Область техники
Настоящее изобретение относится к получению вакцин, которые специфичны к опухолям пациентов и потенциально пригодны для иммунотерапии первичной опухоли, а также метастазов опухоли.
Уровень техники
Злокачественные новообразования являются основной причиной смертности, вызывая 1 из каждых 4 смертей. Лечение злокачественных новообразований традиционно основывалось на законе средних чисел, то есть используют то, что работает лучше у наибольшего количества пациентов. Однако вследствие молекулярной гетерогенности в злокачественных новообразованиях зачастую менее чем 25% подвергнутых лечению индивидуумов получают пользу от одобренных методов лечения. Индивидуализированная медицина, в основе которой лежит специально подобранное лечение пациентов, рассматривается в качестве потенциального решения низкоэффективных и высокозатратных инноваций в области разработки лекарственных средств.
Антигенспецифичная иммунотерапия, имеющая своей целью повышение или индукцию иммунного ответа у пациентов, успешно применяется при контроле онкологических заболеваний. Т-клетки играют центральную роль в клеточно-опосредованном иммунитете человека и животных. Распознавание и связывание конкретного антигена опосредовано Т-клеточными рецепторами (TCR), экспрессирующимися на поверхности Т-клеток. Т-клеточный рецептор (TCR) Т-клетки способен взаимодействовать с иммуногенными пептидами (эпитопами), связанными с молекулами главного комплекса гистосовместимости (МНС) и презентированными на поверхности клеток-мишеней. Специфичное связывание TCR передает каскад сигналов внутрь Т-клетки, приводя к пролиферации и дифференцировке в зрелую эффекторную Т-клетку.
Идентификация растущего количества патоген- и опухолеспецифичных антигенов (ТАА) приводит к получению большой коллекции подходящих мишеней для иммунотерапии. Клетки, презентирующие иммуногенные пептиды (эпитопы), выделенные из этих антигенов, могут быть подвергнуты специфичному направленному воздействию с помощью активной или пассивной стратегии иммунизации. Активная иммунизация может приводить к индукции и экспансии антиген-специфичных Т-клеток у пациентов, которые способны специфично распознавать и уничтожать больные клетки. Для противоопухолевой вакцинации могут использоваться различные антигенные форматы, включающие цельные злокачественные клетки, белки, пептиды или иммунизирующие векторы, такие как РНК-, ДНК- или вирусные векторы, которые могут применяться непосредственно in vivo или in vitro путем стимуляции DC с последующим переносом пациенту.
Злокачественные новообразования могут возникать в результате накопления геномных мутаций и эпигенетических изменений, часть которых может являться причиной заболевания. Дополнительно к опухолеспецифичным антигенам человеческие злокачественные новообразования несут в себе в среднем 100-120 несинонимичных мутаций, из которых многие могут быть мишенями для вакцин. Более чем 95% мутаций в опухоли уникальны и специфичны для пациента (Weide et al. 2008: J. Immunother. 31, 180-188). Количество соматических мутаций, изменяющих белок, которые могут приводить к образованию опухолеспецифичных Т-клеточных эпитопов, находится в диапазоне от 30 до 400. Было предсказано in silico, что на каждого пациента приходится 40-60 эпитопов, ограниченных по HLA класса I, выделенных из опухолеспецифичных соматических мутаций (Azuma et al. 1993: Nature 366, 76-79). Кроме того, также в результате опухолеспецифичных мутаций, вероятно, возникают de novo эпитопы, ограниченные по HLA класса II, однако их количество все еще остается неизвестным.
Следует отметить, что некоторые несинонимичные мутации случайным образом вовлечены в неопластическую трансформацию, критическую для поддержания онкогенного фенотипа (ведущие мутации/driver mutations) и могут представлять собой потенциальную «ахиллесову пяту» раковых клеток. Как таковые несинонимичные мутации не являются объектом для толерантности центральной иммунной системы, и они могут быть идеальными кандидатами для разработки индивидуальных противоопухолевых вакцин. Мутации, обнаруженные в первичной опухоли, также могут присутствовать в метастазах. Однако некоторые исследования продемонстрировали, что в процессе эволюции опухоли метастазирующие опухоли пациента приобретают дополнительные генетические мутации, которые часто являются клинически важными (Suzuki et al. 2007: Mol. Oncol. 1 (2), 172-180; Campbell et al. 2010: Nature 467 (7319), 1109-1113). Кроме того, также молекулярные характеристики многих метастазов имеют значительные отклонения от характеристик первичных опухолей.
Техническая задача, лежащая в основе настоящего изобретения, заключается в получении высокоэффективной индивидуализированной противоопухолевой вакцины.
Настоящее изобретение базируется на идентификации специфичных для пациента опухолевых мутаций и направленном воздействии на индивидуальную для пациента «сигнатуру» опухолевых мутаций. Конкретно, настоящее изобретение, которое включает секвенирование генома, предпочтительно, экзома или транскриптома, базируется на применении подхода индивидуализированной иммунотерапии и имеет своей целью иммунотерапевтическое направленное воздействие на множество индивидуальных опухолевых мутаций. Секвенирование с использованием технологий Секвенирования Следующего Поколения (англ. Next Generation Sequencing) (NGS) позволяет осуществить быструю и не затратную эффективную идентификацию опухолевых мутаций, специфичных для пациента.
Идентификация несинонимичных точечных мутаций, приводящих к аминокислотным заменам, которые будут присутствовать в молекулах главного комплекса гистосовместимости пациента (МНС), обеспечивает новые эпитопы (неоэпитопы), которые специфичны для злокачественного новообразования пациента, но не обнаруживаются в здоровых клетках пациента. Сбор данного набора мутаций из раковых клеток, таких как циркулирующие опухолевые клетки (СТС), позволяет приготовить вакцину, которая индуцирует иммунный ответ с потенциальным направленным воздействием на первичную опухоль, даже если она содержит генетически отличные субпопуляции, а также на опухолевые метастазы. Для вакцинации такие неоэпитопы, идентифицированные согласно настоящему изобретению, предоставляются пациенту в форме полипептида, включающего указанные неоэпитопы, после соответствующего процессирования и презентирования молекулами МНС, неоэпитопы представляются иммунной системе пациента для стимуляции соответствующих Т-клеток.
Предпочтительно, если такой полипептид предоставляется пациенту путем введения РНК, кодирующей полипептид. Стратегия, в которой in vitro транскрибируемая РНК (IVT-RNA) непосредственно инъецируется пациенту различными способами иммунизации, успешно тестировалась на некоторых моделях млекопитающих. РНК может транслироваться в трансфецированных клетках, а экспрессированный белок после процессирования презентируется на молекулах МНС на поверхности клеток для вызова иммунного ответа.
Преимущества использования РНК в качестве варианта обратимой генной терапии включают временную экспрессию и не трансформирующий характер. РНК не нуждается в попадании в ядро для экспрессии и, кроме того, не может интегрироваться в геном хозяина, таким образом, исключая риск канцерогенеза. Степень трансфекции, достигаемая с помощью РНК, является достаточно высокой. Кроме того, количества полученного белка соответствуют количествам при физиологической экспрессии.
Разумное обоснование иммунотерапевтического направленного воздействия на множество индивидуальных мутаций заключается в том, что (i) эти мутации экспрессируются эксклюзивно, (ii) мутированные эпитопы, как ожидается, идеальны для Т-клеточной иммунотерапии, так как Т-клетки, распознающие их, не подвергаются тимусной селекции, (hi) ускользание опухоли от иммунного ответа может быть уменьшено, например, путем направленного воздействия на «ведущие мутации», которые являются в высокой степени существенными для опухолевого фенотипа, и (iv) мультиэпитопный иммунный ответ более вероятно приведет к улучшенному клиническому эффекту.
Сущность изобретения
Настоящее изобретение относится к эффективным способам получения индивидуальных рекомбинантных противоопухолевых вакцин, индуцирующих эффективный и специфичный иммунный ответ у онкопациентов и оказывающих потенциальное направленное воздействие на первичную опухоль, а также на метастазы опухоли. Противоопухолевые вакцины, получаемые согласно изобретению, при введении пациенту дают группу МНС-презентированных эпитопов, специфичных для опухоли пациента и подходящих для стимулирования, инициирования и/или экспансии Т-клеток, направленных против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы. Таким образом, вакцины, описанные в данном документе, предпочтительно способны индуцировать или стимулировать клеточный ответ, предпочтительно, цитотоксическую Т-клеточную активность, против злокачественного новообразования, характеризующегося презентацией одного или нескольких антигенов, экспрессируемых опухолевыми клетками, с помощью молекул МНС класса I. Так как вакцина, получаемая согласно настоящему изобретению, будет направленно воздействовать на опухолеспецифичные мутации, она будет специфичной для опухоли пациента.
В одном аспекте, настоящее изобретение относится к способу получения индивидуализированной противоопухолевой вакцины, включающему стадии:
(a) идентификации опухолеспецифичных соматических мутаций в образце опухоли онкопациента с получением сигнатуры опухолевых мутаций пациента; и
(b) получения вакцины, характерной чертой которой является сигнатура опухолевых мутаций, полученная в стадии (а).
В одном воплощении, способ по изобретению включает следующие стадии:
i) получение образца опухоли из онкопациента и получение неопухолевого образца, который предпочтительно выделен из онкопациента;
ii) идентификацию различий в последовательностях между геномом, экзомом и/или транскриптомом опухолевого образца и геномом, экзомом и/или транскриптомом неопухолевого образца;
iii) дизайн полипептида, включающего эпитопы, включающие различия в последовательностях, определенные в стадии (ii);
iv) получение полипептида, спроектированного в стадии (iii), или нуклеиновой кислоты, предпочтительно, РНК, кодирующей указанный полипептид; и
v) получение вакцины, включающей полипептид или нуклеиновую кислоту, полученные в стадии (iv).
Согласно изобретению, опухолевый образец относится к любому образцу, такому как образец, взятый из организма, выделенный из тканей пациента, содержащий или ожидаемо содержащий опухолевые или раковые клетки. Образец, взятый из организма, может представлять собой любой образец ткани, такой как кровь, образец ткани, полученный из первичной опухоли или из опухолевых метастазов, или любой другой образец, содержащий опухолевые или раковые клетки. Предпочтительно, если образец организма представляет собой кровь, а опухолеспецифичные соматические мутации или различия в последовательностях определяют в одной или в нескольких циркулирующих опухолевых клетках (СТС), содержащихся в крови. В другом воплощении, опухолевый образец относится к одной или нескольким выделенным опухолевым или раковым клеткам, таким как циркулирующие опухолевые клетки (СТС), или к образцу, содержащему одну или несколько выделенных опухолевых или раковых клеток, таким как циркулирующие опухолевые клетки (СТС).
Неопухолевый образец относится к любому образцу, такому как образец из организма, который выделен из тканей пациента или другого индивидуума, который предпочтительно того же вида, что и пациент, предпочтительно, из здорового индивидуума, не содержащего или ожидаемо не содержащего опухолевых или раковых клеток. Образец из организма может представлять собой любой образец ткани, такой как кровь, или образец из нетуморогенной ткани.
Согласно изобретению термин «сигнатура мутаций злокачественных опухолей» может относиться к мутациям злокачественных опухолей, присутствующим в одной или в нескольких раковых клетках пациента, или может относиться только к части мутаций злокачественных опухолей, присутствующих в одной или в нескольких раковых клетках пациента. Соответственно, настоящее изобретение может включать идентификацию всех опухолеспецифичных мутаций, присутствующих в одной или в нескольких раковых клетках пациента, или может включать идентификацию только части опухолеспецифичных мутаций, присутствующих в одной или в нескольких раковых клетках пациента. Как правило, способ по изобретению обеспечивает идентификацию ряда мутаций, которые дают достаточное количество неоэпитопов, включаемых в вакцину. «Мутация злокачественных опухолей» относится к различию в последовательности между нуклеиновой кислотой, содержащейся в раковой клетке, и нуклеиновой кислотой, содержащейся в здоровой клетке.
Предпочтительно, мутации, идентифицированные в способах согласно настоящему изобретению, представляют собой несинонимичные мутации, предпочтительно, несинонимичные мутации белков, экспрессированных в опухолевых или в раковых клетках.
В одном воплощении опухолеспецифичные соматические мутации или различия в последовательности определяются в геноме, предпочтительно, во всем геноме опухолевого образца. Таким образом, способ по изобретению может включать идентификацию сигнатуры опухолевых мутаций генома, предпочтительно, всего генома одной или нескольких раковых клеток. В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций в опухолевом образце онкопациента включает идентификацию полного геномного профиля опухолевых мутаций.
В одном воплощении опухолеспецифичные соматические мутации или различия в последовательности определяются в экзоме, предпочтительно, во всем экзоме опухолевого образца. Экзом представляет собой часть генома организма, и образован экзонами, которые представляют собой кодирующие участки экспрессирующихся генов. Экзом обеспечивает генетический проект, используемый для синтеза белков и других функциональных генных продуктов. Он является наиболее функционально релевантной частью генома и, таким образом, наиболее вероятно вносит вклад в фенотип организма. По оценкам, экзом человеческого генома составляет 1,5% суммарного генома (Ng, PC et al., PLoS Gen., 4(8):1-15, 2008). Таким образом, способ по изобретению может включать идентификацию характерной опухолевой мутации экзома, предпочтительно, всего экзома одной или нескольких раковых клеток. В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций в опухолевом образце онкопациента включает идентификацию полного экзомного профиля опухолевых мутаций.
В одном воплощении опухолеспецифичные соматические мутации или различия в последовательности определяются в транскриптоме, предпочтительно, в полном транскриптоме опухолевого образца. Транскриптом представляет собой набор всех молекул РНК, включающих мРНК, рРНК, тРНК и другие некодирующие РНК, вырабатываемые в одной клетке или в популяции клеток. В контексте настоящего изобретения транскриптом обозначает набор всех молекул РНК, выработанных в одной клетке, в популяции клеток, предпочтительно в популяции раковых клеток, или во всех клетках данного индивидуума в определенный момент времени. Таким образом, способ по изобретению может включать идентификацию сигнатуры опухолевых мутаций транскриптома, предпочтительно, всего транскриптома одной или нескольких раковых клеток. В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций в опухолевом образце онкопациента включает идентификацию полного транскриптомного профиля опухолевых мутаций.
В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций или идентификации различий в последовательностях включает секвенирование генома из одной клетки (single cell sequencing) для одной или нескольких раковых клеток, предпочтительно, из 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или даже более. Таким образом, способ по изобретению может включать идентификацию характерной опухолеспецифичной мутации указанной одной или нескольких раковых клеток. В одном воплощении раковые клетки представляют собой циркулирующие опухолевые клетки. Раковые клетки, такие как циркулирующие опухолевые клетки, могут быть выделены перед секвенированием генома из одной клетки.
В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций или идентификации различий в последовательностях включает применение секвенирования следующего поколения (NGS).
В одном воплощении стадия идентификации опухолеспецифичных соматических мутаций или идентификации различий в последовательностях включает секвенирование геномной ДНК и/или РНК опухолевого образца.
Для обнаружения опухолеспецифичных соматических мутаций или различий в последовательностях информацию о последовательностях, полученную из опухолевого образца, предпочтительно сравнивали с эталонной, такой как информация о последовательностях, полученная в результате секвенирования ДНК или РНК здоровых незлокачественных клеток, таких как клетки зародышевой линии, которые могут быть получены либо из пациента, либо из других индивидуумов. В одном воплощении нормальную геномную зародышевую ДНК получают из мононуклеаров периферической крови (РВМС).
Вакцина, предлагаемая согласно способам настоящего изобретения, относится к вакцине, которая при введении пациенту предпочтительно обеспечивает группу МНС-презентированных эпитопов, например, 2 или более, 5 или более, 10 или более, 15 или более, 20 или более, 25 или более, 30 или более, и предпочтительно, до 60, до 55, до 50, до 45, до 40, до 35 или до 30 МНС-презентированных эпитопов, включающих изменения последовательности на основе идентифицированных мутаций или различий последовательностей. Такие МНС-презентированные эпитопы, включающие изменения последовательности на основе идентифицированных мутаций или различий в последовательностях, также обозначаются в данном документе как «неоэпитопы». Презентация таких эпитопов клетками пациента, в частности антигенпрезентирующими клетками, предпочтительно приводит в результате к направленному воздействию Т-клеток на МНС-связанные эпитопы и, таким образом, на опухоль пациента, предпочтительно, первичную опухоль, а также на опухолевые метастазы, экспрессирующие антигены, из которых выделены МНС-презентированные эпитопы, которые презентируют те же эпитопы, что и на поверхности опухолевых клеток.
Для получения вакцины способ по изобретению может включать произвольное включение в вакцину достаточного количества неоэпитопов (предпочтительно, в форме кодирующей нуклеиновой кислоты) или может включать дополнительную стадию определения применимости идентифицированных мутаций в эпитопах для противоопухолевой вакцинации. Таким образом, дополнительные стадии могут включать одну или несколько из представленных ниже: (i) оценку того, локализованы ли изменения в последовательности известных или прогнозируемых МНС-презентированных эпитопах, (ii) in vitro и/или in silico тестирование того, локализованы ли изменения последовательности в МНС-презентированных эпитопах, например, тестирование того, являются ли изменения последовательности частью пептидных последовательностей, которые процессируются и/или представлены в виде МНС-презентированных эпитопов, и (iii) in vitro тестирование того, способны ли рассматриваемые мутированные эпитопы, конкретно присутствующие в виде их природной последовательности, например, будучи фланкированными аминокислотными последовательностями, которые также фланкируют указанные эпитопы в природном белке, при экспрессии в антигенпрезентирующих клетках стимулировать Т-клетки пациента, обладающие нужной специфичностью. Каждая из таких фланкирующих последовательностей может включать 3 или более аминокислот, 5 или более, 10 или более, 15 или более, 20 или более и, предпочтительно, до 50, до 45, до 40, до 35 или до 30 аминокислот и может фланкировать последовательности эпитопа с N-конца и/или с С-конца.
Мутации или различия в последовательностях, определенные согласно изобретению, могут быть выстроены по их пригодности в качестве эпитопов для противоопухолевой вакцинации. Таким образом, в одном аспекте способ по изобретению включает ручной или компьютерный аналитический процесс, в котором анализируются идентифицированные мутации и отбираются по их пригодности к использованию в соответствующих предлагаемых вакцинах. В предпочтительном воплощении указанный аналитический процесс представляет собой процесс на основе компьютерного алгоритма. Предпочтительно, указанный аналитический процесс включает одну или несколько, а предпочтительно все из следующих стадий:
- идентификацию экспрессирующихся, модифицирующих белок мутаций, например, путем анализа транскриптов;
- идентификацию мутаций, которые потенциально иммуногенны, например, путем сравнения данных, полученных с использованием доступных баз данных подтвержденных иммуногенных эпитопов, например, тех, что содержатся в общедоступных базах иммунных эпитопов, таких как, например, «IMMUNE EPITOPE DATABASE AND ANALYSIS RESOURCE» по адресу http://www.immunoepitope.org
Стадия идентификации мутаций, которые потенциально иммуногенны, может включать определение и/или упорядочивание эпитопов согласно прогнозу их МНС-связывающей способности, предпочтительно связывание с МНС класса-1.
В другом воплощении изобретения эпитопы могут быть отобраны и/или упорядочены путем использования дополнительных параметров, таких как воздействие на белок, ассоциированная экспрессия генов, уникальность последовательности, прогнозированная вероятность презентации и ассоциация с онкогенами.
Анализы множества СТС также позволяют осуществить отбор и приоритезацию мутаций. Например, приоритет мутации, которая обнаружена в большом количестве СТС, может быть выше, чем у мутации, обнаруженной в меньшем количестве СТС.
Коллекция мутаций на основе неоэпитопов, идентифицированных согласно изобретению и обеспечиваемых вакциной по изобретению, предпочтительно присутствует в форме полипептида, включающего указанные неоэпитопы (полиэпитопный полипептид), или в виде нуклеиновой кислоты, конкретно, РНК, кодирующей указанный полипептид. Кроме того, неоэпитопы могут присутствовать в полипептиде в форме вакцинной последовательности, т.е. присутствовать в виде их природной последовательности, например, будучи фланкированными аминокислотными последовательностями, которые также фланкируют указанные эпитопы в природных белках. Каждая из таких фланкирующих последовательностей может включать 5 или более аминокислот, 10 или более, 15 или более, 20 или более и, предпочтительно, до 50, до 45, до 40, до 35 или до 30 аминокислот и может фланкировать эпитопные последовательности с N-конца и/или с С-конца. Таким образом, вакцинная последовательность может включать 20 или большее количество аминокислот, 25 или более, 30 или более, 35 или более, 40 или более, и предпочтительно, до 50, до 45, до 40, до 35 или до 30 аминокислот. В одном воплощении неоэпитопы и/или вакцинные последовательности располагаются в полипептиде голова к хвосту.
В одном воплощении неоэпитопы и/или вакцинные последовательности разделены линкерами, конкретно, нейтральными линкерами. Термин «линкер» согласно изобретению относится к пептидам, добавляемым между двумя пептидными доменами, такими как эпитопы или вакцинные последовательности для связывания указанных пептидных доменов. Нет никакого конкретного ограничения, касающегося линкерной последовательности. Однако предпочтительно, что линкерная последовательность снижает стерические затруднения между двумя пептидными доменами, хорошо транслируется и поддерживает или дает возможность процессирования эпитопов. Кроме того, линкер не должен обладать последовательностями иммуногенных элементов, или обладает ими в небольшом количестве. Линкеры, предпочтительно, не должны создавать неэндогенных неоэпитопов типа тех, которые образуются в области тесного контакта между соседними неоэпитопами, которые могут вызывать нежелательные иммунные реакции. Таким образом, полиэпитопная вакцина предпочтительно содержит линкерные последовательности, которые способны уменьшить количество нежелательных МНС-связывающих эпитопов из мест соединения. Hoyt et al. (EMBO J. 25(8), 1720-9, 2006) и Zhang et al. (J. Biol. Chem., 279(10), 8635-41, 2004) продемонстрировали, что глицин-богатые последовательности способствуют протеасомному процессированию и, таким образом, использование глицин-богатых линкерных последовательностей минимизирует количество линкер-содержащих пептидов, которые могут процессироваться протеасомой. Кроме того, согласно наблюдениям, глицин ингибирует сильное связывание со связывающими бороздками МНС (Abastado et al., J. Immunol. 151(7), 3569-75, 1993). Schlessinger et al. (Proteins, 61(1), 115-26, 2005) обнаружили, что аминокислоты глицин и серии, включенные в аминокислотную последовательность, приводят в результате к получению более гибкого белка, который более эффективно транслируется и процессируется протеасомой, что дает лучший доступ к кодируемым неоэпитопам. Каждый из линкеров может включать 3 или большее количество аминокислот, 6 или более, 10 или более, 15 или более, 20 или более, и предпочтительно, до 50, до 45, до 40, до 35 или до 30 аминокислот. Предпочтительно, линкер обогащен аминокислотами глицином и/или серином. Предпочтительно, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%, по меньшей мере, 80%, по меньшей мере, 90%, или, по меньшей мере, 95% аминокислот линкера представляют собой глицин и/или серин. В одном воплощении линкер по существу состоит из аминокислот глицина и/или серина. В одном воплощении, линкер включает аминокислотную последовательность (GGS)a(GSS)b(GGG)c(SSG)d(GSG)e где a, b, c, d и e независимо представляют собой число, выбранное из 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, или 20 и где a+b+c+d+e отличаются от 0 и предпочтительно составляют 2 или более, 3 или более, 4 или более или 5 или более. В одном воплощении, линкер включает последовательность, описанную в данном документе, включающую линкерные последовательности, описанные в примерах, такие как последовательность GGSGGGGSG.
В другом воплощении настоящего изобретения группу мутаций на основе неоэпитопов, идентифицированных согласно изобретению и представленных с помощью вакцины по изобретению, предпочтительно присутствует в форме группы полипептидов, включающих указанные неоэпитопы, которые также могут перекрываться, или в форме группы нуклеиновых кислот, конкретно, РНК, кодирующих указанные полипептиды.
В одном конкретном предпочтительном воплощении полиэпитопный полипептид согласно настоящему изобретению вводят пациенту в форме нуклеиновой кислоты, предпочтительно, РНК, такой как in vitro транскрибированная или синтетическая РНК, которая может экспрессироваться в клетках пациента, таких как антигенпрезентирующие клетки с получением полипептида. Настоящее изобретение также предусматривает введение одного или нескольких мультиэпитопных полипептидов, которые для целей настоящего изобретения включены с использованием термина «полиэпитопный полипептид», предпочтительно в форме РНК, такой как in vitro транскрибированная или синтетическая РНК, которая может экспрессироваться в клетках пациента, таких как антигенпрезентирующие клетки, с получением одного или нескольких полипептидов. В случае введения более чем одного мультиэпитопного полипептида неоэпитопы, которые представлены с помощью различных мультиэпитопных полипептидов, могут быть различными или частично перекрываться. После того как полипептид по изобретению появляется в клетках пациента, таких как антигенпрезентирующие клетки, полипептид по изобретению процессируется с получением неоэпитопов, идентифицированных согласно изобретению. Введение вакцины, представленной согласно изобретению, может давать эпитопы, презентируемые МНС класса II, которые способны вызывать ответ клеток CD4+ Т-хелперов против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы. В ином случае или дополнительно, введение вакцины, представленной согласно изобретению, может обеспечивать эпитопы, презентируемые МНС класса I, которые способны вызывать ответ CD8+ Т-клеток против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы. Кроме того, введение вакцины, представленной согласно изобретению, может обеспечивать один или несколько неоэпитопов (включая известные неоэпитопы и неоэпитопы, идентифицированные по изобретению), а также один или несколько эпитопов, не содержащих опухолеспецифичных соматических мутаций, но экспрессирующихся опухолевыми клетками и предпочтительно индуцирующими иммунный ответ против опухолевых клеток, предпочтительно опухолеспецифичный иммунный ответ. В одном воплощении введение вакцины, представленной согласно изобретению, обеспечивает неоэпитопы, которые представляют собой эпитопы, презентированные МНС класса II, и/или способны вызывать ответ CD4+ хэлперных Т-клеток против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы, а также эпитопы, не содержащие опухолеспецифичных соматических мутаций, которые представляют собой эпитопы, презентированные МНС класса I, и/или способны вызывать ответ CD8+ Т-клеток против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы. В одном воплощении эпитопы, не содержащие опухолеспецифичных соматических мутаций, выделены из опухолевого антигена. В одном воплощении неоэпитопы и эпитопы, не содержащие опухолеспецифичных соматических мутаций, обладают синергическим эффектом при лечении злокачественного новообразования. Предпочтительно, вакцина, представленная согласно изобретению, применяется для полиэпитопного стимулирования ответа цитотоксических и/или хэлперных Т-клеток.
В следующем аспекте в настоящем изобретении предлагается вакцина, которую получают способом по изобретению. Соответственно, настоящее изобретение относится к вакцине, включающей рекомбинантный полипептид, включающий неоэпитопы на основе мутаций, причем указанные неоэпитопы получены на основании опухолеспецифичных соматических мутаций в опухолевом образце онкопациента, или включающей нуклеиновую кислоту, кодирующую указанный полипептид. Такой рекомбинантный полипептид также может включать эпитопы, не включающие опухолеспецифичных соматических мутаций, обсуждаемых выше. Предпочтительные воплощения такой вакцины описаны выше в контексте способа по изобретению.
Вакцина, представленная согласно изобретению, может включать фармацевтически приемлемый носитель и необязательно может включать один или несколько адъювантов, стабилизаторов и т.д. Вакцина может быть представлена в форме терапевтической или профилактической вакцины.
Другой аспект настоящего изобретения относится к способу индуцирования иммунного ответа у пациента, включающему введение пациенту вакцины, представленной согласно изобретению.
Другой аспект относится к способу лечения онкопациента, включающему стадии:
(a) получение индивидуальной противоопухолевой вакцины с помощью способа по изобретению; и
(b) введение указанной вакцины пациенту.
Другой аспект относится к способу лечения онкопациента, включающему введение пациенту вакцины по изобретению.
В следующих аспектах, в изобретении предлагаются вакцины, описанные в данном документе, для применения в способах лечения, описанных в данном документе, конкретно, для применения при лечении или при предотвращении злокачественного новообразования.
Лечение злокачественного новообразования, описанное в данном документе, может объединяться с хирургическим удалением и/или с традиционной химиотерапией.
Другой аспект изобретения относится к способу определения частоты ложноположительных результатов на основе данных по секвенированию следующего поколения, причем указанный способ включает:
забор первого образца генетического материала от животного или человека;
забор второго образца генетического материала от животного или человека;
забор первого образца генетического материала из опухолевых клеток;
забор второго образца генетического материала из указанных опухолевых клеток;
определение общего покрытия при сравнении с опухолью путем подсчета всех оснований эталонного генома, которые включены как в опухоль, так и, по меньшей мере, в один из числа указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека;
определение общего покрытия при сравнении «сам против себя» путем подсчета всех оснований эталонного генома, которые охватываются обоими из числа указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека;
деление указанного общего покрытия при сравнении с опухолью на указанное общее покрытие при сравнении «сам против себя» с получением нормализации;
определение частоты ложноположительных результатов путем деления 1) количества однонуклеотидных вариаций с баллом качества выше, чем Q при сравнении указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека, на 2) количество однонуклеотидных вариаций с показателем качества выше, чем Q при сравнении указанного первого образца генетического материала из указанных опухолевых клеток и указанного второго образца генетического материала из указанных опухолевых клеток и путем 3) умножения полученного результата на указанную нормализацию.
В одном воплощении, указанный генетический материал является ДНК.
В одном воплощении, Q определяют с помощью:
установления ряда качественных признаков S=(s1,…,sn), где S предпочтительней T=(t1,…,tn), что обозначается через S>T, где si>ti для всех i=1,…,n;
определения частоты ложноположительных результатов путем деления 1) количества однонуклеотидных вариаций с баллом качества выше, чем Q, при сравнении указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека, на 2) количество однонуклеотидных вариаций с показателем качества выше, чем Q, при сравнении указанного первого образца генетического материала из указанных опухолевых клеток и указанного второго образца генетического материала из указанных опухолевых клеток и путем 3) умножения полученного результата на указанную нормализацию:
определения интервала значений для каждого свойства для m мутаций с n всех качественных признаков для каждого;
выборочный отбор до p значений из указанного интервала значений;
создание всех возможных комбинаций из выбранных значений качества, что приводит в результате к получению pn экспериментальных точек;
применения случайного образца указанных экспериментальных точек в качестве прогнозирующего параметра для обучения «случайного леса»;
применения соответствующего промежуточного значения частоты ложноположительных результатов в качестве ответа для указанного обучения «случайного леса»,
где полученный в результате балл регрессии указанного обучения «случайного леса» представляет собой Q.
В одном воплощении, указанный второй образец ДНК из животного или человека является аллогенным по отношению к указанному первому образцу ДНК из животного или человека. В одном воплощении, указанный второй образец ДНК из животного или человека является аутологичным по отношению к указанному первому образцу ДНК из животного или человека. В одном воплощении, указанный второй образец ДНК из животного или человека является ксеногенным по отношению к указанному первому образцу ДНК из животного или человека.
В одном воплощении, указанный генетический материал является РНК.
В одном воплощении, Q определяют с помощью:
установления ряда качественных признаков S=(s1,…,sn), где S предпочтительней T=(t1,…,tn), что обозначается через S>T, где si>ti для всех i=1,…,n;
определения промежуточного частоты ложноположительных результатов путем деления 1) количества однонуклеотидных вариаций с показателем качества S>T при сравнении указанного первого образца РНК из животного или человека и указанного второго образца РНК из животного или человека, на 2) количество однонуклеотидных вариаций с показателем качества S>T при сравнении указанного первого образца РНК из указанных опухолевых клеток и указанного второго образца РНК из указанных опухолевых клеток и путем 3) умножения полученного результата на указанную нормализацию;
определения интервала значений для каждого свойства для m мутаций с n всех качественных признаков;
выборочный отбор до p значений из указанного интервала значений;
создание всех возможных комбинаций из выбранных качественных значений, что приводит в результате к получению pn экспериментальных точек;
применения случайного образца указанных экспериментальных точек в качестве прогнозирующего параметра для обучения «случайного леса»;
применения соответствующего промежуточного значения частоты ложноположительных результатов в качестве ответа на указанное обучение «случайного леса»,
где полученный в результате балл регрессии указанного обучения случайного леса представляет собой Q.
В одном воплощении, указанный второй образец РНК из животного или человека является аллогенным по отношению к указанному первому образцу РНК из животного или человека. В одном воплощении, указанный второй образец РНК из животного или человека является аутологичным по отношению к указанному первому образцу РНК из животного или человека. В одном воплощении, указанный второй образец РНК из животного или человека является ксеногенным по отношению к указанному первому образцу РНК из животного или человека.
В одном воплощении, указанная частота ложноположительных результатов применяется для получения вакцинного состава. В одном воплощении вакцина доставляется внутривенно. В одном воплощении вакцина доставляется внутрикожно. В одном воплощении вакцина доставляется внутримышечно. В одном воплощении вакцина доставляется подкожно. В одном воплощении указанная вакцина специально создана для конкретного пациента.
В одном воплощении один из числа указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека взят у указанного конкретного пациента.
В одном воплощении в указанной стадии определения общего покрытия при сравнении с опухолью путем подсчета всех оснований эталонного генома, который включен как в опухоль, так и, по меньшей мере, в один из числа указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека, применяется автоматизированная система подсчета всех оснований;
В одном воплощении в указанной стадии определения общего покрытия при сравнении «сам против себя» путем подсчета всех оснований эталонного генома, которые перекрываются в обоих из числа указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека, применяется указанная автоматизированная система.
В одном воплощении в указанной стадии деления указанного общего покрытия при сравнении с опухолью на указанное общее покрытие при сравнении «сам против себя» с получением нормализации, применяется указанная автоматизированная система.
В одном воплощении указанная стадия определения частоты ложноположительных результатов путем деления 1) количества однонуклеотидных вариаций с баллом качества выше, чем Q при сравнении указанного первого образца генетического материала из животного или человека и указанного второго образца генетического материала из животного или человека, на 2) количество однонуклеотидных вариаций с баллом качества выше чем Q в сравнении указанного первого образца генетического материала из указанных опухолевых клеток и указанного второго образца генетического материала из указанных опухолевых клеток и путем 3) умножения полученного результата на указанную нормализацию.
Другой аспект изобретения относится к способу определения расчетной кривой соотношений правильного и ложного обнаружения сигналов (ROC), причем указанный способ включает:
получение набора данных мутаций, причем каждая мутация ассоциирована с частотой ложноположительных результатов (FDR); и
для каждой мутации:
определение частоты истинно положительных результатов (TPR) путем вычитания указанного FDR из единицы; и
определение частоты ложноположительных результатов (FPR) путем установления указанного FPR равным указанному FDR; и
получение расчетной ROC путем построения, для каждой мутации, точки при суммарных значениях TPR и FPR до указанной мутации, деленных на сумму всех значений TPR и FPR.
Другие особенности и преимущества настоящего изобретения будут видны из следующего детального описания и из приложенной формулы изобретения.
Подробное описание изобретения
Хотя настоящее изобретение будет описано подробно ниже, следует принять к сведению, что это изобретение не ограничивается описанными здесь конкретными методологией, протоколами и реактивами, поскольку они могут меняться. Также следует принять к сведению, что использованная здесь терминология необходима только для целей описания конкретных воплощений, и не предназначена для ограничения объема настоящего изобретения, который будет ограничен только прилагаемой формулой изобретения. Если иное не указано, все технические и научные термины, использованные здесь, имеют те же смыслы, которые вкладываются в них обычными специалистами в данной области.
Далее будут описаны элементы настоящего изобретения. Эти элементы перечислены с конкретными воплощениями, однако следует понимать, что они могут объединяться любым способом и в любом количестве с созданием дополнительных воплощений. По-разному описанные примеры и предпочтительные воплощения не следует рассматривать как ограничения настоящего изобретения, они предназначены исключительно для ясного описания воплощений. Следует понимать, что описание поддерживает и охватывает воплощения, которые объединяют ясно описанные воплощения с любым количеством раскрытых и/или предпочтительных воплощений. Кроме того, любые перестановки и комбинации всех описанных элементов в данной заявке следует рассматривать как раскрытые описанием настоящей заявки до тех пор, пока из контекста не следует иное. Например, если в предпочтительном воплощении РНК включает поли(А)-хвост, состоящий из 120 нуклеотидов, а в другом предпочтительном воплощении молекула РНК включает аналог 5'-кэпа, то в предпочтительном воплощении РНК включает поли(А)-хвост, состоящий из 120 нуклеотидов и аналог 5'-кэпа.
Предпочтительно, термины, используемые в данном документе, определяются согласно описанию в «A multilingual glossary of biotechnological terms: (IUPAC Recommendations)», H.G.W. Leuenberger, B. Nagel, and H. Kolbl, Eds., (1995) Helvetica Chimica Acta, CH-4010 Basel, Switzerland.
При осуществлении настоящего изобретения будут использоваться, если не указано иное, обычные методы биохимии, клеточной биологии, иммунологии и технологии рекомбинантных ДНК, которые объяснены в публикациях, принадлежащих к данной области техники (см., например, Molecular Cloning: A Laboratory Manual, 2nd Edition, J. Sambrook et al. eds., Cold Spring Harbor Laboratory Press, Cold Spring Harbor 1989).
Во всем этом описании и в формуле изобретения ниже, если контекст не требует иного, слово «включает» и вариации, такие как «содержит» или «содержащий» будут подразумевать включение заявленных установленных компонентов, чисел или стадий, или группы компонентов, чисел или стадий, но не исключение любых других компонентов, чисел или стадий, или группы компонентов, чисел или стадий, хотя в некоторых воплощениях такие другие компоненты, числа или стадии могут быть исключены, т.е. объект изобретения состоят из включения установленных компонентов, чисел или стадий, или группы компонентов, чисел или стадий. Подразумевается, что термины в единственном числе в контексте описания изобретения (и особенно в контексте формулы изобретения) охватывают как форму единственного числа, так и множественного числа до тех пор, пока в данном документе не указано иное или до тех пор пока нет очевидного противоречия контексту. Перечисление интервалов в данном документе предназначено всего лишь для того, чтобы служить в качестве метода сокращения индивидуального обозначения для каждого отдельного значения, попадающего в интервал. До тех пор, пока в данном документе не указано иное, каждое индивидуальное значение включено в описание, как если бы оно было индивидуально перечислено в данном документе.
Все способы, описанные в данном документе, могут осуществляться в любом подходящем порядке, если в данном документе не указано иное или если иное очевидно не следует из контекста. Использование любого из примеров и всех примеров, или типичного выражения (например, «такой как»), представленное в данном документе, предназначено всего лишь для лучшей иллюстрации изобретения и не ограничивает объем изобретения, заявленного иным способом. Никакая формулировка в описании не должна толковаться в качестве обозначения любого не заявленного элемента, существенного для практического применения изобретения.
Несколько документов цитируются по всему тексту данного описания. Каждый из документов, процитированных в данном документе (включая все патенты, патентные заявки, научные публикации, описания производителя, инструкции, и т.д.), процитированные либо выше, либо ниже по тексту, включены таким образом ссылкой в полном объеме. Ничто в данном документе не должно быть истолковано как признание того, что изобретение не имеет право на изменение даты на более раннюю дату по отношению к противопоставленному документу вследствие более раннего создания настоящего изобретения (antedate).
Вакцина, предлагаемая согласно изобретению, представляет собой рекомбинантную вакцину.
Термин «рекомбинантный» в контексте настоящего изобретения означает «сделанный с применением генетической инженерии». Предпочтительно, «рекомбинантный компонент», такой как рекомбинантный полипептид в контексте настоящего изобретения не является природным и предпочтительно является результатом комбинации компонентов, таких как аминокислотные последовательности и последовательности нуклеиновой кислоты, которые не объединены в природе. Например, рекомбинантный полипептид в контексте настоящего изобретения может содержать несколько аминокислотных последовательностей, таких как неоэпитопы или вакцинные последовательности, выделенные из различных белков или различных частей одного белка, сшитые вместе, например, посредством пептидных связей или подходящих линкеров.
Термин «природный» при использовании в данном документе обозначает тот факт, что объект может быть обнаружен в естественной среде. Например, пептид или нуклеиновая кислота, которые присутствуют в организме (включая вирусы) и могут быть выделены из источника в естественной среде и которые не были специально модифицированы человеком в лаборатории, являются природными.
Согласно изобретению термин «вакцина» относится к фармацевтическому препарату (фармацевтической композиции) или к продукту, который при введении индуцирует иммунный ответ, конкретно клеточный иммунный ответ, который распознает и атакует патоген или больную клетку, такую как раковая клетка. Вакцина может использоваться для предотвращения или лечения заболевания. Термин «индивидуальная противоопухолевая вакцина» касается конкретного онкопациента и означает, что противоопухолевая вакцина адаптирована для нужд или особых обстоятельств индивидуального онкопациента.
Термин «иммунный ответ» относится к интегрированному ответу организма на антиген и предпочтительно относится к клеточному иммунному ответу или к клеточному, а также к гуморальному иммунному ответу. Иммунный ответ может быть защитным/превентивным/профилактическим и/или терапевтическим.
«Индукция иммунного ответа» может означать, что перед индукцией не было никакого иммунного ответа против конкретного антигена, но это также может означать, что перед индукцией имелся определенный уровень иммунного ответа против конкретного антигена, и после индукции указанного иммунного ответа он усилился. Таким образом, «индукция иммунного ответа» также включает «усиление иммунного ответа». Предпочтительно, после индукции иммунного ответа у объекта указанный объект защищен от развития заболевания, такого как злокачественное новообразование, или путем индукции иммунного ответа улучшается патологическое состояние. Например, иммунный ответ против антигена, экспрессируемого опухолью, может быть индуцирован у пациента, имеющего злокачественное новообразование, или у объекта с риском развития злокачественного новообразования. Индукция иммунного ответа в данном случае может означать, что патологическое состояние объекта улучшается, что у объекта не разовьются метастазы, или что у объекта с риском развития злокачественного новообразования не разовьется злокачественное новообразование.
Подразумевается, что «клеточный иммунный ответ», «клеточный ответ», «клеточный ответ против антигена» или аналогичный термин включает клеточный ответ, направленный на клетки, отличающиеся презентацией антигена с помощью МНС класса I и класса II. Клеточный ответ относится к клеткам, называемым Т-клетки или Т-лимфоциты, которые действуют либо как «хэлперы», либо как «киллеры». Хэлперные Т-клетки (также обозначаемые CD4+ Т-клетки) играют центральную роль путем регуляции иммунного ответа, и киллерные клетки (также называемые цитотоксическими Т-клетками, цитолитическими Т-клетками, CD8+ Т-клетками или CTL) убивают больные клетки, такие как опухолевые клетки, предотвращая продуцирование большего количества больных клеток. В предпочтительных воплощениях настоящее изобретение включает стимуляцию противоопухолевого CTL-ответа против опухолевых клеток, экспрессирующих один или несколько антигенов, экспрессированных опухолью, и предпочтительно, презентирующих такие антигены, экспрессированные опухолью, с помощью МНС класса I.
«Антиген» согласно изобретению охватывает любое вещество, которое будет вызывать иммунный ответ. Конкретно, «антиген» относится к любому веществу, предпочтительно, к пептиду или белку, который специфично реагирует с антителами или Т-лимфоцитами (Т-клетками). Согласно настоящему изобретению термин «антиген» включает любую молекулу, которая включает, по меньшей мере, один эпитоп. Предпочтительно, антиген в контексте настоящего изобретения представляет собой молекулу, которая необязательно после процессирования индуцирует иммунную реакцию, которая предпочтительно специфична по отношению к антигену (включая клетки, экспрессирующие антиген). Согласно настоящему изобретению может использоваться любой подходящий антиген, который является кандидатом для иммунной реакции, где иммунная реакция предпочтительно представляет собой клеточную иммунную реакцию. В контексте воплощений настоящего изобретения антиген предпочтительно презентируется клеткой, предпочтительно антигенпрезентирующей клеткой, которая включает больную клетку, конкретно опухолевую клетку, в контексте молекул МНС, что приводит в результате к иммунной реакции против антигена. Антиген предпочтительно представляет собой продукт, который соответствует природному антигену или выделен из него. Такие природные антигены включают опухолевые антигены.
