В настоящем изобретении предложены новые производные оксима хромона, который является модулятором рецепторов нервной системы, восприимчивых к нейровозбуждающей аминокислоте глутамату и, кроме того, обеспечивает выгодное высокое экспонирование мозга после перорального введения. Указанные свойства делают производное оксима хромона в соответствии с настоящим изобретением, в частности, пригодным для использования в качестве лекарственного средства, например, при лечении или для профилактики острых и хронических неврологических и/или психиатрических расстройств.
Известно, что глутамат принимает участие во множестве нервных функций. Поэтому важную роль приписывают глутаматергическим рецепторам, в частности, в том, что касается состояния нервных импульсов, синаптической пластичности, развития нервной системы, обучаемости и памяти.
Глутамат также является эндогенным нейротоксином, ответственным за гибель нейронов, наблюдающуюся после ишемии, гипоксии, эпилептических припадков или травматических поражений. Поэтому рецепторы глутамата четко признаются участвующими в различных расстройствах нервной системы и в нейродегенеративных заболеваниях.
Глутаматергическая система включает рецепторы глутамата и транспортеры, также как ферменты метаболизма глутамата. Были охарактеризованы два основные типа глутаматергических рецепторов: ионотропные (iGluRs) и метаботропные (mGluRs) рецепторы. Ионотропные рецепторы глутамата были идентифицированы на основании их фармакологии и затем на основании молекулярной биологии. Семейство iGluR включает NMDA (N-метил-D-аспартат), AMPA (альфа-амино-3-гидрокси-5-метил-4-изоксазолпропионовую кислоту) и подсемейство каинатных рецепторов, названия для химического агониста, который селективно связывается с членами подсемейства. iGluRs представляют собой потенциалозависимые ионные каналы, которые обеспечивают приток катионов после глутаматного связывания. Они непосредственно отвечают за создание потенциалов действия, они инициируют нейропластические изменения в ЦНС и ответственны за многие заболевания, включая хроническую боль. Метаботропные рецепторы глутамата представляют собой семейство, состоящее из семи трансмембранных доменов G-белок-связанных рецепторов (GPCR). До сих пор было идентифицировано восемь подтипов mGluR (mGluR1-mGluR8) и их подразделили на три группы (I-III) на основании гомологии последовательностей, механизма трансдукции и фармакологического профиля. mGluRs принадлежат семейству 3 суперсемейства GPCR, и как таковые, они характеризуются большим внеклеточным аминоконцевым доменом (ATD), в котором расположен сайт связывания глутамата. mGluRs локализованы по всей нервной системе (центральной и периферической), и было показано, что они играют роль в гомеостазе во многих системах органов. Было обнаружено, что они играют важную роль, в частности, в индуцировании длительного потенцирования (LTP) и длительной депрессии (LTD) синаптической передачи, в регулировании барорецепторных рефлексов, пространственном обучении, моторном научении, профессиональной и кинетической интеграции, и также считают, что они принимают участие в острых или хронических или дегенеративных заболеваниях, таких как болезнь Паркинсона, леводопа-индуцированная дискинезия, болезнь Альцгеймера, амиотропный латеральный склероз, спиноцеребеллярная атаксия, эпилепсия или болезнь Хантингтона, также как в нейропсихиатрических расстройствах, таких как беспокойство, депрессия, расстройство аутистического спектра, пост-травматические стрессовые расстройства и шизофрения.
Таким образом, было четко продемонстрировано, что, глутаматергические схемы участвуют в физиопатологии ряда повреждений и поражений нейронов. Многие расстройства нервной системы, включая эпилепсию и хронические или острые дегенеративные процессы, такие, как например, болезнь Альцгеймера, болезнь Хантингтона, болезнь Паркинсона и амиотрофический латеральный склероз (Mattson MP., Neuromolecular Med., 3(2), 65-94, 2003), но также СПИД-индуцированная деменция, рассеянный склероз, спинально-мышечная атрофия, ретинопатия, удар, ишемия, гипоксия, гипогликемию и различные травматические поражения мозга, включают гибель клеток нейронов, вызванную нарушенными уровнями глутамата. Было также показано, что индуцированную лекарственными средствами нейротоксичность, например, нейротоксические эффекты метамфетамина (METH) на стриарные допаминергические нейроны, можно действительно опосредовать за счет избыточной стимуляции рецепторов глутамата (Stagehans SE and Yamamoto BK, Synapse 17(3), 203-9, 1994). Антидепрессантные и подобные анксиолитическим эффекты соединений, воздействующих на глутамат, также наблюдались на мышах, предполагая, что глутаматергическая трансмиссия задействована в патофизиологии аффективных расстройств, таких как глубокая депрессия, шизофрении и беспокойство (Palucha A et al., Pharmacol.Ther. 115(1), 116-47, 2007; Cryan JF et al., Eur. J. Neurosc. 17(11), 2409-17, 2003; Conn PJ et al., Trends Pharmacol. Sci. 30(1), 25-31, 2009). Соответственно, любое соединение, способное модулировать глутаматергическую передачу сигнала или функцию будет представлять собой перспективное терапевтическое соединение для лечения многих расстройств нервной системы.
Кроме того, соединения, модулирующие уровни глутамата, или передачу сигнала могут представлять большую терапевтическую ценность для лечения эпилепсии и/или расстройств, которые прямо не опосредованы уровнями глутамата и/или нарушением функций рецепторов глутамата, но на которые можно влиять, изменяя уровень содержания глутаматов или передачу сигнала.
Аминокислота L-глутамат (в описании именуемая просто как глутамат) представляет собой основной возбуждающий нейротрансмиттер центральной и периферической нервной системы млекопитающих (ЦНС и ПНС, соответственно). Она принимает участие во всех функциях нервной системы и воздействует на развитие нервной системы на всех стадиях, начиная с миграции нейронов, дифференциации и гибели до образования и удаления синапсов. Глутамат повсеместно распределен в высоких концентрациях в нервной системе и вовлечен практически во все физиологические функции, такие как обучение и память, двигательный контроль, развитие синаптической пластичности, сенсорные восприятия, зрение, дыхание и регулирование сердечно-сосудистой деятельности (Meldrum,2000). Известно, что нарушения в глутаматергической системе вызывают нейротоксичность и другие вредные воздействия на нейропередачу, нейроэнергетику и жизнеспособность клеток. Соответственно, было проведено значительное число исследований, чтобы исследовать потенциальные связи между глутаматергической системой и неврологическими или психиатрическими расстройствами.
Глутамат действует через два класса рецепторов (Bräuner-Osborne H et al., J. Med. Chem. 43(14), 2609-45, 2000). Первый класс рецепторов глутамата непосредственно связан с раскрытием катионных каналов в клеточной мембране нейронов. Поэтому их называют ионотропными рецепторами глутамата (iGluRs). iGluRs подразделяют на три подтипа, которые называют в соответствии с их селективными агонистами: N-метил-D-аспартатом (NMDA), α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислотой (AMPA) и каинатом (KA). Второй класс рецепторов глутамата состоит из G-белок-связанных рецепторов (GPCRs), называемых метаботропными рецепторами глутамата (mGluRs). Указанные mGluRs локализованы как пре-, так и постсинаптически. Они объединены со множеством вторичных мессенджеров системы, и их роль состоит в регулировке активности ионных каналов или ферментов, продуцирующих вторичные мессенджеры за счет связывания G-белков с GTP (Conn PJ и Pin JP., Annu. Rev. Pharmacol. Toxicol., 37, 205-37, 1997). Хотя обычно они непосредственно не участвуют в быстрой синаптической передаче, mGluRs модулируют эффективности синапсов за счет регулирования или пост-синаптических каналов и их рецепторов, или пре-синаптического высвобождения или повторного захвата глутамата. Поэтому mGluRs играют важную роль в различных психологических процессах, таких как длительное потенцирование (LTP) и длительная депрессия (LTD), синаптическая передача, регулирование барорецептивных рефлексов, пространственное обучение, моторное научение, и профессиональная и кинетическая интеграция.
К настоящему времени были клонированы и классифицированы три группы mGluRs в соответствии с их гомологией последовательностей, фармакологическими свойствами и механизмами передачи сигналов. Группа I включает mGluR1 и mGluR5, группа II включает mGluR2 и mGluR3 и группа III включает mGluR4, mGluR6, mGluR7 и mGluR8 (Pin JP и Acher F., Curr. Drug Targets CNS Neurol. Disord., 1(3), 297-317, 2002; Schoepp DD et al., Neuropharmacology, 38(10), 1431-76, 1999).
mGluR лиганды/модуляторы можно подразделить на два семейства в зависимости от их сайтов взаимодействия с рецепторами (См. Bräuner-Osborne H et al., J. Med. Chem. 43(14), 2609-45, 2000 для обзора). Первое семейство состоит из ортостерических лигандов (или конкурирующих лигандов), способных взаимодействовать с глутамат связывающим-сайтом mGluRs, который локализован в большой внеклеточной N-терминальной части рецептора (около 560 аминокислот). Примерами ортостерических лигандов являются S-DHPG или LY-367385 для mGluRs группы I, LY-354740 или (2R-4R)-APDC для mGluRs группы II и ACPT-I или L-AP4 для mGluRs группы III. Второе семейство mGluRs лигандов состоит из аллостерических лигандов/модуляторов, которые взаимодействуют с сайтами, отличающимися от внеклеточных активных сайтов рецептора (См. Bridges TM et al., ACS Chem Biol, 3(9), 530-41, 2008 для обзора). Их действие приводит к модуляции эффектов, индуцированных эндогенным лигандом глутаматом. Примерами таких аллостерических модуляторов являются Ro-674853, MPEP или JNJ16259685 для mGluRs группы I и CBiPES, LY181837 или LY487379 для mGluRs группы II.
Примеры аллостерических модуляторов были раскрыты для mGluR подтипа 4 (mGluR4). PHCCC, MPEP и SIB1893 (Maj M et al., Neuropharmacology, 45(7), 895-903, 2003; Mathiesen JM et al., Br. J. Pharmacol. 138(6), 1026-30, 2003) были первыми, описанными в 2003. Позднее в литературе появились сообщения о более эффективных позитивных аллостерических модуляторах (Niswender CM et al., Mol. Pharmacol. 74(5), 1345-58, 2008; Niswender CM et al., Bioorg. Med. Chem. Lett. 18(20), 5626-30, 2008; Williams R et al., Bioorg. Med. Chem. Lett. 19(3), 962-6, 2009; Engers DW et al., J. Med. Chem. May 27 2009) и в двух патентных публикациях, раскрывающих семейства амидо и гетероароматических соединений (WO 2009/010454 и WO 2009/010455).
Многочисленные исследования уже раскрыли потенциальные применения mGluR модуляторов как нейропротекторов (См. Bruno V et al., J. Cereb. Blood Flow Metab., 21(9), 1013-33, 2001 для обзора). Например, антагонисты mGluRs группы I демонстрируют интересные результаты в моделях на животных для изучения беспокойства и пост-ишемических поражений (Pilc A et al., Neuropharmacology, 43(2), 181-7, 2002; Meli E et al., Pharmacol. Biochem. Behav., 73(2), 439-46, 2002), агонисты mGluRs группы II демонстрируют хорошие результаты в моделях на животных болезни Паркинсона и беспокойства (Konieczny J et al., Naunyn-Schmiederbergs Arch. Pharmacol., 358(4), 500-2, 1998).
Модуляторы mGluR группы III демонстрируют положительные результаты в нескольких моделях на животных шизофрении (Palucha-Poniewiera A et al., Neuropharmacology, 55(4), 517-24, 2008) и хронической боли (Goudet C et al., Pain, 137(1), 112-24, 2008; Zhang HM et al., Neuroscience, 158(2), 875-84, 2009).
Было также показано, что mGluR группы III проявляют экситотоксические действия гомоцистеина и гомоцистеиновой кислоты, внося свой вклад в патологию нейронов и старение иммунной системы, которые наблюдаются при болезни Альцгеймера (Boldyrev AA и Johnson P, J. Alzheimers Dis. 11(2), 219-28, 2007).
Кроме того, модуляторы mGluR группы III демонстрируют обнадеживающие результаты в моделях на животных болезни Паркинсона и нейродегенерации (Conn PJ et al., Nat. Rev. Neuroscience, 6(10), 787-98, 2005 для обзора; Vernon AC et al., J. Pharmacol. Exp. Ther., 320(1), 397-409, 2007; Lopez S et al., Neuropharmacology, 55(4), 483-90, 2008; Vernon AC et al., Neuroreport, 19(4), 475-8, 2008; Williams CJ et al., J. Neurochem., 129(1), 4-20, 2014 для обзора). Дополнительно было продемонстрировано с селективными лигандами, что подтипом mGluR, который участвует в таких антипаркинсонических и нейропротекторных эффектах, является mGluR4 (Marino MJ et al., Proc. Natl. Acad. Sci. USA 100(23), 13668-73, 2003; Battaglia G et al., J. Neurosci. 26(27), 7222-9, 2006; Niswender CM et al., Mol. Pharmacol. 74(5), 1345-58, 2008).
Было также показано, что модуляторы mGluR4 проявляют анксиолитическую активность (Stachowicz K et al., Eur. J. Pharmacol., 498(1-3), 153-6, 2004) и обладают антидепрессивными свойствами (Palucha A et al., Neuropharmacology 46(2), 151-9, 2004; Klak K et al., Amino Acids 32(2), 169-72, 2006).
Кроме того, было также показано, что mGluR4 участвуют в ингибировании секреции глюкагона (Uehara S., Diabetes 53(4), 998-1006, 2004). Поэтому, ортостерические или позитивные аллостерические модуляторы mGluR4 обладают потенциалом лечения диабета 2 типа за счет их гипогликемического действия.
Кроме того, было показано, что mGluR4 экспрессируются в клеточной линии рака простаты (Pessimissis N et al., Anticancer Res. 29(1), 371-7, 2009) или колоректальной карциномы (Chang HJ et al., Cli. Cancer Res. 11(9), 3288-95, 2005) и было показано, что их активация за счет PHCCC ингибирует рост медуллобластомы (Iacovelli L et al., J. Neurosci. 26(32) 8388-97, 2006). mGluR4 модуляторы могут поэтому обладать потенциалом для лечения раковых заболеваний.
И наконец, было показано, что рецепторы вкуса глутамата, экспрессируемые в тканях вкуса, являются вариантами mGluR4 рецептора (Eschle BK., Neuroscience, 155(2), 522-9, 2008). Как следствие, mGluR4 модуляторы можно также использовать как вкусовые агенты, ароматизаторы, усилители вкуса или пищевые добавки.
Полученные из хромона ядерные структуры для фармацевтически активных соединений были раскрыты в патентной заявке WO 2004/092154. В этой последней заявке они раскрыты как ингибиторы протеинкиназ.
EP A 0 787723 относится к производным специфической циклопропахроменкарбоновой кислоты, которые, как считают, обладают mGluR антагонистической активностью.
Новый класс лигандов метаботропных рецепторов глутамата раскрыт в WO 2011/051478. Производные оксима хромона, представленные в указанном документе, представляют собой высокоэффективные модуляторы mGluRs, в частности, позитивные аллостерические модуляторы mGluR4, и могут с успехом использоваться в качестве фармацевтических средств, в частности, при лечении или для профилактики острых и хронических неврологических и/или психиатрических расстройств.
В контексте настоящего изобретения было неожиданно обнаружено, что новые производные оксима хромона из класса соединений, раскрытых в WO 2011/051478, не только демонстрируют эффективную активность как позитивные аллостерические модуляторы mGluRs, но также обладают крайне выгодными фармакокинетическими свойствами. В частности, было обнаружено, что такое новое соединение формулы (I), как представлено далее, демонстрирует улучшенное экспонирование мозга после перорального введения по сравнению с соединениями, раскрытыми в WO 2011/051478. Таким образом, настоящее изобретение решает проблему создания новых эффективных модуляторов mGluRs, в частности, позитивных аллостерических модуляторов mGluR4, которые обладают улучшенными фармакокинетическими свойствами, и, в частности, повышенной проницаемостью мозга.
