ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к замещенному производному 2Н-пиразола, выступающему в роли противоопухолевого средства, селективного ингибитора CDK4/6. В частности, настоящее изобретение относится к веществу с формулой (I) или к его фармацевтически приемлемой соли, которая выступает в качестве селективного ингибитора CDK4/6.
УРОВЕНЬ ТЕХНИКИ
Клеточный цикл преимущественно регулируется семейством серин-/треонинкиназ, также известных как циклин-зависимые киназы (cyclin-dependent kinase, CDK). Они способствуют смене фаз клеточного цикла, транскрипции генетической информации, нормальному делению и пролиферации клеток путем связывания с соответствующими циклинами, которые регулируют активность циклин-зависимых киназ. CDK4/6 - критический фактор регуляции клеточного цикла, запускающий переход от фазы роста (G1) к фазе репликации ДНК (S1). Во время пролиферации клеток комплекс циклина D и CDK4/6 фосфорилирует белок гена ретинобластомы (Rb). После фосфорилирования белка Rb - супрессора опухолевого роста, высвобождается фактор транскрипции E2F, который был связан с нефосфорилированным Rb. Начинается транскрипция E2F-зависимых генов, что способствует проходу через контрольную точку, переходу из фазы G1 в фазу S и запуску цикла пролиферации клетки. То есть ингибирование CDK4/6 и последующего образования комплекса циклин D-CDK4/6 препятствует переходу из фазы G1 в фазу S и таким образом подавляет рост опухолевой ткани, что является целью настоящего изобретения. При раке молочной железы (РМЖ) с рецепторами к эстрогенам (ER+) достаточно часто повышена активность CDK4/6, a CDK4/6 представляет собой критическую мишень в сигнальном пути рецепторов к эстрогенам (ER). Доклинические данные позволяют предположить, что одновременное ингибирование CDK4/6 и сигнального пути ER оказывает синергическое действие и подавляет рост клеток ER-положительного рака молочной железы (РМЖ) в фазе G1.
В разработке препаратов, нацеленных на CDK4/6, существует жесткая конкуренция. В 2010 г. Pietzsch опубликовал обзор разработок в этой области (Mini-Rev. Med. Chem. 2010, 10, 527-539). В 2014 г. Malorni также опубликовал обзор последних разработок ингибиторов CDK4/6, проходивших доклинические и клинические исследования применения при раке молочной железы (Curr. Opin. Oncol. 2014, 26, 568-575). Обширные исследования CDK4/6 способствовали разработке ряда ингибиторов CDK с разной степенью селективности и также привели к появлению нескольких высокоселективных эффективных ингибиторов CDK4/6. Палбоциклиб (PD0332991) - один из таких эффективных и высокоселективных ингибиторов CDK4/6, который дошел до стадии клинических исследований и применялся для лечения женщин с раком молочной железы, метастазирующим или на поздней стадии, экспрессирующим рецепторы к эстрогенам (ER+) или не экспрессирующим рецепторы эпидермального фактора роста 2-го типа (HER2-). На основании промежуточных данных исследования PALOMA-1 компания «Пфайзер» подала заявку на регистрацию нового лекарственного средства (NDA) палбоциклиба в Управление по контролю качества пищевых продуктов и лекарственных средств (FDA) США в августе 2014 г. В феврале 2015 г. FDA одобрило вывод палбоциклиба на рынок. Два других ингибитора CDK4/6, абемациклиб (LY2835219) и LEE-011, находятся на стадии набора пациентов в клинические исследования 3-й фазы. Эти низкомолекулярные гетероциклические вещества могут применяться для лечения не только рака молочной железы, но и ряда других злокачественных новообразований. На эти вещества оформлены патенты WO 2012018540, WO 2012129344, WO 2011101409, WO 2011130232, WO 2010075074, WO 2009126584, WO 2008032157 и WO 2003062236.
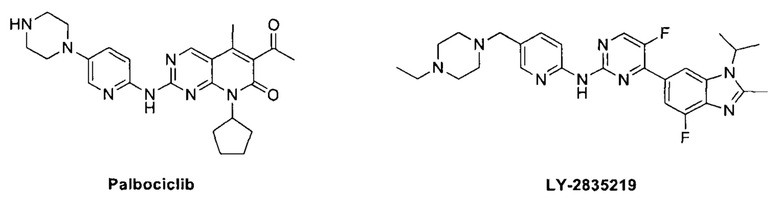
Мы планируем создать новое поколение высокоселективных ингибиторов CDK4/6 с более высокой безопасностью и эффективностью, которые будут лучше отвечать потребностям рынка и обладать большей терапевтической эффективностью. Настоящее изобретение представляет собой селективный ингибитор CDK4/6 с новой структурой, и показано, что вещества с такой структурой обладают превосходным противоопухолевым действием.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
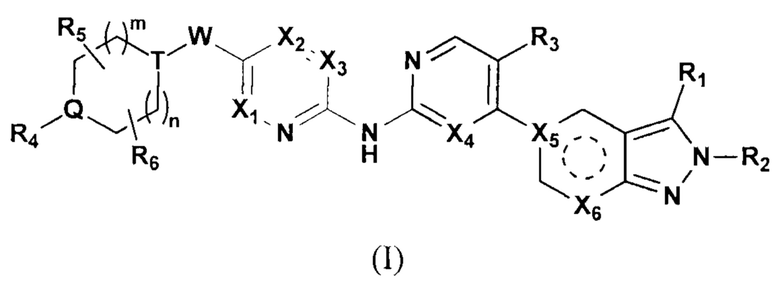
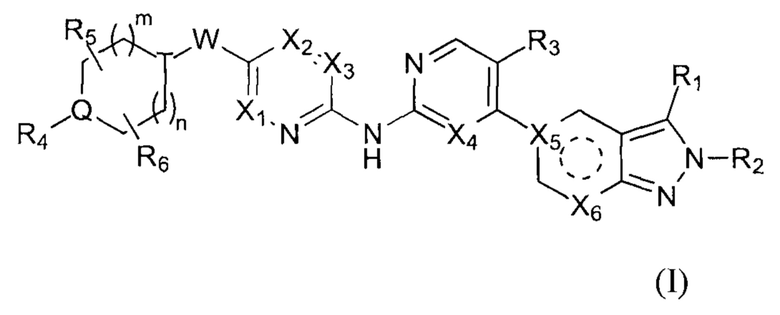
Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли,

где  выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и  ;
;
R1 выбирают из группы, состоящей из Н, галогена, ОН, NH2, C1-8 алкила, С2-8алкенила, С2-8 алкеналкила и циклоалкила С3-7;
R2 выбирают из группы, состоящей из Н, галогена, С1-8 алкила, С3-7 циклоалкила, арильной группы и гетегоарильной группы;
R3 выбирают из группы, состоящей из Н, галогена, -OR8, -SR8, -N(R8)(R9) и С1-3 алкила;
R4, R5 и R6 независимо друг от друга выбирают из группы, состоящей из Н, галогена, ОН, NH2, CN, NO2 и =O, либо выбирают из группы, состоящей из С1-8 алкила, С1-8 алкиламино, N,N-ди(C1-8 алкил)амино, C1-8 алкоксил-С1-8 алкила, C1-8 гидроксиалкила, С2-8 алкенила, С2-8 алкинила, С3-7 циклоалкила и 3-7 членной гетероциклоалкильной группы, каждая из которых необязательно замещена 1, 2 или 3 R;
в некоторых случаях любые два радикала R4, R5 или R6 могу образовать кольцо из 3-7 атомов;
R7 выбирают из группы, состоящей из Н, галогена, -OR8, -SR8, -N(R8)(R9) и С3-7 циклоалкила;
X1, Х2, X3 и Х4 независимо друг от друга выбирают из группы, состоящей из N и C(R10);
Х7 выбирают из группы, состоящей из карбонила и C(R11)(R12);
W выбирают из группы, состоящей из О, S и простой связи;
Т выбирают из группы, состоящей из N и C(R10), и Т не может быть N, когда W - О или S;
Q выбирают из группы, состоящей из N и C(R10);
m и n независимо друг от друга выбирают из группы, состоящей из 0, 1 и 2;
R8 и R9 независимо друг от друга выбирают из группы, состоящей из Н, С1-8 алкила и С3-7 циклоалкила;
R выбирают из группы, состоящей из F, Cl, Br, I, NH2, CN, ОН, CF3, CHF2, CH2F, NHCH3 и N(CH3)2;
в некоторых случаях R8 и R9 присоединяются к одному и тому же атому и образуют 3-7-членное кольцо с 1-4 гетероатомами;
термин «гетеро» или «гетероатом» обозначает О, S, S(=O), S(=O)2 или N;
R10 выбирают из группы, состоящей из Н, галогена, ОН, NH2, CN, C1-6 алкила, C1-6 алкоксила, С3-5 циклоалкила, CN, -OR8, -SR8, -N(R8)(R9), -C(=O)R8, -C(=O)OR8, -C(=O)N(R8)(R9), -S(=O)R8, -S(=O)2R8, -S(=O)N(R8)(R9) и -S(=O)2N(R8)(R9);
R11 и R12 независимо друг от друга выбирают из группы, состоящей из Н, ОН, галогена, C1-8 алкила и С3-7 циклоалкила;
в некоторых случаях R4 и R10 присоединяются к одному и тому же атому и образуют 3-7-членное кольцо; и
в некоторых случаях структурная единица  может быть замещена структурной единицей
может быть замещена структурной единицей  .
.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный R1 выбирают из группы, состоящей из изопропила, 2-пропенила и аллила.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный R2 выбирают из группы, состоящей из метила и фенила.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный R3 представляет собой F.
В некоторых вариантах осуществления настоящего изобретения вышеуказанные R4, R5 и R6 независимо друг от друга выбирают из группы, состоящей из Н, галогена, ОН,  и
и  .
.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный R7 выбирают из группы, состоящей из Н, F и Cl.
В некоторых вариантах осуществления настоящего изобретения структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 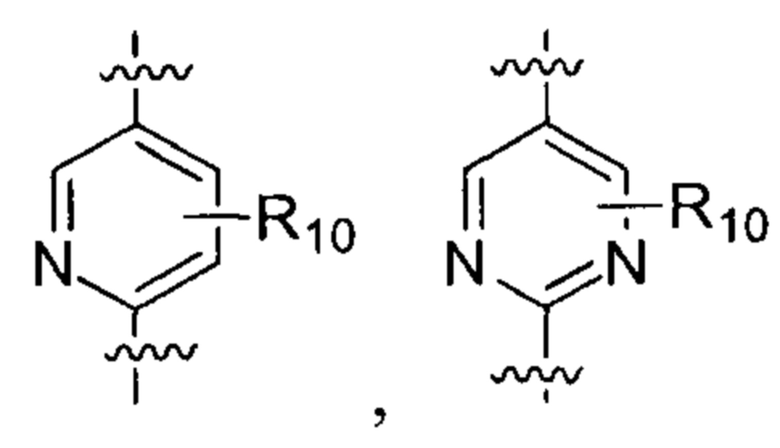 ,
,  и
и 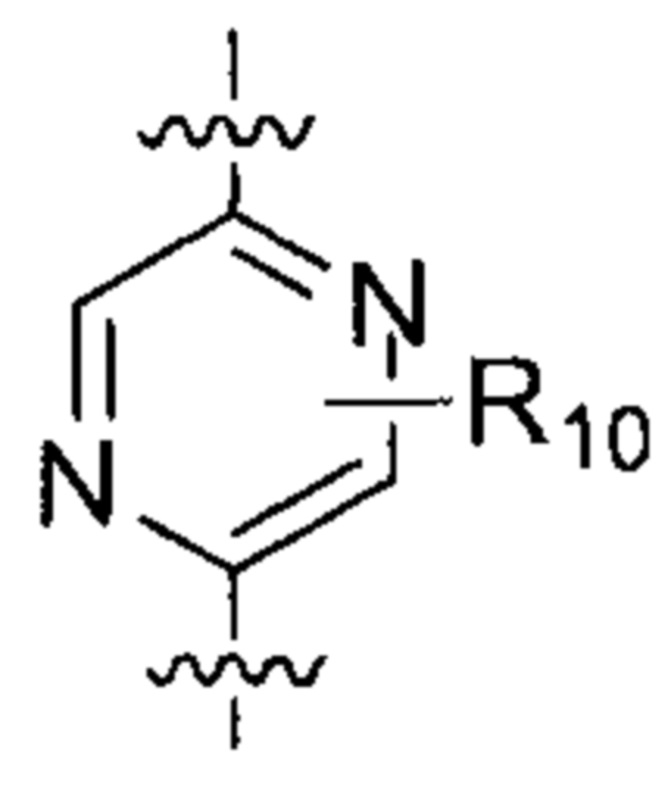 .
.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный R10 выбирают из группы, состоящей из Н, ОН, NH2, F, Cl, CN, 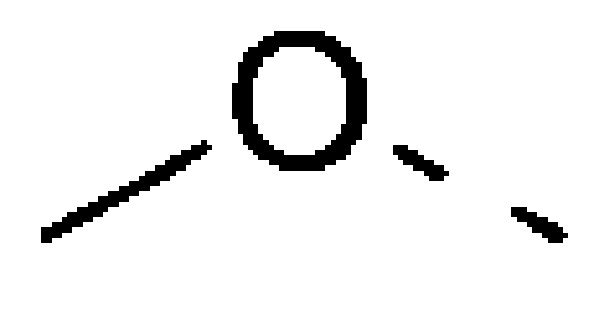 и Me.
и Me.
В некоторых вариантах осуществления настоящего изобретения вышеуказанный Х4 выбирают из группы, состоящей из N и СН.
В некоторых вариантах осуществления настоящего изобретения вышеуказанную структурную единицу 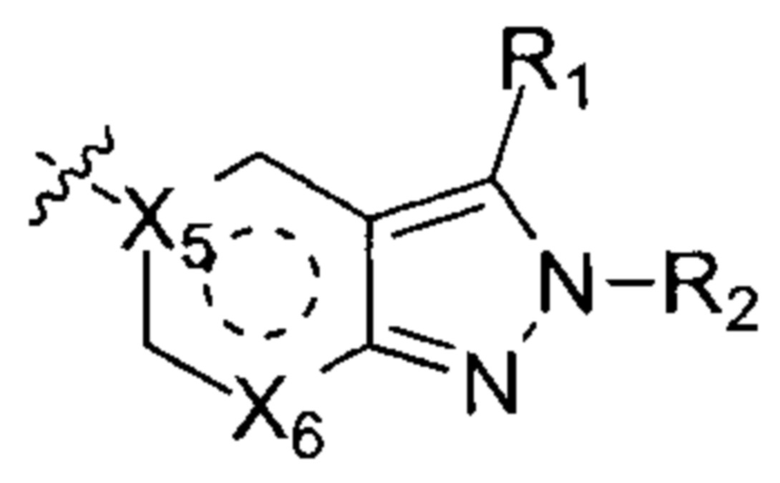 выбирают из группы, состоящей из
выбирают из группы, состоящей из 
 и
и  .
.
В некоторых вариантах осуществления настоящего изобретения вышеуказанную структурную единицу 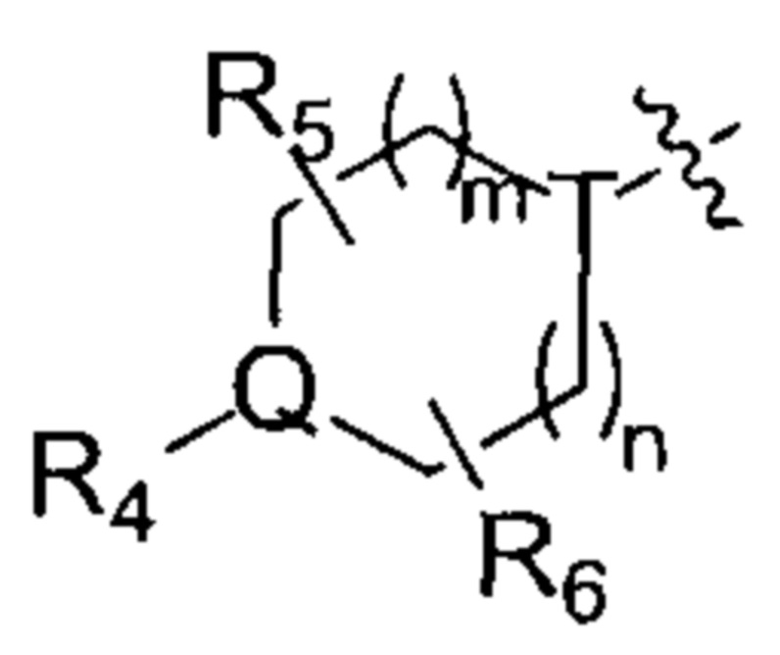 выбирают из группы, состоящей из
выбирают из группы, состоящей из 


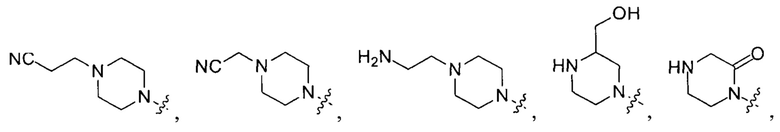
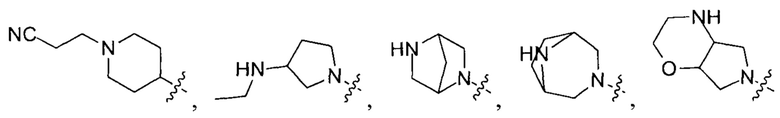 и
и  .
.
Вещество настоящего изобретения выбирают из группы, состоящей из
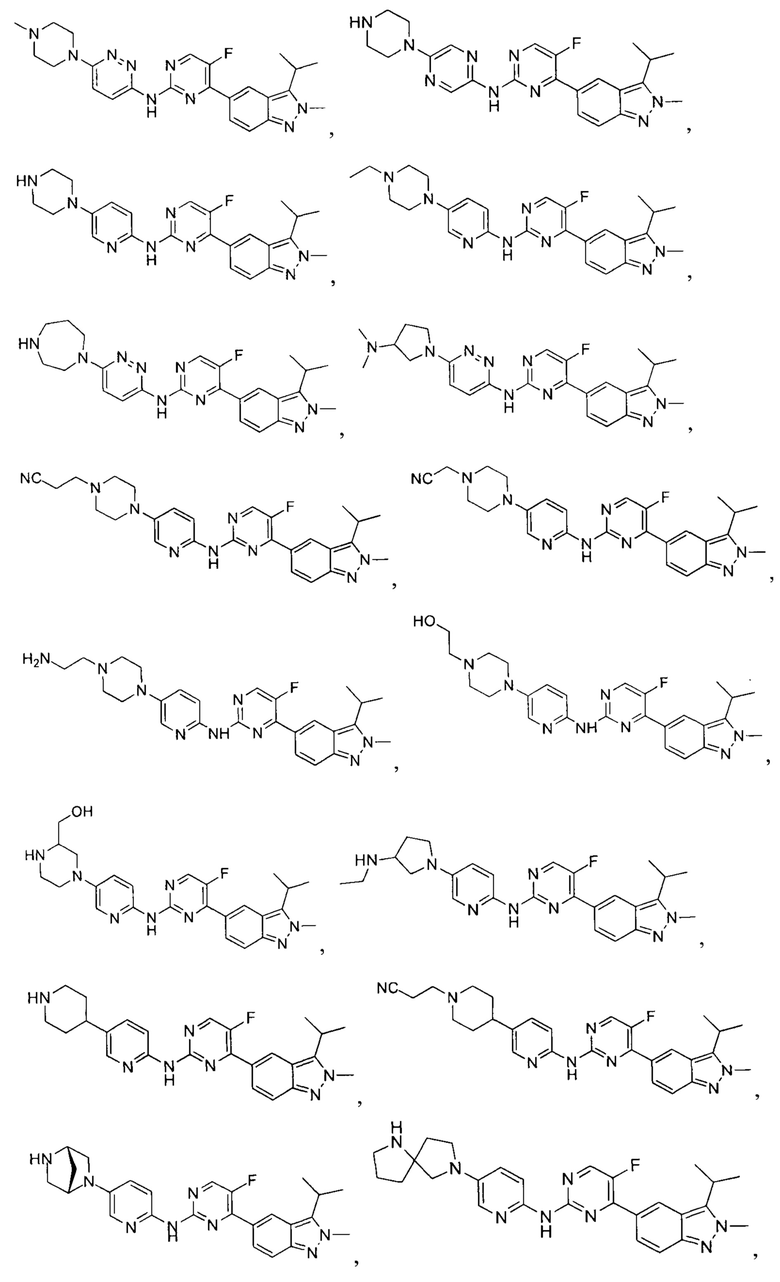

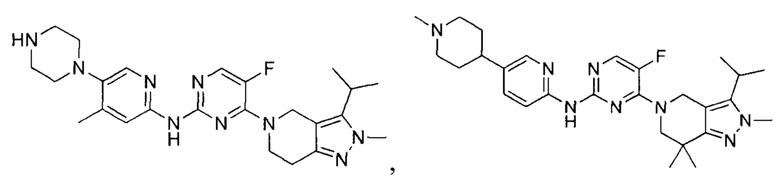 и
и
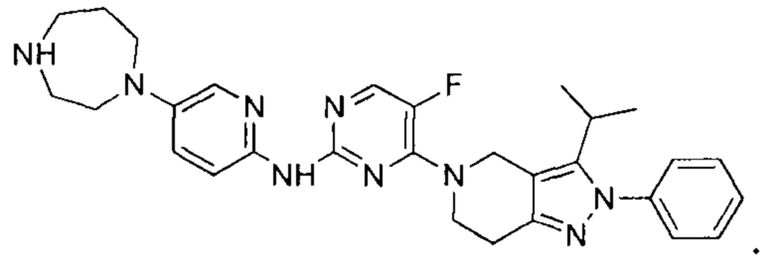
Определения и пояснения
В настоящем документе перечисленные далее термины и фразы имеют следующие значения, если не указано иное. Отдельный термин или фразу, которым не надо определение, не следует считать неопределенными или неясными, а следует понимать в их обычном значении. Торговые наименования в настоящем документе обозначают соответствующий продукт или его активный компонент.
C1-8 обозначает количество атомов углерода в углеводородной группе. Например, C1 обозначает, что в группе содержится только один атом углерода, С2 - два атома углерода, и так далее.
В веществе с формулой (I) термин «C1-8 алкил» обозначает прямую или разветвленную углеводородную группу, содержащую от 1 до 8 атомов углерода, включая, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 2,2-диметилпропил, н-гексил, н-гептил, н-октил и тому подобное.
Термин «С2-8 алкенил» обозначает прямую или разветвленную углеводородную группу, содержащую от 2 до 8 атомов углерода и одну двойную связь, включая этенил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил и тому подобное.
Термин «С2-8 алкинил» обозначает прямую или разветвленную углеводородную группу, содержащую от 2 до 8 атомов углерода и одну тройную связь, включая этинил, пропинил, 1-бутинил, 2-бутинил и тому подобное.
Термин «С3-7 циклоалкил» означает моноциклическую или дициклическую углеводородную группу, содержащую от 3 до 7 атомов углерода, включая циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное.
В настоящем документе термин «фармацевтически приемлемый» означает, применительно к веществам, материалам, составам и (или) лекарственных формам, пригодность к использованию в контакте с тканями человека и животных по результатам достоверной медицинской оценки, без избыточной токсичности, раздражения, анафилаксии и других нарушений и осложнений и с приемлемым соотношением пользы и риска.
Термин «фармацевтически приемлемая соль» означает соли веществ настоящего изобретения, которые получены с использованием веществ настоящего изобретения с указанными замещающими группами и относительно нетоксичных кислот или оснований. Если вещество настоящего изобретения содержит относительно кислотные функциональные группы, его соль присоединения основания можно получить путем взаимодействия достаточно количества основания с нейтральной формой вещества в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, органических аминов, магния и другие. Если вещество настоящего изобретения содержит относительно основные функциональные группы, его соль присоединения кислоты можно получить путем взаимодействия достаточно количества кислоты с нейтральной формой вещества в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли неорганических кислот и соли органических кислот. Под такими неорганическими кислотами подразумеваются, например, соляная кислота, бромоводородная кислота, азотная кислота, угольная кислота, бикарбонат-ион, фосфорная кислота, моногидрофосфат-ион, дигидрофосфат-ион, серная кислота, гидросульфат-ион, гидроиодистая кислота, фосфористая кислота и другие. Под такими органическими кислотами подразумеваются, например, уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, муравьиная кислота, молочная кислота, миндальная кислота, фталевая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, винная кислота, метансульфоновая кислота и другие. Фармацевтически приемлемые соли присоединения кислоты также включают соли аминокислот (например, аргинина и других) и соли органических кислот, таких как глюкуроновая кислота (см. Berge et al., «Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые вещества настоящего изобретения имеют основные или кислотные функциональные группы и поэтому могут быть преобразованы в любые соли присоединения кислоты или основания.
Предпочтительно восстанавливать нейтральную форму вещества путем обычного взаимодействия соли с основанием или кислотой и последующего отделения исходного вещества. Исходная форма вещества отличается от различных солевых форм некоторыми физическими свойствами, например растворимостью в полярных растворителях.
В настоящем документе «фармацевтически приемлемая» соль означает производные веществ настоящего изобретения, полученные путем взаимодействия описанного исходного вещества с кислотой или основанием с образованием соли. Примеры фармацевтически приемлемых солей включают, помимо прочего, соли неорганических или органических кислот с основаниями, такими как амины, щелочные металлы или соли органических кислот, таких как карбоновые кислоты. Фармацевтически приемлемые соли включают обычные нетоксичные соли или соли четвертичного аммония и исходного вещества, такие как соли, образованные нетоксичными неорганическими или органическими кислотами. Обычные нетоксичные соли включают, помимо прочего, соли неорганических или органических кислот. При этом неорганические или органические кислоты выбирают из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбонат-иона, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, муравьиной кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромоводородной кислоты, соляной кислоты, гидроиодистой кислоты, нафтола, 2-гидроксиэтилсульфоновой кислоты, молочной кислоты, лактозы додецилсульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактальдегида, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, фолината, янтарной кислоты, сульфаминовой кислоты, п-аминобензолсульфоновой кислоты, серной кислоты, таннины, винной кислоты и п-толуолсульфоновой кислоты.
Фармацевтически приемлемые соли настоящего изобретения можно получить из исходных веществ с кислотными или основными группами путем обычного процесса химического синтеза. В целом, соли получают следующим образом: проводят реакцию исходных веществ в свободной кислотной или основной форме со стехиометрическим количеством подходящего основания или кислоты в воде, органическом растворителе или их смеси. В целом, предпочтительно использовать неводные среды, такие как эфир, этилацетат, этанол, изопропанол, ацетонитрил и другие.
В дополнение к солевой форме вещество, описанное в настоящем изобретении, может быть представлено в форме пролекарства. Пролекарства веществ, описанных в настоящем документе, легко подвергаются химическим трансформациям в физиологических условиях и таким образом превращаются в вещества настоящего изобретения. Кроме того, пролекарства можно превратить в вещества настоящего изобретения с использованием химических или биохимических методов in vitro.
Некоторые вещества настоящего изобретения могут существовать в несольватированной или сольватированной форме, в том числе в виде гидратов. В целом, сольватированные формы эквивалентны несольватированным формам, и все входят в объем настоящего изобретения.
Некоторые вещества настоящего изобретения могут содержать асимметрические атомы углерода (центры оптической активности) или двойные связи. Все рацематы, диастереомеры, геометрические изомеры и индивидуальные изомеры входят в объем настоящего изобретения.
В настоящем документе рацематы, обогащенные одним или двумя энантиомерами вещества, а также энантиочистые вещества изображены по методу, описанному в Maehr., J. Chem. Ed., 1985, 62: 114-120. Для обозначения абсолютной конфигурации центра хиральности используются клиновидные закрашенные и штриховые связи, если не указано иное. Если описанные в настоящем документе вещества содержат этиленовые двойные связи или другие геометрические центры асимметрии, они включают Е- и Z-геометрические-изомеры, если не указано иное. Аналогичным образом, в объем настоящего изобретения входят все таутомеры.
Вещества настоящего изобретения могут существовать в форме определенных геометрических или стереоизомеров. В объем настоящего изобретения входят вещества настоящего изобретения в любой возможной форме, в том числе транс- и цис-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)- и (L)-энантиомеры, их рацемические смеси и другие смеси, в том числе смеси, богатые энантиомерами и диастереомерами. В состав замещающих групп, например алкильных, могут входить асимметрические атомы углерода. Все их изомеры и смеси изомеров также входят в объем настоящего изобретения.
Оптически активные (R)- и (S)-энантиомеры и (D)- и (L)-энантиомеры могут быть получены путем хирального синтеза, с использованием хиральных реактивов или путем других обычных методик. Энантиомер вещества настоящего изобретения может быть получен путем ассимметрического синтеза или путем образования производного с хиральным вспомогательным элементом, когда смесь диастереоизомеров разделяют, а вспомогательную группу отщепляют для получения желаемого чистого энантиомера. Либо, при наличии в молекуле основной группу (например, аминогруппы) или кислотной группы (например, карбоксильной группы) можно получить соли диастереоизомеров с помощью подходящих оптически активных кислот или оснований. Затем диастереоизомеры разделяют способом, известным специалистам средней квалификации, и получают чистые энантиомеры. Кроме того, обычно энантиомеры и диастереомеры разделяют хроматографическим методом. Такой хроматографический метод подразумевает использование хиральной неподвижной фазы и в некоторых случаях сочетается с химическим методом (например, получение производного карбаминовой кислоты из амина).
Вещества настоящего изобретения могут содержать неестественное соотношение изотопов одного или нескольких атомов в составе веществ. Например, для маркировки веществ могут использоваться радиоактивные изотопы, такие как тритий (3Н), йод-125 (125I) или углерод-14 (14С). Все изотопные варианты веществ настоящего изобретения, радиоактивные или нет, входят в объем настоящего изобретения.
Термин «фармацевтически приемлемый носитель» означает любой состав или носитель, который способен доставить эффективное количество действующего вещества настоящего изобретения, не изменяет биологическую активность действующего вещества и не оказывает токсического побочного действия на живой организм или пациента. Типичные носители включают воду, масла, растения и минералы, основы для кремов, лосьонов мазей и другое. Эти материалы основ включают суспензирующие средства, загустители, усилители проникновения через кожу и другие. Их препараты известны специалистам средней квалификации в области косметических средств или препаратов для местного применения. Подробную информацию о носителя см. в Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005). Приведенный документ полностью включен в настоящий документ посредством ссылки.
Термин «вспомогательное вещество» относится к носителям, разбавителям и (или) среде, необходимой для получения эффективного фармацевтического состава.
В случае лекарственных препаратов или фармакологически активных веществ «эффективное количество» или «терапевтически эффективное количество» означает такое количество препарата, которое не токсично, но способно произвести ожидаемый эффект. Что касается пероральной лекарственной формы в настоящем изобретении, «эффективное количество» действующего вещества в составе означает количество действующего вещества, которое требуется для достижения ожидаемого эффекта при использовании в сочетании с другим действующем веществом в том же составе. Эффективное количество индивидуально и зависит от возраста и общего состояния человека. Оно также зависит от действующего вещества. Специалисты средней квалификации могут определить индивидуальное эффективное количество путем стандартных экспериментов.
Термины «активный компонент», «терапевтическое средство», «активное средство» или «действующее вещество» означают химическое вещество, которое способно эффективно излечить рассматриваемые заболевания, нарушения или состояния.
Термин «замещенный» означает замещение одного или нескольких атомов водорода (в том числе вариантов водорода и дейтерия) на конкретном атоме замещающими группами, при условии, что валентность атома остается в норме и замещенное вещество стабильно. Если замещающая группа представляет собой кетон (т.е. =O), то замещаются два атома водорода. Замещающая группа никогда не располагается на арильной группе. Термин «необязательно замещенный» означает, что замещение может произойти, а может и не произойти. Если не указано иное, допускаются замещающие группы любого химически доступного типа и в любом количестве.
Если в составе или структуре вещества какая-либо переменная (например, R) указана более одного раза, определение переменной зависит от ситуации. Следовательно, например, если группа замещается на 0 или 2 R, то такая группа может быть необязательно замещена не более чем двумя R, и характер R определяется независимо в каждой отдельно взятой ситуации. Кроме того, замещающие группы и (или) их варианты можно сочетать только в том случае, если в результате образуется стабильное вещество.
Если замещающая группа соединяется поперечными связями с двумя атомами кольца, такую группу можно подсоединять к любому атому кольца. Если не указано, что какой-либо атом перечисленной замещающей группы не соединяется с веществом, которое включено в общую формулу, но не конкретизировано, такую группу можно подсоединять через любой из ее атомов. Замещающие группы и (или) их варианты можно сочетать только в том случае, если в результате образуется стабильное вещество. Например, структурная единица  или
или  означает, что замещение может происходить по любому атому циклогексила или циклогексадиена.
означает, что замещение может происходить по любому атому циклогексила или циклогексадиена.
Замещающая алкильная или гетероалкильная группа обычно называется «алкильной замещающей группой», которую выбирают из, помимо прочего, одной или нескольких следующих групп: -R', -OR', =O, =NR', =N-OR', -NR'R'', -SR', галоген -SiR'R''R''', OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', NR'C(O)NR''R''', -NR''C(O)2R', -NR'''''-C(NR'R''R''')=NR'''', NR''''C(NR'R'')=NR''', -S(O)R', -S(O)2R', -S(O)2NR'R'', NR''SO2R', -CN, -NO2, -N3, -CH(Ph)2 и фтор-(С1-С4)алкил. Количество замещающих групп варьирует от 0 до 2m'+1, где m' - общее количество атомов углерода в группе атомов. Предпочтительно, чтобы R', R'', R''', R'''' и R''''' независимо друг от друга представляли собой водород, замещенную или незамещенную гетероалкильную группу, замещенную или незамещенную арильную группу (например, арил, замещенный 1-3 атомами галогенов), замещенную или незамещенную алкильную группу, алкоксильную группу, тиоалкаксильную группу или арилалкильную группу. Если вещество настоящего изобретения содержит один или несколько R, каждый R выбирают независимо друг от друга, как в случае с одним или несколькими R', R'', R''', R'''' и R'''''. Если R' и R'' присоединяются к одному атому азота, их можно сочетать с атомом азота с образованием 5-, 6- или 7-членного кольца. Например, -NR'R'' должен включать, помимо прочего, 1-пирролидинил и 4-морфолинил. Из вышеприведенного обсуждения замещающих групп специалисты средней квалификации могу понять, что термин «алкил» включает группу, атомы углерода которой связаны с группами, отличными от атомов водорода, такими как галогеналкильная группа (например, -CF3, -CH2CF3) и ацильная группа (например, -С(O)СН3, -С(O)CF3, -С(O)СН2ОСН3 и другие).
Аналогично упомянутым замещающим алкильным группам, замещающая арильная или гетероарильная группа обычно именуется «арильной замещающей группой» и ее выбирают из, например R', -OR', -NR'R'', -SR', галогена, -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'C(O)NR''R''', -NR''C(O)2R', -NR'''''-C(NR'R''R''')=NR'''', NR''''C(NR'R'')=NR''', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR''SO2R', -CN, -NO2, -N3, -CH(Ph)2, фтор-(С1-С4)алкоксила и фтор-(С1-С4)алкила. Количество замещающих групп варьирует от 0 до общего количество свободных валентностей арильного кольца, где R', R'', R''', R'''' и R''''' предпочтительно независимо друг от друга выбирать из водорода, замещенной или незамещенной алкильной группы, замещенной или незамещенной гетероалкильной группы, замещенной или незамещенной арильной группы и замещенной или незамещенной гетероарильной группы. Если вещество настоящего изобретения содержит один или несколько R, каждый R выбирают независимо друг от друга, как в случае с одним или несколькими R', R'', R''', R'''' или R'''''.
Две замещающих группы на соседних атомах арильной группы или гетероарильного кольца могут быть необязательно замещены группой с общей формулой -T-C(O)-(CRR')q-U-, где Т и U независимо друг от друга выбирают из -NR-, -О-, CRR'-или простой связи, a q - целое число от 0 до 3. Альтернативно, две замещающих группы на соседних атомах арильной арильной группы или гетероарильного кольца могут быть необязательно замещены группой с общей формулой -А(СН2)rB-, где А и В независимо друг от друга выбирают из -CRR'-, -О-, -NR-, -S-, -S(O)-, S(O)2-, -S(O)2NR'-или простой связи, а r - целое число от 1 до 4. В некоторых случаях простая связь образованного таким образом нового кольца может быть заменена двойной связью. Альтернативно, две замещающих группы на соседних атомах арильной арильной группы или гетероарильного кольца могут быть необязательно замещены группой с общей формулой -А(СН2)rB-, где s и d независимо друг от друга выбирают из целых чисел от О до 3, соответственно. X представляет собой -О-, -NR', -S-, -S(O)-, S(O)2- или S(O)2NR'-. Замещающие группы R, R', R'' и R''' предпочтительно независимо друг от друга выбирать из водорода и замещенной или незамещенной алкильной группы (C1-С6), соответственно.
Термин «гало» или «галоген» сам по себе и в составе другой замещающей группы означает фтор, хлор, бром или йод, если не указано иное. Кроме того, термин «галоалкил» включает моногалоалкил и полигалоалкил. Например, термин «(С1-С4) галоалкил» включает, помимо прочего, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и другие.
Примеры галоалкильных групп включают, помимо прочего, трифторметил, трихлорметил, пентафлорэтил и пентахлорэтил. «Алкоксил» означает вышеупомянутые алкильные группы с определенным количеством атомов углерода и связью через кислородный мостик. Алкоксильная группа C1-6 включает C1 алкоксил, С2 алкоксил, С3 алкоксил, С4 алкоксил, С5 алкоксил и С6 алкоксил. Примеры алкоксилов включают, помимо прочего, метоксил, этоксил, н-пропоксил, изопропоксил, н-бутоксил, втор-бутоксил, трет-бугокснл, н-пентоксил и s-пентоксил. «Циклоалкил» включает насыщенные циклы, такие как циклопропил, циклобутил или циклопентил. Циклоалкил С3-7 включает С3 циклоалкил, С4 циклоалкил, C5 циклоалкил, С6 циклоалкил и С7 циклоалкил. «Алкенил» включает углеводородную группу с прямой или разветвленной цепью с одной или несколькими двойными углеродными связями (например, этенил или пропенил) в любом стабильном участке цепи.
Термин «гало» или «галоген» означает фтор, хлор, бром и йод.
Термин «гетеро» означает гетероатомы или гетероатомные группы (т.е. группы атомов, содержащие гетероатомы, отличные от атомов углерода (С) и водорода (Н), например, кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (В), -О-, -S-, =O, =S, -С(=O)O-, -С(=O)-, -C(=S)-, -S(=O), -S(=O)2-, и необязательно замещенные -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- или -S(=O)N(H)-.
Термин «кольцо» означает замещенный или незамещенный циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил, арил или гетероарил. Так называемые кольца включают простое кольцо, связанное кольцо, спиральное кольцо, комбинированное кольцо или кольцо с внутренним мостиком. Количество атомов в кольце обычно считают по количеству членов кольца. Например, «5-7-членное кольцо» означает, что в кольце от 5 до 7 атомов. Если не указано иное, в некоторых случаях кольцо может содержать от 1 до 3 гетероатомов. То есть «5-7-членное кольцо» включает, например, фенилпиридин и пиперидинил. С другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил и не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую не менее одного кольца, где каждое «кольцо» определяется независимо друг от друга по приведенному выше определению.
Если не указано иное, термин «гетероцикл» или «гетероциклический» означает стабильное моно-, би- или трициклическое кольцо, которое содержит гетероатом(-ы) или гетероатомную(-ые) группу(-ы). Оно может быть насыщенным, частично насыщенным или ненасыщенным (ароматическим) и содержит атомы углерода и 1, 2, 3 или 4 гетероатома в цикле, которые независимо друг от друга выбирают из N, О и S, и при этом любое из вышеуказанных гетероциклических колец может быть конденсировано с бензольным кольцом или представлять собой бициклическую систему. Гетероатомы азота и серы в некоторых случаях могут быть окислены (т.е., NO и S(O)p). Атом азота может быть замещен или не замещен (т.е. N или NR, где R - это Н или другая замещающая группа, которая определяется в настоящем документе). Гетероцикл может быть присоединен к боковым группам любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное вещество стабильно, описанный здесь гетероцикл может иметь замещающую группу по атому углерода или атому азота. Атом(-ы) азота в гетероцикле(-ах) может(могут) необязательно быть кватернизованы. Предпочтительный вариант осуществления - тот, в котором общее количество атомов S и О в гетероцикле превышает 1, и эти атомы не соседние. Другой предпочтительный вариант осуществления - тот, в котором общее количество атомов S и О не превышает 1. Как использовано в данном описании, термин «ароматическая гетероциклическая группа» или «гетероарил» означает стабильное 5-, 6- или 7-членное моноциклическое или бициклическое кольцо, либо 7-, 8-, 9- или 10-членное бициклическое гетероциклическое ароматическое кольцо, которое состоит из атомов углерода и 1, 2, 3 или 4 гетероатомов, которые независимо друг от друга выбирают из N, О и S. Атом азота может быть замещен или не замещен (т.е. N или NR, где R - это Н или другая замещающая группа, определенная в настоящем документе). Гетероатомы азота и серы могут быть необязательно окислены (т.е. NO и S(O)p). Стоит отметить, что общее количество атомов S и О в ароматическом гетероциклическом кольце не превышает 1. В определение гетероцикла также входит кольцо с внутренним мостиком. Если один или несколько атомов (а именно, С, О, N или S) соединяют два не соседних атома углерода или азота с образованием внутреннего мостика в кольце, предпочтительный вариант такого кольца включает, помимо прочего, один атом углерода, два атома углерода, один атом азота, два атома азота и одну углеродно-азотную группу. Следует отметить, что мостик всегда превращает простое кольцо в трициклическое кольцо. В кольце с внутренним мостиком замещающие группы могут быть расположены на любых атомах кольца, в том числе на мостике.
Примеры гетероциклических соединений включают, помимо прочего, акридинил, азоцинил, бензимидазол, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинилдекагдирохинолил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-6]тетрогидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индолил, индоленил, дигидроиндолил, инденил, индолил, 3H-индолил, изатиноил, изобензофуранил, пиран, изоиндолил, изоиндолил, изодигидроиндолил, изоиндолил, индолил, изохинолил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, изоксазолил, оксиндолил, пиримидинил, фенантридил, фенантролинил, феназин, фенотиазин, бнзоксантинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридоксазол, пиридимидазол, пиридотиазол, пиридил, пиримидинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, пиразолил, хиназолинил, хинолинил, 4H-хинолизинил, хиноликсанил, хиноклидинил, тетрагидрофурил, тетрагидроизохинолил, тетрагдирохинолил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенил, тиенилоксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантен. Также включает конденсированные кольца и спиральные соединения.
Если не указано иное, термин «углеводородная группа» или ее конкретное название (например, алкил, алекнил, алкинил, фенил и другие), сам по себе или в составе другой замещающей группы, означает прямую, разветвленную или циклическую углеводородную группу атомов либо их сочетание, которое может быть полностью насыщенным, содержать одну или несколько ненасыщенных связей, может иметь одну, две или более замещающих группу и может быть моновалентным (например, метил), бивалентным (например, метилен) или поливалентным (например, метенил). Она может включать би- или поливалентные группы атомов и может содержать указанное количество атомов углерода (например, C1-С10 означает от 1 до 10 атомов углерода). «Углеводородная группа» включает, помимо прочего, алифатические и ароматические углеводородные группы. Такие алифатические углеводородные группы включают алифатические и циклические углеводородные группы, в том числе, помимо прочего, алкил, алкенил и алкинил. Такие ароматические углеводородные группы включают, помимо прочего, 6-12-членные ароматические углеводородные группы, такие как фенил, нафтил и другие. В некоторых вариантах осуществления термин «алкил» означает прямую или разветвленную группу атомов или группу с сочетанием прямой и разветвленной цепей, которая может быть насыщенной или содержать одну или несколько ненасыщенных связей, а также может включать ди- или поливалентные группы атомов. Примеры насыщенных углеводородных групп атомов включают, помимо прочего, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомологи или изомеры атомных групп, такие как н-пентил, н-гексил, н-гептил, н-октил и другие. Ненасыщенный алкил содержит одну или несколько двойных или тройных связей, например, помимо прочего, винил, 2-пропенил, бутенил, кротонил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и гомологи и изомеры с большим количеством атомов.
Если не указано иное, термин «гетероуглеводородная группа» или ее конкретное название (например, гетероалкил, гетероалкенил, гетероалкинил, гетероарил и другое), сам по себе или в сочетании с другим термином, означает стабильную прямую, разветвленную или циклическую углеводородную группу атомов или их сочетание с определенным количеством атомов углерода и хотя бы одним гетероатомом. В некоторых вариантах осуществления термин «гетероалкил», сам по себе или в сочетании с другим термином, означает стабильную прямую или разветвленную углеводородную группу атомов либо их сочетание с определенным количеством атомов углерода и хотя бы одним гетероатомом. В типичном варианте осуществления гетероатомы выбирают из В, О, N и S, где атомы азота и серы необязательно окислены и гетероатомы азота необязательно кватернизованы. Гетероатомы В, О, N и S могут находиться в любом внутреннем положении (включая положение в остатке молекулы, к которому была прикреплена углеводородная группа) гетероуглеводородной группы. Примеры включают, помимо прочего, -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Подряд могут идти не более двух гетероатомов, например, -CH2-NH-OCH3.
Термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) имеют обычное значение и означают соответствующие алкильные группы, связанные с остатком молекулы через атом кислорода, аминогруппу или атом серы, соответственно.
Если не указано иное, термины «циклическая углеводородная группа», «гетероциклическая углеводородная группа» или соответствующий конкретный термин (такой как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и другие), сами по себе или в сочетании с другими терминами, соответственно означают циклическую «углеводородную группу» или «гетероуглеводородную группу». Кроме того, в гетероуглеводородных группах или гетероциклоуглеводородных группах (таких как гетероалкил, гетероциклоалкил) гетероатомы могут занимать положение в остатке молекулы, к которому прикреплен гетероцикл. Примеры циклоалкилов включат, помимо прочего, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и другие. Некоторые примеры гетероциклических групп включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураниндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если не указано иное, термин «арил» означает ароматическую углеводородную замещающую группу, которая может содержать одну, две или более замещающих группу, может быть моно-, ди- или поливалентной и может быть моно- или полициклической (например, от 1 до 3 колец, хотя бы одно из которых - ароматическое). Все кольца сконденсированы или соединены ковалентными связями. Термин «гетероарил» означает арильную группу (или кольцо) с одним или несколькими гетероатомами. В одном примере варианта осуществления гетероатомы выбирают из В, N, О и S, где атомы азота и серы необязательно окислены и гетероатомы и гетероатомы азота необязательно кватернизованы. Гетероарильная группа может быть присоединения к остатку молекулы через гетероатом. Некоторые примеры арильных и гетероарильных групп включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-бензотиазолил, пуринил, 2-бензимидазолинил, 5-индолил, 1-изохинолинил, 5-изохинолинил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолинил и 6-хинолинил. Замещающие группы любой из вышеуказанных арильных и гетероарильных кольцевых систем выбирают из приемлемых замещающих групп, описанных ниже.
Для простоты термин «арильная группа» при использовании вместе с другими терминами (например, арилоксил, арилтиол, аралкил) включает описанные выше арильные и гетероарильные кольца. То есть термин «аралкил» включает такие группы атомов (например, бензил, фенетил, пиридилметил и другие), в которых арильная группа присоединена к алкильной группе, в том числе такие, в которых атом углерода (например, метилена) был заменен атомом кислорода, например, феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и другие.
Термин «уходящая группа» означает функциональную группу или атом, который может быть замещен другой функциональной группой или атомом путем реакции замещения (например, реакции нуклеофильного замещения). Например, типичные уходящие группы включают трифторметансульфонат; хлор, бром, йод; сульфонатные группы, такие как метансульфонат, тозилат, п-бромбензолсульфонат, п-толуолсульфонат и другие; ацилоксигруппы, такие как ацетокси, трифторацетокси и другие.
Термин «защитная группа» включает, помимо прочего, «аминозащитную группу», «гидроксизащитную группу» и «меркаптозащитную группу». Термин «аминозащитная группа» означает защитную группу, которая способна предотвратить побочные реакции по атому азота. Типичные защитные аминогруппы включают, помимо прочего, формил; ацильные группы, такие как алканоил (такие как ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такие как трет-бутоксикарбонил (Boc); арилметилоксикарбонил, такую как бензилоксикарбонил (Cbz) и 9-флуоренилметилоксикарбонил (Fmoc); арилметил, такую как бензил (Bn), тритил (Tr), 1,1-бис(4'-метоксифенил)метил; силил, такую как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и другие. Термин «гидроксизащитная группа» означает защитные группы, которые способна предотвратить побочные реакции по гидроксилу. Типичные гидроксизащитные группы включают, помимо прочего, алкил, такие как метил, этил и трет-бутил; ацил, такие как алканоил (например, ацетил); арилметил, такие как бензил (Bn), п-метоксибензил (РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такую как триметилсилил (TMS) и трет-бутилдиметилсилил(TBS), и другие.
Вещества настоящего изобретения могут быть получены с помощью различных методов синтеза, хорошо известных специалистам средней квалификации, в том числе путем вариантов осуществления, перечисленных далее, вариантов осуществления, полученных путем сочетания перечисленных вариантов осуществления с другими методами химического синтеза, и путем альтернативных эквивалентов, хорошо известным специалистам средней квалификации. Предпочитаемые варианты осуществления включают, помимо прочего, примеры настоящего изобретения.
Растворители, используемые в настоящем изобретении, имеются на рынке. Обычно реакция проводится в безводном растворителе в инертной атмосфере азота. Данные протонного магнитного резонанса получали на спектрометре Bruker Avance III 400 (400 МГц) с указанием химических сдвигов в ppm относительно сдвига тетраметилсилана. Масс-спектры получали на спектрометре Agilent 1200 Series plus 6110 (& 1956А). ЖХ/МС или МС Shimadzu содержит диодно-матричный детектор (ДМД): SPD-M20A (ЖХ) и детектор Shimadzu Micromass 2020. Масс-спектрометр оснащен источником электрораспылительной ионизации (ESI), работающим в режиме положительного и отрицательного заряда.
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили системе Shimadzu LC20AB, снабженной автосэмплером Shimadzu SIL-20A и диодно-матричным детектором SPD-M20A. Использовали колонку Xtimate С18 (наполнитель 3 мкм, 2,1×300 мм). Способ А 0-60АВ_6 мин. состоял в следующем: в первые 4,2 минуты элюирование с линейным градиентом начали со 100% А (А - 0,0675% ТФУ в воде) и закончили с 60% В (В - 0,0625% ТФУ в MeCN), затем продолжали элюирование еще 1 минуту с 60% В. Затем колонку уравновешивали в течение 0,8 минуты для получения 100:0. Общая продолжительность хроматографирования - 6 минут. Способ А 10-80АВ_6 мин. состоял в следующем: в первые 4,2 минуты элюирование с линейным градиентом начали со 90% А (А - 0,0675% ТФУ в воде) и закончили с 80% В (В - 0,0625% ТФУ в ацетонитриле), затем продолжали элюирование еще 1 минуту с 80% В. Затем колонку уравновешивали в течение 0,8 минуты для получения 90:10. Общая продолжительность хроматографирования - 6 минут. Температура колонки составила 50°C, скорость потока - 0,8 мл/мин. Использованы длины волн диодно-матричного детектора от 200 до 400 нм.
Тонкослойную хроматографию (ТСХ) проводили на пластине с силикагелем GF254 группы компаний Sanpont. Пятна детектировали при УФ-излучении. В некоторых случаях пятна также изучали другими способами. В этих случаях для проявления пластины и обнаружения веществ использовали йод (полученный путем добавления 1 г йода к 10 г силикагеля и тщательного перемешивания), ванилин (полученный путем растворения около 1 г ванилина в 100 мл 10% H2SO4), нингидрин (производства компании Aldrich) или специальное проявляющее средство (полученное путем тщательного перемешивания (NH4)6Mo7O24⋅4H2O, 5 г (NH4)2Ce(IV)(NO3)6, 450 мл Н2О и 50 мл концентрированной H2SO4). Колоночную флэш-хроматографию проводили в колонке с силикагелем Silicycle с размером пор 40-63 мкм (230-400 меш) по методике, сходной с описанной в Still, W.С.; Kahn, М.; and Mitra, М. Journal of Organic Chemistry, 1978, 43, 2923-2925. Обычно используемые в колоночной флэш-хроматографии или тонкослойной хроматографии растворители включают смесь дихлорметана и метанола, смесь этилацетата и метанола и смесь гексана и этилацетата.
Препаративный хроматографический анализ проводили с использованием детектора Gilson UV/VIS-156 на системе Gilson-281 Prep LC 322. Использовали колонки Agella Venusil ASB Prep C18, 5 мкм, 150×21,2 мм; Phenomenex Gemini C18, 5 мкм, 150×30 мм; Boston Symmetrix С18, 5 мкм, 150×30 мм; или Phenomenex Synergi C18, 4 мкм, 150×30 мм. Вещества элюировали со скоростью потока около 25 мл/мин в низком градиенте смеси ацетонитрила с водой, содержащей 0,05% HCl, 0,25% НСООН или 0,5% NH3⋅Н2О в воде. Общая продолжительность анализа составила 8-15 минут.
Селективные ингибиторы CDK4/6, описанные в настоящем изобретении, могут применяться в лечении различных видов рака, в том числе рака молочной железы, немелкоклеточного рака легкого, рака пищевода, рака прямой кишки и острого миелоидного лейкоза. Селективные ингибиторы CDK4/6 можно использовать в монотерапии или в сочетании с другими химиотерапевтическими средствами.
В настоящем изобретении используются следующие сокращения: СВЧ означает сверхвысокочастотный; к.т. означает комнатную температуру; водн. означает водный раствор; ДХМ означает дихлорметан; ТГФ означает тетрагидрофуран; ДМФ означает N,N-диметилформамид; ДМСО означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; МеОН означает метанол; ВОС означает трет-бутоксикарил, аминозащитную группу; Boc2O означает ди-трет-бутилбикарбонат; НОАс означает уксусную кислоту; TEA означает трифторэтиламин; DIPEA означает диизопропилэтиламин; TEA или Et3N означает триэтиламин; BnNH2 означает бензиламин; PMBNH2 означает п-метоксибензиламин; MnO2 означает диоксид марганца; HATU означает O-(7-азабензатриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; POCl3 означает оксихлорид фосфора; NaH означает гидрит натрия; LiAlH4 означает алюмогидрид лития; Pd2(dba)3 означает трис(дибензилиденацетон)дипалладий; Pd(dppf)Cl2 означает [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид; Pd(OAc)2 означает палладия ацетат; Pd(PPh3)4 означает (трифенилфосфин)-палладий; PPh3 означает трифенилфосфин; Xantphos означает 4,5-бис(дифенилфосфино)-9,9-диметилксантен; Xphos означает 2-дициклогексилфосфино-2',4',6'-тризопропилбифенил; BINAP означает (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил; Xphos-PD-G2 означает хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II); NIS означает N-йодсукцинимид; NBS означает N-бромсукцинимид; NCS означает N-хлорсукцинимид; t-BuOK означает калия трет-бутоксид; t-BuONa означает натрия трет-бутоксид; Cs2CO3 означает цезия карбонат; K2CO3 означает калия карбонат; NaBH(OAc)3 означает натрия триацетоксиборгидрид; NaBH3CN означает натрия цианоборгидрид; NaHCO3 означает натрия бикарбонат; Na2SO4 означает натрия сульфат; KOAc означает калия ацетат; Xantphos означает 4,5-бис(дифенилфосфино)-9,9-диметилксантен.
Вещества настоящего изобретения могут быть получены с помощью различных методов синтеза, хорошо известных специалистам средней квалификации, в том числе путем вариантов осуществления, перечисленных далее, вариантов осуществления, полученных путем сочетания перечисленных вариантов осуществления с другими методами химического синтеза, и путем альтернативных эквивалентов, хорошо известным специалистам средней квалификации. Предпочитаемые варианты осуществления включают, помимо прочего, примеры настоящего изобретения.
Вещества настоящего изобретения могут быть получены путем серии этапов синтеза, где R1, R2, R3, R4, R5, R6, R7, X1, Х2, X3, Х4, X5, Х6, X7, m, n, Q, Т и W имеют значение, определенное выше.
Получение вещества с формулой I по схеме реакций 1
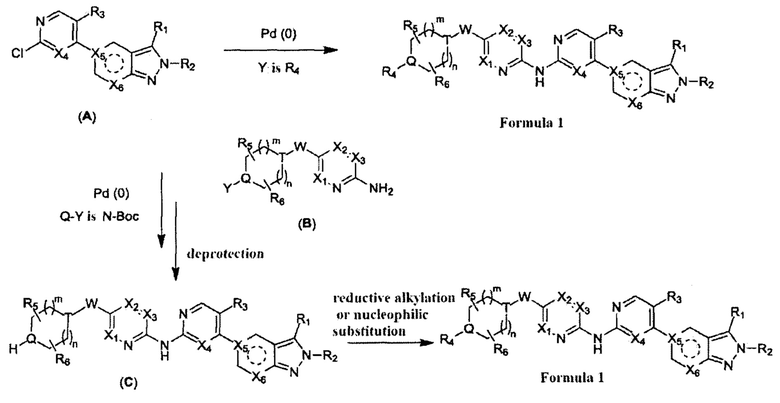
Когда Y=R4, вещество, описываемое формулой I, получают путем взаимодействия производного 2Н-пиразола (А) с 2-ариламином (В) в ходе реакции, описанной выше в схеме реакций 1, с получением вещества, описываемого формулой I. Для проведения реакции нужен подходящий катализатор (например, Pd2(dba)3), подходящий лиганд (например, Xantphos), подходящее основание (например, Cs2CO3) и подходящий растворитель (например, 1,4-диоксан). Согласно схеме реакции 1, реакция с большей вероятностью будет происходить при повышенной температуре.
Когда Q-Y - это N-Boc, вещество, описываемое формулой I, также можно получить путем взаимодействия производного 2Н-пиразола (А) с 2-ариламином (В) в ходе реакции, описанной ниже в схеме реакций 1. Однако, группу Boc нужно удалить сильной кислотой (например, ТФУ) с получением амина (С), после чего амин (С) проходит реакцию алкилирования в условиях восстановительного аминирования или нуклеофильного замещения (например, с NaBH3CN или галоалканом) с получением вещества, описываемого формулой I.
Получение производного 2Н-пиразола (А) по схеме реакции 2
Где  - это
- это 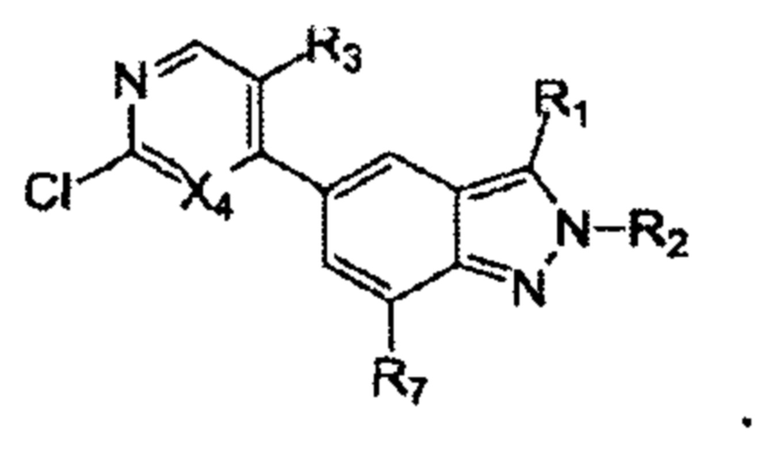

