[Область, к которой относится изобретение]
Настоящее изобретение относится к 1,3,4-оксадиазоламидным производным соединениям, обладающим ингибиторной активностью в отношении гистондеацетилазы 6 (HDAC6), их стереоизомерам или их фармацевтически приемлемым солям; их применениям для получения терапевтических лекарственных средств; способам лечения заболеваний с использованием таких соединений; фармацевтическим композициям, включающим такие соединения; и способам их получения.
[Предпосылки создания изобретения]
Пост-трансляционные модификации, такие как ацетилирование, являются существенно значимыми регуляторными компонентами, лежащими в основе биологических процессов в клетках, и строго регулируются множеством ферментов. Гистоны являются главными белковыми компонентами хроматина и действуют как катушки, вокруг которых нити ДНК, способствуя ДНК конденсации. Также баланс ацетилирования и деацетилирования гистонов играет критическую роль в регуляции генной экспрессии.
Гистондеацетилазы (HDACs) являются ферментами, которые удаляют ацетильные группы из лизиновых остатков на гистоновых белках хроматина, и известно, что они связаны с генным сайленсингом и индуцируют арест клеточного цикла, ангиогенное ингибирование, иммунную регуляцию, гибель клеток и т.д. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщалось о том, что ингибирование ферментативной функции HDACs индуцирует апоптоз раковых клеток in vivo путем снижения активности факторов, ассоциированных с выживанием раковых клеток, и факторов, активирующих апоптоз раковых клеток (Warrell et al, J. Natl. Рак Inst. 1998, 90, 1621-1625).
У человека было идентифицировано 18 HDAC, и они подразделяются на четыре класса на основании их гомологии с HDAC дрожжей. Из них 11 HDAC используют цинк в качестве кофактора, и могут быть подразделены на три группы: Класс I (HDAC1, 2, 3 и 8), Класс II (IIa: HDAC4, 5, 7 и 9; IIb: HDAC6 и 10), Класс IV (HDAC 11). Кроме того, 7 HDAC Класса III (SIRT 1-7) требуют NAD+ вместо цинка в качестве кофактора (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Различные ингибиторы HDAC находятся в стадии в предклинической или клинической разработки, но к настоящему времени были идентифицированы только неселективные ингибиторы HDAC в качестве противораковых средств, и только вориностат (SAHA) и ромидепсин (FK228) были одобрены для лечения кожной T-клеточной лимфомы, и панобиностат (LBH-589) был одобрен для лечения множественной миеломы. Однако известно, что неселективные ингибиторы HDAC вызывают побочные эффекты, такие как слабость и тошнота, обычно при высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Такие побочные эффекты, как сообщалось, возникают в результате ингибирования HDACs класса I. Из-за таких побочных эффектов использование неселективных ингибиторов HDAC в разработке лекарственных средств, отличных от противораковых лекарственных средств, было ограничено (Witt et al., Cancer Letters, 2009, 277, 8-21).
Между тем сообщалось о том, что селективное ингибирование HDACs класса II не будет вызывать токсичность, демонстрируемую при ингибировании HDACs класса I. Также при разработке селективных ингибиторов HDAC побочные эффекты, такие как токсичность, которые вызываются неселективным ингибированием HDAC, могут быть преодолены. Таким образом, селективные ингибиторы HDAC имеют потенциал для разработки в качестве терапевтических средств, эффективных для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, член Класса IIb HDACs, присутствует преимущественно в цитоплазме и вовлечен в деацетилирование ряда не-гистоновых субстратов (HSP90, кортактин и т.д.), включая тубулин (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 содержит два каталитических домена, и домен типа цинкового пальца на C-конце может связываться с убиквитинированными белками. Известно, что HDAC6 имеет ряд не-гистоновых белков в качестве субстратов, и таким образом играет важную роль в различных заболеваниях, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные расстройства (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Общей структурной характеристикой различных ингибиторов HDAC является структура, состоящая из кэп-группы, линкера и цинк-связывающей группы (ZBG), как показано в следующей структуре Вориностата. Многие исследователи изучали ингибиторную активность и селективность в отношении фермента, структурно модифицируя кэп-группу и линкер. Из этих групп, цинк-связывающая группа известна как играющая более важную роль в ингибиторной активности и селективности в отношении фермента (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).

Цинк-связывающая группа, как правило, представляет собой гидроксамовую кислоту или бензамидное производное. При этом, производное гидроксамовой кислоты демонстрирует сильный эффект ингибирования HDAC, но имеет такие проблемы, как низкая биодоступность и сильная ненаправленная активность. Кроме того, бензамидное производное имеет проблему, связанную с тем, что он может продуцировать токсические метаболиты in vivo, поскольку он содержит анилин (Woster et al., Med. Chem. Commun. 2015, онлайн публикация).
Соответственно, существует потребность в разработке селективных ингибиторов HDAC 6 для лечения заболеваний, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания и нейродегенеративные расстройства, которые содержат цинк-связывающую группу, имеют улучшенную биодоступность и в то же время не вызывают побочных эффектов, в отличие от неселективных ингибиторов, которые вызывают побочные эффекты.
[Раскрытие изобретения]
[техническая задача]
Целью настоящего изобретения является обеспечение 1,3,4-оксадиазоламидных производных соединений, обладающих селективной ингибиторной активностью в отношении HDAC6, их стереоизомеров или их фармацевтически приемлемых солей.
Другой целью настоящего изобретения является обеспечение фармацевтических композиций, содержащих 1,3,4-оксадиазоламидные производные соединения, обладающие селективной ингибиторной активностью в отношении HDAC6, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является обеспечение способов для получения новых соединений.
Еще одной целью настоящего изобретения является обеспечение фармацевтических композиций для профилактики или лечения ассоциированных с HDAC6 активностью заболеваний, включая инфекционные заболевания; новообразования; эндокринные, связанные с питанием и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; заболевания глаз и смежных органов; сердечно-сосудистые заболевания; респираторные заболевания; заболевания пищеварительного тракта; заболевания кожной и подкожной ткани; заболевания скелетно-мышечной системы и соединительной ткани; или врожденные дефекты развития, деформации и хромосомные аномалии, которые содержат описанные выше соединения.
Еще одной целью настоящего изобретения является обеспечение применения соединений для получения терапевтических лекарственных средств против ассоциированных с HDAC6 активностью заболеваний.
Еще одной целью настоящего изобретения является обеспечение способов для лечения ассоциированных с HDAC6 активностью заболеваний, которые включают введение терапевтически эффективного количества фармацевтических композиций, содержащих такие соединения.
[Техническое решение]
Авторами настоящего изобретения были обнаружены 1,3,4-оксадиазоламидные производные соединения, которые обладают ингибиторной активностью в отношении гистондеацетилазы 6 (HDAC6), и было обнаружено, что эти соединения можно использовать для ингибирования или лечения ассоциированных с активностью гистондеацетилазы 6 (HDAC6) заболеваний, таким образом, было создано настоящее изобретение.
1,3,4-оксадиазоламидные производные соединения
Для достижения указанных выше целей настоящее изобретение обеспечивает 1,3,4-оксадиазоламидное производное соединение, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемую соль:
[Формула I]

где L1, L2 и L3 каждый независимо представляют собой -(C0-C2 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где три или более из Z1 - Z4 одновременно не могут представлять собой N, и RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой -(C1-C4 алкил), -(C1-C4 алкил)-O(C1-C4 алкил), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -(C3-C6 циклоалкил), -арил, -гетероарил,  или
или  , где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
, где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
Y представляет собой -N-, -O- или -S(=O)2-,
когда Y представляет собой -N-, R4 и R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-(C3-C6 циклоалкил), -C(=O)-O(C1-C4 алкил), -C(=O)-CF3, -S(=O)2-(C1-C4 алкил), -(C2-C6 гетероциклоалкил), бензил или амин защитную группу, где -(C2-C6 гетероциклоалкил) может содержать N, O или S атом в кольце,
и, когда Y представляет собой -O- или -S(=O)2-, R4 и R8 отсутствуют,
R5-R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -OH, -CH2OH или -C(=O)-NH2, и
a - c каждый независимо представляют собой целое число, имеющее значение 1, 2 или 3;
R3 представляет собой -H, -(C1-C4 алкил), -(C1-C4 алкил)-O(C1-C4 алкил), -(C1-C4 алкил)-C(=O)-O(C1-C4 алкил), -(C3-C6 циклоалкил), -арил, -гетероарил, 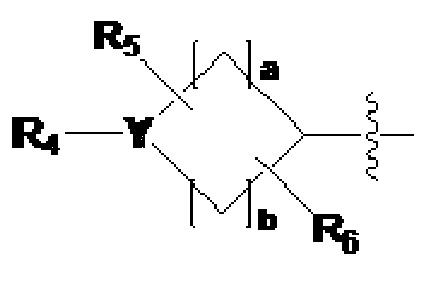 или
или  , где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть независимо замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3, и
, где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть независимо замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3, и
R4, R5, R6, Y, a, b, R1, L1, Z1, Z2, Z3 и Z4 имеют значение, определенное выше; и
X представляет собой F, Cl, Br или I.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения,
L1 и L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1-C2 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где два или более из Z1-Z4 одновременно не могут представлять собой N, и RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой -(C1-C4 алкил), -(C3-C6 циклоалкил), -арил, -гетероарил, или  , где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
, где по меньшей мере один H из -(C3-C6 циклоалкил), -арила или -гетероарила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
Y представляет собой -N-, -O- или -S(=O)2-,
когда Y представляет собой -N-, R4 и R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-CF3, -S(=O)2-(C1-C4 алкил), -(C2-C6 гетероциклоалкил), -C(=O)-(C3-C6 циклоалкил), бензил или амин защитную группу, где -(C2-C6 гетероциклоалкил) может содержать O атом в кольце,
и, когда Y представляет собой -O- или -S(=O)2-, R4 и R8 отсутствуют,
R5-R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -OH, -CH2OH или -C(=O)-NH2, и
a - c каждый независимо представляют собой целое число, имеющее значение 1, 2 или 3;
R3 представляет собой -арил или -гетероарил, где по меньшей мере один H из -арила или -гетероарила может быть независимо замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3; и
X представляет собой F, Cl, Br или I.
В соответствии с более предпочтительным вариантом осуществления настоящего изобретения,
L1 и L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где два или более из Z1-Z4 одновременно не могут представлять собой N, и RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой -(C1-C4 алкил), -пиридинил или , где по меньшей мере один H пиридинила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
Y представляет собой -N-,
R4 представляет собой -(C1-C4 алкил), -C(=O)-(C1-C4 алкил) или -S(=O)2-(C1-C4 алкил),
R5 и R6 каждый независимо представляют собой -H или -(C1-C4 алкил), и
a и b каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой -арил, где по меньшей мере один H арила может быть замещен -X заместителем; и
X представляет собой F, Cl, Br или I.
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения,
L1 и L3 каждый независимо представляют собой -(C0 алкил)-;
L2 представляет собой -(C1 алкил)-;
Z1-Z4 каждый независимо представляют собой N или CRZ, где два или более из Z1 - Z4 одновременно не могут представлять собой N, и RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой -пиридинил или , где по меньшей мере один H пиридинила может быть замещен группой -X, -OH, -(C1-C4 алкил), -O(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-O(C1-C4 алкил) или -CF3,
Y представляет собой -N-,
R4 представляет собой -(C1-C4 алкил), -C(=O)-(C1-C4 алкил) или -S(=O)2-(C1-C4 алкил),
R5 или R6 каждый независимо представляют собой -H, и
a и b каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой -арил, где по меньшей мере один H арила может быть замещен -X заместителем; и
X представляет собой F, Cl, Br или I.
Конкретные соединения, представленные формулой I, показаны в Таблице 1 ниже:
Таблица 1






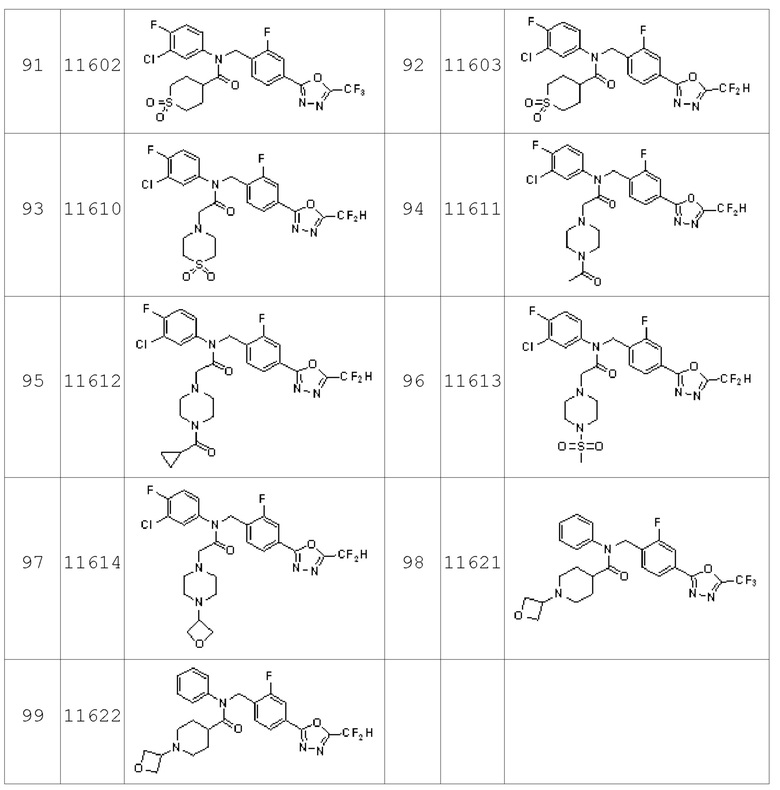
Предпочтительно, соединения, представленные формулой I, их стереоизомеры или их фармацевтически приемлемые соли могут быть выбраны из группы, состоящей из соединений 11110, 11189, 11233, 11237, 11238, 11239, 11240, 11241, 11242, 11243, 11245, 11327, 11332, 11333, 11334, 11339, 11341, 11359, 11360, 11376, 11414, 11418 и 11419. Более предпочтительно, соединения, представленные формулой I, их стереоизомеры или их фармацевтически приемлемые соли могут быть выбраны из группы, состоящей из соединений 11189, 11233, 11239, 11241, 11242, 11243, 11333, 11334, 11341, 11359, 11360, 11376, 11414, 11418 и 11419.
В контексте настоящей заявки термин ''фармацевтически приемлемая соль'' означает любую соль, которую, как правило, используют в фармацевтике. Примеры фармацевтически приемлемой соли включают, но не ограничиваются этим, соли с неорганическими ионами, такими как ионы кальция, калия, натрия или магния, соли с неорганическими кислотами, такими как хлористоводородная кислота, азотная кислота, фосфорная кислота, бромноватая кислота, иодноватая кислота, перхлорная кислота или серная кислота, соли с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, угольная кислота, ванилиновая кислота, иодистоводородная кислота или т.п., соли с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота или нафталинсульфоновая кислота, соли с аминокислотами, такими как глицин, аргинин или лизин, и соли с аминами, такими как триметиламин, триэтиламин, аммиак, пиридин или пиколин.
В настоящем изобретении предпочтительные соли включают гидрохлорид, фосфат, сульфат, трифторацетат, цитрат, бромат, малеат или тартрат, и предпочтительные примеры таких соединений включают 11022, 11136 и 11137, которые раскрыты в настоящей заявке.
Соединения, представленные формулой I, могут содержать один или несколько асимметричных атомов углерода и, таким образом, могут существовать в форме рацематов, рацемических смесей, отдельных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Соединения формулы I можно разделить на такие изомеры способами, известными из уровня техники, например, колоночной хроматографией или ВЭЖХ. Альтернативно, стереоизомеры соединений формулы I можно синтезировать методом стереоспецифического синтеза с использованием оптически чистых исходных веществ и/или реагентов известной конфигурации.
Способы для получения 1,3,4-оксадиазоламидных производных соединений
Настоящее изобретение обеспечивает способы для получения 1,3,4-оксадиазоламидных производных соединений, представленных формулой I, их стереоизомеров или их фармацевтически приемлемых солей.
Предпочтительные способы для получения 1,3,4-оксадиазоламидных производных соединений, представленных формулой I, их стереоизомеров или их фармацевтически приемлемых солей являются такими, как показано на схемах реакций 1-5 ниже, и также включают модификации, очевидные специалистам в данной области.
[Схема реакций 1]

Схема реакций 1 выше показывает способ для синтеза соединений, имеющих амидную структуру. Как показано на схеме реакций 1, соединение формулы 1-1 подвергают восстановительному аминированию при помощи аминового соединения, или соединение формулы 1-2 подвергают реакции замещения аминовым соединением, таким образом получая соединение формулы 1-3. Соединение формулы 1-3 подвергают взаимодействию с ацилхлоридом формулы 1-4 для синтеза соединения формулы 1-5, которое затем подвергают взаимодействию с гидразином, таким образом получая соединение формулы 1-6. Соединение формулы 1-6 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения формулы 1-7. Когда получают соединение формулы 1-8, в котором оксадиазольное кольцо не образовано, его подвергают взаимодействию с 1-метокси-N-триэтиламмониосульфонил-метанимидатом (реагент Бургесса) с получением соединения формулы 1-8.
Соединения, которые синтезированы в соответствии со схемой реакций 1, представляют собой соединения 11022, 11105, 11106, 11107, 11108, 11109, 11110, 11188, 11189, 11246, 11247, 11339, 11340, 11341, 11356, 11357, 11358, 11359, 11360, 11376 и 11584.
[Схема реакций 2]

Схема реакций 2 выше показывает способ для синтеза соединений, имеющих гетероциклоалкиламидную структуру. Как показано на схеме реакций 2, соединение формулы 2-1 подвергают взаимодействию с аминовым соединением для синтеза соединения формулы 2-2, которое затем подвергают реакции замещения, синтезируя таким образом соединение формулы 2-3. Соединение формулы 2-3 подвергают взаимодействию с гидразином с получением соединения формулы 2-4. Соединение формулы 2-4 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения формулы 2-5. Когда получают соединение формулы 2-6, в котором оксадиазольное кольцо не образовано, его подвергают взаимодействию с 1-метокси-N-триэтиламмониосульфонил-метанимидатом (реагент Бургесса) или метансульфонилхлоридом с получением соединения формулы 2-5, у которого затем удаляют защиту, таким образом получая соединение формулы 2-7. Соединение формулы 2-7 подвергают взаимодействию с альдегидом, ацилхлоридом, сульфонилхлоридом, уксусным ангидридом, оксетан-3-оном или т.п., синтезируя таким образом соединение формулы 2-8.
Соединения, который синтезированы в соответствии со схемой реакций 2 выше, представляют собой соединения 11134, 11135, 11136, 11137, 11138, 11139, 11140, 11141, 11142, 11143, 11157, 11158, 11159, 11160, 11161, 11162, 11163, 11164, 11165, 11166, 11187, 11200, 11201, 11202, 11203, 11204, 11205, 11206, 11207, 11208, 11209, 11210, 11211, 11212, 11213, 11214, 11215, 11232, 11233, 11234, 11235, 11236, 11237, 11238, 11239, 11240, 11241, 11242, 11243, 11244, 11245, 11325, 11326, 11327, 11328, 11329, 11330, 11331, 11332, 11333, 11334, 11621 и 11622.
[Схема реакций 3]

Схема реакций 3 выше показывает способ для синтеза соединений, имеющих гетероциклоалкиламидную структуру. Как показано на схеме реакций 3, соединение формулы 2-3 подвергают процедуре удаления защиты с получением соединения формулы 3-4, которое затем подвергают восстановительному аминированию, таким образом получая соединение формулы 3-5. Соединение формулы 3-5 подвергают взаимодействию с гидразином с получением соединения формулы 3-6. Соединение формулы 3-6 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом для синтеза соединения формулы 3-8.
Соединения, которые синтезированы в соответствии со схемой реакций 3 выше, представляют собой соединения 11414, 11418 и 11419.
[Схема реакций 4]

Схема реакций 4 выше показывает способ для синтеза соединений, имеющих гетероциклоалкиламидную структуру. Как показано на схеме реакций 4, соединение формулы 4-1 подвергают взаимодействию с аминовым соединением с получением соединения формулы 4-2, которое затем подвергают реакции замещения, синтезируя таким образом соединение формулы 4-3. Соединение формулы 4-3 подвергают взаимодействию с гидразином с получением соединения формулы 4-4. Соединение формулы 4-4 подвергают взаимодействию с дифторуксусным ангидридом для синтеза соединения формулы 4-5. Соединение формулы 4-5 подвергают процедуре удаления защиты с получением соединения формулы 4-6, которое затем подвергают взаимодействию с метансульфонилхлоридом, таким образом получая соединение формулы 4-7. Соединение формулы 4-7 подвергают взаимодействию с замещенным циклоамином с получением соединения формулы 4-8. Кроме того, соединение формулы 4-7 подвергают взаимодействию с морфолиновым, тиоморфолиновым или пиперазиновым производным для синтеза соединения формулы 4-9. Когда продукт представляет собой незамещенный пиперазин, его подвергают взаимодействию с сульфонилхлоридом, уксусным ангидридом или оксетан-3-оном с получением соединения формулы 4-10.
Соединения, которые синтезированы в соответствии со схемой реакций 4 выше, представляют собой соединения 11534, 11535, 11536, 11537, 11538, 11610, 11611, 11612, 11613 и 11614.
[Схема реакций 5]

Схема реакций 5 выше показывает способ для синтеза соединений, имеющих гетероциклоалкиламидную структуру. Как показано на схеме реакций 5, соединение формулы 5-1 подвергают взаимодействию с аминовым соединением с получением соединения формулы 5-2, которое затем подвергают реакции замещения с получением соединения формулы 5-3. Соединение формулы 5-3 подвергают взаимодействию с гидразином с получением соединения формулы 5-4. Соединение формулы 5-4 подвергают взаимодействию с трифторуксусным ангидридом или дифторуксусным ангидридом для синтеза соединения формулы 5-5, которое затем подвергают взаимодействию с метансульфонилхлоридом, синтезируя таким образом соединение формулы 5-6.
Соединения, которые синтезированы в соответствии со схемой реакций 5 выше, представляют собой соединения 11602 и 11603.
Композиции, включающие 1,3,4-оксадиазоламидные производные соединения, их применение и способ лечения заболеваний с использованием таких композиций
Настоящее изобретение обеспечивает фармацевтическую композицию для профилактики или лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6 (HDAC6), которая содержит, в качестве активного ингредиента, соединение, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемую соль:
[Формула I]

