Область техники, к которой относится изобретение
Настоящее изобретение касается производного алкинилиндазола, которое оказывает ингибирующий эффект на VEGF рецепторную тирозинкиназу, его фармацевтически приемлемой соли и их применения.
Уровень техники
Несмотря на то, что ангиогенез (васкулогенез) клеток и тканей играет значимую роль в процессе развития, заживления ран и т.д., известно, что патологический ангиогенез связан с различными заболеваниями или состояниями, такими как: заболевания сетчатки, такие как возрастная макулярная дегенерация и диабетическая ретинопатия; образование, пролиферация или метастазы опухоли; хроническое воспаление и ревматоидный артрит.
В качестве участвующего в ангиогенезе рецептора известен VEGF рецептор, такой как рецептор 2 фактора пролиферации эндотелия сосудов (также обозначаемого как фактор роста эндотелия сосудов, и далее по тексту именуемого "VEGF"). VEGF рецептор представляет собой вид тиразинкиназного рецептора, и когда VEGF как лиганд связывается с VEGF рецептором, то тиразинкиназный рецептор активируется для передачи сигналов в клетки. В итоге, например, повышается проницаемость сосудов, а также усиливается пролиферация и миграция клеток эндотелия сосудов, что вызывает ангиогенез.
VEGF-2 рецептор участвует не только в ангиогенезе в здоровом организме, но также и в патологическом ангиогенезе, вызванном вышеуказанными заболеваниями или состояниями. Так, например, посредством подавления активности тиразинкиназы VEGF-2 рецептора можно подавлять вызванный киназой ангиогенез, что эффективно при лечении заболеваний или состояний, которые сопровождаются ангиогенезом. Поэтому в целях профилактики или лечения заболеваний или состояний, сопровождающих ангиогенез, были разработаны различные ингибиторы тирозинкиназы VEGF-2 рецептора.
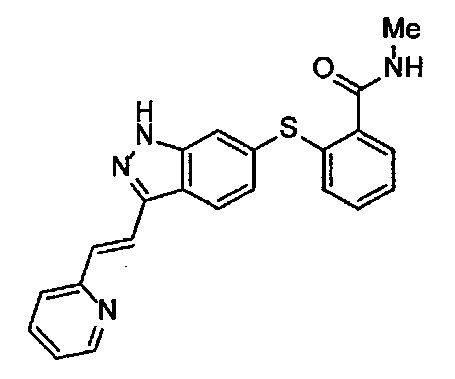
В Патентном документе 1 описаны индазольные соединения, которые ингибируют протеинкиназную активность VEGF рецептора. Например, в Примере 33(а) в Патентном документе 1 продемонстрирован (6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этенил]индазол) (непатентованное наименование: акситиниб), который имеет следующую формулу.
Формула 1
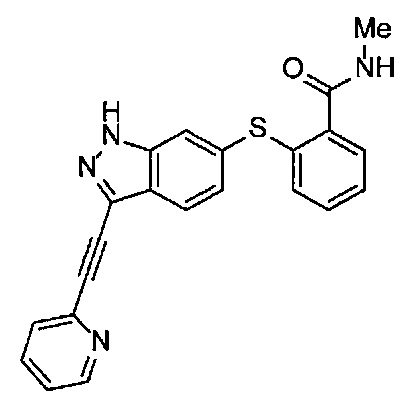
В Патентном документе 2 описаны индазольные соединения, которые могут применяться в качестве модулятора и/или ингибитора протеинкиназы, и способ получения их промежуточного соединения. Например, в Примере 20 продемонстрирован (6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этинил]индазол), который имеет следующую формулу:
Формула 2
В Патентном документе 3 описаны индазольные соединения, которые модулируют или ингибируют активность VEGF-2 рецептора. В Непатентном документе 1 описаны противораковые средства, содержащие ингибитор киназы, стимулируемой VEGF рецептором, например, пазопаниб, акситиниб, сорафениб, сунитиниб.
Также в качестве лекарственных средств для лечения возрастной макулярной дегенерации, которая представляет собой заболевание заднего отрезка глаза, применяют LUCENTIS (зарегистрированная торговая марка), MACUGEN (зарегистрированная торговая марка) и EYLEA (зарегистрированная торговая марка), представляющие собой VEGF ингибиторы. Несмотря на то, что данные лекарственные средства содержат высокомолекулярные соединения, которые специфично связываются с VEGF (антитело или аптамер VEGF), их необходимо вводить в стекловидное тело. Таким образом, например, в офтальмологии, желательна разработка ингибиторов ангиогенеза, которые можно вводить менее инвазивными способами. Однако, ингибитор ангиогенеза, который можно закапывать в глаза, еще не представлен на рынке.
Патентные документы
Патентный документ 1 WO 2001/002369
Патентный документ 2 WO 2006/048745
Патентный документ 3 WO 2004/056806
Непатентные документы
Непатентный документ 1 Current Pharmaceutical Design, 2012, 18, 2921-2935
Краткое описание изобретения
Проблемы, на решение которых направлено настоящее изобретение
Основной целью настоящего изобретения является разработка нового соединения, которое оказывает ингибирующий эффект на VEGF рецепторную тирозинкиназу и может применяться в качестве лекарственного средства для лечения заболеваний, сопровождающих ангиогенез или эдему, например, возрастной макулярной дегенерации и тому подобных.
Кроме того, другой целью настоящего изобретения является разработка соединения, которое оказывает ингибирующий эффект на VEGF рецепторную тирозинкиназу, обладает высокой растворимостью в водном растворе и отличной устойчивостью.
В результате тщательного исследования, направленного на решение указанных проблем, авторы настоящего изобретения обнаружили, что алкинилиндазольное производное, представленное общей формулой (I), и его соли оказывают прекрасный ингибирующий эффект на VEGF рецепторную тирозинкиназу, и сформулировали настоящее изобретение. Алкинилиндазольное производное, представленное общей формулой (I), и его соль обладают высокой растворимостью в водном растворе и отличной фотоустойчивостью в указанном растворе.
Акситиниб, продаваемый в качестве перорального противоопухолевого средства, представляет собой отличный ингибитор VEGF рецептора, оказывающий ингибирующий эффект на VEGF рецепторную тирозинкиназу. Когда авторы настоящего изобретения изучали вопрос, может ли акситиниб применяться в форме жидкого препарата, такого как глазные капли, растворимость данного соединения в водном растворе была низкой, и можно предсказать, что перенос соединения в задний отрезок глаза будет недостаточным. Кроме того, было обнаружено, что акситиниб имеет низкую фотоустойчивость в водном растворе. Ввиду вышеуказанных фактов, затруднительно применять акситиниб в форме жидкого препарата, такого как глазные капли.
Алкинилиндазольное производное, представленное общей формулой (1), и его соль оказывают ингибирующий эффект на VEGF рецепторную тирозинкиназу, как описано выше, и обладают улучшенной растворимостью в водном растворе, кроме того они характеризуются отличной фотоустойчивостью в растворе и, таким образом, вполне могут применяться в форме жидкого препарата, такого как глазные капли.
Настоящее изобретение может включать следующее алкинилиндазольное производное или его фармацевтически приемлемую соль, и содержащее его лекарственное средство:
(1) Алкинилиндазольное производное, представленное следующей общей формулой (I):
Формула 3
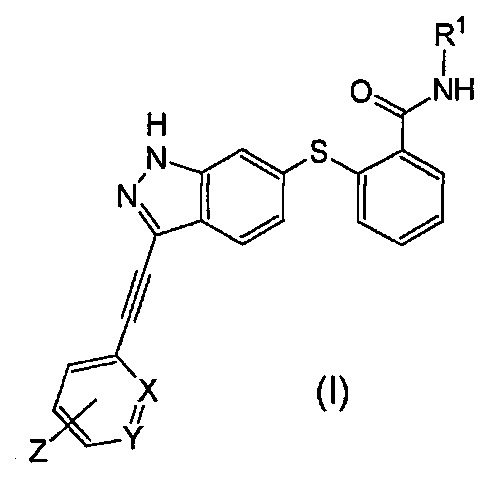
В данной формуле R1 представляет собой низший алкил. X и Y одинаковые или различаются, и каждый представляет собой CH или N, при условии, что X и Y одновременно не являются N. Z представляет собой группу, представленную следующей общей формулой (a):
Формула 4
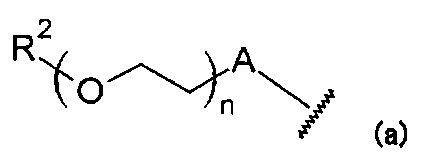
В данной формуле R2 представляет собой низший алкил, который может иметь заместитель, n представляет собой целое число от 1 до 7. A представляет собой подструктуру, представленную следующей Формулой:
Формула 5
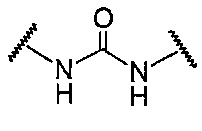 ,
, 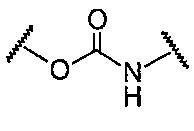 ,
, 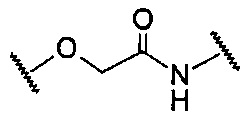 , или
, или 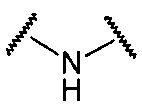 ,
,
или его фармацевтически приемлемая соль.
(2) Алкинилиндазольное производное по приведенному выше п. (1) или его фармацевтически приемлемая соль, где X и Y одновременно представляют собой CH.
(3) Алкинилиндазольное производное по приведенному выше п. (1) или (2), или его фармацевтически приемлемая соль, где Z присоединен в пара-положении.
(4) Алкинилиндазольное производное по любому из приведенных выше пп. (1)-(3) или его фармацевтически приемлемая соль, где A представляет собой подструктуру, представленную следующей Формулой:
Формула 6
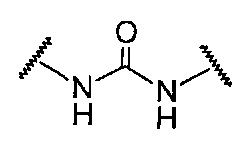
(5) Лекарственное средство, содержащее алкинилиндазольное производное по любому из приведенных выше пп. (1)-(4) или его фармацевтически приемлемую соль.
(6) Лекарственное средство по приведенному выше п. (5), которое представляет собой ингибитор тирозинкиназы рецептора фактора роста эндотелия сосудов (VEGF).
(7) Лекарственное средство по приведенному выше п. (5) или (6), которое применяют для профилактики или лечения заболевания сетчатки, сопровождающего ангиогенез или эдему.
(8) Лекарственное средство по приведенному выше п. (7), где заболевание сетчатки, сопровождающее ангиогенез или эдему, представляет собой возрастную макулярную дегенерацию, макулярный отек, диабетическую ретинопатию, ретинопатию недоношенных, окклюзию вены сетчатки, вторичную катаракту, миопическую хориоидальную неоваскуляризацию или глаукому.
Эффект настоящего изобретения
По настоящему изобретению, можно получить алкинилиндазольное производное, представленное общей Формулой (I), его фармакологически приемлемую соль и лекарственное средство, содержащее указанное алкинилиндазольное производное или его фармакологически приемлемую соль. Так как указанное алкинилиндазольное производное и его фармакологически приемлемая соль по настоящему изобретению оказывают прекрасное подавляющее действие на VEGF рецепторную тирозинкиназу, они эффективны для профилактики или лечения заболеваний или состояний, в которых участвует VEGF рецепторная тирозинкиназа, например, заболеваний или состояний, сопровождающих ангиогенез или эдему. Кроме того, указанное алкинилиндазольное производное и его фармакологически приемлемая соль обладают не только высокой растворимостью в водном растворе, но также отличной проницаемостью в сетчатку или хориоид, что следует из приведенных ниже результатов Тестового примера 5. Также они обладают отличной устойчивостью, особенно фотоустойчивостью в растворе. Таким образом, их можно применять в форме жидких препаратов, например, глазных капель, инъекции и т.д.
Осуществление изобретения
Соединение по настоящему изобретению представляет собой алкинилиндазольное производное, представленное общей Формулой (I), или его фармакологически приемлемую соль. В настоящем тексте, алкинилиндазольное производное, представленное общей Формулой (I), также именуют "соединение (I) по настоящему изобретению".
В настоящем изобретении, фрагмент
Формула 7
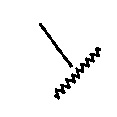
применяют в структурной Формуле для обозначения связи в точке, по которой заместитель или подструктура присоединяется к основному скелету молекулы или к другой подструктуре.
В общей Формуле (I) низшие алкилы, обозначенные R1, могут представлять собой, например, линейные, разветвленные или циклические алкилы, содержащие 1-4 атома углерода. Такие низшие алкилы могут представлять собой, например, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, циклопропил и циклобутил. R1 предпочтительно представляет собой линейный или разветвленный алкил, содержащий 1-3 атома углерода, более предпочтительно алкил, содержащий 1-2 атома углерода (метил или этил), и особенно предпочтительно метил.
X и Y одинаковые или различаются, и каждый представляет собой CH или N, при условии, что X и Y одновременно не являются N. Комбинации X и Y могут включать следующие: (i) X и Y представляют собой CH, (ii) X представляет собой N, и Y представляет собой CH, и (iii) X представляет собой CH, и Y представляет собой N. Предпочтительно, X и Y представляют собой CH.
Положение связывания в шестичленном цикле в группе, обозначенной Z, специальным образом не ограничено, и данное положение может представлять собой любое, выбранное из орто-положения, мета-положения или пара-положения. Следует заметить, что положение связывания представляет собой положение относительно этинильной группы. Предпочтительно, Z присоединен в мета-положении или в пара-положении, и более предпочтительно - в пара-положении.
Z представляет собой группу, имеющую общую Формулу (а). В Формуле (а), низший алкил, который может содержать заместитель, обозначенный R2, может представлять собой, например, линейные, разветвленные или циклические алкилы, содержащие 1-4 атома углерода. Такие низшие алкилы могут представлять собой такие же низшие алкилы, как вышеописанный R1. R2 предпочтительно представляет собой линейный или разветвленный алкил, содержащий 1-3 атома углерода, более предпочтительно, алкил, содержащий 1 или 2 атома углерода (метил или этил). Заместитель в низшем алкиле, который может содержать заместитель, обозначенный R2, может представлять собой, например, гидрокси-группу, амино-группу, диметиламино-группу, ацетиламино-группу и морфолино-группу. Предпочтительные низшие алкилы, которые могут содержать заместитель, обозначенный R2, могут включать, например, метил, 2-гидроксиэтил, 2-аминоэтил, 2-(диметиламино)этил, 2-ацетиламиноэтил и 2-(N-морфолино)этил, и среди перечисленных более предпочтителен метил.
A в общей Формуле (а) представляет собой подструктуру, представленную следующей Формулой.
Формула 8
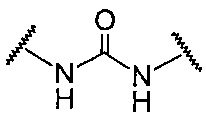 ,
, 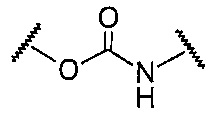 ,
,  , или
, или
Подструктуры, представленные каждой описанной выше Формулой, называют соответственно мочевина, карбамат, α-алкоксиамид и амин. В подструктурах, представленных каждой описанной выше Формулой, их левая часть связана с (поли)этиленгликолевым фрагментом (-(O-CH2CH2)n-, где n такой, как указано выше) в общей Формуле (а), а их правая часть связана с шестичленным циклом. Например, если А представляет собой карбамат или α-алкоксиамид, атомы кислорода и азота в приведенной выше Формуле связаны с (поли)этиленгликолевым фрагментом и шестичленным циклом, соответственно.
В общей Формуле (а), A предпочтительно представляет собой подструктуру (мочевину), представленную следующей Формулой:
Формула 9
В общей Формуле (а), n представляет собой целое число от 1 до 7, предпочтительно целое число от 1 до 5, более предпочтительно целое число от 2 до 5, еще более предпочтительно от 2 до 4, и особенно предпочтительно 3.
Как один из наиболее предпочтительных аспектов соединения (I) по настоящему изобретению, в качестве примера можно привести соединение, в котором в общей Формуле (I) R1 представляет собой метил, X и Y представляют собой CH, и в общей Формуле (a) R2 представляет собой метил, А представляет собой мочевину, n равно 3, и Z присоединен в пара-положении.
Фармакологически приемлемые соли соединения (I) по настоящему изобретению могут включать, например, фармакологически приемлемые кислотно-аддитивные соли, аммониевые соли и соли, образующиеся при добавлении аминокислоты. Можно выбирать и применять отдельно один вид соли, но также для применения можно комбинировать два или более вида солей.
Кислотно-аддитивные соли могут включать соли неорганических кислот или соли органических кислот. Соли неорганических кислот могут включать, например, неорганические соли, представляющие собой гидрохлорид, гидробромида, гидроиодид, нитрат, сульфат, фосфат. В качестве солей неорганических кислот предпочтительны гидрохлорид или гидробромид и т.д., и более предпочтителен гидрохлорид.
Соли органических кислот могут включать, например, органические соли, представляющие собой метансульфонат, бензолсульфонат, п-толуолсульфонат, формиат, ацетат, трифторацетат, оксалат, цитрат, малонат, фумарат, глутарат, адипат, аскорбат, малеат, тартрат, манделат, малат, пантотенат. В качестве солей органических кислот предпочтительны цитрат, фумарат или тартрат.
Аммониевые соли могут включать, например, метилпиридиниевую соль, ацетилпиридиниевую соль. Соли, образующиеся при добавлении аминокислот, могут включать, например, соли, образующиеся при добавлении лизина, глицина, аланина, фенилаланина, глютаминовой кислоты, аспарагиновой кислоты, аргинина.
В качестве фармацевтически приемлемых солей предпочтительны соли неорганических или соли органических кислот, и более предпочтительны соли неорганических кислот. Фармацевтически приемлемые соли могут включать сольваты, такие как гидраты.
Соединение (I) по настоящему изобретению или его фармацевтически приемлемая соль оказывают подавляющее действие на тирозинкиназу. Более конкретно, соединение (I) по настоящему изобретению или его фармацевтически приемлемая соль оказывают подавляющее действие на VEGF-2 рецепторную тирозинкиназу.
Соединение (I) по настоящему изобретению или его фармацевтически приемлемую соль предпочтительно применяют в качестве действующего вещества ингибитора VEGF рецепторной тирозинкиназы. В частности, данное соединение предпочтительно применяют в качестве действующего вещества ингибитора VEGF-2 рецепторной тирозинкиназы.
Поскольку ингибитор VEGF-2 рецепторной тирозинкиназы способен подавлять ангиогенез, его применяют в лечении различных заболеваний или состояний, сопровождающих ангиогенез или эдему. Заболевания или состояния, против которых применяют соединение (I) по настоящему изобретению и его фармакологически приемлемую соль, могут включать, например, заболевания и состояния, для лечения которых эффективно ингибирование VEGF-2 рецепторной тирозинкиназы, и, например, их подходящими примерами являются заболевания, состояния и тому подобные, сопровождающие ангиогенез или эдему.
Заболевания или состояния, сопровождающие ангиогенез или эдему, в частности могут включать, например, рак (опухоль) (например, рак желудка, рак почек, рак толстой кишки, рак легких и т.д.), заболевания сетчатки, сопровождающие ангиогенез или эдему, кератоконъюнктивные заболевания, сопровождающие ангиогенез или эдему (кератоконъюнктивит, нарушение вследствие ношения контактных линз и т.д.), хроническое воспаление, ревматоидный артрит, воспалительные заболевания кожи, псориаз, атеросклероз и инфаркт миокарда. Соединение (I) по настоящему изобретению и его фармакологически приемлемая соль могут применяться в качестве действующих веществ лекарственного средства, применяемого для профилактики или лечения указанных заболеваний или состояний. Среди перечисленных, соединение (I) по настоящему изобретению и его фармакологически приемлемую соль можно применять для профилактики или лечения заболеваний сетчатки, сопровождающих ангиогенез или эдему. Заболевания сетчатки, сопровождающие ангиогенез или эдему, могут включать, например, возрастную макулярную дегенерацию, макулярный отек, диабетическую ретинопатию, ретинопатию недоношенных, окклюзию вены сетчатки, вторичную катаракту, миопическую хориоидальную неоваскуляризацию и глаукому.
Следует обратить внимание, что термин "профилактика" означает замедление или предотвращение наступления состояний или заболеваний и сопровождающих их симптомов, или снижение риска возникновения состояния или заболевания. Кроме того, термин "лечение" означает ослабление или избавление от заболеваний или состояний и/или сопровождающих их симптомов.
Настоящее изобретение также охватывает лекарственное средство, содержащее соединение (I) по настоящему изобретению или его фармакологически приемлемую соль.
Соединение (I) по настоящему изобретению и его фармакологически приемлемую соль можно применять как таковые или в форме различных препаратов, в соответствии с целью введения. Лекарственное средство по настоящему изобретению обычно выпускают в виде фармацевтической композиции, содержащей соединение (I) по настоящему изобретению или его фармакологически приемлемую соль и фармацевтически приемлемый носитель.
Лекарственное средство по настоящему изобретению можно предпочтительно применять в качестве ингибитора VEGF рецепторной тирозинкиназы, и, в частности, его можно предпочтительно применять в качестве ингибитора VEGF-2 рецепторной тирозинкиназы. Кроме того, лекарственное средство по настоящему изобретению предпочтительно применяют для профилактики или лечения различных заболеваний или состояний, сопровождающих ангиогенез или эдему.
В качестве субъекта, которому необходимо ввести дозированное количество лекарственного средства по настоящему изобретению, подходит пациент, страдающий от заболевания или состояния, сопровождающих ангиогенез или эдему. В частности, лекарственное средство подходит для введения пациенту с заболеванием сетчатки, сопровождающим ангиогенез или эдему. Кроме того, в целях профилактики развития заболевания или состояния, соединение (I) по настоящему изобретению или его фармакологически приемлемую соль также можно вводить млекопитающему, у которого могут развиться вышеуказанные заболевания или состояния.
Настоящее изобретение также включает способ подавления VEGF рецепторной тирозинкиназы, в котором соединение (I) по настоящему изобретению или его фармакологически приемлемую соль вводят млекопитающему. Настоящее изобретение также охватывает способ профилактики или лечения заболеваний или состояний, сопровождающих ангиогенез или эдему (предпочтительно, заболеваний сетчатки, сопровождающих ангиогенез или эдему), в котором соединение (I) по настоящему изобретению или его фармакологически приемлемую соль вводят млекопитающему.
Лекарственное средство по настоящему изобретению можно вводить перорально или парентерально людям или млекопитающим, отличным от человека. Млекопитающие, отличные от человека, могут включать, например, мышей, крыс, хомяков, морских свинок, кроликов, кошек, собак, свиней, крупный рогатый скот, лошадей, овец и обезьян.
При применении лекарственного средства по настоящему изобретению в целях профилактики или лечения заболеваний или состояний, сопровождающих ангиогенез или эдему, лекарственное средство можно вводить системно или местно, перорально или парентерально. Что касается способа введения, предпочтительно выбирают способ, наиболее эффективный для лечения. В случае системного введения, помимо перорального введения применяют парентеральное введение, такое как внутривенная инъекция, подкожная инъекция и внутримышечная инъекция. В случае местного введения, препарат вводят, например, в кожу, слизистую оболочку, легкие, бронхи, носовую полость, слизистую оболочку носа, оболочку глаза или в глаза. Препараты для перорального введения могут включать, например, порошки, гранулы, таблетки, капсулы, сиропы и жидкие препараты. Препараты для парентерального введения могут включать, например, инъекции, мази, гели, кремы, припарки, пластыри, лечебные мази, свечи, аэрозоли, ингаляторы, спреи, глазные капли (офтальмологические растворы) и капли для носа. Например, когда лекарственное средство по настоящему изобретению применяют для лечения заболевания глаз, такого как заболевание сетчатки, предпочтительно парентеральное введение, и, в частности, предпочтительно введение в форме глазных капель.
Соединение (I) по настоящему изобретению и его фармакологически приемлемая соль обладают высокой растворимостью в водных растворах. Кроме того, соединение (I) по настоящему изобретению и его фармакологически приемлемая соль обладают отличной устойчивостью в растворах, в частности, отличаются отличной фотоустойчивостью. Соответственно, соединение (I) по настоящему изобретению и его фармакологически приемлемую соль можно применять для приготовления препарата, содержащего водный раствор, предпочтительно жидкого препарата, содержащего водный раствор в качестве основы. Соединение по настоящему изобретению или его фармакологически приемлемую соль можно применять, например, для приготовления указанных выше препаратов, таких как, в частности, сиропы, инъекции, глазные капли, капли для носа и т.д., и среди указанных особенно предпочтительными являются глазные капли.
Лекарственное средство по настоящему изобретению можно получать согласно способу, известному per se в области фармацевтических препаратов, в котором соединение (I) по настоящему изобретению или его фармакологически приемлемую соль обычно смешивают по меньшей мере с одним фармакологически приемлемым носителем и т.п.. Носитель можно выбирать экспертно, в зависимости от формы препарата, предпочтительного для введения. Содержание соединения (I) по настоящему изобретению или его фармакологически приемлемой соли в лекарственном средстве варьируется в зависимости от лекарственной формы, дозировки и т.п., и может выбираться экспертно. Например, содержание обычно может составлять от 0.01 до 99.9 масс. %, предпочтительно от 0.1 до 80 масс. % от общего количества лекарственного средства.
В качестве фармакологически приемлемого носителя можно применять различные органические или неорганические вещества, которые традиционно используют в составе препаратов, и они могут включать, например: наполнители, разрыхлители, связующие средства, пластификаторы, смазывающие средства и т.д. в твердых препаратах; растворители, солюбилизаторы, суспендирующие средства, стабилизаторы, изотонирующие средства, буферные средства, загустители, регуляторы pH, успокоительные и т.д. в жидких препаратах. Кроме того, при необходимости можно применять добавки, такие как консерванты, антиоксиданты, красители и подсластители. Твердый препарат может иметь оболочку из покрывающего средства. Можно применять только один вид или комбинацию двух или более видов носителей и добавок.
Наполнители могут включать, например, лактозу, сахарозу, D-маннит, D-сорбит, крахмал, декстрин, микрокристаллическую целлюлозу, кристаллическую целлюлозу, кармеллозу, кальциевую соль кармеллозы, карбокси-метил-крахмал натрия, малозамещенную гидроксипропилцеллюлозу, смолу акации.
Разрыхлители могут включать, например, кармеллозу, кальциевую соль кармеллозы, натриевую соль кармеллозы, карбокси-метил-крахмал натрия, кроскармеллозу натрия, кросповидон, малозамещенную гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, кристаллическую целлюлозу.
Связующие средства могут включать, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон, кристаллическую целлюлозу, сахарозу, декстрин, крахмал, желатин, кроскармеллозу натрия, смолу акации.
Пластификаторы могут включать, например, легкую безводную кремниевую кислоту, стеарат магния.
Смазывающие средства могут включать, например, стеарат магния, стеарат кальция, тальк.
Покрывающие средства могут включать, например, желатин, сахарозу.
Растворители могут включать, например, очищенную воду, дистиллированную воду для инъекций, физиологический раствор, этанол, пропиленгликоль, макрогол, сезамовое масло, кукурузное масло, оливковое масло.
Солюбилизаторы могут включать, например, пропиленгликоль, D-маннит, бензилбензоат, этанол, триэтаноламин, карбонат натрия, цитрат натрия, полисорбат 80.
Суспендирующие средства могут включать, например, бензалконий хлорид, кармеллозу, гидроксипропилцеллюлозу, пропиленгликоль, повидон, метилцеллюлозу, глицерин моностеарат.
Стабилизаторы могут включать, например, эдетат натрия, гидросульфит натрия, тиосульфат натрия, цитрат натрия, аскорбиновую кислоту, дибутилгидрокситолуол.
Изотонирующие средства могут включать, например, хлорид натрия, хлорид калия, глицерин, маннит, сорбит, борную кислоту, тетраборат натрия, глюкозу, пропиленгликоль.
Буферные средства могут включать, например, гидрофосфат натрия, ацетат натрия, карбонат натрия, цитрат натрия, борную кислоту, тетраборат натрия.
Загустители могут включать, например, гидроксилэтилцеллюлозу, гидроксипропилцеллюлозу, поливиниловый спирт, полиэтиленгликоль.
Регуляторы pH могут включать, например, соляную кислоту, лимонную кислоту, гидроксид натрия, фосфорную кислоту, уксусную кислоту, борную кислоту.
Успокоительные средства могут включать, например, бензиловый спирт.
Консерванты могут включать, например, бензалконий хлорид, метил парагидроксибензоат, этил парагидроксибензоат, пропил парагидроксибензоат, хлорбутанол, бензиловый спирт, дегидроацетат натрия, сорбиновую кислоту.
Антиоксиданты могут включать, например, сульфит натрия, аскорбиновую кислоту.
Красители могут включать, например, пищевой краситель (например, Пищевой Красный №2 или №3), бета-каротин.
Подсластители могут включать, например, сахарин натрия, глицирризинат дикалия, аспартам.
Лекарственное средство по настоящему изобретению может содержать одно или несколько любых других лекарственных средств, если это не ослабляет эффект настоящего изобретения.
Дозировка соединения (I) по настоящему изобретению или его фармакологически приемлемой соли варьируется в зависимости от заболеваний или состояний и субъектов, которые необходимо вылечить, и способов лечения, но, например, при введении взрослым дозировка обычно составляет от 1 нг до 1000 мг, предпочтительно от 1 до 200 мг при пероральном введении. Данную дозировку обычно вводят за один-четыре приема в сутки. В случае парентерального введения дозировка обычно составляет, например, от 1 нг до 1000 мг, предпочтительно от 1 до 200 мг. Данную дозировку обычно вводят за один-четыре приема в день. Кроме того, например, в случае местного введения в глаза, предпочтительно в глаза закапывают одну глазную каплю, обычно содержащую от 0.001 до 10 вес/об. %, предпочтительно от 0.01 до 1 вес./об. % соединения (I) по настоящему изобретению или его фармакологически приемлемой соли, в количестве от 5 до 100 мкл, предпочтительно от 30 до 60 мкл в одной дозе, около 1-6 раз в день.
Далее будет описан способ получения соединения (I) по настоящему изобретению. Описанный далее способ получения является примером способа получения соединения (I) по настоящему изобретению, и способ получения соединения по настоящему изобретению им не ограничивается.
Даже если описанный далее способ получения не содержит конкретное описание, получение можно эффективно осуществлять посредством: введения защитной группы в функциональную группу при необходимости и снятия защиты на последующей стадии; использования функциональной группы в качестве предшественника на каждой стадии и превращения ее в целевую функциональную группу на подходящей стадии; изменения последовательности каждого способа и стадий; и т.д.
Кроме того, на каждой стадии или в каждой реакции можно осуществлять послереакционную обработку традиционным способом. Полученные на каждой стадии или в каждой реакции соединения можно использовать в последующей реакции в виде реакционной смеси или неочищенного продукта. Продукт можно экстрагировать из реакционной смеси по стандартным методикам. Продукт можно экстрагировать или очищать, по необходимости выбирая известный способ, такой как кристаллизация, перекристаллизация, перегонка, разделение и хроматография, и при необходимости комбинируя перечисленные способы.
Соединение (I) по настоящему изобретению можно получать, например, способом, представленным ниже на Схеме реакции 1, или основанным на нем способами.
Схема реакции 1
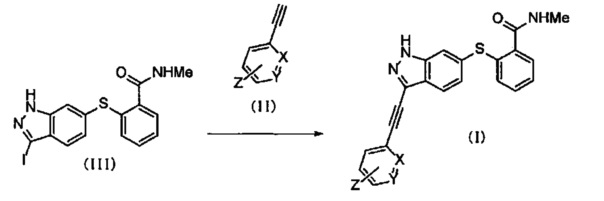
Символы на Схеме имеют такое же значение, как описано в общей Формуле (I). Me представляет собой метил.
Соединение (I) по настоящему изобретению можно получать реакцией соединения, представленного общей Формулой (II) (далее в тексте именуется "соединение (II)") с соединением, представленным Формулой (III) (далее в тексте именуется "соединение (III)"), в присутствии основания и катализатора. Относительно соединения (III), обычно используют 0.5-3 эквивалента, предпочтительно 0.8-2 эквивалента соединения (II).
Основание может включать, например, N,N-диизопропилэтиламин, триэтиламин, диэтиламин, диизопропиламин и т.д., и среди указанных предпочтительны N,N-диизопропилэтиламин или триэтиламин. Относительно соединения (III), обычно используют 1-50 эквивалентов, предпочтительно 3-30 эквивалентов указанного основания.
Катализатор может включать, например, PdCl2(PPh3)2, Pd(PPh3)4, иодид меди, бромид меди и т.д., и среди указанных предпочтительны PdCl2(PPh3)2 и иодид меди. Относительно соединения (III), обычно используют 0.01-0.5 эквивалента, предпочтительно 0.03-0.1 эквивалента указанного катализатора.
Данную реакцию предпочтительно осуществляют в растворителе, инертном к протекающей реакции. Несмотря на то, что такой выбор растворителя особым образом не ограничен, при условии что реакция в нем идет, указанный растворитель может включать, например, N,N-диметилформамид, ацетонитрил, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, толуол, этилацетат и т.д., и среди указанных предпочтителен N,N-диметилформамид или ацетонитрил.
Несмотря на то, что время реакции варьируется в зависимости от используемых реагентов или растворителей, обычно оно составляет 1-24 часа, предпочтительно 2-4 часа. Несмотря на то, что температура реакции варьируется в зависимости от используемых реагентов или растворителей, обычно она составляет 25-160°C, предпочтительно 60-100°C.
Когда A в общей Формуле (а) представляет собой мочевину или карбамат в группе, обозначенной как Z, соединение (II) можно получать способом, представленным ниже на Схеме реакции 2. Соединение, представленное общей Формулой (II-1) (далее в тексте именуется "соединение (II-1)"), и соединение, представленное общей Формулой (II-2) (далее в тексте именуется "соединение (II-2)"), входят в определение соединения (II).
Схема реакции 2
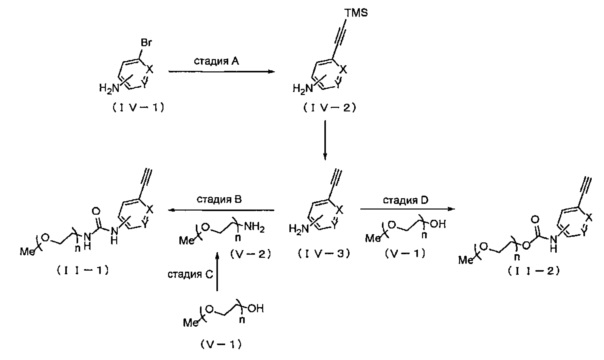
Символы на Схеме имеют такое же значение, как описано в общей Формуле (I). TMS представляет собой триметилсилильную группу. Me представляет собой метил.
Соединение, представленное общей Формулой (IV-3) (далее в тексте именуется "соединение (IV-3)"), коммерчески доступно, и можно применять коммерчески доступные продукты. Кроме того, соединение (IV-3) можно получать согласно известному способу, например, способами, описанными в WO 2013/101184, WO 2011/092197 и т.п., или основанными на них способами, например смотри описанную ниже стадию А.
На стадии A, соединение, представленное общей Формулой (IV-2) (далее в тексте именуется "соединение (IV-2)"), получают реакцией соединения, представленного общей Формулой (IV-1) (далее в тексте именуется "соединение (IV-1)"), с триметилсилил ацетиленом в растворителе в присутствии основания и катализатора, и затем удаляют триметилсилильную защитную группу с соединения (IV-2), получая соединение (IV-3). В реакции, относящейся к стадии A, относительно соединения (IV-1) обычно применяют 1-3 эквивалента, предпочтительно 1.2-1.5 эквивалента триметилсилил ацетилена. Основание может включать те же основания, которые приведены в Формуле реакции 1, предпочтительно триэтиламин. Относительно соединения (IV-1), обычно применяют 1-30 эквивалентов, предпочтительно 10-20 эквивалентов указанного основания. Катализатор может включать те же катализаторы, которые приведены на схеме реакции 1, и предпочтительно применяют PdCl2(PPh3)2 и иодид меди. Относительно соединения (IV-1), обычно применяют 0.01-0.2 эквивалента, предпочтительно 0.02-0.05 эквивалента каждого из указанных катализаторов. Растворитель может представлять собой тот же растворитель, который указан для Схемы реакции 1, предпочтительно тетрагидрофуран. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 3-4 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 25-120°C, предпочтительно 60 -90°C.Кроме того, триметилсилильную защитную группу с полученного соединения (IV-2) снимают раствором метанола с добавлением оснований, получая соединение (IV-3). Концентрация оснований в растворе метанола обычно составляет 5-50% вес/об., предпочтительно 10-15% вес/об.. Данное основание может включать, например, гидроксид натрия, гидроксид калия, гидроксид лития, карбонат калия, карбонат цезия и т.д., и среди указанных предпочтительным является гидроксид натрия. Несмотря на то, что температура реакции и время реакции снятия защиты варьируются в зависимости от применяемых реагентов или растворителей, температура реакции обычно составляет 1-30°C, предпочтительно 15-25°C, и время реакции обычно составляет 1-24 часа, предпочтительно 2-4 часа.
