ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к кристаллической форме II апремиласта, в частности к кристаллической форме II ингибитора фосфодиэстеразы 4 (ФДЭ-4), фармацевтическим композициям, содержащим указанные вещества, способам их получения, и применению кристаллической формы при лечении различных заболеваний и расстройств. Настоящее изобретение относится к области фармацевтической химии.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Как известно из данного уровня техники, различные кристаллические формы могут быть получены из-за различий в кристаллизующих растворителях и способах получения, например, в зависимости от температуры кристаллизации, скорости охлаждения, перемешивания или выдерживания в определенном состоянии, при этом различные кристаллические формы могут иметь разную стабильность и растворимость, иногда даже различную биодоступность в организмах. Соответственно, необходимо получить термодинамически устойчивую кристаллическую форму с высокой степенью чистоты при разработке лекарственного средства, а также легко воспроизводимый способ получения кристаллической формы, подходящий для промышленного производства. Кроме того, в качестве эффективных средств анализа кристаллических форм используются порошковая рентгеновская дифракция (ПРД), инфракрасная спектроскопия (ИК), дифференциальная сканирующая калориметрия (ДСК) и термогравиметрический анализ (ТГ).
Апремиласт (соединение I, химическое название (+)-2-[1-(3-этокси-4-метоксифенил)-2-метилсульфонилэтил]-4-ацетиламиноизоиндолин-1,3-дион) является ингибитором фосфодиэстеразы 4 (ФДЭ-4), который действует на циклический аденозин-3'-5'-монофосфат (цАМФ), где ингибирование ФДЭ-4 может приводить к повышению внутриклеточного уровня цАМФ, тем самым снижая припухлость сустава и улучшая физиологическую функцию суставов. Формула апремиласта показана ниже:
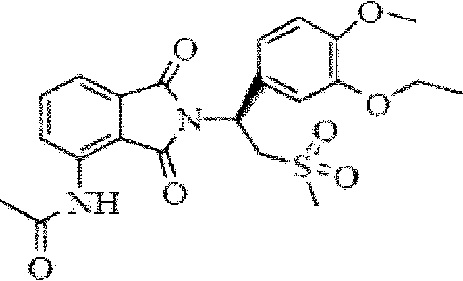
I
Этот препарат был одобрен для лечения псориатического артрита в США в марте 2014 года и одобрен для лечения псориаза в сентябре того же года. Кристаллические формы препарата описаны в патенте CN102702070A; в указанном патенте раскрываются семь твердых форм или кристаллических форм апремиласта, а именно формы A, B, C, D, E, F и G, данные порошковой рентгеновской дифракции (ПРД), дифференциальной сканирующей калориметрии (ДСК) и термогравиметрического анализа (ТГ) представлены ниже в таблице 1.
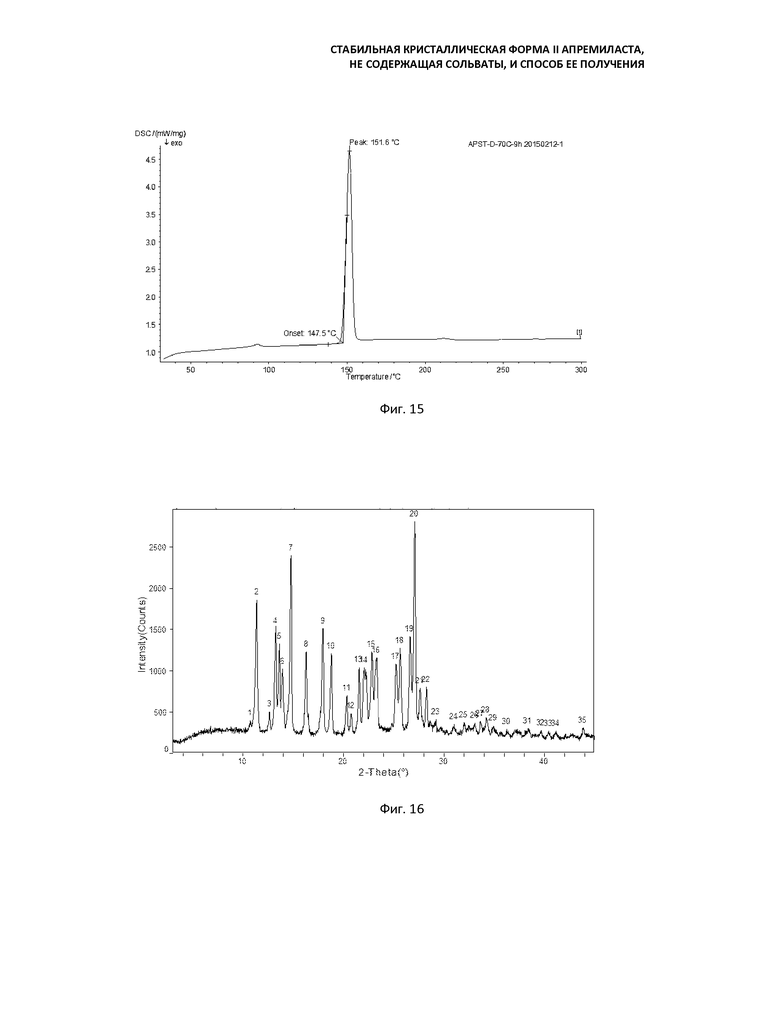
Среди кристаллических форм, формы C, D, E и G являются сольватами и не пригодны для использования в медицине; формы A, B и F являются несольватами или практически не содержат растворителей. В указанном патенте раскрыто замещение между различными кристаллическими формами, но не представлено ни одного действующего примера получения каждой кристаллической формы, поэтому нет возможности воспроизвести варианты осуществления изобретения.
Таблица 1. Данные о кристаллических формах апремиласта, представленные в патенте CN102702070A
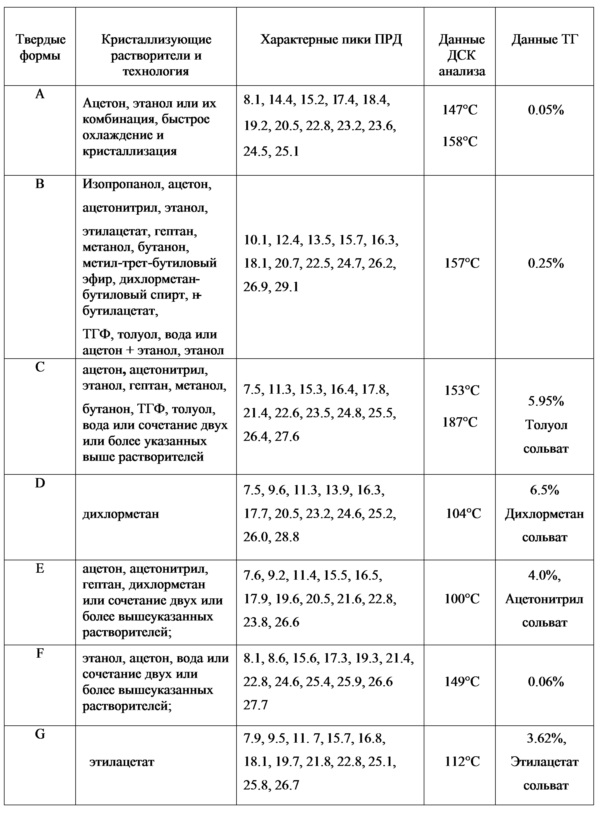
Среди форм A, B, C, D, E, F и G, описанных в патенте CN102702070A и регистрационных файлах, представленных в Европейском Агентстве по оценке лекарственных препаратов компанией Celgene, считается, что форма B является наиболее термодинамически стабильной формой, которая подходит для хранения и обработки. Однако препарат Отесла (Otezla), выпускаемый компанией Celgene, имеет срок годности всего один год, что является недостатком для коммерческого продукта. Таким образом, необходимо получить более термодинамически стабильную кристаллическую форму, пригодную для длительного хранения активного фармацевтического ингредиента и препаратов, кроме того указанная кристаллическая форма не должна влиять на биодоступность в организме или, предпочтительно, имеет лучшую биодоступность, чем известные кристаллические формы.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает новую и стабильную кристаллическую форму II апремиласта, не содержащую сольваты, композиции, содержащие указанное вещество и их применение. Настоящее изобретение также предлагает простые способы получения кристаллической формы II апремиласта, пригодные для промышленного применения. Кроме того, настоящее изобретение относится к способам получения смешанных кристаллов апремиласта, то есть к способам получения смешанных кристаллов апремиласта, включающих кристаллическую форму II апремиласта и кристаллическую форму B апремиласта.
Более того, кристаллическая форма II апремиласта по настоящему изобретению более термодинамически стабильна, чем известные кристаллические формы апремиласта, такие как формы A, B, C, D, E, F или G.
В некоторых вариантах осуществления, кристаллическая форма II апремиласта по настоящему изобретению обладает лучшей биодоступностью в живых организмах, чем известная кристаллическая форма Б апремиласта.
В настоящем изобретении ссылаются на патент CN201410335852.6, в котором раскрываются способы приготовления апремиласта и его промежуточных продуктов; а также на патент CN201410420960.3, в котором раскрывается стабильная кристаллическая форма I апремиласта, не содержащая сольваты, и способы ее получения.
Кристаллическая форма II апремиласта, не содержащая сольваты, по настоящему изобретению отличается тем, что имеет следующую формулу (I):
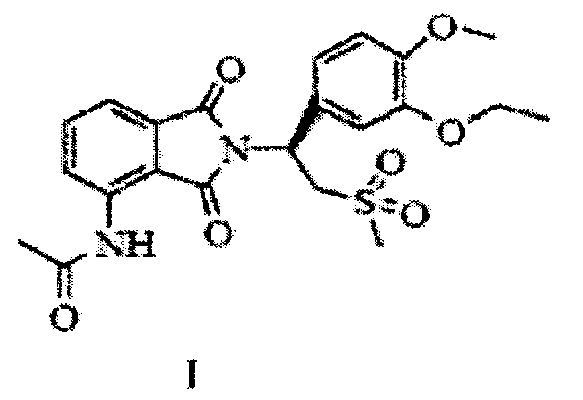
при этом на кривой порошковой рентгеновской дифракции содержатся следующие примерные пики поглощения при угле отражения 2θ ± 0,2:
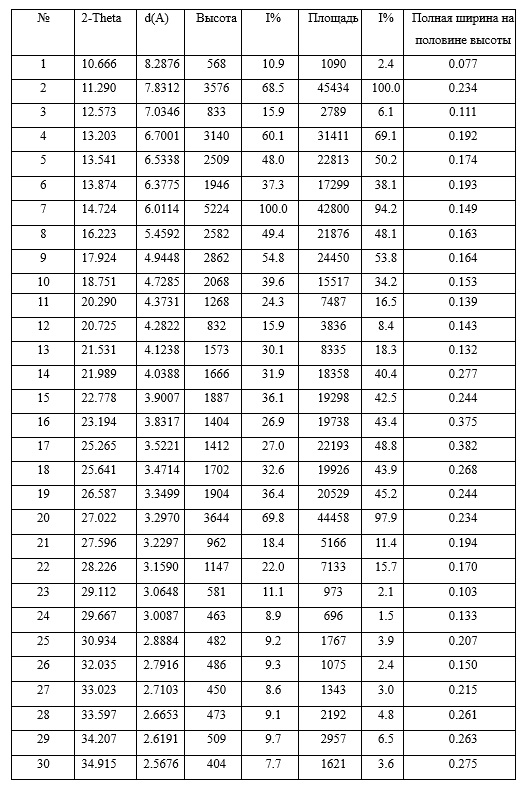
График дифференциальной сканирующей калориметрии (ДСК) показывает один эндотермический пик при температуре 150±3oC в диапазоне 100-180°C;
Термогравиметрический анализ (ТГ) показывает, что вещество не содержит кристаллическую воду или кристаллизующий растворитель;
Температура плавления находится в диапазоне l46-151°C.
