[ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ]
[0001]
Настоящее изобретение относится к кристаллу производного 1,3,5-триазина или его сольвата и содержащей его фармацевтической композиции. Настоящее изобретение относится к способу получения кристалла производного 1,3,5-триазина или его сольвата и содержащей его фармацевтической композиции.
[УРОВЕНЬ ТЕХНИКИ]
[0002]
Аденозинтрифосфат (АТФ) известен как внутриклеточный источник энергии и фосфорилированный субстрат. С другой стороны, также известно, что он действует как передатчик внеклеточной информации. Кроме того, известно, что АТФ высвобождается наружу клеток под действием различных раздражителей, таких как повреждение клеток, воспаление, болевые раздражители и снижение уровня кислорода в крови, и что АТФ высвобождается наружу клеток из первичных чувствительных нервных окончаний одновременно с другими нейромедиаторами. АТФ, высвобождаемый наружу клеток, осуществляет различные виды внеклеточной передачи информации через рецептор АТФ (Непатентный документ 4, Непатентный документ 5).
[0003]
Рецепторы АТФ грубо классифицируются как семейство P2X ионных каналов и семейство P2Y, связанное с G-белком. Сообщается, что семейство рецепторов Р2Х имеет семь подтипов, которые образуют гомотример или гетеротример с другими подтипами Р2Х для функционирования в качестве неселективного катионного канала (Непатентный документ 6).
[0004]
Уже известно, что АТФ вызывает боль, и исследования с использованием методов нокаута и нокдауна P2X3 показали, что рецептор P2X3 участвует в передаче хронической боли. Рецептор P2X3 специфически экспрессируется в периферических сенсорных нервах, образуя гомокомплекс или гетерокомплекс с P2X2 (P2X2/3). (Непатентный документ 1).
[0005]
Впоследствии предполагается, что соединение, обладающее антагонистическим действием в отношении рецепторов Р2Х3 или Р2Х2/3, является полезным для: лечения боли (Патентный документ 1, Непатентный документ 3 и Непатентный документ 7); лечения заболеваний, связанных с нарушением мочеиспускания (Непатентный документ 2); лечения респираторных заболеваний (Непатентный документ 8, Непатентный документ 9, Непатентный документ 10, Патентный документ 2 и Патентный документ 3); лечения хронического кашля (Патентный документ 4, Патентный документ 5 и Непатентный документ 11); лечения гипертензии (Непатентный документ 12); лечения боли, связанной с панкреатитом (Непатентный документ 13); и лечения боли, связанной с эндометриозом (Непатентный документ 14 и Непатентный документ 15).
[0006]
Далее в Патентном документе 6 описано, что производное 1,3,5-триазина, представленное следующей формулой, обладает антагонистическим действием в отношении Р2Х3 и/или Р2Х2/3 и применимо для лечения и/или предотвращения боли:
[Химическая формула 1]
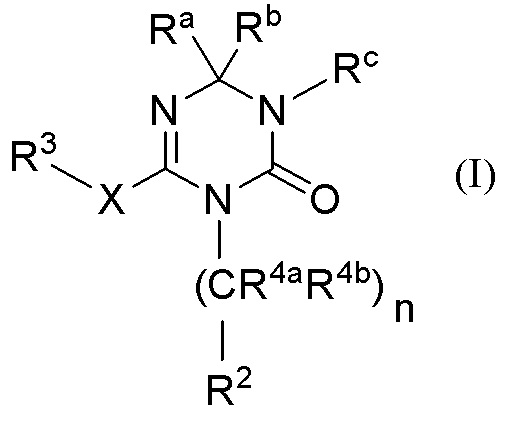
[0007]
Патентный документ 7 описывает, что производное 1,3,5-триазина, представленное следующей формулой, обладает антагонистическим действием в отношении Р2Х3 и/или Р2Х2/3 и применимо для лечения и/или предотвращения боли:
[Химическая формула 2]
[0008]
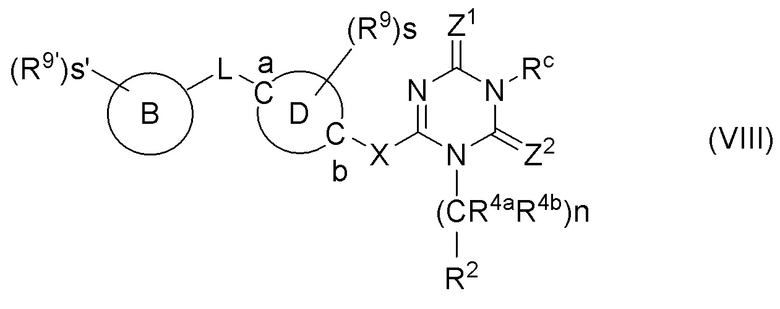
Патентный документ 8 описывает, что производное 1,3,5-триазина, представленное следующей формулой, обладает антагонистическим действием в отношении Р2Х3 и/или Р2Х2/3 и применимо для лечения и/или предотвращения боли:
[Химическая формула 3]
Патентный документ 9 описывает, что производное 1,3,5-триазина, представленное следующей формулой, обладает антагонистическим действием в отношении Р2Х3 и/или Р2Х2/3 и применимо для лечения и/или предотвращения боли:
[Химическая формула 4]
В Примерах Патентного документа 9 раскрыто следующее соединение (I-127), но кристалл соединения не раскрыт.
[Химическая формула 5]
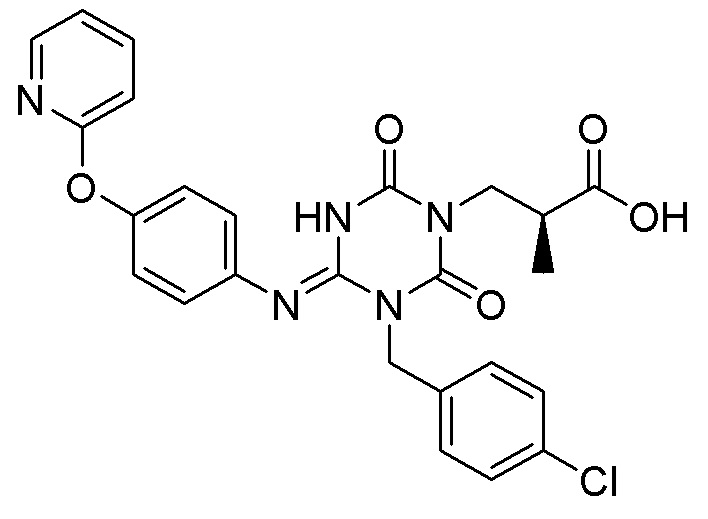
Кроме того, Патентные документы 6, 7, 8 и 9 раскрывают способ получения производного 1,3,5-триазина, но не описывают способ в соответствии с настоящим изобретением и раскрывают только способ получения подобного соединения. Кроме того, Патентный документ 10 раскрывает производное 1,3,5-триазина, проявляющее терапевтический эффект при хроническом кашле, но не описывает кристалл и способ в соответствии с настоящим изобретением.
Кроме того, Непатентный документ 16 раскрывает реакцию аза-присоединения по Михаэлю (S)-1-фенилэтиламина и метилметакрилата.
[ССЫЛКИ НА ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ]
[Патентный документ]
[0009]
[Патентный документ 1] Международная публикация WO 02/094767 A
[Патентный документ 2] Международная публикация WO 2006/012639 A
[Патентный документ 3] Международная публикация WO 2010/149578 A
[Патентный документ 4] Международная публикация WO 2015/027212 A
[Патентный документ 5] Международная публикация WO 2017/058645 A
[Патентный документ 6] Международная публикация WO 2010/092966A
[Патентный документ 7] Международная публикация WO 2012/020749 A
[Патентный документ 8] Международная публикация WO 2013/089212 A
[Патентный документ 9] Международная публикация WO 2014/200078A
[Патентный документ 10] Международная публикация WO 2020/071530 A
[Непатентный документ]
[0010]
[Непатентный документ 1] Neuroscientist 2005, Vol. 11, pp. 345-356
[Непатентный документ 2] J. Physiol. 567.2 2005 pp. 621-639
[Непатентный документ 3] Expert Opin.Ther.Patens 2006 Vol. 16, No. 8, pp. 1113-1127
[Непатентный документ 4] J. Physiol. 554.2 2003 pp. 301-308
[Непатентный документ 5] J. Physiol. 553.3 2003 pp. 683-694
[Непатентный документ 6] Pflungers Arch Eur J physiol 2006, pp. 452, 513-537
[Непатентный документ 7] PNAS 2002, Vol. 99, No. 26, pp. 17179-17184
[Непатентный документ 8] Brouns et al. Am J Respir Cell MoI Biol 2000, Vol. 23, pp. 52-61
[Непатентный документ 9] Basoglu et al. Chest. 2005, Vol. 128, No. 4, pp. 1905-9
[Непатентный документ 10] Adriaensen et al. THE ANATOMICAL RECORD PART A 2003, Vol. 270A, pp. 25-40
[Непатентный документ 11] Lancet, 2015, Vol. 385, pp. 1198-205
[Непатентный документ 12] Nat Med 2016, Vol. 22, pp. 1151-1159
[Непатентный документ 13] Am J Physiol Gastrointest Liver Physiol 2015, Vol. 308, pp 710-719
[Непатентный документ 14] PLoS ONE 2017, Vol. 12, No. 9
[Непатентный документ 15] International Journal of Nanomedicine 2017, Volume 12, 8171-8183
[Непатентный документ 16] Tetrahedron Asymmetry, Vol. 7, No. 3, pp. 699-708, 1996
[СУЩНОСТЬ ИЗОБРЕТЕНИЯ]
[ПРОБЛЕМЫ, РЕШАЕМЫЕ ИЗОБРЕТЕНИЕМ]
[0011]
Фармацевтически активные ингредиенты могут иметь по сушеству различные физические свойства в зависимости от соответствующей твердой формы. Такие различия в физических свойствах могут влиять, например, на способ получения или введения фармацевтически активного ингредиента или на фармацевтическую композицию, содержащую фармацевтически активный ингредиент. Настоящее изобретение относится к кристаллу производного 1,3,5-триазина или его сольвата, который наиболее пригоден по сравнению с другими твердыми формами в способе получения или введения фармацевтически активного ингредиента, или в фармацевтической композиции, содержащей фармацевтически активный ингредиент.
Обычно физические свойства кристалла соединения, пригодного в качестве фармацевтического препарата, оказывают большое влияние на биодоступность лекарственных средств, чистоту лекарственных веществ, состав препаратов и подобное и, таким образом, чрезвычайно важны при разработке фармацевтических продуктов. Следовательно, что касается соединения, представленного Формулой (I), необходимо изучить, какая кристаллическая форма является наиболее превосходной в качестве фармацевтического продукта. То есть, поскольку их физические свойства зависят от свойств отдельных соединений, обычно трудно предсказать кристаллическую форму лекарственного вещества, обладающую хорошими физическими свойствами, и требуется фактически по-разному исследовать каждое соединение.
Следовательно, целью настоящего изобретения является предоставление кристаллической формы, имеющей хорошие физические свойства, в качестве лекарственного вещества для соединения, представленного Формулой (I).
Кроме того, Патентный документ 9 не описывает способ получения соединения I-127, но в качестве подобного соединения Ссылочный пример 3 Патентного документа 9 раскрывает способ получения производного 1,3,5-триазина, как показано в следующей формуле. Однако процесс еще не является достаточным, и его можно дополнительно улучшить.
[Химическая формула 6]
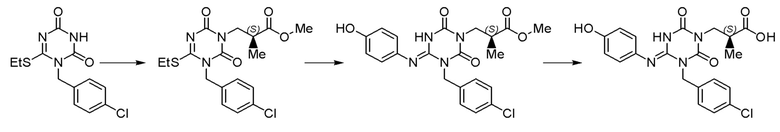
[СРЕДСТВА ДЛЯ РЕШЕНИЯ ПРОБЛЕМЫ]
[0012]
В результате интенсивных исследований авторы настоящего изобретения обнаружили, что в качестве кристаллических форм соединения, представленного Формулой (I), существуют кристаллические формы безводной Формы I, безводной Формы II и дигидрата. Кроме того, они обнаружили, что безводная кристаллическая Форма I и кристаллический дигидрат более стабильны, чем другие кристаллические формы. Кроме того, они обнаружили, что безводная кристаллическая Форма I имеет низкий коэффициент прессуемости (%) кристалла и обладает благоприятной текучестью кристалла по сравнению с другими кристаллическими формами.
Кроме того, авторы настоящего изобретения обнаружили интермедиат, имеющий высокую химическую чистоту и/или оптическую чистоту, способ получения интермедиата и способ получения оптически активного производного 1,3,5-триазина, обладающего антагонистическим действием в отношении P2X3 и/или P2X2/3.
Настоящее изобретение относится к следующим пунктам (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), от (4') до (35'), (3''), (5'') и от (36'') до (42'').
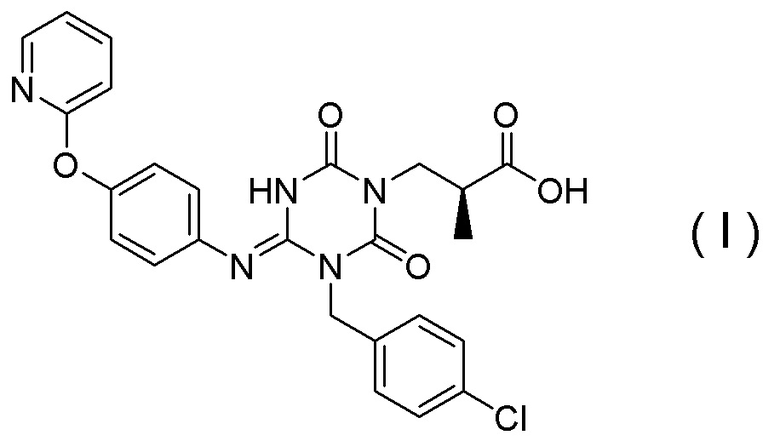
(1') Кристалл соединения, представленного Формулой (I):
[Химическая формула 7]
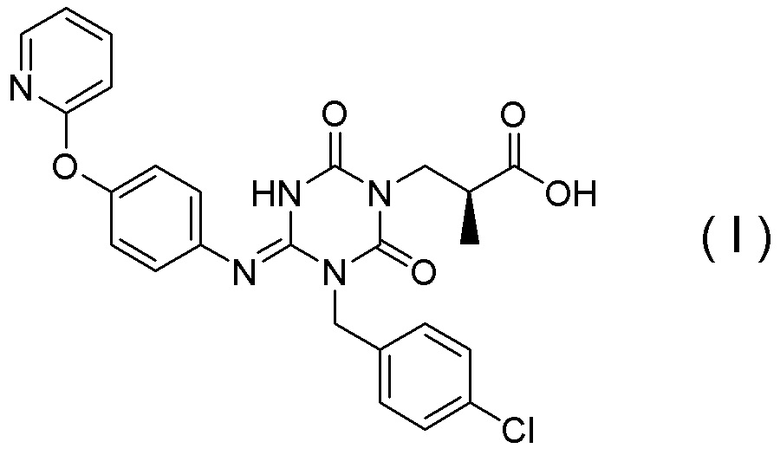
или его сольват.
(2') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая в спектре порошковой рентгеновской дифракции характеристические пики при:
углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2°.
(2'A) Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая характеристические пики при углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2° в спектре порошковой рентгеновской дифракции.
(2'B) Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая характеристические пики при углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2° в спектре порошковой рентгеновской дифракции.
(3') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая в спектре порошковой рентгеновской дифракции характеристические пики при:
углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2°.
(3'A) Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая характеристические пики при углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° в спектре порошковой рентгеновской дифракции.
(3'B) Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая характеристические пики при углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2° в спектре порошковой рентгеновской дифракции.
(3'') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), имеющая пики поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
[0013]
(4') Кристалл дигидрата соединения по вышеуказанному пункту (1'), имеющий характеристические пики при углах дифракции (2θ) 5,7°±0,2°, 7,7°±0,2°, 11,8°±0,2°, 15,2°±0,2° и 17,7°±0,2° в спектре порошковой рентгеновской дифракции.
(5') Кристалл дигидрата соединения по вышеуказанному пункту (1'), имеющий характеристические пики при углах дифракции (2θ) 5,7°±0,2°, 7,7°±0,2°, 11,8°±0,2°, 15,2°±0,2°, 17,7°±0,2°, 20,6°±0,2°, 20,8°±0,2°, 26,5°±0,2°, 27,1°±0,2° и 29,1°±0,2° в спектре порошковой рентгеновской дифракции.
(5'') Кристалл дигидрата соединения по вышеуказанному пункту (1'), имеющий пики поглощения при 871 см-1±2 см-1, 996 см-1±2 см-1, 1114 см-1±2 см-1, 1234 см-1±2см-1, 1340 см-1±2 см-1 и 1577 см-1±2 см-1 в Раман-спектре.
[0014]
(6') Фармацевтическая композиция, содержащая кристалл по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'B), (4'), (5'), (3'') и (5'').
(7') Способ получения кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'B), (4'), (5'), (3'') и (5'').
(8') Фармацевтическая композиция по вышеуказанному пункту (6'), причем фармацевтическая композиция представляет собой антагонист Р2Х3 и/или Р2Х2/3.
(9') Фармацевтическая композиция по вышеуказанному пункту (6'), причем фармацевтическая композиция используется для лечения и/или предотвращения хронического кашля.
(10') Фармацевтическая композиция по вышеуказанному пункту (6'), при этом фармацевтическая композиция используется для лечения и/или предотвращения рефрактерного хронического кашля.
(11') Антагонист Р2Х3 и/или Р2Х2/3, характеризующийся содержанием кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'').
(12') Терапевтическое и/или профилактическое средство при хроническом кашле, характеризующееся содержанием кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'').
(13') Терапевтическое и/или профилактическое средство при рефрактерном хроническом кашле, характеризующееся содержанием кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'').
(14') Способ лечения и/или предотвращения хронического кашля, при этом способ характеризуется введением фармацевтической композиции, содержащей кристалл по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'B), (3'), (3'A), (3'B), (4'), (5'), (3'') и (5'').
(15') Способ лечения и/или предотвращения рефрактерного хронического кашля, при этом способ характеризуется введением фармацевтической композиции, содержащей кристалл по любому из вышеуказанных пунктов (1'), (2'), (2'А), (2'B), (3'), (3'A), (3'B), (4'), (5'), (3'') и (5'').
(16') Применение кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'') для получения лекарственного препарата для лечения и/или предотвращения хронического кашля.
(17') Применение кристалла по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'B), (4'), (5'), (3'') и (5'') для получения лекарственного препарата для лечения и/или предотвращения рефрактерного хронического кашля.
(18') Кристалл по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'') для лечения и/или предотвращения хронического кашля.
(19') Кристалл по любому одному из вышеуказанных пунктов (1'), (2'), (2'А), (2'В), (3'), (3'А), (3'В), (4'), (5'), (3'') и (5'') для лечения и/или предотвращения рефрактерного хронического кашля.
(20') Кристалл по вышеуказанному пункту (1'), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(21') Кристалл по вышеуказанному пункту (2'), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(22') Кристалл по вышеуказанному пункту (2'А), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(23') Кристалл по вышеуказанному пункту (2'B), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(24') Кристалл по вышеуказанному пункту (3'), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(25') Кристалл по вышеуказанному пункту (3'А), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(26') Кристалл по вышеуказанному пункту (3'B), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(27') Кристалл по вышеуказанному пункту (4'), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 4.
(28') Кристалл по вышеуказанному пункту (5'), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 4.
(29') Кристалл по вышеуказанному пункту (1'), характеризующийся Раман-спектром, по существу идентичным спектру, показанному на Фиг. 2.
(30') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие в спектре порошковой рентгеновской дифракции характеристических пиков при:
углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2° и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(31') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(32') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(33') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие в спектре порошковой рентгеновской дифракции характеристических пиков при: углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2° и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(34') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(35') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1 в Раман-спектре.
(36'') Безводная кристаллическая Форма I соединения по вышеуказанному пункту (1'), характеризующаяся одним или более спектрами и/или кривыми, выбранными из группы, состоящей из следующих (а) и (b):
(а) спектр порошковой рентгеновской дифракции, по существу идентичный спектру, показанному на Фиг. 1; и
(b) Раман-спектр, по существу идентичный спектру, показанному на Фиг. 2.
[0015]
(37'') Кристалл по вышеуказанному пункту (1'), характеризующийся Раман-спектром, по существу идентичным спектру, показанному на Фиг. 6.
(38'') Кристалл дигидрата соединения по вышеуказанному пункту (1'), характеризующийся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 5,7°±0,2°, 7,7°±0,2°, 11,8°±0,2°, 15,2°±0,2° и 17,7°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 871 см-1±2 см-1, 996 см-1±2 см-1, 1114 см-1±2 см-1, 1234 см-1±2 см-1, 1340 см-1±2 см-1 и 1577 см-1±2см-1 в Раман-спектре.
(39'') Кристалл дигидрата соединения по вышеуказанному пункту (1'), характеризующийся одним или более физико-химическими свойствами, выбранными из группы, состоящей из следующих (i) и (ii):
(i) наличие характеристических пиков при углах дифракции (2θ) 5,7°±0,2°, 7,7°±0,2°, 11,8°±0,2°, 15,2°±0,2°, 17,7°±0,2°, 20,6°±0,2°, 20,8°±0,2°, 26,5°±0,2°, 27,1°±0,2° и 29,1°±0,2° в спектре порошковой рентгеновской дифракции и
(ii) наличие пиков поглощения при 871 см-1±2 см-1, 996 см-1±2 см-1, 1114 см-1±2 см-1, 1234 см-1±2 см-1, 1340 см-1±2 см-1 и 1577 см-1±2см-1 в Раман-спектре.
(40'') Кристалл дигидрата соединения по вышеуказанному пункту (1'), характеризующийся одним или более спектрами и/или кривыми, выбранными из группы, состоящей из следующих (а) и (b):
(а) спектр порошковой рентгеновской дифракции, по существу идентичный спектру, показанному на Фиг. 4; и
(b) Раман-спектр, по существу идентичный спектру, показанному на Фиг. 6.
(41'') Безводная кристаллическая Форма I соединения вышеуказанного пункта (1'), которая при измерении при 298,15 К по существу соответствует следующим кристаллографическим данным:
Пространственная группа: P1
а=9,8720 (5) Å
b=10,9952 (5) Å
с=12,2781 (6) Å
α=67,712 (4)°
β=80,870 (4)°
γ=80,870 (4)°
(42'') Безводная кристаллическая Форма I соединения вышеуказанного пункта (1'), которая при измерении при 298,15 К характеризуется следующими кристаллографическими данными:
Пространственная группа: P1
а=9,9 ű0,5 Å
b=11,0 ű0,5 Å
с=12,3 ű0,5 Å
α=67,7°±0,5°
β=80,9°±0,5°
γε=86,9°±0,5°
[0016]
Настоящее изобретение также относится к следующим пунктам (1)-(34).
(1) Кристалл соединения, представленного Формулой (I):
[Химическая формула 8]
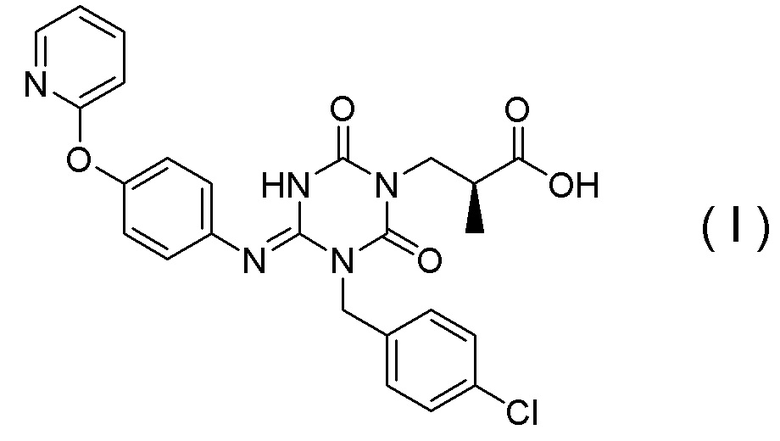
или его сольват.
(2) Кристалл соединения по вышеуказанному пункту (1), имеющий характеристические пики при углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° или 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2° в спектре порошковой рентгеновской дифракции.
(3) Кристалл соединения по вышеуказанному пункту (1), имеющий в спектре порошковой рентгеновской дифракции характеристические пики при:
углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2°.
(4) Фармацевтическая композиция, содержащая кристалл по любому одному из вышеуказанных пунктов (1)-(3).
(5) Способ получения кристалла по любому одному из вышеуказанных пунктов (1)-(3).
