Производные гетероциклические алкильные соединения в качестве селективных ингибиторов гистондеацетилазы и содержащие их фармацевтические композиции
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым гетероциклическим алкильным производным, и более конкретно к новым гетероциклическим алкильным производным, обладающим ингибирующей активностью в отношении гистондеацетилазы (HDAC), к их оптическим изомерам или их фармацевтически приемлемым солям, к их применению для получения лекарственных средств для лечения опосредованных HDAC заболеваний, к содержащим их фармацевтическим композициям, к способам лечения заболеваний с использованием таких фармацевтических композиций, и к способам получения новых гетероциклических алкильных производных.
Уровень техники
Транскрипционная регуляция в клетках представляет собой сложный биологический процесс. Один основной принцип регуляции транскрипции основан на посттрансляционной модификации гистоновых белков, а именно гистоновых белков H2A/B, H3 и H4, образующих октамерные комплексы коровых гистонов. Сложные N-концевые модификации в области лизиновых остатков путем ацетилирования или метилирования и в области сериновых остатков путем фосфорилирования составляют часть так называемого "гистонового кода" (см. Strahl & Ellis, Nature 403, 41-45, 2000).
В простой модели, ацетилирование положительно заряженных остатков лизина снижает сродство к отрицательно заряженной ДНК, и, таким образом, факторы транскрипции могут быть легко введены.
Ацетилирование и деацетилирование гистонов катализируются соответственно гистонацетилтрансферазами (HAT) и гистондеацетилазами (HDAC). HDAC связаны с транскрипционными репрессорными комплексами, переключая хроматитин на молчащую транскрипционно неактивную структуру (см. Marks et al. Nature Cancer Rev. 1, 189-202, 2001). Противоположный результат достигается под воздействием HAT, которые связаны с комплексом транскрипционных активаторов. До настоящего времени были известны три различных класса HDAC, а именно, класс I (HDAC 1-3, 8; мол. вес=42-55 кДа), где HDAC в основном локализованы в ядре и чувствительны к ингибированию трихостатином А (TSA); класс II (HDAC 4-7, 9, 10; мол. вес=120-130 кДа), где HDAC проявляют чувствительность к TSA; и класс III (SIRT 1~7), где HDAC отличаются своей зависимостью от NAD+ и нечувствительностью к TSA.
Ингибиторы гистондеацетилазы (HDAC) образуют новый класс противораковых лекарственных средств, обладающих клеточной дифференцировкой и апоптоз-индуцирующей активностью. Путем направленного воздействия на гистондеацетилазы (HDAC), ингибиторы HDAC воздействуют на структуру хроматина путем ацетилирования гистонов, индуцируя перепрограммирование комплексной транскрипции, например, реактивирование генов-супрессоров опухоли и репрессию онкогенов. Кроме ацетилирования N-концевого остатка лизина в коровом гистоновом белке, ингибиторы HDAC нацелены на негистоновый белок, важный для биологии рака, включая белок теплового шока 90 (HSP90), тубулин или белок-супрессор опухолевого роста р53. Таким образом, ингибиторы HDAC могут быть использованы не только для противораковой терапии, но также для лечения генетических метаболических заболеваний, аутоиммунных заболеваний и тому подобного, так как была показана эффективность на животных моделях воспалительных заболеваний, ревматоидного артрита и нейродегенерации.
Примеры опосредованных гистондеацетилазой заболеваний, связанных с ингибированием HDAC, включают клеточные пролиферативные заболевания, такие как злокачественные опухолевые заболевания, например, рак; воспалительные заболевания, такие как воспалительные заболевания кишечника, болезнь Крона или язвенный энтерит; аутосомные доминантные заболевания, такие как болезнь Хантингтона, синдрома Дауна синдрома Эдвардса или синдрома Патау; генетические метаболические заболевания, такие как диабет, болезнь Ниманна-Пика, болезнь Гоше, фенилкетонурия, болезнь Вильсона или фиброз, например, кистозный фиброз, фиброз печени, фиброз почек, фиброз легких или фиброз кожи; аутоиммунные заболевания, такие как ревматоидный артрит, астма, волчанка, псориаз, псориатический артрит, рассеянный склероз, болезнь Бехчета или отторжение трансплантата органа; острые/хронические неврологические заболевания, такие как удар, или поликистозное заболевание почек; гипертрофия, такая как гипертрофия сердца; сердечная недостаточность, такая как застойная сердечная недостаточность или геморрагическая сердечная недостаточность; глазные заболевания, такие как глаукома, синдром сухого глаза, сухая дегенерация желтого пятна, влажная дегенерация желтого пятна, диабетическая ретинопатия или увеит; нейродегенеративные заболевания, такие как болезнь Альцгеймера, боковой амиотрофический склероз, болезнь Шарко-Мари-Тута, или мышечная атрофия позвоночника, а также состояния и заболевания, вызванные аномальной функцией ферментов HDAC.
Ингибиторы HDAC, известные до настоящего времени, можно классифицировать по своей структуре на четыре категории: 1) жирные кислоты с короткой цепью (масляная кислота и вальпроевая кислота); 2) гидроксамовые кислоты (трихостатин A, SAHA и LBH-589); 3) циклические пептиды (десипептид); и 4) бензамиды (MS-275 и MGCD-0103) (Sonia et al., International Journal of Oncology 33, 637-646, 2008). Эти многие ингибиторы гистондеацетилазы (HDAC) (SAHA, LBH-589, MS-275 и т.п.) ингибируют рост клеток, и эффективно индуцируют дифференцировку клеток и апоптоз различных трансформированных клеток не только в культуральных средах, но также и в животных моделях (Paul A. Marks et. al., Curr Opin. Oncol. 13, 477-483, 2001). Поэтому ингибиторы HDAC, такие как SAHA, LBH-589 и MS-275 были оценены в клинических исследованиях, направленных на лечение различных видов рака (Johnstone R.W, Nat. Rev. Drug. Discov. 1, 287-299, 2002). Представительные соединения, известные в настоящее время как ингибиторы HDAC, включают SAHA (переизданный патент США No. 385069, золинза, вориностат), PXD101 (WO 02/30879, белиностат) и LBH-589 (WO 02/22577, панобиностат), которые представляют собой гидроксаматные соединения, и MS-275 (Европейский патент No. 0847992 энтиностат) и MGCD0103 (WO 04/69823, моцетиностат), которые представляют собой бензамидные соединения. Среди этих соединений SAHA был одобрен в октябре 2006 года, и он был использован в качестве средства для лечения CTCL (кожная Т-клеточная лимфома), и его назначения были дополнительно расширены, но было известно, что SAHA обладает недостатками с точки зрения эффективности и побочных эффектов (Paul A. Marks et al., Cancer Res 66, 5781-5789, 2006).
Различные ингибиторы HDAC находятся на стадиях доклинических или клинических исследований, но до настоящего времени в качестве противоопухолевых агентов были идентифицированы только неселективные ингибиторы HDAC. Известно, что неселективные ингибиторы HDAC вызывают побочные эффекты, такие как усталость и тошнота, обычно при высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщалось, что такие побочные эффекты обусловлены ингибированием HDAC класса I. Из-за таких побочных эффектов использование неселективных ингибиторов HDAC при разработке лекарственных средств, иных, чем противоопухолевые лекарственные средства, было ограничено (Witt et al., Cancer Letters, 2009, 277, 8-21).
В то же время сообщалось, что селективное ингибирование HDAC класса II не показало токсичности, которая имела место при ингибировании HDAC класса I. Кроме того, когда были разработаны селективные ингибиторы HDAC, побочные эффекты, такие как токсичность, вызванная неселективным ингибированием HDAC, можно преодолеть. Таким образом, селективные ингибиторы HDAC имеют потенциал и перспективы для дальнейшей разработки в качестве эффективных терапевтических агентов, предназначенных для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC 6, член HDAC класса IIb, присутствует главным образом в цитоплазме, и она участвует в деацетилировании ряд негистоновых субстратов (HSP90, кортактин и т.п.), включая тубулин (Yao et al., Mol. Cell 2005, 18, 601-607). Кроме того, HDAC 6 имеет два каталитических домена, и C-концевой домен цинкового пальца может связываться с убиквитинированными белками. Известно, что HDAC 6 в качестве субстратов имеет ряд негистоновых белков, и, таким образом, она играет важную роль в различных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные нарушения (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Таким образом, существует потребность в разработке селективных ингибиторов HDAC 6 для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний и нейродегенеративных заболеваний, где такие ингибиторы не вызывают побочных эффектов, в отличие от неселективных ингибиторов.
Сущность изобретения
Решаемая техническая проблема
Целью настоящего изобретения является предоставление новых соединений, обладающих селективной ингибирующей активностью в отношении HDAC, их оптических изомеров или их фармацевтически приемлемых солей.
Другой целью настоящего изобретения является предоставление фармацевтических композиций, содержащих новые соединения, которые обладают высокой ингибирующей активностью в отношении HDAC, их оптические изомеры или их фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предоставление способов получения новых соединений.
Еще одной целью настоящего изобретения является предоставление фармацевтических композиций для лечения заболеваний, связанных с активностью HDAC, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные заболевания, которые содержат указанные выше соединения.
Еще одной целью настоящего изобретения является предоставление применения соединений для получения лекарственных средств для лечения опосредованных HDAC заболеваний, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные заболевания.
Еще одной целью настоящего изобретения является предоставление способов лечения опосредованных HDAC заболеваний, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные заболевания, которые включают введение терапевтически эффективного количества фармацевтических композиций, содержащих указанные выше соединения.
Решение проблемы
Авторы изобретения обнаружили новые соединения, обладающие ингибирующей активностью в отношении HDAC, и использовали эти соединения для подавления развития или для лечения заболевания, опосредованного гистондеацетилазой, получив, таким образом, настоящее изобретение.
Новые ингибиторы HDAC
Для достижения указанных выше целей в настоящем изобретении предложено соединение формулы I, представленной ниже, его оптические изомеры или его фармацевтически приемлемые соли.
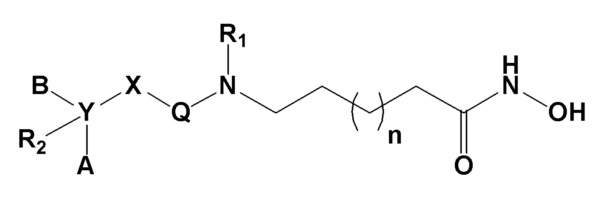
Формула I
где
Х представляет гетероциклический алкил, выбранный из группы, состоящей из
 ,
, 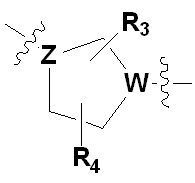 ,
, 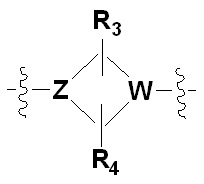 ,
, 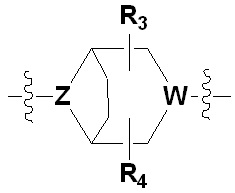 ,
, 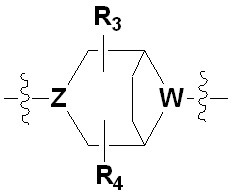 и
и 
где каждый Z и W независимо представляет собой C или N, при этом по меньшей мере один из Z и W представляет собой N,
a, в, с и d независимо равны 1, 2 или 3, и
каждый R3, R4, R5 и R6 независимо представляет собой -Н или -С1-С4-алкил;
Y представляет собой C или N;
каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил, -С3-С12 гетероарил, -С3-С10 циклоалкил, -C2-C10 гетероциклоалкил или -С3-С10 циклоалкенил, где один или несколько атомов водорода в -С1-С4 алкиле могут быть замещены на -OH или галоген, и каждый -С6-С10 арил, -С3-С12 гетероарил, -С3-С10 циклоалкил, -C2-C10 гетероциклоалкил и -С3-С10 циклоалкенил могут быть независимо незамещенными, или один или несколько атомов водорода в них могут быть замещены на -OH, -С1-С4 алкил, -O-С1-С4 алкил, -CF3 или галоген;
Q представляет собой C=O или SO2;
R1 представляет собой -H или -C1-C4 алкил;
R2 представляет собой -H, -OH, -C1-C4 алкил, -C1-C4 алкилгидрокси, галоген или отсутствует, и если Y представляет собой C, то R2 представляет собой -Н, -ОН, -C1-C4 алкил или -C1-C4 алкилгидрокси, и если Y представляет собой N, то R2 отсутствует; и
n равно 1, 2, 3 или 4.
В соответствии с одним из вариантов осуществления настоящего изобретения
Х представляет собой
или
где каждый Z и W независимо представляет собой C или N, и по меньшей мере один из Z и W представляет собой N,
a, в, с и d независимо равны 1, 2 или 3, и
каждый R3, R4, R5 и R6 независимо представляет собой -Н или -C1-C4 алкил;
Y представляет собой C или N;
каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил или -С3-С12 гетероарил, где один или несколько атомов водорода в -С1-С4 алкиле могут быть замещены на -OH или галоген, и -С6-С10 арил или -С3-С12 гетероарил могут быть независимо незамещенными, или один или несколько атомов водорода в них могут быть замещены на -OH, -С1-С4 алкил, -O-С1-С4 алкил, -CF3 или галоген;
Q представляет собой C=O или SO2;
R1 представляет собой -H или -C1-C4 алкил;
R2 представляет собой -H, -OH, галоген или отсутствует, и если Y представляет собой C, то R2 представляет собой -Н, -ОН или галоген, и если Y представляет собой N, то R2 отсутствует; и
n равно 1, 2, 3 или 4.
Согласно другому варианту осуществления настоящего изобретения,
Х представляет собой
где каждый Z и W независимо представляет собой C или N, и по меньшей мере один из Z и W представляет собой N, и
каждый R3 и R4 независимо представляет собой -Н или -С1-С4 алкил;
Y представляет собой C или N;
каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил или -С3-С12 гетероарил, где один или несколько атомов водорода в -С1-С4 алкиле могут быть замещены -OH или галогеном, и -С6-С10 арил и -С3-С12 гетероарил могут быть независимо незамещенными, или один или несколько атомов водорода в них могут быть замещены на -OH, -С1-С4 алкил, -O-С1-С4 алкил, -CF3 или галоген;
Q представляет собой C=O;
R1 представляет собой -H или -C1-C4 алкил;
R2 представляет собой -H, -OH, галоген или отсутствует, и если Y представляет собой C, то R2 представляет собой -Н, -ОН или галоген, и если Y представляет собой N, то R2 отсутствует; и
n равно 3.
Соединения, представленные формулой I, показаны в Таблицах 1-3, приведенных ниже:
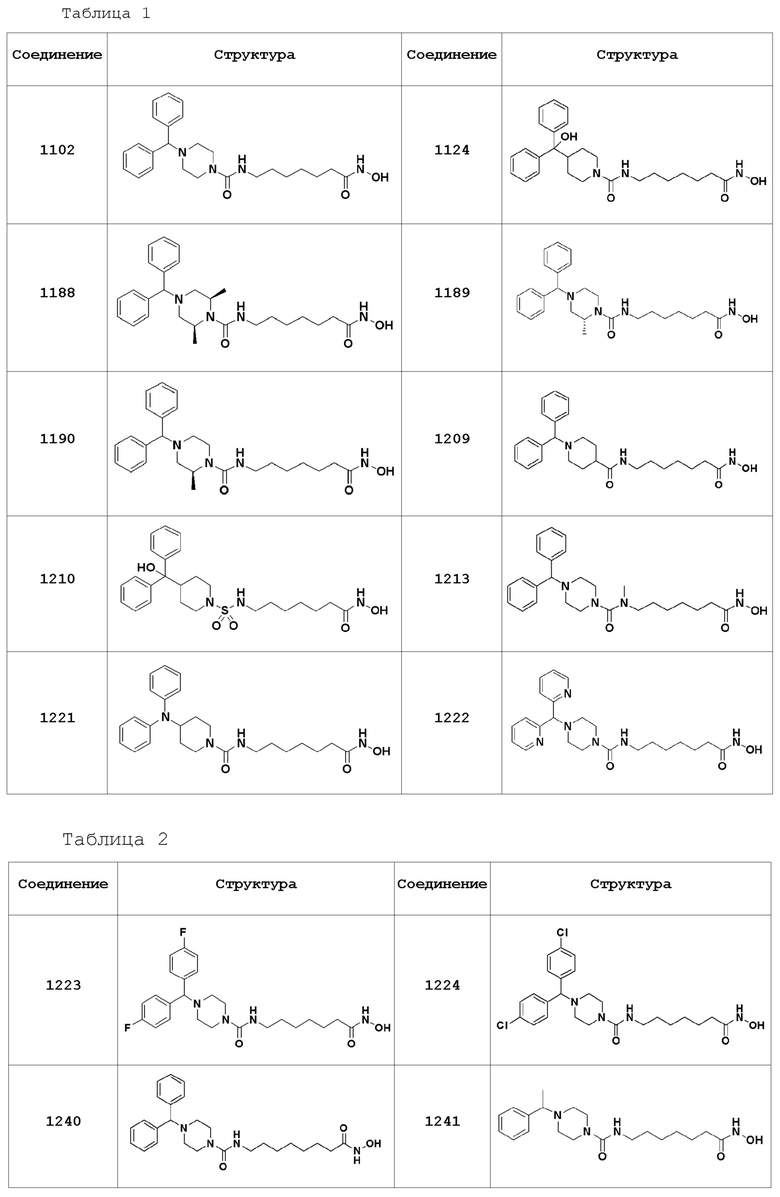
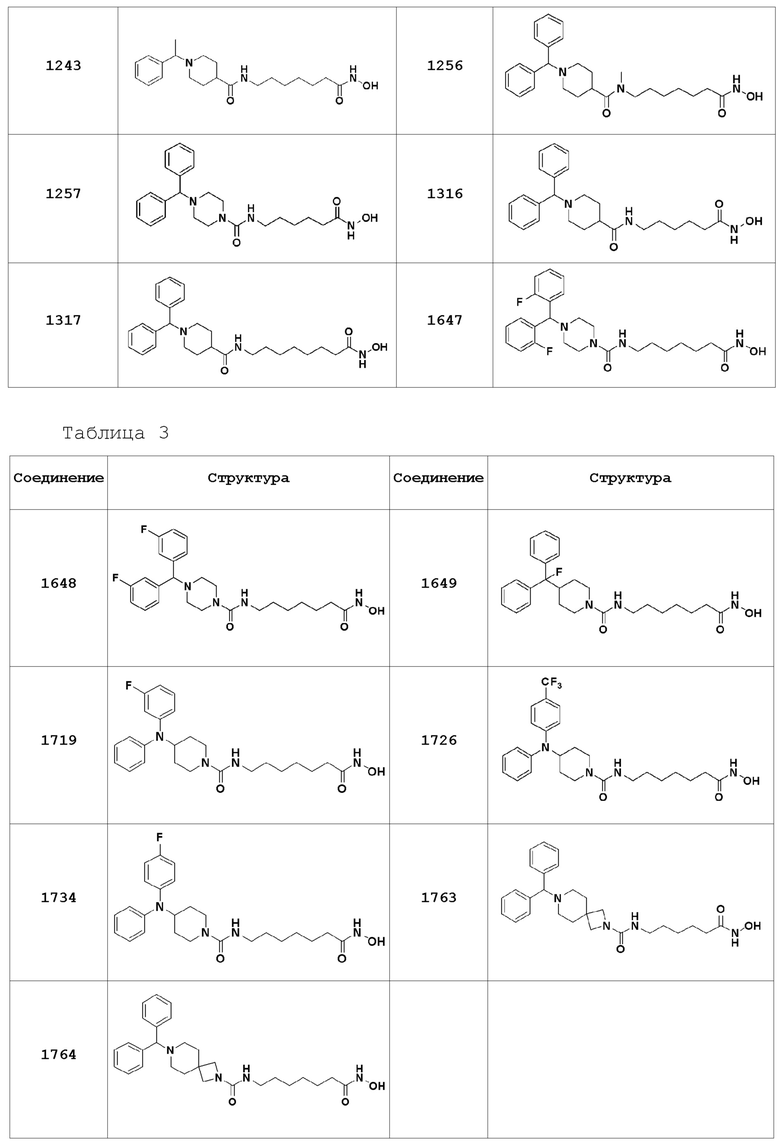
В настоящем изобретении соединения, описанные в представленных выше Таблицах 1-3, или их фармацевтически приемлемые соли предпочтительно выбирают из группы, состоящей из соединений 1102, 1124, 1188, 1189, 1190, 1209, 1221, 1224, 1241 и 1243, и более предпочтительно их выбирают из группы, состоящей из соединений 1102, 1124, 1188 и 1209
Как используется в настоящем описании, термин "фармацевтически приемлемая соль" означает любую соль, которая обычно используется в фармацевтической области. Примеры фармацевтически приемлемой соли включают, но не ограничиваются ими, соли с неорганическими ионами, такими как ионы кальция, калия, натрия или магния, соли с неорганическими кислотами, такими как хлористоводородная кислота, азотная кислота, фосфорная кислота, хлорная кислота, хлорная кислота или серная кислота, соли с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, углекислая кислота, ванилиновая кислота или йодистоводородная кислота, соли с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота или нафталинсульфоновая кислота, соли с аминокислотами, такими как глицин, аргинин или лизин, и соли с аминами, такими как триметиламин, триэтиламин, аммиак, пиридин или пиколин.
Соединения формулы I могут содержать один или несколько асимметричных атомов углерода, и, таким образом, они могут существовать в виде рацематов, рацемических смесей, индивидуальных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Соединения формулы I могут быть разделены на такие изомеры известными в данной области методами, например, колоночной хроматографией или ВЭЖХ. Альтернативно, индивидуальные стереоизомеры соединений формулы I могут быть синтезированы стереоспецифическим синтезом с использованием оптически чистых исходных материалов и/или реагентов с известной конфигурацией.
Композиции, содержащие новое ингибирующее HDAC соединение, их применение и способы лечения заболеваний с их использованием
Настоящее изобретение предоставляет фармацевтическую композицию для профилактики или лечения опосредованного гистондеацетилазой заболевания, где композиция содержит в качестве активного ингредиента соединение, представленное следующей формулой I, его оптический изомер или его фармацевтически приемлемую соль.
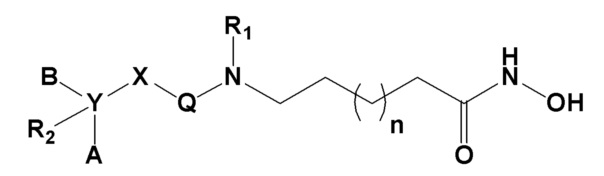
Формула I,
где R1, R2, A, B, X, Y, Q и n определены выше.
