ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому способу получения 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, формы свободного основания определенного лекарственного средства (в форме гидрохлорида), которое способно селективно и эффективно ингибировать устойчивость к лекарственному средству, вызванную ростом раковых клеток и мутациями тирозинкиназы. В соответствии со способом согласно изобретению целевое соединение может быть получено более простым способом по сравнению с обычными способами.
УРОВЕНЬ ТЕХНИКИ
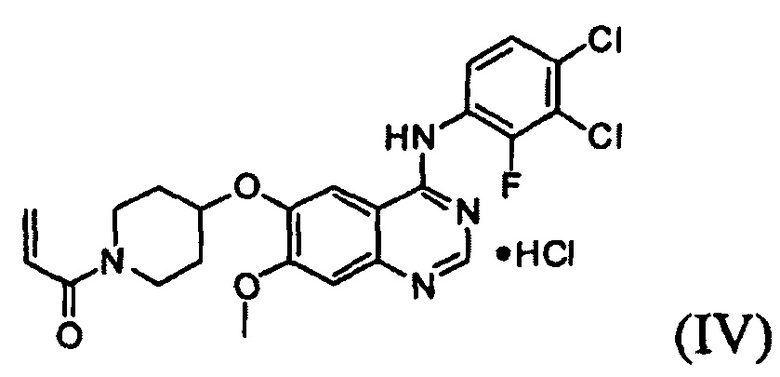
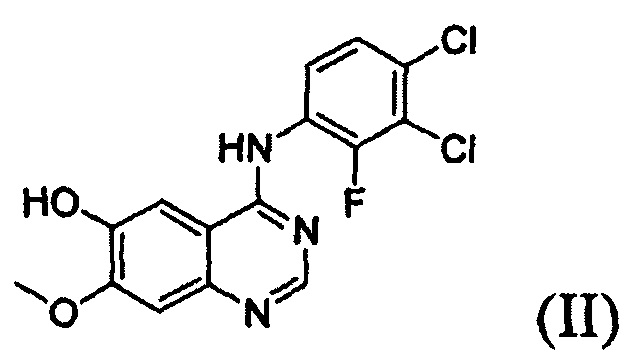
Известно, что 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она гидрохлорид, представленный формулой (IV) ниже, проявляет антипролиферативные свойства, такие как противораковую активность, и считается важным лекарственным средством, которое способно селективно и эффективно ингибировать устойчивость к лекарственному средству, вызванную ростом раковых клеток и мутациями тирозинкиназы. Форма свободного основания соединения формулы (IV), т.е. 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, представленная формулой (I) ниже, также известна под регистрационным номером CAS 1092364-38-9.
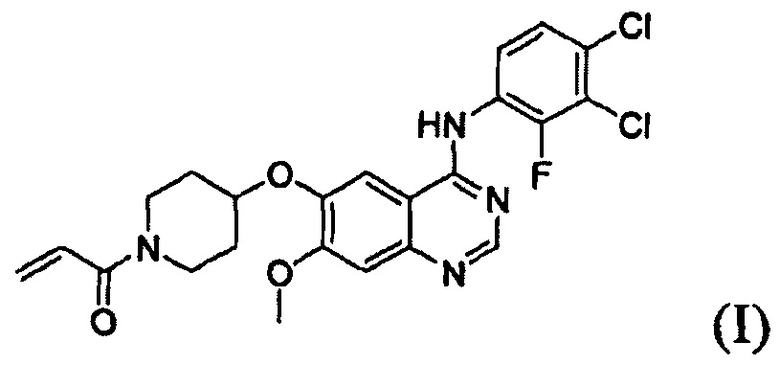
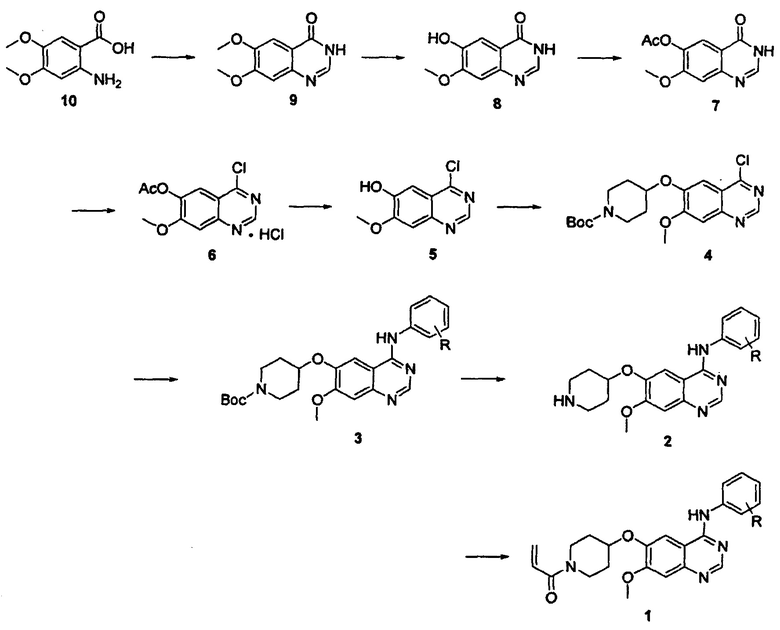
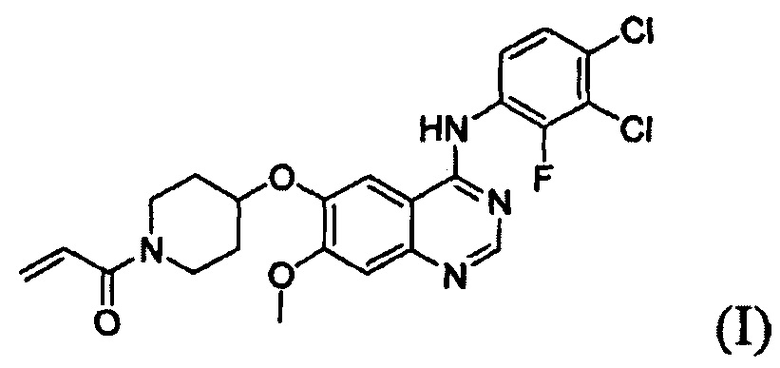
Соединение вышеуказанной формулы (I) может быть получено способом, раскрытым в патенте Республики Корея №1013319, и подробное протекание реакции описано ниже на схеме реакции 1. Соединение формулы (I), полученное в соответствии со схемой реакции 1, можно подвергнуть взаимодействию с соляной кислотой с получением его гидрохлоридной соли, т.е. соединения формулы (IV):
Схема реакции 1
где R представляет собой галоген.
Согласно способу получения, описанному на вышеуказанной схеме реакции 1, соединение 10 подвергают реакции конденсации с гидрохлоридом формамидина при высокой температуре, например, 210°С, с получением соединения 9, которое затем подвергают взаимодействию с L-метионином в органической кислоте, такой как метансульфоновая кислота, в результате чего удаляется метальная группа соединения 9 в положении С-6 с получением соединения 8.
Затем соединение 8 подвергают реакции введения защитной группы в безводной уксусной кислоте и основании, таком как пиридин, с получением соединения 7, которое затем подвергают взаимодействию с неорганическими кислотами, такими как тионилхлорид, оксихлорид фосфора и им подобные, в присутствии каталитического количества N,N-диметилформамида при кипячении с обратным холодильником с получением соединения 6 в форме гидрохлорида.
