Изобретение относится к способу получения алкансульфоновых кислот из диалкилдисульфидов с использованием азотной кислоты и кислорода.
Алкансульфоновые кислоты представляют собой органические производные серной кислоты, от которой они отличаются структурой, в которой гидроксильная группа замещена органическим радикалом. Таким образом, алкансульфоновые кислоты имеют общую структурную формулу R-SO3-H, в которой R означает органический радикал, например, алкил или арил. В зависимости от органического радикала, существует различие между алифатическими, ароматическими или гетероциклическими сульфоновыми кислотами. Свободные сульфоновые кислоты обычно представляют собой бесцветные гигроскопичные вещества, и их сила соответствует силе неорганических кислот. В самом деле, трифторметансульфоновая кислота, имеющая pKa, составляющую -5,5, является одной из самых сильных известных кислот и, таким образом, принадлежит к группе сверхкислот. В отличие от сульфатных солей ртути, свинца и серебра, соответствующие сульфонаты очень хорошо растворимы в воде.
Простейшим представителем алкансульфоновых кислот является метансульфоновая кислота, которую чаще всего обозначают МСК в соответствии с ее названием «метансульфоновая кислота». В то же время, поскольку она имеет множество способов применения, метансульфоновая кислота также является наиболее экономически значимой алкансульфоновой кислотой. Например, метансульфоновая кислота служит в качестве растворителя и катализатора ряда органических реакций, например, алкилирования, этерификации, полимеризации или синтеза гетероциклов. Другая область ее применения заключается в получении кислотно-аддитивных солей основных лекарственных средств с использованием метансульфоновой кислоты в медицине человека. Кроме того, метансульфоновую кислоту все чаще применяют в качестве заместителя чистящих средств, поскольку отсутствие цвета и запаха позволяет легко вводить ее в состав чистящих растворов. С промышленной точки зрения, самыми значимыми являются металлические соли метансульфоновой кислоты, которые применяют в качестве электролитов в ваннах для нанесения гальванических покрытий с использованием метансульфоновой кислоты, конкретно, при производстве электронных плат в электронной промышленности. Другая новая сфера применения метансульфоновой кислоты находится в области нефтедобычи: нефть, находящуюся в нефтеносных пластах, доступных путем бурения, часто удается выделить только в ограниченном количестве, если вообще удается. Таким образом, для улучшения выделения нефти нефтеносные каменные пласты размягчают с использованием метансульфоновой кислоты.
В промышленном масштабе алкансульфоновые кислоты получают путем окисления алкилмеркаптанов и/или диалкилдисульфидов или диалкилполисульфидов.
В патенте WO 98/34914 описан способ окисления алкилмеркаптанов и/или диалкилдисульфидов молекулярным бромом с получением соответствующих алкансульфоновых кислот. В этом способе бромистый водород сначала окисляют кислородом в присутствии каталитических количеств азотной кислоты, либо азотной кислотой в качестве окислителя, с получением молекулярного брома. Оксиды азота, образующиеся в ходе этой реакции, регенерируют кислородом и водой с получением азотной кислоты, которую возвращают на стадию окисления бромистого водорода до молекулярного брома. После этого, с помощью молекулярного брома, полученного таким образом, меркаптан и/или диалкилдисульфид окисляют до соответствующей алкансульфоновой кислоты. В результате применения молекулярного брома, алкансульфоновые кислоты, полученные способом по WO 98/34914, всегда содержат галоген и, следовательно, не подходят для применения в изготовлении электронных плат. Действительно, этом способе применения нельзя допускать присутствия галогена.
Также при получении алкансульфоновых кислот из алкилмеркаптанов или диалкилсульфидов известно применение пероксида водорода в качестве окислителя. Однако эта реакция протекает без осложнений только в присутствии карбоновых кислот. Поэтому считают, что в действительности окислителем является надкарбоновая кислота, образующаяся из карбоновой кислоты и пероксида водорода. Особенно заметный недостаток этого способа состоит в том, что он приводит к образованию смеси алкансульфоновых с карбоновыми и пероксикарбоновыми кислотами, из которой зачастую сложно выделить чистые алкансульфоновые кислоты. Таким образом, требуется затратное в экономическом и энергетическом отношении отделение алкансульфоновых кислот, что делает процесс экономически непривлекательным. Дополнительные затраты создает использование пероксида водорода, который является довольно дорогим окислителем. Другой недостаток заключается в образовании одного моля воды в расчете на один моль прореагировавшего пероксида водорода, что составляет значительное количество воды. Однако этот побочный продукт реакции нельзя превратить обратно в окислитель, и его необходимо отделить от целевой алкансульфоновой кислоты путем дорогостоящей и энергоемкой дистилляции.
В патенте US 4239696 описан альтернативный способ окисления алкилмеркаптанов и диалкилдисульфидов пероксидом водорода. В этом способе алкилмеркаптаны или диалкилдисульфиды окисляют пероксидом водорода в жидкой реакционной среде, содержащей от 1 до 35% алкансульфоновой кислоты в расчете на количество алкилмеркаптанов или диалкилдисульфидов, и не содержащей карбоновых или надкарбоновых кислот. Относительно большая продолжительность реакции, составляющая 3 часа, и применение дорогостоящего пероксида водорода в качестве окислителя являются значимыми недостатками, делающими способ весьма непривлекательным с экономической точки зрения. Что более важно, в патенте US 4239696 предлагается проводить реакцию в две стадии, в то время как способ осуществляют в непрерывном режиме, причем первую стадию осуществляют при температуре до 90°С, а вторую стадию при температурах от 100 до 110°С. Однако алкансульфоновые кислоты, например, метансульфоновая кислота, обладают разъедающим действием при таких температурах, что может привести к сильной коррозии, что делает способ непривлекательным с точки зрения безопасности.
В патентах US 2433395 и 4433396 описано получение алкансульфоновых кислот путем прямого окисления сераорганических соединений с общей формулой RSnR', в которой R и R' представляют собой углеводородные радикалы, а n представляет собой целое число от 1 до 6, конкретно, диалкилдисульфидов, кислородом в присутствии каталитических количеств азотной кислоты. Способ из патента US 2433395 представляет собой одностадийное окисление соединения серы при температуре реакции от примерно 250 до не более чем 300°F, что соответствует температурному диапазону от примерно 121 до примерно 148°С. Для сравнения, способ по патенту US 2433396 включает двухстадийное окисление: на первой стадии окисления соединение серы подвергают воздействию температуры от примерно 20 до примерно 70°С. Когда концентрация сульфоновой кислоты достигает диапазона от 40 до примерно 70%, наблюдается снижение скорости реакции в значимой степени, например, до примерно одной десятой от начальной скорости реакции. Как следствие, реакционную смесь, полученную на первой стадии окисления, подвергают воздействию реакционной температуры от примерно 70 до примерно 150°С на второй стадии окисления с целью увеличения содержания сульфоновой кислоты в реакционной смеси. Еще один недостаток заключается в получении по этим способам бледного красновато-коричневого продукта, что описано в патенте US 2697722. Такое обесцвечивание, по меньшей мере, частично связывают с присутствием веществ, обусловливающих цветность и запах, которые, как считают, образуются в результате неполного окисления соответствующего диалкилдисульфида. Следовательно, необходимо обесцветить неочищенный продукт реакции концентрированной азотной кислотой с целью удаления веществ, обусловливающих цвет и запах, что описано, например, в US 2697722. Еще один недостаток способа по патенту US 2433395 заключается в присутствии в неочищенном продукте других примесей, например, диоксида серы и едких сульфоксидов, удаление которых требует дополнительных стадий очистки. В патенте US 2433395 также описано регулирование температуры реакции путем введения легко испаряющихся жидкостей, например, петролейных эфиров, в реакционную смесь реакции окисления. Такие жидкости, испаряясь при высоком давлении паров, уносят тепло реакции окисления из реакционной смеси, что охлаждает последнюю. Однако в газовой фазе жидкости с высоким давлением паров могут образовывать взрывоопасные газовые смеси с кислородом. Это представляет значительную угрозу безопасности, что делает способ по патенту US 2433395 неподходящим для промышленного применения.
В равной степени неподходящим для промышленного применения является способ получения алкансульфоновых кислот по патенту US 2433396. В этом патенте описан двухстадийный способ получения алкансульфоновых кислот путем окисления диалкилдисульфида в растворе в алкансульфоновой кислоте с использованием кислорода и каталитического количества оксида азота. Однако реакция между смесями, описанными в этом патенте, также представляет проблемы в силу возможности образования взрывоопасных смесей. Более того, алкансульфоновые кислоты, полученные таким способом, также содержат значимые количества оксидов азота, которые нельзя удалить из неочищенной алкансульфоновой кислоты путем удаления легких фракций с помощью газового потока или нагревания. Для получения чистой алкансульфоновой кислоты, подходящей для продажи, необходимо очистить сырую алкансульфоновую кислоту, полученную этим способом, на отдельной стадии, например, как описано в патенте US 2502618, посредством контактирования содержащей алкансульфоновую кислоту фазы с олефином, не способным смешиваться с этой кислотой, более конкретно, моноолефиновым углеводородом, включающим, по меньшей мере, восемь атомов углерода. Еще один недостаток способа по US 2433396 состоит в применении высоких температур реакции на второй стадии окисления, что влечет образование дополнительных веществ, придающих продукту запах и цвет, а также образование продуктов разложения. Соответственно способы по патентам US 2433395 и 2433396 не подходят для получения алкансульфоновых кислот в промышленности.
В патенте US 2697722 описано окисление сульфидов или полисульфидов углеводородов кислородом до соответствующих алкансульфоновых кислот в присутствии, по меньшей мере, стехиометрических количеств азотной кислоты. В таком способе можно избежать уменьшения скорости реакции и использования повышенных температур реакции. Конкретно, в патенте US 2697722 описано окисление сульфидов или полисульфидов углеводородов в жидкой каталитической среде, состоящей из азотной кислоты в концентрации от примерно 10 до 70%. Однако, в итоге, в реакционную смесь также попадают значительные количества воды. Еще одним недостатком способа по US 2697722 является выделение азотной кислоты путем очистки отходящего газа процесса окисления, содержащего оксиды азота, который представляет собой водный раствор оксидов азота, то есть, в сущности, разбавленный раствор азотной кислоты. Такую разбавленную азотную кислоту можно вернуть в систему окисления сульфидов и добавить к азотной кислоте, используемой в процессе. Однако при этом содержание воды в системе окисления постепенно увеличивается, что еще больше увеличивает дороговизну и энергозатратность процесса выделения алкансульфоновой кислоты. В качестве альтернативы, разбавленную азотную кислоту концентрируют перед возвратом в указанную систему. Однако такая альтернатива приводит к дополнительным затратам средств и энергии. Еще один недостаток способа по патенту US 2697722 заключается в образовании больших количеств оксидов азота, которые опасны для здоровья и окружающей среды. Например, оксид азота N2O считается парниковым газом. Соответственно необходимо принятие дорогостоящих и энергозатратных мер во избежание выброса таких газов в окружающую среду и на производственные площадки. Это свидетельствует о том, что патент неперспективен для промышленного применения ни с точки зрения экономики, ни с точки зрения безопасности.
В патенте US 2498318 описан способ окисления диалкилдисульфидов кислородом до алкансульфоновых кислот в присутствии оксидов азота при температуре не более 125°F, что составляет 52°С, с целью предотвращения или, по меньшей мере, уменьшения коксования и коррозии в реакционной зоне. Однако, такие условия реакции не допускают полного превращения диалкилдисульфидов в желаемые алкансульфоновые кислоты. Более того, и в этом способе также образуется бледный красновато-коричневый продукт, который необходимо обесцветить концентрированной азотной кислотой на дополнительной стадии с целью удаления окрашивающих и придающих запах веществ.
В патенте US 2505910 описан еще один способ получения алкансульфоновых кислот путем окисления алкилмеркаптанов кислородом в присутствии каталитических количеств азотной кислоты и небольших количеств воды. В этом способе раствор, включающий алкилмеркаптан и оксид азота в качестве катализатора, насыщают воздухом. Перед тем как воздух поглощается раствором, образуется комплексное соединение меркаптана и оксида азота. Однако, как описано в патенте US 2727920, окисление меркаптана в таком комплексе происходит почти со взрывной силой. Примеры, приведенные в патенте US 2505910, также описывают бурное выделение NO2 при осуществлении этого способа, и это явление также вызывает сильное пенообразование в реакторе. Соответственно, способ по патенту US 2505910 невозможно осуществлять простым и безопасным образом и он, следовательно, не подходит для крупномасштабного получения алкансульфоновых кислот. Кроме того, полученные этим способом алкансульфоновые кислоты содержат окрашенные примеси, которые необходимо удалять путем обработки концентрированной азотной кислотой. Это также делает способ по US 2505910 экономически непривлекательным.
В патенте US 2727920 описан способ одностадийного окисления алкилмеркаптанов водным раствором азотной кислоты и кислородом до соответствующих алкансульфоновых кислот. Но в этом способе водный раствор азотной кислоты вводят в многомолярном избытке, иными словами, в количестве, превышающем стехиометрическое, по отношению к превращаемому меркаптану, что влечет необходимость отделения больших количеств воды и оксидов азота от получаемой алкансульфоновой кислоты. Однако увеличение отношения меркаптана к азотной кислоте не представляется возможным, поскольку, в соответствии с US 2727920, даже небольшие количества алкилмеркаптана вступают в реакцию так бурно, что использование большего соотношения алкилмеркаптана к азотной кислоте даже не рассматривается по причине взрывоопасности. Таким образом, данный способ обеспечивает только низкие объемные производительности. Следовательно, способ по патенту US 2727920 не подходит для промышленного получения алкансульфоновых кислот.
В патенте WO 00/31027 описан способ получения алкансульфоновых кислот окислением алкилмеркаптанов, диалкилдисульфидов и/или диалкилполисульфидов азотной кислотой при температурах от 50 до 150°С. В результате присутствия в реакционной смеси азотной кислоты в большой концентрации в реакцию попадают большие количества воды, которые необходимо в дальнейшем отделить от целевого продукта, что затратно с энергетической и экономической точек зрения. Другой недостаток этого способа заключается в образовании больших количеств оксидов азота, которые опасны для здоровья и представляют угрозу для окружающей среды, a N2O, помимо того, считается парниковым газом. Во избежание выброса оксидов азота необходимо принимать меры, которые также требуют затрат энергии и средств, следовательно способ по WO 00/31027 экономически непривлекателен.
В опубликованной китайской патентной заявке CN-A 101648892 описано получение алкансульфоновых кислот путем окисления диалкилдисульфида с использованием воздуха и азотной кислоты. В этом способе азотная кислота всегда присутствует в избытке по отношению к окисляемому диалкилдисульфиду. Разложение значительных количеств азотной кислоты приводит к обесцвечиванию продукта. Таким образом, для устранения окраски в смесь продуктов необходимо добавить катализатор денитрификации (DeNOx). Более того, применение в способе больших количеств азотной кислоты также невыгодно по причине попадания в процесс больших количеств воды, которые также необходимо удалять путем весьма энергозатратной дистилляции.
Таким образом, целью настоящего изобретения является обеспечение способа получения алкансульфоновых кислот из серосодержащих соединений-предшественников, позволяющего осуществлять недорогое производство алкансульфоновых кислот при высоких выходах с обеспечением мер безопасности.
Цель настоящего изобретения достигается путем окисления диалкилдисульфида, подаваемого в виде раствора с концентрацией не более 20 мас. % в соответствующей алкансульфоновой кислоте, с получением желаемой алкансульфоновой кислоты.
Соответственно настоящее изобретение обеспечивает способ получения алкансульфоновых кислот, соответствующих формуле R-SO3-H, включающий стадию окисления симметричного диалкилдисульфида, соответствующего формуле R-S2-R, в растворе алкансульфоновой кислоты в присутствии каталитических количеств азотной кислоты; в этих формулах причем R обозначает С1-12алкильный радикал, а алкансульфоновая кислота, применяемая в качестве растворителя, идентична алкансульфоновой кислоте, получаемой в ходе окисления указанного диалкилдисульфида; отличительной особенностью данного способа является то, что концентрация диалкилдисульфида в растворе составляет не более 20 процентов по массе (мас. %), отношение диалкилдисульфида к азотной кислоте составляет от 2000:1 (моль/моль) до 1:1 (моль/моль), и концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет более 70 мас. %.
Под выражением «не более 20 массовых процентов» в контексте настоящего изобретения понимают все возможные значения от более чем 0 мас. % до 20 мас. % включительно. Таким образом, выражение «не более 20 массовых процентов» охватывает не только целочисленные значения 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 и 20 мас. %, но также и все величины более 0 мас. % до 20 мас. % включительно, которые можно описать действительными числами.
В контексте настоящего изобретения, раствор диалкилдисульфида в соответствующей алкансульфоновой кислоте также называют реакционной смесью.
Выражение «диалкилдисульфид» в контексте настоящего изобретения применяют в соответствии с понятиями, общеизвестными для лиц, квалифицированных в данной области техники, и под этим выражением понимают группу химических соединений из группы органических дисульфидов, соответствующих общей формуле R1-S2-R1, в которой каждый из R1 и R2 означает углеводородный радикал. При условии, что такие углеводородные радикалы химически не реакционноспособны при окислительных условиях, применяемых в способе по настоящему изобретению, и что указанный диалкилдисульфид растворим или, по меньшей мере, способен в достаточной степени суспендироваться в алкансульфоновой кислоте, способ по настоящему изобретению не подпадает под какие-либо ограничения в отношении длины углеродной цепи или структуры углеводородного радикала в составе диалкилдилсульфида. Радикалы R1 и R2 представляют собой линейные или разветвленные углеводородные радикалы, предпочтительно, линейные, каждый из них содержит С1-12алкильный радикал, предпочтительно, С1-6алкильный радикал, более конкретно, С1-4алкильный радикал, необязательно, замещенный радикалами, не являющимися реакционноспособными при условиях реакций окисления. R1 и R2, предпочтительно, выбирают из группы, включающей метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет-бутил. Поскольку диалкилдисульфид, применяемый в соответствии с настоящим изобретением, является симметричным, радикалы R1 и R2 одинаковые.
