Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения порошка оксида металла, при этом указанным оксидом металла является оксид по меньшей мере одного металла со степенью окисления от (III) до (VI).
Также изобретение относится к способу изготовления таблетки из порошка оксида металла, полученного упомянутым способом.
Изобретение позволяет получать порошок оксида металла с высокой реакционной способностью, а также таблетку из оксида металла. Способность к высокой реакционной способности тесно увязана с гранулометрическим составом частиц, формирующих порошок оксида металла, полученный способом по изобретению, причем этот порошок обладает очень высокой удельной площадью поверхности.
Произведенный порошок может представлять собой порошок оксида одиночного металла, например, оксида урана или оксида церия, или же порошок смешанного оксида металла, например, смешанного оксида урана и церия, смешанного оксида церия и гадолиния или смешанного оксида урана и плутония.
Порошок и таблетка, получаемые способами согласно изобретению, могут найти применение в многочисленных областях, в частности, в ядерной промышленности или же в области ионных проводников.
Уровень техники
Среди оксидов актинидов и/или лантанидов, используемых в ядерной промышленности, диоксид урана UO2 и некоторые смешанные оксиды на основе урана, такие, как смешанный оксид урана и плутония (U, Pu)O2 или смешанный оксид урана и тория (U, Th)O2, входят в число смешанных оксидов металлов, наиболее широко используемых в производстве ядерных видов топлива.
Такие оксиды металлов, одиночные или смешанные, обычно имеют вид спрессованных и спеченных таблеток, которые должны удовлетворять ряду требований со стороны ядерной промышленности. Среди них такое требование, согласно которому таблетки ядерного топлива должны обладать высокой плотностью, обычно превышающей или равной 95%.
Конечная плотность таблетки зависит, в частности, от свойств порошка оксида металла, из которого изготовлена таблетка, а именно от реакционной способности порошка. Она также зависит от других параметров порошка, таких как гомогенность, отсутствие примесей, а также от параметров процесса, например, от параметров спекания.
Поскольку реакционная способность является свойством, определяемым удельной площадью поверхности частиц порошка, формирующих таблетки, то следовательно проводились многочисленные исследования в отношении обеспечения способа производства таблеток ядерного топлива, содержащих частицы оксида металла, средний диаметр которых являлся бы настолько малым, насколько это возможно.
Традиционно способы производства таблеток ядерного топлива включают в себя несколько последовательных стадий, проводимых начиная от водного раствора растворимых солей металла или металлов, присутствующих в порошке оксида металла, входящего в состав таблеток ядерного топлива. Такими растворимыми солями металлов являются, как правило, нитраты или сульфаты упомянутого металла или металлов.
Таким образом в источнике GB 1,128,838, обозначенном в конце данного описания, как [1], раскрыт способ получения топливных элементов из оксидов урана и плутония для ядерных реакторов. В этом способе уран и плутоний быстро соосаждают из водного раствора в таком виде, который обеспечивает их фильтрацию и последующую сушку. В частности, описанный в источнике [1] способ включает последовательно в следующем порядке:
a) приведение в контакт с гидроксидом аммония раствора нитрата уранила и нитрата плутония при соотношении между молярными концентрациями урана и плутония, близком к единице, что обеспечивает соосаждение однородной смеси гидратированных оксидов диураната аммония и плутония,
b) фильтрацию указанного осадка обычным фильтрующим устройством,
c) многократную промывку отфильтрованного осадка водой и ацетоном,
d) воздушную сушку промытого осадка и
e) восстановление высушенного осадка в восстановительной атмосфере, содержащей водород Н2, при температуре от 600 до 900°С для превращения соединений, содержащих уран и плутоний, в порошок оксидов урана и плутония, при этом данный порошок легко спекаем.
Согласно источнику информации [1] такой порошок может быть направлен в шаровую мельницу и затем пропущен через сито, содержащее 325 отверстий на линейный дюйм, что указывает на то, что диаметр частиц полученного порошка оксидов урана и плутония составляет несколько десятков микрометров.
Описанный в источнике [1] способ может также содержать дополнительную стадию прессования при повышенном давлении порошка после стадии е) с последующей стадией спекания для получения таблеток из оксидов урана и плутония.
Основным недостатком способа, раскрытого в источнике [1], является его относительно большое количество последовательных стадий. Кроме того этим способом предусмотрена стадия е) восстановления, которая, будучи проводимой при температурах от 600 до 900°С, является энергоемкой, как и дополнительная стадия измельчения для получения частиц порошка с диаметром настолько малым, насколько возможно.
В источнике информации US 4,314,952, [2] сообщается о способе производства таблеток из диоксида урана UO2 для использования в ядерных реакторах, причем эти таблетки имеют высокий удельный вес, размер образующих их частиц составляет более 50 мкм, предпочтительно от 50 до 100 мкм. Способ, описанный в источнике (2), включает в себя последовательно в следующем порядке:
a) приведение в контакт нитрата уранила с источником серы, как правило, с серной кислотой, при температуре от 300 до 400°С с получением серу со держащего триоксида урана,
b) приведение в контакт указанного триоксида урана с нитратом аммония с получением суспензии, содержащей серусодержащий нерастворимый уранат аммония,
c) приведение в контакт полученной суспензии с гидроксидом аммония для осаждения урана, оставшегося в растворе в виде нерастворимого ураната аммония,
d) извлечение и сушку ураната аммония,
e) восстановление высушенного ураната аммония с получением диоксида урана UO2,
f) прессование диоксида урана с получением таблеток и
g) спекание таблеток в атмосфере водорода Н2 при повышенной температуре.