Полученное таким образом соединение 6 подвергают реакции снятия защиты путем перемешивания в спиртовом растворе, содержащем аммиак (например, 7 н. раствор аммиака в метаноле) с получением соединения 5. Соединение 5 подвергают реакции Мицунобу с соединением трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты с получением соединения 4, которое затем подвергают реакции замещения с анилином в органическом растворителе, таком как 2-пропанол или ацетонитрил, с получением соединения 3. Соединение 3 подвергают взаимодействию с органической кислотой, такой как трифторуксусная кислота, или неорганической кислотой, такой как концентрированная соляная кислота, в органическом растворителе, таком как дихлорметан, в ходе чего происходит снятие трет-бутоксикарбонильной защитной группы с получением соединения 2. В описанной выше реакции Мицунобу могут применяться диизопропиловый эфир азодикарбоновой кислоты, диэтиловый эфир азодикарбоновой кислоты или ди-трет-бутиловый эфир азодикарбоновой кислоты, а также трифенилфосфин.
Соединение 1, т.е. соединение формулы (I) настоящего изобретения, получают, подвергая полученное таким образом соединение 2 реакции ацилирования с акрилоилхлоридом в смеси воды и органического растворителя, такого как тетрагидрофуран и ему подобного, или в дихлорметане в присутствии неорганического основания, такого как бикарбонат натрия, или органического основания, такого как пиридин или триэтиламин. В качестве альтернативы, соединение 2 подвергают реакции конденсации с акриловой кислотой путем применения связывающего агента, например 1-этил-3-(3-диметиламинопропил)-карбодиимида (EDC) или 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата метанаминия (HATU).
Тем не менее, в соответствии с вышеописанным способом, этап получения соединения 9 может быть опасным, так как этот этап проводят при высокой температуре без растворителя, и реакция может протекать неоднородно. Также избыточное количество тионилхлорида, используемое на этапе получения соединения 5, приводит к сложностям на последующих этапах. Поэтому этот способ не подходит для коммерческого применения.
Самый существенный недостаток этого способа получения соединения 1 заключается в очень низком выходе реакции акрилирования, например, 13%, и также в том, что реакция сопровождается множеством побочных реакций, и, таким образом, в указанном способе требуется проведение очистки путем колоночной хроматографии. Также, в том случае, когда соединение 3 получают путем реакции Мицунобу, могут образовываться различные побочные продукты, что требует проведения этапа очистки с использованием колоночной хроматографии, для чего необходимо применение дорогого силикагеля и избыточного количества растворителей подвижной фазы. Таким образом, этот способ не является целесообразным для коммерческого применения.