где формула I определена выше.
Фармацевтическая композиция в соответствии с настоящим изобретением демонстрирует замечательный эффект профилактики или лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6, селективно ингибируя гистондеацетилазу 6.
Заболевания, ассоциированные с активностью гистондеацетилазы 6, включают инфекционные заболевания, такие как прионовая болезнь; новообразования, такие как доброкачественная опухоль (например, миелодиспластический синдром) или злокачественная опухоль (например, множественная миелома, лимфома, лейкоз, рак легкого, рак прямой кишки, рак толстой кишки, рак предстательной железы, уротелиальная карцинома, рак молочной железы, меланома, рак кожи, рак печени, рак головного мозга, рак желудка, рак яичников, рак поджелудочной железы, рак головы и шеи, рак полости рта или глиома); эндокринные, связанные с питанием и метаболические заболевания, такие как болезнь Вильсона, амилоидоз или диабет; психические и поведенческие расстройства, такие как депрессия или синдром Ретта, и тому подобное; неврологические заболевания, такие как атрофия центральной нервной системы (например, болезнь Гентингтона, спинальная мышечная атрофия (SMA), спиноцеребеллярная атаксия (SCA)), нейродегенеративное заболевание (например, болезнь Альцгеймера), двигательное расстройство (например, болезнь Паркинсона), невропатия (например, наследственная невропатия (болезнь Шарко-Мари-Тута)), спорадическая невропатия, воспалительная невропатия, лекарственно-индуцированная невропатия), болезни моторных нейронов (амиотрофический боковой склероз (ALS)) или демиелинизирующие заболевания центральной нервной системы (например, рассеянный склероз (MS)) и тому подобное; заболевания глаз и прилежащих органов, такие как увеит; сердечно-сосудистые заболевания, такие как фибрилляция предсердий или инсульт и тому подобное; респираторные заболевания, такие как астма; заболевания пищеварительного тракта, такие как алкогольная болезнь печени, воспалительное заболевание кишечника, болезнь Крона или язвенная болезнь кишечника и тому подобное; заболевания кожной и подкожной ткани, такие как псориаз; заболевания костно-мышечной системы и соединительной ткани, такие как ревматоидный артрит, остеоартрит или системная красная волчанка (СКВ) и тому подобное; или врожденные пороки развития, деформации и хромосомные аномалии, такие как аутосомно-доминантное поликистозное заболевание почек, а также расстройства или заболевания, связанные с аномальной функцией гистондезацетилазы.
Фармацевтически приемлемая соль представляет собой соль, которая описана выше применительно к фармацевтически приемлемой соли соединения, представленного формулой I в соответствии с настоящим изобретением.
Для введения фармацевтическая композиция в соответствии с настоящим изобретением может дополнительно содержать по меньшей мере один фармацевтически приемлемый носитель в дополнение к соединению формулы I, его стереоизомеру или его фармацевтически приемлемой соли. Фармацевтически приемлемый носитель, который используют в настоящем изобретении, может представлять собой по меньшей мере один из таких носителей, как физиологический раствор, стерильная вода, раствор Рингера, забуференный физиологический раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь двух или более из них. Если необходимо, композиция может содержать другие обычные добавки, такие как антиоксидант, буфер или бактериостатический агент. Кроме того, композиция может быть сформулирована в виде инъекционных препаратов, таких как растворы, суспензии, эмульсии и т.д., пилюль, капсул, гранул или таблеток с использованием разбавителя, диспергирующего агента, поверхностно-активного вещества, связующего и смазывающего вещества. Таким образом, композиция по настоящему изобретению может быть в виде пластырей, жидкостей, пилюль, капсул, гранул, таблеток, суппозиториев и т.д. Эти лекарственные формы можно получить либо обычными способами, которые используются для формулирования в данной области, либо способом, раскрытым в Remington's Pharmaceutical Science (последнее издание), Mack Publishing Company, Easton PA, и они могут быть получены в зависимости от заболеваний или компонентов.
Фармацевтическую композицию по настоящему изобретению можно вводить перорально или парентерально (например, внутривенно, подкожно, интраперитонеально или местно), в зависимости от предполагаемого применения. Доза фармацевтической композиции варьируется в зависимости от массы тела, возраста, пола, состояния здоровья и режима питания пациента, времени введения, способа введения, скорости экскреции, тяжести заболевания и тому подобного. Суточная доза соединения формулы I в соответствии с настоящим изобретением может составлять от 1 до 1000 мг/кг, предпочтительно от 5 до 100 мг/кг, и ее можно вводить от одного до нескольких раз в день.
Фармацевтическая композиция по настоящему изобретению может также содержать, в дополнение к соединению, представленному формулой I, его стереоизомеру или его фармацевтически приемлемой соли, один или несколько активных ингредиентов, которые проявляют лекарственную эффективность, идентичную или сходную с соединением по изобретению.
Настоящее изобретение также обеспечивает способ профилактики или лечения заболевания, ассоциированного с активностью гистондезацетилазы 6, который включает введение терапевтически эффективного количества соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли.
Используемый здесь термин ''терапевтически эффективное количество'' относится к количеству соединения, представленного формулой I, которое эффективно для профилактики или лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6.
Настоящее изобретение также обеспечивает способ селективного ингибирования HDAC6, который включает введение соединения формулы I, его стереоизомера или его фармацевтически приемлемой соли млекопитающим, включая человека.
Способ профилактики или лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6, в соответствии с настоящим изобретением включает ингибирование или предотвращение заболевания, а также устранение самого заболевания до появления симптомов путем введения соединения, представленного формулой I. При лечении заболеваний величина профилактической или терапевтической дозы конкретного активного ингредиента будет варьироваться в зависимости от характера и тяжести заболевания или состояния и может также варьироваться в зависимости от пути, которым вводится активный ингредиент. Доза и частота введения также будут варьироваться в зависимости от возраста, массы тела и реакции отдельного пациента. Подходящие схемы введения легко могут выбрать специалисты в данной области с учетом таких факторов. Кроме того, способ профилактики или лечения заболеваний, связанных с активностью гистондезацетилазы 6, в соответствии с настоящим изобретением может дополнительно включать введение терапевтически эффективного количества дополнительного активного средства, полезного для лечения заболевания, вместе с соединением, представленным формулой I, где дополнительное активное средство может проявлять синергический эффект с соединением формулы I или вспомогательный эффект.
Настоящее изобретение также направлено на обеспечение применения соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболеваний, ассоциированных с активностью гистондеацетилазы 6. Для получения лекарственного средства соединение, представленное формулой I, можно смешать с фармацевтически приемлемым адъювантом, разбавителем, носителем или т.п. и объединить с другими активными средствами таким образом, чтобы активные ингредиенты могли иметь синергические эффекты.
Характерные особенности, относящиеся к применению, композиции и способу лечения по настоящему изобретению, можно подходящим образом объединить, если только они не противоречат друг другу.
[Полезные эффекты]
Соединения, представленные формулой I в соответствии с настоящим изобретением, их стереоизомеры или их фармацевтически приемлемые соли могут селективно ингибировать HDAC6 и, таким образом, проявлять отличные эффекты при профилактике или лечении заболеваний, ассоциированных с активностью гистондеацетилазы 6.
[Способ осуществления изобретения]
Далее настоящее изобретение будет описано более подробно со ссылкой на примеры и экспериментальные примеры. Однако эти примеры представлены для иллюстративных целей только и не предназначены для ограничения объема настоящего изобретения.
Получение 1,3,4-оксадиазоламидных производных соединений
Конкретные способы для получения соединений формулы I являются следующими.
Пример 1: Синтез соединения 11022, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)изоникотинамида
[Стадия 1] Синтез метил 4-((фениламино)метил)бензоата
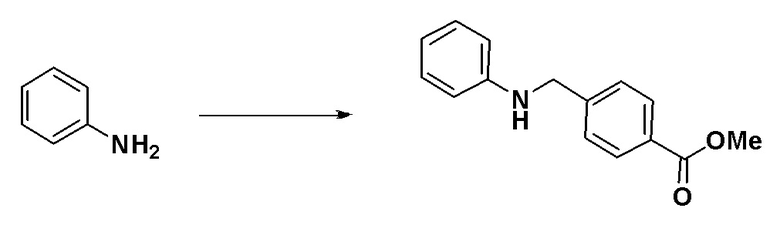
Анилин (1,961 мл, 21,475 ммоль), метил 4-формилбензоат (4,230 г, 25,770 ммоль) и уксусную кислоту (0,614 мл, 10,738 ммоль) растворяли в метиленхлориде (50 мл) и раствор перемешивали при 0°C в течение 10 минут. Затем к перемешиваемому раствору добавляли триацетоксиборогидрид натрия (6,828 г, 32,213 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (4,730 г, 91,3%) в виде бесцветного масла.
[Стадия 2] Синтез метил 4-((N-фенилизоникотинамидо)метил)бензоата

4-((фениламино)метил)бензоат (0,150 г, 0,622 ммоль), синтезированный на стадии  1, гидрохлорид изоникотиноилхлорида (0,221 г, 1,243 ммоль) и N,N-диизопропилэтиламин (0,194 мл, 1,243 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,179 г, 83,1%) в виде белого твердого вещества.
1, гидрохлорид изоникотиноилхлорида (0,221 г, 1,243 ммоль) и N,N-диизопропилэтиламин (0,194 мл, 1,243 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,179 г, 83,1%) в виде белого твердого вещества.
[Стадия 3] Синтез N-(4-(гидразинкарбонил)бензил)-N-фенилизоникотинамида
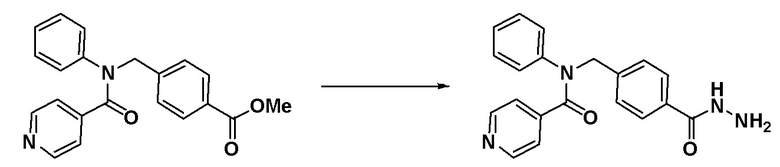
Метил 4-((N-фенилизоникотинамидо)метил)бензоат (0,179 г, 0,517 ммоль), синтезированный на стадии 2, и гидразин гидрат (0,488 мл, 10,335 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,134 г, 74,9%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 11022

N-(4-(гидразинкарбонил)бензил)-N-фенилизоникотинамид (0,105 г, 0,303 ммоль), синтезированный на стадии 3, трифторуксусный ангидрид (0,051 мл, 0,364 ммоль) и триэтиламин (0,084 мл, 0,606 ммоль) растворяли в метиленхлориде (20 мл) при комнатной температуре и раствор перемешивали при этой же температуре. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = от 0% до 30%) и концентрировали с получением указанного в заголовке соединения (0,035 г, 26,1%) в виде белого пенистого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,48 (д, 2H, J=5,8 Гц), 8,06 (д, 2H, J=8,3 Гц), 7,49 (д, 2H, J=8,2 Гц), 7,28-7,14 (м, 5H), 6,98-6,82 (м, 2H), 5,17 (д, 2H, J=19,0 Гц); LRMS(масс-спектрометрия низкого разрешения) (ES) m/z 425,2 (M++1).
[Стадия 5] Синтез соединения 11022 гидрохлорида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)изоникотинамид (0,100 г, 0,236 ммоль), синтезированный на стадии 4, растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (1,00 M раствор в этилацетате, 0,259 мл, 0,259 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли этилацетат (2 мл) с последующим перемешиванием. Осажденное твердое вещество фильтровали, промывали раствором этилацетата и сушили с получением указанного в заголовке соединения (0,108 г, 99,5%) в виде белого твердого вещества.
Пример 2: Синтез соединения 11105, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)ацетамида
[Стадия 1] Синтез метил 4-((N-фенилацетамидо)метил)бензоата

Метил 4-((фениламино)метил)бензоат (0,200 г, 0,829 ммоль) и диизопропилэтиламин (0,290 мл, 1,658 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,088 мл, 1,243 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,220 г, 93,7%) в виде белого твердого вещества.
[Стадия 2] Синтез N-(4-(гидразинкарбонил)бензил)-N-фенилацетамида

Метил 4-((N-фенилацетамидо)метил)бензоат (0,220 г, 0,776 ммоль), синтезированный на стадии 1, и гидразин гидрат (0,733 мл, 15,530 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 2 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/метиленхлорид = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,145 г, 65,9%) в виде белого пенистого твердого вещества.
[Стадия 3] Синтез N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)ацетамида
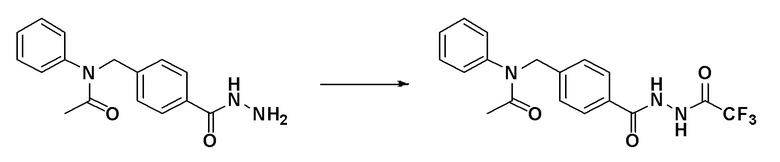
N-(4-(гидразинкарбонил)бензил)-N-фенилацетамид (0,145 г, 0,512 ммоль), синтезированный на стадии 2, и триэтиламин (0,142 мл, 1,024 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,087 мл, 0,614 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,180 г, 92,7%, желтое пенистое твердое вещество).
[Стадия 4] Синтез соединения 11105

N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)ацетамид (0,180 г, 0,475 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,170 г, 0,712 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,088 г, 51,3%) в виде светло-желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,3 Гц), 7,47-7,13 (м, 5H), 7,02 (дд, 2H, J=7,8, 1,5 Гц), 4,98 (с, 2H), 1,93 (с, 3H); LRMS (ES) m/z 362,3 (M++1).
Пример 3: Синтез соединения 11106, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)циклогексанкарбоксамида
[Стадия 1] Синтез 4-((N-фенилциклогексанкарбоксамидо)метил)бензоата

Метил 4-((фениламино)метил)бензоат (0,200 г, 0,829 ммоль) и N,N-диизопропилэтиламин (0,290 мл, 1,658 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли циклогексанкарбонилхлорид (0,166 мл, 1,243 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,285 г, 97,8%) в виде белого твердого вещества.
[Стадия 2] Синтез N-(4-(гидразинкарбонил)бензил)-N-фенилциклогексанкарбоксамида

4-((N-фенилциклогексанкарбоксамидо)метил)бензоат (0,285 г, 0,811 ммоль), синтезированный на стадии 1, и гидразин гидрат (0,766 мл, 16,219 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 2 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,239 г, 83,9%) в виде белого пенистого твердого вещества.
[Стадия 3] Синтез N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)циклогексанкарбоксамида

N-(4-(гидразинкарбонил)бензил)-N-фенилциклогексанкарбоксамид (0,239 г, 0,680 ммоль), синтезированный на стадии 2, и триэтиламин (0,189 мл, 1,360 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,115 мл, 0,816 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,300 г, 98,6%, белое пенистое твердое вещество).
[Стадия 4] Синтез соединения 11106

N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)циклогексанкарбоксамид (0,300 г, 0,670 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,240 г, 1,006 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,096 г, 33,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,3 Гц), 7,44-7,31 (м, 5H), 7,07 (ддд, 2H, J=60,8, 5,1, 4,6 Гц), 4,94 (с, 2H), 2,18 (ддд, 1H, J=11,4, 7,3, 3,1 Гц), 1,74-1,48 (м, 7H), 1,32-1,08 (м, 1H), 1,08-0,40 (м, 2H); LRMS (ES) m/z 430,3 (M++1).
Пример 4: Синтез соединения 11107, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)бензамида
[Стадия 1] Синтез метил 4-((N-фенилбензамидо)метил)бензоата

Метил 4-((фениламино)метил)бензоат (0,200 г, 0,829 ммоль) и N,N-диизопропилэтиламин (0,290 мл, 1,658 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли бензоилхлорид (0,175 г, 1,243 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,264 г, 92,2%) в виде белого твердого вещества.
[Стадия 2] Синтез N-(4-(гидразинкарбонил)бензил)-N-фенилбензамида
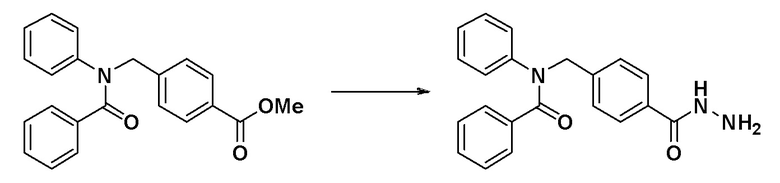
Метил 4-((N-фенилбензамидо)метил)бензоат (0,264 г, 0,764 ммоль), синтезированный на стадии 1, и гидразин гидрат (0,722 мл, 15,287 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 2 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,222 г, 84,1%) в виде белого пенистого твердого вещества.
[Стадия 3] Синтез N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)бензамида

N-(4-(гидразинкарбонил)бензил)-N-фенилбензамид (0,364 г, 1,054 ммоль), синтезированный на стадии 2, и триэтиламин (0,292 мл, 2,108 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,178 мл, 1,265 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,450 г, 96,7%, белое пенистое твердое вещество).
[Стадия 4] Синтез соединения 11107

N-фенил-N-(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)бензамид (0,450 г, 1,019 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,364 г, 1,529 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,250 г, 57,9%) в виде светло-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, 2H, J=8,3 Гц), 7,54 (т, 2H, J=9,9 Гц), 7,38-7,31 (м, 2H), 7,26-7,06 (м, 6H), 6,95 (дд, 2H, J=10,5, 9,1 Гц), 5,23 (с, 2H); LRMS (ES) m/z 430,3 (M++1)
Пример 5: Синтез соединения 11108, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилизоникотинамида
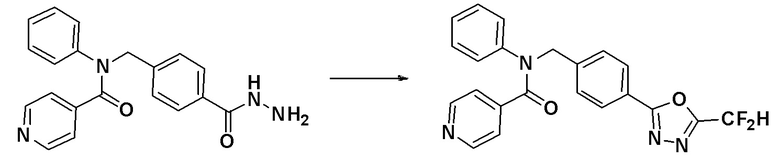
N-(4-(гидразинкарбонил)бензил)-N-фенилизоникотинамид (0,200 г, 0,577 ммоль), синтезированный на стадии 3 Примера 1, 2,2-дифторуксусный ангидрид (0,075 мл, 0,693 ммоль) и триэтиламин (0,160 мл, 1,155 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре и раствор перемешивали при 80°C в течение 1 часа и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,158 г, 67,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,47 (д, 2H, J=4,5 Гц), 8,06 (д, 2H, J=8,2 Гц), 7,47 (д, 2H, J=8,1 Гц), 7,19 (д, 5H, J=5,1 Гц), 7,02 (д, 1H, J=15,5 Гц), 6,90 (д, 3H, J=5,8 Гц), 5,19 (с, 2H); LRMS (ES) m/z 407,3 (M++1).
Пример 6: Синтез соединения 11109, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилизоникотинамида
[Стадия 1] Синтез метил 3-фтор-4-((N-фенилизоникотинамидо)метил)бензоата
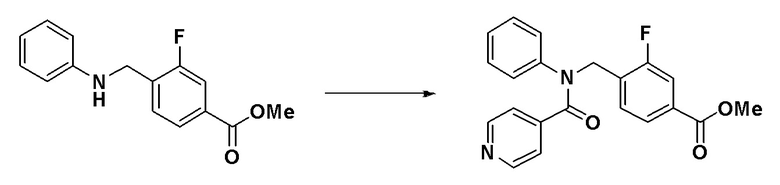
Метил 3-фтор-4-((фениламино)метил)бензоат (0,640 г, 2,468 ммоль) и N,N-диизопропилэтиламин (0,638 г, 4,937 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли гидрохлорид изоникотиноилхлорида (0,879 г, 4,937 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,840 г, 93,4%) в виде желтого пенистого твердого вещества.
[Стадия 2] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилизоникотинамида

3-фтор-4-((N-фенилизоникотинамидо)метил)бензоат (0,840 г, 2,305 ммоль), синтезированный на стадии 1, и гидразин гидрат (2,177 мл, 46,106 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 2 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,814 г, 96,9%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 11109

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилизоникотинамид (0,100 г, 0,274 ммоль), синтезированный на стадии 2, и триэтиламин (0,076 мл, 0,549 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,046 мл, 0,329 ммоль). Смесь перемешивали при 80°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/метиленхлорид = 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,060 г, 49,4%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,49 (с, 2H), 7,89 (дд, 1H, J=8,0, 1,4 Гц), 7,79-7,64 (м, 2H), 7,25 (д, 1H, J=9,0 Гц), 7,29-7,03 (м, 5H), 7,03-6,89 (м, 2H), 5,27 (с, 2H); LRMS (ES) m/z 443,2 (M++1).
Пример 7: Синтез соединения 11110, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилизоникотинамида
[Стадия 1] Синтез N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилизоникотинамида
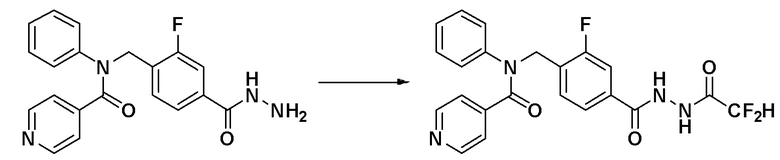
N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилизоникотинамид (0,100 г, 0,274 ммоль), синтезированный на стадии 2 Примера 6, и триэтиламин (0,076 мл, 0,549 ммоль) растворяли в метиленхлориде (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,057 г, 0,329 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,120 г, 98,8%, бесцветное масло).
[Стадия 2] Синтез соединения 11110
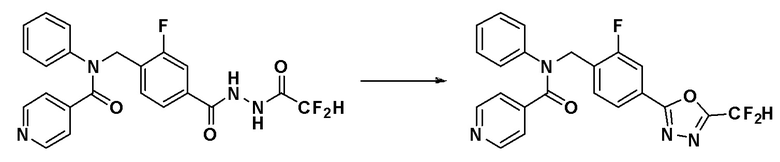
N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилизоникотинамид (0,120 г, 0,271 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,097 г, 0,407 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,027 г, 23,5%) в виде светло-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,49 (д, 2H, J=5,5 Гц), 7,91 (дд, 1H, J=8,0, 1,5 Гц), 7,90-7,57 (м, 2H), 7,29-7,07 (м, 5H), 6,95 (ддд, 3H, J=64,6, 48,3, 41,3 Гц), 5,27 (д, 2H, J=14,0 Гц); LRMS (ES) m/z 425,3 (M++1).
Пример 8: Синтез соединения 11134, трет-бутил 3-(фенил(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамоил)азетидин-1-карбоксилата
[Стадия 1] Синтез трет-бутил 3-(фенилкарбамоил)азетидин-1-карбоксилата

Анилин (1,961 мл, 21,475 ммоль), 1-(трет-бутоксикарбонил)азетидин-3-карбоновую кислоту (4,321 г, 21,475 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC) (6,175 г, 32,213 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt) (4,353 г, 32,213 ммоль) и N,N-диизопропилэтиламин (5,703 мл, 32,213 ммоль) растворяли в метиленхлориде (150 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (4,880 г, 82,2%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил 3-((4-(метоксикарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата

Трет-бутил 3-(фенилкарбамоил)азетидин-1-карбоксилат (1,000 г, 3,619 ммоль), синтезированный на стадии 1, растворяли в тетрагидрофуране (70 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,289 г, 7,237 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут и добавляли метил 4-(бромметил)бензоат (0,829 г, 3,619 ммоль) последующим дополнительным перемешиванием при 45°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем к реакционной смеси добавляли воду (10 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (1,200 г, 78,1%) в виде бесцветного масла.
[Стадия 3] Синтез трет-бутил 3-((4-(гидразинкарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата

Трет-бутил3-((4-(метоксикарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,500 г, 3,534 ммоль), синтезированный на стадии 2, и гидразин моногидрат (3,435 мл, 70,671 ммоль) смешивали в этаноле (15 мл) при комнатной температуре и смесь нагревали с использованием микроволнового излучения при 120°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли воду с последующим экстрагированием метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,400 г, 93,3%, белое твердое вещество).
[Стадия 4] Синтез трет-бутил 3-(фенил(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)карбамоил)азетидин-1-карбоксилата

Трет-бутил 3-((4-(гидразинкарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,800 г, 4,240 ммоль), синтезированный на стадии 3, и триэтиламин (0,710 мл, 5,088 ммоль) растворяли в N,N-диметилформамиде (30 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,649 мл, 4,664 ммоль). Смесь перемешивали при 90°C в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 60%) и концентрировали с получением указанного в заголовке соединения (1,500 г, 68,0%) в виде белого твердого вещества.
[Стадия 5] Синтез соединения 11134

Трет-бутил 3-(фенил(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)карбамоил)азетидин-1-карбоксилат (1,500 г, 2,882 ммоль), синтезированный на стадии 4, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 1,030 г, 4,323 ммоль) смешивали в тетрагидрофуране (15 мл) при комнатной температуре и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (1,200 г, 82,9%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 8,02 (д, 2H, J=8,2 Гц), 7,44-7,31 (м, 5H), 6,97-6,86 (м, 2H), 4,97 (с, 2H), 4,11 (дд, 2H, J=9,9, 4,1 Гц), 3,65 (дд, 2H, J=11,2, 5,8 Гц), 3,34-3,14 (м, 1H), 1,40 (с, 9H); LRMS (ES) m/z 403,4 (M+-100).
Пример 9: Синтез соединения 11135, трет-бутил 4-(фенил(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамоил)пиперидин-1-карбоксилата
[Стадия 1] Синтез трет-бутил 4-(фенилкарбамоил)пиперидин-1-карбоксилата

Анилин (1,961 мл, 21,475 ммоль), 1-(трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту (4,924 г, 21,475 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC) (6,175 г, 32,213 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt) (4,353 г, 32,213 ммоль) и N,N-диизопропилэтиламин (5,703 мл, 32,213 ммоль) растворяли в метиленхлориде (150 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов.
К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (5,040 г, 77,1%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-((4-(метоксикарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-(фенилкарбамоил)пиперидин-1-карбоксилат (1,000 г, 3,285 ммоль), синтезированный на стадии 1, растворяли в тетрагидрофуране (70 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,263 г, 6,571 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут и добавляли метил 4-(бромметил)бензоат (0,753 г, 3,285 ммоль) с последующим дополнительным перемешиванием при 45°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем к реакционной смеси добавляли воду (10 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 120 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (1,300 г, 87,4%) в виде бесцветного масла.
[Стадия 3] Синтез трет-бутил 4-((4-(гидразинкарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((4-(метоксикарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,500 г, 3,315 ммоль), синтезированный на стадии 2, и гидразин моногидрат (3,319 г, 66,291 ммоль) смешивали в этаноле (15 мл) при комнатной температуре и смесь нагревали при 120°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли воду с последующим экстрагированием метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,400 г, 93,3%, белое твердое вещество).
[Стадия 4] Синтез трет-бутил 4-(фенил(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((4-(гидразинкарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,800 г, 3,977 ммоль), синтезированный на стадии 3, и триэтиламин (0,666 мл, 4,773 ммоль) растворяли в N,N-диметилформамиде (30 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,609 мл, 4,375 ммоль). Смесь перемешивали при 90°C в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 60%) и концентрировали с получением указанного в заголовке соединения (1,600 г, 73,3%) в виде белого твердого вещества.
[Стадия 5] Синтез соединения 11135
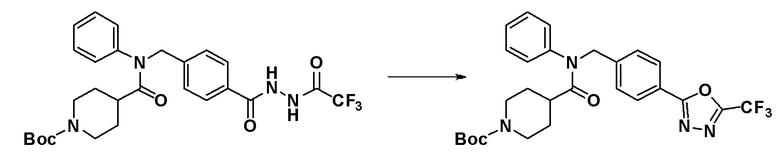
трет-Бутил 4-(фенил(4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)карбамоил)пиперидин-1-карбоксилат (1,600 г, 2,917 ммоль), синтезированный на стадии 4, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 1,043 г, 4,375 ммоль) смешивали в тетрагидрофуране (15 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (1,400 г, 90,5%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 8,03 (д, 2H, J=8,0 Гц), 7,43-7,32 (м, 5H), 7,00 (д, 2H, J=7,1 Гц), 4,96 (д, 2H, J=20,2 Гц), 4,15-3,93 (м, 2H), 2,45 (с, 2H), 2,34 (т, 1H, J=11,3 Гц), 1,77 (кв.д, 2H, J=12,8, 4,0 Гц), 1,60 (д, 2H, J=12,7 Гц), 1,44 (с, 9H); LRMS (ES) m/z 531,4 (M++1).
Пример 10: Синтез соединения 11136, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида гидрохлорида