Помимо использования относительно многочисленных стадий, включая стадию восстановления, осуществляемую при высокой температуре, описанный в источнике [2] способ производства таблеток из диоксида урана, имеет другой существенный недостаток, заключающийся в получении серусодержащих соединений.
В источнике информации US 4,382,885, [3], также сообщается о производстве ядерных видов топлива в виде таблеток, сами же эти таблетки образованы спеченными сферическими элементами. Эти спеченные сферические элементы изготовлены из делящегося материала и имеют диаметр от 100 до 1000 мкм. Этот способ включает не менее десятка стадий, включая стадию формирования капель путем пропускания процеженной суспензии через форсунку. Точнее, суспензию получают растворением одной или нескольких солей актинидов в присутствии реагента, выбранного из гидроксида аммония, оксалата аммония, щавелевой кислоты и смеси из этих соединений. Затем капли суспензии приводят в контакт с газообразным аммиаком, а затем с концентрированным раствором гидроксида аммония для преобразования этих капель желатинированием в сферические элементы. После промывки и сушки в печи при температуре от 150 до 400°С просушенные сферические элементы обжигают при температуре от 400 до 800°С, прессуют в виде таблеток, которые спекают при температуре от 1450 до 1700°С.
Описанный в источнике [3] способ характеризуется избыточно большим количеством стадий, из которых некоторые стадии, например, стадия формирования капель посредством форсунки и приведение их в контакт с газообразным аммиаком для образования желатинированных сферических элементов, имеют особенно сложное осуществление.
В источнике информации US 4,971,734, [4], описан способ получения таблеток ядерного топлива из спеченных оксидов, отвечающих формуле МxОy, где М - один или несколько химических элементов, традиционно используемых в производстве таблеток ядерного топлива, таких, как уран, плутоний, торий, церий, гадолиний или гафний.
Описанный в источнике [4] способ включает в себя последовательно в следующем порядке:
a) обработку перекисью водорода и аммиаком раствора, содержащего одну или несколько солей элемента или элементов М, с получением осадка пероксида,
b) фильтрацию осадка,
c) обжиг отфильтрованного осадка,
d) восстановление в печи подвергнутого обжигу осадка с получением промежуточного порошка из оксидов,
e) прессование промежуточного порошка и
f) спекание прессованного промежуточного порошка с получением таблеток из спеченных оксидов с очень высокой плотностью, превышающей, как правило, 96%.
И хотя в источнике [4] описано получение промежуточного порошка из сферических частиц с контролируемым узким гранулометрическим составом без дополнительной стадии измельчения, грохочения и/или грануляции, тем не менее в нем сказано, что такой промежуточный порошок «освобожден от агломератов, которые образовались при термообработке».
Как и все приведенные выше способы, способ согласно источнику [4] использует особо энергоемкие стадии, в данном случае это стадия обжига и стадия восстановления.
Целью изобретения является следовательно устранение недостатков, присущих уровню техники, и создание способа получения порошка из оксида по меньшей мере одного металла, причем каждый металл обладает степенью окисления от (III) до (VI), в частности, равной (III), (IV) и/или (VI), а именно равной (III) и/или (IV), что позволяет получать порошок с высокой реакционной способностью и настолько тонкой гранулометрией насколько возможно, в частности, при среднем диаметре частиц менее или равном 1 мкм, предпочтительно 100 нм, при этом при сокращенном количестве стадий по сравнению с количеством стадий в способах из уровня техники, например, раскрытых в источниках информации [1] - [3]. В частности, данный способ должен обеспечить такую гранулометрию при отсутствии стадии измельчения.
Кроме того этот способ должен обеспечить получение порошка из оксида по меньшей мере одного металла при отсутствии особо энергоемких термических стадий, таких, как сушка при свыше 100°С, обжиг и/или восстановление.
В целом способ по изобретению должен быть по возможности наиболее непосредственным и обеспечивать промышленное применение, являющееся технически и экономически оптимальным.
Раскрытие изобретения
Названные и другие цели достигаются в первую очередь посредством способа получения порошка из оксида, по меньшей мер, одного металла, причем каждый металл имеет степень окисления от (III) до (VI).
Необходимо уточнить, что выражение «от … до …», употребленное выше и далее по тексту, не следует понимать как только показатель интервала, но также как и предельные показатели этого интервала.
Следовательно способ позволяет получать порошок из оксида по меньшей мере одного металла, причем каждый металл имеет степень окисления, равную (III), (IV), (V) и/или (VI), в частности, равную (III), (IV) и/или (VI), а именно равную (III) и/или (IV).