(6) Способ получения соединения, представленного Формулой (IV):
[Химическая формула 9]
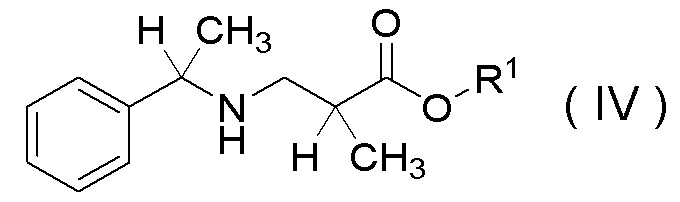
в которой R1 представляет собой C1-C4 алкил,
или его соли,
характеризующийся проведением реакции между соединением, представленным Формулой (II):
[Химическая формула 10]
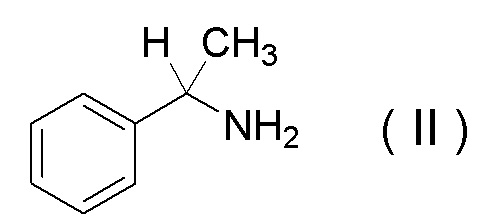
или его солью и соединением, представленным Формулой (III):
[Химическая формула 11]
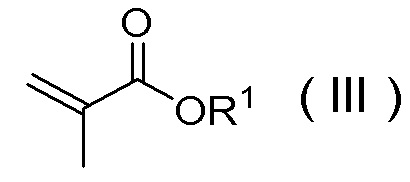
в которой R1 представляет собой C1-C4 алкил,
или его солью
в присутствии одной или более добавок, выбранных из группы, состоящей из хлорида лития, хлорида кальция, хлорида магния, бромида лития, п-толуолсульфокислоты, метансульфокислоты и трифторметансульфокислоты.
(7) Способ по вышеуказанному пункту (6), в котором добавка представляет собой хлорид лития.
(8) Способ получения соли п-толуолсульфокислоты соединения, представленного Формулой (IV-A):
[Химическая формула 12]
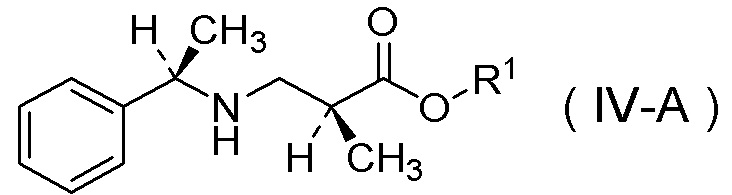
в которой R1 представляет собой C1-C4 алкил,
характеризующийся:
получением соединения, представленного Формулой (IV), или его соли способом по вышеуказанному пункту (6) или (7) и
добавлением к нему п-толуолсульфокислоты.
(9) Способ получения соли п-толуолсульфокислоты соединения, представленного Формулой (IV-A):
[Химическая формула 13]
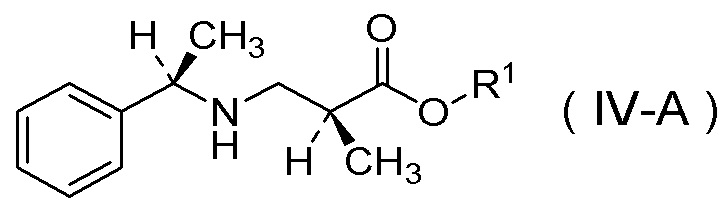
в которой R1 представляет собой C1-C4 алкил,
характеризующийся тем, что добавка представляет собой п-толуолсульфокислоту в способе по пункту (6).
(10) Способ по любому одному из вышеуказанных пунктов (6)-(9), в котором R1 представляет собой метил.
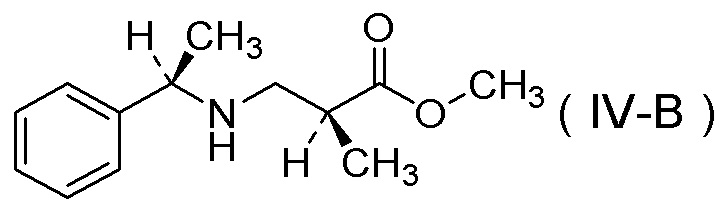
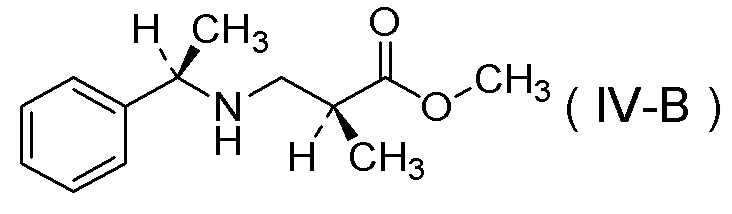
(11) Соль п-толуолсульфокислоты соединения, представленного Формулой (IV-B):
[Химическая формула 14]
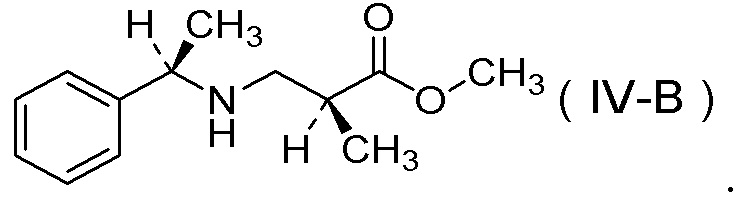
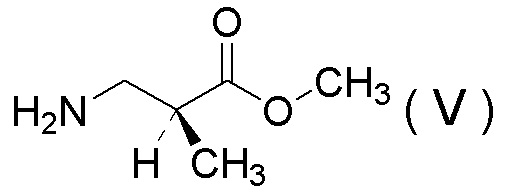
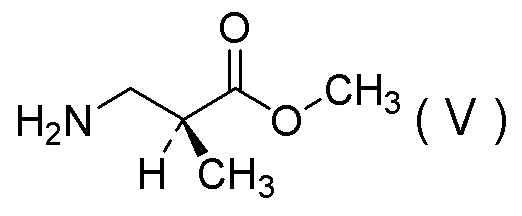
(12) Способ получения соли 1/2 серной кислоты соединения, представленного Формулой (V):
[Химическая формула 15]
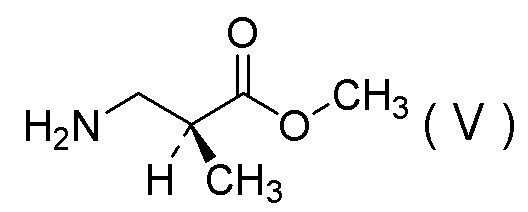
характеризующийся:
подверганием соли п-толуолсульфокислоты соединения, представленного Формулой (IV-B):
[Химическая формула 16]
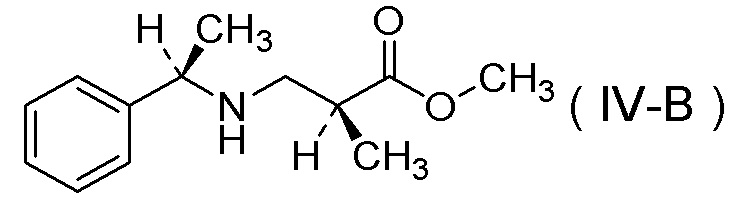
реакции гидрогенолиза и
добавлением к ней серной кислоты.
(13) Соль 1/2 серной кислоты соединения, представленного Формулой (V):
[Химическая формула 17]
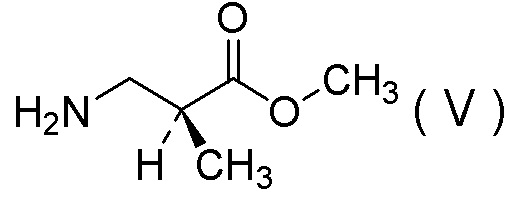 .
.
(14) Способ получения соединения, представленного Формулой (I):
[Химическая формула 18]
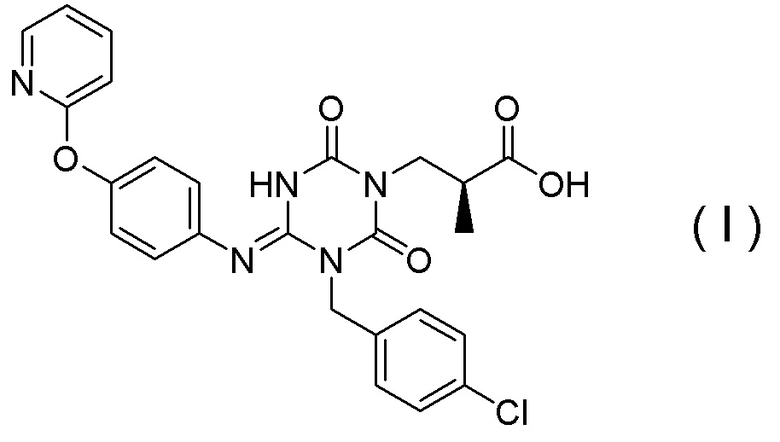
или его соли,
характеризующийся:
подверганием соединения, представленного Формулой (VI):
[Химическая формула 19]
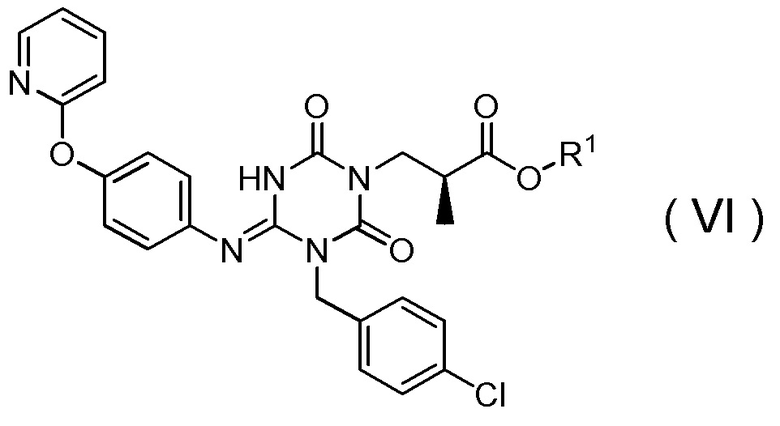
в которой R1 представляет собой C1-C4 алкил,
или его соли
реакции гидролиза в присутствии одного или более растворителей, выбранных из группы, состоящей из изопропилового спирта, тетрагидрофурана и трет-бутанола.
(15) Способ по вышеуказанному пункту (14), в котором R1 представляет собой метил.
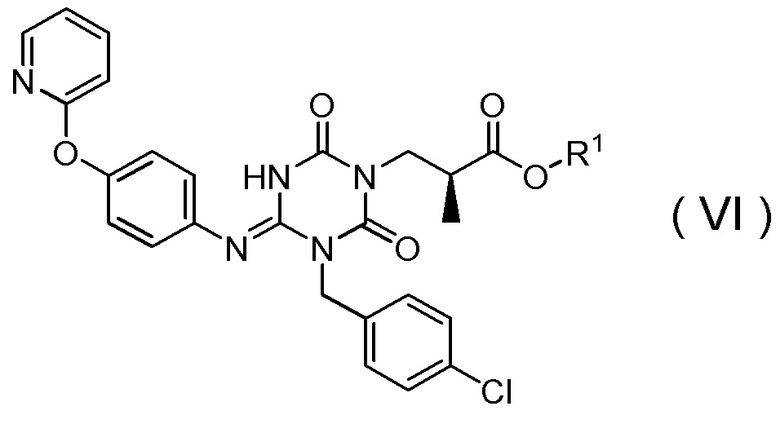
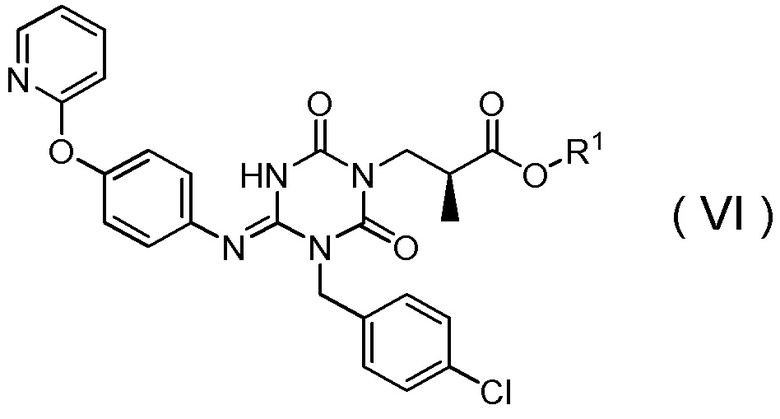
(16) Способ получения соединения, представленного Формулой (VI):
[Химическая формула 20]
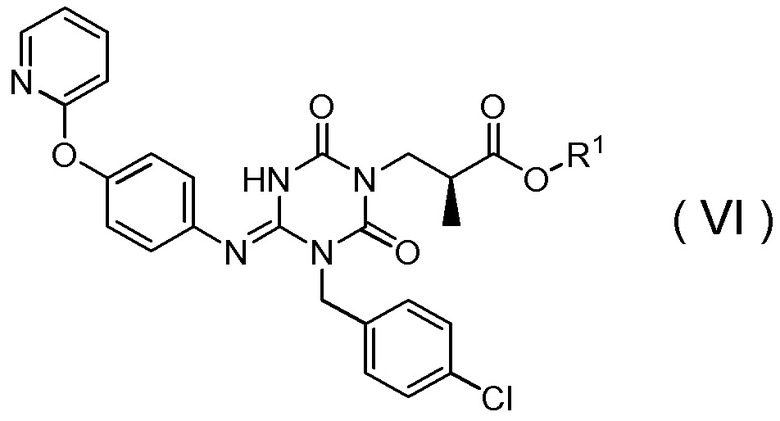
в которой R1 представляет собой метил,
или его соли,
в котором способ включает стадию:
получения соли п-толуолсульфокислоты соединения, представленного Формулой (IV-B):
[Химическая формула 21]
способом по любому одному из вышеуказанных пунктов (6)-(10).
(17) Способ получения соединения, представленного Формулой (VI):
[Химическая формула 22]
в которой R1 представляет собой метил,
или его соли,
в котором способ включает стадию:
получения соли 1/2 серной кислоты соединения, представленного Формулой (V):
[Химическая формула 23]
способом по пункту (12).
(18) Способ получения соединения, представленного Формулой (VI):
[Химическая формула 24]
в которой R1 представляет собой метил,
или его соли,
в котором способ включает стадии:
получения соли п-толуолсульфокислоты соединения, представленного Формулой (IV-B):
[Химическая формула 25]
способом по любому одному из вышеуказанных пунктов (6)-(10) и
получения соли 1/2 серной кислоты соединения, представленного Формулой (V):
[Химическая формула 26]
способом по пункту (12).
(19) Способ получения соединения, представленного Формулой (I):
[Химическая формула 27]
или его соли,
характеризующийся:
получением соединения, представленного Формулой (VI):
[Химическая формула 28]
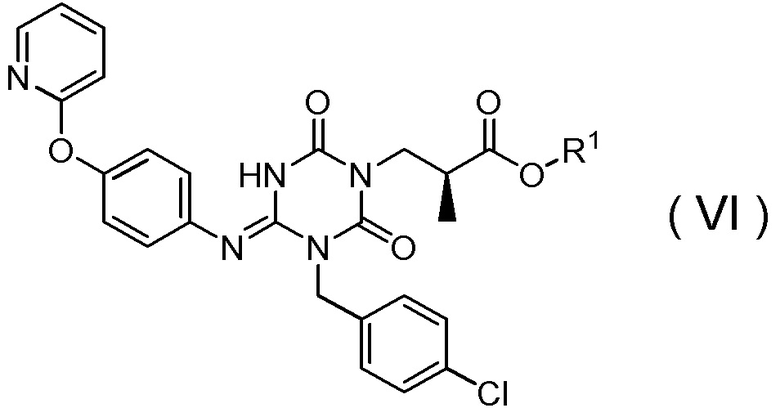
в которой R1 представляет собой метил,
или его соли способом по любому одному из вышеуказанных пунктов (16)-(18) и
подверганием соединения, представленного Формулой (VI), или его соли, полученной таким образом, реакции гидролиза в присутствии одного или более растворителей, выбранных из группы, состоящей из изопропилового спирта, тетрагидрофурана и трет-бутанола.
(20) Фармацевтическая композиция по вышеуказанному пункту (4), при этом фармацевтическая композиция представляет собой антагонист Р2Х3 и/или Р2Х2/3.
(21) Фармацевтическая композиция по вышеуказанному пункту (4), при этом фармацевтическая композиция используется для лечения и/или предотвращения хронического кашля.
(22) Фармацевтическая композиция по вышеуказанному пункту (4), при этом фармацевтическая композиция используется для лечения и/или предотвращения рефрактерного хронического кашля.
(23) Антагонист Р2Х3 и/или Р2Х2/3, характеризующийся содержанием кристалла по любому одному из вышеуказанных пунктов (1)-(3).
(24) Терапевтическое и/или профилактическое средство при хроническом кашле, характеризующееся содержанием кристалла по любому одному из вышеуказанных пунктов (1)-(3).
(25) Терапевтическое и/или профилактическое средство при рефрактерном хроническом кашле, характеризующееся содержанием кристалла по любому одному из вышеуказанных пунктов (1)-(3).
(26) Способ лечения и/или предотвращения хронического кашля, при этом способ характеризуется введением фармацевтической композиции, содержащей кристалл по любому одному из вышеуказанных пунктов (1)-(3).
(27) Способ лечения и/или предотвращения рефрактерного хронического кашля, при этом способ характеризуется введением фармацевтической композиции, содержащей кристалл по любому одному из вышеуказанных пунктов (1)-(3).
(28) Применение кристалла по любому одному из вышеуказанных пунктов (1)-(3) для получения лекарственного препарата для лечения и/или предотвращения хронического кашля.
(29) Применение кристалла по любому одному из вышеуказанных пунктов (1)-(3) для получения лекарственного препарата для лечения и/или предотвращения рефрактерного хронического кашля.
(30) Кристалл по любому одному из вышеуказанных пунктов (1)-(3) для лечения и/или предотвращения хронического кашля.
(31) Кристалл по любому одному из вышеуказанных пунктов (1)-(3) для лечения и/или предотвращения рефрактерного хронического кашля.
(32) Кристалл по вышеуказанному пункту (1), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(33) Кристалл по вышеуказанному пункту (2), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(34) Кристалл по вышеуказанному пункту (3), характеризующийся спектром порошковой рентгеновской дифракции, по существу идентичным спектру, показанному на Фиг. 1.
(35) Кристалл по вышеуказанному пункту (1), характеризующийся Раман-спектром, по существу идентичным спектру, показанному на Фиг. 2.
(36) Кристалл по вышеуказанному пункту (2), характеризующийся Раман-спектром, по существу идентичным спектру, показанному на Фиг. 2.
(37) Кристалл по вышеуказанному пункту (3), характеризующийся Раман-спектром, по существу идентичным спектру, показанному на Фиг. 2.
[ЭФФЕКТ ИЗОБРЕТЕНИЯ]
[0017]
Кристалл настоящего изобретения является пригодным в качестве активного фармацевтического ингредиента соединения, представленного Формулой (I). То есть фармацевтическая композиция, содержащая кристалл настоящего изобретения, является наиболее пригодной в качестве терапевтического средства или профилактического средства при хроническом кашле или рефрактерном хроническом кашле.
Среди кристаллов настоящего изобретения безводная кристаллическая Форма I и кристалл дигидрата являются пригодными в качестве активных фармацевтических ингредиентов.
Кроме того, безводная кристаллическая Форма I имеет следующие характеристики:
(i) наличие низкого коэффициента прессуемости (%) кристалла и наличие благоприятной текучести кристалла;
(ii) наличие кристаллической формы, не содержащей ни одного из остаточных растворителей, перечисленных в руководстве ICH Q3C; и
(iii) наличие высокой стабильности твердого вещества и образование нескольких видов аналогов веществ при хранении активного фармацевтического ингредиента.
Кроме того, кристалл дигидрата имеет характеристики из вышеуказанного (ii) и (iii).
Кроме того, способ процесса настоящего изобретения может предоставлять соединения, пригодные в качестве интермедиатов процесса, представленные Формулой (IV) и Формулой (V), и соединение, представленное Формулой (I), и его кристалл.
Настоящий способ процесса является промышленно превосходным способом процесса, и характеристики способа процесса настоящего изобретения могут включать следующие пункты:
(а) на стадии получения соединения, представленного Формулой (IV), реакцию аза-присоединения по Михаэлю можно ускорить добавлением LiCl или подобного;
(b) на стадии получения соединения, представленного Формулой (V), путем получения продукта в виде 1/2 сульфата продукт может быть получен с высоким выходом и
(c) на стадии получения соединения, представленного Формулой (I), рацемизацию можно подавить, используя изопропиловый спирт или подобное в качестве растворителя реакции.
[КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ]
[0018]
[Фиг. 1] На Фиг. 1 показана порошковая рентгеновская дифрактограмма безводной кристаллической Формы I соединения, представленного Формулой (I). Горизонтальная ось представляет собой 2θ (°), и вертикальная ось представляет собой интенсивность (Число отсчетов).
[Фиг. 2] На Фиг. 2 показан Раман-спектр безводной кристаллической Формы I соединения, представленного Формулой (I). Горизонтальная ось представляет собой рамановский сдвиг (см-1), и вертикальная ось представляет собой пиковую интенсивность.
[Фиг. 3] На Фиг. 3 показаны результаты анализа ДСК безводной кристаллической Формы I соединения, представленного Формулой (I).
[Фиг. 4] На Фиг. 4 показана порошковая рентгеновская дифрактограмма кристалла дигидрата соединения, представленного Формулой (I). Горизонтальная ось представляет собой 2θ (°), и вертикальная ось представляет собой интенсивность (Число отсчетов).
[Фиг. 5] На Фиг. 5 показаны результаты анализа ТГ/ДТА кристалла дигидрата соединения, представленного Формулой (I). Вертикальная ось представляет собой калории (мкВ) или изменение массы (%), и горизонтальная ось представляет собой температуру (°C). «Цел» на фигуре означает градус Цельсия (°C).
[Фиг. 6] На Фиг. 6 показан Раман-спектр кристалла дигидрата соединения, представленного Формулой (I). Горизонтальная ось представляет собой рамановский сдвиг (см-1), и вертикальная ось представляет собой пиковую интенсивность.
[Фиг. 7] На Фиг. 7 показана молекулярная структурная диаграмма безводной кристаллической Формы I соединения, представленного Формулой (I) (показана молекула, содержащая N3).
[Фиг. 8] На Фиг. 8 показана молекулярная структурная диаграмма безводной кристаллической Формы I соединения, представленного Формулой (I) (показана молекула, содержащая N8).
[Фиг. 9] На Фиг. 9 показан ЯМР кристалла сольвата этилацетата/гексана соединения, представленного Формулой (I). Горизонтальная ось представляет собой значение химического сдвига (δ), и вертикальная ось представляет собой относительную интенсивность протонного сигнала.
[Фиг. 10] На Фиг. 10 показана порошковая рентгеновская дифрактограмма кристалла сольвата этилацетата/гексана соединения, представленного Формулой (I). Горизонтальная ось представляет собой 2θ (°), и вертикальная ось представляет собой интенсивность (Число отсчетов).
[Фиг. 11] На Фиг. 11 показан Раман-спектр кристалла сольвата этилацетата/гексана соединения, представленного Формулой (I). Горизонтальная ось представляет собой рамановский сдвиг (см-1), и вертикальная ось представляет собой пиковую интенсивность.
[Фиг. 12] На Фиг. 12 показана порошковая рентгеновская дифрактограмма безводной кристаллической Формы II соединения, представленного Формулой (I). Горизонтальная ось представляет собой 2θ (°), и вертикальная ось представляет собой интенсивность (Число отсчетов).
[Фиг. 13] На Фиг. 13 показаны результаты анализа ТГ/ДТА кристалла сольвата этилацетата/гексана соединения, представленного Формулой (I). Вертикальная ось представляет собой калории (мкВ) или изменение массы (%), и горизонтальная ось представляет собой температуру (°C). «Цел» на фигуре означает градус Цельсия (°C).
[СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ]
[0019]
Выбор и контроль твердой формы важны, особенно для соединения в качестве лекарственного средства. Тщательный выбор и контроль твердой формы могут уменьшить проблемы при получении, составлении или введении, связанные с соединением.
[0020]
Если нет определенной ссылки, числовое значение в настоящем описании и формуле изобретения представляет собой приблизительное значение. Числовое изменение связано с калибровкой устройства, ошибкой устройства, чистотой вещества, размером кристалла, размером образца, температурой и другими факторами.
[0021]
Используемый в настоящем описании термин «кристалл» означает твердое вещество, в котором атомы, ионы, молекулы и подобные составляющие кристалла, расположены трехмерно и правильно, и отличается от аморфного твердого вещества, не имеющего такой регулярной внутренней структуры. Кристалл настоящего изобретения может представлять собой монокристалл, двойной кристалл, поликристалл или подобные.
Кроме того, кристаллические полиморфные модификации могут присутствовать в «кристаллах». В совокупности они называются «кристаллическими формами» и предназначены для включения в настоящее изобретение.
Кроме того, «соединение, представленное Формулой (I)», может образовывать сольват с водой (то есть гидрат) или сольват с обычным органическим растворителем, и такой сольват также, как предполагается, включен в объем настоящего изобретения.