Примеры опосредованных гистондеацетилазой заболеваний, включают клеточные пролиферативные заболевания, такие как злокачественные опухолевые заболевания, например, раки; воспалительные заболевания, такие как воспалительные заболевания кишечника, болезнь Крона или язвенный энтерит; аутосомные доминантные заболевания, такие как болезнь Хантингтона, синдром Дауна, синдром Эдвардса или синдром Патау; генетические метаболические заболевания, такие как диабет, болезнь Ниманна-Пика, болезнь Гоше, фенилкетонурия, болезнь Вильсона или фиброз, например, кистозный фиброз, фиброз печени, фиброз почек, фиброз легких или фиброз кожи; аутоиммунные заболевания, такие как ревматоидный артрит, астма, волчанка, псориаз, псориатический артрит, рассеянный склероз, болезнь Бехчета или отторжение трансплантата органа; острые/хронические неврологические заболевания, такие как удар, или поликистозное заболевание почек; гипертрофия, такая как гипертрофия сердца; сердечная недостаточность, такая как застойная сердечная недостаточность или геморрагическая сердечная недостаточность; глазные заболевания, такие как глаукома, синдром сухого глаза, сухая дегенерация желтого пятна, влажная дегенерация желтого пятна, диабетическая ретинопатия или увеит; нейродегенеративные заболевания, такие как болезнь Альцгеймера, боковой амиотрофический склероз, болезнь Шарко-Мари-Тута, или мышечная атрофия позвоночника, а также состояния и заболевания, вызванные аномальной функцией ферментов HDAC.
Фармацевтически приемлемая соль является такой, как описано выше в отношении фармацевтически приемлемой соли соединения по изобретению, представленного формулой I.
Для введения фармацевтической композиции по изобретению она может дополнительно содержать по меньшей мере один фармацевтически приемлемый носитель в дополнение к соединению формулы I, его изомеру или его фармацевтически приемлемой соли. Фармацевтически приемлемый носитель, который используется в настоящем изобретении, представляет собой по меньшей мере одно из следующего: физиологический раствор, стерильная вода, раствор Рингера, буферированный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь двух или более из них. Если необходимо, композиция может содержать другие обычные добавки, такие как антиоксидант, буфер или бактериостатическое средство. Кроме того, композиция может быть приготовлена в виде препаратов для инъекций, таких как растворы, суспензии, мутная жидкость и т.п., в виде пилюль, капсул, гранул или таблеток, используя для этого разбавитель, диспергирующий агент, поверхностно-активное вещество, связующее и смазывающее вещество. Композиция по изобретению может быть также представлена в форме пластырей, жидкости, пилюль, капсул, гранул, таблеток, суппозиториев и т.п. Эти препараты могут быть получены обычными способами, которые использовались для приготовления препаративных форм в данной области, или способом, описанным в Remington 's Pharmaceutical Science (последняя редакция), Mack Publishing Company, Easton, PA.
Фармацевтическую композицию по изобретению можно вводить перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно), в зависимости от предполагаемого назначения. Доза фармацевтической композиции изменяется в зависимости от массы пациента, возраста, пола, состояния здоровья и принимаемой диеты, времени введения, режима введения, скорости выведения, тяжести заболевания и т.п. Суточная доза соединения формулы I в соответствии с настоящим изобретением может составлять приблизительно от 1 до 500 мг/кг, предпочтительно от 5 до 100 мг/кг, и ее можно вводить один или несколько раз в день.
Фармацевтическая композиция по изобретению может дополнительно содержать, в дополнение к соединению формулы I, его оптическому изомеру или его фармацевтически приемлемой соли, один или несколько активных ингредиентов, которые проявляют идентичную или подобную лекарственную эффективность.
Настоящее изобретение также предоставляет способ предотвращения или лечения опосредованного гистондеацетилазой заболевания, который включает введение терапевтически эффективного количества соединения формулы I, его оптического изомера или его фармацевтически приемлемой соли.
Как используется в настоящем описании, термин "терапевтически эффективное количество" относится к количеству соединения формулы I, которое является эффективным для профилактики или лечения опосредованных гистондеацетилазой заболеваний.
Настоящее изобретение также предоставляет способ ингибирования гистондеацетилазы (HDAC) путем введения соединения, представленного формулой I, его оптического изомера или его фармацевтически приемлемой соли млекопитающим, включая человека.
Способ профилактики или лечения опосредованного гистондеацетилазой заболевания по изобретению включает подавление или предотвращение развития заболевания, также как и воздействие на само заболевание до начала проявления симптомов, путем введения соединения формулы I При лечении заболеваний профилактическая или терапевтическая доза конкретного активного ингредиента будет изменяться в зависимости от природы и тяжести заболевания или состояния, а также она может варьироваться в зависимости от пути введения активного ингредиента. Доза и частота введения дозы также изменяются в зависимости от возраста, массы тела и ответа отдельного пациента. Подходящие режимы дозирования могут быть без труда выбраны специалистами в данной области с учетом таких факторов. Кроме того, способ предотвращения или лечения опосредованного гистондеацетилазой заболевания по изобретению может дополнительно включать введение терапевтически эффективное количество дополнительного активного агента для лечения заболевания вместе с соединением формулы I, где дополнительный активный агент может проявлять вспомогательное действие или синергическое действие с соединением формулы I.
Настоящее изобретение также предназначено для обеспечения применения соединения формулы I, его оптического изомера или его фармацевтически приемлемой соли для получения лекарственного средства для лечения опосредованного гистондеацетилазой заболевания. Для получения лекарственного средства соединение, представленное формулой I, может быть смешано с фармацевтически приемлемым адъювантом, разбавителем, носителем или подобным, а также объединено с другими активными агентами, чтобы активные ингредиенты могли проявлять синергическое действие.
Подробности, упомянутые в части объектов применение, композиция и способ лечения по изобретению могут быть соответствующим образом объединены, если они не противоречат друг другу.
Способы получения новых соединений-ингибиторов HDAC
Настоящее изобретение также предоставляет способы получения соединений формулы I, их оптических изомеров или их фармацевтически приемлемых солей. Далее эти способы получения будут описаны со ссылкой на следующие схемы реакций 1-10.
Схема Реакции 1
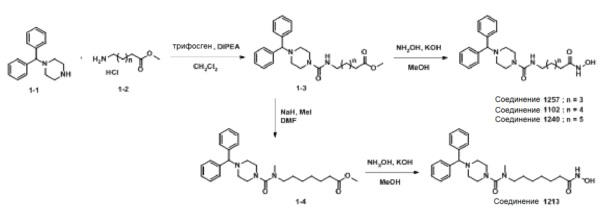
Как показано на приведенной выше схеме реакции 1, соединение формулы 1-1 подвергают взаимодействию с гидрохлоридом метил-6-аминогексаноата, гидрохлоридом метил-7-аминогептаноата или гидрохлоридом метил-8-аминооктаноата (формула 1-2) для синтеза соединения формулы 1-3 в реакции образования мочевины. Гидроксид калия (КОН), метанол и водный раствор гидроксиламина добавляют к соединению формулы 1-3 и подвергают взаимодействию при комнатной температуре, синтезируя, таким образом, конечные соединения 1102, 1240 и 1257.
Кроме того, соединение формулы 1-3, с введенным в него метил-7-аминогептаноатом, подвергают взаимодействию с иодометаном для синтеза соединения формулы 1-4. Гидроксид калия (КОН), метанол и водный раствор гидроксиламина добавляют к соединению формулы 1-4, и реакцию проводят при комнатной температуре комнатной температуре, синтезируя, таким образом, конечное соединение 1213.
Схема Реакции 2
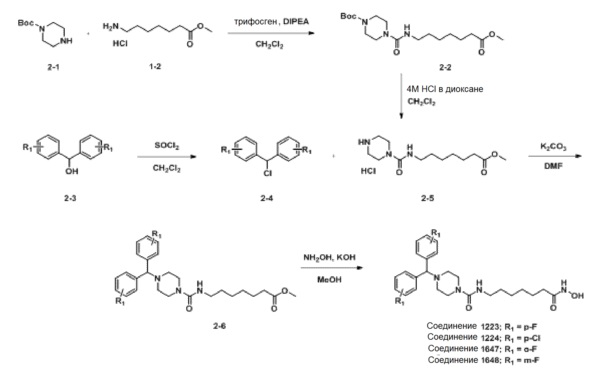
Как показано на приведенной выше схеме реакции 2, соединение формулы 2-1 подвергают взаимодействию с гидрохлоридом метил-7-аминогептаноата (формула 1-2) для синтеза соединения формулы 2-2 в реакции образования мочевины, а затем обрабатывают 4М раствором соляной кислоты для удаления аминозащитной группы (Boc), синтезируя, таким образом, соединение формулы 2-5. Соединение формулы 2-3 подвергают воздействию тионилхлорида с получением соединения формулы 2-4, которое затем подвергают реакции замещения с использованием соединения формулы 2-5 для синтеза соединения формулы 2-6. Гидроксид калия (КОН), метанол и водный раствор гидроксиламина добавляют к соединению формулы 2-6, и реакцию проводят при комнатной температуре комнатной температуре, синтезируя, таким образом, конечные соединения 1223, 1224, 1647 и 1648.
Схема Реакции 3
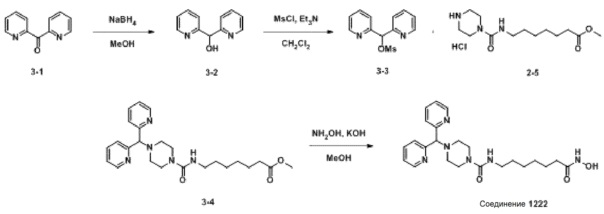
Как показано на приведенной выше схеме реакции 3, соединение формулы 3-1 восстанавливают боргидридом натрия для синтеза соединения формулы 3-2, которое затем подвергают взаимодействию с метансульфонилхлоридом с получением соединения формулы 3-3. Соединение формулы 3-3 подвергают реакции замещения с соединением формулы 2-5 для синтеза соединения формулы 3-4. Затем гидроксид калия (КОН), метанол и водный раствор гидроксиламина добавляют к соединению формулы 3-4, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1222.
Схема Реакции 4
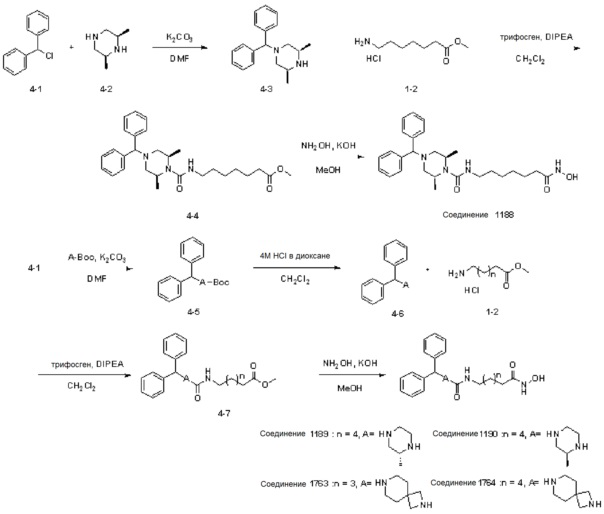
Как показано на приведенной выше схеме реакции 4, соединение формулы 4-1 подвергают реакции замещения с использованием (2S,6R)-2,6-диметилпиперазина (формула 4-2) для синтеза соединения формулы 4-3, которое затем подвергают взаимодействию с гидрохлоридом метил-7-аминогептаноата в реакции образования мочевины для синтеза соединения формулы 4-4. Затем к соединению формулы 4-4 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1188.
Кроме того, соединение формулы 4-1 подвергают взаимодействию с A-Boc-соединением, и затем обрабатывают 4М раствором соляной кислоты для удаления защитной группы (Boc), синтезируя, таким образом, соединение формулы 4-6. Соединение формулы 4-6 подвергают взаимодействию с соединением формулы 1-2 в реакции образования мочевины для синтеза соединения формулы 4-7. Затем к соединению формулы 4-7 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечные соединения 1189, 1190, 1763 и 1764.
Схема Реакции 5
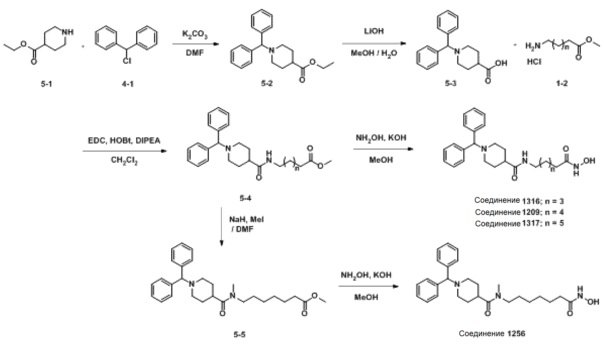
Как показано на приведенной выше схеме реакции 5, соединение формулы 5-1 подвергают реакции замещения с использованием (хлорметилен)дибензола (формула 4-1) для синтеза соединения формулы 5-2, которое затем гидролизуют гидроксидом лития (LiOH) для синтеза соединения формулы 5-3. Соединение формулы 5-3 подвергают амидному взаимодействию с гидрохлоридом метил-6-аминогексаноата, гидрохлоридом метил-7-аминогептаноат или гидрохлоридом метил-8-аминооктаноата для синтеза соединения формулы 5-4. Затем к соединению формулы 5-4 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечные соединения 1209, 1316 и 1317.
Кроме того, соединение формулы 5-4, с введенным него метил-7-аминогептаноатом, подвергают взаимодействию с иодометаном для синтеза соединения формулы 5-5. Затем к соединению формулы 5-5 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1256.
Схема Реакции 6
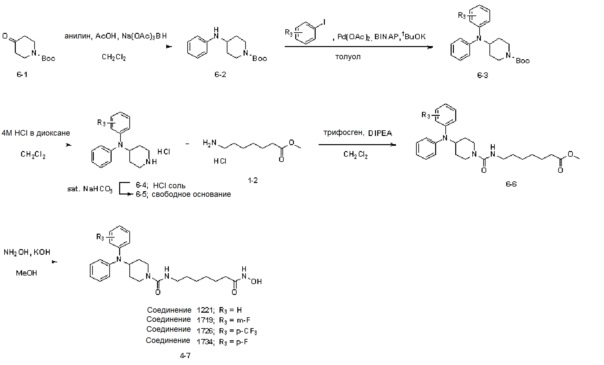
Как показано на приведенной выше схеме реакции 6, соединение формулы 6-1 подвергают восстановительному аминированию с использованием анилина для синтеза соединения формулы 6-2, которое затем подвергают реакции Бухвальда для синтеза соединения формулы 6-3. Соединение формулы 6-3 обрабатывают раствором 4М соляной кислоты для удаления аминозащитной группы (Boc), и затем подвергают взаимодействию с насыщенным раствором бикарбоната натрия для синтеза соединения формулы 6-5. Соединение формулы 6-5 подвергают взаимодействию с гидрохлоридом метил-7-аминогептаноата (формула 1-2) в реакции образования мочевины для синтеза соединения формулы 6-6. Затем к соединению формулы 6-6 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечные соединения 1221, 1719, 1726 и 1734.
Схема Реакции 7
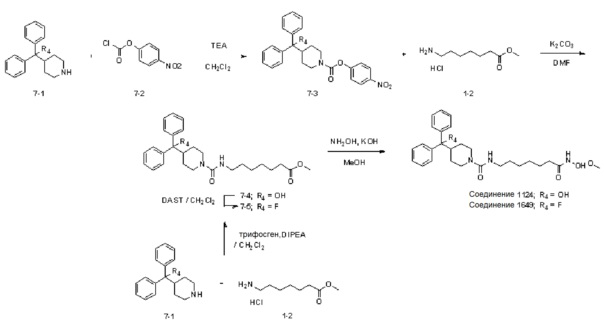
Как показано на схеме реакции 7, соединение формулы 7-1 подвергают взаимодействию с 4-нитрофенилкарбохлоридатом (формула 7-2) для синтеза соединения формулы 7-3, которое затем подвергают реакции замещения с использованием гидрохлорида метил-7-аминогептаноата (формула 1-2) для синтеза соединения формулы 7-4. Затем к соединению формулы 7-4 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1124.
Кроме того, соединение формулы 7-1 подвергают взаимодействию с гидрохлоридом метил-7-аминогептаноата (формула 1-2) в реакции образования мочевины для синтеза соединения формулы 7-4, которое затем подвергают взаимодействию с трифторидом диэтиламиносеры (DAST) для синтеза соединения формулы 7-5. Затем к соединению формулы 7-5 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1649.
Схема Реакции 8
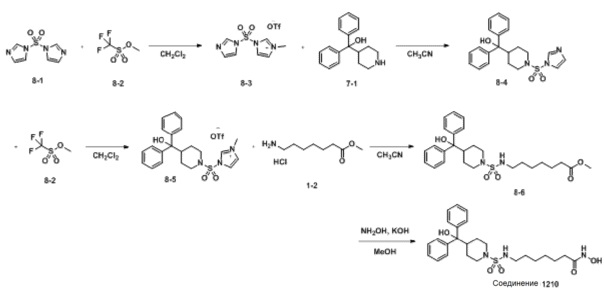
Как показано на приведенной выше схеме реакции 8, соединение формулы 8-1 подвергают взаимодействию с метилтрифторметансульфонатом (формула 8-2) для синтеза соединения формулы 8-3. Соединение формулы 8-3 подвергают взаимодействию с дифенил(пиперидин-4-ил)метанолом (формула 7-1) для синтеза соединения формулы 8-4, которое затем подвергают взаимодействию с метилтрифторметансульфонатом (формула 8-2) для синтеза соединения формулы 8-5. Соединение формулы 8-5 подвергают взаимодействию с гидрохлоридом метил-7-аминогептаноата (формула 1-2) для синтеза соединения формулы 8-6. Затем к соединению формулы 8-6 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1210.
Схема Реакции 9
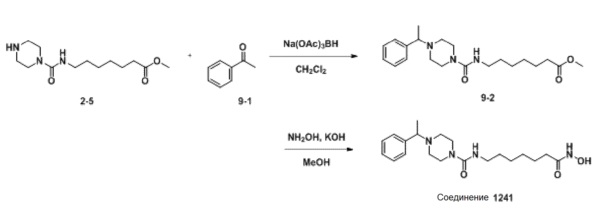
Как показано на приведенной выше схеме реакции 9, соединение формулы 2-5 подвергают восстановительному аминированию с использованием ацетофенона для синтеза соединения формулы 9-2. Затем к соединению формулы 9-2 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1241.
Схема Реакции 10
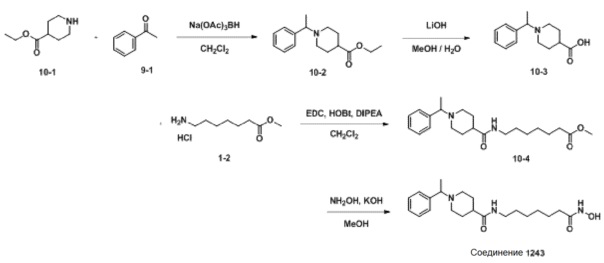
Как показано на приведенной выше схеме реакции 10, соединение формулы 10-1 подвергают восстановительному аминированию с использованием ацетофенона для синтеза соединения формулы 10-2, которое затем гидролизуют с использованием гидроксида лития (LiOH) для синтеза соединения формулы 10-3. Соединение формулы 10-3 подвергают амидному связыванию с гидрохлоридом метил-7-аминогептаноата для синтеза соединения формулы 10-4. Затем к соединению формулы 10-4 добавляют гидроксид калия (КОН), метанол и водный раствор гидроксиламина, и реакцию проводят при комнатной температуре, синтезируя, таким образом, конечное соединение 1243.
Полезные эффекты и технический результат изобретения
Соединения, представленные формулой I в соответствии с настоящим изобретением, их оптические изомеры или их фармацевтически приемлемые соли могут селективно ингибировать HDAC, и таким образом проявляют превосходное действие в отношении предотвращения или лечения опосредованных гистондеацетилазой заболеваний.
Краткое описание фигур
На Фигуре 1 показаны результаты анализа влияния соединения 1102 на облегчение симптомов артрита в моделях индуцированного адъювантом артрита.
Варианты выполнения изобретения
Далее будут представлены предпочтительные примеры, предназначенные для облегчения понимания настоящего изобретения. Однако эти примеры представлены только для лучшего понимания настоящего изобретения, и они не предназначены для ограничения объема притязаний настоящего изобретения.
Реагенты и растворители, упомянутые ниже, были приобретены в Sigma-Aldrich и TCI, если не указано иное, и ВЭЖХ выполняли с использованием установки Waters e2695. В качестве силикагеля для колоночной хроматографии использовали силикагель (230-400 меш) от Merck. Результаты 1H-ЯМР были получены с использованием установки Bruker 400 MHz, и масс-спектры были получены с использованием установки серии Agilent 1100.
Пример 1: Синтез соединения 1102
Стадия 1: Синтез метил-7-(4-бензгидрилпиперазин-1-карбоксамидо)гептаноата (формула 1-3)
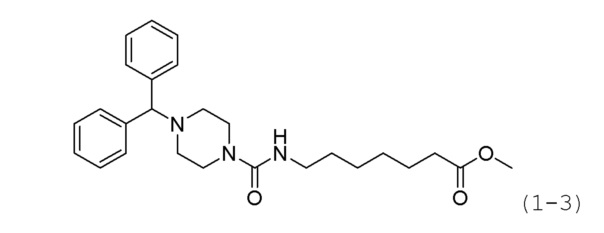
Растворяли 1-бензгидрилпиперазин (0,200 г, 0,793 ммоль), метил-7-аминогептаноат (0,151 г, 0,951 ммоль), трифосген (0,118 г, 0,396 ммоль) и DIPEA (0,415 мл, 2,378 ммоль) в хлористом метилене (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (Waters, C18; водный раствор 1%-муравьиной кислоты (метановая кислота)/ацетонитрил=от 100 до 20%) и концентрировали путем пропускания через картридж SPE (смола PL-HCO3), получая при этом требуемое соединение формулы 1-3 (0,075 г, 21,6%) в виде светло-желтого масла.
