Область техники
Настоящее изобретение относится к новым производным каннабидиол-хинона и синтезу этих соединений. Кроме того, настоящее изобретение относится к применению указанных соединений в качестве лекарственного средства, а также для терапии, в частности, в качестве модуляторов гамма-рецепторов активации пролиферации пероксисом (PPARg), для лечения заболеваний и состояний, восприимчивых к модуляции PPARg. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, и к способу лечения заболеваний с помощью указанных соединений.
Уровень техники
Ядерные рецепторы (NR) представляют большой интерес для исследования и разработки лекарственных средств. NR представляют собой лиганд-зависимые факторы транскрипции, которые обладают способностью непосредственно взаимодействовать с ДНК, регулируя транскрипционную активность генов-мишеней. Данные рецепторы играют важную роль в развитии, клеточном гомеостазе и метаболизме, и они вовлечены в широкий круг заболеваний, в связи с чем прилагаются усилия для их изучения и разработки новых лекарственных средств для фармацевтической промышленности.
В новейшей номенклатуре ядерных рецепторов подсемейство 1 С (NR1C) включает три подтипа рецепторов активации пролиферации пероксисом (PPAR) млекопитающих: PPARα (также называемый NR1C1), PPARβ/δ (также называемый NR1C2) и PPARγ (также называемый PPARg, рецептор глитазона или NR1C3). PPAR контролируют экспрессию генных сетей, участвующих в адипогенезе, липидном обмене, воспалении и поддержание метаболического гомеостаза [Barish et al., 2006]. PPAR активируют транскрипцию генов путем связывания с элементами последовательностей ДНК, известных как элементы ответа пролиферации пероксисом (PPRE) в регуляторной области генов-мишеней PPAR [Poulsen et al., 2012]. Кроме того, PPAR негативно регулируют транскрипцию генов воспалительного ответа посредством антагонизма сигнальных путей белка-активатора-1 (АР-1), ядерного фактора каппа-В (NF-kB), переносчика сигнала и активатора транскрипции 3 (STAT3) и ядерного фактора активированных Т-клеток (NFAT) [Vanden Bergheetal. 2003].
Среди PPAR особый интерес представляет PPARg, так как он участвует в регуляции образования адипоцитов, чувствительности к инсулину и воспалительных процессов [Fievet et al. 2006] [Stienstra et al. 2007] [Tontonoz and Spiegelman, 2008]. PPARg экспрессируется в ряде тканей, включая жировую ткань, клетки скелетной мускулатуры, остеокласты, остеобласты, клетки иммунной системы, а также центральную и периферическую нервную систему. Очевидно, что PPARg является "мастером" или доминирующим регулятором адипогенеза, в связи с тем фактом, что он одновременно является необходимым и достаточным для дифференциации жировых клеток. Регуляторные области большого числа генов, которые играют важную роль в липогенезе и чувствительности к инсулину, такие как аР2, LPL, адипонектин и Glut4, содержат сайты связывания с PPARg [Rosen and MacDougald, 2006]. Следовательно, активация PPARg в жировой ткани воздействует на чувствительность к инсулину во всем организме.
В дополнение к участию этого рецептора в регуляции метаболического гомеостаза, сообщалось о новых возможных механизмах действия PPARg, включая в особенности противовоспалительные, противоопухолевые и антифиброзные свойства [Zhao et al., 2006]. Блокирование сигнализации TGFb/Smad путем активации PPARg приводит к уменьшению отложения коллагена при печеночном, легочном и почечном фиброзе [Ferguson et al., 2009] [Wang et al., 2007] [Zhang et al., 2009]. С другой стороны, активация PPARg оказывает противовоспалительное действие на несколько типов клеток путем ингибирования экспрессии провоспалительных генов, тем самым снижая образование цитокинов, металлопротеиназ и белков острой фазы [Tontonoz and Spiegelman, 2008]. Они также действуют путем повышения противовоспалительных цитокинов и ингибирования экспрессии индуцибельной синтазы окиси азота (iNOS) [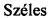 et al., 2007]. Интересно отметить, что агонисты PPARg продемонстрировали противовоспалительное и нейропротекторное действие на нескольких экспериментальных моделях болезни Паркинсона, бокового амиотрофического склероза, рассеянного склероза и инсульта, а также в ряде клинических исследований [Bernardo and Minghetti, 2008]. В этом отношении было показано, что агонисты PPARg имеют высокий уровень экспрессии в предшественниках нейронов (NP), обработанных ретиноевой кислотой, и они участвуют в двух стадиях дифференцировки нейронов эмбриональных стволовых клеток мыши во время и после выработки NP [Ghoochani et al., 2012]. Кроме того, в генетическом смысле необходимо формально рассматривать PPARg как ген-супрессор опухолей. Они экспрессируются в различных опухолевых клетках, а активация PPARg лигандами приводит к ингибированию пролиферации клеток или индукции апоптоза [Tachibana et al., 2008] [Tontonoz and Spiegelman, 2008].
et al., 2007]. Интересно отметить, что агонисты PPARg продемонстрировали противовоспалительное и нейропротекторное действие на нескольких экспериментальных моделях болезни Паркинсона, бокового амиотрофического склероза, рассеянного склероза и инсульта, а также в ряде клинических исследований [Bernardo and Minghetti, 2008]. В этом отношении было показано, что агонисты PPARg имеют высокий уровень экспрессии в предшественниках нейронов (NP), обработанных ретиноевой кислотой, и они участвуют в двух стадиях дифференцировки нейронов эмбриональных стволовых клеток мыши во время и после выработки NP [Ghoochani et al., 2012]. Кроме того, в генетическом смысле необходимо формально рассматривать PPARg как ген-супрессор опухолей. Они экспрессируются в различных опухолевых клетках, а активация PPARg лигандами приводит к ингибированию пролиферации клеток или индукции апоптоза [Tachibana et al., 2008] [Tontonoz and Spiegelman, 2008].
Положительные эффекты активации PPARg с помощью специфических лигандов агонистов можно использовать для лечения некоторых хронических заболеваний, таких как сахарный диабет, атеросклероз, ревматоидный артрит, фиброз печени, воспалительные заболевания кишечника, нефропатия, псориаз, заживление ран на коже, склеродермия (SSc), нейродегенеративные и нейровоспалительные расстройства и рак.
Среди активаторов лигандов PPARg наибольшее клиническое значение имеют тиазолидиндионы (TZD) [Lehmann et al., 1995]. По этой причине в клинической практике до сих пор широко применяют росиглитазон и пиоглитазон. Они оказывают похожее воздействие на гликемический контроль, а также вызывают целый ряд похожих нежелательных явлений, таких как увеличение массы тела, задержка жидкости и повышенный риск сердечной недостаточности, которые, по-видимому, опосредованы активностью PPARg. Действительно, недавно в Европе отменили росиглитазон, а в США ограничили его применение вследствие повышенного риска сердечнососудистых явлений у пациентов с сахарным диабетом 2 типа.
Несмотря на то, что TZD являются сильнодействующими полными агонистами PPARg (PPARg-fa), побочные эффекты, обусловленные механизмом их действия, ограничивают полный терапевтический потенциал данных соединений [Gelman et al., 2007] [Ciudin et al., 2012]. Но физиологическая и терапевтическая значимость пути PPARg способствовала проведению новых исследований, посвященных разработке новых классов молекул, которые уменьшают или устраняют нежелательные явления [Ahmadian et al., 2013]. Таким образом, был достигнут значительный прогресс в исследовании и разработке селективных модуляторов PPARg (PPARg-m), как более безопасной альтернативы PPARg-fa. Доклинические и клинические данные ясно свидетельствуют о том, что селективные PPARg-m в перспективе могут стать следующим поколением агонистов PPARg: эффективных сенсибилизаторов инсулина с превосходным профилем безопасности, по сравнению с PPARg-fa. [Doshi et al.2010].
В этом отношении природные и синтетические каннабиноиды рассматриваются как PPARg-m, которые уменьшают воспалительный процесс посредством активации PPARg. Некоторыми примерами PPARg-m на основе каннабиноидов являются аджулемовая кислота [Liu et al., 2003], [Burstein S. 2005], WIN55212-2 [Sun and Bennett, 2007], 9Δ-ТНС и CBD [O'Sullivan 2007] и CBG [Granja et al., 2012].
Были описаны некоторые хинононовые производные каннабиноидов, такие как CBD-Q (HU-311, в настоящем изобретении также называемый VCE-004) и CBG-Q (VCE-003) [Kogan et al., 2004] [Granja et al., 2012]. Интересно отметить, что VCE-004 (также известный как HU-331) демонстрировал концентрацию ЕС50, составляющую 5 μM, таким образом проявляя в четыре раза более высокую аффинность связывания по сравнению с исходной молекулой CBD (ЕС50 21 μМ), и VCE-003 показал значительное усиление аффинности связывания с PPARg (ЕС50 2,2 μM) по сравнению с исходной молекулой CBG (ЕС50 12,7 μМ) [Granja et al., 2012]. Также были описаны другие CBD-хиноны, такие как CBD-1,4-дигидроксихинон, 4-метил-CBD-хинон и 4-формил-метокси-CBD-хинон, и они проявили более высокую аффинность связывания с PPARg по сравнению с исходной молекулой CDB [WO 2011117429 А1]. Однако выполнение синтеза таких соединений представляется очень сложным, и полученные соединения очень нестабильны, что делает их непригодными для фармацевтической разработки.
Хиноны представляют собой класс токсичных промежуточных соединений, которые могут приводить к различным опасным эффектам in vivo, включая острую цитотоксичность и иммунотоксичность [Bolton et al., 2000]. Механизмы, с помощью которых хиноны вызывают данные эффекты, могут быть достаточно сложными. Хиноны являются акцепторами Михаэля и повреждение клеток может происходить из-за алкилирования важных клеточных белков и/или ДНК. Альтернативно, хиноны являются высоко активными окислительно-восстановительными молекулами, которые могут вступать в окислительно-восстановительный цикл с их полухиноновыми радикалами, приводя к образованию активных форм кислорода (АФК), что может привести к серьезному окислительному стрессу внутри клеток через образование окисленных клеточных макромолекул, включая липиды, белки и ДНК [Monks and Jones, 2012]. Хотя существуют многочисленные примеры соединений на основе хинонов с терапевтическим применением, опасения по поводу неспецифической токсичности и отсутствия селективности, возможности быть акцепторами Михаэля редко закладывают в разработку прототипов лекарственных средств.
Путь Keap1-Nrf2 является главным регулятором цитопротекторных ответов на эндогенный и экзогенный стресс, вызванный активными формами кислорода (АФК) и электрофилами. Ключевыми сигнальными белками пути являются ядерный фактор транскрипции-2 (родственный эритроидному фактору 2) (Nrf2), который связывает вместе небольшие белки Maf с элементом антиоксидантного ответа (ARE) в регуляторных областях генов-мишеней. В обычных условиях Nrf2 сохраняется в цитоплазме благодаря ингибитору Keap1 (Kelch ЕСН-связанный белок 1). Когда клетки подвергаются окислительному стрессу, воздействию электрофилов или химиопрофилактических агентов, Nrf2 освобождается от Keap1-опосредованной репрессии и активирует экспрессию генов, зависящую от элемента антиоксидантного ответа (ARE), с целью поддержания клеточного окислительно-восстановительного гомеостаза [Na and Surh, 2013].
Nrf2 может защищать клетки и ткани от разнообразных токсикантов и канцерогенов путем усиления экспрессии ряда цитопротекторных генов. Исследования показали, что так же как Nrf2 защищает нормальные клетки, Nrf2 также может защищать раковые клетки от химиотерапевтических агентов и способствовать развитию опухолей [Na and Surh 2013]. Раковые клетки переживают постоянный эндогенный окислительный стресс и становятся устойчивыми к воздействию некоторых противораковых агентоа, которые вызывают цитотоксичность за счет образования АФК. В таких условиях, активный путь Nrf2 может поддерживать подходящий окислительно-восстановительный баланс в раковых клетках, сохраняя уровни АФК в пределах диапазона, который способствует их росту и выживанию. Предположительно, длительное накопление или активация Nrf2 обеспечивает для подгруппы предраковых или раковых клеток оптимальные условия для пролиферации, а также возможность избежать апоптоза, метастазировать и переносить терапевтическое вмешательство.
Как известно, ингибирование гиперэкспрессии Nrf2 изменяет фенотипические характеристики раковых клеток, тем самым подтверждая данное предположение [Sporn and Liby, 2012]. Конститутивная гиперактивация Nrf2 наблюдается во многих типах злокачественных новообразований, таких как плоскоклеточный рак, рак легких, рак молочной железы, рак желчного пузыря, рак предстательной железы, рак почки, эпендимомы, эпителиальная карцинома яичников, рак эндометрия и рак поджелудочной железы [Na and Surh, 2013]. У больных раком пациентов с постоянно повышенным уровенем экспрессии Nrf2 в опухолях, в целом наблюдается более низкий уровень выживаемости [Solis et al., 2010]. Таким образом, активация Nrf2 рассматривается как прогностический молекулярный маркер для определения статуса прогрессирования рака и способствует развитию природной и приобретенной устойчивости к химиотерапии. Таким образом, данный антиоксидантный фактор транскрипции также может выступать как протоонкоген, и усиленная активность Nrf2 способствует возникновению солидного рака и устойчивости к химиотерапии [Sporn and Liby, 2012].
С целью улучшения только агонистической активности PPARg, но не допуская активацию Nrf2 во избежание возможных побочных эффектов, в рамках настоящего изобретения была разработана серия новых соединений, начиная с VCE-004 и каннабидиоловой кислоты (CBDA) в качестве опорных соединений. Авторами настоящего изобретения было неожиданно обнаружено, что производные CBD-хинона (производные CBD-Q) с определенными модификациями в положении 3 позволяют получить новые соединения с высоким агонистическим эффектом PPARg, но которые при этом не проявляют электрофильную (активации Nrf2) и цитотоксическую активности. Таким образом, новые соединения подходят для лечения хронических заболеваний, чувствительных к модуляции PPARg.
VCE-004 (соединение I), предшественник производных CBD-Q с II по X согласно настоящему изобретению, представляет собой агонистический лиганд PPARg, который также активирует фактор транскрипции Nrf2, являющийся клеточным сенсором окислительного/электрофильного стресса, отражающим образование АФК в клетках, обработанных VCE-004. Поэтому длительное лечение данным типом производных CBD-Q, которые активируют путь Nrf2, может привести к прогрессированию опухоли, как было объяснено выше. Кроме того, хроменопиразоледионы (chromenopyrazolediones), которые являются структурными аналогами CBD-Q, вызывают цитотоксичность в клетках рака простаты посредством индукции активных форм кислорода (АФК) и PPARg-зависимых механизмов [Morales et al., 2013]. Таким образом, окисление молекулы CBD позволяет получить класс соединений CBD-Q, таких как, например, VCE-004, которые активируют PPARg, а также вызывают АФК-опосредованную активацию Nrf2.
Такие производные CBD-Q согласно настоящему изобретению отличаются от соединений, описанных Коганом и др. [Kogan et al., 2004] и Моралесом и др. [Morales et al., 2013], так как модификации в положении 3 придают соединениям настоящего изобретения способность активировать PPARg и защищают от глутамат-индуцированной цитотоксичности без активации Nrf2. Кроме того, производные CBD-Q с модификациями в положении 3 также ингибируют TGFb-индуцированную транскрипцию и экспрессию гена коллагена. Соединения, описанные в настоящем изобретении, также отличаются от соединений, описанных в WO 20011117429, которые являются нестабильными, их трудно синтезировать, и их никогда не тестировали на активацию Nrf2. Производные CBD-Q, описанные в настоящем изобретении, также показали удивительно низкую цитотоксичность на клеточных линиях нейронного происхождения по сравнению с VCE-004 (соединение I), известного из уровня техники.
Краткое описание изобретения
Исходя из предшествующего уровня техники, задача настоящего изобретения заключается в создании новых производных каннабидиол-хинона (производных CBD-Q), которые проявляют активность при модуляции PPARg, не вызывая активацию Nrf2 и цитотоксичность.
Более конкретно, в настоящем изобретении соединения представляют собой производные каннабидиол-хинона (производные CBD-Q) формулы (I):
где R представляет собой атом углерода линейной или разветвленной группы, представленной: алкильной, арильной, алкенильной, алкинильной, ацильной или алкоксикарбонильной группами; или где R представляет собой атом азота линейной или разветвленной группы, представленной: алкиламинной, ариламинной, алкениламинной или алкиниламинной группами. Кольцо хинона пронумеровано произвольным образом, чтобы показать, в каком положении кольца произведена замена заместителей для визуализации производных CBD-Q настоящего изобретения. Поскольку номенклатура IUPAC могла бы позволить это, нумерацию кольца хинона сохраняли (см. производные формулы II до X, при этом положение 3 указанного кольца хинона являлось положением, где происходили все замены заместителей и номенклатура указанных производных совпадала и отражала данный факт). Однако, когда группы заместителей, связанные в положении 3 кольца хинона, изменяли нумерацию положений вышеуказанного кольца хинона, предписанную номенклатурой IUPAT, применяли результирующую номенклатуру (outcoming nomenclature), хотя только по внешнему виду замена в положении 3 кольца хинона была как будто пропущена, но на самом деле это не так, как это показано с помощью графических формул производных, представленных формулами с XI по XV.
В предпочтительном варианте реализации соединения согласно настоящему изобретению представляют собой соединения формулы (II), (III), (IV), (V), (VI), (VII), (VIII), (X), (XI), (XII), (XIII), (XIV) и (XV).
