Область техники
[0001] Настоящее изобретение относится к пиразолохинолиновым производным, обладающим ингибирующей активностью в отношении фосфодиэстеразы 9 (PDE9), и их фармакологически приемлемым солям, и их фармацевтическим применениям.
Предшествующий уровень техники
[0002] Известно, что циклический гуанозинмонофосфат (далее по тексту называемый cGMP), функционирующий в качестве вторичного мессенджера в клетках, играет важную роль в различных физиологических функциях, в том числе в протекании обучения и развитии памяти.
[0003] На постсинаптическом участке нервных цепей головного мозга монооксид азота (далее по тексту называемый NO), биосинтезируемый синтетазой монооксида азота, активирует гуанилатциклазу, которая является cGMP-синтазой. Активированная гуанилатциклаза биосинтезирует cGMP из гуанозинтрифосфата. cGMP активирует cGMP-зависимую протеинкиназу (далее по тексту называемую PKG), фосфорилирующую различные белки, участвующие в пластичности синапсов. Известно, что активация каскада NO/cGMP/PKG участвует в индукции пластичности синапсов (долговременная потенциация; далее по тексту называемая LTP) гиппокампа, который известен как нейронная основа для протекания обучения и развития памяти (например, см. непатентную литературу 1). Лекарственный препарат, активирующий передачу сигнала по каскаду, известен как улучшающий LTP гиппокампа и протекание обучения у животных, в то же время лекарственный препарат, ингибирующий каскад, известен как оказывающий противоположное действие (непатентная литература 2). Следовательно, исходя из этих фактов, ожидается, что повышение cGMP в головном мозге приведет к улучшению протекания обучения и развитию памяти.
[0004] cGMP метаболизируется до 5′-GMP без PKG-активирующего действия фосфодиэстеразой (далее по тексту называемой PDE). PDE, как известно, имеет 11 семейств, и известно, что PDE9 специфично метаболизирует cGMP и должен экспрессироваться в головном мозге, селезенке, тонком кишечнике и т.п. (например, см. непатентную литературу 3). То есть, ожидается, что ингибирование PDE9 повысит cGMP в головном мозге. Согласно опубликованным данным, ингибитор PDE9 фактически усиливает LTP гиппокампа и улучшает протекание обучения и развитие памяти в тесте по распознанию нового объекта/тесте обучения пассивному избеганию или др. у животных (непатентная литература 4). С клинической точки зрения активность гуанилатциклазы уменьшается, и наблюдают возможность уменьшения уровня cGMP в верхней височной доле у пациентов с болезнью Альцгеймера (непатентная литература 5). Таким образом, есть вероятность, что PDE9 имеет множество тесных взаимосвязей с патологиями нейродегенеративных заболеваний и психиатрических заболеваний, в частности, с патологиями когнитивных дисфункций и др. при болезни Альцгеймера, таких как болезнь Александера, синдром Альперса, болезнь Альцгеймера, латеральный амиотрофический склероз (ALS; известный как болезнь Лу Герига или болезнь двигательного нейрона), атаксия-телеангиэктазия, болезнь Баттена (также известная как болезнь Шпильмайера-Фогта-Баттена), деменция Бинсвангера (субкортикальная ангиосклеротическая энцефалопатия), биполярное расстройство, губчатая энцефалопатия крупного рогатого скота (BSE), болезнь Канавана, индуцированная химиотерапией деменция, синдром Коккейна, кортико-базальная дегенерация, болезнь Крейтцфельда-Якоба, депрессия, синдром Дауна, дегенерация лобно-височной доли (в том числе лобно-височная деменция, семантическая деменция и прогрессирующая экспрессивная афазия), болезнь Герстманна-Штреусслера-Шейнкера, глаукома, болезнь (хорея) Хантингтона, связанная с ВИЧ деменция, гиперкинез, болезнь Кеннеди, корсаковский синдром (конфабуляторный амнестический синдром), болезнь Краббе, деменция с тельцами Леви, прогрессирующая логопеническая афазия, болезнь Мачадо-Джозефа (спинально-церебеллярная атаксия 3 типа), рассеянный склероз, мультисистемная атрофия (оливомостомозжечковая атрофия), тяжелая миастения, болезнь Паркинсона, синдром Пелицеуса-Мерцбахера, синдром Пика, старческий склероз мозга (легкое когнитивное нарушение), первичный латеральный склероз, первичная прогрессирующая афазия, вызванная облучением деменция, синдром Рефсума (заболевание, вызванное накоплением фитановой кислоты), болезнь Сандгоффа, болезнь Шильдера, шизофрения, семантическая деменция, сенильная деменция, синдром Шая-Дрейджера, спинально-церебеллярная атаксия, атрофия остистой мышцы, болезнь Стила-Ричардсона-Ольшевского (прогрессирующий надъядерный паралич), и сосудистый амилоидоз, и сосудистая деменция (мультиинфарктная деменция).
[0005] Недавно стало известно следующее соединение, которое обладает PDE9-ингибирующей активностью и предназначено для предупреждения или терапии болезни Альцгеймера (патентная литература 1).
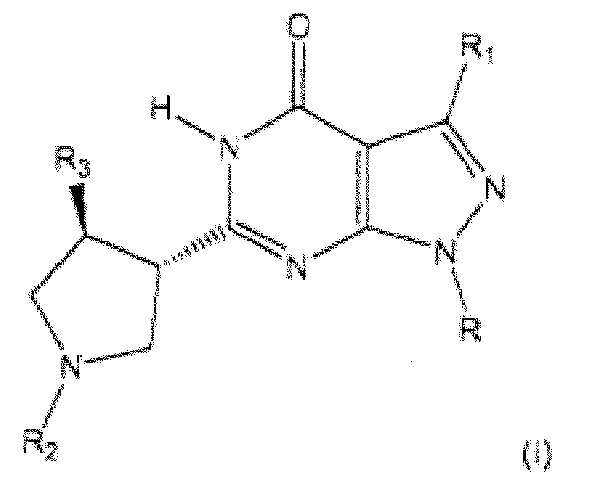
[0006] Приведенное выше соединение представляет собой пиразолопиримидиновое производное, и при этом соединение имеет структуру, абсолютно отличную от пиразолохинолинового каркаса.
[0007] С другой стороны, в качестве соединения с пиразолохинолиновым каркасом известно следующее описанное в патентной литературе 2 соединение:
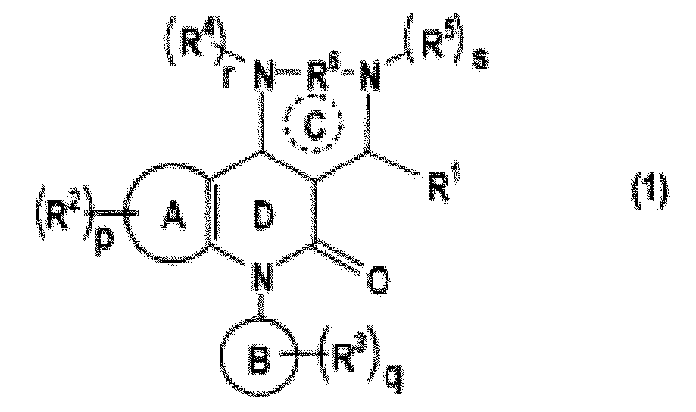 ,
,
где кольцо A является бензольным кольцом или т.п.; а R6 является прямой связью или т.п.
Тем не менее, кольцо B в вышеприведенном соединении означает бензольное кольцо или т.п. Хотя сообщают, что вышеупомянутое соединение обладает ингибирующей активностью в отношении PDE4 и его используют при различных типах воспалительных заболеваний, нет ни описания, ни косвенного указания на ингибирующую активность в отношении PDE9 и т.п.
[0008] В качестве соединений, обладающих PDE9-ингибирующей активностью, известны следующие описанные в патентной литературе 3 и патентной литературе 4 соединения.
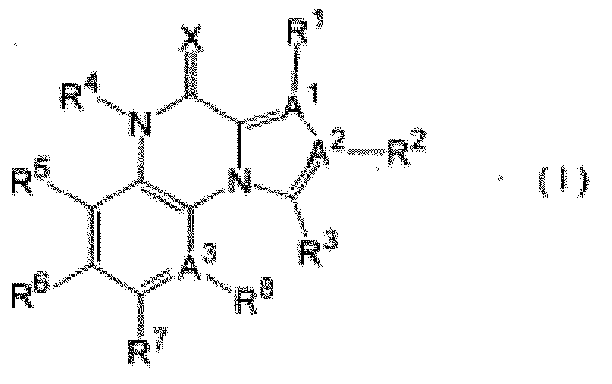
[0009] Любое из вышеупомянутых соединений является хиноксалиновым производным и является соединением со структурой, абсолютно отличной от пиразолохинолинового каркаса.
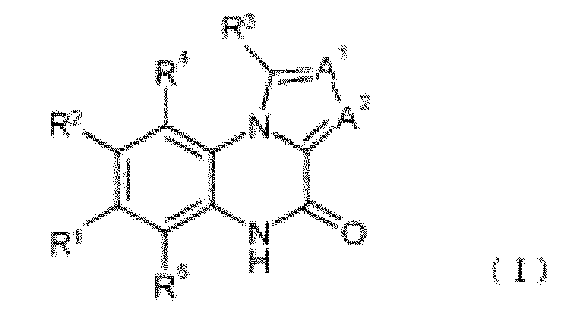
[0010] В качестве соединения с пиразолохинолиновым каркасом и PDE9-ингибирующей активностью известно следующее описанное в патентной литературе 5 соединение:
 ,
,
где либо R1, либо R2 является группой, представленной формулой
[0011] Структура приведенного выше соединения ограничена по R1 и R2, таким образом, соединение является соединением со структурой, абсолютно отличной от соединения по настоящему изобретению.
Список противопоставленных материалов
Патентная литература
[0012]
[патентная литература 1] WO 2008/139293
[патентная литература 2] WO 2007/032466
[патентная литература 3] WO 2008/072779
[патентная литература 4] WO 2010/101230
[патентная литература 5] WO 2012/033144
Непатентная литература
[0013]
[Непатентная литература 1] Domek-Lopacinska et al., "Cyclic GMP metabolism and its role in brain physiology", J Physiol Pharmacol., vol. 56, Suppl 2: pp. 15-34, 2005;
[Непатентная литература 2] Wang X., "Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system", J. Neurocem., vol. 68, pp. 443-456, 1997;
[Непатентная литература 3] Fisher et al., "Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase", J. Biol. Chem., vol. 273: pp. 15559-15564, 1998;
[Непатентная литература 4] van der Staay et al., "The novel selective PDE9 inhibitor BAY 73-6691 improves learning and memory in rodents", Neuropharmacology, vol. 55: pp. 908-918, 2008;
[Непатентная литература 5] Bonkale et al., "Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer′s disease", Neurosci. Lett., vol 187, pp. 5-8, 1995.
Краткое описание изобретения
Техническая задача
[0014] Целью настоящего изобретения является получение нового соединения, или его фармакологически приемлемой соли, которое обладает PDE9-ингибирующим действием, и содержащей его фармацевтической композиции.
Решение задачи
[0015] В результате всесторонних исследований с целью решения вышеупомянутых задач было обнаружено новое пиразолохинолиновое производное, или его фармакологически приемлемая соль, которое обладает PDE9-ингибирующим действием.
[0016] То есть, настоящее изобретение относится к следующим <1> - <20>.
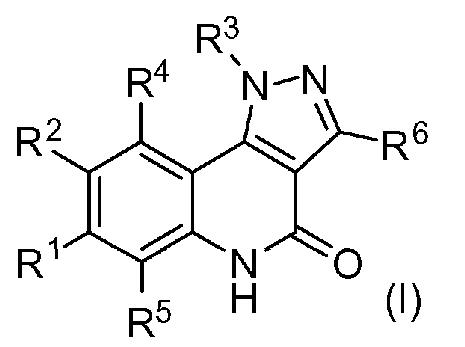
<1> Соединение, или его фармакологически приемлемая соль, представленное формулой (I):
 ,
,
где
R1 является атомом водорода;
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, пиридинильной группы и пиримидинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A1, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B1;
R3 является атомом водорода или атомом фтора;
R4 является атомом водорода;
R5 является оксепанильной группой, диоксепанильной группой, тетрагидропиранильной группой или тетрагидрофуранильной группой, необязательно замещенной метоксигруппой;
R6 является атомом водорода;
группа A1 состоит из атома галогена, C1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, и C1-6-алкоксигруппы; и
группа B1 состоит из атома галогена, цианогруппы, C1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, C1-6-алкокси-C1-6-алкильной группы, C1-6-алкоксигруппы, необязательно замещенной 1-3 атомами галогена, и тетрагидропиранильной группы,
при условии, что если R2 является 3-пиридинильной группой, то заместитель в 4-м положении является атомом галогена или C1-6-алкильной группой, необязательно замещенной 1-3 атомами галогена.
<2> Соединение или его фармакологически приемлемая соль по <1>, где
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы, 4-пиридинильной группы и 5-пиримидинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A2, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B2;
R5 является 4-оксепанильной группой, 1,4-диоксепан-6-ильной группой, 3,4,5,6-тетрагидро-2H-3-пиранильной группой, 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой;
группа A2 состоит из атома хлора и метильной группы, необязательно замещенной 1-2 атомами фтора, этильной группы, метоксигруппы и этоксигруппы; и
группа B2 состоит из атома фтора, атома хлора, цианогруппы, метильной группы, необязательно замещенной 1-3 атомами фтора, этильной группы, метоксиметильной группы, метоксигруппы, необязательно замещенной 1-3 атомами фтора, этоксигруппы, изопропилоксигруппы и 3,4,5,6-тетрагидро-2H-4-пиранильной группы.
<3> Соединение или его фармакологически приемлемая соль по <2>, где R3 является атомом фтора.
<3.1> Соединение или его фармакологически приемлемая соль по <3>, где R5 является 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой.
<4> Соединение или его фармакологически приемлемая соль по <1>, где
R3 является атомом водорода; и
R5 является тетрагидропиранильной группой или тетрагидрофуранильной группой, необязательно замещенной метоксигруппой.
<5> Соединение или его фармакологически приемлемая соль по <2>, где
R3 является атомом водорода; и
R5 является 3,4,5,6-тетрагидро-2H-3-пиранильной группой, 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой.
<6> Соединение или его фармакологически приемлемая соль по <1>, где
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы и 4-пиридинильной группы, где каждый из двух атомов в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A3, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B3;
R3 является атомом водорода;
R4 является атомом водорода;
R5 является 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой;
группа A3 состоит из метильной группы и метоксигруппы; и
группа B3 состоит из метильной группы, метоксигруппы и метоксиметильной группы.
<7> Соединение, или его фармакологически приемлемая соль, выбранное из следующей группы:
1) 7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
2) 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
3) (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
4) 8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
5) 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
6) 1-(1,4-диоксепан-6-ил)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
7) (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
8) 7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
9) (-)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
10) (-)-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
11) (S)-8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
12) (S)-7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он,
13) (S)-8-фтор-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он и
14) (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он.
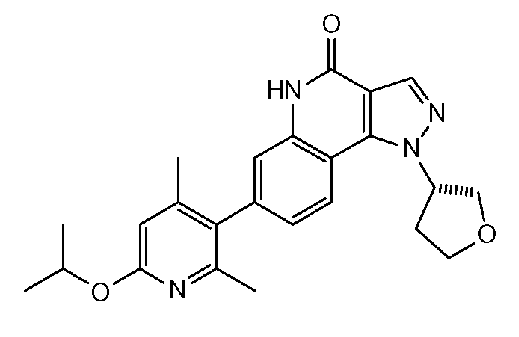
<8> 7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<9> (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль
<10> 8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<11> (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
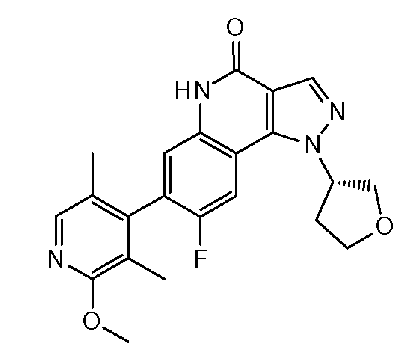
<12> 7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<13> (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
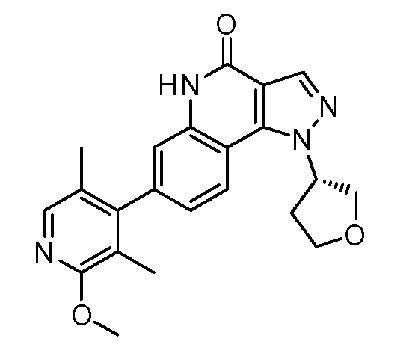
<14> 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
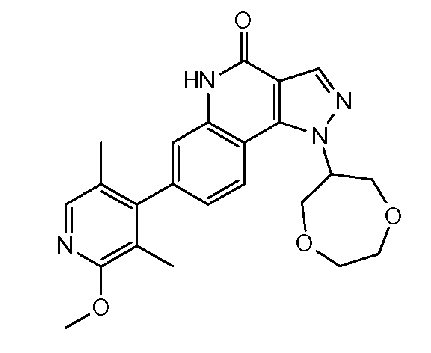
<14.1> 8-фтор-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<14.2> (S)-8-фтор-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
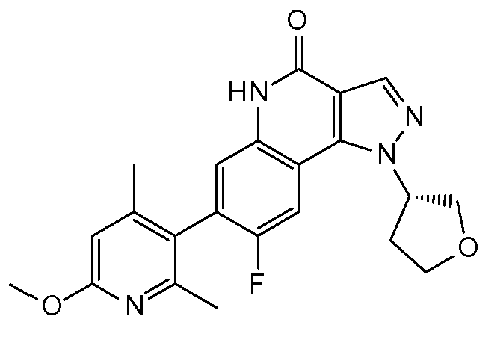
<14.3> 8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<14.4> (S)-8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
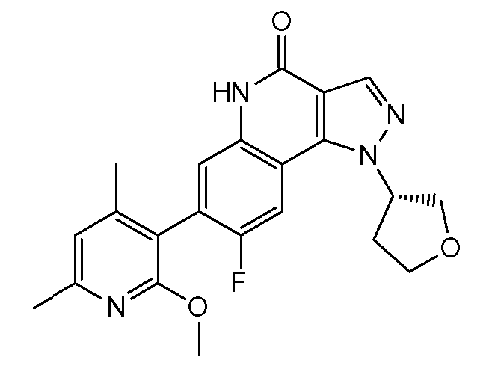
<14.5> 7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль.
<14.6> (S)-7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он или его фармакологически приемлемая соль:
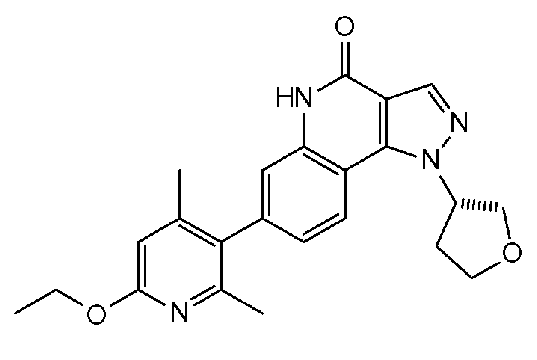
<15> Фармацевтическая композиция, содержащая соединение или его фармакологически приемлемую соль по <1> в качестве активного ингредиента.
<16> Фармацевтическая композиция по <15>, которая является ингибитором PDE9.
<17> Фармацевтическая композиция по <15> для повышения концентрации внутримозгового cGMP.
<18> Средство для облегчения когнитивного нарушения при болезни Альцгеймера, содержащее соединение или его фармакологически приемлемую соль по <1>.
<19> Способ облегчения когнитивного нарушения при болезни Альцгеймера, включающий введение пациенту соединения или его фармакологически приемлемой соли по <1>.
<20> Соединение или его фармакологически приемлемая соль по <1> для применения для облегчения когнитивного нарушения при болезни Альцгеймера.
Полезные эффекты настоящего изобретения
[0017] Пиразолохинолиновое производное (далее по тексту называемое соединение (I)), представленное формулой (I), или его фармакологически приемлемая соль по настоящему изобретению обладают PDE9-ингибирующим действием, как показано в данных по активности в описанном далее примере фармакологического теста. Соединение (I) по настоящему изобретению в основном характеризуется значением IC50, составляющим 1000 нМ или ниже, в качестве PDE9-ингибирующего действия, и предпочтительным является соединение, характеризующееся значением IC50, составляющим 100 нМ или ниже.
[0018] Соединение (I) по настоящему изобретению обладает PDE9-ингибирующим действием, поэтому ожидают повышение концентрации внутримозгового cGMP. PDE9-ингибирующее действие и повышение cGMP приводит к улучшению протекания обучения и развития памяти, и соединение (I) характеризуется возможным применением в качестве терапевтического средства от когнитивных дисфункций и т.п. при болезни Альцгеймера.
Краткое описание графических материалов
[0019] Фиг.1 представляет собой изображение, на котором показана трехмерная структура, полученная в результате рентгеноструктурного анализа соединения, полученного в примере получения 53.
Описание вариантов осуществления
[0020] Далее будет детально описано содержание настоящего изобретения.
[0021] По всему настоящему описанию структурные формулы для соединений для удобства будут демонстрировать только один конкретный изомер, но настоящее изобретение включает все изомеры, такие как геометрические изомеры, оптические изомеры, стереоизомеры и таутомеры, предполагаемые структуры соединения, а также смеси их изомеров, и, таким образом, соединения могут состоять из изомеров и их смесей в любой необходимой пропорции, не ограничиваясь формулами, которые показаны для удобства. Таким образом, например, соединения по настоящему изобретению могут существовать в виде оптически активных форм или рацемических смесей, все из которых включены без ограничения согласно настоящему изобретению, и независимо от того, рацемические ли смеси или оптически активные формы, их можно применять в качестве смесей с оптически активными формами в любой необходимой пропорции. Тем не менее, будет понятно, что некоторые изомеры, или рацематы, или другие смеси изомеров могут проявлять большую активность, чем другие.
[0022] Также могут существовать полиморфные кристаллы, и можно использовать любую кристаллическую форму или их смесь без каких-либо ограничений, а также аморфные формы, и соединения по настоящему изобретению также включают как ангидрат, так и сольват (особенно гидрат).
[0023] Также в настоящее изобретение включены соединения меченного изотопом соединения (I). Меченное изотопом соединение является таким же, как и соединение (I), за исключением того, что один или несколько атомов заменены атомами с атомными массами или массовыми числами, отличными от обычно встречаемых в природе. Изотопы, которые можно включить в соединение по настоящему изобретению, являются изотопами, например, водорода, углерода, азота, кислорода, фтора, фосфора, серы, иода и хлора, и включают 2H, 3H, 11C, 14C, 15N, 18O, 18F, 32P, 35S, 123I и 125I.
[0024] Вышеупомянутые меченные изотопом соединения, например, соединения, в которых включены такие радиоизотопы как 3H и/или 14C, пригодны для анализа распределения в ткани лекарственных препаратов и/или субстратов. Полагают, что 3H и 14C пригодны для облегчения их получения и выявления. Изотопы 11C и 18F считаются пригодными для PET (позитронно-эмиссионной томографии); а изотопы 125I считаются пригодными для SPECT (однофотонной эмиссионной компьютерной томографии); и все они пригодны для получения изображения головного мозга. Замена на более тяжелый изотоп, такой как 2H, послужит причиной для некоторого типа терапевтических преимуществ, включая повышение периода полувыведения in vivo или понижение необходимой дозы вследствие более высокой метаболической стабильности, и, следовательно, считается пригодной в определенной ситуации. Вышеупомянутые меченные изотопами соединения можно аналогично получить путем осуществления процедур, раскрытых в приведенных далее примерах с применением меченных изотопами реагентов, которые можно легко использовать вместо реагентов, которые не мечены изотопом.
[0025] Далее по тексту будут описаны значения терминов, символов и т.п., описываемых в настоящем описании, и будет детально описано настоящее изобретение.
[0026] "Атом галогена" в настоящем описании означает атом фтора, атом хлора, атом брома или атом иода. Подходящие примеры "атома галогена" включают атом фтора и атом хлора.
[0027] "C1-6-алкильная группа" в настоящем описании означает алкильную группу с прямой или разветвленной цепью с 1-6 атомами углерода, и конкретные примеры включают метильную группу, этильную группу, 1-пропильную группу, изопропильную группу, 2-метил-1-пропильную группу, 2-метил-2-пропильную группу, 1-бутильную группу, 2-бутильную группу, 1-пентильную группу, 2-пентильную группу, 3-пентильную группу, 1-гексильную группу, 2-гексильную группу и 3-гексильную группу.
[0028] "C1-6-алкоксигруппа" в настоящем описании означает атом кислорода, к которому прикреплена определенная выше "C1-6-алкильная группа", и конкретные примеры включают метоксигруппу, этоксигруппу, изопропилоксигруппу, 1-пентилоксигруппу и 1-гексилоксигруппу.
[0029] "C1-6-алкокси-C1-6-алкильная группа" в настоящем описании означает определенную выше "C1-6-алкильную группу", к которой прикреплена определенная выше "C1-6-алкоксигруппа", и конкретные примеры включают метоксиметильную группу, 1-метоксиэтильную группу, 2-метоксиэтильную группу, 1-метоксипропильную группу, 2-метоксипропильную группу, 3-метоксипропильную группу, 2-метокси-2-пропильную группу, (1-пропилокси)метильную группу, (изопропилокси)метильную группу, 1-(1-пропилокси)этильную группу, 2-(1-пропилокси)этильную группу, 1-(изопропилокси)этильную группу, 2-(изопропилокси)этильную группу, 1-(1-пропилокси)пропильную группу, 2-(1-пропилокси)пропильную группу, 3-(1-пропилокси)пропильную группу, 2-(1-пропилокси)-2-пропильную группу, 1-(изопропилокси)пропильную группу, 2-(изопропилокси)пропильную группу, 3-(изопропилокси)пропильную группу и 2-(изопропилокси)-2-пропильную группу.
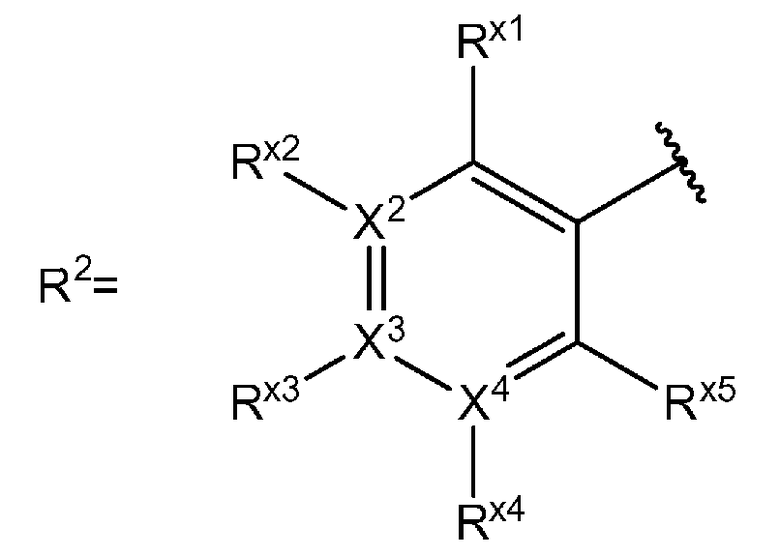
[0030] В определении R2 "ароматическая кольцевая группа, выбранная из группы, состоящей из фенильной группы, пиридинильной группы и пиримидинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A1, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B1" означает:
 ,
,
где
X2 - X4 является атомом углерода или атомом азота, образующим фенильную группу, пиридинильную группу или пиримидинильную группу;
если Xn (n = 2-4) является атомом азота, Rxn отсутствует; и если Xn (n=2-4) является атомом углерода, Rxn является атомом водорода или заместителем, выбранным из группы B1, и Rx1 и Rx5 независимо являются заместителем, выбранным из группы A1.
[0031] Далее по тексту будут описаны определения R1 - R6 соединения, представленного формулой (I), и предпочтительные примеры.
R1 является атомом водорода.
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, пиридинильной группы и пиримидинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A1, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B1.
R2 предпочтительно является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы, 4-пиридинильной группы и 5-пиримидинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A2, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B2;
R2 предпочтительнее является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы и 4-пиридинильной группы, где каждый из двух атомом в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A3, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы B3.
R3 является атомом водорода или атомом фтора.
R4 является атомом водорода.
R5 является оксепанильной группой, диоксепанильной группой, тетрагидропиранильной группой или тетрагидрофуранильной группой необязательно замещенной метоксигруппой.
R5 предпочтительно является 4-оксепанильной группой, 1,4-диоксепан-6-ильной группой, 3,4,5,6-тетрагидро-2H-3-пиранильной группой, 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой и предпочтительнее является 3,4,5,6-тетрагидро-2H-4-пиранильной группой или 3-тетрагидрофуранильной группой.
R6 является атомом водорода.
Группа A1 состоит из атома галогена, C1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, и C1-6-алкоксигруппы.
Группа B1 состоит из атома галогена, цианогруппы, C1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, C1-6-алкокси-C1-6-алкильной группы, C1-6-алкоксигруппы, необязательно замещенной 1-3 атомами галогена, и тетрагидропиранильной группы.
Группа A2 состоит из атома хлора и метильной группы, необязательно замещенной 1-2 атомами фтора, этильной группы, метоксигруппы и этоксигруппы.
Группа B2 состоит из атома фтора, атома хлора, цианогруппы, метильной группы, необязательно замещенной 1-3 атомами фтора, этильной группы, метоксиметильной группы, метоксигруппы, необязательно замещенной 1-3 атомами фтора, этоксигруппы, изопропилоксигруппы и 3,4,5,6-тетрагидро-2H-4-пиранильной группы.
Группа A3 состоит из метильной группы и метоксигруппы.
Группа B3 состоит из метильной группы, метоксигруппы и метоксиметильной группы.
[0032] "Фармакологически приемлемая соль" в настоящем описании конкретно не ограничена при условии, что соль, образованная с соединением по настоящему изобретению, и конкретные примеры включают соли неорганической кислоты, соли органической кислоты, соли неорганического основания, соли органического основания и соли кислых или основных аминокислот.
[0033] Только если "фармакологически приемлемая соль" в настоящем описании является солью, образованной в подходящем соотношении, за исключением любого конкретного ограничивающего описания, то количество молекул кислоты на одну молекулу соединения в образованной соли, даже если конкретно не ограничено, предпочтительно составляет от приблизительно 0,1 до приблизительно 5 молекул, предпочтительнее от приблизительно 0,5 до приблизительно 2 молекул и еще предпочтительнее приблизительно 0,5, приблизительно 1 или приблизительно 2 молекулы на одну молекулу соединения.
[0034] Предпочтительные примеры солей неорганических кислот включают гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты, и предпочтительные примеры солей органических кислот включают ацетаты, сукцинаты, фумараты, малеаты, тартраты, цитраты, лактаты, стеараты, бензоаты, метансульфонаты, п-толуолсульфонаты и бензолсульфонаты.
[0035] Предпочтительные примеры солей неорганических кислот включают соли щелочных металлов, такие как соли натрия и соли калия, соли щелочноземельных металлов, такие как соли кальция и соли магния, соли алюминия и соли аммония, и предпочтительные примеры солей органических оснований включают соли диэтиламина, соли диэтаноламина, соли меглюмина и соли N,N′-дибензилэталендиамина.
[0036] Предпочтительные примеры кислых солей аминокислот включают аспартаты и глютаматы, и предпочтительные примеры солей основных аминокислот включают соли аргинина, соли лизина и соли орнитина.
[0037] [Общие способы получения]
Соединение по настоящему изобретению можно получить с помощью описанных ниже способов. Тем не менее, способы получения соединения по настоящему изобретению ими не ограничены.
[0038] Соединение (I) по настоящему изобретению можно получить с помощью следующих способов получения A, B, C и D.
[0039] <Способ получения A>
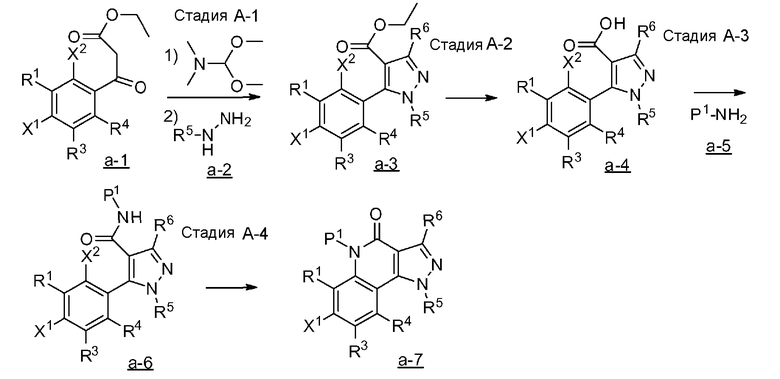
где каждый из R1, R3, R4, R5 и R6 имеет те же определения, что и приведенные выше определения; P1 означает защитную группу группы NH, такую как 2,4-диметоксибензильная группа; и X1 и X2 обозначают атом галогена.
[0040] Стадия A-1
Данная стадия представляет собой стадию реакции конденсации соединения, представленного формулой a-1 (называемого в некоторых случаях соединением a-1; далее по тексту аналогично) с DMF-DMA, а затем посредством хорошо известного способа, предоставляя возможность полученному продукту прореагировать с гидразиновым производным a-2 с образованием структуры пиразольного кольца, таким образом получая соединение a-3. Настоящую реакцию можно осуществлять в потоке газа или в атмосфере инертного газа, такого как азот или аргон.
Соединение a-1 можно синтезировать согласно хорошо известному способу (например, по описанию в Reuman, Michael et al., "Journal of Medicinal Chemistry", 1995, vol. 38, p. 2531-2540, или Wentland Mark P et al., "Journal of Medicinal Chemistry", 1993, vol. 36, p. 1580-1596).
[0041] Данную стадию можно осуществлять точно согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в примерах получения 1, 2, 3, 4, 5, 6, 10 и 11, которые описаны позже, и др.
Поскольку соединение a-2 является коммерчески доступным соединением, то можно использовать его или же его можно синтезировать хорошо известными для специалистов способами. Соединение можно получить путем превращения соответствующего кетонового производного в гидразидимин, и восстановления гидразидимина с помощью боргидрида, цианоборгидрида натрия или др. Соединение a-2 также можно применять в форме соли, такой как гидрохлорид.
[0042] Что касается используемого в настоящей реакции растворителя, то в реакции конденсации соединения a-1 с DMF-DMA, DMF-DMA можно применять в 5-20-кратном молярном эквиваленте в качестве реакционного средства и одновременно растворителя. Растворитель, используемый в последующей реакции образования пиразольного кольца с гидразиновым производным a-2, конкретно не ограничен при условии, что он является растворителем, который до некоторой степени растворяет исходные сырьевые материалы реакции и не подавляет реакцию, но предпочтительно представляет собой метанол, этанол, н-бутанол, трет-бутанол, THF, 1,4-диоксан, воду или смешанный из них растворитель и, предпочтительнее, этанол.
[0043] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции. В реакции конденсации соединения a-1 с DMF-DMA температура реакции предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее является комнатной температурой. В последующей реакции образования пиразольного кольца с гидразиновым производным a-2 температура реакции предпочтительно составляет от комнатной температуры до температуры кипения растворителя (внутренней температуры реакционного сосуда) и предпочтительнее от 70°C до температуры кипения растворителя.
[0044] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции. В реакции конденсации соединения a-1 с DMF-DMA время реакции предпочтительно составляет от 0,5 до 24 часов, и предпочтительнее от 1 до 3 часов, при вышеуказанной температуре после добавления реагентов. В последующей реакции образования пиразольного кольца с гидразиновым производным a-2 время реакции предпочтительно составляет от 0,5 до 24 часов и предпочтительнее от 1 до 8 часов, при вышеуказанной температуре после добавления реагентов.
[0045] Стадия A-2
Данная стадия является стадией гидролиза соединения a-3 в присутствии основания с получением, таким образом, соединения a-4.
[0046] Растворитель, используемый в настоящей реакции, конкретно не ограничен при условии, что он является растворителем, который до некоторой степени растворяет исходные сырьевые материалы и не подавляет реакцию, но предпочтительно включает метанол, этанол, н-бутанол, трет-бутанол, THF, 1,4-диоксан, воду или смешанные из них растворители.
[0047] Основание зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и др., и конкретно не ограничено, но его примеры включают гидроксид натрия, гидроксид лития, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, гидрокарбонат натрия, карбонат калия, карбонат цезия, тетраметилсилилоксид лития (TMSOLi). Основание можно использовать в 1-10-кратном молярном эквиваленте по отношению к a-3.
[0048] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции, и предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее от комнатной температуры до 50°C.
[0049] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции, и предпочтительно составляет от 1 до 48 часов, и предпочтительнее от 2 до 12 часов, при вышеупомянутой температуре после добавления реагентов.
[0050] Стадия A-3
Данная стадия представляет собой стадию предоставления соединению a-4 возможности прореагировать с аминопроизводным a-5 с помощью конденсирующего средства с получением, таким образом, соединения a-6. Настоящую реакцию также можно осуществлять в потоке газа или в атмосфере инертного газа, такого как азот или аргон.
[0051] Данную стадию можно осуществлять точно согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в примере получения 1, 2, 4 и 5, которые описаны позже, и др.
[0052] Конденсирующее средство зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и др. и оно конкретно не ограничено, но можно использовать DCC, EDC, PYBOP, CDI и др. Конденсирующее средство можно использовать в 1-5-кратном молярном эквиваленте, и предпочтительно 1-2-кратном молярном эквиваленте, по отношению к соединению a-4.
[0053] Растворитель, используемый в настоящей реакции, конкретно не ограничен при условии, что он является растворителем, который до некоторой степени растворяет исходные сырьевые материалы и не подавляет реакцию, но предпочтительно включает THF, дихлорметан, DMF или смешанные из них растворители.
[0054] Аминопроизводное a-5 можно использовать в 1-10-кратном молярном эквиваленте, и предпочтительно в 1-2-кратном молярном эквиваленте, по отношению к соединению a-4.
[0055] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции, и предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее от 0°C до комнатной температуры.
[0056] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции. После добавления конденсирующего средства к соединению a-4 реакцию проводят предпочтительно в течение 1-48 часов, и предпочтительнее от 1 до 3 часов, при вышеупомянутой температуре, и после этого добавляют аминопроизводное a-5 и осуществляют реакцию при вышеупомянутой температуре в течение от 1 до 48 часов, и предпочтительнее в течение от 8 до 15 часов.
[0057] Стадия A-4
Данная стадия является стадией внутримолекулярной циклизации соединения a-6 в присутствии основания с получением, таким образом, соединения a-7. Настоящую реакцию также можно осуществлять в потоке газа или в атмосфере инертного газа, такого как азот или аргон.
[0058] Данную стадию можно осуществлять точно согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в примере получения 1, 2, 4 и 5, которые описаны позже, и др.
[0059] Растворитель, используемый в настоящей реакции, конкретно не ограничен при условии, что он является растворителем, который до некоторой степени растворяет исходные сырьевые материалы и не подавляет реакцию, но предпочтительно включает THF, DMF или смешанные из них растворители.
[0060] Основание, в случае применения в реакции, зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и др. и конкретно не ограничено, но его примеры включают такие основания, как гидроксид натрия, KTB, LDA, LHMDS, гидрид натрия и гидрид калия; но предпочтительно представляет собой гидроксид натрия, KTB, гидрид натрия или др. Основание можно использовать в 1-5-кратном молярном эквиваленте, и преимущественно 1-3-кратном молярном эквиваленте, по отношению к соединению a-6.
