Область изобретения
Данное изобретение относится к новым фармацевтически полезным соединениям, в частности к соединениям, которые представляют собой конкурентные ингибиторы трипсиноподобных сериновых протеаз, в частности тромбина, или соединениям, которые метаболизируются до таких конкурентных ингибиторов, к их применению в качестве лекарств, содержащим их фармацевтическим композициям и получению их путем синтеза.
Предпосылки изобретения
Свертывание крови представляет собой ключевой процесс, вовлеченный как в гемостаз (т.е. предотвращение потери крови из поврежденного сосуда), так и в тромбоз (т.е. образование сгустка крови в кровеносном сосуде, иногда приводящее к закупорке сосуда).
Свертывание является результатом комплексной группы ферментативных реакций. Одним из конечных этапов в этой группе реакций является превращение профермента протромбина в активный фермент тромбин.
Известно, что тромбин играет центральную роль в свертывании. Он активирует тромбоциты, приводя к агрегации тромбоцитов, превращает фибриноген в мономеры фибрина, которые спонтанно полимеризуются в полимеры фибрина, и активирует фактор XIII, который, в свою очередь, сшивает эти полимеры с образованием нерастворимого фибрина. Кроме того, тромбин активирует фактор V и фактор VIII, приводя к образованию тромбина из протромбина по механизму "положительной обратной связи".
Следует ожидать, что, ингибируя агрегацию тромбоцитов и образование и сшивку фибрина, эффективные ингибиторы тромбина проявят антитромботическую активность. Кроме того, следует ожидать, что антитромботическая активность усиливается путем эффективного ингибирования механизма положительной обратной связи.
Уровень техники
Более ранняя разработка низкомолекулярных ингибиторов тромбина была описана Claesson в Blood Coagul. Fibrinol. (1994) 5, 411.
Blombäck et al. (в J.Clin. Lab. Invest. 24, suppl. 107, 59, (1969)) сообщали об ингибиторах тромбина на основе аминокислотной последовательности, расположенной вокруг сайта расщепления Аα цепи фибриногена. Относительно обсуждавшихся аминокислотных последовательностей, указанные авторы предположили, что трипептидная последовательность Phe-Val-Arg (P9-P2-P1, далее на нее ссылаются как на последовательность Р3-Р2-Р1) будет наиболее эффективным ингибитором.
Ингибиторы тромбина на основе дипептидильных производных с α,ω-аминоалкилгуанидином в положении Р1 известны из патента США №4346078 и международной заявки на патент WO 93/11152. Сообщалось также о подобных, родственных по структуре дипептидильных производных. Например, международная заявка на патент WO 94/29336 раскрывает соединения, например, с аминометилбензамидинами, циклическими аминоалкиламидинами и циклическими аминоалкилгуанидинами в положении Р1 (международная заявка на патент WO 97/23499 раскрывает пролекарства некоторых из этих соединений); заявка на европейский патент 0648780 раскрывает соединения, например, с циклическими аминоалкилгуанидинами в положении Р1.
Ингибиторы тромбина на основе пептидильных производных, также имеющие в качестве составной части циклические аминоалкилгуанидины (например, или 3-, или 4-аминометил-1-амидинопиперидин) в положении Р1, известны из заявок на европейский патент 0468231, 0559046 и 0641779.
Ингибиторы тромбина на основе трипептидильных производных с аргининовым альдегидом в положении Р1 были впервые раскрыты в заявке на европейский патент 0185390.
Позднее сообщалось о пептидильных производных на основе аргининового альдегида, модифицированных по положению РЗ. Например, международная заявка на патент WO 93/18060 раскрывает оксикислоты, заявка на европейский патент 0526877 - дезаминокислоты и заявка на европейский патент 0542525 - O-метилминдальные кислоты в положении Р3.
Также известны ингибиторы сериновых протеаз (например, тромбина) на основе электрофильных кетонов в положении Р1. Например, заявка на европейский патент 0195212 раскрывает пептидил-α-кетоэфиры и амиды, заявка на европейский патент 0362002 раскрывает кетоны фторалкиламидов, заявка на европейский патент 0364344 раскрывает α,β,δ-трикетосоединения и заявка на европейский патент 0530167 раскрывает α-алкоксикетоновые производные аргинина в положении Р1.
Другие, отличные по структуре ингибиторы трипсиноподобных сериновых протеаз на основе бороновокислых по С-концу производных аргинина и их изотиоурониевых аналогов известны из заявки на европейский патент 0293881.
Позднее ингибиторы тромбина на основе пептидильных производных были описаны в заявке на европейский патент 0669317 и международных заявках на патент WO 95/35309, WO 95/23609, WO 96/25426, WO 97/02284, WO 97/46577, WO 96/32110, WO 96/31504, WO 96/03374, WO 98/06740, WO 97/49404, WO 98/57932, WO 99/29664, WO 00/35869 и WO 00/42059.
В частности, в WO 97/02284 и WO 00/42059 описываются ингибиторы тромбина с замещенными миндальными кислотами по положению Р3.
Тем не менее остается необходимость в эффективных ингибиторах трипсиноподобных сериновых протеаз, таких как тромбин. Существует также потребность в соединениях, которые имеют удобный фармакокинетический профиль и селективны в ингибировании тромбина по сравнению с другими сериновыми протеазами, в частности теми, которые вовлечены в гемостаз. Следует ожидать, что соединения, которые проявляют активность конкурентного ингибитора в отношении тромбина, особенно полезны в качестве антикоагулянтов и, следовательно, в терапевтическом лечении тромбоза и родственных расстройств.
Описание изобретения
Согласно данному изобретению предложено соединение формулы I
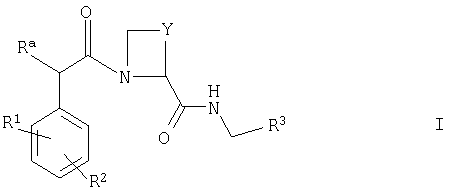
где Ra представляет собой -ОН или -СН2OH;
R1 представляет собой по меньшей мере один возможный галогенозаместитель;
R2 представляет собой один или два C1-3алкоксизаместителя, причем алкильные части этих заместителей сами замещены одним или более чем одним фторозаместителем (то есть R2 представляет собой одну или две фторалкокси(С1-3)группы);
Y представляет собой -СН2- или -(СН2)2- и
R3 представляет собой структурный фрагмент формулы I(1) или I(2)
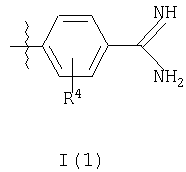
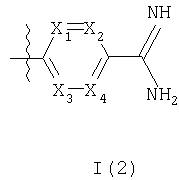
где R4 представляет собой Н или один или более чем один фторозаместитель и
один или двое из X1, X2, Х3 и Х4 представляют собой -N-, а остальные представляют собой -СН-,
или его фармацевтически приемлемое производное.
Термин «фармацевтически приемлемые производные» включает фармацевтически приемлемые соли (например, соли, полученные присоединением кислоты).
Сокращения приведены в конце данного описания изобретения. Волнистые линии на связях во фрагментах формул I(1) и I(2) означают положения присоединения данных фрагментов.
Галогеногруппы, которыми может быть представлен R1, включают в себя фторо, хлоро, бромо и йодо. Во избежание неопределенности, говоря о том, что R1 представляет собой по меньшей мере одну возможную галогеногруппу, имеется в виду, что R1 может либо не присутствовать (и таким образом быть замененным на Н, при этом соблюдены правила валентности), либо он может представлять собой один или более чем один атом галогена.
Когда R3 представляет собой структурный фрагмент формулы I(1), в котором R4 представляет собой один или более чем один фторозаместитель, предпочтительные соединения формулы I включают такие соединения, в которых R4 представляет собой единичный фторозаместитель в положении 2 или 3 либо два фторозаместителя или в положениях 2 и 5, или, более предпочтительно, в положениях 2 и 6 (где положения заместителей определяются относительно точки присоединения структурного фрагмента формулы I(1) к остальной части молекулы (то есть к группе -NHCH2-)).
Когда R3 представляет собой структурный фрагмент формулы I(2), то предпочтительные соединения формулы I включают в себя такие соединения, в которых либо
(а) один из X1, X2, Х3 и Х4 представляет собой -N-, а остальные представляют собой -СН-, либо
(б) оба X1 и Х3 или оба Х2 и Х4 представляют собой -N-, а другие двое (как подходит) представляют собой -СН-.
Предпочтительные соединения формулы I включают в себя те, в которых
R1 представляет собой единичный фторо-, хлоро- или бромозаместитель;
R2 представляет собой С1-2алкокси, замещенный одним или более чем одним фторозаместителем, как, например, -OCHF2, -OCF3, -ОСН2CF3, -OCH2CHF2, -OCH2CH2F или -OCH(CH2F)2;
R3 представляет собой структурный фрагмент формулы I(1);
R4 представляет собой Н.
Более предпочтительные соединения формулы I включают в себя такие соединения, в которых
Ra представляет собой ОН;
R1 представляет собой единичный хлорозаместитель;
R2 представляет собой -OCF2, предпочтительно -OCH2CHF2 или, более предпочтительно, -OCHF2 либо -OCH2CH2F.
Предпочтительные точки замещения R1 и R2 на соответствующей фенильной группе соединения формулы I включают оба мета-положения относительно точки присоединения этой фенильной группы к остальной части молекулы (то есть по положению 3 и/или 5 (предпочтительно 3,5-замещение) относительно углеродного атома, несущего группу α- или β-оксикислоты).
Соединения формулы I, которые можно упомянуть, включают такие соединения, в которых
R2 представляет собой один или два С1-2алкоксизаместителя, алкильные части которых сами замещены одним или более чем одним фторозаместителем, или
R2 представляет собой один или два С3алкоксизаместителя, алкильные части которых сами замещены одним или более чем одним фторозаместителем.
Соединения формулы I могут быть получены в соответствии с методиками, хорошо известными специалистам в данной области техники, например, как описано ниже.
Согласно еще одному аспекту данного изобретения предложен способ получения соединений формулы I, при котором
(1) осуществляют сочетание соединения формулы II
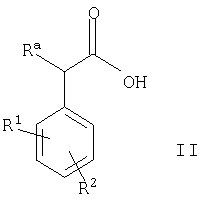
где Ra, R1 и R2 такие, как определено выше, с соединением формулы III
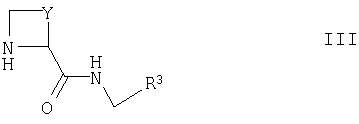
где Y и R3 такие, как определено выше, например, в присутствии агента сочетания (например, оксалилхлорида в DMF, EDC, DCC, HBTU, HATU, РуВОР или TBTU), соответствующего основания (например, пиридина, DMAP, TEA, 2,4,6-коллидина или DIPEA) и подходящего органического растворителя (например, дихлорметана, ацетонитрила, EtOAc или DMF);
(2) осуществляют сочетание соединения формулы IV
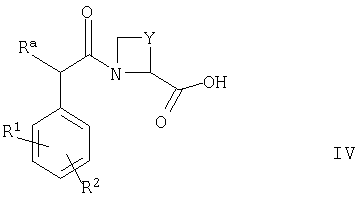
где Ra, R1, R2 и Y такие, как определено выше, с соединением формулы V

где R3 такой, как определено выше, например, в условиях, которые описаны выше в способе (1);
(3) для соединений формулы I, в которых не присутствует R1, восстанавливают соответствующее соединение формулы Ia, как оно определено выше, в котором R5 представляет собой OR6, где R5 и R6 такие, как определено выше, например, путем гидрирования в присутствии подходящего катализатора (например, металлического катализатора на носителе, такого как Pd/C (например, 10% (мас./мас.) Pd/C)) и соответствующего растворителя (например, низшего (например C1-6) алкилового спирта, такого как этанол), и, возможно, в присутствии подходящей кислоты (например, уксусной кислоты), и/или как описано в Synth. Comm. (1998) 4351, или
(4) осуществляют взаимодействие соответствующего соединения формулы XVIA или XVIB, как оно определено ниже, с подходящим источником аммиака (например, ацетатом аммония или газообразным аммиаком) в условиях, известных специалистам в данной области техники, например, путем взаимодействия этилимидоатного промежуточного соединения (образованного в результате взаимодействия соединения формулы XVIA или XVIB с HCl(г) в этаноле) с газообразным аммиаком в этаноле или в условиях, описанных в Tetrahedron Lett. 40, 7067 (1999), причем раскрытие этого документа включено сюда посредством ссылки (например, для получения соединений формулы I, в которых R3 представляет собой структурный фрагмент формулы I(2), в котором X2 или X4 представляет собой N, осуществляют взаимодействие соответствующего соединения формулы XVIB с ацетатом аммония (например, с 1-30 эквивалентами ацетата аммония) в присутствии N-ацетилцистеина (например, 1-30 эквивалентов N-ацетилцистеина) и соответствующего растворителя (например, низшего алкилового (C1-6) спирта, такого как метанол)).
Соединения формулы II доступны благодаря использованию известных и/или стандартных методик.
Например, соединения формулы II, в которых R3 представляет собой ОН, могут быть получены в результате взаимодействия альдегида формулы VI
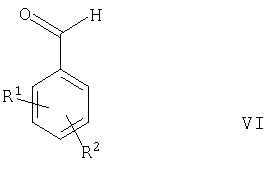
где R1 и R2 такие, как определено выше, с
(а) соединением формулы VII

где R" представляет собой Н или (СН3)3Si, например, при комнатной или повышенной температуре (например, ниже 100°С) в присутствии подходящего органического растворителя (например, хлороформа или метиленхлорида) и, при необходимости, в присутствии подходящего основания (например, TEA) и/или подходящей каталитической системы (например, хлорида бензиламмония или иодида цинка либо с использованием хирального катализатора, например, как описано в Chem. Rev., (1999) 99, 3649) с последующим гидролизом в условиях, которые хорошо известны специалистам в данной области техники (например, как описано ниже);
(б) NaCN или KCN, например, в присутствии NaHSO3 и воды с последующим гидролизом;
(в) хлороформом, например, при повышенной температуре (например, выше комнатной температуры, но ниже 100°С) в присутствии подходящего органического растворителя (например, хлороформа) и при необходимости в присутствии подходящей каталитической системы (например, хлорида бензиламмония) с последующим гидролизом;
(г) соединением формулы VIII

где М представляет собой Mg или Li, с последующим окислительным расщеплением (например, озонолизом или расщеплением, катализируемым осмием либо рутением) в условиях, которые хорошо известны специалистам в данной области техники, или
(д) трис(метилтио)метаном в условиях, которые хорошо известны специалистам в данной области техники, с последующим гидролизом в присутствии, например, HgO и HBF4.
Соединения формулы II, в которых Ra представляет собой -СН2ОН, могут быть получены путем восстановления соединения формулы IX

где R представляет собой С1-6алкил или C1-3алкилфенил и R1 и R2 такие, как определено выше, например, при комнатной или более низкой температуре в присутствии подходящего восстановителя (например, борогидрида натрия) и соответствующего органического растворителя (например, метанола, этанола, THF или их смесей) с последующим гидролизом полученного промежуточного соединения сложного эфира троповой кислоты формулы IXA
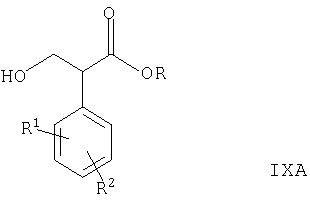
где R, R1 и R2 такие, как определено выше, в условиях, хорошо известных специалистам в данной области техники, например, как описано ниже. Специалисту понятно, что стадии восстановления и гидролиза могут быть выполнены в одной емкости, например, как описано ниже.
Соединения формулы II, в которых Ra представляет собой -ОН, альтернативно могут быть получены путем окисления соединения формулы IXB
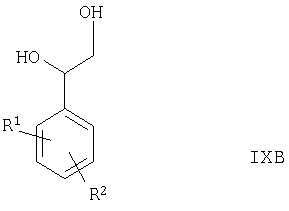
или его производного, которое возможно защищено по вторичной гидроксильной группе, где R1 и R2 такие, как определено выше, в присутствии подходящего окислителя (например, комбинации подходящего свободнорадикального окислителя (такого как TEMPO) и соответствующей гипохлоритной соли (такой как гипохлорит натрия)) в условиях, известных специалистам в данной области техники, например при температуре между -10°С и комнатной температурой в присутствии подходящего растворителя (например, воды, ацетона или их смеси), соответствующей соли (например, галогенида щелочного металла, такого как бромид калия) и подходящего основания (например, карбоната щелочного металла или гидрокарбоната, такого как гидрокарбонат натрия).
Энантиомерные формы соединения формулы II, в котором Ra представляет собой -ОН (то есть те соединения, которые имеют разные конфигурации заместителей вокруг атома С в α-положении относительно группы CO2H), могут быть разделены с помощью стадии образования энантиоспецифичных производных. Это можно осуществить, например, ферментативным способом. Такие ферментативные способы включают в себя, например, трансэтерификацию α-ОН-группы при температуре между комнатной и температурой дефлегмации (например, между 45 и 65°С) в присутствии подходящего фермента (например, Липазы PS Amano), соответствующего сложного эфира (например, винилацетата) и подходящего растворителя (например, метил-трет-бутилового эфира). Изомер, производное которого образуется, затем может быть отделен от не вступившего в реакцию изомера с помощью традиционных методик разделения (например, хроматографии).
Группы, добавленные к соединениям формулы II на такой стадии образования производных, могут быть удалены либо до проведения любых дальнейших реакций, либо на любой более поздней стадии в синтезе соединений формулы I. Дополнительные группы могут быть удалены с использованием традиционных методик (например, для сложных эфиров α-ОН-группы - гидролизом в условиях, известных специалистам в данной области техники (например, при температуре между комнатной и температурой дефлегмации в присутствии подходящего основания (например, NaOH) и соответствующего растворителя (например, МеОН, воды или их смесей))).
Энантиомерные формы соединения формулы II, в котором Rа представляет собой -СН2ОН, могут быть разделены с помощью методик хиральной хроматографии (например, хиральной ВЭЖХ).
Соединения формулы III могут быть получены в результате проведения сочетания соединения формулы X
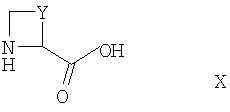
где Y такой, как определено выше, с соединением формулы V, как оно определено выше, например, в условиях, аналогичных описанным здесь для получения соединений формулы I.
Соединения формулы IV могут быть получены в результате проведения сочетания соединения формулы II, как оно определено выше, с соединением формулы X, как оно определено выше, например, в условиях, аналогичных описанным здесь для получения соединений формулы I.
Соединения формулы VI доступны с помощью использования известных и/или стандартных методик. Например, они могут быть получены следующим путем:
(1) металлирования (где металлом может быть, например, щелочной металл, такой как Li, или предпочтительно двухвалентный металл, такой как Mg) соединения формулы XI
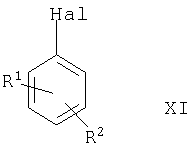
где Hal представляет собой атом галогена, выбранный из Cl, Br и I, и R1 и R2 такие, как определено выше, с последующим взаимодействием с подходящим источником формильной группы (например, N,N-диметилформамидом), например, в условиях, описанных ниже;
(2) восстановления соединения формулы XII
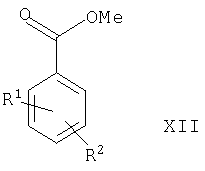
где R1 и R2 такие, как определено выше, в присутствии подходящего восстановителя (например, DIBAL-H), или
(3) окисления соединения формулы XIII
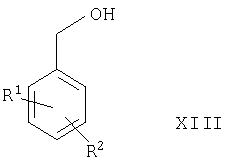
где R1 и R2 такие, как определено выше, в присутствии подходящего окислителя (например, MnO2, пиридинийхлорохромата, комбинации DMSO и оксалилхлорида или SO3-пиридинового комплекса в DMSO).
Соединения формулы IX могут быть получены из соответствующего фенилацетата (который, например, может быть получен из соответствующего ацетофенона, как описано в J. Am. Chem. Soc. 98, 6750 (1976), или из соответствующего бензилцианида с помощью стандартных гидролитических процедур) с применением традиционных методик, например по аналогии с методиками, описанными в J. Org. Chem. 54, 3831 (1989), и/или как описанно ниже.
Соединения формулы IX могут быть получены в результате дигидроксилирования соответствующего соединения формулы XIIIA
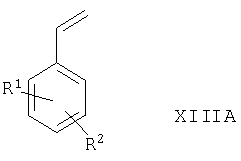
где R1 и R2 такие, как определено выше, в присутствии подходящего дигидроксилирующего агента (например, реагента или смеси реагентов, которые дают OsO4, как например AD-mix-α или, в особенности, AD-mix-β), например в условиях, известных специалистам в данной области техники, например при температуре между -10°С и комнатной в присутствии соответствующего растворителя (например, воды, трет-бутанола или их смеси). Если применяют такие асимметричные окислители как AD-mix-α или AD-mix-β, этот способ можно использовать для получения соединений формулы IXB, имеющих определенные конфигурации (то есть R или S) групп вокруг атомов С, к которым присоединены первичные и вторичные гидроксильные группы.
Соединения формулы XIIIA могут быть получены в результате взаимодействия соответствующего соединения формулы XI, как оно определено выше, с подходящим источником винил-аниона (например, трибутил(винил)олова) в условиях, известных специалистам в данной области техники, например при температуре между комнатной и температурой дефлегмации (например, 50°С) в присутствии соответствующего растворителя (например, толуола), подходящего агента сочетания (например, координационного комплекса на основе палладия(0), такого как тетракис(трифенилфосфин)палладий(0)) и, возможно, в присутствии соответствующего катализатора (например, 2,6-ди-трет-бутил-4-метилфенола).
Соединения формул V, VII, VIII, X, XI, XII и XIII либо имеются в продаже, хорошо известны из литературы, либо могут быть получены по аналогии с описанными здесь способами или с помощью традиционных процедур синтеза согласно стандартным методикам из легкодоступных исходных материалов с использованием соответствующих реагентов и условий реакций. Соединения формул Ia, XVIA и XVIB могут быть получены способами, описанными ниже.
Заместители на фенильном кольце в соединениях формул I, II, III, IV, V, VI, IX, IXA, IXB, XI, XII, XIII и XIIIA могут быть введены и/или взаимопревращены с использованием методик, хорошо известных специалистам в данной области техники, путем стандартных взаимопревращений функциональных групп согласно стандартным методикам из легко доступных исходных материалов с использованием соответствующих реагентов и условий реакций.
Например, соединения формул I, II, IV, VI, IXA, XI, XII и XIII могут быть получены из соответствующих соединений формул XIVA, XIVB, XIVC, XIVD, XIVE, XIVF, XIVG и XIVH соответственно
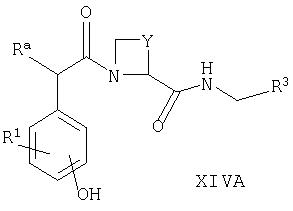
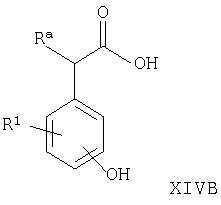
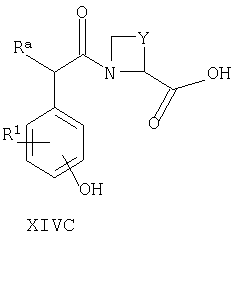
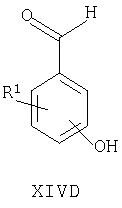
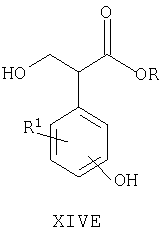
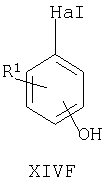
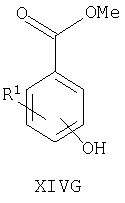
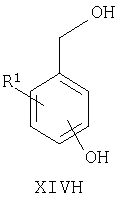
где Ra, R, R1, R3, Y и Hal (как целесообразно) такие, как определено выше, например:
(а) в результате осуществления взаимодействия соответствующего фторированного галогеноалкана (например, фторированного хлоралкана), например, при комнатной или более высокой температуре (например, при температуре дефлегмации) в присутствии подходящего основания (например, трет-бутилата, КОН или NaOH, например, в водном растворе) и соответствующего органического растворителя (например, THF, хлороформа или изопропанола) или
(б) в результате осуществления взаимодействия соединения формулы XIVJ

где Rx представляет собой С1-4алкил, С1-4перфторалкил или фенил (возможно замещенный метилом, нитро или галогено) и Ry представляет собой СН2CF3, CH2CHF2, CH2CH2F или CH(CH2F)2, например, в присутствии подходящего основания (например, K2СО3) и соответствующего растворителя (например, DMF),
например, в обоих случаях, как описано ниже.
Специалисту понятно, что такие превращения с функциональными группами также могут быть осуществлены на более ранней стадии в полной схеме синтеза соединений формул II, IV, VI, IXA, XI, XII и XIII (то есть на соответствующих предшественниках соединений формул XIVB, XIVC, XIVD, XIVE, XIVF, XIVG и XIVH соответственно).
Соединения формул XIVA, XIVB, XIVC, XIVD, XIVE, XIVF, XIVG, XIVH и XIVJ или имеются в продаже, известны из литературы, или могут быть получены или по аналогии с описанными здесь способами либо с помощью традиционных процедур синтеза согласно стандартным методикам из легко доступных исходных материалов с использованием соответствующих реагентов и условий реакций. Например, соединения формул XIVA, XIVB, XIVC, XIVD, XIVE, XIVF, XIVG и XIVH могут быть получены в результате снятия защиты с соответствующих защищенных фенолов (где защитной группой может быть, например, метил, аллил, бензил или трет-бутил) в стандартных условиях. Кроме того, соединения формулы XIVD, в которых R1 является единичным хлорозаместителем, могут быть получены из ди- или тригалогенозамещенного бензола (например, из 1-Br-, 3-Cl-, 5-F-бензола путем замещения атома фтора метоксигруппой (например, путем взаимодействия с NaOMe в смеси 1-метил-2-пирролидинон/метанол при повышенной температуре), замены бромогруппы формильной группой (например, как описано выше для получения соединений формулы VI) и затем деметилирования (например, с использованием PhSH в 1-метил-2-пирролидиноне в присутствии К2СО3)).
Кроме того, соединения формулы I, в которых R1 отсутствует, могут быть получены из соответствующих соединений формулы I (или с использованием их соответствующих предшественников), в которых R1 представляет собой галогено (такой как хлоро), например, путем гидрирования в условиях, известных специалистам в данной области техники.
Соединения формулы I могут быть выделены из своих реакционных смесей с использованием традиционных методик.
Согласно настоящему изобретению фармацевтически приемлемые производные соединений формулы I также включают «защищенные» производные соединений формулы I и/или соединения, которые действуют как пролекарства.
Соединения, которые могут действовать как пролекарства соединений формулы I, которые можно упомянуть, включают соединения формулы Ia
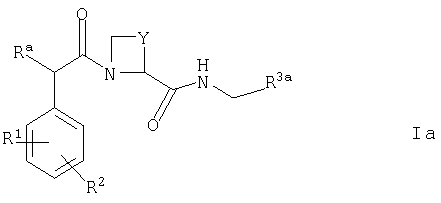
где R3a представляет собой структурный фрагмент формулы I(3) или I(4)
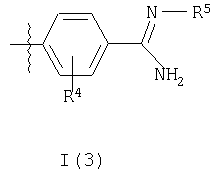
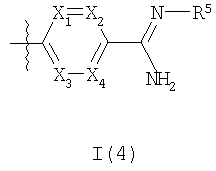
где R5 представляет собой OR6 или C(O)OR7;
R6 представляет собой Н, C1-10алкил, C1-3алкиларил или C1-3алкилоксиарил (причем алкильные части двух последних групп возможно прерваны одним или более чем одним атомом кислорода и арильные части двух последних групп возможно замещены одним или более чем одним заместителем, выбранным из галогено, фенила, метила или метокси, причем последние три группы также возможно замещены одним или более чем одним галогенозаместителем);
R7 представляет собой C1-10алкил (причем последняя группа возможно прервана одним или более чем одним атомом кислорода) или C1-3алкиларил либо C1-3алкилоксиарил (причем алкильные части двух последних групп возможно прерваны одним или более чем одним атомом кислорода и арильные части двух последних групп возможно замещены одним или более чем одним заместителем, выбранным из галогено, фенила, метила или метокси, причем последние три группы также возможно замещены одним или более чем одним галогенозаместителем), и
Ra, R1, R2, Y, R4, X1, Х2, Х3 и Х4 такие, как определено выше,
и их фармацевтически приемлемые производные.
Термин «фармацевтически приемлемые производные» соединений формулы Ia включает в себя фармацевтически приемлемые соли (например, соли, полученные присоединением кислоты).
Волнистые линии на связях во фрагментах формул I(3) и I(4) означают положения присоединения данных фрагментов.
Алкилоксиарильные группы, которыми могут являться R6 и R7, включают в себя алкильную и арильную группу, соединенные через атом кислорода. Алкиларильные и алкилоксиарильные группы присоединены к оставшейся части молекулы через алкильную часть этих групп, причем эти алкильные части (если имеется достаточное количество углеродных атомов, то есть три) могут иметь разветвленную цепь. Арильные части алкиларильных и алкилоксиарильных групп, которыми R6 и R7 могут являться или могут быть замещены, включают карбоциклические и гетероциклические ароматические группы, такие как фенил, нафтил, пиридинил, оксазолил, изоксазолил, тиадиазолил, индолил, бензофуранил и тому подобное.
Алкильные группы, которыми могут являться R6 и R7, могут быть с прямой цепью или, если имеется достаточное количество (то есть минимум три) углеродных атомов, они могут быть с разветвленной цепью и/или циклическими. Кроме того, если имеется достаточное количество (то есть минимум четыре) углеродных атомов, то такие алкильные группы также могут быть частично циклическими/ациклическими. Такие алкильные группы также могут быть насыщенными или, если имеется достаточное количество (то есть минимум два) углеродных атомов, быть ненасыщенными.
Галогеногруппы, которыми R6 и R7 могут быть замещены, включают в себя фторо, хлоро, бромо и иодо.
Когда R5 представляет собой C(O)OR7, предпочтительные группы R7 включают
(а) линейный, разветвленный или циклический С3-6алкил, например С4-6вциклоалкил;
(б) С1-2алкиларильные группы, такие как бензил, возможно замещенные, как указано выше.
Предпочтительные соединения формулы Ia включают в себя те, в которых R5 представляет собой OR6.
Когда R5 представляет собой OR6, предпочтительные группы R6 включают
(а) Н;
(б) незамещенный линейный, разветвленный или циклический С3-8алкил (например, C1-6), такой как линейный С1-3алкил (например, этил или, в особенности, метил), разветвленный С3-8алкил (например, изо-пропил, изо-бутил или 4-гептил) или циклический С4-7алкил (то есть С4-7циклоалкил, например циклобутил или циклогексил);
(в) C1-3алкилоксифенил (например, С2алкилоксифенил), причем фенильная группа возможно замещена одним или более чем одним заместителем, как указано выше (например, трифторметилом);
(г) С1-2алкиларил (например, метиларил), где арильная группа представляет собой фенил, пиридинил, оксазолил или изоксазолил, причем последние три группы возможно замещены одним или более чем одним заместителем, как указано выше (например, метокси, метилом, бромо и/или хлоро).
Предпочтительные соединения формулы Ia включают те, в которых R5 представляет собой OR6, a R6 представляет собой линейный, разветвленный (как подходит) или циклический (как подходит) С1-6алкил (например, C1-4), такой как метил, этил, н-пропил, изо-пропил или циклобутил.
Соединения формулы Ia можно получить с помощью одного или более из следующих способов:
(а) осуществляют взаимодействие соответствующего соединения формулы II, как оно определено выше, с соединением формулы XV
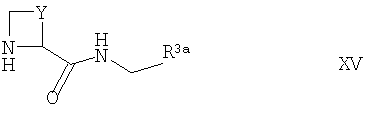
где Y и R3a такие, как определено выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I;
(б) осуществляют взаимодействие соответствующего соединения формулы IV, как оно определено выше, с соединением формулы XVI

где R3a такой, как определено выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I;
(в) для соединений формулы Ia, в которых R5 представляет собой ОН, осуществляют взаимодействие соответствующего соединения формулы XVIA или XVIB
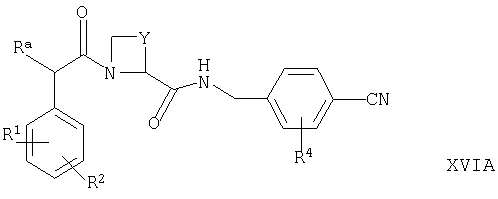
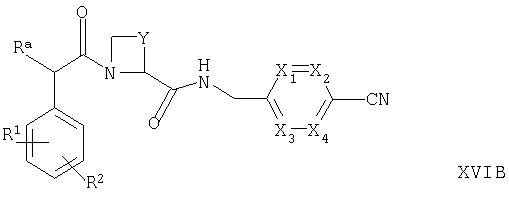
где Ra, R1, R2, R4, Y, X1, Х2, Х3 и Х4 такие, как определено выше, с гидроксиламином, например, в условиях, известных специалистам в данной области техники;
(г) для соединений формулы Ia, в которых R5 представляет собой OR6, осуществляют взаимодействие защищенного производного соответствующего соединения формулы I, которое представляет собой, например, соединение формулы XVII
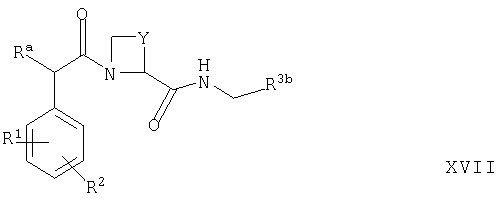
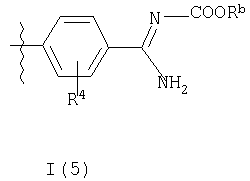
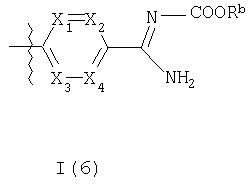
где R3b представляет собой структурный фрагмент формулы I(5) или I(6)
где Rb представляет собой, например, -СН2СН2-Si(СН3)3 или бензил, либо его таутомер, и Ra, R1, R2, Y, R4, X1, Х2, Х3 и Х4 такие, как определено выше, с соединением формулы XVIII

где R6 такой, как определено выше, или его полученной присоединением кислоты солью, например, при температуре между комнатной и температурой дефлегмации в присутствии соответствующего органического растворителя (например, THF, СН3CN, DMF или DMSO) с последующим удалением группы -C(O)ORb в условиях, известных специалистам в данной области техники (например, путем осуществления взаимодействия с QF или TFA (например, как описано ниже));
(д) для соединений формулы Ia, в которых R5 представляет собой ОН, осуществляют взаимодействие соединения формулы XVII, как оно определено выше, в котором Rb представляет собой бензил, с гидроксиламином или его полученной присоединением кислоты солью, например, в условиях, которые известны специалистам в данной области техники;
(е) для соединений формулы Ia, в которых R5 представляет собой COOR7, осуществляют взаимодействие соответствующего соединения формулы I, как оно определено выше, с соединением формулы XIX

где L1 представляет собой подходящую уходящую группу, такую как галогено или нитрофенил (например, 4-нитрофенил), и R7 такой, как определено выше, например, при приблизительно комнатной температуре в присутствии подходящего основания (например, NaOH, например, в водном растворе) и соответствующего органического растворителя (например, метиленхлорида) или
(ж) для соединений формулы Ia, в которых R5 представляет собой ОСН3 или ОСН2СН3, осуществляют взаимодействие соответствующего соединения формулы Ia, в котором R5 представляет собой ОН, с диметилсульфатом или диэтилсульфатом соответственно, например, в присутствии подходящего основания (например, гидроксида щелочного металла, такого как КОН (например, в водном растворе при концентрации, например, 50 мас.%)) и соответствующего катализатора (например, галогенида четвертичного аммония, такого как бензилтриметиламмонийхлорид (например, в растворе СН2Cl2 или THF при концентрации, например, 10 мас.%)).
Волнистые линии на связях во фрагментах формул I(5) и I(6) означают положения связывания данных фрагментов.
Соединения формул XVIA и XVIB могут быть получены путем взаимодействия соответствующего соединения формулы II, как оно определено выше, с соединением формулы XIXA или XIXB
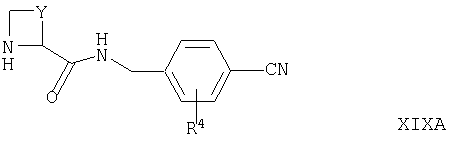
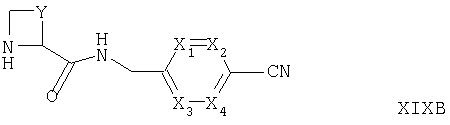
где R4, Y, X1, X2, Х3 и Х4 такие, как они определены выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I.
Соединения формул XVIA и XVIB альтернативно могут быть получены путем взаимодействия соответствующего соединения формулы IV, как оно определено выше, с соединением формулы XIXC или XIXD
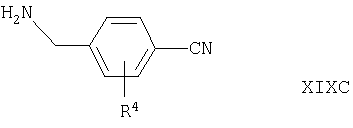
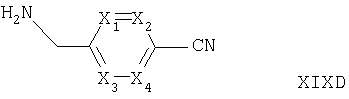
где R4, X1, X2, Х3 и Х4 такие, как они определены выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I.
Соединения формулы XVII могут быть получены путем взаимодействия соответствующего соединения формулы II, как оно определено выше, с соединением формулы XX
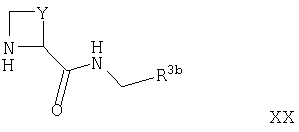
где Y и R3b такие, как они определены выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I.
Альтернативно, соединения формулы XVII могут быть получены путем взаимодействия соответствующего соединения формулы I с соединением, соответствующим соединению формулы XIX, в котором вместо R7 наличествует группа Rb, где Rb такая, как она определена выше, например, в условиях, описанных выше касательно получения соединений формулы Ia.
Соединения формул XV и XX могут быть получены путем взаимодействия соответствующего соединения формулы X, как оно определено выше, соответственно с соединением формулы XVI, как оно определено выше, или с соединением формулы XXI

где R3b такой, как он определен выше, например, в условиях, подобных условиям, описанным выше для синтеза соединений формулы I.
Соединения формулы XVI, XVIII, XIX, XIXA, XIXB, XIXC, XIXD и XXI либо имеются в продаже, хорошо известны из литературы, либо могут быть получены или по аналогии со способами, описанными выше, или с помощью традиционных методик синтеза согласно стандартным методикам из легко доступных исходных материалов с использованием соответствующих реагентов и условий реакций. Например, соединения формул XIXA и XIXB могут быть получены путем взаимодействия соответствующего соединения формулы XIXC или XIXD (как целесообразно) с соединением формулы X, например, в условиях, аналогичных описанным выше.
Соединения формул I и Ia, которые определены выше, и производные их обоих далее здесь упоминаются как «соединения по изобретению».
Таким образом, предпочтительные соединения по изобретению включают в себя соединения из примеров, описанных ниже. В этом отношении соединения по изобретению, которые могут быть упомянуты, включают в себя следующие:
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OEt);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-н-Pr);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-изо-Pr);
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(O-цикло-Bu);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH);
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(СОО-циклопентил);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Z);
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(ОСН2-3-(5-Ме-изоксазол));
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(ОСН2-3-пиридин);
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-изо-Bu);
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OEt);
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn);
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-циклогексил);
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(O-цикло-Bu);
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OCH2CH2OPh(3-CF3));
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(4-Cl));
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(3-MeO));
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(2-Br));
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(4-Me));
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-4-гептил);
Ph(3-Cl)(5-OCHF2)-(S)CH(CH2OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab(OMe);
Ph(3-OCHF2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-OCF3)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-ОСН2CF3)-(R)СН(ОН)С(O)-Aze-Pab;
Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Br)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab;
Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl, 5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH);
Ph(3-Cl, 5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OH);
Ph(3-Cl, 5-OCHF2)-(R)CH(OH)C(O)-Pro-Pab;
Ph(3-Cl, 5-OCHF2)-(R)CH(OH)C(O)-Pro-Pab(OMe);
Ph(3-Cl, 5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-((2-амидино)-5-пиридинил);
Ph(3-Cl, 5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-((2-метоксиамидино)-5-пиридинил);
Ph(3-Cl, 5-OCHF2(R)СН(ОН)С(O)-Aze-NH-СН2-((5-амидино)-2-пиримидинил);
Ph(3-Cl, 5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-((5-метоксиамидино)-2-пиримидинил);
Ph(3-Cl, 5-OCHF2(R)CH(OH)C(O)-Aze-Pab(3-F);
Ph(3-Cl, 5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF);
Ph(3-Cl, 5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe);
Ph(3-Cl, 5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,5-диF)и
Ph(3-Cl, 5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)(OMe).
