменты такие, как сажа, черный пигмент на основе железа-марганца, или шпинель черный; хроматические пигменты такие, как оксид хрома, зеленый оксид хрома, зеленый гидрат кобальта или ультрамарин зеленый, синий кобальт, ультрамарин синий или марганец синий, ультрамарин фиолетовый или кобальт фиолетовый и фиолетовый марганец, красный оксид железа, сульфоселенид кадмия, молибдат красный или ультрамарин красный; коричневый оксид железа, смешанный коричневый, шпинельные фазы и фазы корунда или хром оранжевый; или желтый оксид железа, никель-титан желтый, хром-титан желтый, сульфид кадмия, сульфид кадмия цинка, желтый хром или ванадат висмута.
Содержание пигментов предпочтительно находится в пределах интервала от 1,0 до 40,0% по весу, предпочтительно от 2,0 до 20,0% по весу, более предпочтительно от 5.,0 до 15,0% по весу, в каждом случае из расчета на общий вес пигментного водного материала базового покрытия (b.2.1).
Водный материал базового покрытия (b.2.1) предпочтительно также включает, по крайней мере, один полимер, отличный от сополимеров (CP) и отличный от продуктов реакции (R), в качестве связующего агента, в частности, по крайней мере один полимер, выбранный из группы, которая состоит из полиуретанов, полиэстеров, полиакрилатов и/или сополимеров упомянутых полимеров, в частности, полиуретан-полиакрилат.
Предпочтительные полиуретановые смолы описываются, например, в
- немецкой патентной заявке DE 19948004 А1, стр. 4 строка 19 до стр. 11 строка 29 (полиуретановый преполимер В1),
- европейской патентной заявке ЕР 0228003 А1, стр. 3 строка 24 дл стр. 5 строка 40,
- европейской патентной заявке ЕР 0 634 431 А1, стр. 3 строка 38 до стр. 8 строка 9, или
- международной патентной заявке WO 92/15405, стр. 2 строка 35 до стр. 10 строка 32.
Предпочтительные полиэстеры описываются, например, в DE 4009858 А1 в колонке 6 строка 53 до колонки 7 строка 61 и колонке 10 строка 24 до колонки 13 строка 3.
Предпочтительные сополимеры полиуретана-полиакрилата и их получение описываются, например, в WO 91/15528 А1, стр. 3 строка 21 до стр. 20 строка 33, и в DE 4437535 А1, стр. 2 строка 27 до стр. 6 строка 22.
Описанные полимеры в качестве связующих агентов предпочтительно являются гидрокси-функциональными. Предпочтительно, когда водный материал базового покрытия (b.2.1) включает как, по крайней мере, один сополимер (CP), так и, по крайней мере, один продукт реакции (R), по крайней мере, один сополимер полиуретана-полиакрилата, отличный от сополимеров (CP).
Содержание дополнительных полимеров в качестве связующего агента, предпочтительно, по крайней мере, одного сополимера полиуретана-полиакрилата, предпочтительно находится в интервале от 1,0 дол 20,0% по весу, более предпочтительно от 1,5 до 15,0% по весу, в частности, предпочтительно от 2,0 до 10,0% по весу, в каждом случае из расчета на общий вес пигментного водного материала базового покрытия (b.2.1).
Кроме того, материал базового покрытия (b.2.1) предпочтительно включает, по крайней мере, один типичный сшивающий агент, который является известным как таковой. Материал базового покрытия предпочтительно включает в качестве поперечно сшивающего агента, по крайней мере, одну смолу аминопласта и/или блокированный полиизоцианат, предпочтительно смолу аминопласта. Среди смол аминопласта меламиновые смолы, в частности, являются предпочтительными.
Содержание сшивающих агентов, в частности, смолы аминопласта и/или блокированных полиизоцианатов, более предпочтительно смолы аминопласта, предпочтительно меламиновых смол, желательно находится в интервале от 0,5 до 20,0% по весу, более предпочтительно от 1,0 до 15,0% по весу, в частности, предпочтительно от 1,5 до 10,0% по весу, в каждом случае из расчета на общий вес пигментного водного материала базового покрытия (b.2.1).
Предпочтительно, когда материал базового покрытия (b.2.1) дополнительно включает загуститель. Приемлемые загустители представляют собой неорганические загустители из группы слоистых силикатов. Силикаты лития-алюминия-магния являются особенно приемлемыми. Наряду с неорганическими загустителями, однако, также является возможным использовать один или более органических загустителей. Такие предпочтительно являются выбранными из группы, состоящей из загустителей сополимера (мет)акриловой кислоты-(мет)акрилата, например, коммерческий продукт Rheovis AS S130 (BASF), и загустители полиуретана, например, коммерческий продукт Rheovis PU 1250 (BASF). Используемые загустители являются отличными от описанных выше полимеров, например, предпочтительных связующих агентов. Предпочтение отдается неорганическим загустителям из группы слоистых силикатов.
Содержание загустителей предпочтительно находится в интервале от 0,01 до 5.0% по весу, предпочтительно от 0,02 до 4% по весу, более предпочтительно от 0,05 до 3,0% по весу, в каждом случае из расчета на общий вес пигментного водного материала базового покрытия (b.2.1).
Кроме того, водный материал базового покрытия (b.2.1) может также включать, по крайней мере, одну добавку. Примеры таких добавок представляют собой соли, которые могут подвергаться термическому расщеплению без остатка или существенно без остатка, смолы в качестве связующих веществ, которые отверждаются физически, термически и/или с помощью актиничного излучения, и которые являются отличными от полимеров, которые уже упоминались, дополнительные сшивающие агенты, органические растворители, реактивные разбавители, прозрачные пигменты, наполнители, красители, растворимые в молекулярной дисперсии, наночастицы, светостабилизаторы, антиоксиданты, агенты для удаления воздуха, эмульгаторы, добавки, улучшающие скольжение, ингибиторы полимеризации, инициаторы свободно-радикальной полимеризации, промоторы адгезии, агенты для регуляции подачи, вспомогательные вещества для пленкообразования, агенты для устранения натеков (SCA), ингибиторы горения, ингибиторы коррозии, воски, сиккативы, биоциды и матирующие агенты.
Приемлемые добавки упомянутого выше вида являются известными, например, из
- немецкой патентной заявки DE 19948004 А1, стр. 14 строка 4 до стр. 17 строка 5,
- немецкой патентной заявки DE 10043405 С1, колонка 5, абзацы от [0031] до [0033].
Они используются обычным образом и в известных количествах. Например, содержание их может находиться в диапазоне от 1,0 до 40,0% по весу из расчета на общий вес пигментного материала водного базового покрытия (b.2.1).
Содержание твердых веществ в материале базового покрытия в соответствии с изобретением может изменяться в соответствии с требованиями конкретного случая. Содержание твердых веществ, в первую очередь, определяется вязкостью, необходимой для применения, в частности, для нанесения путем распыления, и, таким образом, может быть скорректировано квалифицированным специалистом на основе его общих знаний в области техники, необязательно, при использовании нескольких предварительных тестов.
Содержание сухих веществ материала базового покрытия (b.2.1) предпочтительно составляет от 5 до 70% по весу, более предпочтительно от 8 до 60% по весу, наиболее предпочтительно от 12 до 55% по весу.
Под содержанием сухих веществ (нелетучая фракция) подразумевают весовую фракцию, которая остается в качестве остатка в результате испарения при определенных условиях. В настоящем описании изобретения содержание сухих веществ определяется в соответствии со стандартом DIN EN ISO 3251. Это делается путем испарения материала базового покрытия при температуре 130°C в течение 60 минут.
