Перекрестная ссылка на родственную заявку
[0001] Настоящая заявка испрашивает приоритет на основании заявки на патент Китая №201310104162.5, поданной 28 марта 2013 года, полное содержание которой включено в настоящую заявку посредством ссылки.
Область техники
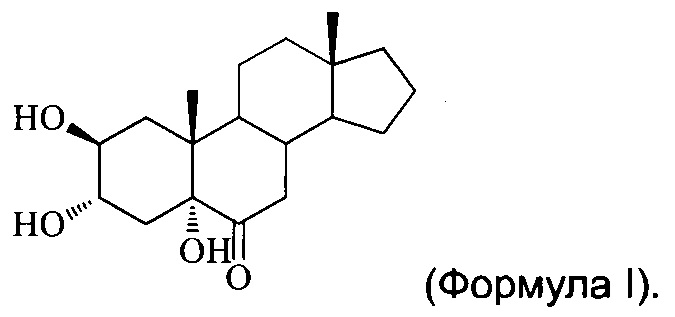
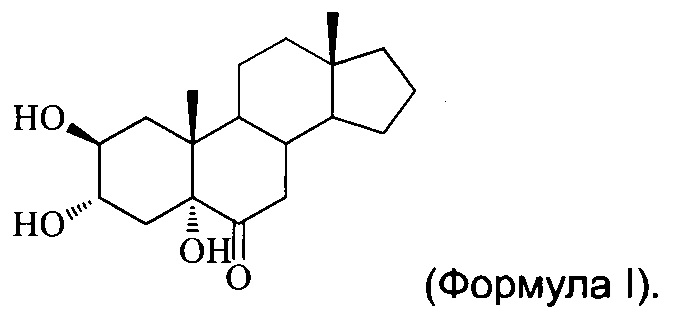
[0002] Настоящее изобретение относится к многоатомному стерону, в частности, 2β,3α,5α-тригидрокси-андрост-6-ону, и способам его получения и медицинским применениям.
Уровень техники
[0003] Многоатомные стероны представляют собой группу важных соединений широко распространенных в природе. Многие многоатомные стероны, выделенные из морских организмов и наземных растений, обладают важными физиологическими функциями, такими как противоопухолевые и повышающие иммунитет свойства. Например, экдистероны и брассиностероиды представляют собой соединения, стимулирующие рост растений.
[0004] Однако распространенные в природе многоатомные стероны содержатся в растениях в крайне малых количествах, в результате чего методики их очистки затруднены и занимают много времени. Кроме того, из-за сложности структуры, например, из-за относительно более длинных и сложных боковых цепей, большинство соединений этой группы невозможно синтезировать, что ограничивает их применение. Если указанные распространенные в природе соединения будут структурно оптимизированы таким образом, чтобы они по существу сохранили присущие им фармацевтические свойства, и в то же время упростить структуру для облегчения синтеза, то это будет иметь большое значение для широкого круга применений.
Краткое описание изобретения
[0005] Согласно настоящему изобретению предложен новый многоатомный стерон, а именно 2β,3α,5α-тригидрокси-андрост-6-он (далее упоминается как YC-10, соединение (I), соединение I, применяемые в данном документе взаимозаменяемо), имеющий структуру формулы (I):
[0006] Соединение формулы (I) было синтезировано авторами настоящего изобретения. Соединение имеет относительно простую структуру в сравнении со многими многоатомными стеронами, распространенными в природе. Например, в нем отсутствуют длинные или сложные боковые цепи, что упрощает синтез. Кроме того, уменьшенная молекулярная масса и относительно простая стереохимическая структура являются полезными для доставки лекарственных средств. Более того, удаление боковых цепей уменьшает возможность взаимодействия соединения с другими веществами. Кроме того, отсутствие боковой цепи в положении 17 в соединении (I) может увеличить in vivo биодоступность соединения и устранить его гормоноподобные действия. Уникальная пространственная конфигурация также может улучшить стереоселективность соединения, с достижением лучшей биологической активности.
[0007] Доказано, что соединение формулы (I) обладает определенными фармакологическими эффектами. Согласно одному аспекту, доказано, что указанное соединение имеет противоопухолевую активность. Согласно другому аспекту, доказано, что указанное соединение обладает нейронопротекторным эффектом, особенно в отношении ганглиозных клеток сетчатки.
[0008] Поэтому, согласно одному аспекту настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемые носители. "Терапевтически эффективное количество" означает количество соединения, которое, при введении объекту для лечения заболевания, является достаточным для осуществления такого лечения заболевания. "Терапевтически эффективное количество" может изменяться в зависимости от соединения, заболевания и его тяжести, а также от возраста, массы и т.д., субъекта, подлежащего лечению. «Фармацевтически приемлемые носители» относятся к разбавителю, вспомогательному веществу, наполнителю или носителю, с которым вводят соединение согласно настоящему изобретению.
[0009] Согласно другому аспекту настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) и второй нейронпротекторный агент. Второй нейронпротекторный агент отличается от соединений, представленных в настоящем изобретении для нейропротекторной цели, но может быть использован в комбинации с ними. В предпочтительных вариантах реализации второй нейронпротекторный агент представляет собой агент, защищающий ганглиозные клетки сетчатки.
[0010] Согласно дополнительному аспекту настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения, имеющего структуру формулы (I), и второе противоопухолевое лекарственное средство. Второе противоопухолевое лекарственное средство отличается от соединений, представленных в настоящем изобретении для противоопухолевых применений, но может быть использовано в комбинации с ними.
[0011] В настоящем документе «опухоль» означает злокачественное или доброкачественное клеточное новообразование кожи или органов тела, например, но не ограничиваясь следующими, молочной железы, простаты, легких, почек, поджелудочной железы, желудка или кишечника. Злокачественные опухоли склонны инвазировать в соседние ткани и диффундировать (метастазировать) в отдаленные органы, такие как кости, печень, легкие или мозг. Термин «опухоль», используемый в настоящем документе, включает тип метастатических опухолевых клеток, например, но не ограничивается следующими, меланому, лимфому, лейкемию, фибросаркому, лейомиосаркому, опухоли тучных клеток и ткани типа карциномы, например, но не ограничиваясь ими, колоректального рака, рака предстательной железы, мелкоклеточного рака легкого и немелкоклеточного рака легкого, рака молочной железы, рака поджелудочной железы, рака мочевого пузыря, рака почки, рака желудка, глиобластомы, первичного рака печени, рака яичников, рака предстательной железы и лейомиосаркома матки.
[0012] Согласно другому аспекту настоящее изобретение обеспечивает применение соединения, имеющего структуру формулы (I), для приготовления нейропротекторных лекарственных средств или противоопухолевых лекарственных средств. Соединения, предлагаемые в настоящем изобретении, как было показано, ингибируют рост опухолевых клеток в зависимости от дозы, вместе со значительным нейропротекторным эффектом.
[0013] Согласно другому дополнительному аспекту настоящее изобретение обеспечивает способ лечения или облегчения заболевания или состояния, таких как заболевания или состояния, связанные с травмой нервных волокон сетчатки или повреждения нейронов центральной нервной системы, вызванных множеством факторов, в том числе офтальмологическими заболеваниями, такими как ишемия сетчатки, травма и повреждение зрительного нерва в результате острой или хронической глаукомы, гипертоническая ретинопатия, диабетическое поражение сетчатки, дегенерация пигмента сетчатки и макулопатия и заболевания центральной нервной системы, такие как инсульт, черепно-мозговая травма, повреждения спинного мозга, болезнь Паркинсона (PD), болезнь Альцгеймера (AD), болезнь Хантингтона (HD), и боковой амиотрофический склероз (БАС). Указанный способ включает введение субъекту терапевтически эффективного количества соединения формулы (I), его пролекарства или сольвата, или фармацевтических композиций, предложенных согласно настоящему изобретению.
[0014] При использовании в настоящем документе «пролекарство» относится к соединениям, в том числе к производным соединений согласно настоящему изобретению, которые имеют расщепляемые группы и в результате сольволиза или в физиологических условиях становятся соединениями согласно настоящему изобретению, которые являются фармацевтически активными in vivo. Такие примеры включают, но не ограничиваются ими, производные сложного эфира холина и т.п., сложные эфиры N-алкилморфолина и т.п.