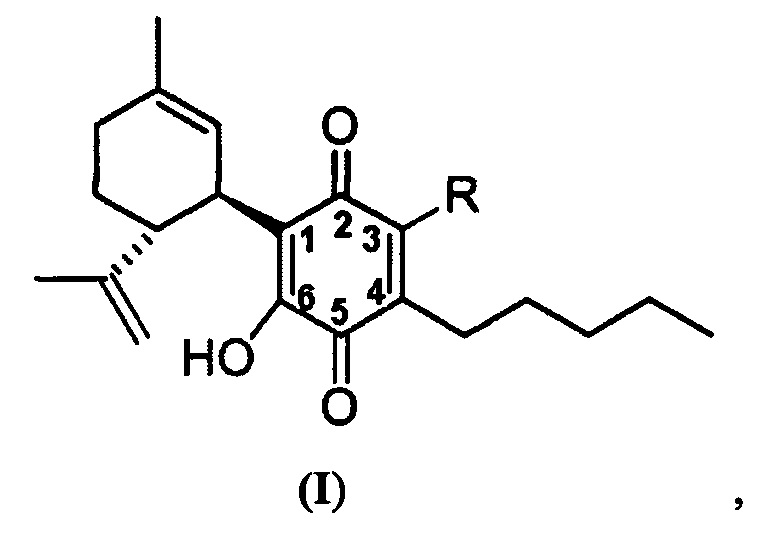
(1'R,6'R)-3-(Этиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
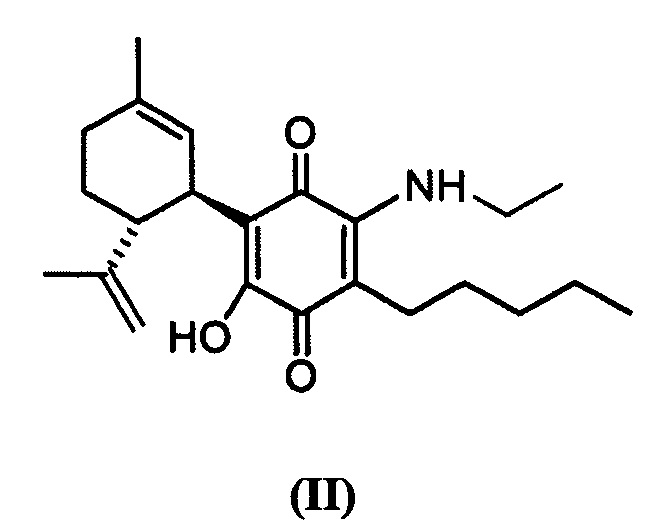
(1'R,6'R)-3-(Пентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
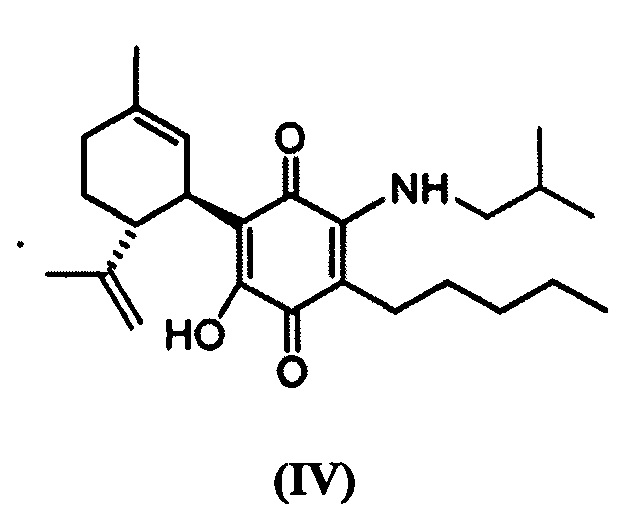
(1'R,6'R)-3-(Изобутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
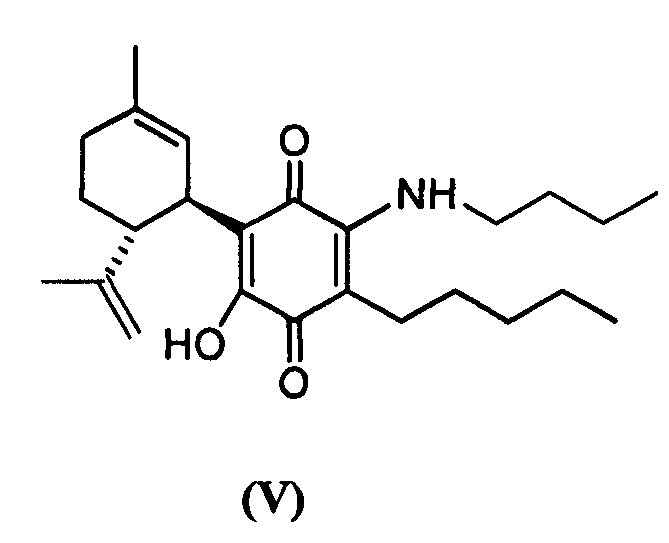
(1'R,6'R)-3-(Бутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
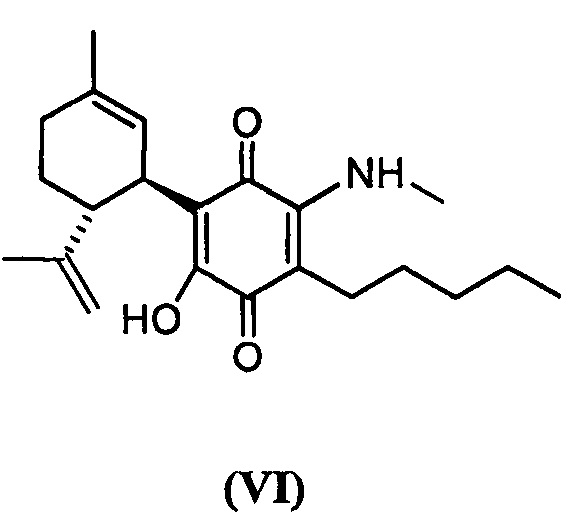
(1'R,6'R)-3-(Метиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
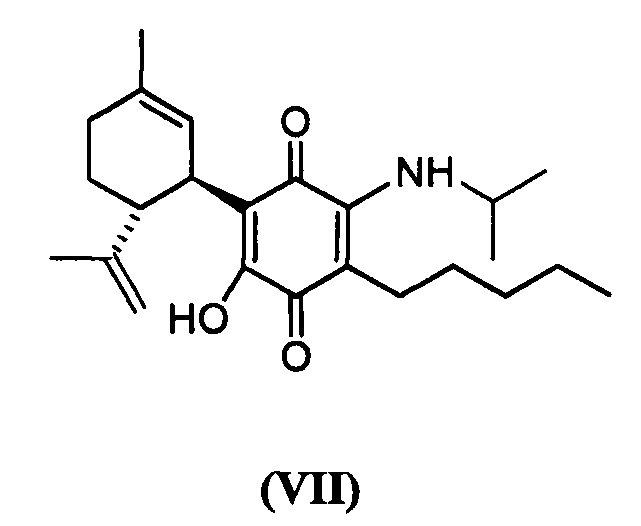
(1'R,6'R)-3-(Изопропиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
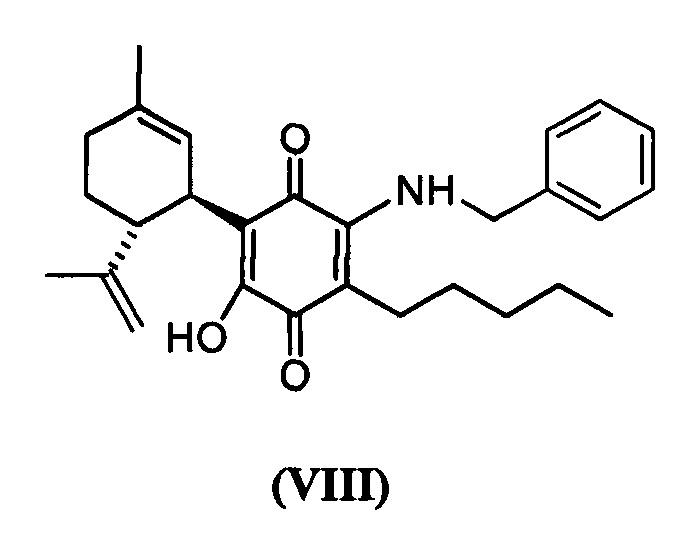
(1'R,6'R)-3-(Бензиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
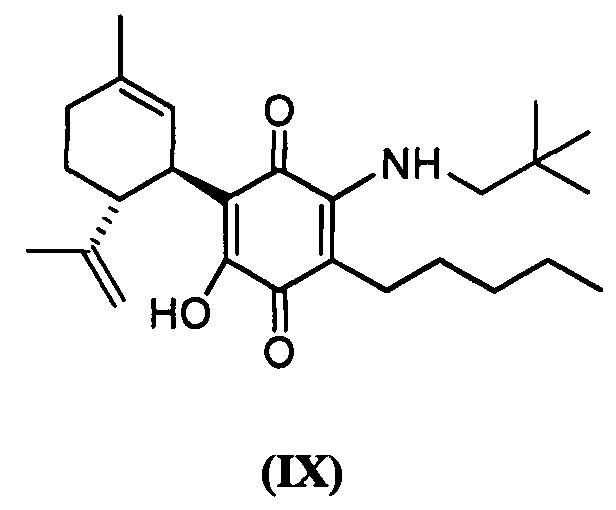
(1'R,6'R)-3-(Неопентиламин)-6-гидрокси-3'-метил-)-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
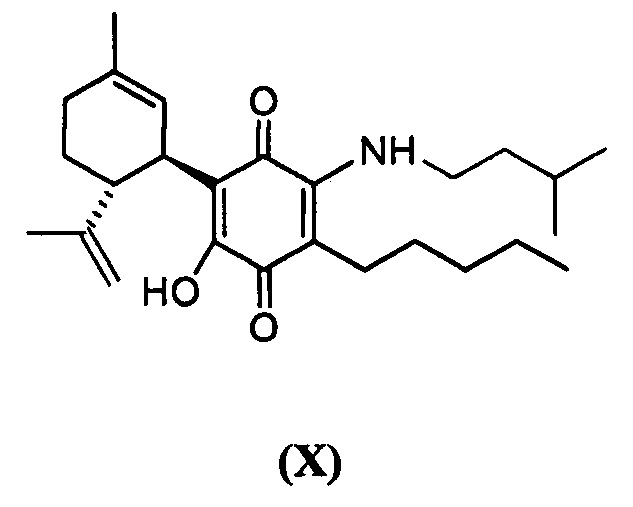
(1'R,6'R) 3-(Изопентиламин)-6-гидроксиамин-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
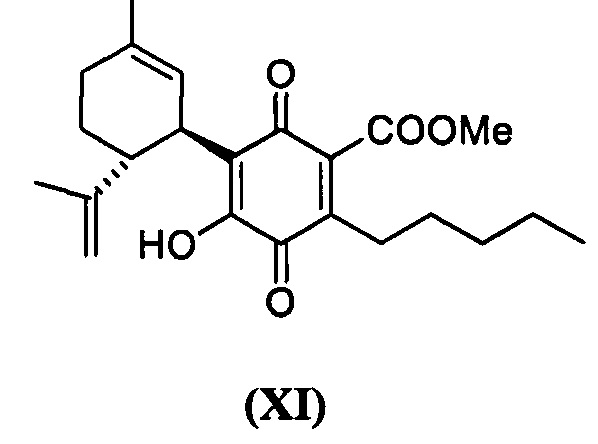
Метил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
Фенилэтил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
(Е)-3,7-Диметилокта-2,6-диенил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат

(1R,4S)-1,7,7-Триметилбицикло[2.2.1]гептан-2-ил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
(1R,2R,4R)-1,5,5-триметилбицикло[2.2.1]гептан-2-ил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
Предшественника VCE-004 (соединение I) для производных CBD-Q с II по X формулы I согласно настоящему изобретению можно легко синтезировать из CBD (ТНС Pharma, Германия; ссылка: THC-1073G-10).
Соединения с XI по XV согласно настоящему изобретению можно синтезировать, используя в качестве исходного вещества природные каннабиноиды CBDA (каннабидиоловая кислота) (ТНС Pharma, Германия; ссылка: ТНС-1232-100), посредством замещения некоторых конкретных радикалов.
Как будет понятно ниже из примеров и графических материалов, модификации в положении 3', содержащиеся в общей формуле I, придают соединениям согласно настоящему изобретению способность активировать PPARg для защиты от глутамат-индуцированной цитотоксичности и ингибировать TGFb-индуцированную выработку коллагена. Данные соединения также показали удивительно низкую цитотоксичность на клеточных линиях нейронного происхождения по сравнению с VCE-004, находящимся на современном уровне техники.
Соединения согласно настоящему изобретению также включают аналоги, производные, таутомерные формы, изомеры, стереоизомеры, полиморфы, фармацевтически приемлемые соли, фармацевтически приемлемые сольваты и их композиции.
В рамках настоящего описания термин "аналог/и" относится к любому объекту, который является структурным производным или гомологичен соединениям формулы (I).
В контексте настоящего изобретения термин "производное/ые" соединений формулы (I) следует интерпретировать как любой аналог CBD-хинона, всегда замещенный в положении 4' и демонстрирующий фармакологические свойства, связанные с этим замещением в положении 4', как это определено в настоящем документе, но при этом указанный аналог также имеет замещенные группы в других положениях молекулы CBD-Q, отличающихся от групп, представленных в указанной формуле (I).
Термин "таутомеры" относится к структурным изомерам органических соединений, которые легко взаимопревращаются химическим способом (таутомеризация).
Термин "изомеры" или "стереоизомеры" относится к соединениям, которые имеют одинаковый химический состав, но различаются расположением атомов или групп в пространстве.
В данном описании термин "полиморф" относится к кристаллическим формам одного и того же химического состава, но с разным пространственным расположением молекул, атомов и/или ионов, образующих кристалл.
Термин "фармацевтически приемлемая соль" относится к любой фармацевтически приемлемой соли, которая при введении пациенту позволяет получить (прямо или косвенно) соединение, как описано в настоящем документе. Такие соли предпочтительно являются кислотно-аддитивными солями с физиологически приемлемыми органическими или неорганическими кислотами. Примеры кислотно-аддитивных солей включают кислотно-аддитивные соли минеральных кислот, такие как, например, гидрохлориды, гидробромиды, гидройодиды, сульфаты, нитраты, фосфаты, и органические кислотно-аддитивные соли, такие как, например, ацетаты, трифторацетаты, малеаты, фумараты, цитраты, оксалаты, сукцинаты, тартраты, малаты, манделаты, метансульфонаты и n-толуолсульфонаты. Примеры аддитивных солей оснований включают неорганические соли, такие как, например, соли натрия, калия, соли кальция и аммония, и органические соли щелочных металлов, такие как, например, этилендиамины, этаноламины, N,N-диалкиленэтаноламины, триэтаноламины и соли основных аминокислот. Тем не менее, следует принимать во внимание, что фармацевтически неприемлемые соли также входят в объем настоящего изобретения, так как их можно применять для получения фармацевтически приемлемых солей. Методики получения солей являются общеизвестными в данной области техники.
Термин "сольват" в соответствии с настоящим изобретением следует понимать как любую форму активного соединения в соответствии с настоящим изобретением, в которой указанное соединение связано нековалентной связью с другой молекулой (обычно с полярным растворителем), включая, в частности, гидраты и алкоголяты.
Еще один вариант реализации настоящего изобретения относится к применению соединений формулы (I) или их производных в качестве лекарственных средств, в частности, как агонистов PPARg для рецепторов PPARg, которые не вызывают активацию Nfr2, в частности, при лечении таких заболеваний, как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатия, псориаз, заживление ран на коже, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертония, ожирение, диабет II типа и других заболеваний, которые можно лечить с помощью агонистов PPARg.
Другой вариант реализации настоящего изобретения относится к применению соединений формулы (I) для получения композиции с более низкой цитотоксичностью для лечения заболеваний, связанных с PPRAg, таких атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатия, псориаз, заживление ран на коже, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертония, ожирение, диабет II типа и других заболеваний, которые можно лечить с помощью агонистов PPARg.
Альтернативный вариант реализации настоящего изобретения относится к применению указанных выше соединений формулы (I) или их производных самих по себе или приготовленных в виде композиций, в частности в виде фармацевтических композиций, которые содержат по меньшей мере одно из соединений согласно настоящему изобретению в сочетании с по меньшей мере другим активным соединением, имеющим кумулятивную или синергическую биологическую активность. В качестве альтернативы, указанные композиции могут быть приготовлены с по меньшей мере одним инертным ингредиентом в качестве носителя или вспомогательного вещества, таким как: сорастворители, поверхностно-активные вещества, масла, увлажняющие вещества, смягчающие вещества, консерванты, стабилизаторы и антиоксиданты. Можно использовать любой фармакологически приемлемый буфер, например, Трис-буфер или фосфатный буфер.
В рамках настоящего описания термины "активное соединение или активный компонент" следует понимать в качестве синонимов, определяющих химическое соединение, которое оказывает терапевтическое воздействие при введении человеку или животному.
Типичные композиции включают в себя соединения согласно настоящему изобретению или их производные, ассоциированные с фармацевтически приемлемыми вспомогательными веществами, которые, в качестве примера, могут быть носителями или разбавителями. Такие композиции могут быть в форме капсулы, саше, бумаги или в другом виде. При изготовлении композиций можно применять обычные способы получения фармацевтических композиций. Например, целевое соединение обычно смешивают с носителем или разбавляют носителем, или заключают в носитель, который может быть в форме ампулы, капсулы, саше, бумаги или в другом виде. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует как основа, вспомогательное вещество или среда для активного соединения. Целевое соединение может быть адсорбировано на гранулированном твердом объекте (container), например, на саше. Некоторые примеры подходящих носителей представляют собой воду, растворы солей, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, арахисовое масло, оливковое масло, лактозу, каолин, сахарозу, циклодекстрин, амилозу, стеарат магния, тальк, желатин, агар, пектин, аравийскую камедь, стеариновую кислоту или низшие алкиловые эфиры целлюлозы, кремниевую кислоту, жирные кислоты, амины жирных кислот, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэритрита и жирных кислот, полиоксиэтилен, гидроксиметилцеллюлозу и поливинилпирролидон. Подобным образом, носитель или разбавитель может содержать любой материал с замедленным высвобождением, известный в данной области техники, такой как глицерилмоностеарат или глицерилдистеарат, по отдельности или в смеси с воском. Составы также могут содержать смачивающие агенты, эмульгаторы и суспендирующие агенты, консервирующие агенты, подслащивающие агенты или вкусовые агенты. Составы согласно настоящему изобретению могут быть приготовлены таким образом, чтобы обеспечить быстрое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту с использованием процедур, хорошо известных в данной области техники.
Фармацевтические композиции могут быть стерилизованы и смешаны, при необходимости, со вспомогательными агентами, эмульгаторами, солями для воздействия на осмотическое давление, буферами и/или красящими веществами и тому подобным, которые не реагируют нежелательным образом с активными соединениями.
Композицию можно применять для лечения заболеваний, таких как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатия, псориаз, заживление ран на коже, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертония, ожирение, диабет II типа и других заболеваний, которые можно лечить с помощью агонистов PPARg.
Один предпочтительный вариант реализации настоящего изобретения относится к способу введения, который может представлять собой любой способ, который эффективно доставляет целевое соединение к подходящей или требуемой области воздействия, например, как пероральный, назальный, местный, легочный, чрескожный или парентеральный способ введения, например, ректальный, подкожный, внутривенный, интрауретральный, внутримышечный, интраназальный способ, а также в виде офтальмологический раствора или мази.
Для назального введения препарат может содержать целевое соединение, растворенное или суспендированное в жидком носителе, в частности, в водном носителе, для аэрозольного применения. Носитель может содержать добавки, такие как солюбилизирующие агенты, например, пропиленгликоль, поверхностно-активные вещества, усилители абсорбции, такие как лецитин (фосфатидилхолин), или циклодекстрин, или консерванты, такие как парабены.
Для получения составов для местного применения, целевое соединение помещают в дерматологический носитель, как известно в данной области техники. Количество целевого соединения для введения и концентрация соединения в составе для местного применения будет зависеть от носителя, системы доставки или выбранного устройства, клинического состояния пациента, побочных эффектов и стабильности соединения в составе. Таким образом, лечащий врач назначает соответствующий препарат, содержащий соответствующую концентрацию целевого соединения, и выбирает количество состава для введения в зависимости от клинического опыта работы с конкретным пациентом или с подобными пациентами.
Для офтальмологического применения целевые соединения готовят в виде растворов, суспензий и мазей, подходящих для применения в качестве глазных лекарственных форм. Как правило, в указанных формах концентрации являются такими, как описано выше для препаратов местного применения.
Для перорального введения можно готовить либо твердые, либо жидкие единичные дозированные формы. Для получения твердых композиций, таких как таблетки, целевое соединение можно смешивать с обычными ингредиентами, такими как тальк, стеарат магния, дикальцийфосфат, алюмосиликат магния, сульфат кальция, крахмал, лактоза, аравийская камедь, метилцеллюлоза и функционально подобными фармацевтическими материалами, такими как разбавители или носители.
Капсулы получают смешиванием целевого соединения с инертным фармацевтическим разбавителем и наполнением твердой желатиновой капсулы подходящего размера этой смесью. Мягкие желатиновые капсулы получают механическим капсулированием суспензии соединения с приемлемым растительным маслом, светлым вазелиновым маслом или другим инертным маслом. Можно получать жидкие единичные дозированные формы для перорального введения, такие как сиропы, эликсиры и суспензии. Водорастворимые формы можно растворять в водном носителе вместе с сахаром, ароматизирующими агентами и консервантами с образованием сиропа. Эликсир готовят с применением водно-спиртового (например, этанола) носителя с подходящими подсластителями, такими как сахар и сахарин вместе с ароматизирующими агентами. Суспензии можно получать на водной основе с помощью суспендирующего агента, такого как аравийская камедь, трагакант, метилцеллюлоза и тому подобных.
Составы, подходящие для парентерального применения, очевидны для среднего специалиста в данной области, например, как применение подходящих инъекционных растворов или суспензий. Стерильный состав подходит для различных местного или парентерального способов введения, включая интрадермальный, внутримышечный, внутрисосудистый и подкожный.
В дополнение к целевому соединению, в зависимости от состава и требуемого способа доставки, композиции могут содержать фармацевтически приемлемые, нетоксичные носители или разбавители, которые включают основы, обычно применяемые для образования фармацевтических композиций для введения животным или людям. Разбавитель выбирают таким образом, чтобы сильно не влиять на биологическую активность комбинации.
Примерами таких разбавителей, которые особенно подходят для инъекционных составов, являются вода, различные солевые растворы, растворы органических или неорганических солей, раствор Рингера, раствор декстрозы и раствор Хэнка. Кроме того, фармацевтическая композиция или состав может содержать добавки, такие как другие носители; адъюванты; или нетоксичные, нетерапевтические, неиммуногенные стабилизаторы и тому подобное.
Кроме того, в композиции могут содержаться вспомогательные вещества. Примеры включают сорастворители, поверхностно-активные вещества, масла, увлажнители, смягчающие вещества, консерванты, стабилизаторы и антиоксиданты. Можно применять любой фармакологически приемлемый буфер, например, Трис-буфер или фосфатный буфер. Эффективными количествами разбавителей, добавок и вспомогательных веществ являются те количества, которые эффективны для получения фармацевтически приемлемого состава с точки зрения растворимости, биологической активности и т.д.
