Настоящее изобретение относится к химическим способам приготовления определенных производных хиназолина или их фармацевтически приемлемых солей. Изобретение также относится к способам приготовления определенных промежуточных соединений, пригодных для приготовления производных хиназолина, и к способам приготовления производных хиназолина, используя указанные промежуточные соединения.
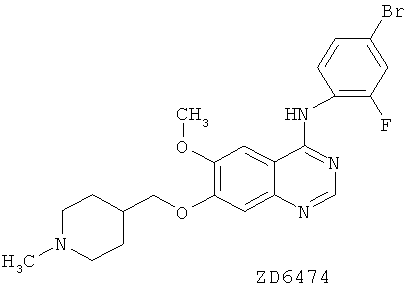
В частности, настоящее изобретение относится к химическим способам и промежуточным соединениям, пригодным для приготовления соединения 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин. Это соединение подпадает под широкое раскрытие в WO 98/13354 и в качестве примеров приведено в WO 01/32651, в примерах 2а, 2b и 2с.
Соединение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин описывается в настоящей заявке с помощью формулы I:

и как ZD6474, кодовый номер, под которым известно соединение. Соединение ZD6474 также известно как Vandetanib и как Zactima™.
Нормальный ангиогенез играет важную роль в разнообразных процессах, включая эмбриональное развитие, заживление ран и некоторых составляющих женской репродуктивной функции. Нежелательный или патологический ангиогенез связан с болезненными состояниями, включая диабетическую ретинопатию, псориаз, злокачественные новообразования, ревматоидный артрит, атерому, саркому Капоши и гемангиому (Fan и др., 1995, Trends Pharmacol. Sci. 16:57-66; Folkman, 1995, Nature Medicine 1:27-31). Полагают, что изменение проницаемости сосудов играет важную роль как в нормальных, так и в патологических физиологических процессах (Cullinan-Bove и др., 1993, Endocrinology 133:829-837; Senger и др., 1993, Cancer and Metastasis Reviews, 12:303-324). Были идентифицированы определенные полипептиды, которые в условиях in vitro обладают стимулирующим действием на рост эндотелиальных клеток, включая кислые и щелочные факторы роста фибробластов (aFGF и bFGF) и фактор роста эндотелия сосудов (VEGF). Активность фактора роста VEGF посредством ограниченной экспрессии его рецепторов в отличие от таковой у FGF является относительно специфичной по отношению к клеткам эндотелия. Исследования, проведенные в последнее время, убедительно свидетельствуют о том, что VEGF является существенным стимулятором как нормального, так и патологического ангиогенеза (Jakeman и др., 1993, Endocrinology, 133:848-859; Kolch и др., 1995, Breast Cancer Research and Treatment, 36: 139-155) и проницаемости сосудов (Connolly и др., 1989, J.Biol.Chem. 264:20017-20024). Антагонистическое действие по отношению к VEGF путем блокирования VEGF при помощи антитела может приводить к ингибированию роста опухолей (Kim и др., 1993, Nature 362:841-844).
Рецепторные тирозинкиназы (RTK) занимают важное место в передаче биохимических сигналов через плазматическую мембрану клеток. Как правило, эти трансмембранные молекулы включают внеклеточный лиганд-связывающий домен, связанный при помощи сегмента в плазматической мембране с внутриклеточным доменом тирозинкиназы. Связывание лиганда с рецептором приводит к стимулированию связанной с рецептором активности тирозинкиназы, что приводит к фосфорилированию остатков тирозина как в рецепторе, так и в других внутриклеточных молекулах. Эти изменения в фосфорилировании тирозина инициируют каскадный путь передачи сигналов, что приводит ко многим ответным реакциям клеток. К настоящему времени было идентифицировано по крайней мере девятнадцать различных подсемейств RTK на основании гомологии аминокислотных последовательностей. Одно из этих подсемейств в настоящее время состоит из fms-подобного рецептора тирозинкиназы Flt-1 (также называется VEGFR-1), рецептора KDR, содержащего домен со вставкой киназы (который также называется VEGFR-2 или Flk-1), и другого fms-подобного рецептора тирозинкиназы, Flt-4. Было показано, что две эти родственные RTK, Fit-1 и KDR связывают VEGF с высоким сродством (De Vries и др., 1992, Science 255:989-991; Terman и др., 1992, Biochem Biophys. Res. Comm. 1992, 187:1579-1586). Связывание VEGF с этими рецепторами, которые экспрессируются в гетерологичных клетках, ассоциировано с изменениями уровня фосфорилирования тирозина белков клетки и потоками кальция.
VEGF является ключевым стимулятором васкулогенеза и ангиогенеза. Этот цитокин вызывает фенотипическое образование отростков сосудов путем стимулирования пролиферации эндотелиальных клеток, экспрессии протеазы и миграции с последующей организацией этих клеток с образованием капилляра (Keck, P.J., Hauser, S.D., Krivi, G., Sanzo, K., Warren, Т., Feder, J. и Connolly, D.T., Science (Washington DC), 246:1309-1312, 1989; Lamoreaux, W.J., Fitzgerald, M.E., Reiner, A., Hasty, K.A. и Charles, S.T., Microvasc. Res., 55:29-42, 1998; Pepper, M.S., Montesano, R., Mandroita, S.J., Orci, L. и Vassalli, J.D., Enzyme Protein, 49:138-162, 1996.). Кроме того, VEGF вызывает значительное повышение проницаемости сосудов (Dvorak, H.F., Detmar, M., Claffey, K.P., Nagy, J.A., van de Water, L., и Senger, D.R., (Int. Arch. Allergy Immunol, 107; 233-235, 1995; Bates, D.O., Heald, R.I., Curry, F.E. и Williams, B.J. Physiol. (Lond.), 533:263-272, 2001), стимулируя формирование сверхпроницаемости недоразвитой сосудистой сети, что является характерной особенностью патологического ангиогенеза.
Было показано, что активирование только KDR достаточно для активизации всех основных фенотипических ответных реакций на VEGF, включая пролиферацию эндотелиальных клеток, миграцию и выживание и индуцирование проницаемости сосудов (Меуеr, М., Clauss, M., Lepple-Wienhues, A., Waltenberger, J., Augustin, H.G., Ziche, M., Lanz, G, Büttner, M., Rziha, H-J. и Dehio, C, EMBO J., 18: 363-374, 1999; Zeng, H., Sanyal, S. и Mukhopadhyay, D., J. Biol. Chem, 276; 32714-32719, 2001; Gille, H., Kowalski, J., Li, B., LeCouter, J., Moffat, B, Zioncheck, T.F., Pelletier, N. и Ferrara, N., J. Biol. Chem, 276:3222-3230, 2001).
ZD6474 является эффективным ингибитором VEGF RTK, а также имеет незначительную активность по отношению к фактору роста эпидермиса (EGF) RTK. ZD6474 ингибирует действия VEGF и представляет интерес в связи с его антиангиогенными действиями и/или действиями на проницаемость сосудов. Ангиогенез и/или повышенная проницаемость сосудов присутствует при многих различных болезненных состояниях, включая злокачественное новообразование (включая лейкоз, множественную миелому и лимфому), диабет, псориаз, ревматоидный артрит, саркому Капоши, гемангиому, острую и хроническую нефропатии, атерому, артериальный рестеноз, аутоиммунные заболевания, острое воспаление, чрезмерное образование рубцов и спаек, лимфатический отек, эндометриоз, дисфункциональное маточное кровотечение и заболевания глаз с пролиферацией сосудов сетчатки, включая дегенерацию желтого пятна, связанную со старением. Было показано, что ZD6474 обладает широким спектром противоопухолевого действия в диапазоне моделей после перорального введения один раз в сутки (Wedge S.R., Ogilvie D.J., Dukes M. и др., Proc. Am. Assoc. Cane. Res. 2001; 42: реферат 3126).
В WO 98/13354 описаны определенные возможные пути для приготовления соединений 4-анилинохиназолина. Однако в документе WO 98/13354 отдельно не описан способ получения соединения формулы I.
В WO 98/10767 также описано несколько возможных путей для приготовления 4-анилинохиназолина. Однако в документе WO 98/10767 отдельно не описан способ получения соединения формулы I.
В WO 01/32651 описаны несколько альтернативных путей получения соединения формулы I.
Путь, который описан в примере 2а заявки WO 01/32651, предусматривает реакцию соединения 4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолина с водным формальдегидом, затем с цианоборогидридом натрия в смеси растворителей тетрагидрофуран и метанол. Продукт очищали с помощью хроматографии и выделяли в виде свободного основания. После этого свободное основание превращали в гидрохлоридную соль путем реакции с хлористым водородом в смеси растворителей метиленхлорид и метанол.
Путь, который описан в примере 2b заявки WO 01/32651, предусматривает реакцию соединения 4-(4-бром-2-фторанилино)-6-метокси-7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)хиназолина с водным формальдегидом в муравьиной кислоте с последующим взаимодействием с гидроксидом натрия в воде и экстрагированием продукта этилацетатом. Продукт находится в виде свободного основания.
Путь, который описан в примере 2 с заявки WO 01/32651, предусматривает реакцию соединения 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина с 4-бром-2-фторанилином и хлористым водородом в изопропаноле. После этого продукт выделяли в виде гидрохлоридной соли. В ЯМР-исследовании гидрохлоридную соль растворяли в диметилсульфоксиде и превращали в свободное основание путем добавления твердого карбоната калия. Затем свободное основание превращали в трифторацетатную соль путем добавления трифторуксусной кислоты. В другом опыте гидрохлоридную соль суспендировали в метиленхлориде и промывали насыщенным гидрокарбонатом натрия для получения свободного основания.
В WO 01/32651 также описаны пути приготовления исходных веществ, которые используются в примерах 2а, 2b и 2с, таких как соединения 4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин, 4-(4-бром-2-фторанилино)-6-метокси-7-(1-(трет-бутоксикарбонил)пиперидин-4-илметокси)хиназолин и 4-хлор-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин. Некоторые из этих путей более подробно обсуждаются ниже.
Пути, описанные в WO 01/32651 для получения ZD6474 (в виде гидрохлоридной соли или свободного основания), также описаны и/или на них ссылаются в публикациях, относящихся к комбинированным лечениям, включающим ZD6474, таких как WO 03/039551, WO 2004/014383, WO 2004/014426, WO 2004/032937, WO 2004/071397 и WO 2005/004870.
Существующие пути для получения соединения формулы I достаточны для синтеза относительно небольших количеств соединения. Тем не менее, для путей, предусматривающих линейный вместо конвергентного синтеза, необходимо использование множественных стадий очистки и выделения большого количества промежуточных соединений. По существу, суммарный выход синтеза не является высоким. Следовательно, существует потребность в более эффективном синтезе соединения формулы I, пригодном для применения для получения больших количеств этого соединения. Также существует потребность в более эффективных синтезах промежуточных соединений, пригодных для синтеза соединения формулы I для применения для получения больших количеств этих промежуточных соединений.
Предпочтительно, новые синтезы должны минимизировать количество промежуточных соединений, которые необходимо выделять, и не должны использовать дорогие и трудоемкие методики очистки. Дополнительно, новые синтезы должны образовывать постоянно высококачественные соединения, в частности, для того, чтобы образовать высококачественное соединение формулы I для соответствия требованиям высокой чистоты для фармацевтического продукта. Новые синтезы также должны использовать методики и реагенты, которые могут безопасно использоваться на производственном предприятии и которые отвечают рекомендациям по охране окружающей среды.
В соответствии с настоящим изобретением сейчас мы обеспечили улучшенные способы приготовления ZD6474, соединения формулы I.
В соответствии с настоящим изобретением обеспечиваются также способы приготовления ключевых промежуточных соединений, которые могут использоваться для приготовления ZD6474.
Новые способы являются благоприятными, поскольку они позволяют получить соединения с высокой чистотой и высоким выходом в большом количестве. Способы позволяют существенно уменьшить количество промежуточных соединений, которые следует выделить, и, в целом, являются более конвергентными, чем предыдущие пути. Такие изменения обеспечивают существенные преимущества относительно времени и затрат.
Для избегания неопределенности термин "ZD6474", как используется в настоящем изобретении, относится к свободному основанию ZD6474, если специально не указано иначе.
Ключевым промежуточным соединением, которое может использоваться для получения ZD6474, является соединение формулы IIа
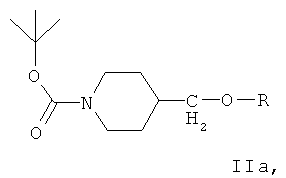
где R представляет собой подходящий сложный сульфонатный эфир, такой как мезилат, эзилат, безилат или тозилат.
В дальнейшем варианте осуществления соединение формулы IIа представляет собой 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин, соединение формулы II:
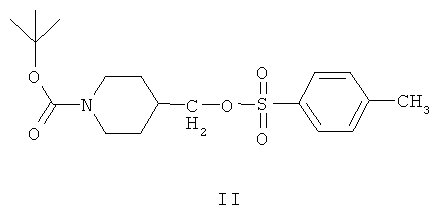
В примере 2 заявки WO 01/32651 описан путь получения соединения формулы II. Этот путь предусматривает реакцию этил 4-пиперидинкарбоксилата с ди-трет-бутилдикарбонатом в этилацетатном растворителе, получая этил 4-(1-(трет-бутоксикарбонил)пиперидин)карбоксилат, который выделяют. После этого этил 4-(1-(трет-бутоксикарбонил)пиперидин)карбоксилат подвергают реакции с литийалюминийгидридом в тетрагидрофуране, получая 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидин, который выделяют. Затем 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидин повергают реакции с 1,4-диазабицикло[2,2,2]октаном и толуолсульфбнилхлоридом в трет-бутилметиловом простом эфирном растворителе, получая соединение формулы II.
В заявке ЕР-А-0317997 описан путь получения соединения формулы II. Этот путь предусматривает реакцию 4-карбоксипиперидина (также известного как изонипекотиновая кислота) с карбонатом натрия и да-трет-бутилдикарбонатом в водном растворителе, получая сложный трет-бутиловый эфир 4-карбокси-пиперидин-1-карбоновой кислоты, который выделяют. После этого сложный трет-бутиловый эфир 4-карбокси-пиперидин-1-карбоновой кислоты подвергают реакции с бораном в тетрагидрофурановом растворителе, получая соединение формулы II.
В заявке WO 94/27965 описан путь получения соединения формулы II. Этот путь предусматривает реакцию 4-гидроксиметилпиперидина с ди-трет-бутилдикарбонатом в тетрагидрофурановом расторителе, получая трет-бутил 4-(гидроксиметил)пиперидин-1-карбоксилат, который выделяют в виде масла. После этого 1-(трет-бутоксикарбонил)-4-гидроксиметилпиперидин подвергают реакции с толуолсульфонилхлоридом и пиридином, получая соединение формулы II.
Пути, описанные в документах предшествующего уровня техники, для получения соединения формулы II достаточны для синтеза относительно небольших количеств соединения. Тем не менее, они все требуют выделения каждого из промежуточных соединений, и следовательно, предусматривают много стадий выделения и/или очистки. Это приводит к удовлетворительному суммарному выходу соединения формулы II, используемого в небольшом масштабе. Однако пути, описанные в документах предшествующего уровня техники, неприемлемы для применения в промышленном масштабе, поскольку они предусматривают много стадий выделения и/или очистки, которые не могут быть эффективно осуществлены в промышленном масштабе. В частности, пути, описанные в документах предшествующего уровня техники, непригодны для применения для приготовления фармацевтического продукта с высокой чистотой.
Следовательно, существует потребность в более эффективном синтезе соединения формулы II, подходящего для применения для получения больших количеств этого соединения. Предпочтительно, новые синтезы не должны предусматривать дорогостоящие и трудоемкие выделения и/или очистки. Таким образом, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, посредством этого уменьшена стоимость и время производства. Предпочтительно, в новом синтезе должно быть минимизировано количество растворителей, используемых при осуществлении метода, что улучшает экологические показатели и предоставляет возможность для восстановления растворителя. Предпочтительно, новый синтез также должен обеспечивать устойчивый и надежный способ выделения соединения формулы II и равным образом обеспечивать высокое качество соединения формулы II, например, для того, чтобы соответствовать нормативным требования для внедрения исходных веществ в получение фармацевтических продуктов.
В соответствии с первым аспектом осуществления настоящего изобретения обеспечивается способ получения соединения формулы IIа из (С1-С6)алкил-4-пиперидинкарбоксилатного соединения формулы III:
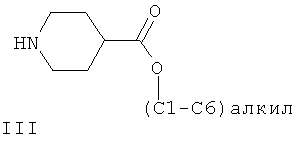
где способ включает стадии:
(а) взаимодействие (С1-С6)алкил-4-пиперидинкарбоксилатного соединения формулы III с ди-трет-бутилдикарбонатом в присутствии толуола или ксилола с образованием первой смеси, содержащей толуол или ксилол, трет-бутанол и соединение формулы IV:
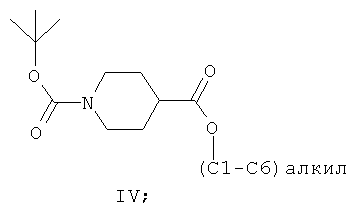
(б) практически полное удаление трет-бутанола из первой смеси;
(в) взаимодействие соединения формулы IV с подходящим восстановителем in situ в присутствии толуола или ксилола с образованием второй смеси, содержащей толуол, восстановленные побочные продукты, включая спиртовые побочные продукты, и соединение формулы V:
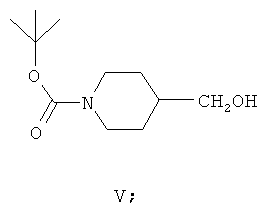
(г) практически полное удаление спиртовых побочных продуктов из второй смеси; и
(д) взаимодействие соединения формулы V с подходящим сульфирующим реагентом in situ с образованием сложного сульфонатного эфира в присутствии подходящего основания и толуола, получая соединение формулы IIа.