[0015] Термин «сольват» относится к формам соединениям, которые ассоциированы с растворителем, обычно с помощью реакции сольволиза. Данная физическая ассоциация включает в себя образование водородных связей. Традиционные растворители включают воду, этанол, уксусную кислоту и т.п. Соединения согласно настоящему изобретению могут быть получены, например, в кристаллической форме и могут быть сольватированы или гидратированы. Подходящие сольваты включают фармацевтически приемлемые сольваты, такие как гидраты, и дополнительно включают как стехиометрические сольваты, так и нестехиометрические сольваты.
[0016] В некоторых случаях сольват может быть выделен, например, когда молекулы одного или более растворителей включены в кристаллическую решетку кристаллического твердого вещества. «Сольват» охватывает как жидкофазные, так и поддающиеся выделению сольваты. Типичные представители сольватов включают гидраты, этаноляты и метаноляты.
[0017] Согласно дополнительному аспекту, предложен способ получения соединения формулы (I). Способ использует андрост-5-ен-3-ол в качестве исходного вещества для получения соединения VI, т.е. 3β-п-толуолсульфонилокси-5α-гидрокси-андрост-6-она; Соединение VI затем подвергают реакции элиминирования с получением соединения IX, т.е. 5α-гидрокси-андрост-2-ен-6-она; Соединение IX затем подвергают окислению по двойной связи в 2-положении и гидролизу с получением соединения I.
[0018] Соединение VI может быть получено с помощью множества способов, примеры которых приведены ниже.
[0019] (1) Исходное вещество: андрост-5-ен-3-ол подвергают окислению смесью Н2О2/муравьиная кислота, щелочному гидролизу, окислению N-бромсукцинимидом и защите п-толуолсульфонилхлоридом.
[0020] В частности, способ включает следующие стадии.
[0021] (1а) В реакционную колбу вносят андрост-5-ен-3-ол и муравьиную кислоту, а затем при низкой температуре добавляют по каплям Н2О2. Реакционную смесь оставляют реагировать в течение 1-2 часов и затем нагревают. К реакционной смеси добавляют воду и перемешивают для диспергирования. Смесь фильтруют и сушат с получением соединения II в виде твердого вещества белого цвета. Исходное вещество : муравьиная кислота : Н2О2 соотносятся как 1:10~30:0,5~3 (масс. : об. : об.);
[0022] (1b) В реакционную колбу добавляют щелочной раствор метанола и соединение II. Реакционную смесь кипятят с обратным холодильником 1-2 ч, и выливают в воду для диспергирования. Смесь фильтруют и сушат с получением соединения III в виде твердого вещества белого цвета. Щелочной раствор метанола выбран из раствора гидроксида калия, гидроксида натрия или метилята натрия в метаноле. Концентрация щелочи в реакционной смеси составляет 2-10%;
[0023] (1с) В реакционную колбу добавляют соединение III, диоксан и воду. Периодически добавляют N-Бромсукцинимид. Смесь оставляют реагировать в течение 2-4 ч, а затем добавляют сульфит натрия. Смесь фильтруют, промывают водой до нейтральной реакции и сушат, получая соединение V в виде твердого вещества белого цвета; и
[0024] (1d) В реакционную колбу добавляют соединение V, пиридин и п-толуолсульфонилхлорид. Реакционную смесь перемешивают в течение 24-36 ч при комнатной температуре и затем выливают в ледяной раствор соляной кислоты. Смесь фильтруют, промывают водой до нейтральной реакции и сушат, получая соединение VI в виде твердого вещества белого цвета.
[0025] (2) Исходное соединение: Андрост-5-ен-3-ол подвергают окислению в присутствии м-хлорпероксибензойной кислоты, ацидолизу, окислению N-бромсукцинимидом и защите п-толуолсульфонилхлоридом.
[0026] В частности, способ синтеза включает следующие стадии.
[0027] (2а) В реакционную колбу добавляют андрост-5-ен-3-ол и CH2Cl2. При перемешивании порциями добавляют м-Хлорпероксибензойную кислоту. Далее смесь перемешивают в течение 2-5 ч в бане со льдом. После окончания реакции смесь промывают Na2CO3, Na2SO3 и водой, высушивают и концентрируют с получением соединения IV;
[0028] (2b) В реакционную колбу добавляют соединение IV и кислый водный раствор ацетона и перемешивают при комнатной температуре в течение нескольких часов. После окончания реакции реакционный раствор доводят до нейтральной среды раствором Na2CO3. Ацетон удаляют и остаток экстрагируют этилацетатом. Органический слой собирают, сушат и концентрируют с получением соединения III. Кислота в кислом водном растворе ацетона представляет собой серную или йодную кислоту. Соединение IV : ацетон : 1 н. кислота соотносятся как 1:20~30:5~10 (масс. : об. : об.);
[0029] (2с) В реакционную колбу добавляют соединение III, диоксан и воду. Периодически добавляют N-Бромсукцинимид. Смесь подвергают взаимодействию в течение 2-4 ч с последующим добавлением сульфита натрия. Смесь фильтруют, промывают водой до нейтральной реакции и сушат, получая соединение V в виде твердого вещества белого цвета; и
[0030] (2d) В реакционную колбу добавляют соединение V, пиридин и п-толуолсульфонилхлорид. Реакционную смесь перемешивают в течение 24-36 ч при комнатной температуре и затем выливают в ледяной раствор соляной кислоты. Смесь фильтруют, промывают водой до нейтральной реакции и сушат, получая соединение VI в виде твердого вещества белого цвета.
[0031] (3) Исходное вещество: Андрост-5-ен-3-ол подвергают введению защиты п-толуолсульфонилхлоридом, окислению в присутствии м-хлорпероксибензойной кислоты, и окислению реагентом Джонса.
[0032] В частности, способ включает следующие этапы.
[0033] (3а) В реакционную колбу добавляют андрост-5-ен-3-ол, безводный пиридин и п-толуолсульфонилхлорид. Реакционную смесь перемешивают при комнатной температуре. После окончания реакции смесь выливают в ледяной раствор соляной кислоты, перемешивают, фильтруют, промывают водой до нейтральной реакции и сушат с получением соединения VII в виде твердого вещества белого цвета;
[0034] (3b) В реакционную колбу добавляют соединение VII и дихлорметан, порциями при перемешивании добавляют м-хлорпероксибензойную кислоту. Далее реакционную смесь перемешивают в течение 2-5 ч в бане со льдом. После окончания реакции, смесь промывают насыщенным раствором сульфита натрия, раствором карбоната натрия и дистиллированной водой. Органический слой собирают, сушат, концентрируют и очищают с помощью колоночной хроматографии на силикагеле, обеспечивая соединение VIII в виде твердого вещества белого цвета;
[0035] (3с) В реакционную колбу добавляют соединение VIII и ацетон, с последующим добавлением при перемешивании реагента Джонса. Реакционную смесь оставляют реагировать в течение нескольких часов при комнатной температуре. После окончания реакции, смесь гасят изопропанолом и доводят до нейтральной реакции среды. Смесь концентрируют при пониженном давлении для удаления ацетона, экстрагируют этилацетатом, промывают, сушат и концентрируют с получением твердого вещества бледно-зеленого цвета. Твердое вещество очищают с помощью колоночной хроматографии на силикагеле для обеспечения соединения VI в виде твердого вещества белого цвета.
[0036] В способах согласно настоящему изобретению, соединение IX получают, например, следующим образом. В реакционную колбу добавляют соединение VI, диметилформамид (DMF), Li2CO3 и LiBr. Реакционную смесь кипятят с обратным холодильником и выливают в ледяной водный раствор соляной кислоты. Смесь перемешивают, фильтруют, промывают водой до нейтральной реакции среды и сушат, получая соединение IX в виде белого твердого вещества. Предпочтительно соединение VI : DMF соотносятся как 1:3~15(масс. : об.); соединение VI : Li2CO3 : LiBr соотносятся как 1:4~12:4~12 (М : М : М).
