Область техники, к которой относится изобретение
Настоящее изобретение относится к усовершенствованному способу получения гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она, который селективно и эффективно ингибирует рост раковых клеток, индуцированный сверхэкспрессией рецептора эпидермального фактора роста, и предупреждает развитие лекарственной резистентности, вызванной мутацией тирозинкиназы, а также к промежуточному соединению, используемому в данном способе.
Предпосылки создания изобретения
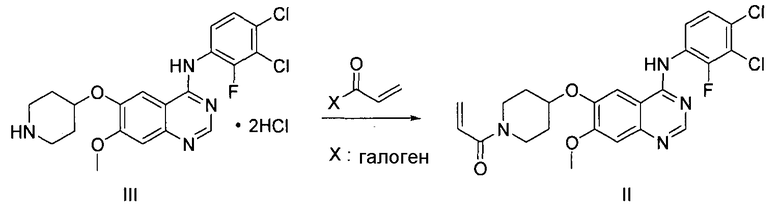
Гидрохлорид 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы I, приведенной ниже, представляет собой важное лекарственное средство, обладающее антипролиферативной активностью, такой как противоопухолевая активность, которое может быть использовано для селективного и эффективного лечения лекарственной резистентности, вызванной мутацией тирозинкиназы. В форме свободного основания, т.е. 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-он, описываемый приведенной ниже формулой II, идентифицирован в CAS регистре под номером 1092364-38-9.
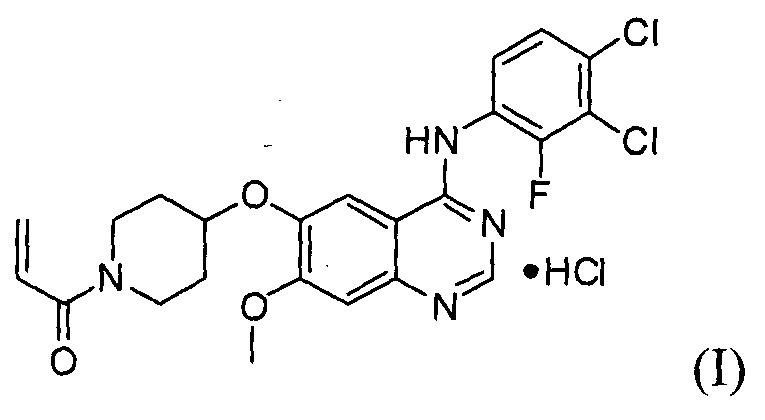
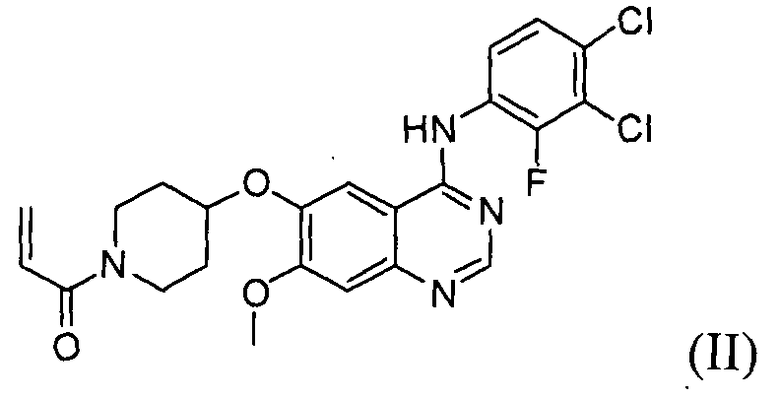
Соединение формулы (II) может быть получено, например, способом, описанным в патенте Кореи № 1013319, а механизм его действия описан на приведенной ниже реакционной схеме I. Соединение формулы (II), получаемое по приведенной ниже реакционной схеме 1, может быть далее подвергнуто реакции с хлористоводородной кислотой с получением соединения формулы (I).
<Схема реакции 1>
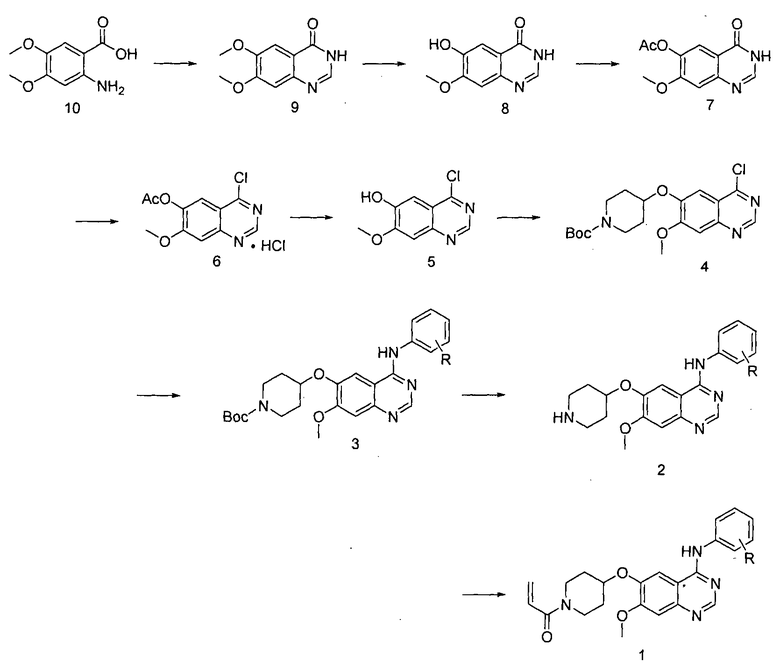
где R означает галоген.
На реакционной схеме I соединение формулы 10 подвергают реакции конденсации с гидрохлоридом формамидина при высокой температуре (например 210°C) с получением соединения формулы 9, которое затем подвергают реакции с L-метионином в органической кислоте (например в метансульфоновой кислоте), в результате которой метильная группа в положении С-6 соединения формулы 9 удаляется с образованием соединения формулы 8.
Далее, соединение формулы 8 защищают посредством реакции в основании (например, в пиридине) и в безводной уксусной кислоте с получением соединения формулы 7, которое затем подвергают реакции с неорганической кислотой (например в тионилхлориде или оксихлориде фосфора) в присутствии каталитического количества N,N-диметилформамида при нагревании с обратным холодильником с получением соединения формулы 6 в виде гидрохлората.