Отношение диалкилдисульфида к азотной кислоте, составляющее от 2000:1 до 1:1 (моль/моль), охватывает все отношения, которые можно выразить целыми и действительными числами, начиная от 2000:1 включительно до 1:1 (моль/моль) включительно. Это отношение целенаправленно включает, не ограничиваясь перечисленным, 2000:1 (моль/моль), 1000:1 (моль/моль), 500:1 (моль/моль), 200:1 (моль/моль), 100:1 (моль/моль), 80:1 (моль/моль), 60:1 (моль/моль), 40:1 (моль/моль), 30:1 (моль/моль), 20:1 (моль/моль), 10:1 (моль/моль), 2:1 (моль/моль) и 1:1 (моль/моль).
В контексте настоящего изобретения, указанные значения концентраций означают концентрации конкретных компонентов в реакционной смеси в момент начала реакции. Например, значение концентрации диалкилдисульфида относится к концентрации, наблюдаемой после введения диалкилдисульфида в реактор и после того, как его затем смешали с алкансульфоновой кислотой, причем количества других присутствующих соединений пренебрежимо малы. Аналогично, значение концентрации алкансульфоновой кислоты относится к концентрации, наблюдаемой после введения алкансульфоновой кислоты в реактор и после того, как ее затем смешали с диалкилдисульфидом, причем количества других присутствующих соединений пренебрежимо малы.
Если способ по настоящему изобретению осуществляют в периодическом режиме, начальная концентрация диалкилдисульфида в реакционной смеси составляет не более 20 мас. %, и непрерывно уменьшается в ходе всего протекания реакции. Начальная концентрация алкансульфоновой кислоты в способе по настоящему изобретению, таким образом, составляет 80 мас. % и в результате непрерывного образования алкансульфоновой кислоты непрерывно растет в ходе всего протекания реакции. Напротив, при непрерывном осуществлении способа по настоящему изобретению наблюдается постоянная концентрация диалкилдисульфида, не превышающая 20 мас. %.
Таким образом обеспечивают безопасное окисление диалкилдисульфида до соответствующей алкансульфоновой кислоты, не представляющее угрозы взрыва даже при осуществлении способа по настоящему изобретению в промышленном масштабе. В самом деле, тот факт, что окисление протекает в растворе с концентрацией алкансульфоновой кислоты более 70 мас. %, обеспечивает поглощение теплоты реакции, выделяющейся особенно сильно в ходе запуска реакции, высококипящим растворителем (температура кипения метансульфоновой кислоты составляет примерно 167°С при 13 гПа). Преимущество этого эффекта заключается в том, что окисление диалкилдисульфидов до алкансульфоновых кислот в способе по настоящему изобретению протекает в плавной, а не взрывной манере. Более того, если способ по настоящему изобретению осуществляют в реакторе периодического действия, по мере протекания реакции количество выделяющейся теплоты уменьшается, поскольку концентрация диалкилдисульфида непрерывно растет.
Применение алкансульфоновой кислоты в качестве высококипящего растворителя обеспечивает дополнительное преимущество в отношении безопасности, поскольку возможность образования газовых смесей органических соединений и кислорода устранена или, по меньшей мере, снижена до такой степени, при которой опасность взрыва отсутствует.
С другой стороны, если концентрация диалкилдисульфида превышает 20 мас. %, что наблюдается в известных способах, теплоту, выделяющуюся в ходе реакции, становится невозможно контролировать в достаточной для промышленных процессов степени.
Применение в качестве растворителя в способе по настоящему изобретению алкансульфоновой кислоты, которая также является целевым продуктом этого процесса, одновременно устраняет необходимость в отделении растворителя от желаемого продукта, которая присутствует в известных способах получения алкансульфоновых кислот. Следовательно, в способе по настоящему изобретению дистилляционная очистка сырого продукта значительно дешевле и проще, что влечет соответствующее снижение капитальных и эксплуатационных затрат.
В соответствии с изложенным в литературе мнением, окисление диалкилдисульфидов до алкансульфоновых кислот протекает с образованием в качестве промежуточных продуктов S-алкилтиоалкансульфоксида R-S-SO-R, затем S-алкилтиоалкантиосульфоната R-S-SO2-R, S-алкилсульфоксидалкантиосульфоната R-SO-SO2-R, и диалкилдисульфона R-SO2-SO2-R, который, в конечном итоге, гидролизуется с образованием желаемой алкансульфоновой кислоты. Совместное использование алкансульфоновой кислоты в качестве растворителя и каталитических количеств азотной кислоты в способе по настоящему изобретению выгодно по сравнению с ранее описанными способами тем, что вода попадает в реакцию только в тех количествах, которые требуются для образования желаемой алкансульфоновой кислоты. Это позволяет получать по существу безводную алкансульфоновую кислоту. Таким образом, заключительная дистилляционная очистка «сырой» алкансульфоновой кислоты служит, главным образом, только для очистки целевой алкансульфоновой кислоты от примесей, присутствующих в следовых количествах, а также от оксидов азота, образовавшихся в ходе термического разложения азотной кислоты. Следовательно, такая дистилляция «сырой» алкансульфоновой кислоты требует не только меньшего количества оборудования, причем менее сложного, но и снижает энергозатраты, поэтому способ по настоящему изобретению позволяет значительно уменьшить капитальные и энергетические затраты, по сравнению со способами, известными ранее в данной области техники.
Способ по настоящему изобретению особенно хорошо подходит для получения метансульфоновой кислоты. С помощью способа по настоящему изобретению можно получать метансульфоновую кислоту чистоты, по меньшей мере, 95%, предпочтительно, по меньшей мере, 97%, более предпочтительно, по меньшей мере, 99%. Значительно сниженное содержание воды приводит к снижению количества нарушений в ходе процесса, конкретно при использовании метансульфоновой кислоты в качестве катализатора в химических реакциях.
Таким образом, в одном из предпочтительных вариантов настоящего изобретения диалкилдисульфид, вступающий в реакцию для получения соответствующей алкансульфоновой кислоты, представляет собой диметилдисульфид, а получаемая алкансульфоновая кислота представляет собой метансульфоновую кислоту.
В соответствии с настоящим изобретением, способ по настоящему изобретению можно осуществлять даже при сравнительно низком отношении диалкилдисульфида к азотной кислоте, составляющем 1000:1 и даже 2000:1 (моль/моль), но также и при любых желаемых отношениях от 1000:1 включительно до 2000:1 (моль/моль) включительно. При увеличении концентрации азотной кислоты до достижения отношения диалкилдисульфида к азотной кислоте, составляющего 500:1 (моль/моль), основная часть диалкилдисульфида превращается в алкансульфоновую кислоту всего за 90 минут.
В одном из предпочтительны вариантов настоящего изобретения отношение диалкилдисульфида к азотной кислоте, таким образом, составляет от 500:1 до 1:1 (моль/моль).
Дальнейшее увеличение концентрации азотной кислоты до достижения отношения диалкилдисульфида к азотной кислоте, составляющего 100:1 (моль/моль), обеспечивает практически полное превращение диалкилдисульфида в алкансульфоновую кислоту всего за 60 минут. В результате еще большего увеличения концентрации азотной кислоты, до отношения диалкилдисульфида к азотной кислоте, составляющего 10:1 (моль/моль), диалкилдисульфид практически полностью окисляется до соответствующей алкансульфоновой кислоты всего за полчаса.
Таким образом, предпочтительно отношение диалкилдисульфида к азотной кислоте составляет от 100:1 до 1:1 (моль/моль).
Отношение диалкилдисульфида к азотной кислоте предпочтительно составляет от 80:1 до 1:1 (моль/моль), от 60:1 до 1:1 (моль/моль), от 40:1 до 1:1 (моль/моль), от 20:1 до 1:1 (моль/моль), или от 10:1 до 1:1 (моль/моль).
В качестве альтернативы, выгодно, чтобы отношение диалкилдисульфида к азотной кислоте всегда превышало 1:1, поскольку количество азотной кислоты можно, таким образом, значительно снизить, что еще больше улучшит эффективность способа по настоящему изобретению. Поскольку в этом способе применяемую азотную кислоту всегда регенерируют при условиях реакции, применение меньших количеств азотной кислоты не оказывает отрицательного воздействия на выход и селективность образования алкансульфоновой кислоты. Более того, при отношении диалкилдисульфида к азотной кислоте, составляющем 2:1 (моль/моль) или более, не наблюдается значимого увеличения продолжительности реакции.
Таким образом, отношение диалкилдисульфида к азотной кислоте, предпочтительно, составляет от 500:1 до 2:1 (моль/моль), от 200:1 (моль/моль) до 2:1 (моль/моль), от 100:1 (моль/моль) до 2:1 (моль/моль), от 80:1 (моль/моль), от 60:1 (моль/моль) до 2:1 (моль/моль), от 40:1 (моль/моль) до 2:1 (моль/моль), от 20:1 (моль/моль) до 2:1 (моль/моль), или 10:1 (моль/моль).
В альтернативном предпочтительном варианте настоящего изобретения отношение диалкилдисульфида к азотной кислоте, таким образом, составляет от 500 до 2:1 (моль/моль).
Способ по настоящему изобретению позволяет получать алкансульфоновые кислоты из соответствующих диалкилдисульфидов с выходами более 90% при соблюдении условий безопасносной работы и при максимальной концентрации диалкилдисульфида в алкансульфоновой кислоте, не превышающей 20 мас. %. Однако, из соображений безопасности еще выгоднее осуществлять способ по настоящему изобретению при концентрации диалкилдисульфида в алкансульфоновой кислоте, не превышающей примерно 10 мас. %. Причина состоит в том, что увеличение температуры и давления при окислении диалкилдисульфида ниже, если концентрация диалкилдисульфида составляет более 10 мас. %, или даже до 20 мас. %. Это позволяет осуществлять еще более эффективное регулирование температуры при осуществлении способа по настоящему изобретению.
В дополнительном предпочтительном варианте настоящего изобретения концентрация диалкилдисульфида в алкансульфоновой кислоте составляет до примерно 10 мас. %.
Концентрация диалкилдисульфида в алкансульфоновой кислоте, предпочтительно, составляет от примерно 1 до примерно 6 мас. %, более предпочтительно от примерно 2 до примерно 6 мас. %, более конкретно, от примерно 4 до примерно 6 мас. %.
Под выражением «примерно» в отношении массовых процентов в контексте настоящего изобретения понимают не только явно приведенное значение, но также значения, отклоняющиеся от явно приведенного значения на ±10%. Таким образом, выражение «примерно 10 мас. %» охватывает не только целые значения 9, 10 и 11 мас. %, но также все значения, которые можно описать действительными числами, и которые находятся между 9 включительно и 11 включительно массовыми процентами. Выражение «примерно 2 мас. %» охватывает не только целочисленное значение 2, но и все значения, которые можно выразить действительными числами, от числа, на 10% меньшего чем 2 мас. %, включительно до числа, большего на 10% чем 2 мас. %, включительно. Выражение «от примерно 4 до примерно 6 мас. %» охватывает не только целые значения 4, 5 и 6 мас. %, но также все значения, которые можно описать действительными числами, от числа, на 10% меньшего чем 4 мас. %, включительно до числа, большего на 10% чем 6 мас. %, включительно.
Что касается температурного режима, было обнаружено, что при окислении диалкилдисульфидов до соответствующих алкансульфоновых кислот в случае, когда температура процесса превышает 90°С, происходит осаждение элементарной серы. Это связывают с недостатком кислорода в реакционной смеси при высоких температурах в результате протекания окисления диалкилдисульфидов: считают, что при дефиците кислорода в реакционной смеси атомы серы в составе диалкилдисульфида не полностью окисляются до серы с положительным формальным зарядом. Иными словами, считают, что диалкилдисульфид не полностью окисляется до S-алкилтиоалкансульфоксида R-S-SO-R, S-алкилтиоалкантиосульфоната R-S-SO2-R, S-алкилсульфоксидалкантиосульфоната R-SO-SO2-R или диалкилдисульфона R-SO2-SO2-R. Также считают, что окисление останавливается при образовании серы с формальным зарядом 0, что, как сообщается, сопровождается разрушением органического соединения, с чем связывают наблюдаемое осаждение элементарной серы. При промышленном получении алкансульфоновой кислоты необходимо не допускать осаждения серы, поскольку осаждение серы отрицательно воздействует на качество продукта, уменьшает выход желаемой алкансульфоновой кислоты, а также может привести к сбоям из-за забитых трубопроводов, насосов, колонн, и т.д. Таким образом, следует избегать постоянных температур реакции, которые составляют более 90°С.
Более того, реакционная температура, не превышающая 90°С, обеспечивает преимущество в способе по настоящему изобретению, заключающееся в отсутствии образования взрывоопасных газовых смесей с кислородом. Причиной служит тот факт, что температуры кипения алкансульфоновых кислот значительно превышают 90°С; например, самая простая из алкансульфоновых кислот, метансульфоновая кислота, кипит при 167°С при 13 гПа. Кроме того, температура кипения диметилдисульфида, являющегося простейшим представителем диалкилдисульфидов, составляет 110°С, то есть превышает максимальную температура процесса в способе по настоящему изобретению. Температура воспламенения диметилдисульфида значительно выше: она составляет 370°С на воздухе при давлении 1 атм.
В еще одном предпочтительном варианте настоящего изобретения, таким образом, способ осуществляют при температурах не более 90°С.
Способ по настоящему изобретению предпочтительно осуществляют при температуре от примерно 30 до 90°С, поскольку, вне зависимости от отношения диалкилдисульфида к азотной кислоте в реакционной смеси, такие температуры позволяют осуществлять практически полное окисление диалкилдисульфида до алкансульфоновой кислоты. Выражение «примерно 30°С» в контексте настоящего изобретения понимают таким образом, что оно также включает значения, отклоняющиеся от 30°С не более чем на -5°С на короткое время, иными словами, на такое время, которое пренебрежимо мало по сравнению с продолжительностью реакции.
Однако, при низких температурах, в силу относительно малого поступления энергии, реакция окисления не завершается довольно длительное время. Например, при температуре реакции, составляющей 30 или 40°С, окисление диметилдисульфида до метансульфоновой кислоты требует примерно 3 или 4 часа для достижения, по существу, полного превращения диметилдисульфида. Осуществление этой же реакции при температуре 70 или 90°С приводит, по существу, к полному превращению диалкилдисульфида меньше чем через 1 час.
В особенно предпочтительном варианте настоящего изобретения способ осуществляют при температуре от примерно 70 до 90°С.
Выражение «примерно 70°С» в контексте настоящего изобретения понимают таким образом, что оно также включает значения, отклоняющиеся от 70°С не более чем на -5°С на время, которое пренебрежимо мало, по сравнению с продолжительностью реакции.
В способе по настоящему изобретению концентрация окисляемого диалкилдисульфида составляет не более 20 мас. %. Массовой долей азотной кислоты в реакционной смеси можно практически пренебречь, поскольку в способе по настоящему изобретению ее применяют в количествах ниже стехиометрического по отношению к количеству диалкилдисульфида.
В контексте настоящего изобретения выражение «стехиометрическое» касательно отношения азотной кислоты к диалкилдисульфиду применяют для обозначения отношения азотной кислоты к диалкилдисульфиду, составляющего 1:1. Соответственно, в контексте настоящего изобретения под выражением «ниже стехиометрического» касательно отношения азотной кислоты к диалкилдисульфиду понимают все отношения азотной кислоты к диалкилдисульфиду, которые меньше 1:1, например, отношения диалкилдисульфида к азотной кислоте, составляющие 80:1, 60:1, 40:1, 20:1 или 10:1 (моль/моль).
Таким образом, в способе по настоящему изобретению реакционная смесь может включать 80 мас. % алкансульфоновой кислоты или более. Кроме того, однако, в реакционной смеси могут присутствовать другие компоненты, выступающие в качестве растворителей и инертные при окислительных условиях. Вспомогательными инертными компонентами считают такие, которые можно отделить от целевой алкансульфоновой кислоты путем дистилляции, и которые имеют низкое давление паров, что означает, что они не образуют взрывоопасных смесей в газовой фазе. Примеры инертных компонентов, приведенные без ограничения, включают сульфоксид и диметилформамид. Доля алкансульфоновой кислоты в реакционной смеси, предпочтительно, как можно более высокая, поскольку алкансульфоновая кислота, применяемая в реакционной смеси в качестве растворителя, не отличается от продукта окисления и, следовательно, при хорошем выходе реакции не требует дистилляционного отделения от смеси продуктов.
В еще одном предпочтительном варианте настоящего изобретения концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, таким образом, составляет, по меньшей мере, 80 мас. %.
В контексте настоящего изобретения, выражение «по меньшей мере 80 мас. %» охватывает все значения от 80 мас. % включительно до менее чем 100 мас. %. Выражение «80 мас. %», таким образом, охватывает целые значения 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 и 99 мас. %, а также все мыслимые значения от 80 мас. % включительно до менее чем 100 мас. %, которые можно выразить действительными числами.
Концентрация алкансульфоновой кислоты в способе по настоящему изобретению предпочтительно составляет, по меньшей мере, примерно 90 мас. %, более конкретно, по меньшей мере, примерно 92 мас. %, и, более предпочтительно, концентрация применяемой алкансульфоновой кислоты составляет от примерно 92 до примерно 96 мас. %. Выражения «примерно 90», «примерно 92» и «примерно 96» мас. % в контексте настоящего изобретения также включают отклонения ±2 мас. % от явно выраженных значений. Таким образом, выражение «по меньшей мере, примерно 90 мас. %» охватывает все значения от 88 мас. % включительно до менее чем 100 мас. %. То есть, например, целые значения 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98 и 99 мас. %, а также все мыслимые значения от 88 мас. % включительно до менее чем 100 мас. %, которые можно выразить действительными числами. Выражение «по меньшей мере, примерно 92 мас. %» охватывает все значения от 90 мас. % включительно до менее чем 100 мас. %. То есть, например, целые значения 90, 91, 92, 93, 94, 95, 96, 97, 98 и 99 мас. %, а также все мыслимые значения от 90 мас. % включительно до менее чем 100 мас. %, которые можно выразить действительными числами. Выражение «от примерно 92 до примерно 96 мас. %» охватывает все значения от 90 мас. % включительно до 98 мас. % включительно, то есть, например, целые значения 90, 91, 92, 93, 94, 95, 96, 97 и 98 мас. %, а также все мыслимые значения от 90 мас. % включительно до 98 мас. % включительно, которые можно выразить действительными числами.