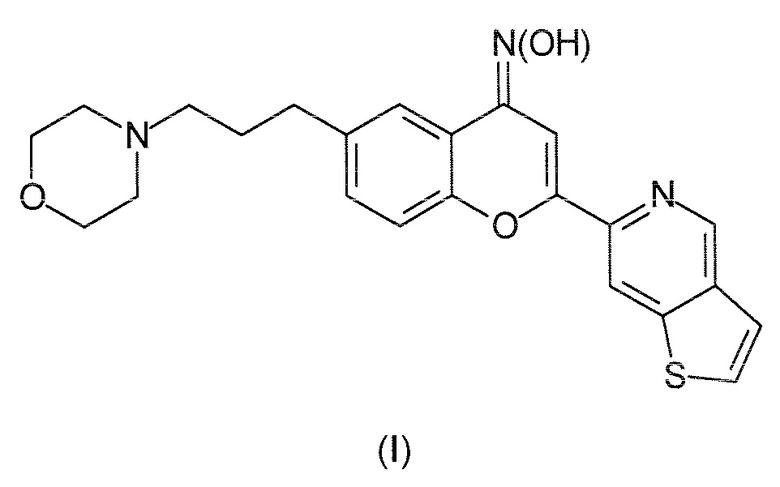
Таким образом, настоящее изобретение относится к соединениям следующей формулы (I):
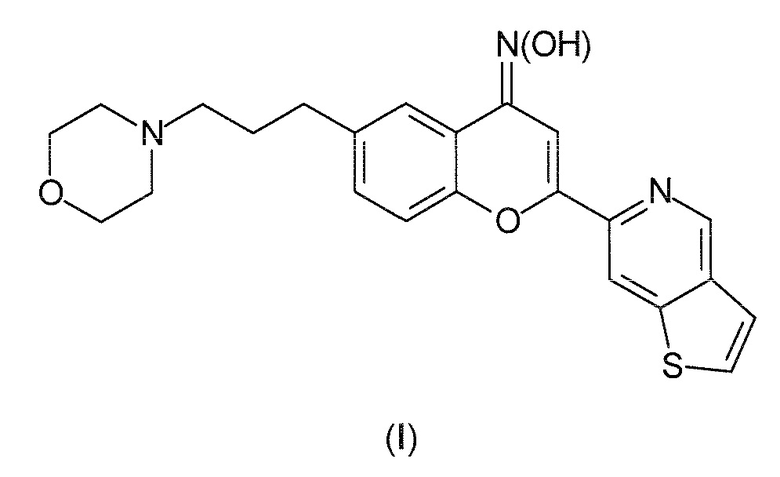
или к их фармацевтически приемлемым солям, сольватам или пролекарствам. В рассматриваемом описании соединение формулы (I) также именуется как ʺPXT002331ʺ.
Соответственно, настоящее изобретение относится к соединению оксиму 6-(3-морфолин-4-илпропил)-2-(тиено[3,2 c]пиридин-6-ил)-4H хромен-4-она или к его фармацевтически приемлемым солям, сольватам или пролекарствам.
Было обнаружено, что соединение формулы (I) в соответствии с настоящим изобретением практически сохраняет эффективную терапевтическую активность структурно родственного соединения в соответствии с примером 127 WO 2011/051478, хотя, неожиданно, демонстрирует улучшенные фармакокинетические характеристики, и, в частности, значительно улучшенное экспонирование мозга, что также продемонстрировано в примере 2.
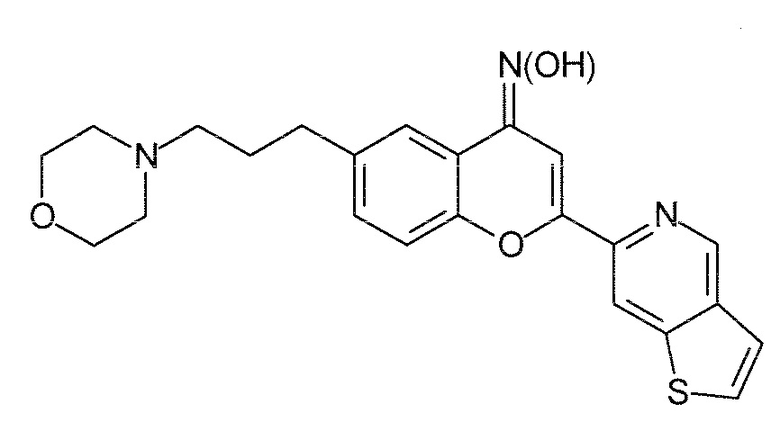
(ʺPXT002331ʺ)
(ʺPXT001858ʺ)
(T=1,5 час)=2,0
Указанные усовершенствованные фармакокинетические характеристики делают соединение формулы (I) крайне выгодным в качестве фармацевтического средства, в частности, в роли фармацевтического пенетранта, например, для медицинского вмешательства при неврологических и/или психиатрических расстройствах. В соответствии с таким выигрышным фармакокинетическим профилем было показано, что соединение формулы (I) обладает эффективной антипаркинсонической активностью в модели MPTP на макаках болезни Паркинсона, в частности, при дозах, которые ниже или равны 25 мг/кг при пероральном введении, что подробно раскрыто в примере 3.
В соответствии с настоящим изобретением соединение формулы (I) можно использовать в качестве модулятора проницаемости мозга mGluRs нервной системы. В частности, соединение формулы (I) можно использовать как аллостерический модулятор проницаемости мозга mGluRs и наболе6е перспективно его использовать в качестве позитивного аллостерического модулятора проницаемости мозга mGluR4.
Настоящее изобретение также относится к фармацевтическим композициям, включающим соединение формулы (I) или его фармацевтически приемлемые соли, сольваты или пролекарства в комбинации с фармацевтически приемлемым эксципиентом. Соответственно, настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемым солям, сольватам или пролекарствам для использования в качестве лекарственного средства.
Настоящее изобретение, кроме того, относится к соединению формулы (I) или к его фармацевтически приемлемым солям, сольватам или пролекарствам или к фармацевтическим композициям, включающим любое из вышеперечисленных в комбинации с фармацевтически приемлемый эксципиентом, для использования при лечении или для профилактики состояния, связанного с нарушенной глутаматергической передачей сигнала и/или функцией, или состояния, на которое можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала. Такое использование соединения формулы (I) или его фармацевтически приемлемых солей, сольватов или пролекарств для получения лекарственных средств для лечения или профилактики состояния, связанного с нарушением глутаматергический передачи сигнала и/или функции, или состояния, на которое можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала, также включено в объем настоящего изобретения.
Кроме того, настоящее изобретение относится к способу лечения или предотвращение состояния, связанного с нарушением глутаматергической передачи сигнала и/или функции, или состояния, на которое можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала у млекопитающего. Соответственно, в настоящем изобретении предложен способ лечения или предотвращения состояния, связанного с нарушением глутаматергической передачи сигнала и/или функции, или состояния, на которое можно воздействовать, изменяя уровень содержания глутамата или передачу сигнала, причем указанный способ включает введение соединения формулы (I) или его фармацевтически приемлемых солей, сольватов или пролекарств или фармацевтических композиций, включающих любое из вышеперечисленных в комбинации с фармацевтически приемлемым эксципиентом, нуждающемуся в этом субъекту (предпочтительно млекопитающему, и более предпочтительно человеку).
Состояния, связанные с нарушением глутаматергической передачи сигнала и/или функции, или состояния, на которые можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала, которые можно лечить и/или предотвратить, используя соединение или фармацевтические композиции в соответствии с настоящим изобретением, включают, например: эпилепсию, включая синдромы новорожденных, младенцев, детей и взрослых, частичные (связанные с локализацией) и генерализованные эпилепсии, с частичными или генерализованными, судорожными и не судорожными припадками, с участием или без участия нарушения сознания, и эпилептический статус; деменцию и связанную с ней эпилепсию, включая деменцию типа Альцгеймера (DAT), болезнь Альцгеймера, болезнь Пика, сосудистую деменцию, болезни телец Леви, деменцию, связанную с метаболической, токсической и дефицитной эпилепсией (включая алкоголизм, гипотиреоидизм и дефицит витамина В12), комплекс СПИД-деменции, болезнь Крейтцфельда-Якоба и атипичную подострую губчатую энцефалопатию; паркинсонизм и двигательные расстройства, включая болезнь Паркинсона, множественную системную атрофию, прогрессирующий супрануклеарный паралич, кортикобазальную дегенерацию, гепатолентикулярную дегенерацию, хорею (включая болезнь Хантингтона и гемибализм), атетоз, дистонию (включая спастическую кривошеесть, профессиональные двигательные расстройства, синдром Жиль де ла Тауретта), Старческую или вызванную лекарственными средствами дискинезии (включая леводопа-индуцированную дискинезию), тремор и миоклонус; двигательные нейрональные заболевания или амиотрофический латеральный склероз (ALS); другие нейродегенеративные и/или наследственные расстройства нервной системы, включая спино-церебеллярные дегенерации, такие как атаксия Фридриха и другие наследственные церебеллярные атаксии, преимущественно спинальные мышечные атрофии, наследственные невропатии и факоматоз; расстройства периферической нервной системы, включая тригеминальную невралгию, расстройство лицевого нерва, расстройство других нервов черепа, расстройство нервных корешков и сплетений, мононевриты, такие как запястный сухожильный синдром и ишиас, наследственные и идиопатические периферические невропатии, воспалительные и токсические невропатии; рассеянный склероз и другие демиелинизирующие эпилепсии нервной системы; церебральную плазию младенцев (спастическую), моноплегическую, параплегическую или тетраплегическую; гемиплегию и гемипарез, вялые или спастические, и другие паралитические синдромы; церебрососудистые расстройства, включая субарахноидальное кровотечение, внутрицеребральное кровотечение, закупорку и стеноз прецеребральных артерий, закупорку церебральных артерий, включая тромбоз и эмболию, ишемию мозга, удар, кратковременные ишемические приступы, атеросклероз, церебрососудистую деменцию, аневризмы, церебральные дефициты, связанные с операциями шунтирования и пересадки сердца; расстройства аутистического спектра, включая аутизм, синдром Аспергера, дезинтегративные расстройства детского возраста и синдром Ретта; мигрени, включая классическую мигрень и варианты, такие как кластерная головная боль; головная боль; мионеврологические расстройства, включая миастению gravis, острые мышечные спазмы, миопатии, включая мышечные дистрофии, миотонии и семейный периодический паралич; расстройства глаз и зрительных путей, включая расстройства сетчатки и нарушения зрения; внутричерепные травмы/поражения и их последствия; травмы/поражения нервов и спинного мозга и их последствия; отравляющие и токсические эффекты не лекарственных веществ; случайные отравления лекарственными средствами, медицинскими веществами и биологическими веществами, действующими на центральную, периферическую и автоиммунную системы; неврологические и психиатрические вредные эффекты лекарственных средств, медицинских веществ и биологических веществ; несостоятельность сфинктера и нарушение половых функций; ментальные расстройства, обычно диагностируемые в младенчестве, детстве или юности, включая: задержку умственного развития, расстройства обучения, расстройства двигательных навыков, коммуникационные расстройства, первазивные расстройства, дефицит внимания и расстройства социального поведения, расстройства питания, тикозные расстройства, расстройство элиминирования; делирий и другие когнитивные расстройства; расстройства, связанные с веществами, включая: расстройства, связанные с алкоголем, расстройства, связанные с никотином, расстройства, связанные с кокаином, опоидами, каннабисом, галлюциногенами и другими наркотиками; шизофрении и другие психотические расстройства; аффективные расстройства, включая депрессивные расстройства и биполярные расстройства; тревожные расстройства, включая панические расстройства, фобии, обсессивно-компульсивные расстройства, стрессовые расстройства (например, пост-травматические стрессовые расстройства), генерализованные стрессовые расстройства; расстройства питания, включая анорексию и булемию; расстройства сна, включая диссомнические расстройства (бессонницу, гиперсомнию, нарколепсию, нарушения сна, связанные с дыханием) и парасомнию; индуцированные лекарственными средствами двигательные расстройства (включая нейролептиком-индуцированный паркинсонизм и старческую дискинезию); эндокринные или метаболические заболевания, включая диабет, нарушения функций эндокринных желез, гипогликемию; острую и хроническую боль; тошноту и рвоту; синдром раздраженного кишечника; или раковые заболевания.
В частности, состояния, связанные с нарушениями глутаматергической передачи сигнала и/или функций, или состояния, на которые можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала, которые необходимо лечить и/или предотвратить, используя соединения или фармацевтические композиции в соответствии с настоящим изобретением, включают: деменцию и связанную с ней эпилепсию, включая деменцию типа Альцгеймера (DAT), болезнь Альцгеймера, болезнь Пика, сосудистую деменцию, болезни телец Леви, деменцию, связанную с метаболической, токсической и дефицитной эпилепсией (включая алкоголизм, гипотиреоидизм и дефицит витамина В12), комплексом СПИД-деменции, болезнью Крейтцфельда-Якоба и атипичной подострой губчатой энцефалопатией; паркинсонизм и двигательные расстройства, включая болезнь Паркинсона, множественную системную атрофию, прогрессирующий супрануклеарный паралича, кортикобазальную дегенерацию, гепатолентикулярную дегенерацию, хорею (включая болезнь Хантингтона и гемибализм), атетоз, дистонию (включая спастическую кривошеесть, профессиональные двигательные расстройства, синдром Жиль де ла Тауретта), старческую или инициированную лекарствами дискинезию (включая леводопа-индуцированную дискинезию), тремор и миоклонус; острую и хроническую боль; тревожные расстройства, включая панические расстройства, фобии, обсессивно-компульсивные расстройства, стрессовые расстройства (включая пост-травматические расстройства) и генерализованные стрессовые расстройства; шизофрению и другие психотические расстройства; аффективные расстройства, включая депрессивные расстройства и биполярные расстройства; эндокринные или метаболические заболевания, включая диабет, нарушения функций эндокринных желез и гипогликемию; или раковые заболевания.
Таким образом, настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемым солям, сольватам или пролекарствам, или к фармацевтическим композициям, включающим любое из вышеуказанных в комбинации с фармацевтически приемлемым эксципиентом, для использования при лечении или для профилактики заболевания/расстройства/состояния, выбранного из: деменции и связанной с ней эпилепсии, включая деменцию типа Альцгеймера (DAT), болезнь Альцгеймера, болезнь Пика, сосудистую деменцию, болезни телец Леви, деменцию, связанную с метаболической, токсической и дефицитной эпилепсией (включая алкоголизм, гипотиреоидизм и дефицит витамина В12), комплекс СПИД-деменции, болезнь Крейтцфельда-Якоба и атипичную подострую губчатую энцефалопатию; паркинсонизм и двигательные расстройства, включая болезнь Паркинсона, множественную системную атрофию, прогрессирующий супрануклеарный паралич, кортикобазальную дегенерацию, гепатолентикулярную дегенерацию, хорею (включая болезнь Хантингтона и гемибализм), атетоз, дистонию (включая спастическую кривошеесть, профессиональные двигательные расстройства, синдром Жиль де ла Тауретта), старческую или вызванную лекарствами дискинезию (включая леводопа-индуцированную дискинезию), тремор и миоклонус; острую и хроническую боль; тревожные расстройства, включая панические расстройства, фобии, обсессивно-компульсивные расстройства, стрессовые расстройства (включая пост-травматические расстройства) и генерализованные стрессовые расстройства; шизофрению и другие психотические расстройства; аффективные расстройства, включая депрессивные расстройства и биполярные расстройства; эндокринные или метаболические заболевания, включая диабет, нарушение функций эндокринных желез и гипогликемию; или раковые заболевания. Настоящее изобретение, в частности, относится к соединению формулы (I) или к его фармацевтически приемлемым солям, сольватам или пролекарствам, или к фармацевтическим композициям, включающим любое из вышеперечисленных в комбинации с фармацевтически приемлемым эксципиентом, для использования при лечении или для профилактики болезни Паркинсона.