Кристаллическую форму и кристалличность можно измерить многими методами, включая, например, порошковую рентгеновскую дифрактометрию, Раман-спектроскопию, инфракрасную абсорбционную спектроскопию, измерение адсорбции/десорбции влаги, дифференциальную сканирующую калориметрию и характеристики растворения.
[0022]
Используемый в настоящем описании термин «соль» означает, например, что «соединение, представленное Формулой (I)», и противоположные молекулы расположены надлежащим образом в одной и той же кристаллической решетке, и может быть включено любое количество противоположных молекул. Термин относится к связи, в которой ионная связь опосредована переносом протона между соединением и противоположной молекулой в кристаллической решетке.
[0023]
Исследования образования солей позволяют изменить физико-химические характеристики агента и, как следствие, биологические характеристики без изменения его химической структуры. Образование солей может существенно повлиять на свойства агента. При выборе подходящей соли гигроскопичность, стабильность, растворимость и технологические свойства соли также являются важными аспектами. Растворимость соли может повлиять на ее пригодность для применения в качестве агента. Если растворимость в воде низкая, скорость растворения при введении in vivo ограничивается процессом поглощения и может привести к низкой биодоступности. Кроме того, низкая растворимость в воде может затруднить введение путем инъекции, и, следовательно, выбор подходящего пути введения может быть ограничен.
[0024]
«Соединение, представленное Формулой (I)», может быть превращено в сольват, фармацевтически приемлемую соль или сольват соли. В одном аспекте настоящего изобретения соединение находится в форме основно-аддитивной соли. Примеры основно-аддитивной соли включают соли, полученные из фармацевтически приемлемых нетоксичных оснований, включая неорганические и органические основания. Примеры соли, полученной из неорганических оснований, включают, но не ограничиваются ими, соли алюминия, кальция, лития, калия, магния, натрия, цинка и соли других металлов. Примеры фармацевтически приемлемой соли на основе нетоксичного основания включают соли первичных, вторичных или третичных аминов и замещенных аминов, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, бензатин, кофеин, холин, хлорпрокаин, циклопрокаин, N'N'-дибензилэтилендиамин, диэтаноламин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, меглумин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, третичный бутиламин (2-метилпропан-2-амин), теобромин, триэтиламин, триметиламин, трипропиламин и трометамин; также как нетоксичный аммоний и четвертичный аммоний и соли катионов, включая, но не ограничиваясь ими, аммоний, тетраметиламмоний и тетраэтиламмоний.
[0025]
Примеры кислотно-аддитивных солей «соединения, представленного Формулой (I)», «соединения, представленного Формулой (II)», «соединения, представленного Формулой (IV)», «соединения, представленного Формулой (IV-A)», «соединения, представленного формулой (IV-B)» и «соединения, представленного Формулой (V)», включают соединение, содержащее:
неорганическую кислоту, такую как хлористоводородная кислота, бромистоводородная кислота, ортофосфорная кислота, азотная кислота, фосфорная кислота или серная кислота; или
органическую кислоту, такую как муравьиная кислота, метансульфокислота, этансульфокислота, п-толуолсульфокислота, уксусная кислота, пропионовая кислота, молочная кислота, лимонная кислота, фумаровая кислота, яблочная кислота, янтарная кислота, салициловая кислота, малеиновая кислота, глицерофосфорная кислота, винная кислота, бензойная кислота, глутаминовая кислота, аспарагиновая кислота, бензолсульфокислота, нафталинсульфокислота, такая как 2-нафталинсульфокислота, гексановая кислота и ацетилсалициловая кислота.
[0026]
Используемый в настоящем описании термин «сольват» относится, например, к сольвату, который надлежащим образом расположен с произвольным числом молекул растворителя по отношению к «соединению, представленному Формулой (I)».
Примеры молекулы растворителя включают ацетонитрил, хлорбензол, хлороформ, циклогексан, 1,2-дихлорэтен, дихлорметан, 1,2-диметоксиэтан, N,N-диметилацетамид, N, N-диметилформамид, 1,4-диоксан, 2-этоксиэтанол, этиленгликоль, формамид, гексан, метанол, 2-метоксиэтанол, метилбутилкетон, метилциклогексан, N-метилпирролидон, нитрометан, пиридин, сульфолан, тетралин, толуол, 1,1,2-трихлорэтен, ксилол, уксусную кислоту, анизол, 1-бутанол, 2-бутанол, трет-бутанол, н-бутилацетат, трет-бутилметиловый эфир, кумол, диметилсульфоксид, этилацетат, диэтиловый эфир, этилформиат, муравьиную кислоту, гептан, изобутилацетат, изопропилацетат, метилацетат, 3-метил-1-бутанол, метилэтилкетон, метилизобутилкетон, 2-метил-1-пропанол, пентан, 1-пентанол, 1-пропанол, 2-пропанол, пропилацетат, тетрагидрофуран, воду (т. е. гидрат), этанол, ацетон, 1,1-диэтоксипропан, 1,1-диметоксиметан, 2,2-диметоксипропан, изооктан, изопропиловый эфир, метилизопропилкетон, метилтетрагидрофуран, петролейный эфир, трихлоруксусную кислоту и трифторуксусную кислоту.
Предпочтительные примеры включают уксусную кислоту, анизол, 1-бутанол, 2-бутанол, н-бутилацетат, трет-бутилметиловый эфир, кумол, диметилсульфоксид, этилацетат, диэтиловый эфир, этилформиат, муравьиную кислоту, гептан, изобутилацетат, изопропилацетат, метилацетат, 3-метил-1-бутанол, метилэтилкетон, метилизобутилкетон, 2-метил-1-пропанол, пентан, 1-пентанол, 1-пропанол, 2-пропанол, пропилацетат, тетрагидрофуран, воду (т. е. гидрат), этанол, ацетон, 1,1-диэтоксипропан, 1,1-диметоксиметан, 2,2-диметоксипропан, изооктан, изопропиловый эфир, метилизопропилкетон, метилтетрагидрофуран, петролейный эфир, трихлоруксусную кислоту и трифторуксусную кислоту.
Более предпочтительные их примеры включают воду (т. е. гидрат), этанол, ацетон, 1,1-диэтоксипропан, 1,1-диметоксиметан, 2,2-диметоксипропан, изооктан, изопропиловый эфир, метилизопропилкетон, метилтетрагидрофуран, петролейный эфир, трихлоруксусную кислоту и трифторуксусную кислоту.
Когда «соединение, представленное Формулой (I)», оставляют в условиях атмосферы, влага поглощается, и к нему может прилипать адсорбированная вода, или может образовываться гидрат.
Кроме того, для «соединения, представленного Формулой (II)», «соединения, представленного Формулой (III)», «соединения, представленного Формулой (IV)», «соединения, представленного Формулой (IV-A)», «соединения, представленного Формулой (IV-B)», «соединения, представленного Формулой (V)» и «соединения, представленного Формулой (VI)», могут быть образованы сольваты.
[0027]
Гидрат настоящего изобретения или его кристалл содержит, например, приблизительно 2 молярных эквивалента молекул воды по отношению к «соединению, представленному Формулой (I)». Предпочтительные примеры кристалла гидрата настоящего изобретения включают дигидрат.
Гидрат настоящего изобретения или его кристалл имеют содержание воды, например, от 4,7 до 9,7 масс.%. Предпочтительно содержание воды составляет приблизительно от 5,6 до 7,6 масс.% (теоретическое значение дигидрата составляет 6,6%, но содержание влаги может увеличиваться вследствие влияния воды, прилипшей к кристаллу, или часть воды в кристалле может быть десорбирована перед измерением, в результате чего содержание влаги может уменьшиться).
[0028]
Кристалл настоящего изобретения может представлять собой продукт конверсии дейтерия. Кристалл настоящего изобретения может быть помечен изотопом (примеры: 3H, 14C, 35S, 125I).
[0029]
Используемый в настоящем описании термин «ангидрид» представляет собой синоним терминов «ансольват», «несольват», «ангидрат» и «негидрат».
[0030]
Соединение, представленное Формулой (I), является антагонистом P2X3 и/или P2X2/3, описанным в Патентном документе 9:
[Химическая формула 29]
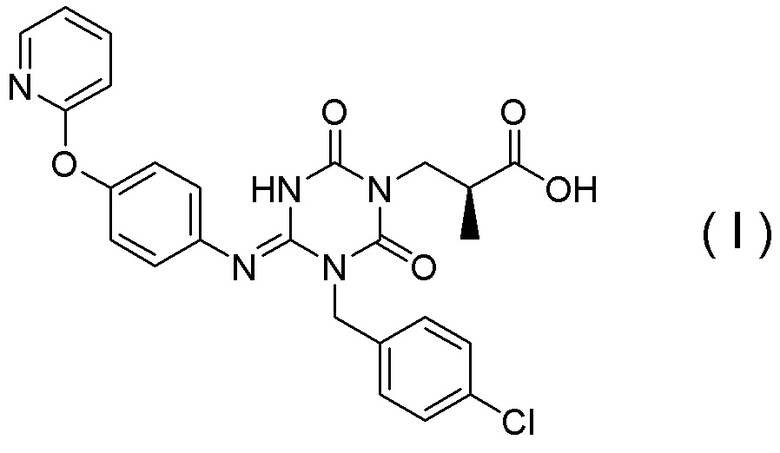
Оно является очень полезным в качестве терапевтического средства или профилактического средства при хроническом кашле. Соединение, представленное Формулой (I), может быть получено в соответствии с Примерами настоящей заявки.
[0031]
Таутомер соединения, представленного Формулой (I), представляет собой соединение (аминоформа), представленное Формулой (I'):
[Химическая формула 30]
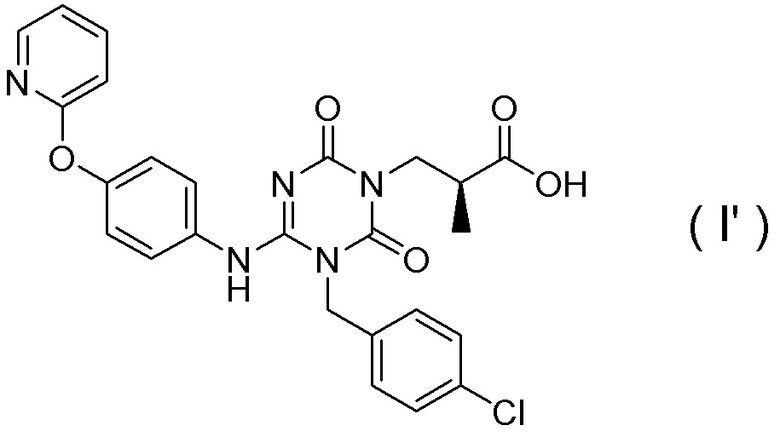
Это соединение обладает антагонистическим действием в отношении рецепторов Р2Х3 и/или Р2Х2/3, подобно соединению, представленному Формулой (I).
Кроме того, соединение, представленное Формулой (VI), также может образовывать таутомеры таким же образом, как описано выше.
[Химическая формула 31]
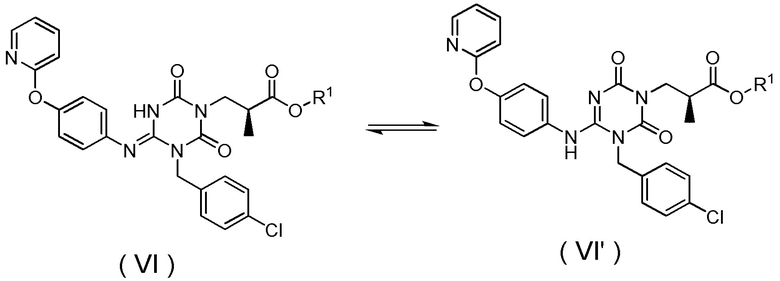
[0032]
Соединение, представленное Формулой (I), может также включать смесь соединения, представленного Формулой (I) (иминоформа), и соединения, представленного Формулой (I') (аминоформа), и они могут быть смешаны в произвольном соотношении. То же самое относится к соединению, представленному Формулой (VI).
В результате анализа структуры монокристалла было подтверждено, что безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет следующую молекулярную структуру (иминоформа) (подробности описаны в Примере 3).
[Химическая формула 32]
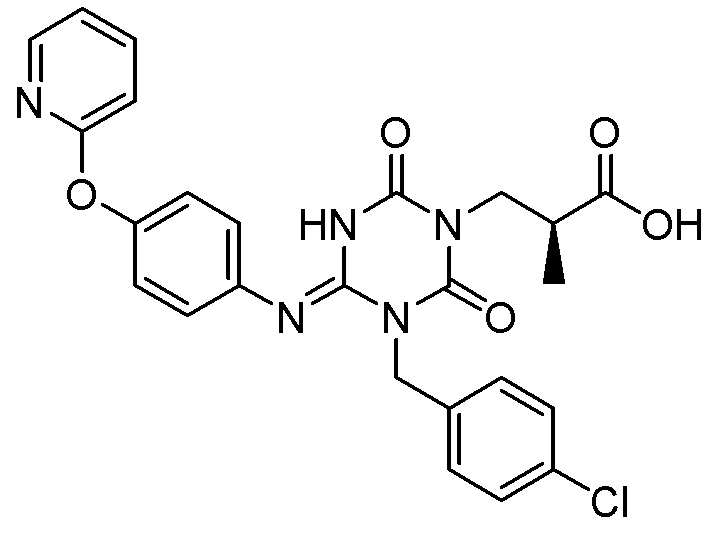
Для кристалла дигидрата соединения, представленного Формулой (I), и безводной кристаллической Формы II соединения, представленного Формулой (I), молекулярная структура (аминоформа/иминоформа) не была идентифицирована.
[0033]
(Порошковая рентгеновская дифракция (XRPD))
Обычно кристаллическое органическое соединение состоит из большого количества молекул, расположенных периодически в трехмерном пространстве. Структурная периодичность обычно развивает физические свойства, которые четко различимы большинством спектроскопических зондов (например, дифракция рентгеновских лучей, инфракрасный спектр, Раман-спектр и ЯМР твердого тела). Среди них порошковая рентгеновская дифракция (XRPD) является одним из наиболее чувствительных методов анализа для измерения кристалличности твердого вещества. При облучении кристалла рентгеновскими лучами рентгеновские лучи отражаются плоскостями кристаллической решетки и интерферируют друг с другом, и только дифракционные лучи в направлении, удовлетворяющем условию, предсказываемому законом Брэгга, увеличиваются по интенсивности, вследствие чего показаны упорядоченные дифракционные лучи, соответствующие периоду структуры. С другой стороны, для аморфного твердого вещества упорядоченный дифракционный луч не наблюдается. Аморфное твердое вещество обычно не имеет упорядоченного периода повторения в своей структуре, поэтому явление дифракции не возникает и показывает нечеткую широкую дифрактограмму XRPD (также называемую картиной гало).
[0034]
Кристаллическая форма ангидрида соединения, представленного Формулой (I), может быть охарактеризована порошковой рентгеновской дифрактограммой и характеристическими пиками. Кристаллическую форму ангидрида соединения, представленного Формулой (I), можно отличить от других кристаллических форм (например, кристалла гидрата) по наличию характеристических дифракционных пиков. Характеристические дифракционные пики, используемые в настоящем описании, выбраны из наблюдаемых дифрактограмм. При различении множества кристаллов пик, который наблюдается в кристалле и не наблюдается в других кристаллах, а не размер пика, является предпочтительным характеристическим пиком при определении кристалла. При таком характеристическом пике даже один или два пика могут характеризовать кристалл. Когда графики, полученные при измерении, сравнивают и эти характеристические пики совпадают друг с другом, можно сказать, что спектры порошковой рентгеновской дифракции по существу совпадают друг с другом.
[0035]
Обычно, поскольку угол дифракции (2θ) при порошковой рентгеновской дифракции может иметь погрешность в пределах диапазона ±0,2°, значение угла дифракции при порошковой рентгеновской дифракции следует понимать как включающее численное значение в пределах диапазона приблизительно ±0,2°. Следовательно, настоящее изобретение охватывает не только кристаллы, у которых углы дифракции пиков при порошковой рентгеновской дифракции полностью совпадают друг с другом, но также кристаллы, у которых углы дифракции пиков совпадают друг с другом с погрешностью приблизительно ±0,2°.
[0036]
Общеизвестно, что интенсивность пиков, отображаемых в следующих таблицах и фигурах, может варьироваться в зависимости от многих факторов, например, влияния селективной ориентации кристалла на рентгеновский пучок, влияния крупных частиц, чистоты анализируемого материала или кристалличности образца. Положение пика также может быть смещено в зависимости от изменения высоты образца. Кроме того, в соответствии с уравнением Брэгга (nλ=2dsinθ) получают разные сдвиги, когда измерения проводят с использованием другой длины волны, и такая другая дифрактограмма, полученная с использованием другой длины волны, также входит в объем настоящего изобретения.
[0037]
Анализ структуры монокристалла (см. Toshio SAKURAI, «X-sen Kozo Kaiseki no Tebiki (Guide to X-ray Structural Analysis)», под издательством Shokabo Co., Ltd. (1983) и Stout & Jensen, «X-Ray Structure Determination: A Practical Guide», Macmillan Co., New York (1968), etc.)) является одним из методов определения кристалла, и с его помощью можно получить кристаллографические параметры в кристалле, координаты атомов (значения, указывающие на пространственно-позиционную взаимосвязь каждого атома) и трехмерную структурную модель. Анализ структуры монокристалла является полезным для идентификации структуры кристалла композита, как в настоящем изобретении.
[0038]
(Раман-спектроскопия)
Раман-спектр показывает колебательные характеристики молекул или сложной системы. Его происхождение лежит в неупругих столкновениях между молекулами и фотонами как частицами света, включая световые лучи. Столкновение молекул с фотонами приводит к обмену энергией, что приводит к изменению энергии, что, в свою очередь, изменяет длину волны фотонов. То есть, поскольку Раман-спектр представляет собой спектральную линию, испускаемую при падении фотонов на молекулу-мишень, и имеет чрезвычайно узкую длину волны, в качестве источника света используется лазер или подобное. Длина волны каждой рамановской линии представлена сдвигом волнового числа от падающего света, который представляет собой разность между обратной величиной длины волны рамановской линии и длины волны падающего света. Раман-спектр используется для измерения колебательного состояния молекулы, которое определяется ее молекулярной структурой.
Обычно, поскольку полоса поглощения (см-1) в Раман-спектре может иметь погрешность в пределах диапазона ±2 см-1, значение пика поглощения следует понимать как включающее численное значение в пределах диапазона приблизительно ±2 см-1. Следовательно, настоящее изобретение охватывает не только кристаллы, у которых пики полос поглощения в Раман-спектрах полностью совпадают друг с другом, но также кристаллы, у которых пики полос поглощения совпадают друг с другом с погрешностью приблизительно ±2 см-1.
[0039]
(Инфракрасная абсорбционная спектроскопия (ИК-метод))
Инфракрасная абсорбционная спектроскопия представляет собой метод измерения для каждого волнового числа степени поглощения инфракрасных лучей при прохождении инфракрасных лучей через образец. Инфракрасный спектр поглощения обычно представляется в виде графика, на котором горизонтальная ось представляет собой волновое число и вертикальная ось представляет собой коэффициент пропускания или коэффициент поглощения. Волновое число и коэффициент пропускания (или коэффициент поглощения) пика поглощения можно прочитать на графике, и можно использовать значения, рассчитанные устройством обработки данных. Инфракрасный спектр поглощения определяется химической структурой вещества. Следовательно, поглощение при различных волновых числах может быть измерено для подтверждения или количественного определения вещества. Различение кристаллической полиморфной модификации может быть выполнено путем сравнения полос поглощения функциональных групп, характеристических для кристаллических полиморфных модификаций, то есть функциональной группы, в основном участвующей в водородной связи в кристаллической структуре, такой как связь С=О, связь ОН и связь NH, также как других характеристических функциональных групп, таких как связь C-X (галоген), связь C=C и связь C≡C. Полосы поглощения характеристической функциональной группы выбраны из приблизительно 20 пиков поглощения, более предпочтительно приблизительно 10 пиков поглощения и наиболее предпочтительно приблизительно 5 пиков поглощения, соответствующих характеристическим функциональным группам. Обычно спектр поглощения образца измеряется в диапазоне волновых чисел от 4000 см-1 до 400 см-1. Спектр поглощения измеряется при тех же рабочих условиях, что и при подтверждении разрешающей способности, шкалы волновых чисел и точности волновых чисел устройства.
[0040]
Обычно, поскольку полоса поглощения (см-1) в инфракрасной абсорбционной спектроскопии может иметь погрешность в пределах диапазона ±2 см-1, значение пика поглощения следует понимать как включающее численное значение в пределах диапазона приблизительно ±2 см-1. Следовательно, настоящее изобретение охватывает не только кристаллы, у которых пики полос поглощения в инфракрасной абсорбционной спектроскопии полностью совпадают друг с другом, но также кристаллы, у которых пики полос поглощения совпадают друг с другом с погрешностью приблизительно ±2 см-1.
[0041]
Примеры метода измерения спектра инфракрасного поглощения включают метод с таблетками бромида калия, метод растворов, метод пасты, метод жидкой пленки, метод тонкой пленки, метод измерения образца газа, метод НПВО и метод диффузного отражения. Среди них метод нарушенного полного внутреннего отражения (НПВО) называется методом измерения полного отражения и является одним из методов отражения. В этом методе образец приводится в тесный контакт с поверхностью призмы, изготовленной из вещества с высоким показателем преломления, такого как KRS-5, свет падает на призму под углом, равным или превышающим критический угол, и свет, полностью отраженный от границы между призмой и образцом, измеряется для получения спектра поглощения. Одним из условий, позволяющих проводить измерения методом НПВО, является то, что показатель преломления призмы больше, чем у образца, и поэтому необходимо менять материал призмы в зависимости от образца. Кроме того, в качестве еще одного условия призма и образец должны находиться в тесном контакте друг с другом. Следовательно, он подходит для измерения жидкости, порошка, пластика, мягкой резины и подобного, и его преимущество заключается в том, что измерение можно выполнять без химической или физической обработки образца. С другой стороны, метод диффузного отражения представляет собой метод измерения образца порошка, поскольку происходит без образования таблетки бромида калия. При воздействии света на образец генерируются свет, который зеркально отражается на поверхности порошка и выходит наружу, и диффузно отраженный свет (рассеянный свет), который входит в образец, повторяет пропускание и диффузию и затем выходит на поверхность. В методе диффузного отражения последний используется для получения спектра поглощения.
[0042]
(13C-ЯМР твердого тела (ядерный магнитный резонанс))
13С-ЯМР твердого тела является пригодным для определения кристаллической формы, поскольку (i) количество спектров совпадает с количеством атомов углерода целевого соединения, (ii) диапазон химического сдвига шире, чем у 1H-ЯМР, (iii) сигнал более резкий, чем у 1H-ЯМР твердого тела, и (iv) даже если добавка содержится, химический сдвиг не изменяется при отсутствии взаимодействия. Следует отметить, что наблюдаемый химический сдвиг, как ожидается, незначительно варьируется в зависимости от конкретного используемого спектрометра и метода пробоподготовки аналитика. Диапазон ошибок в спектре 13С-ЯМР твердого тела составляет приблизительно ±0,5 м.д.
[0043]
(Дифференциальная сканирующая калориметрия (ДСК))
ДСК является одним из основных измерительных методов термического анализа и представляет собой метод измерения тепловых свойств вещества как совокупности атомов и молекул.
Кривую дифференциальной сканирующей калориметрии получают путем измерения изменения количества тепла в зависимости от температуры или времени действия фармацевтически активного ингредиента с помощью ДСК и построения графика полученных данных в зависимости от температуры или времени. Из кривой дифференциальной сканирующей калориметрии можно получить информацию о температуре начала плавления фармацевтически активного ингредиента, максимальном значении кривой эндотермического пика, связанной с плавлением, и энтальпии.
Для ДСК известно, что наблюдаемая температура может зависеть от скорости изменения температуры, также как от метода подготовки образца и конкретного используемого оборудования. Таким образом, «температура плавления» в ДСК относится к начальной температуре, которая менее чувствительна к методам подготовки образца. Диапазон ошибок при начальной температуре, полученный из кривой дифференциальной сканирующей калориметрии, составляет приблизительно ±2°C. При распознавании идентичности кристаллов важна не только точка плавления, но также общая картина, и общая картина может незначительно меняться в зависимости от условий измерения и измерительного прибора.
[0044]
(Термогравиметрия/Дифференциальный термический анализ (ТГ/ДТА))
ТГ/ДТА является одним из основных измерительных методов термического анализа и представляет собой метод измерения массы и тепловых свойств вещества как совокупности атомов и молекул.
ТГ/ДТА представляет собой метод измерения изменений массы и количества тепла фармацевтически активного ингредиента в зависимости от температуры или времени, и кривые ТГ (термогравиметрии) и ДТА (дифференциального термического анализа) получают путем построения графика полученных данных в зависимости от температуры или времени. Из кривых ТГ/ДТА можно получить информацию об изменении массы и количества тепла в отношении разложения, дегидратации, окисления, восстановления, сублимации и испарения фармацевтически активного ингредиента.