Стадия 2: Синтез 4-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1102)

Соединение 1102
Соединение формулы 1-3 (0,075 г, 0,171 ммоль) полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,210 мл, 3,428 ммоль) и гидроксид калия (0,096 г, 1,714 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия (20 мл) с последующим перемешиванием. Осажденное твердое вещество отфильтровывали, промывали водой и сушили, получая, таким образом, целевое соединение 1102 (0,047 г, 62,5%) в виде желтого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6 ) δ 9,39 (уш.с, 1H), 7,42 (д, 4H, J=7,2 Гц), 7,29 (т, 4Н, J=7,5), 7,18 (т, 2H, J=7,3 Гц), 6,40 (т, 1H, J=5,3 Гц), 4,28 (с, 1H), 3,27 (с, 4Н), 2,98-2,93 (м, 2H), 2,08 (с, 4H), 1,89 (т, 2H, J=7,3 Гц), 1,44-1,43 (м, 2Н), 1,34-1,33 (м, 2H), 1,20 (с, 4Н); МS (ESI) m/z 439,6 (M++H).
Пример 2: Синтез соединения 1124
Стадия 1: Синтез 4-нитрофенил-4-(гидроксидифенилметил)пиперидин-1-карбоксилата (формула 7-3)
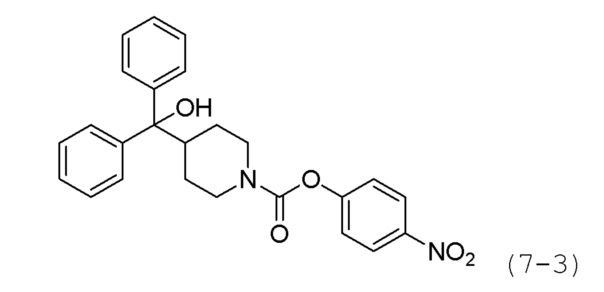
Дифенил (пиперидин-4-ил)метанол (0,100 г, 0,374 ммоль) и триэтиламин (0,104 мл, 0,748 ммоль) растворяли в метиленхлориде (5 мл) при 0°С, и к раствору добавляли 4-нитрофенилхлорформиат (0,083 г, 0,411 ммоль) с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 0 до 20%), и концентрировали с получением целевого соединения формулы 7-3 (0,152 г, 94,0%) в виде бесцветного масла.
Стадия 2: Синтез метил-7-(4-(гидроксидифенилметил)пиперидин-1-карбоксамидо)гептаноата (формула 7-4)
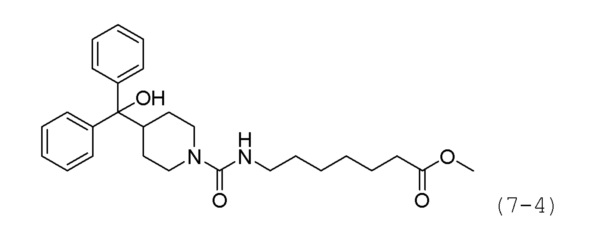
Соединение формулы 7-3 (0,152 г, 0,351 ммоль), полученное на стадии 1, метил-7-аминогептаноат гидрохлорид (0,280 г, 1,757 ммоль) и карбонат калия (0,097 г, 0,703 ммоль) растворяли в N,N-диметилформамиде (5 мл) при комнатной температуре, и раствор перемешивали при 100°С в течение 17 часов. Затем температуру понижали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 10% до 40%) и концентрировали, получая при этом соединение формулы 7-4 (0,075 г, 39,4%) в виде оранжевого масла.
Стадия 3: N-(7-(гидроксиамино)-7-оксогептил)-4-(гидроксидифенилметил)пиперидин-1-карбоксамид (соединение 1124)
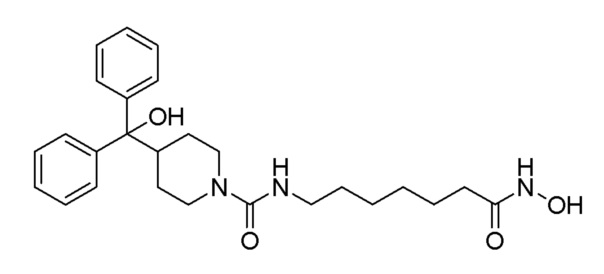
Соединение 1124
Соединение формулы 7-4 (0,075 г, 0,166 ммоль), полученное на стадии 2, гидроксиламин (50,00% водный раствор, 0,203 мл, 3,314 ммоль) и гидроксид калия (0,093 г, 1,657 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивают при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Осажденное твердое вещество отфильтровывали, промывали гексаном, и сушили с получением целевого соединения 1124 (0,007 г, 9,3%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,36 (уш.с, 1H), 7,52 (д, 4Н, J=7,6 Гц), 7,27 (т, 4H, J=7,7 Гц), 7,13 (т, 2H, J=7,3 Гц), 6,30 (т, 1H, J=5,3 Гц), 5,32 (уш.с, 1H), 3,94 (д, 2H, J=13,4 Гц), 2,99-2,94 (м, 2Н), 2,67-2,58 (м, 3H), 1,91 (т, 2H, J=7,4 Гц), 1,48-1,46 (м, 2H), 1,35-1,34 (м, 2H), 1,30-1,25 (м, 6H).
Пример 3: Синтез соединения 1188
Стадия 1: Синтез (3S,5R)-1-бензгидрил-3,5-диметилпиперазина (соединение 4-3)
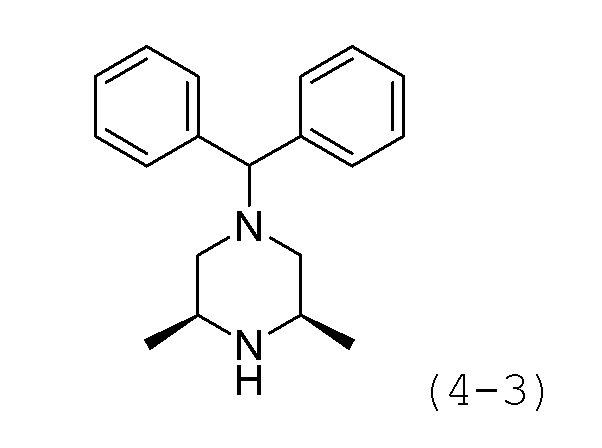
(2R,6S)-2,6-диметилпиперазин (1,000 г, 8,757 ммоль), (хлорметилен)дибензол (3,550 г, 17,515 ммоль) и карбонат калия (6,052 г, 43,787 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; метанол/метиленхлорид=от 0 до 10%) и концентрировали с получением целевого соединения формулы 4-3 (0,798 г, 32,5%) в виде белого твердого вещества.
Стадия 2: Синтез метил-7-((2S,6R)-4-бензгидрил-2,6-диметилпиперазин-1-карбоксамидо)гептаноата (формула 4-4)
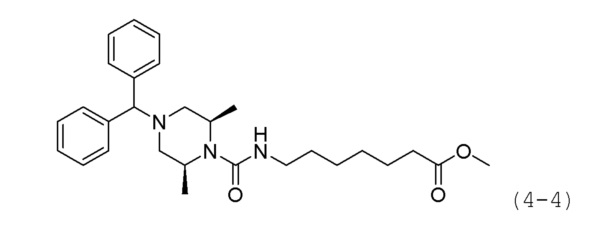
Трифосген (0,159 г, 0,535 ммоль) и диизопропиламин (0,561 мл, 3,210 ммоль) растворяли в метиленхлориде (5 мл) при 0°С и к раствору добавляли гидрохлорид метил-7-аминогептаноата (0,251 г, 1,284 ммоль) с последующим перемешиванием при той же температуре. Соединение формулы 4-3 (0,300 г, 1,070 ммоль) полученное на стадии 1, добавляли к реакционной смеси, с последующим перемешиванием при той же температуре в течение 30 минут. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 30%) и концентрировали с получением целевого соединения формулы 4-4 (0,212 г, 42,6%) в виде белого твердого вещества.
Стадия 3: Синтез (2S,6R)-4-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-2,6-диметилпиперазин-1-карбоксиамида (соединение 1188)
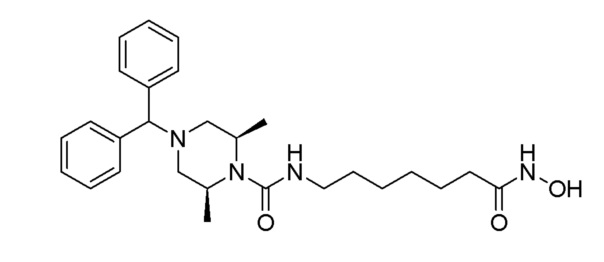
Соединение 1188
Соединение формулы 4-4 (0,100 г, 0,215 ммоль) полученное на стадии 2, гидроксиламин (50,00% водный раствор, 0,263 мл, 4,295 ммоль) и гидроксид калия (0,121 г, 2,148 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1188 (0,099 г, 98,8%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,51 (д, 4H, J=7,5 Гц), 7,30 (т, 4H, J=7,6 Гц), 7,19 (т, 2H, J=7,3 Гц), 6,23 (т, 1H, J=5,3 Гц), 4,23 (с, 1H), 3,94 (уш.с, 2H), 3,03-3,98 (м, 2H), 2,60 (д, 2H, J=10,9 Гц), 1,96-1,90 (м, 4H), 1,46-1,45 (м, 2Н), 1,38-1,36 (м, 2H), 1,26-1,22 (м, 10H).
Пример 4: Синтез соединения 1189
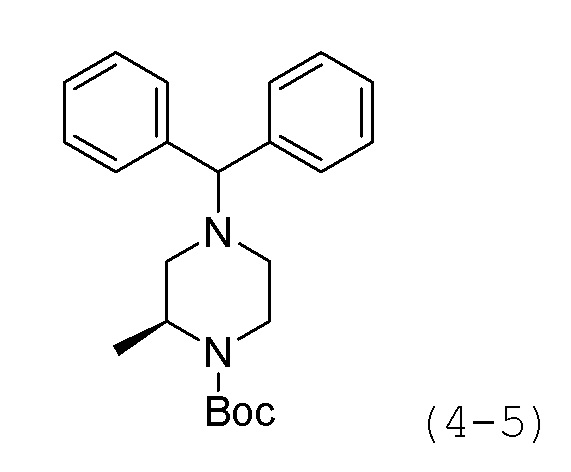
Стадия 1: Синтез трет-бутил-(R)-4-бензгидрил-2-метилпиперазин-1-карбоксилата (формула 4-5)
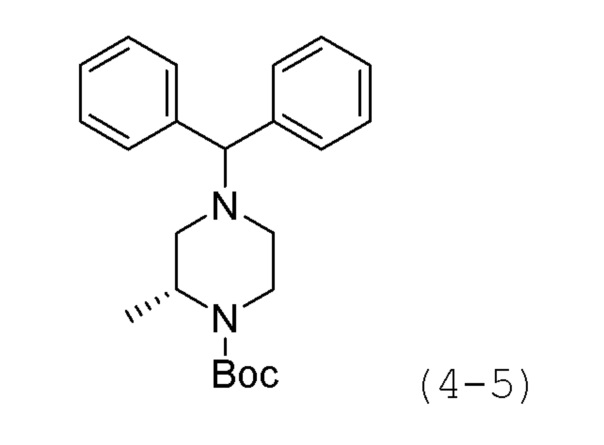
(R)-трет-бутил-2-метилпиперазин-1-карбоксилат (1,000 г, 4,993 ммоль), (хлорметилен)дибензол (2,024 г, 9,986 ммоль) и карбонат калия (3,450 г, 24,965 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре, раствор перемешивали при 80°С в течение 17 часов и затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 10%) и концентрировали с получением целевого соединения формулы 4-5 (0,813 г, 44,4%) в виде белого твердого вещества.
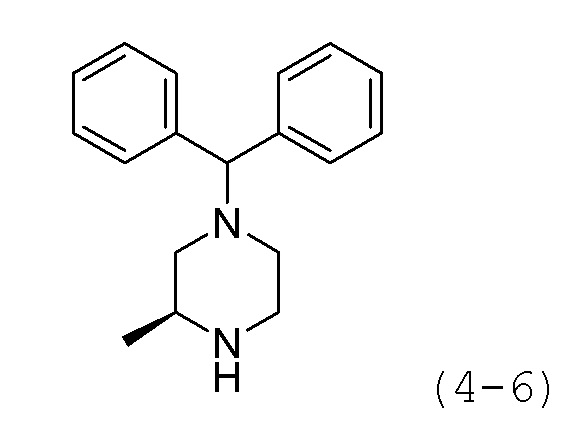
Стадия 2: Синтез (R)-1-бензгидрил-3-метилпиперазина (формула 4-6)
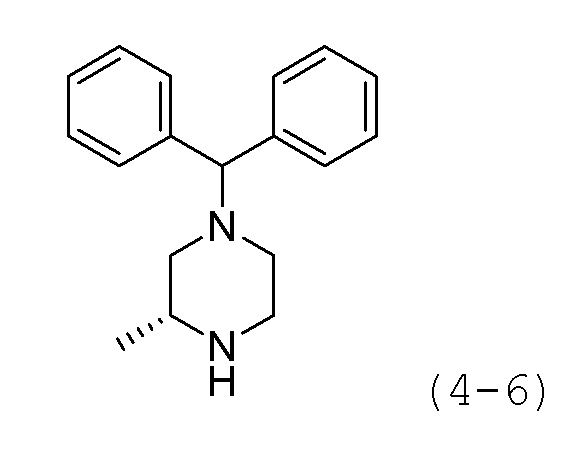
Соединение формулы 4-5 (0,813 г, 2,218 ммоль), полученное на стадии 1, растворяли в метиленхлориде (10 мл) при комнатной температуре, и к раствору добавляли соляную кислоту (4,00М раствор в диоксане, 5,546 мл, 22,183 ммоль), с последующим перемешиванием при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Целевое соединение формулы 4-6 (0,590 г, 99,8%) получали в виде белого твердого вещества без дополнительной очистки.
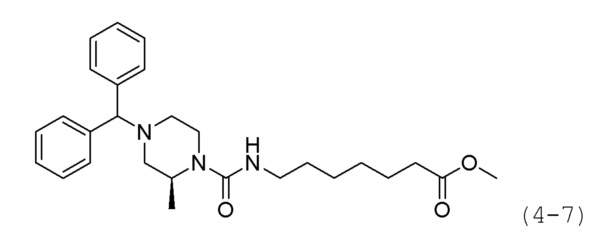
Стадия 3: Синтез метил (R)-7-(4-бензгидрил-2-метилпиперазин-1-карбоксамидо)гептаноата (формула 4-7)
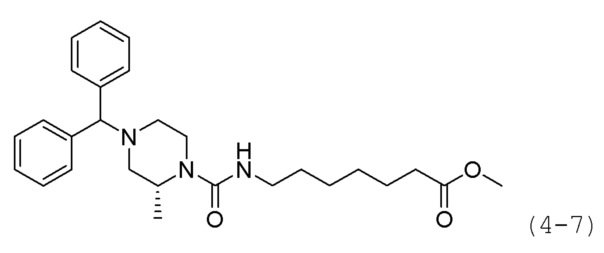
Трифосген (0,167 г, 0,563 ммоль) и DIPEA (1,180 мл, 6,757 ммоль) растворяли в метиленхлориде (5 мл) при 0°С и к раствору добавляли гидрохлорид метил-7-аминогептаноата (0,264 г, 1,351 ммоль) с последующим перемешиванием при той же температуре. Соединение формулы 4-6 (0,300 г, 1,126 ммоль) добавляли к реакционной смеси с последующим перемешиванием при той же температуре в течение 30 минут. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 30%) и концентрировали. Затем концентрат снова очищали с помощью хроматографии (Waters, С18; 1%-муравьиная кислота (метановая кислота) водный раствор/ацетонитрил=от 75% до 5%), и концентрировали путем пропускания через картридж SPE (смола PL-HCO3) с получением целевого соединения формулы 4-7 (0,106 г, 20,8%)
Стадия 4: Синтез (R)-4-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-2-метилпиперазин-1-карбоксамида (соединение 1189)
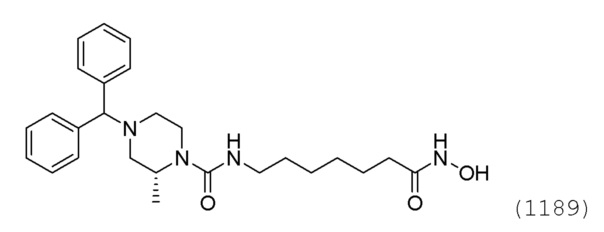
Соединение 1189
Соединение формулы 4-7 (0,100 г, 0,221 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 0,271 мл, 4,429 ммоль) и гидроксид калия (0,124 г, 2,214 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1189 (0,099 г, 98,8%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,42 (уш.с, 2H), 7,45 (т, 4H, J=6,3 Гц), 7,30 (т, 4H, J=7,6 Гц), 7,19 (т, 2H, J=6,8 Гц), 6,36-6,34 (м, 1H), 4,23 (с, 1Н), 4,04 (уш.с, 1H), 3,62 (д, 1H, J=12,4 Гц), 3,01-3,93 (м, 3H), 2,67 (д, 1H, J=9,6 Гц), 2,60 (д, 1H, J=10,8 Гц), 1,95 (дд, 1Н, J=11,0, 3,0 Гц,), 1,88 (т, 2H, J=7,3 Гц), 1,78 (т, 1H, J=10,1 Гц), 1,44-1,43 (м, 2H), 1,36-1,35 (м, 2Н), 1,20-1,18 (м, 7Н).
Пример 5: Синтез соединения 1190
Стадия 1: Синтез трет-бутил-(S)-4-бензгидрил-2-метилпиперазин-1-карбоксилата (формула 4-5)
(S)-трет-бутил-2-метилпиперазин-1-карбоксилат (1,000 г, 4,993 ммоль), (хлорметилен)дибензол (2,024 г, 9,986 ммоль) и карбонат калия (3,450 г, 24,965 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре, и раствор перемешивали при 80°С в течение 17 часов, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 10%) и концентрировали с получением целевого соединения формулы 4-5 (0,742 г, 40,5%) в виде белого твердого вещества.
Стадия 2: Синтез (S)-1-бензгидрил-3-метилпиперазина (формула 4-6)
Соединение формулы 4-5 (0,742 г, 2,025 ммоль), полученное на стадии 1, растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли соляную кислоту (4,00М раствор в диоксане, 5,061 мл, 20,246 ммоль) с последующим перемешиванием при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Продукт (0,530 г, 98,3%, белое твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил-(S)-7-(4-бензгидрил-2-метилпиперазин-1-карбоксамидо)гептаноата (формула 4-7)
Трифосген (0,111 г, 0,375 ммоль) и DIPEA (0,582 г, 4,505 ммоль) растворяли в метиленхлориде (5 мл) при 0°С и к раствору добавляли гидрохлорид метил-7-аминогептаноата (0,176 г, 0,901 ммоль), с последующим перемешиванием при той же температуре. Соединение формулы 4-6 (0,200 г, 0,751 ммоль), полученное на стадии 2, добавляли к реакционной смеси, с последующим перемешиванием при той же температуре в течение 30 минут. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 30%) и концентрировали с получением целевого соединения 4-7 (0,213 г, 62,8%) в виде светло-желтого масла.
Стадия 4: Синтез (S)-4-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-2-метилпиперазин-1-карбоксамида (соединение 1190)
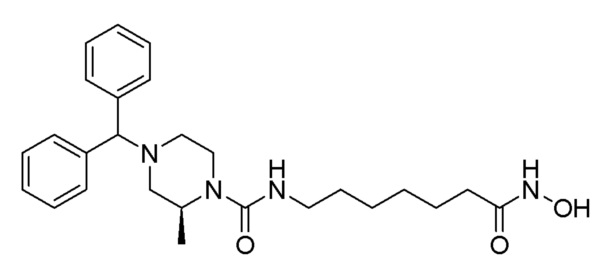
Соединение 1190
Соединение формулы 4-7 (0,100 г, 0,221 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 0,271 мл, 4,429 ммоль) и гидроксид калия (0,124 г, 2,214 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1190 (0,093 г, 92,8%) в виде светло-оранжевого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,43 (уш.с, 2H), 7,45 (т, 4H, J=6,3 Гц), 7,30 (т, 4H, J=7,6 Гц), 6,35 (т, 1H, J=5,5 Гц), 4,23 (с, 1Н), 4,04 (уш.с, 1H), 3,62 (д, 1H, J=12,6 Гц), 3,03-3,92 (м, 3H), 2,67 (д, 1H, J=10,6 Гц), 2,60 (д, 1H, J=11,2 Гц), 1,95 (дд, 1Н, J=11,1, 3,1 Гц,), 1,88 (т, 2H, J=7,4 Гц), 1,80-1,75 (м, 1H), 1,45-1,43 (м, 2Н), 1,36-1,34 (м, 2H), 1,20-1,18 (м, 7H).
Пример 6: Синтез соединения 1209
Стадия 1: Синтез этил-1-бензгидрилпиперидин-4-карбоксилата (формула 5-2)

Этилпиперидин-4-карбоксилат (3,000 г, 19,83 ммоль), (хлорметилен)дибензол (5,802 г, 28,624 ммоль) и карбонат калия (13,187 г, 95,414 ммоль) растворяли в N,N-диметилформамиде (50 мл), и раствор перемешивали при комнатной температуре в течение 17 часов, а затем перемешивали при 80°С в течение 3 часов. Затем раствор охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 40 г; этилацетат/гексан=от 0 до 15%) и концентрировали с получением целевого соединения формулы 5-2 (1,410 г, 22,8%) в виде бесцветного масла.