Целевое соединение может быть включено в микросферы. Целевое соединение может быть помещено в альбуминовые микросферы, из которых можно получить такие микросферы в виде сухого порошка для назального введения. Другие материалы, пригодные для получения микросфер, включают агар, альгинат, хитозан, крахмал, гидроксиэтилкрахмал, альбумин, агарозу, декстран, гиалуроновую кислоту, желатин, коллаген и казеин. Микросферы можно получать различными способами, известными специалистам в данной области техники, например, такими как процесс распылительной сушки или процесс эмульгирования.
Например, альбуминовые микросферы можно получить путем добавления кроличьего сывороточного альбумина в фосфатном буфере в оливковое масло при перемешивании с получением эмульсии вода в масле. Затем в эмульсию добавляют раствор глутарового альдегида и перемешивают эмульсию до сшивки альбумина. Затем микросферы можно выделить с помощью центрифугирования, удалить масло и промыть микросферы, например, с помощью с петролейного эфира, а затем этанола. И, наконец, микросферы можно просеять, собрать и высушить с помощью фильтрации.
Крахмальные микросферы можно получить путем добавления теплого водного раствора крахмала, например, картофельного крахмала, к нагретому раствору полиэтиленгликоля в воде при перемешивании с образованием эмульсии. После образования двухфазной системы (с раствором крахмала в качестве внутренней фазы) далее смесь охлаждают до комнатной температуры при перемешивании, после чего внутренняя фаза превращается в гелевые частицы. Затем эти частицы отфильтровывают при комнатной температуре и суспендируют в растворителе, таком как этанол, после чего эти частицы снова отфильтровывают и оставляют высыхать на воздухе. Микросферы можно делать более твердыми с помощью хорошо известных процедур сшивки, таких как термическая обработка или применение химических сшивающих агентов. Подходящие агенты включают диальдегиды, включая глиоксаль, малоновый диальдегид, янтарный альдегид, адипиновый альдегид, глутаровый альдегид и фталевый альдегид, дикетоны, такие как бутадион, эпихлоргидрин, полифосфаты и бораты. Диальдегиды применяют для поперечной сшивки белков, таких как альбумин, путем взаимодействия с аминогруппами, и дикетонами из оснований Шиффа с аминогруппами. Эпихлоргидрин активирует соединения с нуклеофилами, такими как амино или гидроксил с образованием эпоксидного производного.
Другой предпочтительный вариант реализации настоящего изобретения представляет собой схему приема лекарственного средства. Термин "стандартная лекарственная форма" относится к физически дискретным единицам, пригодным в качестве единичных доз для субъектов, например млекопитающих, таких как люди, собаки, кошки и грызуны, причем каждая единица содержит заданное количество активного вещества, рассчитанное для получения требуемого фармацевтического эффекта, в сочетании с требуемым фармацевтическим разбавителем, носителем или основой. Параметры стандартных лекарственных форм согласно настоящему изобретению определяются и зависят от (а) уникальных характеристик активного вещества и конкретного эффекта, который необходимо достичь, и (b) ограничений, свойственных области compounding, например как активное вещество для применения людьми и животными. Примерами лекарственных форм с однократной дозой являются таблетки, капсулы, пилюли, пакетики с порошком, облатки, суппозитории, гранулы, саше, чайная ложка, столовая ложка, капельницы, ампулы, флаконы, аэрозоли с дозированными порциями, множество отделенных единиц любого из вышеперечисленного, и другие формы, описанные в настоящем документе. Композиции могут быть включены в наборы, которые могут содержать одну или более лекарственных форм с однократной дозой композиции и инструкции по применению для лечения одного или более расстройств, описанных в настоящем документе.
Системы доставки с медленным и пролонгированным высвобождением, включая любой из множества биополимеров (биологических систем), системы с использованием липосом, коллоиды, смолы и другие полимерные системы доставки или компартментые резервуары можно применять с композициями, описанными в настоящем документе, для обеспечения непрерывного или долговременного источника терапевтического соединения. Такие системы с медленным высвобождением применимы к составам для местного, внутриглазного, орального и парентерального способов введения.
В лечении применяют эффективное количество целевого соединения. Дозы соединений, применяемых в соответствии с настоящим изобретением, изменяются в зависимости от соединения и состояния, подлежащего лечению, например, от возраста, веса и клинического состояния пациента-реципиента. Другие факторы включают: способ введения, пациента, историю болезни пациента, тяжесть течения заболевания, а также эффективность конкретного соединения. Доза должна быть достаточной для ослабления симптомов или признаков заболевания, подлежащего лечению, при этом не вызывая неприемлемой токсичности для пациента. В целом, эффективным количеством соединения является то количество, которое обеспечивает субъективное облегчение симптомов или объективно заметное улучшение, регистрируемое практикующим врачом или другим квалифицированным наблюдателем.
И последний вариант реализации настоящего изобретения относится к способу лечения заболеваний, таких как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатия, псориаз, заживление ран на коже, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертония, ожирение и диабет II типа, которые можно лечить с помощью агонистов PPARg; и который включает введение пациенту эффективного количества вышеуказанной композиции.
Сокращения:
АР-1: Белок-активатор-1
ARE: Элемент антиоксидантного ответа
CBD: Каннабидиол.
CBDA: Каннабидиоловая кислота.
CBD-Q: Каннабидиол-хинон.
CBG-Q: Каннабигерол-хинон (также называемый VCE-003).
DCC: Дициклогексилкарбодиимид.
Keap1: Kelch ЕСН-связанный белок 1.
NFAT: Ядерный фактор активированных Т-клеток
NFE2L2 или (Nrf2): Ядерный фактор 2 (эритроидного происхождения 2).
NF-kB: Ядерный фактор каппа-В
NP: Предшественники нейронов
NR1C: Ядерное подсемейство 1 С.
NR: Ядерные рецепторы.
PPAR: Рецепторы активации пролиферации пероксисом.
PPARg: гамма-рецептор активации пролиферации пероксисом, также называемый PPARγ, рецептор глитазона или NR1C3.
PPARg-m: Модуляторы PPARg
PPARg-fa: Полные агонисты PPARg.
PPARα: Альфа-рецептор активации пролиферации пероксисом, также называемый NR1C1.
PPARβ/δ: Бета/дельта-рецептор активации пролиферации пероксисом, также называемый NR1C2.
PPRE: Элемент ответа пролиферации пероксисом.
АФК: Активные формы кислорода
STAT3: Переносчик сигнала и активатор транскрипции 3
TGFb: Трансформирующий фактор роста бета
VCE-004: Соединение каннабидиол-хинон; также называемое HU-331 и соединение I:
HU-331: Соединение каннабидиол-хинон; также называемое VCE-004 и соединение I:
Описание графических материалов
Краткое описание чертежей настоящего изобретения приведено ниже. Более подробное объяснение каждой фигуры включено в каждый соответствующий пример.
Сокращения для фигур:
I: относится к VCE-004 (CBD-Q).
II: относится к соединению формулы (II).
III: относится к соединению формулы (III).
IV: относится к соединению формулы (IV).
V: относится к соединению формулы (V).
VI: относится к соединению формулы (VI).
VII: относится к соединению формулы (VII).
VIII: относится к соединению формулы (VIII).
IX: относится к соединению формулы (IX).
X: относится к соединению формулы (X).
XI: относится к соединению формулы (XI).
XII: относится к соединению формулы (XII).
XIII: относится к соединению формулы (XIII).
XIV: относится к соединению формулы (XIV).
XV: относится к соединению формулы (XV).
Фигура 1. Анализ трансактивации PPARg в клетках НЕК-293
Концентрация тестируемого соединения (μM) показана на оси х, и кратная активация PPARg показана на оси у. На данной фигуре показано воздействие VCE-004 (соединение I) относительно воздействия соединений XI и с II по V (Фигура 1А), и относительно воздействия соединений VI-X (Фигура 1В), и относительно воздействия соединений XII-XV (Фигура 1С) на активность PPARg. В качестве контроля сравнения использовали полный агонист PPARg Росиглитазон (RZG) в концентрации 1 μМ. Кратный уровень активации рассчитывали по отношению к (-) контрольному образцу в качестве эталона без присутствия какого-либо агониста PPARg или активирующего агента. Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов.
Фигура 2. Анализ трансактивации PPARg в клетках фибробластов NIH-3T3.
Значения онцентрации тестируемого соединения (μМ) показана на оси х и кратная активация PPARg показана на оси у. На данной фигуре показано воздействие VCE-004 (соединение I) относительно воздействия соединений III, V, VIII, X и XIII на активность PPARg. В качестве контроля сравнения использовали полный агонист PPARγ Росиглитазон (RZG) в концентрации 1 μМ. Кратный уровень активации рассчитывали по отношению к (-) контрольному образцу в качестве эталона без присутствия какого-либо агониста PPARg или активирующего агента. Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов.
Фигура 3. Производные CBD-хинона ингибируют Росиглитазон-индуцированную активацию PPARg.
(A) Клетки HEK-293 ко-трансфицировали GAL4-PPARg и GAL4-luc. Клетки предварительно инкубировали в течение 30 мин с указанными дозами соединений III, V, VIII, X и XIII, а затем инкубировали в течение 6 часов с 1 μM Росиглитазона (RSZ). Получали белковые лизаты и анализировали их на люциферазную активность. Концентрация тестируемого соединения (μМ) показана на оси х и кратная активация PPARg показана на оси у. На данной фигуре показано воздействие соединений III, V, VIII, X и XIII на RSZ-индуцированную активность PPARg. Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов.
(B) Соединение VIII связывается с сайтом связывания RSZ на PPARg. Особенности связывания соединения VIII (в качестве примера) с PPARg рассчитывали с помощью виртуальной стыковки с применением программного обеспечения AutoDock и настройки алгоритма Vina как системы расчета. Область поиска была установлена таким образом, чтобы найти точки связывания вокруг молекулярной поверхности. Соединение VIII связывается с PPARg в сайте, близко примыкающем к RSZ, но с другой схемой взаимодействия лиганд-рецептор, что приводит к различному конформационному воздействию на рецептор.
Фигура 4. Цитотоксическая активность.
Клеточные линии N2a (4А), НТ22 (4В) и МО3.13 (4С) инкубировали в течение 24 ч с указанными дозами VCE-004 (соединение I) в сравнении с соединениями с II по XV, а жизнеспособность клеток количественно измеряли с помощью анализа на основе МТТ. Результаты представляли в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов и выражали в процентах от жизнеспособности клеток по отношению к (-) контрольному образцу без присутствия какого-либо агониста PPARg или активирующего агента. Контроль принимали за 100% и данные относили к этому значению.
Фигура 5. Транскрипционный анализ Nrf2
Клетки HaCaT-ARE-Luc инкубировали в течение 6 ч с VCE-004 (соединение I) и с соединениями с II по VIII (А) или с соединениями с IX по XV (В) в указанных концентрациях, получали белковые лизаты и анализировали их на люциферазную активность. Прооксидант трет-бутилгидрохинон (tBHQ) в концентрации 20 μМ использовали в качестве положительного контроля. Кратный уровень активации рассчитывали по отношению к (-) контрольному образцу в качестве эталона без присутствия какого-либо агониста PPARg или активирующего агента. Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов.
Фигура 6. Нейропротекторная активность.
Клетки N2a предварительно инкубировали в течение 1 ч с соединениями с I по VIII (5А) и с IX по XV (5В) в указанных концентрациях. Затем клетки обрабатывали 5 мМ глутамата в течение 24 ч для индукции эксайтотоксичности. Жизнеспособность клеток количественно определяли с помощью анализа на основе МТТ. Результаты представляли в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов и выражали в процентах от жизнеспособности клеток по отношению к (-) контрольному образцу без присутствия какого-либо агониста PPARg или активирующего агента и с присутствием (+) или отсутствием (-) глутамата. Контроль принимали за 100% и данные относили к этому значению.
Фигура 7. Ингибирование TGFb-индуцированной транскрипции гена коллагена типа I
Для исследования потенциальной антифиброзной активности CBD-производных, клетки фибробластов NIH-3T3 временно трансфицировали плазмидой COL1A2-Luc с применением Roti©-Fect в соответствии с инструкциями производителя. Конструкт COL1A2-люциферазы содержал последовательность с -353 по +58 п. о. промотора человека COL1A2, слитого с репортерным геном люциферазы (pGL2 basic, Promega, Мэдисон, Висконсин). Двадцать четыре часа спустя клетки инкубировали с соединениями III, V, VIII и X (взяты в качестве демонстративных примеров среди всего семейства производных con CBD-Q, представленных формулами с II по XV) в течение 30 мин и обрабатывали TGFb (50 нг/мл) в течение 6 ч. Получали белковые лизаты и анализировали их на люциферазную активность. Концентрация тестируемого соединения (μМ) показана на оси х и процент активации COL1A2 показан на оси у, считая 100% активацию действием TGFb в отсутствии соединений. Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов.
Фигура 8. Ингибирование TGFb-индуцированного коллагена типа II
Выработку коллагена проводили с использованием метода Sirius Red-Fast Green, предназначенного для количественного определения коллагена и неколлагеновых белков в клеточных гранулах. Выработку коллагена определяли при 540 нм и 605 нм с применением сканирующего спектрофлуорометра Genesis 10 UV (Thermo Fisher Scientific). Для расчета количества коллагена сначала корректировали значение оптической плотности OD 540 путем вычитания вклада Fast Green, который искажает оптическую плотность при длине волны 540 нм. Fast Green вносит 29,1% от значения OD 540. Цветовой эквивалент для коллагена составляет 37,8 и для неколлагеновых белков составляет 2,04 при OD 540 и 640, соответственно.
Коллаген (пг/100 μл клеточного осадка)={[OD 540-(OD 605×0,291)]/37,8×1000}×106.
Эксперименты повторяли три раза и результаты выражали в виде кратной индукции относительно необработанных клеток.
Фигура 9. Влияние производных CBD-Q на образование активных форм кислорода и митохондриальный трансмембранный потенциал
Клетки Jurkat обрабатывали возрастающими концентрациями VCE-004 (HU-311 или соединение I) или соединениями III, V, VII и X (в качестве примера производных соединения I) в течение 2 часов для обнаружения митохондриального мембранного потенциала или в течение 6 часов для обнаружения активных форм кислорода (АФК).
Использовали флуоресцентные зонды H2DCF-DA (20 нМ, зеленая флуоресценция) и MitoTracker Red CMXR (MTR-CMXR) (50 нМ), соответственно, для обнаружения АФК и митохондриального мембранного потенциала (Molecular Probes, Юджин, Орегон, США). После обработки клетки дважды промывали холодным фосфатно-солевым буферным раствором (ФСБ) и инкубировали в ФСБ в течение 20 мин при 37°С с последующим анализом на проточном цитометре FACSCantoII.
Примеры
Примеры в настоящем изобретении, описанные ниже, предназначены для иллюстрации предпочтительных вариантов реализации без ограничения объема его правовой охраны.
Пример 1. Химический синтез и ЯМР-анализ
А) Синтез производных CBD-хинона, начиная с CBD. Синтез соединений с II по Х.
Синтез VCE-004 (также называемое HU-331 или соединение I) из CBD проводили с применением tBuOK в толуоле при комнатной температуре в присутствии воздуха (Схема 1). Синтез производных, замещенных алкиламинами в положении 3, легко осуществить посредством реакции VCE-004 с большим избытком амина при комнатной температуре в реакционной системе с доступом воздуха.
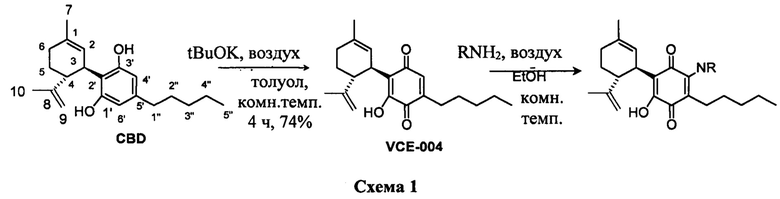
При очистке путем флэш-хроматографии получали продукт с чистотой 90-95%, которую можно дополнительно увеличить до 98% посредством очистки ВЭЖХ. Высокая конверсия достигалась в течение нескольких часов с получением реакций от "точки к точке" (spot-to-spot reactions). Растворитель выпаривали и неочищенный остаток очищали с помощью обращенно-фазовой хроматографии с получением продуктов с чистой примерно 95%.
Получение соединения I.
К раствору CBD (302 мг, 0,960 ммоль) в толуоле (60 мл) добавляли tBuOK (298 мг, 2,656 ммоль) с получением раствора фиолетового цвета. Реакционную смесь перемешивали при комнатной температуре в круглодонной колбе с доступом воздуха, и контроль конверсии осуществляли при помощи ТСХ-анализа (элюент: 10% EtOAc/гексаны). После 4 ч реакционную смесь промывали HCl (5% водный раствор, 100 мл) и водный слой экстрагировали EtOAc (30 мл) (Схема 2). Объединенные органические слои сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток подвергали флэш-хроматографии на SiO2 (0®20% EtOAc/гексаны) с получением 234 мг VCE-004 (соединение I) [твердое вещество коричневого цвета, выход: 74%].

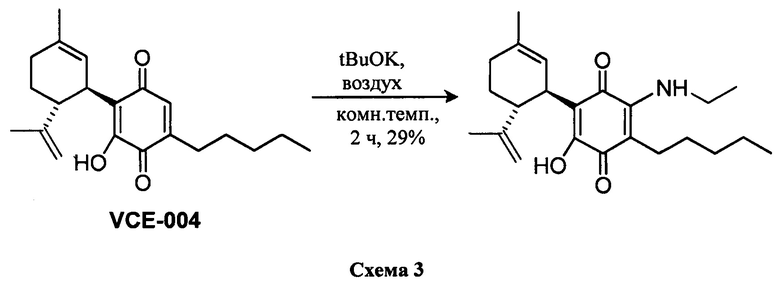
Получение соединения II.
(1'R,6'R)-3-(Этиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-(1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (100 мг, 0,30 ммоль) в EtOH (10 мл) добавляли этиламин (1,0 мл, 70% раствор в Н2О, 12,58 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем обрабатывали, выливая в воду (50 мл), подкисляли до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 3). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30 100% CH3CN/H2O) с получением 33 мг (1'R,6'R)-3-(этиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 29%].
100% CH3CN/H2O) с получением 33 мг (1'R,6'R)-3-(этиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 29%].
1Н ЯМР (CDCl3, 300 МГц) d ppm: 6,35 (bs, 1Н), 5,13 (s, 1H), 4,57 (s, 2H), 3,61 (m, 1H), 3,52 (квинтет, J=13,2; 7,1 Гц, 2H), 2,73 (m, 1H), 2,48 (t, J=7,1 Гц, 2H), 2,26-1,80 (m, 2H), 1,68 (s, 3H), 1,63 (s, 3H), 1,46-1,24 (m, 9H), 0,89 (t, J=6,6 Гц, 3Н).
Получение соединения III.
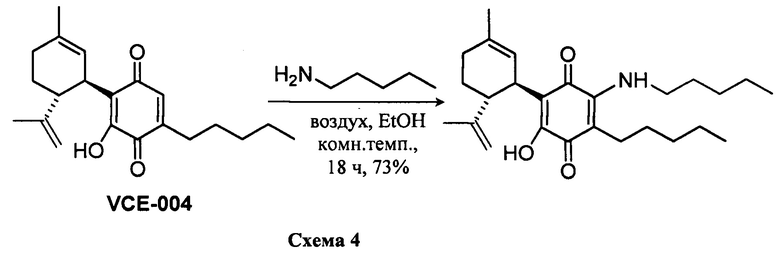
(1'R,6'R)-3-(Пентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (60 мг, 0,155 ммоль) в EtOH (10 мл) добавляли амиламин (0,75 мл, 6,472 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 4). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 47 мг (1'R,6'R)-3-(пентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [твердое вещество фиолетового цвета, выход: 73%].
1Н ЯМР (CDCl3, 300 МГц) d ppm: 6,43 (bs, 1Н), 5,14 (s, 1H), 4,55 (s, 2H), 3,62 (m, 1H), 3,46 (c, J=6,6 Гц, 2H), 2,72 (m, 1H), 2,48 (t, J=7,7 Гц, 2H), 2,31-1,72 (m, 4H), 1,68 (s, 3H), 1,64 (s, 3H), 1,48-1,24 (m, 12H), 0,90 (m, 6H).
