ССЫЛКА НА СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Официальная копия списка последовательностей подана одновременно с описанием в виде форматированного в ASCII текстового файла через EFS-Web с названием файла "23237USPSP-SEQLIST-09MAY2012", датой создания 9 мая 2012 года и размером 4570 байт. Список последовательностей, зарегистрированный через EFS-Web, является частью описания и полностью включен в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ
Настоящее изобретение относится к способу получения индановых и азаиндановых производных пиперидинонкарбоксамида, которые представляют собой антагонисты рецепторов CGRP, пригодные для лечения мигрени. Этот класс соединений описан в патентных заявках США №№ 13/293166, зарегистрированной 10 ноября 2011 года, 13/293177, зарегистрированной 10 ноября 2011 года, и 13/293186, зарегистрированной 10 ноября 2011 года, и международных заявках PCT №№ PCT/US11/60081, зарегистрированной 10 ноября 2011 года, и PCT/US11/60083, зарегистрированной 10 ноября 2011 года.
CGRP (кодируемый геном кальцитонина пептид) представляет собой природный пептид из 37 аминокислот, который образуется посредством тканеспецифического альтернативного процессинга информационной РНК кальцитонина и широко распространен в центральной и периферической нервной системе. CGRP локализован преимущественно в чувствительных афферентных и центральных нейронах и опосредует несколько видов биологического действия, включая вазодилатацию. CGRP экспрессирован в альфа- и бета-формах, которые у крысы и человека отличаются на одну и три аминокислоты, соответственно. CGRP-альфа и CGRP-бета демонстрируют сходные биологические свойства. При высвобождении из клетки CGRP инициирует свой биологический ответ, связываясь со специфическими рецепторами на клеточной поверхности, которые преимущественно связаны с активацией аденилатциклазы. Рецепторы CGRP идентифицированы и фармакологически оценены в нескольких тканях и типах клеток, включая ткани и клетки головного мозга, сердечно-сосудистого, эндотелиального и гладкомышечного происхождения.
На основе фармакологических свойств эти рецепторы подразделяют по меньшей мере на два подтипа, обозначаемых CGRP1 и CGRP2. Селективным антагонистом CGRP1 является α-CGRP-(8-37) человека, фрагмент CGRP, в котором отсутствует семь N-концевых аминокислотных остатков, в то же время селективным агонистом CGRP2 является линейный аналог CGRP, диацетамидометилцистеин CGRP ([Cys(ACM)2,7]CGRP). CGRP представляет собой мощный нейромодулятор, который вовлечен в патологию цереброваскулярных нарушений, таких как мигрень и кластерная головная боль. В клинических исследованиях выявлены повышенные уровни CGRP в яремной вене при приступах мигрени (Goadsby et al., Ann. Neurol., 1990, 28, 183-187), у индивидуумов в промежутках между приступами повышены уровни CGRP в слюне (Bellamy et al., Headache, 2006, 46, 24-33), и показано, что CGRP сам по себе служит причиной возникновения мигренозной головной боли (Lassen et al., Cephalalgia, 2002, 22, 54-61). В клинических испытаниях показано, что антагонист CGRP BIBN4096BS является эффективным при лечении острых приступов мигрени (Olesen et al., New Engl. J. Med., 2004, 350, 1104-1110) и мог предотвращать головную боль, индуцированную инфузией CGRP, в контрольной группе (Petersen et al., Clin. Pharmacol. Ther., 2005, 77, 202-213).
Опосредованная CGRP активация тригеминоваскулярной системы может играть ключевую роль в патогенезе мигрени. Кроме того, CGRP активирует рецепторы на гладких мышцах внутричерепных сосудов, что приводит к увеличенной вазодилатации, которая, как полагают, вносит вклад в головную боль при приступах мигрени (Lance, Headache Pathogenesis: Monoamines, Нейропептиды, Purines and Nitric Oxide, Lippincott-Raven Publishers, 1997, 3-9). Средняя менингеальная артерия, основная артерия в твердой мозговой оболочке, иннервируется чувствительными волокнами из тройничного узла, который содержит несколько нейропептидов, включая CGRP. Стимуляция тройничного узла у кошки приводила к повышенным уровням CGRP, а у людей активация системы тройничного нерва вызывала гиперемию лица и повышение уровней CGRP в наружной яремной вене (Goadsby et al., Ann. Neurol., 1988, 23, 193-196). Электростимуляция твердой мозговой оболочки у крыс увеличивала диаметр средней менингеальной артерии, эффект, который блокировали посредством предшествующего введения CGRP(8-37), антагониста пептида CGRP (Williamson et al., Cephalalgia, 1997, 17, 525-531). Стимуляция тройничного узла увеличивала лицевой кровоток у крысы, который ингибировали CGRP(8-37) (Escott et al., Brain Res. 1995, 669, 93-99). Электростимуляция тройничного узла у игрунковой мартышки приводила к увеличению лицевого кровотока, который можно было блокировать непептидным антагонистом CGRP BIBN4096BS (Doods et al., Br. J. Pharmacol., 2000, 129, 420-423). Таким образом, антагонист CGRP может ослаблять, предотвращать или обращать сосудистое действие CGRP.
Показано, что опосредованная CGRP вазодилатация средней менингеальной артерии крысы повышает чувствительность нейронов каудального ядра тройничного тракта (Williamson et al., The CGRP Family: Calcitonin Gene-Related Peptide (CGRP), Amylin, and Adrenomedullin, Landes Bioscience, 2000, 245-247). Подобным образом, растяжение кровеносных сосудов твердой оболочки при мигренозной головной боли может повышать чувствительность нейронов тройничного тракта. Некоторые из ассоциированных симптомов мигрени, включая внечерепную боль и лицевую аллодинию, могут приводить к повышению чувствительности нейронов тройничного тракта (Burstein et al., Ann. Neurol. 2000, 47, 614-624). Антагонист CGRP может быть полезен для уменьшения, предотвращения или обращения эффектов повышения чувствительности нейронов.
Способность соединений действовать в качестве антагонистов CGRP делает их подходящими фармакологическими средствами при нарушениях, в которые вовлечен CGRP, у людей и животных, особенно у людей. Такие нарушения включают мигрень и кластерную головную боль (Doods, Curr Opin Inves Drugs, 2001, 2(9), 1261-1268; Edvinsson et al., Cephalalgia, 1994, 14, 320-327); хроническую головную боль напряжения (Ashina et al., Neurology, 2000, 14, 1335-1340); боль (Yu et al., Eur. J. Pharm., 1998, 347, 275-282); хроническую боль (Hulsebosch et al., Pain, 2000, 86, 163-175); нейрогенное воспаление и боль при воспалении (Holzer, Neurosci., 1988, 24, 739-768; Delay-Goyet et al., Acta Physiol. Scanda. 1992, 146, 537-538; Salmon et al., Nature Neurosci., 2001, 4(4), 357-358); глазную боль (May et al. Cephalalgia, 2002, 22, 195-196), зубную боль (Awawdeh et al., Int. Endocrin. J., 2002, 35, 30-36), неинсулинозависимый сахарный диабет (Molina et al., Diabetes, 1990, 39, 260-265); сосудистые нарушения; воспаление (Zhang et al., Pain, 2001, 89, 265), артрит, гиперреактивность бронхов, астму, (Foster et al., Ann. NY Acad. Sci., 1992, 657, 397-404; Schini et al., Am. J. Physiol., 1994, 267, H2483-H2490; Zheng et al., J. Virol., 1993, 67, 5786-5791); шок, сепсис (Beer et al., Crit. Care Med., 2002, 30(8), 1794-1798); синдром отмены опиатов (Salmon et al., Nature Neurosci., 2001, 4(4), 357-358); привыкание к морфину (Menard et al., J. Neurosci., 1996, 16 (7), 2342-2351); приступообразные ощущения жара у мужчин и женщин (Chen et al., Lancet, 1993, 342, 49; Spetz et al., J. Urology, 2001, 166, 1720-1723); аллергический дерматит (Wallengren, Contact Dermatitis, 2000, 43(3), 137-143); псориаз; энцефалит, травму головного мозга, ишемию, инсульт, эпилепсию и нейродегенеративные заболевания (Rohrenbeck et al., Neurobiol. of Disease, 1999, 6, 15-34); кожные заболевания (Geppetti and Holzer, Eds., Neurogenic Inflammation, 1996, CRC Press, Boca Raton, FL), нейрогенное покраснение кожи, порозовение и эритему кожи; звон в ушах (Herzog et al., J. Membrane Biology, 2002, 189(3), 225); воспалительное заболевание кишечника, синдром раздраженного кишечника (Hoffman et al. Scandinavian Journal of Gastroenterology, 2002, 37(4) 414-422) и цистит. Особую важность представляет неотложное или профилактическое лечение головной боли, включая мигрень и кластерную головную боль.
В настоящем изобретении описан новый способ получения индановых и азаиндановых производных пиперидинонкарбоксамида, которые представляют собой антагонисты рецепторов CGRP.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому способу получения индановых и азаиндановых производных пиперидинонкарбоксамида, которые представляют собой антагонисты рецепторов CGRP, пригодные для лечения мигрени, с использованием высокоэффективного синтеза спирокислот.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения соединения формулы I:
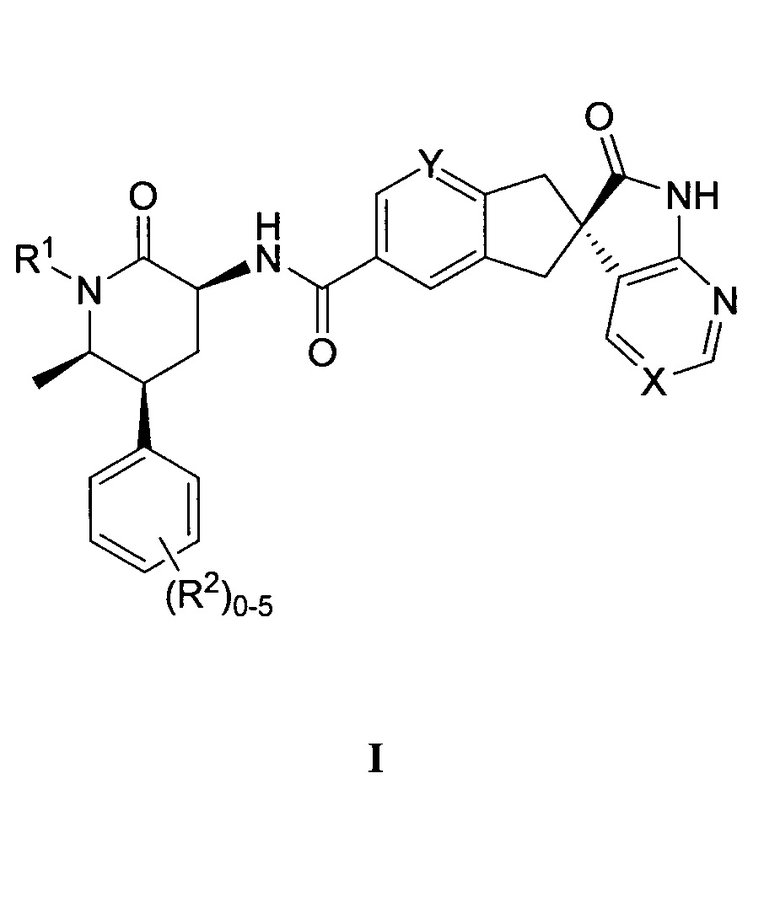 ,
,
или его фармацевтически приемлемой соли, где:
X выбран из -C(R3)= или -N=, где R3 представляет собой водород, F или CN;
Y представляет собой CH или N;
R1 выбран из группы, состоящей из C1-4-алкила, циклопропилметила, циклобутилметила и [1-(трифторметил)циклопропил]метила, каждый из которых необязательно замещен, как допускает валентность, одним или несколькими заместителями, независимо выбранными из группы, состоящей из F и гидрокси; и
R2 представляет собой водород, метил, F, Cl или Br;
включающему инициализацию реакции соединения формулы F:
 ,
,
где
E1 выбран из группы, состоящей из галогена, -C(O)-O-R", -CN, -CONRR', -NRR', -CH2-OR, OR, метила и винила, где каждый из R и R' независимо выбран из группы, состоящей из водорода, C1-6-алкила, арила, гетероарила или бензила, и R" представляет собой водород или углерод, содержащий заместитель, способный к растворению до карбоновой кислоты,
E2 представляет собой функциональную группу со способностью быть уходящей группой, и
PG представляет собой защитную группу азота,
в органической фазе в присутствии моно- или бис-четвертичной соли алкалоида хинного дерева и неорганического основания в водной фазе, с формированием двухфазной среды, содержащей водную фазу и органическую фазу, с получением соединения формулы G: 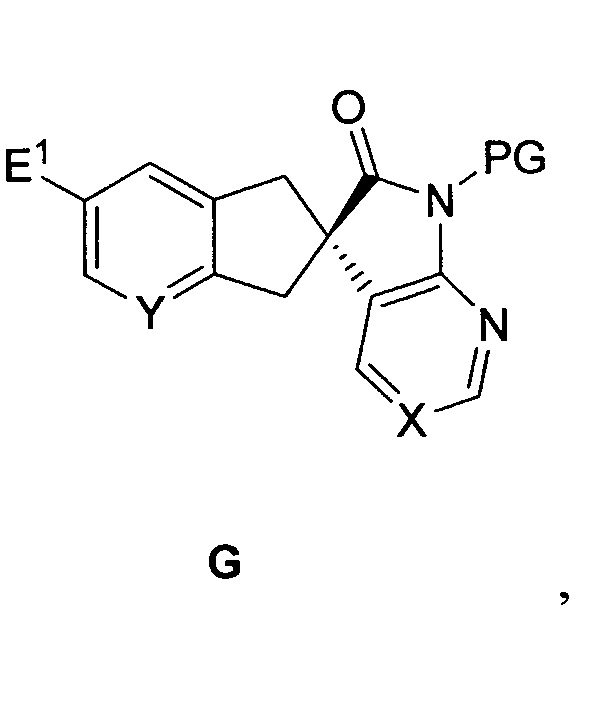
когда E1 отличен от -C(O)-O-H, замену E1 карбоновой кислотой или преобразование E1 в карбоновую кислоту в подходящей химической реакции с выходом соединения формулы H:
 ,
,
снятие защиты с соединения формулы H с выходом соединения формулы C:
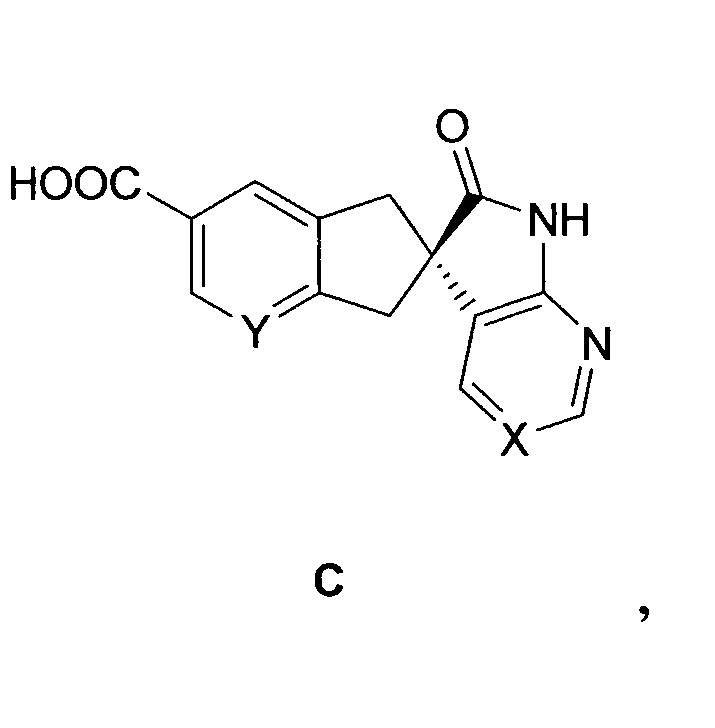
необязательно выделяемого в виде соли, и связывание соединения формулы C с соединением формулы B:
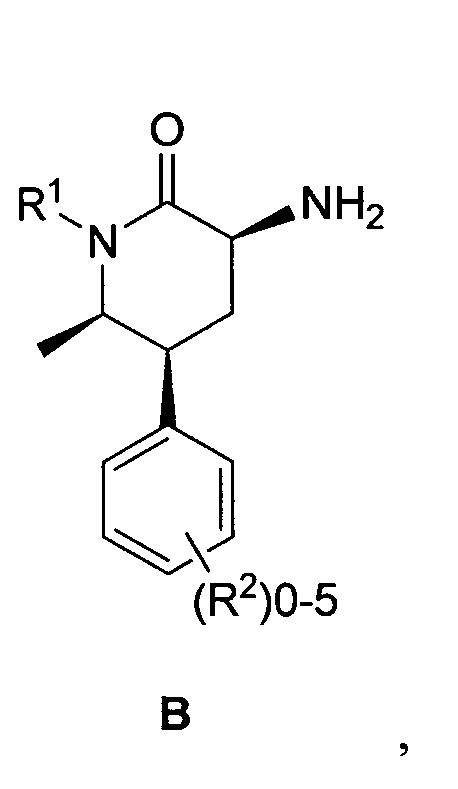
необязательно в виде соли в условиях формирования амидной связи между кислотой и амином с выходом соединения формулы I.
В одном из вариантов осуществления изобретения R1 представляет собой C1-4-алкил, необязательно замещенный 1-3 F или гидрокси или обоими. В одном из классов варианта осуществления R1 выбран из группы, состоящей из изопропила, 2,2,2-трифторэтила, 2,2-дифторэтила, 2-метилпропила, 3,3,3-трифторпропила и 3,3,3-трифтор-2-гидроксипропила. В одном из подклассов варианта осуществления R1 представляет собой 2,2,2-трифторэтил.
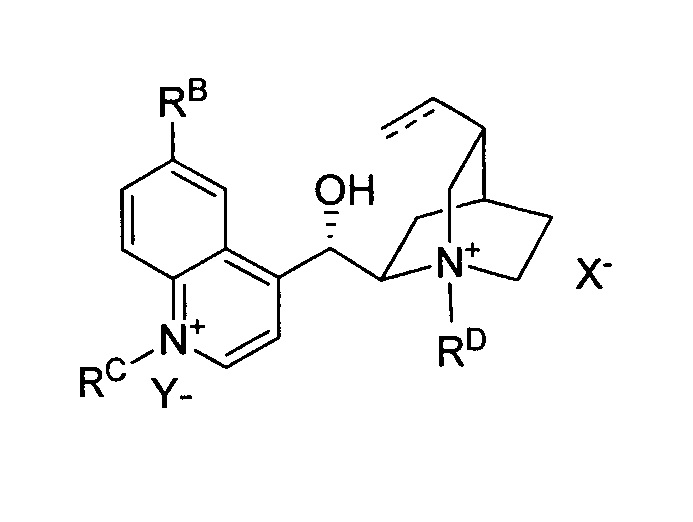
Термин "моно- или бис-четвертичная соль алкалоида хинного дерева" означает соли соединений общей структуры:
 ,
,
где один или оба атома азота являются четвертичными. Если не изображено или не указано иначе, соли алкалоида хинного дерева по изобретению включают все стереоизомеры, включая цинхонин, цинхонидин, хинин, хинидин, дигидроцинхонин, дигидроцинхонидин, дигидрохинидин и дигидрохинин. Изображение