Таким образом, в особенно предпочтительном варианте настоящего изобретения концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет, по меньшей мере, примерно 90 мас. %.
Способ по настоящему изобретению, по существу, не ограничен выбором окислителя, при условии, что окисление осуществляется безопасным образом. В соответствии с настоящим изобретением, кислород, как в свободной, так и в связанной форме, является подходящим окислителем в способе по настоящему изобретению. В контексте настоящего изобретения выражение «кислород в свободной форме» применяют в соответствии с общепринятыми знаниями лиц, квалифицированных в данной области техники, и оно означает кислород, не являющийся частью органического или неорганического соединения, связанной с ними ковалентными связями. Кислород в свободной форме, например, представляет собой молекулярный кислород О2, озон О3 или кислородный радикал. В соответствии с этим, например, молекула кислорода или любой кислородный радикал, являющийся частью комплекса или находящийся в координированной форме, также считается кислородом в свободной форме. Кислород в свободной форме, который применяют в способе по настоящему изобретению, таким образом, может включать чистый кислород или обогащенный кислородом газовый поток, например, обогащенный кислородом воздух или смесь чистого кислорода и газа, не являющегося реакционноспособным при окислительных условиях, который также называют инертным газом, например, азота или аргона. Напротив, под выражением «кислород в связанной форме» понимают любой кислород, который посредством по меньшей мере одной ковалентной связи связан с другими компонентами органического или неорганического соединения. Такие соединения, содержащие кислород в связанной форме, по существу, применяют для переноса атомов кислорода к атомам серы, находящимся в составе диалкилдисульфида, с целью их окисления от формальной степени окисления -1 в диалкилдисульфиде до +3 в диалкилдисульфоне. В качестве альтернативы, в контексте настоящего изобретения для окисления диалкилдисульфида также можно одновременно применять кислород в свободной форме и кислород в связанной форме.
Таким образом, в одном из предпочтительных вариантов настоящего изобретения для окисления в процесс подают воздух, газовый поток, обогащенный кислородом в свободной форме и/или чистый кислород в свободной форме.
Предпочтение отдают применению для окисления кислорода в свободной форме, либо потока газа, обогащенного кислородом в свободной форме. Причина состоит в том, что в результате присутствия в реакционной смеси молекулярного кислорода и воды оксиды азота, образовавшиеся как в результате окисления, так и в результате термического разложения, регенерируются с образованием азотной кислоты. В ходе внедрения способа по настоящему изобретению с азотной кислотой в качестве окислителя такая регенерация происходит сама собой и является сопутствующей реакцией. Соответственно, в том же реакторе, в котором образуются алкансульфоновая кислота и оксиды азота, также происходит и регенерация оксидов азота с участием кислорода и воды с образованием азотной кислоты. Преимущество заключается в том, что предпочтительно отсутствует необходимость в постоянном добавлении азотной кислоты в способ по настоящему изобретению.
Однако, если оксиды азота не полностью регенерируют до азотной кислоты, и поэтому наблюдается потеря азотной кислоты, такую потерю можно восполнить путем добавления свежей азотной кислоты. Такое добавление не регенерированной, то есть потерянной, азотной кислоты можно осуществлять или периодически, или непрерывно, в зависимости от ее конкретных требуемых количеств.
Окисление диалкилдисульфида с помощью газового потока, обогащенного кислородом в свободной форме, обеспечивает преимущество в виде повышенной экономической эффективности при применении газового потока, по сравнению с подачей в реакцию чистого кислорода. Более того, в соответствии с протеканием реакции окисления, содержание кислорода в газовом потоке можно регулировать желаемым образом. В простейшем случае такой газовый поток представляет собой воздух, содержащий более чем 20,942 об. % (объемных процентов) кислорода, что характерно для воздуха.
При подаче в реакционную смесь газового потока, содержащего более 21 об. % кислорода в свободной форме, обеспечивается значительно более полное окисление диалкилдисульфида до соответствующей алкансульфоновой кислоты и регенерация оксидов азота NOx до азотной кислоты.
В особенно предпочтительном варианте настоящего изобретения, таким образом, в процесс с целью окисления подают газовый поток, содержащий кислород в свободной форме в концентрации более чем 21 об. %.
В контексте настоящего изобретения под выражением «более 21 об. % кислорода в свободной форме» понимают все значения, превышающие 21 об. % до 100 об. % включительно, которые можно выразить целыми и действительными числами. В предельном случае, газовый поток с содержанием кислорода в свободной форме, составляющим более 21 об. %, представляет собой чистый кислород в свободной форме, предпочтительно, молекулярный кислород O2.
Способ по настоящему изобретению, по существу, не подвержен каким-либо ограничениям в отношении давления, при котором его осуществляют. Давление в способе по настоящему изобретению задают, как правило, с помощью газового потока, содержащего кислород в свободной форме, который подают в реакционную смесь. В соответствии с настоящим изобретением, это может быть воздух, газовый поток, обогащенный кислородом в свободной форме, более конкретно, газовый поток, содержащий более 21 об. % кислорода в свободной форме, или чистый кислород в свободной форме.
Способ по настоящему изобретению, по существу, не подвержен каким-либо ограничениям в отношении давления. Верхний предел давления определяется стойкостью применяемого реактора к давлению. Поскольку высокое или очень высокое давление требует использования сложных и дорогостоящих реакторов, способ по настоящему изобретению предпочтительно осуществляют при давлении не более 100 бар(абс.). Более того, на практике оказалось, что давление более 20 бар(абс.) не приводит ни к увеличению выхода, ни к ускорению завершения реакции. В контексте настоящего изобретения, выражение «бар(абс.)» синонимично выражению «бар абсолютное» и его применяют в качестве единицы измерения абсолютного давления. В соответствии с общеизвестными для лиц, квалифицированных в данной области техники, сведениями, абсолютное давление измеряют без учета наблюдаемого атмосферного давления, иными словами, только по отношению к нулевому давлению в пустом пространстве. Однако, предпочтительно осуществлять способ по настоящему изобретению при избыточном давлении. В контексте настоящего изобретения выражение «избыточное давление» охватывает все давления, превышающие 1 бар, который можно выразить целыми или действительными числами.
В одном из предпочтительных вариантов настоящего изобретения, таким образом, способ осуществляют при давлении от более чем 1 бар (абс.) до 20 бар (абс.).
В контексте настоящего изобретения выражение «давление примерно 20 бар (абс.)» применяют таким образом, что оно также включает отклонения ±10% от явно приведенного значения. Таким образом, выражение «примерно 20 бар (абс.)» также охватывает все целые значения от 18 бар (абс.) включительно до 22 бар (абс.) включительно, конкретно, значения 18, 19, 20, 21 и 22 бар (абс.), а также все значения, которые можно выразить действительными числами от 18 бар (абс.) включительно до 22 бар (абс.) включительно.
Выгодным образом давления, воздействию которых подвергают реакционную смесь, выбирают таким образом, чтобы увеличить выход продукта. На основании того факта, что выход при образовании метансульфоновой кислоты составляет примерно более 96% при давлении более 2 бар (абс.), например, 3 бар (абс.), стадийное увеличение давления на 3 бар (абс.) в каждом случае приводит к увеличению выхода до более чем 99%, при условии сохранения постоянной температуры.
Таким образом, в особенно предпочтительном варианте настоящего изобретения способ осуществляют при давлении от более чем 2 до примерно 15 бар (абс.).
В контексте настоящего изобретения выражение «давление примерно «15 бар (абс.)» применяют таким образом, что оно также включает отклонения ±2 бар (абс.) от явно приведенного значения. Таким образом, выражение «примерно 15 бар (абс.)» также охватывает все целые значения от 13 бар (абс.) включительно до 17 бар (абс.) включительно, конкретно, значения 13, 14, 15, 16 и 17 бар (абс.), а также все значения, которые можно выразить действительными числами от 13 бар (абс.) включительно до 17 бар (абс.) включительно.
Перед подачей в реактор для реакции окисления реакционную смесь предпочтительно сначала подают в смеситель для увеличения ее гомогенности. Такой дополнительный смеситель предотвращает неоптимальную гомогенность или улучшает ее, или предотвращает разделение реакционной смеси на фазы. Улучшение гомогенности части реакционной смеси, таким образом, способствует улучшению кинетики реакции, что ведет к увеличению выхода желаемой алкансульфоновой кислоты. Реакционную смесь можно гомогенизировать в статическом или динамическом смесителе. Статический смеситель в контексте настоящего изобретения представляет собой смеситель, в котором оптимальное перемешивание текучих сред осуществляют не с помощью движущихся компонентов, например, мешалки или шнека, а только с помощью специального принудительного движения потока смешиваемых текучих сред. В контексте настоящего изобретения динамический смеситель, напротив, представляет собой смеситель, в котором оптимальное смешивание текучих сред протекает посредством воздействия движущихся компонентов. Таким образом, в контексте настоящего изобретения динамический смеситель включает непрерывно работающий бак с мешалкой, в котором не протекает реакции окисления, поскольку, например, энергию, необходимую для начала или поддержания реакции, или каталитически активную азотную кислоту не подают в смеситель.
Гомогенизацию предпочтительно осуществляют в статическом смесителе, поскольку для этого не требуется применение ненадежных и, возможно, требующих частого обслуживания движущихся компонентов, присутствующих в динамическом смесителе. Таким образом, перед, по меньшей мере, одним из реакторов для реакции окисления используют статический смеситель.
Другие факторы, приводящие к увеличению выхода алкансульфоновой кислоты, включают тонкое диспергирование и продолжительное время нахождения кислорода в реакционной смеси. Этого достигают, например, путем подачи кислорода через так называемую распылительную форсунку или перфорированную пластину, путем подходящего перемешивания в реакторе, или путем осуществления реакции в длинном и тонком реакторе с мешалкой, работающем в непрерывном режиме.
Более того, в способе по настоящему изобретению можно применять один или более солюбилизаторов (ожижающий агент) с целью обеспечения улучшенной гомогенности реакционной смеси. В контексте настоящего изобретения выражение «солюбилизатор» применяют в соответствии с общеизвестной информацией для лиц, квалифицированных в данной области техники, и под ним понимают соединение, способствующее растворению плохо растворимого компонента в растворителе. По сути, все соединения подходят в качестве солюбилизатора в способе по настоящему изобретению, при условии, что они позволяют растворять диалкилдисульфид в соответствующей алкансульфоновой кислоте, и сами по себе при условиях окисления диалкилдисульфида до алкансульфоновой кислоты не реагируют ни с диалкилдисульфидом, ни с каким-либо промежуточным продуктом или конечным целевым продуктом. Выбор, по меньшей мере, одного солюбилизатора определяется принципиальным требованием к возможности его легкого отделения от целевой алкансульфоновой кислоты. Если температура кипения желаемой алкансульфоновой кислоты значительно отличается от температуры кипения или температур кипения солюбилизатора или солюбилизаторов, алкансульфоновую кислоту можно отделить от, по меньшей мере, одного солюбилизатора путем дистилляции. Это может потребовать дополнительных капитальных, эксплуатационных и энергетических затрат. Если температуры кипения алкансульфоновой кислоты и, по меньшей мере, одного солюбилизатора недостаточно различаются, или если дистилляционное отделение, по меньшей мере, одного солюбилизатора отрицательно скажется на свойствах целевой алкансульфоновой кислоты, в качестве альтернативы предпочтительно не отделять, по меньшей мере, один солюбилизатор путем дистилляции. Последняя альтернатива не вызывает проблем, если присутствие, по меньшей мере, одного солюбилизатора в алкансульфоновой кислоте не сказывается отрицательно на свойствах алкансульфоновой кислоты и последующих способах ее применения.
Таким образом, в дополнительном предпочтительном варианте настоящего изобретения применяют солюбилизатор для растворения диалкилдисульфида в алкансульфоновой кислоте.
В способе по настоящему изобретению применение S-алкилового сложного эфира алкансульфоновой кислоты, соответствующего формуле R-SO2-S-R, обеспечивает эффективную солюбилизацию диалкилдисульфида в соответствующей алкансульфоновой кислоте. Преимущество этого конкретного солюбилизатора над другими заключается в том, что он образуется в качестве промежуточного продукта окисления диалкилдисульфидов в алкансульфоновые кислоты и, таким образом, при условиях реакции способа по настоящему изобретению способен превращаться в целевой продукт окисления. В сущности, S-алкиловый сложный эфир алкансульфоновой кислоты, образующийся в способе по настоящему изобретению, практически полностью окончательно превращается в алкансульфоновую кислоту не более чем за 2 часа. Таким образом, радикал R в составе S-алкилового сложного эфира алкансульфоновой кислоты идентичен радикалу R в составе алкансульфоновой кислоты, получаемой способом по настоящему изобретению, следовательно, такой же, как описано выше в контексте алкансульфоновой кислоты. Вследствие этого для получения метансульфоновой кислоты из диметилдисульфида особенно предпочтительным солюбилизатором является S-метиловый сложный эфир метансульфоновой кислоты (MMTS). Конкретно, оказалось, что при концентрациях диметилдисульфида в метансульфоновой кислоте, составляющих более чем примерно 7 мас. %, MMTS является особенно хорошим солюбилизатором.
Таким образом, в особенно предпочтительном варианте настоящего изобретения S-алкильный сложный эфир алкансульфоновой кислоты, соответствующий формуле R-SO2-S-R, применяют в качестве солюбилизатора растворения диалкилдисульфида в алкансульфоновой кислоте, причем алкильные радикалы R в составе S-алкилового сложного эфира алкансульфоновой кислоты идентичны алкильным радикалам R в составе превращаемого диалкилдисульфида и алкильным радикалам R в составе алкансульфоновой кислоты.
Особенно предпочтительно, чтобы реакционная смесь, включающая диалкилдисульфид, находилась в одной фазе как до окисления, так и в ходе окисления диалкилдисульфида. Более конкретно, этого достигают путем применения комбинации статического смесителя, установленного выше по потоку от, по меньшей мере, одного реактора для реакции окисления, и применения S-алкилового сложного эфира алкансульфоновой кислоты в качестве солюбилизатора растворения диалкилдисульфида в алкансульфоновой кислоте в реакции окисления.
По сути, способ по настоящему изобретению не подвержен каким-либо ограничениям в отношении типа реактора, применяемого для осуществления способа. Таким образом, способ можно осуществлять в периодическом режиме в реакторе периодического действия, или непрерывно в трубчатом проточном реакторе, либо в реакторе с непрерывным перемешиванием. Предпочтение отдают применению реактора, позволяющего осуществлять способ по настоящему изобретению в непрерывном режиме.
Что касается количества реакторов, способ по настоящему изобретению не подвержен каким-либо ограничениям. Способ по настоящему изобретению можно осуществлять, например, в одном реакторе, например, реакторе с перемешиванием, или в двух или более реакторах, например, в комбинации основного реактора и конечного реактора или пост-реактора. В качестве примера, непрерывно работающий реактор с перемешиванием, выступающий в роли основного реактора, в котором превращается наибольшее количество диалкилдисульфида, можно применять в комбинации с трубчатым проточным реактором в качестве конечного реактора или пост-реактора, служащего для завершения реакции окисления. Для достижения полного превращения диалкилдисульфида такая комбинация требует реакторов лишь сравнительно небольшого объема. Напротив, если способ по настоящему изобретению осуществляют в одиночном реакторе, предпочтительно, в непрерывно работающем реакторе с перемешиванием, полное превращение диалкилдисульфида требует значительно большего объема реактора.
Таким образом, предпочтительно осуществлять способ по настоящему изобретению с использованием комбинации основного реактора и пост-реактора, более конкретно, комбинации непрерывно работающего реактора с перемешиванием и трубчатого проточного реактора.
Если способ по настоящему изобретению осуществляют с использованием комбинации непрерывно работающего реактора с перемешиванием в качестве основного реактора и трубчатого проточного реактора в качестве постреактора, в трубчатый проточный реактор предпочтительно подают дополнительную азотную кислоту и/или кислород с целью обеспечения практически полного окисления остаточной доли диалкилдисульфида в алкансульфоновую кислоту.
Внутренний объем, по меньшей мере, одного реактора, в котором осуществляют реакцию, предпочтительно полностью заполнен реакционной смесью, включающей, по меньшей мере, диалкилдисульфид и алкансульфоновую кислоту. Если над жидкой фазой реакционной смеси образуется газовая фаза, объем этой газовой фазы очень мал, поэтому последствия потенциального взрыва приемлемы. Например, отдельные пузырьки газа могут подняться в область над реакционной смесью. Однако, поскольку объем таких газовых пузырьков пренебрежимо мал по сравнению с объемом реакционной смеси или общим объемом реактора, никакой взрыв в пределах газовых пузырьков не будет заметен.
Если способ по настоящему изобретению осуществляют в двух или более реакторах, внутренний объем, по крайней мере, первого из нескольких реакторов также должен быть полностью заполнен реакционной смесью. Это связано с тем, что концентрация диалкилдисульфида, который может образовать взрывоопасную смесь с кислородом в свободной форме, наиболее высока в первом реакторе. Вероятность образования взрывоопасных смесей, таким образом, тоже наиболее высока в первом реакторе из нескольких. По этой причине внутренний объем, по меньшей мере, первого из нескольких реакторов предпочтительно полностью занят реакционной смесью, включающей, по меньшей мере, диалкилдисульфид и алкансульфоновую кислоту.
