Настоящее изобретение относится к области органической химии, а именно конденсированным производным 1,2-оксазинов общей формулы:
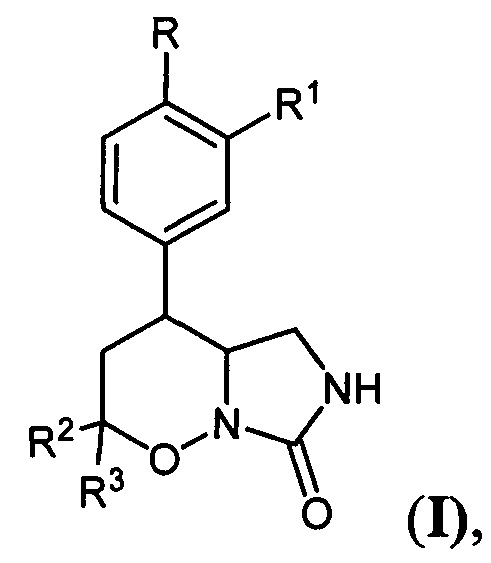 где
где
R=Н, C1-C4-алкокси-группа, либо OCHF2; R1=Н, С1-С4-алкокси-группа, С3-С6-циклоалкокси-группа либо С3-С6 - циклоалкилметокси-группа; R2, R3=Н, С1-С4-алкил либо С1-С4-алкокси-группа, обладающих ингибирующей активностью в отношении фосфодиэстераз подтипа 4, и к способу их получения.
Соединения общей формулы I могут найти применение в качестве фармакологически активных соединений с противораковой, антидепрессантной, противовоспалительной активностью, но, в первую очередь, предназначенных для лечения и/или предотвращения заболеваний, связанных с чрезмерной активностью фосфодиэстераз подтипа 4, таких как хроническая обструктивная болезнь легких, тяжелые формы астмы и бронхита, псориаз, артрит и другие воспалительные заболевания человека. Фосфодиэстеразы (ФДЭ) подтипа 4 (ФДЭ4) - ключевые энзимы, регулирующие метаболизм циклического аденозинмонофосфата (цАМФ) в провоспалительных и иммунных клетках, а также катализирующие переход цАМФ в его неактивную форму - АМФ. Высокое содержание ФДЭ4 в провоспалительных и структурных клетках делает эти ферменты одними из наиболее привлекательных терапевтических целей для воздействия на хроническое воспаление. Ингибиторы ФДЭ4 тормозят разрушение цАМФ и способствуют поддержанию высоких внутриклеточных уровней цАМФ, что снижает активность провоспалительных функций клеток.
Хотя семейство ФДЭ состоит из 11 изоформ, ФДЭ4 является цАМФ-специфической и преобладающей изоформой, которая экспрессируется иммунными и провоспалительными клетками. ФДЭ4 является основным регулятором метаболизма цАМФ практически во всех провоспалительных и структурных клетках, вовлеченных в хроническое воспаление, в частности при хронической обструктивной болезни легких (W.М. Brown, "Treating COPD with PDE 4 inhibitors", International Journal of Chronic Obstructive Pulmonary Disease, 2007, 2, 517-533). Циклический нуклеотид цАМФ является вторичным внутриклеточным мессенджером при осуществлении эффектов гормонов и биологически активных соединений, не проникающих внутрь клетки. Под действием последних в клетках активируются мембранные аденилатциклазы, катализирующие образование цАМФ из аденозинтрифосфата (АТФ), в результате чего происходит накопление циклического нуклеотида.
Уменьшение концентрации циклического нуклеотида, за счет его гидролиза под действием ФДЭ4, сопровождается повышением провоспалительного потенциала клеток, в том числе нейтрофилов, Т-лимфоцитов и макрофагов. Высвобождая различные медиаторы, эти клетки запускают каскад воспалительных реакций, приводящих к развитию системного воспаления. Подавление активности ФДЭ4, напротив, приводит к накоплению в клетках цАМФ и реализации противовоспалительных эффектов гормонов и медиаторов, посредником которых служит циклический нуклеотид цАМФ. В частности, увеличение концентрации внутриклеточного цАМФ ведет к уменьшению дисфункции лейкоцитов, гладкомышечных клеток дыхательных путей и легочных сосудов, эндотелиальных клеток и эпителиальных клеток дыхательных путей, а также фибробластов. Кроме того, ингибиторы ФДЭ4 тормозят высвобождение медиаторов воспаления, таких как ЛТ В4, активные формы кислорода, фактор некроза опухолей α, интерферон-γ и гранзим В.
Таким образом, фармакологическое ингибирование ФДЭ4 позволяет влиять на выраженность хронических воспалительных процессов, задействованных, например, в патогенезе хронической обструктивной болезни легких.
Наиболее известным лекарственным средством указанного типа является Теофиллин - действующее вещество широкого круга доступных препаратов (Теофиллин, Афонилум CP, Вентакс®, Диффумал® 24, Ретафил, Теопэк, Теотард и др.). Теофиллин является неселективным ингибитором практически всех типов ФДЭ и антагонистом аденозиновых рецепторов, вследствие чего обладает большим числом разнообразных эффектов, среди которых следует упомянуть влияние на сократительную активность гладкой мускулатуры, мукоцилиарный клиренс, тонус кровеносных сосудов, частоту и силу сердечных сокращений, кислотность желудочного содержимого и т.д. В то же время, активность теофиллина в отношении ФДЭ4, а значит и его противовоспалительный эффект, выражены в слабой степени. Учитывая его узкий терапевтический диапазон и высокую токсичность при оральном приеме, уровень Теофиллина в крови должен поддерживаться на низком уровне, чтобы избежать побочных эффектов. Применение препарата ассоциируется также с высоким риском развития нежелательных клинически значимых лекарственных взаимодействий. Лечение тяжелой формы хронической обструктивной болезни легких (ХОБЛ) часто требует применения Теофиллина в составе комплексной терапии.
Начиная с 2000-х годов селективные ингибиторы фосфодиэстераз подтипа 4 активно исследуются в качестве лекарственных препаратов для лечения различных воспалительных заболеваний, в частности хронической обструктивной болезни легких, астмы, псориаза и др (J.М. Michalski et al, "PDE4: A Novel Target in the Treatment of Chronic Obstructive Pulmonary Disease", Clin. Pharm. Ther., 2012, 134; S.P. Page et al, "Selective PDE inhibitors as novel treatments for respiratory diseases", Curr. Opin. Pharmacol, 2012, 12, 275; S.J. Bickston et al "Tetomilast: new promise for phosphodiesterase-4 inhibitors", Exp. Opin. Investig. Drugs, 2012, 21, 1845). Золотым стандартом среди ингибиторов ФДЭ4 является препарат Ролипрам (J.-W. Park et al, "The phosphodiesterase 4 inhibitor rolipram protects against cigarette smoke extract-induced apoptosis in human lung fibroblasts", Eur. J. Pharmacol, 2013, 706, 76). Однако частое возникновение тошноты и рвоты, ассоциирующихся с приемом селективных ингибиторов фосфодиэстеразы подтипа 4, существенно снижает их эффективность при использовании в клинической практике (A. Gavalda et al, Exp. Opin. Ther. Patents, 2013, 23, 997; N.S. Tripuraneni et al, J. Theor. Biol, 2016, 394, 117.). Возникновение побочных эффектов ассоциируют с более высокой активностью препаратов в отношении фосфодиэстеразы подтипа 4D (доминирует в рвотном центре головного мозга) по сравнению с терапевтически значимыми фосфодиэстеразами подтипа 4В и 4А (X. Miro et al, "Differential distribution of PDE4D splice variant mRNAs in rat brain suggests association with specific pathways and presynaptical localization", Synapse 2002, 45, 259-269). Известен также аналог препарата Ролипрам - арил-замещенный имидазолидинон Ro-20-1724 (N.A. Saccomano, "Calcium-independent phosphodiesterase inhibitors as putative antidepressants: [3-(bicycloalkyloxy)-4-methoxyphenyl-2-imidazolidinones", Journal of Medicinal Chemistry, 1991, 34, 293), имеющий в своей структуре циклический фрагмент мочевины, как и соединения общей формулы I. Однако, ввиду невысокой терапевтической активности и побочных эффектов, препарат Ro-20-1724 не применяется в клинической практике.
Ниже представлены структурные формулы указанных выше ингибиторов ФДЭ4:
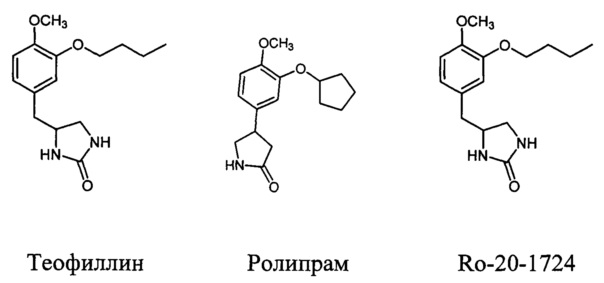
Таким образом, применение ингибиторов фосфодиэстераз подтипа 4 пока ограничено невысокой терапевтической активностью и сильными побочными эффектами, что связывают с их невысокой аффинностью к ФДЭ4, а также отсутствия избирательности в ингибировании различных изотипов ФДЭ4. Основной тенденцией в создании современных лекарственных препаратов против ХОБЛ является разработка молекул с максимальной высокой ингибирующей активностью и селективностью по отношению к ФДЭ подтипов 4В и 4А.
Наиболее близкими по структуре к заявляемым в патенте соединениям являются гексагидро-7H-пирроло[1,2-b][1,2]оксазин-7-оны общей формулы:
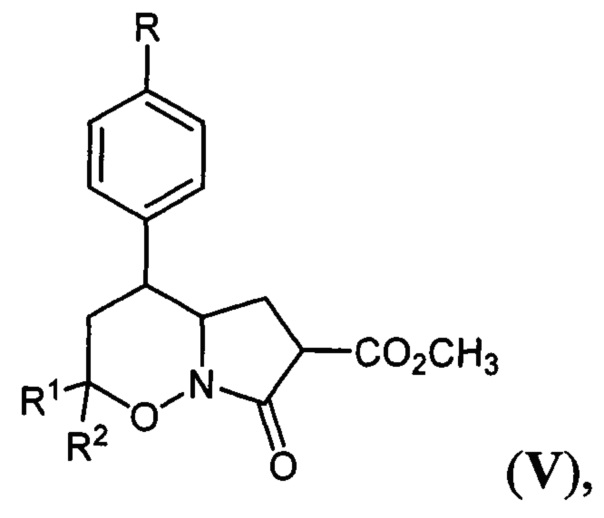
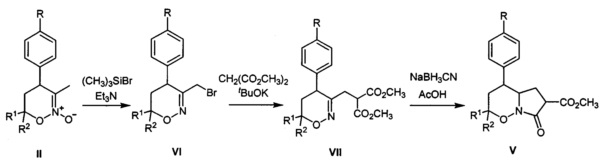
где R=Н, хлор, метокси-группа; R1, R2=Н, метил, этокси-группа. Соединения V были получены по способу, заключающемуся в трансформации известных 1,2-оксазин-N-оксидов (II) в бром-производные (VI) действием избытка триметилсилилбромида и триэтиламина, нуклеофильном замещении брома на остаток диметилмалоната и восстановлении двойной C=N связи в полученных производных (VII) с последующей лактамизацией (A. Yu. Sukhorukov et al, "Diastereoselective Synthesis of γ-Amino Acids and Their Derivatives from Nitroethane via Intermediacy of 5,6-Dihydro-4H-1,2-oxazines Bearing the CH2CH(CO2Me)2 Substituent at C3", Synthesis, 2009, 741-754).
