Область техники, к которой относится изобретение
Настоящее изобретение относится к области синтеза лекарственных средств и относится к способу получения антипаразитарного средства Празиквантела и его промежуточного соединения. В частности, изобретение относится к усовершенствованному способу получения Празиквантела и промежуточных соединений, таких как соединения формулы IV, формулы V, а также к способу получения промежуточных соединений и их использованию.
Предпосылки создания изобретения
Празиквантел представляет собой антипаразитарный препарат широкого спектра действия, полезный для лечения японского шистосомоза, мочеполового шистосомоза, кишечного шистосомоза, парагонимоза, клонорхоза, гидатидоза (эхинококкоз однокамерный), цистицеркоза, спарганоза, фасциолопсидоза, трихомоноза и т.п., в частности, японского шистосомоза и клонорхоза. Празиквантел был сначала коммерчески доступен как «Cesol» в Германии в 1980 году и стал первым выбором для лечения гельминтоза, он имеет химическую структуру формулы I:
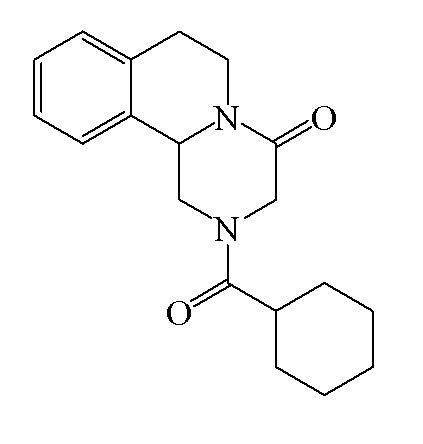
I
DE 2504250 и DE 2508947 раскрывают способ получения Празиквантела с использованием изохинолина в качестве исходного вещества, который широко использовался в мире. Тем не менее, этот способ включает длительную обработку, включающую до 8 стадий, и, таким образом, получают низкий выход около 15%. В процессе обработки используются гипертоксические химические вещества, такие как цианид, и процесс осуществляют под высоким давлением, и, таким образом, этот способ является опасным и высокоаварийным. Помимо этого, этот способ имеет такие недостатки, как выбросы загрязняющих веществ и высокая стоимость защиты окружающей среды, и, таким образом, он значительно ограничен в том, что касается увеличении масштаба производства.
KR 2002076486 раскрывает способ получения Празиквантела с использованием β-фенэтиламина, хлорацетилхлорида, аминоацетальдегид диметилацеталя и т.п. в качестве исходных веществ, который в целом включает меньше стадий реакции без использования цианида. Однако используемый в этом процессе аминоацетальдегид диметилацеталь является дорогостоящим, имеет низкую реакционную способность и селективность и требует высокой температуры реакции, что вызывает побочную реакцию, и, следовательно, не подходит для промышленного производства.
CN 1683346A раскрывает способ получения Празиквантела с использованием β-фенэтиламина, аминоацетилгалогенид гидрохлорида, галогенированного ацетальдегидацеталя и циклогексанкарбонилхлорида в качестве исходных веществ, включающий стадии конденсации, циклизации, ацилирования. Этот способ прост, экологически безвреден и включает меньше стадий, с общим выходом более 50%. Однако исходное вещество галогенированный ацетальдегидацеталь является дорогим с низкой реакционной способностью и селективностью, а исходное вещество аминоацетилгалогенид гидрохлорид нестабилен и имеет тенденцию к ухудшению. По этим причинам этот способ не подходит для промышленного производства.
Сущность изобретения