В предпочтительном воплощении антиген представляет собой опухолевый антиген, т.е. часть опухолевой клетки, такую как белок или пептид, экспрессированный в опухолевой клетке, который может быть выделен из цитоплазмы, с клеточной поверхности или из ядра клетки, в частности, который изначально присутствует во внутриклеточном пространстве ил в виде поверхностных антигенов опухолевых клеток. Например, опухолевые антигены включают карциноэмбриональный антиген, α1-фетопротеин, изоферритин и фетальный сульфогликопротеин, α2-Н-ферропротеин и γ-фетопротеин. Согласно настоящему изобретению опухолевый антиген предпочтительно включает любой антиген, который экспрессируется в определенном типе опухоли и необязательно является характерной чертой по отношению к типу и/или уровню экспрессии для опухолей или злокачественных образований, а также для опухолевых или раковых клеток. В одном воплощении термин «опухолевый антиген» или «опухолеспецифичный антиген» относится к белкам, которые при нормальных условиях специфично экспрессируются в ограниченном количестве тканей и/или органов или на определенных стадиях развития, например, опухолевый антиген может в нормальных условиях специфично экспрессироваться в ткани желудка, предпочтительно, в слизистой желудка, в репродуктивных органах, например, в яичках, в трофобластной ткани, например, в плаценте, или в зародышевых клеточных линиях, и они экспрессируются или аберрантно экспрессируются в одной или в нескольких опухолевых или раковых тканях. В данном контексте, «ограниченное количество» предпочтительно означает не более чем 3, более предпочтительно не более чем 2. Опухолевые антигены в контексте настоящего изобретения включают, например, дифференцировочные антигены, предпочтительно, специфичные для клеточного типа дифференцировочные антигены, т.е. белки, которые при нормальных условиях специфично экспрессируются в определенном клеточном типе на определенной стадии дифференцировки, антигены злокачественного новообразования/яичка, т.е., белки, которые при нормальных условиях специфично экспрессируются в яичке и иногда в плаценте, и антигены, специфичные для зародышевой клеточной линии. Предпочтительно, опухолевый антиген или аберрантная экспрессия опухолевого антигена идентифицирует раковые клетки. В контексте настоящего изобретения опухолевый антиген, который экспрессируется раковой клеткой объекта, например, пациента, страдающего онкологическим заболеванием, предпочтительно представляет собой собственный белок у указанного объекта. В предпочтительных воплощениях опухолевый антиген в контексте настоящего изобретения специфично экспрессируется при нормальных условиях в ткани или в органе, которые не является существенным, т.е. ткань или органы, которые при разрушении с помощью иммунной системы не приводят к смерти объекта, или в органах или структурах организма, которые не доступны или труднодоступны для иммунной системы.
Согласно изобретению термины «опухолевый антиген», «антиген, экспрессированный опухолью», «антиген злокачественного новообразования» и «антиген, экспрессированный клетками злокачественного новообразования» эквивалентны и используются в данном документе взаимозаменяемо.
Термин «иммуногенность» относится к относительной эффективности антигена для индукции иммунной реакции.
«Антигенный пептид» согласно изобретению предпочтительно относится к части или к фрагменту антигена, который способен стимулировать иммунный ответ, предпочтительно, клеточный ответ против антигена или клеток, характеризующихся экспрессией антигена, предпочтительно, с помощью презентации антигена, таких как больные клетки, конкретно, раковые клетки. Предпочтительно, антигенный пептид способен стимулировать клеточный иммунитет против клетки, характеризующейся презентацией антигена с помощью МНС класса I и предпочтительно способен стимулировать антигенреспонсивные цитотоксические Т-лимфоциты (CTL). Предпочтительно, антигенные пептиды согласно изобретению представляют собой пептиды, которые презентируются МНС класса I и/или класса II, или которые могут процессироваться с получением пептидов, презентируемых МНС класса I и/или класса II. Предпочтительно, антигенные пептиды включают аминокислотную последовательность, по существу соответствующую аминокислотной последовательности фрагмента антигена. Предпочтительно, указанный фрагмент антигена представляет собой пептид, презентированный МНС класса I и/или класса II. Предпочтительно, антигенный пептид согласно изобретению включает аминокислотную последовательность, по существу соответствующую аминокислотной последовательности такого фрагмента, и процессируется с получением такого фрагмента, т.е. пептида, презентируемого МНС класса I и/или класса II, который выделен из антигена.
Если пептид будет презентироваться непосредственно, т.е без процессирования, в частности, без расщепления, то он будет иметь длину, которая подходит для связывания с молекулой МНС, конкретно с молекулой МНС класса I, и предпочтительно его длина составляет 7-20 аминокислот, более предпочтительно, 7-12 аминокислот, более предпочтительно, 8-11 аминокислот, конкретно, 9 или 10 аминокислот.
Если пептид является частью более крупного компонента, включающего дополнительные последовательности, например, вакцинной последовательности или полипептида, и презентируется после процессирования, конкретно после расщепления, то пептид, полученный с помощью процессирования, имеет длину, которая подходит для связывания с молекулой МНС, конкретно с молекулой МНС класса I, и предпочтительно, его длина составляет 7-20 аминокислот, более предпочтительно, 7-12 аминокислот, более предпочтительно, 8-11 аминокислот, конкретно, 9 или 10 аминокислот. Предпочтительно, последовательность пептида, который будет презентироваться после процессирования, выделена из аминокислотной последовательности антигена, т.е., его последовательность по существу соответствует и предпочтительно полностью идентична фрагменту антигена. Таким образом, антигенный пептид или вакцинная последовательность согласно изобретению в одном воплощении включает последовательность длиной 7-20 аминокислот, более предпочтительно, 7-12 аминокислот, более предпочтительно, 8-11 аминокислот, конкретно, 9 или 10 аминокислот, которые по существу соответствуют и предпочтительно полностью идентичны фрагменту антигена, а после процессирования антигенного пептида или вакцинной последовательности представляют собой презентированный пептид. Согласно изобретению такой пептид, полученный с помощью процессирования, включает идентифицированное изменение последовательности.
Согласно изобретению антигенный пептид или эпитоп может быть представлен в вакцине в виде части более крупного компонента, такого как вакцинная последовательность и/или полипептид, включающий более чем один антигенный пептид или эпитоп. Присутствующий антигенный пептид или эпитоп получают после соответствующего процессирования.
Пептиды, имеющие аминокислотные последовательности, по существу соответствующие последовательности пептида, которая презентируется молекулой МНС класса I, могут отличаться в одном или в нескольких остатках, которые не существенны для распознавания TCR пептида в качестве презентируемого молекулой МНС класса I, или могут отличаться по связыванию пептида с МНС. Такие по сути соответствующие пептиды также способны стимулировать антиген-респонсивные CTL и могут рассматриваться как иммунологически эквивалентные. Пептиды, имеющие аминокислотные последовательности, отличающиеся от презентируемых пептидов в остатке, который не влияет на распознавание TCR, но улучшает стабильность связывания с МНС, могут улучшить иммуногенность антигенного пептида и могут обозначаться в данном документе как «оптимизированные пептиды». С использованием имеющихся знания по поводу того, какой из этих остатков наиболее вероятно может влиять на связывание либо с МНС, либо с TCR, можно применять рациональный способ дизайна по сути подходящих пептидов. Полученные в результате функциональные пептиды, рассматриваются как антигенные пептиды.
Антигенный пептид при презентации в МНС должен распознаваться Т-клеточным рецептором. Предпочтительно, антигенный пептид, если он распознается Т-клеточным рецептором, способен индуцировать в присутствии соответствующих ко-стимулирующих сигналов клональную экспансию Т-клетки, несущей Т-клеточный рецептор, специфично распознающий антигенный пептид. Предпочтительно, антигенные пептиды, в частности если они презентируются в контексте молекул МНС, способны стимулировать иммунный ответ, предпочтительно клеточный ответ против антигена, от которого они произошли, или против клеток, характеризующихся экспрессией антигена, и, предпочтительно, характеризующихся презентацией антигена. Предпочтительно, антигенный пептид способен стимулировать клеточный иммунитет против клетки, характеризующейся презентацией антигена с помощью МНС класса I и предпочтительно способной стимулировать антиген-респонсивные CTL. Такая клетка предпочтительно является клеткой-мишенью.
«Процессирование антигена» или «процессирование» относится к деградации полипептида или антигена в продукты процессирования, которые представляют собой фрагменты указанного полипептида или антигена (например, деградация полипептида в пептиды) и к ассоциации одного или нескольких из таких фрагментов (например, посредством связывания) с молекулами МНС для презентации с помощью клеток, предпочтительно, антигенпрезентирующих клеток, специфическим Т-клеткам.
«Антигенпрезентирующие клетки» (англ. Antigen presenting cells, APC) представляют собой клетки, которые презентируют пептидные фрагменты белковых антигенов в ассоциации с молекулами МНС на своей клеточной поверхности. Некоторые APC могут активировать антиген-специфичные Т-клетки.
Специализированные антигенпрезентирующие клетки очень эффективны в интернализации антигена или посредством фагоцитоза, или посредством рецептор-опосредованного эндоцитоза, и последующем представлении фрагмента антигена, связанного с молекулой MHC класса II, на своей мембране. Т-клетка распознает и взаимодействует с комплексом антиген-молекула МНС класса II на мембране антигенпрезентирующей клетки. Затем антигенпрезентирующей клеткой вырабатывается дополнительный ко-стимулирующий сигнал, приводя к активации Т-клетки. Экспрессия ко-стимулирующих молекул представляет собой характерную черту специализированных антигенпрезентирующих клеток.
Основные типы специализированных антигенпрезентирующих клеток представляют собой дендритные клетки, которые обладают широким спектром антигенной презентации, и вероятно являются наиболее важными антигенпрезентирующими клетками, макрофаги, В-клетки и определенные активированные эпителиальные клетки.
Дендритные клетки (англ. Dendritic cells, DC) представляют собой популяцию лейкоцитов, которые презентируют захваченные в периферических тканях антигены Т-клеткам посредством обоих антигенпрезентирующих путей, с помощью молекул МНС класса II и I. Хорошо известно, что дендритные клетки являются мощными индукторами иммунного ответа, а активация этих клеток является критической стадией индукции противоопухолевого иммунитета.
Дендритные клетки стандартно классифицируют как «незрелые» и «зрелые» клетки, что может использоваться в качестве простого пути различения между двумя хорошо охарактеризованными фенотипами. Однако эта номенклатура не должна подразумевать исключения всех возможных промежуточных стадий дифференцировки.
Незрелые дендритные клетки характеризуются как антигенпрезентирующие клетки с высокой способностью поглощения и процессирования антигена, которая коррелирует с высокой экспрессией рецептора Fcγ и маннозного рецептора. Зрелый фенотип, как правило, характеризуется более низкой экспрессией этих маркеров, но при этом высокой экспрессией молекул клеточной поверхности, ответственных за активацию Т-клеток, таких как молекулы МНС класса I и II, молекул адгезии (например, CD54 и CD11) и ко-стимулирующих молекул (например, CD40, CD80, CD86 и 4-1 ВВ).
Созревание дендритных клеток обозначается как статус активации дендритной клетки, при котором такие антигенпрезентирующие дендритные клетки приводят к стимуляции Т-клеток, в то время как презентация незрелыми дендритными клетками приводит к устойчивости. Созревание дендритных клеток прежде всего вызвано биомолекулами с микробными свойствами, детектируемыми с помощью рецепторов врожденного иммунитета (бактериальная ДНК, вирусная РНК, эндотоксин и т.д.), про-воспалительных цитокинов (TNF, IL-1, IFN), лигированием CD40 на поверхности дендритных клеток с помощью CD40L, и веществами, высвобождаемыми из клеток в состоянии клеточной смерти под действием стресса. Дендритные клетки могут быть получены культивированием клеток костного мозга in vitro с использованием цитокинов, таких как гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) и фактор некроза опухоли альфа.
Неспециализированные антигенпрезентирующие клетки не экспрессируют конститутивно белки МНС класса II, требуемые для взаимодействия с нативными Т-клетками; они экспрессируются только при стимуляции неспециализированных антигенпрезентирующих клеток с помощью цитокинов, таких как IFNy.
«Антигенпрезентирующие клетки» могут быть нагружены пептидами, презентируемыми молекулами МНС класса I путем трансдукции клеток с использованием нуклеиновой кислоты, предпочтительно РНК, кодирующей пептид или полипептид, включающий презентируемый пептид, например, нуклеиновой кислотой, кодирующей антиген.
В некоторых воплощениях фармацевтическая композиция по изобретению, включающая носитель для доставки гена, целью которого являются дендритные или другие антигенпрезентирующие клетки, может вводиться пациенту, приводя в результате к трансфекции, которая осуществляется in vivo. In vivo трансфекция дендритных клеток может, как правило, осуществляться с использование методов, хорошо известных в данной области, таких как описанные в WO 97/24447, или с помощью метода генной пушки, описанного в Mahvi et al., Immunology and cell Biology 75:456-460, 1997.
Согласно изобретению термин «антигенпрезентирующая клетка» также включает клетки-мишени.
«Клетка-мишень» будет обозначать клетку, которая является мишенью для иммунного ответа, такого как клеточный иммунный ответ. Клетки-мишени включают клетки, которые презентируют антиген или антигенный эпитоп, т.е. пептидный фрагмент, выделенный из антигена, и включают любую нежелательную клетку, такую как раковая клетка. В предпочтительных воплощениях клетка-мишень представляет собой клетку, экспрессирующую антиген, как описано в данном документе, и предпочтительно, презентирующая указанный антиген с помощью молекулы МНС класса I.
Термин «эпитоп» относится к антигенной детерминанте в молекуле, такой как антиген, т.е., к части в нем или к фрагменту молекулы, которая распознается иммунной системой, например, которая распознается Т-клеткой, конкретно при презентации в контексте молекул МНС. Эпитоп белка, такого как опухолевый антиген предпочтительно включает непрерывный или прерывистый участок указанного белка и предпочтительно имеет длину 5-100 аминокислот, предпочтительно 5-50, более предпочтительно 8-30, наиболее предпочтительно 10-25 аминокислот, например, эпитоп может иметь длину предпочтительно 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, или 25 аминокислот. Особенно предпочтительно, если эпитоп в контексте настоящего изобретения представляет собой Т-клеточный эпитоп.
Согласно изобретению эпитоп может связываться с молекулами МНС, такими как молекулы МНС на поверхности клетки и таким образом может представлять собой «МНС-связывающий пептид» или «антигенный пептид». Термин «МНС-связывающий пептид» относится к пептиду, который связывается с молекулой МНС класса I и/или с молекулой МНС класса II. В случае комплексов молекулы МНС класса I/пептид связывающие пептиды, как правило, имеют длину 8-10 аминокислот, хотя могут быть эффективными и более длинные или более короткие пептиды. В случае комплексов молекулы МНС класса II/пептид связывающие пептиды, как правило, имеют длину 10-25 аминокислот, а конкретнее, 13-18 аминокислот, хотя могут быть эффективными и более длинные или более короткие пептиды.
Термины «эпитоп», «антигенный пептид», «антигенный эпитоп», «иммуногенный пептид» и «МНС-связывающий пептид» используются в данном документе взаимозаменяемо и предпочтительно относятся к неполному представлению антигена, который предпочтительно способен вызывать иммунный ответ против антигена или клетки, экспрессирующей или включающей предпочтительно презентированный антиген. Предпочтительно, термины относятся к иммуногенному участку антигена. Предпочтительно, этот участок антигена, которая распознается (т.е., специфично связывается) Т-клеточным рецептором, конкретно если он презентирован в контексте молекул МНС. Такие предпочтительные иммуногенные участки связываются с молекулой МНС класса I или класса II. При использовании в данном документе говорят, что иммуногенный участок «связывается с» молекулой МНС класса I или класса II, если такое связывание детектируется с использованием анализа, известного в данной области.
При использовании в данном документе термин «неоэпитоп» относится к эпитопу, который отсутствует в эталоне, таком как нормальная неопухолевая или клетка зародышевой линии, но который обнаруживается в раковых клетках. В частности к этому относятся ситуации, когда в нормальной не опухолевой или в зародышевой клетке обнаруживается соответствующий эпитоп, однако благодаря одной или нескольким мутациям в опухолевой клетке последовательность эпитопа изменяется, приводя в результате к появлению неоэпитопа.
Термин «участок» относится к части. По отношению к конкретной структуре, такой как аминокислотная последовательность или белок, термин его «участок» может означать непрерывную или дискретную часть указанной структуры. Предпочтительно, участок аминокислотной последовательности включает, по меньшей мере, 1%, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 20%, по меньшей мере, 30%, предпочтительно по меньшей мере, 40%, предпочтительно по меньшей мере, 50%, более предпочтительно по меньшей мере, 60%, более предпочтительно по меньшей мере, 70%, еще более предпочтительно по меньшей мере, 80%, и наиболее предпочтительно по меньшей мере, 90% аминокислот указанной аминокислотной последовательности. Предпочтительно, если участок представляет собой дискретную часть, то указанная дискретная часть состоит из 2, 3, 4, 5, 6, 7, 8, или более частей структуры, причем каждая часть представляет собой непрерывный элемент структуры. Например, дискретная часть аминокислотной последовательности может состоять из 2, 3, 4, 5, 6, 7, 8, или более, предпочтительно, не более чем из 4 частей указанной аминокислотной последовательности, где каждая часть предпочтительно включает, по меньшей мере, 5 непрерывно расположенных аминокислот, по меньшей мере, 10 непрерывно расположенных аминокислот, предпочтительно по меньшей мере, 20 непрерывно расположенных аминокислот, предпочтительно, по меньшей мере, 5 непрерывно расположенных аминокислот из аминокислотной последовательности.
Термины «часть» и «фрагмент» используются в данном документе взаимозаменяемо и относятся к непрерывному элементу. Например, часть структуры, такой как аминокислотная последовательность или белок, относится к непрерывному элементу указанной структуры. Участок, часть или фрагмент структуры предпочтительно включает одно или несколько функциональных свойств указанной структуры. Например, участок, часть или фрагмент эпитопа, пептида или белка предпочтительно является иммунологически эквивалентным эпитопу, пептиду или белку, из которого он выделен. В контексте настоящего изобретения «часть» структуры, такой как аминокислотная последовательность, предпочтительно включает, предпочтительно состоит из, по меньшей мере, 10%, по меньшей мере, 20%, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%, по меньшей мере, 80%, по меньшей мере, 85%, по меньшей мере, 90%, по меньшей мере, 92%, по меньшей мере, 94%, по меньшей мере, 96%, по меньшей мере, 98%, по меньшей мере, 99% цельной структуры аминокислотной последовательности.
Термин «иммунореактивная клетка» в контексте настоящего изобретения относится к клетке, которая оказывает эффекторные функции в процессе иммунной реакции. «Иммуннореактивная клетка» предпочтительно способна связываться с антигеном или с клеткой, характеризующейся презентацией антигена или антигенного пептида, выделенного из антигена, и опосредует иммунный ответ. Например, такие клетки секретируют цитокины и/или хемокины, секретируют антитела, распознают опухолевые клетки и необязательно уничтожают такие клетки. Например, иммунореактивные клетки включают Т-клетки (цитотоксические Т-клетки, хэлперные Т-клетки, опухолепроникающие Т-клетки), В-клетки, натуральные киллеры, нейтрофилы, макрофаги и дендритные клетки. Предпочтительно, в контексте настоящего изобретения «иммунореактивные клетки» представляют собой Т-клетки, предпочтительно CD4+ и/или CD8+ Т-клетки.
Предпочтительно, «иммунореактивная клетка» распознает антиген или антигенный пептид, выделенный из антигена с некоторой степенью специфичности, конкретно, если он презентируется в контексте молекул МНС, как например на поверхности антигенпрезентирующих клеток или больных клеток, таких как опухолевые клетки. Предпочтительно, указанное распознавание дает возможность клетке, которая распознает антиген или антигенный пептид, выделенный из указанного антигена, быть респонсивной или реактивной. Если клетка представляет собой хэлперную Т-клетку (CD4+ Т-клетка), несущую рецепторы, которые распознают антиген или антигенный пептид, выделенный из антигена в контексте молекул МНС класса II, такая респонсивность или реактивность может включать высвобождение цитокинов и/или активацию CD8+ лимфоцитов (CTL) и/или В-клеток. Если клетка представляет собой CTL, то такая респонсивность или реактивность может включать уничтожение клеток, представленных в контексте молекул МНС класса I, т.е., клеток, отличающихся презентацией антигена с использованием молекул МНС класса I, например, посредством апоптоза или перфорин-опосредованного клеточного лизиса. Согласно изобретению респонсивность CTL может включать длительное выделение кальция, клеточное деление, продуцирование цитокинов, таких как IFN-γ и TNF-α, положительную регуляцию активации маркеров, таких как CD44 и CD69, и специфичное цитолитическое уничтожение антиген-экспрессирующих клеток-мишеней. Респонсивность CTL также может определяться с использованием искусственного репортера, который точно выявляет респонсивность CTL. Такие CTL, которые распознают антиген или антигенный пептид, выделенный из антигена, и являются респонсивными или реактивными, также обозначаются в данном документе как «антиген-респонсивные CTL». Если клетка представляет собой В-клетку, то такая респонсивность может включать высвобождение иммуноглобулинов.
Термины «Т-клетка» и «Т-лимфоцит» используются взаимозаменяемо в данном документе и включают Т-хэлперные клетки (CD4+ Т-клетки) и цитотоксические Т-клетки (CTL, CD8+ Т-клетки), которые включают цитолитические Т-клетки.
Т-клетки принадлежат к группе белых кровяных клеток, известных как лимфоциты, и играют центральную роль в клеточно-опосредованном иммунитете. Они могут отличаться от другого типа лимфоцитов, таких как В-клетки и природные киллерные клетки присутствием специального рецептора на их клеточной поверхности, называемого Т-клеточный рецептор (TCR). Тимус является принципиальным органом, отвечающим за созревание Т-клеток. Было обнаружено несколько различных подгрупп Т-клеток, каждая с различной функцией.
Т-хэлперные клетки помогают другим белым кровяным клеткам в иммунологических процессах, включая созревание В-клеток в плазматические клетки и активацию цитотоксических Т-клеток и макрофагов. Эти клетки также известны как CD4+ Т-клетки, так как они экспрессируют белок CD4 на своей поверхности. Хэлперные Т-клетки становятся активированными, когда им презентируются пептидные антигены молекулами МНС класса II, которые экспрессируются на поверхности антигенпрезентирующих клеток (АРС). После активации они быстро делятся и секретируют небольшие белки, называемые цитокинами, которые регулируют или способствуют активному иммунному ответу.
Цитотоксические Т-клетки разрушают инфицированные вирусами клетки и опухолевые клетки, а также вовлечены в отторжение трансплантата. Эти клетки также известны как CD8+ Т-клетки, так как они экспрессируют гликопротеин CD8 на своей поверхности. Эти клетки распознают свои мишени путем связывания с антигеном, ассоциированным с молекулой МНС класса I, которая присутствует на поверхности почти каждой клетки организма.
Большинство Т-клеток имеют Т-клеточный рецептор (TCR), присутствующий в комплексе некоторых белков. Фактически Т-клеточный рецептор состоит из двух отдельных пептидных цепей, которые образуются из независимых генов Т-клеточного рецептора альфа и бета (TCRα и TCRβ) и называются α- и β-TCR-цепями. γδ Т-клетки (гамма дельта Т-клетки) характеризуют небольшую подгруппу Т-клеток, на своей поверхности которых находятся отличающиеся Т-клеточные рецепторы (TCR). При этом в γδ Т-клетках TCR образуется из одной γ-цепи и одной δ-цепи. Эта группа Т-клеток обычно гораздо меньше (2% от суммарных Т-клеток), чем αβ Т-клетки.
Первый сигнал в активации Т-клеток обеспечивается путем связывания Т-клеточного рецептора с коротким пептидом, презентированным главным комплексом гистосовместимости (МНС) на другой клетке. Такое связывание гарантирует, что активируется только Т-клетка с TCR, специфичным к данному пептиду. Клетка-партнер обычно представляет собой специализированную антигенпрезентирующую клетку (АРС), как правило, дендритную клетку в случае наивного ответа, хотя В-клетки и макрофаги могут быть важными АРС. Пептиды, презентированные CD8+ Т-клеткам с помощью молекул МНС класса I, как правило, имеют длину 8-10 аминокислот; пептиды, презентированные CD4+ Т-клеткам с помощью молекул МНС класса II, как правило, длиннее, поскольку концы связывающей бороздки молекулы МНС класса II открыты.
Согласно настоящему изобретению Т-клеточный рецептор способен связываться с определенной мишенью, если он имеет достаточную аффинность к указанной определенной мишени, и связывается с указанной определенной мишенью в стандартных анализах. «Аффинность» или «аффинность связывания» часто измеряют с помощью равновесной константы диссоциации (KD). Т-клеточный рецептор не способен (по существу) к связыванию с мишенью, если он не обладает достаточной аффинностью к указанной мишени, и не связывается существенно с указанной мишенью в стандартных анализах.
Т-клеточный рецептор предпочтительно способен к специфичному связыванию с определенной мишенью. Т-клеточный рецептор специфичен к определенной мишени, если он способен связываться с указанной определенной мишенью, в то время как он (по существу) не способен связываться с другими мишенями, т.е. не обладает достаточной аффинностью к другим мишеням и не связывается в значительной степени с другими мишенями в стандартных анализах.
Образование цитотоксических Т-лимфоцитов может быть вызвано in vivo включением антигена или антигенного пептида в антигенпрезентирующие клетки in vivo. Антиген или антигенный пептид могут быть представлены как в виде белка, так и в виде ДНК (например, внутри вектора) или в виде РНК. Антиген может процессироваться с получением пептида-партнера для молекулы МНС, в то время как его фрагмент может презентироваться без необходимости дополнительного процессирования. Последнее происходит, в частности, если они связываются с молекулами МНС. В целом, возможно введение пациенту с помощью внутрикожной инъекции. Однако инъекция может также проводиться по внутриузловому пути в лимфатический узел (Maloy et al. (2001), Proc Natl Acad Sci USA 98:3299-303). Полученные в результате клетки презентируют представляющий интерес комплекс и распознаются аутологичными цитотоксическими Т-лимфоцитами, которые затем размножаются.
Специфичная активация CD4+ или CD8+ Т-клеток может детектироваться различными путями. Методы детекции специфичной Т-клеточной активации включают детекцию пролиферации Т-клеток, выработки цитокинов (например, лимфокинов), или генерацию цитотоксической активности. Для CD4+ Т-клеток предпочтительным методом детекции специфичной Т-клеточной активации является детекция пролиферации Т-клеток. Для CD8+ Т-клеток предпочтительным методом детекции специфичной Т-клеточной активации является детекция генерации цитотоксической активности.
Термин «главный комплекс гистосовместимости» и аббревиатура «МНС» включает молекулы МНС класса I и МНС класса II и относится к комплексу генов, которые присутствуют у всех позвоночных. Белки или молекулы МНС важны для передачи сигналов между лимфоцитами и антигенпрезентирующими клетками или больными клетками в иммунных реакциях, где белки и молекулы МНС связываются с пептидами и презентируют их для распознавания Т-клеточными рецепторами. Белки, кодируемые МНС, экспрессируются на поверхности клеток и представляют Т-клеткам как свои собственные антигены (пептидные фрагменты из самой клетки), так и чужеродные антигены (например, фрагменты проникших микроорганизмов).
Область МНС делится на три подгруппы, класс I, класс II, и класс III. Белки МНС класса I содержат α-цепь и β2-микроглобулин (не является частью МНС, кодируемого хромосомой 15). Они презентируют антигенные фрагменты цитотоксическим Т-клеткам. На большинстве клеток иммунной системы, особенно на антигенпрезентирующих клетках, белки МНС класса II содержат α- и β-цепи, и они презентируют антигенные фрагменты Т-хэлперным клеткам. Область МНС класса III кодирует другие иммунные компоненты, такие как компоненты комплемента и некоторые компоненты, которые кодируют цитокины.
У человека гены в области МНС, которые кодируют антигенпрезентирующие белки на клеточной поверхности, относятся к генам человеческого лейкоцитарного антигена (HLA). Однако аббревиатура МНС часто используется для обозначения продуктов генов HLA. Гены HLA включают девять так называемых классических генов МНС: HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, и HLA-DRB1.
В одном предпочтительном воплощении всех аспектов изобретения молекула МНС представляет собой молекулу HLA.
Под «клеткой, характеризующейся презентацией антигена» или «клеткой, презентирующей антиген» или под аналогичными выражениями понимают клетку, такую как больная клетка, например, раковая клетка, или антигенпрезентирующая клетка, презентирующую антиген или фрагмент, выделенный из указанного антигена, например, путем процессирования антигена в контексте молекул МНС, конкретно молекул МНС класса I. Аналогично, термин «заболевание, характеризующееся презентацией антигена» обозначает заболевание, в которое вовлечены клетки, характеризующиеся презентацией антигена, в частности, с помощью молекул МНС класса I. На презентацию антигена клеткой может влиять трансфекция клетки нуклеиновой кислотой, такой как РНК, кодирующей антиген.
Под «презентируемым фрагментом антигена» или под аналогичными выражениями понимают, что фрагмент может презентироваться с помощью молекул МНС класса I или класса II, предпочтительно МНС класса I, например, при добавлении непосредственно к антигенпрезентирующим клеткам. В одном воплощении фрагмент представляет собой фрагмент, который в естественной среде презентируется клетками, экспрессирующими антиген.
Термин «иммунологически эквивалентный» означает, что иммунологически эквивалентные молекулы, такие как иммунологически эквивалентные аминокислотные последовательности демонстрируют такие же или по существу такие же иммунологические свойства и/или оказывают такие же или по существу такие же иммунологические эффекты, например, в отношении типа иммунологического эффекта, такого как индуцирование гуморального и/или клеточного иммунного ответа, силы и/или продолжительности индуцированной иммунной реакции или специфичности индуцированной иммунной реакции. В контексте настоящего изобретения термин «иммунологически эквивалентный» предпочтительно используется в отношении иммунологических эффектов или свойств пептида, используемого для иммунизации. Например, аминокислотная последовательность иммунологически эквивалентна эталонной аминокислотной последовательности, если указанная аминокислотная последовательность при экспонировании иммунной системе объекта индуцирует иммунную реакцию, обладающую специфичностью реакции с эталонной аминокислотной последовательностью.
Термин «иммунные эффекторные функции» в контексте настоящего изобретения включает любые функции, опосредованные компонентами иммунной системы, что приводит в результате, например, к уничтожению опухолевых клеток или к ингибированию опухолевого роста и/или к ингибированию развития опухоли, включая ингибирование распространения опухоли и метастазирования. Предпочтительно, иммунные эффекторные функции в контексте настоящего изобретения представляют собой эффекторные функции, опосредованные Т-клетками. Такие функции включают в случае хэлперной Т-клетки (CD4+ Т-клетка) распознавание антигена или антигенного пептида, выделенного из антигена, в контексте молекул МНС класса II с помощью Т-клеточных рецепторов, высвобождение цитокинов и/или активацию CD8+ лимфоцитов (CTL) и/или В-клеток, и в случае CTL распознавание антигена или антигенного пептида, выделенного из антигена, в контексте молекул МНС класса I с помощью Т-клеточных рецепторов, уничтожение клеток, презентированных в контексте молекул МНС класса I, т.е., клеток, характеризующихся презентацией антигена с помощью молекулы МНС класса I, например, посредством апопотоза или перфорин-опосредованного клеточного лизиса, продуцирование цитокинов, таких как IFN-γ и TNF-α, и специфичное цитолитическое уничтожение антиген-экспрессирующих клеток-мишеней.
Термин «геном» относится к суммарному количеству генетической информации в хромосомах организма или клетки. Термин «экзом» относится к кодирующим областям генома. Термин «транскриптом» относится к набору всех молекул РНК.
«Нуклеиновая кислота» согласно изобретению предпочтительно представляет собой дезоксирибонуклеиновую кислоту (ДНК) или рибонуклеиновую кислоту (РНК), более предпочтительно, РНК, наиболее предпочтительно, in vitro транскрибированную РНК (IVT РНК) или синтетическую РНК. Нуклеиновые кислоты включают согласно изобретению геномную ДНК, кДНК, мРНК, рекомбинантно продуцированные и химически синтезированные молекулы. Согласно изобретению нуклеиновая кислота может присутствовать в виде одноцепочечной или двухцепочечной и линейно или ковалентно циклически замкнутой молекулы. Согласно изобретению нуклеиновая кислота может быть изолированной. Термин «изолированная нуклеиновая кислота» означает согласно изобретению, что нуклеиновая кислота (i) была амплифицирована in vitro, например, посредством полимеразной цепной реакции (ПЦР), (ii) была получена рекомбинантно путем клонирования, (iii) была очищена, например, путем расщепления и разделения с помощью гель-электрофореза, или (iv) была синтезирована, например, с помощью химического синтеза. Нуклеиновая кислота может применяться для введения внутрь, т.е. для трансфекции клеток, конкретно, в форме РНК, которая может быть получена путем in vitro транскрипции с ДНК-матрицы. Кроме того, РНК может быть модифицирована перед применением с помощью стабилизирующих последовательностей, кэппирования и полиаденилирования.
Термин «генетический материал» относится к выделенной нуклеиновой кислоте, либо к ДНК, либо к РНК, к участку двойной спирали, к участку хромосомы или к цельному геному организма или клетки, конкретно к экзому или транскриптому.
Термин «мутация» относится к изменению или к различию в последовательности нуклеиновой кислоты (нуклеотидной замене, вставке или делеции) по сравнению с эталоном. «Соматическая мутация» может происходить в любой из клеток организма за исключением зародышевых клеток (спермий и яйцеклетка) и, таким образом, не переходят к потомству. Эти изменения могут (но не всегда) вызывать злокачественное новообразование или другие заболевания. Предпочтительно, мутация представляет собой несинонимичную мутацию. Термин «несинонимичная мутация» относится к мутации, предпочтительно, к нуклеотидной замене, которая приводит в результате к аминокислотной замене, такой как аминокислотная замена в продукте трансляции.
Согласно изобретению термин «мутация» включает точечные мутации, вставки-делеции, сшивки, хромотрипсис и РНК-редактирующие мутации.
Согласно изобретению термин «вставка-делеция» описывает особый класс мутаций, определенный как мутация, приводящая в результате к колокализованным вставке и делеции и к приобретению или к потере нуклеотидов. В кодирующих участках генома, если длина вставки-делеции не кратна 3, они ведут к получению мутации со сдвигом рамки считывания. Вставки-делеции могут противопоставляться точечной мутации; в случае, где вставка-делеция вставляет и удаляет нуклеотиды из последовательности, точечная мутация является формой замены, которая заменяет один нуклеотид на другой.
Сшивки могут приводить к образованию гибридных генов, образованных из двух ранее отдельных генов. Они могут происходить в результате транслокации, интерстициальной делеции, или инверсии сегмента хромосомы. Часто сшитые гены представляют собой онкогены. Онкогенные сшитые гены могут приводить к получению генного продукта с новой или отличной функцией от функции двух партнеров по сшивке. В ином случае, протоонкоген сшивается с сильным промотором и посредством этого включается онкогенная функция путем положительной регуляции, вызванной сильным промотором из 5'-области партнера по сшивке. Получение онкогенных сшитых транскриптов также может быть вызвано транс-сплайсингом или событиями неправильного прочтения.
Согласно изобретению термин «хромотрипсис» относится к генетическому явлению, в ходе которого специфические области генома раскалываются и затем сшиваются вместе в процессе одного разрушительного события.
Согласно изобретению термины «РНК-редакция» или «РНК-редактирование» относятся к молекулярным процессам, в которых информационное содержание в молекуле РНК изменяется посредством химического изменения в составе оснований. РНК-редактирование включает нуклеозидные модификации, такие как дезаминирования цитидина (С) до уридина (U) и аденозина (А) до инозина (I), а также не матричные нуклеотидные добавления и вставки. РНК-редактирование в мРНК изменяет аминокислотную последовательность кодируемого белка, так что он отличается от предсказанного с помощью последовательности геномной ДНК.
Термин «сигнатура мутаций злокачественной опухоли» относится к мутациям, которые присутствуют в раковых клетках по сравнению с неопухолевыми эталонными клетками.
Согласно изобретению, «эталон» может использоваться для корреляции и сравнения результатов, полученных в способах по изобретению из опухолевых образцов. Как правило, «эталон» может быть получен на основе одного или нескольких нормальных образцов, которые не подвержены опухолевому заболеванию, или получены из пациента или из одного или нескольких индивидуумов, предпочтительно, здоровых, в частности, из индивидуумов того же вида. «Эталон» может быть определен эмпирически путем тестирования достаточно большого количества нормальных образцов.
Согласно изобретению может использоваться любой подходящий метод секвенирования, предпочтительными являются технологии Секвенирования Следующего Поколения (NGS). Методы Секвенирования Третьего Поколения могут заменять технологию NGS в будущем для повышения скорости стадии секвенирования метода. С целью прояснения: термины «Секвенирование Следующего Поколения» или «NGS» в контексте настоящего изобретения означают все новые высокопроизводительные технологи секвенирования, которые в отличие от «стандартной» методологии секвенирования, известной как метод Сэнгера, читают матрицы нуклеиновых кислот случайным образом параллельно по всему геному путем разрывов цельного генома на короткие участки. Такие NGS-технологии (также известные как технологии массивно-параллельного секвенирования) могут предоставлять информацию о последовательности нуклеиновых кислот цельного генома, экзома, транскриптома (все транскрибирующиеся последовательности генома) или метилома (все метилированные последовательности генома) в очень короткие сроки, например, в течение 1-2 недель, предпочтительно в течение 1-7 дней или наиболее предпочтительно в течение менее чем 24 часов, и в принципе позволяют осуществлять способы секвенирования отдельной клетки. Множество NGS платформ, которые коммерчески доступны или которые упоминаются в научной литературе, может использоваться в контексте настоящего изобретения, например, те, что описаны в Zhang et al. 2011: The impact of next-generation sequencing on genomics. J. Genet Genomics 38 (3), 95-109; или в Voelkerding et al. 2009: Next generation sequencing: From basic research to diagnostics. Clinical chemistry 55, 641-658. Частными примерами таких NGS-технологий/платформ являются
1) Технология секвенирование - путем-синтеза (sequencing-by-synthesis), известная как пиросеквенирование, выполняемое, например, в «GS-FLX 454 Genome Sequencer™» компании, ассоциированной с «Roche», «454 Life Sciences» (Бренфорд, Коннектикут), впервые описана в Ronaghi et al. 1998: A sequencing method based on real-time pyrophosphate». Science 281 (5375), 363-365. Эта технология использует эмульсионную ПЦР, в которой шарики, связывающие одноцепочечную ДНК, инкапсулируются посредством интенсивного встряхивания в водные мицеллы, содержащие ПЦР-реагенты, окруженные маслом для амплификации с помощью эмульсионной ПЦР. В процессе пиросеквенирования свет, испускаемый из молекул фосфата во время включения нуклеотидов, записывается по мере того, как полимераза синтезирует ДНК-цепь.
2) Способы секвенирование-путем-синтеза, разработанные «Solexa» (теперь часть «Illumina Inc.», Сан Диего, Калифорния), которые базируются на обратимых красителях-терминаторах, и выполняемые, например, в «Illumina/Solexa Genome Analyzer™» и в «Illumina HiSeq 2000 Genome Analyzer™». В данной технологии все четыре нуклеотида добавляются одновременно в кластерные фрагменты с гибридизованными нуклеотидными праймерами в каналы с проточными ячейками, содержащими ДНК-полимеразу. Мостиковая амплификация расширяет кластерные цепи с помощью всех четырех флуоресцентно меченых нуклеотидов для секвенирования.
3) Способы секвенирование-путем-лигирования (Sequencing-by-ligation), например, выполняемые на платформе «SOLid™» «Applied Biosystems» (теперь «Life Technologies Corporation», Карлсбад, Калифорния). В данной технологии пул всех возможных олигонуклеотидов фиксированной длины метят согласно отсеквенированному положению. Олигонуклеотиды отжигаются и лигируются; предпочтительное лигирование с помощью ДНК-лигазы для парных последовательностей приводит в результате к получению сигнала, информативного в отношении нуклеотида в данном положении. Перед секвенированием ДНК амплифицируется с помощью эмульсионной ПЦР. Полученные шарики, каждый из которых содержит только копии одной ДНК-молекулы, располагаются на предметном стекле. В качестве второго примера платформа «Polonator™ G.007» от «Dover Systems» (Салем, Нью-Гемпшир) также применяет способ секвенирование-путем-лигированиея используя случайное расположение, на основе использования шариков, эмульсионной ПЦР для амплификации ДНК-фрагментов для параллельного секвенирования.
4) Технологии секвенирования одиночной молекулы, такие как, например, применяемые в системе «PacBio RS» от «Pacific Biosciences» (Менло-Парк, Калифорния) или в «HeliScope™» на платформе «Helicos Biosciences» (Кембридж, Массачусетс). Отличные характеристики данной технологии заключаются в ее способности секвенировать одиночные молекул ДНК или РНК без амплификации, определенной как секвенирование одиночных молекул ДНК в реальном времени (SMRT). Например, «HeliScope» использует высокочувствительную систему детекции флуоресценции для прямой детекции каждого нуклеотида по мере его синтеза. Аналогичный способ на основе резонансного переноса энергии флуоресценции (FRET) был разработан в «Visigen Biotechnology» (Хьюстон, Техас). Другими технологиями секвенирования одиночных молекул на основе флуоресценции являются «U.S. Genomics» («GeneEngine™») и «Genovoxx» («AnyGene™»).
5) Нанотехнологии для секвенирования одиночных молекул, в которых используются различные наноструктуры, например, расположенные на чипе для отслеживания движения полимеразной молекулы по одиночной цепи в процессе репликации. Частными примерами способов на основе нанотехнологий являются платформа «GridON™» от «Oxford Nanopore Technologies» (Оксфорд, Великобритания), платформы секвенирования нано-пор с вспомогательной гибридизацией («HANS™»), разработанные «Nabsys» (Провидено, Род-Айленд), и запатентованная платформа ДНК-секвенирования на основе лигазы с использованием технологии ДНК-наношариков (DNA nanoball) (DNB), называемая комбинаторным лигированием зонд-якорь («cPAL™»).
6) Технологии секвенирования одиночных молекул на основе электронной микроскопии, например, разработанные «LightSpeed Genomics» (Саннивейл, Калифорния) и «Halcyon Molecular» (Редвуд-Сити, Калифорния).
7) Ионное полупроводниковое секвенирование, которое основано на детекции ионов водорода, которые высвобождаются во время полимеризации ДНК. Например, «Ion Torrent Systems» (Сан-Франциско, Калифорния) используют чип высокой плотности из микро-обработанных лунок для осуществления данного биохимического процесса массивно-параллельным путем. Каждая лунка содержит различную ДНК-матрицу. Дно лунок представляет собой ионочувствительный слой, которое является запатентованным ионным сенсором.
Предпочтительно, ДНК и РНК предпочтительно служат в качестве исходного материала для NGS. Такие нуклеиновые кислоты легко могут быть получены из образцов, таких как биологический материал, например, из свежих, быстрозамороженных или погруженных в парафин фиксированных формалином опухолевых тканей (FFPE) или из свежевыделенных клеток или из СТС, которые присутствуют в периферической крови пациентов. Нормальная не подверженная мутациям геномная ДНК или РНК может быть выделена из нормальной соматической ткани, однако клетки зародышевой линии являются предпочтительными в контексте настоящего изобретения. Зародышевую ДНК или РНК выделяют из мононуклеаров периферической крови (РВМС) пациентов с негематологическими злокачественными опухолями. Хотя нуклеиновые кислоты, выделенные из тканей FFPE или из свежевыделенных одиночных клеток, высоко фрагментированы, они подходят для NGS-применений.