[0037] Согласно способам настоящего изобретения, соединение I можно также получить из соединения IX способами, примеры которых приведены далее.
[0038] (1) Соединение I получают из соединения IX в результате окисления смесью Н2О2/муравьиная кислота и щелочного гидролиза.
[0039] В частности, способ синтеза включает следующие стадии.
[0040] (1а) В реакционную колбу вносят соединение IX и муравьиную кислоту, а затем при низкой температуре добавляют по каплям Н2О2. Реакционную смесь оставляют реагировать в течение 1-2 часов и затем нагревают. К реакционной смеси добавляют воду и перемешивают для диспергирования. Полученную смесь фильтруют с получением белого осадка фильтрования. Осадок сушат с получением соединения X в виде белого твердого вещества. Соединение Х : муравьиная кислота : H2O2 соотносятся как 1:10~30:0,5~3 (масс. : об.: об.);
[0041] (1b) В реакционную колбу добавляют щелочной раствор метанола и соединение X. Затем реакционную смесь кипятят с обратным холодильником в течение 1-2 ч, и выливают в воду для диспергирования. Смесь фильтруют и сушат с получением соединения формулы (I) в виде твердого вещества белого цвета. Раствор щелочи в метаноле выбран из раствора гидроксида калия, гидроксида натрия или метилята натрия в метаноле. Концентрация щелочи в реакционной смеси составляет 2-10%.
[0042] (2) Соединение I получают из соединения IX в результате окисления м-хлорпероксибензойной кислотой и ацидолиза. В частности, способ синтеза включает следующие стадии.
[0043] (2а) В реакционную колбу добавляют соединение IX и CH2Cl2. м-Хлорпероксибензойную кислоту добавляют порциями при перемешивании. Далее смесь перемешивают в течение 2-5 ч в бане со льдом. После окончания реакции смесь промывают Na2CO3, Na2SO3 и водой, высушивают и концентрируют с получением соединения XI;
[0044] (2b) В реакционную колбу добавляют соединение XI и кислый водный раствор ацетона и перемешивают при комнатной температуре в течение нескольких часов. После окончания реакции реакционный раствор доводят до нейтральной реакции среды раствором Na2CO3. Ацетон удаляют, и остаток экстрагируют этилацетатом. Органический слой собирают, сушат и концентрируют с получением соединения I. Кислота в кислом водном растворе ацетона представляет собой серную или йодную кислоту. Соединение XI : ацетон : 1 н. кислота соотносятся как 1:20~30:5~10(масс. : об. : об.).
Краткое описание чертежей
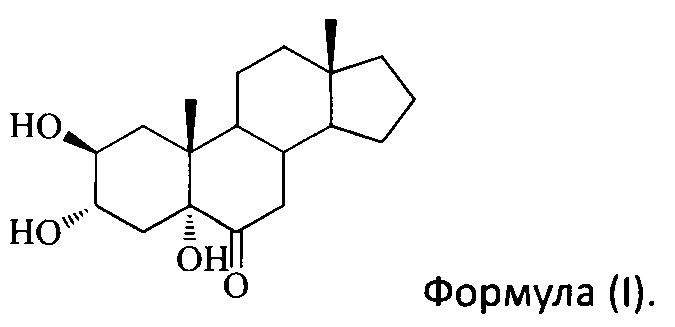
[0045] На Фигуре 1 представлено ингибирование клеток LN18 и DBTRG-50MG соединением I согласно настоящему изобретению (n=3*, р<0,05).
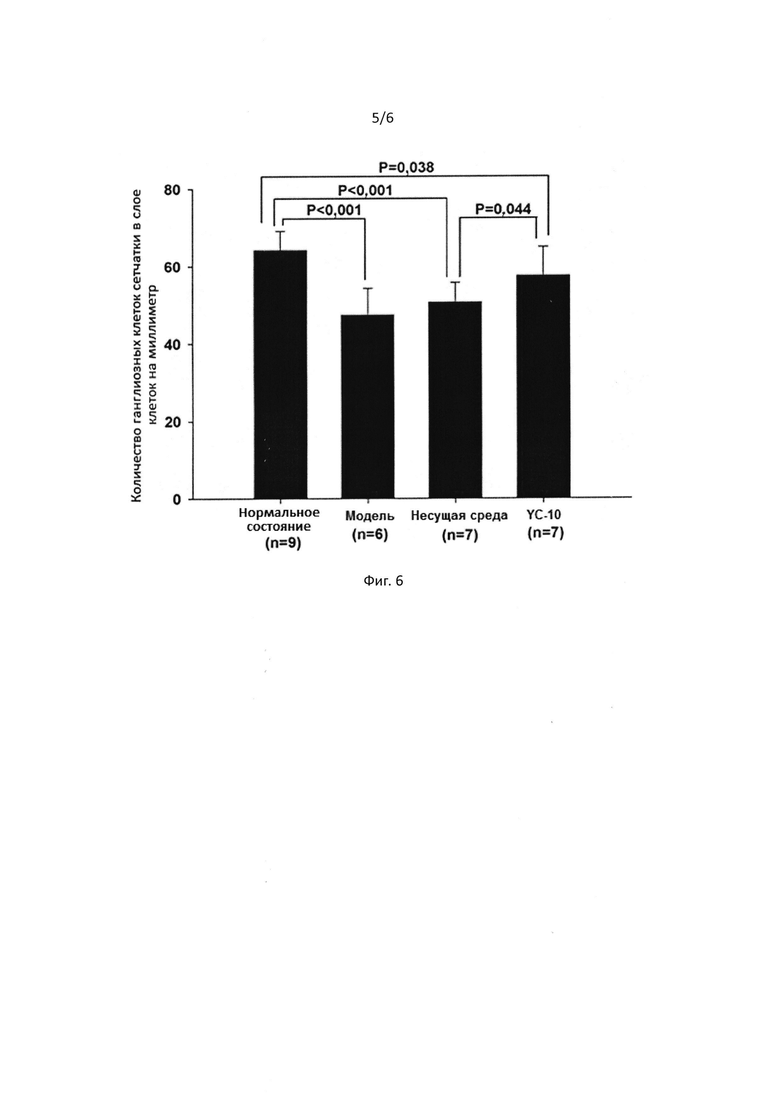
[0046] На Фигуре 2 представлена защита соединением I гранулярных нейронов мозжечка от глутамат-индуцированных повреждений.