Для ТГ/ДТА известно, что наблюдаемые изменения температуры и массы могут зависеть от скорости изменения температуры, также как от метода подготовки образца и конкретного используемого оборудования. Таким образом, «температура плавления» в ТГ/ДТА относится к начальной температуре, которая менее чувствительна к методам подготовки образца. При распознавании идентичности кристаллов важна не только точка плавления, но также общая картина, и общая картина может незначительно меняться в зависимости от условий измерения и измерительного прибора.
[0045]
(Метод измерения изотермы сорбции/десорбции влаги (DVS))
Измерение изотермы адсорбции/десорбции влаги представляет собой метод измерения поведения адсорбции и десорбции влаги путем измерения изменения массы твердого вещества в качестве цели измерения при каждом условии относительной влажности.
В качестве основного метода измерения, основанного на массе сухого вещества при 0% ОВ (относительной влажности 0%), относительная влажность увеличивается на каждые 5% или 10%, и после стабилизации массы при каждой относительной влажности количество адсорбированной воды можно определить по увеличению массы по сравнению с эталонным значением. Подобным образом количество десорбции воды можно измерить, уменьшая относительную влажность на каждые 5% или 10% от 100% ОВ.
При нанесении на график значения изменения массы при каждой относительной влажности можно получить изотерму адсорбции/десорбции. Исходя из этого результата, можно рассматривать явление адсорбции и десорбции налипшей влаги при каждой влажности. Кроме того, когда кристалл ангидрида и кристалл гидрата взаимно подвергаются кристаллическому переходу вследствие влажности, можно рассчитать влажность, при которой происходит кристаллический переход, и количество кристаллической воды.
На сорбцию и десорбцию налипшей воды и кристаллизационной воды влияет размер частиц, кристалличность, габитус кристаллов и подобное, так что результаты измерения могут незначительно изменяться.
[0046]
Фармацевтическая композиция, содержащая кристалл настоящего изобретения, является очень полезной в качестве терапевтического средства или профилактического средства при хроническом кашле.
[0047]
Кристалл настоящего изобретения можно вводить пациенту отдельно или можно вводить в виде фармацевтической композиции, в которой кристалл смешан с подходящим носителем или эксципиентом. Методы составления и введения лекарственного средства могут быть соответствующим образом выбраны и использованы в комбинации с фармацевтическими составами и методами, известными специалисту в данной области техники.
[0048]
Примеры пути введения кристалла настоящего изобретения или фармацевтической композиции, содержащей кристалл, могут включать, но не ограничиваются ими, пероральное, ректальное, чресслизистое или кишечное введение или внутримышечную, подкожную, интраспинальную, интратекальную, прямую внутрижелудочковую, внутривенную, интравитреальную, внутрибрюшинную, интраназальную и интраокулярную инъекцию. Предпочтительным путем введения является пероральное введение.
[0049]
Фармацевтическая композиция настоящего изобретения может быть получена способом, хорошо известным в данной области техники, например, способом стандартного смешивания, растворения, гранулирования, покрытия сахаром, измельчения в порошок, эмульгирования, инкапсулирования, заключения в оболочку или лиофилизации.
[0050]
Кристалл настоящего изобретения или фармацевтическую композицию, содержащую кристалл, можно вводить путем инъекции с использованием водного раствора, предпочтительно физиологически совместимого буфера, такого как раствор Рингера или физиологический раствор.
[0051]
Кристалл настоящего изобретения или фармацевтическую композицию, содержащую кристалл, можно вводить через слизистую оболочку с использованием пенетранта, подходящего для проникновения через барьер. В качестве пенетранта можно использовать вещество, общеизвестное в данной области техники.
[0052]
Кристалл настоящего изобретения или фармацевтическую композицию, содержащую кристалл, можно объединить с фармацевтически приемлемым носителем, хорошо известным в данной области техники, для перорального введения. Носитель позволяет вводить кристалл изобретения в виде таблеток, пилюль, пастилок, таблеток с сахарным покрытием, капсул, раствора, геля, сиропов или суспензий. Фармацевтические композиции для перорального введения могут быть изготовлены путем добавления твердых эксципиентов и при желании других подходящих вспомогательных веществ с последующим измельчением полученной смеси и обработкой смеси гранул с получением таблеток или ядер таблеток, покрытых сахарной оболочкой.
[0053]
Пригодные эксципиенты представляют собой, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит, например, препараты целлюлозы, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал и картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза и/или натрий карбоксиметилцеллюлоза. При необходимости можно добавить разрыхлитель, такой как агар или альгиновая кислота. Также можно использовать соль, такую как альгинат натрия.
[0054]
Примеры фармацевтических композиций, которые можно использовать для перорального введения, включают твердые капсулы, изготовленные из желатина, и герметичные капсулы, изготовленные из желатина и смягчителя, такого как глицерин или сорбит. Твердая капсула может содержать активный ингредиент, смешанный с наполнителем, таким как лактоза, связующим веществом, таким как крахмал, и/или скользящим веществом, таким как тальк или стеарат магния.
[0055]
Фармацевтическая композиция может также содержать подходящий твердый или гелевый носитель или эксципиент. Примеры такого носителя или эксципиента включают карбонат кальция, фосфат кальция, различные сахара, крахмал, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоль.
[0056]
Для кристаллов изобретения или их фармацевтических композиций терапевтически эффективное количество можно сначала оценить с помощью анализа на клеточных культурах. Затем можно составить дозу большого количества для использования на животных моделях для достижения диапазона концентраций в кровотоке, который охватывает IC 50 (то есть концентрация кристалла настоящего изобретения или его фармацевтической композиции, при которой достигается половина максимального ингибирования достигается ФК активности), как определено в клеточной культуре. Затем такую информацию можно использовать для более точного определения пригодного количества того же вещества для людей.
[0057]
Терапевтические эффекты кристалла настоящего изобретения или его фармацевтической композиции можно измерить стандартным фармацевтическим способом в клеточной культуре или на экспериментальных животных. Например, оценка может быть выполнена в соответствии с методом биологических испытаний, описанным в Патентном документе 9. Данные, полученные в результате этих анализов клеточных культур и экспериментов на животных, могут быть использованы для составления ряда дозировок для применения для людей. Дозировка может варьироваться в зависимости от применяемой формы введения и используемого пути введения. Точный путь введения состава и дозировка могут быть выбраны лечащим врачом, учитывая состояние пациента.
[0058]
Также аспектом настоящего изобретения является то, что кристаллы настоящего изобретения или их фармацевтические композиции можно комбинировать с другими средствами для лечения заболеваний и нарушений.
[0059]
Настоящее изобретение предоставляет кристалл ангидрида или кристалл гидрата соединения, представленного Формулой (I). Кристаллическое твердое вещество имеет, по меньшей мере, одну из следующих характеристик:
(1) имеет хорошую стабильность в отношении тепла, влажности, растворителей, света, и подобного, и высокую стабильность при хранении;
(2) имеет хорошую стабильность окрашивания;
(3) имеет хорошую растворимость в воде или органических растворителях;
(4) имеет высокую скорость растворения по отношению к воде или органическим растворителям;
(5) имеет высокую чистоту;
(6) имеет низкий уровень остаточного органического растворителя;
(7) имеет отличные эксплуатационные качества при фильтрации, центрифугировании и составлении;
(8) имеет небольшой удельный объем;
(9) почти не заряжается;
(10) производится с высоким выходом в условиях пониженной нагрузки на окружающую среду и имеет возможность производиться большими партиями;
(11) является пригодным в качестве фармацевтически активного ингредиента для инъекции или активного материала для его получения;
(12) имеет возможность регулирования до диапазона рН, подходящего для внутривенной инъекции без сосудистой боли, что, таким образом, является преимуществом для контроля количества жидкости, уменьшения количества эксципиентов и т. д. во время составления препарата;
(13) имеет хорошую текучесть и
(14) имеет низкий коэффициент прессуемости (%).
В частности, кристаллическое твердое вещество настоящего изобретения обладает высокой стабильностью даже в широком диапазоне влажности (например, от 25 до 99% ОВ или подобного) и при неблагоприятных условиях окружающей среды (например, при высокой влажности).
[0060]
Значение каждого термина, используемого в настоящем описании, будет описано ниже. Если не указано иначе, каждый термин используется в одном и том же значении при использовании отдельно или в комбинации с другим термином.
Термин «состоящий из» означает наличие только компонентов.
Термин «включающий» означает не ограничивающийся компонентами, но не исключающий элементы, которые не описаны.
[0061]
Далее настоящее изобретение будет описано со ссылкой на варианты осуществления. Следует понимать, что во всем описании формы выражения в единственном числе также включают понятие их форм во множественном числе, если не указано иначе. Таким образом, следует понимать, что единственное число также включает его понятие во множественном числе, если не указано иначе.
Также следует понимать, что термины, используемые в настоящем описании, используются в смысле, обычно используемом в данной области техники, если не указано иначе. Таким образом, если не указано иначе, все технические и научные термины, используемые в настоящем описании, имеют те же значения, которые обычно понимаются специалистом в данной области техники, к которой относится данное изобретение. В случае конфликта настоящее описание (включая определения) будет иметь преимущественную силу.
[0062]
Термин «галоген» включает атом фтора, атом хлора, атом брома и атом иода. В частности, атом фтора и атом хлора являются предпочтительными.
[0063]
Термин «алкил» включает линейную или разветвленную углеводородную группу, содержащую от 1 до 15 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода и еще более предпочтительно от 1 до 4 атомов углерода. Его примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил и н-децил.
Предпочтительные варианты осуществления «алкила» включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил и н-пентил. Более предпочтительные варианты осуществления включают метил, этил, н-пропил, изопропил и трет-бутил.
Примеры «C1-C4 алкила» включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
[0064]
Настоящее изобретение включает стадию получения соединения, представленного Формулой (IV):
[Химическая формула 33]
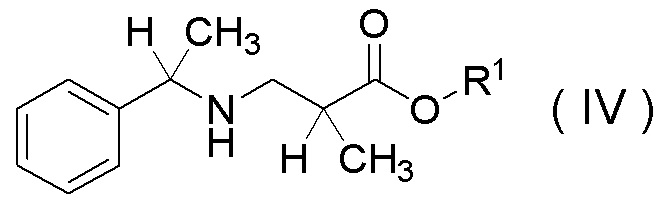
в которой R1 представляет собой C1-C4 алкил,
или его соли,
характеризующуюся проведением реакции между соединением, представленным Формулой (II):
[Химическая формула 34]
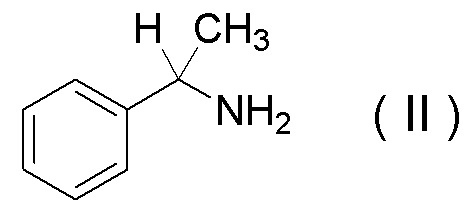
или его солью
и соединением, представленным Формулой (III):
[Химическая формула 35]
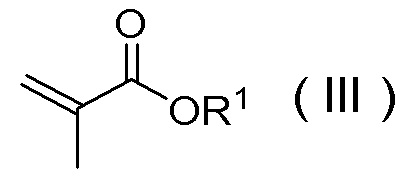
в которой R1 представляет собой C1-C4 алкил,
или его солью
в присутствии одной или более добавок, выбранных из группы, состоящей из хлорида лития, хлорида кальция, хлорида магния, бромида лития, п-толуолсульфокислоты, метансульфокислоты и трифторметансульфокислоты.
Соединение, представленное Формулой (II), или его соль и соединение, представленное Формулой (III), или его соль можно получить в соответствии с известным способом из коммерчески доступных реагентов, или коммерчески доступный продукт может быть использован в качестве этих соединений и солей.
[0065]
Растворитель особым образом не ограничивается, пока реакция не ингибируется, но метанол, этанол, изопропиловый спирт, трет-бутанол или их смешанный растворитель можно использовать в качестве растворителя. Например, можно использовать метанол.
[0066]
В отношении температуры реакции реакцию обычно проводят в диапазоне от комнатной температуры до температуры, при которой растворитель нагревают с обратным холодильником. Например, реакцию можно проводить в диапазоне от -10°С до температуры, при которой растворитель нагревают с обратным холодильником. Например, ее можно проводить при 80°C.
[0067]
Время реакции составляет от 1 до 20 часов, например, от 5 до 7 часов.
[0068]
Используемое количество соединения, представленного Формулой (III), по отношению к соединению, представленному Формулой (II), обычно может составлять от 1,0 до 10,0 эквивалентов, например, от 2,0 до 4,0 эквивалентов, например, 3,0 эквивалента.
[0069]
В качестве добавки можно использовать хлорид лития, хлорид кальция, хлорид магния, бромид лития, п-толуолсульфокислоту, метансульфокислоту, трифторметансульфокислоту или подобные. Совокупность таких добавок можно выбрать и использовать одновременно.
Используемое количество добавки по отношению к соединению, представленному Формулой (II), обычно может составлять от 0,1 до 5,0 эквивалентов, например, от 1,0 до 2,0 эквивалентов, например, от 1,0 до 1,5 эквивалентов.
[0070]
Настоящее изобретение включает стадию получения соли п-толуолсульфокислоты соединения, представленного Формулой (IV-A):
[Химическая формула 36]
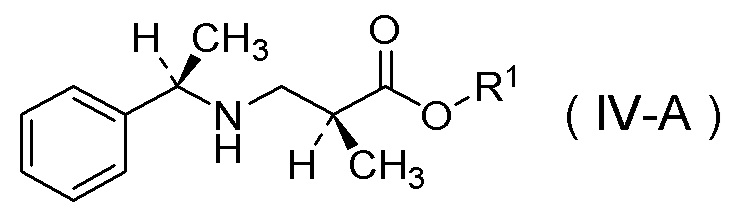
в которой R1 представляет собой C1-C4 алкил,
характеризующуюся:
добавлением п-толуолсульфокислоты к соединению, представленному Формулой (IV):
[Химическая формула 37]
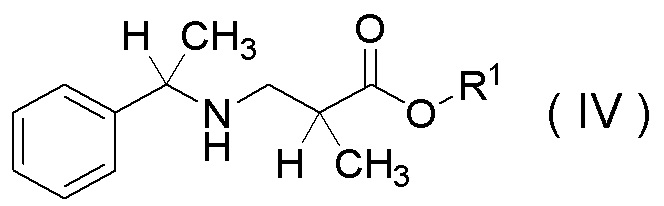
в которой R1 представляет собой C1-C4 алкил,
или его соли.
Используемое количество моногидрата п-толуолсульфокислоты (или водного раствора п-толуолсульфокислоты) по отношению к соединению, представленному Формулой (II), обычно может составлять от 0,5 до 2,0 эквивалентов, например, от 0,8 до 1,0 эквивалента.
[0071]
Настоящее изобретение включает стадию получения соли 1/2 серной кислоты соединения, представленного Формулой (V):
[Химическая формула 38]
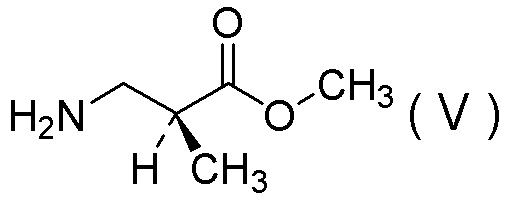
характеризующуюся:
подверганием соли п-толуолсульфокислоты соединения, представленного Формулой (IV-B):
[Химическая формула 39]
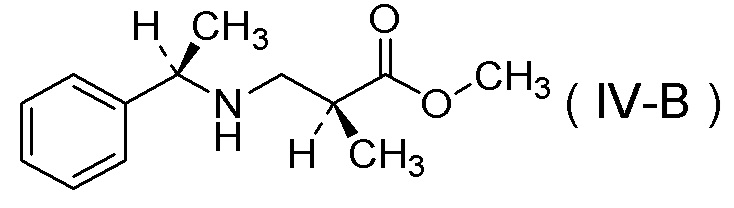
реакции гидрогенолиза и
добавлением к ней серной кислоты.
Соединение, представленное Формулой (IV-B), можно получить в соответствии с вышеописанной стадией.
[0072]
Растворитель особым образом не ограничивается, пока реакция не ингибируется, но можно использовать метанол, этанол, 1-пропанол, изопропиловый спирт, трет-бутанол, тетрагидрофуран или их смешанный растворитель. Например, можно использовать метанол.
[0073]
В отношении температуры реакции реакцию обычно проводят в диапазоне от комнатной температуры до температуры, при которой растворитель нагревают с обратным холодильником. Например, реакцию можно проводить в диапазоне от комнатной температуры до температуры, при которой растворитель нагревают с обратным холодильником. Например, ее можно проводить при от 30 до 50°C.
[0074]
Время реакции составляет от 30 минут до 20 часов, например, от 1 до 3 часов.
[0075]
В качестве катализатора реакции гидролиза можно использовать палладий на угле, гидроксид палладия, палладиевую чернь или подобное.
Используемое количество катализатора реакции гидролиза по отношению к соединению, представленному Формулой (IV-B), обычно может составлять от 0,01 до 1 масс./масс., например, от 0,1 до 0,3 масс./масс.
[0076]
Используемое количество концентрированной серной кислоты по отношению к соединению, представленному Формулой (IV-B), обычно может составлять от 0,01 до 5,0 эквивалентов, например, от 0,3 до 0,4 эквивалента.
[0077]
Настоящий способ включает стадию получения соединения, представленного Формулой (I):
[Химическая формула 40]
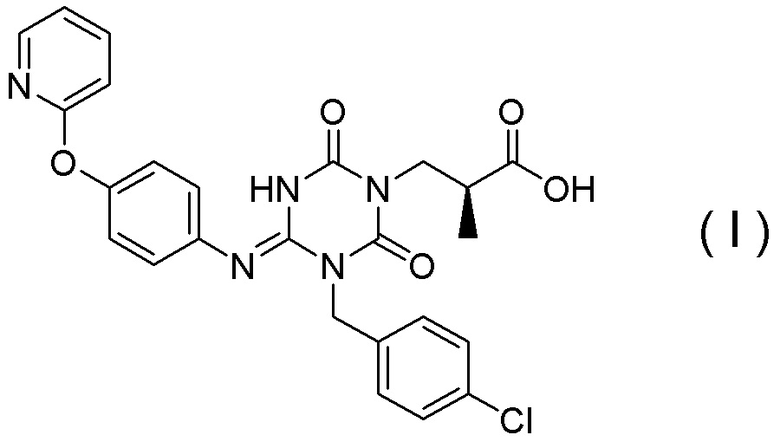
или его соли,
характеризующуюся:
подверганием соединения, представленного Формулой (VI):
[Химическая формула 41]
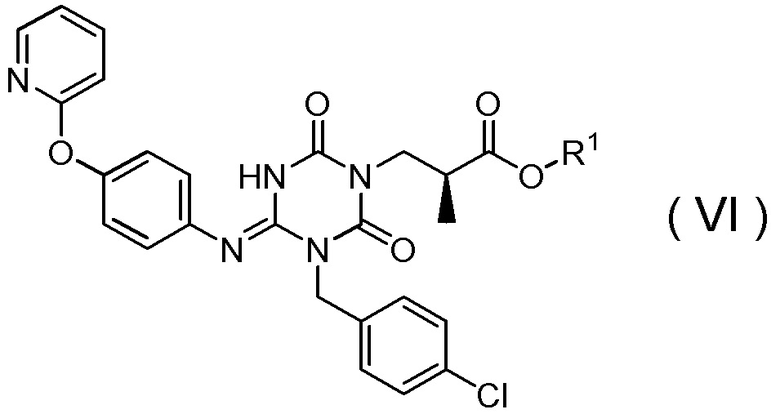
в которой R1 представляет собой C1-C4алкил,
или его соли
реакции гидролиза в присутствии одного или более растворителей, выбранных из группы, состоящей из изопропилового спирта, тетрагидрофурана и трет-бутанола.
Соединение, представленное Формулой (VI), можно получить в соответствии с вышеупомянутыми стадиями и способами, описанными в Патентных документах 6, 7, 8 и 9.
[0078]
Растворитель особым образом не ограничивается, пока реакция не ингибируется, но можно использовать изопропиловый спирт (2-пропанол), тетрагидрофуран и трет-бутанол или их смешанный растворитель. Например, можно использовать изопропиловый спирт (2-пропанол).
[0079]
В отношении температуры реакции реакцию обычно проводят в диапазоне от -10°С до температуры, при которой растворитель нагревают с обратным холодильником. Например, ее можно проводить при от 30°C до 40°C.
[0080]
Время реакции составляет от 0,1 до 20 часов, например, от 1 до 5 часов.
[0081]
В качестве основания можно использовать гидроксид натрия, гидроксид калия, гидроксид лития или подобные. Например, можно использовать гидроксид натрия.
Используемое количество основания по отношению к соединению, представленному Формулой (VI), обычно может составлять от 2,0 до 5,0 эквивалентов, например, от 2,0 до 3,0 эквивалентов.
[ПРИМЕРЫ]
[0082]
Настоящее изобретение будет описано более подробно с помощью следующих примеров. Они не ограничивают настоящее изобретение. Для числовых значений (например, количество, температура и подобные) следует учитывать некоторую погрешность и отклонение.
Если не указано иначе, «%» означает % масс. компонента и % масс. общей массы композиции и «давление» означает давление, равное или близкое к атмосферному давлению.
[0083]
(Измерение порошковой рентгеновской дифрактограммы)
Измерение порошковой рентгеновской дифракции кристалла, полученного в каждом Примере, проводили в соответствии со способом измерения порошковой рентгеновской дифракции, описанным в «Общие испытания, способы и устройства» Японской фармакопеи. Условия измерения показаны ниже.
(Способ 1)
(Устройство)
SmartLab производства Rigaku Corporation
(Способ работы)
Метод измерения: метод отраженных волн
Используемая длина волны: луч CuKα
Ток трубки: 200 мА
Напряжение трубки: 45 кВ
Пластина образца: стекло
Угол падения рентгеновского луча: 2,5°
Ширина выборки: 0,02°
Детектор: HyPix-3000 (режим двухмерного детектирования)
(Способ 2)
(Устройство)
D-8 Discover производства Bruker Corporation
(Способ работы)
Метод измерения: метод отраженных волн
Используемая длина волны: луч CuKα
Ток трубки: 40 мА
Напряжение трубки: 40 кВ
Пластина образца: алюминий
Угол падения рентгеновского луча: 3° и 12°
(Способ 3)
D-8 Discover производства Bruker Corporation
(Способ работы)
Метод измерения: метод отраженных волн
Используемая длина волны: луч CuKα
Ток трубки: 40 мА
Напряжение трубки: 40 кВ
Пластина образца: алюминий
Угол падения рентгеновского луча: 3°
[0084]
(Измерение Раман-спектра)
Измеряли Раман-спектр кристалла, полученного в каждом Примере. Условия измерения показаны ниже.
(Способ 1)
Устройство для измерения: LabRAM ARAMIS (производства HORIBA Jobin Yvon SAS)
Метод измерения: микролазерная Раман-спектроскопия
Длина волны лазера: 633 нм (He-Ne лазер)
Дифракционная решетка: 600 штрихов/мм
Детектор: ПЗС-детектор
Объектив: 20 × (NA 0,25)
Количество интеграций: 5 раз
Время воздействия: 5 секунд
(Способ 2)
Устройство для измерения: RAMANTouch Vis2-NIR-SNU (производства Nanophoton Corporation)
Метод измерения: микролазерная Раман-спектроскопия
Длина волны лазера: 532 нм
Дифракционная решетка: 1200 штрихов/мм
Детектор: ПЗС-детектор
Объектив: 20 × (NA 0,45)
Количество интеграций: 1 раз
Время воздействия: 3 секунды
[0085]
(Измерение дифференциальной сканирующей калориметрии (ДСК))
Измеряли ДСК кристалла, полученного в каждом Примере. Приблизительно 4,199 мг образца взвешивали в алюминиевом тигле и измеряли массу простым запаиванием. Условия измерения показаны ниже. В частности, при измерении с помощью дифференциальной сканирующей калориметрии (ДСК) может возникать ошибка в пределах диапазона ±2°C.
Устройство: TA Instruments Discovery
Диапазон температур измерения: от 0°C до 220°C
Скорость нагревания: 10°C/мин
Атмосфера: N2 50 мл/мин
[0086]
(Измерение ЯМР)
Когда показаны данные ЯМР, все измеренные пики не могут быть описаны.
[0087]
(Измерение ВЭЖХ)
(Способ А)
Колонка: XBridge C18, ϕ 4,6 × 150 мм, 3,5 мкм (Waters)
Колоночный термостат: 40°C
Скорость потока: 1,0 мл в минуту
Длина волны УФ-детектирования: 254 нм
Подвижная фаза А: 0,1% водный раствор трифторуксусной кислоты
Подвижная фаза B: ацетонитрил для жидкостной хроматографии
Программа градиента показана в Таблице 1.