В реакции, изображенной на схеме 2, 5-бром-2Н-индазол (F) можно получить путем взаимодействия 5-бром-2Н-индазола (D) с галоалканом R2X (Е). Для прохождения реакции требуется подходящее основание (например, NaH или MeONa) и подходящий растворитель (например, ТГФ). 5-бром-2Н-индазол (G) можно получить путем галогенирования 5-бром-2Н-индазола (F) в присутствии подходящего галогенирующего реагента (например, Br2, NBS или NIS) и подходящего растворителя (например, DMF или MeCN). 5-бром-2Н-индазол (I) можно получить путем катализируемого палладием конденсации с боратом (Н) в присутствии подходящего катализатора (например, Pd(dppf)Cl2), подходящего основания (например, К2СО3) и подходящего растворителя (например, смеси диоксана и воды). 5-бром-2Н-индазол (K) можно получить путем катализируемой палладием конденсации с бис(пинаколато)дибором (J), в присутствии подходящего катализатора (например, Pd(dppf)Cl2), подходящего основания (например, KOAc) и подходящего катализатора (например, диоксана). Производное 2Н-пиразола (А) можно получить путем катализируемой палладием конденсации с веществом (L) в присутствии подходящего катализатора (например, Pd(dppf)Cl2), подходящего основания (например, K2CO3) и подходящего растворителя (например, диоксана). Согласно схеме реакции 2, реакция с большей вероятностью будет происходить при повышенной температуре. Производное 2Н-пиразола (А') можно получить путем гидрогенизации вещества (А) с подходящим родиевым катализатором (например, Rh(PPh3)3Cl) в присутствии подходящего растворителя (например, тетрагидрофурана). R1' - алкильная группа после восстановления R1.
Получение производного 2Н-пиразола (А) по схеме реакции 3
Где  - это
- это 
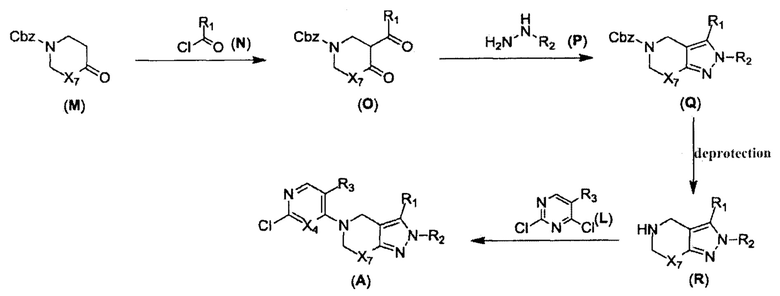
В реакции, показанной на схеме 3, вещество (О) можно получить путем взаимодействия вещества (J) с ацилхлоридом (N). Для прохождения реакции требуется подходящее основание (например, LiHMDS) и подходящий растворитель (например, ТГФ). Вещество (О) может образовать цикл с получением 2Н-пиразола (Q) в присутствии подходящего алкилгидразина (Р) и подходящего растворителя (например, EtOH). 2Н-пиразол (R) можно получить путем гидрогенизации в присутствии подходящего катализатора (например, Pd/C) и подходящего растворителя (например, МеОН). Производное 2Н-пиразола (А) можно получить из вещества (L) в присутствии подходящего основания (например, Et3N) и подходящего растворителя (например, THF). Согласно схеме реакции 3, реакция с большей вероятностью будет происходить при повышенной температуре.
Получение 2-ариламина (В) по схеме реакции 4 (где, W - S или О, Т - это С)

В реакции, показанной на схеме 4, пиридинбромин (U) получают путем взаимодействия 2-бром-5-гидроксипиридина (S) с имеющимся на рынке тиолом или спиртом (Т) в реакции Мицунобу. Пиридинбромид (U) можно затем превратить в 2-ариламин (В) с использованием подходящего палладиевого катализатора (например, Pd2(dba)3), подходящего основания (например, LiHMDS) и подходящего растворителя (например, толуола). Согласно схеме реакции 4, реакция с большей вероятностью будет происходить при повышенной температуре.
Получение 2-ариламина (В) по схеме реакции 5 (где, W - это прямая простая связь)

В реакции, показанной на схеме 5, где W - это прямая простая связь, 2-ариламин (В) можно получить с помощью двух описанных ниже процессов: (1) трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат (X) и нитропиридин (W) сначала проходит реакцию конденсации с палладиевым катализатором, затем восстановление нитрогруппы и двойной связи; и (2) атом брома в нитропиридине (W) замещают имеющимся на рынке амином (V), затем восстанавливают нитрогруппу.
Варианты осуществления
В следующих примерах настоящее изобретение описано более подробно, но объем настоящего изобретения ими не ограничивается.
Подход А
Общий способ получения промежуточного продукта А, промежуточного продукта В и промежуточного продукта С показан ниже.

Этап 1: 5-бром-2(Н)-индазол
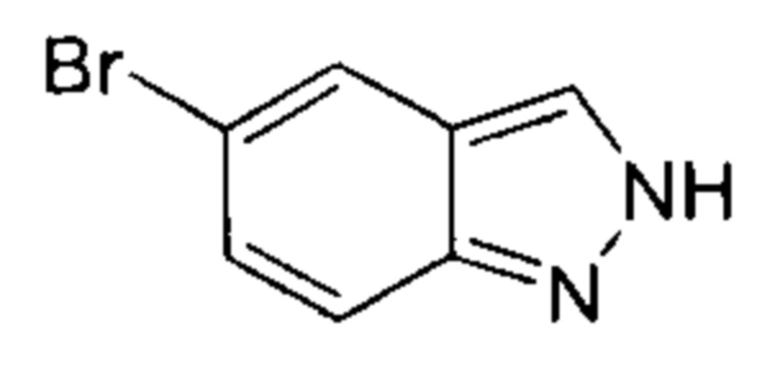
5-бром-2-фторбензальдегид (18,00 г, 88,67 ммоль, 1,00 эквивалент) медленно добавили к 85% водному раствору гидразина гидрата (103,00 г, 2,06 моль, 23,20 эквивалентов), в процессе медленно выпало в осадок белое твердое вещество. Смесь перемешивали при 110°C в течение 16 часов. ЖХ/МС показала, что большая часть полученного осадка представляла собой желаемое вещество. Смесь охладили до 16°C и отфильтровали. Осадок на фильтре промыли водой (100 мл) и получили неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = 3:2) и получили указанное в заголовке соединение (6,50 г, 32,99 ммоль, выход - 37,21%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 197,1 (М+1).
Этап 2: 5-бром-2-метил-2Н-индазол
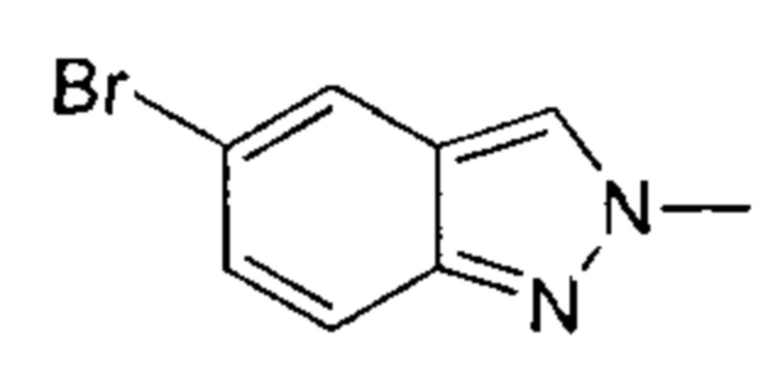
К раствору 5-бром-2Н-индазола (20,00 г, 101,51 ммоль, 1,00 эквивалент) и натрия метоксида (5,48 г, 5,48 ммоль, 5,48 эквивалента) в метаноле (150,00 мл) при 30°C в атмосфере азота добавляли по капле в течение 1 часа метилиодид (57,00 г, 401,58 ммоль, 3,96 эквивалента). Затем смесь нагрели до 85°C и перемешивали в течение 5 часов. ЖХ/МС показала, что исходный материал вступил в реакцию практически полностью, и по данным МС было выявлено желаемое вещество. Смесь охладили до 16°C, упарили и получили неочищенный продукт. Неочищенный продукт разбавили 3% водным раствором NaHCO3 (30 мл) и экстрагировали этилацетатом (80 мл × 2). Органическую фазу упарили под пониженным давлением. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 30:1 до 1:1) и получили указанное в заголовке соединение (8,40 г, 39,80 ммоль, выход - 39,21%), представляющее собой серое твердое вещество. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,33 (с., 1Н), 7,95 (с., 1Н), 7,58 (д., J=8,0 Гц, 1Н), 7,30 (дд, J=1,8 Гц, 8 Гц, 1Н), 4,18 (с., 1Н). ЖХ/МС (ESI) м/з: 210,8 (М+1).
Этап 3: 5-бром-3-йод-2-метил-2Н-индазол

К раствору 5-бром-2-метил-2Н-индазола (8,40 г, 39,80 ммоль, 1,00 эквивалент) в дихлорметане (90 мл) добавили пиридин (4,72 г, 59,70 ммоль, 1,5 эквивалента) и бис(трифторацетокси)йодбензол (20,54 г, 47,76 ммоль, 1,20 эквивалента) при 30°C. Смесь перемешивали в течение 0,5 часа, затем добавили в нее йод (12,12 г, 47,76 ммоль, 1,20 эквивалента) и перемешивали еще 23,5 часа. ЖХ/МС показала, что реакция завершилась. Смесь отфильтровали и получили указанное в заголовке соединение (8,20 г, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 336,9 (М+1).
Этап 4: 5-бром-2-метил-3-изопропенил-2Н-индазол
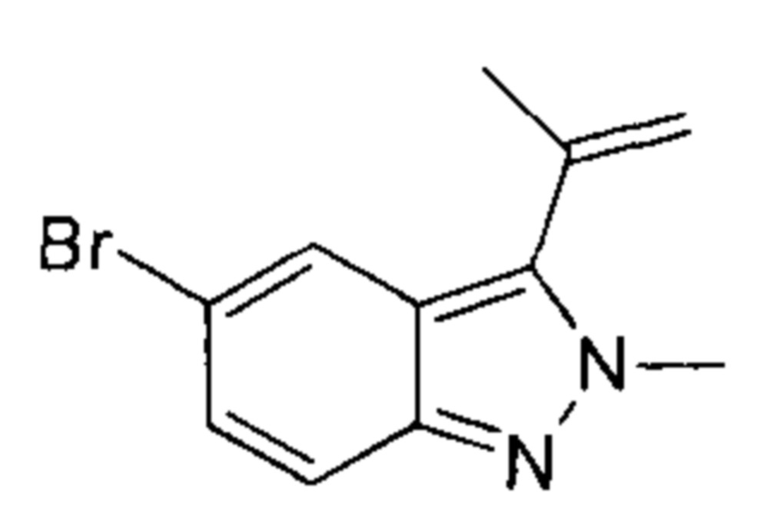
К раствору 5-бром-3-йод-2-метил-2Н-индазола (7,68 г, 22,79 ммоль, 1,00 эквивалент) и изопропенилбората (4,21 г, 25,07 ммоль, 1,11 эквивалента) в диоксане (90,00 мл) добавили насыщенный водный раствор (30 мл) К2СО3 (9,45 г, 68,38 ммоль, 3,00 эквивалента) и Pd(dppf)Cl2⋅CHCl2 (1,86 г, 2,28 ммоль, 0,10 эквивалента.). Смесь перемешивали при 100°C в течение 3 часов. ТСХ показала, что исходный материал вступил в реакцию практически полностью. Смесь охладили до 30°C и отфильтровали, фильтрат подвергли экстракции этилацетатом (100 мл × 3), промыли водой (50 мл × 3), промыли насыщенным солевым раствором (20 мл × 3), высушили над безводным Na2SO4, отфильтровали и упарили. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = 1:1) и получили указанное в заголовке соединение (5,36 г, 21,34 ммоль, выход - 93,66%), представляющее собой желтое масло.
Этап 5: 2-метил-3-изопропенил-5-борат-2Н-индазол
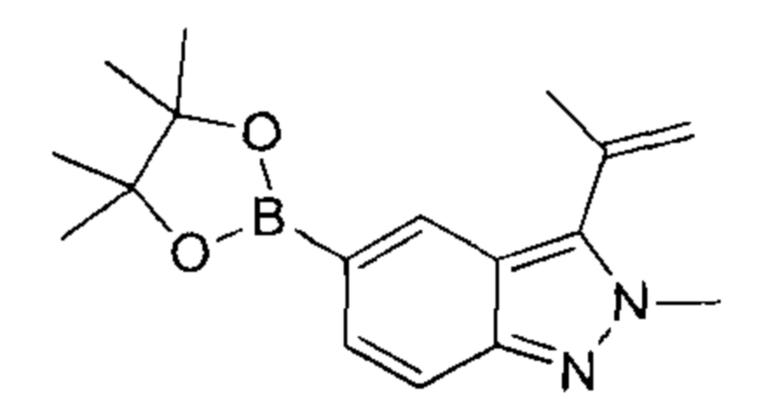
В атмосфере азота к раствору 5-бром-2-метил-3-изопропенил-2Н-индазола (2,80 г, 11,15 ммоль, 1,00 эквивалент) и бис(пинаколато)дибора (3,40 г, 13,38 ммоль, 1,20 эквивалента) в диоксане (56,00 мл) добавили KOAc (3,28 г, 33,45 ммоль, 3,00 эквивалента) и Pd(dppf)Cl2⋅CHCl2 (1,82 г, 2,23 ммоль, 0,20 эквивалента). Смесь перемешивали при 100°C в течение 5 часов. ЖХ/МС показала, что реакция завершилась. Масс-спектр подтвердил наличие желаемого продукта. Смесь охладили до 16°C, разбавили этилацетатом (20 мл) и профильтровали. Фильтрат очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = 1:1) и получили указанный в заголовке продукт (3,30 г, 9,96 ммоль, выход - 89,33%, степень чистоты - 90%), представляющий собой фиолетовое твердое вещество. ЖХ/МС (ESI) м/з: 299,1 (М+1).
Этап 6:
5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-изопропенил-2Н-индазол
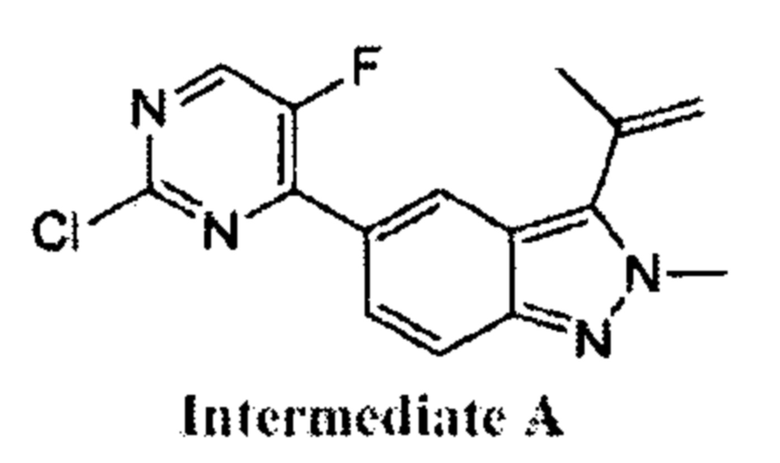
В атмосфере азота к раствору 2,4-дихлор-5-фторпиримидина (147,83 мг, 885,34 мкмоль, 1,20 эквивалента) и 2-метил-3-изопропенил-5-борат-2Н-индазола (220,00 мг, 737,78 мкмоль, 1,00 эквивалент) в диоксане (4 мл) добавили К2СО3 (305,91 мг, 2,21 ммоль, 3,00 эквивалента) и Pd(dppf)Cl2⋅CH2Cl2 (120,50 мг, 147,56 мкмоль, 0,20 эквивалент). Смесь перемешивали при 100°C в течение 3,5 часов. ТСХ показала, что большая часть исходного материала вступила в реакцию, и, по результатам ЖХ/МС, превратилась в желаемый продукт. Смесь охладили до 30°C и профильтровали. Осадок на фильтре промыли этилацетатом (5 мл), фильтрат упарили. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 1:0 до 6:1) и получили желаемое соединение (промежуточный продукт А) (210,0 мг, 693,69 мкмоль, выход - 94,02%), представляющее собой ярко-желтое твердое вещество. ЖХ/МС (ESI) м/з: 303,0 (М+1).
Этап 7:
трет-бутил-5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)пиримидин-2-ил-карбамат

К раствору 5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-(проп-1-ен-2-ил)-2Н-индазола (промежуточный продукт А) (1,00 г, 3,30 ммоль, 1,00 эквивалент) в диоксане (15,00 мл) добавили трет-бутил-карбамат (966,49 мг, 8,25 ммоль, 2,50 эквивалента), калия карбонат (1,37 г, 9,90 ммоль, 3,00 эквивалента), палладия ацетат (74,09 мг, 330,00 мкмоль, 0,10 эквивалент) и Xantphos (381,89 мг, 660,00 мкмоль, 0,20 эквивалент). Через сосуд три раза пропускали азот и перемешивали при 100°C в течение 18 часов. ЖХ/МС показала, что реакция завершилась и образовался желаемый продукт. Раствор охладили до 20°C и профильтровали. Фильтрат упарили под пониженным давлением и получили указанное в заголовке соединение (3,00 г, неочищенный продукт), представляющий собой бледно-желтое твердое вещество. Неочищенный продукт использовали в следующем этапе без очистки. ЖХ/МС (ESI) м/з: 384,1 (М+1).
Этап 8:
5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)пиримидин-2-амин

К раствору трет-бутил-5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)-пиримидин-2-илкар бамата (3,00 г, 3,31 ммоль, 1,00 эквивалент) в дихлорметане (30,00 мл) добавили по капле трифторуксусную кислоту (10,00 мл). Раствор перемешивали при 15°C в течение 1 часа. ТСХ (петролейный эфир : этилацетат = 1:1) показал, что исходный материал полностью вступил в реакцию. Раствор высушили в ротационной вакуумной сушилке под пониженным давлением при 30°C. Неочищенный продукт разбавили дихлорметаном (100 мл) и промыли насыщенным раствором натрия бикарбоната (100 мл) и насыщенным солевым раствором (100 мл). Органическую фазу высушили над безводным натрия сульфатом и упарили. Неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир : этилацетат = от 5:1 до 3:1) и получили указанное в заголовке соединение (770,00 мг, 2,72 ммоль, выход - 82,11%), представляющий собой коричневое твердое вещество.
Этап 9:
5-фтор-4-(3-изопропил-2-метил-2H-индазол-5-ил)пиримидин-2-амин

К раствору 5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)-пиримидин-2-амина (770,00 мг, 2,72 ммоль, 1,00 эквивалент) в метаноле (20,00 мл) добавили палладиевый катализатор на углеродном носителе (250,00 мг). Раствор нагрели до 50°C и перемешивали в течение 24 часов в атмосфере водорода (15 фунт/кв. дюйм). ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционный раствор охладили до 20°C и отфильтровали. Фильтрат упарили и получили указанное в заголовке соединение (промежуточный продукт В) (600,00 мг, 2,10 ммоль, выход - 77,26%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,55 (с., 1Н), 8,20 (д., J=3,9 Гц, 1Н), 7,96 (д., J=9,2 Гц, 1Н), 7,69 (д., J=9,7 Гц, 1Н), 5,06 (ушир. с., 2Н), 4,17 (с, 3H), 3,54-3,45 (м., 1Н), 1,57 (д., J=7,2 Гц, 6Н).
Этап 10:
5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-изопропил-2Н-индазол
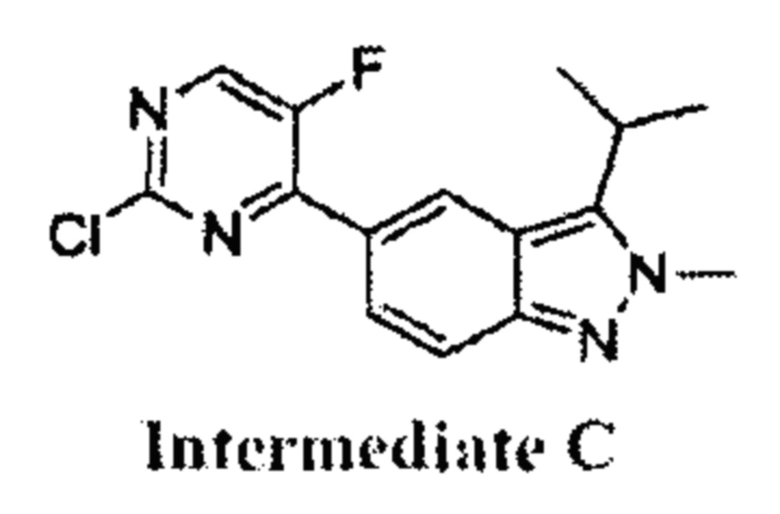
К раствору 5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-изопропенил-2Н-индазола (промежуточный продукт А) (36,50 г, 120,57 ммоль, 1,00 эквивалент) в тетрагидрофуране (182,5 мл) добавили Rh(PPh3)3Cl (11,16 г, 12,06 ммоль, 0,10 эквивалент). Через реакционную смесь несколько раз пропустили водород. Смесь перемешивали при 50°C в атмосфере азота при 50 фунт/кв. дюйм в течение 24 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 25°C и упарили под пониженным давлением. К остатку добавили метанол (100 мл), смесь очистили на шариковой мельнице, перемешивали в течение 16 часов и затем профильтровали. Осадок на фильтре промыли метанолом (15 мл × 3) и высушили под пониженным давлением, получив указанное в заголовке соединение (промежуточный продукт С) (28,00 г, 91,88 ммоль, выход - 76,20%), представляющее собой коричневое желтоватое вещество. 1Н ЯМР (400 МГц, CDCl3) 8,69 (с., 1Н), 8,49 (д., J=3,5 Гц, 1H), 8,06 (д., J=9,3 Гц, 1Н), 7,73 (д., J=9,3 Гц, 1Н), 4,19 (с., 3H), 3,58-3,49 (м., 1Н), 1,59 (д., J=7,2 Гц, 6Н). ЖХ/МС (ESI) м/з: 305,2 (М+1).
Пример 1
N-(5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин

Этап 1: 6-(4-метилпиперазин-1-ил)пиридазин-3-амин

В пробирку для СВЧ-печи поместили 6-хлор-3-аминопиридазин (3,00 г, 23,16 ммоль, 1,00 эквивалент) и 1-метилпиперазин (8,10 г, 80,87 ммоль, 3,49 эквивалента), пробирку запечатали. Смесь нагрели до 170 0°C и перемешивали в течение 1,5 часов под воздействием СВЧ-излучения. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C и упарили под пониженным давлением. Остаток очистили посредством препаративной ВЭЖХ (основная среда), получив указанное в заголовке соединение (3,53 г, 18,27 ммоль, 78,87%), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,11 (д., J=9,6 Гц, 1Н), 6,73 (д., J=9,6 Гц, 1H), 5,63 (с., 2Н), 3,30-3,24 (м., 4Н), 2,42-2,36 (м., 4Н), 2,19 (с., 3H). ЖХ/МС (ESI) м/з: 194,1 (М+1).
Этап 2:
N-(5-фтор-4-(2-метил-3-изопропенил-2Н-индазол-5-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин
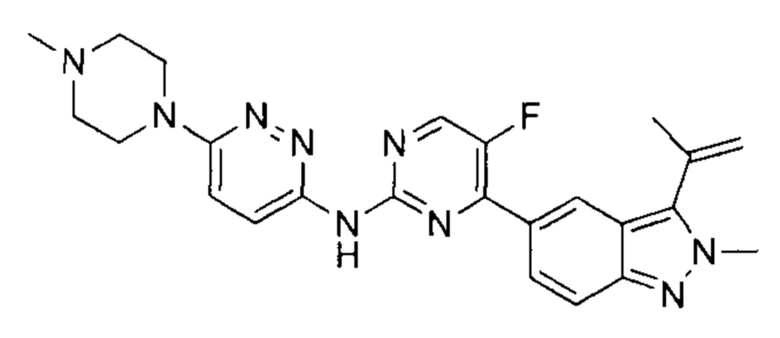
В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-изопропенил-2Н-индазола (промежуточный продукт А) (350,00 мг, 1,16 ммоль, 1,00 эквивалент) и 6-(4-метилпиперазин-1-ил)пиридазин-3-амина (228,65 мг, 1,18 ммоль, 1,02 эквивалента) в диоксане (8 мл) добавили Cs2CO3 (753,39 мг, 2,31 мкмоль, 2,00 эквивалента), Xantphos (267,59 мг, 462,46 мкмоль, 0,40 эквивалент) и Pd2(dba)3 (211,74 мг, 231,21 мкмоль, 0,20 эквивалент). Смесь перемешивали при 100-110°C в течение 16 часов. ТСХ и ЖХ/МС показали, что исходный материал полностью вступил в реакцию. Смесь охладили до 20°C, разбавили этилацетат (20 мл), и профильтровали. Фильтрат упарили и получили неочищенный продукт. Неочищенный продукт добавили к метанолу (10 мл), оставили при 25°C до образования желтого осадка и отфильтровали. Осадок на фильтре промыли небольшим количеством метанола и высушили, получив таким образом указанное в заголовке соединение (120,00 мг, 258,53 мкмоль, выход - 22,29%, степень чистоты - 99%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 460,3 (М+1).
Этап 3:
N-(5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин

К раствору Pd/C (50 мг) в метаноле (10 мл) добавили N-(5-фтор-4-(2-метил-3-изопропенил-2Н-индазол-5-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин (120,00 мг, 261,14 мкмоль, 1,00 эквивалент). Через реакционную систему пропустили водород при давлении 15 фунт/кв. дюйм. Смесь перемешивали при 40-50°C в течение 8 часов. ЖХ/МС показала, что реакция завершилась. Смесь отфильтровали, и фильтрат упарили. Остаток очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (57,00 мг, 123,50 мкмоль, выход - 47,29%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,72 (с., 1Н), 8,46 (д., J=4,0 Гц, 1Н), 8,41 (д., J=9,6 Гц, 1Н), 8,06 (д., J=9,2 Гц, 1H), 7,65 (д., J=9,2 Гц, 1H), 7,40 (д., J=9,2 Гц, 1H), 4,20 (с., 3H), 3,68-3,61 (м., 5Н), 2,63 (т., J=4,8 Гц, 4Н), 2,39 (с., 3H), 1,59 (д., J=7,2 Гц, 6Н). ЖХ/МС (ESI) м/з: 462,2 (М+1).
Пример 2
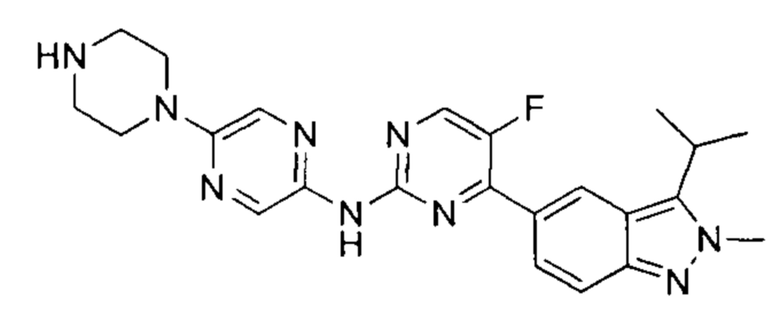
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиразин-2-ил)пидимидин-2-амин
Этап 1:
трет-бутил-4-(5-бромпиразин-2-ил)пиперазин-1-карбоксилат
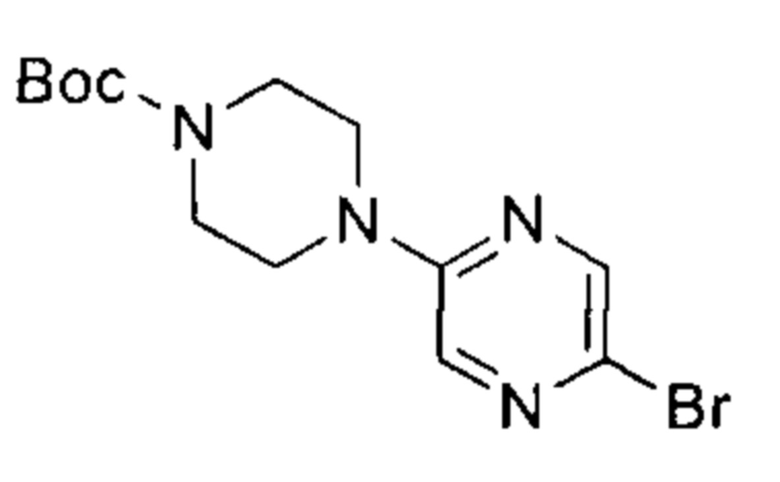
К раствору 2,5-дибромпиразина (10,00 г, 42,04 ммоль, 1,00 эквивалент) в N-метилпирролидоне (100 мл) добавили трет-бутил-пиперазин-1-карбоксилат (7,83 г, 42,04 ммоль, 1,00 эквивалент) и K2CO3 (8,72 г, 63,06 ммоль, 1,50 эквивалента). Смесь нагрели до 100°C и перемешивали в течение 18 часов. ТСХ (петролейный эфир : этилацетат = 10:1) показала, что реакция завершилась. Смесь охладили до 20°C, разбавили водой (200 мл), подвергли экстракции этилацетатом (200 мл × 2), высушили над безводным Na2SO4, профильтровали и упарили. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 5:1) и получили указанное в заголовке соединение (11,00 г, 2,05 ммоль, выход - 76,24%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,15 (д., J=1,38 Гц, 1Н) 7,87 (д., J=1,38 Гц, 1Н) 3,56 (с., 8Н) 1,49 (с., 9Н).
Этап 2:
трет-бутил-4-(5-аминопиразин-2-ил)пиперазин-1-карбоксилат

В атмосфере азота к раствору трет-бутил-4-(5-бром-пиразин-2-ил)пиперазин-1-карбоксилата (10,00 г, 29,14 ммоль, 1,00 эквивалент) и три-трет-бутилфосфина тетрафторборат (2,54 г, 8,74 ммоль, 0,30 эквивалент) в толуоле (100 мл) добавили LHMDS (1М, 60,00 мл, 2,06 эквивалента) и Pd2(dba)3 (2,60 г, 2,84 ммоль, 0,10 эквивалент). Смесь перемешивали при 65°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C, водой остановили реакцию (50 мл), и провели экстракцию этилацетатом (100 мл × 3). Органические фазы объединили и упарили. Остаток очистили в этилацетате (100 мл) и очистили посредством препаративной ВЭЖХ (основная среда), получив указанное в заголовке соединение (5,00 г, 17,90 ммоль, выход - 61,43%), представляющее собой оранжевое твердое вещество. ЖХ/МС (ESI) м/з: 280,1 (М+1).
Этап 3:
трет-бутил-4-(5-((5-фтор-4-(2-метил-3-(изопропенил)-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилат
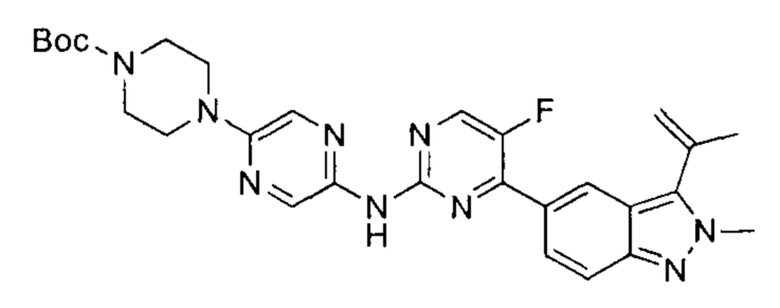
В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-изопропенил-2Н-индазола (200,00 мг, 660,65 мкмоль, 1,00 эквивалент) и трет-бутил-4-(5-аминопиразин-2-ил)пиперазин-1-карбоксилата (246,06 мг, 792,78 мкмоль, 1,20 эквивалента) в диоксане (10 мл) добавили Cs2CO3 (430,51 мг, 1,32 ммоль, 2,00 эквивалента), Xantphos (152,91 мг, 264,26 мкмоль, 0,40 эквивалента), и Pd2(dba)3 (120,99 мг, 132,13 мкмоль, 0,20 эквивалента). Смесь перемешивали при 110-120°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 25°C и профильтровали. Осадок на фильтре промыли этилацетатом, фильтрат упарили. Остаток очистили посредством препаративной ТСХ (дихлорметан : метанол = 20:1), получив указанное в заголовке соединение (150,00 мг, 247,43 мкмоль, выход - 37,45%, степень чистоты - 90%), представляющее собой зеленое твердое вещество. ЖХ/МС (ESI) м/з: 546,2 (М+1).
Этап 4:
трет-бутил-4-(5-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилат
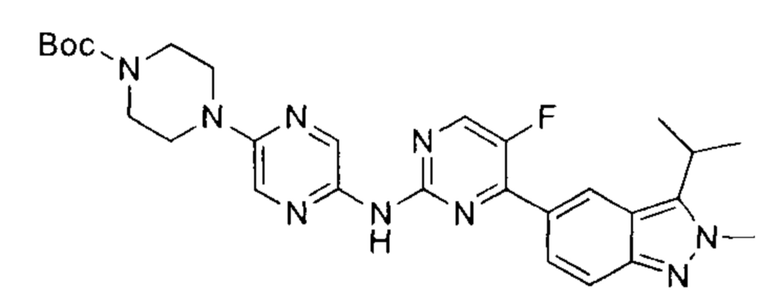
К раствору трет-бутил-4-(5((5-фтор-4-(2-метил-3-(изопропенил)-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилата (90,00 мг, 164,95 мкмоль, 1,00 эквивалент) в тетрагидрофуране (10 мл) добавили Pd/C (100 мг) и уксусную кислоту в каталитическом количестве. Через реакционную систему пропустили водород при давлении 15 фунт/кв. дюйм. Смесь перемешивали при 50-60°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C и профильтровали. Фильтрат упарили, получив указанное в заголовке соединение (90,00 мг, неочищенный продукт), которое сразу использовали в следующем этапе. ЖХ/МС (ESI) м/з: 548,3 (М+1).
Этап 5:
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиразин-2-ил)пидимидин-2-амин
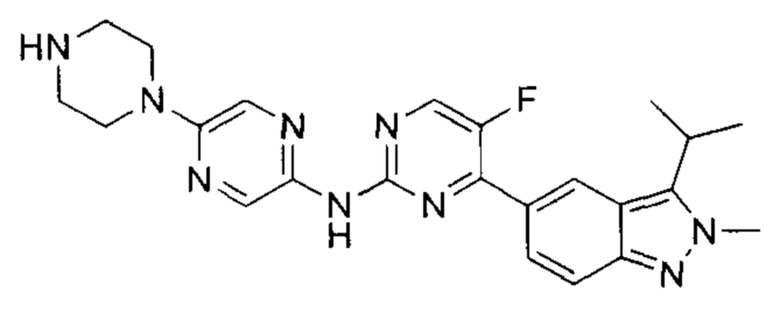
К раствору трет-бутил-4-(5-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилата (90,00 мг, 164,34 мкмоль, 1,00 эквивалент) в метаноле (5 мл) добавили соляную кислоту с метанолом (4М, 1 мл, 20 ммоль, 1,00 эквивалент). Смесь перемешивали при 30-40°C в течение 3 часов. ТСХ показала (дихлорметан : метанол = 30:1), что исходный материал полностью вступил в реакцию. ЖХ/МС показала, что в полученной смеси было 49% желаемого продукта и 44% побочного продукта. Смесь охладили до 25°C и упарили. Остаток очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (6,00 мг, 13,41 мкмоль, выход - 8,16%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,96 (с., 1H), 8,68 (д., J=4,4 Гц, 1Н), 8,31 (д., J=9,2 Гц, 1Н), 8,12 (с., 1Н), 7,79 (д., J=8,8 Гц, 1Н), 4,29 (с., 3H), 3,91 (т., J=4,8 Гц, 4Н), 3,74-3,69 (м., 1Н), 3,43 (т., J=5,2 Гц, 4Н), 1,65 (д., J=7,2 Гц, 6Н). ЖХ/МС (ESI) м/з: 448,1 (М+1).
Пример 3
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пидимидин-2-амин
Этап 1:
трет-бутил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат
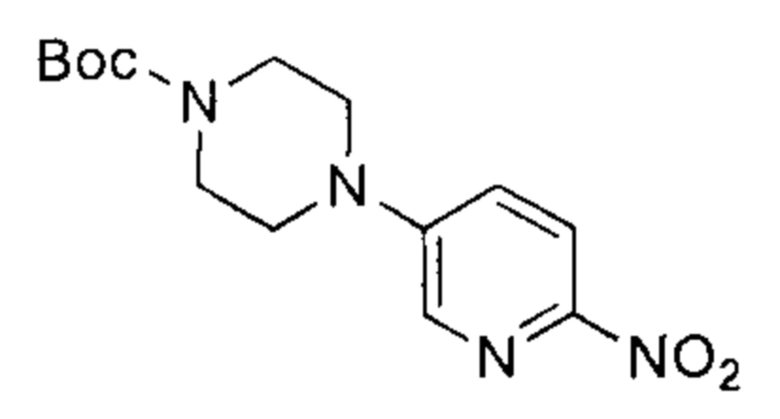
К раствору 5-бром-2-нитропиридина (20,00 г, 98,53 ммоль, 1,00 эквивалент) в диметилсульфоксиде (52 мл) добавили трет-бутил-пиперазин-1-карбоксилат (24,00 г, 128,86 ммоль, 1,31 эквивалента) и триэтиламин (20,00 г, 197,65 ммоль, 2,01 эквивалента). Раствор нагрели до 60°C и перемешивали в течение 18 часов. ТСХ показала (петролейный эфир : этилацетат = 3:1), что реакция завершилась. Раствор разбавили водой (200 мл), перемешивали в течение 30 минут и затем профильтровали. Осадок на фильтре промыли водой и высушили под вакуумом, получив неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир : этилацетат = от 50:1 до 20:1) и получили указанное в заголовке соединение (27,00 г, 87,57 ммоль, выход - 88,87%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (д., J=9,03 Гц, 1Н), 8,13 (д., J=2,89 Гц, 1Н), 7,21 (дд, J=9,10, 2,95 Гц, 1Н), 3,69-3,59 (м., 4Н), 3,51-3,40 (м., 4Н), 1,49 (с., 9Н).
Этап 2:
трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат

В атмосфере азота к раствору трет-бутил-4-(6-нитропиридин-3-ил)-пиперазин-1-карбоксилата (28,00 г, 90,81 ммоль, 1,00 эквивалент) в метаноле (600 мл) добавили палладиевый катализатор на углеродном носителе (6%, 1,7 г). Суспензию откачали и несколько раз насытили водородом. Раствор перемешивали при 50°C в атмосфере азота (50 фунт/кв. дюйм) в течение 18 часов. ТСХ (дихлорметан : метанол = 10:1) показала, что исходный материал полностью вступил в реакцию. Суспензию отфильтровали, фильтрат высушили в ротационной вакуумной сушилке, получив указанное в заголовке соединение (24,13 г, 86,69 ммоль, выход - 95,46%), представляющее собой фиолетовое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,78 (д., J=2,64 Гц, 1Н) 7,18 (дд, J=8,78, 2,89 Гц, 1Н) 6,50 (д., J=8,78 Гц, 1Н) 4,21 (ушир. с., 2Н) 3,60-3,54 (м., 4Н) 3,00-2,92 (м., 4Н) 1,48 (с., 9Н).
Этап 3:
трет-бутил-4-(6-((5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилат

К раствору 5-(2-хлор-5-фторпиримидин-4-ил)-2-метил-3-(проп-1-ен-2-ил)-2Н-индазола (промежуточный продукт А) (200,00 мг, 660,65 мкмоль, 1,00 эквивалент) в диоксане (10,00 мл) добавили трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат (220,67 мг, 792,79 мкмоль, 1,20 эквивалента), Pd2(dba)3 (60,50 мг, 66,07 мкмоль, 0,10 эквивалент), Xantphos (76,45 мг, 132,13 мкмоль, 0,20 эквивалент), и цезия карбонат (430,51 мг, 1,32 ммоль, 2,00 эквивалента). Раствор нагрели до 110°C в атмосфере азота и перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Раствор охладили до 25°C, отфильтровали, упарили и таким образом получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (этилацетат : петролейный эфир = 1:2) и получили указанное в заголовке соединение (320,00 мг, 587,57 мкмоль, выход - 88,94%), представляющее собой бледно-желтое твердое вещество. ЖХ/МС (ESI) м/з: 545,3 (М+1).
Этап 4:
трет-бутил-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилат

В атмосфере азота к раствору трет-бутил-4-(6-((5-фтор-4-(2-метил-3-(проп-1-ен-2-ил)-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата в метаноле (20,00 мл) добавили палладиевый катализатор на углеродном носителе (200,00 мг) и уксусную кислоту (2,10 г, 34,97 ммоль, 59,52 эквивалента). Суспензию откачали и несколько раз насытили водородом. Раствор перемешивали при 50°C в атмосфере азота (32 фунт/кв. дюйм) в течение 96 часов. ЖХ/МС показала, что реакция завершилась. Суспензию охладили до 25°C, профильтровали и упарили, получили указанное в заголовке соединение (500,00 мг, неочищенный продукт), представляющее собой почти белое твердое вещество. ЖХ/МС (ESI) м/з: 547,1 (М+1).
Этап 5:
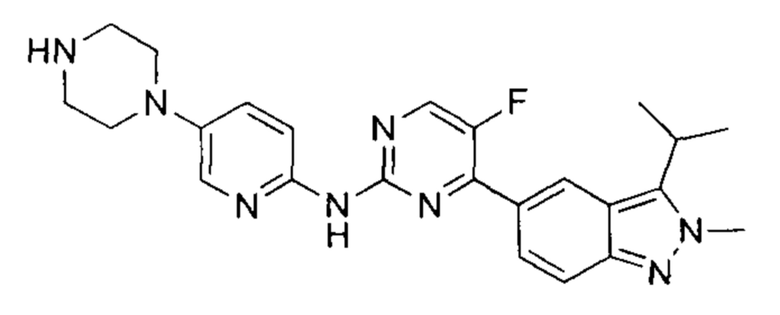
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пидимидин-2-амин

При 25°C к раствору трет-бутил-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (500,00 мг, 914,68 мкмоль, 1,00 эквивалент) в дихлорметане (5,00 мл) добавили одной порцией трифторуксусную кислоту (2,09 г, 18,29 ммоль, 20,00 эквивалента). Раствор перемешивали в течение 0,5 часа. ЖХ/МС показала, что реакция прошла полностью. Раствор упарили под пониженным давлением и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (99,69 мг, 223,26 мкмоль, выход - 24,41%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,98 (с., 1Н), 8,85 (д., J=3,39 Гц, 1Н), 8,53 (д., J=9,03 Гц, 1Н), 8,32 (дд, J=9,72, 2,20 Гц, 1Н), 8,01-7,96 (м., 1Н), 7,93 (д., J=9,03 Гц, 1Н), 7,60 (д., J=9,66 Гц, 1Н), 4,39 (с., 3H), 3,79 (дт, J=13,90, 6,92 Гц, 1Н), 3,65-3,56 (м., 4Н), 3,49 (д., J=5,02 Гц, 4Н), 1,69 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 447,1 (М+1).
Пример 4
N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (210,00 мг, 470,30 мкмоль, 1,00 эквивалент) в метаноле (5,00 мл) добавили ацетальдегид (77,69 мг, 705,46 мкмоль, 1,50 эквивалента), NaBH3CN (59,11 мг, 940,61 мкмоль, 2,00 эквивалента) и уксусную кислоту (14,12 мг, 235,15 мкмоль, 0,50 эквивалент). Раствор перемешивали при 20°C в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Раствор отфильтровали, и фильтрат упарили под пониженным давлением, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (66,50 мг, 128,83 мкмоль, выход - 27,39%, степень чистоты - 99%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8.97 (с., 1Н), 8,84 (д., J=3,6 Гц, 1Н), 8,52 (д., J=9,2 Гц, 1Н), 8,32 (дд, J=9,7, 2,8 Гц, 1Н), 7,98 (д., J=2,8 Гц, 1Н), 7,92 (д., J=9,0 Гц, 1Н), 7,59 (д., J=9,5 Гц, 1Н), 4,38 (с., 3H), 4,02-3,93 (м., 2Н), 3,82-3,74 (м., 3H), 3,38-3,34 (м., 4Н), 3,32-3,28 (м., 2Н), 1,69 (д., J=7,0 Гц, 6Н), 1,46 (т., J=7,3 Гц, 3H). ЖХ/МС (ESI) м/з: 475,2 (М+1).
Пример 5
N-(6-(1,4-диазепан-1-ил)пиридазин-3-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
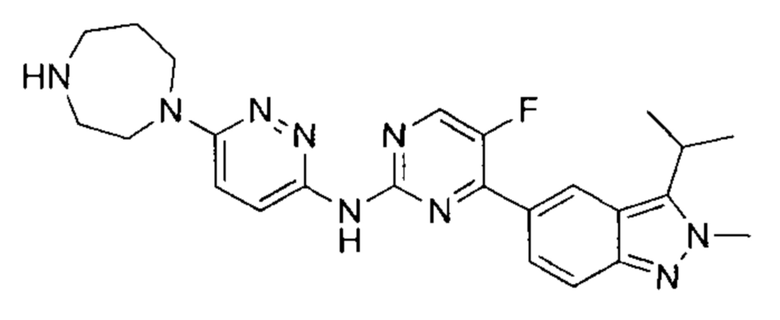
Этап 1:
трет-бутил-4-(6-хлорпиридазин-3-ил)-1,4-диазепан-1-карбоксилат

К раствору 3,6-дихлорпиридазина (2,00 г, 13,42 ммоль, 1,05 эквивалента), трет-бутил-1,4-диазепан-1-карбоксилата (2,56 г, 12,78 ммоль, 1,00 эквивалент) в диметилсульфоксиде (15,00 мл) добавили одной порцией триэтиламин (3,88 г, 38,34 ммоль, 3,00 эквивалента). Раствор нагрели до 80°C и перемешивали в течение 7 часов. ЖХ/МС показала, что реакция завершилась. Масс-спектр подтвердил, что образовался желаемый продукт. Раствор охладили до 25°C, упарили и таким образом получили неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 30:1 до 20:1) и получили указанное в заголовке соединение (1,20 г, неочищенный продукт). Неочищенный продукт представлял собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,18 (д., J=9,4 Гц, 1Н), 6,79 (д., J=9,4 Гц, 1Н), 3,87-3,57 (м., 6Н), 3,39-3,23 (м., 2Н), 1,99-1,93 (м., 2Н), 1,40 (с., 9Н). ЖХ/МС (ESI) м/з: 313,1 (М+1).
Этап 2:
трет-бутил-4-(6-(5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридазин-3-ил)-1,4-диазепан-1-карбоксилат
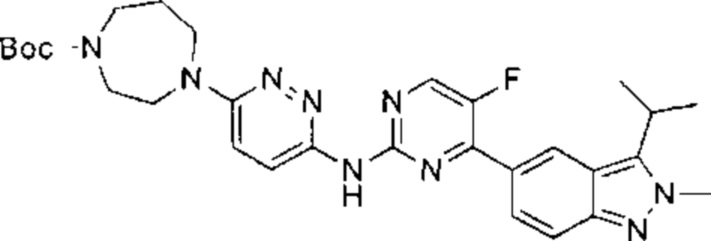
К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амина (промежуточный продукт В) (200,00 мг, 700,97 мкмоль, 1,00 эквивалент) в диоксане (3,00 мл) добавили трет-бутил-4-(6-хлорпиридазин-3-ил)-1,4-диазепан-1-карбоксилат, цезия карбонат (575,00 мк, 1,76 ммоль, 2,52 эквивалента), Pd2(дба)3 (65,00 мг, 70,98 мкмоль, 0,10 эквивалента) и Xantphos (85,00 мг, 146,90 мкмоль, 0,21 эквивалента). Воздух в таре три раза заменяли азотом. Смесь нагрели до 100°С и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Раствор охладили до 20°С, упарили и таким образом получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (этилацетат) и получили указанное в заголовке соединение (120,00 мг, 213,66 мкмоль, выход - 30,48%), представляющее собой бледно-желтое твердое вещество ЖХ/МС (ESI) м/з: 562,2 (М+1).
Этап 3:
N-(6-(1,4-диазепан-1-ил)пиридазин-3-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

К раствору
трет-бутил-4-(6-(5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридазин-3-ил)-1,4-диазепан-1-карбоксилата (120,00 мг, 213,66 мкмоль, 1,00 эквивалент) в дихлорметане (2,00 мл) по капле добавили трифторуксусную кислоту (1,00 мл). Раствор перемешивали при 25°С в течение 1 часа. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Раствор упарили под пониженным давлением и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (45,38 мг, 98,32 мкмоль, выход - 46,02%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,96 (с., 1Н) 8,85 (д., J=3,4 Гц, 1H) 8,48 (д., J=9,2 Гц, 1Н) 8,08 (д., J=10,0 Гц, 1Н) 7,90 (д., J=9,2 Гц, 2Н) 4,36 (с., 3Н) 4,11 (т., J=5,0 Гц, 2Н) 3,89 (т., J=5,9 Гц, 2Н) 3,76 (квинтет, J=7,0 Гц, 1Н) 3,52 (т., J=5,1 Гц, 2Н) 3,48-3,36 (м., 2Н) 2,28 (ушир.с., 2Н) 1,68 (д., J=7,0 Гц, 6Н).
Пример 6
N-(6-(3-(диметиламино)пирролидин-1-ил)пиридазин-3-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

Этап 1:
1-(6-хлорпиридазин-3-ил)-N,N-[-диметилпирролидин-3-амин
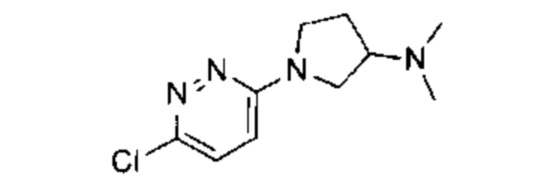
Раствор 3,6-дихлорпиридазина (1,20 г, 8,05 ммоль, 1,00 эквивалент), N,N-диметилпирролидин-3-амина (1,01 г, 8,86 ммоль, 1,10 эквивалента) и триэтиламина (815,06 мг, 8,05 ммоль, 1,00 эквивалент) в N,N-диметилформамиде (15,00 мл) нагрели до 40°C и перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Масс-спектр подтвердил, что образовался желаемый продукт. Раствор охладили до 25°C. Смесь очистили посредством препаративной ВЭЖХ и получили указанное в заголовке соединение (1,40 г, 6,18 ммоль, выход - 76,71%), представляющее собой ярко-фиолетовое твердое вещество. ЖХ/МС (ESI) м/з: 227,1 (М+1).
Этап 2:
N-(6-(3-(диметиламино)пирролидин-1-ил)пиридазин-3-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амина (промежуточный продукт В) (150,00 мг, 525,73 мкмоль, 1,00 эквивалент) в диоксане (3,00 мл) добавили 1-(6-хлорпиридазин-3-ил)-N,N-диметилпирролидин-3-амин (143,03 мг, 630,88 мкмоль, 1,20 эквивалента), цезия карбонат (428,23 мг, 1,31 ммоль, 2,50 эквивалента), Pd2(dba)3 (48,14 мг, 52,57 мкмоль, 0,10 эквивалент), и Xantphos (60,84 мг, 105,15 мкмоль, 0,20 эквивалент). Воздух в таре три раза заменяли азотом. Смесь нагрели до 100°C и перемешивали в течение 18 часов. ЖХ/МС показала, что часть исходного материала не прореагировала, но желаемый продукт был идентифицирован. Раствор охладили до 20°C, разбавили дихлорметаном (10 мл) и профильтровали. Фильтрат упарили и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (15,33 мг, 32,24 мкмоль, выход - 6,13%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,94 (с., 1Н), 8,81 (д., J=3,4 Гц, 1Н), 8,46 (д., J=9,3 Гц, 1Н), 7,99 (ушир. с., 1Н), 7,92-7,78 (м., 2Н), 4,35 (с., 3Н), 4,26-4,09 (м., 2Н), 4,01-3,85 (м., 2Н), 3,84-3,64 (м., 2Н), 3,02 (с., 6Н), 2,66 (ушир. с, 1H), 2,46 (дд, J=7,9, 13,1 Гц, 1H), 1,67 (д., J=7,0 Гц, 6Н).
Пример 7
3-[4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперазин-1-ил]пропаннитрил
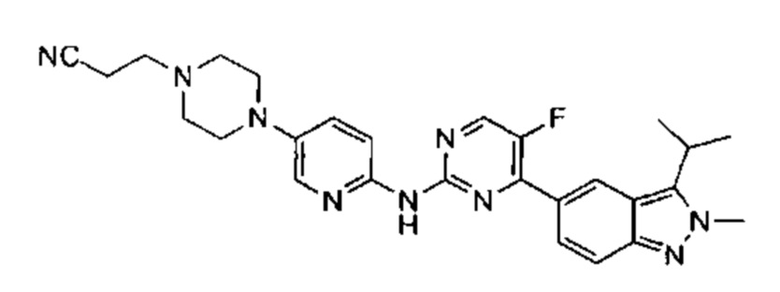
К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (200,00 мг, 447,91 мкмоль, 1,00 эквивалент) в диметилсульфоксиде (4 мл добавили калия карбонат (123,81 мг, 895,82 мкмоль, 2,00 эквивалента) и 3-бромпропионитрил (120,01 мг, 895,82 мкмоль, 73,63 мкл, 2,00 эквивалента). Смесь перемешивали при 25°C в течение 16 часов. ЖХ/МС показала, что в смеси осталось около 50% исходного материала. Затем смесь нагрели до 50°C и перемешивали при 50°C в течение 2 часов. ЖХ/МС показала, что в смеси осталось около 16% исходного материала. Затем в смесь добавили метанол (3 мл). Смесь снова перемешивали при 50°C в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 25°C, выпарили метанол и таким образом получили неочищенный продукт. Последний разбавили водой (15 мл) и профильтровали. Осадок на фильтре очистили в шариковой мельнице с метанолом (5 мл) и получили указанное в заголовке соединение (146,40 мг, 276,05 мкмоль, выход - 61,63%, чистота - 94,2%). 1Н ЯМР (400 МГц, CDCl3) δ 8,68 (с., 1Н), 8,38-8,31 (м., 2Н), 8,07-8,02 (м., 2Н), 7,99 (с., 1Н), 7,73 (д., J=9,16 Гц, 1Н), 7,34 (дд, J=9,03, 3,01 Гц, 1Н), 4,19 (с., 3Н), 3,57-3,46 (м., 1Н), 3,22-3,14 (м., 4Н), 2,82-2,75 (м., 2Н), 2,74-2,67 (м., 4Н), 2,60-2,55 (м., 2Н), 1,59 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 500,3 (М+1).
Пример 8
2-[4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперазин-1-ил]ацетонитрил
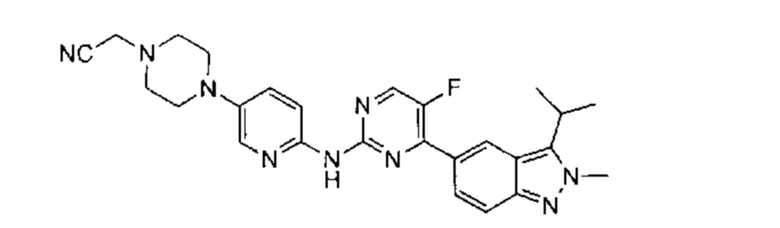
К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (50,00 мг, 111,98 мкмоль, 1,00 эквивалент) в диметилсульфоксиде (3 мл) добавили калия карбонат (30,95 мг, 223,96 мкмоль, 2,00 эквивалента) и бромацетонитрил (26,86 мг, 223,96 мкмоль, 2,00 эквивалента). Смесь перемешивали при 25°C в течение 1 часа. ЖХ/МС показала, что реакция завершилась. Смесь упарили и затем разбавили водой (10 мл). Водную фазу экстрагировали этилацетатом (10 мл×3). Объединенные органические фазы промыли насыщенным солевым раствором (15 мл), высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили в шариковой мельнице с метанолом (5 мл×2) и получили указанное в заголовке соединение (12,40 мг, 24,52 мкмоль), выход - 21,89%, чистота - 96%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,70 (с., 1Н), 8,67 (с., 1Н), 8,58 (д., J=3,96 Гц, 1Н), 8,14-8,00 (м., 2Н), 7,92 (д., J=9,23 Гц, 1Н), 7,66 (д., J=9,23 Гц, 1Н), 7,43 (дд, J=9,04 Гц, 3,01 Гц, 1Н), 4,14 (с., 3Н), 3,81 (с., 2Н), 3,60 (тд, J=13,85, 6,83 Гц, 1Н), 3,22-3,11 (m, 4H), 2,69-2,60 (м., 4Н), 1,50 (д., J=6,97 Гц, 6Н). ЖХ/МС (ESI) м/з: 486,3 (М+1).
Пример 9
N-[5-[4-(2-аминоэтил)пиперазин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
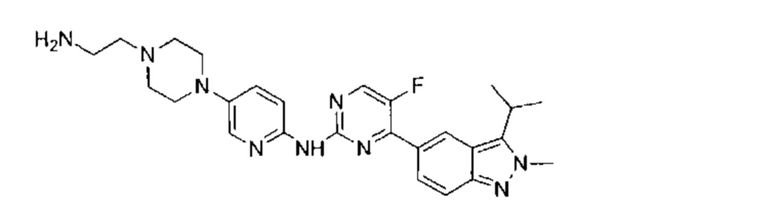
Этап 1:
2-(4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-ил)ацетонитрил
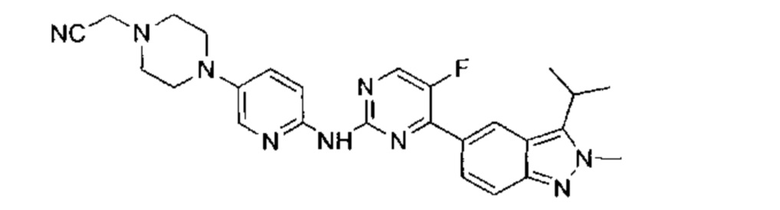
К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (17,60 г, 39,42 ммоль, 1,00 эквивалент) и калия карбоната (10,90 г, 78,84 ммоль, 2,00 эквивалента) в диметилсульфоксиде (176,00 мл) добавили 2-бромацетонитрил (9,46 г, 78,84 ммоль, 5,26 мл, 2,00 эквивалента). Смесь перемешивали при 30°C в течение 1 часа. ЖХ/МС показала, что реакция завершилась. Смесь налили в воду (30 мл) и отфильтровали. Полученный осадок на фильтре промыли водой (5 мл×2), высушили под вакуумом и таким образом получили указанное в заголовке соединение (16,00 г, 32,95 ммоль, выход - 83,59%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 486,3 (М+1)
Этап 2:
N-[5-[4-(2-аминоэтил)пиперазин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
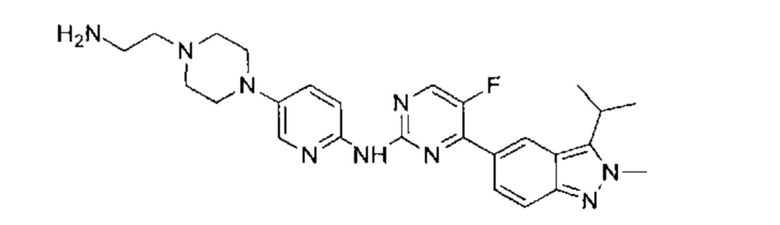
В атмосфере водорода с давлением 50 фунт/кв. дюйм 2-(4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)амино)пиримидин-2-ил)амин)пиридин-3-ил)пиперазин-1-ил)ацетонитрил (6,00 г, 12,36 ммоль, 1,00 эквивалент) и никель Ренея (8,47 г, 98,88 ммоль, 8,00 эквивалентов) в смеси водного аммония (10,00 мл) и тетрагидрофурана (100,00 мл) перемешивали при 50°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь отфильтровали. Полученный осадок на фильтре промыли этанолом (100 мл×3). Объединенные органические слои выпарили под вакуумом и таким образом получили неочищенный продукт. Остаток очистили посредством препаративной ВЭЖХ (соляная кислота) и таким образом получили указанное в заголовке соединение (6,50 г, 10,85 ммоль, выход - 87,80%, чистота -100%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8,90 (с., 1H), 8,79 (д., J=3,6 Гц, 1Н), 8,39-8,22 (м., 2Н), 7,95 (д., J=2,9 Гц, 1Н), 7,82 (д., J=9,2 Гц, 1Н), 7,58 (д., J=9,7 Гц, 1Н), 4,30 (с., 3Н), 3,73 (тд, J=14,0, 7,1 Гц, 3Н), 3,68-3,54 (м., 6Н), 3,50 (ушир. с., 2Н), 3,34 (ушир. с., 2Н), 1,65 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 490,3 (М+1)
Пример 10
2-[4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперазин-1-ил]этанол

Этап 1:
2-[4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперазин-1-ил]этанол
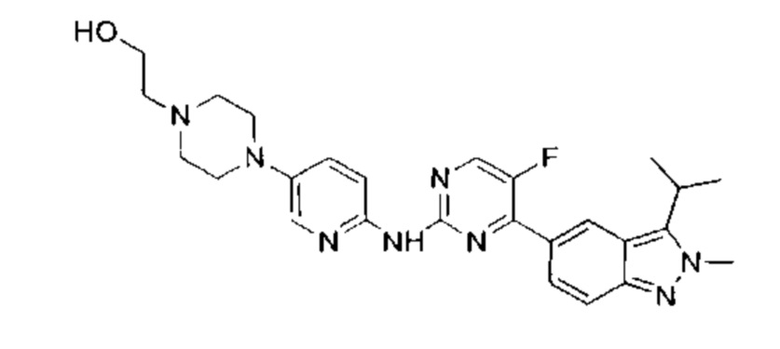
К раствору 5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (2,00 г, 4,48 ммоль, 1,00 эквивалент) и 2-бромэтанола (1,68 г, 13,44 ммоль, 954,55 мкл, 3,00 эквивалента) в этаноле (30,00 мл) добавили диизопропилэтиламин (1,74 г, 13,44 ммоль, 2,35 мл, 3,00 эквивалента). Смесь нагрели до 80°C и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал вступил в реакцию практически полностью, и масс-спектр подтвердил желаемое соединение. Смесь охладили до 25°C и профильтровали. Осадок на фильтре очистили в шаровой мельнице с метанолом (10 мл). Смесь отфильтровали, и осадок на фильтре высушили, получив таким образом указанное в заголовке соединение (1,50 г, 2,97 ммоль, выход - 66,20%, чистота - 97%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,74 (с, 1Н), 8,44 (д., J=3,9 Гц, 1Н), 8,23 (д., J=9,0 Гц, 1Н), 8,07 (д., J=8,9 Гц, 1Н), 8,01 (д., J=2,6 Гц, 1Н), 7,65 (д., J=9,2 Гц, 1Н), 7,49 (дд, J=9,0, 2,9 Гц, 1Н), 4,19 (с., 3Н), 3,75 (т., J=6,0 Гц, 2Н), 3,64 (тд, J=14,1, 7,0 Гц, 1Н), 3,26-3,18 (м., 4Н), 2,80-2,70 (м., 4Н), 2,62 (т., J=6,0 Гц, 2Н), 1,60 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 491,3 (М+1).
Пример 11
(4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-2-ил)метанол