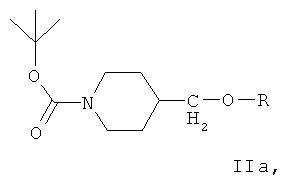
где R представляет собой подходящий сложный сульфонатный эфир, такой как мезилат, эзилат, безилат или тозилат. В одном варианте осуществления изобретения сульфирующий реагент представляет собой тозилхлорид.
Для избежания неопределенности термин 'in situ' обозначает, что взаимодействие осуществляют без выделения реагентов с предыдущей стадии способа.
Способ первого аспекта настоящего изобретения является благоприятным, так как он обеспечивает возможность получения соединения формулы IIа с высокой чистотой и высоким выходом в промышленном масштабе. Обычно каждая из стадий способа первого аспекта настоящего изобретения осуществляется с выходом больше 95%.
Все стадии первого аспекта настоящего изобретения осуществляются в толуоле или ксилоле в качестве растворителя. В другом варианте осуществления все стадии первого аспекта настоящего изобретения осуществляются в толуоле. Это позволяет осуществлять способ в виде непрерывного процесса без выделения и/или очистки промежуточных соединений формул IV и V. Это существенно уменьшает время и стоимость производства формулы IIа в промышленном масштабе. Использование единственного растворителя, такого как толуол или ксилол, также может предоставить возможность повторного использования растворителя, что повышает эффективность способа и обеспечивает преимущества использования для окружающей природной среды. Применение толуола или ксилола в качестве растворителя также позволяет эффективно и удобно удалить реакционно-способные побочные продукты (такие как спирты), например, путем перегонки. Присутствие таких реакционно-способных побочных продуктов может привести к загрязнению соединения формулы IIа, если их своевременно не удалить.
Дополнительно, использование толуола или ксилола в качестве растворителя в способе первого аспекта настоящего изобретения позволяет удобно выделить соединение формулы IIа путем кристаллизации. Соединение формулы IIа, например, может быть выделено с чистотой больше чем 99,5% путем кристаллизации непосредственно из реакционной смеси без необходимости дальнейшей очистки. Это является благоприятным, например, если соединение формулы IIа вводят на поздней стадии в получение фармацевтического продукта, например соединения формулы I, поскольку это минимизирует риск введения загрязнений в фармацевтический продукт.
На стадии (а) способа используют (С1-С6)алкил-4-пиперидинкарбоксилатное соединение формулы III, предпочтительно (С1-С4)алкил-4-пиперидинкарбоксилатное соединение формулы III. В частности, подходящим (С1-С6)алкил-4-пиперидинкарбоксилатным соединением формулы III, которое может применяться на стадии (а), может быть, например, этил 4-пиперидинкарбоксилат. Другим названием для этил 4-пиперидинкарбоксилата является этилизонипекотат.
Взаимодействие на стадии (а) осуществляют при температуре в интервале, например, от 0 до 45°С, подходяще в интервале от 15 до 35°С, более подходяще в интервале от 25 до 30°С.
Исходные вещества (С1-С6)алкил-4-пиперидинкарбоксилатные соединения формулы III и ди-трет-бутилдикарбонат, используемые на стадии (а) способа, являются коммерчески доступными или могут быть получены с помощью общепринятых методов. Например, (С1-С6)алкил-4-пиперидинкарбоксилатные соединения формулы III могут быть получены, как описано в заявке на патент Японии JP 03002162 А2.
Трет-бутанол, который образуется на стадии (а), представляет собой побочный продукт реакции между (С1-С6)алкил-4-пиперидинкарбоксилатным соединением формулы III и ди-трет-бутилдикарбонатом. В процессе согласно настоящему изобретению этот побочный продукт легко и удобно практически полностью удаляется из реакционной смеси, например, путем дистилляции на стадии (б).
Является благоприятным практически полностью удалять трет-бутанольный побочный продукт из реакционной смеси, например, путем дистилляции на стадии (б), поскольку любой трет-бутанольный побочный продукт, который не удаляется, вероятно, реагирует с восстановителем на стадии (в), в связи с этим уменьшается количество восстановителя, доступное для осуществления желательной реакции с соединением формулы IV. Таким образом, удаление трет-бутанольного побочного продукта на стадии (б) предусматривает правильную стехиометрию реагентов на стадии (в) способа и, следовательно, более эффективное осуществление реакции на этой стадии. Это, в свою очередь, обеспечивает высокий выход и чистоту соединения формулы V на стадии (в).
Под термином "практически полностью удаляется" мы подразумеваем, что по меньшей мере 85% побочного трет-бутанола, который образуется на стадии (а), удаляется, например, путем дистилляции. Обычно дистилляцию осуществляют до тех пор, пока внутренняя температура не достигает интервала от 102 до 112°С. Дистилляцию на стадии (б) подходяще осуществляют или при атмосферном или частично пониженном давлении.
Подходящими восстановителями для применения на стадии (в) являются натрий бис(2-метоксиэтокси)алюминийгидрид, литийалюминийгидрид и диизобутилалюминийгидрид. Более предпочтительно, восстановителем, используемым на стадии (в), является натрий бис(2-метоксиэтокси)алюминийгидрид.
Взаимодействие на стадии (в) осуществляют при температуре в интервале, например, от 20 до 55°С, подходяще в интервале от 30 до 50°С, более подходяще в интервале от 35 до 45°С.
Специалист в данной области техники примет во внимание, что реакция стадии (в) обычно обеспечивает восстановленные побочные продукты дополнительно к желательному соединению формулы V. Восстановленные побочные продукты включают спиртовые побочные продукты. Спиртовые побочные продукты имеют происхождение из -O-(С1-С6)алкильной части сложноэфирной группы в соединении формулы IV и также могут иметь происхождение из восстановителя. Например, если соединение формулы IV представляет собой этил 4-(1-трет-бутоксикарбонил)пиперидин)карбоксилат и восстановителем, используемым на стадии (в), является натрий бис(2-метоксиэтокси)алюминийгидрид, то типичные восстановленные побочные продукты включают соли алюминия и спиртовые побочные продукты, такие как этанол и 2-метоксиэтанол. Спиртовые побочные продукты легко и подходяще практически полностью удаляются из реакционной смеси, например, путем дистилляции на стадии (г).
Благоприятным является практически полное удаление спиртовых побочных продуктов на стадии (г), поскольку любые такие побочные продукты, которые не удалены, вероятно, взаимодействуют с сульфирующим реагентом на стадии (д), вследствие этого повышается количество примесей, которые могут загрязнять желательный продукт и уменьшать количество сульфирующего реагента, доступного для желательной реакции с соединением формулы V. Таким образом, удаление спиртовых побочных продуктов позволяет соблюсти правильную стехиометрию реагентов на стадии (д) способа и, следовательно, более эффективно осуществить реакцию на этой стадии. Это, в свою очередь, обеспечивает высокий выход и чистоту соединения формулы II на стадии (д).
Под термином "практически полное удаление" мы подразумеваем, что по меньшей мере 98% спиртовых побочных продуктов, которые образуются на стадии (в), удаляются, например, путем дистилляции. Обычно дистилляцию осуществляют до тех пор, пока внутренняя температура не достигает интервала от 102°С до 112°С. Дистилляцию на стадии (г) подходяще осуществляют или при атмосферном, или частично пониженном давлении.
Дистилляция на стадии (г) также обычно практически полностью удаляет любую присутствующую воду. Это также позволяет соблюсти правильную стехиометрию реагентов на стадии (д) способа, поскольку любая вода, которая не удалена, вероятно, взаимодействует с сульфирующим реагентом на стадии (д), вследствие этого снижается количество сульфирующего реагента, доступного для желательной реакции с соединением формулы V. Под термином "практически полное удаление" мы подразумеваем, что меньше чем 20 мол.% воды остается после дистилляции.
Специалист в данной области техники примет во внимание, что обычно необходимо закаливать реакционную смесь на стадии (в) для удаления любого непрореагировавшего присутствующего восстановителя перед осуществлением реакции на стадии (д). Обычно на стадии закаливания также удаляются некоторые из восстановленных побочных продуктов, перечисленных выше, например соли алюминия и некоторые, но не все, спиртовые побочные продукты. Подходящие гасящие добавки в большинстве случаев могут быть выбраны из любого средства, которое описано в литературе и/или известно специалисту в данной области техники. Например, если восстановитель, используемый на стадии (в), представляет собой натрий бис(2-метоксиэтокси)алюминий гидрид, то гасящая добавка обычно может представлять собой водный раствор виннокислого калия-натрия (также известен как сегнетова соль). Обычно полученную в результате водную фазу (содержащую гасящий восстановитель) затем удаляют путем отделения. Стадию закаливания осуществляют перед дистилляцией на стадии (г).
Подходящим основанием для применения на стадии (д) является третичное аминовое основание, например триэтилендиамин.
Реакцию стадии (д) осуществляют при температуре в интервале, например, от 15 до 45°С, более подходяще в интервале от 25 до 35°С.
Специалист в данной области техники примет во внимание, что типично необходимо закаливать реакционную смесь на стадии (д) для удаления любого непрореагировавшего присутствующего сульфирующего реагента. Подходящие гасящие добавки в большинстве случаев могут быть выбраны из любого средства, которое описано в литературе и/или известно специалисту в данной области техники. Например, подходящей гасящей добавкой может быть основание, такое как гидроксид натрия или карбонат калия.
В одном аспекте способ получения соединения формулы II дополнительно может включать стадию (е) выделения и/или очистки соединения формулы II. Стадия (е) может включать любые подходящие стадии или методики выделения желательного продукта, которые описаны в литературе и/или известны специалисту в данной области техники. Предпочтительные стадии, которые могут применяться, будут обеспечивать высокое качество и высокую чистоту продукта.
Например, стадия (е) может включать стадии промывания соединения формулы II водой и/или разведенной лимонной кислотой. Стадия (е) также, например, может включать кристаллизацию с использованием подходящей системы растворителей. Примером подходящей системы растворителей является система растворителей, содержащая толуол и изогексан, что обеспечивает получение соединения формулы II с высокой чистотой, обычно с чистотой больше чем 98%, подходяще, больше чем 99,5%, и с высоким выходом, обычно с выходом больше чем 80%, подходяще, больше чем 85%. Специалист в данной области техники примет во внимание, что стадия (е) также может включать стадию циклического воздействия температуры (также называемое как "созревание Освальда") соединения формулы II, улучшая таким образом физическую форму продукта, если это является необходимым.
Другим ключевым промежуточным соединением, которое может применяться для получения ZD6474, является защищенное производное 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединение формулы VI:
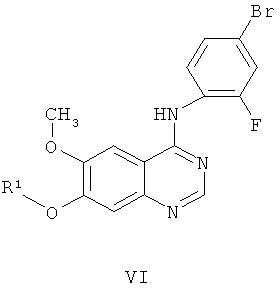
где R1 представляет собой кислотолабильную защитную группу, такую как бензил, замещенный бензил, трет-бутил, аллил или метоксиэтоксиметил.
В примере 2 заявки WO 01/32651 и примере 24 заявки WO 97/32856 описан путь получения гидрохлоридной соли соединения формулы VI, где R' представляет собой бензил. Путь предусматривает реакцию гидрохлоридной соли 7-бензилокси-4-хлор-6-метоксихиназолина с 4-бром-2-фторанилином в 2-пропанольном растворителе с получением гидрохлоридной соли соединения формулы VI, которую выделяют. В примере 2 заявки WO 01/32651 указывается, что гидрохлоридную соль 7-бензилокси-4-хлор-6-метоксихиназолина получают в соответствии с примером 1 заявки WO 97/22596.
В примере 1 заявки WO 97/22596 гидрохлоридную соль 7-бензилокси-4-хлор-6-метоксихиназолина получают путем реакции 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-она с тионилхлоридом в N,N-диметилформамидном растворителе. Аналогичный процесс получения гидрохлоридной соли 7-бензилокси-4-хлор-6-метоксихиназолина описан в примере 4 заявки WO 97/32856.
В заявке WO 98/10767 описан путь получения 6,7-дизамещенных 4-анилинохиназолиновых соединений. Путь предусматривает реакцию 6,7-дизамещенного хиназолинонового соединения с хлорирующим реагентом и катализатором при отсутствии растворителя или с хлорирующим реагентом в присутствии улавливающего средства, получая 6,7-дизамещенное 4-хлорхиназолиновое соединение. Затем 6,7-дизамещенное 4-хлорхиназолиновое соединение подвергают реакции с замещенным анилиновым соединением, необязательно в присутствии подходящего основания, получая гидрохлоридную соль 6,7-дизамещенного 4-анилинохиназолинового соединения, которую затем можно превратить в свободное основание. В заявке WO 98/10767 не описан 7-бензилокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин, также не описан способ его получения.
Пути, описанные в документах предшествующего уровня техники, для получения соединения формулы VI достаточны для синтеза относительно небольших количеств соединения. Однако для всех их необходимо выделение и/или очистка промежуточных соединений. Это приводит к удовлетворительному, но не высокому суммарному выходу соединения формулы VI.
Следовательно, существует потребность в более эффективном синтезе соединения формулы VI, подходящем для получения больших количеств этого соединения. Предпочтительно, в новом синтезе не должны применяться дорогие и трудоемкие методики выделения и/или очистки. Следовательно, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, уменьшая таким образом стоимость и время приготовления. Новый способ также должен обеспечивать эффективное выделение соединения формулы VI в кристаллической форме с высокой чистотой и выходом, где кристаллическая форма будет иметь хорошие фильтрационные характеристики.
В соответствии со вторым аспектом настоящего изобретения обеспечивается способ получения соединения формулы VI:
где R1 представляет собой кислотолабильную защитную группу;
из соединения формулы VII:
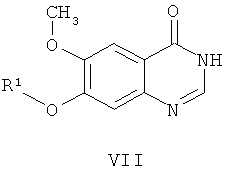
где способ включает стадии:
(ж) взаимодействие соединения формулы VII с подходящим хлорирующим реагентом в присутствии подходящего основания и подходящего растворителя, где реакцию осуществляют путем:
(ж-1) добавления смеси соединения формулы VII и основания в растворителе к смеси хлорирующего реагента в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 60 до 80°С в течение приблизительно 60 минут; или
(ж-2) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в диапазоне от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(ж-3) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 70 до 90°С в течение приблизительно 15 минут, получая соединение формулы VIII:
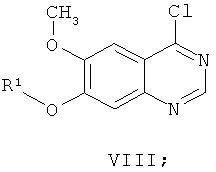 и
и
(з) взаимодействие соединения формулы VIII с 4-бром-2-фторанилином in situ в присутствии растворителя, который используют на стадии (ж), получая гидрохлоридную соль соединения формулы VI;
и после этого соединение формулы VI, полученное в виде гидрохлоридной соли, может быть превращено в свободное основание или в форму альтернативной соли, если это является необходимым.
Термин "кислотолабильная защитная группа" относится к группам, которые легко удаляются в кислых условиях. Подходящими методами защиты являются методы, известные специалисту в данной области техники. Могут применяться общепринятые защитные группы в соответствии со стандартной практикой (для иллюстрации см. T.W.Green, Protective Groups in Organic Synthesis, John Wiley и Sons, 1991). Подходящими защитными группами для R1 являются бензил, замещенный бензил (например, С1-4алкоксибензил и C1-4алкибензил), трет-бутил, 1,1-диметил-1-этилметил, аллил, замещенный аллил (такой как С1-4алкилаллил) или метоксиэтоксиметил. В другом варианте осуществления R1 представляет собой бензил.
Способ второго аспекта изобретения является благоприятным, поскольку он обеспечивает получение соединение формулы VI с высокой чистотой и высоким выходом в большом масштабе. Обычно каждая из стадий способа второго аспекта настоящего изобретения обеспечивает выход больше чем 90%.
Подходящий растворитель для стадии (ж) выбирают из группы, включающей простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как простой 1,2-диметиловый эфир, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол. В одном варианте осуществления изобретения растворитель на стадии (ж) представляет собой анизол или толуол. В другом варианте осуществления изобретения растворитель на стадии (ж) представляет собой толуол.
Обе стадии (ж) и (з) осуществляют в идентичном растворителе, где растворитель выбирают из подходящего растворителя, как описано выше. Это позволяет осуществить способ в виде непрерывного способа без выделения и/или очистки промежуточного соединения формулы VIII. Это существенно уменьшает время и стоимость получения соединения формулы VI в большом масштабе. Дополнительно, применение единственного растворителя может предоставить возможность повторного использования растворителя, что повышает эффективность способа и обеспечивает преимущества использования для окружающей природной среды. Применение толуола или анизола в качестве растворителя реакции является благоприятным, поскольку эти растворители сводят к минимуму образование побочных продуктов, которые могут образовываться вследствие димеризации соединения формулы VII, как обсуждалось выше. Выбор растворителя также позволяет осуществить легкое и удобное выделение соединения формулы VI. Например, если реакционную смесь охлаждают до температуры окружающей среды, то соединение формулы VI обычно образует твердое вещество, где твердое вещество затем может быть собрано с помощью любого общепринятого метода.