[0088]
[Таблица 1]
(об.%)
(об.%)
[0089]
(Способ В)
Колонка: CHIRALPACK AS-RH, ϕ 4,6 × 150 мм, 5 мкм (Daicel Chemical Industries, Ltd.)
Колоночный термостат: 35°C
Скорость потока: 1,0 мл в минуту
Длина волны УФ-детектирования: 254 нм
Подвижная фаза А: очищенная вода для жидкостной хроматографии
Подвижная фаза B: ацетонитрил для жидкостной хроматографии
Программа градиента показана в Таблице 2.
[0090]
[Таблица 2]
(об.%)
(об.%)
[0091]
(Способ С)
Колонка: CHIRALPACK IC, ϕ 4,6 × 250 мм, 5 мкм (Daicel Chemical Industries, Ltd.)
Колоночный термостат: 35°C
Скорость потока: 1,0 мл в минуту
Длина волны УФ-детектирования: 262 нм
Подвижная фаза: 0,1% водный раствор муравьиной кислоты/раствор смеси ацетонитрила для жидкостной хроматографии (3:2)
[0092]
(Способ D)
Колонка: Cadenza CD-C18, ϕ 3,0 × 150 мм, 3 мкм
Колоночный термостат: 50°C
Скорость потока: 0,55 мл в минуту
Длина волны УФ-детектирования: 262 нм
Подвижная фаза А: 0,1% водный раствор ТФУК
Подвижная фаза B: ацетонитрил
Программа градиента показана в Таблице 3.
[0093]
[Таблица 3]
(%)
(%)
Время удерживания ВЭЖХ следует понимать как включающее некоторые ошибки.
[0094]
(Измерение ТГ/ДТА)
Приблизительно 4,4 мг кристалла, полученного в Примере 7, взвешивали, помещали в алюминиевый тигель и измеряли в открытой системе. Условия измерения являются следующими.
Устройство: Hitachi High-Technologies TG/DTA STA7200RV
Диапазон температур измерения: от комнатной температуры до 300°C
Скорость нагревания: 10°C/мин
[0095]
(Метод измерения и анализа для структурного анализа монокристалла)
Условия измерения и методы анализа для структурного анализа монокристалла показаны ниже.
(Устройство)
XtaLAB P 200 MM007 производства Rigaku Corporation
(Условия измерения)
Температура измерения: 25°C
Используемая длина волны: луч CuKα (λ=1,5418 Å)
Программное обеспечение: CrysAlisPro 1.171.39.46e (Rigaku Oxford Diffraction, 2018)
(Обработка данных)
Программное обеспечение: CrysAlisPro 1.171.39.46e (Rigaku Oxford Diffraction, 2018)
Данные были подвергнуты коррекции по Лоренцу и поляризации и поправке на поглощение.
(Структурный анализ кристалла)
Определение фаз проводили с помощью программы прямого метода ShelXT (Sheldrick, G.M., 2015) и уточняли полноматричным методом наименьших квадратов с использованием ShelXL (Sheldrick, G.M., 2015). Все температурные факторы неводородных атомов уточняли с учетом анизотропии. Атом водорода H5 на кислороде O5 получали из разностной карты Фурье и уточняли. Остальные атомы водорода вводили расчетным путем с использованием параметров ShelXL по умолчанию и рассматривали как верхние атомы. Все атомы водорода уточняли с изотропными параметрами. R1 (I˃2,00с(I)) составлял 0,0470, и из конечной разностной карты Фурье подтверждали, что не было ни недостающей, ни ошибочной электронной плотности.
PLUTON (Spek, 1991)/ORTEP (Johnson, 1976) использовали для построения Фиг. 7 и 8 (уровень ВЕРОЯТНОСТИ 50%).
[Пример 1]
[0096]
Синтез соединения (3)
[Химическая формула 42]
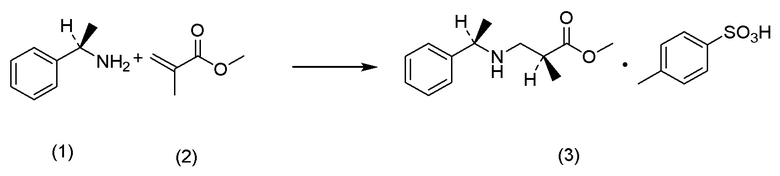
[1] Синтез соединения (3)
Метанол (20 мл) и метилметакрилат (2) (49,64 г, 495,8 ммоль) добавляли к (R)-(+)-1-фенилэтиламину (1) (20,01 г, 165,1 ммоль) при комнатной температуре. После охлаждения до -10°С добавляли хлорид лития (7,07 г, 167 ммоль). Реакционный раствор нагревали до 80°С и перемешивали в течение 6 часов. Реакционный раствор охлаждали до 25°С, добавляли к нему 9,1% водный раствор хлорида натрия (77,03 г) и удаляли водный слой путем разделения жидкостей. К полученному водному слою при комнатной температуре добавляли толуол (61,04 г) и удаляли водный слой путем разделения жидкостей. Полученные два органических слоя объединяли, добавляли толуол (16,99 г) и смесь концентрировали при пониженном давлении при 50°С.
Этанол (16,00 г) и моногидрат п-толуолсульфокислоты (28,91 г, 152,0 ммоль) смешивали вместе с получением этанольного раствора (44,91 г) п-толуолсульфокислоты.
К полученному ранее концентрату добавляли толуол (155,91 г). К смеси при комнатной температуре добавляли этанольный раствор (5,88 г) п-толуолсульфокислоты, полученный выше, и суспензию затравочных кристаллов (19,95 мг, 0,05070 ммоль) в толуоле (63 мкл) с получением суспензии соединения (3). К полученной суспензии добавляли оставшийся этанольный раствор (39,93 г) п-толуолсульфокислоты, полученной выше, и к ней добавляли этанол (10 мл). Полученную смесь перемешивали в течение 2 часов и оставляли на ночь. Смесь охлаждали до 0°С, и перемешивали в течение 2 часов, и твердое вещество собирали фильтрованием с получением соединения (3) (20,75 г, 31,9%) в виде неочищенного продукта.
Толуол (1,92 г), этилацетат (21,70 г) и метанол (2,90 г) добавляли к части (5,00 г) неочищенного продукта соединения (3) и смесь перемешивали при 50°С в течение 3 часов. Смесь охлаждали до 0°С и твердое вещество собирали фильтрованием, в результате чего получали соединение (3) (4,65 г).
Элементный анализ: C 61,22%, H 7,09%, N 3,56%, S 8,12%
1H-ЯМР (ДМСО-d6) δ м.д.: 1,12 (д, J=7,0 Гц, 3H), 1,55 (уш.д, J=6,7 Гц, 3H), 2,29 (с, 3H), 2,50 (с, 2H), 2,83 (уш.дд, J=13,2 Гц, 6,9 Гц, 1H), 2,90 (м, 2H), 3,62 (с, 3H), 7,13 (м, 2H), 7,47 (м, 7H)
[0097]
[2] Синтез затравочного кристалла соединения (3)
(R)-(+)-1-фенилэтиламин (1) (2,00 г, 16,5 ммоль), метанол (1,59 г), метилметакрилат (2) (4,97 г, 49,6 ммоль) и хлорид лития (0,70 г, 17 ммоль) перемешивали при комнатной температуре, и смесь нагревали до 80°С, и перемешивали в течение 4 часов. Реакционный раствор охлаждали до 25°С, добавляли к нему 9,1% водный раствор хлорида натрия (7,70 г) и удаляли водный слой путем разделения жидкостей. После добавления толуола (5,21 г) к полученному органическому слою к нему добавляли метанольный раствор (4,46 г) п-толуолсульфокислоты, полученный растворением моногидрата п-толуолсульфокислоты (2,88 г, 15,1 ммоль) в метаноле (1,58 г). Этот реакционный раствор добавляли к толуолу (6,94 г), охлажденному до 0°С, и смесь перемешивали при 0°С в течение 30 минут. Осажденное твердое вещество собирали фильтрованием с получением затравочного кристалла (1,60 г, 24,6%) соединения (3).
[Пример 1-1]
[0098]
Синтез соединения (3)
[Химическая формула 43]
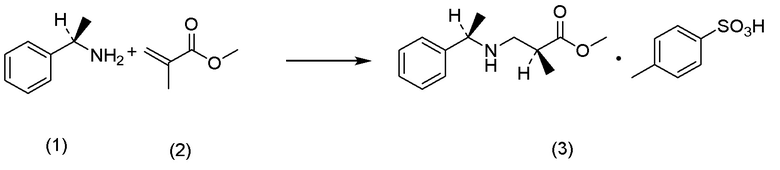
При комнатной температуре моногидрат п-толуолсульфокислоты (1,57 г, 8,25 ммоль) и метилметакрилат (2, 49,57 г, 495,1 ммоль) добавляли к (R)-(+)-1-фенилэтиламину (1) (20,00 г, 165,0 ммоль), и смесь нагревали до 99°С, и перемешивали при 99°С в течение 16 часов. После охлаждения смеси до 25°C к ней добавляли метанол (8 мл) и смешивали с раствором п-толуолсульфокислоты, полученным растворением моногидрата п-толуолсульфокислоты (27,31 г, 143,6 ммоль) в этилацетате (40 мл) и метаноле (4 мл). К этому реакционному раствору добавляли этилацетат (100 мл) при 25°С и выпавшее в осадок твердое вещество собирали фильтрованием с получением соединения (3) (28,14 г) в виде неочищенного продукта.
Этилацетат (15 мл) и метанол (30 мл) добавляли к неочищенному продукту (28,14 г) соединения (3), и смесь нагревали до 60°С, и затем охлаждали до 40°С. Суспензию затравочных кристаллов (20,0 мг, 0,308 ммоль) соединения (3) в этилацетате (60 мкл) добавляли к вышеуказанной смеси при 40°С. Затем полученную суспензию перемешивали при 40°С в течение 30 минут, затем охлаждали до 22°С, к ней добавляли этилацетат (218 мл), и смесь охлаждали до 0°С, и перемешивали в течение 1 часа. Осажденное твердое вещество собирали фильтрованием с получением соединения (3) (22,29 г, 34,32%).
[Пример 2]
[0099]
Синтез соединения (4)
[Химическая формула 44]
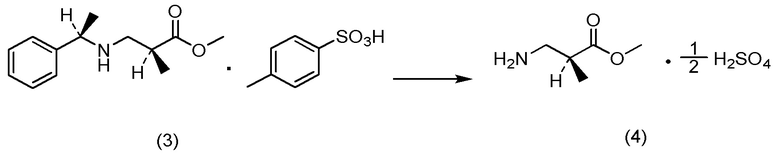
Толуол (95,26 г) и воду (44,00 г) добавляли к соединению (3) (22,00 г, 55,91 ммоль) так, чтобы соединение (3) суспендировалось, к нему добавляли 8% водный раствор гидроксида натрия (27,54 г) и воду (4,40 г) и удаляли водный слой путем разделения жидкостей. К полученному органическому слою добавляли воду (11,00 г) и водный слой удаляли путем разделения жидкостей. Процедуру концентрирования полученного органического слоя при пониженном давлении при 50°С и добавления метанола повторяли так, чтобы растворитель заменился метанолом. К полученному концентрированному раствору добавляли концентрированную серную кислоту (2,04 г, 19,8 ммоль), 10% палладий-на-угле (2,20 г, приблизительно 40% влажности) и метанол (17,41 г). Реакционный раствор нагревали до 40°С и перемешивали в течение 90 минут в атмосфере водорода. Палладий-на-угле удаляли фильтрованием и к полученному фильтрату добавляли метанол (52,24 г) и концентрированную серную кислоту (0,67 г, 6,5 ммоль). Процедуру добавления ацетонитрила к полученному реакционному раствору и концентрирования при пониженном давлении повторяли так, чтобы растворитель заменился ацетонитрилом, и реакционный раствор охлаждали до 0°С. Осажденное твердое вещество собирали фильтрованием с получением соединения (4) (8,58 г, 92,3%).
Элементный анализ: C 35,72%, H 7,18%, N 8,55%, S 9,63%
1H-ЯМР (ДМСО-d6) δ м.д.: 1,10 (д, J=7,1 Гц, 3H), 2,62 (м, 1H), 2,75 (дд, J=12,7 Гц, 5,9 Гц, 1H), 2,89 (дд, J= 12,7 Гц, 7,3 Гц, 1H), 3,63 (с, 3H)
[0100]
(Ссылочный пример 1) Синтез соединения (5)
[Химическая формула 45]
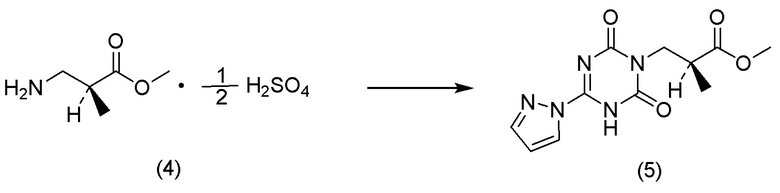
Соединение (4) (19,00 г, 114,3 ммоль) суспендировали в ацетонитриле (45,00 г). При 2°C к нему добавляли 1,8-диазабицикло[5.4.0]-7-ундецен (19,10 г, 125,5 ммоль) и ацетонитрил (3,00 г) и смесь перемешивали при 2°C в течение 30 минут. Реакционный раствор добавляли при 2°С к суспензии, полученной суспендированием N, N-карбонилдиимидазола (21,30 г, 131,4 ммоль) в ацетонитриле (75,00 г). К реакционному раствору добавляли ацетонитрил (15,00 г) и смесь перемешивали при 2°C в течение 1 часа 22 минут. При 2°C к реакционному раствору добавляли 1,8-диазабицикло[5.4.0]-7-ундецен (17,40 г, 114,3 ммоль) и ацетонитрил (3,00 г) и смесь охлаждали до 1°C. К реакционному раствору добавляли 1H-пиразол-1-карбоксамидина гидрохлорид (16,80 г, 114,6 ммоль) и ацетонитрил (3,00 г). Реакционный раствор нагревали до 60°С и перемешивали в течение 2 часов 10 минут. Реакционный раствор охлаждали до 20°С. При 2°С к реакционному раствору добавляли 1,8-диазабицикло[5.4.0]-7-ундецен (27,80 г, 182,6 ммоль) и ацетонитрил (3,00 г) и смесь охлаждали до -10°С. К реакционному раствору добавляли N, N-карбонилдиимидазол (29,70 г, 183,2 ммоль) и ацетонитрил (3,00 г). Реакционный раствор перемешивали при 2°С в течение 1 часа 20 минут. К реакционному раствору добавляли метанол (7,50 г), уксусную кислоту (4,80 г, 79,9 ммоль) и ацетонитрил (3,00 г) при 2°C. Реакционный раствор концентрировали при пониженном давлении при 50°С. К полученному концентрированному раствору добавляли N, N-диметилацетамид (27,00 г), смесь охлаждали до 10°C и к ней добавляли 17%-ный водный раствор серной кислоты (204,1 г) и воду (19,00 г). К реакционному раствору добавляли 17%-ный водный раствор серной кислоты (31,30 г) и воду (2,50 г) при 25°C и смесь перемешивали в течение 1 часа 48 минут. Реакционный раствор концентрировали при пониженном давлении при 50°С. К полученному концентрированному раствору добавляли воду (190 мл), и охлаждали до 2°С, и затем к нему добавляли 17%-ный водный раствор серной кислоты (3,30 г) и воду (1,30 г). Реакционный раствор перемешивали при 2°C в течение 1 часа 15 минут и выпавшее в осадок твердое вещество собирали фильтрованием с получением соединения (5) (27,13 г, 85,0%).
1H-ЯМР (CDCl3) δ м.д.: 1,24 (д, J=7,1 Гц, 3H), 3,02 (м, 1H), 3,68 (с, 3H), 4,02 (дд, J=13,4 Гц, 6,3 Гц, 1H), 4,24 (дд, J=13,3 Гц, 8,4 Гц, 1H), 6,60 (дд, J=2,8 Гц, 1,6 Гц, 1H), 7,85 (д, J=1,6 Гц, 1H), 8,48 (дд, J=2,9 Гц, 0,6 Гц, 1H), 9,70 (уш.с, 1H)
[0101]
(Справочный пример 2) Синтез соединения (8)
[Химическая формула 46]
[1] Синтез соединения (8)
Трет-бутоксид натрия (12,50 г, 130,1 моль) суспендировали в N-метил-2-пирролидоне (64,00 г) и к нему добавляли 4-аминофенол (7) (14,10 г, 129,2 ммоль) и N-метил-2-пирролидон (16,00 г). Реакционный раствор нагревали до 100°С и к нему добавляли 2-бромпиридин (6) (19,50 г, 123,4 ммоль) и N-метил-2-пирролидон (4,00 г). Реакционный раствор перемешивали при 115°С в течение 8 часов 20 минут и охлаждали до 50°С. К реакционному раствору добавляли воду (29,00 г) при 50°С, реакционный раствор охлаждали до 25°С и к нему добавляли воду (107,00 г). К реакционному раствору добавляли затравочный кристалл (8, 20 мг) соединения (8) и воду (195 мг) и смесь перемешивали при 20°С в течение 50 минут. К реакционному раствору добавляли воду (156,00 г) при 25°С, реакционный раствор охлаждали до 5°С и перемешивали в течение 1 часа 30 минут. Осажденное твердое вещество собирали фильтрованием с получением соединения (8) (17,81 г, 77,5%).
1H-ЯМР (CDCl3) δ м.д.: 3,60 (с, 2H), 6,69-6,73 (м, 2H), 6,83 (ддд, J=8,4 Гц, 0,8 Гц, 0,8 Гц, 1H), 6,92-6,96 (м, 3H), 7,63 (ддд, J=8,0 Гц, 7,2 Гц, 2,0 Гц, 1H), 8,18 (ддд, J=5,2 Гц, 2,0 Гц, 0,8 Гц, 1H)
[0102]
[2] Синтез затравочного кристалла соединения (8)
Трет-бутоксид натрия (3,20 г, 33,3 ммоль) суспендировали в N-метил-2-пирролидоне (16,42 г) и к нему добавляли 4-аминофенол (7) (3,64 г, 33,4 ммоль) и N-метил-2-пирролидон (4,12 г). Реакционный раствор нагревали до 100°С и к нему добавляли 2-бромпиридин (6) (5,01 г, 31,7 ммоль) и N-метил-2-пирролидон (1,10 г). Реакционный раствор перемешивали при 115°С в течение 6 часов и к нему добавляли трет-бутоксид натрия (1,07 г, 11,1 ммоль). Смесь перемешивали при 115°С в течение 2 часов 35 минут и затем охлаждали до 50°С. К реакционному раствору добавляли воду (7,50 г) при 50°С и затем реакционный раствор охлаждали до 25°С. К нему добавляли воду (67,54 г) и охлаждали до 1°C для кристаллизации. После перемешивания полученной суспензии при 5°С в течение 30 минут выпавшее в осадок твердое вещество собирали фильтрованием с получением затравочного кристалла (3,84 г, 65,2%) соединения (8).
[0103]
(Ссылочный пример 3) Синтез соединения (9)
[Химическая формула 47]
[1] Синтез соединения (9)
Бромид натрия (4,1 г, 39,9 ммоль) и N, N-диметилацетамид (27,70 г) добавляли к соединению (5) (10,13 г, 36,27 ммоль). К реакционному раствору добавляли N,N-диизопропилэтиламин (5,16 г, 39,9 ммоль) и N,N-диметилацетамид (0,96 г) и смесь нагревали до 75°C. К реакционному раствору добавляли при 75°C раствор 4-хлорбензилхлорида в N,N-диметилацетамиде, полученный растворением 4-хлорбензилхлорида (6,43 г, 39,9 ммоль) в N, N-диметилацетамиде (9,56 г) и добавляли N, N-диметилацетамид (9,56 г). Реакционный раствор перемешивали при 75°С в течение 5 часов 15 минут. Реакционный раствор охлаждали до 25°С, добавляли к нему уксусную кислоту (0,65 г, 11 ммоль) и смесь нагревали до 40°С. К реакционному раствору добавляли раствор соединения (8) в N, N-диметилацетамиде, полученный растворением соединения (8) (7,43 г, 39,9 ммоль) в N,N-диметилацетамиде (9,55 г), и добавляли N,N-диметилацетамид (9,55 г). Реакционный раствор перемешивали при 40°С в течение 3 часов и охлаждали до комнатной температуры. К реакционному раствору добавляли ацетон (27,94 г) и воду (35,46 г). К реакционному раствору добавляли затравочный кристалл (10,13 мг) соединения (9), воду (0,40 г) и ацетон (0,08 г), и смесь перемешивали при комнатной температуре в течение 3 часов 25 минут, и затем оставляли на ночь. После этого реакционный раствор перемешивали при комнатной температуре в течение 1 часа, затем к нему добавляли воду (30,39 г) и смесь перемешивали в течение 3 часов 25 минут. Осажденное твердое вещество собирали фильтрованием с получением соединения (9) (16,72 г, 88,3%).
1H-ЯМР (CDCl3) δ м.д.: 1,19 (д, J=7,1 Гц, 3H), 2,91 (м, 1H), 3,61 (с, 3H), 3,90 (дд, J=13,6 Гц, 6,2 Гц, 1H), 4,12 (дд, J=13,6 Гц, 8,4 Гц, 1H), 5,18 (д, J=14,2 Гц, 1H), 5,22 (д, J=14,2 Гц, 1H), 6,85 (м, 2H), 6,96 (м, 1H), 7,00 (м, 1H), 7,14 (м, 2H), 7,31 (м, 2H), 7,50 (м, 2H), 7,70 (м, 1H), 7,89 (уш.с, 1H), 8,14 (м, 1H)
[0104]
[2] Синтез затравочного кристалла соединения (9)
Бромид натрия (2,00 г, 19,4 ммоль) и N, N-диметилацетамид (13,67 г) добавляли к соединению (5) (5,01 г, 17,9 ммоль). К реакционному раствору добавляли N,N-диизопропилэтиламин (2,55 г, 19,7 ммоль) и N,N-диметилацетамид (0,47 г) и смесь нагревали до 75°C. К реакционному раствору при 75°C добавляли раствор 4-хлорбензилхлорида в N,N-диметилацетамиде, полученный растворением 4-хлорбензилхлорида (3,16 г, 19,6 ммоль) в N,N-диметилацетамиде (4,71 г), и добавляли N,N-диметилацетамид (4,71 г). Реакционный раствор перемешивали при 75°С в течение 4 часов 30 минут. Реакционный раствор охлаждали до 25°С, добавляли к нему уксусную кислоту (0,32 г, 5,3 ммоль) и смесь нагревали до 40°С. К реакционному раствору добавляли раствор соединения (8) в N,N-диметилацетамиде, полученный растворением соединения (8) (3,66 г, 19,7 ммоль) в N,N-диметилацетамиде (4,71 г), и добавляли N,N-диметилацетамид (4,71 г). Реакционный раствор перемешивали при 40°С в течение 3 часов 25 минут и охлаждали до комнатной температуры. К реакционному раствору добавляли ацетон (13,79 г) и воду (17,54 г). Реакционный раствор оставляли стоять в течение ночи при комнатной температуре. После этого реакционный раствор нагревали до 25°С и перемешивали в течение 5 часов, затем к нему добавляли воду (15,00 г) и смесь перемешивали при 25°С в течение 2 часов. Осажденное твердое вещество собирали фильтрованием с получением затравочного кристалла соединения (9) (8,17 г, 87,2%).
[Пример 3]
[0105]
Синтез соединения, представленного Формулой (I)
[Химическая формула 48]
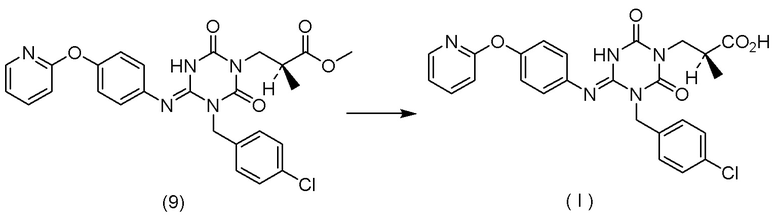
[1] Синтез соединения, представленного Формулой (I)
К соединению (9) (70,00 г, 134,1 ммоль) добавляли 2-пропанол (109,91 г), воду (63,00 г) и 48% водный раствор гидроксида натрия (27,94 г, 335,3 ммоль). Реакционный раствор нагревали до 35°С и перемешивали в течение 4 часов 10 минут. К реакционному раствору добавляли 2-пропанол (32,97 г), метанол (177,30 г) и воду (63,00 г) и смесь нагревали до 50°С. К реакционному раствору добавляли муравьиную кислоту (18,52 г, 402,3 ммоль) и затравочный кристалл соединения, представленного Формулой (I) (70,00 мг), и смесь перемешивали при 50°С в течение 1 часа 10 минут. После этого к нему добавляли воду (280,00 г) и смесь охлаждали до 25°С. Осажденное твердое вещество собирали фильтрованием с получением безводной кристаллической Формы I соединения, представленного Формулой (I) (62,86 г, 92,3%).