Если не указано иное, то этот способ анализа также используется для того, чтобы, например, определить соотношение различных компонентов материала базового покрытия, например, полиуретановой смолы, сополимера полиакрилата и полиуретана, сополимера (CP), продукта реакции (R) или поперечно сшивающего агента в общей массе материала базового покрытия. Определяется также содержание твердых веществ в дисперсии полиуретановой смолы, сополимера полиуретана-полиакрилата, сополимера (CP), продукта реакции (R) или поперечно сшивающего агента, который прибавляется к материалу базового покрытия. Принимая во внимание содержание твердых веществ в дисперсии и количество дисперсии, используемой в материале базового покрытия, можно установить или определить содержание компонента в общей композиции.
Материал базового покрытия в соответствии с изобретением является водным. Выражение «водный» является известным в этом контексте специалисту в данной области техники. Фраза относится в принципе к материалу базового покрытия, который исключительно не основывается на органических растворителях, т.е., не содержит исключительно органические растворители в качестве растворителей материала, но вместо этого и в противовес этому, включает значительную часть воды в качестве растворителя. "Водный раствор" для целей настоящего изобретения предпочтительно следует понимать так, что композиция покрытия, которая рассматривается, в частности, материал базового покрытия, имеет водную фракцию, которая составляет, по крайней мере, 40% по весу, предпочтительно, по крайней мере, 45% по весу, весьма предпочтительно, по крайней мере, 50% по весу, в каждом случае из расчета на общую сумму присутствующих растворителей (то есть, воды и органических растворителей). В свою очередь, является предпочтительным, когда фракция воды составляет от 40 до 95% по весу, более предпочтительно от 45 до 90% по весу, весьма предпочтительно от 50 до 85% по весу, в каждом случае из расчета на общую сумму присутствующих растворителей.
Такое же определение термина "водный", конечно, также применимо ко всем дополнительным системам, которые описываются в контексте настоящего изобретения, например, к водному характеру материалов электрохимического покрытия (е.1) или к водному характеру водных дисперсий сополимеров (CP).
Материалы базового покрытия (b.2.1), используемые в соответствии с изобретением, могут быть получены при использовании смесительных устройств и способов смешивания, которые являются общепринятыми и известными для производства материалов базового покрытия.
По крайней мере, один из материалов базового покрытия (b.2.2.х), который используется в способе в соответствии с изобретением, имеет характеристики, существенные для изобретения, как описывается для материала базового покрытия (b.2.1). Это означает, в частности, что, по крайней мере, один из материалов базового покрытия (b.2.2.х) включает, по крайней мере, одну водную дисперсию, включающую, по крайней мере, один сополимер (CP). Все предпочтительные воплощения и характеристики, представленные в описании материала базового покрытия (b.2.1), предпочтительно применяются, по крайней мере, к одному из материалов базового покрытия (b.2.2.х).
В описанном выше предпочтительном варианте (а) этапа (2.2) способа в соответствии с изобретением, в котором два используемых материала базового покрытия (b.2.2.х) являются идентичными, оба материала базового покрытия (b.2.2.х) очевидным образом имеют характеристики, существенные для изобретения, как описывается для материала базового покрытия (b.2.1). В этом варианте материал базового покрытия (b.2.2.х) предпочтительно включает пигменты эффекта, как описывается выше, в частности, пластинчатые пигменты на основе алюминия. Предпочтительные соотношения составляют от 2 до 10% по весу, предпочтительно от 3 до 8% по весу, в каждом случае из расчета на общий вес материала базового покрытия. Однако материал базового покрытия может включать дополнительные пигменты, то есть, в частности, хроматические пигменты.
В описанном выше предпочтительном варианте (b) этапа (2.2) способа в соответствии с изобретением первый материал базового покрытия (b.2.2.а) предпочтительно наносится первым, этот материал также может называться как материал базового покрытия для подготовки окрашивания. Он служит в качестве грунтовочного слоя для базового пленочного покрытия, которое следует за ним и которое может потом оптимально выполнять свои функции придания цвета и/или эффекта.
В первом особом воплощении варианта (b) материал базового покрытия для подготовки окрашивания такого вида является существенно свободным от хроматических пигментов и пигментов эффекта. В частности, материал базового покрытия (b.2.2.а) такого вида содержит менее чем 2% по весу, предпочтительно менее чем 1% по весу, хроматических пигментов и пигментов эффекта, в каждом случае из расчета на общий вес пигментного водного материала базового покрытия. Он предпочтительно является свободным от таких пигментов. В этом воплощении материал базового покрытия для подготовки окрашивания включает предпочтительно черные и/или белые пигменты, в частности, предпочтительно оба вида таких пигментов. Предпочтительно, он содержит от 5 до 20% по весу, предпочтительно от 8 до 12% по весу белых пигментов и от 0,05 до 1% по весу, предпочтительно от 0,1 до 0.5% по весу черных пигментов, в каждом случае из расчета на общий вес материала базового покрытия. Серый цвет, который получают в результате этого и который может быть задан при различных уровнях яркости при использовании соотношения белых и черных пигментов, представляет собой индивидуально регулируемую основу для нанесения базового покрытия, которая затем следует, таким образом, что цвет и/или эффект, который придается наносимому материалу базового слоя, который следует за ним, может проявляться в оптимальном режиме. Пигменты являются известными квалифицированному специалисту в данной области техники и также описываются выше. Предпочтительный белый пигмент в данном случае представляет собой диоксид титана, а предпочтительный черный пигмент -газовую сажу.
Для материала базового покрытия второго базового покрытия или для второго и третьего базовых покрытий в рамках этого варианта способа (b), предпочтительно применяется то же самое, что указано для материала базового покрытия (b.2.2.х), описанного в варианте (а). В частности, этот материал предпочтительно содержит пигменты эффекта. Как для материала базового покрытия для подготовки к окрашиванию (b.2.2.х), так и для материала второго базового покрытия (b.2.2.х), который предпочтительно включает пигменты эффекта, признаки, существенные для данного изобретения, как описано для материала базового покрытия (b.2.1), должны быть выполнены. Конечно, оба материала базового покрытия (b.2.2.х) также могут выполнять эти функции.
Во втором конкретном варианте осуществления в соответствии с настоящим изобретением также является возможным включение хроматических пигментов в материал базового покрытия (b.2.2.а) для подготовки к окрашиванию. Этот вариант представляет собой предмет выбора, в частности тогда, когда полученная многослойная красочная система должна иметь высокий хроматический цветовой тон, например, темно-красный или желтый цвета. В этом случае материал базового покрытия для подготовки к окрашиванию (b.2.2.а) имеет, например, содержание от 2 до 6% по весу хроматических пигментов, в частности, красных пигментов и/или желтых пигментов, предпочтительно в комбинации с 3-15% по весу, предпочтительно от 4 до 10% по весу белых пигментов. По крайней мере, один дополнительный материал базового покрытия, который затем наносится последовательно, очевидным образом также включает описанные хроматические пигменты так, что первый материал базового покрытия (b.2.2.а) снова служит для подготовки окрашивания. В этом варианте осуществления также любой индивидуальный материал базового покрытия (b.2.2.х), множество таковых или каждый из них могут быть одним из тех, которые выполняют функции, необходимые для осуществления изобретения, как описано для материала базового покрытия (b.2.1).
В описанном выше предпочтительном варианте (с) стадии (2.2) способа в соответствии с изобретением также любой индивидуальный материал базового покрытия (b.2.2.х), множество таковых или каждый из них могут быть одним из тех, которые выполняют функции, необходимые для осуществления изобретения, как описано для материала базового покрытия (b.2.1).
Способ в соответствии с настоящим изобретением позволяет получать многослойные красочные системы без отдельного этапа отверждения. Несмотря на это, применение способа в соответствии с изобретением приводит к получению многослойных красочных систем, которые обладают отличной стойкостью к ударным нагрузкам, в частности, устойчивостью к ударам мелкими камнями.