В другом варианте осуществления, настоящее изобретение относится к способу получения указанной кристаллической формы II апремиласта, не содержащей сольваты, характеризующемуся использованием растворителя, который представляет собой смесь ацетона и воды, смесь ТГФ и воды или смесь ацетона, ТГФ и воды, предпочтительно смесь ацетона и воды. В частности, указанный способ включает в себя:
i) растворение апремиласта или сольвата в ацетоне или ТГФ при повышенной температуре, затем охлаждение до температуры ниже 40°C;
ii) медленное добавление воды, объем которой в 0,5-2 раза превышает объем ацетона или ТГФ при перемешивании, при необходимости, введение кристалла-затравки формы II и перемешивание в течение 30-180 мин;
iii) добавление воды, объем которой в 2-6 раз превышает объем ацетона или ТГФ, перемешивание в течение 1-24 ч при 20°С до температуры кипения в колбе с обратным холодильником; и
iv) фильтрование и сушку с получением кристаллической формы II апремиласта.
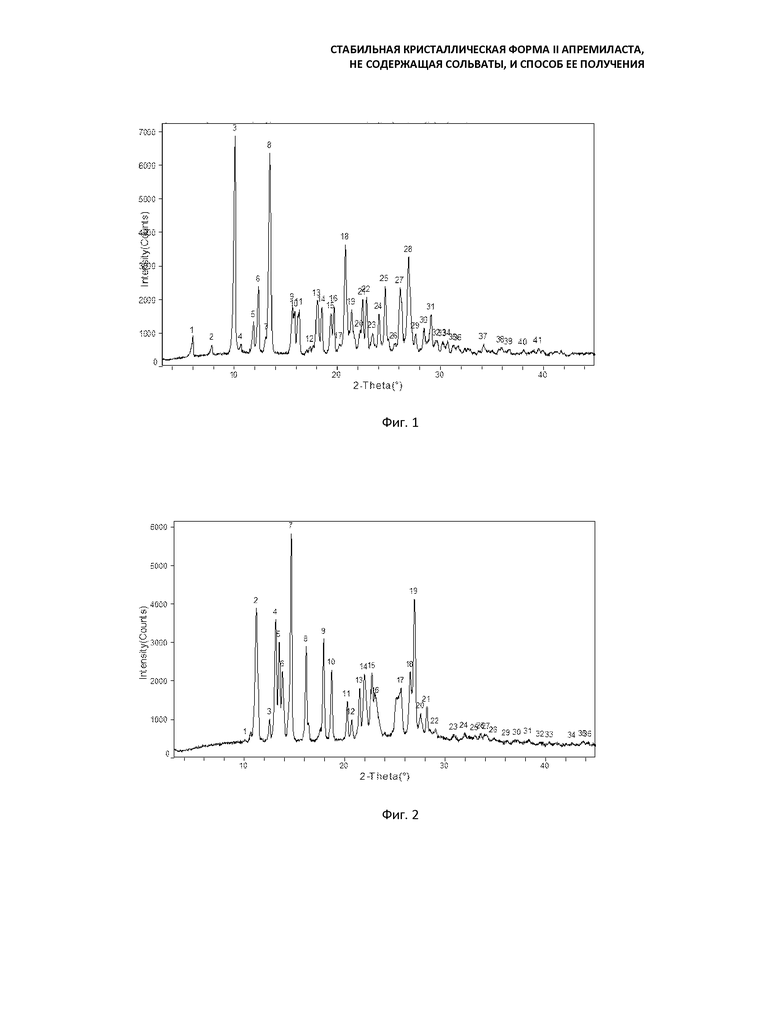
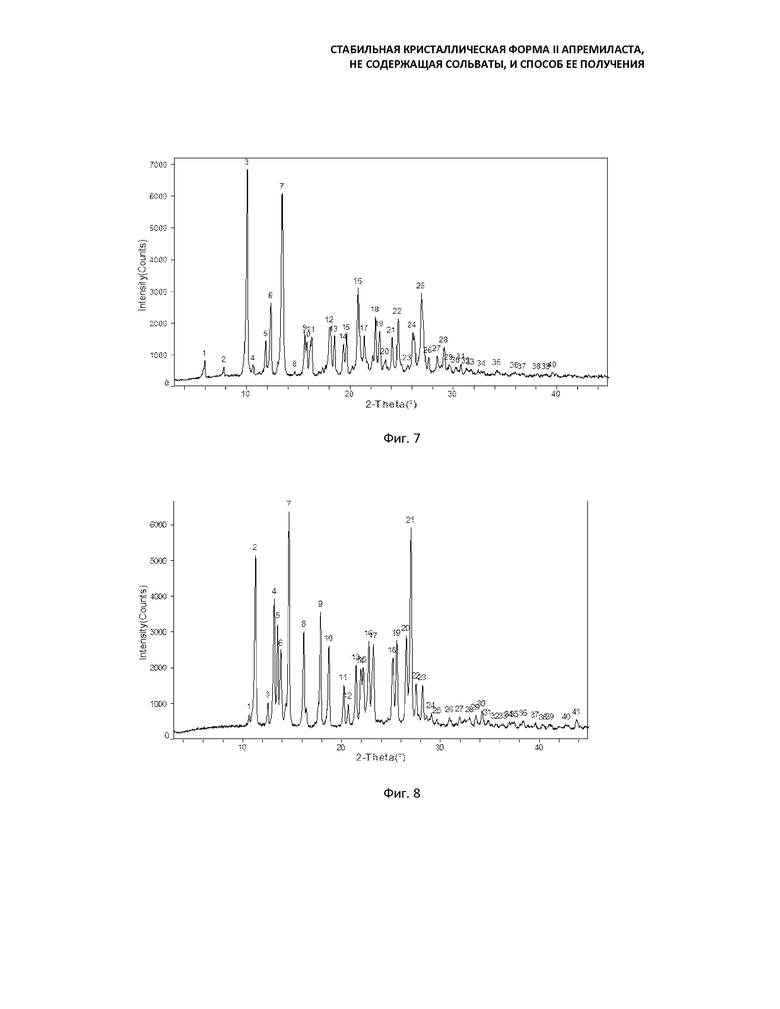
Предпочтительно вышеуказанный способ включает в себя: растворение апремиласта или сольвата при повышенной температуре в ацетоне, концентрация (мл/г) которого в 2-10 раз (предпочтительно в 3-5 раз) превышает концентрацию апремиласта или сольвата, затем охлаждение до температуры ниже 40°C; медленное добавление воды, объем которой в 0,5-2 раза больше объема ацетона при перемешивании, при необходимости, введением кристалла-затравки формы II и перемешивание в течение 0,5-3 часов при той же температуре; добавление воды, объем которой в 2-6 раз больше объема ацетона, перемешивание в течение 1-24 ч при 20°С до температуры кипения в колбе с обратным холодильником; фильтрование, промывку водой и сушку с получением кристаллической формы II апремиласта в виде белого твердого вещества. Кривые порошковой рентгеновской дифракции формы II несколько отличаются друг от друга в зависимости от времени перемешивания после добавления воды, но основные характерные пики при угле отражения 0,2-2θ остаются неизменными (Фиг. 2 и 5), кривые дифференциальной сканирующей калориметрии и термогравиметрического анализа практически идентичны.
Авторы неожиданно обнаружили, что при кристаллизации в смеси ацетона и воды или смеси ТГФ и воды, температура кристаллизации и количество первоначально добавленной воды оказывают значительное влияние на тип полученных кристаллических форм. Температура ниже 40°С более благоприятна для образования формы II. Более предпочтительно, чтобы введение кристалла-затравки формы II способствовало ускорению образования формы II в процессе кристаллизации. После добавления начального количества воды и перемешивания в течение 1-3 часов кристаллическая форма II выпадает в осадок; затем добавляют воду, объем которой в 2-6 раз превышает объем ацетона или ТГФ, перемешивают в течение 1-24 часов при 20°С до температуры кипения в колбе с обратным холодильником, чтобы получить мелкий порошок формы II; затем охлаждают, фильтруют и сушат, чтобы получить кристаллическую форму II апремиласта.
В другом варианте осуществления, настоящее изобретение относится ко второму способу получения кристаллической формы II апремиласта, включающему суспендирование одной или нескольких других кристаллических форм апремиласта с формами с соответствующими размерами частиц (например, формы A, B, C, D, E, F, G или форма I), в смеси ацетона и воды, смеси ТГФ и воды или смеси ТГФ, ацетона и воды (предпочтительно в смеси ацетона и воды), перемешивание в течение 1-72 ч или даже дольше при температуре 30°С в колбе с обратным холодильником, затем охлаждение, фильтрацию и сушку для получения кристаллической формы II. Наше исследование показывает, что чем меньше размер частиц, тем короче время превращения в кристаллическую форму II. Менее устойчивую кристаллическую форму легче превратить в кристаллическую форму II. Соотношение ацетона к воде предпочтительно составляет 1:1-1:4.