Стадия 2: Синтез 1-бензгидрилпиперидин-4-карбоновой кислоты (формула 5-3)
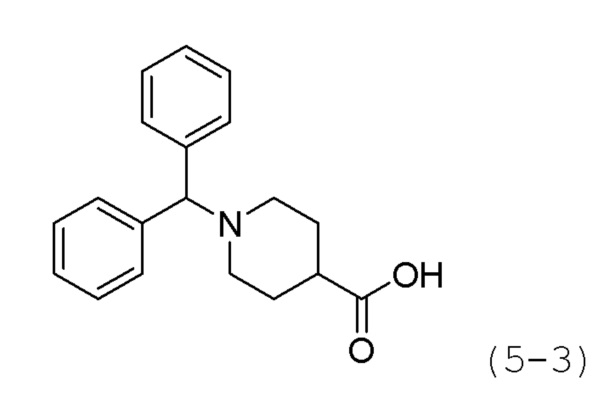
Соединение формулы 5-2 (1,410 г, 4,360 ммоль), полученное на стадии 1, и LiOH (0,209 г, 8,719 ммоль) растворяли в смеси метанол (10 мл)/вода (5 мл) при комнатной температуре, и раствор перемешивали при 60°С в течение 17 часов, а затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, затем нейтрализовали водным раствором 1Н соляной кислоты и концентрировали при пониженном давлении для удаления растворителя. Продукт (1,300 г, 101,0%, белое твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил 7-(1-бензгидрилпиперидин-4-карбоксамидо)гептаноата (формула 5-4)
Соединение формулы 5-3 (1,500 г, 5,078 ммоль), полученное на стадии 2, метил-7-аминогептаноат (1,988 г, 10,156 ммоль), EDC (1,947 г, 10,156 ммоль), HBOt (1,372 г, 10,156 ммоль) и диизопропиламин (4,435 мл, 25,391 ммоль) растворяли в метиленхлориде (30 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 40 г; этилацетат/гексан=от 0 до 40%) и концентрировали с получением целевого соединения формулы 5-4 (1,810 г, 81,6%) в виде бесцветного масла.
Стадия 4: Синтез 1-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)пиперидин-4-карбоксамида (соединение 1209)
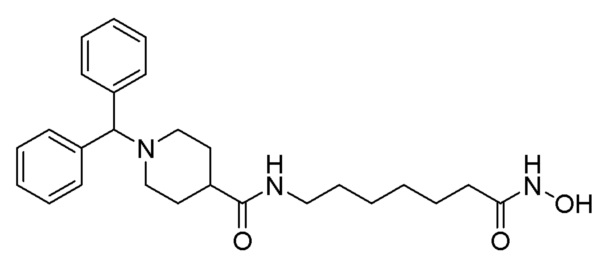
Соединение 1209
Соединение формулы 5-4 (1,000 г, 2,290 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 2,802 мл, 45,809 ммоль) и гидроксид калия (1,285 г, 22,904 ммоль) растворяли в метаноле (15 мл) при 0°С и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении
Целевое соединение 1209 (1,000 г, 99,8%) получали в виде светло-оранжевого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,71 (т, 1Н, J=5,4 Гц), 7,40 (д, 4H, J=7,3 Гц), 7,27 (т, 4 Н, J=7,5 Гц), 7,16 (т, 2H, J=7,3 Гц), 4,25 (с, 1H), 2,98 (кв, 2Н, J=6,4 Гц), 2,79 (d, 2H, J=11,0 Гц), 2,09-2,02 (м, 1H), 1,89 (т, 2H, J=7,3 Гц), 1,77 (т, 2H, J=9,8), 1,66 ~ 1,59 (м, 4Н), 1,45-1,39 (м, 4Н), 1,45-1,39 (м, 2Н) (м, 2H), 1,34 ~ 1,32 (м, 2H), 1,29 ~ 1,27 (м, 4 H); MS (ESI) m/z 438,2 (М++H).
Пример 7: Синтез соединения 1210
Стадия 1: Синтез 1-((1Н-имидазол-1-ил)сульфонила)-3-метил-1H-имидазол-3-ий трифторметансульфоната (формула 8-3)
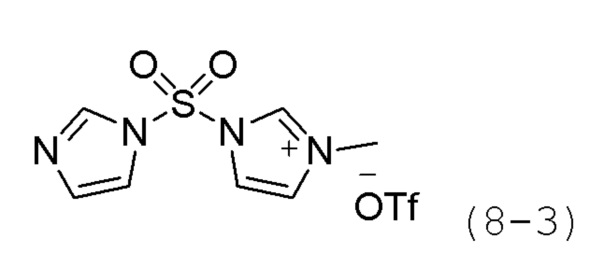
В хлористом метилене (100 мл) при комнатной температуре растворяли 1,1'-сульфонилбис(1Н-имидазол) (5,000 г, 25,227 ммоль) и метилтрифторметансульфонат (2,855 мл, 25,227 ммоль), и раствор перемешивали при той же температуре в течение 3 часов. Осажденное твердое вещество отфильтровывали и сушили, получая целевое соединение формулы 8-3 (5,160 г, 45,3%) в виде светло-желтого масла.
Стадия 2: Синтез (1-(1H-имидазол-1-ил)сульфонил)пиперидин-4-ил)дифенилметанола (формула 8-4)
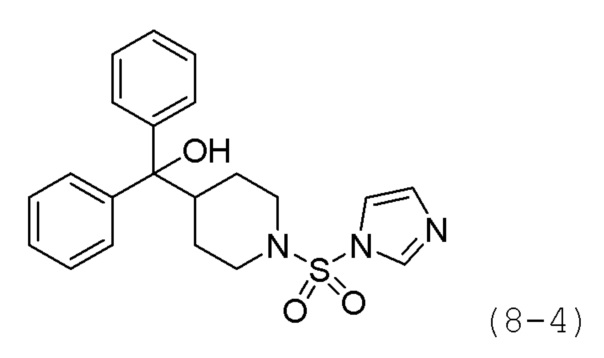
Дифенил(пиперидин-4-ил)метанол (1,000 г, 3,740 ммоль) и соединение формулы 8-3 (2,033 г, 5,610 ммоль), полученное на стадии 1, растворяли в ацетонитриле (20 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении, и концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 40%) и концентрировали с получением целевого соединения формулы 8-4 (0,487 г, 32,8%) в виде белого твердого вещества.
Стадия 3: Синтез (1-((3-метил-1Н-3-иум-имидазол-1-ил)сульфонил)пиперидин-4-ил)дифенилметантрифторметансульфоната (формула 8-5)
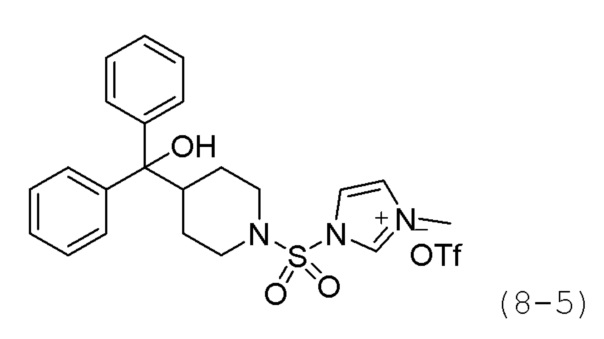
Соединение формулы 8-4 (0,487 г, 1,225 ммоль), полученное на стадии 2, и метилтрифторметансульфонат (0,146 мл, 1,286 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 2 часов. Выпавшее в осадок твердое вещество отфильтровывали, промывали метиленхлоридом и сушили, получая целевое соединение формулы 8-5 (0,670 г, 97,4%) в виде белого твердого вещества.
Стадия 4: Синтез метил-7-((4-(гидроксидифенилметил)пиперидин)-1-сульфонамидо)гепананоата (формула 8-6)
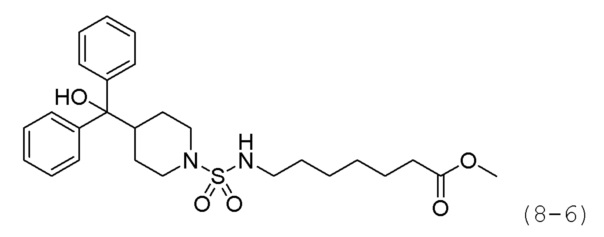
Соединение формулы 8-5 (0,504 г, 0,897 ммоль), полученное на стадии 3, и гидрохлорид метил-7-аминогептаноата (0,228 г, 1,167 ммоль) растворяли в ацетонитриле (3 мл) при 80°С, и раствор перемешивали при той же температуре в течение 12 часов, а затем охлаждали до комнатной температуры для прекращения реакции. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 10% до 60%) и концентрировали с получением целевого соединения формулы 8-6 (0,147 г, 33,5%) в виде белого твердого вещества.
Стадия 5: Синтез N-гидрокси-7-((4-(гидроксидифенилметил) пиперидин)-1-сульфонамидо)гептанамид (соединение 1210)
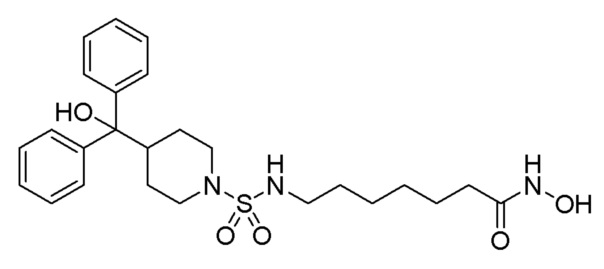
Соединение 1210
Соединение формулы 8-6 (0,150 г, 0,307 ммоль), полученное на стадии 4, гидроксид калия (0,172 г, 3,070 ммоль) и гидроксиламин (50,00% раствор, 0,188 мл, 3,070 ммоль) растворяли в метаноле (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Целевое соединение 1210 (0,067 г, 44,6%) получали в виде белого твердого вещества и использовали без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,34 (с, 1H), 8,68 (с, 1H), 7,52 (д, 4H, J=7,4 Гц), 7,26 (т, 4 Н, J=7,6 Гц,), 7,12 (м, 3H), 3,46 (м, 2Н), 2,82 (м, 2, 2H), 2,63 (м, 3H), 1,93 (т, 2H, J=7,3 Гц), 1,48-1,22 (м, 13H); МS (ESI) m/z 490,6 (M++H).
Пример 8: Синтез соединения 1213
Стадия 1: Синтез метил-7-(4-бензгидрил-N-метилпиперизин-1-карбоксамидо)гептаноата (формула 1-4)
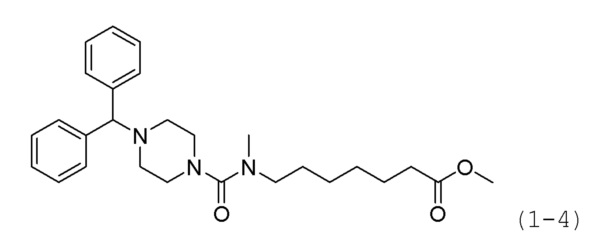
Метил-7-(4-бензгидрилпиперазин-1-карбоксамидо)гептаноат (0,100 г, 0,229 ммоль) и гидрид натрия (60,00%, 0,046 г, 1,143 ммоль) растворяли в N,N-диметилформамиде (3 мл) при 0°С и к раствору добавляли йодметан (0,071 мл, 1,143 ммоль) с последующим перемешиванием при той же температуре в течение 10 минут. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 10% до 40%) и концентрировали с получением целевого соединения формулы 1-4 (0,097 г, 94,0%) в виде бесцветного масла.
Стадия 2: Синтез 4-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-N-метилпиперазин-1-карбоксамида (соединение 1213)
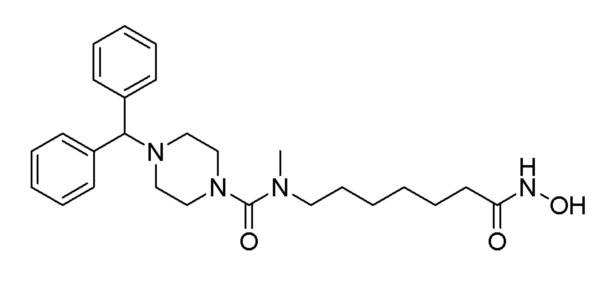
Соединение 1213
Соединение формулы 1-4 (0,097 г, 0,215 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,263 мл, 4,296 ммоль) и гидроксид калия (0,121 г, 2,148 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Целевое соединение 1213 (0,010 г, 10,3%) получали в виде белого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, CD3OD) δ 7,44 (д, 4H, J=7,4 Гц), 7,27 (т, 4Н, J=7,5 Гц), 7,17 (т, 2H, J=7,3 Гц), 4,26 (с, 1H), 3,24-3,22 (м, 4H), 3,17 (т, 2H, J=7,2 Гц), 2,81 (с, 3Н), 2,41-2,38 (м, 4H), 2,07 (т, 2H, J=7,4 Гц), 1,62-1,52 (м, 4H), 1,33-1,24 (м, 4H); MS (ESI) m/z 453,4 (M++H).
Пример 9: Синтез соединения 1221
Стадия 1: Синтез гидрохлорида N,N-дифенилпиперидин-4-амина (формула 6-4)
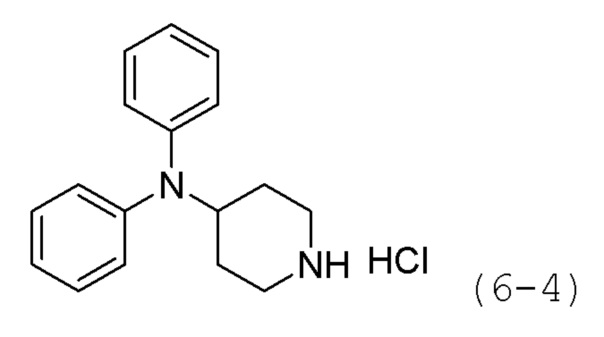
Трет-бутил-4-(дифениламино)пиперидин-1-карбоксилат (1,000 г, 2,837 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли соляную кислоту (4,00 М раствор в 1,4-диоксане, 3,546 мл, 14,185 ммоль) с последующим перемешиванием при той же температуре в течение 17 часов. Выпавшее в осадок твердое вещество отфильтровывали, промывали метиленхлоридом и сушили с получением целевого соединения формулы 6-4 (0,800 г, 97,6%) в виде белого твердого вещества.
Стадия 2: Синтез N,N-дифенилпиперидин-4-амина (формула 6-5)
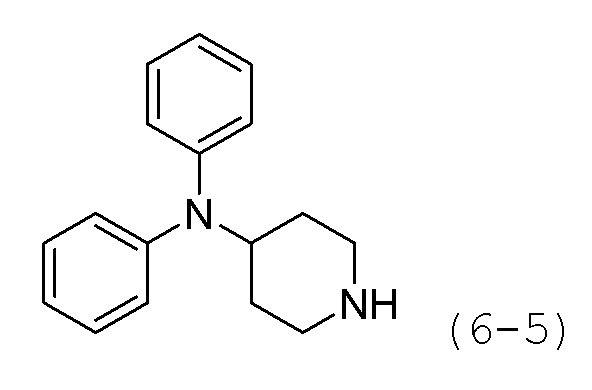
Соединение формулы 6-4 (0,600 г, 2,077 ммоль), полученное на стадии 1, растворяли в воде (5 мл) при комнатной температуре и к раствору добавляли насыщенный водный раствор бикарбоната натрия (50 мл) с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Продукт (0,496 г, 94,6%, бесцветное масло) использовали без дополнительной очистки.
Стадия 3: Синтез метил-7-(4-(дифениламино)иперидин-1-карбоксамидо)гептаноата (формула 6-6)
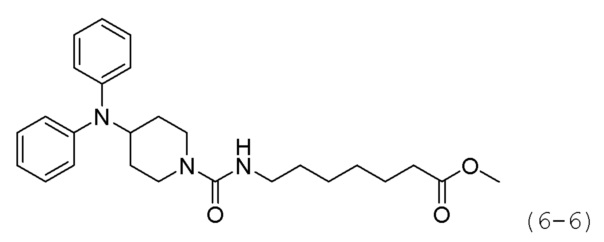
Соединение формулы 6-5 (0,100 г, 0,396 ммоль), полученное на стадии 2, метил-7-аминогептаноат (0,078 г, 0,396 ммоль), трифосген (0,059 г, 0,198 ммоль) и DIPEA (0,415 мл, 2,378 ммоль) растворяли в метиленхлориде (3 мл) при 0°С и раствор перемешивали при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия (50 мл) при 0°С c последующим перемешиванием в течение 10 минут. После завершения реакции к реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 10% до 60%) и концентрировали с получением целевого соединения формулы 6-6 (0,096 г, 55,4%) в виде светло-желтого масла.
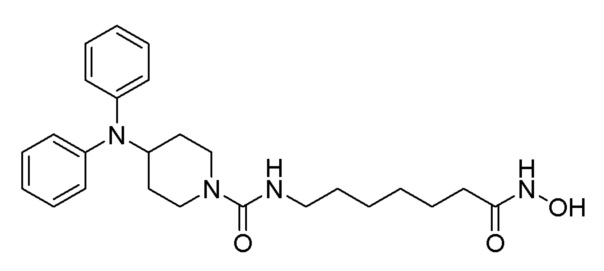
Стадия 4: Синтез 4-(дифениламино)-N-(7-(гидроксиамино)-7-оксогептил)пиперидин-1-карбоксамида (соединение 1221)
Соединение 1221
Соединение формулы 6-6 (0,096 г, 0,219 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 0,268 мл, 4,388 ммоль) и гидроксид калия (0,123 г, 2,194 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия (20 мл) и метиленхлорид (5 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили с получением целевого соединения 1221 (0,076 г, 79,0%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,27 (т, 4H, J=7,8 Гц), 6,97 (т, 2H, J=7,2 Гц), 6,79 (д, 4 Н, J=7,8 Гц), 6,35 (т, 1H, J=5,4 Гц), 4,10-4,04 (м, 1H), 3,97 (д, 2H, J=13,1 Гц), 2,90 (кв, 2H, J=6,4 Гц), 2,78 (т, 2Н, J=12,5 Гц), 1,90 (т, 2H, J=7,3 Гц), 1,84 (д, 2H, J=12,5 Гц), 1,46-1,39 (м, 2H), 1,31-1,27 (м, 2H), 1,17-1,10 (м, 4H), 1,08-1,01 (м, 2H).
Пример 10. Синтез соединения 1222
Стадия 1: Синтез ди(пиридин-2-ил)метанола (формула 3-2)
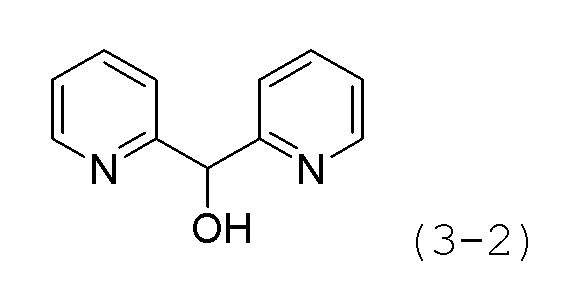
Ди(пиридин-2-ил)метанон (2,000 г, 10,858 ммоль) растворяли в метаноле (20 мл) при 0°С и к раствору добавляли NaBH4 (0,452 г, 11,944 ммоль) с последующим перемешиванием при той же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия (10 мл) при 0°С с последующим перемешиванием в течение 10 минут. После завершения реакции к реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Целевое соединение формулы 3-2 (2,000 г, 98,9%) получали в виде красного масла и использовали без дополнительной очистки.
Стадия 2: Синтез ди(пиридин-2-ил)метилметансульфоната (формула 3-3)
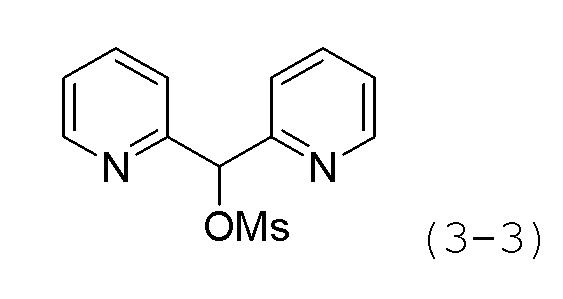
Соединение формулы 3-2 (1,000 г, 5,370 ммоль), полученное на стадии 1, метансульфонилхлорид (0,623 мл, 8,055 ммоль) и триэтиламин (2,246 мл, 16,111 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и раствор перемешивали при той же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 30%) и концентрировали с получением целевого соединения формулы 3-3 (0,670 г, 47,2%) в виде розового твердого вещества.
Стадия 3: Синтез метил-7-(4-(ди(пиридин-2-ил)м-этил)пиперазин-1-карбоксамидо)гептаноата (формула 3-4)
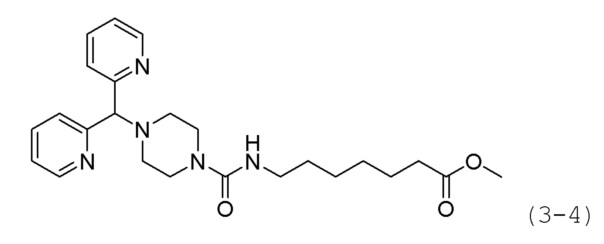
Соединение формулы 3-3 (0,258 г, 0,975 ммоль), полученное на стадии 2, соединение формулы 2-5 (0,200 г, 0,650 ммоль) и карбонат калия (0,449 г, 3,249 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и раствор перемешивали при 80°С в течение 17 часов, а затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0 до 10%) и концентрировали с получением целевого соединения формулы 3-4 (0,255 г, 89,3%) в виде оранжевого масла.
Стадия 4: Синтез 4-(ди(пиридин-2-ил)метил)-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1222)
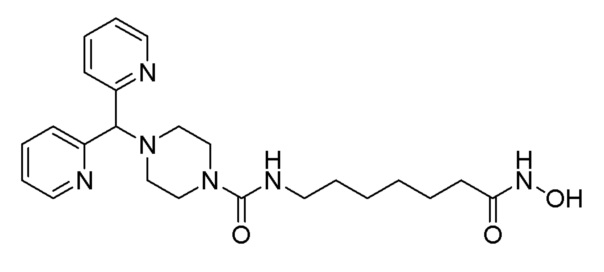
Соединение 1222
Соединение формулы 3-4 (0,255 г, 0,580 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 0,710 мл, 11,603 ммоль) и гидроксид калия (0,326 г, 5,801 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат очищали колоночной хроматографией (Waters, С18; 1%-муравьиная кислота (метановая кислота) водный раствор/водный раствор ацетонитрила=от 70 до 5%) и концентрировали путем пропускания через картридж SPE (смола PL-HCO3), получая при этом целевое соединение 1222 (0,051 г, 20,0%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 8,46 (дт, 2H, J=4,8, 0,8 Гц), 7,77 (тд, 2H, J=7,7, 1,7 Гц), 7,62 (д, 2H, J=7,8 Гц), 7,25-7,22 (м, 2H), 6,40 (т, 1H, J=5,2 Гц), 4,64 (с, 1H), 3,28-3,27 (м, 4H), 2,96 (кв. 2H, J=6,6 Гц), 2,25 (т, 4H, J=4,7 Гц), 1,92 (т, 2H, J=7,3 Гц), 1,48-1,43 (м, 2H), 1,35-1,33 (м, 2H), 1,21-1,20 (м, 4H).