Несколько направленных NGS-методов для секвенирования экзомов описаны в научной литературе (для обзора см., например, Teer and Mullikin 2010: Human Mol Genet 19 (2), R145-51), каждый из которых может использоваться совместно с настоящим изобретением. Многие и этих методов (описанных, например, как захват генома, разделение генома, обогащение генома и т.д.) используют методы гибридизации и включают методы на основе использования чипа (например, Hodges et al. 2007: Nat. Genet. 39, 1522-1527) и методы гибридизации в растворе (например, Choi et al. 2009: Proc. Natl. Acad. Sci USA 106, 19096-19101). Коммерческие наборы для приготовления образца ДНК и последующего захвата экзома также доступны: например, «Illumina Inc.» (Сан-Диего, Калифорния) предлагает «TruSeq™ DNA Sample Preparation Kit» и «Exome Enrichment Kit TruSeq™ Exome Enrichment Kit».
С целью уменьшения количества ложных положительных результатов при детекции опухолеспецифичных соматических мутаций или различий в последовательности при сравнении, например, последовательности опухолевого образца с последовательностью эталонного образца, такого как последовательность образца зародышевой линии, предпочтительно определять последовательность в дубликатах одного или обоих их этих типов образцов. Таким образом, предпочтительно, что последовательность эталонного образца, такая как последовательность образца зародышевой линии, определяется два раза, три раза или более. В ином случае или дополнительно, последовательность опухолевого образца определяется два раза, три раза или более. Также возможно определение последовательности эталонного образца, такой как последовательность образца зародышевой линии и/или последовательности опухолевого образца более чем один раз посредством определения, по меньшей мере, единожды последовательности геномной ДНК и определения, по меньшей мере, единожды последовательности РНК указанного эталонного образца и/или указанного опухолевого образца. Например, путем определения вариаций между репликами эталонного образца, такого как образец зародышевой линии, может быть определена частота ложно положительных (FDR) соматических мутаций в виде статистической величины. Технические повторы образца должны давать идентичные результаты, и любая детектируемая мутация в данном сравнении «сам против себя» является ложно положительной. Конкретно, для определения частоты ложно положительных результатов для детекции соматических мутаций в опухолевом образце относительно эталонного образца, может использоваться технический повтор эталонного образца в качестве эталона для оценки количества ложно положительных результатов. Кроме того, различные связанные с качеством показатели (например, перекрывание или качество SNP) могут быть объединены в единую бальную оценку качества с использованием способа машинного обучения. Для данной соматической вариации могут быть подсчитаны все остальные вариации с превышением бальной оценки качества, что дает возможность упорядочивания всех вариаций в наборе данных.
Согласно изобретению может применяться высокопроизводительный метод полногеномного генотипирования одиночной клетки.
В одном воплощении высокопроизводительного полногеномного генотипирования одиночной клетки может использоваться платформа «Fluidigm». Такой способ может включать следующие стадии:
1. Забор опухолевой ткани/клеток и здоровой ткани от данного пациента.
2. Генетический материал выделяют из опухолевых и из здоровых клеток и затем его экзом (ДНК) секвенируют с использованием стандартных протоколов секвенирования следующего поколения (NGS). Перекрывание NGS таково, что могут детектироваться гетерозиготные аллели, по меньшей мере, с 5%-частотой. Транскриптом (РНК) также выделяют из опухолевых клеток, конвертируют в кДНК и секвенируют для определения того, какие гены экспрессируются опухолевыми клетками.
3. Несинонимичные экспрессирующиеся однонуклеотидные вариации (англ. single nucleotide variation, SNV) идентифицируют, как описано в данном документе. Отфильтровывают участки, которые являются SNP в здоровой ткани.
4. N=96 мутации из (3) отбирают, перекрывая различные частоты. Анализы SNP-генотипирования на основе детектирования флуоресценции разработаны и синтезированы для этих мутаций (примеры таких анализов включают: SNP-анализ на основе «TaqMano» от «Life Technologies» или тесты «SNPtype» от «Fluidigm»). Тесты будут включать праймеры амплификации специфической мишени (STA) для амплификации ампликонов, содержащих данный SNV (это стандарт в тестах «TaqMan» и «SNPtype»).
5. Индивидуальные клетки будут выделены из опухолевой и из здоровой ткани либо с помощью лазерной микродиссекции (LMD) либо с помощью дисагрегации до одноклеточных суспензий с последующей сортировкой, как описано ранее (Dalerba P. et al. (2011) Nature Biotechnology 29:1120-1127). Клетки могут быть отобраны либо без предварительного отбора (т.е., все), или, в ином случае, опухолевые клетки могут быть обогащены. Методы обогащения включают: специфическое окрашивание, сортировка клеток по размеру, гистологический анализ во время LMD, и так далее.
6. Индивидуальные клетки будут выделены в ПЦР-пробирках, содержащих мастер-микс вместе с STA-праймерами, и ампликоны, содержащие SNV, будут амплифицированы. В ином случае, геном одиночной клетки будет амплифицирован амплификацией цельного генома (WGA), как описано ранее (Frumkin D. et al. (2008) Cancer Research 68:5924). Клеточный лизис будет осуществлен либо в стадии нагревания при 95°С, либо посредством специального лизирующего буфера.
7. STA-амплифицированные образцы разводят и загружают на чип для генотипрования «Fluidigm».
8. Образцы из здоровой ткани будут использоваться в качестве положительных контролей для определения кластеров гомозиготных аллелей (без мутации). Так как NGS-данные выявляют, что гомозиготные мутации исключительно редкие, как правило, ожидаются только два кластера: XX и XY, с Х=здоровым.
9. Количество чипов, которые могут быть сделаны, не ограничено, с возможностью практического осуществления анализа до ~1000 одиночных клеток (~10 чипов). Если осуществлять в 384-луночных планшетах, то приготовление образца уменьшится до нескольких дней.
10. Затем определяют SNV для каждой клетки.
В другом воплощении высокопроизводительного полногеномного генотипирования одиночной клетки может использоваться платформа NGS. Такой способ может включать следующие стадии:
1. Стадии 1-6 выше идентичны за исключением того, что N (количество анализируемых SNV) может быть гораздо больше 96. В случае WGA, несколько циклов STA будут осуществлены после. STA-праймеры будут содержать две универсальные последовательности метки на каждом праймере.
2. После STA, праймеры со штрихкодом будут амплифицироваться путем ПЦР в ампликоны. Праймеры со штрихкодом содержат уникальные последовательности «штрихкода» и вышеуказанные универсальные последовательности метки. Каждая клетка, таким образом, будет содержать уникальный штрихкод.
3. Ампликоны из всех клеток будут смешиваться и секвенироваться посредством NGS. Практическое ограничение количества клеток, которые могут быть мультиплексированы, заключается в количестве планшетов, которое может быть приготовлено. Так как образцы могут быть приготовлены в 384-луночных планшетах, то практическое ограничение будет составлять ~5000 клеток.
4. На основе данных о последовательностях детектируют SNV (или друге структурные аномалии) индивидуальных клеток.
Согласно изобретению для приоретизации антигенов можно использовать опухолевую генетическую реконструкцию на основе генотипирования одиночной клетки («филогенетическая антигенная приоритезация»). Кроме антигенной системы приоритетов на основе таких критериев, как экспрессия, тип мутации (несинонимичная против другой), МНС-связывающие характеристики и так далее, можно использовать дополнительное направление для приоритезации, разработанное для того, чтобы справиться с внутриопухолевой и межопухолевой гетерогенностью, и систематической ошибкой биопсии, может использоваться как описано, например, ниже.
1. Идентификация большинства широкораспространенных антигенов
Частоту каждого SNV можно точно оценить на основе анализа одиночной клетки, описанного выше, совместно с высокопроизводительным методом полногеномного генотипирования, и большинство присутствующих широкораспространенных SNV может быть отобрано для получения индивидуальных противоопухолевых вакцин (IVAC).
2. Идентификация первичных базальных антигенов на основе анализа «дерево с корнем» (root tree analysis).
NGS-данные из опухолей предполагают, что гомозиготные мутации (горячие точки в обоих аллелях) представляют собой редкие события. Таким образом, нет необходимости гаплотипирования, и филогенетическое дерево опухолевых соматических мутаций может быть создано на основе данных SNV одиночной клетки. Зародышевая последовательность будет использоваться для корня дерева. С помощью алгоритма для воспроизводства наследственных последовательностей последовательности узлов около корня дерева будут воспроизведены. Эти последовательности содержат самые ранние мутации, которые по прогнозам существуют в первичной опухоли (определенные в данном документе как первичные базальные мутации/антигены). Благодаря низкой вероятности того, что две мутации произойдут на одинаковых аллелях в одном и том же положении генома, мутации в наследственных последовательностях по прогнозам будут фиксированы в опухоли.
Приоритезация первичных базальных антигенов не эквивалентна приоритезации наиболее частых мутаций в биопсии (хотя первичные базальные мутации, как ожидается, будут среди наиболее частых в биопсии). Причина следующая: скажем две SNV присутствуют во всех клетках, выделенных из биопсии (и таким образом имеют такую же частоту - 100%), но одна мутация является базальной, а другая нет, поэтому базальная мутация должна быть отобрана для IVAC. Это происходит из-за того, что базальная мутация вероятно присутствует во всех областях опухоли, тогда как более поздняя мутация может быть более недавней мутацией, которая случайно зафиксировалась в области, где была взята биопсия. Кроме того, базальные антигены, вероятно, существуют в метастазирующих опухолях, полученных из первичной опухоли. Таким образом, путем приоритезации базальных антигенов для IVAC специалист может существенно повысить шанс, что IVAC будет способна ликвидировать всю опухоль, а не только часть опухоли.
Если вторичные опухоли существуют, и они также были отобраны, то можно оценить эволюционное дерево всех опухолей. Такой подход может улучшить ошибкоустойчивость дерева и позволить детектировать мутации, которые являются базальными по отношению ко всем опухолям.
3. Идентификация антигенов, которые максимально охватывают опухоль(и)
Другой способ получения антигенов, который максимально перекрывает все опухолевые участки, заключается в заборе нескольких биопсий из опухоли. Одна стратегия будет заключаться в отборе антигенов, идентифицированных с помощью анализа NGS, присутствующих во всех биопсиях. Для улучшения шансов идентификации базальных мутаций может быть осуществлен филогенетический анализ на основе мутаций одиночной клетки из всех биопсий.
В случае метастазирования, могут быть получены биопсии из всех опухолей, и могут быть отобраны мутации, идентифицированные посредством NGS, которые являются общими для всех опухолей.
4. Использование СТС для приоритезации антигенов, которые ингибируют метастазирование
Считается, что метастазирующие опухоли получаются из одиночных клеток. Таким образом, путем генотипирования индивидуальных клеток, выделенных из различных опухолей данного пациента, совместно с генотипированием циркулирующих опухолевых клеток пациента (СТС), специалист может восстановить эволюционную историю злокачественного новообразования. Ожидание заключается в наблюдении метастазирующих опухолей, эволюционирующих из исходной опухоли посредством филогенетической ветви СТС, выделенных из первичной опухоли.
Ниже (несмещенный метод для идентификации, подсчета и генетического зондирования СТС) мы описываем расширение вышеописанного метода высокопроизводительного полногеномного генотипирования одиночной клетки для несмещенного выделения и геномного анализа СТС. Используя анализ, описанный выше, специалист может в последствие восстановить филогенетическое дерево первичной опухоли, СТС и вторичных опухолей, развившихся из метастазов (если они есть). На основе этого дерева специалист может идентифицировать мутации (ведомые и ведущие), которые появляются в момент или сразу после того, как СТС впервые открепляются от первичной опухоли. Ожидание состоит в том, что геномы СТС, происходящих из первичной опухоли, эволюционно более похожи на геномы первичной опухоли, чем геномы вторичной опухоли. Кроме того, ожидается, что геномы СТС, происходящих из первичной опухоли, будут содержать уникальные мутации, которые фиксируются во вторичных опухолях, или которые вероятно будут фиксироваться, если вторичные опухоли образуются в будущем. Эти уникальные мутации могут быть расположены в порядке приоритета для IVAC для направленного воздействия (или предотвращения) метастазирования.
Преимущество приоритетных мутаций СТС против первичных базальных мутаций заключается в том, что антигены, выделенные из СТС, могут мобилизовать Т-клетки специфично для направленного воздействия на метастазы, и таким образом, они будут независимым крылом направленного воздействия Т-клеток на первичную опухоль (с использованием различных антигенов). Кроме того, если есть мало вторичных опухолей (или их нет), то ожидается, что шанс ускользания от иммунитета от выделенных из СТС антигенов, будет ниже, поскольку вероятность для ускользания опухоли от иммунитета будет коррелировать с количеством опухолевых клеток, несущих данный антиген.
5. Идентификация антигенов, присутствующих в одной и той же клетке ("коктейль» IVAC)
Считается, что опухоль эволюционирует путем подавления мутаций в результате селективного давления иммунной системы и терапии. Противоопухолевые вакцины, направленно воздействующие на множество антигенов, которые сосуществуют на одной и той же клетке и которые также нередки в опухоли, имеют больший шанс преодоления механизма ускользания опухоли от иммунитета, и таким образом, уменьшают шанс рецидива. Такие «вакцины-коктейли» будут аналогичны антиретровирусной комбинированной терапии для HIV+-пациентов. Сосуществующие мутации могут быть идентифицированы с помощью филогенетического анализа или путем проверки SNV-выравнивания всех клеток.
Кроме того, согласно изобретению, может использоваться несмещенный метод идентификации, подсчета и генетического зондирования СТС. Такой способ может включать следующие стадии:
1. Получение биопсии опухоли(ей) и определение атласа соматических мутаций.
2. Операция 1: Отбор N≥96 мутаций для дополнительного исследования на основе ранее разработанной схемы приоритезации.
Операция 2: Осуществление анализа одиночной клетки (см. выше описанный метод высокопроизводительного полногеномного генотипирования одиночной клетки) с последующим филогенетическим анализом для отбора N≥96 первичных базальных мутаций и возможно более недавних мутаций для максимального разнообразия. Первые мутации применимы для идентификации СТС (см. ниже), а последние для осуществления филогенетического анализа (см. раздел «идентификация антигенов, сосуществующих на одной и той же клетке ("коктейль» IVAC»)).
3. Получение цельной крови из онкопациента
4. Лизис эритроцитов
5. Удаление лейкоцитов путем истощения CD45+ клеток (например, посредством сортировки, магнитных микросфер, конъюгированных с анти-CD45 антителом, и т.д.) для обогащения СТС.
6. Удаление свободной ДНК с помощью гидролиза ДНК-азой. Источником свободной ДНК может быть ДНК, присутствующая в крови, или ДНК из мертвых клеток.
7. Сортировка оставшихся клеток в ПЦР-пробирки, осуществление STA (на основе отобранных мутаций) и скрининг на «Fluidigm» (вышеописанный метод высокопроизводительного полногеномного генотипирования одиночной клетки). СТС должны, как правило, быть положительными по множеству SNV.
8. Клетки, идентифицированные как опухолевые (=СТС) могут затем дополнительно анализироваться филогенетически на основе панели скринированных SNV (см. раздел «Идентификация антигенов, сосуществующих на одной и той же клетке («коктейль» IVAC»)).
Также возможно объединить этот способ с ранее созданными способами для выделенных СТС. Например, специалист может отсортировать ЕрСАМ+ клетки, или клетки, положительные по цитокератинам (Rao CG. et al. (2005) International journal of oncology 27:49; Allard WJ. et al. (2004) Clinical Cancer Research 10:6897-6904). Эти предполагаемые СТС могут затем подтверждаться/зондироваться на «Fluidigm»/NGS для выделения их мутаций.
Этот способ может использоваться для подсчета СТС. Так как способ не базируется на конкретном маркере, который может экспрессироваться опухолевыми клетками или не может экспрессироваться опухолевыми клетками, но скорее базируется на профиле мутаций опухолевых соматических мутаций, уникальных для пациента, то он представляет собой несмещенный метод для детекции и подсчета СТС.
Согласно изобретению может быть использован способ, включающий опухолевую филогенетическую реконструкцию на основе генотипирования одиночной клетки для обогащения ведущих мутаций («филогенетический фильтр»).
В одном воплощении данного способа осуществляется пан-опухолевый филогенетический анализ для извлечения ведущих мутаций.
Например, могут быть детектированы ведущие мутации из n=1 опухолей.
В вышеописанном разделе «Идентификация первичных базальных антигенов на основе анализа «дерево с корнем»» мы описали способ выявления исходных последовательностей и/или идентификации клеток, которые имеют последовательности, близкие к «корню дерева». Ожидается, что количество мутаций в этих последовательностях будет значительно меньше, чем количество мутаций в суммарном образце опухоли, так как по определению они представляют собой последовательности, близкие к «корню дерева». Таким образом, с помощью отбора последовательностей, близких к «корню дерева» ожидается, что множество ведомых мутаций будет «филогенетически отфильтровано». Данная процедура имеет потенциал для существенного обогащения ведущих мутаций. Затем ведущие мутации могут использоваться для идентификации/отбора лечения пациента или могут использоваться в качестве ключей для новых терапий.
В другом примере могут быть детектированы ведущие мутации из n>1 опухолей данного типа.
С помощью реконструкции первичных базальных мутаций из множества опухолей конкретного типа специалист может существенно повысить шанс детекции ведущих мутаций. Так как базальные последовательности, близкие к «корню дерева», отфильтровывают множество ведомых мутаций, то соотношение сигнала к фону при детекции ведущих мутаций, как ожидается, существенно повысится. Таким образом, данный способ имеет потенциал детектировать (1) менее частую ведущую мутацию (2) частую ведущую мутацию из меньшего количества образцов.
В другом воплощении способа, включающего опухолевую филогенетическую реконструкцию на основе генотипирования одиночной клетки для обогащения ведущих мутаций («филогенетический фильтр»), осуществляется филогенетический анализ для извлечения ведущих мутаций, вызывающих метастазирование.
В вышеописанном разделе «Использование СТС для приоритизации антигенов, которые ингибируют метастазирование» мы описали способ детекции СТС-ассоциированных мутаций. Этот способ также может использоваться для обогащения ведущих мутаций, приводящих к метастазированию. Например, с помощью картирования комбинированной филогенетики первичной опухоли, вторичной опухоли и СТС, СТС, выделенные из первичной опухоли, должны связываться между филогенетическими ветвями первичной и вторичной опухоли. Такой филогенетический анализ может помочь точно указать мутации, уникальные в данном переходном состоянии между первичной и вторичной опухолями. Фракция этих мутаций может представлять собой ведущие мутации. Кроме того, путем сравнения уникальных СТС-мутаций из различных случаев одной и той же опухоли (т.е., n>1 опухолей), специалист может дополнительно получить обогащение уникальными ведущими мутациями, вызывающими метастазирование.
Согласно изобретению может использоваться филогенетический анализ для идентификации первичных опухолей против вторичных.
В случае метастазирования если делать забор из всех опухолей, то «дерево с корнем» может использоваться для прогноза временного порядка, в котором появляются опухоли: какая опухоль первичная (узлы, ближайшие к «корню дерева») и какие опухоли являются самыми последними. Это может быть полезно в случаях, когда трудно определить, какая опухоль является первичной.
В контексте настоящего изобретения термин «РНК» относится к молекуле, которая включает, по меньшей мере, один рибонуклеотидный остаток и предпочтительно полностью или частично состоит из рибонуклеотидных остатков. «Рибонуклеотид» относится к нуклеотиду, содержащему гидроксильную группу в 2'-положении β-D-рибофуранозильной группы. Термин «РНК» включает двухцепочечную РНК, одноцепочечную РНК, изолированную РНК, такую как частично или полностью очищенная РНК, фактически чистую РНК, синтетическую РНК и рекомбинантно полученную РНК, такую как модифицированная РНК, которая отличается от природной РНК вставкой, делецией, заменой и/или изменением одного или нескольких нуклеотидов. Такие изменения могут включать вставку не нуклеотидного материала, как например на конце(ах) РНК или внутри, например, к одному или нескольким нуклеотидам РНК. Нуклеотиды молекул РНК также могут включать нестандартные нуклеотиды, такие как синтетические нуклеотиды или химически синтезированные нуклеотиды или дезоксинуклеотиды. Эти измененные РНК могут обозначаться как аналоги или аналоги природных РНК.
Согласно настоящему изобретению термин «РНК» включает и предпочтительно относится к «мРНК». Термин «мРНК» означает «матричную РНК» и относится к «транскрипту», который генерируется с использованием ДНК-матрицы и кодирует пептид или полипептид. Как правило, мРНК включает 5'-UTR, область, кодирующую белок, и 3'-UTR. мРНК обладает только ограниченным периодом полужизни в клетках и in vitro. В контексте настоящего изобретения мРНК может быть получена in vitro транскрипцией ДНК-матрицы. Методология in vitro транскрипции известна специалистам. Например, существует множество коммерчески доступных наборов реактивов для in vitro транскрипции.
Согласно изобретению стабильность и эффективность трансляции РНК может, если необходимо, быть модифицирована. Например, РНК может быть стабилизирована и ее трансляция увеличивается с помощью одной или нескольких модификаций, обладающих стабилизирующим эффектом и/или повышением эффективности трансляции РНК. Такие модификации описаны, например, в РСТ/ЕР2006/009448, включенном в данный документ ссылкой. С целью повышения экспрессии РНК, используемой согласно настоящему изобретению, она может быть модифицирована внутри кодирующей области, т.е. последовательности, кодирующей экспрессируемый пептид или белок, предпочтительно без изменения последовательности экспрессируемого пептида или белка, так чтобы повысить GC-содержание для увеличения стабильности мРНК и для осуществления оптимизации кодонов и, таким образом, для усиления трансляции в клетках.
Термин «модификация» в контексте РНК, используемой в настоящем изобретении, включает модификацию РНК, которая в норме не присутствует в указанной РНК.
В одном воплощении изобретения РНК, используемая согласно изобретению, не содержит некэпированные 5'-трифосфаты. Удаление таких некэпированных 5'-трифосфатов может быть достигнуто путем обработки РНК с помощью фосфатазы.
РНК согласно изобретению может содержать модифицированные рибонуклеотиды с целью повышения ее стабильности и/или уменьшения цитотоксичности. Например, в одном воплощении в РНК, использующейся согласно изобретению, 5-метилцитидин частично или полностью замещен, предпочтительно полностью, на цитидин. В ином случае или дополнительно, в одном воплощении в РНК, использующейся согласно изобретению, псевдоуридин частично или полностью замещен, предпочтительно полностью, на уридин.
В одном воплощении термин «модификация» относится к обеспечению РНК 5'-кэп или аналогом 5'-кэп. Термин «5'-кэп» относится к кэпирующей структуре, обнаруженной на 5'-конце молекулы мРНК, и, как правило, состоит из гуанозин-нуклеотида, связанного с мРНК посредством необычной 5'-5' трифосфатной связи. В одном воплощении этот гуанозин метилирован в 7-положении. Термин «стандартный 5'-кэп» относится к природной группе РНК 5'-кэп, предпочтительно к группе кэп 7-метилгуанозина (m7G). В контексте настоящего изобретения термин «5'-кэп» включает аналог 5'-кэп, который напоминает структуру кэп РНК и который модифицирован так, что он обладает способностью стабилизировать РНК и/или усиливать трансляцию РНК при присоединении к ней, предпочтительно in vivo и/или в клетке.
Предпочтительно, 5'-конец РНК включает кэп-структуру, имеющую следующую общую формулу:
где R1 и R2 независимо представляют собой гидрокси или метокси, и W-, X- и Y- независимо представляют собой кислород, серу, селен или ВН3. В предпочтительном воплощении R1 и R2 представляют собой гидрокси, и W-, X- и Y- представляют собой кислород. В следующем предпочтительном воплощении, один из R1 и R2, предпочтительно R1 представляет собой гидрокси, а другой метокси, и W-, X- и Y- представляют собой кислород. В следующем предпочтительном воплощении, R1 и R2 представляют собой гидрокси, и один из W-, X- и Y-, предпочтительно X- представляет собой серу, селен, или ВН3, предпочтительно серу, в то время как другой представляет собой кислород. В следующем предпочтительном воплощении, один из R1 и R2, предпочтительно R2 представляют собой гидрокси, а другой метокси, и один из W-, X- и Y-, предпочтительно X- представляет собой серу, селен, или ВН3, предпочтительно серу, в то время как другой представляет собой кислород.
В вышеописанной формуле нуклеотид с правой стороны связан с цепью РНК через ее 3'-группу.
Те Кэп-структуры, где, по меньшей мере, один из W-, X- и Y- представляет собой серу, т.е., который имеет фосфоротиоатный компонент, существуют в различных диастереомерных формах, каждая из которых охвачена в данном документе. Кроме того, настоящее изобретение охватывает все таутомеры и стереоизомеры вышеописанной формулы.
Например, Кэп-структура, имеющая вышеописанную структуру, где R1 представляет собой метокси, R2 представляет собой гидрокси, X- представляет собой серу, и W- и Y- представляют собой кислород, существуют в двух диастереомерных формах (Rp и Sp). Их можно разрешить на ВЭЖХ с обращенной фазой, и они получили названия D1 и D2 согласно порядку их элюции с колонки ВЭЖХ с обращенной фазой. Согласно изобретению изомер D1 m27,2'-OGppspG является конкретно предпочтительным.
Получение РНК, содержащие 5'-кэп или аналог 5'-кэп, может быть достигнуто с помощью in vitro транскрипции ДНК-матрицы в присутствии указанной 5'-кэп или аналога 5'-кэп, где указанная 5'-кэп котранскрипционно включается в образованную цепь РНК, или РНК может быть получена, например, с помощью in vitro транскрипции, а 5'-кэп может быть присоединен к РНК посттранскрипционно с использованием кэппирующих ферментов, например, кэппирующих ферментов вируса вакцинии.
РНК может включать дополнительные модификации. Например, дополнительная модификация РНК, используемая в настоящем изобретении, может представлять собой удлинение или укорачивание природного поли(А) хвоста или изменение 5'- или 3'-нетранслируемых областей (UTR), как например, введение UTR, который не связан с кодирующей областью указанной РНК, например, вставка или замена имеющейся 3'-UTR на одну или несколько, предпочтительно две копии 3'-UTR, выделенных из гена глобина, такого как альфа2-глобин, альфа1-глобин, бета-глобин, предпочтительно, бета-глобин, более предпочтительно, человеческий бета-глобин.
РНК, содержащая открытую поли-А последовательность, транслируется более эффективно, чем РНК, содержащая скрытую поли-А последовательность. Термин «поли(А) хвост» или «поли-А последовательность» относится к последовательности остатков аденила (А), которые, как правило, локализованы на 3'-конце молекулы РНК, и «открытая поли-А последовательность» означает, что поли-А последовательность на 3'-конце молекулы РНК, содержащая А поли-А-последовательности, за которой не следуют нуклеотиды, отличные от А, локализованные на 3'-конце, т.е. ниже поли-А-последовательности. Кроме того, длинная поли-А-последовательность около 120 пар оснований приводит к получению оптимальной стабильности транскрипта и эффективности трансляции РНК.
Таким образом, с целью повышения стабильности и/или экспрессии РНК, используемой согласно изобретению, она может быть модифицирована так, чтобы присутствовать совместно с поли-А-последовательностью, предпочтительно, имеющей длину 10-500 остатков аденозина, более предпочтительно, 30-300, еще более предпочтительно 65-200 и особенно 100-150 остатков аденозина. В особенно предпочтительном воплощении поли-А-последовательность имеет длину приблизительно 120 остатков аденозина. Для дополнительного повышения стабильности и/или экспрессии РНК, используемой согласно изобретению, поли-А-последовательность может быть открытой.
Кроме того, включение 3'-нетранслируемой области (UTR) в 3'-нетранслируемую область молекулы РНК может привести к повышению эффективности трансляции. Синергический эффект может быть достигнут путем включения двух или нескольких из таких 3'-нетранслируемых областей. 3'-нетранслируемые области могут быть аутологичными или гетерологичными по отношению к РНК, в которую они вводятся. В одном конкретном воплощении 3'-нетранслируемая область выделена из человеческого гена β-глобина.
Комбинация вышеописанных модификаций, т.е. включение поли-А-последовательности, открытие поли-А-последовательности и включение одной или нескольких 3'-нетранслируемых областей, имеет синергическое влияние на стабильность РНК и повышает эффективность трансляции.
Термин «стабильность» РНК относится к «периоду полужизни» РНК. «Период полужизни» относится к периоду времени, который необходим для потери половины активности, количества или числа молекул. В контексте настоящего изобретения, период полужизни РНК является показателем стабильности указанной РНК. Период полужизни РНК может влиять на «продолжительность экспрессии» РНК. Может ожидаться, что РНК, имеющая продолжительный период полужизни, будет экспрессироваться в течение продолжительного периода времени.
Конечно, если согласно изобретению целесообразно уменьшить стабильность и/или эффективность трансляции РНК, то возможна модификация РНК, которая воспрепятствует функции элементов, которые, как описано выше, повышают стабильность и/или эффективность трансляции РНК.
Термин «экспрессия» используется согласно изобретению в его наиболее широком значении, и включает продуцирование РНК и/или пептидов или белков, например, с помощью транскрипции и/или трансляции. По отношению к РНК термин «экспрессия» или «трансляция» относится конкретно к продуцированию пептидов или полипептидов. Также он включает частичную экспрессию нуклеиновых кислот. Кроме того, экспрессия может быть транзиторной или стабильной.
Согласно изобретению термин экспрессия также включает «аберрантную экспрессию» или «аномальную экспрессию». «Абберантная экспрессия» или «аномальная экспрессия» означает согласно изобретению, что экспрессия изменяется, предпочтительно повышается по сравнению с эталоном, например, с состоянием объекта, не имеющего заболевания, ассоциированного с абберантной или аномальной экспрессией определенного белка, например, опухолевого антигена. Повышение экспрессии относится к повышению, по меньшей мере, на 10%, конкретно, по меньшей мере, на 20%, по меньшей мере, на 50% или, по меньшей мере, на 100%, или более. В одном воплощении, экспрессия обнаружена только в больной ткани, в то время как экспрессия в здоровой ткани репрессирована.
Термин «специфично экспрессируется» означает, что белок экспрессируется по существу только в специфической ткани или в органе. Например, то, что опухолевый антиген специфично экспрессируется в слизистой желудка означает, что указанный белок главным образом экспрессируется в слизистой желудка и не экспрессируется в других тканях, или не экспрессируется в значительной степени в других тканях или типах органов. Таким образом, белок, который экспрессируется исключительно в клетках слизистой желудка и в значительно меньшей степени в любой другой ткани, такой как яички, специфично экспрессируется в клетках слизистой желудка. В некоторых воплощениях, опухолевый антиген также может специфично экспрессироваться при нормальных условиях в более чем одном типе ткани, как например, в 2 или 3 типах тканей или органов, но предпочтительно, в не более чем 3 различных типах тканей или органов. В этом случае опухолевый антиген специфично экспрессируется в этих органах. Например, если опухолевый антиген экспрессируется при нормальных условиях предпочтительно в приблизительно равной степени в легком и в желудке, то указанный опухолевый антиген специфично экспрессируется в легком и в желудке.
В контексте настоящего изобретения термин «транскрипция» относится к процессу, в котором генетический код последовательности ДНК транскрибируется в РНК. Затем РНК может транслироваться в белок. Согласно настоящему изобретению термин «транскрипция» включает «in vitro транскрипцию», где термин «in vitro транскрипция» относится к процессу, в котором РНК, конкретно мРНК синтезируется in vitro в бесклеточной системе, предпочтительно с использованием соответствующих клеточных экстрактов. Предпочтительно, клонирующие векторы применяются для генерирования транскриптов. Эти клонирующие векторы, как правило, спроектированы как транскрипционные векторы и согласно изобретением охвачены термином «вектор». Согласно настоящему изобретению, РНК, используемая в настоящем изобретении, предпочтительно является in vitro транскрибированной РНК (IVT-PHK) и может быть получена с помощью in vitro транскрипции соответствующей ДНК-матрицы. Промотор для контроля транскрипции может представлять собой любой промотор для любой РНК-полимеразы. Конкретные примеры РНК-полимераз представляют собой Т7, Т3, и SP6 РНК-полимеразы. Предпочтительно, in vitro транскрипция согласно изобретению контролируется Т7- или SP6-промтором. ДНК-матрица для in vitro транскрипции может быть получена путем клонирования нуклеиновой кислоты, конкретно, кДНК и путем введения в соответствующий вектор для in vitro транскрипции. кДНК может быть получена путем обратной транскрипции РНК.
Термин «трансляция» согласно изобретению относится к процессу в рибосоме клетки, с помощью которой цепь матричной РНК направляет сборку последовательности аминокислот с получением пептида или полипептида.
Последовательности контроля экспрессии или регуляторные последовательности, которые согласно изобретению могут быть функционально связаны с нуклеиновой кислотой, могут быть гомологичными или гетерологичными по отношению к нуклеиновой кислоте. Кодирующая последовательность и регуляторная последовательность связаны вместе «функционально», если они связаны вместе ковалентно, так что транскрипция или трансляция кодирующей последовательности находится под контролем или влиянием регуляторной последовательности. Если кодирующая последовательность транслируется в функциональный белок с функциональной связью регуляторной последовательности с кодирующей последовательностью, то индукция регуляторной последовательности приводит к транскрипции кодирующей последовательности, не вызывая при этом сдвига рамки считывания в кодирующей последовательности или неспособности транслироваться в целевой белок или пептид.
Термин «последовательность, контролирующая экспрессию» или «регуляторная последовательность» включает согласно изобретению промоторы, последовательности связывания с рибосомой и другие элементы контроля, которые контролируют транскрипцию нуклеиновой кислоты или трансляцию полученной РНК. В одном воплощении изобретения могут контролироваться регуляторные последовательности. Точная структура регуляторных последовательностей может варьировать в зависимости от вида или в зависимости от типа клеток, но, как правило, включает 5'-нетранслируемые и 5'- и 3'-нетранслируемые последовательности, которые вовлечены в инициирование транскрипции или трансляции, такие как ТАТА-бокс, кэппирующие последовательности, СААТ-последовательность и так далее. Конкретно, 5'-нетранслируемые регуляторные последовательности включают промоторную область, которая включает последовательность контроля транскрипции функционально связанного гена. Регуляторные последовательности также могут включать энхансерные последовательности или 5'-активаторные последовательности.
Предпочтительно, согласно изобретению, если РНК, экспрессирующаяся в клетке, может вводиться в указанную клетку. В одном воплощении способов по изобретению РНК, которую вводят в клетку, получают с помощью in vitro транскрипции соответствующей ДНК-матрицы.
Согласно изобретению такие термины, как «РНК, способная экспрессироваться» и «кодирующая РНК» используются взаимозаменяемо в данном документе и по отношению к конкретному пептиду или полипептиду означают, что РНК, если она присутствует в соответствующей среде, предпочтительно внутри клетки, может экспрессироваться с получением указанного пептида или полипептида. Предпочтительно, РНК, которая, согласно изобретению, способна взаимодействовать с клеточным аппаратом трансляции с получением пептида или полипептида, способна экспрессироваться.
Термины такие, как «перенос», «введение» или «трансфекция» используются в данном документе взаимозаменяемо и относятся к введению нуклеиновых кислот, конкретно экзогенных или гетерологичных нуклеиновых кислот, в частности, РНК в клетку. Согласно настоящему изобретению клетка может образовывать часть органа, ткани и/или организма. Согласно настоящему изобретению введение нуклеиновой кислоты достигается как с использованием «голой» нуклеиновой кислоты, так и в комбинации с реагентом введения. Предпочтительно, введение нуклеиновых кислот происходит в форме «голых» нуклеиновых кислот. Предпочтительно, РНК вводят в комбинации со стабилизирующими веществами, такими как ингибиторы РНК-азы. Настоящее изобретение также рассматривает повторное введение нуклеиновых кислот в клетки для возможности экспрессии в течение продолжительных периодов времени.
Клетки могут трансфецироваться с помощью любых носителей, которые могут ассоциироваться с РНК, например, путем образования комплексов с РНК или образуя везикулы, в которые заключена или инкапсулирована РНК, приводя в результате к повышенной стабильности РНК по сравнению с «голой» РНК. Носители, используемые согласно изобретению, включают, например, липид-содержащие носители, такие как катионные липиды, липосомы, конкретно катионные липосомы, и мицеллы и наночастицы. Катионные липиды могут образовывать комплексы с отрицательно заряженными нуклеиновыми кислотами. Согласно изобретению может использоваться любой катионный липид.
Предпочтительно, введение РНК, которая кодирует пептид или полипептид, в клетку, конкретно в клетку, присутствующую in vivo, приводит к экспрессии указанного пептида или полипептида в клетке. В конкретных воплощениях, предпочтительно направление нуклеиновых кислот в конкретные клетки. В таких воплощениях, носитель, который применяется для введения нуклеиновой кислоты в клетку (например, ретровирус или липосома), экспонирует направляющую молекулу. Например, молекула, такая как антитело, которое специфично к поверхностному мембранному белку на клетке-мишени, или лиганд к рецептору на клетке-мишени может быть включена в носитель нуклеиновой кислоты или может быть связана с ним. В случае, когда нуклеиновая кислота вводится с помощью липосомы, белки, которые связываются с поверхностным мембранным белком, который ассоциирован с эндоцитозом, могут быть включены в липосомный состав с целью возможности направления и/или поглощения. Такие белки охватывают капсидные белки или их фрагменты, которые специфичны к конкретному типу клеток, антитела к белкам, которые интернализуются, белки, которые имеют внутриклеточную локализацию и т.д.
Согласно настоящему изобретению термин «пептид» относится к веществам, включающим два или более, предпочтительно 3 или более, предпочтительно 4 или более, предпочтительно 6 или более, предпочтительно 8 или более, предпочтительно 10 или более, предпочтительно 13 или более, предпочтительно 16 или более, предпочтительно 21 или более и предпочтительно до 8, 10, 20, 30, 40 или 50, конкретно до 100 аминокислот, соединенных ковалентно пептидными связями. Термин «полипептид» или «белок» относится к крупным пептидам, предпочтительно к пептидам, содержащим более чем 100 аминокислотных остатков, но, в общем, термины «пептид», «полипептид» и «белок» являются синонимами и используются в данном документе взаимозаменяемо.
Согласно изобретению термин «изменение последовательности» по отношению к пептидам или белкам относится к аминокислотным вариантам со вставками, аминокислотным вариантам с добавлениями, аминокислотным вариантам с делениями и аминокислотным вариантам с заменой, предпочтительно, к аминокислотным вариантам с заменой. Все эти изменения последовательности согласно изобретению потенциально могут создавать новые эпитопы.
Аминокислотные варианты со вставкой включают вставки одной или двух или более аминокислот в конкретную аминокислотную последовательность.
Аминокислотные варианты с добавлением включают амино- и/или карбокси-концевые сшивки одной или более аминокислот, как например, 1, 2, 3, 4 или 5, или более аминокислот.
Аминокислотные варианты с делецией характеризуются удалением одной или более аминокислот из последовательности, как например, удалением 1, 2, 3, 4 или 5, или более аминокислот.
Аминокислотные варианты с заменой характеризуются тем, что, по меньшей мере, один остаток в последовательности удаляется, а другой остаток вставляется на его место.
Термин «выделенный» согласно изобретению означает, что конкретный компонент, конкретно, конкретная последовательность присутствует у субъекта, из которого он выделен, конкретно организм или молекула. В случае аминокислотных последовательностей, особенно конкретных областей последовательностей, «выделенный» конкретно означает, что соответствующая аминокислотная последовательность выделена из аминокислотной последовательности, в которой она присутствует.
Термин «клетка» или «клетка-хозяин» предпочтительно представляет собой интактную клетку, т.е. с интактной мембраной, которая не высвобождала свои обычные внутриклеточные компоненты, такие как ферменты, органеллы или генетический материал. Интактная клетка предпочтительно представляет собой жизнеспособную клетку, т.е. живую клетку, способную осуществлять свои обычные метаболические функции. Предпочтительно, указанный термин относится согласно изобретению с любой клетке, которая может быть трансформирована или трансфецирована с использованием экзогенной нуклеиновой кислоты. Термин «клетка» включает согласно изобретению прокариотические клетки (например, Е.coli) или эукариотические клетки (например, дендритные клетки, В-клетки, клетки СНО, клетки COS, клетки K562, клетки HEK293, клетки HELA, дрожжевые клетки, и клетки насекомых). Экзогенная нуклеиновая кислота может быть обнаружена внутри клетки (i) в свободно диспергированном состоянии, как таковом, (ii) включенной в рекомбинантный вектор, или (iii) интегрированной в геном клетки-хозяина или в митохондриальную ДНК. Особенно предпочтительны клетки млекопитающих, такие как клетки человека, мышей, хомяков, свиней, коз и приматов. Клетки могут быть выделены из большого количества типов ткани и включают первичные клетки и клеточные линии. Специфические примеры включают кератиноциты, лейкоциты периферической крови, стволовые клетки костного мозга и эмбриональные стволовые клетки. В следующих воплощениях клетка представляет собой антигенпрезентирующую клетку, в частности, дендритную клетку, моноцит или макрофаг.
Клетка, которая включает молекулу нуклеиновой кислоты, предпочтительно экспрессирует пептид или полипептид, кодируемый нуклеиновой кислотой.
Термин «клональная экспансия» относится к процессу, в котором умножается количество соответствующего компонента. В контексте настоящего изобретения термин предпочтительно используется в контексте иммунологического ответа, в котором лимфоциты стимулируются антигеном, пролиферируют, и специфический лимфоцит, распознающий указанный антиген, умножается. Предпочтительно, если клональная экспансия приводит к дифференцировке лимфоцитов.
Такие термины как «снижение» или «ингибирование» относятся к способности вызывать общее уменьшение уровня, предпочтительно на 5% или более, 10% или более, 20% или более, более предпочтительно, на 50% или более, и наиболее предпочтительно на 75% или более. Термин «ингибирует» или ему подобные выражения включает полное или по существу полное ингибирование, т.е. снижение до ноля или по существу до ноля.
Термины, такие как «увеличение», «усиление», «стимулирование» или «пролонгация» предпочтительно относятся к увеличению, усилению, стимулированию или пролонгации, по меньшей мере, на около 10%, предпочтительно, по меньшей мере, на 20%, предпочтительно, по меньшей мере, на 30%, предпочтительно, по меньшей мере, на 40%, предпочтительно, по меньшей мере, на 50%, предпочтительно, по меньшей мере, на 80%, предпочтительно, по меньшей мере, на 100%, предпочтительно, по меньшей мере, на 200% и конкретно, по меньшей мере, на 300%. Эти термины также могут относиться к увеличению, усилению, стимуляции или пролонгации от ноля или неизмеряемого или недетектируемого уровня до уровня более чем ноль или до уровня, который измеряется или детектируется.
Агенты, композиции и способы, описанные в данном документе, могут использоваться для лечения объекта, имеющего заболевание, например, заболевание, характеризующееся присутствием больных клеток, экспрессирующих антиген и презентирующих антигенный пептид. Особенно предпочтительные заболевания включают злокачественные новообразования. Агенты, композиции и методы, описанные в данном документе, также могут использоваться для иммунизации или вакцинации для предотвращения заболевания, описанного в данном документе.
Согласно изобретению термин «заболевание» относится к любому патологическому состоянию, включая злокачественные новообразования, конкретно те формы злокачественных новообразований, которые описаны в данном документе.
Термин «нормальный» относится к здоровому состоянию или к состояниям у здорового объекта или ткани, т.е. к не патологическим состояниям, где «здоровый» предпочтительно означает не злокачественный.
«Заболевание, включающее клетки, экспрессирующие антиген» согласно изобретению означает, что детектируется экспрессия антигена в клетках больной ткани или органа. Экспрессия в клетках больной ткани или органа может повышаться по сравнению со здоровой тканью или органом. Повышение относится к повышению, по меньшей мере, на 10%, конкретно, по меньшей мере, на 20%, по меньшей мере, на 50%, по меньшей мере, на 100%, по меньшей мере, на 200%, по меньшей мере, на 500%, по меньшей мере, на 1000%, по меньшей мере, на 10000% или даже более. В одном воплощении, экспрессия обнаружена только в больной ткани, в то время как экспрессия в здоровой ткани репрессирована. Согласно изобретению заболевания, включающие или ассоциированные с клетками, экспрессирующими антиген, включают злокачественные новообразования.