Соответственно, авторы настоящего изобретения попытались разработать новый способ получения соединения формулы (I) с высокой чистотой и высоким выходом, который является экономичным, а также подходит для коммерческого применения.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Таким образом, задача настоящего изобретения заключается в обеспечении нового и простого способа получения 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она.
Согласно одному из аспектов настоящего изобретения предложен способ получения соединения формулы (I), который включает этап осуществления взаимодействия соединения формулы (II) с соединением формулы (III) в инертном полярном апротонном растворителе в присутствии основания:
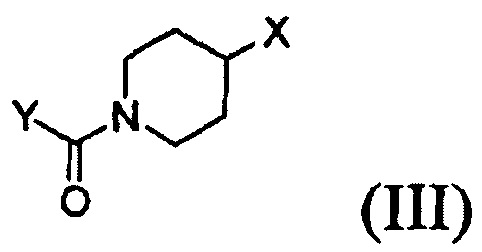
где X представляет собой тозилокси (ОТ), мезилокси (ОМ), трифторметансульфонат, фторсульфонат или галоген; и Y представляет собой этенил или галогенэтил.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии со способом согласно настоящему изобретению, соединение формулы (I), т.е. 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-он, может быть получено путем осуществления взаимодействия соединения формулы (II), т.е. 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола, с соединением формулы (III) в инертном полярном апротонном растворителе в присутствии основания. Механизм этого процесса описан на схеме реакции 2 ниже:
Схема реакции 2
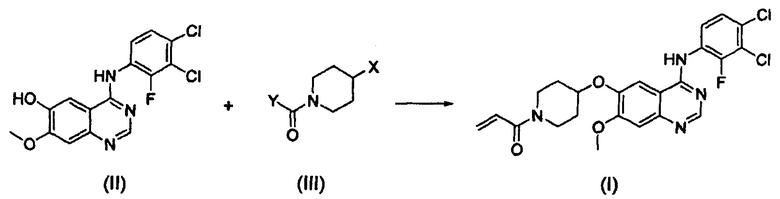
где X и Y являются такими, как определено выше.
Конкретные примеры инертных полярных апротонных растворителей, применяемых в реакции, описанной выше, включают N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он, диметилсульфоксид и их смесь.
Конкретные примеры оснований, применяемых в реакции, описанной выше, включают карбонаты щелочных металлов, такие как бикарбонат натрия, карбонат калия, карбонат цезия и их смесь. Предпочтительно основание используют в количестве от 1 до 5 моль эквивалентов в расчете на 1 моль эквивалент соединения формулы (II).
Вышеописанную реакцию можно проводить при температуре от 60°С до 100°С, предпочтительно от 70°С до 90°С, более предпочтительно от 70°С до 80°С.
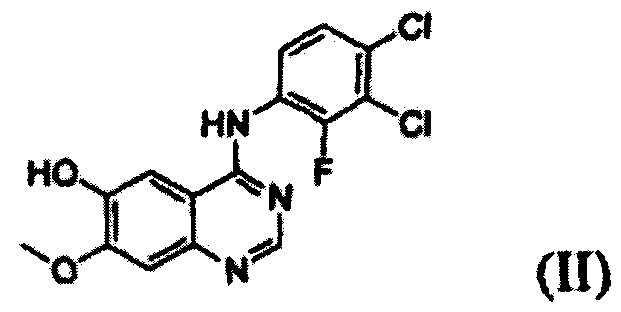
Соединение формулы (II), которое используют как исходный материал в настоящем изобретении, может быть получено с помощью следующих этапов (см. схему реакции 3 ниже):
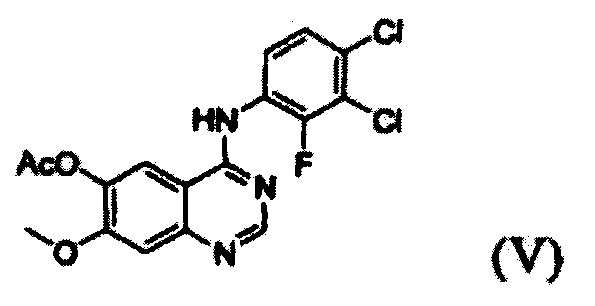
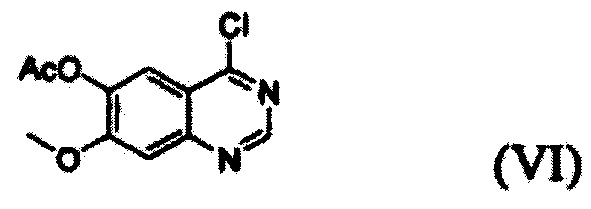
(i) осуществление взаимодействия соединения формулы (VII) с галогенирующим агентом в присутствии органического основания с получением соединения формулы (VI), которое затем подвергают взаимодействию с соединением формулы (VIII) с получением соединения формулы (V), т.е. 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илового эфира уксусной кислоты; и
(ii) осуществление взаимодействия соединения формулы (V) с раствором аммиака в полярном протонном растворителе.