Принимая во внимание недостатки предшествующего уровня техники, в одном аспекте представлен усовершенствованный способ получения Празиквантела. Способ является выгодным, например, он разумно спланирован, прост, экономически эффективен, экологически безвреден и требует умеренных условий реакции, а также дешевых и легко доступных исходных веществ. Более того, промежуточные соединения можно легко получить, общий выход является высоким (≥60%), целевой продукт, соединение формулы I Празиквантел, имеет высокую степень чистоты (чистота в соответствии с ВЭЖХ ≥99,8%). Таким образом, этот способ подходит для промышленного производства в больших масштабах.
В частности, обеспечивается способ получения Празиквантела, включающий следующие стадии:
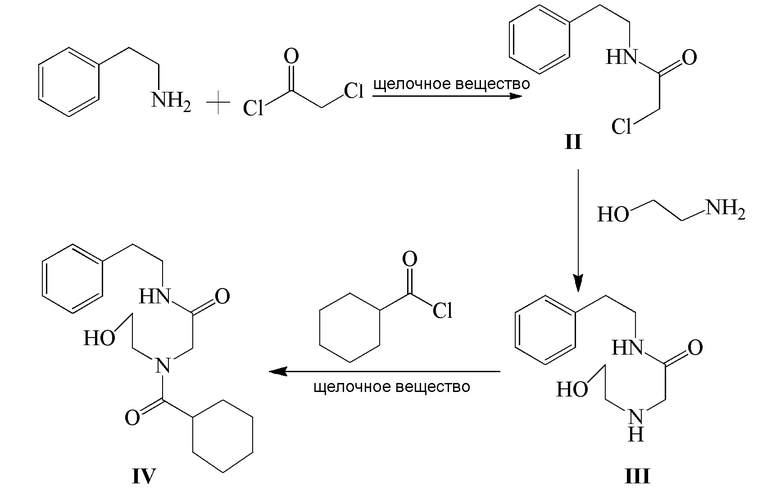
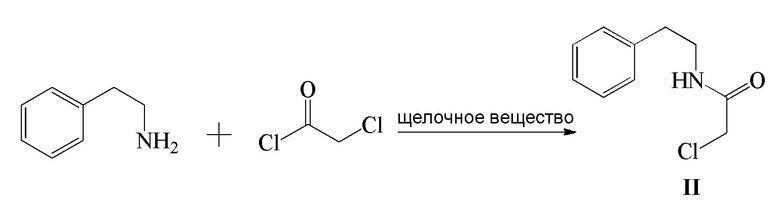
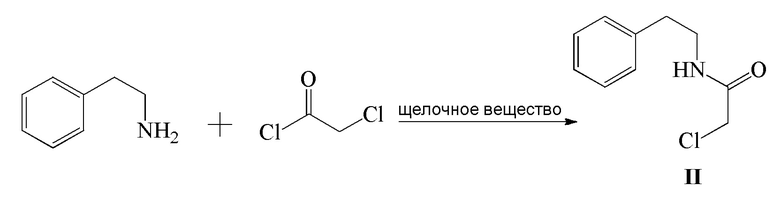
1) β-фенэтиламин и хлорацетилхлорид подвергают реакции конденсации в присутствии щелочного вещества с получением соединения формулы II;
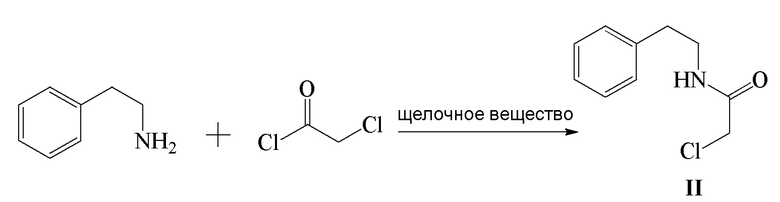
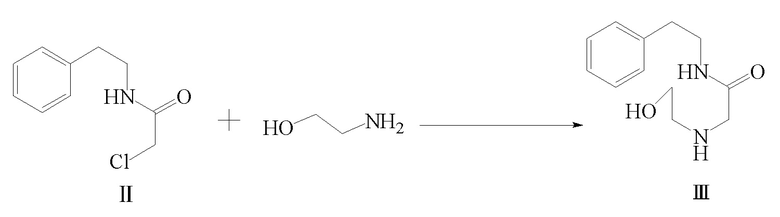
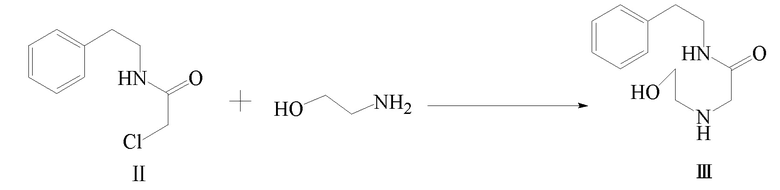
2) соединение формулы II и этаноламин подвергают реакции замещения с получением соединения формулы III;
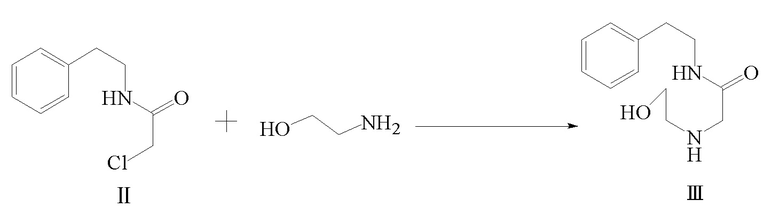
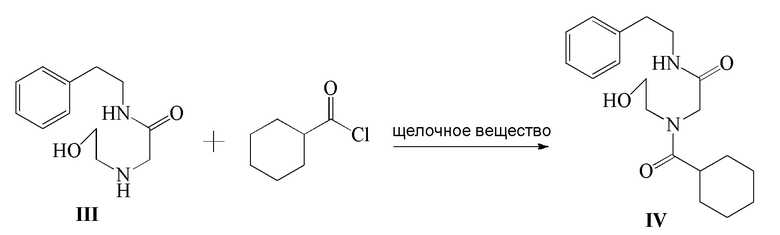
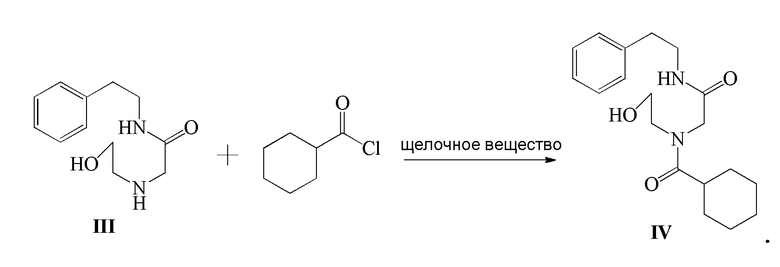
3) соединение формулы III и циклогексанкарбонилхлорид подвергают реакции ацилирования в присутствии щелочного вещества с получением соединения формулы IV;
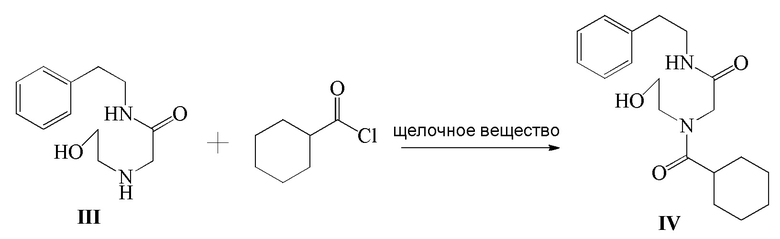
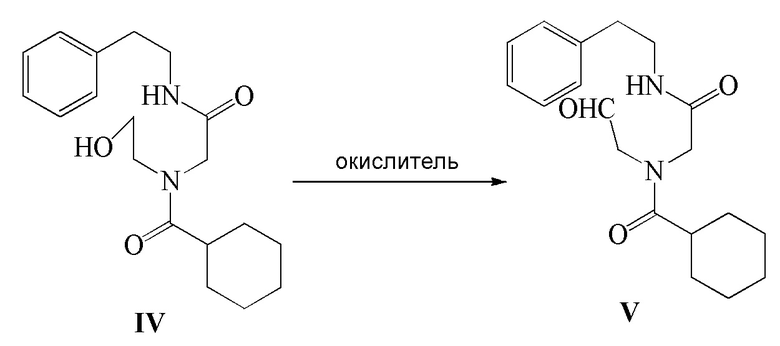
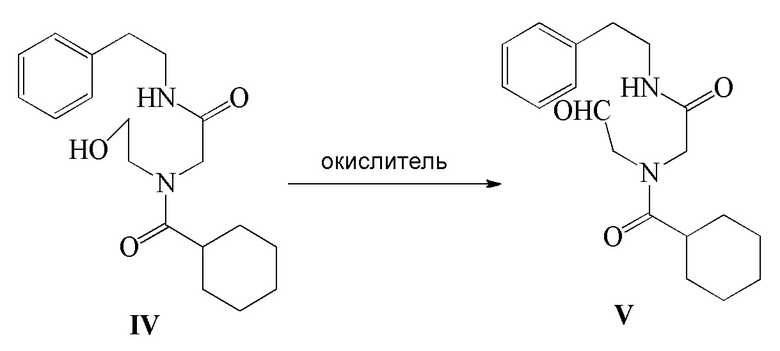
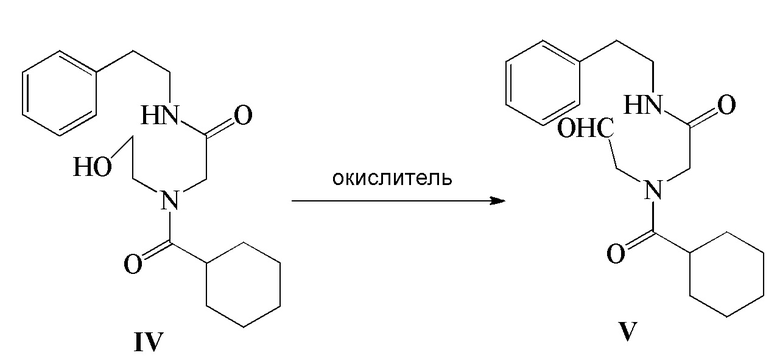
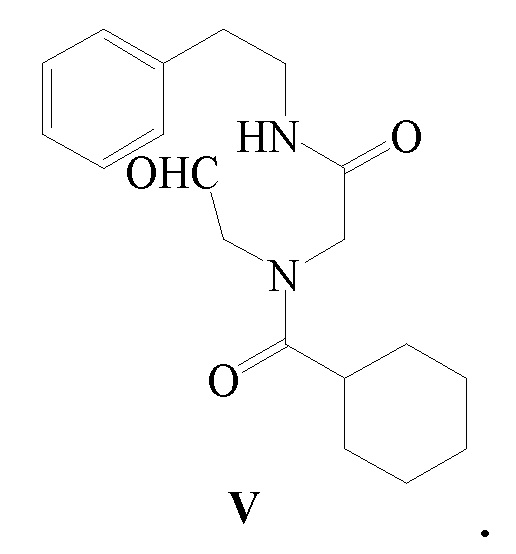
4) соединение формулы IV подвергают реакции окисления в присутствии окислителя с получением соединения формулы V; и
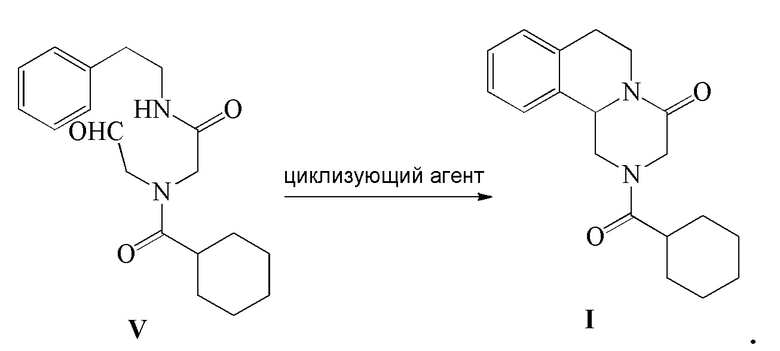
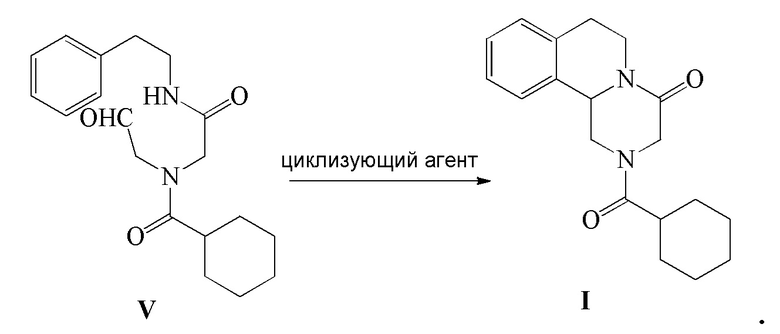
5) соединение формулы V подвергают реакции циклизации в присутствии циклизующего агента с получением Празиквантела в качестве соединения формулы I
В вышеуказанном способе стадию 1), стадию 2), стадию 3), стадию 4) или стадию 5) осуществляют без растворителя или осуществляют по меньшей мере с одним апротонным органическим растворителем в качестве реакционного растворителя. Апротонный органический растворитель представляет собой растворитель, выбранный из группы, состоящей из растворителя, выбранного из простых эфиров, растворителя, выбранного из ароматических углеводородов, растворителя, выбранного из углеводородов или галогенированных углеводородов, или растворителя, выбранного из сложных эфиров; где растворитель, выбранный из простых эфиров, выбран из группы, включающей тетрагидрофуран, диэтиловый эфир, 1,2-диметоксилэтан, метил трет-бутиловый эфир или 2-метилтетрагидрофуран, предпочтительно метил трет-бутиловый эфир; растворитель, выбранный из ароматических углеводородов, выбран из группы, включающей бензол, толуол, этилбензол или ксилол, предпочтительно толуол; растворитель, выбранный из углеводородов или галогенированных углеводородов, выбран из группы, включающей н-гексан, циклогексан, н-гептан, дихлорметан, трихлорметан или дихлорэтан, предпочтительно дихлорметан; растворитель, выбранный из сложных эфиров, выбран из группы, включающей метилформиат, этилформиат, метилацетат, этилацетат или изопропилацетат, предпочтительно этилацетат или изопропилацетат.