Объем настоящего изобретения включает все фармацевтически приемлемые солевые формы соединения формулы (I), которые могут быть образованы, например, в результате протонирования атома, содержащего неподеленную пару электронов, который подвержен протонированию, такие как аминогруппа, с неорганической или органической кислотой, или как соль гидроксигруппы с физиологически приемлемым катионом, так как они хорошо известны специалистам. Примеры солей присоединения оснований включают, например, соли щелочных металлов, такие как соли натрия или калия; соли щелочноземельных металлов, такие как соли кальция или магния; соли аммония; соли алифатических аминов, такие как триметиламин, триэтиламин, дициклогексиламин, этаноламин, диэтаноламин, триэтаноламин, соли прокаина, соли меглумина, соли этаноламина или соли этилендиамина; соли аралкиламина, такие как соли N,N-дибензилэтилендиамина, соли бенетамина; соли гетероциклических ароматических аминов, такие как соли пиридина, соли пиколина, соли хинолина или соли изохинолина; соли четвертичного аммония, такие как соли тетраметиламмония, соли тетраэтиламмония, соли бензилтриметиламмония, соли бензилтриэтиламмония, соли бензилтрибутиламмония, соли метилтриоктиламмония или соли тетрабутиламмония; и соли основных аминокислот, такие как соли аргинина или соли лизина. Примеры солей присоединения кислот включают, например, соли, минеральных кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфатные соли, нитратные соли, фосфатные соли (такие как, например, фосфат, гидрофосфат или дигидрофосфат), карбонатные соли, гидрокарбонатные соли или перхлоратные соли; соли органических кислот, такие как ацетат, пропионат, бутират, пентаноат, гексаноат, гептаноат, октаноат, циклопентанпропионат, ундеканоат, лактат, малеат, оксалат, фумарат, тартрат, малат, цитрат, никотинат, бензоат, салицилат или аскорбат; сульфонатные соли, такие как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, бензолсульфонат, p-толуолсульфонат (тозилат), 2-нафталинсульфонат, 3-фенилсульфонат или камфорсульфонат; и кислые соли аминокислот, такие как соли аспартата или соли глутамата. Предпочтительные фармацевтически приемлемые соли соединения формулы (I) включают гидрохлоридные соли, гидробромидные соли, мезилаты, сульфаты, тартраты, фумараты, ацетаты, цитраты и фосфаты. В частности, предпочтительной фармацевтически приемлемой солью соединения формулы (I) является гидрохлорид. Соответственно, предпочтительно, чтобы соединение формулы (I) было в форме гидрохлорида, гидробромида, мезилата, сульфата, тартрата, фумарата, ацетата, цитрата или фосфата. Более предпочтительно, чтобы соединение формулы (I) было в форме гидрохлорида. Еще более предпочтительно, чтобы соединение формулы (I) было в форме моногидрата бисгидрохлорида (т.е., •2HCl•H2O).
Кроме того, объем настоящего изобретения включат твердые формы соединения формулы (I) в любой сольватированной форме, включая сольваты с водой, например, гидраты, или с органическими растворителями, такими как, например, метанол, этанол или ацетонитрил, т.е. как метанолат, этанолат или ацетонитрилат, соответственно; или в форме любого полиморфа. Следует понимать, что такие сольваты соединения формулы (I) также включают сольваты фармацевтически приемлемых солей соединения формулы (I).
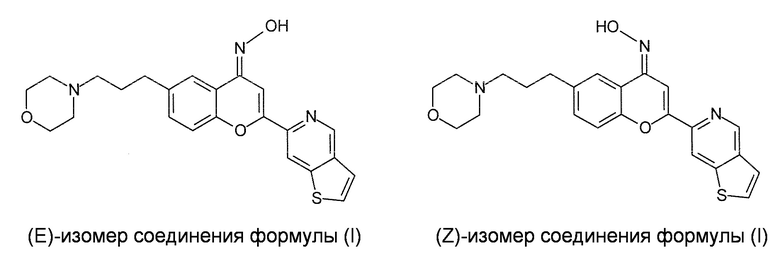
Кроме того, настоящее изобретение включает все возможные изомеры, включая конфигурационные или конформационные изомеры, соединения формулы (I), или в виде смеси, или в чистой или в практически чистой форме. В частности, указанное соединение формулы (I) может иметь (E) конфигурацию или (Z) конфигурацию группы оксима (=N-OH), как представлено далее, и настоящее изобретение включает (E) изомер соединения формулы (I), (Z) изомер соединения формулы (I) и смеси (E) изомера и (Z) изомера соединения формулы (I).
Предпочтительно, чтобы соединение формулы (I) представляло собой (E) изомер, который особенно выгоден с точки зрения его активности. Соответственно, предпочтительно, чтобы по меньшей мере 70 моль%, более предпочтительно по меньшей мере 80 моль%, еще более предпочтительно по меньшей мере 90 моль%, еще более предпочтительно по меньшей мере 95 моль%, еще более предпочтительно по меньшей мере 98 моль%, и еще более предпочтительно по меньшей мере 99 моль% соединения формулы (I) присутствовало в форме (E) изомера. Аналогично, предпочтительно, чтобы по меньшей мере 70 моль%, более предпочтительно по меньшей мере 80 моль%, еще более предпочтительно по меньшей мере 90 моль%, еще более предпочтительно по меньшей мере 95 моль%, еще более предпочтительно по меньшей мере 98 моль%, и еще более предпочтительно по меньшей мере 99 моль% соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства, которые содержатся в фармацевтической композиции настоящего изобретения, находились в форме (E) изомера, т.е., имели (E) конфигурацию группы оксима, присутствующего в соединении формулы (I).
Фармацевтически приемлемые пролекарства соединений формулы (I) являются производными, которые имеют химически или метаболически отщепляемые группы, и в результате сольволиза или в физиологических условиях превращаются в соединение формулы (I), которое является фармацевтически активным in vivo. Пролекарства соединений формулы (I) можно получить обычным способом с функциональной группой соединения, такой как гидроксильная группа. Форма производного пролекарства часто предоставляет преимущества относительно растворимости, тканевой совместимости или отсроченности высвобождения в организме млекопитающих (см., Bundgaard, H., Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985). Такие пролекарства включают, например, ацилоксипроизводные, полученные в результате реакции гидроксильной группы соединения формулы (I) с соответствующим ацилгалогенидом или с соответствующим ангидридом кислоты. Особенно предпочтительными ацилокси производными в качестве пролекарств являются -OC(=O)-CH3, OC(=O) C2H5, OC(=O)-C3H7, OC(=O)-(трет-бутил), -OC(=O)-C15H31, -OC(=O)-CH2CH2COONa, -O(C=O)-CH(NH2)CH3 или -OC(=O)-CH2-N(CH3)2. Соответственно, фармацевтически приемлемым пролекарством может быть соединение формулы (I), где группа оксим OH находится в форме O-ацил-оксим (или ацилокси производного) такой как, например, -OC(=O)-CH3, -OC(=O)-C2H5, -OC(=O)-C3H7, OC(=O)-(трет-бутил), -OC(=O)-C15H31, -OC(=O)-CH2CH2COONa, O(C=O) CH(NH2)CH3 или -OC(=O)-CH2-N(CH3)2. Группа оксим-OH соединения формулы (I) может быть также в форме O-алкил-оксима, такого как, например, -O-CH3, O C2H5, -O-C3H7 или -O-(трет-бутил). Группа оксим-OH соединения формулы (I) может быть также в форме O диалкилфосфинилокси, такой как -O-P(=O)-[O-(CH3)2], -O-P(=O)-[O-(C2-C5)2], -O-P(=O)-[O-(C3-C7)2] или -O-P(=O)-[O-(трет-бутил)2] или в форме O-фосфорная кислота -O-P(=O)-(OH)2 или в форме O-серная кислота -O-SO2-OH. Так, фармацевтически приемлемое пролекарство в соответствии с настоящим изобретением, предпочтительно, представляет собой соединение формулы (I), где указанная OH группа оксима существует в форме O ацил-оксимной группы, O алкил-оксимной группы, O диалкилфосфинилоксигруппы, группы O фосфорной кислоты или группы O серной кислоты.
Соединение формулы (I) можно вводить per se или можно приготовить в форме лекарственного средства. В объем настоящего изобретения входят фармацевтические композиции, включающие в качестве активного ингредиента соединение формулы (I), как определено в описании ранее. Указанные фармацевтические композиции могут необязательно включать один или более из фармацевтически приемлемых эксципиентов, таких как носители, разбавители, наполнители, разрыхлители, смазывающие агенты, связующие, красители, пигменты, стабилизаторы, консерванты или антиоксиданты.
Фармацевтические композиции можно получить, используя известные специалистам в данной области способы, такие как способы, опубликованные в Remington's Pharmaceutical Sciences, 20th Edition. Фармацевтические композиции можно приготовить в виде дозовых форм для перорального, парэнтерального, такого как внутримышечное, внутривенное, подкожное, внутрикожное, интраартериальное, ректальное, назальное, местное, аэрозольное или вагинальное введение. Дозовые формы для перорального введения включают таблетки с нанесенным покрытием или без покрытия, мягкие желатиновые капсулы, твердые желатиновые капсулы, лепешки, пастилки, растворы, эмульсии, суспензии, сиропы, эликсиры, порошки и гранулы для восстановления, диспергируемые порошки и гранулы, лечебные смолы, жевательные таблетки и шипучие таблетки. Дозовые формы для парэнтерального введения включают растворы, эмульсии, суспензии, дисперсии и порошки и гранулы для восстановления. Эмульсии являются предпочтительный дозовой формой для парэнтерального введения. Дозовые формы для ректального и вагинального введения включают суппозитории и средства вагинальной контрацепции. Дозовые формы для назального введения можно вводить путем ингаляции или вдувания, например, используя дозирующий ингалятор. Дозовые формы для местного введение включают кремы, гели, мази, мазевые повязки, пластыри и системы чрескожной доставки.
Соединение формулы (I) в соответствии с изобретением или описанные выше фармацевтические композиции, включающие соединение формулы (I), можно вводить субъекту обычными способами введения, или системно/периферически или в зону желательного воздействия, включая, но ими не ограничиваясь, один или более из следующих способов: пероральный (т.е. в виде таблеток, капсул или в виде растворов для инъекций), местное введение (например, трансдермальное, интраназальное, окулярное, буккальное и сублингвальное), парэнтеральное (например, используя способ инъекций или способ вливаний, и включая, например, инъекции подкожные, внутрикожные, внутримышечные, внутривенные, интраартериальные, внутрисердечные, интратекальные, интраспинальные, интракапсулярные, субкапсулярные, внутриглазничные, внутрибрюшинные, интратрахеальные, подкожные, внутрисуставные, субарахноидальные или внутригрудинные, например, используя депо, например, подкожное или внутримышечное), пульмонарные (например, путем ингаляцонной терапии или способом вдувания, используя, например, аэрозольное, т.е. через рот или нос), гастроинтестинальное, внутриматочное, внутриглазное, подкожное, офтальмологическое (включая интравитреальное или интракамеральное), ректальное, и вагинальное введение. В частности, предпочтительно, чтобы соединение формулы (I) в соответствии с настоящим изобретением или фармацевтическую композицию настоящего изобретения вводили перорально.
Если указанное соединение или фармацевтическую композицию вводят парэнтерально, тогда примеры таких способов введения включают один или более из: внутривенного, интраартериального, внутрибрюшинного, интратекального, интравентрикулярного, внутрисуставного, внутригрудинного, внутричерепного, внутримышечного или подкожного введения соединения или фармацевтической композиции, и/или используя методы вливания. Для парэнтерального введения соединения предпочтительно использовать его в форме стерильного водного раствора, который может содержать другие вещества, например, достаточные количества солей или глюкозы для получения раствора изотоничного крови. Такие водные растворы должны быть значительно буферированы (предпочтительно, до pH от 3 до 9), при необходимости. Препараты в соответствующих парэнтеральных лекарственных формах в стерильных условиях легко получают, используя стандартные фармацевтические методики, хорошо известные специалистам в данной области.
Указанное соединение или фармацевтические композиции можно также вводить перорально в форме таблеток, капсул, средств контрацепции, эликсиров, растворов или суспензий, которые могут содержать вкусовые агенты или красящие агенты для немедленного-, замедленного-, модифицированного-, отложенного-, импульсного- или с контролируемым выделением применения. Пероральное введение соединения или фармацевтической композиции в соответствии с настоящим изобретением является, в частности, предпочтительным.
Таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин, разрыхлители, такие как крахмал (предпочтительно кукурузный, картофельный или тапиоки крахмал), натрийкрахмалгликолят, натрийкроскарамеллоза и некоторые комплексы силикатов, и гранулирующие связующие, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза (HPC), сахароза, желатин и смола акации. Кроме того, могут быть включены смазывающие агенты, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Твердые композиции аналогичного типа также можно использовать в качестве наполнителей в желатиновых капсулах. В этом плане предпочтительные эксципиенты включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров указанный агент может быть объединен с различными подсластителями или отдушками, окрашивающими веществами или красителями, с эмульгаторами и/или суспендирующими агентами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин и с их комбинациями.
Альтернативно, указанное соединение или фармацевтические композиции можно вводить в форме суппозиториев или пессариев, или их можно использовать для местных применений в форме гелей, гидрогелей, лосьонов, растворов, кремов, мазей или порошков для присыпки. Соединение настоящего изобретения можно также вводить накожно или трансдермально, например, используя накожные пластыри.
Указанные соединения или фармацевтические композиции можно также вводить пульмонарно, ректально или в глаза. Для офтальмологического использования их можно приготовить в виде микронизированных суспензий в изотоническом стерильном солевом растворе с соответствующим значением pH, или, предпочтительно, в виде растворов в изотоническом стерильном солевом растворе с соответствующим значением pH, необязательно в комбинации с консервантом, таким как бензалконийхлорид. Альтернативно, их можно приготовить в составе мази, такой как вазелин.
Для местного нанесения на кожу указанное соединение или фармацевтические композиции можно приготовить в виде соответствующей мази, содержащей активное соединение, суспендированное или растворенное в, например, смеси одного или более из следующих: минерального масла, жидкого вазелина, парафина, пропиленгликоля, эмульгирующего воска и воды. Альтернативно, их можно приготовить в виде соответствующих лосьонов или кремов, суспендированных или растворенных в, например, смеси одного или более из следующего: минерального масла, сорбитанмоностеарата, полиэтиленгликоля, жидкого парафина, полисорбата 60, воска цетиловых эфиров, 2-октилдодеканола, бензилового спирта и воды.
Обычно врач определяет реальные дозы, которые будут наиболее подходящими для конкретного субъекта. Конкретные дозы и частота приема для любого конкретного индивидуального субъекта могут варьироваться и будут зависеть от различных факторов, включая активность конкретно используемых соединений, метаболической стабильности и длительности действия указанного соединения, возраста, массы тела, общего состояния здоровья, пола, рациона питания, способа и времени введения, скорости выведения, комбинации лекарственных средств, тяжести конкретного состояния, и конкретного, подлежащего лечению субъекта.
Предполагаемая, еще нелимитирующая доза соединения формулы (I) для введение человеку (приблизительно с массой тела 70 кг) может составлять от 0,05 до 2000 мг, предпочтительно, от 0,1 мг до 1000 мг активного ингредиента в единичной дозе. Указанную единичную дозу можно вводить, например, от 1 до 4 раз в день. Такая доза будет зависеть от способа введения. Далее, в частности, предпочтительная доза соединения формулы (I) для перорального введения млекопитающему (такому как человек) составляет от около 1 до около 25 мг/кг массы тела (например, 1 мг/кг, 2 мг/кг, 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг или 25 мг/кг), причем указанную дозу можно вводить, например, 1, 2, 3 или 4 раза в день (предпочтительно, дважды в день). Еще более предпочтительно, соединение формулы (I) вводить субъекту (например, млекопитающему, предпочтительно, человеку) дважды в день в дозе для каждого введения, от 2 до 25 мг/кг в расчете на массу тела. Предполагается, что может оказаться необходимым осуществлять рутинные вариации дозировок в зависимости от возраста и массы тела пациента/субъекта, также как от тяжести подлежащего лечению заболевания. Точная доза и способ введения будут в конечном счете прописаны соответствующим врачом или ветеринаром.
Соединение формулы (I) в соответствии с настоящим изобретением можно использовать как монотерапию (например, без одновременного введения каких-либо других терапевтических агентов против того же самого, подлежащего лечению или профилактике заболевания, соединением формулы (I), такого как болезнь Паркинсона). Однако, соединение формулы (I) можно также вводить в комбинации с одним или более из других терапевтических агентов. Если соединение формулы (I) используют в комбинации со вторым терапевтическим агентом, активным против того же самого заболевания или состояния, доза каждого соединения может отличаться от дозы, которую используют для введения только одного соответствующего соединения. Комбинация соединения формулы (I) с одним или более из других терапевтических агентов может включать одновременное/сопутствующее введение соединения формулы (I) и другого терапевтического агента (агентов) (или в единой фармацевтической лекарственной форме или в раздельных фармацевтических лекарственных формах), или последовательное/раздельное введение соединения формулы (I) и другого терапевтического агента (агентов). Например, если соединение формулы (I) используют для лечения или профилактики паркинсонизма или двигательного расстройства, в частности для лечения или профилактики болезни Паркинсона, лекарственное средство против болезни Паркинсона (такое как, например, леводопа) можно вводить в комбинации с указанным соединением формулы (I).