Схема реакции 3
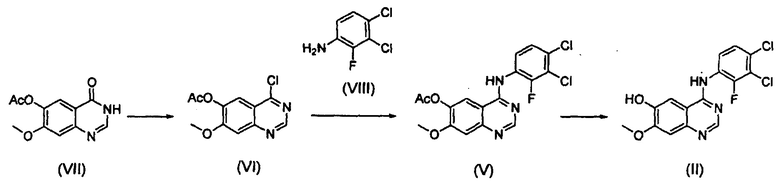
Конкретные примеры органического основания, применяемого на вышеописанном этапе (i), включают диизопропиламин, триэтиламин, диизопропилэтиламин, диэтиламин, пиридин, 4-диметилпиридин, морфолин и их смесь. Конкретные примеры галогенирующего агента включают тионилхлорид, оксихлорид фосфора и их смесь. Вышеописанную реакцию можно проводить при температуре от 50°С до 150°С, предпочтительно от 60°С до 90°С, более предпочтительно при примерно 75°С. На этом этапе соединение формулы (VI) получают в форме раствора, содержащего его в органическом растворителе, а не в изолированной форме. Затем соединение формулы (VI), содержащееся в органическом растворителе, подвергают взаимодействию с соединением формулы (VIII) с получением соединения формулы (V), т.е. 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илового эфира уксусной кислоты.
Соединение формулы (VII), которое применяют как исходный материал в вышеописанной реакции, может быть получено в соответствии со способом, раскрытым в патенте Республики Корея №1013319.
На следующем этапе (ii) соединение формулы (V), полученное на предыдущем этапе (i), подвергают взаимодействию с раствором аммиака или газообразным аммиаком в полярном протонном растворителе (например, метаноле, этаноле, пропаноле и их смеси) при температуре от 0°С до 40°С, предпочтительно от 10°С до 30°С, более предпочтительно при примерно 25°С с получением 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола формулы (II).
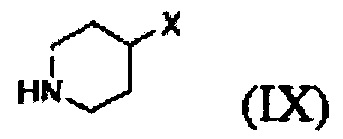
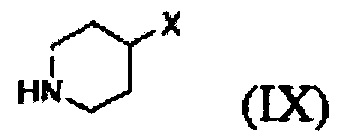
Кроме того, соединение формулы (III), которое используют как исходный материал в настоящем изобретении, может быть получено путем осуществления взаимодействия соединения формулы (IX) или его соли с соединением формулы (X) в присутствии основания или амидного связывающего агента (см. схему реакции 4 ниже).
Схема реакции 4

где X и Y являются такими, как определено выше; и Z представляет собой галоген или гидроксил.
Вышеописанную реакцию можно проводить в органическом растворителе, таком как тетрагидрофуран, этилацетат, ацетон, 1,4-диоксан, ацетонитрил, дихлорметан, четыреххлористый углерод, хлороформ, N,N-диметилформамид или диметилсульфоксид, или в смеси органического растворителя и воды.
Конкретные примеры основания включают неорганическое основание, такое как карбонат натрия, бикарбонат натрия, карбонат кальция, карбонат калия, гидроксид натрия, гидроксид калия и карбонат цезия, органическое основание, такое как диизопропиламин, триэтиламин, диизопропилэтиламин и диэтиламин и их смесь. Конкретные примеры амидных связывающих агентов включают 1-этил-3-(3-диметиламинопропил)карбодиимид, гидроксибензотриазол, O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, N,N'-дициклогексилкарбоимид, 1-гидрокси-7-азабензотриазол, N,N'-диизопропилкарбоимид, (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат и их смесь. Основание или амидный связывающий агент можно использовать в количестве от 3 до 5 моль эквивалентов в расчете на 1 моль эквивалент соединения формулы (IX) или его соли.
Вышеуказанная соль соединения формулы (IX) представляет собой предпочтительно гидрохлоридную соль (2HCl соль) или гидробромидную соль (2HBr соль). Вышеописанную реакцию можно проводить при температуре от -30°С до 30°С, предпочтительно при температуре от примерно 0°С до комнатной температуры при перемешивании в течение подходящего периода времени.
В соответствии со способом согласно настоящему изобретению, целевое соединение формулы (I), 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-он, может быть получено с высокой чистотой и высоким выходом простым способом.
Более того, 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она гидрохлорид, который способен селективно и эффективно ингибировать устойчивость к лекарственному средству, вызванную ростом раковых клеток и мутациями тирозинкиназы, может быть получено путем осуществления взаимодействия соединения формулы (I) с соляной кислотой в органическом растворителе (например, метаноле, этаноле, пропаноле, изопропаноле, бутаноле, этилацетате, ацетоне, тетрагидрофуране, ацетонитриле, 1,4-диоксане их смеси) при температуре от 0°С до 60°С, предпочтительно от 10°С до 40°С, более предпочтительно при примерно 25°С.