[0061] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции, и предпочтительно составляет от -78°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее от -20°C до комнатной температуры.
[0062] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и других реагентов и т.п., используемых в реакции, и предпочтительно составляет от 1 до 48 часов, и предпочтительнее от 1 до 5 часов, при вышеупомянутой температуре.
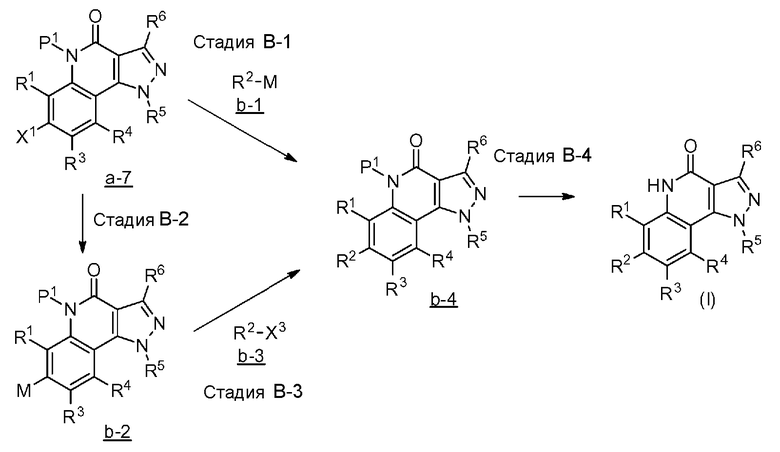
[0063] <Способ получения В>
где каждый из R1, R2, R3, R4, R5, R6 и P1 имеют те же определения, которые приведены выше; X1 и X3 означают атом галогена, и M означает -BF3 -K+, -B(OH)2, группу, представленную формулой:
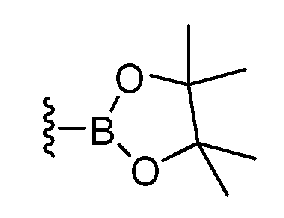 ,
,
-Sn(n-Bu)3, -ZnBr, -ZnCl или т.п.
[0064] Стадия B-1
Данная стадия является стадией проведения с соединением a-7 и соединением b-1 реакции связывания с применением катализатора на основе переходного металла с превращением, таким образом, их в соединение b-4.
[0065] Данную стадию можно осуществлять точно согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в примерах 1, 2 и 3, которые описаны позже, и др.
Соединение a-7 можно получить с помощью <способа получения A> или т.п.
Настоящую реакцию также можно осуществлять в потоке газа или в атмосфере инертного газа, такого как азот или аргон.
[0066] Растворитель, используемый в настоящей реакции, конкретно не ограничен при условии, что он является растворителем, который до некоторой степени растворяет исходные сырьевые материалы и не подавляет реакцию; но их примеры включают спиртовые растворители, такие как метанол или этанол, растворители на основе простых эфиров, такие как THF, DME, MTBE, 1,4-диоксан, простой циклопентилметиловый эфир, простой диэтиловый эфир, простой диизопропиловый эфир, простой дибутиловый эфир и простой дициклопентиловый эфир, растворители на основе ароматических углеводородов, такие как бензол, толуол, ксилол и мезителен, амидные растворители, такие как DMF и NMP, растворители на основе алифатических углеводородов, такие как гептан и гексан, воду или смешанные из них растворители; предпочтительным является растворитель на основе ароматических углеводородов, амидный растворитель, такой как DMF или NMP, растворитель на основе простого эфира, такой как 1,4-диоксан, вода или их смесь, и более предпочтительным является смешанный растворитель из DMF, NMP или 1,4-диоксана и воды.
[0067] Основание зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и др. и конкретно не ограничено, но его примеры включают неорганические основания, такие как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, н-гидрат трикалия фосфата, карбонат цезия, фторид цезия и фторид калия, и органические основания, такие как имидазол, пиридин, TEA и DIPEA; и предпочтительными являются TEA, карбонат цезия и т.п. Также можно добавить гидрофторид калия.
[0068] Катализатор на основе переходного металла зависит от исходных сырьевых материалов, растворителей, которые необходимо использовать, и т.п. и конкретно не ограничен при условии, что он не подавляет реакцию, но предпочтительно включает Pd(PPh3)4, PdCl2(PPh3)2, ацетат/трифенилфосфин палладия (II), ацетат/2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил палладия (II), ацетат/бис[2-(дифенилфосфино)фенил]эфир палладия (II), хлорид палладия (II), Pd2(dba)3/три-трет-бутилфосфин, Pd2(dba)3, Pd(t-Bu3P)2, [(t-Bu)2P(OH)]2PdCl2 и 1,1′-бис(дифенилфосфино)ферроцен дихлорпалладия (II). В зависимости от катализатора на основе переходного металла, который необходимо использовать, использование иодида меди (II), хлорид лития или т.п. в их комбинации в некоторых случаях дает хорошие результаты, такие как повышение выхода и уменьшение времени реакции.
[0069] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей и других реагентов, используемых в реакции, и предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее 60-150°C. Использование микроволнового реактора в некоторых случаях дает хорошие результаты, такие как повышение выхода и уменьшение времени реакции.
[0070] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, других реагентов, используемых в реакции, и температуры реакции и предпочтительно составляет от 1 до 48 часов, и предпочтительнее от 1 до 6 часов, при вышеупомянутой температуре после добавления реагентов.
[0071] Соединение b-1 можно использовать в 1-5-кратном молярном эквиваленте, и предпочтительно в 1-3-кратном молярном эквиваленте, по отношению к соединению a-7.
[0072] Основание можно использовать в 1-10-кратном молярном эквиваленте, и предпочтительно в 2-5-кратном молярном эквиваленте, по отношению к соединению a-7.
[0073] Катализатор на основе переходного металла можно использовать в 0,05-1-кратном молярном эквиваленте, и предпочтительно в 0,05-0,1-кратном молярном эквиваленте, по отношению к соединению a-7.
[0074] Стадия B-2
Данная стадия представляет собой стадию превращения соединения a-7 и бис(пинаколато)дибор или т.п. в соединение b-2 в результате реакции связывания с применением катализатора на основе переходного металла.
[0075] В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах получения 1, 3, 4, 5 и 6 и т.п.
Соединение a-7 можно получить посредством <способа получения A> или т.п.
Данную реакцию можно осуществить в потоке или атмосфере инертного газа, такого как азот или аргон.
[0076] Растворитель, используемый в реакции, особенно не ограничен, если он до определенной степени может растворять исходный материал и не подавляет реакцию. Примеры включают растворители на основе эфиров, такие как THF, DME, MTBE, 1,4-диоксан, простой циклопентилметиловый эфир, простой диэтиловый эфир, простой диизопропиловый эфир, простой дибутиловый эфир и простой дициклопентиловый эфир, растворители на основе ароматических углеводородов, такие как бензол, толуол, ксилол и мезителен, амидные растворители, такие как DMF и NMP, и растворители на основе алифатических углеводородов, таких как гептан и гексан. Предпочтительными являются растворители на основе ароматических углеводородов, амидные растворители, такие как DMF и NMP, или растворители на основе простых эфиров, такие как DME и 1,4-диоксан, или смешанные из них растворители, и более предпочтительными являются DMF, NMP, или 1,4-диоксан, или смешанные из них растворители.
[0077] Основание отличается в зависимости от исходного материала, используемого растворителя и т.п. и особенно не ограничено. Примеры включают неорганические основания, такие как ацетат калия, гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, карбонат цезия, фторид цезия и фторид калия, и органические основания, такие как имидазол, пиридин, TEA и DIPEA. Предпочтительным является ацетат калия или т.п.
[0078] Катализатор на основе переходного металла отличается в зависимости от исходного материала, используемого растворителя и т.п. и особенно не ограничено, если он не подавляет реакцию. Предпочтительные примеры включают Pd(PPh3)4, ацетат/трифенилфосфин палладия (II), ацетат/2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил палладия (II), хлорид палладия (II), Pd2(dba)3/три-трет-бутилфосфин, Pd2(dba)3, Pd(t-Bu3P)2 и 1,1′-бис(дифенилфосфино)ферроцендихлорпалладий (II). Более предпочтительные примеры включают 1,1′-бис(дифенилфосфино)ферроцендихлорпалладий (II).
[0079] Температура реакции обычно отличается в зависимости от исходного материала, растворителя и, помимо прочего, реагента, используемого в реакции, и предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 60 до 150°C. Использование микроволнового реактора в некоторых случаях дает хорошие результаты, такие как повышение выхода и уменьшение времени реакции.
[0080] Время реакции обычно отличается в зависимости от исходного материала, растворителя и, помимо прочего, реагента, используемого в реакции, и температуры реакции, и предпочтительно составляет от 1 до 48 часов, более предпочтительно от 1 до 6 часов, при вышеупомянутой температуре после добавления реагента.
[0081] Бис(пинаколато)дибор можно использовать в количестве от 1 до 5 молярных эквивалентов из расчета по соединению a-7. Количество предпочтительно составляет от 1 до 3 молярных эквивалентов.
[0082] Основание можно использовать в количестве от 1 до 10 молярных эквивалентов из расчета по соединению a-7. Количество предпочтительно составляет от 2 до 5 молярных эквивалентов.
[0083] Катализатор на основании переходного металла можно использовать в количестве от 0,05 до 1 молярных эквивалентов из расчета по соединению a-7. Количество предпочтительно составляет от 0,05 до 0,1 молярного эквивалента.
[0084] Стадия B-3
Данная стадия представляет собой стадию превращения соединения b-3 и соединения b-2 в соединение b-4 в результате реакции связывания с применением катализатора на основе переходного металла.
[0085] Данную стадию можно осуществить при тех же условиях, что и на стадии B-1. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 4, 6 и 25 и т.п.
[0086] Стадия B-4
Данная стадия представляет собой стадию удаления защитной группы P1 соединения b-4 с получением, таким образом, соединения (I). Снятие защитной группы описано во многих хорошо известных литературных источниках, например, T. Greene et al., "Protective Groups in Organic Synthesis" (John Wiley & sons. Inc., New York, 1999) (далее по тексту называемый справочная литература по синтезу 1). Реакция по снятию защитной группы с аминогруппы зависит от типа защитной группы и конкретно не ограничена, но, например, в случае 2,4-диметоксибензильной группы или т.п., снятие защитной группы можно осуществить в кислых условиях.
[0087] В случае, если защитной группой P1 является 2,4-диметоксибензильная группа, то растворитель, используемый в настоящей реакции, может быть любым при условии, что он до некоторой степени растворяет исходные сырьевые материалы и не подавляет реакцию. Растворитель конкретно не ограничен, но его примеры включают спиртовые растворители, такие как метанол и этанол, растворители на основе простых эфиров, такие как THF, DME, MTBE, простой циклопентилметиловый эфир, простой диэтиловый эфир, простой диизопропиловый эфир, простой дибутиловый эфир и простой дициклопентиловый эфир, растворители на основе галогензамещенных углеводородов, таких как дихлорметан и хлороформ, уксусную кислоту или смешанные из них растворители. В качестве растворителя можно использовать кислоту.
[0088] В качестве кислоты можно использовать, например, трифторуксусную кислоту (TFA) и серную кислоту. Предпочтительной является TFA. Кислоту можно использовать в 1-100-кратном объеме по отношению к соединению b-4.
[0089] Температура реакции обычно зависит от исходных сырьевых материалов, растворителей и других реагентов, используемых в реакции, и предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда) и предпочтительнее 40-60°C.
[0090] Время реакции обычно зависит от исходных сырьевых материалов, растворителей, других реагентов, используемых в реакции, и температуры реакции и предпочтительно составляет от 0,5 до 24 часов, и предпочтительнее от 1 до 12 часов, при вышеупомянутой температуре после добавления реагентов.
[0091] <Способ получения C>
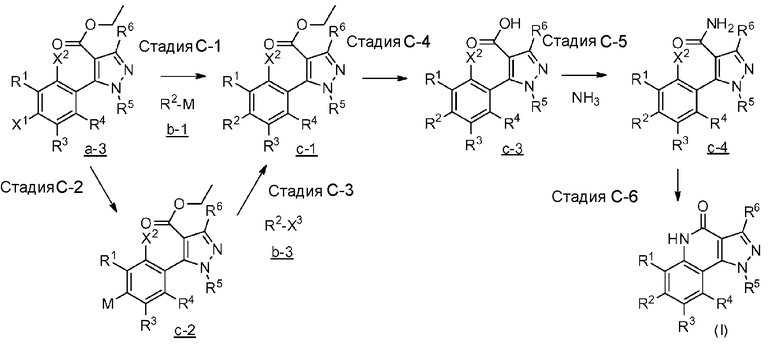
В формулах R1, R2, R3, R4, R5, R6 и M являются такими, как, соответственно, определено выше, и каждый из X1, X2 и X3 представляет собой атом галогена.
[0092] Стадия C-1
Данная стадия представляет собой стадию превращения соединения b-1 и соединения а-3 в соединение с-1 в результате реакции связывания с применением катализатора на основе переходного металла.
[0093] Данную стадию можно осуществить при тех же условиях, что и на стадии B-1 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере 52 и т.п.
[0094] Стадия C-2
Данная стадия представляет собой стадию превращения соединения a-3 и бис(пинаколато)дибор или т.п. в соединение с-2 в результате реакции связывания с применением катализатора на основе переходного металла.
[0095] Данную стадию можно осуществить при тех же условиях, что и на стадии B-2 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах получения 3 и 6 и т.п.
[0096] Стадия C-3
Данная стадия представляет собой стадию превращения соединения b-3 и соединения с-2 в соединение с-1 в результате реакции связывания с применением катализатора на основе переходного металла.
[0097] Данную стадию можно осуществить при тех же условиях, что и на стадии B-3 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере 26 и т.п.
[0098] Стадия C-4
Данная стадия представляет собой стадию получения соединения c-3 в результате гидролиза соединения c-1 в присутствии основания.
[0099] Данную стадию можно осуществить при тех же условиях, что и на стадии А-2 <способа получения А>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере 26 и т.п.
[0100] Стадия C-5
Данная стадия представляет собой стадию получения соединения c-4 в результате осуществления реакции соединения c-3 с водным раствором аммиака с применением конденсирующего средства. Данную реакцию можно осуществить в потоке или атмосфере инертного газа, такого как азот или аргон.
[0101] Данную стадию можно осуществить при тех же условиях, что и на стадии А-3 <способа получения А>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 5, 26, 52, 53, 54 и 55 и т.п.
[0102] Стадия C-6
Данную стадию можно осуществить при тех же условиях, что и на стадии А-4 <способа получения А>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 5, 26, 52, 53, 54 и 55 и т.п.
[0103] <Способ получения D>
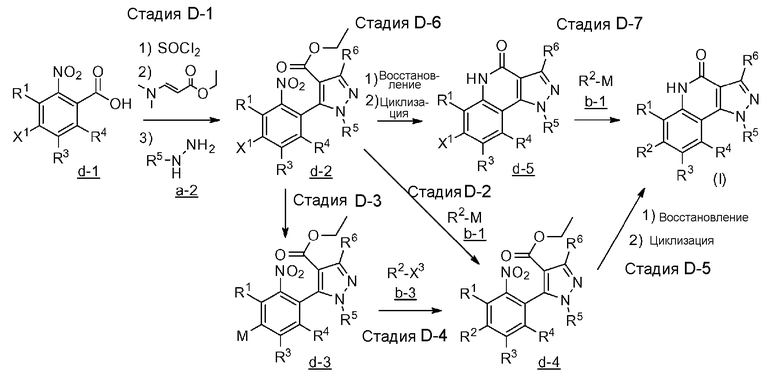
В формулах R1, R2, R3, R4, R5, R6 и M являются такими, как, соответственно, определено выше, и каждый из X1 и X3 представляет собой атом галогена.
[0104] Стадия D-1
Данная стадия представляет собой стадию получения соединения d-2 посредством известного способа проведения реакции соединения d-1 с тионилхлоридом с превращением его в соответствующее хлорангидридное производное, а затем осуществления реакции конденсации с этилдиметиламиноакрилатом и последующего проведения реакции с гидразиновым производным a-2 с образованием пиразольного кольца. Данную реакцию можно осуществить в потоке или атмосфере инертного газа, такого как азот или аргон.
[0105] В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере получения 7 и т.п.
Соединение a-2 может представлять собой коммерчески доступный продукт, используемый как есть, а также его можно синтезировать с помощью известных специалисту в данной области средств. Соединение можно получить путем превращения соответствующего кетонового производного в гидразидимин и восстановления с помощью боргидрида, цианоборгидрида натрия или т.п. Соединение a-2 также можно применять в виде соли, такой как гидрохлорид.
[0106] Растворитель, используемый на стадии проведения реакции соединения d-1 с тионилхлоридом для превращения его в соответствующее хлорангидридное производное, в данной реакции особо не ограничен при условии, что растворитель до некоторой степени может растворять исходный материал реакции и не подавляет реакцию. Растворитель предпочтительно представляет собой THF, ацетонитрил, DMF или DMA, более предпочтительно - ацетонитрил. Растворитель, используемый в следующей реакции конденсации с этилдиметиламиноакрилатом особо не ограничен при условии, что он до некоторой степени растворяет исходный материал реакции и не подавляет реакцию. Растворитель предпочтительно представляет собой THF, ацетонитрил, DMF или DMA, более предпочтительно - ацетонитрил. Растворитель, используемый в последующей реакции образования пиразольного кольца с гидразиновым производным a-2, особо не ограничен при условии, что он до некоторой степени растворяет исходный материал реакции и не подавляет реакцию. Растворитель предпочтительно представляет собой метанол, этанол, н-бутанол, трет-бутанол, THF, 1,4-диоксан, ацетонитрил, воду или смешанный из них растворитель, более предпочтительно - смешанный растворитель из ацетонитрила и воды.
[0107] Температура реакции обычно отличается в зависимости от исходного материала, используемого растворителя и, помимо прочего, реагента, используемого в реакции. Температура реакции на стадии получения соответствующего хлорангидрида из соединения d-1 и тионилхлорида предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 50°C до 80°C. Температура реакции в следующей реакции конденсации с этилдиметиламиноакрилатом предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 20°C до 80°C. Температура реакции в последующей реакции образования пиразольного кольца с гидразиновым производным a-2 предпочтительно составляет от комнатной температуры до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 50°C до температуры кипения растворителя.
[0108] Время реакции обычно отличается в зависимости от исходного материала, используемого растворителя и, помимо прочего, реагента, используемого в реакции. Время реакции на стадии получения соответствующего хлорангидрида в результате реакции соединения d-1 с тионилхлоридом предпочтительно составляет от 0,5 от 24 часов, более предпочтительно от 1 до 3 часов, при вышеупомянутой температуре после добавления реагента. Время реакции в следующей реакции конденсации с этилдиметиламиноакрилатом предпочтительно составляет от 0,5 до 24 часов, более предпочтительно от 1 до 3 часов, при вышеупомянутой температуре после добавления реагента. Время реакции в последующей реакции образования пиразольного кольца с гидразиновым производным a-2 предпочтительно составляет от 0,5 до 60 часов, более предпочтительно от 12 до 24 часов, при вышеупомянутой температуре после добавления реагента.
[0109] Стадия D-2
Данная стадия представляет собой стадию превращения соединения b-1 и соединения d-2 в соединение d-4 в результате реакции связывания с применением катализатора на основе переходного металла.
[0110] Данную стадию можно осуществить при тех же условиях, что и на стадии B-1 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 27 и 43 и т.п.
[0111] Стадия D-3
Данная стадия представляет собой стадию превращения соединения d-2 и бис(пинаколато)дибор или т.п. в соединение d-3 в результате реакции связывания с применением катализатора на основе переходного металла.
[0112] Данную стадию можно осуществить при тех же условиях, что и на стадии B-2 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах получения 7 и 9 и т.п.
[0113] Стадия D-4
Данная стадия представляет собой стадию превращения соединения b-3 и соединения d-3 в соединение d-4 в результате реакции связывания с применением катализатора на основе переходного металла.
[0114] Данную стадию можно осуществить при тех же условиях, что и на стадии B-3 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 45 и 51 и т.п.
[0115] Стадия D-5
Данная стадия представляет собой стадию получения соединения (I) при помощи известного способа путем превращения нитрогруппы соединения d-4 в аминогруппу с применением восстановителя, а затем конденсации аминогруппы со сложным эфиром для осуществления реакции внутримолекулярной циклизации. Данную реакцию можно осуществить в потоке или атмосфере инертного газа, такого как азот или аргон. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемых далее примерах 27, 41, 43, 45, 51 и 62 и т.п.
[0116] Примеры восстановителя на данной стадии включают железо, хлорид олова (II) и гидросульфит натрия. Железо и хлорид олова (II) являются предпочтительными, и более предпочтительным является железо. Реакцию внутримолекулярной циклизации продолжают путем нагревания, в частности, без использования реагента.
[0117] Растворитель, используемый на стадии превращения нитрогруппы соединения d-4 в аминогруппу с помощью восстановителя в данной реакции особо не ограничен при условии, что растворитель до некоторой степени может растворять исходный материал реакции и не подавляет реакцию. Растворителем является метанол, этанол, н-бутанол, трет-бутанол, этилацетат или смешанный из них растворитель, более предпочтительно - метанол или этанол. Растворитель, используемый в последующей реакции внутримолекулярной циклизации, особо не ограничен при условии, что он до некоторой степени растворяет исходный материал реакции и не подавляет реакцию. Растворителем является уксусная кислота, этанол, н-бутанол, трет-бутанол, THF или 1,4-диоксан, предпочтительно уксусная кислота, этанол, н-бутанол или трет-бутанол, более предпочтительно уксусная кислота.
[0118] Температура реакции обычно отличается в зависимости от исходного материала, используемого растворителя и, помимо прочего, реагента, используемого в реакции. Температура реакции на стадии превращения нитрогруппы соединения d-4 в аминогруппу с помощью восстановителя предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 80°C до температуры кипения растворителя (внутренняя температура реакционного сосуда). Температура реакции в последующей реакции внутримолекулярной циклизации предпочтительно составляет от 0°C до температуры кипения растворителя (внутренняя температура реакционного сосуда), более предпочтительно от 50°C до температуры кипения растворителя (внутренняя температура реакционного сосуда).
[0119] Время реакции обычно отличается в зависимости от исходного материала, используемого растворителя и, помимо прочего, реагента, используемого в реакции. Время реакции на стадии превращения нитрогруппы соединения d-4 в аминогруппу с помощью восстановителя предпочтительно составляет от 0,5 до 24 часов, более предпочтительно от 1 до 3 часов, при вышеупомянутой температуре после добавления реагента. Время реакции в последующей реакции внутримолекулярной циклизации предпочтительно составляет от 0,5 до 24 часов, более предпочтительно от 1 до 3 часов, при вышеупомянутой температуре после добавления реагента.
[0120] Стадия D-6
Данная стадия представляет собой стадию получения соединения d-5 при помощи известного способа путем превращения нитрогруппы соединения d-2 в аминогруппу с применением восстановителя, а затем конденсации аминогруппы со сложным эфиром для осуществления реакции внутримолекулярной циклизации.
[0121] Данную стадию можно осуществить при тех же условиях, что и на стадии D-5 <способа получения D>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере 63 и т.п.
[0122] Стадия D-7
Данная стадия представляет собой стадию превращения соединения, представленного соединением b-1, и соединения d-5 в соединение (I) в результате реакции связывания с применением катализатора на основе переходного металла.
[0123] Данную стадию можно осуществить при тех же условиях, что и на стадии B-1 <способа получения B>. В частности, данную стадию можно осуществлять согласно способу, предусматривающему определенные реакционные условия, постреакционные процедуры и очистку и т.п., описанному в описываемом далее примере 63 и т.п.
[0124] После завершения реакции в каждом способе и каждой описанной выше стадии целевое соединение для каждой стадии можно собрать из реакционной смеси согласно традиционному способу.
[0125] Например, в случае, если реакционная смесь полностью представляет собой жидкость, то реакционной смеси, по желанию, давали остыть до комнатной температуры или охлаждают при помощи льда; кислоту, щелочь, окислитель или восстановитель предпочтительно нейтрализуют; добавляют несмешивающийся органический растворитель, по типу воды и этилацетата, и не вступающий в реакцию с целевым соединением; и отделяют слой, содержащий целевое соединение. Затем для промывки слоя, содержащего целевое соединение, добавляют растворитель, не смешивающийся с полученным слоем и не вступающий в реакцию с целевым соединением, и отделяют слой. Кроме того, если слой является органическим слоем, путем сушки слоя с помощью осушителя, такого как безводный сульфат магния или безводный сульфат натрия, и отгонки растворителя можно собрать целевое соединение. Если слой представляет собой водный слой, то путем электрического обессоливания слоя и, таким образом, лиофилизации слоя, можно собрать целевое соединение.
[0126] Если реакционная смесь полностью является жидкостью и по возможности целевое соединение можно собрать путем только отгонки веществ (например, растворителя и реагентов), отличных от целевого соединения при нормальном давлении или уменьшенном давлении.
[0127] Кроме того, в случае, если целевое соединение оседает само по себе в виде твердого вещества, или в случае, если реакционная смесь полностью является жидкостью и только целевое соединение осаждается в виде твердого вещества при осуществлении процедуры сбора, то целевое соединение можно собрать путем сначала сбора на фильтре целевого соединения с помощью способа фильтрации, промывания собранного на фильтре целевого соединения соответствующим органическим или неорганическим растворителем и высушивания целевого соединения, и дополнительно целевое соединение можно собрать путем обработки маточной жидкости аналогично случаю, при котором реакционная смесь полностью является жидкостью.
[0128] Кроме того, в случае, если реагент или катализатор присутствует в виде твердого вещества, или в случае, если реакционная смесь полностью является жидкостью, если реагент или катализатор сам по себе осаждается в виде твердого вещества при осуществлении процедуры сбора, и если целевое соединение растворяется в растворе, то целевое соединение можно получить путем сначала отфильтровывания реагента или катализатора с помощью способа фильтрации, промывания отфильтрованного реагента или катализатора соответствующим органическим или неорганическим растворителем, объединения полученной промытой жидкости с маточной жидкостью и обработки полученной смешанной жидкости аналогично случаю, когда реакционная смесь полностью является жидкостью.
[0129] В частности, в случае, если отличные от целевого соединения вещества, содержащиеся в реакционной смеси, не подавляют реакцию следующей стадии, то реакционную смесь можно использовать в неизменном виде на следующей стадии без отдельного выделения целевого соединения.
[0130] Для повышения чистоты целевого соединения, собранного согласно вышеописанному способу, предпочтительно можно осуществить способ рекристаллизации, различные типа хроматографии и способ перегонки.
[0131] В случае, если собранное целевое соединение является твердым веществом, то чистоту целевого соединения обычно можно повысить с помощью способа рекристаллизации. Согласно способу рекристаллизации можно использовать отдельный растворитель или смешанный растворитель из множества растворителей, который не вступает в реакцию с целевым соединением. В частности, целевое соединение сначала растворяют при комнатной температуре или при нагревании в отдельном растворителе или смешанном растворителе из множества растворителей, который не вступает в реакцию с целевым соединением. Охлаждая смешанную жидкость ледяной водой или т.п. или оставив ее при комнатной температуре, целевое соединение можно кристаллизовать из смешанной жидкости.
[0132] В случае, если собранное целевое соединение является жидкостью, чистоту целевого соединения можно повысить с помощью различных типов хроматографии. В целом можно использовать слабокислые силикагели, такие как силикагель 60 (70-230 меш или 340-400 меш), производимые Merck, или BW-300 (300 меш), производимый Fuji Silysia Chemical Ltd. В случае, если целевое соединение имеет основность и характеризуется слишком интенсивным поглощением вышеупомянутыми силикагелями или в других случаях, то можно использовать покрытый пропиламином силикагель (200-350 меш), производимый Fuji Silysia Chemical Ltd. или т.п. В случае, если целевое соединение обладает биполярностью, в случае, если необходимо элюирование высокополярным растворителем, таким как метанол, или в других случаях можно использовать NAM-200H или NAM-300H, производимые NAM Laboratory. Целевое соединение с повышенной чистотой можно получить путем элюирования целевого соединения отдельным растворителем или множеством растворителей, которые не вступают в реакцию с целевым соединением, с применением данных силикагелей и отгонки растворителя(ей).
[0133] В случае, если собранное целевое соединение является жидкостью, то чистоту целевого соединения можно также повысить с помощью способа перегонки. Согласно способу перегонки можно отогнать целевое соединение путем уменьшения давления у целевого соединения при комнатной температуре или при нагревании.
[0134] Хотя выше приведены типичные примеры способов получения соединения (I) по настоящему изобретению, сырьевые соединения и различные типы реагентов при получении соединения по настоящему изобретению могут образовывать соли, гидраты или сольваты, и любые их соединения и реагенты зависят от исходных сырьевых материалов, растворителей, которые необходимо использовать, и др. и конкретно не ограничены при условии, что они не подавляют реакции. Также, растворитель, который необходимо использовать, зависит от исходных сырьевых материалов, реагентов и др. и, разумеется, конкретно не ограничен при условии, что он не подавляет реакции и до некоторой степени растворяет исходные вещества. В случае если соединение (I) по настоящему изобретению получают в виде свободной массы, то соединение (I) можно превратить в форму соли, которую может принимать соединение (I), или его гидрата с помощью общеизвестного способа.
[0135] В случае, если соединение (I) по настоящему изобретению получают в виде соли соединения (I) или гидрата соединение (I), соль и гидрат можно превратить в свободную массу соединения (I) с помощью общеизвестного способа.
[0136] Различные типы изомеров (например, геометрические изомеры, оптические изомеры, ротационные изомеры, стереоизомеры и таутомеры), получаемые для соединения (I) по настоящему изобретению, можно очистить и выделить с помощью обычных средств разделения, например, рекристаллизации, способа с диастереоизомерными солями, способа ферментативного разрушения и различных типов хроматографии (например, тонкослойная хроматография, колоночная хроматография и газовая хроматография).
[0137] [Фармацевтический препарат] Соединение формулы (I) по настоящему изобретению или его фармацевтически приемлемую соль можно фармацевтически получить с помощью общеизвестного способа, и можно получить лекарственную форму, например, пероральный препарат (таблетка, гранула, порошок, капсула, сироп или др.), инъекционный препарат (для внутривенного введения, для внутримышечного введения, для подкожного введения, для внутрибрюшинного введения и для других целей) и препарат для наружного применения (чрескожный препарат (мазь, пластырь и др.), глазные капли, капли в нос, суппозиторий и др.).
[0138] В случае получения перорального твердого препарата к соединению формулы (I) или его фармацевтически приемлемой соли, при необходимости, добавляют эксципиент, связующее, разрыхлитель, смазывающее вещество, краситель и т.п., и с помощью общеизвестных способов можно получить таблетку, гранулу, порошок и капсулу. При необходимости, таблетку, гранулу, порошок, капсулу и т.п. можно покрыть пленкой.
Примеры эксципиента включают лактозу, кукурузный крахмал и кристаллическую целлюлозу; примеры связующего включают гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу; примеры разрыхлителя включают карбоксиметилцеллюлозу кальция и кроскармеллозу натрия; примеры смазывающего вещества включают стеарат магния и стеарат кальция; примеры красителя включают оксид титана; и примеры пленкообразователя включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, но эти добавки, разумеется, не ограничены данными примерами.
Каждый из этих твердых препаратов, таких как таблетки, капсулы, гранулы и порошки, обычно могут содержать от 0,001 до 99,5% по весу, предпочтительно от 0,01 до 90% по весу и т.п., соединения формулы (I) или его фармацевтически приемлемой соли.
[0139] В случае получения инъекционного препарата (для внутривенного введения, для внутримышечного введения, для подкожного введения, для внутрибрюшинного введения или для других целей) к соединению формулы (I) или его фармацевтически приемлемой соли, при необходимости, добавляют регулятор pH, буферное вещество, суспендирующее средство, солюбилизатор, антиоксидант, консервант (антисептик), изотоническое средство и др., и с помощью общеизвестного способа можно получить инъекционный препарат. Препараты можно лиофилизировать с получением лиофилизированных препаратов по типу растворяемых непосредственно перед приемом.
Примеры регулятора pH и буферного вещества включают органические кислоты или неорганические кислоты и/или их соли; примеры суспендирующего средства включают метилцеллюлозу, полисорбат 80 и карбоксиметилцеллюлозу натрия; примеры солюбилизатора включают полисорбат 80 и полиоксиэтиленсорбитанмонолаурат; примеры антиоксиданта включают α-токоферол; примеры консерванта включают метилпараоксибензоат и этилпараоксибензоат; и примеры изотонического средства включают глюкозу, хлорид натрия и маннит, но эти добавки, разумеется, на ограничены данными примерами.
Каждый из этих инъекционных препаратов обычно может содержать от 0,000001 до 99,5% по весу, предпочтительно от 0,00001 до 90% по весу и т.п., соединения формулы (I) или его фармацевтически приемлемой соли.
[0140] В случае получения препарата для наружного применения основной сырьевой материал добавляют к соединению формулы (I) или его фармацевтически приемлемой соли и, при необходимости, добавляют, например, консервант, стабилизатор, регулятор pH, антиоксидант, краситель и др., и с помощью традиционных способов можно получить, например, чрескожный препарат (мазь, пластырь и др.), глазные капли, капли в нос, суппозиторий и др.
В качестве подлежащих применению основных сырьевых материалов можно использовать различные сырьевые материалы, которые обычно используют, например, для лекарственных препаратов, лечебно-профилактической косметики и косметических средств. Конкретные их примеры включают такие сырьевые материалы, как животные и растительные масла, минеральные масла, сложноэфирные масла, воски, эмульгаторы, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и очищенную воду.
Каждый из этих препаратов для наружного применения обычно может содержать от 0,000001 до 99,5% по весу, предпочтительно от 0,00001 до 90% по весу и т.п., соединения формулы (I) или его фармацевтически приемлемой соли.
[0141] Соединение по настоящему изобретению можно превратить в химический зонд для захвата целевого белка из физиологически активного низкомолекулярного соединения. То есть, соединение по настоящему изобретению можно преобразовать в зонд для аффинной хроматографии, фотоаффинный зонд и т.п. путем введения группы-метки, линкера или т.п. во фрагмент, отличный от структурного фрагмента, необходимого для обнаружения активности соединения, с помощью методики, описанной в J. Mass Spectrum. Soc. Jpn., Vol. 51, No. 5, 2003, p. 492-498, WO2007/139149, или др.
[0142] Примеры группы-метки, линкера или т.п., используемого в химическом зонде, включают группы, показанные в группе, состоящей из приведенных далее (1)-(5):
(1) группы-метки для белков, такие как фотоаффинные группы-метки (например, бензоильная группа, бензофеноновая группа, азидогруппа, карбонильная азидогруппа, диазиридиновая группа, еноновая группа, диазогруппа и нитрогруппа) и хемоаффинные группы (например, кетонные группы, в который атом альфа-углерода заменен атомом галогена, карбамоильная группа, сложноэфирная группа, алкилтиогруппа, α,β-ненасыщенный кетон, акцептор Михаэля для сложного спирта и др. и оксирановая группа);
(2) расщепляемые линкеры, такие как -S-S-, -O-Si-O-, моносахариды (глюкозная группа, галактозная группа и др.), и дисахариды (лактоза и др.), и олигопептидные линкеры, расщепляемые посредством ферментативной реакции;
(3) группы с биотиновой и меткой для фишинга, такие как 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индацен-3-ил)пропионильная группа;
(4) меченные радиоактивным изотопом группы 125I, 32P, 3H, 14C или т.п.; флуоресцентные группы-метки, такие как флуоресцеин, родамин, дансил, умбеллиферон, 7-нитрофуразанил и 3-(4,4-дифтор-5,7-диметил-4H-3a,4a-диаза-4-бора-s-индецен-3-ил)пропионильная группа; хемилюминесцентные группы, такие как люциферин и люминол; и маркеры, способные обнаруживать ионы тяжелых металлов, такие как ионы металлов-лантаноидов и ионы радия; и
(5) группы, связанные с твердофазными носителями, такими как стеклянные шарики, стеклянные пластины, титровальные микропланшеты, агарозные гранулы, агарозные слои, полистирольные гранулы, полистирольные слои, полиамидные гранулы и полиамидные слои.
Зонды, полученные путем введения групп-меток, выбранных из группы, состоящей из (1)-(5) выше или т.п., в соединения согласно способам, описанным в вышеупомянутых документах, или т.п., можно применять в качестве химических зондов для определения меченых белков, что является применимым, например, для поиска и др. новых мишеней для разработки лекарственного средства.
Примеры
[0143] Соединение (I) по настоящему изобретению можно получить, например, с помощью способов, описанных в приведенных далее примерах, и эффекты соединения можно подтверждать с помощью способов, описанных в приведенных далее в тестовых примерах. Однако эти способы являются лишь иллюстративными, и настоящее изобретение так или иначе не ограничено приведенными далее конкретными примерами, и можно осуществить изменения и модификации, не выходя за объем настоящего изобретения.
[0144] Следует отметить, что соединения, для которых описаны названия в литературе или др., получали согласно приведенным в литературе сведениям или т.п.
[0145] Сокращения, используемые в настоящем описании, хорошо известны специалистам в данном области техники. В настоящем описании будут использованы следующие сокращения.
Ac: ацетил
BAST: бис(2-метоксиэтил)аминосеры трифторид
Bn: бензил
Boc: трет-бутоксикарбонил
BOP: бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат
Bu: бутил
CAN: церий-аммоний нитрат
CDI: 1,1′-карбонилдиимидазол
DAST: диэтиламиносеры трифторид
DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен
DCC: 1,3-дициклогексилкарбодиимид
DCM: дихлорметан
DDQ: 2,3-дихлор-5,6-дициано-1,4-бензохинон
DEAD: диэтилазодикарбоксилат
DIAD: диизопропилазодикарбоксилат
DIBAL-H: диизобутилалюминия гидрид
DIPEA: N,N-диизопропилэтиламин
DMAP: 4-(диметиламино)пиридин
DME: 1,2-диметоксиэтан
DMF: N,N-диметилформамид
DMF-DMA: N,N-диметилформамида диметилацеталь
ДМСО: диметилсульфоксид
DTT: дитиотреитол
EDC: 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид
EGTA: гликолевый эфир диаминтетрауксусной кислоты
HATU: O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония гексафторфосфат
HBTU: O-бензотриазол-N,N,N′,N′-тетраметилурония гексафторфосфат
HOBT: 1-гидроксибензотриазол
IPA: изопропиловый спирт
KHMDS: бис(триметилсилил)амид калия
KTB: трет-бутоксид калия
LAH: гидрид лития-алюминия
LDA: диизопропиламид лития
LHMDS: бис(триметилсилил)амид лития
mCPBA: 3-хлорпербензойная кислота
m-: мета
MTBE: простой трет-бутилметиловый эфир
н-: нормальный
NaBH(OAc)3: триацетоксиборгидрид натрия
NaHMDS: бис(триметилсилил)амид натрия
NBS: N-бромсукцинимид
NCS: N-хлорсукцинимид
NIS: N-иодсукцинимид
NMP: N-метил-2-пирролизинон
o-: орто
p-: пара
Pd(t-Bu3P)2: бис(три-трет-бутилфосфин)палладий
Pd2(dba)3: трис(дибензилиденацетон)дипалладий
Комплекс Pd(dppf)Cl2 DCM : комплекс [1,1′-бис(дифенилфосфин)ферроцен]дихлорпалладия(II) DCM
Pd(PPh3)4: тетракис(трифенилфосфин)палладий(0)
PdCl2(PPh3)2: дихлорид бис(трифенилфосфин)палладия(II)
PYBOP: гексафторфосфат бензотриазол-1-илокситрис(пиридино)фосфония
t-: третичный
TBAF: фторид тетрабутиламмония
TEA: триэтиламин
Tf: трифторметансульфонил
TFA: трифторуксусная кислота
TFAA: ангидрид трифторуксусной кислоты
THF: тетрагидрофуран
THP: тетрагидропиран
TMEDA: N,N,N′,N′-тетраметилэтилендиамин
TMS: триметилсилил
Tris: трисгидроксиметиламинометан
Ts: паратолуолсульфонил
1H-ЯМР: спектрометрия на основе протонного ядерного магнитного резонанса
LC-MS: жидкостная хроматография-масс-спектрометрия
Ксантфос: 4,5-бис(дифенилфосфино)-9,9-диметилксантин
Z: бензилоксикарбонил
[0146] "Комнатная температура" в приведенных далее примерах и примерах получения обычно означает от приблизительно 10°C до приблизительно 35°C. % означает весовой процент, если не указано иное.