Согласно изобретению способ включает в себя последовательно в следующем порядке:
a) проведение реакции водного раствора, содержащего, для каждого металла, по меньшей мере одну соль с катионом этого металла, с соединением, содержащим гидроксид, с получением осадка гидратированного оксида указанного по меньшей мере одного металла,
b) отделение полученного осадка,
c) приведение в контакт отделенного осадка с органическим протонным полярным растворителем,
d) удаление органического протонного полярного растворителя путем вакуумной сушки осадка с получением порошка гидратированного оксида указанного по меньшей мере одного металла, при этом указанный порошок образован из частиц, средний диаметр которых менее или равен 1 мкм.
Следовательно, способ согласно изобретению позволяет получать порошок из оксида металла, который обладает сильно выраженной реакционной способностью, и это при резко сокращенном количестве стадий, в противоположность известным из уровня техники способам, например, раскрытым в источниках [1] - [3].
В частности, способ согласно изобретению позволяет получать порошок из оксида металла с особо тонкой гранулометрией при отсутствии дополнительных стадий восстановления, сушки при относительно высокой температуре (обычно свыше 100°С), обжига и/или измельчения.
Получение этого порошка с такой гранулометрией является результатом особой комбинации стадий а) - d) способа, а также, как будет показано ниже, результатом выбора соединения, содержащего гидроксид в качестве реактива, обеспечивающего осаждение гидратированного оксида металла, и стадии вакуумной сушки этого гидратированного оксида металла, предварительно помещенного в органический протонный полярный растворитель.
Как четко следует из подробного, приводимого ниже описания частных вариантов осуществления, авторы изобретения обнаружили, что, когда водный раствор, который содержит, для каждого металла, по меньшей мере одну соль с катионом этого металла, реагирует с соединением, содержащим гидроксид, то образуется сначала осадок гидратированного гидроксида указанного по меньшей мере одного металла. Однако, поскольку этот осадок гидратированного гидроксида металла является особо реакционноспособоным соединением, то он самопроизвольно превращается в осадок гидратированного оксида указанного по меньшей мере одного металла, при этом следует уточнить, что сам этот гидратированный оксид металла постепенно изменяет аморфную структуру на кристаллическую. Такое превращение гидратированного гидроксида металла в гидратированный оксид металла протекает при отсутствии изменения физико-химических свойств осадка, сохраняющего, в частности, свою реакционную способность.
После отделения, например, фильтрацией или центрифугированием осадка, образованного этим гидратированным оксидом металла, он приводится в контакт на стадии с) с органическим протонным полярным растворителем, который затем удаляется вакуумной сушкой. Благодаря особой стадии d) органический протонный полярный растворитель постепенно удаляется. Кроме того, суспензия, образованная осадком гидратированного оксида металла и органическим протонным полярным растворителем подвергается равномерному охлаждению, что позволяет сохранить реакционную способность порошка гидратированного оксида металла и предотвратить агломерацию частиц порошка. Кроме того вакуумная сушка позволяет как быстрее удалить органический протонный полярный растворитель, чем простая сушка на воздухе, так и удалить его при более низких температурах по сравнению с температурами сушки в печи. Однако на стадии d) вакуумной сушки может быть предусмотрена возможность производить легкий нагрев суспензии, образованной из осадка гидратированного оксида металла и органического протонного полярного растворителя. Само собой разумеется, что вакуум, применяемый на данной стадии сушки, приведен в соответствие с выбранным органическим протонным полярным растворителем, в частности, в отношении показателя насыщающей упругости пара.
Также авторами изобретения было выявлено наличие действительной синергии, проявляющейся между выбором реагента, в данном случае соединения, содержащего гидроксид, и выбором стадии с) приведения в контакт и стадии d) особой сушки, так как они позволяют осуществлять сушку порошка гидратированного оксида металла, который не агломерирует. Однако, как будет видно из примеров 1 и 4 ниже, при выборе другого реагента, такого как щавелевая кислота, частицы полученного порошка оксида металла остаются агломерированными между собой, какими бы ни были условия сушки этого порошка.
Согласно предпочтительному варианту осуществления способа по изобретению порошок из гидратированного оксида указанного по меньшей мере одного металла образован из частиц, средний диаметр которых составляет менее или равен 100 нм, предпочтительно менее или равен 20 нм, более предпочтительно менее или равен 10 нм.
Согласно другому предпочтительному варианту осуществления способа по изобретению удельная площадь поверхности порошка гидратированного оксида указанного по меньшей мере одного металла, измеренная методом BET (метод Брунауэра-Эммета-Теллера), составляет более или равна 30 м2/г, предпочтительно более или равна 80 м2/г, более предпочтительно более или равна 100 м2/г.
Эти значения удельной площади поверхности, отражающие реакционную способность порошка гидратированного оксида металла, значительно превосходят те же значения сопоставимого порошка гидратированного оксида металла, который получают при использовании стадий а) - с), аналогичных стадиям способа согласно изобретению, но с использованием стадии d) удаления органического протонного полярного растворителя, проводимой сушкой на воздухе.