Далее настоящее изобретение будет описано более конкретно следующими примерами, но примеры приведены только в иллюстративных целях и не ограничивают настоящее изобретение.
Пример получения 1: Получение 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола, соединения формулы (II)
Этап (i): Получение 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илового эфира уксусной кислоты, соединения формулы (V)
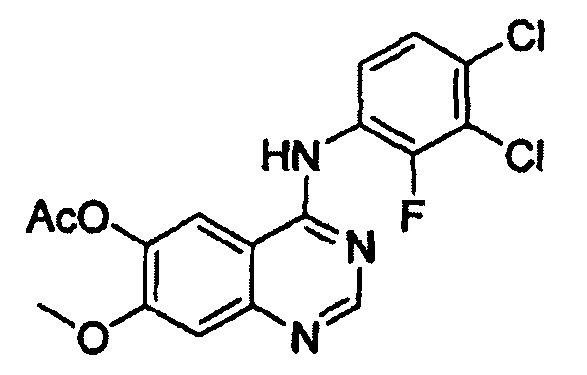
7-метокси-4-оксо-3,4-дигидрохиназолин-6-иловый эфир уксусной кислоты (100 г) добавляли к толуолу (850 мл) и N,N-диизопропилэтиламину (82,5 мл). Оксихлорид фосфора (100 мл) добавляли к реакционной смеси в течение 20 минут при 75°С с последующим перемешиванием в течение 3 часов. Толуол (450 мл) и 3,4-дихлор-2-фторанилин (84,6 г) добавляли к полученной смеси с последующим перемешиванием в течение 2 часов. После завершения реакции полученную смесь охлаждали до 25°С и полученное таким образом твердое вещество отфильтровывали при пониженном давлении и промывали толуолом (400 мл). К твердому веществу добавляли изопропанол (1000 мл) и полученную смесь перемешивали в течение 2 часов. Полученное таким образом твердое вещество отфильтровывали и промывали изопропанолом (400 мл) и затем сушили при 40°С в печи с получением целевого соединения (143 г, выход: 83%).
1Н-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 8,92 (s, 1Н), 8,76 (s, 1Н), 7,69-7,57 (m, 3Н), 4,01 (s, 3Н), 2,38 (s, 3Н).
Этап (ii): Получение 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола. соединения формулы (II)
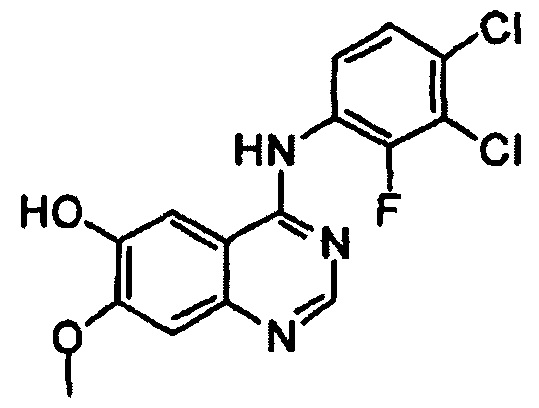
4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-иловый эфир уксусной кислоты (100 г), полученный на этапе (I), смешивали с метанолом (1000 мл). Смесь охлаждали до 10°С-15°С, добавляли раствор аммиака (460 г) и перемешивали в течение 3 часов при 25°С. Полученное таким образом твердое вещество отфильтровывали и промывали смешанным растворителем из метанола (200 мл) и воды (200 мл). Полученное таким образом твердое вещество сушили при 40°С в печи с получением целевого соединения (74 г, выход: 83%).
1Н-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 9,57 (br, 2Н), 8,35 (s, 1Н), 7,68 (s, 3Н), 7,61-7,52 (m, 2Н), 7,21 (s, 1Н), 3,97 (s, 3Н).