Стойкость красочных систем к ударным нагрузкам и устойчивость к ударам мелкими камнями могут быть определены с помощью способов, известных специалисту в данной области техники. Например, один из вариантов представляет собой тест на удары мелкими камнями в соответствии с DIN 55966-1. Оценка надлежащим образом обработанных красочной системой поверхностей с точки зрения степени повреждения и, следовательно, с точки зрения устойчивости к ударам мелкими камнями может осуществляться в соответствии с DIN EN ISO 20567-1.
Способ в соответствии с настоящим изобретением позволяет получать многослойные красочные системы без отдельного этапа отверждения, но, несмотря на это, применение способа в соответствии с изобретением приводит к получению многослойных красочных систем, которые имеют отличную стабильность в отношении микроотверстий так, что большая толщина покрытия соответствующих базовых покрытий может быть достигнута без потери эстетического качества.
Качество устойчивости к микроотверстиям в принципе, может быть определено при использовании предела размера микроотверстий и количества микроотверстий. Предел размера и его определение можно описать следующим образом: при нанесении многослойной красочной системы толщина слоя базового покрытия, расположенного под лаковым покрытием, который дополнительно отверждается не отдельно, а вместе с лаковым покрытием, варьирует. Это пленочное покрытие может, например, представлять собой покрытие, расположенное непосредственно сверху от электрохимического покрытия, и/или покрытие, расположенное непосредственно под лаковым покрытием. Как следует из приведенных деталей, тенденция к образованию микроотверстий должна возрастать с увеличением толщины такого покрытия, поскольку, соответственно, более высокие количества воздуха, органических растворителей и/или воды должны удаляться из покрытия. Толщина этого покрытия, от которой микроотверстия являются различимыми, называется пределом размера микроотверстий. Чем выше предел размера микроотверстий, очевидно, тем лучше качество устойчивости к микроотверстиям. Количество микроотверстий для данной толщины покрытия, конечно, также является показателем качества устойчивости к микроотверстиям.
Настоящее изобретение в данной заявке иллюстрируется примерами, приведенными ниже.
Примеры
Подробное описание отдельных компонентов, которые используются, и способов тестирования
Димерная жирная кислота:
Используемая димерная жирная кислота содержит менее чем 1,5% по весу тримерных молекул, 98% по весу димерных молекул и менее чем 0,3% по весу жирных кислот (мономеры). Ее получают на основе линоленовой кислоты, линолевой кислоты и олеиновой кислоты (Pripol™ 1012-LQ-(GD), от Croda).
Полиэстер (P1):
Его получают в соответствии с примером D DE 4009858 А, колонка 16 строки от 37 до 59. Соответствующий раствор полиэстера имеет содержание сухих веществ, равное 60% по весу, при использовании бутилгликоля вместо бутанола в качестве растворителя, это означает, что присутствующие растворители представляют собой, главным образом, бутилгликоль и воду.
Привитой сополимер (GCP1):
Привитой сополимер на основе полиуретана, полученный в соответствии с DE 19948004 - А1 (стр. 27 - Пример 2), содержание сухих веществ доведено до 32,5% по весу водой.
Определение среднечислового молекулярного веса:
Среднечисловой молекулярный вес был определен с помощью осмоса при давлении насыщенных паров. Измерение осуществляли при использовании парофазного осмометра (модель 10,00 от Knauer) на серии концентраций исследуемого компонента в толуоле при 50°C вместе с бензофеноном в качестве калибровочного вещества для определения константы экспериментальной калибровки используемого прибора (в соответствии с Е.  , G.
, G.  , K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Берлин, стр. 47-54, 1982, в котором бензил использован в качестве калибровочного вещества).
, K.-F. Arndt, "Leitfaden der Polymercharakterisierung", Akademie-Verlag, Берлин, стр. 47-54, 1982, в котором бензил использован в качестве калибровочного вещества).
Получение продукта реакции (R) для использования в соответствии с данным изобретением
В реактор из нержавеющей стали на 4 л, оборудованный лопастной мешалкой, термометром, конденсором, термометром для измерения температуры наверху колонки и отделителем воды, 2000,0 г линейного диольного PolyTHF1000 (2 моль), 579,3 г димерной жирной кислоты (1 моль) и 51 г циклогексана нагревали до 100°C в присутствии 2,1 г оксида ди-н-бутилтина (Axion® CS 2455, от Chemtura). Нагревание продолжали плавно пока не начал образовываться конденсат.При максимальной температуре наверху колонки, равной 85°C, нагревание продолжали поэтапно до 220°C. Динамику реакции отслеживали путем определения кислотного числа. Когда кислотное число достигало ≤ 3 мг KOH/г, еще оставшийся циклогексан удаляли с помощью вакуумной дистилляция. Получали вязкую смолу.
Количество конденсата (воды): 34,9 г
Кислотное число: 2,7 мг KOH/г
Содержание сухих веществ (60 мин. при 130°C): 100,0%
Молекулярный вес (осмос при давлении насыщенных паров):
Среднечисловая молекулярная масса: 2200 г/моль
Вязкость: 5549 мПа,
(измерена при 23°C при использовании ротационного вискозиметра от Brookfield, модель CAP 2000+, ось 3, скорость сдвига: 1333 с-1)
Получение сополимера (CP) для использования в соответствии с данным изобретением
Сополимер (CP) или водную дисперсию, включающую указанный выше полимер, получали так, как представлено ниже.
а) Дисперсию полиуретана, содержащую альфа-метилстирол, получали на основе патента DE 19948004 В4, стр. 27, пример 1, "Herstellung eines  Полиurethans (В)" ["Получения полиуретана (В) в соответствии с изобретением"], за исключением дополнительного использования триметилолпропана, и с содержанием сухих веществ в полученной дисперсии, равным 29%, вместо 35,1% по весу. Основываясь на аддукте (В2), указанном в патенте DE 19948004 В4, Пример получения 1, аддукт получали при использовании моноэтаноламина вместо диэтаноламина:
Полиurethans (В)" ["Получения полиуретана (В) в соответствии с изобретением"], за исключением дополнительного использования триметилолпропана, и с содержанием сухих веществ в полученной дисперсии, равным 29%, вместо 35,1% по весу. Основываясь на аддукте (В2), указанном в патенте DE 19948004 В4, Пример получения 1, аддукт получали при использовании моноэтаноламина вместо диэтаноламина:
С этой целью реакционный резервуар, оснащенный мешалкой, внутренним термометром, противоточным конденсатором и электрическим нагревателем, изначально загружали в токе азот 200,0 частями по весу метилэтил кетона, 800,0 частями по весу N-метилпирролидона и 221,3 частями по весу моноэтаноламина (от BASF SE) при 20°C. К этой смеси добавляли по каплям в течение полутора часов 778,7 частей по весу 1-(1-изоцианат-1-метилэтил)-3-(1-метилвинил)бензола (TMI® (МЕТА) ненасыщенный алифатический изоцианат, от Cytec), который имел содержания изоцианата, равное 20,4% по весу изоцианата, так чтобы не позволить температуре реакционной смеси превысить 40°C. Полученную реакционную смесь подвергали перемешиванию до тех пор, пока свободные изоцианатные группы больше не поддавались обнаружению. После этого реакционную смесь стабилизировали с помощью 200 ч./млн. гидрохинона.
Теоретическое содержание сухих веществ в растворе описанного аддукта, полученного таким образом, было равно 50% по весу.