Соединения по изобретению могут проявлять таутомерию. Все их таутомерные формы и смеси включены в объем данного изобретения. Конкретные таутомерные формы, которые могут быть упомянуты, включают в себя формы, связанные с положением двойной связи в амидиновой функциональной группе в соединении формулы Ia и с положением заместителя R5.
Кроме того, соединения по изобретению содержат один или более чем один асимметрический углеродный атом и поэтому могут проявлять оптическую и/или диастереоизомерию. Диастереоизомеры могут быть разделены с использованием общепринятых методик, например хроматографии. Многие стереоизомеры могут быть выделены разделением рацемической или другой смеси данных соединений с использованием общепринятых, например ВЭЖХ, методик. Альтернативно, желаемые оптические изомеры могут быть получены путем взаимодействия подходящих оптически активных исходных материалов в условиях, которые не приводят к рацемизации либо эпимеризации, или посредством получения производных, например, с гомохиральной кислотой с последующим разделением диастереомерных производных общепринятыми способами (например, ВЭЖХ, хроматографией на диоксиде кремния). Все стереоизомеры включены в объем данного изобретения.
Соединения по изобретению, в которых фрагмент
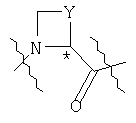
находится в S-конфигурации, являются предпочтительными.
Предпочтительные соединения по изобретению включают в себя таковые, в которых структурный фрагмент
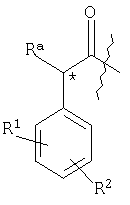
находится в R-конфигурации, когда Rа представляет собой -ОН, или находится в S-конфигурации, когда Ra представляет собой -СН2OH.
Волнистые линии на связях в приведенных выше двух фрагментах означают положения связывания данных фрагментов.
Соединения по изобретению, которые могут быть упомянуты, включают в себя Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab (где в этом случае Aze означает азетидин-2-карбоксилат (то есть в (R)- и/или в (S)-конформациях)) равно как и эквивалентные соединения, в которых вместо водородного атома в амидиновой части в Pab присутствует группа -OR6 (как определено выше), в которой R6 представляет собой С1-3алкил (то есть Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab(OMe), Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab(OEt), Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab(O-н-Pr) или Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab(O-изо-Pr)). Кроме того, соединения по изобретению, которые могут быть упомянуты, включают в себя таковые, которые не являются конкретными соединениями, идентифицированными в предыдущем предложении.
Специалистам в данной области техники очевидно, что в описанных выше и ниже способах функциональным группам промежуточных соединений может потребоваться защита с помощью защитных групп.
Функциональные группы, которые желательно защитить, включают в себя гидрокси-, аминогруппы и группы карбоновой кислоты. Подходящие защитные группы для гидрокси включают в себя возможно замещенные и/или ненасыщенные алкильные группы (например, метил, аллил, бензил или трет-бутил), триалкилсилильные или диарилалкилсилильные группы (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил) и тетрагидропиранил. Подходящие защитные группы для карбоновой кислоты включают в себя С1-6алкиловый или бензиловый эфиры. Подходящие защитные группы для амино и амидино включают в себя трет-бутилоксикарбонил, бензилоксикарбонил или 2-триметилсилилэтоксикарбонил (Теос). Амидинные азоты также могут быть защищены гидрокси- или алкоксигруппами и могут быть либо одно, либо двузащищенными.
Защита и снятие защиты с функциональных групп могут происходить до или после сочетания либо до или после любой другой реакции на вышеупомянутых схемах.
Защитные группы могут быть удалены в соответствии с методиками, которые хорошо известны специалистам в данной области техники и как они описаны ниже.
Специалистам в данной области техники очевидно, что для того чтобы получить соединения по изобретению альтернативным и в некоторых случаях более удобным способом, конкретные стадии способов, упомянутые выше, могут быть выполнены в различном порядке и/или конкретные реакции могут быть выполнены на другой стадии общего способа (то есть в соответствии с конкретной реакцией могут быть добавлены заместители к различным промежуточным соединениям и/или выполнены химические превращения различных промежуточных соединений, упомянутых выше). Это может отрицать или делать необходимой потребность в защитных группах.
Тип применяемой химии будет диктовать необходимость и тип защитных групп, равно как и последовательность выполнения синтеза.
Использование защитных групп основательно описано в "Protective Groups in Organic Chemistry", edited by J.W.F. McOmie, Plenum Press (1973), и "Protective Groups in Organic Synthesis", 3nd edition, T.W.Greene & P.G.M. Wutz, Wiley-lnterscience (1999).
Защищенные производные соединений по изобретению могут быть химически превращены в соединения по изобретению с использованием стандартных методик снятия защиты (например, гидрированием). Специалисту также ясно, что некоторые соединения формулы Ia также могут быть упомянуты как являющиеся «защищенными производными» соединений формулы I.
Медицинское и фармацевтическое применение
Соединения по изобретению могут обладать фармакологической активностью как таковые. Соединения по изобретению, которые могут обладать такой активностью, включают в себя соединения формулы I, но не ограничиваются ими.
Однако другие соединения по изобретению (включая соединения формулы Ia) могут не обладать такой активностью, но могут быть введены парентерально или перорально и после этого могут метаболизироваться в организме с образованием соединений, которые являются фармакологически активными (включая соответствующие соединения формулы I, но не ограничиваясь ими). Такие соединения (также включающие в себя соединения, которые могут обладать некоторой фармакологической активностью, но эта активность существенно ниже, чем таковая у «активных» соединений, до которых они метаболизируются) могут поэтому быть описаны как «пролекарства» активных соединений.
Таким образом, соединения по изобретению являются полезными, поскольку они обладают фармакологической активностью и/или метаболизируются в организме после перорального или парентерального введения с образованием соединений, которые обладают фармакологической активностью. Поэтому соединения по изобретению показаны в качестве фармацевтических средств.
Таким образом, согласно еще одному аспекту данного изобретения предложены соединения по изобретению для применения в качестве фармацевтических средств.
В частности, соединения по изобретению являются сильными ингибиторами тромбина или сами по себе, и/или (например, в случае пролекарств) метаболизируются после введения с образованием сильных ингибиторов тромбина, например, как может быть продемонстрировано в тестах, описанных ниже.
В понятие «пролекарство ингибитора тромбина» авторы включают соединения, которые образуют ингибитор тромбина в экспериментально обнаруживаемом количестве и в пределах предварительно определенного времени (например, приблизительно 1 часа) после перорального или парентерального введения (смотри, например, приведенный ниже Тест Е) или, альтернативно, после инкубирования в присутствии микросом печени (смотри, например, приведенный ниже Тест G).
Таким образом, ожидается, что соединения по изобретению будут полезными при таких состояниях, где требуется ингибирование тромбина, и/или состояниях, где показана антикоагулянтная терапия, включая следующие далее.
Лечение и/или профилактика тромбоза и гиперкоагуляции в крови и/или тканях животных, включая человека
Известно, что гиперкоагуляция может приводить к тромбоэмболическим заболеваниям. Состояния, связанные с гиперкоагуляцией и тромбоэмболическими заболеваниями, которые могут быть упомянуты, включают в себя наследственную или приобретенную резистентность к активированному белку С, такую как мутация фактора V (фактора V Лейдена), и наследственные или приобретенные дефициты антитромбина III, белка С, белка S, гепаринового кофактора II. Другие состояния, которые, как известно, связаны с гиперкоагуляцией и тромбоэмболическим заболеванием, включают в себя циркулирующие антитела против фосфолипидов (волчаночный антикоагулянт), гомоцистеинемию, вызванную гепарином тромбоцитопению и нарушения фибринолиза, а также коагуляционные синдромы (например, диссеминированное внутрисосудистое свертывание крови, ДВС-синдром (DIC)) и сосудистое повреждение в целом (например, в результате хирургического вмешательства).
Лечение состояний, при которых существует нежелательный избыток тромбина без симптомов гиперкоагуляции, например при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера
Конкретные болезненные состояния, которые могут быть упомянуты, включают в себя терапевтическое и/или профилактическое лечение венозного тромбоза (например, тромбоза глубоких вен) и эмболии легких, артериального тромбоза (например, при инфаркте миокарда, нестабильной стенокардии, вызванном тромбозом ударе и тромбозе периферических артерий), и системной эмболии, обычно из предсердия во время фибрилляции предсердий (например, неклапанной фибрилляции предсердий) или из левого желудочка после трансмурального инфаркта миокарда, или вызванной застойной сердечной недостаточностью, профилактику повторной окклюзии (т.е. тромбоза) после тромболиза, чрескожную внутрипросветную ангиопластику (РТА) и операции коронарного шунтирования, предотвращение повторного тромбоза после микрохирургической операции и сосудистой хирургической операции в общем.
Дополнительные показания включают в себя терапевтическое и/или профилактическое лечение диссеминированного внутрисосудистого свертывания крови, вызванного бактериями, множественной травмой, интоксикацией или любым другим механизмом; лечение антикоагулянтами, когда кровь контактирует с чужеродными поверхностями в организме, такими как сосудистые трансплантаты, сосудистые стенты, сосудистые катетеры, механические и биологические искусственные клапаны или любое другое медицинское устройство, и лечение антигоагулянтами, когда кровь контактирует с медицинскими устройствами вне организма, как, например, во время сердечно-сосудистой хирургической операции с использованием аппарата искусственного кровообращения или при гемодиализе, терапевтическое и/или профилактическое лечение идиопатического и респираторного дистресс-синдрома взрослых, фиброза легких после лечения излучением или химиотерапии, септического шока, септицемии, воспалительных реакций, которые включают в себя отек, острый или хронический атеросклероз, как, например, ишемическая болезнь сердца и образование атеросклеротических бляшек, заболевание артерий головного мозга, церебральный инфаркт, церебральный тромбоз, церебральная эмболия, заболевание периферических артерий, ишемию, стенокардию (включая нестабильную стенокардию), реперфузионное повреждение, рестеноз после внутрипросветной ангиопластики (РТА) и хирургическую операцию коронарного шунтирования.
Соединения по данному изобретению, которые ингибируют трипсин и/или тромбин, также могут быть полезны в лечении панкреатита.
Таким образом, соединения по изобретению показаны как для терапевтического, так и/или для профилактического лечения этих состояний.
В соответствии с еще одним аспектом настоящего изобретения предложен способ лечения состояния, при котором требуется ингибирование тромбина, при котором субъекту, страдающему от такого состояния или подверженного ему, вводят терапевтически эффективное количество соединения по данному изобретению.
Соединения по данному изобретению обычно будут вводить перорально, внутривенно, подкожно, трансбуккально, ректально, через кожу, назально, в трахею, в бронхи, любым другим парентеральным путем или посредством ингаляции, в форме фармацевтических препаратов, содержащих соединение по изобретению в виде или свободного основания, или фармацевтически приемлемой нетоксичной соли присоединения органической или неорганической кислоты в фармацевтически приемлемой лекарственной форме.
Предпочтительными способами введения соединений по изобретению являются пероральные. Предпочтительные фармацевтические препараты включают в себя фармацевтические композиции с измененным высвобождением, содержащие соединения по изобретению. Термин фармацевтическая композиция «с измененным высвобождением» хорошо понятен специалисту как включающий в себя любую композицию, для которой начало и/или скорость высвобождения лекарственного средства (то есть соединения по изобретению) изменяют посредством галеновых процедур, и таким образом включает в себя определение, предусмотренное в Фармакопее США (USP XXII) на страницах xIiii и xIiv введения/вводной части, при этом релевантное описание этого документа введено сюда посредством ссылки.
Таким образом, подходящие препараты с измененным высвобождением могут быть приготовлены специалистом согласно стандартным методикам фармации (смотри, например, Pharmaceutisch Weekblad Scientific Edition, 6, 57 (1984); Medical Applications of Controlled Release, Vol II, eds. Langer and Wise (1984) Bocaration, Florida, на страницах 1-34; Industrial Aspects of Pharmaceuticats, ed. Sandel, Swedish Pharmaceutical Press (1993) на страницах 93-104; страницы 191-211 "Pharmaceutics: The Science of Dosage Form Design", ed. M. E. Aulton (1988) (Churchill Livingstone)).
Таким образом, предпочтительные препараты с измененным высвобождением включают в себя таковые, в которых соответствующее соединение по изобретению заключено в полимерной матрице. В этом отношении авторы предпочитают, чтобы препараты, включающие в себя соединения по изобретению, были предложены для перорального введения в форме так называемой "набухающей" системы с измененным высвобождением или системы с измененным высвобождением «с гелеобразующей матрицей», в которых соединение по изобретению предложено вместе с полимером, который набухает в водной среде (то есть с «гидрофильным гелеобразующим компонентом»).
В частности, авторы предпочитают, чтобы соединения по изобретению были приготовлены совместно в композиции с гелеобразующей матрицей, содержащей иота-каррагинан и один или более чем один нейтральный гелеобразующий полимер.
Иота-каррагинан предпочтительно представлен в таком предпочтительном препарате на уровне более чем 15% по массе. Предпочтительные типы иота-каррагинана включают в себя иота-каррагинан "фармацевтический" (имеется у FMC Biopolymer), вязкость которого составляет не менее 5 сантипуаз (сП), предпочтительно в интервале 5-10 сП (для 1,5%-ного раствора, нагретого до 82°С, после чего вязкость измеряют при 75°С с помощью вискозиметра Brookfield LV, оснащенного №1 осью, двигающейся со скоростью 30 об/мин), и иота-каррагинан "технический" (имеется у Fluka Biochemica), вязкость которого предпочтительно составляет не менее 14 мПа·с для 0,3%-ного водного раствора, нагретого до 20°С, после чего вязкость измеряют с помощью вискозиметра с падающим шариком, тип Haake, используемого вместе с термостатом Lauda C3 и Hakke Mess-системой III, и с использованием покрытых золотом шариков из нержавеющей стали с плотностью 7,8 г/см3.
Нейтральный гелеобразующий полимер может быть единичным или представлять собой смесь более чем одного нейтрального разрушаемого полимера, имеющего гелеобразующие свойства и обладающего значительной рН-независимой растворимостью. Уровень нейтрального гелеобразующего полимера в препарате предпочтительно составляет более 10%, но предпочтительно более 20% по массе.
Подходящие нейтральные гелеобразующие полимеры включают в себя полиэтиленоксид (РЕО), производные и члены РЕО-семейства (например, полиэтиленгликоль (ПЭГ), предпочтительно существующий в природе в твердом состоянии, с подходящей молекулярной массой или вязкостью). Если РЕО используется как единичный нейтральный гелеобразующий полимер, то он предпочтительно имеет ММ (молекулярную массу) ≥4 миллиона (4М), соответствуя интервалу вязкости водного раствора 1650-5500 мПа·с (или 1650-5500 сП; измеренной для 1%-ного водного раствора при 25°С с использованием вискозиметра Brookfield RVF с осью №2 при 2 об/мин). Другие примеры подходящих РЕО включают в себя РЕО с ММ приблизительно 5 миллионов (5М), соответствующей интервалу вязкости водного раствора 5500-7500 мПа·с, или РЕО с ММ приблизительно 8 миллионов (8М), соответствующей интервалу вязкости водного раствора 10000-15000 мПа·с. Этот интервал охватывает значение вязкости для типичного раствора (в сП), измеренное при 25°С, гарантированное для этого полимера в Фармакопее США 24/NF (Национальном формуляре) 19, 2000 edition, pp.2285-2286. Если в качестве единичного нейтрального гелеобразующего полимера используется ПЭГ, то он предпочтительно имеет высокую молекулярную массу, например ММ приблизительно 20000, что соответствует интервалу вязкости 2700-3500 мПа·с (или 2700-3500 сП), измеренной с использованием 50%-ного водного раствора (мас./мас.) при 20°С при использовании капиллярного вискозиметра (Ubbelohde или эквивалентного). [Ссылка: European Pharmacopoeia 3rd Ed., 2000, Supplement, pp.908-909.]
Другие подходящие гелеобразующие полимеры включают в себя производные целллюлозы, такие как гидроксипропилметилцеллюлоза (НРМС) или гидроксиэтилцеллюлоза (НЕС), с подходящими высокими значениями вязкостей (например, «НРМС 10000 сП», «НРМС 15000 сП», «НЕС, тип НН» или «НЕС, тип Н»). Если такие гидроксипропилметилцеллюлозные полимеры, как «НРМС 10000 сП» и «НРМС 15000 сП», используются в качестве единичного нейтрального полимера, они имеют соответственно значения кажущейся вязкости 7500-14000 мПа·с (или 7500-14000 сП) и 11250-21000 мПа·с (или 11250-21000 сП) при измерении при 20°С с использованием 2%-ного (мас./мас.) водного раствора, рассчитанные относительно сухого вещества, с использованием капиллярного вискозиметра (Ubbelohde или эквивалентного). Один из типов гидроксиэтилцеллюлозного полимера, например «Natrosol 250 Pharma, тип НН», от Hercules Incorporated (Aqualon), демонстрирует типично вязкость по Brookfield приблизительно в 20000 мПа·с при использовании прибора Brookfield Synchro-Lectric Model LVF в условиях 1%-ной концентрации раствора, с осью №4, скоростью оси 30 об/мин, с фактором 200, 25°С (смотри Natrosol Physical and Chemical Properties booklet, 33.007-E6 (1993), p.21).
Конкретные препараты, которые могут быть упомянуты, включают в себя такие препараты, в которых соединение по изобретению приготовлено совместно с иота-каррагинаном и НРМС (10000 сП) в соотношении 50:50 (мас.%) или совместно с иота-каррагинаном и РЕО 4М в соотношении 50:50 (мас.%).
Предпочтительные дополнительные эксципиенты в таких препаратах включают в себя смазывающие вещества, такие как стеарилфумарат натрия.
В зависимости от расстройства и пациента, которому требуется лечение, и способа введения данные композиции могут быть введены в различных дозах.
Кроме того, соединения по изобретению могут быть комбинированы и/или совместно введены с антитромботическим(и) агентом(ами) с другим механизмом действия, таким как один или более чем один из следующего: с антитромбоцитарными агентами - ацетилсалициловой кислотой, тиклопидином и клопидогрелем; ингибиторами рецепторов и/или синтетазы тромбоксана; антагонистами рецепторов фибриногена; миметиками простациклина; ингибиторами фосфодиэстеразы; антагонистами ADP(аденозиндифосфат)-рецепторов (Р2T) и ингибиторами карбоксипептидазы U (CPU).
Соединения по изобретению могут быть дополнительно комбинированы и/или совместно введены с тромболитическими агентами, такими как один или более чем один тканевый активатор плазминогена (природный, рекомбинантный или измененный), стрептокиназа, урокиназа, проурокиназа, анизоилированный плазминоген-стрептокиназный активаторный комплекс (APSAC), активаторы плазминогена слюнных желез животных и им подобные, в лечении тромботических заболеваний, в частности инфаркта миокарда.
Таким образом, согласно еще одному аспекту данного изобретения предложен фармацевтический препарат, содержащий соединение по изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Подходящие суточные дозы соединений по изобретению для терапевтического лечения людей составляют приблизительно 0,001-100 мг/кг массы тела при пероральном введении и 0,001-50 мг/кг массы тела при парентеральном введении.
Во избежание сомнения термин «лечение», как он использован здесь, включает в себя терапевтическое и/или профилактическое лечение.
Соединения по данному изобретению имеют преимущество перед известными из уровня техники соединениями в том, что они могут быть более эффективными, менее токсичными, быть с более продолжительным действием, иметь более широкий интервал по активности, быть более сильнодействующими, оказывать меньше побочных действий, более легко абсорбироваться и/или иметь лучший фармакокинетический профиль (например, более высокую пероральную биодоступность и/или более низкий клиренс), и/или иметь другие полезные фармакологические, физические или химические свойства. Соединения по изобретению могут иметь дополнительное преимущество перед известными из уровня техники соединениями в том, что их можно вводить реже.
Биологические тесты
Могут быть использованы следующие процедуры анализа.
Тест А
Определение времени свертывания плазмы крови после добавления тромбина (тромбинового времени) (ТТ)
Раствор ингибитора (25 мкл) инкубируют с плазмой (25 мкл) в течение 3 минут. Затем добавляют тромбин человека (Т 6769, Sigma Chem. Co или Hematologic Technologies) в буферном растворе, рН 7,4 (25 мкл, 4,0 NIH (Национальный институт здравоохранения) единицы/мл), и измеряют время свертывания на автоматическом приборе (КС 10, Amelung).
Тромбиновое время (ТТ) выражают в абсолютных значениях (секундах), равно как и в виде отношения ТТ без ингибитора (ТТ0) к ТТ с ингибитором (ТТi). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании), и они соответствуют сигмоидальным кривым доза-ответ согласно уравнению
у=а/[1+(х/IC50)S],
где а = максимальный интервал, то есть 1; s = наклон кривой доза-ответ и IC50 = концентрация ингибитора, которая удваивает время свертывания. Вычисления выполняют на персональном компьютере (ПК) с использованием программного обеспечения GraFit Version 3, задавая уравнение равным сначала 0, определяем конечное значение = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство Великобритании и Северной Ирландии).
Тест В
Определение ингибирования тромбина с помощью хромогенногоавтоматизированного анализа
Эффективность ингибитора тромбина измеряют методом с использованием хромогенного субстрата на автоматизированном процессоре для микропланшетов Plato 3300 (Rosys AG, CH-8634 Hombrechtikon, Швейцария), используя 96-луночные титрационные микропланшеты половинного объема (Costar, Cambridge, MA, США; № по каталогу 3690). Исходные растворы тестируемого вещества в DMSO (72 мкл), 0,1-1 ммоль/л, последовательно разводят DMSO как 1:3 (24+48 мкл) для получения десяти различных концентраций, которые анализируют в качестве образцов для анализа. 2 мкл тестируемого образца разбавляют 124 мкл буфера для анализа, 12 мкл раствора хромогенного субстрата (S-2366, Chromogenix, Mölndal, Швеция) в буфере для анализа и в конце добавляют 12 мкл раствора α-тромбина (α-тромбина человека, Sigma Chemical Co. или Hematologic Technologies) в буфере для анализа, и образцы перемешивают. Конечные анализируемые концентрации составляют: тестируемое вещество 0,00068-13,3 мкмоль/л, S-2366 0,30 ммоль/л, α-тромбин 0,020 единиц NIH/мл. Для вычисления процента ингибирования для тестируемых образцов используют линейное увеличение поглощения в течение 40 минут инкубации при 37°С в сравнении с контрольными пробами без ингибитора. Автоматически регистрируемую величину IC50, соответствующую концентрации ингибитора, вызывающей 50%-ное ингибирование активности тромбина, рассчитывают из кривой зависимости логарифма концентрации от % ингибирования.
Тест С
Определение константы ингибирования Кi для тромбина человека
Определения Кi производят, применяя метод с использованием хромогенного субстрата, при 37°С на центрифужном анализаторе Cobas Bio (Roche, Basel, Швейцария). Остаточную ферментативную активность после инкубации α-тромбина человека с тестируемым соединением, взятым в различных концентрациях, определяют при трех различных концентрациях субстрата и измеряют как изменение оптической плотности при 405 нм.
Растворы тестируемого соединения (100 мкл; обычно в буфере или физиологическом растворе, содержащем BSA 10 г/л) смешивают с 200 мкл α-тромбина человека (Sigma Chemical Co) в буфере для анализа (0,05 моль/л Трис-HCl, рН 7,4, с ионной силой 0,15, доведенной с помощью NaCl), содержащем BSA (10 г/л), и анализируют как образцы на Cobas Bio. К 320 мкл субстрата S-2238 (Chromogenix AB, Mölndal, Швеция) в буфере для анализа добавляют 60 мкл образца вместе с 20 мкл воды и регистрируют изменение плотности (ΔА/мин). Конечные концентрации S-2238 составляют 16, 24 и 50 мкмоль/л, а тромбина 0,125 единиц NIH/мл.
Скорость реакции в стационарном состоянии используют для построения графиков Диксона, то есть диаграмм, показывающих изменение 1/(ΔА/мин) от концентрации ингибитора. Для обратимых конкурентных ингибиторов экспериментальные точки для различных концентраций субстрата обычно образуют прямые линии, которые пересекаются при х=-Кi.
Тест D
Определение частичного активированного тромбопластинового времени (АРТТ)
АРТТ определяют в объединенных образцах нормальной цитратной плазмы человека с помощью реагента РТТ Automated 5, производимого Stago. Ингибиторы добавляют к плазме (10 мкл раствора ингибитора к 90 мкл плазмы), инкубируют с АРТТ-реагентом в течение 3 минут с последующим добавлением 100 мкл раствора хлорида кальция (0,025 М) и определяют АРТТ, используя анализатор свертывания КС10 (Amelung) в соответствии с инструкциями производителя реагента.
Время свертывания выражают в абсолютных значениях (секундах), равно как и в виде отношения АРТТ без ингибитора (АРТТ0) к АРТТ с ингибитором (APTTi). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании), и они соответствуют сигмоидальным кривым доза-ответ согласно уравнению
у=а/[1+(х/IC50)s],
где а = максимальный интервал, то есть 1; s = наклон кривой доза-ответ и IC50 = концентрация ингибитора, которая удваивает время свертывания. Вычисления выполняют на ПК с использованием программного обеспечения GraFit Version 3, задавая уравнение равным сначала 0, определяем конечное значение = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство Великобритании и Северной Ирландии).
IC50APTT определяют как концентрацию ингибитора в плазме человека, которая удваивала частичное активированное тромбопластиновое время.
Тест Е
Определение тромбинового времени ex vivo
Ингибирование тромбина после перорального или парентерального введения соединений по изобретению, растворенных в смеси этанол/Солютол К (SolutolK)/вода (5:5:90) исследуют на находящихся в сознании крысах, которым за один или два дня до эксперимента вводят катетер для взятия оббразцов крови из сонной артерии. В день эксперимента в фиксированное время после введения соединения отбирают образцы крови в пластиковые пробирки, содержащие 1 часть раствора цитрата натрия (0,13 моль на 1 л) и 9 частей крови. Пробирки центрифугируют для получения плазмы, обедненной тромбоцитами.
Образцы плазмы в объеме 50 мкл осаждают 100 мкл охлажденного ацетонитрила. Образцы центрифугируют в течение 10 минут при 4000 об/мин. 75 мкл супернатанта разбавляют 75 мкл 0,2%-ной муравьиной кислоты. Получающиеся растворы в объеме 10 мкл анализируют с помощью LC-MS/MS и, используя калибровочные кривые, определяют концентрации ингибитора тромбина.
Тест F
Определение плазменного клиренса у крысы
Плазменный клиренс оценивают на самцах крыс линии Sprague Dawley. Соединение растворяют в воде и вводят в виде подкожной болюсной инъекции в дозе 4 мкмоль/кг. Собирают образцы крови через повторяющиеся интервалы вплоть до 5 часов после введения лекарственного средства. Образцы крови центрифугируют, плазму отделяют от клеток крови и переносят в пробирки, содержащие цитрат (в конечной концентрации 10%). Образцы плазмы в объеме 50 мкл осаждают 100 мкл охлажденного ацетонитрила. Образцы центрифугируют в течение 10 минут при 4000 об/мин. 75 мкл супернатанта разбавляют 75 мкл 0,2%-ной муравьиной кислоты. Получающиеся растворы в объеме 10 мкл анализируют с помощью LC-MS/MS и, используя калибровочные кривые, определяют концентрации ингибитора тромбина. Площадь под кривой «концентрация плазмы - время» оценивают с использованием log/линейной формулы трапеций и экстраполируют к бесконечному времени. Затем определяют плазменный клиренс (CL) соединения как
CL = доза/AUC (площадь под кривой).
Значения выражают в мл/мин/кг.
Тест G
Определение стабильности in vitro
Микросомы печени получают из образцов печени крыс линии Sprague-Dawley и людей согласно внутренним SOP (стандартным рабочим процедурам). Соединения инкубируют при 37°С при общей концентрации микросомального белка, равной 3 мг/мл, в 0,05 моль/л Трис-буфере при рН 7,4 в присутствии кофакторов NADH (2,5 ммоль/л) и NADPH (0,8 ммоль/л). Начальная концентрация соединения составляет 5 или 10 мкмоль/л. Отбирают образцы для анализа вплоть до 60 минут после начала инкубации. Ферментативную активность в отобранном образце немедленно останавливают добавлением 20%-ной муристиновой кислоты в объеме, соответствующем 3,3% от общего объема образца. Концентрацию соединения, остающуюся в образце через 60 мин (FINAL CONC), определяют с помощью LCMS, используя образец, собранный в нулевой момент времени, в качестве эталонного образца (START CONC).% разрушенного ингибитора тромбина вычисляют как
100%×{[START CONC]-[FINAL CONC]}/[START CONC].
Тест Н
Модель артериального тромбоза
Индуцируют повреждение сосуда путем аппликации хлорида железа (FeCl3) местно на сонную артерию. Крыс анестезируют с помощью внутрибрюшинной инъекции пентобарбитала натрия (80 мг/кг; Apoteksbolaget; Umeå, Швеция) с последующей непрерывной инфузией (12 мг/кг/ч) в продолжение эксперимента. Температуру тела крысы поддерживают равной 38°С в продолжение эксперимента с помощью внешнего обогрева. Эксперимент начинают с 5-минутного контрольного периода. Спустя пять минут внутривенно вводят человеческий 125I-фибриноген (80 кБк; IM53; Amersham International, Buckinghamshire, Соединенное Королевство Великобритании и Северной Ирландии) и используют в качестве маркера для последующего включения фибрин(оген)а в тромб. Проксимальный конец сегмента сонной артерии размещают в пластиковой трубке (6 мм; Silastic®, Dow Corning, MI, США), открытой вдоль, содержащей пропитанную FeCl3 (2 мкл; 55 мас.%; Merck, Дармштадт, Германия) фильтровальную бумагу (диаметр 3 мм; 1F; Munktell, Grycksbo, Швеция). Левую сонную артерию подвергают действию FeCl3 в течение 10 минут и затем удаляют из пластиковой трубки и пропитывают физиологическим раствором. Спустя пятьдесят минут удаляют сонную артерию и промывают в физиологическом растворе. Также отбирают эталонные образцы крови для определения 125I-активности крови, через 10 минут после инъекции 125I-фибриногена и в конце эксперимента. 125I-Активность в эталонных образцах крови и сегменте сосуда измеряют в гамма-счетчике (1282 Compugamma; LKB Wallac Oy, Турку, Финляндия) в день выполнения эксперимента. Размер тромба определяют как количество 125I-активности, включенной в сегмент сосуда, по отношению к 125I-активности в крови (имп/мин/мг).
Общие экспериментальные методики
ТСХ выполняли на силикагеле. Анализ с помощью хиральной ВЭЖХ выполняли с использованием колонки Chiralcel OD размером 46 мм х 250 мм с предколонкой размером 5 см. Температуру колонки поддерживали при 35°С. Использовали скорость потока 1,0 мл/мин. Использовали УФ-детектор Gilson 115 при 228 нм. Подвижная фаза состояла из гексанов, этанола и трифторуксусной кислоты, и для каждого соединения даны соответствующие соотношения. Как правило, продукт растворяли в минимальном количестве этанола и его разбавляли подвижной фазой.
LC-MS/MS осуществляли с использованием прибора HP-1100, оборудованного CTC-PAL-инжектором и колонкой ThermoQuest, Hypersil BDS-С18, 5 мкм, 4×100 мм. Использовали MS-детектор API-3000 (Sciex). Скорость потока составляла 1,2 мл/мин, и подвижная фаза (градиент) состояли из 10-90%-ного ацетонитрила с 90-10%-ным 4 мМ водным ацетатом аммония, каждый из которых содержал 0,2%-ную муравьиную кислоту.
1H ЯМР-спектры записывали с использованием тетраметилсилана в качестве внутреннего стандарта. 13С ЯМР-спектры записывали с использованием номенклатурных дейтерированных растворителей в качестве внутреннего стандарта.
Пример 1
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-цикло-Bu)
(1) 3-Хлор-5-метоксибензальдегид
3,5-Дихлоранизол (74,0 г; 419 ммоль) в THF (200 мл) по каплям добавляли к металлическому магнию (14,2 г; 585 ммоль; предварительно промытому 0,5 н. HCl) в THF (100 мл) при 25°С. После этого добавления по каплям добавляли 1,2-дибромэтан (3,9 г; 20,8 ммоль). Получающуюся темно-коричневую смесь нагревали с обратным холодильником в течение 3 ч. Смесь охлаждали до 0°С и добавляли N,N-диметилформамид (60 мл) в виде одной порции. Смесь распределяли с помощью диэтилового эфира (3×400 мл) и 6 н. HCl (500 мл). Объединенные органические экстракты промывали соляным раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. Посредством флэш-хроматографии (2x) на силикагеле с элюированием смесью Нех/EtOAc (4:1) получили указанное в подзаголовке соединение (38,9 г; 54%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 9.90 (s, 1H), 7.53 (s, 1Н), 7.38 (s, 1H,), 7.15 (s, 1H), 3.87(s, 3H).
(2) 3-Хлор-5-гидроксибензальдегид
Раствор 3-хлор-5-метоксибензальдегида (22,8 г; 134 ммоль; смотри стадию (1) выше) в CH2Cl2 (250 мл) охлаждали до 0°С. По каплям в продолжение 15 мин добавляли трибромид бора (15,8 мл; 167 ммоль). После перемешивания реакционной смеси в течение 2 ч медленно добавляли H2O (50 мл). Затем раствор экстрагировали Et2O (2×100 мл). Органические слои объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Посредством флэш-хроматографии на силикагеле с элюированием смесью Нех/EtOAc (4:1) получили указанное в подзаголовке соединение (5,2 г; 25%).
1H ЯМР (300 МГц; CDCl3) δ 9.85 (s, 1H), 7.35 (s, 1Н), 7.20 (s, 1H), 7.10 (s, 1H), 3.68 (s, 1H).
(3) 3-Хлор-5-дифторметоксибензальдегид
Раствор 3-хлор-5-гидроксибензальдегида (7,5 г; 48 ммоль; смотри стадию (2) выше) в 2-пропаноле (250 мл) и 30%-ном КОН (100 мл) нагревали до температуры дефлегмации. Реакционную смесь при перемешивании в течение 2 ч барботировали CHCIF2. Реакционную смесь охлаждали, подкисляли 1 н. HCl и экстрагировали EtOAc (2×100 мл). Органические экстракты промывали соляным раствором (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Посредством флэш-хроматографии на силикагеле с элюированием смесью Нех/EtOAc (4:1) получили указанное в подзаголовке соединение (4,6 г; 46%).
1H ЯМР (300 МГц; CDCl3) δ 9.95 (s, 1H), 7.72 (s, 1H), 7.52 (s, 1H), 7.40 (s, 1Н), 6.60 (t, JH-F=71.1 Гц, 1Н).
(4) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN
Раствор 3-хлор-5-дифторметоксибензальдегида (4,6 г; 22,3 ммоль; смотри стадию (3) выше) в CH2Cl2 (200 мл) охлаждали до 0°С. Добавляли Znl2 (1,8 г; 5,6 ммоль) и триметилсилилцианид (2,8 г; 27,9 ммоль), реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 15 ч. Данную смесь частично концентрировали в вакууме, получая указанное в подзаголовке соединение в виде жидкости, которую использовали непосредственно на приведенной ниже стадии (5) без дальнейшей очистки или определения характеристик.
(5) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt
Ph(3-Cl)(5-OCHF2-(R,S)CH(OTMS)CN (6,82 г; предположительно 22,3 ммоль; смотри стадию (4) выше) по каплям добавляли к смеси HCl/EtOH (500 мл). Реакционную смесь перемешивали 15 ч, затем частично концентрировали в вакууме, получая указанное в подзаголовке соединение в виде жидкости, которую использовали на приведенной ниже стадии (6) без дальнейшей очистки или определения характеристик.
(6) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt (6,24 г; предположительно 22,3 ммоль; смотри стадию (5) выше) растворяли в THF (250 мл), добавляли 0,5 М H2SO4 (400 мл) и реакционную смесь перемешивали при 40°С в течение 65 ч, охлаждали и затем частично концентрировали в вакууме для удаления большей части THF. Затем реакционную смесь экстрагировали Et2O (3×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения в виде твердого вещества, которое использовали на стадии (7) без дальнейшей очистки или определения характеристик.
(7) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt (6,25 г; предположительно 22,3 ммоль; смотри стадию (6) выше) в 2-пропаноле (175 мл) и 20%-ном КОН (350 мл) перемешивали при комнатной температуре 15 ч. Реакционную смесь затем частично концентрировали в вакууме для удаления большей части 2-пропанола. Оставшуюся смесь подкисляли 1 М H2SO4, экстрагировали Et2O (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества. Посредством флэш-хроматографии на силикагеле с элюированием смесью CHCl3/МеОН/концентрированный NH4OH (6:3:1) получили аммониевую соль указанного в подзаголовке соединения. Эту аммониевую соль затем растворяли в смеси EtOAc (75 мл) и Н2О (75 мл) и подкисляли 2 н. HCl. Органический слой отделяли и промывали соляным раствором (50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением указанного в подзаголовке соединения (3,2 г; 57% со стадий от (4)-(7)).
1H ЯМР (300 МГц; CD3OD) δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71.1 Гц, 1H), 5.16 (s, 1H).
(8) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (а) и Ph(3-Cl)(5-OCHF2)-(S)CH(OAc)C(O)OH (б)
Смесь Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH (3,2 г; 12,7 ммоль; смотри стадию (7) выше) и Липазы PS "Amano" (˜2,0 г) в винилацетате (125 мл) и МТВЕ (125 мл) нагревали с обратным холодильником в течение 48 ч. Реакционную смесь охлаждали, фильтровали через Целит® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле с элюированием смесью CHCl3/МеОН/концентрированный NH4OH (6:3:1), получая аммониевую соль указанных в подзаголовке соединений (а) и (б). Соединение (а) в виде соли растворяли в Н2О, подкисляли 2 н. HCl и экстрагировали EtOAc. Органический слой промывали соляным раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получениме указанного в подзаголовке соединения (а) (1,2 г; 37%).
Для указанного в подзаголовке соединения (а)
1H ЯМР (300 МГц; CD3OD) δ 7.38 (s, 1H), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71.1 Гц, 1Н), 5.17 (s, 1Н).
(9) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (1,1 г; 4,4 ммоль; смотри стадию (8) выше) и H-Aze-Pab(Teoc) (смотри международную патентную заявку WO 00/42059; 2,6 г; 5,7 ммоль) в DMF (50 мл) при 0°С добавляли РуВОР (2,8 г; 5,3 ммоль) и коллидин (1,3 г; 10,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение дополнительных 15 ч. Реакционную смесь концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле (3х) с элюированием сначала смесью CHCl3/EtOH (9:1), затем EtOAc/EtOH (20:1) и окончательно с элюированием смесью СН2Cl2/СН3ОН (95:5) с получением указанного в подзаголовке соединения (1,0 г; 37%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц; CD3OD; смесь ротамеров) δ 7.79-7.85 (d, J=8.7 Гц, 2Н), 7.15-7.48 (m, 5Н), 6.89 и 6.91 (t, jH-F=71.1 Гц, 1H), 5.12 и 5.20 (s, 1H), 4.75-4.85 (m, 1H), 3.97-4.55 (m, 6H), 2.10-2.75 (m, 2H), 1.05-1.15 (m, 2H), 0.09 (s, 9H).
MS (m/z) 611 (M+1)+.
(10) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-цикло-Bu, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,051 г; 0,08 ммоль; смотри стадию (9) выше) растворяли в 3 мл ацетонитрила и добавляли 0,062 г (0,5 ммоль) гидрохлорида O-циклобутилгидроксиламина. Смесь нагревали при 70°С в течение 4,5 ч. Растворитель выпаривали и остаток распределяли между водой и этилацетатом. Водную фазу экстрагировали еще два раза этилацетатом и объединенную органическую фазу промывали водой, соляным раствором, сушили (Na2SO4), фильтровали и упаривали. Выход 0,054 г (95%).
1H-ЯМР (400 МГц; CD3OD): δ 8.66-8.50 (m, 1Н), 7.45 (d, 2H), 7.29 (m, 3Н), 7.15 (m, 2H), 6.88 (t, 1H основной ротамер), 6.85 (t, 1H второстепенный ротамер), 5.18 (s, 1H основной ротамер), 5.12 (s, 1H второстепенный ротамер), 5.16 (m, 1H второстепенный ротамер), 4.78 (m, 1H основной ротамер), 4.70 (m, 1H), 4.50-4.30 (m, 3Н), 4.19-3.93 (m, 3Н), 2.71-2.44 (m, 1H), 2.34-2.11 (m, 5H), 1.78 (m, 1H), 1.62 (m, 1H), 0.96 (m, 2H), 0.01 (s, 9H).
(11) Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(O-цикло-Bu)
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(O-цикло-Bu, Teoc) (0,054 г; 0,08 ммоль; смотри стадию (10) выше) растворяли в 0,5 мл CH2Cl2 и 3 мл TFA. Реакцию оставляли протекать в течение 60 минут. TFA выпаривали и остаток очищали, используя препаративную ВЭЖХ. Интересующие фракции объединяли и сушили вымораживанием (2х), получая 23 мг (54%) указанного в заголовке соединения.