[0147] Химический сдвиг в спектре протонного ядерного магнитного резонанса регистрируют в δ единицах (м.д.) относительно тетраметилсилана; а постоянную взаимодействия регистрируют в герцах (Гц). Сокращения структур расщепления являются следующими: s: синглет, d: дуплет, t: триплет, q: квартет, m: мультиплет, brs: широкий синглет и brd: широкий дуплет.
[0148] В реакции, осуществляемой с помощью микроволнового реактора, в приведенных далее примерах и примерах получения, использовали Emrys™ Liberator, изготовленный Personal chemistry.
[0149] Для разделения оптических изомеров соединения использовали Parallex Flex™, изготовленный Biotage (колонка: одна из CHIRALPAK (R) AD-H, IA, IB и IC, изготовленная Daicel Corp., и CHIRALCEL (R) OD-H и OJ-H, изготовленная Daicel Corp.; размер колонки 2 см Φ × 25 см). Время удерживания в таблицах в примерах означает величину, когда использовали одну из CHIRALPAK (R) AD-H, IA, IB и IC, изготовленная Daicel Corp., и CHIRALCEL (R) OD-H и OJ-H, изготовленная Daicel Corp. (размер колонки: 0,46 см Φ × 15 см или 0,46 см Φ × 25 см), и скорость потока выставляли на 1,00 мл/мин. Вращение плоскости поляризации (+/-) измеряли с помощью хирального детектора OR-2090 (Hg-Xe лампа, 150 Вт), изготовленного JASCO.
Что касается хроматографии, в описании в качестве колоночной хроматографии на силикагеле, использовали Parallel Prep, изготовленный Yamazen Corp., (колонка: колонка Hi-Flash (TM) (силикагелевая), изготовленная Yamazen Corp., размер: один из S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) и 3L (46×130 мм)) или силикагель сферической формы для хроматографии PSQ60B™, изготовленный Fuji Silysia Chemical Ltd., силикагель для хроматографии BW-300™, изготовленный Fuji Silysia Chemical Ltd., Wakogel (R) C-200, изготовленный Wako Pure Chemical Industries, Ltd. или силикагель 60 (R) (70-230 меш), изготовленный Merck Ltd. Japan. В описании в качестве колоночной хроматографии на NH-силикагеле, использовали Parallel Prep, изготовленный Yamazen Corp., (колонка: колонка Hi-Flash (TM) (амино), изготовленная Yamazen Corp., размер: один из S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) и 3L (46×130 мм)) или NH-силикагель (200-350 меш), изготовленный Fuji Silysia Chemical Ltd.
[0150] (±)- означает рацемическую смесь, а (+)- и (-)- означают (+) тип и (-) тип энантиомера, соответственно.
[0151] Названия приведенных далее соединений использовали как указано в “E-notebook” версии 12 (Perkin Elmer), за исключением традиционно используемых реагентов.
[0152] Пример получения 1
Синтез [5-(2,4-диметоксибензил)-4-оксо-1-(тетрагидро-2H-пиран-4-ил)-4,5-дигидро-1H-пиразоло[4,3-c]хинолин-7-ил]бороновой кислоты
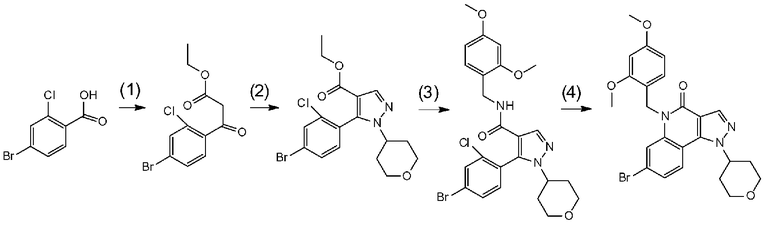
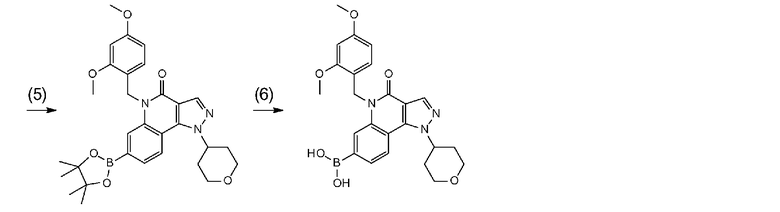
[0153] (1) Синтез этил-3-(4-бром-2-хлорфенил)-3-оксопропионата
4-Бром-2-хлорбензойную кислоту (1 г) суспендировали в DCM (10 мл). CDI (960 мг) добавляли к полученной суспензии и перемешивали при комнатной температуре в течение 4 часов. Данный раствор принимали за "раствор 1". Этилмалонат калия (1,1 г) суспендировали в ацетонитриле (20 мл) в другом сосуде в атмосфере азота и добавляли TEA (1,5 мл). Полученный раствор охлаждали до 0°C и постепенно добавляли хлорид магния (805 мг), а после этого перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь охлаждали до 0°C и добавляли в нее по каплям "раствор 1", приготовленный как указано ранее. По завершению добавления по каплям, реакционную смесь перемешивали при комнатной температуре в течение 17 часов. Реакционную смесь дополнительно перемешивали при 50°C в течение 9 часов. Реакционную смесь концентрировали при пониженном давлении и удаляли DCM. Полученный остаток охлаждали до 0°C и добавляли этилацетат (50 мл) и 2н соляную кислоту (20 мл) и перемешивали при комнатной температуре в течение 1 часа. Полученный органический слой отделяли. Полученный водный слой экстрагировали этилацетатом. Экстракт объединяли с органическим слоем и сушили при помощи безводного сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением, таким образом, указанного в заголовке соединения (1,2 г).
ESI-MS m/z 307 [M+H]+
[0154] (2) Синтез этил-5-(4-бром-2-хлорфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Этил-3-(4-бром-2-хлорфенил)-3-оксопропионат (1,2 г) растворяли в DMF-DMA (4,7 мл) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. К полученному остатку добавляли этанол (23 мл) и гидрохлорид (тетрагидро-2H-пиран-4-ил)гидразина (CAS № 194543-22-1; ChemReach Inc.) (759 мг) и перемешивали при комнатной температуре в течение 15 часов. После этого продукт реакции нагревали до кипения в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и после этого концентрировали при пониженном давлении. Этилацетат и насыщенный водный раствор гидрокарбоната натрия добавляли к полученному остатку и разделяли. Полученный органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили с помощью безводного сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат/н-гептан, 10%-30%-50%) с получением, таким образом, указанного в заголовке соединения (1,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,15 (т, J=7,2 Гц, 3H), 1,63-1,73 (м, 1H), 1,83-1,91 (м, 1H), 2,22-2,45 (м, 2H), 3,29-3,41 (м, 2H), 3,83-3,93 (м, 1H), 3,99-4,10 (м, 2H), 4,09-4,15 (м, 2H), 7,16 (д, J=8,2 Гц, 1H), 7,54 (дд, J=8,2 Гц, 2,0 Гц, 1H), 7,73 (д, J=2,0 Гц, 1H), 8,05 (с, 1H).
ESI-MS m/z 415 [M+H]+
[0155] (3) Синтез 5-(4-бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамида
Этил-5-(4-бром-2-хлорфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилат (1,5 г) добавляли к этанолу (28 мл) и нагревали до 60°C для растворения. 5н водный раствор гидроксида натрия (2,1 мл) добавляли к полученному раствору и перемешивали при 50°C в течение 2 с половиной часов. Реакционную смесь охлаждали до комнатной температуры и после этого добавляли CHCl3 (100 мл), 5н соляную кислоту (12 мл) и насыщенный солевой раствор и разделяли. Полученный органический слой сушили при помощи безводного сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток суспендировали в DCM (31 мл); и добавляли CDI (825 мг), и перемешивали при комнатной температуре. Через 30 минут к полученному раствору добавляли 2,4-диметоксибензиламин (1,0 мл) и перемешивали при комнатной температуре в течение 1 часа. Насыщенный водный раствор гидрокарбоната натрия добавляли к реакционной смеси и разделяли. Полученный водный слой экстрагировали этилацетатом. Экстракт объединяли с полученным органическим слоем и промывали насыщенным водным раствором гидрокарбоната натрия. Полученный органический слой сушили при помощи безводного сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-80%) с получением, таким образом, указанного в заголовке соединения (1,6 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,57-1,64 (м, 1H), 1,83-1,90 (м, 1H), 2,18-2,29 (м, 1H), 2,33-2,44 (м, 1H), 3,27-3,39 (м, 2H), 3,75 (с, 3H), 3,80 (с, 3H), 3,97-4,09 (м, 2H), 4,33-4,26 (м, 2H), 5,72-5,81 (м, 1H), 6,37-6,44 (м, 3H), 7,08 (д, J=8,2 Гц, 1H), 7,17 (д, J=8,4 Гц, 1H), 7,49 (дд, J=8,2 Гц, 2,0 Гц, 1H), 7,70 (д, J=2,0 Гц, 1H), 7,92 (с, 1H).
ESI-MS m/z 536 [M+H]+
[0156] (4) Синтез 7-бром-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-дигидропиразоло[4,3-c]хинолин-4(5H)-она
5-(4-Бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамид (1,6 г) растворяли в THF (29 мл). Раствор охлаждали до 0°C и добавляли KTB (434 г). Смесь перемешивали при комнатной температуре в течение 26 часов. Добавляли насыщенный водный раствор хлорида аммония и метанол к реакционной смеси, которую экстрагировали посредством CHCl3. Полученный органический слой сушили при помощи безводного сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли DMF и воду. Осажденное твердое вещество собирали фильтрованием с получением, таким образом, указанного в заголовке соединения (1,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,10-2,20 (м, 2H), 2,42-2,55 (м, 2H), 3,67 (т, J=11,0 Гц, 2H), 3,68 (с, 3H), 4,02 (с, 3H), 4,19-4,25 (м, 2H), 4,90-5,00 (м, 1H), 5,50 (с, 2H), 6,36 (дд, J=8,2 Гц, 4,2 Гц, 1H), 6,52 (д, J=4,2 Гц, 1H), 7,00 (д, J=8,2 Гц, 1H), 7,39 (д, J=8,4 Гц, 1H), 7,81 (д, J=8,4 Гц, 1H), 7,82 (с, 1H), 8,32 (с, 1H).
ESI-MS m/z 500 [M+H]+
[0157] (5) Синтез 5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-дигидропиразоло[4,3-c]хинолин-4(5H)-она
7-Бром-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-дигидропиразоло[4,3-c]хинолин-4(5H)-он (200 мг) растворяли в 1,4-диоксане (10 мл). Бис(пинаколато)дибор (132 мг), комплекс Pd(dppf)Cl2 DCM (15 мг) и ацетат калия (118 мг) добавляли к полученному раствору и позволяли проходить реакции при 130°C в течение 2 часов с помощью микроволнового реактора. Реакционной смеси давали остыть до комнатной температуры и после этого концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 30%-100%) с получением, таким образом, указанного в заголовке соединения (175 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,24 (с, 6H), 1,34 (с, 6H), 2,13-2,22 (м, 2H), 2,42-2,55 (м, 2H), 3,63-3,77 (м, 2H), 3,74 (с, 3H), 4,02 (с, 3H), 4,19-4,25 (м, 2H), 4,97-5,07 (м, 1H), 5,62 (с, 2H), 6,32 (дд, J=8,2 Гц, 4,2 Гц, 1H), 6,50 (д, J=4,2 Гц, 1H), 7,03 (д, J=8,2 Гц, 1H), 7,68 (д, J=10,0 Гц, 1H), 7,95 (д, J=10,0 Гц, 1H), 8,02 (с, 1H), 8,34 (с, 1H).
ESI-MS m/z 546 [M+H]+
[0158] (6) Синтез [5-(2,4-диметоксибензил)-4-оксо-1-(тетрагидро-2H-пиран-4-ил)-4,5-дигидро-1H-пиразоло[4,3-c]хинолин-7-ил]бороновой кислоты
Синтезированный 5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-дигидропиразоло[4,3-c]хинолин-4(5H)-он (150 мг) растворяли в 1,4-диоксане (10 мл). К раствору добавляли 2н HCl (1 мл) и смесь перемешивали при комнатной температуре. Через 30 минут осажденное твердое вещество собирали при помощи фильтрации. Полученное твердое вещество сушили при пониженном давлении с получением указанного в заголовке соединения (104 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,13-2,25 (м, 2H), 2,42-2,60 (м, 2H), 3,71 (с, 3H), 3,72 (с, 3H), 3,81 (с, 2H), 4,17-4,29 (м, 2H), 4,98-5,09 (м, 1H), 5,62 (с, 2H), 6,32 (дд, J=8,2 Гц, 4,2 Гц, 1H), 6,46 (д, J=4,2 Гц, 1H), 6,94 (д, J=8,2 Гц, 1H), 7,36 (д, J=10,0 Гц, 1H), 7,73 (с, 1H), 8,03 (д, J=10,0 Гц, 1H), 8,36 (с, 1H).
ESI-MS m/z 464 [M+H]+
[0159] Пример получения 2
Синтез 7-хлор-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
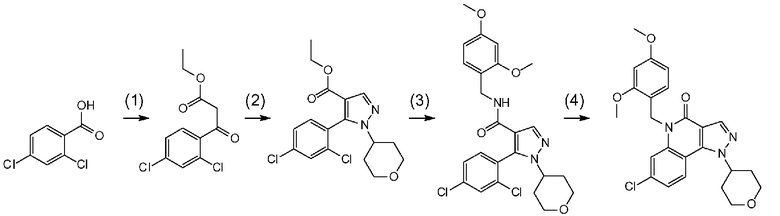
[0160] Указанное в заголовке соединение получали путем проведения реакций (1)-(4) согласно примеру получения 1 с применением 2,4-дихлорбензойной кислоты и гидрохлорида (тетрагидро-2H-пиран-4-ил)гидразина в качестве сырьевых материалов.
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 2,00-2,09 (м, 2H), 2,10-2,24 (м, 2H), 3,62-3,76 (м, 2H), 3,69 (с, 3H), 3,94 (с, 3H), 3,95-4,04 (м, 2H), 5,18-5,27 (м, 1H), 5,36 (ушир.с, 2H), 6,34-6,37 (м, 1H), 6,63-6,65 (м, 2H), 7,37-7,42 (м, 2H), 8,27-8,29 (м, 2H).
ESI-MS m/z 454 [M+H]+
[0161] Пример получения 3
Синтез этил-5-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
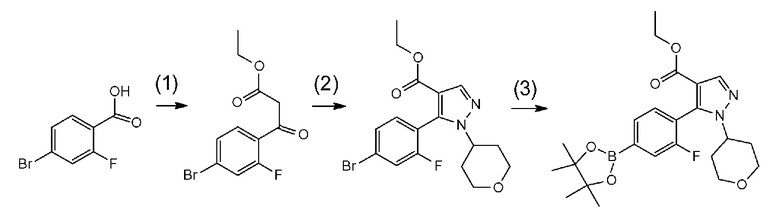
[0162] (1) Синтез этил-3-(4-бром-2-фторфенил)-3-оксопропаноата
CDI (8,88 г) добавляли к суспензии 4-бром-2-фторбензойной кислоты (CAS № 112704-79-7) (10 г) в DCM (97 мл) и смесь перемешивали при комнатной температуре в течение 3,5 часов. Данный раствор называли "раствором 1".
В другом сосуде TEA (15,9 мл) и хлорид магния (10,9 г) последовательно добавляли к суспензии этилмалоната калия (15,5 г) в ацетонитриле (303 мл) и смесь перемешивали при комнатной температуре в течение трех часов и 10 минут. "Раствор 1", приготовленный как описано выше, покапельно добавляли к реакционной смеси в течение 25 минут и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали до половины объема при пониженном давлении. Полученный остаток разбавляли этилацетатом (500 мл) и добавляли 5н соляную кислоту (250 мл) в условиях охлаждения льдом с последующим перемешиванием при комнатной температуре в течение одного часа. Отделяли органический слой. Органический слой промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-20%) с получением указанного в заголовке соединения (12,8 г).
ESI-MS m/z 291 [M+H]+
[0163] (2) Синтез этил-5-(4-бром-2-фторфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Раствор этил-3-(4-бром-2-фторфенил)-3-оксопропаноата (25,6 г) в DMF-DMA (129 мл) перемешивали при комнатной температуре в течение четырех часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли толуол (250 мл). Раствор концентрировали при пониженном давлении. К остатку добавляли этанол (550 мл). Раствор охлаждали в ледяной бане. К раствору добавляли гидрохлорид (тетрагидро-2H-пиран-4-ил)гидразина (15,4 г). Смесь подогревали до комнатной температуры в течение одного часа, а затем нагревали с обратным холодильником в течение двух часов. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали при пониженном давлении. Остаток разделяли путем добавления этилацетата (400 мл) и солевого раствора (200 мл). Органический слой промывали солевым раствором (200 мл), сушили над безводным сульфатом магния и фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-25%). Полученный сырой очищенный продукт суспендировали в смешанном растворе MTBE (30 мл) и н-гептана (50 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Осажденное твердое вещество собирали при помощи фильтрации. Полученное твердое вещество суспендировали в смешанном растворе MTBE (30 мл) и н-гептана (50 мл) с последующим перемешиванием при комнатной температуре в течение ночи. Осажденное твердое вещество собирали при помощи фильтрации. После высушивания получали указанное в заголовке соединение (22,8 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,13-1,23 (м, 3H), 1,63-1,73 (м, 1H), 1,77-1,87 (м, 1H), 2,27-2,44 (м, 2H), 3,29-3,44 (м, 2H), 3,91-4,11 (м, 3H), 4,11-4,20 (м, 2H), 7,16-7,24 (м, 1H), 7,39-7,49 (м, 2H), 8,05 (д, J=0,59 Гц, 1H).
ESI-MS m/z 419 [M+Na]+
[0164] (3) Синтез этил-5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Смесь этил-5-(4-бром-2-фторфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата (2 г), бис(пинаколато)дибора (1,53 г), комплекса Pd(dppf)Cl2-DCM (0,18 г) и ацетата калия (1,48 г) сушили при пониженном давлении с применением вакуумного насоса в течение одного часа. К высушенному остатку добавляли DMF (20 мл) и смесь перемешивали при 85°C в течение шести часов. Реакционной смеси давали остыть до комнатной температуры и затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении. Остаток разделяли путем добавления этилацетата (100 мл) и воды (100 мл). Водный слой экстрагировали этилацетатом (20 мл × 2). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-20%) с получением указанного в заголовке соединения (2,18 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,12-1,17 (м, 3H), 1,37 (с, 12H), 1,64-1,72 (м, 1H), 1,81-1,85 (м, 1H), 2,30-2,39 (м, 2H), 3,28-3,36 (м, 2H), 3,94-4,08 (м, 3H), 4,13 (кв, J=7,0 Гц, 2H), 7,29-7,32 (м, 1H), 7,61-7,64 (м, 1H), 7,68-7,70 (м, 1H), 8,05 (с, 1H).
[0165] Пример получения 4
Синтез (±)-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
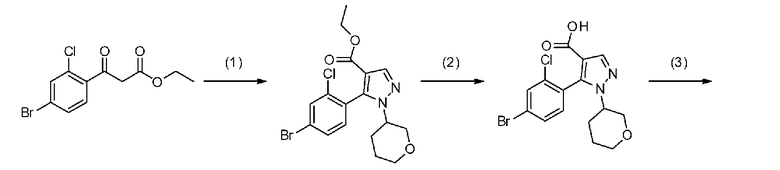
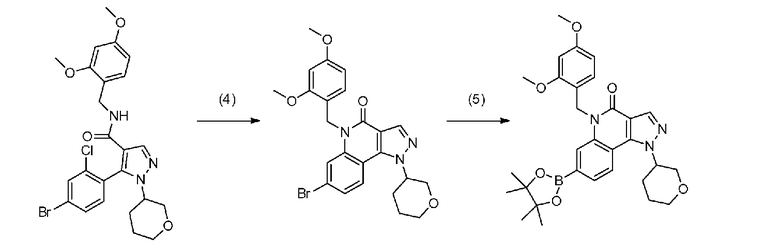
[0166] Указанное в заголовке соединение получали путем проведения реакций (1)-(5) согласно примеру получения 1 с применением этил-3-(4-бром-2-хлорфенил)-3-оксопропаноата, полученного в примере получения 1, и гидрохлорида (±)-(тетрагидро-2H-пиран-3-ил)гидразина, полученного в примере получения 17, в качестве сырьевых материалов.
ESI-MS m/z 546 [M+H]+
[0167] Пример получения 5
Синтез (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
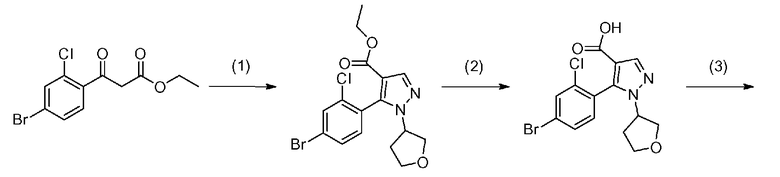
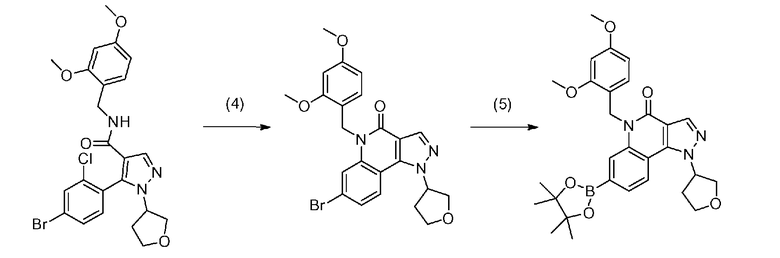
[0168] (1) Синтез (±)-этил-5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата
Этил-3-(4-бром-2-хлорфенил)-3-оксопропаноат, полученный в примере получения 1(1) (2,00 г), растворяли в DMF-DMA (6,96 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в этаноле (40 мл). К раствору добавляли гидрохлорид (±)-(тетрагидрофуран-3-ил)гидразина (998 мг) и смесь нагревали с обратным холодильником в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом и органический слой очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-30%) с получением указанного в заголовке соединения (1,05 г).
ESI-MS m/z 401 [M+H]+
[0169] (2) Синтез (±)-5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновой кислоты
Смесь (±)-этил-5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата (1,05 г) и 5M водного раствора гидроксида натрия (1,58 мл) перемешивали в смешанном растворителе из этанола (20 мл) и воды (5 мл) при 60°C в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли 5M соляную кислоту с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом магния и отфильтровывали осушитель. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1 г).
ESI-MS m/z 371 [M+H]+
[0170] (3) Синтез (±)-5-(4-бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксамида
(±)-5-(4-Бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновую кислоту (1 г) растворяли в DCM (20 мл) и добавляли CDI (611 мг) с последующим перемешиванием при комнатной температуре в течение одного часа. К реакционной смеси добавляли 2,4-диметоксибензиламин (0,809 мл) и смесь перемешивали при комнатной температуре в течение двух часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией при помощи DCM. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-40%) с получением указанного в заголовке соединения (1,26 г).
ESI-MS m/z 522 [M+H]+
[0171] (4) Синтез (±)-7-бром-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(±)-5-(4-Бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксамид (1,26 г) растворяли в THF (25 мл) и добавляли KTB (597 мг) при 0°C. Смесь перемешивали в течение 12 часов при постепенном нагревании до комнатной температуры. Реакционную смесь охлаждали до 0°C и добавляли воду с последующей фильтрацией. Остаток на фильтре хранили отдельно. Фильтрат экстрагировали этилацетатом, а органический слой концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-70%). Полученную фракцию и остаток на фильтре, полученный как описано выше, объединяли и концентрировали с получением указанного в заголовке соединения (488 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,50-2,62 (м, 1H), 2,72-2,82 (м, 1H), 3,76 (с, 3H), 4,02 (с, 3H), 4,07-4,15 (м, 1H), 4,19-4,32 (м, 2H), 4,35-4,42 (м, 1H), 5,46-5,57 (м, 3H), 6,34 (дд, J=8,6 Гц, 2,2 Гц, 1H), 6,52 (д, J=2,2 Гц, 1H), 6,99 (д, J=8,6 Гц, 1H), 7,38 (дд, J=8,6 Гц, 1,8 Гц, 1H), 7,82 (д, J=1,8 Гц, 1H), 7,89 (д, J=8,6 Гц, 1H), 8,32 (с, 1H).
ESI-MS m/z 506 [M+Na]+
[0172] (5) Синтез (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Осуществляли реакцию смеси (±)-7-бром-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (300 мг), бис(пинаколато)дибора (204 мг), комплекса Pd(dppf)Cl2-DCM (13,6 мг) и ацетата калия (182 мг) в смешанном растворителе из 1,4-диоксана (15 мл) и ДМСО (1 мл) с помощью микроволнового реактора при 130°C в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом, а органический слой концентрировали при пониженном давлении. Остаток выливали на силикагелевую подушку и элюировали этилацетатом с получением указанного в заголовке соединения (428 мг) в виде сырого очищенного продукта.
ESI-MS m/z 532 [M+H]+
[0173] Пример получения 6
Синтез этил-5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
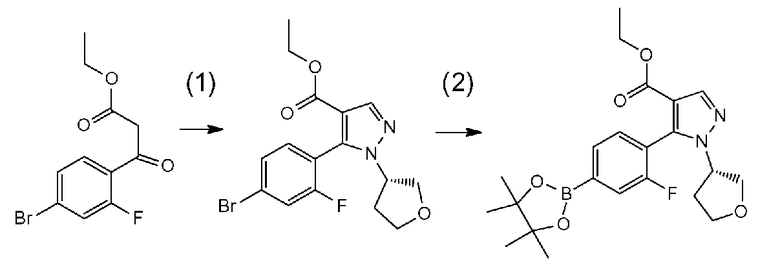
[0174] (1) Синтез этил-5-(4-бром-2-фторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Раствор этил-3-(4-бром-2-фторфенил)-3-оксопропаноата, полученного в примере получения 3(1), (45 г) в DMF-DMA (165 мл) перемешивали при 50°C в течение двух часов и 15 минут. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли толуол (200 мл) и смесь снова концентрировали при пониженном давлении. К остатку добавляли этанол (950 мл) и смесь подогревали до 50°C. К раствору по каплям в течение 35 минут добавляли раствор гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина (21,6 г) в воде (60 мл). Полученную реакционную смесь перемешивали при 50°C в течение двух часов и 10 минут. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали до половины объема при пониженном давлении. К остатку добавляли воду (200 мл) и этанол отгоняли при пониженном давлении. К полученному остатку добавляли этилацетат (500 мл) и отделяли органический слой. Водный слой экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-15%), а затем очищали с помощью хроматографии на короткой колонке NH-силикагеля (этилацетат/н-гептан, 33%) с получением указанного в заголовке соединения (43,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,19 (т, J=7,2 Гц, 3H), 2,19-2,49 (м, 2H), 3,87-4,07 (м, 3H), 4,11-4,25 (м, 3H), 4,58-4,65 (м, 1H), 7,17-7,26 (м, 1H), 7,39-7,47 (м, 2H), 8,06 (с, 1H).
ESI-MS m/z 407 [M+Na]+
[0175] (2) Синтез этил-5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Смесь этил-5-(4-бром-2-фторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (43,1 г), бис(пинаколато)дибора (34,3 г), комплекса Pd(dppf)Cl2-DCM (4,59 г) и ацетата калия (33,1 г) сушили при пониженном давлении с применением вакуумного насоса в течение одного часа. Раствор высушенного остатка в DMF (430 мл) перемешивали при 80°C в течение трех часов и 10 минут. Реакционной смеси давали остыть до комнатной температуры и затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат (430 мл) и солевой раствор (200 мл) с последующим перемешиванием в течение пяти минут. Нерастворимое вещество отфильтровывали через Celite™. Органический слой отделяли от фильтрата. Водный слой повторно экстрагировали этилацетатом (50 мл). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-15%) с получением указанного в заголовке соединения (51,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,16 (т, J=7,2 Гц, 3H), 1,37 (с, 12H), 2,15-2,49 (м, 2H), 3,85-4,06 (м, 3H), 4,14 (кв, J=7,2 Гц, 2H), 4,20 (дд, J=15,6, 8,4 Гц, 1H), 4,57-4,66 (м, 1H), 7,30 (т, J=7,2 Гц, 0,5H), 7,35 (т, J=7,2 Гц, 0,5H), 7,63 (дд, J=5,6, 2,0 Гц, 1H), 7,70 (дд, J=7,2, 2,0 Гц, 1H), 8,06 (с, 1H).
[0176] Пример получения 7
Синтез этил-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
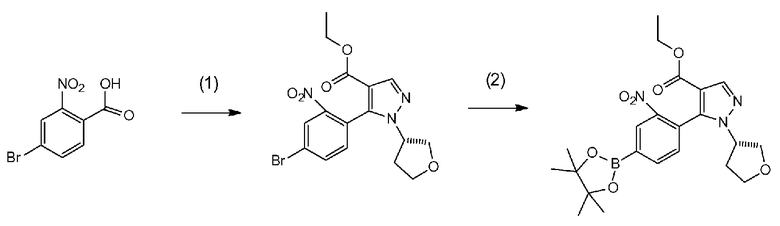
[0177] (1) Синтез этил-5-(4-бром-2-нитрофенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
4-Бром-2-нитробензойную кислоту (10 г) растворяли в ацетонитриле (50 мл). К раствору добавляли тионилхлорид (3,2 мл) и смесь перемешивали в течение трех часов с нагреванием с обратным холодильником. Реакционную смесь охлаждали ледяной водой и по каплям добавляли триэтиламин (11,3 мл). Дополнительно по каплям добавляли этил-3-диметиламинакрилат (6,4 мл). После перемешивания при комнатной температуре в течение трех часов гидрохлорид (S)-(тетрагидрофуран-3-ил)гидразина (6,2 г) растворяли в воде (10 мл) и водный раствор по каплям добавляли в реакционную смесь. После этого смесь перемешивали при комнатной температуре в течение 60 часов. Реакционную смесь разделяли путем добавления воды (50 мл) и этилацетата (200 мл). Органический слой промывали 2н водным раствором гидроксида натрия (100 мл) и солевым раствором (50 мл) и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Этилацетат (5 мл) добавляли к полученному остатку, который растворяли нагреванием с обратным холодильником. Раствор охлаждали ледяной водой. Через один час осажденное твердое вещество собирали при помощи фильтрации с получением сырого очищенного продукта (7,5 г). Дополнительно фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли MTBE (10 мл) и осажденное твердое вещество собирали при помощи фильтрации с получением указанного в заголовке соединения (1,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,13 (тд, J=7,2 Гц, 1,6 Гц, 3H), 2,15-2,34 (м, 1H), 2,39-2,55 (м, 1H), 3,85-4,14 (м, 5H), 4,21 (кв, J=7,7 Гц, 1H), 4,47-4,62 (м, 1H), 7,21 (д, J=8,2 Гц, 0,5H), 7,26 (д, J=8,2 Гц, 0,5H), 7,88 (т, J=2,2 Гц, 0,5H), 7,88 (т, J=2,2 Гц, 0,5H), 8,02 (с, 1H), 8,35 (д, J=2,2 Гц, 0,5H), 8,37 (д, J=2,2 Гц, 0,5H).
ESI-MS m/z 410 [M+H]+
[0178] (2) Синтез этил-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Смесь этил-5-(4-бром-2-нитрофенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (650 мг), бис(пинаколато)дибора (483 мг), комплекса Pd(dppf)Cl2-DCM (64,7 мг) и ацетата калия (467 мг) сушили при пониженном давлении с применением вакуумного насоса в течение одного часа. К высушенному остатку добавляли DMF (6,5 мл) и смесь перемешивали при 80°C в течение четырех часов. Реакционной смеси давали остыть до комнатной температуры и затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении. К остатку добавляли воду с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (417 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,07-1,11 (м, 3H), 1,38 (с, 12H), 2,14-2,31 (м, 1H), 2,41-2,53 (м, 1H), 3,85-4,11 (м, 5H), 4,12-4,24 (м, 1H), 4,49-4,57 (м, 1H), 7,29-7,40 (м, 1H), 8,02-8,03 (м, 1H), 8,13-8,16 (м, 1H), 8,58-8,60 (м, 1H).
[0179] Пример получения 8
Синтез (±)-этил-5-(4-бром-2-нитрофенил)-1-(оксепан-4-ил)-1H-пиразол-4-карбоксилата
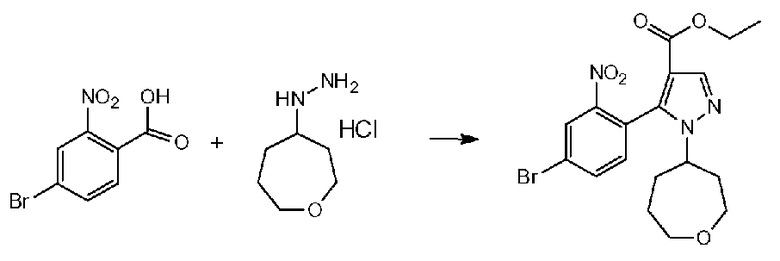
Указанное в заголовке соединение (369 мг) получали с помощью такого же способа, который приведен в примере получения 7, из 4-бром-2-нитробензойной кислоты (2,5 г) и гидрохлорида (±)-оксепан-4-илгидразина, полученного в примере получения 15 (1,69 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,13 (т, J=7,2 Гц, 3H), 1,48-1,65 (м, 1H), 1,76-1,91 (м, 1H), 1,95-2,21 (м, 2H), 2,27-2,51 (м, 2H), 3,54-3,73 (м, 2H), 3,78-3,88 (м, 2H), 4,02-4,13 (м, 3H), 7,20 (д, J=8,0 Гц, 0,5H), 7,21 (д, J=8,0 Гц, 0,5H), 7,87 (дд, J=8,0, 2,0 Гц, 0,5H), 7, 88 (дд, J=8,0, 2,0 Гц, 0,5H), 8,00 (с, 0,5H), 8,01 (с, 0,5H), 8,35 (д, J=2,0 Гц, 0,5H), 8,36 (д, J=2,0 Гц, 0,5H).
ESI-MS m/z 462 [M+Na]+
[0180] Пример получения 9
Синтез этил-1-(1,4-диоксепан-6-ил)-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1H-пиразол-4-карбоксилата
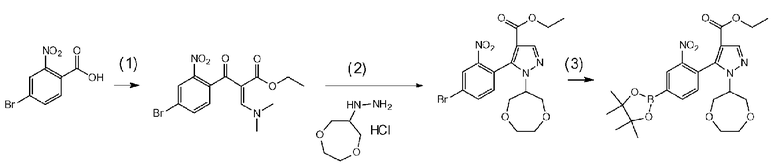
[0181]
(1) Синтез (Z)-этил-2-(4-бром-2-нитробензоил)-3-(диметиламино)акрилата
Раствор 4-бром-2-нитробензойной кислоты (2,5 г) в тионилхлориде (2,93 мл) перемешивали при 80°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли толуол (3 мл) и смесь снова концентрировали при пониженном давлении. Раствор полученного хлорангидрида в ацетонитриле (8 мл) по каплям добавляли в раствор этил-3-диметиламиноакрилата (1,46 г) и TEA (2,83 мл) в ацетонитриле (30 мл) при комнатной температуре в течение 6 минут. Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разделяли путем добавления этилацетата и воды. Водный слой снова экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/гептан, 33-66%) с получением указанного в заголовке соединения (2,55 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,91 (т, J=7,2 Гц, 3H), 3,11 (с, 3H), 3,39 (с, 3H), 3,89 (кв, J=7,2 Гц, 2H), 7,25 (д, J=8,0 Гц, 1H), 7,74 (д, J=8,0, 1,6 Гц, 1H), 8,00 (с, 1H), 8,19 (д, J=1,6 Гц, 1H).
ESI-MS m/z 393 [M+Na]+
[0182]
(2) Синтез этил-5-(4-бром-2-нитрофенил)-1-(1,4-диоксепан-6-ил)-1H-пиразол-4-карбоксилата
К раствору (Z)-этил-2-(4-бром-2-нитробензоил)-3-(диметиламино)акрилата (642 мг) в ацетонитриле (8 мл) добавляли раствор гидрохлорида (1,4-диоксепан-6-ил)гидразина (341 мг), полученный в примере получения 16, в воде (2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи и дополнительно перемешивали при 50°C в течение 9,5 часов. Реакционной смеси давали остыть до комнатной температуры и разделяли путем добавления этилацетата и воды. Водный слой снова экстрагировали этилацетатом. Объединенные органические слои последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором и сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/гептан, 20-33%) с получением указанного в заголовке соединения (408 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,12 (т, J=7,2 Гц, 3H), 3,70-3,83 (м, 2H), 3,87-4,11 (м, 6H), 4,20-4,39 (м, 3H), 7,17 (д, J=8,0 Гц, 1H), 7,88 (дд, J=8,0, 2,0 Гц, 1H), 8,05 (с, 1H), 8,35 (д, J=2,0 Гц, 1H).
ESI-MS m/z 464 [M+Na]+
[0183]
(3) Синтез этил-1-(1,4-диоксепан-6-ил)-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1H-пиразол-4-карбоксилата
Смесь этил-5-(4-бром-2-нитрофенил)-1-(1,4-диоксепан-6-ил)-1H-пиразол-4-карбоксилата (200 мг), бис(пинаколато)дибора (138 мг), комплекса Pd(dppf)Cl2-DCM (19 мг) и ацетата калия (134 мг) сушили при пониженном давлении с применением вакуумного насоса в течение 50 минут. Раствор полученного остатка в DMF (3 мл) перемешивали при 80°C в течение 2 часов и 20 минут. После добавления к реакционной смеси комплекса Pd(dppf)Cl2-DCM (19 мг) реакционную смесь перемешивали при 80°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. Солевой раствор и этилацетат добавляли к полученному остатку и смесь перемешивали при комнатной температуре в течение 5 минут. Отделяли органический слой. Водный слой снова экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/гептан, 33-50%) с получением указанного в заголовке соединения (183 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,08 (т, J=6,8 Гц, 3H), 1,38 (с, 12H), 3,69-3,81 (м, 2H), 3,85-4,10 (м, 6H), 4,22-4,38 (м, 3H), 7,28 (д, J=7,6 Гц, 1H), 8,05 (с, 1H), 8,13 (дд, J=7,6, 1,2 Гц, 1H), 8,57 (д, J=1,2 Гц, 1H).