После окисления диалкилдисульфида до соответствующей алкансульфоновой кислоты смесь продуктов, полученную в результате такого превращения, подвергают дистилляционной очистке. Дистилляционная очистка предпочтительно включает первую дистилляцию и вторую дистилляцию ниже по потоку, причем из алкансульфоновой кислоты в ходе первой дистилляции удаляют низкокипящие вещества, а во второй дистилляции удаляют высококипящие вещества. В простейшем случае дистилляционную очистку осуществляют в двух дистилляционных колоннах. В качестве альтернативы, такая дистилляционная очистка также может протекать в двух термически связанных дистилляционных колоннах или в так называемой колонне с разделителем. Способ по настоящему изобретению, таким образом, предпочтительно, охватывает очистку алкансульфоновой кислоты, полученной в способе по настоящему изобретению, в колонне с разделителем или, по меньшей мере, двух дистилляционных колоннах, предпочтительно, по меньшей мере, в двух термически связанных дистилляционных колоннах.
Способ по настоящему изобретению дополнительно описан следующими параграфами:
1. Способ получения алкансульфоновых кислот, соответствующих формуле R-SO3-H, включающий стадию окисления симметричного диалкилдисульфида, соответствующего формуле R-S2-R, в растворе в алкансульфоновой кислоте в присутствии каталитически активных количеств азотной кислоты, причем R представляет собой С1-12алкильный радикал, и алкансульфоновая кислота, применяемая в качестве растворителя, идентична алкансульфоновой кислоте, получаемой в результате окисления указанного диалкилдисульфида, причем отличительными особенностями указанного способа является следующее: концентрация диалкилдисульфида в растворе не превышает 20 мас. %, отношение диалкилдисульфида к азотной кислоте находится в диапазоне от 2000:1 до 1:1 (моль/моль), а концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет более 70 мас. %.
2. Способ по п. 1, в котором диалкилдисульфид представляет собой димитилдисульфид, а алкансульфоновая кислота представляет собой метансульфоновую кислоту.
3. Способ по п. 1 или 2, в котором отношение диалкилдисульфида к азотной кислоте находится в диапазоне от 500:1 до 1:1 (моль/моль).
4. Способ по п. 1 или 2, в котором отношение диалкилдисульфида к азотной кислоте находится в диапазоне от 500:1 до 2:1 (моль/моль).
5. Способ по любому из пп. с 1 по 4, в котором концентрация диалкилдисульфида в алкансульфоновой кислоте составляет до примерно 10 мас. %.
6. Способ по любому из пп. с 1 по 5, который осуществляют при температуре не более 90°С.
7. Способ по п. 6, который осуществляют при температуре от примерно 70 до примерно 90°С.
8. Способ по любому из пп. с 1 по 7, в котором концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет, по меньшей мере, 80 мас. %.
9. Способ по п. 8, в котором концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет, по меньшей мере, 90 мас. %.
10. Способ по любому из пп. с 1 по 9, в котором в процесс с целью окисления подают воздух, газовый поток, обогащенный кислородом в свободной форме, и/или чистый кислород.
11. Способ по п. 10, в котором в процесс для окисления подают газовый поток, включающий кислород, содержащий более 21 объемн. % кислорода в свободной форме.
12. Способ по любому из пп. с 1 по 11, который осуществляют при давлении от более чем 1 до примерно 20 бар (абс.).
13. Способ по п. 12, который осуществляют при давлении от более чем 2 до примерно 15 бар (абс.).
14. Способ по любому из пп. с 1 по 13, в котором применяют солюбилизатор для растворения диалкилдисульфида в алкансульфоновой кислоте.
15. Способ по п. 14, в котором в качестве солюбилизатора растворения диалкилдисульфида в алкансульфоновой кислоте применяют S-алкильный сложный эфир алкансульфоновой кислоты, соответствующий формуле R-SO2-S-R, причем алкильные радикалы в составе S-алкильного сложного эфира алкансульфоновой кислоты такие же, как алкильные радикалы в составе превращаемого диалкилдисульфида и алкильные радикалы в составе алкансульфоновой кислоты.
Описание чертежей
На фиг. 1 показана зависимость температуры образца в °С от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 23) в адиабатическом калориметре (Phi-TEC II).
На фиг. 2 показана зависимость давления в бар (абс.) от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 23) в адиабатическом калориметре (Phi-TEC II).
На фиг. 3 показана зависимость давления в бар (абс.) от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 23) в адиабатическом калориметре (Phi-TEC II).
На фиг. 4 показана скорость изменения температуры в К/минуту в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 23) в адиабатическом калориметре (Phi-TEC II).
На фиг. 5 показано выделение тепла в Вт/кг в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 23) в адиабатическом калориметре (Phi-TEC II).
На фиг. 6 показана зависимость температуры образца в °С от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 24) в адиабатическом калориметре (Phi-TEC II). Исследуемый материал представлял собой вещество, выгруженное из реакции по эксперименту 23, и имел следующие свойства:
На фиг. 7 показана зависимость давления в бар (абс.) от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 24) в адиабатическом калориметре (Phi-TEC II). Исследуемый материал представлял собой вещество, выгруженное из реакции по эксперименту 23, и имел следующие свойства:
На фиг. 8 показана зависимость давления в бар (абс.) от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 24) в адиабатическом калориметре (Phi-TEC II). Исследуемый материал представлял собой вещество, выгруженное из реакции по эксперименту 23, и имел следующие свойства:
На фиг. 9 показана зависимость температуры образца в°С от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 25) в адиабатическом калориметре (Phi-TEC II).
На фиг. 10 показана зависимость давления в бар (абс.) от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 25) в адиабатическом калориметре (Phi-TEC II).
На фиг. 11 показана зависимость давления в бар (абс.) от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 25) в адиабатическом калориметре (Phi-TEC II).
На фиг. 12 показана скорость изменения температуры в К/мин в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 25) в адиабатическом калориметре (Phi-TEC II).
На фиг. 13 показано выделение тепла в Вт/кг в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 25) в адиабатическом калориметре (Phi-TEC II).
На фиг. 14 показаны зависимости температуры образца в °С (сплошная линия) и давления в бар (абс.) (пунктирная линия) от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 26) в адиабатическом калориметре (Phi-TEC II).
На фиг. 15 показана скорость изменения температуры в К/мин в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 26) в адиабатическом калориметре (Phi-TEC II).
На фиг. 16 показана зависимость давления в бар (абс.) от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 26) в адиабатическом калориметре (Phi-TEC II).
На фиг. 17 показана зависимость температуры образца в °С от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 27) в адиабатическом калориметре (Phi-TEC II).
На фиг. 18 показана зависимость давления в бар (абс.) от времени в секундах в ходе испытания на накопление давления/тепла (эксперимент 27) в адиабатическом калориметре (Phi-TEC II).
На фиг. 19 показана зависимость давления в бар (абс.) от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 27) в адиабатическом калориметре (Phi-TEC II).
На фиг. 20 показана скорость изменения температуры в К/мин в зависимости от температуры образца в °С в ходе испытания на накопление давления/тепла (эксперимент 27) в адиабатическом калориметре (Phi-TEC II).
Примеры
А) Пригодность метансульфоновой кислоты в качестве растворителя при окислении диметилдисульфида
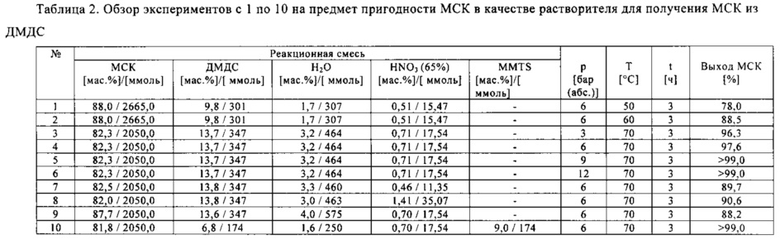
Метансульфоновую кислоту в десяти экспериментах исследовали на применимость в качестве растворителя при получении метансульфоновой кислоты путем окисления диметилдисульфида. Это делали следующим образом: готовили растворы различных количеств диметилдисульфида в смеси метансульфоновой кислоты, азотной кислоты (65 мас. %) и небольших количеств воды, и переносили их в автоклав. Превращение диметилдисульфида в метансульфоновую кислоту протекало при температурах от 50 до 90°С и давлениях от 3 до 12 бар (абс.) в атмосфере кислорода. Для этого кислород вводили в каждый образец с помощью погружной трубки, а для обеспечения оптимального распределения реакционной смеси применяли мешалку. Отдельные составы реакционных смесей в экспериментах с 1 по 10, а также конкретные условия протекающих реакций обобщены в табл. 2.
Результаты экспериментов, показанные в табл. 2, указывают на принципиальную пригодность метансульфоновой кислоты (МСК) в качестве растворителя для окисления диметилдисульфида (ДМДС) с образованием метансульфоновой кислоты. Тем не менее, в зависимости от выбранных условий реакции (давления, температуры и продолжительности), получаются сильно различнающиеся выходы метансульфоновой кислоты. Например, выход метансульфоновой кислоты колеблется от 78,0% (в эксперименте 1) до более чем 99,0% (в экспериментах 5, 6 и 10).
Б) Оптимизация параметров реакции для улучшения производительности
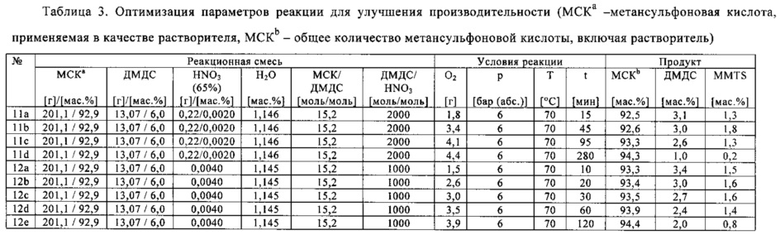
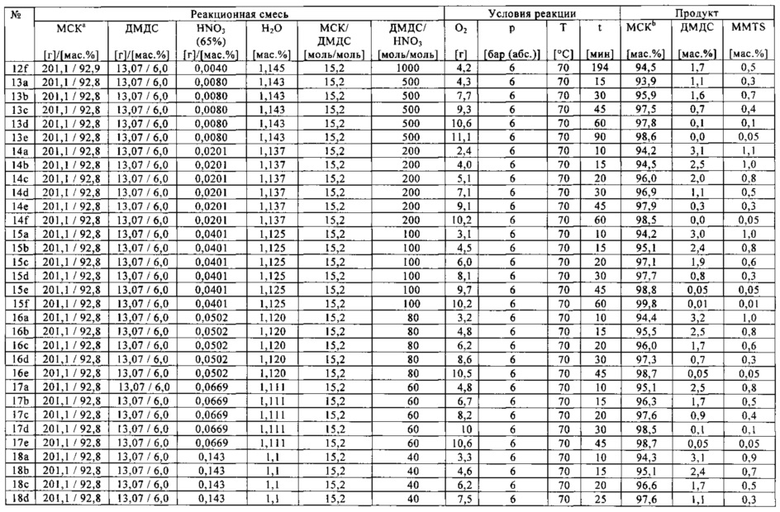
В следующих экспериментах с 11 по 22, осуществленных в автоклаве, параметры реакции оптимизировали с целью обеспечения максимальной производительности (высокого выхода метансульфоновой кислоты и малой продолжительности реакции или продолжительности нахождения веществ в реакторе).
Отдельные составы реакционных смесей с 11 по 22 и конкретные условия реакций в этих экспериментах приведены в табл. 3. В зависимости от конкретной температуры реакции, при отношениях диметилдисульфида к азотной кислоте в диапазоне от 100:1 до 1:1 (моль/моль), по существу, полное превращение диметилдисульфида достигалось всего в течение 1 часа. В самом деле, в эксперименте 21 полное превращение диалкилдисульфида произошло через полчаса.
В) Оптимизация параметров реакции с целью обеспечения безопасности в экспериментах с 23 по 27 протекание реакций с участием различных смесей диметилдисульфида, метансульфоновой кислоты, азотной кислоты и воды исследовали с точки зрения безопасности на производстве, с добавлением чистого кислорода, в основном, при адиабатических условиях. С этой целью проводили несколько экспериментов с различными условиями, в каждом случае применяли образец, закрытый в адиабатическом калориметре (Phitec II).
1. Образцы:
1.1. Метансульфоновая кислота
1.2. Диметилдисульфид
1.3. Азотная кислота, 65%
1.4. Деионизированная вода (т.е. полностью деминерализованная вода)
2. Исследование протекания реакций при адиабатических условиях
2.1. Измерения в адиабатическом калориметре (Phitec II)
2.1.1. Способ измерения
Применяли управляемый с помощью персонального компьютера калориметр Phitec II, способный имитировать условия в крупных реакторах в промышленных установках, даже если используются относительно малые количества образца, например, от 10 до 100 мл.
На калориметре Phitec II применяли метод накопления давления/тепла, в котором (в зависимости от необходимых условий реакции) обеспечивается относительно высокая точность измерений, с учетом тепла, выделяемого реакцией (обычно составляющего примерно от 2 до 5 Вт/кг при использовании закрытого контейнера с образцом). На основании профиля температуры и давления, измеренных с течением времени и отражающих экзотермический эффект реакции и образование газообразных продуктов разложения, можно исследовать термическую устойчивость образца.
Эксперименты проводили с использованием адиабатического калориметра, причем все измерительное оборудование было установлено в автоклаве, устойчивом к действию высокого давления.
В сосуд высокого давления помещали цилиндрический контейнер с образцом, изготовленный из нержавеющей стали, материал №1.457 (в качестве альтернативы, контейнер для образца был изготовлен из сплава Hastelloy), объемом 110 мл; система включала также систему нагрева, причем нагреватели системы нагрева полностью окружали контейнер с образцом, но не находились в механическом контакте с ним. Образец перемешивали с помощью магнитной стержневой мешалки (которую также называют перемешивающим стержнем или якорем), расположенной в основании сосуда с образцом.
Стенки контейнера с образцом были очень тонкими, их толщина обычно составляла всего 0,15 мм. Таким образом, давление, возникающее в закрытом контейнере с образцом в ходе измерения и обусловленное давлением паров и парциальным давлением газообразных продуктов разложения, также устанавливалось и в окружающем контейнер автоклаве посредством отслеживающей и регулирующей системы, во избежание деформации контейнера с образцом.
Температуру среды, содержащей контейнер с образцом, непрерывно адаптировали к температуре образца с целью предотвращения переноса тепла от образца в окружающую его среду. Соответственно, температуру среды регулировали таким образом, чтобы ни в какой момент времени разница температур образца и окружающей среды не составляла 0 К, с целью поддержания преимущественно адиабатических условий.
Желательное отношение теплоемкости контейнера с образцом к теплоемкости образца требует высокой чувствительности измерений, что численно определяют так называемым фактором Ф (Фи), который соответствует следующему уравнению:
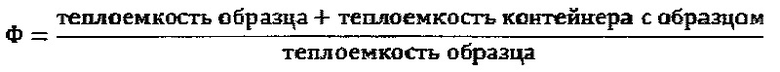
В идеальном случае значение безразмерного фактора Ф составляет немного более 1.
Максимальное адиабатическое повышение температуры, определенное в каждом эксперименте, корректировали с помощью фактора Ф с целью учета энергии, необходимой для нагрева контейнера.
2.1.2 Методика эксперимента и результаты измерений
2.1.2.1 Эксперимент 23
Применяли закрытый контейнер для образца, изготовленный из никель-хром-молибденового сплава Hastelloy С276 объемом 115 мл, оснащенный погружной трубкой для подачи молекулярного кислорода и якорем мешалки.
Состав образца в данном эксперименте кратко описан в таблице ниже:
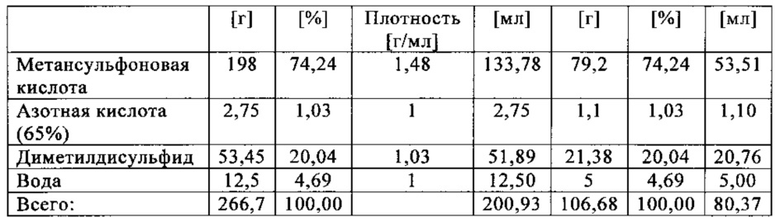
При присущем давлении паров, в предварительно вакуумированный контейнер с образцом сначала загружали метансульфоновую кислоту с добавлением небольших количеств азотной кислоты и воды, а затем добавляли диметилдисульфид. После этого образец нагревали до заданной температуры в 70°С. После выключения нагревателя было очевидным протекание слабо экзотермической реакции, в ходе которой до момента добавления кислорода достигалась температура 72°С. Затем в контейнер с образцом, давление в котором составляло примерно 7,5 бар (абс.), добавляли чистый кислород с целью максимально быстрого увеличения общего давления до 12 бар (абс.). Для этого редуктор давления емкости с кислородом был установлен на целевое давление, что подтверждали показанием манометра. Обратный клапан, установленный в линии подачи, предотвращал обратный ток газа. Сразу после введения кислорода начиналась реакция с сильным экзотермическим эффектом, что сопровождалось очень резким повышением давления.
Поскольку реакция обладает большим экзотермическим эффектом, давление в контейнере с образцом, невзирая на ограничение давления и отключение подачи кислорода по линии с помощью клапана, превысило заданное давление и достигло максимума в 20,4 бар (абс.). Максимальная температура образца составила 119°С. Поскольку система отслеживания температуры не была отслеживать резкий рост температуры, считают, что достижимый максимум температуры мог бы быть еще выше. (В оценку роста температуры и давления не включены эффекты, связанные со скоростью введения кислорода и часть тепла сжатия, возникшего в результате введения кислорода.)
После падения температуры и давления до примерно 84°С и примерно 11,2 бар (абс.) соответственно, кислород снова вводили до достижения давления 12 бар (абс.). Это создавало дополнительный экзотермический эффект, хотя и значительно более слабый, по сравнению с первым добавлением кислорода.
На фиг. 1 и 2 показаны профили изменения температуры образца и давления во времени, а на фиг. 3 показан профиль давления в зависимости от температуры. Погрешность результатов составляет ±1 К для температуры и ±0,4 бар для давления. На фиг. 4 показана скорость изменения температуры в зависимости от температуры образца, а на фиг. 5 показана взаимосвязь температуры и тепловыделения. Для расчета экзотермического эффекта теплоемкость образца оценивали при постоянном давлении (Ср). С этой целью делали допущение о том, что теплоемкость органической составляющей при постоянном давлении (Ср) составляет 2 Дж/(г*К), а Ср неорганической составляющей составляет 4,1 Дж/(г*К).