На стадии B, соединение (IV-3) вводят в реакцию с 4-нитрофенилхлорформиатом в растворителе, получая карбаматный продукт, и затем полученный карбаматный продукт вводят в реакцию с соединением, представленным общей Формулой (V-2) (далее в тексте именуется "соединение (V-2)"), получая соединение (II-1). Относительно соединения (IV-3), обычно применяют 0.8-10 эквивалентов, предпочтительно 1-1.3 эквивалента 4-нитрофенилхлорформиата. Вместо 4-нитрофенилхлорформиата можно применять 2,2,2-трихлорэтилхлорформиат, бис(трихлорметил)карбонат, 1,1'-карбонилдиимидазол, фенилхлорформиат, ди(N-сукцинимидил)карбонат и т.д. Также при необходимости можно использовать основание. Данное основание может включать, например, триэтиламин, трибутиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин и т.д., и среди указанных предпочтителен триэтиламин или пиридин. Относительно соединения (IV-3), обычно применяют 1-30 эквивалентов, предпочтительно 1-5 эквивалентов основания. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, этилацетат, N,N-диметилформамид, диметил сульфоксид, ацетонитрил и т.д., и среди указанных предпочтительны дихлорметан, тетрагидрофуран или 1,4-диоксан. Относительно соединения (IV-3), обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента соединения (V-2). Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-48 часов, предпочтительно 2-24 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-120°C, предпочтительно 25-110°C. Соединение (V-2) коммерчески доступно, и можно применять коммерчески доступные продукты. Кроме того, соединение (V-2) можно получать согласно известному способу, например, способом, описанным в WO 2009/109035 (JP 2011-105735 A), или основанными на нем способами, например смотри описанную ниже стадию C.
На стадии C, соединение (V-2) получают из соединения, представленного общей Формулой (V-1) (далее в тексте именуется "соединение (V-1)"). Сначала соединение (V-1) вводят в реакцию с фталимидом, трифенилфосфином и диэтил азодикарбоксилатом в растворителе (первая стадия). Затем полученный в растворителе продукт можно вводить в реакцию с гидразина моногидратом, получая соединение (V-2) (вторая стадия).
На первой стадии, относительно соединения (V-1), обычно применяют 0.5-2 эквивалента, предпочтительно 1-1.2 эквивалента фталимида, обычно 0.5-2 эквивалента, предпочтительно 1-1.2 эквивалента трифенилфосфина, а также обычно применяют 0.5 - 2 эквивалента, предпочтительно 1-1.2 эквивалента диэтил азодикарбоксилата. Вместо диэтил азодикарбоксилата можно применять бис(2-метоксиэтил)азодикарбоксилат, диизопропилазодикарбоксилат, цианометилен трибутилфосфоран или тому подобные. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, дихлорметан, 1,4-диоксан, тетрагидрофуран, толуол, N,N-диметилформамид и т.д., и среди указанных предпочтителен тетрагидрофуран. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 5-20 часов, предпочтительно 12-18 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На второй стадии, относительно соединения (V-1), применяемого на первой стадии, обычно используют 0.5-3 эквивалента, предпочтительно 1-2.2 эквивалента гидразина моногидрата относительно полученного на первой стадии продукта. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, метанол, этанол, изопропанол и т.д., и среди указанных предпочтителен этанол. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 2-20 часов, предпочтительно 4-18 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 25-120°C, предпочтительно 80-100°C.
На стадии D, соединение (V-1) вводят в реакцию с ди(N-сукцинимидил) карбонатом в растворителе в присутствии основания, получая карбонатный продукт, и затем данный карбонатный продукт конденсируют с соединением (IV-3), получая соединение (II-2). Относительно соединения (V-1), обычно применяют 0.5-5 эквивалентов, предпочтительно 2-3.5 эквивалентов основания, и обычно 0.5-5 эквивалентов, предпочтительно 1-2 эквивалента ди(N-сукцинимидил)карбоната. Вместо ди(N-сукцинимидил)карбоната можно применять бис(трихлорметил)карбонат, 1,1'-карбонилдиимидазол, 4-нитрофенилхлорформиат, фенил хлорформиат и т.д. Основания могут включать, например, триэтиламин, трибутиламин, пиридин, N,N-диизопропилэтиламин, 4-диметиламинопиридин и т.д., и среди указанных предпочтителен триэтиламин. Кроме того, при необходимости можно применять катализатор. Данный катализатор может представлять собой, например, 4-диметиламинопиридин, 4-пирролидинопиридин и т.д., и среди указанных предпочтителен 4-диметиламинопиридин. Относительно соединения (V-1), обычно применяют 0.05-0.5 эквивалента, предпочтительно 0.1-0.2 эквивалента катализатора. Кроме того, относительно соединения (IV-3), обычно применяют 0.5-5 эквивалентов, предпочтительно 1-2 эквивалента соединения (V-1). Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, ацетонитрил, этилацетат, N,N-диметилформамид и т.д., и среди указанных предпочтителен этилацетат. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-48 часов, предпочтительно 10-24 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 60-80°C.
Когда A в общей Формуле (a) представляет собой α-алкоксиамид или амин в группе, обозначенной как Z, соединение (II) можно получать, например, способом, представленным ниже на Схеме реакции 3. Соединение, представленное общей Формулой (II-3) (далее в тексте именуется "соединение (II-3)"), и соединение, представленное общей Формулой (II-4) (далее в тексте именуется "соединение (II-4)"), входят в определение соединения (II).
Схема реакции 3
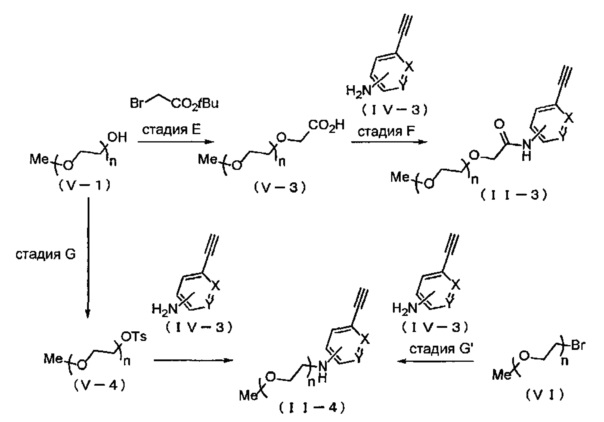
Символы на Схеме имеют такое же значение, как описано в общей Формуле (I). Ts представляет собой п-толуолсульфонильную группу (тозильную группу). Me представляет собой метил, и tBu представляет собой трет-бутил.
На стадии E, соединение (V-1) вводят в реакцию с трет-бутилбромацетатом в подходящем растворителе в присутствии основания, и затем соединение, представленное общей Формулой (V-3) (далее в тексте именуется "соединение (V-3)"), получают гидролизом полученного сложного эфира. Относительно соединения (V-1), обычно применяют 1-5 эквивалентов, предпочтительно 2-3 эквивалента основания, и обычно 1-3 эквивалента, предпочтительно 1-1.2 эквивалента трет-бутилбромацетата. Основание может включать, например, гидрид натрия, диизопропиламид лития, гексаметилдисилазид лития, н-бутиллитий и т.д., и среди указанных предпочтителен гидрид натрия. Вместо трет-бутилбромацетата также можно использовать метил хлорацетат, этил хлорацетат, метил бромацетат, этил бромацетат и т.д. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, N,N-диметилформамид, толуол, диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран и т.д., и среди указанных предпочтителен тетрагидрофуран. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 10-17 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 0-25°C.
Гидролиз сложного эфира осуществляют в подходящем растворителе в присутствии основания. При гидролизе сложного эфира, относительно сложного эфира обычно применяют 1-5 эквивалентов, предпочтительно 2-4 эквивалента основания. Основание может включать, например, гидроксид лития, гидроксид натрия, гидроксид калия и т.д., и среди указанных предпочтителен гидроксид лития. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, смесь органических растворителей (например, ацетонитрил, 1,4-диоксан, тетрагидрофуран и т.д.) и воду, и среди указанных предпочтителен растворитель, представляющий собой смесь тетрагидрофурана и воды. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 3-5 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 60-100°C.
На стадии F, соединение (V-3) вводят в реакцию с соединением (IV-3) в подходящем растворителе в присутствии основания и конденсирующего агента, получая соединение (II-3). Относительно соединения (IV-3), обычно применяют 0.1-3 эквивалента, предпочтительно 0.5-1.2 эквивалента соединения (V-3). Кроме того, относительно соединения (IV-3), обычно применяют 1-3 эквивалента, предпочтительно 1-2 эквивалента конденсирующего агента, и обычно 0.5-5 эквивалентов, предпочтительно 2-4 эквивалента основания. Конденсирующий агент может включать, например, N,N'-дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, 1-этил-3-[3-(диметиламино) пропил] карбодиимид гидрохлорид, бензотриазол-1-илокситрипирролидинофосфония гексафторфосфат, O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, диэтил цианофосфонат, дифенилфосфорил азид, пентафторфенил трифторацетат, изопропил хлорформиат и т.д., и среди указанных предпочтителен 1-этил-3-[3-(диметиламино) пропил] карбодиимид гидрохлорид или O-(7-азабензотриазол-1-ил)-N,N',N'-тетраметилурония гексафторфосфат. Основания могут включать, например, триэтиламин, пиридин, N,N-диизопропилэтиламин, 4-диметиламинопиридин и т.д., и среди указанных предпочтительны триэтиламин, N,N-диизопропилэтиламин или 4-диметиламинопиридин. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, диэтиловый эфир, ацетонитрил, N,N-диметилформамид и т.д., и среди указанных предпочтителен дихлорметан или N,N-диметилформамид. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 3-24 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 25-40°C.
На стадии G, сначала соединение (V-1) вводят в реакцию с п-толуолсульфонил хлоридом в подходящем растворителе в присутствии основания, получая соединение, представленное общей Формулой (V-4) (далее в тексте именуется "соединение (V-4)"). Затем соединение (V-4) вводят в реакцию с соединением (IV-3) в подходящем растворителе в присутствии основания, получая соединение (II-4).
Относительно соединения (V-1), обычно применяют 1-2.2 эквивалента, предпочтительно 1.2-1.5 эквивалентов п-толуолсульфонил хлорида. Относительно соединения (V-1), обычно применяют 1-5 эквивалентов, предпочтительно 1.5-3 эквивалента основания. Основание может включать, например, пиридин, триэтиламин, N,N-диизопропилэтиламин, 4-диметиламинопиридин и т.д., и среди указанных предпочтителен триэтиламин. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и т.д., и среди указанных предпочтителен дихлорметан. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 14-21 час. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 25-50°C.
В реакции соединения (V-4) и соединения (IV-3), относительно соединения (V-3), обычно применяют 0.3-5 эквивалентов, предпочтительно 0.5-4.5 эквивалента соединения (V-4). Относительно соединения (IV-3), обычно применяют 1-5 эквивалентов, предпочтительно 1.5-3 эквивалента основания. Основание может включать, например, гидроксид натрия, гидроксид калия, карбонат цезия, карбонат калия и т.д., и среди указанных предпочтителен карбонат цезия или карбонат калия. В реакции соединения (V-4) и соединения (IV-3), предпочтительно применять катализатор. Относительно соединения (IV-3), обычно применяют 0.1-3 эквивалента, предпочтительно 0.2-1 эквивалент катализатора. Катализатор может включать, например, иодид натрия, иодид калия, иодид тетрабутиламмония и т.д., и среди указанных предпочтителен иодид калия. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, ацетонитрил, тетрагидрофуран, дихлорметан, N,N-диметилформамид, диметилсульфоксид и т.д., и среди указанных предпочтителен ацетонитрил или N,N-диметилформамид. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 9-60 часов, предпочтительно 18-24 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 25-120°C, предпочтительно 80-120°C.
В реакции соединения (V-4) и соединения (IV-3), соединение (II-4) также можно получать, применяя вместо соединения (V-4) соединение, представленное общей Формулой (VI) (далее в тексте именуется "соединение (VI)"), которое изображено на стадии G' в Схеме реакции 3. Предпочтительные условия и т.п. для реакции соединения (VI) и соединения (IV-3) на стадии G' такие же, как для реакции соединения (V-4) и соединения (IV-3) в описанной выше стадии G.
Соединение (III) можно получать известными способами, например, описанными в WO 2006/048745 (JP 2008-518901 A), WO 2001/002369 (Патент Японии №3878849) и т.д. или основанными на них способами, например, согласно приведенной ниже Схеме реакции 4.
Схема реакции 4
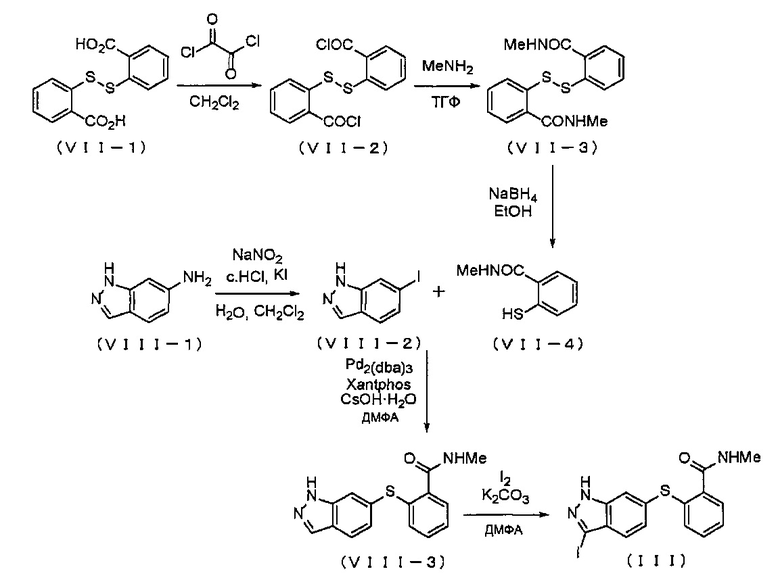
На приведенной Схеме, Me представляет собой метил.
В способе согласно Схеме реакции 4, соединение, представленное Формулой (VII-1) (далее в тексте именуется "соединение (VII-1)"), вводят в реакцию с оксалилхлоридом в подходящем растворителе, получая хлорангидрид, представленный Формулой (VII-2) (далее в тексте именуется "хлорангидрид (VII-2)"), который затем вводят в реакцию с метиламином, получая амидный продукт, представленный Формулой (VII-3) (далее в тексте именуется "амидный продукт (VII-3)"). Амидный продукт, представленный Формулой (VII-3), можно восстанавливать боргидридом натрия, получая тиольный продукт, представленный Формулой (VII-4) (далее в тексте именуется "тиольный продукт (VII-4)").
Относительно соединения (VII-1), обычно применяют 1-5 эквивалентов, предпочтительно 2-4 эквивалента оксалилхлорида. Выбор растворителя особым образом не ограничен, если он не оказывает отрицательного влияния на реакцию, и растворитель может включать, например, тетрагидрофуран, дихлорметан, толуол и т.д., и среди указанных предпочтителен дихлорметан. В реакции соединения (VII-1) с оксалилхлоридом, предпочтительно применять катализатор. Относительно соединения (VII-1), обычно применяют 0.01-0.5 эквивалента, предпочтительно 0.01-0.1 эквивалента данного катализатора. Катализатор может представлять собой N,N-диметилформамид. Несмотря на то, что время реакции варьируется в зависимости от применяемых растворителей, оно обычно составляет 10-60 часов, предпочтительно 15-40 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых растворителей, она обычно составляет 15-100°C, предпочтительно 20-80°C.
Реакцию хлорангидрида (VII-2) с метиламином можно проводить, например, в тетрагидрофуране. Относительно хлорангидрида (VII-2), обычно применяют 1-5 эквивалентов, предпочтительно 2-5 эквивалентов метиламина. Время реакции обычно составляет 6-24 часа, предпочтительно 12-24 часа. Температура реакции обычно составляет 0-100°C, предпочтительно 0-30°C.
При восстановлении амидного продукта (VII-3), относительно амидного продукта (VII-3) обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента боргидрида натрия. Реакцию можно проводить, например, в этаноле. Время реакции обычно составляет 6-24 часа, предпочтительно 12-20 часов. Температура реакции обычно составляет 0-50°C, предпочтительно 0-30°C.
С другой стороны, соединение, представленное Формулой (VIII-1) (далее в тексте именуется "соединение (VIII-1)"), можно вводить в реакцию с нитритом натрия и затем с иодидом калия в кислом водном растворе, получая соединение, представленное формулой (VIII-2) (далее в тексте именуется "соединение (VIII-2)"). Данное соединение (VIII-2) можно вводить в реакцию в присутствии тиольного продукта (VII-4), Pd2(dba)3, 4,5-бис(дифенилфосфино)-9,9-диметилксантена и моногидрата гидроксида цезия, получая соединение, представленное формулой (VIII-3) (далее в тексте именуется "соединение (VIII-3)"). Далее, данное соединение (VIII-3) можно иодировать в присутствии карбоната калия, получая соединение (III).
В реакции получения соединения (VIII-2) из соединения (VIII-1), относительно соединения (VIII-1) обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента нитрита натрия, и обычно 1-5 эквивалентов, предпочтительно 1-3 эквивалента иодида калия. Время реакции соединения (VIII-1) с нитритом натрия обычно составляет от 10 минут до 6 часов, предпочтительно от 0.5 часа до 2 часов. Температура реакции обычно составляет 0-50°C, предпочтительно 0-30°C. Время реакции с иодидом калия обычно составляет от 10 минут до 6 часов, предпочтительно от 0.5 часа до 3 часов. Температура реакции обычно составляет 0-50°C, предпочтительно 0-40°C.
Реакцию получения соединения (VIII-3) из соединения (VIII-2) и тиольного продукта (VII-4) можно осуществлять в атмосфере аргона, например, в N,N-диметилформамиде. Относительно соединения (VIII-2), обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента тиольного продукта (VII-4). Относительно соединения (VIII-2), обычно применяют 0.01-3 эквивалента, предпочтительно 0.03-1 эквивалент Pd2(dba)3. Относительно соединения (VIII-2), обычно применяют 0.05-3 эквивалента, предпочтительно 0.1-2 эквивалента 4,5-бис(дифенилфосфино)-9,9-диметилксантена. Относительно соединения (VIII-2), обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента моногидрата гидроксида цезия. Время реакции обычно составляет 1-24 часа, предпочтительно 2-10 часов. Температура реакции обычно составляет 25-150°C, предпочтительно 80-120°C.
Иодирование соединения (VIII-3) можно проводить, например, в N,N-диметилформамиде. Относительно соединения (VIII-3), обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента карбоната калия. Относительно соединения (VIII-3), обычно применяют 1-5 эквивалентов, предпочтительно 1-3 эквивалента иода. Время реакции обычно составляет 1-24 часа, предпочтительно 1-6 часов. Температура реакции обычно составляет 0-50°C, предпочтительно 0-30°C.
Когда A в общей Формуле (а) представляет собой мочевину, и R2 представляет собой низший алкил, который может быть замещенным, в группе, обозначенной как Z, соединение (II) можно получать способом, проиллюстрированным ниже на Схеме реакции 5. Соединение, представленное формулой (II-5) (далее в тексте именуется "соединение (II-5)"), соединение, представленное формулой (II-6) (далее в тексте именуется "соединение (II-6)"), соединение, представленное формулой (II-7) (далее в тексте именуется "соединение (II-7)"), соединение, представленное формулой (II-8) (далее в тексте именуется "соединение (II-8)"), и соединение, представленное формулой (II-9) (далее в тексте именуется "соединение (II-9)"), входят в определение соединения (II).
Схема реакции 5
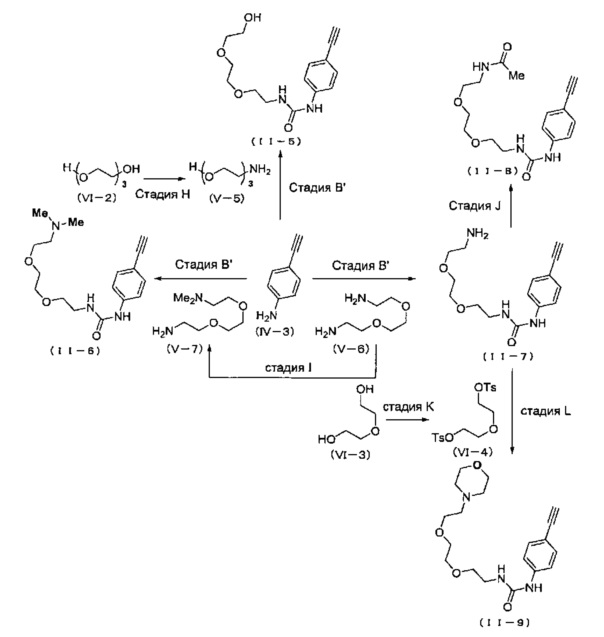
В приведенной схеме, Ts представляет собой п-толуолсульфонильную группу (тозильную группу), и Me представляет собой метил.
На стадии H, соединение, представленное формулой (V-5) (далее в тексте именуется "соединение (V-5)"), получают из соединения, представленного Формулой (VI-2) (далее в тексте именуется "соединение (VI-2)"). Сначала соединение (VI-2) вводят в реакцию с трет-бутил диметилхлорсиланом в присутствии основания в растворителе (первая стадия). Затем продукт, полученный в растворителе, вводят в реакцию с фталимидом, трифенилфосфином и диэтил азодикарбоксилатом (вторая стадия). Далее полученный продукт можно вводить в реакцию с гидразина моногидратом, и затем с соляной кислотой, получая соединение (V-5) (третья стадия).
На первой стадии, относительно соединения (VI-1) обычно применяют 0.8-1.2 эквивалента, предпочтительно 1-1.1 эквивалента основания, и обычно 0.8-2 эквивалента, предпочтительно 1-1.2 эквивалента трет-бутилметилхлорсилана. Основание может включать, например, гидрид натрия, диизопропиламид лития, гексаметилдисилазид лития, н-бутиллитий, и среди указанных предпочтителен гидрид натрия. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, N,N-диметилформамид, толуол, диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран и т.д., и среди указанных предпочтителен тетрагидрофуран. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-48 часов, предпочтительно 15-24 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-100°C, предпочтительно 0-25°C.
На второй стадии, относительно продукта, полученного на первой стадии, обычно применяют 0.5-2 эквивалента, предпочтительно 1-1.6 эквивалента фталимида, обычно 1-2 эквивалента, предпочтительно 1-1.6 эквивалента трифенилфосфина, а также обычно применяют 1-2 эквивалента, предпочтительно 1-1.2 эквивалента диэтил азодикарбоксилата. Вместо диэтил азодикарбоксилата можно применять бис(2-метоксиэтил)азодикарбоксилат, диизопропил азодикарбоксилат, цианометилен трибутилфосфоран или тому подобные. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, дихлорметан, 1,4-диоксан, тетрагидрофуран, толуол, N,N-диметилформамид и т.д., и среди указанных предпочтителен тетрагидрофуран. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 5-20 часов, предпочтительно 12-18 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На третьей стадии, относительно полученного на второй стадии продукта, обычно применяют 1-5 эквивалентов, предпочтительно 1-4.4 эквивалента гидразина моногидрата. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, метанол, этанол, изопропанол и т.д., и среди указанных предпочтителен этанол. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 2-20 часов, предпочтительно 4-18 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 25-120°C, предпочтительно 80-100°C. Затем добавляют 5-10 эквивалентов, предпочтительно 8-10 эквивалентов концентрированной соляной кислоты. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 2-20 часов, предпочтительно 2-4 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-120°C, предпочтительно 90-110°C.
На стадии I, соединение, представленное формулой (V-7) (далее в тексте именуется "соединение (V-7)"), получают из соединения, представленного формулой (V-6) (далее в тексте именуется "соединение (V-6)"). Сначала соединение (V-6) вводят в реакцию с ди-трет-бутил дикарбонатом в растворителе (первая стадия). Затем продукт, полученный в растворителе, вводят в реакцию с формальдегидом, уксусной кислотой и триацетоксиборгидридом натрия (вторая стадия). Далее полученный продукт можно вводить в реакцию с трифторуксусной кислотой, получая соединение (V-7) (третья стадия).
На первой стадии, относительно соединения (V-6), обычно применяют 0.4-0.6 эквивалента, предпочтительно 0.5-0.6 эквивалента ди-трет-бутил дикарбоната (в 30%-ном растворе тетрагидрофурана), и обычно 0.8-1.1 эквивалента, предпочтительно 0.9 -1.0 эквивалента диизопропилэтиламина. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, тетрагидрофуран, дихлорметан, хлороформ, ацетонитрил, 1,4-диоксан и т.д., и среди указанных предпочтителен дихлорметан. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 2-10 часов, предпочтительно 2-4 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На второй стадии, относительно продукта, полученного на первой стадии, обычно применяют 10-40 эквивалентов, предпочтительно 20-30 эквивалентов формальдегида (37%-ный раствор в воде), обычно 10-40 эквивалентов, предпочтительно 20-30 эквивалентов уксусной кислоты, а также обычно 1.5-5 эквивалентов, предпочтительно 1.5-2 эквивалента триацетоксиборгидрида натрия. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, метанол, этанол, изопропанол и т.д., и среди указанных предпочтителен метанол. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 2-10 часов, предпочтительно 2-3 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На третьей стадии, относительно продукта, полученного на второй стадии, обычно применяют 5-20 эквивалентов, предпочтительно 5-17 эквивалентов трифторуксусной кислоты. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, тетрагидрофуран, дихлорметан, хлороформ, ацетонитрил, 1,4-диоксан и т.д., и среди указанных предпочтителен дихлорметан. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет от 30 минут до 2 часов, предпочтительно от 30 минут до 1 часа. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На стадии J, соединение (II-7) вводят в реакцию с ангидридом уксусной кислоты, получая соединение (II-8) в подходящем растворителе. Относительно соединения (II-7), обычно применяют 1-5 эквивалентов, предпочтительно 1.5-3 эквивалента ангидрида уксусной кислоты, и обычно 0.1-0.5 эквивалента, предпочтительно 0.1-0.3 эквивалента 4-диметиламинопиридина. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, тетрагидрофуран, дихлорметан, хлороформ, ацетонитрил, 1,4-диоксан и т.д., и среди указанных предпочтителен ацетонитрил. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 6-10 часов, предпочтительно 6-8 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На стадии K, соединение, представленное Формулой (VI-3) (далее в тексте именуется "соединение (VI-3)"), вводят в реакцию с п-толуолсульфонил хлоридом, получая соединение, представленное Формулой (VI-4) (далее в тексте именуется "соединение (VI-4)"), в подходящем растворителе, в присутствии основания. Относительно соединения (VI-3), обычно применяют 2-3 эквивалента, предпочтительно 2-2.2 эквивалента п-толуолсульфонил хлорида. Относительно соединения (VI-3), обычно применяют 2-8 эквивалентов, предпочтительно 5-8 эквивалентов основания. Основание может включать, например, гидроксид натрия, гидроксид калия, карбонат цезия, карбонат калия, и среди указанных предпочтителен гидроксид натрия или гидроксид калия. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и т.д., и среди указанных предпочтителен дихлорметан. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 1-24 часа, предпочтительно 3-10 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 0-40°C, предпочтительно 0-25°C.
На стадии L, соединение (II-7) вводят в реакцию с соединением (VI-4), получая соединение (II-9) в подходящем растворителе в присутствии основания. Относительно соединения (II-7), обычно применяют 1-3 эквивалента, предпочтительно 1.5-2.0 эквивалента соединения (VI-4). Основание может включать, например, гидроксид натрия, гидроксид калия, карбонат цезия, карбонат калия, и среди указанных предпочтителен карбонат натрия или карбонат калия. Выбор растворителя особым образом не ограничен, и растворитель может включать, например, ацетонитрил, дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан и т.д., и среди указанных предпочтителен ацетонитрил. Несмотря на то, что время реакции варьируется в зависимости от применяемых реагентов или растворителей, оно обычно составляет 10-24 часа, предпочтительно 10-18 часов. Несмотря на то, что температура реакции варьируется в зависимости от применяемых реагентов или растворителей, она обычно составляет 25-120°C, предпочтительно 80-100°C.
На стадии B', соединение (II-5), (II-6) или (II-7) можно получать, применяя соединение (V-5), (V-6) или (V-7) вместо соединения (V-2), таким же образом, как на стадии В.
Полученное соединение (I) по настоящему изобретению можно выделять или очищать в индивидуальном виде или в виде его соли, полученной в результате солеобразующей обработки. Способ выделения или очистки особым образом не ограничен и может быть произвольно выбран из таких традиционных способов, как кристаллизация, перекристаллизация, перегонка, разделение и хроматография, или их комбинация. Сольват соединения (I) по настоящему изобретению можно получать способом, известным per se.
Примеры
Далее настоящее изобретение будет более подробно раскрыто с привлечением Сравнительных примеров, Примеров, Примеров препаратов и Примеров тестирования, но настоящее изобретение не ограничено только ими.
В Сравнительных примерах, Примерах и Примерах тестирования, температуры во всех случаях указаны по шкале Цельсия (°C), если не указано иное. Все количества и процентное содержание указаны из расчета веса, если не указано иное. Реагенты приобретали у таких поставляющих реагенты компаний, как Sigma-Aldrich Corporation, Tokyo Chemical Industry Co., Ltd. или Nacalai Tesque Inc., и применяли без очистки, если не указано иное.
Обычно манипуляции в Сравнительных примерах и Примерах осуществляли в безводном растворителе в атмосфере аргона. Реакционные смеси анализировали методом ТСХ (тонкослойная хроматография), полноту прохождения оценивали по исчезновению исходного вещества, после чего завершали. Для ТСХ применяли силикагель 60F254 (Merck), применяли для проявки подходящий растворитель, и располагали в подходящем положении. Колоночную флэш-хроматографию осуществляли на силикагеле SiliaFlash (зарегистрированная торговая марка) F60 (230-600 меш, Silicycle Inc.).
Для аналитических данных соединений, которые будут описаны в Сравнительных примерах и Примерах, температуры плавления измеряли с помощью МР-500 type V (без корректировки) производства Yanaco Co. 1H-ЯМР спектры записывали на приборе Bruker с рабочей частотой 400 МГц. ЯМР спектры записывали в растворах в CDCl3, применяя в качестве внутреннего стандарта хлороформ (7.26 м.д.) или тетраметилсилан (0.00 м.д.). Другие ЯМР растворители также использовали по необходимости. При описании мультиплетности пиков применяются следующие аббревиатуры: с (синглет), д (дублет), т (триплет), кв (квадруплет), м (мультиплет), ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов), тд (триплет дублетов). При описании констант спин-спинового взаимодействия (величина J), их приводят в Герцах (Гц).
Сравнительный пример 1
Получение 5-[(триметилсилил)этинил]пиридин-3-амина
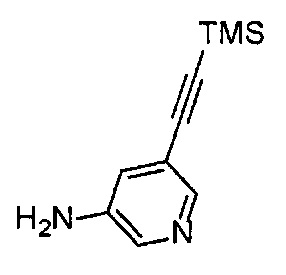
Триэтиламин (20 мл) и триметилсилилацетилен (3.32 мл, 24 ммоль) добавляли в раствор 3-амино-5-бромпиридина (3.46 г, 20 ммоль), PdCl2(PPh3)2 (561.5 мг, 0.80 ммоль) и CuI (76.2 мг, 0.40 ммоль) в тетрагидрофуране (5 мл) при комнатной температуре, перемешивали в течение 5 минут и затем перемешивали при кипячении в течение 21 часа. После фильтрования полученного раствора растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 50/50→0/100), получая указанное в заголовке соединение (2.36 г, 62%) в виде коричневого порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.10 (1Н, д, J=1.6 Гц), 8.01 (1H, д, J=2.8 Гц), 7.03 (1H, дд, J=2.8, 1.6 Гц), 3.68 (2Н, ушир. с), 0.25 (9Н, с).
Сравнительный пример 2
Получение 5-[(триметилсилил)этинил]пиридин-2-амина
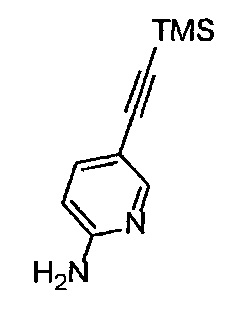
2-Амино-5-бромпиридин (5.2 г, 30 ммоль), PdCl2(PPh3)2 (842.3 мг, 1.2 ммоль), CuI (114.3 мг, 0.60 ммоль), тетрагидрофуран (15 мл), триэтиламин (30 мл) и триметилсилилацетилен (4.98 мл, 36 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 1, получая указанное в заголовке соединение (5.17 г, 91%) в виде желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.21 (1Н, д, J=2.0 Гц), 7.49 (1H, дд, J=8.4, 2.0 Гц), 6.41 (1H, ушир. д, J=8.4 Гц), 4.56 (2Н, ушир. с), 0.24 (9Н, с).
Сравнительный пример 3
Получение 6-[(триметилсилил)этинил]пиридин-3-амина
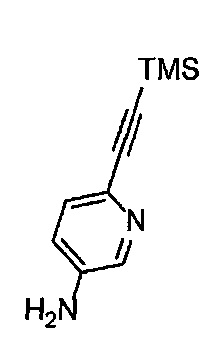
3-Амино-6-бромпиридин (6.50 г, 37.6 ммоль), PdCl2(PPh3)2 (1.05 г, 1.5 ммоль), CuI (142.8 мг, 0.75 ммоль), тетрагидрофуран (19 мл), триэтиламин (38 мл) и триметилсилилацетилен (6.24 мл, 45.1 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 1, получая указанное в заголовке соединение (6.28 г, 88%) в виде коричневого порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.03 (1H, д, J=2.8 Гц), 7.25 (1H, д, J=8.4 Гц), 6.88 (1H, дд, J=8.4, 2.8 Гц), 3.84 (2Н, ушир. с), 0.24 (9Н, с).
Сравнительный пример 4
Получение 5-этинилпиридин-3-амина
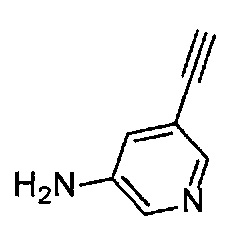
Раствор гидроксида натрия (1.00 г) в метаноле (10 мл) добавляли в раствор 5-[(триметилсилил)этинил]пиридин-3-амина (1.90 г, 10 ммоль) в тетрагидрофуране (10 мл) при комнатной температуре и перемешивали в течение 4 часов. Полученный раствор разбавляли водой и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, сушили безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 50/50→25/75), получая указанное в заголовке соединение (800.1 мг, 68%) в виде коричневого порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.13 (1H, д, J=1.6 Гц), 8.05 (1H, д, J=2.4 Гц), 7.05 (1H, дд, J=2.4, 1.6 Гц), 3.72 (2Н, ушир. с), 3.14 (1Н, с).
Сравнительный пример 5
Получение 5-этинилпиридин-2-амина
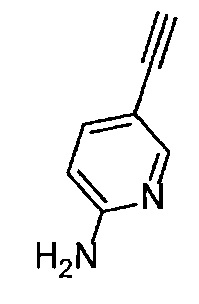
5-[(Триметилсилил)этинил]пиридин-2-амин (4.40 г, 23.2 ммоль), тетрагидрофуран (23 мл), гидроксид натрия (2.30 г) и метанол (23 мл) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 4, получая указанное в заголовке соединение (2.71 г, 99%) в виде коричневого порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.23 (1Н, д, J=2.0 Гц), 7.51 (1H, дд, J=8.8, 2.0 Гц), 6.43 (1H, ушир. д, J=8.8 Гц), 4.59 (2Н, ушир. с), 3.05 (1Н, с).
Сравнительный пример 6
Получение 6-этинилпиридин-3-амина
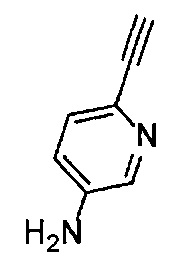
6-[(Триметилсилил)этинил]пиридин-3-амин (6.28 г, 33.0 ммоль), тетрагидрофуран (33 мл), гидроксид натрия (3.30 г) и метанол (33 мл) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 4, получая указанное в заголовке соединение (3.60 г, 92%) в виде черного порошка.
1H ЯМР (400 МГц, CDCl3) δ 8.05 (1Н, д, J=2.8 Гц), 7.27 (1H, д, J=8.4 Гц), 6.89 (1Н, дд, J=8.4, 2.8 Гц), 3.89 (2Н, ушир. с), 3.01 (1H, с).