Пример 1: Получение 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, соединения формулы (I)
Этап (1-1): Получение 1-акрилоилпиперидин-4-илового эфира 4-метилбензолсульфоновой кислоты, соединения формулы (III)
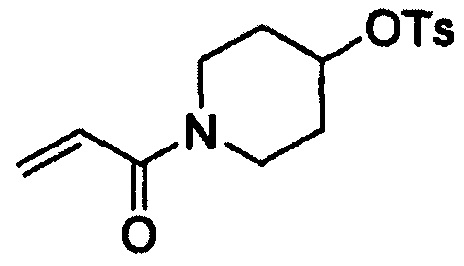
Гидрохлорид пиперидин-4-илового эфира 4-метилбензолсульфоновой кислоты (200 г, 685 ммоль), тетрагидрофуран (ТГФ, 1,6 л) и NaHCO3 (172 г, 2047 ммоль) добавляли в воду (2 л) и смесь охлаждали до 0°С. К реакционной смеси в течение 30 минут добавляли раствор, полученный путем добавления акрилоилхлорида (56 мл, 519 ммоль) к ТГФ (0,4 л), с последующим перемешиванием в течение 1 часа. После завершения реакции к реакционной смеси добавляли МеОН (0,4 л) для гашения реакции. Раствор затем экстрагировали этиловым сложным эфиром (2 л) и промывали водой (2 л). Органический слой отделяли, перегоняли при пониженном давлении и полученный таким образом остаток перекристаллизовывали из смеси дихлорметан-гексан с получением целевого соединения (174 г, выход: 82%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 7,82 (d, 2Н), 7,48 (d, 2Н), 6,80-6,71 (m, 1Н), 6,10-6,13 (m, 1Н), 5,67-5,62 (m, 1Н), 4,76-4,71 (m, 1Н), 3,70-3,68 (m, 2Н), 3,43-3,31 (m, 2Н), 2,42 (s, 3Н), 1,73 (m, 2Н), 1,52 (m, 2Н).
Этап (1-2): Получение 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, соединения формулы (I)
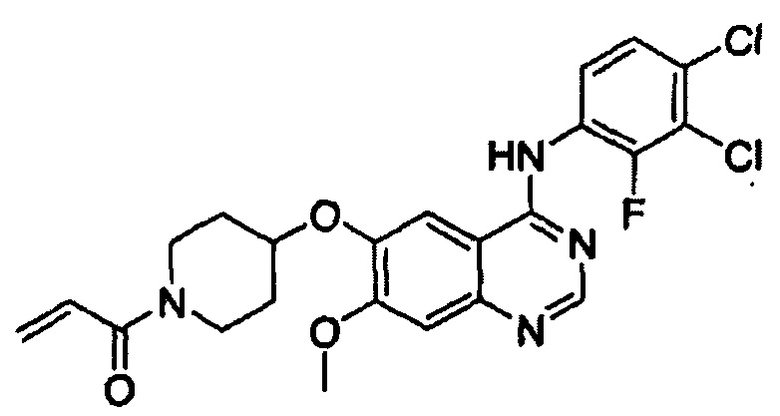
Смешивали 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ол (12 г, 34 ммоль), полученный в примере получения 1, 1-акрилоилпиперидин-4-иловый эфир 4-метилбензолсульфоновой кислоты (16 г, 51 ммоль), полученный на этапе (1-1), K2CO3 (9,4 г, 68 ммоль) и диметилацетамид (ДМАА, 300 мл). Температуру реакции повышали до 70°С и смесь перемешивали в течение 24 часов. После завершения реакции смесь охлаждали до комнатной температуры, экстрагировали этиловым сложным эфиром (300 мл) и затем промывали водой (300 мл). Органический слой отделяли и перегоняли при пониженном давлении. Полученный таким образом остаток отверждали путем добавления этилового сложного эфира, отфильтровывали и сушили с получением целевого соединения (12,8 г, выход: 77%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 9,65 (bs, 1Н), 8,40 (s, 1Н), 7,88 (s, 1Н), 7,64-7,56 (m, 2Н), 7,24 (s, 1H), 6,89-6,80 (m, 1Н), 6,15-6,08 (m, 1Н), 5,70-5,66 (m, 1Н), 4,78 (m, 1Н), 3,94 (s, 3Н), 3,87 (m, 2Н), 3,48 (m, 2Н), 2,03 (m, 2Н), 1,70 (m, 1H).
Пример 2: Получение 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, соединения формулы (I)
Этап (2-1): Получение 1-(3-хлорпропаноил)пиперидин-4-илового эфира 4-метилбензолсульфоновой кислоты, соединения формулы (III)

Смешивали гидрохлорид пиперидин-4-илового эфира 4-метилбензолсульфоновой кислоты (20 г, 68 ммоль) и дихлорметан (200 мл) и смесь охлаждали до 0°С. Триэтиламин (29 мл, 205 ммоль) и 3-хлорпропионилхлорид (7,9 мл, 82 ммоль) добавляли к реакционной смеси с последующим перемешиванием в течение 16 часов при комнатной температуре.
После завершения реакции реакционную смесь экстрагировали этиловым сложным эфиром (200 мл) и затем промывали водой (200 мл). Органический слой отделяли, перегоняли при пониженном давлении и полученный таким образом остаток очищали с получением целевого соединения (18 г, выход: 76%).
1Н-ЯМР (300 МГц CDCl3) δ 7,80 (d, 2Н), 4,76-4,72 (m, 1H), 3,80 (t, 2H), 3,64-3,57 (m, 3Н), 3,40 (m, 1H), 2,77 (t, 2H), 2,46 (s, 3H), 1,85-1,70 (m, 4H).