Пример 11: Синтез соединения 1223
Стадия 1: Синтез трет-бутил-4-(7-метокси-7-оксогептил)карбамоил)пиперазин-1-карбоксилата (формула 2-2)

Трифосген (4,780 г, 16,107 ммоль) и диизопропиламин (16,879 мл, 96,644 ммоль) растворяли в метиленхлориде (100 мл) при 0°С и к раствору добавляли гидрохлорид метил-7-аминогептаноата (6,304 г, 32,215 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли трет-бутилпиперазин-1-карбоксилат (6,000 г, 32,215 ммоль) с последующим перемешиванием при такой же температуре в течение 1 часа. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия (100 мл) при 0°С с последующим перемешиванием в течение 10 минут. После завершения реакции к реакционной смеси добавляли воду, с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 80 г; метанол/метиленхлорид=от 0 до 5%) и концентрировали с получением целевого соединения формулы 2-2 (3,430 г, 28,7%) в виде светло-желтого масла.
Стадия 2: Синтез гидрохлорида метил-7-(пиперазин-1-карбоксамидо)гептаноата (формула 2-5)

Соединение формулы 2-2 (3,430 г, 9,233 ммоль), полученное на стадии 1, растворяли в метиленхлориде (50 мл) при комнатной температуре и к раствору добавляли соляную кислоту (4,00 М раствор в диоксане, 11,542 мл, 46,167 ммоль) с последующим перемешиванием при той же температуре в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли этилацетат (50 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали этилацетатом и сушили, получая целевое соединение формулы 2-5 (2,300 г, 80,9%) в виде белого твердого вещества.
Стадия 3: Синтез 4,4'-(хлорметилен)бис(фторбензол) (формула 2-4)
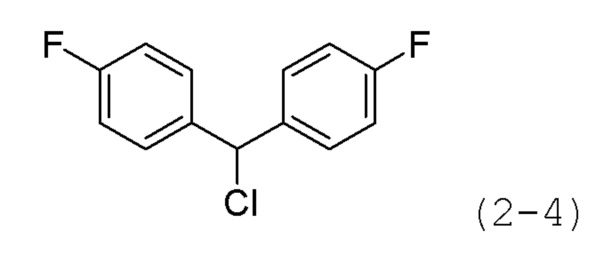
Бис(4-фторфенил)метанол (5,000 г, 22,706 ммоль) растворяли в метиленхлориде (50 мл), раствор перемешивали при комнатной температуре в течение 4 часов и к нему добавляли тионилхлорид (1,812 мл, 24,976 ммоль). Затем раствор перемешивали при 40°С в течение 2 часов, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. В качестве продукта получали целевое соединение формулы 2-4 (5,350 г, 98,7%) в виде оранжевого масла и использовали без дополнительной очистки.
Стадия 4: Синтез метил-7-(4-(бис(4-фторфенил)метил)пиперазин-1-карбоксамидо)гептаноата (формула 2-6)
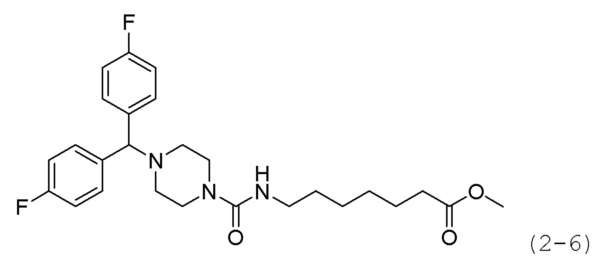
Соединение формулы 2-4 (0,233 г, 0,975 ммоль), полученное на стадии 3, гидрохлорид метил-7-(пиперазин-1-карбоксамидо)гептаноата (0,200 г, 0,650 ммоль) и карбонат калия (0,449 г, 3,249 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и раствор перемешивали при 80°С в течение 17 часов, а затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0 до 10%) и концентрировали с получением целевого соединения формулы 2-6 (0,101 г, 32,8%) в виде светло-коричневого масла.
Стадия 5: Синтез 4-(бис(4-фторфенил)метил)-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1223)
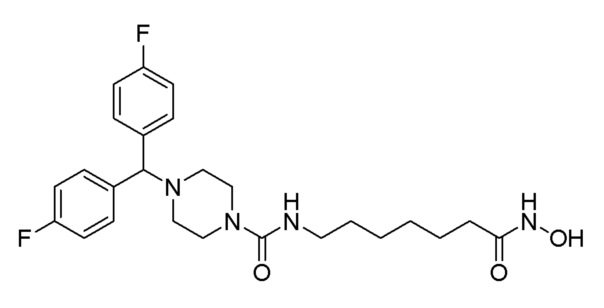
Соединение 1223
Соединение формулы 2-6 (0,101 г, 0,213 ммоль), полученное на стадии 4, гидроксиламин (50,00% водный раствор, 0,261 мл, 4,266 ммоль) и гидроксид калия (0,120 г, 2,133 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат очищали колоночной хроматографией (Waters, С18; 1%-муравьиная кислота (метановая кислота) водный раствор/ацетонитрил=от 70 до 5%) и концентрировали путем пропускания через картридж SPE (смола PL-HCO3) с получением целевого соединения 1223 (0,002 г, 2,0%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, CD3OD) δ 7,48-7,44 (м, 4H), 7,04 (т, 4 Н, J=8,8 Гц), 6,44 (т, 1H, J=5,3 Гц), 4,31 (с, 1H), 3,39 (т, 4H, J=5,0 Гц), 3,16-3,12 (м, 2H), 2,36 (т, 4Н, J=5,0 Гц), 2,09 (т, 4Н, J=7,4 Гц), 1,64-1,61 (м, 2H), 1,51-1,48 (м, 2Н), 1,35-1,33 (м, 4H); MS (ESI) m/z 475,3 (M++H).
Пример 12: Синтез соединения 1224
Стадия 1: Синтез 4,4'-(хлорметилен)бис(хлорбензол) (формула 2-4)

Бис(4-хлорфенил)метанол (10,000 г, 39,507 ммоль) растворяли в метиленхлориде (100 мл) при 0°C и к раствору добавляли тионилхлорид (3,153 мл, 43,458 ммоль) с последующим перемешиванием при комнатной температуре в течение 5 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Целевое соединение формулы 2-4 (10,700 г, 99,7%) получали в виде белого твердого вещества без дополнительной очистки.
Стадия 2: Синтез метил-7-(4-(бис(4-хлорфенил)метил)пиперазин-1-карбоксамидо)гептаноата (формула 2-6)
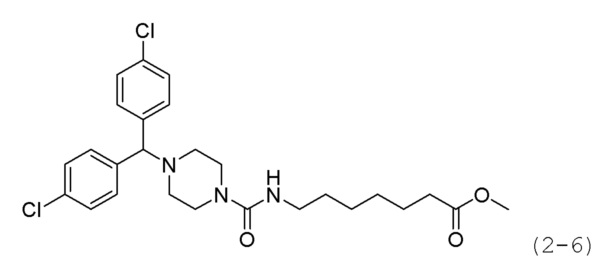
Соединение формулы 2-4 (0,265 г, 0,975 ммоль), полученное на стадии 1, соединение формулы 2-5 (0,200 г, 0,650 ммоль) и карбонат калия (0,449 г, 3,249 ммоль) растворяли в N,N-диметилформамиде (4 мл) при комнатной температуре, и раствор перемешивали при 80°С в течение 17 часов, а затем охлаждали до температуры прекращения реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли воду с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0 до 10%) и концентрировали с получением целевого соединения формулы 2-6 (0,271 г, 82,4%) в виде светло-желтого масла.
Стадия 3: Синтез 4-(бис(4-хлорфенил)метил)-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1224)
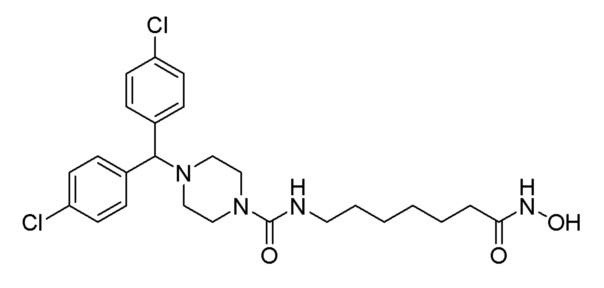
Соединение 1224
Соединение формулы 2-6 (0,271 г, 0,535 ммоль), полученное на стадии 2, гидроксиламин (50,00% водный раствор, 0,655 мл, 10,702 ммоль) и гидроксид калия (0,300 г, 5,351 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и концентрат очищали колоночной хроматографией (Waters, С18; 1%-муравьиная кислота (метановая кислота) водный раствор/ацетонитрил=от 70 до 5%) и концентрировали путем пропускания через картридж SPE (смола PL-HCO3) с получением целевого соединения 1224 (0,035 г, 12,9%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,34 (уш.с, 1H), 8,69 (уш.с, 1H), 7,43 (д, 4 Н, J=8,6 Гц), 7,37 (д, 4H, J=8,4 Гц), 6,41 (т, 1H, J=5,3 Гц), 4,40 (с, 1Н), 3,28-3,27 (м, 4H), 2,96 (кв, 2H, J=6,4 Гц), 2,22-2,21 (м, 4H), 1,92 (т, 2H, J=7,4 Гц), 1,48-1,44 (м, 2H), 1,37-1,35 (м, 2H), 1,24-1,21 (м, 4H); MS (ESI) m/z 507,4 (M++H).
Пример 13. Синтез соединения 1240
Стадия 1: Синтез метил-8-(4-бензгидрилпиперазин-1-карбоксиамидо)октаноата (формула 1-3)
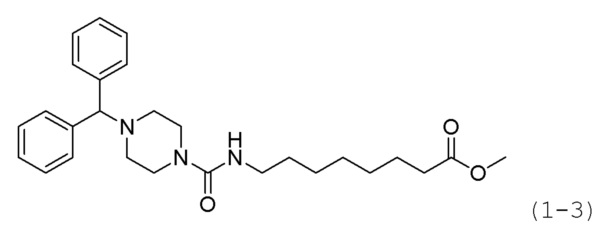
Трифосген (0,118 г, 0,396 ммоль) и диизопропиламин (0,830 мл, 4,755 ммоль) растворяли в метиленхлориде (5 мл) при 0°С, и к раствору добавляли гидрохлорид метил-8-аминооктаноата (0,166 г, 0,793 ммоль) с последующим перемешиванием в течение 1 часа. Исходный материал (0,200 г, 0,793 ммоль) добавляли к реакционной смеси, с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 10% до 70%) и концентрировали с получением целевого соединения формулы 1-3 (0,158 г, 44. 1%) В виде светло-желтого твердого вещества.
Стадия 2: Синтез 4-бензгидрил-N-(8-(гидроксиамино)-8-оксоэтил)пиперазин-1-карбоксамида (соединение 1240)
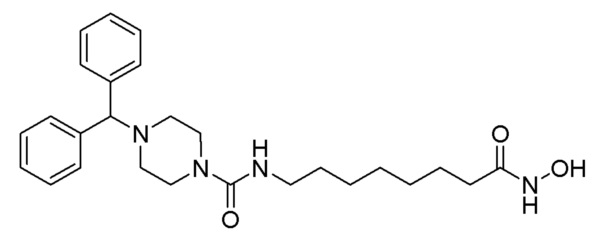
Соединение 1240
Соединение формулы 1-3 (0,158 г, 0,350 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,428 мл, 6,997 ммоль) и гидроксид калия (0,196 г, 3,499 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и насыщенный водный раствор бикарбоната натрия (20 мл) добавляли к концентрату с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили с получением целевого соединения 1240 (0,074 г), 46,7%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,49 (уш.с, 2H), 7,43 (д, 4 Н, J=7,5 Гц), 7,30 (т, 4H, J=7,6 Гц), 7,19 (т, 2H, J=7,3 Гц), 6,42 (т, 1H, J=5,2 Гц), 4,29 (с, 1Н), 3,28-3,27 (м, 4H), 2,97 (кв, 2H, J=6,4 Гц), 2,23-2,22 (м, 4H), 1,90 (т, 2H, J=7,3 Гц), 1,47-1,44 (м, 2H), 1,37-1,34 (м, 2H), 1,22 (уш.с, 4H); MS (ESI) m/z 453,6 (M++H).
Пример 14. Синтез соединения 1241
Стадия 1: Синтез метил-7-(4-(1-фенилэтил)пиперазин-1-карбоксамидо)гептаноата (формула 9-2)
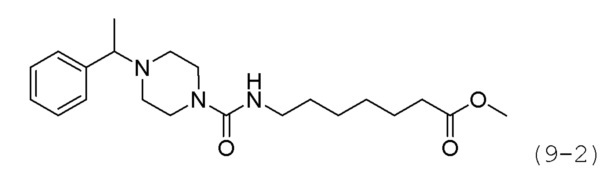
Соединение формулы 2-5 (0,150 г, 0,553 ммоль) и ацетофенон (0,100 г, 0,829 ммоль) растворяли в метиленхлориде (3 мл), и раствор перемешивали при комнатной температуре в течение 10 минут. Затем к раствору добавляли NaBH(OAc)3 (0,234 г, 1,106 ммоль) с последующим перемешиванием при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0 до 5%) и концентрировали с получением целевого соединения формулы 9-2 (0,038 г, 18,3%) в виде бесцветного масла.
Стадия 2: Синтез N-(7-(гидроксиамино)-7-оксогептил)-4-(1-фенилэтил)пиперазин-1-карбоксамида (соединение 1241)
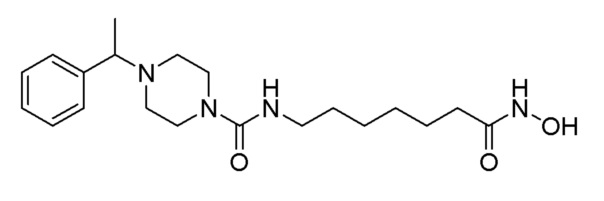
Соединение 1241
Соединение формулы 9-2 (0,038 г, 0,101 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,124 мл, 2,024 ммоль) и гидроксид калия (0,057 г, 1,012 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Целевое соединение 1241 (0,013 г, 34,1%) получали в виде светло-оранжевого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,32-7,30 (м, 4H), 7,26-7,23 (м, 1Н), 3,71-3,34 (м, 5H), 3,11 (т, 2H, J=7,1), 2,50-2,45 (м, 2Н), 2,37-2,32 (м, 2H), 2,05 (т, 2H, J=7,4 Гц), 1,61-1,56 (м, 2H), 1,49-1,44 (м, 2H), 1,37 (д, 3H, J=7,6 Гц), 1,33-1,29 (м, 4H); MS (ESI) m/z 477,2 (M++H).
Пример 15: Синтез соединения 1243
Стадия 1: Синтез этил-1-(1-фенилэтил)пиперидин-4-карбоксилата (формула 10-2)
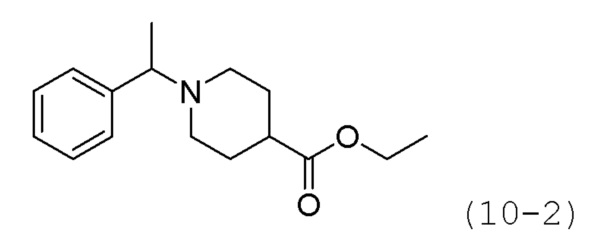
Ацетофенон (1,050 г, 8,739 ммоль) и этилпиперидин-4-карбоксилат (1,751 мл, 11,361 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре, и к раствору добавляли STAB (2,408 г, 11,361 ммоль) с последующим перемешиванием при той же температуре в течение 12 часов. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 10-2 (0,700 г, 30,6%) в виде бесцветного масла.
Стадия 2: Синтез 1-(1-фенилэтил)пиперидин-4-карбоновой кислоты (формула 10-3)
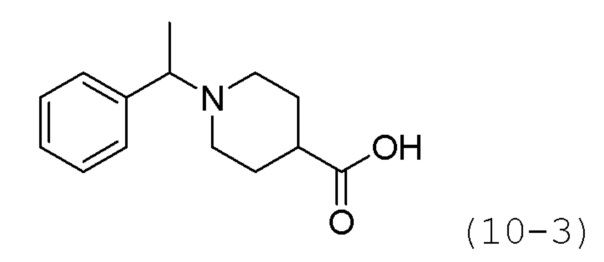
Соединение формулы 10-2 (0,700 г, 2,678 ммоль), полученное на стадии 1, и LiOH (0,096 г, 4,017 ммоль) растворяли в смеси метанол (3 мл)/вода (1 мл) при 40°С, и раствор перемешивали при той же температуре в течение 5 часов, а затем охлаждали до комнатной температуры. Затем к реакционной смеси добавляли 1 М HCl при 0°С, с последующим перемешиванием в течение 10 минут. После завершения реакции к реакционной смеси добавляли воду, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Продукт (0,500 г, 69,2%, белое пенообразное твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил-7-(1-(1-фенилэтил)пиперидин-4-карбоксамидо)гептаноата (формула 10-4)
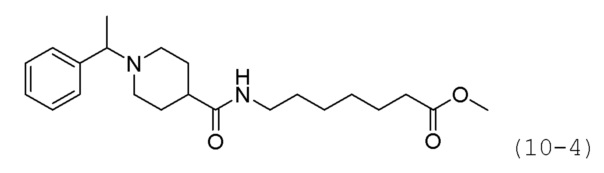
Соединение формулы 10-3 (0,300 г, 1,286 ммоль), полученное на стадии 2, гидрохлорид метил-7-аминогептаноата (0,503 г, 2,572 ммоль), EDC (0,493 г, 2,572 ммоль), HOBt (0,347 г, 2,572 ммоль) и диизопропиламин (1,123 мл, 6,429 ммоль) растворяли в смеси метиленхлорид (4 мл)/N,N-диметилформамид (1 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0 до 10%) и концентрировали с получением целевого соединения формулы 10-4 (0,122 г, 25,3%) в виде коричневого масла.
Стадия 4: Синтез N-(7-(гидроксиамино)-7-оксогептил)-1-(1-фенилэтил)пиперидин-4-карбоксамида (соединение 1243)
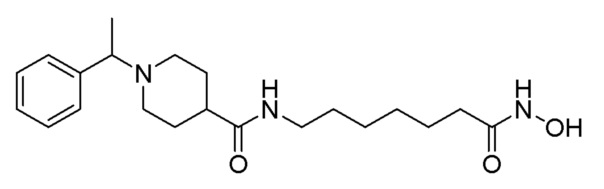
Соединение 1243
Соединение формулы 10-4 (0,122 г, 0,326 ммоль), полученное на стадии 3, гидроксиламин (50,00% водный раствор, 0,398 мл, 6,515 ммоль) и гидроксид калия (0,183 г, 3,257 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Целевое соединение 1243 (0,074 г, 60,5%) получали в виде оранжевого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,19 (уш.с, 1H), 8,71 (уш.с, 1H), 7,64 (т, 1H, J=5,6 Гц), 7,32-7,27 (м, 4H), 7,23-7,20 (м, 1Н), 3,40-3,37 (м, 1H), 3,00-2,95 (м, 3H); MS (ESI) m/z 376,3 (M++H).
Пример 16. Синтез соединения 1256
Стадия 1: Синтез метил-7-(1-бензгидрил-N-метилпиперидин-4-карбоксамидо)гептаноата (формула 5-5)
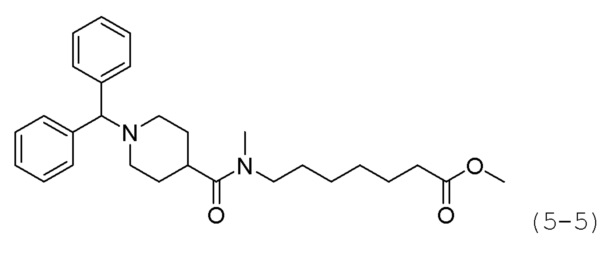
Метил-7-(1-бензгидрилпиперидин-4-карбоксамидо)гептаноат (0,200 г, 0,458 ммоль) и гидрид натрия (60,00%, 0,092 г, 2,290 ммоль) растворяли в N,N-диметилформамиде (5 мл) и раствор перемешивали при комнатной температуре в течение 10 минут. Затем к перемешиваемому раствору добавляли йодметан (0,143 мл, 2,290 ммоль), с последующим перемешиванием при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 10% до 70%) и концентрировали с получением целевого соединения формулы 5-5 (0 089 Г, 43,1%) в виде светло-желтого масла.
Стадия 2: Синтез 1-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-N-метилпиперидин-4-карбоксамида (соединение 1256)
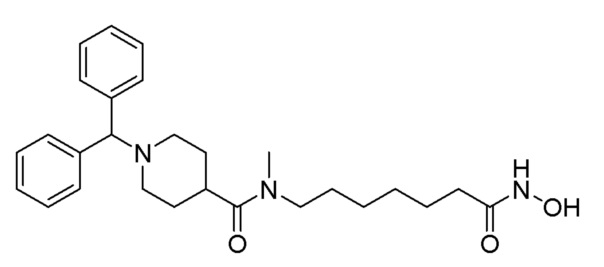
Соединение 1256
Соединение формулы 5-5 (0,089 г, 0,198 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,242 мл, 3,950 ммоль) и гидроксид калия (0,111 г, 1,975 ммоль) растворяли в метаноле (3 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Целевое соединение 1256 (0,089 г, 99,8%) получали в виде белого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,50 (уш.с, 2H), 7,40 (д, 4H, J=7,2 Гц), 7,29 (т, 4 Н, J=7,6 Гц), 7,18 (т, 2H, J=7,3 Гц), 4,30 (с, 1H), 3,23 (кв, 2H, J=7,5 Гц), 2,94 (с, 2H), 2,81 (д, 2H, J=11,4 Гц), 2,76 (с, 1Н), 1,91-1,83 (м, 4H), 1,69-1,53 (м, 4H), 1,46-1,37 (м, 4H), 1,24-1,19 (м, 4H); MS (ESI) m/z 452,6 (M++H).
Пример 17: Синтез соединения 1257
Стадия 1: Синтез метил-6-(4-бензгидрилпиперазин-1-карбоксамидо)гексаноата (формула 1-3)
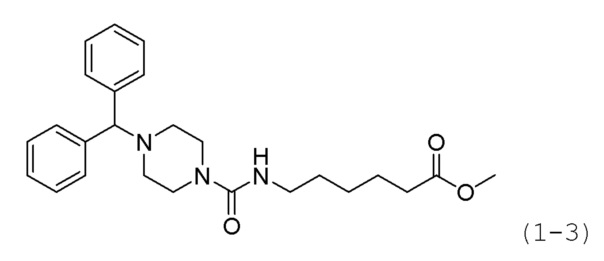
Трифосген (0,294 г, 0,991 ммоль) и диизопропиламин (2,076 мл, 11,888 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и к раствору добавляли гидрохлорид метил-6-аминогексаноата (0,360 г, 1,981 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли 1-бензгидрилпиперазин (0,500 г, 1,981 ммоль), с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 40%) и концентрировали с получением целевого соединения формулы 1-3 (0,320 г, 38,1%) в виде желтого масла.