Злокачественная опухоль (медицинский термин: злокачественное новообразование) представляет собой класс заболеваний, в которых группа клеток проявляет неконтролируемый рост (деление за пределами нормальных границ), инвазию (проникновение внутрь и разрушение соседних тканей), и иногда метастазирование (распространение в другие места в организме через лимфу или кровь). Эти три злокачественных свойства злокачественных образований отличают их от доброкачественных опухолей, которые самоограничиваются и не являются инвазивными или метастазирующими. Большинство злокачественных образований образуют опухоль, но некоторые, подобные лейкозу - нет.
Злокачественная опухоль по существу являются синонимом злокачественному новообразованию. Неоплазия, злокачественное новообразование и злокачественная опухоль по существу являются синонимами.
Согласно изобретению термин «опухоль» или «опухолевое заболевание» относятся к аномальному росту клеток (называемых неопластическими клетками, туморогенными клетками или опухолевыми клетками), предпочтительно образуя опухоль или поражение. Термином «опухолевая клетка» обозначают аномальную клетку, которая растет быстрой неконтролируемой клеточной пролиферацией и продолжает расти после стимулов, которые инициировали прекращение нового роста. Опухоли демонстрируют частичную или полную потерю структурной организации и функциональной координации с нормальной тканью, и обычно образуют отдельную массу ткани, которая может быть доброкачественной, предзлокачественной или злокачественной.
Доброкачественная опухоль представляет собой опухоль, которая лишена всех трех злокачественных свойств злокачественного новообразования. Таким образом, по определению доброкачественная опухоль не растет неограниченно, в агрессивной манере, не проникает в окружающие ее ткани и не распространяется в не соседние ткани (не метастазирует).
Неоплазма представляет собой аномальную массу ткани как результат неоплазии. Неоплазия (новый рост по-гречески) представляет собой аномальную пролиферацию клеток. Рост клеток превышен и не координируется с ростом нормальных тканей вокруг них. Рост продолжается в такой же избыточной манере даже после отмены стимулов. Обычно он вызывает появление узла или опухоли. Неоплазмы могут быть доброкачественными, предзлокачественными или злокачественными.
«Рост опухоли» или «опухолевый рост» согласно изобретению относится к тенденции опухоли увеличиваться в размере и/или к тенденции опухолевых клеток пролиферировать.
Для целей настоящего изобретения термины «злокачественное новообразование» и «заболевание, ассоциированное со злокачественным образованием» используются взаимозаменяемо с терминами «опухоль» и «опухолевое заболевание».
Злокачественные заболевания классифицируют по типу клеток, которые имеют сходство с опухолью, и, таким образом, предполагается, что данная ткань является происхождением опухоли. Это, соответственно, гистология и локализация.
Термины «злокачественное новообразование» или «рак» согласно изобретению включает лейкозы, семиномы, меланомы, тератомы, лифомы, нейробластомы, глиомы, ректальный рак, рак эндометрия, рак почки, рак надпочечника, рак щитовидной железы, злокачественное новообразование крови, рак кожи, рак мозга, рак шейки матки, рак кишечника, рак печени, рак толстой кишки, рак желудка, рак кишечника, рак головы и шеи, желудочно-кишечный рак, рак лимфатического узла, рак пищевода, колоректальный рак, рак поджелудочной железы, рак уха, горла и носа (ENT), рак молочной железы, рак предстательной железы, рак матки, рак яичника, рак легкого и их метастазы. Примеры включают карциному легкого, карциному молочной железы, карциному толстой кишки, почечноклеточную карциному, карциному шейки матки или метастазы типов рака или опухоли, описанные выше. Термин злокачественное новообразование согласно изобретению также включает метастазы злокачественного новообразования и рецидив злокачественного новообразования.
Основные типы рака легкого представляют собой мелкоклеточный рак легкого (SCLC) и немелкоклеточный рак легкого (NSCLC). Существует три основных типа немелкоклеточного рака легкого: плоскоклеточный рак легкого, аденокарцинома и крупноклеточный рак легкого. Аденокарциномы насчитывают приблизительно 10% от всех случаев рака легкого. Данное злокачественное новообразование обычно наблюдается в периферических областях легких в противоположность мелкоклеточному раку легкого и плоскоклеточному раку легкого, которые оба имеют тенденцию к более центральной локализации.
Рак кожи представляет собой злокачественный рост на коже. Наиболее распространенными раками кожи являются базальноклеточный рак, плоскоклеточный рак и меланома. Злокачественная меланома представляет собой серьезный тип рака кожи. Он возникает из-за неконтролируемого роста пигментных клеток, называемых меланоцитами.
Согласно изобретению «карцинома» представляет собой злокачественную опухоль, возникшую из эпителиальных клеток. Данная группа характеризует наиболее распространенные злокачественные новообразования, включающие распространенные формы рака молочной железы, предстательной железы, легкого и толстой кишки.
«Бронхиолярная карцинома» представляет собой карциному легкого, которая, как считают, имеет происхождение из эпителия концевых бронхиол, в которых неопластическая ткань распространяется по альвеолярным стенкам и вырастает в небольшие массы внутри альвеол. Муцин может быть обнаружен в некоторых таких клетках и в веществе альвеол, которые также включают денудированные клетки.
«Аденокарцинома» представляет собой злокачественное новообразование, которое происходит из ткани желез. Эта ткань также является частью крупной категории тканей, известной как эпителиальная ткань. Эпителиальная ткань включает кожу, железы и множество других тканей, которые выстилают полости и органы организма. Эпителий эмбриологически происходит из эктодермы, эндодермы и мезодермы. Чтобы быть отнесенными к аденокарциноме клеткам необязательно быть частью железы, при условии, что они обладают секреторными свойствами. Данная форма карциномы может встречаться у некоторых высших млекопитающих, включая человека. Хорошо дифференцированные аденокарциномы имеют тенденцию к наличию сходства с тканями железы, из которой они происходят, в то время как у плохо дифференцированных такого сходства может не быть. С помощью окрашивания клеток из биопсии, патолог определит, является ли опухоль аденокарциномой или относится к некоторым другим типам злокачественных новообразований. Аденокарциномы могут возникать во множестве тканей организма из-за природы повсеместного присутствия желез внутри организма. Хотя каждая из желез может не секретировать одни и те же вещества, если имеет место экзокринная функция у клетки, то она считается железистой, а ее злокачественная форма, таким образом, называется аденокарциномой. Злокачественные аденокарциномы проникают в другие ткани и часто метастазируют при условии наличия для этого достаточного времени. Аденокарцинома яичника представляет собой наиболее распространенный тип карциномы яичника. Она включает сывороточную и слизистую аденокарциному, светлоклеточную аденокарциному и эндометриоидную аденокарциному.
Почечноклеточная карцинома также известна в качестве почечноклеточного рака, или почечноклеточной аденокарциномы представляет собой рак почки, который происходит из ткани, выстилающей проксимальный извитой канадец, очень небольших каналов в почке, которые фильтруют кровь и удаляют продукты жизнедеятельности. Почечноклеточная карцинома представляет собой гораздо более распространенный тип рака почки у взрослых и наиболее летальный из всех опухолей мочеполовой системы. Различные подтипы почечноклеточных карцином представляют собой светлоклеточную почечноклеточную карциному и папиллярную почечноклеточную карциному. Светлоклеточная почечноклеточная карцинома представляет собой наиболее распространенную форму почечноклеточной карциномы. Под микроскопом клетки, которые составляют светлоклеточную почечноклеточную карциному, кажутся очень светлыми или прозрачными. Папиллярная почечноклеточная карцинома представляет собой второй наиболее распространенный подтип. Эти злокачественные новообразования образуют пальцеподобные отростки (называемые сосочками) в некоторых, если не в большинстве опухолей.
Лимфома и лейкоз представляют собой неоплазии, происходящие из гемопоэтических клеток (кроветворных).
Властная опухоль или бластома представляет собой опухоль (обычно злокачественную), которая имеет сходство с незрелой эмбриональной тканью. Многие из этих опухолей наиболее распространены среди детей.
Под «метастазированием» понимают распространение опухолевых клеток из места своего происхождения в другую часть организма. Образование метастазов представляет собой очень сложный процесс и зависит от отделения злокачественных опухолевых клеток от первичной опухоли, инвазии через внеклеточный матрикс, проникновения через эндотелиальную базальную мембрану для входа в брюшную полость и в сосуды, и затем после осуществления транспортировки кровотоком, от инфильтрации органов-мишеней. Наконец, рост новой опухоли, т.е. вторичной опухоли или метастатической опухоли в месте-мишени зависит от ангиогенеза. Метастазирование опухоли часто происходит даже после удаления первичной опухоли, так как опухолевые клетки или компоненты могут оставаться и развивать метастатический потенциал. В одном воплощении, термин «метастазирование» согласно изобретению относится к «отдаленному метастазированию», которое относится к метастазированию, которое отдалено от первичной опухоли и региональной системы лимфатических узлов.
Клетки вторичной метастатической опухоли имеют сходство с клетками исходной опухоли. Это означает, что если рак яичника метастазирует в печень, то вторичная опухоль будет состоять из аномальных клеток яичника, а не из аномальных клеток печени. Следовательно, опухоль в печени называют метастазирующим раком яичников, а не раком печени.
В раке яичника метастазирование может происходить следующими путями: посредством непосредственного контакта или распространения, он может проникать в близлежащую ткань или органы, расположенные близко или около яичников, такие как фаллопиевы трубы, матка, мочевой пузырь, прямая кишка, и т.д.; путем оседания или выпадения в аномальной полости, которая является наиболее распространенным путем распространения рака яичника. Опухолевые клетки разрывают поверхность массы яичника и «капают» на другие структуры в брюшной полости, такие как печень, желудок, толстая кишка или диафрагма; путем отрыва от массы яичника, проникновения в лимфатические сосуды и затем путем передвижения к другим областям организма или к отдаленным органам, таким как легкое или печень; путем отрыва от массы яичника, проникновения в кровеносную систему и затем путем передвижения к другим областям организма или к отдаленным органам.
Согласно изобретению метастатический рак яичника включает злокачественную опухоль в фаллопиевых трубах, злокачественную опухоль в органах брюшной полости, таких как кишечник, злокачественную опухоль в матке, злокачественную опухоль в мочевом пузыре, злокачественную опухоль в прямой кишке, злокачественную опухоль в печени, злокачественную опухоль в желудке, злокачественную опухоль в толстой кишке, злокачественную опухоль в диафрагме, злокачественную опухоль в легких, злокачественную опухоль в области, выстилающей брюшную или тазовую полость (брюшина), и злокачественную опухоль в мозге. Аналогично, метастатический рак легкого относится к злокачественному образованию, которое распространяется от легких к отдаленным и/или к нескольким местам в организме и включает злокачественную опухоль в печени, злокачественную опухоль в надпочечной железе, злокачественную опухоль в костях и злокачественную опухоль в мозге.
Термин «циркулирующие опухолевые клетки» или «СТС» относится к клеткам, которые открепляются от первичной опухоли или от опухолевых метастазов и циркулируют в кровотоке. СТС могут составлять посевной материал для последующего роста дополнительных опухолей (метастаз) в различных тканях. Циркулирующие опухолевые клетки обнаруживаются с частотой порядка 1-10 СТС на 1 мл цельной крови пациентов с метастазирующим заболеванием. Для выделения СТС были разработаны исследовательские методы. В данной области описано несколько исследовательских методов для выделения СТС, например, методы, которые используют тот факт, что эпителиальные клетки обычно экспрессируют белок клеточной адгезии ЕрСАМ, который отсутствует в нормальных клетках крови. Захват на основе иммуномагнитных микросфер включает в себя обработку образцов крови с помощью антитела к ЕрСАМ, которое конъюгировано с магнитными частицами, с последующим разделением связанных клеток в магнитном поле. Выделенные клетки затем окрашивали с антителом к другому эпителиальному маркеру цитокератину, а также к обычному лейкоцитарному маркеру CD45, так чтобы отличить редкие СТС от контаминирующих лейкоцитов. Этот трудоемкий и полуавтоматизированный способ идентифицирует СТС со средним выходом приблизительно 1 СТС/мл и чистотой 0,1% (Allard et al., 2004: Clin Cancer Res 10, 6897-6904). Второй метод выделения СТС использует микрофлюидное устройство захвата СТС, которое включает течение цельной крови через камеру с 80000 встроенных микростолбиков, которые становятся функциональными из-за покрытия антителом к ЕрСАМ. Затем СТС окрашивают вторичными антителами либо против цитокератина, либо против тканеспецифичных маркеров, таких как PSA в раке предстательной железы или HER2 в раке молочной железы, и визуализируют с помощью автоматизированного сканирования микростолбиков на множестве панелей вдоль трехмерных координат. СТС-чипы способны идентифицировать цитокератин-положительные циркулирующие опухолевые клетки у пациентов со средним выходом 50 клеток/мл и чистотой в интервале 1-80% (Nagrath et al., 2007: Nature 450, 1235-1239). Другая возможность выделения СТС представляет собой использование теста «CellSearch™ Circulating Tumor Cell» (СТС) от «Veridex, LLC» (Раритан, Нью-Джерси), который захватывает, идентифицирует и считает СТС в пробирке крови. Система «CellSearch™» представляет собой методологию, одобренную Управлением по контролю за пищевыми продуктами и лекарственными препаратами США (PDA), для подсчета СТС в цельной крови, который основан на комбинации иммуномагнитного мечения и автоматизированной цифровой микроскопии. Существуют другие методы для выделения СТС, описанные в научной литературе, каждый из которых может использоваться совместно с настоящим изобретением.
Возврат или рецидив происходит, когда человек снова подвергается воздействию состояния, которому он подвергался в прошлом. Например, если пациент страдал от опухолевого заболевания, получил успешное лечение указанного заболевания, и у него снова развилось указанное заболевание, то это указанное вновь развившееся заболевание может рассматриваться как возврат или рецидив. Однако согласно изобретению возврат или рецидив опухолевого заболевания необязательно может происходить в месте исходного опухолевого заболевания. Таким образом, например, если пациент страдал от опухоли яичников и получил успешное лечение, то возврат или рецидив может представлять собой появление опухоли в месте, отличном от яичников. Возврат или рецидив опухоли также включает ситуации, где опухоль появляется в месте, отличном от места исходной опухоли, а также в месте исходной опухоли. Предпочтительно, исходная опухоль, для которой пациент получил лечение, представляет собой первичную опухоль, а опухоль в месте, отличном от места исходной опухоли, представляет собой вторичную или метастатическую опухоль.
Термин «лечить» означает вводить соединение или композицию, как описано в данном документе, объекту с целью предотвращения или устранения заболевания, включая уменьшение размера опухоли или количества опухолей у объекта; прекращение или замедление заболевания у объекта; ингибирование или замедление развития нового заболевания у объекта; уменьшение частоты или тяжести симптомов и/или рецидивов у объекта, который в настоящее время имеет или ранее имел заболевание; и/или продление, т.е. увеличение продолжительности жизни объекта. Конкретно, термин «лечение заболевания» включает излечение, сокращение продолжительности, ослабление, предотвращение, замедление или ингибирование прогрессии или ухудшения, или предотвращение или задержка приступа заболевания или его симптомов.
Термин «имеющий риск» означает объекта, т.е. пациента, который идентифицирован как имеющий более высокие, чем обычные, шансы развития заболевания, конкретно, злокачественного новообразования, по сравнению с общей популяцией. Кроме того, объект, который имел или который имеет в настоящее время заболевание, конкретно злокачественное новообразование, представляет собой объекта, который имеет повышенный риск развития заболевания, как например у объекта может продолжиться развитие заболевания. Объекты, которые в настоящее время имеют или которые имели злокачественное новообразование, также имеют повышенный риск метастазирования злокачественного новообразования.
Термин «иммунотерапия» относится к лечению, включающему активацию специфической иммунной реакции. В контексте настоящего изобретения термины, такие как «защищает», «предотвращает», «профилактический», «превентивный», или «защитный» относятся к предотвращению или лечению или к обоим в отношении появления и/или продолжения заболевания у объекта и конкретно для минимизации шанса, что у объекта разовьется заболевание, или для задержки развития заболевания. Например, человек с риском развития опухоли, как описано выше, будет кандидатом для терапии для предотвращения опухоли.
Профилактическое введение иммунотерапии, например, профилактическое введение композиции по изобретению предпочтительно защищает реципиента от развития заболевания. Терапевтическое введение иммунотерапии, например, терапевтическое введение композиции по изобретению может приводить к ингибированию прогресса/роста заболевания. Ингибирование включает замедление прогресса/роста заболевания, конкретно нарушение прогрессии заболевания, которое предпочтительно приводит к устранению заболевания.
Иммунотерапия может осуществляться с использованием любого и множества способов, в которых агенты, представленные в данном документе, действуют для удаления больных клеток из пациента. Такое удаление может иметь место как результат усиления или индуцирования у пациента иммунного ответа, специфичного к антигену или клетке, экспрессирующей антиген.
В некоторых воплощениях иммунотерапия может быть активной иммунотерапией, в которой лечение основано на in vivo стимулировании эндогенной иммунной системы хозяина для реагирования против больных клеток, с введением агентов, модифицирующих иммунный ответ (таких как полипептиды и нуклеиновые кислоты, представленные в данном документе).
Агенты и композиции, представленные в данном документе, могут использоваться индивидуально или в комбинации со стандартными терапевтическими схемами, такими как хирургическая операция, облучение, химиотерапия и/или трансплантация костного мозга (аутологичная, синергическая, аллогенная или чужеродная).
Термин «иммунизация» или «вакцинация» описывает процесс лечения объекта с целью индуцирования иммунного ответа для терапевтических или профилактических целей.
Термин «in vivo» относится к ситуации в субъекте.
Термины «объект», «индивидуум», «организм» или «пациент» используются взаимозаменяемо и относятся к позвоночным животным, предпочтительно к млекопитающим. Например, млекопитающие в контексте настоящего изобретения представляют собой людей, приматов, отличных от человека, домашних животных, таких как собаки, кошки, овцы, крупный рогатый скот, козы, свиньи, лошади и т.д., лабораторных животных, таких как мыши, крысы, кролики, морские свинки и т.д., а также животных в неволе, таких как животные зоопарков. Термин «животное» при использовании в данном документе включает человека. Термин «объект» также может включать пациента, т.е. животное, предпочтительно человека, имеющего заболевание, предпочтительно, описанное в данном документе.
Термин «аутологичный» используется для описания чего-либо, что выделено из того же объекта. Например, «аутологичный трансплантат» относится к трансплантату ткани или органов, с происхождением из того же объекта. Такие процедуры являются предпочтительными, так как они преодолевают иммунологический барьер, который в ином случае приводит к отторжению.
Термин «гетерологичный» используется для описания чего-то, что состоит из множества различных элементов. Например, костный мозг, перенесенный от одного индивидуума к другому индивидууму, представляет собой гетерологичный трансплантат. Гетерологичный ген представляет собой ген, выделенный из источника, отличного от объекта.
В качестве части композиции для иммунизации или вакцинации, предпочтительно, если один или нескольких агентов, описанных в данном документе, вводятся вместе с одним или несколькими адъювантами для индукции иммунного ответа или для повышения иммунного ответа. Термин «адъювант» относится к соединениям, которые удлиняют или усиливают или ускоряют иммунный ответ. Композиция по настоящему изобретению предпочтительно проявляет свой эффект без добавления адъювантов. Кроме того, композиция по настоящему изобретению может содержать любой известный адъювант. Адъюванты включают гетерологичную группу соединений, таких как эмульсии (например, адъюванты Фрейнда), минеральные соединения (такие как квасцы), бактериальные продукты (такие как Bordetella pertussis токсин), липосомы и иммуно-стимулирующие комплексы. Примеры адъювантов представляют собой монофосфорил-липид-А (MPL, «SmithKline Beecham»). Сапонины, такие как QS21 («SmithKline Beecham»), DQS21 («SmithKline Beecham»; WO 96/33739), QS7, QS17, QS18, и QS-L1 (So et al., 1997, Mol. Cells 7:178-186), неполные адъюванты Фрейнда, полные адъюванты Фрейнда, витамин Е, монтанид, квасцы, CpG-олигнуклеотиды (Krieg et al., 1995, Nature 374:546-549), и различные эмульсии вода-в-масле, которые готовят из биологически деградируемых масел, таких как сквален и/или токоферол.
Другие вещества, которые стимулируют иммунный ответ пациента, также могут быть введены. Например, возможно использование цитокинов в вакцинации, благодаря их регуляторным свойствам в отношении лимфоцитов. Такие цитокины включают, например, интерлейкин-12 (IL-12) который, как было показано, повышает защитное действие вакцин (см. Science 268:1432-1434, 1995), GM-CSF и IL-18.
Существует ряд соединений, которые усиливают иммунный ответ и которые, таким образом, можно использовать в вакцинации. Указанные соединения включают ко-стимулирующие молекулы, представленные в форме белков или нуклеиновых кислот, таких как В7-1 и В7-2 (CD80 и CD86, соответственно).
Согласно изобретению «образец опухоли» представляет собой образец, такой как физический образец, содержащий опухолевые или раковые клетки, такие как циркулирующие опухолевые клетки (СТС), конкретно образец ткани, включающий биологические жидкости и/или клеточный образец. Согласно изобретению «не онкогенный образец» представляет собой такой образец, как физический образец, не содержащий опухолевых или раковых клеток, таких как циркулирующие опухолевые клетки (СТС), конкретно, образец ткани, включающий биологические жидкости и/или клеточный образец. Такие физические образцы могут быть получены стандартным образом, как например, с помощью биопсии ткани, включая пункционную биопсию, и посредством забора крови, бронхиального аспирата, мокроты, мочи, экскрементов и других биологических жидкостей. Согласно изобретению термин «образец» также включает обработанный образец, такой как фракции или изоляты биологических образцов, например, нуклеиновые кислоты или клеточные изоляты.
Терапевтически активные агенты, вакцины и композиции, описанные в данном документе, могут вводиться любым подходящим путем, включая инъекцию или инфузию. Введение может проводиться, например, перорально, внутривенно, внутрибрюшинно, внутримышечно, подкожно или трансдермально, В одном воплощении введение проводится интранодально, как например, посредством инъекции в лимфатический узел. Другие формы введения предусматривают in vitro трансфекцию антигенпрезентирующих клеток, таких как дендритные клетки, нуклеиновыми кислотами, описанными в данном документе, с последующим введением антигенпрезентирующих клеток.
Агенты, описанные в данном документе, вводятся в эффективных количествах. «Эффективное количество» относится к количеству, которое достигает целевой реакции или целевого эффекта индивидуально или совместно с другими дозами. В случае лечения конкретного заболевания или конкретного состояния, целевая реакция предпочтительно относится к ингибированию течения заболевания. Ингибирование включает замедление прогресса заболевания и, в частности, прекращение или реверсию прогресса заболевания. Целевой реакцией при лечении заболевания или состояния также может быть задержка проявления или предотвращение приступа указанного заболевания или указанного состояния.
Эффективное количество агента, описанного в данном документе, будет зависеть от состояния, подвергаемого лечению, от тяжести заболевания, индивидуальных параметров пациента, включающих возраст, физиологическое состояние, размер и массу, продолжительность лечения, тип сопровождающейся терапии (если имеется), специфический путь введения и подобные факторы. Соответственно, вводимые дозы агентов, описанных в данном документе, могут зависеть от множества таких параметров. В случае, когда реакция пациента недостаточна при исходной дозе, могут использоваться более высокие дозы (или эффективно более высокие дозы, полученные с помощью другого более точно локализованного пути введения).
Фармацевтические композиции по изобретению предпочтительно являются стерильными и содержат эффективное количество терапевтически активного вещества для получения целевой реакции или целевого эффекта.
Фармацевтические композиции по изобретению, как правило, вводятся в фармацевтически совместимых количествах и в фармацевтически совместимом препарате. Термин «фармацевтически совместимый» относится к нетоксическому материалу, который не взаимодействует с действием активного компонента фармацевтической композиции. Препараты данного типа обычно могут содержать соли, буферные вещества, консерванты, носители, вспомогательные вещества, усиливающие иммунитет, такие как адъюванты, например, CpG-олигонуклеотиды, цитокины, сапонин, GM-CSF и/или РНК и где это необходимо, терапевтически активные соединения. При использовании в медицине соли должны быть фармацевтически совместимыми. Однако соли, которые не являются фармацевтически совместимыми, могут использоваться для приготовления фармацевтически совместимых солей и включены в изобретение. Фармакологически и фармацевтически совместимые соли данного типа включают в частности соли, приготовленные из следующих кислот: хлороводородной, бромоводородной, серной, азотной, фосфорной, малеиновой, уксусной, салициловой, лимонной, муравьиной, малоновой, янтарной кислоты и так далее. Фармацевтически совместимые соли также могут быть приготовлены в виде солей щелочных металлов или солей щелочноземельных металлов, таких как натриевые соли, калиевые соли или кальциевые соли.
Фармацевтическая композиция может включать фармацевтически совместимый носитель. Термин «носитель» относится к органическому или неорганическому компоненту, природного или синтетического происхождения, где активный компонент объединен с целью облегчения применения. Согласно изобретению термин «фармацевтически совместимый носитель» включает совместимые твердые или жидкие фильтры, разбавители или инкапсулирующие вещества, которые подходят для введения пациенту. Компоненты фармацевтической композиции по изобретению обычно такие, что отсутствует взаимодействие, которое существенно ослабляет фармацевтическую эффективность.
Фармацевтические композиции по изобретению могут содержать подходящие буферные вещества, такие как уксусная кислота в соли, борная кислота в соли и фосфорная кислота в соли.
Фармацевтические композиции, где необходимо, также могут содержать подходящие консерванты, такие как хлорид бензалкония, хлорбутанол, парабен и тимеросал.
Фармацевтические композиции обычно представлены в стандартной лекарственной форме и могут быть приготовлены способом, который известен, как таковой. Фармацевтические композиции по изобретению могут быть представлены в форме капсул, таблеток, пастилок, растворов, суспензий, сиропов, эликсиров или, например, в форме эмульсий.
Композиции, подходящие для парентерального введения, обычно включают стерильный водный или неводный препарат активного соединения, который предпочтительно является изотоническим по отношению к крови реципиента. Примеры совместимых носителей и растворителей это раствор Рингера и изотонический раствор хлорида натрия. Кроме того, обычно используются стерильные фиксированные масла в качестве раствора или суспензионной среды.
Настоящее изобретение описано подробно с помощью чертежей и примеров, представленных ниже, которые используются только для иллюстративных целей и не предназначены для ограничения. Благодаря описанию и примерам, следующие воплощения, которые вероятно включены в изобретение, доступны специалисту в данной области.
Чертежи
Фигура 1.
Верх: Способ обнаружения и приоритезации вероятно иммуногенных соматических мутаций в суммарных опухолевых образцах. Низ: Способ, применяемый к системе В16 и Black6.
Фигура 2: Пример Подтвержденной Мутации в Kif18b
Мутация, идентифицированная в гене Kif18b с помощью NGS секвенирования экзома, которую подтверждали с помощью секвенирования по Сэнгеру. В клетках дикого типа последовательность представляет собой Т/Т. В опухолевых клетках, последовательность представляет собой смесь T/G.
Фигура 3: Иммунологическая реактивность против мутированных последовательностей
Мышей (n=5) иммунизировали дважды (d0, d7) с использованием мутированных пептидных последовательностей (100 мкг + 50 мкг ПолиI:C; подкожно). На 12 день мышей умерщвляли и собирали клетки селезенки. Анализ «IFNγ ELISpot» осуществляли с использованием 5×105 клеток селезенки/на лунку в качестве эффекторов, и 5×104 дендритных клеток костного мозга, загруженных вместе с пептидами (2 мкг/мл в течение 2 ч при 37°С и 5% CO2) в качестве клеток-мишеней. Эффекторные клетки селезенки тестировали против мутированного пептида, пептида дикого типа и контрольного пептида (нуклеопротеина вируса везикулярного стоматита, VSV-NP, ак 52-59). Представлено среднее значение, измеряющее количество пятен, из которых фоновые пятна против VSV-NP вычитались для каждой мыши (незакрашенные круги: мышей иммунизировали пептидом дикого типа; закрашенные боксы: мышей иммунизировали мутированными пептидами). Данные представлены для каждой мыши и изображено среднее значение ± стандартная ошибка среднего.
Фигура 4: Преимущество выживания для мышей, вакцинированных новыми идентифицированными мутированными пептидными последовательностями.
Клетки B16F10 (7,5×104) инокуровали подкожно в d0. Мышей вакцинировали пептидом 30 («Jerini Peptide Technologies» (Берлин); 100 мкг пептид + 50 мкг поли I:С п.к. («Invivogen»)) в день - 4, день + 2 и день + 9. Контрольная группа получала только Поли I:С (50 мкг п.к.). Опухолевой рост отслеживали до дня + 16 *, p<0,05 в логарифмическом ранговом критерии (Mantel-Cox).
Фигура 5:
(А) Примеры повышенной белковой экспрессии (слева eGFP, справа Люцифераза) с РНК, оптимизированной для стабильной и эффективной трансляции (В) Пример полиэпитопной экспансии антиген-специфичных CD8+ и CD4+ Т-клеток с РНК, оптимизированной для эффективного таргетинга антигенов (см. Reference Kreiter, Konrad, Sester et al, Cancer Immunol. Immunother. 56:1577-1587, 2007). Т (С) Пример пре-клинического доказательства противоопухолевой эффективности в модели В16 меланомы с использованием РНК-вакцины, которая кодирует одиночный эпитоп (OVA-SIINFEKL). Данные по выживаемости получали для мышей, обработанных с помощью вакцины индивидуально или с помощью вакцины в комбинации с адъювантом. (D) Дизайн индивидуальных полинеоэпитопных вакцин. Носитель вакцины объединяет функциональные элементы для повышенной экспрессии и оптимизированной иммуногенности. До 30 мутированных эпитопов, которые разделены линкерами, могут быть интегрированы в молекулу в контексте их природных последовательностей.
Фигура 6: Дизайн конструкта
(А) Схематическая диаграмма полиэпитопного РНК-конструкта. Cap: кэп-аналог; 5'UTR: 5'нетранслируемая область; L: Линкер; Seq. 1: РНК-последовательность, кодирующая пептид, содержащий мутированную ак; 3'UTR: 3'нетранслируемая область; poly-А: поли-А хвост. (В) последовательность РНК-конструктов, кодирующих 2 ак последовательности, включающие мутированную ак из B16F10. Старт- и стоп-кодон, а также сигнальный пептид и последовательность MITD не являются частью схематического чертежа, который обозначен с помощью «…».
Фигура 7: Функциональность РНК-полиэпитопа
(А-С) Данные для «IFNγ ELISpot» с использованием 5×105 клеток селезенки на лунку в качестве эффекторов и 5×104 BMDC в качестве клеток-мишеней. BMDC загружали пептидом (2 мкг/мл в течение 2 ч при 37°С и 5% CO2) или трансфецировали РНК (20 мкг) с помощью электропорации. Контрольная РНК представляла собой eGFP (левая панель) или РНК-конструкт, кодирующий 2 несвязанных пептида, содержащих мутированные ак, разделенные линкером. Данные представлены в виде среднего значения ± станд. ош. среднего. (А) Представлены данные для мутированного пептида 30, пептида дикого типа 30 и РНК, кодирующей мутацию 30 и 31. (В) Представлены данные для мутированного пептида 12, пептида дикого типа 12 и РНК, кодирующей мутацию 12 и 39. (С) Изображен характерный ELISpot-скан для считывания данных одной мыши, представленной в (В).
Фигура 8: Два воплощения полинеоэпитопных РНК-вакцин, демонстрирующих соединяющие эпитопы
РНК-вакцина может быть сконструирована вместе с линкерами (верх) или без линкеров (низ) между пептидами, кодирующими мутацию. Хорошие эпитопы включают те, которые включают соматическую мутацию ("*") и связываются с молекулами МНС. Плохие эпитопы включают эпитопы, которые связываются с молекулами МНС, но содержат либо части двух пептидов (низ), либо части пептида и линкерную последовательность (верх).
Фигура 9: Выявление и описание характеристик «Т-клеточного поддающегося воздействию лекарственных средств мутанома (mutanome)»
(А) Блок-схема дает понятие об экспериментальной процедуре, начиная от образцов B16F10 и C57BL/6 до вывода данных «ELISPOT». (В) Представлены количество хитов для каждой стадии оценки и процесс отбора мутаций для подтверждения ДНК и иммуногенное тестирование. Мутации, отобранные для подтверждения и тестирования иммуногенности, это те, которые, согласно прогнозу, являются иммуногенными, и которые находятся в генах, экспрессирующихся при RPKM>10. (С) Т-клеточный поддающийся воздействию лекарственных средств «мутаном» картировали в геноме B16F10. Циклы от внешней к внутренней позиции для следующих подгрупп: (1) присутствие в каждом из трех повторений, (2) FDR<0,05, (3) локализованы в областях, кодирующих белок, (4) вызывают несинонимичные изменения, (5) локализованы в экспрессирующихся генах, и (6) представлены в подтвержденном виде. Мышиные хромосомы (внешний круг), плотность генов (зеленый), экспрессия генов (зеленый(низкая)/желтый/красный(высокая)), и соматические мутации (оранжевый).
Фигура 10: Иммунный ответ, вызванный in vivo с помощью вакцинации мышей длинными синтетическими пептидами, представляющими мутацию
(А,В) «IFN-γ ELISPOT» анализ Т-клеточных эффекторов из мышей, вакцинированных пептидами, кодирующими мутацию. Столбцы характеризуют средние значения (± станд. ош. среднего) 5 мышей на группу. Звездочки указывают статистически значимые различия реактивности против мутации и против пептида дикого типа (критерий Стьюдента; p-значение < 0,05). (А) Спленоциты вакцинированных мышей повторно стимулировали с помощью BMDC, трансфецированных пептидом, кодирующим мутацию, используемым для вакцинации, соответствующим пептидом дикого типа и чужеродным контрольным пептидом (VSV-NP). (В) Для анализа Т-клеточной реактивности против эндогенно процессированных мутаций спленоциты вакцинированных мышей повторно стимулировали с помощью BMDC, трансфецированных контрольной РНК (eGFP) или РНК, кодирующей указанную мутацию. (С) Мутация 30 (ген Kif18B, белок Q6PFD6, мутация p.K739N). Проведение секвенирования по Сэнгеру и последовательность мутации (верх). Локализация белковых доменов и мутации (низ).
Фигура 11: Противоопухолевые эффекты вакцин с мутированным пептидом у мышей с агрессивно растущими опухолями B16F10
(A) C57BL/6 мышам (n=7) инокулировали 7.5×104 В16Р10-клеток п.к. в бок мыши. На 3 и 10 день после инокуляции опухоли, мышей вакцинировали с помощью 100 μг пептида MUT30 или MUT44+50 μг поли(I:С) или с помощью адъюванта отдельно. (В) C57BL/6 мыши (n=5) получали одну иммунизацию 100 μг MUT30 пептида + 50 μг поли(I:С) на - 4 день. В день 0 инокулировали п.к. 7.5×104 клеток B16F10 в бок мыши. Бустерную иммунизацию пептидом MUT30 (+ поли(I:С)) проводили на 2 и на 9 день.
Кривая выживаемости Каплана-Мейера (слева). Кинетика опухолевого роста (справа).
Фигура 12: Вакцинация с помощью РНК, кодирующих мутации, приводит к CD4+ и CD8+ Т-клеточному ответу
Данные анализа окрашивания внутриклеточных цитокинов для IFN-γ в CD4+ и CD8+ Т-клеточных эффекторах, полученных из мышей, вакцинированных РНК, кодирующей мутации. РНК кодировали 1 (Моноэпитоп, верхний ряд), 2 (Биэпитоп, средний ряд), или 16 (Полиэпитоп, нижний ряд) различных мутаций. Пятна характеризуют средние значения для 3 мышей на группу. Звездочки указывают статистически значимые различия реактивности против мутации и против контрольного пептида (VSV-NP) (критерий Стьюдента; p-значение < 0,05). Графики FACS демонстрируют эффекторы из животного с наивысшей секрецией IFN-γ для каждой мутации и выявляют фенотип Т-клеточного ответа.
Фигура 13: Вакцинация с использованием Полиэпитопной РНК, кодирующей мутации, приводит к Т-клеточным ответам против нескольких мутаций
«IFN-γ ELISPOT» анализ Т-клеточных эффекторов из мышей, вакцинированных полиэпитопным пептидом, кодирующим мутации, который несет 16 различных мутаций. Столбцы представляют средние значения (± станд. ош. среднего) для 3 мышей на группу. Фотографии демонстрируют три копии лунок с клетками из одного типичного животного, повторно стимулированными с использованием указанных пептидов.
Фигура 14: Вакцинация с использованием 5 различных модельных эпитопов, кодируемых одной РНК, приводит к иммунному ответу против всех кодируемых эпитопов
A) «IFN-γ ELISPOT» анализ Т-клеточных эффекторов из мышей, вакцинированных модельным полиэпитопным пептидом, кодирующим мутации, который несет 5 различных модельных эпитопов (SIINFEKL, Trp2, VSV-NP, Inf-NP, OVA класса II). Спленоциты повторно стимулировали с использованием указанных пептидов. Пятна характеризуют средние трех копий лунок из 5 мышей на группу. В) Окрашивание пентамером лимфоцитов крови одной контрольной мыши и одной мыши, иммунизированной модельным полиэпитопным пептидом. Окрашенные Inf-NP пентамером клетки CD8+ специфичны к пептиду Inf-NP.
Фигура 15: А CD4+ Т-клетки, индуцирующие мутацию, могут индуцировать мощный противоопухолевый эффект против меланомы B16F10 в синергии со слабым CD8+ Т-клеточным эпитопом
Мышам C57BL/6 (n=8) инокулировали 1×105 клеток B16F10 п.к. в бок. На 3, 10 и 17 день после инокуляции опухоли мышей вакцинировали 100 μг MUT30, Trp2 или с обоими пептидами + 50 μг поли(I:С). А) представлено среднее значение кинетики опухолевого роста для каждой группы. На 28 день средние значения между группами одной обработки и необработанными животными и группой комбинированной обработки статистически различны (тест Манна-Уитни, p-значение < 0,05). В) График выживания Каплана-Мейера для различных групп. Кривые выживания мышей, вакцинированных MUT30 и MUT30 + Trp2, являются статистически различными (логарифмический ранговый критерий, p-значение = 0,0029).
Фигура 16: Обзор способа обнаружения соматических мутаций в В16
Количество индивидуальных стадий дано как пример для одного образца В16, по сравнению с одним образцом black6. «Экзоны» служат для обозначения координат экзонов, определенных всеми белок-кодирующими транскриптами RefSeq.
Фигура 17: Диаграмма Венна, демонстрирует количество соматических вариаций в белок-кодирующих экзонах, обнаруженных одним или двумя или всеми тремя программными инструментами, соответственно.
Количества рассчитывали после фильтрации и представляют собой консенсус всех трех образцов.
Фигура 18: А Примеры обнаруженных однонуклеотидных вариаций: Соматическая мутация, обнаруженная во всех трех образцах В16 (слева), несоматическая мутация, обнаруженная во всех образцах В16 и black6 (посередине) и мутация, обнаруженная только в одном образце black6 (справа). В Вычисленное распределение FDR для набора данных, проверенные мутации которого были отобраны; распределение визуализировано как средняя расчетная кривая ROC с серыми столбиками, указывающими на 95% доверительный интервал для среднего в обоих координатах при единообразно отобранных местоположениях. Среднее получено из распределения расчетных кривых ROC для FDR всех возможных 18 комбинаций (см. текст).
Фигура 19: А Оценочные кривые ROC для сравнения трех различных программных инструментов (дубликаты, 38х покрытие). В Расчетные кривые ROC для сравнения различных средних глубин секвенирования («samtools», без реплик). 38х служит для обозначения покрытия, полученного экспериментом, при этом дискретизация других покрытий была снижена начиная с этих данных. С Расчетные кривые ROC визуализируют эффект воспроизведения эксперимента (38х покрытие, «samtools»). D Расчетные кривые ROC для различных протоколов секвенирования («samtools», без реплик). Кривые оценивали с помощью результатов библиотеки 2×100 нт.
Фигура 20: А Десять проверенных мутаций с наиболее низкими значениями FDR, выбранные с помощью оптимального набора параметров из конечного набора из 2396 вариаций. Ни одна из этих мутаций не присутствует в dbSNP (версия 128; сборка генома mm9). В Относительное количество вариаций, обнаруженное в той же базе данных, что и А, для данного предела FDR, нанесенное на график отдельно для всех вариантов в базе данных и проверенных мутаций. Для визуальной ясности показаны только значения от 0 до 10% FDR.
Фигура 21: Противоопухолевая активность кодирующей мутацию полиэпитопной РНК-вакцины Мыши C57BL/6 (n=10) инокулировали 1×105 клеток B16F10 п.к. в бок мыши. В день 3, 6, 10, 17 и 21 после инокуляции опухоли, мышей вакцинировали полиэпитопной РНК, вместе с липосомным реактивом для трансфекции РНК. Контрольная группа получала липосомы без РНК. На фигуре показан график выживания Каплана-Мейера для различных групп. Кривые выживания статистически отличны (логарифмический ранговый критерий, p-значение = 0,0008).
Примеры
Методы и способы, использованные в данном документе, описаны или осуществляются так, как по существу известно и как описано, например, в Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd Edition (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. Все способы, включая применение наборов и реактивов, осуществляются согласно информации изготовителей, если конкретно не указано иное.
Пример 1. Детекция и приориетизация мутаций
Вначале мы продемонстрировали объективное профилирование образцов опухоли и нормы, которое основано на результатах секвенирования, для идентификации соматических мутаций. Мы продемонстрировали это не только для образцов основной части опухоли, но также, впервые, мы продемонстрировали способность идентифицировать мутации из индивидуальных циркулирующих опухолевых клеток. Затем мы приоритизировали мутации для включения в полинеоэпитопную вакцину на основе спрогнозированной иммуногенности мутаций и продемонстрировали, что идентифицированные мутации действительно являются иммуногенными.
Детекция мутаций
Обоснование использования СТС: детекция циркулирующих опухолевых клеток (СТС) из периферической крови онкопациентов является признанным независимым прогностическим маркером клинического течения опухолевого заболевания (Pantel et al, Trends Mol Med 2010; 16(9):398-406). На протяжении многих лет, клиническое значение СТС были предметом интенсивных научных и клинических исследований в онкологии. Было показано, что детекция СТС в крови пациентов с метастатическим раком молочной железы, предстательной железы и колоректальным раком, имеет прогностическое значение, обеспечивая дополнительную информацию для традиционных методов имеджинга и других прогностических опухолевых биомаркеров. Последовательно забранные образцы крови, взятые у пациента до, в ранней стадии, и после лечения терапевтическим агентом (системным или таргетным) содержат информацию об ответе на лечение/неудаче лечения. Молекулярный анализ обладающих лекарственной устойчивостью СТС может обеспечить дальнейшее понимание механизмов устойчивости (например, мутаций в определенных сигнальных путях или потере целевой экспрессии) у индивидуальных пациентов. Дополнительная возможность профилирования и генетического описания СТС заключается в идентификации новых мишеней злокачественных опухолей для разработки новых таргетных терапий. Эта новая диагностическая стратегия упоминается как «Жидкая Биопсия Опухоли». Поскольку профилирование может быть осуществлено быстро и периодически, с необходимостью получения только крови пациента без хирургического вмешательства, то данный подход может обеспечить обзор состояния опухоли в «реальном времени».
Мутации опухолевых клеток Мы продемонстрировали нашу способность идентифицировать мутации с помощью клеток меланомы В16, захвата экзома для экстракции белок-кодирующих областей, секвенирования следующего поколения с помощью нашего «HiSeq 2000», с последующим биоинформатическим анализом с помощью нашего программного конвейера «iCAM» (фиг.1). Мы идентифицировали 2448 несинонимичных мутаций и отобрали 50 для подтверждения. Мы смогли подтвердить все 50 соматических мутаций.