Кроме того, в соединение формулы (I) можно также ввести радиометку, осуществляя его синтез (например, как раскрыто в примере 1), используя предшественники, включающие по меньшей мере один атом, который представляет собой радиоизотоп. Предпочтительно, используют радиоизотопы атомов углерода, атомов водорода, атомов серы или атомов йода, такие как, например, 14C, 3H, 35S, или 125I. Соединения, меченые 3H (тритием) можно также получить, подвергая соединение формулы (I) реакциям водородного обмена, таким как, например, катализируемая платиной реакция обмена в тритированной уксусной кислоте (т.е., в уксусной кислоте, включающей 3H вместо 1H), катализируемая кислотой реакция обмена в тритированной трифторуксусной кислоте, или гетерогенно-каталитическая реакция обмена с газом тритием. Для специалистов в области синтетической химии будут очевидны различные дополнительные способы введения радиометок в соединение формулы (I) или получения радиомеченных производных указанных соединений. Флуоресцентные метки также можно связать с соединениями формулы (I) в соответствии с известными специалистам способами.
Субъектом или пациентом, таким как субъект, нуждающийся в лечении или предотвращении/профилактике, может быть животное (например, не человек), позвоночное животное, млекопитающее, грызун (например, морская свинка, хомяк, крыса, мышь), mouse (например, мышь), canine (например, собака), feline (например, кошка), equine (например, лошадь), примат, обезьяноподобным (например, обезьяна или ape), monkey (например, мартышка, бабуин), ape (например, горилла, шимпанзе, орангутанг, гиббон) или человек. В контексте настоящего изобретения, в частности, предполагается, что можно лечить животных, которые важны с экономической, сельскохозяйственной или научной точки зрения. Важные с точки зрения науки организмы включают, но ими не ограничиваются, мышей, крыс и кроликов. Низшие организмы, такие как, например, фруктовые мухи, подобные Drosophila melagonaster, и нематоды, подобные Caenorhabditis elegans, можно также использовать в научных экспериментах. Нелимитирующими примерами важных для сельского хозяйства животных служат овцы, крупный рогатый скот и свиньи, хотя, например, кошек и собак можно рассматривать как экономически важных животных. Предпочтительно, чтобы субъект/пациент представлял собой млекопитающее; более предпочтительно, чтобы субъект/пациент был человеком.
Термин ʺлечениеʺ расстройства или заболевания в том смысле, как использован в описании, хорошо известен специалистам в данной области. ʺЛечениеʺ расстройства или заболевания подразумевает, что расстройство или заболевание подразумевается или было диагностировано у пациента/субъекта. Пациент/субъект, который подозревается в том, что он страдает расстройством или заболеванием, обычно демонстрирует специфические клинические и/или патологические симптомы, которые специалисты могут легко отнести к конкретному патологическому состоянию (т.е., могут диагностировать расстройство или заболевание).
ʺЛечениеʺ расстройства или заболевания может, например, привести к остановке развития расстройства или заболевания (например, к отсутствию усиления симптомов) или к задержке развития расстройства или заболевания (в случае, если остановка развития оказывается только кратковременной). ʺЛечениеʺ расстройства или заболевания может также привести к частичной реакции (например, к облегчению симптомов) или к полной реакции (например, исчезновению симптомов) субъекта/пациента, страдающего таким расстройством или заболеванием. Соответственно, ʺлечениеʺ расстройства или заболевания может также относиться к ослаблению расстройства или заболевания, которые могут, например, привести к остановке развития расстройства или заболевания или к замедлению развития расстройства или заболевания. После такой частичной или полной реакции может последовать рецидив. Следует понимать, что субъект/пациент может проявлять широкий круг реакций на лечение (например, примерные реакции, как раскрыто в описании выше).
Лечение расстройства или заболевания может, между прочим, включать радикальное лечение (предпочтительно, приводящее к полной реакции и практически к исцелению расстройства или заболевания) и паллиативное лечение (включая облегчение симптомов).
Также термин ʺпредотвращениеʺ или ʺпрофилактикаʺ расстройства или заболевания в том смысле, как использован в описании, хорошо известен специалистам. Например, пациент/субъект, подразумеваемый в том, что он предрасположен к нарушению или заболеванию, как определено в описании, может, в частности, выиграть от предотвращения/профилактики расстройства или заболевания. Указанный субъект/пациент может быть подвержен или предрасположен к расстройству или заболеванию, включая, но ими не ограничиваясь, наследственную предрасположенность. Такую предрасположенность можно определить с помощью стандартных анализов, используя, например, генетические маркеры или фенотипические индикаторы. Следует понимать, что расстройство или заболевание, которое следует предотвратить в соответствии с настоящим изобретением, может не быть диагностированным или не может быть диагностировано у указанного пациента/субъекта (например, указанный пациент/субъект не проявляет никаких клинических или патологических симптомов). Так, термины ʺпредупреждениеʺ или ʺпрофилактикаʺ включают использование соединения настоящего изобретения до диагностирования или определения любых клинических и/или патологических симптомов или их может диагностировать или определить лечащий врач. Термины ʺпредотвращениеʺ и ʺпрофилактикаʺ в описании используют взаимозаменяемо.
В рассматриваемом описании цитирован ряд документов, включая патентные заявки и научную литературу. Раскрытия указанных документов, хотя и не рассматриваются как относящиеся к патентоспособности настоящего изобретения, включены в описание по ссылке во всей своей полноте. Более конкретно, все ссылочные документы включены в описание по ссылке в той же самой степени, как если бы каждый индивидуальный документ был бы конкретно и индивидуально включен в описание по ссылке.
Настоящее изобретение также раскрыто следующими иллюстративными чертежами. Представленные чертежи демонстрируют:
Фиг. 1: PXT002331 и PXT001858 воздействие на экспонирование мозга после перорального введения крысам (10 мг/кг).
Фиг. 2: PXT002331 и PXT001858 концентрации в плазме после перорального введения крысам (10 мг/кг).
Фиг. 3: Уровни содержания PXT002331 и PXT001858 в мозге после перорального введения крысам (10 мг/кг).
Фиг. 4: Отношение PXT002331 и PXT001858 в мозге/плазме после перорального введения крысам (10 мг/кг).
Фиг. 5: Оценка антипаркинсонической эффективности PXT002331 в модели 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (MPTP) болезни Паркинсона на макаках (см. пример 3). (A) PXT002331 как отдельно действующее лечение; пероральное введение дважды в день в течение 4 дней, определение паркинсонической оценки в день 4; данные представляют собой среднее+s.e.m. после 2 часов наблюдения (n=7 в группе; 1 из 8 в начале эксперимента была исключена); ʺVehʺ=носитель; ʺLDoptʺ =L допа оптимальная доза ʺ; *=P<0,05 от Veh; ***=P<0,001 от Veh; статистический анализ: непараметрический однофакторный дисперсионный анализ ANOVA (тест Фридмана), затем множественное сравнение Даннета. (B) объединенное лечение с использованием PXT002331 (25 мг/кг)+низкая доза L-допа (4-9 мг/кг)- в зависимости от времени; пероральное введение дважды в день в течение 4 дней, оценка в день 4; L-допа оптимальная доза (ʺLDoptʺ):>20 мг/кг; L-допа субоптимальная доза (ʺLDsoʺ): 4-9 мг/кг; комбинированное введение L-допа (субоптимальная доза) и PXT002331: дважды в день в течение 4 дней. (C) Объединенное лечение с использованием PXT002331+низкая доза L допа-отличие паркинсонической оценки обезьян, обработанных низкими дозами L допа и PXT002331 по сравнению с низкими дозами только L допа, и по сравнению с оптимальной дозой L допа; оценка в день 4, между 1 и 2 часами после введения L-допа (т.е., через 2 и 3 часа после введения PXT002331); все обезьяны, обработанные PXT002331+L допа, демонстрируют значительное улучшение паркинсонической оценки. (D) Объединенная обработка с использованием PXT002331+низкая доза L допа-дозозависимая оценка для различных доз PXT002331; определение паркинсонических оценок в день 4; ʺVehʺ=носитель; ʺнизкая LDʺ=низкая доза L допа; ʺLD оптʺ=оптимальная доза L допа; *=P<0,05 от низкой LD; статистический анализ: непараметрический однофакторный дисперсионный анализ ANOVA (тест Фридмана), затем множественное сравнение Даннета; N=7. (E) Компьютеризированная двигательная активность на ранней стадии PD обезьян в модели для PXT002331 в комбинации с L допа (низкая доза или оптимальная доза) после перорального введения; *=P<0,05 от носителя; **=P<0,01 от носителя; ***=P<0,001 от носителя; статистический анализ: дисперсионный анализ Фридмана, затем критерий Даннета; N=5 (6 обезьян/1 исключена). (F) Объединенная обработка с использованием PXT002331+оптимальная доза L допа-оценка несостоятельности и оценка дискинезии.
Настоящее изобретение, в частности, относится к следующим пунктам:
1. Соединение следующей формулы (I):
или его фармацевтически приемлемые соли, сольваты или пролекарства.
2. Соединение по п. 1, где указанное соединение имеет (E) конфигурацию у группы оксима, содержащейся в формуле (I).
3. Соединение по любому одному из пп. 1 или 2, где фармацевтически приемлемой солью является гидрохлоридная соль.
4. Соединение по любому одному из пп. 1-3 для использования в качестве лекарственного средства.
5. Фармацевтическая композиция, включающая соединение по любому одному из пп. 1-3 и фармацевтически приемлемый эксципиент.
6. Соединение по любому одному из пп. 1-3 или фармацевтическая композиция по п. 5 для использования при лечении или для профилактики состояния, связанного с измененной глутаматергической передачей сигнала и/или функцией, или состоянием, на которое можно влиять, изменяя уровень содержания глутамата или передачи сигнала.
7. Использование соединения по любому одному из пп. 1-3 для получения лекарственного средства для лечения или профилактики состояния, связанного с измененной глутаматергической передачей сигнала и/или функцией, или состоянием, на которое можно влиять, изменяя уровень содержания глутамата или передачи сигнала.
8. Способ лечения или предотвращения состояния, связанного с измененной глутаматергической передачей сигнала и/или функцией, или состояния, на которое можно влиять, изменяя уровень содержания глутамата или передачи сигнала, причем указанный способ включает введение соединения по любому одному из пп. 1-3 или фармацевтической композиции по п. 5 нуждающемуся в этом субъекту.
9. Соединение или фармацевтическая композиция для использования в соответствии с п. 6, или использование по п. 7 или способ по п. 8, где указанное состояние, связанное с измененной глутаматергической передачей сигнала и/или функцией, или указанное состояние, на которое можно влиять, изменяя уровень содержания глутамата или передачи сигнала, выбирают из: деменции, паркинсонизма и двигательных расстройств, острой или хронической боли, тревожных расстройств, шизофрении, аффективных расстройств, эндокринных или метаболических заболеваний, диабета, нарушений функций эндокринных желез, гипогликемии или раковых заболеваний.
10. Соединение или фармацевтическая композиция для использования в соответствии с п. 9, или использование по п. 9, или способ по п. 9, где указанную деменцию выбирают из: деменции типа Альцгеймера (DAT); болезни Альцгеймера; болезни Пика; сосудистой деменции; болезни телец Леви; деменции, связанной с метаболическими, токсическими заболеваниями и болезнями дефицита, включая алкоголизм, гипотиреоидизм и дефицит витамина В12; комплекса СПИД-деменции; болезни Крейтцфельда-Якоба; или атипичной подострой губчатой энцефалопатии.
11. Соединение или фармацевтическая композиция для использования в соответствии с п. 9, или использование по п. 9, или способ по п. 9, где указанные паркинсонизм и двигательные расстройства выбирают из: болезни Паркинсона; множественной системной атрофии; прогрессирующего супрануклеарного паралича; кортикобазальной дегенерации; гепатолентикулярной дегенерации; хореи, включая болезнь Хантингтона и гемибализм; атетоза; дистонии, включая спастическую кривошеесть, профессиональные двигательные расстройства и синдром Жиль де ла Тауретта; старческих или связанных с лекарственными средствами дискинезий, включая леводопа-индуцированную дискинезию; тремора; или миоклонуса.
12. Соединение или фармацевтическая композиция для использования по п. 9, или использование по п. 9, или способ по п. 9, где указанные тревожные расстройства выбирают из: панических расстройств; фобий; обсессивно-компульсивных расстройств; стрессовых расстройств, включая пост-травматические стрессовые расстройства; или генерализованных стрессовых расстройств.
13. Соединение или фармацевтическая композиция для использования в соответствии с п. 9, или использование по п. 9, или способ по п. 9, где указанное аффективные расстройства выбирают из депрессивных расстройств или биполярных расстройств.
14. Соединение по любому одному из пп. 1-3 или фармацевтическая композиция по п. 5 для использования при лечении или для профилактики болезни Паркинсона.
15. Использование соединения по любому одному из пп. 1-3 для получения лекарственного средства для лечения или для профилактики болезни Паркинсона.
16. Способ лечения или предотвращения болезни Паркинсона, причем указанный способ включает введение соединения по любому одному из пп. 1-3 или фармацевтической композиции по п. 5 нуждающемуся в этом субъекту.
17. Соединение или фармацевтическая композиция для использования по любому одному из пп. 6 или 9-14 или использование по любому одному из пп. 7, 9-13 или 15, или способ по любому одному из пп. 8-13 или 16, где указанное соединение, указанная фармацевтическая композиция или указанное лекарственное средство вводят перорально.
18. Способ по любому одному из пп. 8-14, 16 или 17, где указанным субъектом является человек.
Далее изобретение будет раскрыто со ссылкой на следующие примеры, которые являются просто иллюстративными и которые не следует рассматривать как ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Пример 1: Получение соединения формулы (I)
1) Общий метод синтеза
Соединение формулы (I) в соответствии с настоящим изобретением (т.е., PXT002331) можно получить из легкодоступных исходных материалов с помощью нескольких подходов к синтезу, используя схемы химических реакций в фазе раствора или в твердой фазе, или в смешанной фазе раствор/твердое вещество. Например, соединение формулы (I) можно получить, используя представленные далее схемы синтеза.
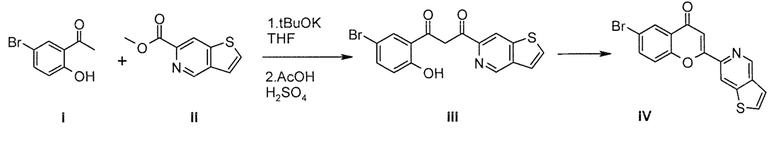
Коммерчески доступный бромацетофенон I подвергают взаимодействию с коммерческим тиено[3,2 c]пиридинметиловым эфиром II в растворителе, таком как тетрагидрофуран (ТГФ), и в присутствии слабого основания, такого как трет-бутоксид калия (tBuOK), до получения промежуточного дикетона III. Этот способ известен как перегруппировка Венкатарамана Бейкера (Baker, W., J.Chem.Soc, 1933, 1381).
Промежуточный дикетон III затем циклизуют в кислотных условиях в присутствии сильного дегидратирующего агента, такого как серная кислота (H2SO4), в кипящей с обратным холодильником уксусной кислоте (AcOH), получая хромон IV.
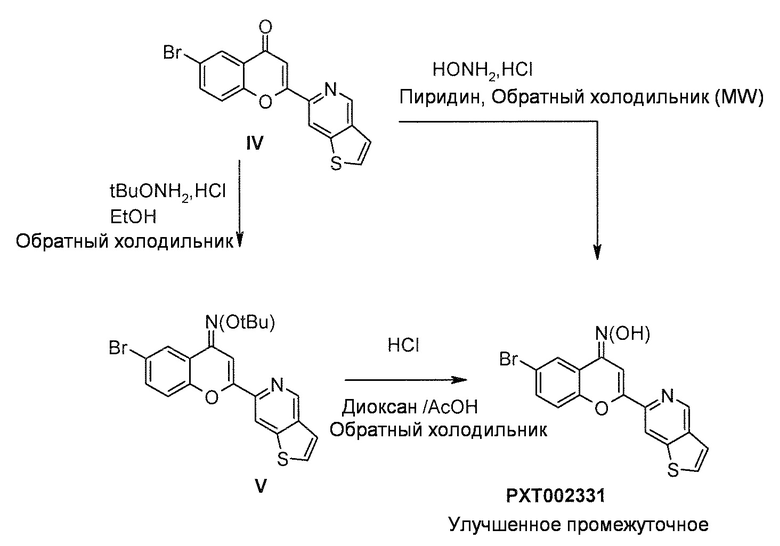
Введение оксима можно обеспечить, осуществляя реакцию производного IV с гидроксиламингидрохлоридом (HONH2, HCl) в пиридине или этаноле в условиях микроволнового облучения до получения непосредственно усовершенствованного промежуточного оксима хромона, которое приводит к PXT002331 в несколько стадий реакции. Указанное усовершенствованное промежуточное соединение, приводящее к PXT002331, можно также получить, используя двухстадийную процедуру, как представлено выше, используя трет-бутилгидроксиламингидрохлорид (tBuONH2, HCl) в этаноле с последующей стадией удаления трет-бутильной группы в кислотных условиях, таких как хлористоводородная кислота (HCl) в смеси полярных растворителей, таких как ТГФ и уксусная кислота.