Сравнительный пример 7
Получение 2-(2-метоксиэтокси)этан-1-амина
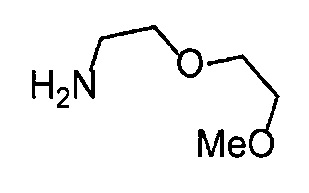
Монометиловый эфир диэтиленгликоля (3.9 мл, 33.3 ммоль) и диэтил азодикарбоксилат (2.2 моль/л раствор в толуоле) (15.5 мл, 34.1 ммоль) добавляли в раствор фталимида (4.90 г, 34.2 ммоль) и трифенилфосфина (9.00 г, 34.2 ммоль) в тетрагидрофуране (180 мл) при комнатной температуре и перемешивали в течение ночи. В полученный раствор затем добавляли этанол (60 мл), перемешивали при комнатной температуре в течение 30 минут, и затем растворитель отгоняли при пониженном давлении. Этилацетат (50 мл) и гексан (50 мл) добавляли в полученный остаток, и отфильтровывали нерастворимый осадок. После упаривания фильтрата при пониженном давлении, добавляли этанол (120 мл) и гидразина моногидрат (2.4 мл, 68.6 ммоль) и перемешивали при кипячении в течение ночи. После охлаждения раствора до комнатной температуры, добавляли концентрированную соляную кислоту (15 мл) и перемешивали при кипячении в течение 1 часа. После охлаждения раствора до комнатной температуры, нерастворимые фракции отфильтровывали, и фильтрат упаривали при пониженном давлении. Добавляли в остаток воду и промывали диэтиловым эфиром, доводили pH водного слоя до 13 с помощью 3н. водного раствора гидроксида натрия, и экстрагировали дихлорметаном. Органический слой сушили безводным сульфатом натрия, растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (1.74 г, 44%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.64-3.61 (2Н, м), 3.57-3.55 (2Н, м), 3.51 (2Н, т, J=5.2 Гц), 3.39 (3H, с), 2.89-2.87 (2Н, м).
Сравнительный пример 8
Получение 2-[2-(2-метоксиэтокси)этокси]этан-1-амина
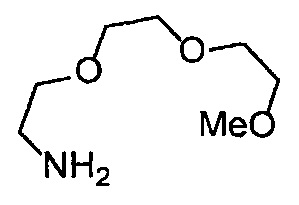
Фталимид (5.59 г, 38 ммоль), трифенилфосфин (10.3 г, 39.2 ммоль), монометиловый эфир триэтиленгликоля (6 мл, 38.4 ммоль), диэтил азодикарбоксилат (2.2 моль/л толуольный раствор) (18.2 мл, 40 ммоль), тетрагидрофуран (150 мл), этанол (220 мл) и гидразина моногидрат (3 мл, 84.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 7, получая указанное в заголовке соединение (4.48 г, 72%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.67-3.64 (6Н, м), 3.57-3.55 (2Н, м), 3.51 (2Н, т, J=5.2 Гц), 3.38 (3H, с), 2.87 (2Н, ушир. т, J=5.2 Гц).
Сравнительный пример 9
Получение 2,5,8,11-тетраоксатридекан-13-амина
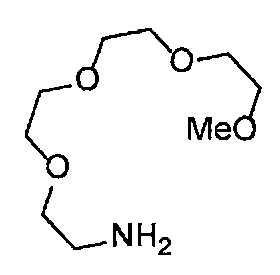
Фталимид (2.28 г, 15.5 ммоль), трифенилфосфин (4.06 г, 15.5 ммоль), монометиловый эфир тетраэтиленгликоля (3 мл, 15 ммоль), диэтил азодикарбоксилат (2.2 моль/л толуольный раствор) (7.2 мл, 15.9 ммоль), тетрагидрофуран (60 мл), этанол (50 мл) и гидразина моногидрат (1.2 мл, 33 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 7, получая указанное в заголовке соединение (2.94 г, 95%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.67-3.62 (10H, м), 3.56-3.54 (2Н, м), 3.51-3.49 (2Н, м), 3.37 (3H, с), 2.88-2.83 (2Н, м).
Сравнительный пример 10
Получение 2,5,8,11,14-пентаоксагексадекан-16-амина
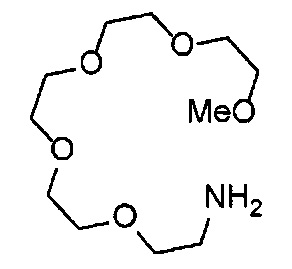
Фталимид (1.34 г, 9.4 ммоль), трифенилфосфин (2.47 г, 9.4 ммоль), монометиловый эфир пентаэтиленгликоля (2 мл, 8.6 ммоль), диэтил азодикарбоксилат (2.2 моль/л толуольный раствор) (4.3 мл, 9.4 ммоль), тетрагидрофуран (50 мл), этанол (100 мл) и гидразина моногидрат (660 мкл, 18.8 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 7, получая указанное в заголовке соединение (1.34 г, 62%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.66-3.64 (14Н, м), 3.56-3.54 (2Н, м), 3.51 (2Н, д, J=5.2 Гц), 3.38 (3H, с), 2.86 (2Н, т, J=5.2 Гц).
Сравнительный пример 11
Получение 1-(3-этинилфенил)-3-(2-метоксиэтил)мочевины
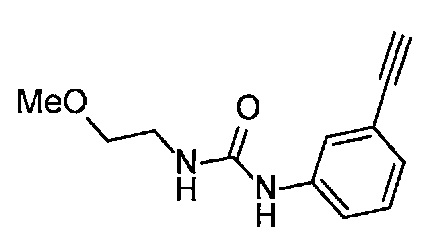
3-Этиниланилин (585.8 мг, 5.0 ммоль) и пиридин (0.44 мл, 5.5 ммоль) добавляли в раствор 4-нитрофенил хлорформиата (1.00 г, 5.0 ммоль) в дихлорметане (50 мл) и перемешивали при комнатной температуре в течение ночи. В полученный раствор затем добавляли 2-метоксиэтиламин (0.94 мл, 11 ммоль), перемешивали при комнатной температуре в течение 5 часов, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 50/50→0/100), получая указанное в заголовке соединение (1.05 г, 96%) в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ 7.42 (1Н, ушир. с), 7.37 (1Н, ушир. д, J=7.6 Гц), 7.23 (1H, т, J=7.6 Гц), 7.17 (1H, ушир. д, J=7.6 Гц), 7.03 (1H, ушир. с), 5.31 (1Н, ушир. с), 3.52 (2Н, ушир. т, J=4.8 Гц), 3.46-3.42 (2Н, м), 3.38 (3H, с), 3.04 (1Н, с).
Сравнительный пример 12
Получение 1-(3-этинилфенил)-3-[2-(2-метоксиэтокси)этил)мочевины
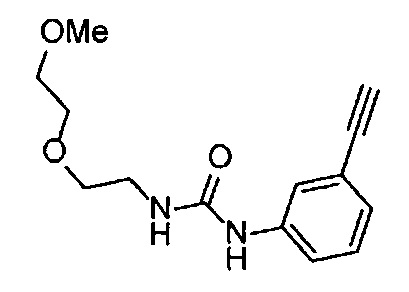
4-Нитрофенил хлорформиат (907.0 мг, 4.5 ммоль), дихлорметан (45 мл), 3-этиниланилин (527.1 мг, 4.5 ммоль), пиридин (0.58 мл, 7.2 ммоль) и 2-(2-метоксиэтокси)этан-1-амин (1.43 г, 12 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 11, получая указанное в заголовке соединение (620.0 мг, 53%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.45-7.44 (1H, м), 7.42 (1H, ушир. д, J=7.6 Гц), 7.23 (1H, т, J=7.6 Гц), 7.15 (1H, ушир. д, J=7.6, 1.2 Гц), 6.98 (1H, ушир. с), 5.31 (1H, ушир. с), 3.67-3.65 (2Н, м), 3.62 (2Н, ушир. т, J=4.8 Гц), 3.58-3.56 (2Н, м), 3.47-3.43 (2Н, м), 3.38 (3H, с), 3.04 (1H, с).
Сравнительный пример 13
Получение 1-(3-этинилфенил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевины
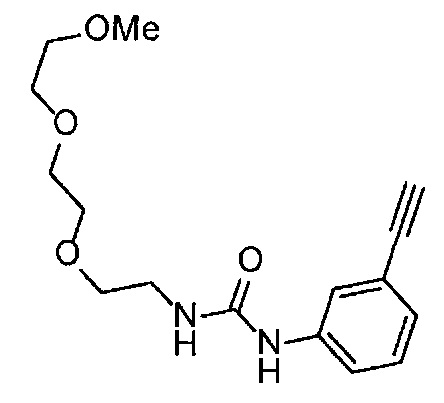
4-Нитрофенил хлорформиат (907.0 мг, 4.5 ммоль), дихлорметан (45 мл), 3-этиниланилин (527.1 мг, 4.5 ммоль), пиридин (0.58 мл, 7.2 ммоль) и 2-[2-(2-метоксиэтокси)этокси]этан-1-амин (1.96 г, 12 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 11, получая указанное в заголовке соединение (819.5 мг, 59%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.58 (1H, ушир. с), 7.51 (1Н, ушир. д, J=7.6 Гц), 7.48 (1Н, т, J=1.2 Гц), 7.21 (1Н, т, J=7.6 Гц), 7.10 (1H, ушир. дт, J=7.6, 1.2 Гц), 5.82 (1H, ушир. с), 3.70-3.67 (6Н, м), 3.64-3.61 (4Н, м), 3.47-3.43 (5Н, м), 3.02 (1Н, с).
Сравнительный пример 14
Получение 1-(3-этинилфенил)-3-(2,5,8,11-тетраоксатридекан-13-ил]мочевины
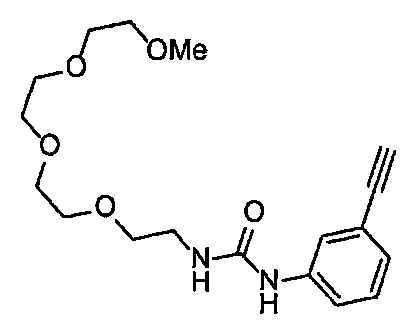
4-Нитрофенил хлорформиат (516.0 мг, 2.6 ммоль), дихлорметан (25 мл), 3-этиниланилин (288 мкл, 2.6 ммоль), пиридин (228 мкл, 2.8 ммоль) и 2,5,8,11-тетраоксатридекан-13-амин (884.5 мг, 4.3 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 11, получая указанное в заголовке соединение (393.0 мг, 44%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.73 (1H, ушир. с), 7.54-7.52 (2Н, м), 7.19 (1H, ушир. т, J=7.1 Гц), 7.09 (1H, ушир. д, J=7.1 Гц), 5.95 (1H, ушир. с), 3.74-3.72 (4Н, м), 3.68-3.65 (4Н, м), 3.62-3.58 (6Н, м), 3.45-3.41 (2Н, м), 3.32 (3H, с), 3.01 (1H, с).
Сравнительный пример 15
Получение 1-(3-этинилфенил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевины
4-Нитрофенил хлорформиат (516.0 мг, 2.6 ммоль), дихлорметан (25 мл), 3-этиниланилин (288 мкл, 2.6 ммоль), пиридин (228 мкл, 2.8 ммоль) и 2,5,8,11,14-пентаоксагексадекан-16-амин (1.00 г, 4.3 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 11, получая указанное в заголовке соединение (965 мг, 96%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.80 (1H, ушир. с), 7.55 (1H, с), 7.54 (1Н, д, J=7.7 Гц), 7.19 (1Н, ушир. т, J=7.7 Гц), 7.08 (1H, ушир. д, J=7.7 Гц), 6.05 (1H, ушир. с), 3.75-3.72 (4Н, м), 3.70-3.64 (6Н, м), 3.62-3.59 (6Н, м), 3.48-3.43 (4Н, м), 3.28 (3H, с), 3.01 (1Н, с).
Сравнительный пример 16
Получение 1-(5-этинилпиридин-3-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевины
5-Этинилпиридин-3-амин (236.3 мг, 2.0 ммоль) добавляли в раствор 4-нитрофенил хлорформиата (403.1 мг, 2.0 ммоль) в тетрагидрофуране (20 мл), перемешивали при комнатной температуре в течение 1 часа, и затем растворитель отгоняли при пониженном давлении. В раствор полученного неочищенного продукта в 1,4-диоксане (20 мл) добавляли 2-[2-(2-метоксиэтокси)этокси]этан-1-амин (359.1 мг, 2.2 ммоль) и триэтиламин (0.62 мл, 4.4 ммоль), перемешивали при кипячении в течение ночи, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол (об./об.) = 91/9), получая указанное в заголовке соединение (447.7 мг, 73%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.40 (1Н, д, J=2.8 Гц), 8.30 (1H, д, J=2.0 Гц), 8.22 (1H, ушир. дд, J=2.8, 2.0 Гц), 7.96 (1H, ушир. с), 6.01 (1Н, ушир. с), 3.72-3.62 (10H, м), 3.46-3.43 (5Н, м), 3.15 (1Н, с).
Сравнительный пример 17
Получение 1-(5-этинилпиридин-3-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевины
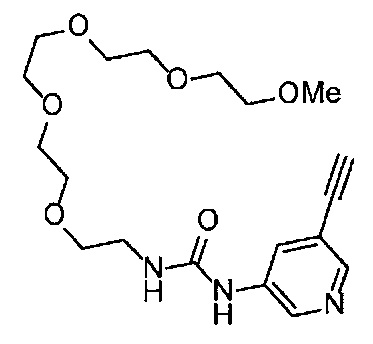
4-Нитрофенил хлорформиат (201.6 мг, 1.0 ммоль), тетрагидрофуран (10 мл), 5-этинилпиридин-3-амин (118.2 мг, 1.0 ммоль), 1,4-диоксан (10 мл), 2,5,8,11,14-пентаоксагексадекан-16-амин (276.5 мг, 1.1 ммоль) и триэтиламин (0.31 мл, 2.2 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (249.5 мг, 63%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.48 (1Н, д, J=2.8 Гц), 8.29 (1H, д, J=1.6 Гц), 8.23 (1H, ушир. дд, J=2.8, 1.6 Гц), 8.06 (1Н, ушир. с), 6.02 (1Н, ушир. с), 3.78-3.75 (4Н, м), 3.72-3.59 (12Н, м), 3.45-3.43 (4Н, м), 3.27 (с, 3H), 3.14 (1H, с).
Сравнительный пример 18
Получение 1-(4-этинилфенил)-3-(2-метоксиэтил)мочевины
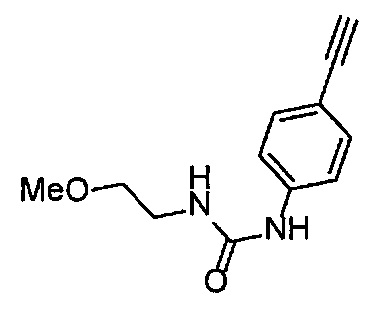
Пиридин (0.42 мл, 5.2 ммоль) и 4-этиниланилин (502.3 мг, 4.3 ммоль) добавляли в раствор 4-нитрофенил хлорформиата (873.4 мг, 4.3 ммоль) в дихлорметане (30 мл) и перемешивали при комнатной температуре в течение 45 минут. В полученный раствор добавляли раствор 2-метоксиэтиламина (664.4 мг, 8.8 ммоль) в дихлорметане (5 мл), перемешивали при комнатной температуре в течение 1 часа, затем разбавляли дихлорметаном и промывали 10%-ным водным раствором лимонной кислоты и насыщенным раствором хлорида натрия. Органический слой сушили безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 1/2), получая указанное в заголовке соединение (759.7 мг, 81%) в виде бледно-оранжевого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.41 (2Н, д, J=8.4 Гц), 7.28 (2Н, д, J=8.4 Гц), 7.14 (1H, ушир. с), 5.31 (1Н, ушир. с), 3.52 (2Н, ушир. т, J=4.8 Гц), 3.46-3.42 (2Н, м), 3.39 (3H, с), 3.02 (1H, с).
Сравнительный пример 19
Получение 1-(4-этинилфенил)-3-[2-(2-метоксиэтокси)этил]мочевины
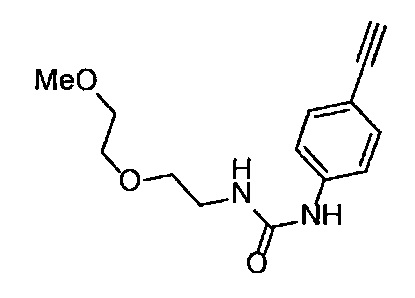
4-Нитрофенил хлорформиат (622.2 мг, 3.1 ммоль), дихлорметан (12 мл), пиридин (0.25 мл, 3.1 ммоль), 4-этиниланилин (300 мг, 2.6 ммоль) и 2-(2-метоксиэтокси)этан-1-амин (613.2 мг, 5.1 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 18, получая указанное в заголовке соединение (485.3 мг, 72%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.38-7.31 (5Н, м), 5.54 (1H, ушир. с), 3.65-3.64 (2Н, м), 3.61 (2Н, т, J=5.2 Гц), 3.57-3.54 (2Н, м), 3.46-3.42 (2Н, м), 3.37 (3H, с), 3.00 (1H, с).
Сравнительный пример 20
Получение 1-(4-этинилфенил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевины
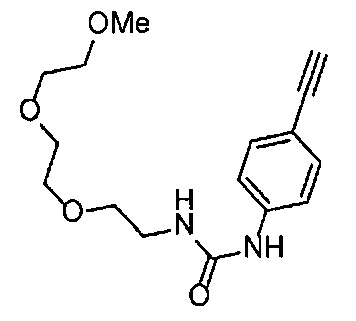
4-Нитрофенил хлорформиат (3.43 г, 17.0 ммоль), дихлорметан (130 мл), пиридин (1.54 мл, 19.0 ммоль), 4-этиниланилин (2.0 г, 17.0 ммоль) и 2-[2-(2-метоксиэтокси)этокси] этан-1-амин (3.60 г, 22.1 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 18, получая указанное в заголовке соединение (3.87 г, 74%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.73 (1H, ушир. с), 7.38 (4Н, м), 5.87 (1Н, ушир. с), 3.69 (6Н, ушир. м), 3.63-3.61 (4Н, м), 3.46-3.45 (5Н, м), 2.99 (1Н, с).
Сравнительный пример 21
Получение 1-(4-этинилфенил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевины
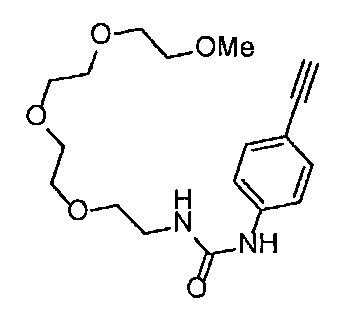
4-Нитрофенил хлорформиат (253.9 мг, 1.3 ммоль), дихлорметан (4 мл), пиридин (0.12 мл, 1.5 ммоль), 4-этиниланилин (120.5 мг, 1.0 ммоль) и 2,5,8,11-тетраоксатридекан-13-амин (417.9 мг, 2.0 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 18, получая указанное в заголовке соединение (348.5 мг, 97%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.82 (1Н, ушир. с), 7.43 (2Н, ушир. д, J=8.8 Гц), 7.37 (2Н, ушир. д, J=8.8 Гц), 5.90 (1H, ушир. т, J=5.2 Гц), 3.77-3.74 (4Н, м), 3.68-3.65 (4Н, м), 3.63-3.58 (6Н, м), 3.45-3.41 (2Н, м), 3.30 (3H, с), 2.99 (1H, с).
Сравнительный пример 22
Получение 1-(4-этинилфенил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевины
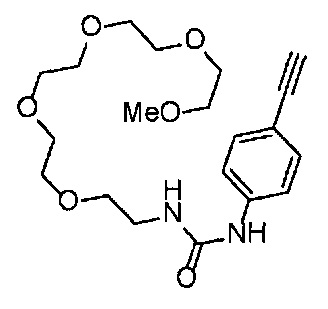
4-Нитрофенил хлорформиат (216.0 мг, 1.1 ммоль), дихлорметан (4 мл), пиридин (0.10 мл, 1.2 ммоль), 4-этиниланилин (103.5 мг, 0.88 ммоль) и 2,5,8,11,14-пентаоксагексадекан-16-амин (431.5 мг, 1.7 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 18, получая указанное в заголовке соединение (335.2 мг, 96%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.93 (1H, ушир. с), 7.44 (2Н, ушир. д, J=8.8 Гц), 7.38 (2Н, ушир. д, J=8.8 Гц), 5.99 (1Н, ушир. т, J=4.4 Гц), 3.78-3.73 (4Н, м), 3.71-3.68 (2Н, м), 3.67-3.64 (4Н, м), 3.63-3.58 (6Н, м), 3.45-3.41 (4Н, м), 3.27 (3H, с), 2.98 (1Н, с).
Сравнительный пример 23
Получение 1-(5-этинилпиридин-2-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевины
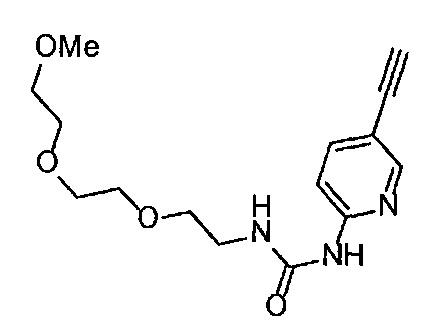
4-Нитрофенил хлорформиат (341.0 мг, 1.7 ммоль), дихлорметан (18 мл), 5-этинилпиридин-2-амин (200 мг, 1.7 ммоль), 1,4-диоксан (15 мл), триэтиламин (1.2 мл, 8.5 ммоль) и 2-[2-(2-метоксиэтокси)этокси]этан-1-амин (408.0 мг, 2.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (168.0 мг, 32%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.31 (1H, д, J=1.8 Гц), 7.71 (1H, дд, J=8.8, 1.8 Гц), 7.11 (1Н, д, J=8.8 Гц), 3.65-3.59 (8Н, м), 3.52-3.47 (4Н, м), 3.34 (4Н, ушир. с).
Сравнительный пример 24
Получение 1-(5-этинилпиридин-2-ил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевины
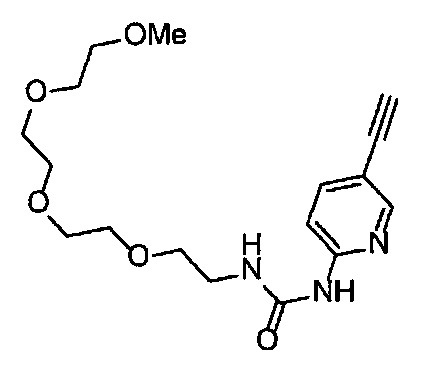
4-Нитрофенил хлорформиат (341.0 мг, 1.7 ммоль), дихлорметан (18 мл), 5-этинилпиридин-2-амин (200 мг, 1.7 ммоль), 1,4-диоксан (15 мл), триэтиламин (520 мкл, 1.9 ммоль) и 2,5,8,11-тетраоксатридекан-13-амин (526.0 мг, 2.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (284.0 мг, 48%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.31 (1Н, д, J=1.8 Гц), 7.72 (1H, дд, J=8.8, 1.8 Гц), 7.12 (1Н, д, J=8.8 Гц), 3.67-3.59 (12Н, м), 3.52-3.46 (4Н, м), 3.35 (1Н, с), 3.34 (3H, с).
Сравнительный пример 25
Получение 1-(5-этинилпиридин-2-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевины
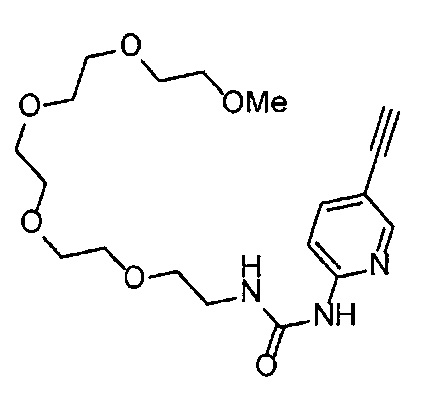
4-Нитрофенил хлорформиат (341.0 мг, 1.7 ммоль), дихлорметан (20 мл), 5-этинилпиридин-2-амин (200 мг, 1.7 ммоль), 1,4-диоксан (15 мл), триэтиламин (260 мкл, 1.9 ммоль) и 2,5,8,11,14-пентаоксагексадекан-16-амин (850.0 мг, 3.4 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (553.0 мг, 83%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.31 (1Н, д, J=1.8 Гц), 7.72 (1H, дд, J=8.8, 1.8 Гц), 7.12 (1Н, д, J=8.8 Гц), 3.66-3.59 (16Н, м), 3.53-3.46 (4Н, м), 3.35 (1H, с), 3.34 (3H, с).
Сравнительный пример 26
Получение 1-(6-этинилпиридин-3-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевины
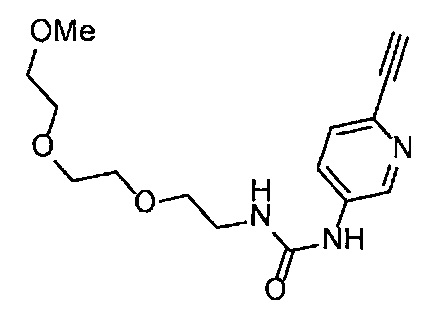
4-Нитрофенил хлорформиат (341.0 мг, 1.7 ммоль), тетрагидрофуран (18 мл), 6-этинилпиридин-3-амин (200 мг, 1.7 ммоль), 1,4-диоксан (15 мл), триэтиламин (260 мкл, 1.9 ммоль) и 2-[2-(2-метоксиэтокси)этокси]этан-1-амин (552.0 мг, 3.4 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (319.7 мг, 62%) в виде бледно-оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.37 (1H, д, J=2.4 Гц), 8.33 (1H, с), 8.12 (1H, дд, J=8.6, 2.4 Гц), 7.40 (1Н, д, J=8.6 Гц), 6.17 (1Н, ушир. с), 3.67-3.58 (10H, м), 3.44-3.43 (2Н, м), 3.40 (3H,с),3.13 (1H, с).
Сравнительный пример 27
Получение 1-(6-этинилпиридин-3-ил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевины
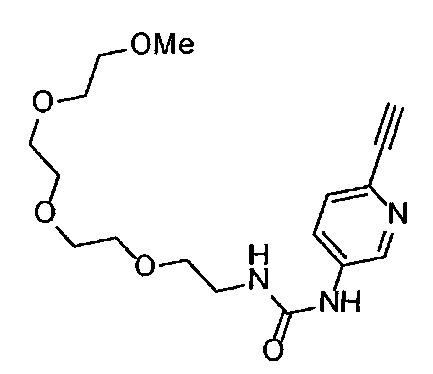
4-Нитрофенил хлорформиат (273.0 мг, 1.4 ммоль), тетрагидрофуран (15 мл), 6-этинилпиридин-3-амин (160 мг, 1.4 ммоль), 1,4-диоксан (15 мл), триэтиламин (260 мкл, 1.9 ммоль) и 2,5,8,11-тетраоксатридекан-13-амин (560.0 мг, 2.7 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (417.3 мг, 88%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.39 (1H, д, J=2.4 Гц), 8.17 (1H, дд, J=8.7, 2.4 Гц), 8.11 (1Н, с), 7.39 (1H, д, J=8.7 Гц), 6.13 (1Н, ушир. с), 3.76-3.74 (4Н, м), 3.68-3.66 (6Н, м), 3.62-3.59 (4Н, м), 3.46-3.42 (2Н, м), 3.31 (3H, с), 3.05 (1Н, с).
Сравнительный пример 28
Получение 1-(6-этинилпиридин-3-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевины
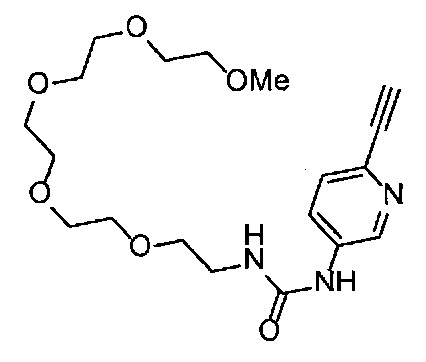
4-Нитрофенил хлорформиат (341.0 мг, 1.7 ммоль), тетрагидрофуран (18 мл), 6-этинилпиридин-3-амин (200.0 мг, 1.7 ммоль), 1,4-диоксан (15 мл), триэтиламин (260 мкл, 1.9 ммоль) и 2,5,8,11,14-пентаоксагексадекан-16-амин (850.0 мг, 3.4 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 16, получая указанное в заголовке соединение (585.0 мг, 88%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.43 (1H, д, J=2.4 Гц), 8.17-8.16 (2Н, м), 7.40 (1Н, д, J=8.4 Гц), 6.13 (1Н, ушир. с), 3.77-3.58 (18Н, м), 3.45-3.44 (2Н, м), 3.26 (3H, с), 3.06 (1Н, с).
Сравнительный пример 29
Получение 2-метоксиэтил (3-этинилфенил)карбамата
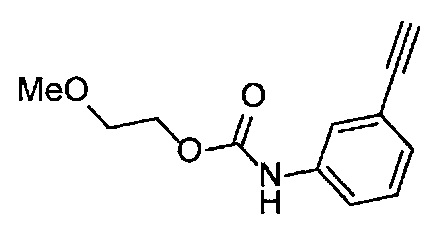
Ди(N-сукцинимидил)карбонат (1.34 г, 5.3 ммоль) и триэтиламин (1.39 мл, 10 ммоль) добавляли в раствор 2-метоксиэтанола (0.39 мл, 5.0 ммоль) в этилацетате (15 мл) при охлаждении на ледяной бане, перемешивали в течение 30 минут, и затем перемешивали при комнатной температуре в течение ночи. В полученный раствор затем добавляли 3-этиниланилин (0.62 мл, 5.5 ммоль) и перемешивали при кипячении в течение 5 часов. Растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 80/20→50/50), получая указанное в заголовке соединение (771.0 мг, 70%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.50 (1H, ушир. с), 7.39 (1Н, ушир. д, J=7.6 Гц), 7.25 (1H, ушир. т, J=7.6 Гц), 7.19 (1Н, ушир. д, J=7.6 Гц), 6.74 (1Н, ушир. с), 4.33 (2Н, ушир. т, J=4.4 Гц), 3.64 (2Н, ушир. т, J=4.4 Гц), 3.42 (3H, с), 3.06 (1Н, с).
Сравнительный пример 30
Получение 2-(2-метоксиэтокси)этил (3-этинилфенил)карбамата
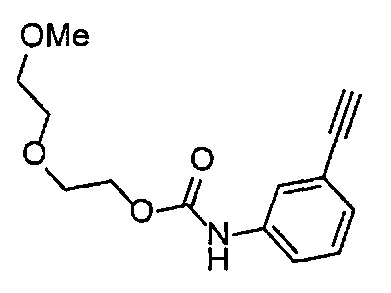
Монометиловый эфир диэтиленгликоля (0.59 мл, 5.0 ммоль), этилацетат (15 мл), ди(N-сукцинимидил)карбонат (1.34 г, 5.3 ммоль), триэтиламин (1.39 мл, 10 ммоль) и 3-этиниланилин (0.62 мл, 5.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (757.5 мг, 57%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.50 (1H, ушир. с), 7.39 (1H, ушир. д, J=7.6 Гц), 7.25 (1H, т, J=7.6 ГЦ), 7.18 (1Н, ушир. дт, J=7.6, 1.2 Гц), 6.75 (1H, ушир. с), 4.34 (2Н, ушир. т, J=4.4 Гц), 3.75 (2Н, ушир. т, J=4.4 Гц), 3.68-3.66 (2Н, м), 3.58-3.56 (2Н, м), 3.39 (3H, с), 3.05 (1H, с).
Сравнительный пример 31
Получение 2-[2-(2-метоксиэтокси)этокси]этил(3-этинилфенил)карбамата
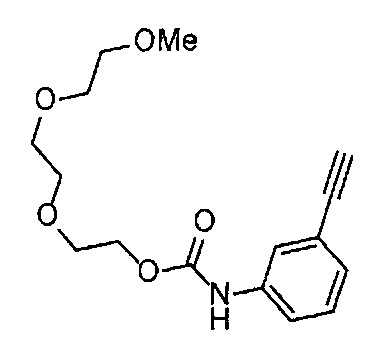
Монометиловый эфир триэтиленгликоля (0.78 мл, 5.0 ммоль), этилацетат (15 мл), ди(N-сукцинимидил)карбонат (1.34 г, 5.3 ммоль), триэтиламин (1.39 мл, 10 ммоль) и 3-этиниланилин (0.62 мл, 5.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (1.00 г, 68%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.51 (1H, ушир. с), 7.40 (1H, ушир. д, J=7.6 Гц), 7.25 (1H, т, J=7.6 Гц), 7.18 (1H, ушир. дт, J=7.6, 1.2 Гц), 6.91 (1H, ушир. с), 4.33 (2Н, ушир. т, J=4.4 Гц), 3.75 (2Н, ушир. т, J=4.4 Гц), 3.70-3.65 (6Н, м), 3.57-3.55 (2Н, м), 3.38 (3H, с), 3.05 (1H, с).
Сравнительный пример 32
Получение 2-метоксиэтил(4-этинилфенил)карбамата
2-Метоксиэтанол (385.9 мг, 5.1 ммоль), этилацетат (4 мл), ди(N-сукцинимидил) карбонат (1.35 г, 5.3 ммоль), триэтиламин (2.0 мл, 14 ммоль) и 4-этиниланилин (704.2 мг, 6.0 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (913.7 мг, 82%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.43 (2Н, ушир. д, J=8.4 Гц), 7.34 (2Н, ушир. д, J=8.4 Гц), 6.79 (1H, ушир. с), 4.34-4.32 (2Н, ушир. т, J=4.4 Гц), 3.65-3.62 (2Н, ушир. т, J=4.4 Гц), 3.41 (3H, с), 3.02 (1Н, с).
Сравнительный пример 33
Получение 2-(2-метоксиэтокси)этил(4-этинилфенил)карбамата
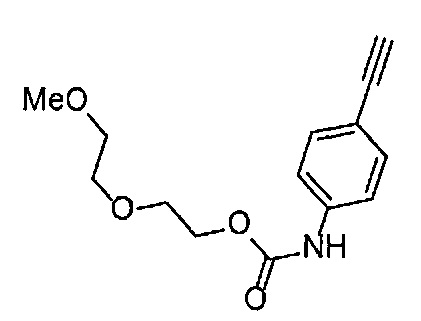
Монометиловый эфир диэтиленгликоля (600.6 мг, 5.0 ммоль), этилацетат (4 мл), ди(N-сукцинимидил)карбонат (1.35 г, 5.3 ммоль), триэтиламин (2 мл, 14 ммоль) и 4-этиниланилин (709.7 мг, 6.1 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (1.06 г, 80%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.44 (2Н, ушир. д, J=8.8 Гц), 7.34 (2Н, д, J=8.8 Гц), 6.77 (1H, ушир. с), 4.34 (2Н, ушир. т, J=4.8 Гц), 3.74 (2Н, ушир. т, J=4.8 Гц), 3.68-3.66 (2Н, м), 3.58-3.56 (2Н, м), 3.39 (3H, с), 3.02 (1H, с).
Сравнительный пример 34
Получение 2-[2-(2-метоксиэтокси)этокси]этил(4-этинилфенил)карбамата
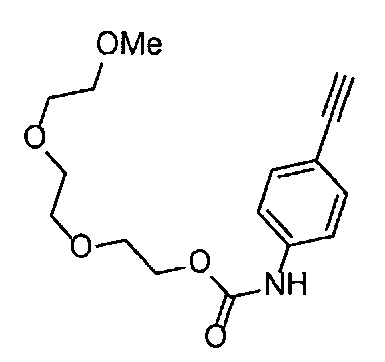
Монометиловый эфир триэтиленгликоля (824.3 мг, 5.0 ммоль), этилацетат (4 мл), ди(N-сукцинимидил)карбонат (1.37 г, 5.3 ммоль), триэтиламин (2.0 мл, 14 ммоль) и 4-этиниланилин (744.2 мг, 6.4 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (1.21 г, 79%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.43 (2Н, ушир. д, J=8.8 Гц), 7.35 (2Н, ушир. д, J=8.8 Гц), 6.93 (1Н, ушир. с), 4.33 (2Н, ушир. т, J=4.8 Гц), 3.74 (2Н, ушир. т, J=4.8 Гц), 3.71-3.69 (6Н, м), 3.57-3.55 (2Н, м), 3.38 (3H, с), 3.02 (1H, с).
Сравнительный пример 35
Получение 2,5,8,11-тетраоксатридекан-13-ил (4-этинилфенил)карбамата
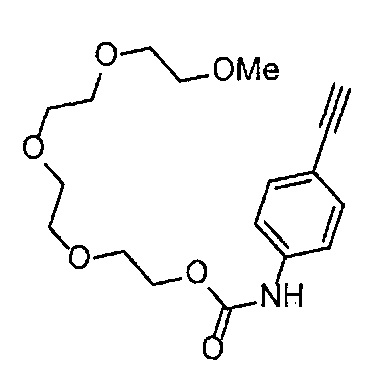
Монометиловый эфир тетраэтиленгликоля (1.53 г, 7.4 ммоль), этилацетат (10 мл), ди(N-сукцинимидил)карбонат (2.11 г, 8.2 ммоль), триэтиламин (3.0 мл, 22 ммоль) и 4-этиниланилин (1.12 г, 9.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (1.54 г, 60%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.43 (2Н, ушир. д, J=8.8 Гц), 7.36 (2Н, ушир. д, J=8.8 Гц), 7.10 (1H, ушир. с), 4.33 (2Н, ушир. т, J=4.8 Гц), 3.74 (2Н, ушир. т, J=4.8 Гц), 3.68-3.64 (10H, м), 3.57-3.54 (2Н, м), 3.37 (3H, с), 3.02 (1H, с).