MS (m/z) 536 (M-1)-; 538 (M+1)+.
1H-ЯМР (400 МГц; CD3OD): δ 7.56 (d, 2H), 7.33 (m, 3Н), 7.15 (m, 2H), 6.89 (t, 1H основной ротамер), 6.86 (t, 1H второстепенный ротамер), 5.18 (s, 1H основной ротамер, и m, 1H второстепенный ротамер), 5.11 (s, 1H второстепенный ротамер), 4.77 (m, 1H основной ротамер), 4.58 (m, 1H), 4.42 (m, 2H), 4.34 (m, 1H основной ротамер), 4.15 (m, 1H основной ротамер), 4.06 (m, 1H второстепенный ротамер), 3.97 (m, 1H второстепенный ротамер), 2.66 (m, 1H второстепенный ротамер), 2.52 (m, 1H основной ротамер), 2.33-2.25 (m, 3Н), 2.01-2.20 (m, 2H), 1.75 (m, 1H), 1.59 (m, 1H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.4, 172.3, 171.9, 171.4, 152.3.
Пример 2
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,148 г; 0,24 ммоль; смотри пример 1(9) выше) растворяли в 9 мл ацетонитрила и добавляли 0,101 г (1,45 ммоль) гидрохлорида гидроксиламина. Смесь нагревали при 70°С в течение 2,5 ч, фильтруют через Целит® и упаривают. Неочищенный продукт (0,145 г; 75%-ной чистоты) непосредственно использовали на следующей стадии без дальнейшей очистки.
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc) (0,145 г; 0,23 ммоль; смотри стадию (1) выше) растворяли в 0,5 мл CH2Cl2 и 9 мл TFA. Реакции позволяли протекать в течение 60 минут. Выпаривали TFA и остаток очищали с использованием препаративной ВЭЖХ. Интересующие фракции собирали и сушили вымораживанием (2х), получая 72 мг (выход за две стадии 62%) указанного в заголовке соединения.
MS (m/z) 482 (М-1)-; 484 (М+1)+.
1H-ЯМР (400 МГц; CD3OD): δ 7.58 (d, 2Н), 7.33 (m, 3H), 7.15 (m, 2H), 6.89 (t, 1H основной ротамер), 6.86 (t, 1H второстепенный ротамер), 5.18 (s, 1H основной ротамер, и m, 1H второстепенный ротамер), 5.12 (s, 1H второстепенный ротамер), 4.77 (m, 1H основной ротамер), 4.42 (m, 2H), 4.34 (m, 1H основной ротамер), 4.14 (m, 1H основной ротамер), 4.06 (m, 1H второстепенный ротамер), 3.95 (m, 1H второстепенный ротамер), 2.66 (m, 1H второстепенный ротамер), 2.50 (m, 1H основной ротамер), 2.27 (m, 1H основной ротамер), 2.14 (m, 1H второстепенный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.4, 172.3, 172.0, 171.4, 152.3, 152.1.
Пример 3
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,045 г; 0,074 ммоль; смотри пример 1(9) выше) растворяли в 3 мл TFA и позволяли протекать реакции в течение 1 ч. Выпаривали TFA и остаток сушили вымораживанием из смеси вода/ацетонитрил с получением 0,043 г (100%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.8-7.75 (m, 2H), 7.55-7.5 (m, 2H), 7.35 (m, 1H, основной ротамер), 7.31 (m, 1H, второстепенный ротамер), 7.19 (m, 1Н, основной ротамер), 7.15 (m, 1H), 7.12 (m, 1H, второстепенный ротамер), 6.89 (t, 1H, основной ротамер), 6.87 (t, 1H, второстепенный ротамер), 5.22 (m, 1H, второстепенный ротамер), 5.20 (s, 1H, основной ротамер), 5.13 (s, 1H, второстепенный ротамер), 4.80 (m, 1H, основной ротамер), 4.6-4.4 (m, 2H), 4.37 (m, 1H, основной ротамер), 4.19 (m, 1H, основной ротамер), 4.07 (m, 1H, второстепенный ротамер), 3.98 (m, 1H, второстепенный ротамер), 2.70 (m, 1H, второстепенный ротамер), 2.55 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.15 (m, 1H, второстепенный ротамер).
13С-ЯМР (100 МГц; CD3OD): (карбонильные и/или амидинные углероды, ротамеры) δ 172.6, 172.5, 172.0, 171.7, 167.0.
MS (m/z) 465 (M-1)-; 467 (M+1)+.
Пример 4
Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(COO-циклопентил)
К раствору Ph(3-Cl)(5-OCHF2)-(R)(OH)C(O)-Aze-Pab × TFA (74 мг; 0,13 ммоль; смотри пример 3 выше) и циклопентилхлорформиата (44 мг; 0,30 ммоль) в метиленхлориде (5 мл) добавляли водный NaOH (0,5 мл; 2 М; 1 ммоль). Смесь перемешивали при комнатной температуре и реакцию контролировали с помощью ВЭЖХ. Через 2,5 часа добавляли воду и отделяли жидкие фазы. Водную фазу дважды экстрагировали метиленхлоридом. Объединенные органические фазы сушили (MgSO4) и очищали на силикагеле (сначала метиленхлорид, затем EtOAc). После удаления растворителей в вакууме твердый остаток растворяли в смеси вода/ацетонитрил и сушили вымораживанием с получением указанного в заголовке соединения в виде твердого вещества белого цвета. Выход 33 мг (44%).
MS (m/z) 579 (M+1)+.
1H ЯМР (400 МГц; CD3OD): δ 7.79 (d, 2H), 7.43-7.30 (т, 5Н), 7.20-7.11 (т, 2H), 6.90 (t, 1H, основной ротамер), 6.87 (t, 1H, второстепенный ротамер), 5.19 (dd, 1H, второстепенный ротамер), 5.18 (s, 1H, основной ротамер), 5.13 (m, 1H), 5.11 (s, 1H, второстепенный ротамер), 4.78 (dd, 1H, основной ротамер), 4.45 (m, 2Н), 4.35 (m, 1H, основной ротамер), 4.16 (s, 1H, основной ротамер), 4.06 (s, 1H, второстепенный ротамер), 3.97 (s, 1H, второстепенный ротамер), 2.68 (m, 1H, второстепенный ротамер), 2.52 (s, 1H, основной ротамер), 2.28 (s, 1H, основной ротамер), 2.16 (s, 1H, второстепенный ротамер), 1.90 (m, 2H), 1.77 (m, 4H), 1.61 (m, 2H).
13С ЯМР (карбонильные и/или амидинные протоны; 100 МГц): δ 173.6, 173.1, 172.6, 170.3, 165.6.
Пример 5
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Z)
Указанное в заголовке соединение получали согласно процедуре, описанной выше в примере 4, начиная с Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab × TFA (73 мг; 0,13 ммоль; смотри пример 3 выше) и бензилхлорформиата (35 мг; 0,21 ммоль). Необходима дополнительная очистка с помощью обращенно-фазовой ВЭЖХ (0,1 М ацетат аммония/MeCN (40:60)). Соответствующие фракции концентрировали в вакууме и экстрагировали EtOAc. Выход 24 мг (32%).
MS (m/z) 602 (M+1)+.
1H ЯМР (400 МГц; CD3OD): δ 7.80 (d, 2H), 7.43-7.25 (m, 8Н), 7.20-7.10 (m, 2H), 6.90 (t, 1H, основной ротамер), 6.88 (t, 1H, второстепенный ротамер), 5.18 (dd, 1H, второстепенный ротамер), 5.18 (s, 2H), 5.17 (s, 1H, ротамер), 5.11 (s, 1H, ротамер), 4.78 (dd, 1H, основной ротамер), 4.45 (m, 2H), 4.34 (m, 1H, основной ротамер), 4.15 (s, 1H, основной ротамер), 4.06 (s, 1H, второстепенный ротамер), 3.97 (s, 1H, второстепенный ротамер), 2.66 (m, 1H, второстепенный ротамер), 2.51 (s, 1H, основной ротамер), 2.27 (s, 1H, основной ротамер), 2.15 (s, 1H, второстепенный ротамер).
13С ЯМР (карбонильные и/или амидинные протоны; 100 МГц): δ 173.6, 173.1, 172.6, 170.5, 164.9.
Пример 6
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab × TFA
(1) 2-Нитро-5-трифторметоксибензойная кислота
К раствору 3-трифторметоксибензойной кислоты (49,0 г; 0,24 моль) в серной кислоте (500 мл) при температуре менее 0°С (баня лед-МеОН) добавляли раствор нитрата калия (31,3 г; 0,31 моль) в серной кислоте (200 мл) в продолжение 20 минут. Получающийся раствор перемешивали при 0°С в течение 2 ч, затем нагревали до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь выливали на лед и получающийся кислотный раствор экстрагировали EtOAc (5х). Объединенные органические экстракты промывали Н2О (1х), соляным раствором (2х), Н2О (1х) и соляным раствором (1х), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (65,7 г) в виде твердого вещества с примесью НОАс. Неочищенное указанное в подзаголовке соединение растворяли в EtOAc и толуоле и концентрировали в вакууме с получением свободного от НОАс твердого вещества (58,4 г; 97%), которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3): δ 10.10 (br s, 1H), 8.02 (d, 1H, J=8 Гц), 7.69 (d, 1H, J=2 Гц), 7.54 (dd, 1Н, J=2 Гц, J=8 Гц).
(2) 2-Амино-5-трифторметоксибензойная кислота
К раствору 2-нитро-5-трифторметоксибензойной кислоты (56,8 г; 0,23 моль; смотри стадию (1) выше) в EtOH (1000 мл) добавляли 10%-ный Pd/C (5,7 г). Через получающийся раствор пропускали струю Н2 в течение 5 ч, фильтровали через Целит® и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (49,7 г; 98%) в виде твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD): δ 7.66 (m, 1H), 7.17 (d, 1H, J=8 Гц), 6.77 (d, 1Н, J=8 Гц).
(3) 2-Амино-3-хлор-5-трифторметоксибензойная кислота
К раствору 2-амино-5-трифторметоксибензойной кислоты (49,0 г; 0,22 моль; смотри стадию (2) выше) в НОАс (1200 мл) медленно добавляли сульфурилхлорид (41,8 г; 0,31 моль). Наблюдали выделение газа. Получающуюся гетерогенную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли дополнительное количество НОАс (300 мл), чтобы способствовать перемешиванию, с последующим добавлением сульфурилхлорида порциями по 5 мл до тех пор, пока исходный материал не был израсходован (по данным ТСХ-анализа). Реакционную смесь концентрировали в вакууме с получением твердых веществ, которые для удаления НОАс промывали на роторном испарителе EtOAc (2х) с последующим промыванием Et2O (1x). Получающиеся твердые вещества далее сушили с получением HCl-соли неочищенного указанного в подзаголовке соединения (60,5 г; 94%), которую использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD): δ 7.72 (s, 1Н), 7.44 (s, 1H), 7.22 (s, взаимозаменяемые).
(4) 3-Хлор-5-трифторметоксибензойная кислота
К раствору 2-амино-3-хлор-5-трифторметоксибензойной кислоты (60,5 г; предположительно 0,22 моль; смотри стадию (3) выше) в 1,4-диоксане (1000 мл) добавляли 6 н. HCl (750 мл). Некоторое количество органических веществ в виде масла выпало из раствора. Диоксановый раствор охлаждали до температуры менее 0°С (баня лед-МеОН). Через капельную воронку добавляли раствор нитрита натрия (18,2 г; 0,26 моль) в Н2O (250 мл) в продолжение 15 минут. Получающийся раствор перемешивали в течение 45 мин. Через капельную воронку медленно добавляли фосфорноватистую кислоту (221,5 мл 50 мас.%-ной в Н2О; 291,2 г; 2,20 моль). Раствор перемешивали при 0°С в течение 1,5 часа, затем нагревали до комнатной температуры (наблюдали выделение газа) и перемешивали в течение 18 часов. Неочищенный раствор переносили в делительную воронку и экстрагировали Et2O (4x). Объединенные органические экстракты экстрагировали водным NaHCO3 (3х). Основный водный слой осторожно подкисляли 6 н. HCl и экстрагировали CH2Cl2 (3х). СН2Cl2-Экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (26,5 г; 46% исходя из 3-трифторметоксибензойной кислоты) в виде твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD): δ 7.98 (s, 1H), 7.83 (s, 1H), 7.58 (s, 1H).
(5) 3-Хлор-5-трифторметоксибензиловый спирт
К раствору 3-хлор-5-трифторметоксибензойной кислоты (22,5 г; 93,5 ммоль; смотри стадию (4) выше) в безводном THF (1200 мл) в атмосфере N2 при комнатной температуре добавляли раствор комплекса BH3·THF (140 мл 1 М в THF; 140,3 ммоль). Раствор кипятили с обратным холодильником в течение 2 ч, охлаждали до комнатной температуры и перемешивали в течение 18 часов, осторожно гасили Н2O и концентрировали в вакууме для удаления как можно большего количества THF. Остаток разбавляли EtOAc и органическую фазу промывали соляным раствором (3х), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (21,2 г; 100%) в виде масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3): δ 7.33 (s, 1H), 7.17 (s, 1Н), 7.14 (s, 1H), 4.72 (s, 2H), 2.05 (br s, 1H).
(6) 3-Хлор-5-трифторметоксибензальдегид
Раствор DMSO (16,1 г; 205,9 ммоль) в безводном CH2Cl2 (300 мл) охлаждали до -78°С. Медленно с помощью шприца добавляли оксалилхлорид (13,1 г; 103,0 ммоль) (наблюдали выделение газа). Получающийся раствор перемешивали при -78°С в течение 15 минут. Через капельную воронку добавляли раствор 3-хлор-5-трифторметоксибензилового спирта (21,2 г; 93,6 ммоль; смотри стадию (5) выше) в CH2Cl2 (200 мл) в продолжение периода времени 15 минут. Непрозрачный раствор перемешивали при -78°С в течение 40 минут и через капельную воронку добавляли DIPEA (60,5 г; 468,0 ммоль) в продолжение 10 минут. Получающийся гомогенный раствор перемешивали при -78°С в течение 1,5 часа, затем нагревали до комнатной температуры и перемешивали 18 часов. Неочищенный раствор концентрировали в вакууме, остаток разбавляли EtOAc и промывали Н2O (1х), 2 н. HCl (1х), соляным раствором (1х), водным NaHCO3 (1x) и соляным раствором (1х). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (19,9 г; 95%), которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3): δ 10.00 (s, 1H), 7.83 (s, 1H), 7.66 (s, 1H), 7.51 (s, 1H).
(7) Ph(3-Cl)(5-OCF3)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-трифторметоксибензальдегида (19,9 г; 88,6 ммоль; смотри стадию (6) выше) в СН2Cl2 (600 мл) при 0°С добавляли Znl2 (1,4 г; 4,4 ммоль) и триметилсилилцианид (9,7 г; 97,5 ммоль). После перемешивания при 0°С в течение 1,5 часа и при комнатной температуре в течение 2 часов ТСХ-анализ показал наличие только исходного материала. Порциями добавляли Znl2 до тех пор, пока реакция не завершилась (в сумме добавили более 30,0 г Znl2). После перемешивания при комнатной температуре в течение 18 часов реакцию гасили водой и отделяли органическую фазу. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (27,7 г; 96%) в виде жидкости, которую использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3): δ 7.43 (s, 1H), 7.28 (s, 1Н), 7.25 (s, 1H), 5.49 (s, 1H), 0.38 (s, 9H).
(8) Ph(3-Cl)(5-OCF3)-(R,S)CH(OH)C(O)OH
Суспензию Ph(3-Cl)(5-OCF3)-(R,S)CH(OTMS)CN (27,7 г; 85,6 ммоль;
смотри стадию (7) выше) в концентрированной HCl (300 мл) кипятили с обратным холодильником в течение 3 часов. Получающуюся коричневую гетерогенную смесь охлаждали до комнатной температуры и экстрагировали Et2O (2x). Первоначальные органические экстракты экстрагировали 2 н. NaOH (2х), затем основный слой подкисляли 2 н. HCl и экстрагировали Et2O. Et2O-экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (4,9 г; 21%). ТСХ-анализ первоначальных органических экстрактов все еще показывал наличие в них указанного в подзаголовке соединения, поэтому процедуру основной экстракции/подкисления повторяли с использованием 6 н. NaOH с получением дополнительного количества неочищенного указанного в подзаголовке соединения (2,8 г; 12%). ТСХ-анализ первоначальных органических экстрактов все еще показывал наличие в них указанного в подзаголовке соединения, поэтому органические экстракты сушили (Na2SO4) и концентрировали в вакууме с получением натриевой соли указанного в подзаголовке соединения (18,3 г) в виде масла. Данную соль затем перерастворяли в Et2O и органическую фазу подкисляли 2 н. HCl и промывали соляным раствором. Получающуюся органическую фазу сушили (Na2SO4), обрабатывали активированным углем, фильтровали через Целит® и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения (14,3 г; 62%) в виде твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD): δ 7.53 (s, 1H), 7.38 (s, 1H), 7.29 (s, 1H), 5.23 (s, 1H).
(9) Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(О)OH (а) и Ph(3-Cl)(5-OCF3)-(S)СН(ОАс)С(О)OH (б)
Смесь Ph(3-Cl)(5-OCF3)-(R,S)CH(OH)C(O)OH (7,7 г; 28,5 ммоль; смотри стадию (8) выше) и Липазы PS "Amano" (3,8 г) в МТВЕ (100 мл) и винилацетате (50 мл) перемешивали при температуре 60°С в течение 26 ч. Реакционную смесь охлаждали, фильтровали через Целит® и осадок на фильтре промывали EtOAc. Объединенные органические фазы концентрировали в вакууме. Посредством флэш-хроматографии на силикагеле с элюированием смесью CHCl3/МеОН/концентрированный NH4OH (6:3:1) получили смесь аммониевых солей указанного в подзаголовке соединения (а) и указанного в подзаголовке соединения (б) (6,7 г) и чистый образец аммониевой соли указанного в подзаголовке соединения (а) (1,2 г) с энантиомерным избытком (е.е.) менее 95%. Соответствующие фракции растворяли в Et2O и промывали 2 н. HCl (1х) и соляным раствором (1х), сушили (Na2SO4), фильтровали и концентрировали с получением соответствующих карбоновых кислот (6,7 г и 1,1 г соответственно). Затем эти фракции по отдельности повторно переводили в условия для проведения разделения и повторно очищали, как необходимо, посредством хроматографии на силикагеле с элюированием смесью CHCl3/МеОН/концентрированный NH4OH (6:3:1, или 75:20:5, или 145:45:10), как необходимо. Очищенное указанное в подзаголовке соединение (а) подкисляли водной HCl или водной лимонной кислотой перед дальнейшим использованием. Аммониевую соль указанного в подзаголовке соединения (б) использовали без определения характеристик.
Для указанного в подзаголовке соединения (а)
1H ЯМР (300 МГц; CD3OD): δ 7.53 (s, 1H), 7.38 (s, 1H), 7.29 (s, 1H), 5.23 (s, 1H).
13С ЯМР (75 МГц; CD3OD): δ 174.9, 150.9, 145.4, 136.3, 126.8, 122.0, 120.6, 118.9, 72.9,
MS (m/z) 269 (M-1)-.
(10) Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
Раствор Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)OH (0,73 г 2,70 ммоль; смотри стадию (9) выше) в DMF (40 мл) в атмосфере азота охлаждали до 0°С. К данному раствору добавляли H-Aze-Pab(Teoc) (1,46 г; 3,24 ммоль), коллидин (0,82 г; 6,75 ммоль) и РуВОР (1,83 г; 3,51 ммоль). Раствор перемешивали при 0°С в течение 2 ч, нагревали до комнатной температуры и перемешивали 18 ч, гасили водой и концентрировали в вакууме. Остаток разбавляли EtOAc и промывали Н2O (1х), водным NaHCO3 (1х), водной лимонной кислотой (1х) и соляным раствором (1х), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного указанного в подзаголовке соединения. Посредством флэш-хроматографии на силикагеле (2х) с элюированием смесью EtOAc/МеОН (30:1), затем CH2Cl2/MeOH (93:7) получили указанное в подзаголовке соединение (0,73 г; 43%) в виде дробимой пены.
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров): δ 7.78-7.82 (d, 2H, J=8 Гц), 7.25-7.54 (m, 5Н), 5.25 и 5.16 (s, 1H), 5.22 и 4.79 (m, 1H), 3.92-4.58 (m, 6H), 2.20-2.76 (m, 2H), 1.04-1.13 (m, 2H), 0.08 (s, 9H).
MS (m/z) 629 (M+1)+.
(11) Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab
Трифторуксусную кислоту (1,0 мл) добавляли к перемешиваемому охлаждаемому на бане лед/вода раствору Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(Teoc) (101 мг; 160 мкмоль; смотри стадию (10) выше) в метиленхлориде (10 мл). Охлаждающую баню удаляли через 1 час. Через 1,5 часа выдерживания при комнатной температуре добавляли ацетонитрил (30 мл) и растворители осторожно удаляли при пониженном давлении. Остаток растворяли в воде и сушили вымораживанием с получением 90 мг (92%) указанного в заголовке соединения в виде его TFA-соли.
MS (m/z) 483 (M-1)-; 485 (M+1)+.
1H ЯМР (300 МГц; CD3OD) (сложный спектр вследствие наличия диастереомеров/ротамеров): δ 7.70-7.80 (m, 2H), 7.45-7.58 (m, 3Н), 7.24-7.38 (m, 2H), 5.26 (s, 1H), 5.17 (m, 1H, второстепенный ротамер), 4.82 (m, 1H, основной ротамер), 4.35-4.6 (m, 3Н), 4.22 (m, 1H, основной ротамер), 3.92-4.12 (m, 2H, второстепенный ротамер), 2.70 (m, 1H, второстепенный ротамер), 2.55 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.16 (m, 1H, второстепенный ротамер).
13С ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры): δ 173.7, 173.4, 173.0, 172.8, 168.1.
Пример 7
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(ОМе)
HATU (71 мг; 0,19 ммоль) добавляли к перемешиваемому охлаждаемому на бане лед/вода раствору Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)ОН (39 мг; 0,14 ммоль; смотри пример 6(9) выше) в DMF (3 мл). Через 30 минут добавляли раствор H-Aze-Pab(OMe)×2HCl (69 мг; 0,21 ммоль; смотри международную патентную заявку WO 00/42059) и 2,4,6-коллидина (0,080 мл; 0,58 ммоль) в DMF (1,5 мл). Реакционную смесь оставляли стоять в течение ночи, позволяя температуре медленно подняться до температуры окружающей среды. Растворители удаляли в вакууме и неочищенный продукт очищали, используя обращенно-фазовую ВЭЖХ (ацетонитрил/0,1 М водн. ацетат аммония), что позволяло получить, после сушки вымораживаем соответствующих фракций, указанное в заголовке соединение (61 мг; 97%) в виде бесцветного твердого вещества.
MS (m/z) 513 (M-1)-; 515 (M+1)+.
1H ЯМР (500 МГц; CD3OD) δ 7.97 (bt, 1H), 7.53 (d, 2H), 7.27 (t, 1H), 7.22 (d, 2Н), 7.19 (t, 1H), 7.11 (t, 2H), 6.77 (s, 1H), 4.92 (s, 1H), 4.9 (bs, 3H), 4.81 (m, 2H), 4.40 (m, 2H), 4.09 (m, 1H), 3.87 (s, 3H), 2.58 (m, 1H), 2.37 (m, 1H).
13С ЯМР (125 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 171.8, 169.9, 156.8.
Пример 8
Параллельный синтез алкоксиамидинов
Этот синтез проводили в 96-луночном блоке Роббинса (Robbins block). В лунки, содержащие соответствующее количество О-замещенного гидроксиламина (конкретизировано ниже; все перечисленное имеется в продаже или получено с использованием хорошо известных из литературы методик), добавляли раствор Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(Теос) (10 мг; 17 мкмоль; смотри пример 6(10) выше) в ацетонитриле (1,0 мл). Блок герметично закрывали и реакционную смесь вращали в течение ночи в сушильном шкафу при 60°С. После охлаждения и фильтрования твердые вещества промывали ацетонитрилом (3×0,3 мл). Объединенные жидкие фракции концентрировали в вакуумной центрифуге. Остаток распределяли между водой (0,4 мл) и этилацетатом (0,4 мл). По окончании жидкостно-жидкостной экстракции все фильтровали через колонку Hydromatrix™. После трехкратного промывания этилацетатом объединенные фильтраты концентрировали в вакуумной центрифуге. Снятие защиты осуществляли путем добавления метиленхлорида (0,1 мл) и трифторуксусной кислоты (0,3 мл). После перемешивания при комнатной температуре в течение 3 часов удаляли растворители в вакууме. Остаток распределяли между водным насыщенным гидрокарбонатом натрия (0,5 мл) и этилацетатом (0,5 мл). После экстракции, фильтрования через Hydromatrix™ и концентрирования (смотри ниже) остаток растворяли в смеси изопропанол/вода (7:3) (1 мл). Отбирали приблизительно 2% этого раствора и разбавляли смесью изопропанол/вода (7:3) (1 мл) для LC-MS-анализа. После удаления растворителей в вакууме твердый остаток переносили в 96-луночный планшет, используя ацетонитрил и этилацетат для растворения соединения. Растворители выпаривали в вакуумной центрифуге, что позволяло получить следующие указанные в заголовке соединения:
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(ОСН2-3-(5-Ме-изоксазол))
(из 3-[(аминоокси)метил]-5-метилизоксазола × HCl (18 мг; 0,11 ммоль)), выход 3,64 мг (35%), MS (m/z) 596 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(ОСН2-3-пиридин)
(из 3-[(аминоокси)метил]пиридина × 2HCl (19 мг; 96 мкмоль), выход 5,14 мг (50%), MS (m/z) 592 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(O-изо-Bu)
(из O-изобутилгидроксиламина × HCl (17 мг; 140 мкмоль), выход 4,4 мг (45%), MS (m/z) 557 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OEt)
(из O-этилгидроксиламина × HCl (14 мг; 140 мкмоль), выход 4,04 мг (42%), MS (m/z) 529 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn)
(из O-бензилгидроксиламина × HCl (17 мг; 110 мкмоль), выход 3,22 мг (29%), MS (m/z) 591 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-циклогексил)
(из O-циклогексилгидроксиламина × HCl (15 мг; 99 мкмоль), выход 2,9 мг (26%), MS (m/z) 583 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-цикло-Bu)
(из O-циклобутилгидроксиламина × HCl (17 мг; 140 мкмоль), выход 3,3 мг (30%), MS (m/z) 555 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OCH2CH2OPh(3-CF3))
(из O-[2-[3-(трифторметил)фенокси]этил]гидроксиламина × HCl (24 мг; 93 мкмоль), выход 6,52 мг (46%), MS (m/z) 689 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(4-Cl))
(из O-(4-хлорбензил)гидроксиламина × HCl (16 мг; 82 мкмоль), выход
3,47 мг (29%), MS (m/z) 625 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(3-MeO))
(из O-(3-метоксибензил)гидроксиламина × HCl (18 мг; 94 мкмоль), выход
4,33 мг (36%), MS (m/z) 621 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab(OBn(2-Br))
(из O-(2-бромбензил)гидроксиламина × HCl (23 мг; 96 мкмоль), выход
3,87 мг (30%), MS (m/z) 671 (М+1)+;
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(OBn(4-Ме))
(из O-(4-метилбензил)гидроксиламина × HCl (14 мг; 81 мкмоль), выход
2,91 мг (25%), MS (m/z) 605 (М+1)+ и
Ph(3-Cl)(5-OCF3)-(R)СН(ОН)С(O)-Aze-Pab(O-4-гептил)
(из O-(4-гептил)гидроксиламина × HCl (15 мг; 89 мкмоль), выход 17 мг (100%), MS (m/z) 599 (М+1)+.
Пример 9
Ph(3-Cl)(5-OCHF2)-(S)CH(CH2OH)C(O)-Aze-Pab×HOAc
(1) 3-Хлор-5-метоксибензойная кислота
Магниевую стружку (Fluka purum для реакций Гриньяра) предварительно обрабатывали следующим образом. Стружку помещали в воронку из пористого стекла и выливали на нее 0,1 М соляную кислоту. Стружку перемешивали с помощью стеклянной палочки в течение нескольких секунд и затем кислоту вымывали 3 порциями воды. Окончательно стружку промывали 2 порциями ацетона и помещали в бутылку. Тетрагидрофуран (100 мл; 99,95%) сушили путем добавления RedAI (1 г, 70 мас.% в толуоле). Предварительно обработанную магниевую стружку (5 г, 200 ммоль) помещали в круглодонную колбу и 3 раза продували азотом. Дихлоранизол (26 г, 146 ммоль) растворяли в THF (100 мл, высушенного с помощью RedAI) и добавляли дибромэтан (1,8 г; 10 ммоль). Реакционную смесь продували азотом и затем кипятили с обратным холодильником в течение 2 часов. Нагревание прерывали и порциями в продолжение 2 минут добавляли сухой лед (10 г). Когда весь сухой лед растворялся, реакционную смесь выливали на лед, содержащий соляную кислоту (400 мл, 2 М). Экстрактивная обработка (диэтиловый эфир, 300 мл) позволила получить 11,2 г (60,2 ммоль; выход 41%) указанного в подзаголовке соединения.
1H-ЯМР (500 МГц; ацетон-d6): δ 7.57 (m, 1Н), 7.49 (m, 1H), 7.23 (m, 1H), 3.91 (s, 3H).
(2) 3-Хлор-5-гидроксибензойная кислота
Оксид алюминия (1,65 г; 60 ммоль) и иод (21 г; 82 ммоль) кипятили с обратным холодильником в толуоле (200 мл) в течение 2 часов. Затем добавляли 3-хлор-5-метоксибензойную кислоту (11,2 г; 60,2 ммоль; смотри стадию (1) выше), растворенную в толуоле (50 мл), вместе с тетрабутиламмонийиодидом (1,5 г; 4 ммоль), и данную смесь кипятили с обратным холодильником в течение еще 2 часов. После охлаждения до температуры окружающей среды экстрактивная обработка позволила получить 8,7 г (50 ммоль; выход 83%) указанного в подзаголовке соединения.
1H-ЯМР (300 МГц; ацетон-d6): δ 9.27 (s, 1H), 7.48 (m, 1H), 7.44 (m, 1H), 7.11 (m, 1H).
(3) 3-Хлор-5-дифторметоксибензойная кислота
3-Хлор-5-гидроксибензойную кислоту (6,4 г; 37,2 ммоль; смотри стадию (2) выше), растворенную в хлороформе (200 мл), переносили в трехгорлую круглодонную колбу вместимостью 500 мл, оборудованную холодильником с сухоледным охлаждением и трубкой для ввода газа. Добавляли гидроксид натрия (100 мл, 5 М) и с энергичным перемешиванием. Порциями через трубку для ввода газа добавляли хлордифторметан (Фреон 22; 25 г, 290 ммоль) при температуре окружающей среды. Через 2 часа реакция завершилась. Экстрактивная обработка позволила получить 6,2 г (28 ммоль; выход 75%) указанного в подзаголовке соединения.
1H-ЯМР (500 МГц; ацетон-d6): δ 7.87 (m, 1Н), 7.74 (m, 1H), 7.54 (m, 1H), 7.19 (t, 1H, JH-F=73 Гц).
(4) 3-Хлор-5-дифторметокси-N-метокси-N-метилбензамид
3-Хлор-5-дифторметоксибензойную кислоту (1,8 г; 8 ммоль; смотри стадию (3) выше) и оксалилхлорид (1,5 г; 11,8 ммоль) растворяли в метиленхлориде (50 мл). Добавляли DMF (2 капли) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Затем добавляли N,O-диметилгидроксиламин (1 г, 10,2 ммоль) и триэтиламин (3 г, 30 ммоль) и после перемешивания в течение еще 10 минут при температуре окружающей среды реакционную смесь концентрировали при пониженном давлении. Остаток переводили в эфир (100 мл) и воду (50 мл). После разделения органическую фазу промывали соляным раствором, высушивали над сульфатом натрия, фильтровали и концентрировали. Этот остаток хроматографировали на диоксиде кремния (гексан/этилацетат (2:1)) с получением 2 г (7,5 ммоль; 93%) указанного в подзаголовке соединения.
1H-ЯМР (400 МГц; CDCl3): δ 7.54 (m, 1H), 7.37 (m, 1H), 7.27 (m, 1H), 6.53 (t, 1H, JH-F=73 Гц).
(5) 3-Хлор-5-дифторметоксиацетофенон
3-Хлор-5-дифторметокси-N-метокси-N-метилбензамид (2 г; 7,5 ммоль; смотри стадию (4) выше) растворяли в эфире (100 мл) и охлаждали в токе азота до -70°С. К перемешиваемой реакционной смеси по каплям с помощью шприца добавляли метиллитий (7 мл, 11 ммоль; 1,6 М в эфире) в течение 1 минуты. Баню из сухого льда удаляли и смесь оставляли для достижения температуры окружающей среды перед тем, как погасить реакцию раствором хлорида аммония (50 мл, 5%-ный NH4Cl в воде). Органическую фазу промывали соляным раствором, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток хроматографировали на диоксиде кремния (гексан/этилацетат (2:1)) с получением 1,5 г (6,8 ммоль; выход 90%) указанного в подзаголовке соединения.
1H-ЯМР (600 МГц; CDCl3): δ 7.77 (m, 1H), 7.59 (m, 1H), 7.35 (m, 1H), 6.56 (t, 1H, JH-F=73 Гц), 2.60 (s, 3Н).
(6) 3-Хлор-5-дифторметоксифенилуксусной кислоты метиловый эфир
3-Хлор-5-дифторметоксиацетофенон (1,5 г; 6,8 ммоль; смотри стадию (5) выше) растворяли в метиленхлориде (200 мл). Добавляли нитрат таллия(III) × 3МеОН на монтмориллоните К-10 (6 г, 10 ммоль (приблизительно 0,6 ммоль/г); смотри J. Am. Chem. Soc., 98, 6750 (1976)) и смесь перемешивали при температуре окружающей среды в течение 20 часов. Смесь фильтровали и фильтрат промывали бикарбонатом натрия (100 мл; 0,5 М), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток хроматографировали на диоксиде кремния (гексан/этилацетат (2:1)) с получением 1 г (4 ммоль; выход 56%) указанного в подзаголовке соединения.
1H-ЯМР (500 МГц; CDCl3): δ 7.14 (m, 1H), 7.06 (m, 1H), 6.96 (m, 1H), 6.50 (t, 1Н, JH-F=73 Гц), 3.72 (s, 3Н), 3.60 (s, 1H).
(7) α-Формил(3-хлор-5-дифторметоксифенил)уксусной кислоты метиловый эфир
3-Хлор-5-дифторметоксифенилуксусной кислоты метиловый эфир (1 г, 4 ммоль; смотри стадию (6) выше) и метилформиат (1 г, 16 ммоль) растворяли в эфире (100 мл) и охлаждали в ледяной бане (приблизительно 2°С). Затем добавляли мелко нарезанный натрий (180 мг; 7,8 ммоль) и метанол (1 мл) и смесь оставляли перемешиваться в ледяной бане в течение ночи. Осторожно добавляли воду (100 мл) и разделяли фазы. Содержащую воду фазу подкисляли соляной кислотой (2 М) до рН 1 и экстрагировали эфиром (2×100 мл). Экстракт высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток хроматографировали на диоксиде кремния (гексан/этилацетат (1:1)) с получением 400 мг (1,4 ммоль; выход 36%) указанного в подзаголовке соединения.
1H-ЯМР (400 МГц): δ 12.10 (d, 1H), 7.32 (d, 1H), 7.11 (m, 1H), 7.07 (m, 1H), 6.94 (m, 1H), 6.51 (t, 1H, JH-F=73 Гц), 3.83 (s, 3Н).
(8) 3-Хлор-5-дифторметокситропная кислота
α-Формил(3-хлор-5-дифторметоксифенил)уксусной кислоты метиловый эфир (400 мг, 1,4 ммоль; смотри стадию (7) выше) растворяли в смеси THF/метанол (50 мл, 9:1). Добавляли борогидрид натрия и смесь перемешивали при температуре окружающей среды в течение 30 минут. Добавляли воду (100 мл) и смесь концентрировали до образования водной суспензии, которую переводили в этилацетат и воду. Фазы разделяли и органическую фазу промывали хлоридом натрия (15%-ным в воде), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в метаноле (30 мл) и подвергали гидролизу с помощью гидроксида натрия (1 мл, 10 М) при температуре окружающей среды в течение 10 минут. Экстрактивная обработка дала 180 мг (0,68 ммоль; выход 48%) указанного в подзаголовке соединения.
1H-ЯМР (500 МГц; CDCl3): δ 7.18 (m, 1Н), 7.10 (m, 1H), 7.00 (m, 1H), 6.50 (t, 1H, JH-F=73 Гц), 4.11 (m, 1H), 3.90 (m, 1H), 3.84 (m, 1H).
(9) Ph(3-Cl)(5-OCHF2)-(S)CH(CH2OH)C(O)-Aze-Pab×HOAc
3-Хлор-5-дифторметокситропную кислоту (180 мг; 0,7 ммоль; смотри стадию (8) выше), H-Aze-Pab(Teoc)×HCl (450 мг, 1 ммоль) и РуВОР (530 мг, 1 ммоль) растворяли в DMF (10 мл), после чего добавляли DIPEA (550 мг; 3,9 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 ч перед тем, как разбавить соляным раствором (20 мл, 15%-ный NaCl) и экстрагировать этилацетатом (40 мл). Экстракт высушивали над сульфатом натрия, фильтровали и упаривали досуха. Остаток растворяли в метиленхлориде (5 мл) и добавляли трифторуксусную кислоту (5 мл). После 1 ч выдерживания при температуре окружающей среды смесь диастереомеров упаривали досуха и остаток хроматографировали на обращенно-фазовой колонке (ацетонитрил/вода (30:70), буфер: 0,1 М ацетат аммония). Сушка вымораживанием дала 36 мг (0,067 ммоль; выход 10,4%) указанного в заголовке соединения.
MS (ES) 481 (M+1)+.
1H-ЯМР (400 МГц; CDCl3): δ 7.77 (d, 2H), 7.57 (d, 2H), 7.30 (m, 1H), 7.13 (m, 2H), 6.87 (t, 1H, JH-F=73 Гц), 4.76 (m, 1H), 4.55 (s, 2H), 4.37 (m, 1H), 4.03 (m, 2H), 3.82 (m, 1H), 3.72 (m, 1H), 2.53 (m, 1H), 2.28 (m, 1H), 1.92 (3, 1,5Н).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 172.3, 171.9, 167.2.
Пример 10
Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab×TFA
(1) 3-Хлор-5-трифторметоксибензилмезилат
К раствору 3-хлор-5-трифторметоксибензилового спирта (6,1 г; 26,9 ммоль; смотри пример 6(5) выше) в CH2Cl2 (250 мл) при 0°С в атмосфере азота добавляли DIPEA (4,2 г; 32,3 ммоль) и метансульфонилхлорид (3,4 г; 29,6 ммоль). Раствор перемешивали при 0°С в течение 1,5 часа и гасили с помощью Н2О. Органические слои отделяли и промывали H2O (1x), 1 н. HCl (1х), Н2О (1х) и водным NaHCO3 (1x) и затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединение (8,2 г; 99%) в виде масла.
1H ЯМР (300 МГц; CDCl3): δ 7.37 (s, 1H), 7.28 (s, 1Н), 7.18 (s, 1H), 5.23 (s, 2Н), 3.07 (s, 3H).
(2) 3-Хлор-5-трифторметоксибензилцианид
К раствору 3-хлор-5-трифторметоксибензилмезилата (8,2 г; 26,8 ммоль; смотри стадию (1) выше) в DMSO (50 мл) добавляли цианид натрия (2,6 г; 53,6 ммоль). Получающийся гетерогенный раствор нагревали до 50°С и обрабатывали ультразвуком в течение 1 часа. Реакционную смесь охлаждали и распределяли между Et2O и Н2О. Органическую фазу промывали Н2O (2х) и соляным раствором (2х). Объединенные водные фазы экстрагировали Et2O (1x). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали при слабом нагревании и частичном вакууме, что позволило получить указанное в подзаголовке соединение (6,3 г; 100%) в виде красноватого летучего масла, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.32 (s, 1H), 7.24 (s, 1H), 7.12 (s, 1H), 3,78 (s, 2H).
(3) 3-Хлор-5-трифторметоксифенилуксусная кислота
К раствору 3-хлор-5-трифторметоксибензилцианида (6,3 г; 26,7 ммоль; смотри стадию (2) выше) в 2-пропаноле (100 мл) добавляли воду (200 мл) и гидроксид натрия (7,5 г; 133,5 ммоль). Раствор кипятили с обратным холодильником в течение 18 ч, охлаждали до комнатной температуры и удаляли 2-пропанол in vacuo. Водную фазу промывали СН2Cl2 (2х) и промывки отбрасывали. Основную водную фазу подкисляли 2 н. HCl и экстрагировали СН2Cl2 (3х). СН2Cl2-Экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволяло получить указанное в подзаголовке соединение (5,2 г; 76%) в виде масла, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3): δ 7.25 (s, 1H), 7.19 (s, 1H), 7.08 (s, 1H), 3.68 (s, 2H).