для RA означает чистый этил, чистый винил или смесь этила и винила. Соли алкалоидов хинного дерева функционируют в качестве катализаторов межфазного переноса в асимметричном синтезе, описываемом в настоящем документе. См. Takashi Ooi and Keiji Maruoka, Recent Advances in Asymmetric Phase-Transfer Catalysis, Angew. Chem. Int. Ed. 2007, 46, 4222-4266.
В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где соль алкалоида хинного дерева является бис-четвертичной и обладает химической структурой формулы II:
 ,
,
где:
RA представляет собой  ,
,
RB выбран из группы, состоящей из водорода и метокси,
каждый из RC и RD независимо выбран из группы, состоящей из C1-6-алкила, C2-6-алкенила, C2-6-алкинила, C3-6-циклоалкила, арила, гетероарила, -C1-4-алкиларила и -C1-4-алкилгетероарила, где C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-6-циклоалкил, арил, гетероарил, и арильные и гетероарильные части -C1-4-алкиларила и -C1-4-алкилгетероарила необязательно замещены одним-пятью заместителями, независимо выбранными из RF,
RE выбран из группы, состоящей из водорода, C(O)R, C(O)OR, CONRR' и C1-6-алкила,
RF выбран из группы, состоящей из C1-4-алкила, арила, C1-4-алкокси, гидрокси, CN, CO2R, CONRR', SR, SO2R, SO3R, PR3, PO(OR)2, PO(OR)(NRR'), PO(NRR')2, P(OR)2, P(OR)(NRR'), P(NRR')2, SiRR'R", B(OR)2, C(O)R, NRR', NO2, галогена и CF3,
каждый из R, R' и R" независимо выбран из группы, состоящей из водорода, C1-6-алкила, гидроксила, C1-6-алкокси, арила, гетероарила, -CH2-арила, -CH2-гетероарила, и
каждый из X1 и X2 представляет собой независимый анион, выбранный из группы, состоящей из галогенида, OH, HSO4, SO4, BF4, SbF6, карбоксилата, карбоната, гидрокарбоната, NO3, сульфоната, гексафторфосфата, фосфата, гидрофосфата и перхлората.
Когда E1 отличен от -C(O)-O-H, E1 можно замещать карбоновой кислотой или преобразовывать в карбоновую кислоту в подходящей химической реакции. Такие реакции хорошо известны специалистам в данной области. Например, когда E1 представляет собой галоген, карбоновую кислоту вводят посредством катализируемого палладием карбонилирования или формирования реактива Гриньяра с последующим добавлением диоксида углерода, или также вводят сложный эфир карбоновой кислоты посредством опосредованного переходным металлом карбонилирования в спиртовых растворителях. Когда E1 представляет собой сложный эфир (-C(O)-O-R"), -CONRR' или -CN, группы можно преобразовывать в карбоновую кислоту посредством сольволиза. Когда E1 представляет собой -NRR', аминогруппу можно преобразовывать в галогены посредством реакции Зандмейера или непосредственно преобразовывать в карбоновую кислоту или сложный эфир через диазоний при опосредованном переходными металлами карбонилировании. Когда E1 представляет собой -CH2-OR, карбоновую кислоту или сложный эфир можно вводить посредством окисления. Когда E1 представляет собой OR, группу OR можно преобразовывать в галогены или O-сульфонаты (такие как тозилат, трифторат), а затем дополнительно преобразовывать в карбоновую кислоту или сложный эфир при опосредованном переходными металлами карбонилировании. Когда E1 представляет собой метил и винил, эти группы можно преобразовывать в карбоновую кислоту посредством окисления (включая KMnO4, O3).
В другом варианте осуществления E1 представляет собой галоген. В этом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где карбонилирование соединения формулы G проводят посредством катализируемой палладием реакции с монооксидом углерода, в соответствии с которой соединение формулы G вступает в реакцию с монооксидом углерода в присутствии палладиевого катализатора, первого основания и лиганда с выходом соединения формулы H. В одном из вариантов осуществления изобретения лиганд представляет собой фосфиновый лиганд. В другом варианте осуществления лиганд выбран из DCPE и DCPP. Основание представляет собой, например, карбонат калия.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где карбонилирование соединения формулы G, где E1 представляет собой галоген, проводят через образование реактива Гриньяра, в результате чего соединение формулы G вступает в реакцию с магнием или литием с последующими CO2 и кислотой с выходом соединения формулы H.
"Содержащий углерод заместитель, способный к солюбилизации до карбоновой кислоты" представляет собой, например, метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, аллил, гексил, октил, циклогексил, бензил и фенил.
Термин "защитная группа азота" означает заместитель, который защищает атом азота в реакции от реагента или химической среды. Защитные группы азота хорошо известны в данной области и включают, например, трет-бутил, винил, фенил, бензил, п-метоксибензил, 3,4-диметоксибензил, п-нитробензил, бензгидрил, тритил, триалкилсилил, простой метоксиметиловый эфир, (2,2,2-трихлорэтокси)метил и 2-(триметилсилил)этокси)метил. Способы снятия защиты с азота также хорошо известны специалистам в данной области. В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где PG выбран из группы, состоящей из C1-6-алкила, винила, C(O)-O-L, C(O)-L, арила, гетероарила, бензила, бензгидрила, тритила, антранила и C1-6-алкоксиметила, где арил, гетероарил, бензил, бензгидрил и тритил необязательно замещены 1-3 заместителями, независимо выбранными из метокси и нитро, C1-6-алкоксиметил необязательно замещен триметилсилилом, а L представляет собой C1-6-алкил, арил или бензил.
Термин "функциональная группа со способностью быть уходящей группой" означает атом или группу атомов, которые уходят из субстрата в реакции замещения или отщепления, т.е. уходящую группу, и включает, например, галоген и сульфонат. В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где E2 выбран из группы, состоящей из галогена, OMs, OTs, OBs, OP(O)(ORi)4, OC(O)Ri, OC(O)ORi и OC(O)NRiRii, где Ri и Rii независимо выбраны из H и C1-6-алкила.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где органическая фаза выбрана из группы, состоящей из бензола, толуола, ксилола, хлорбензола, простого этилового эфира, простого изопропилового эфира, тетрагидрофурана, 2-метилтетрагидрофурана, диоксана, простого метил-трет-бутилового эфира, простого циклопентилметилового эфира, изопропилацетата, этилацетата, гексана, гептана, циклогексана, дихлорметана, дихлорэтана, ацетонитрила, ацетона, метилэтилкетона, бутанола и амилового спирта.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где неорганическое основание выбрано из группы, состоящей из гидроксида натрия, гидроксида лития, гидроксида калия, карбоната натрия, карбоната лития, карбоната калия, гидроксида цезия, карбоната цезия, гидрокарбоната натрия, гидрокарбоната калия, гидрокарбоната лития, фторида лития, фторида натрия, фторида калия, фторида цезия, трет-бутоксида лития, трет-бутоксида натрия, трет-бутоксида калия, фосфата натрия и фосфата калия.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где органическая фаза представляет собой толуол, а неорганическое основание представляет собой гидроксид натрия.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где соединение формулы C выделяют в виде соли, выбранной из соли HCl, H2SO4, Na, K, Li, H3PO4 и HBF4.
Условия формирования амидной связи между кислотой и амином включают, например, реакцию соединений формул B (после разрушения соли) и C со связывающим амиды реагентом и необязательно вспомогательным веществом и основанием в нереакционноспособном растворителе. Связывающие амиды реагенты включают, например, EDC, CDI, SOCl2, (COCl)2, DCC, T3P, DPPA и т.п. Вспомогательные вещества включают HOBT, HOAt, HATU, HOPO и HOSu, пиридин, производные пиридина и т.п. Подходящие основания включают амины формулы N(Rx)3, где каждый из Rx независимо представляет собой водород, алкил и арил, неорганические основания, такие как гидроксид натрия, гидроксид лития, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат лития, карбонат лития, карбонат цезия, фосфат калия и т.п. Условия формирования амидной связи между кислотой или сложным эфиром и амином также включают использование активированного ацила посредством смешанных промежуточных соединений двуокиси углерода. Примеры включают пивалоилхлорид, алкилхлорформиат и основание. Дополнительные примеры связывающих пептиды реагентов в органическом синтезе хорошо известны в данной области и описаны, например, в Han, et al., Tetrahedron 60 (2004) 2447-2467.
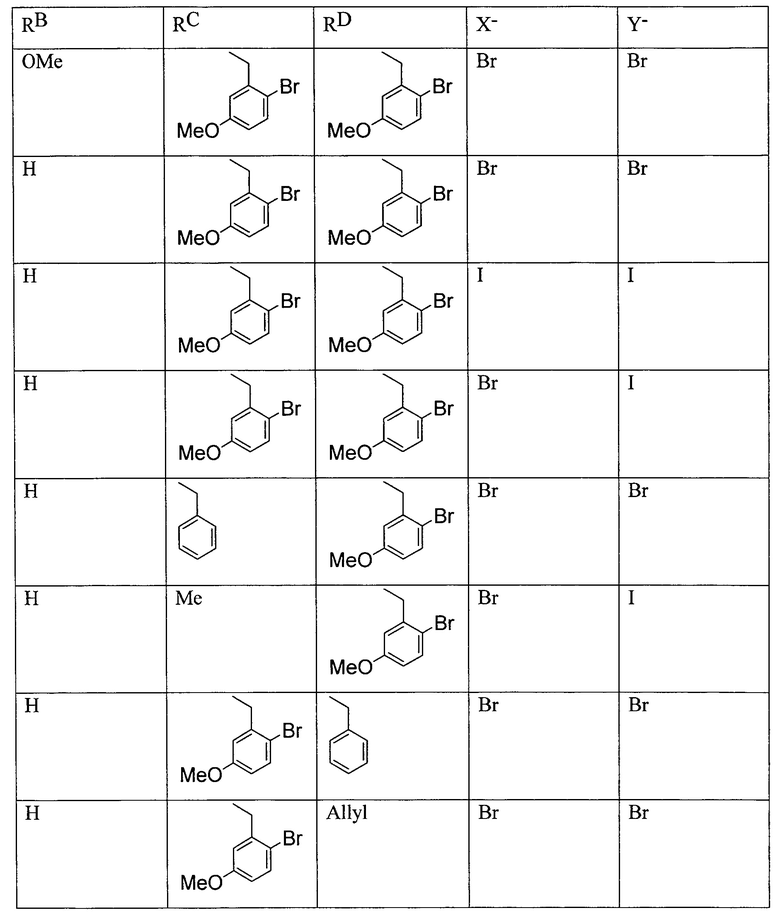
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где в бис-четвертичной соли алкалоида хинного дерева формулы II RC и RD независимо представляют собой C1-6-алкил или бензил, и где алкильные или фенильные части каждого из указанных бензилов необязательно замещены одним-пятью заместителями, независимо выбранными из RF. В одном из вариантов осуществления в бис-четвертичной соли алкалоида хинного дерева формулы II RB представляет собой метокси. В другом варианте осуществления в бис-четвертичной соли алкалоида хинного дерева формулы II RF выбран из группы, состоящей из галогена и метокси.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где бис-четвертичная соль алкалоида хинного дерева представляет собой формулу:
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, дополнительно включающему получение соединения формулы F посредством реакции соединения формулы J:
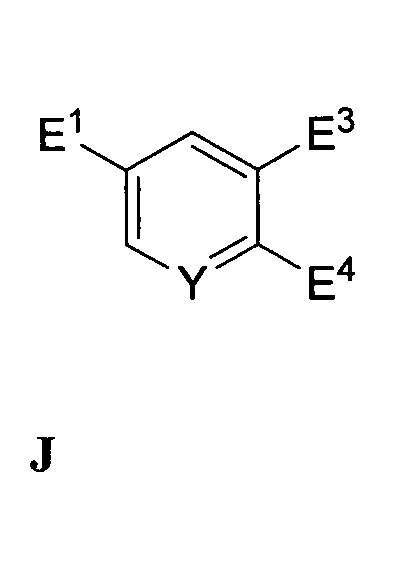 ,
,
где E3 и E4 независимо представляют собой функциональные группы со способностью быть уходящей группой,
с R4-Z1 или его солью и HC(O)-N(R5)2, где R4 представляет собой C1-6-алкил, Z1 представляет собой галогенид магния или лития, и каждый R5 независимо представляет собой H или C1-6-алкил, с выходом соединения формулы K:
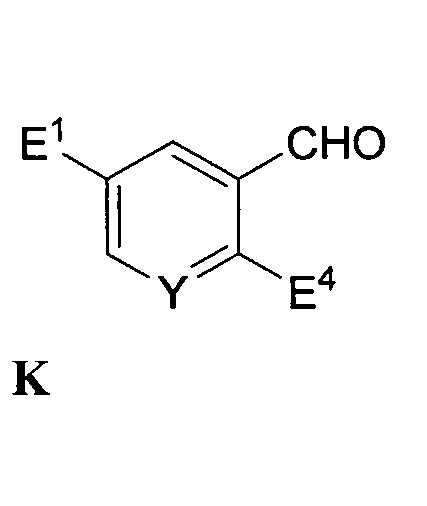 ,
,
реакции соединения формулы K с первым восстановителем с выходом соединения формулы L:
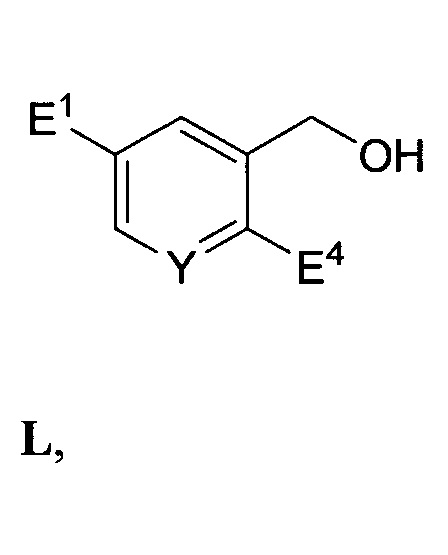
введения защитной группы спирта (APG) в соединение формулы L с выходом соединения формулы M:
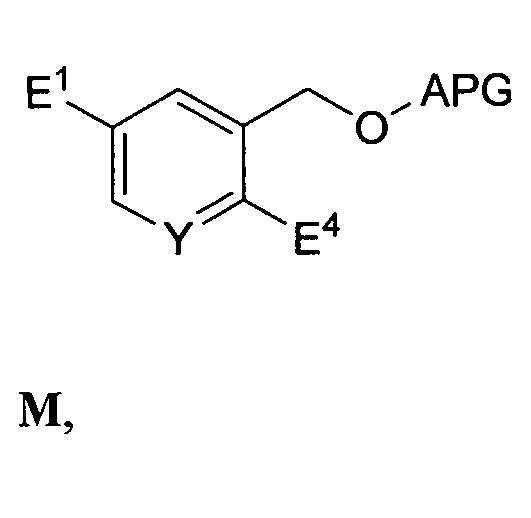
(i) восстановительного карбонилирования соединения формулы M посредством катализируемой палладием реакции с монооксидом углерода и водородом или (ii) реакции соединения формулы M с R6-Z2 или его солью и HC(O)-N(R7)2, где R6 представляет собой C1-6-алкил, Z2 представляет собой галогенид магния или лития, и каждый R7 независимо представляет собой H или C1-6-алкил, с выходом соединения формулы N:
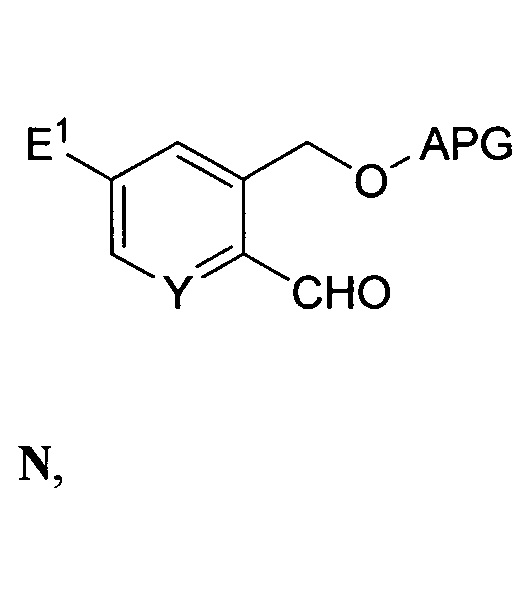
и приведенных ниже этапа (a) или этапа (b) с выходом соединения формулы F:
Этап (a) - связывание соединения формулы N с соединением формулы O:
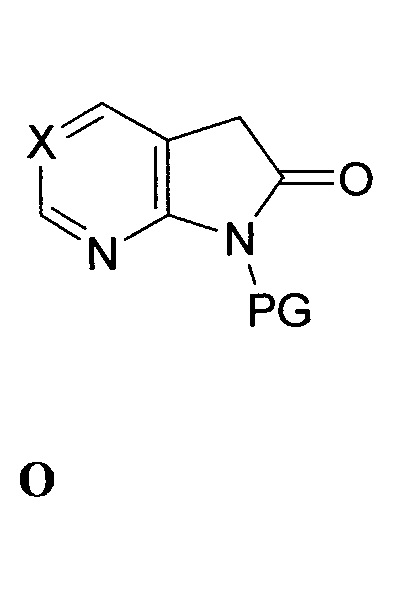 ,
,
в присутствии второго основания с выходом соединения формулы P:
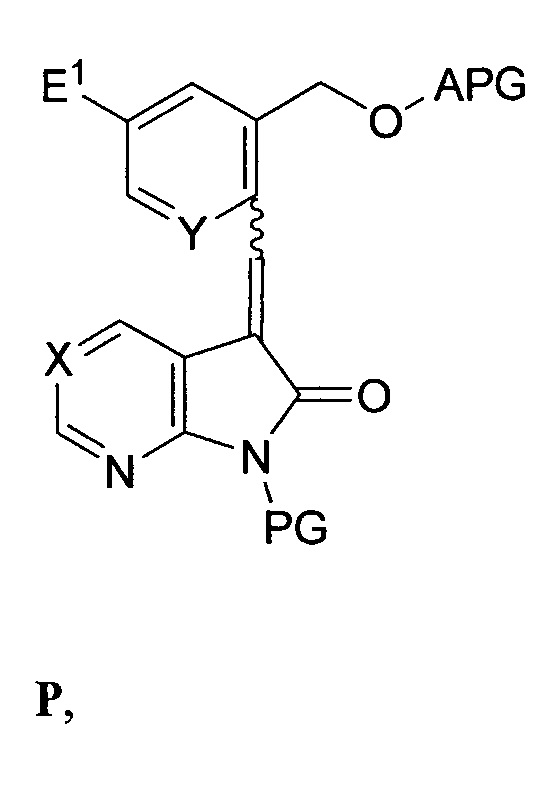
и реакция соединения формулы P со вторым восстановителем с последующим снятием защиты со спиртовой группы и замещением спиртовой группы подходящей уходящей группой E2 с выходом соединения формулы F; или
Этап (b) - восстановление альдегида в соединении формулы N в спирт с получением соединения формулы Q:
 ,
,
реакция соединения формулы Q с активатором с выходом соединения формулы R:
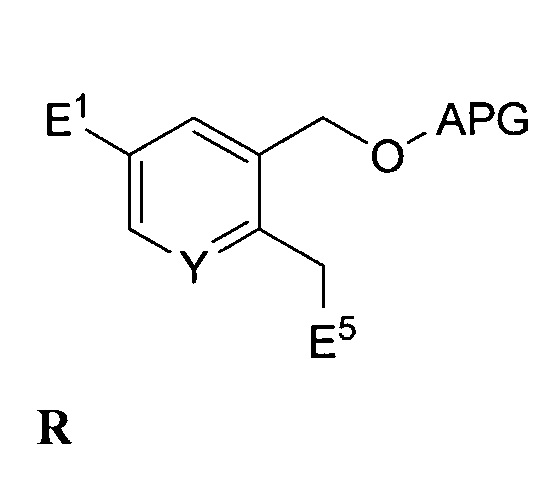 ,
,
где E5 независимо представляет собой функциональную группу со способностью быть уходящей группой, и связывающая соединение формулы R с соединением формулы O в присутствии второго основания с выходом соединения формулы S:
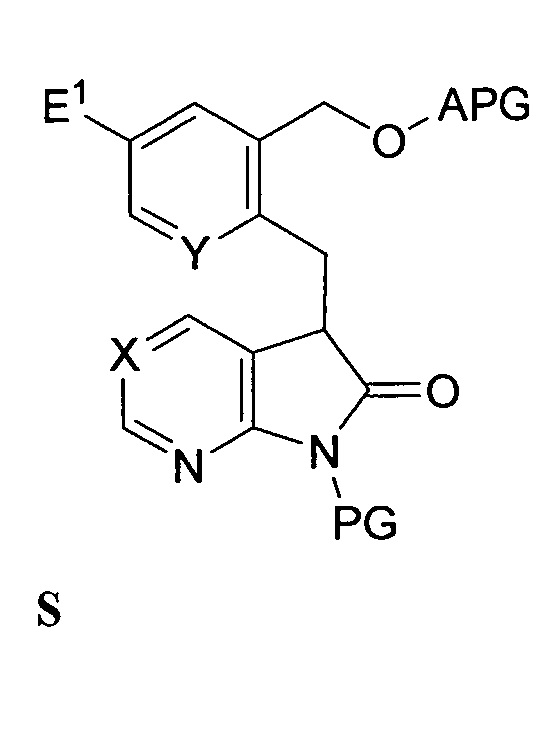 ,
,
с последующим снятием защиты со спиртовой группы и замещением спиртовой группы подходящей уходящей группой E2 с выходом соединения формулы F. В одном из вариантов осуществления изобретения E3 и E4 независимо представляют собой галоген.
В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где соединение формулы N можно получать реакцией соединения формулы M с R6-Z2 или его солью и HC(O)-N(R7)2, где R6 представляет собой C1-6-алкил, Z2 представляет собой галогенид магния или лития, и каждый R7 независимо представляет собой H или C1-6-алкил, с выходом соединения формулы N. В одном из вариантов осуществления изобретения R4 и R6 независимо представляют собой изопропил или втор-бутил, где необязательная соль R4-Z1 и R6-Z2 в каждом случае представляет собой литий, и каждый R5 и R7 представляет собой метил.
Термин "защитная группа спиртовой группы" означает заместитель, защищающий атом кислорода спиртовой группы в реакции от реагента или химической среды. Защитные группы спиртовых групп хорошо известны в данной области. Введение и удаление защитных групп спиртовых групп хорошо известно специалисту в данной области. См., например, Philip J. Koceienski, Protecting Groups, 3rd Ed., Thieme, 2005. В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где APG выбрана из группы, состоящей из THP, тетрагидрофурила, SEM, MOM, BOM, TMS, TES, TBDMS, TIPS и бензила, который необязательно замещен 1-3 заместителями, независимо выбранными из галогена, метила, метокси и нитро.
Термин "восстановитель" означает реагент, способный поставлять водород, и хорошо известен специалисту в данной области. В одном из вариантов осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где первый восстановитель и второй восстановитель независимо выбраны из группы, состоящей из H2, HCO2H, HCO2NH4, NaBH4, LiBH4 и LiAlH4.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где второе основание выбрано из группы, состоящей из DBU, N,N-диизопропилэтиламина, TEA, морфолина и N-метилморфолина.
В другом варианте осуществления настоящее изобретение относится к способу, описываемому в настоящем документе, где второе основание представляет собой неорганическое основание.
Замещение спиртовой группы подходящей уходящей группой E2 можно проводить способами, хорошо известными специалистам в данной области. Например, спиртовую группу можно замещать хлоридом в реакции с тионилхлоридом. Термин "активатор" означает реагент, способный замещать спиртовую группу уходящей группой, например, мезилхлорид, тозилхлорид, (PhO)2POCl, оксалилхлорид, SOCl2 и фосген.
В другом варианте осуществления настоящее изобретение относится к соединению, выбранному из следующей группы:
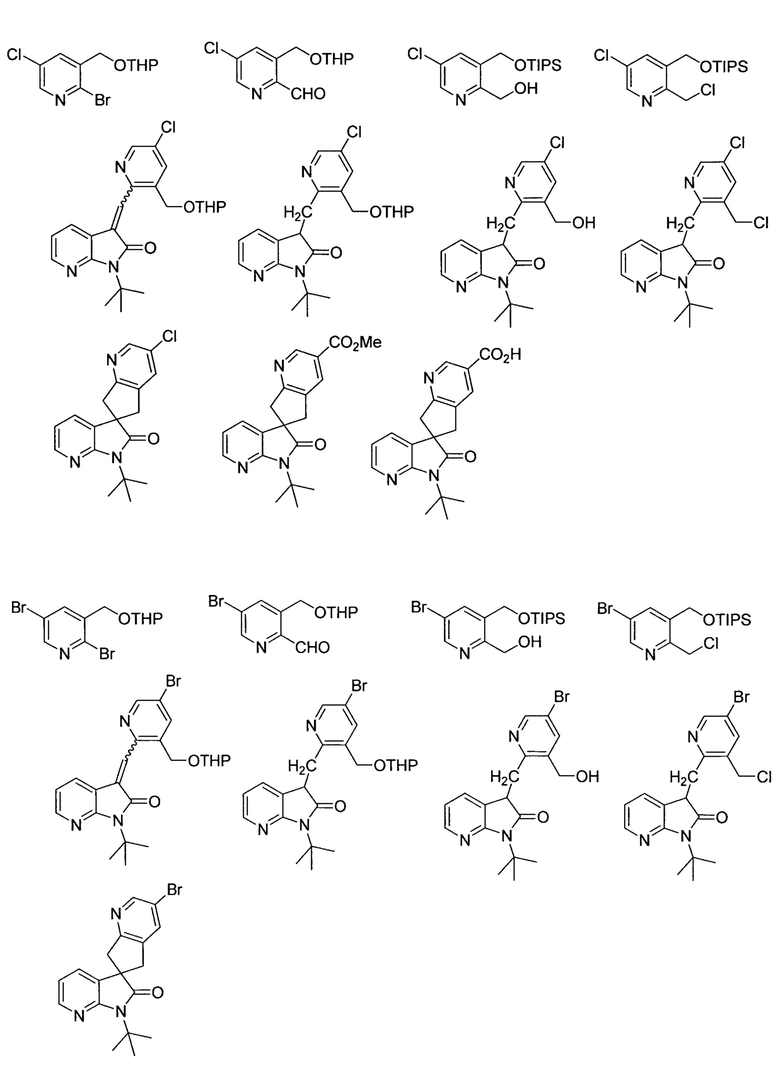
или соли любого из указанного выше.
Как используют в настоящем документе, термин "алкил" относится к одновалентной неразветвленной или разветвленной цепи, насыщенному алифатическому углеводородному радикалу с количеством атомов углерода в указанном диапазоне. Таким образом, например, "C1-6 алкил" (или "C1-C6-алкил") относится к любому из изомеров гексилалкила и пентилалкила, а также н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера, "C1-4-алкил" относится к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера, "C1-3-алкил" относится к н-пропилу, изопропилу, этилу и метилу.
Термин "ацил" означает -C(O)-алкил, где алкил является таким, как определено выше.
Термин "алкокси" означает -O-алкил, где алкил является таким, как определено выше.
Термин "алкенил" относится к одновалентной неразветвленной или разветвленной цепи, насыщенному алифатическому углеводородному радикалу с количеством атомов углерода в указанном диапазоне и по меньшей мере с одной двойной связью углерод-углерод и одинарными связями углерод-углерод в других положениях. Алкенил включает, например, этенил, 1-метилэтинил, 2-пропенил, 2-бутенил, 1,4-пентадиенил и т.п.
Термин "алкинил" относится к одновалентной неразветвленной или разветвленной цепи, насыщенному алифатическому углеводородному радикалу с количеством атомов углерода в указанном диапазоне и по меньшей мере с одной тройной связью углерод-углерод и одинарными и двойными связями углерод-углерод в других положениях. Алкинил включает, например, 2-пропинил, 1-бутинил, 3-гексен-5-инил и т.п.
Термин "циклоалкил" относится к любому моноциклическому кольцу алкана с количеством атомов углерода в указанном диапазоне. Таким образом, например, "C3-6-циклоалкил" (или "C3-C6-циклоалкил") относится к циклопропилу, циклобутилу, циклопентилу и циклогексилу, а "C3-5-циклоалкил" относится к циклопропилу, циклобутилу и циклопентилу.
Термин "галоген" (или "гало") относится к фтору, хлору, брому и йоду (альтернативно, обозначаемым как фторо, хлоро, бромо и йодо).
Термин "арил" относится к фенилу, нафтилу и антранилу.
Термин "гетероарил" относится к (i) 5- или 6-членному гетероароматическому кольцу, содержащему от 1 до 3 гетероатомов, независимо выбранных из N, O и S, или (ii) представляет собой гетеробициклическое кольцо, выбранное из индолила, хинолинила, изохинолинила и хиноксалинила. Подходящие 5- и 6-членные гетероароматические кольца включают, например, пиридил (также обозначаемый как пиридинил), пирролил, пиразинил, пиримидинил, пиридазинил, триазинил, тиенил, фуранил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, оксадиазолил, оксатриазолил, тиазолил, изотиазолил и тиадиазолил. Гетероарилами, представляющими особый интерес, являются пирролил, имидазолил, пиридил, пиразинил, хинолинил (или хинолил), изохинолинил (или изохинолил) и хиноксалинил.
Примеры 4-7-членных, насыщенных гетероциклических колец в объеме настоящего изобретения включают, например, азетидинил, пиперидинил, морфолинил, тиоморфолинил, тиазолидинил, изотиазолидинил, оксазолидинил, изоксазолидинил, пирролидинил, имидазолидинил, пиперазинил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, гексагидропиримидинил, тиазинанил, тиазепанил, азепанил, диазепанил, тетрагидропиранил, тетрагидротиопиранил и диоксанил. Примеры 4-7-членных, ненасыщенных гетероциклических колец в объеме настоящего изобретения (см. HetB) включают мононенасыщенные гетероциклические кольца, соответствующие насыщенным гетероциклическим кольцам, перечисленным в предыдущем предложении, в которых одинарная связь замещена двойной связью (например, одинарная связь углерод-углерод замещена двойной связью углерод-углерод).
Следует понимать, что конкретные кольца, перечисленные выше, не являются ограничением колец, которые можно использовать в настоящем изобретении. Эти кольца являются исключительно иллюстративными.
Если в конкретном случае явно не указано противоположного, любое из различных циклических колец и циклических систем, описываемых в настоящем документе, можно связывать с остальной частью соединений по любому кольцевому атому (т.е. любому атому углерода или любому гетероатому) при условии, что образуется стабильное соединение.
Если явно не указано противоположного, все диапазоны, приводимые в настоящем документе, являются включительными. Например, гетероароматическое кольцо, описанное как содержащее "от 1 до 4 гетероатомов", означает кольцо, которое может содержать 1, 2, 3 или 4 гетероатома. Также следует понимать, что любой диапазон, указанный в настоящем документе, включает в свой объем все поддиапазоны в этом диапазоне. Таким образом, например, гетероциклическое кольцо, описанное как содержащее "от 1 до 4 гетероатомов", предназначено для включения в качестве его составляющий гетероциклических колец, содержащих от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома и 4 гетероатома. В качестве другого примера, арил или гетероарил, описанные как необязательно замещенные "1-4 заместителями", предназначены для включения в качестве их составляющих арила или гетероарила, замещенных 1-4 заместителями, 2-4 заместителями, 3-4 заместителями, 4 заместителями, 1-3 заместителями, 2-3 заместителями, 3 заместителями, 1-2 заместителями, 2 заместителями и 1 заместителем.
Когда любая переменная встречается более одного раза в любом компоненте или в любой другой формуле, изображающей и описывающей соединения по настоящему изобретению, ее определение в каждом случае не зависит от ее определения в любом другом случае. Также комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям.
Если явно не указано противоположного, замещение указанным заместителем допустимо по любому атому в кольце (например, циклоалкиле, ариле или гетероариле) при условии, что такое замещение в кольце химически допустимо и приводит к стабильному соединению.
Соединения по изобретению содержат хиральные центры и, в результате выбора заместителей и комбинаций заместителей, могут содержать дополнительные хиральные центры и, таким образом, могут находиться в виде смесей стереоизомеров, или в виде отдельных диастереомеров или энантиомеров. Все изомерные формы этих соединений, отдельно или в смесях, входят в объем настоящего изобретения.
В тех случаях, когда заместители и комбинации заместителей обеспечивают существование в соединениях по изобретению таутомеров (например, кето-енольных таутомеров), все таутомерные формы этих соединений, находящиеся отдельно или в смесях, входят в объем настоящего изобретения. Следует понимать, что соединения по настоящему изобретению с гидроксильными заместителями на атоме углерода гетероароматического кольца включают соединения, у которых присутствует только гидроксигруппа, соединения, у которых присутствует только таутомерная кето-форма (т.е. оксо-заместитель), и соединения, у которых присутствуют обе кето- и енольная формы.
СОКРАЩЕНИЯ
На всем протяжении описания используют приведенные ниже сокращения.
На схемах 1-15, описанных ниже, представлены существующие ранее способы синтеза соединений формулы I.
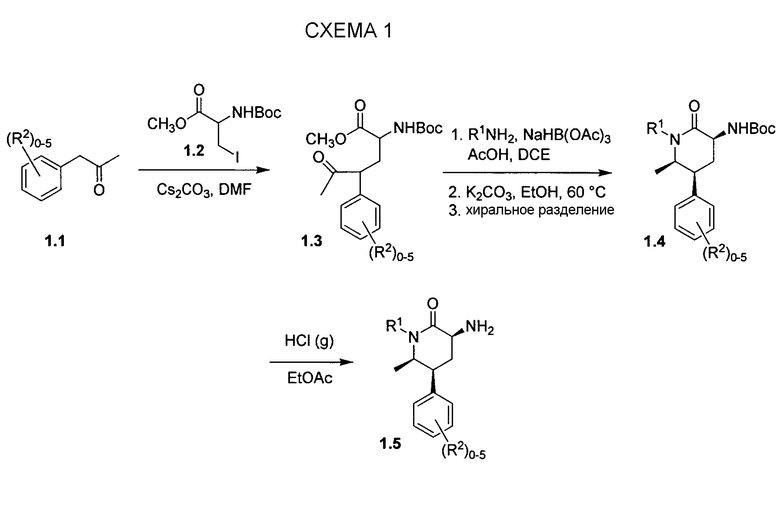
Схема 1 иллюстрирует путь до промежуточных соединений 3-аминопиперидинона типа 1.5, которые можно использовать для получения соединений по настоящему изобретению. Арилацетон 1.1 можно алкилировать с использованием йодаланинового производного 1.2 в основных условиях с получением сложного кетоэфира 1.3. Восстановительное аминирование с последующими реакцией замыкания цикла и эпимеризацией позволяет получать замещенный преимущественно в цис-положении лактам 1.4 в виде рацемической смеси. Хиральное разделение с использованием, например, жидкостной хроматографии с нормальными фазами и удаление защитной группы Boc посредством HCl в EtOAc обеспечивает получение 3-аминопиперидинона 1.5 в виде гидрохлоридной соли.
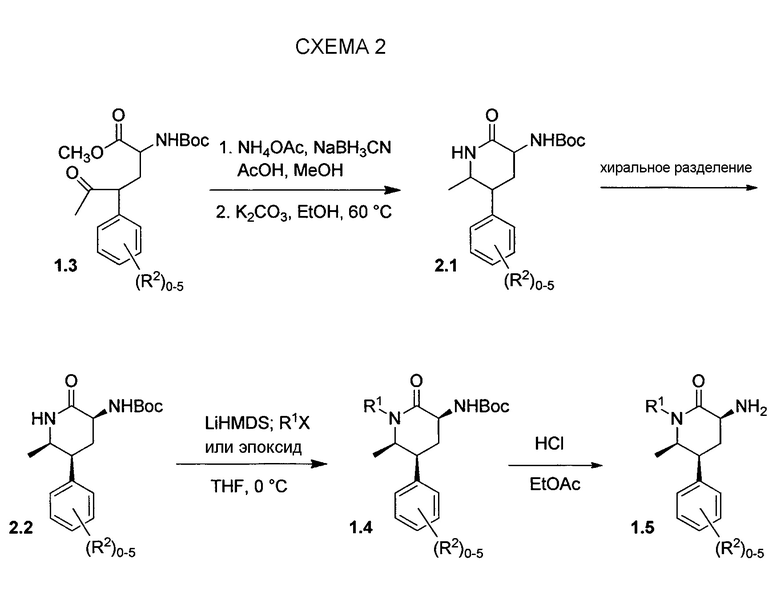
На схеме 2 представлена альтернативная последовательность этапов до промежуточных соединений 3-аминопиперидинона типа 1.5. Восстановительное аминирование сложного кетоэфира 1.3 аммиаком с последующей эпимеризацией позволяет получать 2.1 в виде замещенной в основном в цис-положении рацемической смеси. Хиральное разделение энантиомеров позволяет получать 2.2. N-алкилирование LiHMDS, например, в виде основания и алкилгалогенидом или эпоксидом позволяет получать 1.4. Затем удаление защитной группы Boc посредством HCl позволяет получать 1.5 в виде гидрохлоридной соли.
Третий способ получения промежуточных соединений 3-аминопиперидинона типа 1.5 представлен на схеме 3. N-алкилирование 5-бром-6-метилпиридин-2(1H)-она (3.1) с использованием карбоната цезия в качестве основания и алкилгалогенида с последующим нитрированием позволяет получать 3.2. Затем катализируемое палладием перекрестное связывание с арилбороновой кислотой позволяет получать 3.3. Гидрирование с использованием оксида платины в кислых условиях и хиральное разделение смеси замещенного в основном в цис-положении рацемического продукта позволяет получать 1.5 в виде одного энантиомера.