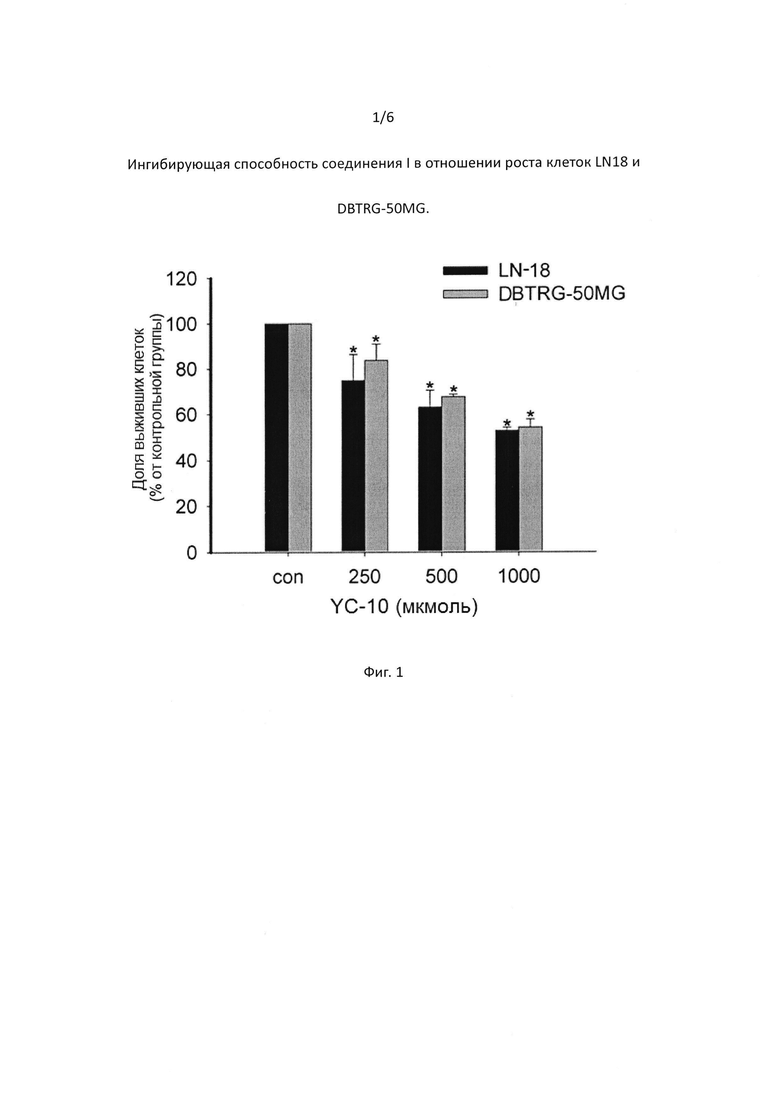
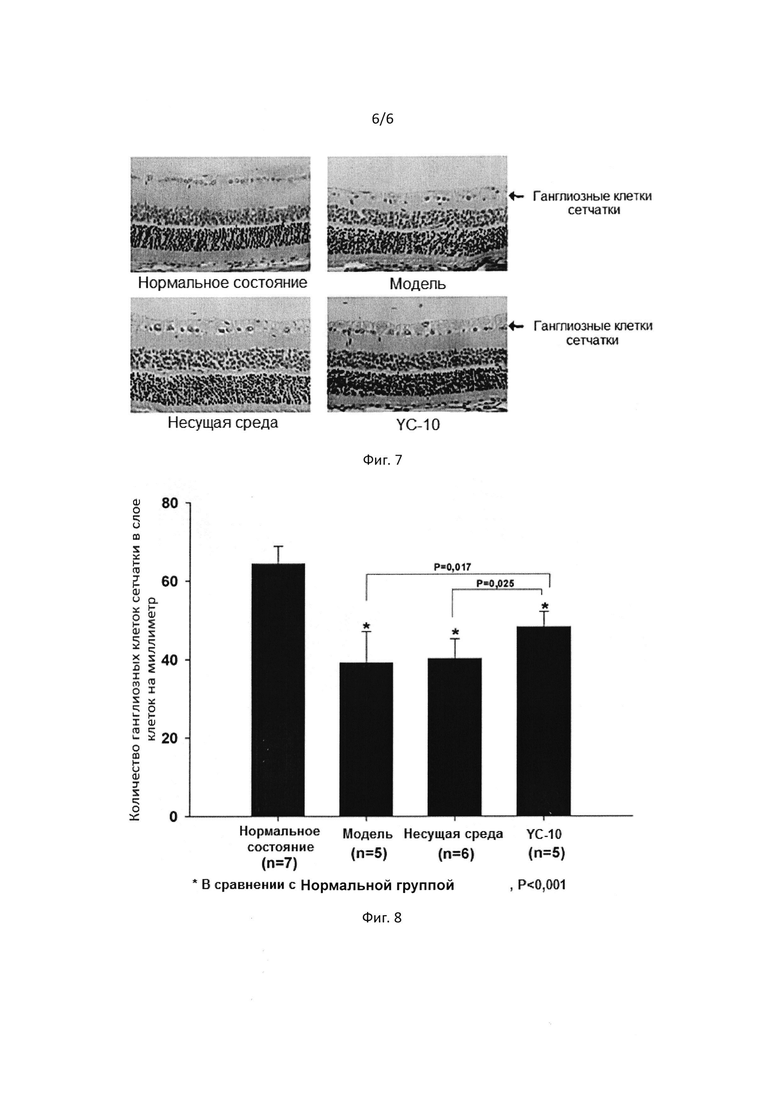
[0047] На Фигуре 3 показано, что соединение I дозозависимым образом увеличивает выживаемость гранулярных нейронов мозжечка.
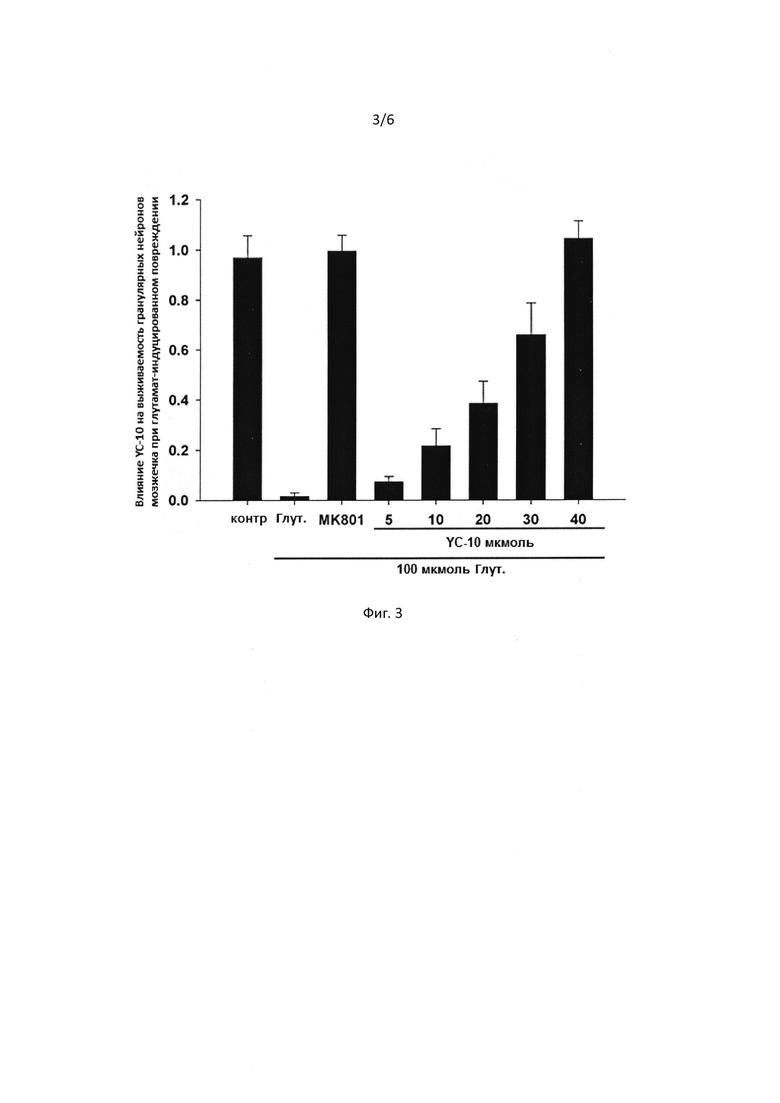
[0048] На Фигуре 4 представлена защита соединением I гранулярных нейронов мозжечка от гибели вызванной низким содержанием калия.