Затем в следующем реакционном резервуаре, оснащенном мешалкой, внутренним термометром, противоточным конденсатором и электрическим нагревателем, растворяли 431,7 частей по весу линейного полиэстер полиола и 69,7 частей по весу диметилолпропионовой кислоты (от GEO Specialty Chemicals) в 355,8 частях по весу метилэтил кетона и 61,6 частях по весу N-метилпирролидона в токе азота. Линейный полиэстер полиола предварительно получали из димеризованной жирной кислоты (Pripol® 1012, от Uniqema), изофталевой кислоты (от BP Chemicals) и гексан-1,6-диола (от BASF SE) (весовое соотношение изначальных компонентов: димеризованной жирной кислоты к изофталевой кислоте к гексан-1,6-диолу=54,00:30,02:15,98) и имеет гидроксильное число, равное 73 мг KOH/г твердой фазы, и среднечисловую молярную массу, равную 1379 г/моль. К полученному раствору при 45°C добавляли 288,6 частей по весу изофорон диизоцианата (Basonat® I, от BASF SE), который имеет содержание изоцианата, равное 37,75% по весу. После того, как экзотермическая реакция приостанавливалась, реакционную смесь постепенно нагревали до 80°C при перемешивании. Перемешивание продолжалось при этой температуре до тех пор, пока содержание изоцианата в растворе не становилось постоянным и равным 3,2% по весу. После этого реакционную смесь охлаждали до 65°C, и 85,2 частей по весу описанного выше аддукта добавляли вместе с 21,8 частями по весу триметилолпропана (от BASF SE). Полученную реакционную смесь перемешивали при 65°C до тех пор, пока содержание изоцианата в растворе не снижалось до 1,0% по весу. В то же время прибавляли 22,2% по весу диэтаноламина (от BASF SE), и содержание изоцианатных групп отслеживали до тех пор, пока свободные изоцианатные группы больше не поддавались обнаружению. Полученный растворимый полиуретан смешивали с 139,7 частями по весу метоксипропанола и 43,3 частями по весу триэтиламина (от BASF SE). Через 30 минут после добавления амина температура раствора была снижена до 60°C, после чего прибавляли 1981 часть по весу деионизированной воды при перемешивании в течение периода времени 30 минут. Метилэтил кетон отгоняли из полученной дисперсии при 60°C при сниженном давлении. После этого любые потери растворителя и воды были компенсированы.
Таким образом, полученная дисперсия полиуретана, содержащего альфа-метилстирол, имела содержание сухих веществ, равное 29,0% по весу, кислотное число составляло 34,0 мг KOH/г твердой фазы, а значение pH составляло 7,0 (измерено при 23°C).
b) Для получения первичной водной дисперсии сополимера (CP) в соответствии с изобретением в атмосфера азота 1961,2 частей по весу дисперсии полиуретана, содержащего альфа-метилстирол, в соответствии с а) разбавляли 40,0 частями по весу метоксипропанола (0,07% на основе полиуретана) и 686,5 частями по весу деионизированной воды, и нагревали до 80°C.Потом содержание реактора нагревали до 80°C, 0,6 части по весу пероксодисульфата аммония, растворенного в 35,7 частях по весу деионизированной воды, добавляли в реактор при атмосферном давлении. После этого при непрерывном перемешивании смесь из 301,6 частей по весу метилметакрилата, 261,6 частей по весу н-бутил акрилата, 5,6 частей по весу аллил метакрилата (0,87 моль % на основе общего содержания винилового мономера) и 134,9 частей по весу N-метилпирролидона равномерно прибавляли в течение периода времени пять часов. Вместе с началом добавления мономерной смеси раствор 1,1 части по весу пероксодисульфата аммония в 71,3 частях по весу деионизированной воды, аналогично, добавляли в течение пяти часов.
Во время осуществления свободнорадикальной полимеризации содержание свободных мономеров определяли каждые 30 минут с помощью газовой хроматографии (ГХ) (ГХ: один раз с помощью 50 м кремнеземной капиллярной колонки с полиэтиленгликолевой фазой и один раз с помощью 50 м кремнеземной капиллярной колонки с полидиметилсилоксановой фазой, газ-носитель: гелий, устройство ввода пробы с делением потока при 150°C, температурный режим термостата 40-220°C, пламенно-ионизационный датчик, температура детектора 275°C, внутренний стандарт: изобутил акрилат), и самое высокое суммарное содержание мономера, основываясь на дисперсии, равное 0,5% по весу, было обнаружено через 30 мин. (3,1% по весу из расчета на общее количество олефиновых ненасыщенных мономеров, которые использовались для полимеризации). После одновременного окончания добавления определенных количеств мономера и инициатора, полученную реакционную смесь перемешивали при 80°C в течение последующего часа и потом охлаждали до комнатной температуры.
Полученная первичная дисперсия сополимера имела очень хорошую стабильность при хранении. Содержание сухих веществ таковой составляло 32,5% по весу, кислотное число было равно 18,8 мг KOH/г твердой фазы, и pH таковой было 7,0. Размер частиц (z среднее), определенный с помощью фотонной корреляционной спектроскопии был равен 96 нм. С помощью газовой хроматографии (ГХ: один раз с помощью 50 м кремнеземной капиллярной колонки с полиэтиленгликолевой фазой и один раз с помощью 50 м кремнеземной капиллярной колонки с полидиметилсилоксановой фазой, газ-носитель: гелий, устройство ввода пробы с делением потока при 250°C, температурный режим термостата 40-220°C, пламенно-ионизационный датчик, температура детектора 275°C, внутренний стандарт: н-пропилгликоль), определяли содержание метоксипропанола, равное 2,7% по весу, и содержание N-метилпирролидона, равное 5,7% по весу.
После экстракции лиофилизированного полимера с помощью тетрагидрофурана, содержание геля было определено гравиметрически как такое, равное 80,3% по весу. По этой причине дисперсию подвергали лиофилизации и определяли лиофилизированный полимер, и затем полимер экстрагировали при использовании избытка тетрагидрофурана (соотношения тетрагидрофурана к лиофилизированному сополимеру=300:1) при 25°C в течение 24 часов. Изолировали нерастворенные вещества (содержание геля), высушивали при 50°C в печи с циркуляцией воздуха в течение 4 часов и после этого повторно взвешивали.
Получение водорастворимого материала базового покрытия 1 (не в соответствии с изобретением), которое может непосредственно наносится на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Компоненты, перечисленные как "водная фаза" в Таблице А, смешивали вместе в указанном порядке для образования водной смеси. На следующем этапе органическую смесь получали из компонентов, перечисленных как "органическая фаза". Органическую смесь прибавляли к водной смеси. Потом суммарную смесь перемешивали в течение 10 минут и доводили при использовании деионизированной воды и диметилэтаноламина до значения pH, равного 8, и до вязкости готового к нанесению материала, равной 58 мПа при сдвигающей нагрузке 1000 с-1, как измерено с помощью ротационного вискозиметра (прибор Rheomat RM 180 от Mettle-Toledo) при 23°C.

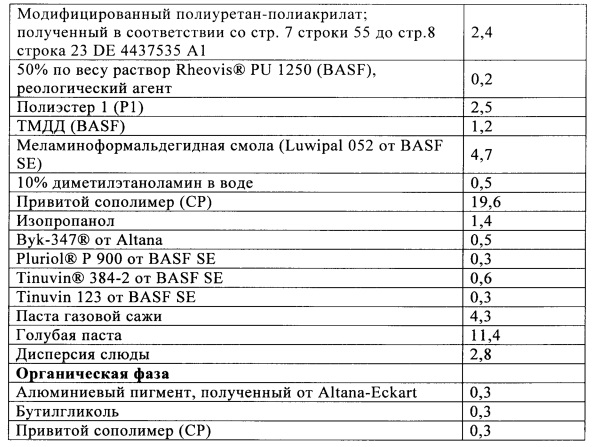
Получение голубой пасты:
Голубую пасту получали из 69,8 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 12,5 частей по весу Paliogen® Blue L 6482, 1,5 частей по весу диметилэтаноламина (10% в деминерализованной воде), 1,2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 15 частей по весу деионизированной воды.