1H-ЯМР (CDCl3) δ м.д.: 1,13 (д, J=7,0 Гц, 3H), 2,76 (м, 1H), 3,83 (дд, J=13,5 Гц, 6,1 Гц, 1H), 4,03 (дд, J=13,5 Гц, 8,5 Гц, 1H), 5,14 (м, 1H), 5,25 (д, J=14,4 Гц, 1H), 6,82 (д, J=8,6 Гц, 2H), 7,00 (м, 2H), 7,08 (м, 2H), 7,25 (м, 2H), 7,43 (д, J=8,3 Гц, 2H), 7,72 (м, 1H), 8,06 (дд, J=5,4 Гц, 1,8 Гц, 1H), 8,67 (уш.с, 1H)
[0106]
[2] Синтез затравочного кристалла соединения, представленного Формулой (I)
К соединению (9) (1,50 г, 2,87 ммоль) добавляли метанол (5,95 г), воду (3,00 г) и 48% водный раствор гидроксида натрия (0,60 г, 7,20 ммоль). Реакционный раствор нагревали до 40°С и перемешивали в течение 1 часа 30 минут. Реакционный раствор охлаждали до комнатной температуры, добавляли к нему при комнатной температуре муравьиную кислоту (0,40 г, 8,62 ммоль), этилацетат (10,5 мл) и воду (9 мл) и удаляли водный слой путем разделения жидкостей. К полученному органическому слою добавляли воду (3 мл), водный слой удаляли путем разделения жидкостей, к органическому слою добавляли 2-пропанол (90 мл) и смесь концентрировали при пониженном давлении при 40°С. К полученному концентрированному остатку добавляли воду (7,5 мл) и 2-пропанол (7,5 мл) и смесь перемешивали при 25°C в течение 1 часа 30 минут. Добавляли воду (7,5 мл) и метанол (7,5 мл), затем смесь нагревали до 60°С, и перемешивали в течение 2 часов, и охлаждали до 25°С. Осажденное твердое вещество собирали фильтрованием с получением затравочного кристалла (10, 1,25 г, 85,6%) безводной кристаллической Формы I кристаллического вещества соединения, представленного Формулой (I).
[0107]
Результаты порошковой рентгеновской дифракции безводной кристаллической Формы I соединения, представленного Формулой (I), показаны на Фиг. 1 (Способ 1).
В спектре порошковой рентгеновской дифракции пики распознаются при:
углах дифракции (2θ): 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2° или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2°.
В спектре порошковой рентгеновской дифракции пики при следующих углах дифракции (2θ) являются в частности характеристическими для безводной кристаллической Формы I соединения, представленного Формулой (I):
15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и
25,4°±0,2° или
7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2°.
[0108]
Результаты Раман-спектра безводной кристаллической Формы I соединения, представленного Формулой (I), показаны на Фиг. 2 (Способ 1).
Основные пики поглощения распознаются при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1093 см-1±2 см-1, 1128 см-1±2 см-1, 1243 см-1±2 см-1, 1370 см-1±2 см-1, 1599 см-1±2 см-1, 1659 см-1±2 см-1, 1735 см-1±2 см-1, 2938 см-1±2 см-1 и 3067 см-1±2 см-1.
[0109]
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пики поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пик поглощения при 829 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пик поглощения при 989 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пик поглощения при 1013 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пик поглощения при 1128 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет пик поглощения при 1370 см-1±2 см-1.
В одном варианте осуществления безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет один или более пиков поглощения, выбранных из группы, состоящей из пика поглощения при 829 см-1±2 см-1, пика поглощения при 989 см-1±2 см-1, пика поглощения при 1013 см-1±2 см-1, пика поглощения при 1128 см-1±2 см-1 и пика поглощения при 1370 см-1±2 см-1.
[0110]
Результаты ДСК анализа безводной кристаллической Формы I соединения, представленного Формулой (I), показаны на Фиг. 3. Начальная температура составляла приблизительно 196°С.
[0111]
Результаты анализа структуры монокристалла безводной кристаллической Формы I соединения, представленного Формулой (I), показаны ниже.
Кристаллографические данные представлены в Таблице 4.
[Таблица 4]
В настоящем описании V представляет собой объем элементарной решетки, и Z представляет собой количество молекул в элементарной решетке.
[0112]
Атомные координаты неводородных атомов показаны в Таблицах 5-7. В настоящем описании U (экв) означает эквивалентный изотропный температурный фактор.
[Таблица 5]
[Таблица 6]
[0114]
[Таблица 7]
[0115]
Атомные координаты атомов водорода приведены в Таблицах 8 и 9. В настоящем описании U (изо) означает изотропный температурный фактор. Количества атомов водорода в Таблицах 8 и 9 были присвоены по отношению к количеству неводородных атомов, связанных с ними.
[Таблица 8]
[0116]
[Таблица 9]
[0117]
Кроме того, расстояния между атомными связями (единица измерения: ангстрем) показаны в Таблицах 10-11.
[Таблица 10]
[0118]
[Таблица 11]
[0119]
В безводной кристаллической Форме I соединения, представленного Формулой (I), две молекулы соединения, представленного Формулой (I), находились в виде асимметричной структуры. Схемы молекулярной структуры соединения, представленного Формулой (I), показаны на Фиг. 7 и 8, соответственно.
Количество неводородных атомов в Таблицах 5-7 и Таблицах 10 и 11 соответствует количеству, показанному на Фиг. 7 и 8, соответственно.
[0120]
Как показано в Таблицах 10-11, длина связи C12-N2 составляла приблизительно 1,26 Å и длина связи C37-N7 составляла приблизительно 1,27 Å.
Поскольку длина связи C12-N2 и длина связи C37-N7 короче, чем длина связи C12-N3 (приблизительно 1,39 Å) и длина связи C37-N8 (приблизительно 1,37 Å), соединения, представленные Формулой (I), в безводной кристаллической Форме I были идентифицированы наличием иминоструктуры:
[Химическая формула 49]
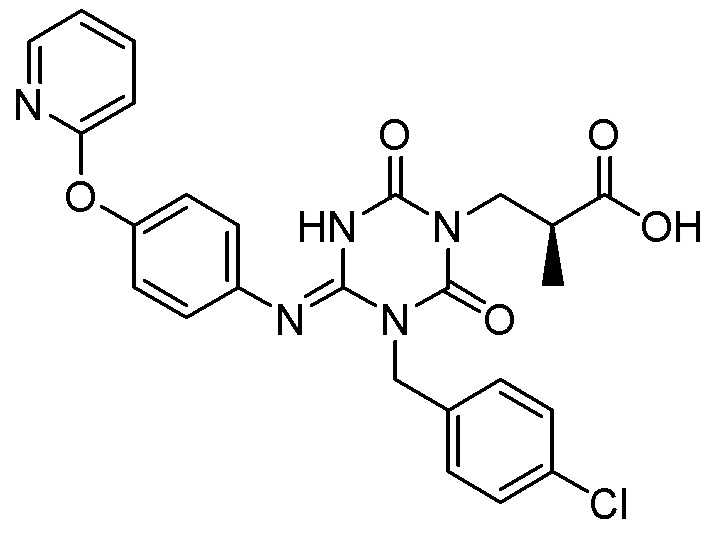
[Пример 4]
[0121]
(Эффект, достигаемый добавкой для ускорения реакции аза-присоединения по Михаэлю)
[Химическая формула 50]
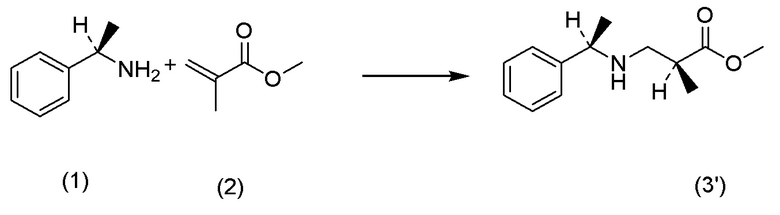
Реакция, подобная вышеприведенной схеме, описана в Tetrahedron Asymmetry, Vol. 7, No. 3, pp. 699-708, 1996 (Непатентный документ 16). В этой литературе продукт реакции аза-присоединения по Михаэлю получают путем нагревания и кипячения с обратным холодильником в течение 9 дней с использованием метанола в качестве растворителя реакции (выход 74%, диастереомерная смесь 1:1). Метилметакрилат (соединение 2), который является исходным материалом, представляет собой соединение, используемое в качестве исходного материала для синтеза полимера, и может полимеризоваться при воздействии реакции при высокой температуре в течение длительного времени. Следовательно, эти условия реакции не подходят для промышленного способа.
С другой стороны, было обнаружено, что в вышеописанной реакции аза-присоединения по Михаэлю, когда хлорид лития, хлорид кальция, хлорид магния, бромид лития, п-толуолсульфокислоту, метансульфокислоту или трифторметансульфокислоту использовали в качестве добавки, реакция была ускорена, как показано в Таблице 12 ниже.
[Таблица 12]
(ч)
(%)⃰
Таким же образом, как в Примере 1, реакции проводили при соответствующих условиях, указанных в Таблице 12 выше. Каждый реакционный раствор отбирали при перемешивании, взвешивали приблизительно 100 мг реакционного раствора, добавляли метанол для жидкостной хроматографии для разбавления раствора до 100 мл, вводили 10 мкл разбавленного раствора и проводили ВЭЖХ (Способ А).
⃰ Превр. (%) рассчитывали по следующей формуле.
[Математическая формула 1]

В настоящем описании A1, A3' и A3" представляют собой площади пиков при измерении ВЭЖХ соответственно.
A1: Соединение (1)
A3': Соединение (3')
A3'': соединение (3'')
Соединение (3'') имеет следующую химическую структурную формулу.
[Химическая формула 51]
До сих пор были известны только условия реакции при нагревании и кипячении с обратным холодильником в течение 9 дней, как описано в Непатентном документе 16, но реакция ускоряется в присутствии этих добавок и завершается за короткое время от 1 до 7 часов, так что можно сказать, что настоящий технологический способ является промышленно превосходным технологическим способом.
[0122]
В Непатентном документе 16 после завершения реакции аза-присоединения по Михаэлю (S)-1-фенилэтиламина ((S)-α-метилбензиламина) и метилметакрилата добавляют п-толуолсульфокислоту с получением соли п-толуолсульфокислоты. С другой стороны, в настоящей заявке после завершения реакции аза-присоединения по Михаэлю (R)-1-фенилэтиламина ((R)-(+)-1-фенилэтиламина) и метилметакрилата добавляют п-толуолсульфокислоту для получения п-толуолсульфокислоты. То есть в Непатентном документе 16 получена соль п-толуолсульфокислоты энантиомера Формулы (IV-B) настоящей заявки, и таким образом настоящее изобретение не описано в Непатентном документе 16:
[Химическая формула 52]
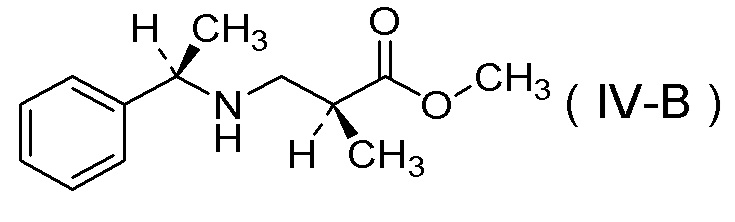
[Пример 5]
[0123]
(Сравнение выходов выделения сульфата и гидрохлорида соединения (4'))
[Химическая формула 53]
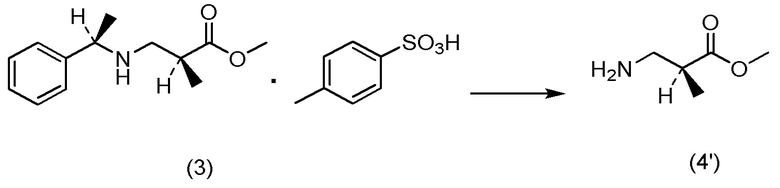
К фильтрату после удаления палладия-на-угле добавляли метанол и 4 моль/л раствор хлористоводородной кислоты-этилацетата таким же образом, как в технологическом способе, описанном в Примере 2. В качестве кристаллизационного растворителя добавляли этилацетат и повторяли процедуру концентрации при пониженном давлении. Растворитель заменяли этилацетатом и смесь охлаждали до 0°С. Осажденное твердое вещество собирали фильтрованием с получением гидрохлорида соединения (4'). Сульфат соединения (4') синтезировали в соответствии с Примером 2, описанным выше.
В настоящем описании, что касается выхода при выделении гидрохлорида соединения (4') или сульфата соединения (4') и фильтратов после сбора осажденного твердого вещества путем фильтрации, взвешивали приблизительно 200 мг каждого и добавляли 15 мл ацетонитрила и 45 мкл триэтиламина для жидкостной хроматографии, затем добавляли 15 мкл бензоилхлорида и добавляли ацетонитрил для жидкостной хроматографии, чтобы разбавить каждую смесь до 20 мл, затем вводили 10 мкл смеси и проводили ВЭЖХ (Способ B). Отдельно в качестве стандартного раствора для расчета концентрации взвешивали приблизительно 10 мг выделенного твердого вещества гидрохлорида соединения (4') или выделенного твердого вещества сульфата соединения (4') и добавляли 15 мл ацетонитрила и 45 мкл триэтиламина для жидкостной хроматографии, затем добавляли 15 мкл бензоилхлорида и добавляли ацетонитрил для жидкостной хроматографии, чтобы разбавить смесь до 20 мл, затем вводили 10 мкл смеси, проводили ВЭЖХ (Способ B) и концентрацию соединения (4'), содержащегося в фильтрате, рассчитывали по следующей формуле.
[Математическая формула 2]

В настоящем описании MS, ML, AS и AL представляют собой площади пиков при измерении ВЭЖХ, соответственно.
MS: взвешенное количество (мг) выделенного твердого вещества гидрохлорида соединения (4') или сульфата соединения (4') при измерении ВЭЖХ
ML: взвешенное количество (мг) фильтрата при измерении ВЭЖХ
AS: площадь пика выделенного твердого вещества гидрохлорида соединения (4') или сульфата соединения (4') при измерении ВЭЖХ
AL: площадь пика фильтрата при измерении ВЭЖХ
[Таблица 13]
Как видно из Таблицы 13, при выделении соединения (4') в виде гидрохлорида (ввод 1-6) выход при выделении варьировался от приблизительно 77% до приблизительно 88%. Кроме того, было подтверждено, что в фильтрате элюируется от приблизительно 3% до приблизительно 10% свободной формы (соединение (4')).
С другой стороны, когда соединение (4') было выделено в виде сульфата (ввод 7-10), был достигнут высокий выход при выделении от приблизительно 92% до приблизительно 94%. Доля свободной формы, элюируемой в фильтрат, составляла от приблизительно 2 до приблизительно 3%.
Из вышеприведенных результатов можно сказать, что способ получения соединения (4') в виде сульфата представляет собой промышленно превосходный технологический способ, поскольку количество свободной формы, элюируемой в фильтрате, невелико, а это означает, что потери во время способа являются незначительными.
[Пример 6]
[0124]
(Эффект подавления рацемизации)
[Химическая формула 54]
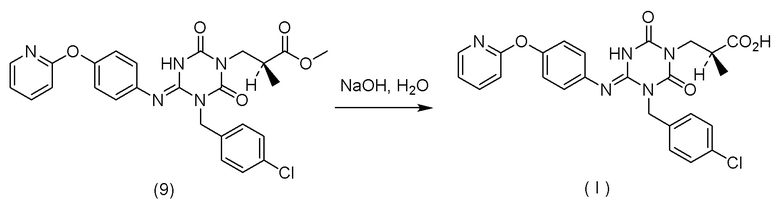
Патентные документы 6, 7, 8 и 9 раскрывают способ получения карбоновой кислоты путем гидролиза сложного эфира, и в качестве растворителя используют диоксан, ТГФ, ДМСО, смешанный раствор МеОН-ТГФ, смешанный раствор ТГФ-EtOH-вода, смешанный раствор МеОН-вода, смешанный раствор МеОН-ТГФ-вода или подобные.
С другой стороны, когда изопропиловый спирт, ТГФ или трет-BuOH использовали в качестве реакционного растворителя в реакции гидролиза, было обнаружено, что рацемизация продукта подавляется, как показано в Таблице 14 ниже.
[Таблица 14]
(ч)
(°C)
Таким же образом, как в Примере 3, реакции проводили при соответствующих условиях, указанных в таблице выше. Взвешивали приблизительно 100 мг каждого реакционного раствора, к нему добавляли метанол для жидкостной хроматографии, чтобы разбавить раствор до 20 мл, вводили 10 мкл разбавленного раствора и проводили ВЭЖХ (Способ С).
⃰Количество R-формы соединения (9) рассчитывали по следующей формуле. Единица представляет собой площадь пика (%).
[Математическая формула 3]
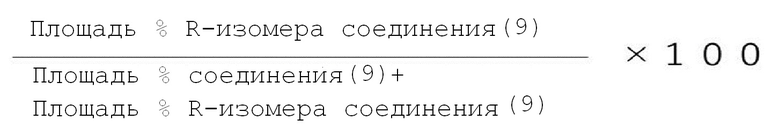
⃰⃰Количество R-изомера соединения, представленного Формулой (I), рассчитывали по следующей формуле. Единица представляет собой площадь пика (%).
[Математическая формула 4]
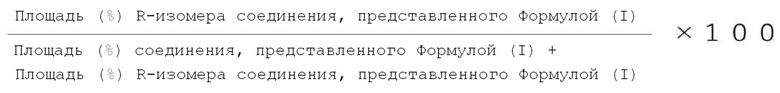
Структурные формулы R-изомера соединения (9) и R-изомера соединения, представленного Формулой (I), являются следующими. Молекулярные структуры (аминоформа/иминоформа) R-изомера соединения (9) и R-изомера соединения, представленного Формулой (I), не определены.
[Химическая формула 55]
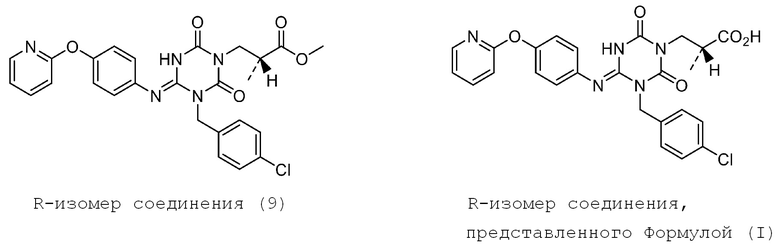
Как описано в Таблице 14 выше, было обнаружено, что при использовании метанола в качестве реакционного растворителя происходила рацемизация и количество оптических изомеров увеличивалось от 0,43% до 0,93%. Также в случае этанола количество оптических изомеров подобным образом увеличивалось от 0,43% до приблизительно 0,7%. С другой стороны, при использовании изопропилового спирта, тетрагидрофурана и трет-бутанола оно увеличивалось до приблизительно 0,5%.
Из вышеприведенных результатов стало ясно, что при использовании изопропилового спирта, тетрагидрофурана и трет-бутанола соотношение немного увеличивалось по сравнению с соотношением R-изомера, изначально содержащегося в соединении (9) в качестве исходного вещества, и подавлялась рацемизация по сравнению со случаем, когда в качестве реакционных растворителей использовали метанол и этанол. Таким образом, можно сказать, что настоящий технологический способ является промышленно превосходным технологическим способом.
[Пример 7]
[0125]
[1] Синтез кристалла дигидрата соединения, представленного Формулой (I)
К безводной кристаллической Форме I (50,00 г, 98,43 ммоль) соединения, представленного Формулой (I), добавляли 2-пропанол (314,02 г), воду (150,00 г) и 48% гидроксид натрия (20,51 г, 246,1 ммоль) и растворяли. К полученному раствору добавляли 35% хлористоводородную кислоту (25,64 г, 246,1 ммоль) и затравочный кристалл дигидрата соединения, представленного Формулой (I) (50,00 мг), затем смесь перемешивали при комнатной температуре в течение 1 часа, и добавляли воду (250,00 г), и перемешивали в течение 2 часов. Полученную суспензию охлаждали до 5°С и фильтровали с получением кристалла дигидрата (49,23 г) соединения, представленного Формулой (I).
Кристалл дигидрата подтверждали термогравиметрическим/дифференциально-термическим анализом (ТГ/ДТА), измерением поглощения и десорбции влаги (DVS) и измерением порошковой рентгеновской дифракции.
[0126]
Порошковая рентгеновская дифрактограмма кристалла дигидрата соединения, представленного Формулой (I), показана на Фиг. 4 (Способ 1).
Пики при углах дифракции 5,7°±0,2°, 7,7°±0,2°, 11,8°±0,2°, 15,2°±0,2°, 17,7°±0,2°, 20,6°±0,2°, 20,8°±0,2°, 26,5°±0,2°, 27,1°±0,2° и 29,1°±0,2° наиболее характерны для кристалла дигидрата соединения, представленного Формулой (I).
[0127]
Результаты Раман-спектра кристалла дигидрата соединения, представленного Формулой (I), показаны на Фиг. 6 (Способ 2).
Основные пики поглощения распознаются при 871 см-1±2 см-1, 996 см-1±2 см-1, 1093 см-1±2 см-1, 1114 см-1±2 см-1, 1234 см-1±2 см-1, 1248 см-1±2 см-1, 1340 см-1±2 см-1, 1577 см-1±2 см-1, 1603 см-1±2 см-1, 1662 см-1±2 см-1, 1738 см-1±2 см-1, 2971 см-1±2 см-1 и 3073 см-1±2 см-1.
[0128]
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пики поглощения при 871 см-1±2 см-1, 996 см-1±2 см-1, 1114 см-1±2 см-1, 1234 см-1±2 см-1, 1340 см-1±2 см-1 и 1577 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 871 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 996 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 1114 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 1234 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 1340 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет пик поглощения при 1577 см-1±2 см-1.
В одном варианте осуществления кристалл дигидрата соединения, представленного Формулой (I), имеет один или более пиков поглощения, выбранных из группы, состоящей из пика поглощения при 871 см-1±2 см-1, пика поглощения при 996 см-1±2 см-1, пика поглощения при 1114 см-1±2 см-1, пика поглощения при 1234 см-1±2 см-1, пика поглощения при 1340 см-1±2 см-1 и пика поглощения при 1577 см-1±2 см-1.
[0129]
Результаты термогравиметрического/дифференциального термического анализа (ТГ/ДТА) кристалла дигидрата соединения, представленного Формулой (I), показаны на Фиг. 5. В результате от приблизительно 55°C до приблизительно 85°C была подтверждена потеря массы 6,4% с эндотермическим пиком. Поскольку теоретическое значение содержания влаги кристалла дигидрата соединения, представленного Формулой (I), составляло 6,6%, было подтверждено, что оно представляет собой кристалл дигидрата соединения, представленного Формулой (I).
[0130]
[2] Синтез затравочного кристалла дигидрата соединения, представленного Формулой (I)
К соединению (9) (70,00 г, 134,1 ммоль) добавляли 2-пропанол (109,91 г), воду (63,02 г) и 48% гидроксид натрия (27,95 г, 335,4 ммоль) и смесь перемешивали при 25°С в течение 4 часов. К полученному реакционному раствору добавляли 2-пропанол (16,49 г), метанол (127,43 г) и воду (217,00 г), затем к нему добавляли муравьиную кислоту (9,88 г, 215 ммоль) при 25°С и смесь перемешивали при 25°С в течение 35 минут. К полученной суспензии по каплям добавляли водный раствор муравьиной кислоты, полученный смешиванием муравьиной кислоты (8,64 г, 188 ммоль) и воды (70,00 г) при 25°C, и затем добавляли воду (7,00 г) и метанол (27,70 г). Полученную суспензию отфильтровывали с получением затравочного кристалла дигидрата (64,04 г) соединения, представленного Формулой (I).
[0131]
(Ссылочный пример 4)
(Синтез Соединения I-127 описан в Патентном документе 9)
Как описано выше, каждая стадия получения соединения I-127 конкретно не описана в Патентном документе 9. Соединение I-127 синтезировали тем же способом, что и аналогичное соединение соединения I-127 (Ссылочный пример 3 Патентного документа 9). Ниже показана только конечная стадия.