Сравнительный пример 36
Получение 2,5,8,11,14-пентаоксагексадекан-16-ил (4-этинилфенил)карбамата
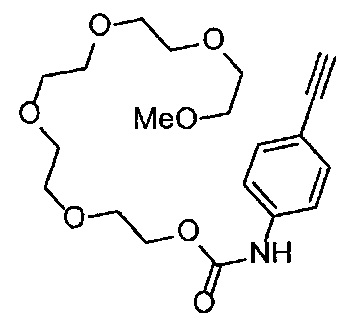
Монометиловый эфир пентаэтиленгликоля (1.54 г, 6.1 ммоль), этилацетат (10 мл), ди(N-сукцинимидил)карбонат (1.77 г, 6.9 ммоль), триэтиламин (2.6 мл, 19 ммоль), 4-этиниланилин (962.2 мг, 8.2 ммоль) и 4-диметиламинопиридин (78.5 мг, 0.64 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 29, получая указанное в заголовке соединение (1.20 г, 50%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.43 (2Н, ушир. д, J=8.8 Гц), 7.37 (2Н, ушир. д, J=8.8 Гц), 7.14 (1Н, ушир. с), 4.33 (2Н, ушир. т, J=4.8 Гц), 3.74 (2Н, ушир. т, J=4.8 Гц), 3.67-3.63 (14Н, м), 3.55-3.52 (2Н, м), 3.36 (3H, с), 3.02 (1H, с).
Сравнительный пример 37
Получение 3,6,9,12-тетраоксатридекановой кислоты
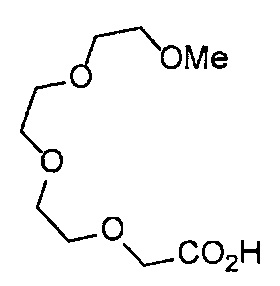
Монометиловый эфир тетраэтиленгликоля (3.04 г, 19 ммоль) добавляли в раствор 60%-ного гидрида натрия (1.37 г, 34 ммоль) в тетрагидрофуране (10 мл) при охлаждении на ледяной бане и перемешивали в течение 20 минут. В полученный раствор прикапывали трет-бутил бромацетат (4.31 г, 22 ммоль) при охлаждении на ледяной бане и перемешивали при комнатной температуре в течение 13 часов. В полученный раствор затем добавляли гидроксид лития (1.73 г, 72 ммоль) и воду (15 мл) и перемешивали при кипячении в течение 4 часов. Полученный раствор разбавляли водой и промывали диэтиловым эфиром. Доводили pH водного слоя до 1 6н. раствором соляной кислоты, и экстрагировали смесью дихлорметан/метанол (об./об.) = 10/1. Органические слои объединяли, затем сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (2.13 г, 52%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 4.16 (2Н, с), 3.78-3.75 (2Н, м), 3.70-3.63 (8Н, м), 3.59-3.57 (2Н, м), 3.39 (3H, с).
Сравнительный пример 38
Получение 3,6,9,12,15-пентаоксагексадекановой кислоты
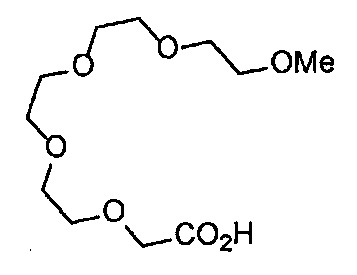
60%-ный гидрид натрия (2.20 г, 54.6 ммоль), тетрагидрофуран (29 мл), монометиловый эфир тетраэтиленгликоля (3.80 г, 18.2 ммоль), трет-бутил бромацетат (2.93 мл, 20.0 ммоль), гидроксид лития (1.30 г, 54.6 ммоль) и воду (3.6 мл) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 37, получая указанное в заголовке соединение (2.79 г, 58%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 4.16 (2Н, с), 3.77-3.56 (16Н, м), 3.39 (3H, с).
Сравнительный пример 39
Получение 3,6,9,12,15,18-гексаоксанонадекановой кислоты
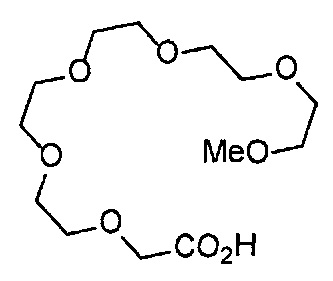
60%-ный гидрид натрия (600 мг, 15 ммоль), тетрагидрофуран (8 мл), монометиловый эфир пентаэтиленгликоля (1.26 г, 5.0 ммоль), трет-бутил бромацетат (0.81 мл, 5.5 ммоль), гидроксид лития (359.3 мг, 15 ммоль) и воду (1 мл) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 37, получая указанное в заголовке соединение (1.38 г, 89%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 4.17 (2Н, с), 3.75-3.55 (20Н, м), 3.38 (3H, с).
Сравнительный пример 40
Получение N-(3-этинилфенил)-2,5,8,11,14,17-гексаоксанонадекан-19-амида
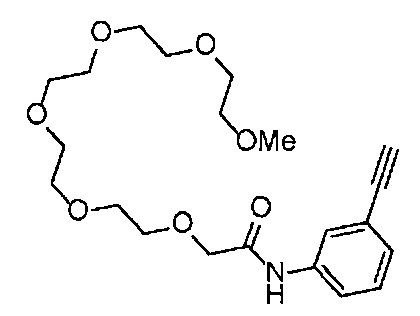
1-Этил-3-[3-(диметиламино)пропил]карбодиимид гидрохлорид (238.5 мг, 1.2 ммоль), 4-диметиламинопиридин (161.1 мг, 1.3 ммоль) и 3-этиниланилин (116.3 мг, 0.99 ммоль) добавляли в раствор 3,6,9,12,15,18-гексаоксанонадекановой кислоты (214.4 мг, 0.70 ммоль) в дихлорметане (3 мл) и перемешивали при комнатной температуре в течение 3 часов. Полученную реакционную смесь разбавляли этилацетатом, промывали 1н. раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, и затем сушили безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (218.2 мг, 77%) в виде оранжевого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.89 (1Н, с), 7.74 (1Н, т, J=1.6 Гц), 7.67 (1H, дт, J=8.0, 1.6 Гц), 7.28 (1Н, дт, J=8.0 Гц), 7.24 (1Н, дт, J=8.0, 1.6 Гц), 4.10 (2Н, с), 3.77-3.75 (2Н, м), 3.72-3.71 (4Н, м), 3.69-3.67 (2Н, м), 3.63-3.59 (10H, м), 3.53-3.51 (2Н, м), 3.36 (3H, с), 3.10 (1Н, с).
Сравнительный пример 41
Получение N-(4-этинилфенил)-2,5,8,11-тетраоксатридекан-13-амида
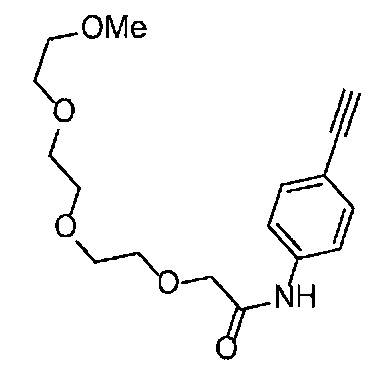
3,6,9,12-Тетраоксатридекановую кислоту (488.9 мг, 2.2 ммоль), дихлорметан (3 мл), 1-этил-3-[3-(диметиламино)пропил]карбодиимид гидрохлорид (766.8 мг, 4.0 ммоль), 4-диметиламинопиридин (366.4 мг, 6.0 ммоль) и 4-этиниланилин (234.3 мг, 2.0 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 40, получая указанное в заголовке соединение (257.8 мг, 40%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.88 (1H, ушир. с), 7.61 (2Н, ушир. д, J=8.4 Гц), 7.45 (2Н, ушир. д, J=8.4 Гц), 4.11 (2Н, с), 3.77-3.60 (10H, м), 3.52-3.49 (2Н, м), 3.34 (3H, с), 3.04 (1Н, с).
Сравнительный пример 42
Получение N-(4-этинилфенил)-2,5,8,11,14-пентаоксагексадекан-16-амида
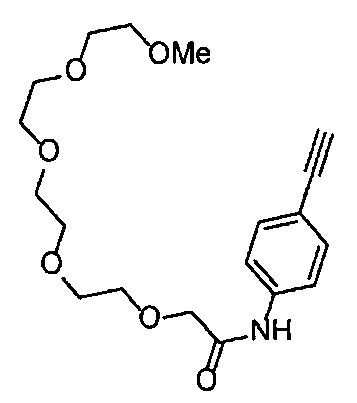
3,6,9,12,15-Пентаоксагексадекановую кислоту (878.8 мг, 3.3 ммоль), дихлорметан (5 мл), 1-этил-3-[3-(диметиламино)пропил]карбодиимид гидрохлорид (1.15 г, 6.0 ммоль), 4-диметиламинопиридин (1.47 г, 12 ммоль) и 4-этиниланилин (351.5 мг, 3.0 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 40, получая указанное в заголовке соединение (408.3 мг, 37%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.88 (1Н, ушир. с), 7.61 (2Н, ушир. д, J=8.4 Гц), 7.45 (2Н, ушир. д, J=8.4 Гц), 4.11 (2Н, с), 3.77-3.59 (14Н, м), 3.52-3.50 (2Н, м), 3.36 (3H, с), 3.04 (1Н, с).
Сравнительный пример 43
Получение N-(4-этинилфенил)-2,5,8,11,14,17-гексаоксанонадекан-19-амида
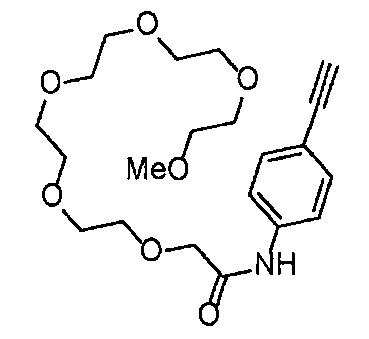
3,6,9,12,15,18-Гексаоксанонадекановую кислоту (220.3 мг, 0.71 ммоль), дихлорметан (3 мл), 4-этиниланилин (109.8 мг, 0.94 ммоль), 1-этил-3-[3-(диметиламино) пропил]карбодиимид гидрохлорид (284.4 мг, 1.5 ммоль) и 4-диметиламинопиридин (173.5 мг, 1.4 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 40, получая указанное в заголовке соединение (199.6 мг, 69%) в виде красного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.02 (1H, ушир. с), 7.64 (2Н, ушир. д, J=8.4 Гц), 7.44 (2Н, ушир. д, J=8.4 Гц), 4.10 (2Н, с), 3.77-3.74 (2Н, м), 3.72-3.70 (4Н, м), 3.68-3.66 (2Н, м), 3.62-3.59 (10H, м), 3.52-3.50 (2Н, м), 3.35 (3H, с), 3.11 (1Н, с).
Сравнительный пример 44
Получение N-(5-этинилпиридин-2-ил)-2,5,8,11-тетраоксатридекан-13-амида
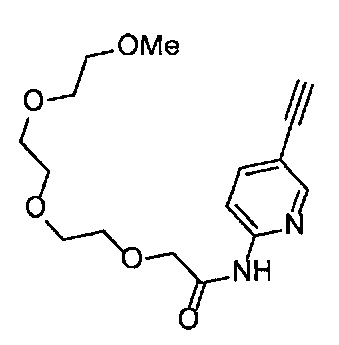
1-Этил-3-[3-(диметиламино)пропил]карбодиимид гидрохлорид (526.1 мг, 2.7 ммоль), 4-диметиламинопиридин (664.6 мг, 5.4 ммоль), дихлорметан (5 мл) и 5-этинилпиридин-2-амин (239.6 мг, 2.0 ммоль) добавляли в 3,6,9,12-тетраоксатридекановую кислоту (320.1 мг, 1.4 ммоль) и перемешивали при комнатной температуре в течение ночи. Полученную реакционную смесь разбавляли этилацетатом, промывали 10%-ным водным раствором лимонной кислоты, сушили безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 1/4), получая указанное в заголовке соединение (199.4 мг, 43%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.24 (1Н, ушир. с), 8.42 (1Н, ушир. д, J=2.4 Гц), 8.23 (1H, д, J=8.8 Гц), 7.79 (1Н, дд, J=8.8, 2.4 Гц), 4.16 (2Н, с), 3.79-3.77 (2Н, м), 3.74-3.72 (6Н, м), 3.66-3.63 (2Н, м), 3.55-3.52 (2Н, м), 3.37 (3H, с), 3.16 (1Н, с).
Сравнительный пример 45
Получение N-(5-этинилпиридин-2-ил)-2,5,8,11,14-пентаоксагексадекан-16-амида
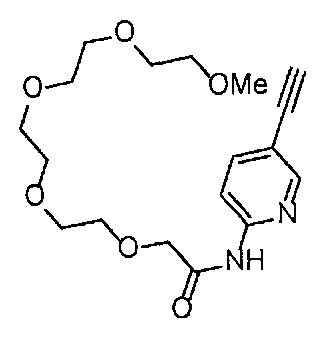
3,6,9,12,15-Пентаоксангексадекановую кислоту (427.8 мг, 1.6 ммоль), N,N-диметилформамид (5 мл), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1.24 г, 3.3 ммоль), N,N-диизопропилэтиламин (1.2 мл, 6.9 ммоль) и 5-этинилпиридин-2-амин (181 мг, 1.5 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 44, получая указанное в заголовке соединение (140.2 мг, 25%) в виде красного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.28 (1Н, ушир. с), 8.41 (1Н, с), 8.21 (1Н, д, J=8.4 Гц), 7.78 (1H, ушир. д, J=8.4 Гц), 4.18 (2Н, с), 3.79-3.77 (2Н, м), 3.74-3.71 (6Н, м), 3.66-3.63 (6Н, м), 3.55-3.52 (2Н, м), 3.36 (3H, с), 3.22 (1Н, с).
Сравнительный пример 46
Получение N-(5-этинилпиридин-2-ил)-2,5,8,11,14,17-гексаоксанонадекан-19-амида
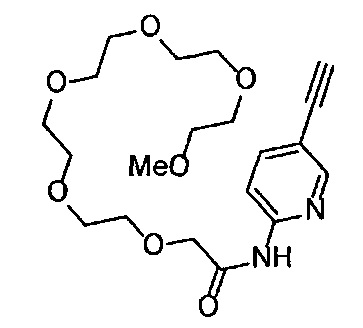
3,6,9,12,15,18-Гексаоксанонадекановую кислоту (315.0 мг, 1.0 ммоль), дихлорметан (1 мл), N,N-диметилформамид (3 мл), 1-этил-3-[3-(диметиламино)пропил] карбодиимид гидрохлорид (413.0 мг, 2.2 ммоль), 4-диметиламинопиридин (497.0 мг, 4.1 ммоль) и 5-этинилпиридин-2-амин (239.0 мг, 2.0 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 44, получая указанное в заголовке соединение (266.6 мг, 64%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.27 (1Н, ушир. с), 8.41 (1H, ушир. д, J=2.0 Гц), 8.23 (1H, ушир. д, J=8.8 Гц), 7.79 (1Н, дд, J=8.8, 2.0 Гц), 4.16 (2Н, с), 3.78-3.63 (18Н, м), 3.55-3.54 (2Н, м), 3.37 (3H, с), 3.16 (1Н, с).
Сравнительный пример 47
Получение N-(6-этинилпиридин-3-ил)-2,5,8,11-тетраоксатридекан-13-амида
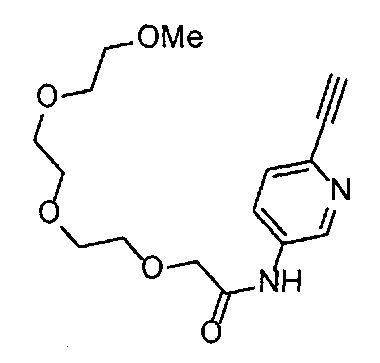
3,6,9,12-Тетраоксатридекановую кислоту (446.5 мг, 2.0 ммоль), 1-этил-3-[3-(диметиламино)пропил]карбодиимид гидрохлорид (776.0 мг, 4.0 ммоль), дихлорметан (10 мл), триэтиламин (1.1 мл, 7.9 ммоль), 6-этинилпиридин-3-амин (255.2 мг, 2.2 ммоль) и 4-диметиламинопиридин (30.5 мг, 0.25 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 44, получая указанное в заголовке соединение (96.7 мг, 15%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.16 (1Н, ушир. с), 8.66 (1Н, ушир. с), 8.28 (1H, дд, J=8.4, 2.4 Гц), 7.47 (1H, ушир. д, J=8.4 Гц), 4.14 (2Н, с), 3.77-3.63 (10H, м), 3.51-3.49 (2Н, м), 3.32 (3H, с), 3.14 (1Н, с).
Сравнительный пример 48
Получение 2-[2-(2-метоксиэтокси)этокси]этил-4-метилбензолсульфоната
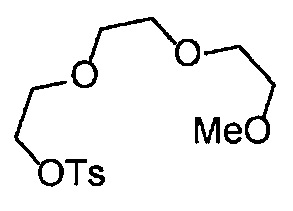
Триэтиламин (1.27 мл, 9.15 ммоль) и монометиловый эфир триэтиленгликоля (525 мкл, 3.05 ммоль) добавляли в раствор п-толуолсульфонил хлорида (872.2 мг, 4.58 ммоль) в дихлорметане (3 мл) и перемешивали при комнатной температуре в течение ночи. В полученный раствор затем добавляли метанол (0.5 мл), перемешивали при комнатной температуре в течение 1 часа, и растворитель отгоняли при пониженном давлении. Добавляли в полученный остаток диэтиловый эфир, нерастворимые фракции отфильтровывали и промывали диэтиловым эфиром и изопропанолом. Фильтрат упаривали при пониженном давлении, добавляли в остаток воду, и экстрагировали дихлорметаном. Органические слои объединяли, затем сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (796.9 мг, 82%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.80 (2Н, д, J=8.2 Гц), 7.34 (2Н, д, J=8.2 Гц), 4.16 (2Н, ушир. т, J=4.8 Гц), 3.70-3.68 (2Н, м), 3.62-3.59 (6Н, м), 3.54-3.52 (2Н, м), 3.37 (3H, с), 2.45 (3H, с).
Сравнительный пример 49
Получение 2,5,8,11-тетраоксатридекан-13-ил 4-метилбензолсульфоната
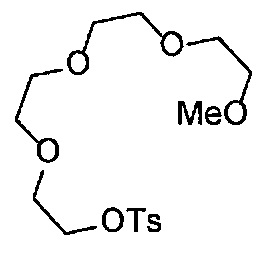
п-Толуолсульфонил хлорид (7.19 г, 37.7 ммоль), дихлорметан (25 мл), триэтиламин (10.5 мл, 75.3 ммоль) и монометиловый эфир тетраэтиленгликоля (5 мл, 25.1 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 48, получая указанное в заголовке соединение (8.24 г, 90%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.80 (2Н, д, J=8.2 Гц), 7.34 (2Н, д, J=8.2 Гц), 4.16 (2Н, ушир. т, J=5.0 Гц), 3.70-3.68 (2Н, м), 3.65-3.62 (6Н, м), 3.59-3.57 (4Н, м), 3.55-3.53 (2Н, м), 3.37 (3H, с), 2.45 (3H, с).
Сравнительный пример 50
Получение 2,5,8,11,14-пентаоксагексадекан-16-ил 4-метилбензолсульфоната
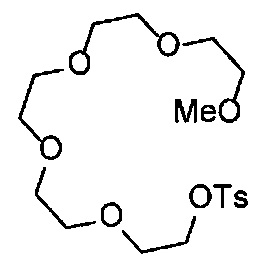
п-Толуолсульфонил хлорид (3.06 г, 16.1 ммоль), дихлорметан (12 мл), триэтиламин (4.5 мл, 32.1 ммоль) и монометиловый эфир пентаэтиленгликоля (2.5 мл, 10.7 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 48, получая указанное в заголовке соединение (3.14 г, 72%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.80 (2Н, д, J=8.2 Гц), 7.34 (2Н, д, J=8.2 Гц), 4.16 (2Н, ушир. т, J=4.4 Гц), 3.70-3.58 (16Н, м), 3.55-3.54 (2Н, м), 3.37 (3H, ушир. с), 2.45 (3H, с).
Сравнительный пример 51
Получение 3-этинил-N-(2-метоксиэтил)анилина
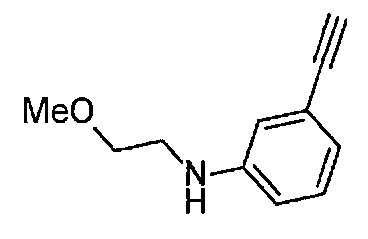
3-Этиниланилин (288 мкл, 2.6 ммоль) и 1-бром-2-метоксиэтан (360 мкл, 3.8 ммоль) добавляли в раствор карбоната цезия (1.37 г, 4.2 ммоль) и иодида калия (84.0 мг, 0.52 ммоль) в N,N-диметилформамиде (8 мл) и перемешивали при 100°C в течение ночи. Полученный раствор разбавляли водой, экстрагировали этилацетатом. Органический слой промывали водой, сушили безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан→гексан/этилацетат (об./об.) = 96/4), получая указанное в заголовке соединение (132.2 мг, 29%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.11 (1Н, т, J=7.5 Гц), 6.85 (1Н, ушир. д, J=7.5 Гц), 6.74 (1H, т, J=2.2 Гц), 6.62 (1Н, т, J=7.5, 2.2 Гц), 4.06 (1H, ушир. с), 3.59 (2Н, т, J=5.2 Гц), 3.38 (3H, с), 3.27 (2Н, т, J=5.2 Гц), 3.00 (1Н, с).
Сравнительный пример 52
Получение 3-этинил-N-[2-(2-метоксиэтокси)этил]анилина
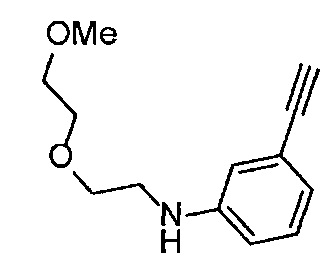
Карбонат цезия (1.37 г, 4.2 ммоль), иодид калия (84.0 мг, 0.52 ммоль), N,N-диметилформамид (8 мл), 3-этиниланилин (288 мкл, 2.6 ммоль) и 1-бром-2-(2-метоксиэтокси)этан (517 мкл, 3.8 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 51, получая указанное в заголовке соединение (155.0 мг, 28%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.10 (1H, т, J=7.6 Гц), 6.84 (1H, ушир. д, J=7.6 Гц), 6.74 (1H, т, J=2.2 Гц), 6.62 (1H, ушир. дд, J=7.6, 2.2 Гц), 4.16 (1H, ушир. с), 3.70 (2Н, т, J=5.2 Гц), 3.65-3.63 (2Н, м), 3.57-3.54 (2Н, м), 3.40 (3H, с), 3.31-3.27 (2Н, м), 3.00 (1H, с).
Сравнительный пример 53
Получение 3-этинил-N-{2-[2-(2-метоксиэтокси)этокси]этил}анилина
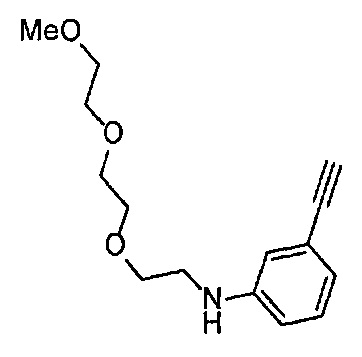
Карбонат калия (194.0 мг, 1.4 ммоль), иодид калия (29.0 мг, 0.18 ммоль) и 3-этиниланилин (96 мкл, 0.85 ммоль) добавляли в раствор 2-[2-(2-метоксиэтокси)этокси] этил-4-метилбензолсульфоната (223.0 мг, 0.7 ммоль) в ацетонитриле (4 мл) и перемешивали при кипячении в течение ночи. Нерастворимые фракции отфильтровывали из полученного раствора, и затем фильтрат упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 60/40→40/60), получая указанное в заголовке соединение (100.2 мг, 54%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.10 (1H, т, J=7.9 Гц), 6.84 (1Н, ушир. д, J=7.9 Гц), 6.74 (1Н, т, J=2.2 Гц), 6.62 (1H, ушир. дд, J=7.9, 2.2 Гц), 4.20 (1Н, ушир. с), 3.70 (2Н, т, J=5.2 Гц), 3.67-3.64 (6Н, м), 3.57-3.55 (2Н, м), 3.39 (3H, с), 3.28 (2Н, ушир. м), 3.00 (1H, с).
Сравнительный пример 54
Получение 4-этинил-N-[2-(2-метоксиэтокси)этил]анилина
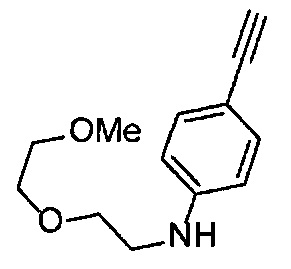
Карбонат цезия (1.37 г, 4.2 ммоль), иодид калия (84.0 мг, 0.52 ммоль), N,N-диметилформамид (8 мл), 4-этиниланилин (300.0 мг, 2.6 ммоль) и 1-бром-2-(2-метоксиэтокси)этан (777 мкл, 5.8 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 51, получая указанное в заголовке соединение (100.0 мг, 18%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.30 (2Н, д, J=8.8 Гц), 6.52 (1H, ушир. с=8.8 Гц), 4.33 (1Н, ушир. с), 3.69 (2Н, т, J=5.2 Гц), 3.64-3.62 (2Н, м), 3.56-3.54 (2Н, м), 3.39 (3H, с), 3.30-3.29 (2Н, м), 2.95 (1Н, с).
Сравнительный пример 55
Получение 4-этинил-N-{2-[2-(2-метоксиэтокси)этокси]этил}анилина
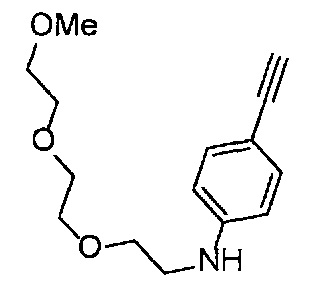
2-[2-(2-Метоксиэтокси)этокси]этил 4-метилбензолсульфонат (669.0 мг, 2.1 ммоль), ацетонитрил (12 мл), карбонат калия (582.0 мг, 4.2 ммоль), иодид калия (87.0 мг, 0.52 ммоль) и 4-этиниланилин (300.0 мг, 2.6 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 53, получая указанное в заголовке соединение (224.9 мг, 40%) в виде коричневого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.30 (2Н, д, J=8.8 Гц), 6.53 (1H, ушир. с=8.8 Гц), 4.36 (1Н, ушир. с), 3.70 (2Н, т, J=5.2 Гц), 3.65-3.64 (6Н, м), 3.57-3.54 (2Н, м), 3.38 (3H, с), 3.32-3.28 (2Н, м), 2.95 (1H, с).
Сравнительный пример 56
Получение N-(4-этинилфенил)-2,5,8,11-тетраоксатридекан-13-амина
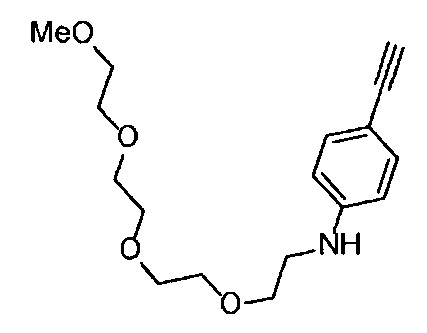
2,5,8,11-Тетраоксатридекан-13-ил 4-метилбензолсульфонат (1.00 г, 2.8 ммоль), ацетонитрил (20 мл), карбонат калия (1.40 г, 10 ммоль), иодид калия (227 мг, 1.4 ммоль) и 4-этиниланилин (800.0 мг, 6.8 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 53, получая указанное в заголовке соединение (446.0 мг, 53%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.30 (2Н, д, J=8.8 Гц), 6.53 (1H, д, J=8.8 Гц), 4.36 (1H, ушир. с), 3.70 (2Н, т, J=5.2 Гц), 3.66-3.63 (10H, м), 3.55-3.53 (2Н, м), 3.37 (3H, с), 3.32-3.28 (2Н, м), 2.95 (1Н, с).
Сравнительный пример 57
Получение N-(4-этинилфенил)-2,5,8,11,14-пентаоксагексадекан-16-амина
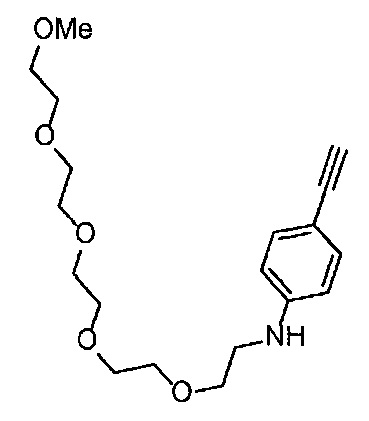
2,5,8,11,14-Пентаоксагексадекан-16-ил 4-метилбензолсульфонат (694.3 мг, 1.7 ммоль), ацетонитрил (12 мл), карбонат калия (885.0 мг, 6.4 ммоль), иодид калия (141.8 мг, 0.85 ммоль) и 4-этиниланилин (500.0 мг, 4.3 ммоль) использовали в качестве исходных веществ и проводили реакцию таким же образом, как в Сравнительном примере 53, получая указанное в заголовке соединение (104.0 мг, 17%) в виде желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.30 (2Н, д, J=8.8 Гц), 6.53 (2Н, д, J=8.8 Гц), 4.39 (1H, ушир. с), 3.70 (2Н, т, J=5.2 Гц), 3.66-3.62 (14Н, м), 3.55-3.52 (2Н, м), 3.37 (3H, с), 3.31-3.28 (2Н, м), 2.95 (1H, с).
Сравнительный пример 58
Получение 2,2'-дитиосалицил дихлорида
Оксалилхлорид (25 мл, 291.5 ммоль) и N,N-диметилформамид (150 мкл, 1.94 ммоль) добавляли в раствор 2,2'-дитиосалициловой кислоты (25.0 г, 81.6 ммоль) в дихлорметане (220 мл) и перемешивали при комнатной температуре в течение 18 часов. Полученный раствор перемешивали при 50°C в течение 24 часов, и затем растворитель отгоняли при пониженном давлении. Полученное твердое вещество промывали гексаном при 0°C и сушили, получая указанное в заголовке соединение (24.9 г, 89%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.40 (2Н, дд, J=8.0, 1.4 Гц), 7.77 (2Н, дд, J=8.0, 1.1 Гц), 7.55 (2Н, тд, J=8.0, 1.4 Гц), 7.24 (2Н, тд, J=8.0, 1.1 Гц).
Сравнительный пример 59
Получение 2,2'-дитиобис(N-метилбензамида)
Метиламин (2М раствор в тетрагидрофуране) (80 мл, 160.2 ммоль) добавляли в раствор 2,2'-дитиосалицил дихлорида (12.5 г, 36.4 ммоль) в тетрагидрофуране (56 мл) при 0°C и перемешивали при комнатной температуре в течение 16 часов. В полученный раствор добавляли воду, перемешивали 30 минут, и затем растворитель отгоняли при пониженном давлении. Полученное твердое вещество промывали водой и сушили, получая указанное в заголовке соединение (11.7 г, 96%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.75 (2Н, ушир. д, J=7.6 Гц), 7.47 (2Н, дд, J=7.6, 1.2 Гц), 7.36 (2Н, тд, J=7.6, 1.2 Гц), 7.24 (2Н, тд, J=7.6, 1.2 Гц), 6.13 (2Н, ушир. м), 2.97 (6Н, д, J=4.8 Гц).
Сравнительный пример 60
Получение 2-меркапто-N-метилбензамида
Боргидрид натрия (3.05 г, 80.7 ммоль) добавляли в раствор 2,2'-дитиобис(N-метилбензамида) (11.7 г, 35.1 ммоль) в этаноле (110 мл) при 0°C и перемешивали при комнатной температуре в течение 16 часов. Полученный раствор разбавляли водой, доводили значение pH до 1 2н. раствором соляной кислоты, и затем растворитель отгоняли при пониженном давлении. Добавляли в остаток воду и экстрагировали этилацетатом. Органический слой сушили безводным сульфатом магния, растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (7.72 г, 66%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.42 (1Н, дд, J=7.8, 1.5 Гц), 7.32 (1H, дд, J=7.8, 1.3 Гц), 7.25 (1Н, тд, J=7.8, 1.5 Гц), 7.13 (1Н, тд, J=7.8, 1.3 Гц), 6.12 (1H, ушир. м), 4.77 (1H, с), 2.99 (3H, д, J=4.8 Гц).
Сравнительный пример 61
Получение 6-иод-1Н-индазола
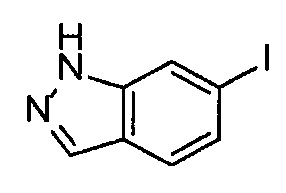
Концентрированную соляную кислоту (35 мл, 420 ммоль) и водный раствор (30 мл) нитрита натрия (6.64 г, 96 ммоль) добавляли в суспензию, полученную путем добавления воды (30 мл) к 6-аминоиндазолу (10.4 г, 78 ммоль) при 0°C, и перемешивали при 0°C в течение 30 минут. Затем в результирующий раствор добавляли водный раствор (30 мл) иодида калия (15.91 г, 96 ммоль) при 0°C, перемешивали при комнатной температуре в течение 30 минут, в полученный раствор затем добавляли дихлорметан (80 мл) и перемешивали при 40°C в течение 2 часов. Полученную реакционную смесь охлаждали до 0°C, затем доводили pH до 14 3н. водным раствором гидроксида натрия, и осадок отделяли фильтрованием. Полученный осадок промывали 10%-ным раствором тиосульфата натрия, растворенного в тетрагидрофуране, и затем добавляли силикагель. После перемешивания при комнатной температуре в течение 1 часа добавляли гексан (600 мл) и фильтровали. Остаток дважды промывали раствором ТГФ/гексан (1/3 (об./об.)), затем растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (15.23 г, 80%) в виде оранжевого порошка.
1H ЯМР (400 МГц, CDCl3) δ 10.24 (1Н, ушир. с), 8.04 (1H, ушир. с), 7.92 (1Н, ушир. с), 7.51 (1H, ушир. д, J=8.4 Гц), 7.46 (1H, дд, J=8.4, 1.2 Гц).
Сравнительный пример 62
Получение 2-{(1H-индазол-6-ил)тио}-N-метилбензамида
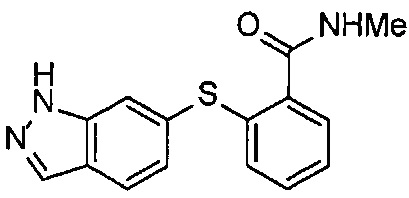
N,N-Диметилформамид (25 мл) добавляли к 6-иод-1Н-индазолу (5.51 г, 22 ммоль), 2-меркапто-N-метилбензамиду (5.16 г, 31 ммоль), Pd2(dba)3 (1.02 г, 1.1 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантену (1.46 г, 2.5 ммоль) и моногидрату гидроксида цезия (5.67 г, 33 ммоль) и перемешивали в атмосфере аргона при 100°C в течение 4.5 часов. Растворитель отгоняли из реакционной смеси при пониженном давлении, остаток растворяли в этилацетате и промывали водой и насыщенным раствором хлорида натрия. Органический слой сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 1/3), получая указанное в заголовке соединение (6.14 г, 96%») в виде бледно-оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.13 (1Н, ушир. с), 8.36 (1Н, ушир. кв, J=4.4 Гц), 8.10 (1Н, с), 7.78 (1Н, ушир. д, J=8.4 Гц), 7.59 (1Н, ушир. с), 7.48-7.46 (1Н, м), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.25 (1H, тд, J=7.6, 1.6 Гц), 7.08 (1Н, дд, J=8.4, 1.6 Гц), 6.99-6.97 (1Н, м), 2.76 (3H, д, J=4.4 Гц).