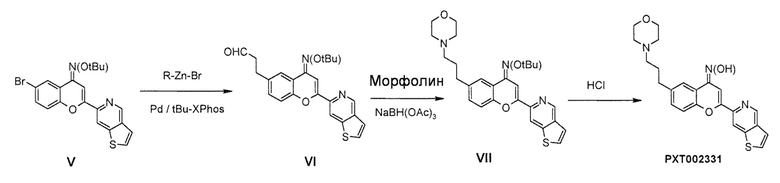
Введение алкиленовой боковой цепи осуществляют, используя катализируемую палладием реакцию перекрестного сочетания, такую как реакция перекрестного сочетания Негиши, используя коммерчески доступный реагент цинка и соответствующую каталитическую систему лиганд/палладий. Затем функционализация с последующим стандартным восстановительным аминированием с использованием слабых восстанавливающих агентов, таких как триацетоксиборгидрид, приводит к получению усовершенствованного промежуточного соединения VII с хорошими выходом. Конечное удаление защитной группы оксима в кислотных условиях приводит к соединению формулы (I), т.е. PXT002331.
2) Синтез соединения формулы (I)
Коммерчески доступные исходные материалы, используемые в следующем экспериментальном описании, закупают у Aldrich, Sigma, ACROS или ABCR, если не указано иначе.
Раскрытые далее соединения названы в соответствии со стандартами, использованными в программе AutoNom v1.0.1.1 (MDL Information Systems, Inc.).
1H ЯМР анализ проводят, используя прибор BRUKER NMR, модель DPX-400 МГц FT-NMR (с Фурье преобразованием). В качестве внутреннего стандарта используют остаточный сигнал дейтерированного растворителя. Химические сдвиги (δ) выражают как мд относительно сигнала остаточного растворителя (δ=2,50 для 1H ЯМР в ДМСО-d6, и 7,26 в CDCl3). Обозначения s=с (синглет), d=д (дублет), t=т (триплет), q=кв (квадруплет), br=шир (широкий). Некоторые соединения в экспериментальной части существуют в виде смеси E/Z изомеров в различных отношениях. Отношение E/Z изомеров четко определено для конечного соединения PXT002331.
Представленные в описании данные МС получают следующим образом: Масс-спектр: прибор ЖХ/МС GH/MS Waters ZMD (ESI).
ВЭЖХ анализ осуществляют, используя Waters X-bridge TM C8 50 мм×4,6 мм колонку при скорости потока 2 мл/мин; 8 мин, градиент H2O:CH3CN:TFA с 100:0:0,1% до 0:100:0,05%, с УФ детектором (254 нм).
Масс-управляемую препаративную ВЭЖХ очистку осуществляют, используя масс-управляемый автоматический прибор Fraction lynx от Waters, снабженный Sunfire Prep C18 OBD колонкой 19×100 мм 5 мкм, если не указано иначе. Все стадии очистки осуществляют с градиентом ACN/H2O или ACN/H2O/HCOOH (0,1%).
Соединение формулы (I) получают в соответствии со следующей схемой реакции:
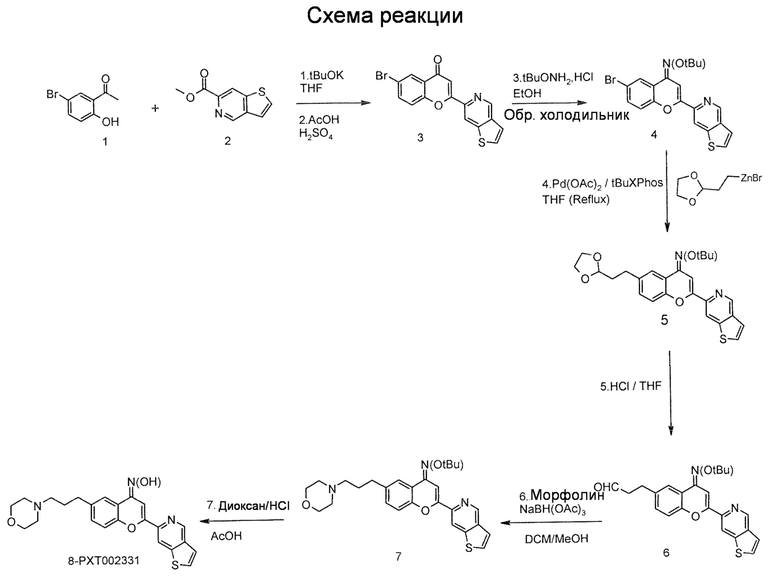
Стадии 1 и 2: 6-бром-2-(тиено[3,2-c]пиридин-6-ил)-4H хромен-4-он (3)
К суспензии трет-бутоксида калия (156,0 г, 1,39 моль, 3,0 экв) в ТГФ (500 мл) при 0°C добавляют раствор 5-бром-2-гидроксиацетофенона (100,0 г, 0,47 моль, 1 экв) в ТГФ (500 мл). Реакционную смесь интенсивно перемешивают в течение 10 минут. Раствор тиенопиридинового эфира (98 г, 0,51 моль, 1,1 экв) в ТГФ (1,0 л) добавляют к реакционной смеси. Полученную красноватую суспензию кипятят с обратных холодильником в течение 1 часа, причем по данным ЖХ/МС анализов к этому времени реакция завершается. Реакционную смесь охлаждают до комнатной температуры (КТ), получая густую суспензию оранжевого цвета, которую выливают в ледяную воду (5,0 л). Водный слой нейтрализуют, добавляя водный раствор HCl (1,5 н) при интенсивном перемешивании. Полученное твердое вещество желтого цвета собирают фильтрованием, промывают водой и сушат с отсосом. Полученную сухую массу снова дополнительно сушат в течение ночи под давлением при 45°C в течение 16 час, получая 156 г твердого вещества желтого цвета.
Затем твердое желтое вещество (156 г) суспендируют при КТ в ледяной уксусной кислоте (1,0 л) и концентрированной H2SO4 (10 мл). Полученную смесь нагревают при 110°C в течение 2 часов. Реакционная смесь превращается в суспензию коричневого цвета. После подтверждения завершения реакции (по данным ЖХ/МС), полученную сухую массу суспендируют в ледяной воде (2,0 л) и нейтрализуют, добавляя водный раствор NaOH (1 н). Полученный твердый осадок бежевого цвета собирают фильтрованием, промывают водой и сушат с отсосом. Полученный материал дополнительно сушат в течение одной ночи при 50°C в условиях высокого вакуума до получения 140,0 г указанного в заголовке соединения в виде твердого вещества бежевого цвета.
Выход: 83%.
ЖХ/МС: Найденная масса (m/z, M+1, 358,0), Площадь 94,78%.
1H ЯМР (ДМСО-d6, 400 МГц) δ 9,32 (с, 1H), 9,06 (с, 1H), 8,15 (м, 2H), 8,06 (м, 1H), 7,84 (д, J 5,4 Гц, 1H), 7,77 (д, J 5,4 Гц, 1H), 7,32 (с, 1H).
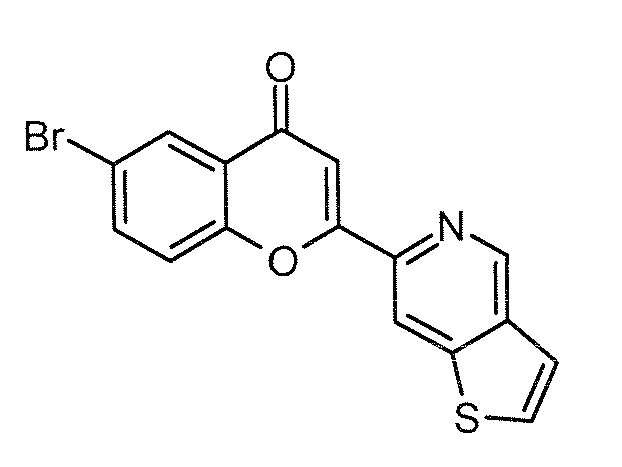
Стадия 3: O-трет-бутилоксим 6-бром-2-(тиено[3,2-c]пиридин-6-ил)-4H хромен-4-она (4)
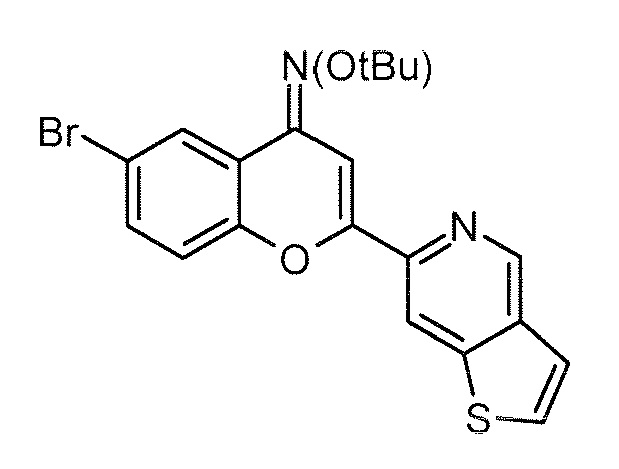
В запаянной ампуле суспензию 6-бром-2-(тиено[3,2-c]пиридин-6-ил)-4H хромен-4-она (20,0 г, 56 ммоль, 1 экв) и O-трет-бутилгидроксиламингидрохлорида (14,0 г, 112 ммоль, 2 экв) в безводном EtOH (300 мл) нагревают при 115°C в течение 20 часов. После подтверждения завершения реакции по данным ТСХ, реакционную смесь фильтруют, и твердое вещество желтого цвета дважды промывают холодным EtOH (50 мл) и сушат в условиях вакуума, получая 20 г указанного в заголовке соединения в виде твердого вещества желтого цвета.
Выход: 83%.
ЖХ/МС: Найденная масса (m/z, M+1, 429,0), Площадь 97,83%.
1H ЯМР (ДМСО-d6, 400 МГц) δ 9,25 (с, 1H), 8,78 (с, 1H), 8,05 (м, 2H), 7,71 (м, 2H), 7,59 (с, 1H), 7,48 (с, 1H), 1,40 (с, 9H).
Стадия 4: O-трет-бутилоксим 6-(2-[1,3]диоксолан-2-илэтил)-2-(тиено[3,2-c]пиридин-6-ил)-4H хромен-4-она (5)
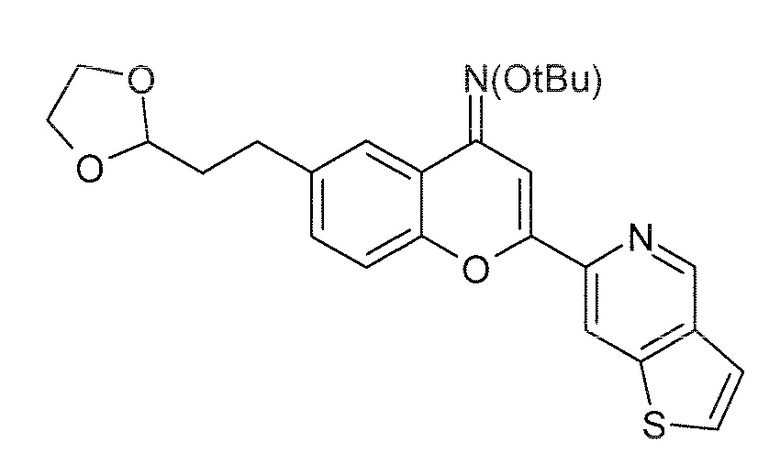
К дегазированному раствору O-трет-бутилоксима 6-бром-2-(тиено[3,2-c]пиридин-6-ил)-4H хромен-4-она (100,0 г, 233 ммоль, 1 экв) и 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенила (4,9 г, 11,6 ммоль, 0,05 экв) в безводном ТГФ (500 мл) добавляют ацетат палладия(II) (2,6 г, 11,6 ммоль, 0,05 экв), затем 2-(1,3-диоксолан-2-ил)этилцинкбромидный раствор (0,5 M в ТГФ, 652 мл, 362 ммоль, 1,5 экв). Реакционную смесь нагревают при 100°C в течение 14 часов. После завершения реакции, подтвержденного результатами ЖХ/МС, реакционную смесь гасят водой (20 мл) и концентрируют в вакууме. Полученное сырое масло желтого цвета очищают хроматографически на силикагеле, используя в качестве элюента циклогексан/этилацетат (80/20), и получая 85 г указанного в заголовке соединения в виде твердого вещества желтого цвета.
Выход: 82%
ВЭЖХ: 93,00% (254 нм), RT: 2,50 мин.
ЖХ/МС: Найденная масса (m/z, M+1, 451,0), Площадь 93,96%.
1H ЯМР (ДМСО-d6, 400 МГц) δ 9,27 (с, 1H), 8,77 (с, 1H), 8,05 (д, J 5,4 Гц, 1H), 7,78 (с, 1H), 7,73 (д, J 5,4 Гц, 1H), 7,62 (с, 1H), 7,43 (м, 2H), 4,85 (м, 1H), 3,93 (м, 2H), 3,80 (м, 2H), 2,75 (м, 2H), 1,90 (с, 2H), 1,39 (с, 9H).
Стадия 5: 3-(4-трет-бутоксиимино-2-(тиено[3,2-c]пиридин-6-ил)-4H-хромен-6-ил)пропиональдегид (6)
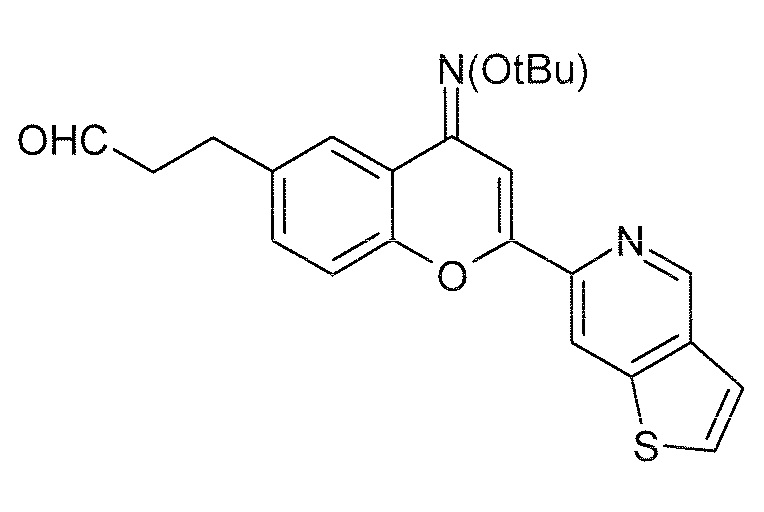
К раствору O-трет-бутилоксима 6-(2-[1,3]диоксолан-2-илэтил)-2-(тиено[3,2-c]пиридин-6-ил)-4H-хромен-4-она (100,0 г, 222 ммоль, 1 экв) в ТГФ (1,0 л) медленно добавляют водный раствор HCl (3 н, 1,0 л). Полученную смесь желтого цвета перемешивают при комнатной температуре в течение 24 часов, получая густую желтую эмульсию. После завершения реакции (ЖХ/МС), реакционную смесь нейтрализуют, добавляя водный насыщенный раствор NaHCO3, и экстрагируют CH2Cl2 (2×5,0 л). Объединенные органические экстракты промывают солевым раствором (2,0 л), сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 89 г указанного в заголовке соединения в виде твердого вещества желтого цвета. Полученное твердое вещество желтого цвета переносят сырым на следующие стадии без дополнительной очистки.
Выход: 92%
ЖХ/МС: Найденная масса (m/z, M+1, 407,3), Площадь 91%.
1H ЯМР (CDCl3, 400 МГц) δ 9,79 (с, 1H), 9,09 (с, 1H), 8,39 (с, 1H), 7,82 (с, 1H), 7,67 (с, 1H), 7,52 (д, J 5,4 Гц, 1H), 7,43 (д, J 5,4 Гц, 1H), 7,17 (м, 2H), 2,93 (м, 2H), 2,77 (с, 2H), 1,37 (с, 9H).