Ниже представлен пример влияния на белок обнаруженной соматической мутации в клетках меланомы В16:
Kif18b, NM_197959, экзон 3
Мутация (+15 ак) SPSKPSFQEFVDWENVSPELNSTDQPFLPS
Дикий тип (+15 ак) SPSKPSFQEFVDWEKVSPELNSTDQPFLPS
Мутации индивидуальных циркулирующих опухолевых клеток (СТС): Затем мы смогли идентифицировать опухоль-специфичные соматические мутации NGS-профилированием РНК из одиночных СТС. Меченные клетки меланомы В16 внутривенно инъецировали в мышиные хвосты, мышей умерщвляли, кровь собирали из сердец, клетки сортировали для извлечения меченных циркулирующих клеток В16 (СТС), экстрагировали РНК, осуществляли «SMART» кДНК-синтез и неспецифическую амплификацию, а затем NGS-анализ RNA-Seq и последующий анализ данных (ниже).
Мы профилировали восемь индивидуальных СТС и идентифицировали соматические мутации. Более того, в восьми из восьми клеток, идентифицировали ранее идентифицированные мутации. Во множестве случаев, данные демонстрируют гетерогенность на уровне индивидуальных клеток. Например, в положении 144078227 на хромосоме 2 (сборка mm9) в гене Snx15, в двух клетках был эталонный нуклеотид (С), тогда как в двух других клетках был мутированный нуклеотид (Т).
Это говорит о том, что мы способны профилировать индивидуальные СТС для идентификации соматических мутаций, фундаментальный путь к iVAC (индивидуализированной вакцине) «в реальном времени», в которых пациенты профилированы соответствующим образом и результаты отражают текущий статус пациента, а не статус в более раннюю временную точку. Более того, это говорит о том, что мы способны идентифицировать гетерогенные соматические мутации, которые присутствуют в поднаборе опухолевых клеток, что позволяет оценить частоту мутаций, например, для идентификации основных мутаций и редких мутаций.
Способы
Образцы: Для эксперимента профилирования, образцы включают 5-10 мм образцы хвостов мышей C57BL/6 («Black6») и сильноагрессивные мышиные клетки меланомы («В16»), которые изначально были получены из мышей Black6.
Циркулирующие опухолевые клетки (СТС) были созданы с использованием флуоресцентно меченых клеток меланомы В16. Клетки В16 ресуспендировали в PBS и добавляли к ним равный объем свежеприготовленного раствора CFSE (5 μМ в PBS). Образец осторожно перемешивали вибрационным смесителем, с последующей инкубацией в течение 10 минут при комнатной температуре. Для остановки реакции мечения, равное количество PBS, содержащего 20% FSC добавляли к образцу и осторожно смешивали вибрационным смесителем. После 20 минутной инкубации при комнатной температуре клетки дважды отмывали с помощью PBS. В конце клетки ресуспендировали в PBS и инъецировали внутривенно (в.в.) мышам. Через 3 минуты мышей умерщвляли и собирали кровь.
Эритроциты из образцов крови лизировали добавлением 1,5 мл свежеприготовленного раствора «PharmLyse» («Beckton Dickinson») на 100 мкл крови. После одной стадии отмывки добавляли к образцу 7-AAD и инкубировали в течение 5 минут при комнатной температуре. За инкубацией следовали две стадии отмывки и образец ресуспендировали в 500 мкл PBS.
Меченные CFSE циркулирующие клетки В16 сортировали с помощью клеточного сортера «Aria I» («BD»). Одиночные клетки сортировали в 96-луночные планшеты с v-образным дном, в которые был добавлен буфер «RLT» («Qiagen»), в объеме 50 мкл/лунку. После окончания сортировки планшеты хранили при -80°С до начала экстракции нуклеиновых кислот и приготовления образцов.
Экстракция нуклеиновых кислот и приготовление образцов: нуклеиновые кислоты из клеток В16 (ДНК и РНК) и ткани хвоста Black6 (ДНК) экстрагировали с помощью набора «Qiagen DNeasy Blood and Tissue kit» (для ДНК) и набора «Qiagen RNeasy Micro kit» (для РНК).
Из индивидуальных отсортированных СТС экстрагировали РНК и осуществляли SMART-синтез кДНК и неспецифическую амплификацию. РНК из сортированных СТС-клеток экстрагировали с помощью набора «RNeasy Micro Kit» («Qiagen», Хильден, Германия) согласно инструкциям поставщика. Модифицированный протокол «BD SMART» использовали для синтеза кДНК: Обратную транскриптазу «Mint» («Evrogen», Москва, Россия) объединяли с олиго(dT)-T-праймером long для праймирования реакции синтеза первой цепи и TS-short ("Eurogentec S.A.», Сераинг, Бельгия), вводящим олиго(рибо0) последовательность, для обеспечения возможности создание удлиненной матрицы посредством активности терминальной трансферазы у обратной транскриптазы и для переключения матрицы [Chenchik, A., Y. et al. 1998. Generation and use of high quality cDNA from small amounts of total RNA by SMART PCR. In Gene Cloning and Analysis by RT-PCR. P. L. J. Siebert, ed. BioTechniques Books, MA, Natick. 305-319]. Первая цепь кДНК, синтезированная согласно инструкциям производителя подверглась амплификации в течение 35 циклов с 5 Ед. полимеразы «PfuUltra Hotstart High-Fidelity DNA Polymerase» ("Stratagene», Ла-Хойя, Калифорния) и 0,48 μM праймера TS-PCR в присутствии 200 μM dNTP (условия амплификации: 2 мин. при 95°С в течение, 30 с при 94°С, 30 с при 65°С, 1 мин. при 72°С в течение, конечное удлинение в течение 6 мин. при 72°С). Успешную амплификацию СТС-генов контролировали с помощью специфических праймеров для отслеживания актина и GAPDH.
Секвенирование следующего поколения, ДНК-секвенирование: Захват экзома для ресеквенирования ДНК осуществляли с помощью теста для захвата на основе раствора «Agilent Sure-Select» [Gnirke A et al: Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009, 27:182-189], в данном случае спроектированном для захвата всех областей, кодирующих белки мыши.
Вкратце, 3 мкг очищенной геномной ДНК фрагментировали до 150-200 п.о. с помощью ультразвукового устройства «Covaris S2». Концы фрагментов гДНК достраивали с помощью Т4 ДНК-полимеразы, фрагмента Кленова и фосфорилировали по 5' с помощью Т4 полинуклеотидкиназы. гДНК-фрагменты с тупыми концами 3'-аденилировали с использованием фрагмента Кленова (от 3' к 5' экзо минус). 3'-концевые одиночные Т-перекрывающиеся парные концевые адаптеры «Illumina» лигировали к гДНК-фрагментам с помощью Т4 ДНК-лигазы в молярном соотношении адаптера к вставке геномной ДНК 10:1. Лигированные с адаптерами фрагменты гДНК обогащали, добавляли последовательности, специфичные для предварительного захвата и проточной кюветы, с помощью праймеров «Illumina РЕ PCR 1.0» и «2.0» и полимеразы «Herculase II» («Agilent») в течение 4 циклов ПЦР.
500 нг дотированных с адаптером обогащенных ПЦР фрагментов гДНК гибридизовали с РНК-библиотекой биотинилированных приманок цельного мышиного экзома «SureSelect» фирмы «Agilent» в течение 24 часов при 65°С. Гибридизованные комплексы гДНК/РНК-приманка удаляли с помощью покрытых стрептавидином магнитных микросфер. Комплексы гДНК/РНК-приманка отмывали и РНК-приманки отщепляли в ходе элюции в элюционном буфере «SureSelect», оставляя захваченные обогащенные ПЦР гДНК фрагменты с дотированными адаптерами. Фрагменты гДНК амплифицировали ПЦР после захвата с помощью ДНК-полимеразы «Herculase II» (Agilent) и праймеров «SureSelect GA» в течение 10 циклов.
Все очистки осуществили в 1,8х объеме магнитных микросфер «AMPure ХР» («Agencourt»). Все контроли качества осуществляли с помощью теста «Qubit HS» от «Invitrogen», а размер фрагментов определяли с помощью теста «HS DNA» на «Bioanalyzer 2100» от «Agilent».
Библиотеки гДНК с обогащенным экзомом кластеризовали на «cBot» с помощью набора «Truseq SR cluster kit v2.5» при использовании 7 рМ, и 50 п.о. секвенировали на «Illumina HiSeq2000» с помощью набора «Truseq SBS kit-HS 50 bp».
Секввнирование следующего поколения, Секвенирование РНК (RNA-Seq): mRNA-seq кДНК-библиотеки с «штрих-кодами» получали из 5 мкг общей РНК с помощью модифицированной версии протокола «Illumina mRNA-seq». мРНК выделяли с помощью магнитных микросфер «Seramag Oligo(dT)» («Thermo Scientific»). Выделенную мРНК фрагментировали с помощью двухвалентных катионов и нагревания, что приводило к получению фрагментов в диапазоне 160-220 нуклеотидов. Фрагментиро ванную мРНК преобразовывали в кДНК с помощью случайных праймеров и SuperScriptII (Invitrogen) с последующим синтезом второй цепи с использованием полимеразы I и РНКазы Н. Концы кДНК достраивали с помощью Т4 ДНК-полимеразы, фрагмента Кленова и фосфорилировали по 5' с помощью Т4 полинуклеотидкиназы. кДНК-фрагменты с тупыми концами 3'-аденилировали с использованием фрагмента Кленова (от 3' к 5' экзо минус). 3'-концевые одиночные Т-перекрывающие, специфичные для «Illumina multiplex» адаптеры лигировали с помощью Т4 ДНК-лигазы в молярном соотношении адаптера к кДНК-вставке 10:1.
кДНК-библиотеки очищали и отбирали по размеру 200-220 п.о. с помощью геля «E-Gel 2% SizeSelect gel» («Invitrogen»). Обогащение, добавление последовательностей «Illumina» для индексирования из шести оснований и последовательностей для проточной кюветы осуществляли ПЦР с использованием ДНК-полимеразы «Phusion» («Finnzymes»). Все очистки осуществляли с помощью 1,8х объема магнитных микросфер «AgencourtAMPure ХР». Все контроли качества осуществляли с помощью теста «Qubit HS» от «Invitrogen» и размер фрагментов определяли с помощью теста «HS DNA» на «Bioanalyzer 2100» от «Agilent».
Библиотеки RNA-Seq с штрих-кодами кластеризовали на «cBot» с помощью набора «Truseq SR cluster kit v2.5» при использовании 7 рМ, и секвенировали по 50 п.о. на «Illumina HiSeq2000» с помощью набора «Truseq SBS kit-HS 50 bp".
СТС: Для RNA-Seq-профилирования СТС, использовали модифицированную версию данного протокола в котором использовали 500-700 нг SMART-амплифицированной кДНК, двухконцевые адаптеры были лигированы а обогащение ПЦР осуществлено с помощью праймеров «Illumina РЕ PCR primers» 1.0 и 2.0.
Анализ данных NGS, генная экспрессия: Для определения значений экспрессии, выходные ряды последовательностей для образцов РНК, полученные с помощью «Illumina HiSeq 2000», предварительно обрабатывали согласно стандартному протоколу «Illumina». Анализ включает фильтрование низкокачественных ридов и разуплотнение. Для анализа транскриптома RNA-Seq риды последовательностей выравнивали относительно референсной геномной последовательности [Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature, 420, 520-562 (2002)] с использованием «bowtie» (версия 0.12.5) [Langmead В. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25] с использованием параметров «-v2 -best» для выравниваний генома и параметров по умолчанию для выравниваний транскриптов. Координаты выравнивания сравнивали с координатами экзонов транскриптов RefSeq [Pruitt KD. et al. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005 Jan 1;33 (Database issue):D501-4} и для каждого транскрипта количество перекрывающих выравниваний записывали. Риды последовательностей, которые не поддаются выравниванию с геномном последовательностью, выравнивали относительно базы данных всех возможных экзон-экзонных соединяющих последовательностей транскриптов RefSeq. Количество ридов, выровненных относительно границ сплайсинга, объединяли с количествами соответствующего транскрипта, полученного в предыдущей стадии и нормализовали по RPKM (количество ридов, на килобазу экзонной модели на миллион картированных ридов) (Mortazavi A et al., Nat Methods 2008; 5:621-8). (2008). Mapping and quantifying mammalian transcriptomes by rna-seq. Nat Methods, 5(7):621-628]) для каждого транскрипта. Как значения экспрессии гена, так и значения экспрессии экзона рассчитывали на основании нормализованного количества ридов, перекрывающих каждый ген или экзон, соответственно.
Обнаружение мутаций, основанная часть опухоль: 50 нт одноконцевые риды «Illumina HiSeq 2000» выравнивали с помощью «bwa» (версия 0.5.8 с) [Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25:1754-60] с использованием настроек по умолчанию с эталонной сборкой мышиного генома mm9. Неопределенные риды - это те из ридов, которые картируются во множестве мест генома - удаляли, оставшиеся выравнивания сортировали, индексировали и конвертировали в бинарный и сжатый формат (ВАМ), а баллы качества ридов преобразовали из стандарта «Illumina» «phred+64» в баллы качества стандарта Сенгера с помощью сценариев оболочки.
Для каждой из секвенируемых дорожек идентифицировали мутации с помощью трех компьютерных программ: «samtools» (версия 0.1.8) [Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011 Apr 15; 27(8):1157-8. Epub 2011 Feb 13], GATK (version 1.0.4418) [McKenna A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep; 20(9):1297-303. Epub 2010 Jul 19], и «SomaticSniper» (http://genome.wustl.edu/software/somaticsniper). Для «samtools» использовали рекомендованные авторами настройки и критерии фильтра, включая фильтрацию первого раунда, максимально покрытие 200. Для второго раунда фильтрации «samtools» минимальный балл качества вставки-делеции был равен 50, минимальное качество точечной мутации было равно 30. Для прогноза мутаций «GATK», мы следовали разработанным авторами указаниям с наработанными рекомендациями, представленными в пользовательском руководстве «GATK» (http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit). Стадия рекалибровки балла качества варианта была опущена и заменена жесткой фильтрацией. Для присваивания мутаций с помощью «SomaticSniper» использовали параметры по умолчанию, и только прогнозируемые мутации с «соматическим баллом» равным 30 или больше рассматривали в дальнейшем.
Обнаружение мутаций, СТС: Согласно iCAM-способу для основной части опухоли, 50 нт одноконцевые риды «Illumina HiSeq 2000» выравнивали с помощью «bwa» (версия 0.5.8 с) [5]) с использованием настроек по умолчанию по референсной сборке мышиного генома mm9. Поскольку риды СТС NGS получали из теста RNA-Seq, риды также выравнивали по последовательностям транскриптома, включая экзон-экзонные соединения, с использованием «bowtie» (выше). С использованием всех нуклеотидных последовательностей из ридов мы сравнили как референсный геном, так и мутации, полученные из основной части опухоли В16. Для отображения результатов идентифицированные мутации оценивали как с помощью скриптов Perl, так и вручную с помощью программного обеспечения «samtools» и «IGV» (Integrated Genome Viewer).
Выходным результатом «обнаружения мутаций» является идентификация соматических мутаций в опухолевых клетках, из образца - данные NGS - список мутаций. В образцах В16 мы идентифицировали 2448 соматических мутаций с помощью повторного секвенирования экзома.
Приоритизация мутации
Затем мы продемонстрировали возможность конвейерной приоритезации мутаций для включения в вакцину. Этот способ, названный «конвейер детекции индивидуальных мутаций в злокачественной опухоли» (англ. individual cancer mutation detection pipeline, iCAM) идентифицирует и приоритезирует соматические мутации посредством серии стадий включения множества новейших алгоритмов и биоинформатических методов. Выходным результатом данного способа является перечень соматических мутаций, приоритезированных на основании вероятной иммуногенности.
Идентификация соматических мутаций: Мутации идентифицированы с помощью трех различных алгоритмов, как для образца В16, таки для образца Black6 (Обнаружение мутаций, выше). Первая стадия iCAM заключается в объединении выходных перечней каждого алгоритма для получения высокодостоверного перечня соматических мутаций. Варианты выдаваемые «GATK» и «samtools» в одном образце относительно референсного генома. Для выбора высокодостоверных мутаций с незначительным количеством ложноположительных результатов для данного образца (т.е. опухоли или нормы), мутации отбирали так, чтобы они были идентифицированы во всех репликах. Затем отбирали варианты, которые присутствуют в опухолевом образце, но не присутствуют в нормальном образце. «SomaticSniper» автоматически сообщает о потенциальных соматических вариациях из парных данных опухоли и нормы. Мы дополнительно отфильтровали результаты по пересечению результатов, полученных из реплик. Для удаления максимально возможного количества ложноположительных прогнозов, мы осуществили пересечение перечней мутаций, полученных путем применения всех алгоритмов ко всем репликам. Конечная стадия для каждой соматической мутации заключается в присвоении значения достоверности (p-значение) для каждой мутации на основании глубины покрытия, качества SNP, качества консенсуса и качества картирования.
Влияние мутации: влияние фильтрованных, консенсусных, соматических мутаций определяется скриптом в пределах конвейера детекции мутаций «iCaM». Во-первых, мутации, которые происходят в генетических областях, которые не являются уникальными в геноме, например, такие которые происходят в паралогах белков и в псевдогенах, исключены из анализа, поскольку риды последовательностей, которые выравниваются во множестве мест, удаляются. Во-вторых, определяется, происходит ли мутация в транскрипте. В-третьих, определяется происходит ли мутация в белок-кодирующей области. В-четвертых, последовательность транскрипта транслируется с или без мутации для определения, того, есть ли изменение в аминокислотной последовательности.
Экспрессия мутации: конвейер iCAM выбирает соматические мутации, которые обнаружены в генах и экзонах, которые экспрессируются в опухолевых клетках. Уровни экспрессии определяются по NGS-данным RNA-Seq опухолевых клеток (выше). Количество ридов, которые перекрывают ген и экзон указывает на уровни экспрессии. Эти количества нормализуются относительно RPKM (количество ридов, на килобазу экзонной модели на миллион картированных ридов) [Mortazavi A. et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008 Jul; 5(7):621-8. Epub 2008 May 30]) и те, которые экспрессируются выше 10 RPKM, отбираются.
Связывание МНС: для определения вероятности того, что эпитоп, содержащий мутированный пептид, связывается с молекулой МНС, конвейер iCAM запускается в модифицированной версии программного обеспечения для МНС-прогноза из «Immune Epitope Database» (http://www.iedb.org/). Локальная установка включает модификации для оптимизации потока данных через алгоритм. Для данных В16 и Black6, прогноз был запущен с помощью всех доступных аллелей black6 МНС класса I и всех эпитопов для соответствующих длин пептидов. Мутации выбирают таким образом, чтобы они находились в эпитопе, который входит в 95% процентиль распределения балла прогноза тренировочных данных IEDB (http://mhcbindingpredictions.immuneepitope.org/dataset.html), учитывая все аллели МНС и все потенциальные эпитопы, перекрывающие мутацию.
Критерий выбора мутации: соматические мутации выбирают по следующим критериям: а) имеющие уникальное содержимое последовательности, b) идентифицированные всеми тремя программами, с) высокая достоверность мутации, d) несинонимичное изменение белка, е) высокий уровень экспрессии транскрипта, f) и прогноз связывания с подходящим МНС класса I.
Выходным результатом данного способа является перечень соматических мутаций, приоритезированных на основании вероятной иммуногенности. В клетках меланомы В16 присутствует 2448 соматических мутаций. 1247 из этих мутаций обнаружены в генных транскриптах. Их них 734 вызывают несинонимичные изменения в белках. Из них 149 находятся в генах, экспрессируемых в опухолевых клетках. Из этих экспрессирующих несинонимичные мутации, 102, согласно прогнозу, присутствуют на молекулах МНС. Эти 102 вероятные иммуногенные мутации затем проходят через стадию подтверждения мутаций (ниже).
Подтверждение мутаций
Соматические мутации из повторно секвенированного ДНК-экзома подтверждали любым из двух способов, повторного секвенирования мутированной области и анализ RNA-Seq.
Для подтверждения мутаций повторным секвенированием, геномную область, содержащую мутацию, амплифицировали стандартным ПЦР из 50 нг как опухолевой ДНК, так и нормальной контрольной ДНК. Размер амплифицированных продуктов находился в диапазоне 150-400 нт. Специфичность реакции контролировали загрузкой продукта ПЦР на устройство «Qiaxel» («Qiagen»). Продукты ПЦР очищали с помощью набора для очистки «minElute PCR purification kit» («Qiagen»). Специфичные продукты ПЦР секвенировали стандартным способом секвенирования по Сэнгеру («Eurofins»), с последующим анализом электрофореграмм.
Подтверждение мутации также было выполнено проверкой опухолевой РНК. Значения экспрессии опухолевых генов и экзонов получали из RNA-Seq (NGS-секвенирование РНК), путем картирования. нуклеотидных последовательностей на транскриптах и их подсчета. Мы проверили сами данные последовательностей для идентификации мутаций в опухолевом образце [Berger MF. et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010 Apr; 20(4):413-27. Epub 2010 Feb 23], тем самым обеспечив независимое подтверждение идентифицированных соматических мутаций, полученных из ДНК.
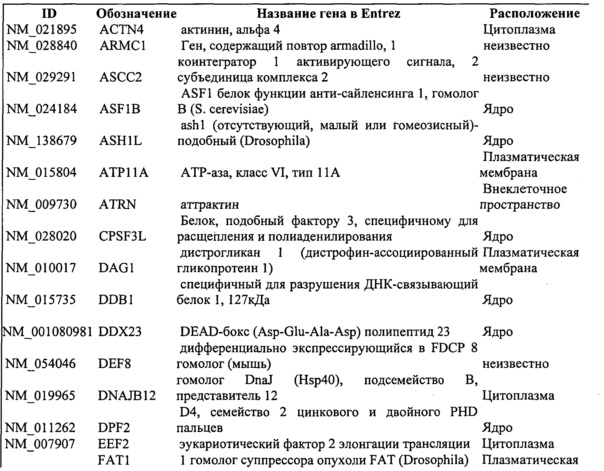
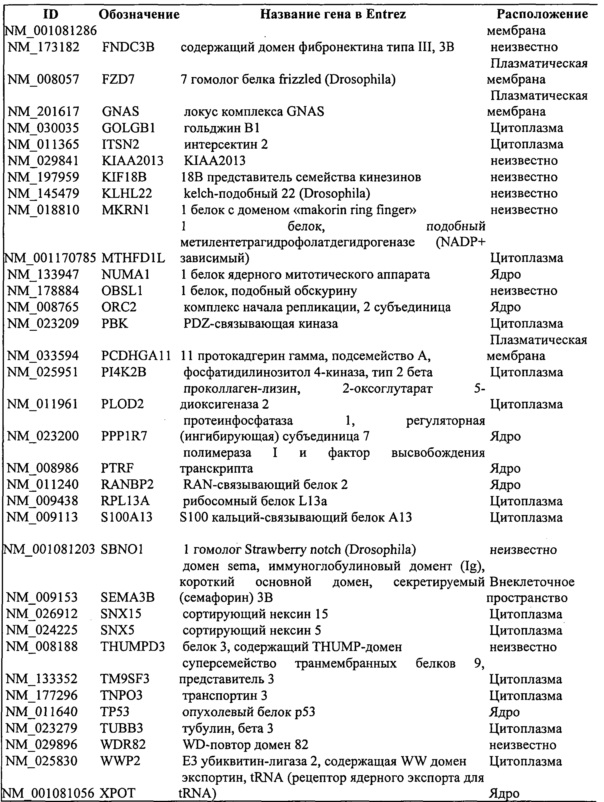
Пример 2. Алгоритм отбора IVAC позволяет осуществить детекцию иммуногенных мутаций
Для того, чтобы исследовать, можно ли индуцировать специфичные Т-клеточные ответы против подтвержденных мутаций клеток меланомы B16F10, не подвергавшиеся обработкам мыши C57BL/6 (n=5/пептид) иммунизировали дважды (d0, d7) подкожно 100 мкг пептида (+50 мкг полиI:С в качестве адъюванта), содержащего либо мутированную ак последовательность, либо ак последовательность дикого типа (см. таблицу 2). Все пептиды имеют длину 27 ак с мутированной ак/ак дикого типа в центральном положении. На 12 день мышей умерщвляли и собирали клетки селезенки. Для считывания данных использовали способ «IFNγ ELISpot» с 5×105 клетками селезенки/лунку в качестве эффекторов, и 5×104 дендритных клеток костного мозга, загруженных пептидами (2 мкг/мл) в качестве клеток-мишеней. Эффекторные клетки селезенки тестировали против мутированного пептида, пептида дикого типа и контрольного пептида (нуклеопротеина вируса везикулярного стоматита, VSV-NP).
На 44 протестированных последовательностях мы наблюдали, что 6 из них индуцируют Т-клеточный иммунитет, направленный только против мутированной последовательности, но не против пептида дикого типа (фиг.3).
Данные доказывают, что идентифицированные и приоритезированные мутации могут быть использованы для индукции опухоль-специфичного Т-клеточного иммунитета после применения в качестве пептидной вакцины в необработанных антигеном мышах.

Пример 3. Идентифицированные мутации могут обеспечивать терапевтический противоопухолевый иммунитет
Для проверки наличия у идентифицированных мутаций способности наделять противоопухолевым иммунитетом после вакцинации интактных мышей мы исследовали этот вопрос с пептидом №30, который, как было показано, индуцирует селективную в отношении мутации Т-клеточную реактивность. Клетки B16F10 (7,5×104) инокуровали подкожно в d0. Мышей вакцинировали пептидом 30 (см. таблицу 1; 100 мкг пептид + 50 мкг ПолиI:С п.к.) в день - 4, день + 2 и день + 9. Контрольная группа получала только ПолиI:С (50 мкг п.к.). Рост опухоли отслеживали каждый второй день. На день + 16 мы обнаружили, что только у 1 из 5 мышей в группе пептидной вакцины развилась опухоль, тогда как в контрольной группе у 4 из 5 мышей, был показан рост опухоли.
Данные доказывают, что пептидная последовательность, включающая B16F10-специфичную мутацию, может наделять противоопухолевым иммунитетом, который эффективно способен разрушать опухолевые клетки (см. фиг.4). Поскольку B16F10 является сильноагрессивной опухолевой клеточной линией, то обнаружение того, что методология, примененная для идентификации и приоритетизации мутаций в конечном счете приводит к отбору мутации, которая сама по себе уже является эффективной в качестве вакцины, является важным доказательством концепции всего этого процесса.
Пример 4. Данные поддерживающие полиэпитопную антигенную презентацию
Проверенные мутации из белок-кодирующих областей пациента, составляют пул, из которого могут быть выбраны кандидаты для сборки шаблона вакцины с полинеоэпитопом для использования в качестве предшественника для GMP-изготовления РНК-вакцины. Подходящие в качестве вакцинного каркаса векторные кассеты уже были описаны (Holtkamp, S. et al., Blood, 108:4009-4017, 2006; Kreiter, S. et al., Cancer Immunol. Immunother., 56:1577-1587, 2007; Kreiter, S. et al., J.Immunol., 180:309-318, 2008). Предпочтительные векторные кассеты модифицированы в их кодирующих и нетранслируемых областях (UTR) и обеспечивают максимизированную трансляцию кодируемого белка в течение длительных периодов времени (Holtkamp, S. et al., Blood, 108:4009-4017, 2006; Kuhn, A. N. et al., Gene Ther., 17:961-971, 2010). Кроме того, векторный каркас содержит антиген-направляющие модули для одновременной экспансии как цитотоксичных, так и хэлперных Т-клеток (Kreiter, S. et al., Cancer Immunol. Immunother., 56:1577-1587, 2007; Kreiter, S. et al., J. Immunol., 180:309-318, 2008; Kreiter, S. et al., Cancer Research, 70 (22), 9031-9040, 2010 (фиг.5). Важно отметить, что мы доказали, что такая РНК-вакцина может быть использована для презентации множества эпитопов МНС класса I и II одновременно.
IVAC последовательности РНК-вакцины с полинеоэпитопом построены с участка вплоть до 30 аминокислот, которые включают мутации в центре. Эти последовательности соединены голова-к-хвосту через короткие линкеры для образования вакцины с полинеоэпитопом, кодирующей вплоть до 30 или большего количества выбранных мутаций и фланкирующие их области. Эти пациент-специфичные индивидуально оптимизированные вставки являются кодон-оптимизированными и клонированными в РНК-каркас, описанный выше. Контроль качества таких конструктов включает in vitro транскрипцию и экспрессию в клетках для проверки функциональной транскрипции и трансляции. Анализ трансляции будет осуществлен с антителами против с-концевого направляющего домена.
Пример 5. Научная проверка концепции для РНК-конструкта с поли-нео эпитопом
Концепция РНК с поли-нео эпитопом основана на длинной мРНК, транскрибированной in vitro, которая состоит из последовательно соединенных последовательностей, кодирующих мутированные пептиды, соединенные линкерными последовательностями (см. фиг.6). Кодирующие последовательности выбирают из несинонимичных мутаций и в них всегда встроен кодон для мутированной аминокислоты, фланкированной областями от 30 до 75 пар оснований из контекста исходной последовательности. Линкерная последовательность кодирует аминокислоты, которые предпочтительно не обрабатываются клеточной машинерией процессирования антигена.
In vitro транскрибируемые конструкты основаны на векторе pST1-А120, содержащем Т7-промотор, тандем бета-глобулиновой 3' UTR последовательности и 120 п.о. поли(А) хвост, которые был показаны как увеличивающие стабильность и трансляционную эффективность РНК, способность кодируемого антигена стимулировать Т-клетки (Holtkamp S. et al., Blood 2006; PMID: 16940422). Кроме того, были вставлены фрагмент сигнального пептида МНС класса I и трансмембранный и цитозольный домены, включая стоп-кодон (сигнал направленной миграции МНС класса I или MITD), фланкированные полилинкерной последовательностью для клонирования эпитопов (Kreiter S. et al., J. Immunol., 180:309-318, 2008). Было показано, что последние увеличивают презентацию антигена, тем самым усиливая экспансию антигенспецифичных CD8+ и CD4+ Т-клеток и улучшая эффекторные функции.
Для первой проверки концепции, использовались биэпитопные векторы, т.е. кодирующие один полипептид, содержащий два мутированных эпитопа. Кодон-оптимизированные последовательности, кодирующие (i) мутированный эпитоп из 20-50 аминокислот, (ii) глицин/серин-богатый линкер, (iii) второй мутированный эпитоп из 20-50 аминокислот, и (iv) дополнительный глицин/серин-богатый линкер, фланкированный соответствующими участками распознавания рестрикционными эндонуклеазами, для клонирования в конструкт на основе pSTI, описанный выше - были спроектированы и синтезированы коммерческим поставщиком («Geneart», Регенсбург, Германия). После проверки последовательности, их клонировали в векторный каркас на основе pSTI для получения конструктов, как изображено на фиг.6.
Плазмиды на основе pST1-А120, описанные выше, были линеаризованы рестрикционной эндонуклеазой класса Us. ДНК линеаризованных плазмид очищали экстракцией фенол/хлороформом и осаждением этанолом. Количество линеаризованной векторной ДНК оценивали спектрофотометрически и подвергали in vitro транскрипции фактически также, как описано у Pokrovskaya and Gurevich (1994, Anal. Biochem. 220:420-423). Для получения РНК с соответствующим образом модифицированными 5'-кэпирующими структурами в реакцию транскрипции был добавлен копирующий аналог. В реакциях GTP присутствовал в концентрации 1,5 мМ, тогда как кэпирующий аналог присутствовал в концентрации 6,0 мМ. Все другие NTP присутствовали в концентрации 7,5 мМ. В конце реакции транскрипции линеаризованную векторную ДНК расщепляли 0,1 Ед./мкл TURBO ДНКазы («Ambion», Аустин/Техас, США) в течение 15 минут при 37°С. РНК очищали из этих реакций с помощью набора «MEGAclear Kit» («Ambion», Аустин/Техас, США) согласно протоколу изготовителя. Концентрацию и количество РНК оценивали спектрофотометрически и анализировали на «Bioanalyzer 2100» («Agilent», Санта-Клара, Калифорния, США).
Для проверки того, что последовательность, в которую включена мутированная аминокислота, и которая фланкирована как по 5'- так и по 3'-концу линкерной последовательностью, может быть процессирована, презентирована и распознана антиген-специфичными Т-клетками, мы использовали Т-клетки из вакцинированных пептидом мышей в качестве эффекторных клеток. В «IFNγ ELISpot» мы протестировали способность Т-клеток, индуцированных пептидной вакцинацией, как описано выше, распознавать клетки-мишени (дендритные клетки костного мозга, BMDC), активированные либо пептидом (2 мкг/мл, в течение 2 часов при 37°С и 5% CO2) либо трансфецированные РНК (20 мкг, полученной, как описано выше) с помощью электропорации. Как пояснено примером на фиг.7, для мутации 12 и 30 (см. таблицу 2) мы можем наблюдать, что РНК-конструкт способен привести к возникновению эпитопа, распознаваемого специфичными по мутации Т-клетками.
С представленными данными мы могли бы продемонстрировать, что РНК, кодирующая полинеоэпитоп, включая глицин/серин богатый линкер может быть транслирована и процессирована в антигенпрезентирующих клетках, что приводит к презентации корректного эпитопа, который распознается антиген-специфичными Т-клетками.
Пример 6. Проектирование вакцины с полинеоэпитопом - Необходимость линкера
РНК-конструкт с полинеоэпитопом содержит каркасный конструкт, в который помещено множество пептидов, кодируемых соматическими мутациями, соединенных с последовательностью линкерного пептида. В дополнение к оптимизации кодонов и повышению стабильности РНК и трансляционной эффективности каркасом, одно воплощение РНК вакцины с полинеоэпитопом включает линкеры, спроектированные для увеличения презентации МНС I и II класса антигенных пептидов и уменьшения презентации вредных эпитопов.
Линкер: последовательность линкера была спроектирована для соединения множества содержащих мутации пептидов. Линкер должен позволять образование и презентацию мутированного эпитопа, при этом мешая образованию вредных эпитопов, таких как те, что образованы стыком между соседними пептидами или между линкерной последовательностью и эндогенными пептидами. Эти «соединительные» эпитопы могут не только конкурировать с целевыми эпитопами за презентацию на клеточной поверхности, снижая эффективность вакцины, но также могут приводить к нежелательной аутоиммунной реакции. Таким образом, мы разработали линкерную последовательность для того, чтобы а) избежать образования «соединительных» пептидов, которые связываются с молекулами МНС, b) избежать образования «соединительных» пептидов протеасомальным процессированием, с) осуществить эффективную трансляцию и процессирование протеосомой.
Для того чтобы избежать образования «соединительных» пептидов, которые связываются с молекулами МНС, мы сравнивали различные линкерные последовательности. Глицин, например, ингибирует сильное связывание в связывающей бороздке МНС [Abastado JP. et al., J Immunol. 1993 Oct 1; 151(7):3569-75]. Мы проверили множество последовательностей линкеров и множество длин линкеров и рассчитали количество «соединительных» пептидов, которые связывают молекулы МНС.Мы использовали инструменты программного обеспечения «Immune Epitope Database» (IEDB, http://www.immuneepitope.org/) для расчета вероятности того, что данная пептидная последовательность содержит лиганд, который будет связывать молекулы МНС класса I.
В модели В16, мы идентифицировали 102 экспрессируемых несинонимичных соматических мутации, которые, согласно прогнозу, будут презентироваться на молекуле МНС класса I. Используя 50 подтвержденных мутаций, мы путем вычислений спроектировали различные вакцинные продукты, при этом либо не использовали линкеры, либо использовали линкеры различных длин, и рассчитали количество вредных «соединительных» пептидов с помощью алгоритма IEDB (фиг.8).
В таблице 5 показаны результаты нескольких различных линкеров, различных длин линкеров, и результаты без линкера и с пятью линкерами. Количество МНС-связывающих соединяющих пептидов находится в диапазоне от 2 до 91 для прогнозируемых данных для 9 ак и 10 ак эпитопов (вверху и внизу). Размер линкера влияет на количество соединяющих пептидов (внизу). Для этой последовательности наименьшие 9 ак эпитопы спрогнозированы для 7 ак линкерной последовательности GGSGGGG.
Линкер 1 и Линкер 2, используемые в конструктах полинеоэпитопной РНК-вакцины, протестированной экспериментально (см. ниже) также имеют выгодно низкое количество прогнозируемых соединяющих неоэпитопов. Это справедливо для прогнозов 9-меров и 10-меров.
Это говорит о том, что последовательность линкера критически важна для образования плохих МНС-связывающих эпитопов. Более того, длина линкерной последовательности влияет на количество плохих МНС-связывающих эпитопов. Мы обнаружили, что последовательности, которые являются G-богатыми, препятствуют образованию МНС-связывающих лигандов.
Для того, чтобы избежать протеосомального процессирования, в результате которой могут образоваться «соединительные» пептиды, мы исследовали применение различных аминокислот в линкере. Глицин-богатые последовательности ухудшают протеосомальное процессирование [Hoyt МА et al. (2006). EMBO J 25 (8):1720-9; Zhang M. and Coffino P. (2004) J Biol Chem 279 (10):8635-41]. Таким образом, действие глицин-богатых линкерных последовательностей основано на минимизации количества линкер-содержащих пептидов, которые могут быть процессированы протеасомой.
Линкер должен позволять содержащим мутации пептидам эффективно транслироваться и процессироваться протеасомой. Аминокислоты глицин и серин являются гибкими [Schlessinger A and Rost В., Proteins. 2005 Oct 1; 61(1):115-26]; их включение в линкеры приводит к получению более гибкого белка. Мы включили глицин и серин в линкер для увеличения гибкости белка, который должен позволить более эффективно транслировать и процессировать протеасомой, что в свою очередь позволяет дать лучший доступ к кодируемым антигенным пептидам.
Таким образом, линкер должен быть глицин богатым для того, чтобы препятствовать связыванию с МНС плохих эпитопов; должен препятствовать способности протеасомы процессировать линкерные пептиды, что может быть осуществлено включением глицина; и должен быть гибким для увеличения доступа к пептидам, содержащим мутации, что может быть осуществлено через комбинацию аминокислот глицина и серина. Следовательно, в одном воплощении вакцинного конструкта изобретения, последовательности GGSGGGGSGG и GGSGGGSGGS предпочтительно включены в качестве линкерных последовательностей.
Пример 7. РНК-вакцина с полинеоэпитопом
РНК-конструкты с полинеоэпитопом основаны на векторе pST1-A120, содержащем Т7 промотор, тандем 3'UTR последовательности бета-глобина и 120 п.о. поли(А) хвоста, которые, как было показано, увеличивают стабильность и трансляционную эффективность РНК, тем самым усиливая способность кодируемого антигена стимулировать Т-клетки ((Holtkamp S. et al., Blood 2006; PMID: 16940422). Кроме того, были вставлены фрагмент сигнального пептида МНС класса I и трансмембранных и цитозольных доменов включая стоп-кодон (сигнал направленной миграции МНС класса I или MITD) фланкированный полилинкерной последовательностью для клонирования эпитопов (Kreiter S. et al., J. Immunol., 180:309-318, 2008). Было показано, что последний увеличивает презентацию антигена, тем самым усиливая экспансию антигенспецифичных CD8+ и CD4+ Т-клеток и улучшая эффекторные функции.
Для получения РНК-конструктов с полинеоэпитопом для 50 идентифицированных и проверенных мутаций B16F10, были созданы 3 РНК-конструкта. Конструкты состоят из кодоноптимизированных последовательностей, кодирующих (i) мутированный эпитоп из 25 аминокислот, (ii) глицин/серин-богатый линкер, (iii) повторы последовательности мутированного эпитопа, за которыми следуют глицин/серин-богатый линкер. Цепь последовательностей, содержащих мутированный эпитоп, и линкеры фланкированы подходящими участками распознавания эндонуклеазами рестрикции для клонирования в конструкт на основе pST1, как описано выше. Вакцинные конструкты были спроектированы и синтезированы «GENEART». После проверки последовательности, их клонировали в векторный каркас на основе pST1 для получения вакцинных конструктов с РНК полинеоэпитопом.
Описание клинического метода
Клиническое применение будет охватывать следующие стадии:
- Подходящие пациенты должны дать согласие на ДНК-анализ секвенированием следующего поколения.
- Образец опухоли, полученный в ходе рутинных диагностических процедур (залитая в парафин, фиксированная формалином ткань) и клетки периферической крови будут получены и использованы для анализа мутаций, как описано.
- Обнаруженные мутации будут подтверждены
- На основании этого будет спроектирована приоритетизированная вакцина. Для РНК-вакцин синезом генов и клонированием будет получен эталонный плазмидный шаблон.
- Плазмиды будут использованы для получения РНК клинической чистоты, контроля качества и получения РНК-вакцины.
- Вакцинный лекарственный продукт будет послан в соответствующий центр проведения испытаний для клинического применения.
- РНК-вакцина может быть использована в качестве неупакованной вакцины в буферной смеси или может быть инкапсулирована в наночастицы или липосомы для прямой инъекции, например, в лимфоузлы, п.к., в.в., в.м. В ином случае РНК-вакцина может быть использована для трансфекции in vitro например, дендритных клеток для адоптивного переноса.
Весь клинический процесс займет меньше чем 6 недель. «Лаг-фаза» между информированным согласием пациента и доступностью лекарственного средства будет тщательно урегулирована протоколом клинического исследования, включая разрешение на продолжение проведения режима стандартного лечения до того, как исследовательский продукт будет доступен.
Пример 8. Идентификация опухолевых мутаций и использование их для опухолевой вакцинации
Мы применили NGS-ресеквенирование экзома для обнаружения мутаций в клеточной линии мышиной меланомы B16F10 и идентифицировали 962 несинонимичные соматических точечных мутации, 563 - в экспрессируемых генах. Потенциальные ведущие мутации происходят в генах классических опухолевых супрессоров (Pten, Trp53, Тр63, Pml) и генах, вовлеченных в протоонкогенные сигнальные пути, которые контролируют клеточную пролиферацию (например, Mdm1, Pdgfra), клеточную адгезию и миграцию (e.g. Fdz7, Fat1) или апоптоз (Casp9). Более того, B16F10 накапливает мутации в Aim1 и Trrap, которые ранее были описаны как часто изменяемые в человеческой меланоме.
Иммуногенность и специфичность 50 проверенных мутаций были проверены с помощью мышей C57BL/6, иммунизированных длинными пептидами, кодирующими мутированные эпитопы. Одна треть (16/50) их была показана как иммуногенная. Их них, 60% проявляет иммунные ответы предпочтительно направленные против мутированной последовательности по сравнению с последовательностью дикого типа.
Мы протестировали гипотезу в моделях опухолевых трансплантов. Иммунизация пептидами наделяет in vivo опухолевым контролем в защитных и терапевтических условиях, квалифицируя мутированные эпитопы, содержащие одиночные аминокислотные замены, в качестве эффективных вакцин.
Животные
Мыши C57BL/6 («Jackson Laboratories») содержались в соответствии с федеральной политикой и политикой штата о проведении исследований на животных при университете Майнца.
Клетки
Клеточная линия меланомы B16F10 была приобретена в 2010 году в Американской коллекции типовых культур (номер продукта: АТСС CRL-6475, номер лота: 58078645). Ранние (3-й, 4-й) пассажи клеток использовали в опухолевых экспериментах. Клетки тестировали стандартным образом на Micoplasma. Повторную аутентификацию клеток после получения не проводили.
Секвенирование следующего поколения
Экстракция нуклеиновых кислот и приготовление образцов: ДНК и РНК из большого количества клеток B16F10 и ДНК и ткани хвоста C57BL/6 экстрагировали трижды с помощью набора «Qiagen DNeasy Blood and Tissue kit» (для ДНК) и набора «Qiagen RNeasy Micro kit» (для РНК).