На схеме 4 представлен путь синтеза до промежуточных соединений 3-аминопиперидинона типа 4.4. Арилацетонитрил 4.1 можно алкилировать с использованием йодаланинового производного 1.2 в основных условиях с получением сложного цианоэфира 4.2. Восстановительная реакция замыкания цикла с использованием водорода и гидроксида палладия на углероде или никеля Ренея, эпимеризация и хиральное разделение позволяют получать цис-лактам 4.3 в виде одного энантиомера. Затем N-алкилирование и удаление защитной группы Boc позволяет получать 4.4 в виде гидрохлоридной соли.
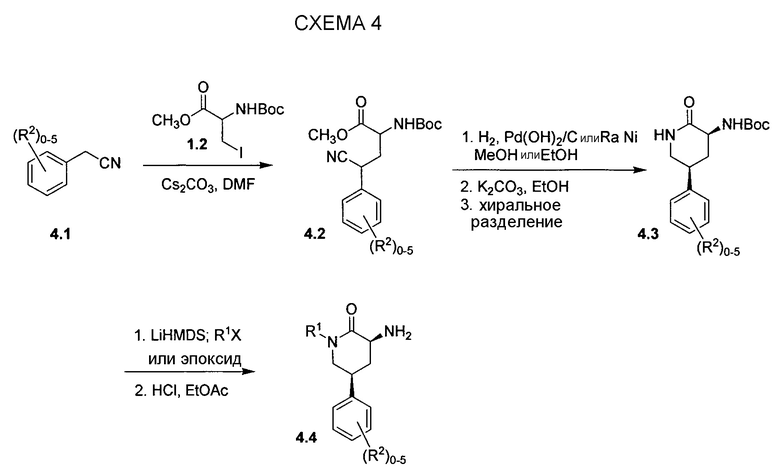
На схеме 5 проиллюстрирован альтернативный путь к промежуточным соединениям 3-аминопиперидинона типа 4.4. Арилацетонитрил 5.1 можно конденсировать с акрилатом 5.2 при повышенной температуре с получением сложного эфира 4-цианобутаноата 5.3. Гидрирование нитрила 5.3 с использованием катализатора никеля Ренея и раствора аммиака в этаноле позволяет получать соответствующий аминовый продукт, который, как правило, преобразуется в цикл in situ с получением пиперидинона 5.4. N-алкилирование лактама 5.4 можно проводить рядом способов, известным специалистам в области органического синтеза, а точный выбор условий зависит от характера алкилирующего средства, R1X. Электрофильное азидирование полученного замещенного лактама 5.5 можно проводить способом, сходным со способом, описанным Эвансом и коллегами (Evans et al. (1990) J. Am. Chem. Soc. 112, 4011-4030), с получением азида 5.6 в виде смеси диастереоизомеров, которые можно разделять посредством хроматографии. Желаемый цис-диастереомер азида 5.6 можно восстанавливать посредством каталитического гидрирования в присутствии ди-трет-бутилдикарбоната с получением соответствующего защищенного Boc амина 5.7, и разделение энантиомеров с использованием хиральной ВЭЖХ или SFC приводит к (3S,5S)-изомеру 5.8. В заключение, стандартное снятие защиты позволяет получать желаемое промежуточное соединение 3-аминопиперидинона 4.4 в виде гидрохлоридной соли.
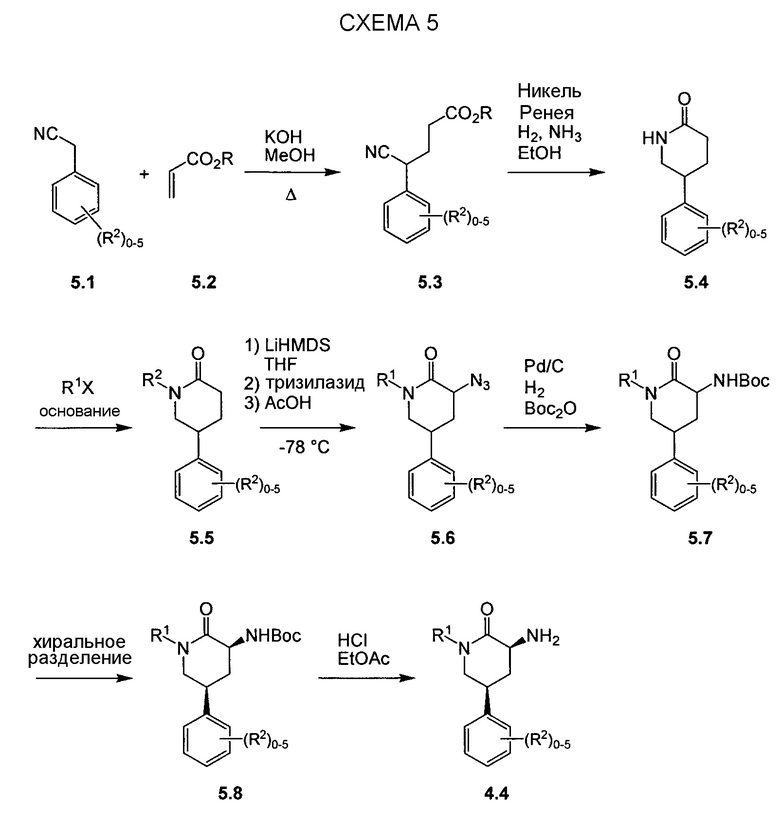
Другим подходом к получению представляющих интерес промежуточных соединений 3-аминопиперидинона, которые особенно пригодны для получения 3-амино-6-метил-5-арилпиперидин-2-онов, таких как 1.5, приведен на схеме 6. Пиридин-2(1H)-он 3.1 можно преобразовывать в N-замещенный пиридинон 6.1 посредством обработки подходящим электрофилом (R1X) в основных условиях. Затем пиридинон 6.1 можно подвергать реакции связывания Сузуки-Мияура с бороновой кислотой 6.2, и полученный 5-арилпиридинон 6.3 можно гидрогенизировать с использованием, например, катализатора оксида платины(IV) с получением соответствующего 5-арилпиперидинона 6.4, который, как правило, получают преимущественно в виде цис-изомера. Дальнейшую обработку пиперидинона 6.4 можно проводить способом, аналогичным способу, описанному на схеме 5. В частности, электрофильное азидирование с последующим одностадийным восстановлением и защитой Boc приводит к карбамату 6.6, а желаемый энантиомер можно получать с использованием хиральной хроматографии. В некоторых случаях желаемый диастереомер азида 6.5 можно выделять в виде рацемической смеси (3S,5S,6R)- и (3R,5R,6S)-изомеров с последующей хроматографией неочищенного продукта на силикагеле, и эту смесь можно обрабатывать, как описано на схеме 6. В других случаях предпочтительным может являться преобразовывать смесь диастереомеров азида 6.5 в соответствующий карбамат 6.6. Смесь диастереомеров карбамата 6.6 можно эпимеризовать в основных условиях, таких как карбонат калия в EtOH, с получением смеси, которая в значительной степени обогащена желаемыми (3S,5S,6R)- и (3R,5R,6S)-изомерами, для получения представляющего интерес энантиомера можно проводить дополнительную очистку, как описано в настоящем документе.
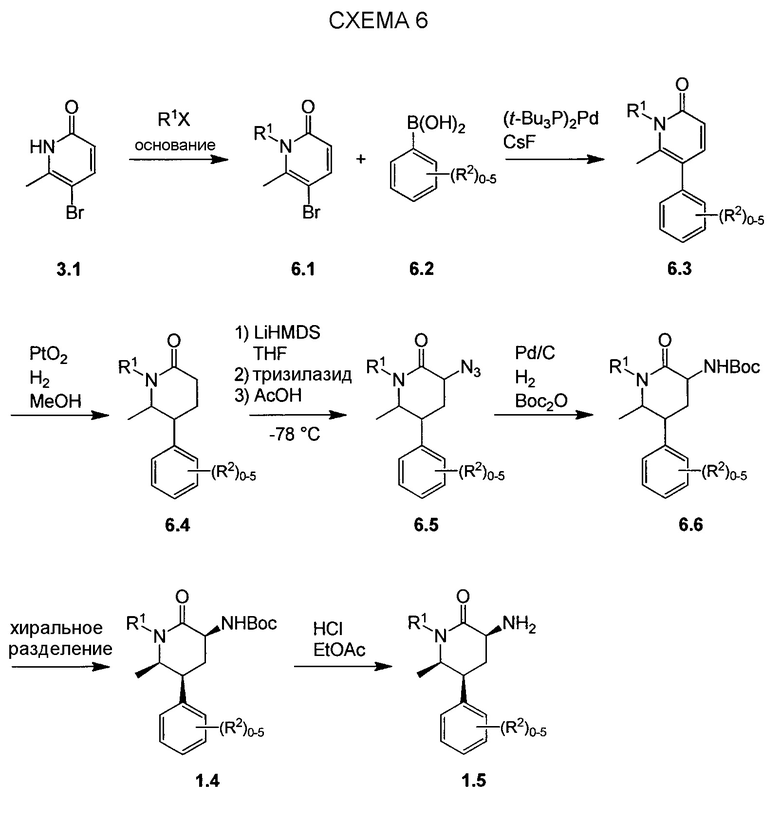
На схеме 7 представлен путь синтеза до промежуточного соединения азаоксиндолпиридиновой кислоты 7.4. Диазотирование аминопиридина 7.1, получение которого описано в WO 2008/020902, с последующей обработкой йодидом калия в присутствии NaNO2 позволяет получать йодид 7.2. Затем катализируемое палладием карбонилирование в метаноле позволяет получать сложный эфир 7.3, который можно омылять гидроксидом натрия с получением 7.4.
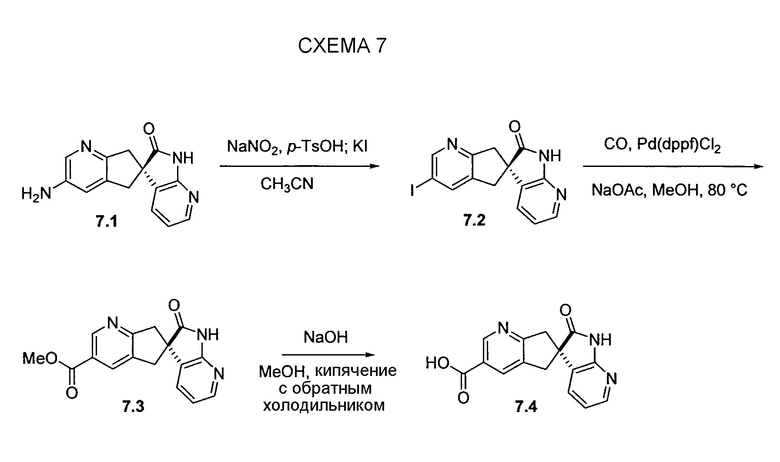
На схеме 8 представлен альтернативный синтез промежуточного соединения азаоксиндолпиридиновой кислоты 7.4. Этерификация двухосновной кислоты 8.1 с последующим бромированием позволяет получать 8.2. Затем восстановление борогидридом натрия обеспечивает получение диола 8.3. Алкилирование защищенного азаоксиндола 8.4 бис-мезилатом, полученным из 8.3, позволяет получать спироцикл 8.5. Катализируемое палладием карбонилирование в метаноле с последующим хиральным разделением позволяет получать сложный эфир 8.6 в виде одного энантиомера. Затем удаление защитной группы SEM в кислых условиях и гидролиз сложного эфира с использованием гидроксида натрия позволяет получать 7.4.
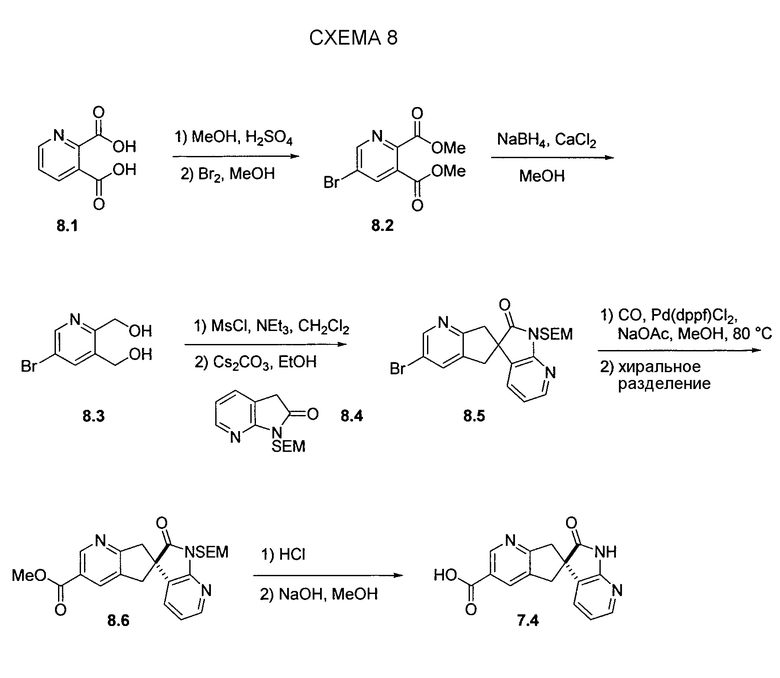
На схеме 9 представлен путь синтеза до промежуточного соединения диазаоксиндолкарбоновой кислоты 9.7. Проводят этерификацию кислоты 9.1 с последующим винилированием в условиях катализа палладием с получением дивинилпиридина 9.2. Затем озонолиз с восстановительной обработкой борогидридом позволяет получать диол 9.3. После мезилирования и обработки хлоридом натрия, полученное дихлорное промежуточное соединение 9.4 можно алкилировать оксиндолом 9.5 в основных условиях с получением спироцикла 9.6 с последующим хиральным разделением энантиомеров. Дехлорирование в условиях буферизированного гидрирования и снятие защиты в кислых условиях позволяет получать кислоту 9.7.
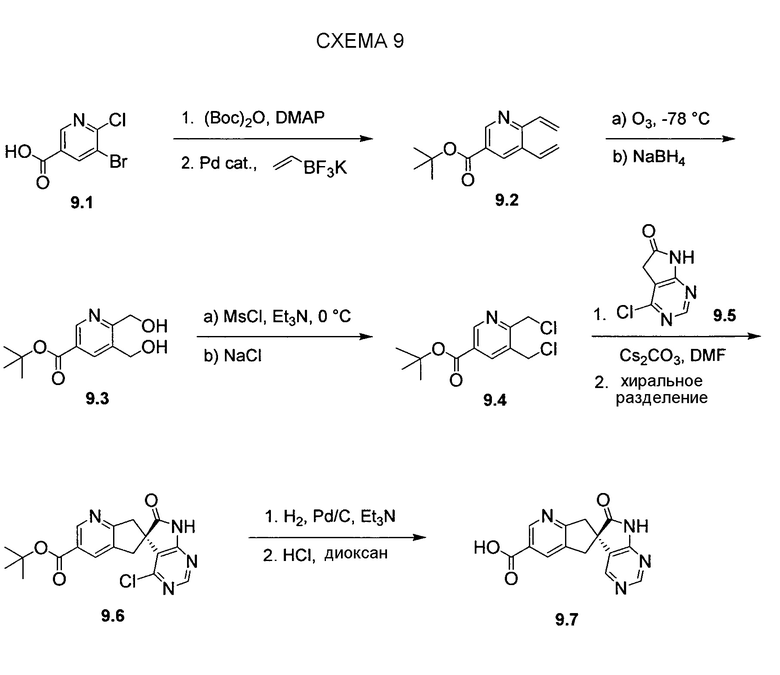
Пригодные производные промежуточных соединений, описываемых в настоящем документе, можно получать хорошо известными способами. Один такой пример проиллюстрирован на схеме 10, на которой промежуточное соединение азаоксиндол 7.4 преобразуют в соответствующее нитриловое производное 10.2, которое можно использовать для получения соединений по настоящему изобретению. Бромирование 7.4 N-бромсукцинимидом в трифториддигидрат бора позволяет получать бромпроизводное 10.1, которое можно преобразовывать в желаемый нитрил 10.2 с использованием цианида цинка и палладиевого катализатора, как показано.
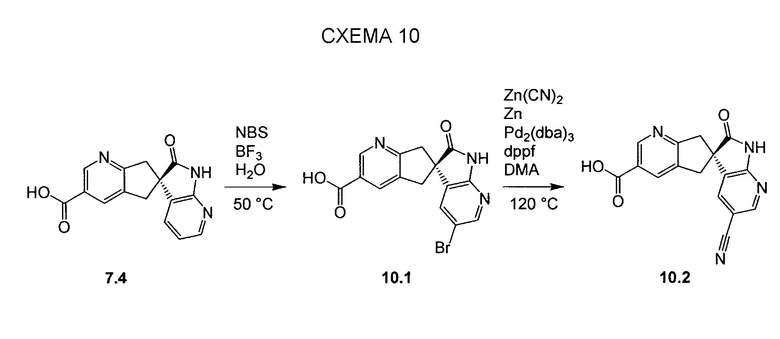
На схеме 11 представлен путь синтеза до промежуточного соединения азаоксиндолиндановой кислоты 11.17. Этерификация двухосновной кислоты 11.1 с последующим гидрированием с использованием в качестве катализатора палладия на углероде позволяет получать анилин 11.2. Дибензилирование в основных условиях с нагреванием позволяет получать 11.3, а восстановление сложного диэфира посредством LiAlH4 обеспечивает получение диола 11.4. Хлорирование тионилхлоридом позволяет получать бензилхлорид 11.5. Катализируемое палладием аминирование бромида 11.6 трет-бутиламином позволяет получать 11.7. Последовательная обработка н-гексиллитием и метилхлорформиатом (2×) позволяет получать сложный азаоксиндоловый эфир 11.8. Алкилирование бензилхлоридом 11.5 в основных условиях в присутствии получаемого из цинхонидина катализатора 11.12 (получаемого посредством алкилирования цинхонидина 11.10 бензилбромидом 11.11) позволяет получать спироцикл 11.13. Снятие защиты азаоксиндола с использованием метансульфоновой кислоты с нагреванием и дибензилирование в стандартных условиях гидрирования позволяет получать анилин 11.14. Диазотирование с последующей обработкой йодидом калия позволяет получать йодид 11.15. Затем катализируемое палладием карбонилирование в метаноле позволяет получать сложный эфир 11.16, который можно омылять гидроксидом натрия с получением 11.17.
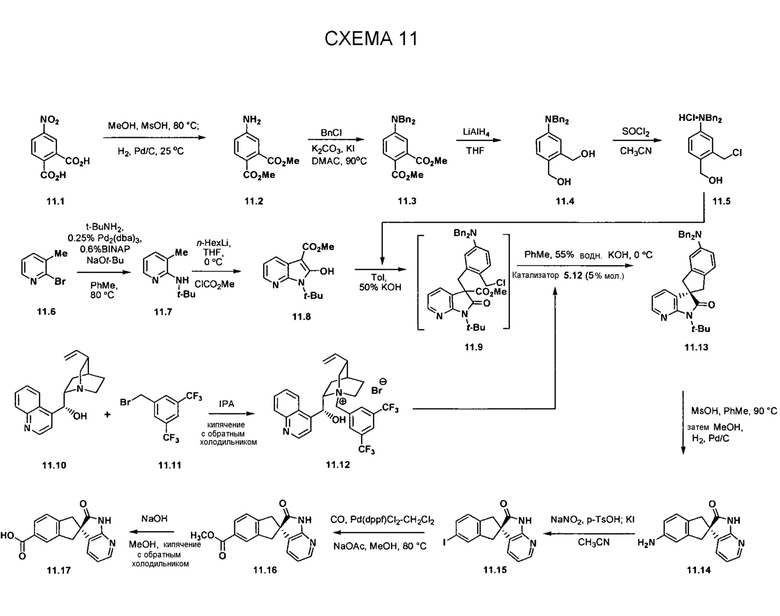
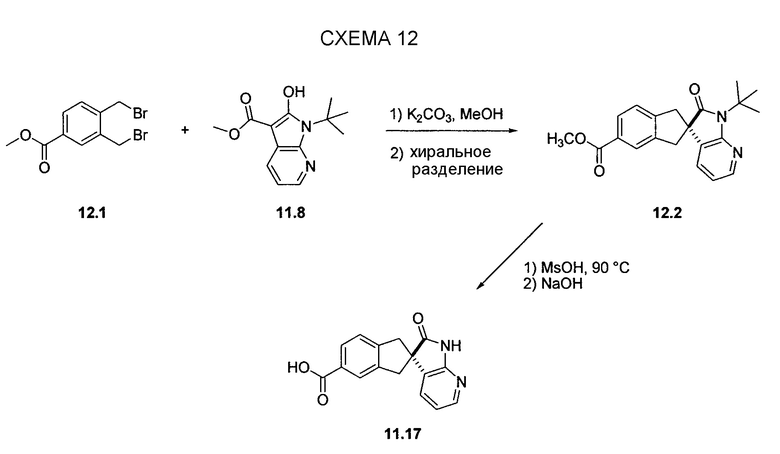
На схеме 12 представлен альтернативный синтез промежуточного соединения азаоксиндолпиридиновой кислоты 11.17. Алкилирование сложного азаоксиндолового эфира 11.8 дибензилбромидом 12.1 с последующим хиральным разделением энантиомеров позволяет получать сложный эфир 12.2. Последовательное снятие защиты азаоксиндола с использованием метансульфоновой кислоты с нагреванием и гидролизом сложного эфира позволяет получать 11.17.
На схеме 13 представлен путь синтеза до промежуточного соединения диазаоксиндолкарбоновой кислоты 13.4. Алкилирование дибромида 12.1 оксиндолом 9.5 в основных условиях и последующее хиральное разделение позволяет получать спироцикл 13.2. Затем дехлорирование в условиях буферизированного гидрирования и гидролиз сложного эфира позволяет получать кислоту 13.4.

Пригодные производные промежуточных соединений, описываемых в настоящем документе, можно получать хорошо известными способами. Один такой пример проиллюстрирован на схеме 14, на которой промежуточное соединение азаоксиндола 11.17 преобразуют в соответствующее нитрильное производное 14.2, которое можно использовать для получения соединений по настоящему изобретению. Обработка 11.17 бромом в уксусной кислоте позволяет получать бромпроизводное 14.1, которое можно преобразовывать в желаемый нитрил 14.2 с использованием цианида цинка и палладиевого катализатора, как показано.
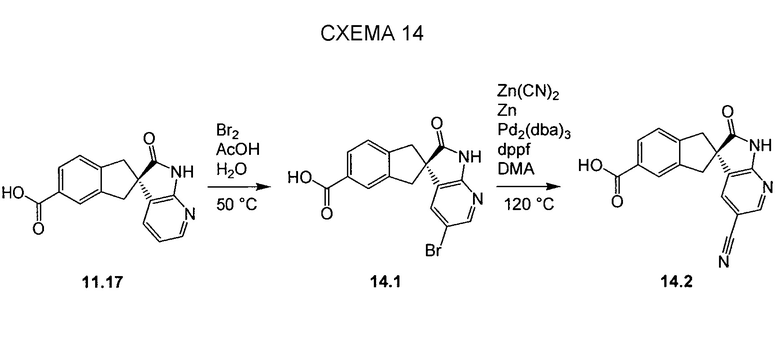
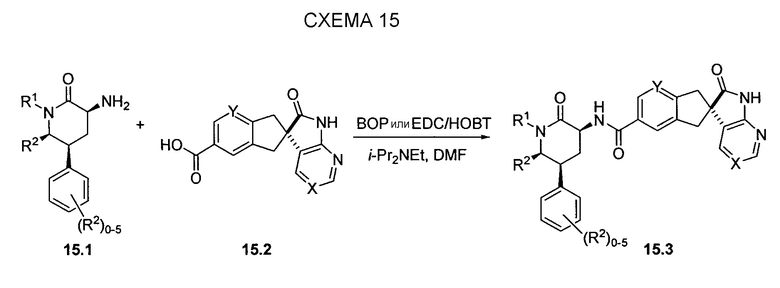
Схема 15 иллюстрирует условия, которые можно использовать для связывания промежуточных соединений 3-аминопиперидинона, таких как 15.1, и промежуточного соединения карбоновой кислоты 15.2 с получением, в данном случае, амидов 15.3. Эти стандартные условия связывания являются характерными для способов получения соединений по настоящему изобретению.
У описанных выше способов синтеза лактамного промежуточного соединения существуют один или несколько недостатков: разделение рацемической смеси посредством хиральной ВЭЖХ, разделение смеси диастереомеров посредством кристаллизации и/или использование дорогого PtO2. В способе по настоящему изобретению используют индуцированное трансаминазой динамическое кинетическое разделение, обеспечивающее высокую диастереоселективность в положениях C5 и C6. Открыто и разработано N-монотрифторэтилирование. Получение цис- и транс-изомеров в альфа-положении амина успешно контролировали посредством кристаллизации в присутствии аральдегидных производных. В целом, этапы синтеза являются менее продолжительными, практичными и эффективными, и происходит значительное увеличение выхода.
ПРИМЕР 1
Изопропил-2-(трет-бутоксикарбониламино)-3-(метилсульфонилокси)пропаноат (2)

К раствору сложного изопропилового эфира N-трет-бутил-L-серина 1 (12 г, 48,5 ммоль)* и метансульфонилхлорида (4,0 мл) в дихлорметане (100 мл) медленно добавляли триэтиламин (7,2 мл) в ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли 1Н HCl (40 мл) с перемешиванием. Органический слой отделяли, промывали 1Н HCl (40 мл) и насыщенным солевым раствором (40 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 2 (14,5 г, 91,9%) в виде твердого вещества. 1H ЯМР (CDCl3, 500 МГц): δ 5,45 (с, уш., 1H), 5,13 (м, 1H), 4,62-4,47 (м, 3Н), 3,04 (с, 3Н), 1,48 (с, 9Н), 1,31 (д, J=6,4 Гц, 6Н); 13C ЯМР (CDCl3, 100 МГц): δ 168,0, 135,1, 80,6, 70,5, 69,1, 53,3, 37,4, 28,3, 21,7, 21,6; масса/заряд в HRMS, рассчитанное для C12H23NO7S, составляет 348,1087 (M+Na); выявленное составляет 348,1097
*получение 1 опубликовано в J. Med. Chem., 2010, 53, 6825-6837 6825
Изопропил-2-(трет-бутоксикарбониламино)-3-йодопропаноат (3)

К раствору 2 (392 г) в ацетоне (3,14 л) добавляли йодид натрия (542 г). Температуру реакции поднимали с 17°C до 29°C. Реакционную смесь поддерживали при комнатной температуре в течение выходных. Смесь фильтровали и промывали MTBE. Фильтрат и смывы комбинировали и концентрировали. Остаток обрабатывали MTBE и водой с небольшим количеством тиосульфата натрия. Органический слой промывали водой и концентрировали до масла. Масло медленно выливали в смесь воды (2 л) и DMF (300 мл) с небольшим количеством затравки при 5°C. Кристаллы отфильтровывали и сушили с получением 3 (400 г, выход 93%).
Изопропил-4-(4-бромфенил)-2-(трет-бутоксикарбониламино)-5-оксогексаноат (5) и изопропил-4-фенил-2-(трет-бутоксикарбониламино)-5-оксогексаноат (6)
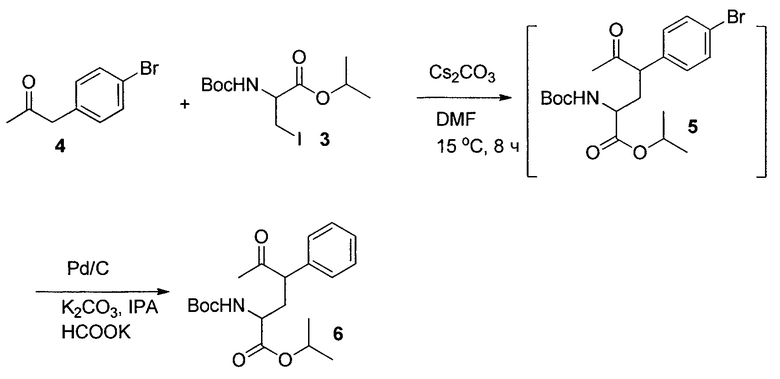
К раствору 4 (51,7 г, 243 ммоль) в DMF (850 мл) добавляли 3 (88 г, 246 ммоль). Полученный раствор охлаждали до 5°C и одной порцией добавляли Cs2CO3 (240 г). Суспензию нагревали до 15°C и перемешивали при этой температуре в течение 2,5 часов. Дополнительно добавляли Cs2CO3 (25 г) и смесь перемешивали в течение дополнительных 8 часов или до того, как анализ ВЭЖХ указывал на преобразование более 95%. Затем партию медленно гасили в смеси 2Н HCl (850 мл) и MTBE (900 мл) при 5-20°C. Органический слой отделяли, и водный слой экстрагировали MTBE (400 мл). Объединенные органические слои дважды отмывали 5% раствором NaHCO3 (400 мл). Полученный раствор, содержащий желаемый продукт 5 (чистота в LC 90%), концентрировали в вакууме. Остаток растворяли в изопропаноле (1 л). К раствору добавляли K2CO3 (25 г), формиат калия (34 г) и 10% Pd/C (20 г). Смесь нагревали до 60°C и перемешивали в течение 2 часов. После охлаждения до комнатной температуры смесь фильтровали. Анализ ВЭЖХ фильтрата показал, что раствор содержал 6 (54,7 г, 95% масс., выход 62%). Неочищенный продукт непосредственно использовали на следующем этапе без дополнительной очистки. Соединение 6 представляет собой смесь двух пар диастереомеров 6-1 и 6-2, частично разделяемых посредством флэш-хроматографии на силикагеле с этилацетатом и гептаном в качестве элюента (1:10). 6-1: 1H ЯМР (CDCl3, 500 МГц): δ 7,35 (м, 2H), 7,30 (м, 1H), 7,20 (м, 2H), 5,17 (уш., 1H), 4,95 (м, 1H), 4,76 (уш., 1H), 3,73 (м, 1H), 2,70 (уш., 1H), 2,07 (с, 1H), 1,45 (с, 9H), 1,29 (д, J=6,6 Гц, 3Н), 1,28 (д, J=6,6 Гц, 3H); 6-2: 1H ЯМР (CDCl3, 500 МГц): δ 5,12 (м, 1H), 4,70 (м, 1H), 3,27 (м, 1H), 2,80 (м, 1H), 2,34 (с, 3H), 1,50 (с, 9H), 1,26 (д, J=6,6 Гц, 3H), 1,25 (д, J=6,6 Гц, 3H); масса/заряд в HRMS: рассчитано для 6-1: C20H29NO5 386,1938 (M+Na); выявлено 386,1947.
Изопропил-2-((трет-бутоксикарбонил)амино)акрилат (7)
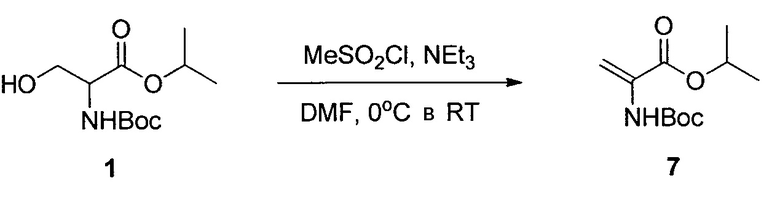
К раствору 1 (10,05 г, 40,6 ммоль) в DMF (100 мл) добавляли MsCl (4,12 мл, 52,8 ммоль) при охлаждении на льду. Затем посредством капельной воронки капельно добавляли триэтиламин (14,16 мл, 102,0 ммоль) в течение 30 минут, при поддержании температуры реакции в пределах 0-5°C. После завершения добавления охлаждающую баню удаляли и желтую гетерогенную реакционную смесь выдерживали при комнатной температуре в атмосфере N2 в течение ночи. Реакционную смесь разбавляли ледяной водой (1 л) и MTBE (1 л). Слои разделяли, и водный слой подвергали обратной экстракции MTBE (500 мл). Органические слои комбинировали и промывали 1 M лимонной кислотой (750 мл), водой (1 л), а затем 10% водным NaCl (1 л). Органический раствор содержал 7 (8,652 г, выход 93%). Растворитель заменяли на DMSO при <40°C, и раствор непосредственно использовали на следующем этапе.
Изопропил-4-фенил-2-(трет-бутоксикарбониламино)-5-оксогексаноат (6)
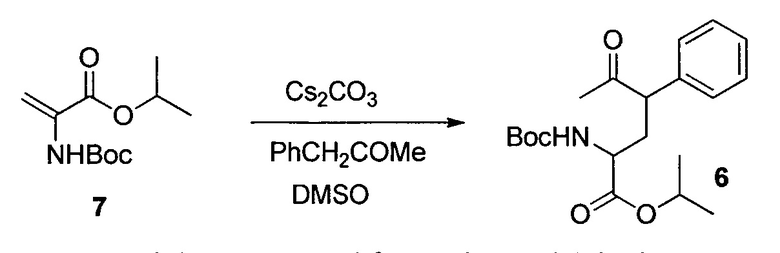
Соединение 6 получали из 7 в DMSO в присутствии 0,5 эквивалента Cs2CO3 с 1,05 эквивалента фенилацетона при комнатной температуре с выходом 79%.
трет-Бутил (5S,6R)-6-метил-2-оксо-5-фенилпиперидин-3-илкарбамат (8)