Сравнительный пример 63
Получение 2-{(3-иод-1H-индазол-6-ил)тио}-N-метилбензамида
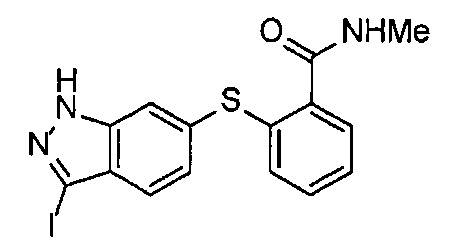
Раствор иода (11.09 г, 46 ммоль) в N,N-диметилформамиде (20 мл) добавляли в раствор 2-{(1Н-индазол-6-ил)тио}-N-метилбензамида (7.32 г, 26 ммоль) и карбоната калия (7.36 г, 53 ммоль) в N,N-диметилформамиде (30 мл) при 0°C в течение 30 минут и перемешивали при комнатной температуре в течение 3.5 часов. Растворитель отгоняли из реакционной смеси при пониженном давлении, затем остаток растворяли в этилацетате и промывали 10%-ным водным раствором тиосульфата натрия, насыщенным водным раствором бикарбоната натрия и насыщенным раствором хлорида натрия. Органический слой сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученное твердое вещество промывали раствором гексан/этилацетат (1/1 (об./об.)) и сушили, получая указанное в заголовке соединение (7.60 г, 72%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.55 (1H, ушир. с), 8.37 (1H, ушир. кв, J=4.4 Гц), 7.56 (1Н, ушир. с), 7.49-7.47 (1H, м), 7.44 (1Н, ушир. д, J=8.8 Гц), 7.31 (1H, тд, J=7.6, 2.0 Гц), 7.28 (1Н, тд, J=7.6, 2.0 Гц), 7.14 (1H, дд, J=8.8, 1.6 Гц), 7.04-7.02 (1Н, м), 2.76 (3H, д, J=4.4 Гц).
Сравнительный пример 64
Получение моно-трет-бутилдиметилсилильного простого эфира триэтиленгликоля
Триэтиленгликоль (2.2 мл, 16.5 ммоль) добавляли в раствор 60%-ного гидрида натрия (713 мг, 17.8 ммоль) в тетрагидрофуране (88 мл) при 0°C и перемешивали при комнатной температуре в течение 40 минут. В полученный раствор добавляли трет-бутилдиметилхлорсилан (2.74 г, 18.2 ммоль), перемешивали при комнатной температуре в течение 23 часов, затем добавляли воду, и экстрагировали дихлорметаном. Органический слой сушили безводным сульфатом натрия, затем растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (4.19 г, колич.) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.79-3.72 (4Н, м), 3.67-3.54 (8Н, м), 0.90 (9Н, с), 0.07 (6Н, с).
Сравнительный пример 65
Получение 2-(2,2,3,3-тетраметил-4,7,10-триокса-3-силадодекан-12-ил)изоиндолин-1,3-диона
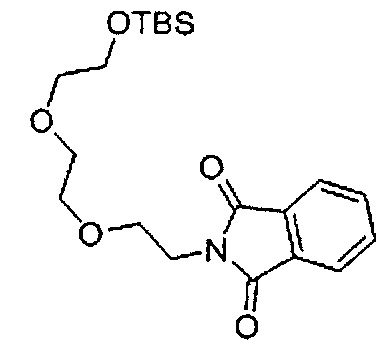
Моно-трет-бутилдиметилсилильный простой эфир триэтиленгликоля (1.3 г, 4.92 ммоль) и диэтил азодикарбоксилат (2.2 моль/л, толуольный раствор) (2.4 мл, 5.3 ммоль) добавляли в раствор фталимида (1.15 г, 7.82 ммоль) и трифенилфосфина (2.06 г, 7.85 ммоль) в тетрагидрофуране (50 мл) при комнатной температуре и перемешивали в течение ночи. Затем добавляли этанол (20 мл), перемешивали при комнатной температуре в течение 30 минут, и затем растворитель отгоняли при пониженном давлении. В полученный остаток добавляли этилацетат (12.5 мл) и гексан (12.5 мл), и нерастворимые фракции- отфильтровывали. Фильтрат упаривали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат (об./об.) = 5/95→0/100), получая указанное в заголовке соединение (822 мг, 42%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.84 (2Н, дд, J=5.2, 3.2 Гц), 7.71 (2Н, дд, J=5.2, 3.2 Гц), 3.90 (2Н, т, J=6.0 Гц), 3.74 (2Н, т, J=6.0 Гц), 3.70 (2Н, т, J=5.6 Гц), 3.65-3.59 (4Н, м), 3.50 (2Н, т, J=5.6 Гц), 0.87 (с, 9Н), 0.04 (с, 6Н).
Сравнительный пример 66
Получение 2-[2-(2-аминоэтокси)этокси] этанола
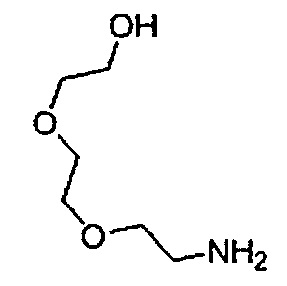
Гидразина моногидрат (322.6 мкл, 9.2 ммоль) добавляли в раствор 2-(2,2,3,3-тетраметил-4,7,10-триокса-3-силадодекан-12-ил)изоиндолин-1,3-диона (822 мг, 2.09 ммоль) в этаноле (120 мл) и перемешивали при кипячении в течение ночи. После охлаждения до комнатной температуры, добавляли концентрированную соляную кислоту (2 мл) и перемешивали при кипячении в течение 2 часов. Полученный раствор охлаждали до комнатной температуры, затем нерастворимые фракции отфильтровывали, и фильтрат упаривали при пониженном давлении. Добавляли в остаток воду, промывали этил ацетатом. pH водного слоя доводили до 9 3н. водным раствором гидроксида натрия и промывали смесью дихлорметан/метанол (об./об.) = 10/1. Растворитель в водном слое отгоняли при пониженном давлении, и затем в остаток добавляли дихлорметан. Нерастворимые фракции отфильтровывали, и растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (239.1 мг, 77%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.73 (2Н, т, J=4.4 Гц), 3.69-3.64 (6Н, м), 3.59 (2Н, т, J=4.4 Гц), 3.03 (2Н, ушир. т, J=4.4 Гц).
Сравнительный пример 67
Получение 1-(4-этинилфенил)-3-{2-[2-(2-гидроксиэтокси)этокси]этил}мочевины
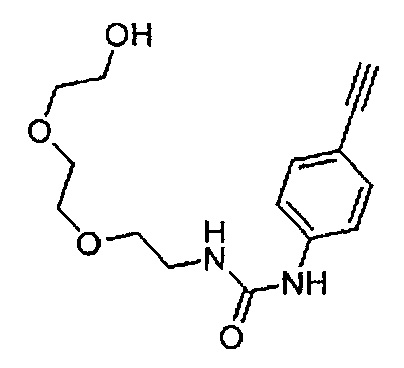
4-Этиниланилин (170.4 мг, 1.5 ммоль) добавляли в раствор 4-нитрофенил хлорформиата (289.3 мг, 1.4 ммоль) в тетрагидрофуране (6.8 мл), перемешивали при комнатной температуре в течение 3 часов, и затем растворитель отгоняли при пониженном давлении. В раствор полученного неочищенного продукта в дихлорметане (4 мл) добавляли раствор 2-[2-(2-аминоэтокси)этокси]этанола (239 мг, 1.6 ммоль) в дихлорметане (4 мл) и триэтиламин (303 мкл, 2.2 ммоль), перемешивали при комнатной температуре в течение ночи, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (этилацетат/метанол (об./об.) = 100/0→90/10), получая указанное в заголовке соединение (325.4 мг, 77%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.30 (1Н, ушир. с), 7.32 (4Н, м), 6.22 (1H, ушир. т, J=4.4 Гц), 4.37 (1Н, ушир. с), 3.72 (2Н, ушир. т, J=4.4 Гц), 3.55-3.53 (6Н, м), 3.49 (2Н, т, J=4.4 Гц), 3.36-3.34 (2Н, м), 3.05 (1Н, с).
Сравнительный пример 68
Получение 1-{2-[2-(2-аминоэтокси)этокси]этил}-3-(4-этинилфенил)мочевины
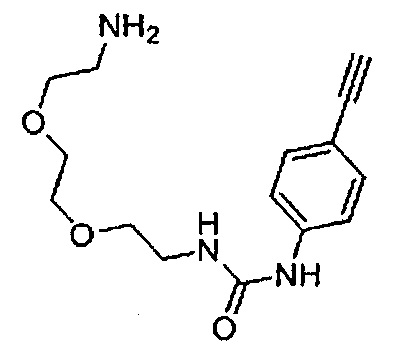
4-Этиниланилин (200 мг, 1.7 ммоль) добавляли в раствор 4-нитрофенил хлорформиата (341 мг, 1.7 ммоль) в тетрагидрофуране (7.5 мл), перемешивали при комнатной температуре в течение 3 часов, и затем растворитель отгоняли при пониженном давлении. Раствор полученного неочищенного продукта в дихлорметане (10 мл) прикапывали в раствор 2,2'-[1,2-этан-диил-бис(окси)]бис-этанамина (496 мкл, 3.4 ммоль) в дихлорметане (20 мл) при 0°C, перемешивали при комнатной температуре в течение 3 часов, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол (об./об.) = 99/1→90/10), получая указанное в заголовке соединение (417 мг, 84%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.71 (1H, ушир. с), 7.37 (4Н, м), 5.85 (1Н, ушир. с), 3.68-3.58 (8Н, м), 3.49-3.46 (2Н, м), 3.00-2.98 (3H, м), 1.73 (2Н, ушир. с).
Сравнительный пример 69
Получение трет-бутил N-{2-[2-(2-аминоэтокси)этокси]этил}карбамата
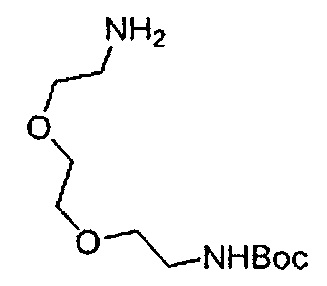
2,2'-[1,2-Этан-диил-бис(окси)]бис-этанамин (2 мл, 13.6 ммоль) и диизопропилэтиламин (2.4 мл, 13.8 ммоль) добавляли в раствор ди-трет-бутил дикарбоната (30%-ный раствор в тетрагидрофуране) (5.1 мл, 6.2 ммоль) в дихлорметане (29 мл), перемешивали при комнатной температуре в течение 4 часов, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол/триэтиламин (об./об./v) = 18/1/1), получая указанное в заголовке соединение (810 мг, 53%) в виде бледно-желтого маслянистого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 5.13 (1Н, ушир. с), 3.62-3.49 (10H, м), 3.33 (2Н, ушир. с), 2.88 (2Н, т, J=5.2 Гц), 1.45 (9Н, с).
Сравнительный пример 70
Получение трет-бутил (2-{2-[2-(диметиламино)этокси]этокси}этил)карбамата
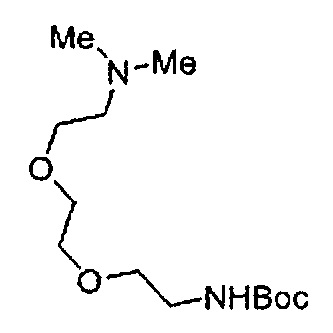
Триацетоксиборгидрид натрия (1.04 г, 4.9 ммоль) добавляли в раствор трет-бутил {2-[2-(2-аминоэтокси)этокси]этил}карбамата (810 мг, 3.3 ммоль), формальдегида (37% водный раствор) (8 мл, 98.6 ммоль) и уксусной кислоты (5.6 мл, 97.9 ммоль) в метаноле (20 мл) при 0°C, перемешивали при комнатной температуре в течение 3 часов, и затем добавляли насыщенный водный раствор карбоната калия. Полученный раствор экстрагировали смесью дихлорметан/метанол (об./об.) = 10/1, органический слой сушили безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол (об./об.) = 4/1), получая указанное в заголовке соединение (277.5 мг, 31%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 5.22 (1Н, ушир. с), 3.63-3.61 (6Н, м), 3.54 (2Н, ушир. т, J=4.8 Гц), 3.32-3.29 (2Н, м), 2.60 (2Н, ушир. т, J=5.6 Гц), 2.33 (6Н, с), 1.44 (9Н, с).
Сравнительный пример 71
Получение 2-[2-(2-аминоэтокси)этокси]-N,N-диметил-этанамина
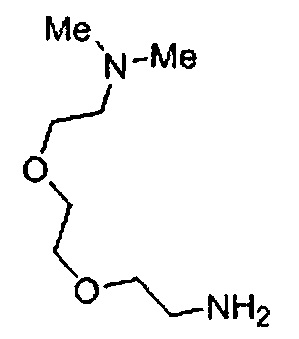
Трифторуксусную кислоту (2.5 мл, 14.7 ммоль) добавляли в раствор трет-бутил (2-{2-[2-(диметиламино)этокси]этокси}этил)карбамата (236 мг, 0.85 ммоль) в дихлорметане (2.5 мл), перемешивали при комнатной температуре в течение 1 часа, и затем растворитель отгоняли при пониженном давлении. Остаток растворяли в метаноле и подщелачивали 3н водным раствором гидроксида натрия. Полученный раствор экстрагировали смесью дихлорметан/метанол (об./об.) = 10/1, органический слой сушили безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (137.2 мг, 91%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 3.63-3.60 (4Н, м), 3.58 (2Н, т, J=5.6 Гц), 3.51 (2Н, т, J=5.2 Гц), 2.87-2.86 (2Н, м), 2.52 (2Н, т, J=5.6 Гц), 2.26 (с, 6Н).
Сравнительный пример 72
Получение 1-(2-{2-[2-(диметиламино)этокси]этокси}этил)-3-(4-этинилфенил)мочевины
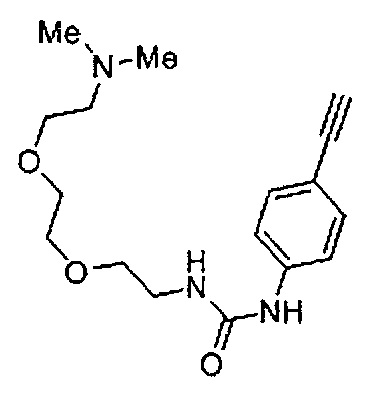
Объединяли 4-нитрофенил хлорформиат (516 мг, 2.6 ммоль), дихлорметан (25 мл), 4-этиниланилин (300 мг, 2.6 ммоль) и пиридин (206 мкл, 2.6 ммоль), перемешивали при комнатной температуре в течение 3 часов, и затем растворитель отгоняли при пониженном давлении. В раствор полученного неочищенного продукта в дихлорметане (4 мл), добавляли раствор 2-[2-(2-аминоэтокси)этокси]-N,N-диметилэтанамина (600 мг, 3.4 ммоль) в дихлорметане (25 мл) и триэтиламин (356.8 мкл, 2.6 ммоль), перемешивали при комнатной температуре в течение 3 часов, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/ метанол (об./об.) = 99/1→90/10), получая указанное в заголовке соединение (201 мг, 26%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9.17 (1H, ушир. с), 7.55 (2Н, ушир. д, J=8.4 Гц), 7.35 (2Н, ушир. д, J=8.4 Гц), 6.70 (1H, ушир. т, J=4.8 Гц), 4.01-3.99 (2Н, м), 3.68-3.66 (2Н, м), 3.63-3.61 (2Н, м), 3.58 (2Н, ушир. т, J=4.8 Гц), 3.47-3.43 (2Н, м), 3.27-3.25 (2Н, м), 2.99 (1H, с), 2.88 (6Н, с).
Сравнительный пример 73
Получение N-[2-(2-{2-[3-(4-этинилфенил)уреидо]этокси}этокси)этил]ацетамида
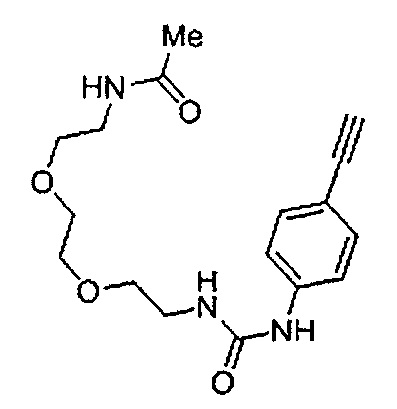
Безводную уксусную кислоту (48.2 мкл, 0.51 ммоль) и 4-диметиламинопиридин (8.3 мг, 0.068 ммоль) добавляли в раствор 1-{2-[2-(2-аминоэтокси)этокси]этил}-3-(4-этинилфенил)мочевины (100 мг, 0.34 ммоль) в ацетонитриле (2 мл), перемешивали при комнатной температуре в течение 8 часов, и затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/ метанол (об./об.) = 98/2→90/10), получая указанное в заголовке соединение (75.2 мг, 66%) в виде бледно-желтого маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8.51 (1Н, ушир. с), 7.43 (2Н, ушир. д, J=8.8 Гц), 7.38 (2Н, ушир. д, J=8.8 Гц), 5.94 (1Н, ушир. с), 5.69 (1H, ушир. с), 3.68-3.55 (8Н, м), 3.49-3.46 (4Н, м), 2.99 (1H, с), 2.04 (3H, с).
Сравнительный пример 74
Получение оксибис(этан-2,1-диил)бис(4-метилбензолсульфонат)
Диэтиленгликоль (980 мкл, 9.4 ммоль) и п-толуолсульфонил хлорид (3.58 г, 18.8 ммоль) добавляли в раствор гидроксида калия (4.23 г, 75.4 ммоль) в дихлорметане (20 мл) при 0°C, и перемешивали при 0°C в течение 3 часов. Полученный раствор разбавляли водой и экстрагировали дихлорметаном. Органический слой промывали водой, затем сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая указанное в заголовке соединение (3.38 г, 87%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.78 (4Н, д, J=8.4 Гц), 7.35 (4Н, д, J=8.4 Гц), 4.09 (4Н, т, J=4.8 Гц), 3.61 (4Н, т, J=4.8 Гц), 2.45 (6Н, с).
Сравнительный пример 75
Получение 1-(4-этинилфенил)-3-{2-[2-(2-морфолиноэтокси)этокси]этил}мочевины
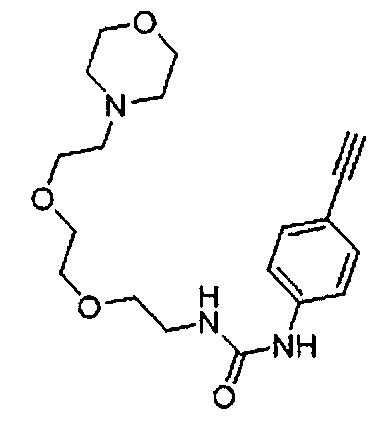
Оксибис(этан-2,1-диил) бис(4-метилбензолсульфонат) (233.8 мг, 0.56 ммоль) и карбонат калия (97.4 мг, 0.71 ммоль) добавляли в раствор 1-{2-[2-(2-аминоэтокси)этокси] этил}-3-(4-этинилфенил)мочевины (136 мг, 0.47 ммоль) в ацетонитриле (5 мл), перемешивали при кипячении в течение 16 часов, и затем растворитель отгоняли при пониженном давлении. В остаток добавляли воду, и экстрагировали раствором дихлорметан/метанол (об./об.) = 10/1. Органический слой сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол (об./об.) = 99/1→90/10), получая указанное в заголовке соединение (116.1 мг, 68%) в виде бесцветного маслянистого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7.89 (1Н, ушир. с), 7.38 (2Н, ушир. д, J=8.8 Гц), 7.34 (2Н, д, J=8.8 Гц), 5.90 (1H, ушир. с), 3.71-3.69 (4Н, м), 3.62-3.60 (6Н, м), 3.55 (2Н, т, J=4.4 Гц), 3.42-3.41 (2Н, м), 3.01 (1Н, с), 2.62-2.60 (2Н, м), 2.53-2.52 (4Н, м).
Пример 1
Получение 2-{[3-({3-[3-(2-метоксиэтил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 1)
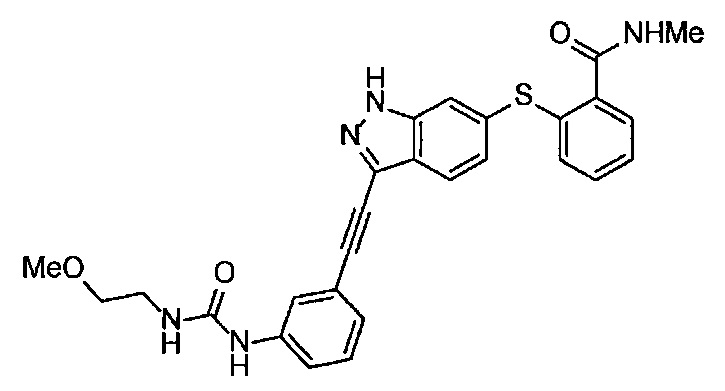
N,N-Диизопропилэтиламин (0.13 мл, 0.72 ммоль) добавляли в раствор 2-{(3-иод-1Н-индазол-6-ил)тио}-N-метилбензамида (100.0 мг, 0.24 ммоль), 1-(3-этинилфенил)-3-(2-метоксиэтил)мочевины (63.3 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль) и CuI (4.6 мг, 0.024 ммоль) в N,N-диметилформамиде (2 мл) при комнатной температуре, перемешивали в течение 5 минут и затем перемешивали при 80°C в течение 2 часов. Полученный раствор разбавляли этилацетатом, промывали насыщенным раствором хлорида натрия, сушили безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол (об./об.) = 95/5→90/10), получая указанное в заголовке соединение (74.3 мг, 61%) в виде коричневого порошка.
1H ЯМР (400 МГц, CD3OD) δ 7.78 (1H, ушир. д, J=8.4 Гц), 7.69 (1Н, т, J=1.6 Гц), 7.59 (1H, ушир. с), 7.48-7.46 (1H, м), 7.40-7.19 (7Н, м), 3.49 (2Н, т, J=5.6 Гц), 3.40-3.37 (5Н, м), 2.85 (3H, с).
Пример 2
Получение 2-({3-[(3-{3-[2-(2-метоксиэтокси)этил]уреидо}фенил)этинил]-1Н-индазол-6-ил}тио)-N-метилбензамида (Соединение 2)
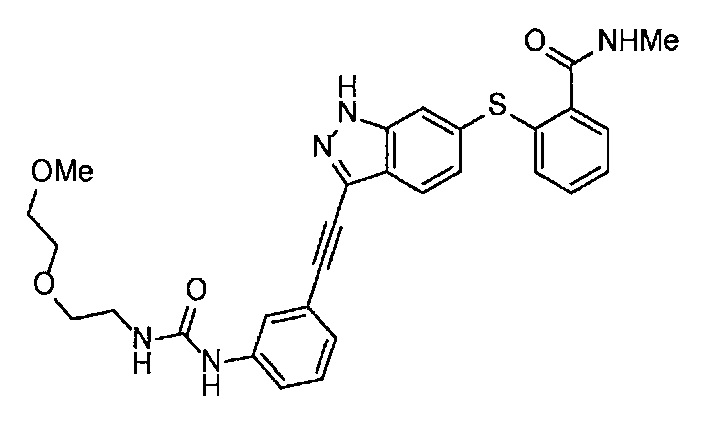
2-{(3-Иод-1H-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(3-этинилфенил)-3-[2-(2-метоксиэтокси)этил]мочевину (76.1 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (72.6 мг, 55%) в виде бледно-оранжевого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.55 (1Н, ушир. с), 8.73 (1H, ушир. с), 8.36 (1Н, ушир. кв, J=4.8 Гц), 7.81 (1Н, ушир. д, J=8.4 Гц), 7.78 (1Н, ушир. с), 7.61 (1H, ушир. с), 7.49 (1H, дд, J=7.6, 2.0 Гц), 7.37-7.27 (4Н, м), 7.21-7.17 (2Н, м), 7.05 (1Н, ушир. дд, J=7.6, 2.0 Гц), 6.25 (1Н, ушир. т, J=5.2 Гц), 3.56-3.53 (2Н, м), 3.48-3.45 (4Н, м), 3.28-3.24 (5Н, м), 2.76 (3H,д,1=4.8 Гц).
Пример 3
Получение 2-[(3-{[3-(3-{2-[2-(2-метоксиэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 3)
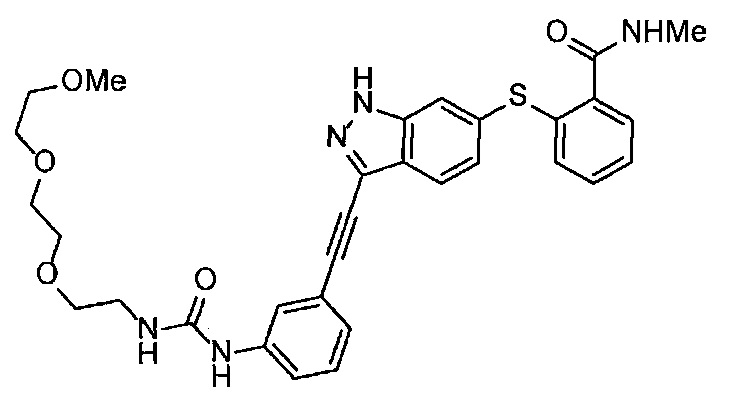
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(3-этинилфенил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевину (88.8 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (58.5 мг, 41%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.55 (1Н, ушир. с), 8.72 (1H, ушир. с), 8.35 (1Н, ушир. кв, J=4.8 Гц), 7.81 (1H, ушир. д, J=8.4 Гц), 7.78 (1Н, ушир. с), 7.61 (1Н, ушир. с), 7.49 (1H, дд, J=7.6, 2.0 Гц), 7.37-7.27 (4Н, м), 7.21-7.17 (2Н, м), 7.05 (1Н, дд, J=7.6, 2.0 Гц), 6.25 (1Н, ушир. т, J=5.2 Гц), 3.54-3.51 (6Н, м), 3.47-3.43 (4Н, м), 3.29-3.24 (5Н, м), 2.76 (3H, д, J=4.8 Гц).
Пример 4
Получение 2-{[(3-({3-[3-(2,5,8,11-тетраоксатридекан-13-ил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 4)
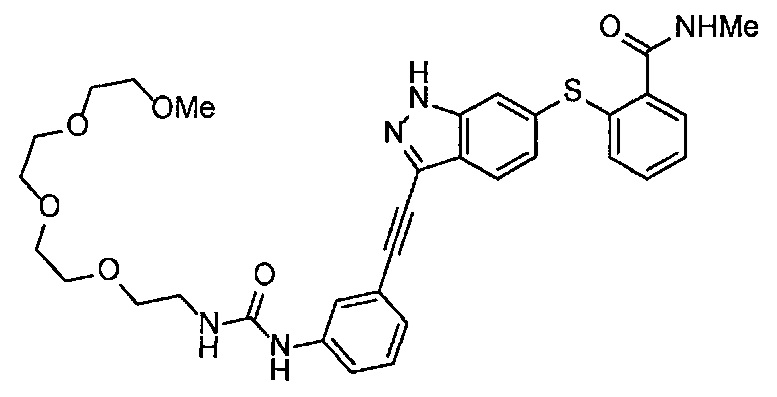
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(3-этинилфенил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевину (102.6 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (29.5 мг, 19%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.78 (1H, д, J=8.8 Гц), 7.70 (1Н, ушир. с), 7.59 (1H, ушир. с), 7.47 (1Н, ушир. д, J=7.2 Гц), 7.40 (1H, ушир. д, J=8.0 Гц), 7.35-7.23 (6Н, м), 3.65-3.58 (12Н, м), 3.54-3.51 (2Н, м), 3.39 (2Н, т, J=5.2 Гц), 3.33 (3H, с), 2.85 (3H, с).
Пример 5
Получение 2-{[3-({3-[3-(2,5,8,11,14-пентаоксагексадекан-16-ил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 5)
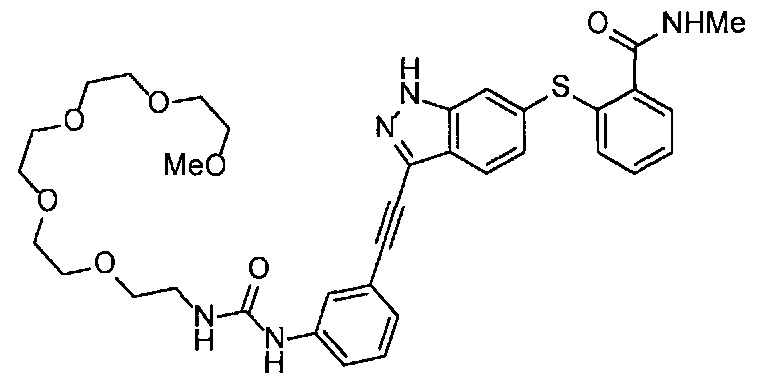
2-{(3-Иод-1H-индазол-6-ил)тио}-N-метилбензамид (150.0 мг, 0.37 ммоль), 1-(3-этинилфенил)-3-(2,5,8,11,14- пентаоксагексадекан-16-ил)мочевину (173.0 мг, 0.44 ммоль), PdCl2(PPh3)2 (13.0 мг, 0.019 ммоль), CuI (6.9 мг, 0.036 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.19 мл, 1.1 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (43.6 мг, 18%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 10.48 (1Н, ушир. с), 7.83 (1H, ушир. с), 7.81 (1Н, д, J=8.4 Гц), 7.69 (1H, ушир. с), 7.65-7.63 (1H, м), 7.56 (1Н, дт, J=8.0, 1.8 Гц), 7.51 (1Н, ушир. с), 7.32-7.19 (6Н, м), 6.34 (1H, ушир. м), 6.02 (1H, ушир. с), 3.77-3.74 (4Н, м), 3.70-3.63 (6Н, м), 3.61-3.59 (6Н, м), 3.50-3.44 (4Н, м), 3.27 (3H, с), 2.95 (3H, д, J=4.8 Гц).
Пример 6
Получение 2-{[3-({4-[3-(2-метоксиэтил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 6)
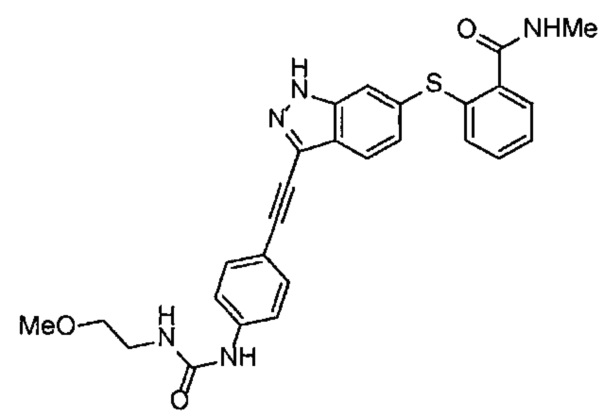
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (104.6 мг, 0.26 ммоль), 1-(4-этинилфенил)-3-(2-метоксиэтил)мочевину (108.5 мг, 0.50 ммоль), PdCl2(PPh3)2 (5.8 мг, 0.0083 ммоль), CuI (2.7 мг, 0.014 ммоль), N,N-диметилформамид (1 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (77.8 мг, 61%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.46 (1Н, ушир. с), 8.82 (1H, с), 8.36 (1Н, кв, J=4.8 Гц), 7.82 (1Н, д, J=8.8 ГЦ), 7.60 (1Н, ушир. с), 7.51 (2Н, ушир. д, J=8.8 Гц), 7.49-7.46 (3H, м), 7.32 (1H, тд, J=7.2, 1.6 Гц), 7.29 (1Н, тд, J=7.2, 1.6 Гц), 7.18 (1H, дд, J=8.8, 1.6 Гц), 7.05 (1H, дд, J=7.2, 1.6 Гц), 6.30 (1H, т, J=5.6 Гц), 3.39 (2Н, т, J=5.6 Гц), 3.29 (3H, с), 3.26 (2Н, м), 2.76 (3H, д, J=4.8 Гц).
Пример 7
Получение 2-({3-[(4-{3-[2-(2-метоксиэтокси)этил]уреидо}фенил)этинил]-1Н-индазол-6-ил}тио)-N-метилбензамида (Соединение 7)
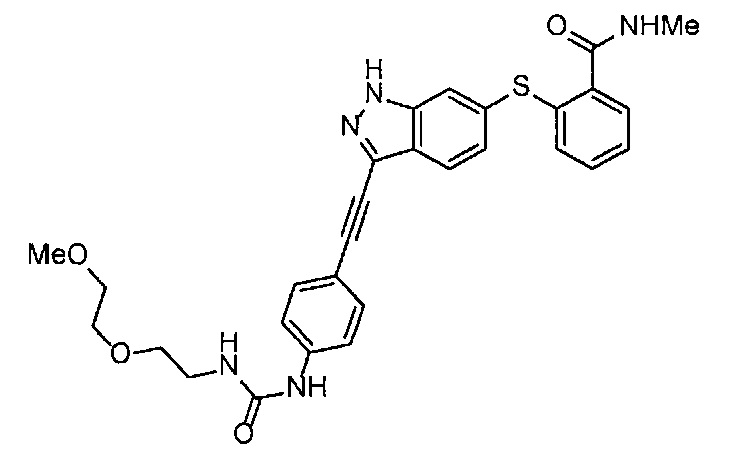
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (102.5 мг, 0.25 ммоль), 1-(4-этинилфенил)-3-[2-(2-метоксиэтокси)этил]мочевину (132.5 мг, 0.51 ммоль), PdCl2(PPh3)2 (5.3 мг, 0.0076 ммоль), CuI (3.1 мг, 0.016 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.086 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (51.2 мг, 38%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.47 (1H, ушир. с), 8.86 (1H, с), 8.36 (1Н, кв, J=4.8 Гц), 7.82 (1H, д, J=8.4 ГЦ), 7.60 (1Н, ушир. с), 7.53-7.44 (5Н, м), 7.32 (1Н, тд, J=7.6, 1.2 Гц), 7.29 (1H, тд, J=7.6, 1.2 Гц), 7.18 (1Н, дд, J=8.4, 1.2 Гц), 7.05 (1Н, дд, J=7.6, 1.2 Гц), 6.29 (1H, т, J=5.6 Гц), 3.56-3.54 (2Н, м), 3.48-3.45 (4Н, м), 3.27 (2Н, м), 3.26 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 8
Получение 2-[(3-{[4-(3-{2-[2-(2-метоксиэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 8)
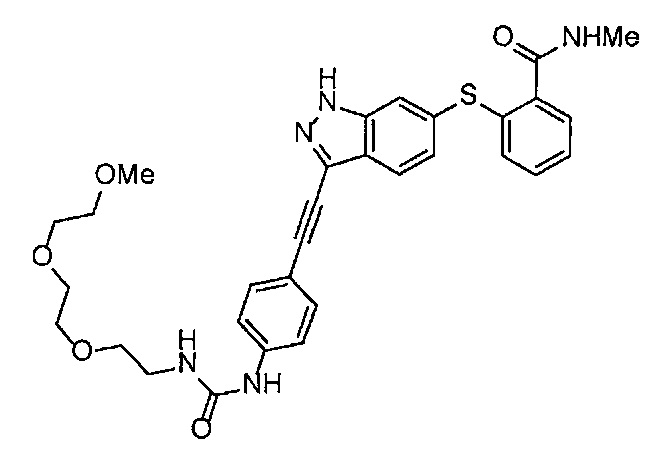
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (104.4 мг, 0.26 ммоль), 1-(4-этинилфенил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевину (118.6 мг, 0.39 ммоль), PdCl2(PPh3)2 (5.6 мг, 0.0080 ммоль), CuI (3.0 мг, 0.016 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (49.9 мг, 33%) в виде белого порошка. Температура плавления: 95-96°C
1H ЯМР (400 МГц, ДМСО-d6) δ 13.46 (1Н, ушир. с), 8.83 (1H, с), 8.36 (1H, ушир. кв, J=4.4 Гц), 7.81 (1Н, д, J=8.4 ГЦ), 7.60 (1Н, ушир. с), 7.52-7.47 (5Н, м), 7.32 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.18 (1H, дд, J=8.4, 1.2 Гц), 7.05 (1Н, дд, J=7.6, 1.6 Гц), 6.28 (1H, т, J=5.6 Гц), 3.54-3.52 (6Н, м), 3.47 (2Н, т, J=5.6 Гц), 3.45-3.42 (2Н, м), 3.26 (2Н, м), 3.24 (3H, с), 2.76 (3H, д, J=4.4 Гц).