Соединение формулы 6 добавляют при перемешивании к аммиаксодержащему спиртовому раствору (например к 7N аммиаксодержащему раствору метанола), в результате чего удаляется ацетильная группа с образованием соединения формулы 5. Соединение формулы 5 подвергают реакции Мицунобу, используя трет-бутил-4-гидроксипиперидин-1-карбоксилат, с получением соединения формулы 4, которое затем подвергают реакции замещения с анилином в органическом растворителе (например в 2-пропаноле или ацетонитриле) с получением соединения формулы 3. В реакции Мицунобу может быть использован диизопропилазодикарбоксилат, диэтилазодикарбоксилат или ди-трет-бутилазодикарбоксилат и трифенилфосфин. Соединение формулы 3 подвергают реакции с органической или неорганической кислотой (например трифторуксусной кислотой или концентрированной хлористоводородной кислотой) в органическом растворителе (например в дихлорметане), в результате которой трет-бутоксикарбонильная группа удаляется с образованием соединения формулы 2.
Далее, для получения соединения формулы I (т.е. соединения формулы II по настоящему изобретению) соединение формулы 2 подвергают реакции ацилирования с акрилоилхлоридом в смеси органического растворителя (например тетрагидрофурана) и воды, или в дихлорметане, в присутствии неорганического или органического основания (например бикарбоната натрия, пиридина или триэтиламина). Альтернативно, соединение формулы 2 подвергают реакции конденсации с акриловой кислотой в присутствии связующего вещества (например такого как 1-этил-3-(3-диметиламинопропил)карбодиимид ((EDC)) или 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметил европий гексафторфосфат метанаминий (HATU)).
Однако в указанном выше способе стадия получения соединения формулы 9 является потенциально опасной, поскольку проводится при высокой температуре, без растворителя, и не всегда эта реакция проводится одинаково. Кроме того, на стадии получения соединения формулы 5 используется избыточное количество тионилхлорида, что может создавать трудности при проведении следующих стадий процесса. Соответственно, данный способ трудно масштабировать до варианта коммерческого применения.
Основной недостаток описанного выше способа получения соединения формулы (I) заключается в том, что выход готового продукта в реакции с акриловой кислотой очень низкий (т.е. 13%), и данная реакция сопряжена с проведением множества побочных реакций, для которых требуется стадия очистки методом колоночной хроматографии. Кроме того, когда получают соединение формулы 3 по реакции Мицунобу, образуются различные побочные продукты, в связи с чем возникает необходимость в стадии очистки методом колоночной хроматографии. Поскольку в данном случае требуются избыточные количества силикагеля и избыточные количества растворителей для подвижной фазы, указанный выше метод не представляется подходящим для его коммерциализации.