Стадия 6: O-трет-бутилоксим 6-(3-морфолин-4-илпропил)-2-(тиено[3,2-c]пиридин-6-ил)-4H-хромен-4-она (7)
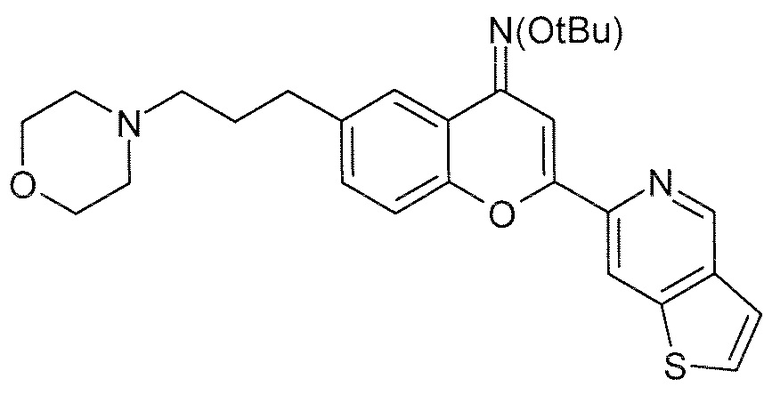
К смеси 3-(4-трет-бутоксиимино-(2-тиено[3,2-c]пиридин-6-ил)-4H-хромен-6-ил)пропиональдегида (100,0 г, 246 ммоль, 1 экв), морфолина (50 мл, 492 ммоль, 2 экв) в CH2Cl2 (1,0 л) и метанола (500 мл) добавляют триацетоксиборгидрид натрия (104 г, 492 ммоль, 2 экв) в атмосфере N2. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов. После завершения реакции (по данным ЖХ/МС), полученную смесь нейтрализуют, добавляя водный насыщенный раствор NaHCO3, и экстрагируют CH2Cl2 (2×5,0 л). Объединенные органические экстракты промывают солевым раствором (2,0 л), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая твердое густое вещество коричневого цвета. Полученное сырое коричневое вещество очищают, используя хроматографическую обработку на силикагеле, получая 73,0 г указанного в заголовке соединения в виде твердого вещества желтого цвета.
Выход: 63%.
ВЭЖХ: 95,97% (254 нм).
ЖХ/МС: Найденная масса (m/z, M+1, 478,3), Площадь 96,62%.
1H ЯМР (ДМСО-d6, 400 МГц) δ 9,24 (с, 1H), 8,74 (с, 1H), 8,03 (д, J 5,4 Гц, 1H), 7,76 (с, 1H), 7,70 (д, J 5,4 Гц, 1H), 7,59 (м, 1H), 7,39 (м, 2H), 3,56 (м, 4H), 2,65 (м, 2H), 2,28 (м, 6H), 1,73 (м, 2H), 1,36 (с, 9H).
Стадия 7: Оксим 6-(3-морфолин-4-илпропил)-2-(тиено[3,2-c]пиридин-6-ил)-4H-хромен-4-она
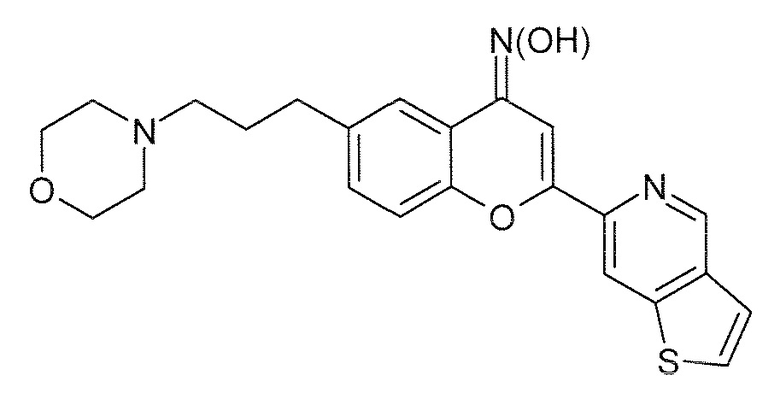
К перемешиваемому раствору O-трет-бутилоксима 6-(3-морфолин-4-илпропил)-2-(тиено[3,2-c]пиридин-6-ил)-4H-хромен-4-она (10,0 г, 21 ммоль, 1 экв) в уксусной кислоте (100 мл) добавляют раствор диоксан-HCl (4 M, 150 мл, 3 экв) при комнатной температуре в инертной атмосфере. Реакционную смесь нагревают при 80°C в течение 14 часов (контроль с помощью ЖХ/МС указывает на 100% конверсию). Органические растворители концентрируют в вакууме, при этом твердая масса начинает осаждаться. Твердое вещество желтого цвета отфильтровывают, промывают диоксаном (200 мл), Et2O (2×50 мл), получая 8 г твердого вещества желтого цвета в виде HCl соли.
Выход: 90%
ВЭЖХ чистота: 98,44% (254 нм). E/Z отношение=97,54%/1,75%.
ЖХ/МС: Найденная масса (m/z, M+, 422,3), Площадь 97,3%.
1H ЯМР (ДМСО, 400 МГц) δ 11,06 (шир, 1H), 10,72 (шир, 1H), 9,28 (с, 1H), 8,80 (с, 1H), 8,07 (д, J 5,4 Гц, 1H), 7,76-7,70 (м, 3H), 7,47-7,41 (м, 2H), 3,95 (м, 2H), 3,80 (м, 2H), 3,42, (м, 2H), 3,08 (м, 4H), 2,71 (м, 2H), 2,10 (м, 2H).
Пример 2: Биологическая оценка соединения формулы (I)
Соединение формулы (I) в соответствии с настоящим изобретением (т.е., PXT002331) тестируют в отношении его агонистической и/или позитивной аллостерической модуляторной активности в отношении человеческих mGluR4, используя анализ кальция, раскрытый в 171 WO 2011/051478. Было обнаружено, что PXT002331 обладает эффективностью pEC50=7,12 (что соответствует EC50 около 0,076 мМ), что сравнимо с эффективностью соединения примера 127 WO 2011/051478 (т.е., ʺPXT001858ʺ), эффективность которого pEC50=7,44 (что соответствует EC50 около 0,036 мкМ).
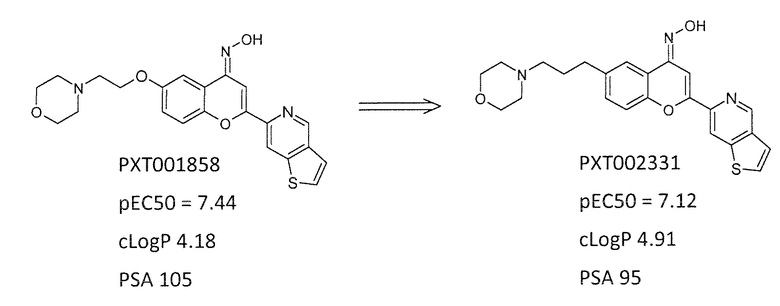
In vitro ADME профиль PXT002331 также весьма сходен со ссылкой на метаболическую стабильность фазы I: CL (час/r):55/101 мкл/мин/мг белка и кишечную абсорбцию: CaCo-2 (A-B, pgp):4,11.10-6 см/сек, без эффлюкса.
В обоих случаях, т.е. PXT002331 и PXT001858, связывание плазменного белка было высоким, причем менее чем 1% свободной фракции и соединений не страдают от недостатка растворимости (s > 10 мг/мл в воде) в виде гидрохлоридной соли.
Однако, несмотря на весьма сходные физико-химические свойства и ADME профили, было обнаружено, что PXT002331 демонстрирует неожиданный, крайне выгодный пероральный in vivo PK профиль при сравнении с PXT001858, как раскрыто далее.
In vivo фармакокинетические оценки:
PXT002331 и PXT001858 вводят перорально (p.o.) в дозе 10 мг/кг самцам крыс штамма Sprague-Dawley. Вводимый объем составляет 10 мл/кг. Параллельно PXT002331 вводят внутривенно (в.в.) в дозе 1 мг/кг, при объеме введения 2 мл/кг. Образцы крови (200 мкл) отбирают в моменты времени от 15 минут до 24 часов для p.o. введения и от 5 минут до 24 часов для в.в. введения в охлажденные льдом ампулы, содержащие 0,2% K2EDTA. Ампулы центрифугируют при скорости 10000 об/мин в течение 5 минут при 4°C. Плазму (надосадочную жидкость) выделяют в другую ампулу и хранят при -80°C до анализа. Две группы по 3 животных используют для каждого способа введения: в одной группе образцы крови отбирают для определения кинетики плазмы, экспонированной за 24 часовой период, и во второй группе кровь и образцы мозга отбирают в один конечный момент времени (0,5, 1,0, 1,5, 2,0, 4,0 часа) для определения кинетики экспонирования мозга и отношения мозг/плазма.
Анализ соединения:
Соответствующее исходное соединение (свободное основание) анализируют в образцах плазмы и в гомогенате мозга, используя метод ЖХ-МС/МС. Концентрации выражают в нг/мл плазмы или в нг/г ткани мозга.
Результаты:
В дозе 10 мг/кг, с тем же самым носителем (твин-80/этанол/ 30% HPBCD (2/10/88)), PXT002331 демонстрирует сопоставимое с PXT001858 экспонирование плазмы, что отражается по его AUC (1,1-кратно) и Cмакс (0,7-кратно). Пероральная биодоступность PXT002331 в этом эксперименте составляет 39%. Несмотря на аналогичную пероральную абсорбцию, PXT002331 имеет более высокое отношение мозг/плазма (6,5 против 2,0 при T=1,5 час; см. Фиг. 4), что приводит к 3-кратному улучшению AUC мозга при сравнении с PXT001858. Апостериори, одна потенциальная гипотеза может основываться на различии конфигураций фазы II в кишечнике и печени во время пероральной абсорбции. Когда оба соединения анализируют in vitro в присутствии UGT (UDP-глюкуронозилтрансфераза), PXT002331 демонстрирует гораздо более низкий уровень глюкуронизации по сравнению с PXT001858 (См. таблицу ниже). Тем не менее, такое различие, наблюдаемое in vitro, само по себе не может объяснить неожиданные преимущества PK результатов, полученных для PXT002331. Результаты, полученные в указанных экспериментах, дополнительно суммированы в таблицах 1-3 ниже и на Фиг. 1-4.
Таблица 1: PK параметры PXT002331 и PXT001858 после перорального введения крысам в дозе 10 мг/кг.
(г/G выход)
Таблица 2: Глюкуронизация PXT002331 и PXT001858 in vitro (площадь пиков) в микросомах печени крыс.
Таблица 3: Глюкуронизация PXT002331 и PXT001858 in vitro (площади пиков) в микросомах кишечника крыс
Полученные результаты показывают, что соединение формулы (I) в соответствии с настоящим изобретением, т.е. PXT002331, обладает крайне выгодными фармакокинетическими свойствами и демонстрирует значительно улучшенное экспонирование мозга по сравнению с соединением примера 127 из WO 2011/051478 (ʺPXT001858ʺ). Указанные свойства делают соединение формулы (I), в частности, подходящим в качестве терапевтического агента, например, для лечения или для профилактики неврологических и/или психиатрических расстройств.
Пример 3: In vivo оценка соединения формулы (I) в модели болезни Паркинсона на MPTP обезьянах
Антипаркинсоническую эффективность соединения формулы (I) в соответствии с настоящим изобретением (т.е., PXT002331) оценивают в модели 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (MPTP) против болезни Паркинсон на макаках (используют макак вида Macaca fascicularis), которые воспроизводят большинство клинических и патологических признаков болезни Паркинсона и считаются ʺзолотым стандартомʺ (См. Porras G et al., Cold Spring Harb Perspect Med., 2(3):009308, 2012 и цитированные там ссылки для общего описания MPTP модели).
Результаты указанных экспериментов суммированы на Фиг. 5A до 5F. В частности, было обнаружено, что PXT002331 при только одной обработке демонстрирует эффективную антипаркинсоническую активность на MPTP обработанных макаках, с оптимальным улучшением паркинсонической оценки при дозах введения 2-25 мг/кг перорально (p.o.) дважды в день (См. Фиг. 5A). В указанном эксперименте, PXT002331 вводят перорально дважды в день в течение 4 дней, и паркинсоническую оценку определяют в день 4 в течение 2 часов наблюдения (результаты представлены как среднее значение+стандартная средняя ошибка (s.e.m.); n=7 обезьян в группе).
Антипаркинсоническую эффективность PXT002331 (25 мг/кг) дополнительно оценивают в комбинации с низкими (субоптимальными) дозами L допа (леводопа; 4-9 мг/кг) (См. Фиг. 5B и 5C). Дозы перорально вводят дважды в день в течение 4 дней, и определение паркинсонической оценки проводят в день 4 (между 1 и 2 часами после введения L-допа, т.е., между 2 и 3 часами после введения PXT002331). Как также представлено на Фиг. 5B, было обнаружено, что совместное введение PXT002331 и субоптимальной дозы L допа дает значительное усовершенствование паркинсонической оценки по сравнению с введением только L допа (субоптимальная доза). Указанные результаты кроме того указывают на увеличение ʺonʺ-времени, достигаемого PXT002331 в комбинации с L допа, что является клинически высоко релевантным преимуществом, как также отражено тем фактом, что ʺonʺ-время является конечной точкой для оценки клинической эффективности фазы 3 болезни Паркинсона у пациентов. Примечательно, что все обработанные обезьяны демонстрируют значительное повышение паркинсонической оценки, что указывает на высокую устойчивость антипаркинсонического эффекта PXT002331 (См. Фиг. 5C). Полученные результаты подтверждают тот факт, что PXT002331 можно с успехом использовать как дополнительную обработку вместе с L допа (леводопа).
Результаты дозозависимой оценки комбинации PXT002331 (в дозах от 2 мг/кг до 100 мг/кг) с L допа (низкая доза) представлены на Фиг. 5D (оценку паркинсонических оценок проводят в день 4). Было обнаружено, что PXT002331 в комбинации с L допа вызывает чрезвычайно сильный антипаркинсонический эффект после перорального введения в интервале различных доз. Оптимальную антипаркинсоническую эффективность достигают при введении PXT002331в дозах от 2 мг/кг до 25 мг/кг.
Значительное улучшение двигательной активности можно дополнительно продемонстрировать для PXT002331 (25 мг/кг) при пероральном введении в комбинации или с низкой дозой L допа, или с оптимальной дозой L допа, что также представлено на Фиг. 5E (ранняя стадия модели PD обезьян; N=5). В указанном эксперименте каждую из обезьян снабжают детектором движений и сигналов, которые накапливаются, используя 24 световых пучка и видеорегистратор перемещений для дифференцирования всех типов движений. Двигательную активность измеряют в течение 1 часа.
Как представлено на Фиг. 5F, кроме того было обнаружено, что повышающиеся дозы PXT002331 в комбинации с L допа (оптимальная доза) обеспечивают улучшение оценки недееспособности, без индуцирования дискинезии. В частности, значительное улучшение оценки несостоятельности можно достичь, используя 25 мг/кг PXT002331 в комбинации с L допа (оптимальная доза). Кроме того, ни одна из тестированных комбинаций PXT002331 (в дозах от 25 мг/кг до 100 мг/кг) с оптимальной (высокой) дозой L допа не приводит к индуцированию дискинезии, которая является нежелательным побочным эффектом, который обычно наблюдается при лечении с использованием L допа.