В силу концентрации диметилдисульфида, при осуществлении этого эксперимента наблюдали очень резкий рост температуры и давления. Однако, при условиях эксперимента не произошло повреждения и тем более разрушения контейнера с образцом. Следовательно, достигнутое в этом эксперименте повышение температуры и давления не было критическим. Таким образом, концентрация 20 мас. % представляет граничную область способа по настоящему изобретению, при которой окисление диалкилдисульфида до соответствующей алкансульфоновой кислоты все еще можно осуществлять безопасным и простым образом. Следовательно, для контролируемого осуществления окисления диалкилдисульфида до алкансульфоновой кислоты концентрация диалкилдисульфида в реакционной смеси не должна превышать 20 мас. %, предпочтительно, она должна составлять менее чем 20 мас. %.
2.1.2.2. Эксперимент 24
В этом эксперименте применяли реакционную среду, выгруженную из эксперимента 23, и исследовали воздействие добавления кислорода при различных температурах. Для этого контейнер для образца наполняли реакционной средой, полученной в опыте по эксперименту 23, и образец сначала нагревали при перемешивании до заданной температуры, составляющей 50°С. После выключения нагревателя давление составляло примерно 0,7 бар (абс.), а температура сохранялась на постоянном уровне. Затем вводили чистый кислород через погружную трубку в контейнер с образцом с целью достижения конечного заданного давления 12 бар (абс.). Для этого редуктор давления емкости с кислородом был установлен на это давление, что подтверждали показанием манометра. Обратный клапан, установленный в линии подачи, предотвращал обратный ток газа.
Даже при относительно быстром введении кислорода было очевидным резкое повышение температуры, сначала до примерно 60°С. В этот момент давление в контейнере с образцом, несмотря на ограничение давления и отключение клапана линии подачи кислорода, возрастало выше заданного и достигало значения примерно 13 бар (абс.). После небольшого падения температура образца возрастала до примерно 105°С без сообщения энергии извне, исключительно за счет тепла, выделяемого экзотермической реакцией. Вместе с этим наблюдали падение давления от 13 до 3 бар (абс.). После нагревания до 111°С и короткой переходной фазы кислород снова вводили до заданного давления 12 бар (абс.), при этом устанавливалось давление 12,1 бар (абс.). Как в ходе введения кислорода, так и после этого наблюдали лишь небольшое изменение температуры, составляющее примерно 1 К. После этого осуществляли еще две стадии нагревания до конечной температуры 121°С, но протекания экзотермической реакции не наблюдали. Эксперимент, таким образом, завершали. После охлаждения до комнатной температуры и сброса давления в контейнере с образцом остаток образца выгружали.
На фиг. 6 показан профиль изменения температуры образца во времени, а на фиг. 7 и 8 показаны профили давления в зависимости от времени и от температуры соответственно. Погрешность результатов составляет ±1 К для температуры и ±0,4 бар для давления.
Профили температуры и давления, показанные на фиг. 6 и 7, свидетельствуют о том, что в ходе осуществления последующей реакции с участием реакционной среды, выгруженной в эксперименте 23, по-прежнему наблюдается некоторое превращение. Однако, в этой последующей реакции рост как давления, так и температуры значительно ниже, чем в ходе предшествующего превращения. Эксперименты 23 и 24 свидетельствуют о возможности регулируемого окисления диалкилдисульфида до соответствующей алкансульфоновой кислоты в двух последовательных реакторах.
2.1.2.3. Эксперимент 25
В данном эксперименте кислород добавляли при 50°С (в отличие от соответствующей температуры 70°С в эксперименте 23), а заданное давление составляло 12 бар (абс.). С этой целью применяли закрытый контейнер для образца, изготовленный из сплава нержавеющей стали Hastelloy С276 объемом 115 мл, оснащенный погружной трубкой и якорем мешалки.
Состав образца в данном эксперименте был следующим:
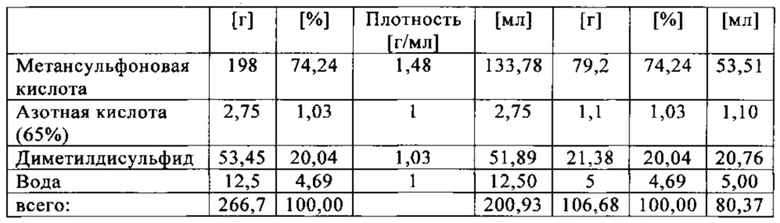
При присущем давлении паров в предварительно вакуумированный контейнер для образца сначала загружали метансульфоновую кислоту с добавлением требуемых количеств азотной кислоты и воды. Затем в контейнер для образца загружали необходимое количество диметилдисульфида. После этого образец нагревали до заданной температуры в 50°С при перемешивании.
После выключения нагревателя, было очевидно протекание слабо экзотермической реакции, в ходе которой до момента добавления кислорода достигалась температура 51,4°С. Затем в контейнер с образцом, давление в котором составляло примерно 6,7 бар (абс.), через погружную трубку добавляли чистый кислород с целью максимально быстрого увеличения общего давления до 12 бар (абс.). Для этого редуктор давления емкости с кислородом был установлен на соответствующее давление, что подтверждали показанием манометра. Обратный клапан, установленный в линии подачи, предотвращал обратный ток газа. Сразу после введения кислорода начиналась реакция с сильным экзотермическим эффектом, что сопровождалось очень резким повышением давления. Несмотря на ограничение давления и отключение подачи кислорода по линии с помощью клапана, давление в контейнере с образцом достигло примерно 18,5 бар (абс.). Максимальная температура образца составила примерно 98°С. Поскольку система отслеживания температуры не была отслеживать резкий рост температуры, считают, что достижимый максимум температуры мог бы быть еще выше. (В оценку роста температуры и давления не включены эффекты, связанные со скоростью введения кислорода и часть тепла сжатия, возникшего в результате введения кислорода.) После достижения максимальной температуры и отключения подачи кислорода, а также падения давления до примерно 8,9 бар (абс.), эксперимент заканчивали.
На фиг. 9 показан профиль изменения температуры образца во времени. На фиг. 10 и 11 показаны профили давления в зависимости от времени и от температуры соответственно. Погрешность измерений составляла ±1 К для температуры и ±0,4 бар для давления. На фиг. 12 показана скорость изменения температуры в зависимости от температуры образца, а на фиг. 13 показано выделение тепла в зависимости от температуры образца. Для расчета экзотермического эффекта теплоемкость образца оценивали при постоянном давлении (Ср). С этой целью делали допущение о том, что теплоемкость органической составляющей при постоянном давлении (Ср) составляет 2 Дж/(г*К), а Ср неорганической составляющей составляет 4,1 Дж/(г*К).
В эксперименте 25 окисление диметилдисульфида до соответствующей метансульфоновой кислоты начинали путем подачи кислорода при температуре 50°С, что на 20°С ниже соответствующей температуры в эксперименте 23. Соответственно, кривые изменения температуры и выделения тепла реакции во времени также сдвинуты на эту температурную разницу в эксперименте 25, по сравнению с соответствующими профилями в эксперименте 23. Не считая этого сдвига, профиль изменения температуры во времени и профиль выделения тепла во времени в эксперименте 25 параллельны соответствующим профилям в эксперименте 23 (ср. фиг. 4 и фиг. 5 с фиг. 12 и фиг. 13). Аналогичные комментарии также применимы к изменению температуры и давления в экспериментах 23 и 25 (ср. фиг. 1 и фиг. 2 с фиг. 9-11).
Результаты экспериментов 23 и 25 демонстрируют, что окисление диалкилдисульфида протекает в обоих экспериментах со сравнимым экзотермическим эффектом, вне зависимости от соответствующей начальной температуры.
2.1.2.4. Эксперимент 26
В отличие от экспериментов с 23 по 25, в которых применяли контейнер для образца, изготовленный из сплава Hastelloy С276, оснащенный погружной трубкой, данный эксперимент проводили в контейнере для образца, изготовленном из нержавеющей стали, материал 1.4571. Закрытый контейнер для образца в данном эксперименте имел объем 110 мл и был оснащен якорем мешалки, но не оснащен погружной трубкой.
Начальная заданная температура перед добавлением кислорода составляла 50°С, а заданное давление для добавления кислорода составляло 12 бар (абс.).
Состав образца в данном эксперименте приведен в таблице ниже:
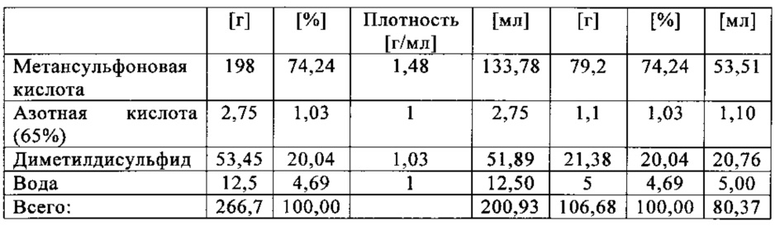
При присущем давлении паров в предварительно вакуумированный контейнер для образца сначала загружали метансульфоновую кислоту с добавлением требуемых количеств азотной кислоты и воды. Затем в контейнер для образца загружали необходимое количество диметилдисульфида. После этого образец нагревали до заданной температуры 50°С при перемешивании.
Профили зависимости температуры (сплошная линия) и давления (пунктирная линия) образца от времени показаны совместно на фиг. 14. На фиг. 15 показана скорость изменения температуры как функция от температуры образца, а на фиг. 16 показан профиль зависимости давления от температуры.
После выключения нагревателя, когда была достигнута заданная температура 50°С, температура возрастала до примерно 51,5°С, после чего начинали добавление кислорода. Начинали введение чистого кислорода при давлении в контейнере примерно 71,2 бар (абс.) с целью максимально быстрого достижения общего давления 12 бар (абс.) путем подачи чистого кислорода. Для этого редуктор давления емкости с кислородом был установлен на целевое давление, что подтверждали показанием манометра. Обратный клапан, установленный в линии подачи, предотвращал обратный ток газа. Через некоторое время после введения кислорода начиналась реакция с сильным экзотермическим эффектом, достигнутый температурный максимум составил примерно 350°С. Задержку связывают с тем, что, в отличие от экспериментов с 23 по 25, кислород вводили в контейнер с образцом не через погружную трубку, а сверху над жидкой фазой.
В ходе добавления кислорода был слышен стук в корпусе автоклава, в котором для осуществления экспериментов находился калориметр. После этого эксперимент заканчивали путем выключения системы нагревания. Регистрировали рост фактического давления, составляющий до примерно 5,5 бар (абс.), но последующий профиль зависимости давления от времени не регистрировался. Это связано с разрушением барометрического датчика, максимально допустимое давление которого составляло 100 бар.
Пары диметилдисульфида способны образовать взрывоопасную смесь с воздухом или кислородом; соответствующая температура воспламенения составляет 370°С. Что касается температуры воспламенения смесей взрывоопасных газов и паров с воздухом или окисляющим газом, известно, что температура падает очень резко при росте давления (см., например, Hirsch, W., Brandes, Е., "
 Gemische bei
Gemische bei 
 ", Physikalisch Technische Bundesanstalt, Braunschweig, 2005). При резком увеличении температуры и повышенном начальном давлении при добавлении кислорода создаются условия для достижения температуры воспламенения диметилдисульфида. Несмотря на относительно малый объем свободного газа в контейнере для образца, с большой вероятностью можно заключить, что произошло самовоспламенение газовой фазы, содержащей кислород и диметилдисульфид.
", Physikalisch Technische Bundesanstalt, Braunschweig, 2005). При резком увеличении температуры и повышенном начальном давлении при добавлении кислорода создаются условия для достижения температуры воспламенения диметилдисульфида. Несмотря на относительно малый объем свободного газа в контейнере для образца, с большой вероятностью можно заключить, что произошло самовоспламенение газовой фазы, содержащей кислород и диметилдисульфид.
Данный эксперимент четко демонстрирует, что образования взрывоопасных смесей горючего диалкилдисульфида и кислорода необходимо избегать.
2.1.2.5. Эксперимент 27
Применяли контейнер для образца, изготовленный из сплава Hastelloy С276. Закрытый контейнер для образца имел объем 115 мл и был оснащен погружной трубкой и якорем мешалки.
Начальная заданная температура перед добавлением кислорода составляла 50°С, а заданное давление для добавления кислорода составляло 12 бар (абс.).
Состав образца в данном эксперименте кратко описан в таблице ниже:
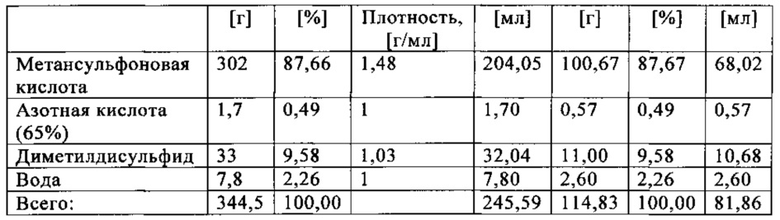
При присущем давлении паров в предварительно вакуумированный контейнер с образцом сначала загружали метансульфоновую кислоту с добавлением небольших количеств азотной кислоты и воды, а затем загружали диметилдисульфид. После этого образец нагревали до заданной температуры 70°С при перемешивании.
Профили зависимости температуры образца и давления от времени показаны на фиг. 17 и 18 соответственно. На фиг. 19 показан профиль зависимости давления от температуры, а на фиг. 20 показана скорость изменения температуры образца в зависимости от температуры образца.
Экзотермическая реакция начиналась сразу после выключения нагрева, после достижения температуры 70°С. При температуре примерно 71,3°С в контейнер с образцом быстро добавляли кислород с целью достижения общего давления 12 бар (абс.). После достижения этого давления и первого закрытия впускного клапана температура образца составляла примерно 93°С. После дальнейшего падения давления кислород снова вводили с целью восстановления давления 12 бар (абс.). Однако сопутствующее увеличение температуры уже не было спонтанным, а происходило довольно медленно. В течение периода продолжительностью приблизительно 4900 с при дальнейших повторных введениях кислорода максимальная достигнутая температура образца составила примерно 110°С.