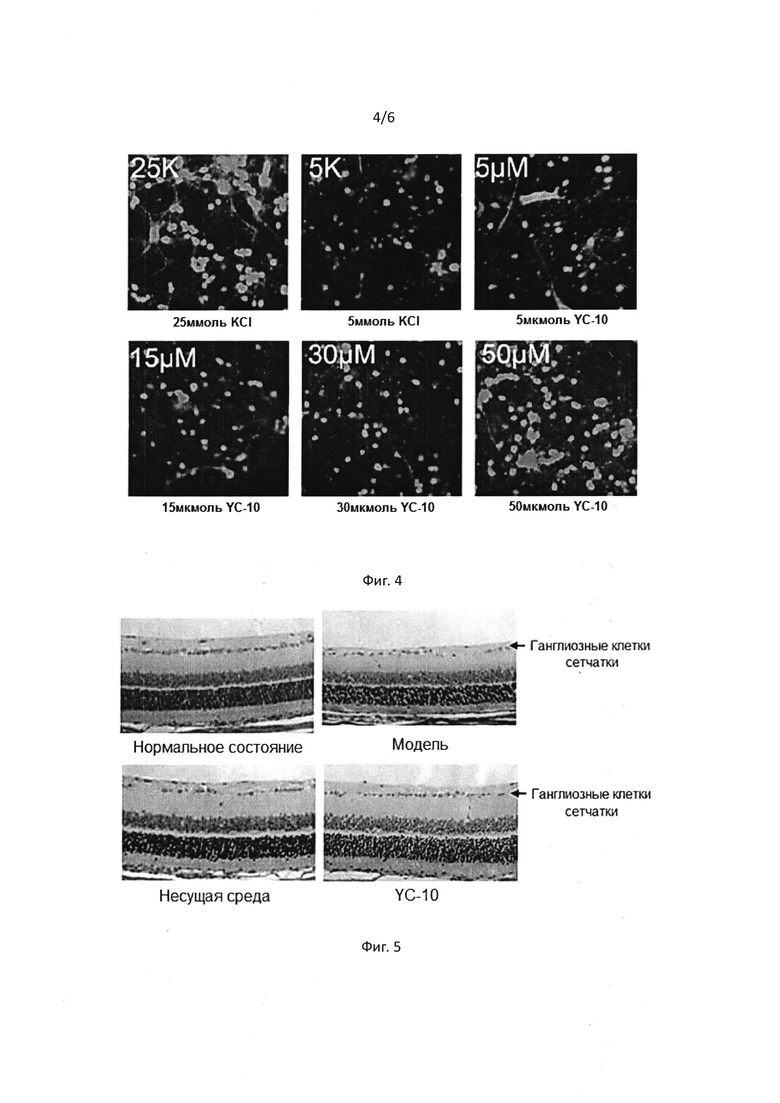
[0049] На Фигуре 5 показано, что количество ганглиозных клеток сетчатки значительно снизилось в модели пережатия оптического нерва, в то же время показано, что соединение I предотвращает снижение количества ганглиозных клеток сетчатки.
[0050] На Фигуре 6 представлен график, демонстрирующий статистику содержания ганглиозных клеток сетчатки в различных группах образцов модели пережатия оптического нерва.
[0051] На Фигуре 7 показано, что количество ганглиозных клеток сетчатки значительно уменьшается в модели высокого глазного давления и ишемии, в то время как показано, что соединение I предотвращает снижение количества ганглиозных клеток сетчатки.
[0052] На Фигуре 8 представлен график, демонстрирующий статистику содержания ганглиозных клеток сетчатки в различных группах образцов модели высокого глазного давления и ишемии.
Подробное описание изобретения
[0053] Нижеследующее приведено только для наглядности. При этом объем изобретения не должен быть ограничен примерами, приведенными ниже. В следующих примерах, соединение I относится к 2β,3α,5α-андрост-тригидрокси-6-ону; соединение II относится к 3β,6β-диформилокси-5-α-андрост-5-олу; соединение III относится к андрост-3β,5α,6β-триолу; соединение IV относится к 3β-гидрокси-андрост-5α,6β-эпоксиду; соединение V относится к 3β,5α-дигидрокси-андрост-6-ону; соединение VI относится к 3β-п-толуолсульфонилокси-5α-гидрокси-андрост-6-ону; соединение VII относится к 3β-п-толуолсульфонилокси-андрост-5-ену; соединение VIII относится к 3β-п-толуолсульфонилокси-андрост-5β,6β-эпокси; соединение IX относится к 5α-гидрокси-андрост-2-ен-6-ону; соединение X относится к 2β,3α-5α-diformyloxy-гидрокси-андрост-6-ону; соединение XI относится к 2β,3β-эпокси-5α-гидрокси-андрост-6-ону.
Получение соединения I - Пример 1
[0054] Стадия 1 - В двухлитровую реакционную колбу добавляли соединение андрост-5-ен-3-ол (54,5 г) и муравьиную кислоту (1 л, 88%). Реакционную смесь охлаждали до 25°С и медленно добавляли пероксид водорода (82,5 мл, 30%). После окончания реакции, которое отслеживали с помощью тонкослойной хроматографии (ТСХ), реакционную смесь нагревали до 75°С, чтобы удалить избыток пероксида водорода. Добавляли воду (1 л) и перемешивали для диспергирования. Полученную смесь фильтровали с получением осадка фильтрования белого цвета. Осадок погружали в насыщенный раствор NaHCO3 и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения II (62,4 г) в виде твердого вещества белого цвета.
[0055] Стадия 2 - В двухлитровую реакционную колбу добавляли гидроксид калия (45 г), метанол (1500 мл), воду (300 мл) и соединение II (60 г). Реакционную смесь кипятили с обратным холодильником. Отсутствие остатков соединения II подтверждали с помощью ТСХ. Реакционную смесь охлаждали до комнатной температуры, и выливали в воду (3 л) для диспергирования. Смесь доводили концентрированной соляной кислотой до рН=7 и оставляли до выпадения осадка. Полученную смесь фильтровали и осадок на фильтре промывали до нейтральной реакции и сушили с получением соединения III (49,6 г) в виде твердого вещества белого цвета.
[0056] Стадия 3 - В литровую реакционную колбу добавляли соединение III (49 г), диоксан (600 мл) и воду (200 мл). После полного растворения соединения III, добавляли в четыре приема N-бромсукцинимид (42,5 г). После исчерпания соединения III, о чем свидетельствовала ТСХ, реакцию останавливали. Добавляли сульфит натрия (11 г) для удаления избытка окислителя. Полученную смесь растворяли в воде (4 л) и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения V (47,8 г) в виде твердого вещества белого цвета.
[0057] Стадия 4 - В реакционную колбу объемом 500 мл добавляли пиридин (135 мл), соединение V (44,3 г) и п-толуолсульфонилхлорид (45 г). Полученную смесь перемешивали при комнатной температуре. После исчерпания соединения V, о чем свидетельствовала ТСХ, реакцию останавливали. Смесь выливали в ледяной водный раствор соляной кислоты (300 мл, 1:1 (по объему)), перемешивали и фильтровали. Осадок на фильтре промывали до рН=7 и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения VI (61,7 г) в виде твердого вещества белого цвета.
[0058] Стадия 5 - В литровую реакционную колбу добавляли N,N-диметилформамид (325 мл), соединение VI (54 г), Li2CO3 (52,1 г) и LiBr (60,5 г). Полученную смесь кипятили с обратным холодильником. После исчерпания соединения VI, о чем свидетельствовала ТСХ, реакцию останавливали. Смесь выливали в ледяной водный раствор соляной кислоты (2 л, 1:1 (об. : об.)), перемешивали и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения IX (32 г) в виде твердого вещества белого цвета.
[0059] Стадия 6 - В реакционную колбу объемом 500 мл добавляли соединение IX (30 г) и муравьиную кислоту (600 мл, 88%). Полученную смесь нагревали до растворения твердого вещества и затем охлаждали до 25°С. Медленно добавляли пероксид водорода (24 мл, 30%). После исчерпания соединения IX, что отслеживали с помощью ТСХ, реакционную смесь нагревали до 75°С и выдерживали так в течение 10 минут, чтобы удалить избыток пероксида водорода. Добавляли воду (3 л) и перемешивали для диспергирования. Полученную смесь фильтровали с получением осадка фильтрования белого цвета. Осадок погружали в насыщенный раствор NaHCO3 и затем фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения X (26 г) в виде твердого вещества белого цвета.
[0060] Стадия 7 - В реакционную колбу объемом 500 мл добавляли гидроксид калия (27 г), метанол (600 мл), воду (72 мл) и соединение X (23,4 г). Реакционную смесь кипятили с обратным холодильником. С помощью ТСХ подтверждали отсутствие соединения X. Реакционную смесь охлаждали до комнатной температуры, и выливали в воду (3 л) для диспергирования. Смесь доводили концентрированной соляной кислотой до рН=7 и оставляли до выпадения осадка. Полученную смесь фильтровали и осадок на фильтре промывали до нейтральной реакции среды с последующей пререкристаллизацией из ацетона, и сушили с получением соединения I (16 г) в виде белого твердого вещества.