В 5 л RBF с перемешиванием сверху, с контролем температуры, зондом pH и основной линией добавления добавляли декагидрат тетрабората натрия (26,7 г) и DI воду (1,4 л). После растворения всех твердых веществ добавляли изопропиламин (82,8 г). pH буфера доводили до pH 10,5 с использованием 6Н HCl. Буфер охлаждали до комнатной температуры. Затем добавляли пиридоксаль-5-фосфат (2,8 г) и SEQ ID NO: 1 (70 г) и медленно растворяли при комнатной температуре.
Масло (197,9 г), содержащее 70,7% масс. сложного кетоэфира 6 (140 г, 0,385 моль), растворяли в DMSO (1,4 л). Раствор добавляли в колбу в течение 5-10 мин, и реакционную смесь нагревали до 55°C. pH доводили до 10,5 по показаниям ручного pH-метра и контролировали в течение ночи с использованием автоматического контроллера pH с использованием 8 M водного изопропиламина. Реакционную смесь выдерживали в течение 24 часов.
После подтверждения преобразования >95A% посредством ВЭЖХ реакционную смесь экстрагировали посредством первой добавляемой смеси iPA:IPAc (3:4, 2,8 л) и перемешивания в течение 20 мин. Фазы разделяли, и водный слой подвергали обратной экстракции смесью iPA:IPAc (2:8, 2,8 л). Фазы разделяли, органические слои комбинировали и промывали DI водой (0,5 л). Аналитический выход в органическом слое по ВЭЖХ представлял собой 8 (114,6 г) с >60:1 dr в положениях C5 и C6. Отношение стереоизомеров в положении C2 составляло ~1:1. Экстракт концентрировали и растворяли в CH2Cl2. Органический раствор отмывали водой, затем насыщенным водным NaCl, концентрировали и кристаллизовали из MTBE/н-гексана (2:3). Кристалл отфильтровывали при комнатной температуре и промывали MTBE/н-гексаном (2:3) и сушили с получением смеси цис- и транс-изомеров (~1:1,2) лактама 8 (99,6 г, 80,0%) в виде кристаллов.
Цис:транс (~1: 1,2) смесь, но интеграция ЯМР характеризовалась как 1:1 (при подсчете атомных номеров) Мр 87-90,9°C; 1H ЯМР (CDCl3, 400 МГц):δ 7,40-7,20 (м, 8H, цис или транс), 7,16-7,12 (м, 2H, цис или транс); 6,56 (уш. с, 1H, транс), 6,35 (уш. с, 1H, цис), 5,57 (уш. д, J=4,6 Гц, 1H, цис), 5,34 (уш. д, J=5,7 Гц, 1H, транс), 4,33-4,15 (м, 2H, цис или транс), 3,93 (м, 1H, транс), 3,81 (м, 1H, цис), 3,41 (дт, J=11,8, 5,0 Гц, 1H, цис), 3,29 (дт, J=8,0, 4,4 Гц, 1H, транс), 2,74 (м, 1H, цис), 2,57 (м, 1H, транс), 2,23 (ддд, J=13,5, 8,0, 4,4 Гц, транс), 2,07 (кв., J=11,8 Гц, 1H, цис), 1,46 (с, 9H, цис), 1,42 (с, 9H, транс), 1,05 (д, J=6,9 Гц, 3Н, транс), 0,89 (д, J=6,9 Гц, 3Н, цис); 13C ЯМР (CDCl3, 100 МГц): δ 171,52 (цис), 171,46 (транс), 156,04 (цис или транс), 155,93 (цис или транс), 140,8 (цис), 139,9 (транс), 128,8 (транс), 128,7 (цис), 128,6 (транс), 128,1 (цис), 127,25 (транс), 127,18 (цис), 79,98 (транс), 79,91 (цис), 52,4 (транс), 51,8 (уш., цис), 51,7 (цис), 49,0 (уш., транс), 42,1 (цис), 41,9 (транс), 32,4 (уш., транс), 30,1 (цис), 28,57 (цис или транс), 28,53 (цис или транс), 18,3 (цис), 18,1 (уш., транс); масса/заряд в HRMS, рассчитанное для C17H24N2O3, составляло 327,1679 (M+Na); выявлено 327,1696.
трет-Бутил-(5S,6R)-6-метил-2-оксо-5-фенил-1-(2,2,2-трифторэтил)пиперидин-3-илкарбамат (9) и трет-бутил-(5S,6R)-6-метил-2-оксо-5-фенил-1-(2,2,2-трифторэтил)пиперидин-3-ил(2,2,2-трифторэтил)карбамат (10)
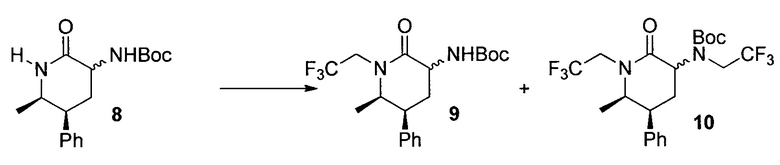
К раствору 8 (480 г, 1,58 моль) в безводном THF (3,8 л) добавляли раствор трет-амоксида лития в гептане (512 мл, 3,1 M, 1,58 моль) приблизительно в течение 15 мин при поддержании температуры реакции от 15 до 20°C. Затем полученный раствор охлаждали до температуры от 0 до 2°C. Затем в течение 15 мин добавляли 2,2,2-трифторэтилтрифторметансульфонат (368 г, 1,58 моль) при поддержании температуры реакции от 0 до 3°C. Раствор перемешивали при 0°C в течение 15 мин. В смесь через капельную воронку в течение 30 мин добавляли DMPU (300 мл) при поддержании температуры реакции от 0 до 3°C. Полученный раствор перемешивали при 0°C в течение 2,5 часов. В смесь течение 10 мин добавляли дополнительный 2,2,2-трифторэтилтрифторметансульфонат (182 г, 0,79 моль) с последующим дополнительным 3,1 M раствором трет-амоксида лития (104 мл) при поддержании температуры реакции от 0 до 3°C. Партию перемешивали в течение дополнительных 2,5 часов при 0°C. Смесь гасили в смеси гептана (4,8 л), воды (3,4 л) и 2Н раствора HCl (280 мл) при температуре ниже 15°C. Фазы разделяли. Водную фазу экстрагировали гептаном (4 л). Комбинированную органическую фазу отмывали водой (2 л). Раствор концентрировали при пониженном давлении до объема приблизительно 1 л при температурах от 25 до 50°C. Неочищенный материал пропускали через порцию силикагеля небольшого слоя с гептаном/этилацетатом. Полученный раствор концентрировали при пониженном давлении до остановки возгонки при температуре ниже 50°C, растворяли в IPAc (2 л) и использовали на следующем этапе обработки. Аналитический выход 9 для цис- и транс-изомеров составлял 85% с отношением ≈8:1.
Аналитически чистые цис- и транс-изомеры 9 выделяли посредством хроматографии на силикагеле с этилацетатом и гептаном в качестве элюента. 9 (цис): 1H ЯМР (CDCl3, 500 МГц): δ 7,30 (м, 5H), 5,75 (с, уш., 1H), 4,35 (м, 1H), 4,15 (м, 1H), 3,80 (м, 1H), 3,50 (м, 1H), 3,17 (м, 1H), 2,45 (м, 2H), 1,45 (с, 9H), 0,93 (д, J=6,7 Гц, 3H); 13C ЯМР (CDCl3, 100 МГц): δ 170,3, 155,9, 140,0, 128,6, 127,6, 127,1, 124,6 (кв., J=279 Гц), 79,7, 58,7, 52,2, 45,3 (кв., J=33,7 Гц), 41,9, 28,3, 27,4, 13,4; HRMS: масса/заряд, рассчитанное для C19H25F3N2O3, составляет 387,1890 (M+H); выявлено: 387,1899. 9 (транс): 1H ЯМР (CDCl3, 500 МГц): δ 7,40 (м, 2H), 7,30 (м, 3H), 5,55 (уш., 1H), 4,53 (уш., 1H), 4,45 (м, 1H), 3,78 (м,2H), 3,45 (м, 1H), 3,0 (м, 1H), 2,12 (м, 1H), 1,46 (с, 9H), 1,12 (д, J=7,0 Гц, 3H); 13C ЯМР (CDCl3, 100 МГц): δ 170,2, 155,9, 139,6, 128,7, 127,9, 127,4, 124,3 (кв., J=279 Гц), 80,0, 59,6, 49,1, 46,9 (кв., J=34,0 Гц), 42,1, 28,3, 25,3, 13,4; HRMS: масса/заряд, рассчитанное для C19H25F3N2O3, составляло 387,1890 (M+H); выявлено 387,1901.
4-Нитробензоат (3S,5S,6R)-6-метил-2-оксо-5-фенил-1-(2,2,2-трифторэтил)пиперидин-3-аминия (11)
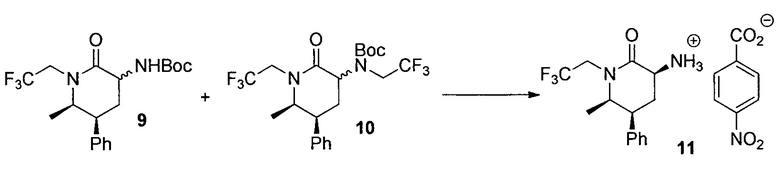
К раствору неочищенного 9, полученного в эксперименте выше, (аналитический образец 10 г, 25,9 ммоль) в iPAC (8 мл), добавляли моногидрат п-толуолсульфоновой кислоты (6,7 г, 35,2 ммоль) и смесь перемешивали при 50-60°C в течение 3 часов до завершения реакции (>99%). Раствор охлаждали до 15-20°C и промывали 10% водным K2CO3 с последующей водой. Водные слои повторно экстрагировали iPAc (5 мл). Органические слои комбинировали и нагревали до 55-60°C. Медленно добавляли 4-нитробензойную кислоту (3,9 г, 23,2 ммоль) в течение 20 мин. Смесь медленно охлаждали до комнатной температуры. Добавляли 5-нитро-2-гидроксилбензальдегид (50 мг) и партию перемешивали по меньшей мере в течение 12 часов. Смесь фильтровали и промывали MeCN с получением 11 в виде кристаллов. Необязательно, получали взвесь в MeCN для дополнительной очистки 11. Фактический выход составлял 90%. Mp 205-208°C; 1H ЯМР (DMSO-d6, 400 МГц): δ 8,21 (дд, J=9,0, 2,1 Гц, 2H), 8,08 (дд, J=9,0, 2,1 Гц, 2H), 7,37 (т, J=7,4 Гц, 2H), 7,28 (т, J=7,4 Гц, 1H), 7,24 (д, J=7,4 Гц, 2H), 4,65 (ддд, J=15,1, 9,7, 7,7 Гц, 1H), 3,72-3,98 (м, 3H), 3,57 (м, 1H), 2,46 (кв., J=12,6 Гц, 1H), 2,25 (м, 1H), 0,90 (д, J=6,4 Гц, 3H); 19F ЯМР (DMSO-d6, 376 МГц): δ 69 (с); 13C ЯМР (DMSO-d6, 100 МГц): δ 168,7, 167,3, 148,3, 143,8, 140,1, 130,1, 128,6, 127,4, 127,0, 124,9 (кв., J=280,9 Гц), 122,8, 58,7, 49,8, 44,5 (кв., J=32,7 Гц), 40,6, 25,3, 13,2.
(5S,6R)-3-Амино-6-метил-5-фенил-1-(2,2,2-трифторэтил)пиперидин-2-он (12)
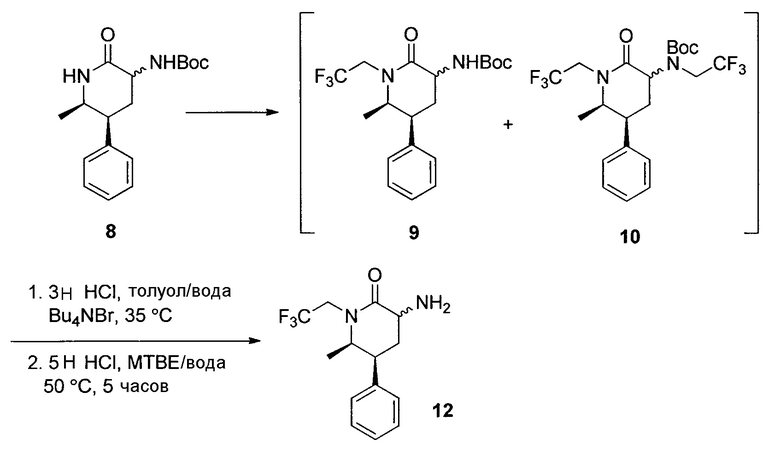
К смеси 8 (20,0 г, 65,7 ммоль) и Na2S2O3 (0,52 г, 3,3 ммоль) в THF (200 мл) добавляли трет-BuOLi (6,8 г, 85 ммоль) при 16°C. Смесь перемешивали при 16°C в течение 15 мин с последующим добавлением трифторэтилтрифторметансульфоната (20,6 г, 89 ммоль) одной порцией. Полученную смесь перемешивали в течение 18 часов при 16°C. Реакционную смесь гасили посредством добавления толуола (70 мл) с последующим 0,5Н раствором HCl (50 мл). Водный слой разделяли и экстрагировали толуолом (20 мл). Объединенный органический слой содержал 87% 9, 6% 10 и 6% 8 по ВЭЖХ, и выход желаемого продукта 9 составлял 87%. Затем органический слой перемешивали с 3Н раствором HCl (80 мл) и бромидом тетрабутиламмония (0,8 г) приблизительно в течение 3 часов до тех пор, пока анализ ВЭЖХ не указывал на завершение селективного удаления группы Boc в непрореагировавшем 8. Водный слой удаляли. Затем органический слой, содержащий 9 и 10, концентрировали при пониженном давлении при 60°C с удалением большей части растворителя. Остаток растворяли в MTBE (60 мл), и добавляли 5Н раствор HCl (65 мл). Двухфазный раствор энергично перемешивали при 50°C приблизительно в течение 5 часов до завершения снятия защиты 9, при этом 10 оставался в основном в неизменном состоянии. После добавление к смеси гептана (30 мл), Органический слой отделяли при 45°C. Водный слой разбавляли водой (60 мл) и полученный водный раствор промывали гептаном (30 мл) при 45°C. Затем водный раствор смешивали с MTBE (100 мл) и повышали основность 10Н раствором NaOH до получения pH смеси приблизительно 10. Органический слой отделяли, и водный слой подвергали обратной экстракции MTBE (60 мл). Объединенные органические слои отмывали насыщенным солевым раствором (60 мл). Полученный органический раствор подходил для следующей реакции. Раствор содержал 12 (15,6 г, 83% из 8) с 97% чистоты в LC в виде смеси двух диастереомеров (цис- и транс-) в отношении 4:1.
4-Метилбензоат (3S,5S,6R)-6-метил-2-оксо-5-фенил-1-(2,2,2-трифторэтил)пиперидин-3-аминия (13)
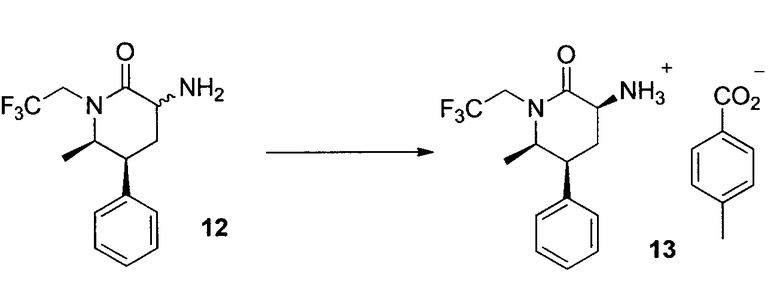
К суспензии 4-метилбензойной кислоты (6,8 г, 49,9 ммоль) и 3,5-дихлорсалицилового альдегида (93 мг, 0,49 ммоль) в MTBE (40 мл) добавляли раствор 12 (13,9 г, 48,5 ммоль) в MTBE (приблизительно 150 мл) в течение 1 часа при 50°C. Полученную суспензию перемешивали приблизительно в течение 3 часов при 50°C. Твердые вещества собирали посредством фильтрации после охлаждения до -5°C в течение 1 часа. Осадок отмывали MTBE (50 мл). Твердые вещества сушили в вакуумной печи с получением 13 (17,6 г, 86%) в виде кристаллов с 99,5% чистоты в LC и 99,6% de. 1H ЯМР (DMSO-d6, 400 МГц): δ 7,85 (д, J=8,1 Гц, 2H), 7,40 (м, 2H), 7,25 (м, 5H), 6,0 (уш., 3H), 4,65 (м, 1H), 3,65-3,80 (м, 2H), 3,45-3,65 (м, 2H), 2,35 (с, 3H), 2,30 (м, 1H), 2,15 (м, 1H), 0,88 (д, J=6,5 Гц, 3H); 13C ЯМР (DMSO-d6, 100 МГц): δ 172,4, 168,5, 142,1, 141,1, 130,9, 129,7, 129,2, 129,0, 128,0, 125,5 (кв., J=279 Гц), 59,1, 51,6, 45,1 (кв., J=32 Гц), 41,6, 28,0, 21,5, 13,9.
Тригидрат (S)-N-((3S,5S,6R)-6-метил-2-оксо-5-фенил-1-(2,2,2-трифторэтил)пиперидин-3-ил)-2'-оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоксамида (15)
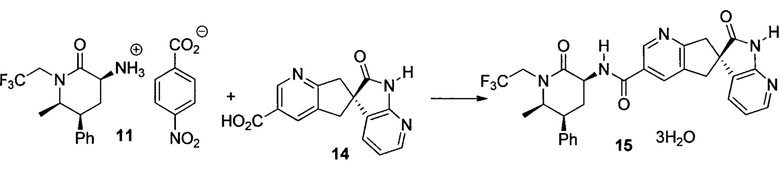
К суспензии 11 (465 г, 96% масс., 0,99 моль) в iPAC (4,6 л) добавляли 5% водный K3PO4 (4,6 л). Смесь перемешивали в течение 5 мин. Органический слой отделяли и дважды промывали 5% водным K3PO4 (4,6 л) и концентрировали при пониженном давлении и растворяли в ацетонитриле (1,8 л).
В другую колбу добавляли 14 (303 г, 91,4% масс.), ацетонитрил (1,8 л) и воду (1,8 л) с последующим 10Н NaOH (99 мл). Полученный раствор перемешивали в течение 5 мин при комнатной температуре, и к смеси добавляли полученный выше раствор хирального амина, и контейнер промывали ацетонитрилом (900 мл). Добавляли гидрат HOBT (164 г) с последующим гидрохлоридом EDC (283 г). Смесь перемешивали при комнатной температуре в течение 2,5 часов. К смеси добавляли iPAc (4,6 л), и органический слой отделяли, промывали 5% водным NaHCO3 (2,3 л) с последующей смесью 15% водной лимонной кислоты (3,2 л) и насыщенного водного NaCl (1,2 л). В заключение, полученный органический слой промывали 5% водным NaHCO3 (2,3 л). Органический раствор концентрировали при температуре ниже 50°C и растворяли в метаноле (2,3 л). Раствор медленно добавляли к смеси воды (6 л) и метанола (600 мл) приблизительно с 2 г затравочного кристалла. И полученную суспензию перемешивали в течение ночи при комнатной температуре. Кристаллы отфильтровывали, промывали водой/метанолом (4 л, 10:1) и сушили в потоке азота при комнатной температуре с получением 15 (576 г, выход 97%) в виде тригидрата. 1H ЯМР (500 МГц, CDCl3): δ 10,15 (уш. с, 1Н), 8,91 (уш. с, 1Н), 8,21 (д, J=6,0 Гц, 1Н), 8,16 (дд, J=5,3, 1,5 Гц, 1Н), 8,01 (уш. с, 1Н), 7,39-7,33 (м, 2Н), 7,31-7,25 (м, 1Н), 7,22-7,20 (м, 2Н), 7,17 (дд, J=7,4, 1,6 Гц, 1Н), 6,88 (дд, J=7,4, 5,3 Гц, 1Н), 4,94 (дкв., J=9,3, 7,6 Гц, 1Н), 4,45-4,37 (м, 1Н), 3,94-3,87 (м, 1Н), 3,72 (д, J=17,2 Гц, 1Н), 3,63-3,56 (м, 2Н), 3,38-3,26 (м, 1Н), 3,24 (д, J=17,3 Гц, 1Н), 3,13 (д, J=16,5 Гц, 1Н), 2,78 (кв., J=12,5 Гц, 1Н), 2,62-2,56 (м, 1Н), 1,11 (д, J=6,5 Гц, 3Н); 13C ЯМР (126 МГц, CD3CN): δ 181,42, 170,63, 166,73, 166,63, 156,90, 148,55, 148,08, 141,74, 135,77, 132,08, 131,09, 130,08, 129,66, 129,56, 128,78, 128,07, 126,25 (кв., J=280,1 Гц), 119,41, 60,14, 53,07, 52,00, 46,41 (кв., J=33,3 Гц), 45,18, 42,80, 41,72, 27,79, 13,46; масса/заряд в HRMS: рассчитано для C29H26F3N5O3 550,2061 (M+H): выявлено 550,2059.
Альтернативный способ для 15:
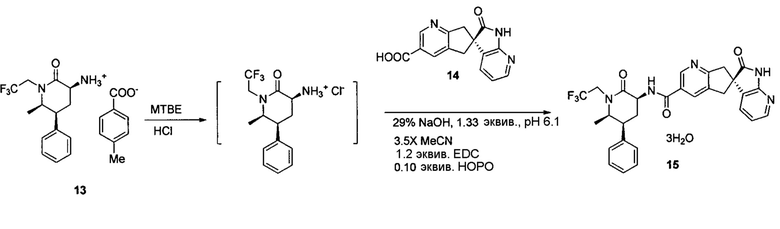
К суспензии 13 (10 г, 98% масс., 23,2 ммоль) в MTBE (70 мл) добавляли 0,6Н HCl (42 мл). Органический слой отделяли и экстрагировали дополнительной 0,6Н HCl (8 мл). Комбинированный водный раствор промывали MTBE (10 мл×3). К полученному водному раствору добавляли ацетонитрил (35 мл) и 14 (6,66 г, 99% масс.). Полученную суспензию нейтрализовали 29% раствором NaOH до pH 6. Добавляли HOPO (0,26 г) с последующим гидрохлоридом EDC (5,34 г). Смесь перемешивали при комнатной температуре в течение 6-12 часов до завершения преобразования (>99%). Добавляли этанол (30 мл) и смесь нагревали до 35°C. Полученный раствор добавляли в течение 2 часов в другую трехгорлую колбу, содержащую этанол (10 мл), воду (30 мл) и затравочный кристаллы 15 (0,4 г). Одновременно в смесь также добавляли воду (70 мл). Затем суспензию охлаждали до 5°C в течение 30 мин и фильтровали. Осадок промывали смесью этанола/воды (1:3, 40 мл). Осадок сушили в вакуумной печи при 40°C с получением тригидрата 15 (13,7 г, 95%) в виде кристаллов.
ПРИМЕР 2
N-Метокси-N-метил-2-(2,3,6-трифторфенил)ацетамид (17)
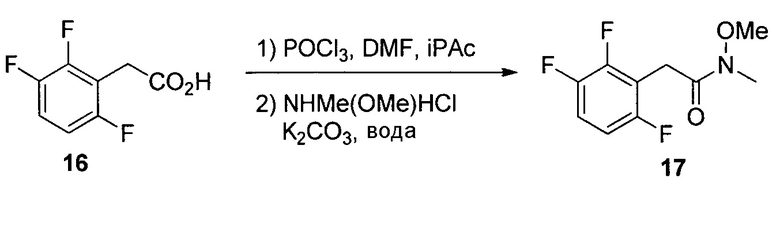
К раствору DMF (58,1 мл, 750 ммоль) в iPAC (951 мл) добавляли POCl3 (55,9 мл, 600 ммоль) при охлаждении на льду. После выдерживания в течение 1 часа в ледяной бане добавляли кислоту 16 (95 г, 500 ммоль) при охлаждении на льду. Раствор перемешивали при охлаждении на льду в течение 30 мин. Раствор в течение 30 мин добавляли в раствор K2CO3 (254 г, 1,835 моль) и NHMe(OMe)HCl (73,2 г, 750 ммоль) в воде (951 мл) при температуре ниже 8°C. После выдерживания в течение 30 мин при температуре ниже 8°C органический слой отделяли, дважды промывали водой (500 мл) и однократно насыщенным водным NaCl (100 мл) и концентрировали при пониженном давлении с получением 17 в виде масла (117,9 г, 97,7% масс., выход 99%). 1H ЯМР (CDCl3, 400 МГц); δ 7,05 (м, 1H), 6,82 (м, 1H), 3,86 (с, 2H), 3,76 (с, 3H), 3,22 (с, 3H); 19F ЯМР (CDCl3, 376,6 МГц); δ -120,4 (дд, J=15,1, 2,7 Гц), -137,9 (дд, J=20,8, 2,7 Гц), -143,5 (дд, J=20,8, 15,1 Гц); 13C ЯМР (CDCl3, 100 МГц); δ (ддд, J=244, 6,2, 2,7 Гц), 149,3 (ддд, J=249, 14,4, 169,4, 156,98,4 Гц), 147,1 (ддд, J=244, 13,1, 3,5 Гц), 115,5 (ддд, J=19,4, 9,9, 1,5 Гц), 133,4 (дд, J=22,3, 16,4 Гц), 110,2 (ддд, J=24,8, 6,7, 4,1 Гц), 32,4 (уш.), 26,6 (м); масса/заряд в HRMS рассчитано для C10H10F3NO2 234,0736 (M+H); выявлено 234,0746.
1-(2,3,6-Трифторфенил)пропан-2-он (18)

Смесь CeCl3 (438 г, 1779 ммоль) и THF (12 л) нагревали при 40°C приблизительно в течение 2 часов, затем охлаждали до 5°C. Добавляли хлорид метилмагния в THF (3 M, 3,4 л) при 5-9°C, а затем смесь нагревали до 16°C и выдерживали в течение 1 часа. Суспензию повторно охлаждали до температур от -10 до -15°C. В суспензию в течение 15 мин добавляли раствор 17 (1,19 кг) в THF (2,4 л). После подтверждения завершения реакции реакционную смесь переносили в холодный раствор соляной кислоты (2 N, 8,4 л) и MTBE (5 л) при 5-10°C. Водную фазу отделяли, и органический слой промывали водным 5% K2CO3 (6 л), а затем 10% водным NaCl (5 л). Органический слой сушили над Na2SO4, концентрировали с получением неочищенного 18 (917 г, >99% масс.) c выходом 95%. Неочищенный 18 использовали на следующем этапе без дополнительной очистки. Аналитически чистый 18 получали посредством колонки с силикагелем. 1H ЯМР (CDCl3, 400 МГц); δ 7,07 (м, 1H), 6,84 (м, 1H), 3,82 (с, 2H), 2,28 (с, 3H); 19F ЯМР (CDCl3, 376,6 МГц); δ -120,3 (дд, J=15,3, 2,5 Гц), -137,8 (дд, J=21,2, 2,5 Гц), -143,0 (дд, J=20,2, 15,3 Гц); 13C ЯМР (CDCl3, 100 МГц); δ 202,2, 156,5 (ддд, J=244, 6,3, 2,9 Гц), 148,9 (ддд, J=249, 14,4, 8,6 Гц), 147,0 (ддд, J=244, 13,1, 3,5 Гц), 115,7 (ддд, J=19,4, 10,5, 1,2 Гц), 112,8 (дд, J=22,7, 17,0 Гц), 110,3 (ддд, J=24,8, 6,7, 4,1 Гц), 37,2 (д, J=1,2 Гц), 29,3.
Изопропил-2-((трет-бутоксикарбонил)амино)-5-оксо-4-(2,3,6-трифторфенил)гексаноат (19)
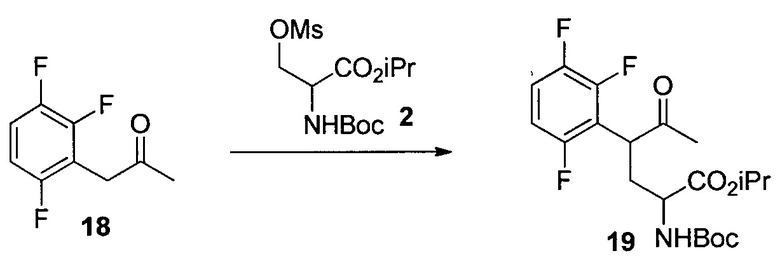
К раствору 18 (195 г, 1,03 моль) в MTBE (1,8 л) добавляли бромид цинка (67 г, 0,30 моль) с последующим 2 (390 г, 1,2 моль). Затем несколькими порциями добавляли трет-BuOLi (290 г, 3,6 моль) при поддержании температуры реакции при температуре ниже 40°C. Полученную смесь перемешивали при 35°C в течение 24 часов и гасили в смеси 2Н HCl (5,6 л) и гептана (5 л) при 0°C. Органический слой отделяли и дважды промывали 5% водным NaHCO3 (5 л). Полученный органический раствор концентрировали при пониженном давлении. Остаток растворяли в гептане (2 л), и раствор снова концентрировали при пониженном давлении. Полученное масло растворяли в DMSO (2,5 л), и раствор использовали на следующем этапе без дополнительной очистки. Анализ ВЭЖХ показал, что раствор содержал желаемый продукт 19 (290 г, выход 67%) в качестве основного компонента вместе с 5% исходного вещества 18. Аналитически чистый продукт 19 в виде одной пары диастереомеров выделяли посредством хроматографии на силикагеле со смесью этилацетата и гептана в качестве элюента. HRMS: масса/заряд, рассчитанное для C20H26F3NO5, 418,1836 (M+H); выявлено 418,1849.
трет-Бутил-((5S,6R)-6-метил-2-оксо-5-(2,3,6-трифторфенил)пиперидин-3-ил)карбамат (20)

В 0,5-л цилиндрический реактор Sixfors с перемешиванием сверху, контролем температуры, зондом pH и основной линией добавления добавляли декагидрат тетрабората натрия (3,12 г) и DI воду (163 мл). После растворения всех твердых веществ добавляли изопропиламин (9,63 г). pH буфера доводили до pH 10,5 с использованием 6Н HCl. Буфер охлаждали до комнатной температуры. Затем добавляли пиридоксаль-5-фосфат (0,33 г) и SEQ ID NO: 1 (8,15 г) и медленно растворяли при комнатной температуре.
Неочищенный сложный кетоэфир 19 (23,6 г, 69% масс., аналитический образец 16,3 г, 39 ммоль) растворяли в DMSO (163 мл), и раствор добавляли в реактор в течение 5-10 мин. Затем реакционную смесь нагревали до 55°C. pH доводили до 10,5 по показаниям ручного pH-метра и контролировали в течение ночи с использованием автоматического контроллера pH с использованием 8 M водного изопропиламина. Реакционную смесь выдерживали в течение 27,5 часов.
После подтверждения преобразования >95A% посредством ВЭЖХ реакционную смесь экстрагировали посредством первой добавляемой смеси iPA:iPAc (3:4, 350 мл) и перемешивания в течение 20 мин. Фазы разделяли, и водный слой подвергали обратной экстракции смесью iPA:iPAc (2:8, 350 мл). Фазы разделяли. Органические слои комбинировали и промывали DI водой (90 мл). Аналитический выход в органическом слое на основе ВЭЖХ составлял 20 (9,86 г, 70,5% аналитический выход) с >60:1 dr в положениях C5 и C6.
трет-Бутил-((3S,5S,6R)-6-метил-2-оксо-5-(2,3,6-трифторфенил)пиперидин-3-ил)карбамат (21)
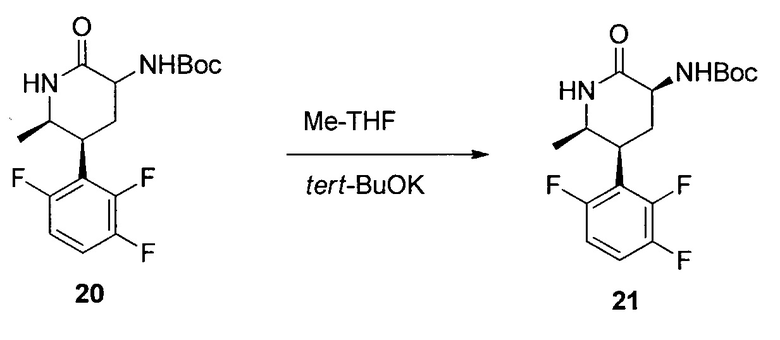
Раствор неочищенной смеси цис- и транс-изомеров 20 в смеси iPAc и iPA (1,83% масс., 9,9 кг; аналитический образец 181 г в виде смеси) концентрировали при пониженном давлении и растворяли в 2-Me-THF (3,6 л). К раствору добавляли трет-BuOK (66,6 г, 0,594 моль) при комнатной температуре. Суспензию перемешивали при комнатной температуре в течение 2 часов. Смесь выливали в воду (3,5 л), и органический слой отделяли, промывали 15% масс. водного NaCl (3,5 л), сушили над Na2SO4 и концентрировали досуха. Остаток суспендировали iPAc (275 мл) и гептаном (900 мл) при 60°C. Суспензию медленно охлаждали до 1°C. Твердое вещество фильтровали и промывали iPAc и гептаном (1:3), сушили с получением 21 (166 г, 93% масс.; 85%) в виде кристаллов. Mp 176-179°C; 1H ЯМР (CDCl3, 500 МГц): δ 7,06 (м, 1H), 6,84 (м, 1H), 5,83 (уш. с, 1H), 5,58 (уш. с, 1H), 4,22 (м, 1H), 3,88-3,79 (м, 2H), 2,77 (м, 1H), 2,25 (м, 1H), 1,46 (с, 9H), 1,08 (д, J=6,4 Гц, 3H); 19F ЯМР (CDCl3, 376 МГц): δ -117 (д, J=14 Гц), -135 (д, J=20 Гц), -142 (дд, J=20, 14 Гц); 13C ЯМР (CDCl3, 100 МГц): δ 171,1, 156,6 (ддд, J=245, 6,4, 2,8 Гц), 155,8, 149,3 (ддд, J=248, 14,4, 8,8 Гц), 147,4 (ддд, J=245, 14,2, 3,8 Гц), 118,0 (дд, J=19,3, 14,5 Гц), 115,9 (дд, J=19,2, 10,4 Гц), 111,0 (ддд, J=26,4, 6,0, 4,3 Гц), 79,8, 51,4, 49,5, 34,1, 29,3, 28,3, 18,0; HRMS: масса/заряд, рассчитанное для C17H21F3N2O3, составляет 381,1396 (M+Na); выявлено 381,1410.
трет-Бутил-((5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-3-ил)карбамат (22)
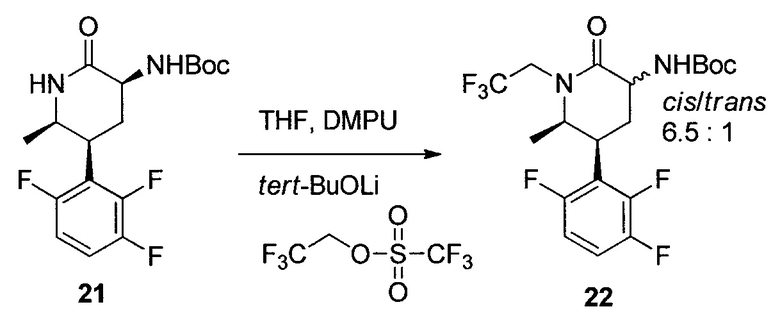
К раствору 21 (10 г, чистота 87%, 24,3 ммоль) в THF (70 мл) одной порцией добавляли трет-BuOLi (2,5 г, 31,2 ммоль) при 5°C. Раствор охлаждали до температуры от 0 до 5°C и одной порцией добавляли трифторэтилтрифторметансульфонат (10,0 г, 43 ммоль). Медленно в течение 15 мин добавляли DMPU (7 мл) при поддержании температуры реакции ниже 5°C. После перемешивания смеси при 0°C в течение 3 часов, добавляли дополнительный трет-BuOLi (0,9 г, 11,2 ммоль). Смесь выдерживали в течение дополнительных 90 мин. Смесь гасили 0,2Н HCl (70 мл) с последующим добавлением гептана (80 мл). Органический слой отделяли, и водный слой экстрагировали гептаном (30 мл). Объединенные органические слои промывали 15% водной лимонной кислотой (50 мл) и 5% водным NaHCO3 (50 мл). Раствор концентрировали при пониженном давлении при 40°C, и полученное масло растворяли в iPAC (30 мл). Раствор непосредственно использовали на следующем этапе без дополнительной очистки. Анализ ВЭЖХ показал, что раствор содержал 22 (9,8 г, 92% в виде смеси цис- и транс-изомеров в отношении 6,5:1) вместе с 4% исходного вещества 21 и 8% N,N'-алкилированного соединения. Аналитически чистый 22 (цис-изомер) выделяли посредством хроматографии на силикагеле с этилацетатом и гептаном в качестве элюента. 1H ЯМР (CDCl3, 500 МГц): δ 7,15 (м, 1H), 6,85 (м, 1H), 5,45 (уш., с, 1H), 4,90 (м, H), 4,20 (м, 1H), 3,92 (м, 2H), 3,28 (м, 1H), 2,70 (м, 2H), 1,48 (с, 9H), 1,20 (д, J=5,9 Гц, 3H); 13C ЯМР (CDCl3, 100 МГц): δ 170,2, 156,9 (ддд, J=245, 6,3, 2,7 Гц), 156,0, 149,6 (ддд, J=251, 14,8, 8,8 Гц), 147,6 (ддд, J=246, 13,9, 3,6 Гц), 124,5 (кв., J=281 Гц), 117,6 (дд, J=19,2, 3,7 Гц), 116,4 (дд, J=19,1, 10,4 Гц), 111,4 (ддд, J=25,8, 6,4, 4,1Гц), 56,6, 52,8, 45,3 (кв., J=34,2 Гц), 35,2, 28,7, 28,3 (уш. т, J=4 Гц), 14,6; HRMS: масса/заряд, рассчитанное для C19H22F6N2O3 (M+H): 441,1607; выявлено 441,1617.
(S)-2-Ацетамидо-3-фенилпропаноат (3S,5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-3-аминия (23)
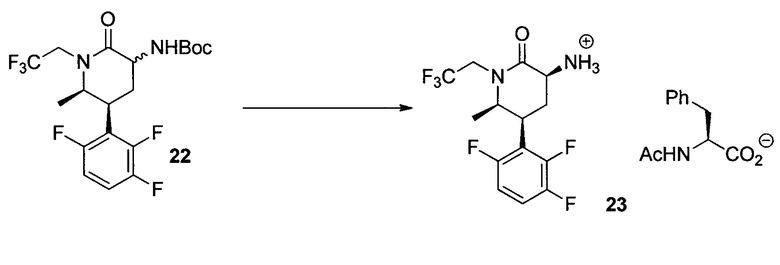
Раствор 22 iPAc (529 г аналитического образца, 1,2 моль), полученный на предыдущем этапе, разбавляли до 6 л с использованием iPAc, добавляли моногидрид п-толуолсульфоновой кислоты (343 г, 1,8 моль) и раствор нагревали до 55°C. Через 4 часа реакцию завершали (>99% преобразования). В раствор после охлаждения до 15-25°C добавляли водный K2CO3 (530 г в 3 л воды). Водный слой отделяли и подвергали обратной экстракции iPAc (2 л). Растворы в iPAc комбинировали, и общий объем доводили до 10 л, добавляя iPAc. Раствор нагревали до 50-60°C. Добавляли приблизительно 20 г N-ацетил-L-фенилаланина и раствор перемешивали в течение 15 мин или до осаждения твердых веществ. Медленно добавляли оставшийся N-ацетил-L-фенилаланин (всего 250 г, 1,2 моль) и добавляли 2-гидрокси-5-нитробензальдегид (2 г). Суспензию перемешивали в течение 12 часов при 20°C, а затем охлаждали до 0°C в течение 3 часов. Суспензию фильтровали, трижды промывали iPAc и сушили с получением 23 (583 г, выход 89%) в виде кристаллов. Mp 188-190°C; 1H ЯМР (DMSO-d6, 400 МГц): δ 7,96 (д, J=8,0 Гц, 1H) , 7,48 (м, 1H), 7,15-7,25 (м, 6H), 4,65 (ддд, J=19,4, 15,3, 9,6 Гц, 1H), 4,33 (ддд, J=8,7, 8,4, 4,9 Гц, 1H), 3,70-3,87 (м, 3H), 3,57 (дд, J=11,5, 6,6 Гц, 1H), 3,04 (дд, J=13,7, 4,9 Гц, 1H), 2,82 (дд, J=13,7, 8,9 Гц, 1H), 2,59 (м, 1H), 2,24 (м, 1H), 2,95 (с, 3H), 1,10 (д, J=6,4 Гц, 1H); 19F ЯМР (DMSO-d6, 376 МГц): δ -69 (с), -118 (д, J=15 Гц), -137 (д, J=21 Гц), -142 (дд, J=21, 15 Гц); 13C ЯМР (DMSO-d6, 100 МГц): δ 173,6, 171,1, 168,7, 156,3 (ддд, J=243,5, 7,0, 3,1 Гц), 148,7 (ддд, J=249, 14,4, 9,1 Гц), 146,8 (ддд, J=245, 13,7, 3,1 Гц), 138,5, 129,2, 128,0, 126,1, 124,9 (кв., J=280,9 Гц), 117,4,0 (дд, J=19,3, 13,8 Гц), 116,7 (дд, J=19,3, 10,6 Гц), 111,8 (ддд, J=26,0, 6,7, 3,6 Гц), 56,6, 54,3, 51,2, 44,3 (кв., J=32,5 Гц), 37,2, 34,8, 26,9 (уш. т, J=4 Гц), 22,5, 14,1.
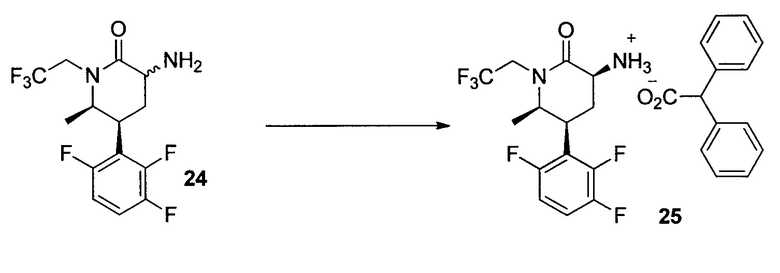
2,2-Дифенилацетат (3S,5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-3-аминия (25)
К смеси неочищенного вещества, содержащей (5S,6R)-3-амино-6-метил-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-2-он (24, 2,00 г, 5,88 ммоль), полученной тем же способом, что и в предыдущем примере, и 3,5-дихлор-2-гидроксибензальдегида (0,011 г, 0,059 ммоль) в изопропилацетате (15,0 мл) при 55-60°C в атмосфере азота медленно добавляли раствор дифенилуксусной кислоты (1,26 г, 5,88 ммоль) в THF (10,0 мл) в течение 2 часов. После завершения добавления кислоты густую суспензию соли перемешивали при 55-60°C в течение дополнительных 18 часов, а затем позволяли охлаждаться до температуры окружающей среды. Соль фильтровали и промывали изопропилацетатом. После сушки при 60°C в вакуумной печи с продувкой азота в течение 8 часов получали 25 (2,97 г, 91,4%) в виде кристаллов. 1H ЯМР (500 МГц, DMSO-d6): δ 7,48 (кв. д, J=9,4, 4,9 Гц, 1Н), 7,32 (д, J=7,7 Гц, 4Н), 7,25-7,26 (м, 4Н), 7,19-7,17 (м, 3Н), 6,79 (уш., 3H), 4,95 (с, 1Н), 4,67 (дкв., J=15,3, 9,7 Гц, 1Н), 3,81-3,79 (м, 3Н), 3,62 (дд, J=11,6, 6,5 Гц, 1Н), 2,66-2,62 (м, 1Н), 2,25 (дд, J=12,9, 6,4 Гц, 1Н), 1,11 (д, J=6,5 Гц, 3Н); 13C ЯМР (100 МГц, DMSO-d6): δ 174,4, 171,8, 156,9 (ддд, J=244, 7,0, 2,5 Гц), 149,1 (ддд, J=249, 14,4, 8,5 Гц), 147,2 (ддд, J=246, 13,9, 3,2 Гц), 141,4, 129,0, 128,5, 126,7, 125,5 (кв., J=281 Гц), 118,0 (дд, J=19,8, 13,8 Гц), 117,1 (дд, J=19,2, 10,6 Гц), 112,3 (ддд, J=26,1, 6,7, 3,3 Гц), 58,5, 57,1, 51,7, 44,8 (кв., J=32,7 Гц), 35,3, 27,5 (уш. т, J=4,6 Гц), 14,5.
1H-Индол-2-карбоксилат (3S,5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-3-аминия (26)