Процесс протекает по следующей схеме:
Этот способ не пригоден для получения заявляемых в патенте 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-онов общей формулы I.
В литературе отсутствуют данные о биологической активности соединений общей формулы V, в частности какие-либо сведения об их ингибирующей активности в отношении фосфодиэстераз подтипа 4.
Технической задачей данного изобретения является расширение ассортимента новых высокоактивных и селективных ингибиторов фосфодиэстеразы подтипа 4 и разработка способов их получения с целью дальнейшего создания лекарственных препаратов на их основе.
Поставленная техническая задача достигается новыми замещенными 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-онами общей формулы:
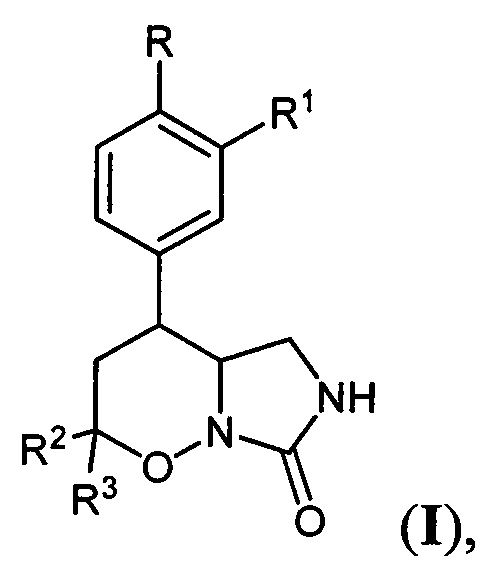
где R=Н, С1-С4 - алкокси-группа, либо OCHF2; R1=Н, С1-С4-алкокси-группа, либо C3-C6 - циклоалкокси-группа либо C3-C6 - циклоалкилметокси-группа; R2, R3=Н, С1-С4-алкил, либо С1-С4-алкокси-группа.
Соединения общей формулы I обладают повышенной эффективностью и селективностью в ингибировании фосфодиэстераз подтипа 4, и являются перспективными для применения в терапии хронических воспалительных заболеваний. Наиболее предпочтительными оказались соединения, выбранные из группы 4-[3-(циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-он, 4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2,2-диметилгексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-он, 4-(3-(циклопентилокси)-4-метоксифенил)гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-он.
Предложен также способ получения соединений общей формулы I, где R=Н, С1-С4-алкокси-группа либо OCHF2; R1=Н, С1-С4-алкокси-группа, C3-C6 - циклоалкокси-группа либо C3-C6-циклоалкилметокси-группа; R2, R3=Н, С1-С4-алкил либо С1-С4-алкокси-группа, заключающийся в том, что 5,6-дигидро-4Н-1,2-оксазин-N-оксиды общей формулы:
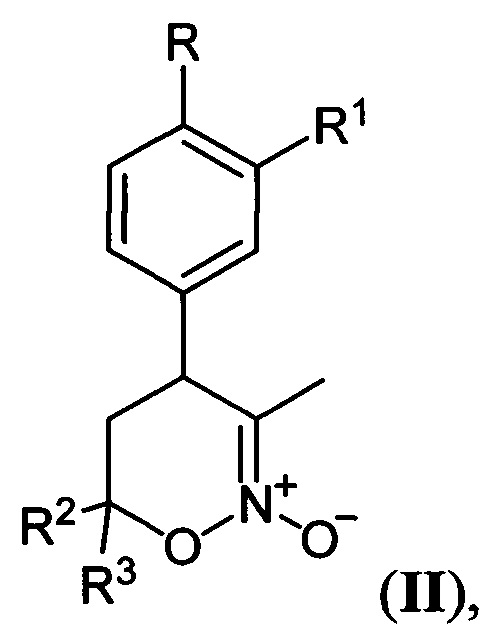
где R, R1, R2 и R3 имеют вышеуказанные значения, подвергают последовательной обработке триметилсилилбромидом с азотистым основанием и бромидом металла в среде апротонного растворителя при пониженной температуре с последующей обработкой образующихся при этом соответствующих бромидов азидом натрия при повышенной температуре в среде апротонного растворителя, полученные при этом соответствующие 3-азидометил-5,6-дигидро-4Н-1,2-оксазины общей формулы:
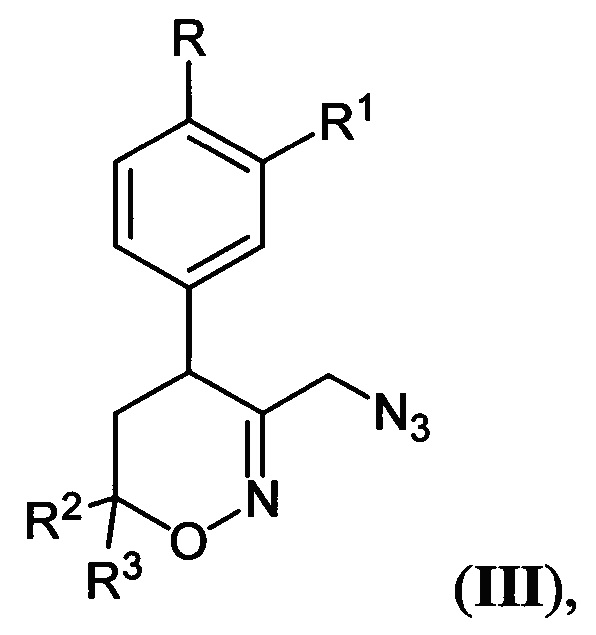
где R, R1, R2 и R3 имеют вышеуказанные значения, подвергают взаимодействию с цианоборгидридом натрия в протонной кислоте с последующим каталитическим восстановлением полученных при этом соответствующих 3-азидометил-1,2-оксазинанов общей формулы:
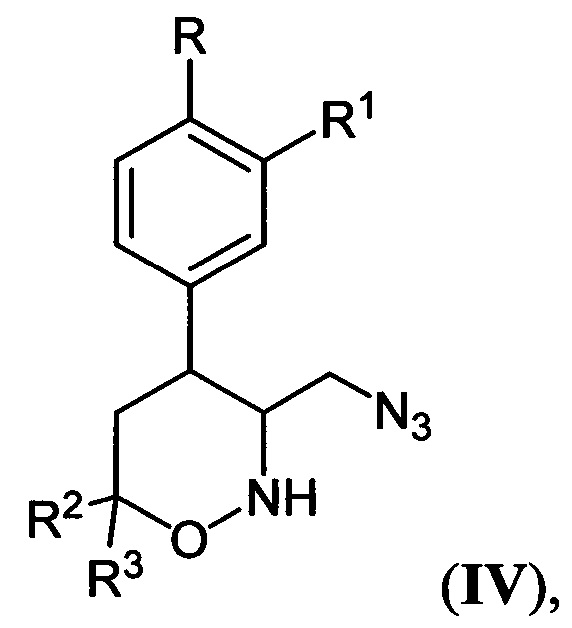
где R, R1, R2 и R3 имеют вышеуказанные значения, водородом в присутствии металлического катализатора в среде низкомолекулярного спирта и обработкой реакционной смеси карбомоилирующим реагентом с азотистым основанием в апротонном растворителе.
В качестве азотистого основания используют, например, триэтиламин, в качестве бромида металла используют преимущественно бромид кобальта, в качестве металлического катализатора используют преимущественно никель Ренея, в качестве карбомоилирующего агента используют, например, трифосген, в качестве протонной кислоты используют преимущественно уксусную кислоту, а в качестве апротонного растворителя используют, например, ДМФА, ТГФ или хлористый метилен.
Заявленные в патенте производные ряда гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-она общей формулы I в литературе не описаны. Не было также никаких данных, позволяющих предположить, что вещества общей формулы I могут ингибировать фосфодиэстеразу подтипа 4 и, что их можно использовать в качестве противовопалительных средств или каких-либо других лекарственных препаратов. Кроме того, моноциклический аналог предлагаемых соединений общей формулы I - соединение структурной формулы:
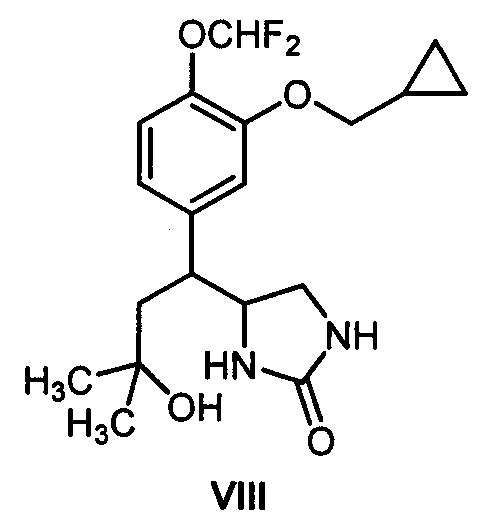
не содержащее шестичленного 1,2-оксазинового цикла (синтезированное авторами данного изобретения) - практически не проявляет активность как ингибитор фосфодиэстеразы подтипа 4 (см. таблицу). Это демонстрирует принципиальную роль предложенной бициклической гетероциклической системы I, способной ингибировать фосфодиэстеразу подтипа 4, для проявления целевого фармакологического эффекта.
Таким образом, заявляемое изобретение соответствует критерию «новизна» и «изобретательский уровень».
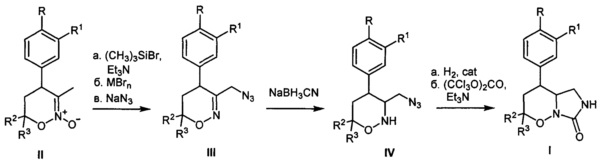
Для получения соединений общей формулы I предложен новый способ, который заключается в последовательности следующих химических стадий: 1) превращение известных 5,6-дигидро-4Н-1,2-оксазин-N-оксидов II в 3-азидометил-5,6-дигидро-4Н-1,2-оксазины III; 2) восстановление соединений III до насыщенных 3-азидометил-1,2-оксазинанов IV цианоборогидридом натрия в присутствии протонной кислоты; 3) превращение 3-азидометил-1,2-оксазинанов IV в целевые гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-оны I в две операции, включающие восстановление азидо-группы и последующее карбомоилирование.