[0061] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(C); ИК (KBr, cm-1) v: 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 2
[0062] Стадия 1 - В реакционную колбу объемом 2 л добавляли соединение андрост-5-ен-3-ол (70 г) и CH2Cl2 (1200 мл). К полученной смеси при перемешивании порциями добавляли м-хлорпероксмбензойную кислоту (105 г). Реакционную смесь затем перемешивали на бане со льдом в течение 5 ч. После окончания реакции, смесь промывали насыщенным раствором сульфита натрия, раствором карбоната натрия и дистиллированной водой. Органический слой собирали, сушили и концентрировали с получением соединения IV (62 г) в виде твердого вещества желтого цвета.
[0063] Стадия 2 - Соединение IV (60 г) растворяли в ацетоне (3 л). К раствору добавляли 1 н. раствор H2SO4 (400 мл). Смесь перемешивали при комнатной температуре в течение 3 ч. После окончания реакции, смесь нейтрализовали раствором Na2CO3 и концентрировали при пониженном давлении для удаления ацетона. Смесь экстрагировали этилацетатом, органический слой собирали и сушили над безводным сульфатом натрия с получением твердого вещества желтого цвета (48 г). Твердое вещество перекристаллизовывали в ацетоне с получением соединения III (30,5 г).
[0064] Стадии 3-7 идентичны стадиям 3-7 в Примере 1.
[0065] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1H ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v: 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 3
[0066] Стадии 1-5 идентичны стадиям 1-5 в Примере 1.
[0067] Стадия 6 - В реакционной колбе объемом 500 мл растворяли соединение IX (23 г) в CH2Cl2 (600 мл). к полученной смеси при перемешивании порциями добавляли м-хлорпероксибензойную кислоту (34,6 г). Реакционную смесь затем перемешивали на бане со льдом в течение 5 ч. После окончания реакции, смесь промывали насыщенным раствором сульфита натрия, раствором карбоната натрия и дистиллированной водой. Органический слой собирали, сушили и концентрировали с получением соединения IX (20,7 г) в виде твердого вещества желтого цвета.
[0068] Стадия 7 - В реакционной колбе объемом 2 л соединение XI (17,4 г) растворяли в ацетоне (900 мл). К смеси добавляли 1 н. раствор H2SO4 (180 мл). Смесь перемешивали при комнатной температуре в течение 3 ч. После окончания реакции, смесь нейтрализовали раствором Na2CO3 и концентрировали при пониженном давлении для удаления ацетона. Смесь экстрагировали этилацетатом, органический слой собирали и сушили над безводным сульфатом натрия с получением твердого вещества желтого цвета (14,4 г). Твердое вещество перекристаллизовывали из ацетона и получали соединение I в виде твердого вещества белого цвета.
[0069] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-CH3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН ); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v. 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 4
[0070] Стадия 1 - В реакционную колбу объемом 250 мл добавляли соединение андрост-5-ен-3-ол (14,64 г) и безводный пиридин (125 мл). К указанной смеси добавляли порциями п-толуолсульфонилхлорид (26,05 г). Смесь оставляли реагировать при комнатной температуре в течение 24 ч. После исчерпания исходных ингредиентов, которое подтверждали с помощью ТСХ, реакцию останавливали. Смесь выливали в ледяной раствор HCl (2000 мл, 17%) при интенсивном перемешивании и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили в вакууме с получением соединения VII (22,48 г) в виде твердого вещества белого цвета.
[0071] Стадия 2 - В реакционную колбу объемом 250 мл добавляли соединение VII (15,00 г) и CH2Cl2 (200 мл). К полученной смеси при перемешивании порциями добавляли м-хлорпероксибензойную кислоту (15,12 г). Реакционную смесь дополнительно перемешивали на бане со льдом в течение 5 ч. После окончания реакции, смесь промывали насыщенным раствором сульфита натрия, раствором карбоната натрия и дистиллированной водой. Органический слой собирали и сушили над безводным сульфатом натрия. Органические растворители выпаривали. Остаток сушили в вакууме с получением неочищенного продукта (14,07 г). Твердое вещество очищали с помощью колоночной хроматографии на силикагеле для обеспечения соединения VIII (12,3 г) в виде твердого вещества белого цвета.
[0072] Стадия 3 - В реакционную колбу объемом 1000 мл добавляли соединение VIII (14,07 г) и ацетон (750 мл). К указанной смеси при перемешивании добавляли реагент Джонса (30 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. После окончания реакции, что подтверждали с помощью ТСХ, смесь гасили изопропанолом и доводили до нейтральной реакции среды раствором Na2CO3. Смесь концентрировали при пониженном давлении для удаления ацетона и экстрагировали этилацетатом. Органический слой собирали и промывали несколько раз дистиллированной водой, сушили над безводным сульфатом натрия и концентрировали с получением твердого вещества бледно-зеленого цвета. Твердое вещество очищали с помощью колоночной хроматографии на силикагеле для обеспечения соединения VI (12,89 г) в виде твердого вещества белого цвета.
[0073] Стадии 4-6 идентичны стадиям 5-7 в Примере 1.
[0074] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(0; ИК (KBr, cm-1) v. 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 5
[0075] Стадии 1-4 идентичны стадиям 1-4 в Примере 1.
[0076] Стадия 5 - В литровую реакционную колбу добавляли N,N-диметилформамид (325 мл), соединение VI (54 г), N2CO3 (54 г) и LiBr (69,5 г). Реакционную смесь кипятили с обратным холодильником. После исчерпания соединения VI, о чем свидетельствовала ТСХ, реакцию останавливали. Смесь выливали в ледяной водный раствор соляной кислоты (2 л, 1:1 (по объему)), перемешивали и фильтровали. Осадок на фильтре промывали до нейтральной реакции и сушили с получением соединения IX (32,5 г) в виде твердого вещества белого цвета.
[0077] Стадии 6-7 идентичны стадиям 6-7 в Примере 1.
[0078] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v: 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 6
[0079] Стадии 1-4 идентичны стадиям 1-4 в Примере 1.
[0080] Стадия 5 - В литровую реакционную колбу добавляли N,N-диметилформамид (325 мл), соединение VI (54 г), сухой N2CO3 (54 г) и LiBr (34,7 г). Реакционную смесь кипятили с обратным холодильником. После исчерпания соединения VI, о чем свидетельствовала ТСХ, реакцию останавливали. Смесь выливали в ледяной водный раствор соляной кислоты (2 л, 1:1 (об. : об.)), перемешивали и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения IX (30,2 г) в виде твердого вещества белого цвета.
[0081] Стадии 6-7 идентичны стадиям 6-7 в Примере 1.
[0082] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1H ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v. 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 7
[0083] Стадии 1-4 идентичны стадиям 1-4 в Примере 1.
[0084] Стадия 5 - В литровую реакционную колбу добавляли N,N-диметилформамид (432 мл), соединение VI (54 г), сухой Li2CO3 (52,1 г) и LiBr (60,5 г). Реакционную смесь киятили с обратным холодильником. После исчерпания соединения VI, что отслеживали с помощью ТСХ, реакцию останавливали. Смесь выливали в ледяной водный раствор соляной кислоты (2 л, 1:1 (об. : об.)), перемешивали и фильтровали. Осадок на фильтре промывали до нейтральной реакции и сушили с получением соединения IX (30,5 г) в виде твердого вещества белого цвета.
[0085] Стадия 6 - В реакционную колбу объемом 500 мл добавляли соединение IX (30 г) и муравьиную кислоту (600 мл, 88%). Реакционную смесь нагревали до растворения твердого вещества и затем охлаждали до 25°С. Медленно добавляли пероксид водорода (20 мл, 30%). После исчерпания соединения IX, что отслеживали с помощью ТСХ, реакционную смесь нагревали до 75°С и выдерживали так в течение 10 минут, чтобы удалить избыток пероксида водорода. Добавляли воду (3 л) и перемешивали для диспергирования. Полученную смесь фильтровали с получением осадка фильтрования белого цвета. Осадок погружали в насыщенный раствор NaHCO3 и фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения X (28 г) в виде твердого вещества белого цвета.