Получение соединения IV.
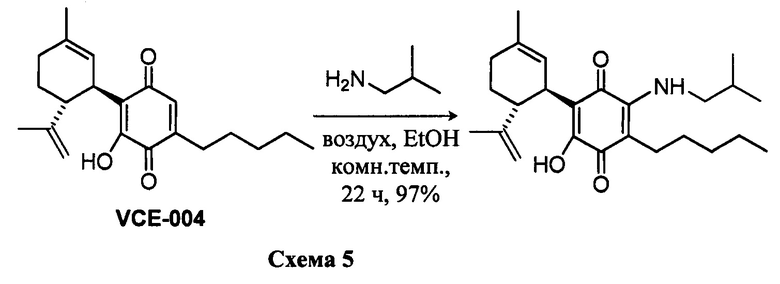
(1'R,6'R)-3-(Изобутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (100 мг, 0,304 ммоль) в EtOH (12 мл) добавляли изобутиламин (1,2 мл, 12,075 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 22 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 5). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 119 мг (1'R,6'R)-3-(изобутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [твердое вещество фиолетового цвета, выход: 97%].
1H ЯМР (CDCl3, 300 МГц) d ppm: 6,53 (bs, 1H), 5,15 (s, 1H), 4,56 (s, 2H), 3,62 (m, 1H), 3,27 (t, J=6,6 Гц, 2H), 2,73 (dt, J=12,0 Гц, 2,8 Гц, 1H), 2,47 (t, J=7,1 Гц, 2H), 2,27-1,72 (m, 4H), 1,68 (s, 3H), 1,64 (s, 3H), 1,47-1,29 (m, 7H), 1,00 (s, 3H), 0,97 (s, 3H), 0,89 (t, J=6,6 Гц, 3H).
Получение соединения V.
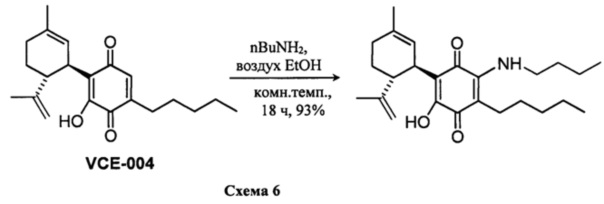
(1'R,6'R)-3-(Бутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (109 мг, 0,332 ммоль) в EtOH (12 мл) добавляли н-бутиламин (1,2 мл, 12,143 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 6). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 115 мг (1'R,6'R)-3-(бутиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [твердое вещество фиолетового цвета, выход: 93%].
1Н ЯМР (CDCl3, 300 МГц) d ppm: 6,44 (bs, 1Н), 5,14 (s, 1H), 4,56 (s, 2H), 3,61 (m, 1H), 3,46 (q, J=6,6 Гц, 2H), 2,73 (m, 1H), 2,48 (t, J=7,1 Гц, 2H), 2,19 (m, 1H), 1,98 (m, 1H), 1,78-1,57 (m, 8H), 1,49-1,25 (m, 10H), 0,96 (t, J=7,1 Гц, 3H), 0,89 (m, 3H).
Получение соединения VI.
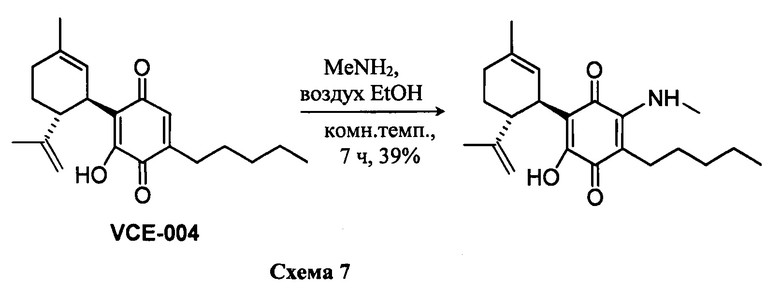
(1'R,6'R)-3-(Метиламин)-6-гидрокси-3'-метил--4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (266 мг, 0,810 ммоль) в EtOH (20 мл) добавляли метиламин (4,0 мл, 8 М раствор в EtOH, 32,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 7 ч. Смесь вливали в Н2О (100 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (70 мл) (Схема 7). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 114 мг (1'R,6'R)-3-(Метиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [твердое вещество фиолетового цвета, выход: 39%].
1Н ЯМР (CDCl3l2, 300 МГц) d ppm: 8,38 (bs, 1H), 6,54 (m, 1H), 5,12 (s, 1H), 4,56 (s, 2H), 3,63 (m, 1H), 3,19 (d, J=6,0 Гц, 3Н), 2,71 (dt, J=11,5 Гц, 2,7 Гц, 1H), 2,54 (t, J=7,1 Гц, 2H), 2,28-1,71 (m, 3H), 1,67 (s, 3H), 1,63 (s, 3H), 1,51-1,25 (m, 6H), 0,89 (t, J=7,1 Гц, 3H).
Получение соединения VII.
(1'R,6'R)-3-(Изопропиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-ил)-1,1'-би(циклогексан)]-2',3,б-триен-2,5-дион
К раствору VCE-004 (104 мг, 0,317 ммоль) в EtOH (10 мл) добавляли изопропиламин (1,0 мл, 11,639 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 22 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 8). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30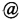 100% CH3CN/H2O) с получением 92 мг (1'R,6'R)-3-(изопропиламино)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 75%].
100% CH3CN/H2O) с получением 92 мг (1'R,6'R)-3-(изопропиламино)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 75%].
1Н ЯМР (CDCl2, 300 МГц) d ppm: 6,40 (m, 1H), 5,14 (s, 1H), 4,56 (s, 2H), 3,95 (m, 1H), 3,61 (m, 1H), 2,73 (m, 1H), 2,45 (t, J=6,6 Гц, 2H), 2,21 (m, 1H), 1,92 (m, 1H), 1,77 (m, 2H), 1,67 (s, 3H), 1,63 (s, 3H), 1,45-1,28 (m, 6H), 1,26 (s, 3H), 1,24 (s, 3H), 0,89 (t, J=7,1 Гц, 3H).
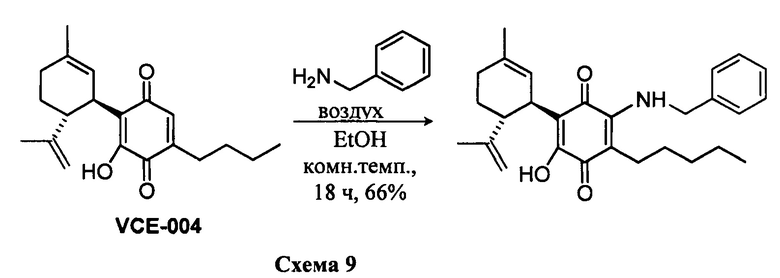
Получение соединения VIII.
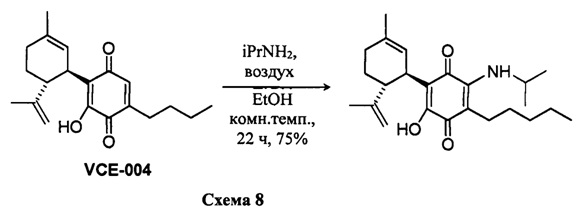
(1'R,6'R)-3-(Бензиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (117 мг, 0,303 ммоль) в EtOH (13 мл) добавляли бензиламин (1,3 мл, 11,913 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 9). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30100% CH3CN/H2O) с получением 87 мг (1'R,6'R)-3-(бензиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [твердое вещество фиолетового цвета, выход: 66%].
1Н ЯМР (CDCl2, 300 МГц) d ppm: 8,30 (bs, 1Н), 7,44-7,26 (m, 5H), 6,64 (m, 1H), 5,15 (s, 1H), 4,65 (d, J=6,0 Гц, 2H), 4,59 (m, 2H), 3,64 (m, 1H), 2,73 (m, 1H), 2,47 (t, J=7,7 Гц, 2H), 2,30-1,76 (m, 4H), 1,68 (s, 3H), 1,64 (s, 3H), 1,54-1,23 (m, 6H), 0,88 (m, 3H).
Получение соединения IX.
(1'R,6'R)-3-(Неопентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (47 мг, 0,143 ммоль) в EtOH (7 мл) добавляли неопентиламин (0,7 мл, 6,031 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 10). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 57 мг (1'R,6'R)-3-(Неопентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 97%].
1Н ЯМР (CDCl2, 300 МГц) d ppm: 6,59 (m, 1H), 5,15 (s, 1H), 4,56 (s, 2H), 3,63 (m, 1H), 3,26 (d, J=5,5 Гц, 2H), 2,74 (dt, J=12,0 Гц, 3,3 Гц, 1H), 2,49 (t, J=7,1 Гц, 2H), 2,26-1,83 (m, 3H), 1,68 (s, 3H), 1,63 (s, 3H), 1,50-1,23 (m, 7H), 1,00 (s, 9H), 0,90 (t, J=6,6 Гц, 3H
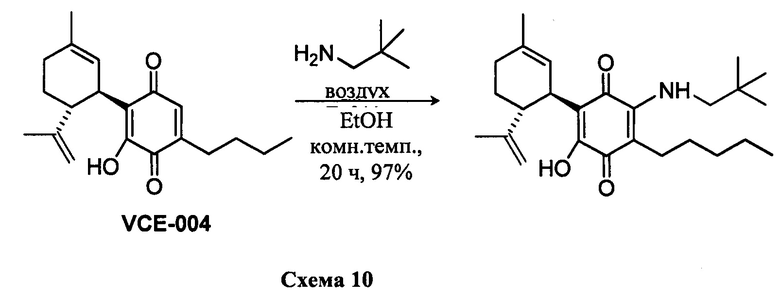
Получение соединения X.
(1'R,6'R)-3-(Изопентиламин-6-гидрокси)-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-дион
К раствору VCE-004 (101 мг, 0,307 ммоль) в EtOH (15 мл) добавляли изопентиламин (1,5 мл, 12,735 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 22 ч. Смесь вливали в Н2О (50 мл), доводили до рН=2 с помощью HCl (10% водный раствор) и экстрагировали CH2Cl2 (30 мл) (Схема 11). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии (30®100% CH3CN/H2O) с получением 125 мг (1'R,6'R)-3-(изопентиламин)-6-гидрокси-3'-метил-4-пентил-6'-(проп-1-ен-2-ил)-[1,1'-би(циклогексан)]-2',3,6-триен-2,5-диона [масло фиолетового цвета, выход: 98%].
1Н ЯМР (CDCl3, 300 МГц) d ppm: 6,38 (bs, 1H), 5,13 (s, 1H), 4,55 (s, 2H), 3,61 (m, 1H), 3,48 (q, J=6,0 Гц, 2H), 2,72 (m, 1H), 2,48 (t, J=7,1 Гц, 2H), 2,21 (m, 1H), 2,00-1,60 (m, 8H), 1,54 (q, J=7,1 Гц, 2H), 1,46-1,23 (m, 8H), 0,95 (s, 3H), 0,93 (s, 3H), 0,88 (t, J=6,6 Гц, 3H).
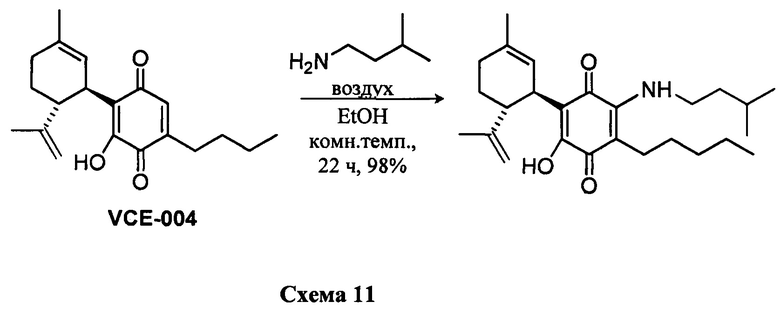
В) Синтез производных CBD-хинона из каннабидиоловой кислоты CBDA. Синтез соединений с XI по XV.
Синтез предшественника соединения XI
Мегил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат (CBDA-метиловый эфир)

a) К раствору каннабидиоловой кислоты (CBDA) (180 мг, 0,40 ммоль) в метаноле (5 мл) добавляли дициклогексилкарбодиимид (DCC) (163 мг, 1,6 ммоль) и катализатор п-толуолсульфоновую кислоту (приблизительно 5 мг) (Схема 12). После перемешивания в течение 40 мин. реакционную смесь обрабатывали путем упаривания. Остаток растворяли в толуоле (приблизительно 10 мл) и охлаждали (-18°С) для осаждения мочевины. Через 1 ч раствор фильтровали на фильтре из пористого стекла, и остаток очищали с помощью флэш-хроматографии на RP С-18 силикагеле с получением 140 мг метил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилата [бесцветная пена, выход: 75%].
b) К раствору каннабидиоловой кислоты (CBDA) (200 мг, 0,54 ммоль) в метаноле (8 мл) добавляли триметилсилилдиазометан (3,0 мл, 2M в гексане) (Схема 12). После перемешивания в течение 5 мин при комнатной температуре, реакционную смесь обрабатывали путем упаривания. Продукт был достаточно чистыми для непосредственно использоваться на стадии окисления.
1Н ЯМР (CDCl3, 300 МГц) d ppm 11,97 (s, 1Н), 6,40 (bs, 1H), 6,21 (s, 1H), 5,54 (bs, 1H), 4,51 (bs, 1H), 4,38 (bs, 1H), 3,90 (s, 3H), 2,77 (m, 2H), 1,81 (bs, 3H), 1,70 (bs, 3H), 0,89 (t, J=6,6 Гц, 3H).
Получение соединения XI
Метил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
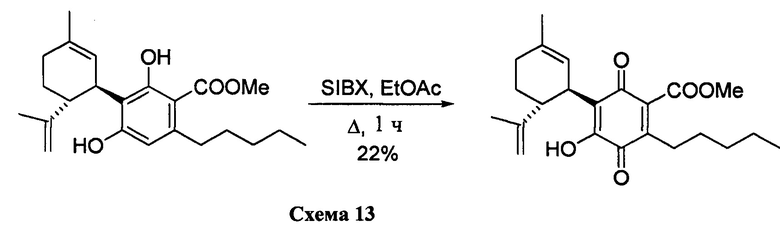
К раствору 100 мг (0,27 ммоль) метил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилата (CBDA-метиловый эфир) в 4 мл EtOAc добавляли SIBX (460 мг, 0,77 ммоль, 3 моль эквив.) и реакционную смесь нагревали с обратным холодильником в течение 1 ч (Схема 13). После охлаждения и фильтрации через целит, фильтрат последовательно промывали 5% NaHCO3 и насыщенным солевым раствором. После высушивания (Na2SO4) и выпаривания, остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир-CH2Cl2 8:5 в качестве элюента) с получением 24 мг соединения XI [твердое вещество коричневого цвета, выход: 22%].
1Н ЯМР (CDCl3, 300 МГц) d ppm 7,00 (bs, 1H), 5,13 (bs, 1Н), 4,57 (s, 1H), 4,53 (s, 1H), 3,89 (s, 3H), 3,73 (bd, J=7,0 Гц, 1H), 2,74 (td, J=9,1, 9,1, 1,5 Гц, 1H), 2,36 (t, J=7,5 Гц, 2H), 1,72 (bs, 3H), 1,64 (bs, 3H), 0,88 (t, J=6,6 Гц, 3H).
Синтез предшественника соединения ХII
Фенетил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоат (CBDA-фенетиловый эфир)
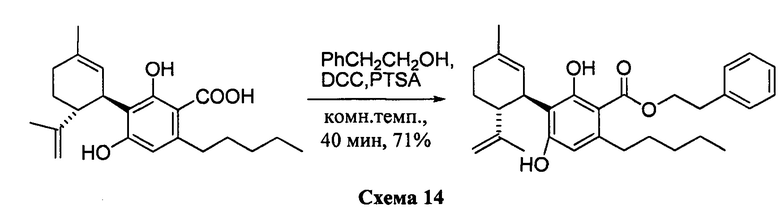
К раствору каннабидиоловой кислоты (CBDA) (2,15 г, 6,0 ммоль) в CH2Cl2 (20 мл) добавляли фенетиловый спирт (0,860 мл), а затем DCC (2,550 г, 12 ммоль, 2 моль эквив.) и катализатор PTSA (30 мг). После 1 ч, реакционную смесь обрабатывали путем упаривания и остаток растворяли в толуоле, охлаждали при -18°С в течение 20 мин для осаждения дициклогексилмочевины. После фильтрации фильтрат упаривали, и остаток очищали с помощью флэш-хроматографии на RP18 силикагеле с применением градиента смеси метанол-вода (от 6:4 до чистого метанола) в качестве элюента. Было получено 1,52 г (71%) масла.
1Н ЯМР (CDCl3, 300 МГц) d ppm 12,0 (s, 1H), 7,35-7,24 m, 5H), 6,51 (bs, 1H), 6,21 (s, 1H), 5,55 (bs, 1H), 4,55 (t, J=7,5 Гц, 1H), 4,53 (bs, 1H), 4,38 (bs, 1H), 4,10 (bs, 1H), 3,10 (t, J=7,5 Гц, 2H), 2,70 (m, 2H), 1,79 (bs, 3H), 1,71 (bs, 3H), 0,88 (t, J=6,6 Гц, 3H).
Получение соединения XII
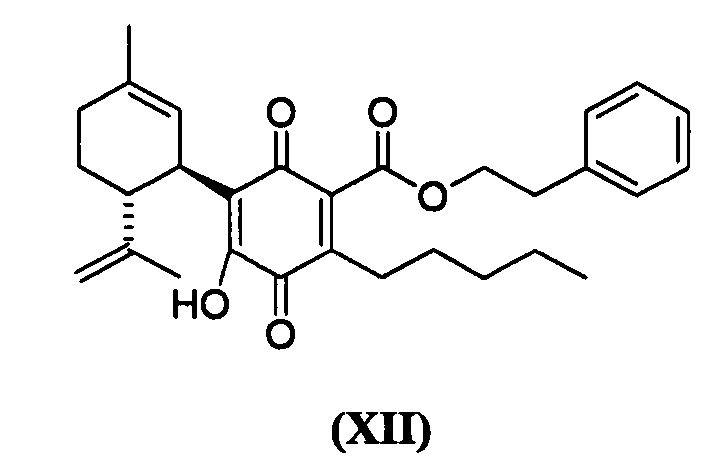
Фенетил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
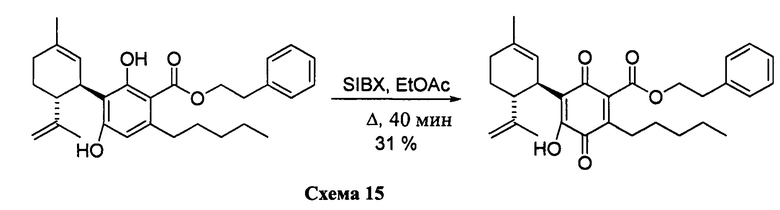
К раствору 302 мг (0,65 ммоль) фенетил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоата в 4 мл EtOAc добавляли SIBX (1,10 г, 39,1 ммоль, 6 моль эквив.) и реакционную смесь нагревали с обратным холодильником в течение 1 ч (Схема 15). После охлаждения и фильтрации через целит, фильтрат последовательно промывали 5% NaHCO3 и насыщенным солевым раствором. После высушивания (Na2SO4) и выпаривания, остаток очищали с помощью флэш-хроматографии на RP-18 силикагеле с применением градиента смеси метанол-вода (от 6:4 до чистого метанола) в качестве элюента, в результате получали 94 мг (31%) соединения XII. 1Н ЯМР (CDCl3, 300 МГц) d ppm 7,00 (bs, 1Н), 5,14 (bs, 1H), 4,54 (s, 1H), 4,52 (s, 1H), 4,51 (t, J=7,5 Гц), 3,74 (bd, J=7,0 Гц, 1H), 3,02 (t, J=7,5 Гц, 2H), 2,75 (br t, J=9,1 1,5 Гц, 1H), 2,26 (t, J=7,5 Гц, 2H), 1,74 (bs, 3H), 1,67 (bs, 3H), 0,86 (t, J=6,6 Гц, 3H).