Этап 1:
(пиперазин-2-ил)метанол

При 0°C к суспензии лития алюминия тетрагидрата (5,61 г, 147,74 ммоль, 1,5 эквивалента) в тетрагидрофуране (300 мл) добавили несколькими порциями пиперазин-2-карбоновую кислоту (20,00 г, 98,49 ммоль, 1,00 эквивалент, 2 гидрохлорид). Смесь нагрели до 70°C и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционную смесь охладили до 0°C и остановили реакцию водой (5 мл) и водным раствором натрия гидроксида (15%, 5 мл). Смесь отфильтровали, осадок на фильтре промыли дихлорметаном (100 мл). Фильтрат высушили над безводным натрия сульфатом и упарили, получив указанное в заголовке соединение (2,4 г, 20,66 ммоль, выход - 20,98%), представляющее собой светло-желтое масло. 1H ЯМР (400 МГц, CDCl3) δ 3,54 (дд, J=4,1, 10,7 Гц, 1Н), 3,40 (д., J=7,2 Гц, 1Н), 3,02-2,95 (м., 1Н), 2,90 (дд, J=11,9, 2,6 Гц, 3Н), 2,81-2,75 (м., 4Н), 2,47(дд, J=11,8, 10,3 Гц, 1Н), 1,41 (с., 1Н).
Этап 2:
(4-(6-нитропиридин-3-ил)пиперазин-2-ил)метанол
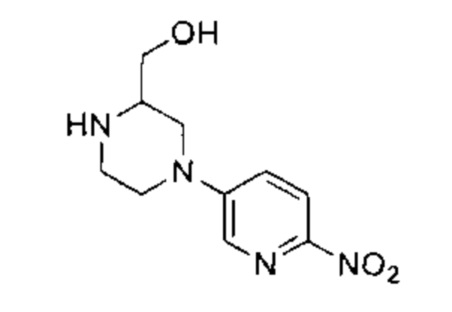
К раствору (пиперазин-2-ил)метанола (2,4 г, 20,66 ммоль, 1,00 эквивалент) в диметилсульфоксиде (20,00 мл) добавили 5-бром-2-нитропиридин (4,19 г, 20,66 ммоль, 1,00 эквивалент) и триэтиламин (4,18 г, 41,32 ммоль, 2,00 эквивалента). Реакционную смесь нагрели до 50°C и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Полученный раствор использовали на следующем этапе без предварительной обработки. ЖХ/МС (ESI) м/з: 293,1 (М+1).
Этап 3:
трет-бутил-2-(((трет-бутоксикарбонил)окси)метил)-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат

К раствору (4-(6-нитропиридин-3-ил)пиперазин-2-ил)метанола (4,92 г, 20,65 ммоль, 1,00 эквивалент) в дихлорметане (40,00 мл) добавили триэтиламин (6,27 г, 61,95 ммоль, 3,00 эквивалента) и ди-трет-бутилбикарбонат (9,01 г, 41,3 ммоль, 2,00 эквивалента). Смесь перемешивали при 15°C в течение 18 часов. ТСХ (петролейный эфир: этилацетат = 1:1) показал, что исходный материал полностью вступил в реакцию. Реакционный раствор разбавили водой (100 мл), и под вакуумом удалили дихлорметан. Водную фазу экстрагировали этилацетатом (50 мл×3). Объединенные органические фазы промыли последовательно водой (50 мл) и насыщенным солевым раствором (30 мл), высушили над безводным натрия сульфатом и упарили до сухого остатка. Остаток очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = от 10:1 до 3:1) и получили указанное в заголовке соединение (1,5 г, 3,42 ммоль, выход - 16,57%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (д., J=9,03 Гц, 1Н), 8,11 (д., J=3,01 Гц, 1Н), 7,20 (дд, J=9,16, 3,14 Гц, 1H), 4,46 (ушир. с., 1Н), 4,12-4,25 (м., 2Н), 4,02 (ушир. с., 1Н), 3,93 (д., J=13,30 Гц, 1H), 3,70-3,80 (м., 1Н), 3,28-3,43 (м., 2Н), 3,14-3,25 (м., 1H), 1,50 (с., 9Н), 1,46 (с., 9Н).
Этап 4:
трет-бутил-4-(6-амино-3-пиридил)-2-(трет-бутоксикарбонилоксиметил)пиперазин-1-карбоксилат
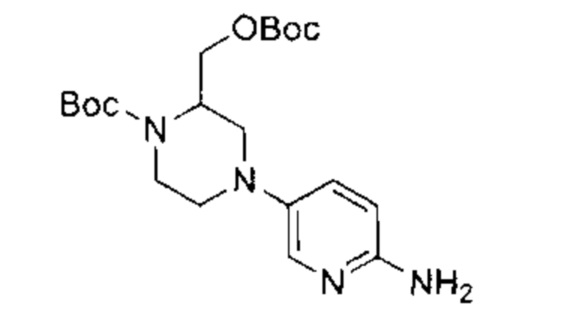
К раствору
трет-бутил-2-(((трет-бутоксикарбонил)окси)метил)-4-(6-нитропиридин-3-ил)-пиперазин -1-карбоксилата (500 мг, 1,14 ммоль, 1,00 эквивалент) в метаноле (30,00 мл) добавили влажный палладиевый катализатор на углеродном носителе (200,00 мг). Через колбу с реакционной смесью три раза пропускали аргон и водород. Смесь перемешивали при 15°C в течение 18 часов в атмосфере водорода с давлением 15 фунт/кв.дюйм. ТСХ (петролейный эфир: этилацетат = 1:1) показал, что исходный материал полностью вступил в реакцию. Реакционную смесь отфильтровали, и осадок на фильтре промыли метанолом (10 мл). Осадок, полученный путем упаривания фильтрата, очистили посредством препаративной ТСХ (этилацетат), получив указанное в заголовке соединение (200,00 мг, 489,61 мкмоль, выход - 42,95%), представляющее собой коричневое твердое вещество. ЖХ/МС (ESI) м/з: 409,2 (М+1).
Этап 5:
трет-бутил-2-(((трет-бутоксикарбонил)окси)метил)-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилат
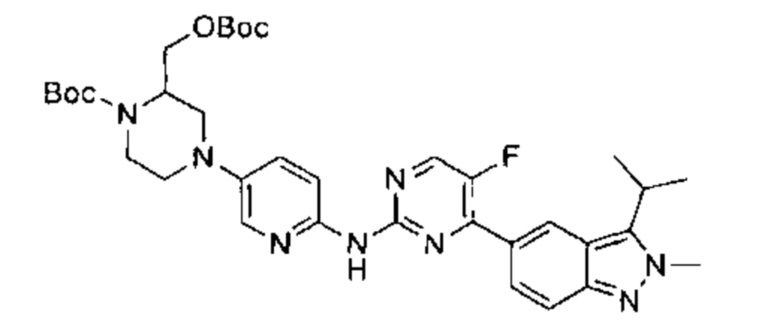
В атмосфере азота к раствору трет-бутил-4-(6-амино-3-пиридил)-2-(трет-бутоксикарбонилоксиметил)пиперазин-1-карбоксилата (200,00 мг, 489,61 мкмоль, 1,00 эквивалент) в диоксане (5 мл) добавили 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-2Н-индазол (промежуточный продукт С) (150,70 мг, 494,51 мкмоль, 1,01 эквивалента), цезия карбонат (319,05 мг, 979,22 мкмоль, 2,00 эквивалента), Pd2(дба)3 (44,83 мг, 48,96 мкмоль, 0,10 эквивалента) и Xantphos (56,66 мг, 97,92 мкмоль, 0,20 эквивалента). Через колбу с реакционной смесью три раза пропускали азот. Смесь нагрели до 100°C и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционный раствор охладили до 15°C и отфильтровали. Осадок на фильтре промыли этилацетатом (5 мл). Осадок, полученный путем упаривания фильтрата, очистили посредством препаративной ТСХ (петролейный эфир : этилацетат = 1:1), получив указанное в заголовке соединение (104,00 мг, 153,67 мкмоль, выход - 31,39%), представляющее собой коричневое твердое вещество. 1Н ЯМР (300 МГц, CDCl3) δ 8,69 (с., 1Н), 8,37 (д., J=4,0 Гц, 1H), 8,07-7,98 (м., 2Н), 7,73 (д., J=9,2 Гц, 1Н), 7,35 (дд, J=2,9, 9,1 Гц, 1Н), 4,49 (ушир. с., 1Н), 4,46-4,25 (м., 2Н), 4,19 (с., 3Н), 4,03 (ушир. с., 1Н), 3,57-3,50 (м., 2Н), 3,39 (д., J=11,5 Гц, 1Н), 3,23 (т., J=9,6 Гц, 1Н), 2,94-2,69 (м., 2Н), 1,60 (д., J=7,0 Гц, 6Н), 1,50 (с., 9Н), 1,50 (с., 9Н).
Этап 6:
(4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-2-ил)метанол

К раствору трет-бутил-2-((t-бутоксикарбонил)окси)метил)-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (100 мг, 147,76 мкмоль, 1,00 эквивалент) в дихлорметане (2,00 мл) добавили трифторуксусную кислоту (1,00 мл). Смесь перемешивали при 15°C в течение 1 часа. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Осадок, полученный путем упаривания реакционного раствора, очистили посредством препаративной ВЭЖХ (соляная кислота) и таким образом получили указанное в заголовке соединение (43,08 мг, 78,41 ммоль, выход - 53,07%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8,92 (с., 1Н), 8,80 (д., J=3,5 Гц, 1Н), 8,41 (д., J=9,2 Гц, 1Н), 8,30 (дд, J=2,5, 9,7 Гц, 1Н), 7,96 (д., J=2,5 Гц, 1Н), 7,85 (д., J=9,0 Гц, 1H), 7,57 (д., J=9,5 Гц, 1Н), 4,33 (с., 3Н), 3,96-3,84 (м., 3Н), 3,84-3,69 (м., 2Н), 3,57 (д., J=12,9 Гц, 2Н), 3,39 (дт, J=3,0, 12,1 Гц, 1Н), 3,28-3,09 (м, 2Н), 1,65 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 477,2 (М+1).
Пример 12
N-[5-[3-(этиламино)пирролидин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пидимидин-2-амин
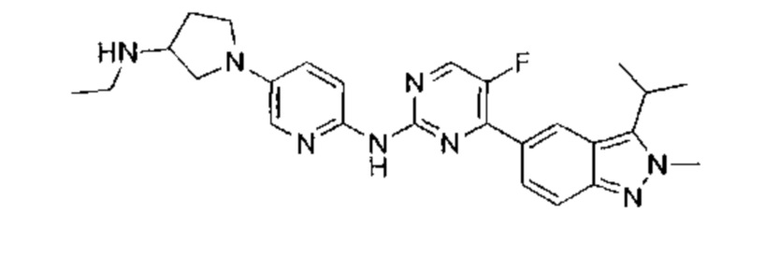
Этап 1:
трет-бутил-(3S)-3-гидроксипирролидин-1-карбоксилат
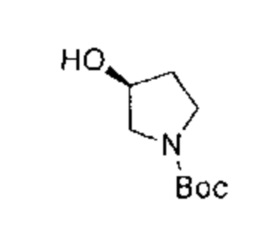
К раствору (3S)-пирролидин-3-ола (3,50 г, 28,32 ммоль, 1,00 эквивалент, гидрохлорид) в дихлорметане (30,00 мл) добавили триэтиламин (11,46 г, 113,28 ммоль, 4,00 эквивалента) и ди-трет-бутилбикарбонат (8,04 г, 36,82 ммоль, 1,30 эквивалента). Смесь перемешивали при 20°C в течение 18 часов. ТСХ показала, что реакция завершилась. Смесь разбавили водой (10 мл), затем добавили водный раствор лимонной кислоты (10%, 20 мл). Водную фазу экстрагировали дихлорметаном (10 мл×2). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл×1), высушили над безводным натрия сульфатом, профильтровали и упарили до указанного в заголовке соединения (4,00 г, неочищенный продукт), представляющее собой желтое масло. 1Н ЯМР (400 МГц, метанол-d4) δ 4,42-4,32 (м., 1Н), 3,49-3,35 (м., 3Н), 3,31-3,19 (м., 1Н), 2,08-1,82 (м., 2Н), 1,51-1,42 (м., 9Н).
Этап 2:
трет-бутил-3-оксопирролидин-1-карбоксилат
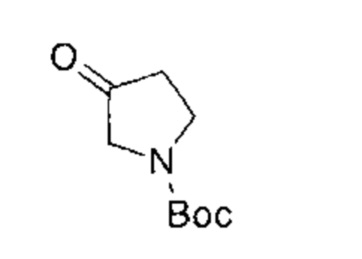
При 0°C к раствору тертрет-бутил-(3S)-3-гидроксипирролидин-1-карбоксилата (500,00 мг, 2,67 ммоль, 1,00 эквивалент) в дихлорметане (10,00 мл) добавили несколькими порциями реактив Десса-Мартина (1,70 г, 4,01 ммоль, 1,50 эквивалента). Смесь нагрели до 20°C и перемешивали при 20°C в течение 18 часов. ТСХ показала, что реакция завершилась. Реакцию остановили, добавив в раствор 10 мл 30% раствора натрия сульфата. Затем добавили раствор натрия бикарбоната (5 мл). Водную фазу экстрагировали дихлорметаном (5 мл×3). Объединенные органические фазы промыли насыщенным солевым раствором (15 мл×1), высушили над безводным натрия сульфатом, профильтровали и упарили до указанного в заголовке соединения (453,00 мг, неочищенный продукт), представляющее собой желтое масло. 1Н ЯМР (400 МГц, CDCl3) δ 3,85-3,65 (м., 4Н), 2,66-2,52 (м., 2Н), 1,51-1,44 (м., 9Н).
Этап 3:
трет-бутил-3-[бензил(этил)амино]пирролидин-1-карбоксилат

При 0°C к раствору трет-бутил-3-оксопирролидин-1-карбоксилата (353,00 мг, 1,91 ммоль, 1,00 эквивалент) в дихлорметане (8,00 мл) добавили бензилэтиламин (309,90 мг, 2,29 ммоль, 1,20 эквивалента) и уксусную кислоту (1,15 мг, 19,10 мкмоль, 0,01 эквивалент). Смесь перемешивали при 0°C в течение 0,5 часа. Затем добавили натрия цианоборгидрид (647,69 мг, 3,06 ммоль, 1,60 эквивалента). Смесь перемешивали при 25°C в течение 3 часов. ЖХ/МС показала, что реакция завершилась. Реакцию остановили, добавив в смесь воду (10 мл). Водную фазу экстрагировали дихлорметаном (10 мл×2). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл×1), высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = от 20:1 до 10:1) и получили указанное в заголовке соединение (211,00 мг, 693,10 мкмоль, выход - 36,29%), представляющее собой бесцветное масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,38-7,28 (м., 4Н), 7,25 (д., J=6,27 Гц, 1Н), 3,71-3,06 (м., 7Н), 2,59 (кв., J=7,15 Гц, 2Н), 2,06-1,98 (м., 1Н), 1,90-1,77 (м., 1Н), 1,46 (с., 9Н), 1,04-0,97 (м., 3Н). ЖХ/МС (ESI) м/з: 305,3 (М+1).
Этап 4:
N-бензил-N-этилпирролидин-3-амин
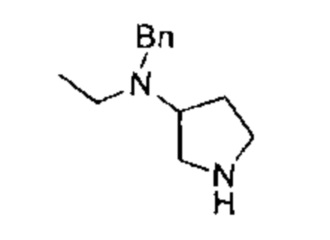
К раствору трет-бутил-3-[бензил(этил)амино]пирролидин-1-карбоксилата (2,00 г, 6,57 ммоль, 1,00 эквивалент) в дихлорметане (12 мл) добавили трифторуксусную кислоту (4,59 г, 40,27 ммоль, 6,13 эквивалента). Смесь перемешивали при 20°C в течение 30 минут. ТСХ показала, что реакция завершилась. Реакционный раствор упарили, получив указанное в заголовке соединение (2,00 г, неочищенный продукт, трифторацетат), представляющее собой желтое масло. ЖХ/МС (ESI) м/з: 205,3 (М+1).
Этап 5:
N-бензил-N-этил-1-(6-нитро-3-пиридил)пирролидин-3-амин

К раствору N-бензил-N-этилпирролидин-3-амина (2,00 г, 4,63 ммоль, 1,00 эквивалент, трифторацетат) в диметилсульфоксиде (10,00 мл) добавили триэтиламин (2,74 г, 27,06 ммоль, 5,85 эквивалента) и 5-бром-2-нитропиридин (1,13 г, 5,55 ммоль, 1,20 эквивалента). Смесь перемешивали при 90°C в течение 18 часов. ЖХ/МС показала, что реакция завершилась. Смесь разбавили водой (20 мл), и водную фазу подвергли экстракции этилацетат (15 мл×4). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл×1), высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = от 10:1 до 3:1) и получили указанное в заголовке соединение (700,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,16 (д., J=9,04 Гц, 1Н), 7,77 (д., J=3,01 Гц, 1Н), 7,41-7,28 (м., 4Н), 6,81 (дд, J=9,14, 2,92 Гц, 1Н), 3,85-3,25 (м., 8Н), 2,68 (кв., J=7,03 Гц, 2Н), 2,36-1,97 (м., 2Н), 1,09 (т., J=7,06 Гц, 3Н). ЖХ/МС (ESI) м/з: 327,2 (М+1).
Этап 6:
[5-[3-[бензил(этил)амино]пирролидин-1-ил]пиридин-2-амин
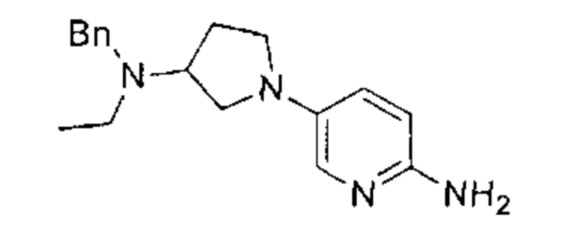
К раствору N-бензил-N-этил-1-(6-нитро-3-пиридил)пирролидин-3-амина (450,00 мг, 1,38 ммоль, 1,00 эквивалент) в этаноле (10 мл) добавили цинковую пыль (360,62 мг, 5,52 ммоль, 4,00 эквивалента) и аммония хлорид (737,48 мг, 13,80 ммоль, 10,00 эквивалента). Смесь нагрели до 70°C и перемешивали в течение 3 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, предварительно охладив до 25°C. Фильтрат упарили и получили указанное в заголовке соединение (216,00 мг, неочищенный продукт), представляющее собой фиолетовое твердое вещество. ЖХ/МС (ESI) м/з: 297,2 (М+1).
Этап 7:
N-[5-[3-[бензил(этил)амино]пирролидин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
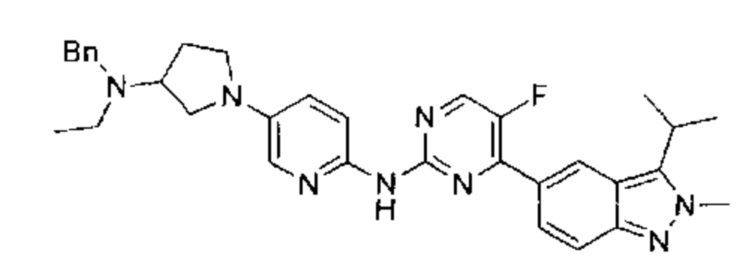
В атмосфере азота к раствору 5-[3-[бензил(этил)амино]пирролидин-1-ил]пиридин-2-амина (129,00 мг, 435,21 мкмоль, 1,00 эквивалент) в диоксане (4,00 мл) добавили 5-(2-хлор-5-фтор-пиримидин-4-ил)-3-изопропил-2-метил-2Н-индазол (промежуточный продукт С) (159,16 мг, 522,25 мкмоль, 1,20 эквивалента), Pd2(dba)3 (39,85 мг, 43,52 мкмоль, 0,10 эквивалент), Xantphos (50,36 мг, 87,04 мкмоль, 0,20 эквивалент), и цезия карбонат (354,50 мг, 1,09 ммоль, 2,50 эквивалента). Смесь перемешивали при 110°C в течение 18 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 25°C, упарили и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (этилацетат) и получили указанное в заголовке соединение (145,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 565,3 (М+1).
Этап 8:
N-[5-[3-(этиламино)пирролидин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

К раствору N-[5-[3-[бензил(этил)амино]пирролидин-1-ил]-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амина (181,00 мг, 320,52 мкмоль, 1,00 эквивалент) в тетрагидрофуране (5 мл) добавили влажный палладиевый катализатор на углеродном носителе (400,00 мг) и формамид (2,02 г, 32,05 ммоль, 100,00 эквивалента). Смесь перемешивали при 60°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 25°C и затем профильтровали. Фильтрат упарили, получив неочищенный продукт, который затем очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (34,64 мг, 59,47 мкмоль, выход - 18,56%, степень чистоты - 94%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8,89 (с., 1H), 8,75 (д., J=3,64 Гц, 1Н), 8,35 (д., J=9,29 Гц, 1Н), 7,92 (дд, J=9,60, 2,95 Гц, 1Н), 7,81 (д., J=9,16 Гц, 1H), 7,64 (д., J=2,89 Гц, 1Н), 7,53 (д., J=9,54 Гц, 1Н), 4,30 (с., 3Н), 4,18-4,02 (м., 1Н), 3,61-3,85 (м., 4Н), 3,54-3,41 (м., 1H), 3,23 (кв., J=7,24 Гц, 2Н), 2,69-2,52 (м., 1H), 2,44-2,27 (м., 1Н), 1,65 (д., J=7,03 Гц, 6Н), 1,40 (т., J=7,28 Гц, 3Н). ЖХ/МС (ESI) м/з: 475,3 (М+1).
Пример 13
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-[5-(пиперидин-4-ил)-2-пиридил]пиримидин-2-амин
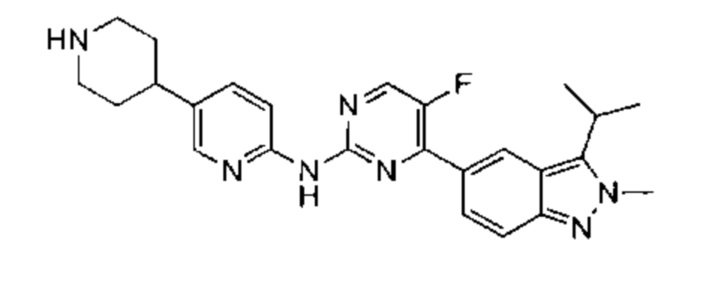
Этап 1:
трет-бутил-4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперидин-1-карбоксилат

К раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-2Н-индазола (1,00 г, 3,28 ммоль, 1,00 эквивалент) в диоксане (10 ил) добавили трет-бутил-4-(6-амино-3-пиридил)пиперидин-1-карбоксилат (1,09 г, 3,94 ммоль, 1,20 эквивалента), Pd2(dba)3 (150,18 мг, 164,00 мкмоль, 0,05 эквивалента), Xantphos (189,79 мг, 328,00 мкмоль, 0,10 эквивалент) и цезия карбонат (2,14 г, 6,56 мкмоль, 2,00 эквивалента) в атмосфере азота. Смесь перемешивали при 110°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь упарили и получили неочищенный продукт. Последний разбавили дихлорметаном (10 мл) и профильтровали. Фильтрат упарили и получили неочищенный продукт. Неочищенный продукт очистили в шариковой мельнице с метанолом (10 мл) и получили указанное в заголовке соединение (1,11 г, неочищенный продукт). ЖХ/МС (ESI) м/з: 546,2 (М+1).
Этап 2:
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-[5-(пиперидин-4-ил)-2-пиридил]пиримидин-2-амин
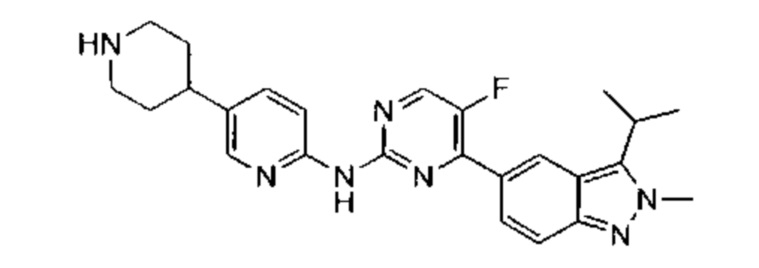
К раствору
трет-бутил-4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]пиперидин-1-карбоксилата (1,08 г, 1,98 мкмоль, 1,00 эквивалент) в дихлорметане (12 мл) добавили трифторуксусную кислоту (5,36 г, 46,97 ммоль, 3,48 мл, 23,72 эквивалента). Смесь перемешивали при 25°C в течение 1 часа. ЖХ/МС показала, что реакция завершилась. Смесь упарили и получили неочищенный продукт. 0,7 г неочищенного продукта очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (183,40 мг, 350,38 мкмоль, выход - 17,70%, чистота - 99,05%), представляющее собой желтое твердое вещество. 1H ЯМР (400 МГц, метанол-d4) δ 8.93 (с, 1Н), 8,83 (d, J=3,51 Гц, 1Н), 8,32-8,45 (м., 3Н), 7,85 (д., J=9,03 Гц, 1Н), 7,62 (д., J=9,16 Гц, 1H), 4,33 (с., 3Н), 3,74 (д., J=6,99 Гц, 1Н), 3,57 (д., J=12,67 Гц, 2Н), 3,10-3,27 (м., 3Н), 2,20 (д., J=13,68 Гц, 2Н), 1,96-2,10 (м., 2Н), 1,66 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 446,2 (М+1).
Пример 14
3-[4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]1-пиперидинил]пропаннитрил
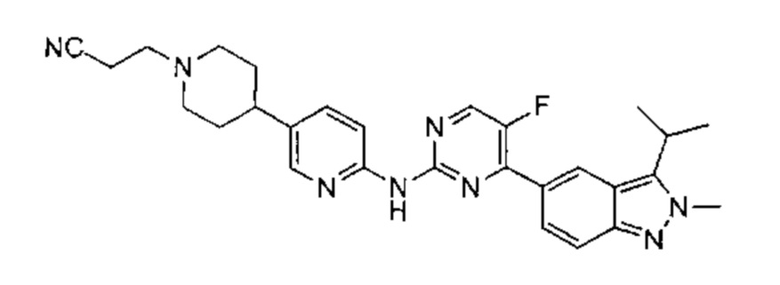
К раствору
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-[5-(4-пиперидинил)-2-пиридил]пиримидин-2-амина (1,25 г, 1,86 ммоль, 1,00 эквивалент, трифторацетат) в метаноле (10 мл) добавили триэтиламин (1,10 г, 10,82 ммоль, 1,50 мл, 5,82 эквивалента). Смесь перемешивали при 25°C в течение 30 минут и затем добавили в нее калия карботнат (514,14 мг, 3,72 ммоль, 2,00 эквивалента) и 3-бромпропионитрил (498,37 мг, 3,72 ммоль, 305,75 мкл, 2,00 эквивалента). Смесь перемешивали при 25°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь упарили, разбавили водой (15 мл) и затем профильтровали. Осадок на фильтре очистили на шариковой мельнице с метанолом (8 мл) и получили указанное в заголовке соединение (217,00 мг, 412,59 мкмоль, выход - 22,18%, чистота - 94,8%). 1Н ЯМР (400 МГц, CDCl3) δ 8,70 (с., 1Н), 8,42-8,36 (м., 2Н), 8,19 (д., J=2,13 Гц, 1Н), 8,02-8,09 (м., 2Н), 7,74 (д., J=9,16 Гц, 1H), 7,58 (дд, J=8,66 Гц, 2,38 Гц, 1Н), 4,19 (с., 3Н), 3,59-3,45 (м., 1Н), 3,04 (д., J=11,29 Гц, 2Н), 2,91-2,73 (м., 2Н), 2,60-2,47 (м., 3Н), 2,23 (дт, J=11,48 Гц, 2,51 Гц, 2Н), 1,93-1,72 (м., 4Н), 1,59 (с., 6Н). ЖХ/МС (ESI) м/з: 499,3 (М+1).
Пример 15
N-(5-((1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

Этап 1:
трет-бутил-5-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридин-3-ил)(1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-карбоксилат
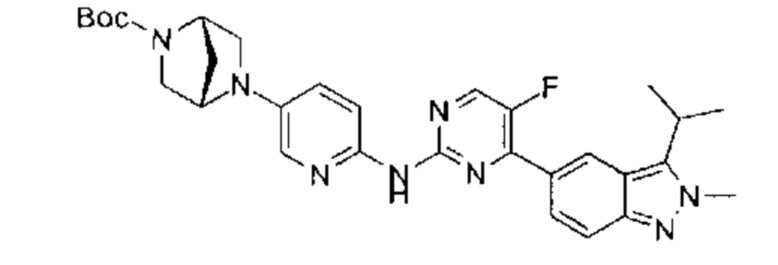
В атмосфере азота через раствор 5-(2-хлор-5-фтор-пиримидин-4-ил)-3-изопропил-2-метил-2Н-индазол (промежуточный продукт С) (376,67 мг, 1,24 ммоль, 1,20 эквивалента),
трет-бутил-5-(6-амино-3-пиридил)-(1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-карбоксила та (300,00 мг, 1,03 ммоль, 1,00 эквивалент), цезия карбоната (671,19 мг, 2,06 ммоль, 2,00 эквивалента), Pd(OAc)2 (46,25 мг, 206,00 мкмоль, 0,20 эквивалента) и XPhos (196,41 мг, 412,00 мкмоль, 0,40 эквивалента) в диоксане (10,00 мл) три раза пропускали азот. Смесь перемешивали при 100°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь упаривали до сухого остатка при пониженном давлении, и остаток очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 1:2), получив указанное в заголовке соединение (350,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 559,3 (М+1).
Этап 2:
N-(5-((1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
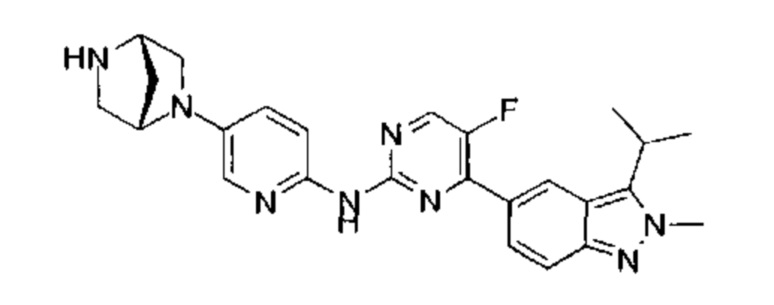
К раствору
трет-бутил-5-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)ам ино)пиридин-3-ил)-(1S,4S)-2,5-диазабицикло[2.2.1]гептан-2-карбоксилата (365,00 мг, 653,36 мкмоль, 1,00 эквивалент) в дихлорметане (8,00 мл) по капле добавляли трифторуксусную кислоту (4,00 мл) при 0°C в течение 15 минут. Реакционный раствор оставили при 15°C на 80 минут для прохождения реакции. ЖХ/МС показала, что реакция завершилась. Смесь упаривали до сухого остатка при пониженном давлении, и остаток очистили посредством препаративной ВЭЖХ (соляная кислота), получив указанное в заголовке соединение (110,00 мг, 239,90 мкмоль, выход - 36,72%, чистота - 100%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,92 (с., 1Н), 8,78 (д., J=3,51 Гц, 1Н), 8,43 (д., J=9,29 Гц, 1Н), 7,98 (дд, J=9,66, 2,89 Гц, 1Н), 7,88-7,83 (м., 1Н), 7,72 (д, J=2,89 Гц, 1Н), 7,54 (д., J=9,54 Гц, 1Н), 4,83 (с., 1Н), 4,62 (с., 1Н), 4,34 (с., 3Н), 3,80 (дд, J=10,60, 2,32 Гц, 1Н), 3,77-3,70 (м., 1H), 3,55 (д., J=10,54 Гц, 1Н), 3,50-3,41 (м., 2Н), 2,36 (д., J=11,29 Гц, 1Н), 2,17 (д., J=11,42 Гц, 1H), 1,66 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 459,3 (М+1).
Пример 16
N-(5-(1,7-диазаспиро[4.4]нонан-7-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

Этап 1: (S)-1-бензоилпролина метиловый эфир
К раствору L-пролина метилового эфира гидрохлорида (20,00 г, 120,76 ммоль, 1,00 эквивалент) в дихлорметане (300,00 мл) добавили триэтиламин (36,66 г, 362,28 ммоль, 3,00 эквивалента) и бензоилхлорид (16,98 г, 120,76 ммоль, 1,00 эквивалент). Реакционную смесь перемешивали при 15°C в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Осадок, полученный путем упаривания реакционного раствора, разбавили этилацетатом (200 мл) и последовательно промыли водой (100 мл), безводным раствором лимонной кислоты (2,5%, 100 мл) и насыщенным натрия бикарбонатом (100 мл). Органическую фазу высушили над безводным натрия сульфатом и упарили, получив указанное в заголовке соединение (24,00 г., 102,89 ммоль, выход - 85,20%), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,63-7,52 (м., 2Н), 7,45-7,38 (м., 3Н), 4,68 (дд, J=5,2, 8,2 Гц, 1Н), 3,82-3,75 (м., 3Н), 3,66 (тд, J=7,0, 10,2 Гц, 1H), 3,57-3,53 (м., 1Н), 2,40-2,17 (м., 1H), 2,07-1,98 (м., 2Н), 1,95-1,82 (м, 1Н).
Этап 2:
метил-1-бензоил-2-(нитрилометил)пирролидин-2-карбоксилат

При -78°C к раствору метил-(S)1-бензоилпирролидин-2-карбоксилата (10,00 г, 42,87 ммоль, 1,00 эквивалент) в тетрагидрофуране (150,00 мл) добавили раствор лития диизопропиламида в тетрагидрофуране (2 моль/л, 25,72 мл, 1,20 эквивалента) и перемешивали смесь в течение 1 часа. К смеси по капле добавили раствор 2-бромацетонитрила (6,17 г, 51,44 ммоль, 1,20 эквивалента) в тетрагидрофуране (30,00 мл). Смесь перемешивали при 15°C в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакцию остановили, добавив в смесь насыщенный водный раствор аммония хлорида (100 мл), и провели разделение фаз. Затем водную фазу подвергли экстракции этилацетатом (100 мл). Органические фазы объединили, высушили над безводным натрия сульфатом и упарили, получив указанное в заголовке соединение (12,00 г, неочищенный продукт), представляющее собой коричневое масло. Неочищенный продукт использовали в следующем этапе без предварительной обработки. 1H ЯМР (400 МГц, CDCl3) δ 7,60-7,54 (м., 2Н), 7,50-7,40 (м., 3Н), 3,81 (с., 3Н), 3,80-3,77 (м., 1Н), 3,77-3,73 (м., 1Н), 3,71-3,64 (м., 1H), 3,15 (д., J=17,2 Гц, 1H), 2,44-2,27 (м., 2Н), 2,17-2,05 (м., 2Н).
Этап 3: 1-бензил-1,7-диазаспиро[4,4]нонан-6-он

К раствору метил 1-бензоил-2-(нитрилметил)пирролидин-2-карбоксилата (5,00 г, 18,36 ммоль, 1,00 эквивалент) и водного аммония (5,00 мл) в метаноле (100,00 мл) добавили никель Ренея (5,00 г). Через колбу с реакционной смесью три раза пропускали аргон и водород. Смесь нагрели до 70°C под давлением водорода, равным 55 фунт/кв.дюйм, и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционную смесь охладили до 25°C и профильтровали. Остаток, полученный путем упаривания фильтрата, очистили посредством колоночной хроматографии с силикагелем (петролейный эфир : этилацетат = 3:1 либо дихлорметан : метанол = 30:1) и получили указанное в заголовке соединение (2,25 г, выход - 50,17%), представляющее собой светло-коричневое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,62-7,50 (м., 2Н), 7,45-7,35 (м., 3Н), 5,84 (ушир. с., 1Н), 3,71-3,53 (м., 3Н), 3,42-3,34 (м., 1Н), 2,95 (ддд, J=12.8, 9,7, 6,8 Гц, 1Н), 2,34 (тд., J=6,7, 12,5 Гц, 1Н), 2,20-1,96 (м., 3Н), 1,94-1,82 (м., 1Н).
Этап 4: 1-бензил-1,7-диазаспиро[4,4]нонан

При 0°C к суспензии алюмогидрида лития (7,13 г, 187,99 ммоль, 5,60 эквивалента) в тетрагидрофуране (300,00 мл) добавили несколькими порциями 1-бензил-1,7-диазаспиро[4,4]нонан-6-он (8,2 г, 33,57 ммоль, 1,00 эквивалент). После добавления реактивов смесь нагрели до 70°C и перемешивали в течение 2 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционную смесь охладили до 15°C, остановили реакцию водой (7 мл), затем добавили водный раствор натрия гидроксида (15%, 7 мл) и воду (21 мл). Смесь отфильтровали. Фильтрат высушили над безводным натрия сульфатом, и остаток, полученный путем упаривания, очистили посредством колоночной хроматографии с силикагелем (дихлорметан: метанол = от 30:1 до 10:1), получив указанное в заголовке соединение (3,67 г, 16,97 ммоль, выход - 50,54%), представляющее собой бледно-желтое масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,36-7,28 (м., 4Н), 7,26-7,20 (м., 1Н), 3,69 (д., J=13,2 Гц, 1Н), 3,59 (д., J=13,2 Гц, 1Н), 3,13-3,04 (м., 2Н), 2,99 (тд., J=10,8, 7,7 Гц, 1Н), 2,77 (д., J=11,2 Гц, 1Н), 2,70-2,62 (м., 1Н), 2,59-2,51 (м., 1Н), 2,01 (тд., J=12.9, 8,1 Гц, 2Н), 1,92-1,85 (м., 2Н), 1,80-1,71 (м., 2Н), 1,59 (ддд, J=12,6, 7,6, 4,6 Гц, 1Н).
Этап 5:
1-бензил-7-(6-нитропиридин-3-ил)-1,7-диазаспиро[4,4]нонан
К раствору 1-бензил-1,7-диазаспиро[4,4]нонана (1,80 г, 8,32 ммоль, 1,00 эквивалент) и 5-бром-2-нитропиридина (1,81 г, 8,90 ммоль, 1,07 эквивалента) в N,N-диметилформамиде (20,00 мл) добавили триэтиламин (1,80 г, 17,80 ммоль, 2,14 эквивалента). Смесь нагрели до 100°C и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционную смесь охладили до 15°C, разбавили этилацетатом (40 мл), затем дважды промыли водой (50 мл×2). Водную фазу экстрагировали этилацетатом (20 мл). Объединенные органические фазы высушили над безводным натрия сульфатом, и остаток, полученный путем упаривания, очистили посредством колоночной хроматографии с силикагелем (петролейный эфир : этилацетат = от 10:1 до 1:1), получив указанное в заголовке соединение (1,65 г, 4,88 ммоль, выход - 58,60%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (д., J=9,0 Гц, 1Н), 7,83 (д., J=2,9 Гц, 1Н), 7,37-7,29 (м., 5Н), 7,27-7,22 (м., 1Н), 6,86 (дд, J=9,1, 2,9 Гц, 1Н), 3,78-3,70 (м., 1Н), 3,70-3,61 (м., 2Н), 3,57-3,45 (м., 2Н), 3,27 (д., J=10,3 Гц, 1Н), 2,83-2,74 (м., 1Н), 2,69 (тд., J=7,4, 9,3 Гц, 1Н), 2,35 (тд., J=12,5, 9,0 Гц, 1Н), 1,98-1,82 (м., 5Н).
Этап 6:
5-(1-бензил-1,7-диазаспиро[4,4]нонан-7-ил)пиридин-2-амин