Согласно другому предпочтительному варианту осуществления способа по изобретению способ дополнительно включает после стадии b) и перед стадией с) стадию промывки осадка указанного гидратированного оксида по меньшей мере одного металла, который был отделен, например, фильтрацией или центрифугированием, на стадии b).
Такая промывка может осуществляться один или несколько раз одним и тем же или разными растворителями, причем этот растворитель или эти растворители предпочтительно являются протонными, при необходимости в смеси с водой.
Такая промывка может проводиться, в частности, этанолом или же смесью из воды и этанола. Если на стадии промывки осадка гидратированного оксида металла используется вода, то эта вода предпочтительно является деионизированной.
Согласно другому предпочтительному варианту способа по изобретению способ дополнительно включает после стадии d):
е) термообработку порошка гидратированного оксида указанного по меньшей мере одного металла с получением порошка безводного оксида указанного по меньшей мере одного металла.
Следовательно, на стадии е) под действием термообработки порошка, полученного на стадии d), происходит полная дегидратация гидратированного оксида металла с образованием соответствующего безводного оксида металла.
Следует уточнить, что частичная дегидратация гидратированного оксида металла происходит собственно также на стадии d), т.е. во время удаления органического протонного полярного растворителя вакуумной сушкой.
Как уже упоминалось, согласно способу получения порошка из оксида по меньшей мере одного металла согласно изобретению каждый металл обладает степенью окисления, составляющей от (III) до (VI), в частности, равной (III), (IV) и/или (VI), а именно равной (III) и/или (IV).
Согласно предпочтительному варианту изобретения каждый металл выбран из актинидов, лантанидов и переходных металлов, причем эти металлы обязательно имеет степень окисления, составляющую от (III) до (VI), в частности, равную (III), (IV) и/или (VI), а именно, равную (III) и/или (IV).
Если металл представляет собой актинид, то он предпочтительно выбран из химических элементов, входящих в группу, состоящую из: урана U, тория Th, плутония Pu, нептуния Np, америция Am и кюрия Cm.
Если металл является лантанидом, то он предпочтительно выбран из химических элементов, входящих в группу, состоящую из: церия Се, гадолиния Gd, неодима Nd, самария Sm и европия Eu.
Если металл представляет собой переходный металл, то он предпочтительно выбран из химических элементов, входящих в группу, состоящую из: титана Ti, хрома Cr, циркония Zr, скандия Sc, иттрия Y и гафния Нf.
В качестве примера, металл может быть предпочтительно выбран из химических элементов, входящих в группу, состоящую из: U(IV), U(VI), Th(IV), Pu(III), Pu(IV), Pu(VI), Am(III), Np(IV), Np(VI), Ce(III), Ce(IV), Gd(III), Nd(III), Zr(IV).
Способ согласно изобретению относится к получению порошка оксида по меньшей мере одного металла, при этом каждый металл обладает степенью окисления от (III) до (VI), в частности, равным (III), (IV) и/или (VI), а именно равным (III) и/или (IV).
Следовательно, согласно первому варианту осуществления изобретения способ позволяет получать порошок оксида одного металла, называемого простым оксидом. Таким образом этот порошок оксида одного металла может быть, в частности, порошком оксида актинида, лантанида или переходного металла.
Простой оксид предпочтительно выбран из диоксида урана UO2+δ, триоксида урана UO3, октаоксида триурана U3O8, диоксида церия СеО2-δ, диоксида тория ThO2, диоксида плутония PuO2-δ, диоксида нептуния NpO2+δ, диоксида циркония ZrO2 и диоксида гафния HfO2. δ - величина, переменная в зависимости от образующего соответствующий оксид металла. Обычно δ составляет от 0 до 0,5, при этом значение 0 включается, а значение 0,5 исключается (0≤δ<0,5).
Согласно второму варианту осуществления изобретения способ позволяет также получать порошок оксида двух, трех и даже более металлов, называемого также смешанным оксидом. Такой порошок оксида двух, трех и более металлов может быть следовательно порошком смешанного оксида актинидов, смешанного оксида лантанидов или смешанного оксида переходных металлов. Он может быть также порошком смешанного оксида актинида (актинидов) и лантанида (лантанидов), смешанного оксида актинида (актинидов) и переходного металла (переходных металлов), смешанного оксида лантанида (лантанидов) и переходного металла (переходных металлов) или же смешанного оксида актинида (актинидов), лантанида (лантанидов) и переходного металла (переходных металлов).
Такой смешанный оксид предпочтительно выбран из смешанного оксида урана и церия (U, Се)O2±δ, смешанного оксида урана и плутония (U, Pu)О2±δ, смешанного оксида урана и америция (U, Am)O2±δ, смешанного оксида урана и тория (U, Th)O2+δ, смешанного оксида церия и гадолиния (Се, Gd)O2-δ, смешанного оксида урана и гадолиния (U, Gd)O2±δ, смешанного оксида тория и плутония (Th, Pu)О2-δ, смешанного оксида тория и иттрия (Th, Y)O2-δ и смешанного оксида урана, плутония и америция (U, Pu, Am)O2±δ. Величина δ является переменной и зависит от металлов, образующих соответствующий оксид. Обычно δ составляет от 0 до 0,5, при этом значение 0 включается, значение 0,5 исключается (0≤δ<0,5).