В этом эксперименте не только рост температуры, но и повышение давления оказались значительно меньше, чем в экспериментах 23 и 25 (см. фиг. 17 и 18). Таким образом, в отличие от экспериментов 23 и 25, выбранная начальная концентрация диметилдисульфида в этом эксперименте позволяет осуществлять периодическое добавление кислорода в течение продолжительного промежутка времени без заметного превышения заданного давления 12 бар (абс.). Параметры процесса в эксперименте 27, конкретно выбранная концентрация диметилдисульфида, позволяют осуществлять соответствующий способ получения метансульфоновой кислоты без проблем в отношении безопасности.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МЕТАНСУЛЬФОКИСЛОТЫ | 2016 |
|
RU2648245C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНСУЛЬФОКИСЛОТ | 1999 |
|
RU2230735C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНОГО РАСТВОРА ГЛИОКСИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2481322C2 |
| РЕГЕНЕРАЦИЯ СОДЕРЖАЩИХ МЕТАЛЛ КАТАЛИЗАТОРОВ | 2011 |
|
RU2579147C2 |
| Способ получения уксусной кислоты | 1990 |
|
SU1808826A1 |
| ОКИСЛИТЕЛЬНОЕ ДЕГИДРИРОВАНИЕ ЭТАНА И ИЗВЛЕЧЕНИЕ УКСУСНОЙ КИСЛОТЫ | 2016 |
|
RU2725646C2 |
| ПРИМЕНЕНИЕ АЛКАНСУЛЬФОНОВОЙ КИСЛОТЫ ДЛЯ ЧИСТКИ В САХАРНОЙ ПРОМЫШЛЕННОСТИ | 2016 |
|
RU2695848C2 |
| СПОСОБ ПОЛУЧЕНИЯ УКСУСНОЙ КИСЛОТЫ И КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ПОЛУЧЕНИЯ УКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2132840C1 |
| ПОЛУЧЕНИЕ АМИНОКСИДА ОКИСЛЕНИЕМ ТРЕТИЧНОГО АМИНА | 2017 |
|
RU2754032C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТОПРОПИНОВОГО АЛЬДЕГИДА | 2011 |
|
RU2595039C2 |
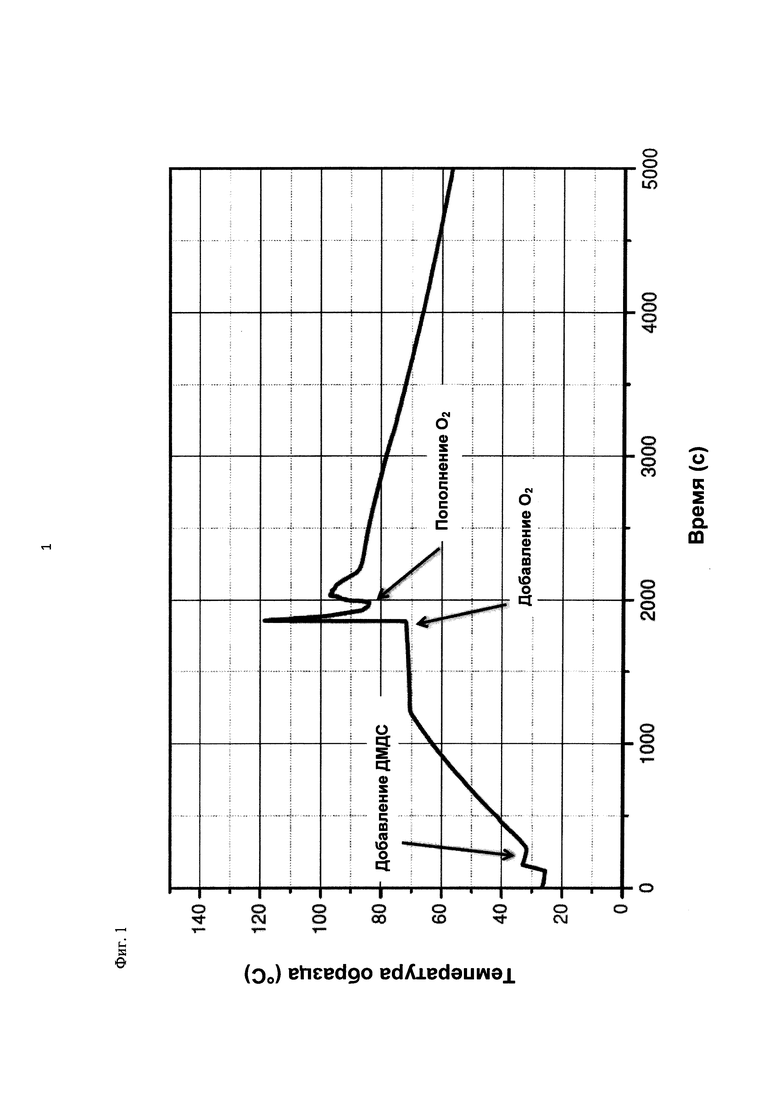
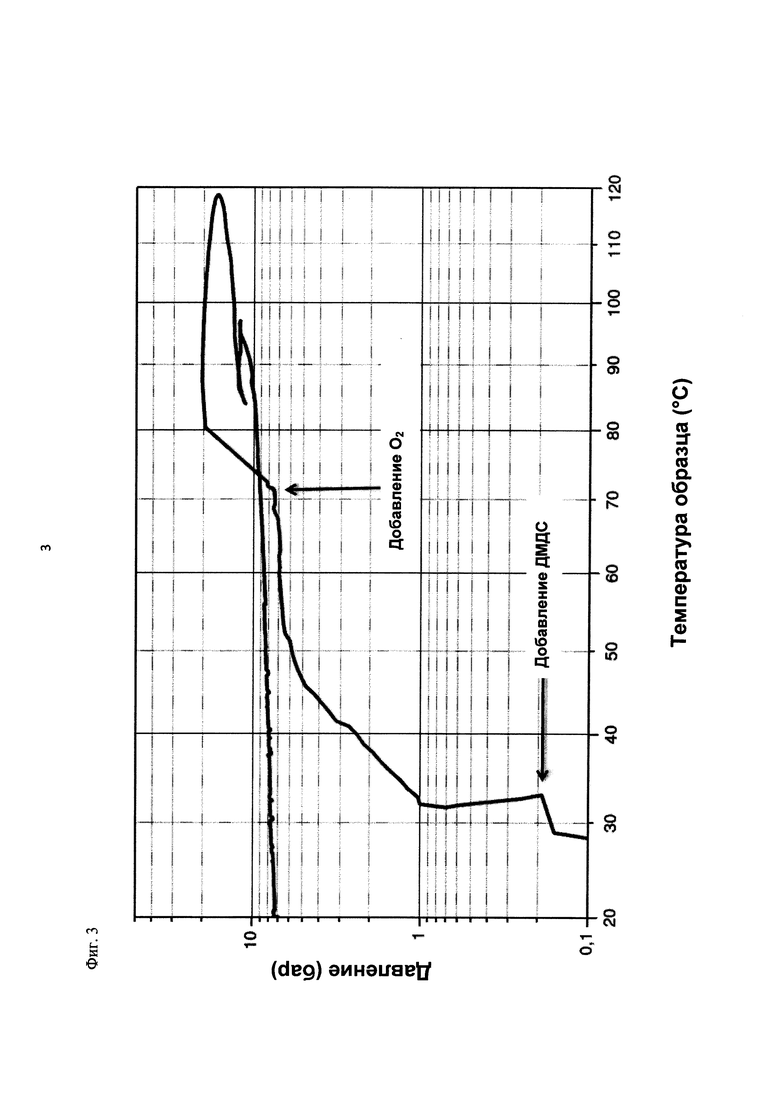
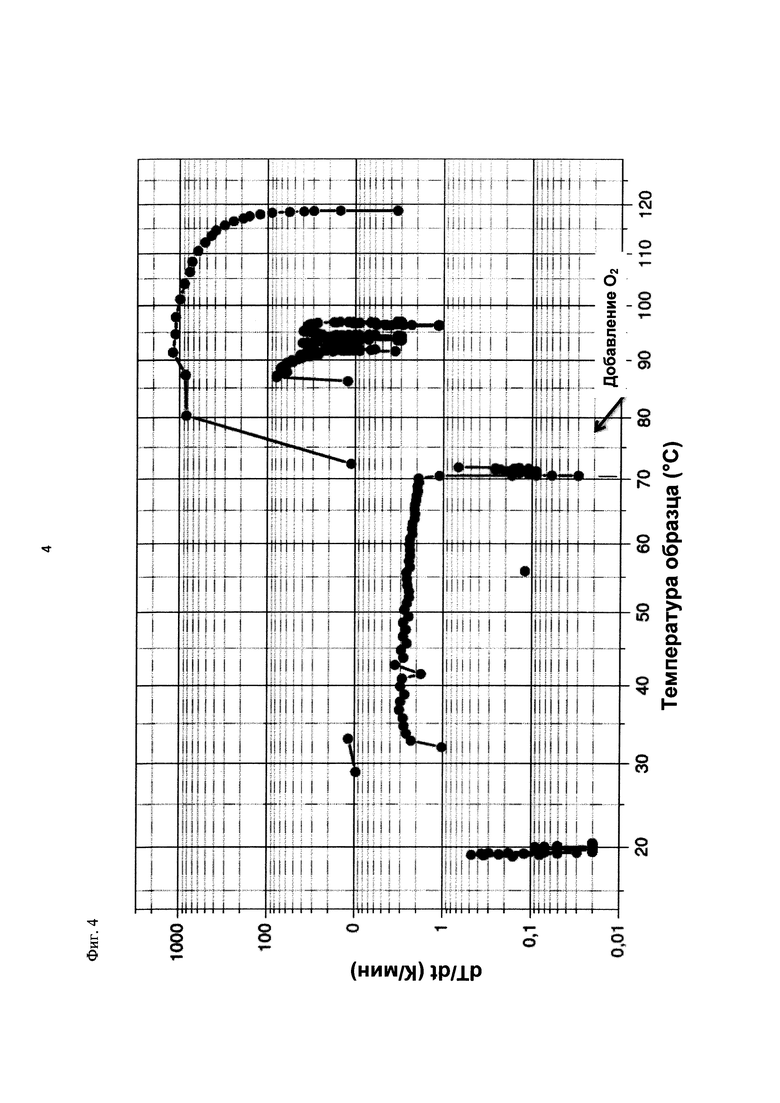
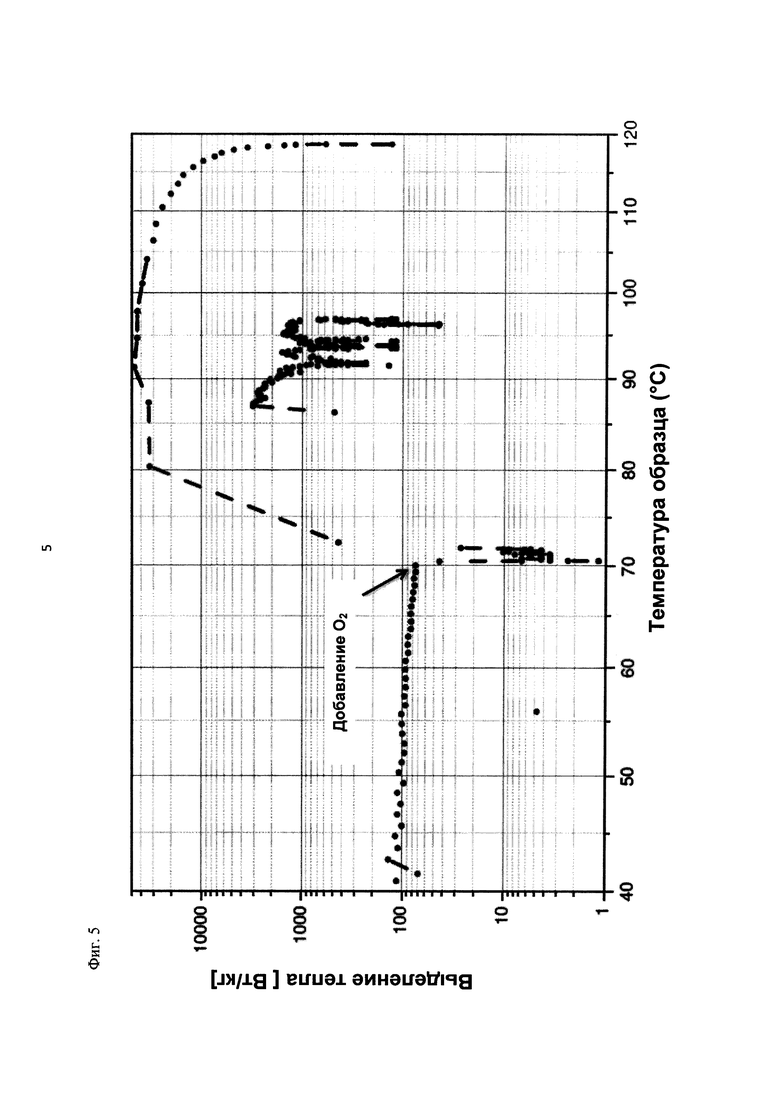
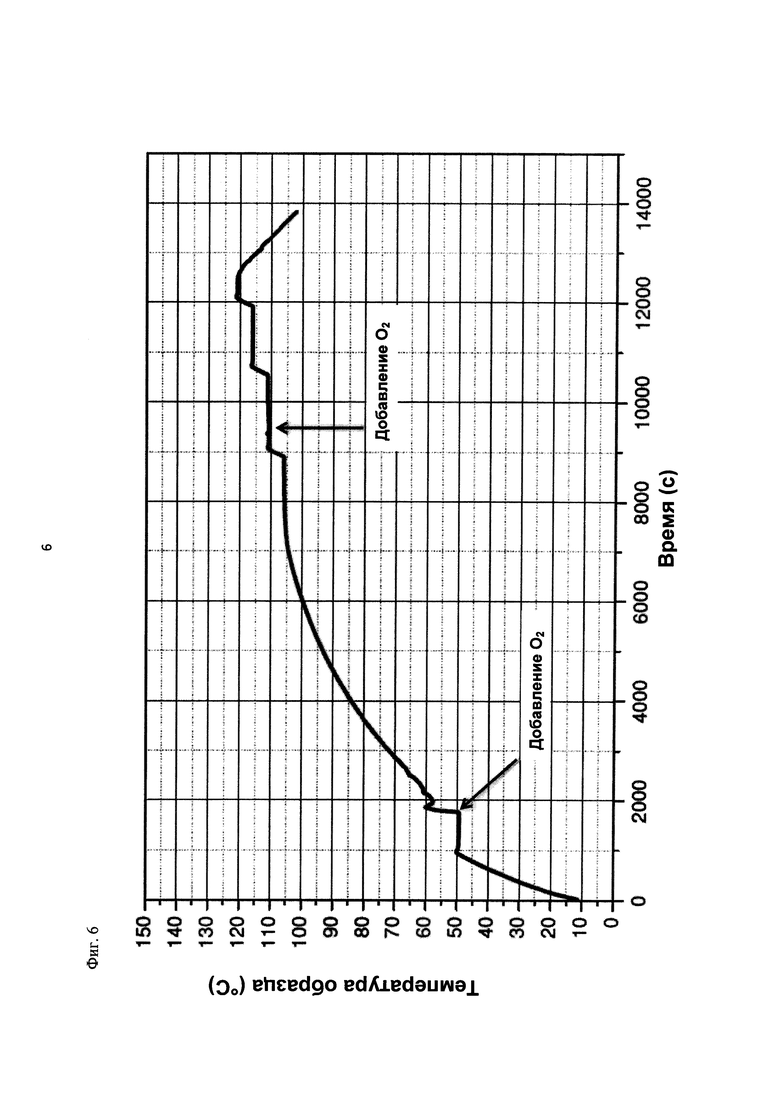
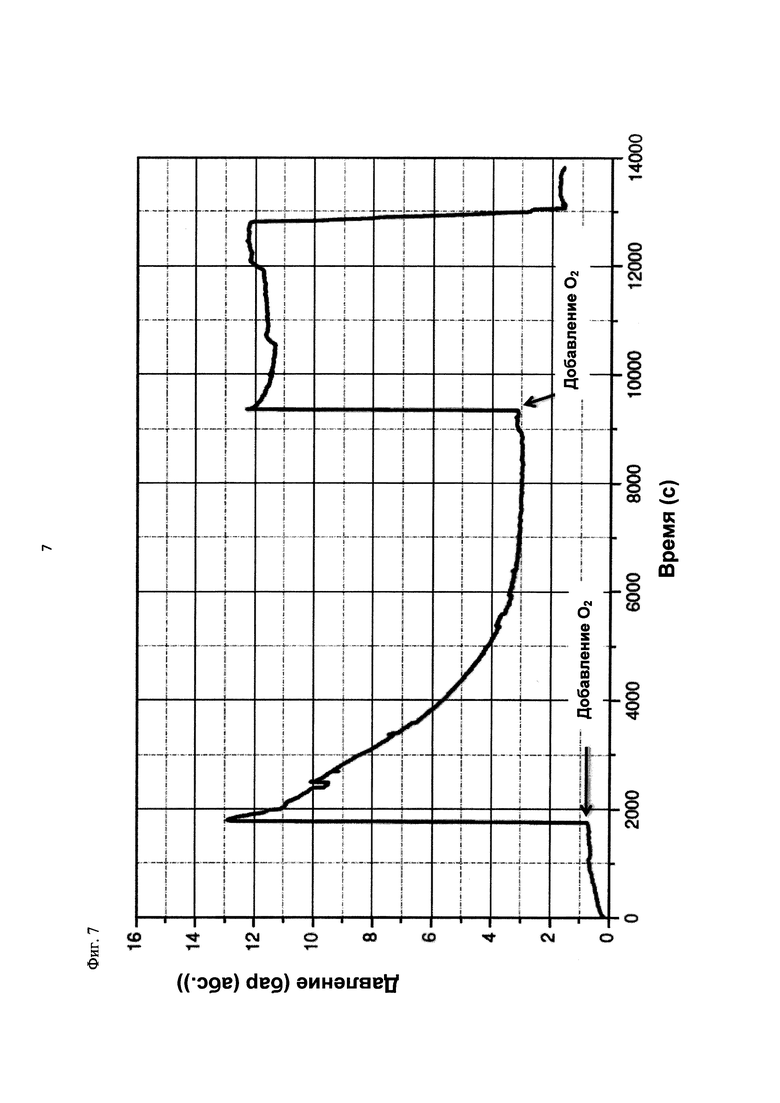
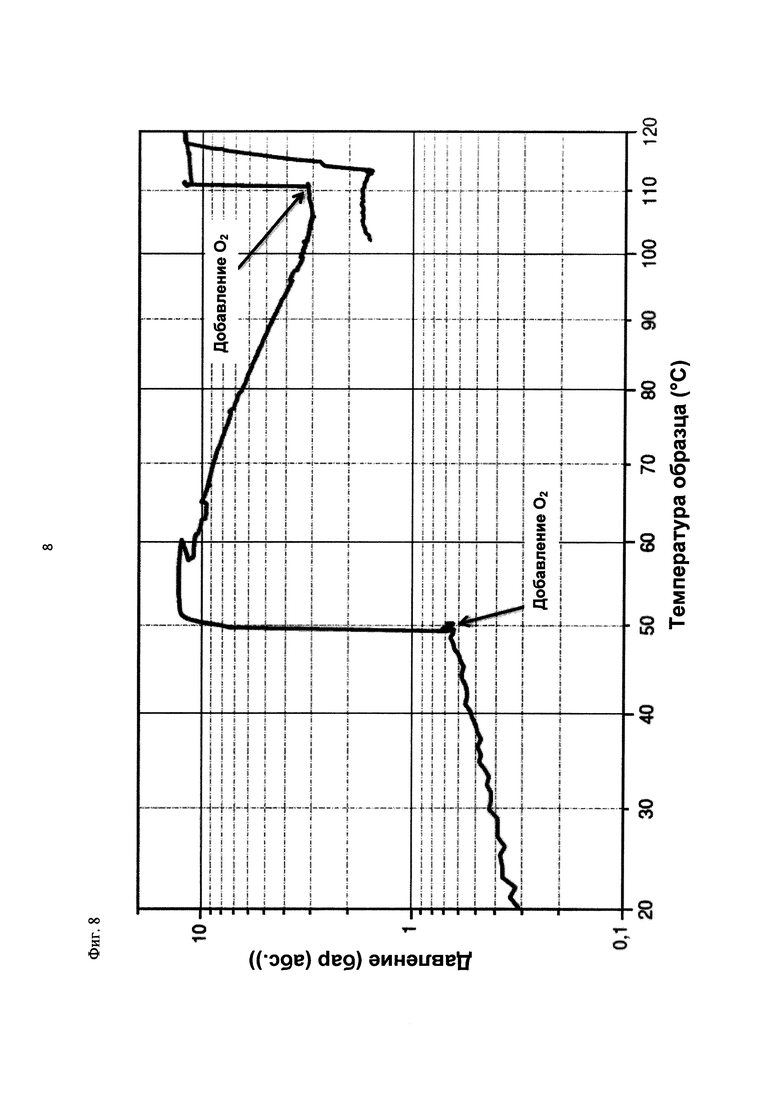
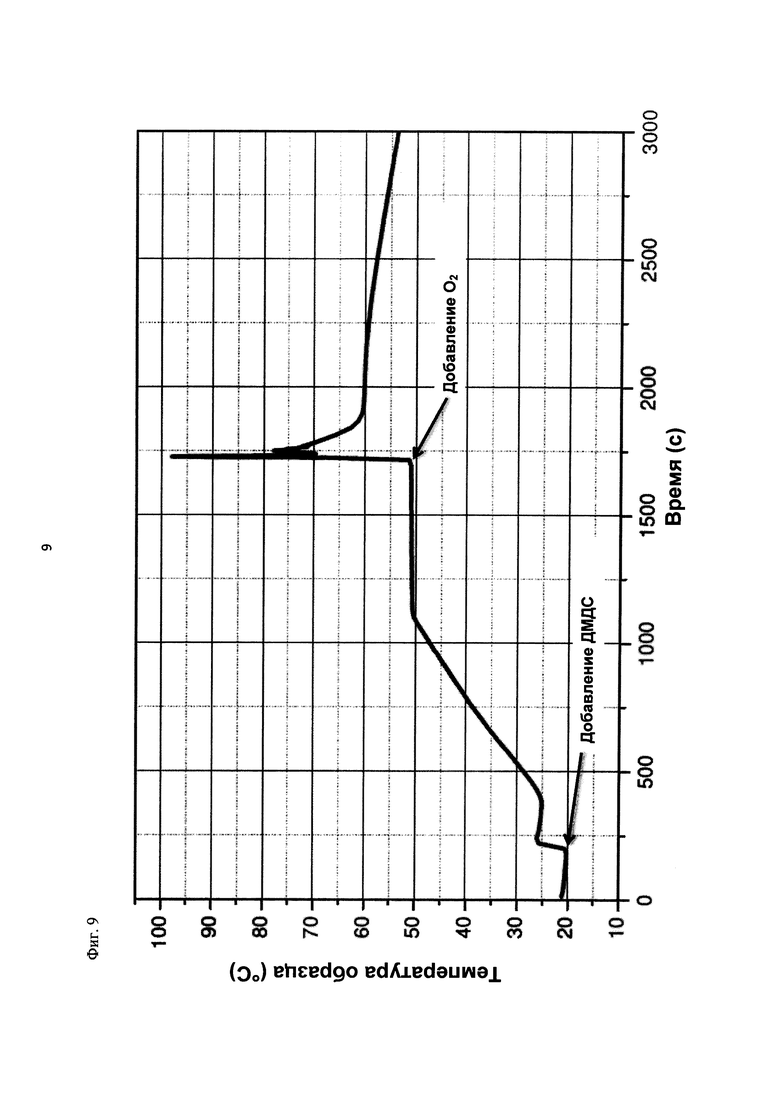
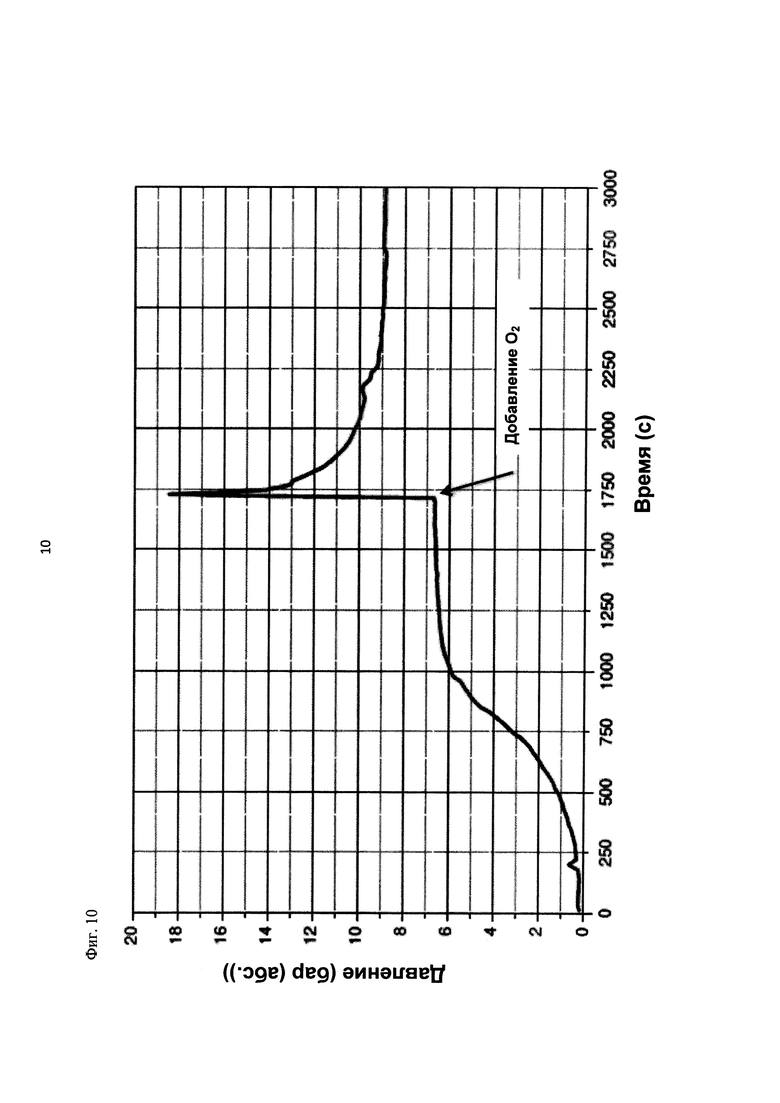
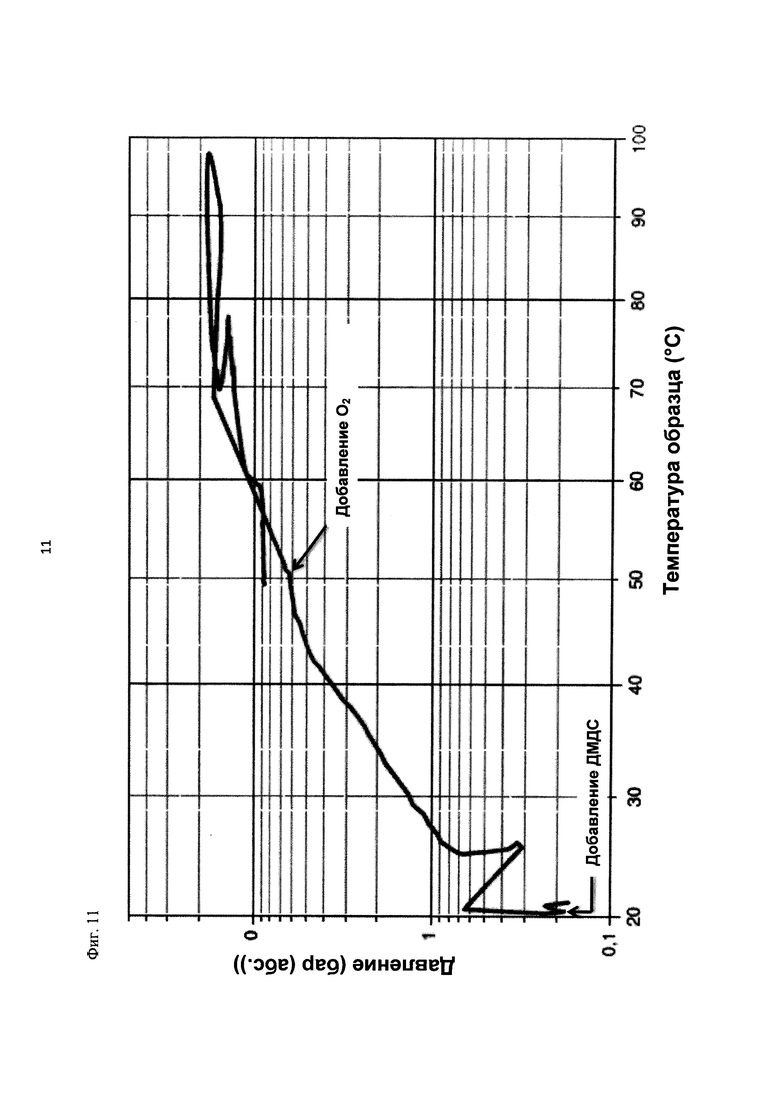