Получение пасты газовой сажи:
Пасту газовой сажи получали из 25 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 10 частей по весу газовой сажи, 0,1 части по весу метил изобутилкетона, 1,36 частей по весу диметилэтаноламина (10% в деминерализованной воде), 2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 61,45 частей по весу деионизированной воды.
Получение дисперсии слюды:
Дисперсию слюды получали путем смешивания при использовании блока для перемешивания 1,5 частей по весу привитого сополимера (CP) и 1,3 частей по весу коммерческой слюды Mearlin Ext. Fine Violet 539V от Merck.
Получение водорастворимого материала базового покрытия 2 (не в соответствии с изобретением), который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 2 получали аналогично Таблице А, за исключением использования привитого сополимера (GCP1) вместо привитого сополимера (CP). Кроме того, при получении используемой дисперсии слюды привитой сополимер (GCP1) использовали вместо привитого сополимера (CP).
Получение водорастворимого материала базового покрытия 3 (не в соответствии с изобретением), который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 3 получали аналогично Таблице А, за исключением использования привитого сополимера (GCP1) вместо привитого сополимера (CP) и использования продукта реакции (R) вместо полиэстера Р1. Кроме того, при получении используемой дисперсии слюды привитой сополимер (GCP1) использовали вместо привитого сополимера (CP).
Получение водорастворимого материала базового покрытия II в соответствии с настоящим изобретением, который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия I1 получали аналогично Таблице А, за исключением использования продукта реакции (R) вместо полиэстера Р1.

Сравнение между водорастворимыми материалами базового покрытия 1-3 и водорастворимым материалом базового покрытия II
Для определения предельного размера микроотверстий и количества микроотверстий многослойные красочные системы получали с помощью следующего общего способа:
На стальной лист с катодным электрохимическим покрытием размером 30×50 см наносили липкую ленту по одной продольной кромке, чтобы было возможно определить разницу толщины после покрытия. Водорастворимый материал базового покрытия наносили электростатически клинообразным образом. Полученный водорастворимый слой базового покрытия испаряли при комнатной температуре в течение одной минуты и потом высушивали в сушильной печи с циркуляцией воздуха при 70°C в течение 10 минут. Общепринятый двухкомпонентный материал лакового покрытия наносили на слой высушенного водорастворимого базового покрытия. Полученный слой лакового покрытия испаряли при комнатной температуре в течение 20 минут. После этого слой водорастворимого базового покрытия и слой лакового покрытия отверждали в сушильной печи с циркуляцией воздуха при 140°C в течение 20 минут. После визуальной оценки микроотверстий в полученной многослойной красочной системе, нанесенной клинообразно, определяли толщину покрытия предельного размера микроотверстия. Результаты могут быть найдены в Таблице 1.

Результаты подтверждают, что применение комбинации связующих агентов в соответствии с предлагаемым изобретением заметно повышает предельный размер микроотверстий по сравнению с водорастворимыми материалами базовых покрытий 1-3 при одновременном уменьшении количества микроотверстий.
Получение водорастворимого материала базового покрытия 4 (не в соответствии с изобретением), который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Компоненты, перечисленные как "водная фаза" в Таблице В, смешивали вместе в указанном порядке для образования водной смеси. На следующем этапе органическую смесь получали из компонентов, перечисленных как "органическая фаза". Органическую смесь прибавляли к водной смеси. Потом суммарную смесь перемешивали в течение 10 минут и доводили при использовании деионизированной воды и диметилэтаноламина до значения pH, равного 8, и до вязкости готового к нанесению материала, равной 58 мПа при сдвигающей нагрузке 1000 с-1, как измерено с помощью ротационного вискозиметра (прибор Rheomat RM 180 от Mettle-Toledo) при 23°C.

Получение белой пасты:
Белую пасту получали из 43 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 50 частей по весу рутила титана 2310, 3 частей по весу 1-пропокси-2-пропанола и 4 частей по весу деионизированной воды.
Получение пасты газовой сажи:
Пасту газовой сажи получали из 25 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 10 частей по весу газовой сажи, 0,1 части по весу метил изобутилкетона, 1,36 частей по весу диметилэтаноламина (10% в деминерализованной воде), 2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 61,45 частей по весу деионизированной воды.
Получение водорастворимого материала базового покрытия 5 (не в соответствии с изобретением), который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 5 получали аналогично Таблице С за исключением того, что использовался привитой сополимер (GCP1) вместо привитого сополимера (CP).
Получение водорастворимого материала базового покрытия 6 (не в соответствии с изобретением), который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 6 получали аналогично Таблице С за исключением того, что использовался привитой сополимер (GCP1) вместо привитого сополимера (CP) и продукт реакции (R) вместо полиэстера Р1.
Получение материала базового покрытия I2 в соответствии с изобретением, который может быть нанесен непосредственно на катодное электрохимическое покрытие в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия I2 получали аналогично Таблице С за исключением того, что использовался продукт реакции (R) вместо полиэстера (Р1).

Получение водорастворимого материала базового покрытия 7, которое может быть нанесено непосредственно на водорастворимые материалы базовых покрытий 4-6 и 12 в качестве придающего цвет покрытия

Получение голубой пасты:
Голубую пасту получали из 69,8 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 12,5 частей по весу Paliogen® Blue L 6482, 1,5 частей по весу диметилэтаноламина (10% в деминерализованной воде), 1,2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 15 частей по весу деионизированной воды.
Сравнение между водорастворимыми материалами базового покрытия 4-6 и водорастворимым материалом базового покрытия I2
Для того чтобы определить предельный размер микроотверстий и количество микроотверстий, многослойные красочные системы получали с помощью следующего общего способа:
На стальной лист с катодным электрохимическим покрытием размером 30×50 см наносили липкую ленту по одной продольной кромке, чтобы было возможно определить разницу толщины после покрытия. Водорастворимый материал базового покрытия наносили электростатически клинообразным образом. Полученный водорастворимый слой базового покрытия испаряли при комнатной температуре в течение 4 минут. После этого водорастворимый материал базового покрытия 7 наносили слоем покрытия с толщиной 12-14 мкм, испаряли при комнатной температуре в течение 4 минут и потом был отверждали в сушильной печи с циркуляцией воздуха при 70°C в течение 10 минут. Общепринятый двухкомпонентный материал лакового покрытия наносили на слой высушенного водорастворимого базового покрытия. Полученный слой лакового покрытия испаряли при комнатной температуре в течение 20 минут. После этого слой водорастворимого базового покрытия и слой лакового покрытия отверждали в сушильной печи с циркуляцией воздуха при 140°C в течение 20 минут. После визуальной оценки микроотверстий в полученной многослойной красочной системе, нанесенной клинообразно, определяли толщину покрытия предельного размера микроотверстия. Результаты могут быть найдены в Таблице 2.


Результаты показывают, что применение комбинации связующих агентов в соответствии с предлагаемым изобретением заметно повышает предельный размер микроотверстия по сравнению с водорастворимыми материалами базовых покрытий 4-6 при одновременном уменьшении количества микроотверстий и предотвращает появление микроотверстий вплоть до максимальной полученной толщины клина.