(4) Этил-3-хлор-5-трифторметоксифенилацетат
К раствору 3-хлор-5-трифторметоксифенилуксусной кислоты (5,2 г; 20,4 ммоль; смотри стадию (3) выше) в EtOH (600 мл) добавляли серную кислоту (несколько капель). Раствор кипятили с обратным холодильником в течение 18 ч, охлаждали до комнатной температуры, нейтрализовали твердым NaHCO3 и удаляли EtOH in vacuo. Остаток разбавляли EtOAc, затем промывали H2O (1x), водным NaHCO3 (1x) и соляным раствором (1x). Органические фазы высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (5,5 г; 96%) в виде масла, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.24 (s, 1H), 7.16 (s, 1H), 7.07 (s, 1H), 4.13-4.22 (q, J=8 Гц, 2H), 3.63 (s, 2H), 1.24-1.32 (t, J=8 Гц, 3Н).
(5) Ph(3-Cl)(5-OCF3)-(R,S)CH(CHO)C(O)OEt
К раствору этил-3-хлор-5-трифторметоксифенилацетата (4,5 г; 15,9 ммоль; смотри стадию (4) выше) в безводном THF (400 мл) в атмосфере азота при температуре менее 0°С (в бане лед-МеОН) добавляли этилат натрия (4,5 г; 63,6 ммоль). Охлажденный раствор перемешивали в течение 40 минут и добавляли этилформиат (8,1 г; 111,3 ммоль). Раствор перемешивали при 0°С в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение 2 часов. Затем THF удаляли in vacuo. Остаток разбавляли Et2O и экстрагировали Н2O (1x) и 0,5 М NaOH (3х). Водные экстракты подкисляли 2 н. HCl и экстрагировали CH2Cl2 (3х). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить неочищенное указанное в подзаголовке соединение (3,9 г). Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (4:1) позволила получить указанное в подзаголовке соединение (3,0 г; 61%) в виде масла.
1H ЯМР (300 МГц; CDCl3; смесь изомеров): δ 12.30 и 12.25 (s, 1H), 7.39 и 7.34 (s, 1H), 7.21 (s, 1H), 7.17 (s, 1H), 7.08 (s, 1H), 4.27-4.37 (q, J=8 Гц, 2Н), 1.28-1.38 (t, J=8 Гц, 3H).
(6) Ph(3-Cl)(5-OCF3)-(R,S)CH(CH2OH)C(O)OEt
К раствору Ph(3-Cl)(5-OCF3)-(R,S)CH(CHO)C(O)OEt (3,0 г; 9,66 ммоль; смотри стадию (5) выше) в МеОН (200 мл) при -10°С (в бане лед-МеОН) порциями в течение 5 мин добавляли борогидрид натрия (0,7 г; 19,32 ммоль). Раствор перемешивали при -10°С в течение 45 минут и добавляли дополнительное количество борогидрида натрия (0,4 г). Еще через 15 минут реакцию гасили водным хлоридом аммония, смесь делали слабокислой с помощью 2 н. HCl и МеОН удаляли in vacuo. Остаток разбавляли EtOAc и промывали H2O (1x), водным NaHCO3 (1x) и соляным раствором (1х). Органические фазы высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволяло получить неочищенное указанное в подзаголовке соединение. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (5:1) позволила получить указанное в подзаголовке соединение (2,0 г; 66%) в виде масла.
1H ЯМР (300 МГц; CDCl3): δ 7.26 (s, 1H), 7.19 (s, 1H), 7.07 (s, 1H), 4.16-4.28 (m, 2Н), 4.04-4.15 (m, 1H), 3.76-3.94 (m, 2H), 2.33 (t, J=6 Гц, 1H), 1.18-1.30 (t, J=8 Гц, 3Н).
(7) Ph(3-Cl)-OCF3)-(R,S)CH(CH2OH)C(O)OH
К раствору Ph(3-Cl)(5-OCF3)-(R,S)CH(CH2OH)C(O)OEt (2,0 г; 6,24 ммоль; смотри стадию (6) выше) в THF (50 мл) и Н2О (25 мл) добавляли гидроксид лития моногидрат (0,5 г; 12,48 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа и THF удаляли in vacuo. Остаток разбавляли H2O, затем промывали CHCl3 (2х) и промывки отбрасывали. Основный водный слой подкисляли 2 н. HCl и экстрагировали CHCl3 (4х). CHCl3-Экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить неочищенное указанное в подзаголовке соединение (1,5 г) в виде масла. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/МеОН/концентрированный NH4OH (градиент от 7,0:2,5:0,5 до 6:3:1) позволила получить аммониевую соль указанного в подзаголовке соединения (1,1 г). Аммониевую соль распределяли между 1 н. HCl и CHCl3. Органические фазы высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (также известное как 3-хлор-5-трифторметокситропная кислота) в виде масла (1,1 г; 62%).
1H ЯМР (300 МГц; CD3OD): δ 7.41 (s, 1H), 7.27 (s, 1H), 7.24 (s, 1H), 4.03 (m, 1Н), 3.75-3.87 (m, 2H).
(8) Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab(Teoc) (а) и Ph(3-Cl)(5-OCF3)-(R)CH(CH2OH)C(O)-Aze-Pab(Teoc) (б)
К раствору Ph(3-Cl)(5-OCF3)-(R,S)CH(CH2OH)C(O)OH (0,65 г; 2,28 ммоль; смотри стадию (7) выше) в DMF (40 мл) при температуре ниже 0°С (в бане лед-МеОН) добавляли H-Aze-Pab(Teoc) (0,90 г; 2,39 ммоль), коллидин (0,71 г; 5,70 ммоль) и РуВОР (1,31 г; 2,51 ммоль). Получающийся раствор перемешивали при температуре ниже 0°С в течение 1 ч, нагревали до комнатной температуры и перемешивали 1 ч. Затем DMF удаляли in vacuo. Остаток разбавляли EtOAc и промывали разбавленной водной HCl (1х), соляным раствором (1х), водным NaHCO3 (1x) и соляным раствором (1х). Органические слои высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить неочищенное указанное в подзаголовке соединение (2,1 г) в виде смеси диастереомеров. Флэш-хроматография (3х) на силикагеле с элюированием сначала смесью EtOAc/МеОН (95:5), затем CH2Cl2/MeOH (97:3) и окончательно СН2Cl2/МеОН (95:5) позволила получить указанные в подзаголовке соединения - диастереомер (а) (0,51 г; 35%) и диастереомер (б) (0,45 г; 31%) в виде дробимой пены.
Для указанного в подзаголовке соединения диастереомера (а)
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.79-7.85 (d, J=8 Гц, 2Н), 7,22-7.49 (m, 5Н), 5.17-4.77 (m, 1H), 4.53-4.18 (m, 4Н), 3.58-4.11 (m, 5H), 2.47-2.73 (m, 1H), 2.11-2.34 (m, 1H), 1.08-1.12 (m, 2H), 0.07 (s, 9H).
MS (m/z) 643 (M+1)+.
(9) Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab(Teoc) (78 мг; 0,121 ммоль; смотри стадию (8) выше - диастереомер (а)) растворяли в 5 мл трифторуксусной кислоты. Через 10 минут реакция завершилась, и растворитель выпаривали. Остаток высушивали вымораживанием из воды и ацетонитрила с получением желаемого продукта. Выход 70 мг (94%).
MS (m/z) 483 (M-1)-; 485 (М+1)+
1H-ЯМР (400 МГц, D2O) ротамеры (1:1): δ 8.83 (bt, 1H), 7.79 (d, 1H), 7.72 (d, 1H), 7.54 (d, 1H), 7.43 (d, 2H), 7.35 (m, 1Н, ротамер), 7.28 (m, 1H, ротамер), 7.20 (m, 1H, ротамер), 7.05 (m, 1H, ротамер), 5.22 (m, 1H, ротамер), 4.83 (m, 1H, ротамер), 4.57 (m, 2H, ротамер), 4.38 (m, 2H, ротамер), 4.3-3.7 (m, 5H), 2.77 (m, 1H, ротамер), 2.55 (m, 1H, ротамер), 2.27 (m, 1H).
13С-ЯМР (100 МГц; D2O) (карбонильные и/или амидинные углероды, ротамеры) δ 172.9, 172.2, 172.0, 171.8, 166.9.
Пример 11
Ph(3-Cl)(5-OCF3)-(3)СН(СН2OH)С(O)-Aze-Pab(ОМе)
(1) Ph(3-Cl)(5-OCF3)-(S)СН(СН2OH)С(O)-Aze-Pab(Оме, Teoc)
Ph(3-Cl)(5-OCF3)-(S)СН(СН2OH)С(O)-Aze-Pab(Теос) (100 мг; 0,155 ммоль; смотри пример 10(8) выше) растворяли в 12 мл тетрагидрофурана. Добавляли О-метилгидроксиламина гидрохлорид (44 мг; 0,53 ммоль) и реакционную смесь нагревали при 50°С в течение ночи. Реакционную смесь упаривали и остаток очищали препаративной ВЭЖХ (СН3CN/0,1 М NH4OAC (70:30)). Подходящие фракции упаривали, остаток растворяли в небольшом количестве ацетонитрила и воды и высушивали вымораживанием. Сушку вымораживанием повторяли снова. Выход 80 мг (76%) чистого материала.
1H-ЯМР (400 МГц; CD3OD; ротамеры): δ 7.5-7.4 (m, 3Н), 7.35-7.2 (m, 4H), 5.15 (m, 1H, минорный ротамер), 4.74 (m, 1H, основной ротамер), 4.5-4.25 (m, 3Н), 4.2-3.95 (m, 4H), 3.91 (b, 3Н), 3.9-3.6 (m, 2H), 2.63 (m, 1H, минорный ротамер), 2.50 (m, 1H, основной ротамер), 2.3-2.1 (m, 1H), 0.95 (m, 2H), 0.02 (s, 9Н, основной ротамер), 0.01 (s, 9H, минорный ротамер).
(2) Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab(OMe)
Ph(3-Cl)(5-OCF3)-(S)CH(CH2OH)C(O)-Aze-Pab(OMe, Teoc) (80 мг; 0,12 ммоль; смотри стадию (1) выше) растворяли в 1 мл метиленхлорида и охлаждали на ледяной бане. Добавляли 3 мл трифторуксусной кислоты и реакционный сосуд выдерживали в ледяной бане в течение двух часов. Смесь упаривали, растворяли в этилацетате и промывали три раза NaHCO3 (водн.), затем водой и соляным раствором. Органическую фазу высушивали (Na2SO4), фильтровали и упаривали. Остаток высушивали вымораживанием из небольшого количества ацетонитрила и воды. Выход 60 мг (95%) чистого указанного в заголовке продукта.
MS (m/z) 528 (М-1)-; 531 (М+1)+.
1H-ЯМР (500 МГц; CD3OD; ротамеры): δ 7.65-7.55 (m, 3H, ротамеры), 7.45 (m, 1Н, основной ротамер), 7.4-7.2 (m, 4Н), 5.15 (m, 1H, минорный ротамер), 4.74 (m, 1H, основной ротамер), 4.5-4.3 (m, 3H), 4.05-3.95 (m, 2H), 3.85 (m, 1H, основной ротамер), 3.82 (s, 3H, основной ротамер), 3.81 (s, 3H, минорный ротамер), 3.73 (m, 1H, основной ротамер), 3.67 (m, 1H, минорный ротамер), 3.62 (m, 1H, минорный ротамер), 2.63 (m, 1H, минорный ротамер), 2.50 (m, 1H, основной ротамер), 2.24 (m, 1H, основной ротамер), 2.16 (m, 1H, минорный ротамер).
13С-ЯМР (125 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 174.0, 173.2, 172.7, 172.6, 155.1.
Пример 12
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe. Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,40 г; 0,65 ммоль; смотри пример 1(9) выше) растворяли в 20 мл ацетонитрила и добавляли 0,50 г (6,0 ммоль) O-метилгидроксиламина гидрохлорида. Смесь нагревали при 70°С в течение 2 ч. Растворитель выпаривали и остаток распределяли между водой и этилацетатом. Водную фазу еще дважды экстрагировали этилацетатом, объединенную органическую фазу промывали водой, соляным раствором, высушивали (Na2SO4), фильтровали и упаривали. Выход 0,41 г (91%).
1H-ЯМР (400 МГц; CDCl3): δ 7.83 (bt, 1H), 7.57 (bs, 1H), 7.47 (d, 2H), 7.30 (d, 2H), 7.20 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.53 (t, 1H), 4.89 (s, 1H), 4.87 (m, 1H), 4.47 (m, 2H), 4.4-4.2 (b, 1H), 4.17-4.1 (m, 3H), 3.95 (s, 3H), 3.67 (m, 1H), 2.68 (m, 1H), 2.42 (m, 1H), 0.97 (m, 2H), 0.01 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe, Teoc) (0,40 г; 0,62 ммоль; смотри стадию (1) выше) растворяли в 5 мл TFA и оставляли взаимодействовать в течение 30 минут. TFA выпаривали и остаток распределяли между этилацетатом и NaHCO3 (водн.). Водную фазу еще дважды экстрагировали этилацетатом, объединенную органическую фазу промывали водой, соляным раствором, высушивали (Na2SO4), фильтровали и упаривали. Продукт высушивали вымораживанием из смеси вода/ацетонитрил. Никакой очистки не требовалось. Выход 0,28 г (85%).
1H-ЯМР (600 МГц; CDCl3): δ 7.89 (bt, 1H), 7.57 (d, 2H), 7.28 (d, 2H), 7.18 (m, 1Н), 7.13 (m, 1H), 6.99 (m, 1H), 6.51 (t, 1H), 4.88 (s, 1H), 4.87 (m, 1H), 4.80 (bs, 2H), 4.48 (dd, 1H), 4.43 (dd, 1H), 4.10 (m, 1H), 3.89 (s, 3H), 3.68 (m, 1H), 2.68 (m, 1H), 2.40 (m, 1H).
13С-ЯМР (125 МГц; CDCl3) (карбонильные и/или амидинные углероды, ротамеры) δ 172.9, 170.8, 152.7, 152.6.
MS (m/z) 495 (M-1)-, 497 (M+1)+.
Пример 13
Ph(3-OCHF2)-(R)CH(OH)C(O)-Aze-Pab×HOAc
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) (13 мг; 0,026 ммоль; смотри пример 12 выше) растворяли в абсолютном этаноле (5 мл) и добавляли 30 мг 10%-ного Pd/C. В конце добавляли уксусную кислоту (5 мкл) и смесь гидрировали при атмосферном давлении в течение 20 ч. Смесь фильтровали через Целит®, упаривали и очищали с помощью обращенно-фазовой ВЭЖХ (0,1 М водный ацетат аммония/MeCN). Соответствующие фракции высушивали вымораживанием, что позволило получить указанное в заголовке соединение в виде твердого вещества белого цвета (8,5 мг, 66%).
1H-ЯМР (400 МГц; CD3OD, ротамеры): δ 7.73-7.78 (m, 2H), 7.55 (d, 2H), 7.19-7.43 (m, 3H), 7.06-7.13 (m, 1H), 6.83 (t, 1H, JH-F=74 Гц, основной ротамер), 6.81 (t, 1H, основной ротамер), 5.20 (s, 1H, основной ротамер), 5.19 (m, 1H, минорный ротамер), 5.15 (s, 1H, минорный ротамер), 4.78 (m, 1H, основной ротамер), 4.4-4.6 (несколько пиков, 2H), 4.35 (m, 1H, основной ротамер), 4.08 (m, 1H), 3.99 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер), 1.89 (s, 3H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 173.7, 172.9, 168.3.
MS (m/z) 433 (M+1)+; 431 (M-1)-.
Пример 14
Ph(3-OCF3)-(R)СН(ОН)С(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCF3)-(R)CH(OH)C(O)-Aze-Pab×TFA (34 мг; 0,057 ммоль; из примера 6) растворяли в 5 мл этанола и добавляли 20 мг 10%-ного Pd/C. Смесь гидрировали при атмосферном давлении в течение ночи. Смесь фильтровали через Целит®, упаривали и высушивали вымораживанием из смеси вода/ацетонитрил.
1H-ЯМР (400 МГц; CD3OD; ротамеры): δ 7.8-7.7 (m, 2H), 7.55 (m, 2H), 7.5-7.2 (m, 4H), 5.24 (s, 1H, основной ротамер), 5.23 (m, 1H, минорный ротамер), 5.18 (s, 1H, минорный ротамер), 4.77 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.36 (m, 1H, основной ротамер), 4.08 (m, 1H), 3.99 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 174.1, 173.9, 173.5, 172.9, 168.2.
19F-ЯМР (282 МГц; CD3OD): -59.8 и -59.9 (3F, минорный и основной ротамер соответственно), -77.4 (3F) показывает, что это соль TFA.
MS (m/z) 451.3 (M+1)+.
Пример 15
Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 3-Хлор-5-трифторэтоксибензальдегид
К перемешиваемому на магнитной мешалке раствору 3-хлор-5-гидроксибензальдегида (2,0 г; 12,8 ммоль; смотри пример 1(2) выше) и карбоната калия (2,3 г; 16,6 ммоль) в DMF (35 мл) в атмосфере азота добавляли 2,2,2-трифторэтил-п-толуолсульфонат (4,2 г; 16,6 ммоль) при комнатной температуре. Смесь нагревали до 110°С в течение 7 ч и затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, выливали в охлажденную во льду 2 н. HCl (100 мл) и экстрагировали EtOAc (2×75 мл). Объединенные органические экстракты промывали 0,5 н. HCl (2×50 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Коричневое масло хроматографировали на силикагеле с элюированием смесью Нех/EtOAc (6:1), что позволяло получить указанное в подзаголовке соединение (1,9 г; 61%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 9.44 (s, 1H), 7.56 (s, 1H), 7.33 (s, 1H), 7.28 (s, 1H), 4.42 (q, J=8 Гц, 2H).
(2) Ph(3-Cl)(5-OCH2CF3)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-трифторэтоксибензальдегида (5,2 г; 21,7 ммоль; смотри стадию (1) выше) и иодида цинка (1,7 г; 5,4 ммоль) в CH2Cl2 (200 мл) в атмосфере азота по каплям с помощью шприца при 0°С добавляли триметилсилилцианид (4,3 г; 43,3 ммоль). Смесь перемешивали при 0°С в течение 3 ч, затем разбавляли Н2О (150 мл). Органический слой отделяли, высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (6,9 г; 95%) в виде желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.27 (s, 1H), 6.98 (s, 2H), 5.44 (s, 1H), 4.38 (q, J=8 Гц, 2H), 0.30 (s, 9H).
(3) Ph(3-Cl)(5-OCH2CF3)-(R,S)CH(OH)C(O)OH
Концентрированную соляную кислоту (170 мл) добавляли к Ph(3-Cl)(5-OCH2CF3)-(R,S)CH(OTMS)CN (6,9 г; 20,4 ммоль; смотри стадию (2) выше) и перемешивали при 100°С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь далее охлаждали до 0°С и медленно подщелачивали 3 н. NaOH (300 мл). Эту смесь промывали Et2O (2×100 мл) и водный слой подкисляли 2 н. HCl (50 мл). Затем водный слой экстрагировали EtOAc (2×100 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (5,3 г; 92%) в виде бледно-желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.18 (s, 1H), 7.07 (s, 1H), 7.02 (s, 1H), 5.13 (s, 1H), 4.58 (q, J=8 Гц, 2H).
(4) Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)OH (а) и Ph(3-Cl)(5-ОСН2CF3)-(S)CH(OAc)C(O)OH (б)
Раствор Ph(3-Cl)(5-OCH2CF3)-(R,S)CH(OH)C(O)OH (7,06 г; 24,8 ммоль; смотри стадию (3) выше) и Липазы PS "Amano" (4,30 г) в винилацетате (250 мл) и МТВЕ (250 мл) нагревали при 70°С в атмосфере азота в течение 40 ч. Реакционную смесь охлаждали до комнатной температуры, фермент удаляли посредством фильтрования с промыванием EtOAc и фильтрат концентрировали in vacuo. Хроматография на силикагеле с элюированием смесью CHCl3/MeOH/Et3N (92:6:2) позволяла получить указанную в подзаголовке триэтиламинную соль указанного в подзаголовке соединения (а) (3,02 г) в виде желтого масла. Соль указанного в подзаголовке соединения (а) растворяли в Н2O (150 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (2×75 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения (а) (2,18 г) в виде беловатого твердого вещества. В дополнение к этому после проведения упомянутой выше колоночной хроматографии получали триэтиламинную соль указанного в подзаголовке соединения (б) (4,73 г).
Данные для указанного в подзаголовке соединения (а)
Т.пл. 98-103°С.
1H ЯМР (300 МГц; CD3OD) δ 7.18 (s, 1H), 7.07 (s, 1Н), 7.02 (s, 1H), 5.13 (s, 1H), 4.58 (q, J=8 Гц, 2H).
13C ЯМР (75 МГц; CD3OD) δ 175.4, 159.6, 144.6, 136.2, 125.0 (q, J=277 Гц), 121.8, 115.9, 113.1, 73.3, 67.0 (q, J=35 Гц).
ВЭЖХ-анализ: 98,6%, >99% е.е., колонка Chiralcel OD (подвижная фаза Нех/EtOH/TFA (97:3:0,5)).
[α]25 D = -81.5°(с=1.0, МеОН).
APCl-MS: (M-1)=283 m/z.
(5)Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-ОСН2CF3)-(R)СН(ОН)С(O)ОН (0,50 г; 1,8 ммоль; смотри стадию (4) выше (соединение (а))) в DMF (20 мл) в атмосфере азота добавляли H-Aze-Pab(Teoc)×HCl (1,03 г; 2,3 ммоль), РуВОР (1,01 г; 1,9 ммоль) и DIPEA (0,57 г; 4,4 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 20 ч. Смесь концентрировали in vacuo, остаток дважды хроматографировали на силикагеле с элюированием сначала смесью CHCl3/EtOH (10:1) и затем EtOAc/EtOH (10:1), что позволило получить указанное в подзаголовке соединение (0,55 г; 48%) в виде дробимой белой пены.
Т.пл. 90-95°С.
Rf=0,42 (CHCl3/EtOH (10:1)).
1Н ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.78-7.81 (m, 2H), 7.38-7.41 (m, 2H), 7.12-7.16 (m, 1H), 7.00-7.06 (m, 2H), 5.09-5.22 и 4.75-4.79 (m, 2Н), 3.94-4.61 (m, 8Н), 2.09-2.75 (m, 2H), 1.04-1.11 (m, 2H), 0.70 (s, 9H).
APCl-MS: (M+1)=643 m/z.
(6) Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,066 г; 0,103 ммоль; смотри стадию (5) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в течение 30 мин. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил с получением 0,060 г (94%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.8-7.7 (m, 2H), 7.6-7.5 (m, 2H), 7.2-7.0 (m, 3Н), 5.21 (m, 1H, минорный ротамер), 5.17 (s, 1H, основной ротамер), 5.11 (s, 1H, минорный ротамер), 4.81 (m, 1H, основной ротамер), 4.6-4.4 (m, 4H), 4.37 (m, 1H, основной ротамер), 4.16 (m, 1H, основной ротамер), 4.06 (m, 1H, минорный ротамер), 3.99 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.54 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.2, 171.8, 171.7, 167.0.
MS (m/z) 499.3 (M+1)+.
Пример 16
Ph(3-Cl)(5-OCH2CF3)-(R)CH(OH)C(O)-Aze-Pab(OMe)
К раствору Ph(3-Cl)(5-ОСН2CF3)-(R)СН(ОН)С(O)ОН (0,48 г; 1,7 ммоль; смотри пример 15(4) выше (соединение (а))) в DMF (20 мл) в атмосфере азота добавляли H-Aze-Pab(OMe)×2HCl (0,74 г; 2,2 ммоль), РуВОР (0,97 г; 1,9 ммоль) и DIPEA (0,55 г; 4,2 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 20 ч. Смесь концентрировали in vacuo, остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (10:1) и второй раз EtOAc/EtOH (10:1), что позволило получить указанное в заголовке соединение (0,62 г; 69%) в виде дробимой белой пены.
Т.пл. 75-80°С.
Rf=0,43 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.57-7.60 (m, 2H), 7.32-7.36 (m, 2H), 7.13-7.17 (m, 1H), 7.00-7.06 (m, 2H), 5.09-5.19 и 4.74-4.80 (m, 2H), 3.93-4.62 (m, 6H), 3.81 (s, 3H), 2.10-2.73 (m, 2H).
APCl-MS: (M+1)=529 m/z.
Пример 17
Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 2,2-Дифторэтиловый эфир метансульфокислоты
К перемешиваемому на магнитной мешалке раствору 2,2-дифторэтанола (1,52 г; 18,5 ммоль) в CH2Cl2 (20 мл) в атмосфере азота добавляли триэтиламин (5,61 г; 55,5 ммоль) и метансульфонилхлорид (2,54 г; 22,2 ммоль) при 0°С. Смесь перемешивали при 0°С в течение 1,5 ч, разбавляли CH2Cl2 (50 мл) и промывали 2 н. HCl (50 мл). Водный слой экстрагировали CH2Cl2 (30 мл), объединенные органические экстракты промывали соляным раствором (30 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (2,52 г; 85%) в виде желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 6.02 (tt, J=3 Гц, J=55 Гц, 1H), 4.39 (dt, J=3 Гц, J=13 Гц, 2Н), 3.13 (s, 3Н).
(2) 3-Хлор-5-дифторэтоксибензальдегид
К раствору 3-хлор-5-гидроксибензальдегида (1,50 г; 9,6 ммоль; смотри пример 1(2) выше) и карбоната калия (1,72 г; 12,5 ммоль) в DMF (10 мл) в атмосфере азота по каплям добавляли раствор 2,2-дифторэтилового эфира метансульфокислоты (2,0 г; 12,5 ммоль; смотри стадию (1) выше) в DMF (10 мл) при комнатной температуре. Смесь нагревали до 100°С в течение 6 ч, затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, выливали в охлажденную во льду 2 н. HCl (100 мл) и экстрагировали EtOAc (2×75 мл). Объединенные органические экстракты промывали 0,5 н. HCl (2×50 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Коричневое масло хроматографировали на силикагеле с элюированием смесью Нех/EtOAc (5:1), что позволяло получить указанное в подзаголовке соединение (1,35 г; 64%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 9.92 (s, 1H), 7.52 (s, 1H), 7.31 (s, 1H), 7.22 (s, 1 Н), 6.12 (tt, J=3 Гц, J=55 Гц, 1Н), 4.26 (dt, J=3 Гц, J=15 Гц, 2Н).
(3) Ph(3-Cl)(5-OCH2CHF2)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-дифторэтоксибензальдегида (1,35 г; 6,1 ммоль; смотри стадию (2) выше) и иодида цинка (0,48 г; 1,5 ммоль) в CH2Cl2 (50 мл) в атмосфере азота по каплям при 0°С добавляли триметилсилилцианид (1,21 г; 12,2 ммоль). Смесь перемешивали при 0°С в течение 3 ч, затем разбавляли Н2O (50 мл). Органический слой отделяли, высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (1,85 г; 95%) в виде коричневого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.13 (s, 1H), 6.94 (s, 2H), 6.10 (tt, J=3 Гц, J=55 Гц, 1H), 5.43 (s, 1H), 4.20 (dt, J=3 Гц, J=15 Гц, 2H), 0.28 (s. 9H).
(4) Ph(3-Cl)(5-OCH2CHF2)-(R,S)CH(OH)C(O)OH
Концентрированную соляную кислоту (60 мл) добавляли к Ph(3-Cl)(5-OCH2CHF3)-(R,S)CH(OTMS)CN (1,85 г; 5,8 ммоль; смотри стадию (3) выше) и перемешивали при 100°С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь далее охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (˜180 мл) и промывали Et2O (2×75 мл). Водный слой подкисляли 2 н. HCl (20 мл) и экстрагировали EtOAc (2×75 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (1,50 г; 97%) в виде бледно-желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.15 (s, 1H), 7.05 (s, 1H), 6.98 (s, 1H), 6.19 (tt, J=4 Гц, J=55 Гц, 1 Н), 5.12 (s, 1 Н), 4.25 (dt, J=4 Гц, J=17 Гц, 2H).
(5) Ph(3-Cl)(5-OCH2CHF2)-(S)CH(OAc)C(O)OH (а) и Ph(3-Cl)(5-OCH2CHF2)-(R)СН(OH)C(О)OH (б)
Раствор Ph(3-Cl)(5-OCH2CHF2)-(R,S)CH(OH)C(O)OH (3,90 г; 14,6 ммоль; смотри стадию (4) выше) и Липазы PS "Amano" (2,50 г) в винилацетате (140 мл) и МТВЕ (140 мл) нагревали при 70°С в атмосфере азота в течение 40 ч. Реакционную смесь охлаждали до комнатной температуры, фермент удаляли посредством фильтрования с промыванием EtOAc и фильтрат концентрировали in vacuo. Хроматография на силикагеле с элюированием смесью CHCl3/MeOH/Et2N (92:6:2) позволила получить триэтиламинную соль указанного в подзаголовке соединения (а) в виде желтого масла. В дополнение к этому получали триэтиламинную соль указанного в подзаголовке соединения (б) (1,47 г), эту соль растворяли в Н2О (100 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (2×75 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения (б) (1,00 г) в виде беловатого твердого вещества.
Данные для указанного в подзаголовке соединения (б)
Т.пл. 103-106°С.
Rf=0,39 (CHCl3/MeOH/Et2N (90:8:2)).
1H ЯМР (300 МГц; CD3OD) δ 7.13 (s, 1H), 7.04 (s, 1H), 6.97 (s, 1H), 6.17 (tt, J=4 Гц, J=55 Гц, 1H), 5.12 (s, 1 Н), 4.24 (dt, J=4 Гц, J=8 Гц, 2Н).
13С ЯМР (75 МГц; CD3OD) δ 175.5, 160.3, 144.5, 136.1, 121.3, 115.7, 115.3 (t, J=240 Гц), 112.9, 73.4, 68.6 (t, J=29 Гц).
ВЭЖХ-анализ: 96,2%, >95,0% е.е., колонка ChiralPak AD (подвижная фаза Нех/EtOH/TFA (95:5:0,5)).
[α]25 D = -84.0°(с=0.85, МеОН).
APCl-MS: (M-1)=265 m/z.
(6)Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (0,35 г; 1,3 ммоль; смотри стадию (5) выше (соединение (б))) в DMF (18 мл) в атмосфере азота добавляли H-Aze-Pab(Teoc)×HCl (0,76 г; 1,7 ммоль), РуВОР (0,75 г; 1,4 ммоль) и DIPEA (0,43 г; 3,3 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 20 ч. Смесь концентрировали in vacuo и остаток дважды хроматографировали на силикагеле с элюированием сначала смесью CHCl3/EtOH (10:1) и затем EtOAc/EtOH (10:1), что позволило получить указанное в подзаголовке соединение (0,69 г; 84%) в виде дробимой белой пены.
Т.пл. 108-118°С.
Rf=0,48 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.78-7.81 (m, 2H), 7.40-7.43 (m, 2H), 7.09-7.12 (m, 1H), 6.96-7.02 (m, 2H), 6.16 (t, J=57 Гц, 1Н), 5,09-5.20 и 4.75-4.80 (m, 2H), 3.95-4.55 (m, 8H), 2.10-2.75 (m, 2H), 1.04-1.11 (m, 2H), 0.07 (s, 9H).
APCl-MS: (M+1)=625 m/z.
(7) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,086 г; 0,138 ммоль; смотри стадию (6) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в течение 1 ч. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил с получением 0,080 г (98%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (300 МГц; CD3OD) ротамеры: δ 7.8-7.7 (m, 2H), 7.6-7.5 (m, 2H), 7.15-6.95 (m, 3Н), 6.35-5.95 (m, 1H), 5.20 (m, 1H, минорный ротамер), 5.14 (s, 1H, основной ротамер), 5.10 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.6-4.0 (m, 6H), 2.70 (m, 1H, минорный ротамер), 2.53 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 174.0, 173.8, 173.4, 172.9, 168.2.
MS (m/z) 481.2 (M+1)+.
Пример 18
Ph(3-Cl)(5-OCH2CHF2)(R)CH(OH)C(O)-Aze-Pab(OMe)
К раствору Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (0,30 г; 1,7 ммоль; смотри пример 17(5) выше (соединение (б))) в DMF (15 мл) в атмосфере азота добавляли H-Aze-Pab(OMe)×2HCl (0,49 г; 1,5 ммоль), РуВОР (0,65 г; 1,2 ммоль) и DIPEA (0,36 г; 2,8 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 20 ч. Смесь концентрировали in vacuo, остаток трижды хроматографировали на силикагеле с элюированием сначала смесью CHCl3/EtOH (10:1), затем EtOAc/EtOH (10:1) и окончательно CHCl3/МеОН (20:1), что позволило получить указанное в заголовке соединение (0,47 г; 81%) в виде дробимой белой пены.
Т.пл. 65-75°С.
Rf=0,37 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.58-7.60 (m, 2H), 7.32-7.35 (m, 2H), 7.09-7.12 (m, 1H), 6.96-7.02 (m, 2H), 6.16 (t, J=55 Гц, 1Н), 5.08-5.18 и 4.74-4.80 (m, 2H), 3.96-4.50 (m, 6H), 3.80 (s, 3H), 2.10-2.75 (m, 2H).
APCl-MS: (M+1)=511 m/z.
Пример 19
Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1)Ph(3-Cl)(5-TMSO)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-гидроксибензальдегида (9,8 г; 62,6 ммоль; смотри пример 1(2) выше) и Znl2 (5,0 г; 15,7 ммоль) в безводном CH2Cl2 (500 мл) при 0°С добавляли триметилсилилцианид (13,7 г; 138 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Добавляли воду (250 мл) и разделяли слои. Водный слой экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (16,9 г; 83%) в виде желтого масла, которое использовали без дальнейшей очистки.
Rf=0,42 (Нех/EtOAc (3:1)).
1H ЯМР (300 МГц; CDCl3) δ 7.06 (s, 1H), 6.86 (s, 2H), 5.40 (s, 1H), 0.30 (s, 9Н), 0.24 (s, 9H).
(2) Ph(3-Cl)(5-OH)-(R,S)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OTMS)-(R,S)CH(OTMS)CN (22,6 г; 68,8 ммоль; смотри стадию (1) выше) в концентрированной HCl (200 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 ч. Реакционную смесь охлаждали до 0°С и медленно подщелачивали 2 н. NaOH. Смесь промывали Et2O (3×100 мл) для удаления органических примесей. Водный слой подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (9,3 г; 67%) в виде коричневого масла, которое использовали без дальнейшей очистки.
Rf=0,23 (CHCl3/МеОН/концентрированный NH4OH (6:3:1)).
1Н ЯМР (300 МГц; CD3OD) δ 7.05 (s, 1H), 6.94 (s, 1H), 6.73 (s, 1H), 5.03 (s, 1H).
(3) Ph(3-Cl)(5-OH)-(R,S)CH(OH)C(O)OEt
К раствору Ph(3-Cl)(5-OH)-(R,S)CH(OH)C(O)OH (9,3 г; 46,0 ммоль; смотри стадию (2) выше) в абсолютном EtOH (200 мл) добавляли концентрированную серную кислоту (0,25 мл) и реакционную смесь кипятили с обратным холодильником в атмосфере азота в течение 4 ч. Реакционную смесь охлаждали до 0°С и добавляли твердый NaHCO3 (0,2 г). Реакционную смесь концентрировали in vacuo и распределяли между насыщенным NaHCO3 (100 мл) и Et2O (3×50 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения (6,9 г; 65%) в виде желтого масла, которое использовали без дальнейшей очистки.
Rf=0,62 (CHCl3/МеОН/концентрированный NH4ОН (6:3:1)).
1H ЯМР (300 МГц; CDCl3) δ 6.99 (s, 1H), 6.81 (s, 2H), 5.07 (s, 1H), 4.16-4.32 (m, 2H), 1.23 (t, J=7 Гц, 3H).
(4) Ph(3-Cl)(5-OCH2F)-(R,S)CH(OH)C(O)OEt
К раствору Ph(3-Cl)(5-OH)-(R,S)CH(OH)C(O)OEt (6,1 г; 26,8 ммоль; смотри стадию (3) выше) в DMF (100 мл) в герметично закрытой колбе в атмосфере азота при 0°С добавляли карбонат цезия (13,1 г; 40,2 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 минут с последующим добавлением иодида калия (0,5 г; 2,7 ммоль). Реакционную смесь охлаждали до -78°С и сосуд барботировали хлорфторметаном (18,4 г; 268 ммоль). Затем герметично закрытую колбу оставляли нагреваться до комнатной температуры и перемешивали в течение 18 ч. Реакционную смесь охлаждали до 0°С, осторожно вентилировали, чтобы удалить любой избыток хлорфторметана и распределяли между Н2O (20 мл) и Et2O (3×50 мл). Объединенные органические фазы промывали соляным раствором (2×50 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (градиент от 9:1 до 3:1) позволила получить указанное в подзаголовке соединение (2,4 г; 35%) в виде светло-желтого масла. Примечание: данное соединение слабо различимо в УФ на ТСХ. Его можно сделать видимым с помощью окрашивания ТСХ-пластины бромкрезоловым зеленым.
Rf=0,46 (Нех/EtOAc (2:1)).
1H ЯМР (300 МГц; CDCl3) δ 7.21 (s, 1H), 7.08 (s, 1H), 7.05 (s, 1H), 5.70 (d, JH-F=54 Гц, 2Н), 5.12 (d, J=5 Гц, 1H), 3.80-4.35 (m, 2Н), 3.50 (d, J=5 Гц, 1H), 1.26 (t, J=7 Гц, 3H).
(5) Ph(3-Cl(5-ОСН2F)-(R,S)СН(OH)С(O)OH
К раствору Ph(3-Cl)(5-OCH2F)-(R,S)CH(OH)C(O)OEt (1,8 г; 6,8 ммоль; смотри стадию (4) выше) в смеси H2О/THF (30 мл, 1:2) при 0°С в атмосфере азота добавляли гидроксид лития моногидрат (0,40 г; 10,3 ммоль). Смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь концентрировали in vacuo и распределяли между Н2О (5 мл) и Et2O (2×20 мл). Водный слой осторожно подкисляли 0,2 н. HCl при 0°С и экстрагировали EtOAc (3×30 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (1,4 г; 87%) в виде бесцветного масла, которое отвердевало при стоянии до твердого вещества белого цвета.
Rf=0,43 (CHCl3/MeOH/Et3N (6:2:1)).
1H ЯМР (300 МГц; CD3OD) δ 7.24 (s, 1H), 7.17 (s, 1H), 7.07 (s, 1H), 5.78 (d, JH-F=54 Гц, 2Н), 5.13 (s, 1H).
(6) Ph(3-Cl(5-ОСН2F)-(R)СН(OH)С(О)OH (а) и Ph(3-Cl)(5-ОСН2F)-(S)CH(OAc)C(O)OH (б)
Смесь Ph(3-Cl)(5-OCH2F)-(R,S)CH(OH)C(O)OH (3,2 г; 13,9 ммоль; смотри стадию (5) выше) и Липазы PS "Атапо" (1,9 г) в винилацетате (150 мл) и МТВЕ (150 мл) нагревали при 70°С в атмосфере азота в течение 3 сут. Реакционную смесь охлаждали, фильтровали через Целит® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали in vacuo и подвергали флэш-хроматографии на силикагеле с элюированием смесью CHCl3/МеОН/Et3N (15:1:0,5), что позволило получить триэтиламинную соль указанного в подзаголовке соединения (а) (0,50 г; 21%), которую использовали без нейтрализации. В дополнение к этому получали триэтиламинную соль указанного в подзаголовке соединения (б) (0,46 г; 20%).
Данные для указанного в подзаголовке соединения (а)
Rf=0,19 (CHCl3/МеОН/Et3N (15:1:0,5)).
1H ЯМР (300 МГц; CD3OD) δ 7.26 (s, 1H), 7.18 (s, 1H), 6.97 (s, 1H), 5.74 (d, JH-F=54 Гц, 2Н), 4.81 (s, 1H), 3.17 (q, J=7 Гц, 6Н), 1.28 (t, J=7 Гц, 9Н).
Данные для указанного в подзаголовке соединения (б)
Rf=0,33 (CHCl3/МеОН/Et3N(15:1:0,5)).
1H ЯМР (300 МГц; CD3OD) δ 7.28 (s, 1H), 7.19 (s, 1H), 7.09 (s, 1H), 5.76 (d, JH-F=54 Гц, 2Н), 5.75 (s, 1H), 3.17 (q, J=7 Гц, 6Н), 2.16 (s, 3Н), 1.28 (t, J=7 Гц, 9Н).