В этой связи, авторы настоящего изобретения предприняли попытку разработать новый способ получения соединения формулы (I) с высокой степенью чистоты и высоким выходом, причем указанный способ является экономически рентабельным и подходящим для его коммерческого применения.
Краткое описание сущности изобретения
Соответственно, настоящее изобретение относится к способу получения гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она.
В контексте своего второго объекта настоящее изобретение относится к промежуточным соединениям, используемым при получении гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она.
Согласно одному аспекту осуществления настоящего изобретения предлагается способ получения гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I), который включает следующие стадии:
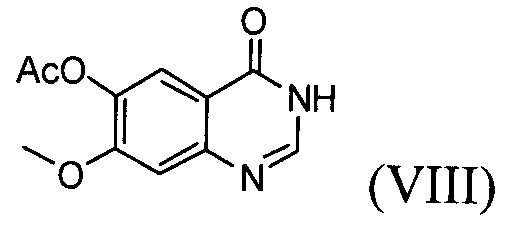
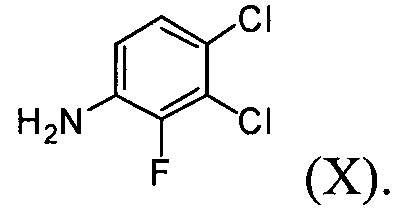
(1) проведение реакции соединения формулы (VIII) с галогенирующим агентом в присутствии органического основания, с последующим проведением реакции с соединением формулы (Х), с получением соединения формулы (VI);
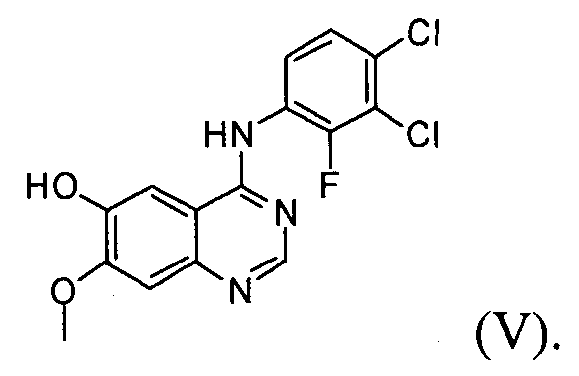
(2) проведение реакции соединения формулы (VI) с раствором аммиака в полярном протонном растворителе с получением соединения формулы (V);
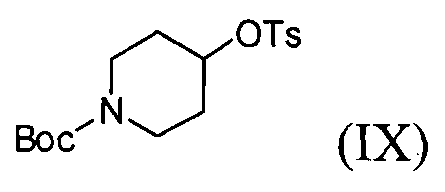
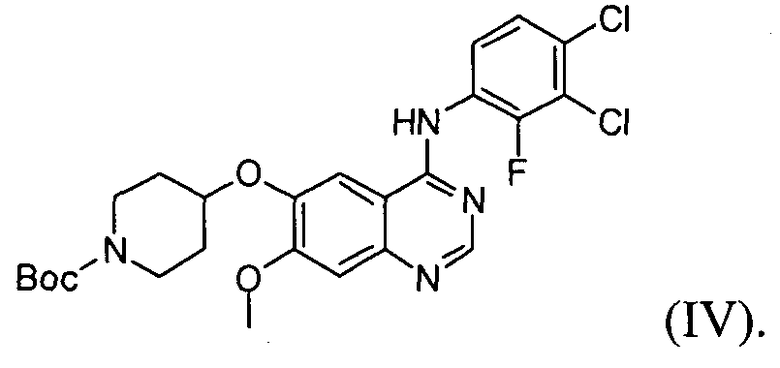
(3) проведение реакции соединения формулы (V) с соединением формулы (IX) в инертном полярном протонном растворителе в присутствии основания с получением соединения формулы (IV);
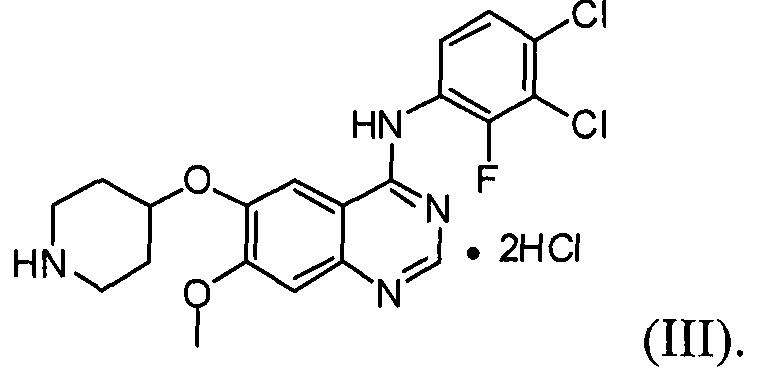
(4) проведение реакции соединения формулы (IV) с хлористоводородной кислотой в инертном растворителе с получением соединения формулы (III);
(5) проведение реакции акрилирования соединения формулы (III) с 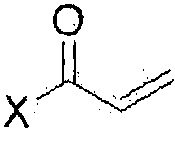 (где Х означает галоген) в присутствии основания с получением соединения формулы (II); и
(где Х означает галоген) в присутствии основания с получением соединения формулы (II); и
(6) проведение реакции соединения формулы (II) с хлористоводородной кислотой с получением соединения формулы (I):
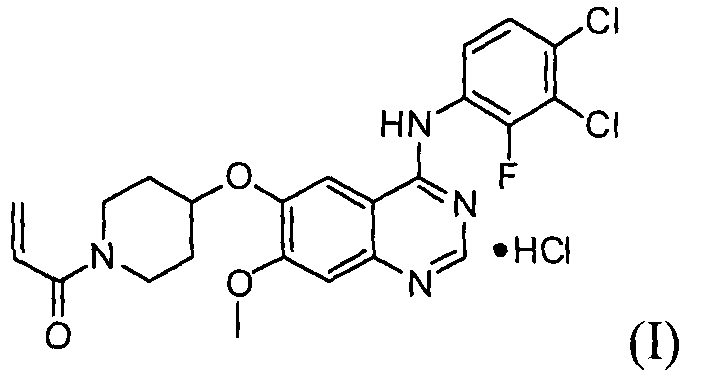
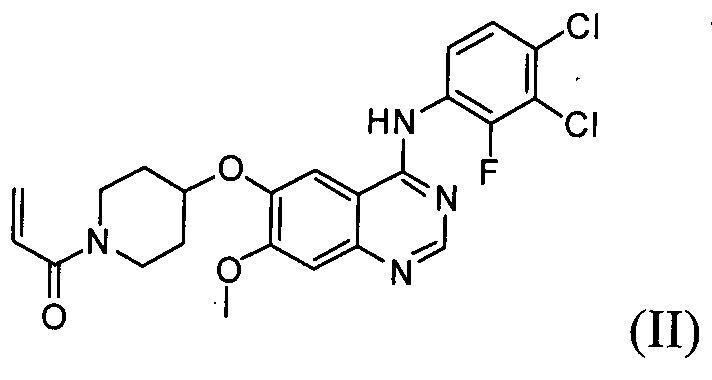
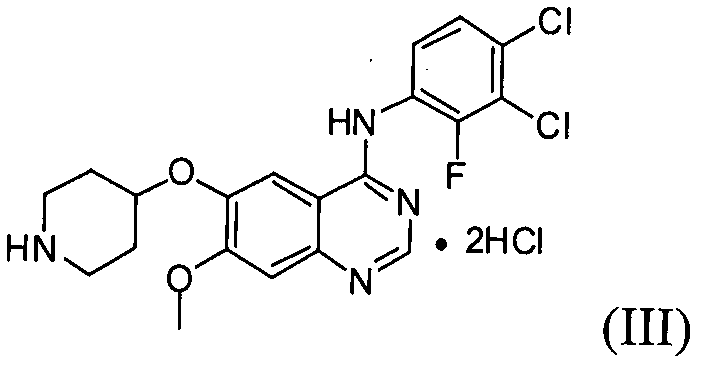
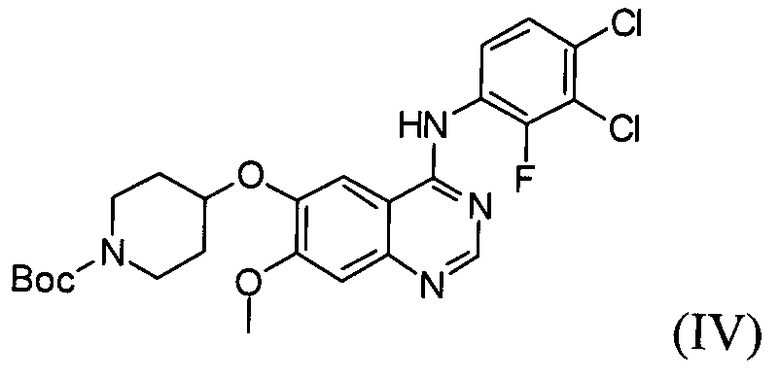
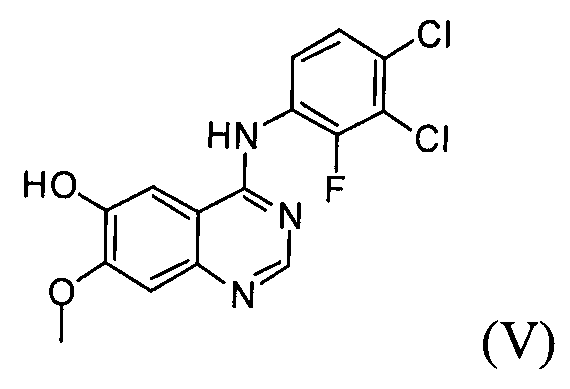
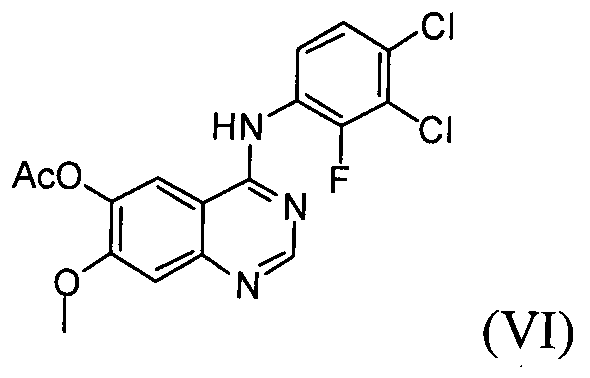
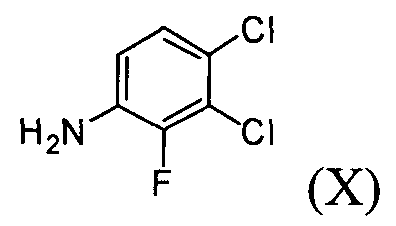
Настоящее изобретение, в соответствии с другим своим аспектом, относится к дигидрохлориду N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина формулы (III), трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилату формулы (IV) и 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-олу формулы (V), которые могут быть использованы в качестве промежуточных соединений для получения соединения формулы (I).