К раствору 1-бензил-7-(6-нитро-3-пиридил)-1,7-диазаспиро[4,4]нонана (2,60 г, 7,68 ммоль, 1,00 эквивалент) в этаноле (40,00 мл) добавили цинковую пыль (2,01 г, 30,73 ммоль, 4,00 эквивалента) и аммония хлорид (4,11 г, 76,83 ммоль, 10,00 эквивалентов). Смесь нагрели до 80°C и перемешивали в течение 3 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционную смесь охладили до 15°C, разбавили этанолом (30 мл) и затем профильтровали. Осадок на фильтре промыли этанолом (30 мл). Неочищенный продукт, полученный путем упаривания фильтрата, очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (560,00 мг, 1,72 ммоль, выход - 22,34%, степень чистоты - 94,5%), представляющее собой красное масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,52 (д., J=2,8 Гц, 1Н), 7,36-7,28 (м., 4Н), 7,21-7,14 (м., 1Н), 6,86 (дд, J=8,8, 3,0 Гц, 1Н), 6,51 (д., J=8,7 Гц, 1Н), 3,98 (ушир. с, 2Н), 3,74 (д., J=13,3 Гц, 1Н), 3,63 (д., J=13,2 Гц, 1Н), 3,47-3,38 (м., 2Н), 3,24 (кв., J=8,6 Гц, 1Н), 3,04 (д., J=9,4 Гц, 1H), 2,77-2,59 (м., 2Н), 2,37 (с., 1Н), 2,27 (тд., J=12,5, 8,6 Гц, 1Н), 2,02-1,75 (м., 6Н).
Этап 7:
N-(5-(1-бензил-1,7-диазаспиро[4,4]нонан-7-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
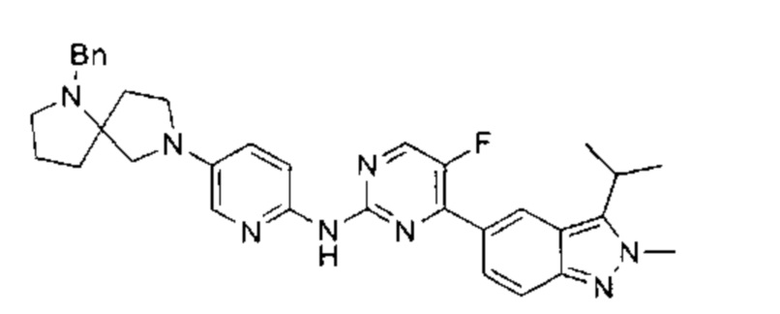
В атмосфере азота к раствору 5-(1-бензил-1,7-диазаспиро[4,4]нонан-7-ил)пиридин-2-амина (370 мг, 1,20 ммоль, 1,00 эквивалент) и 5-(2-хлор-5-фтор- пиримидин-4-ил)-3-изопропил-2-метил-2Н-индазола (438,84 мг, 1,44 ммоль, 1,20 эквивалента) в тетрагидрофуране (20,00 мл) добавили калия трет-бутоксид (403,84 мг, 3,60 ммоль, 3,00 эквивалента) и хлор[2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил][2-(2-аминоэтил)фенил]палладий(II) (88,63 мг, 120,00 ммоль, 0,10 эквивалента). Смесь нагрели до 80°C и перемешивали в течение 18 часов. ЖХ/МС показала, что осталось 9,9% исходного материала и образовалось 33,6% желаемого продукта. Реакционную смесь охладили до 15°C, разбавили водой (20 мл) и этилацетатом (20 мл) и профильтровали. После разделения фаз фильтрата водную фазу дважды подвергли экстракции этилацетатом (20 мл×2). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл) и упарили. Полученный остаток очистили посредством препаративной ТСХ (этилацетат: метанол = 10:1) и получили указанное в заголовке соединение (220,00 мг, 381,47 мкмоль, степень чистоты - 31,79%), представляющее собой коричневое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,69 (с., 1Н), 8,34 (д., J=4,0 Гц, 1Н), 8,22 (ушир. с., 1Н), 8,06 (д., J=9,3 Гц, 1Н), 7,89 (ушир. с., 1Н), 7,78-7,69 (м., 2Н), 7,40-7,29 (м., 4Н), 7,27-7,22 (м., 1Н), 7,00 (дд, J=2,9, 9,0 Гц, 1Н), 4,17 (с., 3Н), 3,74 (ушир. с., 1Н), 3,70-3,59 (м., 1Н), 3,56-3,49 (м., 3Н), 3,33 (кв., J=8,4 Гц, 1Н), 3,13 (д., J=9,2 Гц, 1Н), 2,75 (ушир. с., 1Н), 2,69 (ушир. с., 1Н), 2,31 (ушир. с., 1Н), 1,95-1,84 (м., 5Н), 1,60 (д., J=7,0 Гц, 6Н).
Этап 8:
N-(5-(1,7-диазаспиро[4.4]нонан-7-ил)пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
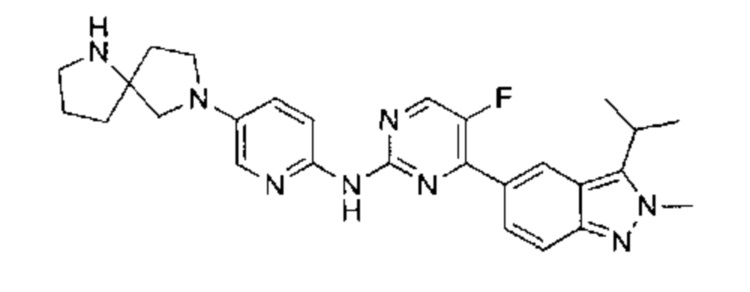
При 80°C N-(5-(1-бензил-1,7-диазаспиро[4,4]нонан-7-ил)-2-пиридил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин (220,00 мг, 381,47 мкмоль, 1,00 эквивалент), аммония формиат (481,12 мг, 7,63 ммоль, 20,00 эквивалента) и влажный Pd/C (220,00 мг) в смеси (22,00 мл) и метанола (22,00 мл) перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (30,00 мг, 80,56 мкмоль, выход - 21,12%, степень чистоты - 98%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,92 (с., 1Н), 8,78 (д., J=3,5 Гц, 1Н), 8,41 (д., J=9,2 Гц, 1Н), 8,00-7,81 (м., 2Н), 7,66 (д., J=2,5 Гц, 1Н), 7,55 (д., J=9,5 Гц, 1Н), 4,34 (с., 3Н), 3,93 (д., J=10,9 Гц, 1Н), 3,80-3,67 (м., 2Н), 3,65-3,47 (м., 4Н), 2,64-2,54 (м., 1Н), 2,53-2,43 (м., 1Н), 2,35-2,18 (м., 4Н), 1,67 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 487,4 (М+1).
Пример 17
5-фтор-N-(5-(гексагидропирроло[3,4-b][1,4]оксазин-6(2Н)-ил)пиридин-2-ил)-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

Этап 1:
трет-бутил-3-бром-4-(2-гидроксиэтокси)пирролидин-1-карбоксилат

К раствору трет-бутил-2,5-дигидропиррол-1-карбоксилата (8,00 г, 47,28 ммоль, 1,00 эквивалент) в этиленгликоле (30,00 мл) добавили несколькими порциями N-бромсукцинимид (9,26 г, 52,01 ммоль, 1,10 эквивалента). Смесь перемешивали при 15°C в течение 16 часов. ЖХ/МС и ТСХ показали, что реакция завершилась. К смеси добавили воду (100 мл). Полученную смесь перемешивали при 15°C в течение 0,5 часа и экстрагировали этилацетатом (100 мл×3). Объединенные органические слои высушили над безводным натрия сульфатом и упарили под вакуумом, получив указанное в заголовке соединение (14,00 г, неочищенный продукт), представляющее собой белое масло. 1Н ЯМР (400 МГц, CDCl3) δ 4,31 (ушир. с., 1Н), 4,17 (д., J=5,5 Гц, 1Н), 3,98-3,92 (м., 1Н), 3,87-3,82 (м., 1H), 3,77-3,72 (м., 2Н), 3,71-3,58 (м., 2Н), 3,57-3,39 (м., 1Н), 2,06-2,01 (м., 1Н), 1,49 (д., J=6,7 Гц, 9Н). ЖХ/МС (ESI) м/з: 264,0 (М+1-56).
Этап 2:
трет-бутил-3-бром-4-(2-(тозилокси)этокси)пирролидин-1-карбоксилат

При 0°C к раствору
трет-бутил-3-бром-4-(2-гидроксиэтокси)пирролидин-1-карбоксилата (14,00 г, 45,14 ммоль, 1,00 эквивалент), триэтиламина (6,85 г, 67,71 ммоль, 1,50 эквивалента) и 4-диметиламинопиридина (551,48 мг, 4,51 ммоль, 0,10 эквивалент) в толуоле (100,00 мл) добавили п-толуолсульфонилхлорид (11,19 г, 58,68 ммоль, 1,30 эквивалента). Смесь перемешивали при 10°C в течение 16 часов. ЖХ/МС и ТСХ показали, что реакция завершилась. Смесь разбавили водой (100 мл), и водный слой подвергли экстракции этилацетат (100 мл). Объединенные органические слои промыли насыщенным водным раствором натрия хлорида (30 мл×2), высушили над натрия сульфатом и упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = 4:1), получив указанное в заголовке соединение (11,10 г, 23,90 ммоль, выход - 52,95%), представляющее собой белое масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,81 (д., J=8,3 Гц, 2Н), 7,38 (д., J=8,0 Гц, 2Н), 4,19-4,11 (м., 3Н), 4,10-4,02 (м., 1Н), 3,90-3,66 (м., 5Н), 3,45-3,29 (м., 1Н), 2,48 (с, 3Н), 1,49 (с., 9Н). ЖХ/МС (ESI) м/з: 408,0 (М+1-56).
Этап 3:
трет-бутил-4-бензилгексагидропиролло[3,4-b][1,4]оксазин-6(2Н)-карбоксилат
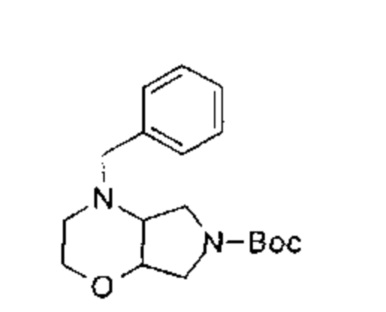
При 140°C трет-бутил-3-бром-4-(2-(тозилокси)этокси)пирролидин-1-карбоксилат (11,10 г, 23,90 ммоль, 1,00 эквивалент) и бензиламин (7,68 г, 71,70 ммоль, 3,00 эквивалента) в ксилене (150,00 мл) перемешивали в течение 16 часов. ЖХ/МС и ТСХ показали, что реакция завершилась. Смесь упарили под вакуумом и получили сухой остаток. Остаток разбавили этилацетатом (100 мл) и водой (100 мл). Водный слой экстрагировали этилацетатом (100 мл×2). Объединенные органические слои высушили над натрия сульфатом и упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством колоночной хроматографии (ПЭ : ЭА = от 20:1 до 4:1), получив указанное в заголовке соединение (5,80 г, 14,21 ммоль, выход - 59,45%, степень чистоты - 78%), представляющее собой желтое масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,81 (д., J=8,3 Гц, 2Н), 7,38 (д., J=8,0 Гц, 2Н), 4,19-4,11 (м., 3Н), 4,10-4,02 (м., 1Н), 3,90-3,66 (м., 5Н), 3,45-3,29 (м., 1Н), 2,48 (с, 3Н), 1,49 (с, 9Н). ЖХ/МС (ESI) м/з: 319,1 (М+1).
Этап 4: 4-бензилгексагидропирроло[3,4-b][1,4]оксазин

При 0°C к раствору
трет-бутил-4-бензилгексагидропирроло[3,4-b][1,4]оксазин-6(2Н)-карбоксилата (2,90 г, 9,11 ммоль, 1,00 эквивалент) в этилацетате (30,00 мл) добавили соляную кислоту/этилацетат (30,00 мл). Смесь перемешивали при 10°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь упарили под вакуумом и получили указанное в заголовке соединение (2,60 г, 8,93 ммоль, выход - 98,00%, гидрохлорид), представляющее собой твердое белое вещество. ЖХ/МС (ESI) м/з: 219,3 (М+1).
Этап 5:
4-бензил-6-(6-нитропиридин-3-ил)октагидропиролло [3,4-b][1,4]-оксазин
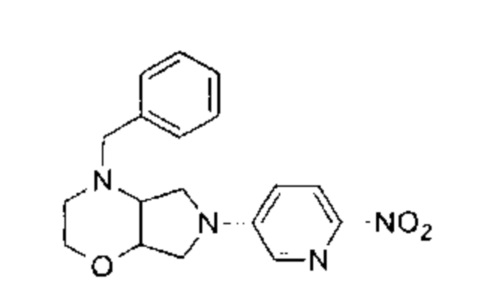
При 80°C смесь 4-бензилгексагидропиролло[3,4-b][1,4]оксазина (2,60 г, 8,93 ммоль, 1,00 эквивалент, 2HCl), 5-бром-2-нитропиридина (1,81 г, 8,93 ммоль, 1,00 эквивалент) и триэтиламина (3,61 г, 35,72 ммоль, 4,00 эквивалента) в N,N-диметилформамиде (30,00 мл) перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь разбавили этилацетат (40 мл) и водой (120 мл) и подвергли экстракции этилацетатом (40 мл×3). Объединенные органические слои высушили над натрия сульфатом и упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 3:1 до 1:1), получив указанное в заголовке соединение (2,00 г, 5,88 ммоль, выход - 65,80%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 341,1 (М+1).
Этап 6:
5-(4-бензилгексагидропиролло[3,4-b][1,4]оксазин-6(2Н)-ил)пиридин-2-амин
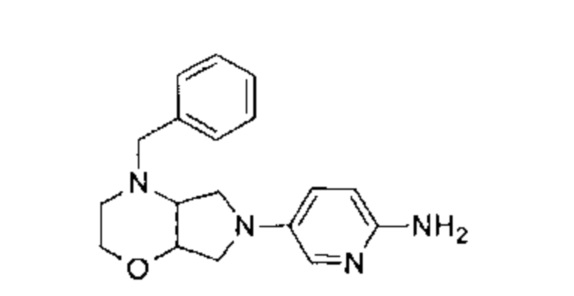
При 80°C смесь
4-бензил-6-(6-нитропиридин-3-ил)октагидропиролло[3,4-b][1,4]-оксазина (2,00 г, 5,88 ммоль, 1,00 эквивалент), железных опилок (1,64 г, 29,40 ммоль, 5,00 эквивалента) и аммония хлорида (3,15 г, 58,80 ммоль, 10,00 эквивалентов) в этаноле (50,00 мл) и воде (10,00 мл) перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством препаративной ВЭЖХ (трифторуксусная кислота) и получили указанное в заголовке соединение (1,00 г, 3,13 ммоль, выход - 53,15%, степень чистоты - 97%), представляющее собой коричневое масло. ЖХ/МС (ESI) м/з: 311,2 (М+1).
Этап 7:
N-(5-(4-бензилгексагидропиролло[3,4-b][1,4]оксазин-6(2Н)-ил)-пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин

В атмосфере азота раствор 5-(4-бензилгексагидропиролло[3,4-b][1,4]оксазин-6(2Н)-ил)пиридин-2-амина (300,00 мг, 966,53 мкмоль, 1,00 эквивалент), 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метилиндазола (промежуточный продукт С) (294,55 мг, 966,53 мкмоль, 1,00 эквивалент),
хлор-[2-(дициклогексилфосфино)-2'-4'-6'-триизопропил-1,1'-бифенил][2-(2-аминоэтил)фенил]палладия(II) (71,40 мг, 96,65 мкмоль, 0,10 эквивалент) и калия трет-бутоксида (325,36 мг, 2,90 ммоль, 3,00 эквивалента) в тетрагидрофуране (30,00 мл) перемешивали при 80°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. К смеси последовательно добавили воду (30 мл) и этилацетат (30 мл). Полученную смесь перемешивали при 20°C в течение 15 минут, отфильтровали и подвергли разделению фаз. Водный слой экстрагировали этилацетатом (20 мл×2). Объединенные органические слои высушили над безводным натрия сульфатом и упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством препаративной ТСХ (дихлорметан : метанол = 10:1), получив указанное в заголовке соединение (190,00 мг, 265,95 мкмоль, выход - 27,52%, степень чистоты - 81%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 579,4 (М+1).
Этап 8:
5-фтор-N-(5-(гексагидропирроло[3,4-b][1,4]оксазин-6(2Н)-ил)пиридин-2-ил)-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
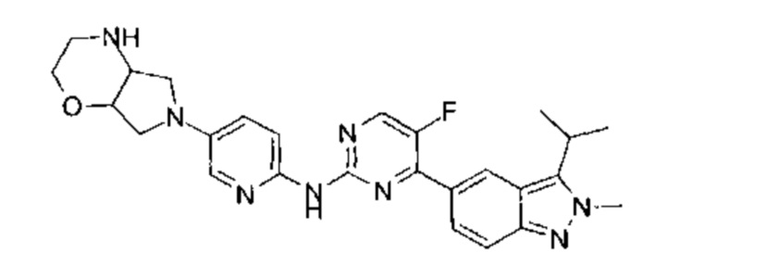
Раствор смеси N-(5-(4-бензилгексагидропиролло[3,4-b][1,4]оксазин-6(2Н)-ил) пиридин-2-ил)-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амина (160,00 мг, 276,49 мкмоль, 1,00 эквивалент), формиата аммония (348,38 мг, 5,53 ммоль, 20,00 эквивалентов) и влажного Pd/C (15,00 мг) в тетрагидрофуране (16,00 мл) и метаноле (16,00 мл) перемешивали при 80°C в течение 16 часов. ЖХ/МС показала, что образовался желаемый продукт. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (20,00 мг, 40,94 мкмоль, выход - 14,81%, степень чистоты - 100%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,89 (с, 1Н), 8,75 (д., J=3,6 Гц, 1Н), 8,33 (д., J=9,0 Гц, 1Н), 7,90 (д., J=1,1 Гц, 1Н), 7,81 (д., J=9,0 Гц, 1Н), 7,62 (с., 1Н), 7,54 (д., J=9,4 Гц, 1Н), 4,59 (ушир. с., 1H), 4,30 (с., 3Н), 4,19 (ушир. с., 1Н), 4,11 (д., J=11,9 Гц, 1Н), 3,97-3,79 (м., 3Н), 3,79-3,67 (м., 2Н), 3,58 (д., J=11,0 Гц, 1Н), 3,51 (ушир. с., 1Н), 3,28 (д., J=13,1 Гц, 1Н), 1,65 (д., J=6,9 Гц, 6Н). ЖХ/МС (ESI) м/з: 489,4 (М+1).
Пример 18
1-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-2-он

Этап 1:
трет-бутилN-(5-бром-2-пиридил)-N-трет-бутоксикарбонилкарбамат
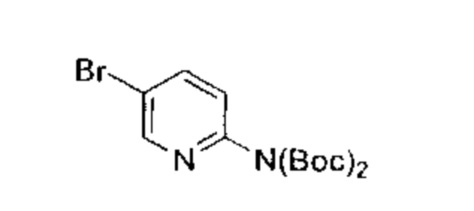
При 0°C к раствору 5-бромпиридин-2-амина (2,00 г, 11,56 ммоль, 1,00 эквивалент), N,N-диизопропилэтиленамина (4,48 г, 34,68 ммоль, 6,05 мл, 3,00 эквивалента) и диметиламинопиридина (282,46 мг, 2,31 ммоль, 0,20 эквивалент) в дихлорметане (30,00 мл) добавили ди-трет-бутилкарбонат (7,57 г, 34,68 ммоль, 7,97 мл, 3,00 эквивалента). Реакционную смесь перемешивали при 30°C в течение 16 часов. ЖХ/МС и ТСХ показали, что реакция завершилась. Смесь упарили под вакуумом и получили неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (этилацетат : петролейный эфир = 50:1) и получили указанное в заголовке соединение (2,00 г, 5,36 ммоль, выход - 46,35%), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,55 (д., J=2,3 Гц, 1Н), 7,86 (дд, J=2,5, 8,4 Гц, 1Н), 7,20 (д., J=8,5 Гц, 1Н), 1,47 (с., 18Н). ЖХ/МС (ESI) м/з: 373,1 (М+1).
Этап 2: бензил-3-оксопиперазин-1-карбоксилат

При 0°C к раствору смеси натрия карбоната (24,77 г, 233,73 ммоль, 3,00 эквивалента) и пиперазин-2-она (7,80 г, 77,91 ммоль, 1,00 эквивалент) в этилацетате (70,00 мл) и воды (70,00 мл) добавили бензилхлорформиат (16,79 г, 93,49 ммоль, 13,99 мл, степень чистоты - 95%, 1,20 эквивалента). Реакционную смесь перемешивали при 30°C в течение 16 часов. ТСХ показала, что реакция завершилась. Смесь подвергли экстракции этилацетатом (80 мл×3). Объединенные органические слои промыли насыщенным водным раствором натрия хлорида (80 мл×3), высушили над натрия сульфатом и упарили под вакуумом, получив неочищенный продукт. Неочищенный продукт очистили в шаровой мельнице в смеси петролейный эфир : этилацетат (20:1, 80 мл). Полученную смесь перемешивали при 30°C в течение 15 минут и профильтровали. Твердое вещество высушили под вакуумом и получили указанное в заголовке соединение (15,50 г, 66,17 ммоль, выход - 84,93%), представляющее собой белое твердое вещество.
Этап 3:
бензил-4-[6-[бис(трет-бутоксикарбонил)амино]-3-пиридил]-3-оксопиперазин-1-карбоксилат
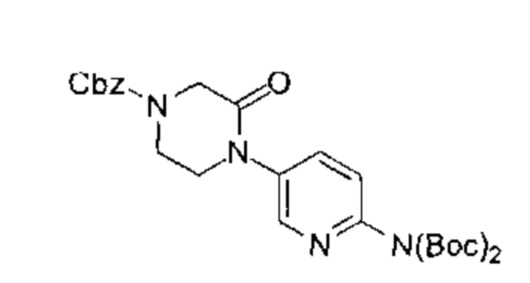
В атмосфере азота смесь бензил-3-оксопиперазин-1-карбоксилата (4,00 г, 17,08 ммоль, 1,00 эквивалент), трет-бутил-N-бром-2-пиридил)-N-трет-бутоксикарбонилкарбамата (6,37 г, 17,08 ммоль, 1,00 эквивалент),
N,N-диметилэтан-1,2-диамина (602,09 мг, 6,83 ммоль, 734,26 мкл, 0,40 эквивалент), калия карбонат (7,08 г, 51,24 ммоль, 3,00 эквивалента) и CuI (650,42 мг, 3,42 ммоль, 0,20 эквивалент) в диоксане (80,00 мл) перемешивали при 100°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт, который затем очистили посредством препаративной ТСХ (петролейный эфир : этилацетат = 1:1) с получением смеси указанного в заголовке соединения и моно-Вос-продукта (3,20 г, неочищенный продукт). ЖХ/МС (ESI) м/з: 527,2 (М+1).
Этап 4:
бензил-4-(6-аминопиридин-3-ил)-3-оксопиперазин-1-карбоксилат
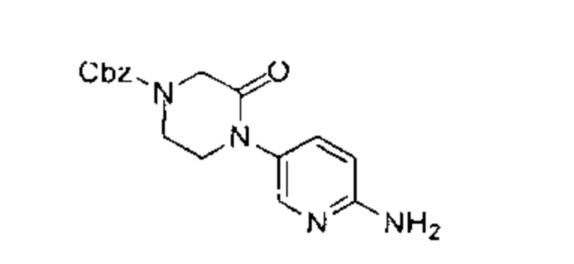
К раствору
бензил-4-[6-[бис(трет-бутоксикарбонил)амино]-3-пиридил]-3-оксопиперазин-1-карбоксилата (3,20 г, 6,08 ммоль, 1,00 эквивалент) в дихлорметане (20,00 мл) добавили трифторуксусную кислоту (10,00 мл) при 0°C. Реакционную смесь перемешивали при 30°C в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь упарили под вакуумом, получив неочищенный продукт, который разбавили дихлорметаном. К смеси добавили калия карбонат (10 г). Полученную смесь перемешивали при 30°C в течение 15 минут и профильтровали. Фильтрат упарили под вакуумом и получили указанное в заголовке соединение (2,00 г, неочищенный продукт), представляющее собой коричневое масло. 1Н ЯМР (400 МГц, метанол-d4) δ 7,87 (д., J=2,5 Гц, 1H), 7,42-7,36 (м., 4Н), 7,25-7,13 (м., 2Н), 6,63 (д., J=8,8 Гц, 1Н), 5,21 (с., 2Н), 4.28 (ушир. с., 2Н), 3,88 (ушир. с., 2Н), 3,73 (т., J=5,3 Гц, 2Н). ЖХ/МС (ESI) м/з: 327,1 (М+1).
Этап 5:
бензил-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)-3-оксопиперазин-1-карбоксилат

В атмосфере азота смесь 5-(2-хлор-5-фтор-пиримидин-4-ил)-3-изопропил-2-метинидазола (промежуточный продукт С) (1,60 г, 5,25 ммоль, 1,00 эквивалент), бензил-4-(6-аминопиридин-3-ил)-3-оксопиперазин-1-карбоксилата (2,06 г, 6,30 ммоль, 1,20 эквивалента), цезия карбоната (5,13 г, 15,75 ммоль, 3,00 эквивалента), XPhos (500,58 мг, 1,05 ммоль, 0,20 эквивалент) и Pd(OAc)2 (117,87 мг, 525,00 мкмоль, 0,10 эквивалент) в диоксане (50,00 мл) перемешивали при 110°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (дихлорметан : метанол = 20:1), получив указанное в заголовке соединение (2,40 г, 3,59 ммоль, выход - 68,42%, степень чистоты - 89%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,65 (с., 1H), 8,70 (с., 1Н), 8,57 (д., J=9,0 Гц, 1H), 8,45 (д., J=3,8 Гц, 1Н), 8,33 (д., J=2,5 Гц, 1Н), 8,05 (д., J=9,2 Гц, 1Н), 7,75 (д., J=9,2 Гц, 1Н), 7,67 (дд, J=8,9, 2,6 Гц, 1Н), 7,47-7,31 (м., 5Н), 5,22 (с., 2Н), 4,37 (с., 2Н), 4,20 (с., 3Н), 3,93-3,86 (м., 2Н), 3,82-3,72 (м., 2Н), 3,58-3,46 (м., 1Н), 1,61 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 595,2 (М+1).
Этап 6:
1-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-2-он

В атмосфере водорода смесь бензил-4-(6-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-
-2-ил)амино)пиридин-3-ил)-3-оксопиперазин-1-карбоксилата (2,40 г, 3,59 ммоль, 1,00 эквивалент) и влажного Pd/C (400,00 мг, чистота - 10%) в метаноле (20,00 мл) и тетрагидрофуран (20,00 мл) перемешивали при 30°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, и фильтрат упарили под вакуумом, получив неочищенный продукт, который разбавили дихрометаном (20 мл). К смеси по капле добавили соляную кислоту/этилацетат (5 мл, 4 М). Затем полученную смесь упарили под вакуумом и получили неочищенный продукт, который затем очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (1,25 г, 2,32 ммоль, выход - 64,62%, степень чистоты - 99%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8,92 (с., 1Н), 8,83 (д., J=3,6 Гц, 1Н), 8,63 (д., J=2,1 Гц, 1Н), 8,45 (дд, J=2,5, 9,4 Гц, 1Н), 8,36 (д., J=9,3 Гц, 1Н), 7,86-7,78 (м., 1H), 7,67 (д., J=9,5 Гц, 1Н), 4,31 (с., 3Н), 4,20-4,10 (м., 4Н), 3,83-3,68 (м., 3Н), 1,66 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 461,2 (М+1).
Пример 19
N-[5-(3,8-диазабицикло[3.2.1]октан-3-ил)-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
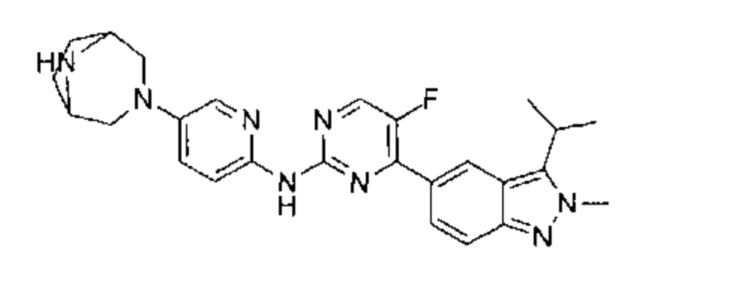
Этап 1:
трет-бутил-3-(6-нитро-3-пиридил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат

К раствору трет-бутил-3,8-диазабицикло[3,2,1]октан-8-карбоксилата (1,00 г, 4,71 ммоль, 1,00 эквивалент) в диметилсульфоксиде (20,00 мл) добавили триэтиламин (1,43 г, 14,13 ммоль, 3,00 эквивалента) и 5-бром-2-нитропиридин (1,15 г, 5,65 ммоль, 1,20 эквивалента). Смесь перемешивали при 100°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. К смеси добавили 40 мл воды, чтобы смесь охладилась до 20°C. Водную фазу подвергли экстракции дихлорметаном (50 мл×2), промыли насыщенным солевым раствором (50 мл×1), высушили над безводным натрия сульфатом, профильтровали и упарили. Неочищенный продукт очистили в шариковой мельнице с метанолом (2 мл×3) и получили указанное в заголовке соединение (639,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (д., J=9,2 Гц, 1Н), 8,09 (д., J=3,0 Гц, 1Н), 7,15 (дд, J=9,2 Гц, 3,0 Гц, 1Н), 4,47 (ушир. с., 2Н), 3,58 (д., J=10,0 Гц, 2Н), 3,27 (ушир. с., 2Н), 2,12-1,99 (м., 2Н), 1,86-1,76 (м., 2Н), 1,49 (с., 9Н). ЖХ/МС (ESI) м/з: 335,2 (М+1).
Этап 2:
трет-бутил-3-(6-амино-3-пиридил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат
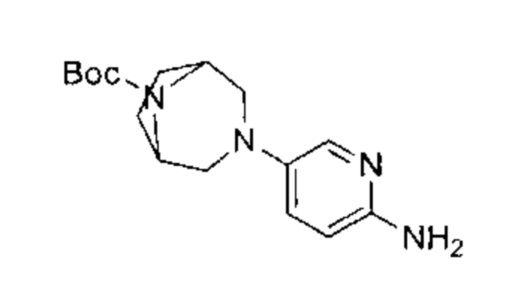
В атмосфере водорода (15 фунт/кв.дюйм) к раствору трет-бутил-3-(6-нитро-3-пиридил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилата (339,00 мг, 1,01 ммоль, 1,00 эквивалент) в метаноле (11,00 мл) добавили палладиевый катализатор на углеродном носителе (50,00 мг). Реакционную смесь перемешивали при 20°C в течение 18 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали и упарили, получив указанное в заголовке соединение (287,00 мг, неочищенный продукт), представляющее собой фиолетовое твердое вещество. ЖХ/МС (ESI) м/з: 305,2 (М+1).
Этап 3:
трет-бутил-3-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридил]-3,8-диазабицикло[3,2,1]октан-8-карбоксилат
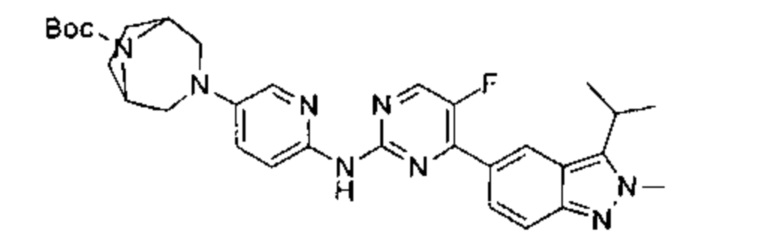
К раствору трет-бутил-3-(6-амино-3-пиридил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилата (140,00 мг, 459,94 мкмоль, 1,00 эквивалент) в диоксане добавили 5-(2-хлор-5-фтор-пиримидин-4-ил)-3-изопропил-2-метил-2Н-индазола (промежуточный продукт С) (249,50 мг, 818,69 мкмоль, 1,78 эквивалента), Xantphos (79,84 мг, 137,98 мкмоль, 0,30 эквивалент), цезия карбонат (446,58 мг, 1,37 ммоль, 2,98 эквивалента) и Pd2(dba)3 (63,18 мг, 68,99 мкмоль, 0,15 эквивалент) в атмосфере азота. Смесь перемешивали при 110°C в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию, но желаемый продукт не образовался. ТСХ показала, что исходный материал полностью вступил в реакцию и было получено два новых соединения. Реакционную смесь профильтровали, и осадок на фильтре растворили в 10 мл воды. Водную фазу экстрагировали дихлорметаном (10 мл×2). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над безводным натрия сульфатом, профильтровали и упарили до указанного в заголовке соединения (25,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 573,3 (М+1).
Этап 4:
N-[5-(3,8-диазабицикло[3.2.1]октан-3-ил)-2-пиридил]-5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-амин
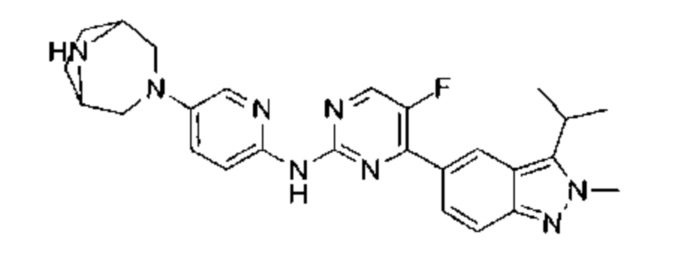
К раствору
трет-бутил-3-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]ам ино]-3-пиридил]-3,8-диазабицикло[3,2,1]октан-8-карбоксилат (25,00 мг, 43,66 мкмоль, 1,00 эквивалент) в дихлорметане (2 мл) добавили трифторуксусную кислоту (765,00 мг, 6,71 ммоль, 153,67 эквивалента). Смесь перемешивали при 20°C в течение 0,5 часа. ЖХ/МС показала, что реакция завершилась. Реакционную смесь упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (11,80 мг, 22,82 мкмоль, выход - 52,27%, степень чистоты - 91,39%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,89 (с., 1Н), 8,77 (д., J=3,5 Гц, 1H), 8,35 (д, J=9,2 Гц, 1Н), 8,25 (дд, J=9,5, 2,1 Гц, 1Н), 7,87 (д., J=1,9 Гц, 1Н), 7,81 (д., J=9,0 Гц, 1Н), 7,55 (д., J=9,5 Гц, 1Н), 4,30 (с., 5Н), 3,84-3,67 (м., 3Н), 3,36-3,32 (м., 2Н), 2,20 (с., 4Н), 1,64 (д., J=6,9 Гц, 6Н). ЖХ/МС (ESI) м/з: 473,3 (М+1).
Пример 20
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(4-метил-5-пиперазин-1-ил-2-пиридил)пидимидин-2-амин
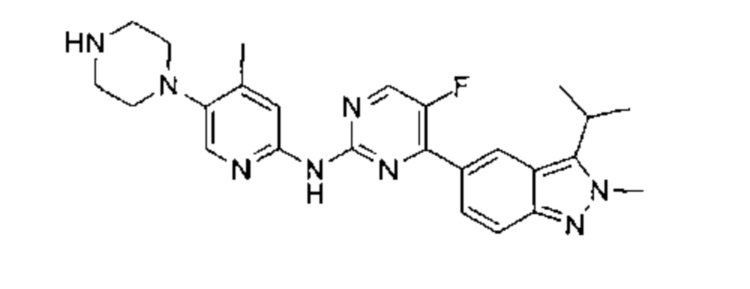
Этап 1:
трет-бутил-4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-4-метил-3-пиридил]пиперазин-1-карбоксилат
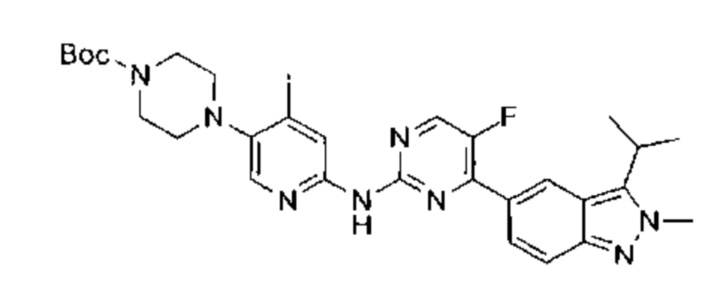
К раствору трет-бутил-4-(6-амино-4-метил-3-пиридил)пиперазин-1-карбоксилата (200,00 мг, 684,04 мкмоль, 1,00 эквивалент) в диоксане (5,00 мл) в атмосфере азота добавили 5-(2-хлор-5-фтор-пиримидин-4-ил)-3-изопропил-2-метил-2Н-индазол (промежуточный продукт С) (250,15 мг, 820,85 мкмоль, 1,20 эквивалента), Pd2(dba)3 (62,64 мг, 68,40 мкмоль, 0,10 эквивалент), Xantphos (79,16 мг, 136,81 мкмоль, 0,20 эквивалент) и цезия карбонат (445,75 мг, 1,37 ммоль, 2,00 эквивалента). Смесь перемешивали при 110°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь упарили, неочищенный продукт очистили посредством препаративной TLC (этилацетат : петролейный эфир = 2:1) и получили указанное в заголовке соединение (150,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 561,4 (М+1).
Этап 2:
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(4-метил-5-пиперазин-1-ил-2-пиридил)пидимидин-2-амин

К раствору
трет-бутил-4-[6-[[5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-4-метил-3-пиридил]пиперазин-1-карбоксилата (150,00 мг, 267,54 мкмоль, 1,00 эквивалент) в дихлорметане (4 мл) добавили трифторуксусную кислоту (1,53 г, 13,42 ммоль, 50,16 эквивалента) при 20°C. Реакционную смесь перемешивали в течение 0,5 часа. ЖХ/МС показала, что реакция завершилась. Смесь упарили, получив неочищенный продукт, который затем очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (111,60 мг, 224,54 мкмоль, выход - 83,93%, степень чистоты - 100%, гидрохлорид). 1Н ЯМР (400 МГц, метанол-d4) δ 8,93 (с., 1Н), 8,83 (d, J=3,51 Гц, 1Н), 8,32-8,45 (м., 3Н), 7,85 (д., J=9,03 Гц, 1Н), 7,62 (д., J=9,16 Гц, 1Н), 4,33 (с., 3Н), 3,74 (д., J=6,99 Гц, 1Н), 3,57 (д., J=12,67 Гц, 2Н), 3,10-3,27 (м., 3Н), 2,20 (д., J=13,68 Гц, 2Н), 1,96-2,10 (м., 2Н), 1,66 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 446,2 (М+1).
Подход В
Получение
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиридин-2-амина
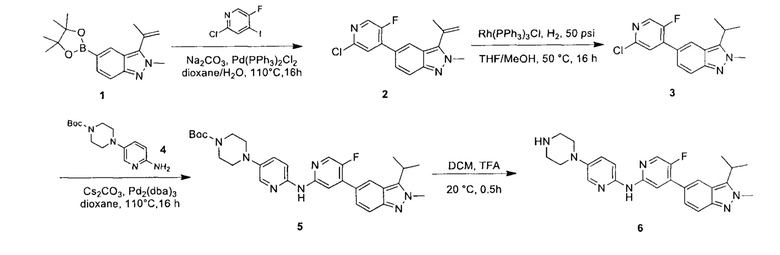
Пример 21
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиридин-2-амин

Этап 1:
5-(2-хлор-5-фторпиридин-4-ил)-2-метил-3-изопропенил-2Н-индазол
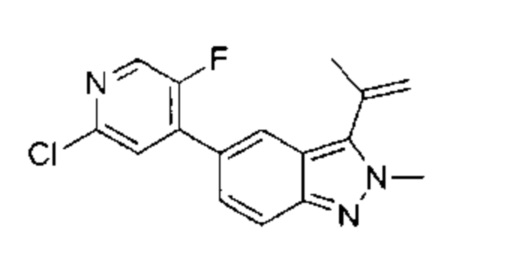
В атмосфере азота к раствору 2-метил-3-изопропенил-5-борат-2Н-индазола (500,00 мг, 1,68 ммоль, 1,00 эквивалент) в диоксане (10 мл) и воды (2 мл) добавили 2-хлор-5-фтор-4-йодпиридин (518,98 мг, 2,02 ммоль, 1,20 эквивалента), K2CO3 (696,58 мг, 5,04 ммоль, 3,00 эквивалента) и Pd(dppf)Cl2 (122,93 мг, 168,00 мкмоль, 0,10 эквивалент). Смесь перемешивали при 110°C в течение 3 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C и добавили в нее воду (10 мл). Провели экстракцию этилацетатом (15 мл×3). Органические фазы объединили и промыли солевым раствором (15 мл×2), высушили над безводным натрия сульфатом и профильтровали. Остаток, полученный путем упаривания фильтрата, очистили посредством колоночной хроматографии с силикагелем (петролейный эфир : этилацетат = от 30:1 до 5:1) и получили желаемое вещество (500 мг, 1,66 ммоль, выход - 98,63%), представляющее собой желтое масло. 1Н ЯМР (400 МГц, CDCl3) δ 8,32 (д., J=2,1 Гц, 1H), 7,89 (с., 1Н), 7,77 (дд, J=9,0, 0,8 Гц, 1H), 7,52-7,47 (м., 2Н), 5,69 (т., J=1,5 Гц, 1H), 5,35 (с., 1Н), 4,22 (с., 3Н), 2,30 (д., J=1,0 Гц, 3Н). ЖХ/МС (ESI) м/з: 302,0 (М+1).
Этап 2:
5-(2-хлор-5-фторпиридин-4-ил)-3-изопропил-2-метил-2Н-индазол

К смеси 5-(2-хлор-5-фторпиридин-4-ил)-2-метил-3-изопропенил-2Н-индазола (500 мг, 1,66 ммоль, 1,00 эквивалент) в метаноле (5 мл) и тетрагидрофуране (5 мл) добавили Rh(PPh3)3Cl (153,59 мг, 166,00 мкмоль, 0,1 эквивалента). Через систему пропустили водород под давлением 50 фунт/кв.дюйм. Смесь перемешивали при 50°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C и упарили, получив осадок. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 5:1) и получили указанное в заголовке соединение (460,00 мг, 1,51 ммоль, выход - 91,23%), представляющее собой белое масло. 1Н ЯМР (400 МГц, CDCl3) δ 8,33 (д., J=2,3 Гц, 1Н), 8,05 (с., 1Н), 7,75 (дд, J=9,0, 0,8 Гц, 1Н), 7,50 (д., J=5,6 Гц, 1Н), 7,45 (тд., J=9,0, 1,7 Гц, 1H), 4,20 (с., 3Н), 3,55-3,48 (м., 1Н), 1,58 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 304,0 (М+1).
Этап 3:
трет-бутил-4-(5-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)пиридин-2-ил)амино)пиридин-2-ил)пиперазин-1-карбоксилат
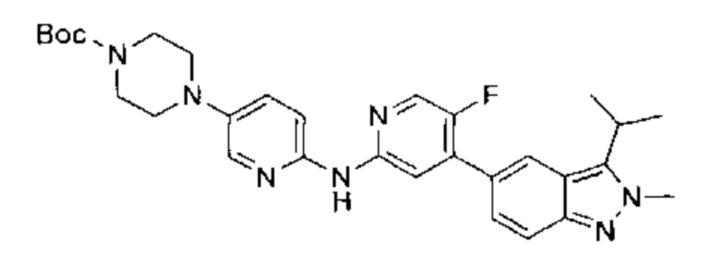
В атмосфере азота к раствору 5-(2-хлор-5-фторпиридин-4-ил)-3-изопропил-2-метил-2Н-индазола (200,00 мг, 658,41 мкмоль, 1,00 эквивалент) и
трет-бутил-4-(6-амино-3-пиридин)пиперазин-1-карбоксилата (274,90 мг, 987,62 мкмоль, 1,50 эквивалента) в диоксане (8 мл) добавили Cs2CO3 (643,57 мг, 1,98 ммоль, 3,00 эквивалента), Xantphos (76,19 мг, 131,68 мкмоль, 0,20 эквивалент), и Pd2(dba)3 (60,29 мг, 65,84 мкмоль, 0,10 эквивалент). Смесь перемешивали при 110°C в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Смесь охладили до 20°C и профильтровали. Остаток на фильтре промыли этилацетатом. Фильтрат упарили, получив указанное в заголовке соединение (350,00 мг, неочищенный продукт), представляющее собой черное масло, который использовали в следующем этапе без дальнейшей очистки. ЖХ/МС (ESI) м/з: 546,4 (М+1).
Этап 4:
5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиридин-2-амин