Предпочтительно в вышеуказанном способе стадию 2) осуществляют без растворителя. Предпочтительно стадию 1), стадию 3), стадию 4) или стадию 5) осуществляют с использованием по меньшей мере одного апротонного органического растворителя в качестве реакционного растворителя.
В вышеуказанном способе температура реакции стадии 1), стадии 2), стадии 3), стадии 4) или стадии 5) составляет от -10°C до 100°C, предпочтительно от 0°C до 40°C, более предпочтительно от 5°C до 15°C, наиболее предпочтительно от 10°C до 15°C.
Предпочтительно в вышеуказанном способе стадию 1), стадию 2), стадию 3), стадию 4) или стадию 5) осуществляют в ледяной бане, при комнатной температуре или температуре от 0°C до 40°C.
В вышеуказанном способе стадию 1), стадию 3) или стадию 5) предпочтительно осуществляют в ледяной бане, стадию 2) предпочтительно осуществляют при комнатной температуре, стадию 4) предпочтительно осуществляют при температуре от 0°C до 40°C, более предпочтительно от 5°C до 15°C, наиболее предпочтительно от 10°C до 15°C.
В вышеуказанном способе щелочное вещество на стадии 1), стадии 3) представляет собой одно или несколько щелочных веществ, выбранных из группы, включающей триэтиламин, имидазол, пиридин, 2-метилпиридин, 2,6-диметилпиридин, 4-диметиламинопиридин, диизопропиламин, диметилизопропиламин, диизопропилэтиламин, NaOH, Na2CO3, NaHCO3, KOH или K2CO3, предпочтительно щелочное вещество, выбранное из группы, включающей триэтиламин, NaOH, Na2CO3, NaHCO3, KOH или K2CO3.
В вышеуказанном способе молярное отношение соединения формулы II на стадии 2) к этаноламину составляет 1:2-1:15, предпочтительно 1:3-1:8.
В вышеуказанном способе окислитель на стадии 4) представляет собой по меньшей мере одну группу, выбранную из группы, включающей: NaClO/TEMPO/NaBr, Ca(ClO)2/TEMPO/NaBr, TCCA/TEMPO, NaNO2/FeCl3/TEMPO/воздух, NaNO2/FeCl3/TEMPO/O2 или DMSO/SO3-Py/Et3N. ʺNaClO/TEMPO/NaBrʺ относится к комбинации NaClO, TEMPO и NaBr, и другие выражения должны быть объяснены аналогичным образом. Компонент TEMPO в окислителе, используемом в реакции окисления, относится к TEMPO и его производному, например, одному или нескольким, выбранным из группы, включающей TEMPO, 4-OH-TEMPO, 4-(4-метилбензолсульфонилокси)-TEMPO, 4-ацетиламино-TEMPO, 4-бензоилокси-TEMPO, 4-NH2-TEMPO, 4-окси-TEMPO или 4-метансульфонилокси-TEMPO.
В вышеуказанном способе циклизующий агент на стадии 5) представляет собой один или несколько агентов, выбранных из группы, включающей: муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, пара-толуолсульфоновую кислоту, бензолсульфоновую кислоту, хлорную кислоту или концентрированную серную кислоту, предпочтительно концентрированную серную кислоту или метансульфоновую кислоту.
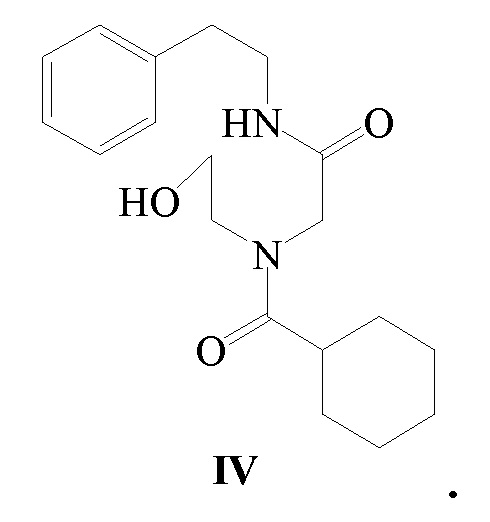
В другом аспекте, обеспечивается ключевое промежуточное соединение формулы IV для получения Празиквантела.