(7) Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору триэтиламинной соли Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)OH (0,50 г; 1,50 ммоль; смотри стадию (6) выше) и HAze-Pab(Teoc)·HCl (0,87 г; 1,90 ммоль) в безводном DMF (15 мл) в атмосфере азота при 0°С добавляли РуВОР (0,85 г; 2,60 ммоль) и DIPEA (0,48 г; 3,70 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали in vacuo и дважды подвергали флэш-хроматографии на силикагеле с элюированием первоначально смесью CHCl3/EtOH (9:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в подзаголовке соединение (0,23 г; 26%) в виде дробимой белой пены.
Т.пл. 88-92°С.
Rf=0,61 (CHCl3/EtOH (9:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.81 (d, J=8 Гц, 2Н), 7.40-7.42 (m, 2H), 7.06-7.23 (m, 3Н), 5.76 (d, JH-F=51 Гц, 2Н), 5.10-5.16 и 4.77-4.83 (m, 2H), 3.80-4.49 (m, 6H), 2.30-2.53 (m, 2H), 1.08 (t, J=7 Гц, 2H), 0.08 (s, 9H).
APCl-MS: (M+1)=593 m/z.
(8) Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,051 г; 0,086 ммоль; смотри стадию (7) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в течение 20 мин. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил. Чистота продукта составляла 95% с 5% дефторметилированного материала. Попытки очистить его с помощью препаративной ОФЖХ (СН3CN/0,1 М NH4OAc) не увенчались успехом, и материал, частично в виде ацетата, растворяли в 5 мл TFA, упаривали и высушивали вымораживанием с получением 26 мг (51%) указанного в заголовке соединения в виде его TFA-соли. Чистота 95%.
1H-ЯМР (600 МГц; CD3OD) ротамеры: δ 7.8-7.7 (m, 2H), 7.6-7.5 (m, 2H), 7.21 (s, 1H, основной ротамер), 7.17 (s, 1H, минорный ротамер), 7.13 (s, 1H, основной ротамер), 7.09 (s, 1H, минорный ротамер), 7.07 (m, 1H, основной ротамер), 7.04 (m, 1H, минорный ротамер), 5.73 (d, 2H), 5.18 (m, 1H, минорный ротамер), 5.16 (s, 1H, основной ротамер), 5.09 (s, 1H, минорный ротамер), 4.78 (m, 1H, минорный ротамер), 4.56 (d, 1H, основной ротамер), 4.50 (d, 1H, минорный ротамер), 4.46 (d, 1H, минорный ротамер), 4.45 (d, 1H, основной ротамер), 4.35 (m, 1H, основной ротамер), 4.14 (m, 1H, основной ротамер), 4.05 (m, 1H, минорный ротамер), 3.97 (m, 1H, минорный ротамер), 2.68 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.28 (m, 1H, основной ротамер), 2.19 (m, 1H, минорный ротамер).
13С-ЯМР (150 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 173.9, 173.3, 172.9, 168.2.
ESI-MS+: (M+1)=449 (m/z).
Пример 20
Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(OMe)
К раствору триэтиламинной соли Ph(3-Cl)(5-OCH2F)-(R)CH(OH)C(O)OH (0,60 г; 1,80 ммоль; смотри пример 19(6) выше) и HAze-Pab(OMe)·2HCl (0,79 г; 2,30 ммоль) в DMF (15 мл) в атмосфере азота при 0°С добавляли РуВОР (1,04 г; 1,90 ммоль) и DIPEA (0,58 г; 4,50 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали in vacuo и трижды подвергали флэш-хроматографии на силикагеле с элюированием сначала смесью CHCl3/EtOH (9:1) и затем дважды EtOAc/EtOH (20:1), что позволило получить указанное в заголовке соединение (0,22 г; 26%) в виде дробимой белой пены.
Т.пл. 66-70°С.
Rf=0,45 (CHCl3/EtOH (9:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.59 (d, J=8 Гц, 2H), 7.32 (d, J=7 Гц, 2H), 7.06-7.23 (m, 3H), 5.75 (s, JH-F=54 Гц, 1H), 5.10-5.16 и 4.78-4.84 (m, 2H), 4.11-4.45 (m, 4H), 3.80 (s, 3H), 2.10-2.75 (m, 2H).
13С-ЯМР (150 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 173.0, 170.8, 170.7, 152.5.
APCl-MS: (M+1)=479 m/z.
Пример 21
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) (2-Монофторэтил)метансульфонат
К перемешиваемому на магнитной мешалке раствору 2-фторэтанола (5,0 г; 78,0 ммоль) в СН2Cl2 (90 мл) в атмосфере азота при 0°С добавляли триэтиламин (23,7 г; 234 ммоль) и метансульфонилхлорид (10,7 г; 93,7 ммоль). Смесь перемешивали при 0°С в течение 1,5 ч, разбавляли СН2Cl2 (100 мл) и промывали 2 н. HCl (100 мл). Водный слой экстрагировали CH2Cl2 (50 мл), объединенные органические экстракты промывали соляным раствором (75 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (9,7 г; 88%) в виде желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 4.76 (t, J=4 Гц, 1Н), 4.64 (t, J=4 Гц, 1Н), 4.52 (t, J=4 Гц, 1Н), 4.43 (t, J=4 Гц, 1Н), 3.09 (s, 3Н).
(2) 3-Хлор-5-монофторэтоксибензальдегид
К раствору 3-хлор-5-гидроксибензальдегида (8,2 г; 52,5 ммоль; смотри пример 1(2) выше) и карбоната калия (9,4 г; 68,2 ммоль) в DMF (10 мл) в атмосфере азота по каплям добавляли раствор (2-монофторэтил)метансульфоната (9,7 г; 68,2 ммоль; смотри стадию (1) выше) в DMF (120 мл) при комнатной температуре. Смесь нагревали до 100°С в течение 5 ч, затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, выливали в охлажденную во льду 2 н. HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали соляным раствором, высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Коричневое масло хроматографировали на силикагеле с элюированием смесью Нех/EtOAc (4:1), что позволило получить указанное в подзаголовке соединение (7,6 г; 71%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 9.92 (s, 1Н), 7.48 (s, 1Н), 7.32 (s, 1Н), 7.21 (s, 1Н), 4.87 (t, J=4 Гц, 1Н), 4.71 (t, J=3 Гц, 1Н), 4.33 (t, J=3 Гц, 1Н), 4.24 (t, J=3 Гц, 1Н).
(3) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-монофторэтоксибензальдегида (7,6 г; 37,5 ммоль; смотри стадию (2) выше) и иодида цинка (3,0 г; 9,38 ммоль) в CH2Cl2 (310 мл) по каплям добавляли триметилсилилцианид (7,4 г; 75,0 ммоль) при 0°С в атмосфере азота. Смесь перемешивали при 0°С в течение 3 ч и при комнатной температуре в течение ночи. Реакционную смесь разбавляли Н2О (300 мл), органический слой отделяли, высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (10,6 г; 94%) в виде коричневого масла, которое использовали без дальнейшей очистки или определения характеристик.
(4) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH
Концентрированную соляную кислоту (100 мл) добавляли к Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN (10,6 г; 5,8 ммоль; смотри стадию (3) выше), и раствор перемешивали при 100°С в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь далее охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (˜300 мл) и промывали Et2O (3×200 мл). Водный слой подкисляли 2 н. HCl (80 мл) и экстрагировали EtOAc (3×300 мл) Объединенные EtOAc-экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (8,6 г; 98%) в виде твердого вещества бледно-желтого цвета, которое использовали без дальнейшей очистки.
Rf=0,28 (CHCl3/МеОН/концентрированный NH4OH (90:8:2)).
1H ЯМР (300 МГц; CD3OD) δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
(5) Ph(3-Cl)(5-OCH2CH2F)-(S)CH(OAc)C(O)OH (а) и Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (б)
Раствор Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH (8,6 г; 34,5 ммоль; смотри стадию (4) выше) и Липазы PS "Amano" (4,0 г) в винилацетате (250 мл) и МТВЕ (250 мл) нагревали при 70°С в атмосфере азота в течение 3 сут. Реакционную смесь охлаждали до комнатной температуры и фермент удаляли посредством фильтрования через Целит®. Осадок на фильтре промывали EtOAc и фильтрат концентрировали in vacuo. Хроматография на силикагеле с элюированием смесью CHCl3/MeOH/Et3N (90:8:2) позволила получить триэтиламинную соль указанного в подзаголовке соединения (а) в виде желтого масла. В дополнение к этому получали триэтиламинную соль указанного в подзаголовке соединения (б) (4,0 г). Соль указанного в подзаголовке соединения (б) растворяли в H2O (250 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения (б) (2,8 г; 32%) в виде желтого масла.
Данные для указанного в подзаголовке соединения (б):
Rf=0,28 (CHCl3/МеОН/концентрированный NH4OH (90:8:2)).
1H ЯМР (300 МГц; CD3OD) δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
(6) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (940 мг; 3,78 ммоль; смотри стадию (5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (2,21 г; 4,91 ммоль), РуВОР (2,16 г; 4,15 ммоль) и DIPEA (1,22 г; 9,45 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 4 ч. Смесь концентрировали in vacuo и остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в подзаголовке соединение (450 мг; 20%) в виде дробимой белой пены.
Т.пл. 80-88°С.
Rf=0,60 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; СО3CD; сложная смесь ротамеров) δ 7.79 (d, J=8 Гц, 2Н), 7.42 (d, J=8 Гц, 2Н), 7.05-7.08 (m, 1H), 6.93-6.99 (m, 2H), 5.08-5.13 (m, 1H), 4.75-4.80 (m, 2H), 4.60-4.68 (m, 1H), 3.95-4.55 (m, 8H), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
APCl-MS: (M+1)=607 m/z.
(7) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,357 г; 0,589 ммоль; смотри стадию (6) выше) растворяли в 10 мл TFA и оставляли взаимодействовать в течение 40 мин. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил с получением 0,33 г (93%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (600 МГц; CD3OD, ротамеры): δ 7.8-7.7 (m, 2H), 7.54 (d, 2H), 7.08 (s, 1H, основной ротамер), 7.04 (s, 1H, минорный ротамер), 6.99 (s, 1H, основной ротамер), 6.95 (s, 1H), 6.92 (s, 1H, минорный ротамер), 5.18 (m, 1H, минорный ротамер), 5.14 (s, 1H, основной ротамер), 5.08 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.73 (m, 1H), 4.65 (m, 1H), 4.6-4.4 (m, 2Н), 4.35 (m, 1H, основной ротамер), 4.21 (дублет мультиплетов, 2Н), 4.12 (m, 1H, основной ротамер), 4.06 (m, 1H, минорный ротамер), 3.99 (m, 1H, минорный ротамер), 2.69 (m, 1H, минорный ротамер), 2.53 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.14 (m, 1H, минорный ротамер).
13С-ЯМР (150 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 172.8, 172.1, 167.4.
ESI-MS+: (M+1)=463 (m/z).
Пример 22
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OMe)
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (818 мг; 3,29 ммоль; смотри пример 21(5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCI (1,43 г; 4,27 ммоль), РуВОР (1,89 г; 3,68 ммоль) и DIPEA (1,06 г; 8,23 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo, и остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в заголовке соединение (880 мг; 54%) в виде дробимой белой пены.
Т.пл. 65-72°С.
Rf=0,60 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; СО3CD; сложная смесь ротамеров) δ 7.58-7.60 (d, J=8 Гц, 2Н), 7.34 (d, J=7 Гц, 2Н), 7.05-7.08 (m, 2H), 6.95-6.99 (m, 1H), 5.08-5.13 (m, 1H), 4.77-4.82 (m, 1H), 4.60-4.68 (m, 1H), 3.99-4.51 (m, 7H), 3.82 (s, 3H), 2.10-2.75 (m, 2H).
13С-ЯМР (150 МГц; CD3CD) (карбонильные и/или амидинные углероды) δ 173.3, 170.8, 152.5.
APCI-MS: (M+1)=493 m/z.
Пример 23
Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 1,3-Дифторизопропилметансульфонат
К перемешиваемому на магнитной мешалке раствору 1,3-дифтор-2-пропанола (7,0 г; 72,8 ммоль) в CH2Cl2 (100 мл) в атмосфере азота при 0°С добавляли триэтиламин (22,1 г; 219 ммоль) и метансульфонилхлорид (10,0 г; 87,4 ммоль). Смесь перемешивали при 0°С в течение 3 ч. Смесь промывали 2 н. HCl (150 мл) и слои разделяли. Водный слой экстрагировали CH2Cl2 (200 мл), объединенные органические экстракты промывали соляным раствором (100 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (11,5 г; 91%) в виде желтого масла, которое используют без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 4.97-5.08 (m, 1H), 4.75-4.77 (m, 2H), 4.59-4.61 (m, 2H), 3.12 (s, 3H).
(2) Ph(3-Cl)(5-OCH(CH2F)2)CHO
К раствору 3-хлор-5-гидроксибензальдегида (8,0 г; 50,7 ммоль; смотри пример 1(2) выше) и карбоната калия (9,1 г; 66,0 ммоль) в DMF (75 мл) в атмосфере азота по каплям добавляли раствор 1,3-дифторизопропилметансульфоната (11,5 г; 66,0 ммоль; смотри стадию (1) выше) в DMF (75 мл) при комнатной температуре. Смесь нагревали до 110°С в течение 18 ч. Реакционную смесь охлаждали до 0°С, выливали в охлажденную во льду 2 н. HCl (200 мл) и экстрагировали EtOAc (3×250 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Коричневое масло хроматографировали на силикагеле с элюированием смесью Нех/EtOAc (4:1), что позволило получить указанное в подзаголовке соединение (4,4 г; 37%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 9.92 (s, 1H), 7.51 (s, 1H), 7.36 (s, 1H), 7.26 (s, 1H), 4.70-4.89 (m, 3Н), 4.63-4.68 (m, 2H).
(3) Ph(3-Cl)(5-OCH(CH2F)2)-(R,S)CH(OTMS)CN
К раствору Ph(3-Cl)(5-OCH(CH2F)2)CHO (4,4 г; 18,7 ммоль; смотри стадию (2) выше) и иодида цинка (1,5 г; 4,67 ммоль) в CH2Cl2 (200 мл) при 0°С в атмосфере азота по каплям добавляли триметилсилилцианид (3,7 г; 37,3 ммоль). Смесь перемешивали при 0°С в течение 3 ч и при комнатной температуре в течение ночи, затем разбавляли Н2O (200 мл). Органический слой отделяли, высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (5,5 г; 87%) в виде коричневого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.12 (s, 1H), 7.00 (s, 2H), 5.42 (s, 1H), 4.70-4.80 (m, 3Н), 4.59-4.64 (m, 2H), 0.26 (s, 9H).
(4)Ph(3-Cl)(5-OCH(CH2F)2)-(R,S)CH(OH)C(O)OH
Концентрированную соляную кислоту (50 мл) добавляли к Ph(3-Cl)(5-OCH(CH2F)2)-(R,S)CH(OTMS)CN (5,5 г; 16,3 ммоль; смотри стадию (3) выше), и раствор перемешивали при 100°С в течение 1,5 ч. После охлаждения до комнатной температуры реакционную смесь далее охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (˜200 мл) и промывали Et2O (3×200 мл). Водный слой подкисляли 2 н. HCl (75 мл) и экстрагировали EtOAc (3×200 мл). Объединенные EtOAc-экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (4,6 г; 100%) в виде коричневого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.14 (s, 1H), 7.08 (s, 1H), 7.02 (s, 1H), 5.12 (s, 1H), 4.70-4.90 (m, 3Н), 4.52-4.67 (m, 2H).
(5) Ph(3-Cl)(5-ОСН(СН2F)2)-(S)СН(ОАс)С(O)OH (а) и Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)OH (б)
Раствор Ph(3-Cl)(5-OCH(CH2F)2)-(R,S)CH(OH)C(O)OH (4,6 г; 16,4 ммоль; смотри стадию (4) выше) и Липазы PS "Amano" (3,0 г) в винилацетате (150 мл) и МТВЕ (150 мл) нагревали при 70°С в атмосфере азота в течение 2,5 сут. Реакционную смесь охлаждали до комнатной температуры, фермент удаляли посредством фильтрования через Целит®. Осадок на фильтре промывали EtOAc и фильтрат концентрировали in vacuo. Хроматография на силикагеле с элюированием смесью CHCl3/MeOH/Et3N (90:8:2) позволила получить триэтиламинную соль указанного в подзаголовке соединения (а) в виде желтого масла. В дополнение к этому получали триэтиламинную соль указанного в подзаголовке соединения (б) (2,2 г), эту соль растворяли в H2O (100 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением указанного в подзаголовке соединения (б) (1,4 г; 29%) в виде желтого масла.
Данные для указанного в подзаголовке соединения (б)
1H ЯМР (300 МГц; CD3OD) δ 7.14 (s, 1H), 7.08 (s, 1H), 7.02 (s, 1H), 5.12 (s, 1H), 4.70-4.90 (m, 3Н), 4.52-4.67 (m, 2H).
(6)Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)OH (824 мг; 2,94 ммоль; смотри стадию (5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (1,71 г; 3,81 ммоль), РуВОР (1,68 г; 3,23 ммоль) и DIPEA (949 мг; 7,34 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo, остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в подзаголовке соединение (720 мг; 38%) в виде дробимой белой пены.
Т.пл. 78-84°С.
Rf=0,62 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; CD3CD; сложная смесь ротамеров) δ 7.79 (d, J=8 Гц, 2H), 7.42 (d, J=8 Гц, 2H), 7.00-7.12 (m, 3Н), 5.08-5.20 (m, 1H), 3.97-4.80 (m, 12H), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
APCI-MS: (M+1)=639 m/z.
(7) Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,129 г; 0,202 ммоль; смотри стадию (6) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в течение 20 мин. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил, что дает 0,123 г (100%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (400 МГц; CD3CD) ротамеры: δ 7.8-7.7 (m, 2H), 7.55 (d, 2H), 7.2-7.0 (m, 3Н), 5.18 (m, 1H, минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.08 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер, частично закрытый пиком CD3OH), 4.75-4.4 (m, 7H), 4.38 (m, 1H, основной ротамер), 4.15 (m, 1H, основной ротамер), 4.1-3.9 (m, 2H, 2 сигнала от минорного ротамера), 2.70 (m, 1H, минорный ротамер), 2.53 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.9, 172.6, 172.2, 171.7, 167.1.
ESI-MS+: (M+1)=495 (m/z).
Пример 24
Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)Aze-Pab(OMe)
К раствору Ph(3-Cl)(5-OCH(CH2F)2)-(R)CH(OH)C(O)OH (513 мг; 1,83 ммоль; смотри пример 23(5) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCl (797 мг; 2,38 ммоль), РуВОР (1,04 г; 2,01 ммоль) и DIPEA (591 мг; 4,57 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo, остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в заголовке соединение (370 мг; 39%) в виде дробимой белой пены.
Т.пл. 58-63°С.
Rf=0,66 (CHCl3/EtOH (10:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.58-7.60 (d, J=8 Гц, 2H), 7.34 (d, J=8 Гц, 2Н), 7.00-7.12 (m, 3Н), 5.08-5.20 (m, 1H), 4.65-4.82 (m, 3Н), 4.28-4.65 (m, 5H), 3.92-4.18 (m, 2H), 3.82 (s, 3Н), 2.10-2.75 (m, 2H).
13С-ЯМР (150 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 173.2, 170.8, 152.5.
APCI-MS: (M+1)=525 m/z.
Пример 25
Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 1-Бром-3-фтор-5-бензилоксибензол
Гидрид натрия (60%-ную дисперсию в масле; 24,0 г, 0,48 моль) добавляли порциями к перемешиваемому раствору безводного бензилового спирта (64,5 г; 0,60 моль) в THF (1,0 л). После перемешивания смеси в течение 1 ч по каплям добавляли раствор 1-бром-3,5-дифторбензола (76,8 г; 0,40 моль) в THF (100 мл) в продолжение периода времени 1 ч. Реакционную смесь перемешивали при комнатной температуре в течение 2 сут. Добавляли воду (400 мл) и THF удаляли in vacuo. Водный слой экстрагировали гексаном (3×150 мл). Объединенные органические экстракты промывали 2 н. NaOH (2×100 мл), затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволяло получить указанное в подзаголовке соединение (110,7 г; 98%) в виде светло-желтого масла, которое использовали без дальнейшей очистки.
Rf=0,47 (Hex).
1H ЯМР (300 МГц; CDCl3) δ 7.36-7.41 (m, 5H), 6.94 (bs, 1H), 6.87 (d, JH-F=8 Гц, 1H), 6.63 (d, JH-F=10 Гц, 1H), 5.03 (s, 2H).
(2) 3-Бром-5-фторфенол
К раствору 1-бром-3-фтор-5-бензилоксибензола (110,0 г; 0,39 моль; смотри стадию (1) выше) и N,N-диметланилина (474,0 г; 3,92 моль) в безводном CH2Cl2 (1,0 л) при 0°С добавляли хлорид алюминия (156,0 г; 1,17 моль). Через 10 мин ледяную баню удаляли и перемешивание продолжали в течение 2 ч. Реакцию гасили путем осторожного добавления 3 н. HCl (600 мл). Слои разделяли и водный слой экстрагировали CH2Cl2 (2×150 мл). Объединенные органические экстракты промывали 2 н. HCl (250 мл) и H2O (3×250 мл). К органическому слою добавляли 15%-ный КОН (500 мл) и слои разделяли. Органический слой еще раз экстрагировали 2 н. HCl (2×70 мл). Объединенные водные слои промывали CH2Cl2 (3×100 мл) и затем подкисляли 4 н. HCl. Затем водный слой экстрагировали Et2O (3×125 мл), объединенные Et2O-экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (69,0 г; 92%) в виде коричневого масла, которое использовали без дальнейшей очистки.
Т.пл. 33-35°С.
Rf=0,25 (CHCl3).
1H ЯМР (300 МГц; DMSO-d6) δ 10.38 (s, 1H), 6.90 (dd, JH-F=11 Гц, J=2 Гц, 1Н), 6.81 (s, 1Н), 6.59 (dt, JH-F=11 Гц, J=2 Гц, 1Н).
APCI-MS: (M-1)=189 m/z.
(3) 1-Бром-3-фтор-5-дифторметоксибензол
Смесь 3-бром-5-фторфенола (6,1 г; 31,0 ммоль; смотри стадию (2) выше) и хлордифторметана (13,0 г; 150,0 ммоль) в изо-PrOH (100 мл) и 30%-ном КОН (80 мл) нагревали в герметично закрытой колбе в течение 18 ч при 80-85°С. Реакционную смесь охлаждали до комнатной температуры и слои разделяли. Органический слой концентрировали in vacuo, что позволило получить бесцветное масло. Водный слой экстрагировали Et2O (3×30 мл). Неочищенное масло и объединенные органические экстракты промывали 2 н. NaOH (3×30 мл) и Н2O (3×30 мл). Органическую фазу затем высушивали (Na2SO4), фильтровали через небольшую набивку из силикагеля и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (6,1 г; 79%) в виде бесцветного масла, которое используют без дальнейшей очистки.
1H ЯМР (300 МГц; CDCl3) δ 7.11-7.14 (m, 2H), 6.84 (dt, J=9 Гц, J=2 Гц, 1Н), 6.50 (t, JH-F=72 Гц, 1Н).
(4) 1-Фтор-3-дифторметокси-5-винилбензол
Три(бутил)винилстаннан (7,0 г; 22,2 ммоль) добавляли к суспензии 1-бром-3-фтор-5-дифторметоксибензола (4,9 г; 20,2 ммоль; смотри стадию (3) выше), дихлорбис(трифенилфосфин)палладия(II) (1,42 г; 2,02 ммоль) и безводного хлорида лития (0,90 г; 20,2 ммоль) в THF (40 мл) в атмосфере азота при 65°С и смесь перемешивали в течение 5 ч. Реакционную смесь охлаждали до 0°С и добавляли 1 н. NaOH (90 мл). Бифазную смесь энергично перемешивали в течение 1 часа, затем слои разделяли. Водный слой экстрагировали Et2O (3×70 мл). Объединенные органические слои промывали 2 н. NaOH (2×40 мл) и Н2О (40 мл), затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием гексаном позволила получить указанное в подзаголовке соединение (2,2 г; 57%) в виде бесцветного масла.
Rf=0,47 (Hex).
1H ЯМР (300 МГц; CDCl3) δ 6.93-6.99 (m, 2H), 6.73-6.78 (m, 1H), 6.67 (dd, J=18 Гц, J=11 Гц, 1H), 6.51 (t, JH-F=73 Гц, 1H), 5.77 (d,J=18 Гц, 1H), 5.36 (d, J=11 Гц, 1Н).
(5) Ph(3-F)(5-OCHF2)-(R)CH(OH)СН2OH
2-Метил-2-пропанол (140 мл), Н2О (140 мл) и AD-mix-β (39,2 г) объединяли вместе и охлаждали до 0°С. 1-Фтор-3-дифторметокси-5-винилбензол (5,0 г; 26,4 ммоль; смотри стадию (4) выше) немедленно растворяли в небольшом количестве 2-метил-2-пропанола и гетерогенную смесь энергично перемешивали при 0°С до тех пор, пока ТСХ не показывала отсутствие исходного материала. Реакцию гасили при 0°С путем добавления сульфита натрия (42,0 г) и затем нагревали до комнатной температуры и перемешивали в течение 60 мин. Реакционную смесь экстрагировали Et2O (3×120 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOAc (3:2) позволила получить указанное в подзаголовке соединение (5,8 г; 98%) в виде бесцветного масла.
Rf=0,41 (CHCl3/EtOAc (3:2)).
1H ЯМР (300 МГц; COCl3) δ 6.96-6.99 (m, 2H), 6.77-6.82 (m, 1H), 6.51 (t, JH-F=73 Гц, 1H), 4.79-4.85 (m, 1H), 3.76-3.84 (m, 1H), 3.58-3.66 (m, 1H), 2.66 (d, J=3 Гц, 1H),2.00 (t, J=6 Гц, 1H).
ВЭЖХ-анализ: 89,2%, >99% е.е., колонка ChiralPak AD (подвижная фаза Нех/EtOH (95:5)).
(6) Ph(3-F)(5-OCHF2)-(R)CH(OH)CH2OTBS
Раствор Ph(3-F)(5-OCHF2)-(R)CH(OH)CH2OH (5,5 г; 24,7 ммоль; смотри стадию (5) выше), 4-(диметиламино)пиридина (121 мг; 1,0 ммоль) и триэтиламина (3,0 г; 29,6 ммоль) в безводном CH2Cl2 (100 мл) охлаждали до 0°С. По каплям добавляли 1,0 М раствор трет-бутилдиметилсилилхлорида в СН2Cl2 (26,0 мл; 26,0 ммоль), реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Добавляли насыщенный раствор хлорида аммония (60 мл) и слои разделяли. Органический слой промывали насыщенным раствором хлорида аммония (60 мл) и Н2O (2×35 мл), затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/Нех (3:1) позволила получить указанное в подзаголовке соединение (7,9 г; 85%) в виде желтого масла.
Rf=0,47 (CHCl3/Hex (3:1)).
1H ЯМР (300 МГц; CDCl3) δ 6.95-6.98 (m, 2H), 6.76-6.79 (m, 1H), 6.51 (t, JH-F=73 Гц, 1H), 4.71-4.74 (m, 1H), 3.75-3.80 (m, 1H), 3.48-3.54 (m, 1H), 2.99 (bs, 1H), 0.91 (s, 9Н), 0.05 (s, 3H), 0.00 (s, 3Н).
(7) Ph(3-F)(5-OCHF2)-(R)CH(OMEM)CH2OTBS
К раствору Ph(3-F)(5-OCHF2)-(R)CH(OH)CH2OTBS (7,9 г; 0,51 ммоль; смотри стадию (6) выше) и DIPEA (4,9 г; 48,1 ммоль) в безводном CH2Cl2 (50 мл) при 0°С в атмосфере азота по каплям добавляли 2-метоксиэтоксиметилхлорид (6,6 г; 48,1 ммоль). Смесь перемешивали в течение 24 ч. Добавляли насыщенный раствор хлорида аммония (70 мл) и слои разделяли.
Органический слой промывали насыщенным раствором хлорида аммония (70 мл) и Н2O (3×60 мл), затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (8,8 г; 99%) в виде желтого масла, которое использовали без дальнейшей очистки.
Rf=0,41 (CHCl3/EtOAc(4:1)).
1H ЯМР (300 МГц; CDCl3) δ 7.20 (s, 1H), 7.06 (s, 1H), 7.02 (s, 1H), 6,50 (t, JH-F=73 Гц, 1H), 4.79-4.81 (m, 1H), 4.66-4.68 (m, 2Н), 3.47-3.82 (m, 6H), 3.36 (s, 3Н), 0.85 (s, 9H), 0.01 (s, 3Н), 0.00 (s, 3H).
(8) Ph(3-F)(5-OCHF2)(R)CH(OMEM)CH2OH
К раствору Ph(3-F)(5-OCHF2)-(R)CH(OMEM)CH2OTBS (9,3 г; 21,9 ммоль; смотри стадию (7) выше) в THF (60 мл) при комнатной температуре добавляли 1,0 М раствор тетрабутиламмонийфторида в THF (70,0 мл; 70,0 ммоль) и смесь перемешивали в течение ночи в атмосфере азота. Реакционную смесь концентрировали in vacuo. Остаток желтого цвета растворяли в Et2O (100 мл) и гексане (100 мл) и последовательно промывали насыщенным раствором хлорида аммония (2×150 мл) и Н2O (3×70 мл). Органический слой высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (1:1) позволила получить указанное в подзаголовке соединение (4,2 г; 62%) в виде желтого масла.
Rf=0,42 (Нех/EtOAc(1:1)).
1H ЯМР (300 МГц; CDCl3) δ 6.91-6.95 (m, 2H), 6.75-6.81 (m, 1H), 6.51 (t, JH-F=73 Гц, 1H), 4.80-4.82 (m, 1H), 4.70-4.74 (m, 2H), 3.88-3.93 (m, 1H), 3.67-3.71 (m, 3Н), 3.53-3.56 (m, 2H), 3.39 (s, 3Н), 2.96-2.99 (m, 1H).
(9) Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)OH
Раствор Ph(3-F)(5-OCHF2)-(R)СН(ОМЕМ)СН2OH (4,2 г; 13,4 ммоль; смотри стадию (8) выше) в ацетоне (100 мл) добавляли к водному 5%-ному раствору NaHCO3 (35 мл). Эту перемешиваемую на магнитной мешалке гетерогенную смесь охлаждали до 0°С и добавляли бромид калия (159 мг; 1,3 ммоль) и 2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал (2,2 г; 14,1 ммоль). Затем по каплям в продолжение периода времени 20 мин добавляли гипохлорит натрия (5,25%-ный; 30 мл) при энергичном перемешивании смеси и поддержании температуры 0°С. Через 1 ч добавляли дополнительное количество гипохлорита натрия (30 мл) и 5%-ный раствор NaHCO3 (35 мл), перемешивание продолжали при 0°С в течение 2 ч. Ацетон удаляли in vacuo. Водный слой промывали Et2O (4×40 мл). Водный слой подкисляли до рН 3,5 10%-ной лимонной кислотой и экстрагировали EtOAc (4×50 мл). Затем объединенные EtOAc-экстракты последовательно промывали H2O (4×30 мл) и соляным раствором (60 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (4,3 г; 98%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
Rf=0,74 (CHCl3/МеОН/Et3N (8,0:1,5:0,5)).
1H ЯМР (300 МГц; ацетон-d6) δ 7.16-7.18 (m, 2Н), 7.16 (t, JH-F=89 Гц, 1Н), 7.00-7.03 (m, 1Н), 5.30 (s, 1H), 4.88 (d, J=7 Гц, 1Н), 4.80 (d, J=7 Гц, 1Н), 3.54-3.75 (m, 2H), 3.46-3.49 (m, 2H), 3.28 (s, 3Н).
(10)Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)OH (1,1 г; 3,4 ммоль; смотри стадию (9) выше) в DMF (20 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (2,0 г; 4,4 ммоль), РуВОР (1,9 г; 3,7 ммоль) и DIPEA (1,1 г; 8,4 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo и остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc/EtOH (20:1), что позволило получить указанное в подзаголовке соединение (1,3 г; 56%) в виде дробимой белой пены.
Rf=0,65 (CHCl3/EtOH (15:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.80-7.84 (m, 2H), 7.40-7.46 (m, 2H), 6.95-7.16 (m, 3Н), 6.92 и 6.88 (t, JH-F=73 Гц, 1H), 5.28 и 5.08 (s, 1H), 5.18-5.22 и 4.70-4.78 (m, 1H), 4.50-4.75 (m, 1H), 4.30-4.49 (m, 2H), 4.21-4.26 (m, 1H), 3.97-4.08 (m, 1H), 3.35-3.72 (m, 6H), 3.30 (s, 3Н), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
(11) Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
Смесь Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc) (590 мг; 0,87 ммоль; смотри стадию (10) выше) и четырехбромистого углерода (287 мг; 0,87 ммоль) в 2-пропаноле (20 мл) кипятили с обратным холодильником в течение 1,5 ч. Затем данную смесь концентрировали in vacuo, распределяли между Н2О (50 мл) и EtOAc (3×50 мл). Водный слой экстрагировали дополнительным количеством EtOAc (2×10 мл). Объединенные органические экстракты промывали соляным раствором (30 мл), затем высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOH (15:1) позволила получить указанное в подзаголовке соединение (60 мг; 12%) в виде дробимой белой пены. Rf=0,46 (CHCl3/EtOH (15:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.74 (d, J=8 Гц, 2Н), 7.35-7.37 (m, 2Н), 6.97-7.07 (m, 2H), 6.80-6.84 (m, 1H), 6.82 и 6.80 (t, JH-F=73 Гц, 1H), 5.10 и 5.06 (s, 1H), 4.68-4.70 (m, 1H), 3.97-4.60 (m, 6H), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9Н).
APCI-MS: (M+1)=595 m/z.
(12) Ph(3-F)(5-OCHF2)(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,053 г; 0,089 ммоль; смотри стадию (11) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в течение 80 мин при охлаждении в ледяной бане. TFA выпаривали и остаток высушивали вымораживанием из смеси вода/ацетонитрил с получением 0,042 г (80%) указанного в заголовке соединения в виде его TFA-соли.
1H-ЯМР (300 МГц; CD3OD) ротамеры: δ 7.7-7.6 (m, 2H), 7.5-7.4 (m, 2H), 7.1-6.6 (m, 4H), 5.2-5.0 (m, 1H плюс минорный ротамер 1H), приблизительно 4.8 (основной ротамер предыдущего сигнала, перекрытый сигналом от CD3ОН), 4.6-4.3 (m, 2H), 4.26 (m, 1H, основной ротамер), 4.10 (m, 1H, основной ротамер), 3.96 (m, 1H, минорный ротамер), 3.89 (m, 1H, минорный ротамер), 2.60 (m, 1H, минорный ротамер), 2.44 (m, 1H, основной ротамер), 2.19 (m, 1H, основной ротамер), 2.05 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 172.8, 172.0, 167.0.
ESI-MS+: (M+1)=451 (m/z).
Пример 26
Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
(1) Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(OMe)
К раствору Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)OH (1,0 г; 3,1 ммоль; смотри пример 25(9) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCl (1,4 г; 4,1 ммоль), РуВОР (1,8 г; 3,4 ммоль) и DIPEA (1,0 г; 7,8 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo, остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1) и второй раз EtOAc, что позволило получить указанное в подзаголовке соединение (1,5 г; 79%) в виде дробимой белой пены.
Rf=0,24 (EtOAc).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.58-7.62 (m, 2H), 7.32-7.38 (m, 2H), 7.03-7.16 (m, 3Н), 6.92 и 6.88 (d, JH-F=73 Гц, 1Н), 5.27 и 5.08 (s, 1Н), 5.22-5.15 и 4.75-4.80 (m, 1H), 4.38-4.65 (m, 5Н), 3.92-4.27 (m, 1H), 3.82 (s, 3Н), 3.43-3.68 (m, 4H), 3.29 (s, 3Н), 2.28-2.85 (m, 2H).
(2)Ph(3-F)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
Смесь Ph(3-F)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(OMe) (828 мг; 2,33 ммоль; смотри стадию (1) выше) и четырехбромистого углерода (525 мг; 2,33 ммоль) в 2-пропаноле (20 мл) кипятили с обратным холодильником в течение 8 ч и затем перемешивали при комнатной температуре в течение ночи. Смесь концентрировали in vacuo и остаток распределяли между H2O (70 мл) и EtOAc (50 мл). Водный слой экстрагировали EtOAc (2×25 мл). Затем объединенные органические экстракты промывали соляным раствором (35 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOH (15:1) позволила получить указанное в заголовке соединение (520 мг; 74%) в виде дробимой белой пены.
Т.пл. 73-81°С.
Rf=0,43 (CHCl3/EtOH(15:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.59 (d, J=8 Гц, 2H), 7.32-7.37 (m, 2H), 7.05-7.14 (m, 2H), 6.87-6.92 (m, 1H), 6.90 и 6.86 (t, JH-F= 73 Гц, 1H), 5.13-5.18 и 4.75-4.85 (m, 2H), 4.15-4.45 (m, 4H), 3.81 (s, 3Н), 2.10-2.75 (m, 2H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 172.0, 171.4, 153.9.
APCI-MS: (M+1)=481 m/z.
Пример 27
Ph(3-Br)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 1,3-Дибром-5-бензилоксибензол
Гидрид натрия (9,9 г; 0,414 моль, 95%-ный, безводный) добавляли порциями к перемешиваемому раствору бензилового спирта (41,0 г; 0,394 моль) в THF (1,0 л) при комнатной температуре в атмосфере азота и перемешивали в течение 1 ч. К этому раствору по каплям добавляли 1,3-дибром-5-фторбензол (100,0 г; 0,394 моль). После перемешивания в течение ночи смесь распределяли между Н2O (600 мл) и EtOAc (4×600 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием гексанами позволила получить указанное в подзаголовке соединение (101,3 г; 75%) в виде желтого масла.
1H ЯМР (300 МГц; CDCl3) δ 7.30-7.48 (m, 5H), 7.18 (s, 1H), 7.06 (s, 2H), 4.99 (s, 2H).
(2) 3,5-Дибромфенол
Хлорид алюминия (11,7 г; 87,6 ммоль) добавляли порциями к раствору 1,3-дибром-5-бензилоксибензола (10,0 г; 29,2 ммоль; смотри стадию (1) выше) и N,N-диметиланилина (35,4 г; 292 ммоль) в CH2Cl2 (100 мл) при комнатной температуре в атмосфере азота. Через 30 мин смесь распределяли между 1 н. HCl (300 мл) и EtOAc (5×150 мл). Объединенные органические экстракты промывали насыщенным NaHCOз (150 мл) и затем соляным раствором (150 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (9:1) позволила получить указанное в подзаголовке соединение (6,1 г; 82%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц; CDCl3) δ 7.21 (s, 1Н), 6.97 (s, 2H), 5.88 (bs, 1 Н).
(3) 1,3-Дибром-5-монофторметоксибензол
В тарированную, герметично закрытую, находящуюся под давлением круглодонную колбу вместимостью 350 мл, содержащую суспензию 3,5-дибромфенола (10,0 г; 39,7 ммоль; смотри стадию (2) выше) и Cs2СО3 (20,7 г; 63,5 ммоль) в DMF (150 мл), при -78°С добавляли хлорфторметан посредством барботирования в течение 5 мин через мембрану. Мембрану заменяли на тефлоновую пробку, колбу затем герметично закрывали и оставляли нагреваться до комнатной температуры, затем колбу взвешивали и определяли, что в ней содержится 9,0 г (131 ммоль) хлорфторметана. Данный раствор нагревали на масляной бане, установленной на 70°С, в течение ночи. Колбу охлаждали до комнатной температуры, осторожно сбрасывали давление и содержимое разбавляли водой (100 мл). Затем водный слой экстрагировали Et2O (3×100 мл), объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием гексанами позволила получить указанное в подзаголовке соединение (7,9 г; 71%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц; CDCl3) δ 7.40 (s, 1H), 7.18 (s, 2H), 5.67 (d, JH-F=53 Гц, 2Н).
(4) 1-Бром-3-монофторметокси-5-винилбензол
Три(бутил)винилолово (10,0 г; 31,4 ммоль) по каплям добавляли к раствору 1,3-дибром-5-монофторметоксибензола (8,5 г; 29,9 ммоль; смотри стадию (3) выше), тетракис(трифенилфосфин)палладия(0) (690 мг; 0,599 ммоль) и 2,6-ди-трет-бутил-4-метилфенола (на кончике шпателя) в толуоле (100 мл) в атмосфере азота. Смесь перемешивали при 70°С в течение 8 ч. Смесь охлаждали до 0°С и добавляли 1 н. NaOH (70 мл). Через 1 ч смесь затем экстрагировали с помощью СН2Cl2 (3×300 мл), объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием гексанами позволила получить указанное в подзаголовке соединение (4,3 г; 57%) в виде бесцветного масла.