Следует уточнить, что после стадии d) полученный порошок оксида указанного по меньшей мере одного металла, будь этот оксид простым или сложным, представляет собой порошок оксида металла в гидратированном виде, обозначаемого обычно как nН2О.
Согласно предпочтительному варианту изобретения на стадии а), для каждого металла, соль с катионом этого металла выбрана из сульфата, нитрата, галогенида, при этом следует уточнить, что этот катион может быть трехвалентным, четырехвалентным, пятивалентным и/или шестивалентным. Если соль представляет собой галогенид, то предпочтительно используется хлорид или бромид.
В контексте настоящего изобретения соединение, содержащее гидроксид, используемое в качестве реагента на стадии а) способа, представляет собой соединение, содержащее по меньшей мере один анион гидроксида ОН- и по меньшей мере один катион для обеспечения электронейтральности упомянутого соединения.
В качестве примера, катион соединения, содержащего гидроксид, может быть первичным, вторичным или третичным аммонием или просто катионом аммония NH4+. Катион может быть также катионом гидразиния N2H5+.
Катион соединения, содержащего гидроксид, может быть также катионом металла, в частности, катионом щелочного металла, например, натрия Na или калия К, или катионом щелочноземельного металла, например, кальция Са или магния Mg.
Соединение, содержащее гидроксид, может быть выбрано из гидроксида аммония NH4OH, гидроксида гидразиния N2H5OH, гидроксида натрия NaOH, гидроксида калия КОН, гидроксида кальция Са(ОН)2 или же гидроксида магния Mg(OH)2.
Соединение, содержащее гидроксид, может также происходить от соединения, способного образовывать анион ОН- в водном растворе. В качестве примера, соединение, содержащее катион аммония NH4+ и анион гидроксида ОН-, может происходить либо от гидроксида аммония NH4OH в виде соли, либо от продукта реакции аммиака NH3 с водой.
Согласно предпочтительному варианту изобретения на стадии а) в качестве соединения, содержащего гидроксид, используют гидроксид аммония NH4OH или гидроксид гидразиния N2H5OH.
Согласно предпочтительному варианту изобретения на стадии а) молярное содержание соединения, содержащего гидроксид, является избыточным по отношению к общему молярному содержанию катиона (катионов) указанного по меньшей мере одного металла со степенью окисления от (III) до (VI). Следует уточнить, что этот или эти катионы являются трехвалентными, четырехвалентными, пятивалентными и/или шестивалентными в зависимости от степень окисления соответствующего металла. Такое молярное содержание соединения, содержащего гидроксид, предпочтительно составляет от 150 до 600%, предпочтительно от 300 до 500%, от общего молярного содержания катиона (катионов) указанного по меньшей мере одного металла.
Согласно предпочтительному варианту изобретения на стадии с) органический протонный полярный растворитель выбран из карбоновой кислоты, первичного амина и спирта.
В том случае, когда органический протонный полярный растворитель представляет собой карбоновую кислоту, то она может быть выбрана, в частности, из муравьиной, уксусной и пропионовой кислот.
Если органический протонный полярный растворитель представляет собой первичный амин, то он может быть выбран, в частности, из метиламина, этиламина и изопропиламина.
Если органический протонный полярный растворитель представляет собой спирт, то он может быть, в частности, одноатомным или многоатомным спиртом. Такой спирт предпочтительно выбран из группы, состоящей из метанола, этанола и этандиола.
Согласно предпочтительному варианту изобретения на стадии d) вакуумная сушка проводится посредством вакуумного распределительного устройства, причем это устройство позволяет одновременно создавать вакуум внутри баллона, содержащего суспензию из осадка гидратированного оксида металла и органического протонного полярного растворителя, и нарушать вакуум путем введения газа в этот баллон, в частности, инертного, например, диазота, аргона или гелия. В качестве альтернативы можно вводить также восстановительный газ.
Для содействия выпариванию органического протонного полярного растворителя из суспензии и для получения порошка гидратированного оксида металла по возможности в наиболее сухом состоянии, в частности, предпочтительно нагреть и/или поддерживать в состоянии перемешивания суспензию, образованную из осадка гидратированного оксида металла и органического протонного полярного растворителя.
Во-вторых, изобретение относится к порошку оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI), в частности, равную (III), (IV) и/или (VI), а именно равную (III), и/или (IV), полученному охарактеризованным выше способом, при этом оптимальные признаки данного способа могут быть взяты раздельно или в сочетании.
В-третьих, изобретение относится к разным видам применения такого порошка оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI), в частности, равную (III), (IV) и/или (VI), а именно равную (III), и/или (VI).
Согласно изобретению такой порошок оксида по меньшей мере одного металла может применяться в производстве ядерного топлива.
Согласно изобретению порошок оксида по меньшей мере одного металла может также применяться в качестве носителя катализатора.
Согласно изобретению порошок оксида по меньшей мере одного металла, если этим металлом выступает уран, может также применяться для получения октаоксида триурана U3O8.