К смеси неочищенного вещества, содержащей 24 (2,00 г, 5,88 ммоль) и 3,5-дихлор-2-гидроксибензальдегид (0,011 г, 0,059 ммоль), в изопропилацетате (15,0 мл) при 55-60°C в атмосфере азота медленно в течение 2 часов добавляли раствор 1H-индол-2-карбоновой кислоты (0,96 г, 5,88 ммоль) в THF (10,0 мл). После завершения добавления кислоты густую суспензию соли перемешивали при 55-60°C в течение дополнительных 18 часов, а затем позволяли охлаждаться до температуры окружающей среды. Соль фильтровали и промывали изопропилацетатом. После сушки при 60°C в вакуумной печи с продувкой азотом в течение 8 часов выделяли 26 (2,33 г, 79,0%) в виде кристаллов. 1H ЯМР (500 МГц, DMSO): δ 11,40 (с, 1Н), 7,56 (д, J=8,0 Гц, 1Н), 7,45 (уш., 3Н), 7,47 (ддд, J=14,8, 10,1, 8,3 Гц, 1Н), 7,41-7,40 (м, 1Н), 7,16-7,14 (м, 2Н), 6,98-6,97 (м, 1Н), 6,87 (с, 1Н), 4,69 (дкв., J=15,3, 9,6 Гц, 1Н), 3,84-3,81 (м, 4Н), 2,76-2,71 (м, 1Н), 2,34 (дд, J=12,7, 6,3 Гц, 1Н), 1,13 (д, J=6,5 Гц, 3Н); 13C ЯМР (100 МГц, DMSO-d6): δ 170,9, 164,8, 156,8 (ддд, J=244, 7,0, 2,5 Гц), 149,1 (ддд, J=249, 14,4, 8,5 Гц), 147,2 (ддд, J=246, 13,9, 3,2 Гц), 137,0, 133,5, 127,8, 125,4 (кв., J=282 Гц), 123,3, 121,8, 119,7, 117,8 (дд, J=19,8, 13,8 Гц), 117,2 (дд, J=19,2, 10,6 Гц), 112,7, 112,3 (ддд, J=26,1, 6,7, 3,3 Гц), 105,1, 57,1, 51,3, 44,8 (кв., J=32,7 Гц), 35,2, 26,9, 14,5.
Моногидрат N-((3S,5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,6-трифторфенил)пиперидин-3-ил)-2'-оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоксамида (28)
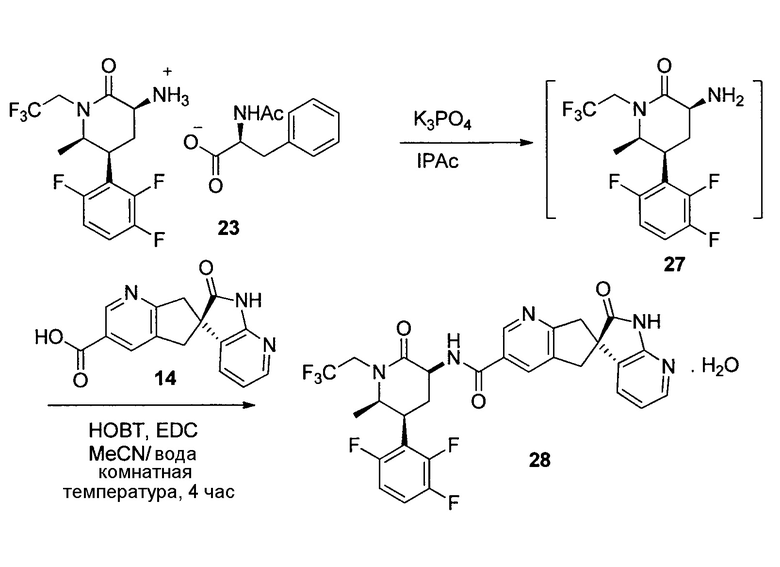
К суспензии 23 (5,0 г, 9,1 ммоль) в изопропилацетате (50 мл) добавляли 5% водный K3PO4 (50 мл). Смесь перемешивали в течение 5 мин. Органический слой отделяли и промывали водным K3PO4 (50 мл). Растворитель удаляли при пониженном давлении и полученное масло (27) растворяли в ацетонитриле (20 мл). В другую колбу добавляли 14 (2,57 г), ацетонитрил (40 мл), воду (20 мл) и раствор NaOH (10Н, 0,9 мл). К смеси добавляли раствор 27 в ацетонитриле с последующим моногидратом HOBT (1,5 г) и гидрохлоридом EDC (2,6 г). Смесь перемешивали при комнатной температуре в течение 4 часов, и анализ ВЭЖХ показал полное преобразование. Реакционную смесь перемешивали с изопропилацетатом (60 мл), и водный слой удаляли. Органический слой промывали 5% водным NaHCO3 (40 мл) с последующим смешиванием с 15% водной лимонной кислотой (40 мл) и насыщенным водным NaCl (10 мл). В заключение полученный органический слой промывали 5% водным NaHCO3 (40 мл). Растворитель удаляли при пониженном давлении, и остаток растворяли в метаноле (20 мл). В смесь воды (50 мл) и метанола (5 мл) в течение 30 мин медленно добавляли раствор метанола с хорошим перемешиванием, с последующим добавлением воды (50 мл) в течение 30 мин. Суспензию перемешивали в течение ночи при комнатной температуре. Смесь фильтровали, и кристаллы сушили в вакуумной печи в течение 5 часов при 50°C с получением 28 (5,4 г, 95%) в виде моногидрата. ¹H ЯМР (500 МГц, CD3OD): δ 8,88 (т, J=1,2 Гц, 1Н), 8,15 (т, J=1,2 Гц, 1Н), 8,09 (дд, J=5,3, 1,5 Гц, 1Н), 7,36 (дд, J=7,4, 1,5 Гц, 1Н), 7,28 (кв. д, J=9,3, 4,7 Гц, 1Н), 7,01 (тдд, J=9,7, 3,6, 1,9 Гц, 1Н), 6,96 (дд, J=7,4, 5,3 Гц, 1Н), 4,80 (дкв., J=15,2, 9,2 Гц, 1Н), 4,56 (дд, J=11,7, 6,8 Гц, 1Н), 4,03 (ддд, J=13,6, 4,2, 2,6 Гц, 1Н), 3,97-3,90 (м, 1Н), 3,68 (дкв., J=15,3, 8,8 Гц, 1Н), 3,59 (т, J=16,2 Гц, 2Н), 3,35 (д, J=4,4 Гц, 1Н), 3,32 (д, J=3,5 Гц, 1Н), 3,21 (кв. т, J=12,7, 3,1 Гц, 1Н), 2,38-2,32 (м, 1Н), 1,34 (д, J=6,5 Гц, 3Н); 13C ЯМР (126 МГц, CD3OD): δ 182,79, 171,48, 168,03, 166,71, 159,37 (ддд, J=244,1, 6,5, 2,1 Гц), 157,43, 150,88 (ддд, J=249,4, 14,4, 8,7 Гц), 148,96 (ддд, J=243,8, 13,7, 3,1 Гц), 148,67, 148,15, 136,84, 133,43, 131,63, 130,83, 130,48, 126,41 (кв., J=280,0 Гц), 119,85, 118,89 (дд, J=19,0, 13,5 Гц), 117,77 (дд, J=19,8, 10,8 Гц), 112,80 (ддд, J=26,5, 6,5, 4,2 Гц), 58,86, 53,67, 52,87, 46,56 (кв., J=33,3 Гц), 45,18, 42,06, 36,95, 27,76 (т, J=4,8 Гц), 14,11.
ПРИМЕР 3
3-Гидрокси-3-(2,3,6-трифторфенил)бутан-2-он (30)
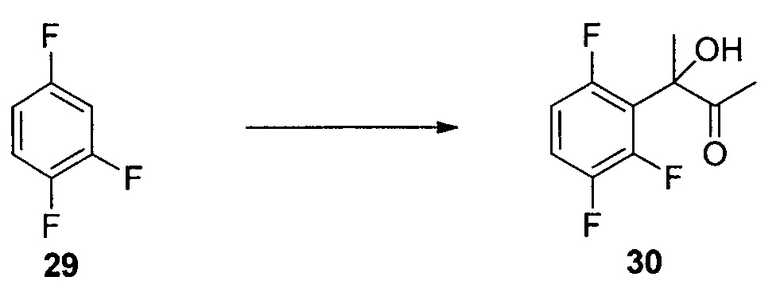
К раствору 1,2,4-трифторбензола (29, 49,00 г, 371 ммоль) и диизопропиламина (4,23 мл, 29,7 ммоль) в THF (750 мл) при -70°C медленно добавляли 2,5 M н-BuLi (156,0 мл, 390 ммоль) с поддержанием температуры от -45 до -40°C. Партию перемешивали в течение 30 мин. В другой колбе получали раствор 2,3-бутадиона (37,7 мл, 427 ммоль) в THF (150 мл) и охлаждали до -70°C. Полученный ранее раствор трифторбензола лития переносили во вторую колбу при температурах от -70 до -45°C. Реакционную смесь перемешивали в течение 1 часа при температурах от -55 до -45, а затем гасили, добавляя AcOH (25,7 мл, 445 ммоль), а затем воду (150 мл). После нагревания до комнатной температуры водный слой отделяли. Водный раствор экстрагировали MTBE (200 мл×1), и объединенные органические слои промывали насыщенным солевым раствором (100 мл×1). Органический слой концентрировали при 25-35°C. Через остаток пропускали гептан (100 мл×1) и концентрировали досуха, и получали 30 (87,94 г, 90,2% масс., выход 98% и >99% чистоты по ВЭЖХ) в виде масла. 1H ЯМР (CDCl3, 400 МГц): δ 7,16 (м, 1H), 6,86 (м, 1H), 6,88 (с, 1H), 4,59 (с, 1H), 2,22 (с, 3H), 1,84 (дд, J=4,0, 2,8 Гц, 3H); 19F ЯМР (CDCl3, 376,6 МГц): δ -114,6 (дд, J=14,5, 1,4 Гц), -133,6 (д, J=19,9 Гц), -141,3 (дд, J=19,9, 14,5 Гц); 13C ЯМР (CDCl3, 100 МГц): δ 207,4, 156,4 (ддд, J=247, 6,2, 2,9 Гц), 149,4 (ддд, J=253, 15,0, 9,0 Гц), 147,5 (ддд, J=245, 14,4, 3,3 Гц), 119,4 (дд, J=17,3, 11,7 Гц), 117,0 (ддд, J=19,3, 11,1, 1,4 Гц), 116,6 (ддд, J=26,6, 6,5, 4,1 Гц), 77,9, 25,0 (дд, J=6,5, 4,9 Гц), 23,3.
3-(2,3,6-Трифторфенил)бут-3-ен-2-он (31)
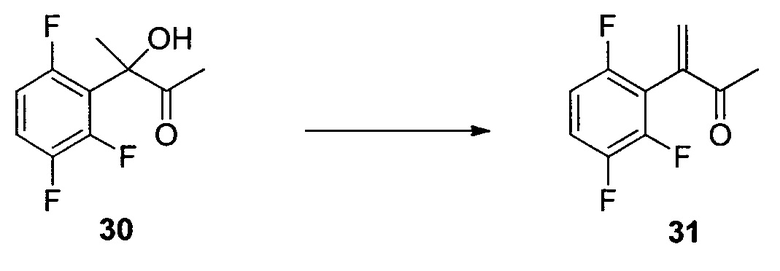
Гидроксикетон 30 (7,69 г, 35,2 ммоль) и 95% H2SO4 (26,2 мл, 492,8 ммоль) нагнетали при 2,3 и 9,2 мл/мин, соответственно, в проточный реактор. Температуру при смешивании поддерживали при 22-25°C, помещая реактор в водяную баню (21°C). Сток гасили в смеси холодной воды (106 г) и гептана/IPAc (1:1, 92 мл) в реакторе с рубашкой, охлаждаемом до 0°C; внутренняя температура гасимого раствора в течение реакции составляла ~7°C. Слои в реакторе гашения разделяли, и органический слой промывали 10% NaH2PO4/Na2HPO4 (1:1, 50 мл). pH конечной промывки составлял 5-6. В органический раствор добавляли Solka Flock (3,85 г, 50% масс.). Полученную взвесь концентрировали и сменяли растворители на гептан при 25-30°C. Смесь фильтровали, промывали гептаном (50 мл×1). Комбинированные фильтраты концентрировали при пониженном давлении с получением 31 в виде светло-желтого масла (6,86 г, 90% масс., выход 87%), которое отверждали в холодильнике. 1H ЯМР (CDCl3, 400 МГц): δ 7,13 (м, 1H), 6,86 (м, 1H), 6,60 (с, 1H), 6,15 (с, 1H), 2,46 (с, 3H); 19F ЯМР (CDCl3, 376,6 МГц): δ -117,7 (дд, J=15,0, 1,4 Гц), -135,4 (дд, J=21,4, 1,4 Гц), -42,7 (дд, J=21,4, 15,0 Гц); 13C ЯМР (CDCl3, 100 МГц): δ 196,3, 155,3 (ддд, J=245, 5,1, 2,9 Гц), 147,9 (ддд, J=250, 14,5, 7,8 Гц), 147,0 (ддд, J=245, 13,4, 3,7 Гц), 137,5 (д, J=1,3 Гц), 131,7, 116,6 (ддд, J=19,9, 9,7, 1,2 Гц), 116,2 (дд, J=22,6, 16,5 Гц), 110,6 (ддд, J=24,8, 6,5, 4,1 Гц), 25,8.
Альтернативный синтез 3-(2,3,6-трифторфенил)бут-3-ен-2-она (31)
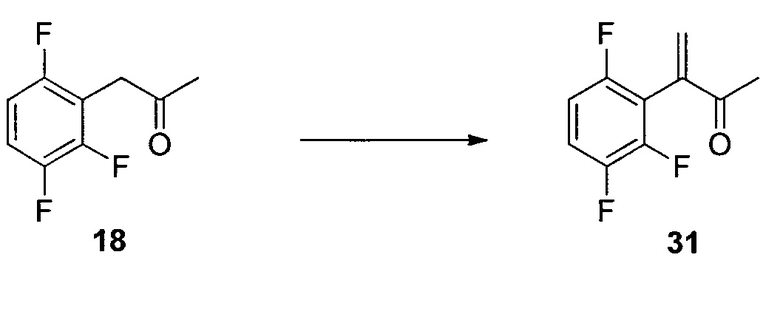
Раствор 18 (3,5 г, 18,6 ммоль), уксусную кислоту (0,34 мл, 5,58 ммоль), пиперидин (0,37 мл, 3,72 ммоль), формальдегид (6,0 г, 37% водный раствор) в MeCN (20 мл) нагревали в течение выходных. Преобразование происходило приблизительно на 60%. Реакционную смесь нагревали до 70°C в течение ночи. Смесь концентрировали и экстрагировали MTBE и HCl (0,5Н). Органический слой промывали водным K2CO3 (0,5Н) и водой по очереди. Органический слой концентрировали. Продукт выделяли посредством хроматографии на колонке (гексан и EtOAc) с выходом 31 (2,29 г, 61,5%).
Изопропил-2-((дифенилметилен)амино)-5-оксо-4-(2,3,6-трифторфенил)гексаноат (32)