Режим добавления реагентов на стадии (ж) (то есть, как описано на стадиях (ж-1), (ж-2) и (ж-3)) является благоприятным, так как это сводит к минимуму образование побочных продуктов/примесей на этой стадии. Обычно любые такие побочные продукты/примеси преимущественно образуются путем димеризации соединения формулы VII. Уменьшение образования побочных продуктов/примесей предоставляет возможность использовать промежуточное соединение формулы VIII, полученное на стадии (ж), на стадии (з) без выделения и/или очистки. Уменьшение образования побочных продуктов/примесей на стадии (ж) также предоставляет возможность правильной стехиометрии для реагентов на стадии (з) способа и, следовательно, более эффективное осуществление реакции на этой стадии. В свою очередь, это обеспечивает высокий выход и высокую чистоту соединения формулы VI на стадии (з).
В одном аспекте изобретения обе стадии (ж) и (з) осуществляют в толуоле в качестве растворителя. В другом аспекте изобретения обе стадии (ж) и (з) осуществляют в анизоле в качестве растворителя. В еще другом аспекте изобретения стадии (ж) и (з) осуществляют в смеси растворителей толуол и анизол.
Подходящий хлорирующий реагент для применения на стадии (ж) представляет собой оксихлорид фосфора. Обычно на стадии (ж) применяют молярный избыток хлорирующего реагента по отношению к соединению формулы VII. Например, можно использовать молярный избыток в диапазоне от 1,3 до 2,0, подходяще в интервале от 1,7 до 1,8.
Подходящим основанием для применения на стадии (ж) является основание, выбранное из триэтиламина и N,N-диизопропилэтиламина. В частности, основание представляет собой N,N-диизопропилэтиламин. Применение N,N-диизопропилэтиламина в качестве основания на стадии (ж) является благоприятным, так как это сводит к минимуму образование побочных продуктов, которые могут образовываться вследствие димеризации соединения формулы VII, как обсуждалось выше (например, по сравнению с применением триэтиламина в качестве основания на стадии (ж)). Добавление к реакционной смеси источника хлорида (такого как, например, гидрохлорид триэтиламина) также может уменьшить образование таких побочных продуктов.
На стадии (ж-1) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 60 до 80°С, подходяще в интервале от 65 до 80°С, более подходяще в интервале от 70 до 75°С.
На стадии (ж-2) добавление реагентов осуществляют при температуре окружающей среды. Под термином "температура окружающей среды" мы подразумеваем температуру в интервале от 10 до 30°С, в особенности температуру в интервале от 15 до 25°С, более предпочтительно температуру около 20°С. После этого реакционную смесь нагревают до температуры в диапазоне от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж-3) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж) термин "приблизительно" используется в выражениях "приблизительно 60 минут", "приблизительно 15 минут", "приблизительно 90 минут и "приблизительно 1 час" для указания того, что указанные промежутки времени не должны истолковываться как абсолютные значения, поскольку, что следует принять во внимание специалисту в данной области техники, периоды времени могут значительно отличаться. Например, указанные периоды времени могут отличаться на ±50%, предпочтительно на ±15%, предпочтительно на ±10% от значений, указанных на стадии (ж).
Специалист в данной области техники примет во внимание, что на стадии (ж) смесь соединения формулы VII и основания в подходящем растворителе обычно будет находиться в виде суспензии. Смесь хлорирующего реагента в растворителе, выбранном из толуола и анизола, обычно будет находиться в виде раствора. Однако многие факторы могут оказывать влияние на изменение этих форм. Такими факторами могут являться, например, количество каждого из реагентов, добавляемых к растворителю, предпочтительное основание или хлорирующий реагент, выбранный для применения на стадии (ж), и/или температура, выбранная для применения на стадии (ж).
Реакцию стадии (з) осуществляют при температуре в интервале от 60 до 85°С, подходяще в интервале от 65 до 80°С, более подходяще в интервале от 70 до 75°С.
В одном аспекте изобретения после осуществления стадии (з) способа соединение формулы VI сразу используют в другом способе (например, в способе получения 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина, как обсуждается далее). В другом аспекте изобретения после осуществления стадии (з) способа соединение формулы VI выделяют и/или очищают, например, перед хранением, транспортировкой и/или дальнейшей реакцией. Таким образом, в одном аспекте изобретения способ получения соединения формулы VI дополнительно включает стадию (и) выделения соединения формулы VI. Стадия (и) может включать любые подходящие стадии или методики для выделения желательного продукта, которые описаны в литературе и/или которые известны специалисту в данной области техники.
Предпочтительные стадии, которые будут применяться, будут обеспечивать высокое качество и высокую чистоту продукта. Реакционная смесь может быть охлаждена до температуры окружающей среды, при этой температуре соединение формулы VI обычно образует твердое вещество, и твердое вещество, образованное таким образом, может быть собрано с помощью любого общепринятого способа, например путем фильтрации.
Оба соединения - и соединение формулы VII, и 4-бром-2-фторанилиновое исходное вещество, являются коммерчески доступными или могут быть получены с помощью общепринятых методов. Например, соединение формулы VII, где R1 представляет собой бензил, может быть получено, как описано в примере 2 ниже, приготовление исходных веществ.
Другим ключевым промежуточным соединением, которое может применяться для получения ZD6474, является 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин, соединение формулы IX:
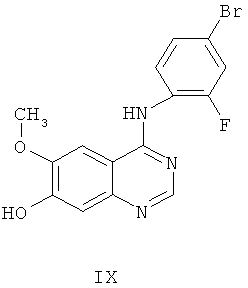
В примере 2 заявки WO 01/32651 и в примере 24 заявки WO 97/32856 описан путь получения гидрохлоридной соли соединения формулы IX. Путь предусматривает реакцию гидрохлоридной соли 7-бензилокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина с трифторуксусной кислотой, получая соединение формулы IX.
Как обсуждалось выше, в заявке WO 98/10767 описан путь получения 6,7-дизамещенных 4-анилинохиназолиновых соединений. В заявке WO 98/10767 не описан 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин, также не описан способ его получения.
Пути, описанные в документах предшествующего уровня техники, для получения соединения формулы IX, достаточны для синтеза относительно небольших количеств соединения. Однако для всех них необходимо выделение и/или очистка промежуточных соединений. Это приводит к удовлетворительному, но не высокому суммарному выходу соединения формулы IX.
Следовательно, существует потребность в более эффективном синтезе соединения формулы IX, подходящем для получения больших количеств этого соединения. Предпочтительно, в новом синтезе не должны применяться дорогие и трудоемкие методики очистки. Следовательно, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, уменьшая таким образом стоимость и время приготовления. Предпочтительно, в новом синтезе должно быть сведено к минимуму количество растворителей, используемых при осуществлении способа, что улучшает экологические показатели и предоставляет возможность для восстановления растворителя. Новый синтез также должен предоставить возможность эффективной кристаллизации соединения формулы IX в кристаллической форме с хорошими фильтрационными характеристиками и с высокой чистотой и выходом.
В соответствии с третьим аспектом настоящего изобретения обеспечивается способ приготовления 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединения формулы IX:
из соединения формулы VII:
где способ включает стадии:
(ж) взаимодействие соединения формулы VII с подходящим хлорирующим реагентом в присутствии подходящего основания и подходящего растворителя, где реакцию осуществляют путем:
(ж-1) добавления смеси соединения формулы VII и основания в растворителе к смеси хлорирующего реагента в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 60 до 80°С в течение приблизительно 60 минут; или
(ж-2) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в диапазоне от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(ж-3) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 70 до 90°С в течение приблизительно 15 минут, получая соединение формулы VIII:
(з) взаимодействия соединения формулы VIII с 4-бром-2-фторанилином in situ в присутствии растворителя, используемого на стадии (ж), получая соединение формулы VI;
(к) удаления R1 из соединения формулы VI in situ в присутствии растворителя, используемого на стадиях (ж) и (з), получая соединение формулы IX или его соль;
и после этого соединение формулы IX, полученное в форме свободного основания, может быть превращено в солевую форму и соединение формулы IX, полученное в форме соли, может быть превращено в свободное основание или в форму альтернативной соли, если это является необходимым.
Способ согласно третьему аспекту изобретения является благоприятным, поскольку он предоставляет возможность получения соединения формулы IX с высокой чистотой и высоким выходом в большом масштабе. Обычно каждая из стадий способа третьего аспекта настоящего изобретения протекает с выходом по крайней мере 95%. Обычно в способе согласно третьему аспекту настоящего изобретения синтезируют соединение формулы IX с выходом по меньшей мере 85%.
Все стадии (ж), (з) и (к) осуществляются в идентичном растворителе, где растворитель выбирают из группы, включающей простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как простой 1,2-диметиловый эфир, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол. В одном варианте осуществления изобретения растворитель для стадии (ж), (з) и (к) представляет собой анизол или толуол. В другом варианте осуществления изобретения растворитель для стадии (ж), (з) и (к) представляет собой толуол. Это позволяет осуществлять способ в виде непрерывного способа без выделения и/или очистки промежуточных соединений формул VIII и VI. Это существенно уменьшает время и стоимость приготовления соединения формулы IX в большом масштабе. Использование единственного растворителя может предоставить возможность повторного использования растворителя, что повышает эффективность способа и обеспечивает преимущества использования для окружающей природной среды. Применение этих растворителей в качестве растворителя реакции является благоприятным, поскольку эти растворители сводят к минимуму образование побочных продуктов, которые могут образовываться вследствие димеризации соединения формулы VII, как обсуждалось выше. Выбор растворителя также позволяет осуществить легкое и удобное выделение соединения формулы VI. Например, если реакционную смесь охлаждают до температуры окружающей среды, то соединение формулы VI обычно образует твердое вещество, которое затем может быть собрано с помощью любого общепринятого способа.
Как обсуждалось выше, режим добавления реагентов на стадии (ж) (то есть, как описано на стадиях (ж-1), (ж-2) и (ж-3)) является благоприятным, так как это сводит к минимуму образование побочных продуктов/примесей на этой стадии (эти побочные продукты/примеси обычно преимущественно образуются путем димеризации соединения формулы VII). Это предоставляет возможность использовать промежуточное соединение формулы VIII, полученное на стадии (ж), на стадии (з) без выделения и/или очистки. Уменьшение образования побочных продуктов/примесей на стадии (ж) позволяет соблюсти правильную стехиометрию реагентов на стадии (з) способа и, следовательно, более эффективно осуществить реакцию на этой стадии. В свою очередь, это обеспечивает высокий выход и высокую чистоту соединения формулы VI на стадии (з).
В одном аспекте изобретения стадии (ж), (з) и (к) все осуществляют в толуоле в качестве растворителя. Применение толуола в качестве растворителя на стадии (к), где R1 представляет собой бензил, является благоприятным, поскольку толуол захватывает катион бензила, который образуется при реакции снятия защиты. Это помогает уменьшить бензилированные примеси, которые потенциально могут образовываться на стадии (к) способа. Толуол также обеспечивает более эффективную кристаллизацию соединения IX и кристаллическую форму соединения IX с улучшенными фильтрационными характеристиками.
В другом аспекте изобретения стадии (ж), (з) и (к) все осуществляют в единственном растворителе, таком как анизол хлорбензол, трифтортолуол, ксилол или этилбензол.
Подходящий хлорирующий реагент для применения на стадии (ж) представляет собой оксихлорид фосфора. Обычно на стадии (ж) применяют молярный избыток хлорирующего реагента по отношению к соединению формулы VII. Например, можно использовать молярный избыток в диапазоне от 1,3 до 2,0, подходяще в интервале от 1,7 до 1,8.
Подходящим основанием для применения на стадии (ж) является основание, выбранное из триэтиламина, трипропиламина и N,N-диизопропилэтиламина. В частности, основание представляет собой триэтиламин. Применение триэтиламина в качестве основания на стадии (ж) является благоприятным, поскольку предоставляет возможность более эффективной кристаллизации соединения IX и получения кристаллической формы соединения IX с улучшенными фильтрационными характеристиками.
На стадии (ж-1) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 60 до 80°С, подходяще в интервале от 65 до 75°С, более подходяще в интервале от 70 до 75°С.
На стадии (ж-2) добавление реагентов осуществляют при температуре окружающей среды. Под термином "температура окружающей среды" мы подразумеваем температуру в интервале от 10 до 30°С, в особенности температуру в интервале от 15 до 25°С, более предпочтительно температуру около 20°С.После этого реакционную смесь нагревают до температуры в диапазоне от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж-3) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж) термин "приблизительно" используется в выражениях "приблизительно 60 минут", "приблизительно 15 минут", "приблизительно 90 минут" и "приблизительно 1 час" для указания того, что указанные промежутки времени не должны истолковываться как абсолютные значения, поскольку, что следует принять во внимание специалисту в данной области техники, периоды времени могут значительно отличаться. Например, указанные периоды времени могут отличаться на ±50%, предпочтительно ±15%, предпочтительно на ±10% от значений, указанных на стадии (ж).
Специалист в данной области техники примет во внимание, что на стадии (ж) смесь соединения формулы VII и основания в подходящем растворителе обычно будет находиться в виде суспензии. Смесь хлорирующего реагента в растворителе, выбранном из толуола и анизола, обычно будет находиться в виде раствора. Однако многие факторы могут оказывать влияние на изменение этих форм. Такими факторами могут являться, например, количество каждого из реагентов, добавляемых к растворителю и предпочтительное основание или хлорирующий реагент, выбранный для применения на стадии (ж).
Реакцию стадии (з) осуществляют при температуре в интервале от 60 до 90°С, подходяще от 60 до 85°С, подходяще в интервале от 65 до 80°С, более подходяще в интервале от 70 до 75°С.
В этом аспекте изобретения, после приготовления соединения формулы VI на стадии (з) соединение сразу используют на стадии (к) для приготовления соединения формулы IX. Другими словами, соединение формулы VI как таковое не выделяют, но используют в виде раствора или взвеси в растворителе, выбранном из группы, включающей простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как 1,2-диметоксиэтан, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол. В одном варианте осуществления изобретения растворитель для стадии (к) представляет собой анизол или толуол. В другом варианте осуществления изобретения растворитель для стадии (к) представляет собой толуол. Таким образом, соединение формулы IX может быть приготовлено из соединения формулы VII методикой в одном сосуде.
Подходящим методом удаления кислото-лабильной защитной группы in situ на стадии (к) является взаимодействие с кислотой, такой как трифторуксусная кислота. Необязательно, дополнительно к трифторуксусной кислоте или вместо нее можно использовать вторую кислоту (такую, как хлористый водород или бромистый водород). Если для удаления R1 на стадии (к) используется кислота, то после этого соединение формулы IX получают в форме соли. Применение трифторуксусной кислоты на стадии (к) является благоприятным, поскольку это предоставляет возможность легкого выделения соединения формулы IX, например, путем кристаллизации из трифторуксусной кислоты путем добавления воды и охлаждения или путем добавления водного основания щелочного металла, такого как гидроксид калия, гидроксид натрия, ацетат натрия, ацетат калия, более предпочтительно гидроксид калия, с последующим добавлением воды и охлаждением. Кристаллическое твердое вещество, которое при этом образуется, может быть собрано с помощью любого общепринятого способа, например путем фильтрации.
Реакцию стадии (к) осуществляют при температуре в интервале от 60 до 90°С, подходяще от 60 до 80°С, более подходяще в интервале от 70 до 75°С.
В одном аспекте изобретения, после осуществления стадии (к) способа соединение формулы IX выделяют и/или очищают. Можно использовать любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или которые известны специалисту в данной области техники. Предпочтительные стадии, которые будут применяться, будут обеспечивать высокое качество и высокую чистоту продукта. Например, соединение формулы IX может быть выделено из трифторуксусной кислоты путем добавления воды и охлаждения или более предпочтительно путем добавления водного основания щелочного металла, такого как гидроксид калия, и воды и охлаждения, как обсуждалось выше.
В соответствии с четвертым аспектом настоящего изобретения обеспечивается способ приготовления 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединения формулы IX:
из соединения формулы VII:
где способ включает стадии:
(ж) взаимодействие соединения формулы VII с подходящим хлорирующим реагентом в присутствии подходящего основания и подходящего растворителя, выбранного из толуола и анизола, где реакцию осуществляют путем:
(ж-1) добавления смеси соединения формулы VII и основания в растворителе к смеси хлорирующего реагента в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 60 до 80°С в течение приблизительно 60 минут; или
(ж-2) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в диапазоне от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(ж-3) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 70 до 90°С в течение приблизительно 15 минут, получая соединение формулы VIII:
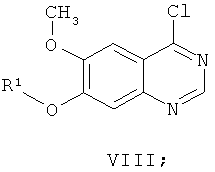
(з) взаимодействия соединения формулы VIII с 4-бром-2-фторанилином in situ в присутствии растворителя, который используют на стадии (ж), получая соединение формулы VI:
;
(и) выделения соединения формулы VI; и
(л) удаления R1 из соединения формулы VI, получая соединение формулы IX
или его соль;
и после этого соединение формулы IX, полученное в форме свободного основания, может быть превращено в солевую форму, и соединение формулы IX, полученное в форме соли, может быть превращено в свободное основание или в форму альтернативной соли, такой как соль трифторуксусной кислоты или гидрохлорид, если это является необходимым.
Способ в соответствии с четвертым аспектом изобретения является благоприятным, поскольку он предоставляет возможность получения соединения формулы IX с высокой чистотой и высоким выходом в большом масштабе.