Получение водорастворимого материала базового покрытия 8, которое может быть нанесено непосредственно ниже водорастворимых материалов базовых покрытий 9-11 и I3 в качестве не придающего цвет покрытия
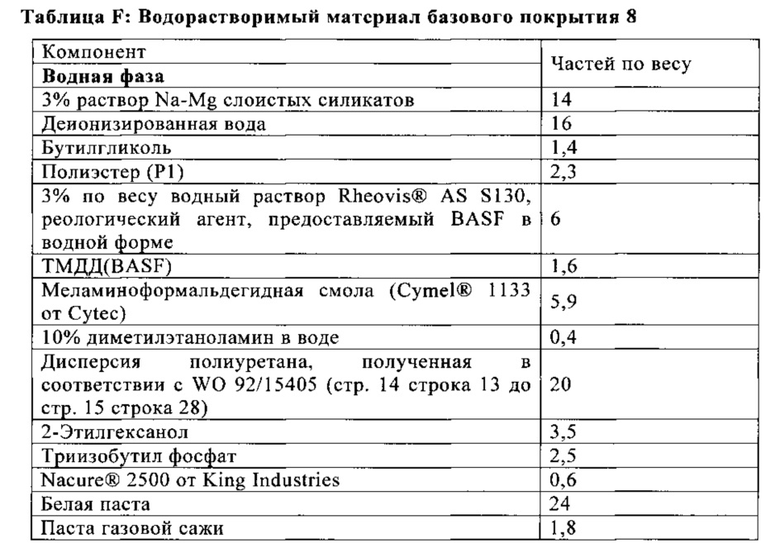
Получение пасты газовой сажи:
Пасту газовой сажи получали из 25 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 10 частей по весу газовой сажи, 0,1 части по весу метил изобутилкетона, 1,36 частей по весу диметилэтаноламина (10% в деминерализованной воде), 2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 61,45 частей по весу деионизированной воды.
Получение белой пасты:
Белую пасту получали из 43 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 50 частей по весу рутила титана 2310, 3 частей по весу 1-пропокси-2-пропанола и 4 частей по весу деионизированной воды.
Получение водорастворимого материала базового покрытия 9 (не в соответствии с изобретением), которое может быть нанесено непосредственно на водорастворимый материал базового покрытия 8 в качестве придающего цвет покрытия

Получение голубой пасты:
Голубую пасту получали из 69,8 частей по весу дисперсии полиуретана, модифицированного акриловой смолой, полученной в соответствии с международной заявкой WO 91/15528, дисперсии связующего агента А, 12,5 частей по весу Paliogen® Blue L 6482, 1,5 частей по весу диметилэтаноламина (10% в деминерализованной воде), 1,2 частей по весу коммерческого полиэфира (Pluriol® Р900 от BASF SE) и 15 частей по весу деионизированной воды.
Получение водорастворимого материала базового покрытия 10 (не в соответствии с изобретением), которое может быть нанесено непосредственно на водорастворимый материал базового покрытия 8 в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 10 получали аналогично Таблице G за исключением того, что использовали привитой сополимер (GCP1) вместо привитого сополимер (CP).
Получение водорастворимого материала базового покрытия 11 (не в соответствии с изобретением), которое может быть нанесено непосредственно на водорастворимый материал базового покрытия 8 в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия 11 получали аналогично Таблице G за исключением того, что использовался привитой сополимер (GCP1) вместо привитого сополимера (CP), и использовали продукт реакции (R) вместо полиэстера Р1.
Получение водорастворимого материала базового покрытия 13 (в соответствии с изобретением), которое может быть нанесено непосредственно на водорастворимый материал базового покрытия 8 в качестве придающего цвет покрытия
Водорастворимый материал базового покрытия I3 получали аналогично Таблице G за исключением того, что использовали продукт реакции (R) вместо полиэстера Р1.

Сравнение между водорастворимыми материалами базовых покрытий 9-11 и водорастворимым материалом базового покрытия I3
Для того чтобы определить предельный размер микроотверстий и количество микроотверстий, многослойные красочные системы получали с помощью следующего общего способа:
На стальной лист с катодным электрохимическим покрытием размером 30×50 см наносили липкую ленту по одной продольной кромке, чтобы было возможно определить разницу толщины после покрытия. Водорастворимый материал базового покрытия 8 наносили слоем покрытия с толщиной 16-18 мкм. После этого полученную конструкцию подвергали испарению при комнатной температуре в течение 4 минут.
Водорастворимые материалы базовых покрытий 9-11 и 13 наносили электростатически клинообразно, подвергали испарению при комнатной температуре в течение 4 минут и отверждали в сушильной печи с циркуляцией воздуха при 70°C в течение 10 минут. Общепринятый двухкомпонентный материал лакового покрытия наносили на высушенный слой водорастворимого базового покрытия. Полученный слой лакового покрытия испаряли при комнатной температуре в течение 20 минут. После этого слой водорастворимого базового покрытия и слой лакового покрытия отверждали в сушильной печи с циркуляцией воздуха при 140°C в течение 20 минут. После визуальной оценки микроотверстий в полученной многослойной красочной системе, нанесенной клинообразно, определяли толщину покрытия предельного размера микроотверстия. Результаты (предоставлены только для толщины слоя водорастворимых базовых покрытий 9-11 и 13, в которых микроотверстия могут быть обнаружены) могут быть найдены в Таблице 3.

Результаты показывают, что применение комбинации связующих агентов в соответствии с предлагаемым изобретением заметно повышает предельный размер микроотверстий по сравнению с водорастворимыми материалами базовых покрытий 9-11 при одновременном уменьшении количества микроотверстий и предотвращает появление микроотверстий вплоть до максимальной полученной толщины клина.
Краткое описание чертежей
Фигура 1:
Схематическое образование многослойной красочной системы (М) в соответствии с изобретением, которая размещается на металлическом субстрате (S) и включает отвержденное электрохимическое покрытие (Е.1) и базовое покрытие (В.2.1), а также лаковое покрытие (К), которые были отверждены совместно.
Фигура 2:
Схематическое образование многослойной красочной системы (М) в соответствии с изобретением, которая размещается на металлическом субстрате (S) и включает отвержденное электрохимическое покрытие (Е.1), два базовых покрытия (В.2.2.x), в частности, первое базовое покрытие (В.2.2.а) и самое верхнее базовое покрытие (B.2.2.Z), которое размещается на нем, а также лаковое покрытие (K), которые были отверждены совместно.