[0184] Пример получения 10
Синтез этил-5-(4-бром-2,5-дифторфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
[0185] (1) Синтез этил-3-(4-бром-2,5-дифторфенил)-3-оксопропаноата
4-Бром-2,5-дифторбензойную кислоту (395 мг) суспендировали в DCM (3,6 мл). К раствору добавляли CDI (378 мг) и смесь перемешивали при комнатной температуре в течение приблизительно трех часов. Данный раствор называли "раствором 1". В другом сосуде суспендировали этилмалонат калия (567 мг) в ацетонитриле (11 мл) в атмосфере азота, последовательно добавляли TEA (0,58 мл) и хлорид магния (397 мг) и затем смесь перемешивали при комнатной температуре в течение приблизительно трех часов. Полученный как описано выше "раствор 1" по каплям добавляли в реакционную смесь. По завершении покапельного добавления смесь перемешивали при комнатной температуре в течение приблизительно 20 часов. Этилацетат (50 мл) добавляли в реакционную смесь, которую охлаждали до 0°C. Добавляли 5н соляную кислоту (25 мл) и смесь перемешивали при комнатной температуре в течение одного часа. Отделяли органический слой. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-7%) с получением указанного в заголовке соединения (420 мг).
ESI-MS m/z 329, 331 [M+Na]+
[0186] (2) Синтез этил-5-(4-бром-2,5-дифторфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Этил-3-(4-бром-2,5-дифторфенил)-3-оксопропаноат (420 мг) растворяли в DMF-DMA (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 1,5 часа и дополнительно перемешивали при 45°C в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении. Этанол (6 мл) и гидрохлорид (тетрагидро-2H-пиран-4-ил)гидразина (250 мг) добавляли к полученному остатку и смесь перемешивали при 90°C в течение 40 минут. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток разделяли путем добавления этилацетата и солевого раствора. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (этилацетат/н-гептан, 14%-35%-52%) с получением указанного в заголовке соединения (400 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,21 (т, J=7,1 Гц, 3H), 1,63-1,73 (м, 1H), 1,78-1,87 (м, 1H), 2,27-2,44 (м, 2H), 3,33-3,43 (м, 2H), 3,92-4,22 (м, 5H), 7,09-7,14 (м, 1H), 7,44-7,50 (м, 1H), 8,05 (с, 1H).
[0187] Пример получения 11-1
Синтез этил-5-(4-бром-2,5-дифторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
[0188] Этил-3-(4-бром-2,5-дифторфенил)-3-оксопропаноат, полученный в примере получения 10(1) (4 г), растворяли в DMF-DMA (18 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и к полученному остатку (5,9 г) добавляли этанол (80 мл) с последующим подогревом до 60°C. Раствор гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина (2,17 г) в воде (4,5 мл) добавляли к раствору в течение двух минут и смесь перемешивали при 60°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток разделяли путем добавления этилацетата и солевого раствора. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на NH-силикагеле (первый раз: этилацетат/н-гептан, 10%-30%, второй раз: этилацетат/н-гептан, 40%) с получением указанного в заголовке соединения (4,31 г).
ESI-MS m/z 423 [M+Na]+
[0189] Пример получения 11-2
Синтез этил-5-(4-бром-2,5-дифторфенил)-1-((R)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата
Указанное в заголовке соединение синтезировали согласно примеру получения 11-1 из гидрохлорида (R)-(тетрагидрофуран-3-ил)гидразина.
ESI-MS m/z 423 [M+Na]+
[0190] Пример получения 12
Синтез гидрохлорида (±)-(тетрагидрофуран-3-ил)гидразина (способ A)
[0191] (1) Синтез бензил 2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилата
3-Оксотетрагидрофуран (5,70 г) растворяли в метаноле (150 мл) и к раствору добавляли бензилкарбазат (10 г). Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали. Получали 14,8 г остатка в виде сырого очищенного продукта. Его использовали для следующей реакции без дополнительной очистки.
[0192] (2) Синтез (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата
Бензил 2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилат (14,8 г) суспендировали в воде (96 мл). К суспензии добавляли уксусную кислоту (42,1 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа. Суспензия превращалась в раствор. К раствору малыми порциями добавляли цианоборгидрид натрия (4,0 г). Смешанный раствор перемешивали при комнатной температуре в течение двух часов. Реакционную смесь охлаждали до 0°C. Реакционную смесь нейтрализовали путем добавления 5н водного раствора гидроксида натрия. Смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (метанол/этилацетат, 5%). Получали указанное в заголовке соединение (13,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,73-1,80 (м, 1H), 1,92-2,06 (м, 1H), 3,66-3,82 (м, 3H), 3,82-4,03 (м, 2H), 5,14 (с, 2H), 7,31-7,40 (м, 5H).
Было обнаружено, что указанное в заголовке соединение можно разделить на оптические изомеры с помощью хиральной ВЭЖХ в приведенных далее условиях. Условия разделения оптических изомеров [CHIRALPAC (R) OD-H, изготовленная Daicel Corporation, 10% этанол/н-гексан, время удерживания = 12,39 мин, 13,5 мин]
[0193] (3) Синтез гидрохлорида (±)-(тетрагидрофуран-3-ил)гидразина
Бензил-2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (32,3 мг) растворяли в метаноле (3 мл). К раствору добавляли 10% палладированный уголь (50% влаги) (17 мг) и смесь перемешивали при комнатной температуре в течение двух часов в атмосфере водорода. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (1 мл). К раствору добавляли 4н раствор хлороводорода-1,4-диоксана (3 мл). Смесь перемешивали при комнатной температуре в течение трех часов. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,9 мг).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 1,90-2,10 (м, 1H), 2,19-2,32 (м, 1H), 3,53-4,35 (м, 5H).
[0194] Пример получения 13
Синтез гидрохлорида (±)-(тетрагидрофуран-3-ил)гидразина (способ B)
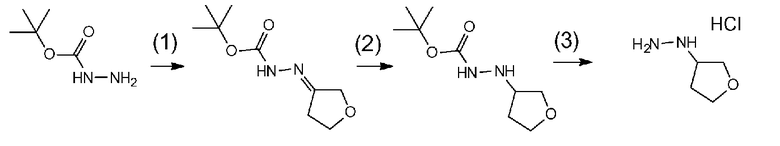
[0195] (1) Синтез трет-бутил-2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилата
3-Оксотетрагидрофуран (10,38 г) растворяли в метаноле (200 мл) и к раствору добавляли трет-бутилкарбазат (17,53 г). Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали с получением указанного в заголовке соединения (27,3 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 2,46 (т, J=6,9 Гц, 2H), 4,10 (т, J=6,9 Гц, 2H), 4,33 (с, 2H).
[0196] (2) Синтез (±)-трет-бутил-2-(тетрагидрофуран-3-ил)гидразинкарбоксилата
трет-Бутил-2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилат (17,26 г) суспендировали в воде (130 мл). К суспензии добавляли уксусную кислоту (57,2 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение одного часа. К раствору малыми порциями добавляли цианоборгидрид натрия (5,36 г). Смешанный раствор перемешивали при комнатной температуре в течение двух часов. Реакционную смесь охлаждали до 0°C. Реакционную смесь нейтрализовали путем добавления 5н водного раствора гидроксида натрия. Смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (5% метанол/этилацетат). Получали указанное в заголовке соединение (15,3 г).
[0197] (3) Синтез гидрохлорида (±)-(тетрагидрофуран-3-ил)гидразина
(±)-трет-бутил-2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (5 г) растворяли в метаноле (40 мл). К раствору добавляли 4н раствор хлороводорода-1,4-диоксана (40 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении. Остаток растирали в этилацетате, воде и метаноле. Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (2,09 г).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 1,92-2,02 (м, 1H), 2,19-2,30 (м, 1H), 3,70-3,84 (м, 3H).
[0198] Пример получения 14
Синтез гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина
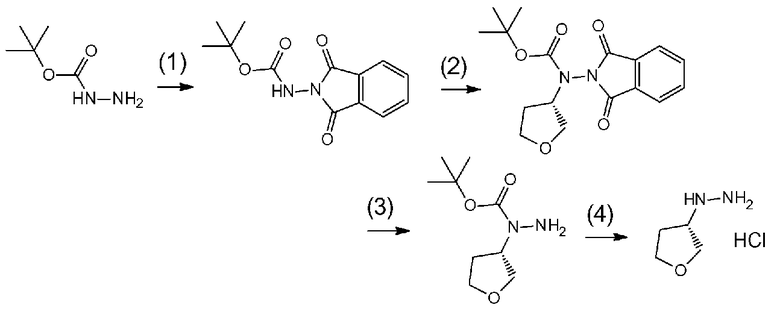
[0199] (1) Синтез трет-бутил-(1,3-диоксоизоиндолин-2-ил)карбамата
Суспензию фталиевого ангидрида (30,0 г) и трет-бутилкарбазата (CAS № 870-46-2) (26,8 г) в толуоле (600 мл) азеотропно кипятили с обратным холодильником с применением уловителя Дина-Старка в течение 3,25 часов. Нерастворимое вещество удаляли с помощью высокотемпературной очистки. Фильтрат концентрировали до приблизительно одной трети объема при пониженном давлении и затем охлаждали на льду. Осажденное твердое вещество собирали при помощи фильтрации. Полученное вещество растворяли в этилацетате (750 мл) и очищали с помощью хроматографии на короткой колонке NH-силикагеля (100% этилацетат). Целевую фракцию концентрировали и остаток затем растирали в этилацетате (20 мл). Полученное твердое вещество собирали с помощью фильтрации и сушили при пониженном давлении с получением указанного в заголовке соединения (16,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,52 (с, 9H), 6,55 (ушир.с, 1H), 7,79 (дд, J=5,6, 3,2 Гц, 2H), 7,91 (дд, J=5,6, 3,2 Гц, 2H).
[0200] (2) Синтез (S)-трет-бутил-(1,3-диоксоизоиндолин-2-ил)(тетрагидрофуран-3-ил)карбамата
DEAD (11,5 мл) добавляли по каплям к раствору (R)-(-)-3-гидрокситетрагидрофурана (CAS № 86087-24-3) (4,84 г), трет-бутил-(1,3-диоксоизоиндолин-2-ил)карбамата (12 г) и трифенилфосфина (18,0 г) в THF (160 мл) с охлаждением на льду в течение пяти минут. Реакционную смесь перемешивали при 0°C в течение трех минут, а затем при комнатной температуре в течение семи часов и 40 минут. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 20%) с получением указанного в заголовке соединения (12,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,29 (с, 6H), 1,53 (с, 3H), 2,12-2,33 (м, 2H), 3,63-3,97 (м, 4H), 4,84-4,94 (м, 0,33H), 5,04-5,14 (м, 0,67H), 7,75-7,84 (м, 2H), 7,87-7,94 (м, 2H).
ESI-MS m/z 355 [M+Na]+
Результаты анализа оптической чистоты >98% э.и. [IC, 10% этанол/н-гексан, время удерживания = 9,7 мин]
[0201] (3) Синтез (S)-трет-бутил-1-(тетрагидрофуран-3-ил)гидразинкарбоксилата
Метилгидразин (3,94 мл) по каплям добавляли к раствору (S)-трет-бутил-(1,3-диоксоизоиндолин-2-ил)(тетрагидрофуран-3-ил)карбамата (12,3 г) в THF (125 мл) с охлаждением на льду в течение двух минут. Реакционную смесь перемешивали при 0°C в течение 30 минут, при комнатной температуре в течение трех дней, а затем при 50°C в течение четырех часов. Реакционную смесь охлаждали на льду, а затем при помощи фильтрации из реакционной смеси удаляли нерастворимое вещество. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-14%) с получением указанного в заголовке соединения (7,04 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,48 (с, 9H), 2,00-2,11 (м, 2H), 3,67-3,82 (м, 4H), 3,87 (дд, J=8,8, 7,2 Гц, 1H), 3,97 (дд, J=15,2, 7,2 Гц, 1H), 4,67-4,80 (м, 1H).
ESI-MS m/z 225 [M+Na]+
[0202] (4) Синтез гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина
(S)-трет-бутил-(тетрагидрофуран-3-ил)гидразинкарбоксилат (7,04 г) растворяли в 4н растворе хлороводорода-1,4-диоксана (60 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 25 минут, а затем при 50°C в течение двух часов. Реакционную смесь концентрировали при пониженном давлении. Остаток растирали в MTBE и этаноле. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,85 г).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 1,90-2,04 (м, 1H), 2,19-2,32 (м, 1H), 3,70-3,84 (м, 3H), 3,86-4,02 (м, 2H).
[0203] Пример получения 15
Синтез гидрохлорида (±)-оксепан-4-илгидразина
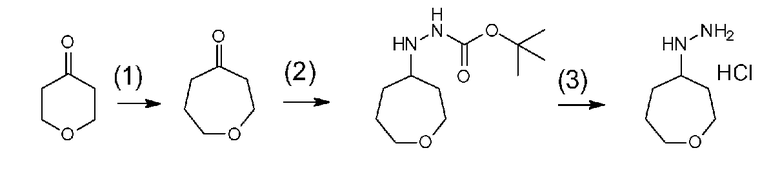
[0204] (1) Синтез оксепан-4-она
Комплекс трифторид бора-простой диэтиловый эфир (13,8 мл) добавляли к раствору тетрагидро-4H-пиран-4-она (CAS № 29943-42-8) (10,0 г) в DCM (400 мл) при комнатной температуре. Реакционную смесь охлаждали до -25°C. Триметилсилилдиазометан (2M раствор в н-гексане, 55 мл) по каплям добавляли в реакционную смесь в течение 40 минут и затем смесь перемешивали при такой же температуре в течение 2,5 часов. К реакционной смеси добавляли воду (40 мл) с последующим перемешиванием при комнатной температуре. Отделяли органический слой. Органический слой промывали насыщенным водным раствором хлорида аммония: 28% водный аммоний = 10:1 (55 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-14%) с получением указанного в заголовке соединения (3,80 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,82-1,89 (м, 2H), 2,65-2,72 (м, 4H), 3,85-3,94 (м, 4H).
[0205] (2) Синтез (±)-трет-бутил-2-(оксепан-4-ил)гидразинкарбоксилата
Указанное в заголовке соединение (4,60 г) получали тем же способом, который описан в примерах получения 13-(1) и 13-(2), из оксепан-4-она (3,80 г) и трет-бутилкарбазата (3,61 г).
ESI-MS m/z 253 [M+Na]+
[0206] (3) Синтез гидрохлорида (±)-оксепан-4-илгидразина
Указанное в заголовке соединение (3,72 г) получали тем же способом, который описан в примере получения 13-(3), из (±)-трет-бутил 2-(оксепан-4-ил)гидразинкарбоксилата (4,60 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,50-1,82 (м, 4H), 2,02-2,34 (м, 2H), 3,08-3,18 (м, 1H), 3,47-3,57 (м, 2H), 3,61-3,74 (м, 2H).
[0207] Пример получения 16
Синтез (1,4-диоксепан-6-ил)гидразина гидрохлорида
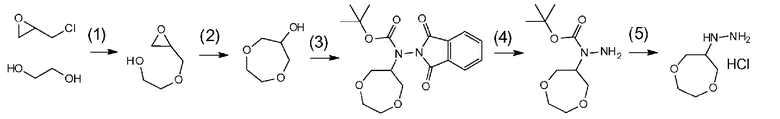
[0208] (1) Синтез 2-(оксиран-2-илметокси)этанола
Эпихлоргидрин (31 г) по каплям добавляли к смеси этиленгликоля (20,8 г) и комплекса трифторид бора-простой диэтиловый эфир (0,255 мл) с охлаждением на льду в течение одного часа. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и 10 минут и затем при 80°C в течение одного часа. Реакционной смеси давали остыть до комнатной температуры. Реакционную смесь по каплям добавляли к раствору охлажденного на льду порошка гидроксида калия (20,7 г) в 1,4-диоксане (110 мл) в течение 45 минут. Полученную реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Нерастворимое вещество в реакционной смеси удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Остаток очищали путем перегонки с получением фракции с температурой кипения, составляющей 58-62°C при давлении 0,3 мм рт.ст. Продукт очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-75%) с получением указанного в заголовке соединения (3,11 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,10 (т, J=6,4 Гц, 1H), 2,65 (дд, J=4,8, 2,8 Гц, 1H), 2,82 (т, J=4,8 Гц, 1H), 3,16-3,21 (м, 1H), 3,46 (дд, J=12,0, 6,0 Гц, 1H), 3,57-3,78 (м, 3H), 3,81-3,89 (м, 2H).
[0209] (2) Синтез 1,4-диоксепан-6-ола
Раствор 2-(оксиран-2-илметокси)этанола (3,11 г) в 1,4-диоксане (200 мл) по каплям добавляли в течение четырех часов и 20 минут к раствору тетрафторборат лития (415 мг) и гидроксида лития (69 мг) в 1,4-диоксане (200 мл), подогретом при 55°C. Реакционную смесь перемешивали при 50°C в течение 50 минут и затем при комнатной температуре в течение 10 минут. Нерастворимое вещество в реакционной смеси удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 40%-50%). Затем его еще раз очищали с помощью колоночной хроматографии на силикагеле (простой диэтиловый эфир/н-гексан 50%-100%) с получением указанного в заголовке соединения (56 мг).
Дополнительно, содержащую примеси фракцию еще раз очищали с помощью колоночной хроматографии на силикагеле (простой диэтиловый эфир, 100%) с получением указанного в заголовке соединения (212 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,57 (ушир.д, J=8,8 Гц, 1H), 3,70-3,77 (м, 2H), 3,82-3,91 (м, 6H), 3,96 (ушир.с, 1H).
[0210] (3) Синтез трет-бутил-1,4-диоксепан-6-ил-(1,3-диоксоизоиндолин-2-ил)карбамата
DEAD (2,2M в толуоле, 1,55 мл) по каплям добавляли к раствору 1,4-диоксепан-6-ола (265 мг), трет-бутил-(1,3-диоксоизоиндолин-2-ил)карбамата (560 мг), полученного в примере получения 14-(1), и трифенилфосфина (840 мг) в THF (10 мл) с охлаждением на льду в течение 3 минут. Реакционную смесь перемешивали при 0°C в течение 6 минут и дополнительно перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении. После добавления толуола (2,5 мл) к полученному остатку осажденное твердое вещество удаляли при помощи фильтрации. Фильтрат очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 20%) с получением указанного в заголовке соединения (713 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,28 (с, 5,4H), 1,50 (с, 3,6H), 3,60-3,72 (м, 4H), 4,02-4,11 (м, 2H), 4,13-4,21 (м, 2H), 4,64-4,71 (м, 0,4H), 4,83-4,92 (м, 0,6H), 7,77-7,83 (м, 2H), 7,89-7,96 (м, 2H).
ESI-MS m/z 385 [M+Na]+
[0211] (4) Синтез трет-бутил-1-(1,4-диоксепан-6-ил)гидразинкарбоксилата
Метилгидразин (0,21 мл) по каплям добавляли к раствору трет-бутил 1,4-диоксепан-6-ил(1,3-диоксоизоиндолин-2-ил)карбамата (710 мг) в THF (7 мл) в течение 1 минуты. Реакционную смесь перемешивали при комнатной температуре в течение 3 дней и дополнительно перемешивали при 50°C в течение 11 часов. После остывания реакционной смеси до комнатной температуры нерастворимое вещество удаляли из реакционной смеси при помощи фильтрации. Фильтрат концентрировали при пониженном давлении. После добавления толуола к остатку осажденное твердое вещество удаляли при помощи фильтрации. Фильтрат очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 15%-25%) с получением указанного в заголовке соединения (393 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47 (с, 9H), 3,67-3,88 (м, 6H), 3,94 (д, J=6,8 Гц, 4H), 4,40-4,60 (м, 1H).
ESI-MS m/z 255 [M+Na]+
[0212] (5) Синтез гидрохлорида (1,4-диоксепан-6-ил)гидразина
4M раствор хлороводород-1,4-диоксана (3 мл) добавляли к раствору трет-бутил-1-(1,4-диоксепан-6-ил)гидразинкарбоксилата (392 мг) в диоксане (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и дополнительно перемешивали при 50°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (341 мг).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 3,38 (квинтет, J=4,4 Гц, 1H), 3,62-3,74 (м, 4H), 3,80 (дд, J=12,8, 4,4 Гц, 2H), 3,86 (дд, J=12,8, 4,4 Гц, 2H).
[0213] Пример получения 17
Синтез гидрохлорида (±)-(тетрагидро-2H-пиран-3-ил)гидразина
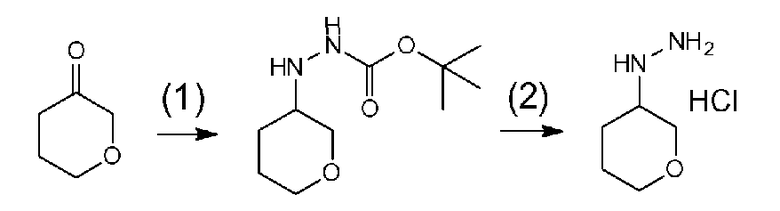
[0214] Указанное в заголовке соединение получали путем проведения реакций (1)-(2) согласно примеру получения 13 с применением дигидро-пиран-3-она в качестве сырьевого материала.
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 1,53-1,64 (м, 1H), 1,72-1,87 (м, 2H), 1,98-2,09 (м, 1H), 3,06-3,15 (м, 1H), 3,59-3,72 (м, 3H), 3,81-3,90 (м, 1H).
[0215] Пример получения 18
Синтез гидрохлорида (3SR,4RS)-4-гидразинилтетрагидрофуран-3-ола
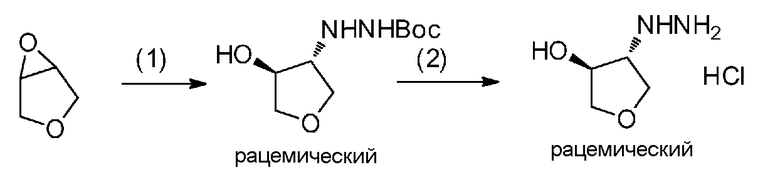
[0216] (1) Синтез трет-бутил-2-[(3RS,4SR)-4-гидрокситетрагидрофуран-3-ил]гидразинкарбоксилата
3,4-Эпокситетрагидрофуран (3,33 мл) и трет-бутилкарбазат (6,14 г) растворяли в 2-пропаноле (15 мл) и раствор нагревали до 90°C. Через три дня дополнительно добавляли трет-бутилкарбазат (6,3 г). После нагревания при перемешивании в течение двух дополнительных дней реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли ксилол и смесь снова концентрировали при пониженном давлении. Остаток разделяли путем добавления хлороформа и солевого раствора. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (5,78 г).
ESI-MS m/z 241 [M+Na]+
[0217] (2) Синтез гидрохлорида (3SR,4RS)-4-гидразинилтетрагидрофуран-3-ола
4M раствор хлороводорода-1,4-диоксана (50 мл) добавляли к раствору трет-бутил-2-((3RS,4SR)-4-гидрокситетрагидрофуран-3-ил)гидразинкарбоксилата (5,78 г) в метаноле (30 мл) с охлаждением льдом, а затем смесь подогревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали с получением указанного в заголовке соединения (5 г).
1H-ЯМР (400 МГц, CD3OD) δ (м.д.): 3,49-3,54 (м, 1H), 3,57-3,63 (м, 1H), 3,65 (дд, J=9,67, 2,64 Гц, 1H), 3,70-3,76 (м, 1H), 3,96-4,08 (м, 2H), 4,28-4,32 (м, 1H).
[0218] Пример получения 19
Синтез (2,4,6-триметилпиридин-3-ил)бороновой кислоты
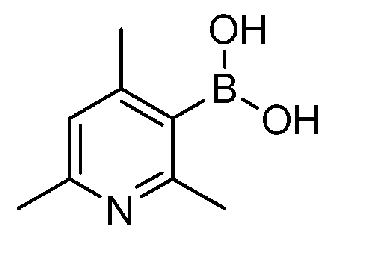
[0219] 3-Бром-2,4,6-триметилпиридин (CAS № 23079-73-4; Prasenjit MaI etc., Journal of Organic Chemistry, 68(9), pp.3446-3453) (1 г) добавляли в THF (20 мл). Раствор охлаждали до -78°C добавляли н-бутиллитий (1,63M раствор в н-гексане, 3,37 мл) с последующим перемешиванием при той же температуре в течение 30 минут. К реакционной смеси добавляли триметилборат (0,78 мл) и смесь перемешивали при -78°C в течение 10 минут и при комнатной температуре в течение 50 минут. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и концентрировали реакционную смесь при пониженном давлении. Полученный остаток разделяли на масло и воду путем добавления воды и DCM. Водный слой концентрировали при пониженном давлении. К полученному остатку добавляли DCM и этанол. Нерастворимое вещество фильтровали, а фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (242 мг).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 2,50 (с, 3H), 2,63 (с, 3H), 2,67 (с, 3H), 7,52 (с, 1H).
[0220] Пример получения 20
Синтез 2-бром-5-(метоксиметил)-1,3-диметилбензол
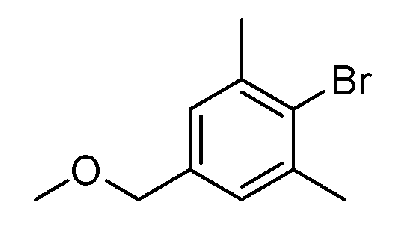
[0221] 2-Броммезитилен (5,00 г) растворяли в четыреххлористый углерод (50 мл). К раствору добавляли NBS (4,45 г) и перекись бензоила (182 мг) и смесь перемешивали при 80°C в течение трех часов. Реакционной смеси давали остыть до комнатной температуры и фильтровали. Собранное при помощи фильтрации твердое вещество промывали н-гептаном. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан). Полученную фракцию концентрировали при пониженном давлении. Остаток растворяли в THF (120 мл). К раствору добавляли метоксид натрия (28% раствор в метаноле, 9,35 мл) и смесь перемешивали при 80°C в течение четырех часов. Реакционной смеси давали остыть до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду с последующей экстракцией посредством DCM. Органический слой концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-5%). Полученную фракцию концентрировали при пониженном давлении и остаток еще раз очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан) с получением указанного в заголовке соединения (880 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,41 (с, 6H), 3,38 (с, 3H), 4,35 (с, 2H), 7,05 (с, 2H).
[0222] Пример получения 21
Синтез 3-бром-6-хлор-2,4-диметилпиридина
[0223] 5-Бром-4,6-диметилпиридин-2-амин (CAS № 89856-44-0; Aldrich) (4,00 г) добавляли к смешанному раствору концентрированной соляной кислоты (24 мл) и воды (24 мл). Раствор охлаждали до 0°C и добавляли нитрит натрия (3,57 г) с последующим перемешиванием при той же температуре в течение 10 минут. К раствору добавляли хлорид меди(I) (5,91 г) и смесь перемешивали при 0°C в течение пяти минут и при комнатной температуре в течение четырех часов и 15 минут. Реакционную смесь охлаждали до 0°C и добавляли 5н водный раствор гидроксида натрия для подщелачивания реакционной смеси. К реакционной смеси добавляли этилацетат с последующей фильтрацией. Органический слой в фильтрате отделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%) с получением указанного в заголовке соединения (1,79 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,39 (с, 3H), 2,65 (с, 3H), 7,06 (с, 1H).
[0224] Пример получения 22
Синтез 3-бром-6-метокси-2,4-диметилпиридина
[0225] 3-Бром-6-хлор-2,4-диметилпиридин, полученный в примере получения 21 (200 мг), добавляли к DMF (1 мл). К раствору добавляли метоксид натрия (28% раствор в метаноле, 0,741 мл) и смесь перемешивали при 60°C в течение 15 часов. К реакционной смеси добавляли воду с последующей экстракцией простым диэтиловым эфиром. Органический слой концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (172 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,34 (с, 3H), 2,57 (с, 3H), 3,88 (с, 3H), 6,46 (с, 1H).
[0226] Пример получения 23
Синтез 3-бром-6-метокси-2,4-диметилпиридина
[0227] (1) Синтез 5-бром-4,6-диметилпиридин-2-ола
2-Амино-5-бром-4,6-диметилпиридин (15 г) растворяли в смешанном растворе серной кислоты (14,2 мл) и воды (212 мл). К раствору добавляли раствор нитрита натрия (6,18 г) в воде (31 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа с последующей экстракцией хлороформом. Органический слой сушили над безводным сульфатом магния и отфильтровывали осушитель. Фильтрат концентрировали при пониженном давлении. К остатку добавляли MTBE для осаждения твердого вещества с последующей фильтрацией. Остаток на фильтре промывали MTBE с получением указанного в заголовке соединения (13,7 г).
ESI-MS m/z 204 [M+H]+
[0228] (2) Синтез 3-бром-6-метокси-2,4-диметилпиридина
Смесь 5-бром-4,6-диметилпиридин-2-ола (7 г), метилиодида (21,6 мл) и карбоната серебра (19,1 г) перемешивали в растворителе-хлороформе (140 мл) при комнатной температуре в течение 36 часов. Реакционную смесь выливали на силикагелевую подушку и элюировали смешанным растворителем (этилацетат:н-гептан=2:8). Полученный раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения (6,98 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,32-2,35 (м, 3H), 2,56-2,58 (м, 3H), 3,88 (с, 3H), 6,43-6,48 (м, 1H).
ESI-MS m/z 216 [M+H]+
[0229] Пример получения 24
Синтез (6-метокси-2,4-диметилпиридин-3-ил)бороновой кислоты
[0230] 3-Бром-6-метокси-2,4-диметилпиридина (150 мг) добавляли к THF (3 мл). Раствор охлаждали до -78°C добавляли н-бутиллитий (1,63 M раствор в н-гексане, 0,468 мл) с последующим перемешиванием при той же температуре в течение 30 минут. К реакционной смеси добавляли триметилборат (0,108 мл) и смесь перемешивали при -78°C в течение 10 минут и при комнатной температуре в течение 50 минут. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и концентрировали реакционную смесь при пониженном давлении. Отгоняли THF. Фильтровали полученный остаток. Собранное фильтрацией твердое вещество промывали водой и н-гептаном с получением указанного в заголовке соединения (41 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,31 (с, 3H), 2,48 (с, 3H), 3,89 (с, 3H), 4,77 (ушир.с, 2H), 6,35 (с, 1H).
[0231] Пример получения 25
Синтез 3-хлор-2-метокси-4,6-диметилпиридина
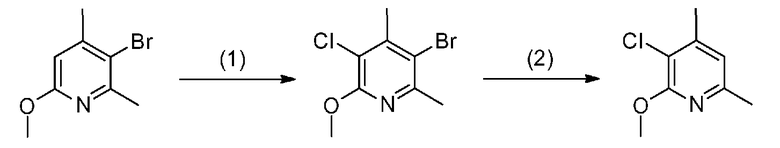
[0232] (1) Синтез 3-бром-5-хлор-6-метокси-2,4-диметилпиридина
3-Бром-6-метокси-2,4-диметилпиридин, полученный в примере получения 22 (800 мг), добавляли к DMF (4 мл). К раствору добавляли NCS (494 мг) и смесь перемешивали при 80°C в течение 14 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%). Получали указанное в заголовке соединение (930 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,51 (с, 3H), 2,56 (с, 3H), 3,98 (с, 3H).
[0233] (2) Синтез 3-хлор-2-метокси-4,6-диметилпиридина
3-Бром-5-хлор-6-метокси-2,4-диметилпиридин (930 мг) добавляли к THF (10 мл). Раствор охлаждали до -78°C и добавляли н-бутиллитий (2,6M раствор в н-гексане, 1,428 мл) с последующим перемешиванием при такой же температуре в течение одного часа. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией посредством DCM. Органический слой промывали солевым раствором и сушили над сульфатом натрия. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (300 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,31 (с, 3H), 2,38 (с, 3H), 3,99 (с, 3H), 6,62 (с, 1H).
[0234] Пример получения 26
Синтез 3-бром-2-метокси-4,6-диметилпиридина
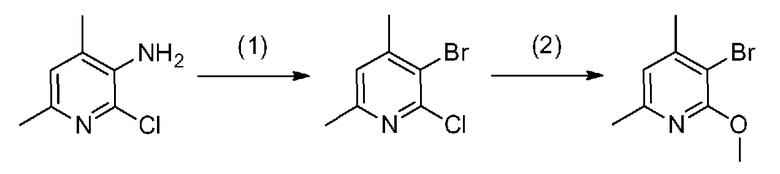
[0235] (1) Синтез 3-бром-2-хлор-4,6-диметилпиридина
2-Хлор-4,6-диметилпиридин-3-амин (2,85 г) растворяли в бромистоводородной кислоте (15 мл, 48% водный раствор) и раствор охлаждали до 0°C. Раствор нитрита натрия (1,51 г) в воде (2 мл) медленно по каплям добавляли к раствору и смесь перемешивали при 0°C в течение 15 минут. К раствору по каплям добавляли суспензию бромида меди(I) (4,18 г) в бромистоводородной кислоте (5 мл, 48% водный раствор) и смесь перемешивали при 0°C в течение 10 минут, а затем при 60°C в течение одного часа. Реакционную смесь охлаждали до комнатной температуры с последующей экстракцией этилацетатом. Органический слой выливали непосредственно на NH-силикагелевую подушку и элюировали этилацетатом. Полученный раствор концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 0%-30%) с получением указанного в заголовке соединения (2,97 г).
ESI-MS m/z 220 [M+H]+
[0236] (2) Синтез 3-бром-2-метокси-4,6-диметилпиридина
Смесь 3-бром-2-хлор-4,6-диметилпиридина (2,97 г) и метоксида натрия (11,0 мл, 28% раствор в метаноле) перемешивали в DMF-растворителе (30 мл) при 80°C в течение 36 часов. К реакционной смеси добавляли воду с последующей экстракцией простым диэтиловым эфиром. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (2,33 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,33-2,34 (м, 3H), 2,36-2,38 (м, 3H), 3,98 (с, 3H), 6,61-6,64 (м, 1H).
ESI-MS m/z 216 [M+H]+
[0237] Пример получения 27
Синтез (2-метокси-4,6-диметилпиридин-3-ил)бороновой кислоты
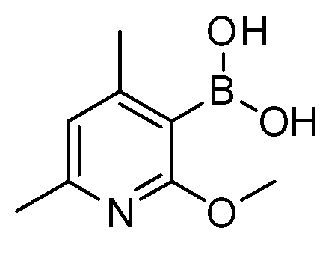
[0238] Указанное в заголовке соединение синтезировали согласно примеру получения 24 с применением 3-бром-2-метокси-4,6-диметилпиридина.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,37-2,42 (с, 3H), 2,47-2,52 (с, 3H), 3,99 (с, 3H), 5,91 (с, 2H), 6,60-6,67 (с, 1H).
[0239] Пример получения 28
Синтез 4-бром-2-метокси-3,5-диметилпиридина
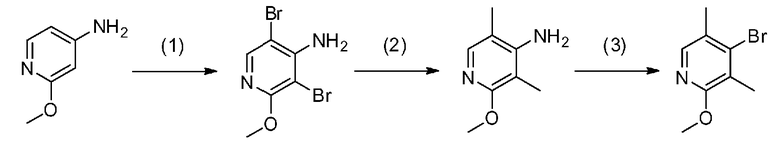
[0240] (1) Синтез 3,5-дибром-2-метоксипиридин-4-амина
Смесь 2-метокси-пиридин-4-иламина (15 г) и NBS (47,3 г) перемешивали в уксусной кислоте-растворителе (150 мл) при комнатной температуре в течение трех часов. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли 5M водный раствор гидроксида натрия (200 мл) при 0°C с последующей экстракцией простым диэтиловым эфиром. Органический слой непосредственно очищали с помощью силикагелевой подушки (этилацетат/н-гептан, 10%) с получением указанного в заголовке соединения (32,4 г).
ESI-MS m/z 283 [M+H]+
[0241] (2) Синтез 2-метокси-3,5-диметилпиридин-4-амина
Смесь 3,5-дибром-2-метоксипиридин-4-амина (16 г), триметилбороксина (19,8 мл), комплекса Pd(dppf)Cl2-DCM (4,15 г) и карбоната калия (23,5 г) нагревали с обратным холодильником в смешанном растворителе из 1,4-диоксана (320 мл) и воды (32 мл) в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли воду и этилацетат с последующей фильтрацией через Celite™. Фильтрат экстрагировали этилацетатом и органический слой выливали на силикагелевую подушку (NH-силикагель) и элюировали этилацетатом. К полученному раствору добавляли NH-силикагель (30 г) и смесь концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 0%-30%) с получением указанного в заголовке соединения (4,43 г).
ESI-MS m/z 153 [M+H]+
[0242] (3) Синтез 4-бром-2-метокси-3,5-диметилпиридина
Смесь бромида меди(I) (12,1 г) и трет-бутилнитрита (7,07 мл) перемешивали в ацетонитрильном растворителе (80 мл) при 70°C в течение 10 минут. К реакционной смеси по каплям добавляли раствор 2-метокси-3,5-диметилпиридин-4-амина (3,9 г) в ацетонитриле (40 мл) при такой же температуре и смесь перемешивали при 70°C в течение одного часа. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный водный раствор бикарбоната натрия, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь фильтровали через Celite™ и фильтрат экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан, 100%, затем NH-силикагелевая подушка, н-гептан, 100%) с получением указанного в заголовке соединения (4,3 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,28-2,29 (м, 3H), 2,29-2,31 (м, 3H), 3,93 (с, 3H), 7,77-7,84 (м, 1H).
ESI-MS m/z 216 [M+H]+
[0243] Пример получения 29
Синтез (2-метокси-3,5-диметилпиридин-4-ил)бороновой кислоты
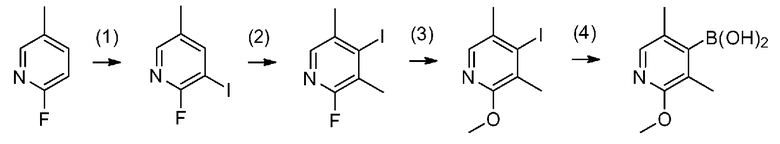
[0244] (1) Синтез 2-фтор-3-иод-5-метилпиридина
Диизопропиламин (92 мл) добавляли к THF (1,2 л) и смесь охлаждали до -18°C в атмосфере азота. К раствору по каплям добавляли 2,69M раствор н-бутиллития в гексане (224 мл). По завершении покапельного добавления смесь подогревали до -5°C с перемешиванием в течение 20 минут. Реакционную смесь охлаждали до -73°C. К реакционной смеси по каплям добавляли 2-фтор-5-метилпиридин (61 г) в THF (240 мл). Реакционную смесь перемешивали при -75°C в течение 3,5 часов. К реакционной смеси по каплям добавляли раствор иода (139 г) в THF (24 мл). Реакционную смесь перемешивали при -75°C в течение одного часа и 55 минут. По завершении реакции к реакционной смеси добавляли воду (220 мл) при такой же температуре. Смесь перемешивали при такой же температуре в течение пяти минут. Реакционной смеси давали остыть до комнатной температуры, а затем добавляли воду (1,2 л). К смеси добавляли раствор пентагидрата тиосульфата натрия (136 г) в воде (300 мл) и воду (300 мл) с последующим перемешиванием в течение 10 минут. Смесь экстрагировали при помощи MTBE (1,2 л). Органический слой промывали солевым раствором (500 мл). Объединенные водные слои экстрагировали при помощи MTBE (1 л). Объединенные органические слои сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан с последующим охлаждением. Осажденное твердое вещество собирали при помощи фильтрации. Остаток промывали н-гептаном. Фильтрат охлаждали и осажденное твердое вещество собирали при помощи фильтрации. Данную процедуру повторяли пять раз с получением указанного в заголовке соединения (109,69 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,29-2,31 (м, 3H), 7,93-8,14 (м, 2H).