К раствору трет-бутил-4-(5-((5-фтор-4-(3-изопропил-2-метил-2Н-индазол-5-ил) пиридин-2-ил)амино)пиридин-2-ил)пиперазин-1-карбоксилата (350,00 мг, 641,44 мкмоль, 1,00 эквивалент) в дихлорметане (5 мл) добавили трифторуксусную кислоту (2,0 мл). Смесь перемешивали при 20°C в течение 0,5 часа. ЖХ/МС показала, что реакция прошла полностью. Смесь упарили под пониженным давлением, чтобы удалить дихлорметан и трифторуксусную кислоту и получить остаток. Остаток очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (250,00 мг, 558,27 мкмоль, выход - 87,03%, чистота - 99,49%). 1H ЯМР (400 МГц, метанол-d4) δ 8,47 (д., J=2,6 Гц, 1Н), 8,39 (с., 1H), 8,21 (дд, J=9,7, 2,9 Гц, 1Н), 7,89 (д., J=2,8 Гц, 1Н), 7,87-7,80 (м., 2Н), 7,44 (д., J=9,5 Гц, 1Н), 7,40 (д., J=5,5 Гц, 1Н), 4,31 (с., 3Н), 3,76-3,69 (м., 1Н), 3,56-3,51 (м., 4Н), 3,50-3,45 (м., 4Н), 1,64 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 304,0 (М+1).
Подход С
Общий способ получения промежуточного продукта D показан ниже.
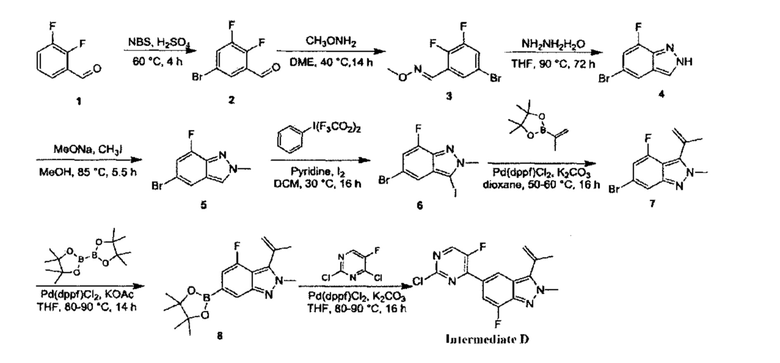
Этап 1: 5-бром-2,3-дифторбензальдегид
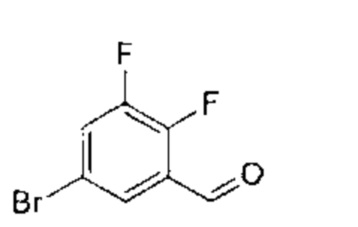
2,3-дифторбензальдегид (3,00 г, 21,22 ммоль, 1,00 эквивалент) растворили в соляной кислоте (18,4 моль/л, 10,20 мл, 8,89 эквивалента) и нагревали до 60°C в течение 40 минут. После этого добавили в течение 20 минут тремя порциями
1-бромпирролидин-2,5-дион (4,51 г, 25,33 ммоль, 1,20 эквивалента). Смесь нагревали в течение 3 часов в атмосфере азота. ТСХ и ВЭЖХ показали, что реакция завершилась. Реакционную смесь налили в ледяную воду, подвергли экстракции петролейным эфиром (30 мл×2), промыли водой (30 мл×2) и насыщенным солевым раствором (30 мл×2) и затем упарили под пониженным давлением. Упаренный остаток очистили посредством колоночной хроматографии (петролейный эфир) и получили указанное в заголовке соединение (2,10 г, 9,50 ммоль, выход - 45,00%), представляющее собой желтую жидкость. 1Н ЯМР (400 МГц, CDCl3) δ 10,29 (с, 1Н), 7,77 (ушир. с., 1H), 7,65-7,54 (м., 1Н).
Этап 2:
(транс)-5-бром-2,3-дифторбензальдегидоксометилоксим

Смесь 5-бром-2,3-дифторбензальдегида (1,50 г, 6,79 ммоль, 1,00 эквивалент), О-метилгидроксиламина (680,52 мг, 8,15 ммоль, 1,20 эквивалента) и калия карбоната (1,13 г, 8,15 ммоль, 1,2 эквивалента) в диметиловом эфире (20,00 мл) нагрели до 40°C и перемешивали в течение 14 часов. ТСХ показала, что около 1% исходного материала не вступило в реакцию. Реакционную смесь профильтровали, и фильтрат упарили под пониженным давлением и получили указанное в заголовке соединение (1,00 г, неочищенный продукт), которое использовали в следующем этапе без дальнейшей очистки. 1Н ЯМР (400 МГц, CDCl3) δ 8,22 (с, 1Н), 7,77-7,75 (м., 1Н), 7,35-7,27 (м., 1Н), 4,04 (с., 3Н).
Этап 3: 5-бром-7-фтор-2Н-индазол
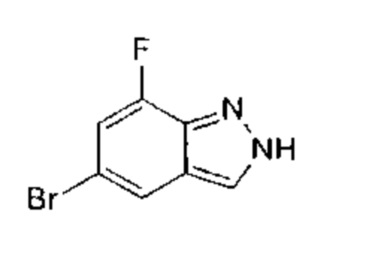
Раствор (транс)-5-бром-2,3-дифторбензальдегидоксометилоксима (4,90 г, 19,60 ммоль, 1,00 эквивалент) в тетрагидрофуране (35,00 мл) и гидразин гидрате (20,60 г, 411,51 ммоль, 21,00 эквивалента, концентрация - 85%) нагрели до 90°C и перемешивали в течение 72 часов. ЖХ/МС показала, что 20% исходного материала не вступило в реакцию. Органический растворитель выпарили, и полученную смесь разбавили этилацетатом (20 мл), промыли водой (20 мл×2) и затем упарили под пониженным давлением до сухого остатка. Остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 60:1 до 40:1) и получили указанное в заголовке соединение (3,25 г, 12,09 ммоль, выход - 61,69%, степень чистоты - 80%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 215,0 (М+1).
Этап 4: 5-бром-7-фтор-2-метил-2Н-индазол

В атмосфере азота к раствору 5-бром-7-фтор-2Н-индазола (1,10 г, 5,12 ммоль, 1,00 эквивалент) и натрия метоксида (552,71 мг, 10,23 ммоль, 2,00 эквивалента) в метаноле (20,00 мл) добавляли иодметан (1,09 г, 7,67 ммоль, 1,50 эквивалента) по капле при 30°C в течение 30 минут. Смесь нагревали до 85°C в течение 30 минут и перемешивали в течение 5 часов. ЖХ/МС показала, что 3% исходного материала не вступило в реакцию. Реакционную смесь охладили до 30°C, упарили под пониженным давлением, разбавили 3% водным раствором натрия бикарбоната и затем подвергли экстракции этилацетатом (20 мл×2). Органическую фазу упарили под пониженным давлением, остаток очистили посредством колоночной хроматографии (петролейный эфир : этилацетат = от 20:1 до 5:1) и получили указанное в заголовке соединение (360, мг, 1,57 ммоль, выход - 30,66%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 229,0 (М+1).
Этап 5: 5-бром-7-фтор-3-йод-2-метил-2Н-индазол

При 30°C к раствору 5-бром-7-фтор-2-метил-2Н-индазола (500,00 мг, 2,18 ммоль, 1,00 эквивалент) в дихлорметане (5,00 мл) добавили пиридин (259,00 мл, 3,27 ммоль, 1,50 эквивалента) и бис(трифторацетокси)йодбензол (1,13 г, 2,62 ммоль, 1,20 эквивалента) и перемешивали смесь в течение 30 минут. При 30°C добавили йод (664,86 мг, 2,62 ммоль, 1,20 эквивалента) и перемешивали в течение 23,5 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию. Реакционный раствор отфильтровали. Осадок на фильтре промыли растворителем (петролейный эфир : дихлорметан = 2:1, 10 мл) и получив указанное в заголовке соединение (300,00 мг, 828,31 мкмоль, выход - 38,00%, степень чистоты - 98%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 354,8 (М+1).
Этап 6:
6-бром-4-фтор-2-метил-3-(пропен-2-ил)-2Н-индазол
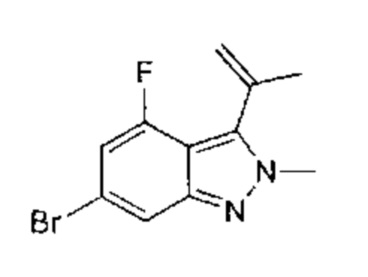
В атмосфере азота к раствору 5-бром-7-фтор-3-йод-2-метил-2Н-индазола (320,00 мг, 901,56 мкмоль, 1,00 эквивалент) и 2-изопропенил-4,4,5,5-тетраметил-1,3,2-диоксаборолана (181,80 мг, 1,08 ммоль, 1,20 эквивалента) в тетрагидрофуране (8,00 мл) и воде (5,00 мкл) добавили калия карбонат (373,81 мг, 2,70 ммоль, 3,00 эквивалента) и Pd(dppf)Cl2 (131,93 мг, 180,31 мкмоль, 0,20 эквивалента). Реакционную смесь перемешивали при 50-60°C в течение 16 часов. ЖХ/МС показала, что образовалось 55% желаемого продукта. ТСХ показала, что исходный материал полностью вступил в реакцию. Смесь профильтровали, и фильтрат упарили досуха. Упаренный остаток очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = 10:1) и получили указанное в заголовке соединение (210,00 мг, 780,35 мкмоль, выход - 86,56%), представляющее собой светло-коричневую жидкость. ЖХ/МС (ESI) м/з: 268,9 (М+1).
Этап 7:
4-фтор-2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-(пропен-2-ил)-2Н-индазол
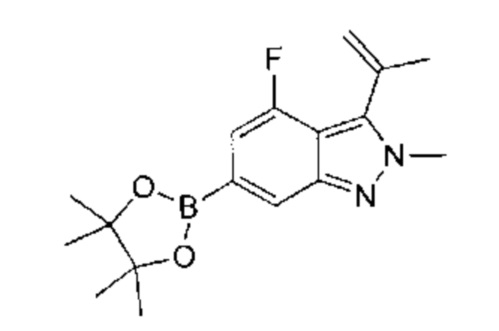
В атмосфере азота к раствору 5-бром-7-фтор-3-изопропенил-2-метил-2Н-индазола (302,00 мг, 1,12 ммоль, 1,00 эквивалент) и бис(пинаколато)диборона (341,30 мг, 1,34 ммоль, 1,20 эквивалента) в тетрагидрофуране (10,00 мл) добавили 3 капли воды, Pd(dppf)Cl2 (163,90 мг, 224,00 мкмоль, 0,20 эквивалент) и калия ацетат (329,75 мг, 3,36 ммоль, 3,00 эквивалента). Смесь перемешивали при 80-90°С в течение 14 часов. ТСХ показала (петролейный эфир: этилацетат = 5:1), что большая часть исходного материала вступила в реакцию. Смесь охладили до 25°С и профильтровали. Фильтрат упарили досуха. Упаренный остаток очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = 8:1) и получили указанное в заголовке соединение (306,00 мг, 967,80 мкмоль, выход - 86,41%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 317,2 (М+1).
Этап 8:
5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2-метил-3-(пропен-2-ил)-2Н-индазол
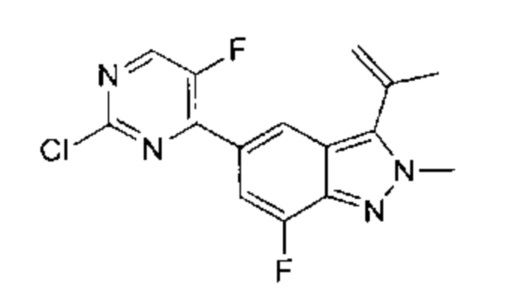
В атмосфере азота к раствору 2,4-дихлор-5-фтор-пиримидина (177,44 мг, 1,06 ммоль, 1,20 эквивалента) и 7-фтор-3-изопропенил-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индазола (280,00 мг, 885,57 мкмоль, 1,00 эквивалент) в тетрагидрофуране добавили калия карбонат (367,18 мг, 2,66 ммоль, 3,00 эквивалента), Pd(dppf)Cl2 (129,59 мг, 177,11 мкмоль, 0,20 эквивалент) и 3 капли воды. Смесь перемешивали при 80-90°С в течение 16 часов. ТСХ показала (петролейный эфир: этилацетат = 5:1), что прореагировала большая часть исходного материала. Смесь охладили до 25°С и профильтровали. Осадок на фильтре промыли этилацетатом (3 мл × 2), фильтрат упарили досуха. Сухой остаток очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = 7:1) и получили указанное в заголовке соединение (промежуточный продукт D) (260,0 мг, 802,57 мкмоль, выход - 90,63%, степень чистоты - 99%), представляющее собой ярко-желтое твердое вещество. ЖХ/МС (ESI) м/з: 320,9 (М+1).
Пример 22
5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин

Этап 1:
трет-бутил-4-(6-((5-фтор-4-(7-фтор-2-метил-3-(изопропен-2-ил)-2Н-индазол-5-ил)пиримидин-2-ил) амино)пиридин-3-ил)пиперазин-1-карбоксилат
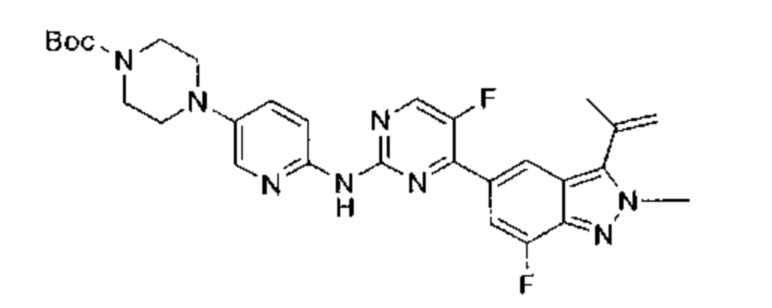
В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2-метил-3-(изопропен-2-ил)-2Н-индазола (промежуточный продукт D) (100 мг, 311,80 ммоль, 1,00 эквивалент) в диоксане (3 мл) добавили
трет-бутил-4-(6-амино-3-пиридил)пиперазин-1-карбоксилат (99,81 мг, 358,57 ммоль, 1,15 эквивалента), цезия карбонат (203,18 мг, 623,6 ммоль, 2,00 эквивалента), Pd2(dba)3 (57,1 мг, 62,36 мкмоль, 0,2 эквивалента) и Xantphos (72,17 мг, 124,72 мкмоль, 0,40 эквивалента). Через смесь три раза пропустили азот, затем ее нагрели до 100°С и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционный раствор охладили до 25°С, разбавили дихлорметаном (5 мл) и профильтровали. Остаток, полученный путем упаривания фильтрата, очистили посредством препаративной ТСХ (этилацетат) и получили указанное в заголовке соединение (100,00 мг, 177,74 мкмоль, выход - 57,01%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,38 (д., J=3,9 Гц, 1Н), 8,30 (с, 1Н), 8,03 (д., J=2,8 Гц, 1H), 7,80 (д., J=12,7 Гц, 1Н), 7,38 (дд, J=9,0, 2,9 Гц, 1H), 5,75-5,69 (м., 1Н), 5,37 (с, 1Н), 4,23 (с, 3Н), 3,67-3,56 (м., 4Н), 3,17-3,04 (м., 4Н), 2,31 (с, 3Н), 1,50 (с, 9Н).
Этап 2:
трет-бутил-4-(6-((5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилат
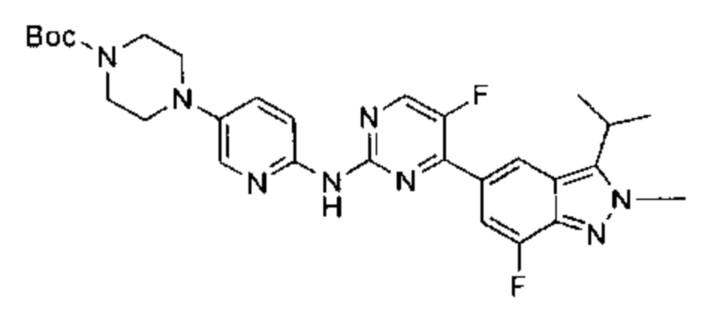
К раствору-смеси трет-бутил-4-(6-((5-фтор-4-(7-фтор-2-метил-3-(изопропен-2-ил)-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (100,00 мг, 177,74 мкмоль, 1,00 эквивалент) в метаноле (10,00 мл) и уксусной кислоте (500 мкл) добавили трис(трифенилфосфин)родия хлорид (49,33 мг, 53,32 мкмоль, 0,3 эквивалента). Через колбу с реакционной смесью три раза пропускали аргон и водород. Под давлением водорода (50 фунт/кв.дюйм) смесь нагрели до 50°С и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Реакционный раствор охладили до 25°С и отфильтровали. Остаток, полученный путем упаривания фильтрата, очистили посредством препаративной ТСХ (этилацетат) и получили указанное в заголовке соединение (15 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 565,3 (М+1).
Этап 3:
5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин
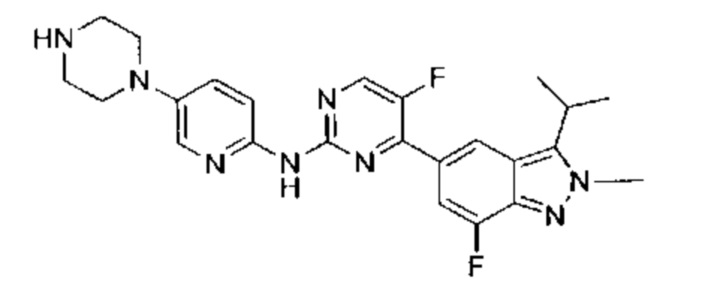
К раствору трет-бутил-4-(6-((5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1 -карбоксилата (15 мг, 26,57 мкмоль, 1,00 эквивалент) в дихлорметане (1 мл) добавили трифторуксусную кислоту (500 мкл). Смесь перемешивали при 25°С в течение 1 часа. ЖХ/МС показала, что исходный материал полностью вступил в реакцию с образованием желаемого продукта. Остаток, полученный путем упаривания реакционного раствора, очистили посредством препаративной ВЭЖХ (соляная кислота) и таким образом получили указанное в заголовке соединение (3,88 мг, 8,27 мкмоль, выход - 31,12%, степень чистоты - 99%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,73 (д., J=3,9 Гц, 1H), 8,63 (с., 1Н), 8,27 (дд, J=2,6, 9,7 Гц, 1Н), 7,91 (д., J=2,4 Гц, 1Н), 7,84 (д., J=13,2 Гц, 1Н), 7,55 (д., J=9,7 Гц, 1Н), 4,22 (с, 3Н), 3,71-3,61 (м., 1H), 3,60-3,51 (м., 4Н), 3,49-3,41 (м., 4Н), 1,59 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 465,2 (М+1).
Пример 23
2-[4-[6-[[5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридинил]пиперазин-1-ил]этанол

Этап 1:
2-[4-[6-[[5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)пиримидин-2-ил]амино]-3-пиридинил]пиперазин-1-ил]этанол
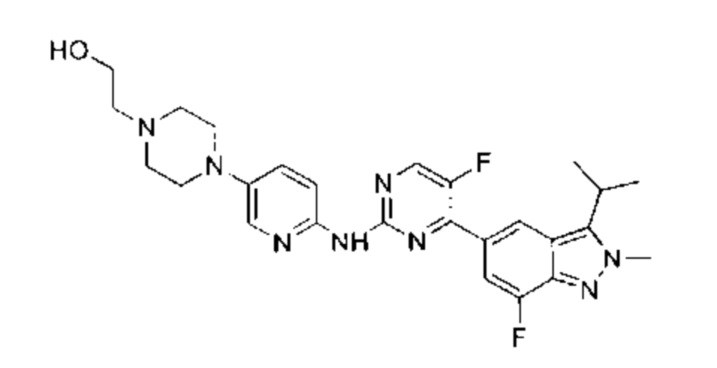
К раствору
5-фтор-4-(7-фтор-3-изопропил-2-метил-2Н-индазол-5-ил)-N-(5-пиперазин-1-ил-2-пиридил)пиримидин-2-амина (80,00 мг, 172,22 мкмоль, 1,00 эквивалент) и 2-бромэтанола (64,56 мг, 516,66 мкмоль, 3,00 эквивалента) в ацетонитриле (5,00 мл) добавили диизопропилэтиламин (66,77 мг, 516,66 мкмоль, 3,00 эквивалента). Смесь нагрели до 70°С и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал практически полностью вступил в реакцию. Масс-спектр подтвердил наличие желаемого вещества. Реакционную смесь упарили под пониженным давлением и получили сухой остаток. Остаток разбавили этилацетатом (10 мл) и промыли последовательно водой (5×3 мл) и солевым раствором (3×5 мл). Органическую фазу высушили над безводным натрия сульфатом и отфильтровали. Фильтрат упарили, и остаток очистили посредством препаративной ВЭЖХ (основная среда), получив указанное в заголовке соединение (20,31 мг, 39,14 мкмоль, выход - 22,72%, степень чистоты - 98%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,59 (с, 1H), 8,48 (д., J=3,5 Гц, 1Н), 8,18 (д., J=8,8 Гц, 1Н), 8,01 (ушир. с. 1Н), 7,80 (д., J=12,5 Гц, 1Н), 7,73-7,63 (м., 1Н), 4,22 (с, 3Н), 3,76 (т., J=5,8 Гц, 2Н), 3,71-3,60 (м., 1Н), 3,24 (ушир. с, 5Н), 2,76 (ушир. с. 4Н), 2,64 (т., J=5,5 Гц, 2Н), 1,60 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 509,3 (М+1).
Подход D
Далее описано общий способ получения промежуточного продукта Е.
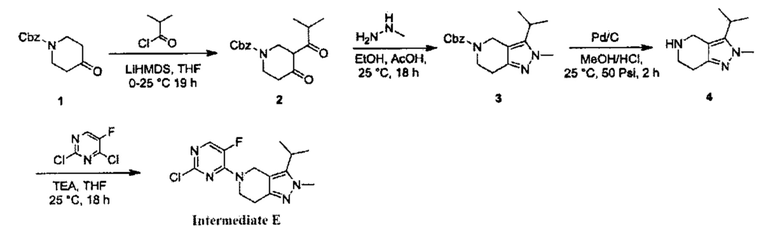
Этап 1: бензил-3-изобутирил-4-оксопиперидин-1-карбоксилат

При 0°С к раствору бензил-4-оксопиперидин-1-карбоксилата (128,07 г, 549,04 ммоль, 1,95 эквивалента) в толуоле (600,00 мл) медленно по капле добавили LiHMDS (550 мл, 550,00 ммоль, 1,95 эквивалента). Смесь перемешивали при 0°С в течение 1 часа. Затем в реакционную смесь при 0°С медленно по капле добавили 2-метилпропионилхлорид (30,00 г, 281,56 ммоль, 1,00 эквивалент), после чего смесь нагрели до 25°С и перемешивали в течение 18 часов. ТСХ показала (петролейный эфир: этилацетат = 5:1), что исходный материал прореагировал полностью. Реакцию остановили, добавив в реакционную смесь 10% водный раствор уксусной кислоты (100 мл), Полученную смесь подвергли разделению фаз. Органический слой последовательно промыли водой (500 мл) и насыщенным солевым раствором (500 мл). Полученный органический слой высушили над безводным натрия сульфатом и отфильтровали. Фильтрат упарили, и полученный остаток очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = от 50:1 до 20:1), получив указанное в заголовке соединение (57,00 г, неочищенный продукт), представляющее собой бледно-желтое масло. 1Н ЯМР (400 МГц, CDCl3) δ 16,10 (с., 1H), 7,38-7,36 (м., 5Н), 5,18 (с, 2Н), 4,33 (с, 2Н), 3,69-3,66 (м., 2Н), 2,68-2,57 (м., 1Н), 2,48 (ушир. с, 2Н), 1,14 (д., J=6,4 Гц, 6Н).
Этап 2:
бензил-6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-карбоксилат

При 25°С к раствору бензил-3-изобутирил-4-оксопиперидин-1-карбоксилата (57,00 г, 187,90 ммоль, 1,00 эквивалент) в этаноле (600,00 мл) добавили 40% водный раствор метилгидразина (110,00 г, 954,53 ммоль, 5,08 эквивалента). Смесь перемешивали в течение 1 часа. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционный раствор упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (трифторуксусная кислота) и получили гомогенный раствор. Из раствора выпарили ацетонитрил. Значение рН водной фазы довели до 8 с помощью натрия бикарбоната, и затем водную фазу подвергли двукратной экстракции дихлорметаном (500 мл × 2). Объединенные органические слои высушили над натрия сульфатом и отфильтровали. Фильтрат упарили и получили указанное в заголовке соединение (6,50 г, 20,74 ммоль, выход- 11,4%), представляющее собой бесцветное масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,43-7,29 (м., 5Н), 5,18 (с, 2Н), 4,58 (с, 2Н), 3,82-3,71 (м., 5Н),3,03 (квин., J=7,0 Гц, 1H), 2,82-2,67 (м., 2Н), 1,28 (д., J=6,4 Гц, 6Н).
Этап 3:
3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридин

При 25°С к раствору
бензил-6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-карбоксилат а (6,50 г, 20,74 ммоль, 1,00 эквивалент) в метаноле (100,00 мл) добавили Pd/C (1,00 г) и раствор соляной кислоты (1,00 мл) с концентрацией 12 моль/л. Через смесь три раза пропускали по очереди азот и водород. Через реакционный раствор пропустили водород, и смесь перемешивали под давлением 50 фунт/кв.дюйм в течение 2 часов. ТСХ показала (петролейный эфир: этилацетат = 1:1), что исходный материал прореагировал полностью. Реакционную смесь профильтровали, и фильтрат упарили и получили указанное в заголовке соединение (5,25 г, неочищенный продукт), представляющее собой почти белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 9,93 (ушир. с, 2Н), 4,31 (ушир. с., 2Н), 3,76 (с., 3Н), 3,09-2,98 (м., 3Н), 2,10 (ушир. с., 2Н), 1,26 (д., J=7,0 Гц, 6Н).
Этап 4:
5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридин
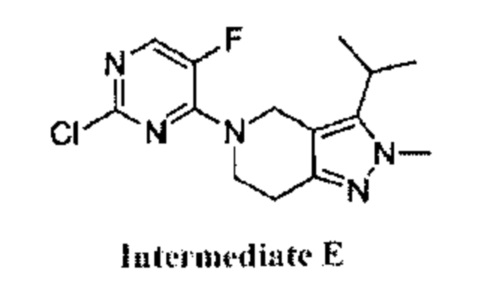
При 25°С к раствору 3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (5,25 г, 20,75 ммоль, 1,00 эквивалент) в тетрагидрофуране (60,00 мл) добавили 2,4-дихлор-5-фторпиримидин (3,50 г, 20,96 ммоль, 1,01 эквивалента) и триэтиламин (9,51 г, 94,01 ммоль, 4,53 эквивалента). Реакционную смесь перемешивали в течение 18 часов. ТСХ показала (петролейный эфир: этилацетат = 3:1), что исходный материал прореагировал полностью. Реакционный раствор разбавили водой (200 мл) и затем подвергли экстракции этилацетатом (200 мл × 2). Объединенные органические слои высушили над безводным натрия сульфатом, отфильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = от 5:1 до 2:1) и получили указанное в заголовке соединение (промежуточный продукт Е) (5,60 г, 18,08 ммоль, выход - 87,12%), представляющее собой почти белое твердое вещество. 1Н ЯМР (300 МГц, CDCl3) δ 7,94 (д., J=6,0 Гц, 1Н), 4,84 (с, 2Н), 4,03 (т., J=5,8 Гц, 2Н), 3,79 (с., 3Н), 3,07 (септ., J=7,1 Гц, 1H), 2,88 (т., J=5,7 Гц, 2Н), 1,32 (д., J=7,2 Гц, 6Н).
Пример 24
5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиридин-2-амин
Этап 1:
трет-бутил-6-нитро-5',6'-дигидро-[3,4'-бипиридил]-1'(2'Н)карбоксилат
В атмосфере азота к раствору 5-бром-2-нитропиридина (10,00 г, 49,26 ммоль, 1,02 эквивалента) в диоксане (120 мл) и воде (10 мл) добавили
трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоксилат (15,00 г, 48,51 ммоль, 1,00 эквивалент), калия карбонат (5,9 г, 72,77 ммоль, 1,50 эквивалента) и Pd(dppf)Cl2 (1,77 г, 2,43 ммоль, 0,05 эквивалента). Реакционную смесь нагрели до 80°С и перемешивали в течение 18 часов. ТСХ показала (петролейный эфир: этилацетат = 3:1), что исходный материал прореагировал полностью. Реакционный раствор профильтровали, высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = 3:1) и получили указанное в заголовке соединение (18,70 г, неочищенный продукт), представляющее собой светло-желтое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 8,64 (д., J=2,0 Гц, 1Н) 8,24 (д., J=8,4 Гц, 1Н) 7,95 (дд., J=8,5, 2,3 Гц, 1Н) 6,33 (ушир. с., 1Н) 4,16 (д., J=2,6 Гц, 2Н) 3,69 (т., J=5,6 Гц, 2Н) 2,57 (ушир. с., 2Н) 1,50 (с., 9Н).
Этап 2:
трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат

К раствору
трет-бутил-4-(6-нитро-3-пиридил)-3,6-дигидро-2Н-пиридин-1-карбоксилата (18,70 г, 48,51 ммоль, 1,00 эквивалент) в метаноле (500 мл) добавили Pd/C (2,00 г). Через реакционную смесь три раза пропускали по очереди азот и водород. Через реакционный раствор пропустили водород, чтобы создать давление 50 фунт/кв. дюйм. Реакционную смесь нагрели до 50°С и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Смесь профильтровали, и фильтрат упарили и получили указанное в заголовке соединение (10,40 г, 37,50 ммоль, выход - 77,31%), представляющее собой почти белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,92 (д., J=2,4 Гц, 1Н) 7,31-7,27 (м., 1Н) 6,48 (д., J=8,5 Гц, 1Н) 4,35 (ушир. с., 2Н) 4,23 (ушир. с., 2Н) 2,78 (т., J=12,1 Гц, 2Н) 2,60-2,48 (м., 1Н) 1,78 (ушир. с., 2H) 1,61-1,52 (м., 2Н) 1,48 (с., 9Н).
Этап 3: 5-(1-метилпиперидин-4-ил)пиридин-2-амин

При 0°С к раствору алюмогидрид лития (2,05 г, 54,09 ммоль, 3,00 эквивалента) в тетрагидрофуране (100 мл) медленно по капле добавили раствор трет-бутил-4-(6-амино-3-пиридинил)пиперидин-1-карбоксилата (5,00 г, 3,18 ммоль, 1,00 эквивалент) в тетрагидрофуране (50 мл). После добавления реактивов смесь нагрели до 70°С и перемешивали в течение 2 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. При 0°С, реакцию остановили, добавив в реакционную смесь 20% водный раствор калия гидроксида (4 мл), и получили большое количество твердого вещества. Реакционную смесь профильтровали, фильтрат упарили и получили указанное в заголовке соединение (4,00 г, неочищенный продукт), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,93 (д., J=2,3 Гц, 1H) 7,31 (дд, J=8,4, 2,4 Гц, 1Н) 6,46 (д., J=8,4 Гц, 1Н) 4,33 (ушир. с., 2Н) 2,95 (д., J=11,7 Гц, 2Н) 2,41-2,32 (м., 1Н) 2,31 (с., 3Н) 2,04-1,98 (м., 2Н) 1,80-1,67 (м., 4Н).
Этап 4:
5-фтор-4-(3-изопропил-2-метил-6,7-дигидро-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амин

В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (2,00 г, 6,46 ммоль, 1,00 эквивалент) в толуоле (20,00 мл) добавили 5-(1-метил-4-пиперидинил)пиридин-2-амин (1,36 г, 7,10 ммоль, 1,10 эквивалента), цезия карбонат (4,21 г, 12,91 ммоль, 2,00 эквивалента), Pd2(dba)3 (400,00 мг, 436,81 мкмоль, 0,07 эквивалент) и Xantphos (520,00 мг, 898,69 мкмоль, 0,14 эквивалент). Смесь нагрели до 110°С и перемешивали в течение 18 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционную смесь охладили до 20°С и упарили до сухого остатка Остаток очистили посредством колоночной хроматографии (этилацетат, затем дихлорметан: метанол = 10:1) и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (2,48 г, 4,61 ммоль, выход - 71,42%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,27 (д., J=1,9 Гц, 1Н), 8,13 (д., J=7,2 Гц, 1Н), 8,01 (д., J=8,5 Гц, 1Н), 7,45 (д., J=8,8 Гц, 1Н), 5,01 (с., 2Н), 4,16 (т., J=5,8 Гц, 2Н), 3,77 (с., 3Н), 3,65 (д., J=12,3 Гц, 2Н), 3,26-3,12 (м., 3Н), 3,09-2,98 (м., 1Н), 2,93 (с., 3Н), 2,86 (т., J=5,8 Гц, 2Н), 2,22-1,98 (м., 4Н), 1,34 (д., J=7,0 Гц, 6Н).
Пример 25
N-5-фтор-4-(3-изопропил-2-метил-6,7-дигидро-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин

В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (200,00 мг, 645,64 мкмоль, 1,00 эквивалент) и 6-(4-метилпиперазин-1-ил)пиридазин-3-амина (124,77 мг, 645,64 мкмоль, 1,00 эквивалент) в диоксане (5,00 мл) добавили цезия карбонат (420,72 мг, 1,29 ммоль, 2,00 эквивалента), Pd2(dba)3 (59,12 мг, 64,56 мкмоль, 0,10 эквивалент) и Xantphos (74,72 мг, 129,13 мкмоль, 0,20 эквивалент). Реакционную смесь затем нагрели до 110°С и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционную смесь охладили до 25°С, профильтровали и упарили под пониженным давлением, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (132,29 мг, 283,54 мкмоль, выход - 43,92%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,23 (д., J=9,91 Гц, 1Н), 7,93 (д., J=6,90 Гц, 1Н), 7,34 (д., J=9,91 Гц, 1Н), 4,85 (с., 2Н), 4,02 (т., J=5,84 Гц, 2Н), 3,77 (с., 3Н), 3,63-3,57 (м., 4Н), 3,17 (кв., J=7,00 Гц, 1Н), 2,81 (т., J=5,77 Гц, 2Н), 2,65-2,59 (м., 4Н), 2,38 (с., 3Н), 1,32 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 467,2 (М+1).
Пример 26
5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(4-метил-1,4-диазепан-1-ил)пиридин-2-ил)пиримидин-2-амин