Процесс протекает по следующей схеме:
(MBrn - бромид металла, Cat - металлический катализатор)
5,6-Дигидро-4Н-1,2-оксазин-N-оксиды II превращают в 3-азидометил-5,6-дигидро-4Н-1,2-оксазины III через промежуточное получение соответствующих бромидов с помощью модифицированного по сравнению с литературным метода, заключающегося в последовательной обработке соединений II галогенидом кремния, например, триметилсилилбромидом с азотистым основанием, например, триэтиламином и бромидом металла, например, кобальта. Полученные соответствующие бромиды без дополнительной очистки трансформируют в 3-азидометил-5,6-дигидро-4Н-1,2-оксазины III действием азида натрия в подходящем растворителе, например, ДМФА. На следующей стадии 3-азидометил-5,6-дигидро-4Н-1,2-оксазины III восстанавливают действием цианоборогидридом натрия в протонной кислоте, например, уксусной в 3-азидометил-1,2-оксазинаны IV. Трансформацию 3-азидометил-1,2-оксазинанов IV в целевые гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-оны I осуществляют в две операции, включающие восстановление азидо-группы водородом на металлическом катализаторе, например, никеле Ренея в низкомолекулярном спирте, например, метаноле и последующую обработку получающегося амина карбомоилирующим реагентом, например, трифосгеном с азотистым основанием, например, триэтиламином в апротонном растворителе, например, хлористом метилене.
Исходными веществами для предлагаемого способа получения замещенных 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-онов общей I являются известные 1,2-оксазин-N-оксиды II, которые могут быть получены из коммерчески доступных ароматических альдегидов IX и нитроэтана известными способами в граммовых и килограммовых количествах как показано на схемах ниже:
путь 1: синтез 1,2-оксазин-N-оксидов II не замещенных по положению С-6 (S. Kanemasa, "Cycloaddition/Ring opening of 3-unsubstituted cyclic nitronates, isoxazoline and 5,6-dihydro-4H-1,2-oxazineN-oxides, as synthetic equivalents of functionalized nitrile oxides" Tetrahedron Letters, 1998, 39, 8869-8872).
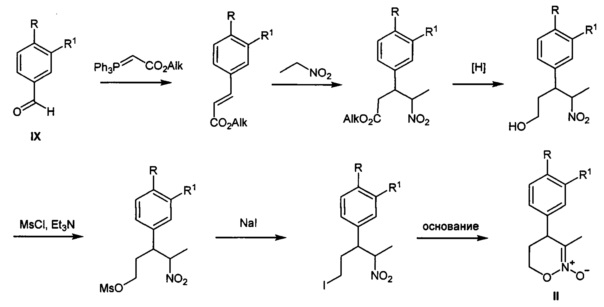
путь 2: синтез 1,2-оксазин-N-оксидов II замещенных по положению С-6 (А.A. Tishkov, "2-Silyloxy-1,2-oxazines, a New Type of Acetals of Conjugated Nitroso Alkenes", J. Org. Chem., 2003, 68, 9477-9480)
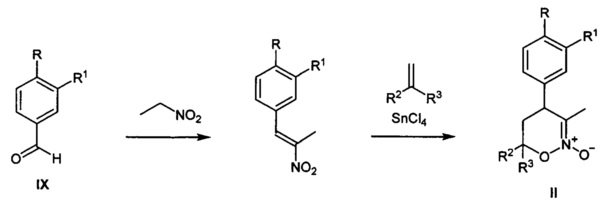
Найденные способы превращения II в III и IV в I новые и являются одним из предметов данного изобретения.
Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций.
Техническим результатом изобретения являются новые вещества - замещенные 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-оны общей формулы I, проявляющие высокую ингибирующую активность в отношении терапевтической мишени - фосфодиэстеразы подтипа 4. По результатам биологических испытаний in vitro из соединений формулы I наиболее активным является 4-[3-(циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-он (Ia), превосходящий по активности ближайший аналог - эталонный селективный ингибитор фосфодиэстеразы подтипа 4 Ro-20-1724. Кроме того, соединения формулы I, например, соединения Ia, Iб и Iг существенно превосходят применяемый в клиническом практике лекарственный препарат Теофиллин. Эти результаты подтверждают промышленную применимость настоящего изобретения. Вещество Ia формулы:
было дополнительно исследовано на более широком круге фосфодиэстераз 4-ого типа (ФДЭ 4А, 4В, 4С и 4D), являющихся терапевтическими мишенями для различных заболеваний.
Изобретение иллюстрируется примерами, не ограничивающими его объем.
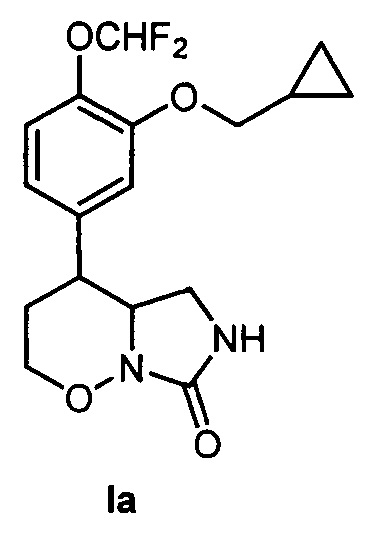
Пример 1. Получение 4-[3-(Циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-она (Ia) по следующей схеме:
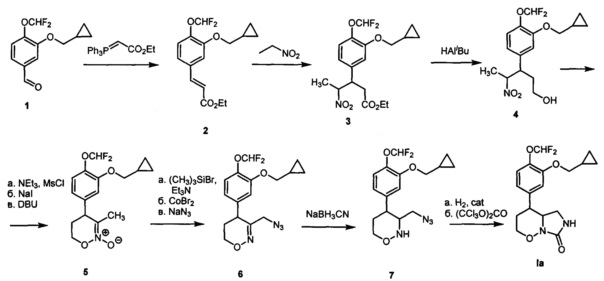
(DBU - 1,8-диазабицикло[5.4.0]ундец-7-ен, Ms - метансульфонил, cat - катализатор)
1) 3-Циклопропилметокси-4-(дифторметокси)бензальдегид 1 (Y. Lin, "А convenient method for the synthesis of roflumilast", Research on Chemical Intermediates, 2013, 39, 2107-2113) (518 мг, 2,140 ммоль) растворяли в сухом дихлорметане (5 мл) в атмосфере аргона. Добавляли илид Ph3P=CHCO2Et (785 мг, 2,256 ммоль) и перемешивали при комнатной температуре в течение 6 часов, затем выдерживали 48 часов. Упаривали дихлорметан при пониженном давлении, добавляли смесь гексана и диэтилового эфира (9:1). Выпавший белый осадок отфильтровывали, а раствор упаривали при пониженном давлении. Продукт очищали при помощи колоночной хроматографии с использованием смеси гексана и этилацетата (1:1) в качестве элюента, с получением этил (Е)-3-(3-(циклопропилметокси)-4-(дифторметокси)фенио)акрилата 2 в виде твердого белого вещества с выходом 648 мг (97%). Т. пл. 48.5-50.5°C (перекристаллизован из пентана). 1Н ЯМР (300 МГц, CDCl3) δ 7.56 (д, J=16.0 Гц, 1H), 7.11 (д, J=7.9 Гц, 1Н), 7.05 (с, 1Н), 7.04 (д, J=7.9 Гц, 1H), 6.64 (т, J=75.3 Гц, 1Н), 6.32 (д, J=16.0 Гц, 1Н), 4.22 (кв, J=7.0 Гц, 2Н), 3.86 (д, J=6.9 Гц, 2Н), 1.30 (т, J=7.0 Гц, 3H), 1.35-1.18 (м, 1H), 0.62 (м, 2Н), 0.33 (м, 2Н). Масс-спектр (EI): m/z 312 (M+).
2) Алкен 2 (648 мг, 2,078 ммоль) растворяли в нитроэтане (1,5 мл), добавляли диазабициклоундецен (0,31 мл, 2,078 ммоль). Перемешивали при комнатной температуре в течение 16 часов. Гасили реакцию добавлением 2М водного раствора соляной кислоты, разбавляли раствор водой (20 мл) и экстрагировали метилтретбутиловым эфиром (30 мл и 2 раза по 15 мл). Экстракт промывали насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (9:1 → 6:1) в качестве элюента, с получением этил 3-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-4-нитропентаноата 3 в виде прозрачного маслообразного вещества с выходом 729 мг (91%, смесь изомеров). 1Н ЯМР (300 МГц, CDCl3) δ 7.04 (д, J=8.2 Гц, 1H), 6.76 (с, 1Н), 6.72 (д, J=8.2 Гц, 1Н), 6.56 (т, J=75.5 Гц, 1Н), 4.73 (дкв, J=9.2, 6.6 Гц, 1Н), 3.91 (кв, J=7.1, 2Н), 3.81 (д, J=6.9 Гц, 2Н), 3.59 (ддд, J=9.7, 9.2, 5.0 Гц, 1Н), 2.68 (дд, J=15.8, 9.7 Гц, 1Н), 2.58 (дд, J=15.8, 5.0 Гц, 1Н), 1.28 (д, J=6.6 Гц, 3H), 1.24-1.12 (м, 1Н), 1.02 (т, J=7.1, 3H), 0.56 (м, 2Н), 0.28 (м, 2Н) (изомер 1). 1Н ЯМР (300 МГц, CDCl3) δ 7.03 (д, J=8.9 Гц, 1Н), 6.74 (с, 1Н), 6.72 (д, J=8.9 Гц, 1Н), 6.56 (т, J=75.5 Гц, 1Н), 4.84 (дкв, J=8.4, 6.7 Гц, 1Н), 4.00 (кв, J=7.2, 2Н), 3.80 (д, J=6.9 Гц, 2Н), 3.63 (ддд, J=9.1, 8.4, 6.0 Гц, 1Н), 2.78 (дд, J=16.0, 6.0 Гц, 1Н), 2.66 (дд, J=16.0, 9.1 Гц, 1H), 1.52 (д, J=6.7 Гц, 3H), 1.27-1.15 (м, 1H), 1.08 (т, J=7.2 Гц, 3H), 0.58 (м, 2Н), 0.30 (м, 2Н) (изомер 2). Масс-спектр (EI): m/z 387 (М+).