Обе стадии (ж) и (з) осуществляют в идентичном растворителе, где растворитель выбирают из группы, включающей простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как простой 1,2-диметиловый эфир, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол. В одном варианте осуществления изобретения растворитель для стадии (ж) и (з) представляет собой анизол или толуол. В другом варианте осуществления изобретения растворитель для стадии (ж) и (з) представляет собой толуол. Это позволяет осуществлять способ в виде непрерывного способа без выделения и/или очистки промежуточного соединения формулы VIII. Это существенно уменьшает время и стоимость приготовления соединения формулы IX в большом масштабе. Применение единственного растворителя на стадиях (ж) и (з) предоставляет возможность повторного использования растворителя, что повышает эффективность способа и обеспечивает преимущества использования для окружающей природной среды. Применение толуола или анизола в качестве растворителя реакции на стадиях (ж) и (з) является благоприятным, поскольку эти растворители сводят к минимуму образование побочных продуктов, которые могут образовываться вследствие димеризации соединения формулы VII, как обсуждалось выше. Выбор растворителя также позволяет осуществить легкое и удобное выделение соединения формулы VI. Например, если реакционную смесь охлаждают до температуры окружающей среды, то соединение формулы VI обычно образует твердое вещество, где твердое вещество затем может быть собрано с помощью любого общепринятого метода.
Как обсуждалось выше, режим добавления реагентов на стадии (ж) (то есть, как описано на стадиях (ж-1), (ж-2) и (ж-3)) является благоприятным, так как это сводит к минимуму образование побочных продуктов/примесей на этой стадии (эти побочные продукты/примеси обычно преимущественно образуются путем димеризации соединения формулы VII). Это предоставляет возможность использовать промежуточное соединение формулы VIII, полученное на стадии (ж), на стадии (з) без выделения и/или очистки. Уменьшение образования побочных продуктов/примесей на стадии (ж) позволяет соблюсти правильную стехиометрию реагентов на стадии (з) способа и, следовательно, более эффективное осуществление реакции на этой стадии. Это, в свою очередь, обеспечивает высокий выход и чистоту соединения формулы VI на стадии (з).
В одном аспекте изобретения обе стадии (ж) и (з) осуществляют в толуоле в качестве растворителя. В другом аспекте изобретения обе стадии (ж) и (з) осуществляют в анизоле в качестве растворителя.
Подходящий хлорирующий реагент для применения на стадии (ж) представляет собой оксихлорид фосфора. Обычно на стадии (ж) применяют молярный избыток хлорирующего реагента по отношению к соединению формулы VII. Например, можно использовать молярный избыток в диапазоне от 1,3 до 2,0, подходяще в интервале от 1,7 до 1,8.
Подходящим основанием для применения на стадии (ж) является основание, выбранное из триэтиламина и N,N-диизопропилэтиламина. В одном варианте осуществления изобретения основание представляет собой триэтиламин. Применение триэтиламина в качестве основания на стадии (ж) является благоприятным, поскольку предоставляет возможность более эффективной кристаллизации соединения IX и получения кристаллической формы соединения IX с улучшенными фильтрационными характеристиками.
В другом варианте осуществления основание представляет собой N,N-диизопропилэтиламин. Применение N,N-диизопропилэтиламина в качестве основания на стадии (ж) является благоприятным, так как это сводит к минимуму образование побочных продуктов, которые могут образовываться вследствие димеризации соединения формулы VII, как обсуждалось выше (например, по сравнению с применением триэтиламина в качестве основания на стадии (ж)). Добавление к реакционной смеси источника хлорида (такого, как, например, триэтиламин гидрохлорид) также может уменьшить образование таких побочных продуктов.
На стадии (ж-1) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 60 до 80°С, подходяще в интервале от 65 до 75°С, более подходяще в интервале от 70 до 75°С.
На стадии (ж-2) добавление реагентов осуществляют при температуре окружающей среды. Под термином "температура окружающей среды" мы подразумеваем температуру в интервале от 10 до 30°С, в особенности температуру в интервале от 15 до 25°С, более предпочтительно температуру около 20°С.После этого реакционную смесь нагревают до температуры в диапазоне от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж-3) реакцию осуществляют при температуре в интервале от 60 до 110°С, подходяще от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще в интервале от 80 до 85°С.
На стадии (ж) термин "приблизительно" используется в выражениях "приблизительно 60 минут", "приблизительно 15 минут", "приблизительно 90 минут" и "приблизительно 1 час" для указания того, что указанные промежутки времени не должны истолковываться как абсолютные значения, поскольку, что следует принять во внимание специалисту в данной области техники, периоды времени могут значительно отличаться. Например, указанные периоды времени могут отличаться на ±50%, предпочтительно ±15%, предпочтительно на ±10% от значений, указанных на стадии (ж).
Специалист в данной области техники примет во внимание, что на стадии (ж) смесь соединения формулы VII и основания в подходящем растворителе обычно будет находиться в виде суспензии. Смесь хлорирующего реагента в растворителе, выбранном из толуола и анизола, обычно будет находиться в виде раствора. Однако многие факторы могут оказывать влияние на изменение этих форм. Такими факторами могут являться, например, количество каждого из реагентов, добавляемых к растворителю, и предпочтительное основание или хлорирующий реагент, выбранный для применения на стадии (ж).
Реакцию стадии (з) осуществляют при температуре в интервале от 60 до 90°С, подходяще от 60 до 90°С, подходяще в интервале от 65 до 80°С, более подходяще в интервале от 70 до 75°С.
В этом аспекте изобретения после приготовления соединения формулы VI на стадии (з) соединение выделяют и, необязательно, очищают на стадии (и) способа. Затем выделенное соединение формулы VI используется на стадии (л) для приготовления соединения формулы IX или сразу или после хранения в течение подходящего периода времени. Выделение соединения формулы VI на стадии (и), где R1 представляет собой бензил, является благоприятным, поскольку это предоставляет возможность широкого выбора методов для удаления бензильной группы из соединения формулы VI на стадии (л), например, по сравнению с тем, когда эта стадия осуществляется in situ.
Стадия (л), где R1 представляет собой бензил, может включать любые подходящие стадии или методики для удаления бензильной группы, которые описаны в литературе и/или которые известны специалисту в данной области техники. Предпочтительные стадии, которые будут применяться, будут обеспечивать высокое качество и высокую чистоту продукта. Например, на стадии (л) бензильная группа может быть удалена путем взаимодействия с подходящим гидрирующим средством, таким как палладий на угле, например, в присутствии подходящего замедляющего средства, такого как бромид цинка или йодид цинка. Применение гидрирующего средства является благоприятным, поскольку это обеспечивает высокую эффективность способа удаления бензильной группы на стадии (л) и поскольку это предоставляет возможность эффективного удаления побочных продуктов из потока отходов.
Другим подходящим методом удаления кислотолабильной защитной группы, где R1 представляет собой бензильную группу, на стадии (л) является взаимодействие с кислотой, такой как трифторуксусная кислота. Необязательно, дополнительно к трифторуксусной кислоте или вместо нее можно использовать вторую кислоту (такую, как хлористый водород или бромистый водород). Если кислота используется для удаления бензильной группы на стадии (л), то после этого соединение формулы IX получают в форме соли. Применение трифторуксусной кислоты на стадии (л) является благоприятным, поскольку это предоставляет возможность легкого выделения соединения формулы IX, например, путем кристаллизации из трифторуксусной кислоты путем добавления воды и охлаждения или более предпочтительно путем добавления водного основания щелочного металла, такого как гидроксид калия, гидроксид натрия, ацетат натрия, ацетат калия, более предпочтительно гидроксид калия с последующим добавлением воды и охлаждением. Кристаллическое твердое вещество, которое при этом образуется, может быть собрано с помощью любого общепринятого способа, например путем фильтрации.
Реакцию стадии (л), где R1 представляет собой бензил, можно осуществлять при любой температуре и в любом растворителе, подходящем для предпочтительного метода удаления бензильной группы, который используется. Примерами подходящих растворителей для кислотоосновного удаления бензильной группы является этанол, простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как простой 1,2-диметиловый эфир, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол или дихлорметан.
В одном аспекте изобретения после осуществления стадии (л) способа соединение формулы IX выделяют и/или очищают. Могут использоваться любые подходящие стадии или методики для выделения и/или очистки желательного продукта, которые описаны в литературе и/или которые известны специалисту в данной области техники. Предпочтительные стадии, которые будут применяться, будут обеспечивать высокое качество и высокую чистоту продукта.
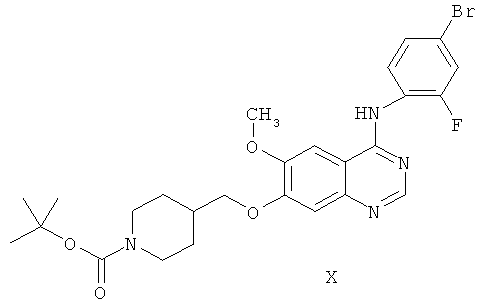
Другим ключевым промежуточным соединением, которое может применяться для получения ZD6474, является 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин, соединение формулы X:
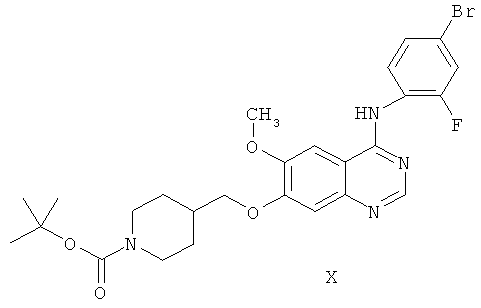
В примере 2 заявки WO 01/32651 описан путь получения соединения формулы X. Путь предусматривает реакцию 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина с карбонатом калия и 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидином в N,N-диметилформамидном растворителе, получая соединение формулы X.
Как обсуждалось выше, в заявке WO 98/10767 описан путь получения 6,7-дизамещенных 4-анилинохиназолиновых соединений. В заявке WO 98/10767 не описан 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин, также не описан способ его получения.
Пути, описанные в документах предшествующего уровня техники для получения соединения формулы X, достаточны для синтеза относительно небольших количеств соединения. Тем не менее, существует потребность в более эффективном синтезе соединения формулы X, подходящем для получения больших количеств этого соединения. Предпочтительно, в новом синтезе не должны применяться дорогие и трудоемкие методики очистки. Следовательно, в новом синтезе должно быть уменьшено количество необходимых методик выделения и/или очистки, уменьшая таким образом стоимость и время приготовления. Предпочтительно, в новом синтезе должно быть сведено к минимуму количество растворителей, используемых при осуществлении способа, что улучшает экологические показатели и предоставляет возможность для восстановления растворителя. Новый синтез также обеспечивает соединение формулы Х с высокой чистотой и высоким выходом.
В соответствии с пятым аспектом настоящего изобретения обеспечивается способ приготовления 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединения формулы X:
из соединения формулы VII:
;
где способ включает стадии превращения соединения формулы VII в соединение формулы IX:
путем осуществления способа, как обсуждалось выше, для третьего или четвертого аспекта изобретения; и
(м) взаимодействие соединения формулы IX с соединением формулы II, как определено выше, в присутствии подходящего основания, получая соединение формулы Х или его соль;
и после этого соединение формулы X, полученное в форме свободного основания или в сольватированной или несольватированной форме, может быть превращено в солевую форму, и соединение формулы X, полученное в форме соли, может быть превращено в свободное основание или в форму альтернативной соли, если это является необходимым.
Способ в соответствии с пятым аспектом изобретения является благоприятным, поскольку он позволяет получить соединение формулы Х с высокой чистотой и высоким выходом в большом масштабе. Обычно способ в соответствии с пятым аспектом настоящего изобретения осуществляется с выходом более, чем 80%. Способ в соответствии с пятым аспектом изобретения также является благоприятным с учетом по меньшей мере тех аргументов, которые обсуждались выше для третьего и четвертого аспектов изобретения.
Обычно соединение формулы IX выделяют и/или очищают перед осуществлением стадии (м), например, используя любые подходящие стадии или методики, которые описаны в литературе и/или которые известны специалисту в данной области техники, как обсуждалось выше.
В другом варианте осуществления изобретения после приготовления соединения формулы IX на стадии (к), где R1 представляет собой бензил (или замещенный бензил), если в качестве способа снятия защиты с бензильной группы используется гидрирование, то соединение сразу используют на стадии (м) для приготовления соединения формулы X. Другими словами, соединение формулы IX как таковое не выделяют, но используют в виде раствора или взвеси в подходящем растворителе, таком как N-метилпирролидон, диметилформамид или диметилацетамид. В одном варианте осуществления изобретения растворитель для стадии (к) представляет собой N-метилпирролидон. Таким образом, соединение формулы Х может быть приготовлено из соединения формулы VIII методикой в одном сосуде.
Подходящее основание для применения на стадии (м) выбирают из карбоната натрия, бикарбоната натрия, карбоната калия, гидроксида натрия, трет-бутанола калия и гидроксида калия.
Стадия (м) может осуществляться в любом подходящем растворителе и при любой подходящей температуре.
Если основание, которое используют на стадии (м), выбирают из карбоната натрия и карбоната калия, то подходящими растворителями являются, например, N-метилпирролидон, N-этилпирролидон, диметилацетамид, диметилсульфоксид, сульфолин, метилэтилкетон и N,N-диметилформамид. В этом аспекте стадию (м) обычно можно осуществлять при температуре в диапазоне от 60 до 120°С, подходяще от 70 до 105°С, подходяще в интервале от 80 до 100°С, подходяще в интервале 70-90°С, подходяще в интервале от 90 до 95°С. В дальнейшем варианте осуществления в интервале 75-85°С.
Если основание, которое используют на стадии (м), выбирают из гидроксида натрия и гидроксида калия, то подходящими растворителями являются, например, простой арилалкиловый эфир, такой как анизол, простой диалкиловый эфир, такой как 1,2-диметоксиэтан, бензол, замещенный галогеном, такой как хлорбензол или трифтортолуол, или бензол, замещенный алкилом, такой как ксилол, этилбензол или толуол или ацетонитрил. В одном варианте осуществления изобретения растворитель для стадии (м) представляет собой анизол или толуол. В другом варианте осуществления изобретения растворитель для стадии (м) представляет собой толуол. В этом аспекте стадию (м) обычно можно осуществлять при температуре в диапазоне от 60 до 90°С, подходяще в интервале от 65 до 85°С, подходяще в интервале от 70 до 80°С. В этом аспекте стадию (м) подходяще можно осуществлять путем добавления воды, основания (такого, как гидроксид натрия или гидроксид калия) и подходящего межфазного катализатора в толуоле к реакционной смеси. Подходящими межфазными катализаторами являются, например, бромид тетрабутиламмония и Adogen® 464 (хлорид метилтриалкил(С8-10) аммония, CAS 63393-96-4).
В одном аспекте способ в соответствии с пятым аспектом изобретения может включать стадию (н) выделения соединения формулы X. Стадия (н) может включать любые подходящие стадии или методики для выделения соединения формулы X, которые описаны в литературе и/или которые известны специалисту в данной области техники.
Например, если основание, которое используют на стадии (м), выбирают из карбоната натрия и карбоната калия, стадия (н) может включать стадии:
(н-1) добавление воды и предоставление возможности осуществления кристаллизации соединения формулы X, собирание соединения формулы Х и промывка соединения формулы Х водой, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила, при температуре в диапазоне от 25 до 55°С, подходяще от 45 до 55°С; или
(н-2) добавление воды и спирта, выбранного из метанола, этанола, изопропанола и н-пропанола (предпочтительно изопропанола) и предоставление возможности осуществления кристаллизации соединения формулы X, собирание соединения формулы Х и промывка соединения формулы Х смесью воды и спирта, выбранного из метанола, этанола, изопропанола и н-пропанола, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила, при температуре в диапазоне от 25 до 55°С, подходяще от 45 до 55°С.
Стадии (н-1) и (н-2) являются благоприятными, поскольку они эффективны для удаления непрореагировавшего соединения формулы IX, а также примесей, которые обычно образуются при осуществлении стадии (м) способа. Такие примеси включают те примеси, которые образуются при реакции соединения формулы II с атомом азота в 1-ом положении в хиназолиновом кольце вместо желательного положения в гидроксизаместителе.
Если основание, которое используют на стадии (м) выбирают из гидроксида натрия и гидроксида калия, то стадия (н) может включать стадии предоставления возможности осуществления кристаллизации соединения формулы Х (например, кристаллизации из толуольной фазы) и собирания соединения формулы Х при помощи любого общепринятого способа. Этот аспект является благоприятным, поскольку соединение формулы Х кристаллизуется непосредственно из реакционной смеси с высоким выходом (например, по меньшей мере с выходом 80%) и с высокой чистотой без необходимости дальнейшей очистки продукта.
На стадиях (н) соединение формулы X, которое при этом образуется (например, которое выделяют в виде кристаллического твердого вещества), может быть собрано с помощью любого общепринятого способа, например путем фильтрации. После этого собранное кристаллическое твердое вещество, если это является необходимым, может быть промыто подходящим растворителем и затем может быть высушено.
В соответствии с шестым аспектом настоящего изобретения обеспечивается способ приготовления 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединения формулы X:
из 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина, соединения формулы IX:
(м) взаимодействие соединения формулы IX с соединением формулы II, как определено выше, в присутствии подходящего основания, получая соединение формулы Х или его соль; и
(н) выделение соединения формулы Х путем:
(н-1) добавления воды и предоставления возможности осуществления кристаллизации соединения формулы X, собирания соединения формулы Х и промывки соединения формулы Х водой, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила, при температуре в диапазоне от 25 до 55°С, подходяще от 45 до 55°С; или (н-2) добавления воды и спирта, выбранного из метанола, этанола, изопропанола и н-пропанола (предпочтительно изопропанола) и предоставления возможности осуществления кристаллизации соединения формулы X, собирания соединения формулы Х и промывки соединения формулы Х смесью воды и спиртом, выбранным из метанола, этанола, изопропанола и н-пропанола, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила, при температуре в диапазоне от 25 до 55°С, подходяще от 25 до 55°С;
и после этого соединение формулы X, полученное в форме свободного основания или в сольватированной или в несольватированной форме (или сольвата растворителей из NMP, этилацетата или смеси обоих растворителей), может быть превращено в солевую форму, и соединение формулы X, полученное в форме соли, может быть превращено в свободное основание или в форму альтернативной соли, если это является необходимым.