Этап (2-2): Получение 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, соединения формулы (I)

Повторяли процедуру этапа (1-2) примера 1 за исключением того, что вместо 1-акрилоилпиперидин-4-илового эфира 4-метилбензолсульфоновой кислоты (16 г, 51 ммоль), полученного на этапе (1-1), использовали 1-(3-хлорпропаноил)пиперидин-4-иловый эфир 4-метилбензолсульфоновой кислоты (13 г, 35 ммоль), полученный на вышеописанном этапе (2-1), с получением целевого соединения (7,4 г, выход: 58%).
1Н-ЯМР (300 МГц, ДМСО-d6) δ 9,65 (bs, 1H), 8,40 (s, 1Н), 7,88 (s, 1Н), 7,64-7,56 (m, 2Н), 7,24 (s, 1Н), 6,89-6,80 (m, 1Н), 6,15-6,08 (m, 1Н), 5,70-5,66 (m, 1Н), 4,78 (m, 1Н), 3,94 (s, 3Н), 3,87 (m, 2Н), 3,48 (m, 2Н), 2,03 (m, 2Н), 1,70 (m, 1Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА 1-(3,4-ДИХЛОР-2-ФТОРФЕНИЛАМИНО)-7-МЕТОКСИХИНАЗОЛИН-6-ИЛОКСИ)ПИПЕРИДИН-1-ИЛ)ПРОП-2-ЕН-1-ОНА И ИСПОЛЬЗУЕМЫХ В НЕМ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2012 |
|
RU2563630C1 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(4-БРОМ-2-ФТОРАНИЛИНО)-6-МЕТОКСИ-7(1-МЕТИЛПИПЕРИДИН-4-ИЛМЕТОКСИ)ХИНАЗОЛИНА(ZD6474) И СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2448102C2 |
| 4-ФЕНОКСИ-НИКОТИНАМИДЫ ИЛИ 4-ФЕНОКСИ-ПИРИМИДИН-5-КАРБОКСАМИДЫ | 2011 |
|
RU2565077C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2267489C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| ПРОИЗВОДНЫЕ 2-(4-ГИДРОКСИПИПЕРИДИНО)-1-АЛКАНОЛА И ПРОИЗВОДНЫЕ 2-(4-ГИДРОКСИПИПЕРИДИНО)-1-АЛКАНОНА | 1992 |
|
RU2065859C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-[2-(БЕНЗИМИДАЗОЛ-1-ИЛ)ХИНОЛИН-8-ИЛ]ПИПЕРИДИН-4-ИЛАМИНА | 2004 |
|
RU2323214C2 |
| ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОПУХОЛЕЙ | 2001 |
|
RU2276151C2 |
Настоящее изобретение относится к новому способу получения 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I). Соединение обладает действием селективного ингибитора устойчивости к лекарственному средству, вызванному ростом раковых клеток и мутациями тирозинкиназы. Способ заключается во взаимодействии соединения формулы (II) с соединением формулы (III) в инертном полярном апротонном растворителе в присутствии основания,

 .
.
Исходное соединение формулы (II) получают (i) путем осуществления взаимодействия соединения формулы (VII) с галогенирующим агентом в присутствии органического основания с получением соединения формулы (VI), которое затем подвергают взаимодействию с соединением формулы (VIII) с получением соединения формулы (V); и путем (ii) осуществления взаимодействия соединения формулы (V) с раствором аммиака в полярном протонном растворителе



 Соединение формулы (III) получают взаимодействием соединения формулы (IX) или его соли с соединением формулы (X) в присутствии основания или амидного связывающего агента
Соединение формулы (III) получают взаимодействием соединения формулы (IX) или его соли с соединением формулы (X) в присутствии основания или амидного связывающего агента
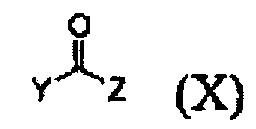 . Способ позволяет получить промежуточные продукты и целевое соединение с высоким выходом и чистотой и является более простым по сравнению с ранее известными способами, поскольку не требует дополнительных стадий для получения двойной связи в боковой цепи соединения. 12 з.п. ф-лы, 2 пр.
. Способ позволяет получить промежуточные продукты и целевое соединение с высоким выходом и чистотой и является более простым по сравнению с ранее известными способами, поскольку не требует дополнительных стадий для получения двойной связи в боковой цепи соединения. 12 з.п. ф-лы, 2 пр.
1. Способ получения соединения формулы (I), включающий осуществление взаимодействия соединения формулы (II) с соединением формулы (III) в инертном полярном апротонном растворителе в присутствии основания
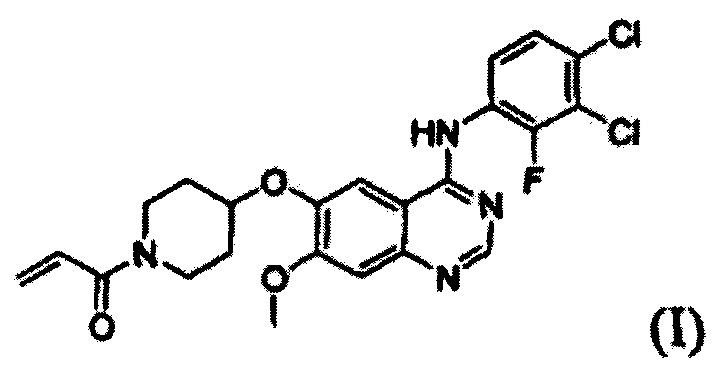


где X представляет собой тозилокси (ОТ), мезилокси (ОМ), трифторметансульфонат, фторсульфонат или галоген; и Y представляет собой этенил или галогенэтил.