3) Нитроэфир 3 (560 мг, 1,444 ммоль) растворяли в сухом дихлорметане (24 мл) в атмосфере аргона. Реакционную смесь охлаждали до -78°C. Добавляли раствор 1,2М диизобутилалюминийгидрида в толуоле (4,8 мл, 5,78 ммоль). Перемешивали 5 минут при -78°C, затем 16 часов при комнатной температуре. Охлаждали до -78°C и добавляли раствор 1,2М диизобутилалюминийгидрида в толуоле (2,4 мл, 2,88 ммоль). Перемешивали в течение 8,5 часов. Гасили реакцию добавлением 1М водного раствора соляной кислоты (20 мл), разбавляли водой (100 мл) и экстрагировали дихлорметаном (50 мл и 2 раза по 20 мл). Экстракт промывали водой (100 мл) и насыщенным раствором хлорида натрия в воде (100 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (7:1 → 5:1 → 3:1 → 1:1) в качестве элюента, с получением 3-(3-(циклопропилметокси)-4-(дифторметокси)phenyl)-4-нитропентан-1-ола 4 в виде прозрачного маслообразного вещества с выходом 278 мг (56%, смесь изомеров). 1Н ЯМР (300 МГц, CDCl3) δ 7.13 (д, J=7.5 Гц, 1Н), 6.75 (с, 1Н), 6.73 (д, J=7.5 Гц, 1Н), 6.62 (т, J=75.4 Гц, 1Н), 4.78-4.62 (м, 1Н), 3.86 (д, J=6.9 Гц, 2Н), 3.51 (дд, J=10.6, 5.3 Гц, 1Н), 3.40-3.25 (м, 2Н), 1.86 (дд, J=13.6, 6.5 Гц, 1Н), 1.32 (д, J=6.6 Гц, 3H), 1.30-1.23 (м, 1Н), 0.96 (дд, J=13.6, 6.7 Гц, 1H), 0.65 (м, 2Н), 0.35 (м, 2Н) (изомер 1). 1H ЯМР (300 МГц, CDCl3) δ 7.09 (д, J=7.6 Гц, 1H), 6.74 (д, J=7.6 Гц, 1Н), 6.74 (д, J=1.7 Гц, 1Н), 6.59 (т, J=75.4 Гц, 1Н), 4.78 (дкв, J=12.0, 6.6 Гц, 1Н), 3.84 (д, J=6.8 Гц, 2Н), 3.57 (ддд, J=12.0, 6.1, 4.9 Гц, 1Н), 3.35 (м, 2Н), 2.10-1.95 (м, 1Н), 1.81 (ддд, J=14.0, 11.1, 4.9 Гц, 1Н), 1.61 (д, J=6.6 Гц, 3H), 1.29-1.15 (м, 1Н), 0.63 (м, 2Н), 0.34 (м, 2Н) (изомер 2). Масс-спектр (EI): m/z 345 (М+).
4) Полученный на предыдущей стадии нитроспирт 4 (244 мг, 0,707 ммоль) растворяли в сухом дихлорметане (2,2 мл) в атмосфере аргона, охлаждали до 0°C и добавляли триэтиламин (160 мкл, 1,13 ммоль) и метансульфонилхлорид (70 мкл, 0,848 ммоль). Перемешивали при 0°C в течение 15 минут, затем выдерживали при 5°C в течение 5,5 часов. Гасили реакцию добавлением воды (10 мл), экстрагировали дихлорметаном (3 по 30 мл). Экстракт промывали насыщенным раствором хлорида аммония в воде (50 мл) и насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. К остатку добавляли 1М раствор иодида натрия в ацетоне (1,2 мл, 1,2 ммоль). Выдерживали при комнатной температуре в течение 48 часов. Добавляли в реакционную смесь воду (30 мл), экстрагировали этилацетатом (30 мл). Экстракт сушили сульфатом натрия, упаривали при пониженном давлении и остаток сушили в вакууме. Полученный продукт растворяли в сухом дихлорметане (2,3 мл), добавляли диазабициклоундецен (85 мкл, 0,571 ммоль). Перемешивали при комнатной температуре в течение 3,5 часов. В реакционную смесь добавляли этилацетат (40 мл) и 0,25М раствором гидросульфата натрия в воде (40 мл). Экстрагировали этилацетатом (2 раза по 20 мл), экстракт промывали насыщенным раствором хлорида натрия в воде (30 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:1 → 0:1) в качестве элюента, с получением 4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-3-метил-5,6-дигидро-4Н-1,2-оксазин 2-оксида 5 в виде прозрачного маслообразного вещества с выходом 146 мг (78%). 1Н ЯМР (300 МГц, CDCl3) δ 7.12 (д, J=8.3 Гц, 1Н), 6.73 (д, J=8.3 Гц, 1Н), 6.70 (с, 1Н), 6.60 (т, J=75.4 Гц, 1Н), 4.41 (м, 2Н), 3.83 (д, J=7.0 Гц, 2Н), 3.80-3.60 (м, 1Н), 2.36 (дт, J=13.5, 6.3 Гц, 1Н), 2.01 (ддд, J=13.5, 11.9, 7.7 Гц, 1Н), 1.89 (с, 3H), 1.43-1.07 (м, 1Н), 0.64 (м, 2Н), 0.34 (м, 2Н). 13С ЯМР (75 МГц, JMOD, CDCl3) δ 150.95, 139.54, 123.36, 122.24, 120.64, 115.85 (т, J=255.5 Гц), 113.64, 74.24, 69.27, 43.65, 30.38, 18.23, 10.20, 3.28. 19F ЯМР (282 МГц, CDCl3) δ -82.51 (д, J=75.4 Гц). Масс-спектр высокого разрешения (ESI): m/z 328.1350 (МН+); рассчитано для [C16H20F2NO4+]: 328.1355.
5) 5,6-Дигидро-4Н-1,2-оксазин-N-оксид 5 (146 мг, 0,446 ммоль) растворяли в сухом дихлорметане (0,9 мл) в атмосфере аргона, добавляли перегнанный триэтиламин (95 мкл, 0,699 ммоль). Реакционную смесь охлаждали до -78°C, затем добавляли триметилсилилбромид (82 мкл, 0,624 ммоль). Перемешивали смесь при -78°C в течение 30 минут, затем выдерживали при -20°C в течение 28 часов. Нагревали смесь до 0°C, добавляли в токе аргона раствор бромида кобальта (195 мг, 0,891 ммоль) в сухом тетрагидрофуране (2 мл). Перемешивали смесь в течение 3 часов при комнатной температуре, затем выдерживали 15 часов. Реакцию гасили добавлением 0,25М водного раствора гидросульфата натрия (30 мл), полученный раствор экстрагировали этилацетатом (40 мл и 2 раза по 30 мл). Экстракт промывали водой (50 мл) и насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Остаток растворяли в диметилформамиде (6 мл), добавляли азид натрия (145 мг, 2.23 ммоль) и перемешивали при 60°C в атмосфере аргона в течение 4,5 часов. Реакцию гасили добавлением воды (40 мл), смесь экстрагировали метилтретбутиловым эфиром (40 мл и 2 раза по 30 мл), экстракт промывали насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (10:1 → 5:1) в качестве элюента, с получением 3-(азидометил)-4-(3 -(циклопропилметокси)-4-(дифторметокси)фенил)-5,6-дигидро-4Н-1,2-оксазина 6 в виде прозрачного маслообразного вещества с выходом 119 мг (76%). 1Н ЯМР (300 МГц, CDCl3) δ 7.13 (д, J=7.7 Гц, 1H), 6.74 (с, 1Н), 6.73 (д, J=7.7 Гц,
1Н), 6.60 (т, J=75.4 Гц, 1Н), 4.06 (т, J=5.1 Гц, 2Н), 3.88 (д, J=14.6 Гц, 1H), 3.84 (д, J=7.2 Гц, 2Н), 3.62 (д, J=14.6 Гц, 1H), 3.55 (дд, J=10.2, 6.1 Гц, 1Н), 2.30 (дд, J=13.2, 6.1 Гц, 1Н), 1.98 (ддд, J=13.2, 10.2, 5.1 Гц, 1Н), 1.35-1.14 (м, 1Н), 0.63 (м, 2Н), 0.34 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 154.22, 151.11, 139.77, 138.68, 123.28, 120.77, 116.14 (т, J=260.1 Гц), 114.18, 74.06, 64.02, 52.99, 36.77, 28.36, 10.13, 3.22. 19F ЯМР (282 МГц, CDCl3) δ -82.47 (д, J=75.4 Гц). Масс-спектр высокого разрешения (ESI): m/z 353.1414 (МН*); рассчитано для [C16H19F2N4O3+]: 353.1420.
6) Азид 6 (89 мг, 0,253 ммоль) растворяли в ледяной уксусной кислоте (2,2 мл), добавляли цианоборгидрид натрия (143 мг, 2,28 ммоль). Перемешивали реакционную смесь в течение 1 часа. Реакционную смесь гасили избытком насыщенного водного раствора карбоната натрия (10 мл), раствор экстрагировали этилацетатом (3 раза по 30 мл). Экстракт промывали насыщенным раствором хлорида натрия в воде (30 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (10:0 → 5:1 → 1:1) в качестве элюента, с получением 3-(азидометил)-4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-1,2-оксазинана 7 в виде прозрачного маслообразного вещества с выходом 98 мг (89%). 1Н ЯМР (300 МГц, CDCl3) δ 1Н ЯМР (300 МГц, CDCl3) δ 7.12 (д, J=8.9 Гц, 1H), 6.79 (д, J=2.4 Гц, 1Н), 6.77 (дд, J=8.9, 2.4 Гц, 1H), 6.61 (т, J=75.6 Гц, 1Н), 5.49 (с, ушир., 1Н), 4.12 (м, 1Н), 3.87 (д, J=6.9 Гц, 2Н), 3.83 (ддд, J=11.8, 2.9, 2.6 Гц, 1H), 3.35-3.25 (м, 1Н), 3.30 (дд, J=13.0, 6.9 Гц, 1Н), 3.10 (дд, J=13.0, 8.0 Гц, 1Н), 2.67 (ддд, J=11.8, 10.1, 4.5 Гц, 1Н), 2.02-1.89 (м, 1Н), 1.84 (дддд, J=13.5, 4.5, 2.6, 2.2 Гц, 1H), 1.34-1.20 (м, 1H), 0.65 (м, 2Н), 0.36 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 150.95, 140.22, 139.57, 123.24, 120.19, 116.31 (т, J=259.7 Гц), 113.89, 74.11, 70.85, 61.92, 51.50, 44.23, 33.53, 10.28, 3.29. 19F ЯМР (282 МГц, CDCl3) δ -81.59 (д, J=75.6 Гц). Масс-спектр высокого разрешения (ESI): m/z 355.1571 (MH+); рассчитано для [C6H21F2N4O3+]: 355.1576.
7) Азид 7 (61 мг, 0.172 ммоль) растворяли в метаноле (1.5 мл), добавляли раствор дитретбутилдикарбоната (37,5 мг, 0,172 ммоль) в метаноле (0,5 мл) и взвесь никеля Ренея в метаноле (1 мл). Гидрировали в автоклаве при комнатной температуре и давлении водорода 1 бар в течение 10 минут. Отделяли никель Ренея фильтрованием, промывали метанолом (5 раз по 3 мл), и объединенный раствор упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (5:1 → 1:1) в качестве элюента, с получением прозрачного маслообразного вещества, которое растворяли в сухом дихлорметане (1 мл), охлаждали до 0°C и добавляли трифторуксусную кислоту (0,5 мл). Перемешивали в течение 1 часа, затем упаривали реакционную смесь при пониженном давлении. Растворяли продукт в сухом дихлорметане (0,5 мл). Охлаждали до 0°C и добавляли раствор трифосгена (17,5 мг, 0,059 ммоль) в сухом дихлорметане и триэтиламин (90 мкл, 0,625 ммоль). перемешивали при 0°C в атмосфере аргона в течение 1 часа, затем упаривали реакционную смесь при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:1 → 0:1) в качестве элюента, с получением 4-[3-(циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-она Ia в виде прозрачного маслообразного вещества с выходом 14 мг (23%). 1Н ЯМР (300 МГц, CDCl3) δ 7.14 (д, J=7.8 Гц, 1Н), 6.80 (с, 1Н), 6.79 (д, J=7.8 Гц, 1Н), 6.61 (т, J=75.5 Гц, 1H), 5.47 (с, 1Н), 4.09 (ддд, J=10.7, 4.8, 2.3 Гц, 1Н), 4.02 (ддд, J=12.0, 10.7, 2.1 Гц, 1Н), 3.87 (д, J=6.9 Гц, 2Н), 3.86-3.81 (м, 1Н), 3.29 (дд, J=9.0, 6.7 Гц, 1Н), 3.02 (ддд, J=9.0, 2.5 Гц, 1Н), 2.87 (ддд, J=12.3, 10.5, 4.1 Гц, 1Н), 2.14 (дддд, J=13.8, 12.3, 12.0, 4.8 Гц, 1Н), 1.90 (дддд, J=13.8, 4.1, 2.3, 2.1 Гц, 1H), 1.36-1.20 (м, 1Н), 0.70-0.61 (м, 2Н), 0.40-0.33 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 160.22, 151.04, 139.85, 139.14, 123.32, 120.38, 116.22 (т, J=260.0 Гц), 114.13, 74.24, 69.73, 60.30, 41.79, 40.60, 31.38, 10.30, 3.34. 19F ЯМР (282 МГц, CDCl3) δ -81.69 (д, J=75.5 Гц). Масс-спектр высокого разрешения (ESI): m/z 355.1459 (MH+); рассчитано для [C17H21F2N2O4+]: 355.1464.