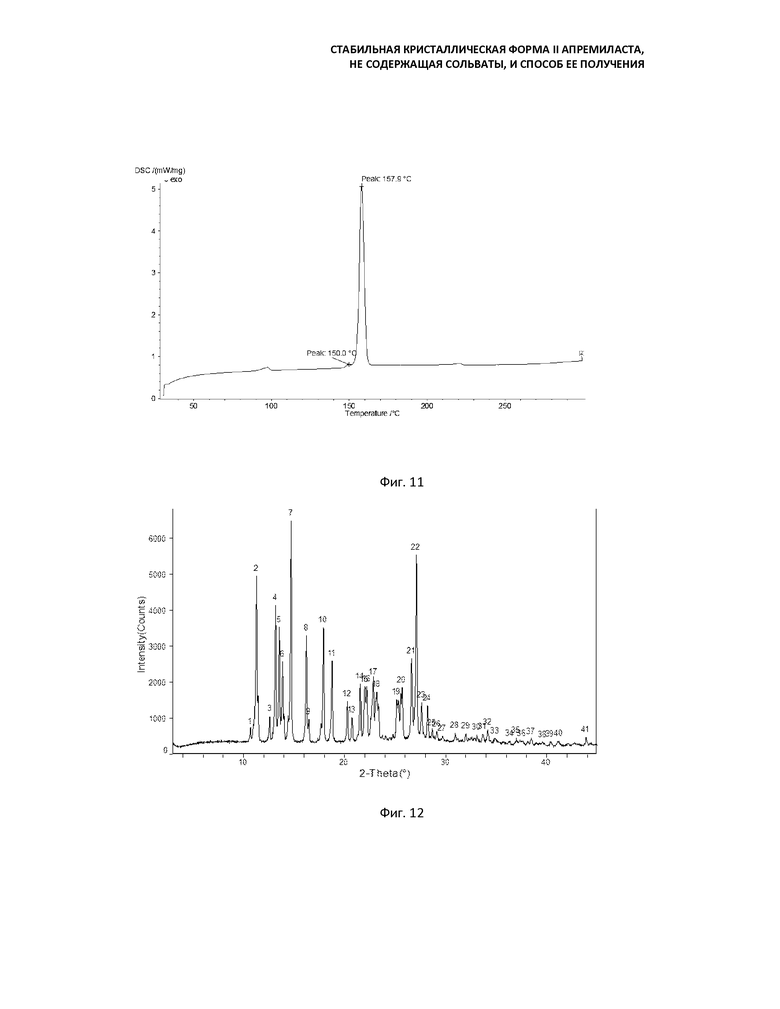
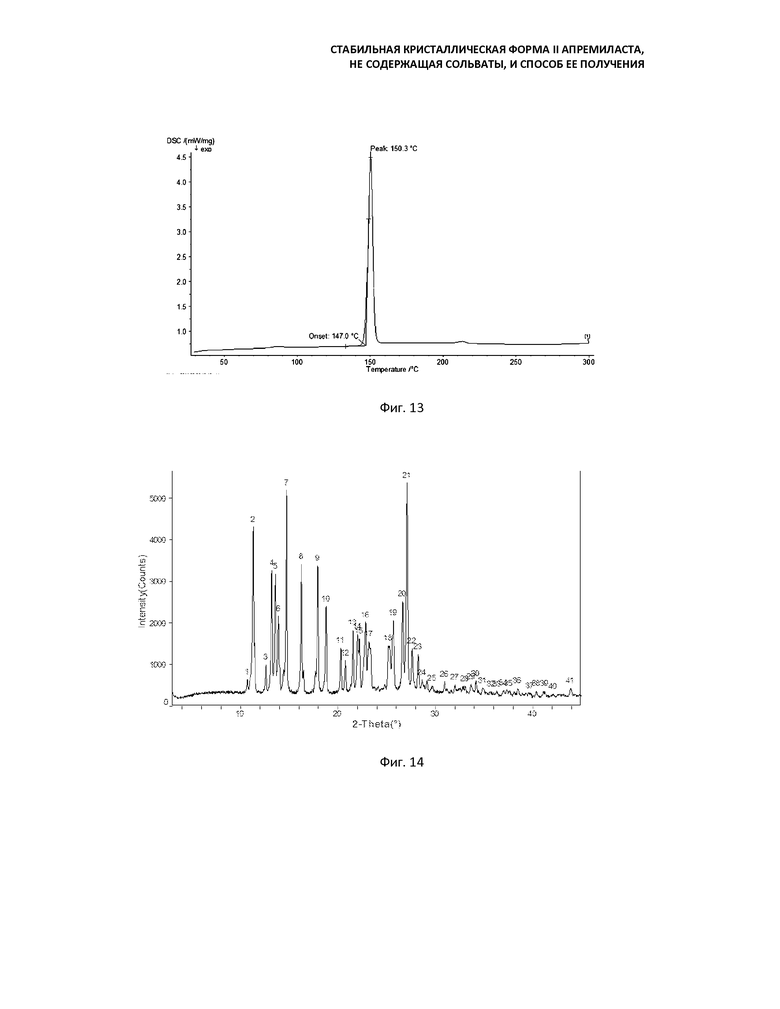
Кроме того, кристаллизация в смеси ацетона и воды может приводить к образованию смешанных кристаллов апремиласта, то есть смешанных кристаллов, включающих кристаллическую форму II апремиласта и кристаллическую форму В апремиласта; соотношение формы В к форме II в смешанных кристаллах можно регулироваться путем изменения соотношения ацетона к воде, а также времени и скорости добавления воды; другими словами, смешанные кристаллы могут содержать 0-100% формы II. Кривые порошковой рентгеновской дифракции смешанных кристаллов могут значительно меняться из-за различного соотношения двух форм, и на графиках ДСК будут показаны два пика поглощения при температурах 150 ± 3°С и 157 ± 3°С, соответственно, значения которых могут варьироваться в зависимости от соотношения двух форм. Таким образом, независимо от того, присутствует ли форма II, можно определить наличие характерных пиков поглощения формы II порошковой рентгеновской дифракцией, а также дополнительно подтвердить результаты наличием пика поглощения при температуре 150 ± 3 ° C на кривой ДСК.
Кроме того, смешанные кристаллы апремиласта также могут быть получены способом, включающим: суспендирование других кристаллических форм апремиласта с соответствующими размерами частиц (например, формы A, B, C, D, E, F или G) в смеси ацетона и воды, TГФ и воды или смеси ТГФ, ацетона и воды, перемешивание при температуре 20°С и нагрев до температуры кипения в колбе с обратным холодильником и перемешивание в течение разных промежутков времени; меняя время перемешивания и температуру можно получить смешанные кристаллы с различным соотношением.
Смешанные кристаллы апремиласта по настоящему изобретению содержат, по меньшей мере, две кристаллические формы, где, по меньшей мере, одна из кристаллических форм представляет собой кристаллическую форму II апремиласта, а другие кристаллические формы могут быть кристаллическими формами, указанными в ссылках на предшествующий уровень техники, например, формы A, B, C, D, E, F или G, предпочтительны кристаллическая форма B или F.
Кроме того, смешанные кристаллы апремиласта по настоящему изобретению предпочтительно состоят из кристаллической формы II апремиласта и кристаллической формы B апремиласта в любом соотношении. Очевидно, что смешанные кристаллы кристаллической формы II и кристаллической формы В могут использоваться в качестве активных фармацевтических ингредиентов. Однако кристаллическая форма II благодаря своей термодинамической стабильности наиболее предпочтительна для использования в качестве активного фармацевтического ингредиента.
Преимущественно, при кристаллизации в смеси ацетона и воды можно получить кристаллическую форму II апремиласта в виде белого или почти белого вещества с высокой чистотой до 99,8% и отдельной примесью, содержанием менее 0,1%, оптическая изомерия апремиласта по существу остается неизменной. Тем не менее, кристаллическая форма B апремиласта, полученная кристаллизацией в ацетоне или в смеси ацетона с этиловым спиртом, представляет собой светло-желтое вещество, и наиболее неблагоприятным образом количество R-изомера конечного продукта увеличивается после каждой кристаллизации. В патенте CN102702070 указывается, что оптическая чистота конечного продукта составляет 98%, промежуточный продукт имеет оптическую чистоту, равную 99,2%, и продукт имеет тенденцию к агломерации при сушке; при использовании большого количества этанола будут получены кристаллические формы с большим размером частиц, нелегко поддающимися измельчению и вызывающими трудности при использовании непосредственно при подготовке препарата.
Более предпочтительно, кристаллическая форма II апремиласта согласно настоящему изобретению получается в виде порошка; в некоторых вариантах осуществления, размер частиц D90 составляет менее 15 мкм. Продукт может быть непосредственно использован в процессе подготовки без измельчения.
Более того, кристаллическая форма II апремиласта, полученная в настоящем изобретении, может быть проанализирована порошковой рентгеновской дифракцией (ПРД), инфракрасной спектроскопией (ИК) (таблетки с KBr), дифференциальной сканирующей калориметрией (ДСК) и термогравиметрическим анализом (ТГ). Термогравиметрический анализ (ТГ) показывает, что в кристаллической форме II практически не содержится сольвата; кривая порошковой рентгеновской дифракцией кристаллической формы II отличается от кривых форм A, B, C, D, E, F и G, описанных в китайском патенте CN102702070, поэтому данная форма является новой кристаллической формой. Результат анализа показан ниже.
На кривой порошковой рентгеновской дифракции показаны характерные пики поглощения при угле отражения 2θ ± 0,2:
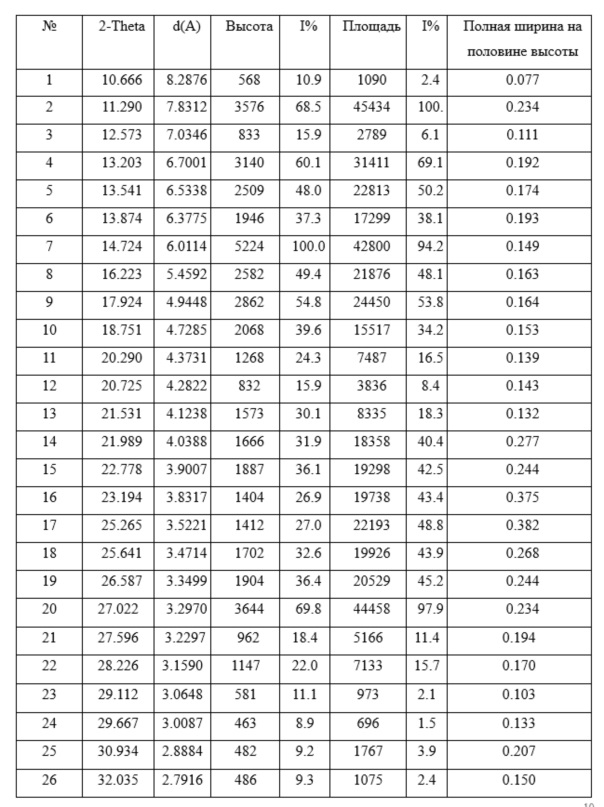
Кривая порошковой рентгеновской дифракции кристаллической формы II апремиласта содержит следующие основные характерные пики поглощения при угле отражения 2θ±0.2: 11,2, 13,2, 13,5, 13,8, 14,7, 16,2, 17,9, 18,7, 20,2, 20,7, 27,0.
Более того, кривая порошковой рентгеновской дифракции кристаллической формы II апремиласта содержит следующие пять характерных пики поглощения при угле отражения 2θ±0.2: 11,2, 13,2, 13,5, 13,8, 14,7.
Наконец, кривая порошковой рентгеновской дифракции кристаллической формы II апремиласта содержит следующие два характерных пика поглощения при угле отражения 2θ ± 0,2: 11,2, 14,7.