Стадия 2: Синтез 4-бензгидрил-N-(6-(гидроксиамино)-6-оксогексил)пиперазин-1-карбоксамида (соединение 1257)
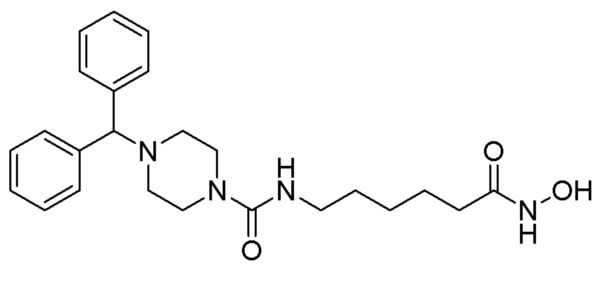
Соединение 1257
Соединение формулы 1-3 (0,200 г, 0,472 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,578 мл, 9,444 ммоль) и гидроксид калия (0,265 г, 4,722 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Целевое соединение 1257 (0,049 г, 24,4%) получали в виде светло-желтого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,43 (д, 4H, J=7,2 Гц), 7,30 (т, 4H, J=7,6 Гц), 7,19 (т, 2H, J=7,3 Гц), 6,42 (т, 1H, J=5,4 Гц), 4,29 (с, 1H), 3,27 (т, 4H, J=4,5 Гц), 2,96 (кв, 2H, J=6,4 Гц), 2,23 (т, 4Н, J=4,6 Гц), 1,90 (т, 2H, J=7,4 Гц), 1,47-1,44 (м, 2H), 1,38-1,34 (м, 2H), 1,20-1,16 (м, 2H); MS (ESI) m/z 425,5 (M++H).
Пример 18: Синтез соединения 1316
Стадия 1: Синтез метил-6-(1-бензгидрилпиперидин-4-карбоксамидо)гексаноата (формула 5-4)
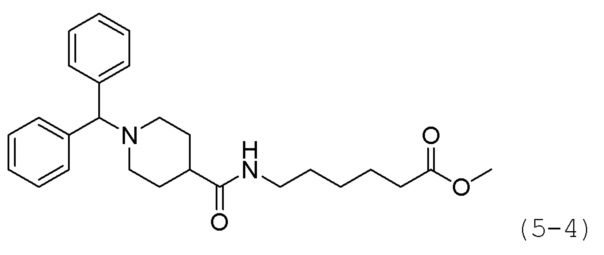
Соединение формулы 5-3 (0,300 г, 1,016 ммоль), гидрохлорид метил-6-аминогексаноата (0,369 г, 2,031 ммоль), EDC (0,389 г, 2,031 ммоль), HOBt (0,274 г, 2,031 ммоль) и диизопропиламин (0,887 мл, 5,078 ммоль) растворяли в смеси метиленхлорид (3 мл)/N,N-диметилформамид (0,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 30%) и концентрировали с получением целевого соединения формулы 5-4 (0 161 Г, 37,5%) в виде светло-желтого масла.
Стадия 2: Синтез 1-бензгидрил-N-(6-(гидроксиамино)-6-оксогексил)пиперидин-4-карбоксамида (соединение 1316)
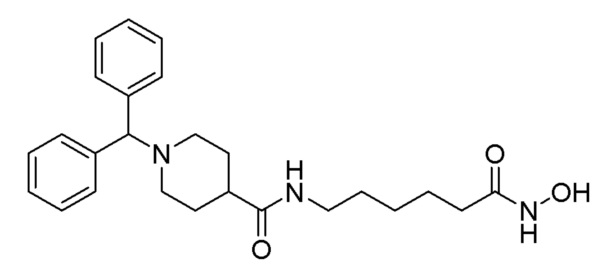
Соединение 1316
Соединение формулы 5-4 (0,161 г, 0,381 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,466 мл, 7,620 ммоль) и гидроксид калия (0,214 г, 3,810 ммоль) растворяли в метаноле (3 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Целевое соединение 1316 (0,056 г, 34,7%) получали в виде белого твердого вещества без дополнительной очистки.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,33 (уш.с, 1H), 8,66 (уш.с, 1H), 7,70 (т, 1H, J=5,6 Гц), 7,41 (д, 4 Н, J=7,4 Гц), 7,17 (т, 2H, J=7,4 Гц), 4,26 (с, 2H), 2,99-2,28 (м, 2H), 2,80 (д, 2H, J=11,5 Гц), 2,09 ~ 2,02 (м, 1H), 1,91 (т, 2H, J=7,5 Гц), 1,80 ~ 1,75 (м, 2H), 1,68 ~ 1,59 (м, 4H).
Пример 19. Синтез соединения 1317
Стадия 1: Синтез метил-8-(1-бензгидрилпиперидин-4-карбоксамидо)октаноата (формула 5-4)
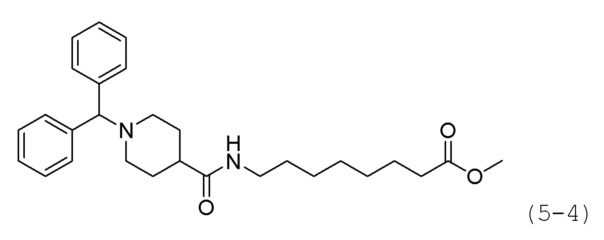
Соединение формулы 5-3 (0,300 г, 1,016 ммоль), гидрохлорид метил-8-аминооктаноата (0,426 г, 2,031 ммоль), EDC (0,389 г, 2,031 ммоль), HOBt (0,274 г, 2,031 ммоль) и диизопропиламин (0,887 мл, 5,078 ммоль) растворяли в смеси метиленхлорид (3 мл)/N,N-диметилформамид (0,5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 17 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 30%) и концентрировали с получением целевого соединения формулы 5-4 (0,220 г, 49,6%) в виде бесцветного масла.
Стадия 2: Синтез 1-бензгидрил-N-(8-(гидроксиамино)-8-оксоэтил)пиперидин-4-карбоксамида (соединение 1317)
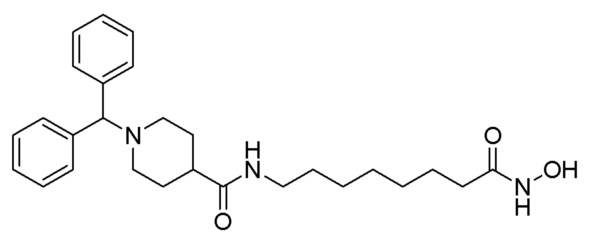
Соединение 1317
Соединение формулы 5-4 (0,220 г, 0,488 ммоль), полученное на стадии 1, гидроксиламин (50,00% водный раствор, 0,597 мл, 9,764 ммоль) и гидроксид калия (0,274 г, 4,882 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и насыщенный водный раствор бикарбоната натрия (20 мл) добавляли к концентрату с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили с получением целевого соединения 1317 (0,189 г, 85,7%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 9,46 (уш.с, 2H), 7,73 (т, 1H, J=5,3 Гц), 7,41 (д, 4 Н, J=8,0 Гц), 7,28 (т, 4H, J=7,5 Гц), 7,17 (т, 2H, J=7,3 Гц), 4,26 (с, 1H), 2,99 (кв, 2H, J=6,4 Гц, J=6,4 Гц), 2,80 (д, 2H, J=11,1 Гц), 2,10-2,04 (м, 1H), 1,87 (т, 2H, J=7,3 Гц), 1,78 (т, 2H, J=10,0 Гц), 1,67-1,56 (м, 4H), 1,46-1,42 (м, 2H), 1,36-1,33 (м, 2H), 1,21 (уш.с, 6H).
Пример 20. Синтез соединения 1647
Стадия 1: Синтез 2,2'-(хлорметилен)бис(фторбензол) (формула 2-4)
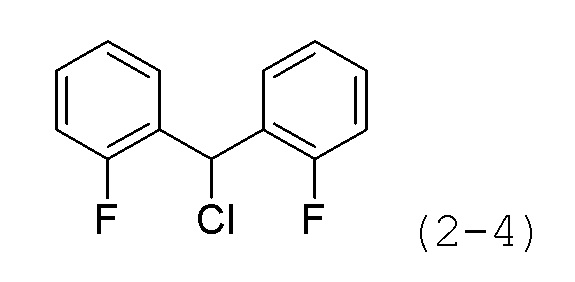
Бис(2-фторфенил)метанол (0,500 г, 2,270 ммоль) и триэтиламин (0,348 мл, 2,498 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре, и к раствору добавляли метансульфонилхлорид (0,193 мл, 2,498 ммоль) с последующим перемешиванием при той же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 5%) и концентрировали с получением целевого соединения формулы 2-4 (0,290 г, 53,5%) в виде бесцветного масла.
Стадия 2: Синтез метил-7-(4-(бис(2-фторфенил)метил)пиперазин-1-карбоксамидо)гептаноата (формула 2-6)
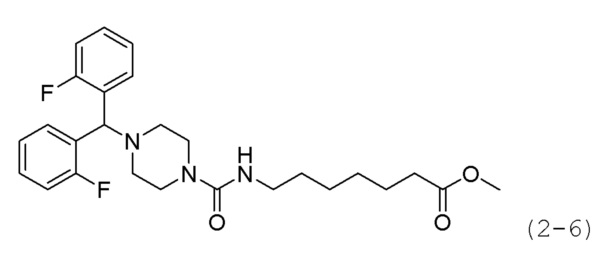
Соединение формулы 2-4 (0,448 г, 1,877 ммоль), полученное на стадии 1, гидрохлорид метил-7-(пиперазин-1-ил)гептаноата (0,746 г, 2,816 ммоль) и карбонат калия (1,297 г, 9,386 ммоль) растворяли в N,N-диметилформамиде (8 мл) при 80°С, и раствор перемешивали при той же температуре в течение 16 часов, а затем охлаждали до комнатной температуры для прекращения реакции. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 60%) и концентрировали с получением целевого соединения формулы 2-6 (0,170 г, 19,1%) в виде ярко-желтого твердого вещества.
Стадия 3: Синтез 4-(бис(2-фторфенил)метил)-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1647)
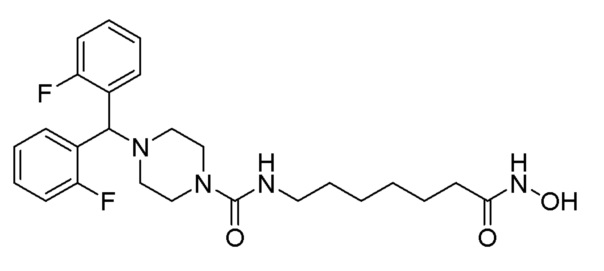
Соединение1647
Соединение формулы 2-6 (0,200 г, 0,422 ммоль), полученное на стадии 2, и гидроксиламин (50,00% водный раствор, 0,258 мл, 4,223 ммоль) растворяли в метаноле (5 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли метанол (10 мл) и насыщенный водный раствор бикарбоната натрия (90 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1647 (0,200 г, 99,8%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,59-7,55 (м, 2H), 7,31-7,26 (м, 2H), 7,23-7,19 (м, 2H), 7,16-7,11 (м, 2H), 6,42 (т, 1H, J=5,5 Гц), 4,96 (с, 1Н), 3,29-3,28 (м, 4H), 2,99-2,94 (м, 2Н), 2,28-2,26 (м, 4H), 1,93-1,89 (м, 2H), 1,47-1,43 (м, 2H), 1,37-1,33 (м, 2Н), 1,19-1,20 (м, 4H); MS (ESI) m/z 475,4 (M++H).
Пример 21: Синтез соединения 1648
Стадия 1: Синтез 3,3'-(хлорметилен)бис(фторбензол) (формула 2-4)
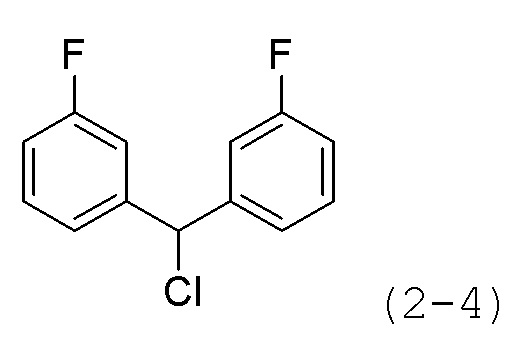
Бис(3-фторфенил)метанол (1,000 г, 4,541 ммоль) и триэтиламин (0,696 мл, 4,995 ммоль) растворяли в метиленхлориде (10 мл), и к раствору добавляли метансульфонилхлорид (0,387 мл, 4,995 ммоль) с последующим перемешиванием при той же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 5%) и концентрировали с получением целевого соединения формулы 2-4 (0,670 г, 61,8%) в виде бесцветного масла.
Стадия 2: Синтез 7-(4-(бис(3-фторфенил)метил)пиперазин-1-карбоксамидо)гептаноата (формула 2-6)

Соединение формулы 2-4 (0,670 г, 2,807 ммоль), полученное на стадии 1, гидрохлорид метил-7-(пиперазин-1-ил)гептаноата (1,115 г, 4,211 ммоль) и карбонат калия (1,940 г, 14,037 ммоль) растворяли в N,N-диметилформамиде (10 мл) при 80°С, и раствор перемешивали при той же температуре в течение 16 часов, а затем охлаждали до комнатной температуры для прекращения реакции. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 24 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 2-6 (0,294 г, 22,1%) в виде белого твердого вещества.
Стадия 3: Синтез 4-(бис(3-фторфенил)метил)-N-(7-(гидроксиамино)-7-оксогептил)пиперазин-1-карбоксамида (соединение 1648)
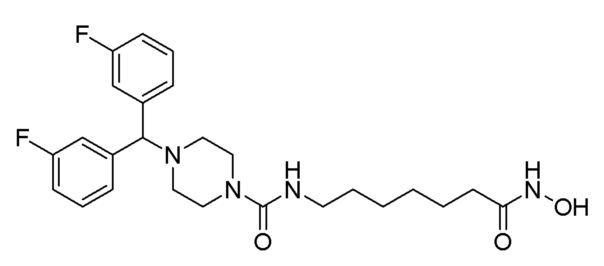
Соединение 1648
Соединение формулы 2-6 (0,100 г, 0,211 ммоль), полученное на стадии 2, и гидроксиламин (50,00% водный раствор, 0,129 мл, 2,112 ммоль) растворяли в метаноле (3 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли метанол (10 мл) и насыщенный водный раствор бикарбоната натрия (90 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1648 (0,097 г, 97,1%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,34 (с, 1H), 8,72 (с, 1Н), 7,38-7,33 (м, 2H), 7,28-7,25 (м, 4H), 7,06-7,01 (м, 2H), 6,40 (т, 1H, J=5,5 Гц), 4,41 (с, 1H), 3,29-3,27 (м, 4H), 2,99-2,94 (м, 2H), 2,24-2,22 (м, 4H), 1,93-1,90 (м, 2H), 1,47-1,44 (м, 2H), 1,37-1,33 (м, 2H), 1,21-1,20 (м, 4H); MS (ESI) m/z 475,4 (M++H).
Пример 22: Синтез соединения 1649
Стадия 1: Синтез метил-7-(4-(гидроксидифенилметил)пиперидин-1-карбоксамидо)гептаноата (формула 7-4)
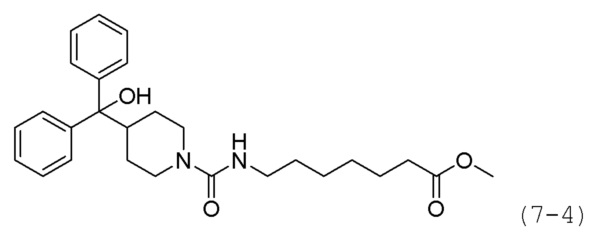
Гидрохлорид метил-7-аминогептаноата (0,366 г, 1,870 ммоль) и трифосген (0,277 г, 0,935 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,977 мл, 5,610 ммоль) с последующим перемешиванием в течение 1 часа. К реакционной смеси добавляли дифенил(пиперидин-4-ил)метанол (0,500 г, 1,870 ммоль), с последующим перемешиванием при той же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 7-4 (0,609 г, 71,9%) в виде бесцветного масла.
Стадия 2: Синтез метил-7-(4-(фтордифенилметил)пиперидин-1-карбоксамидо)гептаноата (формула 7-5)
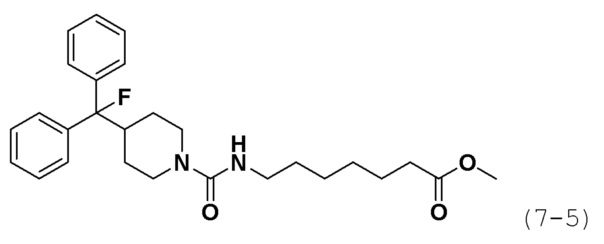
Соединение формулы 7-4 (0,300 г, 0,663 ммоль), полученное на стадии 1, растворяли в метиленхлориде (5 мл) при 0°С, и к раствору добавляли трифторид диэтиламиносеры (DAST, 0,114 мл, 0,862 ммоль) с последующим перемешиванием при комнатной температуре в течение 16 часов. К реакционной смеси добавляли воду с последующей экстракцией метиленхлоридом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя, и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 7-5 (0,143 г, 47,5%) в виде белого твердого вещества.
Стадия 3: Синтез 4-(фтордифенилметил)-N-(7-(гидроксиамино)-7-оксогептил)пиперидин-1-карбоксамида (соединение 1649)
Соединение 1649
Соединение формулы 7-5 (0,140 г, 0,308 ммоль), полученное на стадии 2, и гидроксиламин (50,00% водный раствор, 0,188 мл, 3,080 ммоль) растворяли в метаноле (3 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли метанол (10 мл) и насыщенный водный раствор бикарбоната натрия (90 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили, получая целевое соединение 1649 (0,122 г, 87,0%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,49-7,47 (м, 4H), 7,37-7,33 (м, 4H), 7,26-7,22 (м, 2H), 6,36 (т, 1H, J=5,5 Гц), 3,95-3,92 (м, 2Н), 2,98-2,79 (м, 3H), 2,65-2,59 (м, 2Н), 1,93-1,89 (м, 2H), 1,47-1,44 (м, 2H), 1,36-1,33 (м, 2Н), 1,29-1,20 (м, 8H); MS (ESI) m/z 456,6 (M++H).
Пример 23: Синтез соединения 1719
Стадия 1: Синтез трет-бутил-4-(фениламино)пиперидин-1-карбоксилата (формула 6-2)
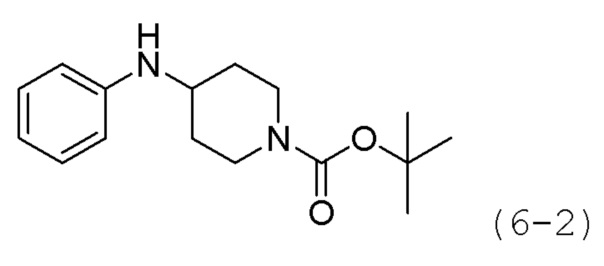
Трет-бутил-4-оксопиперидин-1-карбоксилат (5,000 г, 25,094 ммоль), анилин (2,749 мл, 30,113 ммоль) и уксусную кислоту (2,155 мл, 37,641 ммоль) растворяли в метиленхлориде (50 мл) при комнатной температуре, и к раствору добавляли триацетоксиборгидрид натрия (5,850 г, 27,604 ммоль) с последующим перемешиванием при той же температуре в течение 16 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. К концентрату добавляли этилацетат (100 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали гексаном и сушили с получением целевого соединения формулы 6-2 (4,640 г, 66,9%) в виде белого твердого вещества.
Стадия 2: Синтез трет-бутил-4-(3-фторфенил)(фенил)амино)пиперидин-1-карбоксилата (формула 6-3)
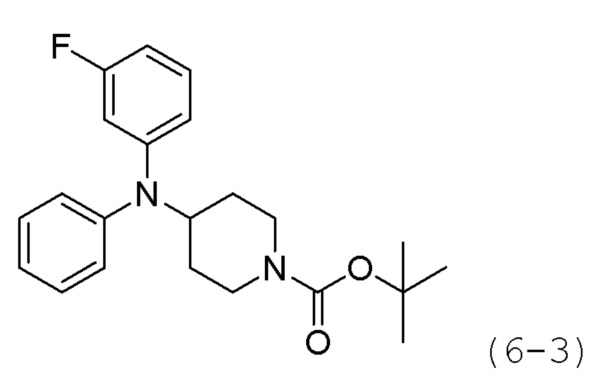
Соединение формулы 6-2 (0,500 г, 1,809 ммоль), полученное на стадии 1, 1-фтор-3-йодбензол (0,422 г, 1,900 ммоль), ацетат палладия (II, 0,016 г, 0,072 ммоль) 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (0,051 г, 0,081 ммоль) и трет-бутоксид калия (0,254 г, 2,261 ммоль) растворяли в толуоле (5 мл) при 110°С, и раствор перемешивали при той же температуре в течение 16 часов, затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, и к фильтрату добавляли насыщенный водный раствор хлорида натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 10%) и концентрировали с получением целевого соединения формулы 6-3 (0,292 г, 43,6%) в виде желтого твердого вещества.
Стадия 3: Синтез гидрохлорида N-(3-фторфенил)-N-фенилпиперидин-4-амина (формула 6-4)
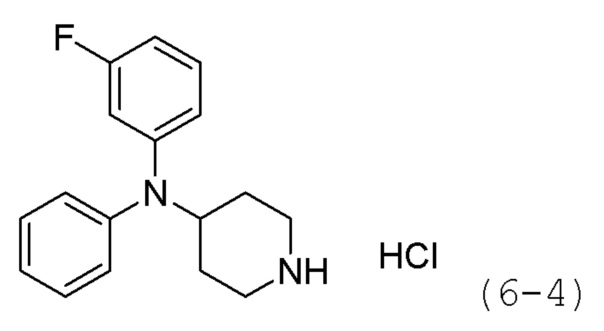
Соединение формулы 6-3 (0,285 г, 0,769 ммоль), полученное на стадии 2, растворяли в метиленхлориде (10 мл) при той же температуре, и к раствору добавляли соляную кислоту (4,00 М раствор в диоксане, 0,962 мл, 3,846 ммоль) с последующим перемешиванием при той же температуре в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Продукт (0,212 г, 89,8%, желтое твердое вещество) использовали без дополнительной очистки.