Секвенирование ДНК экзома: Захват экзома для ресеквенирования ДНК осуществляли трижды с помощью теста для захвата на основе раствора «Agilent Sure-Select mouse» (Gnirke A et al., Nat Biotechnol 2009; 27:182-9), разработанном для захвата всех кодирующих мышиные белки областей. 3 μг очищенной геномной ДНК (гДНК) фрагментировали до 150-200 п.о. с помощью ультразвукового устройства «Covaris S2». Концы фрагментов достраивали, фосфорилировали по 5' и аденилировали по 3' согласно инструкциям производителя. Парные концевые адаптеры «Illumina» лигировали к гДНК фрагментам с использованием молярного соотношения адаптеров к гДНК 10:1. Обогащенные последовательности для предварительного захвата и проточной кюветы добавляли с помощью праймеров «Illumina РЕ PCR 1.0» и «2.0» в ходе 4 циклов ПЦР. 500 нг дотированных с адаптером обогащенных ПЦР фрагментов гДНК гибридизовали с РНК-библиотекой биотинилированных приманок цельного мышиного экзома «SureS elect» фирмы «Agilent» в течение 24 часов при 65°С. Гибридизованные комплексы гДНК/РНК-приманка удаляли с помощью покрытых стрептавидином магнитных микросфер, и РНК-приманки отщепляли в ходе элюции в элюционном буфере «SureSelect». Эти элюированные гДНК фрагменты амплифицировали ПЦР после захвата в течение 10 циклов. Библиотеки гДНК с обогащенным экзомом кластеризовали на «cBot» с помощью набора «Truseq SR cluster kit v2.5» при использовании 7 рМ, и 50 п.о. секвенировали на «Illumina HiSeq2000» с помощью набора «Truseq SBS kit-HS 50 bp».
Генная экспрессия на основе РНК, профилирование «транскриптома» (RNA-Seq): Библиотеки «mRNA-seq cDNA» с штрих-кодом готовили в трех повторах, из 5 мкг общей РНК (модифицированный протокол «Illumina mRNA-seq»). мРНК выделяли с помощью магнитных микросфер «Seramag Oligo(dT)» («Thermo Scientific») и фрагментировали с помощью дивалентных катионов и нагревания. Полученные фрагменты (160-220 н.о.) преобразовывали в кДНК с помощью случайных праймеров и «SuperScriptII» («Invitrogen») с последующим синтезом второй цепи при использовании ДНК-полимеразы I и РНКазы Н. Проводили репарацию концов кДНК в, и затем их фосфорилировали по 5' и аденилировали по 3' согласно инструкциям производителя. 3'-концевые одиночные Т-перекрывающие, специфичные для «Illumina multiplex» адаптеры лигировали с помощью Т4 ДНК-лигазы в молярном соотношении адаптера к кДНК-вставке 10:1. кДНК-библиотеки очищали и отбирали по размеру 200-220 п.о. (гель «E-Gel 2% SizeSelect», «Invitrogen»). Обогащение, добавление последовательностей «Illumina» для индексирования из шести оснований и последовательностей для проточной кюветы осуществляли ПЦР с использованием ДНК-полимеразы «Phusion» («Finnzymes»). Все очистки вплоть до этой стадии осуществляли в 1,8х объеме магнитных микросфер «AgencourtAMPure ХР». Все контроли качества осуществляли с помощью теста «Qubit HS» от «Invitrogen» и размер фрагментов определяли с помощью теста «HS DNA» на «Bioanalyzer 2100» от «Agilent». Библиотеки RNA-Seq с штрих-кодами кластеризовали и секвенировали как описано выше.
Анализ данных NGS, экспрессия генов: Считанные выходные данные последовательностей образцов РНК предварительно обрабатывали согласно стандартному протоколу «Illumina», включая фильтрование для считанных данных низкого качества. Считанные данные последовательностей выравнивали по эталонной геномной последовательности mm9 (Waterston RH et al., Nature 2002; 420:520-б2) с помощью «bowtie» (версия 0.12.5) (Langmead В et al., Genome Biol 2009; 10:R25). Для выравнивания генома разрешались два несовпадения и записывалось только лучшее выравнивание ("-v2 - best"); для выравниваний транскриптома использовали параметры по умолчанию. Считанные данные, невыравниваемые по геномной последовательности, выравнивали по базе данных последовательностей всех возможных экзон-экзонных соединений транскриптов RefSeq (Pruitt KD et al., Nucleic Acids Res 2007; 35:D61-D65). Значения экспрессии определяли по пересечению координат считывания с теми из транскриптов RefSeq, на которые приходились считанные данные по перекрывающемуся экзону и соединению, и нормализовали по единицам экспрессии RPKM (количество ридов, на килобазу экзонной модели на миллион картированных ридов) (Mortazavi A et al., Nat Methods 2008; 5:621-8).
Анализ данных NGS, обнаружение соматических мутаций: Соматические мутации идентифицировали, как описано в Примере 9. 50 нуклеотидные односторонние риды выравнивали по референсному мышиному геному mm9 с помощью «bwa» (параметры по умолчанию, версия 0.5.8 с) (Li Н and Durbin R, Bioinformatics 2009; 25:1754-60). Неоднозначные риды, картированные во многих местах генома, удаляли. Мутации идентифицировали с помощью трех программ: «samtools» (версия 0.1.8) (Li H, Bioinformatics 2011; 27:1157-8), «GATK» (версия 1.0.4418) (McKenna A et al, Genome Res 2010; 20:1297-303), и «SomaticSniper» (http://genome.wustl.edu/software/somaticsniper) (Ding L et al., Hum Mol Genet 2010; 19:R188-R196). Потенциальным вариациям, идентифицированным во всех трех повторах B16F10, присваивалось доверительное значение «частоты ложных результатов» (FDR) (см. также пример 9).
Отбор, проверка и функция мутаций.
Выбор: Для отбора мутации должны удовлетворять следующим критериям: (i) присутствуют во всех трех повторах B16F10 и отсутствуют во всех трех повторах C57BL/6, (ii) FDR≤0,05, (iii) гомогенность в C57BL/6, (iv) встречаются в транскрипте RefSeq, и (v) вызывают несинонимичные изменения, которые могут быть подсчитаны как достоверная мутация. Отбор для проверки и тестирования иммуногенности требует, чтобы мутации являлись экспрессируемыми генами (медианное значение RPKM по репликам > 10).
Проверка: Мутации в ДНК классифицировали как проверенные, если они подтверждались секвенированием по Сенгеру или ридами B16F10 RNA-Seq. Все отобранные варианты амплифицировали из 50 нг ДНК клеток B16F10 или ткани хвоста C57BL/6 с помощью фланкирующих праймеров, продукты визуализировали («QIAxcel system», «Qiagen») и очищали (набор для очистки «QIAquick PCR Purification Kit», «Qiagen»). Ампликон ожидаемого размера вырезали из геля, очищали («QIAquick Gel Extraction Kit», «Qiagen») и секвенировали по Сенгеру («Eurofins MWG Operon», Эберсберг, Германия), с прямым праймером, использованным для амплификации ПЦР.
Функциональное воздействие: Программы «SIFT» (Kumar P et al., Nat Protoc 2009; 4:1073-81) и «POLYPHEN-2» (Adzhubei IA et al., Nat Methods 2010; 7:248-9), которые прогнозируют функциональную значимость аминокислоты в функции белка на основании расположения белковых доменов и межвидовой консервативности последовательностей, использовали для оценки воздействия выбранных мутаций. Инструменты «Ingenuity IPA» использовали для логического выведения функции гена.
Синтетические пептиды и адъюванты
Все пептиды, включая овальбумин класса I (OVA258-265), класса II (OVA class II330-338), нуклеопротеин гриппа (Inf-NP366-374), нуклеопротеин вируса везикулярного стоматита (VSV-NP52-59) и родственный тирозиназе белок 2 (Trp2180-188) приобрели в «Jerini Peptide Technologies» (Берлин, Германия). Синтетические пептиды были длиной 27 аминокислот с мутированной аминокислотой (MUT) или аминокислотой дикого типа (WT) в положении 14. Полиинозиновую:полицитидиловую кислоту (поли(I:С), InvivoGen) использовали в качестве подкожно инъецируемого адъюванта. МНС-Пентамер специфичный к пептиду Inf-NP366-374, приобрели у «ProImmune Ltd.».
Иммунизация мышей
Самок мышей C57BL/6 одного возраста инъецировали подкожно 100 мкг пептида и 50 мкг поли(I:С), приготовленных в PBS (200 мкл общий объем) в бок между последним ребром и тазом (5 мышей на группу). Каждую группу иммунизировали в день 0 и в день 7 пептидами, кодирующими два различных пептида, один пептид в каждый бок. Двенадцать дней после исходной инъекции мышей забивали и выделяли спленоциты для иммунологического тестирования.
В ином случае, совпадающих по возрасту самок мышей C57BL/6инъецировали внутривенно 20 мкг in vitro транскрибированной РНК, приготовленной с 20 мкл 2Lipofectamine™ RNAiMAX» («Invitrogen») в PBS в общем инъецируемом объеме 200 мкл (3 мыши на группу). Каждую группу иммунизировали в день 0, 3, 7, 14 и 18. Через двадцать три дня после первой инъекции мышей забивали и выделяли спленоциты для иммунологического тестирования. ДНК-последовательности, представляющие одну (моноэпитоп), две (биэпитоп) или 16 мутаций (полиэпитоп) конструировали с использованием 50 аминокислот (ак) с мутацией в положении 25 ак (биэпитоп) или с использованием 27 ак с мутацией в положении 14 (моно- и полиэпитоп), разделяли глицин/сериновым линкером из 9 ак и клонировали в каркас pST1-2BgUTR-A120 (Holtkamp et al., Blood 2006; 108:4009-17). In vitro транскрипция с этой матрицы и очистка ранее были описаны (Kreiter et al., Cancer Immunol Immunother 2007; 56:1577-87).
Иммуноферментный спот-анализ
Иммуноферментный спот-анализ (ELISPOT) (Kreiter S et al., Cancer Res 2010; 70:9031-40) и образование сингенных дендритных клеток из костного мозга (BMDC) в качестве стимуляторов ранее были описаны (Lutz MB et al., J Immunol Methods 1999; 223:77-92). BMDC либо активировали пептидом (2 мкг/мл), либо трансфицировали in vitro транскрибированной (IVT) РНК, кодирующей указанную мутацию или контрольную РНК (eGFP-RNA). Последовательности, представляющие две мутации, каждая содержащая 50 аминокислот с мутацией в положении 25 и отделенная глицин/сериновым линкером из 9 ак, клонировали в каркас pST1-2BgUTR-A120 (Holtkamp S et al., Blood 2006; 108:4009-17). In vitro транскрипция с этого матрицы и очистка ранее были описаны (Kreiter S et al., Cancer Immunol Immunother 2007; 56:1577-87). Для анализа 5×104 BMDC, моделированных пептидом или РНК, коинкубировали с 5×105 свежевыделенных спленоцитов в титрационном микропланшете, покрытом анти-IFN-γ антителом (10 μг/мл, клон AN18; «Mabtech»). Через 18 часов при 37°С, секрецию цитокинов детектировали с помощью анти-IFN-γ антитела (клон R4-6A2; «Mabtech»). Количество пятен подсчитывали и анализировали с помощью «ImmunoSpot® S5 Versa ELISPOT Analyzer», программного обеспечения «IImmunoCaptureTM Image Acquisition» и программного обеспечения «ImmunoSpot® Analysis» версии 5. Статистический анализ осуществляли с помощью критерия Стьюдента и критерия Манна-Уинтни (непараметрический критерий). Ответы считались значимыми, если либо тест давал p-значение <0,05, либо среднее количество пятен составляло >30 пятен/5×105 эффекторных клеток. Реактивности оценивали по среднему количество пятен (-: <30; +: >30; ++: >50; +++>200 пятен/лунку).
Анализ внутриклеточных цитокинов
Аликвоты спленоцитов, полученных для анализа «ELISPOT» подвергали анализу выработки цитокинов посредством внутриклеточной проточной цитометрии. С этой целью 2×106 спленоцитов на образец помещали в 96-луночный планшет в культуральную среду (RPMI+10% FCS), обогащенную ингибитором аппарата Гольджи Брефелдином А (10 μг/мл). Клетки из каждого животного повторно стимулировали в течение 5 часов при 37°С 2×105 BMDC, активированных пептидом. После инкубации клетки отмывали-PBS, ресуспендировали в 50 мкл PBS и окрашивали внеклеточно следующими анти-мышиными антителами в течение 20 минут при 4°С: анти-CD4 FITC, анти-CD8 АРС-Су7 («BD Pharmingen»). После инкубации клетки отмывали PBS и затем ресуспендировали в 100 мкл раствора «Cytofix/Cytoperm» («BD Bioscience») в течение 20 мин. при 4°С для пермеабилизации внешней мембраны. После пермеабилизации клетки омывали буфером «Perm/Wash-Buffer» («BD Bioscience»), ресуспендировали в 50 мкл/образец в буфере «Perm/Wash-Buffer» («BD Bioscience»), и окрашивали внутриклеточного следующими анти-мышиными антителами в течение 30 минут при 4°С: анти-IFN-γ РЕ, анти-TNF-α РЕ-Су7, анти-IL2 АРС («BD Pharmingen»). После отмывки буфером «Perm/Wash-Buffer» клетки ресуспендировали в PBS, содержащем 1% параформальдегида для анализа проточной цитометрией. Образцы анализировали с помощью цитометра «BD FACSCanto™ II» и «FlowJo» (версия 7.6.3).
Модель опухоли меланомы В16
Для экспериментов по опухолевой вакцинации 7,5×104 клеток меланомы B16F10 инокулировали п.к. в бока мышей C57BL/6. В профилактическом режиме, иммунизацию пептидом, специфичным для мутации, осуществляли за 4 дня перед и через 2 и 9 дней после инокуляции опухоли. В терапевтическом эксперименте пептидную вакцину вводили через 3 и 10 дней после инъекции опухоли. Размеры опухолей измеряли каждые три дня и умерщвляли мышей, при достижении опухолями диаметра 15 мм.
В ином случае для экспериментов по вакцинации против опухоли в бока одновозрастных самок мышей C57BL/6 инокулировали п.к. 1×105 клеток меланомы B16F10. Вакцинацию пептидом осуществляли на 3, 10 и 17 день после инокуляции опухоли 100 мкг пептида и 50 мкг поли(I:С) приготовленных в PBS (общий объем 200 мкл), инъецированных подкожно в бок межу последним ребром и тазом. Иммунизации РНК осуществляли с помощью 20 мкг in vitro траскрибированных кодирующих мутацию РНК, приготовленных с 20 мкл «Lipofectamine™ RNAiMAX» («Invitrogen») в PBS с общим инъецируемым объемом 200 мкл. В качестве контроля одну группу животных инъецировали «RNAiMAX» («Invitrogen») в PBS. Животных иммунизировали на 3, 6, 10, 17 и 21 день после инокуляцию опухоли. Размеры опухолей измеряли каждые три дня с помощью штангенциркуля и умерщвляли мышей, когда диаметр опухолей достигал 15 мм.
Идентификация несинонимичных мутаций в мышиной меланоме B16F10.
Наша задача заключалась в идентификации потенциально иммуногенных соматических точечных мутаций в мышиной меланоме B16F10 с помощью NGS, и в тестировании их на предмет in vivo иммуногенности пептидной вакцинацией мышей и измерением вызванных Т-клеточных ответов с помощью анализа «ELISPOT» (фиг.9А). Мы секвенировали экзомы фонового генома дикого типа C57BL/6 и клеток B16F10, каждый трижды выделенный и захваченный. Для каждого образца было получено больше чем 100 миллионов односторонних ридов длиной 50 нуклеотидов. Из них 80% выровнялись уникально на мышином геноме mm9, a 49% выровнялись на мишени, что демонстрирует удачное обогащение мишени и приводит к более чем 20 кратному перекрытию 70% нуклеотидов-мишеней в каждом из трех образцов. RNA-Seq клеток B16F10, также профилированные в трех повторах, образовали 30 миллионов односторонних ридов длиной 50 нуклеотидов, из которых 80% выровнялись на мышином транскриптоме.
Риды ДНК (захват экзома) из B16F10 и C57BL/6 анализировали для идентификации соматических мутаций. Анализ вариации числа копий (Sathirapongsasuti JF et al., Bioinformatics 2011; 27:2648-54) продемонстрировал амплификации и делеции ДНК в B16F10, включая гомозиготоную делецию опухолевого супрессора Cdkn2a (ингибитор 2А циклин-зависимой киназы, p16Ink4A). Фокусируясь на точечных мутациях для идентификации возможных иммуногенных мутаций, мы идентифицировали 3570 соматических точечных мутаций при FDR≤0,05 (фиг.9В). Наиболее частым классом мутаций являлись транзиции C>Т/G>A, как правило, являющиеся результатом воздействия ультрафиолетового излучения (Pfeifer GP et al., Mutat Res 2005; 571:19-31). Из этих соматических мутаций, 1392 произошли в транскриптах, со 126 мутациями в нетранслированных областях. Из 1266 мутаций в кодирующих областях, 962 вызвали несинонимичные изменения в белках, а 563 из них произошли в экспрессируемых генах (фиг.9В).
Присвоение идентифицированных мутаций генам-носителям и проверка
Примечательно, что многие из мутировавших генов (962 гена, содержащих несинонимичные соматические точечные мутации), ранее были ассоциированы с фенотипами злокачественных опухолей. Мутации были обнаружены в известных генах-опухолевых супрессорах, включая Pten, Trp53 (также называемом р53), и Tp63. В Trp53, наиболее известном опухолевом супрессоре (Zilfou JT et al., Cold Spring Harb Perspect Biol 2009; 1:a001883), мутация аспарагина в аспарагиновую кислоту в положении 127 (p.N127D) расположена в ДНК-связывающем домене и согласно прогнозу «SIFT» изменяет функцию. Pten содержит две мутации (p.A39V, p.T131P), которые обе согласно прогнозу оказывают вредное воздействие на функцию белка. Показано, что мутация p.T131P, соседствующая с мутацией (p.R130M), снижает активность фосфатазы (Dey N et al., Cancer Res 2008; 68:1862-71). Более того, мутации были обнаружены в генах, ассоциированных с сигнальными путями восстановления ДНК, таких как Brca2 (белок рака молочной железы 2, раннее возникновение), Atm (белок мутирующий при телеангиоэктатической атаксии), Ddb1 (специфичный для повреждений ДНК-связывающий белок 1) и Rad9b (RAD9, гомолог В). Более того, мутации происходят в других ассоциированных с опухолями генах, включая Aim1 (опухолевый супрессор «Отсутствующий при меланоме 1»), Flt1 (онкоген Vegr1, fms-родственная тирозинкиназа 1), Pml (опухолевый супрессор «промиелоцитарный лейкоз»), Fat1 («опухолевый супрессор FAT, гомолог 1»), Mdm1 (TP53 связывающий ядерный белок), Mta3 (ассоциированное с метастазированием семейство 1, представитель 3), и Alk (рецепторная тирозинкиназа анапластической лимфомы). Мы обнаружили мутацию p.S144F в Pdgfra (рецепторе тромбоцитарного фактора роста, альфа полипептиде), связанной с клеточной мембраной рецепторной тирозинкиназе сигнального пути MAPK/ERK, ранее идентифицированной в опухолях (Verhaak RG et al., Cancer Cell 2010; 17:98-110). Мутация произошла в p.L222V в Casp9 (касапаза 9, апоптоз-связанная цистеиновая пептидаза). CASP9 протеолитически расщепляет поли(АВР-рибоза)полимеразу (PARP), регулирует апоптоз, и был связана с нескольких злокачественными новообразованиями (Hajra KM et al., Apoptosis 2004; 9:691-704). Мутации, которые мы обнаружили, потенциально могут вносить вклад в передачу сигнала PARP и апоптотическую передачу сигнала. Наиболее интересно то, что мутации не были обнаружены в Braf, c-Kit, Kras или Nras. Однако мутации были идентифицированы в Rassf7 (RAS-ассоциированном белке) (p.S90R), Ksr1 (киназа-супрессор ras 1) (p.L301V) и Atm (PI3K сигнальный путь) (р.K91T), которые все согласно прогнозу оказывают значимое воздействие на функцию белка. Trrap (белок, ассоциированный с доменом трансформации/транскрипции) был идентифицирован ранее в этом году в образцах человеческой меланомы в качестве новой потенциальной мишени в меланомах (Wei X et al., Nat Genet 2011; 43:442-6). В B16F10, мутация в Trrap происходит в p.K2783R и согласно прогнозу нарушает перекрывание FAT домена киназы, родственной фоссфатидилинозитокиназе (PIK).
Из 962 несинонимичных мутаций, идентифицированных с помощью NGS, мы выбрали 50 мутаций, включая 41 с FDR<0,05, для проверки на основе ПЦР и тестирования иммуногенности. Критериями отбора были расположение в экспрессируемом гене (RPKM>10) и прогнозируемая иммуногенность. Примечательно, что мы были способны проверить все 50 мутаций (таблица 6, фиг.9В).


На фиг.9С показано расположение хромосом B16F10, плотность генов, экспрессия генов, мутации и отфильтрованные мутации (внутренние кольца).
In vivo тестирование иммуногенности с помощью представляющими мутации длинными пептидами
Для обеспечения антигенов для тестирования иммуногенности этих мутаций мы использовали длинные пептиды, которые имеют много различных преимуществ по сравнению с другими пептидами для иммунизации (Melief CJ and van der Burg SH, Nat Rev Cancer 2008; 8:351-60). Длинные пептиды способны индуцировать антигенспецифичные CD8+ а также CD4+ Т-клетки (Zwaveling S et al., Cancer Res 2002; 62:6187-93; Bijker MS et al., J Immunol 2007; 179:5033-40). Более того, длинные пептиды требуют процессирования для того, чтобы быть представленными на молекулах МНС. Такое поглощение наиболее эффективно осуществляется дендритными клетками, которые являются оптимальными для праймирования мощного Т-клеточного ответа. Подготовленные пептиды, напротив, не требуют обрезания и экзогенно загружаются на все клетки, экспрессирующие молекулы МНС, включая неактивированные В- и Т-клетки, что приводит к индукции толерантности и фратрицида (Toes RE et al., J Immunol 1996; 156:3911-8; Su MW et al., J Immunol 1993; 151:658-67). Для каждой из 50 проверенных мутаций мы разработали пептиды длиной 27 аминокислот, в которых в центре расположена мутантная аминокислота или аминокислота дикого типа. Таким образом, любой потенциальный эпитоп МНС класса I и II, длиной 8-14 аминокислот, несущий мутацию, может быть процессирован из этого пептида-предшественника. В качестве адъюванта для вакцинации пептидом мы использовали поли(I:С), который как известно стимулирует перекрестную презентацию и увеличивает эффективность вакцины (Datta SK et al., J Immunol 2003; 170:4102-10; Schuiz О et al., Nature 2005; 433:887-92). 50 мутаций тестировали in vivo в мышах для индукции Т-клеток. Интересно, что 16 из 50 кодирующих мутацию пептидов были обнаружены как вызывающие иммунные ответы в иммунизированных мышах. Индуцированные Т-клетки, демонстрировали различные паттерны реактивности (таблица 7).
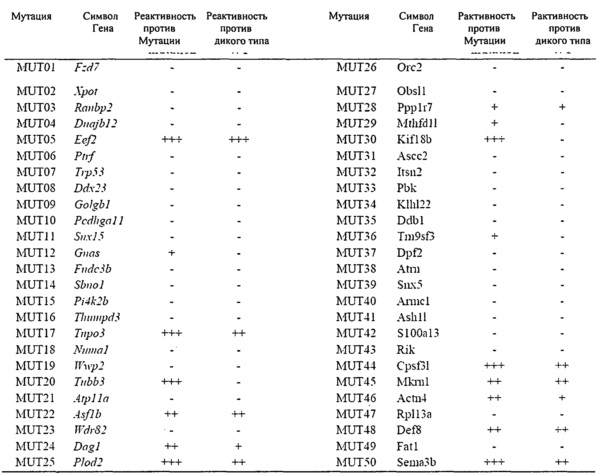
Двенадцать пептидов индуцирующих иммунных ответ, преимущественно являются мутантным эпитопом. Это подтверждено на примере мышей, иммунизированных мутациями 30 (MUT30, Kif18b) и 36 (MUT36, Plod2) (фиг.10А). Анализ «ELISPOT» выявил сильные зависимые от мутации иммунные ответы без перекрестной реактивности против пептида дикого типа или несвязанного контрольного пептида (VSV-NP). С пятью пептидами включая мутации 05 (MUT05, EefZ) и 25 (MUT25, Plod2) (фиг.10А), были получены иммунные ответы со сравнимым распознаванием как мутантного пептида, так и дикого типа пептида. Основная часть мутантных пептидов была не способна индуцировать значимые Т-клеточные ответы, что подтверждается примером мутаций 01 (MUT01, Fzd7), 02 (MUT02, Xpot), и 07 (MUT07, Trp53). Иммунные ответы, индуцированные некоторыми из обнаруженных мутаций, находились в диапазоне иммуногенности (500 пятен/5×105 клеток), полученном на мышах, иммунизированных в качестве положительного контроля описанным эпитопом МНС класса I из опухолевого антигена мышиной меланомы, родственного тирозиназе белка 2 (Trp2180-188, фиг.10А) (Bloom MB et al., Exp Med 1997; 185:453-9; Schreurs MW et al. Cancer Res 2000; 60:6995-7001). Для отобранных пептидов, которые индуцируют сильный зависимый от мутации Т-клеточный ответ, мы подтвердили иммунное распознавание независимым подходом. Вместо длинных пептидов, для считывания иммунологических данных использовали in vitro транскрибированную РНК (IVT RNA), кодирующую фрагменты мутантных пептидов MUT17, MLJT30 и MUT44. BMDC, трансфецированные кодирующей мутацию РНК или нерелевантной РНК, выполняли функцию антигенпрезентирующих клеток (АРС) в анализе «ELISPOT», при этом спленоциты иммунизированных мышей выполняли функцию популяции эффекторных клеток. BMDC, трансфецированные мРНК, кодирующими MUT17, MUT30 и MUT44, специфично и сильно распознавались спленоцитами мышей, иммунизированными соответствующими длинными пептидами (фиг.10В). Была задокументирована значимо более низкая реактивность против контрольных РНК-трансфецированных BMDC, что вероятно обусловлено неспецифической активацией BMDC одноцепочечной РНК (критерий Стьюдента; MUT17: р=0,0024, MUT30: р=0,0122, MUT44: р=0,0075). Эти данные подтверждают, что индуцированные специфичные относительно мутации Т-клетки эффективно распознают эндогенно процессированные эпитопы. Две мутации, которые индуцируют предпочтительное распознавание мутантных эпитопов, находятся в генах Actn4 и Kif18b. Соматическая мутация в ACTN4 (актинин, альфа 4) p.F835V находится в кальций-связывающем белковом домене «EF-hand». Хотя как «SIFT» так и «POLYPHEN» прогнозируют значительное влияние этой мутации на функцию белка, ген не является известным онкогеном. Однако специфичные к мутации Т-клетки против ACTN4 недавно были ассоциированы с положительным исходом для пациентов (Echchakir H et al., Cancer Res 2001; 61:4078-83). KIF18B (представитель семейства кинезина, 18В) является кинезином со способностью передвижения по микротрубочкам и способностью связывать АТФ и нуклеотиды, который вовлечен в регуляцию деления клеток (Lee YM et al., Gene 2010; 466:16-25) (фиг.10С). Последовательность ДНК в положении, кодируемом р.K739 является гомогенной в референсной C57BL/6, при этом риды ДНК из B16F10 выявили гетерозиготную соматическую мутацию. Оба нуклеотида детектировались в ридах 16F10 RNA-Seq и были подтверждены секвенированием по Сэнгеру. KIF18B ранее не был ассоциирован со злокачественным опухолевым фенотипом. Мутация p.K739N не локализована в известном функциональном или консервативном белковом домене (фиг.10С, внизу) и таким образом, наиболее вероятно является ведомой, чем ведущей мутацией. Эти примеры подтверждают утрату корреляции между способностью индуцировать мутационно-распознаваемый иммунный ответ и функциональную или иммунологическую релевантность.
In vivo оценка противоопухолевой активности вакцинных кандидатов
Для оценки будут ли иммунные ответы, вызываемые in vivo транслироваться в противоопухолевые эффекты в мышах, несущих опухоли, мы выбрали в качестве примеров MUT30 (мутация в Kif18b) и MUT44. Было показано, что эти мутации индуцируют сильную иммунную реакцию, предпочтительно против мутированного пептида и могут быть эндогенно процессированы (фиг.10А, В). Терапевтический потенциал вакцинации мутантными пептидами исследовали иммунизацией мышей либо MUT30, либо MUT44 и адъювантом через 3 и 10 дней после прививки 7,5×105 B16F10. Рост опухолей ингибировался вакцинациями обоими пептидами, по сравнению с контрольной группой (фиг.11А). Поскольку B16F10 является очень агрессивно растущей опухолью, мы также протестировали защитные иммунные ответы. Мышей иммунизировали пептидом MUT30, инокулировали п.к. 7,5×105 клеток B16F10 через 4 дня и повторно стимулировали MUT30 через 2 и 9 дней после заражения опухолью. Наблюдали полную защиту от опухоли и выживание 40% мышей, обработанных MUT30, при этом все мыши в контрольной группе умерли в течение 44 дней (фиг.11В, слева). У тех из мышей, у которых опухоли развились, несмотря на иммунизацию MUT30, рост опухолей был медленней, из-за удлинения медианы выживания на 6 дней по сравнению с контрольной группой (фиг.11В, справа). Эти данные предполагают, что вакцинация всего лишь против одной мутации способна наделить противоопухолевыми эффектами.
Иммунизация РНК, кодирующими мутации.
50 проверенных мутаций из клеточной линии меланомы B16F10 использовали для конструирования различных РНК-вакцин. ДНК-последовательности, представляющие одну (моноэпитоп), две (биэпитоп) или 16 различных мутаций (полиэпитоп), конструировали с использованием 50 аминокислот (ак) с мутацией в положении 25 (биэпитоп) или с использованием 27 ак с мутацией в положении 14 (моно- и полиэпитоп) и отделяли с помощью глицин/серинового линкера длиной 9 ак. Эти конструкты клонировали в каркас pST1-2BgUTR-A120 для in vitro транскрипции мРНК (Holtkamp et al., Blood 2006; 108:4009-17).
Для проверки in vivo способности индуцировать Т-клеточные ответы против различных РНК-вакцин группы из трех мышей C57BL/6 иммунизировали составом РНК с липофектамином «RNAiMAX» с последующей внутривенной инъекцией. После 5 иммунизации мышей умерщвляли и анализировали спленоциты на предмет специфичных к мутациям Т-клеточным ответам путем окрашивания внутриклеточных цитокинов и анализа «IFN-γ ELISPOT» после повторной стимуляции соответствующим пептидом, кодирующим мутацию или контрольным пептидом (VSV-NP).
На фиг.12 показан один пример для каждого вакцинного дизайна. В верхней строке мышей вакцинировали РНК с моноэпитопом, кодирующим MUT30 (мутация в Kif18b), который индуцирует MUT30-специфичные CD4+ Т-клетки (см. примерный график FACS). В средней строке диаграмма и график FACS демонстрируют индукцию MUT08-специфичных (мутация в Ddx23) CD4+ Т-клеток после иммунизации биэпитопом, кодирующим MUT33 и MUT08. В нижней строке мышей иммунизировали с полиэпитопом, кодирующим 16 различных мутаций, включая MUT08, MUT33 и MUT27 (см. таблицу 8). Диаграмма и график FACS иллюстрируют, что MUT27 реактивные Т-клетки имеют CD8 фенотип.
Такой же полиэпитоп использовали для получения данных, представленных на фиг.13. Диаграмма демонстрирует данные ELISPOT после повторной стимуляции спленоцитов контрольным (VSV-NP), MUT08, MUT27 и MUT33 пептидами, доказывая, что вакцина с полиэпитопом может индуцировать специфический Т-клеточные ответы против некоторых различных мутаций.
В совокупности данные демонстрируют возможность индукции специфичных к мутации Т-клеток с помощью моно-, би- и полиэпитопов. Более того, данные демонстрируют индукцию CD4+ и CD8+ Т-клеток и индукцию некоторых различных специфичностей из одного конструкта.
Иммунизация модельными эпитопами
Для дальнейшего описания дизайна полиэпитопной РНК-вакцины конструировали последовательности ДНК, в которые включены пять различных известных модельных эпитопов, в том числе один эпитоп МНС класса II (овальбумин класса I (SIINFEKL), класса II (OVA класса II), нуклеопротеин гриппа (Inf-NP), нуклеопротеин вируса везикулярного стоматита (VSV-NP) и родственный тирозиназе белок 2 (Trp2)). Эпитопы были разделены таким же глицин/сериновым линкером длиной 9 ак, что и использованный для мутантного полиэпитопа. Эти конструкты клонировали в каркас pST1-2BgUTR-A120 для in vitro транскрипции мРНК.
In vitro транскрибированные РНК использовали для вакцинации пяти мышей C57BL/6 внутриузловой иммунизацией (четыре иммунизации по 20 мкг РНК в паховые лимфоузлы). Через пять дней после иммунизации собирали для анализа из мышей образцы крови и спленоцитов. Фиг.14А демонстрирует анализ «IFN-γ ELISPOT» спленоцитов, повторно стимулированных указанными пептидами. Ясно видно, что все три эпитопа МНС класса I (SIINFEKL, Trp2 и VSV-NP) индуцируют очень большое количество антиген-специфичных CD8+ Т-клеток. Также эпитоп OVA класса II (эпитоп МНС класса II) индуцирует сильный CD4+ Т-клеточный ответ. Четвертый эпитоп МНС класса I анализировали окрашиванием Inf-NP-специфичных CD8+ Т-клеток флуоресцентно меченным пентамерным МНС-пептидным комплексом (Пентамером) (фиг.14В).
Эти данные доказывают, что полиэпитопный дизайн с использованием глицин/серинового линкера для отделения различных иммуногенных эпитопов МНС класса I и класса II, способен индуцировать специфичные Т-клетки против каждого кодируемого эпитопа, вне зависимости от иммунодоминирования.
Противоопухолевый ответ после терапии кодирующей мутации полиэпитопной РНК-вакциной
Такой же полиэпитоп, иммуногенность которого была проанализирована на фиг.13, был использован для исследования противоопухолевой активности кодирующих мутации РНК против опухолевых клеток B16F10. В частности, группы мышей C57BL/6 (n=10) подкожно инокулировали 1×105 клеток меланомы B16F10 в бок. На 3, 6, 10, 17 и 21 дни мышей иммунизировали полиэпитопной РНК с помощью реактива для липосомальной трансфекции. Контрольную группу инъецировали только липосомами.
Фиг.21 демонстрирует кривые выживания групп, раскрывая значимо улучшенное медианное выживание в течение 27 дней, с 1 из 10 мышей, выжившей без опухоли, по сравнению с 18, 5 днями медианного выживания в контрольной группе.
Противоопухолевый ответ после терапии комбинацией мутантного и нормального пептида
Противоопухолевую активность проверенных мутаций оценивали в терапевтическом in vivo опухолевом эксперименте с использованием MUT30 в качестве пептидной вакцины. В частности, группы мышей C57BL/6 (n=8) подкожно инокулировали 1×105 клеток меланомы B16F10 в бок. В дни 3, 10 и 17 мышей иммунизировали пептидами MUT30, родственным тирозиназе белком 2 (Trp2180-188) с использованием полиI:С в качестве адъюванта, или комбинацией обоих пептидов. Trp2 является известным эпитопом CD8+, экспрессируемым клетками меланомы B16F10.
Фиг.15А демонстрирует средний рост опухолей в группах. Можно ясно увидеть, что до дня 28 рост опухолей почти полностью ингибирован в группе, которая была иммунизирована комбинацией известного CD8+ Т-клеточного эпитопа и индуцирующего CD4+ Т-клетки MUT30. Известный эпитоп Trp2 отдельно не достаточен для обеспечения хорошего противоопухолевого эффекта в данных условиях, но в начале эксперимента обе группы с одиночной терапией (MUT30 и Trp2) все еще обеспечивают ингибирование опухолевого роста в сравнении с необработанной группой вплоть до 25 дня. Эти данные подкрепляются кривыми выживания, показанными на фиг.15В. Ясно видно, что медиана выживания повышается у мышей, инъецированных одиночными пептидами, с 1/8 мышей выживших в группе, вакцинированных Trp2. Кроме того, группа, обработанная обоими пептидами демонстрирует еще более лучшую медиану выживания с 2/8 выживших мышей.
В совокупности оба эпитопа действуют синергично при обеспечении сильного противоопухолевого эффекта.
Пример 9. Среда для достоверной детекции соматических мутаций и применение к клеткам меланомы B16-F10.
NGS объективен, поскольку позволяет осуществить высокопроизводительное обнаружение вариаций в целом геноме или целевых областях, таких как кодирующие белки экзоны.
Однако, несмотря на революционность, платформа NGS все еще склонна к ошибкам, ведущим к ошибочному прогнозу вариаций. Более того, качество результатов зависит от параметров экспериментального дизайна и методологий анализа. Хотя прогнозы вариаций, как правило, включают баллы, разработанные для того, чтобы отличить истинные вариации от ошибок, практичность этих баллов не полностью понятна, как и их интерпретация в отношении оптимизации экспериментов. Это особенно верно при сравнении состояния тканей, например, при сравнении опухоли и нормальной ткани, на предмет соматических мутаций. Как следствие, исследователи вынуждены полагаться на личный опыт, чтобы определить параметры эксперимента и произвольные пороги фильтрации для выбора мутаций.
Наше исследование нацелено на а) создание среды для сопоставления параметров и способов идентификации соматических мутаций и b), назначения значения достоверности выявленным мутациям. Мы трижды отсеквенировали образцы из мышей C57BL/6 и клеточной линии меланомы B16-F10. С помощью этих данных мы получили частоту ложноположительных результатов для соматических мутаций, меру, которую мы затем использовали для оценки существующего программного обеспечения для обнаружения мутаций и для оценки лабораторных протоколов.
Различные экспериментальные и алгоритмические факторы вносят вклад в частоту ложноположительных результатов для вариаций, обнаруженных NGS [Nothnagel, M. et al., Hum. Genet. 2011 Feb 23 [Epub ahead of print]]. Источники ошибок включают артефакты ПЦР, ошибки при затравке [Hansen, K.D., et al., Nucleic. Acids. Res. 38, e131 (2010); Taub, M.A. et al., Genome Med. 2, 87 (2010)] и направленное обогащение [Bainbridge, M.N. et al., Genome Biol. 11, R62 (2010)], эффекты последовательностей [Nakamura, K. et al., Acids Res. (2011) впервые опубликовано онлайн 16 мая 2011 года doi:10.1093/nar/gkr344], прогнозы оснований, вызывающие ошибки последовательности [Kircher, M. et al., Genome Biol. 10, R83 (2009). Epub 2009 Aug 14] и выравнивание ридов [Lassmann, T. et al., Bioinformatics 27, 130-131 (2011)], вызывая изменчивость в покрытии и ошибки секвенирования, которые влияют на дальнейший анализ, например, прогноз вариантов вокруг вставок-делеций [Li, H., Bioinformatics 27, 1157-1158 (2011)].
Для указания вклада различных источников ошибок в прогноз соматических мутаций общая статистическая модель не была описана; только индивидуальные аспекты охвачены без удаления всех систематических ошибок. Недавние вычислительные способы измерения ожидаемого количества прогнозов ложно положительных мутаций включают использование соотношения перехода/трансверсии набора вариаций [Zhang, Z., Gerstein, M., Nucleic Acids Res 31, 5338-5348 (2003); DePristo, M.A. et al., Nature Genetics 43, 491-498 (2011)], машинное обучение [DePristo, M.A. et al., Nature Genetics 43, 491-498 (2011)] и ошибки наследования при работе с семейными геномами [Ewen, K.R. et al., Am. J. Hum. Genet. 67, 727-736 (2000)] или объединенными образцами [Druley, Т.Е. et al., Nature Methods 6, 263-265 (2009); Bansal, V., Bioinformatics 26, 318-324 (2010)]. В целях оптимизации Druley et al. [Druley, Т.Е. et al., Nature Methods 6, 263-265 (2009)] полагались на короткие фрагменты плазмидной последовательности, которые, однако, могут не быть репрезентативными для образца. Для набора однонуклеотидных вариаций (SNV) и выбранных экспериментов, сравнение с SNV, идентифицированными другими методами осуществимо [Van Tassell, C.P. et al., Nature Methods 5, 247-252 (2008)] но трудно оценимо в рамках новых соматических мутаций.
Используя проект секвенирования экзома в качестве примера, мы предлагаем рассчитать частоту ложноположительных результатов (FDR) только на основании данных NGS. Способ не только применим к выбору и приоритизации диагностических и терапевтических мишеней, но также поддерживает разработку алгоритмов и способов, позволяя нам определить достоверные рекомендации для похожих экспериментов.
Для обнаружения мутаций, ДНК из ткани хвоста трех мышей C57BL/6 (black6) (из одного выводка) и ДНК из клеток меланомы B16-F10 (В 16), в трех повторах, индивидуально обогащали по белок-кодирующим экзонам («Agilent Sure Select Whole Mouse Exome»), с получением в итоге 6 образцов. РНК экстрагировали из клеток В16 трижды. Одноконцевые 50 нт (1×50 нт) и двухконцевые 100 нт (2×100 нт) риды получали на «Illumina HiSeq 2000». Каждый образец загружали в индивидуальную дорожку, что в среднем дает 104 миллиона ридов на дорожку. ДНК-риды выравнивали по референсному мышиному геному с помощью «bwa» [Li, H. Durbin, R., Bioinformatics 25, 1754-1760 (2009)], а РНК-риды выравнивали с помощью «bowtie» [Langmead, В. et al., Genome Biol. 10, R25 (2009)]. Среднее покрытие для 97% целевых областей в библиотеках 1×50 нт составило 38, а эксперименты 2×100 нт дали среднее покрытие для 98% целевых областей равное 165.
Соматические вариации независимо идентифицировали с помощью пакетов программного обеспечения «SAMtools» [Li, H. et al., Bioinformatics 25, 2078-2079 (2009)], «GATK» [DePristo, M.A. et al., Nature Genetics 43, 491-498 (2011)] и «SomaticSNiPer» [Ding, L. et al., Hum. Mol. Genet (2010) впервые опубликовано он-лайн 15 сентября 2010 года] (фиг.16) сравнением однонуклеотидных вариаций, обнаруженных в образцах В16, с соответствующими локусами в образцах black6 (клетки В16 изначально были получены из мышей black6). Потенциальные мутации фильтровали согласно рекомендациям авторов соответствующего программного обеспечения («SAMtools» и «GATK.») или отбором соответствующего нижнего порога для соматического балла «SomaticSNiPer», соответственно.
Для получения частоты ложноположительных результатов (FDR) при обнаружении мутаций, вначале мы осуществили пересечение участков мутаций и получили 1355 высококачественных соматических мутаций в качестве консенсуса среди всех трех программ (фиг.17). Однако наблюдаемые различия в результатах примененных программных инструментов являются существенными. Для того чтобы избежать ошибочных выводов, мы разработали способ назначения FDR каждой мутации с помощью реплик. Технические повторы образца должны давать идентичные результаты, и любая детектируемая мутация в данном сравнении «сам против себя» является ложно положительной. Таким образом, для определения частоты ложноположительных результатов при детекции соматических мутаций в опухолевом образце относительно нормального образца («сравнение с опухолью»), мы может использовать технический повтор нормального образца в качестве эталона для оценки количества ложно положительных результатов.
На фиг.18А продемонстрированы примеры вариаций, обнаруженных в данных black6/B16, включая соматическую мутацию (слева), несоматическую вариацию относительно эталона (посередине), и возможные ложно положительные результаты (справа). Каждая соматическая мутация может быть ассоциирована с баллом качества Q. Количество ложно положительных результатов при сравнении с опухолью указывает на несколько ложно положительных результатов при сравнении «сам против себя». Таким образом, для данной мутации с баллом качества Q, детектируемым при сравнении с опухолью, мы оцениваем частоту ложноположительных результатов вычислением соотношения мутаций, обнаруженных при сравнении «сам против себя», с баллом Q или лучше к общему количеству мутаций, обнаруженных при сравнении с опухолью, с баллом Q или лучше.