Полученные результаты подтверждают, что PXT002331 чрезвычайно выгоден для использования при лечении или для профилактики болезни Паркинсона, как в монотерапии (без сопутствующего использования дополнительных антипаркинсонических лекарственных средств) и в совместной терапии с использованием дополнительных анипаркинсонических лекарственных средств, таких как L-допа (леводопа). В указанных экспериментах было обнаружено, что дозы от 2 мг/кг до 25 мг/кг PXT002331 при пероральном введении дважды в день являются эффективными.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2709786C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ТЕЛЕЦ ЛЕВИ, СОДЕРЖАЩЕЕ ПРОИЗВОДНОЕ ПИРАЗОЛОХИНОЛИНА | 2018 |
|
RU2802212C2 |
| 3-АЗАБИЦИКЛО[3.1.0]ГЕКСИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2510396C2 |
| ПРОИЗВОДНЫЕ ЭТИНИЛА | 2017 |
|
RU2745068C2 |
| ПРОИЗВОДНЫЕ ЭТИНИЛА КАК МОДУЛЯТОРЫ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2016 |
|
RU2721776C2 |
| КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2017 |
|
RU2764716C2 |
| 1,2,4-ТРИАЗОЛО[4,3-a]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПОЛОЖИТЕЛЬНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ MGLUR2 | 2015 |
|
RU2695651C2 |
| СОЛЬ СУКЦИНАТ 2-((4-(1-МЕТИЛ-4-(ПИРИДИН-4-ИЛ)-1Н-ПИРАЗОЛ-3-ИЛ)ФЕНОКСИ)МЕТИЛ)ХИНОЛИНА | 2007 |
|
RU2430918C2 |
| 1-[1-(4-БЕНЗИЛОКСИ-3,5-ДИФТОР-БЕНЗОИЛ)-4-ФТОР-ПИРРОЛИДИН-2-КАРБОНИЛ]-ПИРРОЛИДИН-2-КАРБОНИТРИЛ | 2021 |
|
RU2841064C1 |
| НОВОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ЛИГНАНА | 2006 |
|
RU2354370C1 |
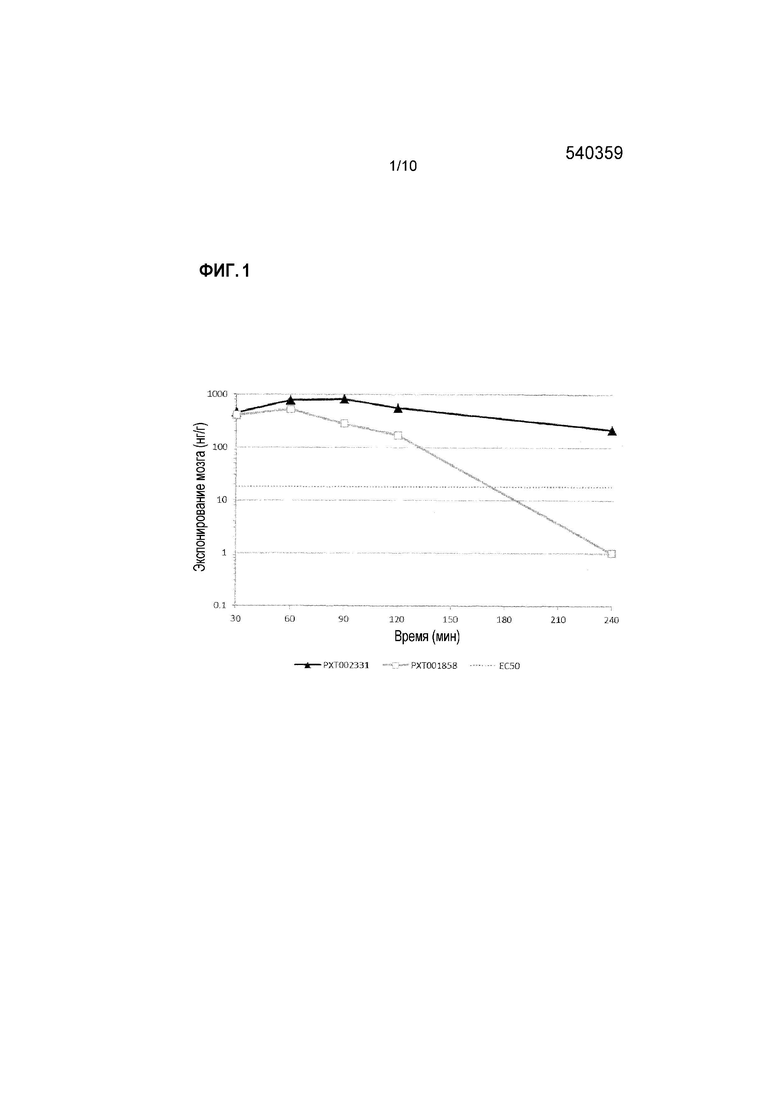
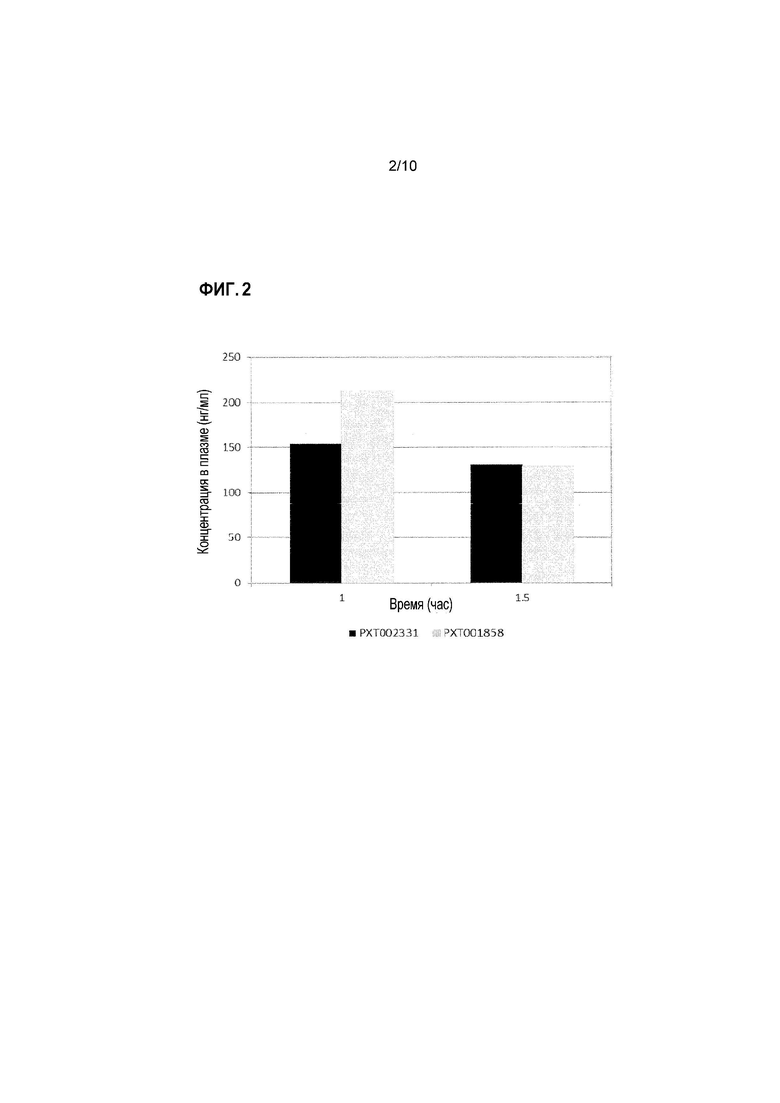
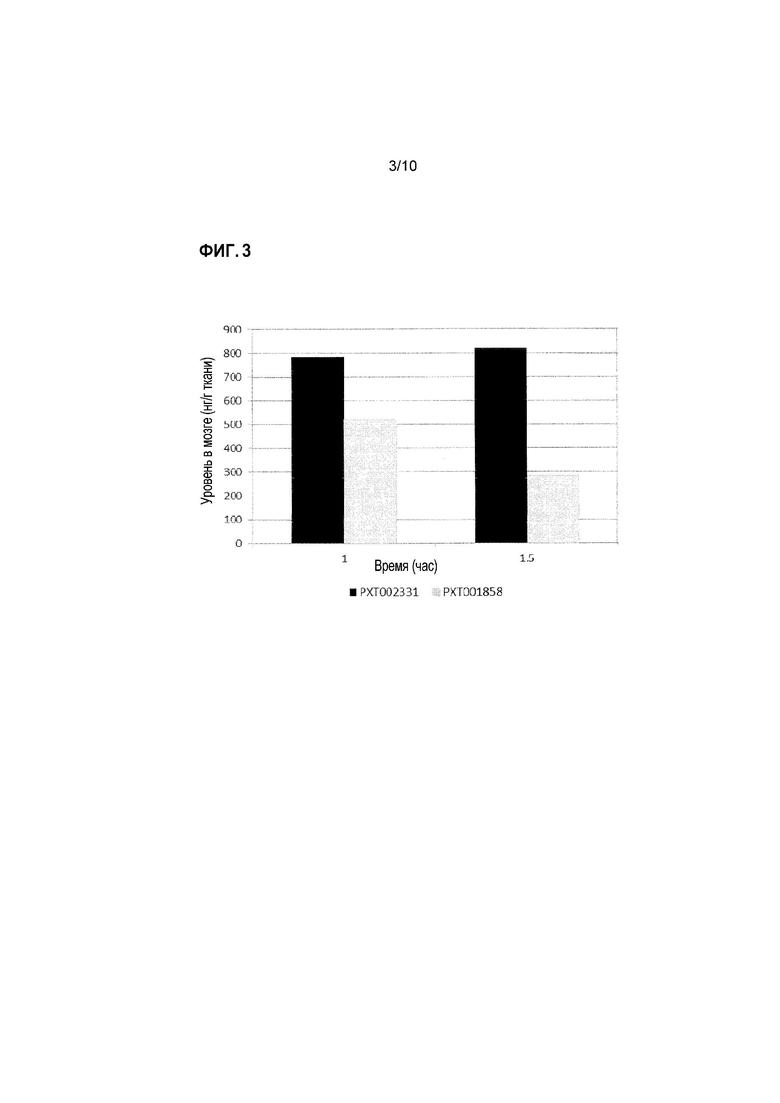
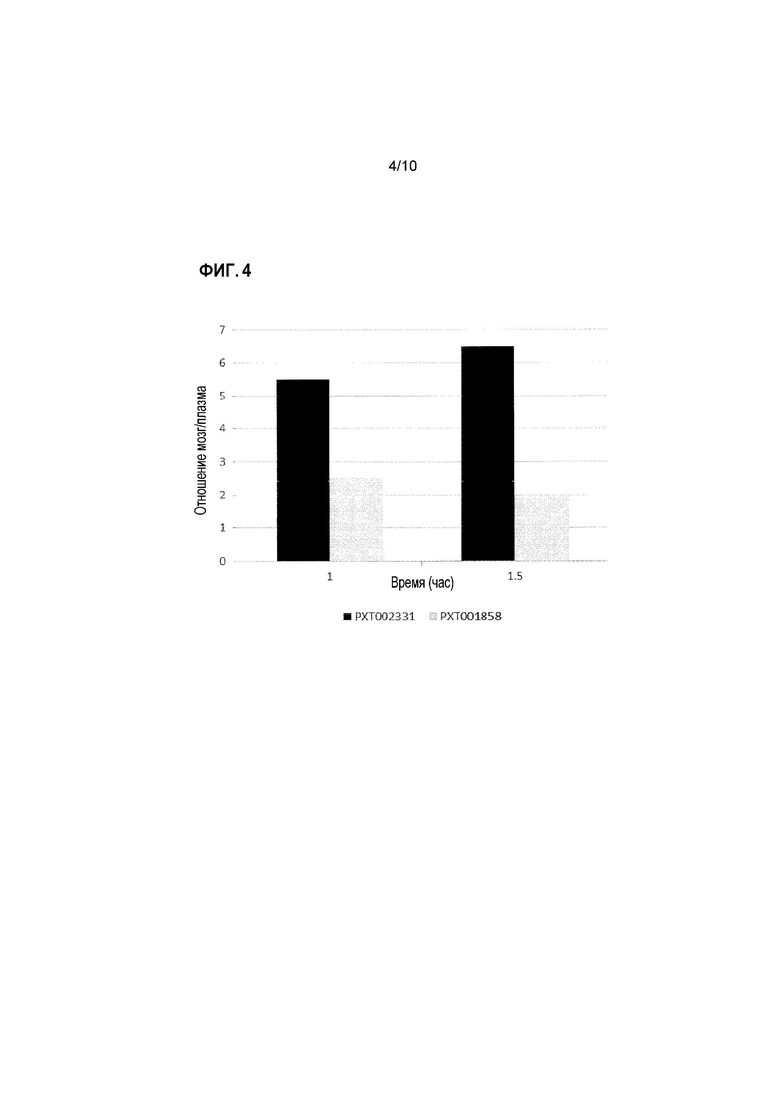
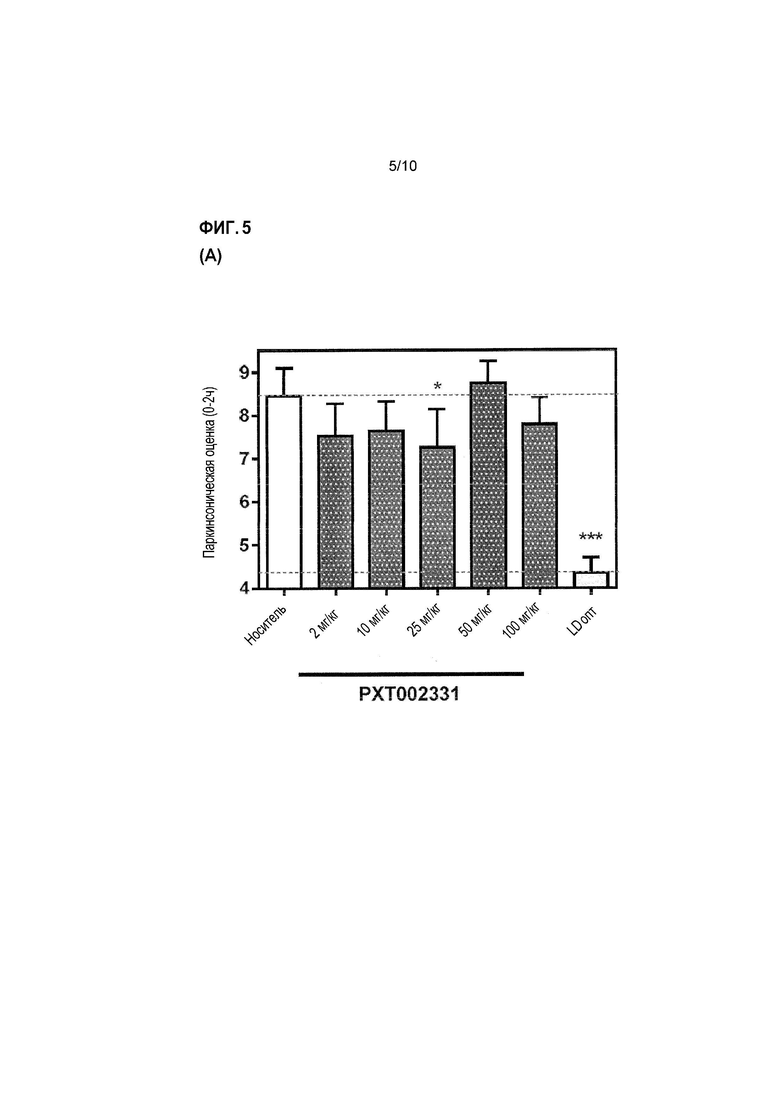
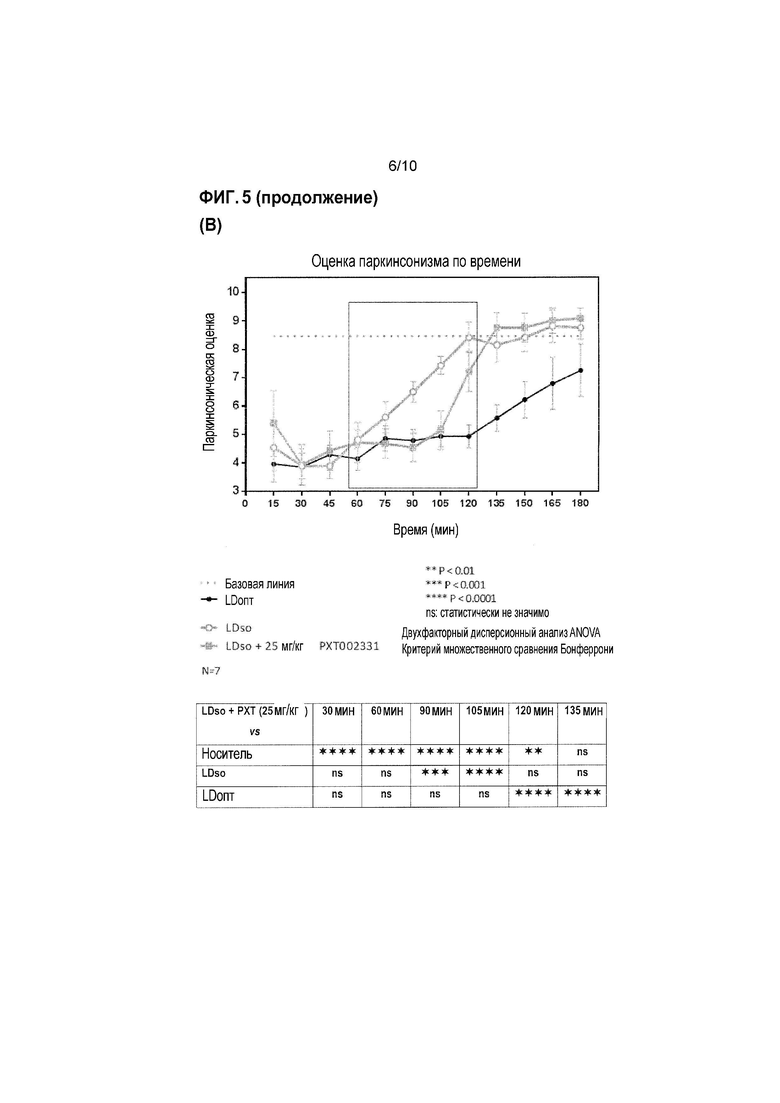
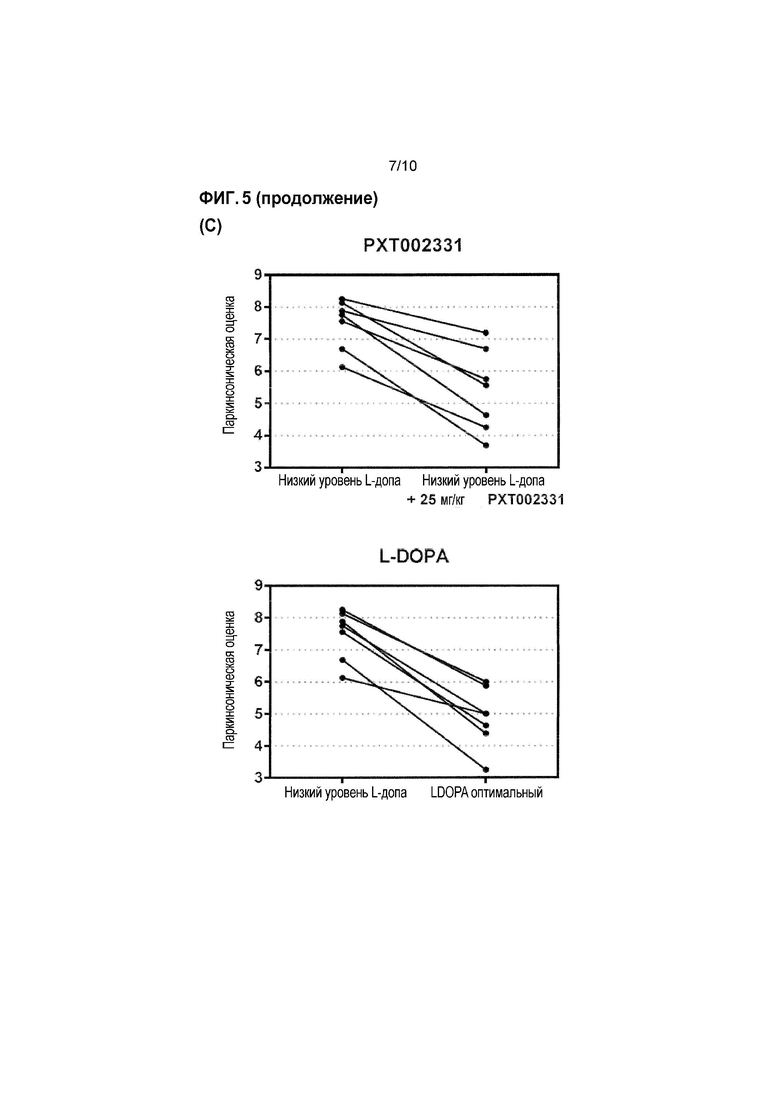
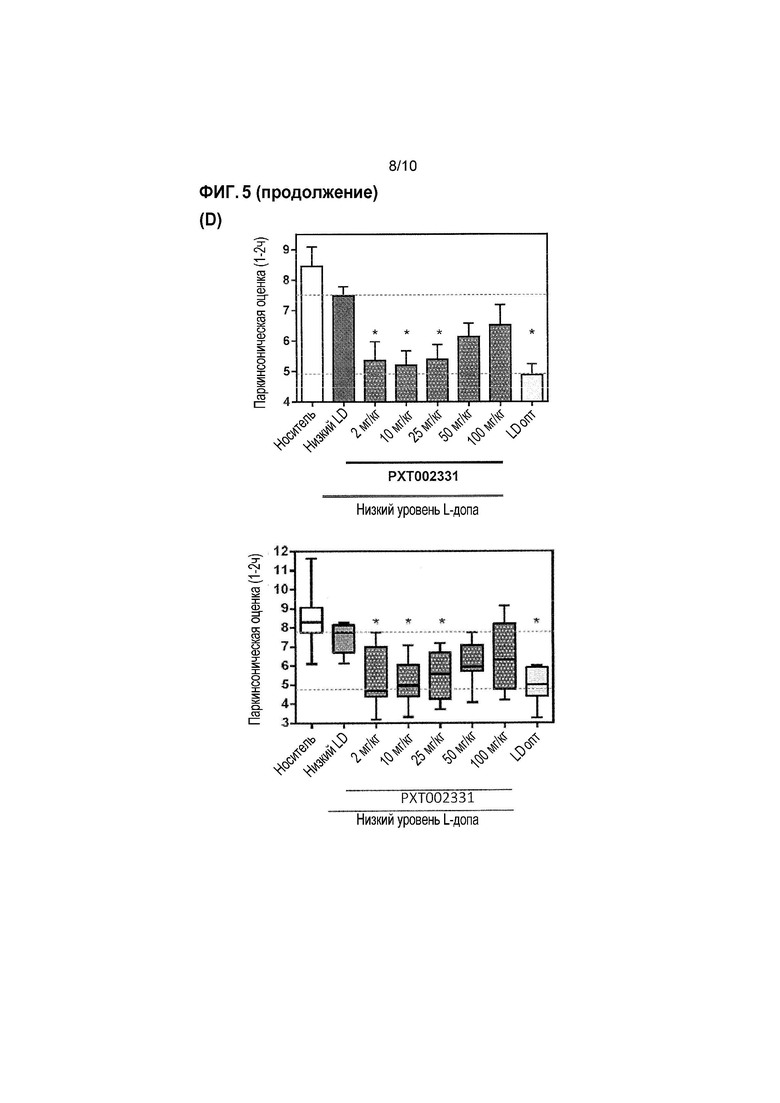
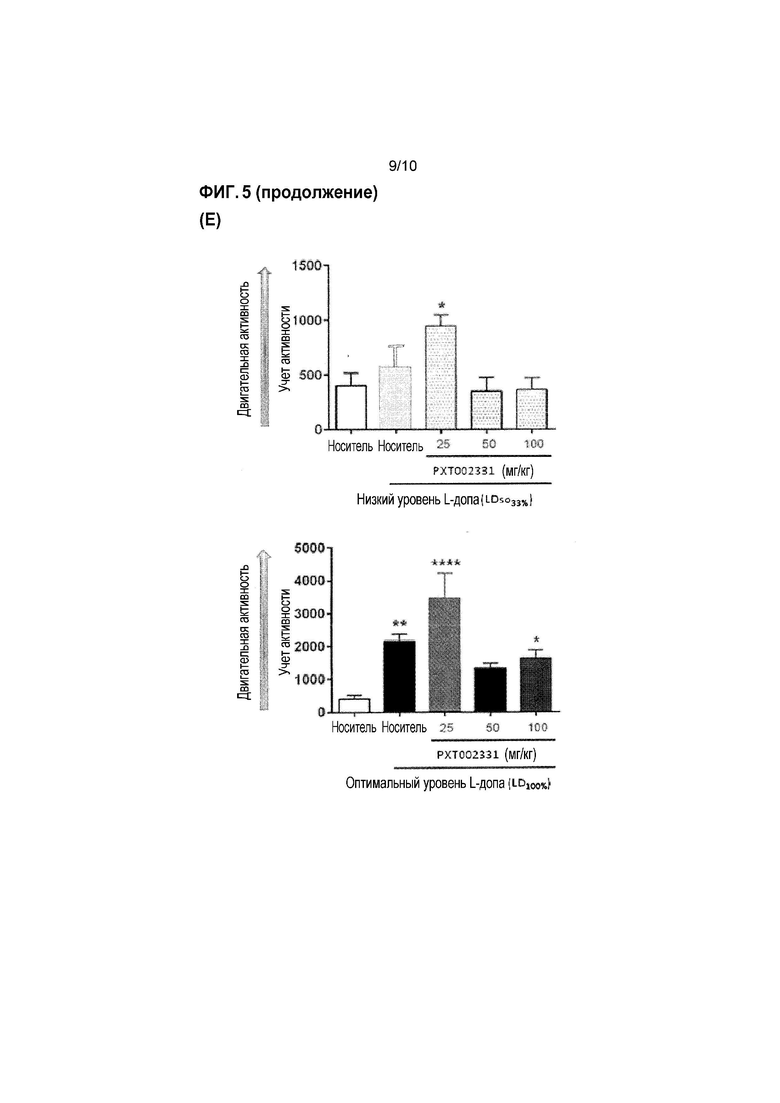
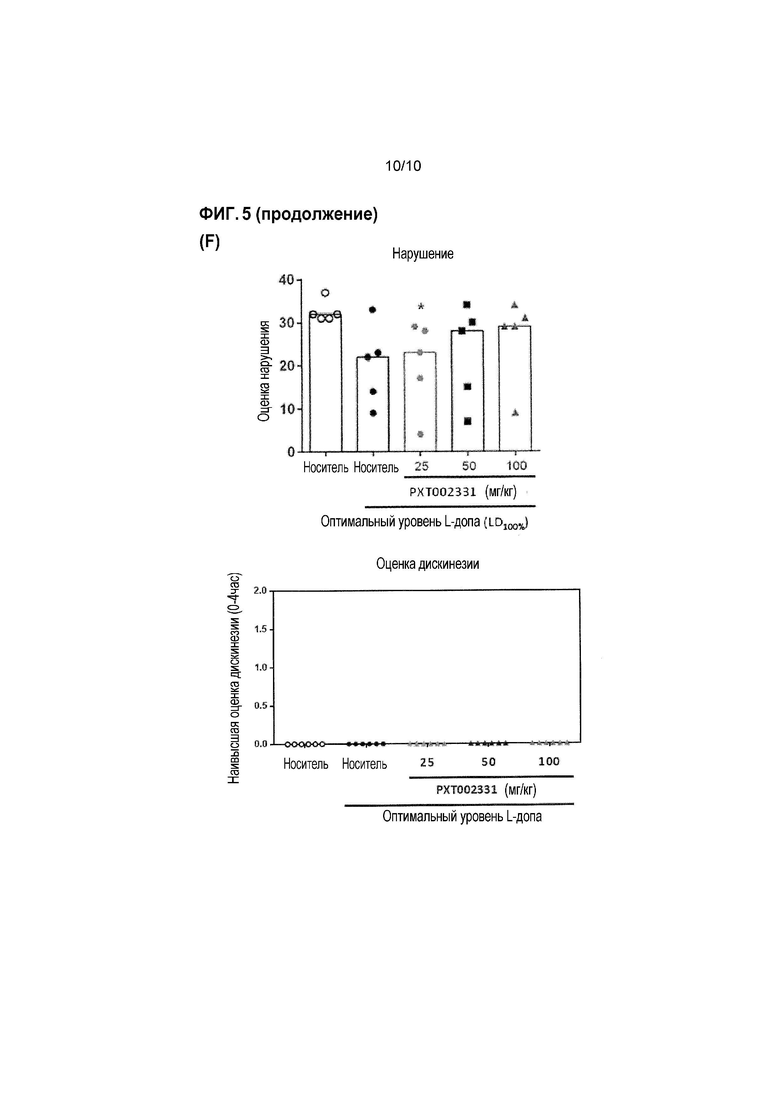
Изобретение относится к новому производному оксима хромона формулы (I), также относится к фармацевтическим композициям, содержащим указанное соединение, и к их использованию для лечения или профилактики состояний, связанных с измененной глутаматергической передачей сигнала и/или функцией, или состояний, на которые можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала, в частности болезни Паркинсона. Технический результат: получено новое соединение формулы (I), которое представляет собой модулятор проницаемости мозга mGluRs нервной системы и, кроме того, обеспечивает выгодную высокую проницаемость мозга после перорального введения. 6 н. и 8 з.п. ф-лы, 10 ил., 3 табл., 3 пр.