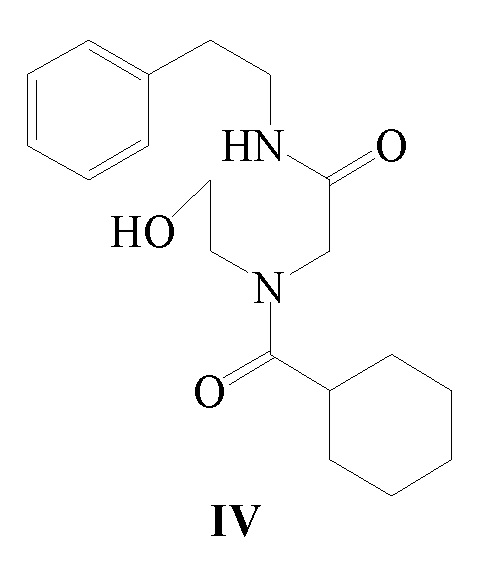
Соединение формулы IV можно получить следующим способом: используя β-фенэтиламин в качестве исходного вещества, соединение формулы IV получают при помощи реакций конденсации, замещения и ацилирования. В этом способе щелочное вещество, температура реакции, реакционный растворитель, молярные соотношения реагирующих веществ могут быть использованы в соответствии со способом получения Празиквантела, как указано выше, и не будут повторяться. Специалист в данной области техники сможет осуществить модификацию или усовершенствование способа получения согласно известному уровню техники или для получения ключевого промежуточного соединения, то есть соединения формулы IV, с использованием другого способа синтеза.
В другом аспекте, обеспечивается ключевое промежуточное соединение формулы V для получения Празиквантела:
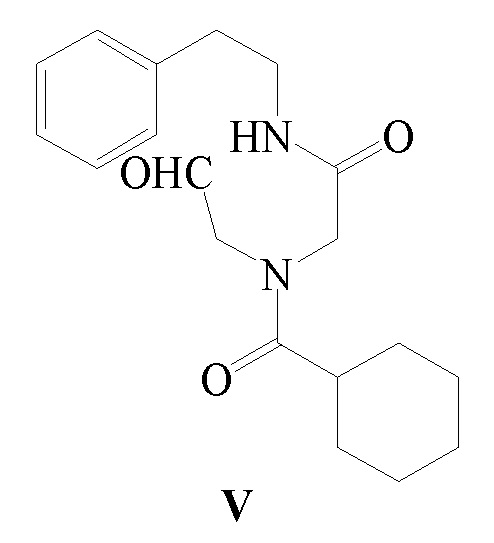
Соединение формулы V можно получить следующим способом: соединение формулы V получают путем реакции окисления вышеуказанного соединения формулы IV в присутствии окислителя. В этом способе окислитель, температура реакции, растворитель реакции или т.п. можно использовать в соответствии со способом получения Празиквантела, как описано выше. Получение соединения формулы IV можно осуществить в соответствии с представленным выше описанием, и оно не будет повторяться.
В следующем аспекте, обеспечивается применение соединения формулы IV для получения антипаразитарного средства Празиквантела.
В еще одном аспекте, обеспечивается применение соединения формулы V для получения антипаразитарного средства Празиквантела.
Способ получения Празиквантела в соответствии с изобретением разумно спланирован, экономичен, экологичен и требует дешевых и легко доступных исходных материалов. В частности, промежуточные соединения формулы IV и формулы V могут быть легко получены с высоким суммарным выходом (≥60%), целевой продукт Празиквантел имеет высокую степень чистоты (чистота в соответствии с ВЭЖХ ≥99,8%). Таким образом, этот способ подходит для промышленного производства в больших масштабах.
Подробное описание изобретения
Изобретение будет далее проиллюстрировано следующими Примерами, которые, однако, не следует понимать как какое-либо его ограничение. Специалист в данной области техники сможет осуществить модификацию или усовершенствование способа получения согласно известному уровню техники, которые охватываются объемом изобретения. Объем защиты и сущность изобретения определяются формулой изобретения и эквивалентными ей техническими решениями.
1H ЯМР регистрировали при помощи ядерного магнитно-резонансного спектрометра AM 400 с химическим сдвигом, показанным как δ (млн.д.).
Масс-спектр определяли при помощи Shimadzu ЖХМС-2010 ВЭЖХ-МС.
Термины, используемые в описании и формуле изобретения, представлены следующим образом. Если не указано иное, другие неопределенные термины имеют общепринятое в данной области техники значение.
TEMPO: 2,2,6,6-тетраметилпиперидин-1-оксил свободный радикал
TCCA: трихлоризоциануровая кислота
SO3-Py: триоксид серы - пиридин
Вышеуказанные реагенты приобретены у Sinopharm Chemical Reagent Co., Ltd.
Пример 1
Стадия 1): в реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), CH2Cl2 (150 мл) и NaOH (7,30 г, 182,50 ммоль), затем добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. Затем к реакционной жидкости добавляли 150 мл воды и смесь перемешивали и давали выстояться и органический слой отделяли. Органический слой промывали разбавленным водным раствором хлористоводородной кислоты (50 мл) и затем водой (100 мл ×2), сушили над безводным сульфатом магния, фильтровали и концентрировали с получением 24,73 г соединения формулы II в виде белого твердого вещества (выход: 98,7%).
1H ЯМР (CDCl3) δ: 2,86(т, 2H), 3,57(кв., 2H), 4,02(с, 2H), 6,59(с, 1H), 7,20-7,35(м, 5H).
МС (ESI) m/z: 198,07([M+1]+), 220,05([M+Na]+)
Стадия 2): В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы II (22,90 г, 115,85 ммоль) и этаноламин (42,60 г, 697,45 ммоль) и смесь перемешивали при комнатной температуре в течение 12 часов. Этаноламин отгоняли при помощи дистилляции при пониженном давлении с получением 23,67 г соединения формулы III в виде желтого масла (выход 91,9%).
1H ЯМР(CDCl3) δ: 2,37(с, 2H), 2,64(т, 2H), 2,83(т, 2H), 3,23(с, 2H), 3,44(с, 1H), 3,51-3,58(м, 4H), 7,20-7,32(м, 5H).
МС (ESI) m/z: 223,15([M+1]+), 245,13([M+Na]+)
Стадия 3): В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы III (18,50 г, 83,23 ммоль), CH2Cl2 (150 мл) и триэтиламин (12,64 г, 124,91 ммоль), к смеси добавляли по каплям циклогексанкарбонилхлорид (12,77 г, 87,10 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 2 часов. К реакционной жидкости добавляли разбавленный водный раствор хлористоводородной кислоты (150 мл), затем перемешивали и давали выстояться. Органический слой отделяли, промывали водой (100 мл ×2), сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученное твердое вещество суспендировали с метил трет-бутиловым эфиром с получением 25,62 г соединения формулы IV в виде белого твердого вещества (выход 92,6%).