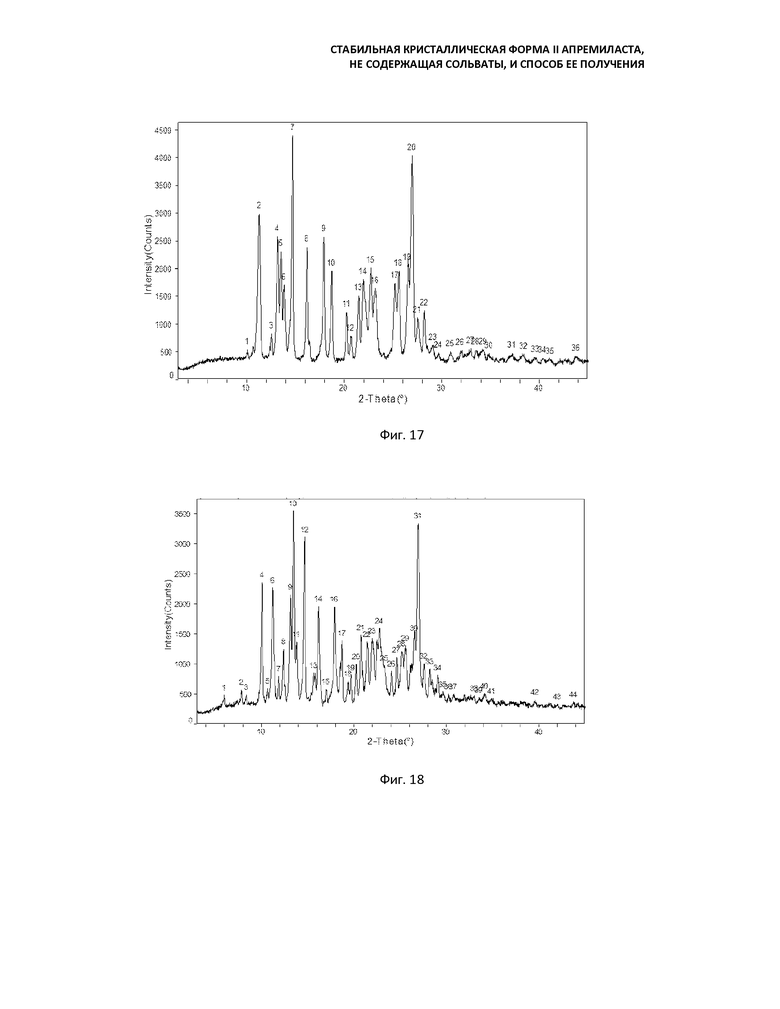
В другом варианте осуществления, кривая порошковой рентгеновской дифракции кристаллической формы II, полученной в смеси ацетона и воды, может иметь разные характерные пики поглощения между 20-30° при угле отражения 2θ±0,2 из-за ошибки измерения, но присутствие кристаллической формы II может быть определено в полученном продукте основным характерным пиком поглощения при угле отражения 2θ±0,2. Что более важно, характерные пики поглощения между 10-20° при угле отражения 2θ ± 0,2 практически одинаковы (Фиг. 2, 5, 6, 8, 12, 14, 16 и 17). Нет никаких существенных различий между кривыми ДСК, которые показывают один пик поглощения при температуре 150 ± 3°C в диапазоне 100-180 ° C.
По сравнению с пробным эксципиентом, препараты, содержащие кристаллическую форму II апремиласта в качестве активного фармацевтического ингредиента, показывают вышеупомянутые основные характерные пики поглощения, например, 11,2, 13,2, 13,5, 13,8, 14,7 и т. д. или 11,2, 14,7.
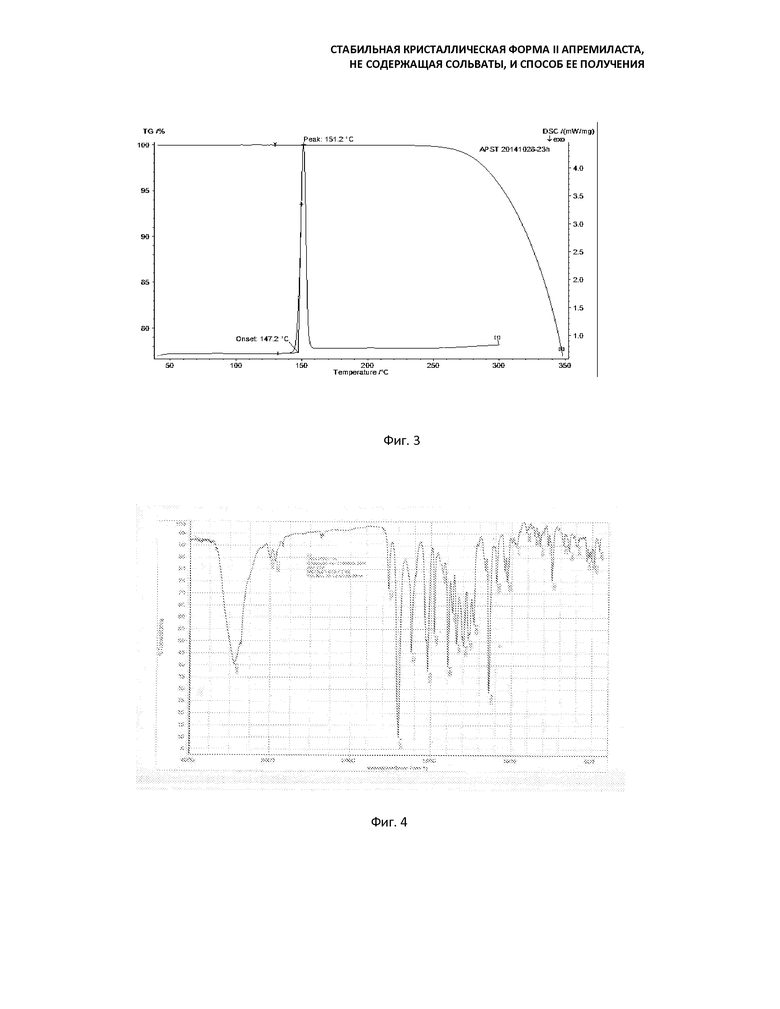
Кривая ДСК кристаллической формы II апремиласта содержит один пик поглощения при 150±3°C в диапазоне 100-180°C. График термогравиметрического анализа (ТГ) показывает, что в кристаллической форме II практически не содержится кристаллизующих растворителей или воды. Потеря веса и разложение наблюдались примерно с 250°C (Фиг. 3). Температура плавления формы II составляет 146-151° С.
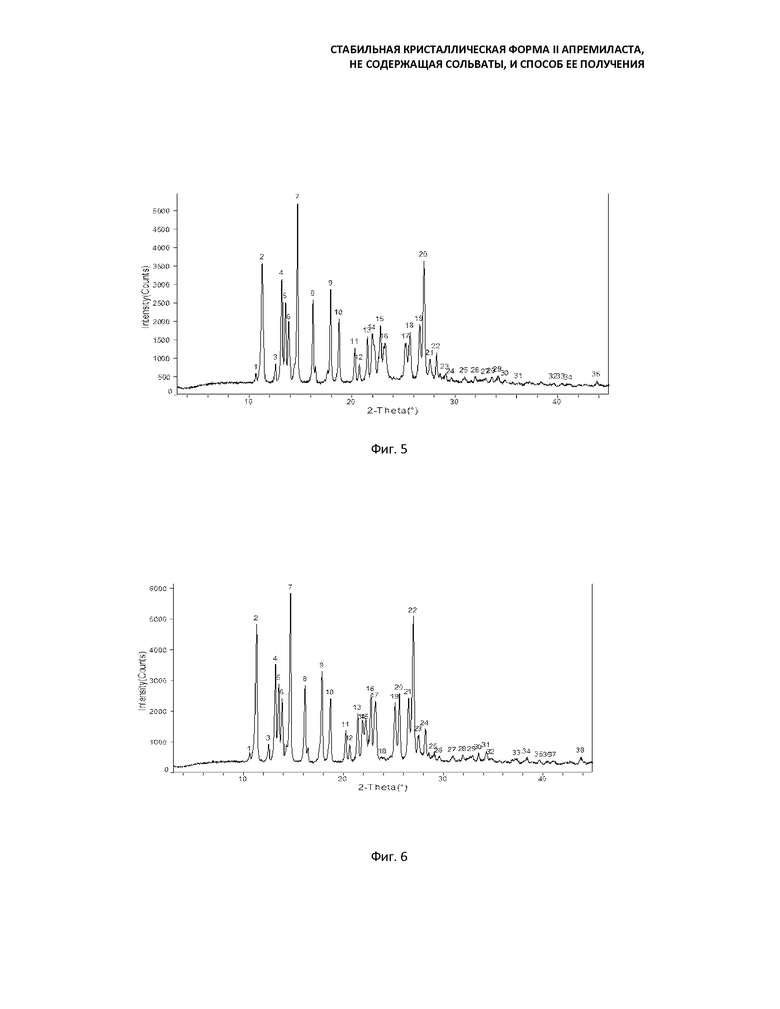
Инфракрасная спектроскопия (ИК) показывает, что форма II имеет следующие характерные пики поглощения: 3002, 2932, 1763, 1697, 1621, 1519, 1480, 1428, 1394, 1367, 1340, 1297, 1269, 1234, 1163, 1139, 1095, 1044, 1028, 971, 908, 859, 826, 774, 750.
Ниже приведена серия сравнительных исследований формы I, формы II и формы В.
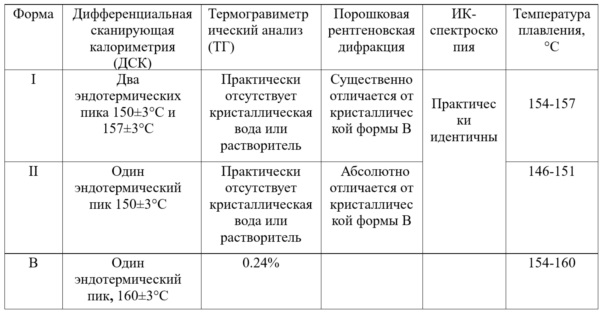
1. Анализ формы I, формы II и формы B: сравнение кристаллической формы I, формы II и формы B апремиласта показано в таблице 2.
Таблица 2. Сравнение кристаллической формы I, формы II и формы B апремиласта
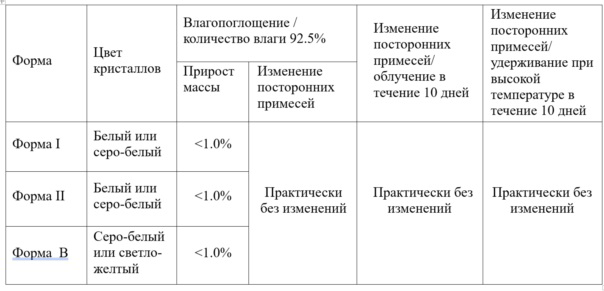
2. Эксперименты с факторами, влияющими на формы I, формы II и формы B:
Кроме того, ту же самую партию сырья кристаллизовали в смеси ацетона и этанола для получения формы В и в смеси ацетона и воды для получения формы I и формы II, а затем сравнивали внешний вид и цвет продуктов. Эксперименты с факторами, влияющими на указанные три формы, показали, что кристаллической формы I, формы II и формы B апремиласта были стабильными в условиях высокой температуры и высокой влажности, кристаллические формы показали идентичную стабильность при облучении.