ESI-MS m/z 238 [M+H]+
[0245] (2) Синтез 2-фтор-4-иод-3,5-диметилпиридина
Диизопропиламин (88 мл) добавляли к THF (1,2 л) и смесь охлаждали до -18°C в атмосфере азота. К раствору по каплям добавляли 2,69M раствор н-бутиллития в гексане (215 мл). По завершении покапельного добавления смесь подогревали до -5°C с перемешиванием в течение 30 минут. Реакционную смесь охлаждали до -72°C. К реакционной смеси по каплям добавляли раствор 2-фтор-3-иод-5-метилпиридина (109,69 г) в THF (240 мл). Реакционную смесь перемешивали при -74°C в течение 1,5 часа. К реакционной смеси по каплям добавляли раствор метилиодида (36 мл) в THF (160 мл). Реакционную смесь перемешивали при температуре от -70°C до -74°C в течение двух часов. По завершении реакции к реакционной смеси добавляли воду (200 мл) при такой же температуре. Смесь перемешивали при той же температуре в течение двух минут. Реакционной смеси давали остыть до комнатной температуры, а затем добавляли воду (1,2 л). Смешанный раствор перемешивали в течение трех минут. Дополнительно добавляли воду (300 мл). Смесь экстрагировали при помощи MTBE (1,2 л). Органический слой промывали солевым раствором (500 мл). Объединенные водные слои экстрагировали при помощи MTBE (1 л). Объединенные органические слои сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (100 мл) с последующим охлаждением. Осажденное твердое вещество собирали при помощи фильтрации. Остаток промывали н-гептаном. Фильтрат охлаждали и осажденное твердое вещество собирали при помощи фильтрации. Данную процедуру повторяли дважды с получением указанного в заголовке соединения (86,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,39-2,40 (м, 6H), 7,80-7,82 (м, 1H).
ESI-MS m/z 252 [M+H]+
[0246] (3) Синтез 4-иод-2-метокси-3,5-диметилпиридина
28% Раствор метоксида натрия в метаноле (185 мл) добавляли к 2-фтор-4-иод-3,5-диметилпиридину (97,4 г) в THF (954 мл) при 20°C. Смесь перемешивали при температуре от 55°C до 65°C в течение двух часов. Реакционную смесь охлаждали, а затем разделяли путем добавления MTBE (1 л) и воды (1 л). Органический слой промывали солевым раствором. Объединенные водные слои экстрагировали при помощи MTBE (500 мл × 2). Объединенные органические слои сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (50 мл) и смесь перемешивали при 0°C в течение одного часа. Осажденное твердое вещество собирали при помощи фильтрации. Твердое вещество промывали охлажденным н-гептаном (10 мл). Получали указанное в заголовке соединение (42,6 г). Фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (5 мл) и смесь перемешивали при 0°C в течение 30 минут. Осажденное твердое вещество собирали при помощи фильтрации. Твердое вещество промывали охлажденным н-гептаном (2 мл). Получали указанное в заголовке соединение (20,2 г). Фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (5 мл) и смесь перемешивали при 0°C в течение 30 минут. Осажденное твердое вещество собирали при помощи фильтрации. Твердое вещество промывали охлажденным н-гептаном (2 мл). Получали указанное в заголовке соединение (10,7 г). Получали объединенное указанное в заголовке соединение (73,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,33-2,34 (м, 3H), 2,36-2,38 (м, 3H), 3,92 (с, 3H), 7,76 (с, 1H).
ESI-MS m/z 264 [M+H]+
[0247] (4) Синтез (2-метокси-3,5-диметилпиридин-4-ил)бороновой кислоты
4-Иод-2-метокси-3,5-диметилпиридин (2,0 г) в THF (40 мл) охлаждали до -78°C. 2,69M раствор н-бутиллития в гексане (6,5 мл) по каплям добавляли к раствору в течение 10 минут. Смесь перемешивали при -78°C в течение 20 минут. К смеси по каплям добавляли триизопропилборат (5,26 мл) в течение пяти минут. Смесь перемешивали с подогревом до 20°C в течение 1,5 часа. К реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Водный слой нейтрализовали лимонной кислотой. Водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией, а затем фильтрат концентрировали при пониженном давлении. Остаток растирали с добавлением MTBE. Осажденное твердое вещество собирали при помощи фильтрации. Твердое вещество называли первой партией. Фильтрат концентрировали при пониженном давлении. Остаток растирали с добавлением MTBE. Осажденное указанное в заголовке соединение (551 мг) собирали с помощью фильтрации. Первую партию суспендировали в этилацетате. Растирание осуществляли с добавлением небольшого количества MTBE. Осажденное указанное в заголовке соединение (553,3 мг) собирали с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Остаток растирали с добавлением MTBE. Осажденное указанное в заголовке соединение (121,1 мг) собирали с помощью фильтрации. Получали объединенное указанное в заголовке соединение (1,23 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,19-2,20 (м, 3H), 2,23-2,24 (м, 3H), 3,91 (с, 3H), 4,94 (ушир.с, 2H), 7,74 (с, 1H).
ESI-MS m/z 182 [M+H]+
[0248] Пример получения 30
Синтез 3-бром-6-(дифторметил)-2,4-диметилпиридина
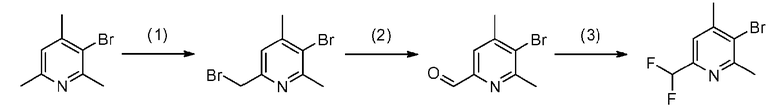
[0249] (1) Синтез 3-бром-6-(бромметил)-2,4-диметилпиридина
Смесь 3-бром-2,4,6-триметилпиридина (15,6 г), NBS (13,9 г) и перекиси бензоила (567 мг) нагревали с обратным холодильником в растворе четыреххлористого углерода (300 мл) в течение двух часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали и остаток на фильтре промывали четыреххлористым углеродом. Полученный фильтрат концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (8,00 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,39-2,42 (м, 3H), 2,66-2,69 (м, 3H), 4,44 (с, 2H), 7,15 (с, 1H).
[0250] (2) Синтез 5-бром-4,6-диметилпиколинальдегида
Метоксид натрия (1,16 г) добавляли к раствору 2-нитропропана (1,96 мл) в метаноле (40 мл) при комнатной температуре и смесь перемешивали при такой же температуре в течение 20 минут. К реакционной смеси добавляли 3-бром-6-(бромметил)-2,4-диметилпиридин (2,00 г) и смесь перемешивали при 50°C в течение пяти часов. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли воду с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-50%) с получением указанного в заголовке соединения (565 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,42-2,55 (м, 3H), 2,72-2,85 (м, 3H), 7,60-7,70 (м, 1H), 10,00 (с, 1H).
[0251] (3) Синтез 3-бром-6-(дифторметил)-2,4-диметилпиридина
BAST (1,07 мл) добавляли к раствору 5-бром-4,6-диметилпиколинальдегида (565 мг) в DCM (10 мл) при 0°C и смесь перемешивали при постепенном подогреве до комнатной температуры в течение 12 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией при помощи DCM. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-50%) с получением указанного в заголовке соединения (415 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,47 (с, 3H), 2,71 (с, 3H), 6,39-6,70 (м, 1H), 7,33 (с, 1H).
[0252] Пример получения 31
Синтез 3-бром-(6-фторметил)-2-метокси-4-метилпиридина
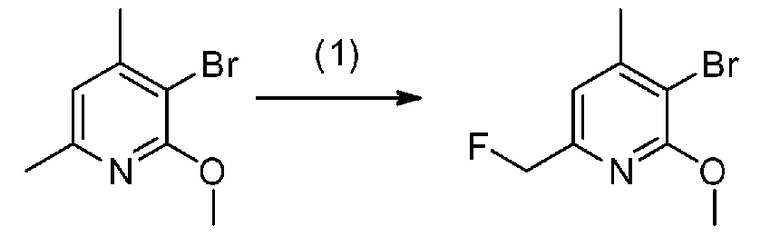
[0253] (1) Синтез 3-бром-(6-фторметил)-2-метокси-4-метилпиридина
Смесь 3-бром-2-метокси-4,6-диметилпиридина, полученного в примере получения 26(2) (300 мг), NBS (247 мг) и перекиси бензоила (10,1 мг) нагревали с обратным холодильником в растворителе, представляющем собой четыреххлористый углерод (6 мл), в течение двух часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали. Полученный фильтрат концентрировали при пониженном давлении. Остаток растворяли в TBAF (5,55 мл, 1M раствор в THF) и смесь перемешивали при комнатной температуре в течение двух часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-5%) и затем при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 0%-5%) с получением указанного в заголовке соединения (136 мг).
ESI-MS m/z 234 [M+H]+
[0254] Пример получения 32
Синтез 3-бром-6-(фторметил)-2,4-диметилпиридина
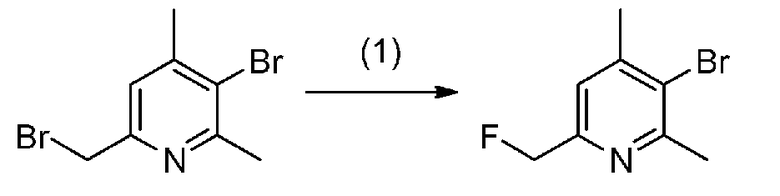
[0255] (1) Синтез 3-бром-6-(фторметил)-2,4-диметилпиридина
Смесь 3-бром-6-(бромметил)-2,4-диметилпиридина, полученного в примере получения 30(1) (2,00 г), и TBAF (35,8 мл, 1M раствор в THF) перемешивали при комнатной температуре в течение двух часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-50%) с получением указанного в заголовке соединения (572 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,44 (с, 3H), 2,67 (с, 3H), 5,28-5,47 (м, 2H), 7,14-7,19 (м, 1H).
[0256] Пример получения 33
Синтез 3-бром-2-(фторметил)-4,6-диметилпиридина
[0257] (1) Синтез 3-бром-2-(бромметил)-4,6-диметилпиридина
Смесь 3-бром-2,4,6-триметилпиридина (15,6 г), NBS (13,9 г) и перекиси бензоила (567 мг) нагревали с обратным холодильником в растворителе, представляющем собой четыреххлористый углерод (300 мл) в течение двух часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали и остаток на фильтре промывали четыреххлористым углеродом. Полученный фильтрат концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (3,51 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,37-2,41 (м, 3H), 2,47 (с, 3H), 4,72 (с, 2H), 6,97 (с, 1H).
[0258] (2) Синтез 3-бром-2-(фторметил)-4,6-диметилпиридина
Смесь 3-бром-2-(бромметил)-4,6-диметилпиридина (1,00 г) и TBAF (17,9 мл, 1M раствор в THF) перемешивали при комнатной температуре в течение двух часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-30%) с получением указанного в заголовке соединения (651 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,40 (с, 3H), 2,51 (с, 3H), 5,49-5,67 (м, 2H), 7,05 (с, 1H).
[0259] Пример получения 34
Синтез 3-бром-2-(дифторметил)-4,6-диметилпиридина
[0260] (1) Синтез 3-бром-4,6-диметилпиколинальдегида
Метоксид натрия (581 мг) добавляли к раствору 2-нитропропана (0,982 мл) в метаноле (20 мл) при комнатной температуре и смесь перемешивали при такой же температуре в течение 20 минут. К реакционной смеси добавляли 3-бром-2-(бромметил)-4,6-диметилпиридин, полученный в примере получения 33(1) (1,00 г), и смесь перемешивали при 50°C в течение пяти часов. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли воду с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-50%) с получением указанного в заголовке соединения (467 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,45-2,48 (м, 3H), 2,58 (с, 3H), 7,23-7,25 (м, 1H), 10,32 (с, 1H).
[0261] (2) Синтез 3-бром-2-(дифторметил)-4,6-диметилпиридина
BAST (0,884 мл) добавляли к раствору 3-бром-4,6-диметилпиколинальдегида (467 мг) в DCM (10 мл) при 0°C и смесь перемешивали при постепенном подогреве до комнатной температуры в течение 12 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией при помощи DCM. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-50%) с получением указанного в заголовке соединения (362 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,43 (с, 3H), 2,54 (с, 3H), 6,81-7,10 (м, 1H), 7,16 (с, 1H).
[0262] Пример получения 35
Синтез 3-бром-2-(фторметил)-6-метокси-4-метилпиридина
[0263] (1) Синтез 3-бром-2-(бромметил)-6-метокси-4-метилпиридина
Смесь 3-бром-6-метокси-2,4-диметилпиридина, полученного в примере получения 22 (200 мг), NBS (165 мг) и перекиси бензоила (6,73 мг) нагревали с обратным холодильником в растворителе, представляющем собой четыреххлористый углерод (4 мл), в течение двух часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали. Полученный фильтрат концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-5%) с получением указанного в заголовке соединения (126 мг).
ESI-MS m/z 296 [M+H]+
[0264] (2) Синтез 3-бром-2-(фторметил)-6-метокси-4-метилпиридина
Смесь 3-бром-2-(бромметил)-6-метокси-4-метилпиридина (126 мг) и TBAF (1,71 мл, 1M раствора в THF) перемешивали при комнатной температуре в течение двух часов. Реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (37 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,35-2,42 (м, 3H), 3,89-3,97 (м, 3H), 5,42-5,59 (м, 2H), 6,65 (с, 1H).
ESI-MS m/z 234 [M+H]+
[0265] Пример получения 36
Синтез 3-бром-4-(фторметил)-6-метокси-2-метилпиридина
[0266] (1) Синтез (2-хлор-6-метилпиридин-4-ил)метанола
Комплекс боргидрид-ТГФ (16,5 мл, 1,06M раствор в THF) добавляли к раствору 2-хлор-6-метилпиридин-4-карбоновой кислоты (2 г) в THF (10 мл) и смесь нагревали с обратным холодильником в течение 12 часов. К реакционной смеси добавляли 5M соляную кислоту и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь нейтрализовали путем добавления насыщенного водного раствора бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-50%) с получением указанного в заголовке соединения (1,75 г).
ESI-MS m/z 158 [M+H]+
[0267] (2) Синтез (2-метокси-6-метилпиридин-4-ил)метанола
Метоксид натрия (11,3 мл, 28% раствор в метаноле) добавляли к раствору (2-хлор-6-метилпиридин-4-ил)метанола (1,75 г) в DMF (18 мл) и смесь перемешивали при 80°C в течение 12 часов. После этого реакционную смесь перемешивали при 120°C в течение семи часов. Реакционную смесь концентрировали при пониженном давлении и к остатку добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-70%) с получением указанного в заголовке соединения (1,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,76 (т, J=6,1 Гц, 1H), 2,45 (с, 3H), 3,92 (с, 3H), 4,64 (д, J=6,1 Гц, 2H), 6,50-6,56 (м, 1H), 6,68-6,73 (м, 1H).
[0268] (3) Синтез (3-бром-6-метокси-2-метилпиридин-4-ил)метанола
Смесь (2-метокси-6-метилпиридин-4-ил)метанола (1,1 г) и NBS (1,34 г) перемешивали в уксусной кислоте-растворителе (22 мл) при комнатной температуре в течение 12 часов. К реакционной смеси добавляли 5M водный раствор гидроксида натрия с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-50%) с получением указанного в заголовке соединения (1,32 г).
ESI-MS m/z 234 [M+H]+
[0269] (4) Синтез 3-бром-4-(фторметил)-6-метокси-2-метилпиридина
BAST (0,89 мл) добавляли к раствору (3-бром-6-метокси-2-метилпиридин-4-ил)метанола (800 мг) в DCM (16 мл) при -60°C и смесь перемешивали при постепенном подогреве до комнатной температуры в течение двух часов и перемешивали при комнатной температуре в течение еще одного часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией при помощи DCM. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%, затем NH-силикагель, этилацетат/н-гептан, 0%-5%) с получением указанного в заголовке соединения (632 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,57 (с, 3H), 3,91 (с, 3H), 5,29-5,47 (м, 2H), 6,70 (с, 1H).
ESI-MS m/z 234 [M+H]+
[0270] Пример получения 37
Синтез 3-бром-5-хлор-2-метокси-4-метилпиридина
[0271] (1) Синтез 3-бром-2-хлор-4-метилпиридина
3-амино-2-хлор-4-метилпиридин (2 г) добавляли к смешанному растворителю из 48% водного раствора бромоводорода (17 мл) и воды (12 мл). К раствору добавляли нитрит натрия (2,5 г) при 0°C. Дополнительно добавляли бром (2,2 мл). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь разделяли путем добавления 5н водного раствора гидроксида натрия и этилацетата. Органический слой промывали солевым раствором и затем сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,7 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,51 (с, 3H), 7,01-7,24 (м, 1H), 8,06-8,35 (м, 1H).
[0272] (2) Синтез 3-бром-2-метокси-4-метилпиридина
3-Бром-2-хлор-4-метилпиридин (1 г) добавляли к DMF (5,6 мл). К раствору добавляли метоксид натрия (28% раствор в метаноле, 4,6 мл) и смесь перемешивали при 100°C в течение 12 часов. Реакционную смесь разделяли путем добавления этилацетата и воды. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (1,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,40 (с, 3H), 4,00 (с, 3H), 6,77 (д, J=5,1 Гц, 1H), 7,94 (д, J=5,1 Гц, 1H).
[0273] (3) Синтез 3-бром-5-хлор-2-метокси-4-метилпиридина
3-Бром-2-метокси-4-метилпиридин (100 мг) добавляли к DMF (575 мкл). К раствору добавляли NCS (72,5 мг) и смесь перемешивали при 80°C в течение трех часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (100 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,51 (с, 3H), 3,98 (с, 3H), 8,02 (с, 1H).
[0274] Пример получения 38
Синтез 3-бром-6-фтор-2,4-диметилпиридина
[0275] 2-Амино-5-бром-4,6-диметилпиридин (2 г) суспендировали во фтороборной кислоте (48% водный раствор, 7,5 мл). К раствору добавляли нитрит натрия (890 мг), растворенный в воде (3 мл), при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 минут. Осажденное твердое вещество собирали путем фильтрации и суспендировали в н-гептане (100 мл). Раствор перемешивали с нагреванием с обратным холодильником в течение двух часов. После охлаждения до комнатной температуры осажденное твердое вещество собирали путем фильтрации. Полученное твердое вещество сушили при пониженном давлении с получением указанного в заголовке соединения (500 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,43 (с, 3H), 2,62 (с, 3H), 6,67 (с, 1H).
[0276] Пример получения 39
Синтез 3-бром-4-хлор-2,6-диметилпиридина
[0277] (1) Синтез 4-хлор-2,6-диметилпиридина
2,6-Диметил-4-гидроксипиридин (1 г) добавляли к хлористому фосфорилу (5 мл). Раствор перемешивали при 100°C в течение шести часов. Реакционную смесь разделяли путем добавления воды, 5н водного раствора гидроксида натрия и этилацетата. Органический слой промывали солевым раствором и затем сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,15 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,51 (с, 6H), 6,99 (с, 2H).
[0278] (2) Синтез 3-бром-4-хлор-2,6-диметилпиридина
4-Хлор-2,6-диметилпиридин (1,5 г) добавляли к смешанному растворителю из трифторуксусной кислоты (3 мл) и концентрированной серной кислоты (6 мл). К раствору добавляли NBS (2,2 г) и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционной смеси добавляли 5н водный раствор гидроксида натрия с последующим разделением при помощи этилацетата. Органический слой промывали солевым раствором и затем сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (500 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,46 (с, 3H), 2,49 (с, 3H), 7,11 (с, 1H).
ESI-MS m/z 222 [M+H]+
[0279] Пример получения 40
Синтез 3-бром-5-хлор-2-метокси-4,6-диметилпиридина
[0280] (1) Синтез 2-метокси-4,6-диметилпиридина
2-Хлор-4,6-диметилпиридин (CAS №: 30838-93-8) (400 мг) добавляли к DMF (3,3 мл). К раствору добавляли метоксид натрия (28% раствор в метаноле, 2,6 мл) и смесь перемешивали при 100°C в течение 12 часов. Реакционную смесь разделяли путем добавления этилацетата и воды. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (380 мг) в виде 50% раствора в DMF.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,24 (с, 3H), 2,40 (с, 3H), 3,89 (с, 3H), 6,35 (с, 1H), 6,56 (с, 1H).
[0281] (2) Синтез 3-бром-5-хлор-2-метокси-4,6-диметилпиридина
2-Метокси-4,6-диметилпиридин (380 мг) добавляли к DMF (3 мл). К раствору добавляли NCS (407 мг) и смесь перемешивали при 80°C в течение одного часа. После этого к раствору добавляли NBS (542 мг) с последующим перемешиванием в течение одного часа. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (600 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,50 (с, 3H), 2,51 (с, 3H), 3,97 (с, 3H).
ESI-MS m/z 252 [M+H]+
[0282] Пример получения 41
Синтез 3-бром-5-фтор-2-метокси-4-метилпиридина
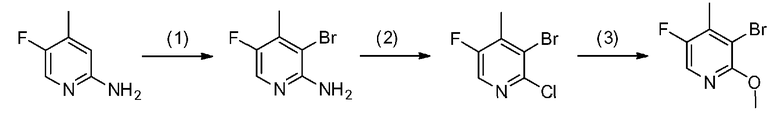
[0283] (1) Синтез 3-бром-5-фтор-4-метилпиридил-2-амина
К ацетонитрилу (14 мл) добавляли 5-фтор-4-метилпиридил-2-амин (2 г). К раствору добавляли NBS (3,1 г). Реакционную смесь перемешивали при комнатной температуре в течение пяти часов. Реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (2,4 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,33 (с, 3H), 4,82 (ушир.с, 2H), 7,84 (с, 1H).
ESI-MS m/z 207 [M+H]+
[0284] (2) Синтез 3-бром-2-хлор-5-фтор-4-метилпиридина
3-Бром-5-фтор-4-метилпиридил-2-амин (2,4 г) добавляли к смешанному растворителю из концентрированной соляной кислоты (11 мл) и воды (11 мл). К раствору добавляли нитрит натрия (2,1 г) и хлорид меди(I) (3,5 г) и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционной смеси добавляли 5н водный раствор гидроксида натрия и этилацетата и нерастворимое вещество удаляли путем фильтрации через стеклянный фильтр. Отделяли фильтрат. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (340 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,44 (с, 3H), 8,16 (с, 1H).
ESI-MS m/z 226 [M+H]+
[0285] (3) Синтез 3-бром-5-фтор-2-метокси-4-метилпиридина
3-Бром-2-хлор-5-фтор-4-метилпиридин (340 мг) добавляли к DMF (1,8 мл). К раствору добавляли метоксид натрия (28% раствор в метаноле, 5,4 мл) и смесь перемешивали при 80°C в течение двух часов. К реакционной смеси добавляли воду. Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (240 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,38 (с, 3H), 3,92 (с, 3H), 7,86 (с, 1H).
ESI-MS m/z 222 [M+H]+
[0286] Пример получения 42
Синтез 5-бром-4,6-диметилпиколинонитрила
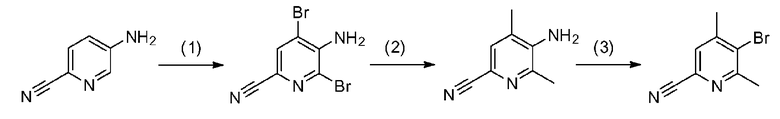
[0287] (1) Синтез 5-амино-4,6-дибромпиколинонитрила
К 48% водному раствору бромоводорода (14 мл) добавляли 5-амино-2-цианопиридин (2 г). К раствору добавляли бром (2,2 мл) при 0°C. Реакционную смесь подогревали до комнатной температуры и перемешивали в течение шести часов. Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (4,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 5,09 (ушир.с, 2H), 7,69 (с, 1H).
ESI-MS m/z 278 [M+H]+
[0288] (2) Синтез 5-амино-4,6-диметилпиколинонитрила
4,6-Дибром-5-амино-2-цианопиридин (1 г) растворяли в смешанном растворителе из 1,4-диоксана (10 мл) и воды (1 мл). К раствору добавляли триметилбороксин (1,3 г), комплекс Pd(dppf)Cl2-DCM (264 мг) и карбонат калия (1,5 мг) и в смеси проводили реакцию с помощью микроволнового реактора при 140°C в течение четырех часов. Реакционной смеси давали остыть до комнатной температуры и затем разделяли путем добавления этилацетата и воды. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-100%) с получением указанного в заголовке соединения (390 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,18 (с, 3H), 2,44 (с, 3H), 4,05 (ушир.с, 2H), 7,28 (с, 1H).
[0289] (3) Синтез 5-бром-4,6-диметилпиколинонитрила
К водному бромоводороду (2,9 мл) добавляли 5-амино-4,6-диметилпиколинонитрил (390 мг). К раствору добавляли бром (164 мкл) и нитрит натрия (467 мг) при 0°C. Раствор подогревали до комнатной температуры и перемешивали в течение четырех часов. К реакционной смеси добавляли 5н водный раствор гидроксида натрия с последующим разделением при помощи этилацетата. Органический слой промывали солевым раствором и затем сушили с помощью сульфата магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-30%) с получением указанного в заголовке соединения (300 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,47 (с, 3H), 2,72 (с, 3H), 7,40 (с, 1H).
ESI-MS m/z 213 [M+H]+
[0290] Пример получения 43
Синтез 3-бром-6-(дифторметокси)-2,4-диметилпиридина
[0291] (1) Синтез 3-бром-6-(дифторметокси)-2,4-диметилпиридина
Смесь 5-бром-4,6-диметилпиридин-2-ола, полученного в примере получения 23(1) (500 мг), 2-(фторсульфонил)дифторуксусной кислоты (0,307 мл) и сульфат натрия (70,3 мг) перемешивали в ацетонитрильном растворителе (10 мл) при комнатной температуре в течение 3,5 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и затем смесь концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом, а органический слой концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 0%-10%) с получением указанного в заголовке соединения (68,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,38-2,41 (м, 3H), 2,57-2,60 (м, 3H), 6,61-6,64 (м, 1H), 7,25-7,63 (м, 1H).
ESI-MS m/z 252 [M+H]+
[0292] Пример получения 44
Синтез 3-бром-2-этокси-4-метилпиридина
3-Бром-2-хлор-4-метилпиридин, полученный в примере получения 37(1) (1 г), добавляли к смешанному растворителю из этанола (2 мл) и DMF (5,6 мл). К раствору добавляли гидрид натрия (60% масляная дисперсия, 58 мг) и смесь перемешивали при 100°C в течение пяти часов. Реакционную смесь разделяли путем добавления этилацетата и воды. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%-30%) с получением указанного в заголовке соединения (40% раствор в н-гептане, 250 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43 (т, J=7,0 Гц, 3H), 2,39 (с, 3H), 4,41 (кв, J=7,0 Гц, 2H), 6,57-6,88 (м, 1H), 7,80-8,04 (м, 1H).
[0293] Пример получения 45
Синтез 2-(дифторметокси)-4-иод-3,5-диметилпиридина
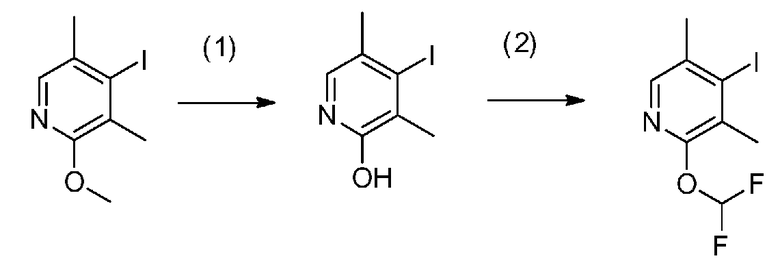
[0294] (1) Синтез 4-иод-3,5-диметилпиридин-2-ола
4-Иод-2-метокси-3,5-метилпиридин, полученный в примере получения 29(3) (3 г), и иодид натрия (4,27 г) добавляли к ацетонитрилу (132 мл) и смесь перемешивали при комнатной температуре в течение одного часа. К смешанному раствору добавляли хлортриметилсилан (3,61 мл) и смесь перемешивали при комнатной температуре в течение 30 минут и затем при 70°C в течение пяти часов. Реакционную смесь охлаждали до комнатной температуры, а затем добавляли воду и хлороформ. Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (2,33 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 2,10 (с, 3H), 2,20 (с, 3H), 7,15 (с, 1H), 11,59 (ушир.с, 1H).
ESI-MS m/z 250 [M+H]+
[0295] (2) Синтез 2-(дифторметокси)-4-иод-3,5-диметилпиридина
К ацетонитрилу (5,7 мл) добавляли 4-иод-3,5-диметилпиридин-2-ол (350 мг), 2-(фторсульфонил)дифторуксусную кислоту (0,17 мл) и сульфат натрия (39,9 мг). Смесь перемешивали при комнатной температуре в течение 3,5 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (378,6 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,39 (с, 3H), 2,43 (с, 3H), 7,42 (т, J=72,0 Гц, 1H), 7,79 (с, 1H).
ESI-MS m/z 300 [M+H]+
[0296] Пример получения 46
Синтез 2-этокси-4-иод-3,5-диметилпиридина
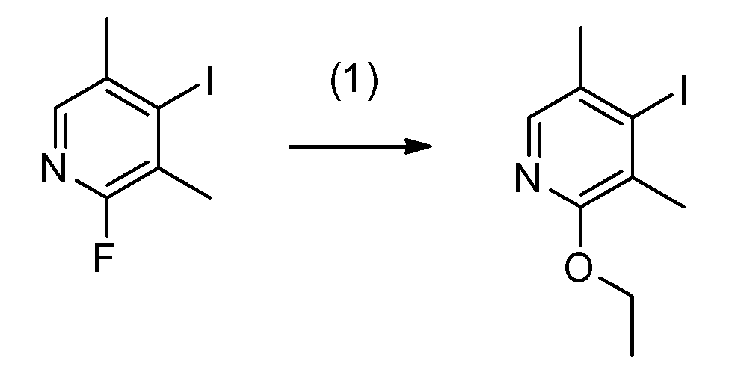
[0297] 20% Раствор этоксида натрия в этаноле (1,23 мл) добавляли к раствору 2-фтор-4-иод-3,5-диметилпиридина, полученного в примере получения 29(2) (400 мг), в THF (5 мл) и смешанный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали при 0°C, а затем добавляли MTBE (20 мл) и воду (20 мл). Отделяли органический слой. Органический слой промывали солевым раствором. Объединенные водные слои экстрагировали посредством MTBE. Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (423,8 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38 (т, J=7,0 Гц, 3H), 2,32 (с, 3H), 2,37 (с, 3H), 4,33 (кв, J=7,0 Гц, 2H), 7,74 (с, 1H).
ESI-MS m/z 278 [M+H]+
[0298] Пример получения 47
Синтез 4-иод-2-изопропилокси-3,5-диметилпиридина
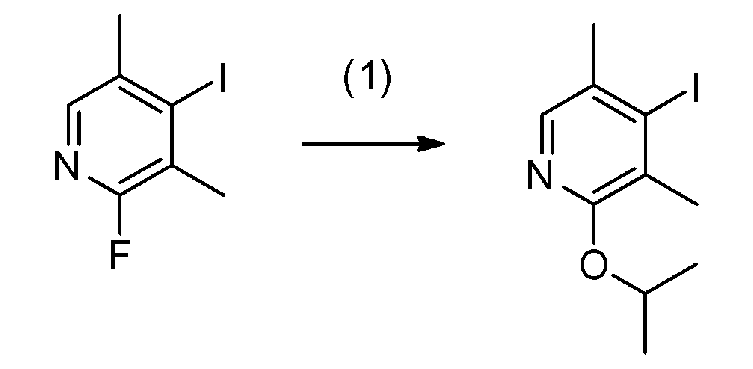
[0299] Гидрид натрия (60% масляную дисперсию, 191 мг) добавляли к раствору IPA (0,77 мл) в THF (5 мл). После прекращения образования пены к раствору добавляли раствор 2-фтор-4-иод-3,5-диметилпиридина, полученного в примере получения 29 (500 мг), в THF (5 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Смесь перемешивали при 50°C в течение двух часов, а затем реакционную смесь охлаждали до комнатной температуры. Реакционную смесь охлаждали при 0°C, а затем добавляли MTBE (20 мл) и воду (20 мл). Отделяли органический слой. Органический слой промывали солевым раствором. Объединенные водные слои экстрагировали посредством MTBE. Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (490 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,33 (д, J=6,3 Гц, 6H), 2,31-2,32 (м, 3H), 2,34-2,35 (м, 3H), 5,21-5,27 (м, 1H), 7,73-7,75 (м, 1H).
ESI-MS m/z 292 [M+H]+
[0300] Пример получения 48
Синтез 3-бром-6-изопропилокси-2,4-диметилпиридина
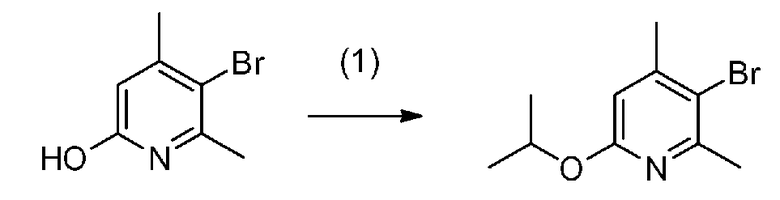
[0301] KTB (222 мг) добавляли к суспензии 5-бром-4,6-диметилпиридин-2-ола, полученного в примере получения 23(1) (400 мг) в DME (2 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли карбонат калия (192 мг) и 2-иодпропан (572 мг). Смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры, а нерастворимое вещество удаляли путем фильтрации и промывки посредством DME. Фильтрат концентрировали при пониженном давлении. К остатку добавляли хлороформ. Раствор промывали 0,1н водным раствором соляной кислоты. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-50%) с получением указанного в заголовке соединения (133,9 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,31 (д, J=6,25 Гц, 6H), 2,32 (с, 3H), 2,25 (с, 3H), 5,17-5,27 (м, 1H), 6,37-6,46 (м, 1H).
ESI-MS m/z 244 [M+H]+
[0302] Пример получения 49
Синтез 3-этил-4-иод-2-метокси-5-метилпиридина
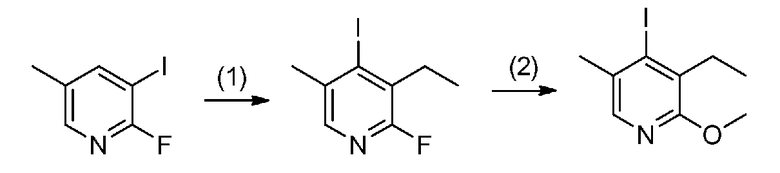
[0303] (1) Синтез 3-этил-2-фтор-4-иод-5-метилпиридина
Указанное в заголовке соединение синтезировали согласно примерам получения 29(2) и 29(3) с применением 2-фтор-3-иод-5-метилпиридина и этилиодида в качестве сырьевых материалов. Тем не менее, после добавления этилиодида температуру постепенно повышали до -17°C.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,11-1,22 (м, 3H), 2,35-2,45 (м, 3H), 2,80-2,91 (м, 2H), 7,81 (с, 1H).
[0304] (2) Синтез 3-этил-4-иод-2-метокси-5-метилпиридина
Указанное в заголовке соединение синтезировали согласно примеру получения 29(3) с применением 3-этил-2-фтор-4-иод-5-метилпиридина.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,04-1,13 (м, 3H), 2,29-2,37 (м, 3H), 2,83 (кв, J=7,42 Гц, 2H), 3,89-3,93 (м, 3H), 7,76 (с, 1H).
[0305] Пример получения 50
Синтез 4-(4-бром-3,5-диметилфенил)-3,6-дигидро-2H-пирана
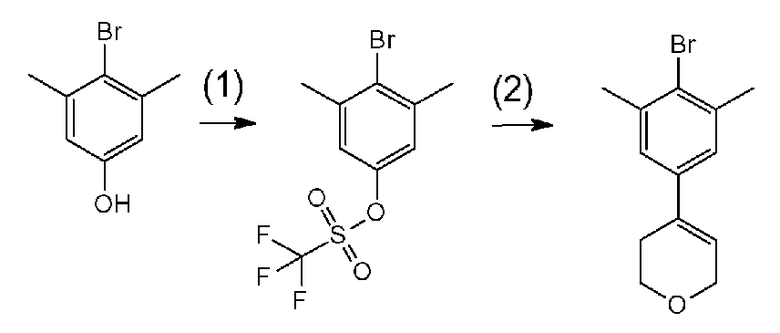
[0306] (1) Синтез трифторметансульфоната 4-бром-3,5-диметилфенила
Трифторметансульфоновый ангидрид (2,0 мл) по каплям добавляли к раствору 4-бром-3,5-диметилфенола (CAS № 7463-51-6) (2,0 г) и TEA (1,94 мл) в DCM (20 мл) с охлаждением на льду в течение трех минут. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли лед и этилацетат и отделяли органический слой. Органический слой последовательно промывали 1н соляной кислотой, водой, насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5%) с получением указанного в заголовке соединения (3,20 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,45 (с, 6H), 7,01 (с, 2H).
[0307] (2) Синтез 4-(4-бром-3,5-диметилфенил)-3,6-дигидро-2H-пирана
Карбонат калия (1,99 г) и комплекс Pd(dppf)Cl2-DCM (196 мг) добавляли к раствору трифторметансульфоната 4-бром-3,5-диметилфенила (1,6 г) и пинаколового эфира 3,6-дигидро-2H-пиран-4-бороновой кислоты (CAS № 287944-16-5) (1,11 г) в DMF (16 мл). Реакционную смесь перемешивали при 85°C в течение четырех часов. Реакционной смеси давали остыть до комнатной температуры, а затем реакционную смесь концентрировали при пониженном давлении. К остатку добавляли MTBE, воду и солевой раствор и отделяли органический слой. Органический слой промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 2%) с получением указанного в заголовке соединения (747 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,42 (с, 6H), 2,45-2,51 (м, 2H), 3,92 (т, J=5,6 Гц, 2H), 4,30 (дд, J=6,0, 2,8 Гц, 2H), 6,08-6,12 (м, 1H), 7,09 (с, 2H).
ESI-MS m/z 267, 269 [M+H]+
[0308] Пример получения 51
Синтез 3-бром-6-этокси-2,4-диметилпиридина
Смесь 5-бром-4,6-диметилпиридин-2-ола, полученная в примере получения 23(1) (50 мг), этилиодида (2,0 мл) и карбоната серебра (1,4 г) перемешивали в растворителе-хлороформе (10 мл) при комнатной температуре в течение 36 часов. Реакционную смесь выливали на силикагелевую подушку и элюировали смешанным растворителем (этилацетат/н-гептан, 10%). Полученный раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения (550 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,36 (т, J=7,0 Гц, 3H), 2,33 (с, 3H), 2,56 (с, 3H), 4,27 (кв, J=7,0 Гц, 2H), 6,44 (с, 1H).