Подробное описание изобретения
Способ по настоящему изобретению может быть осуществлен так, как показано на приведенных ниже схемах реакции 2-6. Стадии (1) и (2) в данном способе могут быть проведены в соответствии со схемой реакции 2, приведенной ниже:
<Схема реакции 2>
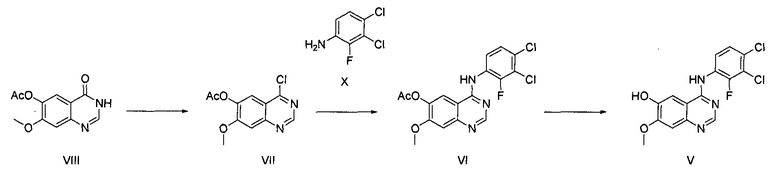
На стадии (1) проводят реакцию соединения формулы (VIII), взятого в качестве исходного соединения, с галогенирующим агентом в растворителе, таком как толуол или бензол, в присутствии органического основания, с последующей реакцией с соединением формулы (Х), получая ацетат 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ила формулы (VI).
Соединение формулы (VIII) может быть получено способом, описанным в патенте Кореи № 1013319.
Органическое основание, используемое на стадии (1) в способе по настоящему изобретению, может быть выбрано из группы, состоящей из диизопропиламина, триэтиламина, диизопропилэтиламина, диэтиламина, пиридина, 4-диметилпиридина, морфолина и их смеси; и галогенирующий агент может быть выбран из группы, состоящей из тионилхлорида, оксихлорида фосфора и их смеси.
Реакцию можно проводить при температуре от 50°C до 150°C, предпочтительно от 60°C до 90°C и более предпочтительно при температуре примерно 75°C. В результате реакции с галогенирующим агентом может быть получено соединение формулы (VII) в органическом растворителе, от которого оно не может быть легко отделено. Далее, соединение формулы (VII) в органическом растворителе подвергают реакции с соединением формулы (Х) с получением ацетата 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ила формулы (VI).
На стадии (2) соединение формулы (VI), полученное на стадии (1), подвергают реакции с раствором аммиака или газообразным аммиаком в полярном протонном растворителе (таком как, например, метанол, этанол и пропанол) при температуре от 0°C до 40°C, предпочтительно от 10°C до 30°C, более предпочтительно примерно при температуре 25°C, с получением 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола формулы (V).
На стадии (3), как показано на схеме реакции 3, соединение формулы (V) подвергают реакции с трет-бутил-4-(тозилокси)пиперидин-1-карбоксилатом формулы (IX) в инертном полярном растворителе, в присутствии основания, с получением трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилата формулы (IV).
<Схема реакции 3>
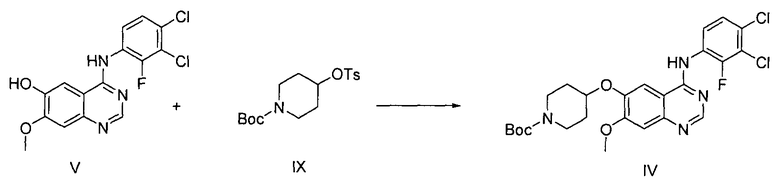
Инертный полярный растворитель, используемый на стадии (3) в способе по настоящему изобретению, может быть выбран из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, N-метилпирролидин-2-она, диметилсульфоксида и их смеси. Основание может представлять собой соль, такую как карбонат щелочного металла, выбранную из группы, состоящей из гидрокарбоната натрия, карбоната калия, карбоната цезия и их смеси. Указанное основание используется в количестве от 1 до 5 молярных эквивалентов на 1 молярный эквивалент соединения формулы (V). Реакцию можно проводить при температуре от 60°C до 100°C, предпочтительно от 70°C до 90°C, более предпочтительно при температуре примерно 80°C.
Согласно одному варианту осуществления настоящего изобретения соединение формулы (IV) может быть получено с высокой степенью чистоты и высоким выходом путем простой перекристаллизации с использованием К2СО3 на стадии (3) в способе по настоящему изобретению. Напротив, согласно стандартному способу, описанному в патенте Кореи № 1013319, необходимо в качестве основного реагента использовать дорогостоящий диизопропилазодикарбоксилат (DIAD) и проводить очистку продукта колоночной хроматографией. Соответственно, стандартный способ является не только неэкономичным, но и также неэффективным, по сравнению со способом по настоящему изобретению, с точки зрения чистоты продукта и его выхода (см. таблицу 1).
На стадии (4), как показано на схеме реакции 4, соединение формулы (IV) подвергают реакции с хлористоводородной кислотой в инертном растворителе с получением дигидрохлорида N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина формулы (III).
<Схема реакции 4>
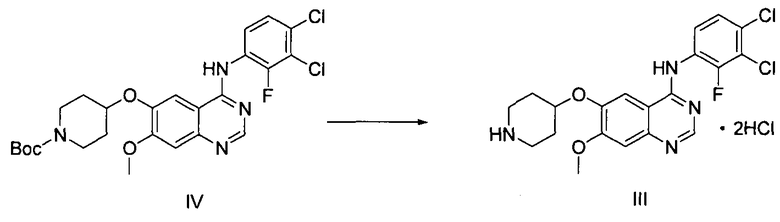
Инертный растворитель, используемый на стадии 4 в способе по настоящему изобретению, может быть выбран из группы, состоящей из метанола, этанола, пропанола, этилацетата, метилацетата, ацетона и их смеси. Хлористоводородная кислота может быть использована в количестве от 3 до 10 молярных эквивалентов на 1 молярный эквивалент соединения формулы (IV). Реакция может быть проведена при перемешивании в течение 1-24 часов при температуре от 0°C до 60°C, предпочтительно от 10°C до 40°C, более предпочтительно при температуре примерно 25°C.