Таблица 3. Результаты экспериментов по стабилизации формы I, формы II и формы B
3. Эксперименты по термостабильности формы II и формы B:
Кристаллическую форму II и форму В апремиласта суспендировали в воде и перемешивали в течение 24-48 ч при температуре 60°С и 100°С соответственно, затем охлаждали, фильтровали и сушили, а также были определены кривые порошковой рентгеновской дифракции, кривые ДСК, температура плавления и наличие посторонних примесей (См. Таблицу 4).
Таблица 4. Эксперименты по термостабильности формы II и формы B
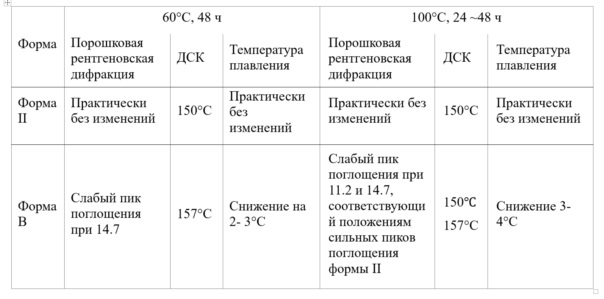
Вышеприведенные результаты экспериментов показали, что кристаллическая форма B апремиласта менее термодинамически стабильна, чем кристаллическая форма II апремиласта; кривые рентгеновской порошковой дифракции и кривые ДСК нагретой формы В показывают характерные пики поглощения формы II, а кривые формы II не демонстрируют существенных изменений. Более предпочтительно, при том же условии мелкого помола форма II проявляла слабый электростатический эффект или вообще не проявляла электростатический эффект, тогда как форма В проявляла более высокий электростатический эффект. Кроме того, когда форму В суспендировали в воде при 100°С и перемешивали в течение 24 ч, фильтровали и сушили, наблюдался серьезный электростатический эффект, а форма II практически не оказывала электростатического эффекта. Большой электростатический эффект отрицательно влияет на обработку препарата. При перемешивании при 100°С в течение 48 ч обе формы не показали значительного изменения оптической изомерии; количество посторонних примесей, N-деацетилатов, увеличилось, причем прирост формы B несколько больше, чем у формы II (то есть 0,059% и 0,046% соответственно), разница незначительна.
4. Преобразование форм A, B, C, D, E, F, G в форму II
Кроме того, чистую форму II можно получить суспендированием молотых форм A, B, C, D, E, F и/или G в смеси ацетона и воды и перемешиванием при температуре от 50°C до температуры кипения в колбе с обратным холодильником в течение 1-48 часов. Это снова показывает, что форма II более термодинамически устойчива, чем форма B.
Среди форм A, B, C, D, E, F и G, представленных компанией Celgene, форма B считается самой термодинамически стабильной формой и, следовательно, пригодна для использования в медицине. Наши эксперименты показывают, что форма II еще более стабильна, чем форма В, поэтому она лучше подходит для длительного хранения и может быть использована в качестве лекарственного кристалла при обработке препарата.
5. Эксперименты на растворимость формы B и формы II
Испытывали и сравнивали растворимость формы II и формы В в обычных растворителях, таких как ацетон, бутанон, этиловый спирт, метанол, этилацетат, ацетонитрил, дихлорметан, тетрагидрофуран, петролейный эфир, н-гексан, вода. Результаты эксперимента показывают, что по существу нет разницы между формой II и формой B.
6. Исследование фармакокинетических свойств формы B и формы II на животных
1) Физиологические тесты на реакцию абсорбции на крысах
Исследование фармакокинетических свойств кристаллической формы B и формы II апремиласта с аналогичными размерами частиц на крысах Спрега-Доули показало, что как кристаллическая форма II апремиласта по настоящему изобретению, так и кристаллическая форма B апремиласта по разному воздействуют на крыс, в зависимости от их половой принадлежности; обе формы показали аналогичные Tmax, Cmax, T1/2. Воздействие формы II на самок крыс было в 1,5 раза дольше, чем воздействие формы В, поэтому форма II обладает более сильной биологической активностью.
7. Фармацевтическая композиция
В одном варианте осуществления настоящего изобретения предлагаются фармацевтические композиции, содержащие указанную кристаллическую форму II апремиласта, не содержащую сольваты, в качестве активного фармацевтического ингредиента и фармацевтически приемлемый носитель, где активный фармацевтический ингредиент содержит 1-100% кристаллической формы II апремиласта.
Очевидно, что подобно применению и показаниям формы В, кристаллическая форма II апремиласта, не содержащая сольваты, полученная в соответствии с настоящим изобретением, может быть использована для лечения заболевания или расстройства, которое может быть улучшено ингибированием TNF-α, где заболевание или расстройство включают в себя псориаз, псориатический артрит, анкилозирующий спондилит, ревматоидный артрит, атопический дерматит, язву языка у пациентов с болезнью Бехчета, хронический кожный саркоидоз, гигантоклеточный артериит, болезнь Паркинсона, узелковую почесуху, лишай Вильсона, сложные заболевания полости рта, волчанку, гепатит, увеит, синдром сухого глаза, депрессию, интерстициальный цистит, вульводинию, простатит, остеоартрит, диффузную В-крупноклеточную лимфому, полимиозит, дерматомиозит, миозит с включенными тельцами, эрозивный остеоартроз, эндометриоз, корешковую невропатию и гангренозную пиодермию или хроническую обструктивную болезнь легких.
В другом варианте осуществления изобретения кристаллическая форма II апремиласта, не содержащая сольваты, полученная в соответствии с настоящим изобретением, может быть использована для лечения заболевания или расстройства, которое может быть улучшено ингибированием ФДЭ-4, где заболевание или расстройство включают в себя ВИЧ, гепатит, синдром расстройства дыхания у взрослых, заболевания костей, хроническую обструктивную болезнь легких, хроническую воспалительную болезнь легких, дерматит, воспалительное заболевание кожи, атопический дерматит, кистозный фиброз, септический шок, сепсис, эндотоксиновый шок, гемодинамический шок, септический синдром, постишемический реперфузионный синдром, менингит, фиброз, псориаз, псориатический артрит, кахексию, отторжение трансплантата, реакцию "трансплантата против хозяина", аутоиммунное заболевание, ревматоидный артрит, спондилит, анкилозирующий спондилит, ревматоидный артрит, остеоартрит, остеопороз, болезнь Крона, язвенный колит, воспаление кишечника, рассеянный склероз, системную красную волчанку, проказы в узловатой лепрозной эритеме, радиационные повреждения и гипероксию.
Кроме того, кристаллическая форма II апремиласта, не содержащая сольваты, полученная в соответствии с настоящим изобретением, может использоваться для лечения рака, где рак может представлять собой миеломную болень, злокачественную меланому, спонгиобластому, лейкемию и твердую опухоль.
Наконец, кристаллическая форма II апремиласта, не содержащая сольваты, полученная согласно настоящему изобретению, может использоваться для лечения саркоидоза, при этом саркоидоз может представлять собой саркоидоз сердца, кожный саркоидоз, саркоидоз печени, саркоидоз полости рта, саркоидоз нервной системы, саркоидоз придаточной пазухи носа, синдром Лефгрена, ознобленную волчанку, увеит или хронический кожный саркоидоз.
Очевидно, что кристаллическая форма II апремиласта, не содержащая сольваты, может использоваться в качестве активного фармацевтического ингредиента для лечения вышеупомянутых заболеваний и расстройств. Соответствующие лекарственные формы включают таблетки, капсулы, диспергируемые таблетки, таблетки для рассасывания и т. д.; Вспомогательные материалы могут быть выбраны из, но не ограничиваются ими, лактозы, маннитола, поливинилполипирролидона, аэросила, стеарата магния, микрокристаллической целлюлозы, карбоксиметилцеллюлозы натрия, гидроксипропилцеллюлозы, кроскамеллозы натрия, предпочтительно выбранны из лактозы, микрокристаллической целлюлозы, кроскармеллозы натрия, аэросила и стеарата магния.
Апремиласт, указанный в настоящем изобретении, может быть легко получен в соответствии со способами, описанными в предшествующих уровнях техники в патентах US6020358, US6962940 или CN201410335852.6.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
Рисунки, включенные в настоящую заявку, составляют части спецификации и могут быть использованы для иллюстрации настоящего изобретения вместе со спецификацией и формулой изобретения.