Стадия 4: Синтез метил-7-(4-((3-фторфенил)(фенил)амино)пиперидин-1-карбоксамидо)гептаноата (формула 6-6)
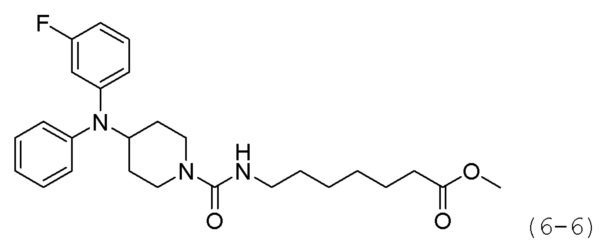
Гидрохлорид метил-7-аминогептаноата (0,135 г, 0,691 ммоль) и трифосген (0,103 г, 0,345 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,361 мл, 2,073 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли соединение формулы 6-4 (0,212 г, 0,691 ммоль), полученное на стадии 3, с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г); этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 6-6 (0,219 г, 69,6%) в виде бесцветного масла.
Стадия 5: Синтез 4-((3-фторфенил)(фенил)амино)-N-(7-(гидроксиамино)-7-оксогептил)пиперидин-1-карбоксамида (соединение 1719)
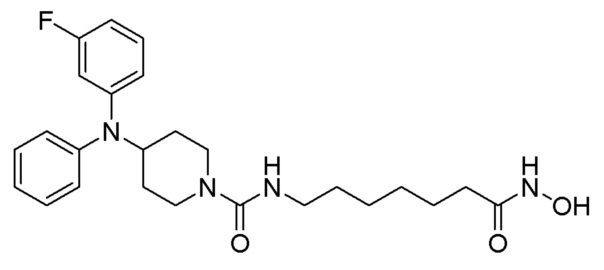
Соединение 1719
Соединение формулы 6-6 (0,219 г, 0,481 ммоль), полученное на стадии 4, гидроксиламин (50,00% водный раствор, 0,294 мл, 4,807 ммоль) и гидроксид калия (0,270 г, 4,807 ммоль) растворяли в метаноле (5 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли метанол (1 мл) и насыщенный водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Выпавшее в осадок твердое вещество отфильтровывали, промывали гексаном и сушили, получая целевое соединение 1719 (0,196 г, 89,3%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,44-7,42 (м, 2H), 7,32-7,28 (м, 1Н), 7,16-7,10 (м, 1H), 7,05-7,03 (м, 2Н), 6,51-6,46 (м, 1H), 6,37-6,32 (м, 3H), 4,10-4,08 (м, 1Н), 3,97-3,94 (м, 2H), 2,92-2,87 (м, 2Н), 2,83-2,77 (м, 2H), 1,92-1,88 (м, 2H), 1,85-1,52 (м, 2H), 1,45-1,41 (м, 2H), 1,30-1,27 (м, 2Н), 1,16-1,08 (м, 4H), 1,06-1,00 (м, 2H); MS (ESI) m/z 457,5 (M++H).
Пример 24: Синтез соединения 1726
Стадия 1: Синтез трет-бутил-4-(фенил(4-(трифторметил)фенил)амино)пиперидин-1-карбоксилата (формула 6-3)
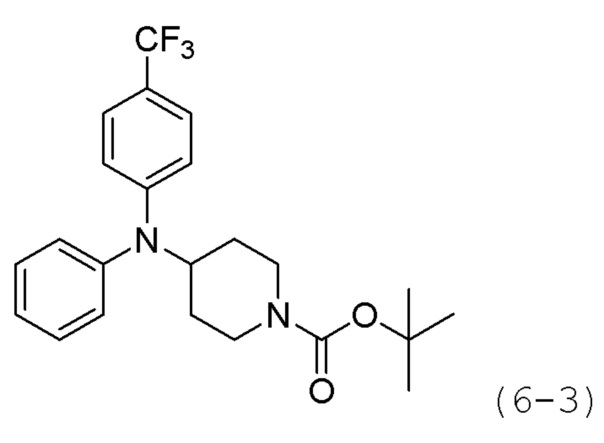
Трет-бутил-4-(фениламино)пиперидин-1-карбоксилат (1,000 г, 3,618 ммоль), 1-йод-4-(трифторметил)бензол (1,033 г, 3,799 ммоль), ацетат палладия (II, 0,032 г, 0,145 ммоль), 2,2'-бис (дифенилфосфино)-1,1'-бинафтил (0,101 г, 0,163 ммоль) и трет-бутоксид калия (0,507 г, 4,523 ммоль) растворяли в толуоле (5 мл) при 110°С и раствор перемешивали при той же температуре в течение 16 часов, а затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, и к фильтрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 10%) и концентрировали с получением целевого соединения формулы 6-3 (0,040 г, 2,9%) в виде коричневого масла.
Стадия 2: Синтез гидрохлорида N-фенил-N-(4-(трифторметил)фенил)пиперидин-4-амина (формула 6-4)
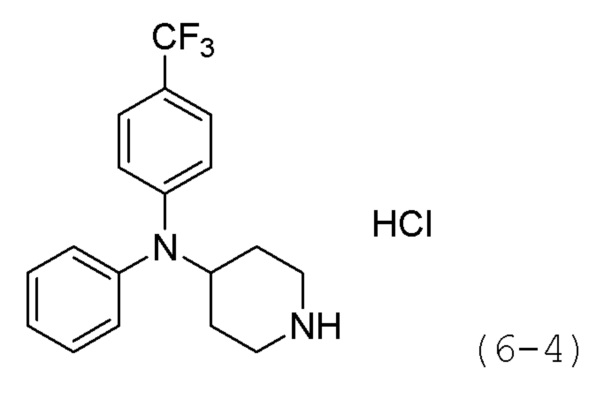
Соединение формулы 6-3 (0,890 г, 2,494 ммоль), полученное на стадии 1, растворяли в метиленхлориде (20 мл) при комнатной температуре, и к раствору добавляли соляную кислоту (4,00 М раствор, 3,118 мл, 12,471 ммоль) с последующим перемешиванием при той же температуре в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Продукт (0,890 г, 100,0%, желтое твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил-7-(4-(фенил(4-(трифторметил)фенил)амино)пиперидин-1-карбоксамидо)гептаноата (формула 6-6)
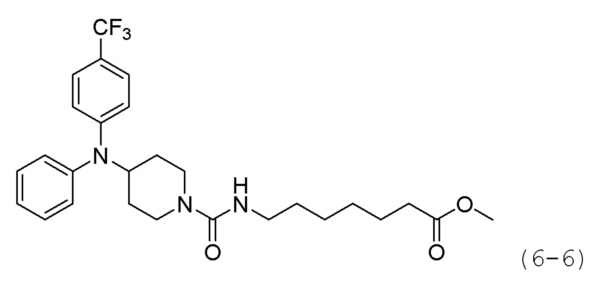
Гидрохлорид метил-7-аминогептаноата (0,219 г, 1,121 ммоль) и трифосген (0,166 г, 0,561 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,586 мл, 3,363 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли соединение формулы 6-4 (0,400 г, 1,121 ммоль) с последующим перемешиванием при комнатной температуре в течение 3 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 6-6 (0 277 Г, 48,9%) в виде бесцветного масла.
Стадия 4: Синтез N-(7-(гидроксиамино)-7-оксогептил)-4-(фенил(4-(трифторметил)фенил)амино)пиперидин-1-карбоксамида (соединение 1726)
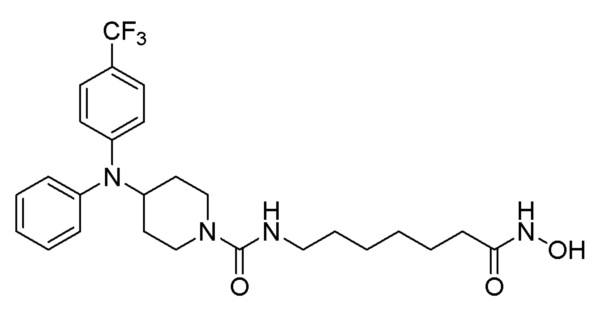
Соединение 1726
Соединение формулы 6-6 (0,170 г, 0,336 ммоль), полученное на стадии 3, и гидроксиламин (50,00%-ный водный раствор, 0,205 мл, 3,356 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и к раствору добавляли гидроксид калия (0,188 г, 3,356 ммоль) с последующим перемешиванием при той же температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, осажденное твердое вещество отфильтровывали, промывали гексаном и сушили, получая целевое соединение 1726 (0,139 г, 81,8%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,33 (с, 1H), 8,66 (с, 1H), 7,21-7,16 (м, 4H), 6,97-6,93 (м, 2H), 6,84-6,80 (м, 1Н), 6,67-6,65 (м, 2H), 6,36-6,34 (м, 1H), 4,07-4,02 (м, 1Н), 3,98-3,95 (м, 2H), 2,93-2,88 (м, 2Н), 2,81-2,75 (м, 2H), 1,93-1,89 (м, 2H), 1,85-1,82 (м, 2H), 1,45-1,43 (м, 2H), 1,31-1,27 (м, 2H), 1,18-1,15 (м, 4H), 1,05-1,01 (м, 2Н); МS (ESI) m/z 457,5 (M++H).
Пример 25: Синтез соединения 1734
Стадия 1: Синтез трет-бутил-4-((4-фторфенил)(фенил)амино)пиперидин-1-карбоксилата (формула 6-3)
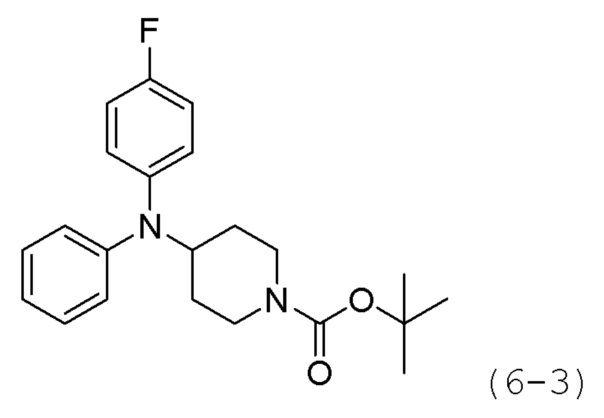
Трет-бутил-4-(фениламино)пиперидин-1-карбоксилат (0,820 г, 2,967 ммоль), 1-фтор-4-йодбензол (0,358 мл, 3,115 ммоль), ацетат палладия (II, 0,027 г, 0,119 ммоль) 2,2'-бис(дифенилфосфино)-1,1'-бинафтал (0,083 г, 0,134 ммоль) и трет-бутоксид калия (0,416 г, 3,709 ммоль) растворяли в толуоле (5 мл) при 110°С, и раствор перемешивали при той же температуре в течение 16 часов, а затем охлаждали до комнатной температуры для прекращения реакции. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, и к фильтрату добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 12 г; этилацетат/гексан=от 0 до 10%) и концентрировали с получением целевого соединения формулы 6-3 (0,352 г, 32,0%) в виде ярко-желтого твердого вещества.
Стадия 2: Синтез гидрохлорида N-(4-фторфенил)-N-фенилпиперидин-4-амина (формула 6-4)
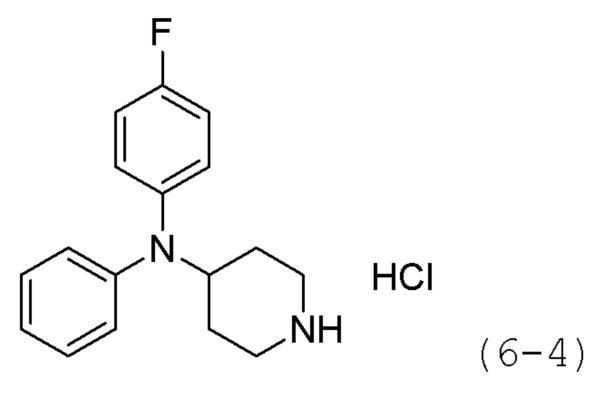
Соединение формулы 6-3 (0,340 г, 0,918 ммоль), полученное на стадии 1, и соляную кислоту (4,00 М раствор, 1,147 мл, 4,589 ммоль) растворяли в метиленхлориде (5 мл) при комнатной температуре и раствор перемешивали при той же температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Продукт (0,281 г, 99,8%, желтое твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил 7-(4-(4-фторфенил)(фенил)амино)пиперидин-1-карбоксамидо)гептаноата (формула 6-6)
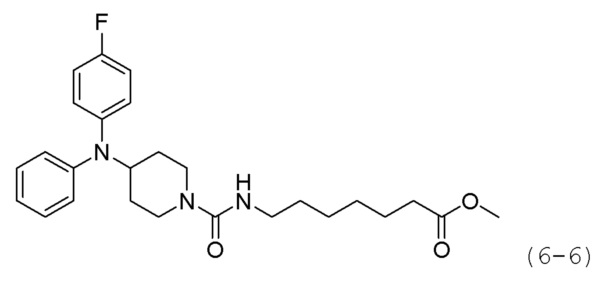
Метил-7-аминогептаноат гидрохлорид (0,179 г, 0,913 ммоль) и трифосген (0,135 г, 0,456 ммоль) растворяли в метиленхлориде (10 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,477 мл, 2,738 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли соединение формулы 6-4 (0,280 г, 0,913 ммоль), полученное на стадии 2, с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; этилацетат/гексан=от 0% до 50%) и концентрировали с получением целевого соединения формулы 6-6 (0,185 г, 44,5%) в виде бесцветного масла.
Стадия 4: Синтез 4-((4-фторфенил)(фенил)амино)-N-(7-(гидроксиамино)-7-оксогептил)пиперидин-1-карбоксамида (соединение 1734)
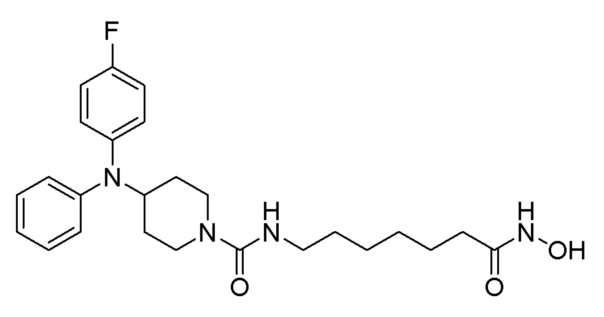
Соединение 1734
Соединение формулы 6-6 (0,260 г, 0,569 ммоль), полученное на стадии 3, и гидроксиламин (50,00% водный раствор, 0,348 мл, 5,695 ммоль) растворяли в метаноле (5 мл) при 0°С, и раствор перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и осажденное твердое вещество отфильтровывали, промывали гексаном и сушили, получая целевое соединение 1734 (0,185 г, 71,2%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 10,33 (с, 1H), 8,66 (с, 1H), 7,21-7,16 (м, 4H), 6,97-6,93 (м, 2H), 6,84-6,80 (м, 1Н), 6,67-6,65 (м, 2H), 6,36-6,34 (м, 1H), 4,07-4,02 (м, 1Н), 3,98-3,95 (м, 2H), 2,93-2,88 (м, 2Н), 2,81-2,75 (м, 2H), 1,93-1,89 (м, 2H), 1,85-1,82 (м, 2H), 1,45-1,43 (м, 2H), 1,31-1,27 (м, 2H), 1,18-1,15 (м, 4H), 1,05-1,01 (м, 2Н); МS (ESI) m/z 457,5 (M++H).
Пример 26. Синтез соединения 1763
Стадия 1: Синтез трет-бутил 7-бензгидрил-2,7-диазаспиро[3,5]нонан-2-карбоксилата (формула 4-5)

(Хлорметилен)дибензол (0,439 мл, 2,467 ммоль), трет-бутил 2,7-диазаспиро[3,5]нонан-2-карбоксилат (0,614 г, 2,714 ммоль) и карбонат калия (1,705 г, 12,335 ммоль) растворяли в N,N-диметилформамиде (10 мл) при 80°C, и раствор перемешивали при той же температуре в течение 16 часов, а затем охлаждали до комнатной температуры для прекращения реакции. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Затем к концентрату добавляли этилацетат (100 мл) с последующим перемешиванием, и выпавшее в осадок твердое вещество отфильтровывали, промывали этилацетатом и сушили, получая целевое соединение формулы 4-5 (0,411 г, 42,4%) в виде белого твердого вещества.
Стадия 2: Синтез гидрохлорида 7-бензгидрил-2,7-диазаспиро[3,5]нонана (формула 4-6)
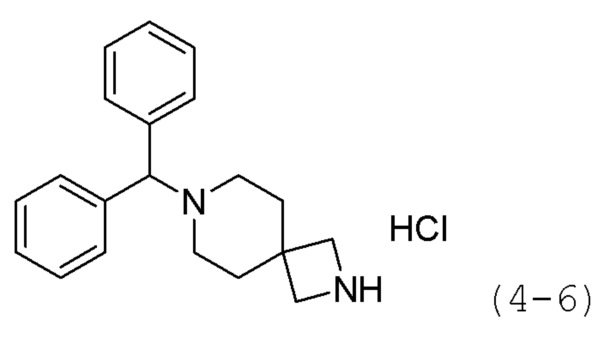
Соединение формулы 4-5 (0,411 г, 1,047 ммоль), полученное на стадии 1, растворяли в метиленхлориде (8 мл) при комнатной температуре, и к раствору добавляли соляную кислоту (4,00 М раствор в диоксане, 1,309 мл, 5,235 ммоль) с последующим перемешиванием при той же температуре в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и продукт (0,344 г, 99,9% белое твердое вещество) использовали без дополнительной очистки.
Стадия 3: Синтез метил-6-(7-бензгидрил-2,7-диазаспиро[3,5]нонан-2-карбоксамидо)гексаноата (формула 4-7)
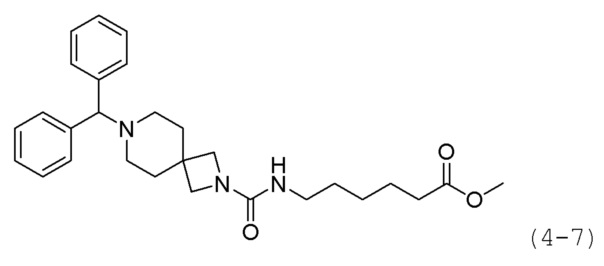
Гидрохлорид метил-6-аминогексаноата (0,100 г, 0,549 ммоль) и трифосген (0,078 г, 0,261 ммоль) растворяли в метиленхлориде (5 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,273 мл, 1,569 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли соединение формулы 4-6 (0,172 г, 0,523 ммоль), полученное на стадии 2, с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0% до 3%) и концентрировали с получением целевого соединения формулы 4-7 (0,148 г, 61,0%) в виде ярко-красного твердого вещества.
Стадия 4: Синтез 7-бензгидрил-N-(6-(гидроксиамино)-6-оксогексил)-2,7-диазаспиро[3,5]нонан-2-карбоксамида (соединение 1763)
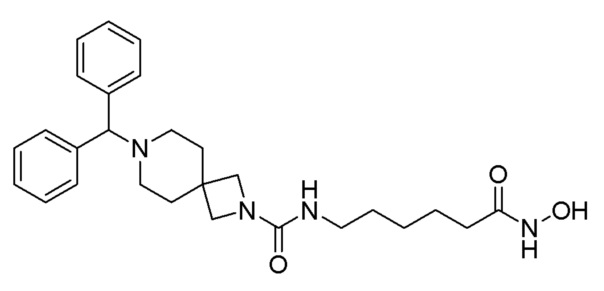
Соединение 1763
Соединение формулы 4-7 (0,148 г, 0,319 ммоль), полученное на стадии 3, и гидроксиламин (50,00% водный раствор, 0,195 мл, 3,192 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, к концентрату добавляли метанол (1 мл) и насыщенный водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Осажденное твердое вещество отфильтровывали, промывали гексаном и сушили, и полученный материал перекристаллизовывали из этилацетата (10 мл) при 25°С и фильтровали. Полученное твердое вещество промывали гексаном и сушили с получением целевого соединения 1763 (0,044 г, 29,7%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,40-7,38 (м, 4H), 7,29-7,25 (м, 4H), 7,18-7,14 (м, 2H), 6,18-6,17 (м, 1H), 4,26 (с, 1H), 3,42-3,33 (м, 4H), 2,91-2,90 (м, 2H), 2,20-2,19 (м, 4H), 1,85-1,82 (м, 2H), 1,65-1,64 (м, 4H), 1,43-1,40 (м, 2H), 1,33-1,30 (м, 2H), 1,19-1,15 (м, 2H); MS (ESI) m/z 465,3 (M++H).
Пример 27: Синтез соединения 1764
Стадия 1: Синтез метил-7-(7-бензгидрил-2,7-диазаспиро[3,5] нонан-2-карбоксамидо)гептаноата (формула 4-7)

Гидрохлорид метил-7-аминогептаноата (0,107 г, 0,549 ммоль) и трифосген (0,078 г, 0,261 ммоль) растворяли в метиленхлориде (5 мл) при 0°С, и к раствору добавляли N,N-диизопропилэтиламин (0,273 мл, 1,569 ммоль) с последующим перемешиванием при той же температуре. К реакционной смеси добавляли соединение формулы 4-6 (0,172 г, 0,523 ммоль), полученное на стадии 2 Примера 26, с последующим перемешиванием при комнатной температуре в течение 3 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили безводным сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (картридж SiO2, 4 г; метанол/метиленхлорид=от 0% до 3%) и концентрировали с получением целевого соединения формулы 4-7 (0,136 г, 54,4%) в виде ярко-красного твердого вещества.