На стадии 5, как показано на схеме реакции 5, соединение формулы (III) подвергают реакции акрилирования с 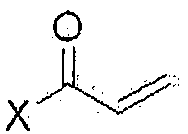 (где Х означает галоген), например, с акрилоилхлоридом в присутствии основания с получением 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (II).
(где Х означает галоген), например, с акрилоилхлоридом в присутствии основания с получением 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (II).
<Схема реакции 5>
Стадия (5) в способе по настоящему изобретению может быть проведена в органическом растворителе, таком как тетрагидрофуран, этилацетат, ацетон, 1,4-диоксан, ацетонитрил, дихлорметан, тетрахлорид углерода, хлороформ, N,N-диметилформамид или диметилсульфоксид, или в смеси указанного органического растворителя и воды. Предпочтительно использовать смесь органического растворителя, выбранного из группы, состоящей из тетрагидрофурана, этилацетата, ацетона, 1,4-диоксана и ацетонитрила, и воды.
Основание, используемое на стадии (5), может быть выбрано из группы, состоящей из неорганического основания, такого как карбонат натрия, карбонат кальция, карбонат калия, гидроксид натрия, гидроксид калия или карбонат цезия, или органического основания, такого как диизопропиламин, триэтиламин, диизопропилэтиламин или диэтиламин. В этой реакции основание можно использовать в количестве от 3 до 5 молярных эквивалентов на 1 молярный эквивалент соединения формулы (III). Реакция акрилирования может быть проведена при перемешивании в течение от 20 минут до 3 часов при температуре от -30°C до 20°C, предпочтительно при температуре примерно 0°C.
После завершения реакции полученную смесь подвергают перекристаллизации в водном растворе ацетона, взятом в количестве от 15 до 30 (вес./об.) раз превышающем количество соединения формулы (III).
В соответствии с одним вариантом осуществления настоящего изобретения соединение формулы (II) может быть получено с высокой степенью чистоты и высоким выходом путем простой перекристаллизации, проводимой на стадии (5) в способе по настоящему изобретению. В то же время, согласно стандартному способу, описанному в патенте Кореи № 1013319, для этого необходима очистка продукта методом колоночной хроматографии. В этой связи, стандартный способ является неэффективным по сравнению со способом по настоящему изобретению с точки зрения выхода и чистоты получаемого продукта (см. таблицу 2).
На стадии (6), как показано на схеме реакции 6, соединение формулы (II) подвергают реакции с хлористоводородной кислотой в органическом растворителе с получением гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I).
<Схема реакции 6>
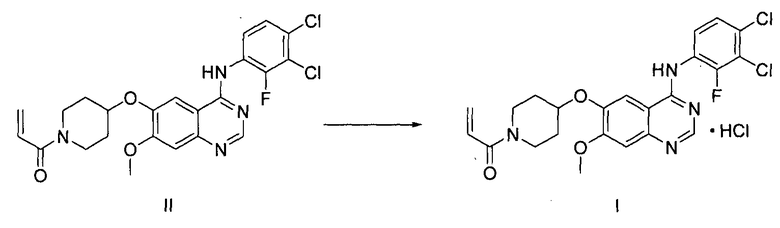
Органический растворитель, используемый на стадии (6) в способе по настоящему изобретению, может быть выбран из группы, состоящей из метанола, этанола, пропанола, изопропанола, бутанола, этилацетата, ацетона, тетрагидрофурана, ацетонитрила, 1,4-диоксана и их смеси. Указанная реакция может быть проведена при температуре от 0°C до 60°C, предпочтительно от 10°C до 40°C, более предпочтительно при температуре примерно 25°C.
Настоящее изобретение относится к получению новых соединений, таких как дигидрохлорид N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина формулы (III), трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилат формулы (IV) и 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ол формулы (V), которые являются ключевыми промежуточными соединениями, используемыми в способе по настоящему изобретению. Эти соединения могут быть использованы при получении гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метосихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I), который селективно и эффективно ингибирует рост раковых клеток, индуцированный сверхэкспрессией рецептора эпидермального фактора роста, и предотвращает развитие резистентности к лекарственному средству, вызванной мутацией тирозинкиназы.
В соответствии с настоящим изобретением соединения формул (I) и (II) могут быть получены с высоким выходом простым и недорогим способом. Согласно способу по настоящему изобретению соединение формулы (VIII) может быть просто преобразовано в соединение формулы (VI) in situ, а соединение формулы (V) может быть получено без использования какой-либо стадии очистки. Кроме того, традиционный способ получения соединений формул (II), (III) и (IV) требует проведения дополнительной стадии очистки или экстракции, например, методом колоночной хроматографии, что делает его менее удобным для коммерческого применения. В то же время, способ по настоящему изобретению позволяет получать готовый продукт с высокой степенью чистоты и с высоким выходом путем добавления растворителя к реакционной смеси с получением продукта в твердой фазе и с последующими перекристаллизацией и фильтрованием продукта.
Приведенные ниже примеры предназначены для дальнейшего пояснения настоящего изобретения, но они никоим образом не ограничивают его объем.