ESI-MS m/z 232 [M+H]+
[0309] Пример получения 52
Синтез (-)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата и (+)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата
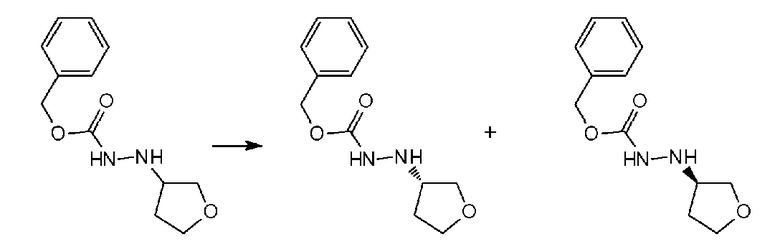
[0310] Насыщенный водный раствор бикарбоната натрия (30 мл) добавляли к раствору (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата, полученному в примере получения 12-(2) (11,5 г), в MTBE (110 мл). Смесь перемешивали в течение 10 минут при комнатной температуре, а затем отделяли органический слой. Полученный органический слой последовательно промывали насыщенным бикарбонатом натрия и солевым раствором, и сушили над безводным сульфатом магния, и осушитель удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/гексан, 25-50%) и концентрировали конечную фракцию. К остатку добавляли простой диэтиловый эфир (30 мл) и гексан (15 мл). Осажденное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением чистого (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата (6,17 г).
Данный продукт растворяли в этаноле и фильтровали через микропористый фильтр. Полученный фильтрат разделяли на оптические изомеры в двух условиях. Условие 1: OD-H (20 мм Φ × 250 мм длина), 20% IPA-гексан, 25 мл/мин. Условие 2: AD-H (20 мм Φ × 250 мм длина), 20% IPA-гексан, 24 мл/мин. Целевую фракцию концентрировали с получением указанного в заголовке соединения с коротким временем удерживания и (-) вращением плоскости поляризации (2,60 г, >99% э.и. [OD-H, 20% IPA/гексан, время удерживания = 11,2 мин]) и указанного в заголовке соединения с длительным временем удерживания и (+) вращением плоскости поляризации (2,59 г, 97,2% э.и. [OD-H, 20% IPA/гексан, время удерживания = 12,4 мин]).
[0311] Пример получения 53
Синтез гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина
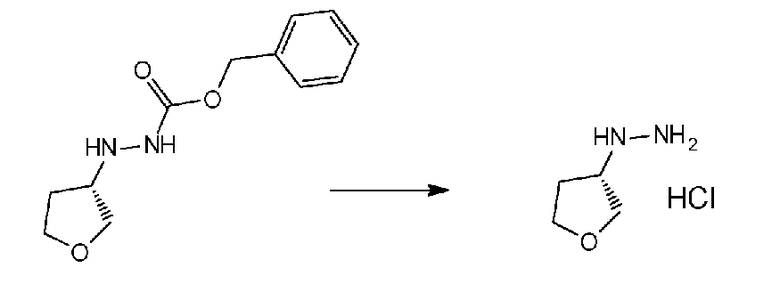
[0312] (-)-Бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (50 г) растворяли в метаноле (500 мл) и добавляли ди-трет-бутилдикарбонат (92,4 г) и палладированный уголь (50% влаги) (5 г). Смесь перемешивали при 25°C и 15 фунт/кв.дюйм в течение 48 часов в атмосфере водорода. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в простом диизопропиловом эфире (300 мл). После охлаждения при 0°C к раствору добавляли соляную кислоту/простой диизопропиловый эфир (500 мл). Смесь перемешивали при 10°C в течение 14 часов. Осажденное твердое вещество собирали при помощи фильтрации. Девять раз проводили такую же процедуру из (-)-бензил-2-(тетрагидрофуран-3-ил)гидразинкарбоксилата (70 г) и один раз проводили такую же процедуру из (-)-бензил-2-(тетрагидрофуран-3-ил)гидразинкарбоксилата (50 г). Полученное твердое вещество растирали с DCM/этанолом (10/1) (1 л) в течение двух часов. Осажденное твердое вещество собирали при помощи фильтрации. Полученное твердое вещество сушили при пониженном давлении с получением указанного в заголовке соединения (235 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,87-2,09 (м, 2H), 3,55-3,71 (м, 2H), 3,71-3,84 (м, 3H).
Как вращение плоскости поляризации Z-производного указанного в заголовке соединения, так и вращение плоскости поляризации Z-производного гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина, полученного в примере получения 14, являются отрицательными. Согласно хиральному ВЭЖХ-анализу время удерживания обоих соединений были идентичными.
По результатам рентгеноструктурного анализа подтверждали, что абсолютная конфигурация полученного указанного в заголовке соединения была (S)-формой. Результат показан на фиг.1 в виде его ORTEP-изображения (параметр Флека = -0,05).
[0313] Пример получения 54
Синтез (R)-(тетрагидрофуран-3-ил)гидразина гидрохлорида
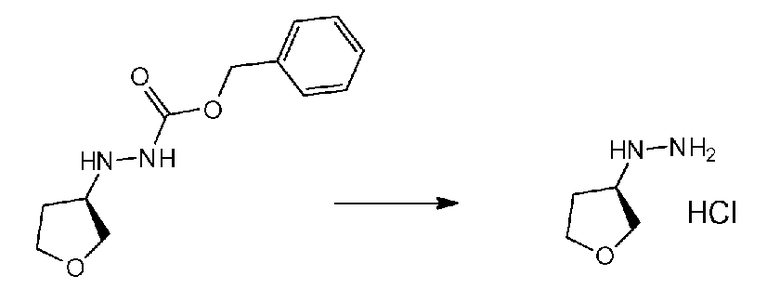
[0314] Указанное в заголовке соединение получали тем же способом, который описан в примере получения 53, из (+)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата.
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 1,85-2,07 (м, 2H), 3,55-3,71 (м, 2H), 3,71-3,80 (м, 3H).
[0315] Пример 1
Синтез 7-(2,6-диметилфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
[0316] 7-Хлор-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он, полученный в примере получения 2 (100 мг), растворяли в DMF (3,3 мл). К раствору добавляли 2,6-диметилфенилбороновую кислоту (33 мг), Pd(PPh3)4 (13 мг), карбонат калия (91 мг) и воду (0,7 мл) и в смеси проводили реакцию с помощью микроволнового реактора при 150°C в течение двух часов. Реакционной смеси давали остыть до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 30%-50%-80%) с получением 5-(2,4-диметоксибензил)-7-(2,6-диметилфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (80 мг). 5-(2,4-Диметоксибензил)-7-(2,6-диметилфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (75 мг) растворяли в TFA (1 мл) и смесь перемешивали при 65°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток нейтрализовали путем добавления насыщенного водного раствора бикарбоната натрия. Водный раствор экстрагировали посредством DCM. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-70%-80%) с получением указанного в заголовке соединения (10 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,06 (с, 6H), 2,18-2,22 (м, 2H), 2,42-2,60 (м, 2H), 3,69-3,78 (м, 2H), 4,19-4,26 (м, 2H), 5,00-5,10 (м, 1H), 7,09-7,26 (м, 5H), 8,03 (д, J=8,4 Гц, 1H), 8,31 (с, 1H), 8,83 (с, 1H).
ESI-MS m/z 374 [M+H]+
[0317] Пример 2
Синтез 7-(2,4,6-триметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
[0318] Указанное в заголовке соединение получали тем же способом, который описан в примере 1, из 7-хлор-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного в примере получения 2, и (2,4,6-триметилпиридин-3-ил)бороновой кислоты, полученной в примере получения 19.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,05 (с, 3H), 2,17-2,21 (м, 2H), 2,29 (с, 3H), 2,48-2,60 (м, 2H), 2,57 (с, 3H), 3,67-3,76 (м, 2H), 4,20-4,28 (м, 2H), 5,01-5,11 (м, 1H), 6,99 (с, 1H), 7,13 (дд, J=8,2 Гц, 1,6 Гц, 1H), 7,27 (д, J=1,6 Гц, 1H), 8,05 (д, J=8,2 Гц, 1H), 8,31 (с, 1H), 10,60 (с, 1H).
ESI-MS m/z 389 [M+H]+
[0319] Пример 3
Синтез 7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
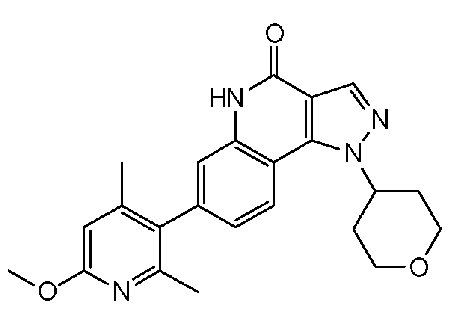
[0320] 7-Бром-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он, полученный в примере получения 1(4) (41 мг), растворяли в DMF (2 мл). К раствору добавляли (6-метокси-2,4-диметилпиридин-3-ил)бороновую кислоту, полученную в примере получения 24 (15 мг), Pd(PPh3)4 (4,8 мг), карбонат цезия (54 мг) и воду (0,5 мл) и в смеси проводили реакцию с помощью микроволнового реактора при 150°C в течение двух часов. Реакционной смеси давали остыть до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток растворяли в TFA (1 мл) и смесь перемешивали при 65°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток нейтрализовали путем добавления 5н водного раствора гидроксида натрия. Водный раствор экстрагировали посредством DCM. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (3,7 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,03 (с, 3H), 2,17-2,21 (м, 2H), 2,22 (с, 3H), 2,48-2,60 (м, 2H), 3,67-3,76 (м, 2H), 3,97 (с, 3H), 4,20-4,28 (м, 2H), 5,01-5,11 (м, 1H), 6,55 (с, 1H), 7,13 (дд, J=8,2 Гц, 1,6 Гц, 1H), 7,19 (д, J=1,6 Гц, 1H), 8,03 (д, J=8,2 Гц, 1H), 8,32 (с, 1H), 10,60 (с, 1H).
ESI-MS m/z 405 [M+H]+
[0321] Пример 4
Синтез 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
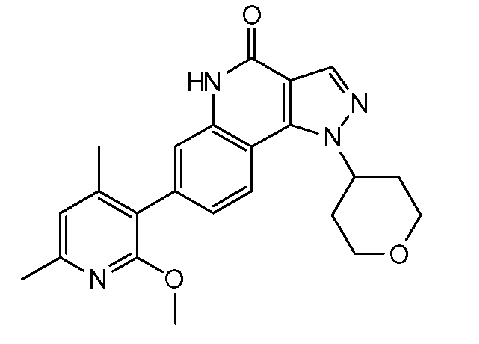
5-(2,4-Диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он, полученный в примере получения 1(5) (100 мг), растворяли в 1,4-диоксане (4 мл). К раствору добавляли 3-хлор-2-метокси-4,6-диметилпиридин, полученный в примере получения 25 (47,2 мг), [(t-Bu)2P(OH)]2PdCl2 (4,6 мг), карбонат цезия (119 мг) и воду (1 мл) и в смеси проводили реакцию с помощью микроволнового реактора при 130°C в течение четырех часов. Реакционную смесь экстрагировали посредством DCM. Органический слой промывали солевым раствором и сушили над сульфатом натрия. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-100%) с получением 1:1 смеси 5-(2,4-диметоксибензил)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и 5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (76 мг). Смесь (76 мг) растворяли в TFA (1,5 мл) и смесь перемешивали при 65°C в течение трех часов. Реакционную смесь концентрировали при пониженном давлении. К полученному остатку добавляли DCM и насыщенный водный раствор бикарбоната натрия с последующей экстракцией при помощи DCM. Органический слой промывали солевым раствором и сушили над сульфатом натрия. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (22 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,10 (с, 3H), 2,15-2,23 (м, 2H), 2,47-2,59 (м, 2H), 2,48 (с, 3H), 3,65-3,73 (м, 2H), 3,86 (с, 3H), 4,20-4,26 (м, 2H), 5,01-5,10 (м, 1H), 6,74 (с, 1H), 7,20 (дд, J=8,4 Гц, 1,6 Гц, 1H), 7,27 (д, J=1,6 Гц, 1H), 8,01 (д, J=8,4 Гц, 1H), 8,30 (с, 1H), 10,01 (с, 1H).
ESI-MS m/z 405 [M+H]+
[0322] Пример 5
Синтез 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
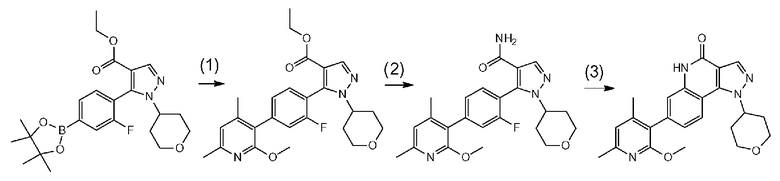
[0323] (1) Синтез этил-5-[2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Воду (5 мл), 3-бром-2-метокси-4,6-диметилпиридин, полученный в примере получения 26 (784 мг), Pd(PPh3)4 (380 мг) и карбонат цезия (2,36 г) добавляли к раствору этил-5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата, полученного в примере получения 3 (2,04 г), в 1,4-диоксане (20 мл) и реакционной смеси давали прореагировать при 110°C в течение двух часов в атмосфере азота. Реакционной смеси давали остыть до комнатной температуры и затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат (100 мл) и воду (100 мл). Водный слой экстрагировали этилацетатом (50 мл × 2). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 10%-23%). Полученное тем же способом указанное в заголовке соединение (578 мг) объединяли и объединенный продукт еще раз очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-70%) с получением указанного в заголовке соединения (2,07 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,11-1,18 (м, 3H), 1,72-1,80 (м, 1H), 1,85-1,92 (м, 1H), 2,14 (с, 3H), 2,30-2,48 (м, 5H), 3,35-3,46 (м, 2H), 3,88 (с, 3H), 4,03-4,18 (м, 5H), 6,71-6,73 (м, 1H), 7,08-7,15 (м, 2H), 7,30-7,35 (м, 1H), 8,09-8,10 (м, 1H).
ESI-MS m/z 454 [M+H]+
[0324] (2) Синтез 5-[2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамида
К этанолу (30 мл) добавляли этил-5-[2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил]-1H-пиразол-4-карбоксилат (2,06 г). После перемешания суспензии при 60°C в течение трех минут добавляли 5н водный раствор гидроксида натрия (3,6 мл) и смесь перемешивали при 60°C-70°C в течение одного часа. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли хлороформ (20 мл) и 5н соляную кислоту (6 мл). Осажденное твердое вещество собирали при помощи фильтрации. К полученному твердому веществу добавляли толуол и смесь концентрировали при пониженном давлении. Полученный остаток растворяли в DMF (15 мл). К раствору добавляли CDI (935 мг) и смесь перемешивали при комнатной температуре в течение одного часа в атмосфере азота. К реакционной смеси добавляли 28% водный раствор аммиака (1,4 мл) и смесь перемешивали при комнатной температуре в течение пяти часов. Реакционную смесь концентрировали при пониженном давлении. Остаток разделяли путем добавления хлороформа (100 мл) и воды (50 мл). Водный слой экстрагировали хлороформом (50 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (50 мл). Смывы экстрагировали хлороформом (5 мл). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток растирали с добавлением MTBE (5 мл). Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (1,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,68-1,78 (м, 1H), 1,86-1,95 (м, 1H), 2,13 (с, 3H), 2,29-2,45 (м, 2H), 2,47 (с, 3H), 3,32-3,47 (м, 2H), 3,69 (с, 3H), 4,00-4,15 (м, 3H), 5,29 (ушир.с, 2H), 6,72 (с, 1H), 7,14-7,26 (м, 2H), 7,37-7,43 (м, 1H), 8,07 (с, 1H).
ESI-MS m/z 447 [M+Na]+
[0325] (3) Синтез 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
KTB (655 мг) добавляли к раствору 5-[2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамида (1,52 г) в NMP (15 мл) и смесь перемешивали при 90°C в течение 30 минут. К реакционной смеси добавляли KTB (40 мг) и смесь перемешивали при 90°C в течение 30 минут. Дополнительно к реакционной смеси добавляли KTB (40 мг) и смесь перемешивали при 90°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры. К реакционной смеси добавляли воду (3 мл). В осадок выпадало твердое вещество. После непрерывного перемешания в течение одного часа осажденное твердое вещество собирали путем фильтрации. Остаток промывали водой (1 мл). Полученное твердое вещество суспендировали в 1-пропаноле/воде (9/1) (2 мл) и растворяли путем нагревания с обратным холодильником. Раствор охлаждали до комнатной температуры в течение одного часа. Осажденное твердое вещество собирали при помощи фильтрации. Полученное твердое вещество сушили при пониженном давлении при 50°C с получением указанного в заголовке соединения (872 мг). Показания приборов были идентичны показаниям для указанного в заголовке соединения, синтезированного в примере 4.
[0326] Пример 6
Синтез 7-(2-метокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
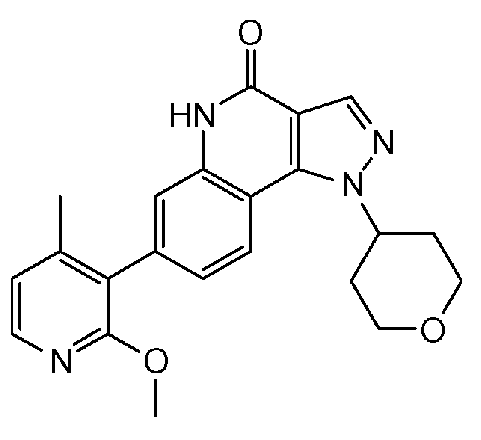
5-(2,4-Диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он, полученный в примере получения 1(5) (100 мг), растворяли в 1,4-диоксане (4 мл). К раствору добавляли 3-бром-2-метокси-4-метилпиридин, полученный в примере получения 37(2) (55,6 мг), Pd(PPh3)4 (10,6 мг), карбонат цезия (179 мг) и воду (1 мл) и в смеси проводили реакцию с помощью микроволнового реактора при 130°C в течение трех часов. Реакционной смеси давали остыть до комнатной температуры и затем разделяли путем добавления этилацетата. Органический слой промывали солевым раствором и затем сушили над сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 30%-50%-80%) с получением 5-(2,4-диметоксибензил)-7-(2-метокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (78 мг). 5-(2,4-Диметоксибензил)-7-(2-метокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (78 мг) растворяли в TFA (1 мл) и смесь перемешивали при 65°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток нейтрализовали путем добавления насыщенного водного раствора бикарбоната натрия. Водный раствор экстрагировали посредством DCM. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-70%-80%-100%) с получением указанного в заголовке соединения (30 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,15 (с, 3H), 2,15-2,24 (м, 2H), 2,45-2,59 (м, 2H), 3,70 (т, J=12,0 Гц, 2H), 3,87 (с, 3H), 4,20-4,27 (м, 2H), 5,02-5,11 (м, 1H), 6,89 (д, J=5,1 Гц, 1H), 7,21 (дд, J=8,2 Гц, 1,6 Гц, 1H), 7,34 (д, J=1,6 Гц, 1H), 8,03 (д, J=8,6 Гц, 1H), 8,11 (д, J=5,1 Гц, 1H), 8,31 (с, 1H), 10,57 (ушир.с, 1H).
ESI-MS m/z 391 [M+H]+
[0327] Соединения примеров 7-22 синтезировали как в примере 6.
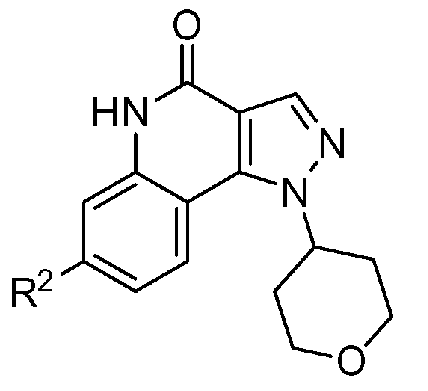
ESI-MS m/z 418 [M+H]+
ESI-MS m/z 393 [M+H]+
ESI-MS m/z 409 [M+H]+
ESI-MS m/z 425 [M+H]+
ESI-MS m/z 439 [M+H]+
ESI-MS m/z 409 [M+H]+
ESI-MS m/z 400 [M+H]+
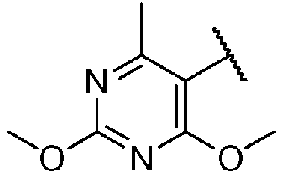
ESI-MS m/z 422 [M+H]+
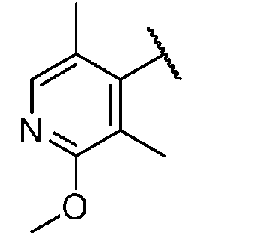
ESI-MS m/z 405 [M+H]+
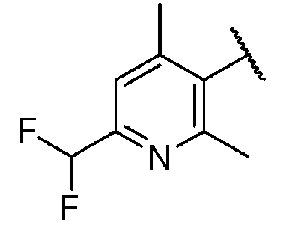
ESI-MS m/z 425 [M+H]+
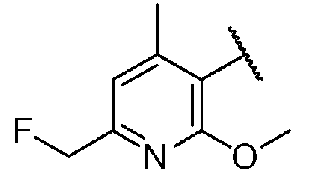
ESI-MS m/z 423 [M+H]+
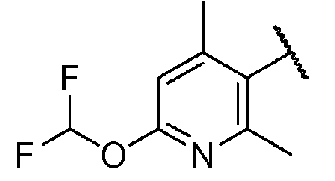
ESI-MS m/z 441 [M+H]+
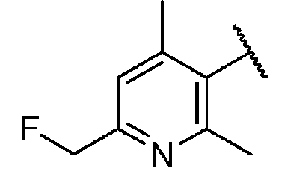
ESI-MS m/z 407 [M+H]+
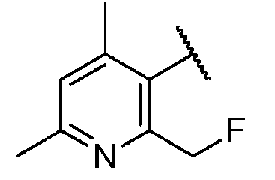
ESI-MS m/z 407 [M+H]+
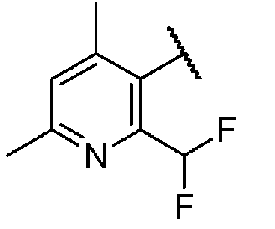
ESI-MS m/z 425 [M+H]+
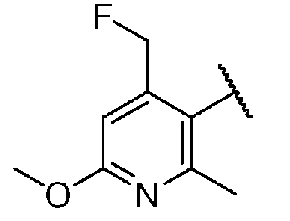
ESI-MS m/z 423 [M+H]+
[0328] Пример 23
Синтез 7-(2-этокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
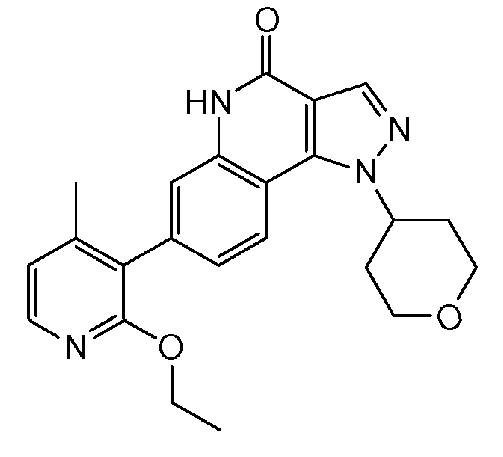
[5-(2,4-диметоксибензил)-4-оксо-1-(тетрагидро-2H-пиран-4-ил)-4,5-дигидро-1H-пиразоло[4,3-c]хинолин-7-ил]бороновую кислоту, полученную в примере получения 1 (70 мг), растворяли в 1,4-диоксане (4 мл). К раствору добавляли 3-бром-2-этокси-4-метилпиридин, полученный в примере получения 44 (49 мг), Pd(PPh3)4 (8,7 мг), карбонат цезия (148 мг) и воду (1 мл) и в смеси проводили реакцию с помощью микроволнового реактора при 130°C в течение двух часов. Реакционной смеси давали остыть до комнатной температуры и затем разделяли путем добавления этилацетата. Органический слой промывали солевым раствором и затем сушили над сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 30%-50%-80%) с получением 5-(2,4-диметоксибензил)-7-(2-этокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (55 мг). В TFA (1 мл) растворяли 5-(2,4-диметоксибензил)-7-(2-этокси-4-метилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (55 мг) и смесь перемешивали при 65°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток нейтрализовали путем добавления насыщенного водного раствора бикарбоната натрия. Водный раствор экстрагировали посредством DCM. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли с помощью фильтрации. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-70%-80%-100%) с получением указанного в заголовке соединения (17 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,25 (т, J=7,5 Гц, 3H), 2,15 (с, 3H), 2,15-2,25 (м, 2H), 2,46-2,60 (м, 2H), 3,65-3,77 (м, 2H), 4,20-4,29 (м, 2H), 4,35 (кв, J=7,5 Гц, 2H), 5,03-5,12 (м, 1H), 6,86 (д, J=5,5 Гц, 1H), 7,21 (дд, J=1,6 Гц, 8,2 Гц, 1H), 7,35 (с, 1H), 8,01 (д, J=8,2 Гц, 1H), 8,08 (д, J=5,5 Гц, 1H), 8,30 (с, 1H), 10,51 (ушир.с, 1H).
ESI-MS m/z 405 [M+H]+
[0329] Соединение из примера 24 синтезировали как описано в примере 23.
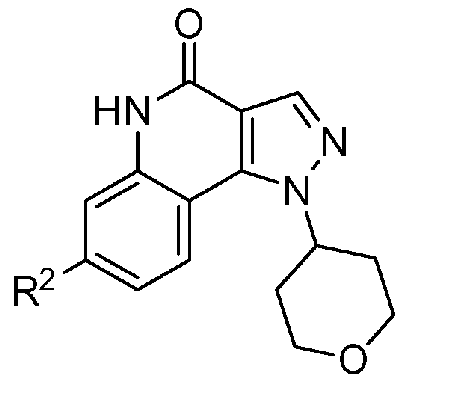
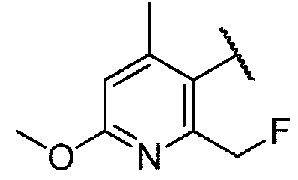
ESI-MS m/z 423 [M+H]+
[0330] Пример 25
Синтез (+)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
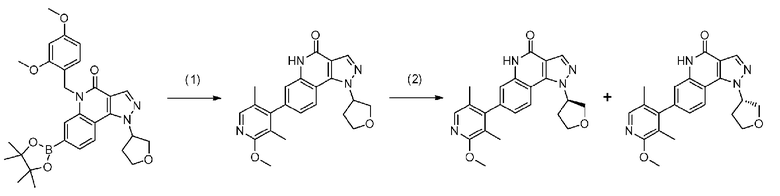
[0331] (1) Синтез (±)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Осуществляли реакцию в смеси из (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного в примере получения 5 (219 мг), 4-бром-2-метокси-3,5-диметилпиридина, полученного в примере получения 28 (134 мг), Pd(PPh3)4 (23,8 мг) и карбоната цезия (403 мг) в смешанном растворителе из 1,4-диоксана (8 мл) и воды (2 мл) с помощью микроволнового реактора при 130°C в течение 70 минут. Реакционную смесь охлаждали до комнатной температуры, а затем сразу очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10%-90%). Полученный продукт реакции сочетания растворяли в TFA (4 мл) и смесь перемешивали при 70°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия с последующей экстракцией этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали при помощи колоночной хроматографии на силикагеле (DCM, 100%, затем этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (78 мг).
ESI-MS m/z 391 [M+H]+
[0332] (2) Синтез (+)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(±)-7-(2-Метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он анализировали с помощью колонки для хиральной хроматографии [AD-H (0,46 см Φ × 15 см), подвижная фаза; 100% этанол] для выявления (+)-формы на 7,8 мин и (-)-формы на 9,7 мин и подтверждения возможности разделения оптических изомеров. (±)-7-(2-Метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (78 мг) растворяли в смешанном растворителе из этанола (12 мл) и метанола (12 мл) и раствор фильтровали через ватную пробку. Фильтрат разделяли по оптическим изомерам с помощью хиральной колоночной хроматографии [колонка для хиральной хроматографии: колонка AD-H, растворитель для элюирования: 100% этанол, скорость потока: 10 мл/мин, время элюирования: 80 минут/элюирование, инъекция: 2 мл/инъекция, короткое время удерживания: (+)-форма, длительное время удерживания: (-)-форма] с получением 26,4 мг (+)-формы и 25,2 мг (-)-формы указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,92-1,94 (м, 3H), 1,94-1,96 (м, 3H), 2,55-2,66 (м, 1H), 2,76-2,86 (м, 1H), 4,00 (с, 3H), 4,09-4,16 (м, 1H), 4,24-4,37 (м, 2H), 4,39-4,45 (м, 1H), 5,61-5,68 (м, 1H), 7,04 (д, J=1,5 Гц, 1H), 7,08 (дд, J=1,5 Гц, 8,3 Гц, 1H), 7,94 (с, 1H), 8,13 (д, J=8,3 Гц, 1H), 8,31 (с, 1H), 8,86 (с, 1H).
ESI-MS m/z 391 [M+H]+
[0333] Пример 26
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
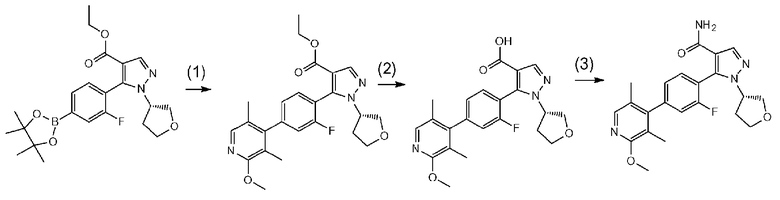

[0334] (1) Синтез этил-5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Воду (170 мл), 4-иод-2-метокси-3,5-диметилпиридин, полученный в примере получения 29(3) (35,6 г), Pd(PPh3)4 (6,52 г) и карбонат цезия (110 г) добавляли к раствору этил-5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата, полученного в примере получения 6 (51,9 г), в 1,4-диоксане (500 мл) и реакционной смеси давали прореагировать при 110°C в течение шести часов. Реакционной смеси давали остыть до комнатной температуры, а затем отделяли органический слой. Органический слой концентрировали при пониженном давлении. К полученному остатку добавляли водный слой, этилацетат (700 мл) и воду (100 мл) и отделяли органический слой. Водный слой повторно экстрагировали этилацетатом (50 мл). Объединенные органические слои последовательно промывали водой и солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 5%-14%). Затем продукт еще раз очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, 2%-10%) с получением указанного в заголовке соединения (43,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,16 (т, J=7,2 Гц, 1,5H), 1,17 (т, J=7,2 Гц, 1,5H), 1,97 (с, 1,5H), 1,98 (с, 1,5H), 1,99 (с, 1,5H), 2,00 (с, 1,5H), 2,25-2,55 (м, 2H), 3,92-4,27 (м, 6H), 3,99 (с, 1,5H), 4,00 (с, 1,5H), 4,65-4,75 (м, 1H), 7,01 (д, J=9,2 Гц, 1H), 7,05 (д, J=7,2 Гц, 1H), 7,39 (т, J=7,2 Гц, 0,5H), 7,45 (т, J=7,2 Гц, 0,5H), 7,93 (с, 1H), 8,12 (с, 1H).
ESI-MS m/z 440 [M+H]+
[0335] (2) Синтез 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоновой кислоты
5н водный раствор гидроксида натрия (79 мл) добавляли к раствору этил 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (43,2 г) в этаноле (574 мл) при комнатной температуре и реакционную смесь перемешивали при 60°C в течение двух часов и 10 минут. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали до половины объема при пониженном давлении. К остатку добавляли воду (300 мл) и этанол отгоняли при пониженном давлении. К полученному остатку добавляли MTBE (130 мл) и отделяли водный слой. Органический слой экстрагировали водой (30 мл). Объединенные водные слои подкисляли при помощи 5н соляной кислоты (78 мл) с охлаждением на льду и дважды экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (39,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,91 (с, 1,5H), 1,94 (с, 1,5H), 1,98 (с, 1,5H), 2,01 (с, 1,5H), 2,25-2,56 (м, 2H), 3,92-4,17 (м, 3H), 3,96 (с, 1,5H), 4,00 (с, 1,5H), 4,23 (дд, J=16,0, 8,0 Гц, 1H), 4,65-4,77 (м, 1H), 6,99 (ушир.д, J=10,0 Гц, 1H), 7,03 (ушир.д, J=7,6 Гц, 1H), 7,38 (т, J=7,6 Гц, 0,5H), 7,44 (т, J=7,6 Гц, 0,5H), 7,90 (с, 0,5H), 7,94 (с, 0,5H), 8,14 (с, 1H).
ESI-MS m/z 434 [M+Na]+
[0336] (3) Синтез 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида
CDI (21,4 г) за один раз добавляли к раствору 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоновой кислоты (38,7 г) в DMF (290 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 95 минут. К реакционной смеси добавляли 28% водный раствор аммиака (95 мл) и смесь перемешивали при комнатной температуре в течение 35 минут. К реакционной смеси еще раз добавляли 28% водный раствор аммиака (95 мл) и смесь перемешивали при комнатной температуре в течение 90 минут. Реакционную смесь концентрировали при пониженном давлении. К полученному остатку добавляли хлороформ (250 мл) и воду (80 мл) и отделяли органический слой. Водный слой повторно экстрагировали хлороформом (50 мл). Объединенные органические слои последовательно промывали насыщенным водным раствором хлорида аммония (60 мл × 3) и солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат пропускали через силикагелевую подушку (NH-силикагель). Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (37,2 г).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98 (ушир.с, 6H), 2,24-2,60 (м, 2H), 3,90-4,20 (м, 3H), 3,99 (с, 3H), 4,23 (дд, J=16,0, 8,0 Гц, 1H), 4,62-4,71 (м, 1H), 5,32 (ушир.с, 2H), 7,05 (ушир.д, J=10,0 Гц, 1H), 7,10 (дд, J=7,6, 1,2 Гц, 1H), 7,42-7,56 (м, 1H), 7,94 (ушир.с, 1H), 8,03 (с, 1H).
ESI-MS m/z 411 [M+H]+
[0337] (4) Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Порошок гидроксида натрия (9,43 г) за один раз добавляли к раствору 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида (37,2 г) в ДМСО (186 мл) при комнатной температуре. Реакционную смесь перемешивали при той же температуре в течение 50 минут, а затем при 70°C в течение 45 минут. В условиях водяного охлаждения к реакционной смеси по каплям добавляли воду (600 мл), а затем по каплям добавляли уксусную кислоту (13,5 мл). Осажденный порошок собирали при помощи фильтрации. Собранную субстанцию промывали водой и MTBE, а затем сушили при пониженном давлении с получением указанного в заголовке соединения (34,0 г).
1H-ЯМР и ESI-MS указанного в заголовке соединения были идентичны приведенным в примере 25. Указанное в заголовке соединение демонстрировало (-) вращение плоскости поляризации и характеризовалось >99% э.и. оптической чистоты [AD-H, 100% этанол, время удерживания: 9,7 мин].
[0338] Пример 27
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
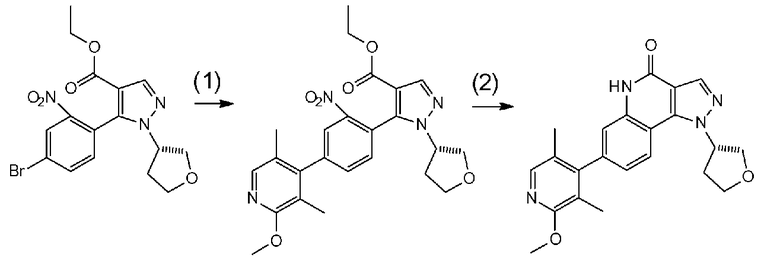
[0339] (1) Синтез этил-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-(4-бром-2-нитрофенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат, полученный в примере получения 7(1) (1,5 г), растворяли в толуоле (50 мл). К раствору добавляли (2-метокси-3,5-диметилпиридин-4-ил)бороновую кислоту, полученную в примере получения 29 (728 мг), бис(трифенилфосфин)дихлорпалладий(II) (128 мг), карбонат натрия (1,16 г) и воду (10 мл) и в смеси проводили реакцию при 100°C в течение четырех часов. После охлаждения реакционной смеси до комнатной температуры добавляли этилацетат (50 мл) и воду (50 мл) и реакционную смесь фильтровали через Celite™. Фильтрат разделяли путем добавления этилацетата (100 мл). Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Этанол (2 мл) добавляли к полученному остатку, который растворяли нагреванием с обратным холодильником. Раствор охлаждали ледяной водой. Через один час осажденное твердое вещество собирали путем фильтрации с получением указанного в заголовке соединения (750 мг). Фильтрат концентрировали при пониженном давлении. Этанол (1 мл) добавляли к полученному остатку, который растворяли нагреванием с обратным холодильником. Раствор охлаждали ледяной водой. Через один час осажденное твердое вещество собирали путем фильтрации с получением указанного в заголовке соединения (450 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,07-1,14 (м, 3H), 1,98 (д, J=3,9 Гц, 3H), 2,01 (д, J=3,9 Гц, 3H), 2,21-2,40 (м, 1H), 2,47-2,58 (м, 1H), 3,92-4,00 (м, 1H), 4,00 (с, 3H), 4,02-4,18 (м, 4H), 4,23 (кв, J=7,7 Гц, 1H), 4,56-4,66 (м, 1H), 7,43 (д, J=8,2 Гц, 0,67H), 7,48 (д, J=8,2 Гц, 0,33H), 7,51-7,56 (м, 1H), 7,96-8,02 (м, 2H), 8,08 (с, 1H).
ESI-MS m/z 467 [M+H]+
[0340] (2) Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Этил-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат (1,1 г) суспендировали в этаноле (13 мл). К раствору добавляли железный порошок (280 мг) и насыщенный водный раствор хлорида аммония (3 мл) и смесь перемешивали при 100°C в течение 3,5 часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали через Celite™. Фильтрат разделяли путем добавления этилацетата (100 мл) и воды (50 мл). Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в уксусной кислоте (2 мл) с последующим перемешиванием при 50°C. Через четыре часа реакционную смесь охлаждали до комнатной температуры и добавляли воду (20 мл). Осажденное твердое вещество собирали при помощи фильтрации. Воду (1,5 мл) и 1-пропанол (10 мл) добавляли к полученному твердому веществу, которое растворяли нагреванием с обратным холодильником. Раствор охлаждали ледяной водой. Через один час осажденное твердое вещество собирали путем фильтрации и промывали посредством MTBE (5 мл) с получением указанного в заголовке соединения (780 мг).
[0341] Соединения примеров 28-32 синтезировали как описано в примере 25.