трет-Бутил 3-(фенил(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамоил)азетидин-1-карбоксилат (1,100 г, 2,189 ммоль), синтезированный в Примере 8, растворяли в дихлорметане (50 мл) и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 2,736 мл, 10,945 ммоль) при 0°C с последующим перемешиванием при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,920 г, 95,8%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3+MeOD) δ 7,96 (дд, 2H, J=45,0, 36,1 Гц), 7,35 (ддд, 5H, J=40,2, 37,9, 10,0 Гц), 6,99 (д, 2H, J=77,6 Гц), 5,12-4,80 (м, 1H), 4,33 (с, 2H), 3,78 (д, 2H, J=25,5 Гц), 3,30 (д, 1H, J=120,8 Гц), 2,37 (с, 2H); LRMS (ES) m/z 403,0 (M++1).
Пример 11: Синтез соединения 11137, N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида гидрохлорида

трет-Бутил 4-(фенил(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)карбамоил)пиперидин-1-карбоксилат (1,300 г, 2,450 ммоль), синтезированный в Примере 9, растворяли в дихлорметане (50 мл) и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 3,063 мл, 12,251 ммоль) при 0°C с последующим перемешиванием при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (1,080 г, 94,4%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3+MeOD) δ 7,91 (дд, 2H, J=103,5, 50,3 Гц), 7,72-7,19 (м, 5H), 6,95 (с, 2H), 5,24-4,68 (м, 2H), 4,03-3,27 (м, 2H), 3,04-2,64 (м, 2H), 2,49 (с, 2H), 2,09 (с, 2H), 1,78 (д, 2H, J=93,2 Гц); LRMS (ES) m/z 431,4 (M++1).
Пример 12: Синтез соединения 11138, 1-метил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,100 г, 0,228 ммоль), синтезированный в Примере 10, и формальдегид (37,00% раствор в воде, 0,025 мл, 0,342 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,072 г, 0,342 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,038 г, 40,0%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,2 Гц), 7,52-7,30 (м, 6H), 6,90 (дд, 2H, J=6,5, 2,8 Гц), 4,92 (д, 2H, J=19,3 Гц), 3,51-3,14 (м, 5H), 2,35 (с, 3H); LRMS (ES) m/z 417,3 (M++1).
Пример 13: Синтез соединения 11139, 1-этил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида
[Стадия 1] Синтез соединения 11139
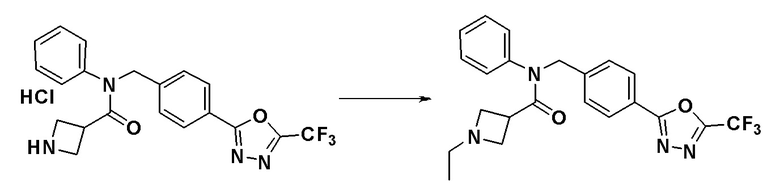
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,100 г, 0,228 ммоль), синтезированный в Примере 10, и ацетальдегид (0,019 мл, 0,342 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,072 г, 0,342 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,042 г, 42,8%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,07-7,97 (м, 2H), 7,34 (дт, 5H, J=22,3, 14,0 Гц), 6,95-6,83 (м, 2H), 4,92 (д, 2H, J=19,5 Гц), 3,55-3,08 (м, 5H), 2,58 (кв., 2H, J=7,2 Гц), 0,96 (т, 3H, J=7,2 Гц); LRMS (ES) m/z 431,3 (M++1).
Пример 14: Синтез соединения 11140, 1-метил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,100 г, 0,214 ммоль), синтезированный в Примере 11, и формальдегид (37,00% раствор в воде, 0,024 мл, 0,321 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,068 г, 0,321 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,072 г, 75,6%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,3 Гц), 7,43-7,30 (м, 5H), 6,97 (дд, 2H, J=6,4, 3,2 Гц), 4,94 (с, 2H), 2,78 (д, 2H, J=113,6 Гц), 2,16 (дд, 4H, J=68,5, 23,5 Гц), 1,96 (дт, 3H, J=20,3, 13,8 Гц), 1,73 (с, 2H); LRMS (ES) m/z 431,3 (M++1).
Пример 15: Синтез соединения 11141, 1-этил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида
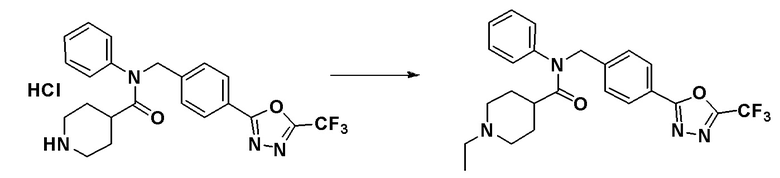
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,100 г, 0,214 ммоль), синтезированный в Примере 11, и ацетальдегид (0,018 мл, 0,321 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,068 г, 0,321 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,065 г, 66,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,3 Гц), 7,43-7,31 (м, 5H), 6,97 (дд, 2H, J=6,6, 2,9 Гц), 4,94 (с, 2H), 3,04 (с, 2H), 2,40 (д, 3H, J=75,4 Гц), 2,02-1,66 (м, 6H), 1,15 (дд, 3H, J=32,3, 25,8 Гц); LRMS (ES) m/z 459,34 (M++1)
Пример 16: Синтез соединения 11142, 1-изопропил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида
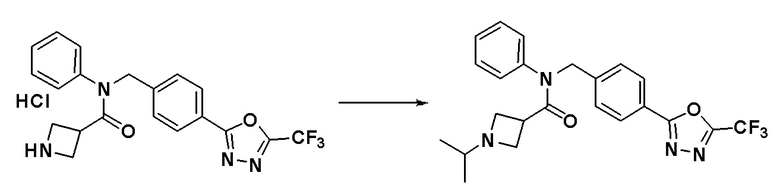
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,100 г, 0,228 ммоль), синтезированный в Примере 10, и ацетон (0,025 мл, 0,342 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,072 г, 0,342 ммоль) с последующим перемешиванием при этой же температуре. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,056 г, 55,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,2 Гц), 7,42-7,28 (м, 5H), 6,91 (дд, 2H, J=6,4, 3,1 Гц), 4,94 (с, 2H), 3,31 (д, 5H, J=21,1 Гц), 2,50 (с, 1H), 0,94 (д, 6H, J=6,1 Гц); LRMS (ES) m/z 445,3 (M++1).
Пример 17: Синтез соединения 11143, 1-изопропил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,100 г, 0,214 ммоль), синтезированный в Примере 11, и ацетон (0,024 мл, 0,321 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,068 г, 0,321 ммоль) с последующим перемешиванием при этой же температуре. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,021 г, 20,8%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,3 Гц), 7,36 (дд, 5H, J=7,4, 4,2 Гц), 6,95 (дд, 2H, J=6,5, 3,1 Гц), 4,92 (с, 2H), 3,37 (д, 3H, J=63,0 Гц), 2,75 (д, 3H, J=67,4 Гц), 2,22 (с, 1H), 1,96 (с, 2H, J=30,4 Гц), 1,25 (с, 6H, J=169,3 Гц); LRMS (ES) m/z 473,3 (M++1).
Пример 18: Синтез соединения 11157, N-фенил-1-пропионил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,080 г, 0,182 ммоль), синтезированный в Примере 10, и N,N-диизопропилэтиламин (0,063 мл, 0,365 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,018 мл, 0,201 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,008 г, 9,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,2 Гц), 7,45-7,29 (м, 6H), 6,92 (дд, 2H, J=6,5, 2,8 Гц), 4,98 (с, 2H), 4,37-4,08 (м, 2H), 3,79 (д, 2H, J=6,7 Гц), 3,30 (ддд, 1H, J=15,1, 8,8, 6,4 Гц), 2,15-1,94 (м, 3H), 1,25 (с, 1H, J=20,0 Гц), 1,09 (т, 3H, J=7,5 Гц); LRMS (ES) m/z 459,3 (M++1).
Пример 19: Синтез соединения 11158, 1-изобутирил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,080 г, 0,182 ммоль), синтезированный в Примере 10, и N,N-диизопропилэтиламин (0,063 мл, 0,365 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,021 мл, 0,201 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,025 г, 29,0%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,3 Гц), 7,45-7,28 (м, 5H), 6,92 (дд, 2H, J=6,3, 3,2 Гц), 5,04-4,87 (м, 2H), 4,53-4,16 (м, 1H), 3,95-3,59 (м, 2H), 3,36-3,20 (м, 1H), 2,39 (тд, 2H, J=13,6, 6,8 Гц), 1,07 (дд, 6H, J=16,3, 6,8 Гц); LRMS (ES) m/z 473,3 (M++1).
Пример 20: Синтез соединения 11159, N-фенил-1-(2,2,2-трифторацетил)-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,080 г, 0,182 ммоль), синтезированный в Примере 10, и N,N-диизопропилэтиламин (0,063 мл, 0,365 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2,2-трифторуксусный ангидрид (0,028 мл, 0,201 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,013 г, 14,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, 2H, J=8,2 Гц), 7,44-7,31 (м, 5H), 6,96-6,83 (м, 2H), 4,97 (т, 2H, J=8,2 Гц), 4,72-4,62 (м, 1H), 4,14 (дт, 2H, J=14,4, 8,2 Гц), 3,89-3,76 (м, 1H), 3,50-3,36 (м, 1H); LRMS (ES) m/z 499,3 (M++1).
Пример 21: Синтез соединения 11160, 1-(метилсульфонил)-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,080 г, 0,182 ммоль), синтезированный в Примере 10, и N,N-диизопропилэтиламин (0,064 мл, 0,365 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,016 мл, 0,201 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,018 г, 20,6%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,08-7,98 (м, 2H), 7,43-7,31 (м, 5H), 6,91 (ддд, 2H, J=5,5, 4,6, 2,9 Гц), 4,96 (с, 2H), 4,12 (дд, 2H, J=15,2, 7,3 Гц), 3,72-3,62 (м, 2H), 3,38-3,26 (м, 1H), 2,89 (д, 3H, J=4,0 Гц); LRMS (ES) m/z 481,2 (M++1).
Пример 22: Синтез соединения 11161, 1-ацетил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,059 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,013 мл, 0,188 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,052 г, 64,2%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,3 Гц), 7,37 (дд, 5H, J=5,4, 3,0 Гц), 6,99 (дд, 2H, J=6,6, 2,9 Гц), 4,94 (с, 2H), 4,51 (с, 1H), 3,77 (с, 1H), 2,80 (с, 1H), 2,38 (ддд, 2H, J=30,5, 20,3, 9,3 Гц), 2,04 (с, 3H, J=9,5, 4,9 Гц), 1,88-1,53 (м, 4H); LRMS (ES) m/z 473,3 (M++1).
Пример 23: Синтез соединения 11162, N-фенил-1-пропионил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,059 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,016 мл, 0,188 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,061 г, 73,2%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,0 Гц), 7,37 (дд, 5H, J=5,0, 3,0 Гц), 6,99 (дд, 2H, J=6,2, 2,6 Гц), 4,94 (с, 2H), 4,51 (с, 1H), 3,85 (с, 1H), 2,36 (ддд, 4H, J=21,9, 14,8, 9,1 Гц), 1,90-1,52 (м, 5H), 1,12 (т, 3H, J=7,5 Гц); LRMS (ES) m/z 487,4 (M++1).
Пример 24: Синтез соединения 11163, 1-изобутирил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,059 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,020 мл, 0,188 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,064 г, 74,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,3 Гц), 7,37 (дд, 5H, J=5,3, 3,1 Гц), 6,99 (дд, 2H, J=6,2, 3,2 Гц), 4,94 (с, 2H), 2,74 (дт, 2H, J=13,4, 6,7 Гц), 2,48-2,26 (м, 2H), 1,90-1,49 (м, 6H), 1,10 (т, 6H, J=11,3 Гц); LRMS (ES) m/z 501,3 (M++1).
Пример 25: Синтез соединения 11164, N-фенил-1-(2,2,2-трифторацетил)-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида
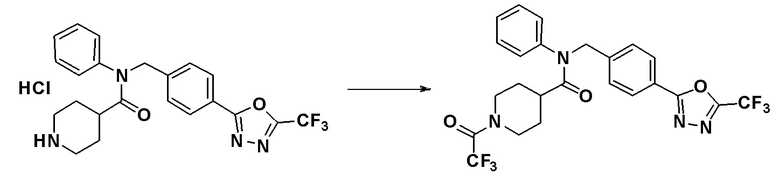
N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,059 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2,2-трифторуксусный ангидрид (0,027 мл, 0,188 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,061 г, 67,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,2 Гц), 7,38 (т, 5H, J=7,1 Гц), 7,05-6,87 (м, 2H), 4,94 (кв., 2H, J=14,5 Гц), 4,42 (д, 1H, J=13,2 Гц), 3,97 (д, 1H, J=14,3 Гц), 2,93 (т, 1H, J=13,1 Гц), 2,62 (т, 1H, J=12,0 Гц), 2,50 (дд, 1H, J=12,5, 8,5 Гц), 1,89 (дд, 2H, J=24,8, 13,1 Гц), 1,72 (д, 2H, J=14,0 Гц); LRMS (ES) m/z 527,3 (M++1).
Пример 26: Синтез соединения 11165, 1-(метилсульфонил)-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,060 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,015 мл, 0,188 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,070 г, 80,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, 2H, J=8,1 Гц), 7,37 (дд, 5H, J=5,3, 2,8 Гц), 6,98 (дд, 2H, J=6,3, 2,7 Гц), 4,94 (с, 2H), 3,80-3,62 (м, 2H), 2,72 (с, 3H), 2,51 (дд, 2H, J=16,4, 7,1 Гц), 2,38-2,22 (м, 1H), 2,02-1,84 (м, 2H), 1,78-1,66 (м, 2H); LRMS (ES) m/z 509,2 (M++1).
Пример 27: Синтез соединения 11166, 1-бензил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамида
[Стадия 1] Синтез соединения 11166

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)пиперидин-4-карбоксамид гидрохлорид (0,080 г, 0,171 ммоль), синтезированный в Примере 11, и N,N-диизопропилэтиламин (0,059 мл, 0,343 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли (бромметил)бензол (0,024 мл, 0,206 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,060 г, 67,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,01 (д, 2H, J=8,0 Гц), 7,44-7,24 (м, 10H), 7,02-6,84 (м, 2H), 4,94 (с, 2H), 3,41 (с, 2H), 2,83 (с, 2H), 2,17 (с, 1H), 1,93 (д, 2H, J=10,8 Гц), 1,64 (д, 5H, J=36,5 Гц); LRMS (ES) m/z 521,4 (M++1).
Пример 28: Синтез соединения 11187, N1-ацетил-N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамида

N-фенил-N-(4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)азетидин-3-карбоксамид гидрохлорид (0,080 г, 0,182 ммоль), синтезированный в Примере 10, и N,N-диизопропилэтиламин (0,063 мл, 0,365 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,014 мл, 0,201 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2 пластина, 20×20×1 мм; 100%-водный раствор этилацетата/гексан = 100%)) и концентрировали с получением указанного в заголовке соединения (0,020 г, 24,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 8,03 (д, 2H, J=8,1 Гц), 7,44-7,28 (м, 5H), 6,91 (дд, 2H, J=6,5, 2,4 Гц), 4,97 (с, 2H), 4,44 (с, 1H), 3,92 (дд, 3H, J=107,3, 55,1 Гц), 3,29 (ддд, 1H, J=15,2, 8,8, 6,3 Гц), 1,81 (д, 3H, J=6,8 Гц); LRMS (ES) m/z 445,3 (M++1).
Пример 29: Синтез соединения 11188, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)изоникотинамида
[Стадия 1] Синтез метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоата

3-Фторанилин (0,200 г, 1,800 ммоль), метил 4-(бромметил)-3-фторбензоат (0,445 г, 1,800 ммоль) и карбонат кальция (0,497 г, 3,600 ммоль) растворяли в ацетонитриле (15 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,324 г, 64,9%) в виде бесцветного масла.
[Стадия 2] Синтез метил 3-фтор-4-((N-(3-фторфенил)изоникотинамидо)метил)бензоата

Метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоат (0,320 г, 1,154 ммоль), синтезированный на стадии 1, изоникотиноил гидрохлорид (0,247 г, 1,385 ммоль) и N,N-диизопропилэтиламин (0,398 мл, 2,308 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (0,300 г, 68,0%) в виде желтого пенистого твердого вещества.
[Стадия 3] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)изоникотинамида

Метил 3-фтор-4-((N-(3-фторфенил)изоникотинамидо)метил)бензоат (0,300 г, 0,785 ммоль), синтезированный на стадии 2, и гидразин гидрат (0,786 г, 15,692 ммоль) смешивали в этаноле (20 мл) и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,250 г, 83,3%) в виде желтого пенистого твердого вещества.
[Стадия 4] Синтез N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)изоникотинамида

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)изоникотинамид (0,125 г, 0,327 ммоль), синтезированный на стадии 3, и триэтиламин (0,091 мл, 0,654 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,055 мл, 0,392 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,145 г, 92,7%) в виде желтого пенистого твердого вещества.
[Стадия 5] Синтез соединения 11188
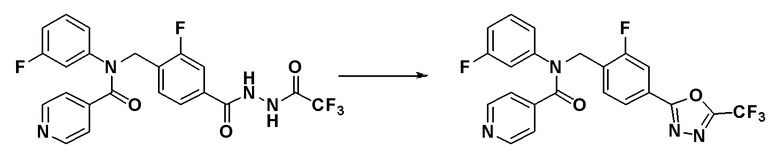
N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)изоникотинамид (0,160 г, 0,334 ммоль), синтезированный на стадии 4, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,120 г, 0,502 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,058 г, 37,7%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,53 (с, 2H), 7,91 (дд, 1H, J=8,0, 1,5 Гц), 7,83-7,57 (м, 2H), 7,37-7,27 (м, 2H), 7,17 (дд, 1H, J=8,1, 6,3 Гц), 6,94 (тд, 1H, J=8,1, 2,3 Гц), 6,78-6,62 (м, 2H), 5,23 (д, 2H, J=20,2 Гц); LRMS (ES) m/z 461,3 (M++1).
Пример 30: Синтез соединения 11189, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)изоникотинамида
[Стадия 1] Синтез N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)изоникотинамида

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)изоникотинамид (0,125 г, 0,327 ммоль), синтезированный на стадии 3 Примера 29, и триэтиламин (0,091 мл, 0,654 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,043 мл, 0,392 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,135 г, 89,7%) в виде желтого пенистого твердого вещества.
[Стадия 2] Синтез соединения 11189

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)изоникотинамид (0,160 г, 0,348 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,124 г, 0,521 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,089 г, 57,9%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,54 (д, 2H, J=4,8 Гц), 7,91 (д, 1H, J=7,8 Гц), 7,77 (д, 1H, J=9,9 Гц), 7,68 (т, 1H, J=7,6 Гц), 7,32 (д, 2H, J=2,9 Гц), 7,17 (дд, 1H, J=14,7, 7,3 Гц), 7,05-6,83 (м, 2H), 6,83-6,61 (м, 2H), 5,33-5,12 (м, 2H); LRMS (ES) m/z 442,9 (M++1).
Пример 31: Синтез соединения 11200, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-метил-N-фенилпиперидин-4-карбоксамида
[Стадия 1] Синтез трет-бутил 4-((2-фтор-4-(метоксикарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-(фенилкарбамоил)пиперидин-1-карбоксилат (2,000 г, 6,571 ммоль), синтезированный на стадии 1 Примера 9, растворяли в тетрагидрофуране (80 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,526 г, 13,141 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут и добавляли метил 4-(бромметил)-3-фторбензоат (1,948 г, 7,885 ммоль) с последующим дополнительным перемешиванием при 50°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем к реакционной смеси добавляли воду (20 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (2,600 г, 84,1%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 4-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((2-фтор-4-(метоксикарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (2,500 г, 5,313 ммоль), синтезированный на стадии 1, и гидразин моногидрат (5,154 мл, 106,261 ммоль) смешивали в этаноле (100 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и охлаждали до комнатной температуры. Затем реакционную смесь концентрировали при пониженном давлении для удаления растворителя. К концентрату добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (2,400 г, 96,0%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-((2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,200 г, 2,550 ммоль), синтезированный на стадии 2, и триэтиламин (0,427 мл, 3,060 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,390 мл, 2,805 ммоль) с последующим перемешиванием при этой же температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,400 г, 96,9%) в виде бесцветного масла.
[Стадия 4] Синтез трет-бутил 4-((2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,400 г, 2,471 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,883 г, 3,707 ммоль) смешивали в тетрагидрофуране (100 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = 10% до 30%) и концентрировали с получением указанного в заголовке соединения (0,740 г, 54,6%) в виде белого твердого вещества.
[Стадия 5] Синтез N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамида гидрохлорида

трет-Бутил 4-((2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (0,740 г, 1,349 ммоль), синтезированный на стадии 4, растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 1,686 мл, 6,745 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,610 г, 93,3%) в виде белого твердого вещества.
[Стадия 6] Синтез соединения 11200