[Химическая формула 56]
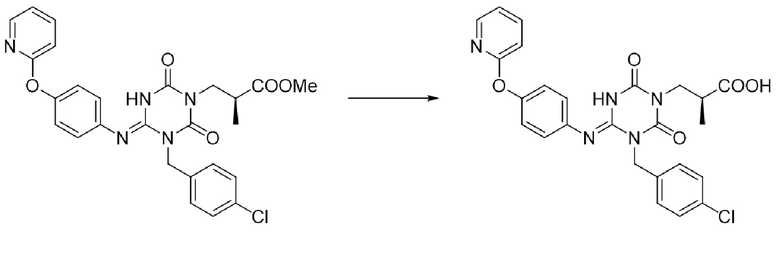
Метил-(S,E)-3-(3-(4-хлорбензил)-2,6-диоксо-4-((4-(пиридин-2-илокси)фенил)имино)-1,3,5-триазинан-1-ил)-2-метилпропаноат (0,365 г, 0,7 ммоль), MeOH (1 мл), ТГФ (1 мл), H2O (1 мл) и 4 моль/л водный раствор LiOH (0,7 мл, 2,80 ммоль) смешивали и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор добавляли к полунасыщенному солевому раствору (100 мл) и 5% раствору лимонной кислоты и смесь экстрагировали этилацетатом (100 мл), затем органический слой промывали полунасыщенным солевым раствором (100 мл). Органический слой высушивали над сульфатом магния и затем отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат/гексан=50:50 до этилацетат/гексан=80:20), затем перекристаллизовывали со смешанным растворителем этилацетат/гексан, собирали фильтрованием и затем высушивали при пониженном давлении при 80°С в течение 3 часов с получением белого порошка.
1H ЯМР (ДМСО-d6)δ: 0,86 (1,5H, т, J=6,8 Гц, гексан), 1,03 (3H, д, J=6,8 Гц), 1,18 (0,75H, т, J=7,4 Гц, AcOEt), 1,25 (2H, уш.с, гексан), 1,99 (0,75H, с, AcOEt), 2,76 (1H, т, J=7,2 Гц), 3,81 (1H, т, J=10,5 Гц), 3,95 (1H, т, J= 10,2 Гц), 4,03 (0,5H, кв, J=7,4 Гц, AcOEt), 5,29 (2H, с), 7,02-7,12 (4H, м), 7,36-7,45 (6H, м), 7,85 (1H, т, J=7,5 Гц), 8,16 (1H, с), 9,34 (1H, уш.с).
ЖХ/МС m/z 508 [M+] Время удерживания 2,02 мин.
В настоящем описании ЖХ/МС измеряли в условиях Способа 1 Патентного документа 9.
[0132]
Результаты 1H-ЯМР показаны на Фиг. 9.
На диаграмме спектра ЯМР распознаются пики этилацетата и гексана. Поскольку полученный кристалл высушивали при пониженном давлении при 80°С в течение 3 часов, предполагалось, что полученный кристалл представлял собой не адгезионный растворитель на поверхности кристалла, а этилацетат и гексан, содержащиеся в кристаллической решетке. В ТГ/ДТА (Фиг. 13) была подтверждена потеря массы, сопровождающаяся эндотермией, и таким образом было предположено, что полученный кристалл представляет собой кристалл сольвата этилацетата и гексана.
Кроме того, из соотношения интегрирования 1H-ЯМР молярное соотношение содержания соединения, представленного формулой:
[Химическая формула 57]
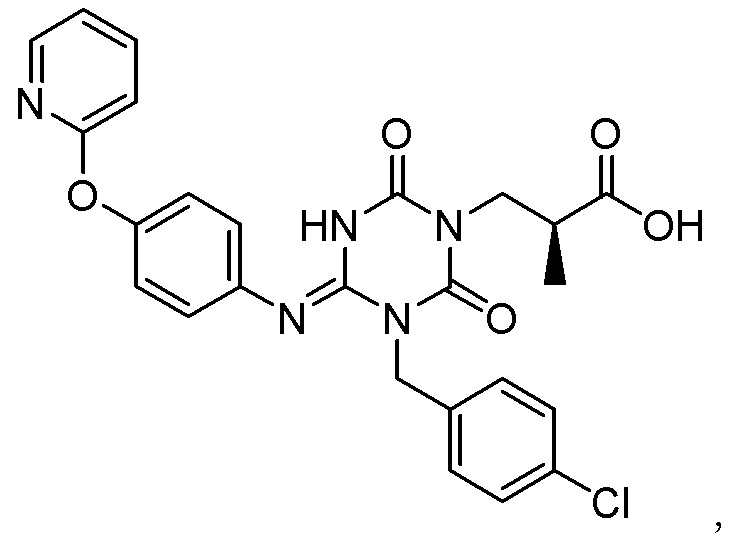
этилацетата и гексана составляло приблизительно 4:1:1.
Следовательно, что касается соединения I-127, описанного в Патентном документе 9, кристалл сольвата этилацетат/гексан соединения, представленного формулой:
[Химическая формула 58]
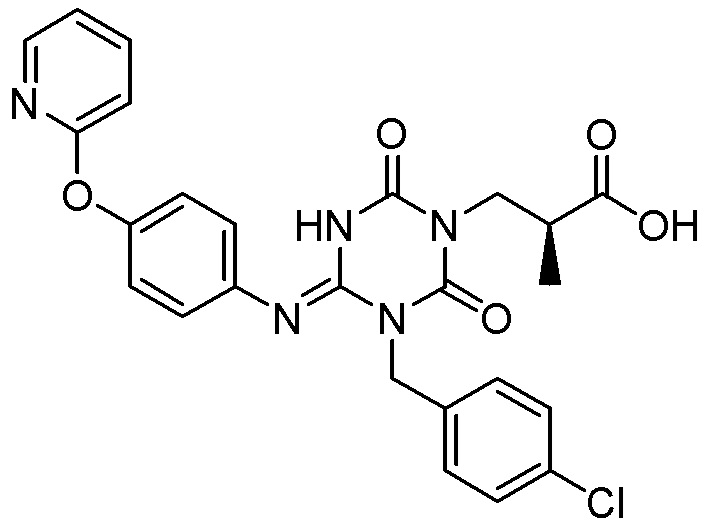
предположительно было получено (однако молекулярная структура (аминоформа/иминоформа) является неизвестной).
[0133]
Далее на Фиг. 10 показана порошковая рентгеновская дифрактограмма полученного белого порошка (Способ 2).
В спектре порошковой рентгеновской дифракции пики распознаются при углах дифракции (2θ): 8,3°±0,2°, 10,8°±0,2°, 13,4°±0,2°, 17,9°±0,2°, 19,4°±0,2°, 21,7°±0,2° и 25,6°±0,2°.
[0134]
Результат Раман-спектра полученного кристалла показан на Фиг. 11 (Способ 2).
Основные пики поглощения распознаются при 821 см-1±2 см-1, 856 см-1±2 см-1, 893 см-1±2 см-1, 1002 см-1±2 см-1, 1094 см-1±2 см-1, 1221 см-1±2 см-1, 1269 см-1±2 см-1, 1575 см-1±2 см-1, 1604 см-1±2 см-1, 1614 см-1±2 см-1, 1649 см-1±2 см-1, 1725 см-1±2 см-1 и 3073 см-1±2 см-1.
Основные пики поглощения распознаются при 821 см-1±2 см-1, 1002 см-1±2 см-1, 1269 см-1±2 см-1, 1575 см-1±2 см-1, 1614 см-1±2 см-1, 1649 см-1±2 см-1 и 1725 см-1±2 см-1.
Основные пики поглощения распознаются при 1575 см-1±2 см-1, 1614 см-1±2 см-1 и 1649 см-1±2 см-1.
Основные пики поглощения распознаются при 1614 см-1±2 см-1 и 1649 см-1±2 см-1.
[Пример 8]
[0135]
(Синтез безводной кристаллической Формы II соединения, представленного Формулой (I))
Безводную кристаллическую Форму I (приблизительно 10 мг) соединения, представленного Формулой (I), взвешивали в пробирке и к ней добавляли 200 мкл CHCl3. Смесь перемешивали c магнитной мешалкой при 400 об/мин при 25°С (процесс в течение ночи). Через 7 дней смесь отфильтровывали и полученный порошок подтверждали методом порошковой рентгеновской дифракции.
Порошковая рентгеновская дифрактограмма безводной кристаллической Формы II соединения, представленного Формулой (I), показана на Фиг. 12 (Способ 3).
В спектре порошковой рентгеновской дифракции пики распознаются при углах дифракции (2θ) 8,5°±0,2°, 12,1°±0,2°, 16,4°±0,2°, 17,1°±0,2°, 18,1°±0,2°, 18,5°±0,2°, 20,3°±0,2°, 23,0°±0,2° и 24,7°±0,2°.
В спектре порошковой рентгеновской дифракции пики распознаются при углах дифракции (2θ) 8,5°±0,2°, 12,1°±0,2°, 16,4°±0,2°, 17,1°±0,2° и 18,1°±0,2°.
[Пример 9]
[0136]
(Испытание на сыпучесть (Коэффициент прессуемости и коэффициент Хауснера))
(Способ измерения)
1) Приблизительно 10 г образца осторожно помещали в 50 мл мерный цилиндр и измеряли количество образца.
2) Верхнюю поверхность порошкового слоя тщательно выравнивали без уплотнения и считывали свободный насыпной объем (V0).
3) Измерительный цилиндр устанавливали на опорную стойку порошкового тестера (типа PT-X, Hosokawa Micron Corporation).
4) Образец порошка постукивали 10 раз, 500 раз, 1250 раз, 2500 раз и считывали соответствующие насыпные объемы (V10, V500, V1250, V2500).
5) Процесс завершали, когда разница объемов была равна или меньше минимальной шкалы (0,5 мл), и конечный общий объем определяли как конечный объем встряхивания (Vf).
6) Стадии 1)-5) повторяли три раза, и измеряли, и средние значения принимали для расчета коэффициента прессуемости и коэффициента Хауснера.
[0137]
(Способ расчета коэффициента прессуемости и коэффициента Хауснера)
Коэффициент прессуемости=(V0 - Vf)/V0 × 100
Коэффициент Хауснера=V0/Vf
[0138]
(Результаты)
Результаты представлены в Таблице 15.
[Таблица 15]
Как видно из Таблицы 15, было обнаружено, что безводная кристаллическая Форма I соединения, представленного Формулой (I), имеет самый низкий коэффициент прессуемости (%) и лучшую сыпучесть по сравнению с двумя другими кристаллическими формами (ссылка: материал PMDA; сыпучесть порошка; измерения коэффициента прессуемости и Хауснера, выбранные в Таблице 16).
[Таблица 16]
В целом известно, что стабильное получение может быть осуществлено, когда сыпучесть фармацевтического порошка является высокой в процессе получения твердого фармацевтического вещества. Низкая сыпучесть фармацевтических порошков может вызвать явление мостика в воронке в устройстве для обработки порошка, например, таблеточном прессе, и увеличить отклонение массы таблетки. Для получения стабильного фармацевтического порошка требуется кристаллическая форма, обладающая высокой сыпучестью, и было обнаружено, что безводная кристаллическая Форма I соединения, представленного Формулой (I), является наиболее предпочтительной кристаллической формой в процессе получения фармацевтических препаратов.
[Пример 10]
[0139]
(Количество остаточного растворителя, указанного в руководстве ICH Q3C)
Гексан представляет собой растворитель (класс 2), остаточное количество которого в активном фармацевтическом ингредиенте должно регулироваться, и этилацетат представляет собой малотоксичный растворитель (класс 3). Следовательно, необходимо довести остаточное количество гексана в активном фармацевтическом ингредиенте до заданного значения или менее. В Таблице 17 ниже показаны растворители класса 2, перечисленные в руководстве ICH Q3C.
[Таблица 17]
(мг/день)