Трудности возникают при определении Q, так как большинство сред детекции мутаций вычисляют несколько баллов качества. Здесь мы применяем классификатор «случайный лес» [Breiman, L., Statist. Sci. 16, 199-231 (2001)] для объединения множества баллов в один балл качества Q. Мы ссылаемся на раздел методов для подробностей относительно деталей балла качества и вычислений FDR.
Потенциальная систематическая ошибка оценки в сравнении способов заключается в разном покрытии; мы, таким образом, нормализуем частоту ложноположительных результатов для покрытия:
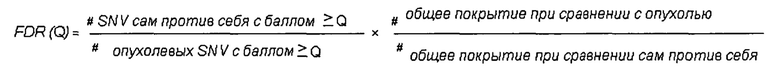
Мы вычисляем общее покрытие путем подсчета всех оснований референсного генома, которые покрываются как опухолевым так и нормальным образцом или обоими образцами «сам против себя», соответственно.
Путем оценки количества ложноположительных и положительных результатов при каждой частоте ложноположительных результатов FDR (см. способы), мы получили кривые соотношений правильного и ложного обнаружения сигналов (ROC) и рассчитали AUC (площадь под кривой) для каждой способа обнаружения мутаций, что, таким образом, позволило осуществить сравнение стратегий выявления мутаций (фиг.18В).
Более того, отбор эталонных данных может повлиять на расчет FDR. С помощью доступных данных black6/B16 возможно создать 18 триплетов (комбинаций black6 vs. black6 и black6 vs. b16). При сравнении полученных распределений FDR для наборов соматических мутаций, результаты являются согласуемые (фиг.18В).
Используя это определение частоты ложноположительных результатов мы получили стандартную среду для оценки влияния множества экспериментальных и алгоритмических параметров на полученный набор соматических мутаций. Затем мы применили эту среду для изучения влияния программных инструментов, покрытия, двухконцевого секвенирования и количества технических реплик на идентификацию соматических мутаций.
Во-первых, выбор программного инструмента оказывает четкое воздействие на идентифицированные соматические мутации (фиг.19А). В протестированных данных «SAMtools» дал наиболее сильное обогащение истинных положительных результатов в наборе соматических мутаций, ранжированных по FDR. Однако следует отметить, что все инструменты предлагают много параметров и баллов качества для индивидуальных мутаций. Здесь, мы использовали настройки по умолчанию, указанные разработчиками алгоритма; мы ожидаем, что параметры могут быть оптимизированы и подчеркиваем, что определенная здесь среда FDR разработана для проведения и оценки такой оптимизации.
Для описанного эксперимента секвенирования B16 мы секвенировали каждый образец в индивидуальной проточной кювете и достигли 38-кратного среднего покрытия оснований целевой области для индивидуальных образцов. Однако это покрытие не нужно для получения одинаково хорошего набора соматических мутаций, что может снизить затраты. К тому же влияние глубины покрытия на полногеномную детекцию SNV недавно обсуждалось [Ajay, S.S. et al., Genome Res. 21, 1498-1505 (2011)]. Для изучения эффекта покрытия на данных захвата экзонов, мы уменьшили количество выровненных по последовательностям ридов для каждой библиотеки 1/50 нт для получения примерного покрытия 5, 10 и 20 раз, соответственно, и затем повторно применили алгоритмы прогнозирования мутаций. Как и ожидалось, более высокое покрытие приводит к лучшему (т.е. с меньшим количеством ложноположительных результатов) набору соматических мутаций, несмотря на то, что улучшение с 20-кратного покрытия до максимального покрытия является незначительным (фиг.19В).
Несложно смоделировать и оценить различные экспериментальные параметры с помощью доступных данных и среды. При сравнении дубликатов с трипликатами, оказалось, что трипликаты не несут пользы по сравнению с дубликатами (фиг.19С), хотя дубликаты несут явное улучшение по сравнению с исследованием вообще без реплик. По показателю соотношения соматических мутаций в данных наборах, мы видим обогащение при FDR 5% с 24,2% для прогона без реплик до 71,2% для дубликатов и 85,8% для трипликатов. Несмотря на обогащение, использование перекрытия трипликатов удаляет больше мутаций с низким FDR, чем с высоким FDR, на что указывает более низкий ROC AUC и смещение кривой влево (фиг.19С): специфичность немного повышается за счет более низкой чувствительности.
Дополнительно секвенированная 2×100 нт библиотека была использована для моделирования 1×100, двух 2×50, и двух 1×50 нт библиотек, соответственно, путем in silicio удаления второго рида и/или 3' и 5' концов ридов, что дает, в общем, 5 смоделированных библиотек. Эти библиотеки сравнивали с использованием вычисленных FDR спрогнозированных мутаций (фиг.19D). Несмотря на намного более высокое среднее покрытие (больше чем 77 против 38), соматические мутации, обнаруженные с помощью 2×50 5' и 1×100 нт библиотек имеют более низкие ROC AUC и, таким образом, худшее распределение FDR, чем у 1×50 нт библиотеки. Это происходит из-за накопления мутаций с высоким FDR в областях с низким уровнем покрытия, поскольку наборы мутаций с низким FDR являются очень похожими. Следствие заключается в том, что оптимальная длина секвенирования либо должна быть незначительной, чтобы секвенируемые основания концентрировались вокруг последовательностей зондов захвата (что, тем не менее, потенциально приводит к потере информации о соматическом статусе мутаций в непокрытых областях), либо должна быть близкой к длине фрагмента (2×100 нт = 200 нт общая длина для ~250 нт фрагментов в нашем случае), эффективно заполняя просветы в покрытии. Этот вывод также поддержан ROC AUC 2×50 нт 3' библиотеки (смоделированной с помощью только 3'-концов 2×100 нт библиотеки), которое выше, чем ROC AUC 2×50 нт 5' библиотеки (смоделированной с помощью только 5' концов 2×100 нт библиотеки), несмотря на более низкое качество оснований в 3'-концах ридов.
Эти наблюдения позволяют нам определить лучшие практические процедуры для обнаружения соматических мутаций. Среди всех оцененных параметров, 20-кратное покрытие в обоих образцах, и использование технического дубликата, позволяют достигнуть близких к оптимальным результатов в этих относительно гомогенных образцах, и это с учетом затрат. 1×50 нт библиотека, дающая примерно 100 миллионов ридов, по всей видимости, является наиболее прагматичным выбором для достижения данного покрытия. Это верно для всех возможных пар наборов данных. Мы ретроспективно применили эти настройки параметров, без использования дополнительного фильтрования для прогноза сырых вариантов, и рассчитали FDR для 50 выбранных мутаций из пересечения всех трех способов, как показано на фиг.17. Все мутации были подтверждены комбинацией повторного секвенирования по Сэнгеру и ридами последовательностей B16-RNA-Seq. 44 из числа этих мутаций могли бы быть обнаружены с помощью 5% порога FDR (фиг.20). В качестве отрицательного контроля мы повторно секвенировали локусы 44 спрогнозированных мутаций с высокими значениями FDR (>50%) и проверили соответствующие последовательности в данных RNA-Seq. Мы обнаружили, что 37 из этих мутаций не подтверждаются, помимо того, что оставшиеся семь локусов потенциальных мутаций не покрывались ридами RNA-Seq и не были получены в реакции секвенирования.
Хотя мы показали применение среды по четырем конкретным вопросам, она отнюдь не ограничивается этими параметрами, и может быть применена для изучения влияния всех экспериментальных или алгоритмических параметров, например, влияния программного обеспечения для выравнивания, выбора системы мер для мутаций, или выбора оборудования для отбора экзома.
Мы осуществили все эксперименты на наборе экспериментов с клетками меланомы В16; однако, способ не ограничен этими данными. Единственное требование заключается в доступности эталонного набора данных «сам против себя», что означает, что, по меньшей мере, один технический повтор неопухолевого образца должен быть получен для каждого нового протокола. Хотя наши эксперименты указывают на то, что способ надежен относительно выбора технического повтора в определенных пределах, что означает, что повтор не обязательно необходим в каждом одиночном эксперименте. Однако способ требует, чтобы различные показатели качества были сравнимыми между эталонным набором данных и оставшимися наборами данных.
В рамках данного вклада мы инициировали использование статистической среды для детекции соматических мутаций с учетом частоты ложноположительных результатов. Эта среда не только применима для выбора диагностических или терапевтических мишеней, но также позволяет осуществить общее сравнение экспериментальных и расчетных стадий протокола на сгенерированных квазидостоверных данных. Здесь мы применили эту идею для того, чтобы осуществлять протокольные решения в отношении программных инструментов, покрытия, реплик, а также секвенирования парных концов.
Способы
Захват и секвенирование библиотеки.
Секвенирование следующего поколения, ДНК-секвенирование: Захват экзома для ресеквенирования ДНК осуществляли с помощью теста для захвата на основе раствора «Agilent Sure-Select» [Gnirke, A., et al., Nat. Biotechnol. 27, 182-189 (2009)], в данном случае настроенном на захват всех известных мышиных экзонов.
3 мкг очищенной геномной ДНК фрагментировали до 150-200 н.о. с помощью ультразвукового устройства «Covaris S2». Концы фрагментов гДНК достраивали с помощью Т4 ДНК-полимеразы, фрагмента Кленова и фосфорилировали по 5' с помощью Т4-полинуклеотидкиназы. гДНК-фрагменты с тупыми концами 3'-аденилировали с использованием фрагмента Кленова (от 3' к 5', экзо минус). С помощью Т4 ДНК-лигазы 3'-концевые одиночные Т-перекрывающиеся парные концевые адаптеры «Illumina» лигировали с гДНК-фрагментами при молярном соотношении адаптера к вставке геномной ДНК 10:1. Лигированные с адаптерами фрагменты гДНК обогащали, добавляли последовательности, специфичные для предварительного захвата и проточной кюветы, с помощью праймеров «Illumina РЕ PCR 1.0» и «2.0» и полимеразы «Herculase II» («Agilent») в течение 4 циклов ПЦР.
500 нг дотированных с адаптером обогащенных ПЦР-фрагментов гДНК гибридизовали с РНК-библиотекой биотинилированных приманок цельного мышиного экзома «SureSelect» фирмы «Agilent» в течение 24 часов при 65°С. Гибридизованные комплексы гДНК/РНК-приманка удаляли с помощью покрытых стрептавидином магнитных микросфер. Комплексы гДНК/РНК-приманка отмывали и РНК-приманки отщепляли в ходе элюции в элюционном буфере «SureSelect», оставляя захваченные обогащенные ПЦР фрагменты гДНК с дотированными адаптерами. Фрагменты гДНК амплифицировали ПЦР после захвата с помощью ДНК-полимеразы «Herculase II» (Agilent) и праймеров «SureSelect GA» в течение 10 циклов.
Очистки осуществляли в 1,8× объеме магнитных микросфер «AMPure ХР» («Agencourt»). Для контролей качества мы использовали тест «Qubit HS» от «Invitrogen», a размер фрагментов определяли с помощью теста «HS DNA» на «Bioanalyzer 2100» от «Agilent».
Обогащенные экземами библиотеки гДНК кластеризовали на «cBot» с помощью набора «Truseq SR cluster kit v2.5» с использованием 7 рМ и секвенировали на «Illumina HiSeq200» с помощью набора «Truseq SBS».
Анализ данных экзома
Риды последовательностей выравнивали с помощью «bwa» (версия 0.5.8 с) [Li, Н. Durbin, R., Bioinformatics 25, 1754-1760 (2009)] при использовании настроек по умолчанию на референсной сборке мышиного генома mm9 [Mouse Genome Sequencing Consortium, Nature 420, 520-562 (2002)]. Неоднозначные риды - риды, которые картируются во многих местах генома согласно выходным данным «bwa» - были удалены. Оставшиеся выравнивания сортировали, индексировали и преобразовывали в бинарный и сжатый формат (ВАМ), и баллы качества ридов преобразовывали из стандартного для «Illumina» «phred+64» в стандартные баллы качества Сэнгера с помощью сценариев оболочки.
Для каждой из секвенируемых дорожек идентифицировали мутации с помощью трех компьютерных программ: «SAMtools pileup» (версия 0.1.8) [Li, H. et al., Bioinformatics 25, 2078-2079 (2009)], «GATK» (версия 1.0.4418) [DePristo, M.A. et al., Nature Genetics 43, 491-498 (2011)], и «SomaticSniper» [Ding, L. et al.. Hum. Mol. Genet (2010) впервые опубликовано онлайн 15 сентября 2010 года]. Для «SAMtools» использовали рекомендованные авторами настройки и критерии фильтра (http://sourceforge.net/apps/mediawiki/SAMtools/index.php?title=SAM_FAQ: обращение в сентябре 2011), включая фильтрацию первого круга, максимальное покрытие 200. Для второго круга фильтрации «SAMtools» минимальный балл качества вставки-делеции составлял 50, минимальное качество точечной мутации составляло 30. Для присваивания мутаций с помощью «GATK» мы следовали разработанным авторами указаниям с наработанными рекомендациями, которые представлены в пользовательском руководстве «GATK» (http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit; обращение в октябре 2010 года). Для каждого образца осуществляли локальное повторное выравнивание вокруг участков вставки-делеции с последующей рекалибровкой качества оснований. Модуль «UnifiedGenotyper» применяли к полученным в результате файлам с данными выравнивания. При необходимости известные полиморфизмы dbSNP [Sherry, S.T. et al., Nucleic Acids Res. 29, 308-311 (2009)] (версия 128 для mm9) дополняли индивидуальные стадии. Стадия рекалибровки балла варианта была опущена и заменена жесткой фильтрацией. Для присваивания мутаций с помощью «SomaticSniper» использовали параметры по умолчанию, а в дальнейшем рассматривали только прогнозируемые мутации с «соматическим баллом» равным 30 или больше. Кроме того, для каждого потенциально мутированного локуса мы требовали ненулевого покрытия в нормальной ткани и удаляли все мутации, расположенные в повторных последовательностях, как определено «RepeatMasker» из «UCSC Genome Browser» для сборки мышиного генома mm9 [Fujita, P.A. et al., Nucleic Acids Res. 39, 876-882 (2011)].
PHK-Seq
Библиотеки mRNA-seqcDNA с штрихкодами получали из 5 мкг общей РНК с помощью модифицированной версии протокола «Illumina mRNA-seq». мРНК выделяли с помощью магнитных микросфер «SeramagOligo(dT)» («Thermo Scientific»). Выделенную мРНК фрагментировали с помощью дивалентных катионов и нагревания, что приводило к получению фрагментов в диапазоне 160-200 нуклеотидов. Фрагментированную мРНК преобразовывали в кДНК с помощью случайных праймеров и «SuperScriptII» («Invitrogen») с последующим синтезом второй цепи с использованием полимеразы I и РНКазы Н. Концы кДНК достраивали с помощью Т4 ДНК-полимеразы, фрагмента Кленова и фосфорилировали по 5'-концам с помощью Т4 полинуклеотидкиназы. кДНК-фрагменты с тупыми концами 3'-аденилировали с использованием фрагмента Кленова (от 3' к 5' экзо минус). С помощью Т4 ДНК-лигазы 3'-концевые одиночные Т-перекрывающие мультиплекс-специфичные адаптеры «Illumina» лигировали с кДНК-фрагментами. кДНК-библиотеки очищали и отбирали по размеру 300 п.о. с помощью геля «E-Gel 2% SizeSelect gel» («Invitrogen»). Обогащение, добавление последовательностей «Illumina» для индексирования из шести оснований и последовательностей для проточной кюветы осуществляли ПЦР с использованием ДНК-полимеразы «Phusion» («Finnzymes»). Очистки осуществляли в 1,8× объеме магнитных микросфер «AMPure ХР».