Пример 2. Получение 4-(3-(Циклопропилметокси)-4-(дифторметокси)фенил)-2,2-диметилгексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-она (Iб) по следующей схеме:
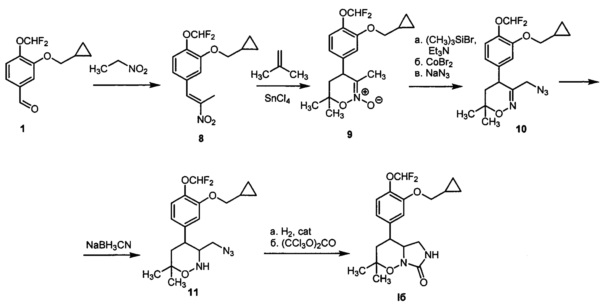
1) К раствору 3-циклопропилметокси-4-(дифторметокси)бензальдегида 1 (Y. Lin, "A convenient method for the synthesis of roflumilast", Research on Chemical Intermediates, 2013, 39, 2107-2113) (0.811 r, 3.35 ммоль) в нитроэтане (6.7 мл) добавляли ацетат аммония (0.258 г, 3.35 ммоль) и уксусную кислоту (3 мл). Смесь интенсивно нагревали с перемешиванием при 90°C в атмосфере аргона в течении 9 часов, после чего упаривали в вакууме. Остаток хроматографировали на силикагеле с помощью смеси гексана и этилацетата в качестве элюента (1:1 → 0:1). Получали 0.542 г (54%) 2-(циклопропилметокси)-1-(дифторметокси)-4-(2-нитропроп-1-ен-1 -ил)бензола 8 в виде желтого масла. 1Н ЯМР (300 МГц, CDCl3) δ 7.98 (с, 1Н), 7.20 (д, J=7.9 Гц, 1H), 7.00 (д, J=7.9 Гц, 1H), 6.99 (с, 1Н), 6.68 (т, J=75.1 Гц, 1H), 3.89 (д, J=6.9 Гц, 2Н), 2.42 (с, 3H), 1.38-1.18 (м, 1Н), 0.65 (м, 2Н), 0.36 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 150.66, 148.01, 141.54, 132.60, 130.85, 122.90, 122.67, 116.07, 115.98 (т, J=260.5 Гц), 74.17, 14.00, 10.11, 3.25. 19F ЯМР (282 МГц, CDCl3) δ -82.59 (д, J=74.9 Гц). Масс-спектр высокого разрешения (ESI): m/z 300.1048 (МН+); рассчитано для [C14H16F2NO4]+ 300.1042.
2) Нитроалкен 8 (628 мг, 2,100 ммоль) растворяли в сухом дихлорметане (18 мл) в токе аргона. Охлаждали смесь до -78°C, добавляли тетрахлорид олова (270 мкл, 2,310 ммоль). Полученную смесь перемешивали при -78°C в течение 5 минут, затем добавляли раствор 2-метилпропена (1 г, 17,86 ммоль) в сухом дихлорметане (13 мл). Перемешивали полученную смесь при -78°C в течение 1 часа. Реакцию гасили добавлением насыщенного раствора карбоната натрия в воде (70 мл) к реакционной смеси. Полученный раствор экстрагировали этилацетатом (3 раза по 50 мл), экстракт промывали водой (100 мл) и насыщенным раствором хлорида натрия в воде (100 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:1 → 0:1) в качестве элюента. Получали 584 мг (78%) 4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-3,6,6-триметил-5,6-дигидро-4Н-1,2-оксазин 2-оксида 9 в виде маслообразного вещества. 1Н ЯМР (300 МГц, CDCl3) δ 7.13 (д, J=8.1 Гц, 1Н), 6.74 (д, J=8.1 Гц, 1Н), 6.69 (с, 1H), 6.62 (т, J=75.4 Гц, 1Н), 3.84 (д, J=6.8 Гц, 2Н), 3.66 (дд, J=10.4, 8.0 Гц, 1Н), 2.11 (дд, J=13.9, 8.0 Гц, 1Н), 1.97-1.84 (м, 1H), 1.88 (с, 3H), 1.45 (с, 3H), 1.42 (с, 3H), 1.33-1.18 (м, 1Н), 0.66 (м, 2Н), 0.36 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 151.36, 139.92, 139.06, 123.37, 121.45, 120.77, 116.19 (т, J=260.0 Гц), 113.57, 81.39, 74.26, 43.22, 41.76, 27.89, 22.21, 17.35, 10.22, 3.31. 19F ЯМР (282 МГц, CDCl3) δ -82.53 (д, J=75.4 Гц). Масс-спектр высокого разрешения (ESI): m/z 356.1658 (MH+); рассчитано для [C18H24F2NO4+]: 356.1668.
3) 1,2-Оксазин-N-оксид 9 (552 мг, 1,555 ммоль) растворяли в сухом дихлорметане (9 мл) в атмосфере аргона, добавляли перегнанный триэтиламин (325 мкл, 2,332 ммоль). Реакционную смесь охлаждали до -78°C, затем добавляли триметилсилилбромид (290 мкл, 2,199 ммоль). Перемешивали смесь при -78°C в течение 1 часа, затем выдерживали при -40°C в течение 48 часов. Нагревали смесь до 0°C, добавляли в токе аргона раствор бромида кобальта (688 мг, 3,142 ммоль) в сухом тетрагидрофуране (6 мл). Перемешивали смесь в течение 5,5 часов при комнатной температуре. Реакцию гасили добавлением 0,25М водного раствора гидросульфата натрия (50 мл), полученный раствор экстрагировали этилацетатом (50 мл и 2 раза по 25 мл). Экстракт промывали водой (50 мл) и насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Остаток растворяли в диметилформамиде (20 мл), добавляли азид натрия (505 мг, 7,775 ммоль) и перемешивали при 60°C в атмосфере аргона в течение 5,5 часов. Реакцию гасили добавлением воды (70 мл), смесь экстрагировали метилтретбутиловым эфиром (70 мл и 3 по 30 мл), экстракт промывали водой (60 мл) и насыщенным раствором хлорида натрия в воде (60 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:0 → 10:1 → 5:1) в качестве элюента, с получением 3-(азидометил)-4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-6,6-диметил-5,6-дигидро-4Н-1,2-оксазина 10 в виде желтоватого прозрачного маслообразного вещества с выходом 353 мг (72%). 1Н ЯМР (300 МГц, CDCl3) δ 7.14 (д, J=8.0 Гц, 1Н), 6.76 (д, J=8.0 Гц, 1Н), 6.74 (с, 1Н), 6.62 (т, J=75.4 Гц, 1Н), 3.89 (д, J=14.5 Гц, 1H), 3.85 (д, J=6.6 Гц, 2Н), 3.55 (дд, J=12.0, 7.7 Гц, 1Н), 3.52 (д, J=14.5 Гц, 1Н), 2.11 (дд, J=13.6, 7.7 Гц, 1Н), 1.90 (дд, J=13.6, 12.0 Гц, 1H), 1.39 (с, 3H), 1.31 (с, 3H), 1.29-1.19 (м, 1Н), 0.65 (м, 2Н), 0.36 (м, 2Н). 13С ЯМР (75 МГц, JMOD, CDCl3) δ 154.13, 151.31, 139.62, 137.97, 123.53, 119.98, 117.89 (т, J=260.0 Гц), 114.15, 75.25, 74.20, 52.85, 40.15, 37.67, 28.46, 22.54, 10.24, 3.33. 19F ЯМР (282 МГц, CDCl3) δ -82.49 (д, J=75.4 Гц). Масс-спектр высокого разрешения (ESI): m/z 381.1729 (МН+); рассчитано для [C18H23F2N4O3+]: 381.1733.
4) 1,2-Оксазин 10 (293 мг, 0,771 ммоль) растворяли в ледяной уксусной кислоте (2,5 мл), добавляли цианоборгидрид натрия (435 мг, 6,905 ммоль). Перемешивали реакционную смесь в течение 4 часов. Реакционную смесь гасили избытком насыщенного водного раствора карбоната натрия (60 мл), раствор экстрагировали этилацетатом (60 мл и 2 раза по 40 мл). Экстракт промывали водой (70 мл) и насыщенным раствором хлорида натрия в воде (70 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (10:0 → 5:1 → 3:1) в качестве элюента, с получением 3-(азидометил)-4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-6,6-диметил-1,2-оксазинана 11 в виде прозрачного маслообразного вещества с выходом 180 мг (61%). 1Н ЯМР (300 МГц, CDCl3) 7.11 (д, J=7.7 Гц, 1Н), 6.77 (с, 1Н), 6.76 (д, J=7.7 Гц, 1Н), 6.61 (т, J=75.6 Гц, 1Н), 5.33 (с, ушир., 1Н), 3.87 (д, J=6.9 Гц, 2Н), 3.32 (д, J=10.8 Гц, 1Н), 3.21-3.06 (м, 1Н), 3.13 (д, J=10.8 Гц, 1H), 2.83 (тд, J=10.1, 7.4 Гц, 1Н), 1.78-1.68 (м, 2Н), 1.40 (с, 3H), 1.33-1.20 (м, 1H, 18-СН), 1.24 (с, 3H), 0.65 (м, 2Н), 0.36 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 149.51, 140.44, 139.45, 123.22, 120.21, 115.36 (т, J=256.9 Гц), 113.92, 74.84, 74.12, 61.51, 51.38, 43.85, 40.93, 29.29, 22.07, 10.31, 3.31. 19F ЯМР (282 МГц, CDCl3) δ -82.39 (д, J=75.6 Гц). Масс-спектр высокого разрешения (ESI): m/z 383.1877 (МН+); рассчитано для [C18H25F2N4O3+]: 383.1889.