Способ в соответствии с шестым аспектом осуществления изобретения является благоприятным, поскольку он позволяет получить соединение формулы Х с высокой чистотой и высоким выходом в большом масштабе. Обычно каждая из стадий способа согласно шестому аспекту настоящего изобретения осуществляется с выходом больше чем 80%.
Способ обеспечивает эффективное удаление любого непрореагировавшего соединения формулы IX, а также любых примесей, которые обычно образуются при осуществлении стадии (м) способа. Такие примеси включают те примеси, которые образуются при реакции соединения формулы II с атомом азота в 1-ом положении в хиназолиновом кольце вместо желательного положения в гидроксизаместителе.
Подходящее основание для применения на стадии (м) выбирают из карбоната натрия, гидроксида натрия, гидроксида калия и карбоната калия.
Стадия (м) может осуществляться в любом подходящем растворителе или при любой подходящей температуре.
Если основание, которое используют на стадии (м), выбирают из карбоната натрия и карбоната калия, то подходящими растворителями являются, например, N-метилпирролидон, N-этилпирролидон и N,N-диметилформамид. В этом аспекте стадию (м) обычно можно осуществлять при температуре в диапазоне от 70 до 105°С, подходяще от 80 до 100°С, подходяще от 90 до 95°С.
Стадии (н-1) и (н-2) являются благоприятными, поскольку они эффективны для удаления непрореагировавшего соединения формулы IX, а также примесей, которые обычно образуются при осуществлении стадии (м) способа. Такие примеси включают те примеси, которые образуются при реакции соединения формулы II с атомом азота в 1-ом положении в хиназолиновом кольце вместо желательного положения в гидроксизаместителе.
На стадиях (н-1) и (н-2) образованное таким образом кристаллическое твердое вещество может быть собрано с помощью любого общепринятого способа, например путем фильтрации. После этого собранное кристаллическое твердое вещество, если это является необходимым, может быть промыто подходящим растворителем и затем может быть высушено.
Соединение формулы II, которое используется на стадии (м) способов согласно пятому и шестому аспектам осуществления изобретения, может быть получено при помощи любого способа, описанного в литературе, или общепринятого способа. В одном аспекте изобретения соединение формулы II, которое используется на стадии (м) согласно пятому и шестому аспектам осуществления изобретения, получают в соответствии со способом согласно первому аспекту осуществления изобретения, как обсуждалось выше.
В соответствии с седьмым аспектом изобретения обеспечивается способ приготовления ZD6474:
из соединения формулы X:
где способ включает стадии:
(о) взаимодействие соединения формулы Х с муравьиной кислотой и формальдегидом или полимером формальдегида, подходяще в воде при температуре в диапазоне от 70 до 95°С, подходяще от 70 до 90°С с образованием соли муравьиной кислоты с ZD6474;
(п) добавление инертного органического растворителя, выбранного из тетрагидрофурана, бутиронитрила и метанола, и подходящего основания, получая таким образом свободное основание ZD6474;
после этого ZD6474, полученное в форме свободного основания, может быть превращено в фармацевтически приемлемую соль, если это является необходимым.
На стадии (о) способа согласно седьмому аспекту осуществления изобретения, реакцию осуществляют с помощью переходного промежуточного соединения, где промежуточное соединение представляет собой 4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин, соединение формулы XI:
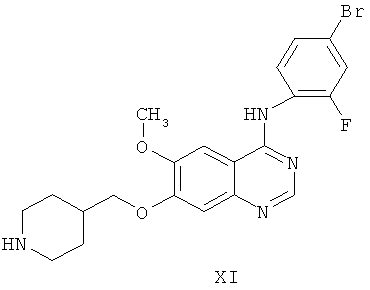
Способ согласно седьмому аспекту осуществления изобретения является благоприятным, поскольку он позволяет получить ZD6474 с высокой чистотой и высоким выходом в большом масштабе. Обычно каждая из стадий способа согласно седьмому аспекту настоящего изобретения обеспечивает выход больше чем 90%.
Соединение формулы X, которое используют на стадии (о) способа согласно седьмому аспекту осуществления изобретения, может быть получено при помощи любого способа, описанного в литературе, или общепринятого способа (например, как описано в WO 01/32651, что обсуждалось ранее). Альтернативно, в одном аспекте изобретения соединение формулы X, которое используют на стадии (о) согласно седьмому аспекту осуществления изобретения, получают в соответствии со способом согласно пятому или шестому аспекту осуществления изобретения, как обсуждалось выше.
Стадию (о) осуществляют при температуре в диапазоне от 70 до 95°С, подходяще от 70 до 90°С, подходяще в интервале от 75 до 85°С, более подходяще приблизительно при 80°С.
Предпочтительно, стадию (о) осуществляют в инертной атмосфере, например в атмосфере азота. Это является благоприятным, поскольку способ стадии (о) может образовывать газообразный водород и угарный газ в виде побочного продукта, где газообразный водород необходимо удалить из реакционного сосуда безопасным и эффективным образом.
На стадии (о) получают соль муравьиной кислоты с ZD6474. Эту соль превращают в свободное основание ZD6474 на стадии (п) способа.
На стадии (о) примерами полимеров формальдегида являются параформальдегид и s-триоксан (1,3,5 триоксан).
Подходящий инертный органический растворитель для применения на стадии (п) выбирают из тетрагидрофурана, бутиронитрила и метанола (предпочтительно тетрагидрофурана или метанола). Инертный органический растворитель добавляют к реакционной смеси после завершения реакции на стадии (о). Специалист в данной области техники примет во внимание, что может быть необходимо охладить реакционную смесь перед добавлением инертного органического растворителя.
Подходящим основанием для применения на стадии (п) является гидроксид натрия или гидроксид калия (предпочтительно гидроксид калия). Добавление основания на стадии (п) превращает соль муравьиной кислоты с ZD6474 в свободное основание ZD6474.
Если инертный органический растворитель, который используют на стадии (п), выбирают из тетрагидрофурана и бутиронитрила, то ZD6474 продукт эффективно переносят из водной фазы в органическую фазу. Это происходит в связи с тем, что после образования свободное основание ZD6474 преимущественно растворимо в инертном органическом растворителе (тогда как соль муравьиной кислоты с ZD6474 растворима в водной фазе). Если инертный органический растворитель, который используют на стадии (п), представляет собой метанол, то свободное основание ZD6474 обычно кристаллизуется непосредственно из реакционной смеси. Если основание представляет собой гидроксид калия, то это является предпочтительно благоприятным, поскольку соль муравьиной кислоты полностью растворяется в метанольном растворителе и не загрязняет выделенное соединение ZD6474. Это также обеспечивает кристаллическое соединение с хорошими фильтрационными характеристиками (которое может быть выделено или в виде ангидратной формы, метаноатной формы или смешанной метаноатногидратной). Таким образом, стадия (п) способа является благоприятной, поскольку она способствует и облегчает выделение и очистку ZD6474 продукта, предпочтительно, если способ осуществляют в большом масштабе.
Стадию (п) осуществляют при температуре в диапазоне от 30 до 70°С, подходяще в интервале от 40 до 65°С, более подходяще в интервале от 40 до 60°С.
В одном аспекте способ согласно седьмому аспекту осуществления изобретения может включать стадию (р) выделения и/или очистки свободного основания ZD6474. Стадия (р) может включать любые подходящие стадии или методики выделения и/или очистки свободного основания ZD6474, которые описаны в литературе и/или которые известны специалисту в данной области техники. Альтернативно, например, если инертный органический растворитель, который используют на стадии (п), выбирают из тетрагидрофурана и бутиронитрила, то стадия (р) может включать стадии:
(р-1) отделение и удаление водной фазы из органической фазы;
(р-2) загрузка н-бутилацетата в органическую фазу;
(р-3) промывка органической фазы водой и отделение и удаление водной фазы из органической фазы;
(р-4) добавление тетрагидрофурана и н-бутилацетата к органической фазе;
(р-5) дистилляция органической фазы таким образом, чтобы практически полностью удалить воду и тетрагидрофуран и получить суспензию ZD6474 преимущественно в н-бутилацетате;
(р-6) предоставление возможности завершения кристаллизации ZD6474;
и
(р-7) сбор ZD6474.
Стадии (р-1), (р-2) и (р-3) являются благоприятными, поскольку в них легко и удобно удаляются соли муравьиной кислоты и остатки формальдегида или полимера формальдегида из ZD6474 продукта, растворенного в органической фазе.
В одном аспекте стадии (р-1), (р-2), (р-3) и (р-4) каждая осуществляется при температуре в диапазоне от 50 до 65°С, подходяще в интервале от 55 до б5°С, более подходяще приблизительно 60°С.
Обычно стадии (р-1), (р-2) и (р-3) каждая может повторяться два раза перед осуществлением стадии (р-4).
На стадии (р-5) практически полностью удаляется любая вода и тетрагидрофуран, которые присутствуют в органической фазе, которая была отделена от водной фазы на стадиях (р-1) и (р-3). Дистилляцию осуществляют таким образом, чтобы обеспечить композицию растворителей, содержащую около 90 мас.% н-бутилацетата. Другими словами, раствор ZD6474 преимущественно в н-бутилацетате представляет собой раствор ZD6474 в композиции растворителей, которая содержит около 90 мас.% н-бутилацетата. Обычно дистилляцию осуществляют при внутренней температуре в интервале от 90 до 110°С, подходяще от 90 до 104°С, достигают подходяще в интервале 100-110°С. Дистилляцию на стадии (р-5) подходяще осуществляют при атмосферном давлении (или пониженном давлении, но более подходяще при давлении окружающей среды).
Для избежания неопределенности в (р-6) выражение 'предоставление возможности завершения кристаллизации ZD6474' обозначает, что процесс кристаллизации завершается в используемых условиях, это не означает, что 100% ZD6474 кристаллизовалось в реакционной смеси.
Альтернативная стадия (р) выделения и/или очистки свободного основания ZD6474, если инертный органический растворитель, который используется на стадии (п), представляет собой тетрагидрофуран, может включать стадии:
(р-8) добавление воды к раствору ZD6474 в органической фазе, полученной после стадии (р-1), таким образом, чтобы позволить осуществиться кристаллизации ZD6474; и
(р-9) сбор ZD6474.
В каждой из вышеописанных стадий образованное таким образом кристаллическое твердое вещество может быть собрано с помощью любого общепринятого способа, например путем фильтрации. После этого собранное кристаллическое твердое вещество, если это является необходимым, дополнительно может быть очищено и затем может быть высушено.
Стадия (р) выделения ZD6474 продукта является благоприятной, поскольку она обеспечивает ZD6474 с высоким выходом (например, обычно с выходом больше чем 90%) и высокой чистотой (например, обычно чистотой больше чем 99%). Дополнительно, стадия (р) обеспечивает форму ZD6474, которая легко поддается фильтрованию в большом масштабе.
В другом аспекте настоящего изобретения ZD6474, полученное в соответствии со способом согласно седьмому аспекту осуществления изобретения, как обсуждалось выше, дополнительно может быть очищено. Дальнейшая очистка ZD6474 может включать любые подходящие стадии или методики для выделения и/или очистки ZD6474, которые описаны в литературе и/или которые известны специалисту в данной области техники. Альтернативно, дальнейшая очистка ZD6474 может включать стадии нагревания суспензии ZD6474, полученной в способе согласно седьмому аспекту настоящего изобретения в смеси тетрагидрофурана, воды и бутилацетата до температуры флегмы, охлаждения полученной смеси до температуры в диапазоне от 50 до 65°С (подходяще приблизительно 60°С), разделения водной и органических фаз и фильтрования органической фазы. После этого фильтрат можно объединять с дополнительным количеством тетрагидрофурана и бутилацетата и полученную смесь нагревать до температуры в диапазоне от 90 до 110°С, подходяще от 90 до 110°С (подходяще в диапазоне от 100 до 110°С), затем охлаждать до температуры в диапазоне от 40 до -10°С, подходяще от 25 до 0°С (подходяще в интервале от 0 до 10°С, более подходяще приблизительно 5°С, в дальнейшем варианте осуществления при температуре около 25°С), получая взвесь ZD6474. После этого ZD6474 может быть собран при помощи любого общепринятого способа, например путем фильтрации, и необязательно промыт этилацетатом. Это является благоприятным, поскольку в описанном процессе уменьшается количество воды при окончании дистилляции ниже 1%, обеспечивая таким образом, что образуется безводная форма ZD6474.
Альтернативно, например, если инертный органический растворитель, который используется на стадии (п), представляет собой тетрагидрофуран, то стадия (р) может включать стадии:
(р-1) отделение и удаление водной фазы из органической фазы;
(р-2) фильтрование органической фазы;
(р-3) загрузка н-бутилацетата в органическую фазу;
(р-4) промывка органической фазы водой и отделение и удаление водной фазы из органической фазы;
(р-5) добавление тетрагидрофурана и н-бутилацетата к органической фазе;
(р-6) дистилляция органической фазы таким образом, чтобы практически полностью удалить воду и тетрагидрофуран и получить суспензию ZD6474 преимущественно в н-бутилацетате;
(р-7) охлаждение и загрузка дополнительного количества тетрагидрофурана; и
(р-8) предоставление возможности завершения кристаллизации ZD6474;
и
(р-9) сбор ZD6474.
Стадия (р-7) является благоприятной, поскольку она улучшает качество продукта, полученного путем солюбилизации примесей в маточных растворах. Это предоставляет возможность телескопирования продукции неочищенного API (активного фармацевтического компонента) с выделением очищенного API за единственную стадию.
В соответствии с восьмым аспектом настоящего изобретения обеспечивается способ приготовления ZD6474 из соединения формулы VII:
где способ включает стадии:
(ж) взаимодействие соединения формулы VII с подходящим хлорирующим реагентом в присутствии подходящего основания и растворителя, выбранного из хлорбензола, трифтортолуола, ксилола, этилбензола, толуола и анизола, более предпочтительно анизола и толуола, где реакцию осуществляют путем:
(ж-1) добавления смеси соединения формулы VII и основания в растворителе к смеси хлорирующего реагента в растворителе при температуре в диапазоне от 60 до 90°С, подходяще от 60 до 80°С в течение приблизительно 60 минут; или
(ж-2) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре окружающей среды в течение приблизительно 15 минут и после этого нагревания реакционной смеси в течение приблизительно 90 минут до температуры в диапазоне от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 часа; или
(ж-3) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре в диапазоне от 60 до 110°С, подходяще от 70 до 90°С в течение приблизительно 15 минут, получая соединение формулы VIII:
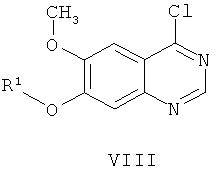 ;
;
(з) взаимодействие соединения формулы VIII с 4-бром-2-фторанилином in situ в присутствии растворителя, который используют на стадии (ж), получая соединение формулы VI:
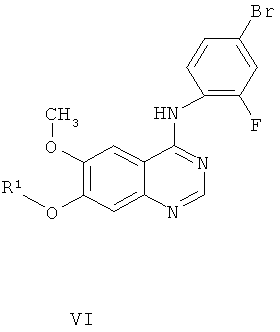
(к) удаление R1 из соединения формулы VI in situ в присутствии растворителя, который используют на стадиях (ж) и (з), получая соединение формулы IX:
(м) взаимодействие соединения формулы IX с соединением формулы II, как определено выше, в присутствии подходящего основания, получая соединение формулы X;
(о) взаимодействие соединения формулы Х с муравьиной кислотой и формальдегидом или полимером формальдегида подходяще в воде при температуре в диапазоне от 70 до 90°С, получая соль муравьиной кислоты с ZD6474; и
(п) добавление инертного органического растворителя, выбранного из тетрагидрофурана, бутиронитрила и метанола и подходящего основания, получая таким образом свободное основание ZD6474; и необязательно
(р) дальнейшая очистка ZD6474 в смеси тетрагидрофурана, воды и бутилацетата, получая требуемую кристаллическую безводную форму, пригодную для производства таблеток.
после этого ZD6474, полученное в форме свободного основания, может быть превращено в фармацевтически приемлемую соль, если это является необходимым.
Способ согласно восьмому аспекту осуществления изобретения является благоприятным, так как он предоставляет возможность получения ZD6474 с высокой чистотой и высоким выходом в большом масштабе. Обычно каждая из стадий способа согласно седьмому аспекту настоящего изобретения обеспечивает выход больше чем 90%.
Предпочтительные аспекты способа согласно восьмому аспекту осуществления изобретения являются такими, как указано выше при описании характерных стадий, как описано в первом, втором, третьем, четвертом, пятом, шестом и седьмом аспектах настоящего изобретения. В частности, предпочтительные аспекты способа согласно восьмому аспекту осуществления изобретения являются такими, как указано выше, например, при описании характерных стадий третьего, пятого, шестого и/или седьмого аспектов настоящего изобретения.
Подходяще, стадии (ж), (з) и (к) способа согласно восьмому аспекту настоящего изобретения все осуществляют в толуоле в качестве растворителя и триэтиламине в качестве основания.
Подходяще, приемлемый способ удаления бензильной группы in situ на стадии (к) способа согласно восьмому аспекту настоящего изобретения, где R1 представляет собой бензил, осуществляется путем взаимодействия с трифторуксусной кислотой.