Синтез предшественника соединения XIII
(Е)-3,7-диметилокта-2,6-диенил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоат (CBDA- гераниловый эфир)
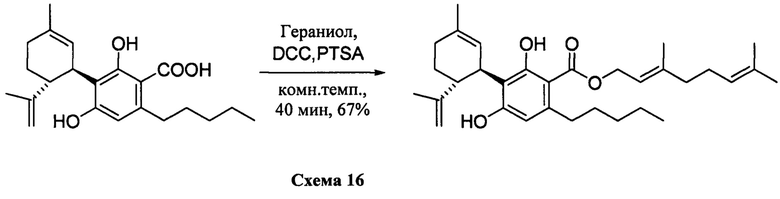
К раствору каннабидиоловой кислоты (CBDA) (300 мг, 0,84 ммоль) в CH2Cl2 (4 мл) добавляли гераниол (0,18 мл, 10,1 ммоль, 1,2 моль эквив.), а затем DCC (345 мг, 1,68 ммоль, 2 моль эквив.) и катализатор PTSA (30 мг). После 25 мин, реакционную смесь обрабатывали путем упаривания и остаток растворяли в толуоле, охлаждали при -18°С в течение 20 мин. для осаждения дициклогексилмочевины. После фильтрации фильтрат упаривали, и остаток очищали с помощью флэш-хроматографии с "гравитационным элюированием" на силикагеле с применением петролейного эфира-EtOAc 95:5 в качестве элюента. Получали 200 мг (67%) бесцветного масла. 1Н ЯМР (CDCl3, 300 МГц) d ppm 12,1 (s, 1Н), 6,48 (bs, 1H), 6,20 (s, 1H), 5,54 (bs, 1H), 5,45 (brt, J=6,7 Гц, 1H), 5,08 ((br s, 1H), 4,81 (d, J=6,7 Гц, 2H), 4,51 (bs, 1H), 4,38 (bs, 1H), 4,08 (bs, 1H), 2,74 (m, 2H), 1,78 (bs, 3H), 1,75 (bs, 3H), 1,71 (bs, 3H), 1,67 (bs, 3H), 0,88 (t, J=6,6 Гц, 3H).
Получение соединения XIII
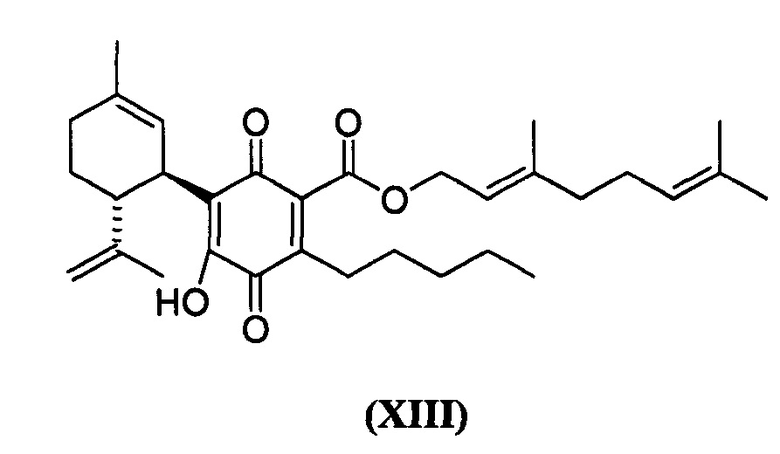
(Е)-3,7-диметилокта-2,6-диенил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат

К раствору 200 мг (0,40 ммоль) (Е)-3,7-диметилокта-2,6-диенил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентил-бензоата в 4 мл EtOAc добавляли SIBX (680 мг, 2,4 ммоль, 6 моль эквив.) и реакционную смесь нагревали с обратным холодильником в течение 40 мин (Схема 17). После охлаждения и фильтрации через целит, фильтрат последовательно промывали 5% NaHCO3 и насыщенным солевым раствором. После высушивания (Na2SC4) и выпаривания, остаток очищали с помощью флэш-хроматографии на RP-18 силикагеле с применением градиента смеси метанол-вода (от 6:4 до чистого метанола) в качестве элюента, в результате получая 18 мг (9%) соединения XIII.
1Н ЯМР (CDCl3, 300 МГц) d ppm 6,99 (bs, 1Н), 5,38 (bt, J=6,8 Гц, 1H), 5,12 (bs, 1H), 5,07 (bs, 1H), 4,81 (bs, 1H), 4,80 (bs, 1H), 4,56 (bs, 1H), 3,97 (d, J=6,8 Гц, 2H), 2,73 (m, 1H), 2,37 (m, 2H), 1,73 (bs, 3H), 1,70 (bs, 3H), 1,67 (bs, 3H), 1,62 (bs, 3H), 0,86 (t, J=6,9, 3H).
Синтез предшественника соединения XIV
(1S,2S,4R)-1,7,7-триметилбицикло[2.2.1]гептан-2-ил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоат (CBDA борниловый эфир)
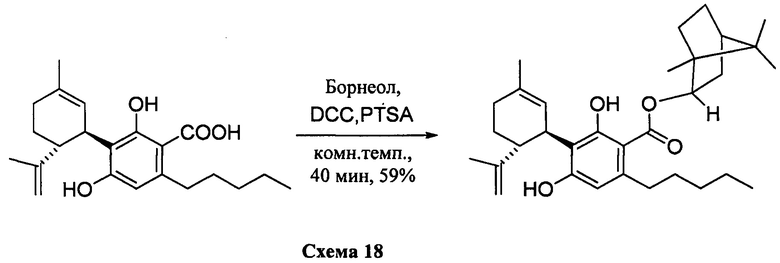
К раствору каннабидиоловой кислоты (CBDA) (302 мг, 0,84 ммоль) в CH2Cl2 (4 мл) добавляли (-) (S)-борнеол (157 мг, 1,2 моль эквив.), а затем DCC (350 мг, 2 моль эквив.) и катализатор PTSA (30 мг). После 40 мин, реакционную смесь обрабатывали путем упаривания и остаток растворяли в толуоле, охлаждали при -18°С в течение 20 мин для осаждения дициклогексилмочевины. После фильтрации фильтрат упаривали, и остаток очищали с помощью флэш-хроматографии на RP18-силикагеле с применением градиента смеси метанол-вода (от 6:4 до чистого метанола) в качестве элюента. В результате получали 178 мг (59%) бесцветного масла.
1Н ЯМР (CDCl3, 300 МГц) d ppm 12,2 (s, 1H), 6,48 (bs, 1H), 6,23 (s, 1H), 5,54 (bs, 1H), 5,54 (bs, 1H), 5,19 ((br s, 1H), 4,52 (bs, 1H), 4,40 (bs, 1H), 4,12 (bs, 1H), 2,91 (m, 2H), 1,80 (bs, 3H), 1,71 (bs, 3H), 0,96 (s, 3H), 0,89 (s, 6H), 0,88 (t, J=6,6 Гц, 3H).
Получение соединения XIV
((1S,2S,4R)-)-1,7,7-триметилбицикло[2.2.1]гептан-2-ил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
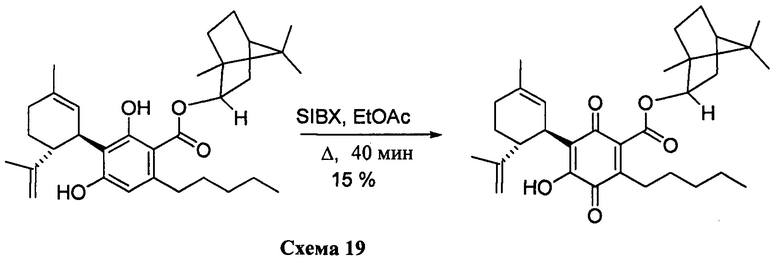
К раствору 170 мг (0,34 ммоль) (1S, 2S, 4К)-1,7,7-триметилбицикло[2.2.1]гептан-2-ил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоата в 4 мл EtOAc добавляли SIBX (578 мг, 2,1 ммоль, 6 моль эквив.) и реакционную смесь нагревали с обратным холодильником в течение 40 мин (Схема 19). После охлаждения и фильтрации через целит, фильтрат последовательно промывали 5% NaHCO3 и насыщенным солевым раствором. После высушивания (Na2SO4) и выпаривания, остаток очищали с помощью колоночночной хроматографии с "гравитационным элюированием" на силикагеле с применением петролейного эфира-EtOAc 98:2 в качестве элюента с получением 25 мг (15%) соединения XIV.
1Н ЯМР (CDCl3, 300 МГц) d ppm 6,98 (bs, 1Н), 5,16 (bs, 1H), 5,10 (bd, J=10 Гц, 1H), 4,58 (bs, 1H), 4,56 (bs, 1H), 3,75 (bd, J=6,8 Гц, 1H), 2,73 (m, 1H), 2,37 (m, 2H), 1,61 (bs, 3H), 0,92 (s, 3H), 0,90 (s, 3H), 0,88 (s, 3H), 0,86 (t, J=6,9, 3H).
Синтез предшественника соединения XV
(1R,2R,4R)-1,5,5-Триметилбицикло[2.2.1]гептан-2-ил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоат (CBDA фенхиловый эфир)

К раствору каннабидиоловой кислоты (CBDA) (550 мг, 1,54 ммоль) в CH2Cl2 (4 мл) добавляли (+) (R)-фенхол (284 мг, 1,2 моль эквив.), а затем DCC (634 мг, 2 моль эквив.) и катализатор PTSA (30 мг). Спустя 40 мин реакционную смесь обрабатывали путем упаривания, и остаток растворяли в толуоле, охлаждали при -18°С в течение 20 мин для осаждения дициклогексилмочевины. После фильтрации фильтрат упаривали, и остаток очищали с помощью колоночночной хроматографии с "гравитационным элюированием" на силикагеле с получением 350 мг (64%) бесцветного масла. 1Н ЯМР (CDCl3, 300 МГц) d ppm 12,34 (s, 1H), 6,50 (bs, 1H), 6,24 (s, 1H), 5,57 (bs, 1H), 4,64 (bs, 1H), 4,52 (bs, 1H), 4,39 (bs, 1H), 4,10 (bs, 1H), 2,97 (m, 2H), 1,71 (bs, 3H), 1,20 (s, 3H), 1,14 (s, 3H), 0,96 (s, 3H), 0,89 (s, 6H), 0,89 (t, J=6,6 Гц, 3H), 0,79 (s, 3H).
Получение соединения XV
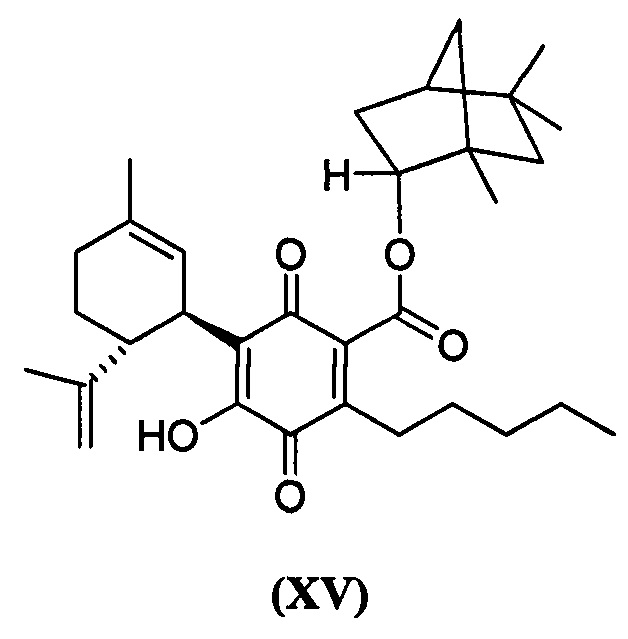
(1R,2R,4R)-1,5,5-триметилбицикло[2.2.1]гептан-2-ил-4-гидрокси-5-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-3,6-диоксо-2-пентилциклогекса-1,4-диенкарбоксилат
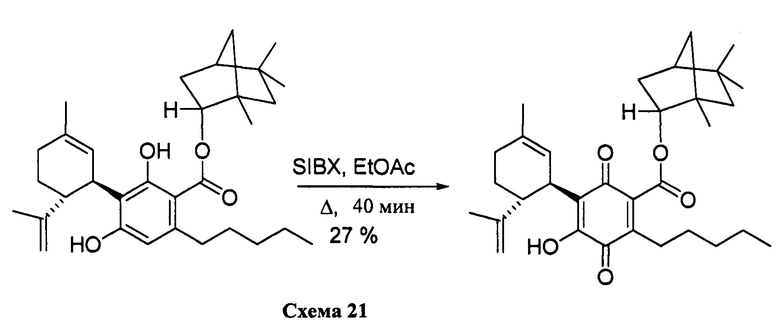
К раствору 300 мг (0,61 ммоль) (1R, 2R, 4R)-1,5,5-триметилбицикло[2.2.1]-гептан-2-ил-2,4-дигидрокси-3-((1R,6R)-3-метил-6-(проп-1-ен-2-ил)циклогекс-2-енил)-6-пентилбензоат в 4 мл EtOAc добавляли SIBX (1,019 г, 6 моль эквив.) и реакционную смесь нагревали с обратным холодильником в течение 40 мин (схема 21). После охлаждения и фильтрации через целит полученный фильтрат последовательно промывали 5% NaHCO3 и насыщенным солевым раствором. После высушивания (Na2SO4) и выпаривания, остаток очищали с помощью колоночночной хроматографии с "гравитационным элюированием" на силикагеле с применением петролейного эфира-EtOAc 98:2 в качестве элюента с получением 81 мг (27%) соединения XV.
1Н ЯМР (CDCl3, 300 МГц) d ppm 6,98 (bs, 1Н), 5,16 (bs, 1H), 5,10 (bd, J=10 Гц, 1H), 4,60 (bs, 1H), 4,57 (bs, 1H), 4,55 (bs, 1H), 3,73 (bd, J=10 Гц, 1H), 2,73 (m, 1H), 2,38 (m, 2H), 1,67 (bs, 3H), 1,15 (s, 3H), 1,10 (s, 3H), 0,86 (s, 3H), 0,86 (t, J=6,9, 3H).
Анализы in vitro
Пример 2. Агонистическая активность PPARg.
Для исследования биологической активности новых соединений авторы проводили трансактивацию PPARg в клетках НЕК-293 и в клетках фибробластов NIH-3T3.
Клетки HEK293T и клетки первичных фибробластов человека культивировали при 37°С во влажной атмосфере, содержащей 5% СО2, на среде DMEM с добавлением 10% фетальной телячьей сыворотки (FBS) и 1% (об./об.) пенициллина/стрептомицина. Росиглитазон приобретали у Cayman Chemical Company (Анн-Арбор, Мичиган, США). Все другие реагенты были приобретены у Sigma Со (Сент-Луис, Миссури, США). Клетки HEK293T (2×103/на лунку) (фигуры 1А, 1В и 1С) или клетки NIH-3T3 (5×103/на лунку) (фигура 2) высевали в белые 96-луночные планшеты с прозрачным дном BD Falcon™ White with Clear Bottom 96-well Microtest™ Optilux™ Plate на 24 часа. После этого клетки временно ко-трансфицировали экспрессионным вектором GAL4-PPARy и вектором с люциферазным репортером GAL4-luc с применением Roti©-Fect (Carl Roth, Карлсруэ, Германия) в соответствии с инструкциями производителя. Через двадцать четыре часа после трансфекции, клетки предварительно обрабатывали возрастающими дозами соединений в течение 6 часов. Затем клетки лизировали в 25 мМ Трис-фосфате с рН 7,8, 8 мМ MgCl2, 1 мМ ДТТ, 1% Тритона Х-100 и 7% глицерина. В лизате клеток измеряли активность люциферазы с применением мультирежимного микропланшетного ридера TriStar LB 941 (Berthold) и следуя инструкциям набора для анализа люциферазы (Promega, Мэдисон, Висконсин, США). Концентрацию белка определяли по методу Бредфорда (Bio-Rad, Ричмонд, Калифорния, США). Из каждой экспериментальной величины вычитали значения фона, полученные для лизирующего буфера, а удельную трансактивацию выражали в виде кратной индукции относительно необработанных клеток. Все эксперименты проводили по меньшей мере в трех повторностях. Использовали плазмиду Gal4-hPPARgamma (название плазмиды: pCMV-BD-hPPARg, лаборатория Sinal, кафедра фармакологии, Университет Далхаузи) и репортерную плазмиду Gal4 luc, которая содержала пять ДНК-связывающих сайтов Gal4, сшитых с геном люциферазы. Приведенный выше метод анализа проиллюстрирован на фигуре 1 (А, В и С) и фигуре 2, которые демонстрируют влияние VCE-004 (соединение I) и аналогов на активность PPARg посредством метода трансактивации, выполненного в клетках временно гиперэкспрессирующих PPARg в сочетании с репортерным геном люциферазы (PPARg-GAL4/GAL4-LUC) и обработанных соединениями в течение 6 часов. Данные приведены как средние значения с величинами среднеквадратичных погрешностей для трех повторностей. Значимое увеличение активности люциферазы наблюдали для хиноновых производных по сравнению с необработанными клетками. Данный результат подтверждает, что соединения с II по XIV являются значительно более активными, чем соединение VCE-004 (соединение I) для активации PPARg в концентрациях от 5 до 50 μМ. Соединения с II по X усиливают трансактивацию PPARg дозозависимым образом, и соединения II, III, IV, V, VII и VIII являются наиболее активными соединениями. Кроме того, в более высоких концентрациях (10, 25 и 50 μМ) эти соединения являются высокоактивными для активации PPARg, по сравнению с VCE-004 (соединение I). RZG, который является полным агонистом PPARg, увеличивает активность PPARg более чем в 100 раз в концентрации 1 μМ. В противоположность этому, максимальная индукция активности PPARg, вызванная введением соединений согласно настоящему изобретению в концентрации 1 μМ, никогда не увеличивалась более чем в 5 раз, что свидетельствует о том, что данные новые соединения являются модуляторами PPARg, а не полными агонистами PPARg.
Пример 3. Производные каннабидиол-хинона и Росиглитазон связываются с одним и тем же сайтом в белке PPARg.
(A) Клетки HEK293T культивировали при 37°С во влажной атмосфере, содержащей 5% СО2, в среде DMEM с добавлением 10% фетальной телячьей сыворотки (FBS) и 1% (об./об.) пенициллина/стрептомицина. Росиглитазон приобретали у Cayman Chemical Company (Анн-Арбор, Мичиган, США). Клетки НЕК293Т (2×103/на лунку) (фигура 3А) высевали в белые 96-луночные планшеты с прозрачным дном BD Falcon™ White with Clear Bottom 96-well Microtest™ Optilux™ Plate на 24 часа. После этого клетки временно ко-трансфицировали экспрессионным вектором GAL4-PPARγ и вектором с люциферазным репортером GAL4-luc с применением Roti©-Fect (Carl Roth, Карлсруэ, Германия) в соответствии с инструкциями производителя. Через двадцать четыре часа после трансфекции, клетки предварительно обрабатывали возрастающими дозами соединений в течение 30 мин и затем стимулировали RSZ (1 μМ) в течение 6 часов. Измеряли транскрипционную активность PPARg в соответствии с примером 2, тем самым подтверждая, что соединения III, V, VIII, X и XIII способны уменьшать RSZ-индуцированную трансактивацию PPARg. Таким образом можно предположить, что соединения, III, V, VIII, X и XIII и RSZ могут связываться с одним тем же сайтом связывания на PPARg.
(B) Особенности связывания соединения VIII (в качестве примера) с PPARg рассчитывали с помощью виртуальной стыковки с применением программного обеспечения AutoDock и настройки алгоритма Vina как системы расчета. Область поиска была установлена таким образом, чтобы найти точки связывания вокруг молекулярной поверхности. Для обеспечения эффективности метода стыковки так же рассчитывали особенности стандартного лиганда RSZ для PPARg, чтобы использовать эти результаты в качестве контроля. AutoDock показал 10 стабильных конформаций для каждого лиганда (RSZ и соединения VIII). Шесть из этих конформаций для обоих RSZ и соединения VIII совпали с сайтом связывания RSZ, о котором ранее сообщалось [Liberato et al. 2012]. Остатки Y473, Н323, I326, S289 и Н449 в PPARg определены как анкерные позиции и являются частью группы из десяти аминокислот с близким пространственным расположением, которые образуют сайт связывания для лигандов PPARg [Nolte et al. 1998], [Itot et al. 2008], [Li et al. 2008]. Сайт связывания RSZ показал большую термодинамическую стабильность для соединения VIII, чем для RSZ (фигура 3В), что дает возможность предположить о более высокой аффинности вышеуказанного соединения для данного рецептора. Действительно, конформация соединения VIII с наиболее высокой аффинностью показала аффинность связывания -8,0 ккал/моль, тогда как лучшая конформация RSZ показала -6,9 ккал/моль. Тем не менее, только два из 10 связывающих остатков RSZ, I341 и R288 в PPARg вероятно взаимодействуют с соединением VIII. В целом, данные результаты свидетельствуют о том, что соединение VIII может сильнее связываться с PPARg, чем с RSZ в близко примыкающем сайте, но с другой схемой взаимодействия лиганд-рецептор, что приводит к различному конформационному воздействию на рецептор. Кроме того, блокирования I341 и R288 будет достаточно, чтобы избежать проникновения RSZ, тем самым снижая действие данного лекарственного средства.