1H ЯМР (300 МГц; CDCl3) δ 7.30 (s, 1H), 7.16 (s, 1H), 7.01 (s, 1H), 6.60 (dd. J=6 Гц, J=11 Гц, 1H), 5.74 (d, J=16 Гц, 1H), 5.67 (d, JH-F=53 Гц, 2H), 5.32 (d, J=8 Гц, 1H).
(5) Ph(3-Br)(5-OCH2F)-(R)CH(OH)CH2OH
2-Метил-2-пропанол (100 мл), Н2О (100 мл) и AD-mix-β (27,5 г) объединяли вместе и охлаждали до 0°С. Немедленно добавляли 1-бром-3-монофторметокси-5-винилбензол (4,3 г; 17,3 ммоль; смотри стадию (4) выше), и данную гетерогенную смесь энергично перемешивали при 0°С до подтверждения с помощью ТСХ отсутствия исходного материала. Реакцию гасили при 0°С добавлением насыщенного сульфита натрия (200 мл), затем нагревали до комнатной температуры и перемешивали в течение 60 мин. Реакционную смесь экстрагировали EtOAc (3×150 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (4,9 г; 100%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.30 (s, 1Н), 7.15 (s, 1H), 7.11 (s, 1H), 5.70 (d, JH-F=53 Гц, 2Н), 4.62-4.70 (m, 1H), 3.52-3.70 (m, 2H).
ВЭЖХ-анализ: 92,1%; 96,9% е.е., колонка ChiralPak AD (подвижная фаза Нех/EtOH(95:5)).
(6) Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)CH2OTBS
К раствору Ph(3-Br)(5-OCH2F)-(R)CH(OH)CH2OH (4,9 г; 18,6 ммоль; смотри стадию (5) выше), 4-(диметиламино)пиридина (453 мг; 3,71 ммоль) и DIPEA (8,9 г; 93,0 ммоль) в безводном CH2Cl2 (200 мл) по каплям добавляли 1,0 М раствор трет-бутилдиметилсилилхлорида в CH2Cl2 (22,3 мл; 22,3 ммоль). Реакционную смесь перемешивали 10 ч при комнатной температуре. К смеси добавляли DIPEA (8,9 г; 93,0 ммоль) и 2-метоксиэтоксиметилхлорид (13,9 г; 111 ммоль) по каплям. Через 16 ч добавляли дополнительное количество 2-метоксиэтоксиметилхлорида (2,2 г) и реакционную смесь перемешивали в течение ночи. Смесь разбавляли Н2O (100 мл) и слои разделяли. Затем водный слой экстрагировали CH2Cl2 (3×200 мл), объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (5:1) позволила получить указанное в подзаголовке соединение (4,8 г; 55%) в виде бесцветного масла.
1H ЯМР (300 МГц; CDCl3) δ 7.29 (s, 1H), 7.22 (s, 1H), 7.05 (s, 1H), 5.74 (d, JH-F=53 Гц, 2H), 4.84 (d, J=7 Гц, 1H), 4.70-4.74 (m, 2H), 3.50-3.91 (m, 6H), 3.42 (s, 3Н), 0,90 (s, 9H), 0.05 (s, 3Н), 0.01 (s, 3H).
(7) Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)CH2OH
К раствору Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)CH2OTBS (4,7 г; 10,1 ммоль; смотри стадию (6) выше) в THF (100 мл) добавляли 1,0 М раствор тетрабутиламмонийфторида в THF (13,1 мл; 13,1 ммоль) при комнатной температуре и смесь перемешивали 1 ч. Затем смесь распределяли между Н2O (100 мл) и EtOAc (3×100 мл), объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (3,3 г; 92%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.22 (s, 1Н), 7.14 (s, 1H), 7.03 (s, 1H), 5.71 (d, JH-F=53 Гц, 2Н), 4.80-4.82 (m, 1H), 4.58-4.66 (m, 2H), 3.71-3.77 (m, 1H), 3.39-3.65 (m, 5Н), 3.27 (s, 3H).
(8)Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)C(O)OH
Раствор Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)CH2OH (2,1 г; 6,0 ммоль; смотри стадию (7) выше) в ацетоне (40 мл) добавляли к водному 5%-ному раствору NaHCO3 (15 мл). Эту перемешиваемую с помощью магнитной мешалки гетерогенную смесь охлаждали до 0°С, к ней добавляли бромид калия (70 мг; 0,60 ммоль) и 2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал (976 мг; 5,8 ммоль). Затем по каплям добавляли гипохлорит натрия (5,25%-ный; 15 мл) в течение периода 10 мин при энергичном перемешивании смеси и поддержании температуры 0°С. Через 1 ч добавляли дополнительное количество гипохлорита натрия (10 мл) и раствора NaHCO3 (20 мл) и перемешивание продолжали при 0°С в течение дополнительных 4 ч. Ацетон удаляли на роторном испарителе. Водный слой разбавляли 10%-ным раствором NaHCO3 (30 мл) и промывали Et2O (3×20 мл). Водный слой подкисляли до рН 3,5 10%-ной лимонной кислотой и экстрагировали EtOAc (3×40 мл). Затем объединенные EtOAc-экстракты промывали Н2O (3×50 мл) и соляным раствором (50 мл), высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (1,7 г; 78%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.38 (s, 1H), 7.25 (s, 1H), 7.18 (s, 1H), 5.76 (d, JH-F=53 Гц, 2H), 5.21 (s, 1H), 4.83 (d, J=7 Гц, 1H), 4.75 (d, J=7 Гц, 1H), 3.62-3.78 (m, 2H), 3.48-3.52 (m, 2H), 3.32 (s, 3H).
(9) Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)C(O)OH (1,0 г; 2,72 ммоль; смотри стадию (8) выше) в DMF (20 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (1,6 г; 3,5 ммоль), РуВОР (1,6 г; 3,0 ммоль) и DIPEA (880 мг; 6,81 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo и остаток дважды хроматографировали на силикагеле с элюированием первоначально смесью CHCl3/EtOH (15:1), а во второй раз смесью EtOAc/EtOH (20:1), что позволило получить указанное в подзаголовке соединение (1,2 г; 62%) в виде дробимой белой пены.
1H ЯМР (300 МГц; CD3OD, сложная смесь ротамеров) δ 7.80-7.84 (m, 2H), 7.40-7.46 (m, 2H), 7.13-7.32 (m, 3Н), 5.84-5.87 (m, 1H), 5.67-5.69 (m, 1H), 5.25 и 5.07 (s, 1H), 5.18-5.23 и 4.80-4.88 (m, 1H), 3.97-4.79 (m, 8H), 3.60-3.71 (m, 2H), 3.40-3.53 (m, 2H), 3.32 (s, 3Н), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9Н).
(10) Ph(3-Br)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
Смесь Ph(3-Br)(5-OCH2F)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc) (347 мг; 0,478 ммоль; смотри стадию (9) выше) и четырехбромистого углерода (159 мг; 0,478 ммоль) в 2-пропаноле (10 мл) кипятили с обратным холодильником в течение 1,5 ч. Смесь концентрировали in vacuo, затем распределяли с помощью Н2O (20 мл) и EtOAc (3×30 мл). Объединенные органические слои высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOH (15:1) позволила получить указанное в подзаголовке соединение (59 мг; 19%) в виде дробимой белой пены.
Т.пл. 81-87°С.
Rf=0,58 (CHCl3/EtOH (9:1)).
1H ЯМР (300 МГц; CD3OD, сложная смесь ротамеров) δ 7.84 (d, J=8 Гц, 2H), 7.40-7.48 (m, 2H), 7.18-7.30 (m, 3Н), 5.80 (d, JH-F=53 Гц, 2H), 5.21 и 5.15 (s, 1H), 5.18-5.24 и 4.80-4.88 (m. 1H), 3.98-4.54 (m, 6H), 2.10-2.70 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
APCI-MS: (M+1)=637 m/z.
(11) Ph(3-Br)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Br)(5-OCH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,073 г; 0,11 ммоль; смотри стадию (10) выше) растворяли в 5 мл TFA и оставляли взаимодействовать в течение 90 мин, одновременно охлаждая на ледяной бане. TFA выпаривали и остаток очищали препаративной ВЭЖХ с использованием СН3CN/0,1 М NH4OAc (30:70). Подходящие фракции упаривали и высушивали вымораживанием из смеси вода/ацетонитрил с получением 49 мг (77%) указанного в заголовке соединения в виде его ацетатной соли.
1H-ЯМР (300 МГц; CD3OD; ротамеры) δ 7.8-7.7 (m, 2H), 7.54 (m, 2H), 7.37 (s, 1H, основной ротамер), 7.33 (s, 1H, минорный ротамер), 7.25-7.1 (m, 2H), 5.75 (d, 2H), 5.22 (m, 1H, минорный ротамер), 5.18 (s, 1H, основной ротамер), 5.11 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.6-4.4 (m, 2H), 4.37 (m, 1H, основной ротамер), 4.16 (m, 1H, основной ротамер), 4.1-3.9 (m, 2H, два сигнала от минорного ротамера), 2.70 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер), 1.89 (s, 3Н).
ESI-MS+: (М+1)=493/495 (m/z).
Пример 28
Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab×TFA
(1) 1,3-Дибром-5-дифторметоксибензол
В тарированную, герметично закрытую, находящуюся под давлением круглодонную колбу вместимостью 350 мл, содержащую раствор 3,5-дибромфенола (10,0 г; 39,7 ммоль; смотри пример 27(2) выше) в 2-пропаноле (100 мл) и 30%-ном КОН (80 мл), при -78°С добавляли хлордифторметан посредством барботирования в течение 15 мин через мембрану. Мембрану меняли на тефлоновую пробку, затем колбу герметично закрывали и оставляли нагреваться до комнатной температуры, тогда колбу взвешивали и определяли, что в ней содержится 12,0 г (138 ммоль) хлордифторметана. Данный раствор кипятили с обратным холодильником в течение ночи на масляной бане, установленной на 80°С. Колбу охлаждали до комнатной температуры, осторожно сбрасывали давление и содержимое разбавляли Н2O (200 мл). Водный слой экстрагировали CHCl3 (2×150 мл), затем объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Остаток очищали дистилляцией с использованием трубки с шаровым расширением (Kugelrohr) при 80°С и 26,7 Па (0,2 мм рт.ст.), что позволило получить указанное в подзаголовке соединение (9,6 г; 80%) в виде прозрачной жидкости.
1H ЯМР (300 МГц; CDCl3) δ 7.55 (s, 1H), 7.26 (s, 2H), 6.52 (t, JH-F=68 Гц, 1Н).
(2) 1-Бром-3-дифторметокси-5-винилбензол
Три(бутил)винилолово (10,5 г; 33,1 ммоль) по каплям добавляли к раствору 1,3-дибром-5-дифторметоксибензола (9,1 г; 30,1 ммоль; смотри стадию (1) выше), тетракис(трифенилфосфин)палладия(0) (700 мг; 0,60 ммоль) и 2,6-ди-трет-бутил-4-метилфенола (на кончике шпателя) в толуоле (125 мл) в атмосфере азота. Смесь перемешивали при 50°С в течение ночи. Смесь охлаждали до 0°С и добавляли 1 н. NaOH (70 мл). Через 1 ч смесь затем экстрагировали CH2Cl2 (3×300 мл), объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием гексанами позволила получить указанное в подзаголовке соединение (5,1 г; 68%) в виде бесцветного масла.
1H ЯМР (300 МГц; CDCl3) δ 7.53 (s, 1H), 7.18 (s, 1H), 7.08 (s, 1H), 6.60 (dd, J=6 Гц, J=11 Гц, 1H), 6.57 (t, JH-F=68 Гц, 1H), 5.77 (d, J=11 Гц, 1H), 5.36 (d, J=8 Гц, 1Н).
(3) Ph(3-Br)(5-OCHF2)-(R)CH(OH)CH2OH
2-Метил-2-пропанол (150 мл), H2O (150 мл) и AD-mix-β (27,8 г) объединяли вместе и охлаждали до 0°С. Немедленно добавляли 1-бром-3-дифторметокси-5-винилбензол (4,6 г; 18,6 ммоль; смотри стадию (2) выше), и данную гетерогенную взвесь энергично перемешивали при 0°С до подтверждения с помощью ТСХ отсутствия исходного материала, затем раствор нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили при 0°С добавлением насыщенного сульфита натрия (300 мл) и затем нагревали до комнатной температуры и перемешивали в течение 60 мин. Реакционную смесь экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (5,0 г; 95%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.43 (s, 1Н), 7.23 (s, 1H), 7.16 (s, 1H), 6.86 (t, JH-F=75 Гц, 1H), 4.64-4.67 (m, 1H), 3.54-3.59 (m, 2H).
ВЭЖХ-анализ: 88,6%; 96,3% е.е., колонка ChiralPak AD (подвижная фаза Нех/EtOH (95:5)).
(4) Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)CH2OTBS
К раствору Ph(3-Br)(5-OCHF2)-(R)CH(OH)CH2OH (4,9 г; 17,3 ммоль; смотри стадию (3) выше), 4-(диметиламино)пиридина (420 мг; 3,5 ммоль) и DIPEA (11,2 г; 86,3 ммоль) в безводном CH2Cl2 (250 мл) по каплям добавляли 1,0 М раствор трет-бутилдиметилсилилхлорида в CH2Cl2 (20,7 мл; 20,7 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. К смеси по каплям добавляли DIPEA (11,2 г; 86,3 ммоль) и 2-метоксиэтоксиметилхлорид (12,9 г; 104 ммоль). Через 3 сут. добавляли дополнительное количество 2-метоксиэтоксиметилхлорида (3,3 г) и реакционную смесь перемешивали в течение ночи. Смесь разбавляли водой (250 мл) и слои разделяли. Водный слой экстрагировали CH2Cl2 (2×250 мл), затем объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью Нех/EtOAc (4:1) позволила получить указанное в подзаголовке соединение (4,3 г; 51%) в виде бесцветного масла.
1H ЯМР (300 МГц; CDCl3) δ 7.40 (s, 1H), 7.25 (s, 1H), 7.08 (s, 1H), 6.58 (t, JH-F=75 Гц, 1H), 4.84 (d, J=7 Гц, 1H), 4.70-4.74 (m, 2H), 3.50-3.91 (m, 6H), 3.42 (s, 3Н), 0,90 (s, 9H), 0.12 (s, 3Н), 0.05 (s, 3H).
(5) Ph(3-Br)(5-OCHF2)-(R)СН(OMEM)СН2ОН
К раствору Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)CH2OTBS (3,3 г; 6,9 ммоль; смотри стадию (4) выше) в THF (60 мл) добавляли 1,0 М раствор тетрабутиламмонийфторида в THF (9,0 мл; 9,0 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 45 мин, затем смесь распределяли между водой (150 мл) и EtOAc (2×120 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (2,5 г; 98%) в виде желтого масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.35 (s, 1Н), 7.21 (s, 1H), 7.08 (s, 1H), 6.83 (t, JH-F=73 Гц, 1H), 4.73 (d, J=7 Гц, 1H), 4.59-4.68 (m, 2H), 3.40-3.80 (m, 6H), 3.26 (s, 3H).
(6) Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)OH
Раствор Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)CH2OH (3,0 г; 8,1 ммоль; смотри стадию (5) выше) в ацетоне (60 мл) добавляли к водному 5%-ному раствору NaHCO3 (25 мл). Эту перемешиваемую с помощью магнитной мешалки гетерогенную смесь охлаждали до 0°С, затем добавляли бромид калия (100 мг; 0,81 ммоль) и 2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал (1,3 г; 8,5 ммоль). Затем по каплям добавляли гипохлорит натрия (5,25%-ный; 19 мл) в течение периода 10 мин при энергичном перемешивании смеси и поддержании температуры 0°С. Через 1 ч добавляли дополнительное количество гипохлорита натрия (17 мл) и раствора NaHCO3 (34 мл), перемешивание продолжали при 0°С в течение дополнительных 4 ч. Ацетон удаляли на роторном испарителе. Водный слой разбавляли 10%-ным раствором NaHCO3 (30 мл) и промывали Et2O (3×20 мл). Водный слой подкисляли до рН 3,5 10%-ной лимонной кислотой и экстрагировали EtOAc (3×40 мл). Объединенные EtOAc-слои промывали Н2O (3×50 мл) и соляным раствором (50 мл) и затем сушили (Na2SO4), фильтровали и концентрировали in vacuo, что позволило получить указанное в подзаголовке соединение (2,1 г; 66%) в виде бесцветного масла, которое использовали без дальнейшей очистки.
1H ЯМР (300 МГц; CD3OD) δ 7.51 (s, 1H), 7.32 (s, 1H), 7.24 (s, 1H), 6.88 (t, JH-F=73 Гц, 1H), 5.21 (s, 1H), 4.84 (d, J=7 Гц, 1H), 4.76 (d, J=7 Гц, 1Н), 3.62-3.80 (m, 2H), 3.48-3.52 (m, 2H), 3.32 (s, 3H).
(7) Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)OH (1,0 г; 2,62 ммоль; смотри стадию (6) выше) в DMF (50 мл) в атмосфере азота при 0°С добавляли HAze-Pab(Teoc)·HCl (1,5 г; 3,38 ммоль), РуВОР (1,5 г; 2,9 ммоль) и DIPEA (840 мг; 6,50 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo и остаток хроматографировали на силикагеле с элюированием смесью CHCl3/EtOH (15:1), что позволило получить указанное в подзаголовке соединение (1,1 г; 59%) в виде дробимой белой пены.
1H ЯМР (300 МГц; CD3OD, сложная смесь ротамеров) δ 7.79-7.83 (m, 2H), 7.26-7.52 (m, 5Н), 6.94 и 6.91 (t, JH-F=73 Гц, 1Н), 5.27 и 5.07 (s, 1H), 5.20-5.23 и 4.80-4.88 (m, 1H), 4.01-4.79 (m, 8Н), 3.60-3.71 (m, 2H), 3.40-3.53 (m, 2H), 3.32 (s, 3Н), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
(8) Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
Смесь Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(Teoc) (369 мг; 0,496 ммоль; смотри стадию (7) выше) и четырехбромистого углерода (165 мг; 0,496 ммоль) в 2-пропаноле (10 мл) кипятили с обратным холодильником в течение 12 ч. Смесь концентрировали in vacuo, затем распределяли с помощью H2O (15 мл) и EtOAc (5×20 мл). Объединенные органические фазы высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOH (15:1) позволяла получить указанное в подзаголовке соединение (134 мг; 41%) в виде дробимой белой пены.
Т.пл.92-98°С.
Rf=0,37 (CHCl3/EtOH (9:1)).
1H ЯМР (300 МГц; CD3OD, сложная смесь ротамеров) δ 7.80-7.86 (m, 2H), 7.40-7.48 (m, 2H), 7.10-7.33 (m, 3Н), 6.92 и 6.88 (t, JH-F=73 Гц, 1H), 5.18 и 5.11 (s, 1H), 5.18-5.24 и 4.76-4.80 (m, 1H), 3.98-4.54 (m, 6H), 2.10-2.70 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
APCI-MS: (M+1)=655 m/z.
(9) Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab×TFA
Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,081 г; 0,124 ммоль; смотри стадию (8) выше) растворяли в 5 мл TFA и оставляли взаимодействовать в течение 80 мин с одновременным охлаждением на ледяной бане. TFA выпаривали и остаток очищали препаративной ВЭЖХ с использованием СН3CN/0,1 М NH4OAc (30:70). Подходящие фракции упаривали и высушивали вымораживанием из смеси вода/ацетонитрил с получением 59 мг (83%) указанного в заголовке соединения в виде его ацетатной соли.
1H-ЯМР (300 МГц; CD3OD; ротамеры): δ 7.8-7.7 (m, 2H), 7.6-7.4 (m, 3Н), 7.3-7.2 (m, 2H), 6.89 (t, 1H, основной ротамер), 6.87 (t, 1H, минорный ротамер), 5.23 (m, 1H, минорный ротамер), 5.21 (s, 1H, основной ротамер), 5.13 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.6-4.4 (m, 2H), 4.38 (m, 1Н, основной ротамер), 4.20 (m, 1H, основной ротамер), 4.1-3.9 (m, 2H, два сигнала от минорного ротамера), 2.70 (m, 1H, минорный ротамер), 2.54 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер), 1.89 (s, 3Н).
13С-ЯМР (75 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 172.0, 171.7, 167.0.
MS (m/z) 511/513 (M+1)+.
Пример 29
Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
(1) Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(OMe)
К раствору Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)OH (957 мг; 2,48 ммоль; смотри пример 28(6) выше) в DMF (30 мл) в атмосфере азота при 0°С добавляли HAze-Pab(OMe)·2HCl (1,1 г; 3,2 ммоль), РуВОР (1,4 г; 2,7 ммоль) и DIPEA (804 мг; 6,2 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали in vacuo, остаток дважды подвергали хроматографии на силикагеле с элюированием первоначально смесью CHCl3/EtOH (9:1) и во второй раз EtOAc/EtOH (15:1), что позволило получить указанное в подзаголовке соединение (1,1 г; 72%) в виде дробимой белой пены.
1H ЯМР (300 МГц; CD3OD, сложная смесь ротамеров) δ 7.59-7.65 (m, 2H), 7.20-7.55 (m, 5H), 6.95 и 6.91 (t, JH-F=73 Гц, 1H), 5.27 и 5.07 (s, 1H), 5.18-5.23 и 4.75-4.84 (m, 1H), 3.87-4.89 (m, 6H), 3.84 (s, 3Н), 3.60-3.71 (m, 2H), 3.40-3.53 (m, 2H), 3.32 (s, 3Н), 2.10-2.75 (m, 2H).
(2) Ph(3-Br)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe)
Смесь Ph(3-Br)(5-OCHF2)-(R)CH(OMEM)C(O)-Aze-Pab(OMe) (1,1 г; 1,8 ммоль; смотри стадию (1) выше) и четырехбромистого углерода (583 мг; 1,8 ммоль) в 2-пропаноле (30 мл) кипятили с обратным холодильником в течение 2,5 сут. В продолжение этого времени для полного завершения реакции добавляли дополнительное количество четырехбромистого углерода (5 порций по 50 мг с интервалами для дополнительных 0,90 ммоль). Смесь концентрировали in vacuo, затем распределяли с помощью Н2О (50 мл) и EtOAc (5×25 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Флэш-хроматография на силикагеле с элюированием смесью CHCl3/EtOH (15:1) позволила получить указанное в заголовке соединение (460 мг; 50%) в виде дробимой белой пены.
Т.пл. 71-75°С.
Rf=0,63 (CHCl3/EtOH (9:1)).
1H ЯМР (300 МГц; CD3OD; сложная смесь ротамеров) δ 7.59 (d, J=8 Гц, 2Н), 7.20-7.54 (m, 5Н), 6.90 и 6.87 (t, JH-F=73 Гц, 1Н), 5.18 и 5.11 (s, 1H), 4.76-4.80 (m, 1H), 3.98-4.54 (m, 4H), 3.82 (s, 3H), 2.10-2.70 (m, 2H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.5, 172.1, 171.6, 154.1.
APCI-MS: (M+1)=542 m/z.
Пример 30
Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
(1) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z)
Boc-Aze-Pab(Z) (смотри заявку на международный патент WO 97/02284, 92 мг; 0,197 ммоль) растворяли в 10 мл EtOAc, насыщенного HCl (г), и оставляли взаимодействовать в течение 10 мин. Растворитель выпаривали, остаток смешивали с Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (50 мг; 0,188 ммоль; смотри пример 17(5) выше), РуВОР (109 мг; 0,209 ммоль) и окончательно с диизопропилэтиламином (96 мг; 0,75 ммоль) в 2 мл DMF. Смесь перемешивали в течение 2 ч, затем выливали в 50 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле с элюированием смесью EtOAc/МеОН (9:1). Выход 100 мг (87%).
1H ЯМР (300 МГц; CD3OD, смесь ротамеров) δ 7.85-7.75 (m, 2H), 7.45-7.25 (m, 7H), 7.11 (m, 1H, основной ротамер), 7.08 (m, 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.13 (bt, 1H), 5.25-5.05 (m, 3H), 4.77 (m, 1H, частично невидим за сигналом от CD3ОН), 4.5-3.9 (m, 7H), 2.64 (m, 1H, минорный ротамер), 2.47 (m, 1H, основной ротамер), 2.25 (m, 1H, основной ротамер), 2.13 (m, 1H, минорный ротамер).
(2) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Гидроксиламина гидрохлорид (65 мг; 0,94 ммоль) и триэтиламин (0,319 г; 3,16 ммоль) смешивали в 8 мл THF и обрабатывали ультразвуком в течение 1 ч при 40°С. Добавляли Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z) (96 мг; 0,156 ммоль; смотри стадию (1) выше) с дополнительными 8 мл THF. Смесь перемешивали при 40°С в течение 4,5 суток. Растворитель выпаривали и неочищенный продукт очищали препаративной ОФЖХ в смеси СН3CN/0,1 М NH4OAC (40:60). Выход 30 мг (38%). Чистота 99%.
1H ЯМР (300 МГц; CD3OD, смесь ротамеров) δ 7.6-7.55 (m, 2H), 7.35-7.3 (m, 2H), 7.12 (m, 1Н, основной ротамер), 7.09 (m, 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.15 (триплет мультиплетов, 1H), 5.15 (m, 1H, минорный ротамер), 5.13 (s, 1H, основной ротамер), 5.08 (s, 1H, минорный ротамер), 4.77 (m, 1H, основной ротамер), 4.5-4.2 (m, 5H), 4.08 (m, 1H, основной ротамер), 3.97 (m, 1H, минорный ротамер), 2.66 (m, 1H, минорный ротамер), 2.50 (m, 1H, основной ротамер), 2.27 (m, 1H, основной ротамер), 2.14 (m, 1H, минорный ротамер),
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 172.8, 172.2, 171.4, 159.1, 158.9, 154.2.
APCI-MS: (M+1)=497/499 m/z.
Пример 31
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OH)
(1) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Z)
Boc-Aze-Pab(Z) (130 мг; 0,279 ммоль) растворяли в 15 мл EtOAc, насыщенного HCl (г), и оставляли взаимодействовать в течение 10 мин. Растворитель выпаривали и остаток смешивали с Ph(3-Cl)(5-OCH2CH2F)-(R)СН(ОН)С(O)ОН (63 мг; 0,188 ммоль; смотри пример 21(5) выше) в 3 мл DMF, РуВОР (147 мг; 0,279 ммоль) и окончательно с диизопропилэтиламином (134 мг; 1,03 ммоль). Смесь перемешивали в течение 130 мин и затем выливали в 75 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в смеси EtOAc/МеОН (95:5). Выход 119 мг (79%).
1H ЯМР (400 МГц; CDCl3) δ 8.06 (bt, 1H), 7.67 (d, 2H), 7.45-7.25 (m, 5H), 7.18 (d, 2H), 6.89 (m, 1H), 6.84 (m, 1H), 6.76 (m, 1H), 5.16 (s, 2H), 4.84 (s, 1H), 4.79 (m, 1H), 4.66 (дублет мультиплетов, 2H), 4.4-4.3 (m, 2H), 4.10 (дублет мультиплетов, 2H), 4.02 (m, 1H), 3.67 (m, 1H), 2.46 (m, 1H), 2.28 (m, 1H).
(2) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(OH)
Гидроксиламина гидрохлорид (80 мг; 1,16 ммоль) и триэтиламин (0,392 г; 3,87 ммоль) смешивали в 9 мл THF и обрабатывали ультразвуком в течение 1 ч при 40°С. Добавляли Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Z) (96 мг; 0,156 ммоль; смотри стадию (1) выше) с дополнительными 9 мл THF. Смесь перемешивали при 40°С в течение 48 ч и 3 суток при комнатной температуре. Растворитель выпаривали и неочищенный продукт очищали препаративной ОФЖХ в смеси СН3CN/0,1 М NH4OAc (30:70). Выход 72 мг (78%). Чистота 100%.
1H ЯМР (400 МГц; CD3OD, смесь ротамеров) δ 7.6-7.55 (m, 2H), 7.35-7.25 (m, 4H), 7.07 (m, 1H, основной ротамер), 7.04 (m, 1H, минорный ротамер), 7.0-6.9 (m, 2H), 5.12 (m, 1H, минорный ротамер), 5.08 (s, 1H, минорный ротамер), 5.04 (s, 1H), 4.78 (m, 1H, основной ротамер), 4.68 (дублет мультиплетов, 2H), 4.5-4.25 (m, 3Н), 4.20 (дублет мультиплетов, 2H), 4.06 (m, 1H, основной ротамер), 3.97 (m, 1H, минорный ротамер), 2.65 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.27 (m, 1H, основной ротамер), 2.14 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 172.3, 171.5, 159.8, 154.3.
APCI-MS: (M+1)=479/481 m/z.
Пример 32
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Pro-Pab
(1) Boc-Pro-Pab(Teoc)
Boc-Pro-Pab(Z) (смотри заявку на международный патент WO 97/02284; 15,0 г; 0,0321 моль) растворяли в 150 мл этанола и добавляли 200 мг 10%-ного Pd/C (50%-ной влажности). Смесь перемешивали и гидрировали при атмосферном давлении в течение 2 ч, фильтровали через Hyflo и концентрировали. Продукт использовали без дальнейшей очистки. 10 г этого продукта (0,029 моль) растворяли в 300 мл THF. Добавляли Теос-n-нитрофенилкарбонат (10 г; 0,035 моль). В продолжение 3 мин добавляли раствор карбоната калия (5,2 г; 0,038 моль) в 50 мл воды, получающийся раствор перемешивали в течение 3 суток, концентрировали, и остаток трижды экстрагировали EtOAc. Объединенный органический слой промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш- хроматографии на силикагеле с использованием смеси метиленхлорид/ацетон (4:1). Выход 9,8 г (69%).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Pro-Pab(Teoc)
Boc-Pro-Pab(Teoc) (107 мг; 0,218 ммоль; смотри стадию (1) выше) растворяли в 10 мл EtOAc, насыщенного HCl (г), и оставляли взаимодействовать в течение 10 мин. Растворитель выпаривали, остаток смешивали с Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (50 мг; 0,198 ммоль; смотри пример 1(8) выше) в 3 мл DMF, РуВОР (115 мг; 0,218 ммоль) и окончательно с диизопропилэтиламином (104 мг; 0,80 ммоль). Смесь перемешивали в течение 2 ч, затем выливали в 75 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в смеси EtOAc/МеОН (95:5). Выход 89 мг (72%).
1H ЯМР (400 МГц; CDCl3) δ 7.54 (bt, 1H), 7.47 (d, 2H), 7.12 (m, 1H), 7.08 (d, 2Н), 7.02 (m, 1H), 6.95 (m, 1H), 6.50 (t, 1H), 5.21 (s, 1H), 4.42 (m, 1H), 4.35-4.15 (m, 3Н), 3.59 (m, 1H), 2.94 (m, 1H), 2.1-1.7 (m, 4H), 1.06 (m, 2H), 0.04 (s, 9H).
(3) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Pro-Pab×TFA
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Pro-Pab(Teoc) (85 мг; 0,136 ммоль; смотри стадию (2) выше) растворяли в 1 мл метиленхлорида и охлаждали на ледяной бане. Добавляли TFA (4 мл) и реакционную смесь перемешивали в течение 90 мин. TFA выпаривали и остаток высушивали вымораживанием из воды и ацетонитрила. Выход 72 мг (92%). Чистота 94%.
1H ЯМР (400 МГц; CD3OD, смесь ротамеров) δ 7.85-7.7 (m, 2H), 7.58 (d, 2H, основной ротамер), 7.47 (d, 2H, минорный ротамер), 7.35 (m, 1H, основной ротамер), 7.27 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.88 (t, 1H), 5.38 (s, 1H, основной ротамер), 5.22 (s, 1H, минорный ротамер), 4.58 (d, 1H), 4.5-4.2 (m, 2H), 3.8-3.5 (m, 1H), 3.35 (m, 1H), 2.2-1.8 (m, 4H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды) δ 173.6, 171.1, 167.0.
APCI-MS: (M+1)=481/483 m/z.
Пример 33
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Pro-Pab(OMe)
(1) 4-Азидометил-N-метоксибензамидин
4-Азидометилбензнитрил (17,3 г; 0,109 моль; Nishiyama et al.; Chem. Lett. (1982) 1477) растворяли в 500 мл толуола и 200 мл абсолютного этанола. Раствор охлаждали до -10°С и через него барботировали HCl (г) до насыщения. Смесь выдерживали в холодильнике в течение 2 суток, в продолжение которых испарялась большая часть растворителей. Добавляли диэтиловый эфир и осуществляли декантирование. Продукт перерастворяли в растворе O-метилгидроксиламина (10,5 г; 0,125 моль) и триэтиламина (56 мл) в 200 мл метанола. Смесь оставляли стоять в течение 3 суток, после чего метанол выпаривали с добавлением EtOAc. Органическую фазу промывали водой, разбавляли НОАс и водным бикарбонатом натрия, высушивали (Na2SO4) и снова разбавляли EtOAc до суммарного объема в 500 мл. Образец в объеме 25 мл упаривали досуха. Остаток составлял 932 мг. Суммарный выход 18,6 г (83%).
(2) 4-Аминометил-N-метоксибензамидин
К раствору 4-азидометил-N-метоксибензамидина (11,3 г; 0,055 моль; смотри стадию (1) выше) в 200 мл этанола добавляли 200 мг PtO2. Смесь гидрировали при постоянном барботировании водородом в течение 4 ч и потом фильтровали через Целит® и упаривали. Выход 7,34 г (74%).
(3) Boc-Pro-Pab(OMe)
К суспензии Вос-Pro-ОН (9,7 г; 0,045 моль), 4-аминометил-N-метоксибензамидина (7,33 г; 0,041 моль; смотри стадию (2) выше) и диметиламинопиридина (7,8 г; 0,064 моль) в 300 мл ацетонитрила добавляли EDC, основание (11,7 мл; 0,068 моль). Смесь перемешивали в течение 18 ч, концентрировали и распределяли между водой и EtOAc. Органический слой промывали водой, водным бикарбонатом натрия, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в EtOAc. Выход 9,73 г (63%).
(4) Н-Pro-Pab(OMe)×2HCl
Boc-Pro-Pab(OMe) (9,7 г; 0,026 моль; смотри стадию (3) выше) растворяли в 250 мл EtOAc. Этот охлажденный во льду раствор насыщали HCl (г) посредством барботирования в течение 5 мин. Продукт немедленно выпадал в осадок, и добавляли 125 мл абсолютного этанола. Смесь обрабатывали ультразвуком до тех пор, пока большая часть материала не переходила в твердое состояние. Добавляли диэтиловый эфир (200 мл) и суспензию фильтровали. Несколько комков, которые не переходили в твердое состояние, снова обрабатывали абсолютным этанолом и диэтиловым эфиром. Твердое вещество высушивали. Выход 7,57 г (86%).
1H ЯМР (400 МГц; CD3OD) δ 7.74 (d, 2Н), 7.58 (d, 2H), 4.55 (s, 2H), 4.38 (m, 1H), 3.98 (s, 3Н), 3.45-3.3 (m, 2H), 2.50 (m, 1H), 2.15-2.0 (m, 3H).
(5) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Pro-Pab(OMe)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (50 мг; 0,198 ммоль; смотри пример 1(8) выше), H-Pro-Pab(OMe) (76 мг; 0,218 ммоль; смотри стадию (4) выше) и РуВОР (115 мг; 0,218 ммоль) растворяли в 2 мл DMF. Добавляли диизопропилэтиламин (104 мг; 0,80 ммоль) и смесь перемешивали в течение 2,5 ч. Смесь выливали в 50 мл воды, трижды экстрагировали EtOAc, объединенную органическую фазу промывали соляным раствором, высушивали (Na2SO4) и упаривали. Остаток подвергали флэш-хроматографии на силикагеле в смеси EtOAc/МеОН (95:5). Выход 37 мг (36%). Чистота 98%.
1H ЯМР (400 МГц; CD3OD, смесь ротамеров) δ 7.60 (d, 2H, основной ротамер), 7.57 (d, 2H, минорный ротамер), 7.4-7.1 (m, 5H), 6.89 (t, 1H, основной ротамер), 6.87 (t, 1H, минорный ротамер), 5.35 (s, 1H, основной ротамер), 5.21 (s, 1H, минорный ротамер), 4.72 (m, 1H, минорный ротамер), 4.5-4.35 (m, 1H и 2H, основной ротамер), 4.3-4.25 (m, 2H, минорный ротамер), 3.814 (s, 3Н, основной ротамер), 3.807 (s, 3Н, минорный ротамер), 3.75-3.5 (m, 1H), 3.35 (m, 1H), 2.2-1.8 (m, 4H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 173.3, 173.2, 171.3, 171.0, 153.9, 152.4.
APCI-MS: (M+1)=511/513 m/z.
Пример 34
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)С(O)-Aze-NH-СН2-((2-амидино)-5-
пиридинил)
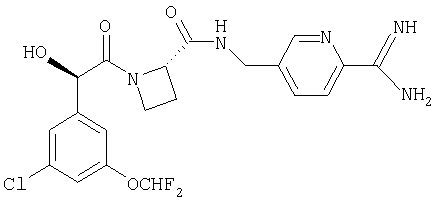
(1) 6-Цианоникотиновая кислота
К раствору N-оксида никотиновой кислоты (51 г; 0,37 моль) в 1,2 л DMF добавляли NaCN (54 г; 1,1 моль) с последующим добавлением триэтиламина (255 мл; 1,83 моль) и TMSCl (185 мл). Реакционную смесь перемешивали при 110°С в течение 10 ч, фильтровали и фильтрат концентрировали. Остаток растворяли в 100 мл 2 н. HCl и экстрагировали метиленхлоридом. Органические слои объединяли, концентрировали и перекристаллизовывали из воды с получением 12 г (22%) продукта.
(2) 5-(Гидроксиметил)пиридин-2-карбонитрил
К раствору 6-цианоникотиновой кислоты (12 г; 0,081 моль; смотри стадию (1) выше) в THF при 0°С добавляли Et3N (12,4 мл; 0,0892 моль) с последующим добавлением этилхлорформиата (8,53 мл; 0,0892 моль). Реакционную смесь перемешивали в течение 15 мин и добавляли NaBH4 (6,14 г; 0,162 моль). Затем смесь перемешивали при КТ в течение ночи, гасили водой и экстрагировали метиленхлоридом. Органический слой концентрировали и очищали колоночной хроматографией с получением 4 г (20%) спирта.
(3) 5-(Азидометил)пиридин-2-карбонитрил
5-(Гидроксиметил)пиридин-2-карбонитрил (4 г; 0,03 моль; смотри стадию (2) выше) растворяли в 25 мл метиленхлорида и охлаждали в ледяной бане. По каплям добавляли мезилхлорид (2,32 мл; 0,0300 моль) и затем триэтиламин (4,6 мл; 0,033 моль). Реакционную смесь перемешивали и после смешивания неочищенный мезилат обрабатывали NaN3 (7,35 г; 0,113 моль) в 20 мл DMF. Реакционную смесь перемешивали при 40°С в течение 2 ч, разбавляли водой и экстрагировали этилацетатом. Органический слой концентрировали с получением 3,95 г (83%) неочищенного азида.
(4) 5-(трет-Бутоксикарбониламинометил)пиридин-2-карбонитрил
К раствору 5-(азидометил)пиридин-2-карбонитрила (3,95 г; 0,0248 моль; смотри стадию (3) выше) в 30 мл THF и 10 мл воды добавляли трифенилфосфин (7,8 г; 0,0298 моль) и полученную смесь перемешивали в течение 24 ч. Затем добавляли триэтиламин (3,8 мл; 0,027 моль) с последующим добавлением Вос-ангидрида (5,4 г; 0,025 моль) с перемешиванием в течение 2 ч. Реакционную смесь распределяли между водой и этилацетатом. Органический слой концентрировали и очищали колоночной хроматографией с получением 2,1 г (36%) указанного в подзаголовке соединения.
1H ЯМР (300 МГц; CDCl3) δ 8.6 (s, 1H), 8.0 (d, 1Н), 8.9 (d, 1H), 4.1 (m, 2H), 1.4 (s, 9H).
(5) 5-(Аминометил)пиридин-2-карбонитрил × 2HCl
5-(трет-Бутоксикарбониламинометил)пиридин-2-карбонитрил (0,200 г; 0,86 ммоль; смотри стадию (4) выше) растворяли в 10 мл EtOAc, насыщенного HCl (г), и перемешивали в течение 30 мин. Растворитель выпаривали и получали 0,175 г (99%) указанного в подзаголовке соединения в виде его дигидрохлоридной соли.
1H ЯМР (500 МГц; D2O) δ 8.79 (s, 1H), 8.17 (d, 1H), 8.05 (d, 1H), 4.38 (s, 2H).