Пример 1: Получение ацетата 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ила (соединение формулы VI))
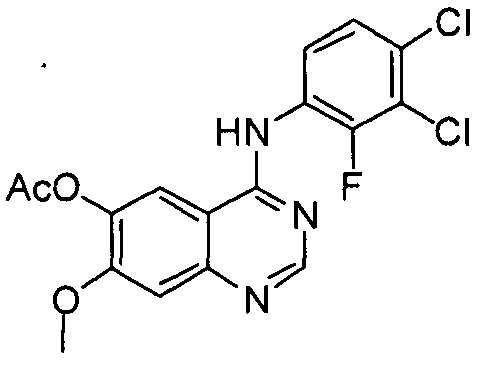
Добавляют ацетат 7-метокси-4-оксо-3,4-дигидрохиназолин-ила (100 г) к толуолу (850 мл) и N,N-диизопропилэтиламину (82,5 мл). В эту смесь в течение 20 минут при температуре 75°C добавляют оксихлорид фосфора (100 мл) с последующим перемешиванием в течение 3 часов. К полученной смеси добавляют толуол (450 мл) и 3,4-дихлор-2-фторанилин (84,6 г) с последующим перемешиванием в течение 2 часов. После завершения реакции полученную смесь охлаждают до температуры 25°C. Полученное при этом твердое вещество отфильтровывают при пониженном давлении и промывают толуолом (400 мл). К указанному твердому веществу добавляют изопропанол (1000 мл) и смесь затем перемешивают в течение 2 часов. Полученное твердое вещество отфильтровывают и промывают изопропанолом (400 мл). Твердое вещество сушат в сушильном шкафу при температуре 40°C с получением соединения формулы (VI) (143 г, выход: 83%).
1H-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 8,92 (с, 1H), 8,76 (с, 1H), 7,69-7,57 (м, 3H), 4,01 (с, 3H), 2,38 (с, 3H).
Пример 2: Получение 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ола (соединение формулы (V)
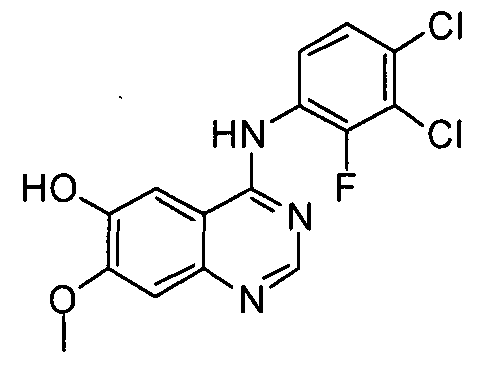
Ацетат 4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-ила (100 г) перемешивают с метанолом (1000 мл). Смесь охлаждают до 10-15°C, добавляют раствор аммиака (460 г) и перемешивают смесь в течение 3 часов при температуре 25°C. Полученное таким образом твердое вещество отфильтровывают и промывают смешанным растворителем, состоящим из метанола (200 мл) и воды (200 мл). Полученное твердое вещество сушат в сушильном шкафу при температуре 40°C с получением соединения формулы (V) (74 г, выход: 83%).
1H-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 9,57 (уш., 2H), 8,35 (с, 1H), 7,68 (с, 1H), 7,61-7,52 (м, 2H), 7,21 (с, 1H), 3,97 (с, 3H).
Пример 3: Получение трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилата (соединение формулы (IV))

4-(3,4-Дихлор-2-фторфениламино)-7-метоксихиназолин-6-ол (60 г) смешивают с N-диметилформамидом (360 мл) при перемешивании, после чего к полученной смеси добавляют трет-бутил-4-(тозилокси)пиперидин-1-карбоксилат (120 г) и карбонат калия (72 г). Температуру реакционной смеси повышают до 70°C и перемешивают смесь еще в течение 14 часов. Температуру полученного раствора понижают до 25°C и медленно добавляют к смеси воду (480 мл). Полученное таким образом твердое вещество отфильтровывают и сушат. Твердое вещество растворяют в смешанном растворителе (600 мл), состоящем из дихлорметана и метанола. К смеси добавляют активированный уголь (6 г), затем перемешивают смесь в течение 30 минут. Полученную смесь фильтруют через слой целита, перегоняют при пониженном давлении, добавляют ацетон (300 мл) и перемешивают смесь в течение 2 часов. Полученное твердое вещество отфильтровывают и промывают ацетоном (100 мл). Указанное твердое вещество сушат в сушильном шкафу при температуре 40°C с получением соединения формулы (IV) (75 г, выход: 83%).
1H-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 8,69 (с, 1H), 8,47 (т, 1H), 7,34-7,29 (м, 2H), 7,20 (с, 1H), 4,63-4,60 (м, 1H), 3,82 (с, 3H), 3,83-3,76 (м, 2H), 3,37-3,29 (м, 2H), 1,99-1,96 (м, 2H), 1,90-1,84 (м, 2H), 1,48 (с, 9H).
Пример 4: Получение дигидрохлорида N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина (соединение формулы (III))
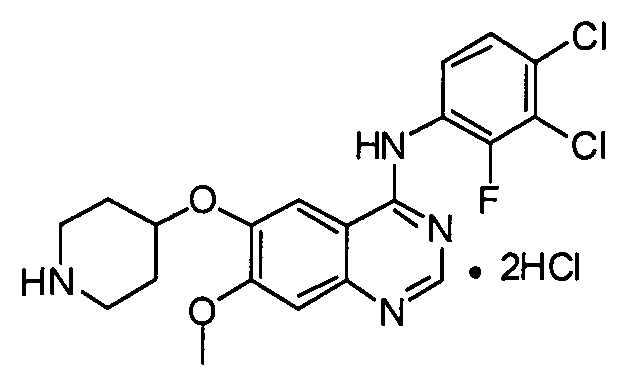
К трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилату (75 г) добавляют ацетон (740 мл) и затем смесь перемешивают. В течение 10 минут к смеси добавляют хлористоводородную кислоту (145 мл) и смесь перемешивают в течение 5 часов. После завершения реакции полученную смесь фильтруют и полученное таким образом твердое вещество промывают ацетоном (73 мл). Твердое вещество сушат в сушильном шкафу при температуре 30°C с получением соединения формулы (III) (71 г, выход: 99%).
1H-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 12,95 (уш.с, 1H), 9,42 (уш.с, 1H), 9,18 (уш.с, 1H), 9,01 (с, 1H), 8,86 (с, 1H), 7,69-7,56 (м, 2H), 7,45 (с, 1H), 5,11-5,08 (м, 1H), 4,03 (с, 3H), 3,29-3,20 (м, 4H), 2,33-2,30 (м, 2H), 1,96-1,93 (м, 2H).