Пример 4. Тесты на цитотоксичность.
Электрофильные хиноны индуцируют цитотоксичность и активируют путь Nrf2, клеточный сенсор образования активных форм кислорода. На фигуре 4 проанализирована индуцированная клеточная гибель для трех различных типов клеток N2a (А), НТ22 (В) и МО3.13 (С), вызванная введением соединения VCE-004 (соединение I) и соединений с II по XV.
Три клеточные линии МО3.13, N2A и НТ22 культивировали при 37°С во влажной атмосфере, содержащей 5% СО2, в среде DMEM с добавлением 10% фетальной телячьей сыворотки (FBS) и 1% (об./об.) пенициллина/стрептомицина. Жизнеспособность клеток N2A, НТ22 и МО3.13 определяли с помощью анализа на основе МТТ. В кратком изложении, клетки высевали с плотностью 104 клеток/на лунку в 96-луночные планшеты в количестве 200 μл клеточной суспензии на лунку и культивировали в течение 24 часов. Затем клетки инкубировали с несколькими концентрациями соединений в течение 24 часов. После этого 100 μл МТТ (5 мг/мл) из смеси раствора МТТ: DMEM (1:2) добавляли в каждую лунку и клетки инкубировали в течение 4 ч при 37°С в темноте. Затем реакцию останавливали, удаляли супернатант и в каждую лунку добавляли 100 μл ДМСО, затем инкубировали в течение 10 минут при осторожном встряхивании. В результате измеряли оптическую плотность при 550 нм с использованием TriStar LB 941 (Berthold Technologies, GmbH & Co. KG). В качестве 100% были взяты контрольные клетки и данные были отнесены к этому значению. Клеточные линии N2a (фигура 4А), НТ22 (фигура 4В) и МО3.13 (фигура 4С) инкубировали в течение 24 ч с указанными дозами соединений VCE-004 (соединение I) и соединений с II по XV, а жизнеспособность клеток количественно измеряли с помощью анализа на основе МТТ. Результаты представляли в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов и выражали в процентах от жизнеспособности клеток по отношению к (-) контрольному образцу. Контроль принимали за 100%, и полученные данные сопоставляли с этим значением. Результаты показали, что цитотоксическая активность, связанная с VCE-004 (соединение I), коррелировала с его способностью индуцировать активацию Nrf2. В этом же смысле, отсутствие цитотоксической активности, описанной в настоящем изобретении для соединений с II по XV, которые являются производными от VCE-004 в положении 3', коррелирует с их неспособностью активировать Nrf2.
Пример 5. Транскрипционная активность Nrf2.
Для изучения активности соединений на пути Nrf2 получали клеточную линию HaCaT-ARE-Luc. Клетки HaCat ко-трансфицировали репортерной плазмидой Nqol ARE-Luc и плазмидой pPGK-Puro с применением реагента для трансфекции Lipofectamine© 2000 (Life Technologies, Карлсбад, Калифорния, США). Стабильных трансформантов выделяли и поддерживали на среде RPMI 1640, содержащей 10% FBS, 1% пенициллина/стрептомицина и 10 μл/мл пуромицина. Клетки НаСаТ-ARE-Luc инкубировали в течение 6 ч с VCE-004 (соединение I) и с соединениями с II по VIII (А) или с соединениями с IX по XV (В) в указанных концентрациях, получали белковые лизаты и анализировали их на люциферазную активность, как описано в примере 1. Прооксидант трет-бутилгидрохинон (tBHQ) в концентрации 20 μМ использовали в качестве положительного контроля. Кратный уровень активации рассчитывали по отношению к (-) контрольному образцу в качестве эталона (фигура 5А и 5В). Результаты представлены в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов. Результаты подтвердили, что отсутствует реактивная электрофильная активность VCE-004 (соединение I) во всех соединениях (производных в положении 4), описанных в настоящем изобретении.
Пример 6. Анализ нейропротекции.
Активация ядерных рецепторов PPARg играет важную роль в нейропротекции, и известно, что агонисты PPARg предотвращают глутамат-индуцированную цитотоксичность в нейронных клетках.
Клетки N2a предварительно инкубировали с соединениями с I по VIII (фигура 6А) и с IX по XV (фигура 6В) в указанных концентрациях в течение 1 ч и затем обрабатывали 5 мМ глутамата в течение 24 часов для индукции эксайтотоксичности. Цитотоксичность определяли с помощью анализа на основе МТТ, как описано в примере 4. Результаты представляли в виде среднего значения ± стандартное отклонение для по меньшей мере трех независимых экспериментов и выражали в процентах от жизнеспособности клеток по отношению к (-) контрольному образцу. Контроль принимали за 100% и данные относили к этому значению.
Данные результаты показали заметные различия между соединением I и соединениями с II по XV, которые являются модуляторами PPARg, а также защищают нейронные клетки от глутамат-индуцированной гибели клеток.
Пример 7. Влияние производных CBD-хинона на транскрипцию гена коллагена.
Лиганды PPARg, как сообщалось, оказывают антифиброзное действие, а блокада TGFb-сигнализации посредством активации PPARg приводит к снижению выработки коллагена в фибробластах.
Клетки фибробластов NIH-3T3 временно трансфицировали плазмидой COL1A2-Luc, которая содержала последовательность с -353 по +58 п. о. промотора человека COL1A2, слитого с репортерным геном люциферазы. Двадцать четыре часа спустя клетки инкубировали с соединениями III, V, VIII и X (в качестве примеров) в течение 30 мин и обрабатывали TGFb (50 нг/мл) в течение 6 ч. Получали белковые лизаты и анализировали их на люциферазную активность. Было показано, что соединения III, V, VIII и X заметно ингибируют TGFb-индуцированную транскрипцию гена коллагена типа I (фигура 7).
Пример 8. Влияние производных CBD-хинона на выработку коллагена.
Выработку коллагена осуществляли с использованием метода Sirius Red-Fast Green, предназначенного для количественного определения коллагена и неколлагеновых белков в клеточных гранулах. Клетки NIH-3T3 высевали с плотностью 5×104/на лунку в 24-луночные планшеты и инкубировали в течение ночи при 37°С для обеспечения прикрепления клеток. Далее, клетки предварительно инкубировали 1 час с указанными концентрациями соединений III, V, VIII и X, и TGFb (50 нг/мл) в течение 24 часов. После обработки, клеточные осадки экстрагировали в течение ночи при 4°С с 100 μл 0,5 М уксусной кислоты. Затем к клеточным осадкам добавляли 1 мл раствора красителя (0,1% Sirius Red и 0,1% Fast Green, растворенных в насыщенной пикриновой кислоте) и осторожно перемешивали при комнатной температуре в течение 30 минут. Затем образцы центрифугировали при 10000 об/мин в течение 5 минут, чтобы осадить коллаген. Супернатанты осторожно удаляли, не нарушая осадок, и в каждую пробирку добавляли 1 мл 0,1 М хлористоводородной кислоты для удаления несвязанного красителя. Образцы центрифугировали при 10000 об/мин в течение 5 минут, затем в каждую пробирку добавляли 1 мл 0,5 М натрия хлорида и энергично перемешивали на вортексе для освобождения связанного красителя. Образцы центрифугировали при 2500 об/мин в течение 5 минут для повторного осаждения любого клеточного дебриса.
Определяли выработку коллагена и результаты выражали в виде кратной индукции относительно необработанных клеток. Было показано, что соединения III, V, VIII и X заметно ингибируют TGFb-индуцированную выработку коллагена в фибробластах (фигура 8). Цитотоксичность, связанная с VCE-004 (HU-331), не позволяла исследовать влияние данного соединения на TGFb-индуцированную выработку коллагена.
Пример 9. Влияние VCE-004 и производных CDB-хинона на образование активных форм кислорода (АФК) и на митохондриальный трансмембранный потенциал.
Митохондриальный мембранный потенциал имеет решающее значение для поддержания физиологических функций дыхательной цепи для образования АТФ. Значительная потеря митохондриального мембранного потенциала в клетках приводит к недостатку энергии с последующей гибелью. Таким образом, возможность определять митохондриальный мембранный потенциал и АФК позволяет собирать важные сведения о физиологическом состоянии клетки и функционировании митохондрий в ответ на электрофильные и реактивные молекулы.
На фигуре 5 показано, что VCE-004 (соединение I) представляет собой реакционноспособное соединение, которое активирует путь Nrf2. Для дальнейшего подтверждения влияния на выработку внутриклеточных АФК и на нарушение митохондриального мембранного потенциала, изобретатели непосредственно анализировали HU-311 и соединения согласно настоящему изобретению.
Клетки Jurkat выращивали при 37°С и 5% СО2 в питательной среде RPMI 1640 с добавками, содержащей 10% FCS, инактивированной нагреванием, 2 мМ глутамина и антибиотики. Для оценки митохондриального трансмембранного потенциала и образования активных форм кислорода (АФК), клетки (5×105/мл) обрабатывали возрастающими концентрациями VCE-004 (соединение I) или соединениями III, V, VII и X (в качестве примеров производных соединения I) в течение 2 часов для обнаружения митохондриального мембранного потенциала или в течение 6 часов для обнаружения АФК. После обработки клетки дважды промывали холодным фосфатно-солевым буферным раствором (ФСБ) и инкубировали в ФСБ с флуоресцентными зондами H2DCF-DA (зеленая флуоресценция) (20 нМ) для обнаружения АФК, и MitoTracker Red CMXR (MTR-CMXR) (50 нМ) для обнаружения митохондриального мембранного потенциала (Molecular Probes, Юджин, Орегон, США) в течение 20 мин при 37°С с последующим анализом на проточном цитометре FACSCantoII. Авторы обнаружили, что VCE-004 (соединение I) индуцирует заметное увеличение уровней внутриклеточных АФК и нарушение митохондриального мембранного потенциала. В противоположность этому, соединения III, V, VII и X не являются активными (увеличение уровня АФК) и не вызывают уменьшение митохондриального мембранного потенциала.
На фигуре 9А показано, что соединения формулы I вызывают заметное увеличение доли клеток с гиперэкспрессией АФК в зависимости от концентрации. В противоположность этому, соединения III, V, VIII и X неспособны значительно индуцировать накопление АФК в обработанных клетках. Образование АФК коррелирует с нарушением митохондриального мембранного потенциала, как показано на фигуре 9 В.
Пример 10. Сравнение взаимодействия VCE-004 и соединения XI с цистеамином.
Десять мг VCE-004 (соединение I) и соединения XI (в качестве примера CBD-производных согласно изобретению, применимо и к другим членам семейства соединений вышеуказанных производных с II по X и с XII по XV) независимо друг от друга растворяли в 1 мл ДМСО, и раствор обрабатывали избытком (4 моль эквивалентов) цистеамина. После перемешивания при комнатной температуре в течение 1 ч, растворы разбавляли водой (2 мл) и экстрагировали петролейным эфиром-эфиром 9:1. После выпаривания остаток растворяли в CDCl3 и анализировали с помощью 1Н-ЯМР. В то время как соединение XI было извлечено без потерь, VCE-004 (I) полностью вступало в реакцию и не было обнаружено в остатках, указывая на то, что VCE-004 необратимо связывалось с цистеамином.
Представленные результаты обосновывают терапевтическое применение соединений, описанных в настоящем изобретении, в частности, соединений III, V, VIII, X и XIII при лечении нейродегенеративных заболеваний и мозговых нарушений, связанных с травмами, где нейровоспаления и нейротоксичность играют существенную роль. Кроме того, соединения согласно настоящему изобретению особенно подходят для применения в качестве модулятора PPARg, в частности, для лечения заболеваний и состояний, восприимчивых к модуляции PPARg.
Ссылки:
- Ahmadian М, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. 2013. PPARy signaling and metabolism: the good, the bad and the future. Nat Med. 19:557-66
- Barish GD, Narkar VA, Evans RM. 2006. PPARδ: a dagger in the heart of the metabolic syndrome. J Clin Invest. 116:590-597
- Bernardo A, Minghetti L. 2008. Regulation of Glial Cell Functions by PPAR- gamma natural and Synthetic Agonists. PPAR Res. 864140.
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. 2000. Role of quinones in toxicology. Chem Res Toxicol. 3:135-60.
- Burstein S. 2005. PPAR-gamma: a nuclear receptor with affinity for cannabinoids. Life Sci. 77:1674-84.
- Ciudin A, Hernandez C,  R. 2012. Update on cardiovascular safety of PPARgamma agonists and relevance to medicinal chemistry and clinical pharmacology. Curr Top Med Chem. 12: 585-604.
R. 2012. Update on cardiovascular safety of PPARgamma agonists and relevance to medicinal chemistry and clinical pharmacology. Curr Top Med Chem. 12: 585-604.
- Doshi LS, Brahma MK, Bahirat UA, Dixit AV, Nemmani KV. 2012. Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin Investig Drugs. 19:489-512.
- Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, Huxlin KR. et al. 2009. Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 41:722-30.
- Fievet C, Fruchart JC, Staels B. 2006. PPAR alpha and PPAR gamma dual agonists for the treatment of type2 diabetes and the metabolicsyndrome. Curr. Opin. Pharmacol. 6: 606-614.
- Gelman, L., Feige, J.N., Desvergne, B. 2007. Molecular basis of selective PPARgamma modulation for the treatment of type 2 diabetes. Biochim. Biophys.Acta 1771: 1094-1107.
- Ghoochani A, Shabani K, Peymani M, Ghaedi K, Karamali F, Karbalaei K, Tanhaie S, Salamian A, Esmaeili A, Valian-Borujeni S, Hashemi M, Nasr-Esfahani MH, Baharvand H. 2012. The influence of peroxisome proliferator-activated receptor g(l) during differentiation of mouse embryonic stem cells to neural cells. Differentiation. 83: 60-67
- Granja AG, Carrillo-Salinas F, Pagani A,  M, Negri R, Navarrete C, Mecha M, Mestre L, Fiebich BL, Cantarero I, Calzado MA, Bellido ML, Fernandez-Ruiz J, Appendino G, Guaza C,
M, Negri R, Navarrete C, Mecha M, Mestre L, Fiebich BL, Cantarero I, Calzado MA, Bellido ML, Fernandez-Ruiz J, Appendino G, Guaza C,  E. 2012. A cannabigerol quinone alleviates neuroinflammation in a chronic model of multiple sclerosis. J Neuroimmune Pharmacol. 4:1002-1016
E. 2012. A cannabigerol quinone alleviates neuroinflammation in a chronic model of multiple sclerosis. J Neuroimmune Pharmacol. 4:1002-1016
- Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, Nagy L, Yamamoto K, Schwabe JW. 2008. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol 15:924-931.
- Kogan NM, Rabinowitz R, Levi P, Gibson D, Sandor P, Schlesinger M, and Mechoulam R. 2004. Synthesis and antitumor activity of quinonoid derivatives of cannabinoids. J Med Chem 47: 3800-3806
- Lehmann JM, Moore LB, Smith-Oliver ТА, Wilkison WO, Willson TM, Kliewer SA. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 270:12953-12956.
- Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A, Suino-Powell K, Baker PR, Freeman BA, Chen YE, Xu FIE. 2008. Molecular recognition of nitrated fatty acids by PPAR gamma. Nat Struct Mol Biol 15: 865-867
- Liberato MV, Nascimento AS, Ayers SD, Lin JZ, Cvoro A, Silveira RL,  L, Souza PC, Saidemberg D, Deng T, Amato AA, Togashi M, Hsueh WA, Phillips K, Palma MS, Neves FA, Skaf MS, Webb P, Polikarpov I. 2012. Medium Chain Fatty Acids Are Selective Peroxisome Proliferator activated Receptor (PPAR) с Activators and Pan-PPAR Partial Agonists. Plos One 7 e36297.
L, Souza PC, Saidemberg D, Deng T, Amato AA, Togashi M, Hsueh WA, Phillips K, Palma MS, Neves FA, Skaf MS, Webb P, Polikarpov I. 2012. Medium Chain Fatty Acids Are Selective Peroxisome Proliferator activated Receptor (PPAR) с Activators and Pan-PPAR Partial Agonists. Plos One 7 e36297.
- Liu J, Li H, Burstein SH, Zurier RB, Chen JD. 2003. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol. Pharmacol. 63: 983-992.
- Monks TJ, Jones DC. 2002. The metabolism and toxicity of quinones, quinonimines, quinone methides, and quinone-thioethers. Curr Drug Metab. 4:425-38.
- Morales P, Vara D,  M,
M,  MC, Olea-Azar C, Goya P,
MC, Olea-Azar C, Goya P,  J,
J,  I, Jagerovic N. 2013. Synthetic cannabinoid quinones: preparation, in vitro antiproliferative effects and in vivo prostate antitumor activity. Eur J Med Chem. 70: 111-119.
I, Jagerovic N. 2013. Synthetic cannabinoid quinones: preparation, in vitro antiproliferative effects and in vivo prostate antitumor activity. Eur J Med Chem. 70: 111-119.
- Na HK, Surh YJ. 2013. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic Biol Med. 67:353-365
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV.1998. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395: 137-143.
- O'Sullivan SE. 2007. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 152:576-82.
- Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. 2012. Semin Cell Dev Biol. 23:631-639.
- Rosen ED, MacDougald OA. 2006. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 7:885-96.
- Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. 2010. Nrf2 and Keapl abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 16:3743-53
- M.B. Sporn, K.T. Liby. 2012. NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer. 12: 564-57
- Stienstra R, Duval C, Muller M, Kersten S, 2007. PPARs, obesity, and inflammation. PPAR Res. 95974.
- Sun Y, Bennett A. 2007. Cannabinoids: A New Group of Agonists of PPARs. PPAR Res. 23 513.
-  , L.,
, L.,  , D., Nagy, L., 2007. PPARgamma in immunity and inflammation: cell types and diseases. Biochim. Biophys. Acta 1771: 1014-1030.
, D., Nagy, L., 2007. PPARgamma in immunity and inflammation: cell types and diseases. Biochim. Biophys. Acta 1771: 1014-1030.
- Tachibana K, Yamasaki D, Ishimoto K, Doi T. 2008. The Role of PPARs in Cancer. PPAR Res. 102737.
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 77: 289-312.
- Vanden Berghe W, Vermeulen L, Delerive P, DeBosscher К Staels B, Haegeman G. 2003. A paradigm for gene regulation: inflammation, NF-kB and PPAR. Adv.Exp.Med.Biol. 544:181-196.
- Vivacell Biotechnology Espana, S.L. 2011. Cannabinoid quinone derivatives. WO 2011117429 A1.
- Wang W, Liu F, Chen N. 2007. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-betal in renal interstitial fibroblasts. Mediators Inflamm: 62641
- Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. 2006. PPARgamma agonists prevent TGFbetal/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 350: 385-391.