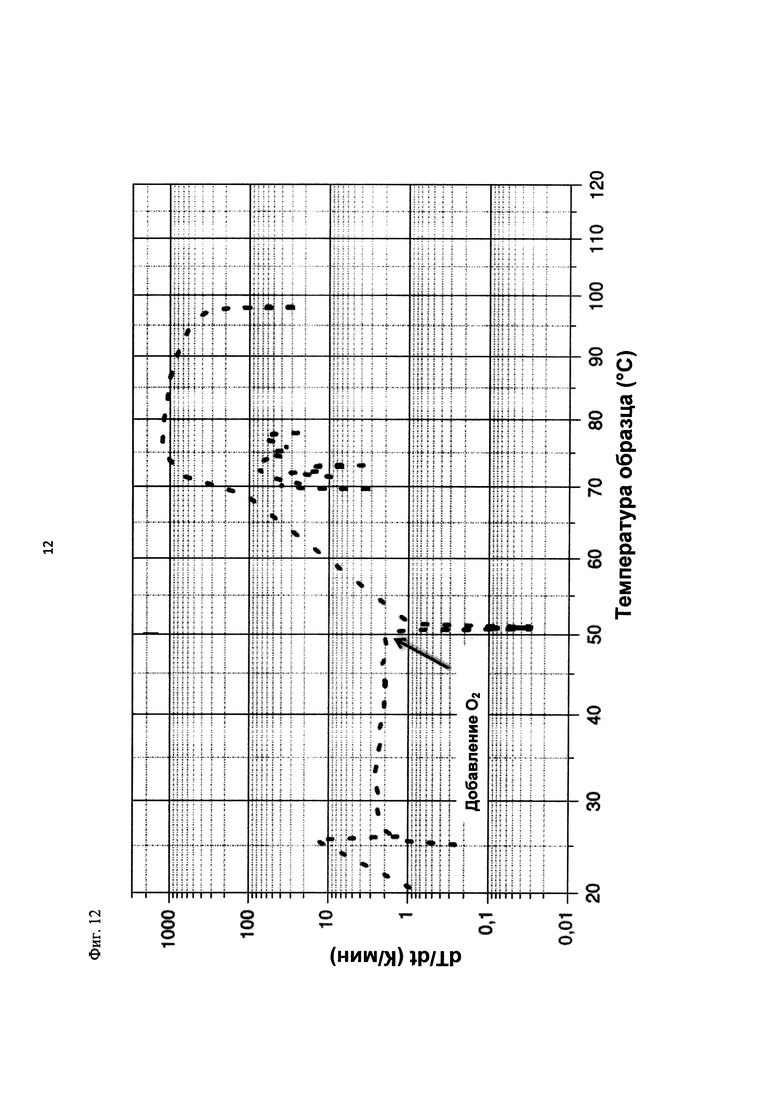
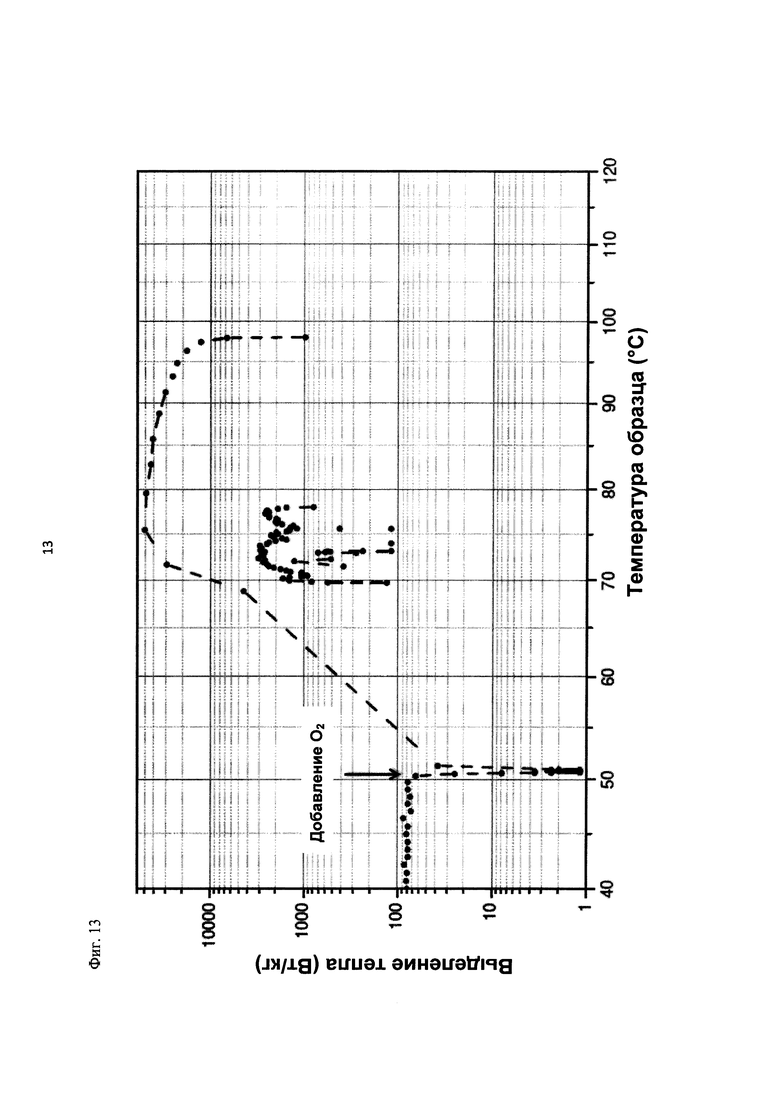
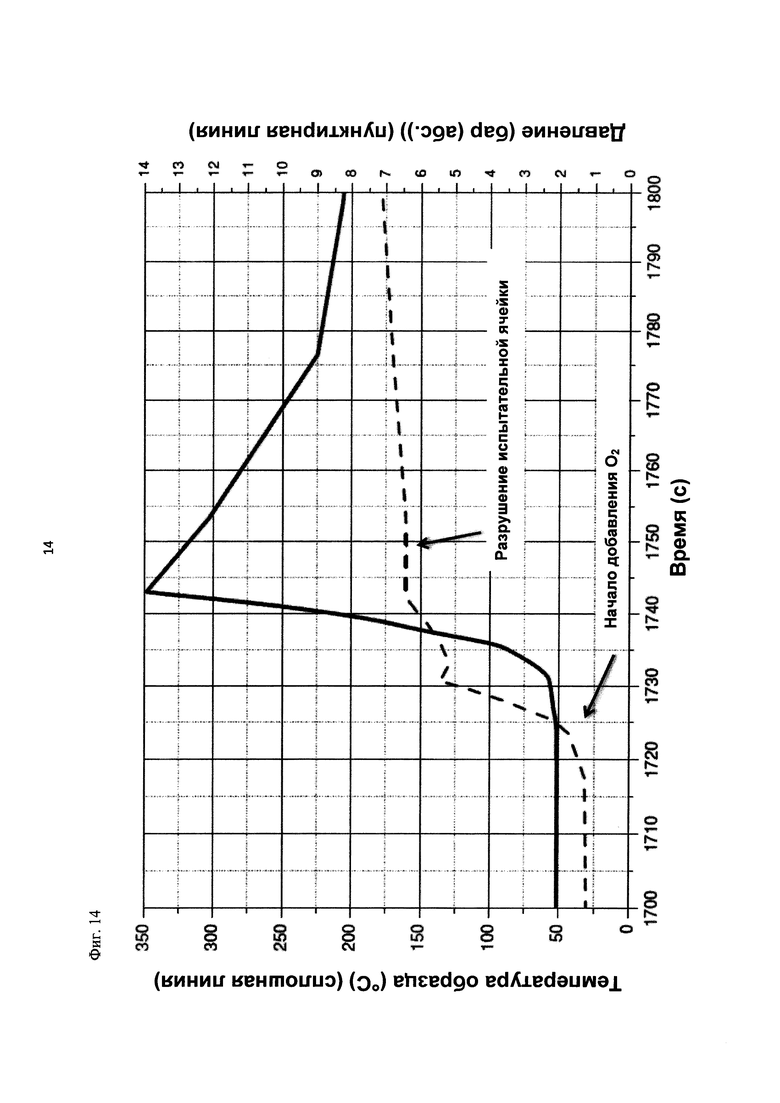
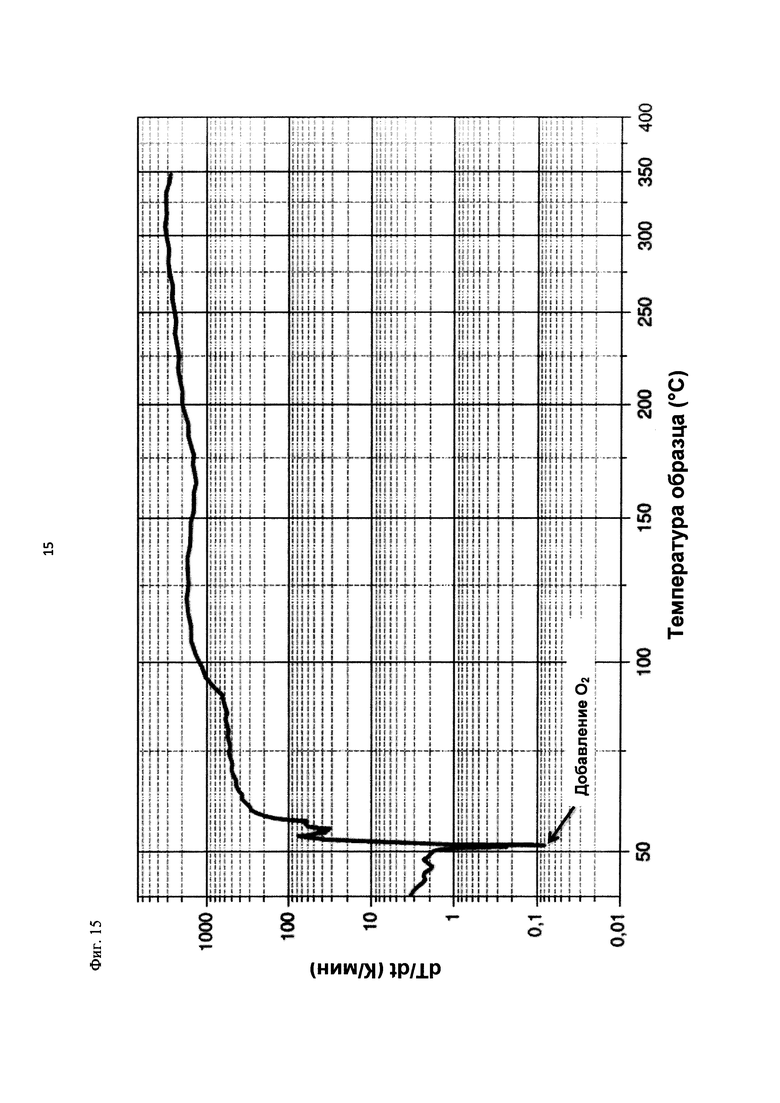
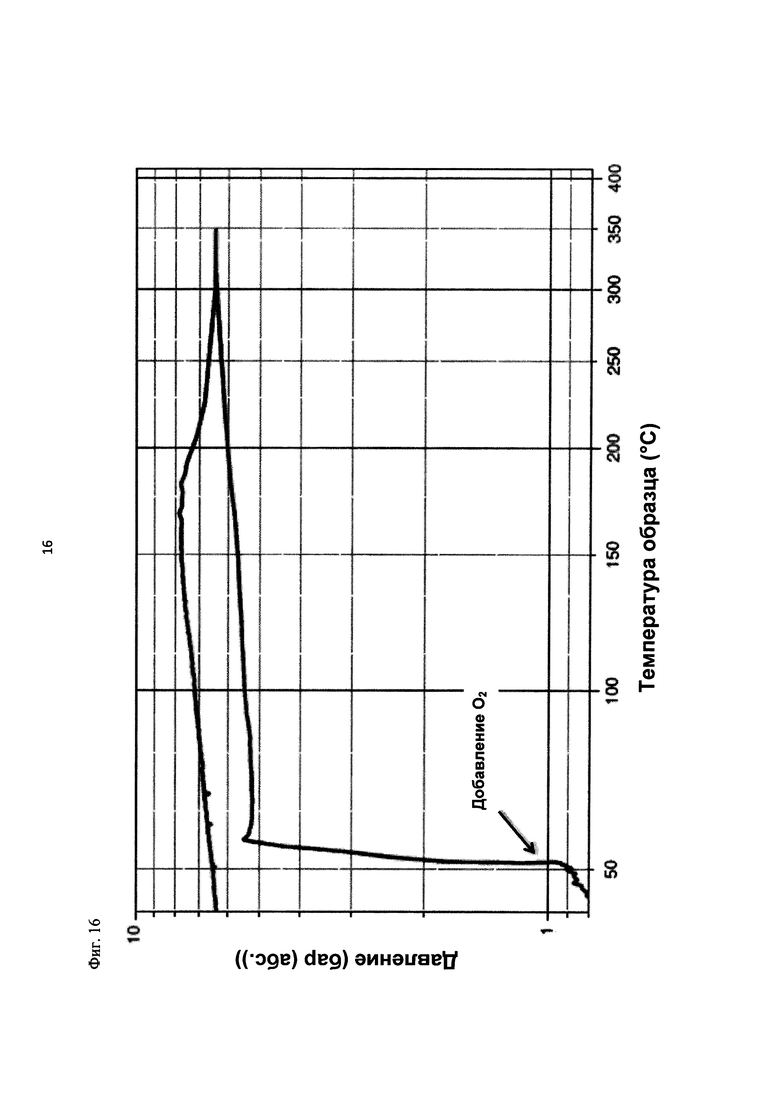
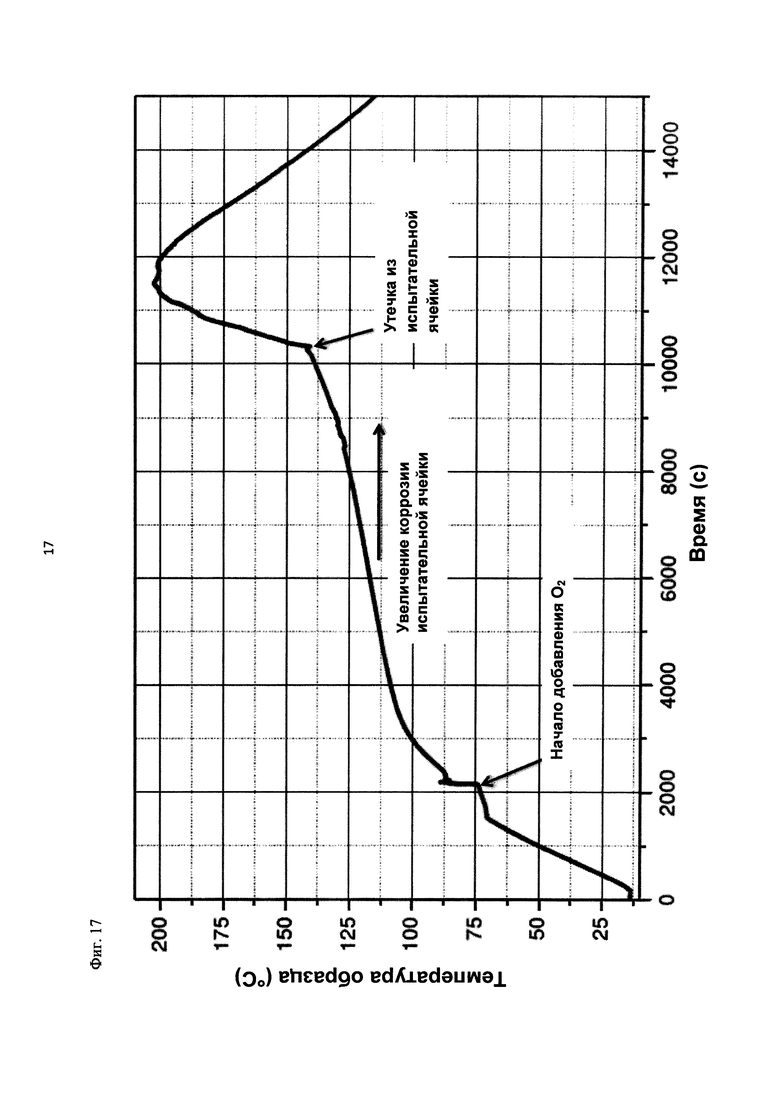
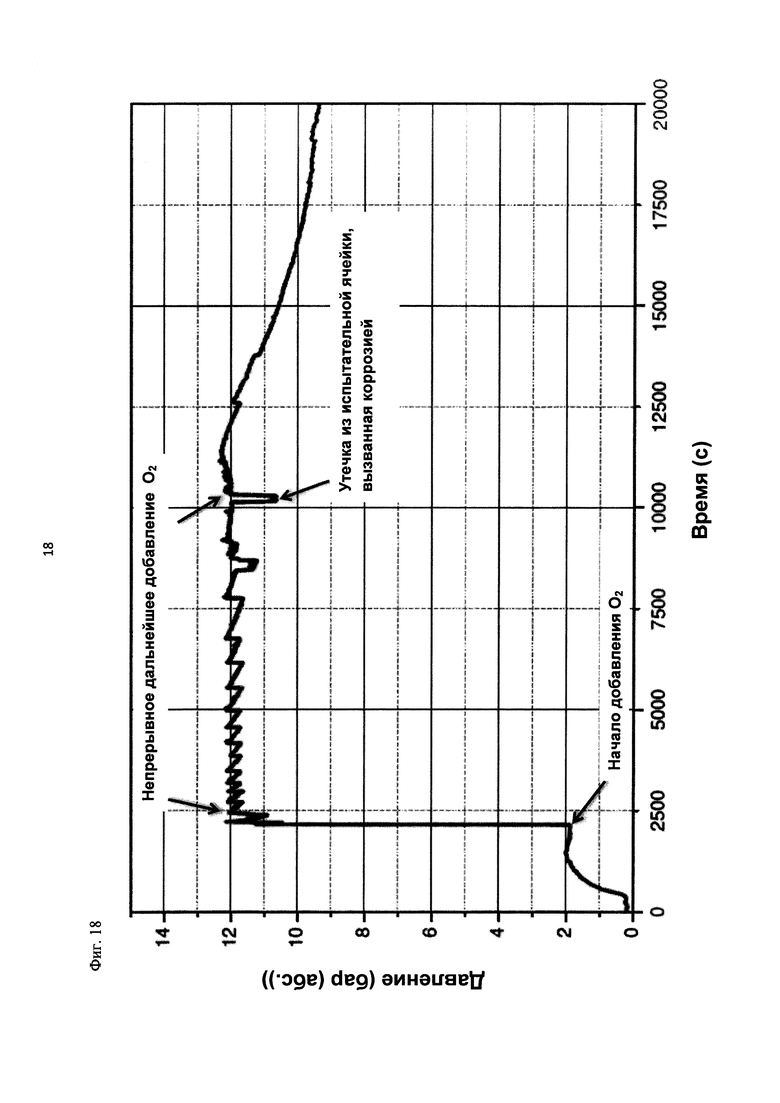
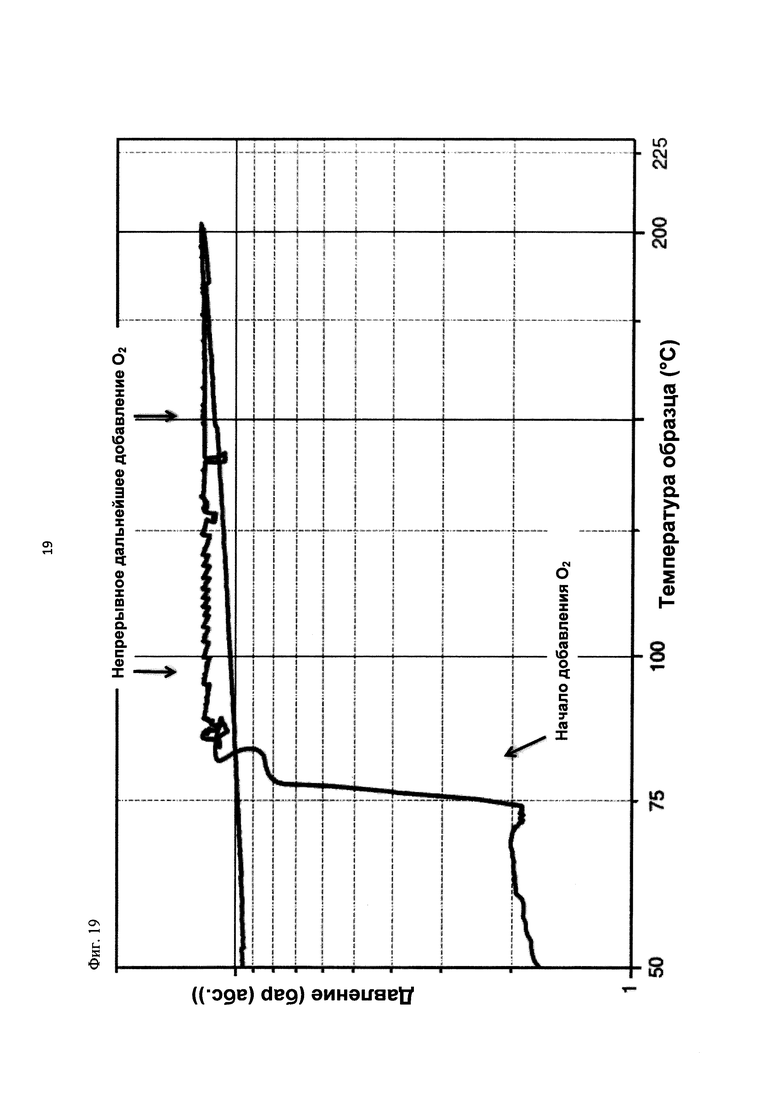
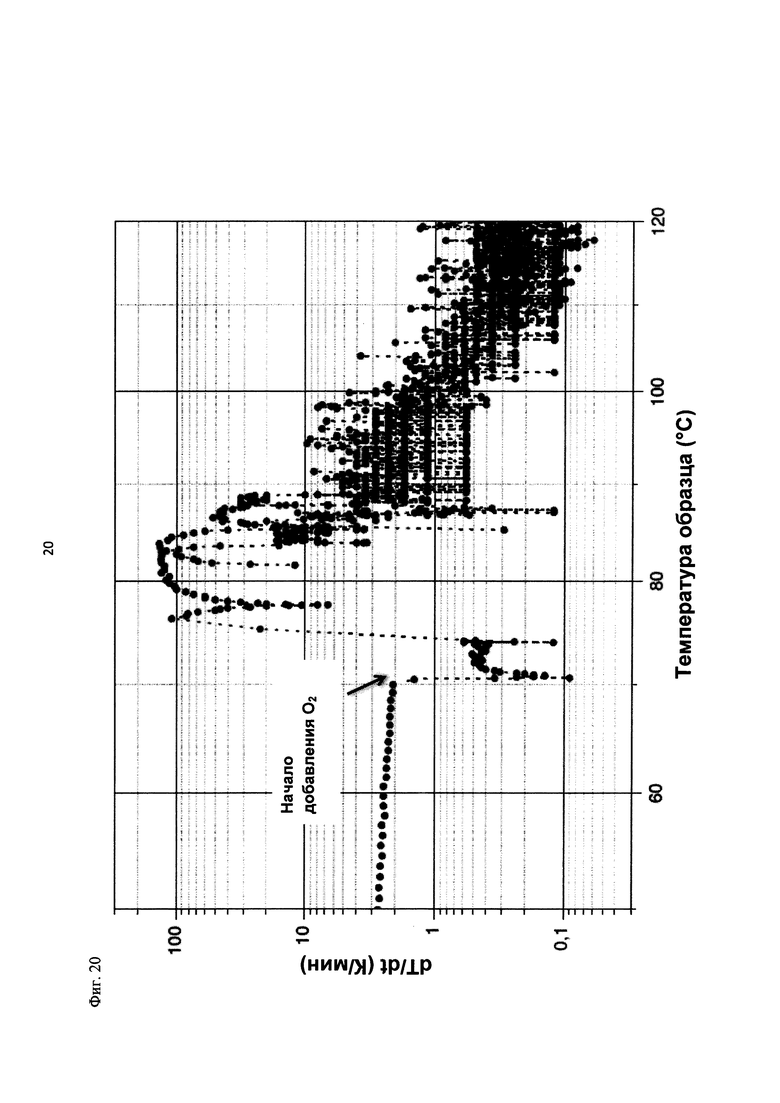
Изобретение представляет собой способ получения алкансульфоновых кислот формулы R-SO3-H, включающий стадию окисления симметричного диалкилдисульфида формулы R-S2-R, в растворе алкансульфоновой кислоты, в присутствии каталитических количеств азотной кислоты, причем R обозначает С1-12алкильный радикал, а алкансульфоновая кислота, применяемая в качестве растворителя, идентична алкансульфоновой кислоте, получаемой в ходе окисления указанного диалкилдисульфида. Согласно изобретению концентрация диалкилдисульфида в растворе составляет не более 20 мас.%, отношение диалкилдисульфида к азотной кислоте составляет от 2000:1 до 1:1 (моль/моль), а концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет более 70 мас.%. 14 з.п. ф-лы, 20 ил., 3 табл.
1. Способ получения алкансульфоновых кислот формулы R-SO3-H, включающий стадию окисления симметричного диалкилдисульфида формулы R-S2-R, в растворе алкансульфоновой кислоты, в присутствии каталитических количеств азотной кислоты, причем R обозначает С1-12алкильный радикал, а алкансульфоновая кислота, применяемая в качестве растворителя, идентична алкансульфоновой кислоте, получаемой в ходе окисления указанного диалкилдисульфида, отличающийся тем, что концентрация диалкилдисульфида в растворе составляет не более 20 мас. %, отношение диалкилдисульфида к азотной кислоте составляет от 2000:1 до 1:1 (моль/моль), а концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет более 70 мас. %.
2. Способ по п. 1, в котором диалкилдисульфид представляет собой диметилдисульфид, а алкансульфоновая кислота представляет собой метансульфоновую кислоту.
3. Способ по п. 1 или 2, в котором отношение диалкилдисульфида к азотной кислоте находится в диапазоне от 500:1 до 1:1 (моль/моль).