Подходяще, основание, которое используют на стадии (м) способа согласно восьмому аспекту настоящего изобретения, представляет собой карбонат калия, и подходящий растворитель представляет собой N-метилпирролидон.
Способ согласно восьмому аспекту настоящего изобретения обычно может включать стадию (н) выделения соединения формулы Х перед осуществлениями стадий (о) и (п). Подходяще, стадия (н) может быть осуществлена, как описано в данной заявке выше.
Подходяще, приемлемым основанием для применения на стадии (п) согласно восьмому аспекту настоящего изобретения является гидроксид калия.
Подходяще, приемлемым растворителем для применения на стадии (п) согласно восьмому аспекту настоящего изобретения является метанол. Способ согласно восьмому аспекту настоящего изобретения может включать стадию (р) выделения и/или очистки свободного основания ZD6474. Стадия (р) может быть осуществлена, как описано в данной заявке выше.
Далее изобретение иллюстрируется следующими примерами и данными, которые не ограничивают его объем, в которых, если специально не указано иначе:
(i) упаривания осуществляют на роторном испарителе в вакууме, и процедуры обработки осуществляют после удаления остатков твердых веществ, таких как осушители, путем фильтрации;
(ii) выходы представлены только с целью иллюстрации и необязательно, что могут быть достигнуты максимальные значения;
(iii) точки плавления являются нескорректированными и их определяли с помощью Mettler DSC820e;
(iv) структуры конечных продуктов подтверждали с помощью ядерного (обычно протонного) магнитного резонанса (ЯМР) и масс-спектральных методик; значения химических сдвигов протонного магнитного резонанса измеряли на дельта-шкале и пики мультиплетности представляли следующим образом: s, синглет; d, дублет; t, триплет; т, мультиплет; br, широкий; q, квартет, quin, квинтет; все образцы анализировали на Bruker DPX 400 МГц при 300К в указанном растворителе, 16 сканирований, время повторения импульсов 10 секунд;
(v) промежуточные соединения обычно не были полностью охарактеризованы и их чистоту оценивали с помощью ЯМР-анализа;
(vi) химические символы имеют их обычное значение; используются единицы и символы СИ; и
(vii) использованы следующие сокращения:
Пример 1
Получение 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)-пиперидина (соединение формулы II)
Ди-трет-бутил дикарбонат (88,63 г) в толуоле (296 мл) добавляли к перемешиваемому раствору этилизонипекотата (62,88 г) в толуоле (317 мл). Затем реакционную смесь дистиллировали при атмосферном давлении, удаляли около 130 мл дистиллята при конечной температуре дистилляции 112°С. После этого к реакционной смеси добавляли натрий бис(2-метоксиэтокси)алюминий гидрид (Red-Al, 65% мас./мас. раствор в толуоле, 161 г) в толуоле (220 мл) в течение приблизительно 60 минут. К реакционной смеси добавляли раствор 0,5 молярной сегнетовой соли (191 мл) и водную фазу отделяли при 40°С. Органическую фазу промывали 15% об./мас. соляным раствором (3×136 мл) и водой (136 мл). Раствор дистиллировали при температуре воздуха, удаляли около 400 мл дистиллята при конечной температуре дистилляции 112°С. К реакционной смеси добавляли триэтилендиамин (51,62 г), затем добавляли тозилхлорид (87,90 г) в толуоле (416 мл) в течение приблизительно 60 минут. К реакционной смеси добавляли гидроксид натрия (2 н., 160 мл) и органический слой отделяли и промывали последовательно водой (80 мл), лимонной кислотой (0,5М, 80 мл) и водой (80 мл). Органическую фазу концентрировали при пониженном давлении при максимальной внутренней температуре 70°С, собирали около 600 мл дистиллята. Раствор охлаждали до 20°С и добавляли изогексан (160 мл). После осуществления кристаллизации добавляли дополнительное количество изогексана (320 мл). Продукт подвергали циклическому воздействию температуры до 40°С, суспензию охлаждали до 5°С и продукт выделяли путем фильтрации и высушивали при 40°С. Выход: 127,9 г, 86,5%; ЯМР-спектр (СDСl3) 1,0-1,2 (m, 2H), 1,45 (s, 9H), 1,65 (d, 2H), 1,75-1,9 (m, 2H), 2,45 (s, 3Н), 2,55-2,75 (m, 2H) 3,85 (d, 1H), 4,0-4,2 (br s, 2H), 7,35 (d, 2H), 7,8 (d, 2H); Масс-спектр [ESI]: (MNa)+=392.
Пример 2
Получение гидрохлоридной соли 7-бензилокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина (гидрохлоридная соль соединения формулы VI)
7-Бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (20,00 г) смешивали с анизолом (190 мл) и N,N-диизопропилэтиламином (13,74 г). Реакционную смесь инертировали азотом и охлаждали до 15°С. К реакционной смеси загружали оксихлорид фосфора (14,12 г) в течение 15 минут, затем добавляли анизол (10 мл) в качестве промывки. Реакционную смесь перемешивали в течение 15 минут при 15°С и затем нагревали до 80°С в течение 90 минут. После этого реакционную смесь перемешивали при 80°С в течение одного часа. К реакционной смеси добавляли раствор 4-бром-2-фторанилина (16,8 г) в анизоле (20 мл) в течение 40 минут. Реакционную смесь перемешивали при 80°С в течение 90 минут. После этого реакционную смесь охлаждали до 25°С и продукт выделяли путем фильтрации. Выход: 26,9 г, 84%; ЯМР-спектр (ДМСОd6, CD3COOD) 4,0 (s, 3Н), 5,37 (s, 2H), 7,35-7,5 (m, 4H), 7,52-7,62 (m, 4H), 7,8 (d, 1H), 8,14 (s, 1H), 8,79 (s, 1H); Масс-спектр [ESI] (N+H)+=454,0591.
Исходное вещество 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он получали следующим образом:
Смесь ванилиновой кислоты (200 г), ацетонитрила (600 мл) и N-этилдиизопропиламина (580 мл) нагревали в колбе с обратным холодильником. После этого добавляли бензилбромид (347 мл) в течение 3 часов. Реакционную смесь выдерживали в колбе с обратным холодильником в течение 15 часов. Добавляли триэтиламин (50 мл) и реакционную смесь выдерживали в колбе с обратным холодильником дополнительно в течение 30 минут. Добавляли ацетонитрил (400 мл) и реакционную смесь нагревали до 81°С. Добавляли воду (300 мл) и реакционную смесь охлаждали до 45°С. Реакционную смесь выдерживали при 45°С в течение 30 минут до тех пор, пока не осуществлялась кристаллизация. После этого реакционной смеси позволяли охладиться до 24°С и затем дополнительно охлаждали до 8°С и продукт (бензил 4-(бензилокси)-3-метоксибензоат) выделяли путем фильтрации. Твердое вещество промывали водой (3×500 мл) и затем высушивали в вакууме при 45°С. Выход: 387 г, 93,4%; ЯМР-спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2H), 5,3 (s, 2H), 6,9 (d, 1H), 7,2-7,4 (m, 10Н), 7,6-7,7 (m, 2H); Масс-спектр (M+H)+=349,2.
Бензил 4-(бензилокси)-3-метоксибензоат (78 г) смешивали с дихлорметаном (580 мл), водой (72 мл) и ледяной уксусной кислотой (288 мл). Смесь охлаждали до 10°С. Добавляли концентрированную серную кислоту (108 мл) контролируемым образом, поддерживая температуру реакционной смеси ниже 25°С. После этого добавляли концентрированную азотную кислоту (17,5 мл), поддерживая температуру реакционной смеси ниже 20°С. После этого реакционную смесь перемешивали при 20°С в течение 23 часов. Нижний водный слой удаляли и органический слой промывали водой (290 мл). Органический слой отделяли и дистиллировали до 270 мл при атмосферном давлении. К реакционной смеси добавляли изопропанол (750 мл) при 45°С. Затем реакционную смесь нагревали до 40°С и перемешивали при этой температуре в течение 15 минут. После этого полученную суспензию охлаждали до 20°С, затем до 5°С и выдерживали при этой температуре в течение одного часа. Продукт (бензил 4-(бензилокси)-5-метокси-2-нитробензоат) выделяли путем фильтрации, промывали изопропанолом (200 мл) и высушивали при температуре ниже 25°С. Выход: 78,4 г, 89,6%; ЯМР-спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2H), 5,3 (s, 2H), 7,1 (s, 1H), 7,3-7,4 (m, 10 Н), 7,5 (s, 1H); Масс-спектр (M+H)+ 394,1.
Бензил 4-(бензилокси)-5-метокси-2-нитробензоат (77 г) растворяли в ацетонитриле (882 мл). К раствору добавляли дитионит натрия (160,5 г) и температуру доводили до 25°С. После этого добавляли воду (588 мл), поддерживая температуру равной 25°С. Значение рН поддерживали равным 6, используя 8,8 М гидроксид натрия при восстановлении. После этого взвесь нагревали до 65°С и нижнюю водную фазу удаляли. После этого добавляли концентрированную соляную кислоту (35% мас./мас., 7,25 мл). Взвеси позволяли охладиться до 40°С и затем до 20°С. Добавляли раствор гидроксида натрия (47% мас./мас., 12,4 мл) и взвесь охлаждали до 0°С. Продукт (бензил 2-амино-4-(бензилокси)-5-метоксибензоат) выделяли путем фильтрации, промывали водой (2×196 мл) и после этого высушивали при 40°С в вакууме. Выход: 66,2 г, 92,4%; ЯМР-спектр (CDCl3) 3,8 (s, 3Н), 5,1 (s, 2H), 5,3 (s, 2H), 6,2 (s, 1H), 7,3-7,4 (m, 10Н); Масс-спектр (M+Н)+=364,1.
Смешивали бензил 2-амино-4-(бензилокси)-5-метоксибензоат (5,55 кг), формамидин ацетат (2,2 кг) и изобутанол (33,3 л). Затем реакционную смесь нагревали до 97°С и перемешивали при этой температуре в течение 6 часов. После этого реакционную смесь охлаждали до 25°С в течение по меньшей мере часа и затем перемешивали при этой температуре в течение 30 минут. Продукт (7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он) выделяли путем фильтрации, промывали изобутанолом (6,1 л) и высушивали в вакуум-сушильном шкафу при температуре от 40 до 45°С. Выход: 4,25 кг, 98%; ЯМР-спектр (ДМСОd6) 3,9 (s, 3Н), 5,3 (s, 2H), 7,3 (s, 1H), 7,3-7,5 (m, 6H), 8,0 (s, 1H); Масс-спектр (М+H)+=283,1.
Исходное вещество 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он дополнительно получали следующим образом:
Смесь ванилиновой кислоты (20 г), ацетонитрила (60 мл) и N-этилдиизопропиламина (58 мл) нагревали в колбе с обратным холодильником. После этого добавляли бензилбромид (34,7 мл) в течение 15 минут. Реакционную смесь выдерживали в колбе с обратным холодильником в течение приблизительно 10 часов. Добавляли триэтиламин (5 мл) и реакционную смесь выдерживали в колбе с обратным холодильником дополнительно в течение 30 минут. Добавляли ацетонитрил (40 мл) и воду (30 мл) и реакционную смесь охлаждали до 45°С.Реакционную смесь выдерживали при 45°С до тех пор, пока не осуществлялась кристаллизация. После этого реакционной смеси позволяли охладиться до 24°С и затем дополнительно охлаждали до 8°С и продукт (бензил 4-(бензилокси)-3-метоксибензоат) выделяли путем фильтрации. Твердое вещество промывали водой (3×50 мл) и затем высушивали в вакууме при 45°С. Выход: 38,7 г, 93%; ЯМР-спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2H), 5,3 (s, 2H), 6,9 (d, 1H), 7,2-7,4 (m, 10Н), 7,6-7,7 (m, 2H); Масс-спектр (M+H)+=349,2.
Бензил 4-(бензилокси)-3-метоксибензоат (135 г) растворяли в дихлорметане (339 мл). Добавляли ледяную уксусную кислоту (175,5 г) и смесь охлаждали до 10°С. Добавляли концентрированную серную кислоту (151,6 г) контролируемым образом, поддерживая температуру реакционной смеси ниже 25°С. После этого добавляли концентрированную азотную кислоту (61,6 г) в течение 15 минут, поддерживая температуру реакционной смеси ниже 25°С. Затем реакционную смесь нагревали до 40°С и перемешивали в течение 3 часов. Нижний водный слой удаляли и органический слой два раза промывали водой (2×168 мл). Органический слой дистиллировали при атмосферном давлении для удаления дихлорметана (186 мл). К реакционной смеси добавляли изопропанол (339 мл) при 40°С. Реакционную смесь выдерживали при 40°С в течение 15 минут. После этого полученную суспензию охлаждали до 20°С в течение 30 минут, затем до 5°С и выдерживали при этой температуре в течение одного часа. Продукт (бензил 4-(бензилокси)-5-метокси-2-нитробензоат) выделяли путем фильтрации, промывали изопропанолом (336 мл) и высушивали при температуре ниже 25°С. Выход: 135,7 г, 89,6%; ЯМР-спектр (CDCl3) 3,9 (s, 3Н), 5,2 (s, 2H), 5,3 (s, 2H), 7,1 (s, 1H), 7,3-7,4 (m, 10Н), 7,5 (s, 1H); Масс-спектр (M+H)+ 394.1.
Бензил 4-(бензилокси)-5-метокси-2-нитробензоат (90 г) загружали к ацетонитрилу (660 г). К раствору добавляли 85% дитионит натрия (75 г) и температуру доводили до 20°С. После этого добавляли воду (516 г), поддерживая температуру равной 20°С. После этого взвесь нагревали до 65°С и перемешивали в течение 30 минут. Добавляли дитионит натрия (75 г) и смесь перемешивали дополнительно в течение 30 минут. Нижнюю водную фазу удаляли. После этого добавляли концентрированную соляную кислоту (33% мас./мас., 12,48 г) для установления значения рН<1. Суспензию выдерживали в течение 1 часа. Взвесь охлаждали до 20°С в течение 30 минут. Добавляли раствор гидроксида натрия (20% мас./мас., 59,29 г) для получения значения рН 10. Взвесь охлаждали до 0°С и перемешивали в течение одного часа. Продукт (бензил 2-амино-4-(бензилокси)-5-метоксибензоат) выделяли путем фильтрации, два раза промывали водой (2×222 мл) и после этого высушивали при 60°С в вакууме. Выход: 78,81 г, 95%; ЯМР-спектр (СDСl3) 3,8 (s, 3Н), 5,1 (s, 2H), 5,3 (s, 2H), 6,2 (s, 1H), 7,3-7,4 (m, 10Н); Масс-спектр (M+H)+=364,1.
Смешивали бензил 2-амино-4-(бензилокси)-5-метоксибензоат (80,0 г), формамидин ацетат (32,0 г) и изобутанол (480 мл). Затем реакционную смесь нагревали до 97°С и перемешивали при этой температуре в течение 6 часов. После этого реакционную смесь охлаждали до 25°С в течение по меньшей мере часа и затем перемешивали при этой температуре в течение 30 минут. Продукт (7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он) выделяли путем фильтрации, промывали изобутанолом (64,2 г) и высушивали в вакуум-сушильном шкафу при температуре от 40 до 45°С. Выход: 60,8 г, 98%; ЯМР-спектр (ДМСОd6) 3,9 (s, 3Н). 5,3 (s, 2H), 7,3 (s, 1H), 7,3-7,5 (m, 6H), 8,0 (s, 1H); Масс-спектр (М+Н)+=283,1.
Пример 3
Получение гидрохлоридной соли 7-бензилокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина (гидрохлоридная соль соединения формулы VI)
7-Бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (20,00 г) смешивали с толуолом (190 мл) и N,N-диизопропилэтиламином (13,74 г). Реакционную смесь инертировали азотом и охлаждали до 15°С. К реакционной смеси загружали оксихлорид фосфора (19,8 г) в течение 15 минут, затем добавляли толуол (10 мл) в качестве промывки. Реакционную смесь перемешивали в течение 15 минут при 15°С и затем нагревали до 80°С в течение 90 минут. После этого реакционную смесь перемешивали при 80°С в течение двух часов. К реакционной смеси добавляли раствор 4-бром-2-фторанилина (16,8 г) в толуоле (40 мл) в течение 40 минут, затем добавляли толуол (10 мл) в качестве промывки. После этого реакционную смесь перемешивали при 80°С в течение 4 часов. После этого реакционную смесь охлаждали до 25°С и продукт выделяли путем фильтрации. Фильтровальный осадок два раза промывали водой (2×40 мл). Выход: 34,37 г, 87%; ЯМР-спектр (ДМСОd6, CD3COOD) 4,0 (s, 3Н), 5,37 (s, 2H), 7,35-7,5 (m, 4H), 7,52-7,62 (m, 4H), 7,8 (d, 1H), 8,14 (s, 1H), 8,79 (s, 1H); Масс-спектр [ESI] (M+H)+=454,0591.