Согласно изобретению порошок оксида по меньшей мере одного металла может также применяться в способе гидрофторирования.
Согласно изобретению порошок оксида по меньшей мере одного металла может также применяться в производстве ионных проводников, например, твердого электролита для топливного элемента на твердом оксиде (Solid Oxide Fuel Cell: SOFC) или для зонда для измерения кислорода.
Согласно изобретению порошок оксида по меньшей мере одного металла может также применяться в производстве керамики. Такое керамическое изделие, называемое также оксидным керамическим изделием, может, в частности, применяться в виде таблетки ядерного топлива или в качестве ионного проводника, причем последний может быть твердым электролитом для топливного элемента SOFC или зондом для измерения кислорода, как упомянуто выше.
В-четвертых, изобретение относится к способу производства таблетки из оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI), в частности, равную (III), (IV) и/или (VI), а именно равную (III), и/или (IV).
Согласно изобретению данный способ включает в себя последовательно в следующем порядке:
1) получение порошка оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI), охарактеризованным выше способом, при этом оптимальные признаки могут быть взяты раздельно или в сочетании,
2) прессование указанного порошка и
3) термообработку спрессованного порошка с получением таблетки из оксида по меньшей мере одного металла.
Другими словами, способ производства таблеток из оксида по меньшей мере одного металла, причем каждый металл имеет степень окисления от (III) до (VI), включает в себя последовательно в следующем порядке:
a) проведение реакции водного раствора, содержащего, для каждого металла, по меньшей мере одну соль с катионом этого металла с соединением, содержащим гидроксид, с получением осадка гидратированного оксида указанного по меньшей мере одного металла,
b) отделение полученного осадка,
c) приведение в контакт отделенного осадка с органическим протонным полярным растворителем,
d) удаление органического протонного полярного растворителя вакуумной сушкой осадка с получением порошка гидратированного оксида указанного по меньшей мере одного металла, причем указанный порошок образован из частиц, средний диаметр которых составляет менее или равен 1 мкм,
e) прессование указанного порошка и
f) термообработку спрессованного порошка с получением таблетки из оксида по меньшей мере одного металла.
Следовательно, способ согласно изобретению позволяет получать таблетку из оксида металла, имеющую высокую плотность, при этом значительно снизилось количество стадий в противоположность известным из уровня техники способам, например, описанным в источниках [1] - [3].
Кроме того такая таблетка из оксида металла, полученная способом согласно изобретению, обладает очень высокой механической прочностью, что является неоспоримым преимуществом при ее последующем использовании.
Согласно предпочтительному варианту способа по изобретению таблетка из оксида по меньшей мере одного металла обладает плотностью, составляющей по меньшей мере 90%, предпочтительно по меньшей мере 95%.
Согласно другому предпочтительному варианту способа по изобретению на стадии 3) или f) термообработка проводится с применением температурного градиента в диапазоне от комнатной температуры до температуры менее или равной 1600°С, предпочтительно менее или равной 1400°С. Таким образом обеспечивается спекание частиц в спрессованной таблетке.
Важно отметить, что используемая на стадии 3) или f) максимальная температура для спекания спрессованного порошка, предварительно приготовленного способом согласно изобретению, меньше по меньшей мере на 100°С, даже по меньшей мере на 300°С, чем температура, необходимая для спекания спрессованного порошка, приготовленного известными из уровня техники способами. Следовательно, способ изготовления таблетки из оксида по меньшей мере одного металла согласно изобретению характеризуется неоспоримым преимуществом благодаря дополнительному снижению расхода тепловой энергии в дополнение к упомянутому выше, что связано со способом получения порошка оксида по меньшей мере одного металла, у которого, например, резко сокращено количества стадий.
В-пятых, изобретение относится к таблетке из оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI), полученной описанный выше способом, при этом предпочтительные признаки этого способа могут применяться раздельно или в сочетании.
В-шестых, изобретение относится к разным видам применения таблетки из оксида по меньшей мере одного металла, при этом каждый металл имеет степень окисления от (III) до (VI).
Согласно изобретению таблетка из оксида по меньшей мере одного металла может применяться в качестве ядерного топлива.
Согласно изобретению таблетка из оксида по меньшей мере одного металла может также применяться в качестве ионного проводника, который может быть, в частности, твердым электролитом для топливного элемента с твердым оксидом (SOFC) или зондом для измерения кислорода, как упоминалось выше.
Другие признаки и преимущества изобретения станут более понятными из приводимого ниже описания и примеров на синтез оксидов металлов согласно изобретению, которые могут быть простыми и смешанными, гидратированными и/или безводными.
Само собой разумеется, что примеры приведены только для пояснения объекта изобретения и совершенно его не ограничивают.