Пример 9
Получение 2-{[3-({4-[3-(2,5,8,11-тетраоксатридекан-13-ил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 9)
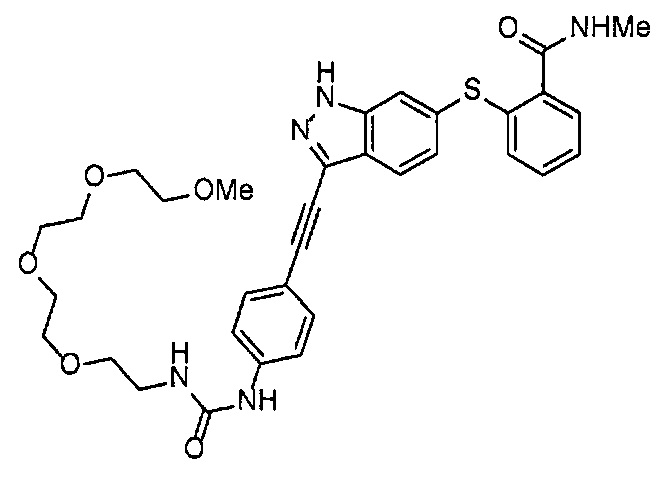
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (103.8 мг, 0.25 ммоль), 1-(4-этинилфенил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевину (125.3 мг, 0.32 ммоль), PdCl2(PPh3)2 (5.6 мг, 0.0080 ммоль), CuI (2.8 мг, 0.015 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (54.8 мг, 34%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.46 (1H, ушир. с), 8.83 (1Н, с), 8.36 (1H, кв, J=4.4 Гц), 7.82 (1Н, д, J=8.4 Гц), 7.60 (1Н, ушир. с), 7.53-7.47 (5Н, м), 7.32 (1H, тд, J=7.6, 1.6 Гц), 7.27 (1H, тд, J=7.6, 1.6 Гц), 7.17 (1Н, дд, J=8.4, 1.2 Гц), 7.05 (1H, дд, J=7.6, 1.6 Гц), 6.28 (1Н, т, J=5.6 Гц), 3.55-3.50 (10H, м), 3.47 (2Н, ушир. т, J=5.6 Гц), 3.43-3.41 (2Н, м), 3.26 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.4 Гц).
Пример 10
Получение 2-{[3-({4-[3-(2,5,8,11,14-пентаоксагексадекан-16-ил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 10)
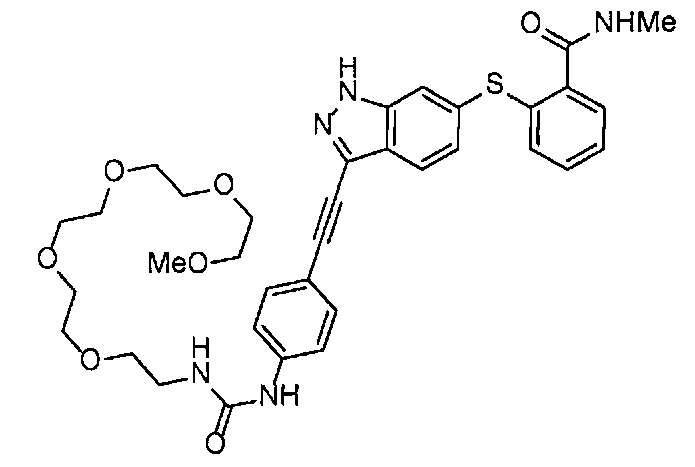
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (101.7 мг, 0.25 ммоль), 1-(4-этинилфенил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевину (114.4 мг, 0.33 ммоль), PdCl2(PPh3)2 (5.8 мг, 0.0083 ммоль), CuI (2.8 мг, 0.015 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (48.2 мг, 29%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.46 (1H, ушир. с), 8.83 (1H, с), 8.36 (1H, кв, J=4.8 Гц), 7.82 (1H, д, J=8.4 Гц), 7.60 (1Н, ушир. с), 7.52-7.47 (5Н, м), 7.31 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1Н, тд, J=7.6, 1.6 Гц), 7.18 (1Н, дд, J=8.4, 1.2 Гц), 7.05 (1H, дд, J=7.6, 1.6 Гц), 6.28 (1H, т, J=5.6 Гц), 3.55-3.45 (16Н, м), 3.43-3.41 (2Н, м), 3.27 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 11
Получение 2-метоксиэтил {3-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 11)
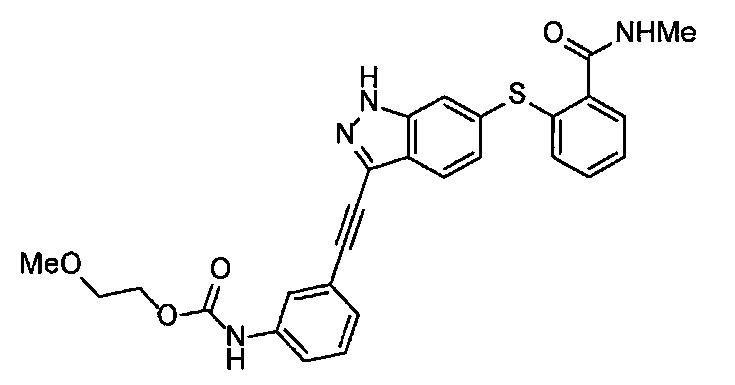
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 2-метоксиэтил (3-этинилфенил)карбамат (63.6 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (72.8 мг, 60%) в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ 11.39 (1H, ушир. с), 7.85 (1H, ушир. с), 7.78 (1H, д, J=8.4 Гц), 7.69-7.53 (5Н, м), 7.48-7.46 (1H, м), 7.31-7.25 (2Н, м), 7.21-7.17 (2Н, м), 6.35 (1H, ушир. кв, J=4.8 Гц), 4.36 (2Н, ушир. т, J=4.4 Гц), 3.69 (2Н, ушир. т, J=4.4 Гц), 3.44 (3H, с), 2.96 (3H, ушир. д, J=4.8 Гц).
Пример 12
Получение 2-(2-метоксиэтокси)этил{3-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 12)
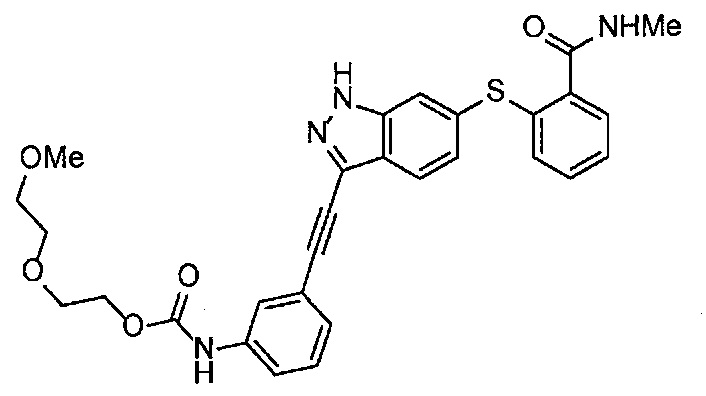
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 2-(2-метоксиэтокси)этил (3-этинилфенил)карбамат (76.4 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (70.1 мг, 53%) в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ 10.97 (1Н, ушир. с), 7.78 (1H, д, J=8.4 Гц), 7.64-7.52 (5Н, м), 7.31-7.18 (6Н, м), 6.33 (1Н, ушир. м), 4.34 (2Н, ушир. т, J=4.4 Гц), 3.78 (2Н, ушир. т, J=4.4 Гц), 3.72-3.69 (2Н, м), 3.59-3.57 (2Н, м), 3.36 (3H, с), 2.96 (3H, ушир. д, J=4.8 Гц).
Пример 13
Получение 2-[2-(2-метоксиэтокси)этокси]этил{3-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 13)
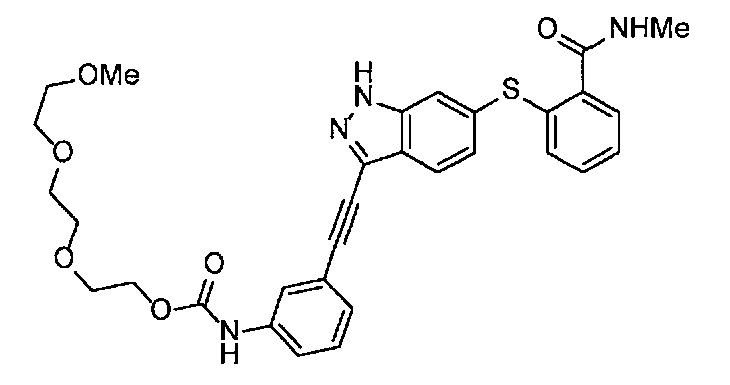
2-{(3-Иод-1H-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 2-[2-(2-метоксиэтокси)этокси]этил(3-этинилфенил)карбамат (89.1 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (57.7 мг, 40%) в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ 11.62 (1Н, ушир. с), 7.97 (1H, ушир. с), 7.75 (1H, ушир. д, J=8.4 Гц), 7.62-7.59 (4Н, м), 7.28-7.16 (6Н, м), 6.43 (1Н, ушир. м), 4.31 (2Н, ушир. т, J=4.4 Гц), 3.76 (2Н, ушир. т, J=4.4 Гц), 3.72-3.70 (2Н, м), 3.68-3.66 (2Н, м), 3.62-3.60 (2Н, м), 3.49-3.47 (2Н, м), 3.29 (3H, с), 2.96 (3H, ушир. д, J=4.8 Гц).
Пример 14
Получение 2-метоксиэтил {4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 14)
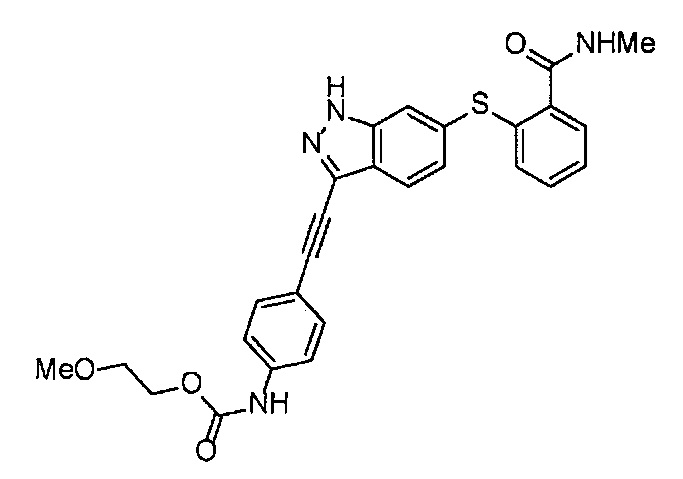
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (101.2 мг, 0.25 ммоль), 2-метоксиэтил (4-этинилфенил)карбамат (88.8 мг, 0.41 ммоль), PdCl2(PPh3)2 (5.9 мг, 0.0084 ммоль), CuI (2.7 мг, 0.014 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (69.8 мг, 56%) в виде бледно-желтого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.49 (1H, ушир. с), 10.02 (1H, с), 8.36 (1H, кв, J=4.8 Гц), 7.82 (1H, д, J=8.4 Гц), 7.60-7.55 (5Н, м), 7.49-7.48 (1Н, м), 7.32 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.18 (1H, дд, J=8.4, 1.6 Гц), 7.05 (1H, дд, J=7.6, 1.6 Гц), 4.23 (2Н, ушир. т, J=4.8 Гц), 3.58 (2Н, ушир. т, J=4.8 Гц), 3.29 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 15
Получение 2-(2-метоксиэтокси)этил{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 15)
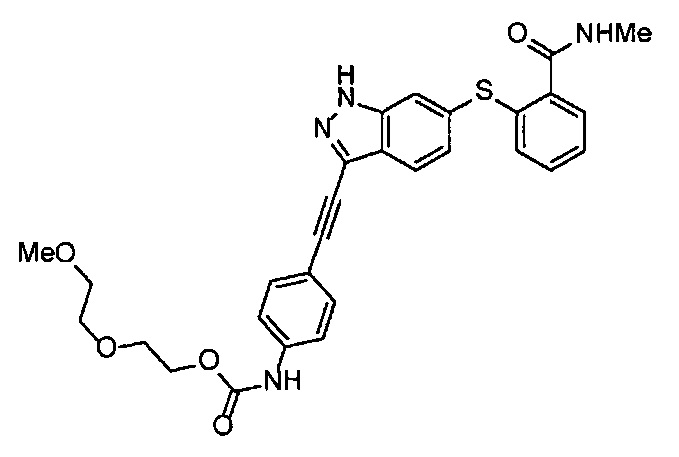
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (101.2 мг, 0.25 ммоль), 2-(2-метоксиэтокси)этил(4-этинилфенил)карбамат (102.4 мг, 0.39 ммоль), PdCl2(PPh3)2 (5.3 мг, 0.0076 ммоль), CuI (2.8 мг, 0.015 ммоль), N,N-диметилформамид (1.0 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (79.4 мг, 59%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.48 (1H, ушир. с), 10.01 (1H, с), 8.35 (1H, кв, J=4.4 Гц), 7.81 (1H, д, J=8.4 Гц), 7.59-7.54 (5Н, м), 7.49-7.46 (1Н, м), 7.32 (1Н, тд, J=7.6, 1.6 Гц), 7.27 (1H, тд, J=7.6, 1.6 Гц), 7.17 (1H, дд, J=8.4, 1.2 Гц), 7.04 (1Н, дд, J=7.6, 1.6 Гц), 4.22 (2Н, ушир. т, J=4.8 Гц), 3.65 (2Н, ушир. т, J=4.8 Гц), 3.57-3.54 (2Н, м), 3.46-3.43 (2Н, м), 3.24 (3H, с), 2.75 (3H, д, J=4.4 Гц).
Пример 16
Получение 2-[2-(2-метоксиэтокси)этокси]этил{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 16)
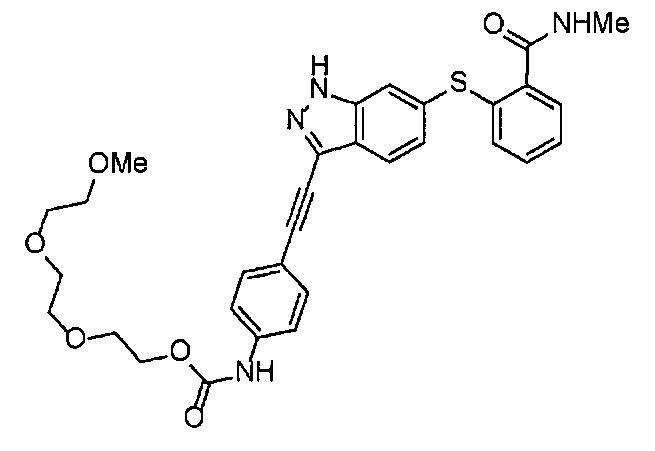
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (101.5 мг, 0.25 ммоль), 2-[2-(2-метоксиэтокси)этокси]этил(4-этинилфенил)карбамат (122.7 мг, 0.40 ммоль), PdCl2(PPh3)2 (5.7 мг, 0.0081 ммоль), CuI (2.9 мг, 0.015 ммоль), N,N-диметилформамид (1.0 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (90.9 мг, 62%) в виде белого порошка.
Температура плавления: 103-104°C
1H ЯМР (400 МГц, ДМСО-d6) δ 13.48 (1Н, ушир. с), 10.02 (1H, с), 8.35 (1H, кв, J=4.8 Гц), 7.81 (1H, д, J=8.4 Гц), 7.59 (1Н, ушир. с), 7.56 (4Н, ушир. м), 7.48 (1Н, дд, J=7.6, 1.6 Гц), 7.31 (1H, тд, J=7.6, 1.6 Гц), 7.27 (1H, тд, J=7.6. 1.6 Гц), 7.17 (1H, дд, J=8.4, 1.2 Гц), 7.04 (1Н, дд, J=7.6, 1.6 Гц), 4.22 (2Н, ушир. т, J=4.4 Гц), 3.65 (2Н, ушир. т, J=4.4 Гц), 3.57-3.49 (6Н, м), 3.42-3.39 (2Н, м), 3.22 (3H, с), 2.75 (3H, д, J=4.8 Гц).
Пример 17
Получение 2,5,8,11-тетраоксатридекан-13-ил{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 17)
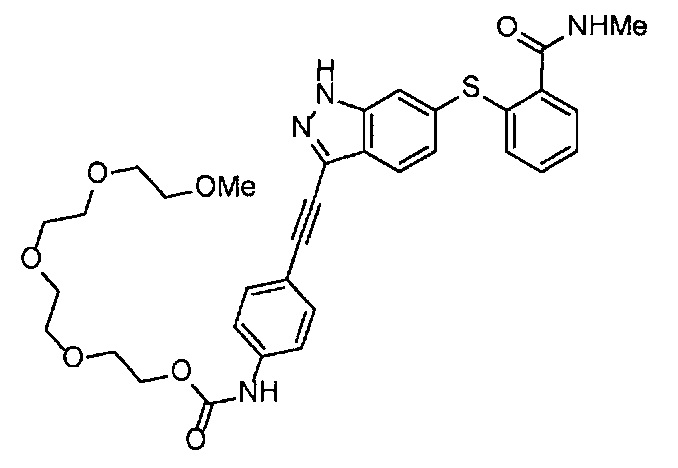
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (103.1 мг, 0.25 ммоль), 2,5,8,11-тетраоксатридекан-13-ил(4-этинилфенил)карбамат (129.8 мг, 0.37 ммоль), PdCl2(PPh3)2 (5.6 мг, 0.0080 ммоль), CuI (3.1 мг, 0.016 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (77.6 мг, 48%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.49 (1H, ушир. с), 10.04 (1Н, с), 8.36 (1Н, кв, J=4.8 Гц), 7.82 (1H, ушир. д, J=8.4 Гц), 7.60 (1Н, ушир. с), 7.57 (4Н, м), 7.50-7.48 (1Н, м), 7.35-7.27 (2Н, м), 7.18 (1H, дд, J=8.4, 1.2 Гц), 7.05 (1H, дд, J=7.6, 1.6 Гц), 4.24-4.22 (2Н, м), 3.68-3.66 (2Н, м), 3.57-3.49 (10H, м), 3.43-3.40 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 18
Получение 2,5,8,11,14-пентаоксагексадекан-16-ил{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}карбамата (Соединение 18)
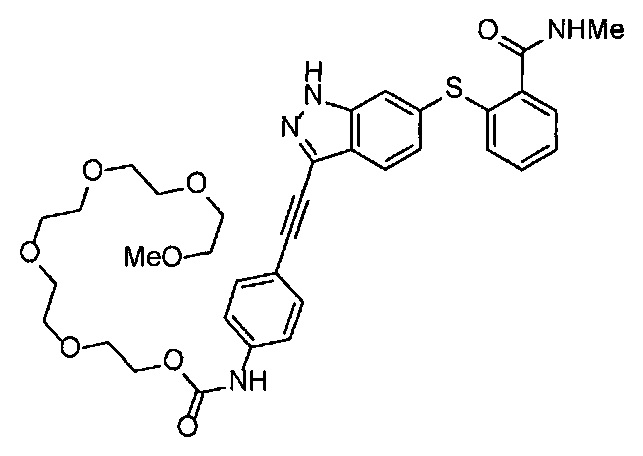
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (107.0 мг, 0.26 ммоль), 2,5,8,11,14-пентаоксагексадекан-16-ил (4-этинилфенил)карбамат (160.4 мг, 0.41 ммоль), PdCl2(PPh3)2 (5.2 мг, 0.0074 ммоль), CuI (3.1 мг, 0.016 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (77.9 мг, 44%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.49 (1H, ушир. с), 10.04 (1H, с), 8.36 (1Н, кв, J=4.8 Гц), 7.82 (1Н, д, J=8.4 Гц), 7.60 (1Н, ушир. с), 7.57 (4Н, м), 7.49-7.48 (1H, м), 7.32 (1H, тд, J=7.6, 2.0 Гц), 7.29 (1Н, тд, J=7.6, 2.0 Гц), 7.18 (1Н, дд, J=8.4, 1.6 Гц), 7.05 (1H, дд, J=7.6, 2.0 Гц), 4.23 (2Н, ушир. т, J=4.4 Гц), 3.67 (2Н, ушир. т, J=4.4 Гц), 3.58-3.48 (14Н, м), 3.43-3.40 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 19
Получение N-{3-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}-2,5,8,11,14,17-гексаоксанонадекан-19-амида (Соединение 19)
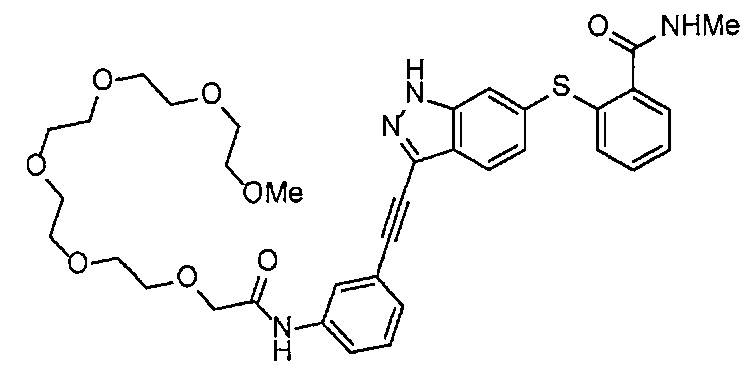
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.5 мг, 0.25 ммоль), PdCl2(PPh3)2 (5.2 мг, 0.0074 ммоль), CuI (2.6 мг, 0.014 ммоль), N,N-диметилформамид (1.7 мл), N-(3-этинилфенил)-2,5,8,11,14,17-гексаоксанонадекан-19-амид (140.1 мг, 0.34 ммоль) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (33.0 мг, 19%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.58 (1H, ушир. с), 9.77 (1Н, с), 8.37 (1Н, кв, J=4.4 Гц), 7.99 (1Н, ушир. т, J=1.6 Гц), 7.82 (1Н, д, J=8.4 Гц), 7.70 (1Н, дт, J=7.6, 1.6 Гц), 7.62 (1Н, ушир. с), 7.50-7.48 (1Н, м), 7.42 (1Н, т, J=7.6 Гц), 7.38 (1H, дт, J=7.6, 1.6 Гц), 7.33 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.20 (1Н, дд, J=8.4, 1.6 Гц), 7.07-7.05 (1H, м), 4.11 (2Н, с), 3.70-3.68 (2Н, м), 3.64-3.62 (2Н, м), 3.58-3.54 (4Н, м), 3.51-3.47 (10H, м), 3.41-3.39 (2Н, м), 3.22 (3H, с), 2.76 (3H, д, J=4.4 Гц).
Пример 20
Получение N-{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}-2,5,8,11-тетраоксатридекан-13-амида (Соединение 20)
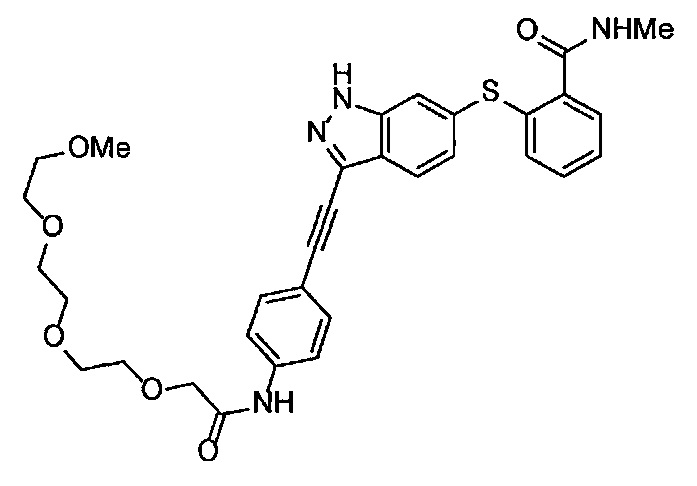
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), N-(4-этинилфенил)-2,5,8,11-тетраоксатридекан-13-амид (93.2 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), ацетонитрил (1 мл) и триэтиламин (1 мл) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (91.1 мг, 62%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.50 (1H, с), 9.84 (1Н, с), 8.35 (1H, ушир. кв, J=4.4 Гц), 7.83 (1H, д, J=8.4 Гц), 7.75 (2Н, ушир. д, J=8.8 Гц), 7.61 (2Н, ушир. д, J=8.8 Гц), 7.60 (1Н, ушир. с), 7.48 (1Н, дд, J=7.2, 2.0 Гц), 7.34-7.26 (2Н, м), 7.18 (1Н, дд, J=8.4, 1.6 Гц), 7.05 (1Н, ушир. д, J=7.2 Гц), 4.11 (2Н, с), 3.70-3.67 (2Н, м), 3.63-3.61 (2Н, м), 3.57-3.51 (6Н, м), 3.43-3.41 (2Н, м), 3.23 (3H, с), 2.76 (3H, ушир. д, J=4.4 Гц).
Пример 21
Получение N-{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}-2,5,8,11,14-пентаоксагексадекан-16-амида (Соединение 21)
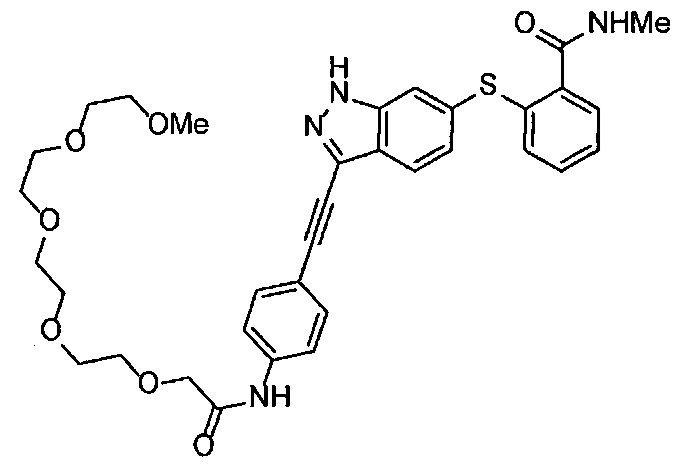
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), N-(4-этинилфенил)-2,5,8,11,14-пентаоксагексадекан-16-амид (105.9 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), ацетонитрил (1 мл) и триэтиламин (1 мл) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (75.0 мг, 47%) в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.50 (1H, с), 9.83 (1Н, с), 8.35 (1H, ушир. кв, J=4.4 Гц), 7.83 (1Н, ушир. д, J=8.4 Гц), 7.75 (2Н, ушир. д, J=8.8 Гц), 7.61 (2Н, ушир. д, J=8.8 Гц), 7.60 (1H, ушир. с), 7.49 (1H, дд, J=7.2, 2.0 Гц), 7.34-7.26 (2Н, м), 7.18 (1H, дд, J=8.4, 1.6 Гц), 7.05 (1Н, ушир. д, J=7.2 Гц), 4.11 (2Н, с), 3.70-3.67 (2Н, м), 3.64-3.61 (2Н, м), 3.57-3.49 (10H, м), 3.42-3.40 (2Н, м), 3.22 (3H, с), 2.76 (3H, ушир. д, J=4.4 Гц).
Пример 22
Получение N-{4-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]фенил}-2,5,8,11,14,17-гексаоксанонадекан-19-амида (Соединение 22)
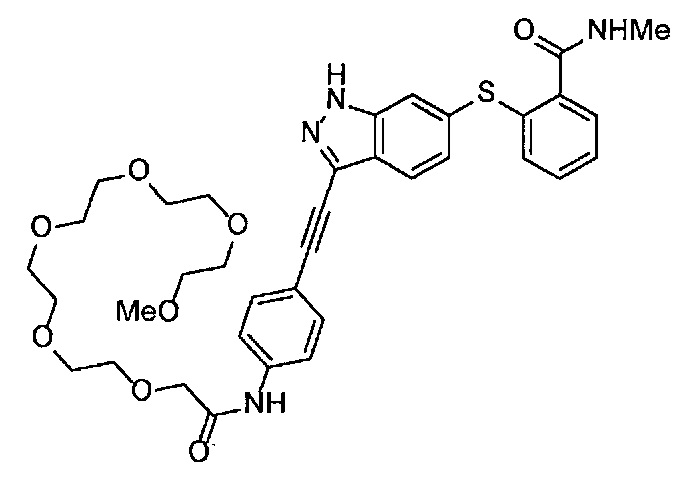
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.6 мг, 0.25 ммоль), PdCl2(PPh3)2 (5.4 мг, 0.0077 ммоль), CuI (2.5 мг, 0.013 ммоль), N,N-диметилформамид (0.8 мл), N-(4-этинилфенил)-2,5,8,11,14,17-гексаоксанонадекан-19-амид (142.1 мг, 0.35 ммоль) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (54.7 мг, 32%) в виде бледно-коричневого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.51 (1Н, ушир. с), 9.84 (1Н, с), 8.36 (1H, кв, J=4.8 Гц), 7.83 (1H, д, J=8.4 Гц), 7.76 (2Н, ушир. д, J=8.8 Гц), 7.62-7.60 (3H, м), 7.50-7.48 (1Н, м), 7.32 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.18 (1Н, дд, J=8.4, 1.6 Гц), 7.06-7.04 (1H, м), 4.11 (2Н, с), 3.69-3.67 (2Н, м), 3.63-3.61 (2Н, м), 3.58-3.53 (4Н, м), 3.51-3.48 (10H, м), 3.42-3.39 (2Н, м), 3.22 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 23
Получение 2-{[3-({3-[(2-метоксиэтил)амино]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 23)
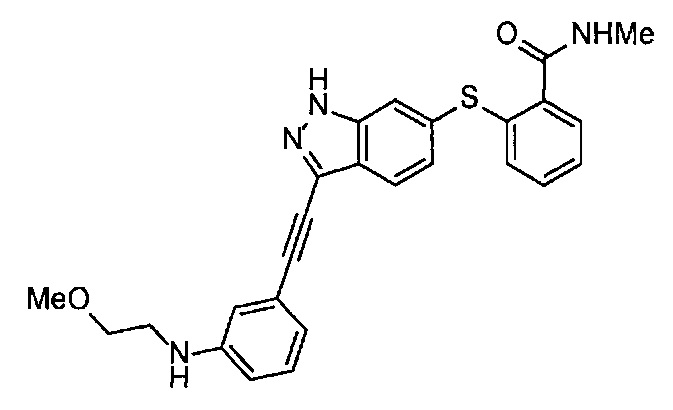
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 3-этинил-N-(2-метоксиэтил)анилин (50.8 мг, 0.29 мг), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (15 мг, 10%) в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ 10.08 (1Н, ушир. с), 7.82 (1H, д, J=8.4 Гц), 7.66-7.64 (1H, м), 7.48 (1H, ушир. с), 7.33-7.31 (3H, м), 7.22 (1Н, дд, J=8.4, 1.6 Гц), 7.17 (1Н, т, J=8.0 Гц), 6.99 (1Н, ушир. д, J=8.0 Гц), 6.88 (1Н, т, J=2.0 Гц), 6.66 (1Н, дд, J=8.0, 2.0 Гц), 6.29 (1Н, ушир. м), 4.11 (1H, ушир. м), 3.62 (2Н, т, J=5.6 Гц), 3.40 (3H, с), 3.34-3.33 (2Н, м), 2.95 (3H, д, J=4.8 Гц).
Пример 24
Получение 2-({3-[(3-{[2-(2-метоксиэтокси)этил]амино}фенил)этинил]-1Н-индазол-6-ил}тио)-N-метилбензамида (Соединение 24)
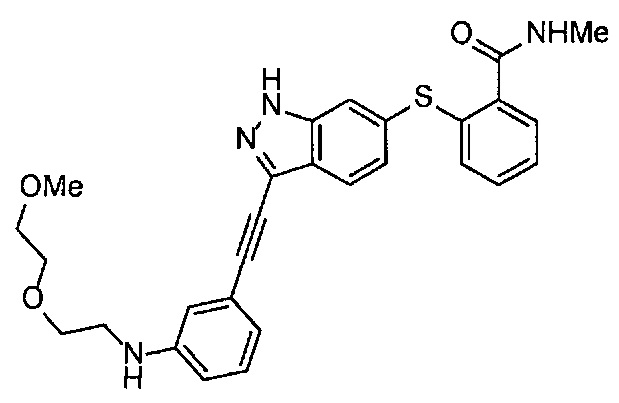
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 3-этинил-N-[2-(2-метоксиэтокси)этил]анилин (64.2 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (51.4 мг, 42%) в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ 10.14 (1H, ушир. с), 7.82 (1Н, д, J=8.4 Гц), 7.66-7.64 (1H, м),
7.49 (1Н, ушир. с), 7.33-7.30 (3H, м), 7.22 (1H, дд, J=8.4, 1.2 Гц), 7.17 (1H, т, J=8.0 Гц), 6.98 (1Н, д, J=8.0 Гц), 6.88 (1H, т, J=2.0 Гц), 6.66 (1Н, ушир. д, J=8.0 Гц), 6.30 (1H, ушир. м), 4.22 (1H, ушир. м), 3.73 (2Н, т, J=5.2 Гц), 3.67-3.65 (2Н, м), 3.58-3.56 (2Н, м), 3.41 (3H, с), 3.34-3.33 (2Н, м), 2.95 (3H, д, J=5.2 Гц).
Пример 25
Получение 2-[(3-{[3-({2-[2-(2-метоксиэтокси)этокси]этил}амино)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 25)
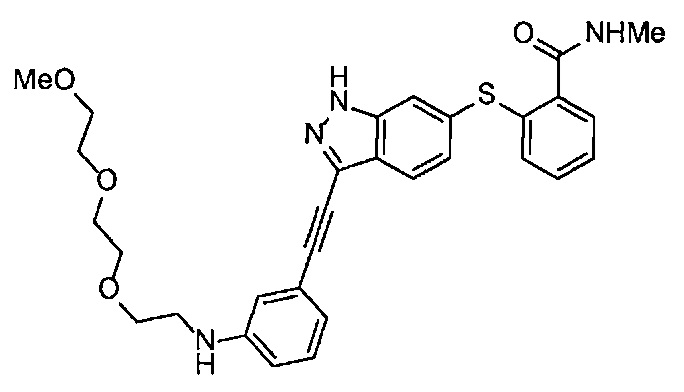
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 3-этинил-N-{2-[2-(2-метоксиэтокси)этокси]этил} анилин (76.4 мг, 0.29 мг), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (47.3 мг, 36%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 10.70 (1H, ушир. с), 7.81 (1Н, д, J=8.4 Гц), 7.63-7.61 (1H, м), 7.59 (1H, ушир. с), 7.29-7.26 (3H, м), 7.22-7.19 (1Н, м), 7.16 (1Н, т, J=8.0 Гц), 6.97 (1H, д, J=8.0 Гц), 6.87 (1Н, т, J=2.0 Гц), 6.65 (1Н, дд, J=8.0, 2.0 Гц), 6.32 (1H, ушир. м), 4.26 (1H, ушир. м), 3.72 (2Н, д, J=5.2 Гц), 3.67-3.66 (6Н, м), 3.57-3.55 (2Н, м), 3.38 (3H, с), 3.32-3.31 (2Н, м), 2.96 (3H, д, J=4.8 Гц).
Пример 26
Получение 2-({3-[(4-{[2-(2-метоксиэтокси)этил]амино}фенил)этинил]-1Н-индазол-6-ил}тио)-N-метилбензамида (Соединение 26)
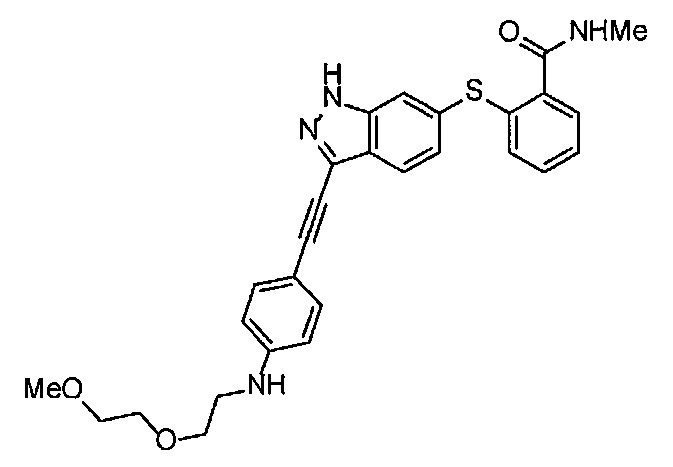
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (120.0 мг, 0.29 ммоль), 4-этинил-N-[2-(2-метоксиэтокси)этил]анилин (77.0 мг, 0.35 мг), PdCl2(PPh3)2 (10.3 мг, 0.015 ммоль), CuI (5.5 мг, 0.029 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.88 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (11.5 мг, 8%) в виде бледно-желтого порошка.
1H ЯМР (400 МГц, CDCl3) δ 10.36 (1H, ушир. с), 7.81 (1Н, ушир. д, J=8.4 Гц), 7.64-7.62 (1Н, м), 7.52 (1H, ушир. с), 7.44 (2Н, ушир. д, J=8.4 Гц), 7.30-7.28 (2Н, м), 7.22-7.18 (2Н, м), 6.59 (2Н, ушир. д, J=8.4 Гц), 6.31 (1H, ушир. м), 4.40 (1H, ушир. м), 3.72 (2Н, т, J=5.2 Гц), 3.67-3.65 (2Н, м), 3.58-3.56 (2Н, м), 3.41 (3H, с), 3.36-3.34 (2Н, м), 2.95 (3H, д, J=4.8 Гц).