Пример 4
Получение соли трифторуксусной кислоты с 7-гидрокси-4-(4-бром-2-(фторанилино)-6-метоксихиназолином (соль трифторуксусной кислоты с соединением формулы IX)
7-Бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (100 г), триэтиламин (59,3 мл) и толуол (650 мл) загружали в сосуд и инертировали азотом. Содержимое нагревали до 40°С и загружали в течение приблизительно 40 минут к раствору оксихлорида фосфора (97,7 г) в толуоле (400 мл), выдерживали при 73°С в сосуде, инертированном азотом. После этого реакционную смесь выдерживали при температуре около 73°С в течение приблизительно 90 минут. 4-Бром-2-фторанилин (84,1 г) растворяли в толуоле (250 мл) и загружали к реакционной смеси при 73°С и продолжали перемешивать при этой температуре приблизительно в течение 4 часов. После этого к реакционной смеси добавляли трифторуксусную кислоту (350 мл) при 73°С и реакционную смесь перемешивали при 73°С в течение 6 часов и затем охлаждали до 60°С. К реакционной смеси добавляли воду (1750 мл) и температуру поддерживали при 60°С приблизительно в течение 30 минут и затем нагревали до 70°С и перемешивали при 70°С приблизительно в течение 22 часов. После этого реакционную смесь охлаждали до 20°С и продукт выделяли путем фильтрации, промывали водой (200 мл) и высушивали при 50°С. Выход: 120 г, 93%; ЯМР-спектр (ДМСOd6) 4,0 (s, 3Н), 7,24 (s, 1H), 7,56 (m, 2H), 7,78 (d, 1H), 8,02 (s, 1H), 8,73 (s, 1H); Масс-спектр (М+H)+=454,0591.
Пример 5
Получение соли трифторуксусной кислоты с 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолином (соль трифторуксусной кислоты с соединением формулы IX)
7-Бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (15 г), триэтиламин (9,0 мл) и толуол (90 мл) загружали в сосуд и инертировали азотом. Содержимое выдерживали при температуре окружающей среды и загружали в течение приблизительно 40 минут к раствору оксихлорида фосфора (14,7 г) в толуоле (60 мл), выдерживали при 73°С в сосуде, инертированном азотом. Затем добавляли толуольную (7,5 мл) промывочную линию. После этого реакционную смесь выдерживали при температуре около 73°С в течение приблизительно 90 минут. 4-Бром-2-фторанилин (12,6 г) растворяли в толуоле (30 мл) и загружали к реакционной смеси при 73°С и продолжали перемешивать при этой температуре приблизительно в течение 4 часов. После этого к реакционной смеси добавляли трифторуксусную кислоту (60 мл) при 73°С и реакционную смесь перемешивали при 73°С в течение 6 часов и затем охлаждали до 60°С. Гидроксид калия (48-50% мас./мас., 16,1 мл) в воде (10,5 мл) загружали приблизительно в течение 30 минут, затем в течение часа выдерживали при 60°С. К реакционной смеси добавляли воду (180 мл) приблизительно в течение 70 минут, затем добавляли затравку соли 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина с трифторуксусной кислотой (0,13 г). Партию выдерживали при 60°С приблизительно в течение 60 минут и затем добавляли воду (60 мл) приблизительно в течение 20 минут. Реакционную смесь выдерживали приблизительно в течение двух часов, затем охлаждали до 20°С и продукт выделяли путем фильтрации, промывали толуолом (50 мл) и метанолом/водой (1:10, 50 мл) и высушивали при 50°С. Выход: 22 г, 89%; ЯМР-спектр (ДМСОd6) 4,0 (s, 3Н), 7,24 (s, 1H), 7,56 (m, 2H), 7,78 (d, 1H), 8,02 (s, 1H), 8,73 (s, 1H); Масс-спектр (М+Н)+=454,0591.
Пример 6
Получение хлористоводородной соли 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина (хлористоводородная соль соединения формулы IX)
7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-он (30,00 г) смешивали с гидрохлоридом триэтиламина (2,99 г), анизолом (285 мл) и N,N-диизопропилэтиламином (20,71 г). Реакционную смесь инертировали азотом и охлаждали до 15°С. К реакционной смеси добавляли оксихлорид фосфора (21,4 г) в течение 15 минут, затем добавляли анизольную (30 мл) промывку. После этого реакционную смесь перемешивали в течение 15 минут при 15°С и затем нагревали до 80°С в течение 90 минут. Реакционную смесь перемешивали при 80°С в течение одного часа. К реакционной смеси добавляли раствор 4-бром-2-фторанилина (25,2 г) в анизоле (15 мл) в течение 25 минут. Реакционную смесь перемешивали в течение 4 часов при 80°С. Водный хлористый водород (35% мас./мас., 122 мл) и уксусную кислоту (198 мл) загружали к реакционной смеси. Реакционную смесь перемешивали в течение 3 часов и затем анизольный слой удаляли. Реакционную смесь охлаждали до 25°С и твердое вещество выделяли путем фильтрации. Выход: 13,9 г, 54%; ЯМР-спектр (ДМСОd6) 4,0 (s, 3H), 7,43 (s, 1Н), 7,5 (m, 2H), 7,7 (d, 1H), 8,37 (s, 1H), 8,72 (s, 1H); Масс-спектр (M+H)+=454,0591.
Пример 7
Получение хлористоводородной соли 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина (хлористоводородная соль соединения формулы IX)
Оксихлорид фосфора (6,0 мл) добавляли в течение 60 минут к перемешиваемой взвеси 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-она (10,0 г) и N,N-диизопропилэтиламина (7,45 мл) в толуоле (105 мл) при 20°С. После перемешивания реакционной смеси в течение 30 минут при 20°С реакционную смесь нагревали в течение 90 минут до 73°С и затем перемешивали дополнительно в течение 3 часов при этой температуре. К реакционной смеси добавляли 4-бром-2-фторанилин (8,4 г) в толуоле (20 мл) при 73°С, затем добавляли толуольную промывку (5 мл). К реакционной смеси добавляли трифторуксусную кислоту (35 мл, 3,5 об.) в течение 10 минут при 73°С и после этого реакционную смесь перемешивали при этой температуре в течение 5 часов. После этого реакционную смесь охлаждали до 60°С и добавляли воду (175 мл) в течение 15 минут. После этого реакционную смесь нагревали до 68°С и перемешивали при этой температуре в течение 8 часов. После этого реакционную смесь охлаждали до 20°С в течение 1 часа и продукт отфильтровывали и промывали водой (20 мл). Выход: 11,56 г, 90%; ЯМР-спектр (ДМСОd6) 4,0 (s, 3Н), 7,43 (s, 1Н), 7,5 (m, 2H), 7,7 (d, 1H), 8,37 (s, 1H), 8,72 (s, 1H); Масс-спектр (М+Н)+=454,0591.
Пример 8
Получение соли трифторуксусной кислоты с 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолином (соль трифторуксусной кислоты с соединением формулы IX)
Оксихлорид фосфора (6,0 мл) добавляли в течение 15 минут к перемешиваемой взвеси 7-бензилокси-6-метокси-3,4-дигидрохиназолин-4-она (10,0 г) и триэтиламина (5,9 мл) в толуоле (105 мл) при 73°С и реакционную смесь перемешивали дополнительно в течение 3 часов. К реакционной смеси добавляли 4-бром-2-фторанилин (8,4 г) в толуоле (20 мл) при 73°С, затем добавляли толуольную промывку (5 мл). После этого к реакционной смеси добавляли трифторуксусную кислоту (35 мл, 3,5 об.) в течение 10 минут при 73°С и после этого реакционную смесь перемешивали при этой температуре дополнительно в течение 5 часов. Реакционную смесь охлаждали до 60°С и добавляли воду (175 мл) в течение 15 минут. После этого реакционную смесь нагревали до 68°С и перемешивали при этой температуре в течение 8 часов. Взвесь охлаждали до 20°С в течение 1 часа и продукт отфильтровывали и промывали водой (20 мл). Выход: 11,24 г, 87%; ЯМР-спектр (ДМСОd6) 8,72 (1H, s), 8,02 (1H, s), 7,76-7,73 (1H, m), 7,56-7,50 (2H, m), 7,25 (1H, s), 3,97 (3Н, s); Масс-спектр (М+Н)+=454,0591.
Пример 9
Получение 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина (соединение формулы X)
7-Гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин (100 г) и карбонат калия (113,8 г) суспендировали в N-метилпирролидиноне (1070 мл) и перемешивали в течение 10 минут перед добавлением 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидина (152,2 г).
Затем реакционную смесь нагревали до 95°С в течение 4 часов, после этого снова охлаждали до 70°С. После этого добавляли воду (1922 мл) в течение 15 минут. Реакционную смесь выдерживали при 73°С в течение 1 часа, затем охлаждали до 40°С и продукт выделяли путем фильтрации. Продукт промывали водой (549 мл), взвесь промывали этилацетатом (549 мл) при 50°С в течение 1 часа и затем промывали этилацетатом (275 мл) и высушивали при 50°С. Выход: 137 г, 86%; ЯМР-спектр (ДМСОd6) 1,15-1,3 (m, 2H), 1,46 (s, 9H), 1,8 (d, 2H), 2,0-2,1 (m, 1Н), 2,65-2,9 (m, 2H) 3,95 (s, 3H), 4,02 (br s, 2H), 4,05 (d, 2H), 7,2 (s, 1H), 7,48 (d, 1H), 7,55 (t, 1H), 7,65 (d, 1H), 7,8 (d, 1H), 8,35 (s, 1H), 9,55 (br s, 1H); Масс-спектр [ESI] (М+H)+=561-563.
Пример 10
Получение 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина (соединение формулы X)
7-Гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин (5,0 г) и карбонат калия (5,7 г) суспендировали в N-метилпирролидиноне (53,5 мл) и перемешивали в течение 10 минут. После этого добавляли 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин (7,6 г). Затем реакционную смесь нагревали до 95°С и перемешивали при этой температуре в течение 3,5 часов, после этого снова охлаждали до 70°С. Добавляли изопропанол (25 мл) и затем добавляли воду (75 мл) в течение 15 минут. После этого реакционную смесь перемешивали при 73°С в течение 1 часа, затем охлаждали до 40°С и продукт выделяли путем фильтрации. Продукт промывали водой (27,4 мл) и высушивали при 50°С. Выход: 6,72 г, 87,2%; ЯМР-спектр (ДMCOd6) 1,15-1,3 (m, 2H), 1,46 (s, 9H), 1,8 (d, 2H), 2,0-2,1 (m, 1H), 2,65-2,9 (m, 2H) 3,95 (s, 3H), 4,02 (br s, 2H), 4,05 (d, 2H), 7,2 (s, 1H), 7,48 (d, 1H), 7,55 (t, 1H), 7,65 (d, 1H), 7,8 (d, 1H), 8,35 (s, 1H), 9,55 (br s, 1H); Масс-спектр [ESI] (М+Н)+ 561-563.
Пример 11
Получение 7-(1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина (соединение формулы X)
7-Гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолин (9,7 г), гидроксид натрия (47% мас./мас., 5,0 мл) и Adogen® 464 (1,5 г) добавляли к воде (50 мл) при перемешивании. После этого к реакционной смеси добавляли 1-(трет-бутоксикарбонил)-4-(4-метилфенилсульфонилоксиметил)пиперидин (10,0 г) в виде раствора в толуоле (35 мл) и нагревали до 70°С в течение 18 часов. После этого реакционную смесь охлаждали до 20°С и продукт выделяли путем фильтрации. Затем продукт промывали толуолом (20 мл) и высушивали при 50°С. Выход: 8,72 г, 77%; ЯМР-спектр (ДМСОd6 1,15-1,3 (m, 2H), 1,46 (s, 9H), 1,8 (d, 2H), 2,0-2,1 (m, 1H), 2,65-2,9 (m, 2H) 3,95 (s, 3H), 4,02 (br s, 2H), 4,05 (d, 2H), 7,2 (s, 1H), 7,48 (d, 1H), 7,55 (t, 1H), 7,65 (d, 1H), 7,8 (d, 1H), 8,35 (s, 1H), 9,55 (br s, 1H); Масс-спектр [ESI] (M+H)+=561-563.
Пример 12
Получение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (100 г), воду (80 мл), муравьиную кислоту (120 мл) и водный формальдегид (38% мас./мас., 28,2 г) добавляли в сосуд, оборудованный подвесной мешалкой, дефлегматором и продуваемый азотом. Реакционную смесь нагревали до 80°С в течение 90 минут и перемешивали при этой температуре в течение 5 часов. После этого реакционную смесь охлаждали до 20°С и добавляли тетрагидрофуран (500 мл). Реакционную смесь нагревали до 40°С и добавляли гидроксид натрия (47% мас./мас., 265 мл), затем добавляли воду (60 мл). Водную фазу отделяли и отбрасывали. Органическую фазу доводили до 60°С и добавляли воду (300 мл) и бутилацетат (300 мл). Полученную смесь перемешивали при 60°С в течение 15 минут и затем водную фазу отделяли и отбрасывали. После этого к органической фазе добавляли воду (400 мл), ее перемешивали при 60°С в течение 15 минут и затем водную фазу отделяли и отбрасывали. К органической фазе добавляли бутилацетат (300 мл) и тетрагидрофуран (50 мл) и устанавливали для дистилляции при давлении воздуха. Дистилляцию останавливали, когда температура содержимого достигала 104°С. После этого взвесь охлаждали до 20°С и выдерживали в течение 2 часов, затем продукт выделяли путем фильтрации. Продукт промывали бутилацетатом (300 мл) и высушивали при 50°С. Выход: 76,7 г, 90,6%; ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5H, m), 2,15 (3H, s), 2,76 (2Н, m), 3,63 (3H, s), 3,97 (2Н, d), 7,38 (1H, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37 (1H, s); Масс-спектр (M+H)+=475.
Пример 13
Получение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (35,0 г), воду (28 мл), муравьиную кислоту (42 мл) и водный формальдегид (37% мас./мас., 8,2 г) добавляли в сосуд, оборудованный подвесной мешалкой, дефлегматором и продуваемый азотом. Реакционную смесь нагревали до 80°С и перемешивали при этой температуре в течение 5 часов. После этого реакционную смесь охлаждали до 40°С и добавляли тетрагидрофуран (175 мл). Добавляли гидроксид натрия (47% мас./мас., 61,9 мл) при 40°С, затем добавляли воду (21 мл). Затем водную фазу отделяли и отбрасывали. К органической фазе добавляли воду (420 мл) при 40°С в течение 30 минут. После этого взвесь охлаждали до 20°С, затем продукт выделяли путем фильтрации. Продукт промывали водой (175 мл) и высушивали при 50°С. Выход: 27,1 г, 91,4%; ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5H, m), 2,15 (3H, s), 2,76 (2Н, m), 3,63 (3H, s), 3,97 (2Н, d), 7,38 (1H, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37 (1H, s); Масс-спектр (М+Н)+=475.
Пример 14
Получение 4-(4-бром-2-(фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (100 г), воду (80 мл), муравьиную кислоту (120 мл) и водный формальдегид (37% мас./мас., 26,7 г) добавляли в сосуд, оборудованный подвесной мешалкой, дефлегматором и продуваемый азотом. Реакционную смесь нагревали до 80°С в течение 90 минут и перемешивали при этой температуре в течение 5 часов. После этого реакционную смесь охлаждали до 60°С и добавляли метанол (800 мл), затем добавляли гидроксид калия (49% мас./мас., 228 мл) в течение 2 часов. Взвесь охлаждали до 20°С в течение 2 часов, затем продукт выделяли путем фильтрации. Продукт два раза промывали водным метанолом (2:1 метанол:вода, 300 мл) и высушивали при 50°С. Выход: 79,6 г, 94%: ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5Н, m), 2,15 (3Н, s), 2,76 (2Н, m), 3,63 (3Н, s), 3,97 (2Н, d), 7,38 (1H, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37 (1H, s); Масс-спектр (M+H)+=475.
Пример 15
Получение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (100 г), воду (45 мл), муравьиную кислоту (120 мл) и водный формальдегид (37% мас./мас., 101,8 г) добавляли в сосуд, оборудованный подвесной мешалкой и дефлегматором и продуваемый азотом. Реакционную смесь нагревали до 80°С в течение 90 минут и перемешивали при этой температуре в течение 5 часов. После этого реакционную смесь охлаждали до 60°С и метанол (800 мл) добавляли, затем добавляли гидроксид калия (49% мас./мас., 228 мл) в течение 2 часов. Взвесь охлаждали до 20°С в течение 2 часов, затем продукт выделяли путем фильтрации. Продукт два раза промывали водным метанолом (2:1 метанол:вода, 300 мл) и высушивали при 50°С. Выход: 79,6 г, 94%: ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5Н, m), 2,15 (3Н, s), 2,76 (2Н, m), 3,63 (3Н, s), 3,97 (2Н, d), 7,38 (1H, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37 (1H, s); Масс-спектр (M+H)+=475.
Пример 16
Получение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (36 г @ 100% мас./мас.), воду (16 мл), муравьиную кислоту (44 мл) и водный формальдегид (37% мас./мас., 36,4 г) добавляли в сосуд, оборудованный подвесной мешалкой и дефлегматором и продуваемый азотом. Реакционную смесь нагревали до 80°С в течение 90 минут и перемешивали при этой температуре в течение 7 часов. После этого реакционную смесь охлаждали до 60°С и добавляли метанол (376 мл), затем добавляли гидроксид калия (49% мас./мас., 86 мл) в течение 2 часов. Взвесь затравливали ZD6474 (метанолатная форма, 300 мг) и охлаждали до 20°С в течение 2 часов, затем продукт выделяли путем фильтрации. Продукт два раза промывали водным метанолом (80:20 метанол:вода, 67 мл) и высушивали при температуре окружающей среды. Выход: 32,4 г, 95%: ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5Н, m), 2,15 (3Н, s), 2,76 (2Н, m), 3,63 (3Н, s), 3,97 (2Н, d), 7,38 (1Н, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37(1H, s); Масс-спектр (M+H)+=475.