Дифенилиденизопропилглицинат (2,0 г, 7,0 ммоль) и 31 (1,4 г, 7,0 ммоль) растворяли в THF (10 мл). Раствор охлаждали до -10°C. В раствор несколькими порциями добавляли трет-BuOLi (0,56 г, 7,0 ммоль). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. После гашения посредством добавления водного NH4Cl, растворители удаляли посредством возгонки при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем с элюцией гексаном и EtOAc с выходом 32 (3,0 г, 89%) в виде масла, которое непосредственно использовали на следующем этапе.
Изопропил-2-((трет-бутоксикарбонил)амино)-5-оксо-4-(2,3,6-трифторфенил)гексаноат (19)
Соединение 32 (100 мг, 0,21 ммоль) растворяли в THF (2 мл) и раствор охлаждали до -10°C. Добавляли соляную кислоту (2Н, 1 мл) и перемешивали до исчезновения всего исходного вещества на TLC. Доводили pH реакционной смеси (pH>10), добавляя водный K2CO3. В смесь добавляли Boc2O (68 мг, 0,31 ммоль) и перемешивали в течение ночи. Реакцию завершали, проверяя на TLC, и продукт был идентичен продукту, получаемому путем связывания соединений йода.
Изопропил-2-((трет-бутоксикарбонил)амино)-5-оксо-4-(2,3,6-трифторфенил)гексаноат (19)
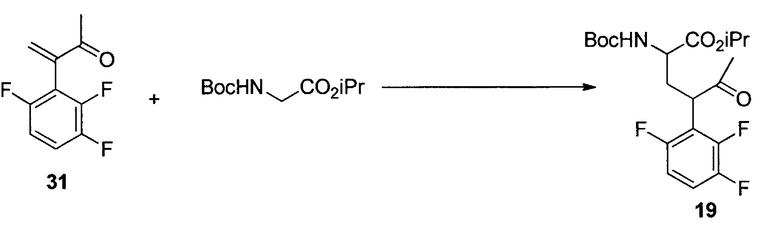
В 100-мл круглодонную колбу добавляли 2-метил THF (43,7 мл) и диизопропиламин (4,92 мл, 34,2 ммоль) и раствор охлаждали до -70°C. Капельно добавляли н-BuLi (13,08 мл, 32,7 ммоль) в течение чего температуру поддерживали при температуре ниже -45°C. Смесь перемешивали при -45°C в течение 0,5 часа. Капельно добавляли сложный N-Boc-глициновый эфир (3,58 г), поддерживая температуру при значениях от -45 до -40°C и выдерживали при этой же температуре в течение 1 часа.
Затем тем же способом при температурах от -45 до -40°C капельно добавляли раствор 31 (2,91 г, 14,5 ммоль) в 2-метил THF (2,9 мл). Через 0,5-1 час выдерживания анализ LC демонстрировал почти полное завершение реакции. Реакцию гасили посредством добавления HOAc (3,83 мл) и смесь нагревали до -10°C и добавляли воду (11,6 мл, 4 об.) при <20°C. Фазы разделяли, и органический слой промывали 16% водным раствором NaCl (11,6 мл). Желаемый в анализе продукт 19 в виде смеси диастереомеров в органическом растворе составлял 5,40 г (выход 89%). Органический слой концентрировали с получением неочищенного продукта 19, который непосредственно использовали на следующем этапе реакции. С целью характеристики, малый образец очищали посредством флэш-хроматографии (силикагель, EtOAc/гексан=1:10) с получением двух диастереомеров 19A и 19B. 19A в виде бесцветного масла, 1H ЯМР (CD3CN, 400 МГц) δ: 7,29 (м, 1Н), 7,02 (м, 1Н), 5,58 (д, J=6,1 Гц, 1Н), 4,91 (м, 1Н), 4,19-4,05 (м, 2Н), 2,79 (м, 1Н), 2,05 (с, 3Н), 1,84 (м, 1Н), 1,41 (с, 9Н), 1,23 (д, J=6,7 Гц, 3Н), 1,22 (д, J=6,7 Гц, 3Н); 13C ЯМР (CD3CN, 100 МГц) δ: 204,7, 172,4, 158,6 (ддд, J=244, 6, 3 Гц), 156,3, 149,8 (ддд, J=248, 15, 9 Гц), 148,5 (ддд, J=242, 14, 3 Гц), 118,3 (дд, J=21, 16 Гц), 117,7 (ддд, J=19, 10, 2 Гц), 112,6 (ддд, J=26, 7, 4 Гц), 80,2, 70,0, 53,5, 46,0, 32,0, 28,5, 22,0, 21,9. 19B в виде бесцветных кристаллов, Mp 91,5-92,0°C, 1H ЯМР (CD3CN, 400 МГц) δ: 7,31 (м, 1Н), 7,03 (м, 1Н), 5,61 (д, J=8,2 Гц, 1Н), 4,95 (м, 1Н), 4,19 (дд, J=10,2, 5,1 Гц, 1Н), 3,72 (м, 1Н), 2,45-2,29 (м, 2Н), 2,09 (с, 3Н), 1,41 (с, 9Н), 1,21 (д, J=6,3 Гц, 3Н), 1,20 (д, J=6,3 Гц, 3Н); 13C ЯМР (CD3CN, 100 МГц) δ: 205,0, 172,8, 157,9 (ддд, J=244, 7, 3 Гц), 156,5, 150,3 (ддд, J=248, 149, 9 Гц), 148,5 (ддд, J=242, 13, 4 Гц), 117,9 (дд, J=19, 10 Гц), 115,9 (дд, J=21, 15 Гц), 111,5 (ддд, J=25, 8, 4 Гц), 80,1, 69,9, 52,9, 46,5, 31,1, 28,5, 22,0, 21,9.
ПРИМЕР 4
N-Метокси-N-метил-2-(о-толил)ацетамид (34)
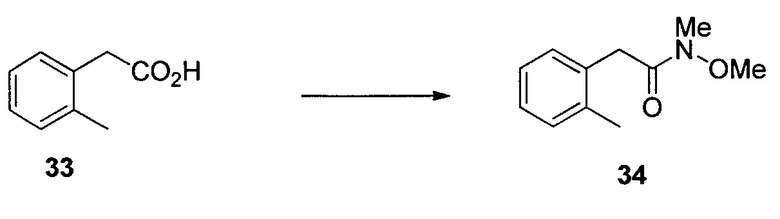
К раствору NHMe(OMe)·HCl (203 г, 2,1 моль) в THF (1 л), H2O (400 мл) и TEA (263 г, 2,2 моль) добавляли 33 (200 г, 1,3 моль) и CDI (243 г, 1,5 моль) при 0-10°C. Реакционную смесь перемешивали при 0-10°C в течение 5 часов. После демонстрации прохождения реакции посредством ВЭЖХ смесь фильтровали через целит, и фильтрат разделяли водой и EtOAc. Органический раствор сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-10% EtOAc/PE) с получением 34 (200 г, выход 78%). 1H ЯМР (CDCl3, 400 МГц): δ 7,17-7,13 (м, 4Н), 3,75 (м, 2Н), 3,66 (д, 3Н), 3,11 (с, 3H), 2,20 (с, 3Н), 1,63-1,55 (м, 1Н); MS (ESI) m/e [M+H]+: 194,1.
1-(o-Толил)пропан-2-он (35)
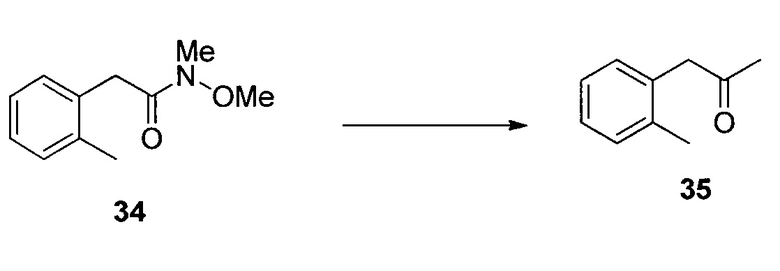
Раствор CeCl3 (114,4 г, 0,45 моль) в THF (4 л) дегазировали в течение 1 часа и нагревали до 45-50°C в течение 5 часов. Когда раствор охлаждали до -10~-5°C, добавляли MeMgCl (193,2 г, 2,6 моль) в THF и смесь перемешивали в течение 1 часа при -10~-5°C. После добавления амида 34 (256 г, 1,3 моль) в реакционную смесь при -10~-5°C, смесь перемешивали в течение 5 часов при 10-20°C. После завершения реакции по данным мониторинга LCMS смесь гасили посредством 1 M HCl, а затем разделяли водой и EtOAc. Органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (2-10% EtOAc/PE) с получением 35 (157 г, выход 80%).1H ЯМР (CDCl3, 400 МГц): δ 7,1-6,91 (д, 4Н), 3,55 (с, 3Н), 2,25 (с, 3Н), 2,05 (с, 3Н); MS (ESI) m/e [M+H]+: 149,05.
Изопропил-2-((трет-бутоксикарбонил)амино)-5-оксо-4-(o-толил)гексаноат (36)
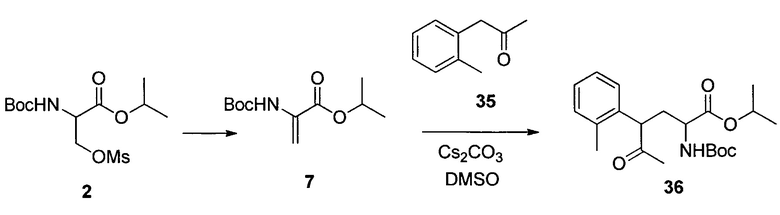
К раствору 2 (181,2 г, 0,557 моль) в THF (1 л) частями добавляли TEA (84,6 г, 0,836 моль) при 15-20°C. Смесь перемешивали в течение 30 часов. После завершения реакции раствор концентрировали с получением неочищенного 7. К раствору 35 (82,5 г, 0,557 моль) и Cs2CO3 (91 г, 0,279 моль) в DMSO (1 л) медленно добавляли неочищенный 7 в DMSO (500 мл) в течение 30 мин при 15-20°C. Смесь перемешивали в течение 1 часа. После завершения реакции смесь разделяли водой и MTBE (5 л) и дважды экстрагировали MTBE. Объединенный органический слой сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-10% EtOAc/PE) с получением 36 (138 г, выход 65%). 1H ЯМР (DMSO-d6, 400 МГц): δ 7,14-7,09 (м, 3H), 7,10-6,91 (д, 1Н), 4,93-4,89 (м, 1H), 4,05-3,98 (с, 3H), 2,39-2,37 (д, 3H), 1,98-1,92 (д, 3H), 1,20-1,19 (м, 9H), 1,18-1,15 (м, 6H); MS (ESI) m/e [M+H]+: 364,2.
трет-Бутил-((5S,6R)-6-метил-2-оксо-5-(o-толил)пиперидин-3-ил)карбамат (37)
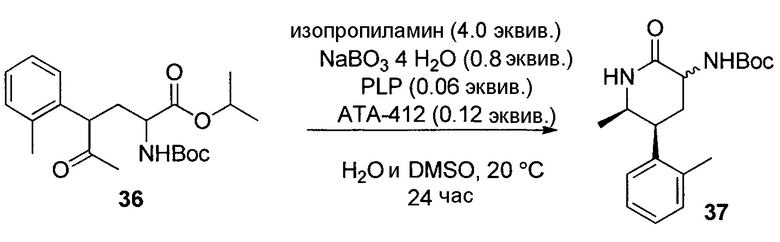
К раствору NaBO3·4H2O (10 г, 0,026 моль) в H2O (180 г) капельно добавляли изопропиламин (30 г, 0,25 ммоль) при 20°C. После перемешивания смеси в течение 30 мин pH доводили до 10,3-10,5 посредством 6M HCl. К раствору добавляли PLP (1 г, 6,4 ммоль) и ATA-412 (25 г) при 20°C. После перемешивания указанной выше смеси в течение 1 часа медленно добавляли раствор 36 (50 г, 4,7 ммоль) в DMSO (250 мл) при 20°C. Раствор нагревали до 55°C и перемешивали в течение 24 часов. После завершения реакции, реакцию гасили изопропиловым спиртом (100 мл), а затем разделяли водой и IPAc. Органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-20% EtOAc/PE) с получением 37 (16,5 г, выход 40%). 1H (DMSO-d6, 400 МГц): δ 7,83 (с, 1H), 7,18-7,13 (м, 4H), 4,06 (уш., 1H), 3,67-3,51 (м, 2H), 2,30 (д, 3H), 1,99 (т, 1H), 1,36 (с, 9H), 0,82 (м, 3H); MS (ESI) m/e [M+H]+: 319,2.
трет-Бутил-((5S,6R)-6-метил-2-оксо-5-(o-толил)-1-(2,2,2-трифторэтил)пиперидин-3-ил)карбамат (38)
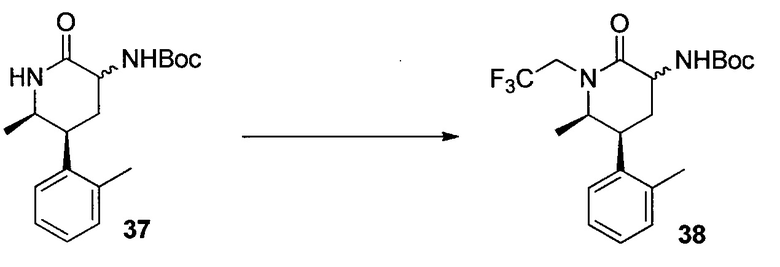
К раствору 37 (50 г, 0,164 моль) и DMPU (25 г, 0,2 моль) в THF (500 мл) частями добавляли трет-BuOLi (16,5 г, 0,2 моль) при 20°C в течение 0,5 часов. После дегазации смеси в течение 30 мин при 20°C добавляли CF3CH2OTf (45,8 г, 0,2 моль) при 20-25°C. Реакционную смесь перемешивали в течение 24 часов при 20-25°C. После завершения реакции смесь гасили водой, а затем разделяли водой и EtOAc. Органический раствор сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (2-10% EtOAc/PE) с получением продукта 38 (50 г, выход 78%). 1H ЯМР (DMSO-d6, 400 МГц): δ 7,18-7,13 (м, 4H), 4,17 (м, 1H), 4,11 (уш., 1H), 3,67 (м, 3H), 2,66 (с, 1H), 2,33 (д, 3H), 1,83 (т, 1H), 1,41 (с, 9H), 0,96 (д, 3H); MS (ESI) m/e [M+H]+ 405,17.
4-Метилбензоат (3S,5S,6R)-6-метил-2-оксо-5-(o-толил)-1-(2,2,2-трифторэтил)пиперидин-3-аминия (39)
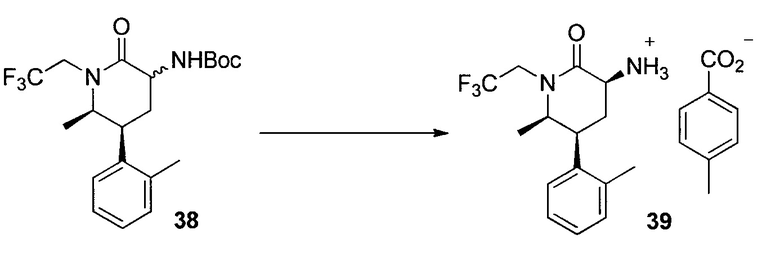
К раствору 38 (2,0 г, 5,0 моль) в THF (10 мл) капельно добавляли 6Н HCl (10 мл, 59,9 ммоль). Реакционную смесь выдерживали при 20-25°C в течение 5 часов, затем концентрировали с удалением THF. Остаток разбавляли MTBE и повышали основность K2CO3. Добавляли всего 3 мл H2O для растворения всех твердых веществ. Органический слой отделяли, промывали насыщенным солевым раствором, и заменяли растворитель на IPAC. К трети органического слоя добавляли 4-метилбензойную кислоту (0,27 г, 2,00 ммоль). Раствор нагревали до 50°C и добавляли 2-гидрокси-5-нитробензальдегид (0,0028 г, 0,017 ммоль). Реакционную смесь выдерживали при 20-25°C в течение 16 часов. Полученную взвесь охлаждали в ледяной бане до 2°C и фильтровали. Твердые вещества промывали iPAc и сушили с получением продукта 39 (0,38 г, 52%) в виде кристаллов. 1H ЯМР (500 МГц, DMSO-d6): δ 7,82 (д, J=8,0 Гц, 2Н), 7,26 (д, J=7,9 Гц, 2Н), 7,18-7,19 (м, 3Н), 7,12 (д, J=7,5 Гц, 1Н), 4,67 (дкв., J=15,2, 9,7 Гц, 1Н), 3,72-3,74 (м, 2Н), 3,61-3,63 (м, 1Н), 3,55 (дд, J=11,3, 6,7 Гц, 1Н), 2,40 (дд, J=25,2, 12,9 Гц, 1Н), 2,35 (с, 3Н), 2,32 (с, 3Н), 2,02 (дд, J=12,6, 6,7 Гц, 1Н), 0,91 (д, J=6,4 Гц; 3Н); 13C ЯМР (100 МГц, DMSO-d6): δ 172,7, 168,3, 142,4, 139,1, 136,1, 130,9, 130,5, 129,7, 129,3, 127,4, 127,2, 126,5, 125,6 (кв., J=281 Гц), 56,2, 52,1, 45,0 (кв., J=32,3 Гц), 38,5, 29,1, 21,5, 18,7, 14,2.
(S)-N-((3S,5S,6R)-6-метил-2-оксо-5-(o-толил)-1-(2,2,2-трифторэтил)пиперидин-3-ил)-2'-оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоксамид (41)
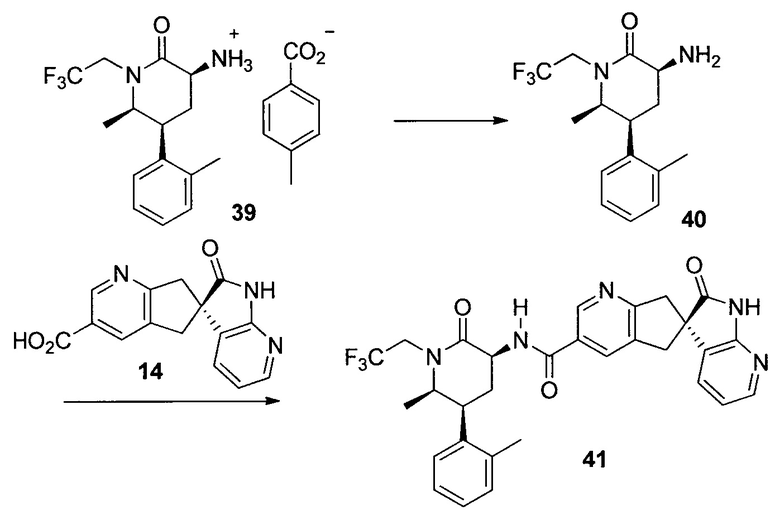
Соль 39 (0,25 г, 0,58 ммоль) разделяли между IPAC (2,5 мл) и 5% масс. водным раствором K3PO4 (2,5 мл) и дважды промывали 5% масс. водным K3PO4. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного 40. Неочищенный 40 растворяли в MeCN (1,75 мл) и H2O (1,0 мл). К этой смеси добавляли кислоту 14 (0,17 г, 0,53 ммоль), HOBT (0,11 г, 0,70 ммоль) и EDC.HCl (0,17 г, 0,87 ммоль). Гетерогенную смесь выдерживали при 20-25°C в течение 16 часов. Гомогенную реакционную смесь разделяли между IPAC и насыщенным водным NaHCO3 и дважды промывали насыщенным водным NaHCO3. Органический слой промывали 15% масс. водным раствором лимонной кислоты, насыщенным водным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над Na2SO4 и концентрировали с получением продукта 41 (0,29 г, выход 89%). 1H ЯМР (400 МГц, CD3OD): δ 8,88 (д, J=1,9 Гц, 1Н), 8,15 (д, J=1,9 Гц, 1Н), 8,08 (дд, J=5,3, 1,6 Гц, 1Н), 7,35 (дд, J=7,4, 1,6 Гц, 1Н), 7,15-7,18 (м, 4Н), 6,95 (дд, J=7,4, 5,3 Гц, 1Н), 4,59 (дд, J=11,5, 7,0 Гц, 1Н), 3,89-3,92 (м, 1Н), 3,81 (дт, J=13,4, 3,2 Гц, 1Н), 3,61-3,63 (м, 4Н), 3,31-3,32 (м, 2Н), 2,93-2,95 (м, 1Н), 2,40 (с, 3Н), 2,14-2,17 (м, 1Н); 1,14 (д, J=6,5 Гц, 3Н); 1H ЯМР (500 МГц, CDCl3): δ 8,91 (с, 1H), 8,56 (с, 1H), 8,17 (дд, J=5,0, 1,5 Гц, 1H), 8,05 (с, 1H), 7,30 (д, J=5,0 Гц, 1H), 7,20 (м, 3H), 7,12 (м, 2H), 6,89 (дд, J=7,5, 5,0 Гц, 1H), 5,00-4,94 (м, 1H), 4,55 (м, 1H), 3,93-3,90 (м, 1H), 3,81-3,76 (м, 2H), 3,68 (д, J=16,5 Гц, 1H), 3,31-3,23 (м, 2H), 3,17 (д, J=16,5 Гц, 1H), 2,75-2,67 (м, 2H), 2,41 (с, 3H), 1,11 (д, J=6,5 Гц, 3H); 13C ЯМР (100 МГц, CD3OD): δ 182,8, 171,8, 168,0, 166,7, 157,5, 148,7, 148,2, 139,5, 137,5, 136,8, 133,4, 132,0, 131,6, 130,8, 130,6, 128,4, 128,3, 127,4, 126,6 (кв., J=283 Гц), 119,9, 58,1, 53,7, 53,0, 46,7 (кв., J=33,4 Гц), 45,2, 42,1, 40,2, 28,8, 19,0, 13,6; HRMS: масса/заряд=564,2219 (M+1), рассчитанное масса/заряд для C30H28F3N5O35=64,2234.
ПРИМЕР 5
N-Метокси-N-метил-2-(2,3,5-трифторфенил)ацетамид (43)
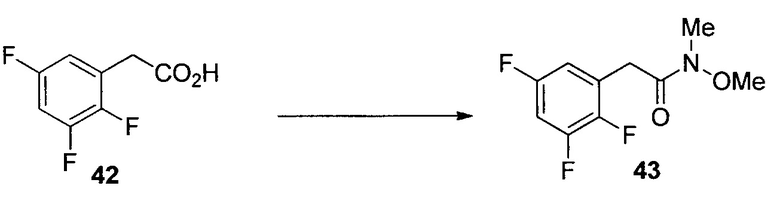
К раствору NHMe(OMe)·HCl (20,3 г, 0,21 моль) в THF (110 мл), H2O (40 мл) и TEA (26,3 г, 0,22 моль) добавляли 2,3,5-трифторфенилуксусную кислоту (42, 24,7 г, 0,13 моль) и CDI (24,3 г, 0,15 моль) при 0-10°C. Реакционную смесь перемешивали при 0-10°C в течение 5 часов. После демонстрации прохождения реакции посредством ВЭЖХ смесь фильтровали через целит, и фильтрат разделяли водой и EtOAc. Органический раствор сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-10% EtOAc/PE) с получением 43 (24,0 г, выход 80%). 1H ЯМР (CDCl3, 400 МГц): δ 6,76-6,72 (м, 2Н), 3,72 (м, 2Н), 3,66 (д, 3Н), 3,15 (д, 3Н); MS (ESI) m/e [M+H]+: 234,07.
1-(2,3,5-Трифторфенил)пропан-2-он (44)
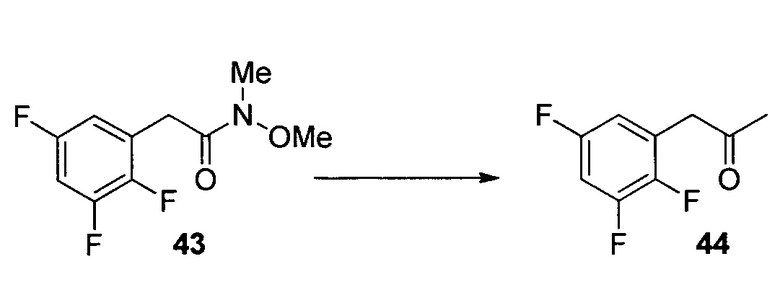
Раствор CeCl3 (11,44 г, 0,045 моль) в THF (350 мл) дегазировали в течение 1 часа и нагревали до 45-50°C в течение 5 часов. Когда раствор охлаждали до -10~5°C, добавляли MeMgCl (19,44 г, 0,26 моль) в THF и смесь перемешивали в течение 1 часа при -10~-5°C. После добавления в реакционную смесь амида 43 (30,3 г, 0,13 моль) при -10~-5°C смесь перемешивали в течение 5 часов при 10-20°C. После завершения реакции смесь гасили 1M HCl, а затем разделяли водой и EtOAc. Органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (2-10% EtOAc/PE) с получением 44 (20,8 г, выход 85%). 1H ЯМР (CDCl3, 400 МГц): δ 6,91-6,78 (м, 1Н), 6,69 (дд, 1Н), 3,77 (д, 2Н), 2,25 (с, 3Н); MS (ESI) m/e [M+H]+: 189,05.
Изопропил-2-((трет-бутоксикарбонил)амино)-5-оксо-4-(2,3,5-трифторфенил)гексаноат (45)

К раствору 2 (10,98 г, 33,7 ммоль) в THF (50 мл) частями добавляли TEA (4,8 г, 47,4 ммоль) при 15-20°C. Смесь перемешивали в течение 30 часов. После завершения реакции раствор концентрировали с получением неочищенного 7. К раствору 44 (6,3 г, 33,7 ммоль) и Cs2CO3 (5,0 г, 15,3 ммоль) в DMSO (35 мл) медленно добавляли неочищенный 7 в DMSO (35 мл) в течение 30 мин при 15-20°C. Смесь перемешивали в течение 1 часа. После завершения реакции смесь разделяли водой и MTBE (50 мл) и экстрагировали дважды MTBE. Объединенный органический слой сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-10% EtOAc/PE) с получением 45 (8,4 г, выход 60%). 1H ЯМР (DMSO-d6, 400 МГц): δ 6,77 (д, 1H), 6,59 (д, 1Н), 5,11 (м, 1H), 4,93-4,89 (м, 1H), 4,12 (с, 2H), 2,66 (д, 1H), 2,05-2,01 (д, 3H), 1,38 (м, 9H), 1,18-1,15 (м, 6H); MS (ESI) m/e [M+H]+: 418,18.
трет-Бутил-((5S,6R)-6-метил-2-оксо-5-(2,3,5-трифторфенил)пиперидин-3-ил)карбамат (46)

К раствору NaBO3·4H2O (1,5 г, 3,9 ммоль) в H2O (18,2 г) капельно добавляли изопропиламин (1,16 г, 19,6 ммоль) при 20°C. После перемешивания смеси в течение 30 мин pH доводили до 10,2-10,3 посредством 6Н HCl. К раствору при 20°C добавляли PLP (0,042 г, 0,27 ммоль) и ATA-412 (1,0 г). После перемешивания указанной выше смеси в течение 1 часа медленно добавляли раствор 45 (2 г, 4,7 ммоль) в DMSO (10 мл) при 20°C. Раствор нагревали до 55°C и перемешивали в течение 24 часов. После завершения реакции реакцию гасили изопропиловым спиртом (10 мл), а затем разделяли водой и IPAc. Органический раствор сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (5-20% EtOAc/PE) с получением 46 (1,45 г, выход 85%). 1H ЯМР (DMSO-d6, 400 МГц): δ 7,95-7,78 (м, 1H), 6,95 (м, 1H), 3,01 (т, 1H), 1,36 (с, 9H), 1,17-1,10 (уш., 4H), 1,12 (м, 3H); MS (ESI) m/e [M+H]+: 359,15.
трет-Бутил-((5S,6R)-6-метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,5-трифторфенил)пиперидин-3-ил)карбамат (47)
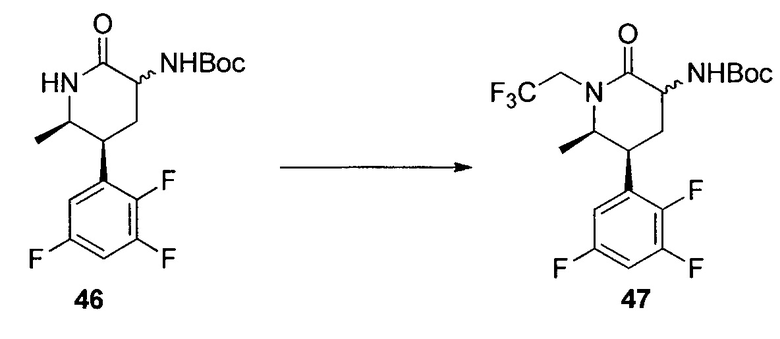
К раствору 46 (50 г, 0,14 моль) и DMPU (7,2 г, 0,06 моль) в THF (500 мл) частями добавляли трет-BuOLi (8,6 г, 0,11 моль) при 20°C в течение 0,5 часов. После дегазации смеси в течение 30 мин при 20°C добавляли CF3CH2OTf (37,5 г, 0,14 моль) при 20-25°C. Раствор перемешивали в течение 24 часов при 20-25°C. После завершения реакции смесь гасили водой, а затем разделяли водой и EtOAc. Органический раствор сушили над Na2SO4 и концентрировали. Неочищенный остаток дополнительно очищали посредством флэш-хроматографии на силикагеле (2-10% EtOAc/PE) с получением продукта 47 (50 г, выход 82%). 1H (DMSO-d6, 400 МГц): δ 7,52 (м, 1Н), 7,22-6,91 (м, 1Н), 4,65 (м, 1Н), 3,85 (уш., 1H), 3,38-3,37 (м, 2Н), 1,40 (с, 9Н), 90 (м, 3Н) , 3Н) 7,00 (уш.с, 0,90 (м, 3Н); MS (ESI) m/e [M+H]+: 441,15.
(5S,6R)-3-Амино-6-метил-1-(2,2,2-трифторэтил)-5-(2,3,5-трифторфенил)пиперидин-2-он (48)
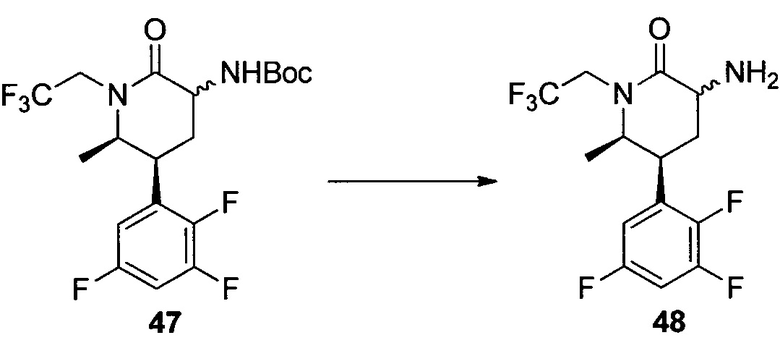
К раствору 47 (2,0 г, 4,5 ммоль) в iPAC (20 мл) добавляли моногидрат п-толуолсульфоновой кислоты (1,3 г, 6,8 ммоль). Смесь выдерживали при 55°C в течение 4 часов. После завершения реакции взвесь охлаждали в ледяной бане до 5°C и добавляли раствор карбоната калия (1,9 г, 13,6 ммоль) в H2O (10 мл). Водный слой (pH=10) отделяли, и органический слой последовательно промывали насыщенным водным NaHCO3, водой и насыщенным солевым раствором и сушили над Na2SO4. Посредством концентрации получали 48 (1,3 г, выход 86%) в виде смеси диастереомеров 4:1.
(3S,5S,6R)-3-Амино-6-метил-1-(2,2,2-трифторэтил)-5-(2,3,5-трифторфенил)пиперидин-2-он (S)-2-гидроксисукцинат (49)
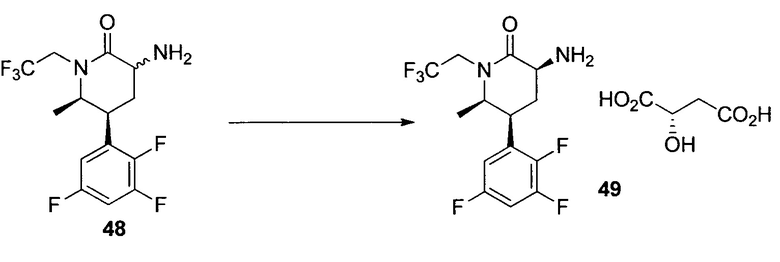
К раствору 48 (0,24 г, 0,72 ммоль) в THF (3,7 мл) добавляли L-(-)-яблочную кислоту (0,10 г, 0,75 ммоль). Гомогенную реакционную смесь нагревали до 58°C и выдерживали в течение 3 часов. После завершения реакции взвесь охлаждали до 20-25°C и выдерживали в течение 16 часов. Твердые вещества фильтровали, дважды промывали ледяным THF и сушили с получением продукта 49 (0,25 г, выход 73%) в виде кристаллов. 1H ЯМР (400 МГц, DMSO-d6): δ 7,50-7,54 (м, 1Н), 7,01-7,06 (м, 1Н), 4,68 (дкв., J=15,3, 9,6 Гц, 1Н), 4,05 (дд, J=11,6, 6,7 Гц, 1Н), 3,91-3,92 (м, 2Н), 3,84-3,87 (м, 2Н), 2,51 (м, 1H), 2,45 (м, 1H), 2,33 (дд, J=15,6, 4,4 Гц, 1Н), 2,15 (дд, J=12,3, 6,7 Гц, 1Н), 0,97 (д, J=6,4 Гц, 3Н); 13C ЯМР (100 МГц, DSMO-d6): δ 176,3, 172,0, 168,4, 157,4 (ддд, J=243, 11, 2 Гц), 150,0 (дт, J=248, 14 Гц), 144,7 (ддд, J=242, 13, 4 Гц), 130,4 (дд, J=13, 9 Гц), 124,8 (кв., J=281 Гц), 110,9 (дт, J=26, 3 Гц), 105,1 (дд, J=28, 21 Гц), 66,1, 56,0, 49,6, 44,5 (кв., J=33 Гц), 41,3, 35,1, 25,1, 13,8; 19F ЯМР (377 МГц, DMSO-d6): δ -69,3, -114,6 (д, J=14,9 Гц), -134,7 (д, J=21,8 Гц), -148,8 (дд, J=21,8, 14,9 Гц).
(S)-N-((3S,5S,6R)-6-Метил-2-оксо-1-(2,2,2-трифторэтил)-5-(2,3,5-трифторфенил)пиперидин-3-ил)-2'-оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоксамид (51)
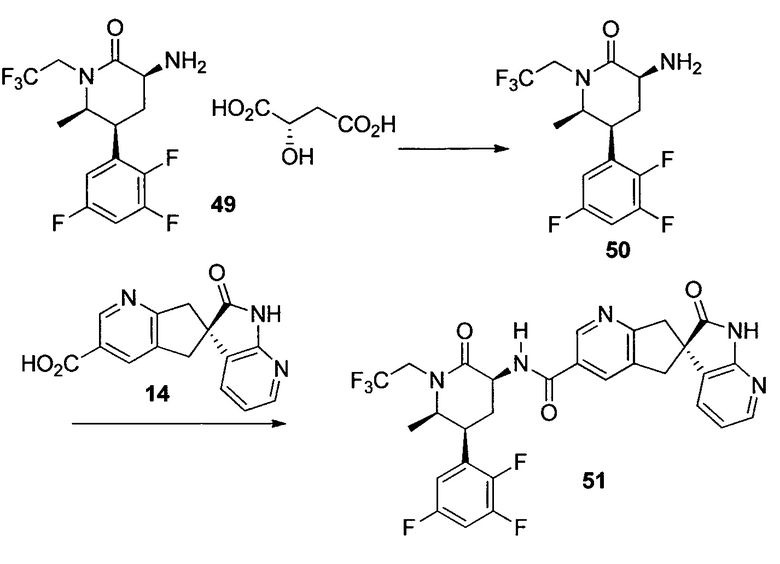
Соль 49 (0,60 г, 1,27 ммоль) разделяли в IPAC (6 мл) и 5% масс. водном K3PO4 (6 мл) и дважды промывали 5% масс. водным K3PO4. Органический слой промывали насыщенным солевым раствором, сушили Na2SO4 и концентрировали с получением неочищенного 50. Затем неочищенный 50 растворяли в MeCN (4,2 мл) и H2O (2,4 мл). К этому раствору добавляли кислоту 14 (0,32 г, 1,12 ммоль), HOBT (0,22 г, 1,42 ммоль) и EDC.HCl (0,34 г, 1,77 ммоль). Гетерогенную смесь выдерживали при 20-25°C в течение 16 часов. Реакционную смесь разделяли между IPAC и насыщенным водным NaHCO3 и дважды промывали насыщенным водным NaHCO3. Затем органический слой промывали 5% масс. водной лимонной кислотой, насыщенным водным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над Na2SO4 и концентрировали с получением продукта 51. Соединение 51 кристаллизовали из раствора этанола посредством добавления воды. 1H ЯМР (500 МГц, CD3OD): δ 9,15 (с, 1H), 8,82 (с, 1H), 8,22 (дд, J=6,1, 1,2 Гц, 1H), 8,13 (дд, J=7,3, 1,2 Гц, 1H), 7,37 (дд, J=7,3, 6,1 Гц, 1H), 7,16 (м, 1H), 6,94 (м, 1H), 4,79 (м, 1H), 4,67 (дд, J=11,5, 7,1 Гц, 1H), 4,06 (м, 1H), 4,01 (д, J=14,2 Гц, 1H), 3,90 (с, 2H), 3,79 (д, J=18,3 Гц, 1H), 3,73 (м, 1H), 3,69 (д, J=16,6 Гц, 1H), 2,89 (кв., J=12,5 Гц, 1H), 2,28 (м, 1H), 1,20 (д, J=6,4 Гц, 3H); 13C ЯМР (400 МГц, CD3OD): δ 182,8, 171,4, 168,1, 166,7, 159,6 (ддд, J=245, 10,5, 2,8 Гц), 157,5, 151,9 (дт, J=250, 14,2 Гц), 148,7, 148,2, 146,9 (ддд, J=243, 12,6, 3,9 Гц), 136,8, 133,4, 132,3 (дд, J=13,5, 8,5 Гц), 131,6, 130,8, 130,6, 126,4 (кв., J=280 Гц), 119,8, 111,7 (уш.д, J=24,8 Гц), 105,7 (дд, J=28,1, 21,8 Гц), 69,2, 58,0, 53,7, 52,5, 46,7 (кв., J=33,6 Гц), 45,2, 42,1, 37,5, 27,6, 13,7; 19F ЯМР (400 МГц, CD3OD): δ -71,96, -116,67 (д, J=14,7 Гц), -136,41 (д, J=20,0 Гц), -150,47 (дд, J=19,5, 15,2 Гц); HRMS: масса/заряд=604,1778 (M+1), рассчитанное для C29H24F6N5O3 масса/заряд=604,1778.
ПРИМЕР 6
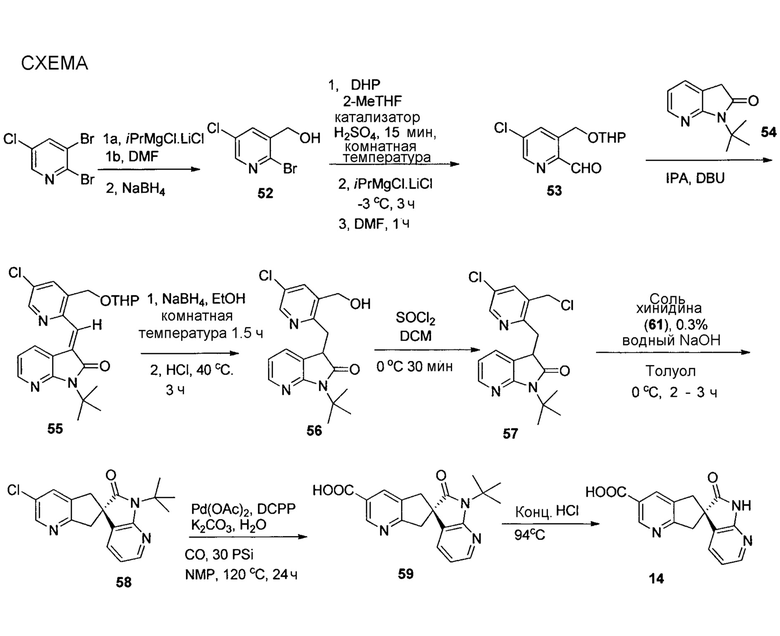
(2-Бром-5-хлорпиридин-3-ил)метанол (52)
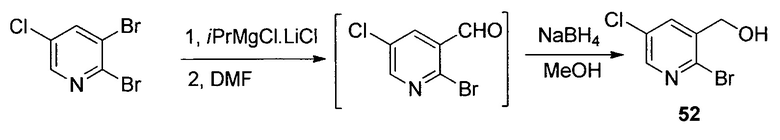
К раствору 2,3-дибром-5-хлорпиридина (60 г, 221 ммоль) в THF (500 мл) добавляли раствор раствора изопропилхлорида магния в хлориде лития в THF (1,3M, 185 мл) при -40°C приблизительно в течение 30 мин. Раствор перемешивали в течение 30 мин при -40°C и добавляли DMF (50 мл). Полученный раствор нагревали до комнатной температуры и перемешивали в течение 30 мин. Реакцию гасили 1Н HCl (400 мл) и добавляли MTBE (200 мл). Органический слой отделяли и дважды промывали 5% водным NaHCO3 (200 мл). Растворитель удаляли при пониженном давлении при 50°C. Полученные твердые вещества (альдегидное промежуточное соединение) растворяли в метаноле (400 мл). Раствор охлаждали до 5°C в ледяной бане. Медленно добавляли NaBH4 (3,6 г) в течение 30 мин при поддержании температуры реакционной смеси ниже комнатной температуры. Реакционную смесь перемешивали в течение дополнительных 30 мин с последующим добавлением воды (125 мл). Полученную смесь концентрировали при пониженном давлении приблизительно до 150 мл. При концентрировании осаждались твердые вещества. Суспензию энергично перемешивали при комнатной температуре в течение 1 часа, и твердые вещества собирали посредством фильтрации. Влажный осадок сушили в вакуумной печи в течение ночи при 60°C с получением 52 (45,6 г, 93%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,26 (д, J=2,5 Гц, 1H), 7,88 (д, J=2,5 Гц, 1H), 4,73 (д, J=5,8 Гц, 2H), 2,33 (т, J=11,4 Гц, 1H); 13C ЯМР (CDCl3, 100 МГц): δ 147,12, 138,48, 138,39, 136,14, 132,06, 62,76.
5-Хлор-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиколиновый альдегид (53)
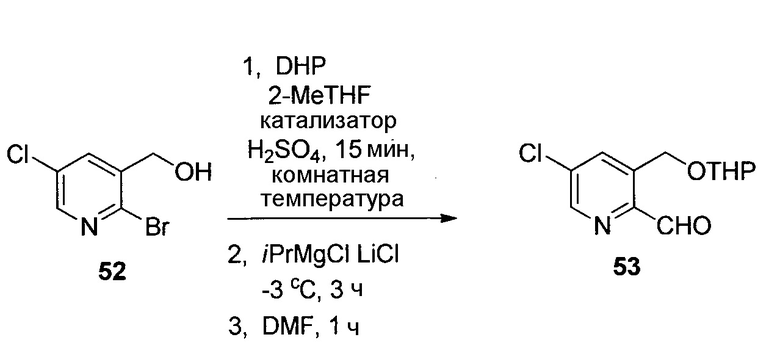
К раствору 52 (5,0 г, 22,5 ммоль) в 2-MeTHF (15 мл) добавляли 3,4-дигидро-2H-пиран (2,7 мл, 29,6 ммоль) и концентрированную серную кислоту (125 мг) при комнатной температуре. Раствор перемешивали в течение 10 мин, а затем охлаждали до -3°C. Медленно добавляли раствор изопропилхлорида магния в хлориде лития (1,3 M, 30 мл, 39 ммоль) при температурах от -3 до 3°C. Полученный раствор перемешивали при -3°C в течение 3 часов до демонстрации ВЭЖХ прохождения преобразования более 97%. Добавляли DMF (5 мл) в течение 15 мин при температуре ниже 5°C. Полученный раствор перемешивали в течение дополнительного 1 часа при этой температуре. Реакционную смесь гасили добавлением MTBE (50 мл), 15% водной лимонной кислоты (25 мл) и воды (15 мл). Органический слой отделяли и дважды промывали 5% водным NaCl (50 мл). Органический раствор концентрировали при пониженном давлении при 50°C с получением 53 в виде масла (6,2 г, 68% масс., 16,6 ммоль, выход 74%). Неочищенный продукт непосредственно использовали на следующем этапе без дополнительной очистки. Чистый образец выделяли посредством флэш-хроматографии на силикагеле с 5% этилацетатом в гексане в качестве элюента. 1H ЯМР (CDCl3, 400 МГц): δ 10,13 (с, 1H), 8,65 (с, 1H), 8,20 (с, 1H), 5,25 (д, J=16,6 Гц, 1H), 5,01 (д, J=16,6 Гц, 1H), 4,80 (м, 1H), 3,88 (м, 1H), 3,58 (м, 1H), 1,7 (м, 6H); 13C ЯМР (CDCl3, 100 МГц): δ 194,20, 147,06, 146,32, 138,98, 136,41, 134,87, 99,04, 64,42, 62,72, 30,53, 25,30, 19,66.
(E)-1-(трет-бутил)-3-((5-хлор-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридин-2-ил)метилен)-1H-пирроло[2,3-b]пиридин-2(3H)-он (55)
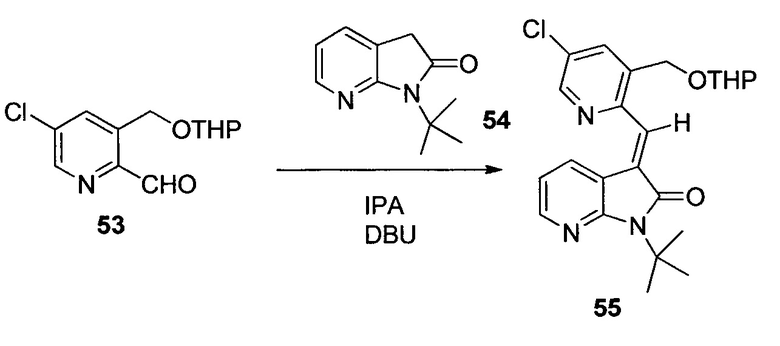
К раствору неочищенного 53 (6,2 г, 68% масс., 16,6 ммоль) и 54 (3,46 г, 18,3 ммоль) в изопропаноле (40 мл) добавляли DBU (0,12 г, 0,83 ммоль) при -2°C. После перемешивания при -2°C в течение 2 часов раствор нагревали до 10°C и перемешивали при этой температуре в течение 3 часов. Из раствора осаждались желтые твердые вещества. Суспензию перемешивали в течение ночи, позволяя партии медленно нагреваться до комнатной температуры. В заключение, суспензию нагревали до 50°C и перемешивали в течение 4 часов при этой температуре. После охлаждения до 30°C из капельной воронки капельно добавляли воду (35 мл) в течение 30 мин. Суспензию охлаждали до комнатной температуры и фильтровали. Осадок промывали смесью изопропанола (3 мл) и воды (3 мл). Осадки собирали и сушили в вакуумной печи в течение ночи при 50°C с получением 55 (6,2 г, 87%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,72 (дд, J=7,5, 1,8 Гц), 8,66 (д, J=2,4 Гц, 1H), 8,18 (дд, J=5,1, 1,8 Гц, 1H), 7,94 (д, J=2,4 Гц, 1H), 7,78 (с, 1H, 1H), 6,89 (дд, J=7,5, 5,1 Гц, 1H), 4,99 (д, J=13,8 Гц, 1H), 4,80 (м, 1H), 4,70 (д, J=13,8 Гц, 1H), 3,90 (м, 1H), 3,60 (м, 1H), 1,83 (с, 9H), 2,0-1,5 (м, 6H). Подтверждение двойной связи в качестве транс-изомера проводили в эксперименте NOE. 13C ЯМР (CDCl3, 100 МГц): δ 168,75, 159,64, 148,99, 147,85, 146,65, 137,01, 135,29, 133,56, 132,41, 129,50, 129,37, 117,27, 116,32, 98,77, 64,80, 62,49, 58,62, 30,39, 29,01, 25,26, 19,34.
1-(трет-бутил)-3-((5-хлор-3-(гидроксиметил)пиридин-2-ил)метил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (56)
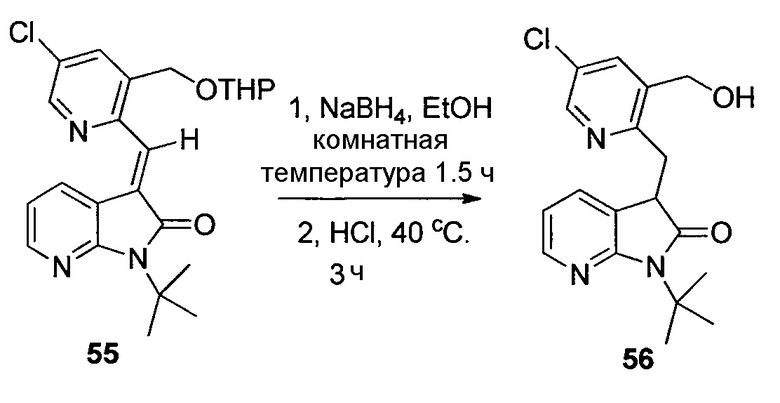
К суспензии 55 (3,0 г, 7,0 ммоль) в этаноле (25 мл) одной порцией добавляли NaBH4 (0,37 г). Полученную суспензию перемешивали при комнатной температуре в течение 1 часа. Реакцию гасили, медленно добавляя воду (10 мл), с последующим 6Н раствором HCl в изопропаноле (5 мл). Раствор нагревали до 40°C и перемешивали в течение 3 часов. Реакционную смесь смешивали с MTBE (50 мл) и насыщенным водным NaCl (50 мл). Органические вещества отделяли и промывали водой (50 мл). Раствор концентрировали при пониженном давлении при 50°C, и остаток растирали с гексаном (30 мл). Полученную суспензию перемешивали при комнатной температуре в течение 30 мин. Осадки собирали посредством фильтрации с получением 56 (2,2 г, 86%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,34 (с, 1H), 8,15 (д, J=4,9 Гц, 1H), 7,74 (с, 1H), 7,30 (д, J=7,1 Гц, 1H), 6,83 (т, J=5,7 Гц, 1H), 4,73 (дд, J=13,4, 4,9 Гц, 1H), 4,63 (дд, J=13,4, 5,7 Гц, 1H), 4,01 (т, J=6,1 Гц, 1H), 3,44 (дд, J=15,4, 5,2 Гц, 1H), 3,17 (дд, J=15,4, 7,2 Гц, 1H), 2,94 (т, J=5,5 Гц, 1H), 1,79 (с, 9H); 13C ЯМР (CDCl3, 100 МГц): δ 178,72, 159,12, 153,82, 146,45, 145,83, 135,72, 135,32, 130,63, 130,27, 124,04, 117,33, 61,40, 58,70, 44,12, 34,01, 28,81.
1-(трет-бутил)-3-((5-хлор-3-(хлорметил)пиридин-2-ил)метил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (57)
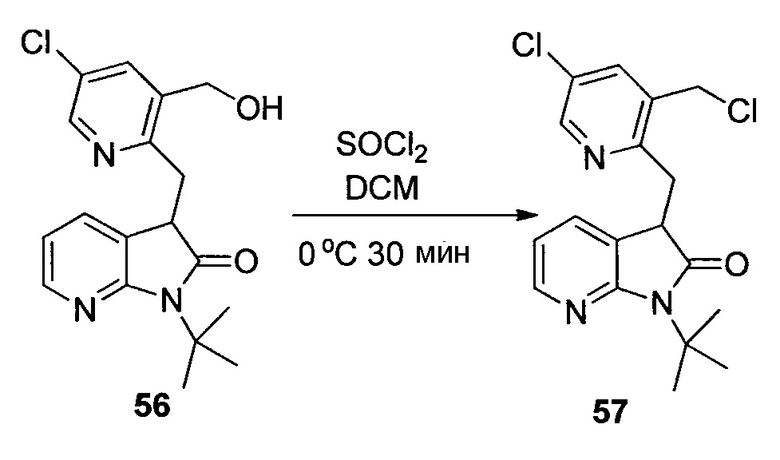
К раствору 56 (5,8 г, 16,8 ммоль) в дихлорметане (30 мл) добавляли DMF (60 мкл) и тионилхлорид (2,2 г) при 5°C. Смесь перемешивали в течение 30 мин при этой температуре с последующим добавлением 5% водного NaCl (30 мл). Органический слой отделяли и промывали 5% водным NaCl (30 мл). Растворитель удаляли, и остаток растворяли в гептане (20 мл). Раствор перемешивали в течение 30 мин и продукт осаждали. Суспензию охлаждали до 0°C и фильтровали с получением 57 (5,8 г, 93%) в виде твердого вещества: 1H ЯМР (CDCl3, 400 МГц): δ 8,36 (д, J=2,3 Гц, 1H), 8,13 (дд, J=5,1, 1,4 Гц, 1H), 7,65 (д, J=2,3 Гц, 1H), 7,19 (ом, 1H), 6,78 (дд, J=7,3, 5,2 Гц, 1H), 4,58 (м, 2H), 4,06 (м, 1H), 3,66 (дд, J=16,3, 4,6 Гц, 1H), 3,32 (дд, J=16,3, 7,5 Гц, 1H), 1,75 (с, 9H); 13C ЯМР (CDCl3, 100 МГц): δ 178,06, 159,45, 154,58, 147,39, 145,73, 136,87, 132,47, 130,42, 130,11, 123,77, 117,03, 58,51, 43,37, 42,25, 33,69, 28,82.
(S)-1'-(трет-бутил)-3-хлор-5,7-дигидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-2'(1'H)-он (58)
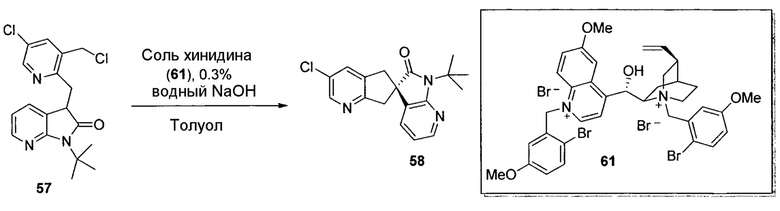
Раствор 57 (2,39 г, 6,56 ммоль) в толуоле (50 мл) охлаждали до -2,5°C в атмосфере азота. Добавляли соединение 61 (17 мг, 0,020 ммоль) и полученный раствор выдерживали приблизительно в течение 15 мин при охлаждении до -3,3°C. Добавляли предварительно охлажденный (-1°C) водный NaOH (26,2 мл, 0,3Н) в течение 4 мин при температуре ниже -0,6°C. Реакционную смесь выдерживали при -1,3°C в течение 3 часов. Реакцию гасили водой (10 мл). Органический слой промывали водой (10 мл), концентрировали, промывали IPA с получением неочищенного продукта 58 (2,59 г, 94,4% э.и., 83% масс. при ЯМР в сравнении с 1,3,5-триметоксибензолом в качестве внутреннего стандарта).
Неочищенный продукт перекристаллизовывали из IPA и воды, фильтровали и сушили в печи при 50°C с получением 58 (1,95 г, 95,7% масс., 99% э.и., выход 87%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,42 (с, 1H), 8,19 (д, J=5,2 Гц, 1H), 7,56 (с, 1H), 7,10 (д, J=7,3 Гц, 1H), 6,83 (дд, J=7,3, 5,2 Гц, 1H), 3,60 (дд, J=24,9, 16,8 Гц, 2Н), 3,09 (дд, J=28,6, 16,8 Гц, 2H); 13C ЯМР (CDCl3, 100 Гц): δ 179,43, 160,54, 157,82, 147,44, 146,54, 135,80, 132,17, 130,62, 129,33, 128,36, 117,69, 58,83, 51,94, 44,35, 41,57, 28,83.
Бромид (1S,2R,4S,5R)-1-(2-бром-5-метоксибензил)-2-((S)-(1-(2-бром-5-метоксибензил)-6-метоксихинолин-1-ий-4-ил)(гидрокси)метил)-5-винилхинуклидин-1-ия (61)