1H ЯМР(CDCl3) δ: 1,20-1,79(м, 10H), 2,58(м, 1H), 2,81(т, 2H), 3,49-3,58 (м, 4H), 3,70(т, 2H), 3,83(с, 2H), 3,98(с, 1H), 6,48(с, 1H), 7,18-7,31(м, 5H).
МС (ESI) m/z: 333,23([M+1]+), 355,21([M+Na]+)
Стадия 4): В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы IV (10,20 г, 30,68 ммоль), CH2Cl2 (150 мл), водный раствор 15 масс.% NaBr (10,53 г, 15,35 ммоль) и TEMPO (0,05 г, 0,32 ммоль) и температуру в сосуде контролировали на уровне 5-10°C и затем добавляли по каплям водный раствор NaClO (180 г, 32,24 ммоль), pH которого доводили до 8-9 насыщенным водным раствором NaHCO3. Реакцию осуществляли в течение 20 часов. Водный слой отделяли и экстрагировали при помощи 30 мл CH2Cl2. Органический слой объединяли, промывали водным раствором тиосульфата натрия (100 мл×2), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 8,92 г соединения формулы V в виде светло-желтого твердого вещества (выход 88,0%).
1H ЯМР(CDCl3) δ: 1,21-1,76(м, 10H), 2,43(м, 1H), 2,88-2,99(м, 2H), 3,21-3,65(м, 2H), 3,88-3,99(м, 2H), 4,32-4,80(м, 3H), 7,19-7,31(м, 5H)
МС (ESI) m/z: 331,21([M+1]+), 353,19([M+Na]+)
Стадия 5): В реакционный сосуд объемом 100 мл добавляли концентрированную серную кислоту (15 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,60 г, 16,95 ммоль) в CH2Cl2 (15 мл) в ледяной бане. Реакцию осуществляли в течение 8 часов. Реакционную жидкость выливали в 150 мл ледяной воды и экстрагировали при помощи CH2Cl2 (50 мл ×2). Органический слой объединяли, промывали насыщенным водным раствором Na2CO3 (50 мл), промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола с получением 4,45 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,92%, выход 84,0%).
1HЯМР(CDCl3) δ: 1,46-1,79 (м, 10H), 2,45(м, 1H), 2,75-3,01(м, 4H), 4,06(д, 1H), 4,45(д, 1H), 4,78-4,80(м, 2H), 5,15(дд, 1H), 7,16-7,26(м, 4H)
МС (ESI) m/z: 313,21([M+1]+), 335,19([M+Na]+)
Пример 2
За исключением того, что соединение формулы II на стадии 1) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), CH2Cl2 (150 мл) и NaHCO3 (21,30 г, 253,54 ммоль), к ним добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. К реакционной жидкости добавляли 150 мл воды, раствор перемешивали и давали выстояться. Органический слой отделяли и органическую фазу промывали разбавленным водным раствором хлористоводородной кислоты (50 мл), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 24,85 г соединения формулы II в виде белого твердого вещества (выход 99,2%).
Пример 3
За исключением того, что соединение формулы II на стадии 1) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), CH2Cl2 (150 мл) и Na2CO3 (20,15 г, 190,11 ммоль), к ним добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. Затем к реакционной жидкости добавляли 150 мл воды, смесь перемешивали и давали выстояться. Органический слой отделяли и органическую фазу промывали разбавленным водным раствором хлористоводородной кислоты (50 мл), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 24,38 г соединения формулы II в виде белого твердого вещества (выход 97,3%).
Пример 4
За исключением того, что соединение формулы II на стадии 1) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), CH2Cl2 (150 мл) и K2CO3(26,27 г, 190,07 ммоль), к ним добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. Затем к реакционной жидкости добавляли 150 мл воды, перемешивали и давали выстояться. Органический слой отделяли и органическую фазу промывали разбавленным водным раствором хлористоводородной кислоты (50 мл), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 24,45 г соединения формулы II в виде белого твердого вещества (выход 97,6%).
Пример 5
За исключением того, что соединение формулы II на стадии 1) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), толуол (150 мл) и NaOH (7,30 г, 182,50 ммоль), затем добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. Затем к реакционной жидкости добавляли 150 мл воды, перемешивали и давали выстояться. Органический слой отделяли и органическую фазу промывали разбавленным водным раствором хлористоводородной кислоты (50 мл), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 24,43 г соединения формулы II в виде белого твердого вещества (выход 97,5%).
Пример 6
За исключением того, что соединение формулы II на стадии 1) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли β-фенэтиламин (15,36 г, 126,75 ммоль), метил трет-бутиловый эфир (150 мл) и NaOH (7,30 г, 182,50 ммоль), затем добавляли по каплям хлорацетилхлорид (15,0 г, 132,80 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 1 часа. Затем к реакционной жидкости добавляли 150 мл воды, перемешивали и давали выстояться. Органический слой отделяли и органическую фазу промывали разбавленным водным раствором хлористоводородной кислоты (50 мл), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 24,25 г соединения формулы II в виде белого твердого вещества (выход 96,8%).
Пример 7
За исключением того, что соединение формулы III на стадии 2) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы II (24,50 г, 123,95 ммоль) и этаноламин (30,28 г, 495,74 ммоль) и перемешивали при комнатной температуре в течение 12 часов. Этаноламин отгоняли при помощи дистилляции при пониженном давлении с получением 24,66 г соединения формулы III в виде желтого масла (выход 89,5%).
Пример 8
За исключением того, что соединение формулы III на стадии 2) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы II (12,70 г, 64,25 ммоль) и этаноламин (31,40 г, 514,08 ммоль) и перемешивали при комнатной температуре в течение 12 часов. Этаноламин отгоняли при помощи дистилляции при пониженном давлении с получением 13,25 г соединения формулы III в виде желтого масла (выход 92,8%).
Пример 9
За исключением того, что соединение формулы IV на стадии 3) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы III (28,60 г, 128,67 ммоль), CH2Cl2 (300 мл) и NaOH (7,72 г, 193,0 ммоль), затем добавляли по каплям циклогексанкарбонилхлорид (19,81 г, 135,12 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 2 часов. К реакционной жидкости добавляли 150 мл разбавленного водного раствора хлористоводородной кислоты, перемешивали и давали выстояться. Органический слой отделяли, промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали. Полученное твердое вещество суспендировали с метил трет-бутиловым эфиром с получением 40,42 г соединения формулы IV в виде белого твердого вещества (выход 94,5%).
Пример 10
За исключением того, что соединение формулы IV на стадии 3) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы III (7,50 г, 33,74 ммоль), CH2Cl2 (100 мл) и NaHCO3 (5,67 г, 67,49 ммоль), затем добавляли по каплям циклогексанкарбонилхлорид (5,20 г, 35,47 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 2 часов. К реакционной жидкости добавляли 50 мл разбавленного водного раствора хлористоводородной кислоты, перемешивали и давали выстояться. Органический слой отделяли, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали. Полученное твердое вещество суспендировали с метил трет-бутиловым эфиром с получением 10,04 г соединения формулы IV в виде белого твердого вещества (выход 89,5%).