(6) Boc-Aze-NH-CH2-5-Py(2-CN)
К смеси 5-(аминометил)пиридин-2-карбонитрила × 2HCl (0,175 г; 0,85 ммоль; смотри стадию (5) выше), Boc-Aze-OH (0,201 г; 1,00 ммоль) и TBTU (0,321 г; 1,00 ммоль) в 5 мл DMF добавляли диметиламинопиридин (0,367 г; 3,00 ммоль). Смесь перемешивали в течение ночи и потом выливали в воду и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водным бикарбонатом натрия, высушивали (Na2SO4) и упаривали. Неочищенный продукт, начинающий кристаллизоваться, использовали как таковой на следующей стадии. Выход 0,23 г (73%).
1H ЯМР (500 МГц; CDCl3) δ 8.66 (s, 1H), 8.2-7.8 (уширенный, 1H), 7.79 (d, 1H), 7.67 (d, 1H), 4.73 (m, 1H), 4.65-4.5 (m, 2H), 3.94 (m, 1H), 3.81 (m, 1H), 2.6-2.35 (m, 2H), 1.8 (уширенный, 1H), 1.45 (s, 9H).
(7) H-Aze-NH-CH2-5-Py(2-CN)×2HCl
Boc-Aze-NH-CH2-5-Py(2-CN) (0,23 г; 0,73 ммоль; смотри стадию (6) выше) растворяли в 10 мл EtOAc, насыщенного HCl (г), и перемешивали в течение 30 мин. Растворитель выпаривали и получали 0,21 г (100%) указанного в подзаголовке соединения в виде его дигидрохлоридной соли.
1H ЯМР (500 МГц; D2O) δ 8.64 (s, 1H), 8.0-7.9 (m, 2H), 5.19 (m, 1H), 4.65-4.55 (m, 2H), 4.20 (m, 1H), 4.03 (m, 1H), 2.88 (m, 1H), 2.64 (m, 1H).
(8) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Aze-NH-CH2-5-Py(2-CN)
К смеси H-Aze-NH-CH2-5-Py(2-CN)×2HCl (0,206 г; 0,713 ммоль; смотри стадию (7) выше), Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,180 г; 0,713 ммоль; смотри пример 1(8) выше) и РуВОР (0,408 г; 0,784 ммоль) в 5 мл DMF добавляли диметиламинопиридин (0,367 г; 3,00 ммоль). Смесь перемешивали в течение ночи, потом выливали в воду и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водным бикарбонатом натрия, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в EtOAc с получением чистого продукта. Выход 0,197 г (61%).
1H ЯМР (500 МГц; CDCl3) δ 8.63 (m, 1Н), 8.22 (bt, 1H), 7.78 (m, 1H), 7.67 (m, 1H), 7.21 (m, 1H), 7.16 (m, 1H), 7.04 (m, 1H), 6.56 (t, 1H), 4.97 (bd, 1H), 4.92 (m, 1H), 4.6-4.5 (m, 2H), 4.40 (bd, 1H), 4.18 (m, 1H), 3.80 (m, 1H), 2.69 (m, 1H), 2.46 (m, 1H), 1.92 (s, 1H).
APCI-MS: (M+1)=451/453 m/z.
(9) Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)-С(O)-Aze-NH-СН2-((2-амидино)-5-пиридинил)×HOAc
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Aze-NH-CH2-5-Py(2-CN) (0,200 г; 0,444 ммоль; смотри стадию (8) выше), ацетат аммония (1,00 г; 0,0130 моль) и N-ацетилцистеин (2,0 г; 0,0122 моль) в 10 мл метанола нагревали при 50°С в течение 2 суток. Препаративная ОФЖХ в смеси СН3CN/0,1 М NH4OAc (30:70) и повторное пропускание соответствующих фракций в смеси СН3CN/0,1 М NH4OAc (5:95-40:60) позволили получить 60 мг (26%) чистого указанного в заголовке соединения в виде его ацетатной соли после высушивания вымораживанием из воды и ацетонитрила. Чистота 100%.
1H ЯМР (500 МГц; D2O, смесь ротамеров) δ 8.68 (s, 1H, основной ротамер), 8.62 (s, 1H, минорный ротамер), 8.05-7.9 (m, 2H), 7.33 (m, 1H, ротамер), 7.27 (m, 1H, ротамер), 7.22 (m, 1H, ротамер), 7.17 (m, 1H, ротамер), 7.01 (m, 1H, ротамер), 6.84 (t, 1H), 5.32 (s, 1H, основной ротамер), 5.20 (m, 1H, минорный ротамер), 5.13 (s, 1H, минорный ротамер), 4.88 (m, 1H, основной ротамер), 4.65-4.55 (m, 2H, основной ротамер), 4.45-4.35 (m, 1H, ротамер плюс 1H, минорный ротамер), 4.31 (d, 1H, минорный ротамер), 4.2-4.05 (m, 1H плюс 1H, ротамер), 2.80 (m, 1H, минорный ротамер), 2.61 (m, 1H, основной ротамер), 2.33 (m, 1H, основной ротамер), 2.24 (m, 1H, минорный ротамер), 1.93 (s, 3Н).
13С-ЯМР (100 МГц; D2O) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 181.6, 173.3, 172.7, 172.6, 172.3, 162.6, 162.3.
APCI-MS: (M+1)=468/470 m/z.
Пример 35
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)С(O)-Aze-NH-СН2-((2-метоксиамидино)-5-пиридинил)
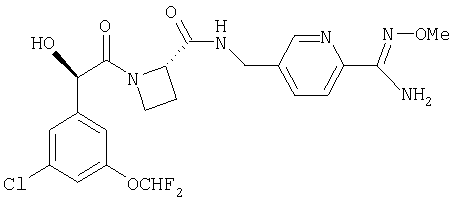
(1) Вос-NH-СН2-[(2-(амино(гидроксилимино)метил))-5-пиридинил]
5-(трет-Бутоксикарбониламинометил)пиридин-2-карбонитрил (1,00 г; 4,29 ммоль; смотри пример 34(4) выше) растворяли в 10 мл этанола и добавляли гидроксиламина гидрохлорид (0,894 г; 0,0129 моль) и триэтиламин (1,30 г; 0,0129 моль). Смесь перемешивали при комнатной температуре в течение 6 суток. Смесь распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Выход 0,96 г (84%).
1H ЯМР (400 МГц; ацетон-d6) δ 9.01 (bs, 1Н), 8.50 (bs, 1H), 7.87 (m, 1H), 7.70 (m, 1H), 6.58 (уширенный, 1H), 5.70 (уширенный, 2Н), 4.31 (d, 2H), 1.41 (s, 1H).
(2) Вос-Aze-NH-СН2-(2-(амидино)-5-пиридинил)×НОАс
Эту реакцию осуществляли согласно способу, описанному в Judkins et al., Synth. Comm. (1998) 4351. Суспензию Вос-NH-СН2-[(2-(амино(гидроксилимино)метил))-5-пиридинила] (0,910 г; 3,42 ммоль; смотри стадию (1) выше), уксусного ангидрида (0,35 мл; 3,7 ммоль) и 0,35 г 10%-ного Pd/C (50%-ной влажности) в 100 мл уксусной кислоты гидрировали под давлением 490 кПа (5 атм) в течение 5 ч. Смесь фильтровали через Целит и концентрировали. Остаток высушивали вымораживанием из воды и ацетонитрила с получением 0,97 г (92%) указанного в подзаголовке соединения.
1H ЯМР (500 МГц; CD3OD) δ 8.74 (s, 1H), 8.12 (d, 1H), 7.98 (d, 1H), 4.38 (s, 2H), 1.92 (s, 3H), 1.46 (s, 9H).
(3) Вос-NH-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил)
К суспензии Вос-NH-СН2-(2-(амидино)-5-пиридинила)×НОАс (0,96 г; 3,1 ммоль; смотри стадию (2) выше) в 75 мл THF добавляли раствор карбоната калия (1,07 г; 7,7 ммоль) и Теос-п-нитрофенилкарбоната (1,14 г; 4,02 ммоль) в 15 мл воды. Смесь перемешивали в течение ночи. Добавляли избыток глицина и карбоната калия и реакцию продолжали в течение 2 ч. THF выпаривали и остаток трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Продукт можно было использовать без дальнейшей очистки.
1H ЯМР (500 МГц; CDCl3) δ 9.31 (уширенный, 1H), 8.52 (s, 1H), 8.41 (d, 1H), 8.35 (уширенный, 1H), 7.74 (d, 1H), 4.97 (уширенный, 1H), 4.39 (m, 2H), 4.26 (m, 2H), 1.46 (s, 9Н), 1.14 (m, 2H), 0.07 (s, 9H).
(4) Н2N-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил)×2HCl
Вос-NH-СН2-(2-(амино(триметилсилилэтилимино)метил-5-пиридинил) (0,23 г; 0,58 ммоль; смотри стадию (3) выше) растворяли в 25 мл EtOAc, насыщенного HCl (г), и перемешивали в течение 30 мин. Растворитель выпаривали и продукт использовали без дальнейшей очистки. Выход 0,21 г (98%).
1H ЯМР (500 МГц; D2O) δ 8.89 (s, 1H), 8.25 (s, 2H), 4.55 (m, 2H), 4.42 (s, 2H), 1.20 (m, 2H), 0.09 (s, 9H).
(5) Вос-Aze-HN-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил)
К раствору Н2N-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинила)×2HCl (0,21 г; 0,57 ммоль; смотри стадию (4) выше), Boc-Aze-OH (0,127 г; 0,631 ммоль) и TBTU (233 мг; 0,726 ммоль) в 5 мл DMF добавляли диметиламинопиридин (269 мг; 2,20 ммоль). Смесь перемешивали в течение ночи, выливали в 100 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водным бикарбонатом натрия и водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш- хроматографии на силикагеле в EtOAc с получением 170 мг (56%) желаемого продукта.
1H ЯМР (500 МГц; CDCl3) δ 9.33 (уширенный, 1Н), 8.54 (s, 1H), 8.41 (d, 1Н), 8.36 (уширенный, 1H), 7.75 (m, 1H), 4.72 (m, 1H), 4.56 (m, 2H), 4.26 (m, 2H), 3.93 (m, 1H), 3.80 (m, 1H), 2.6-2.4 (m, 2H), 1.42 (s, 9Н), 1.14 (m, 2H), 0.07 (s, 9Н).
(6) Н-Aze-NH-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил)×2HCl
Вос-Aze-NH-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил) (170 мг; 0,356 ммоль; смотри стадию (5) выше) растворяли в 25 мл EtOAc, насыщенного HCl (г), и перемешивали в течение 30 мин. Растворитель выпаривали и продукт использовали без дальнейшей очистки. Выход 160 мг (100%).
1H ЯМР (500 МГц; CD3OD) δ 9.00 (m, 1H), 8.84 (m, 1H), 8.23 (d, 2H), 8.10 (m, 1H), 5.09 (m, 1H), 4.7-4.6 (m, 2H), 4.51 (m, 2H), 4.14 (m, 1H), 3.97 (m, 1H), 2.86 (m, 1H), 2.58 (m, 1H), 1.22 (m, 2H), 0.11 (s, 9H).
(7) Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинил)
К раствору Н-Aze-NH-СН2-(2-(амино(триметилсилилэтилимино)метил)-5-пиридинила)×2HCl (160 мг; 0,462 ммоль; смотри стадию (6) выше), Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (131 мг; 0,462 ммоль; смотри пример 1(8) выше) и РуВОР (263 мг; 0,505 ммоль) в 5 мл DMF добавляли диизопропилэтиламин (0,30 мл; 1,71 ммоль). Смесь перемешивали в течение ночи, выливали в 100 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водным бикарбонатом натрия и водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в смеси EtOAc/МеОН (95:5) с получением 148 мг (52%) желаемого продукта.
(8) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-NH-CH2-(2-(метоксиамино-(триметилсилилэтилимино)метил)-5-пиридинил)
Суспензию Ph(3-Cl)(5-OCHF2)-(R)CH(OH)-C(O)-Aze-NH-CH2-(2-(метоксиамино-(триметилсилилэтилимино)метил)-5-пиридинила) (148 мг; 0,242 ммоль; смотри стадию (7) выше) и O-метилгидроксиламина (202 мг; 2,42 ммоль) в 10 мл ацетонитрила нагревали при 70°С в течение 3 ч. Смесь распределяли между водой и EtOAc. Водный слой дважды экстрагировали EtOAc и объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный материал подвергали флэш-хроматографии на силикагеле в смеси EtOAc/МеОН (95:5) с получением 44 мг (28%) чистого материала.
1H ЯМР (500 МГц; CDCl3) δ 8.55 (m, 1H), 8.05 (bt, 1Н), 7.70 (m, 1H), 7.58 (s, 1Н), 7.56 (d, 1H), 7.22 (m, 1H), 7.16 (m, 1H), 7.03 (m, 1H), 6.50 (t, 1H), 4.92 (s, 1H), 4.89 (m, 1H), 4.55-4.45 (m, 2Н), 4.38 (уширенный, 1H), 4.2-4.1 (m, 3H), 4.00 (s, 3Н), 3.73 (m, 1H), 2.69 (m, 1H), 2.44 (m, 1H), 0.97 (m, 2H), 0.02 (s, 9H).
(9) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-NH-СН2-((2-метоксиамидино)-5-пиридинил)
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-(2-(метоксиамино(три-метилсилилэтилимино)метил)-5-пиридинил) (44 мг; 0,069 ммоль; смотри стадию (8) выше) растворяют в 2 мл TFA и оставляют взаимодействовать в течение 1 ч. TFA выпаривают и остаток распределяют между EtOAc и водным бикарбонатом натрия. Водный слой экстрагируют EtOAc и объединенную органическую фазу промывают водой, высушивают (Na2SO4) и упаривают. Выход 30 мг (88%). Чистота >95%.
1H ЯМР (500 МГц; CDCl3) δ 8.44 (m, 1H), 8.03 (bt, 1H), 7.91 (m, 1H), 7.60 (m, 1H), 7.19 (m, 1H), 7.13 (m, 1H), 7.00 (m, 1H), 6.52 (t, 1H), 5.6-5.45 (уширенный, 2H), 4.90 (s, 1H), 4.89 (m, 1H), 4.55-4.4 (m, 2H), 4.27 (уширенный, 1Н), 4.12 (m, 1H), 3.92 (s, 3H), 2.68 (m, 1H), 2.41 (m, 1H).
13С-ЯМР (100 МГц; CDCl3) (карбонильные и/или амидинные углероды) δ 173.0, 170.9, 152.6.
APCI-MS: (M+1)=498/500 m/z.
Пример 36
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-((5-амидино)-2-пиримидинил)

(1) 2-Амино-2-иминоэтилкарбамат·АсОН
N-Boc-аминоацетонитрил (40,2 г; 257,4 ммоль) и N-ацетилцистеин (42,0 г; 257,4 ммоль) растворяли в метаноле (300 мл) при 60°С и в течение 18 ч пропускали аммиак. Растворитель удаляли in vacuo. После ионообменной хроматографии (Амберлит IRA-400 (АсОН)) и перекристаллизации из ацетона получали 28,4 г (53%) указанного в подзаголовке соединения в виде твердого вещества белого цвета.
1H ЯМР (300 МГц; CD3OD) δ 4.41 (t, J=4.9 Гц, 1 Н), 4.01 (s, 2H), 2.91 (d, J=5.0 Гц, 2H), 2.01 (s, 3Н), 1.46 (s, 9H).
(2) 1,3-Бис(диметиламино)-2-цианотриметинперхлорат
Раствор 3-диметиламиноакрилонитрила (25,0 г; 260,0 ммоль) в хлороформе (75 мл) по каплям добавляли к раствору (хлорметилен)диметиламмонийхлорида (50,0 г; 390,1 ммоль) в хлороформе (175 мл) при 0°С. Реакционную смесь перемешивали дополнительно 2 ч при 0°С, затем оставляли нагреваться до комнатной температуры в течение ночи, потом нагревали в течение 8 ч при температуре дефлегмации. Растворитель удаляли in vacuo. Остаток добавляли к смеси перхлората натрия (110 мг; 0,898 ммоль) в воде (150 мл) и этаноле (300 мл). Смесь нагревали при температуре дефлегмации в течение 15 мин, затем охлаждали и оставляли стоять в течение ночи в холодильнике. Осадок собирали и перекристаллизовывали из этанола с получением 23,8 г (52%) указанного в подзаголовке соединения в виде бесцветных игольчатых кристаллов.
Т.пл. 140-141°С.
1H ЯМР (300 МГц; CDCl3) δ 8.24 (s, 2H), 3.59 (s, 6H), 3.51 (s, 6H).
(3) Вос-NH-СН2-(5-циано)-2-пиримидин
Смесь трет-бутил-2-амино-2-иминоэтилкарбамата·АсОН (5,0 г; 23,8 ммоль; смотри стадию (1) выше) и 1,3-бис(диметиламино)-2-цианотриметинперхлората (6,0 г; 23,8 ммоль; смотри стадию (2) выше) в пиридине (300 мл) перемешивали в атмосфере азота при 70-75°С в течение 16 ч и затем нагревали при температуре дефлегмации в течение 6 ч. Смесь охлаждали до комнатной температуры и растворитель удаляли in vacuo. Остаток экстрагировали горячей смесью (1:1) этилацетата и хлороформа, фильтровали через небольшую набивку диоксида кремния и концентрировали с получением неочищенного продукта. Флэш-хроматография на диоксиде кремния с элюированием хлороформом дала 4,0 г (71%) указанного в подзаголовке соединения в виде бесцветного масла, которое при стоянии перешло в твердое состояние.
Т.пл. 86-87°С.
Rf=0,77 (диоксид кремния, этилацетат/хлороформ (3:2)).
1H ЯМР (300 МГц; DMSO-d6) δ 9.25 (s, 2H), 7.39 (bt, 1H), 4.39 (d, J=6 Гц, 2Н), 1.38 (s, 9H).
13С ЯМР (750 МГц; DMSO-d6) δ 170.4, 160.3, 155.8, 115.2, 106.9, 80.0, 46.3, 28.1.
APCl-MS: (M+1)=235 m/z.
(4) Вос-Aze-NH-СН2-((5-циано)-2-пиримидинил)
Вос-NH-СН2-(5-циано)-2-пиримидин (1,14 г; 4,87 ммоль; смотри стадию (3) выше) растворяли в 50 мл EtOAc, насыщенного HCl (г), оставили взаимодействовать в течение 1 ч и концентрировали. Остаток растворяли в 20 мл DMF и охлаждали в ледяной бане. Добавляли диизопропилэтиламин (3,5 мл; 0,020 моль), Boc-Aze-OH (1,08 г; 5,37 ммоль) и HATU (2,80 г; 5,38 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали и продукт очищали препаративной ОФЖХ с использованием СН3CN/0,1 М NH4OAc (40:60). Ацетонитрил выпаривали и водный слой трижды экстрагировали EtOAc. Объединенный органический слой высушивали (MgSO4) и упаривали. Выход 1,12 г (72%).
1H ЯМР (400 МГц; CDCl3) δ 8.95 (s, 2H), 4.82 (d, 2H), 4.74 (m, 1H), 3.95 (m, 1H), 3.84 (m, 1H), 2.6-2.4 (m, 2H), 1.47 (s, 9H).
(5) Вос-Aze-NH-СН2-((5-амидино)-2-пиримидинил)×НОАс
Раствор Вос-Aze-NH-СН2-((5-циано)-2-пиримидинила) (0,83 г; 2,6 ммоль; смотри стадию (4) выше), N-ацетилцистеина (0,43 г; 2,6 ммоль) и ацетата аммония (0,60 г; 7,8 ммоль) в 10 мл метанола нагревали при 60°С в атмосфере азота в течение 2 суток. Растворитель выпаривали и неочищенный материал очищали препаративной ОФЖХ с использованием градиента СН3CN/0,1 М NH4OAc (от 5:95 до 100:0). Интересующие фракции высушивали вымораживанием с получением 1,0 г (93%) желаемого материала.
1H ЯМР (300 МГц; D2O; сигналы, перекрываемые сигналом от HDO) δ 9.17 (s, 2Н), 4.1-3.9 (m, 2H), 2.60 (m, 1Н), 2.29 (m, 1H), 1.93 (s, 3Н), 1.44 (s, 9H).
(6) Вос-Aze-NH-СН2-[(5-(амино(триметилсилилэтилимино)метил))-2-пиримидинил]
К суспензии Вос-Aze-NH-СН2-((5-амидино)-2-пиримидинила)×НОАс (0,95 г; 2,41 ммоль; смотри стадию (5) выше) в 50 мл THF добавляли раствор Теос-п-нитрофенилкарбоната (0,85 г; 3,0 ммоль) и карбоната калия (1,0 г; 7,2 ммоль) в 10 мл воды. Смесь перемешивали в течение 24 ч, концентрировали и распределяли между водой и метиленхлоридом. Органический слой дважды промывали насыщенным водным бикарбонатом натрия, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле в смеси гептан/EtOAc (1:1). Выход 1,04 г (90%).
1H ЯМР (300 МГц; CDCl3) δ 9.16 (s, 2H), 4.80 (d, 2H), 4.73 (m, 1H), 4.26 (m, 2H), 4.0-3.8 (m, 2H), 2.6-2.4 (m, 2H), 1.47 (s, 9H), 1.12 (m, 2H), 0.07 (s, 9H).
(7) Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-[(5-(амино(триметилсилилэтилимино)метил))-2-пиримидинил]
Вос-Aze-NH-СН2-[(5-(амино(триметилсилилэтилимино)метил))-2-пиримидинил] (0,209 г; 0,437 ммоль; смотри стадию (6) выше) растворяли в 25 мл EtOAc, насыщенного HCl (г), и оставляли взаимодействовать в течение 15 мин. Растворитель выпаривали и остаток растворяли в 4 мл DMF. Добавляли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,100 г; 0,396 ммоль; смотри пример 1(8) выше), РуВОР (0,231 г; 0,444 ммоль) и диизопропилэтиламин (0,208 г; 1,61 ммоль) и смесь перемешивали в течение 80 мин. Реакционную смесь выливали в 100 мл воды и трижды экстрагировали EtOAc. Объединенный органический слой промывали соляным раствором, высушивали (Na2SO4) и упаривали. Неочищенный продукт очищали препаративной ОФЖХ с использованием смеси СН3CN/0,1 М NH4OAc (1:1). Выход 63 мг (26%).
1H ЯМР (400 МГц; CDCl3; смесь ротамеров) δ 9.3 (уширенный, 1H), 9.03 (s, 2H, минорный ротамер), 9.00 (s, 2H, основной ротамер), 8.25 (m, 1H, основной ротамер), 7.9 (уширенный, 1H), 7.80 (m, 1H, минорный ротамер), 7.2-6.9 (m, 3Н), 6.50 (t, 1H), 5.14 (s, 1H, минорный ротамер), 5.08 (m, 1H, минорный ротамер), 4.94 (s, 1H, основной ротамер), 4.80 (m, 1H, основной ротамер), 4.7-4.4 (m, 2H), 4.3-3.9 (m, 3H), 3.74 (m, 1H, основной ротамер), 2.7-2.1 (m, 2H), 1.03 (m, 2H), 0.01 (s, 9H).
(8) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-NH-СН2-[(5-амидино)-2-пиримидинил)×TFA
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-[(5-(амино(триметилсилилэтилимино)метил))-2-пиримидинил] (21 мг; 0,034 ммоль; смотри стадию (7) выше) растворяли в 0,5 мл метиленхлорида и охлаждали в ледяной бане. Добавляли TFA (2 мл), смесь перемешивали в течение 60 мин и затем концентрировали. Продукт высушивали вымораживанием из воды и ацетонитрила. Выход 20 мг (100%). Чистота 100%.
1H ЯМР (400 МГц; CD3OD; смесь ротамерных сигналов, перекрываемых сигналом от HDO) δ 9.08 (s, 2H), 7.4-7.1 (m, 3H), 6.88 (t, 1H, основной ротамер), 6.85 (t, 1H, минорный ротамер), 5.30 (m, 1H, минорный ротамер), 5.22 (s, 1H, минорный ротамер), 5.20 (s, 1H, основной ротамер), 4.73 (m, 1H, основной ротамер), 4.34 (m, 1H, ротамер), 4.21 (m, 1H, ротамер), 4.15-3.95 (m, 2H, ротамеры), 2.73 (m, 1H, ротамер), 2.57 (m, 1H, ротамер), 2.45-2.25 (m, 2H, ротамеры).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 173.0, 172.6, 172.1, 171.0, 163.4.
APCl-MS: (M+1)=469/471 m/z.
Пример 37
Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-NH-СН2-((5-метоксиамидино)-2-пиримидинил)
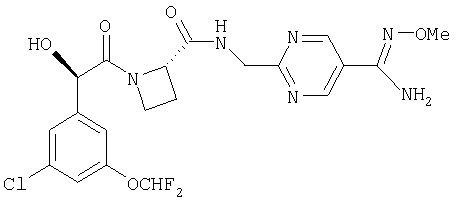
(1) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-NH-СН2-[(5-метоксиамино(триметилсилилэтилимино)метил))-2-пиримидинил]
Суспензию Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-[(5-амино(триметилсилилэтилимино)метил))-2-пиримидинила] (40 мг; 0,065 ммоль; смотри пример 36(7) выше) и O-метилгидроксиламина (33 мг; 0,40 ммоль) в 3 мл ацетонитрила нагревали при 70°С в течение 3 ч. Смесь распределяли между водой и EtOAc. Водный слой дважды экстрагировали EtOAc, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Выход 33 мг (79%).
1H ЯМР (400 МГц; CDCl3; смесь ротамеров) δ 8.76 (s, 2H, основной ротамер), 8.70 (s, 2H, ротамер), 8.18 (m, 1Н), 7.62 (s, 1H), 7.4-6.9 (m, 4H), 6.50 (bt, 1H), 5.3-4.5 (m, 4H), 4.2-4.05 (m, 3Н), 3.96 (s, 3Н), 3.68 (m, 1H), 2.8-2.2 (m, 2H), 2.1 (уширенный, 1H), 0.96 (m, 2H), 0,01 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-NH-СН2-((5-метоксиамидино)-2-пиримидинил)
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-NH-СН2-[(5-метоксиамино(триметилсилилэтилимино)метил))-2-пиримидинил] (33 мг; 0,052 ммоль; смотри стадию (1) выше) растворяли в 0,5 мл метиленхлорида и охлаждали в ледяной бане. Добавляли TFA (2 мл), смесь перемешивали в течение 2 ч и затем концентрировали. Продукт высушивали вымораживанием из воды и ацетонитрила. Выход 31 мг(81%). Чистота 100%.
1H ЯМР (400 МГц; CD3OD; смесь ротамеров, сигналы перекрываются сигналом от HDO) δ 8.96 (s, 2H, ротамер), 8.94 (s, 2H, ротамер), 7.4-7.3 (m, 1H), 7.2-7.1 (m, 2H), 6.88 (t, 1H, ротамер), 6.85 (t, 1H, ротамер), 5.29 (m, 1H, ротамер), 5.24 (s, 1H, ротамер), 5.20 (s, 1H, ротамер), 4.75-4.55 (m, 2H), 4.33 (m, 1H, ротамер), 4.19 (m, 1H, ротамер), 4.15-3.95 (m, 2H, ротамеры), 3.88 (s, 3Н, ротамер), 3.86 (s, 3Н, ротамер), 2.72 (m, 1H, ротамер), 2.56 (m, 1H, ротамер), 2.45-2.25 (m, 2H, ротамеры).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 172.8, 172.6, 172.1, 171.8, 167.8, 167.7, 155.1, 152.3, 152.1.
APCl-MS: (M+1)=499/501 m/z.
Пример 38
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(3-F)
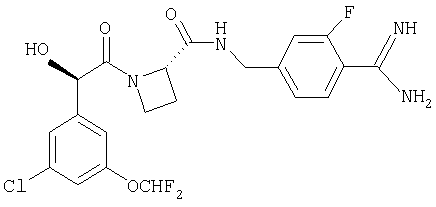
(1) 2-Фтор-4-винилбензнитрил
Раствор 4-бром-2-фторбензнитрила (4,92 г; 0,0246 моль), винилтрибутилолова (0,78 г; 0,246 моль) и тетракистрифенилфосфина (0,67 г; 0,58 ммоль) в 250 мл толуола кипятили с обратным холодильником в атмосфере азота в течение ночи. Растворитель выпаривали и остаток подвергали флэш-хроматографии на силикагеле в системе растворителей от смеси гептан/СН2Cl2 (1:1) до чистого CH2Cl2. Получали бесцветное масло, которое кристаллизуется. Выход 3,0 г (82%).
1H ЯМР (300 МГц; CDCl3) δ 7.56 (m, 1H), 7.3-7.2 (m, 2H), 6.69 (m, 1H), 5.89 (d, 1H), 5.51 (d, 1H).
(2) 2-Фтор-4-гидроксиметилбензнитрил
Охлажденный раствор (-78°С) 2-фтор-4-винилбензнитрила (1,3 г; 8,8 ммоль; смотри стадию (1) выше) в 40 мл CH2Cl2 и 5 мл метанола барботировали озоном (50 л/ч; 29 г/м3) в течение 30 мин. Для удаления избытка озона вслед за этим смесь барботировали аргоном. Добавляли борогидрид натрия (0,67 г; 0,018 моль) и охлаждающую баню убирали. Смесь перемешивали и реакции позволяли протекать в течение 1 ч. Смесь упаривали и добавляли 2 М HCl. Смесь дважды экстрагировали диэтиловым эфиром и объединенную эфирную фракцию высушивали (Na2SO4) и упаривали. Неочищенный продукт кристаллизовали. Выход 1,1 г (81%).
1H ЯМР (300 МГц; CDCl3) δ 7.59 (m, 1H), 7.3-7.2 (m, 2H), 4.79 (d, 2H), 2.26 (t, 1H).
(3) 4-Циано-3-фторбензилметансульфонат
2-Фтор-4-гидроксиметилбензнитрил (1,3 г; 8,6 ммоль; смотри стадию (2) выше) растворяли в 50 мл CH2Cl2 и охлаждали в ледяной бане. Добавляли триэтиламин (0,87 г; 8,6 ммоль) и метансульфонилхлорид (0,99 г; 8,7 ммоль). После перемешивания в течение 1,5 ч реакционную смесь промывали 1 М HCl. Органическую фазу высушивали (Na2SO4) и упаривали. Продукт можно было использовать без очистки. Выход бесцветного масла 1,8 г (92%).
1H ЯМР (400 МГц; CDCl3) δ 7.66 (m, 1Н), 7.35-7.3 (m, 2H), 5.26 (s, 2H), 3.07 (s, 3H).
(4) Азидометил-2-фторбензнитрил
К охлажденному во льду раствору 4-циано-3-фторбензилметансульфоната (1,8 г; 7,9 ммоль; смотри стадию (3) выше) добавляли азид натрия (0,8 г; 0,012 моль). Смесь перемешивали в течение ночи и затем выливали в 200 мл воды и трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу пять раз промывали водой, высушивали (Na2SO4) и упаривали. Неочищенное бесцветное масло можно было использовать без дальнейшей очистки. Выход 1,2 г (87%).
1H ЯМР (300 МГц; CDCl3) δ 7.64 (m, 1H), 7.25-7.18 (m, 2H), 4.47 (s, 2H).
(5) 4-Аминометил-2-фторбензнитрил
К суспензии хлорида двухвалентного олова дигидрата (0,45 г; 2,4 ммоль) в 20 мл ацетонитрила при перемешивании добавляли тиофенол (1,07 г; 9,7 ммоль) и триэтиламин (0,726 г; 7,17 ммоль). После этого добавляли раствор 4-азидометил-2-фторбензнитрила (0,279 г; 1,58 ммоль; смотри стадию (4) выше) в нескольких миллилитрах ацетонитрила. Через 1,5 ч азид израсходовался, и растворитель выпаривали. Остаток растворяли в метиленхлориде и трижды промывали 2 М NaOH. Органическую фазу дважды экстрагировали 1 М HCl. Объединенную кислотную водную фазу промывали метиленхлоридом и затем подщелачивали 2 М NaOH и трижды экстрагировали метиленхлоридом. Органическую фазу высушивали (Na2SO4) и упаривали с получением 0,172 г (72%) желаемого указанного в подзаголовке соединения, которое можно было использовать без очистки.
1H ЯМР (400 МГц; CDCl3) δ 7.58 (m, 1H), 7.3-7.2 (m, 2H), 3.98 (s, 2H), 1.55-1.35 (уширенный, 2Н).
(6) Boc-Aze-NHCH2-Ph(3-F, 4-CN)
К охлажденному во льду раствору Boc-Aze-OH (0,194 г; 0,96 ммоль) в 5 мл DMF добавляли TBTU (0,50 г; 9,6 ммоль). Через 30 мин добавляли другой раствор, содержащий 4-аминометил-2-фторбензнитрил (0,17 г; 0,81 ммоль; смотри стадию (5) выше) и диизопропилэтиламин (0,326 г; 2,53 ммоль) в 7 мл DMF. Полученный раствор перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали и продукт очищали с помощью препаративной ОФЖХ, используя смесь СН3CN/0,1 М NH4OAc (50:50). Высушивание вымораживанием давало 0,237 г (74%) желаемого указанного в подзаголовке соединения.
1H ЯМР (300 МГц; CD3OD) δ 7.70 (m, 1H), 7.35-7.25 (m, 2H), 4.65-4.35 (m, 3Н), 4.0-3.85 (m, 2H), 2.51 (m, 1H), 2.19 (m, 1H), 1.40 (s, 9H).
(7) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-NHCH2-Ph(3-F, 4-CN)
Boc-Aze-NHCH2-Ph(3-F, 4-CN) (0,118 г; 0,354 ммоль; со стадии (6) выше) растворяют в 30 мл EtOAc, насыщенного HCl (г). Реакционную смесь перемешивали в течение 20 мин и упаривали. Полученный дигидрохлорид и HATU (0,152 г; 0,400 ммоль) растворяли в 5 мл DMF. Данный раствор добавляли к охлажденному во льду раствору Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)ОН (0,101 г; 0,400 ммоль; смотри пример 1(8) выше) в 5 мл DMF. Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Растворитель выпаривали и продукт очищали с помощью препаративной ОФЖХ в смеси СН3CN/0,1 М NH4OAc (50:50). Высушивание вымораживанием давало 0,130 г (77%) желаемого указанного в подзаголовке соединения.
1H ЯМР (500 МГц; CD3OD, смесь ротамеров) δ 7.7-7.6 (m, 1H), 7.35-7.1 (m, 5Н), 6.88 (t, 1H, ротамер), 6.86 (t, 1H, ротамер), 5.25-5.1 (m, 1H плюс минорный ротамер от следующего протона), 4.80 (m, 1H, основной ротамер), 4.6-4.4 (m, 2H), 4.36 (m, 1H, основной ротамер), 4.18 (m, 1H, основной ротамер), 4.07 (m, 1H, минорный ротамер), 3.98 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.53 (m, 1H, основной ротамер), 2.29 (m, 1H, основной ротамер), 2.16 (m, 1H, минорный ротамер).
(8) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(3-F)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-NHCH2-Ph(3-F, 4-CN) (0,130 г; 0,278 ммоль; смотри стадию (7) выше) растворяли в 80 мл этанола, насыщенного HCl (г). Смесь оставляли взаимодействовать при комнатной температуре в течение ночи. Растворитель выпаривали и остаток перерастворяли в 100 мл этанола, насыщенного NH3 (г). Реакцию оставляли медленно протекать при комнатной температуре в течение двух суток. Температуру повышали до 50°С и реакцию продолжали в течение следующих 3 суток. После расходования исходного материала растворитель выпаривали. Продукт очищали с помощью препаративной ОФЖХ и высушивание вымораживанием давало 17 мг (13%) указанного в заголовке соединения в виде его НОАс-соли.
1H ЯМР (600 МГц; CD3OD, смесь ротамеров) δ 7.65-7.6 (m, 1H), 7.4-7.3 (m, 3Н), 7.25-7.1 (m, 2H), 7.15-6.7 (m, 1H), 5.25-5.1 (m, 1H плюс минорный ротамер от следующего протона), 4.8 (m, 1H, основной ротамер, частично невидимый из-за CD3ОН), 4.6-3.95 (m, 4H), 2.69 (m, 1H, минорный ротамер), 2.56 (m, 1H, основной ротамер), 2.28 (m, 1H, основной ротамер), 2.14 (m, 1H, минорный ротамер), 1.90 (s, 3Н).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 180.6, 173.4, 173.1, 172.9, 164.5, 162.3, 159.8.
APCl-MS: (M+1)=485/487 m/z.
Пример 39
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)
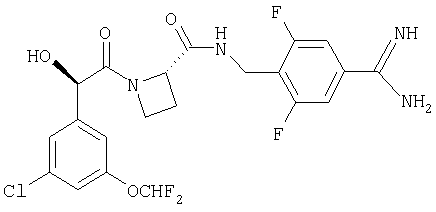
(1) 2,6-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензнитрил
(Метилсульфинил)(метилтио)метан (7,26 г; 0,0584 моль) растворяли в 100 мл безводного THF в атмосфере аргона и охлаждали до -78°С. По каплям с перемешиванием добавляли бутиллитий в гексане (16 мл; 1,6 М; 0,0256 моль). Смесь перемешивали в течение 15 мин. Тем временем раствор 3,4,5-трифторбензнитрила (4,0 г; 0,025 ммоль) в 100 мл безводного THF охлаждали до -78°С в атмосфере аргона и упомянутый первым раствор с помощью канюли в течение периода времени 35 мин добавляли в последний раствор. Через 30 мин охлаждающую баню убирали и, когда температура реакционной смеси достигала комнатной, реакционную смесь выливали в 400 мл воды. THF выпаривали и оставшийся водный слой трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу промывали водой, высушивали (Na2SO4) и упаривали. Выход 2,0 г (30%).
1H ЯМР (500 МГц; CDCl3) δ 7.4-7.25 (m, 2H), 5.01 (s, 1H, диастереомер), 4.91 (s, 1H, диастереомер), 2.88 (s, 3Н, диастереомер), 2.52 (s, 3Н, диастереомер), 2.49 (s, 3Н, диастереомер), 2.34 (s, 3Н, диастереомер), 1.72 (уширенный, 1H).
(2) 2,6-Дифтор-4-формилбензнитрил
2,6-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензнитрил (2,17 г; 8,32 ммоль; смотри стадию (1) выше) растворяли в 90 мл THF и добавляли 3,5 мл концентрированной серной кислоты. Смесь оставляли при комнатной температуре на 3 суток и затем выливали в 450 мл воды. Далее следовала трехкратная экстракция с помощью EtOAc, и объединенную эфирную фазу дважды промывали водным бикарбонатом натрия и соляным раствором, высушивали (Na2SO4) и упаривали. Выход 1,36 г (98%). Положение формильной группы устанавливали с помощью 13С ЯМР. Сигнал от фторированных углеродов при 162,7 млн-1 демонстрировал ожидаемый раздвоенный вид с двумя постоянными связи порядка 260 и 6,3 Гц соответственно, относящимся к ipso- и мета-связанным атомам фтора.
1H ЯМР (400 МГц; CDCl3) δ 10.35 (s, 1H), 7.33 (m, 2H).
(3) 2,6-Дифтор-4-гидроксиметилбензнитрил
2,6-Дифтор-4-формилбензнитрил (1,36 г; 8,13 ммоль; смотри стадию (2) выше) растворяли в 25 мл метанола и охлаждали в ледяной бане. Порциями с перемешиванием добавляли борогидрид натрия (0,307 г; 8,12 ммоль) и реакционную смесь оставляли на 65 мин. Растворитель выпаривали и остаток распределяли между диэтиловым эфиром и водным бикарбонатом натрия. Эфирный слой промывали дополнительным количеством водного бикарбоната натрия и соляным раствором, высушивали (Na2SO4) и упаривали.
Неочищенный продукт быстро кристаллизовался и мог быть использован без дальнейшей очистки. Выход 1,24 г (90%).
1H ЯМР (400 МГц; CDCl3) δ 7.24 (m, 2H), 4.81 (s, 2H), 2.10 (уширенный, 1Н).
(4) 4-Циано-2,6-дифторбензилметансульфонат
К охлажденному во льду раствору 2,6-дифтор-4-гидроксиметилбензнитрила (1,24 г; 7,32 ммоль; смотри стадию (3) выше) и метансульфонилхлорида (0,93 г; 8,1 ммоль) в 60 мл метиленхлорида с перемешиванием добавляли триэтиламин (0,81 г; 8,1 ммоль). Через 3 ч при 0°С смесь дважды промывали 1 М HCl и один раз водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 1,61 г (89%).
1H ЯМР (300 МГц; CDCl3) δ 7.29 (m, 2H), 5.33 (s, 2H), 3.07 (s, 3Н).