Взвесь хинидина (62, 8,1 г, 23,7 ммоль, содержащая ≈14% дигидрохинидина) и 2-бром-5-метоксибензилбромида (63, 16,59 г, 59,3 ммоль) в IPA (4,0 мл) и DMF (28,4 мл) дегазировали в вакууме и продували N2, затем нагревали до 70°C в течение 7 часов. Реакционную смесь охлаждали до 22°C, этот реакционный раствор добавляли в AcOEt (320 мл) при 22°C в течение 10 мин при перемешивании. Полученную взвесь выдерживали при 22°C в течение периода от 1 до 2 часов, фильтровали, промывали AcOEt (2×24 мл), затем гексаном (2×24 мл). Твердое вещество сушили в вакууме с получением порошка в виде смеси бис-соли (бис-хинидиновая соль 61 и бис-дигидрохинидиновая соль). (Всего 19,7 г, выход 94%). Очищали аутентичный образец 61 посредством SFC (колонка IC, 20×250 мм, 60% MeOH/CO2, 50 мл/мин, 10 мПа, 35°C, 220 нм, концентрация образца: 133 мг/мл в MeOH; желаемый пик: от 3 до 4,5 мин). 1H ЯМР (CDCl3, 500 МГц): δ 9,34 (д, J=6,1 Гц, 1H), 8,46 (д, J=6,1 Гц, 1H), 8,38 (д, J=9,7 Гц, 1H), 8,0 (дд, J=9,7, 2,1 Гц, 1H), 7,86 (с, 1H), 7,79 (д, J=8,9 Гц, 1H), 7,74 (д, J=8,9 Гц, 1H), 7,60 (д, J=2,5 Гц, 1H), 7,42 (д, J=2,3 Гц, 1H), 7,17 (дд, J=8,8, 2,8 Гц, 1H), 7,03 (дд, J=8,8, 2,7 Гц, 1H), 6,93 (с, 1H), 6,50 (д, J=2,4 Гц, 1Н), 6,06 (м, 1H), 5,24 (м, 3H), 4,95 (д, J=12,9 Гц, 1H), 4,37 (м, 1H), 4,23 (м, 4H), 4,12 (м, 1H), 3,88 (с, 3H), 3,69 (с, 3H), 3,54 (м, 1H), 3,32 (с, 2H), 3,23 (м, 1H), 2,71 (м, 1H), 2,51 (с, 2H), 2,33 (м, 1H), 1,94 (уш., 1H), 1,83 (уш., 2H), 1,17 (уш., 1H); 13C ЯМР (DMSO-d6, 100 Гц): δ 159,45, 159,07, 158,67, 156,12, 146,01, 137,08, 134,68, 134,30, 133,21, 132,98, 128,18, 128,03, 127,45, 122,13, 121,89, 121,22, 118,08, 117,5, 117,07, 116,73, 116,20, 115,81, 112,67, 105,09, 66,81, 65,51, 62,43, 56,75, 56,06, 55,91, 55,52, 54,80, 36,84, 25,91, 23,10, 20,75.
(S)-1'-(трет-бутил)-2'-оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоновая кислота (59)
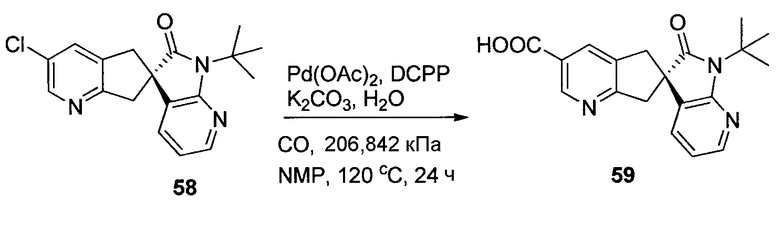
Смесь 58 (5,0 г, 14,5 ммоль), K2CO3 (5,01 г, 36,2 ммоль), Pd(OAc)2 (33 мг, 0,145 ммоль), 1,3-бис(дициклогексилфосфино)пропана (DCPP, 127 мг, 0,290 ммоль) и воды (0,522 мл, 29,0 ммоль) в NMP (32 мл) нагревали при 120°C при 206,842 кПа CO в течение 24 часов. После охлаждения до комнатной температуры полученную взвесь разбавляли водой (100 мл). pH медленно доводили до 3-4 2Н HCl. Взвесь выдерживали при комнатной температуре в течение 1 часа, фильтровали, промывали водой (40 до 50 мл), сушили в печи при 60°C с получением 59 (4,64 г, 95%) в виде твердого вещества. 1H ЯМР (DMSO-d6, 500 МГц): δ 8,90 (с, 1H), 8,19 (д, J=5,2 Гц, 1H), 7,54 (д, J=7,3 Гц, 1H), 6,99 (дд, J=7,3, 5,2 Гц, 1H), 3,33 (м, 4H), 1,72 (с, 9H); 13C ЯМР (DMSO-d6, 125 МГц): δ 180,16, 167,44, 166,97, 158,07, 149,76, 146,61, 135,39, 133,09, 130,36, 128,81, 125,48, 118,44, 58,19, 51,12, 44,56, 41,24, 28,91.
(S)-2'-Оксо-1',2',5,7-тетрагидроспиро[циклопента[b]пиридин-6,3'-пирроло[2,3-b]пиридин]-3-карбоновая кислота (14)
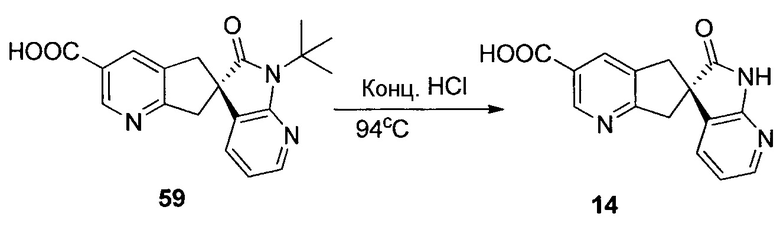
К 59 (4 г, 97% масс.) добавляли 37% HCl (от 40 до 44 мл). Взвесь нагревали при 94°C в течение периода до 48 часов, охлаждали до комнатной температуры. Растворитель частично удаляли, снижая давление, приблизительно до 2 объемов (оставалось ~4 мл воды). Остаток разбавляли водой (20 мл) с последующим доведением pH до 2,6 посредством NaOH (3,5Н, 4,5 мл). Густую взвесь выдерживали в течение периода от 1 до 2 часов, фильтровали, промывали водой (2×8 мл), с последующей водой/ацетоном (1:1, 8 мл). Влажный осадок сушили с получением соединения 14 (3,1 г, 98% масс., 94%) в виде кристаллов. 1H ЯМР (DMSO-d6, 500 МГц): δ 13,31 (уш., 1H), 11,14 (с, 1H), 8,91 (с, 1H), 8,11 (м, 2H), 7,49 (дд, J=7,3, 1,3 Гц, 1H), 6,93 (дд, J=7,3, 5,3 Гц, 1H), 3,36 (м, 4H); 13C ЯМР (DMSO-d6, 125 МГц): δ 181,06, 167,36, 166,95, 156,80, 149,79, 147,32, 135,37, 133,19, 130,73, 128,88, 125,50, 118,46, 51,78, 44,12, 40,70.
ПРИМЕР 7
1-(трет-бутил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (54)
Смесь соединения 54a (10,0 г, 40,3 ммоль), NaCl (2,9 г) и воды (2 мл) в DMSO (50 мл) нагревали при 120°C в течение 30 мин. Смесь охлаждали до 30°C с последующим добавлением MTBE (200 мл) и воды (50 мл). Органический слой отделяли, и водный слой экстрагировали дополнительным MTBE (50 мл). Объединенный органический слой трижды промывали водой (50 мл). Растворитель удаляли в вакууме, и полученное твердое вещество сушили в вакуумной печи при 30°C с получением 54 (7,0 г, 92%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 8,15 (дд, J=5,2, 1,4 Гц, 1H), 7,40 (дд, J=7,2, 1,4 Гц, 1H), 6,88 (дд, J=7,2, 5,2 Гц, 1H), 3,45 (с, 2H), 1,78 (с, 9H); 13C ЯМР (CDCl3, 100 МГц): δ 174,99, 160,06, 145,82, 130,80, 119,51, 117,15, 58,53, 35,98, 28,80.
ПРИМЕР 8
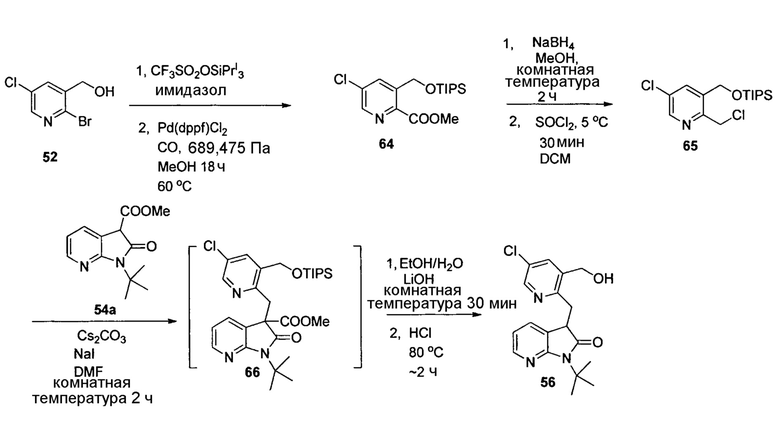
Метил-5-хлор-3-(((триизопропилсилил)окси)метил)пиколинат (64)
К раствору 52 (15,0 г, 67,4 ммоль) в дихлорметане (60 мл) добавляли триизопропилсилилтрифторметансульфонат (29,0 г, 94 ммоль). Раствор охлаждали до 5°C и несколькими частями добавляли имидазол (12,0 г, 176 ммоль) при температуре ниже 20°C. Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и добавляли 5% насыщенный солевой раствор (50 мл). Органический слой отделяли, и растворитель удаляли в вакууме при 50°C. Полученное масло растворяли в метаноле (100 мл). Раствор содержали в CO (689,475 Па) при 60°C в течение 18 часов в присутствии 5 мол.% Pd(dppf)Cl2. Растворитель удаляли и остаток переносили на силикагель (60 г) в фильтровальную воронку. Смесь промывали смесью 10% этилацетата в гексане (400 мл). Полученный раствор концентрировали с получением неочищенного 64 (29,2 г, 98% LCAP, 84% масс., выход 100%) в виде масла, которое непосредственно использовали на следующем этапе без дополнительной очистки. 1H ЯМР (CDCl3, 400 МГц): δ 8,56 (д, J=2,4 Гц, 1H), 8,30 (д, J=2,4 Гц, 1H), 5,23 (с, 2H), 4,01 (с, 3H), 1,25 (м, 3H), 1,12 (д, J=6,8 Гц, 18H).
5-Хлор-2-(хлорметил)-3-(((триизопропилсилил)окси)метил)пиридин (65)
К раствору неочищенного 64 (29,2 г, 84% масс., 67,4 ммоль) в метаноле (120 мл) порционно добавляли NaBH4 (11,6 г) приблизительно в течение 1 часа при 5°C. Реакционную смесь гасили водой (150 мл), и смесь экстрагировали MTBE (150 мл). Органический раствор промывали водой (100 мл). Растворитель удаляли в вакууме при 50°C, и остаток растворяли в дихлорметане (60 мл). Раствор концентрировали в вакууме при 60°C. Полученный остаток растворяли в дихлорметане (100 мл). Раствор охлаждали до 0°C и капельно добавляли DMF (0,5 г) с последующим тионилхлоридом (11,1 г). Затем реакционную смесь перемешивали в течение 30 мин при 0°C и гасили 5% насыщенным солевым раствором (100 мл). Органический слой отделяли и промывали насыщенным солевым раствором (100 мл). Растворитель удаляли в вакууме при 60°C с получением 65 в виде масла (25 г, 92% LCAP, 70% масс., выход 72%), которое использовали на следующем этапе без дополнительной очистки. 1H ЯМР (CDCl3, 400 МГц): δ 8,44 (д, J=2,4 Гц, 1H), 7,94 (д, J=2,4 Гц, 1H), 4,96 (с, 2H), 4,65 (с, 2H), 1,22 (м, 3H), 1,12 (д, J=6,7 Гц, 18H).
1-(трет-бутил)-3-((5-хлор-3-(гидроксиметил)пиридин-2-ил)метил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (56)
К раствору 65 (8 г, 70% масс., 16,1 ммоль) и 54a (4,15 г) в DMF (30 мл) добавляли Cs2CO3 (5,76 г) и NaI (2,4 г). Смесь перемешивали при комнатной температуре в течение 1 часа, и реакционную смесь смешивали с MTBE (100 мл) и 15% водной лимонной кислотой (80 мл). Органические вещества дважды промывали водой (80 мл), и удаляли растворитель. Остаток (66, 90% LCAP) растворяли в смеси этанола (70 мл) и воды (20 мл). После добавления LiOH (2,8 г) раствор перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь подкисляли 6Н раствором HCl в IPA (17 мл). Полученный раствор нагревали при 80°C в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляли MTBE (100 мл) и 5% насыщенным солевым раствором (50 мл). Органический слой промывали водой (50 мл) и сушили над MgSO4. Раствор концентрировали в вакууме при 50°C, и остаток кристаллизовали из гексана (30 мл) с получением 56 (3,75 г, 67% из 65) в виде кристаллов.
ПРИМЕР 9
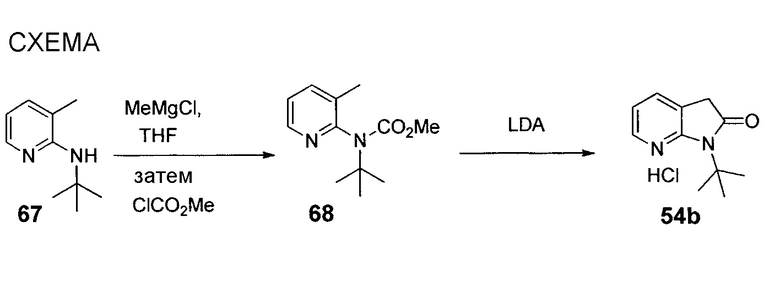
Метил-трет-бутил(3-метилпиридин-2-ил)карбамат (68)
К N-(трет-бутил)-3-метилпиридин-2-амину (67) (16,78 г, 92% масс., 102 ммоль) в THF (100 мл) добавляли MeMgCl (44,3 мл, 3M, 133 ммoль) в THF при -10°C в течение 5 мин. Реакционную смесь нагревали до комнатной температуры и выдерживали в течение 80 мин, затем охлаждали до -20 - -15°C и добавляли метилхлорформиат (8,7 мл, 112 моль) в течение 10 мин при -8°C. Реакционную смесь постепенно нагревали и выдерживали в течение ночи при комнатной температуре. Реакционную смесь гасили, добавляя 15% водную лимонную кислоту (13 мл), воду (40 мл) и MTBE (33 мл) при 0°C. Органический слой отделяли и последовательно промывали водой (50 мл), насыщенным NaHCO3/водой (1:3, 50 мл), насыщенным солевым раствором (50 мл) и водой (50 мл). Органический слой концентрировали и промывали THF с получением 68 (19,3 г, 92%).
1-(трет-бутил)-1H-пирроло[2,3-b]пиридин-2(3H)-он гидрохлорид (54b)
Соединение 68 (5 г, 97% масс., 21,8 ммоль) в THF (30 мл) или толуоле (50 мл) дегазировали, охлаждали до -45°C. Добавляли LDA (45,8 мл, 1,0Н) при температурах от -45 до -40°C в течение 13 мин. (Примечание: LDA получали отдельно в другой колбе с использованием 1,0 эквив. н-BuLi и 1,1 эквив. диизопропиламина в THF при температурах от -35°C до 0 до 12°C, затем охлаждали до 0°C.)
Указанную выше реакционную смесь постепенно нагревали до 13°C в течение 4,5 часов, охлаждали в ледяной бане и гасили 2Н HCl (~40 мл) (pH~4) и толуолом (15 мл) при температуре ниже 20°C. Органический слой отделяли и последовательно промывали водой (15 мл), насыщенным солевым раствором (15 мл) и водой (15 мл). Органический раствор, содержащий 54 (3,79 г, аналитический выход 91%), концентрировали, промывали толуолом с удалением воды. К раствору остатка в толуоле (7,6 мл), добавляли 2Н HCl в простом эфире (12 мл) в течение 30 мин. К смеси добавляли гексан (8 мл) и выдерживали 1 час. Осадки фильтровали, промывали толуолом/гексаном (1:2, 8 мл), затем гексаном (8 мл) и сушили в вакууме с потоком азота с получением соли 54b (3,92 г, 87%) в виде твердого вещества. 1H ЯМР (CDCl3, 400 МГц): δ 11,9 (уш., 1H), 8,13 (дд, J=5,2, 0,8 Гц, 1H), 7,54 (дд, J=7,2, 1,1 Гц, 1H), 6,96 (дд, J=7,2, 5,2 Гц, 1H), 3,52 (с, 2H), 1,69 (с, 9H); 13C ЯМР (DMSO-d6, 100 МГц): δ 174,99, 159,8, 145,67, 131,77, 120,54, 117,68, 57,89, 35,80, 28,99.
ПРИМЕР 10
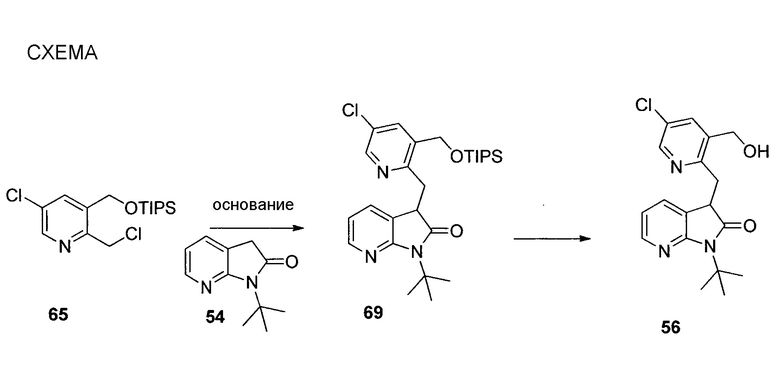
1-(трет-бутил)-3-((5-хлор-3-(((триизопропилсилил)окси)метил)пиридин-2-ил)метил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (69)
Раствор 65 (23,84 г, 60,7 ммоль) и 54 (17,32 г, 91 ммоль) в THF (200 мл) дегазировали продувки N2 в вакууме при температуре ниже 5°C. К раствору добавляли раствор амоксида лития в гептене (27,4 мл, 40%, 85 ммоль) с поддержанием температуры ниже 7°C. Реакционную смесь выдерживали при температуре ниже 5°C в течение 40 мин, затем гасили насыщенным водным NH4Cl (10 мл), разбавляли гексаном (120 мл). Органический слой отделяли, промывали насыщенным водным NH4Cl (100 мл) и водой (150 мл), концентрировали и очищали посредством колонки с силикагелем (от 0 до 5% AcOEt/гексана) с получением 69, который использовали в следующей реакции без дополнительной очистки.
Масса/заряд в HRMS, рассчитанное для C28H41ClN3O2Si, составляло 502,2651 (M+H); выявлено 502,2641.
1-(трет-бутил)-3-((5-хлор-3-(гидроксиметил)пиридин-2-ил)метил)-1H-пирроло[2,3-b]пиридин-2(3H)-он (56)
Раствор 69 (макс. 60,7 ммоль) в THF (50 мл), полученный на предыдущем этапе, дегазировали посредством продувки N2 в вакууме, охлаждали при температуре ниже 5°C с последующим добавлением TBAF (79 мл, 1,0Н в THF). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа 20 мин, охлаждали при температуре ниже 10°C, гасили водой (100 мл) и разбавляли AcOEt (200 мл). Водный слой экстрагировали AcOEt (100 мл). Объединенный органический слой сушили MgSO4, фильтровали, концентрировали и очищали посредством колонки с силикагелем (от 50 до 100% AcOEt/гексана) с получением 56 (16,3 г, 78% из 65) в виде твердого вещества.
РЕЗУЛЬТАТЫ
Как представлено в таблице 1, бис-четвертичный катализатор для представленной реакции спироциклизации является намного более активным и эффективным по сравнению с моно-четвертичным катализатором. Для этого эксперимента бис-четвертичные катализаторы содержали от ~12 до 15% насыщенных по двойным связям соединений гидрохинидина или гидроцинхонина.
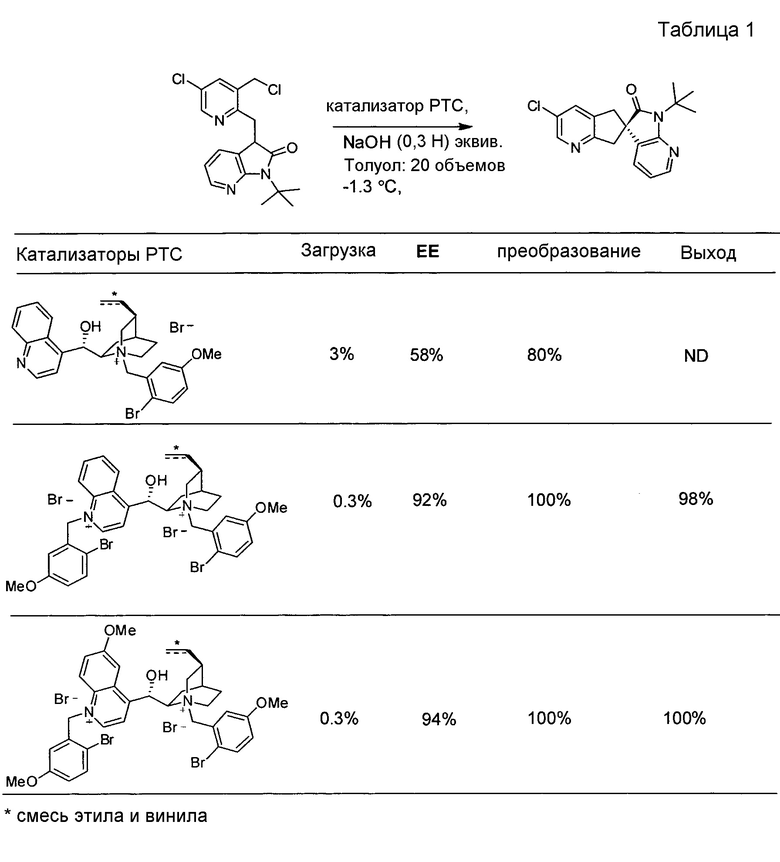
В таблицах 2 и 3 представлено исследование SAR бис-четвертичных катализаторов PTC для спироциклизации. Оба бис-четвертичных катализатора - хинидин и цинхонин - являются очень эффективными (записи 2-5). Бис-четвертичный катализатор хинидин являлся относительно лучшим, чем бис-четвертичный катализатор цинхонин (записи 2 и 3). Одной из самых эффективных групп для этой реакции является 2-бром-5-метоксибензильная группа.
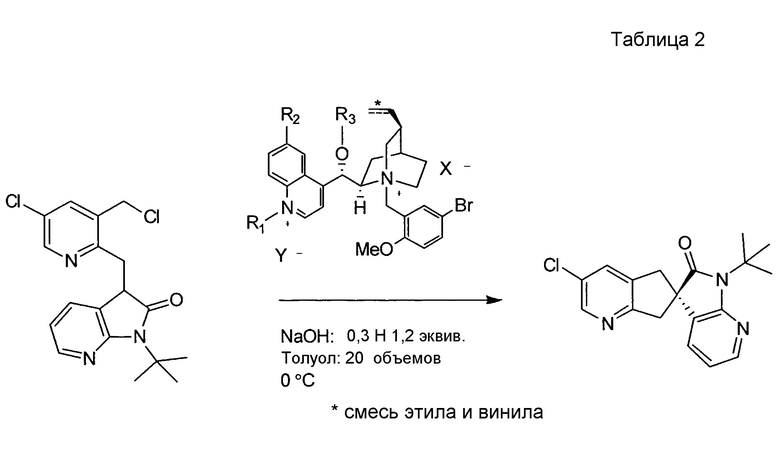
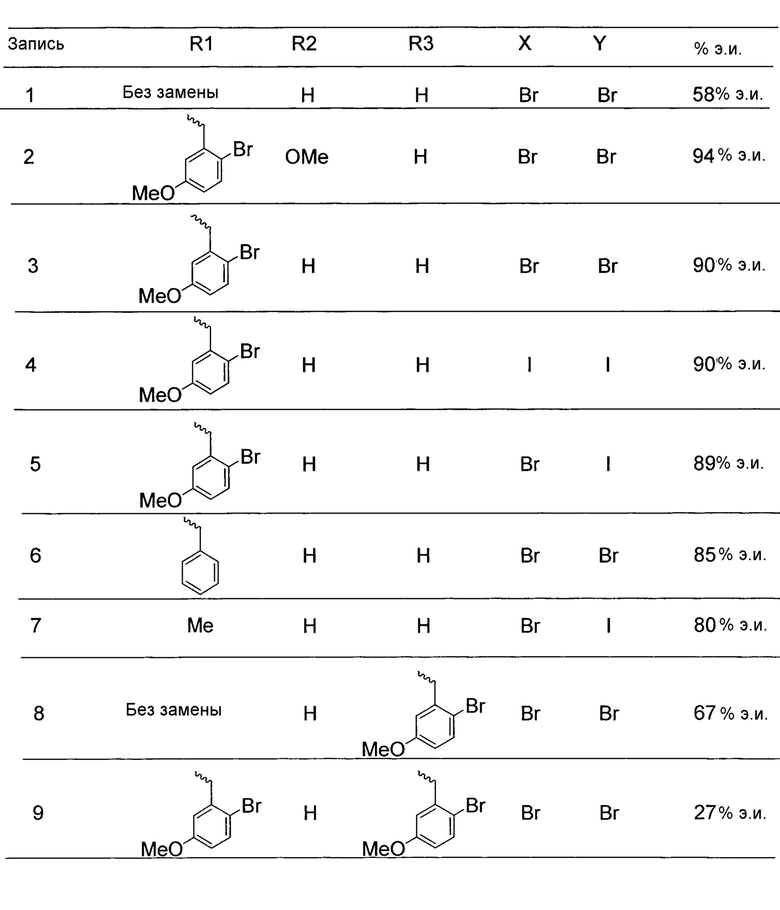
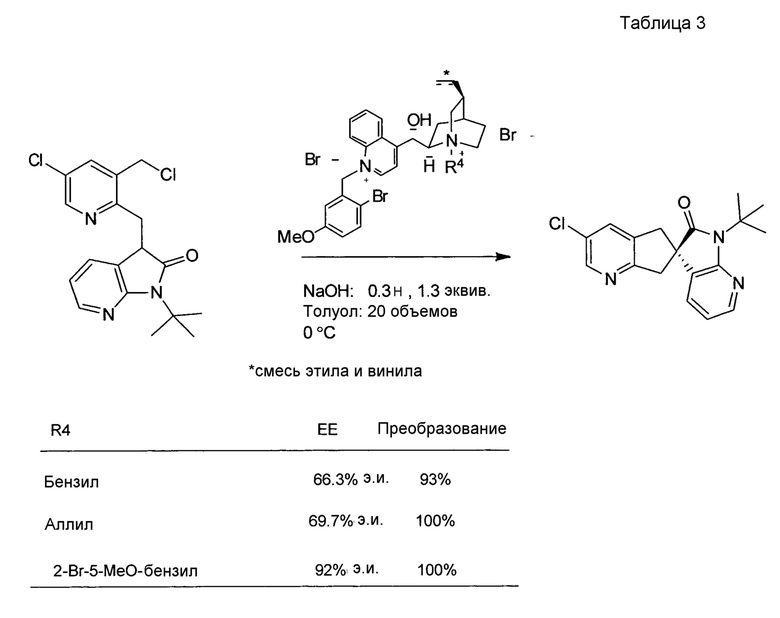
В таблице 4 представлено исследование различных функциональных групп.
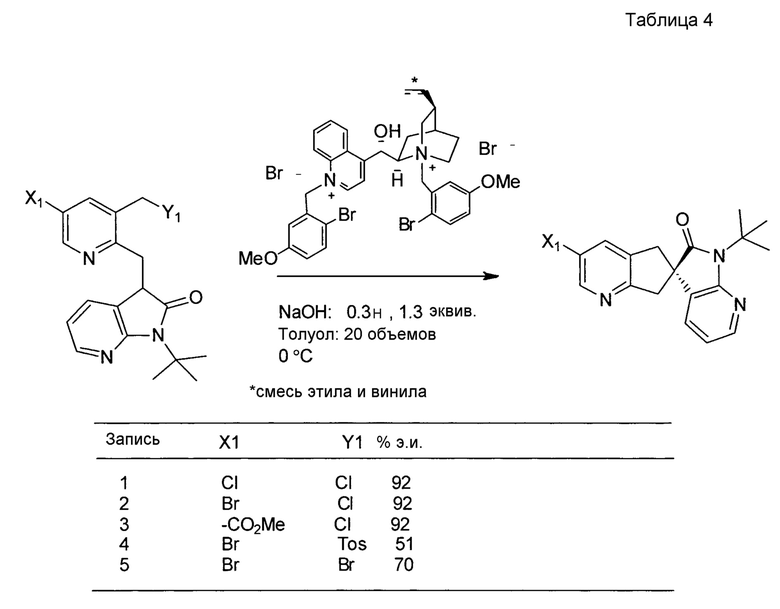
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2651369C1 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2627654C2 |
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2012 |
|
RU2605537C2 |
| ОБЩИЙ СИНТЕЗ МИРИАПОРОНОВ | 2003 |
|
RU2328492C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИМЕЮЩИЕ СРОДСТВО К МУСКАРИНОВЫМ РЕЦЕПТОРАМ | 2008 |
|
RU2446166C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ ДИГИДРОПИРАН-2-ОНА | 2007 |
|
RU2444519C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 1-О-АЦИЛ-2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗ | 2010 |
|
RU2559364C2 |