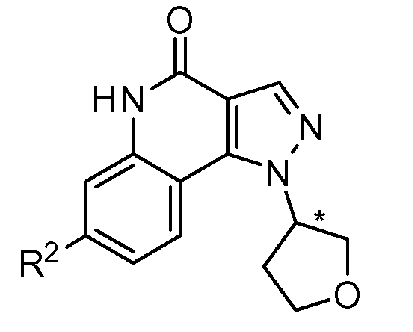
Подвижная фаза
Вращение плоскости поляризации (+/-)
Время удерживания (мин)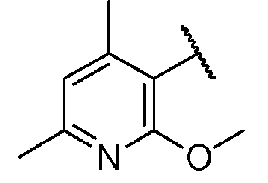
ESI-MS m/z 391 [M+H]+
100% этанол
(+): 7,0
(-): 6,0
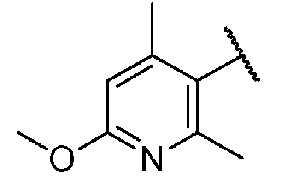
ESI-MS m/z 391 [M+H]+
100% этанол
(+): 8,5
(-): 6,2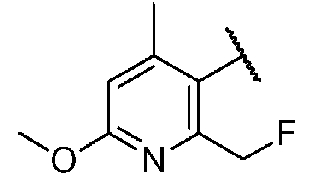
ESI-MS m/z 409 [M+H]+
100% этанол
(+): 9,0
(-): 9,7
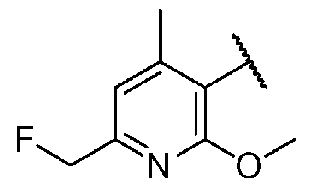
ESI-MS m/z 409 [M+H]+
100% этанол
(+): 6,0
(-): 7,1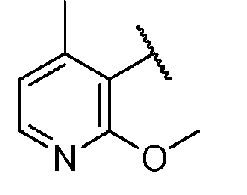
ESI-MS m/z 377 [M+H]+
100% этанол
(+): 5,8
(-): 7,5
[0342] Пример 33
Синтез (+)-7-(2,6-диметилфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-4-метилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
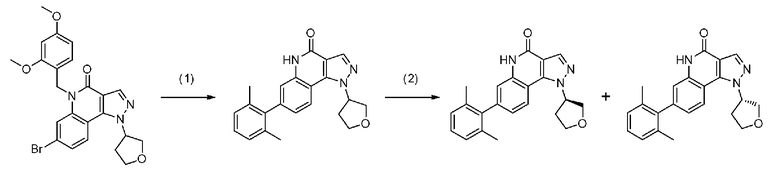
[0343] Указанное в заголовке соединение получали в результате осуществления реакций (1)-(2) тем же способом, который описано в примере 25, с применением (±)-7-бром-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного в примере получения 5(4), и 2,6-диметилфенилбороновой кислоты в качестве сырьевых материалов. Разделение оптических изомеров (2) в условиях колонки для хиральной хроматографии: IB, подвижная фаза: 100% этанол и скорость потока: 1,00 мл/мин выявляло (-)-форму в момент 4,0 мин и (+)-форму в момент 4,4 мин. Таким образом, разделение оптических изомеров осуществляли с помощью IB-колонки для разделения оптических изомеров в следующих условиях подвижной фазы: 100% этанол, скорость потока: 10,0 мл/мин, время элюирования: 60 мин/цикл и введение: 1,5 мл/цикл и получали (-)-форму с более коротким временем удерживания и (+)-форму с более длительным временем удерживания.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,06 (с, 6H), 2,60-2,64 (м, 1H), 2,79-2,83 (м, 1H), 4,12-4,13 (м, 1H), 4,24-4,37 (м, 2H), 4,40-4,45 (м, 1H), 5,62-5,70 (м, 1H), 7,10-7,19 (м, 4H), 7,21-7,24 (м, 1H), 8,10-8,12 (м, 1H), 8,30 (с, 1H), 9,57 (ушир.с, 1H).
ESI-MS m/z 360 [M+H]+
[0344] Пример 34
Синтез (+)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
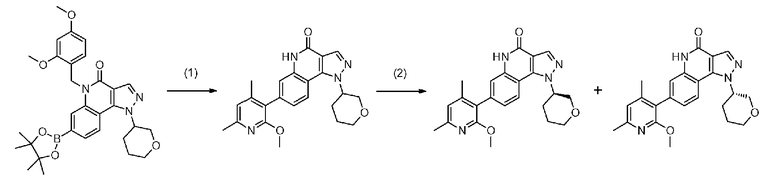
[0345] Указанное в заголовке соединение получали в результате осуществления реакций (1)-(2) тем же способом, который описан в примере 25, с применением в качестве сырьевых материалов (±)-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного в примере получения 4, и 3-бром-2-метокси-4,6-диметилпиридина, полученного в примере получения 26. Разделение оптических изомеров (2) в условиях колонки для хиральной хроматографии: OD-H, подвижная фаза: 100% этанол и скорость потока: 1,00 мл/мин выявляло (+)-форму в момент времени 4,8 мин и (-)-форму в момент времени 5,2 мин. Таким образом, разделение оптических изомеров проводили с помощью колонки OD-H для разделения оптических изомеров и с помощью растворителя для элюирования 100% этанола и получали (+)-форму с более коротким временем удерживания и (-)-форму с более длительным временем удерживания.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,92-2,00 (м, 2H), 2,10 (с, 3H), 2,38-2,54 (м, 5H), 3,53-3,61 (м, 1H), 3,86 (с, 3H), 3,88-3,95 (м, 1H), 4,04-4,10 (м, 1H), 4,28-4,35 (м, 1H), 4,95-5,05 (м, 1H), 6,72-6,75 (м, 1H), 7,18-7,21 (м, 1H), 7,22 (д, J=1,4 Гц, 1H), 8,09 (д, J=8,4 Гц, 1H), 8,28 (с, 1H), 9,63 (с, 1H).
ESI-MS m/z 405 [M+H]+
[0346] Соединения примеров 35-39 синтезировали как описано в примере 34.
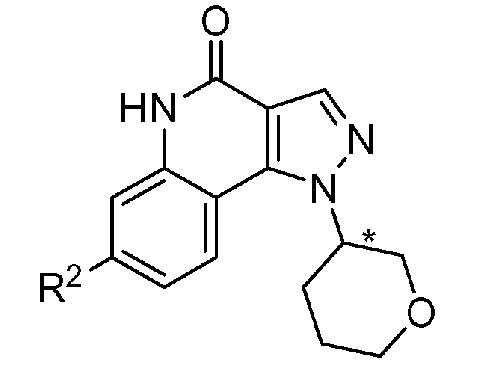
Подвижная фаза
Вращение плоскости поляризации (+/-)
Время удерживания (мин)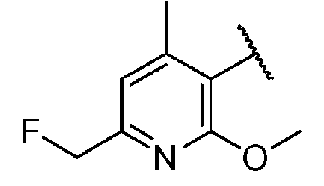
ESI-MS m/z 423 [M+H]+
100% этанол
(+): 5,8
(-): 6,4
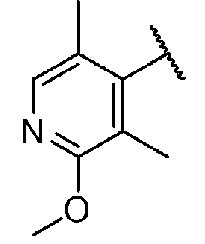
ESI-MS m/z 405 [M+H]+
100% этанол
(+): 4,6
(-): 5,1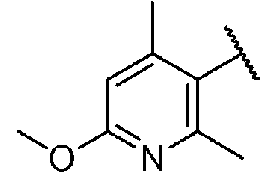
ESI-MS m/z 405 [M+H]+
100% этанол
(+): 8,0
(-): 10,7
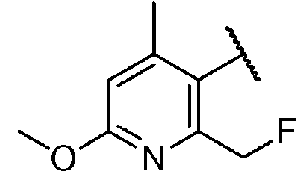
ESI-MS m/z 423 [M+H]+
100% этанол
(+): 5,2
(-): 5,8
ESI-MS m/z 391 [M+H]+
100% этанол
(+): 6,0
(-): 6,6
[0347] Пример 40
Синтез 7-[2,6-диметил-4-(тетрагидро-2H-пиран-4-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
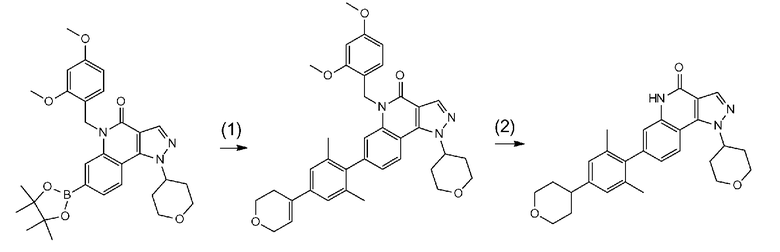
[0348] (1) Синтез 7-[4-(3,6-дигидро-2H-пиран-4-ил)-2,6-диметилфенил]-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Воду (0,2 мл), 4-(4-бром-3,5-диметилфенил)-3,6-дигидро-2H-пиран (44,1 мг), полученный в примере получения 50, Pd(PPh3)4 (12,7 мг) и карбонат цезия (108 мг) добавляли к раствору 5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного в примере получения 1(5) (60 мг), в 1,4-диоксане (1,5 мл). Реакционную смесь перемешивали при 100°C в течение ночи. После остывания реакционной смеси до комнатной температуры к реакционной смеси добавляли этилацетат и воду и отделяли органический слой. Органический слой промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматография на NH-силикагеле (этилацетат/н-гептан, 20-50%) с получением указанного в заголовке соединения (44 мг).
ESI-MS m/z 606 [M+H]+
[0349] (2) Синтез 7-[2,6-диметил-4-(тетрагидро-2H-пиран-4-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
10% Палладированный уголь (50% влаги, 15 мг) добавляли к раствору 7-[4-(3,6-дигидро-2H-пиран-4-ил)-2,6-диметилфенил]-5-(2,4-диметоксибензил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (44 мг) в этаноле (2 мл)-ТГФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение четырех часов и 35 минут в атмосфере водорода. Катализатор удаляли из реакционной смеси путем фильтрации и затем фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли TFA (1,5 мл). Реакционную смесь перемешивали при 60°C в течение 14 часов. Реакционной смеси давали остыть до комнатной температуры, а затем реакционную смесь концентрировали при пониженном давлении. К остатку добавляли хлороформ и насыщенный водный раствор бикарбоната натрия и отделяли органический слой. Водный слой повторно экстрагировали хлороформом. Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ 100%, затем этилацетат 100%). Целевую фракцию собирали и концентрировали. К полученному остатку добавляли этилацетат и MTBE. Осажденное твердое вещество собирали фильтрацией и сушили при пониженном давлении с получением указанного в заголовке соединения (14,8 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,79-1,95 (м, 4H), 2,07 (с, 6H), 2,20 (д, J=12,8 Гц, 2H), 2,53 (ддд, J=15,6, 11,6, 4,0 Гц, 2H), 2,72-2,81 (м, 1H), 3,56 (тд, J=10,8, 2,8 Гц, 2H), 3,71 (т, J=10,4 Гц, 2H), 4, 12 (дд, J=10,4, 2,8 Гц, 2H), 4,24 (д, J=10,8 Гц, 2H), 5,03-5,11 (м, 1H), 7,03 (с, 2H), 7,13 (дд, J=8,0, 1,2 Гц, 1H), 7,25 (д, J=1,2 Гц, 1H), 8,01 (д, J=8,0 Гц, 1H), 8,31 (с, 1H), 10,26 (ушир.с, 1H).
ESI-MS m/z 458 [M+H]+
[0350] Пример 41
Синтез (+)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
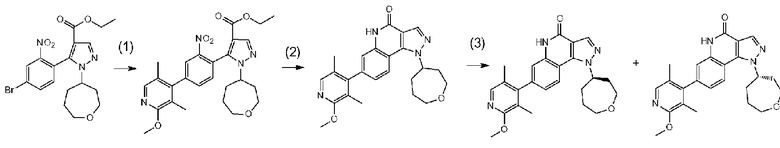
[0351] (1) Синтез (±)-этил-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-(оксепан-4-ил)-1H-пиразол-4-карбоксилата
Указанное в заголовке соединение (80 мг) получали тем же способом, который описан в примере 27-(1), из (±) этил-5-(4-бром-2-нитрофенил)-1-(оксепан-4-ил)-1H-пиразол-4-карбоксилата, полученного в примере получения 8 (85 мг), и (2-метокси-3,5-диметилпиридин-4-ил)бороновой кислоты, полученной в примере получения 29 (42,1 мг).
ESI-MS m/z 495 [M+H]+
[0352] (2) Синтез (±)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Указанное в заголовке соединение (53 мг) получали тем же способом, который описан в примере 45-(2), из (±)-этил-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-(оксепан-4-ил)-1H-пиразол-4-карбоксилата (80 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,90-2,05 (м, 2H), 1,94, 1,95, 1,96 (с, 3H), 1,97 (с, 3H), 2,33-2,70 (м, 4H), 3,70-3,80 (м, 1H), 3,92-4,08 (м, 3H), 4,01 (с, 3H), 5,20-5,29 (м, 1H), 7,08 (дд, J=8,4, 1,6 Гц, 1H), 7,19 (д, J=1,6 Гц, 1H), 7,95 (с, 1H), 8,11 (д, J=8,4 Гц, 1H), 8,30 (с, 1H), 10,30 (ушир.с, 1H).
ESI-MS m/z 419 [M+H]+
[0353] (3) Синтез (+)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(оксепан-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (53 мг) растворяли в этаноле (5 мл) и раствор фильтровали через микропористый фильтр. Фильтрат разделяли на оптические изомеры с помощью OD-H CIRALCEL(R), произведенной DAICEL Corporation (20 мм диаметром × 250 мм длиной) в условиях, представляющих собой этанол 100% и 10 мл/мин. Получали указанное в заголовке соединение со временем удерживания 11 минут и (+) вращением плоскости поляризации (15,9 мг, >98% э.и. [OD-H CIRALCEL (R) (0,46 см Φ × 25 см), 20% этанол/гексан, время удерживания = 7,3 мин]) и указанное в заголовке соединение со временем удерживания 12 минут и (-) вращением плоскости поляризации (16,7 мг, >98% э.и. [OD-H CIRALCEL (R) (0,46 см Φ × 25 см), 20% этанол/гексан, время удерживания = 7,9 мин]).
[0354] Соединение из примера 42 синтезировали как описано в примере 41.
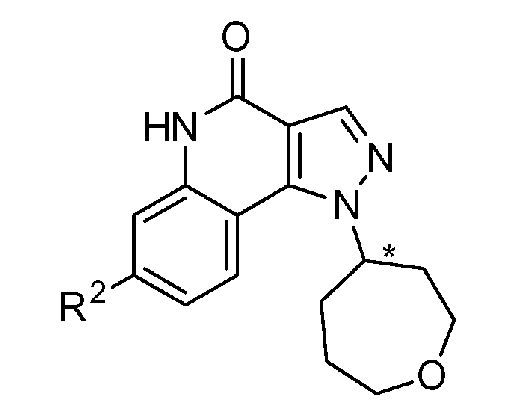
Подвижная фаза
Вращение плоскости поляризации (+/-)
Время удерживания (мин)
ESI-MS m/z 418 [M+H]+
100% IPA
(+): 8,3
(-): 9,4
[0355] Пример 43
Синтез 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
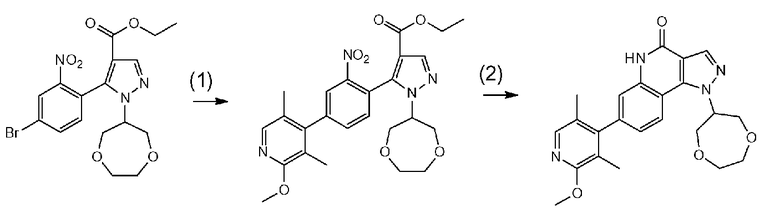
[0356] (1) Синтез этил-1-(1,4-диоксепан-6-ил)-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1H-пиразол-4-карбоксилата
Воду (0,2 мл), (2-метокси-3,5-диметилпиридин-4-ил)бороновую кислоту, полученную в примере получения 29 (39,5 мг), Pd(PPh3)4 (10,5 мг) и карбонат цезия (178 мг) добавляли к раствору этил-5-(4-бром-2-нитрофенил)-1-(1,4-диоксепан-6-ил)-1H-пиразол-4-карбоксилата, полученного в примере получения 9-(2) (80 мг), в 1,4-диоксане (1,3 мл) и реакционную смесь перемешивали при 100°C в течение 6,75 часа. К реакционной смеси добавляли (2-метокси-3,5-диметилпиридин-4-ил)бороновую кислоту (15 мг) и реакционную смесь перемешивали при 100°C в течение 2,5 часа. После остывания реакционной смеси до комнатной температуры к реакционной смеси добавляли этилацетат и воду и отделяли органический слой. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (силикагель, этилацетат/н-гептан, 20-33%) с получением указанного в заголовке соединения (64 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,09 (т, J=7,2 Гц, 1,5H), 1,11 (т, J=7,2 Гц, 1,5H), 1,98 (с, 1,5H), 1,99 (с, 1,5H), 2,01 (с, 1,5H), 2,02 (с, 1,5H), 3,73-3,87 (м, 2H), 3,90-4,02 (м, 2H), 4,00 (с, 3H), 4,03-4,17 (м, 4H), 4,30-4,40 (м, 2H), 4,41-4,49 (м, 1H), 7,39 (д, J=7,6 Гц, 1H), 7,52 (дд, J=7,6, 1,6 Гц, 1H), 7,95-8,01 (м, 2H), 8,11 (с, 1H).
ESI-MS m/z 519 [M+Na]+
[0357] (2) Синтез 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Железный порошок (28,8 мг) добавляли к раствору этил-1-(1,4-диоксепан-6-ил)-5-[4-(2-метокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1H-пиразол-4-карбоксилата (64 мг) в уксусной кислоте (2 мл)-воде (0,1 мл) и реакционную смесь перемешивали при 80°C в течение 2,5 часов в атмосфере азота. Реакционной смеси давали остыть до комнатной температуры и к реакционной смеси добавляли этилацетат (10 мл). Нерастворимое вещество удаляли путем фильтрации через Celite™. Фильтрат концентрировали при пониженном давлении. Раствор остатка в этилацетате последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом магния, фильтровали. Фильтрат пропускали через NH-силикагелевую подушку. Полученный раствор концентрировали при пониженном давлении. К остатку добавляли этилацетат (0,3 мл) и МТВЕ (0,3 мл). Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (29,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,93 (с, 3H), 1,96 (с, 3H), 3,90-4,07 (м, 4H), 4,00 (с, 3H), 4,38 (дд, J=12,0, 6,0 Гц, 2H), 4,40 (дд, J=12,0, 6,8 Гц, 2H), 5,50 (тт, J=6,8, 6,0 Гц, 1H), 7,08 (д, J=8,0 Гц, 1H), 7,17 (с, 1H), 7,94 (с, 1H), 8,13 (д, J=8,0 Гц, 1H), 8,36 (с, 1H), 10,23 (ушир.с, 1H).
ESI-MS m/z 421 [M+H]+
[0358] Соединение из примера 44 синтезировали как описано в примере 43.
ESI-MS m/z 420 [M+H]+
[0359] Пример 45
Синтез 1-(1,4-диоксепан-6-ил)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
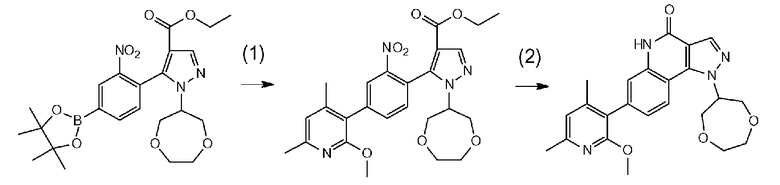
[0360] (1) Синтез этил-1-(1,4-диоксепан-6-ил)-5-{4-(2-метокси-4,6-диметилпиридин-3-ил)-2-нитрофенил}-1H-пиразол-4-карбоксилата
Воду (0,3 мл), 3-бром-2-метокси-4,6-диметилпиридин, полученный в примере получения 26 (32,5 мг), Pd(PPh3)4 (7,2 мг) и карбонат цезия (122 мг) добавляли к раствору этил-1-(1,4-диоксепан-6-ил)-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1H-пиразол-4-карбоксилата (61 мг) в 1,4-диоксане (1,2 мл) и реакционную смесь перемешивали при 100°C в течение 4 часов. К реакционной смеси добавляли Pd(PPh3)4 (7,2 мг) и реакционную смесь перемешивали при 100°C в течение 1 часа и 10 минут. Реакционной смеси давали остыть до комнатной температуры и разделяли путем добавления этилацетата и воды и отделяли органический слой. Полученный органический слой последовательно промывали водой и солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 15-20%) с получением указанного в заголовке соединения (15 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,06 (т, J=7,2 Гц, 3H), 2,16 (с, 3H), 2,48 (с, 3H), 3,74-3,88 (м, 2H), 3,88 (с, 3H), 3,91-4,16 (м, 6H), 4,28-4,37 (м, 2H), 4,47-4,55 (м, 1H), 6,74 (с, 1H), 7,29 (д, J=7,6 Гц, 1H), 7,60 (дд, J=7,6, 1,6 Гц, 1H), 8,07 (д, J=1,6 Гц, 1H), 8,10 (с, 1H).
ESI-MS m/z 519[M+Na]+
[0361] (2) Синтез 1-(1,4-диоксепан-6-ил)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Железный порошок (17 мг) добавляли к раствору этил-1-(1,4-диоксепан-6-ил)-5-{4-(2-метокси-4,6-диметилпиридин-3-ил)-2-нитрофенил}-1H-пиразол-4-карбоксилата (15 мг) в уксусной кислоте (1 мл)-воде (0,05 мл) и смесь перемешивали при 80°C в течение 4,25 часа в атмосфере азота. Реакционной смеси давали остыть до комнатной температуры и к реакционной смеси добавляли этилацетат (5 мл). Нерастворимое вещество удаляли путем фильтрации через Celite™. Фильтрат концентрировали при пониженном давлении. Раствор остатка в этилацетате последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали. Остаток очищали препаративной тонкослойной хроматографией (силикагель, этилацетат/н-гептан, 66%) с получением указанного в заголовке соединения (1,5 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,09 (с, 3H), 2,48 (с, 3H), 3,85 (с, 3H), 3,91-3,99 (м, 2H), 4,00-4,08 (м, 2H), 4,36 (дд, J=12,8, 6,4 Гц, 2H), 4,42 (дд, J=12,8, 6,4 Гц, 2H), 5,49 (тт, J=6,8, 6,4 Гц, 1H), 6,72 (с, 1H), 7,15-7,21 (м, 2H), 8,08 (д, J=8,0 Гц, 1H), 8,34 (с, 1H), 9,17 (ушир.с, 1H).
ESI-MS m/z 421 [M+H]+
[0362] Соединения из примеров 46 и 47 синтезировали как описано в примере 45.
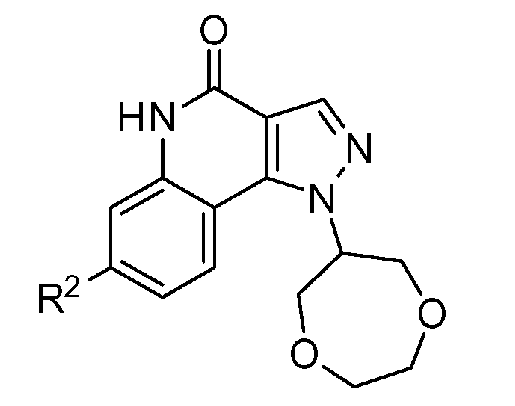
ESI-MS m/z 421 [M+H]+
ESI-MS m/z 407 [M+H]+
[0363] Пример 48
Синтез (S)-7-(2-изопропилокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
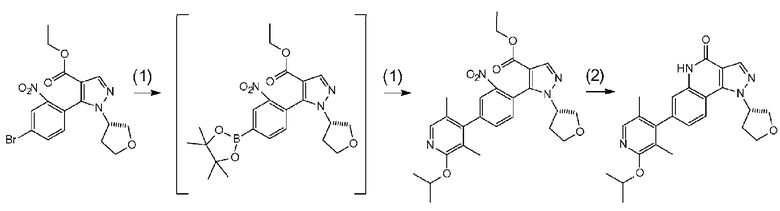
[0364] (1) Синтез этил-5-[4-(2-изопропилокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-(4-бром-2-нитрофенил)-1-[(S)-тетрагидрофуран-3-ил]- 1H-пиразол-4-карбоксилат, полученный в примере получения 7-(1) (200 мг), превращали в этил-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат тем же способом, который описан в примере получения 7-(2). К реакционной смеси добавляли раствор 4-иод-2-изопропилокси-3,5-диметилпиридина, полученного в примере получения 47 (142 мг), в DMF (0,5 мл) и воду (0,5 мл) и смесь перемешивали при 110°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем разделяли путем добавления этилацетата и воды. Водный слой экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 50%-100%) с получением указанного в заголовке соединения (138,1 мг).
ESI-MS m/z 517 [M+Na]+
[0365] (2) Синтез (S)-7-(2-изопропилокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Указанное в заголовке соединение (67,1 мг) получали тем же способом, который описан в примере 45-(2), из этил-5-[4-(2-изопропилокси-3,5-диметилпиридин-4-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (138,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,37-1,41 (м, 6H), 1,91 (с, 3H), 1,95 (с, 3H), 2,59-2,64 (м, 1H), 2,79-2,83 (м, 1H), 4,12-4,29 (м, 1H), 4,23-4,27 (м, 2H), 4,39-4,46 (м, 1H), 5,29-5,40 (м, 1H), 5,62-5,69 (м, 1H), 7,07-7,09 (м, 1H), 7,19-7,20 (м, 1H), 7,90-7,92 (м, 1H), 8,13 (д, J=8,40 Гц, 1H), 8,31 (с, 1H), 10,18 (с, 1H).
ESI-MS m/z 419 [M+H]+
[0366] Соединения из примеров 49 и 50 синтезировали как описано в примере 48.
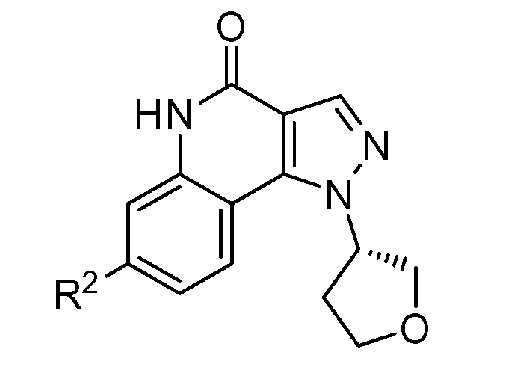
ESI-MS m/z 449 [M+Na]+
ESI-MS m/z 405 [M+H]+
[0367] Пример 51
Синтез (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
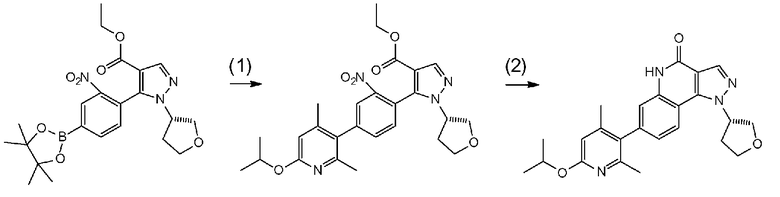
[0368] (1) Синтез этил-5-[4-(6-изопропилокси-2,4-диметилпиридин-3-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат, полученный в примере получения 7-(2) (70 мг), растворяли в смешанном растворе из 1,4-диоксана (1 мл) и воды (0,2 мл) и добавляли 3-бром-6-изопропилокси-2,4-диметилпиридин (41,1 мг), Pd(PPh3)4 (17,7 мг) и карбонат цезия (150 мг). Реакционную смесь перемешивали при 110°C в течение ночи. После остывания реакционной смеси до комнатной температуры реакционную смесь очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10-50%-100%) с получением указанного в заголовке соединения (66,5 мг).
ESI-MS m/z 495 [M+H]+
[0369] (2) Синтез (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Раствор этил-5-[4-(6-изопропилокси-2,4-диметилпиридин-3-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (65,1 мг) в уксусной кислоте (1,5 мл)-воде (0,15 мл) перемешивали при 80°C в течение 15 минут. К раствору добавляли железный порошок (45,1 мг) и смесь перемешивали при такой же температуре в течение двух часов в атмосфере азота. Реакционной смеси давали остыть до комнатной температуры и к реакционной смеси добавляли этилацетат (5 мл). Нерастворимое вещество удаляли путем фильтрации через Celite™. Фильтрат концентрировали при пониженном давлении. Раствор остатка в этилацетате промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток суспендировали и растирали с добавлением MTBE. Осажденное твердое вещество собирали фильтрацией с получением указанного в заголовке соединения (35,1 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,38 (д, J=6,25 Гц, 6H), 2,01 (с, 3H), 2,20 (с, 3H), 2,55-2,67 (м, 1H), 2,76-2,87 (м, 1H), 4,12-4,14 (м, 1H), 4,23-4,37 (м, 2H), 4,39-4,45 (м, 1H), 5,30-5,35 (м, 1H), 5,61-5,69 (м, 1H), 6,48 (с, 1H), 7,11-7,13 (м, 1H), 7,16-7,17 (м, 1H), 8,09-8,11 (м, 1H), 8,31 (с, 1H), 9,58 (ушир.с, 1H).
ESI-MS m/z 419 [M+H]+
[0370] Пример 52
Синтез 8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
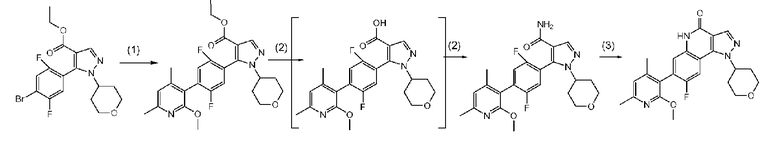
[0371] (1) Синтез этил-5-[2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата
Pd(PPh3)4 (50 мг), карбонат цезия (282 мг) и воду (0,5 мл) добавляли к смешанному раствору этил-5-(4-бром-2,5-дифторфенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата, полученного в примере получения 10 (180 мг), (2-метокси-4,6-диметилпиридин-3-ил)бороновой кислоты, полученной в примере получения 27 (90 мг) и 1,4-диоксана (2 мл) и смесь перемешивали при 110°C в течение шести часов. После охлаждения реакционной смеси до комнатной температуры добавляли этилацетат и солевой раствор и смесь фильтровали через ватную пробку. Органический слой отделяли и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (этилацетат/н-гептан, 25%-46%-53%) с получением указанного в заголовке соединения (157 мг).
ESI-MS m/z 494 [M+Na]+
[0372] (2) Синтез 5-[2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамида
5н водный раствор гидроксида натрия (0,3 мл) добавляли к раствору этил-5-[2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксилата (157 мг) в этаноле (3 мл) и смесь перемешивали при 55°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток разделяли путем добавления хлороформа, 5н соляной кислоты и насыщенного водного раствора хлорида аммония. Органический слой сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией, а фильтрат концентрировали при пониженном давлении с получением 5-[2,5-дифтор-4-(2-метокси-4,6-диметил-пиридин-3-ил)фенил]-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоновой кислоты (155 мг) в качестве сырого очищенного продукта. Карбоновую кислоту (155 мг) растворяли в DMF (1 мл) и THF (3 мл). Затем добавляли CDI (108 мг) и смесь перемешивали при комнатной температуре в течение приблизительно 1,5 часа. К реакционной смеси добавляли 28% водный раствор аммиака (0,35 мл) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток разделяли путем добавления этилацетата и солевого раствора. Органический слой промывали насыщенным водным раствором бикарбоната натрия и сушили над безводным сульфатом магния. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток отверждали путем добавления н-гептана/MTBE (1/9) с получением указанного в заголовке соединения (82 мг). Указанное в заголовке соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS m/z 465 [M+Na]+
[0373] (3) Синтез 8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
KTB (41 мг) добавляли к раствору 5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-карбоксамида (81 мг) в NMP (0,4 мл) и смесь нагревали до 90°C. Через один час дополнительно добавляли KTB (20 мг) с последующим перемешиванием в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры с последующим добавлением насыщенного водного раствора хлорида аммония (2 мл) и воды (1 мл). Образовавшееся твердое вещество отфильтровывали, промывали водой (2 мл) и сушили при пониженном давлении при 60°C с получением указанного в заголовке соединения (57 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,11 (с, 3H), 2,14-2,24 (м, 2H), 2,50 (с, 3H), 2,42-2,63 (м, 2H), 3,65-3,79 (м, 2H), 3,87 (с, 3H), 4,20-4,28 (м, 2H), 4,93-5,03 (м, 1H), 6,76 (с, 1H), 7,35 (д, J=6,44 Гц, 1H), 7,71 (д, J=10,35 Гц, 1H), 8,30 (с, 1H), 10,93 (ушир.с, 1H).
ESI-MS m/z 423 [M+H]+
[0374] Пример 53
Синтез (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
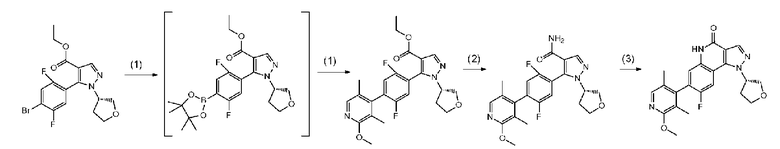
[0375] (1) Синтез этил-5-(2,5-дифтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-(4-бром-2,5-дифторфенил)-1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилат, полученный в примере получения 11-1 (4,31 г), бис(пинаколато)дибор (3,27 г), ацетат калия (3,16 г) и комплекс Pd(dppf)Cl2-DCM (439 мг) добавляли к DMF (41,6 мл) и смесь перемешивали при 95°C в атмосфере азота. Через два часа реакционную смесь перемешивали при 105°C в течение четырех часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении, к остатку добавляли солевой раствор и этилацетат и смесь перемешивали при комнатной температуре в течение пяти минут. Смесь повторно фильтровали через Celite™ и фильтрат экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали через Celite™. Фильтрат концентрировали. Остаток очищали с помощью хроматографии на силикагеле (н-гептан/этилацетат, 20%-30%-80%) с получением этил-5-(2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата (2,95 г). Полученный этил-5-(2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилат (900 мг), 4-иод-2-метокси-3,5-диметилпиридин, полученный в примере получения 29(3) (634 мг), Pd(PPh3)4 (116 мг) и карбонат цезия (1,96 г) добавляли к смешанному растворителю из 1,4-диоксана (9,3 мл) и воды (3,1 мл) и смесь нагревали с обратным холодильником в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры и разделяли путем добавления этилацетата и солевого раствора. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат/н-гептан, первый раз: 15%-36%-47%, второй раз: 10%-30%-35%) с получением указанного в заголовке соединения (280 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,17-1,23 (м, 3H), 1,99-2,06 (м, 6H), 2,26-2,55 (м, 2H), 3,92-4,29 (м, 9H), 4,65-4,75 (м, 1H), 6,95-7,03 (м, 1H), 7,14-7,25 (м, 1H), 7,96 (с, 1H), 8,12 (с, 1H).
ESI-MS m/z 480 [M+Na]+
[0376] (2) Синтез 5-(2,5-дифтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида
5н гидроксид натрия (0,5 мл) добавляли к раствору этил-5-[2,5-дифтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата (280 мг) в этаноле (3,6 мл) и смесь перемешивали при 65°C в течение трех часов. После охлаждения реакционной смеси до комнатной температуры добавляли хлороформ и солевой раствор и смесь доводили до pH 6 посредством 5н соляной кислоты и насыщенного раствора хлорида аммония. Органический слой сушили над безводным сульфатом магния и осушитель удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении с получением 5-(2,5-дифтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил)-1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновой кислоты (238 мг) в виде сырого очищенного продукта. К раствору карбоновой кислоты (238 мг) в DMF (3 мл) добавляли CDI (121 мг) и смесь перемешивали при комнатной температуре в течение одного часа. К реакционной смеси добавляли 28% водный раствор аммиака (0,6 мл) с последующим перемешиванием в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток разделяли путем добавления хлороформа и насыщенного водного раствора бикарбоната натрия. Органический слой промывали солевым раствором и затем сушили над безводным сульфатом магния, а осушитель удаляли фильтрацией. Фильтрат пропускали через силикагелевую подушку (NH-силикагель; элюирование этилацетатом) и полученный фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (186 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,95-2,10 (м, 6H), 2,25-2,57 (м, 2H), 3,91-4,29 (м, 7H), 4,69 (ушир.с, 1H), 5,24-5,57 (м, 2H), 7,01 (дд, J=8,79, 5,66 Гц, 1H), 7,17-7,26 (м, 1H), 7,94-7,99 (м, 2H).
ESI-MS m/z 451 [M+Na]+
[0377] (3) Синтез (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Гидроксид натрия (порошок, 82 мг) добавляли к раствору 5-(2,5-дифтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида (186 мг) в ДМСО (1,5 мл) и смесь перемешивали при 75°C в течение 1,5 часа. Реакционную смесь охлаждали до комнатной температуры и затем добавляли воду (5,5 мл) при перемешивании. Дополнительно добавляли уксусную кислоту (0,12 мл) с последующим перемешиванием в течение 30 минут. Образовавшееся твердое вещество отфильтровывали, промывали водой (5 мл) и затем сушили при пониженном давлении при 60°C в течение одного часа с получением указанного в заголовке соединения (155 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98 (с, 3H), 2,00 (с, 3H), 2,55-2,72 (м, 1H), 2,73-2,86 (м, 1H), 4,01 (с, 3H), 4,13 (тд, J=8,40, 4,69 Гц, 1H), 4,20-4,39 (м, 2H), 4,43 (дт, J=9,57, 3,03 Гц, 1H), 5,52-5,62 (м, 1H), 7,23 (д, J=6,25 Гц, 1H), 7,84 (д, J=9,96 Гц, 1H), 7,98 (с, 1H), 8,32 (с, 1H), 10,82 (ушир.с, 1H).
ESI-MS m/z 409 [M+1]+
[0378] Пример 54
Синтез (S)-8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
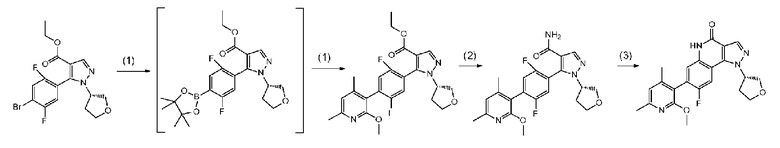
[0379] (1) Синтез этил-5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-(4-бром-2,5-дифторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат (4,31 г), бис(пинаколато)дибор (3,27 г), ацетат калия (3,16 г), комплекс Pd(dppf)Cl2-DCM (439 мг) добавляли к DMF (41,6 мл) и реакционную смесь перемешивали при 95°C в атмосфере азота. После перемешания реакционной смеси в течение приблизительно 2 часов реакционную смесь перемешивали при 105°C в течение приблизительно 4 часов. Реакционную смесь охлаждали до комнатной температуры, а затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении и после этого к остатку добавляли солевой раствор и этилацетат, раствор перемешивали при комнатной температуре в течение 5 минут. Реакционную смесь еще раз фильтровали через Celite™ и фильтрат экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали через Celite™. Концентрировали фильтрат и остаток очищали с помощью хроматографии на силикагеле (гептан/этилацетат, 20%-30%-80%) с получением этил-5-(2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (2,95 г). Этил-5-(2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат (812 мг), 3-бром-2-метокси-4,6-диметилпиридин (490 мг), Pd(PPh3)4 (130 мг) и карбонат цезия (1,77 г) добавляли к смешанному растворителю из 1,4-диоксана (9,00 мл) и воды (3,00 мл) и реакционную смесь нагревали с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и разделяли путем добавления этилацетата и солевого раствора. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат/н-гептан, 11%-30%-50%) с получением указанного в заголовке соединения (397 мг). Указанное в заголовке соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS m/z 480 [M+Na]+
[0380] (2) Синтез 5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида
5н водный раствор гидроксида натрия (0,8 мл) добавляли к раствору этил-5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (397 мг) в этаноле (5 мл) и реакционную смесь перемешивали при 70°C в течение 1 часа. После охлаждения реакционной смеси до комнатной температуры добавляли хлороформ и солевой раствор и смесь доводили до pH 6 посредством 5н соляной кислоты и насыщенного водного раствора хлорида аммония. Органический слой сушили над безводным сульфатом магния и осушитель удаляли фильтрацией. Фильтраты концентрировали при пониженном давлении с получением 5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоновой кислоты (457 мг) в виде сырого продукта. К раствору карбоновой кислоты (457 мг) в DMF (6 мл) добавляли CDI (211 мг) и реакционную смесь перемешивали при комнатной температуре. Через 75 минут к реакционной смеси добавляли 28% водный раствор аммиака (0,88 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток разделяли путем добавления этилацетата и насыщенного водного раствора хлорида аммония. Органический слой промывали насыщенным водным раствором бикарбоната натрия, сушили над безводным сульфатом магния, а осушитель удаляли фильтрацией. После концентрирования фильтрата при пониженном давлении осажденное твердое вещество удаляли путем фильтрации и промывали дихлорметаном и этилацетатом. Фильтрат концентрировали при пониженном давлении и остаток очищали при помощи хроматографии на силикагеле (этилацетат/н-гептан, 60%-80%-85%) с получением указанного в заголовке соединения (199 мг). Данное указанное в заголовке соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS m/z 451 [M+Na]+
[0381] (3) Синтез (S)-8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Гидроксид натрия (порошок, 74 мг) добавляли к раствору 5-(2,5-дифтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида (199 мг) в ДМСО (2 мл) и реакционную смесь перемешивали при 75°C в течение 1,5 часа. После охлаждения реакционной смеси до комнатной температуры к реакционной смеси при перемешивании добавляли воду, уксусную кислоту (0,106 мл) и этилацетат. После фильтрации осажденного твердого вещества органический слой промывали водой, насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом магния и осушитель удаляли фильтрацией. После концентрирования фильтрата при пониженном давлении остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 55%-90%-96%) и полученный сырой продукт отверждали путем добавления н-гептана/MTBE с получением указанного в заголовке соединения (33 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,11 (с, 3H), 2,49 (с, 3H), 2,56-2,67 (м, 1H), 2,71-2,84 (м, 1H), 3,86 (с, 3H), 4,07-4,40 (м, 4H), 5,49-5,62 (м, 1H), 6,68-6,78 (м, 1H), 7,24-7,31 (м, 1H), 7,74-7,84 (м, 1H), 8,25-8,32 (м, 1H), 10,16 (ушир.с, 1H).