Пример 11
За исключением того, что соединение формулы IV на стадии 3) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы III (7,50 г, 33,74 ммоль), толуол (250 мл) и NaOH (2,70 г, 67,49 ммоль), затем добавляли по каплям циклогексанкарбонилхлорид (5,20 г, 35,47 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 2 часов. К реакционной жидкости добавляли 50 мл разбавленного водного раствора хлористоводородной кислоты, перемешивали и давали выстояться. Органический слой отделяли, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали. Полученное твердое вещество суспендировали с метил трет-бутиловым эфиром с получением 9,60 г соединения формулы IV в виде белого твердого вещества (выход 85,6%).
Пример 12
За исключением того, что соединение формулы IV на стадии 3) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы III (7,50 г, 33,74 ммоль), изопропилацетат (150 мл) и NaOH (2,70 г, 67,49 ммоль), затем добавляли по каплям циклогексанкарбонилхлорид (5,20 г, 35,47 ммоль) в ледяной бане. После добавления реакцию осуществляли в течение 2 часов. К реакционной жидкости добавляли 50 мл разбавленного водного раствора хлористоводородной кислоты, перемешивали и давали выстояться. Органический слой отделяли, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали. Полученное твердое вещество суспендировали с метил трет-бутиловым эфиром с получением 9,28 г соединения формулы IV в виде белого твердого вещества (выход 82,7%).
Пример 13
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 250 мл последовательно добавляли полученное ранее соединение формулы IV (6,10 г, 18,35 ммоль), CH2Cl2 (100 мл) и TEMPO (0,03 г, 0,19 ммоль), затем добавляли TCCA (4,30 г, 18,50 ммоль), температуру в сосуде контролировали на уровне 5-10°C. Температуру повышали до комнатной температуры и реакцию осуществляли в течение 24 часов при перемешивании. Реакционную смесь фильтровали и фильтровальный слой промывали при помощи 30 мл CH2Cl2. Фильтрат промывали при помощи 200 мл насыщенного водного раствора Na2CO3, промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 5,16 г соединения формулы V в виде светло-желтого твердого вещества (выход 85,1%).
Пример 14
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 1 л последовательно добавляли полученное ранее соединение формулы IV (10,50 г, 31,58 ммоль), DMSO (60 мл) и Et3N (31,96 г, 315,84 ммоль) и температуру в сосуде контролировали на уровне 10-15°C, затем добавляли по каплям раствор SO3-Py (30,16 г, 189,50 ммоль) в DMSO (110 мл) и реакцию осуществляли в течение 10 часов. К реакционной жидкости добавляли 300 мл воды и экстрагировали при помощи CH2Cl2 (100 мл ×2). Органическую фазу объединяли, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 10,0 г соединения формулы V в виде желтого твердого вещества (выход 95,8%).
Пример 15
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы IV (10,20 г, 30,68 ммоль), CH2Cl2 (150 мл), водный раствор 15 масс.% NaBr (10,53 г, 15,35 ммоль) и TEMPO (0,05 г, 0,32 ммоль) и температуру в сосуде контролировали на уровне 5-10°C, затем к смеси добавляли по каплям водный раствор Ca(ClO)2 (4,61 г, 32,24 ммоль) и реакцию осуществляли в течение 20 часов. Водный слой отделяли и экстрагировали при помощи 30 мл CH2Cl2. Органический слой объединяли, промывали водным раствором тиосульфата натрия (100 мл ×2), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 7,91 г соединения формулы V в виде светло-желтого твердого вещества (выход 78,0%).
Пример 16
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы IV (10,20 г, 30,68 ммоль), изопропилацетат (250 мл), водный раствор 15 масс.% NaBr (10,53 г, 15,35 ммоль) и TEMPO (0,05 г, 0,32 ммоль) и температуру в сосуде контролировали на уровне 5-10°C, затем к смеси добавляли по каплям водный раствор NaClO (180 г, 32,24 ммоль), pH которого доводили до 8-9 насыщенным водным раствором NaHCO3, и реакцию осуществляли в течение 20 часов. Водный слой отделяли и экстрагировали при помощи 50 мл изопропилацетата. Органический слой объединяли, промывали водным раствором тиосульфата натрия (100 мл ×2), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 7,65 г соединения формулы V в виде светло-желтого твердого вещества (выход 75,5%).
Пример 17
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 500 мл последовательно добавляли полученное ранее соединение формулы IV (10,20 г, 30,68 ммоль), метил трет-бутиловый эфир (300 мл), 15 масс.% водный раствор NaBr (10,53 г, 15,35 ммоль) и TEMPO (0,05 г, 0,32 ммоль) и температуру в сосуде контролировали на уровне 5-10°C, затем к смеси добавляли по каплям водный раствор NaClO (180 г, 32,24 ммоль), pH которого доводили до 8-9 насыщенным водным раствором NaHCO3, и реакцию осуществляли в течение 20 часов. Водный слой отделяли и экстрагировали при помощи 50 мл метил трет-бутилового эфира. Органический слой объединяли, промывали водным раствором тиосульфата натрия (100 мл ×2), промывали водой (100 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 8,19 г соединения формулы V в виде светло-желтого твердого вещества (выход 80,8%).
Пример 18
За исключением того, что соединение формулы V на стадии 4) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реактор объемом 500 мл последовательно добавляли полученное ранее соединение формулы IV (10,20 г, 30,68 ммоль), CH2Cl2 (100 мл), NaNO2 (0,21 г, 3,04 ммоль), FeCl3 (0,50 г, 3,08 ммоль) и TEMPO (0,10 г, 0,64 ммоль) и реакцию осуществляли в течение 10 часов при комнатной температуре при давлении O2 0,3 MПa. Реакционную жидкость промывали водным раствором тиосульфата натрия (50 мл ×2), промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением 9,94 г соединения формулы V в виде светло-желтого твердого вещества (выход 98,0%).
Пример 19
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли метансульфоновую кислоту (20 мл), затем добавляли по каплям полученный ранее раствор соединения формулы V (5,40 г, 16,34 ммоль) в CH2Cl2 (10 мл) в ледяной бане. После добавления температуру повышали до комнатной температуры и реакцию осуществляли в течение 10 часов. Реакционную жидкость выливали в 100 мл ледяной воды и экстрагировали при помощи CH2Cl2 (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 4,27 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,83%, выход 83,6%).
Пример 20
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли трифторуксусную кислоту (15 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,40 г, 16,34 ммоль) в CH2Cl2 (10 мл) в ледяной бане. После добавления температуру повышали до комнатной температуры и реакцию осуществляли в течение 10 часов. Реакционную жидкость выливали в 100 мл ледяной воды и экстрагировали при помощи CH2Cl2 (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 3,92 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,93%, выход 76,8%).
Пример 21
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли трифторметансульфоновую кислоту (20 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,40 г, 16,34 ммоль) в CH2Cl2 (10 мл) в ледяной бане. После добавления температуру повышали до комнатной температуры и реакцию осуществляли в течение 10 часов. Реакционную жидкость выливали в 100 мл ледяной воды и экстрагировали при помощи CH2Cl2 (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 3,60 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,86%, выход 70,5%).
Пример 22
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли бензолсульфоновую кислоту (20 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,40 г, 16,34 ммоль) в CH2Cl2 (10 мл) в ледяной бане. После добавления температуру повышали до комнатной температуры и реакцию осуществляли в течение 10 часов. Реакционную жидкость выливали в 100 мл ледяной воды и экстрагировали при помощи CH2Cl2 (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 3,71 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,85%, выход 72,7%).