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5, формальдегид (37,00% раствор в воде, 0,012 мл, 0,155 ммоль) и триацетоксиборогидрид натрия (0,033 г, 0,155 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 41,9%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,86 (д, 1H, J=7,8 Гц), 7,72 (д, 1H, J=9,4 Гц), 7,56 (т, 1H, J=7,0 Гц), 7,36 (д, 3H, J=12,4 Гц), 7,03 (т, 2H, J=13,6 Гц), 5,01 (д, 2H, J=22,8 Гц), 3,32-3,03 (м, 2H), 2,53-2,38 (м, 4H), 2,03 (дд, 4H, J=43,3, 40,1 Гц), 1,86 (д, 2H, J=48,6 Гц); LRMS (ES) m/z 463,3 (M++1).
Пример 32: Синтез соединения 11201, 1-этил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, ацетальдегид (0,009 мл, 0,155 ммоль) и триацетоксиборогидрид натрия (0,033 г, 0,155 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,025 г, 50,9%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,85 (д, 1H, J=6,8 Гц), 7,71 (д, 1H, J=8,8 Гц), 7,53 (с, 1H), 7,36 (с, 3H), 7,01 (д, 2H, J=26,5 Гц), 5,11-4,94 (м, 2H), 3,26 (с, 3H), 2,77 (с, 2H), 2,48 (с, 3H), 2,05-2,00 (м, 3H, J=57,5 Гц), 1,22 (д, 3H, J=5,3 Гц); LRMS (ES) m/z 477,3 (M++1).
Пример 33: Синтез соединения 11202, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-изопропил-N-фенилпиперидин-4-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, ацетон (0,011 мл, 0,155 ммоль) и триацетоксиборогидрид натрия (0,033 г, 0,155 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 39,5%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,86 (д, 1H, J=7,9 Гц), 7,73 (д, 1H, J=9,5 Гц), 7,55-7,48 (м, 1H), 7,40-7,33 (м, 3H), 7,05-6,98 (м, 2H), 5,02 (с, 2H), 3,40 (т, 3H, J=58,2 Гц), 2,78 (д, 2H, J=8,9 Гц), 2,60 (с, 1H), 2,11 (д, 2H, J=36,1 Гц), 2,00 (д, 4H, J=9,5 Гц), 1,30 (д, 4H, J=5,9 Гц); LRMS (ES) m/z 491,0 (M++1).
Пример 34: Синтез соединения 11203, 1-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, и N,N-диизопропилэтиламин (0,036 мл, 0,206 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,008 мл, 0,113 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,032 г, 63,3%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=7,9 Гц), 7,72 (д, 1H, J=9,4 Гц), 7,62-7,55 (м, 1H), 7,39 (с, 3H), 7,12-7,02 (м, 2H), 5,04 (с, 2H), 4,53 (с, 1H), 3,79 (с, 1H), 2,84 (с, 1H), 2,51-2,42 (м, 1H), 2,34 (с, 1H), 2,06 (т, 4H, J=4,7 Гц), 1,78 (д, 3H, J=69,3 Гц); LRMS (ES) m/z 491,1 (M++1).
Пример 35: Синтез соединения 11204, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-1-пропионилпиперидин-4-карбоксамида
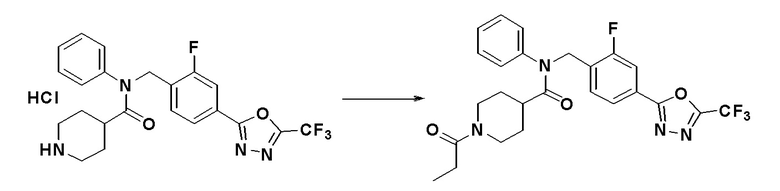
N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, и N,N-диизопропилэтиламин (0,036 мл, 0,206 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,010 мл, 0,113 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 57,7%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (т, 1H, J=6,2 Гц), 7,74-7,70 (м, 1H), 7,58 (д, 1H, J=6,7 Гц), 7,39 (д, 3H, J=4,5 Гц), 7,06 (с, 2H), 5,04 (с, 3H), 4,54 (с, 1H), 3,83 (с, 1H), 2,78 (с, 1H), 2,31 (с, 4H), 1,77 (с, 2H, J=67,0 Гц), 1,19-1,02 (м, 4H); LRMS (ES) m/z 505,3 (M++1).
Пример 36: Синтез соединения 11205, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-изобутирил-N-фенилпиперидин-4-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, и N,N-диизопропилэтиламин (0,036 мл, 0,206 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,012 мл, 0,113 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 56,1%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=8,0 Гц), 7,71 (т, 1H, J=11,3 Гц), 7,58 (т, 1H, J=7,5 Гц), 7,42-7,37 (м, 3H), 7,07 (д, 2H, J=7,7 Гц), 5,00 (д, 2H, J=39,7 Гц), 4,56 (с, 1H), 4,04-3,80 (м, 1H), 2,94-2,66 (м, 2H), 2,53-2,41 (м, 1H), 2,31 (дт, 1H, J=45,9, 23,1 Гц), 1,76 (дд, 2H, J=36,0, 30,1 Гц), 1,10 (д, 8H, J=6,8 Гц); LRMS (ES) m/z 519,5 (M++1).
Пример 37: Синтез соединения 11206, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-1-(2,2,2-трифторацетил)пиперидин-4-карбоксамида
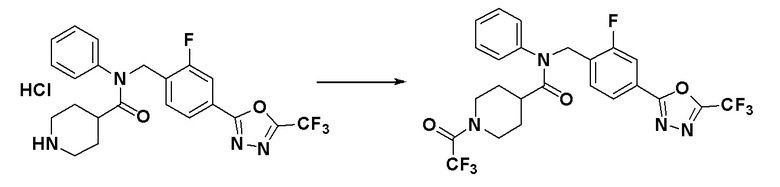
N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, и N,N-диизопропилэтиламин (0,036 мл, 0,206 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,016 мл, 0,113 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 53,4%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=8,0 Гц), 7,73 (д, 1H, J=9,6 Гц), 7,57 (т, 1H, J=7,4 Гц), 7,41 (д, 3H, J=5,4 Гц), 7,07 (д, 2H, J=6,9 Гц), 5,10-4,98 (м, 2H), 4,43 (д, 1H, J=13,4 Гц), 3,98 (д, 1H, J=13,8 Гц), 2,96 (т, 1H, J=12,8 Гц), 2,74-2,58 (м, 1H), 2,54 (т, 1H, J=10,5 Гц), 1,96-1,81 (м, 2H), 1,75 (д, 2H, J=13,0 Гц); LRMS (ES) m/z 545,4 (M++1).
Пример 38: Синтез соединения 11207, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(метилсульфонил)-N-фенилпиперидин-4-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,103 ммоль), синтезированный на стадии 5 Примера 31, и N,N-диизопропилэтиламин (0,036 мл, 0,206 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,009 мл, 0,113 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 55,3%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=7,8 Гц), 7,73 (д, 1H, J=9,4 Гц), 7,58 (т, 1H, J=6,4 Гц), 7,39 (с, 3H), 7,06 (д, 2H, J=5,7 Гц), 5,04 (с, 2H), 3,74 (д, 2H, J=10,1 Гц), 2,74 (д, 3H, J=2,1 Гц), 2,54 (т, 2H, J=11,7 Гц), 2,36 (дд, 1H, J=10,1, 7,7 Гц), 1,95 (дд, 2H, J=23,6, 11,6 Гц), 1,75 (д, 2H, J=13,2 Гц); LRMS (ES) m/z 527,3 (M++1).
Пример 39: Синтез соединения 11208, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-N-фенилпиперидин-4-карбоксамида
[Стадия 1] Синтез трет-бутил 4-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(фенил)карбамоил)пиперидин-1-карбоксилата
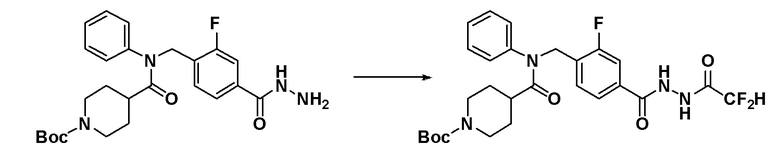
трет-Бутил 4-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,200 г, 2,550 ммоль), синтезированный на стадии 2 Примера 31, и триэтиламин (0,427 мл, 3,060 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли дифторуксусный ангидрид (0,349 мл, 2,805 ммоль). Смесь перемешивали при этой же температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,350 г, 96,5%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 4-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)пиперидин-1-карбоксилата

трет-Бутил 4-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(фенил)карбамоил)пиперидин-1-карбоксилат (1,350 г, 2,461 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,880 г, 3,691 ммоль) смешивали в тетрагидрофуране (100 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 10% до 30%) и концентрировали с получением указанного в заголовке соединения (0,800 г, 61,3%) в виде белого твердого вещества.
[Стадия 3] Синтез N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамида гидрохлорида

трет-Бутил 4-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)пиперидин-1-карбоксилат (0,800 г, 1,508 ммоль), синтезированный на стадии 2, растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 1,885 мл, 7,539 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,660 г, 93,7%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 11208
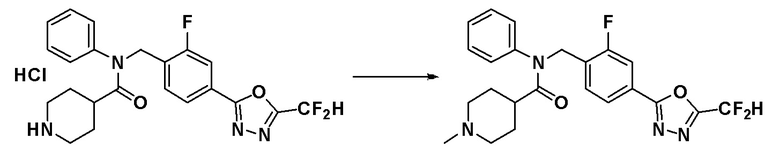
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3, формальдегид (37,00% раствор в воде, 0,012 мл, 0,161 ммоль) и триацетоксиборогидрид натрия (0,034 г, 0,161 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,022 г, 46,2%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,86 (т, 1H, J=13,4 Гц), 7,72 (д, 1H, J=9,7 Гц), 7,55 (т, 1H, J=7,5 Гц), 7,41-7,33 (м, 3H), 7,09-7,02 (м, 2H), 6,92 (т, 1H, J=51,7 Гц), 5,07-4,94 (м, 2H), 3,16-3,02 (м, 2H), 2,38 (с, 1H), 2,24 (с, 3H), 1,97 (д, 3H, J=9,6 Гц), 1,81 (д, 3H, J=9,7 Гц); LRMS (ES) m/z 445,3 (M++1).
Пример 40: Синтез соединения 11209, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-этил-N-фенилпиперидин-4-карбоксамида
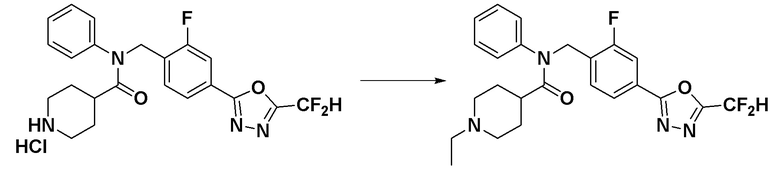
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, ацетальдегид (0,009 мл, 0,161 ммоль) и триацетоксиборогидрид натрия (0,034 г, 0,161 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,025 г, 50,9%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,86 (д, 1H, J=6,0 Гц), 7,73 (д, 1H, J=6,7 Гц), 7,52 (с, 1H), 7,37 (с, 3H), 7,03 (с, 2H), 6,88 (д, 1H, J=51,6 Гц), 5,02 (с, 2H), 3,48 (д, 2H, J=5,4 Гц), 3,25 (с, 3H), 2,77 (с, 3H), 2,50 (с, 2H), 1,25 (с, 4H); LRMS (ES) m/z 459,2 (M++1).
Пример 41: Синтез соединения 11210, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-изопропил-N-фенилпиперидин-4-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, ацетон (0,012 мл, 0,161 ммоль) и триацетоксиборогидрид натрия (0,034 г, 0,161 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 39,5%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,85 (т, 1H, J=17,4 Гц), 7,75 (д, 1H, J=9,5 Гц), 7,46 (д, 1H, J=25,1 Гц), 7,38 (д, 3H, J=2,9 Гц), 7,01 (д, 2H, J=2,6 Гц), 6,97-6,81 (м, 1H), 5,08-4,98 (м, 2H), 3,59 (с, 1H), 3,38 (д, 1H, J=17,5 Гц), 2,87 (д, 2H, J=199,2 Гц), 2,30 (д, 3H, J=82,5 Гц), 1,95 (д, 3H, J=47,9 Гц), 1,40 (с, 6H); LRMS (ES) m/z 473,1 (M++1).
Пример 42: Синтез соединения 11211, 1-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамида
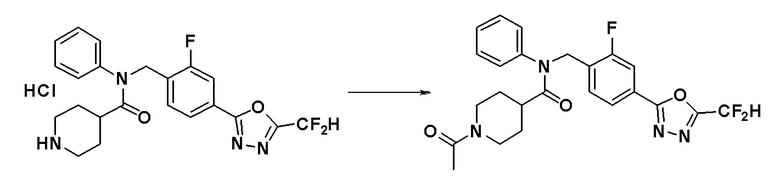
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, и N,N-диизопропилэтиламин (0,037 мл, 0,214 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,008 мл, 0,118 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 59,3%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=7,8 Гц), 7,72 (д, 1H, J=9,5 Гц), 7,56 (т, 1H, J=7,2 Гц), 7,39 (с, 3H), 7,06 (д, 2H, J=6,1 Гц), 6,92 (т, 1H, J=51,6 Гц), 5,03 (т, 2H, J=11,0 Гц), 4,54 (д, 1H, J=11,3 Гц), 3,75 (т, 1H, J=41,8 Гц), 2,83 (д, 1H, J=11,1 Гц), 2,41 (ддд, 3H, J=79,6, 32,3, 14,8 Гц), 1,85 (д, 2H, J=10,1 Гц), 1,79-1,62 (м, 4H); LRMS (ES) m/z 473,4 (M++1).
Пример 43: Синтез соединения 11212, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-1-пропионилпиперидин-4-карбоксамида
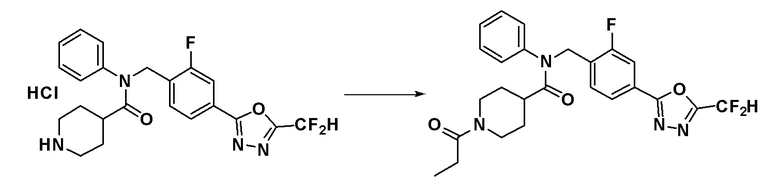
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, и N,N-диизопропилэтиламин (0,037 мл, 0,214 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,010 мл, 0,118 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 57,6%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,87 (д, 1H, J=7,8 Гц), 7,72 (д, 1H, J=9,5 Гц), 7,56 (т, 1H, J=7,1 Гц), 7,39 (д, 3H, J=5,5 Гц), 7,05 (т, 2H, J=13,9 Гц), 6,92 (т, 1H, J=51,7 Гц), 5,02 (д, 3H, J=27,7 Гц), 4,55 (с, 1H), 3,92-3,68 (м, 1H), 2,96-2,66 (м, 2H), 2,45 (т, 2H, J=10,5 Гц), 1,78 (д, 4H, J=56,0 Гц), 1,13 (т, 3H, J=7,1 Гц); LRMS (ES) m/z 487,2 (M++1).
Пример 44: Синтез соединения 11213, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-изобутирил-N-фенилпиперидин-4-карбоксамида
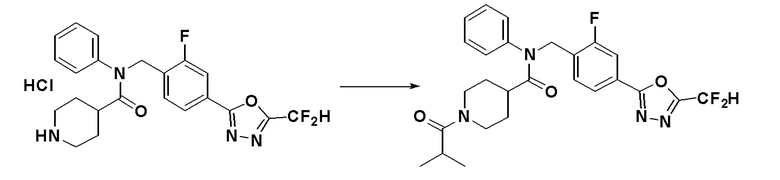
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, и N,N-диизопропилэтиламин (0,037 мл, 0,214 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,012 мл, 0,118 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 56,0%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,90-7,82 (м, 1H), 7,72 (т, 1H, J=10,3 Гц), 7,56 (д, 1H, J=6,6 Гц), 7,39 (д, 3H, J=4,7 Гц), 7,06 (д, 2H, J=3,0 Гц), 6,99-6,82 (м, 1H), 5,01 (д, 3H, J=32,5 Гц), 4,56 (с, 1H), 3,91 (с, 1H), 2,76 (дд, 3H, J=23,9, 18,5 Гц), 2,46 (д, 2H, J=6,7 Гц), 2,31 (с, 1H), 1,82 (с, 1H), 1,29-1,20 (м, 2H), 1,13-1,09 (м, 4H); LRMS (ES) m/z 501,3 (M++1).
Пример 45: Синтез соединения 11214, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-1-(2,2,2-трифторацетил)пиперидин-4-карбоксамида
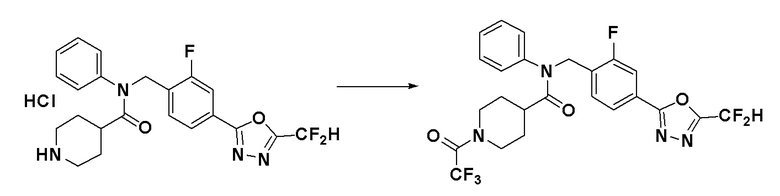
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, и N,N-диизопропилэтиламин (0,037 мл, 0,214 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,017 мл, 0,118 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 53,2%) в виде бесцветного масла.
1H ЯМР (700 МГц, CDCl3) δ 7,88 (д, 1H, J=8,0 Гц), 7,72 (т, 1H, J=20,1 Гц), 7,55 (дд, 1H, J=20,6, 13,1 Гц), 7,42 (дд, 3H, J=16,5, 7,5 Гц), 7,07 (д, 2H, J=7,6 Гц), 7,01-6,85 (м, 1H), 5,04 (кв., 2H, J=14,8 Гц), 4,43 (д, 1H, J=13,5 Гц), 3,97 (т, 1H, J=24,0 Гц), 2,96 (т, 1H, J=12,7 Гц), 2,65 (т, 1H, J=12,3 Гц), 2,54 (дт, 1H, J=16,1, 7,8 Гц), 1,97-1,83 (м, 2H), 1,75 (д, 2H, J=13,3 Гц); LRMS (ES) m/z 527,3 (M++1).
Пример 46: Синтез соединения 11215, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(метилсульфонил)-N-фенилпиперидин-4-карбоксамид

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,050 г, 0,107 ммоль), синтезированный на стадии 3 Примера 39, и N,N-диизопропилэтиламин (0,037 мл, 0,214 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,009 мл, 0,118 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = 5% до 70%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 55,1%) в виде белого твердого вещества.
1H ЯМР (700 МГц, CDCl3) δ 7,88 (д, 1H, J=7,2 Гц), 7,73 (д, 1H, J=9,2 Гц), 7,56 (т, 1H, J=6,1 Гц), 7,39 (с, 3H), 7,06 (с, 2H), 6,93 (тд, 1H, J=51,6, 2,2 Гц), 5,03 (д, 2H, J=14,7 Гц), 3,75 (д, 2H, J=9,6 Гц), 2,74 (д, 3H, J=2,3 Гц), 2,55 (т, 2H, J=11,7 Гц), 2,36 (дд, 1H, J=10,0, 7,3 Гц), 1,95 (дд, 2H, J=23,5, 11,5 Гц), 1,75 (д, 2H, J=13,3 Гц); LRMS (ES) m/z 509,3 (M++1).
Пример 47: Синтез соединения 11232, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-метил-N-фенилазетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил 3-((2-фтор-4-(метоксикарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата

трет-Бутил 3-(фенилкарбамоил)азетидин-1-карбоксилат (2,000 г, 7,237 ммоль), синтезированный на стадии 1 Примера 8, растворяли в тетрагидрофуране (80 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,579 г, 14,475 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут и затем добавляли метил 4-(бромметил)-3-фторбензоат (2,146 г, 8,685 ммоль) с последующим дополнительным перемешиванием при 50°C в течение 12 часов. Затем реакционную смесь охлаждали до комнатной температуры и к реакционной смеси добавляли воду (20 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (2,800 г, 87,4%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 3-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата

трет-Бутил 3-((2-фтор-4-(метоксикарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (2,500 г, 5,650 ммоль), синтезированный на стадии 1, и гидразин моногидрат (5,481 мл, 112,997 ммоль) смешивали в этаноле (100 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (2,400 г, 96,0%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 3-((2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата
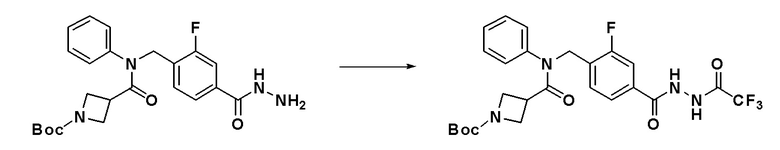
трет-Бутил 3-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,200 г, 2,712 ммоль), синтезированный на стадии 2, и триэтиламин (0,454 мл, 3,254 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,415 мл, 2,983 ммоль). Смесь перемешивали при этой же температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,400 г, 95,9%) в виде бесцветного масла.
[Стадия 4] Синтез трет-бутил 3-((2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамоил)азетидин-1-карбоксилата
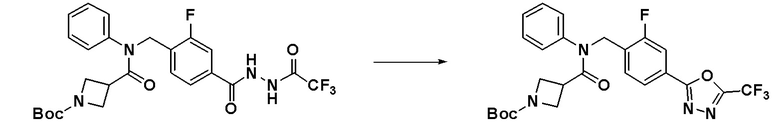
трет-Бутил 3-((2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,400 г, 2,600 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,929 г, 3,900 ммоль) смешивали в тетрагидрофуране (100 мл) при комнатной температуре. Смесь нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 10% до 30%) и концентрировали с получением указанного в заголовке соединения (0,800 г, 59,1%) в виде белого твердого вещества.
[Стадия 5] Синтез N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамида гидрохлорида

трет-Бутил 3-((2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (0,800 г, 1,537 ммоль), синтезированный на стадии 4, растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 1,921 мл, 7,685 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,600 г, 85,4%) в виде белого твердого вещества.
[Стадия 6] Синтез соединения 11232

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5, и формальдегид (37,00% раствор в воде, 0,012 мл, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,017 г, 35,8%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,92-7,84 (м, 1H), 7,73 (т, 1H, J=15,4 Гц), 7,60-7,51 (м, 1H), 7,37 (д, 3H, J=14,7 Гц), 6,93 (дд, 2H, J=32,2, 3,1 Гц), 5,11-5,00 (м, 2H), 3,84-3,58 (м, 4H), 3,49-3,40 (м, 1H), 2,66-2,51 (м, 3H); LRMS (ES) m/z 435,2 (M++1).
Пример 48: Синтез соединения 11233, 1-этил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 47, и ацетальдегид (0,009 мл, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,015 г, 30,6%) в виде коричневого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (д, 1H, J=7,7 Гц), 7,72 (т, 1H, J=15,3 Гц), 7,58 (т, 1H, J=6,9 Гц), 7,40 (д, 3H, J=39,7 Гц), 6,96 (т, 2H, J=20,0 Гц), 5,11-4,95 (м, 2H), 3,40 (д, 5H, J=67,9 Гц), 2,62 (д, 2H, J=4,5 Гц), 0,95 (д, 3H, J=71,5 Гц); LRMS (ES) m/z 449,3 (M++1).
Пример 49: Синтез соединения 11234, 1-ацетил-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамида
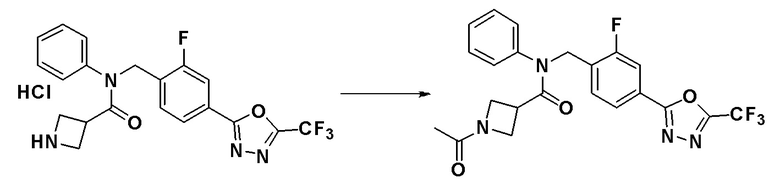
N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 47, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,009 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол = 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,027 г, 53,3%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,94-7,83 (м, 1H), 7,75 (т, 1H, J=9,4 Гц), 7,62-7,54 (м, 1H), 7,39 (с, 3H), 6,99 (д, 2H, J=5,4 Гц), 5,08 (с, 2H), 4,47 (т, 1H, J=7,0 Гц), 4,08-4,01 (м, 1H), 3,94 (т, 1H, J=8,4 Гц), 3,70 (т, 1H, J=9,5 Гц), 3,38-3,27 (м, 1H), 1,84 (с, 3H); LRMS (ES) m/z 463,3 (M++1).
Пример 50: Синтез соединения 11235, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенил-1-пропионилазетидин-3-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 47, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,011 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,012 г, 23,0%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,87 (дд, 1H, J=23,0, 10,1 Гц), 7,75 (д, 1H, J=9,5 Гц), 7,61 (дт, 1H, J=15,0, 7,6 Гц), 7,44-7,34 (м, 3H), 7,00 (д, 2H, J=5,7 Гц), 5,13-5,02 (м, 2H), 4,41 (д, 1H, J=76,8 Гц), 3,92 (дд, 2H, J=97,6, 59,0 Гц), 3,69 (дд, 1H, J=59,2, 21,2 Гц), 3,40-3,28 (м, 1H), 2,11 (д, 2H, J=51,5 Гц), 1,13-1,05 (м, 3H); LRMS (ES) m/z 477,2 (M++1).
Пример 51: Синтез соединения 11236, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-изобутирил-N-фенилазетидин-3-карбоксамида

N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 47, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,013 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,036 г, 67,1%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,87 (т, 1H, J=10,4 Гц), 7,74 (д, 1H, J=9,6 Гц), 7,61 (дт, 1H, J=29,5, 7,6 Гц), 7,45-7,33 (м, 3H), 6,98 (дд, 2H, J=28,1, 7,6 Гц), 5,15-4,97 (м, 2H), 4,38 (дд, 1H, J=60,8, 56,4 Гц), 4,14-3,57 (м, 3H), 3,39-3,28 (м, 1H), 2,45-2,32 (м, 1H), 1,07 (с, 6H); LRMS (ES) m/z 491,3 (M++1).
Пример 52: Синтез соединения 11237, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(метансульфонил)-N-фенилазетидин-3-карбоксамида
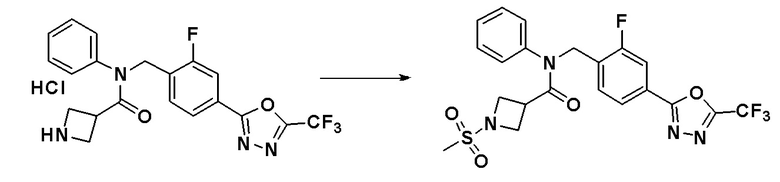
N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 47, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,009 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,026 г, 47,7%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,89 (т, 1H, J=9,1 Гц), 7,75 (д, 1H, J=9,4 Гц), 7,57 (т, 1H, J=7,5 Гц), 7,40 (д, 3H, J=1,5 Гц), 6,98 (д, 2H, J=3,7 Гц), 5,08 (с, 2H), 4,13 (т, 2H, J=7,5 Гц), 3,70 (т, 2H, J=8,3 Гц), 3,36 (п, 1H, J=7,9 Гц), 2,91 (с, 3H); LRMS (ES) m/z 499,2 (M++1).
Пример 53: Синтез соединения 11238, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-метил-N-фенилазетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил 3-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(фенил)карбамоил)азетидин-1-карбоксилата

трет-Бутил 3-((2-фтор-4-(гидразинкарбонил)бензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,200 г, 2,712 ммоль), синтезированный на стадии 2 Примера 47, и триэтиламин (0,454 мл, 3,254 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли дифторуксусный ангидрид (0,371 мл, 2,983 ммоль). Смесь перемешивали при этой же температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,400 г, 99,2%) в виде бесцветного масла.
[Стадия 2] Синтез трет-бутил 3-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)азетидин-1-карбоксилата
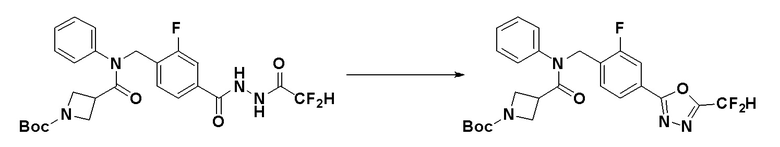
трет-Бутил 3-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(фенил)карбамоил)азетидин-1-карбоксилат (1,400 г, 2,690 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,961 г, 4,035 ммоль) смешивали в тетрагидрофуране (100 мл) и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 10% до 30%) с получением указанного в заголовке соединения (0,700 г, 51,8%) в виде белого твердого вещества.
[Стадия 3] Синтез N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамида гидрохлорида