Этап 1: 1-метил-4-(6-нитропиридин-3-ил)-1,4-диазепан
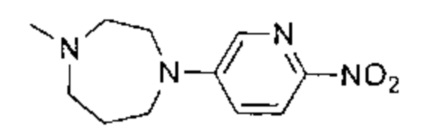
К раствору 5-бром-2-нитро-пиридина (2,00 г, 9,85 ммоль, 1,00 эквивалент) и 1-метил-1,4-диазепана (1,69 г, 14,78 ммоль, 1,50 эквивалента) в диметилсульфоксиде (20,00 мл) добавили калия карбонат (2,72 г, 19,70 ммоль, 2,00 эквивалента). Смесь нагрели до 80°С и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционную смесь охладили до 25°С и добавили воду (50 мл). Смесь затем подвергли экстракции этилацетатом (100 мл × 3). Объединенные органические фазы промыли насыщенным солевым раствором (100 мл × 3), высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = от 30:1 до 1:3) и получили указанное в заголовке соединение (2,00 г, 8,46 ммоль, выход - 85,94%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 237,1 (М+1).
Этап 2: 5-(4-метил-1,4-диазепан-1-ил)пиридин-2-амин
При 25°С к раствору 1-метил-4-(6-нитропиридин-3-ил)-1,4-диазепана (2,00 г, 8,46 ммоль, 1,00 эквивалент) в метаноле (20,00 мл) добавили Pd/C (10%, 500 мг). Через реакционную смесь три раза пропускали по очереди азот и водород. Через реакционный раствор пропустили водород, чтобы создать давление 15 фунт/кв. дюйм. Реакционный раствор перемешивали в течение 2 часов. ТСХ показала, что исходный материал полностью вступил в реакцию. Реакционную смесь профильтровали, упарили под пониженным давлением и получили указанное в заголовке соединение (1,70 г, 8,24 ммоль, выход - 97,41%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 207,1 (М+1).
Этап 3: 5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(4-метил-1,4-диазепан-1-ил)пиридин-2-ил)пиримидин-2-амин
В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло [4,3-с]пиридина (промежуточный продукт Е) (200,00 мг, 645,64 мкмоль, 1,00 эквивалент) и 5-(4-метил-1,4-диазепан-1-ил)пиридин-2-амина (159,83 мг, 774,77 мкмоль, 1,20 эквивалента) в диоксане (5,00 мл) добавили цезия карбонат (420,72 мг, 1,29 ммоль, 2,00 эквивалента), Pd2(dba)3 (59,12 мг, 64,56 мкмоль, 0,10 эквивалент) и Xantphos (74,72 мг, 129,13 мкмоль, 0,20 эквивалент). Реакционную смесь затем нагрели до 110°С и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционную смесь охладили до 25°С, профильтровали и упарили под пониженным давлением, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (93,10 мг, 194,12 мкмоль, выход - 30,07%). 1Н ЯМР (400 МГц, метанол-d4) δ 7,91-7,84 (м., 2Н), 7,77 (д., J=3,01 Гц, 1Н), 7,22 (дд, J=9,16, 3,14 Гц, 1Н), 4,85 (с., 2Н), 4,01 (т., J=5,83 Гц, 2Н), 3,77 (с., 3Н), 3,62-3,57 (м., 2Н), 3,52 (т., J=6,27 Гц, 2Н), 3,17 (кв., J=7,03 Гц, 1Н), 2,83-2,75 (м., 4Н), 2,67-2,61 (м., 2Н), 2,40 (с., 3Н), 2,06 (дт, J=11,51, 5,98 Гц, 2Н), 1,36-1,29 (м., 6Н). ЖХ/МС (ESI) м/з: 480,2 (М+1).
Пример 27
N-(5-(4-(диметиламино)пиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин
Этап 1:
N,N-диметил-1-(6-нитропиридин-3-ил)пиперидин-4-амин
В атмосфере азота к раствору N,N-диметилпиперидин-4-амина (100,00 мг, 779,97 мкмоль, 1,00 эквивалент) и 5-бром-2-нитропиридина (158,33 мг, 779,97 мкмоль, 1,00 эквивалент) в диоксане (5 мл) добавили BINAP (48,57 мг, 78,00 мкмоль, 0,10 эквивалента), цезия карбонат (508,26 мг, 1,56 ммоль, 2,00 эквивалента) и Pd(OAc)2 (17,51 мг, 78,00 мкмоль, 0,10 эквивалента). Реакционную смесь затем нагрели до 90°С и перемешивали в течение 16 часов. ТСХ показала, что исходный материал полностью вступил в реакцию. Реакционную смесь профильтровали и упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (дихлорметан: метанол = 10:1) и получили указанное в заголовке соединение (125,00 мг, 499,40 мкмоль, выход - 64,03%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц. CDCl3) δ 8,20-8,14 (м., 2Н), 7,21 (дд, J=9,2, 3,2 Гц, 1Н), 3,99 (д., J=13,2 Гц, 2Н), 3,12-3,02 (м., 2Н), 2,47-2,39 (м., 1Н), 2,33 (с., 6Н), 2,01 (д., J=12,4 Гц, 2Н), 1,70-1,64 (м., 2Н). ЖХ/МС (ESI) м/з: 251,1 (М+1).
Этап 2:
5-(4-(диметиламино)-пиперидин-1-ил)пиридин-2-амин
К раствору N,N-диметил-1-(6-нитропиридин-3-ил)пиперидин-4-амина (525,00 мг, 2,10 ммоль, 1,00 эквивалент) в метаноле (10 мл) добавили Pd-C (10%, 100 мг). Через реакционную смесь три раза пропускали по очереди азот и водород. Через реакционный раствор пропустили водород, чтобы создать давление 15 фунт/кв.дюйм. Реакционный раствор перемешивали в течение 4 часов. ТСХ показала, что исходный материал полностью вступил в реакцию. Реакционную смесь профильтровали, фильтрат упарили и получили указанное в заголовке соединение (450,00 мг, 2,04 ммоль, выход - 97,27%), представляющее собой желтое твердое вещество. 1Н ЯМР (400 МГц, метанол-d6) δ 7,65-7,61 (м., 1Н), 7,34 (дд, J=9,2, 2,4 Гц, 1Н), 6,59 (дд, J=8,8, 0,8 Гц, 1Н), 3,52-3,44 (м., 2Н), 2,65 (дт, J=12,4, 2,4 Гц, 2Н), 2,42-2,38 (м., 6Н), 2,05-1,98 (м., 2Н), 1,67 (дк, J=12,4, 4,4 Гц, 2Н). ЖХ/МС (ESI) м/з: 221,0 (М+1).
Этап 3:
N-(5-(4-(диметиламино)пиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин
В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (150,00 мг, 484,23 мкмоль, 1,00 эквивалент) и
5-[4-(диметиламино)-1-пиперидинил]пиридин-2-амина (128,02 мг, 581,08 мкмоль, 1,20 эквивалента) в диоксане (5,00 мл) добавили Pd2(dba)3 (44,34 мг, 48,42 мкмоль, 0,10 эквивалент), цезия карбонат (315,54 мг, 968,46 мкмоль, 2,00 эквивалента) и Xantphos (56,04 мг, 96,85 мкмоль, 0,20 эквивалент). Реакционную смесь затем нагрели до 110°С и перемешивали в течение 16 часов. ЖХ/МС показала, что исходный материал прореагировал полностью. Реакционную смесь охладили до 25°С, профильтровали и упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (муравьиная кислота), получив желаемую смесь (81,64 мг, 165,39 мкмоль, выход - 34,16%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,01-7,86 (м., 3Н), 7,51 (дд, J=9,16, 2,76 Гц, 1Н), 4,85 (с., 2Н), 4,06-3,96 (м., 2Н), 3,84-3,72 (м., 5Н), 3,38-3,33 (м., 1H), 3,17 (дт, J=14,02, 6,98 Гц, 1Н), 2,90 (с., 6Н), 2,85-2,77 (м., 4Н), 2,22 (д., J=12,05 Гц, 2Н), 1,90 (кд, J=12,03, 3,70 Гц, 2Н), 1,32 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 494,2 (М+1).
Пример 28
N5-(2-(диметиламино)этил)-N2-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-N5-метилпиридин-2,5-диамин
Этап 1:
N-(2-(диметиламино)этил)-N-метил-6-нитропиридин-3-амин
К раствору 5-бром-2-нитро-пиридина (2,00 г, 9,85 ммоль, 1,00 эквивалент) и N,N,N-триметилэтан-1,2-диамина (1,51 г, 14,78 ммоль, 1,50 эквивалента) в диметилформамиде (20,00 мл) добавили одной порцией калия карбонат (2,72 г, 19,70 ммоль, 2,00 эквивалента). Реакционную смесь нагрели до 90°С и перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Реакционный раствор охладили до 25°С и добавили в него воду (50 мл). Реакционную смесь подвергли экстракции этилацетатом (100 мл × 3). Органические фазы объединили и промыли насыщенным солевым раствором (100 мл × 3), затем высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = от 30:1 до 1:3) и получили указанное в заголовке соединение (1,50 г, 6,69 ммоль, выход - 67,91%), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3): δ, 8,19-8,15 (м., 1Н), 7,99 (д., J=3,14 Гц, 1Н), 7,02 (дд., J=9,29, 3,14 Гц, 1Н), 3,63-3,57 (м., 2Н), 3,16 (с., 3Н), 2,57-2,51 (м., 2Н), 2,31 (с., 6Н). ЖХ/МС (ESI) м/з: 225,1 (М+1).
Этап 2:
N3-(2-(диметиламино)этил)-N3-метилпиридин-3,6-диамин
В атмосфере азота к раствору N,N,N'-триметил-N'-(6-нитро-3-пиридил)этан-1,2-диамина (1,50 г, 6,69 ммоль, 1,00 эквивалент) в метаноле (15,00 мл) добавили Pd/C (300 мг). Через суспензию несколько раз пропустили H2 и затем перемешивали при 25°С в атмосфере H2 (1 атмосфера) в течение 2 часов. ТСХ показала, что реакция завершилась. Реакционный раствор отфильтровали и упарили, получив указанное в заголовке соединение (1,20 г, 6,18 ммоль, выход - 92,33%), представляющее собой белое твердое вещество, которое не требовало дальнейшей очистки. ЖХ/МС (ESI) м/з: 195,1 (М+1).
Этап 3:
N5-(2-(диметиламино)этил)-N2-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-N5-метилпиридин-2,5-диамин
В пробирку для СВЧ-печи добавили раствор 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (150,00 мг, 484,23 мкмоль, 1,00 эквивалент),
N3-(2-(диметиламино)этил)-N3-метилпиридин-3,6-диамина (112,89 мг, 581,08 мкмоль, 1,20 эквивалента), цезия карбоната (315,54 мг, 968,46 мкмоль, 2,00 эквивалента), Pd2(dba)3 (88,68 мг, 96,85 мкмоль, 0,20 эквивалента) и Xantphos (56,04 мг, 96,85 мкмоль, 0,20 эквивалента) в диоксане (5,00 мл). Через реакционный раствор несколько раз пропустили азот, нагрели до 130°С и перемешивали в течение 2,5 часов. ЖХ/МС показала, что вещество не полностью вступило в реакцию и образовалось около 14% продукта. Реакционный раствор охладили до 25°С, отфильтровали, упарили и получили неочищенный продукт. Неочищенные продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (91,63 мг, 7 мкмоль, выход - 47%). 1Н ЯМР (400 МГц, метанол-d4): δ, 8,25 (д., J=7,03 Гц, 1Н), 8,03 (дд., J=9,54, 3,01 Гц, 1Н), 7,95 (д., J=2,89 Гц, 1Н), 7,46 (д., J=9,54 Гц, 1Н), 5,08 (с., 2Н), 4,25 (т., J=5,58 Гц, 2Н), 4,06 (с., 3Н), 3,88 (т., J=7,22 Гц, 2Н), 3,45 (т., J=7,22 Гц, 2Н), 3,40-3,35 (м., 1Н), 3,15-3,07 (м., 5Н), 3,0-22,97 (м., 6Н), 1,43 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 468,3 (М+1).
Пример 29
4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-илпиримидин-2-ил)амино)пиридин-3-ил)пиперидин-4-ол
Этап 1:
трет-бутил-4-(6-аминопиридин-3-ил)-4-гидроксипиперидин-карбоксилат
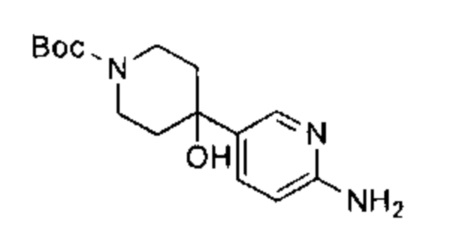
При -70°С к раствору 5-бромпиридин-2-амина (1,00 г, 5,78 ммоль, 1,00 эквивалент) в тетрагидрофуране (13,00 мл) медленно по капле добавили н-бутиллитий (2,5 М, 7,07 мл, 3,06 эквивалента). После перемешивания смеси при -70°С в течение 1,5 часов реакционный раствор нагрели до 25°С и затем перемешивали 2 часа. ЖХ/МС показала, что 19% полученной смеси представляли собой исходное вещество, также образовалось несколько новых соединений и около 8% продукта. Реакцию остановили, добавив в раствор воду и насыщенный раствор аммония хлорида при -70°С, затем нагрели до 25°С. Смесь подвергли экстракции этилацетатом (50 мл × 3). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл × 3), высушили над безводным натрия сульфатом, профильтровали и упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (300,00 мг, 1,02 ммоль, выход - 17,69%), представляющее собой желтое масло. 1Н ЯМР (300 МГц, ДМСО-d6): δ, 7,98 (д., J=2,07 Гц, 1Н), 7,43 (дд., J=8,67, 2,45 Гц, 1Н), 6,38 (д., J=8,67 Гц, 1Н), 5,75 (с., 2Н), 4,89 (с., 1Н), 3,79 (д., J=9,80 Гц, 2Н), 3,11 (ушир. с., 2Н), 1,77-1,63 (м., 2Н), 1,62-1,52 (м., 2Н), 1,41 (с., 9Н). ЖХ/МС (ESI) м/з: 294,0 (М+1).
Этап 2:
трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил-аминопиридин-3-ил)-4-гидроксипиперидин-карбоксилат
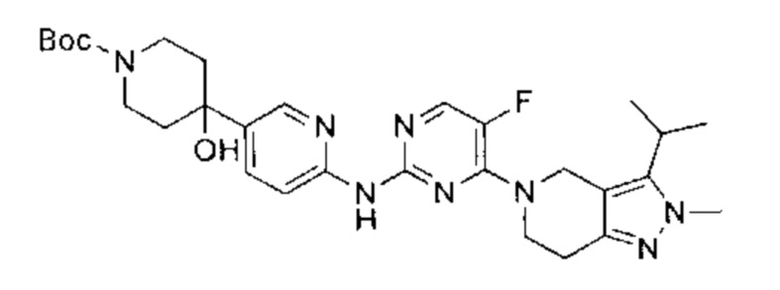
В атмосфере азота к раствору трет-бутил-4-(6-амино-3-пиридил)-4-гидрокси-пиперидин-1-карбоксилата (130,00 мг, 443,14 мкмоль, 1,00 эквивалент) в диоксане (5,00 мл) добавили 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетргидро-2Н-пиразоло[4,3-с]пиридин (промежуточный продукт Е) (164,73 мг, 531,77 мкмоль, 1,20 эквивалента), Pd2(dba)3 (40,58 мг, 44,31 мкмоль, 0,10 эквивалента), Xantphos (51,28 мг, 88,62 мкмоль, 0,20 эквивалента) и цезия карбонат (288,77 мг, 886,28 мкмоль, 2,00 эквивалента). Реакционную смесь перемешивали при 110°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор охладили до 25°С и подвергли фильтрованию с отсасыванием. Фильтрат упарили и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (дихлорметан: метанол = 10:1) и получили указанное в заголовке соединение (220,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 567,2 (М+1).
Этап 3:
4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-илпиримидин-2-ил-аминопиридин-3-ил)пиперидин-4-ол
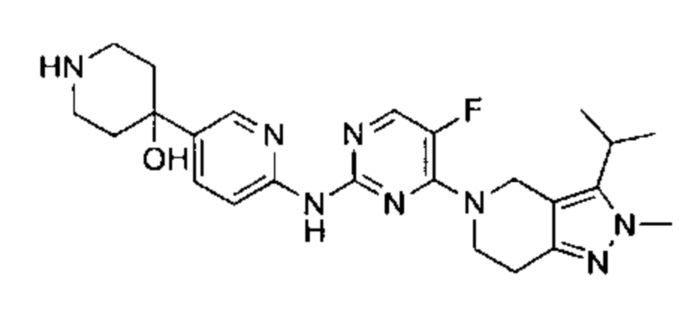
При 0°С к раствору трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил-аминопиридин-3-ил)-4-гидроксипиперидинкарбоксилата (220,00 мг, 388,23 мкмоль, 1,00 эквивалент) в дихлорметане (4,00 мл) добавили трифторуксусную кислоту (2,00 мл). Реакционную смесь нагрели до 25°С и перемешивали в течение 1 часа. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (115,01 мг, 246,51 мкмоль, выход - 63,50%). 1Н ЯМР (300 МГц, метанол-d4) δ 8,53 (д., J=1,9 Гц, 1Н), 8,35 (дд, J=9,0, 2,3 Гц, 1Н), 8,29 (д., J=6,6 Гц, 1H), 7,50 (д., J=9,0 Гц, 1Н), 4,26 (т., J=5 Гц, 2Н), 4,07 (с., 3Н), 3,60-3,32 (м., 7Н), 3,16-3,06 (м., 2Н), 2,47-2,30 (м., 2Н), 2,02 (д., J=13,8 Гц, 2Н), 1,49-1,40 (м., 6Н). ЖХ/МС (ESI) м/з: 467,2 (М+1).
Пример 30
N-(5-(1,4-диазепан-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин
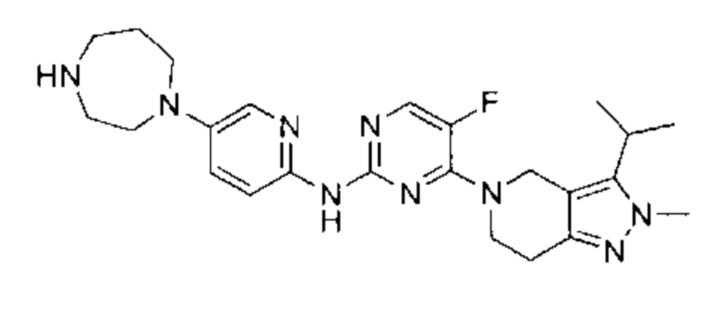
Этап 1:
трет-бутил-4-(6-нитропиридин-3-ил)-1,4-диазепан-1-карбоксилат
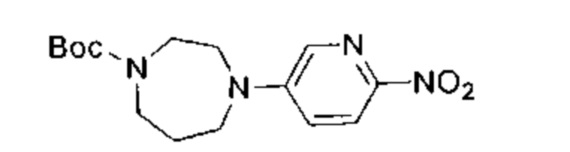
К раствору 5-бром-2-нитропиридина (2,00 г, 9,85 ммоль, 1,00 эквивалент) и трет-бутил-1,4-диазепан-1-карбоксилата (2,37 г, 11,82 ммоль, 1,20 эквивалента) в диметилсульфоксиде (20,00 мл) одной порцией добавили калия карбонат (2,72 г, 19,70 ммоль, 2,00 эквивалента). Реакционную смесь нагрели до 80°С и перемешивали в течение 16 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор охладили до 25°С и добавили в него воду (50 мл). Полученную смесь подвергли экстракции этилацетатом (100 мл × 3). Объединенные органические фазы промыли насыщенным солевым раствором (100 мл × 3), высушили над безводным натрия сульфатом, профильтровали и упарили, получив таким образом неочищенный продукт. Неочищенный продукт очистили посредством колоночной хроматографии (петролейный эфир: этилацетат = от 30:1 до 1:3) и получили указанное в заголовке соединение (2,00 г, 6,20 ммоль, выход - 62,99%), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 323,1 (М+1).
Этап 2:
трет-бутил-4-(6-аминопиридин-3-ил)-1,4-диазепан-1-карбоксилат
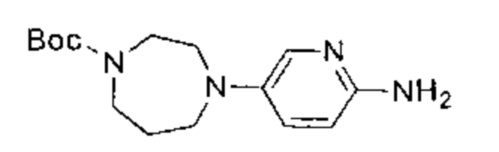
В атмосфере азота к раствору трет-бутил-4-(6-нитропиридин-3-ил)-1,4-диазепан-1-карбоксилата (1,80 г, 5,58 ммоль, 1,00 эквивалент) в метаноле (20,00 мл) добавили Pd/C (300 мг). Через суспензию несколько раз пропустили H2 и затем перемешивали при 25°С в атмосфере Н2 (1 атмосфера) в течение 2 часов. ТСХ показала, что реакция завершилась с образованием нового соединения. Реакционную смесь профильтровали, фильтрат упарили и получили неочищенный продукт (1,70 г, неочищенный продукт), представляющее собой белое твердое вещество. Неочищенный продукт использовали в следующем этапе без предварительной очистки. ЖХ/МС (ESI) м/з: 293,2 (М+1).
Этап 3:
трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)амино)пиридин-3-ил)-1,4-диазепан-карбоксилат

В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-6,7-дигидро-4Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (200,00 мг, 645,64 мкмоль, 1,00 эквивалент) в диоксане (5,00 мл) добавили трет-бутил-4-(6-аминопиридин-3-ил)-1,4-диазепан-1-карбоксилат (226,53 мг, 774,77 мкмоль, 1,20 эквивалента), Pd2(dba)3 (59,12 мг, 164,56 мкмоль, 0,01 эквивалент), цезия карбонат (420,72 мг, 1,29 ммоль, 2,00 эквивалента) и Xantphos (74,72 мг, 129,13 мкмоль, 0,20 эквивалент). Реакционную смесь перемешивали при 110°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор охладили до 25°С и подвергли фильтрованию с отсасыванием. Фильтрат упарили и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (этилацетат) и получили указанное в заголовке соединение (250,00 мг, неочищенный продукт), представляющее собой белое твердое вещество. ЖХ/МС (ESI) м/з: 566,2 (М+1).
Этап 4:
N-(5-(1,4-диазепан-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин
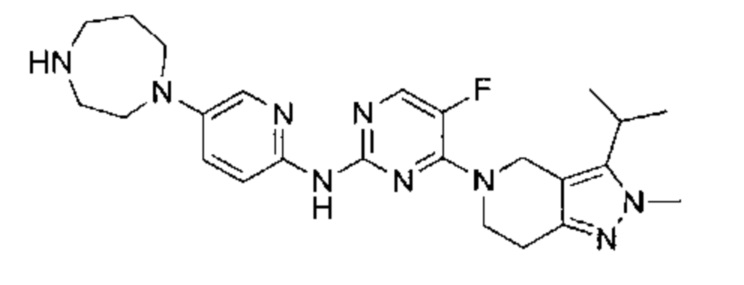
В атмосфере азота при 25°С к раствору трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил-амино)пиридин-3-ил)-1,4-диазепан-1-карбоксилата (250,00 мг, 441,95 мкмоль, 1,00 эквивалент) в дихлорметане (5,00 мл) добавили трифторуксусную кислоту (3 мл). Реакционную смесь перемешивали при 25°С в течение получаса. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор упарили, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (127,94 мг, 274,80 мкмоль, выход - 62,18%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,23 (д., J=6,78 Гц, 1Н), 7,94 (дд, J=9,47, 2,70 Гц, 1Н), 7,86 (д., J=2,64 Гц, 1Н), 7,41 (д., J=9,41 Гц, 1Н), 5,04-5,14 (м., 2Н), 4,24 (т., J=5,21 Гц, 2Н), 4,06 (с., 3Н), 3,83-3,94 (м., 2Н), 3,65 (т., J=6,02 Гц, 2Н), 3,45-3,53 (м., 2Н), 3,34-3,42 (м., 3Н), 3,06-3,13 (м., 2Н), 2,24-2,33 (м., 2Н), 1,44 (д., J=7,03 Гц, 6Н). ЖХ/МС (ESI) м/з: 466,2 (М+1).
Пример 31
N-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-5-(пиперазин-1-ил)пиразин-2-амин

Этап 1:
трет-бутил-4-(5-аминопиразин-2-ил)пиперазин-1-карбоксилат

В атмосфере азота к раствору трет-бутил-4-(5-бромпиразин-2-ил)пиперазин-1-карбоксилата (10,00 г, 29,14 ммоль, 1,00 эквивалент) и три-трет-бутилфосфония тетрафторбората (2,54 г, 8,74 ммоль, 0,30 эквивалента) в толуоле (100,00 мл) добавили LiHMDS (1М, 60,00 мл, 2,06 эквивалента) и Pd2(dba)3 (2,60 г, 2,84 ммоль, 0,10 эквивалент). Реакционный раствор перемешивали при 65°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционную смесь охладили до 25°С, и остановили реакцию, добавив в раствор воду (50 мл). Полученную смесь подвергли экстракции этилацетатом (100 мл × 3). Объединенные органические фазы упарили досуха и получили неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (основная среда) и получили указанное в заголовке соединение (5,00 г, 17,90 ммоль, выход - 61,43%), представляющее собой оранжевое твердое вещество. ЖХ/МС (ESI) м/з: 280,1 (М+1).
Этап 2:
трет-бутил-4-(5-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилат
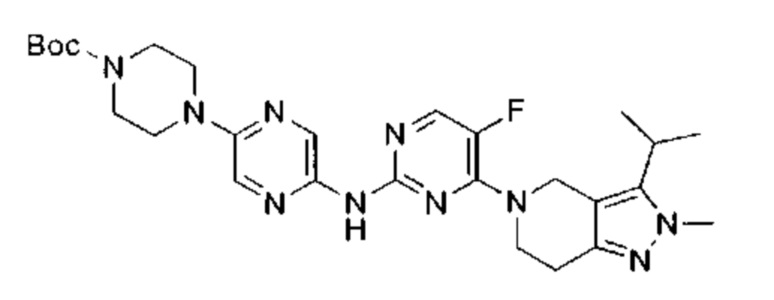
В пробирке для СВЧ-печи к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (500,00 мг, 1,61 ммоль, 1,00 эквивалент) и трет-бутил-4-(5-аминопиразин-2-ил)пиперазин-1-карбоксилата (495,97 мг, 1,78 ммоль, 1,10 эквивалента) в диоксане (16,00 мл) добавили Pd2(dba)3 (147,81 мг, 161,41 мкмоль, 0,10 эквивалента), BINАР (201,01 мг, 322,82 мкмоль, 0,20 эквивалента), натрия трет-бутоксид (232,67 мг, 2,42 ммоль, 1,50 эквивалента) и воду (400,00 мкл). Через реакционную систему в течение 2 минут пропускали азот, затем пробирку запечатали, нагрели до 140°С, и перемешивали смесь в течение 4 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор отфильтровали через диатомит (промытый этилацетатом), и фильтрат промыли водой (20 мл). Органическую фазу упарили досуха, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 2:1) и получили указанное в заголовке соединение (800,00 мг, неочищенный продукт), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 553,3 (М+1).
Этап 3: N-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-5-(пиперазин-1-ил)пиразин-2-амин

К раствору трет-бутил-4-(5-((5-фтор-4-(3-изопропил-2-метил-6,7-дигидро-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-амино)пиразин-2-ил)пиперазин-1-карбоксилата (800,00 мг, 1,45 ммоль, 1,00 эквивалент) в дихлорметане (15,00 мл) добавили трифторуксусную кислоту (23,03 г, 201,95 ммоль, 139,28 эквивалента). Реакционную смесь перемешивали при 30°С в течение 7 часов. ЖХ/МС показала, что реакция завершилась с образованием желаемого продукта. Реакционный раствор упарили досуха, получив неочищенный продукт. Неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (213,05 мг, 435,69 мкмоль, выход - 30,05%). 1Н ЯМР (400 МГц, метанол-d4): δ 8,26 (с., 1Н), 8,16-8,24 (м., 2Н), 5,20 (с., 2Н), 4,36 (ушир. с., 2Н), 4,07 (с., 3Н), 3,84-3,98 (м., 4Н), 3,35-3,44 (м., 5Н), 3,15 (ушир. с., 2Н), 1,33-1,53 (м., 6Н). ЖХ/МС (ESI) м/з: 453,2 (М+1).
Пример 32
N-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил)-5-(4-метилпиперазин-1-ил)пиразин-2-амин
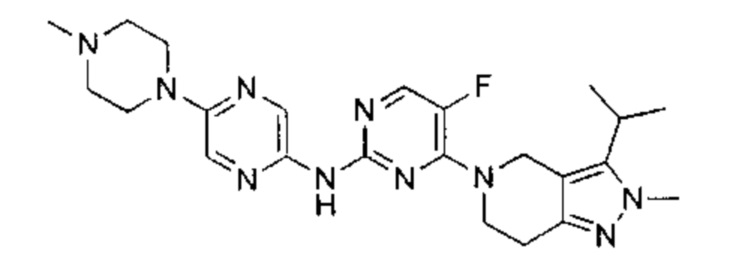
При 30°С к раствору 5-фтор-4-(3-изопропил-2-метил-6,7-дигидро-2Н-пиразоло [4,3-с]пиридин-5(4Н)-ил)-N-(5-(пиперазин-1-ил)пиразин-2-ил)пиримидин-2-амина (250,00 мг, 552,45 мкмоль, 1,00 эквивалент) в метаноле (25,00 мл) добавили NaBH3CN (69,43 мг, 1,10 ммоль, 2,00 эквивалента), НСНО (24,89 мг, 828,68 мкмоль, 1,50 эквивалента) и трифторуксусную кислоту (62,99 мг, 552,45 мкмоль, 1,00 эквивалент).Реакционную смесь перемешивали при 30°С в течение 4 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь упарили и высушили. Полученный неочищенный продукт очистили посредством препаративной ВЭЖХ (соляная кислота) и затем посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = 1:1), получив указанное в заголовке соединение (35,03 мг, 75,08 мкмоль, выход-13,59%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,88 (с., 1Н), 7,97 (д., J=1,13 Гц, 1Н), 7,93 (д., J=6,90 Гц, 1Н), 4,04 (т., J=5,84 Гц, 2Н), 3,75-3,79 (м., 3Н), 3,57-3,67 (м., 4Н), 3,12-3,22 (м., 1Н), 2,77-2,85 (м., 6Н), 2,52 (с., 3Н), 2,03 (с., 1Н), 1,33 (д., J=7,03 Гц, 6Н), 1,28-1,32 (м., 1Н). ЖХ/МС (ESI) м/з: 467,2 (М+1).
Пример 33
5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(4-метил-5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин

Этап 1: 5-бром-4-метил-2-нитропиридин
К H2SO4 (184,00 г, 1,88 моль, 23,39 эквивалента), охлажденной до 0°С, медленно по капле при температуре ниже -20°С добавили Н2О2 (58,06 г, 1,71 моль, 21,28 эквивалента). Добавили раствор 5-бром-4-метилпиридин-2-амина (15,00 г, 80,20 ммоль, 1,00 эквивалент) в H2SO4 (100,00 мл). Реакционную смесь перемешивали в течение 45 минут на ледяной бане и затем нагрели до 30°С. Через три часа цвет реакционного раствора постепенно изменился с травянистого зеленого на ярко-желтый. К реакционной смеси добавили лед (500 мл). Осажденное твердое вещество отфильтровали, промыли водой, затем высушили под вакуумом и таким образом получили указанное в заголовке соединение (12,00 г, 55,29 ммоль, выход - 68,95%), представляющее собой ярко-оранжевое твердое вещество, которое больше не очищали. ЖХ/МС (ESI) м/з: 216,9 (М+1), 218,9 (М+3).
Этап 2:
трет-бутил-4-(4-метил-6-нитропиридин-3-ил)пиперазин-1-карбоксилат
В атмосфере азота к раствору 5-бром-4-метил-2-нитропиридина (500,00 мг, 2,30 ммоль, 1,00 эквивалент), трет-бутип пиперазин-1-карбоксилата (514,05 мг, 2,76 ммоль, 1,20 эквивалента), BINAP (286,43 мг, 460,00 мкмоль, 0,20 эквивалент) и Cs2CO3 (1,05 г, 3,22 ммоль, 1,40 эквивалента) в диоксане (20,00 мл) добавили Pd2(dba)3 (210,62 мг, 230,00 мкмоль, 0,10 эквивалента). Реакционную смесь перемешивали при 110°С в течение 16 часов в атмосфере азота. ЖХ/МС показала, что реакция завершилась. Реакционную смесь охладили до 20°С и профильтровали. Фильтрат упарили, высушили и затем очистили посредством ТСХ (петролейный эфир: этилацетат = 4:1) и получили указанное в заголовке соединение (420,00 мг, 1,30 ммоль, выход - 56,65%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 323,0 (М+1).
Этап 3:
трет-бутил-4-(6-амино-4-метилпиридин-3-ил)пиперазин-1-карбоксилат
В атмосфере азота к раствору трет-бутил-4-(4-метил-6-нитропиридин-3-ил) пиперазин-1-карбоксилата (350,00 мг, 651,45 мкмоль, 1,00 эквивалент) в метаноле (15,00 мл) добавили 10% Pd-C (200 мг). Суспензию откачали и насытили водородом, затем перемешивали при 30°С в атмосфере Н2 (15 фунт/кв. дюйм) в течение 1,5 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, упарили и высушили. Полученный неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = 1:1) и получили указанное в заголовке соединение (125,00 мг, 427,53 мкмоль, выход - 65,63%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 293,2 (М+1).
Этап 4:
трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил-амино)-4-метилпиридин-3-ил)пиперазин-1-карбоксилат
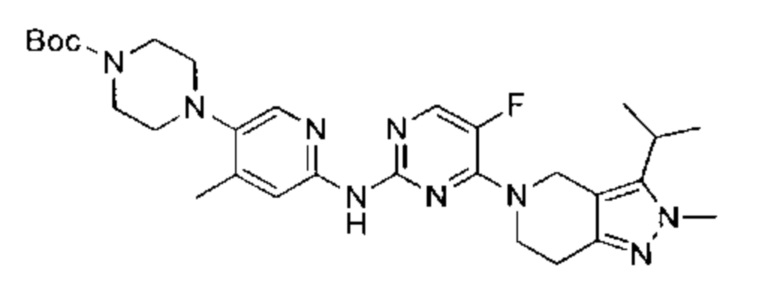
Смесь трет-бутил-4-(6-амино-4-метилпиридин-3-ил)пиперазин-1-карбоксилата (124,59 мг, 426,12 мкмоль, 1,10 эквивалента), 5-(2-хлор-5-фторпиримидин-4-ил)-3-изопропил-2-метил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридина (промежуточный продукт Е) (120,00 мг, 387,38 мкмоль, 1,00 эквивалент), Pd2(dba)3 (35,47 мг, 38,74 мкмоль, 0,10 эквивалента), Xantphos (44,83 мг, 77,48 мкмоль, 0,20 эквивалента) и Cs2CO3 (252,43 мг, 774,76 мкмоль, 2,00 эквивалента) в диоксане (20,00 мл) насытили N2. Реакционную смесь затем перемешивали при 110°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь профильтровали, упарили и высушили. Полученный неочищенный продукт разбавили этилацетатом (50 мл) и промыли водой (20 мл). Органическую фазу высушили в ротационной вакуумной сушилке и очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 1:1), получив указанное в заголовке соединение (150,00 мг, 265,17 мкмоль, выход - 68,45%), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 566,3 (М+1).
Этап 5: 5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(4-метил-5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин

К раствору трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-метил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-ил-амино)-4-метилпиридин-3-ил)пиперазин-1-карбоксилата (150,00 мг, 265,17 мкмоль, 1,00 эквивалент) в дихлорметане (7,50 мл) добавили трифторуксусную кислоту (11,52 г, 101,00 ммоль, 380,87 эквивалента). Реакционную смесь перемешивали при 30°С в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Смесь упарили и высушили. Полученный неочищенный продукт очистили посредством препаративной ВЭЖХ (муравьиная кислота) и получили указанное в заголовке соединение (121,00 мг, 236,52 мкмоль, выход - 89,19%), представляющее собой желтое масло. 1Н ЯМР (400 МГц, метанол-d4) δ 8,15 (д., J=7,2 Гц, 1Н), 8,05 (с., 1Н), 7,24 (с., 1H), 5,00 (с., 2Н), 4,16 (т., J=5,8 Гц, 2Н), 3,81 (с., 3Н), 3,51-3,39 (м., 4Н), 3,31-3,24 (м., 4Н), 3,24-3,16 (м., 1Н), 2,89 (т., J=5,8 Гц, 2Н), 2,55-2,45 (м., 3Н), 1,36 (д., J=7,2 Гц, 6Н). ЖХ/МС (ESI) м/з: 466,2 (М+1).
Подход Е
Синтез
5-фтор-4-(6,7-дигидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амина: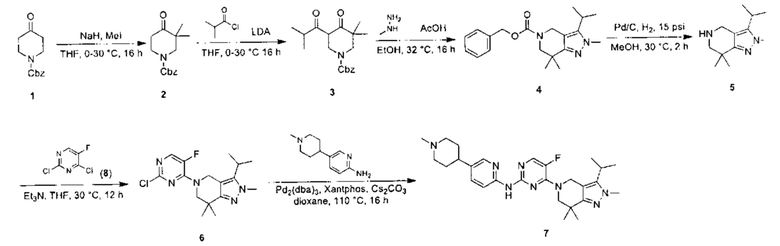
Пример 34
5-фтор-4-(6,7-дигидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амин
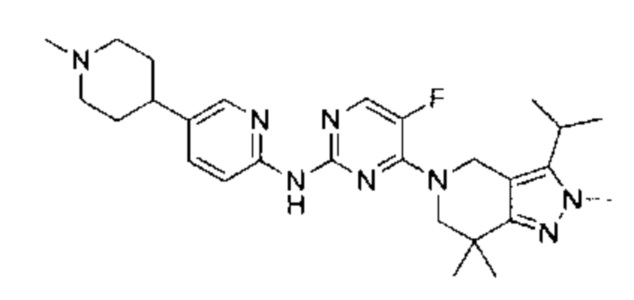
Этап 1: бензил-3,3-диметил-4-оксопиперидин-1-карбоксилат

При 0°С к раствору бензил-4-оксопиперидин-1-карбоксилата (2,00 г, 8,57 ммоль, 1,00 эквивалент) в тетрагидрофуране (25 мл) добавили одной порцией натрия гидрид (719,88 мг, 18,00 ммоль, 2,10 эквивалента) и калия йодид (3,80 г, 26,74 ммоль, 3,12 эквивалента). Смесь перемешивали при 30°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась. В реакционную смесь добавили раствор аммония хлорида (50 мл) и затем подвергли экстракции этилацетатом (50 мл × 2). Органическую фазу упарили и высушили. Полученный остаток очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 6:1) и получили указанное в заголовке соединение (870,00 мг, 3,33 ммоль, выход - 38,86%), представляющее собой оранжевое масло. 1Н ЯМР (400 МГц, метанол-d4) δ 7,38-7,33 (м., 5Н), 5,19 (с., 2Н), 3,81-3,77 (т., J=6,4 Гц, 2Н), 3,50-3,46 (м., 2Н), 2,50 (с., 2Н), 1,11-1,04 (м., 6Н). ЖХ/МС (ESI) м/з: 284,1 (М+1).
Этап 2:
бензил-5-(изобутирил)-3,3-диметил-4-оксопиперидин-1-карбоксилат

При 0°С к раствору бензил-3,3-диметил-4-оксопиперидин-1-карбоксилата (6,00 г, 22,96 ммоль, 1,00 эквивалент) в толуоле (150,00 мл) одной порцией добавили LDA (2М, 22,96 мл, 2,00 эквивалента). Затем реакционную смесь перемешивали при той же температуре в течение 30 минут и добавили изобутилхлорид (3,67 г, 34,44 ммоль, 1,50 эквивалента). Полученную смесь медленно нагрели до 30°С и перемешивали в течение 16 часов. ТСХ показала (петролейный эфир: этилацетат = 6:1), что реакция завершилась. Реакцию остановили раствором аммония хлорида (80 мл), затем провели экстракцию этилацетатом (100 мл × 2). Органические фазы объединили, упарили и высушили. Полученный неочищенный продукт очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = 50: 1) и получили указанное в заголовке соединение (5,60 г, 16,90 ммоль, выход - 73,60%), представляющее собой желтое масло.
Этап 3:
Бензил-6,7-дигидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-карбоксилат

К раствору бензил-5-(изобутирил)-3,3-диметил-4-оксопиперидин-1-карбоксилата (3,90 г, 11,77 ммоль, 1,00 эквивалент) и 40% водного раствора метилгидразина (13,99 г, 121,47 ммоль, 10,32 эквивалента) в этаноле (35,00 мл) одной порцией добавили уксусную кислоту (706,66 мг, 11,77 ммоль, 1,00 эквивалент). Реакционную смесь перемешивали при 32°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь упарили и высушили. Полученный остаток очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 5:1) и получили указанное в заголовке соединение (600,00 мг, 1,76 ммоль, выход - 14,93%), представляющее собой прозрачное масло. ЖХ/МС (ESI) м/з: 342,2 (М+1).
Этап 4:
3-изопропил-2,7,7-триметил-4,5,6,7-тетрагидро-2Н-пиразоло[4,3-с]пиридин
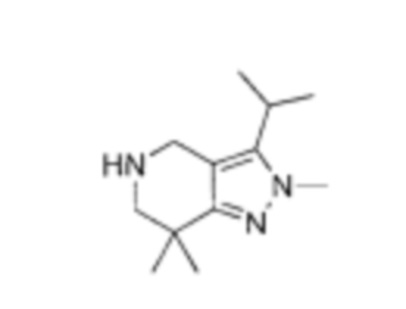
В атмосфере азота к раствору бензил-6,7-дигидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-карбоксилата (600,00 мг, 1,76 ммоль, 1,00 эквивалент) в метаноле (20,00 мл) добавили 10% Pd/C (0,4 г). Суспензию откачали и насытили водородом, затем перемешивали при 30°С в атмосфере Н2 (15 фунт/кв.дюйм) в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Смесь профильтровали, упарили и высушили, и получили указанное в заголовке соединение (323,00 мг, 1,56 ммоль, выход - 88,53%), представляющее собой желтое масло. ЖХ/МС (ESI) м/з: 208,2 (М+1).
Этап 5:
5-(2-хлор-5-фторпиримидин-4-ил)-4,5,6,7-тетрагидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин
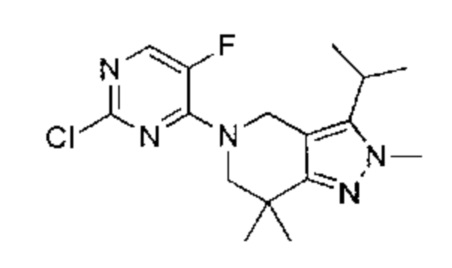
При 30°С к раствору 3-изопропил-2,7,7-триметил-4,5,6,7-тетрагидро-2Н-пиразоло-[4,3-с]пиридина (323,00 мг, 1,56 ммоль, 1,00 эквивалент) и 2,4-дихлор-5-фторпиримидина (338,62 мг, 2,03 ммоль, 1,30 эквивалента) в тетрагидрофуране (25,00 мл) добавили триэтиламин (1,58 г, 15,58 ммоль, 10,00 эквивалента). Реакционную смесь перемешивали при 30°С в течение 12 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь разбавили насыщенным солевым раствором (20 мл) и затем подвергли экстракции этилацетатом (50 мл × 2). Органические фазы объединили и высушили. Полученный остаток очистили посредством препаративной ТСХ (петролейный эфир: этилацетат = 4:1) и получили указанное в заголовке соединение (380,00 мг, 1,12 ммоль, выход - 72,11%), представляющее собой желтое масло. ЖХ/МС (ESI) м/з: 338,1 (М+1).
Этап 6:
5-фтор-4-(6,7-дигидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амин
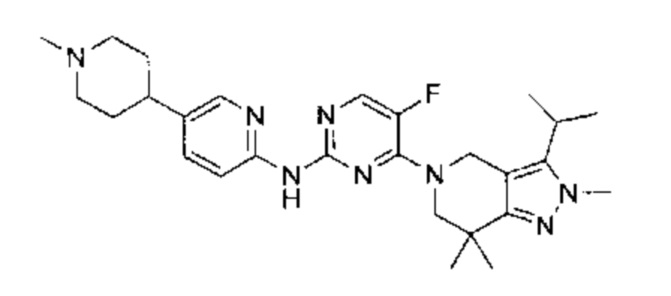
В атмосфере азота к смеси 5-(2-хлор-5-фторпиримидин-4-ил)-4,5,6,7-тетрагидро-3-изопропил-2,7,7-триметил-2Н-пиразоло[4,3-с]пиридина (380,00 мг, 1,12 ммоль, 1,00 эквивалент), 5-(1-метилпиперидин-4-ил)пиридин-2-амина (257,07 мг, 1,34 ммоль, 1,20 эквивалента) и Cs2CO3 (729,84 мг, 2,24 ммоль, 2,00 эквивалента) в диоксане (25,00 мл) добавили одной порцией Pd2(dba)3 (102,56 мг, 112,00 ммоль, 0,10 эквивалента) и Xantphos (129,61 мг, 224,00 мкмоль, 0,20 эквивалента). Реакционную смесь насытили N2 и и затем перемешивали при 110°С в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь разбавили водой (10 мл) и затем подвергли экстракции этилацетатом (50 мл ×2). Органические фазы объединили и высушили. Полученный остаток очистили посредством препаративной ВЭЖХ (соляная кислота), затем очистили посредством сверхкритической флюидной хроматографии (СФХ) (AD-3S_5_40_3 мл, колонка: Chiralpak AD-3100×4,6 мм, внутр. диам. 3 мкм, подвижная фаза: 40% этанола (0,05% диаэтаноламина) в СО2, скорость потока: 3 мл/мин, длина волны: 220 нм) и получили указанное в заголовке соединение (60,00 мг, 121,80 мкмоль, выход - 10,87%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,36 (с., 1Н), 8,30 (д., J=6,8 Гц, 1Н), 8,27-8,19 (м., 1H), 7,56-7,48 (м., 1Н), 5,09 (с., 2Н), 4,12 (с., 3Н), 4,05 (с., 2Н), 3,73-3,60 (м., 2Н), 3,44-3,35 (м., 1Н), 3,31-3,25 (м., 1Н), 3,21-3,09 (м., 1Н), 2,96 (с., 3Н), 2.35 (с., 1Н), 2,26-2,08 (м., 4Н), 1,51-1,44 (м., 12Н). ЖХ/МС (ESI) м/з: 493,3 (М+1).
Подход F
Синтез
N-(5-(1,4-диазепан-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амина