Фигура 3:
Схематическое образование многослойной красочной системы (М) в соответствии с изобретением, которая размещается на металлическом субстрате (S) и включает отвержденное электрохимическое покрытие (Е.1), три базовых покрытия (В.2.2.x), в частности, первое базовое покрытие (В.2.2.а), базовое покрытие (B.2.2.b), которое размещается над ним, и самое верхнее базовое покрытие (B.2.2.z), а также лаковое покрытие (K), которые были отверждены совместно.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2675915C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2665510C1 |
| СОСТАВ ДЛЯ ПОКРЫТИЯ НА ВОДНОЙ ОСНОВЕ И ИЗГОТОВЛЕНИЕ МНОГОСЛОЙНЫХ КРАСОЧНЫХ СИСТЕМ С ПРИМЕНЕНИЕМ УКАЗАННОГО СОСТАВА ДЛЯ ПОКРЫТИЯ | 2014 |
|
RU2677334C1 |
| ВОДНАЯ ДИСПЕРСИЯ СОПОЛИМЕРА | 2014 |
|
RU2666537C2 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2016 |
|
RU2700867C1 |
| ВОДНЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ ПОЛИМЕРЫ, ПОЛУЧЕННЫЕ МНОГОСТАДИЙНЫМ СПОСОБОМ, И КОМПОЗИЦИИ МАТЕРИАЛА ПОКРЫТИЯ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2743013C2 |
| АГЕНТ ДЛЯ ПИГМЕНТИРОВАННОГО ПОКРЫТИЯ И ПОЛУЧЕННЫЕ ИЗ НЕГО ПОКРЫТИЯ | 2015 |
|
RU2677477C2 |
| АГЕНТ ДЛЯ ПИГМЕНТИРОВАННОГО ПОКРЫТИЯ И ПОЛУЧЕННЫЕ ИЗ НЕГО ПОКРЫТИЯ | 2015 |
|
RU2676633C1 |
| СОПОЛИМЕР И ПИГМЕНТИРОВАННЫЙ АГЕНТ ДЛЯ ПОКРЫТИЯ, СОДЕРЖАЩИЙ ТАКОЙ ПОЛИМЕР | 2015 |
|
RU2676084C1 |
| КАРБОКСИФУНКЦИОНАЛЬНЫЕ ПРОДУКТЫ РЕАКЦИИ НА ОСНОВЕ ПРОСТОГО ПОЛИЭФИРА И ВОДНЫЕ ГРУНТОВОЧНЫЕ МАТЕРИАЛЫ, ВКЛЮЧАЮЩИЕ ПРОДУКТЫ РЕАКЦИИ | 2016 |
|
RU2708390C1 |
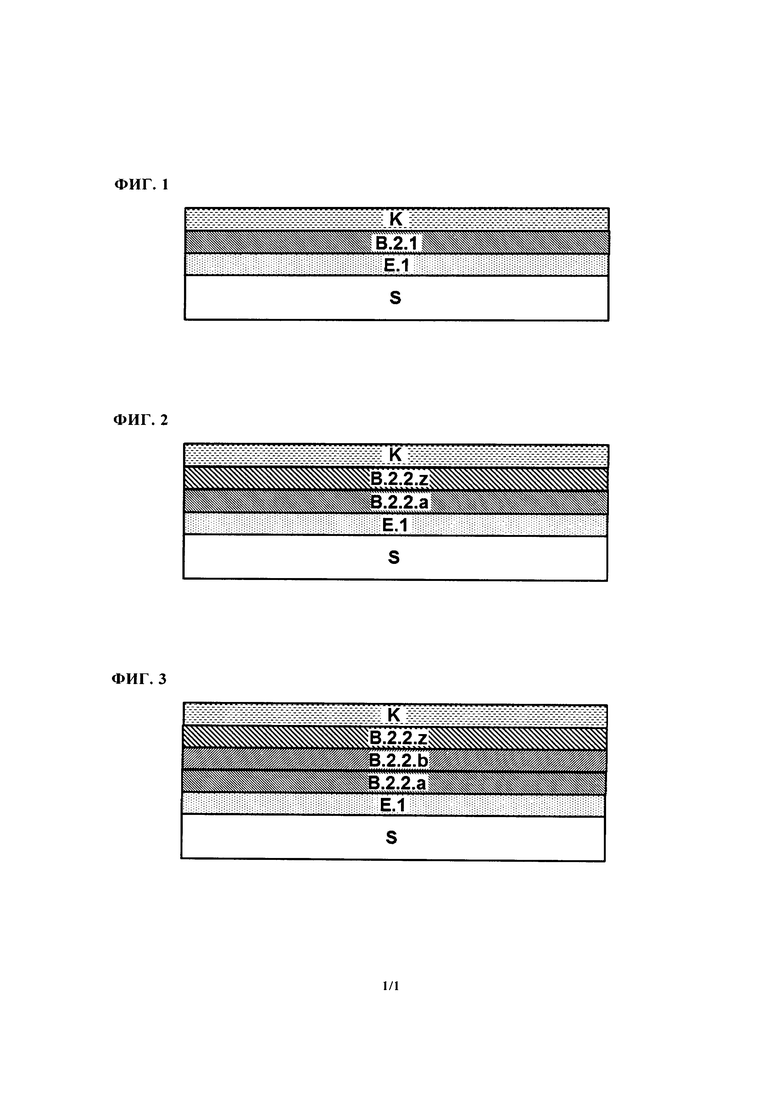
Изобретение относится к способу получения многослойной красочной системы на металлической основе, в которой базовое покрытие или непосредственно последовательные слои базового покрытия получают непосредственно на металлической основе, покрытой отвержденным электрохимическим покрытием, лаковое покрытие получают непосредственно на одном базовом покрытии или на самом верхнем из множества базовых покрытий, а потом одно или более базовых покрытий и лаковое покрытие совместно отверждают, при этом по крайней мере один материал базового покрытия, который используется для получения базовых покрытий, включает по крайней мере одну водную дисперсию, содержащую по крайней мере один сополимер (CP), где указанный сополимер (CP) получают путем первоначальной загрузки водной дисперсия по крайней мере одного полиуретана, и последующей полимеризации смеси ненасыщенных олефиновых мономеров в присутствии полиуретана из (i), где используют растворимый в воде активатор, ненасыщенные олефиновые мономеры дозируют таким образом, что концентрация 6,0% по весу из расчета на общее количество ненасыщенных олефиновых мономеров, которые используются для полимеризации, в реакционном растворе не превышается в течение всего времени реакции, и смесь ненасыщенных олефиновых мономеров включает по крайней мере один ненасыщенный полиолефиновый мономер, а также включает по крайней мере один линейный гидрокси-функциональный продукт реакции (R), который имеет кислотное число менее чем 20 мг KOH/г, получение которого предусматривает применение по крайней мере одного соединения (v), содержащего две функциональные группы (v.1), и алифатического или аралифатического радикала гидрокарбила (v.2), который размещается между функциональными группами и содержит от 12 до 70 атомов углерода. Техническим результатом изобретения является повышение качества покрытий. 2 н. и 16 з.п. ф-лы, 3 ил., 11 табл.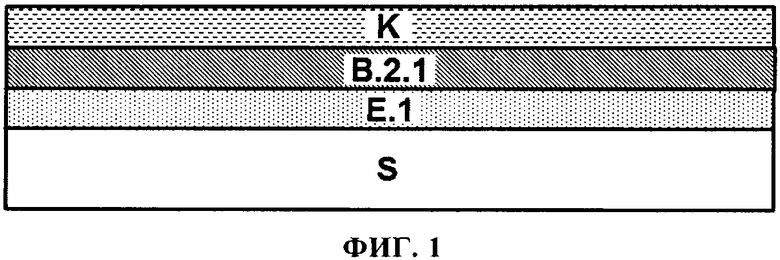
1. Способ получения многослойной красочной системы (М) на металлическом субстрате (S), включающий:
(1) получение отвержденного электрохимического покрытия (Е.1) на металлической основе (S) путем электрофоретического нанесения электрохимического покрытия (е.1) на основу (S) и последующее отверждение электрохимического покрытия (е.1),
(2) получение (2.1) базового покрытия (В.2.1) или (2.2) непосредственно последовательных базовых слоев (В.2.2.x) непосредственно на отвержденном электрохимическом покрытии (Е.1) путем (2.1) нанесения водного материала базового покрытия (b.2.1) непосредственно на электрохимическое покрытие (Е.1) или (2.2) нанесения множества материалов базового покрытия (b.2.2.х) в непосредственной последовательности на электрохимическое покрытие (Е.1),
(3) получение лакового покрытия (К) непосредственно на (3.1) базовом покрытии (В.2.1) или (3.2) на самом верхнем базовом покрытии (В.2.2.x) путем нанесения материала лакового покрытия (k) непосредственно на (3.1) базовое покрытие (В.2.1) или (3.2) самое верхнее базовое покрытие (В.2.2.x),
(4) совместное отверждение (4.1) базового покрытия (В.2.1) и лакового покрытия (К) или (4.2) базовых покрытий (В.2.2.x) и лакового покрытия (К),
где
материал базового покрытия (b.2.1) или по крайней мере один из материалов базового покрытия (b.2.2.х) включает
по крайней мере одну водную дисперсию, содержащую по крайней мере один сополимер (CP), где указанный сополимер (CP) получают путем
(i) первоначальной загрузки водной дисперсии по крайней мере одного полиуретана, последующей
(ii) полимеризации смеси ненасыщенных олефиновых мономеров в присутствии полиуретана из этапа (i),
где
(a) используют растворимый в воде активатор,
(b) ненасыщенные олефиновые мономеры дозируют таким образом, что концентрация 6,0% по весу из расчета на общее количество ненасыщенных олефиновых мономеров, которые используются для полимеризации, в реакционном растворе не превышается в течение всего времени реакции, и
(с) смесь ненасыщенных олефиновых мономеров включает по крайней мере один ненасыщенный полиолефиновый мономер,
и включает
по крайней мере один линейный продукт реакции с гидроксильной функциональной группой (R), имеющий кислотное число менее чем 20 мг КОН/г, получение которого вовлекает применение по крайней мере одного соединения (v), содержащего две функциональные группы (v.1) и алифатический или аралифатический радикал гидрокарбила (v.2), который размещается между функциональными группами и имеет от 12 до 70 атомов углерода.