Пример 23
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли концентрированную серную кислоту (15 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,60 г, 16,95 ммоль) в толуоле (15 мл) в ледяной бане, и реакцию осуществляли в течение 8 часов. Реакционную жидкость выливали в 150 мл ледяной воды и экстрагировали толуолом (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 4,54 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,87%, выход 85,7%).
Пример 24
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли концентрированную серную кислоту (15 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,60 г, 16,95 ммоль) в метил трет-бутиловом эфире (15 мл) в ледяной бане, и реакцию осуществляли в течение 8 часов. Реакционную жидкость выливали в 150 мл ледяной воды и экстрагировали метил трет-бутиловым эфиром (50 мл ×2). Органическую фазу объединяли, промывали при помощи 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 4,30 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,85%, выход 81,3%).
Пример 25
За исключением того, что соединение формулы I Празиквантел на стадии 5) получали в соответствии со следующими процедурами, другие стадии были такими же, как в Примере 1.
В реакционный сосуд объемом 100 мл добавляли концентрированную серную кислоту (15 мл), к которой добавляли по каплям полученный ранее раствор соединения формулы V (5,60 г, 16,95 ммоль) в изопропилацетате (15 мл) в ледяной бане, и реакцию осуществляли в течение 8 часов. Реакционную жидкость выливали в 150 мл ледяной воды и экстрагировали изопропилацетатом (50 мл ×2). Органическую фазу объединяли, промывали 50 мл насыщенного водного раствора Na2CO3, промывали водой (50 мл ×2), сушили безводным сульфатом магния, фильтровали и концентрировали с получением светло-желтого твердого вещества, которое перекристаллизовывали из этанола, с получением 4,27 г соединения формулы I Празиквантела в виде белого твердого вещества (чистота в соответствии с ВЭЖХ 99,91%, выход 80,6%).
Хотя в настоящей заявке проиллюстрированы типичные варианты осуществления, изобретение не следует ограничивать подробным описанием, приведенным выше. Возможные изменения и замены могут быть осуществлены в изобретении без отступления от его сути. Соответственно, специалист в данной области техники может обдумать модификации и эквиваленты с использованием обычных экспериментов, и такие модификации и эквиваленты охватываются сутью и объемом изобретения, определенного прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| СПОСОБ ПОЛУЧЕНИЯ УЛИПРИСТАЛА АЦЕТАТА И ЕГО ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2012 |
|
RU2624007C2 |
| ПРОИЗВОДНЫЕ ДИЦИКЛОАЗААЛКАНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2008 |
|
RU2487866C2 |
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛАМИНА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695133C1 |
| СОЕДИНЕНИЯ (R)-5-КАРБАМОИЛ-8-ФТОР-3-N,N-ДИЗАМЕЩЕННЫЕ-АМИНО-3,4-ДИГИДРО-2H-1-БЕНЗОПИРАНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1994 |
|
RU2142951C1 |
| ИНГИБИТОРЫ Р38 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2357957C2 |
| 2β,3α,5α-ТРИГИДРОКСИ-АНДРОСТ-6-ОН, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2014 |
|
RU2629929C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АЛКИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2683022C1 |
Изобретение относится к способу получения Празиквантела. Способ включает следующие стадии: 1) β-фенэтиламин и хлорацетилхлорид подвергают реакции конденсации в присутствии щелочного вещества с получением соединения формулы II; 2) соединение формулы II и этаноламин подвергают реакции замещения с получением соединения формулы III; 3) соединение формулы III и циклогексанкарбонилхлорид подвергают реакции ацилирования в присутствии щелочного вещества с получением соединения формулы IV; 4) соединение формулы IV подвергают реакции окисления в присутствии окислителя с получением соединения формулы V; и 5) соединение формулы V подвергают реакции циклизации в присутствии циклизующего агента с получением Празиквантела в качестве соединения формулы I. Формулы I, II, III, IV и V приведены в формуле изобретения. Стадию 2) осуществляют без растворителя. Стадию 1), стадию 3), стадию 4) или стадию 5) осуществляют по меньшей мере с одним апротонным органическим растворителем в качестве растворителя. Температура реакции стадии 1), стадии 2), стадии 3), стадии 4) или стадии 5) составляет от -10°C до 100°C. Также предложены промежуточные соединения, способы их получения и применение. Изобретение предлагает простой, экономически эффективный и экологически безопасный способ получения Празиквантела с высокими выходом и чистотой. 7 н. и 13 з.п. ф-лы, 25 пр.
1. Способ получения Празиквантела, включающий следующие стадии:
1) β-фенэтиламин и хлорацетилхлорид подвергают реакции конденсации в присутствии щелочного вещества с получением соединения формулы II;
2) соединение формулы II и этаноламин подвергают реакции замещения с получением соединения формулы III;
3) соединение формулы III и циклогексанкарбонилхлорид подвергают реакции ацилирования в присутствии щелочного вещества с получением соединения формулы IV;
4) соединение формулы IV подвергают реакции окисления в присутствии окислителя с получением соединения формулы V; и
5) соединение формулы V подвергают реакции циклизации в присутствии циклизующего агента с получением Празиквантела в качестве соединения формулы I
 ,
,
причем стадию 2) осуществляют без растворителя и стадию 1), стадию 3), стадию 4) или стадию 5) осуществляют по меньшей мере с одним апротонным органическим растворителем в качестве растворителя; где температура реакции стадии 1), стадии 2), стадии 3), стадии 4) или стадии 5) составляет от -10°C до 100°C, предпочтительно от 0°C до 40°C, более предпочтительно от 5°C до 15°C, наиболее предпочтительно от 10°C до 15°C.
2. Способ по п. 1, отличающийся тем, что
апротонный органический растворитель представляет собой по меньшей мере один растворитель, выбранный из группы, состоящей из растворителя на основе простых эфиров, растворителя на основе ароматических углеводородов, растворителя на основе углеводородов или галогенированных углеводородов или растворителя на основе сложных эфиров.
3. Способ по п. 2, отличающийся тем, что
растворитель на основе простых эфиров выбран из группы, состоящей из тетрагидрофурана, диэтилового эфира, 1,2-диметоксилэтана, метил трет-бутилового эфира или 2-метилтетрагидрофурана, предпочтительно метил трет-бутилового эфира;
растворитель на основе ароматических углеводородов выбран из группы, состоящей из бензола, толуола, этилбензола или ксилола, предпочтительно толуола;
растворитель на основе углеводородов или галогенированных углеводородов выбран из группы, состоящей из н-гексана, циклогексана, н-гептана, дихлорметана, трихлорметана или дихлорэтана, предпочтительно дихлорметана;
растворитель на основе сложных эфиров выбран из группы, состоящей из метилформиата, этилформиата, метилацетата, этилацетата или изопропилацетата, предпочтительно этилацетата или изопропилацетата.
4. Способ по п. 1, отличающийся тем, что
стадию 1), стадию 2), стадию 3), стадию 4) или стадию 5) осуществляют в ледяной бане, при комнатной температуре или температуре от 0°C до 40°C.