Допустимая суточная доза (ДСД) гексана составляет 2,9 мг/сутки. Когда в качестве активного фармацевтического ингредиента используют кристалл сольвата этилацетат/гексан соединения, представленного Формулой (I), ДСД гексана может быть равной или превышать нормативное значение в зависимости от дозы. С другой стороны, безводная кристаллическая Форма I и кристалл дигидрата соединения, представленного Формулой (I), не содержало гексан в качестве остаточного растворителя, и было обнаружено, что является превосходным в качестве кристаллической формы, используемой для активного фармацевтического ингредиента.
[Пример 11]
[0140]
(Исследование стабильности твердого вещества)
(Способ измерения)
Образец массой 5 мг (n=4) точно взвешивали в 2 мл виале, закрывали крышкой и хранили при предварительно определенной температуре (герметичное хранение). Затем каждый образец вынимали после хранения в течение предварительно определенного периода и оценивали стабильность (содержание).
(Условия хранения)
80°C герметично закрытый
(Период хранения)
одна неделя
(Условия приготовления исследуемого раствора)
Каждую виалу вымывали в 25 мл мерную колбу.
Разбавляющая среда: ацетонитрил/вода=1/1
(Условия измерения ВЭЖХ)
Способ D
(Результаты)
Результаты представлены в Таблице 18.
[Таблица 18]
В любой из кристаллических форм остаточное соотношение составляло 99% или более, и было обнаружено, что она является стабильной кристаллической формой. В кристалле сольвата этилацетат/гексан соединения, представленного Формулой (I), образовалось пять типов аналогичных веществ. В безводной кристаллической Форме I соединения, представленного Формулой (I), образовалось три типа аналогичных веществ, и в кристалле дигидрата соединения, представленного Формулой (I), образовалось два типа аналогичных веществ. Из вышеизложенного было обнаружено, что в безводной кристаллической Форме I и в кристалле дигидрата соединения, представленного Формулой (I), можно получить несколько типов аналогичного вещества.
[0141]
Следующие примеры составов являются просто примерами и никоим образом не предназначены для ограничения объема изобретения.
Соединение изобретения можно вводить в виде фармацевтической композиции любым стандартным путем, в частности, энтерально, например, перорально, например, в форме таблетки или капсулы, или парентерально, например, в форме инъекции или суспензии; местно, например, в форме лосьона, геля, мази или крема; или в назальной форме или форме суппозитория. Фармацевтическая композиция, содержащая соединение изобретения в свободной форме или в форме фармацевтически приемлемой соли вместе, по меньшей мере, с одним фармацевтически приемлемым носителем или разбавителем, может быть получена стандартным способом путем смешивания, гранулирования или нанесения покрытия. Например, пероральная композиция может представлять собой таблетку, гранулу или капсулу, содержащую эксципиент, разрыхлитель, связующее вещество, скользящее вещество или подобные, также как активный ингредиент или подобные. Композиция для инъекций может представлять собой раствор или суспензию, может быть стерилизована или может содержать консервант, стабилизатор, буферный агент или подобные.
[ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ]
[0142]
Кристалл соединения, представленного Формулой (I) настоящего изобретения, является пригодным в качестве активного фармацевтического ингредиента. Фармацевтическая композиция, содержащая кристалл соединения, представленного Формулой (I), является наиболее пригодной в качестве терапевтического или профилактического средства при хроническом кашле.
Кроме того, настоящее изобретение является пригодным в качестве способа получения соединения, представленного Формулой (I).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЯНАЯ ФОРМА ИНГИБИТОРА ДИПЕПТИДИЛПЕПДИДАЗЫ IV И СПОСОБ ПОЛУЧЕНИЯ СОЛЕВОЙ ФОРМЫ | 2017 |
|
RU2734549C2 |
| СОЛЬ ИНГИБИТОРА SYK И ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2019 |
|
RU2818103C2 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| СОЛЬ ПЕНТАЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И ЕГО КРИСТАЛЛ | 2020 |
|
RU2820938C2 |
| КРИСТАЛЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2397173C2 |
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2640417C2 |
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2674441C1 |
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| СОЛЬ И ПОЛИМОРФ ФЕНИЛ-ПИРИМИДОНОВОГО СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2761213C2 |
| КРИСТАЛЛИЧЕСКОЕ ИНДОЛСУЛЬФОНАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2336269C2 |
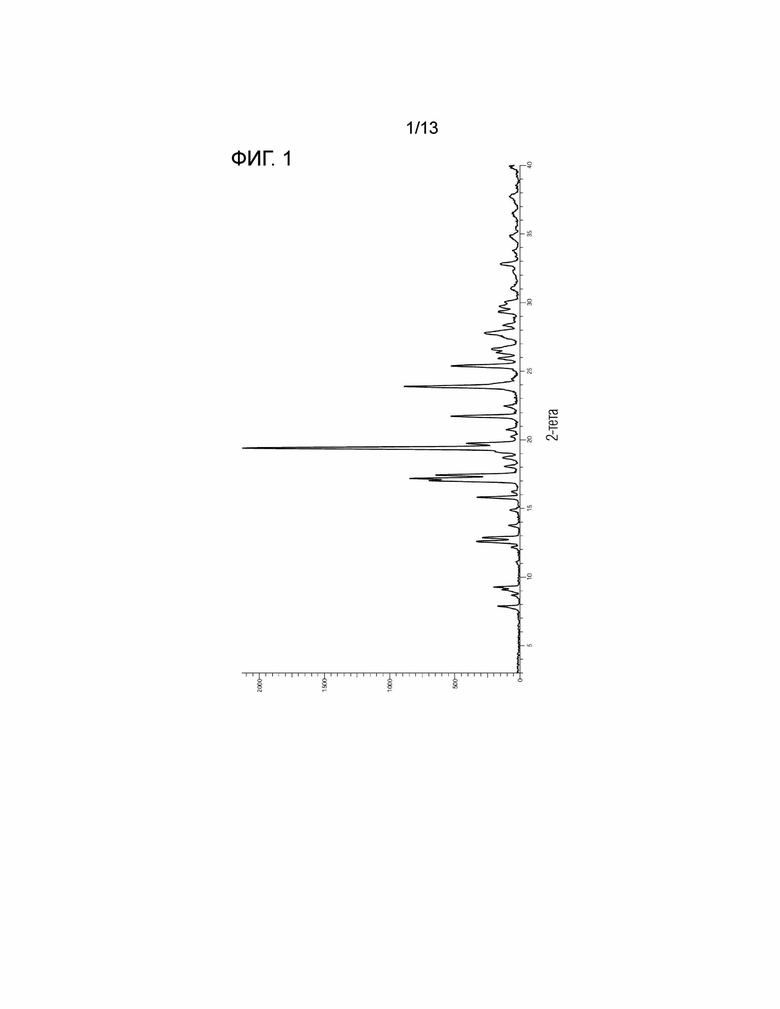
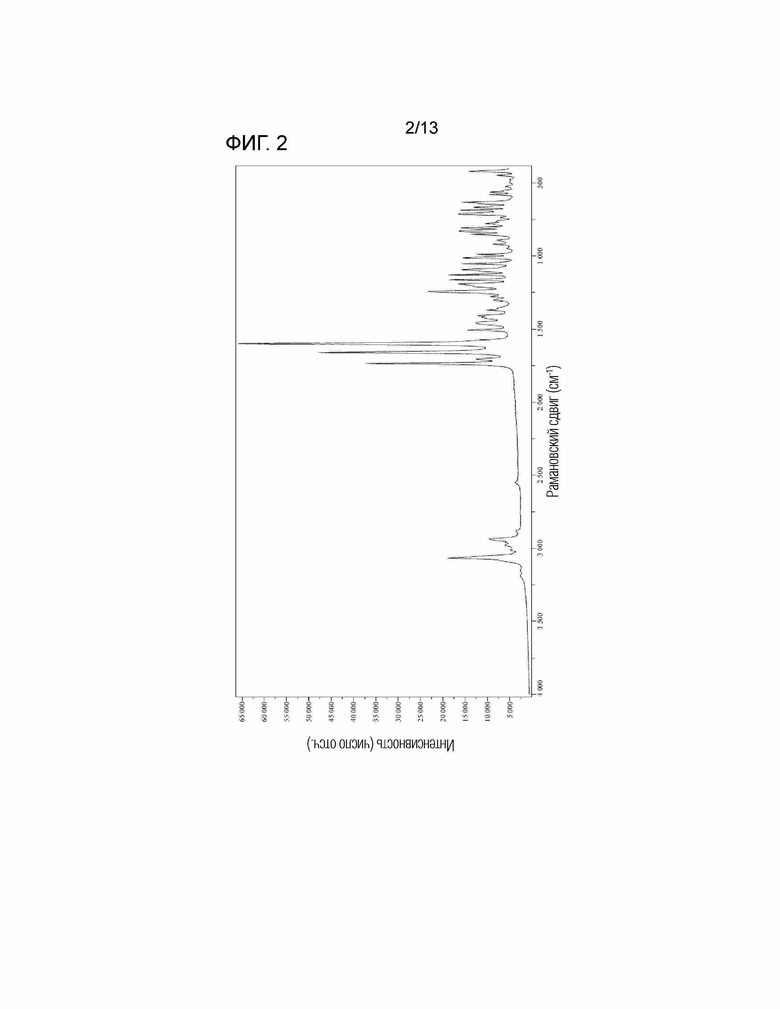
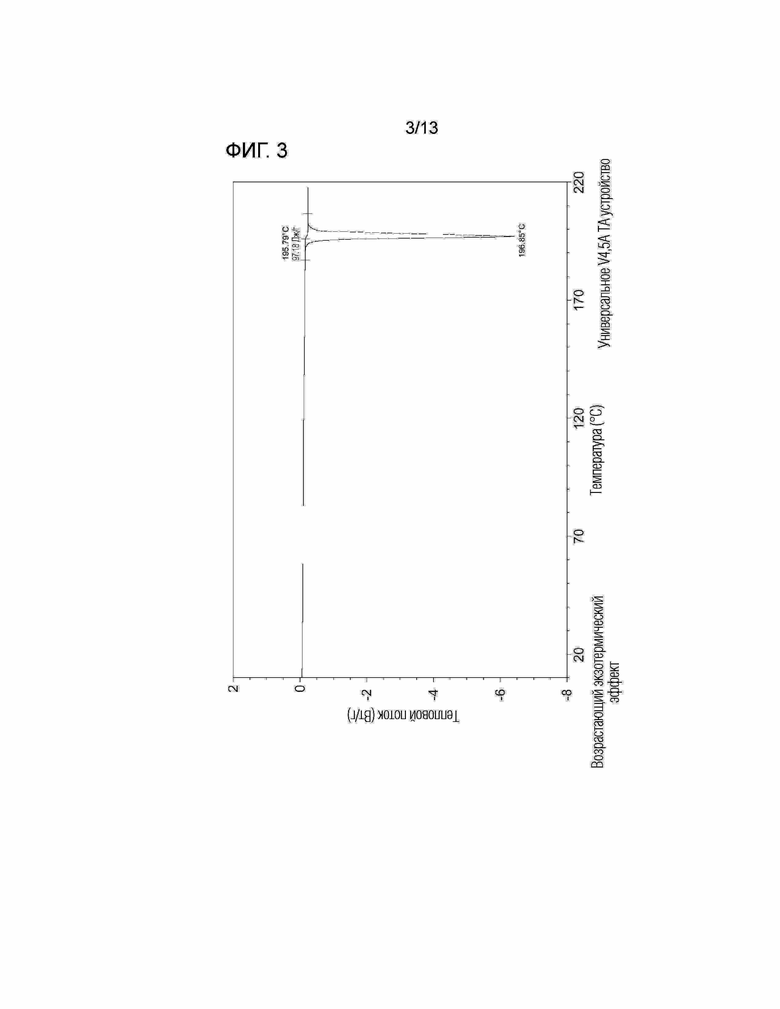
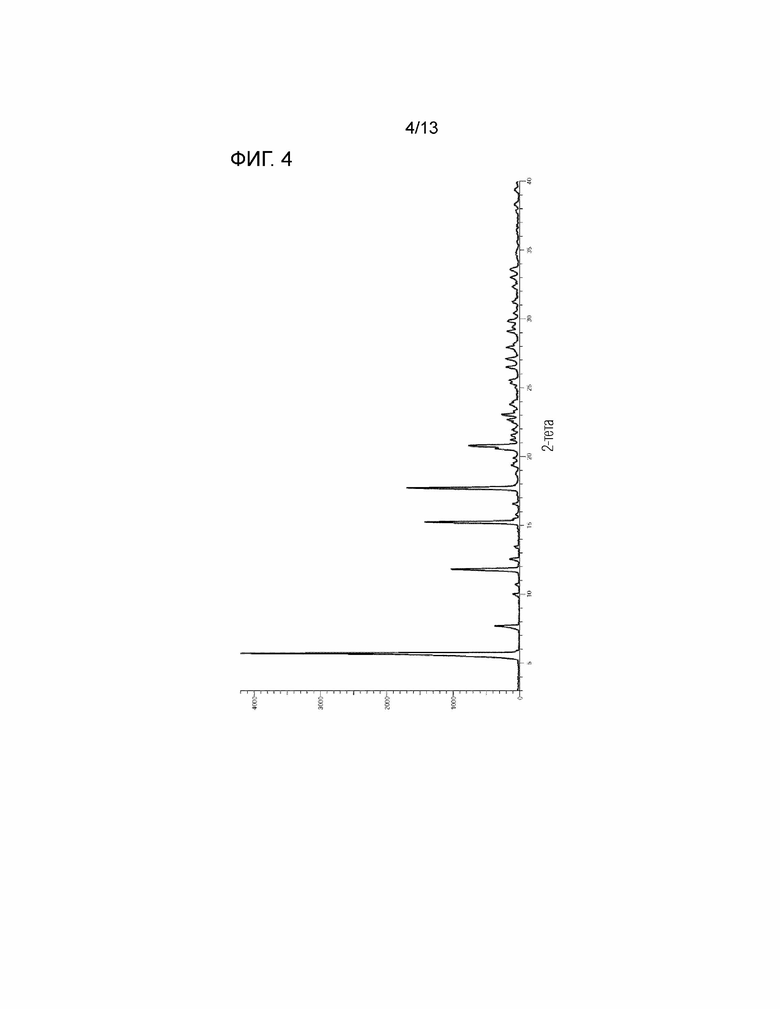
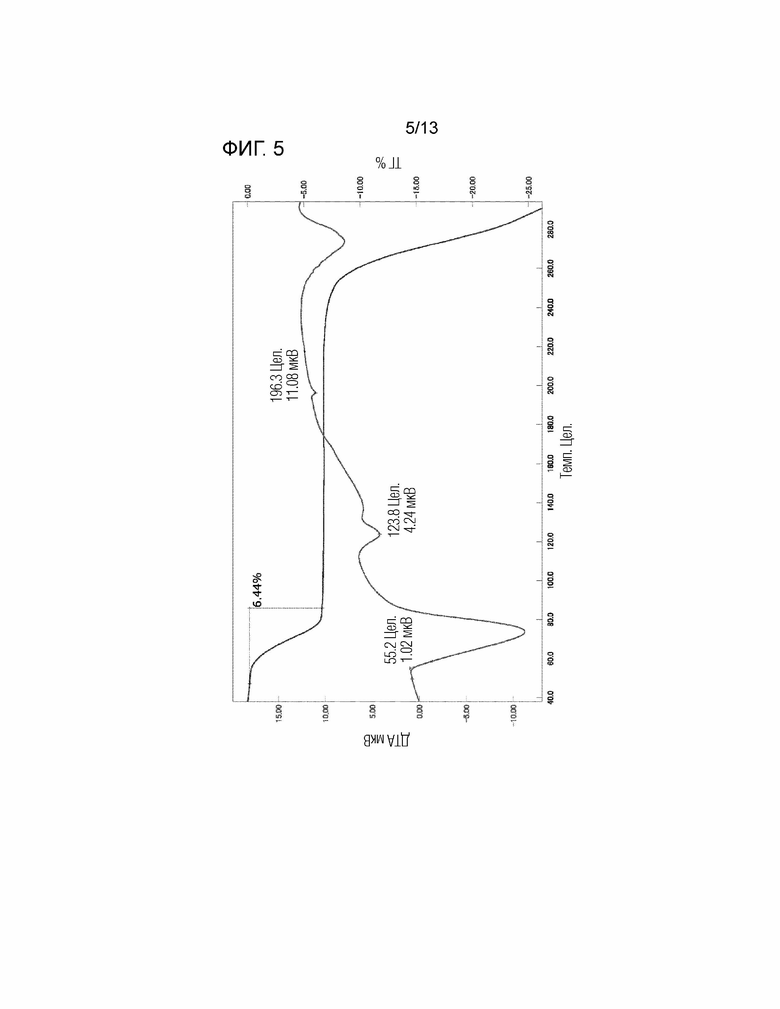
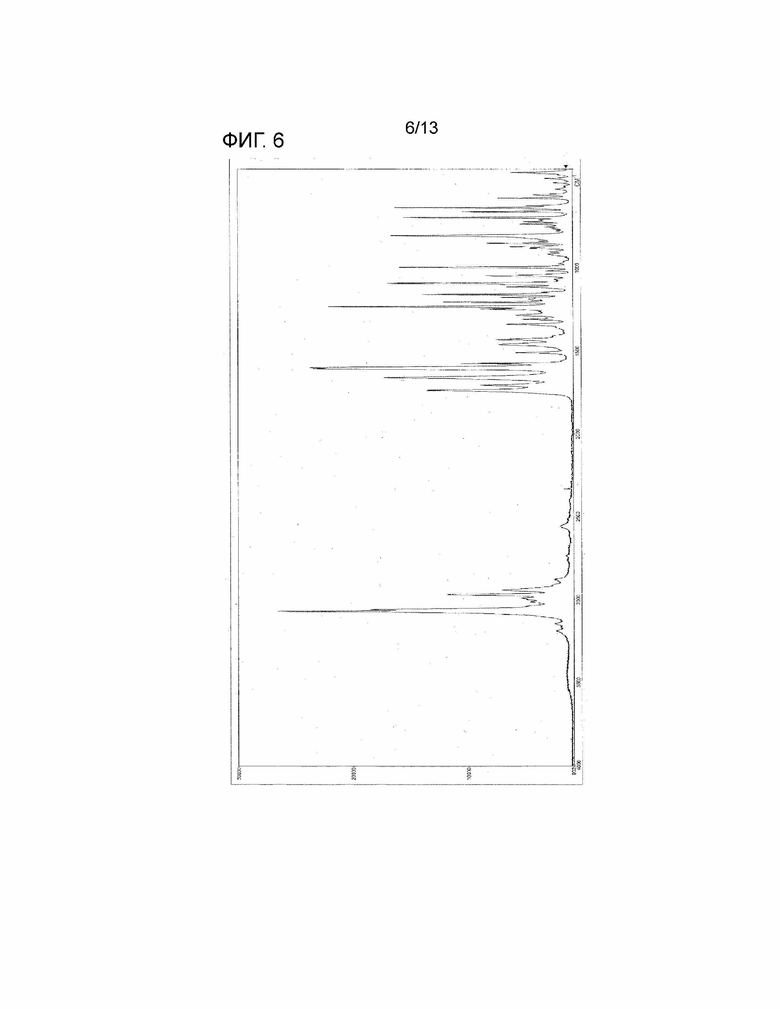
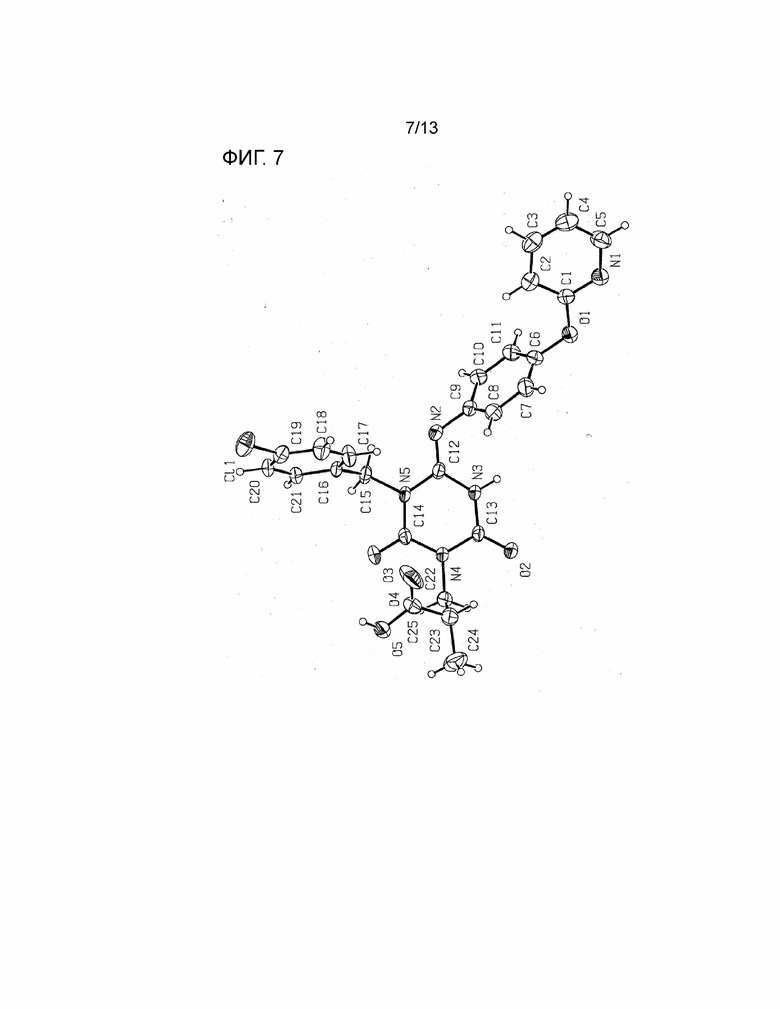
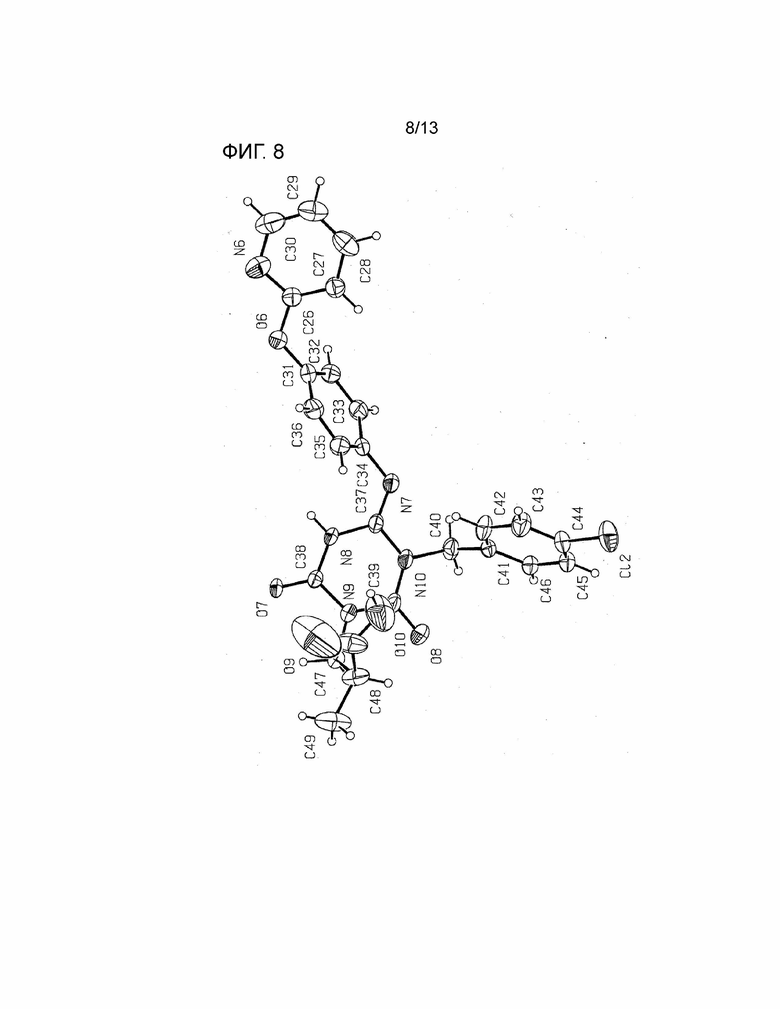

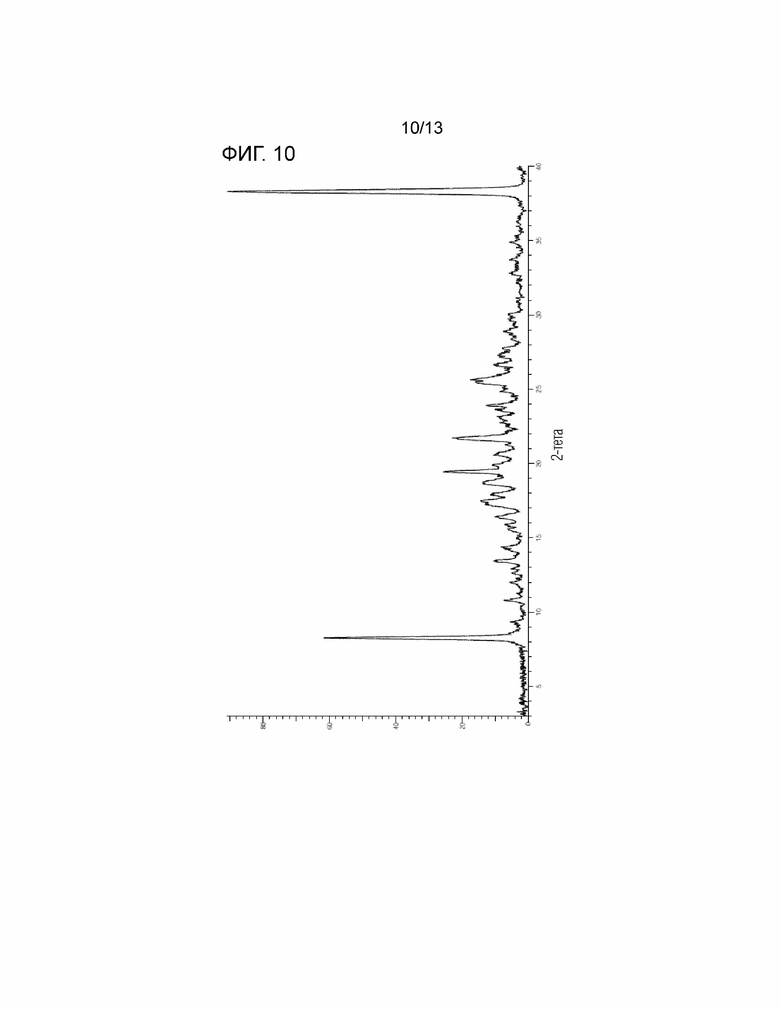
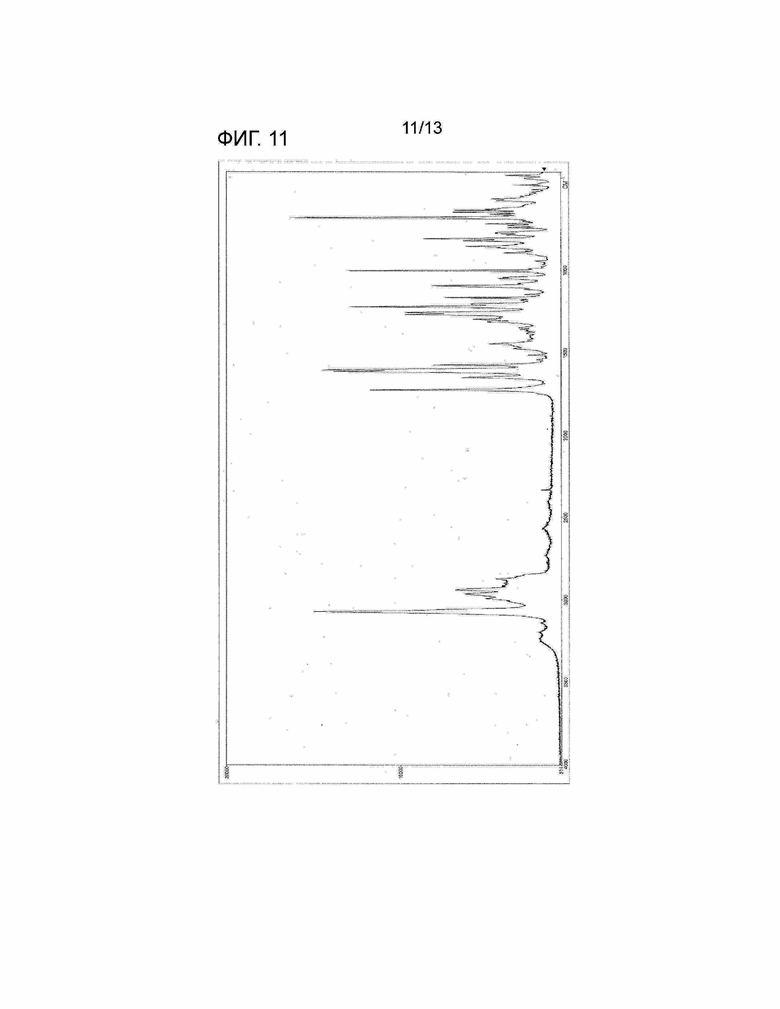
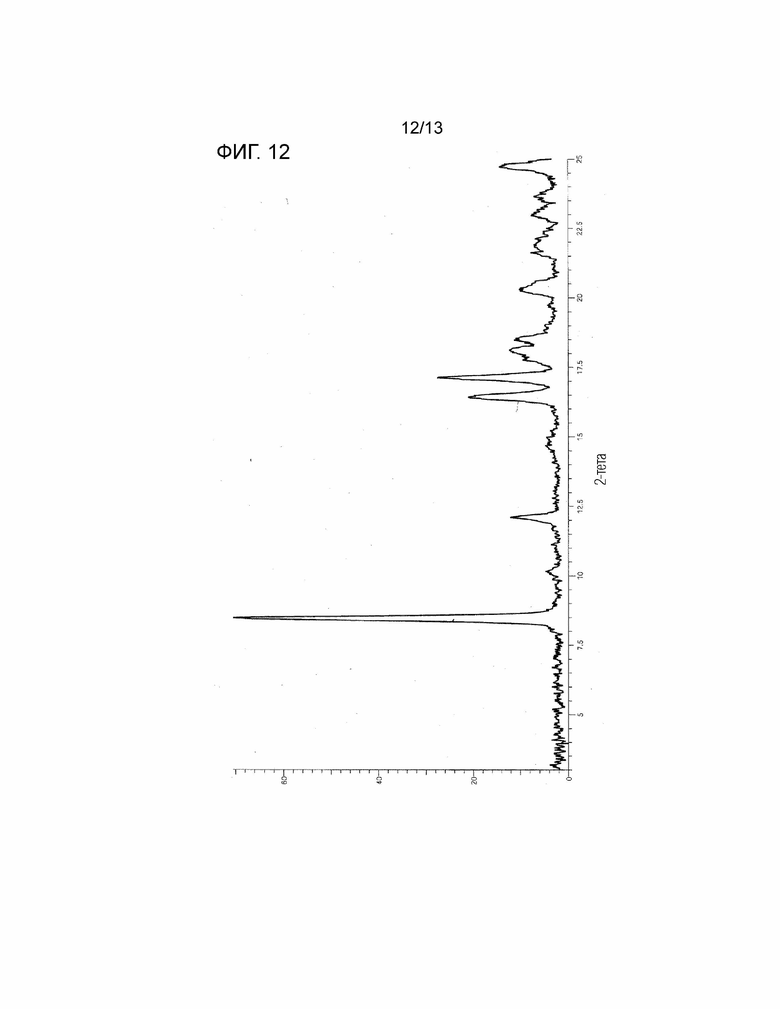
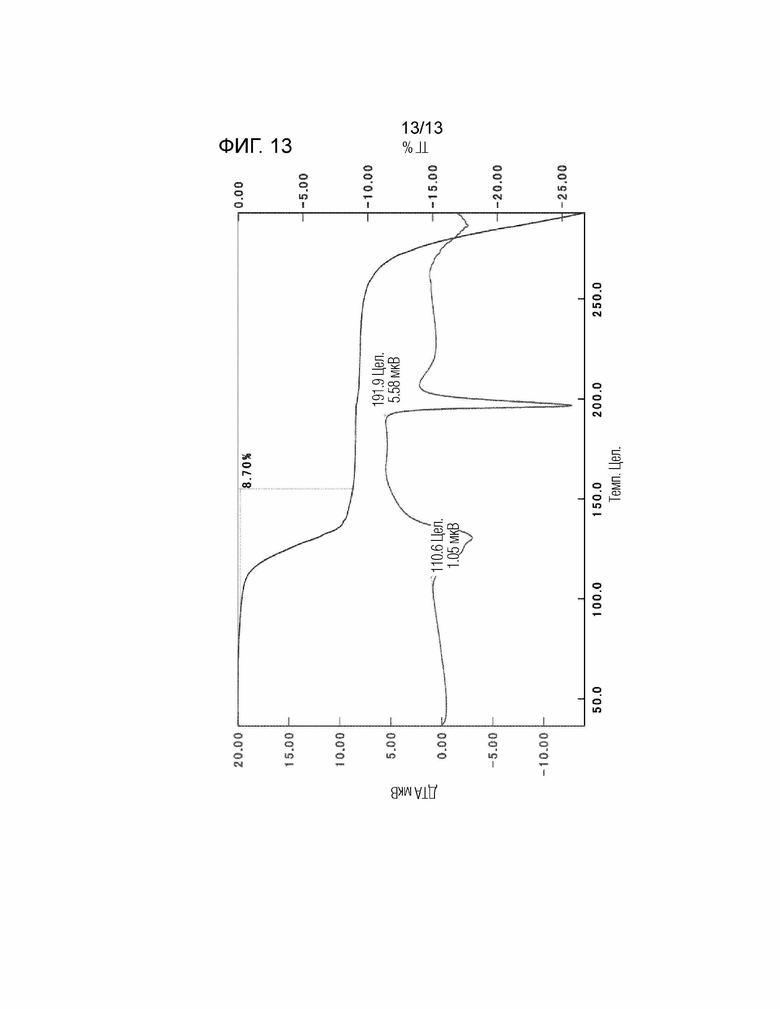
Изобретение относится к способу получения Формы I кристалла ангидрата соединения, представленного формулой (I), характеризующейся тем, что она обладает спектром порошковой рентгеновской дифракции, содержащим характеристические пики при углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2°; или углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2°. Способ осуществляют путем нагревания раствора соединения, представленного формулой (I), в 2-пропаноле с водой и метанолом до 50°С с последующим добавлением воды, охлаждением и фильтрованием полученного осадка. Технический результат - кристаллическая форма ангидрата соединения формулы (I), обладающая свойствами стабильности, низким коэффициентом прессуемости (%) кристалла и благоприятной текучестью кристалла. 2 з.п. ф-лы, 13 ил., 18 табл., 11 пр.
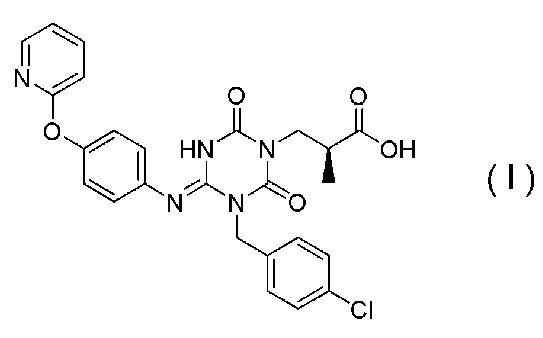
1. Способ получения Формы I кристалла ангидрата соединения, представленного Формулой (I):
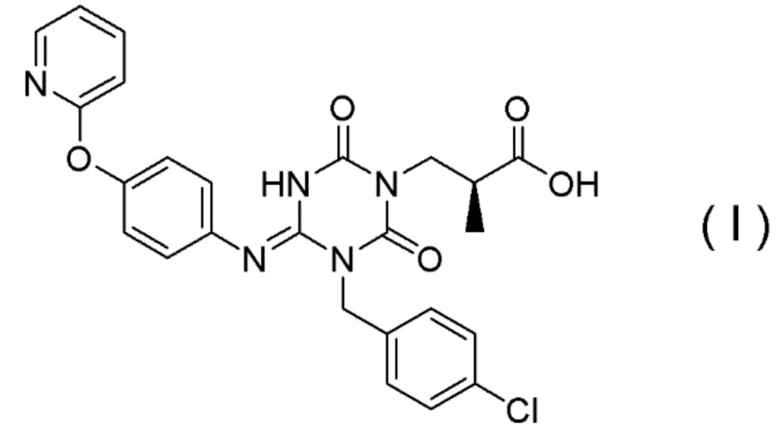
характеризующейся тем, что она обладает спектром порошковой рентгеновской дифракции, содержащим характеристические пики при:
углах дифракции (2θ) 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2° и 25,4°±0,2°; или
углах дифракции (2θ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2° и 19,4°±0,2°;
включающий нагревание раствора соединения, представленного формулой (I), в 2-пропаноле с водой и метанолом до 50°С с последующим добавлением воды, охлаждением и фильтрованием полученного осадка.
2. Способ по п.1, отличающийся тем, что Форма I кристалла ангидрата обладает спектром порошковой рентгеновской дифракции, содержащим характеристические пики при:
углах дифракции (2θ) 12,6°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2°, 26,6°±0,2°, 27,8°±0,2° и 32,8°±0,2°; или
углах дифракции (2Ɵ) 7,9°±0,2°, 9,3°±0,2°, 12,9°±0,2°, 15,8°±0,2°, 17,2°±0,2°, 19,4°±0,2°, 21,7°±0,2°, 23,9°±0,2°, 25,4°±0,2° и 27,8°±0,2°.
3. Способ по п.1, отличающийся тем, что Форма I кристалла ангидрата обладает Раман-спектром, содержащим пики поглощения при 829 см-1±2 см-1, 989 см-1±2 см-1, 1013 см-1±2 см-1, 1128 см-1±2 см-1 и 1370 см-1±2 см-1.
| УСТРОЙСТВО ДЛЯ ПРЕДОХРАНЕНИЯ БУКСИРНЫХ ТРОСОВ ИЛИ КАНАТОВ ОТ ИСТИРАНИЯ | 1931 |
|
SU30198A1 |
| MINO R | |||
| CAIRA | |||
| Crystalline Polymorphism of Organic Compounds, 1998, pp.163-208 | |||
| SHERRY L | |||
| MORISSETTE et al | |||
| High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids, ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, pp.275-300 | |||
| Клиническая фармакокинетика: теоретические, прикладные и | |||