2. Способ в соответствии с п. 1, где смесь ненасыщенных олефиновых мономеров, которые используются на этапе (ii), включает от 0,1 до 6,0 мол. % ненасыщенных полиолефиновых мономеров.
3. Способ в соответствии с п. 1 или 2, где смесь ненасыщенных олефиновых мономеров, которые используются на этапе (ii), включает аллил метакрилат и не содержит никакого дополнительного ненасыщенного полиолефинового мономера.
4. Способ в соответствии с п. 1 или 2, где ненасыщенные олефиновые мономеры на этапе (ii) (b) дозируют таким образом, что концентрация 4,0% по весу из расчета на общее количество ненасыщенных олефиновых мономеров, которые используются для полимеризации, в реакционном растворе не превышается в течение всего времени реакции.
5. Способ в соответствии с п. 1 или 2, где функциональные группы (v.1) по крайней мере одного соединения (v) являются выбранными из группы, состоящей из групп гидроксила и групп карбоксила.
6. Способ в соответствии с п. 1 или 2, где димерные жирные кислоты и/или димерные диолы используются в качестве соединений (v) при получении продукта реакции (R).
7. Способ в соответствии с п. 1 или 2, где продукт реакции (R) получают путем реакции димерных жирных кислот с алифатическими, аралифатическими и/или ароматическими дигидрокси-функциональными соединениями, которые имеют среднечисловой молекулярный вес от 120 до 6000 г/моль.
8. Способ в соответствии с п. 7, где используемые алифатические, аралифатические и/или ароматические дигидрокси-функциональные соединения представляют собой полиэтердиолы, полиэстердиолы и/или димерные диолы.
9. Способ в соответствии с любым из пп. 1, 2 или 8, где по крайней мере один продукт реакции (R) является выбранным из группы, которая состоит из:
- продуктов реакции, которые получают путем реакции димерных жирных кислот по крайней мере с одним алифатическим дигидрокси-функциональным соединением общей структурной формулы (I):

где R представляет собой С3-С6 алкиленовый радикал, а n выбирают таким образом, что соединение формулы (I) имеет среднечисловую молекулярную массу от 120 до 6000 г/моль, димерные жирные кислоты и соединения формулы (I) используют в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловую молекулярную массу от 600 до 40000 г/моль и кислотное число менее чем 10 мг КОН/г,
- продуктов реакции, которые получают путем реакции димерных жирных кислот по крайней мере с одним дигидрокси-функциональным соединением общей структурной формулы (II):

где
R представляет собой бивалентный органический радикал, включающий от 2 до 10 атомов углерода,
R1 и R2 каждый независимо представляет собой линейные или разветвленные радикалы алкилена, содержащие от 2 до 10 атомов углерода,
X и Y каждый независимо представляет собой О, S или NR3, где R3 представляет собой водород или алкильный радикал, содержащий от 1 до 6 атомов углерода, и
m и n соответствующим образом являются выбранными таким образом, что соединение формулы (II) имеет среднечисловой молекулярный вес от 450 до 2200 г/моль,
где компоненты (а) и (b) используются в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловой молекулярный вес от 1200 до 5000 г/моль и кислотное число менее чем 10 мг КОН/г,
- продуктов реакции, которые получают путем реакции димерных жирных кислот с димерными диолами, где димерные жирные кислоты и димерные диолы используются в молярном соотношении от 0,7/2,3 до 1,6/1,7, а полученный продукт реакции имеет среднечисловой молекулярный вес от 1200 до 5000 г/моль и кислотное число менее чем 10 мг КОН/г.
10. Способ в соответствии с любым из пп. 1, 2 или 8, где материал базового покрытия (b.2.1) или по крайней мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), дополнительно включает(ют) по крайней мере один гидрокси-функциональный полимер, отличный от (CP) и (R), в качестве связующего агента, выбранный из группы, которая состоит из полиуретанов, полиэстеров, полиакрилатов и сополимеров этих полимеров, и меламиновой смолы в качестве поперечно сшивающего агента.
11. Способ в соответствии с любым из пп.в 1, 2 или 8, где материал базового покрытия (b.2.1) или по крайней мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), включает(ют) по крайней мере один цветной пигмент и/или пигмент эффекта.
12. Способ в соответствии с любым из пп. 1, 2 или 8, где материал базового покрытия (b.2.1) или по крайней мере один из материалов базового покрытия (b.2.2.х) включает пигмент эффекта на основе металла, предпочтительно пластинчатый пигмент на основе алюминия.
13. Способ в соответствии с любым из пп. 1, 2 или 8, где материал базового покрытия (b.2.1) или по крайней мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), представляет(ют) собой однокомпонентную композиции покрытия.
14. Способ в соответствии с любым из пп. 1, 2 или 8, где совместное отверждение (4) осуществляют при температурах от 100 до 250°C в течение периода времени от 5 до 60 мин.
15. Способ в соответствии с любым из пп. 1, 2 или 8, где получают (2.2) два базовых покрытия (В.2.2.а) и (B.2.2.z), для которых используемые материалы водного базового покрытия (b.2.2.а) и (b.2.2.z) являются идентичными и включают пигменты эффекта.
16. Способ в соответствии с п. 15, где материал базового покрытия (b.2.2.а) наносится путем электростатического распыления, а материал базового покрытия (b.2.2.z) наносится путем пневматического нанесения.
17. Способ в соответствии с любым из пп. 1, 2 или 8, где (2.2) получают по крайней мере два базовых покрытия, первое базовое покрытие (В.2.2.а) непосредственно над электрохимическим покрытием (Е.1), включающее белые пигменты и черные пигменты, и дополнительные базовые покрытия (В.2.2.x), включающие пигменты эффекта.
18. Многослойная красочная система (М), которая получена в соответствии со способом в соответствии с любым из пп. 1-17.
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Роликовый конвейер | 1979 |
|
SU839885A1 |
| ВОДНАЯ КОМПОЗИЦИЯ ПОКРЫТИЯ, СОДЕРЖАЩАЯ ПОЛИМЕР, ПОЛУЧЕННЫЙ СТУПЕНЧАТОЙ ПОЛИМЕРИЗАЦИЕЙ, И ПОЛИУРЕТАН | 2000 |
|
RU2254351C2 |
| ВОДНОЕ ДВУХКОМПОНЕНТНОЕ ПОЛИУРЕТАНОВОЕ СРЕДСТВО ДЛЯ ПОКРЫТИЙ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В СПОСОБЕ ПОЛУЧЕНИЯ МНОГОСЛОЙНОГО ЛАКОКРАСОЧНОГО ПОКРЫТИЯ | 1994 |
|
RU2136713C1 |