трет-Бутил 3-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(фенил)карбамоил)азетидин-1-карбоксилат (0,700 г, 1,393 ммоль), синтезированный на стадии 2, растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 1,741 мл, 6,965 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,600 г, 98,1%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 11238

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3, и формальдегид (37,00% раствор в воде, 0,013 мл, 0,171 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,036 г, 0,171 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,010 г, 21,1%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (д, 1H, J=7,8 Гц), 7,73 (д, 1H, J=9,6 Гц), 7,61-7,53 (м, 1H), 7,35 (т, 3H, J=9,7 Гц), 7,15 (д, 1H, J=6,8 Гц), 6,97 (дд, 2H, J=29,0, 22,5 Гц), 5,11-4,97 (м, 2H), 3,24 (дд, 4H, J=67,4, 30,6 Гц), 2,32 (с, 3H), 2,00-1,68 (м, 1H); LRMS (ES) m/z 417,3 (M++1).
Пример 54: Синтез соединения 11239, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-этил-N-фенилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и ацетальдегид (0,010 мл, 0,171 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,036 г, 0,171 ммоль) добавляли. Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,017 г, 34,7%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,91-7,83 (м, 1H), 7,74 (д, 1H, J=9,7 Гц), 7,57-7,49 (м, 1H), 7,44-7,34 (м, 3H), 7,02-6,83 (м, 3H), 5,10-4,99 (м, 2H), 3,71-3,59 (м, 2H), 3,52 (дд, 2H, J=19,3, 11,2 Гц), 3,49-3,37 (м, 1H), 2,84-2,72 (м, 2H), 1,13-1,00 (м, 3H); LRMS (ES) m/z 431,3 (M++1).
Пример 55: Синтез соединения 11240, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-изопропил-N-фенилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и ацетон (0,013 мл, 0,171 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,036 г, 0,171 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,025 г, 49,4%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,92-7,82 (м, 1H), 7,73 (т, 1H, J=8,8 Гц), 7,54 (т, 1H, J=7,5 Гц), 7,38 (д, 3H, J=5,3 Гц), 7,04-6,82 (м, 3H), 5,03 (д, 2H, J=21,2 Гц), 3,63-3,34 (м, 5H), 2,80-2,67 (м, 1H), 1,03 (т, 6H, J=7,8 Гц); LRMS (ES) m/z 445,3 (M++1).
Пример 56: Синтез соединения 11241, 1-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамида
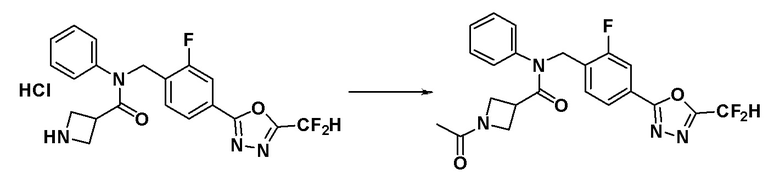
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и N,N-диизопропилэтиламин (0,039 мл, 0,228 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,009 мл, 0,125 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,014 г, 27,6%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (д, 1H, J=7,9 Гц), 7,73 (т, 1H, J=12,4 Гц), 7,58 (т, 1H, J=7,5 Гц), 7,39 (д, 3H, J=5,2 Гц), 7,01-6,82 (м, 3H), 5,10-5,05 (м, 2H), 4,48 (т, 1H, J=7,1 Гц), 4,09-4,01 (м, 1H), 3,94 (т, 1H, J=8,3 Гц), 3,70 (т, 1H, J=9,3 Гц), 3,36-3,27 (м, 1H), 1,50-1,43 (м, 3H); LRMS (ES) m/z 445,3 (M++1).
Пример 57: Синтез соединения 11242, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-1-пропионилазетидин-3-карбоксамида
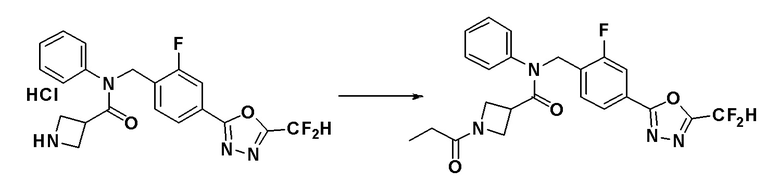
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и N,N-диизопропилэтиламин (0,039 мл, 0,228 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,011 мл, 0,125 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,017 г, 32,5%) в виде желтого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (д, 1H, J=7,9 Гц), 7,74 (д, 1H, J=9,7 Гц), 7,57 (дд, 1H, J=19,2, 11,7 Гц), 7,40 (дд, 3H, J=16,9, 7,9 Гц), 7,03-6,83 (м, 3H), 5,08 (с, 2H), 4,56-4,32 (м, 1H), 4,03 (ддд, 4H, J=153,6, 33,8, 26,6 Гц), 3,36-3,28 (м, 1H), 2,89-2,80 (м, 1H), 2,07 (д, 2H, J=8,1 Гц), 1,10 (т, 3H, J=7,6 Гц); LRMS (ES) m/z 459,3 (M++1).
Пример 58: Синтез соединения 11243, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-изобутирил-N-фенилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и N,N-диизопропилэтиламин (0,039 мл, 0,228 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,013 мл, 0,125 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,023 г, 42,7%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (дд, 1H, J=8,0, 1,2 Гц), 7,74 (дд, 1H, J=9,7, 1,1 Гц), 7,61-7,54 (м, 1H), 7,39 (дт, 3H, J=11,8, 4,2 Гц), 7,02-6,83 (м, 3H), 5,14-4,97 (м, 2H), 4,50 (с, 1H), 4,00 (д, 2H, J=56,0 Гц), 3,74-3,61 (м, 1H), 3,38-3,24 (м, 1H), 2,43-2,34 (м, 1H), 1,07 (д, 6H, J=25,4 Гц); LRMS (ES) m/z 473,3 (M++1).
Пример 59: Синтез соединения 11244, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенил-1-(2,2,2-трифторацетил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и N,N-диизопропилэтиламин (0,039 мл, 0,228 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2,2-трифторуксусный ангидрид (0,018 мл, 0,125 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2 пластина, 20×20×1 мм; метанол/дихлорметан = 10%) и концентрировали с получением указанного в заголовке соединения (0,005 г, 8,8%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,89 (д, 1H, J=7,9 Гц), 7,76 (д, 1H, J=9,6 Гц), 7,57 (дд, 1H, J=15,6, 8,0 Гц), 7,42 (д, 3H, J=3,2 Гц), 7,02-6,83 (м, 3H), 5,13-5,07 (м, 2H), 4,74-4,67 (м, 1H), 4,30-4,24 (м, 1H), 4,19 (т, 1H, J=9,1 Гц), 3,87 (т, 1H, J=9,8 Гц), 3,47 (дт, 1H, J=22,3, 7,9 Гц); LRMS (ES) m/z 499,1 (M++1).
Пример 60: Синтез соединения 11245, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(метилсульфонил)-N-фенилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилазетидин-3-карбоксамид гидрохлорид (0,050 г, 0,114 ммоль), синтезированный на стадии 3 Примера 53, и N,N-диизопропилэтиламин (0,040 мл, 0,228 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,010 мл, 0,125 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,013 г, 23,7%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 700 МГц) δ 7,88 (д, 1H, J=7,9 Гц), 7,75 (д, 1H, J=9,6 Гц), 7,55 (дд, 1H, J=18,1, 10,7 Гц), 7,40 (д, 3H, J=1,6 Гц), 7,02-6,84 (м, 3H), 5,07 (д, 2H, J=14,4 Гц), 4,14 (т, 2H, J=7,5 Гц), 3,71 (т, 2H, J=8,3 Гц), 3,39-3,32 (м, 1H), 2,91 (с, 3H); LRMS (ES) m/z 481,2 (M++1).
Пример 61: Синтез соединения 11246, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилацетамида
[Стадия 1] Синтез метил 3-фтор-4-((N-фенилацетамидо)метил)бензоата

Метил 3-фтор-4-((фениламино)метил)бензоат (0,150 г, 0,579 ммоль) и N,N-диизопропиламин (0,199 мл, 1,157 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,045 мл, 0,636 ммоль) с последующим перемешиванием при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,158 г, 90,6%) в виде бесцветного масла.
[Стадия 2] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилацетамида

Метил 3-фтор-4-((N-фенилацетамидо)метил)бензоат (0,158 г, 0,524 ммоль), синтезированный на стадии 1, и оксид азота (0,495 мл, 10,487 ммоль) смешивали в этаноле (10 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 5 часов и охлаждали до комнатной температуры. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,150 г, 94,9%) в виде бесцветного масла.
[Стадия 3] Синтез N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-фенилацетамида

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилацетамид (0,075 г, 0,249 ммоль), синтезированный на стадии 2, и триэтиламин (0,069 мл, 0,498 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,042 мл, 0,299 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,090 г, 91,0%) в виде бесцветного масла.
[Стадия 4] Синтез соединения 11246

N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-фенилацетамид (0,090 г, 0,227 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,081 г, 0,340 ммоль) смешивали с дихлорметаном (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,004 г, 4,7%) в виде бесцветного масла.
1H ЯМР (CDCl3, 400 МГц) δ 7,84 (дд, 1H, J=8,0, 1,6 Гц), 7,68 (дд, 1H J=9,7, 1,4 Гц), 7,60 (т, J=7,6 Гц, 1H), 7,36-7,28 (м, 3H), 7,05 (дд, 2H, J=7,9, 1,4 Гц), 5,03 (с, 2H), 1,91 (с, 3H); LRMS (ES) m/z 380,2 (M++1).
Пример 62: Синтез соединения 11247, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилацетамида
[Стадия 1] N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилацетамид

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-фенилацетамид (0,075 г, 0,249 ммоль), синтезированный на стадии 2 Примера 61, и триэтиламин (0,069 мл, 0,498 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,032 мл, 0,299 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,093 г, 98,5%) в виде бесцветного масла.
[Стадия 2] Синтез соединения 11247

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-фенилацетамид (0,093 г, 0,245 ммоль), синтезированный на стадии 1, 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,088 г, 0,368 ммоль) смешивали в дихлорметане (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,005 г, 5,6%) в виде бесцветного масла.
1H ЯМР (CDCl3, 400 МГц) δ 7,84 (дд, J=8,0, 1,6 Гц, 1H), 7,69 (дд, J=9,8, 1,5 Гц, 1H), 7,58 (т, J=7,6 Гц, 1H), 7,37-7,30 (м, 3H),7,06-7,03 (м, 2H), 6,90 (т, J=48,6 Гц, 1H), 5,04 (с, 2H), 1,93 (с, 3H); LRMS (ES) m/z 362,2 (M++1).
Пример 63: Синтез соединения 11325, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-метилазетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил 3-((2-фтор-4-(метоксикарбонил)бензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилата
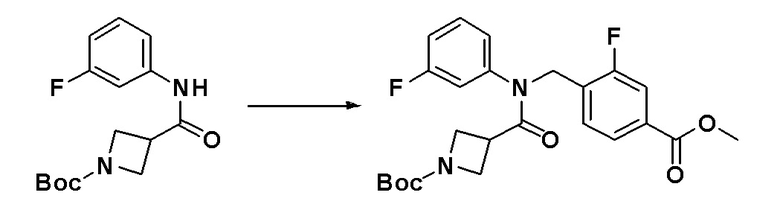
трет-Бутил 3-((3-фторфенил)карбамоил)азетидин-1-карбоксилат (0,550 г, 1,869 ммоль) растворяли в тетрагидрофуране (80 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,149 г, 3,737 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут и затем добавляли метил 4-(бромметил)-3-фторбензоат (0,508 г, 2,056 ммоль). Реакционную смесь продолжали перемешивать при 50°C в течение 12 часов и охлаждали до комнатной температуры. Затем к реакционной смеси добавляли воду (20 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 5% до 50%) и концентрировали с получением указанного в заголовке соединения (0,700 г, 81,4%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил 3-((2-фтор-4-(гидразинкарбонил)бензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилата
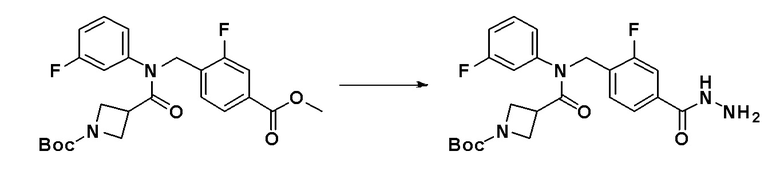
трет-Бутил 3-((2-фтор-4-(метоксикарбонил)бензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилат (0,700 г, 1,520 ммоль), синтезированный на стадии 1, и гидразин моногидрат (1,475 мл, 30,403 ммоль) смешивали в этаноле (50 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и к концентрату добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,600 г, 85,7%) в виде белого твердого вещества.
[Стадия 3] Синтез трет-бутил 3-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилата

трет-Бутил 3-((2-фтор-4-(гидразинкарбонил)бензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилат (0,600 г, 1,303 ммоль), синтезированный на стадии 2, и триэтиламин (0,218 мл, 1,564 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли дифторуксусный ангидрид (0,178 мл, 1,433 ммоль). Смесь перемешивали при этой же температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,600 г, 85,5%) в виде бесцветного масла.
[Стадия 4] Синтез трет-бутил 3-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилата

трет-Бутил 3-((4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилат (0,600 г, 1,114 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,398 г, 1,671 ммоль) смешивали в тетрагидрофуране (15 мл) при комнатной температуре. Смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (0,500 г, 86,2%) в виде белого твердого вещества.
[Стадия 5] Синтез N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамида гидрохлорида
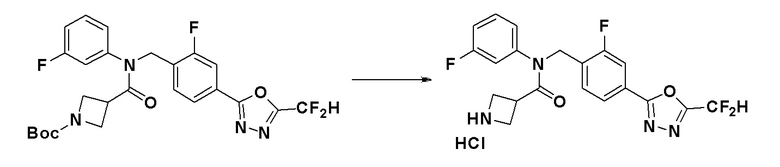
трет-Бутил 3-((4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)(3-фторфенил)карбамоил)азетидин-1-карбоксилат (0,500 г, 0,961 ммоль), синтезированный на стадии 4, растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 1,201 мл, 4,803 ммоль). Смесь перемешивали при этой же температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат суспендировали в диэтиловом эфире (50 мл) и фильтровали. Полученное твердое вещество промывали диэтиловым эфиром и сушили с получением указанного в заголовке соединения (0,430 г, 98,0%) в виде белого твердого вещества.
[Стадия 6] Синтез соединения 11325

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5, и формальдегид (37,00% раствор в воде, 0,012 мл, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,007 г, 14,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,6 Гц), 7,74 (дд, 1H, J=9,8, 1,5 Гц), 7,53 (т, 1H, J=7,6 Гц), 7,34 (тд, 1H, J=8,2, 6,4 Гц), 7,09 (тд, 1H, J=8,3, 2,3 Гц), 6,93 (дд, 1H, J=68,9, 34,4 Гц), 6,76-6,66 (м, 2H), 4,99 (д, 2H, J=19,7 Гц), 3,60 (дд, 2H, J=18,8, 10,8 Гц), 3,46 (т, 2H, J=8,0 Гц), 3,35 (дт, 1H, J=16,2, 8,1 Гц), 2,44 (д, 3H, J=13,4 Гц); LRMS (ES) m/z 435,2 (M++1).
Пример 64: Синтез соединения 11326, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-этил-N-(3-фторфенил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и ацетальдегид (0,009 мл, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,007 г, 14,3%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,5 Гц), 7,73 (дд, 1H, J=9,8, 1,5 Гц), 7,54 (т, 1H, J=7,6 Гц), 7,33 (тд, 1H, J=8,1, 6,4 Гц), 7,08 (тд, 1H, J=8,3, 1,9 Гц), 6,93 (дд, 1H, J=69,2, 34,1 Гц), 6,78-6,69 (м, 2H), 4,99 (д, 2H, J=19,8 Гц), 3,50-3,18 (м, 5H), 2,59 (кв., 2H, J=7,2 Гц), 0,96 (т, 3H, J=7,2 Гц); LRMS (ES) m/z 449,3 (M++1).
Пример 65: Синтез соединения 11327, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-пропилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и пропиональдегид (0,010 г, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,017 г, 33,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, 1H, J=8,0, 1,5 Гц), 7,72 (дд, 1H, J=9,8, 1,5 Гц), 7,51 (дд, 1H, J=15,7, 8,0 Гц), 7,32 (тт, 1H, J=11,9, 5,9 Гц), 7,07 (тд, 1H, J=8,1, 2,1 Гц), 6,90 (дд, 1H, J=58,6, 44,7 Гц), 6,77-6,68 (м, 2H), 4,99 (д, 2H, J=16,3 Гц), 3,61-3,47 (м, 2H), 3,47-3,31 (м, 3H), 2,57 (дд, 2H, J=18,4, 10,5 Гц), 1,39 (дкв., 2H, J=14,9, 7,4 Гц), 0,90-0,81 (м, 3H); LRMS (ES) m/z 463,2 (M++1).
Пример 66: Синтез соединения 11328, 1-бутил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и бутиральдегид (0,012 г, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 38,4%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, 1H, J=8,0, 1,5 Гц), 7,73 (дд, 1H, J=9,8, 1,5 Гц), 7,51 (т, 1H, J=7,6 Гц), 7,34 (дт, 1H, J=14,2, 6,5 Гц), 7,13-7,05 (м, 1H), 6,90 (дд, 1H, J=58,6, 44,7 Гц), 6,77-6,64 (м, 2H), 4,99 (д, 2H, J=13,6 Гц), 3,71-3,56 (м, 2H), 3,56-3,38 (м, 3H), 2,67 (дд, 2H, J=19,4, 11,4 Гц), 1,42-1,21 (м, 4H), 0,92-0,79 (м, 3H); LRMS (ES) m/z 477,3 (M++1).
Пример 67: Синтез соединения 11329, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-изопропилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и ацетон (0,012 мл, 0,164 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (0,035 г, 0,164 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 20%) и концентрировали с получением указанного в заголовке соединения (0,010 г, 19,8%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,5 Гц), 7,73 (дд, 1H, J=9,8, 1,5 Гц), 7,54 (т, 1H, J=7,5 Гц), 7,33 (тт, 1H, J=12,4, 6,2 Гц), 7,07 (тд, 1H, J=8,2, 2,0 Гц), 7,05-6,77 (м, 1H), 6,78-6,68 (м, 2H), 4,98 (д, 2H, J=19,6 Гц), 3,43 (с, 2H), 3,37-3,22 (м, 3H), 2,61-2,48 (м, 1H), 1,02-0,92 (м, 6H); LRMS (ES) m/z 463,2 (M++1).
Пример 68: Синтез соединения 11330, 1-ацетил-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,009 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 90%) и концентрировали с получением указанного в заголовке соединения (0,016 г, 31,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,6 Гц), 7,74 (дд, 1H, J=9,8, 1,6 Гц), 7,60-7,51 (м, 1H), 7,40-7,30 (м, 1H), 7,09 (тд, 1H, J=8,2, 2,4 Гц), 7,06-6,78 (м, 1H), 6,74 (дт, 2H, J=7,9, 6,8 Гц), 5,02 (д, 2H, J=19,1 Гц), 4,45 (с, 1H), 3,99 (д, 2H, J=33,8 Гц), 3,79-3,68 (м, 1H), 3,31 (тт, 1H, J=8,8, 6,3 Гц), 1,82 (с, 3H); LRMS (ES) m/z 463,2 (M++1).
Пример 69: Синтез соединения 11331, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-пропионилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,011 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 90%) и концентрировали с получением указанного в заголовке соединения (0,018 г, 34,5%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,5 Гц), 7,73 (дд, 1H, J=9,8, 1,4 Гц), 7,54 (дд, 1H, J=16,0, 8,3 Гц), 7,35 (дд, 1H, J=14,4, 8,1 Гц), 7,13-7,05 (м, 1H), 7,05-6,77 (м, 1H), 6,74 (дд, 2H, J=7,5, 5,3 Гц), 5,07-4,99 (м, 2H), 4,35 (дд, 1H, J=24,2, 19,7 Гц), 4,14-3,79 (м, 2H), 3,79-3,65 (м, 1H), 3,32 (тт, 1H, J=8,9, 6,4 Гц), 2,11-1,94 (м, 2H), 1,08 (дд, 3H, J=10,0, 5,1 Гц); LRMS (ES) m/z 477,3 (M++1).
Пример 70: Синтез соединения 11332, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-изобутирилазетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изобутирилхлорид (0,013 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 90%) и концентрировали с получением указанного в заголовке соединения (0,021 г, 39,1%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,4 Гц), 7,72 (дт, 1H, J=15,7, 7,8 Гц), 7,54 (дд, 1H, J=15,6, 8,0 Гц), 7,36 (дд, 1H, J=8,3, 6,3 Гц), 7,15-7,05 (м, 1H), 7,05-6,78 (м, 1H), 6,78-6,68 (м, 2H), 5,02 (д, 2H, J=19,0 Гц), 4,47 (с, 1H), 4,07-3,62 (м, 3H), 3,38-3,25 (м, 1H), 2,48-2,30 (м, 1H), 1,11-0,95 (м, 6H); LRMS (ES) m/z 491,2 (M++1).
Пример 71: Синтез соединения 11333, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-1-(метилсульфонил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,009 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 90%) и концентрировали с получением указанного в заголовке соединения (0,012 г, 22,0%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,5 Гц), 7,74 (дд, 1H, J=9,8, 1,4 Гц), 7,52 (дд, 1H, J=15,1, 7,5 Гц), 7,36 (дд, 1H, J=11,1, 5,0 Гц), 7,14-7,07 (м, 1H), 7,05-6,78 (м, 1H), 6,78-6,69 (м, 2H), 5,08-4,98 (м, 2H), 4,16-4,07 (м, 2H), 3,77-3,68 (м, 2H), 3,41-3,28 (м, 1H), 2,89 (д, 3H, J=5,3 Гц); LRMS (ES) m/z 499,2 (M++1).
Пример 72: Синтез соединения 11334, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(этилсульфонил)-N-(3-фторфенил)азетидин-3-карбоксамида

N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)азетидин-3-карбоксамид гидрохлорид (0,050 г, 0,109 ммоль), синтезированный на стадии 5 Примера 63, и N,N-диизопропилэтиламин (0,038 мл, 0,219 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли этансульфонилхлорид (0,015 мл, 0,120 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 90%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 35,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,6 Гц), 7,79-7,70 (м, 1H), 7,56 (дд, 1H, J=15,7, 8,1 Гц), 7,35 (тд, 1H, J=8,1, 6,4 Гц), 7,10 (тд, 1H, J=8,1, 2,1 Гц), 7,07-6,78 (м, 1H), 6,78-6,70 (м, 2H), 5,10-4,98 (м, 2H), 4,22-4,10 (м, 2H), 3,74-3,61 (м, 2H), 3,41-3,30 (м, 1H), 3,00-2,90 (м, 2H), 1,34 (кв., 3H, J=7,4 Гц); LRMS (ES) m/z 513,2 (M++1).
Пример 73: Синтез соединения 11339, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)ацетамида
[Стадия 1] Синтез метил 4-(бромметил)-3-фторбензоата
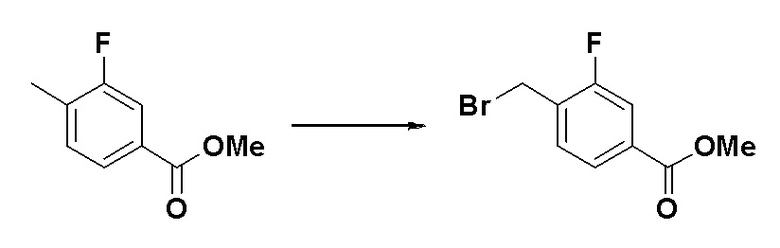
Метил 3-фтор-4-метилбензоат (8,500 г, 50,544 ммоль), 1-бромпирролидин-2,5-он (NBS, 9,446 г, 53,071 ммоль) и изобисизобутиронитрил (AIBN, 0,415 г, 2,527 ммоль) смешивали в дихлорметане (150 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (7,600 г, 60,9%) в виде белого твердого вещества.
[Стадия 2] Синтез метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоата

3-Фторанилин (1,000 г, 8,999 ммоль), метил 4-(бромметил)-3-фторбензоат (2,446 г, 9,899 ммоль), синтезированный на стадии 1, и N,N-диизопропилэтиламин (3,102 мл, 17,999 ммоль) растворяли в ацетонитриле (50 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (2,170 г, 87,0%) в виде бесцветного масла.
[Стадия 3] Синтез метил 3-фтор-4-((N-(3-фторфенил)ацетамидо)метил)бензоата

Метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоат (0,200 г, 0,721 ммоль), синтезированный на стадии 2, и N,N-диизопропилэтиламин (0,251 мл, 1,443 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли ацетилхлорид (0,061 мл, 0,866 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,210 г, 91,2%) в виде желтого масла.
[Стадия 4] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)ацетамида

Метил 3-фтор-4-((N-(3-фторфенил)ацетамидо)метил)бензоат (0,210 г, 0,658 ммоль), синтезированный на стадии 3, и гидразин гидрат (0,658 г, 13,153 ммоль) смешивали в этаноле (10 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. Затем к реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,187 г, 89,0%) в виде белого пенистого твердого вещества.
[Стадия 5] Синтез соединения 11339
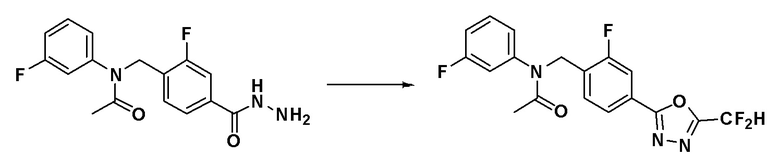
N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)ацетамид (0,090 г, 0,282 ммоль), синтезированный на стадии 4, и триэтиламин (0,079 мл, 0,564 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,037 мл, 0,338 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,071 г, 66,4%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,85 (дт, 1H, J=13,6, 6,8 Гц), 7,75-7,66 (м, 1H), 7,63-7,52 (м, 1H), 7,32 (тт, 1H, J=12,0, 6,0 Гц), 7,10-7,01 (м, 1H), 6,94-6,75 (м, 3H), 5,02 (с, 2H), 1,95 (с, 3H); LRMS (ES) m/z 380,2 (M++1).
Пример 74: Синтез соединения 11340, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-(3-фторфенил)ацетамида
[Стадия 1] N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)ацетамид

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)ацетамид (0,090 г, 0,282 ммоль), синтезированный на стадии 4 Примера 73, и триэтиламин (0,079 мл, 0,564 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,048 мл, 0,338 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,112 г, 95,7%) в виде желтого масла.
[Стадия 2] Синтез соединения 11340
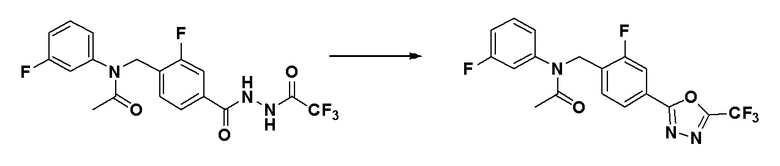
N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)-N-(3-фторфенил)ацетамид (0,120 г, 0,289 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,103 г, 0,433 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при комнатной температуре в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,029 г, 25,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,4 Гц), 7,73 (д, 1H, J=9,7 Гц), 7,61 (т, 1H, J=7,6 Гц), 7,34 (тд, 1H, J=8,1, 6,5 Гц), 7,05 (тд, 1H, J=8,3, 2,1 Гц), 6,84 (дд, 2H, J=22,0, 8,5 Гц), 5,03 (с, 2H), 1,95 (с, 3H); LRMS (ES) m/z 398,1 (M++1).
Пример 75: Синтез соединения 11341, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-фенилизоникотинамида
[Стадия 1] Синтез метил 6-((N-фенилизоникотинамидо)метил)никотината

Метил 6-((фениламино)метил)никотинат (0,050 г, 0,206 ммоль) и триэтиламин (0,058 мл, 0,413 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изоникотиноилхлорид гидрохлорид (0,044 г, 0,248 ммоль) с последующим перемешиванием при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,071 г, 99,0%) в виде бесцветного масла.
[Стадия 2] Синтез N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилизоникотинамида
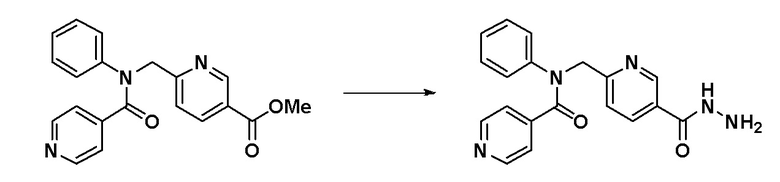
Метил 6-((N-фенилизоникотинамидо)метил)никотинат (0,071 г, 0,204 ммоль), синтезированный на стадии 1, и гидразин гидрат (0,199 мл, 4,088 ммоль) растворяли в этаноле (10 мл) при 90°C и раствор перемешивали при этой же температуре в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,070 г, 98,6%) в виде бесцветного масла.
[Стадия 3] Синтез соединения 11341

N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилизоникотинамид (0,070 г, 0,202 ммоль), синтезированный на стадии 2, и триэтиламин (0,056 мл, 0,403 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,028 мл, 0,262 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (0,045 г, 54,8%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 9,29 (д, 1H, J=1,7 Гц), 8,47 (д, 2H, J=5,3 Гц), 8,38 (дд, 1H, J=8,2, 2,2 Гц), 7,63 (д, 1H, J=8,2 Гц), 7,25-7,16 (м, 5H), 7,16-7,09 (м, 2H), 7,09-6,79 (м, 1H), 5,28 (д, 2H, J=21,7 Гц); LRMS (ES) m/z 408,3 (M++1).
Пример 76: Синтез соединения 11356, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)пропионамида
[Sep 1] Синтез метил 3-фтор-4-((N-(3-фторфенил)пропионамидо)метил)бензоата

Метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоат (0,100 г, 0,361 ммоль), синтезированный на стадии 2 Примера 73, и N,N-диизопропилэтиламин (0,126 мл, 0,721 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли пропионилхлорид (0,041 мл, 0,469 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 83,2%) в виде бесцветного масла.
[Стадия 2] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)пропионамида

Метил 3-фтор-4-((N-(3-фторфенил)пропионамидо)метил)бензоат (0,100 г, 0,300 ммоль), синтезированный на стадии 1, и гидразин моногидрат (0,292 мл, 6,000 ммоль) смешивали в этаноле (6 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,098 г, 98,0%) в виде бесцветного масла.
[Стадия 3] Синтез N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)пропионамида
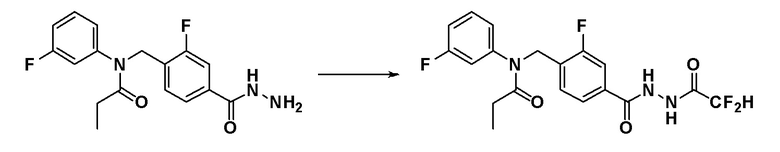
N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)пропионамид (0,098 г, 0,294 ммоль), синтезированный на стадии 2, и триэтиламин (0,082 мл, 0,588 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,038 мл, 0,353 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,120 г, 99,2%) в виде бесцветного масла.
[Стадия 4] Синтез соединения 11356

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)пропионамид (0,120 г, 0,292 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,104 г, 0,438 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,018 г, 15,7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 9,29 (д, 1H, J=1,7 Гц), 8,47 (д, 2H, J=5,3 Гц), 8,38 (дд, 1H, J=8,2, 2,2 Гц), 7,63 (д, 1H, J=8,2 Гц), 7,25-7,16 (м, 5H), 7,16-7,09 (м, 2H), 7,09-6,79 (м, 1H), 5,28 (д, 2H, J=21,7 Гц); LRMS (ES) m/z 408,3 (M++1).
Пример 77: Синтез соединения 11357, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)бутирамида
[Стадия 1] Синтез метил 3-фтор-4-((N-(3-фторфенил)бутирамидо)метил)бензоата
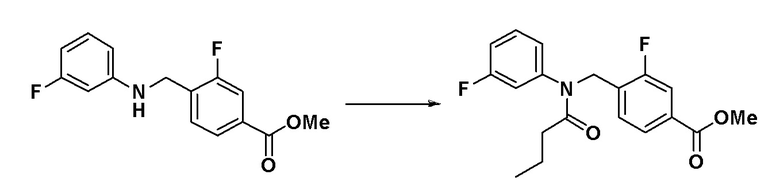
Метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоат (0,100 г, 0,361 ммоль), синтезированный на стадии 2 Примера 73, и N,N-диизопропилэтиламин (0,126 мл, 0,721 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли бутирилхлорид (0,049 мл, 0,469 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,103 г, 82,2%) в виде бесцветного масла.
[Стадия 2] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)бутирамида

Метил 3-фтор-4-((N-(3-фторфенил)бутирамидо)метил)бензоат (0,103 г, 0,297 ммоль), синтезированный на стадии 1, и гидразин моногидрат (0,288 мл, 5,930 ммоль) смешивали в этаноле (6 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,100 г, 97,1%) в виде бесцветного масла.
[Стадия 3] Синтез N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)бутирамида
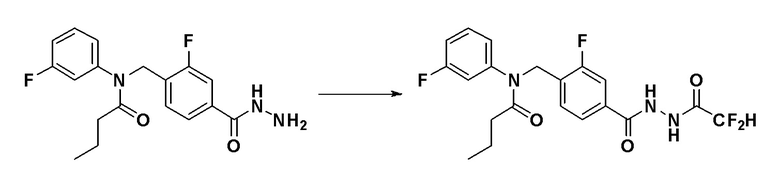
N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)бутирамид (0,100 г, 0,288 ммоль), синтезированный на стадии 2, и триэтиламин (0,080 мл, 0,576 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,038 мл, 0,345 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,120 г, 98,0%) в виде бесцветного масла.
[Стадия 4] Синтез соединения 11357

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)бутирамид (0,120 г, 0,282 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,101 г, 0,423 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,012 г, 10,4%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,1, 1,7 Гц), 7,72 (дд, 1H, J=9,8, 1,7 Гц), 7,59 (т, 1H, J=7,6 Гц), 7,33 (тд, 1H, J=8,1, 6,3 Гц), 7,11-7,01 (м, 1H), 6,93-6,75 (м, 3H), 5,02 (с, 2H), 2,10 (т, 2H, J=7,4 Гц), 1,64 (г, 2H, J=7,4 Гц), 0,86 (т, 3H, J=7,4 Гц); LRMS (ES) m/z 408,2(M++1).
Пример 78: Синтез соединения 11358, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-(3-фторфенил)-3-метилбутанамида
[Стадия 1] Синтез метил 3-фтор-4-((N-(3-фторфенил)-3-метилбутанамидо)метил)бензоата

Метил 3-фтор-4-(((3-фторфенил)амино)метил)бензоат (0,100 г, 0,361 ммоль), синтезированный на стадии 2 Примера 73, и N,N-диизопропилэтиламин (0,126 мл, 0,721 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 3-метилбутаноилхлорид (0,057 мл, 0,469 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,115 г, 88,2%) в виде бесцветного масла.
[Стадия 2] Синтез N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)-3-метилбутанамида
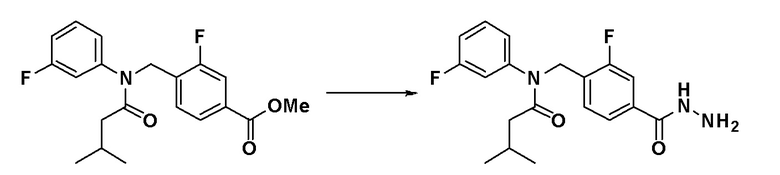
Метил 3-фтор-4-((N-(3-фторфенил)-3-метилбутанамидо)метил)бензоат (0,115 г, 0,318 ммоль), синтезированный на стадии 1, и гидразин моногидрат (0,309 мл, 6,364 ммоль) смешивали в этаноле (6 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,105 г, 91,3%) в виде бесцветного масла.
[Стадия 3] N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)-3-метилбутанамид

N-(2-фтор-4-(гидразинкарбонил)бензил)-N-(3-фторфенил)-3-метилбутанамид (0,105 г, 0,291 ммоль), синтезированный на стадии 2, и триэтиламин (0,081 мл, 0,581 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,038 мл, 0,349 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,122 г, 95,6%) в виде бесцветного масла.
[Стадия 4] Синтез соединения 11358

N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-N-(3-фторфенил)-3-метилбутанамид (0,140 г, 0,319 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,114 г, 0,478 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,011 г, 8,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, 1H, J=8,0, 1,7 Гц), 7,72 (дд, 1H, J=9,8, 1,7 Гц), 7,60 (т, 1H, J=7,6 Гц), 7,33 (тд, 1H, J=8,1, 6,3 Гц), 7,10-7,02 (м, 1H), 6,93-6,74 (м, 3H), 5,03 (с, 2H), 2,18 (дкв., 1H, J=13,5, 6,7 Гц), 2,01 (д, 2H, J=7,0 Гц), 0,87 (д, 6H, J=6,6 Гц); LRMS (ES) m/z 422,3 (M++1).
Пример 79: Синтез соединения 11359, N-(3-фторфенил)-N-((5-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)изоникотинамида
[Стадия 1] Синтез метил 6-((N-(3-фторфенил)изоникотинамидо)метил)никотината

Метил 6-(((3-фторфенил)амино)метил)никотинат (0,200 г, 0,768 ммоль) и N,N-диизопропилэтиламин (0,268 мл, 1,537 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли изоникотиноилхлорид гидрохлорид (0,178 г, 0,999 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (0,277 г, 98,7%) в виде бесцветного масла.
[Стадия 2] Синтез N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)изоникотинамида

Метил 6-((N-(3-фторфенил)изоникотинамидо)метил)никотинат (0,277 г, 0,758 ммоль), синтезированный на стадии 1, и гидразин моногидрат (0,737 мл, 15,163 ммоль) смешивали в этаноле (10 мл) и смесь нагревали с использованием микроволнового излучения при 120°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,220 г, 79,4%) в виде белого пенистого твердого вещества.
[Стадия 3] Синтез N-(3-фторфенил)-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)изоникотинамида
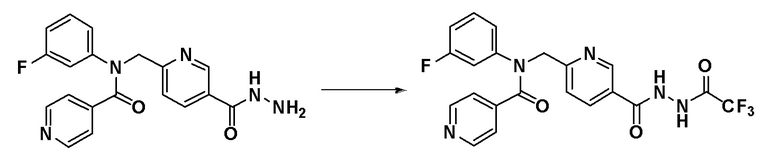
N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)изоникотинамид (0,100 г, 0,274 ммоль), синтезированный на стадии 2, и триэтиламин (0,076 мл, 0,547 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,050 мл, 0,356 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,121 г, 95,8%) в виде бесцветного масла.
[Стадия 4] Синтез соединения 11359

N-(3-фторфенил)-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)изоникотинамид (0,130 г, 0,282 ммоль), синтезированный на стадии 3, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,101 г, 0,423 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (0,024 г, 19,2%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 9,37-9,26 (м, 1H), 8,61-8,48 (м, 2H), 8,40 (дд, 1H, J=8,2, 2,2 Гц), 7,63 (д, 1H, J=8,2 Гц), 7,32-7,27 (м, 2H), 7,18 (тд, 1H, J=8,2, 6,2 Гц), 7,02 (дт, 1H, J=9,4, 2,3 Гц), 6,96-6,87 (м, 2H), 5,28 (с, 2H); LRMS (ES) m/z 444,3 (M++1).
Пример 80: Синтез соединения 11360, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-(3-фторфенил)изоникотинамида
[Стадия 1] Синтез N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-(3-фторфенил)изоникотинамида

N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)изоникотинамид (0,120 г, 0,328 ммоль), синтезированный на стадии 2 Примера 79, и триэтиламин (0,092 мл, 0,657 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,046 мл, 0,427 ммоль). Смесь перемешивали при этой же температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,140 г, 96,1%) в виде бесцветного масла.
[Стадия 2] Синтез соединения 11360
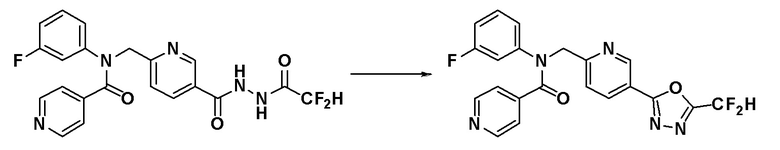
N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-(3-фторфенил)изоникотинамид (0,140 г, 0,316 ммоль), синтезированный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,113 г, 0,474 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (0,021 г, 15,6%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3) δ 9,35-9,24 (м, 1H), 8,57-8,48 (м, 2H), 8,40 (дд, 1H, J=8,2, 2,2 Гц), 7,62 (д, 1H, J=8,2 Гц), 7,24 (д, 2H, J=1,5 Гц), 7,18 (тд, 1H, J=8,2, 6,3 Гц), 7,08-6,98 (м, 1H), 6,95-6,80 (м, 3H), 5,27 (с, 2H); LRMS (ES) m/z 426,3 (M++1).
Пример 81: Синтез соединения 11376, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-(3-фторфенил)никотинамида
[Стадия 1] Синтез метил 6-(((3-фторфенил)амино)метил)никотината

3-Фторанилин (1,500 г, 13,499 ммоль) и метил 6-формилникотинат (2,452 г, 14,849 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли триацетоксиборогидрид натрия (4,291 г, 20,248 ммоль) добавляли к раствору. Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (2,830 г, 80,5%) в виде желтого твердого вещества.
[Стадия 2] Синтез метил 6-((N-(3-фторфенил)никотинамидо)метил)никотината

Метил 6-(((3-фторфенил)амино)метил)никотинат (0,100 г, 0,384 ммоль), синтезированный на стадии 1, и триэтиламин (0,107 мл, 0,768 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и к раствору добавляли никотиноилхлорид гидрохлорид (0,103 г, 0,576 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,111 г, 79,1%) в виде желтого масла.
[Стадия 3] Синтез N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)никотинамида

Метил 6-((N-(3-фторфенил)никотинамидо)метил)никотинат (0,111 г, 0,304 ммоль), синтезированный на стадии 2, и гидразин моногидрат (0,295 мл, 6,076 ммоль) смешивали в этаноле (10 мл) при комнатной температуре и смесь нагревали при кипячении с обратным холодильником в течение 18 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,108 г, 97,3%) в виде желтого масла.
[Стадия 4] Синтез N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-(3-фторфенил)никотинамида

N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)никотинамид (0,111 г, 0,304 ммоль), синтезированный на стадии 3, и триэтиламин (0,085 мл, 0,608 ммоль) растворяли в тетрагидрофуране (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,050 мл, 0,456 ммоль). Смесь перемешивали при 80°C в течение 18 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,133 г, 98,7%) в виде бесцветного масла.
[Стадия 5] Синтез соединения 11376

N-((5-(2-(2,2-дифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)-N-(3-фторфенил)никотинамид (0,140 г, 0,316 ммоль), синтезированный на стадии 4, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,226 г, 0,947 ммоль) смешивали в тетрагидрофуране (10 мл) и смесь нагревали с использованием микроволнового излучения при 150°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,018 г, 13,4%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3) δ 9,35-9,24 (м, 2H), 8,81 (д, 1H, J=5,1 Гц), 8,64-8,51 (м, 2H), 8,41 (ддд, 1H, J=8,3, 3,6, 2,3 Гц), 7,81-7,62 (м, 2H), 7,26-7,15 (м, 2H), 6,88-6,42 (м, 2H), 5,32 (д, 2H, J=2,8 Гц); LRMS (ES) m/z 426,4 (M++1).
Пример 82: Синтез соединения 11414, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-N-фенилпиперидин-4-карбоксамида
[Стадия 1] Синтез метил 6-((1-(трет-бутоксикарбонил)-N-фенилпиперидин-4-карбоксамидо)метил)никотината

трет-Бутил 4-(фенилкарбамоил)пиперидин-1-карбоксилат (0,400 г, 1,314 ммоль) растворяли в тетрагидрофуране (30 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,079 г, 1,971 ммоль), поддерживая температуру при 0°C. Смесь перемешивали в течение 20 минут. К реакционному раствору добавляли метил 6-(бромметил)никотинат (0,333 г, 1,446 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду (2 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 40%) и концентрировали с получением указанного в заголовке соединения (0,390 г, 65,4%) в виде пенистого твердого вещества.
[Стадия 2] Синтез метил 6-((N-фенилпиперидин-4-карбоксамидо)метил)никотината гидрохлорида

Метил 6-((1-(трет-бутоксикарбонил)-N-фенилпиперидин-4-карбоксамидо)метил)никотинат (0,390 г, 0,860 ммоль), синтезированный на стадии 1, растворяли в дихлорметане (30 мл) и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 2,150 мл, 8,599 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,330 г, 98,4%) в виде белого твердого вещества.
[Стадия 3] Синтез метил 6-((1-метил-N-фенилпиперидин-4-карбоксамидо)метил)никотината

Метил 6-((N-фенилпиперидин-4-карбоксамидо)метил)никотинат гидрохлорид (0,150 г, 0,385 ммоль), синтезированный на стадии 2, формальдегид (37,00% раствор в воде, 0,143 мл, 1,924 ммоль) и триацетоксиборогидрид натрия (0,204 г, 0,962 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 10%) и концентрировали с получением указанного в заголовке соединения (0,120 г, 84,9%) в виде пенистого твердого вещества.
[Стадия 4] Синтез N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1-метил-N-фенилпиперидин-4-карбоксамида

Метил 6-((1-метил-N-фенилпиперидин-4-карбоксамидо)метил)никотинат (0,120 г, 0,327 ммоль), синтезированный на стадии 3, и гидразин моногидрат (0,079 мл, 1,633 ммоль) растворяли в этаноле (10 мл) при комнатной температуре и раствор нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (0,115 г, 95,8%) в виде пенистого твердого вещества.
[Стадия 5] Синтез соединения 11414

N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1-метил-N-фенилпиперидин-4-карбоксамид (0,200 г, 0,544 ммоль), синтезированный на стадии 4, 2,2-дифторуксусный ангидрид (0,178 мл, 1,633 ммоль) и триэтиламин (0,152 мл, 1,089 ммоль) растворяли в тетрагидрофуране (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2 пластина, 20×20×1 мм; метанол/дихлорметан = 20%) и концентрировали с получением указанного в заголовке соединения (0,002 г, 0,9%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,30-9,24 (м, 1H), 8,36 (дд, 1H, J=8,2, 2,2 Гц), 7,58-7,33 (м, 5H), 7,25-7,22 (м, 1H), 7,10-6,80 (м, 1H), 5,05 (с, 2H), 3,46 (д, 2H, J=11,1 Гц), 3,26 (д, 2H, J=11,8 Гц), 2,91 (с, 2H), 2,74 (д, 3H, J=4,1 Гц), 2,36 (с, 1H), 1,96 (д, 2H, J=14,7 Гц); LRMS (ES) m/z 428,5 (M++1).
Пример 83: Синтез соединения 11418, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-(3-фторфенил)-1-метилпиперидин-4-карбоксамида
[Стадия 1] Синтез метил 6-((1-(трет-бутоксикарбонил)-N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотината

трет-Бутил 4-((3-фторфенил)карбамоил)пиперидин-1-карбоксилат (0,400 г, 1,241 ммоль) растворяли в тетрагидрофуране (30 мл) и к раствору медленно добавляли гидрид натрия (60,00%, 0,074 г, 1,861 ммоль), поддерживая температуру при 0°C. Реакционную смесь перемешивали в течение 20 минут. Затем к реакционному раствору добавляли метил 6-(бромметил)никотинат (0,314 г, 1,365 ммоль) с последующим дополнительным перемешиванием при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду (2 мл) при 0°C с последующим перемешиванием в течение 5 минут. После завершения реакции к реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 5% до 40%) и концентрировали с получением указанного в заголовке соединения (0,400 г, 68,4%) в виде пенистого твердого вещества.
[Стадия 2] Синтез метил 6-((N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотината гидрохлорида

Метил 6-((1-(трет-бутоксикарбонил)-N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотинат (0,400 г, 0,848 ммоль), синтезированный на стадии 1, растворяли в дихлорметане (30 мл) и к раствору добавляли хлористоводородную кислоту (4,00 M раствор в диоксане, 2,121 мл, 8,483 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (0,340 г, 98,3%) в виде белого твердого вещества.
[Стадия 3] Синтез метил 6-((N-(3-фторфенил)-1-метилпиперидин-4-карбоксамидо)метил)никотината

Метил 6-((N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотинат гидрохлорид (0,150 г, 0,368 ммоль), синтезированный на стадии 2, формальдегид (37,00% раствор в воде, 0,137 мл, 1,839 ммоль) и триацетоксиборогидрид натрия (0,156 г, 0,736 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 10%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 91,7%) в виде пенистого твердого вещества.
[Стадия 4] Синтез N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1-метилпиперидин-4-карбоксамида

Метил 6-((N-(3-фторфенил)-1-метилпиперидин-4-карбоксамидо)метил)никотинат (0,130 г, 0,337 ммоль), синтезированный на стадии 3, и гидразин моногидрат (0,082 мл, 1,686 ммоль) растворяли в этаноле (10 мл) при комнатной температуре и раствор нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (0,120 г, 92,3%) в виде пенистого твердого вещества.
[Стадия 5] Синтез соединения 11418