- Zhang GY, Yi CG, Li X, Ma B, Li ZJ, Chen XL. Guo SZ, Gao WY. 2009. Troglitazone suppresses transforming growth factor-beta 1-induced collagen type I expression in keloid fibroblasts. Br J Dermatol. 160:762-70
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ КАННАБИГЕРОЛА | 2015 |
|
RU2684913C2 |
| ГИДРОКСИПИРИДОКСАЗЕПИНЫ В КАЧЕСТВЕ АКТИВАТОРОВ NRF2 | 2020 |
|
RU2812931C2 |
| ПРОИЗВОДНЫЕ БЕНЗОПИПЕРИДИНА, БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2142945C1 |
| ТИЕНОПИРИДОН КАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2374250C2 |
| ПРОИЗВОДНОЕ 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2804127C2 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| АНАЛОГИ ВИТАМИНА D, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ УКАЗАННЫЕ АНАЛОГИ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2320644C2 |
| ТРИТЕРПЕНОВЫЕ ПРОИЗВОДНЫЕ ЛУПЕОЛЬНОГО ТИПА КАК ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ | 2010 |
|
RU2561604C2 |
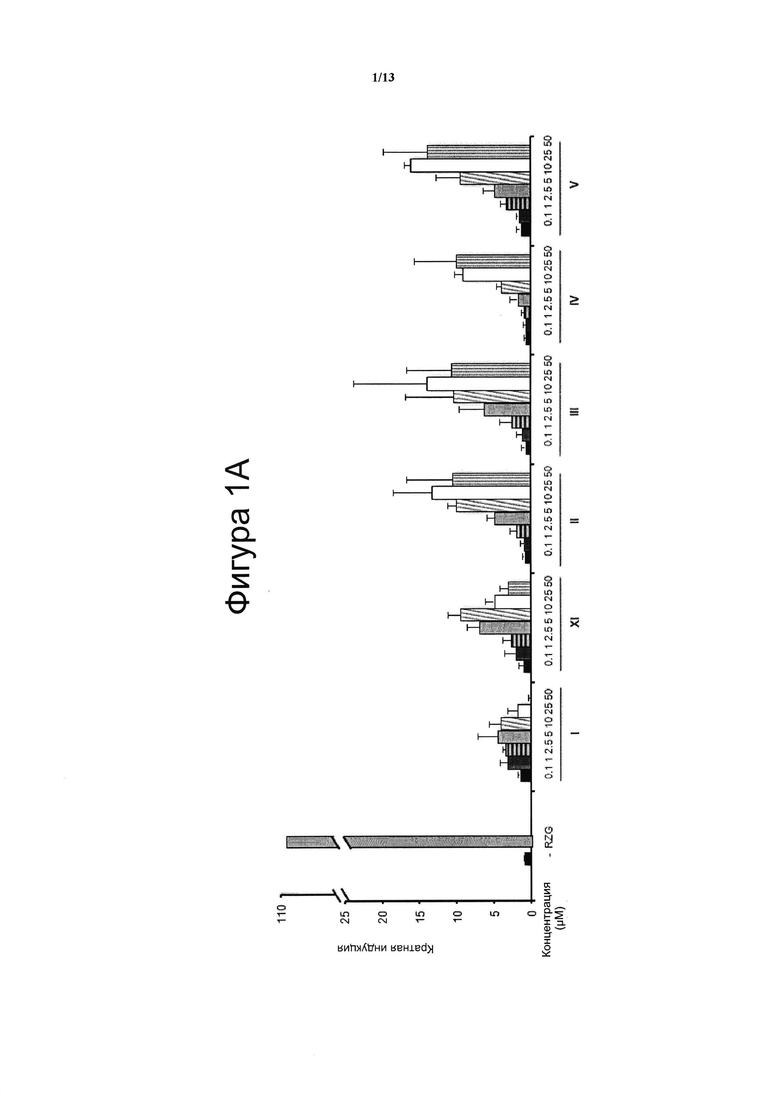
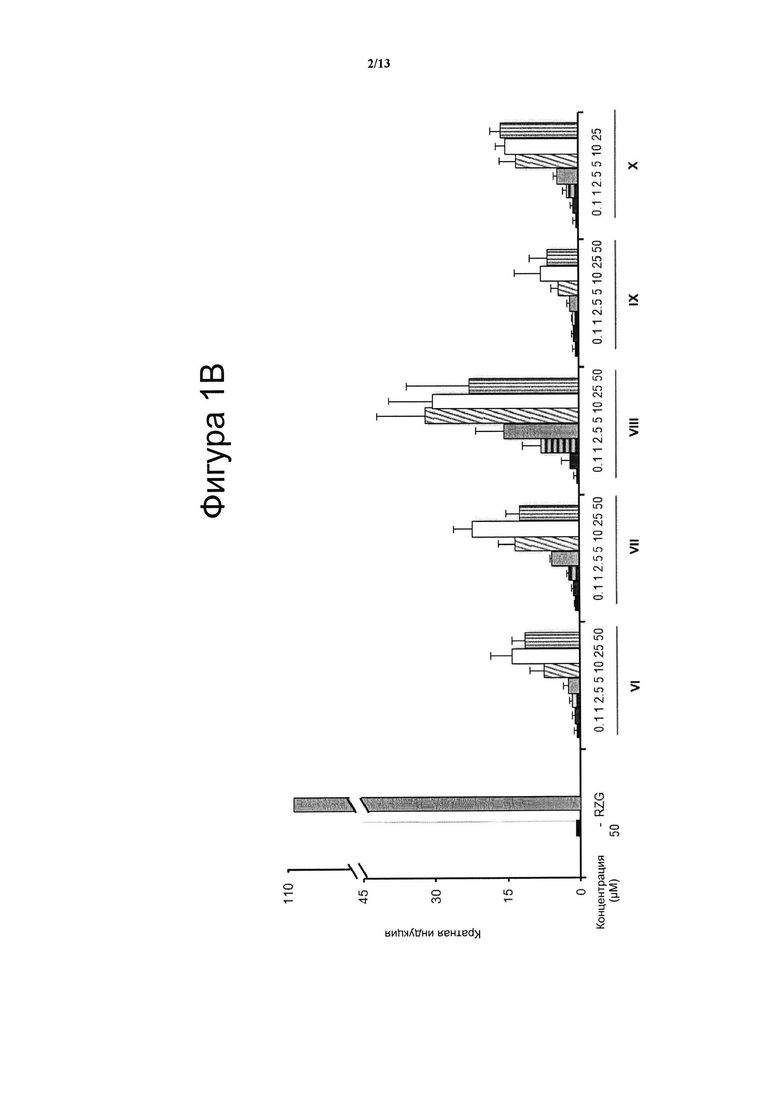
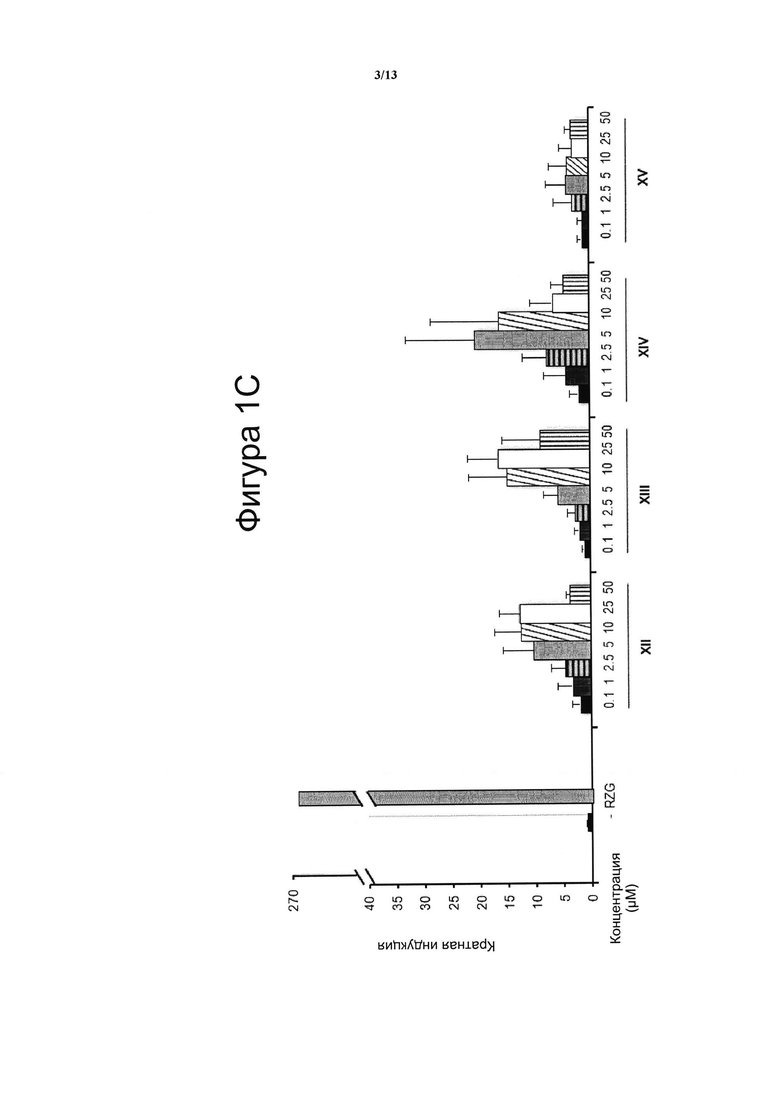
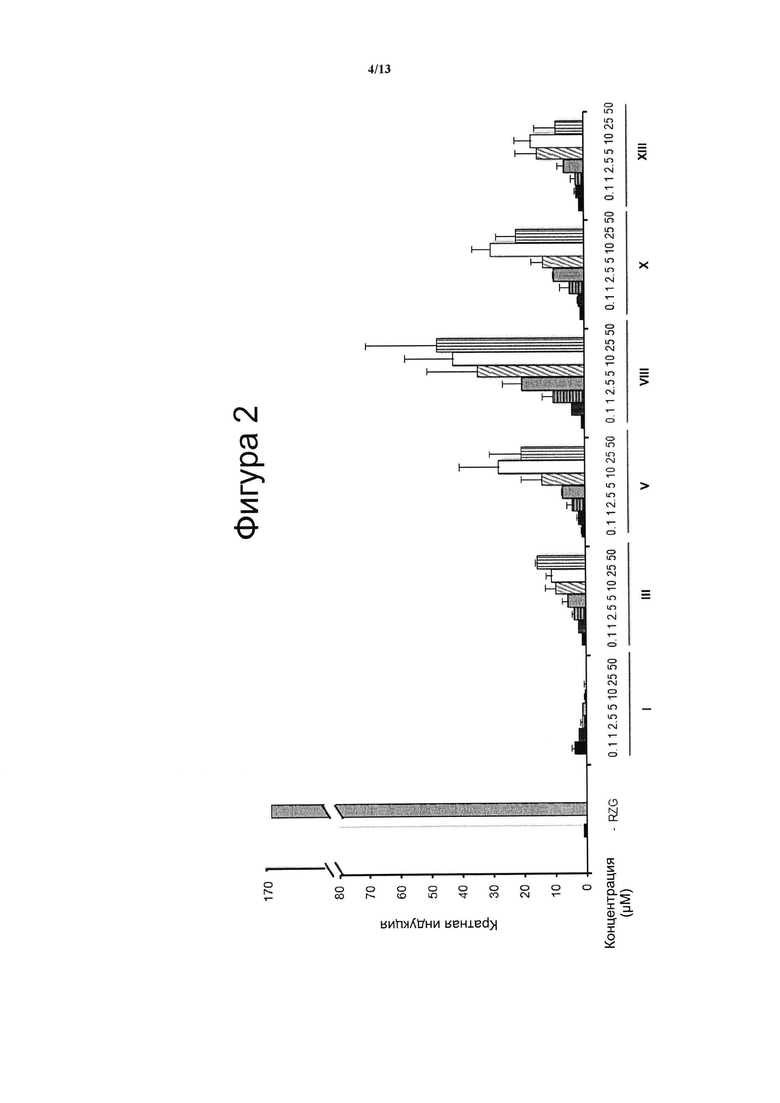
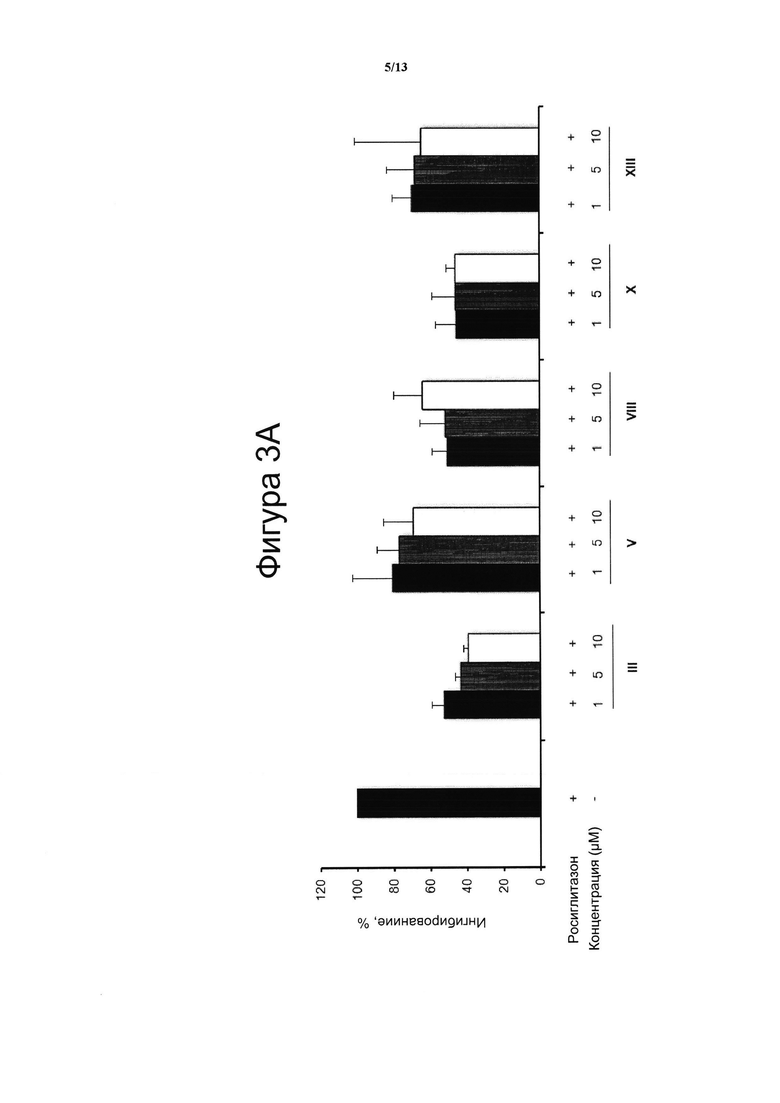
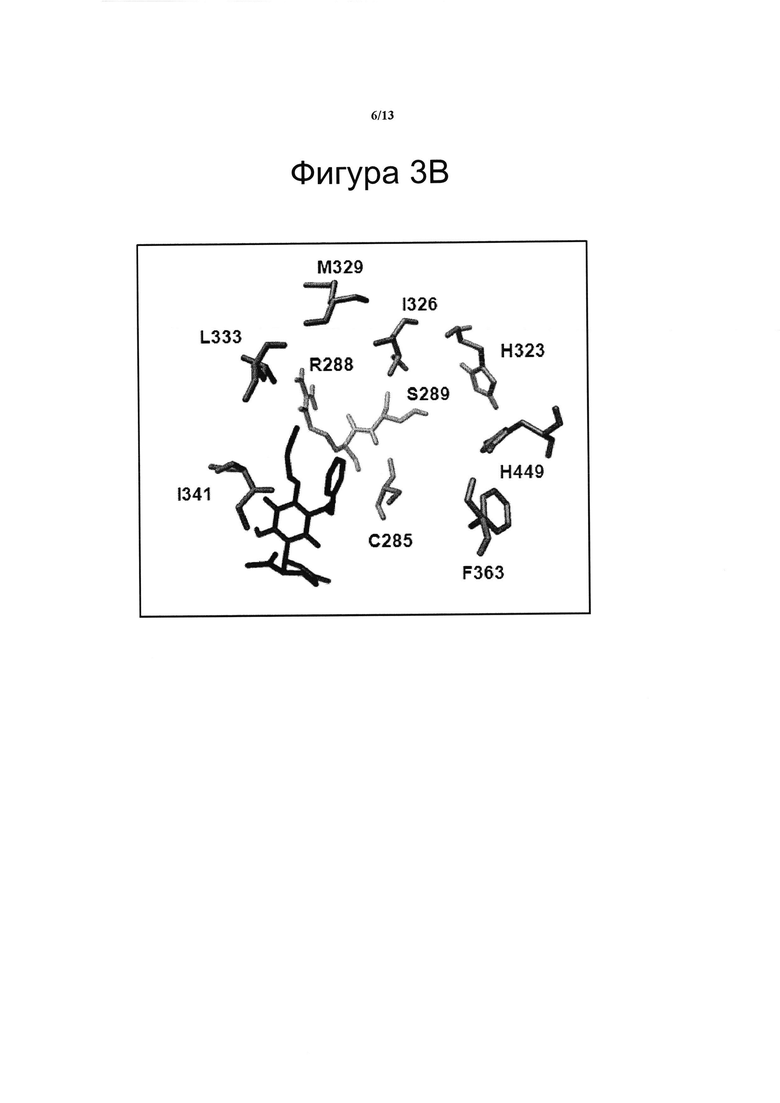
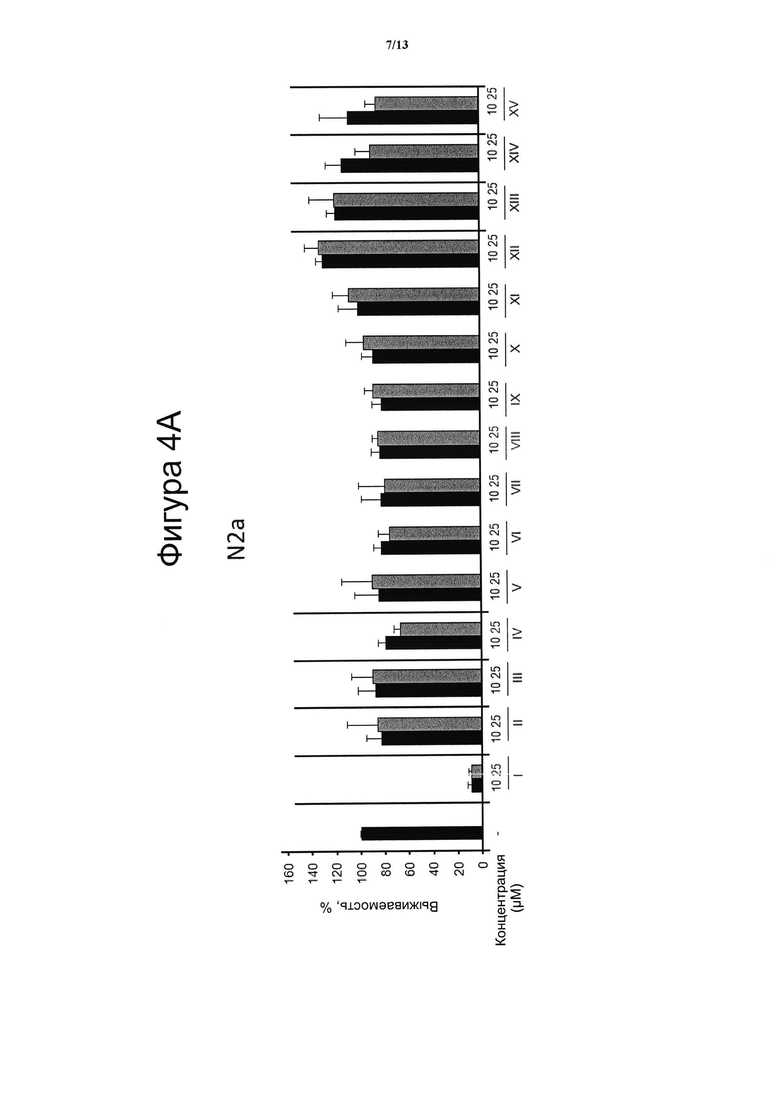

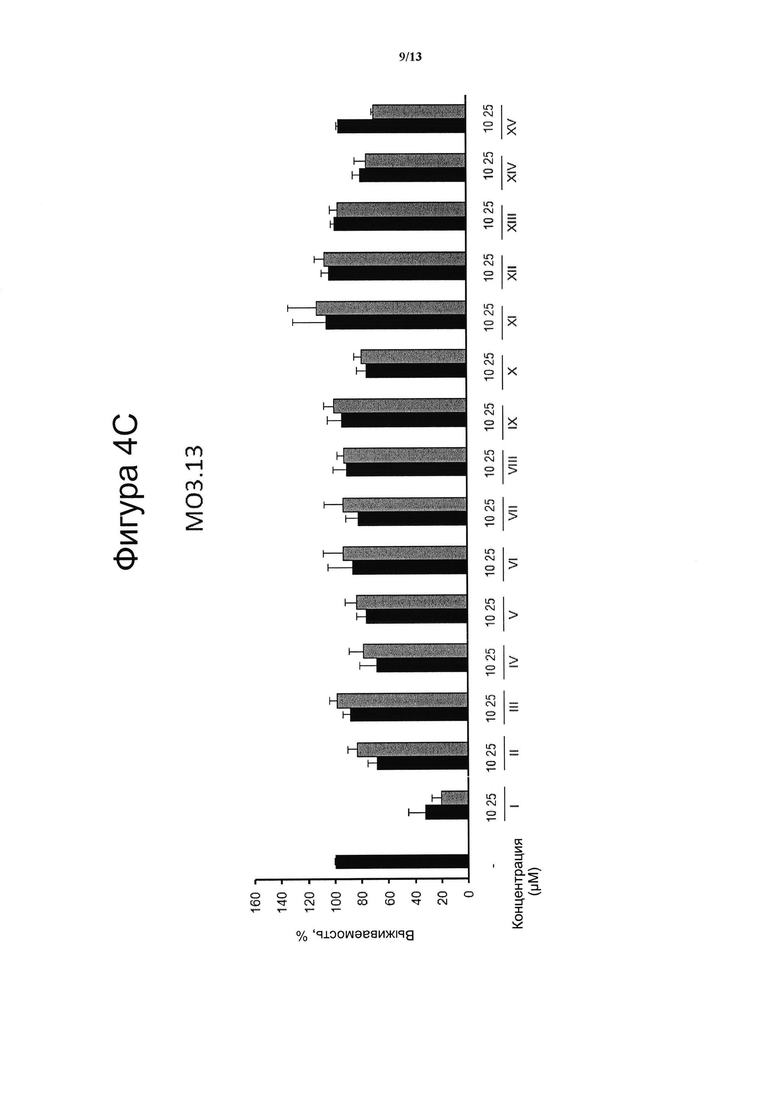
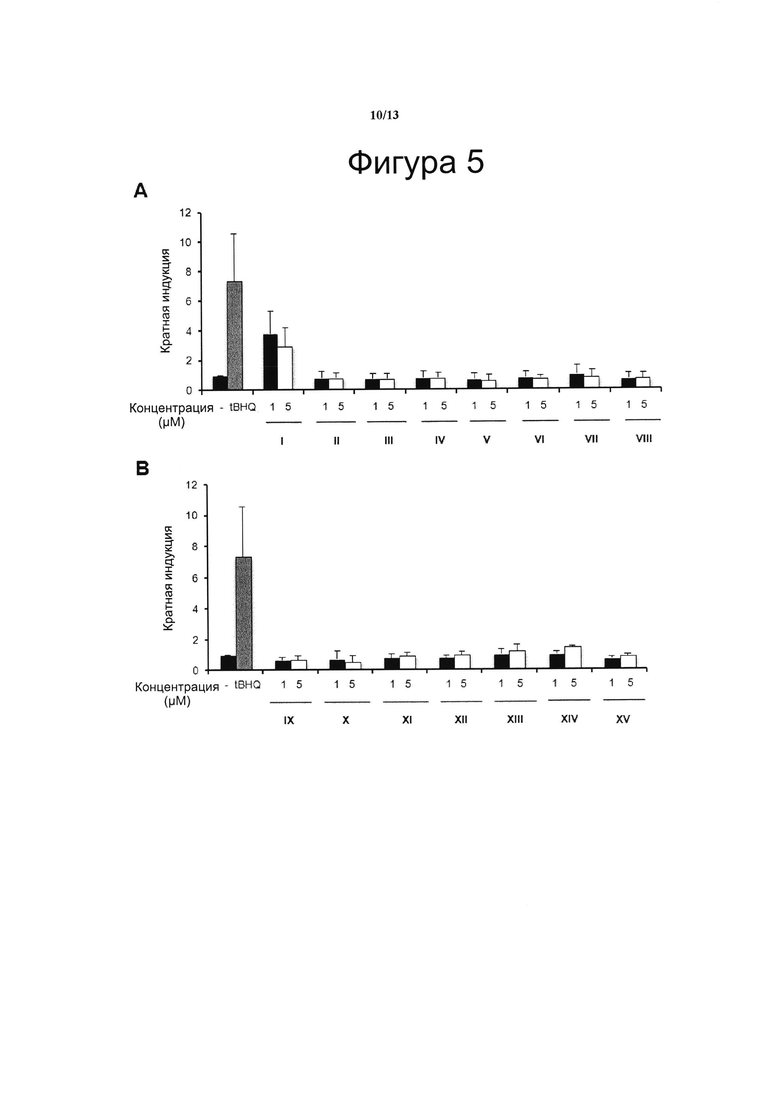
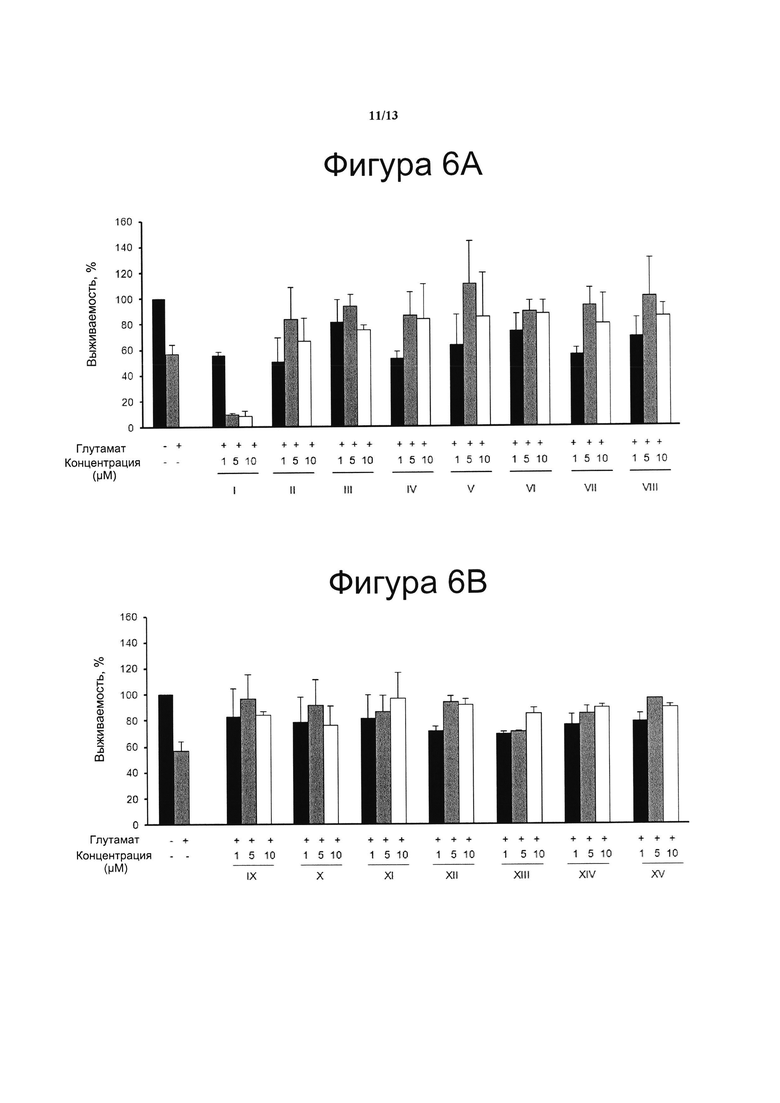
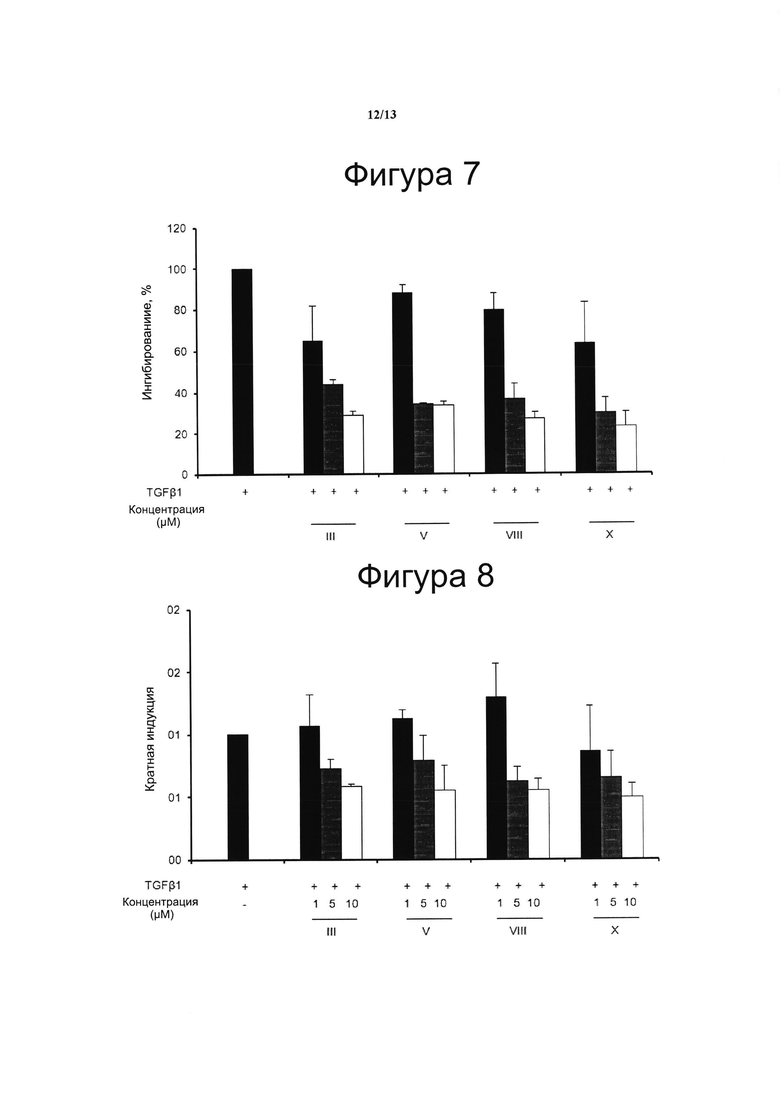
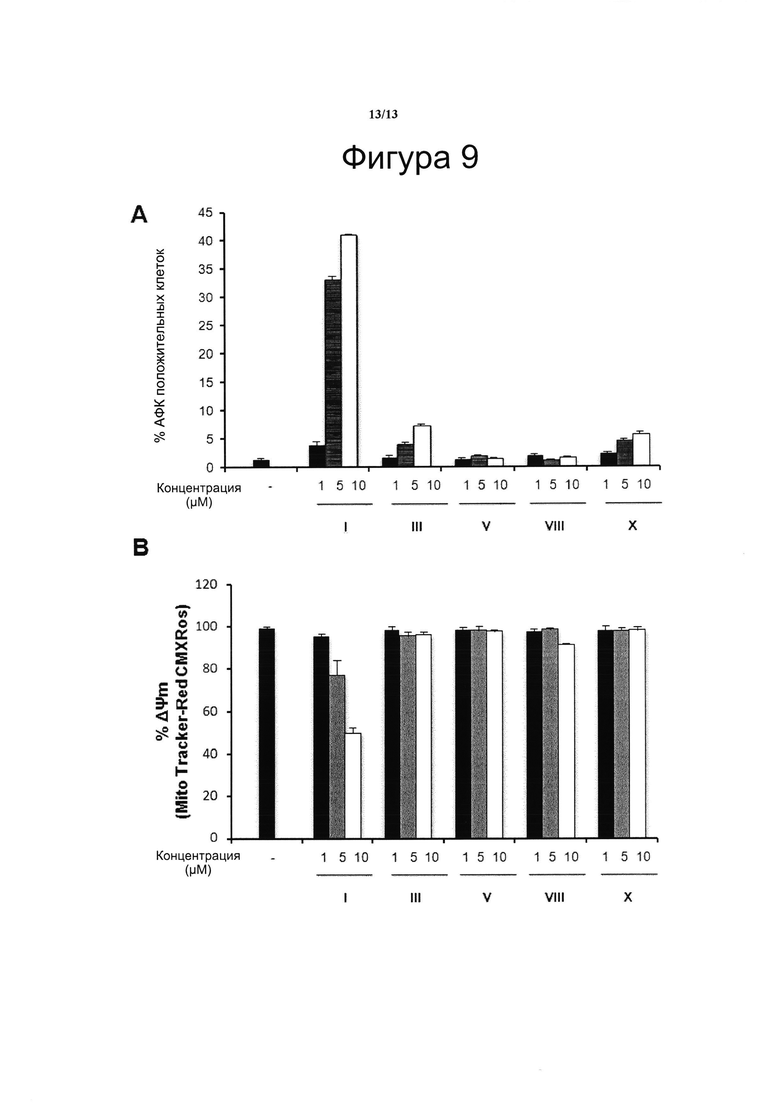
Настоящее изобретение относится к новым производным каннабидиол-хинона формулы (I) или к их фармацевтически приемлемым солям
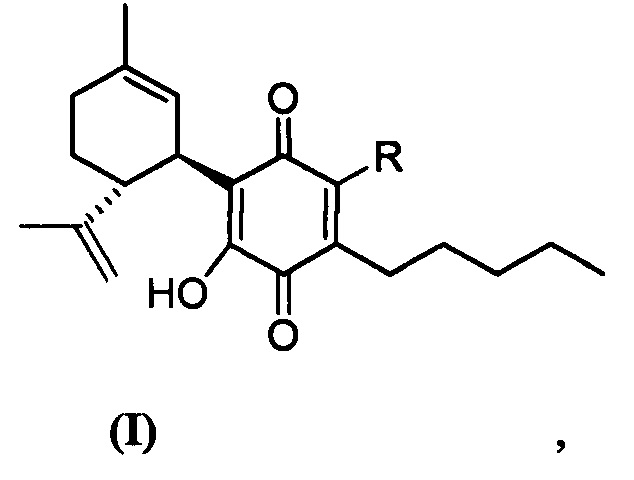
где R представляет собой атом углерода линейной или разветвленной группы, представленной алкоксикарбонильной группой; или где R представляет собой атом азота линейной или разветвленной группы, представленной алкиламинной или бензиламинной группами, применяемыми для лечения заболеваний, опосредованных PPRAg, а также к содержащей предлагаемые соединения композиции. 2 н. и 6 з.п. ф-лы, 15 ил., 10 пр.
1. Соединения формулы (I) или их фармацевтически приемлемые соли
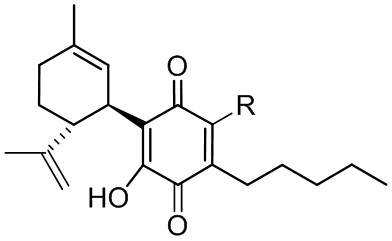
(I)
где R представляет собой атом углерода линейной или разветвленной группы, представленной алкоксикарбонильной группой; или где R представляет собой атом азота линейной или разветвленной группы, представленной алкиламинной или бензиламинной группами.