Пример 27
Получение 2-[(3-{[4-({2-[2-(2-метоксиэтокси)этокси]этил}амино)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 27)
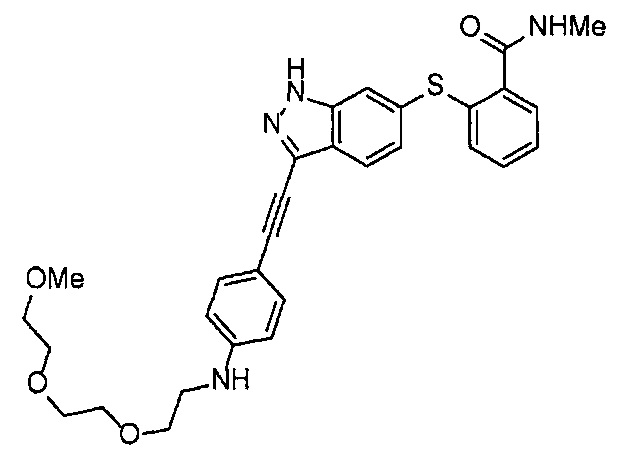
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 4-этинил-N-{2-[2-(2-метоксиэтокси)этокси]этил}анилин (77.0 мг, 0.29 мг), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (41.6 мг, 31%) в виде бледно-желтого твердого вещества. Температура плавления; 66-67°C
1H ЯМР (400 МГц, CDCl3) δ 10.12 (1Н, ушир. с), 7.81 (1H, ушир. д, J=8.4 Гц), 7.65-7.63 (1Н, м), 7.48 (1Н, ушир. с), 7.44 (2Н, ушир. д, J=8.8 Гц), 7.32-7.29 (2Н, м), 7.24-7.22 (1Н, м), 7.20 (1H, дд, J=8.4, 1.2 Гц), 6.60 (2Н, ушир. д, J=8.8 Гц), 6.30 (1H, ушир. м), 4.44 (1Н, ушир. м), 3.72 (2Н, т, J=5.2 Гц), 3.67-3.65 (6Н, м), 3.58-3.56 (2Н, м), 3.40 (3H, с), 3.35-3.31 (2Н, м), 2.95 (3H, д, J=4.8 Гц).
Пример 28
Получение 2-{[3-({4-[(2,5,8,11-тетраоксатридекан-13-ил)амино]фенил }этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 28)
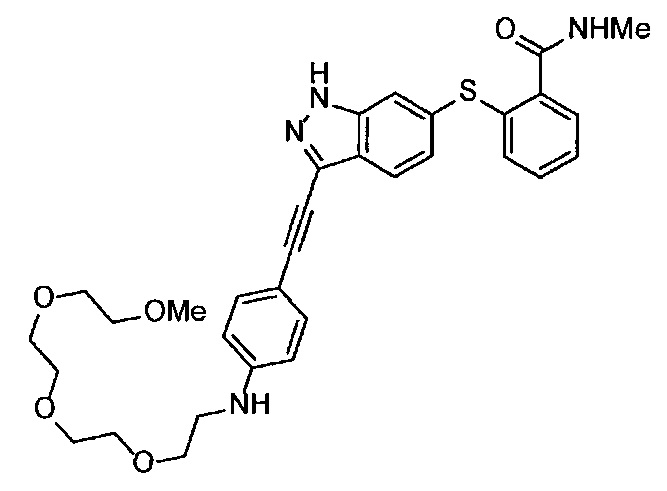
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), N-(4-этинилфенил)-2,5,8,11-тетраоксатридекан-13-амин (100.0 мг, 0.33 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (25.4 мг, 18%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 11.36 (1H, ушир. с), 7.80 (1H, ушир. д, J=8.4 Гц), 7.68 (1H, ушир. с), 7.59-7.57 (1Н, м), 7.42 (2Н, ушир. д, J=8.6 Гц), 7.25-7.22 (2Н, м), 7.18 (1H, дд, J=8.4, 1.2 Гц), 7.13-7.11 (1H, м), 6.57 (2Н, ушир. д, J=8.6 Гц), 6.38 (1Н, ушир. м), 4.48 (1Н, ушир. м), 3.70 (2Н, т, J=5.2 Гц), 3.68-3.63 (10H, м), 3.55-3.53 (2Н, м), 3.36 (3H, с), 3.31 (2Н, м), 2.97 (3H, д, J=4.8 Гц).
Пример 29
Получение 2-{[3-({4-[(2,5,8,11,14-пентаоксагексадекан-16-ил)амино]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 29)
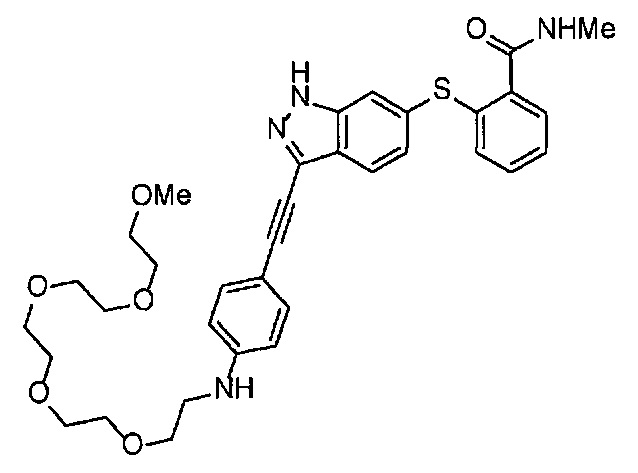
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), N-(4-этинилфенил)-2,5,8,11,14-пентаоксагексадекан-16-амин (100.0 мг, 0.28 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (13.8 мг, 9%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 11.77 (1Н, ушир. с), 7.78 (1Н, ушир. д, J=8.4 Гц), 7.72 (1Н, ушир. с), 7.57-7.54 (1H, м), 7.41 (2Н, ушир. д, J=8.6 Гц), 7.23-7.17 (2Н, м), 7.16 (1Н, дд, J=8.4, 1.6 Гц), 7.09-7.07 (1H, м), 6.56 (2Н, ушир. д, J=8.6 Гц), 6.46 (1H, кв, J=4.8 Гц), 4.49 (1H, ушир. м), 3.69 (2Н, т, J=5.2 Гц), 3.66-3.60 (14Н, м), 3.53-3.51 (2Н, м), 3.35 (3H, с), 3.30-3.28 (2Н, м), 2.96 (3H, д, J=4.8 Гц).
Пример 30
Получение 2-[(3-{[5-(3-{2-[2-(2-метоксиэтокси)этокси]этил}уреидо)пиридин-3-ил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 30)
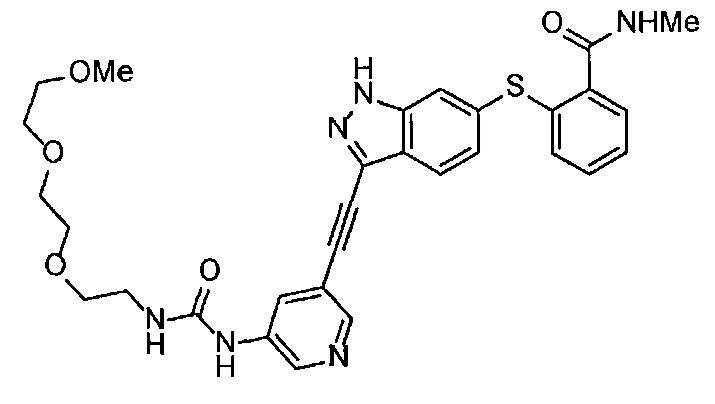
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(5-этинилпиридин-3-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевину (89.1 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилзтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (40.1 мг, 28%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.63 (1Н, ушир. с), 8.94 (1H, ушир. с), 8.51 (1H, д, J=2.4 Гц), 8.39 (1Н, д, J=2.4 Гц), 8.35 (1H, ушир. кв, J=4.4 Гц), 8.21 (1H, ушир. д, J=2.4 Гц), 7.86 (1Н, д, J=8.4 Гц), 7.63 (1Н, с), 7.49 (1Н, дд, J=7.4, 1.6 Гц), 7.35-7.28 (2Н, м), 7.21 (1Н, ушир. д, J=8.4 Гц), 7.07 (1Н, ушир. д, J=7.4 Гц), 6.44 (1Н, ушир. т, J=5.2 Гц), 3.54-3.42 (10H, м), 3.30-3.28 (2Н, м), 3.24 (3H, с), 2.76 (3H, д, J=4.4 Гц).
Пример 31
Получение 2-{[3-({5-[3-(2,5,8,11,14-пентаоксагексадекан-16-ил)уреидо]пиридин-3-ил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 31)
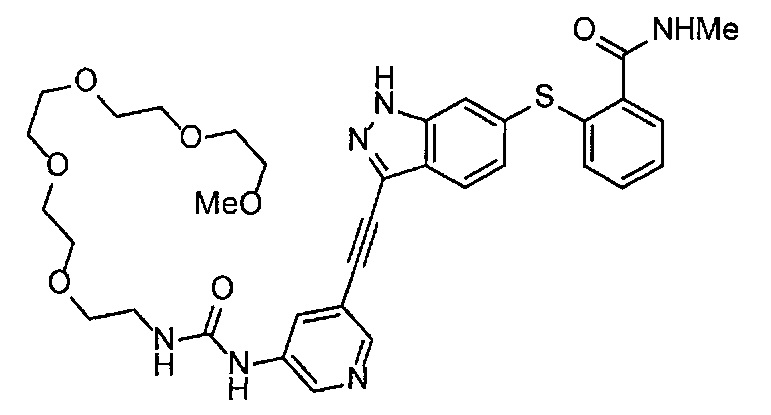
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(5-этинилпиридин-3-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевину (114.7 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (83.8 мг, 51%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8.54 (1H, ушир. с), 8.36 (1Н, ушир. с), 8.19 (1Н, т, J=2.0 Гц), 7.90 (1H, д, J=8.4 Гц), 7.59 (1Н, с), 7.49-7.47 (1Н, м), 7.37-7.30 (2Н, м), 7.27-7.24 (1H, м), 7.21 (1Н, дд, J=8.4, 1.6 Гц), 3.65-3.58 (16Н, м), 3.51-3.49 (2Н, м), 3.41 (2Н, ушир. т, J=5.2 Гц), 3.31 (3H,с), 2.85 (3H, с).
Пример 32
Получение 2-[(3-{[6-(3-{2-[2-(2-метоксиэтокси)этокси]этил}уреидо)пиридин-3-ил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 32)
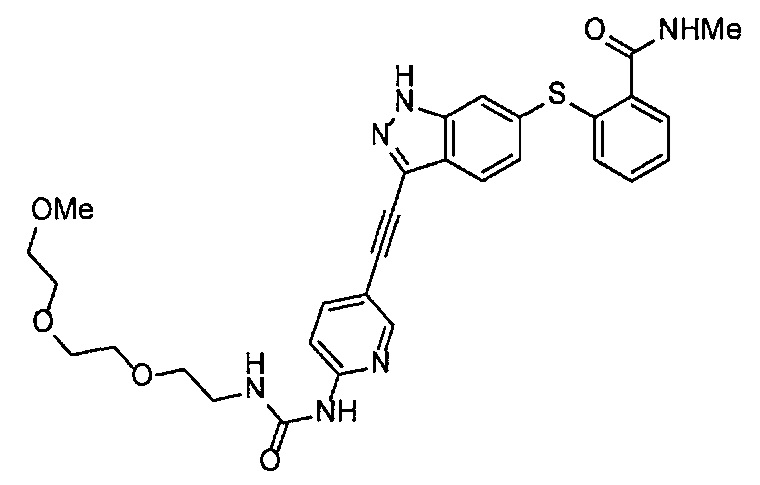
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(5-этинилпиридин-2-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевину (82.4 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (52.9 мг, 37%) в виде белого порошка.
1H ЯМР (400 МГц, CDCl3) δ 11.24 (1Н, ушир. с), 8.85 (1Н, ушир. с), 8.40 (1Н, д, J=2.0 Гц), 8.16 (1Н, ушир. с), 7.75 (1H, д, J=8.4 Гц), 7.73 (1H, дд, J=8.4, 2.0 Гц), 7.63-7.61 (1Н, м), 7.57 (1H, ушир. с), 7.31-7.29 (2Н, м), 7.24-7.22 (1H, м), 7.19 (1H, дд, J=8.4, 1.4 Гц), 6.99 (1H, ушир. д, J=8.4 Гц), 6.52-6.51 (1Н, ушир. м), 3.70-3.66 (8Н, м), 3.58-3.55 (4Н, м), 3.37 (3H, с), 2.98 (3H, д, J=4.8 Гц).
Пример 33
Получение 2-{[3-({6-[3-(2,5,8,11-тетраоксатридекан-13-ил)уреидо]пиридин-3-ил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 33)
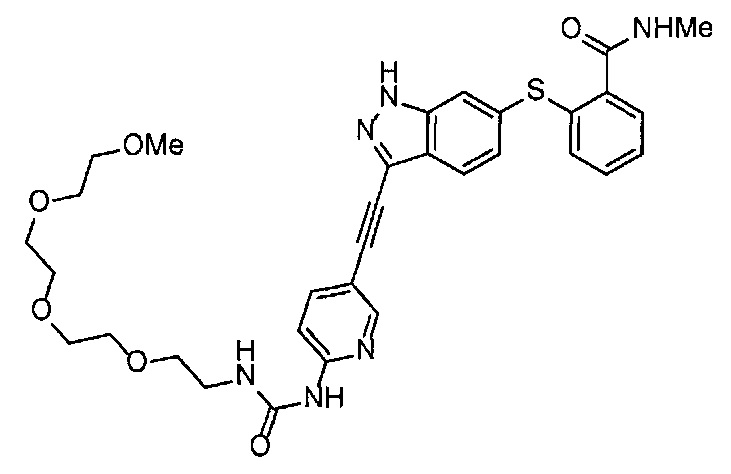
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(5-этинилпиридин-2-ил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевину (94.2 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (23.3 мг, 15%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8.44 (1Н, ушир. д, J=2.4 Гц), 7.84 (1Н, дд, J=8.8, 2.4 Гц), 7.76 (1H, ушир. д, J=8.4 Гц), 7.57 (1H, ушир. с), 7.48-7.46 (1Н, м), 7.34-7.31 (2Н, м), 7.25-7.22 (1Н, м), 7.18 (1Н, дд, J=8.4, 1.6 Гц), 7.17 (1H, д, J=8.8 Гц), 3.65-3.57 (12Н, м), 3.50-3.47 (4Н, м), 3.29 (3H, м), 2.85 (3H, с).
Пример 34
Получение 2-{[3-({6-[3-(2,5,8,11,14-пентаоксагексадекан-16-ил)уреидо]пиридин-3-ил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 34)
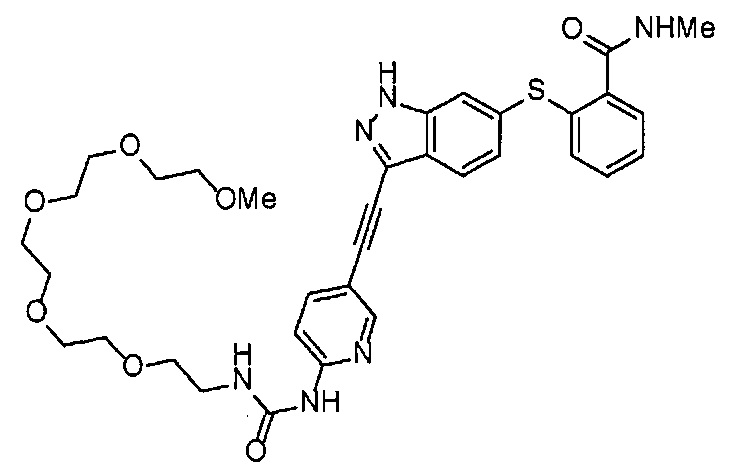
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(5-этинилпиридин-2-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевину (106.0 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (16.9 мг, 10%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 11.93 (1Н, с), 8.95 (1H, ушир. с), 8.75 (1Н, с), 8.38 (1Н, д, J=2.0 Гц), 7.74 (1Н, д, J=8.0 Гц), 7.71 (1Н, дд, J=8.8, 2.0 Гц), 7.65 (1H, ушир. с), 7.59-7.57 (1H, м), 7.26-7.23 (2Н, м), 7.18-7.16 (2Н, м), 7.03 (1H, д, J=8.8 Гц), 6.54 (1Н, ушир. кв, J=4.8 Гц), 3.64-3.56 (18Н, м), 3.52-3.50 (2Н, м), 3.33 (3H, м), 2.98 (3H, д, J=4.8 Гц).
Пример 35
Получение 2-[(3-{[5-(3-{2-[2-(2-метоксиэтокси)этокси]этил}уреидо)пиридин-2-ил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 35)
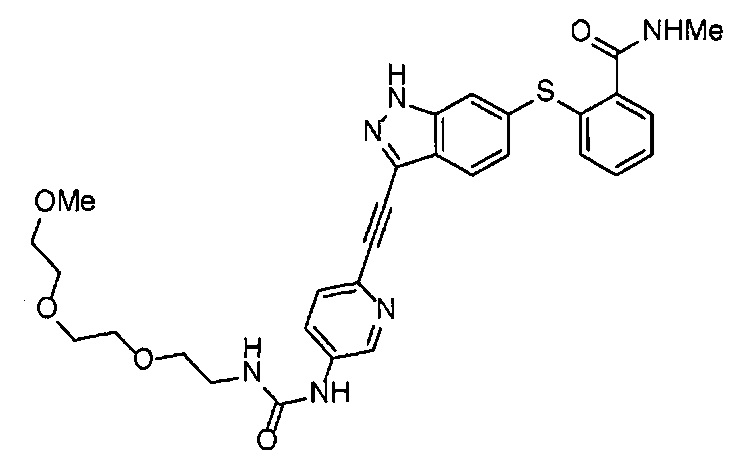
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(6-этинилпиридин-3-ил)-3-{2-[2-(2-метоксиэтокси)этокси]этил}мочевину (82.4 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (18.3 мг, 13%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 12.47 (1Н, ушир. с), 8.39 (1Н, ушир. с), 8.33 (1Н, ушир. с), 8.04 (1H, ушир. дд, J=8.4, 2.0 Гц), 7.70 (1Н, ушир. д, J=8.4 Гц), 7.63 (1Н, ушир. с), 7.51-7.49 (1H, м), 7.38 (1Н, ушир. д, J=8.4 Гц), 7.13-7.08 (3H, ушир. м), 7.03-7.01 (1H, м), 6.97-6.96 (1Н, ушир. м), 6.21-6.20 (1Н, ушир. м), 3.65-3.58 (10H, м), 3.44-3.43 (2Н, м), 3.36 (3H, с), 2.92 (3H, ушир. д, J=4.8 Гц).
Пример 36
Получение 2-{[3-({5-[3-(2,5,8,11-тетраоксатридекан-13-ил)уреидо]пиридин-2-ил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 36)
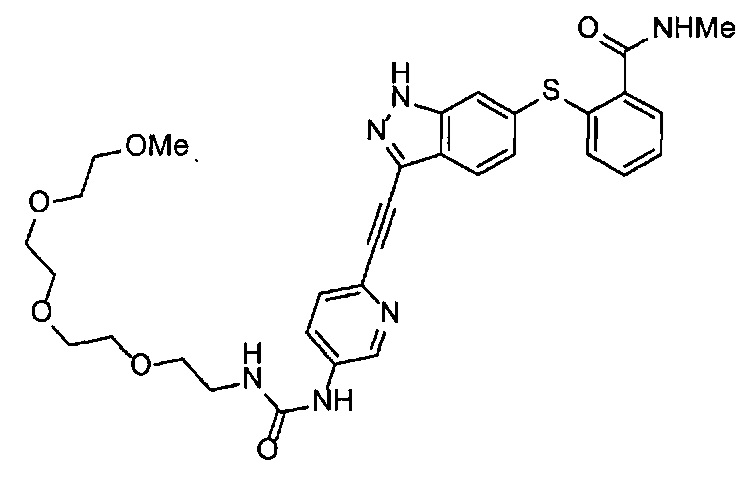
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(6-этинилпиридин-3-ил)-3-(2,5,8,11-тетраоксатридекан-13-ил)мочевину (94.2 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (27.9 мг, 18%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8.57 (1Н, ушир. д, J=2.4 Гц), 8.00 (1Н, дд, J=8.8, 2.4 Гц), 7.82 (1H, ушир. д, J=8.4 Гц), 7.60 (1H, д, J=8.8 Гц), 7.59 (1Н, ушир. с), 7.48-7.46 (1Н, м), 7.35-7.29 (2Н, м), 7.24-7.22 (1Н, м), 7.19 (1Н, дд, J=8.4, 1.2 Гц), 3.64-3.57 (12Н, м), 3.53-3.51 (2Н, м), 3.40 (2Н, т, J=5.2 Гц), 3.32 (3H, с), 2.92 (3H, с).
Пример 37
Получение 2-{[3-({5-[3-(2,5,8,11,14-пентаоксагексадекан-16-ил)уреидо]пиридин-2-ил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 37)
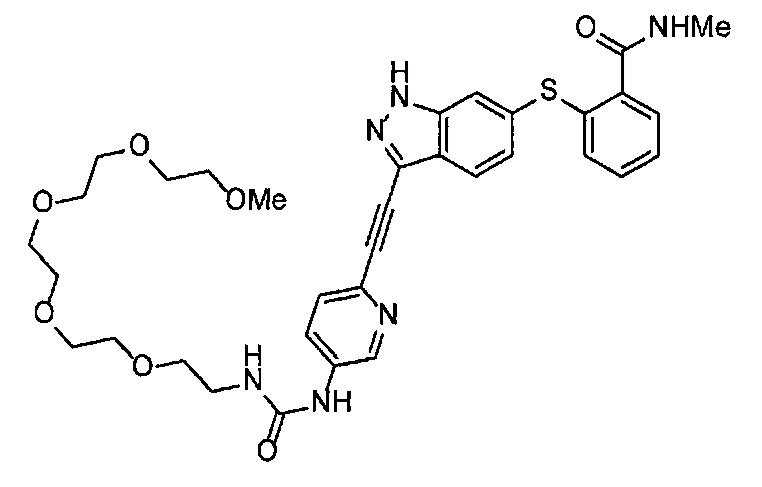
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-(6-этинилпиридин-3-ил)-3-(2,5,8,11,14-пентаоксагексадекан-16-ил)мочевину (106.0 мг, 0.27 ммоль), PdCl2(PPh3)2 (8.4 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), N,N-диметилформамид (2 мл) и N,N-диизопропилэтиламин (0.13 мл, 0.72 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (33.5 мг, 20%) в виде бледно-желтого твердого вещества.
Температура плавления: 77-78°C
1H ЯМР (400 МГц, CDCl3) δ 12.00 (1Н, ушир. с), 8.50 (1H, д, J=2.4 Гц), 8.32 (1H, с), 8.12 (1Н, дд, J=8.4, 2.4 Гц), 7.80 (1H, д, J=8.4 Гц), 7.67 (1Н, ушир. с), 7.58-7.55 (1Н, м), 7.45 (1Н, д, J=8.4 Гц), 7.22-7.19 (2Н, м), 7.16 (1H, дд, J=8.4, 1.6 Гц), 7.11-7.10 (1Н, м), 6.69 (1Н, ушир. кв, J=4.8 Гц), 6.24 (1H, ушир. м), 3.72-3.70 (4Н, м), 3.68-3.64 (6Н, м), 3.62-3.58 (6Н, м), 3.47-3.45 (4Н, м), 3.27 (3H, с), 2.96 (3H, д, J=4.8 Гц).
Пример 38
Получение N-{5-[(6-{[2-(метилкарбамоил)фенил]тио}-1H-индазол-3-ил)этинил]пиридин-2-ил}-2,5,8,11-тетраоксатридекан-13-амида (Соединение 38)
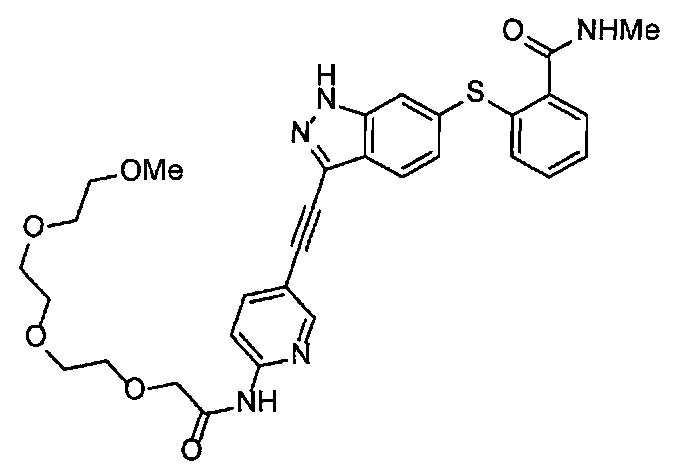
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (120.5 мг, 0.29 ммоль), N-(5-этинилпиридин-2-ил)-2,5,8,11-тетраоксатридекан-13-амид (162.3 мг, 0.50 ммоль), PdCl2(PPh3)2 (6.5 мг, 0.0093 ммоль), CuI (3.5 мг, 0.018 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (127.0 г, 71%) в виде желтого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.59 (1H, ушир. с), 10.15 (1Н, с), 8.63 (1Н, дд, J=2.4, 0.8 Гц), 8.36 (1H, кв, J=4.8 Гц), 8.18 (1H, дд, J=8.4, 0.8 Гц), 8.10 (1Н, дд, J=8.4, 2.4 Гц), 7.87 (1Н, дд, J=8.4, 0.8 Гц), 7.61 (1H, ушир. с), 7.50-7.48 (1H, м), 7.33 (1H, тд, J=7.6, 2.0 Гц), 7.30 (1H, тд, J=7.6, 2.0 Гц), 7.20 (1H, дд, J=8.4, 1.2 Гц), 7.06 (1H, дд, J=7.6, 2.0 Гц), 4.19 (2Н, с), 3.70-3.68 (2Н, м), 3.62-3.59 (2Н, м), 3.58-3.54 (4Н, м), 3.53-3.51 (2Н, м), 3.43-3.41 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 39
Получение N-{5-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]пиридин-2-ил}-2,5,8,11,14-пентаоксагексадекан-16-амида (Соединение 39)
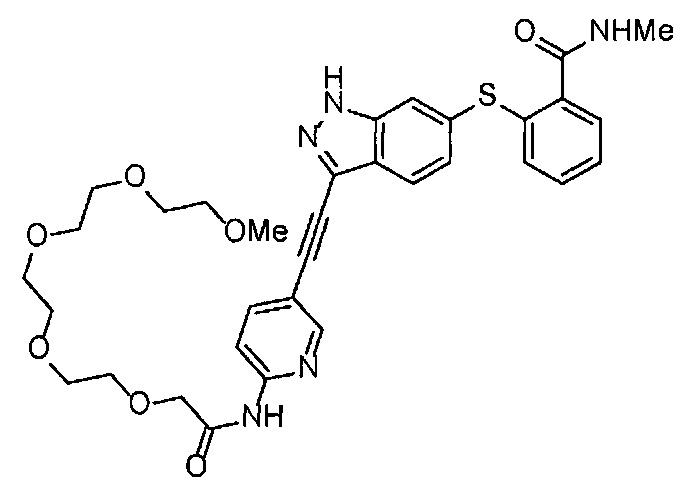
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (119.7 мг, 0.29 ммоль), N-(5-этинилпиридин-2-ил)-2,5,8,11,14-пентаоксагексадекан-16-амид (140.2 мг, 0.38 ммоль), PdCl2(PPh3)2 (5.6 мг, 0.0080 ммоль), CuI (3.0 мг, 0.016 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (95 мг, 50%) в виде белого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.59 (1Н, ушир. с), 10.15 (1Н, с), 8.64 (1Н, дд, J=2.4, 0.8 Гц), 8.36 (1Н, кв, J=4.8 Гц), 8.18 (1H, дд, J=8.4, 0.8 Гц), 8.10 (1H, дд, J=8.4, 2.4 Гц), 7.88 (1Н, ушир. д, J=8.4 Гц), 7.62 (1Н, ушир. с), 7.51-7.48 (1Н, м), 7.33 (1H, тд, J=7.6, 1.6 Гц), 7.30 (1H, тд, J=7.6, 1.6 Гц), 7.20 (1H, дд, J=8.4, 1.6 Гц), 7.06 (1H, дд, J=7.6, 1.6 Гц), 4.19 (2Н, с), 3.71-3.68 (2Н, м), 3.62-3.60 (2Н, м), 3.59-3.56 (4Н, м), 3.54-3.47 (6Н, м), 3.43-3.41 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 40
Получение N-{5-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]пиридин-2-ил}-2,5,8,11,14,17-гексаоксанонадекан-19-амида (Соединение 40)
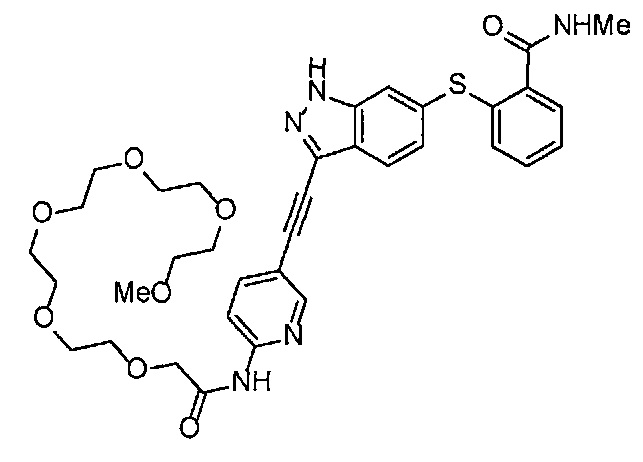
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (108.6 мг, 0.27 ммоль), N-(5-этинилпиридин-2-ил)-2,5,8,11,14,17-гексаоксанонадекан-19-амид (130.2 мг, 0.32 ммоль), PdCl2(PPh3)2 (5.4 мг, 0.0077 ммоль), CuI (2.7 мг, 0.014 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (36.0 мг, 20%) в виде бледно-коричневого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.59 (1Н, ушир. с), 10.16 (1H, с), 8.64 (1H, дд, J=2.4, 0.8 Гц), 8.37 (1H, кв, J=4.8 Гц), 8.18 (1H, дд, J=8.8, 0.8 Гц), 8.10 (1Н, дд, J=8.8, 2.4 Гц), 7.88 (1H, ушир. д, J=8.4 Гц), 7.61 (1Н, ушир. с), 7.50-7.48 (1H, м), 7.33 (1Н, тд, J=7.6, 1.6 Гц), 7.29 (1Н, тд, J=7.6, 1.6 Гц), 7.20 (1H, дд, J=8.4, 1.6 Гц), 7.07-7.05 (1Н, м), 4.19 (2Н, с), 3.70-3.68 (2Н, м), 3.62-3.60 (2Н, м), 3.58-3.55 (4Н, м), 3.53-3.48 (10H, м), 3.42-3.40 (2Н, м), 3.22 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 41
Получение N-{6-[(6-{[2-(метилкарбамоил)фенил]тио}-1Н-индазол-3-ил)этинил]пиридин-2-ил}-2,5,8,11-тетраоксатридекан-13-амида (Соединение 41)
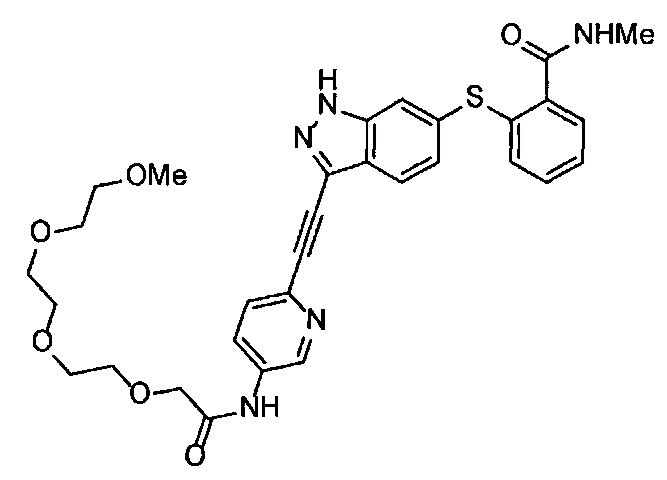
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (122.4 мг, 0.30 ммоль), N-(6-этинилпиридин-3-ил)-2,5,8,11-тетраоксатридекан-13-амид (80 мг, 0.25 ммоль), PdCl2(PPh3)2 (6.7 мг, 0.0095 ммоль), CuI (3.5 мг, 0.018 ммоль), N,N-диметилформамид (0.8 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (70.4 мг, 39%) в виде бледно-коричневого порошка.
1H ЯМР (400 МГц, ДМСО-d6) δ 13.63 (1Н, ушир. с), 10.08 (1Н, с), 8.80 (1Н, ушир. д, J=2.4 Гц), 8.36 (1Н, кв, J=4.8 Гц), 8.20 (1H, дд, J=8.8, 2.4 Гц), 7.83 (1H, дд, J=8.4, 2.4 Гц), 7.73 (1H, д, J=8.8 Гц), 7.62 (1H, ушир. с), 7.50-7.48 (1H, м), 7.33 (1H, тд, J=7.6, 2.0 Гц), 7.30 (1Н, тд, J=7.6, 2.0 Гц), 7.21 (1Н, дд, J=8.4, 1.2 Гц), 7.09-7.07 (1H, м), 4.16 (2Н, с), 3.71-3.68 (2Н, м), 3.65-3.62 (2Н, м), 3.58-3.50 (6Н, м), 3.43-3.41 (2Н, м), 3.23 (3H, с), 2.76 (3H, д, J=4.8 Гц).
Пример 42
Получение 2-[(3-{[4-(3-{2-[2-(2-гидроксиэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 42)
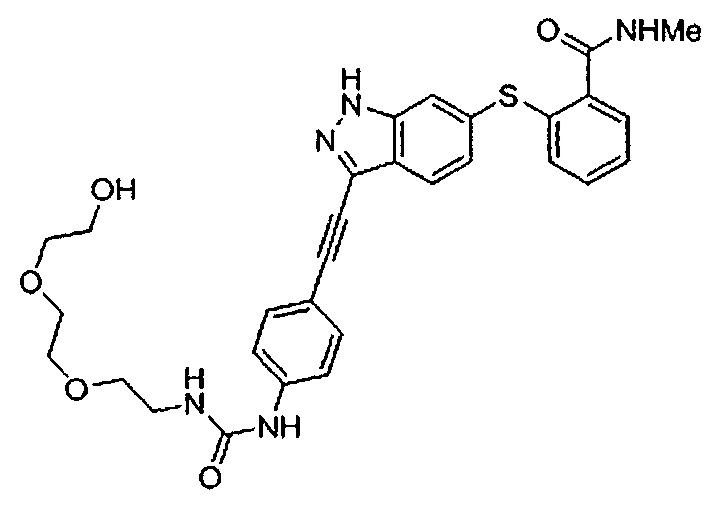
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (119.5 мг, 0.29 ммоль), 1-(4-этинилфенил)-3-{2-[2-(2-гидроксиэтокси)этокси]этил}мочевину (93.9 мг, 0.32 ммоль), PdCl2(PPh3)2 (10.5 мг, 0.015 ммоль), CuI (5.5 мг, 0.029 ммоль), ацетонитрил (1 мл) и триэтиламин (1 мл) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (36.6 мг, 22%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.75 (1H, ушир. д, J=8.4 Гц), 7.57 (1H, ушир. с), 7.50-7.43 (5Н, м), 7.33 (1H, тд, J=7.6, 1.6 Гц), 7.29 (1Н, тд, J=7.6, 1.6 Гц), 7.22 (1Н, ушир. д, J=7.6 Гц), 7.16 (1H, дд, J=8.4, 1.2 Гц), 3.70-3.64 (6Н, м), 3.60-3.56 (4Н, м), 3.39 (2Н, т, J=5.2 Гц), 2.85 (3H, с).
Пример 43
Получение 2-[(3-{[4-(3-{2-[2-(2-аминоэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 43)
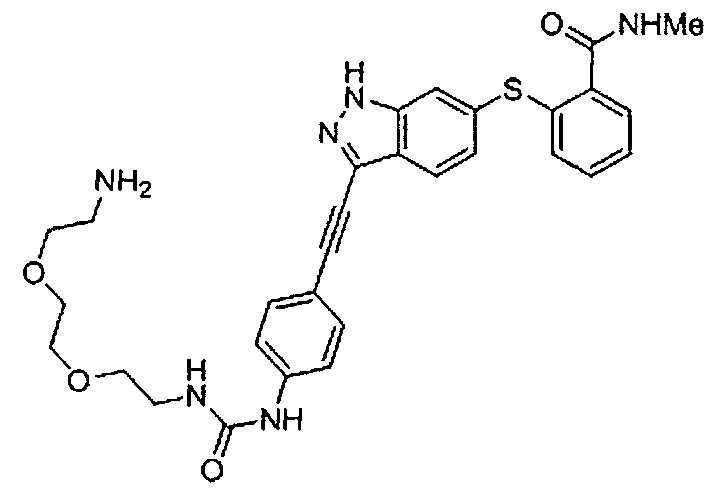
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (100.0 мг, 0.24 ммоль), 1-{2-[2-(2-аминоэтокси)этокси]этил}-3-(4-этинилфенил)мочевину (84.5 мг, 0.29 ммоль), PdCl2(PPh3)2 (8.6 мг, 0.012 ммоль), CuI (4.6 мг, 0.024 ммоль), ацетонитрил (1 мл) и триэтиламин (1 мл) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (50.4 мг, 37%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.74 (1H, ушир. д, J=8.4 Гц), 7.57 (1Н, ушир. с), 7.49-7.41 (5Н, м), 7.32 (1Н, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.21 (1H, ушир. д, J=7.6 Гц), 7.16 (1Н, дд, J=8.4, 1.2 Гц), 3.64-3.56 (6Н, м), 3.51 (2Н, т, J=5.2 Гц), 3.39 (2Н, т, J=5.2 Гц), 2.85 (3H, с), 2.78 (2Н, ушир. т, J=5.2 Гц).