[0086] Стадия 7 - В реакционную колбу объемом 500 мл добавляли гидроксид калия (18 г), метанол (600 мл), воду (72 мл) и соединение X (23,4 г). Реакционную смесь кипятили с обратным холодильником. С помощью ТСХ подтверждали отсутствие соединения X. Реакционную смесь охлаждали до комнатной температуры, и выливали в воду (3 л) для диспергирования. Смесь доводили концентрированной соляной кислотой до рН=7 и оставляли до выпадения осадка. Полученную смесь фильтровали и осадок на фильтре промывали до нейтральной реакции, перекристаллизовывали из ацетона и сушили с получением соединения I (8,8 г) в виде твердого вещества белого цвета.
[0087] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН ); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(C), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(C), 210,65(C); ИК (KBr, cm-1) v: 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 8
[0088] Стадии 1-5 идентичны стадиям 1-5 в Примере 7.
[0089] Стадия 6 - В реакционную колбу объемом 500 мл добавляли соединение IX (30 г) и муравьиную кислоту (600 мл, 88%). Реакционную смесь нагревали до растворения твердого вещества и затем охлаждали до температуры ниже 25°С. Медленно добавляли пероксид водорода (36 мл, 30%). После исчерпания соединения IX, что отслеживали с помощью тонкослойной хроматографии, реакционную смесь нагревали до 75°С и выдерживали так в течение 10 минут, чтобы удалить избыток пероксида водорода. К реакционной смеси добавляли воду (3 л) и перемешивали для диспергирования. Полученную смесь фильтровали с получением осадка фильтрования белого цвета. Осадок погружали в насыщенный раствор NaHCO3 до тех пор, пока не прекратилось выделение пузырьков, а затем фильтровали. Осадок на фильтре промывали до нейтральной реакции среды и сушили с получением соединения X (24 г) в виде белого твердого вещества.
[0090] Стадия 7 идентична стадии 7 в Примере 7.
[0091] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН ); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v. 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Получение соединения I - Пример 9
[0092] Стадии 1-5 идентичны стадиям 1-5 в Примере 7.
[0093] Стадия 6 - В реакционную колбу объемом 500 мл добавляли соединение IX (30 г) и муравьиную кислоту (600 мл, 88%). Реакционную смесь нагревали до растворения твердого соединения и затем охлаждали до 25°С. Медленно добавляли пероксид водорода (45 мл, 30%). После исчерпания соединения IX, что отслеживали с помощью тонкослойной хроматографии, реакционную смесь нагревали до 75°С и выдерживали так в течение 10 минут, чтобы удалить избыток пероксида водорода. К реакционной смеси добавляли воду (3 л) и перемешивали для диспергирования. Полученную смесь фильтровали с получением осадка фильтрования белого цвета. Осадок погружали в насыщенный раствор NaHCO3 до тех пор, пока не прекратилось выделение пузырьков, а затем фильтровали. Осадок на фильтре промывали до нейтральной реакции и сушили с получением соединения X (23 г) в виде белого твердого вещества.
[0094] Стадия 7 - В реакционную колбу объемом 500 мл добавляли гидроксид калия (27 г), метанол (300 мл), воду (36 мл) и соединение X (12,5 г). Реакционную смесь кипятили с обратным холодильником. С помощью ТСХ подтверждали отсутствие соединения X. Реакционную смесь охлаждали до комнатной температуры, и выливали в воду (3 л) для диспергирования. Смесь доводили концентрированной соляной кислотой до рН=7 и оставляли до выпадения осадка. Полученную смесь фильтровали и осадок на фильтре промывали до нейтральной реакции среды, перекристаллизовывали из ацетона и сушили с получением соединения I (8,0 г) в виде твердого вещества белого цвета.
[0095] Т. пл: 197~201°С; удельное вращение: -50° (2 мг/мл абсолютного этанола); 1Н ЯМР (CDCl3, 400 МГц) δ: 0,65 (с, 3Н, 18-СН3), 0,87 (с, 3Н, 19-СН3), 3,76~3,83 (д, J=28Hz, 2Н, 2-СН и 3-СН ); 13С ЯМР (CDCl3, 400 МГц) δ: 15,52 (СН3), 17,27(СН3), 19,94(СН2), 20,53(СН2), 24,68(СН2), 26,90(СН2), 33,02(СН2), 36,45(СН), 39,72(СН2), 40,92(С), 41,26(СН2), 42,54(С), 44,99(СН), 54,04(СН), 68,94(СН), 70,19(СН), 79,71(С), 210,65(С); ИК (KBr, cm-1) v: 3296, 2937, 1726, 1064; MC(APCI)m/z: 319(М-3).
Противоопухолевая активность Соединения I
[0096] Посев клеток и лечение: Клетки LN18 и DBTRG-50MG, находящиеся в логарифмической фазе роста, были приготовлены в виде клеточных суспензий с питательной средой. Клетки высевали в 96-луночный планшет при плотности 100 мкл на лунку, 3×104/мл. Через 12 ч после посева, наблюдали полную адгезию клеток. В лунки добавляли YC-10 до конечной концентрации YC-10 равной 250, 500 и 1000 мкМ, при этом каждая концентрационная группа включает 5 одинаковых образцов.
[0097] Реакция МТТ с сукцинатдегидрогеназой: На 24-ом часу культивирования, добавили 10 мкл (5 мг/мл) МТТ в каждую лунку, затем инкубировали еще 4 ч. В это время, в живых клетках можно наблюдать с помощью микроскопии гранулят фиолетового кристаллического формазана.
[0098] Растворение частиц формазана: Аккуратно слили надосадочную жидкость. В лунки добавили ДМСО в количестве 100 мкл/лунка, чтобы растворить кристалличность. Смесь встряхивали на мини-шейкере в течение 5 мин и измеряли оптическую плотность (значение OD) при 570 нм для каждой лунки методом иммунометрического анализа, связанного с ферментом.
[0099] Каждую группу экспериментов повторяли 3 раза.
[00100] Выживаемость (%) = значение OD в группе лечения / значение OD в контрольной группе * 100%.
[00101] Все данные представлены как среднее значение ± стандартное отклонение. Был использован статистический программный пакет SPSS 13.0. Односторонний дисперсионный анализ и t-тест были использованы для анализа данных. Для создания Фигуры 1 было использовано программное обеспечение Sigmaplot. Как видно из Фигуры 1, после 24 ч обработки с помощью YC-10 с концентрацией 250, 500, 1000 мкМ, выживаемость клеток из группы лечения была статистически значимой по сравнению с контрольной группой (р<0,05). YC-10 дозозависимым образом убил опухолевые клетки.
Нейропротекторная активность Соединения I
[00102] Была изучена in vivo и in vitro токсичность и фармакологические функции YC-10, чтобы оценить его нейропротекторную активность и возможность стать потенциальным клиническим препаратом. В целом, результаты показали, что у мышей, которым вводили большие дозы YC-10 (250 мг/кг) никаких очевидных аномалий не наблюдалось. Исследования показали, что YC-10 был достаточно эффективным в увеличении выживаемости гранулярных нейронов мозжечка в обеих моделях повреждения: глутамат-индуцированной модели и модели дефицита калия. Было также показано на модели животных, страдающих и от повреждения зрительного нерва, и от ишемии сетчатки, что YC-10 значительно увеличивает долю выживших ганглиозных клеток сетчатки. Представленные результаты демонстрируют, что YC-10 имеет нейронпротекторную активность без очевидных токсических или побочных эффектов.
1. Токсикологическое исследование
Тест на максимально переносимую дозу
[00103] Инъекции YC-10 с концентрацией 25 мг/мл готовили с 40% гидроксипропилциклодекстрином, и вводили через хвостовую вену 30 мышам вида КунМинг (половина самцов и половина самок, массой 18-22 г) при дозировке 0,1 мл/10 г.
[00104] Наблюдение за мышами проводилось непрерывно. Все мыши вели себя и ели как обычно и имели яркий окрас меха и подшерстка. Аномальных выделений в ротовой полости, глазах, носу или ушах не наблюдалось. Мыши испражнялись нормально. Масса мышей немного возросла. Ни одна мышь не умерла. Мышей умерщвляли через 14 дней, препарировали и визуально осматривали важные органы, такие как сердце, печень, селезенка, почки, желудочно-кишечный тракт. Никаких аномальных изменений не было обнаружено. Представленные результаты показали, что YC-10 в количестве в 250 мг/кг не был токсичен для мышей.