N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1-метилпиперидин-4-карбоксамид (0,200 г, 0,519 ммоль), синтезированный на стадии 4, и триэтиламин (0,145 мл, 1,038 ммоль) растворяли в тетрагидрофуране (13 мл) и к раствору добавляли 2,2-дифторуксусный ангидрид (0,085 мл, 0,778 ммоль) при комнатной температуре. Реакционный раствор нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали и затем полученное вещество снова очищали хроматографией (SiO2 пластина, 20×20×1 мм; метанол/дихлорметан = 10%) и концентрировали с получением указанного в заголовке соединения (0,015 г, 6,5%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,25 (м, 1H), 8,38 (м, 1H), 7,54 (м, 1H), 7,35 (м, 1H), 7,10-6,82 (м, 4H), 5,05 (с, 2H), 2,95 (м, 2H), 2,42-2,32 (м, 5H), 1,99-1,78 (м, 5H); LRMS (ES) m/z 446,4 (M++1).
Пример 84: Синтез соединения 11419, N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-этил-N-(3-фторфенил)пиперидин-4-карбоксамида
[Стадия 1] Синтез метил 6-((1-этил-N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотината
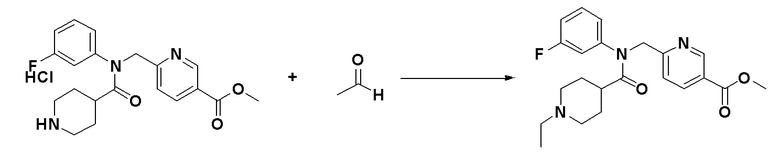
Метил 6-((N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотинат гидрохлорид (0,150 г, 0,368 ммоль), синтезированный на стадии 2 Примера 83, ацетальдегид (0,104 мл, 1,839 ммоль) и триацетоксиборогидрид натрия (0,156 г, 0,736 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 10%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 88,5%) в виде пенистого твердого вещества.
[Стадия 2] Синтез 1-этил-N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперидин-4-карбоксамида

Метил 6-((1-этил-N-(3-фторфенил)пиперидин-4-карбоксамидо)метил)никотинат (0,130 г, 0,325 ммоль), синтезированный на стадии 1, и гидразин моногидрат (0,079 мл, 1,627 ммоль) растворяли в этаноле (10 мл) при комнатной температуре и раствор нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 5% до 30%) и концентрировали с получением указанного в заголовке соединения (0,120 г, 92,3%) в виде пенистого твердого вещества.
[Стадия 3] Синтез соединения 11419

1-этил-N-(3-фторфенил)-N-((5-(гидразинкарбонил)пиридин-2-ил)метил)пиперидин-4-карбоксамид (0,200 г, 0,501 ммоль), синтезированный на стадии 2, и триэтиламин (0,140 мл, 1,001 ммоль) растворяли в тетрагидрофуране (13 мл) и к раствору добавляли 2,2-дифторуксусный ангидрид (0,082 мл, 0,751 ммоль) при комнатной температуре. Реакционный раствор нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали и затем полученное вещество снова очищали хроматографией (SiO2 пластина, 20×20×1 мм; метанол/дихлорметан = 10%) и концентрировали с получением указанного в заголовке соединения (0,015 г, 6,5%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,25 (м, 1H), 8,37 (м, 1H), 7,52 (м, 1H), 7,36 (м, 1H), 7,12-6,82 (м, 4H), 5,04 (с, 2H), 3,11 (м, 2H), 2,63-2,16 (м, 4H), 2,01-1,90 (м, 5H), 1,18 (м, 3H); LRMS (ES) m/z 460,5 (M++1).
Пример 85: Синтез соединения 11534, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(пирролидин-1-ил)ацетамида
[Стадия 1] Синтез 2-(бензилокси)-N-(4-хлор-3-фторфенил)ацетамида

4-хлор-3-фторанилин (1,000 г, 6,870 ммоль), 2-(бензилокси)уксусную кислоту (1,179 мл, 8,244 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC, 2,133 г, 13,740 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 1,857 г, 13,740 ммоль) и N,N-диизопропилэтиламин (2,393 мл, 13,740 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (1,880 г, 93,2%) в виде желтого твердого вещества.
[Стадия 2] Синтез метил 4-((2-(бензилокси)-N-(3-хлор-4-фторфенил)ацетамидо)метил)-3-фторбензоата

2-(Бензилокси)-N-(4-хлор-3-фторфенил)ацетамид (1,880 г, 6,401 ммоль), синтезированный на стадии 1, и карбонат кальция (1,327 г, 9,601 ммоль) растворяли в тетрагидрофуране (50 мл) при 0°C и к раствору добавляли метил 4-(бромметил)-3-фторбензоат (1,898 г, 7,681 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (1,780 г, 60,5%) в виде желтого масла.
[Стадия 3] Синтез 2-(бензилокси)-N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(гидразинкарбонил)бензил)ацетамида

Метил 4-((2-(бензилокси)-N-(3-хлор-4-фторфенил)ацетамидо)метил)-3-фторбензоат (1,780 г, 3,871 ммоль), синтезированный на стадии 2, и гидразин моногидрат (5,644 мл, 116,120 ммоль) растворяли в этаноле (8 мл)/воде (2 мл) при комнатной температуре и раствор перемешивали при 80°C в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (1,580 г, 88,8%, желтое твердое вещество).
[Стадия 4] Синтез 2-(бензилокси)-N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)ацетамида

2-(Бензилокси)-N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(гидразинкарбонил)бензил)ацетамид (1,580 г, 3,436 ммоль), синтезированный на стадии 3, и триэтиламин (0,958 мл, 6,871 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,513 мл, 4,123 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (1,120 г, 60,6%) в виде бесцветного масла.
[Стадия 5] Синтез N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-2-гидроксиацетамида

2-(Бензилокси)-N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)ацетамид (0,880 г, 1,636 ммоль), синтезированный на стадии 4, растворяли в метаноле (50 мл) при комнатной температуре и к раствору медленно добавляли 10%-Pd/C (600 мг). Смесь перемешивали при этой же температуре в течение 5 часов под баллоном водорода. Реакционную смесь фильтровали через слой целита для удаления твердых веществ и к фильтрату добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,658 г, 89,8%) в виде желтого масла.
[Стадия 6] Синтез 2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфоната

N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)-2-гидроксиацетамид (0,658 г, 1,470 ммоль), синтезированный на стадии 5, и N,N-диизопропилэтиламин (0,768 мл, 4,409 ммоль) растворяли в дихлорметане (50 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,284 мл, 3,674 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 50%) и концентрировали с получением указанного в заголовке соединения (0,254 г, 34,0%) в виде желтого масла.
[Стадия 7] Синтез соединения 11534

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6, пирролидин (0,011 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,013 г, 27,3%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,91 (дд, 1H, J=8,0, 1,7 Гц), 7,76 (дд, 1H, J=9,9, 1,7 Гц), 7,65 (т, 1H, J=7,6 Гц), 7,29 (дд, 1H, J=6,2, 2,6 Гц), 7,22-7,07 (м, 2H), 7,06-6,75 (м, 1H), 5,01 (с, 2H), 3,88-3,73 (м, 4H), 3,20 (т, 2H, J=8,7 Гц), 2,17 (д, 4H, J=6,2 Гц); LRMS (ES) m/z 483,4 (M++1).
Пример 86: Синтез соединения 11535, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-морфолиноацетамида

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, морфолин (0,013 мл, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,042 г, 85,5%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,88 (ддд, 1H, J=8,0, 4,9, 1,7 Гц), 7,73 (ддд, 1H, J=9,8, 8,0, 1,7 Гц), 7,62 (тд, 1H, J=7,6, 2,3 Гц), 7,27-7,23 (м, 1H), 7,18-7,05 (м, 2H), 7,05-6,76 (м, 1H), 5,03-4,90 (м, 2H), 3,82 (дт, 4H, J=23,4, 4,8 Гц), 3,27-3,04 (м, 2H), 2,77 (д, 4H, J=55,3 Гц); LRMS (ES) m/z 499,5 (M++1).
Пример 87: Синтез соединения 11536, (S)-N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(3-гидроксипирролидин-1-ил)ацетамида

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, (S)-пирролидин-3-ол (0,013 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 30%) и концентрировали с получением указанного в заголовке соединения (0,044 г, 89,6%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,88 (т, 1H, J=7,0 Гц), 7,79-7,51 (м, 2H), 7,24-7,05 (м, 3H), 6,92 (т, 1H, J=51,8 Гц), 5,02 (с, 2H), 4,21-3,43 (м, 4H), 3,27 (д, 1H, J=47,5 Гц), 2,49 (с, 5H); LRMS (ES) m/z 499,5 (M++1).
Пример 88: Синтез соединения 11537, (R)-N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(2-(гидроксиметил)пирролидин-1-ил)ацетамида

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, (R)-пирролидин-2-илметанол (0,015 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/гексан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,019 г, 37,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,89 (ддд, 1H, J=8,2, 6,9, 1,7 Гц), 7,75 (ддд, 1H, J=9,9, 6,1, 1,7 Гц), 7,60 (дт, 1H, J=14,8, 7,6 Гц), 7,25-7,05 (м, 3H), 7,03-6,77 (м, 1H), 5,08-4,92 (м, 2H), 3,92-2,66 (м, 7H), 2,29-1,47 (м, 4H); LRMS (ES) m/z 513,5 (M++1).
Пример 89: Синтез соединения 11538, (S)-1-(2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтил)пирролидин-2-карбоксамида
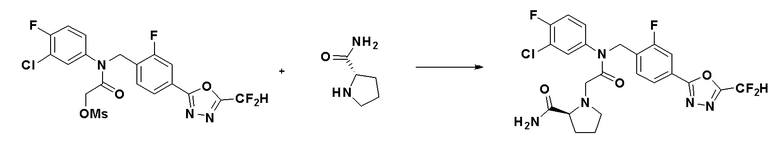
2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, (S)-пирролидин-2-карбоксамид (0,017 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,041 г, 79,2%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,89 (тд, 1H, J=7,4, 1,6 Гц), 7,75 (ддд, 1H, J=9,8, 6,0, 1,6 Гц), 7,60 (дт, 1H, J=14,8, 7,5 Гц), 7,25-7,05 (м, 2H), 7,06-6,76 (м, 2H), 5,09-4,91 (м, 2H), 3,85-2,98 (м, 6H), 3,18-3,02 (м, 1H), 2,22-1,68 (м, 4H); LRMS (ES) m/z 526,5 (M++1).
Пример 90: Синтез соединения 11584, N-фенил-N-((5-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)изоникотинамида
[Стадия 1] Синтез N-фенил-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)изоникотинамида

N-((5-(гидразинкарбонил)пиридин-2-ил)метил)-N-фенилизоникотинамид (1,000 г, 2,879 ммоль), синтезированный на стадии 2 Примера 75, и триэтиламин (0,802 мл, 5,757 ммоль) растворяли в тетрагидрофуране (30 мл) и к раствору добавляли трифторуксусный ангидрид (0,813 мл, 5,757 ммоль) при комнатной температуре. Смесь нагревали при кипячении с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующим экстрагированием дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Указанное в заголовке соединение использовали без дополнительной очистки (0,750 г, 61,2%) в виде бесцветного масла.
[Стадия 2] Синтез соединения 11584

N-фенил-N-((5-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)пиридин-2-ил)метил)изоникотинамид (0,900 г, 2,030 ммоль), синтезированный на стадии 1, 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,484 г, 2,030 ммоль) смешивали в тетрагидрофуране (15 мл) при комнатной температуре и смесь нагревали с использованием микроволнового излучения при 150°C в течение 30 минут и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием дихлорметаном. Экстракт фильтровали через пластиковый фильтр для удаления твердого остатка и водного слоя и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 80 г картридж; этилацетат/гексан = от 10% до 60%) и концентрировали и затем полученный продукт снова очищали хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 10% до 60%) и концентрировали с получением указанного в заголовке соединения (0,470 г, 54,4%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (м, 1H), 8,54 (м, 2H), 8,40 (м, 1H), 7,65 (м, 1H), 7,40 (м, 2H), 7,25 (м, 3H), 7,17 (м, 2H), 5,32 (с, 2H); LRMS (ES) m/z 426,4 (M++1).
Пример 91: Синтез соединения 11602, N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида
[Стадия 1] Синтез N-(3-хлор-4-фторфенил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида
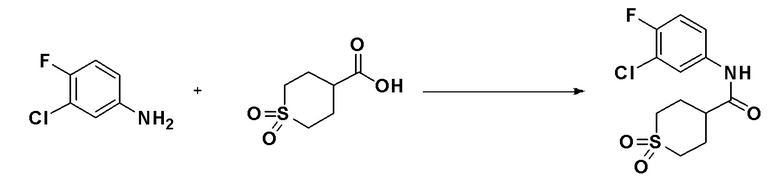
3-Хлор-4-фторанилин (0,700 г, 4,809 ммоль), тетрагидро-2H-тиопиран-4-карбоновую кислоту 1,1-диоксид (0,943 г, 5,290 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (EDC-HCl, 1,844 г, 9,618 ммоль), 1H-бензо[d][1,2,3]триазол-1-ол (HOBt, 1,300 г, 9,618 ммоль) и N,N-диизопропилэтиламин (1,675 мл, 9,618 ммоль) растворяли в дихлорметане (100 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,988 г, 67,2%) в виде белого твердого вещества.
[Стадия 2] Синтез метил 4-((N-(3-хлор-4-фторфенил)-1,1-диоксидотетрагидро-2H-тиопиран-4-карбоксамидо)метил)-3-фторбензоата

N-(3-хлор-4-фторфенил)тетрагидро-2H-тиопиран-4-карбоксамид 1,1-диоксид (1,000 г, 3,271 ммоль), синтезированный на стадии 1, и гидрид натрия (60,00%, 0,262 г, 6,541 ммоль) растворяли в N,N-диметилформамиде (50 мл) при комнатной температуре и к раствору добавляли метил 4-(бромметил)-3-фторбензоат (1,212 г, 4,906 ммоль). Смесь перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 100%) и концентрировали с получением указанного в заголовке соединения (1,240 г, 80,3%) в виде белого твердого вещества.
[Стадия 3] Синтез N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(гидразинкарбонил)бензил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида

Метил 4-((N-(3-хлор-4-фторфенил)-1,1-диоксидотетрагидро-2H-тиопиран-4-карбоксамидо)метил)-3-фторбензоат (1,240 г, 2,628 ммоль), синтезированный на стадии 2, и гидразин моногидрат (2,554 мл, 52,554 ммоль) растворяли в этаноле (20 мл)/воде (5 мл) при комнатной температуре и раствор перемешивали при 80°C в течение 5 часов и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Указанное в заголовке соединение использовали без дополнительной очистки (1,180 г, 95,2%) в виде желтого твердого вещества.
[Стадия 4] Синтез N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида
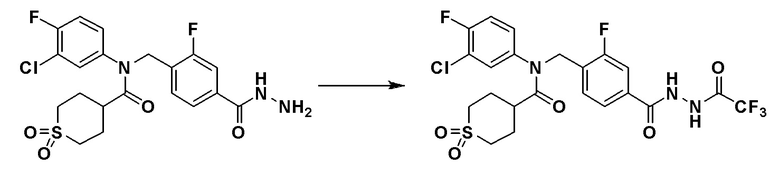
N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(гидразинкарбонил)бензил)тетрагидро-2H-тиопиран-4-карбоксамид 1,1-диоксид (0,200 г, 0,424 ммоль), синтезированный на стадии 3, 1,1-диоксид (0,200 г, 0,424 ммоль) и триэтиламин (0,118 мл, 0,848 ммоль) растворяли в тетрагидрофуране (10 мл) при комнатной температуре и к раствору добавляли трифторуксусный ангидрид (0,180 мл, 1,271 ммоль). Смесь перемешивали при 80°C в течение 1 часа и затем охлаждали до комнатной температуры для остановки реакции. Реакционную смесь фильтровали для удаления твердых частиц и фильтрат концентрировали при пониженном давлении для удаления растворителя. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,160 г, 66,5%) в виде бесцветного масла.
[Стадия 5] Синтез соединения 11602

N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(2-(2,2,2-трифторацетил)гидразин-1-карбонил)бензил)тетрагидро-2H-тиопиран-4-карбоксамид 1,1-диоксид (0,160 г, 0,282 ммоль), синтезированный на стадии 4, и триэтиламин (0,079 мл, 0,563 ммоль) растворяли в дихлорметане (15 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,033 мл, 0,423 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 19,4%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,89 (дд, 1H, J=8,0, 1,7 Гц), 7,76 (дд, 1H, J=9,8, 1,7 Гц), 7,54 (т, 1H, J=7,6 Гц), 7,21-7,12 (м, 2H), 6,91 (ддд, 1H, J=8,7, 4,1, 2,7 Гц), 4,98 (с, 2H), 3,38-3,25 (м, 1H), 2,79 (ддд, 2H, J=13,9, 9,9, 3,7 Гц), 2,50 (тт, 1H, J=7,8, 3,6 Гц), 2,43-2,29 (м, 2H), 2,18-2,06 (м, 3H); LRMS (ES) 550,4 m/z (M++1).
Пример 92: Синтез соединения 11603, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида
[Стадия 1] Синтез N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)тетрагидро-2H-тиопиран-4-карбоксамида 1,1-диоксида
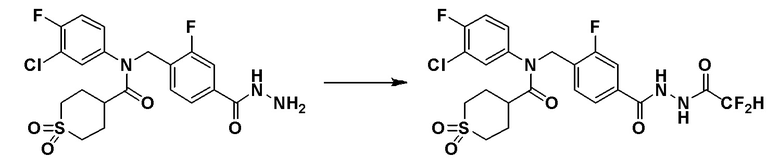
N-(3-хлор-4-фторфенил)-N-(2-фтор-4-(гидразинкарбонил)бензил)тетрагидро-2H-тиопиран-4-карбоксамид 1,1-диоксид (0,200 г, 0,424 ммоль), синтезированный на стадии 3 Примера 91, и триэтиламин (0,118 мл, 0,848 ммоль) растворяли в тетрагидрофуране (10 мл) при комнатной температуре и к раствору добавляли 2,2-дифторуксусный ангидрид (0,158 мл, 1,271 ммоль). Смесь перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры для остановки реакции. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,170 г, 72,9%) в виде бесцветного масла.
[Стадия 2] Синтез соединения 11603
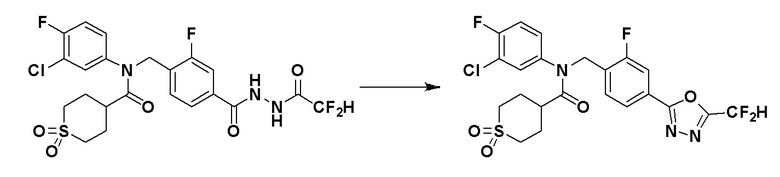
N-(3-хлор-4-фторфенил)-N-(4-(2-(2,2-дифторацетил)гидразин-1-карбонил)-2-фторбензил)тетрагидро-2H-тиопиран-4-карбоксамид 1,1-диоксид (0,170 г, 0,309 ммоль), синтезированный на стадии 1, и триэтиламин (0,086 мл, 0,618 ммоль) растворяли в дихлорметане (15 мл) при комнатной температуре и к раствору добавляли метансульфонилхлорид (0,036 мл, 0,464 ммоль). Смесь перемешивали при этой же температуре в течение 2 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 80%) и концентрировали с получением указанного в заголовке соединения (0,015 г, 9,1%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,88 (дд, 1H, J=8,0, 1,7 Гц), 7,75 (дд, 1H, J=9,9, 1,7 Гц), 7,52 (т, 1H, J=7,6 Гц), 7,21-7,10 (м, 2H), 7,06-6,76 (м, 2H), 4,97 (с, 2H), 3,37-3,26 (м, 2H), 2,79 (ддд, 2H, J=13,9, 9,8, 3,6 Гц), 2,50 (тт, 1H, J=7,9, 3,6 Гц), 2,42-2,28 (м, 2H), 2,19-2,05 (м, 2H); LRMS (ES) m/z 532,3 (M++1).
Пример 93: Синтез соединения 11610, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(1,1-диоксидотиоморфолино)ацетамида
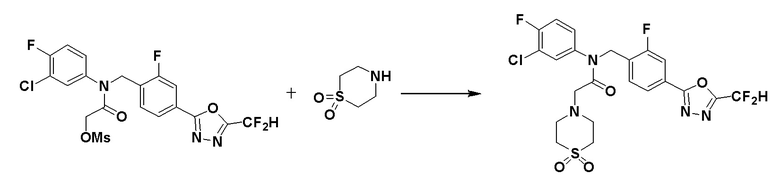
2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, тиоморфолин 1,1-диоксид (0,020 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 5%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 55,7%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (тд, 1H, J=7,7, 1,7 Гц), 7,74 (тд, 1H, J=9,8, 1,7 Гц), 7,56 (т, 1H, J=7,5 Гц), 7,19 (дд, 1H, J=6,4, 2,6 Гц), 7,15 (т, 1H, J=8,5 Гц), 7,09-6,99 (м, 1H), 6,97-6,77 (м, 1H), 4,99 (д, 2H, J=5,7 Гц), 3,15-3,06 (м, 10H); LRMS (ES) m/z 547,4 (M++1).
Пример 94: Синтез соединения 11611, 2-(4-ацетилпиперазин-1-ил)-N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)ацетамида
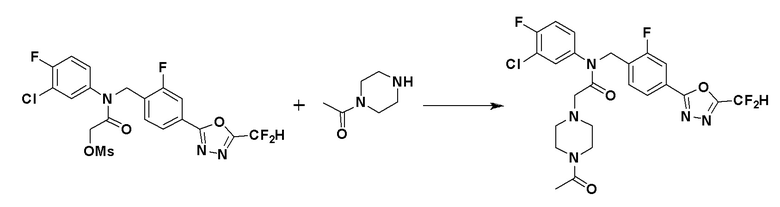
2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, 1-(пиперазин-1-ил)этан (0,019 г, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,046 г, 86,5%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,85 (ддд, 1H, J=8,0, 6,3, 1,7 Гц), 7,72 (тд, 1H, J=9,7, 1,7 Гц), 7,63-7,54 (м, 1H), 7,22 (дд, 1H, J=6,4, 2,6 Гц), 7,12 (т, 1H, J=8,5 Гц), 7,07-7,00 (м, 1H), 6,99-6,75 (м, 1H), 4,98 (д, 2H, J=4,5 Гц), 3,62 (дт, 2H, J=10,1, 4,9 Гц), 3,48 (дт, 2H, J=9,9, 4,9 Гц), 3,00 (д, 2H, J=8,3 Гц), 2,64-2,38 (м, 4H), 2,04 (д, 3H, J=1,1 Гц); LRMS (ES) m/z 540,4 (M++1).
Пример 95: Синтез соединения 11612, N-(3-хлор-4-фторфенил)-2-(4-(циклопропанкарбонил)пиперазин-1-ил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)ацетамида

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,050 г, 0,098 ммоль), синтезированный на стадии 6 Примера 85, циклопропил(пиперазин-1-ил)метанон (0,021 мл, 0,148 ммоль) и N,N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяли в ацетонитриле (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,045 г, 80,8%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (тд, 1H, J=7,1, 6,2, 1,7 Гц), 7,73 (т, 1H, J=9,6 Гц), 7,60 (т, 1H, J=7,6 Гц), 7,25-7,20 (м, 1H), 7,12 (т, 1H, J=8,5 Гц), 7,04 (д, 1H, J=6,4 Гц), 7,01-6,74 (м, 1H), 4,99 (д, 2H, J=4,6 Гц), 3,68 (с, 4H), 3,03 (д, 2H, J=12,1 Гц), 2,54 (д, 4H, J=35,4 Гц), 1,68 (тт, 1H, J=8,1, 4,6 Гц), 0,95 (дт, 2H, J=6,5, 3,4 Гц), 0,74 (дкв., 2H, J=7,2, 3,8 Гц); LRMS (ES) m/z 566,4 (M++1).
Пример 96: Синтез соединения 11613, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(4-(метилсульфонил)пиперазин-1-ил)ацетамида
[Стадия 1] Синтез N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(пиперазин-1-ил)ацетамида

2-((3-хлор-4-фторфенил)(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)амино)-2-оксоэтилметансульфонат (0,200 г, 0,394 ммоль), синтезированный на стадии 6 Примера 85, пиперазин (0,051 г, 0,591 ммоль) и N,N-диизопропилэтиламин (0,137 мл, 0,788 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан = от 0% до 15%) и концентрировали с получением указанного в заголовке соединения (0,158 г, 80,6%) в виде белого твердого вещества.
[Стадия 2] Синтез соединения 11613