4. Способ по п. 1 или 2, в котором отношение диалкилдисульфида к азотной кислоте находится в диапазоне от 500:1 до 2:1 (моль/моль).
5. Способ по п. 1 или 2, в котором концентрация диалкилдисульфида в алкансульфоновой кислоте составляет до примерно 10 мас. %.
6. Способ по п. 1 или 2, который осуществляют при температурах не более примерно 90°С.
7. Способ по п. 6, который осуществляют при температурах от примерно 70 до примерно 90°С.
8. Способ по пп. 1, 2 и 7, в котором концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет по меньшей мере 80 мас. %.
9. Способ по п. 8, в котором концентрация алкансульфоновой кислоты, применяемой в качестве растворителя, составляет по меньшей мере примерно 90 мас. %.
10. Способ по пп. 1, 2, 7 и 9, в котором с целью осуществления окисления в процесс подают воздух, газовый поток, обогащенный кислородом в свободной форме и/или чистый кислород в свободной форме.
11. Способ по п. 10, в котором с целью осуществления окисления в процесс подают газовый поток, содержащий более чем 21 об. % кислорода в свободной форме.
12. Способ по пп. 1, 2, 7, 9 и 11, который осуществляют при давлении от более чем 1 до примерно 20 бар (абс).
13. Способ по п. 12, который осуществляют при давлении от более чем примерно 2 до примерно 15 бар (абс).
14. Способ по пп. 1, 2, 7, 9, 11 и 13, в котором применяют солюбилизатор для растворения диалкилдисульфида в алкансульфоновой кислоте.
15. Способ по п. 14, в котором в качестве солюбилизатора для растворения диалкилдисульфида в алкансульфоновой кислоте применяют S-алкильный сложный эфир алкансульфоновой кислоты формулы R-SO2-S-R, причем алкильные радикалы в составе S-алкилового сложного эфира алкансульфоновой кислоты идентичны алкильным радикалам в составе превращаемого диалкилдисульфида и алкильным радикалам в составе алкансульфоновой кислоты.
| WO 1998034914 A1, 13.08.1998 | |||
| US 4239696 A, 15.12.1980 | |||
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНСУЛЬФОКИСЛОТ | 1999 |
|
RU2230735C2 |