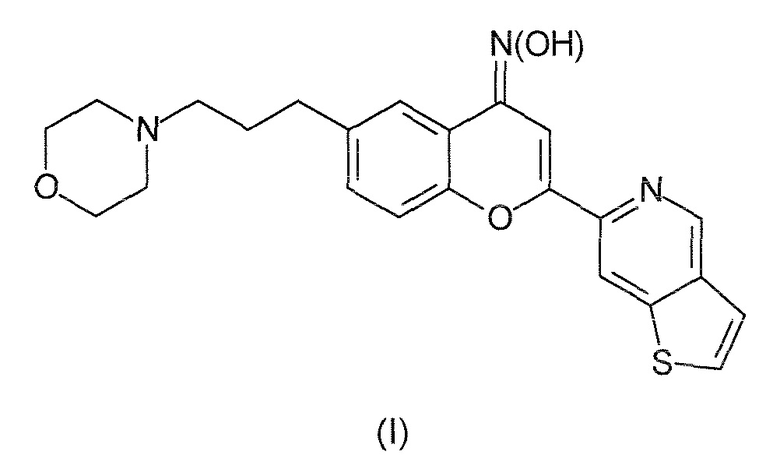
1. Соединение следующей формулы (I):
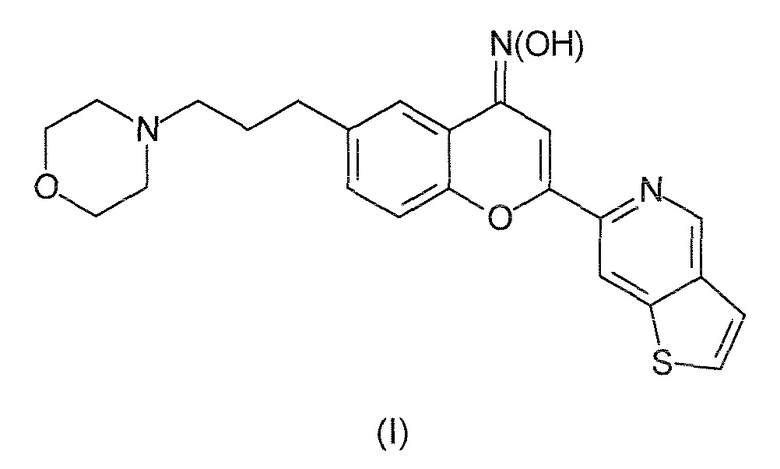
или его фармацевтически приемлемые соли.
2. Соединение по п. 1, где указанное соединение имеет (E) конфигурацию у группы оксима, содержащейся в формуле (I).
3. Соединение по любому из пп. 1 или 2, где фармацевтически приемлемой солью является гидрохлоридная соль.
4. Соединение по любому из пп. 1-3 для применения в качестве лекарственного средства, представляющего собой модулятор проницаемости мозга mGluRs нервной системы.
5. Фармацевтическая композиция, представляющая собой модулятор проницаемости мозга mGluRs нервной системы, содержащая эффективное количество соединения по любому из пп. 1-3, и фармацевтически приемлемый эксципиент.
6. Применение соединения по любому из пп. 1-3 для получения лекарственного средства для лечения или профилактики состояния, связанного с измененной глутаматергической передачей сигнала и/или функцией, или состоянием, на которое можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала.
7. Применение соединения по любому из пп. 1-3 для получения лекарственного средства для лечения или профилактики болезни Паркинсона.
8. Применение по п. 7, где указанное лекарственное средство вводят перорально.
9. Способ лечения состояния, связанного с измененной глутаматергической передачей сигнала и/или функцией, или состояния, на которое можно воздействовать, изменяя уровень содержания глутамата или передачи сигнала, включающий введение соединения по п. 1 нуждающемуся в этом субъекту.
10. Способ по п. 9, где указанный способ включает пероральное введение указанного соединения указанному субъекту.
11. Способ по п. 9, где указанным субъектом является человек.
12. Способ лечения болезни Паркинсона, включающий введение соединения по п. 1 нуждающемуся в этом субъекту.
13. Способ по п. 12, где указанный способ включает пероральное введение указанного соединения указанному субъекту.
14. Способ по п. 12, где указанным субъектом является человек.
| WO 2011051478 A1, 05.05.2011 | |||
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2557059C2 |