ESI-MS m/z 409 [M+1]+
[0382] Пример 55
Синтез (S)-8-фтор-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
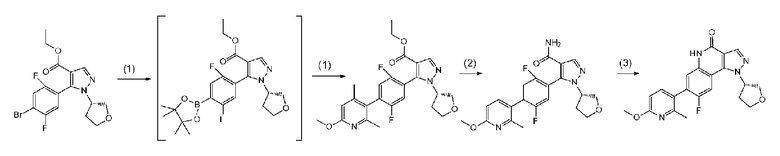
[0383] (1) Синтез этил-5-(2,5-дифтор-4-(6-метокси-2,4-диметилпиридин-3-ил)фенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Смесь из этил-5-[2,5-дифтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата, синтезированного в соответствии с примером 53 (430 мг), 3-бром-6-метокси-2,4-диметилпиридина, полученного в примере получения 23 (223 мг), гидрофторид калия (254 мг), Pd(PPh3)4 (90 мг) и н-гидрат трикалийфосфата (400 мг) в DME (8 мл) и воде (2 мл) нагревали с обратным холодильником при 110°C в течение семи часов. Реакционную смесь охлаждали до комнатной температуры и разделяли путем добавления этилацетата и солевого раствора. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат/н-гептан: 16%-37%-46%) и колоночной хроматографией на силикагеле (этилацетат/н-гептан: 28%-49%-54%) с получением указанного в заголовке соединения (144 мг). Данное соединение использовали для следующей реакции без дополнительной очистки.
ESI-MS m/z 458 [M+H]+
Реакции (2)-(3) проводили в соответствии с примером 53. Тем не менее, в реакции (3) указанное в заголовке соединение, полученное в виде сырого очищенного продукта, подвергали колоночной хроматографии на силикагеле (этилацетат/н-гептан, 60%-95%) и затем очищали путем затвердевания из MTBE с получением указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,02-2,10 (м, 3H), 2,23-2,27 (м, 3H), 2,56-2,69 (м, 1H), 2,72-2,87 (м, 1H), 3,97 (с, 3H), 4,13 (тд, J=8,44, 4,59 Гц, 1H), 4,20-4,37 (м, 2H), 4,43 (дд, J=9,86, 3,22 Гц, 1H), 5,51-5,63 (м, 1H), 6,57 (д, J=0,59 Гц, 1H), 7,20-7,25 (м, 1H), 7,82 (д, J=10,15 Гц, 1H), 8,31 (с, 1H), 10,38 (ушир.с, 1H).
ESI-MS m/z 409 [M+H]+
[0384] Пример 56
Синтез (S)-7-(3-этил-2-метокси-5-метилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
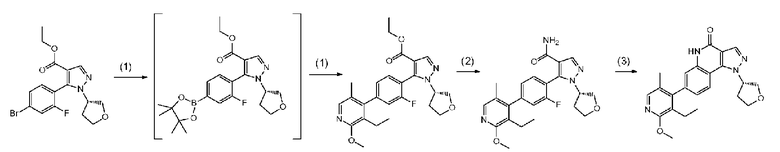
[0385] Указанное в заголовке соединение получали путем проведения реакций (1)-(3) в соответствии с примером 53 с применением этил-5-(4-бром-2-фторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата, полученного в примере получения 6(1), и 3-этил-4-иод-2-метокси-5-метилпиридина, полученного в примере получения 49.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 0,94-1,06 (м, 3H), 1,88-1,96 (м, 3H), 2,35 (кв, J=7,48 Гц, 2H), 2,55-2,69 (м, 1H), 2,75-2,88 (м, 1H), 3,96-4,05 (м, 3H), 4,08-4,19 (м, 1H), 4,22-4,38 (м, 2H), 4,38-4,48 (м, 1H), 5,60-5,72 (м, 1H), 7,09 (дд, J=8,30, 1,66 Гц, 1H), 7,20-7,25 (м, 1H), 7,93-7,95 (м, 1H), 8,12 (д, J=8,20 Гц, 1H), 8,30 (с, 1H), 10,38 (ушир.с, 1H).
ESI-MS m/z 405 [M+H]+
[0386] Пример 57
Синтез (R)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
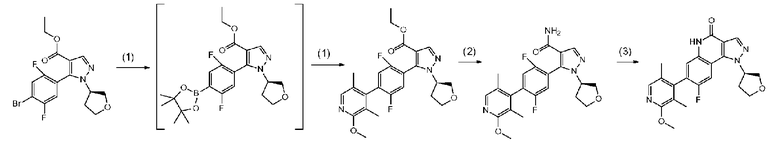
[0387] Указанное в заголовке соединение получали путем проведения реакций (1)-(3) в соответствии с примером 53 с применением этил-5-(4-бром-2,5-дифторфенил)-1-((R)-тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата, полученного в примере получения 11-2, и 4-иод-2-метокси-3,5-диметилпиридина, полученного в примере получения 29(3). Тем не менее, в реакции (3) указанное в заголовке соединение, полученное в виде сырого очищенного продукта, очищали путем промывания 1-пропанолом.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,98 (с, 3H), 2,00 (с, 3H), 2,56-2,71 (м, 1H), 2,73-2,89 (м, 1H), 4,01 (с, 3H), 4,13 (тд, J=8,40, 4,69 Гц, 1H), 4,18-4,38 (м, 2H), 4,40-4,48 (м, 1H), 5,51-5,64 (м, 1H), 7,21-7,30 (м, 1H), 7,84 (д, J=10,15 Гц, 1H), 7,98 (д, J=0,78 Гц, 1H), 8,32 (с, 1H), 11,05 (ушир.с, 1H).
ESI-MS m/z 409 [M+H]+
[0388] Соединения из примеров 58 и 59 синтезировали как описано в примере 57.
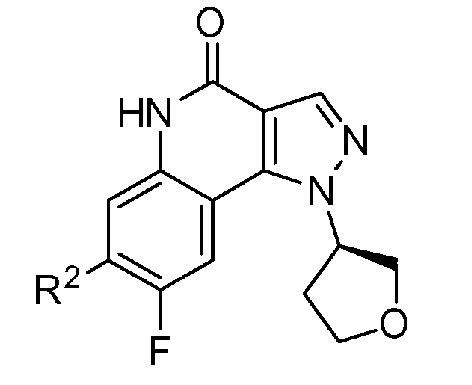
ESI-MS m/z 409 [M+H]+
ESI-MS m/z 409 [M+H]+
[0389] Пример 60
Синтез 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-[(3RS,4SR)-4-метокситетрагидрофуран-3-ил]-1H-пиразоло[4,3-c]хинолин-4(5H)-она
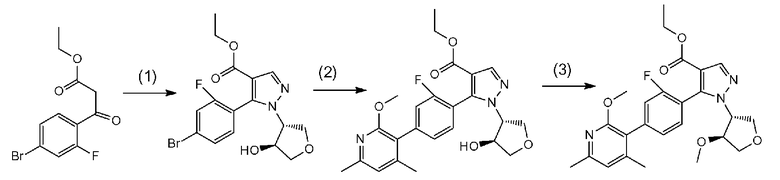
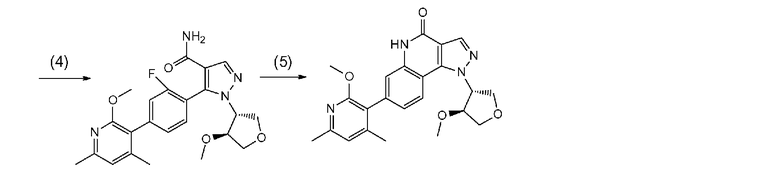
[0390] (1) Синтез этил-5-(4-бром-2-фторфенил)-1-[(3RS,4SR)-4-гидрокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Указанное в заголовке соединение синтезировали в соответствии с примером получения 7 с применением гидрохлорида (3SR,4RS)-4-гидразинилтетрагидрофуран-3-ола, полученного в примере получения 18, вместо гидрохлорида (S)-(тетрагидрофуран-3-ил)гидразина.
ESI-MS m/z 421 [M+Na]+
[0391] (2) Синтез этил-5-(2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(3RS,4SR)-4-гидрокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-(4-бром-2-фторфенил)-1-[(3RS,4SR)-4-гидрокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат (3,8 г), бис(пинаколато)дибор (2,90 г), ацетат калия (2,80 г) и комплекс Pd(dppf)Cl2-DCM (480 мг) добавляли к DMF (38,3 мл) и смесь перемешивали при 90°C в атмосфере азота. После перемешания реакционной смеси в течение приблизительно двух часов добавляли раствор 3-бром-2-метокси-4,6-диметилпиридина, полученного в примере получения 26 (3,09 г), в DMF (15 мл) и воду (22 мл) и смесь подогревали до 120°C и дополнительно перемешивали в течение приблизительно пяти часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении, а остаток пропускали через силикагелевую подушку (NH-силикагель, элюирование этилацетатом). Фильтрат концентрировали до приблизительно 200 мл и затем разделяли путем добавления солевого раствора. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат/н-гептан, 70%-90%) с получением указанного в заголовке соединения (2,26 г).
ESI-MS m/z 456 [M+H]+
[0392] (3) Синтез этил-5-(2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(3RS,4SR)-4-метокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Гидрид натрия (60% масляная дисперсия, 86 мг) добавляли к раствору этил-5-(2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(3RS,4SR)-4-гидрокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (612 мг) в THF (5 мл) с охлаждением на льду с последующим перемешиванием в течение трех минут. К реакционной смеси добавляли метилиодид (0,142 мл) и смесь перемешивали при такой же температуре в течение пяти минут и затем подогревали до комнатной температуры и перемешивали в течение двух часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и остаток очищали колоночной хроматографией на силикагеле (этилацетат/н-гептан, 27%-48%) с получением указанного в заголовке соединения (379 мг).
ESI-MS m/z 492 [M+Na]+
[0393] (4) Синтез 5-(2-фтор-4-(2-метокси-4,6-диметилпиридин-3-ил)фенил)-1-[(3RS,4SR)-4-метокситетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида
Указанное в заголовке соединение синтезировали согласно примеру 53(2).
ESI-MS m/z 463 [M+Na]+
[0394] (5) Синтез 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-[(3RS,4SR)-4-метокситетрагидрофуран-3-ил]-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Указанное в заголовке соединение получали согласно примеру 53. Тем не менее, указанное в заголовке соединение, полученное в виде сырого очищенного продукта, очищали путем промывания MTBE.
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 2,10 (с, 3H), 2,49 (с, 3H), 3,42 (с, 3H), 3,86 (с, 3H), 4,10 (дд, J=10,15, 2,15 Гц, 1H), 4,24-4,37 (м, 2H), 4,47 (дд, J=9,47, 6,35 Гц, 1H), 4,64-4,70 (м, 1H), 5,47-5,51 (м, 1H), 6,73-6,74 (м, 1H), 7,17-7,23 (м, 1H), 7,27-7,30 (м, 1H), 8,16 (д, J=8,40 Гц, 1H), 8,27-8,32 (м, 1H), 10,11 (с, 1H).
ESI-MS m/z 421 [M+H]+
[0395] Пример 61
Синтез 7-(2-метокси-3,5-диметилпиридин-4-ил)-1-[(3RS,4SR)-4-метокситетрагидрофуран-3-ил]-1H-пиразоло[4,3-c]хинолин-4(5H)-она
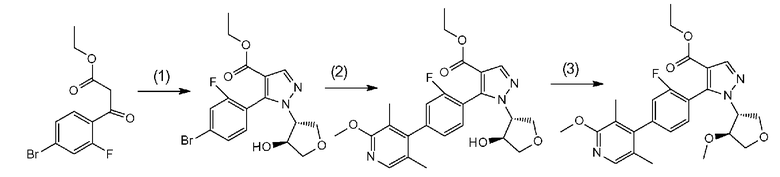
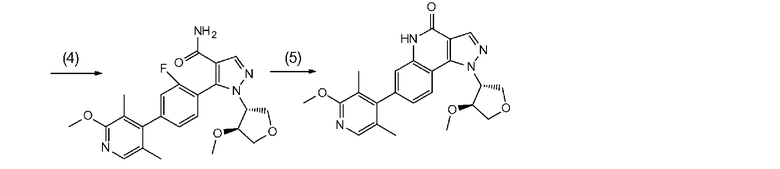
[0396] Указанное в заголовке соединение получали путем проведения реакций (1)-(5) в соответствии с примером 60 с применением 4-иод-2-метокси-3,5-диметилпиридина, полученного в примере получения 29(3).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,89-2,00 (м, 6H), 3,42 (д, J=0,59 Гц, 3H), 4,00 (с, 3H), 4,07-4,16 (м, 1H), 4,25-4,39 (м, 2H), 4,48 (дд, J=9,47, 6,35 Гц, 1H), 4,68 (дд, J=4,69, 1,95 Гц, 1H), 5,50 (ддд, J=6,20, 4,25, 1,86 Гц, 1H), 7,06-7,14 (м, 1H), 7,16-7,21 (м, 1H), 7,95 (с, 1H), 8,22 (д, J=8,40 Гц, 1H), 8,29-8,34 (м, 1H), 10,11 (с, 1H).
ESI-MS m/z 421 [M+H]+
[0397] Пример 62
Синтез (S)-7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
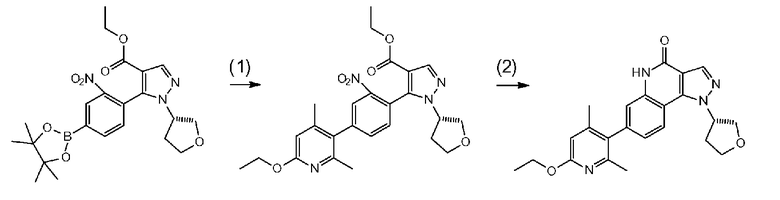
[0398] (1) Синтез этил-5-[4-(6-этокси-2,4-диметилпиридин-3-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Этил-5-[2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат, полученный в примере получения 7-(2) (200 мг), растворяли в смешанном растворе из 1,4-диоксана (4 мл) и воды (1 мл). Добавляли 3-бром-6-этокси-2,4-диметилпиридин, полученный в примере получения 51 (121 мг), Pd(PPh3)4 (25 мг) и карбонат цезия (428 мг) и в смеси проводили реакцию с помощью микроволнового реактора при 130°C в течение трех часов. Реакционной смеси давали остыть до комнатной температуры с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия. Осушитель удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (этилацетат/н-гептан, 30%-100%) с получением указанного в заголовке соединения (97 мг).
ESI-MS m/z 481 [M+H]+
[0399] (2) Синтез (S)-7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Этил-5-[4-(6-этокси-2,4-диметилпиридин-3-ил)-2-нитрофенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилат (97 мг) растворяли в уксусной кислоте (1 мл). К раствору добавляли железный порошок (56 мг) и смесь перемешивали при 90°C в течение четырех часов. Реакционной смеси давали остыть до комнатной температуры и к реакционной смеси добавляли воду (2 мл). Осажденное твердое вещество собирали фильтрацией и промывали водой. Полученное твердое вещество растворяли в этаноле (1 мл) при 90°C. Раствор охлаждали льдом, и осажденное твердое вещество собирали при помощи фильтрации. Полученное твердое вещество промывали посредством MTBE с получением указанного в заголовке соединения (16 мг).
1H-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,43 (т, J=7,0 Гц, 3H), 2,03 (с, 3H), 2,21 (с, 3H), 2,55-2,67 (м, 1H), 2,76-2,87 (м, 1H), 4,07-4,17 (м, 2H), 4,23-4,47 (м, 4H), 5,62-5,70 (м, 1H), 6,53 (с, 1H), 7,12 (дд, J=8,2 Гц, 1,6 Гц, 1H), 7,32 (д, J=1,6 Гц, 1H), 8,10 (д, J=8,6 Гц, 1H), 8,31 (с, 1H), 11,02 (ушир.с, 1H).
ESI-MS m/z 405 [M+H]+
[0400] Пример 63
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
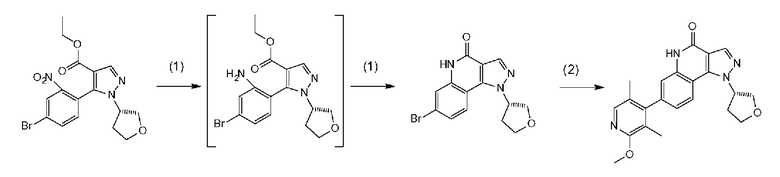
[0401] (1) Синтез (S)-7-бром-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Гидросульфит натрия (265 мг) добавляли к раствору этил-5-(4-бром-2-нитрофенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата, полученного в примере получения 7(1) (100 мг), в THF (1 мл) и воде (0,5 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 46 часов. Реакционную смесь охлаждали при 0°C и затем добавляли 5н соляную кислоту (0,25 мл). Смесь перемешивали при комнатной температуре в течение трех часов. После охлаждения при 0°C к реакционной смеси добавляли 5н водный раствор гидроксида натрия (0,25 мл). Смесь экстрагировали изопропилацетатом. Органический слой промывали водой и солевым раствором и затем концентрировали при пониженном давлении. Получали этил-5-(2-амино-4-бромфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразоло-4-карбоксилат (71 мг) в виде сырого очищенного продукта. Его использовали для следующей стадии без дополнительной очистки. Этил-5-(2-амино-4-бромфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразоло-4-карбоксилат (50 мг), полученный в виде сырого очищенного продукта, добавляли к уксусной кислоте (1 мл). Смесь перемешивали при 60°C в течение двух часов. После охлаждения реакционной смеси до комнатной температуры добавляли воду (1 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Осажденное твердое вещество собирали при помощи фильтрации. Твердое вещество промывали этанолом (1 мл) и затем сушили при пониженном давлении. Получали указанное в заголовке соединение (42 мг).
1H-ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 2,41-2,56 (м, 2H), 3,89-4,03 (м, 2H), 4,10-4,19 (м, 2H), 5,78 (м, 1H), 7,41-7,44 (м, 1H), 7,64-7,65 (м, 1H), 8,16-8,18 (м, 2H), 11,53 (с, 1H).
ESI-MS m/z 336 [M+H]+
[0402] (2) Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(S)-7-Бром-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он (100 мг), (2-метокси-3,5-диметил-пиридин-4-ил)бороновую кислоту, полученную в примере получения 29 (4) (65 мг), и карбонат цезия (293 мг) добавляли к смешанному раствору из DMF (5 мл) и воды (1 мл) при комнатной температуре. К смеси добавляли PdCl2(PPh3)2 (10,5 мг) в потоке газообразного азота. Смесь перемешивали при 80°C в течение одного часа и при 100°C в течение 4,5 часа. После охлаждения реакционной смеси до комнатной температуры добавляли воду (5 мл) и смесь экстрагировали изопропилацетатом. Органический слой промывали водой и солевым раствором и затем концентрировали при пониженном давлении. Получали сырой очищенный продукт (64,7 мг) в качестве указанного в заголовке соединения. Показания приборов для данного соединения были идентичны показаниям для (-)-формы из примера 25.
[0403] [Примеры фармакологических тестов]
Пример теста PDE9-ингибиторной активности
1) Получение рекомбинантного белка PDE9 человека
Амплифицировали кДНК-фрагмент hsPDE9A1, исходя из последовательности оснований (№ доступа: AF048837) hsPDE9A1, зарегистрированной в базе данных GenBank, и с применением следующих последовательностей (Hokkaido System Science Co., Ltd.) в качестве праймера, и кДНК-библиотеки гиппокампа человека (Clontech Laboratories, Inc.) в качестве матричной ДНК, и с применением ДНК-полимеразы Pfu50 (Invitrogen Corp.), и с помощью полимеразной цепной реакции (ПЦР) с приведенными далее условиями.
Праймер hPDE9-1: AGGATGGGATCCGGCTCCTCCA (SEQ No. 1).
Праймер hPDE9A-3: CAGGCACAGTCTCCTTCACTG (SEQ No. 2).
Условия ПЦР: [96°C, 5 мин] × 1 цикл, [(96°C, 10 секунд), (57°C, 5 секунд), (72°C, 2 мин)] × 30 циклов
[0404] Полученный кДНК-фрагмент hsPDE9A1 встраивали в вектор клонирования TOPO-TA (Invitrogen Corp.) и проверяли последовательностью оснований; и после этого продукт реакции трансфицировали в вектор pcDNA 3.1/myc His-метка (Invitrogen Corp.), таким образом получая вектор экспрессии PDE9 человека для клеток млекопитающих. Вектор экспрессии PDE9 человека для клеток млекопитающих трансфицировали с временной экспрессией в клетку HEK293 с помощью реактива LIPOFETAMINE 2000 (Gibco). С помощью способа вестерн-блоттинга подтверждали, что PDE9A экспрессировалась в клетке HEK293, а затем кДНК-фрагмент PDE9A1 трансфицировали в вектор pYNG (Katakura Industries Co., Ltd.), таким образом получая вектор экспрессии для клеток насекомых. Супернатант гомогенизированного шелкопряда, у которого экспрессировалось большое количество PDE9, очищали с помощью уравновешенной Ni-колонки с применением буфера A (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT, 10 ммоль/л имидазола). Через 1 час после перемешания супернатанта и Ni-колонки проводили очистку с помощью буфера B (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT) и осуществляли элюирование с помощью буфера C (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT, 100 ммоль/л имидазола). Препаративно собирали фракцию элюирования, таким образом получая раствор фермента PDE9.
[0405] 2) Измерение PDE9-ингибирующего действия
К 100 мкл раствора буфера D (40 ммоль/л Tris-HCl, pH: 7,4, 10 ммоль/л MgCl2, 1 мМ DTT, 2 мкМ cGMP), содержащего [3H]-cGMP (0,5 мкКи/мл), 10 мкл раствора соединения для оценки (раствора, в котором соединение растворяли в ДМСО и разбавляли так, чтобы концентрация ДМСО становилась 5%) и 90 мкл раствора, приготовленного путем разбавления приготовленного как описано выше раствора фермента PDE9 буфером E (40 ммоль/л Tris-HCl, pH: 7,4, 10 ммоль/л MgCl2, 1 мм DTT, 1 ммоль/л EGTA) добавляли в условиях охлаждения льдом. Полученный смешанный раствор инкубировали при 30°C в течение 10 мин, а после этого нагревали в течение 2 мин в кипяченой воде для остановки ферментативной реакции PDE9. Затем полученному раствору давали остыть до комнатной температуры; к нему добавляли 50 мкл 5′-нуклеотидазы (Biomol GmbH, 10 единиц/мл); и полученный раствор инкубировали при 30°C в течение 10 мин с превращением, таким образом, образовавшегося в результате предыдущей реакции [3H]-5′-GMP в [3H]-гуанозин. 500 мкл анионообменной смолы (смолы Bio-Rad AG1-X2, размер ячейки: 200-400, H2O:смола = 2:1) добавляли к полученной жидкости и позволяли отстаиваться в течение 10 мин, а после этого центрифугировали (2000 об/мин, 10 мин); и супернатант, в котором присутствовал [3H]-гуанозин, переносили в LumaPlate (PerkinElmer, Inc.) и измеряли радиоактивность с помощью счетчика люминесцентных вспышек для микропланшета TopCount NXT (PerkinElmer, Inc.).
[0406] Процент ингибирования у оцениваемого соединения рассчитывали с помощью приведенной далее формулы, принимая радиоактивность контроля, не содержащего оцениваемое соединение, за (A), радиоактивность холостой пробы, не содержащей фермент, за (B) и радиоактивность оцениваемого соединения за (C).
Процент ингибирования = 100-{[(C)-(B)]/[(A)-(B)]}×100(%)
Величину IC50 для PDE9 у оцениваемого соединения определяли из процента ингибирования для различных концентраций. Величина IC50 у каждого оцениваемого соединения показана в таблице 10.
[0407]
[0408] 3) Влияние на cGMP цереброспинальной жидкости грызунов
Тестируемое соединение вводили самцам мышей ICR (Charles River Laboratories Japan, Inc.), самцам крыс Sprague-Dawley (SD) (Charles River Laboratories Japan, Inc.) или самцам крыс Long-Evans (LE) (Institute for Animal Reproduction), а затем в условиях анестезии пентобарбиталом собирали цереброспинальную жидкость и хранили при -20°C. cGMP в цереброспинальной жидкости измеряли согласно EIA-процедуре с ацетилированием в наборе cGMP EIA (GE Healthcare) или процедуре без ацетилирования в наборе cGMP EIA (Cayman). Результатом было повышение (C) количества cGMP у группы, которой вводили тестируемое соединение (B), по отношению к количеству cGMP у группы, которой вводили носитель (A), и рассчитывали с помощью следующей формулы:
Повышение cGMP (C) = [(B)-(A)]/(A)×100(%)
Результаты показаны в приведенной далее таблице.
или (R/S)
[0409] 4) Влияние на cGMP гиппокампа грызунов
Тестируемое соединение вводили самцам крыс Sprague-Dawley (Charles River Laboratories Japan, Inc.) или самцам крыс Long-Evans (Institute for Animal Reproduction), а затем животных умерщвляли с помощью микроволн под анестезией пентобарбиталом и извлекали гиппокамп. После измерения сырого веса гиппокамп замораживали с помощью жидкого азота и хранили при -80°C. При измерении cGMP в гиппокампе добавляли раствор 0,5M перхлорной кислоты/1 мм EDTA в количестве 5% (вес/объем) по сырому весу и гомогенизировали смесь. После гомогенизации гомогенат центрифугировали (10000 об/мин, 15 мин) и собирали супернатант. Собранный супернатант нейтрализовали посредством 2М раствора бикарбоната калия и центрифугировали (13000 об/мин, 10 мин). Концентрацию cGMP в супернатанте измеряли согласно EIA-процедуре без ацетилирования в наборе cGMP EIA (GE Healthcare). Результатом было повышение (C) количества cGMP у группы, которой вводили тестируемое соединение (B), по отношению к количеству cGMP у группы, которой вводили носитель (A), и рассчитывали с помощью следующей формулы:
Повышение cGMP (C) = [(B)-(A)]/(A)×100(%)
Результаты показаны в приведенной далее таблице.
или
(R/S)
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЬ ПРОИЗВОДНОГО ПИРАЗОЛОХИНОЛИНА И ЕЕ КРИСТАЛЛ | 2014 |
|
RU2655171C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛОПИРИМИДИНА | 2007 |
|
RU2420530C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2016 |
|
RU2687245C1 |
| ПИРИДИНИЛПИРАЗОЛОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2655172C2 |
| МЕСТНОЕ ПРОТИВОГРИБКОВОЕ СРЕДСТВО | 2012 |
|
RU2593990C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2006 |
|
RU2409573C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА | 2004 |
|
RU2332412C2 |
| ПРОИЗВОДНОЕ ЦИКЛОГЕКСАНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2478621C2 |
| НОВОЕ ИМИДАЗООКСАЗИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2012 |
|
RU2578608C2 |

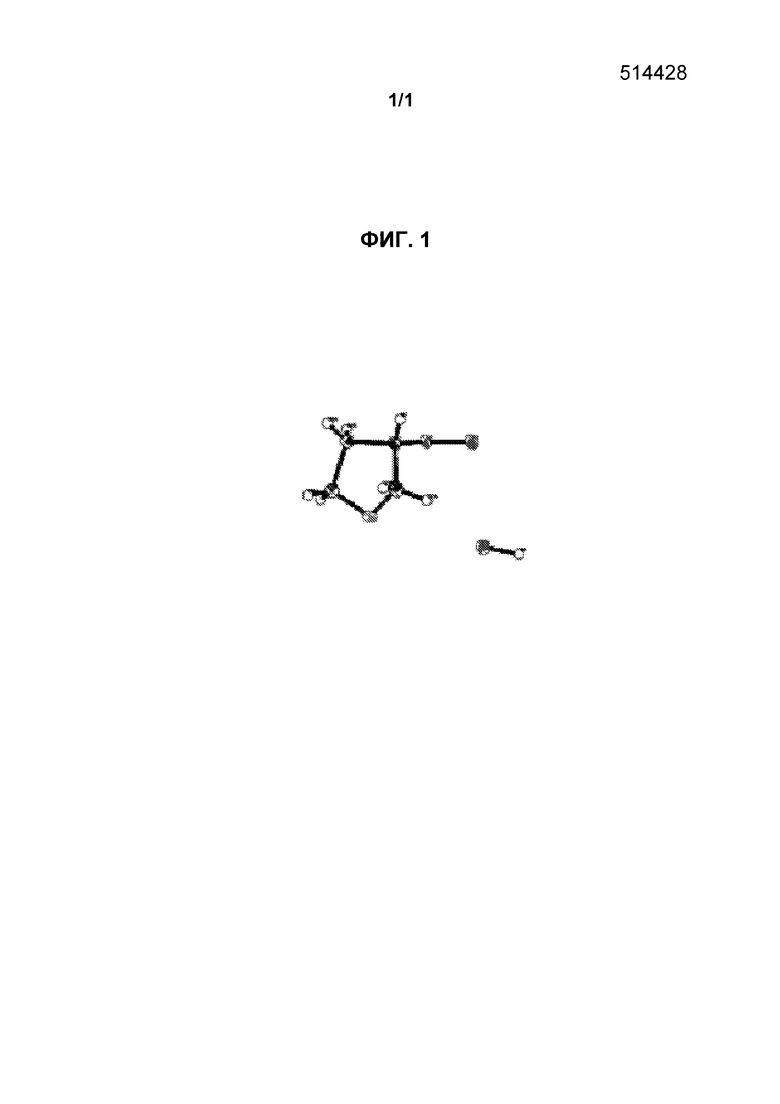
Изобретение относится к соединению, представленному формулой (I), или к его фармакологически приемлемой соли, где R1 является атомом водорода; R2 является ароматической кольцевой группой, определенной в формуле изобретения, R3 является атомом водорода или фтора, R4 является атомом водорода; R5 является оксепанильной группой и т.д.; R6 является атомом водорода. Заявленное соединение обладает PDE9-ингибирующим действием. Поэтому возможно повышение концентрации внутримозгового cGMP. PDE9-ингибирующее действие и повышение cGMP приводит к улучшению протекания обучения и развития памяти. Соединение формулы (I) может применяться в качестве терапевтического средства от когнитивных дисфункций при болезни Альцгеймера. 12 н. и 7 з.п. ф-лы, 1 ил., 12 табл., 63 пр.
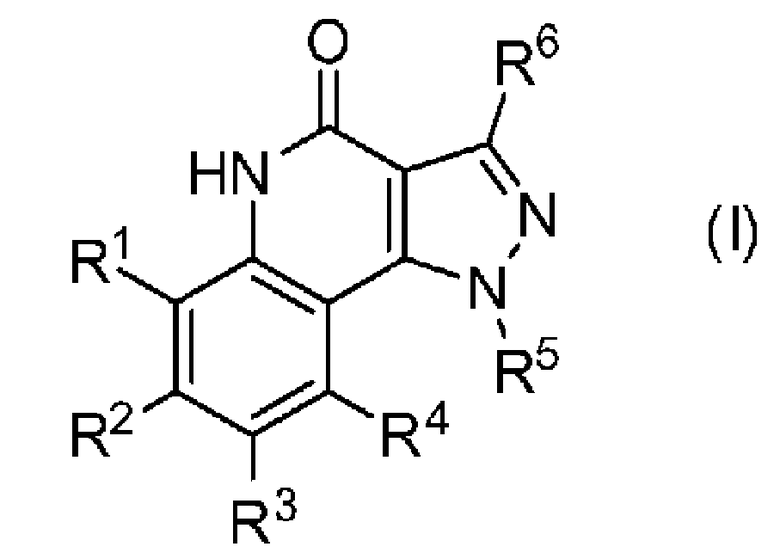
1. Соединение, представленное формулой (I):
или его фармакологически приемлемая соль,
где R1 является атомом водорода;
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, пиридинильной группы и пиримидинильной группы, где каждый из двух атомов в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы А1, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы В1;
R3 является атомом водорода или атомом фтора;
R4 является атомом водорода;
R5 является оксепанильной группой, диоксепанильной группой, тетрагидропиранильной группой или тетрагидрофуранильной группой, необязательно замещенной метоксигруппой;
R6 является атомом водорода;
группа А1 состоит из атома галогена, С1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, и С1-6-алкоксигруппы; и
группа В1 состоит из атома галогена, цианогруппы, С1-6-алкильной группы, необязательно замещенной 1-3 атомами галогена, С1-6-алкокси-С1-6-алкильной группы, C1-6-алкоксигруппы, необязательно замещенной 1-3 атомами галогена, и тетрагидропиранильной группы,
при условии, что если R2 является 3-пиридинильной группой, то заместитель в 4-м положении является атомом галогена или С1-6-алкильной группой, необязательно замещенной 1-3 атомами галогена.
2. Соединение или его фармакологически приемлемая соль по п. 1, где
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы, 4-пиридинильной группы и 5-пиримидинильной группы, где каждый из двух атомов в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-c]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы А2, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы В2;
R5 является 4-оксепанильной группой, 1,4-диоксепан-6-ильной группой, 3,4,5,6-тетрагидро-2Н-3-пиранильной группой, 3,4,5,6-тетрагидро-2Н-4-пиранильной группой или 3-тетрагидрофуранильной группой;
группа А2 состоит из атома хлора и метильной группы, необязательно замещенной 1-2 атомами фтора, этильной группы, метоксигруппы и этоксигруппы; и
группа В2 состоит из атома фтора, атома хлора, цианогруппы, метильной группы, необязательно замещенной 1-3 атомами фтора, этильной группы, метоксиметильной группы, метоксигруппы, необязательно замещенной 1-3 атомами фтора, этоксигруппы, изопропилоксигруппы и 3,4,5,6-тетрагидро-2Н-4-пиранильной группы.
3. Соединение или его фармакологически приемлемая соль по п. 2, где R3 является атомом фтора.
4. Соединение или его фармакологически приемлемая соль по п. 1, где
R3 является атомом водорода; и
R5 является тетрагидропиранильной группой или тетрагидрофуранильной группой, необязательно замещенной метоксигруппой.
5. Соединение или его фармакологически приемлемая соль по п. 2, где
R3 является атомом водорода; и
R5 является 3,4,5,6-тетрагидро-2Н-3-пиранильной группой, 3,4,5,6-тетрагидро-2Н-4-пиранильной группой или 3-тетрагидрофуранильной группой.
6. Соединение или его фармакологически приемлемая соль по п. 1, где
R2 является ароматической кольцевой группой, выбранной из группы, состоящей из фенильной группы, 3-пиридинильной группы и 4-пиридинильной группы, где каждый из двух атомов в ароматическом кольце, которые находятся рядом с атомом углерода, соединенным с пиразоло[4,3-с]хинолиновым кольцом, независимо имеют заместитель, выбранный из группы A3, а другие атомы в ароматическом кольце независимо необязательно имеют заместитель, выбранный из группы В3;
R3 является атомом водорода;
R4 является атомом водорода;
R5 является 3,4,5,6-тетрагидро-2Н-4-пиранильной группой или 3-тетрагидрофуранильной группой;
группа A3 состоит из метильной группы и метоксигруппы; и
группа В3 состоит из метильной группы, метоксигруппы и метоксиметильной группы.
7. Соединение или его фармакологически приемлемая соль, выбранные из следующей группы:
1) 7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
2) 7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
3) (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
4) 8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
5) 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
6) 1-(1,4-диоксепан-6-ил)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
7) (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
8) 7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
9) (-)-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
10) (-)-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
11) (S)-8-фтор-7-(2-метокси-4,6-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
12) (S)-7-(6-этокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он,
13) (S)-8-фтор-7-(6-метокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он и
14) (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он.
8. 7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль.
9. (S)-7-(6-изопропилокси-2,4-диметилпиридин-3-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль:
10. 8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль.
11. (S)-8-фтор-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль:

12. 7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль.
13. (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль:
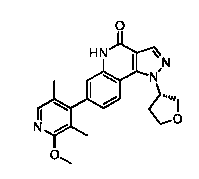
14. 1-(1,4-диоксепан-6-ил)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1Н-пиразоло[4,3-с]хинолин-4(5Н)-он или его фармакологически приемлемая соль:
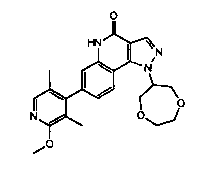
15. Фармацевтическая композиция, ингибирующая PDE9, содержащая соединение или его фармакологически приемлемую соль по п. 1 в качестве активного ингредиента.
16. Фармацевтическая композиция, ингибирующая PDE9, по п. 15 для повышения концентрации внутримозгового cGMP.
17. Средство для облегчения когнитивного нарушения при болезни Альцгеймера, содержащее соединение или его фармакологически приемлемую соль по п. 1.
18. Способ облегчения когнитивного нарушения при болезни Альцгеймера, включающий введение пациенту соединения или его фармакологически приемлемой соли по п. 1.
19. Соединение или его фармакологически приемлемая соль по п. 1 для применения для облегчения когнитивного нарушения при болезни Альцгеймера.
| УСТРОЙСТВО ДЛЯ ОБОГРЕВА ПОМЕЩЕНИЙ | 1996 |
|
RU2103613C1 |
| ХРУСТЯЩИЙ НАПОЛНИТЕЛЬ ДЛЯ МОЛОКА И/ИЛИ МОЛОЧНОГО ПРОДУКТА, КОТОРЫЙ ПРЕДСТАВЛЯЕТ СОБОЙ СМЕСЬ ОРЕХОВ, КАРАМЕЛИ, ЭКСТРУЗИОННЫХ ШАРИКОВ, ШОКОЛАДА И ПЕЧЕНЬЯ | 2020 |
|
RU2754457C1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| EP 1925617 A1, 28.05.2008 | |||
| ПИРАЗОЛОПИРИДИНЫ И ИХ АНАЛОГИ | 2004 |
|
RU2426734C2 |