5) Азид 11 (152 мг, 0,398 ммоль) растворяли в метаноле (2 мл), добавляли взвесь никеля Ренея в метаноле (0,5 мл). Гидрировали в автоклаве при комнатной температуре и давлении водорода 10 бар в течение 1 часа, затем добавляли никель Ренея и гидрировали в течение 40 минут при тех же условиях. Отделяли никель Ренея фильтрованием, промывали метанолом (6 раз по 3 мл), и объединенный раствор упаривали при пониженном давлении. Растворяли полученное вещество в сухом дихлорметане (2,8 мл), охлаждали раствор до 0°C. Добавляли раствор трифосгена (59 мг, 0,199 ммоль) в сухом дихлорметане (1 мл) и перегнанный триэтиламин (170 мкл, 1,222 ммоль). Перемешивали в атмосфере аргона в течение 2 часов. Гасили реакцию добавлением 10% водного раствора соляной кислоты (10 мл), разбавляли водой (50 мл) и экстрагировали дихлорметаном (50 мл и 2 раза по 30 мл). Экстракт промывали водой (2 раза по 50 мл) и насыщенным раствором хлорида натрия в воде (50 мл), сушили сульфатом натрия и упаривали при пониженном давлении. Продукт очищали колоночной хроматографией с помощью смеси гексана и этилацетата (1:1 → 0:1) в качестве элюента, с получением 4-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2,2-диметилгексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-она Iб в виде твердого белого вещества с выходом 78 мг (51%). Т. пл. 163-165°C. 1Н ЯМР (400 МГц, CDCl3) δ 7.13 (д, J=8.7 Гц, 1Н), 6.78 (дд, J=8.7, 2.0 Гц, 1Н), 6.77 (д, J=2.0 Гц, 1Н), 6.61 (т, J=75.5 Гц, 1Н), 4.62 (с, 1H), 3.88 (д, J=6.9 Гц, 2Н), 3.67 (ддд, J=10.9, 7.3, 6.2 Гц, 1Н), 3.30 (дд, J=9.3, 7.3 Гц, 1Н), 3.06 (дд, J=9.3, 6.2 Гц, 1H), 3.03 (дд, J=10.9, 6.2 Гц, 1Н), 1.86-1.80 (м, 2Н), 1.48 (с, 3H), 1.38 (с, 3H), 1.31-1.23 (м, 1Н), 0.69-0.63 (м, 2Н), 0.41-0.32 (м, 2Н). 13С ЯМР (101 МГц, CDCl3) δ 160.13, 151.09, 139.88, 138.90, 123.34, 120.18, 116.25 (т, J=260.0 Гц), 113.93, 79.81, 74.30, 60.32, 42.98, 41.25, 40.96, 28.51, 23.00, 10.34, 3.36. 19F ЯМР (282 МГц, CDCl3) δ -82.48 (д, J=75.7 Гц). Масс-спектр высокого разрешения (ESI): m/z 383.1775 (МН+); рассчитано для [C19H25F2N2O4+]: 383.1777.
Пример 3. Получение 4-фенилгексагидро-7Н-имидазо[1,5-b][1,2]оксазин-7-она (Iв).
Аналогично примеру 1 из бензальдегида получено соединение Iв с выходом 11% (0.05 г). Маслообразное вещество. 1Н ЯМР (300 МГц, CDCl3) δ 7.30-7.10 (м, 5Н), 5.45 (с, 1H), 4.11 (ддд, J=10.6, 4.5, 2.4 Гц, 1H), 4.02 (ддд, J=12.0, 10.6, 2.4 Гц, 1H), 3.84 (м, 1H), 3.31 (дд, J=9.1, 6.6 Гц, 1H), 3.04 (м, 1Н), 2.98 (ддд, J=12.0, 10.6, 3.9 Гц, 1H), 2.14 (м, 1Н), 1.91 (м, 1Н). Масс-спектр высокого разрешения (ESI): m/z 219.1141 (МН+); рассчитано для [C12H15N2O2+]: 219.1134. Элементный анализ: найдено, %: С, 66.35, Н 6.27; N 12.80. Вычислено для C12H14N2O2, %: С, 66.04; Н, 6.47; N, 12.84.
Пример 4. Получение 4-(3-(циклопентилокси)-4-метоксифенил)гексагидро-7Н-имидазо[1,5-b][1,2]оксазин-7-она (Iг).
Аналогично примеру 1 из 3-(циклопентилокси)-4-метоксибензальдегида получено соединение Iг с выходом 13% (0.03 г). Маслообразное вещество. 1Н ЯМР (300 МГц, CDCl3) δ 6.83 (д, J=7.3 Гц, 1Н), 6.73 (д, J=7.3 Гц, 1Н), 6.72 (с, 1Н), 5.40 (с, 1Н), 4.76 (м, 1Н), 4.09 (м, 1Н), 4.04 (м, 1Н), 3.85-3.81 (с и м, 3Н и 1Н), 3.33 (дд, J=9.2, 6.7 Гц, 1Н), 3.01 (ддд, J=9.2, 2.4 Гц, 1H), 2.85 (ддд, J=12.5, 10.2, 4.5 Гц, 1Н), 2.12 (м, 1Н), 1.92 (м, 1Н), 1.56-1.71 (м, 2Н), 1.79-1.94 (м, 6Н). Масс-спектр высокого разрешения (ESI): m/z 333.1813 (МН+); рассчитано для [C18H25N2O4+]: 333.1814. Элементный анализ: найдено, %: С, 65.37; Н, 7.01; N, 8.25. Вычислено для C18H24N2O4, %: С, 65.04; Н, 7.28; N, 8.43.
Пример 5. Получение 4-(4-метоксифенил)гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-она (Iд).
Аналогично примеру 1 из анисового альдегида получено соединение Iд с выходом 9% (0.03 г). Маслообразное вещество. 1Н ЯМР (300 МГц, CDCl3) δ 7.08 (д, J=8.5 Гц, 2Н), 6.80 (д, J=8.5 Гц, 2Н), 5.51 (с, 1Н), 4.07 (м, 1Н), 4.01 (м, 1Н), 3.72 (с, 3H), 3.82 (м, 1H), 3.30 (дд, J=9.3, 6.9 Гц, 1Н), 3.01 (дд, J=9.3, 2.1 Гц, 1Н), 2.92 (ддд, J=12.1, 10.1, 4.3 Гц, 1Н), 2.13 (м, 1H), 1.90 (м, 1Н). Масс-спектр высокого разрешения (ESI): m/z 249.1238 (МН+); рассчитано для [C13H17N2O3+]: 249.1239. Элементный анализ: найдено, %: С, 62.80, Н 6.47; N 11.30. Вычислено для C13H16N2O3, %: С, 62.89; Н, 6.50; N, 11.28.
Пример 6. Получение 4-(3,4-диметоксифенил)гексагидро-7Н-имидазо[1,5-b][1,2]оксазин-7-она (Ie).
Аналогично примеру 1 из 3,4-диметоксибензальдегида получено соединение Ie с выходом 8% (0.03 г). Маслообразное вещество. 1Н ЯМР (300 МГц, CDCl3) δ 6.73 (д, J=7.6 Гц, 1Н), 6.64 (с, 1Н), 6.44 (д, J=7.6 Гц, 1Н), 5.54 (с, 1H), 4.10 (м, 1Н), 4.02 (м, 1Н), 3.82 и 3.83 (2 с и м, 3Н, 3Н и 1Н), 3.28 (дд, J=8.9, 6.7 Гц, 1Н), 3.02 (дд, J=8.9, 2.6 Гц, 1Н), 2.88 (ддд, J=12.5, 10.4, 3.9 Гц, 1Н), 2.13 (м, 1Н), 1.91 (м, 1Н). Масс-спектр высокого разрешения (ESI): m/z 279.1353 (МН+); рассчитано для [C14H19N2O4+]: 279.1345. Элементный анализ: найдено, %: С, 60.57; Н, 6.41; N, 9.89. Вычислено для C14H18N2O4, %: С, 60.42; Н, 6.52; N, 10.07.
Пример 7. Получение 2,2-диметил-4-фенилгексагидро-7Н-имидазо[1,5-b][1,2]оксазин-7-она (Iж).
Аналогично примеру 2 из 3,6,6-триметил-4-фенил-5,6-дигидро-4Н-1,2-оксазин 2-оксида (A.A. Tishkov, "2-Silyloxy-1,2-oxazines, a New Type of Acetals of Conjugated Nitroso Alkenes", J. Org. Chem., 2003, 68, 9477-9480) получено соединение Iж с выходом 57% (0.32 г). Маслообразное вещество. 1Н ЯМР (300 МГц, CDCl3) δ 7.45-7.12 (м, 5Н), 5.30 (с, 1H), 3.69 (ддд, J=10.9, 6.8, 6.8 Гц, 1Н), 3.28 (т, J=8.1 Гц, 1Н), 3.09 (м, 2Н), 2.00-1.75 (м, 2Н), 1.49 (с, 3H), 1.38 (с, 3H). Масс-спектр высокого разрешения (ESI): m/z 247.1453 (МН+); рассчитано для [C14H19N2O2+]: 247.1446. Элементный анализ: найдено, %: С, 68.55; Н, 7.23; N, 11.15. Вычислено для C14H18N2O2, %: С, 68.27; Н, 7.37; N, 11.37.
Пример 8. Получение 4-(4-метоксифенил)-2,2-диметилгексагидро-7Н-имидазо[1,5-b][1,2]оксазин-7-она (Iз).
Аналогично примеру 2 из 4-(4-метоксифенил)-3,6,6-триметил-5,6-дигидро-4Н-1,2-оксазин 2-оксида (A. Yu. Sukhorukov, "A Convenient procedure for the Synthesis of 3-substituted 5,6-Dihydro-4H-1,2-oxazines from Nitroethane", Synthesis, 2007, 97-107.) получено соединение Iз с выходом 45% (0.29 г). Т. пл. 153°C (с разложением).
1Н ЯМР (300 МГц, CDCl3) δ 7.09 (д, J=8.4 Гц, 2Н), 6.81 (д, J=8.4 Гц, 2Н), 5.05 (с, 1H), 3.82 (м, 3H), 3.71 (ддд, J=10.7, 7.3, 6.8 Гц, 1Н), 3.25 (м, 1Н), 3.15-2.95 (м, 2Н), 1.99-1.72 (м, 2Н), 1.48 (с, 3H), 1.37 (с, 3H). Масс-спектр высокого разрешения (ESI): m/z 277.1547 (МН+); рассчитано для [C15H21N2O3]: 277.1552. Элементный анализ: найдено, %: С, 65.41; Н, 7.11; N, 10.33. Вычислено для C15H20N2O3, %: С, 65.20; Н, 7.30; N, 10.14.
Пример 9. Получение 2-этокси-4-(4-метоксифенил)гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-она (Iи).
Аналогично примеру 2 из 6-этокси-4-(4-метоксифенил)-3-метил-5,6-дигидро-4Н-1,2-оксазин 2-оксида (A.A. Tishkov, "2-Silyloxy-1,2-oxazines, а New Type of Acetals of Conjugated Nitroso Alkenes", J. Org. Chem., 2003, 68, 9477-9480) получено соединение Iи с выходом 11% (0.09 г). Маслообразное вещество.