2. Фармакологические исследования
2.1 Защита гранулярных нейронов мозжечка с помощью YC-10 от глутамат-индуцированных повреждений.
[00105] Гранулярные нейроны мозжечка культивированные in vitro в течение 8 дней были разбиты на группы. Группы лечения получали МК801 или YC-10 в различных концентрациях с последующим инкубированием в течение 30 мин. После этого модельная группа и все группы лечения были заменены свободным от Mg2+ буфером Локка, и добавляли глутамат (конечная концентрация 100 мкМ), глицин (конечная концентрация 10 мкМ) и лекарственные средства в соответствующих концентрациях. Клетки инкубировали при 37°С в течение 30 минут, заменяли первоначальную среду, инкубировали в течение еще 24 ч, с последующим окрашиванием флуоресцеин диацетатом. Полученные результаты приведены на Фигуре 2.
[00106] Результаты показали, что глутамат может вызывать повреждение и гибель гранулярных нейронов мозжечка. МК801 смог предотвратить глутамат-индуцированное повреждение гранулярных нейронов мозжечка. YC-10 был также эффективен в предотвращении глутамат-индуцированного повреждения экзотоксином гранулярных нейронов мозжечка дозозависимым образом. YC-10 защитил гранулярные нейроны мозжечка от глутамат-индуцированных повреждений (Фиг. 3).
2.2 Гранулярные нейроны мозжечка защищенные с помощью YC-10 от гибели вызванной низким содержанием калия.
[00107] Гранулярные нейроны мозжечка, культивированные in vitro в течение 8 дней, были разбиты на группы. Группы лечения получали YC-10 в различных концентрациях с последующим инкубированием в течение 30 мин. После этого модельная группа и все группы лечения были замещены с помощью 5K (т.е. 5 ммоль KCl) питательной среды Игла (25K питательной среды Игла для контрольной группы), с добавлением YC-10 в соответствующих концентрациях. Клетки инкубировали при 37°С в течение 24 ч, с последующим окрашиванием флуоресцеин диацетатом. Результаты показаны на рисунке 4.
[00108] Как показано на Фиг. 4, среда с низким содержанием калия может снизить смертность гранулярных нейронов мозжечка. YC-10 (50 мкмоль) может защитить нейроны от гибели, связанной с низким содержанием калия. YC-10 защитил гранулярные нейроны мозжечка от гибели, связанной с низким содержанием калия.
2.3 Защита ганглиозных клеток сетчатки с помощью YC-10 от гибели в результате пережатия оптического нерва.
[00109] Для анестезирования крыс был использован 10% хлоралгидрат. YC-10 (20 мг/кг) или растворители вводили в хвостовую вену за 20 мин до операции. Глаза были подвергнуты местной анестезии. Конъюнктива была разрезана вдоль лимба роговицы с помощью ножниц для роговицы и интраокулярных микрозажимов. Боковая прямая мышца была рассечена, чтобы полностью оголить зрительный нерв. Поперечные щипцы были использованы, чтобы пережать оптический нерв на 5 секунд на 2 мм позади глазного яблока, после чего его отпустили. После операции нанесли противомикробную глазную мазь, чтобы предотвратить инфекцию. Лекарства вводили через два часа после операции, на второй и на третий день. На седьмой день был проведен осмотр глазных яблок на наличие патологий.
Как показано на Фиг. 5, осмотр на наличие патологий показал, что пережатие оптического нерва может вызывать гибель ганглиозных клеток сетчатки. Было показано, что YC-10 замедляет или предотвращает гибель в результате пережатия, то есть, YC-10 может предотвратить гибель ганглиозных клеток сетчатки при травме зрительного нерва в результате пережатия. Количества ганглиозных клеток сетчатки в каждой группе были зарегистрированы и представлены на Фиг. 6.
2.4 Защита ганглиозных клеток сетчатки с помощью YC-10 от гибели вследствие повышенного глазного давления и ишемии.
[00110] Для анестезирования крыс был использован 10% хлоралгидрат. Глаза были подвергнуты местной анестезии. Перфузионный аппарат был размещен на 176 см выше глазных яблок крысы, (что обеспечивает внутриглазное давление 130 мм.рт.ст.). Игла шприца 30G 1/2 осторожно вставляли в переднюю камеру глаза. Глазные яблоки стали белыми и было записано время начала ишемии. Спустя 1 ч после начала ишемии иглу шприца быстро извлекали и обрабатывали глаза глазными каплями с антибиотиком. Крысы были возвращены в клетку. Препараты вводили 20 перед моделированием для группы растворителей и YC-10 группы (20 мг/кг). Через два часа, на второй и на третий день после моделирования крысы были подвергнуты лечению путем введения YC-10 через хвостовую вену. На седьмой день после моделирования глазные яблоки были подвергнуты проверке на наличие патологий.
[00111] Как показано на Фиг. 7, осмотр на наличие патологий показал, что высокое глазное давление и ишемия могут вызывать гибель ганглиозных клеток сетчатки. Было показано, что YC-10 замедляет или предотвращает гибель в результате ишемии, то есть, YC-10 может предотвратить гибель ганглиозных клеток сетчатки от высокого глазного давления и ишемии. Количества ганглиозных клеток сетчатки в каждой группе были зарегистрированы и представлены на Фиг. 8.

Изобретение относится к соединению 2β,3α,5α-тригидрокси-андрост-6-ону, имеющему структуру формулы I
Изобретение также относится к фармацевтической композиции и к способу получения соединения формулы I. Технический результат: получено новое соединение, обладающее нейронопротекторным эффектом, особенно в отношении ганглиозных клеток сетчатки, и может также применяться при лечении глиобластомы. 3 н. и 1 з.п. ф-лы, 8 ил., 9 пр.
1. Соединение 2β,3α,5α-тригидрокси-андрост-6-он, имеющее структуру формулы I:
2. Фармацевтическая композиция для протекции нейронов и/или лечения глиобластомы, содержащая терапевтически эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель.
3. Фармацевтическая композиция по п. 2, в которой нейроны представляют собой ганглиозные клетки сетчатки.
4. Способ получения соединения по п. 1, включающий:
(a) применение андрост-5-ен-3-ола в качестве исходного соединения с получением соединения VI, т.е. 3β-п-толуолсульфонилокси-5α-гидрокси-андрост-6-она;
(b) проведение реакции элиминирования соединения VI с получением соединения IX, т.е. 5α-гидрокси-андрост-2-ен-6-она; и
(c) окисление соединения IX по двойной связи в положении 2 и гидролиз с получением соединения I, причем
стадию (а) осуществляют по одному из следующих вариантов:
(i) осуществление последовательно: окисления андрост-5-ен-3-ола смесью Н2О2/муравьиная кислота, щелочного гидролиза, окисления N-бромсукцинимидом и защиты п-толуолсульфонилхлоридом с получением соединения VI;
(ii) осуществление последовательно: окисления андрост-5-ен-3-ола в присутствии м-хлорпероксибензойной кислоты, ацидолиза, окисления N-бромсукцинимидом и защиты п-толуолсульфонилхлоридом с получением соединения VI; или
(iii) осуществление последовательно: введения защиты в андрост-5-ен-3-ол п-толуолсульфонилхлоридом, окисления в присутствии м-хлорпероксибензойной кислоты, и окисления реагентом Джонса с получением соединения VI.
| J | |||
| A | |||
| Ramirez et al | |||
| "Synthesis and bioactivity evaluation of brassinosteroid analogs" Steroids, vol | |||
| Разборное приспособление для накатки на рельсы сошедших с них колес подвижного состава | 1920 |
|
SU65A1 |
| Y | |||
| Suarez et al | |||
| "Sterol stringency of proliferation and cell cycle progression in human cells" Biochimica et Biophysica Acta, vol | |||
| Предохранитель для запальной свечи в двигателе внутреннего горения | 1922 |
|
SU1734A1 |
| 6,7-ОКИСЛЕННЫЕ СТЕРОИДЫ | 1997 |
|
RU2196143C2 |
| CN 101884638 A, 17.11.2010 | |||
| WO 2006002907 A1, 12.01.2006 | |||
| FR 2967914 A1, 01.06.2012. | |||