5. Способ по п. 1, отличающийся тем, что
стадию 1), стадию 3) или стадию 5) осуществляют в ледяной бане, стадию 2) осуществляют при комнатной температуре, стадию 4) осуществляют при температуре от 0°C до 40°C, предпочтительно при температуре от 5°C до 15°C, более предпочтительно при температуре от 10°C до 15°C.
6. Способ по п. 1, отличающийся тем, что
щелочное вещество на стадии 1) и стадии 3) представляет собой одно или несколько веществ, каждое из которых независимо выбрано из группы, состоящей из триэтиламина, имидазола, пиридина, 2-метилпиридина, 2,6-диметилпиридина, 4-диметиламинопиридина, диизопропиламина, диметилизопропиламина, диизопропилэтиламина, NaOH, Na2CO3, NaHCO3, KOH или K2CO3, предпочтительно одно или несколько веществ, выбранных из группы, состоящей из триэтиламина, NaOH, Na2CO3, NaHCO3, KOH или K2CO3.
7. Способ по п. 1, отличающийся тем, что
молярное отношение соединения формулы II к этаноламину на стадии 2) составляет 1:2-1:15, предпочтительно 1:3-1:8.
8. Способ по п. 1, отличающийся тем, что
окислитель на стадии 4) представляет собой по меньшей мере одну группу, выбранную из группы, состоящей из: NaClO/TEMPO/NaBr, Ca(ClO)2/TEMPO/NaBr, TCCA/TEMPO, DMSO/SO3-Py/Et3N, NaNO2/FeCl3/TEMPO/воздух, NaNO2/FeCl3/TEMPO/O2.
9. Способ по п. 1, отличающийся тем, что
циклизующий агент на стадии 5) представляет собой один или несколько агентов, выбранных из группы, состоящей из муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, метансульфоновой кислоты, трифторметансульфоновой кислоты, пара-толуолсульфоновой кислоты, бензолсульфоновой кислоты, перхлорной кислоты или концентрированной серной кислоты, предпочтительно концентрированной серной кислоты или метансульфоновой кислоты.
10. Промежуточное соединение для получения Празиквантела, имеющее структуру формулы IV:
11. Способ получения промежуточного соединения по п. 10, включающий следующие стадии:
1) β-фенэтиламин и хлорацетилхлорид подвергают реакции конденсации в присутствии щелочного вещества с получением соединения формулы II;
2) соединение формулы II и этаноламин подвергают реакции замещения с получением соединения формулы III; и
3) соединение формулы III и циклогексанкарбонилхлорид подвергают реакции ацилирования в присутствии щелочного вещества с получением соединения формулы IV
 ,
,
причем стадию 1), стадию 2) или стадию 3) осуществляют без растворителя или осуществляют по меньшей мере с одним апротонным органическим растворителем в качестве реакционного растворителя, где температура реакции стадии 1), стадии 2) или стадии 3) составляет от -10°C до 100°C, предпочтительно от 0°C до 40°C, более предпочтительно от 5°C до 15°C, наиболее предпочтительно от 10°C до 15°C.
12. Способ получения по п. 11, отличающийся тем, что
щелочное вещество на стадии 1) и стадии 3) представляет собой одно или несколько веществ, каждое из которых независимо выбрано из группы, состоящей из триэтиламина, имидазола, пиридина, 2-метилпиридина, 2,6-диметилпиридина, 4-диметиламинопиридина, диизопропиламина, диметилизопропиламина, диизопропилэтиламина, NaOH, Na2CO3, NaHCO3, KOH или K2CO3, предпочтительно одно или несколько веществ, выбранных из группы, состоящей из триэтиламина, NaOH, Na2CO3, NaHCO3, KOH или K2CO3.
13. Способ получения по п. 11, отличающийся тем, что
молярное отношение соединения формулы II к этаноламину на стадии 2) составляет 1:2-1:15, предпочтительно 1:3-1:8.
14. Способ получения по п. 11, отличающийся тем, что
апротонный органический растворитель представляет собой по меньшей мере один растворитель, выбранный из группы, состоящей из растворителя на основе простых эфиров, растворителя на основе ароматических углеводородов, растворителя на основе углеводородов или галогенированных углеводородов или растворителя на основе сложных эфиров.
15. Способ получения по п. 14, отличающийся тем, что
растворитель на основе простых эфиров выбран из группы, состоящей из тетрагидрофурана, диэтилового эфира, 1,2-диметоксилэтана, метил трет-бутилового эфира или 2-метилтетрагидрофурана, предпочтительно метил трет-бутилового эфира;
растворитель на основе ароматических углеводородов выбран из группы, состоящей из бензола, толуола, этилбензола или ксилола, предпочтительно толуола;
растворитель на основе углеводородов или галогенированных углеводородов выбран из группы, состоящей из н-гексана, циклогексана, н-гептана, дихлорметана, трихлорметана или дихлорэтана, предпочтительно дихлорметана;
растворитель на основе сложных эфиров выбран из группы, состоящей из метилформиата, этилформиата, метилацетата, этилацетата или изопропилацетата, предпочтительно этилацетата или изопропилацетата.
16. Применение промежуточного соединения формулы IV по п. 10 в получении антипаразитарного средства Празиквантела.
17. Промежуточное соединение для получения Празиквантела, имеющее структуру формулы V:
18. Способ получения промежуточного соединения по п. 17, включающий реакцию окисления соединения формулы IV в присутствии окислителя с получением соединения формулы V
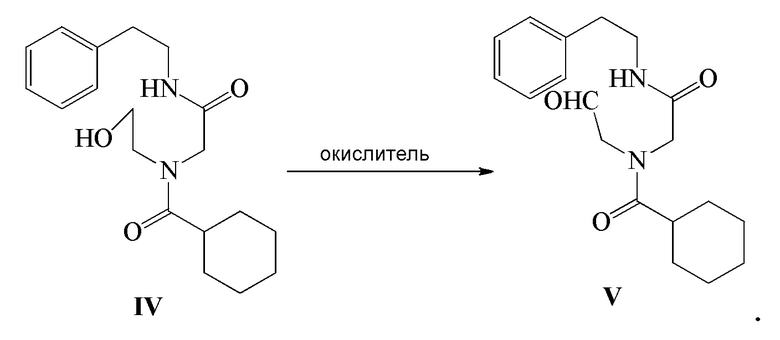 ,
,
причем реакцию осуществляют по меньшей мере с одним апротонным органическим растворителем в качестве реакционного растворителя, где температура реакции составляет от -10°C до 100°C, предпочтительно от 0°C до 40°C, более предпочтительно от 5°C до 15°C, наиболее предпочтительно от 10°C до 15°C.
19. Способ получения по п. 18, отличающийся тем, что
окислитель представляет собой по меньшей мере одну группу, выбранную из группы, состоящей из NaClO/TEMPO/NaBr, Ca(ClO)2/TEMPO/NaBr, TCCA/TEMPO и DMSO/SO3-Py/Et3N, NaNO2/FeCl3/TEMPO/воздух, NaNO2/FeCl3/TEMPO/O2.
20. Применение промежуточного соединения формулы V по п. 17 в получении антипаразитарного средства Празиквантела.
| KR 20020076486 A, 11.10.2002 | |||
| CN 103044422 A, 17.04.2013 | |||
| US 4362875 A1, 07.12.1982 | |||
| CN 1683346 A, 19.10.2005 | |||
| АМИДОАЦЕТОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2008 |
|
RU2425825C2 |