1Н ЯМР (300 МГц, CDCl3) δ 7.08 (д, J=8.5 Гц, 2Н), 6.82 (д, J=8.5 Гц, 2Н), 5.01 (с, 1Н), 4.81 (т, J=2.0 Гц, 1 Н), 3.82 (м, 3H), 3.78-3.71 (с и 2 м, 3Н, 1Н и 1H), 3.51 (м, 1H), 3.26-3.23 (м, 1Н), 3.15-2.97 (м, 2Н), 1.98-1.91 (2 м, 2Н), 1.25 (т, J=7.3 Гц, 3H). Масс-спектр высокого разрешения (ESI): m/z 293.1512 (МН*); рассчитано для [C15H21N2O4+]: 293.1501. Элементный анализ: найдено, %: С, 61.89; Н, 6.77; N, 9.65. Вычислено для C15H20N2O3, %: С, 61.63; Н, 6.90; N, 9.58.
Пример 10. Получение 2-метокси-4-(4-метоксифенил)-2-метилгексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-она (Iк).
Аналогично примеру 2 из 6-метокси-4-(4-метоксифенил)-3,6-диметил-5,6-дигидро-4Н-1,2-оксазин 2-оксида (A.A. Tishkov, "2-Silyloxy-1,2-oxazines, а New Type of Acetals of Conjugated Nitroso Alkenes", J. Org. Chem., 2003, 68, 9477-9480) получено соединение Iк с выходом 15% (0.1 г). Маслообразное вещество.
1Н ЯМР (300 МГц, CDCl3) δ 6.82 (д, J=8.4 Гц, 2Н), 7.09 (д, J=8.4 Гц, 2Н), 5.32 (с, 1Н), 3.82 (м, 3H), 3.73 (ддд, J=10.9, 7.0, 7.0 Гц, 1Н), 3.29-3.22 (с и м, 3Н и 1Н), 3.14-2.94 (м, 2Н), 1.98-1.70 (м, 2Н), 1.31 (с, 3H). Масс-спектр высокого разрешения (ESI): m/z 293.1495 (МН*); рассчитано для [C15H21N2O4+]: 293.1501. Элементный анализ: найдено, %: С, 61.48; Н, 6.92; N, 9.35. Вычислено для C15H20N2O3, %: С, 61.63; Н, 6.90; N, 9.58.
Пример 11. (Сравнительный) Получение 4-(1-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-3-гидрокси-3-метилбутил)имидазолидин-2-он (VIII).
Продукт Iб (39 мг, 0,102 ммоль) растворяли в метаноле (1 мл), добавляли взвесь никеля Ренея в метаноле (0,25 мл). Гидрировали в автоклаве при 70°C и давлении водорода 40 бар в течение 5.5 часов. Отделяли никель Ренея фильтрованием, промывали метанолом (6 раз по 3 мл), и объединенный раствор упаривали при пониженном давлении. Остаток хроматографировали на силикагеле. Получали 34 мг (87%) имидазолидин-2-она VIII в виде бесцветного масла. 1Н ЯМР (300 МГц, CDCl3) δ 7.26 (с, 1H), 7.08 (д, J=8.4 Гц, 1Н), 6.75 (с, 1Н), 6.71 (д, J=8.4 Гц, 1Н), 6.59 (т, J=75.5 Гц, 1Н), 4.86 (с, 1Н), 3.85 (д, J=6.7 Гц, 2Н), 3.10 (дд, J=8.9, 8.6 Гц, 1Н), 3.00 (т, J=8.6, 8.2 Гц, 1Н), 2.96-2.82 (м, 2Н), 1.99 (дд, J=14.7, 4.3 Гц, 1Н), 1.87 (дд, J=14.7, 7.1 Гц, 1H), 1.32-1.22 (м, 1Н), 1.25 (с, ушир., 1H), 1.19 (с, 3H), 1.10 (с, 3H), 0.64 (м, 2Н), 0.35 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ 164.38, 150.78, 141.81, 139.37 (т, J=2.9 Гц), 123.05, 120.65, 116.32 (т, J=259.6 Гц), 114.54, 74.17, 70.64, 58.87, 47.81, 47.02, 45.61, 31.90, 28.36, 10.29, 3.30. 19F ЯМР (282 МГц, CDCl3) δ -82.36 (д, J=75.5 Гц). Масс-спектр высокого разрешения (ESI): m/z 385.1925 (МН*); рассчитано для [C19H26F2N2O4+]: 385.1933.
Пример 12. Исследование активности полученных соединений в ингибировании фосфодиэстеразы подтипа 4В1 in vitro
Испытание соединений на ингибирование процесса гидролиза циклического аденозинмонофосфата под действием ФДЭ4В1 осуществляли следующим образом, основанным на известной процедуре (P.Н. Schafer, "Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity", Cellular Signalling, 2014, 26, 2016-2029). В экспериментах проводили энзиматические реакции между флуоресцентным субстратом ФАМ-цАМФ (2-(6-[флуоресценил]аминогексил-карбомоил)аденозин-3,5-циклический монофосфат), ферментом ФДЭ4В1 и исследуемым ингибитором в буферном растворе (диапазон изученных концентраций ингибитора от 10 мкМ до 0.3 нМ, 10 измерений). Определение ингибирующей активности лиганда (концентрации полу-ингибирования IC50) производилось методом поляризации флуоресценции (возбуждение при 485 нм, испускание при 528 нм) в ходе энзиматической реакции. Результаты исследования суммированы в Таблице.
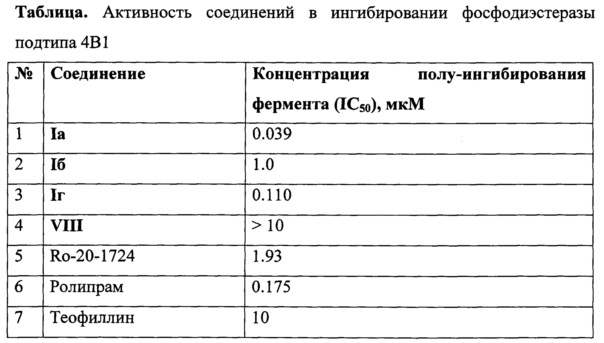
Из таблицы на примере соединений Ia, Iб и Iг видно, что они подавляют активность фермента ФДЭ4В1 на 50% в микро- и наномолярных концентрациях. Активность этих соединений на порядки превышает активность штатного препарата сравнения Теофиллин.
Пример 13. Исследование активности соединения Ia в ингибировании различных изотипов фосфодиэстераз подтипа 4 in vitro.
Для определения селективности соединения Ia проводились аналогичные исследования in vitro на панели из 8 изотопных фосфодиэстераз подтипа 4 (изотипы ФДЭ4А1А, ФДЭ4А4В, ФДЭ4А10, ФДЭ4В1, ФДЭ4В2, ФДЭ4С1, ФДЭ4D2, ФДЭ4D3). Эти исследования проводились при одной концентрации соединения (0.100 мкМ) и измерялась его ингибирующая способность в % от протекания реакции гидролиза субстрата ФАМ-цАМФ. Результаты исследования суммированы на Фиг.
Фиг. Активность соединения Ia в ингибировании различных изотипов фосфодиэстераз подтипа 4 in vitro:
(i) фосфодиэстераза ФДЭ4А1А; (ii) фосфодиэстераза ФДЭ4А4В; (iii) фосфодиэстераза ФДЭ4А10; (iv) фосфодиэстераза ФДЭ4В1; (v) фосфодиэстераза ФДЭ4В2; (vi) фосфодиэстераза ФДЭ4С1; (vii) фосфодиэстераза ФДЭ4Б2; (viii) фосфодиэстераза ФДЭ4D3.
На Фиг. на примере соединения Ia показано, что 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-оны проявляет селективность в отношении наиболее терапевтически значимых фосфодиэстераз подтипов 4В и 4А. Оно проявляет меньшую активность в отношении фосфодиэстераз подтипа 4D, ингибирование которых ассоциируют с возникновением побочных эффектов.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она | 2022 |
|
RU2789599C1 |
| ПРОТИВОВОСПАЛИТЕЛЬНОЕ СОЕДИНЕНИЕ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2019 |
|
RU2759626C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ РЕЦЕПТОРОВ CGRP | 2013 |
|
RU2672056C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| ПРОИЗВОДНЫЕ ГАЛАНТАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2241001C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МУСКАРИНОВЫЕ АГОНИСТЫ И КОМПОЗИЦИИ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2002 |
|
RU2292346C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2012 |
|
RU2605537C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| СОЕДИНЕНИЕ ОКСАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2418793C2 |

Настоящее изобретение относится к способу получения замещенных 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-онов общей формулы:
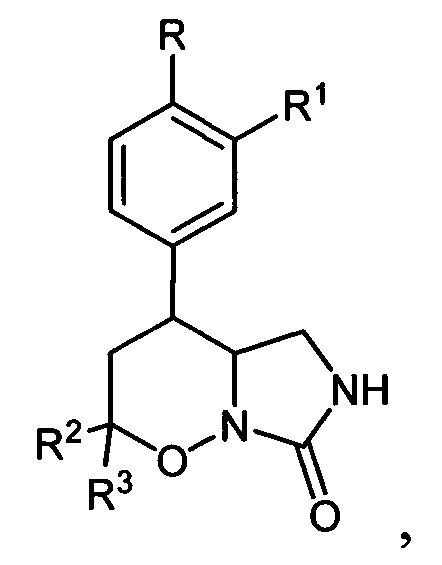 проявляющих ингибирующую активность в отношении фосфодиэстеразы подтипа 4. Способ заключается в том, что 5,6-дигидро-4Н-1,2-оксазин-N-оксиды общей формулы:
проявляющих ингибирующую активность в отношении фосфодиэстеразы подтипа 4. Способ заключается в том, что 5,6-дигидро-4Н-1,2-оксазин-N-оксиды общей формулы:
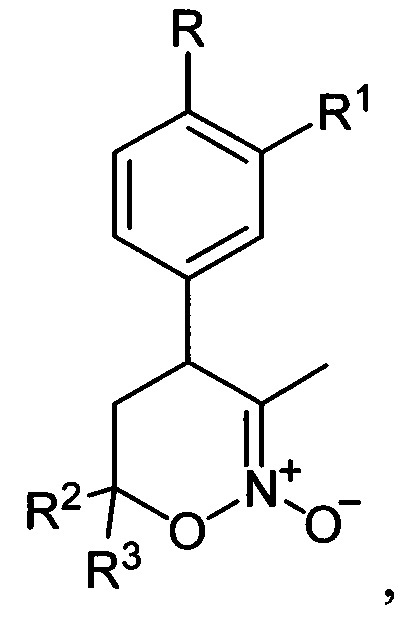 подвергают последовательной обработке триметилсилилбромидом с азотистым основанием и бромидом металла в среде апротонного растворителя при пониженной температуре с последующей обработкой образующихся при этом соответствующих бромидов азидом натрия при повышенной температуре в среде апротонного растворителя с получением полученные соответствующих 3-азидометил-5,6-дигидро-4Н-1,2-оксазины общей формулы:
подвергают последовательной обработке триметилсилилбромидом с азотистым основанием и бромидом металла в среде апротонного растворителя при пониженной температуре с последующей обработкой образующихся при этом соответствующих бромидов азидом натрия при повышенной температуре в среде апротонного растворителя с получением полученные соответствующих 3-азидометил-5,6-дигидро-4Н-1,2-оксазины общей формулы:
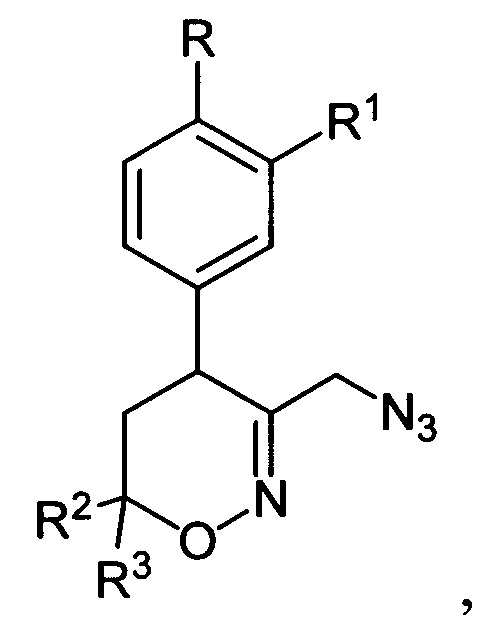 которые подвергают взаимодействию с цианоборгидридом натрия в протонной кислоте с последующим каталитическим восстановлением полученных при этом соответствующих 3-азидометил-1,2-оксазинанов общей формулы:
которые подвергают взаимодействию с цианоборгидридом натрия в протонной кислоте с последующим каталитическим восстановлением полученных при этом соответствующих 3-азидометил-1,2-оксазинанов общей формулы:
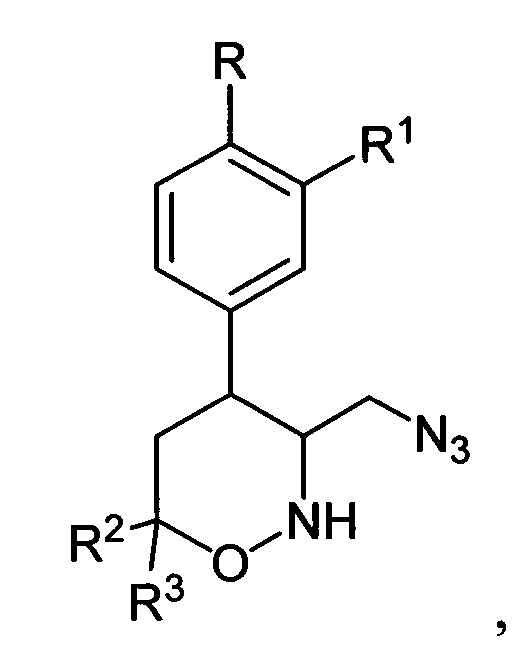 водородом в присутствии металлического катализатора в среде низкомолекулярного спирта и обработкой реакционной смеси карбомоилирующим реагентом с азотистым основанием в апротонном растворителе. Также изобретение относится к новому соединению - 4-[3-(циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-ону. 2 н. и 2 з.п. ф-лы, 1 ил., 1 табл., 13 пр.