Пример 35
N-(5-(1,4-диазепан-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин
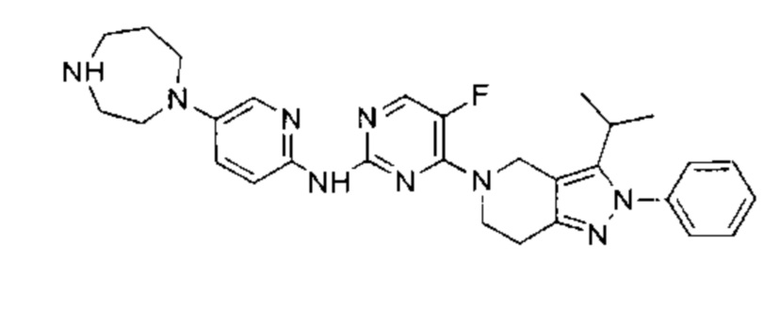
Этап 1:
Бензил-6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-карбоксилат
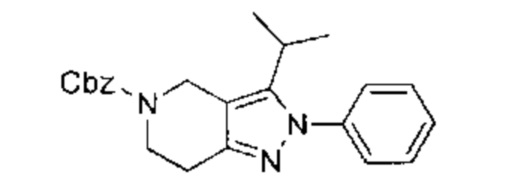
К раствору бензил-3-(2-метилпропионил)-4-оксопиперидин-1-карбоксилата (500,00 мг, 1,65 ммоль, 1,00 эквивалент) в этаноле (5,00 мл) добавили фенилгидразин (178,24 мг, 1,65 ммоль, 1,00 эквивалент). Реакционную смесь нагрели до 80°С и перемешивали при этой температуре в течение 4 часов. ТСХ (петролейный эфир: этилацетат = 2:1) и ЖХ/МС показали, что реакция завершилась. Реакционную смесь охладили до 25°С и затем упарили под пониженным давлением, чтобы удалить растворитель. Полученный остаток очистили посредством колоночной хроматографии с силикагелем (петролейный эфир: этилацетат = 5:1) и получили указанное в заголовке соединение (330,00 мг, 878,92 мкмоль, выход - 53,27%), представляющее собой красное масло. 1Н ЯМР (400 МГц, CDCl3) δ 7,50-7,36 (м., 10Н), 5,23 (с., 2Н), 4,70 (с., 2Н), 3,83 (ушир. с., 2Н), 3,07 (кв., J=7,0 Гц, 1Н), 2,86 (ушир. с., 2Н), 1,28-1,20 (м., 6Н). ЖХ/МС (ESI) м/з: 376,1 (М+1).
Этап 2:
4,5,6,7-тетрагидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин
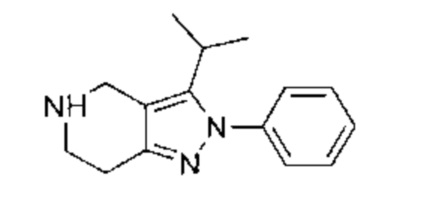
К раствору бензил-6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло-[4,3-с]пиридин-5(4Н)-карбоксила та (330,0 мг, 878,92 мкмоль, 1,00 эквивалент) в метаноле (5,00 мл) добавили 10% палладиевый катализатор на углеродном носителе (50 мг). Реакционную смесь перемешивали при 25°С в течение 2 часов в атмосфере Н2 (15 фунт/кв.дюйм). ТСХ показала (дихлорметан: метанол = 10:1), что реакция завершилась. Реакционную смесь профильтровали, фильтрат упарили и получили указанное в заголовке соединение (200,00 мг, неочищенный продукт), представляющее собой желтое масло.
Этап 3:
5-(2-хлор-5-фторпиримидин-4-ил)-4,5,6,7-тетрагидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин

К раствору 4,5,6,7-тетрагидро-3-изопропил-2-фенил-2Н-пиразоло-[4,3-с]пиридина (200,00 мг, 828,74 мкмоль, 1,00 эквивалент) в тетрагидрофуране (5,00 мл) добавили триэтиламин (167,72 мг, 1,66 ммоль, 2,00 эквивалента) и 2,4-дихлор-5-фтор-пиримидин (166,05 мг, 994,49 мкмоль, 1,20 эквивалента). Реакционную смесь перемешивали при 25°С в течение 2 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь упарили и высушили. Полученный остаток очистили посредством препаративной ТСХ (петоолейный эфир: этилацетат = 2:1) и получили указанное в заголовке соединение (190,00 мг, 510,97 мкмоль, выход - 61,66%), представляющее собой белое твердое вещество. 1Н ЯМР (400 МГц, CDCl3) δ 7,99 (д., J=6,0 Гц, 1Н), 7,52-7,43 (м., 3Н), 7,40-7,36 (м., 2Н), 4,97 (с., 2Н), 4,12 (т., J=5,9 Гц, 2Н), 3,10 (кв., J=7,1 Гц, 1Н), 2,99 (т., J=5,9 Гц, 2Н), 1,30 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 372,0 (М+1).
Этап 4:
трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-аминопиридин-3-ил)-1,4-диазепан-1-карбоксилат

В атмосфере азота к раствору 5-(2-хлор-5-фторпиримидин-4-ил)-4,5,6,7-тетрагидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридина (190,00 мг, 510,97 мкмоль, 1,00 эквивалент), и трет-бутил
4-(6-амино-3-пиридил)-1,4-диазепан-1-карбоксилата (224,10 мг, 766,46 мкмоль, 1,50 эквивалента) в диоксане (5,00 мл) добавили Cs2CO3 (499,45 мг, 1,53 ммоль, 3,00 эквивалента), Xantphos (59,13 мг, 102,19 мкмоль, 0,20 эквивалента), и Pd2(dba)3 (46,79 мг, 51,10 мкмоль, 0,10 эквивалента). Реакционную смесь нагрели до 120°С и перемешивали при этой температуре в течение 16 часов. ЖХ/МС показала, что реакция завершилась. Реакционную смесь профильтровали, и фильтрат упарили. Полученный остаток очистили посредством препаративной ТСХ (чистый этилацетат) и получили указанное в заголовке соединение (100,00 мг, неочищенный продукта), представляющее собой желтое твердое вещество. ЖХ/МС (ESI) м/з: 628,3 (М+1).
Этап 5:
N-(5-(1,4-диазепан-1-ил)пиридин-2-ил)-5-фтор-4-(6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-амин

К раствору трет-бутил-4-(6-(5-фтор-4-(6,7-дигидро-3-изопропил-2-фенил-2Н-пиразоло-[4,3-с]пиридин-5(4Н)-ил)пиримидин-2-аминопиридин-3-ил)-1,4-диазепан-1-карбоксилата (100,00 мг, 159,30 мкмоль, 1,00 эквивалент) в дихлорметане (3,00 мл) добавили трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при 25°С в течение 0,5 часа. ЖХ/МС показала, что реакция прошла полностью. Реакционную смесь упарили под пониженным давлением, чтобы удалить растворитель. Полученный остаток очистили посредством препаративной ВЭЖХ (соляная кислота) и получили указанное в заголовке соединение (50,00 мг, 94,76 мкмоль, выход - 59,49%). 1Н ЯМР (400 МГц, метанол-d4) δ 8,24 (д., J=6,8 Гц, 1Н), 7,92 (дд, J=9,4, 2,8 Гц, 1H), 7,86 (д., J=2,6 Гц, 1Н), 7,73-7,68 (м., 3Н), 7,65-7,57 (м., 2Н), 7,39 (д., J=9,5 Гц, 1Н), 5,17 (с., 2Н), 4,31 (т., J=5,0 Гц, 2Н), 3,92-3,84 (м., 2Н), 3,65 (т., J=6,0 Гц, 2Н), 3,52-3,45 (м., 2Н), 3,41-3,36 (м., 2Н), 3,14 (т., J=5,1 Гц, 2Н), 3,06 (тд., J=14,0, 6,9 Гц, 1Н), 2,33-2,24 (м., 2Н), 1,37 (д., J=7,0 Гц, 6Н). ЖХ/МС (ESI) м/з: 528,2 (М+1)
Фармакологические эксперименты
Вещества настоящего изобретения представляют собой селективные ингибиторы CDK4/6, например, их активность против CDK2 меньше, чем против CDK4/6 (по результатам сравнения ИК50). Результаты следующих экспериментов подтверждают, что вещества, перечисленные в настоящем изобретении, являются специфическими ингибиторами CDK4/6 и потенциально могут использоваться в качестве противоопухолевых средств. В настоящем документе ИК50 означает концентрацию средства, при котором проявляется 50% его максимальной ингибирующей способности. Вещества настоящего изобретения также обладают непредсказуемой биологической активностью. Например, вещество из Примера 13 не было субстратом эффлюксного транспортера, в то время как доказано, что палбоциклиб и LY2835219 являются таковыми. Кроме того, воздействие вещества из Примера 13 на ткани головного мозга было выражено намного больше, чем таковое палбоциклиба или LY2835219. Можно предположить, что вещество из Примера 13, в сравнении с палбоциклибом и LY2835219, представляет собой более перспективное средство лечения рака молочной железы с метастазами в головной мозг, что составляет около 15% случаев рака молочной железы. Перечисленные здесь биологические активности представляют собой биологическую активность лишь некоторых отдельных примеров.
Биохимические эксперименты
Экспериментальные материалы:
Экспериментальные материалы включали CDK2/циклин А, CDK4/циклин D1, CDK6/циклин D1 (Life Technologies); ULight-меченые полипептидные субстраты, а именно, ULight-4E-BP1 и ULight-MBP (PerkinElmer); меченые европием антитела к основному белку миелина и меченые европием кроличьи антитела (PerkinElmer); и Envision® многоканальный планшет-ридер (PerkinElmer) для детектирования сингала.
Экспериментальный метод:
Испытуемые вещества разбавляли три раза, чтобы получить десять постепенно уменьшающихся концентраций от 5 мкМ до 0,25 нМ.
• Ферментная реакционная система CDK2/циклин А
В ферментной реакционной системе объемом 10 мкл, содержащей 0,5 нМ белка CDK2/циклин А, 100 нМ полипептида ULight-MBP (Myelin Basic Protein, основной белок миелина) и 25 мкМ АТФ, выполнили анализ по стандартному методу Lance Ultra. В буферный раствор с ферментом добавляли последовательные разведения изучаемых веществ. Буферный раствор содержал 50 мМ раствора гидроксиэтилпиперазинэтансульфоновой кислоты (рН=7,5), 1 мМ этилендиаминтетрауксусной кислоты, 10 мМ магния хлорида, 0,01% Brij-35 и 2 мМ дитиотреитола. После начала реакции планшет OptiPlate на 384 лунок закрыли сверху самоклеющейся пленкой TopSeal-A и инкубировали при комнатной температуре в течение 60 минут.
• Ферментная реакционная система CDK4/циклин D1
В ферментной реакционной системе объемом 10 мкл, содержащей 0,3 нМ белка CDK4/циклин D1, 50 нМ полипептида ULight-4E-BP1 и 350 мкМ АТФ, выполнили анализ по стандартному методу Lance Ultra. В буферный раствор с ферментом добавляли разведения изучаемых веществ. Буферный раствор содержал 50 мМ раствора гидроксиэтилпиперазинэтансульфоновой кислоты (рН=7,5), 1 мМ этилендиаминтетрауксусной кислоты, 10 мМ магния хлорида, 0,01% Brij-35 и 2 мМ дитиотреитола. После начала реакции планшет OptiPlate на 384 лунок закрыли сверху самоклеющейся пленкой TopSeal-A и инкубировали при комнатной температуре в течение 180 минут.
• Ферментная реакционная система CDK6/циклин D1
В ферментной реакционной системе объемом 10 мкл, содержащей 0,8 нМ белка CDK6/циклин D1, 50 нМ полипептида ULight-4E-BP1 и 250 мкМ АТФ, выполнили анализ по стандартному методу Lance Ultra. В буферный раствор с ферментом добавляли разведения изучаемых веществ. Буферный раствор содержал 50 мМ раствора гидроксиэтилпиперазинэтансульфоновой кислоты (рН=7,5), 1 мМ этилендиаминтетрауксусной кислоты, 10 мМ магния хлорида, 0,01% Brij-35 и 2 мМ дитиотреитола. После начала реакции планшет OptiPlate на 384 лунок закрыли сверху самоклеющейся пленкой TopSeal-A и инкубировали при комнатной температуре в течение 180 минут.
Буфер для остановки ферментативной реакции приготовили путем растворения ЭДТА в однократном разведении буфера для анализа. Реакцию останавливали при комнатной температуре в течение 5 минут. К каждой из реакционной систем CDK2/циклин А, CDK-4/циклин D1 и CDK6/циклин D1 добавили по 5 мкл смеси для анализа (содержащей антитела к основному белку миелина, меченые европием, и кроличьи антитела, меченые европием, соответственно). Инкубировали при комнатной температуре в течение 60 минут. Сигналы прохождения реакции детектировали инструментом En Vision на основании принципа времяразрешенного флуоресцентного индуктивно-резонансного переноса энергии.
Анализ данных:
Исходные данные перевели в степень ингибирования по уравнению (макс.-отношение)/(макс.-мин.)*100%. Значения IC50 получили путем подбора аппроксимирующей кривой по четырем параметрам (полученным в режиме 205 в XLFIT5, iDBS). В таблице 1 представлена ингибирующая активность веществ изобретения по отношению к киназам CDK2, CDK4 и CDK6.
Эксперименты с культурами клеток
Экспериментальные материалы:
Среда RPMI 1640, эмбриональная телячья сыворотка, пенициллин/стептомицин приобрели в компании Promega (г. Мадисон, шт. Висконсин). Линии клеток MCF-7 и MDA-MB-436 приобрели в Европейской коллекции культур клеток (ЕСАСС). Использовали многоканальный планшет-ридер En Vision® (PerkinElmer).
Экспериментальный метод:
Клетки MCF-7 инокулировали в черный планшет на 384 лунок, добавив в каждую лунку по 45 мкл суспензии клеток (содержащей 200 клеток MCF-7). Планшет инкубировали в течение ночи в СО2-инкубаторе.
Испытуемые вещества три раза разводили Bravo до получения десятой концентрации, т.е. разбавили от 10 мкМ до 0,508 нМ. В отдельных лунках провели контрольное испытание. В центральный планшет добавили по 49 мкл среды на лунку. В центральный планшет в соответствующие положения добавили по 1 мкл последовательных разведений веществ, и в планшет с клетками добавили перемешанную смесь в количестве 5 мкл на лунку. Планшет с клетками инкубировали в CO2-инкубаторе в течение 6 дней.
Также в каждую лунку планшета с клетками добавили по 25 мкл реактива Promega CellTiter-Glo. Планшет с клетками инкубировали при комнатной температуре в течение 10 минут, чтобы стабилизировать люминисцентный сигнал. Для детектирования сигнала использовали многоканальный планшет-ридер En Vision® (PerkinElmer).
Анализ данных:
Исходные данные перевели в степень ингибирования по уравнению (макс.-отношение)/(макс.-мин.)*100%. Значения IC50 получили путем подбора аппроксимирующей кривой по четырем параметрам (полученным в режиме 205 в XLFIT5, iDBS). В таблице 1 представлена ингибирующая активность веществ изобретения по отношению к пролиферации клеток MCF-7 и MDA-MB-436.
Вывод по результатам эксперимента: Вещества настоящего изобретения обладают значимой ингибирующей активностью по отношению к CDK4 и CDK6 и высокоселективны по отношению к CDK2. Кроме того, вещества настоящего изобретения обладают значимой ингибирующей активностью по отношению к пролиферации клеток рака молочной железы MCF-7, экспрессирующим рецепторы к эстрогенам, и обладают низкой ингибирующей активностью по отношению к пролиферации не экпрессирующих такие рецепторы клеток MDA-MB-436. Вещества из примеров 3, 9, 10 и 15 обладали более высокой ингибирующей активность по отношению к CDK4 и CDK6, чем эталонные вещества палбоциклиб и LY2835219. Вещества из примеров 3, 7, 9, 10 и 15 обладали более высокой ингибирующей активность по отношению к пролиферации клеток MCF-7, чем эталонные вещества палбоциклиб и LY2835219. Вещество из примера 7 обладало более селективной способностью подавлять пролиферацию клеток рака молочной железы, экспрессирующих рецепторы к эстрогенам, чем эталонное вещество LY2835219 (MCF-7/MDA-MB-436).

Эксперимент по оценке двустороннего проникновения через мембрану клеток Сасо-2
Цель эксперимента:
Клетки рака толстой кишки человека Сасо-2 - модель in vitro, широко применяемая для изучения всасывания в тонком кишечнике. Модель монослоя клеток Сасо-2 широко используется для оценки пассивного и активного транспорта в процессе всасывания в тонком кишечнике. GF120918A - сильный ингибитор эффлюксных транспортеров, которые содержат Р-гликопротеин (P-gp), белок резистентности рака молочной железы (BCRP) и другие. В этом эксперименте определяли способность вещества из примера 7, а также эталонных веществ палбоциклиба и LY2835219 проникать в обоих направлениях через мембрану клеток Сасо-2 и определяли эффлюксные транспортеры испытуемых веществ путем добавления GF120918A.
Методика эксперимента:
Стандартные условия проведения эксперимента:
- испытуемая концентрация: 2 мкМ (ДМСО ≥ 1%);
- количество повторностей: n=2;
- направление: в обоих направлениях, т.е. А→В и В→А;
- продолжительность инкубации: одна временная точка, 2 часа;
- транспортный буфер: HBSS (сбалансированный солевой раствор Хэнкса), рН=7,4;
- условия инкубации: 37°С, 5% СО2.
После инкубации образцы растворов, отобранных из лунок для дозирования и принимающих лунок, немедленно смешали с холодным раствором ацетонитрила, содержащим внутренний стандарт. Концентрации испытуемых веществ во всех образцах (в том числе исходных дозируемых растворов, супернатанта из лунок для дозирования и растворов из принимающих лунок) определяли по методу ЖХ/МС/МС. Рассчитали кажущиеся коэффициенты проницаемости, коэффициенты двусторонней проницаемости и другие параметры.
Вывод по результатам эксперимента:
В таблице 2 указаны коэффициенты проницаемости вещества из примера 7 и эталонных веществ палбоциклиба и LY2835219 в монослое клеток Сасо-2. Вещество из примера 7 обладало большей проницаемостью, чем эталонные вещества палбоциклиб и LY2835219; к тому же, менее вероятно, что на его всасывание и транспорт in vivo окажут влияние эффлюксные транспортеры. Более высокая проницаемость может способствовать тому, что вещество из примера 7 будет лучше распределяться в ткани организма (например, в ткани легкого и молочной железы) и будет обладать более эффективным противоопухолевым действием in vivo. Возможно, благодаря более высокой проницаемости вещество из примера 7 также будет лучше проникать через гематоэнцефалический барьер и таким образом способствовать лечению метастазов рака молочной железы или легкого.

Исследования проникновения в головной мозг
Цель эксперимента:
В качестве испытуемых животных взяли крыс Спраг-Доули. Для оценки распределения вещества из примера 7 и эталонных веществ палбоциклиба и LY2835219 в тканях головного мозга измеряли их концентрации в головном мозге и в плазме крови после перорального введения в различных временных точках определяли с помощью ЖХ/МС/МС.
Методика эксперимента:
В компании Shanghai Slack Laboratory Animal Co., Ltd. приобрели двенадцать здоровых самцов крыс Спраг-Доули возрастом от 7 до 9 месяцев. Приготовили суспензию испытуемого вещества в 0,5% метилцеллюлозе с концентрацией 1 мг/мл, взяв соответствующую навеску. Крысы получили перорально разовую дозу 10 мг/кг. Через 0,5 ч, 2 ч, 8 ч и 24 ч после введения умерщвляли по 3 крысы, у которых отбирали цельную кровь и ткани головного мозга. Плазму получали путем центрифугирования цельной крови (3000 г, 15 минут, 4°С). Образцы гомогената тканей головного мозга готовили путем смешивания тканей со смесью метанола и фосфатного буфера (метанол: фосфатный буфер = 1:2) в соотношении 5: 1 и гомогенизации. Концентрацию лекарственных средств в каждом образце определяли с помощью ЖХ/МС/МС. Для оценки воздействия рассчитывали площадь под кривой (AUC).
Вывод по результатам эксперимента:
В таблице 3 показано воздействие вещества из примера 7 и эталонных веществ палбоциклиба и LY2835219 на ткань головного мозга при пероральной дозе 10 мг/кг. Видно, что воздействие вещества из примера 7 на ткани головного мозга крыс превышало таковое эталонных веществ палбоциклиба и LY2835219 в двух временных точках, 0,5 часа и 2 часа, т.е. вещество из примера 7 потенциально обладает лучшей возможностью лечения раковых метастазов в головной мозг. В то же время, воздействие вещества из примера 7 настоящего изобретения на головной мозг значимо снизилось через 8 часов и в числовом выражении находилось между воздействием эталонных веществ палбоциклиба и LY2835219, т.е. вещество из примера 7 настоящего изобретения не будет надолго задерживаться в головном мозге и более безопасно.

Исследование фармакодинамики in vivo
Исследование фармакодинамики in vivo проводили у голых мышей BALB/c ксенотрансплантатом, полученным из человеческой линии опухолевых клеток, которым под кожу имплантировали клетки рака молочной железы человека линии MCF-7.
Методика эксперимента:
Самок голых мышей BALB/c возрастом от 6 до 8 недель и весом 18-22 г содержали в отдельных вентилируемых клетках (по 10 мышей в клетке) в специальной стерильной среде. Во всех клетках перед использованием дезинфицировали подстилки и воду. У всех животных был свободный доступ к стандартному сертифицированному лабораторному корму, имеющемуся на рынке. Всего в компании Shanghai Slack Laboratory Animal Co., LTD для исследования приобрели 100 мышей. Каждой мыши имплантировали таблетку с эстрогенами (0,36 г, замедленное высвобождение в течение 60 дней) под кожу левой части живота. Через 3 дня каждой мыши имплантировали опухолевые клетки (10×106 в 0,2 мл фосфатного буфера) под кожу правой части живота для роста опухоли. Препараты начали вводить, когда средний объем опухоли достиг около 150-200 мм3. Испытуемые вещества вводили перорально ежедневно в дозах, показанных в таблице 5. Объем опухоли измеряли с помощью двухкоординатного штангенциркуля каждые три дня, выражали в кубических миллиметрах и рассчитывали по формуле V=0,5a×b2, где а и b - наибольший и наименьший диаметр опухоли, соответственно. Эффективность противоопухолевого действия определяли путем деления среднего увеличения объема опухоли животных, получавших вещества, на увеличение объема опухоли контрольных животных.
Вывод по результатам эксперимента:
Вещества из примеров 3 и 7 настоящего изобретения оказывали значимую противоопухолевую активность в модели рака молочной железы у мышей MCF-7 с ксенотрансплантатом, полученным из человеческой линии опухолевых клеток. Как показано в таблице 4, через 21 день после начала эксперимента объем опухоли в контрольной группе животных быстро вырос с исходных 187 мм3 до 1443 мм3, в то время как объем опухоли в группе животных, которые получали вещество из примера 3, вырос всего лишь с 187 мм3 до 432 мм3, и в группе, получавшей вещество из примера 3, опухоли росли значимо медленнее, чем в группах, получавших эталонные вещества палбоциклиб и LY2835219. На основании того, что использованная доза вещества из примера 3 (25 мг/кг) была вполовину меньше таковой LY2835219 (50 мг/кг) и почти вполовину меньше таковой палбоциклиба (45 мг/кг) (было показано, что животные не переносят более высокие дозы палбоциклиба), мы считаем, что противоопухолевая активность вещества из примера 3 в значимой степени превосходит таковую двух эталонных веществ. В то же время через 21 после введения объем опухоли в группе, которая получала вещество из примера 7, медленно увеличился с 187 мм3 до 350 м3, и опухоли росли значимо медленнее, чем в группе, которая получала ту же дозу эталонного вещества LY2835219, то есть вещество из примера 7 обладает более выраженной противоопухолевой активностью, чем эталонное вещество LY2835219 в той же дозе.
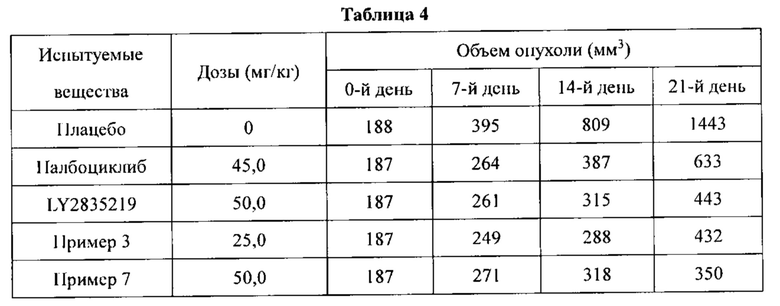
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ГИРАЗЫ | 2012 |
|
RU2626979C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА ИЛИ ПИРРОЛОПИРИДИН/ПИРИМИДИН-2-ОНА | 2014 |
|
RU2666532C2 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА | 2020 |
|
RU2803284C1 |
Изобретение относится к новому замещенному производному 2Н-пиразолу, выступающему в роли селективного ингибитора CDK4/6. В частности, раскрытое изобретение представляет собой вещество с формулой (I) или его фармацевтически приемлемую соль, которая выступает в качестве селективного ингибитора CDK4/6.
2 н. и 16 з.п. ф-лы, 4 табл., 35 пр.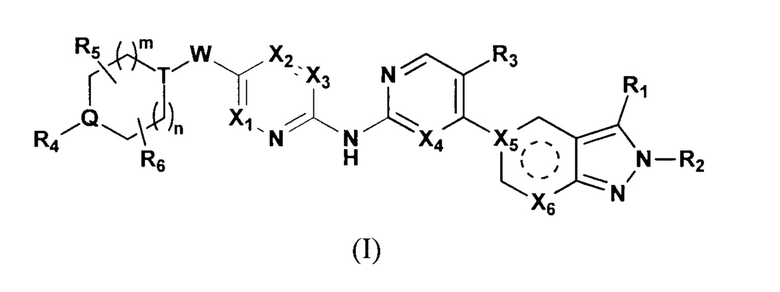
1. Замещенное производное 2Н-пиразола, имеющее формулу (I) или его фармацевтически приемлемая соль
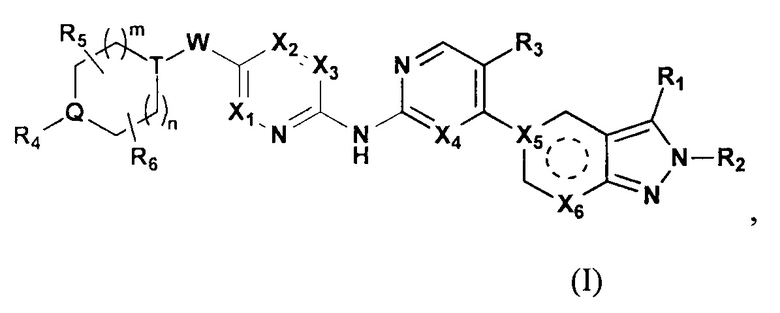
где 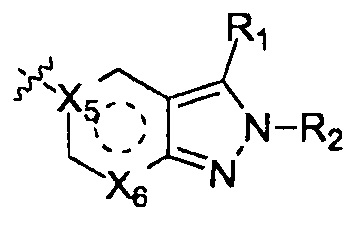 выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и 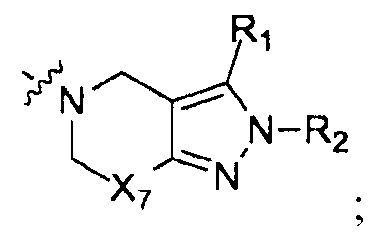
R1 выбирают из группы, состоящей из C1-8 алкила и С3 алкенила;
R2 выбирают из группы, состоящей из C1-8 алкила и фенила;
R3 выбирают из группы, состоящей из Н, галогена, -N(R8)(R9) и C1-3 алкила;
R4, R5 и R6 независимо друг от друга выбирают из группы, состоящей из Н, галогена, ОH, NH2, CN и =O, или выбирают из группы, состоящей из C1-8 алкила, C1-8 алкиламино, N,N-ди(C1-8 алкил)амино и 3-7-членной гетероциклоалкильной группы, каждый из который необязательно замещен одним, двумя или тремя R;
в некоторых случаях любые два радикала R4, R5 или R6 могут образовать кольцо из 3-7 атомов;
R7 выбирают из группы, состоящей из Н и галогена;
X1, Х2, Х3 и Х4 независимо друг от друга выбирают из группы, состоящей из N и C(R10);
Х7 выбирают из группы, состоящей из карбонила и C(R11)(R12);
W представляет собой простую связь;
Т выбирают из группы, состоящей из N и C(R10);
Q выбирают из группы, состоящей из N и C(R10);
m и n независимо друг от друга выбирают из группы, состоящей из 0, 1 и 2;
R8 и R9 независимо друг от друга выбирают из группы, состоящей из Н, С1-8 алкила;
R выбирают из группы, состоящей из F, Cl, Br, I, NH2, CN, ОН, CF3, CHF2, CH2F, NHCH3 и N(СН3)2;
R10 выбирают из группы, состоящей из Н, галогена, ОН, NH2, CN, C1-6 алкила и С1-6 алкоксила;
R11 и R12 независимо друг от друга выбирают из группы, состоящей из Н и C1-8 алкила;
в некоторых случаях R4 и R10 присоединяются к одному и тому же атому и образуют 3-7-членное кольцо, и
в некоторых случаях структурная единица  может быть замещена
может быть замещена 
2. Производное по п. 1 или его фармацевтически приемлемая соль, в которых структурная единица  замещена
замещена 
3. Производное по п. 1 или его фармацевтически приемлемая соль, в которых R1 выбирают из группы, состоящей из изопропила, 2-пропенила и аллила.
4. Производное по п. 1 или его фармацевтически приемлемая соль, в которых R1 выбирают из группы, состоящей из изопропила.
5. Производное по п. 1 или его фармацевтически приемлемая соль, в которых R2 представляет собой метил.
6. Производное по п. 1 или его фармацевтически приемлемая соль, в котором R3 - это F.
7. Производное по п. 1 или его фармацевтически приемлемая соль, в которых
 представляет собой
представляет собой 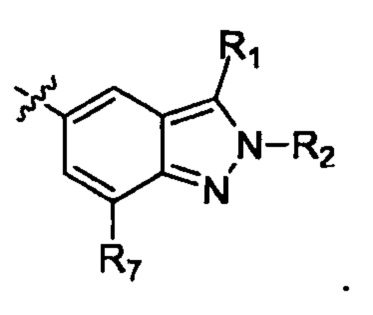
8. Производное по п. 1 или его фармацевтически приемлемая соль, где R4, R5 и R6 независимо друг от друга выбирают из группы, состоящей из Н, галогена, ОН, NH2, 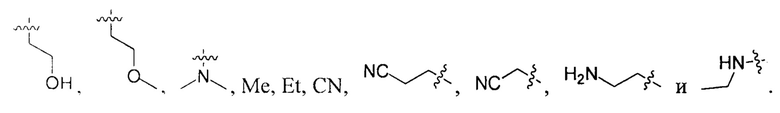
9. Производное по п. 1 или его фармацевтически приемлемая соль, в которых R7 выбирают из группы, состоящей из Н, F и Cl.
10. Производное по п. 1 или его фармацевтически приемлемая соль, в которых структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
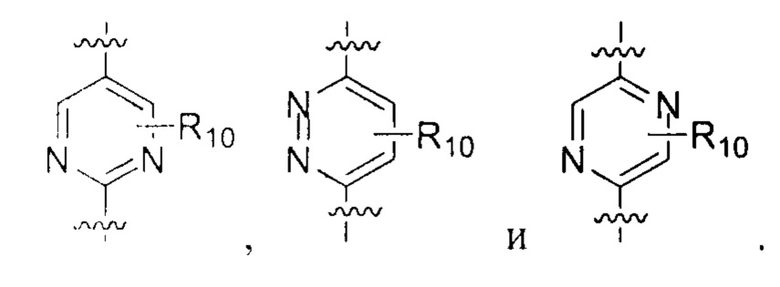
11. Производное по п. 1 или его фармацевтически приемлемая соль, в которых R10 выбирают из группы, состоящей из Н, ОН, NH2, F, Cl, CN,  и Me.
и Me.
12. Производное по п. 1 или его фармацевтически приемлемая соль, в которых Х4 выбирают из группы, состоящей из N и СН.
13. Производное по п. 1 или его фармацевтически приемлемая соль, в которых структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 

14. Производное по п. 1 или его фармацевтически приемлемая соль, в которых структурную единицу 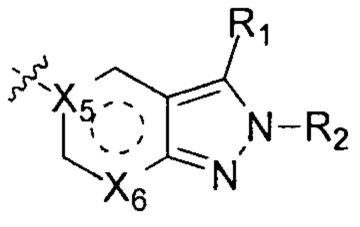 выбирают из группы, состоящей из
выбирают из группы, состоящей из 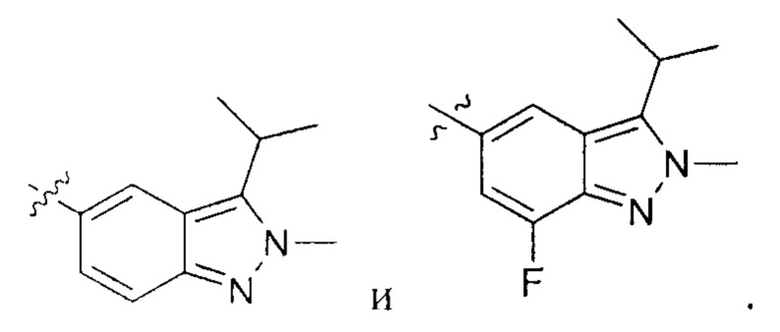
15. Производное по п. 1 или его фармацевтически приемлемая соль, в которых структурную единицу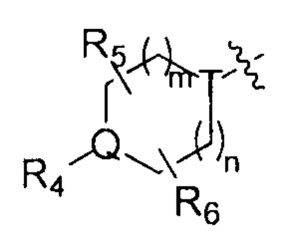 выбирают из группы, состоящей из
выбирают из группы, состоящей из 

16. Производное по п. 1 или его фармацевтически приемлемая соль, в которых
структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 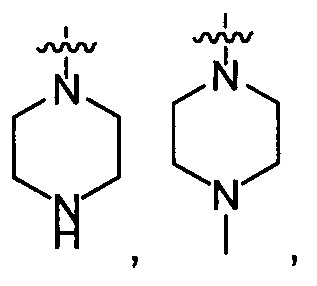
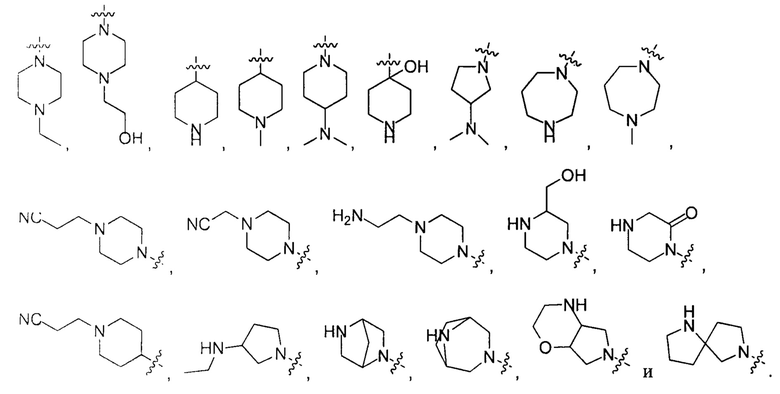
17. Производное по п. 1 или его фармацевтически приемлемая соль, выбранная из группы, состоящей из
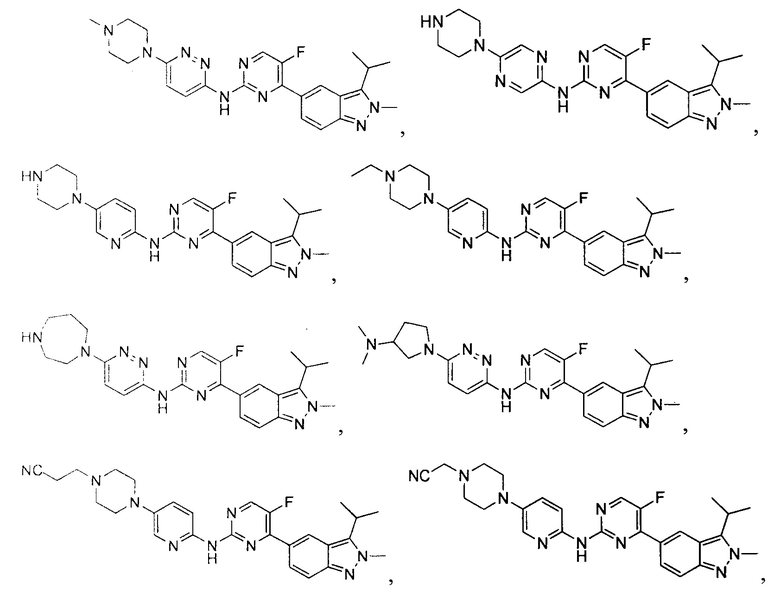

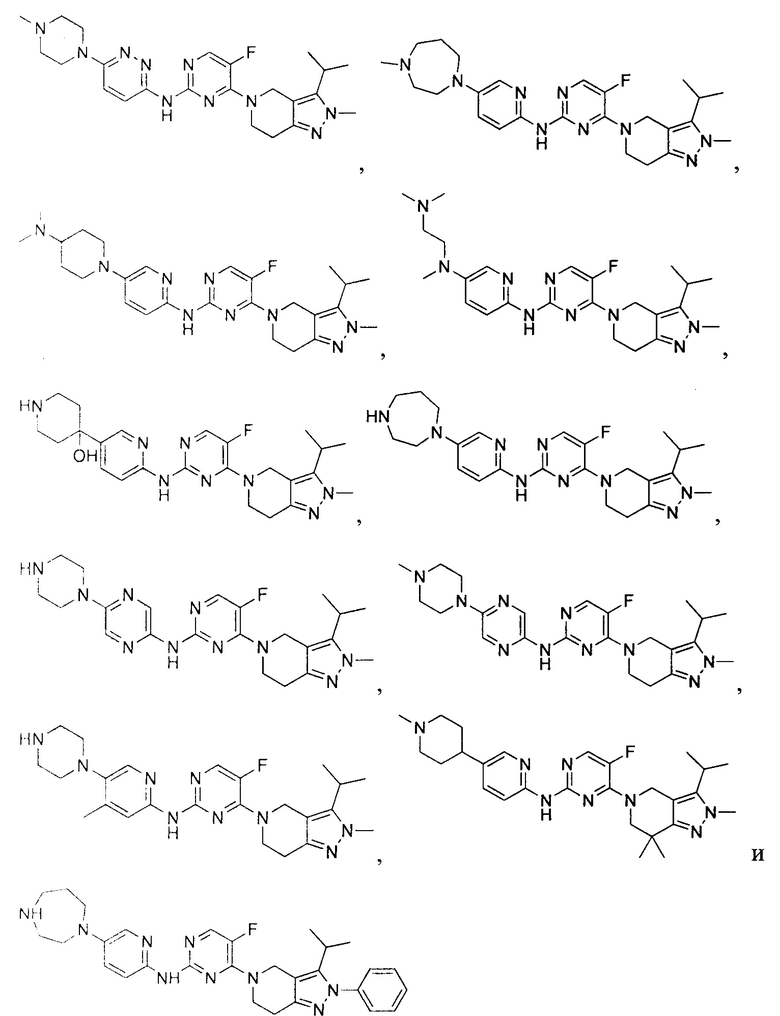
18. Замещенное производное 2Н-пиразола, имеющее формулу (I) или его фармацевтически приемлемая соль
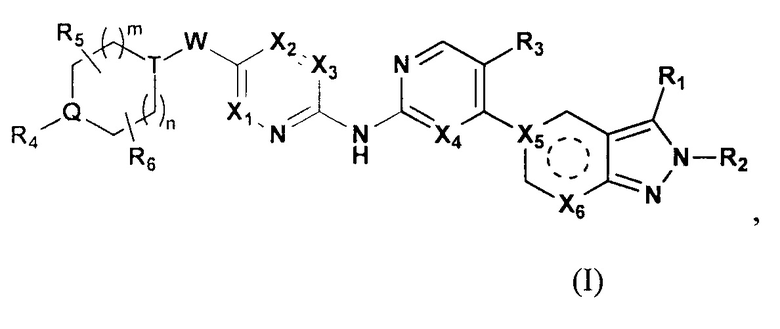
где 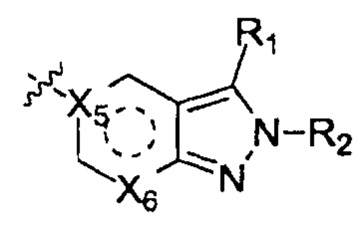 выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и 
R1 выбирают из группы, состоящей из изопропила, 2-пропенила и аллила;
R2 выбирают из группы, состоящей из метила и фенила;
R3 выбирают из группы, состоящей из Н и галогена;
R4, R5 и R6 независимо друг от друга выбирают из группы, состоящей из Н, галогена, OH, NH2, CN и =О, или выбирают из группы, состоящей из C1-8 алкила, C1-8 алкиламино, N,N-ди(С1-8 алкил)амино и 3-7-членной гетероциклоалкильной группы, каждый из который необязательно замещен одним, двумя или тремя R;
в некоторых случаях любые два радикала R4, R5 или R6 могут образовать кольцо из 3-7 атомов;
R7 выбирают из группы, состоящей из Н и галогена;
X1, Х2, Х3 и X4 независимо друг от друга выбирают из группы, состоящей из N и C(R10):
Х7 выбирают из группы, состоящей из карбонила и C(R11)(R12);
W представляет собой простую связь;
Т выбирают из группы, состоящей из N и C(R10);
Q выбирают из группы, состоящей из N и C(R10);
m и n независимо друг от друга выбирают из группы, состоящей из 0, 1 и 2;
R выбирают из группы, состоящей из F, Cl, Br, I, NH2, CN, ОН, CF3, CHF2, CH2F, NHCH3 и N(СH3)2;
R10 выбирают из группы, состоящей из Н, галогена, ОН, NH2, CN, C1-6 алкила и С1-6 алкоксила;
R11 и R12 независимо друг от друга выбирают из группы, состоящей из Н и C1-8 алкила;
в некоторых случаях R4 и R10 присоединяются к одному и тому же атому и
образуют 3-7-членное кольцо.
| EA 201170872 A1, 30.12.2011 | |||
| WO 2009061345 A2, 14.05.2009 | |||
| WO 2011101409 A1, 25.08.2011 | |||
| WO 2011130232 A1, 20.10.2011. |