Пример 5: Получение 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она (соединение формулы (II))
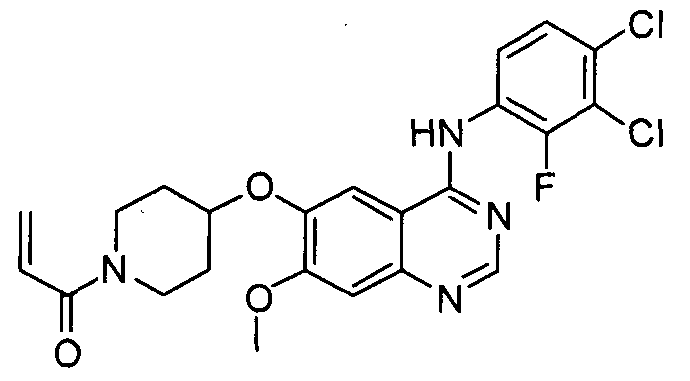
Дигидрохлорид N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина (100 г) и гидрокарбонат натрия (66 г) добавляют к смешанному растворителю, состоящему из тетрагидрофурана (630 мл) и воды (1 л), и ледяной водой охлаждают реакционную смесь до температуры 0°C. К реакционной смеси в течение 30 минут медленно добавляют акрилоилхлорид (24 мл), разбавленный тетрагидрофураном (370 мл), затем смесь перемешивают в течение 30 минут при температуре 0°C. После завершения реакции к полученной смеси добавляют водный раствор ацетона (2,0 л), смесь перемешивают в течение 12 часов и затем фильтруют с получением 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она (72 г. выход: 75%). Полученное таким образом твердое вещество растворяют в смешанном растворителе, состоящем из дихлорметана (200 мл) и метанола (100 мл), добавляют этилацетат (1,2 л) и смесь перемешивают в течение 12 часов. Полученное твердое вещество отфильтровывают и промывают этилацетатом (100 мл). Затем твердое вещество сушат в сушильном шкафу при температуре 40°C с получением соединения формулы (II) (55 г, выход: 76%, общий выход=57%).
1H-ЯМР (CDC13, 300 МГц, м.д.) δ 8,68 (с, 1H), 8,39 (т, 3H), 7,3l (м, 3H), 6,61 (м, 1H), 6,29 (м, 1H), 5,72 (м, 1H), 4,75 (м, 1H), 4,02 (с, 3H), 3,89 (м, 2H), 3,60 (м, 2H), 1,86 (м, 4H).
Пример 6: Получение гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она (соединение формулы (I))
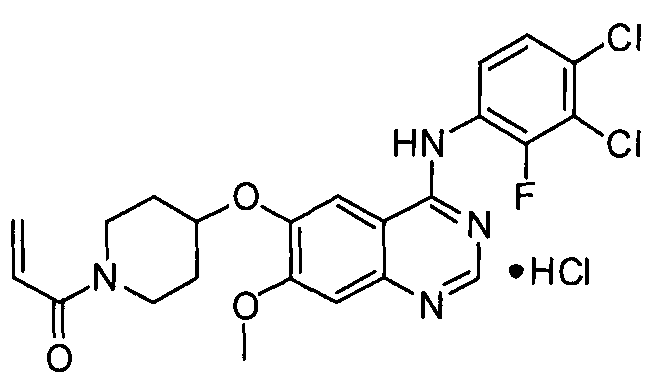
1-(4-(4-(3,4-Дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-он (150 г) добавляют к метанолу (700 мл). В эту же смесь добавляют хлористоводородную кислоту (38,2 мл), разбавленную метанолом (300 мл), затем смесь перемешивают в течение 24 часов. Полученное таким образом твердое вещество отфильтровывают и промывают ацетоном (100 мл). Указанное твердое вещество сушат в сушильном шкафу при температуре 40°C в течение 24 часов с получением соединения формулы (I) (131 г, выход: 81%).
1H-ЯМР (ДМСО-d6, 300 МГц, м.д.) δ 12,31 (уш.с, 1H), 8,83 (с, 1H), 8,67 (с, 1H), 7,64-7,55 (м, 2H), 7,39 (с, 1H), 6,87-6,78 (м, 1H), 6,12-6,06 (м, 1H), 5,68-5,64 (м, 1H), 5,07-5,01 (м, 1H), 4,06-3,88 (м, 5H), 3,51 (т, 1H), 3,32 (т, 1H), 2,10 (т, 1H), 1,60 (т, 1H).
Несмотря на то, что настоящее изобретение описано применительно к указанным выше конкретным вариантам его осуществления, следует понимать, что специалисты в данной области могут внести различные изменения и модификации в настоящее изобретение, которые входят в объем настоящего изобретения, как определено прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1-(4-(4-(3,4-ДИХЛОР-2-ФТОРФЕНИЛАМИНО)-7-МЕТОКСИХИНАЗОЛИН-6-ИЛОКСИ)ПИПЕРИДИН-1-ИЛ)ПРОП-2-ЕН-1-ОНА | 2014 |
|
RU2671843C2 |
| НОВОЕ АМИДНОЕ ПРОИЗВОДНОЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК | 2008 |
|
RU2434010C2 |
| ПРОИЗВОДНЫЕ 5-АМИНОЦИКЛИЛМЕТИЛОКСАЗОЛИДИН-2-ОНА | 2008 |
|
RU2492169C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АНГИОГЕНЕЗА | 2000 |
|
RU2262935C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(4-БРОМ-2-ФТОРАНИЛИНО)-6-МЕТОКСИ-7(1-МЕТИЛПИПЕРИДИН-4-ИЛМЕТОКСИ)ХИНАЗОЛИНА(ZD6474) И СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2448102C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ДОСТИЖЕНИЯ АНТИПРОЛИФЕРАТИВНОГО ЭФФЕКТА | 1996 |
|
RU2153495C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ТЕТРАЗОЛМЕТАНСУЛЬФОНОВОЙ КИСЛОТЫ И НОВОЕ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ В НЕМ | 2011 |
|
RU2509769C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2267489C2 |
| СПОСОБ ПОЛУЧЕНИЯ (6R)-3-ГЕКСИЛ-4-ГИДРОКСИ-6-УНДЕЦИЛ-5,6-ДИГИДРОПИРАН-2-ОНА И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ, ПРИМЕНЯЕМОГО В ДАННОМ СПОСОБЕ | 2008 |
|
RU2434860C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
Настоящее изобретение относится к улучшенному способу получения гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I). Указанное соединение селективно и эффективно ингибирует рост раковых клеток, индуцированный сверхэкспрессией рецептора эпидермального фактора роста (EGFR), и предотвращает развитие резистентности к лекарственному средству, вызванное мутацией тирозинкиназы. Способ включает следующие стадии: (1) проведение реакции соединения формулы (VIII) с галогенирующим агентом в присутствии органического основания, с последующим проведением реакции с соединением формулы (Х) с получением соединения формулы (VI); (2) проведение реакции соединения формулы (VI) с раствором аммиака в полярном протонном растворителе с получением соединения формулы (V); (3) проведение реакции соединения формулы (V) с соединением формулы (IX) в инертном полярном протонном растворителе в присутствии основания с получением соединения формулы (IV); (4) проведение реакции соединения формулы (IV) с хлористоводородной кислотой в инертном растворителе с получением соединения формулы (III); (5) акрилирование соединения формулы (III) соединением  (где Х означает галоген) в присутствии основания с получением соединении формулы (II); и (6) проведение реакции соединения формулы (II) с хлористоводородной кислотой с получением соединения формулы (I).