Пример 44
Получение 2-{[3-({4-[3-(2-{2-[2-(диметиламино)этокси]этокси}этил)уреидо]фенил}этинил)-1Н-индазол-6-ил]тио}-N-метилбензамида (Соединение 44)
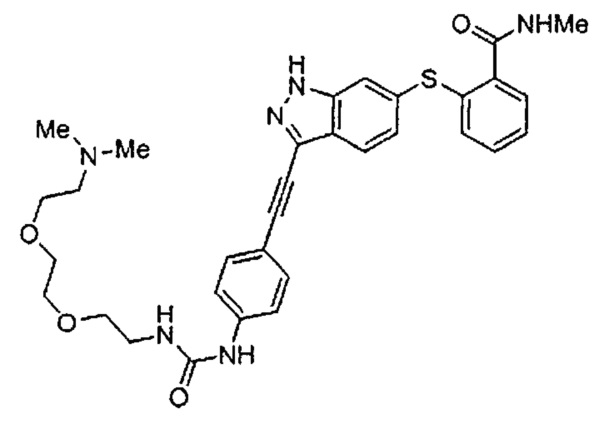
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (119.4 мг, 0.29 ммоль), 1-(2-{2-[2-(диметиламино)этокси]этокси}этил)-3-(4-этинилфенил)мочевину (111.9 мг, 0.35 ммоль), PdCl2(PPh3)2 (10.5 мг, 0.015 ммоль), CuI (5.5 мг, 0.029 ммоль), N,N-диметилформамид (1.2 мл) и N,N-диизопропилэтиламин (0.15 мл, 0.86 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (36.4 мг, 21%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.75 (1Н, ушир. д, J=8.4 Гц), 7.57 (1H, ушир. с), 7.50-7.42 (5Н, м), 7.32 (1Н, тд, J=7.6, 1.6 Гц), 7.29 (1H, тд, J=7.6, 1.6 Гц), 7.22 (1H, ушир. д, J=7.6 Гц), 7.17 (1H, дд, J=8.4, 1.2 Гц), 3.62-3.55 (8Н, м), 3.38 (2Н, т, J=5.6 Гц), 2.85 (3H, с), 2.53 (2Н, т, J=5.6 Гц), 2.25 (6Н, с).
Пример 45
Получение 2-[(3-{[4-(3-{2-[2-(2-ацетамидэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]-N-метилбензамида (Соединение 45)
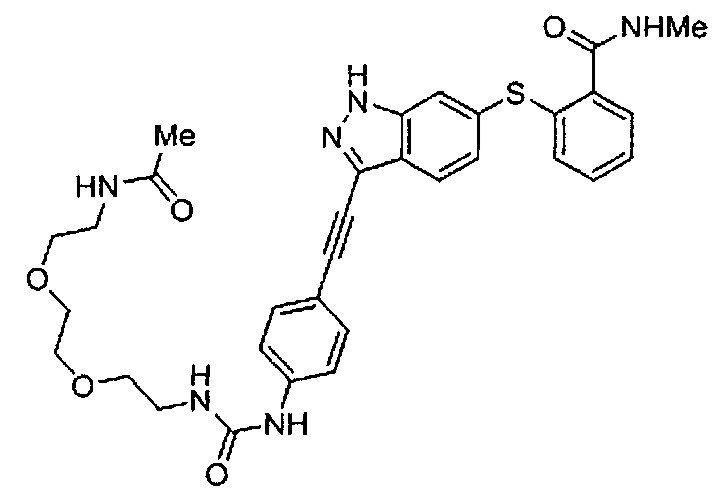
2-{(3-Иод-1H-индазол-6-ил)тио}-N-метилбензамид (90 мг, 0.22 ммоль), N-[2-(2-{2-[3-(4-этинилфенил)уреидо]этокси}этокси)этил]ацетамид (74 мг, 0.22 ммоль), PdCl2(PPh3)2 (7.7 мг, 0.011 ммоль), CuI (4.2 мг, 0.022 ммоль), ацетонитрил (1 мл) и триэтиламин (1 мл) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (31.8 мг, 24%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.76 (1H, д, J=8.4 Гц), 7.57 (1H, с), 7.50-7.43 (5Н, м), 7.33 (1H, тд, J=7.6, 1.6 Гц), 7.30 (1H, тд, J=7.6, 1.6 Гц), 7.23 (1Н, ушир. д, J=7.6 Гц), 7.18 (1Н, дд, J=8.4, 1.2 Гц), 3.63-3.53 (10H, м), 3.41-3.40 (2Н, м), 2.85 (3H, с), 1.94 (3H, с).
Пример 46
Получение N-метил-2-[(3-{[4-(3-{2-[2-(2-морфолиноэтокси)этокси]этил}уреидо)фенил]этинил}-1Н-индазол-6-ил)тио]бензамида (Соединение 46)
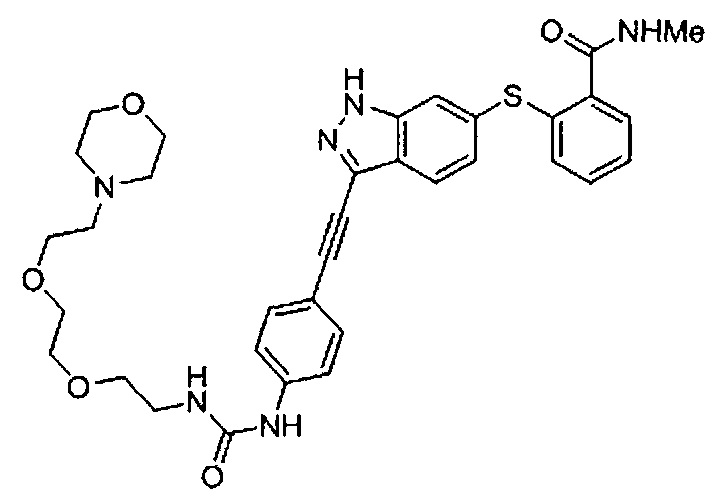
2-{(3-Иод-1Н-индазол-6-ил)тио}-N-метилбензамид (108.4 мг, 0.27 ммоль), 1-(4-этинилфенил)-3-{2-[2-(2-морфолиноэтокси)этокси]этил}мочевину (105.2 мг, 0.29 ммоль), PdCl2(PPh3)2 (9.3 мг, 0.013 ммоль), CuI (5.1 мг, 0.027 ммоль), диметилформамид (1.5 мл) и N,N-диизопропилэтиламин (0.14 мл, 0.80 ммоль) использовали в качестве исходных веществ, и проводили реакцию таким же образом, как в Примере 1, получая указанное в заголовке соединение (48.1 мг, 28%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.75 (1Н, ушир. д, J=8.4 Гц), 7.57 (1H, ушир. с), 7.50-7.42 (5Н, м), 7.32 (1Н, тд, J=7.6, 1.6 Гц), 7.29 (1Н, тд, J=7.6, 1.6 Гц), 7.21 (1H, ушир. д, J=7.6 Гц), 7.17 (1Н, дд, J=8.4, 1.2 Гц), 3.66-3.59 (10H, м), 3.56 (2Н, т, J=5.2 Гц), 3.38 (2Н, т, J=5.2 Гц), 2.85 (3H, с), 2.55 (2Н, т, J=5.2 Гц), 2.49-2.47 (4Н, м).
Примеры препаратов
Лекарственное средство, содержащее соединение по настоящему изобретению в качестве действующего вещества, можно выпускать, например, в форме следующих препаратов
1. Капсула
Полное количество компонентов (1), (2) и (3) и 1/2 количества компонента (4) смешивали и гранулировали, затем добавляли оставшееся количество компонента (4), и полученную массу помещали в желатиновую капсулу.
2. Таблетка
Полное количество компонентов (1), (2) и (3), 2/3 п. (4) и 1/2 количества компонента (5) смешивали и гранулировали. В полученную гранулу добавляли оставшееся количество компонентов (4) и (5), и прессовали в таблетку на таблеточном прессе.
3. Глазные капли
Полное количество компонентов (1), (2), (3), (4), (5) и 90 мл компонента (6) смешивали в асептических условиях и затем добавляли оставшийся объем компонента (6), получая глазные капли.
Пример тестирования 1 Оценка соединений в тесте ингибирования активности киназы VEGF-2 рецептора
В качестве испытуемых веществ брали Соединения 1-41, получение которых описано выше в Примерах.
Определение активности ингибирования киназы для каждого из соединений, описанных выше в Примерах, проводили методом Off-chip Mobility Shift Assay. Для этого анализа готовили человеческий рекомбинантный рецептор VEGF-2 в экспрессирующей бакуловирусной системе. Рекомбинантный белок экспрессировали как GST гибридный белок, используя аминокислоты 790-1356 цитозольного домена рецептора VEGF-2 (NP_002244.1) и присоединяя глутатион-S-трансферазу (GST) к N-концу. Экспрессируемый GST-VEGF-2 гибридный белок очищали хроматографией на глутатион-сефарозе. Кроме того, испытуемое соединение растворяли в диметилсульфоксиде, получая раствор с концентрацией примерно в 100 раз выше, чем тестируемая концентрация. Полученный раствор разбавляли буфером для проведения анализа (20 мМ HEPES, 0.01% Triton X-100 и 2 мМ DTT, Ph 7.5) в 25 раз, получая раствор с концентрацией в 4 выше, чем тестируемая. В тесте на ингибирование киназы, в качестве субстрата применяли CSK-тиду. Для проведения киназной реакции, 10 мл 2х концентрированного раствора VEGF-2 рецепторной киназы, 5 мл 4х концентрированного раствора испытуемого соединения, приготовленного в буфере для анализа, и 5 мл 4х концентрированного раствора субстрат/АТФ/металл смешивали в лунках полипропиленового 384-луночного планшета и проводили реакцию при комнатной температуре в течение 1 часа (концентрация субстрата: CSK-тида 1000 нМ, концентрация АТФ: 75 мкМ, магний: 5 мМ). Через 1 час добавляли 60 мл терминирующего буфера (QuickScout Screening Assist MSA) для остановки реакции. После этого разделяли пептид-субстрат и фосфорилированный пептид, содержащиеся в реакционной системе, на системе LabChip3000 (Caliper Life Science), и определяли количество обоих пептидов. Эффективность киназной реакции определяли как соотношение продуктов (P/(P+S)), вычисленное по высоте пика пептида-субстрата (S) и высоте пика фосфорилированного пептида (P). Усредненный сигнал контрольных лунок, содержащих все компоненты для реакции, принимали за 0% ингибирования, а усредненный сигнал фоновых лунок (без добавления фермента) принимали за 100% ингибирования, и степень ингибирования вычисляли по усредненному сигналу тестовых лунок для каждого испытуемого соединения. Значение IC50 определяли путем аппроксимации логистической кривой для 4 параметров, по концентрации испытуемого соединения и соответствующей степени ингибирования, с применением нелинейного метода наименьших квадратов.
Ингибирующая VEGF-2 рецептор активность соединений 1-41, полученных в Примерах, приведена в Таблице 1 и Таблице 2. Полученные в указанных Примерах соединения показали высокую степень ингибирования киназной активности VEGF-2 рецептора.
Кроме того, в качестве сравнительного примера исследовали подавление активности VEGF-2 рецепторной киназы аналогично описанному выше способу, применяя указанные ниже соединения A и B. Результаты приведены в Таблице 2.
Соединение A: 6-[2-(метилкарбамоил)фенилсульфанил]-3-Е-[2-(пиридин-2-ил)этенил] индазол (непатентованное название: Акситиниб)
Соединение B: 6-[2-(метилкарбамоил)фенилсульфанил]-3-[2-(пиридин-2-ил)этинил] индазол.
Что касается соединения A, то приобретали и использовали Акситиниб производства Tocris Bioscience (номер по каталогу: 4350). Соединение B синтезировали согласно способу, описанному в Примере 20 в WO 2006/048745.
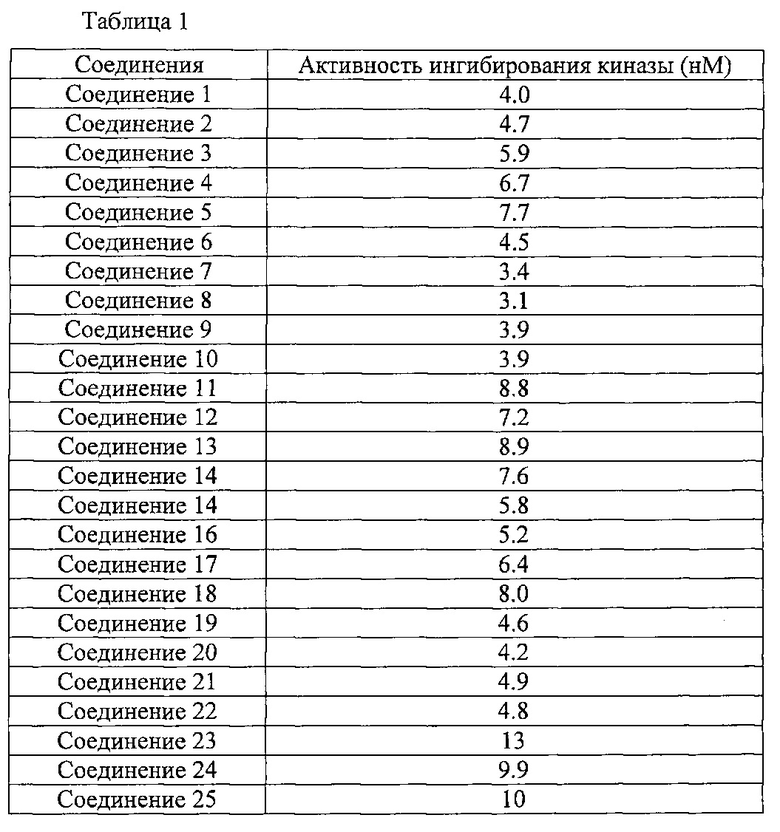
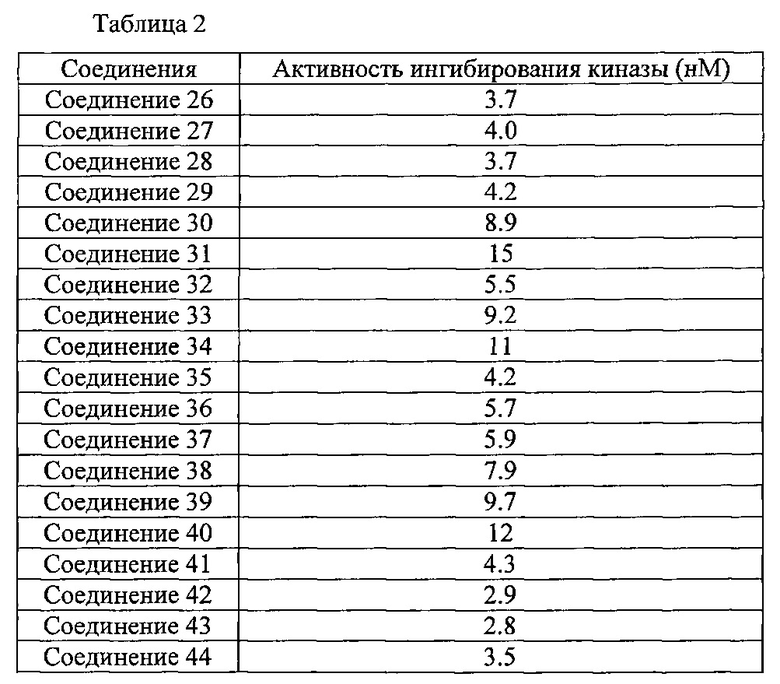
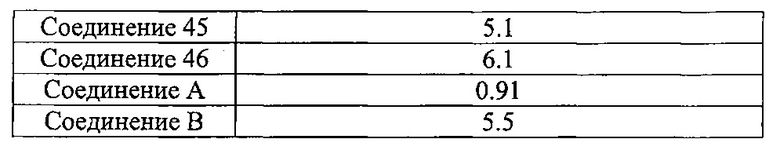
Пример тестирования 2. Эффект подавления роста, оказываемый соединениями на рост ретинальных капиллярных эндотелиальных (hRMVE) клеток человека (hRMVEC) посредством стимуляции VEGF.
Выращивание и перенос человеческих ретинальных капиллярных эндотелиальных клеток (Cell Systems Corporation, Human Retinal Microvascular Endothelial Cells, каталожный номер: ACBRI 181) осуществляли согласно прилагающимся инструкциям. Раствор фактора прикрепления (Cell Systems Corporation, Фактор прикрепления) помещали в 96-луночный планшет (Iwaki Glass Co., Ltd.) до заполнения лунок и затем удаляли, так что он покрывал поверхность лунок. После этого клетки (hRMVEC), суспендированные в среде CS-C (Cell Systems Corporation, CS-C medium kit, каталожный номер: CS-4Z0-500R), содержащей плазму, факторы роста и Culture Boost, высевали с плотностью 2×103 клеток/100 мкл/лунку, и инкубировали при 37°C в атмосфере с 5% CO2 в течение 1 дня. После этого удаляли среду из hRMVEC, дважды промывали раствором PBS и затем инкубировали в среде CS-C (Cell Systems Corporation, CS-C medium kit, каталожный номер: CS-4Z3-500R), содержащей 1% FBS и фактор роста, в течение 6 часов. Затем снова удаляли среду из hRMVEC и один раз промывали раствором PBS, затем добавляли среду CS-C (Cell Systems Corporation, CS-C medium kit, каталожный номер: CS-4ZO-500S), содержащую рекомбинантный человеческий VEGF (Becton Dickinson and Company), в которую добавляли раствор, полученный разбавлением каждого соединения в 3 раза, в количестве 100 мкл/лунку, и инкубировали hRMVEC в течение 48 часов.
Число клеток определяли с помощью МТТ-теста (применяли набор Cell Counting Kit-8 от DOJINDO LABORATORIES), и степень подавления роста hRMVEC испытуемым соединением вычисляли по следующему уравнению:
Степень подавления роста клеток (%) = 100-100 × (поглощение лунки с добавлением VEGF и соединения - поглощение лунки без добавления VEGF) / (поглощение лунки с добавлением VEGF-поглощение лунки без добавления VEGF).
Кроме того, зная степень подавления роста клеток, вычисляли IC50 (M) как концентрацию, при которой наблюдается 50%-ное подавление, используя для вычисления следующую формулу:
IC50(M)=10∧(LOG(А/В)×(50-С)/(D-С)+LOG(В))
A: Более высокая из 2 концентраций, являющихся наиболее близкими к 50%-ному подавлению более высокими и низкими значениями.
В: Более низкая из 2 концентраций, являющихся наиболее близкими к 50%-ному подавлению более высокими и низкими значениями
C: Степень подавления при концентрации B
D: Степень подавления при концентрации A
Активность репрезентативных соединений, относящихся к полученным в Примерах соединениям, по подавлению роста hRMVE клеток представлена в Таблице 3. Кроме того, в качестве сравнительного примера аналогично измеряли активность подавления роста hRMVE клеток для соединения A и соединения В, применявшихся в Примере тестирования 1, и результаты также помещали в Таблицу 3.
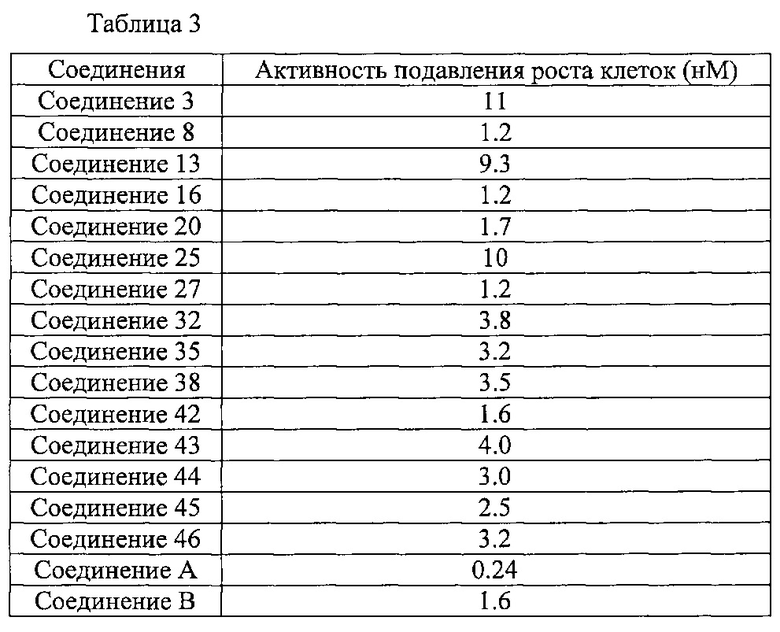
Пример тестирования 3. Исследование растворимости
1.0 мл 0.1% фосфатного буферного раствора (pH 7.0) добавляли к 1.00 мг соединений, полученных в Примерах. Полученную смесь подвергали ультразвуковой обработке в течение 30 секунд и затем встряхивали при комнатной температуре в течение ночи. Полученную суспензию оставляли при комнатной температуре на 30 минут, затем фильтровали через хроматографический диск (0.20 мкм), получая тестируемый раствор. Тестируемый раствор и стандартный раствор анализировали методом LCMS (приборы: ACQUITY UPLC H-CLASS SYSTEM и Xevo TQ-S, производство Nihon Waters K.K.), и растворимость вычисляли методом внешнего стандарта по площади полученного пика.
В качестве сравнительного примера, исследование растворимости аналогичным образом проводили для соединения A и соединения B, использовавшихся в Примере тестирования 1.
Условия анализа методом СВЭЖХ
Колонка: ACQUITY UPLC (зарегистрированная торговая марка) BEH С18,1.7 мкм, 2.1×50 мм (Waters)
Подвижная фаза: 0.006% водный раствор муравьиной кислоты: метанол = от 40:60 до 60:40
Длина волны детектора: 354 нм
Температура колонки: 40°C
Скорость потока: 0.25 мл/мин
Объем ввода: 1.0 мкл
Стандартный раствор: 1.00 мг полученных в Примерах соединений растворяли в метаноле (10 мл), и затем разбавляли в 1000 раз метанолом, получая стандартный раствор с концентрацией 100 нг/мл.
Результаты измерения растворимости в 0.1% фосфатном буферном растворе (pH 7.0) приведены в Таблице 4.
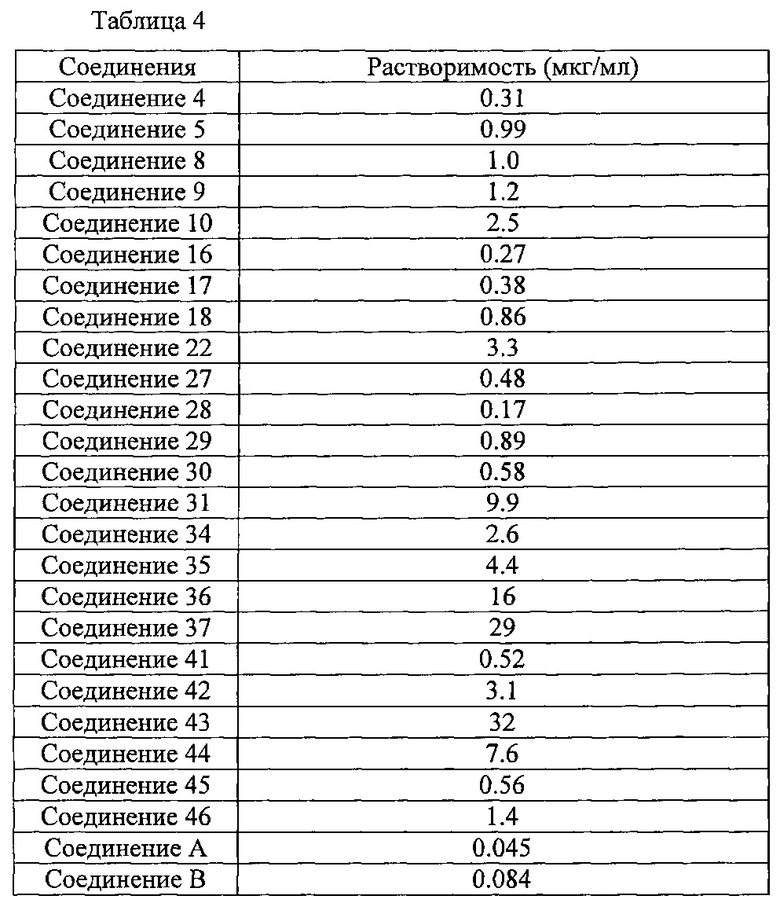
Пример тестирования 4. Исследование фотоустойчивости
1.00 мг полученных в Примерах соединений растворяли в 10 мл метанола, затем разбавляли смесью вода/метанол (об./об. = 1:1), получая тестируемые растворы с концентрацией 10 мкг/мл. Приготовленный образец оставляли под белой флуоресцентной лампой (500 люкс) на 6 часов, и затем анализировали методом ВЭЖХ, определяя остаточное содержание (%) соединений.
Условия анализа методом ВЭЖХ приведены ниже. В качестве сравнительного примера, такое же исследование проводили для соединения A, использовавшегося в Примере тестирования 1.
Остаточные количества (светоустойчивость) приведены в Таблице 5. Полученные в Примерах соединения были устойчивы к свету в растворе.
Условия анализа методом ВЭЖХ
Колонка: YMC-Pack ODS-A AA12S05-1506WT, 6.0×150 мм, 5 мкм (YMC CO., LTD.)
Подвижная фаза: Раствор А: 0.01 об./об. % водный раствор муравьиной кислоты, Раствор В: 0.01 об./об. % раствор муравьиной кислоты - метанол, соотношение (A:B)=4:6
Длина волны детектора: 323 нм (соединение 8), 330 нм (соединение 37), 354/363 нм (Z форма/E форма соединения A)
Температура колонки: 40°C
Скорость потока: 1.0 мл/мин
Объем ввода: 20 мкл
Стандартный раствор: Тестируемый раствор непосредственно после получения хранили при температуре 4°C и применяли в качестве стандартного раствора.

Пример тестирования 5. Проникновение лекарственного средства в сетчатку/хороид
Для полученных в Примерах соединений 8, 27 и B определяли концентрацию, необходимую для проникновения лекарственного средства в ткани сетчатки/хороида после закапывания. Применяемый в данном исследовании офтальмологический раствор получали общеизвестным способом суспендированием соединения 8, соединения 27 или соединения A, соответственно, в фосфатном буферном растворе (pH 7.0) таким образом, что концентрация лекарственного средства составляла 0.25%. Для измерения концентрации лекарственного средства в тканях сетчатки/хороида, глазные капли вводили кроликам (японский белый кролик) в дозировке 50 мкл на глаз, кроликов усыпляли через 2 часа после введения и удаляли глазные яблоки. Удаленные глазные яблоки замораживали жидким азотом, рассекали на две части посередине, и затем брали образцы тканей сетчатки/хороида из заднего отрезка глаза. Для экстрагирования соединения из образцов тканей, ткани мелко измельчали в метаноле, получая лизат, и затем лизат центрифугировали, собирая супернатант. В экстрагированный раствор соединения из супернатанта добавляли эквивалентное количество ультрачистой воды, раствор фильтровали через мембранный фильтр (0.22 мкм), и полученный фильтрат служил финальным раствором для измерения. Концентрацию лекарственного средства в фильтрате определяли методом жидкостной хроматографии-масс-спектрометрии (LC/MS/MS), и концентрацию соединения в тканях сетчатки/хороида вычисляли по площади пика, полученной методом внешнего стандарта. Условия проведения LC/MS/MS анализа приведены ниже.
Условия проведения анализа методом LC/MS/MS
Прибор: ACQUITY UPLC (зарегистрированная торговая марка) H-Class system (Waters)
Колонка: ACQUITY UPLC (зарегистрированная торговая марка) ВЕН С18, 1.7 мкм, 2.1×50 мм (Waters)
Подвижная фаза: 0.006% водный раствор муравьиной кислоты: метанол = 40:60 (соединение 27), 0.006% водный раствор муравьиной кислоты: метанол = 47:53 (соединения 8 и B)
Детектор: тандемный квадрупольный масс-спектрометр Xevo (зарегистрированная торговая марка) TQ-S (Waters) с датчиком ESI
Способ ионизации: регистрация положительно заряженных ионов методом ESI
Режим анализа: MRM
Напряжение на конусе: (соединение 8) 6 B, (соединение 27) 72 B, (соединение В) 2 B
Напряжение в ячейке соударений: 26 эВ (соединение 8); 26 эВ (соединение 27); 18 эВ (соединение B)
Превращение ионов: m/z 588.12→367.97 (соединение 8); m/z 545.18→393.99 (соединение 27); m/z 385.07→353.95 (соединение В)
Средние концентрации соединений в сетчатке/хороиде приведены в Таблице 6. Для определения концентрации соединения в сетчатке/хороиде, проводили тест нулевой гипотезы с помощью JMP (зарегистрированная торговая марка) (SAS Institute Inc.), исключали выброс, и затем использовали среднее значение ± стандартное отклонение.

Проникающие концентрации соединений 8 и 27 в сетчатку/хороид были значительно более высокими, чем у соединения B (соединения B и 8: n=6, соединение 27: n=4, p≤0.05). Из приведенных в Таблице 6 результатов видно, что соединение (I) по настоящему изобретению обладает отличной проникающей способностью в ткани сетчатки/хороида.
Промышленное применение
Соединение (I) по настоящему изобретению и его фармакологически приемлемая соль могут применяться в качестве ингибитора VEGF рецептора и пригодны для профилактики или лечения разных заболеваний или состояний, в которых задействован данный рецептор. Кроме того, соединение (I) по настоящему изобретению и его фармакологически приемлемая соль обладают высокой растворимостью в водных растворах и отличной устойчивостью в растворенном состоянии, и поэтому могут применяться, например, в форме жидких препаратов, таких как глазные капли. Соединение (I) по настоящему изобретению и его фармакологически приемлемая соль могут применяться в качестве лекарственных средств и т.п. для профилактики или лечения заболеваний сетчатки, сопровождающих ангиогенез или эдему.
| название | год | авторы | номер документа |
|---|---|---|---|
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2008 |
|
RU2450001C2 |
| АГОНИСТЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО СРЕДСТВА | 2014 |
|
RU2654483C2 |
| АГОНИСТ СФИНГОЗИН-1-ФОСФАТНЫХ РЕЦЕПТОРОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2020 |
|
RU2798605C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА | 2020 |
|
RU2803284C1 |
Изобретение относится к алкинилиндазольному производному, имеющему общую формулу (I):
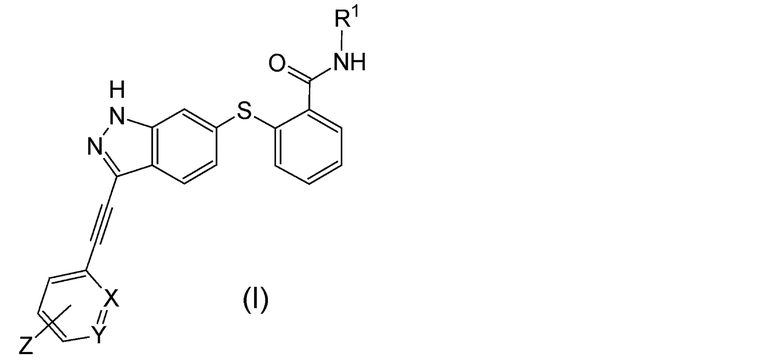 ,
,
или его фармацевтически приемлемой соли. В формуле (I) R1 представляет собой низший алкил, X и Y одинаковые или разные и каждый представляет собой CH или N, при условии, что X и Y одновременно не являются N, Z представляет собой группу, имеющую общую Формулу (a):
 ,
,
в которой R2 представляет собой низший алкил, который может иметь заместитель, выбранный из группы, состоящей из: гидрокси, амино, диметиламино, ацетиламино, или морфолино, n представляет собой целое число от 1 до 7, A представляет собой подструктуру, представленную следующей формулой:
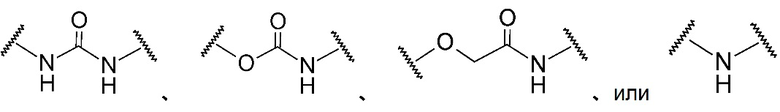 .
.
Также предложено лекарственное средство, содержащее алкинилиндазольное производное. Алкинилиндазольное производное формулы (I) представляет собой ингибитор VEGF рецептора тирозинкиназы и применяется для лечения или профилактики заболевания сетчатки, сопровождающего ангиогенез или эдему. 2 н. и 5 з.п. ф-лы, 6 табл., 126 пр.
1. Алкинилиндазольное производное, имеющее общую формулу (I):
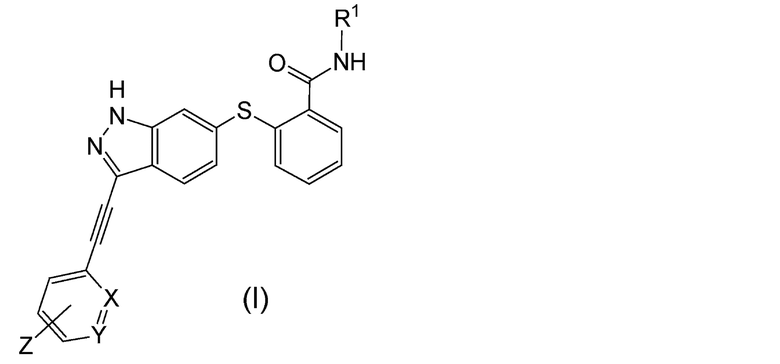 ,
,
в которой R1 представляет собой низший алкил, X и Y одинаковые или разные и каждый представляет собой CH или N, при условии, что X и Y одновременно не являются N, Z представляет собой группу, имеющую общую Формулу (a):
 ,
,
в которой R2 представляет собой низший алкил, который может иметь заместитель, выбранный из группы, состоящей из: гидрокси, амино, диметиламино, ацетиламино, или морфолино, n представляет собой целое число от 1 до 7, A представляет собой подструктуру, представленную следующей формулой:
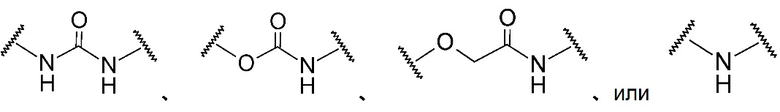 ,
,
или его фармацевтически приемлемая соль.
2. Алкинилиндазольное производное по п. 1 или его фармацевтически приемлемая соль, где X и Y одновременно представляют собой CH.
3. Алкинилиндазольное производное по п. 1 или 2 или его фармацевтически приемлемая соль, где Z присоединен в пара-положении.
4. Алкинилиндазольное производное по любому из пп. 1 - 3 или его фармацевтически приемлемая соль, где A представляет собой подструктуру, имеющую следующую формулу:

5. Лекарственное средство, которое представляет собой ингибитор VEGF рецептора (фактора роста эндотелия сосудов) тирозинкиназы, отличающееся содержанием алкинилиндазольного производного по любому из пп. 1 - 4 или его фармацевтически приемлемой соли от 0,001 до 10 вес./об.% в лекарственном средстве.
6. Лекарственное средство по п. 5, применяющееся для лечения или профилактики заболевания сетчатки, сопровождающего ангиогенез или эдему.
7. Лекарственное средство по п. 6, где заболевание сетчатки, сопровождающее ангиогенез или эдему, представляет собой возрастную макулярную дегенерацию, макулярную эдему, диабетическую ретинопатию, ретинопатию недоношенных, окклюзию вены сетчатки, вторичную катаракту, миопическую хориоидальную неоваскуляризацию или глаукому.
| WO 2006048745 A1, 11.05.2006 | |||
| WO 2006048744 A1, 11.05.2006 | |||
| WO 2004094388 A2, 04.11.2004 | |||
| WO 2003024931 A1, 27.03.2003 | |||
| WO 2004056806 A1, 08.07.2004 | |||
| WO 2001002369 A2, 11.01.2001 | |||
| US 20110008421 A1, 13.01.2011 | |||
| WO 2013138346 A1, 19.09.2013 | |||
| WO 2013138343 A1, 19.09.2013 | |||
| RU 2009136593 A, 10.04.2011. |