N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(пиперазин-1-ил)ацетамид (0,050 г, 0,100 ммоль), синтезированный на стадии 1, метансульфонилхлорид (0,012 мл, 0,151 ммоль) и N,N-диизопропилэтиламин (0,035 мл, 0,201 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли 1,0N водный раствор хлористоводородной кислоты с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 5%) и концентрировали с получением указанного в заголовке соединения (0,054 г, 93,4%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,85 (ддд, 1H, J=8,2, 6,4, 1,7 Гц), 7,71 (тд, 1H, J=9,8, 1,7 Гц), 7,57 (тд, 1H, J=7,7, 7,2, 1,7 Гц), 7,20 (дд, 1H, J=6,4, 2,6 Гц), 7,12 (т, 1H, J=8,5 Гц), 7,03 (тд, 1H, J=5,3, 4,7, 1,7 Гц), 6,84 (дд, 1H, J=51,7, 1,4 Гц), 4,98 (д, 2H, J=4,6 Гц), 3,28-3,19 (м, 4H), 2,98 (с, 2H), 2,75 (д, 3H, J=1,4 Гц), 2,59 (кв., 4H, J=5,7, 4,9 Гц); LRMS (ES) m/z 576,4 (M++1).
Пример 97: Синтез соединения 11614, N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(4-(оксетан-3-ил)пиперазин-1-ил)ацетамида

N-(3-хлор-4-фторфенил)-N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-2-(пиперазин-1-ил)ацетамид (0,050 г, 0,100 ммоль), синтезированный на стадии 1 Примера 96, триацетоксиборогидрид натрия (0,043 г, 0,201 ммоль) и оксетан-3-он (0,011 г, 0,151 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли 1,0N водный раствор хлористоводородной кислоты с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 5%) и концентрировали с получением указанного в заголовке соединения (0,046 г, 82,7%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 7,85 (ддд, 1H, J=7,9, 6,2, 1,7 Гц), 7,71 (тд, 1H, J=9,9, 1,7 Гц), 7,59 (тд, 1H, J=7,6, 5,4 Гц), 7,22 (дд, 1H, J=6,5, 2,6 Гц), 7,10 (т, 1H, J=8,5 Гц), 7,03 (с, 1H), 7,03-6,75 (м, 1H), 4,98 (д, 2H, J=4,3 Гц), 4,67-4,52 (м, 4H), 3,57-3,44 (м, 1H), 2,94 (с, 2H), 2,44 (д, 8H, J=60,9 Гц); LRMS (ES) m/z 554,5 (M++1).
Пример 98: Синтез соединения 11621, N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(оксетан-3-ил)-N-фенилпиперидин-4-карбоксамида
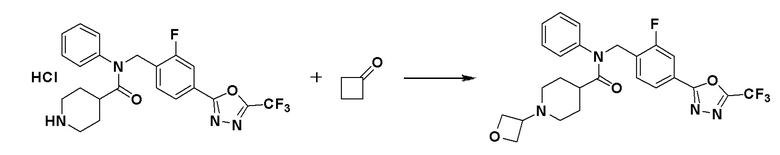
N-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,150 г, 0,309 ммоль), синтезированный на стадии 5 Примера 31, триацетоксиборогидрид натрия (0,131 г, 0,619 ммоль) и циклобутанон (0,026 г, 0,371 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(оксетан-3-ил)-N-фенилпиперидин-4-карбоксамида (0,130 г, 83,3%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ=7,85 (дд, 1H, J=8,0, 1,7 Гц), 7,71 (дд, 1H, J=9,7, 1,7 Гц), 7,56 (т, 1H, J=7,6 Гц), 7,41-7,31 (м, 3H), 7,06-6,99 (м, 2H), 5,02 (с, 2H), 4,66 (с, 2H), 4,59 (т, 2H, J=6,6 Гц), 3,46 (с, 1H), 2,79 (с, 2H), 2,28 (с, 1H), 1,91 (с, 1H), 1,73 (с, 5H); LRMS (ES) m/z 505,2 (M++1).
Пример 99: Синтез соединения 11622, N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(оксетан-3-ил)-N-фенилпиперидин-4-карбоксамида
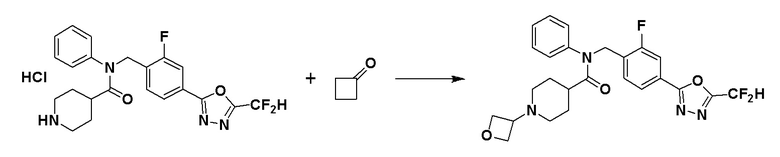
N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-N-фенилпиперидин-4-карбоксамид гидрохлорид (0,150 г, 0,321 ммоль), синтезированный на стадии 3 Примера 39, триацетоксиборогидрид натрия (0,136 г, 0,643 ммоль) и циклобутанон (0,027 г, 0,386 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре и раствор перемешивали при этой же температуре в течение 18 часов. К реакционной смеси добавляли воду с последующим экстрагированием этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили при помощи безводного сульфата магния, фильтровали и затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан = от 0% до 15%) и концентрировали с получением N-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(оксетан-3-ил)-N-фенилпиперидин-4-карбоксамида (0,130 г, 83,2%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,85 (дд, 1H, J=8,0, 1,7 Гц), 7,70 (дд, 1H, J=9,8, 1,7 Гц), 7,54 (т, 1H, J=7,6 Гц), 7,35 (дд, 3H, J=4,9, 2,0 Гц), 7,06-6,99 (м, 2H), 6,84 (д, 1H, J=51,7 Гц), 5,02 (с, 2H), 4,65 (с, 2H), 4,59 (т, 2H, J=6,5 Гц), 3,45 (с, 1H), 2,78 (с, 2H), 2,28 (с, 1H), 2,03-1,85 (м, 2H), 1,73 (с, 4H); LRMS (ES) m/z 487,2 (M++1).
Измерение активности соединений по настоящему изобретению и протокол анализа
Экспериментальный пример 1: анализы ингибирования ферментативной активности HDAC (in vitro)
Для исследования селективности соединений формулы I по настоящему изобретению в отношении HDAC6 с использованием анализов ингибирования ферментативной активности HDAC1 и HDAC6 осуществляли эксперимент с использованием обычно используемого вещества в качестве контроля.
Ферментативную активность HDAC измеряли с использованием набора HDAC Fluorimetric Drug Discovery Kit (BML-AK511, 516) от Enzo Life Science. Для испытания ферментативной активности HDAC1 человеческий рекомбинантный HDAC1 (BML-SE456) использовали в качестве источника фермента и Fluor de Lys®-ʺSIRT1 (BNL-KI177) использовали в качестве субстрата. 5-кратное разведение соединения использовали для посева в 96-луночный планшет и затем 0,3 мкг фермента и 10 мкМ субстрата добавляли в каждую лунку планшета и давали прореагировать при 30°C в течение 60 минут. Затем добавляли Fluor de Lys®-Developer II (BML-KI176) и давали прореагировать в течение 30 минут, после этого измеряли флуоресценцию (возб. 360, эм. 460) с использованием планшет-ридера (Flexstation 3, Molecular Device). HDAC6 фермент испытывали с использованием человеческого рекомбинантного HDAC6 (382180) от Calbiochem в соответствии с тем же протоколом, который использовали для испытания ферментативной активности HDAC1. На основании полученных значений, каждое IC50 значение рассчитывали с использованием программы GraphPad Prism4.0.
Результаты анализов ингибирования ферментативной активности HDAC
Как можно видеть в Таблице 2 выше, 1,3,4-оксадиазоламидные производные соединения, их стереоизомеры или их фармацевтически приемлемые соли в соответствии с настоящим изобретением показали от примерно в 36 до примерно в 3846 раз бóльшую селективную ингибиторную активность в отношении HDAC6 в анализах ингибирования активности HDAC1 и HDAC6.
Экспериментальный Пример 2: Анализ эффекта HDAC6-специфических ингибиторов на аксональный транспорт митохондрий (in vitro)
Анализировали эффект HDAC6-специфических ингибиторов на аксональный транспорт митохондрий. В частности, для исследования, действительно ли соединения, представленные формулой I в соответствии с настоящим изобретением, селективно ингибируют активность HDAC6 для повышения ацетилирования тубулина, который является основным субстратом HDAC6, улучшая таким образом скорость аксонального транспорта митохондрий, которая уменьшается при лечении амилоидом-бета в нейрональных аксонах, осуществляли сравнительный экспермент с использованием соединения, которое уже было разработано, в качестве контроля.
Нейроны гиппокампа от эмбрионов крыс Sprague-Dawley (SD) на 17-18 день развития эмбриона (E17-18) культивировали в покрытой внеклеточным матриксом чаше для визуализации в течение 7 дней и затем обрабатывали при помощи 1 мкМ амилоид-бета пептидов. Через 24 часа нейроны обрабатывали соединениями на 8-й день in vitro и спустя 3 часа, обрабатывали при помощи MitoTracker Red CMXRos (Life Technologies, NY, USA) в течение последних 5 минут для окрашивания митохондрий. Аксональный транспорт окрашенных митохондрий наблюдали визуально с использованием конфокального микроскопа (Leica SP8; Leica Microsystems, UK) с 1-секундными интервалами в течение 1 минуты и скорость транспорта в секунду каждой митохондрии определяли с использованием программы анализа IMARIS (BITPLANE, Zurich, Switzerland).
В результате, было обнаружено, что 1,3,4-оксадиазоламидные производные соединения, их стереоизомеры или фармацевтически приемлемые соли в соответствии с настоящим изобретением улучшали скорость митохондриального аксонального транспорта.
Изобретение относится к 1,3,4-оксадиазоламидному производному соединению, представленному следующей формулой I, его стереоизомеру или его фармацевтически приемлемой соли, где L1, L2 или L3 каждый независимо представляют собой связь или -(C1-C2 алкилен)-; Z1 - Z4 каждый независимо представляют собой N или CRZ, где три или четыре из Z1 - Z4 каждый независимо представляет собой CRZ, и RZ представляет собой -H или -X; R1 представляет собой -CX2H или -CX3; R2 представляет собой -(C1-C4 алкил), -(C3-C6 циклоалкил), -фенил, пиридинил, 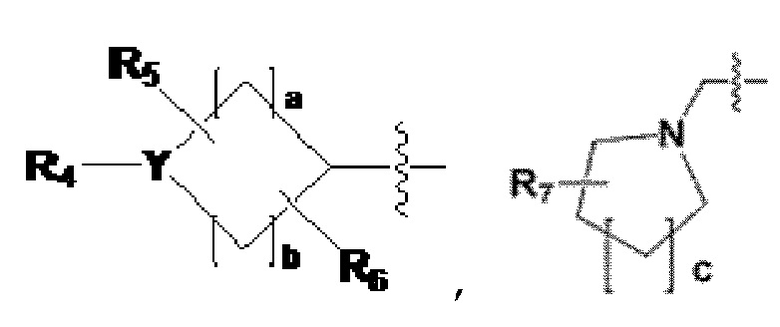 или
или 
Y представляет собой -N-, -O- или -S(=O)2-, когда Y представляет собой -N-, R4 и R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-(C3-C6 циклоалкил), -C(=O)-О(C1-C4 алкил), -C(=O)-CF3, -S(=O)2-(C1-C4 алкил), -оксетанил или бензил, и, когда Y представляет собой -O- или -S(=O)2-, R4 и R8 отсутствуют; R5 - R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -OH, -CH2OH или -C(=O)-NH2, и a - c каждый независимо представляют собой целое число, имеющее значение 1 или 2; R3 представляет собой фенил, -фенил, который может быть замещен группой -X, и X представляет собой F, Cl, Br или I. Соединения по изобретению предназначены для получения лекарственного средства для лечения гистондеацетилаза 6-опосредованного заболевания. 4 н. и 6 з.п. ф-лы, 2 табл., 101 пр.
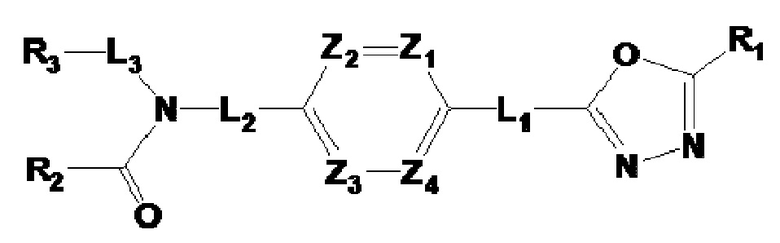 (I)
(I)
1. 1,3,4-Оксадиазоламидное производное соединение, представленное следующей формулой I, его стереоизомер или его фармацевтически приемлемая соль:
[Формула I]

где L1, L2 или L3 каждый независимо представляют собой связь или -(C1-C2 алкилен)-;
Z1 - Z4 каждый независимо представляют собой N или CRZ, где три или четыре из Z1 - Z4 каждый независимо представляет собой CRZ, и RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой -(C1-C4 алкил), -(C3-C6 циклоалкил), -фенил, пиридинил,  или
или 
Y представляет собой -N-, -O- или -S(=O)2-,
когда Y представляет собой -N-, R4 и R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-(C3-C6 циклоалкил), -C(=O)-О(C1-C4 алкил), -C(=O)-CF3, -S(=O)2-(C1-C4 алкил), -оксетанил или бензил,
и, когда Y представляет собой -O- или -S(=O)2-, R4 и R8 отсутствуют,
R5 - R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -OH, -CH2OH или -C(=O)-NH2, и
a - c каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой фенил, -фенил, который может быть замещен группой -X, и
X представляет собой F, Cl, Br или I.
2. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где
L1 и L3 представляют собой связь;
L2 представляет собой -(C1-C2 алкилен)-;
Z1 - Z4 каждый независимо представляют собой N или CRZ, где три или четыре из Z1 - Z4 каждый независимо представляет собой CRZ, и RZ представляет собой -H или -X;
R1 представляет собой -CX2H или -CX3;
R2 представляет собой -(C1-C4 алкил), -(C3-C6 циклоалкил), -фенил, -пиридинил, 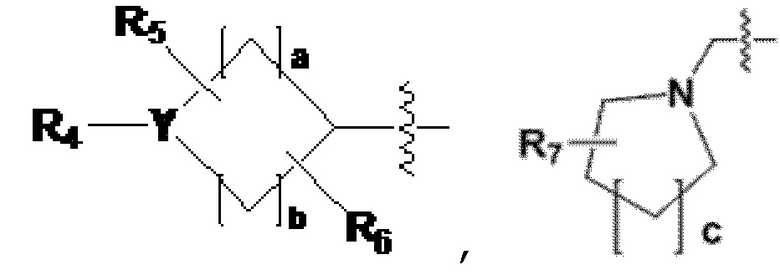 или
или  ,
,
Y представляет собой -N-, -O- или -S(=O)2-,
когда Y представляет собой -N-, R4 и R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -C(=O)-(C1-C4 алкил), -C(=O)-CF3, -S(=O)2-(C1-C4 алкил), -оксетанил, -C(=O)-(C3-C6 циклоалкил) или бензил,
и, когда Y представляет собой -O- или -S(=O)2-, R4 и R8 отсутствуют,
R5 - R8 каждый независимо представляют собой -H, -(C1-C4 алкил), -OH, -CH2OH или -C(=O)-NH2, и
a - c каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой –фенил –, который может быть замещен группой -X; и
X представляет собой F, Cl, Br или I.
3. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 2,
где
L1 и L3 представляют собой связь;
L2 представляет собой -(C1 алкилен)-;
Z1 - Z4 каждый независимо представляют собой N или CRZ, где три или четыре из Z1 - Z4 каждый независимо представляют собой CRZ, и RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой -(C1-C4 алкил), -пиридинил или 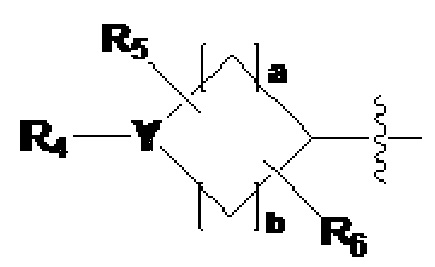 ,
,
Y представляет собой -N-,
R4 представляет собой -(C1-C4 алкил), -C(=O)-(C1-C4 алкил) или -S(=O)2-(C1-C4 алкил),
R5 или R6 каждый независимо представляют собой -H или -(C1-C4 алкил), и
a и b каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой -фенил, который может быть замещен группой -X; и
X представляет собой F, Cl, Br или I.
4. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 3, где
L1 и L3 представляют собой связь;
L2 представляет собой -(C1 алкилен)-;
Z1 - Z4 каждый независимо представляют собой N или CRZ, где три или четыре из Z1 - Z4 каждый независимо представляет собой CRZ, и RZ представляет собой -H или -X;
R1 представляет собой -CF2H или -CF3;
R2 представляет собой -пиридинил или ,
Y представляет собой -N-,
R4 представляет собой -(C1-C4 алкил), -C(=O)-(C1-C4 алкил) или -S(=O)2-(C1-C4 алкил),
R5 или R6 каждый независимо представляют собой -H, и
a и b каждый независимо представляют собой целое число, имеющее значение 1 или 2;
R3 представляет собой -фенил, который может быть замещен группой -X; и
X представляет собой F, Cl, Br или I.
5. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:



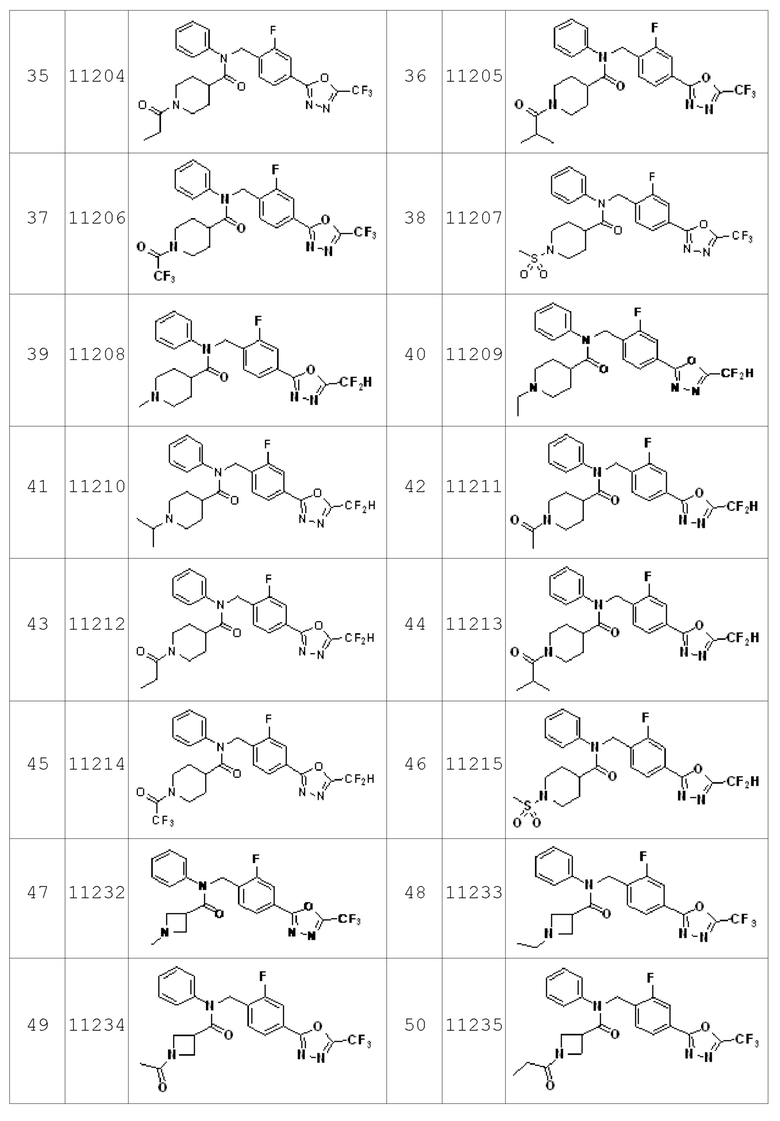
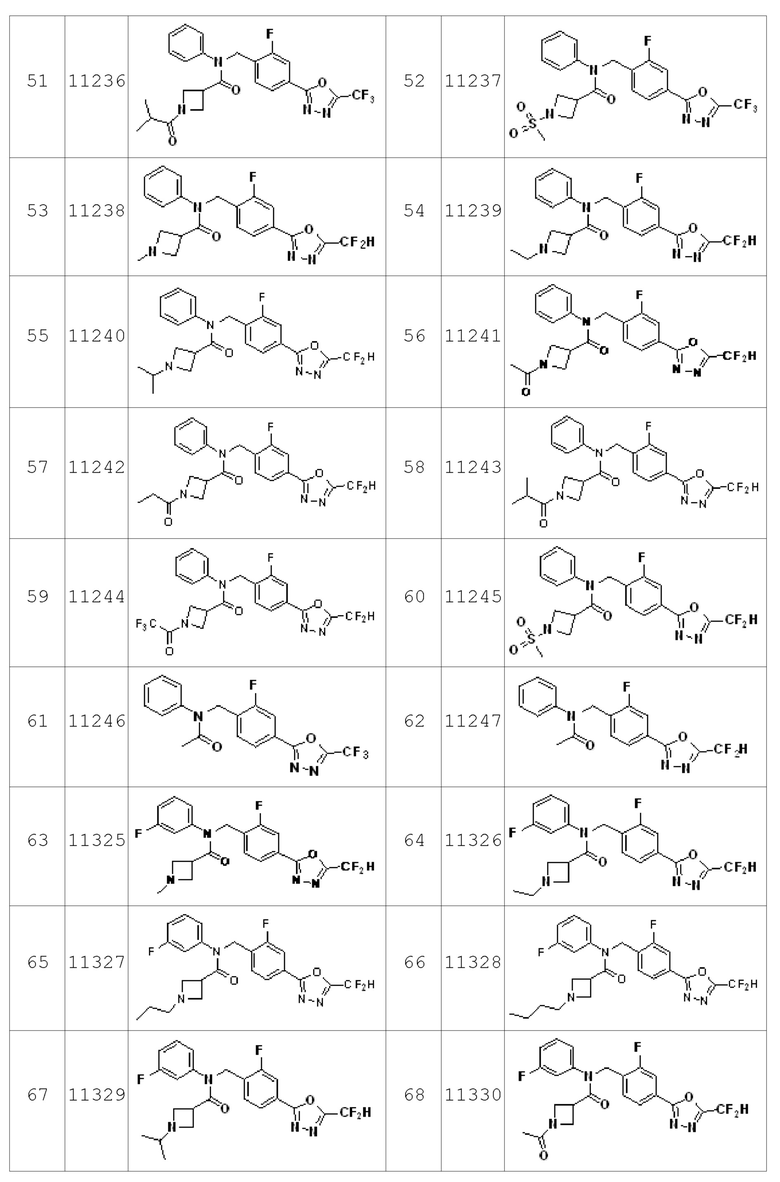


6. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 5, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:

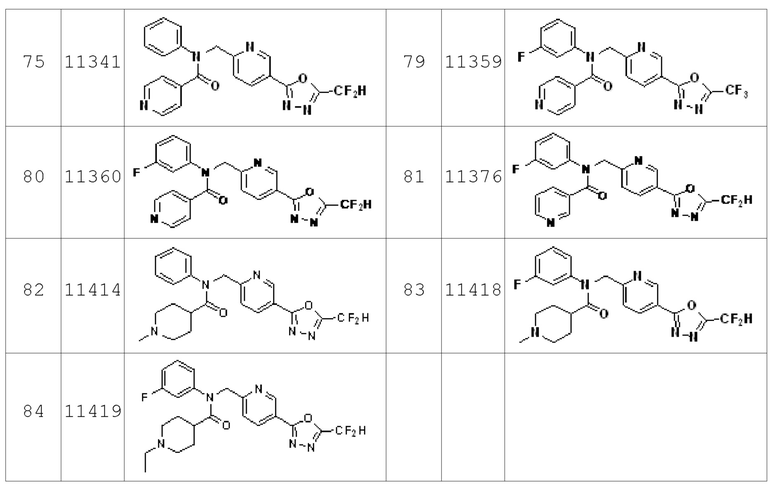
7. 1,3,4-Оксадиазоламидное производное соединение, представленное формулой I, его стереоизомер или его фармацевтически приемлемая соль по п. 6, где соединение, представленное формулой I, выбрано из группы, состоящей из соединений, описанных в следующей таблице:


8. Фармацевтическая композиция для профилактики или лечения гистондеацетилаза 6-опосредованного заболевания, содержащая, в качестве активного ингредиента, эффективное количество соединения, представленного формулой I, его стереоизомер или его фармацевтически приемлемую соль по любому из пп. 1-7 и фармацевтически приемлемый носитель.
9. Способ лечения гистондеацетилаза 6-опосредованного заболевания, включающий введение терапевтически эффективного количества соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-7.
10. Применение соединения, представленного формулой I, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-7 для получения лекарственного средства для лечения гистондеацетилаза 6-опосредованного заболевания.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| EA 201491060 A, 30.09.2014 | |||
| Устройство для обрушивания семян | 1980 |
|
SU904653A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