(где Х означает галоген) в присутствии основания с получением соединении формулы (II); и (6) проведение реакции соединения формулы (II) с хлористоводородной кислотой с получением соединения формулы (I).








Промежуточные соединения (III), (IV) и (V) являются новыми. Способ позволяет упростить процесс, например, за счет исключения потенциально опасных стадий процесса получения 6,7-диметоксихиназолин-4-она, избытка тионилхлорида, колоночной хроматографии. При этом исключается ряд побочных реакций и значительно повышается общий выход (с 13% до 95%) и качество целевого продукта. 4 н. и 14 з.п.ф-лы., 2 табл.,6 прим.
1. Способ получения гидрохлорида 1-(4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она формулы (I), который включает следующие стадии:
(1) проведение реакции соединения формулы (VIII) с галогенирующим агентом в присутствии органического основания с последующим проведением реакции с соединением формулы (Х) с получением соединения формулы (VI);
(2) проведение реакции соединения формулы (VI) с раствором аммиака в полярном протонном растворителе с получением соединения формулы (V);
(3) проведение реакции соединения формулы (V) с соединением формулы (IX) в инертном полярном протонном растворителе в присутствии основания с получением соединения формулы (IV);
(4) проведение реакции соединения формулы (IV) с хлористоводородной кислотой в инертном растворителе с получением соединения формулы (III);
(5) проведение реакции акрилирования соединения формулы (III) с (где Х означает галоген) в присутствии основания с получением соединении формулы (II); и
(6) проведение реакции соединения формулы (II) с хлористоводородной кислотой с получением соединения формулы (I):
2. Способ по п. 1, отличающийся тем, что реакцию на стадии (1) проводят в растворителе, выбранном из группы, состоящей из толуола, бензола и их смеси.
3. Способ по п. 1, отличающийся тем, что указанное органическое основание, используемое на стадии (1), выбрано из группы, состоящей из диизопропиламина, триэтиламина, диизопропилэтиламина, диэтиламина, пиридина, 4-диметилпиридина, морфолина и их смеси.
4. Способ по п. 1, отличающийся тем, что указанный галогенирующий агент, используемый на стадии (1), выбран из группы, состоящей из тионилхлорида, оксихлорида фосфора и их смеси.
5. Способ по п. 1, отличающийся тем, что указанный полярный протонный растворитель, используемый на стадии (2), выбран из группы, состоящей из метанола, этанола, пропанола и их смеси.
6. Способ по п. 1, отличающийся тем, что указанный инертный полярный протонный растворитель, используемый на стадии (3), выбран из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, N-метилпиррролидин-2-она, диметилсульфоксида и их смеси.
7. Способ по п. 1, отличающийся тем, что указанное основание, используемое на стадии (3), представляет собой щелочную соль угольной кислоты, выбранную из группы, состоящей из гидрокарбоната натрия, карбоната калия, карбоната цезия и их
смеси.
8. Способ по п. 7, отличающийся тем, что указанное основание используется в количестве от 1 до 5 молярных эквивалентов на 1 молярный эквивалент соединения формулы (V).
9. Способ по п. 1, отличающийся тем, что указанный инертный растворитель, используемый на стадии (4), выбран из группы, состоящей из метанола, этанола, пропанола, этилацетата, метилацетата, ацетона и их смеси.
10. Способ по п. 1, отличающийся тем, что указанная хлористоводородная кислота на стадии (4) используется в количестве от 3 до 10 молярных эквивалентов на 1 молярный эквивалент соединения формулы (IV).
11. Способ по п. 1, отличающийся тем, что указанная стадия (5) проводится в органическом растворителе, выбранном из группы, состоящей из тетрагидрофурана, этилацетата, ацетона, 1,4-диоксана, ацетонитрила, дихлорметана, тетрахлорида углерода, хлороформа, N,N-диметилформамида и диметилсульфоксида, или в смеси указанного органического растворителя и воды.
12. Способ по п. 1, отличающийся тем, что указанное основание, используемое на стадии (5), выбрано из группы, состоящей из карбоната натрия, карбоната кальция, карбоната калия, гидроксида натрия, гидроксида калия, карбоната цезия, диизопропиламина, триэтиламина, диизопропилэтиламина и диэтиламина.
13. Способ по п. 1, отличающийся тем, что указанное основание на стадии (5) используется в количестве от 3 до 5 молярных эквивалентов на 1 молярный эквивалент соединения формулы (III).
14. Способ по п. 1, отличающийся тем, что указанная стадия (5) далее включает проведение перекристаллизации соединения формулы (II) с использованием водного раствора ацетона, взятого в количестве, от 15 до 30 (вес./об.) раз превышающем количество соединения формулы (III).
15. Способ по п. 1, отличающийся тем, что указанная стадия (6) проводится в органическом растворителе, выбранном из группы, состоящей из метанола, этанола, пропанола, изопропанола, бутанола, этилацетата, ацетона, тетрагидрофурана, ацетонитрила, 1,4-диоксана и их смеси.
16. Дигидрохлорид N-(3,4-дихлор-2-фторфенил)-7-метокси-6-(пиперидин-4-илокси)хиназолин-4-амина формулы (III):
17. Трет-бутил-4-(4-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-карбоксилат формулы (IV):
18. 4-(3,4-Дихлор-2-фторфениламино)-7-метоксихиназолин-6-ол формулы (V):
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Гаечный ключ | 1927 |
|
SU9064A1 |
| NAM, HYUN-JIN et al., Antitumor activity of HM781-36B, an irreversible Pan-HER inhibitor, alone or in combination with cytotoxic chemotherapeutic agents in gastric cancer, Cancer | |||