2. Способ по п. 1, отличающийся тем, что инертный полярный апротонный растворитель выбран из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, N-метилпирролидин-2-она, диметилсульфоксида и их смеси.
3. Способ по п. 1, отличающийся тем, что основание представляет собой карбонат щелочного металла, выбранный из группы, состоящей из бикарбоната натрия, карбоната калия, карбоната цезия и их смеси.
4. Способ по п. 1, отличающийся тем, что основание применяют в количестве от 1 до 5 моль эквивалентов в расчете на 1 моль эквивалент соединения формулы (II).
5. Способ по п. 1, отличающийся тем, что соединение формулы (II) получают (i) путем осуществления взаимодействия соединения формулы (VII) с галогенирующим агентом в присутствии органического основания с получением соединения формулы (VI), которое затем подвергают взаимодействию с соединением формулы (VIII) с получением соединения формулы (V); и путем (ii) осуществления взаимодействия соединения формулы (V) с раствором аммиака в полярном протонном растворителе


6. Способ по п. 5, отличающийся тем, что органическое основание выбрано из группы, состоящей из диизопропиламина, триэтиламина, диизопропилэтиламина, диэтиламина, пиридина, 4-диметилпиридина, морфолина и их смеси.
7. Способ по п. 5, отличающийся тем, что галогенирующий агент выбран из группы, состоящей из тионилхлорида, оксихлорида фосфора и их смеси.
8. Способ по п. 5, отличающийся тем, что полярный протонный растворитель выбран из группы, состоящей из метанола, этанола, пропанола и их смеси.
9. Способ по п. 1, отличающийся тем, что соединение формулы (III) получают путем осуществления взаимодействия соединения формулы (IX) или его соли с соединением формулы (X) в присутствии основания или амидного связывающего агента
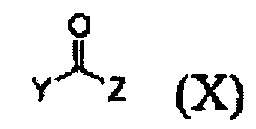
где X и Y являются такими, как определено в п. 1; и Z представляет собой галоген или гидроксил.
10. Способ по п. 9, отличающийся тем, что взаимодействие между соединением формулы (IX) или его солью и соединением формулы (X) проводят в органическом растворителе или в смеси органического растворителя и воды; и органический растворитель представляет собой тетрагидрофуран, этилацетат, ацетон, 1,4-диоксан, ацетонитрил, дихлорметан, четыреххлористый углерод, хлороформ, N,N-диметилформамид или диметилсульфоксид.
11. Способ по п. 9, отличающийся тем, что основание, применяемое в реакции между соединением формулы (IX) или его солью и соединением формулы (X), выбрано из группы, состоящей из карбоната натрия, бикарбоната натрия, карбоната кальция, карбоната калия, гидроксида натрия, гидроксида калия, карбоната цезия, диизопропиламина, триэтиламина, диизопропилэтиламина, диэтиламина и их смеси.
12. Способ по п. 9, отличающийся тем, что амидный связывающий агент выбран из следующей группы: 1-этил-3-(3-диметиламинопропил)карбодиимида, гидроксибензотриазола, O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата, N,N'-дициклогексилкарбоимида, 1-гидрокси-7-азабензотриазола, N,N'-диизопропилкарбоимида, (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфата и их смеси.
13. Способ по п. 9, отличающийся тем, что соль соединения формулы (IX) представляет собой гидрохлоридную или гидробромидную соль.
| WO 2008150118 A2, 11.12.2008 & RU 2434010, 20.11.2011 | |||
| WO 2011155793 A2,15.12.2011 & RU 2575829 C2, 20.02.2016 | |||
| WO 2012169733 A1,13.12.2012 | |||
| Mi Young Cha; ET AL.,Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models International Journal of Cancer, 2012, Vol:130, Nr:10, Page(s):2445 - 2454 (реферат | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| HONNORATY, ANNE-MARIE ET AL., Deracemization process of α-aminoesters via pyridoxal | |||
| II.- Study of copolymerization of polymerizable forms of pyridoxal | |||
| Reactivity of corresponding polymers, Bulletin de la Societe Chimique de France, 1995, vol | |||
| Способ получения нерастворимых лаков основных красителей в субстанции и на волокнах | 1923 |
|
SU132A1 |
| РУЧНОЙ ПИТАТЕЛЬНЫЙ НАСОС | 1921 |
|
SU721A1 |