Фиг. 1: кривая порошковой рентгеновской дифракции формы B;
Фиг. 2: кривая порошковой рентгеновской дифракции формы II (перемешивание в течение 4 часов после добавления воды);
Фиг. 3: кривая термогравиметрического анализа и кривая ДСК формы II (перемешивание в течение 23 часов после добавления воды);
Фиг. 4: кривая ИК-спектроскопии формы II (перемешивание в течение 23 часов после добавления воды);
Фиг. 5: кривая порошковой рентгеновской дифракции формы II (перемешивание в течение 23 часов после добавления воды);
Фиг. 6: кривая порошковой рентгеновской дифракции формы II (перемешивание в течение 48 ч при 60°C);
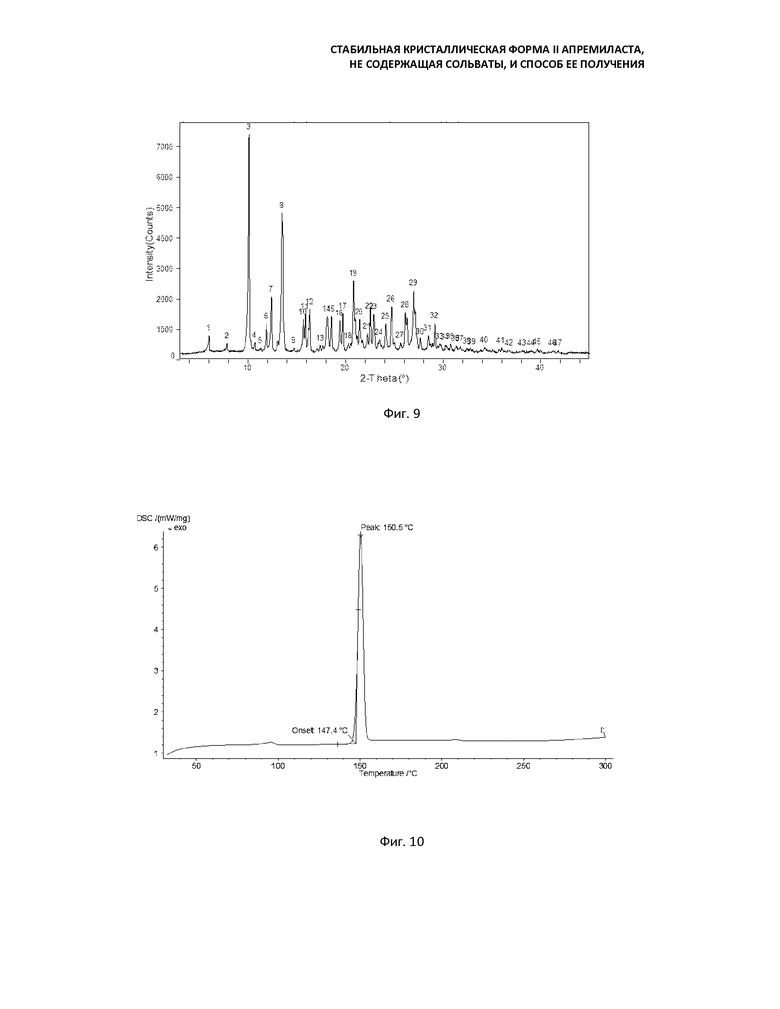
Фиг. 7: кривая порошковой рентгеновской дифракции формы B (перемешивание в течение 48 ч при 60°C);
Фиг. 8: кривая порошковой рентгеновской дифракции формы II (перемешивание в течение 24 ч при 100°C);
Фиг. 9: кривая порошковой рентгеновской дифракции формы B (перемешивание в течение 24 ч при 100°C);
Фиг. 10: кривая ДСК формы II (перемешивание в течение 24 ч при 100°C);
Фиг. 11: кривая ДСК формы B (перемешивание в течение 24 ч при 100°C);
Фиг. 12: кривая порошковой рентгеновской дифракции продукта, полученного суспендированием формы В в смеси ацетона и воды и перемешиванием в течение 36 ч при 70°С;
Фиг. 13: кривая ДСК продукта, полученного суспендированием формы В в смеси ацетона и воды и перемешиванием в течение 36 ч при 70°С;
Фиг. 14: кривая порошковой рентгеновской дифракции продукта, полученного суспендированием формы D в смеси ацетона и воды и перемешиванием в течение 10 ч при 70°C;
Фиг. 15: кривая ДСК продукта, полученного суспендированием формы D в смеси ацетона и воды и перемешиванием в течение 10 ч при 70°С;
Фиг. 16: кривая порошковой рентгеновской дифракции формы II;
Фиг. 17: кривая порошковой рентгеновской дифракции формы II;
Фиг. 18: кривая порошковой рентгеновской дифракции смеси формы II и формы B (соотношение примерно 1:1);
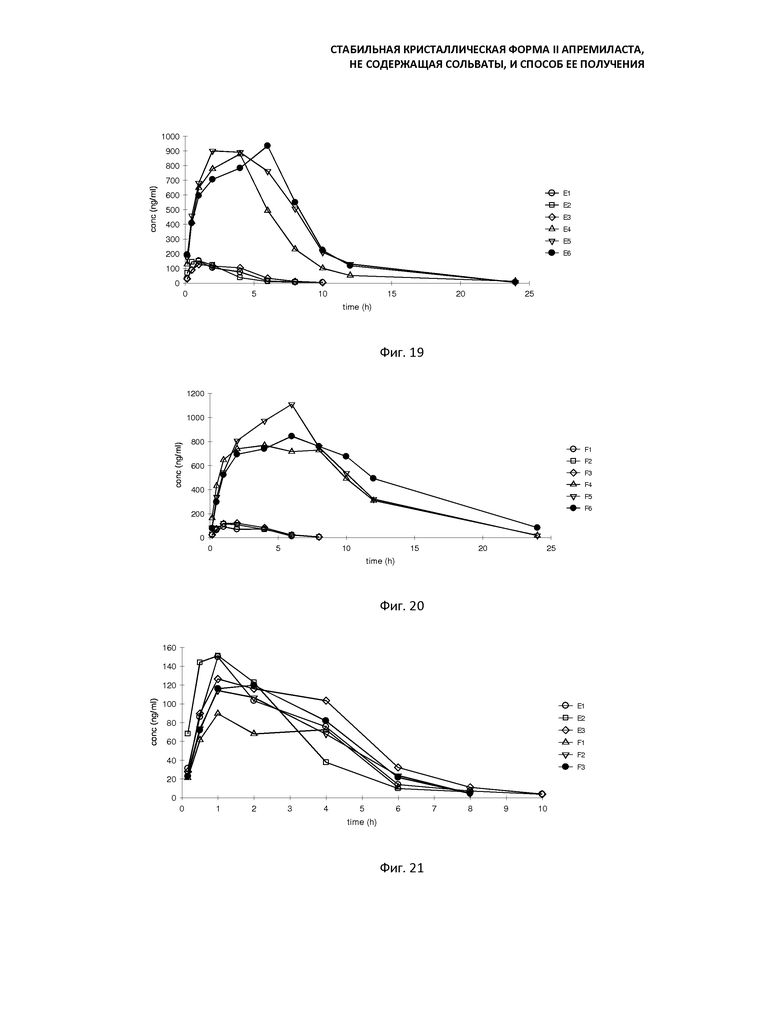
Фиг. 19: График зависимости концентрации плазмы от времени у крыс, после введения через желудочный зонд 10 мг/кг кристаллической формы В апремиласта;
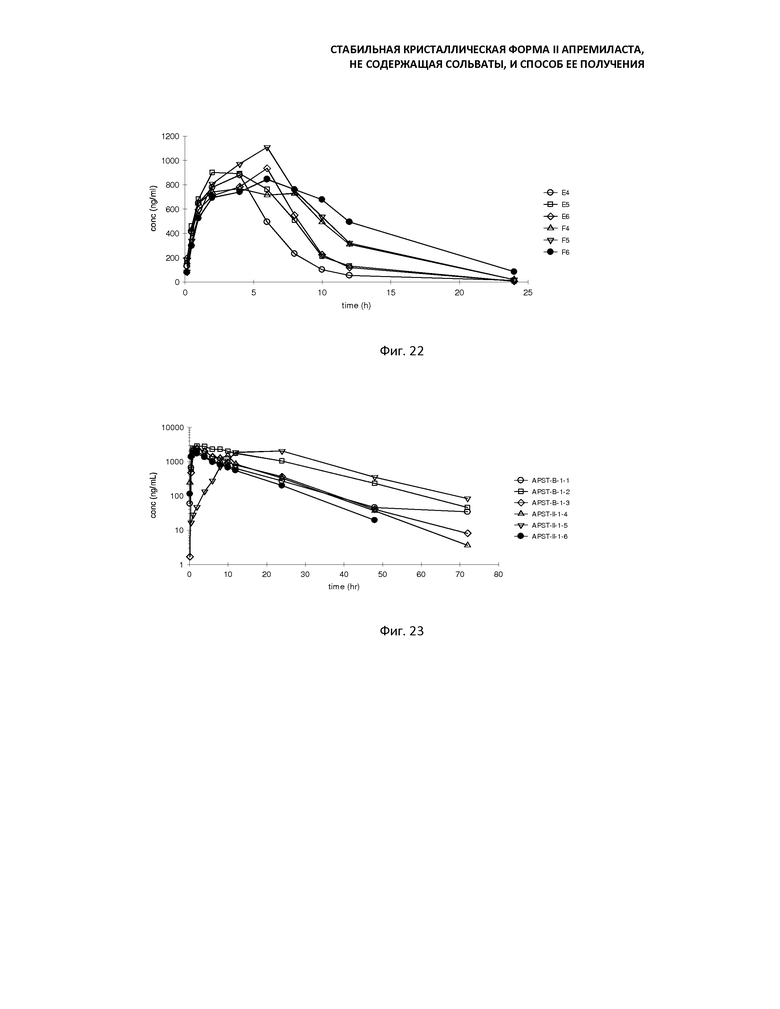
Фиг. 20: График зависимости концентрации плазмы от времени у крыс, после введения через желудочный зонд 10 мг/кг кристаллической формы II апремиласта;
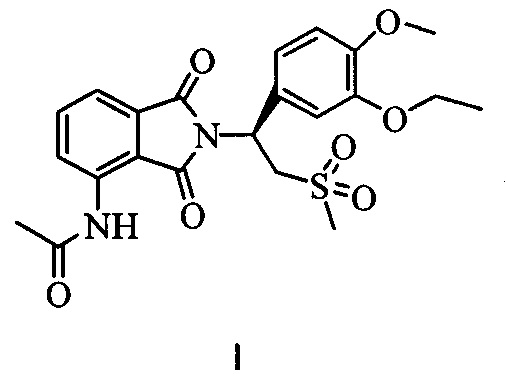
Фиг. 21: График зависимости концентрации плазмы от времени у самцов крыс, после введения через желудочный зонд 10 мг/кг кристаллической формы В и формы II апремиласта, соответственно;
Фиг. 22: График зависимости концентрации плазмы от времени у самок крыс, после введения через желудочный зонд 10 мг/кг кристаллической формы В и формы II апремиласта, соответственно;
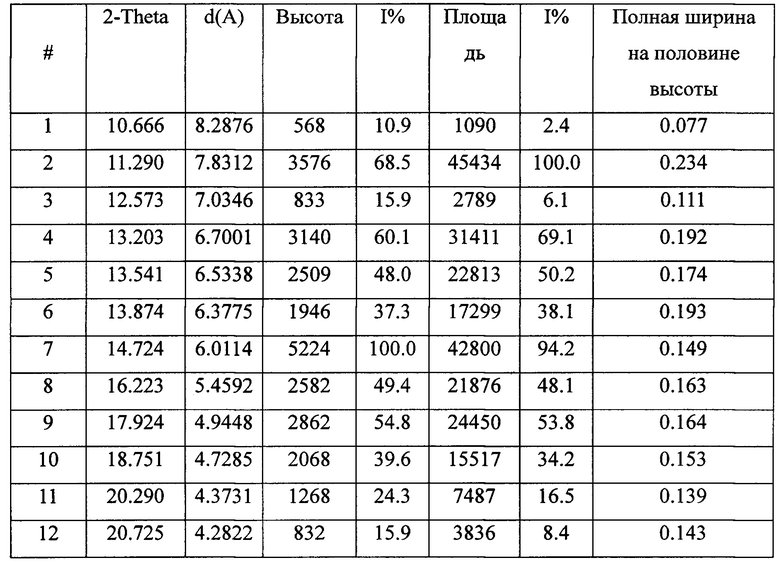
Фиг. 23: График зависимости концентрации плазмы от времени у собак породы бигль после перорального введения 30 мг за один период кристаллической формы В и формы II апремиласта в виде капсул с содержанием порошка исходного материала. (Изображение выше - постоянная координата, ниже - полулогарифмическая координата).
Устройства и методы тестирования:
1.Порошковая рентгеновская дифракция:
Модель устройства: Bruker D8 ADVANCE, порошковый рентгеновский дифрактометр
Условия эксперимента: оптический источник: CuKα 40кВ 40мА; щель расходимости: 1мм; щель Соллера: 0,4мм; режим сканирования: непрерывное сканирование; зона сканирования: 3o~45o; интервал измерения: 0,02°; скорость сканирования: 8°/мин.