Библиотеки RNA-seq с штрихкодами кластеризовали на «cBot» с помощью набора «Truseq SR cluster kit v2.5» с использованием 7 рМ и секвенировали на «Illumina HiSeq200» с помощью набора «Truseq SBS».
Сырые выходные данные HiSeq обрабатывали согласно стандартному протоколу «Illumina», включая удаление ридов низкого качества и разуплотнение. Риды последовательностей затем выравнивали по референсной геномной последовательности [Mouse Genome Sequencing Consortium, Nature 420, 520-562 (2002)] с помощью «bowtie» [Langmead, B. et al., Genome Biol. 10, R25 (2009)]. Координаты выравнивания сравнивали с координатами экзонов транскриптов RefSeq [Pruitt, K.D. et al., Nucleic Acids Res. 33, 501-504 (2005)] и для каждого транскрипта записывали количества перекрывающих выравниваний. Риды последовательностей, которые не выравнивались с геномной последовательностью, выравнивали относительно базы данных всех возможных экзон-экзонных соединяющих последовательностей транскриптов RefSeq [Pruitt, K.D. et al., Nucleic Acids Res. 33, 501-504 (2005)]. Координаты выравнивания сравнивали с координатами экзонов RefSeq и координатами соединений, риды подсчитывали, и нормализовали относительно RPKM (количество ридов, которые картируются на килобазу нуклеотидов транскрипта на миллион картированных ридов) [Mortazavi, A. et al., Nat. Methods 5, 621-628 (2008)]) для каждого транскрипта.
Проверка SNV
Мы выбрали SNV для проверки ресеквенированием по Сэнгеру и по РНК. Идентифицировали SNV, которые были спрогнозированы всеми тремя программами, которые были несинонимичными и которые были обнаружены в транскриптах, имеющих минимум 10 RPKM. Среди них мы выбрали 50 с наиболее высокими баллами качества SNP, предоставляемые программами. В качестве отрицательного контроля, выбрали 44 SNV, которые имели FDR 50% или больше, которые присутствовали только на одном образце клеточной линии, и которые прогнозировались только одной программой, присваивающей мутации. С помощью ДНК выбранные варианты проверяли амплификацией ПЦР областей, с использованием 50 нг ДНК, с последующим секвенированием по Сенгеру («Eurofins MWG Operon», Эберсберг, Германия). Реакции были удачны для 50 и 32 локусов положительных и отрицательных контролей, соответственно. Проверку также осуществили изучением опухолевых ридов PHK-Seq.
Расчет FDR и машинное самообучение
Вычисления Балла Качества «Случайный Лес»: Широко применяемые алгоритмы обозначения мутаций (DePristo, M.A. et al., Nature Genetics 43, 491-498 (2011), Li, H. et al., Bioinformatics 25, 2078-2079 (2009), Ding, L. et al., Hum. Mol. Genet (2010) впервые опубликовано он-лайн 15 сентября 2010 года) выдают множественные баллы, которые все потенциально влияют на качество прогноза мутаций. Они включают, без ограничения перечисленным, качество представляющего интерес основания, присвоенное инструментом, качество выравнивания по этой позиции, количество ридов, охватывающих эту позицию, или балл различия между двумя геномами, сравниваемыми в этой позиции. Для вычисления частоты ложноположительных результатов нам требуется упорядочивание мутаций, однако это неосуществимо напрямую для всех мутаций, поскольку мы можем иметь противоречащую информацию из различных баллов качества.
Мы используем следующую стратегию для достижения полного упорядочивания. На первом этапе, мы применяем очень строгое определение превосходства, предполагая, что мутация имеет лучшее качество, чем другая, если и только если она превосходит во всех категориях. Так что набор качественных признаков S=(s1,…,sn) предпочтительней Т=(t1,…,tn), что обозначается как S>T, если si>ti для всех i=1,…,n. Мы определили промежуточный FDR (IFDR) следующим образом
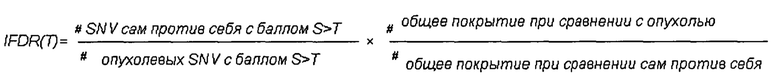
Тем не менее, мы рассматриваем IFDR только как промежуточный шаг, поскольку во многих тесно связанных случаях сравнение не является возможным, и мы, таким образом, не извлекаем выгоду из огромного количества доступных данных. Таким образом, мы воспользовались преимуществом хорошего обобщающего свойства регрессии «случайный лес» [Breiman, L., Statist. Sci. 16, 199-231 (2001)] и натренировали «случайный лес», реализованный в R (R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2010, Liaw, A., Wiener, M., R News 2, 18-22 (2002)).
Для m мутаций на входе с n свойствами качества каждая, был определен диапазон значений для каждого свойства и вплоть до p значений были отобраны с равномерным интервалом из этого диапазона; если набор значений свойства качества меньше, чем p, то этот набор использовался вместо отобранного набора. Затем создаются все возможные комбинации отобранных или отсортированных значений качества, что приводит к максимуму pn точек данных в n-мерном пространстве качества. Случайный образец, включающий 1% этих точек, и соответствующие значения IFDR использовали в качестве прогностического фактора и ответа, соответственно, для тренировки «случайного леса».
Полученный балл регрессии является нашим обобщенным баллом качества Q; его можно рассматривать как локально взвешенную комбинацией отдельных баллов качества. Он позволяет провести прямое сравнение по одному значению любых двух мутаций и вычислить актуальную частоту ложноположительных результатов:
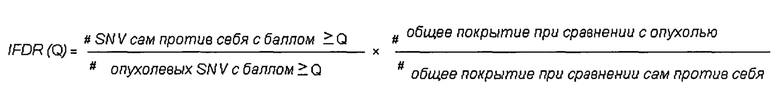
Для тренировки модели «случайный лес», использованной для получения результатов в данном исследовании, мы рассчитали IFDR образца по соматическим мутациям всех образцов перед отбором случайного 1% подмножества. Это позволяет картировать общее доступное пространство качества до значений FDR. Мы использовали свойства качества «качество SNP», «глубина покрытия», «качество консенсуса» и «качество картирования RMS» («SAMtools», P=20); «качество SNP», «глубина покрытия», «достоверность варианта/нефильтрованная глубина» и «качество картирования RMS» («GATK», Р=20); или «качество SNP», «глубина покрытия», «качество консенсуса», «качество картирования RMS» и «соматический балл» («SomaticSNiPer», P=12), соответственно. Различные значения p обеспечивают размер набора сравнимой величины.
Вычисление общего покрытия: Количество возможных прогнозов мутаций может ввести основную системную ошибку в определение частоты ложноположительных результатов. Только если у нас есть такое же количество возможных мест мутаций, происходящих при нашем сравнении опухоли и нашем сравнении «сам против себя», количество прогнозируемых мутаций сопоставимо и может служить в качестве основы для вычисления частоты ложноположительных результатов. Для коррекции этой потенциальной систематической ошибки мы применили соотношение общего покрытия. В качестве общего покрытия мы определили количество оснований с покрытием, по меньшей мере, в одном из двух образцов, которые используются для назначения мутаций. Мы рассчитали общее покрытие индивидуально для опухолевого сравнения, а также для сравнения «сам против себя».
Оценка ROC
Кривые соотношений правильного и ложного обнаружения сигналов (Receiver operating characteristic, ROC) и соответствующая площадь под кривой (area under curve, AUC) пригодны для организации классификаторов и визуализации их эффективности [Fawcett, Т., Pattern Recogn. Lett. 27, 861-874 (2006)]. Мы расширили эту концепцию для оценки эффективности экспериментальных и вычислительных процедур. Однако построение диаграмм ROC требует знания всех истинных положительных и ложноположительных (ТР и FP) примеров в наборе данных, информации, которая, как правило, не представлена, и которую трудно получить для данных, полученных высокопроизводительным подходом (таких, как данные NGS). Таким образом, мы используем вычисленные FDR для оценки соответствующих уровней ТР и FP и для построения графика ROC и для расчета AUC. Центральная идея заключается в том, что FDR одиночной мутации в наборе данных дает долю того, сколько данная мутация вносит в сумму мутаций TP/FP, соответственно. Также для перечня случайных назначений ТР и FP, полученные значения ROC AUC будут равны 0,5 в нашем способе, что указывает на полностью случайный прогноз. Мы начали с двумя условиями:
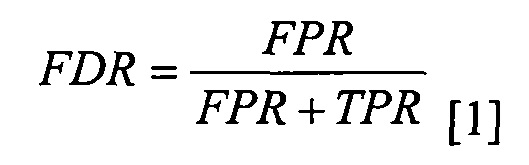
и
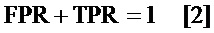
с FPR и TPR представляющие собой необходимые ложноположительные и истинно положительные соотношения, соответственно, для данной мутации, определяя соответствующую точку в пространстве ROC. [1] и [2] могут быть перегруппированы в
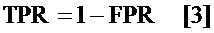
и
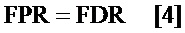
Для получения расчетной кривой ROC, мутации в наборе данных сортировали по FDR и для каждой мутации наносилась точка на график в суммарных значениях в пределах этой мутации, разделенных на сумму все значений TPR и TPR, соответственно. AUC рассчитана суммированием площадей всех идущих подряд трапезоидов между кривой и осью x.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРЕДСКАЗАНИЕ ИММУНОГЕННОСТИ Т-КЛЕТОЧНЫХ ЭПИТОПОВ | 2014 |
|
RU2724370C2 |
| ИНДИВИДУАЛИЗИРОВАННЫЕ ПРОТИВООПУХОЛЕВЫЕ ВАКЦИНЫ | 2012 |
|
RU2779946C2 |
| СПОСОБЫ ПРОГНОЗИРОВАНИЯ ПРИМЕНИМОСТИ БЕЛКОВ ИЛИ БЕЛКОВЫХ ФРАГМЕНТОВ ДЛЯ ИММУНОТЕРАПИИ | 2017 |
|
RU2782336C2 |
| СПОСОБЫ ПРОГНОЗИРОВАНИЯ ПРИМЕНИМОСТИ СПЕЦИФИЧНЫХ ДЛЯ ЗАБОЛЕВАНИЯ АМИНОКИСЛОТНЫХ МОДИФИКАЦИЙ ДЛЯ ИММУНОТЕРАПИИ | 2018 |
|
RU2799341C2 |
| ОПРЕДЕЛЕНИЕ ДОЗЫ ДЛЯ ИММУНОТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2017 |
|
RU2771717C2 |
| АНТИГЕННЫЕ РЕЦЕПТОРЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2794945C2 |
| АНТИТЕЛА ПРОТИВ КЛАУДИНА 18.2 ДЛЯ ДИАГНОСТИКИ РАКА | 2013 |
|
RU2661772C2 |
| АНТИГЕННЫЕ РЕЦЕПТОРЫ И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2792042C2 |
| ИДЕНТИФИКАЦИЯ ОПУХОЛЕАССОЦИИРОВАННЫХ АНТИГЕНОВ ДЛЯ ДИАГНОСТИКИ И ТЕРАПИИ | 2013 |
|
RU2644686C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АНТИТЕЛ ПРОТИВ КЛАУДИНА 18.2 ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2699549C2 |











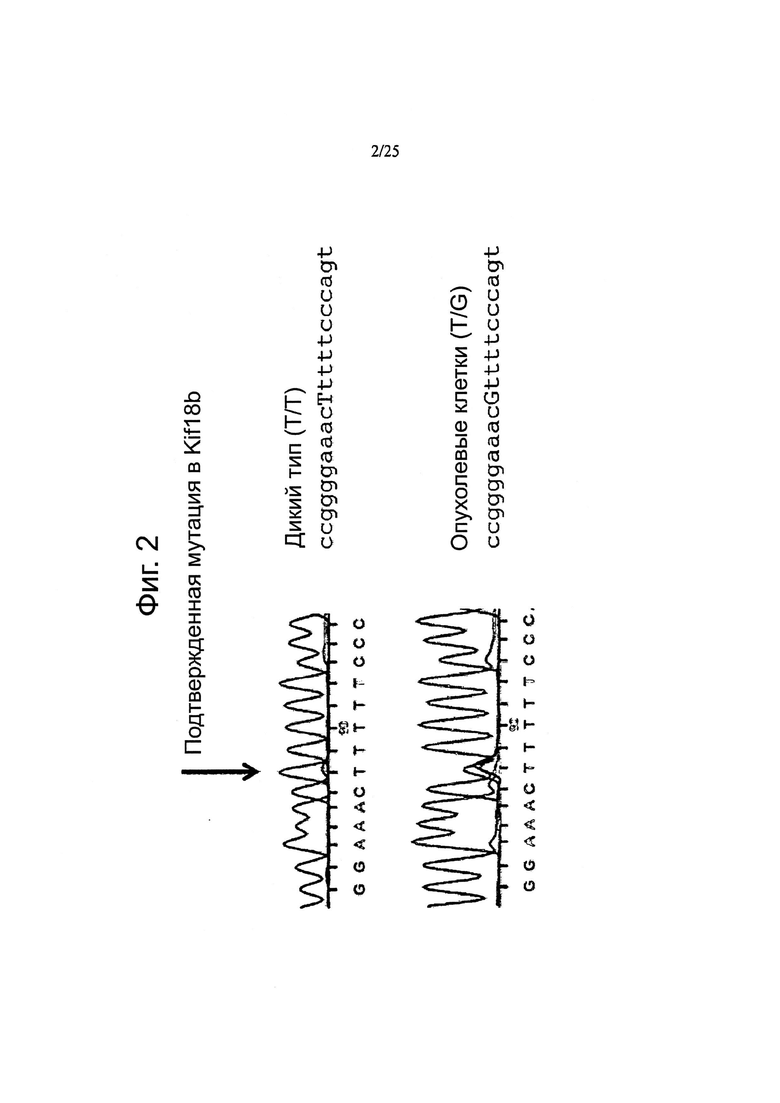
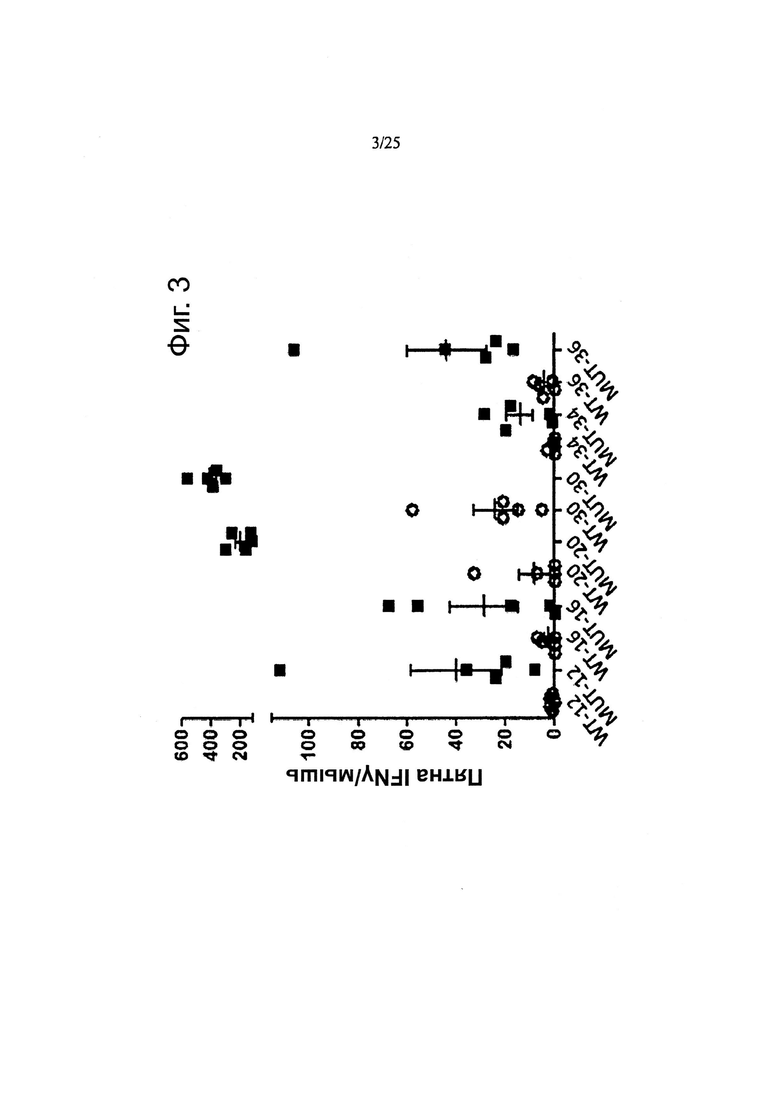
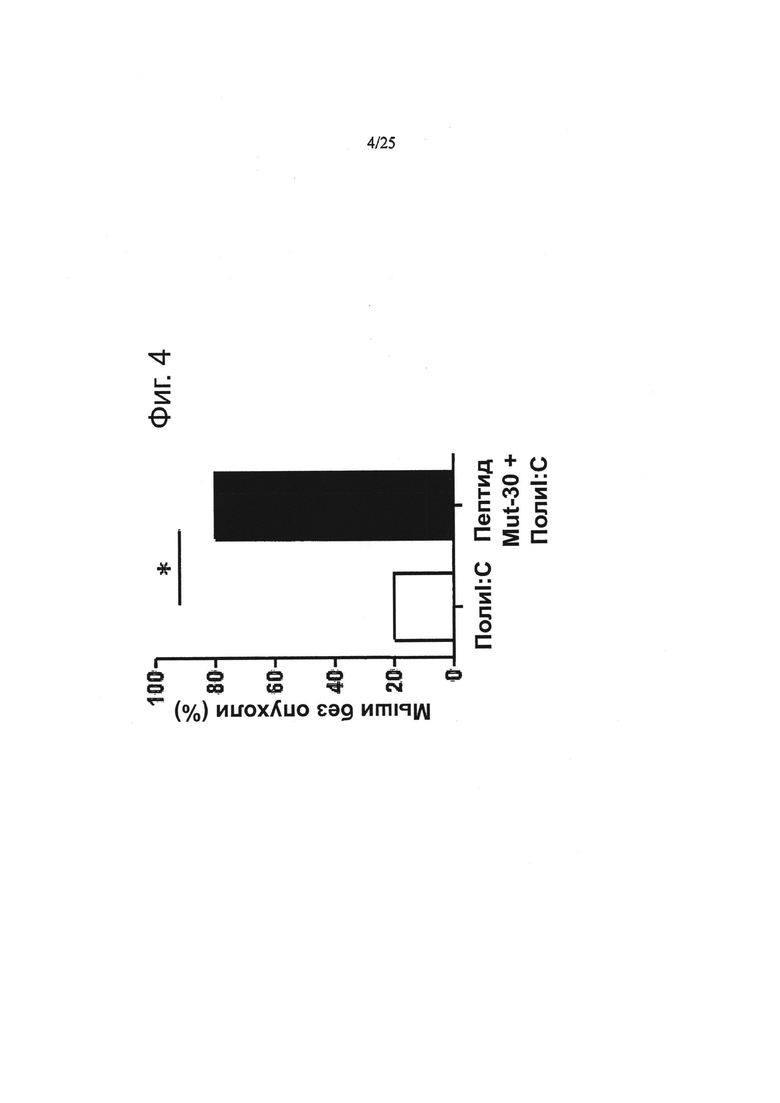
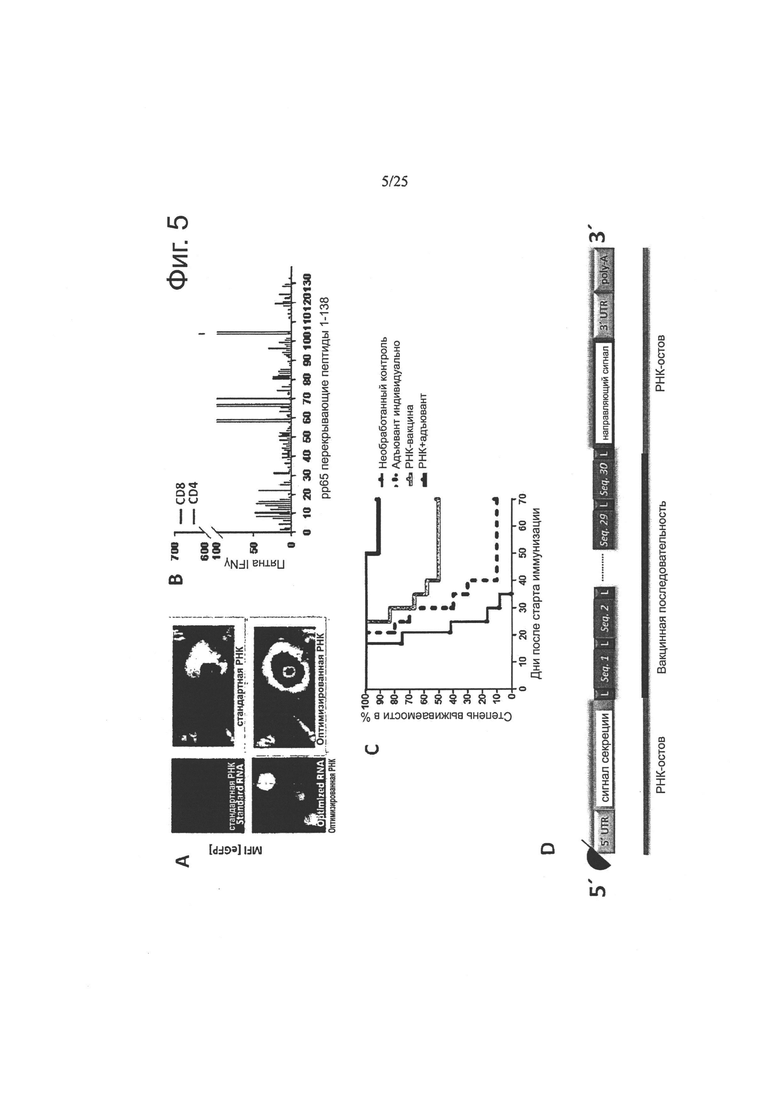

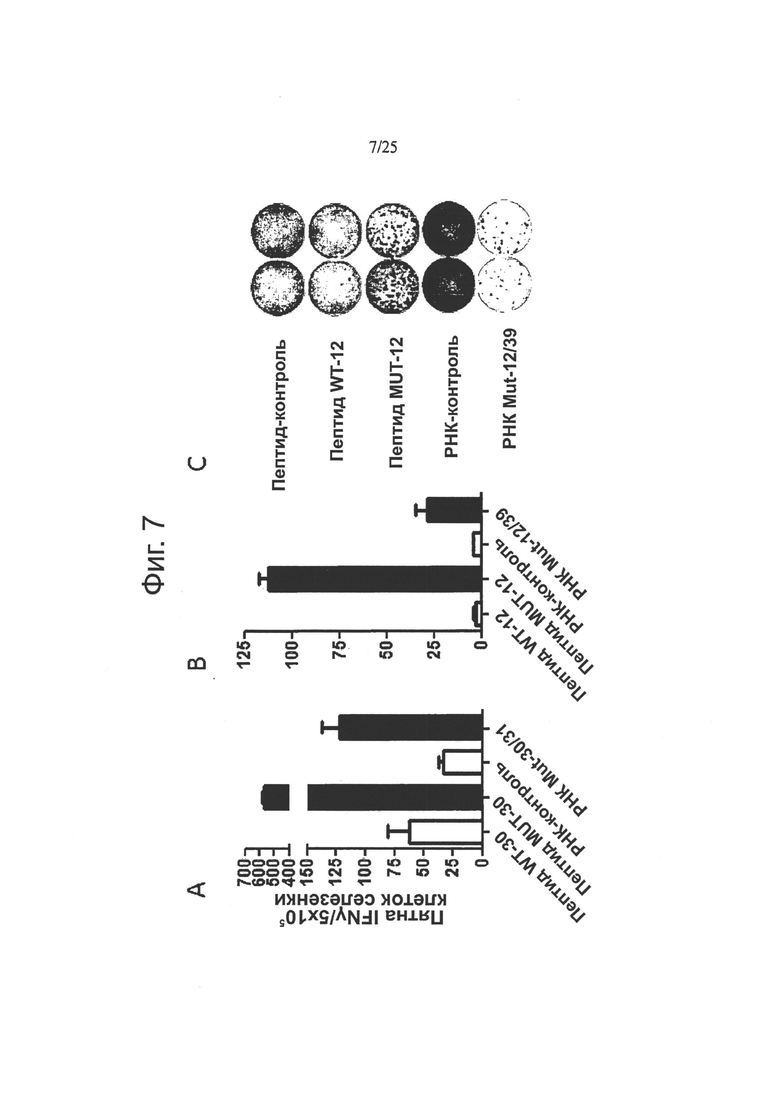
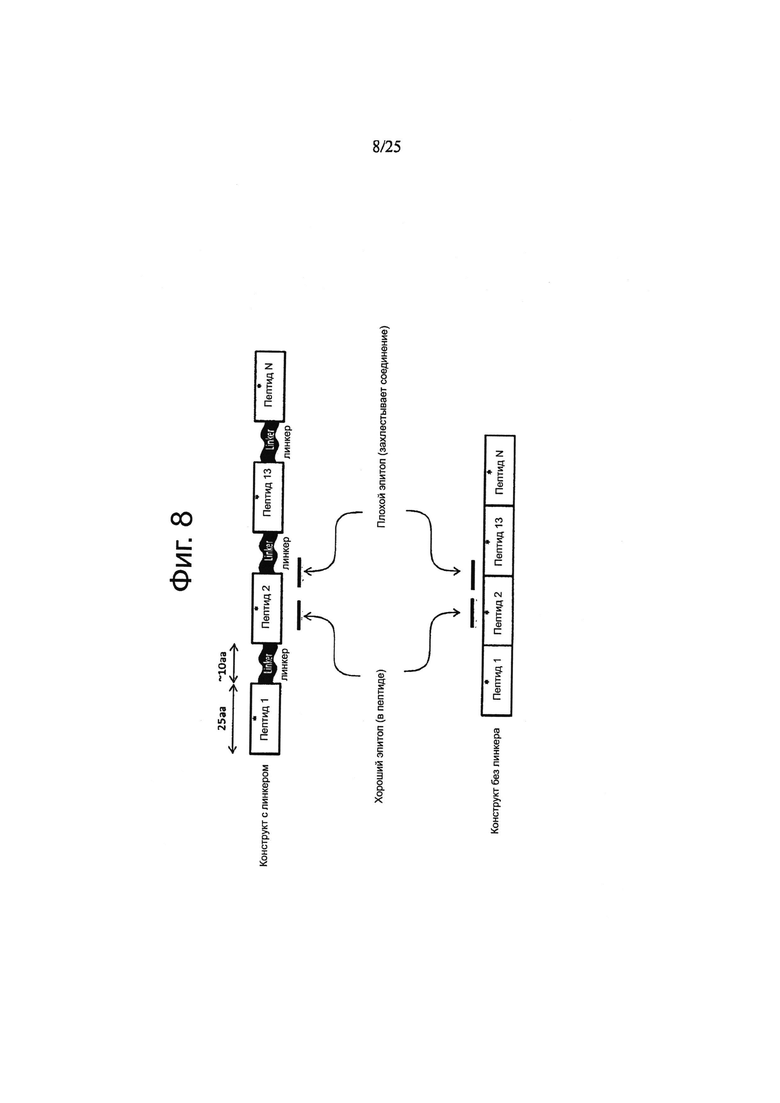
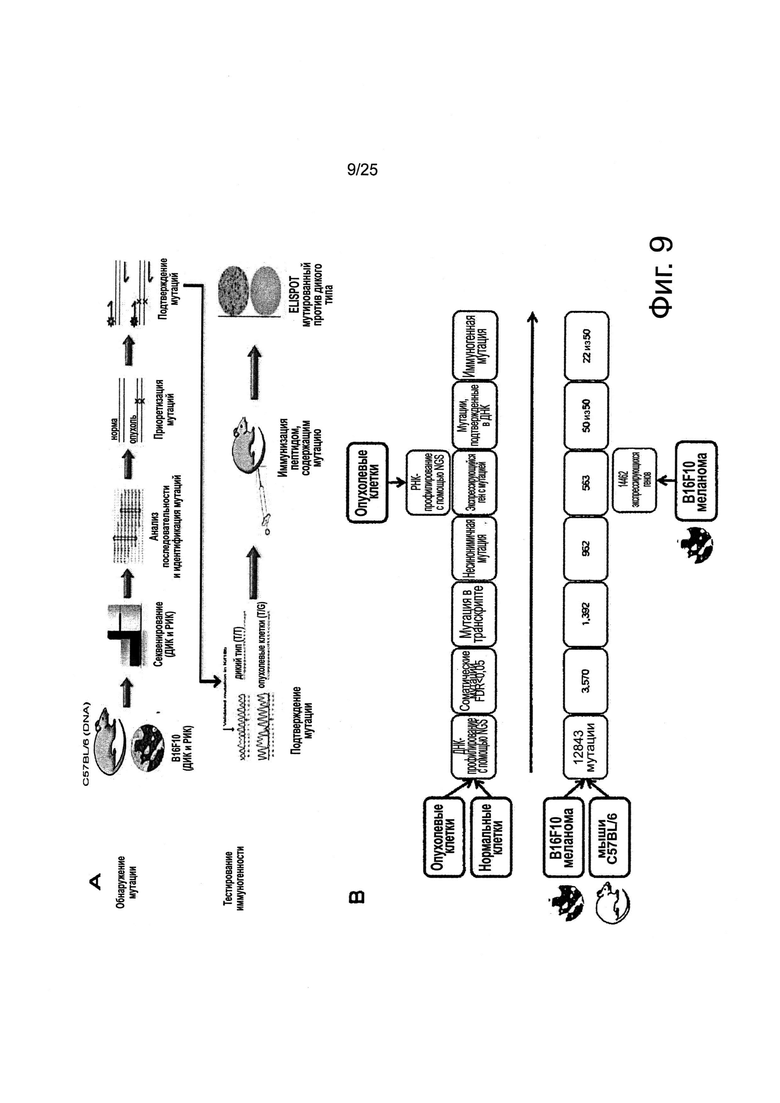
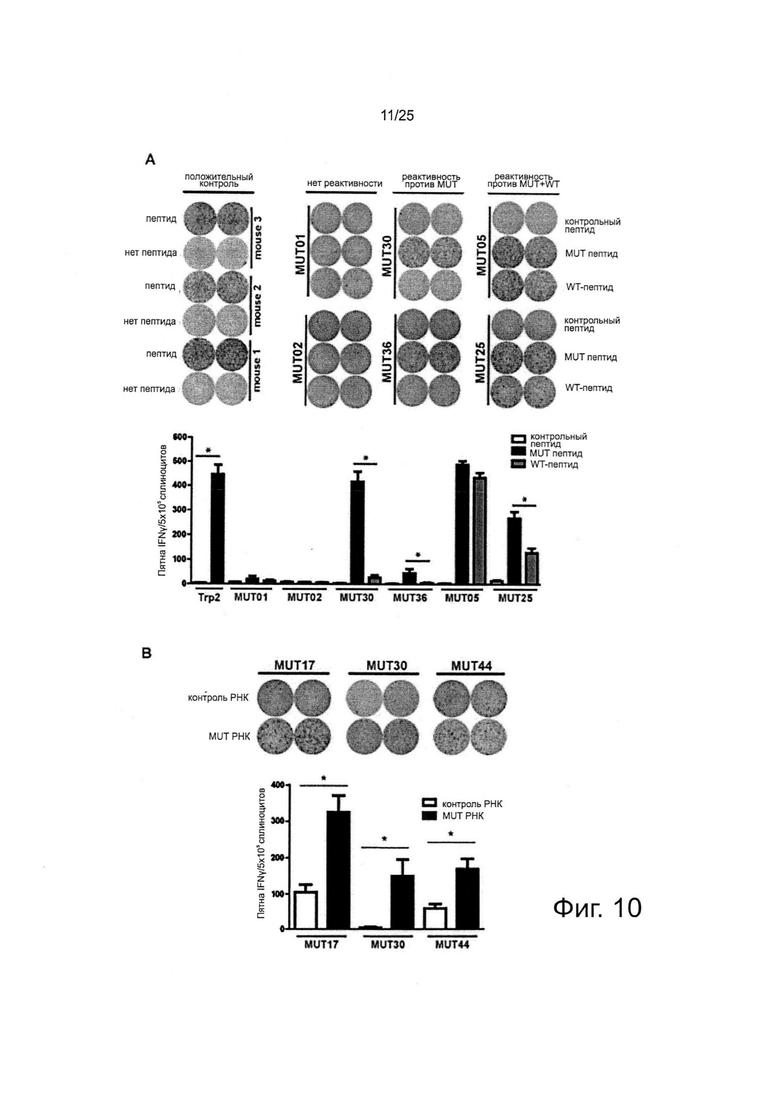

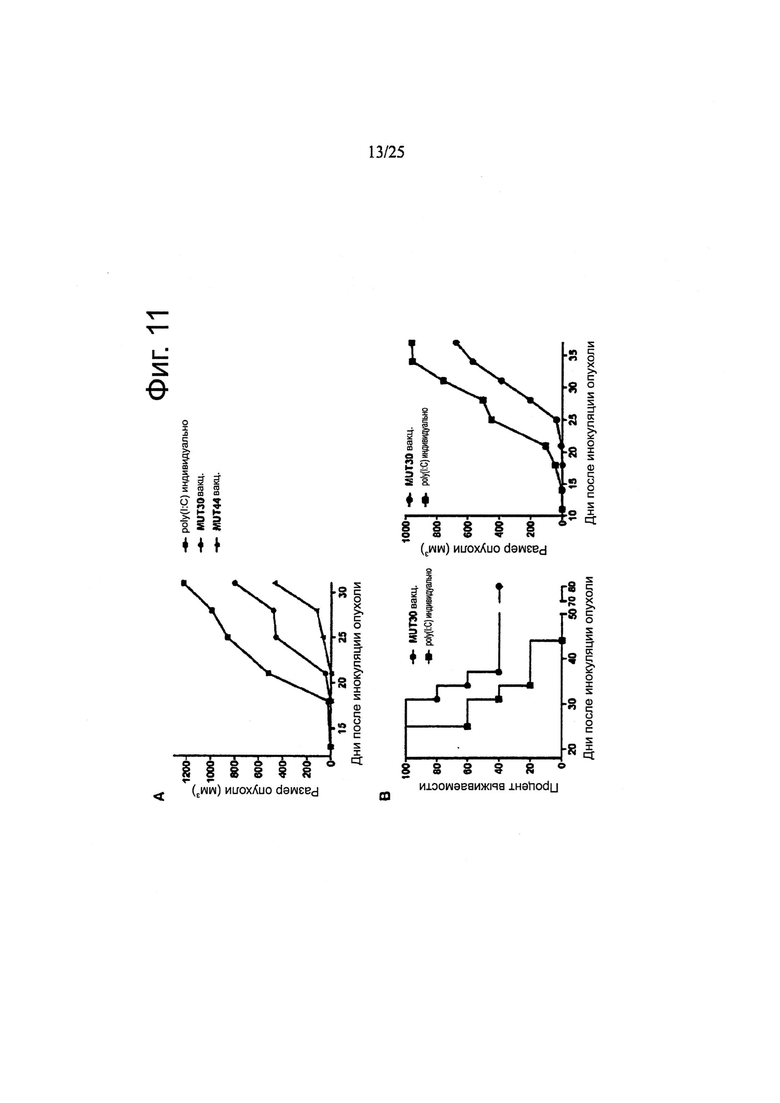
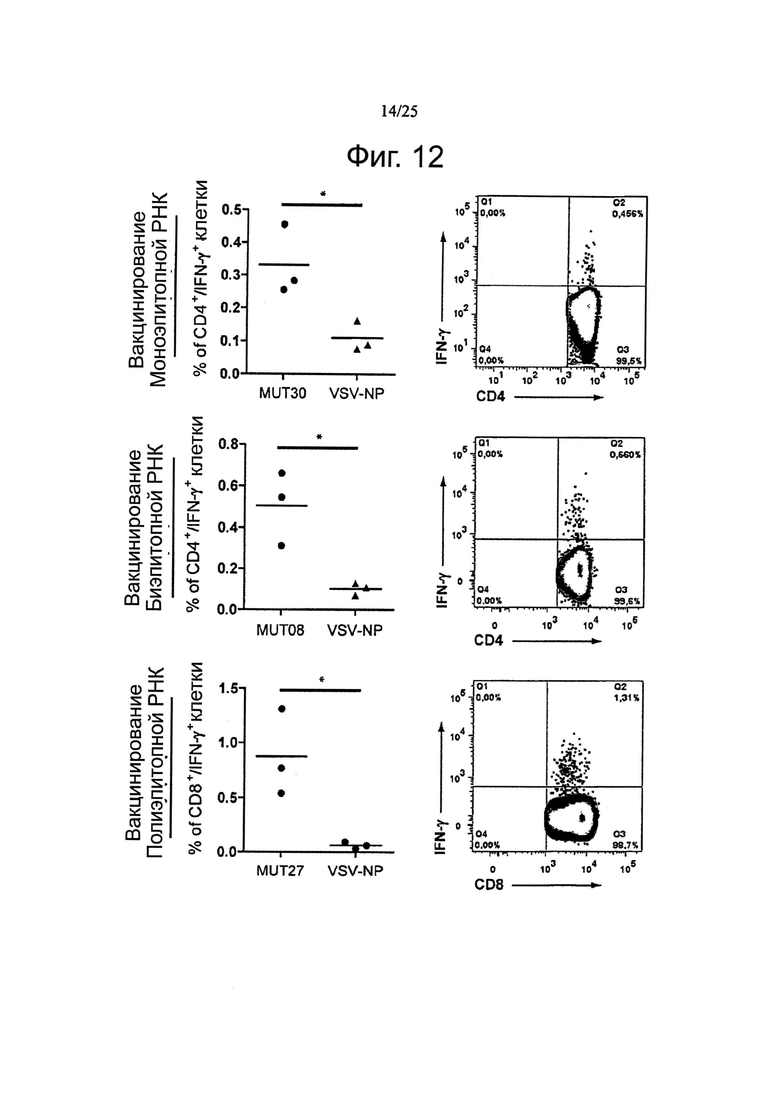
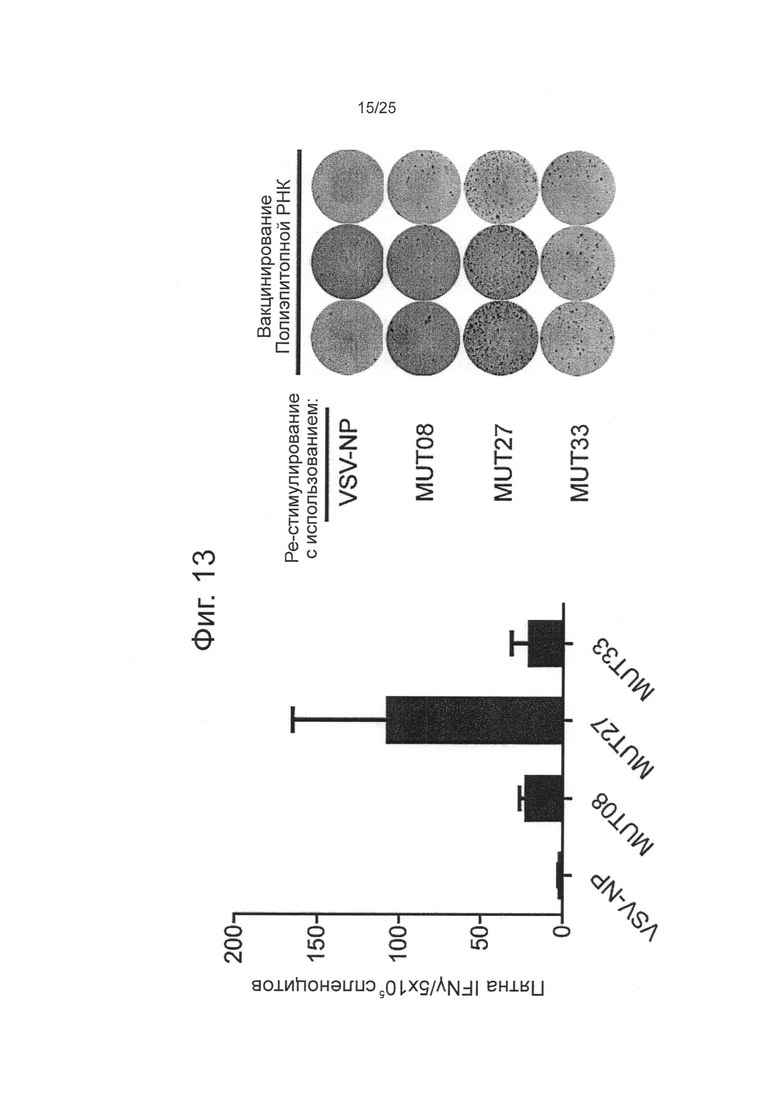
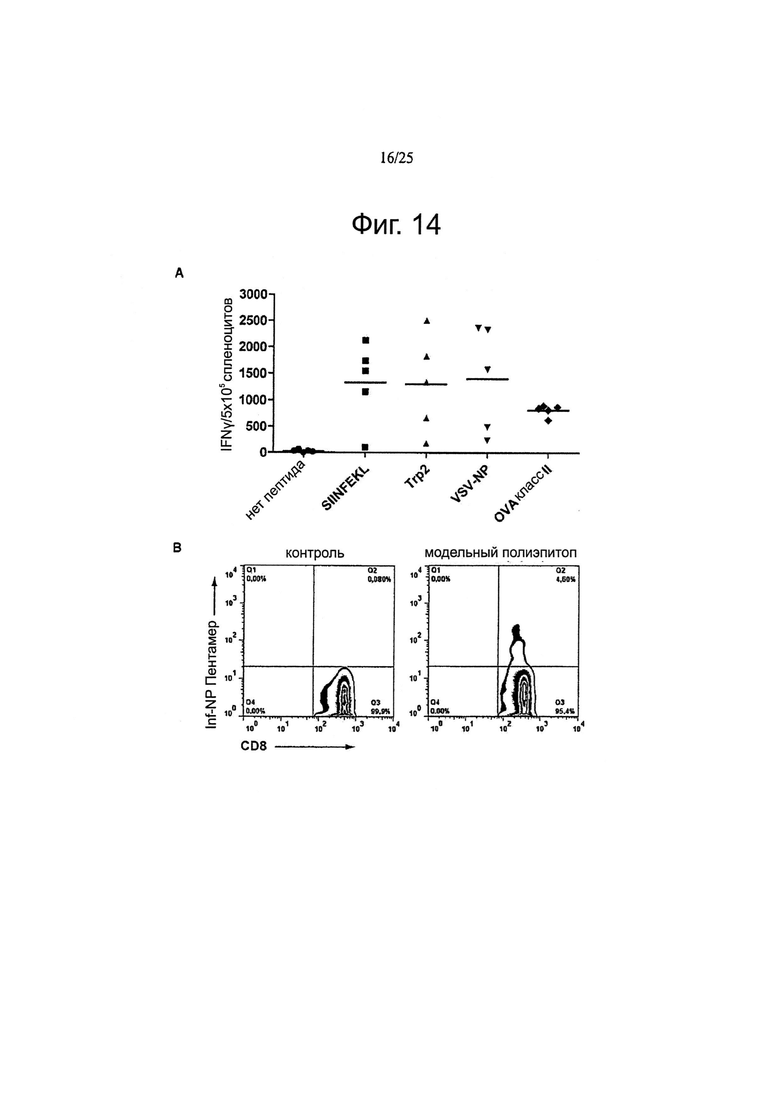
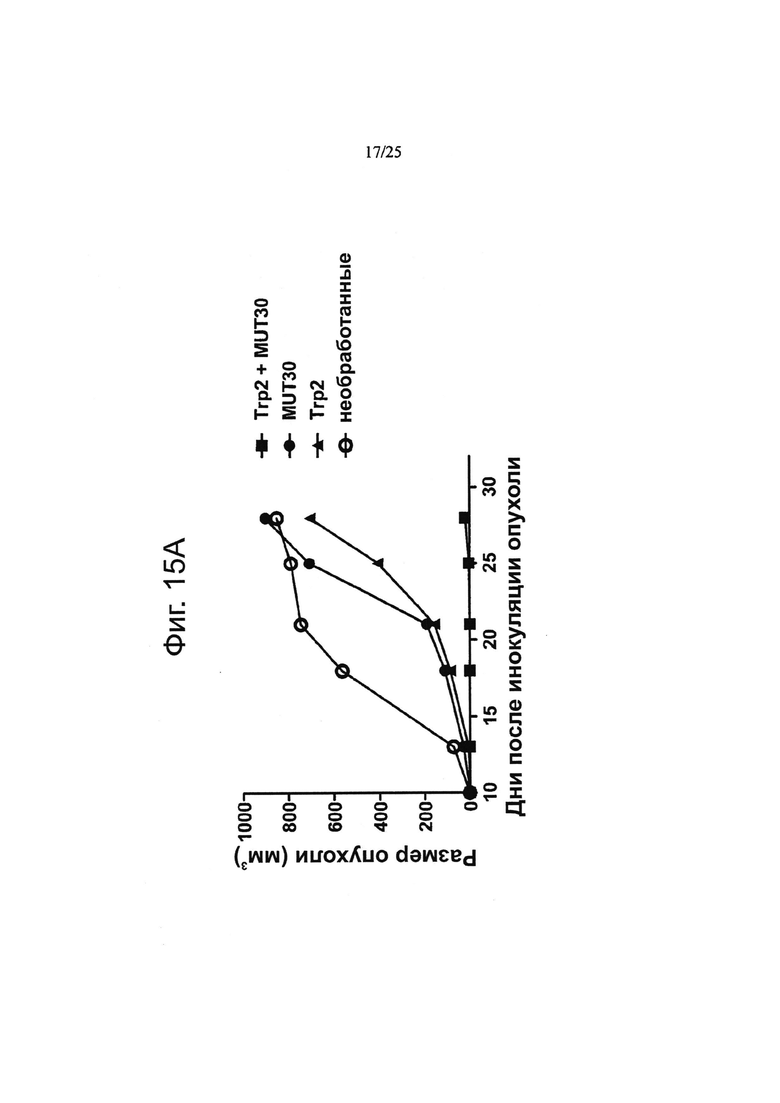
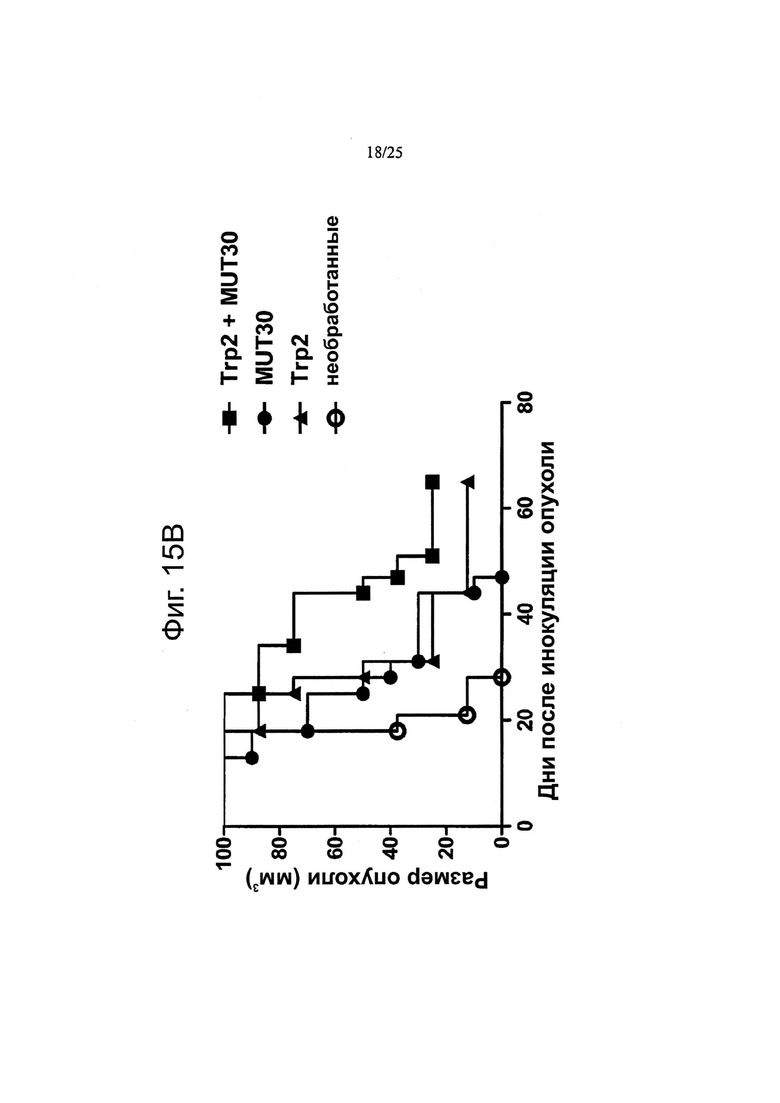
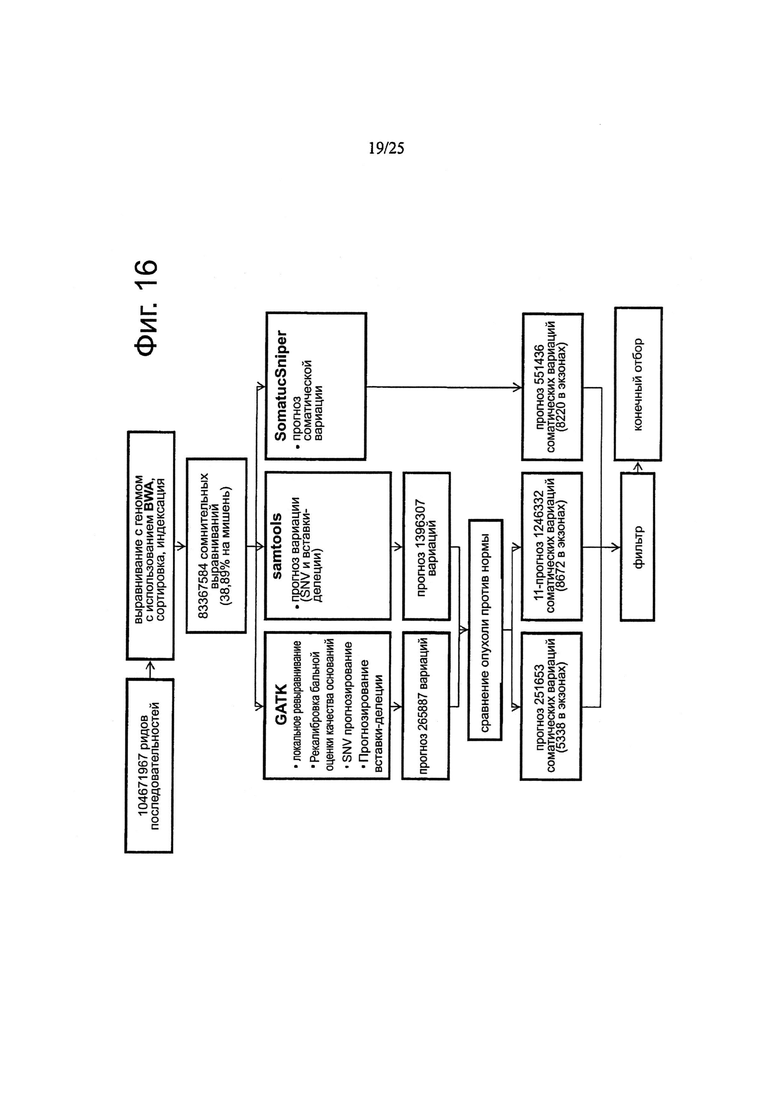
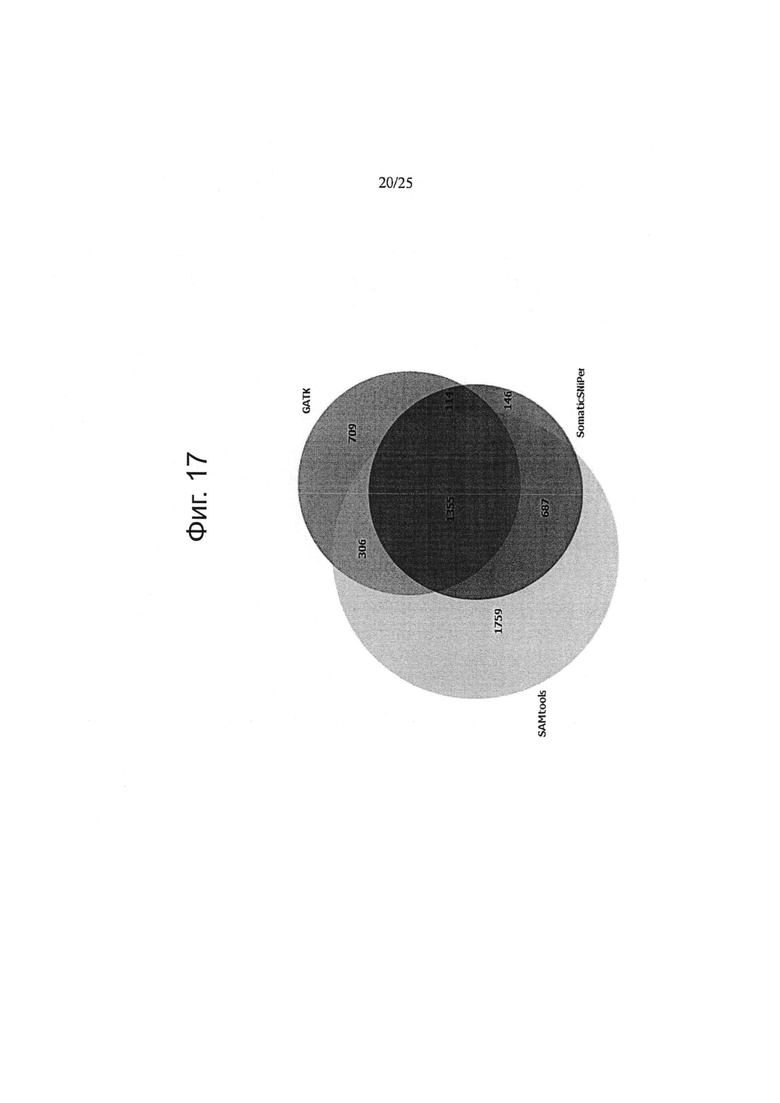
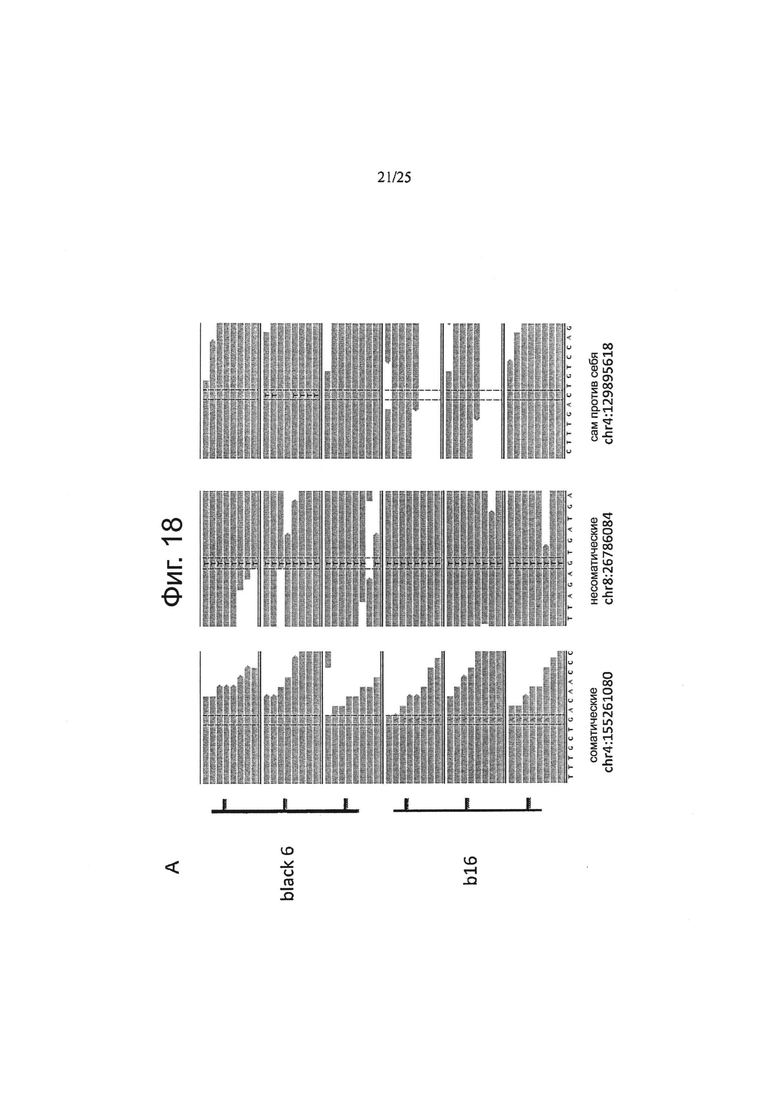
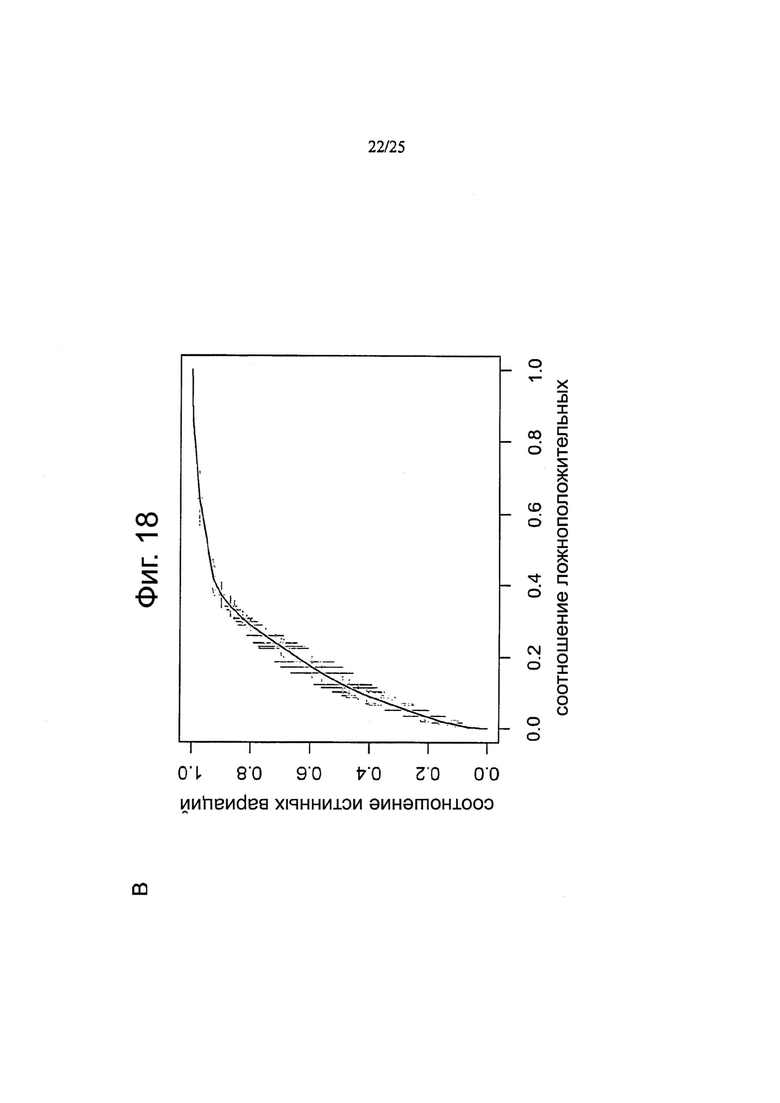
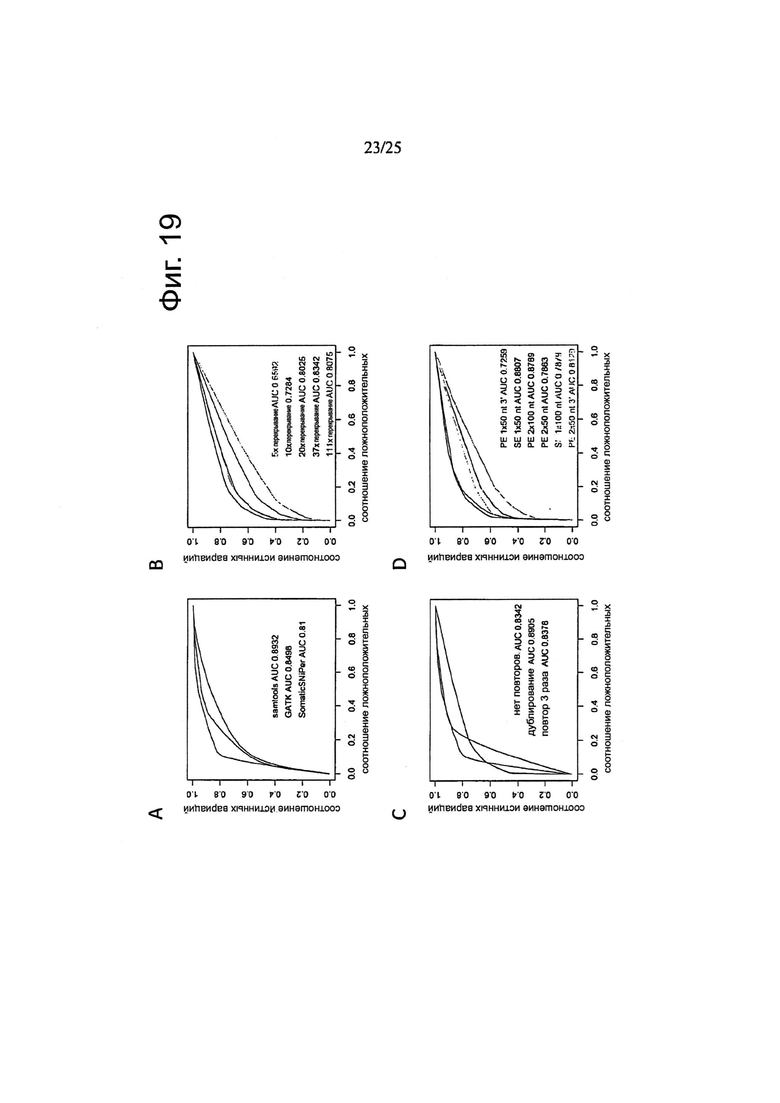
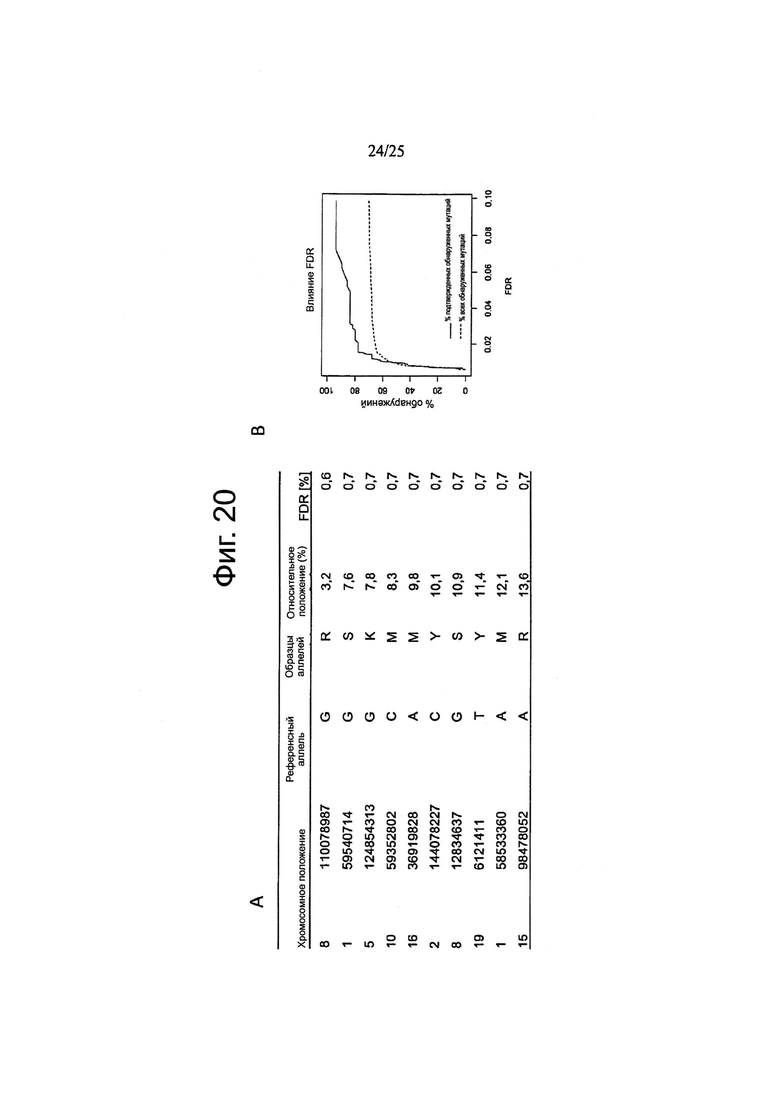
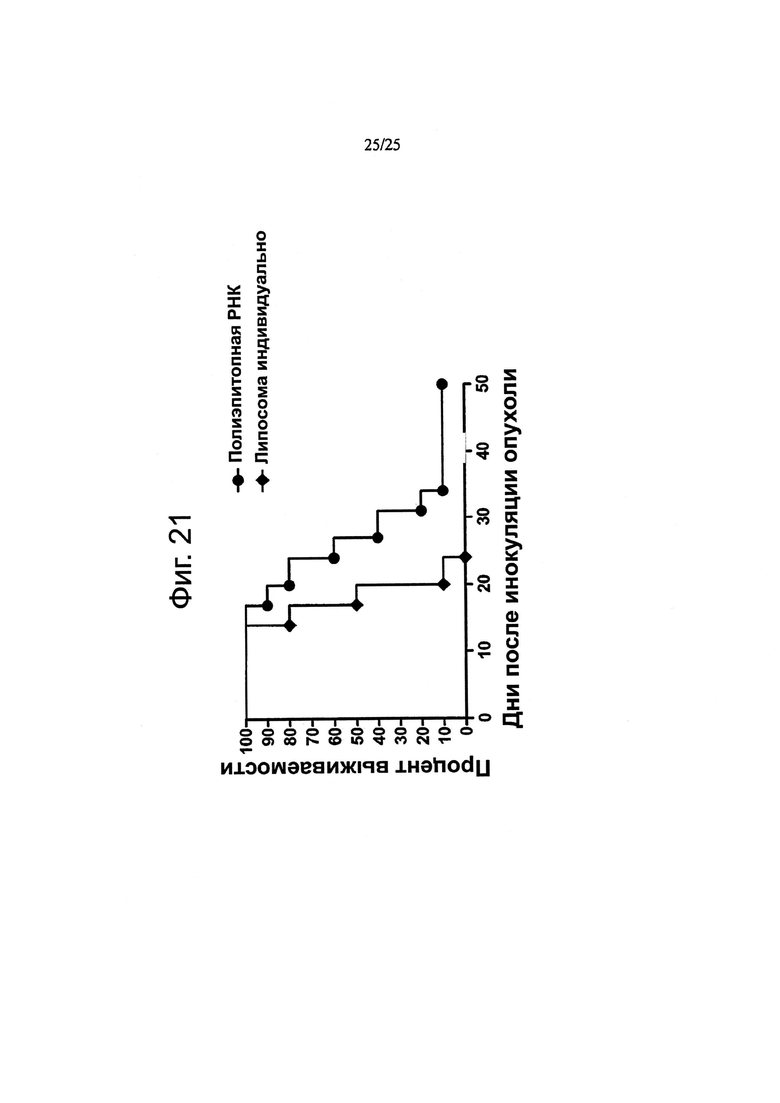
Настоящее изобретение относится к предоставлению вакцин, которые специфичны к опухолям пациентов и потенциально применимы для иммунотерапии первичной опухоли, а также метастазов опухоли. В одном аспекте настоящее изобретение относится к способу получения индивидуальной противоопухолевой вакцины, включающему стадии: (а) идентификации опухолеспецифичных соматических мутаций в образце опухоли онкопациента с получением сигнатуры опухолевых мутаций пациента; и (b) получения РНК вакцины, характерной чертой которой является сигнатура опухолевых мутаций, полученная на стадии (а). В следующем аспекте настоящее изобретение относится к вакцине, которую можно получить указанным способом. 4 н. и 15 з.п. ф-лы, 21 ил., 8 табл., 9 пр.
1. Способ получения индивидуальной противоопухолевой вакцины, включающий стадии:
(a) идентификации опухолеспецифичных соматических мутаций в образце опухоли онкопациента с получением сигнатуры опухолевых мутаций пациента; и
(b) получения РНК вакцины, характерной чертой которой является сигнатура опухолевых мутаций, полученная на стадии (а),
где РНК вакцина содержит РНК, кодирующую рекомбинантный полиэпитопный полипептид, включающий неоэпитопы на основе мутаций.
2. Способ по п. 1, где стадия идентификации опухолеспецифичных соматических мутаций включает идентификацию сигнатуры опухолевых мутаций экзома одной или нескольких раковых клеток.
3. Способ по п. 1, где стадия идентификации опухолеспецифичных соматических мутаций включает секвенирование генома одной клетки из одной или нескольких раковых клеток.
4. Способ по п. 3, где раковые клетки представляют собой циркулирующие опухолевые клетки.
5. Способ по п. 1, где стадия идентификации опухолеспецифичных соматических мутаций включает применение секвенирования следующего поколения (NGS).
6. Способ по п. 1, где стадия идентификации опухолеспецифичных соматических мутаций включает секвенирование геномной ДНК и/или РНК опухолевого образца.
7. Способ по п. 6, где стадия идентификации опухолеспецифичных соматических мутаций повторяется по меньшей мере дважды.
8. Способ по п. 1, включающий дополнительную стадию определения пригодности идентифицированных мутаций в эпитопах для противоопухолевой вакцинации.
9. Способ по п. 1, где рекомбинантный полиэпитопный полипептид включает до 30 неоэпитопов, основанных на мутации.
10. Способ по п. 1, где рекомбинантный полиэпитопный полипептид дополнительно включает эпитопы, не содержащие опухолеспецифичных соматических мутаций, которые экспрессируются опухолевыми клетками.
11. Способ по п. 1, где эпитопы при образовании вакцинной последовательности представлены в контексте своей природной последовательности.
12. Способ по п. 11, где вакцинная последовательность имеет длину около 30 аминокислот.
13. Способ по п. 1, где неоэпитопы, эпитопы и/или вакцинные последовательности располагаются по системе голова к хвосту.
14. Способ по п. 1, где неоэпитопы, эпитопы и/или вакцинные последовательности разделены линкерами.
15. Способ по любому из пп. 1-14, где вакцина представляет собой профилактическую и/или терапевтическую вакцину.
16. Индивидуальная противоопухолевая РНК вакцина, полученная способом по любому из пп. 1-15, где РНК вакцина содержит РНК, кодирующую рекомбинантный полиэпитопный полипептид, содержащий основанные на мутациях неоэпитопы, и где РНК вакцина, будучи введенной пациенту, обеспечивает группу МНС-презентированных эпитопов, включающих изменения последовательности на основе идентифицированных мутаций.
17. Вакцина по п. 16, где МНС-презентируемые эпитопы являются эпитопами, презентируемыми МНС класса II, которые способны вызывать ответ клеток CD4+ Т-хелперов против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы и, необязательно, эпитопы, презентируемые МНС класса I, которые способны вызывать ответ CD8+ Т-клеток против клеток, экспрессирующих антигены, из которых выделены МНС-презентированные эпитопы.
18. Способ лечения рака, включающий стадии:
(a) получения индивидуальной противоопухолевой вакцины способом по любому из пп. 1-15; и
(b) введения указанной вакцины пациенту.
19. Способ лечения рака, включающий введение пациенту вакцины по любому из пп. 16 или 17.
| WO 9934015 A2, 08.07.1999 | |||
| WO 2010037395 A2, 08.04.2010 | |||
| RAMMENSEE H.G | |||
| et al | |||
| Towards patient-specific tumor antigen selection for vaccination | |||
| Immunol Rev | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