водородом в присутствии металлического катализатора в среде низкомолекулярного спирта и обработкой реакционной смеси карбомоилирующим реагентом с азотистым основанием в апротонном растворителе. Также изобретение относится к новому соединению - 4-[3-(циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-ону. 2 н. и 2 з.п. ф-лы, 1 ил., 1 табл., 13 пр.
1. 4-[3-(Циклопропилметокси)-4-(дифторметокси)фенил]гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-он формулы:
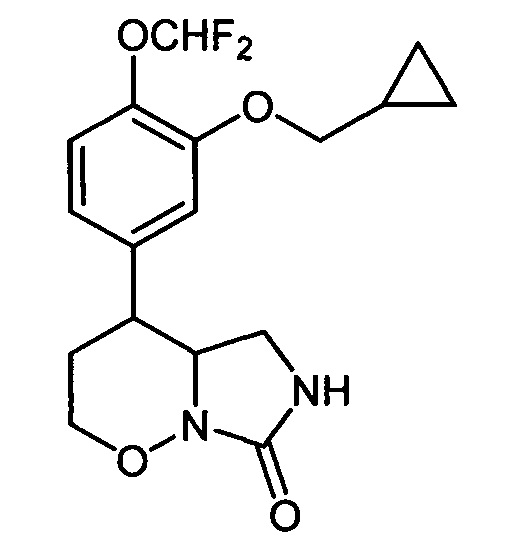
2. Соединение по п. 1, обладающее ингибирующей активностью в отношении фосфодиэстераз подтипа 4.
3. Способ получения замещенных 4-арил-гексагидро-7H-имидазоло[1,5-b][1,2]оксазин-7-онов общей формулы:
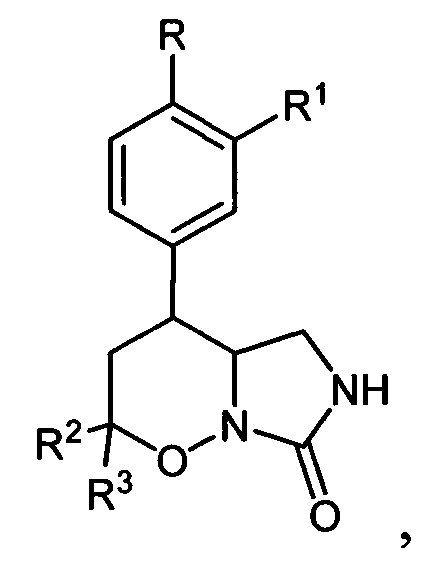
где R=Н, С1-С4 - алкокси-группа либо OCHF2; R1 = Н, С1-С4 - алкокси-группа, С3-С6 - циклоалкокси-группа либо С3-С6 - циклоалкилметокси-группа; R2, R3 = Н, С1-С4 - алкил либо С1-С4 - алкокси-группа, заключающийся в том, что 5,6-дигидро-4Н-1,2-оксазин-N-оксиды общей формулы:
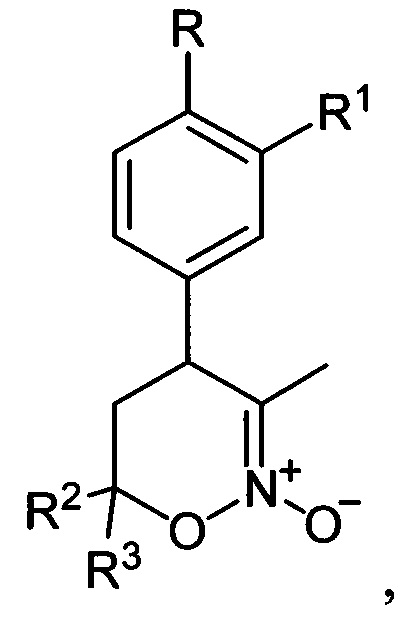
где R, R1, R2 и R3 имеют вышеуказанные значения, подвергают последовательной обработке триметилсилилбромидом с азотистым основанием и бромидом металла в среде апротонного растворителя при пониженной температуре с последующей обработкой образующихся при этом соответствующих бромидов азидом натрия при повышенной температуре в среде апротонного растворителя, полученные при этом соответствующие 3-азидометил-5,6-дигидро-4Н-1,2-оксазины общей формулы:
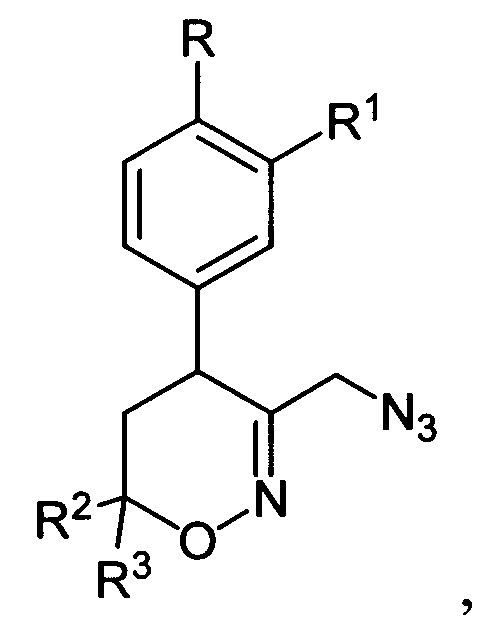
где R, R1, R2 и R3 имеют вышеуказанные значения, подвергают взаимодействию с цианоборгидридом натрия в протонной кислоте с последующим каталитическим восстановлением полученных при этом соответствующих 3-азидометил-1,2-оксазинанов общей формулы:
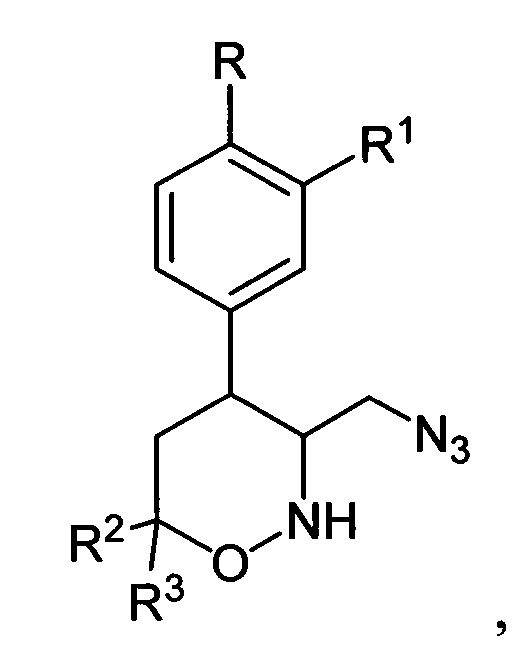
где R, R1, R2 и R3 имеют вышеуказанные значения, водородом в присутствии металлического катализатора в среде низкомолекулярного спирта и обработкой реакционной смеси карбомоилирующим реагентом с азотистым основанием в апротонном растворителе.
4. Способ получения соединений по п. 3, отличающийся тем, что в качестве азотистого основания используют триэтиламин, в качестве бромида металла используют бромид кобальта, в качестве металлического катализатора используют никель Ренея, в качестве карбомоилирующего агента используют трифосген, в качестве протонной кислоты используют уксусную кислоту, а в качестве апротонного растворителя используют ДМФА, ТГФ или хлористый метилен.
| Dorokhov V.S | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Москва, РУДН, 24-28 апреля 2017г | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЖМУРОВ П.А., и др."СИНТЕЗ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ IVB | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| A | |||
| Yu | |||
| Sukhorukov et al, "Diastereoselective Synthesis of γ-Amino Acids and Their Derivatives from Nitroethane via Intermediacy of 5,6-Dihydro-4H-1,2-oxazines Bearing the CH 2 CH(CO 2 Me) 2 Substituent at C3", Synthesis, No.5, 2009, 741-754. | |||