2. ИК-спектроскопия
Модель устройства: NICOLET 670-FTIR
Условия эксперимента: таблетки KBr
3. ДСК:
Модель устройства: NETZSCH DSC 204 F1
Тип тигля: алюминиевый тигель (иглопробивной)
Продувочный газ: азот высокой степени чистоты, 20мл/ мин
Защитный газ: азот высокой степени чистоты, 60мл/ мин
Скорость повышения температуры: 10°C/ мин
4. Термогравиметрический анализ
Модель устройства: NETZSCH TG 209 F1
Материал тигля: оксид алюминия
Продувочный газ: азот высокой степени чистоты, 20мл/ мин
Защитный газ: азот высокой степени чистоты, 10мл/ мин
Скорость повышения температуры: 10°C/ мин
5. Температура плавления:
Тестер точки плавления RD-1 компании Tianjin Xuyang Scientific Instruments Equipment Co., Ltd.
6. Определение размера частиц:
Анализатор размера частиц Mastersizer 2000 компании Malvern Instruments Ltd.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение дополнительно описано примерами ниже. Однако примеры не должны толковаться как любые ограничения по настоящему изобретению. Любые изменения температуры или соотношения растворителей попадают в сферу защиты настоящего патента.
Пример 1: кристаллическая форма II апремиласта
Апремиласт (10,0 г) и ацетон 35 мл добавляли в трехгорлую колбу и нагревали до растворения, затем охлаждали до температуры ниже 35°C. Медленно добавляли 0,5-3,0 объема очищенной воды, небольшое количество формы II в качестве затравочного кристалла, перемешивали в течение 1 часа до осаждения продукта, затем 2 раза добавляли очищенную воду объемом 70 мл, перемешивали при 15-20°С в течение ночи (всего около 24 ч), фильтровали и промывали водой, сушили при 60°С, чтобы получить около 9,32 г кристаллической формы II апремиласта, температура плавления составляла 147,2-149,8 ° С.
Пример 2: кристаллическая форма II апремиласта
Апремиласт (400,0 г) и ацетон 1200 мл добавляли в трехгорлую колбу и нагревали до растворения. Медленно добавляли 0,5-3,0 объема очищенной воды, небольшое количество формы II в качестве затравочного кристалла, перемешивали в течение 1 часа до осаждения продукта, затем 2 раза добавляли очищенную воду объемом 2,4 л, перемешивали при 10-60°С в течение ночи (всего около 18 ч), фильтровали и промывали водой, сушили при 60°С, чтобы получить около 392,3 г кристаллической формы II апремиласта, температура плавления составляла 147,2-150,2°C.
Пример 3: кристаллическая форма B апремиласта
Апремиласт (10,0 г) и ацетон 30 мл добавляли в трехгорлую колбу и нагревали до растворения, затем охлаждали до температуры ниже 30°C. Медленно добавляли 10 мл очищенной воды, охлаждали и перемешивали до осаждения продукта, перемешивали в течение 2 часов, затем медленно добавляли воду (100 мл), нагревали и перемешивали в течение ночи (около 24 часов), фильтровали и промывали водой, сушили при 60°C, чтобы получить около 9,45 г кристаллической формы B, температура плавления: 156,2-157,8 ° C.
Пример 4:
Основное лекарственное средство пропускали через сито с размером ячеек 200 меш, наполнитель и средство для улучшения распадаемости таблеток пропускали через сито с размером ячеек 80 меш; предписанные количества наполнителя и средства для улучшения распадаемости таблеток взвешивали и смешивали, затем смесь и основное лекарственное средство смешивали по методу приращения на равное количество, затем добавляли заданные количества глидантов и лубрикантов, смешивали до однородного состояния и таблетировали.
Пример 5:
Основное лекарственное средство пропускали через сито с размером ячеек 200 меш, наполнитель и средство для улучшения распадаемости таблеток пропускали через сито с размером ячеек 80 меш; предписанные количества наполнителя и средства для улучшения распадаемости таблеток взвешивали и смешивали, затем смесь и основное лекарственное средство смешивали по методу приращения на равное количество, затем добавляли заданные количества глидантов и лубрикантов, смешивали до однородного состояния и таблетировали.
Пример 6: Исследования термодинамической стабильности формы II и формы B
Кристаллические формы II и формы B апремиласта суспендировали в воде, соответственно, перемешивали при 60-100°C в течение 48 часов, отбирали в течение определенного интервала времени и фильтровали, сушили для определения изменения температуры плавления; отбирали в течение 24 ч, 48 ч, фильтровали и сушили для определения изменения кривых порошковой рентгеновской дифракции, кривых ДСК, температуры плавления, электростатических эффектов и наличия посторонних примесей.
Пример 7: Исследование конверсии кристаллической формы В апремиласта
Кристаллическую форму В апремиласта (5,0 г, размер частиц менее 300 меш) суспендировали в 60 мл смеси ацетона и воды (1:3), нагревали до температуры кипения в колбе с обратным холодильником и перемешивали в течение 36 ч, затем охлаждали при перемешивании, фильтровали, сушили при 60°С для получения около 4,88 г кристаллической формы II апремиласта, температура плавления: 148,3-150,3 ° С.
Кривая порошковой рентгеновской дифракции и кривая ДСК продемонстрировали полное преобразование в форму II.
Пример 8: Исследование конверсии кристаллической формы D апремиласта
Кристаллическую форму D апремиласта (5,0 г, размер частиц менее 200 меш) суспендировали в 60 мл смеси ацетона и воды (1:3), нагревали до температуры кипения в колбе с обратным холодильником и перемешивали в течение 8-10 ч, затем охлаждали при перемешивании, фильтровали, сушили при 60°С для получения около 4,60 г кристаллической формы II апремиласта, температура плавления: 148,3-150,5 ° С.
Кривая порошковой рентгеновской дифракции и кривая ДСК продемонстрировали полное преобразование в форму II.
Пример 9: Фармакокинетическое сравнительное исследование кристаллической формы В и формы II апремиласта на крысах Спрега-Доули
1. Контроль размеров частиц кристаллов
Размеры частиц двух форм контролируются микронизацией (см. ниже):
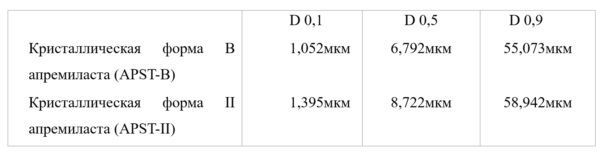
2. Метод:
Двенадцать крыс Спрега-Доули весом 200-220 г, среди которых имелись 6 самцов и 6 самок, были случайным образом разделены на две группы (называемые группой А и группой В).
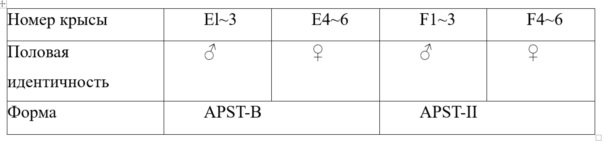
В соответствии со способом введения препарата и размером дозировки, указанными в ссылках Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (Food and Drug Administration, FDA), две кристаллические формы были введены через желудочный зонд для исследования их фармакокинетического поведения у крыс. Размеры дозировки двух форм составляли 10 мг/кг (растворитель представляет собой 1% карбоксиметилцеллюлозы). Крысам не давали пищу в течение 12 часов перед исследованием, и кормили только через 4 часа после введения препарата, ограничения приема жидкости не требовалось во время всего исследования.
Забор крови осуществлялся из венозного сплетения спустя 10 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч, 12 ч и 24 ч после введения препарата. После центрифугирования в течение 5 мин при 8000 об/мин, плазму отделяли и помещали в холодильник при температуре -20°С. Для определения концентрации апремиласта в плазме применяли метод жидкостной хроматографии/тандемной масс-спектрометрии. Диапазон линейности апремиласта в плазме составлял 2~1000 нг/мл, линейность хорошая.
3. Результаты экспериментов
График зависимости концентрации плазмы от времени у крыс после введения через желудочный зонд 10 мг/кг суспензий APST-B и APST-II, соответственно, показаны на фиг. 19-22.
4. Результаты анализов
Основные фармакокинетические параметры апремиласта, включающего в себя кристаллическую форму В и кристаллическую форму II, введенного крысам через желудочный зонд, в количестве 10 мг/кг, приведены ниже; время до достижения пика Tmax составляет 1-6 ч (в среднем 2,5 ч) и 1-6 ч (в среднем 3 ч), соответственно; максимальные концентрации Cmax равны 523,05±417,46 и 506,90±451,89 нг/мл, соответственно, площадь под кривой AUC0-t на графике зависимости концентрации плазмы от времени составляла 3766,48±3617,82 и 5533,11±5613,02 нг·ч/мл, соответственно, воздействие 10 мг/кг кристаллической формы В апремиласта на крыс составляет около 68,07% от воздействия кристаллической формы II апремиласта, разница между этими двумя группами, а также разница воздействий на самок и самцов была довольно большой, разница воздействий между этими двумя группами не была статистически значимой из-за большого стандартного отклонения. Таким образом, фармакокинетику у самок и самцов анализировали отдельно ниже.
Ниже приведены основные фармакокинетические параметры апремиласта, включающего в себя кристаллическую форму В и кристаллическую форму II, введенного самцам крыс через желудочный зонд, в количестве 10 мг/кг: время до достижения пика Tmax составляла 1 ч (в среднем 1 ч) и 1-2 ч (в среднем 1 ч); максимальные концентрации Cmax соответственно равны 142,4 ± 13,96 и 107,63 ± 16,05 нг/мл, площадь под кривой AUC0-t на графике зависимости концентрации плазмы от времени составляла соответственно 530,44 ± 70,05 и 445,59 ± 81,25 нг·ч/мл, Cmax и площадь под кривой AUC0-t 10 мг/кг кристаллической формы B апремиласта у самцов крыс составляли около 132,3% и 119,0%, соответственно, от кристаллической формы II апремиласта; результаты показали, что резорбция кристаллической формы B апремиласта у самцов крыс лучше, чем резорбция кристаллической формы II апремиласта, но разница не была статистически значимой.
Ниже приведены основные фармакокинетические параметры апремиласта, включающего в себя кристаллическую форму В и кристаллическую форму II, введенного самкам крыс через желудочный зонд, в количестве 10 мг/кг: время до достижения пика Tmax составляло 2-6 ч (в среднем 4 ч) и 4-6 ч (в среднем 6 ч); максимальные концентрации Cmax составляли соответственно 903,7 ± 28,47 и 906,17 ± 178,89 нг/мл, площадь под кривой AUC0-t на графике зависимости концентрации плазмы от времени составляла, соответственно, 7002,52 ± 1140,54 и 10620,62 ± 1053,56 нг·ч/мл, площадь под кривой AUC0-t у 10 мг/кг кристаллической формы B апремиласта у самок крыс составляла 65,9% от кристаллической формы II апремиласта; результаты показали, что резорбция кристаллической формы B апремиласта у самки ниже, чем резорбция кристаллической формы II апремиласта, и различие между кристаллической формой В апремиласта (APST-B) и кристаллической формой II апремиласта (APST-II) статистически значимо (P <0,05).