Изобретение относится к области органической химии, а именно к способу получения соединения формулы I или его фармацевтически приемлемой соли, включающему взаимодействие соединения формулы F в органической фазе в присутствии бис-четвертичной соли алкалоида хинного дерева и неорганического основания в водной фазе, с формированием двухфазной среды, содержащей водную фазу и органическую фазу, с получением соединения формулы G, когда E1 отличен от -C(O)-O-H, замену E1 карбоновой кислотой или преобразование E1 в карбоновую кислоту в подходящей химической реакции с выходом соединения формулы H, снятие защиты с соединения формулы H с выходом соединения формулы C, необязательно выделяемого в виде соли, и связывание соединения формулы C с соединением формулы B, необязательно в виде соли, в условиях формирования амидной связи между кислотой и амином с выходом соединения формулы I. Также изобретение относится к промежуточным соединениям. Технический результат: разработан способ получения индановых и азаиндановых производных пиперидинонкарбоксамида, которые являются антагонистами рецепторов CGRP, пригодных для лечения мигрени, с использованием высокоэффективного синтеза спирокислот. 2 н. и 16 з.п. ф-лы, 4 табл., 10 пр.
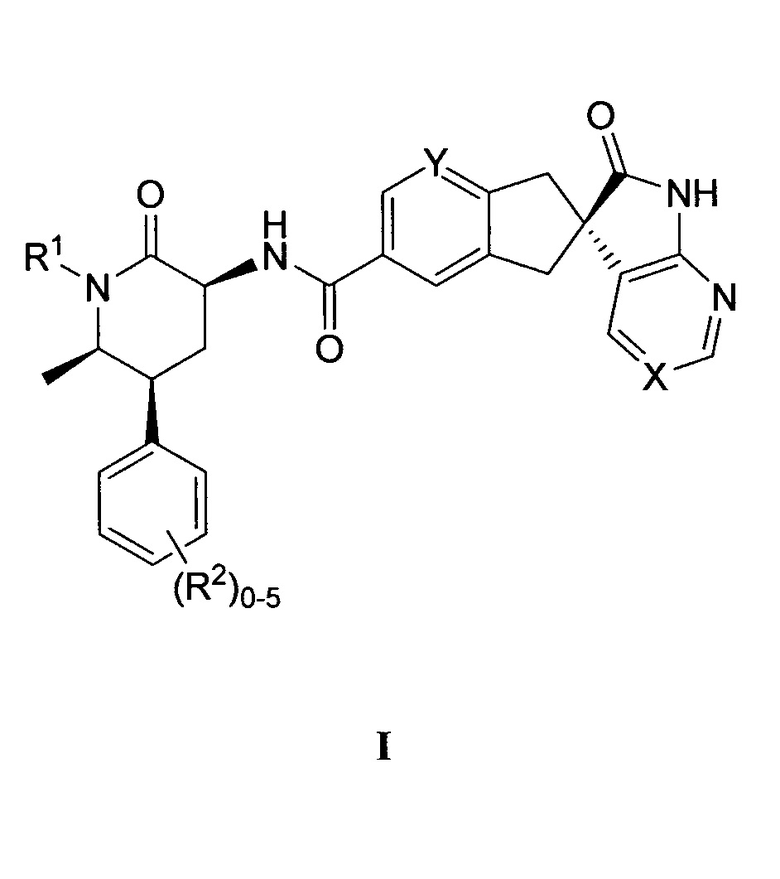 ,
,  ,
, 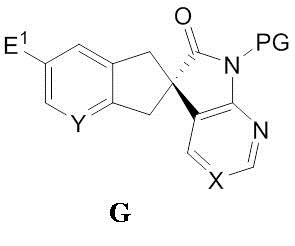
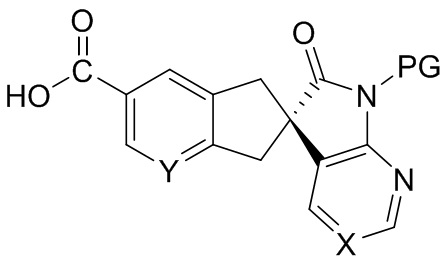 H,
H,  ,
, 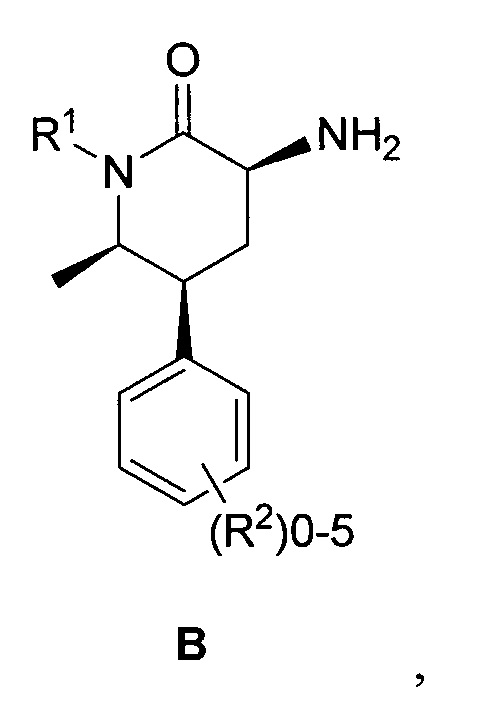
1. Способ получения соединения формулы I:
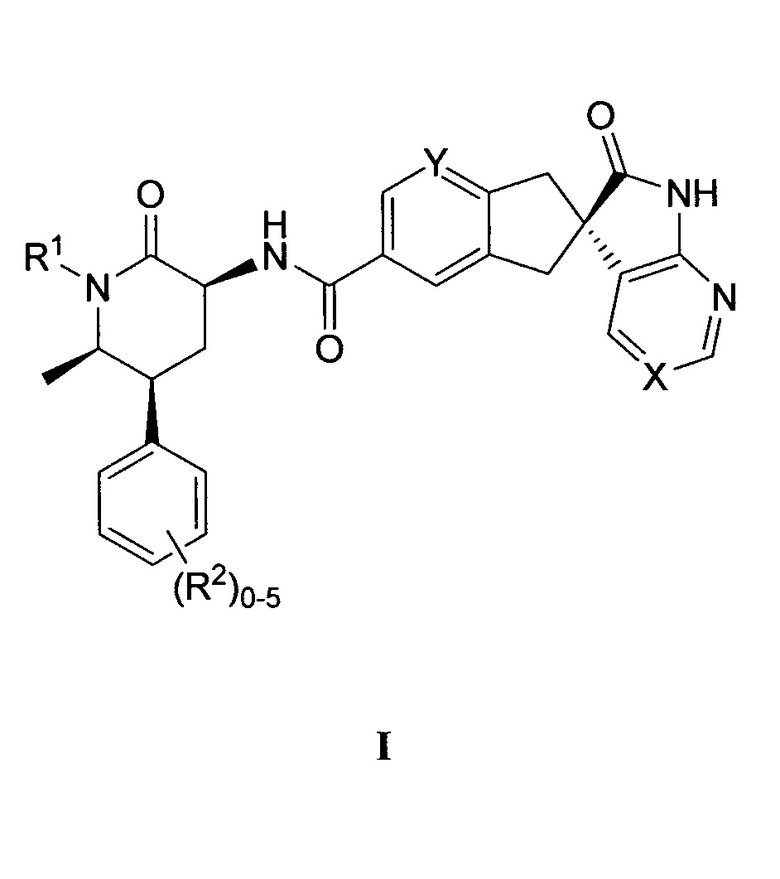 ,
,
или его фармацевтически приемлемой соли, где:
X выбран из -C(R3)=, где R3 представляет собой водород, F или CN;
Y представляет собой N;
R1 выбран из группы, состоящей из C1-4-алкила, циклопропилметила, циклобутилметила и [1-(трифторметил)циклопропил]метила, каждый из которых необязательно, как позволяет валентность, замещен одним или несколькими заместителями F; и
R2 представляет собой водород, метил, F, Cl или Br;
включающий инициализацию реакции соединения формулы F:
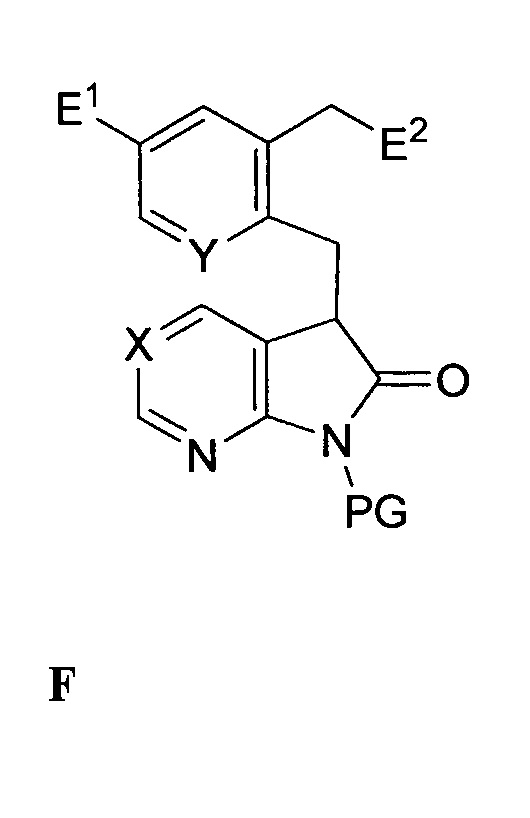 ,
,
где E1 выбран из галогена и -C(O)-O-R", где R" представляет собой содержащий углерод заместитель, способный к растворению до карбоновой кислоты,
E2 представляет собой функциональную группу со способностью быть уходящей группой, и
PG представляет собой защитную группу азота, где PG выбран из группы, состоящей из C1-6-алкила, винила, C(O)-O-L, C(O)-L, арила, гетероарила, бензила, бензгидрила, тритила, антранила и C1-6-алкоксиметила, где арил, гетероарил, бензил, бензгидрил и тритил необязательно замещены 1-3 заместителями, независимо выбранными из метокси и нитро, C1-6-алкоксиметил необязательно замещен триметилсилилом, а L представляет собой C1-6-алкил, арил или бензил,
в органической фазе в присутствии бис-четвертичной соли алкалоида хинного дерева и неорганического основания в водной фазе, с формированием двухфазной среды, содержащей водную фазу и органическую фазу, с получением соединения формулы G:
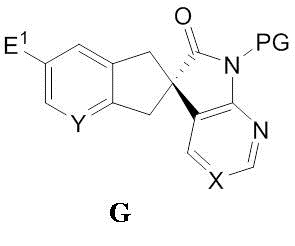 ,
,
когда E1 отличен от -C(O)-O-H, замену E1 карбоновой кислотой или преобразование E1 в карбоновую кислоту в подходящей химической реакции с выходом соединения формулы H:
 H,
H,
снятие защиты с соединения формулы H с выходом соединения формулы C:
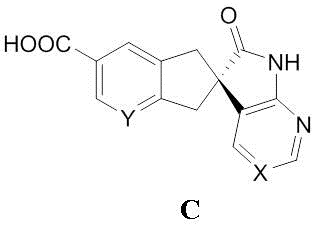 ,
,
необязательно выделяемого в виде соли, и связывание соединения формулы C с соединением формулы B:
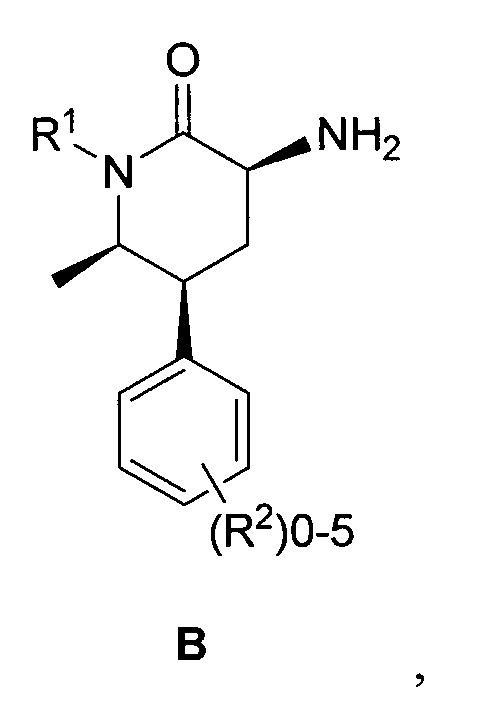
необязательно в виде соли, в условиях формирования амидной связи между кислотой и амином с выходом соединения формулы I.
2. Способ по п. 1, где бис-четвертичная соль алкалоида хинного дерева обладает химической структурой формулы II:
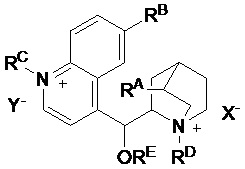 II ,
II ,
где:
RA представляет собой 
RB выбран из группы, состоящей из водорода и метокси,
каждый из RC и RD независимо выбран из группы, состоящей из C1-6-алкила, C2-6-алкенила, арила и -C1-4-алкиларила, где арильная часть -C1-4-алкиларила необязательно замещена одним - пятью заместителями, независимо выбранными из RF,
RE выбран из группы, состоящей из водорода и -C1-4-алкиларила, где арильная часть -C1-4-алкиларила необязательно замещена одним - пятью заместителями, независимо выбранными из RF,
RF выбран из группы, состоящей из водорода, C1-4-алкокси, галогена и CF3, и
каждый из X и Y представляет собой независимый анион, выбранный из галогенида.
3. Способ по п. 1, где E1 представляет собой галоген, и карбонилирование соединения формулы G проводят посредством катализируемой палладием реакции с монооксидом углерода, при этом соединение формулы G вступает в реакцию с монооксидом углерода в присутствии палладиевого катализатора, первого основания и лиганда с выходом соединения формулы H.
4. Способ по п. 3, где лиганд представляет собой фосфиновый лиганд.
5. Способ по п. 1, где E1 представляет собой галоген, и карбонилирование соединения формулы G проводят посредством формирования реактива Гриньяра, при этом соединение формулы G вступает в реакцию с магнием или литием, затем с CO2 и кислотой, с выходом соединения формулы H.
6. Способ по п. 1, где E2 выбран из группы, состоящей из галогена, OMs, OTs, OBs, OP(O)(ORi)4, OC(O)Ri, OC(O)ORi и OC(O)NRiRii, где Ri и Rii независимо выбраны из группы, состоящей из водорода и C1-6-алкила.
7. Способ по п. 1, где органическая фаза выбрана из группы, состоящей из бензола, толуола, ксилола, хлорбензола, простого этилового эфира, простого изопропилового эфира, тетрагидрофурана, 2-метилтетрагидрофурана, диоксана, простого метил-трет-бутилового эфира, простого циклопентилметилового эфира, изопропилацетата, этилацетата, гексана, гептана, циклогексана, дихлорметана, дихлорэтана, ацетонитрила, ацетона, метилэтилкетона, бутанола и амилового спирта.
8. Способ по п. 1, где неорганическое основание выбрано из группы, состоящей из гидроксида натрия, гидроксида лития, гидроксида калия, карбоната натрия, карбоната лития, карбоната калия, гидроксида цезия, карбоната цезия, гидрокарбоната натрия, гидрокарбоната калия, гидрокарбоната лития, фторида лития, фторида натрия, фторида калия, фторида цезия, трет-бутоксида лития, трет-бутоксида натрия, трет-бутоксида калия, фосфата натрия и фосфата калия.
9. Способ по п. 1, где органическая фаза представляет собой толуол, а неорганическое основание представляет собой гидроксид натрия.
10. Способ по п. 1, где соединение формулы C выделяют в виде соли HCl.
11. Способ по п. 2, где в бис-четвертичной соли алкалоида хинного дерева формулы II Rc и RD независимо выбраны из группы, состоящей из C1-6-алкила и бензила, и где C1-6-алкильная или фенильная части каждого из указанного бензила необязательно замещены одним - пятью заместителями, независимо выбранными из RF.
12. Способ по п. 11, где в бис-четвертичной соли алкалоида хинного дерева формулы II RB представляет собой метокси.
13. Способ по п. 12, где в бис-четвертичной соли алкалоида хинного дерева формулы II RF выбран из группы, состоящей из галогена и метокси.
14. Способ по п. 1, где бис-четвертичная соль алкалоида хинного дерева представляет собой формулу

15. Способ по п. 1, дополнительно включающий получение соединения формулы F посредством реакции соединения формулы J:
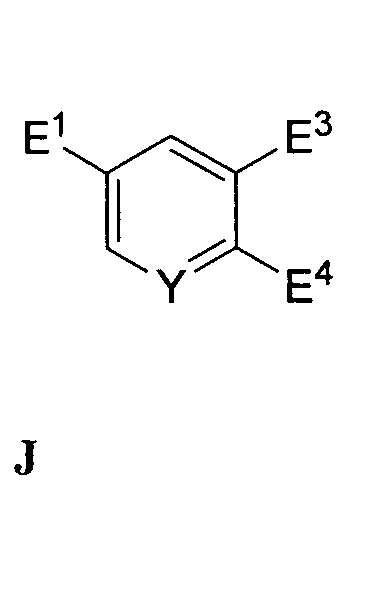 ,
,
где E3 и E4 независимо представляют собой функциональные группы со способностью быть уходящей группой,
с R4-Z1 или его солью и HC(O)-N(R5)2, где R4 представляет собой C1-6-алкил, Z1 представляет собой галогенид магния или литий, и каждый R5 независимо представляет собой H или C1-6-алкил, с выходом промежуточного соединения формулы K:
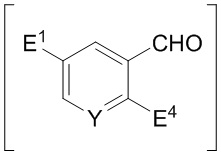 K,
K,
реакции промежуточного соединения формулы K с первым восстановителем с выходом соединения формулы L, где указанный первый восстановитель независимо представляет собой: H2, HCO2H, HCO2NH4, NaBH4, LiBH4 или LiAlH4:
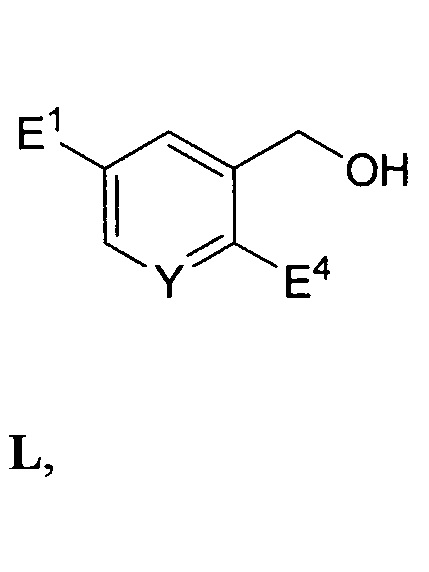
введения защитной группы спирта (APG) в соединение формулы L с выходом соединения формулы M, где APG представляет собой: THP, тетрагидрофурил, SEM, MOM, BOM, TMS, TES, TBDMS, TIPS или бензил, который необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, метила, метокси и нитро:
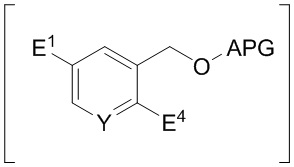 M,
M,
(i) реакции промежуточного соединения формулы M с R6-Z2 или его солью и HC(O)-N(R7)2, где R6 представляет собой C1-6-алкил, Z2 представляет собой галогенид магния или литий, и каждый R7 независимо представляет собой H или C1-6-алкил, с выходом соединения формулы N:
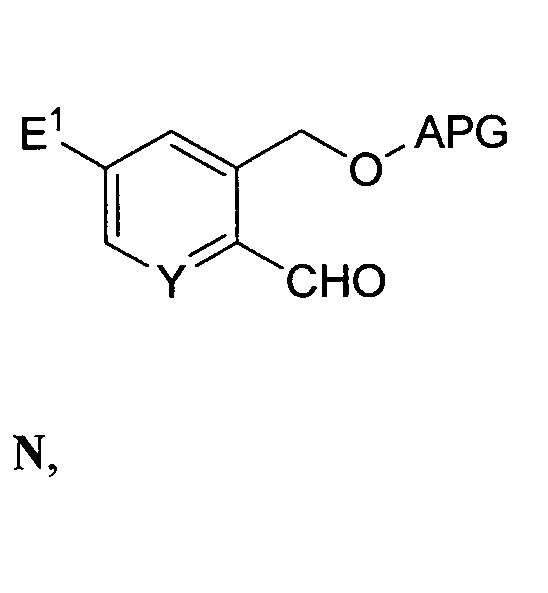
и приведенных ниже этапа (a) с выходом соединения формулы F;
Этап (a) - связывание соединения формулы N с соединением формулы O:
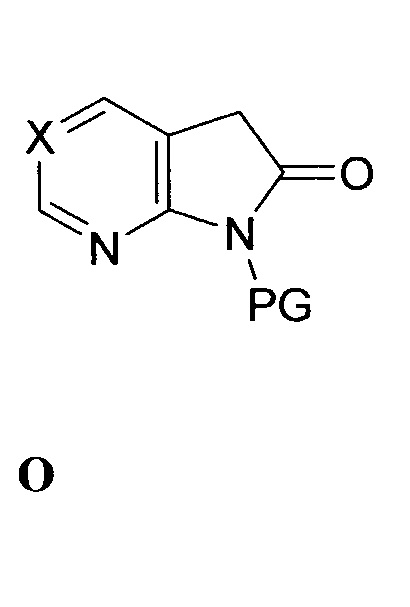 ,
,
в присутствии второго основания с выходом соединения формулы P:
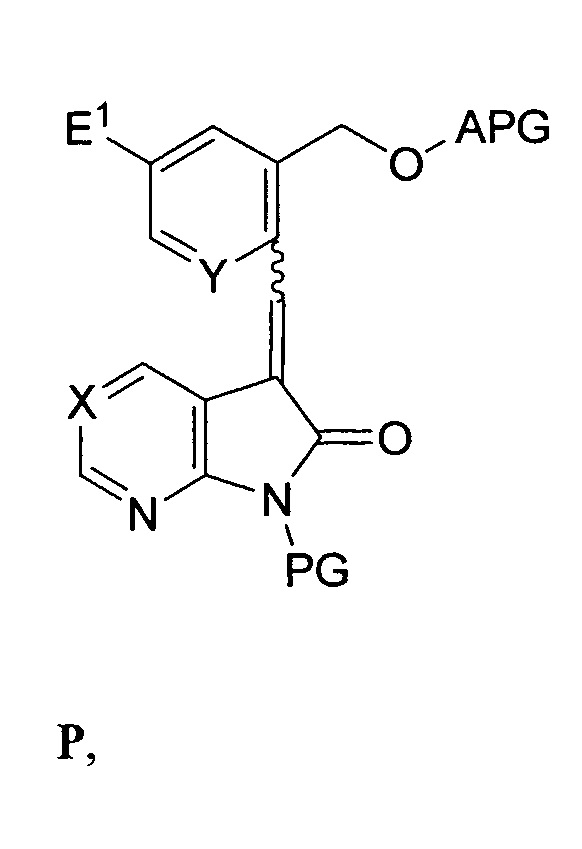
и реакция соединения формулы P со вторым восстановителем, где указанный второй восстановитель независимо представляет собой: H2, HCO2H, HCO2NH4, NaBH4, LiBH4 и LiAlH4, с последующим снятием защиты со спиртовой группы и замещением спиртовой группы подходящей уходящей группой E2 с выходом соединения формулы F.
16. Способ по п. 15, где E3 и E4 независимо представляют собой галоген.
17. Способ по п. 15, где второе основание представляет собой неорганическое основание.
18. Соединение, выбранное из следующей группы:
или соли любого из указанного выше.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| WO 20101399717 A1, 09.12.2010. | |||