(5) 4-Азидометил-2,6-дифторбензнитрил
Смесь 4-циано-2,6-дифторбензилметансульфоната (1,61 г; 6,51 ммоль; смотри стадию (4) выше) и азида натрия (0,72 г; 0,0111 моль) в 10 мл воды и 20 мл DMF перемешивали при комнатной температуре в течение ночи. Полученный материал далее выливали в 200 мл воды и трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу пять раз промывали водой, высушивали (Na2SO4) и упаривали. Небольшой образец упаривали для нужд ЯМР и продукт кристаллизовали. Остаток аккуратно упаривали, но не полностью досуха. Выход (теоретически 1,26 г) считался полностью количественным на основании данных ЯМР и аналитической ВЭЖХ.
1H ЯМР (400 МГц; CDCl3) δ 7.29 (m, 2H), 4.46 (s, 2H).
(6) 4-Аминометил-2,6-дифторбензнитрил
Это взаимодействие осуществляли в соответствии с методикой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 520 мг 10%-ного Pd/C (50%-ной влажности) в 20 мл воды добавляли раствор борогидрида натрия (0,834 г; 0,0221 моль) в 20 мл воды. В результате выделялся газ. 4-Азидометил-2,6-дифторбензнитрил (1,26 г; 6,49 ммоль; смотри стадию (5) выше) растворяли в 50 мл THF и добавляли к водной смеси в ледяной бане в течение 15 мин. Смесь перемешивали в течение 4 ч, после чего в нее добавляли 20 мл 2 М HCl и смесь фильтровали через Целит. Целит промывали дополнительным количеством воды, объединенную водную фазу промывали EtOAc, затем подщелачивали 2 М NaOH. Далее следовала трехкратная экстракция метиленхлоридом и объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Выход 0,87 г (80%).
1H ЯМР (400 МГц; CDCl3) δ 7.20 (m, 2H), 3.96 (s, 2H), 1.51 (уширенный, 2Н).
(7) 2,6-Дифтор-4-трет-бутоксикарбониламинометилбензнитрил
Раствор 4-аминометил-2,6-дифторбензнитрила (0,876 г; 5,21 ммоль; смотри стадию (6) выше) растворяли в 50 мл THF и добавляли ди-трет-бутилдикарбонат (1,14 г; 5,22 ммоль) в 10 мл THF. Смесь перемешивали в течение 3,5 ч. THF выпаривали и остаток распределяли между водой и EtOAc. Органический слой трижды промывали 0,5 М HCl и водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 1,38 г (99%).
1H ЯМР (300 МГц; CDCl3) δ 7.21 (m, 2H), 4.95 (уширенный, 1Н), 4.43 (уширенный, 2H), 1.52 (s, 9H).
(8) Вос-Pab(2,6-диF)(OH)
Смесь 2,6-дифтор-4-трет-бутоксикарбониламинометилбензнитрила (1,38 г; 5,16 ммоль; смотри стадию (7) выше), гидроксиламина гидрохлорида (1,08 г; 0,0155 моль) и триэтиламина (1,57 г; 0,0155 моль) в 20 мл этанола перемешивали при комнатной температуре в течение 36 ч. Растворитель выпаривали и остаток распределяли между водой и метиленхлоридом. Органический слой промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 1,43 г (92%).
1H ЯМР (500 МГц; CD3OD) δ 7.14 (m, 2H), 4.97 (уширенный, 1Н), 4.84 (уширенный, 2H), 4.40 (уширенный, 2H), 1.43 (s, 9H).
(9) Вос-Pab(2,6-диF)×НОАс
Это взаимодействие осуществляли в соответствии с методикой, описанной Judkins et al., Synth. Comm. (1998) 4351. Вос-Pab(2,6-диF)(ОН) (1,32 г; 4,37 ммоль; смотри стадию (8) выше), уксусный ангидрид (0,477 г; 4,68 ммоль) и 442 мг 10%-ного Pd/C (50%-ной влажности) в 100 мл уксусной кислоты гидрировали при 490 кПа (5 атм) в течение 3,5 ч. Смесь фильтровали через Целит, промывали этанолом и упаривали. Остаток высушивали вымораживанием из ацетонитрила, воды и нескольких капель этанола. Указанный в подзаголовке продукт мог быть использован без дальнейшей очистки. Выход 0,149 г (99%).
1H ЯМР (400 МГц; CD3OD) δ 7.45 (m, 2Н), 4.34 (s, 2H), 1.90 (s, 3Н), 1.40 (s, 9H).
(10) Вос-Pab(2,6-диF)(Teoc)
К раствору Вос-Pab(2,6-диF)×НОАс (1,56 г; 5,49 ммоль; смотри стадию (9) выше) в 100 мл THF и 1 мл воды добавляли 2-(триметилсилил)этил-п-нитрофенилкарбонат (1,67 г; 5,89 ммоль). В течение 5 мин по каплям добавляли раствор карбоната калия (1,57 г; 0,0114 моль) в 20 мл воды. Смесь перемешивали в течение ночи. THF выпаривали и остаток распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом, объединенную органическую фазу дважды промывали водным бикарбонатом натрия, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в смеси гептан/EtOAc (2:1) дает 1,71 г (73%) чистого соединения.
1H ЯМР (400 МГц; COCl3) δ 7.43 (m, 2H), 4.97 (уширенный, 1Н), 4.41 (уширенный, 2H), 4.24 (m, 2H), 1.41 (s, 9H), 1.11 (m, 2H), 0,06 (s, 9H).
(11) Вос-Aze-Pab(2,6-диF)(Теос)
Вос-Pab(2,6-диF)(Теос) (1,009 г; 2,35 ммоль; смотри стадию (10) выше) растворяли в 50 мл EtOAc, насыщенного HCl (г). Смесь оставляли на 10 мин, упаривали, растворяли в 18 мл DMF и затем охлаждали в ледяной бане. Добавляли Boc-Aze-OH (0,450 г; 2,24 ммоль), РуВОР (1,24 г; 2,35 ммоль) и, наконец, диизопропилэтиламин (1,158 г; 8,96 ммоль). Реакционную смесь перемешивали в течение 2 ч, затем выливали в 350 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали соляным раствором, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в смеси гептан/EtOAc (1:3) дала 1,097 г (96%) желаемого соединения.
1H ЯМР (500 МГц; CDCl3) δ 7.46 (m, 2H), 4.65-4.5 (m, 3Н), 4.23 (m, 2H), 3.87 (m, 1H), 3.74 (m, 1H), 2.45-2.3 (m, 2H), 1.40 (s, 9H), 1.10 (m, 2H), 0,05 (s, 9H).
(12) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc)
Вос-Aze-Pab(2,6-диF)(Теос) (0,256 г; 0,500 ммоль; смотри стадию (11) выше) растворяли в 20 мл EtOAc, насыщенного HCl (г). Смесь оставляли на 10 мин, упаривали и растворяли в 5 мл DMF. Добавляли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,120 г; 0,475 ммоль; смотри пример 1(8) выше), РуВОР (0,263 г; 0,498 ммоль) и, наконец, диизопропилэтиламин (0,245 г; 1,89 ммоль). Реакционную смесь перемешивали в течение 2 ч, затем выливали в 350 мл воды и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали соляным раствором, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в EtOAc дала 0,184 г (60%) желаемого указанного в подзаголовке соединения.
1H ЯМР (400 МГц; CD3OD, смесь ротамеров) δ 7.55-7.45 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.27 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1Н, основной ротамер), 6.86 (t, 1H, минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.12 (m, 1H, минорный ротамер), 5.06 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.24 (m, 2H), 4.13 (m, 1H, основной ротамер), 4.04 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.22 (m, 1H, основной ротамер), 2.10 (m, 1H, минорный ротамер), 1.07 (m, 2H), 0,07 (m, 9H).
(13) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(2,6-диF)
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,6-диF)(Теос) (81 мг; 0,127 ммоль; смотри стадию (12) выше) растворяли в 0,5 мл метиленхлорида и охлаждали в ледяной бане. Добавляли TFA (3 мл) и реакционную смесь оставляли на 75 мин. TFA выпаривали и остаток высушивали вымораживанием из воды и ацетонитрила. Неочищенный продукт очищали препаративной ОФЖХ в смеси СН3CN/0,1 М NH4OAC (35:65), получая 39 мг (55%) указанного в заголовке соединения в виде его НОАс-соли, чистота 99%.
1H ЯМР (400 МГц; CD3OD, смесь ротамеров) δ 7.5-7.4 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.28 (m, 1H, минорный ротамер), 7.2-7.1 (m, 3Н), 6.90 (t, 1H, основной ротамер), 6.86 (t, минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.14 (m, 1H, минорный ротамер), 5.07 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.65-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.16 (m, 1H, основной ротамер), 4.03 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.63 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.21 (m, 1H, основной ротамер), 2.07 (m, 1H, минорный ротамер), 1.89 (s, 3Н).
13С-ЯМР (75 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 171.9, 171.2, 165.0, 162.8, 160.4.
APCl-MS: (M+1)=503/505 m/z.
Пример 40
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe)

(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)С(O)-Aze-Pab(2,6-диF)(Оме, Теос)
Смесь Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,6-диF)(Теос) (64 мг; 0,099 ммоль; смотри пример 39(12) выше) и O-метилгидроксиламина гидрохлорида (50 мг; 0,60 ммоль) в 4 мл ацетонитрила нагревали при 70°С в течение 3 ч. Растворитель выпаривали и остаток распределяли между водой и EtOAc. Водный слой дважды экстрагировали EtOAc, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 58 мг (87%).
1H ЯМР (400 МГц; CDCl3) δ 7.90 (bt, 1H), 7.46 (m, 1H), 7.25-6.95 (m, 5H), 6.51 (t, 1H), 4.88 (s, 1H), 4.83 (m, 1H), 4.6-4.5 (m, 2H), 4.4-3.9 (m, 4H), 3.95 (s, 3H), 3.63 (m, 1H), 2.67 (m, 1H), 2.38 (m, 1H), 1.87 (уширенный, 1H), 0,98 (m, 2H), 0.01 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe)
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,6-диF)(ОМе, Теос) (58 мг; 0,086 ммоль; смотри стадию (1) выше) растворяли в 3 мл TFA, охлаждали в ледяной бане и оставляли взаимодействовать на 2 ч. TFA выпаривали и остаток растворяли в EtOAc. Органический слой дважды промывали водным карбонатом натрия и водой, высушивали (Na2SO4) и упаривали. Остаток высушивали вымораживанием из воды и ацетонитрила с получением 42 мг (92%) указанного в заголовке соединения. Чистота 94%.
1H ЯМР (300 МГц; CDCl3) δ 7.95 (bt, 1H), 7.2-7.1 (m, 4H), 6.99 (m, 1H), 6.52 (t, 1H), 4.88 (s, 1H), 4.85-4.75 (m, ЗН), 4.6-4.45 (m, 2H), 4.29 (уширенный, 1H), 4.09 (m, 1H), 3.89 (s, ЗН), 3.69 (m, 1H), 2.64 (m, 1H), 2.38 (m, 1H), 1.85 (уширенный, 1H).
13С-ЯМР (100 МГц; CDCl3) (карбонильные и/или амидинные углероды) δ 172.1, 169.8, 151.9.
APCl-MS: (M+1)=533/535 m/z.
Пример 41
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)
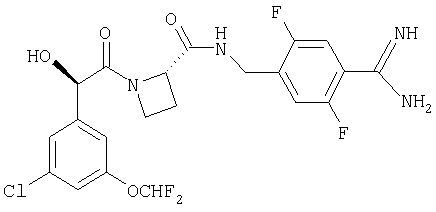
(1) 2,5-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензнитрил
(Метилсульфинил)(метилтио)метан (3,16 г; 0,0255 моль) растворяли в 50 мл безводного THF в атмосфере аргона и затем охлаждали до -78°С. По каплям с помощью шприца добавляли бутиллитий в гексане (16 мл; 1,6 М; 0,0256 моль). Смесь перемешивали в течение 15 мин. Тем временем раствор 2,4,5-трифторбензнитрила (2,0 г; 0,013 моль) в 50 мл безводного THF охлаждали до -78°С в атмосфере аргона и упомянутый первым раствор с помощью канюли в течение периода времени 3-5 мин добавляли в последний раствор. Через 30 мин охлаждающую баню убирали и, когда температура реакционной смеси достигала комнатной, реакционную смесь выливали в 200 мл воды. THF выпаривали и оставшийся водный слой трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт начинал кристаллизоваться и мог быть использован в том виде, в каком он есть, на следующей стадии. Выход 2,8 г (84%).
1H ЯМР (500 МГц; CDCl3) δ 7.51-7.44 (m, 2H, основной диастереомер), 7.39 (dd, 1H, минорный диастереомер), 5.00 (s, 1H, минорный диастереомер), 4.92 (s, 1H, основной диастереомер), 2.59 (s, ЗН, минорный диастереомер), 2.56 (s, 1H, основной диастереомер), 2.46 (s, 1H, минорный диастереомер), 2.40 (s, 1H, основной диастереомер).
(2) 2,5-Дифтор-4-формилбензнитрил
2,5-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензнитрил (2,8 г; 0,0107 моль; смотри стадию (1) выше) растворяли в 100 мл THF и добавляли 6,5 г концентрированной серной кислоты. Смесь оставляли при комнатной температуре на 6 суток и затем выливали в 500 мл воды. Далее следовала трехкратная экстракция диэтиловым эфиром, объединенную эфирную фазу несколько раз промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле, используя смесь гептан/EtOAc (8:2). Выход 1,2 г (67%). Положение формильной группы устанавливали с помощью 13С ЯМР. Углеродные сигналы от фторированных углеродов при 160.1 и 158.4 соответственно являлись дублетами, а не квартетами, которыми они были бы, если бы формильная группа находилась в положении 2.
1H ЯМР (300 МГц; CDCl3) δ 10.36 (d, 1H), 7.72 (dd, 1H), 7.54 (dd, 1H).
(3) 2,5-Дифтор-4-гидроксиметилбензнитрил
2,5-Дифтор-4-формилбензнитрил (3,60 г; 0,0215 моль; смотри стадию (2) выше) растворяли в 50 мл метанола и охлаждали в ледяной бане. Порциями с перемешиванием добавляли борогидрид натрия (0,815 г; 0,0215 моль) и реакционную смесь оставляли на 45 мин. Добавляли воду (300 мл) и после этого осторожно добавляли 2 М HCl до достижения кислого значения рН. Смесь трижды экстрагировали диэтиловым эфиром, объединенную эфирную фазу промывали водой, высушивали (Na2SO4) и упаривали. Неочищенный продукт быстро кристаллизовался и мог быть использован без дальнейшей очистки. Выход 3,1 г (85%).
1H ЯМР (300 МГц; CDCl3) δ 7.45 (dd, 1H), 7.30 (dd, 1H), 4.85 (s, 2H), 2.10 (уширенный, 1H).
(4) 4-Циано-2,5-дифторбензилметансульфонат
К охлажденному во льду раствору 2,5-дифтор-4-гидроксиметилбензнитрила (3,10 г; 0,0183 моль; смотри стадию (3) выше) и метансульфонилхлорида (2,21 г; 0,0192 моль) в 60 мл метиленхлорида с перемешиванием добавляли триэтиламин (1,95 г; 0,0192 моль). Через 1,5 ч при 0°С смесь промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 4,5 г (99%).
1Н ЯМР (300 МГц; CDCl3) δ 7.45-7.35 (m, 2H), 5.32 (s, 2H), 3.13 (s, 3H).
(5) 4-Азидометил-2,5-дифторбензнитрил
Смесь 4-циано-2,5-дифторбензилметансульфоната (4,5 г; 0,0182 моль; смотри стадию (4) выше) и азида натрия (2,0 г; 0,031 моль) в 20 мл воды и 40 мл DMF перемешивали при комнатной температуре в течение 2 ч. Далее ее выливали в 300 мл воды и трижды экстрагировали диэтиловым эфиром. Объединенную эфирную фазу несколько раз промывали водой, высушивали (Na2SO4) и упаривали. Небольшой образец упаривали для нужд ЯМР и продукт кристаллизовали. Остаток осторожно упаривали, но не полностью досуха. Выход (теоретически 3,5 г) считался полностью количественным на основании данных ЯМР и аналитической ВЭЖХ.
1H ЯМР (500 МГц; CDCl3) δ.38 (dd, 1H), 7.32 (dd, 1H), 4.54 (s, 2H).
(6) 4-Аминометил-2,5-дифторбензнитрил
Это взаимодействие осуществляли в соответствии с методикой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 300 мг 10%-ного Pd/C (50%-ной влажности) в 20 мл воды добавляли раствор борогидрида натрия (0,779 г; 0,0206 моль) в 20 мл воды. В результате выделялся газ. 4-Азидометил-2,5-дифторбензнитрил (1,00 г; 5,15 ммоль; со стадии (5) выше) растворяли 60 мл THF и добавляли к водной смеси в ледяной бане. Смесь перемешивали в течение 1,5 ч, после чего в нее добавляли 10 мл 2 М HCl и смесь фильтровали через Целит. Целит промывали дополнительным количеством воды, объединенную водную фазу промывали EtOAc, затем подщелачивали 2 М NaOH. Далее следовала трехкратная экстракция метиленхлоридом, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Выход 0,47 г (54%).
1Н ЯМР (300 МГц; CDCl3) δ 7.39 (dd, 1H), 7.29 (dd, 1H), 3.99 (s, 2H), 1.45 (уширенный, 2H).
(7) 2,5-Дифтор-4-трет-бутоксикарбониламинометилбензнитрил
Раствор 4-аминометил-2,5-дифторбензнитрила (0,46 г; 2,7 ммоль; смотри стадию (6) выше) и ди-трет-бутилдикарбоната (0,60 г; 2,7 ммоль) в 10 мл THF перемешивали в течение ночи. THF выпаривали и остаток распределяли между водой и EtOAc. Органический слой промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 0,71 г (97%).
1H ЯМР (300 МГц; CDCl3) δ 7.35-7.2 (m, 2H), 5.11 (уширенный триплет, 1H), 4.38 (d, 2H), 1.45 (s, 9H).
(8) Вос-Pab(2,5-диF)(ОН)
Смесь 2,5-дифтор-4-трет-бутоксикарбониламинометилбензнитрила (0,70 г; 2,6 ммоль; смотри стадию (7) выше), гидроксиламина гидрохлорида (0,54 г; 7,8 ммоль) и триэтиламина (0,79 г; 7,8 ммоль) в 10 мл этанола перемешивали при комнатной температуре в течение 6 суток. Далее ее распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 0,72 г (92%).
1H ЯМР (500 МГц; CD3OD) δ 7.27 (dd, 1H), 7.12 (dd, 1H), 4.29 (s, 2H), 1.47 (s, 9H).
(9) Вос-Pab(2,5-диF)×НОАс
Это взаимодействие осуществляли в соответствии с методикой, описанной Judkins et al., Synth. Comm. (1998) 4351. Вос-Pab(2,5-диF)(ОН) (0,70 г; 2,3 ммоль; смотри стадию (8) выше), уксусный ангидрид (0,25 г; 2,4 ммоль) и 230 мг 10%-ного Pd/C (50%-ной влажности) в 70 мл уксусной кислоты гидрировали при 490 кПа (5 атм) в течение 2,5 ч. Смесь фильтровали через Целит и упаривали. Остаток высушивали вымораживанием из ацетонитрила и воды. Продукт мог быть использован без дальнейшей очистки на следующей стадии. Выход 0,80 г (100%).
1H ЯМР (500 МГц; CD3OD) δ 7.49 (dd, 1H), 7.31 (dd, 1H), 4.33 (s, 2H), 1.91 (s, 3H), 1.46 (s, 9H).
(10) Boc-Pab(2,5-диF)(Teoc)
К суспензии Вос-Pab(2,5-диF)×НОАс (0,80 г; 2,3 ммоль; смотри стадию (9) выше) в 50 мл THF добавляли 2-(триметилсилил)этил-п-нитрофенилкарбонат (0,85 г; 3,0 ммоль). По каплям добавляли раствор карбоната калия (0,80 г; 5,8 ммоль) в 10 мл воды. Смесь перемешивали в течение ночи. Избыток Теос-реагента разрушали, добавляя к раствору глицин (0,100 г) и карбонат калия (0,75 г) и оставляя его взаимодействовать в течение дополнительных 2 ч. THF выпаривали и остаток распределяли между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в смеси гептан/EtOAc (2:1) дает 0,72 г (72%) чистого соединения.
1H ЯМР (400 МГц; CDCl3) δ 8.00 (dd, 1H), 7.15 (dd, 1H), 4.98 (уширенный, 1Н), 4.36 (bd, 2Н), 4.24 (m, 2H), 1.45 (s, 9Н), 1.12 (m, 2H), 0,07 (s, 9H).
(11) Н-Pab(2,5-диF)(Teoc)×2HCl
Вос-Pab(2,5-диF)(Теос) (0,38 г; 0,88 ммоль; смотри стадию (10) выше) растворяли в 50 мл EtOAc, насыщенного HCl (г). Смесь оставляли на 30 мин и упаривали.
1H ЯМР (500 МГц; CD3OD) δ 7.75-7.6 (m, 2H), 4.46 (m, 2H), 4.3 (s, 2H), 1.15 (m, 2H), 0,07 (s, 9H).
(12) Вос-Aze-Pab(2,5-диF)(Теос)
К перемешиваемому раствору Boc-Aze-OH (0,189 г; 0,94 ммоль), Н-Pab(2,5-диF)(Теос)×2HCl (0,36 г; 0,89 ммоль; смотри стадию (11) выше) и РуВОР (0,54 г; 1,03 ммоль) в 5 мл DMF добавляли диизопропилэтиламин (0,49 г; 3,8 ммоль) и смесь оставляли взаимодействовать в течение ночи. Полученный продукт выливали в водный бикарбонат натрия и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в смеси гептан/EtOAc (3:7) дала достаточно чистое соединение. Выход 0,25 г (48%).
1H ЯМР (500 МГц; CDCl3) δ 7.98 (dd, 1H), 7.13 (dd, 1H), 4.69 (m, 1H), 4.53 (m, 2H), 4.22 (m, 2H), 3.92 (m, 1H), 3.79 (m, 1H), 2.55-2.35 (m, 2H), 1.44 (s, 9H), 1.11 (m, 2H), 0.06 (s, 9H).
(13) Н-Aze-Pab(2,5-диF)(Teoc)×2HCl
Вос-Aze-Pab(2,5-диF)(Теос) (0,25 г; 0,49 ммоль; смотри стадию (12) выше) растворяли в 50 мл EtOAc, насыщенного HCl (г). Смесь оставляли на 30 мин и упаривали. Продукт использовали на следующей стадии без дальнейшей очистки. Выход 0,23 г (97%).
1H ЯМР (400 МГц; CD3OD) δ 7.59 (dd, 1H), 7.47 (dd, 1H), 5.14 (m, 1H), 4.54 (m, 2Н), 4.48 (m, 2Н), 4.15 (m, 1H), 3.96 (m, 1H), 2.87 (m, 1H), 2.56 (m, 1H), 1.17 (m, 2H), 0.05 (s, 9H).
(14) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)(Teoc)
К раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,12 г; 0,47 ммоль; смотри пример 1(8) выше), Н-Aze-Pab(2,5-диF)(Теос)×2HCl (0,23 г; 0,47 ммоль; смотри стадию (13) выше) и РуВОР (0,27 г; 0,52 ммоль) в 10 мл DMF добавляли диизопропилэтиламин (0,245 г; 1,90 ммоль) и смесь перемешивали в течение ночи. Полученный продукт выливали в воду и трижды экстрагировали EtOAc. Объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Флэш-хроматография на силикагеле в EtOAc дала 100 мг чистой фракции и 30 мг фракции с 90%-ной чистотой. Общий выход 0,13 г (41%).
1H ЯМР (400 МГц; CDCl3) δ 9.80 (уширенный, 1H), 8.05 (bt, 1H), 7.94 (dd, 1H), 7.20 (m, 1H), 7.2-7.1 (m, 2H), 7.02 (m, 1H), 6.54 (t, 1H), 4.93 (s, 1H), 4.91 (m, 1H), 4.51 (m, 2H), 4.28 (уширенный, 1H), 4.23 (m, 2H), 4.13 (m, 1H), 3.74 (m, 1H), 2.69 (m, 1H),2.43 (m, 1H), 1.73 (уширенный, 1H), 1.11 (m, 2H), 1.11 (s, 9H).
(15) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5ди-F)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)(Teoc) (60 мг; 0,093 ммоль; чистая фракция со стадии (14) выше) растворяли в 3 мл TFA и оставляли на 1 ч при комнатной температуре. TFA выпаривали и остаток высушивали вымораживанием из воды и ацетонитрила, получая 55 мг (96%) указанного в заголовке соединения в виде его TFA-соли, чистота >99%.
1H ЯМР (500 МГц; CD3OD, смесь ротамеров) δ 7.55-7.3 (m, 3Н), 7.2-7.1 (m, 2H), 6.88 (t, 1H, основной ротамер), 6.86 (t, 1H, минорный ротамер), 5.22 (m, 1H, минорный ротамер), 5.20 (s, 1H, основной ротамер), 5.13 (s, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.36 (m, 1H, основной ротамер), 4.19 (m, 1H, основной ротамер), 4.07 (m, 1H, минорный ротамер), 3.98 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.54 (m, 1H, основной ротамер), 2.28 (m, 1H, основной ротамер), 2.14 (m, 1H, минорный ротамер).
13С-ЯМР (75 МГц; CD3OD) (карбонильные и/или амидинные углероды, смесь ротамеров) δ 173.0, 172.6, 172.1, 172.0, 162.4.
APCl-MS: (M+1)=503/505 m/z.
Пример 42
Ph(3-Ch(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,5-диF)(ОМе)
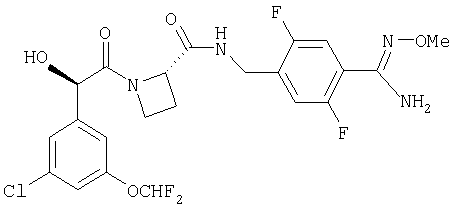
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)(OMe, Teoc)
Смесь Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc) (40 мг; 0,062 ммоль; смотри пример 41(14) выше) и О-метилгидроксиламина гидрохлорида (58 мг; 0,70 ммоль) в 5 мл ацетонитрила нагревали при 70°С в течение 2 ч. Растворитель выпаривали и остаток распределяли между водой и EtOAc. Водный слой экстрагировали EtOAc, объединенную органическую фазу промывали водой, высушивали (Na2SO4) и упаривали. Продукт мог быть использован без дальнейшей очистки. Выход 35 мг (84%).
1H ЯМР (600 МГц; CDCl3) δ 7.99 (bt, 1H), 7.72 (s, 1H), 7.20 (m, 1H), 7.15-7.1 (m, 1H), 7.07 (dd, 1H), 7.01 (m, 1H), 6.53 (t, 1H), 4.90 (s, 1H), 4.88 (m, 1H), 4.48 (m, 2Н), 4.2-4.1 (m, 3H), 3.95 (s, 3Н), 3.67 (m, 1H), 2.68 (m, 1H), 2.41 (m, 1H), 0,97 (m, 2H), 0.07 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)С(O)-Aze-Pab(2.5-диF(OMe)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,5-диF)(OMe, Teoc) (35 мг; 0,052 ммоль; смотри стадию (1) выше) растворяли в 3 мл TFA и оставляли взаимодействовать в продолжение 30 мин. TFA выпаривали и остаток высушивали вымораживанием из воды и ацетонитрила с получением 29 мг (99%) указанного в заголовке соединения. Чистота 97%.
1H ЯМР (300 МГц; CDCl3) δ 8.01 (bt, 1H), 7.45 (dd, 1H), 7.20 (m, 1H), 7.15 (m, 1H), 7.09 (dd, 1H), 7.02 (m, 1H), 6.54 (t, 1H), 5.2-5.0 (m, 2H), 4.95-4.85 (m, 2H), 4.6-4.4 (m, 2H), 4.25 (уширенный, 1H), 4.13 (m, 1H), 3.90 (s, 3H), 3.71 (m, 1H), 2.69 (m, 1H), 2.43 (m, 1H).
13С-ЯМР (75 МГц; CDCl3) (карбонильные и/или амидинные углероды) δ 173,0, 170.9, 152.6.
APCl-MS: (M+1)=533/535 m/z.
Пример 43
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OEt)
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Oet, Теос)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (55 мг; 0,090 ммоль; смотри пример 1(9) выше) и O-этилгидроксиламина гидрохлорид (53 мг; 0,54 ммоль) растворяли в 4 мл THF. Смесь перемешивали при 60°С в течение 5 ч. Растворитель выпаривали. Остаток хроматографировали на силикагеле, элюируя смесью метиленхлорид/метанол (95:5), что позволило получить 55 мг (93%) указанного в подзаголовке соединения.
1H-ЯМР (400 МГц; CDCl3): δ 7.84 (bt, 1H), 7.59 (bs, 1H), 7.47 (bd, 1H), 7.29 (bd, 1H), 7.21 (m, 1H), 7.14 (m, 1H), 7.02 (m, 1H), 6.53 (t, 1H), 4.90 (s, 1H), 4.86 (m, 1H), 4.55-4.4 (m, 2H), 4.25-4.1 (m, 5H), 3.69 (m, 1H), 2.66 (m, 1H), 2.41 (m, 1H), 1.33 (t, 3H), 0.98 (m, 2H), 0.02 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OEt)
К охлажденному во льду раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OEt, Teoc) (55 мг; 0,084 ммоль; смотри стадию (1) выше) в 0,5 мл метиленхлорида добавляли 3 мл TFA. Смесь перемешивали (ледяная баня) в течение 160 минут. Материал очищали, используя препаративную ВЭЖХ. Представляющие интерес фракции объединяли и высушивали вымораживанием (2х), собирая 20 мг (47%) указанного в заголовке соединения.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.59 (bd, 2H), 7.35 (m, 1H), 7.32 (bd, 2H), 7.25-7.1 (m, 2H), 6.89 (t, 1H, основной ротамер), 6.86 (t, 1H, минорный ротамер), 5.18 (s, 1H, основной ротамер), 5.18 (m, 1H, минорный ротамер), 5.11 (s, 1H, минорный ротамер), 4.77 (m, 1H), 4.5-4.3 (m, 3H), 4.2-3.9 (m, 3H), 2.67 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.28 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер), 1.28 (t, 3H).
13С-ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 172.4, 171.9, 171.4, 153.8, 152.3.
MS (m/z) 509 (М-1)-, 511 (М+1)+.
Пример 44
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-н-Pr)
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-н-Pr, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (53 мг; 0,087 ммоль; смотри пример 1(9) выше) и O-н-пропилгидроксиламина гидрохлорид (58 мг, 0,52 ммоль) растворяли в 4 мл THF. Смесь перемешивали при 60°С в течение 5 ч. Растворитель выпаривали. Остаток хроматографировали на силикагеле, элюируя смесью метиленхлорид/метанол (95:5), что позволило получить 51 мг (88%) указанного в подзаголовке соединения.
1H-ЯМР (400 МГц; CDCl3): δ 7.84 (m, 1H), 7.59 (bs, 1H), 7.47 (bd, 2H), 7.28 (bd, 2H), 7.21 (m, 1H), 7.14 (m, 1H), 7.02 (m, 1H), 6.53 (t, 1H), 4.90 (s, 1H), 4.85 (m, 1H), 4.55-4.4 (m, 2H), 4.2-4.05 (m, 5H), 3.69 (m, 1H), 2.65 (m, 1H), 2.41 (m, 1H), 1.74 (m, 2H), 1.05-0.95 (m, 5H), 0.03 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-н-Pr)
К охлажденному во льду раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-н-Pr, Teoc) (51 мг; 0,078 ммоль; смотри стадию (1) выше) в 0,5 мл метиленхлорида добавляли 3 мл TFA. Смесь перемешивали (ледяная баня) в течение 110 минут. Материал очищали, используя препаративную ВЭЖХ. Представляющую интерес фракцию упаривали и высушивали вымораживанием, собирая 20 мг (47%) указанного в заголовке соединения.
1H-ЯМР (500 МГц; CD3OD; ротамеры) δ 7.61 (bd, 2H), 7.38 (m, 1H), 7.35 (bd, 2H), 7.22 (m, 1H, основной ротамер), 7.18 (m, 1H), 7.15 (m, 1H, минорный ротамер), 6.92 (t, 1H, основной ротамер), 6.89 (t, 1H, минорный ротамер), 5.20 (s, 1H, основной ротамер), 5.20 (m, 1H, минорный ротамер), 4.80 (m, 1H, основной ротамер), 4.5-4.4 (m, 2H, включая минорный ротамер, соответствующий основному при 4.37), 4.37 (m, 1H, основной ротамер), 4.18 (m, 1H, основной ротамер), 4.09 (m, 1H, минорный ротамер), 3.99 (m, 2H), 2.70 (m, 1H, минорный ротамер), 2.54 (m, 1H, основной ротамер), 2.30 (m, 1H, основной ротамер), 2.18 (m, 1H, минорный ротамер), 1.73 (m, 2H), 1.01 (t, 3Н).
13С-ЯМР (125 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 171.4, 153.8, 152.3.
MS (m/z) 523 (М-1)-, 525 (М+1)+.
Пример 45
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-изо-Pr)
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)С(O)-Aze-Pab(O-изо-Pr, Teoc)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (50 мг; 0,082 ммоль; смотри пример 1(9) выше) и O-изо-пропилгидроксиламина гидрохлорид (55 мг; 0,49 ммоль) растворяли в 4 мл THF. Смесь перемешивали при 60°С в течение 5 ч. Растворитель выпаривали. Остаток хроматографировали на силикагеле с элюированием смесью метиленхлорид/метанол (95:5), что позволило получить 46 мг (84%) указанного в подзаголовке соединения.
1H-ЯМР (400 МГц; CDCl3): δ 7.84 (m, 1H), 7.57 (bs, 1H), 7.48 (bd, 2H), 7.29 (bd, 2H), 7.21 (m, 1H), 7.14 (m, 1H), 7.02 (m, 1H), 6.53 (t, 1H), 4.91 (s, 1H), 4.87 (m, 1H), 4.55-4.45 (m, 2H), 4.42 (m, 1H), 4.2-4.1 (m, 3H), 3.69 (m, 1H), 2.66 (m, 1H), 2.42 (m, 1H), 1.30 (d, 6H), 0.98 (m, 2H), 0.02 (s, 9H).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-изо-Pr)
К охлажденному во льду раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(O-изо-Pr, Teoc) (46 мг; 0,069 ммоль; смотри стадию (1) выше) в 0,5 мл метиленхлорида добавляли 3 мл TFA. Смесь перемешивали (ледяная баня) в течение 150 минут. Материал очищали, используя препаративную ВЭЖХ. Представляющую интерес фракцию упаривали и высушивали вымораживанием (2х) с получением 22 мг (58%) указанного в заголовке соединения.
1H-ЯМР (400 МГц; CD3OD; ротамеры): δ 7.59 (d, 2H), 7.35 (m, 1H), 7.32 (d, 2H), 7.19 (m, 1H, основной ротамер), 7.15 (m, 1H), 7.12 (m, 1H, минорный ротамер), 6.89 (t, 1H, основной ротамер), 6.86 (t, 1H, минорный ротамер), 5.18 (s, 1H, основной ротамер), 5.18 (m, 1H, минорный ротамер), 5.12 (s, 1H, минорный ротамер), 4.78 (m, 1H, основной ротамер), 4.5-3.9 (m, 5H), 2.67 (m, 1H, минорный ротамер), 2.52 (m, 1H, основной ротамер), 2.28 (m, 1H, основной ротамер), 2.15 (m, 1H, минорный ротамер), 1.26 (d, 6H).
13С ЯМР (100 МГц; CD3OD) (карбонильные и/или амидинные углероды, ротамеры) δ 171.9, 171.4, 153.6.
MS (m/z) 523 (M-1)-, 525 (M+1)+.
Пример 46
Указанные в заголовках соединения из примеров 3, 6, 9, 10, 13-15, 17, 19, 21, 23, 25, 27, 28, 32, 34, 36, 38, 39 и 41 были проверены в соответствии с вышеприведенным Тестом А и обнаружено, что для всех из них значения IC50ТТ меньше 3,5 мкМ. Обнаружено, что соединения из примеров 3, 6, 9, 10, 13,15, 17, 19, 21, 23, 27, 32, 34 и 39 показывают значения меньше 0,02 мкМ; соединения из примеров 25 и 28 - меньше 0,03 мкМ, соединение из примера 14 - меньше 0,04 мкМ и соединения из примеров 38 и 41 - меньше 0,15 мкМ.
Пример 47
Указанные в заголовках соединения из примеров 3, 6, 13, 15, 17, 19, 21, 23, 25, 27, 28, 32 и 34 проверены в соответствии с вышеприведенным Тестом D и обнаружено, что для всех из них значение IC50APTT меньше 1 мкМ.
Пример 48
Указанные в заголовках соединения из примеров 1, 2, 4, 5, 7, 12, 16, 18, 20, 22, 24, 26, 29, 30, 33 и 43-45 проверены в соответствии в вышеприведенным Тестом Е и обнаружено, что они проявляют пероральную и/или парентеральную биодоступность у крысы как соответствующий активный ингибитор (свободный амидин).
Пример 49
Указанные в заголовках соединения из примеров 1, 2, 7, 8, 11, 12, 16, 18, 22, 24, 26, 29, 33, 37, 40, 43 и 45 проверены в соответствии в вышеприведенным Тестом G и обнаружено, что они превращаются в соответствующий активный ингибитор (свободный амидин) в микросомах печени людей и крыс.
Сокращения
Ас = ацетил
АсОН = уксусная кислота
APCl = химическая ионизация при атмосферном давлении (в отношении MS)
API = ионизация при атмосферном давлении (в отношении MS)
водн. = водный
AUC = площадь под кривой
Aze = (S)-азетидин-2-карбоксилат (если не определено особо)
AzeOH = азетидин-2-карбоновая кислота
Bn = бензил
Вос = трет-бутилоксикарбонил
BSA = бычий сывороточный альбумин
Bu = бутил
Bzl = бензил
Cl = химическая ионизация (в отношении MS)
сут. = сутки
DCC = дициклогексилкарбодиимид
DIBAL-H = диизобутилалюминийгидрид
DIPEA = диизопропилэтиламин
DMAP = 4-(N,N-диметиламино)пиридин
DMF = диметилформамид
DMSO = диметилсульфоксид
DVT = тромбоз глубоких вен
EDC = 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид
е.е. = энантиомерный избыток
Et = этил
эфир = диэтиловый эфир
EtOAc = этилацетат
EtOH = этанол
Et2O = диэтиловый эфир
ч = час (часы)
HATU = O-(азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
HBTU = [N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония гексафторфосфат]
HCl = соляная кислота, газообразный хлористый водород или соль гидрохлорид (в зависимости от контекста)
Hex = гексаны
НОАс = уксусная кислота
ВЭЖХ = высокоэффективная жидкостная хроматография
ЖХ = жидкостная хроматография
Me = метил
MEM = метоксиэтоксиметил
МеОН = метанол
мин = минута(ы)
MS = масс-спектроскопия
МТВЕ = метил-трет-бутиловый эфир
NADH = никотинамидадениндинуклеотид, восстановленная форма
NADPH = никотинамидадениндинуклеотидфосфат, восстановленная форма
NIH = Национальный институт здравоохранения (США)
NIHU = единица тромбина Национального института здравоохранения
ЯМР = ядерный магнитный резонанс
ОАс = ацетат
Pab = п-амидинобензиламино
Н-Pab = п-амидинобензиламин
Ph = фенил
Pr = пропил
Pro = (S)-пролинил
РуВОР = (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат
QF = тетрабутиламмонийфторид
RedAI = натрия бис(2-метоксиэтокси)алюминийгидрид
ОФЖХ = обращенно-фазовая высокоэффективная жидкостная хроматография
КТ = комнатная температура
SOP = стандартная рабочая процедура(ы)
TBTU = [N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония тетрафторборат]
TEA = триэтиламин
Теос = 2-(триметилсилил)этоксикарбонил
TEMPO = 2,2,6,6-тетраметил-1-пиперидинилокси, свободный радикал
TFA = трифторуксусная кислота
THF = тетрагидрофуран
ТНР = тетрагидропиранил
ТСХ = тонкослойная хроматография
TMSCl = триметилсилилхлорид
TMSCN = триметилсилилцианид
УФ = ультрафиолет
Z = бензилоксикарбонил
Префиксы н-, втор-, изо- и трет- имеют свои обычные значения: нормальный, вторичный, изо- и третичный. Префикс с означает цикло.
Изобретение относится к новым соединениям, выбранным из
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab,
или их фармацевтически приемлемым солям. Соединения ингибируют тромбин и могут быть использованы в фармацевтической композиции для лечения состояния, в котором требуется ингибирование тромбина. 5 н. и 11 з.п. ф-лы.
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(ОН) или
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab, или фармацевтически приемлемая соль любого из этих соединений.
Приоритет по пунктам и признакам:
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| WO 00/42059 A1, 20.07.2000 | |||
| ЦИКЛИЧЕСКИЕ АМИНОПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ И ПРЕДУПРЕЖДЕНИЯ ЗАБОЛЕВАНИЙ | 1997 |
|
RU2163596C2 |