Пример 10: Исследование по сопоставлению фармакокинетического исследования кристаллической формы B и формы II апремиласта у собак породы бигль.
1. Контроль размера частиц кристаллической формы
Так же, как в примере 9.
2. Метод
Шесть здоровых собак породы бигль весом 6-8 кг, среди которых имеются три самки и три самца, были случайным образом разделены на 2 группы, в каждой по три собаки.
В соответствии со способом введения и дозировками лекарство вводится перорально, дозировка составляет 30 мг. После того, как порошок лекарственного средства, включающий две кристаллические формы, взвешивают массой примерно 30 мг соответственно, его вводят в оболочку капсулы и используют 10 мл воды для введения лекарства. Собакам породы бигль не давали пищу в течение 12 часов до начала исследования, и кормили только через 4 часа после введения препарата, ограничения приема жидкости не требовалось во время всего исследования. Конкретные примеры показано в таблице ниже.
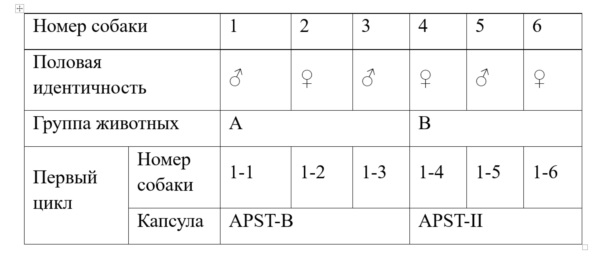
Забор венозной крови в количестве 0,5 мл осуществлялся из малой подкожной вены перед введением препарата (0ч) или спустя 10 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч, 12 ч, 24 ч, 48 ч и 72 ч после введения препарата. После центрифугирования в течение 5 мин при 8000 об/мин, плазму отделяли и помещали в незамерзающую пластиковую пробирку для хранения в холодильнике при -20°C.
Для определения концентрации апремиласта в плазме применяли метод жидкостной хроматографии/тандемной масс-спектрометрии. Диапазон линейности апремиласта в плазме составлял 5~5000 нг/мл, линейность хорошая.
1. Результаты эксперимента
График зависимости концентрации плазмы от времени у собак породы бигль после перорального введения 30 мг/кг капсулы, содержащей кристаллическую форму В и кристаллическую форму II апремиласта (APST-B и APST-II), соответственно, показаны на фиг. 23.
4. Результаты анализов
Основные фармакокинетические параметры апремиласта у собак породы бигль после перорального введения капсулы, включающей в себя кристаллическую форму В и кристаллическую форму II апремиласта, в количестве 30 мг/кг, приведены ниже:
После логарифмической конверсии фармакокинетических параметров Cmax и AUC0-t проводилась статистическая проверка гипотез (t-статистика), основанная на распределении Стьюдента. Существенного различия между кристаллической формой В апремиласта (APST-B) и кристаллической формой II апремиласта (APST-II) не обнаружено (p>0,05).
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЬ 3-(3,5-ДИХЛОР-4-ГИДРОКСИБЕНЗОИЛ)- 1,1-ДИОКСО-2,3-ДИГИДРО-1,3-БЕНЗОТИАЗОЛА | 2018 |
|
RU2772043C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО 1,3,5-ТРИАЗИНА ИЛИ ЕГО СОЛЬВАТА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2837449C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 6-(1Н-ИМИДАЗОЛ-1-ИЛ)-2-ФЕНИЛХИНАЗОЛИНА И ЕГО СОЛЕЙ | 2010 |
|
RU2557547C2 |
| ТВЕРДЫЕ ФОРМЫ ИНГИБИТОРА TTK | 2017 |
|
RU2753905C2 |
| СОЛЯНАЯ ФОРМА ИНГИБИТОРА ДИПЕПТИДИЛПЕПДИДАЗЫ IV И СПОСОБ ПОЛУЧЕНИЯ СОЛЕВОЙ ФОРМЫ | 2017 |
|
RU2734549C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПИРИДО[3,4-D]ПИРИМИДИНОВОГО ПРОИЗВОДНОГО | 2017 |
|
RU2793759C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ { [1-ЦИАНО-5-(4-ХЛОРОФЕНОКСИ)-4-ГИДРОКСИИЗОХИНОЛИН-3-КАРБОНИЛ]-АМИНО} -УКСУСНОЙ КИСЛОТЫ | 2014 |
|
RU2666144C2 |
| ФУМАРАТ ПИРИДИЛАМИНА И ЕГО КРИСТАЛЛЫ | 2015 |
|
RU2684278C1 |
| КРИСТАЛЛЫ ГИДРОБРОМАТА ПРАСУГРЕЛЯ | 2010 |
|
RU2484094C1 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ СОЛЕВАЯ ФОРМА 3-(1,2,4-ТРИАЗОЛО[4,3-А]ПИРИДИН-3-ИЛЭТИНИЛ)-4-МЕТИЛ-N-(4-((4-МЕТИЛПИПЕРАЗИН-1-ИЛ)МЕТИЛ)-3-ТРИФТОРМЕТИЛФЕНИЛ)БЕНЗАМИДА ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ | 2016 |
|
RU2652992C2 |
Изобретение относится к стабильной кристаллической форме II апремиласта, не содержащей сольваты, способу ее приготовления, фармацевтической композиции и фармацевтическому применению. При угле отражения 2θ±0,2 кривая порошковой рентгеновской дифракции кристаллической формы II апремиласта имеет сдедующие характерные пики поглощения: 11,2, 13,2, 13,5, 13,8, 14,7, 16,2, 17,9, 18,7, 20,2, 20,7, 27,0; термограмма дифференциальной сканирующей калориметрии (ДСК) показывает один пик поглощения при температуре 150±3ºC. Активность кристаллической формы II не ниже, чем у кристаллической формы В, а термодинамическая стабильность кристаллической формы II превосходит термодинамическую стабильность кристаллической формы В. 3 н. и 4 з.п. ф-лы, 23 ил., 4 табл., 10 пр.
1. Кристаллическая форма II апремиласта, представленная формулой (I)
характеризующаяся тем, что:
i) на термограмме дифференциальной сканирующей калориметрии (ДСК) показан только один эндотермический пик при температуре 150±3°С в диапазоне 100~180°С;
ii) термогравиметрический анализ (ТГ) позволяет сделать вывод об отсутствии кристаллизующего растворителя;
iii) температура плавления составляет 146-151°С;
iv) картина дифракции рентгеновских лучей на порошке (ПРД) кристаллической формы II из апремиласта имеет следующие характерные пики поглощения при 2θ±0,2: 11.2, 13.2, 13.5, 13.8, 14.7, 16.2, 17.9, 18.7, 20.2, 20.7, 27.0.
2. Кристаллическая форма II апремиласта по п. 1, отличающаяся тем, что согласно данным порошковой рентгеновской дифракции кристаллической формы II апремиласта имеются следующие характерные пики поглощения при угле отражения 2θ±0,2:
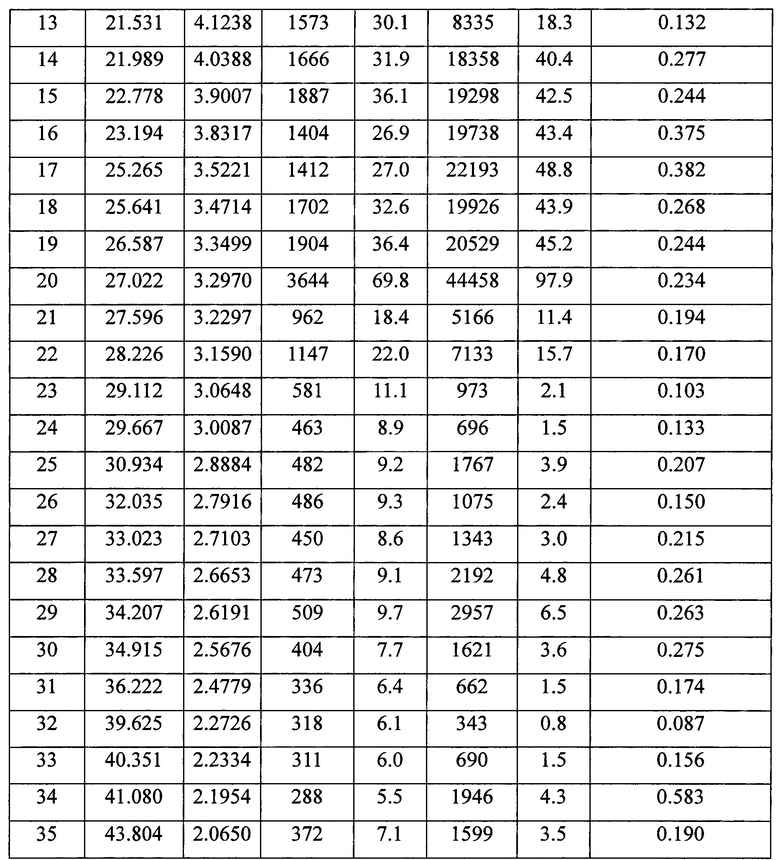
3. Кристаллическая форма II апремиласта по п. 1, отличающаяся тем, что кристаллическая форма II апремиласта соответствует данным порошковой рентгеновской дифракции, показанным на прилагаемых графиках: фиг. 2, фиг. 5, фиг. 6, фиг. 8, фиг. 12, фиг. 14, фиг. 16 или фиг. 17.
4. Способ получения кристаллической формы II апремиласта по пп. 1-3, включающий следующие стадии:
i) нагревание апремиласта или сольватов, растворение в ацетоне или ТГФ, охлаждение до температуры ниже 40°С;
ii) медленное помешивание и добавление по каплям воды, объем которой в 0,5-3 раза превышает объем ацетона или ТГФ, при необходимости, введение кристалла-затравки формы II, непрерывное перемешивание в течение 30-180 мин;
iii) добавление воды, объем которой в 2-6 раз превышает объем ацетона или ТГФ, перемешивание в течение 1-24 ч при 20°С до температуры кипения в колбе с обратным холодильником; и
iv) фильтрование и сушка с получением кристаллической формы II апремиласта.
5. Фармацевтическая композиция, обладающая активностью ингибитора фосфодиэстеразы 4 (ФДЭ-4), характеризующаяся тем, что включает кристаллическую форму II апремиласта по пп. 1-3 в качестве активного фармацевтического ингредиента и фармацевтически приемлемый носитель.
6. Фармацевтическая композиция по п. 5, отличающаяся тем, что массовое содержание кристаллической формы II апремиласта составляет 1-100%.
7. Кристаллическая форма II апремиласта по пп. 1-3, применяемая для изготовления лекарств для лечения заболеваний или расстройств, которые могут быть улучшены путем ингибирования продуцирования TNF-α, где описанные заболевания или расстройства включают в себя псориаз, псориатический артрит.
| RU 2010143907 A, 10.05.2012 | |||
| CN 104496886 A, 08.04.2015 | |||
| US 6020358 A1, 01.02.2000. |