Пример 17
Очистка 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина (ZD6474)
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин, полученный по любому из Примеров 12-16 (100 г), суспендировали в тетрагидрофуране (500 мл), воде (250 мл) и бутилацетате (400 мл) и нагревали в колбе с обратным холодильником для осуществления растворения. Затем смесь охлаждали до 60°С и водную фазу отделяли и отбрасывали. Органическую фазу фильтровали. К органическим фильтратам добавляли тетрагидрофуран (50 мл) и бутилацетат (600 мл) и затем нагревали до перегонки при давлении воздуха до достижения внутренней температуры 106°С. После этого взвесь охлаждали до 5°С, фильтровали и промывали этилацетатом (200 мл). Продукт высушивали при 50°С. Выход: 91,8 г, 91,8%: ЯМР-спектр (пиридин-d5) 1,49 (2Н, m), 1,75-1,90 (5Н, m), 2,15 (3Н, s), 2,76 (2Н, m), 3,63 (3Н, s), 3,97 (2Н, d), 7,38 (1H, ddd), 7,49 (1H, dd), 7,64 (1H, s), 7,88 (1H, t), 7,89 (1H, s), 9,01 (1H, s), 10,37 (1H, s); Масс-спектр (M+H)+=475.
Пример 18
Получение 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)1-хиназолина (ZD6474)
7-(1-Трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолин (40 г), воду (16 мл), муравьиную кислоту (43 мл) и водный формальдегид (37% мас./мас., 33 мл) добавляли в сосуд, оборудованный подвесной мешалкой, дефлегматором и термометром.
Реакционную смесь нагревали до 81°С и перемешивали при этой температуре в течение 5 часов. Реакционную смесь охлаждали до 60°С и добавляли тетрагидрофуран (178 мл). Температуру реакционной смеси доводили до 40°С и добавляли гидроксид калия (49% мас./мас., 84 мл), затем добавляли воду (22 мл). Водную фазу отделяли и отбрасывали. Органическую фазу доводили до 60°С и добавляли воду (107 мл) и бутилацетат (107 мл). Водную фазу отделяли и отбрасывали. Органическую фазу фильтровали, затем через тетрагидрофурановую промывку (18 мл). Температуру фильтратов доводили до 60°С и добавляли бутилацетат (107 мл). Реакционную смесь устанавливали для дистилляции при давлении воздуха. Дистилляцию останавливали, когда температура содержимого достигала 106°С. Взвесь охлаждали до 65°С и добавляли тетрагидрофуран (107 мл). Взвесь охлаждали до 0-5°С и выдерживали в течение 1 часа, затем продукт выделяли путем фильтрации. Продукт промывали этилацетатом (72 мл) и высушивали при 50°С. Выход: 24,82 г, 80,3%.
Пример 19
Порошковая дифракция рентгеновских лучей безводного ZD6474
В способах согласно настоящему изобретению синтезируется безводная форма ZD6474. Безводная форма ZD6474 характеризуется порошковой дифракцией рентгеновских лучей и характеризуется таким образом, что по меньшей мере одно из следующих значений 2-тета, измеренное с помощью СuКα излучения: 15,0° и 21,4°. Безводная форма ZD6474 характеризуется с помощью СuКα порошковой рентгенограммы, приведенной на фигуре 1. Десять наиболее видных пиков приведены в таблице 1.
10-25
3-10
1-3
с(сильная)
ср (средняя)
сл (слабая)
Аналитический прибор: Siemens D5000, калиброванный с помощью кварцев.
Спектры порошковой дифракции рентгеновских лучей определяли путем установки образца кристаллического вещества ZD6474 на тонкую пластинку единичного кремниевого кристалла (SSC) Siemens и распыления образца тонким слоем с помощью предметного стекла. Образец вращали при 30 оборотов в минуту (для улучшения статистики подсчетов) и облучали рентгеновскими лучами, генерируемыми медной длинноострофокусной трубкой, которая работает при 40 кВ и 40 мА, используя СuКα облучение при длине волны 1,5406 ангстрем. Коллимированный источник рентгеновских лучей пропускали через автоматически изменяемую дивергентую щель, установленную при V20, и отраженное излучение пропускали через противорассеивающую щель 2 мм и щель детектора 0,2 мм. Образец экспонировали в течение 1 секунды с увеличением 2-тета на 0,02 градуса (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Время испытания составляло 31 минуту и 41 секунду. Прибор оборудован сцинтилляционным счетчиком в качестве детектора. Контрольные и опытные данные записывали с помощью рабочей станции Dell Optiplex 686 NT 4,0 Workstation, используя программное обеспечение Diffract+. Специалист в области порошковой дифракции рентгеновских лучей примет во внимание, что на относительную интенсивность пиков могут оказывать влияние, например, частицы размером более 30 микрон и неединичные характеристические отношения, которые могут влиять на анализ образцов. Специалист в данной области техники также примет во внимание, что на положения отражений могут влиять определенная высота установки образца в дифрактометре и калибровка нуля дифрактометра. Планарность поверхности образца также может оказывать небольшое влияние. Следовательно, приведенные данные порошковой рентгенограммы не должны толковаться как абсолютные значения. Более подробная информация о порошковой дифракции рентгеновских лучей приведена в следующих источниках: Jenkins, R & Snyder, R.L. 'Introduction to X-Ray Powder Diffractometry' John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. & Alexander, L.E. (1974), X-Ray Diffraction Procedures.
Краткое описание фигур
Фигура 1: Порошковая рентгенограмма для безводного ZD6474 - значения 2-тета указаны на горизонтальной оси, а относительные интенсивности спектральных линий (импульсы) указаны на вертикальной оси.
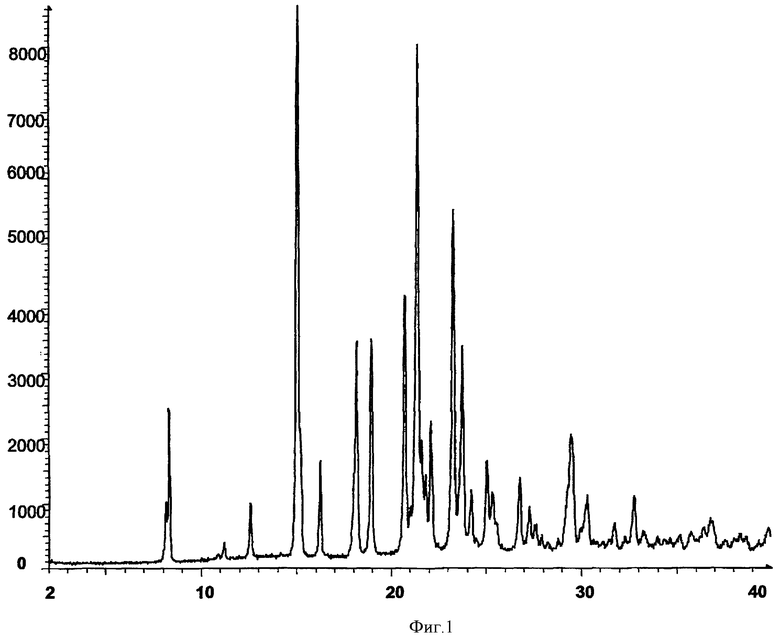
Настоящее изобретения относится к способу получения 4-(4-бром-2-фторанилино)-6-метокси-7(1-метилпиперидин-4-илметокси)хиназолина (ZD6474), имеющего формулу, приведенную ниже, и его солей из соединения формулы X. Изобретение также относится к способу получения промежуточного соединения формулы Х и его солей, а также к способам получения других определенных промежуточных соединений, которые пригодны для получения (ZD6474). 6 н. и 21 з.п. ф-лы, 1 фиг., 2 таб., 19 пр.

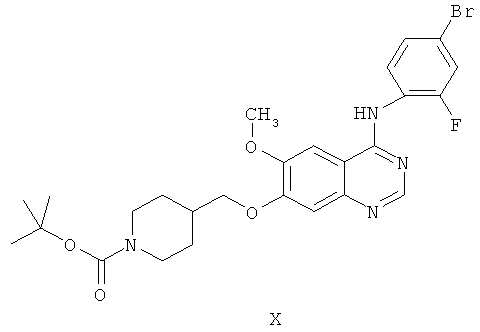
1. Способ получения соединения формулы VI
где R1 представляет собой кислотно-лабильную защитную группу, из соединения формулы VII
включающий следующие стадии:
(ж) взаимодействия соединения формулы VII с подходящим хлорирующим реагентом в присутствии подходящего основания и подходящего растворителя, где реакцию осуществляют путем:
(ж-1) добавления смеси соединения формулы VII и основания в растворителе к смеси хлорирующего реагента в растворителе при температуре в диапазоне от 60 до 90°С в течение приблизительно 60 мин; или
(ж-2) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре окружающей среды в течение приблизительно 15 мин и после этого нагревания реакционной смеси в течение приблизительно 90 мин до температуры в диапазоне от 70 до 90°С и перемешивания реакционной смеси при этой температуре приблизительно в течение 1 ч; или
(ж-3) добавления хлорирующего реагента к смеси соединения формулы VII и основания в растворителе при температуре в диапазоне от 60 до 110°С в течение приблизительно 15 мин, с получением соединения формулы VIII
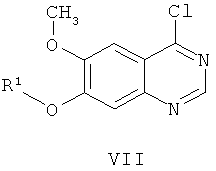 ; и
; и
(з) взаимодействия соединения формулы VIII с 4-бром-2-фторанилином in situ в присутствии растворителя, который используют на стадии (ж), с получением гидрохлоридной соли соединения формулы VI;
и после этого соединение формулы VI, полученное в виде гидрохлоридной соли, при необходимости превращают в свободное основание или в форму другой соли.
2. Способ по п.1, в котором обе стадии (ж) и (з) осуществляют в толуоле.
3. Способ по п.1 или 2, в котором хлорирующий реагент, который используют на стадии (ж), представляет собой оксихлорид фосфора.
4. Способ по п.1 или 2, в котором основание, которое используют на стадии (ж), выбирают из триэтиламина и N,N-диизопропилэтиламина.
5. Способ по п.1 или 2, который дополнительно включает стадию (и) выделения соединения формулы VI.
6. Способ получения 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина соединения формулы IX
из соединения формулы VII
в котором R1 представляет собой кислотно-лабильную защитную группу, включающий стадии превращения соединения формулы VII в соединение формулы VI
путем осуществления способа по одному или более из пп.1-4; и (к) удаления R1 из соединения формулы VI in situ в присутствии растворителя, который используют на стадиях (ж) и (з), с получением соединения формулы IX или его соли;
и после этого соединение формулы IX, полученное в форме свободного основания, при необходимости превращают в солевую форму, и соединение формулы IX, полученное в форме соли, при необходимости превращают в свободное основание или в форму другой соли.
7. Способ по п.6, где R1 представляет собой бензил и на стадии (к) бензильную группу удаляют in situ путем взаимодействия с трифторуксусной кислотой при температуре в диапазоне от 60 до 80°С.
8. Способ по п.6, где R1 представляет собой бензил и бензильную группу удаляют в присутствии трифторуксусной кислоты и соединение формулы IX превращают в соль трифторуксусной кислоты путем добавления гидроксида калия или добавления гидроксида натрия и воды.
9. Способ получения 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина соединения формулы IX
из соединения формулы VII
в котором R1 представляет собой кислотно-лабильную защитную группу,
включающий стадии превращения соединения формулы VII в соединение формулы VI
путем осуществления способа по п.5; и
(л) удаления R1 из соединения формулы VI, получая соединение формулы IX или его соль;
и после этого соединение формулы IX, полученное в форме свободного основания, при необходимости превращают в солевую форму, и соединение формулы IX, полученное в форме соли, при необходимости превращают в свободное основание или в форму другой соли.
10. Способ по п.9, где R1 представляет собой бензил и на стадии (л) бензильную группу удаляют путем реакции с подходящим гидрирующим средством.
11. Способ получения 7-((1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина соединения формулы X
из соединения формулы VII
в котором R1 представляет собой кислотно-лабильную защитную группу, включающий стадии превращения соединения формулы VII в соединение формулы IX
путем осуществления способа по одному или более из пп.6-9; и
(м) взаимодействия соединения формулы IX с соединением формулы II
в присутствии подходящего основания с получением соединения формулы Х или его соли;
и после этого соединение формулы X, полученное в форме свободного основания, при необходимости превращают в солевую форму, и соединение формулы X, полученное в форме соли, при необходимости превращают в свободное основание или в форму другой соли.
12. Способ по п.11, в котором основание, которое используют на стадии (м), выбирают из карбоната натрия, карбоната калия, гидроксида натрия и гидроксида калия.
13. Способ по п.11 или 12, который дополнительно включает стадию (н) выделения соединения формулы X.
14. Способ получения 7-((1-трет-бутоксикарбонил)пиперидин-4-илметокси)-4-(4-бром-2-фторанилино)-6-метоксихиназолина соединения формулы X
из 7-гидрокси-4-(4-бром-2-фторанилино)-6-метоксихиназолина соединения формулы IX
включающий стадии получения соединения формулы IX по одному или более из пп.6-10 и далее
(м) взаимодействия соединения формулы IX с соединением формулы II
в присутствии подходящего основания с получением соединения формулы Х или его соли; и
(н) выделения соединения формулы Х путем:
(н-1) добавления воды и осуществления кристаллизации соединения формулы X, собирания соединения формулы Х и промывки соединения формулы Х водой, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила при температуре в диапазоне от 25 до 55°С;
или
(н-2) добавления воды и спирта, выбранного из метанола, этанола, изопропанола и н-пропанола, и осуществления кристаллизации соединения формулы X, собирания соединения формулы Х и промывки соединения формулы Х смесью воды и спирта, выбранного из метанола, этанола, изопропанола и н-пропанола, затем растворителем, выбранным из этилацетата, бутилацетата и ацетонитрила, при температуре в диапазоне от 25 до 55°С;
и после этого соединение формулы X, полученное в форме свободного основания, при необходимости превращают в солевую форму, и соединение формулы X, полученное в форме соли, при необходимости превращают в свободное основание или в форму другой соли.
15. Способ по п.14, в котором основание, которое используют на стадии (м), выбирают из карбоната натрия и карбоната калия.
16. Способ по любому из пп.11, 12, 14 или 15, в котором соединение формулы II, которое используют на стадии (м), получают посредством
(а) взаимодействия (С1-С6)алкил-4-пиперидинкарбоксилатного соединения формулы III
с ди-трет-бутил дикарбонатом в присутствии толуола или ксилола с образованием первой смеси, содержащей толуол или ксилол, трет-бутанол и соединение формулы IV
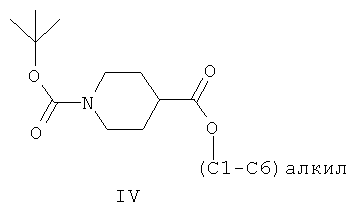
(б) практически полного удаления трет-бутанола из первой смеси;
(в) взаимодействия соединения формулы IV с подходящим восстановителем in situ в присутствии толуола или ксилола с образованием второй смеси, содержащей толуол, восстановленные побочные продукты, включая спиртовые побочные продукты, и соединение формулы V
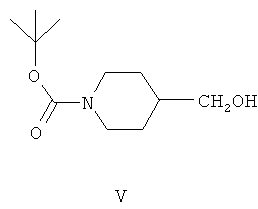
(г) практически полного удаления спиртовых побочных продуктов из второй смеси; и
(д) взаимодействия соединения формулы V с подходящим сульфирующим реагентом in situ с образованием сложного сульфонатного эфира в присутствии подходящего основания и толуола с получением соединения формулы IIа
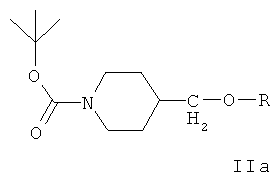
где R представляет собой подходящий сложный сульфонатный эфир;
которое при использовании на стадии д) тозилхлорида в качестве сульфирующего реагента представляет собой соединение формулы II
17. Способ по п.16, в котором (С1-С6)алкил-4-пиперидинкарбоксилатное соединение формулы III представляет собой этил 4-пиперидинкарбоксилат.
18. Способ по п.16, в котором на стадии (в) восстановитель выбирают из натрий бис(2-метоксиэтокси)алюминий гидрида, литийалюминийгидрида и диизобутилалюминий гидрида.
19. Способ по п.18, в котором на стадии (в) восстановитель представляет собой натрий бис(2-метоксиэтокси)алюминий гидрид.
20. Способ по п.16, в котором на стадии (д) основание представляет собой триэтилендиамин.
21. Способ по п.16, который дополнительно включает стадию (е) выделения соединения формулы IIа.
22. Способ по п.21, в котором стадия (е) включает кристаллизацию с использованием системы растворителей толуол и изогексан.
23. Способ получения 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолина, ZD6474:
из соединения формулы X
включающий следующие стадии:
(о) взаимодействия соединения формулы X, полученного способом по любому из пп.11-16, с муравьиной кислотой и формальдегидом или полимером формальдегида с образованием соли муравьиной кислоты с ZD6474;
(п) добавления инертного органического растворителя и подходящего основания с получением свободного основания ZD6474, которое при необходимости превращают в фармацевтически приемлемую соль.
24. Способ по п.23, в котором стадию (о) осуществляют в воде при температуре в диапазоне от 70 до 90°С.
25. Способ по п.23 или 24, в котором инертный органический растворитель, используемый на стадии (п), выбирают из тетрагидрофурана, бутиронитрила и метанола.
26. Способ по п.23 или 24, в котором основание, используемое на стадии (п), выбирают из гидроксида натрия и гидроксида калия.
27. Способ по п.23 или 24, который дополнительно включает дальнейшую очистку ZD6474 из смеси тетрагидрофурана, воды и бутилацетата с получением кристаллической безводной формы.