2. Соединение по п. 1, выбранное из:
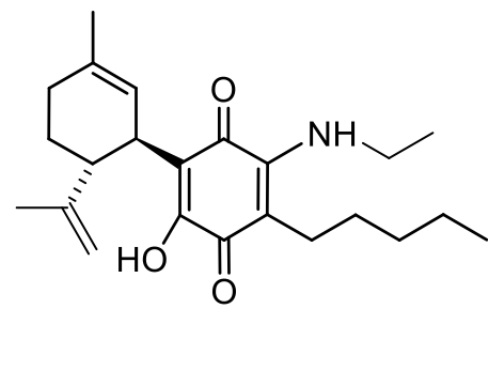
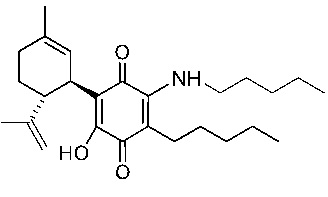 (II)
(II)
(III)
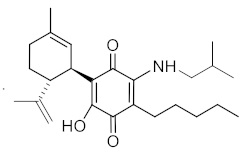
(IV)
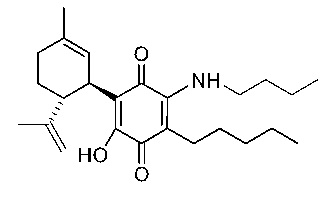
(V)
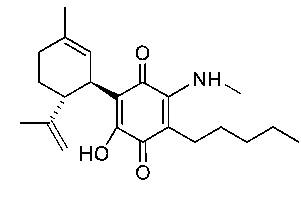
(VI)
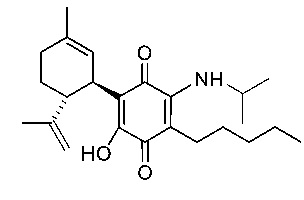
(VII)
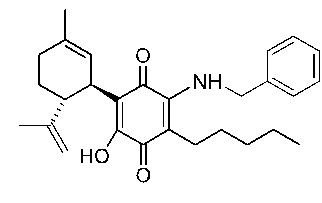
(VIII)

(IX)
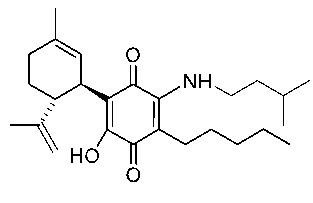
(X)

(XI)
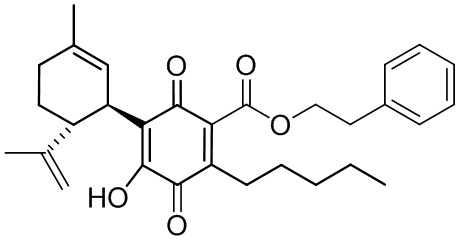
(XII)
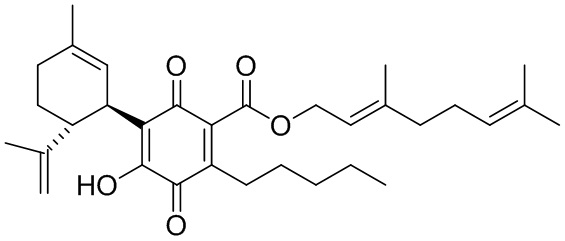
(XIII)
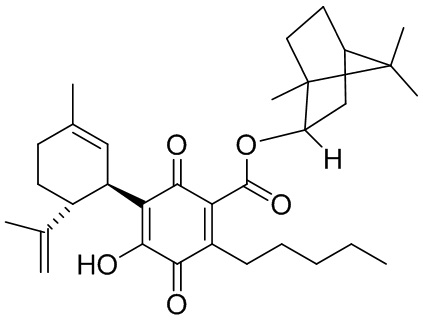
(XIV)
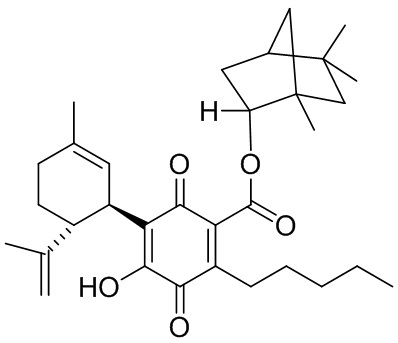
(XV)
1. Композиции для лечения заболевания, опосредованного PPRAg, содержащие эффективное количество соединения формулы (I)
(I)
где R представляет собой атом углерода линейной или разветвленной группы, представленной алкоксикарбонильной группой; или где R представляет собой атом азота линейной или разветвленной группы, представленной алкиламинной или бензиламинной группами и фармацевтически инертный ингредиент, такой как вспомогательное вещество и/или носитель.
4. Композиция по п. 3, отличающаяся тем, что соединение формулы (I) выбрано из:
(II)
(III)
(IV)
(V)
(VI)
(VII)
(VIII)
(IX)
(X)
(XI)
(XII)
(XIII)
(XIV)
(XV)
5. Соединение по любому из пп. 1 или 2 для применения в качестве лекарственного средства.
6. Соединение по любому из пп. 1 или 2 для применения для лечения заболеваний, опосредованных PPRAg.
7. Соединение по п. 6 для лечения заболевания, выбранного из: атеросклероза, воспалительных заболеваний кишечника, ревматоидного артрита, фиброза печени, нефропатии, псориаза, заживления кожных ран, регенерации кожи, панкреатита, гастрита, нейродегенеративных расстройств, нейровоспалительных расстройств, склеродермии, рака, гипертонии, ожирения или диабета II типа.
8. Композиция по пп. 3, 4 для применения для лечения заболевания, выбранного из: атеросклероза, воспалительных заболеваний кишечника, ревматоидного артрита, фиброза печени, нефропатии, псориаза, заживления кожных ран, регенерации кожи, панкреатита, гастрита, нейродегенеративных расстройств, нейровоспалительных расстройств, склеродермии, рака, гипертонии, ожирения или диабета II типа.
| ВИНТОВОЙ НАГНЕТАТЕЛЬ | 2013 |
|
RU2551255C2 |
| WO 2005067917 A1, 28.07.2005 | |||
| WO 2008107878 A1, 12.09.2008 | |||
| EA 200800044 A1, 30.06.2008. | |||