Стадия 2: Синтез 7-бензгидрил-N-(7-(гидроксиамино)-7-оксогептил)-2,7-диазаспиро[3,5]нонан-2-карбоксамида (соединение 1764)
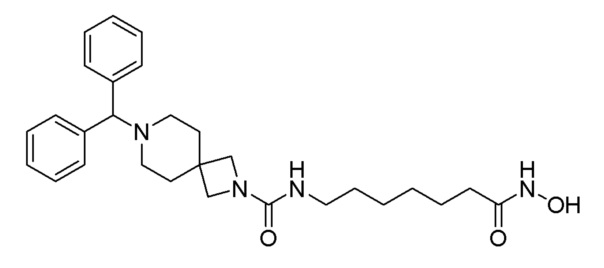
Соединение 1764
Соединение формулы 4-7 (0,136 г, 0,285 ммоль), полученное на стадии 1, и гидроксиламин (50,00%, 0,188 г, 2,847 ммоль) растворяли в метаноле (5 мл) при комнатной температуре, и раствор перемешивали при той же температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, и к концентрату добавляли метанол (1 мл) и насыщенный водный раствор бикарбоната натрия (30 мл) с последующим перемешиванием. Осажденное твердое вещество отфильтровывали, промывали гексаном и сушили, и полученный материал перекристаллизовывали из этилацетата (10 мл) при 25°С и фильтровали. Полученное твердое вещество промывали гексаном и сушили с получением целевого соединения 1764 (0,021 г, 15,4%) в виде белого твердого вещества.
1H ЯМР (400 Мгц, DMSO-d6) δ 7,40-7,38 (м, 4H), 7,29-7,25 (м, 4H), 7,18-7,14 (м, 2H), 6,16 (т, 1H, J=5,5 Гц), 4,26 (с, 1H), 3,42-3,41 (м, 4H), 2,92-2,88 (м, 2H), 2,19-2,18 (м, 4H), 1,88-1,84 (м, 2H), 1,65-1,64 (м, 4H), 1,43-1,42 (м, 2H), 1,32-1,31 (м, 2H), 1,19-1,18 (м, 4H); MS (ESI) m/z 479,6 (M++H).
Измерение активностей соединений по изобретению и аналитический протокол
Экспериментальный Пример 1: Подтверждение ингибирования ферментативной активности HDAC (in vitro)
Поскольку селективные ингибиторы HDAC6 важны из-за селективности ингибирования HDAC1, который вызывает побочные эффекты, была проанализирована селективность в отношении ферментов HDAC1/6 и селективность в отношении клеток (HDAC1: ацетилирование гистонов; HDAC6: ацетилирование тубулина).
1. Экспериментальный метод
С использованием флуориметрического набора для анализа лекарственных препаратов HDAC1 (Enzolifescities: BML-AK511) и рекомбинантного HDAC6 человека (Calbiochem: 382180) определяли способность испытуемых соединений ингибировать активности HDAC. Для анализа HDAC1 ее обрабатывали концентрациями 100, 1000 и 10000 нМ, и для анализа HDAC6 ее обрабатывали концентрациями 0,1, 1, 10, 100 и 1000 нМ, оставляли взаимодействовать при 37°С в течение 60 минут, а затем обрабатывали проявителем, и оставляли взаимодействовать при 37°С в течение 30 минут, после чего измеряли интенсивность флуоресценции (возбуждение - 390 нм; излучение - 460 нм) с использованием прибора FlexStatin3 (Molecular Device).
2. Экспериментальные данные и результаты
Результаты эксперимента представлены в Таблице 4.
Таблица 4
Способность ингибирования активности ферментов HDAC (HDAC 1 и HDAC 6)
Как показано в Таблице 4, приведенной выше, контрольное соединение ACY-1215 показало 48-кратную селективность (0,01 мкМ для HDAC6 и 0,48 мкМ для HDAC1), соединение 1102 показало 960-кратную селективность (0,004 мкМ для HDAC6 и 3,84 мкМ для HDAC1), соединение 1124 показало 170-кратную селективность (0,024 мкМ для HDAC6 и 4,09 мкМ для HDAC1) и соединение 1209 показало 193-кратную селективность (0,006 мкМ для HDAC6 и 1,16 мкМ для HDAC1), из чего следует, что новые производные по изобретению обладают превосходной селективностью в отношении ферментов HDAC1/HDAC6.
Экспериментальный Пример 2: Эффекты действия соединения 1102 в модели артрита, индуцированного адъювантом
1. Экспериментальный метод
100 мкл полного адъюванта Фрейнда (Chondrex) вводили внутрикожно в хвост каждой крысы Льюиса для индуцирования модели заболевания на животных. Крыс за один день до индуцирования делили на группы на основе массы тела, и испытуемое соединение вводили перорально крысам в различных дозах один раз в день с последующей оценкой результатов.
Со дня первого введения тестируемого соединения два раза в неделю проводили клиническую оценку состояния животных и регистрировали массу тела. Клиническую оценку выполняли по бальной системе (0-4 балла), а общую клиническую оценку регистрировали после наблюдения за лапами каждой крысы (0: нормальное состояние; и 16: очень тяжелый отек)
2. Экспериментальные результаты
Результаты эксперимента показаны на Фигуре 1. Лечебное действие испытуемого соединения в модели артрита оценивали на основе степени тяжести отека сустава, и более высокая клиническая оценка указывала на более тяжелую степень отека.
Как показано на Фигуре 1, группа, которая не получала соединение по изобретению (получавшая только носитель) имела показатель оценки 9-11 (тяжелый отек); в то время как группа, получавшая соединения 1102 в дозе 1 мг/кг, имела показатель оценки 6-8; группа, получавшая 10 мг/кг соединения 1102, имела показатель оценки 4-6; и группа, получавшая 50 мг/кг соединения 1102, имела показатель оценки 1-3, что свидетельствует о том, что соединение 1102 по изобретению облегчает симптомы артрита.
Промышленная применимость
Соединения по изобретению, представленные формулой I, их оптические изомеры или их фармацевтически приемлемые соли могут селективно ингибировать HDAC, и, таким образом, их можно эффективно использовать для профилактики или лечения опосредованных гистондеацетилазой заболеваний.
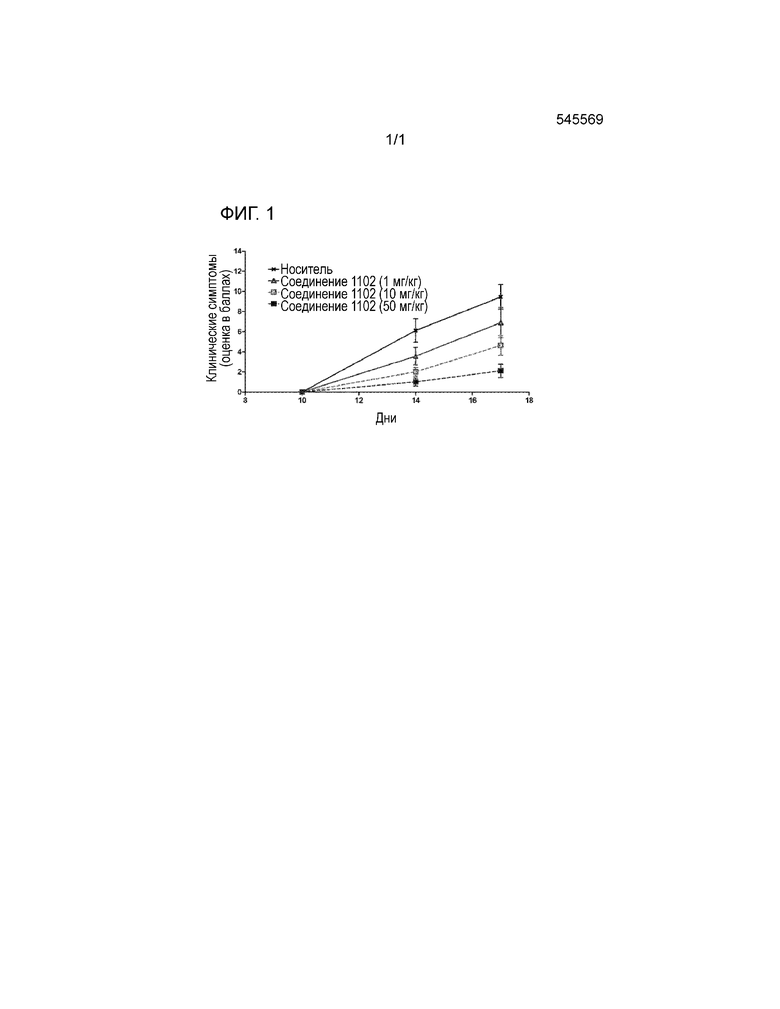
Изобретение относится к соединению, представленному формулой I, его оптическому изомеру или его фармацевтически приемлемой соли, которые обладают ингибирующей активностью в отношении гистондеацетилазы (HDAC). В формуле I Х представляет гетероциклический алкил, выбранный из группы, состоящей из
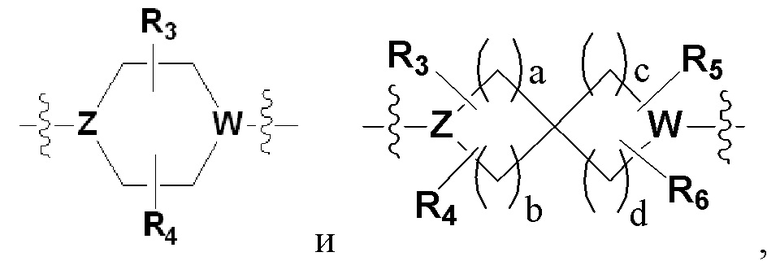
где каждый Z и W независимо представляет собой CH или N, при этом по меньшей мере один из Z и W представляет собой N, каждый a, b, с и d независимо равен 1, 2 или 3, каждый R3, R4, R5 и R6 независимо представляет собой -Н или -С1-С4-алкил; Y представляет собой C или N; каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил, -С3-С12 N-содержащий гетероарил, где -С6-С10 арил может быть независимо незамещенным или один или несколько атомов водорода в нем могут быть замещены на -CF3 или галоген; Q представляет собой C=O или SO2; R1 представляет собой -H или -C1-C4 алкил; R2 представляет собой -H, -OH, галоген или отсутствует; n равно 1, 2, 3 или 4. Изобретение относится также к фармацевтической композиции, способу лечения опосредованного гистондеацетилазой заболевания и к применению указанного соединения для изготовления лекарственного средства для лечения опосредованного гистондеацетилазой заболевания. 4 н. и 4 з.п. ф-лы, 1 ил., 4 табл., 27 пр.
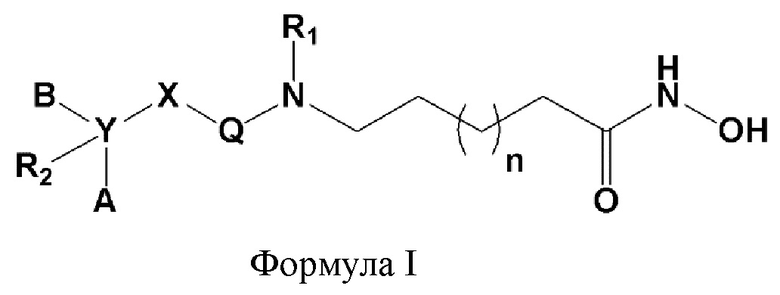
1. Соединение, представленное следующей формулой I, его оптический изомер или его фармацевтически приемлемая соль:
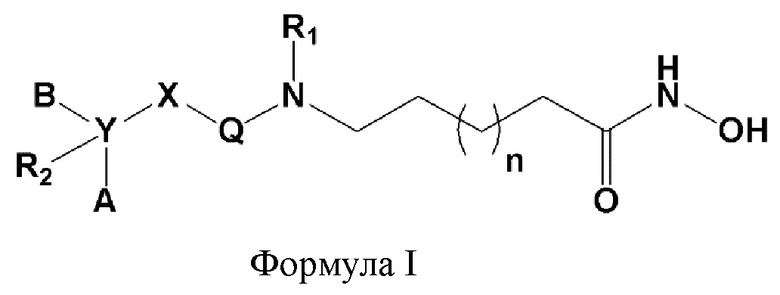
где Х представляет гетероциклический алкил, выбранный из группы, состоящей из
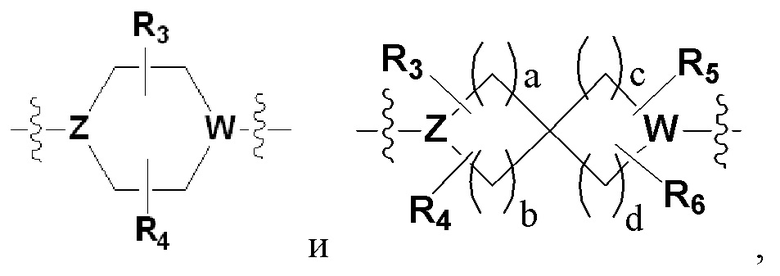
где каждый Z и W независимо представляет собой CH или N, при этом по меньшей мере один из Z и W представляет собой N,
каждый a, b, с и d независимо равен 1, 2 или 3 и
каждый R3, R4, R5 и R6 независимо представляет собой -Н или -С1-С4-алкил;
Y представляет собой C или N;
каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил, -С3-С12 N-содержащий гетероарил, где -С6-С10 арил может быть независимо незамещенным или один или несколько атомов водорода в нем могут быть замещены на -CF3 или галоген;
Q представляет собой C=O или SO2;
R1 представляет собой -H или -C1-C4 алкил;
R2 представляет собой -H, -OH, галоген или отсутствует, и если Y представляет собой C, то R2 представляет собой -Н, -ОН или галоген, и если Y представляет собой N, то R2 отсутствует; и
n равно 1, 2, 3 или 4.
2. Соединение, представленное формулой I, его оптический изомер или его фармацевтически приемлемая соль по п.1,
где Х представляет собой
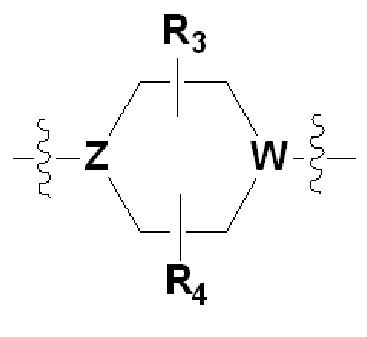 ,
,
где каждый Z и W независимо представляет собой CH или N и по меньшей мере один из Z и W представляет собой N, и
каждый R3 и R4 независимо представляет собой -Н или -С1-С4 алкил;
Y представляет собой C или N;
каждый А и В независимо представляет собой -С1-С4 алкил, -С6-С10 арил или -С3-С12 N-содержащий гетероарил, где -С6-С10 арил может быть независимо незамещенным или один или несколько атомов водорода в нем могут быть замещены на -CF3 или галоген;
Q представляет собой C=O;
R1 представляет собой -H или -C1-C4 алкил;
если Y представляет собой C, то R2 представляет собой -Н, -ОН или галоген, и если Y представляет собой N, то R2 отсутствует; и
n равно 3.
3. Соединение, представленное формулой I, его оптический изомер или его фармацевтически приемлемая соль по п.1, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, представленных в следующей таблице:
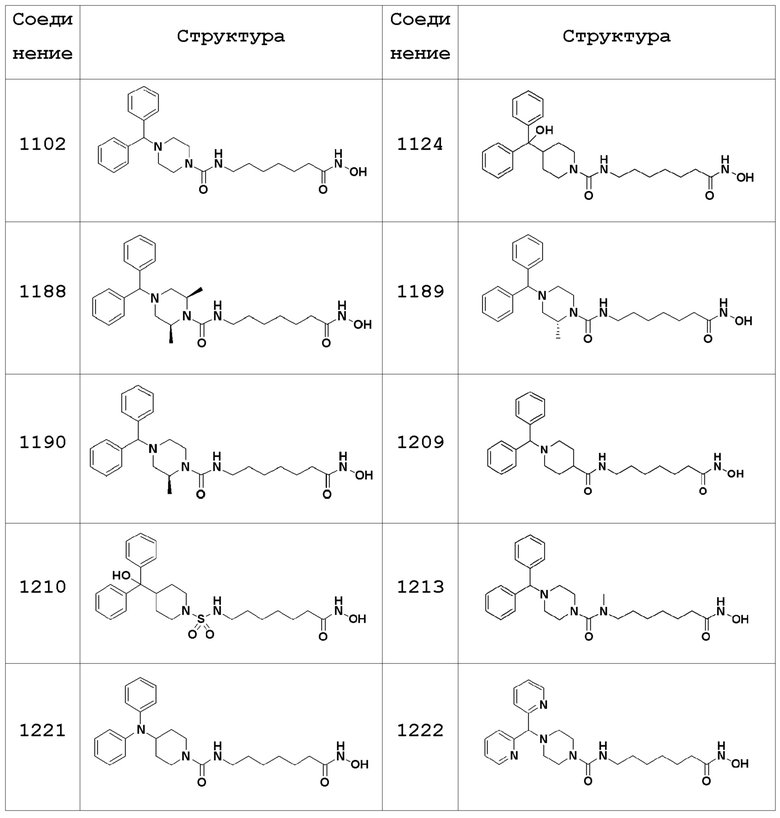
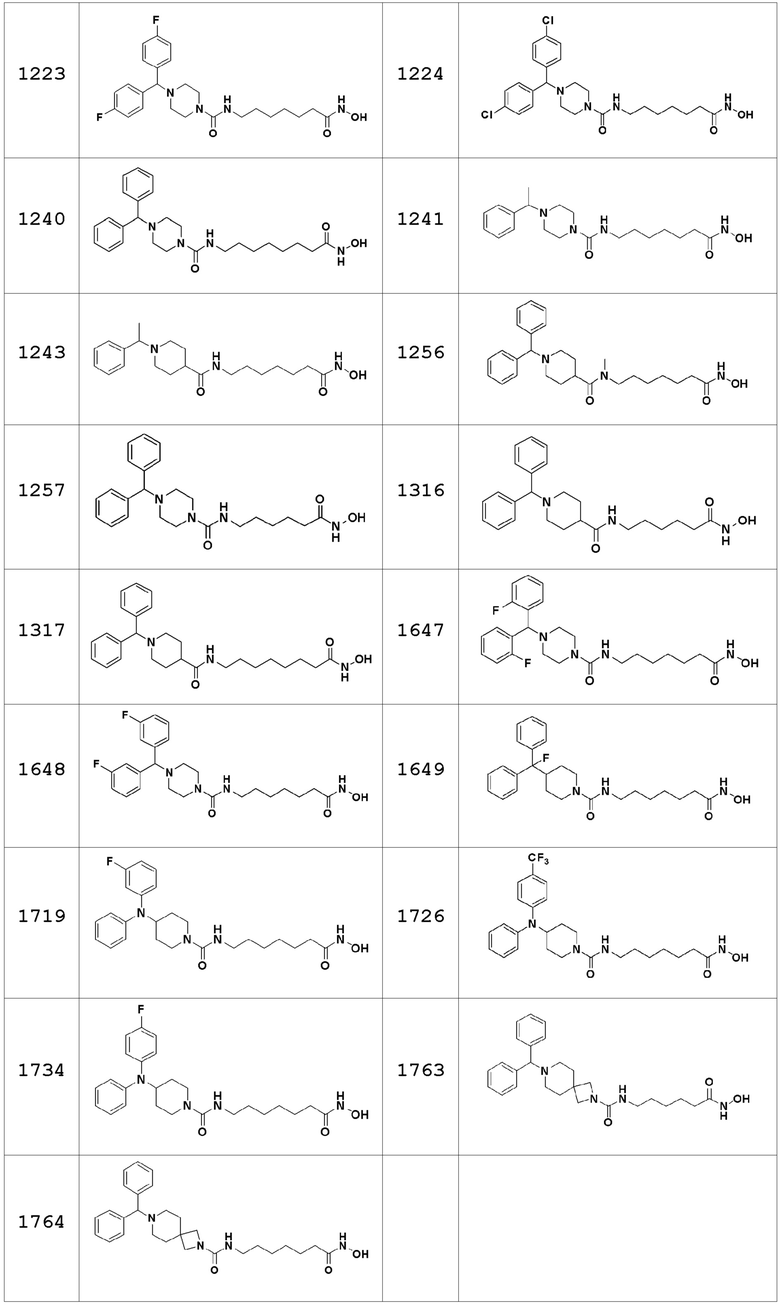
4. Соединение, представленное формулой I, его оптический изомер или его фармацевтически приемлемая соль по п.3, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, представленных в следующей таблице:
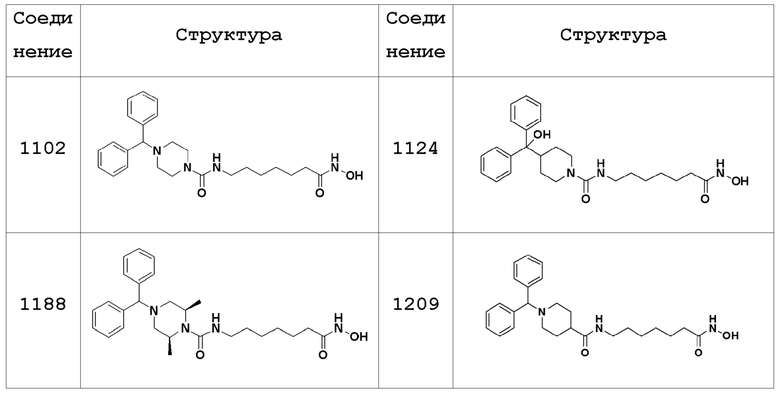
5. Фармацевтическая композиция для профилактики или лечения опосредуемого гистондеацетилазой заболевания, содержащая в качестве активного ингредиента соединение формулы I, его оптический изомер или его фармацевтически приемлемую соль по любому из пп.1-4.
6. Фармацевтическая композиция по п.5, где опосредованное гистондеацетилазой заболевание представляет собой клеточную пролиферативную болезнь, воспалительное заболевание, аутосомное доминантное заболевание, генетическое метаболическое заболевание, аутоиммунное заболевание, острое/хроническое неврологическое заболевание, гипертрофию, сердечную недостаточность, глазное заболевание или нейродегенеративное заболевание.
7. Способ лечения опосредованного гистондеацетилазой заболевания, включающий введение терапевтически эффективного количества соединения формулы I, его оптического изомера или его фармацевтически приемлемой соли по любому из пп.1-4.
8. Применение соединения формулы I, его оптического изомера или его фармацевтически приемлемой соли по любому из пп.1-4 для изготовления лекарственного средства для лечения опосредованного гистондеацетилазой заболевания.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| M | |||
| JUNG ET AL., Analogues of trichostatin A and trapoxin B as histone deacetylase inhibitors, BIOORG | |||
| MED | |||
| CHEM | |||
| LETT., 1997, Vol | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Насос | 1917 |
|
SU13A1 |
| Счетный прибор | 1924 |
|
SU1655A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА С ИНГИБИРУЮЩЕЙ ГИСТОНДЕАЦЕТИЛАЗУ АКТИВНОСТЬЮ | 2005 |
|
RU2416600C2 |