Краткое описание чертежей
Фиг. 1 - снимок сравнительного порошка в примере 1, полученный сканирующим электронным микроскопом;
фиг. 2А, 2В - снимки порошка согласно изобретению в примере 1, полученные соответственно с помощью сканирующего электронного микроскопа (фиг. 2А) и просвечивающего электронного микроскопа (фиг. 2В);
фиг. 3 - дифрактограмма порошка согласно изобретению в примере 1, полученная методом рентгеновской дифрактометрии и отображающая изменение интенсивности дифрагированных рентгеновских лучей (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка упомянутых рентгеновских лучей (обозначенного 2θ и выраженного в °);
фиг. 4 - дифрактограммы порошка согласно изобретению в примере 1, полученные при температуре от 30 до 1100°С методом рентгеновской дифрактометрии, отображающие обнаруженное изменение интенсивности дифрагированных рентгеновских лучей, (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка упомянутых рентгеновских лучей (обозначенного 2θ и выраженного в °);
фиг. 5 - кривая, отображающая протекание относительной потери массы (обозначенной Δm и выраженной в %) пробы порошка гидратированного оксида урана (IV) согласно изобретению в примере 1 в зависимости от применяемой температуры (обозначенной Т и выраженной в °С);
фиг. 6 - кривая, отображающая протекание относительной линейной усадки (обозначенной ΔL/L0 и выраженной в %) сравнительной таблетки и таблетки, полученной после прессования порошка гидратированного оксида урана (IV) согласно изобретению в примере 1, в зависимости от применяемой температуры (обозначенной Т и выраженной в °С);
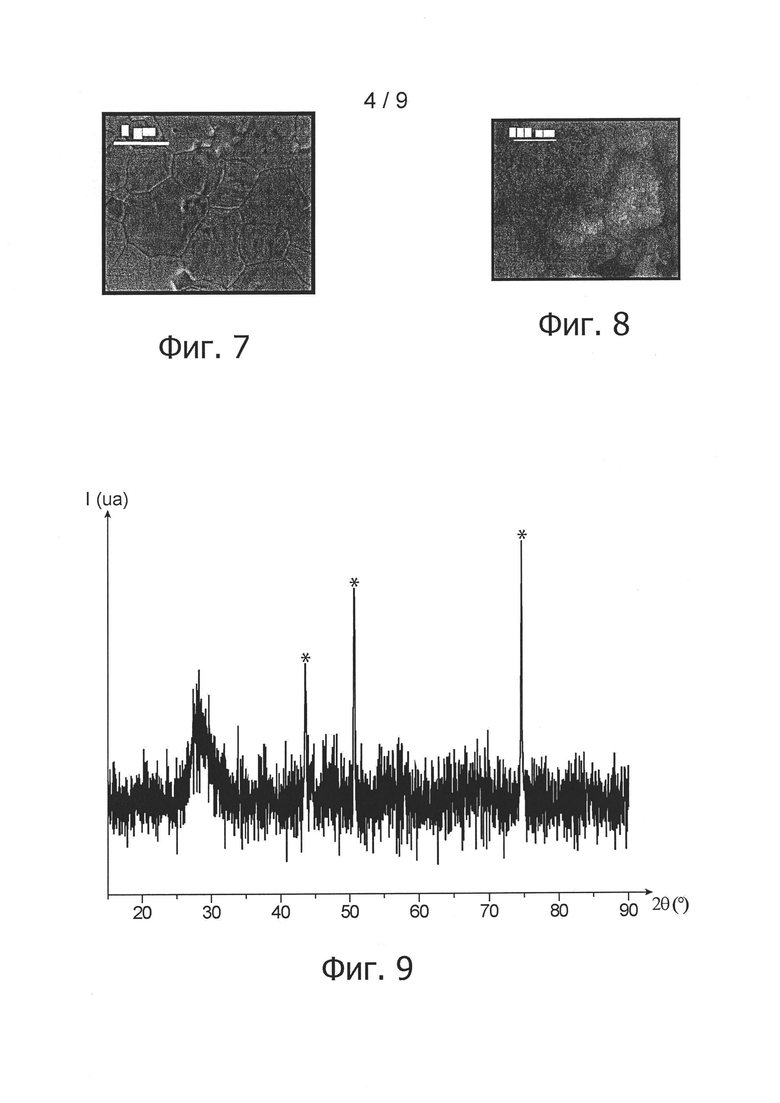
фиг. 7 - снимок спеченной таблетки согласно изобретению в примере 1, полученный сканирующим электронным микроскопом;
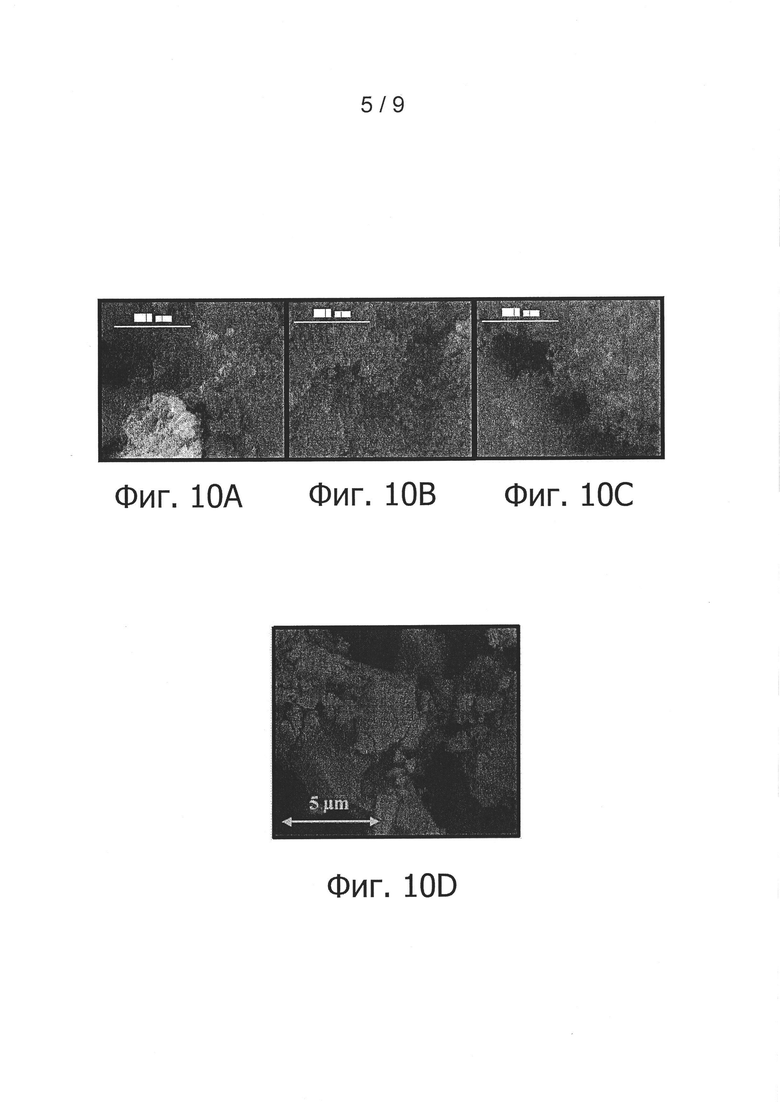
фиг. 8 - снимок порошка согласно изобретению в примере 2, полученный сканирующим электронным микроскопом;
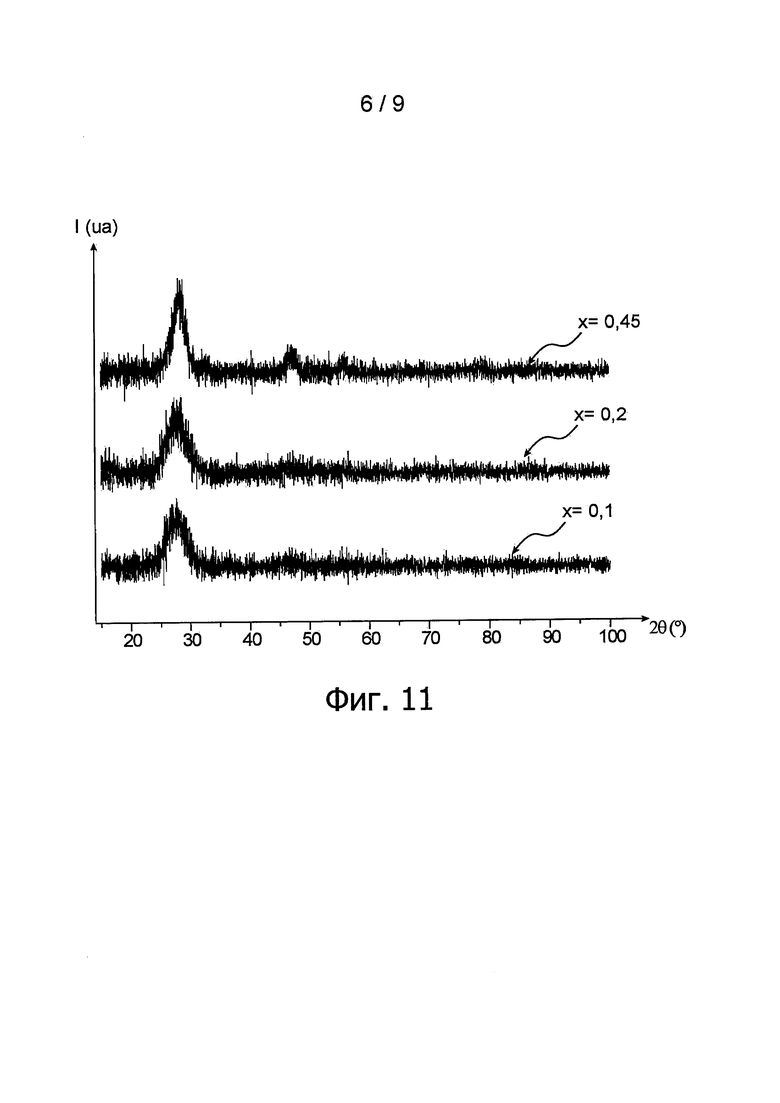
фиг. 9 - дифрактограмма порошка согласно изобретению в примере 2, полученная методом рентгеновской дифрактометрии и отображающая обнаруженное изменение интенсивности дифрагированных рентгеновских лучей (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка рентгеновских лучей (обозначенного 2θ и выраженного в °);
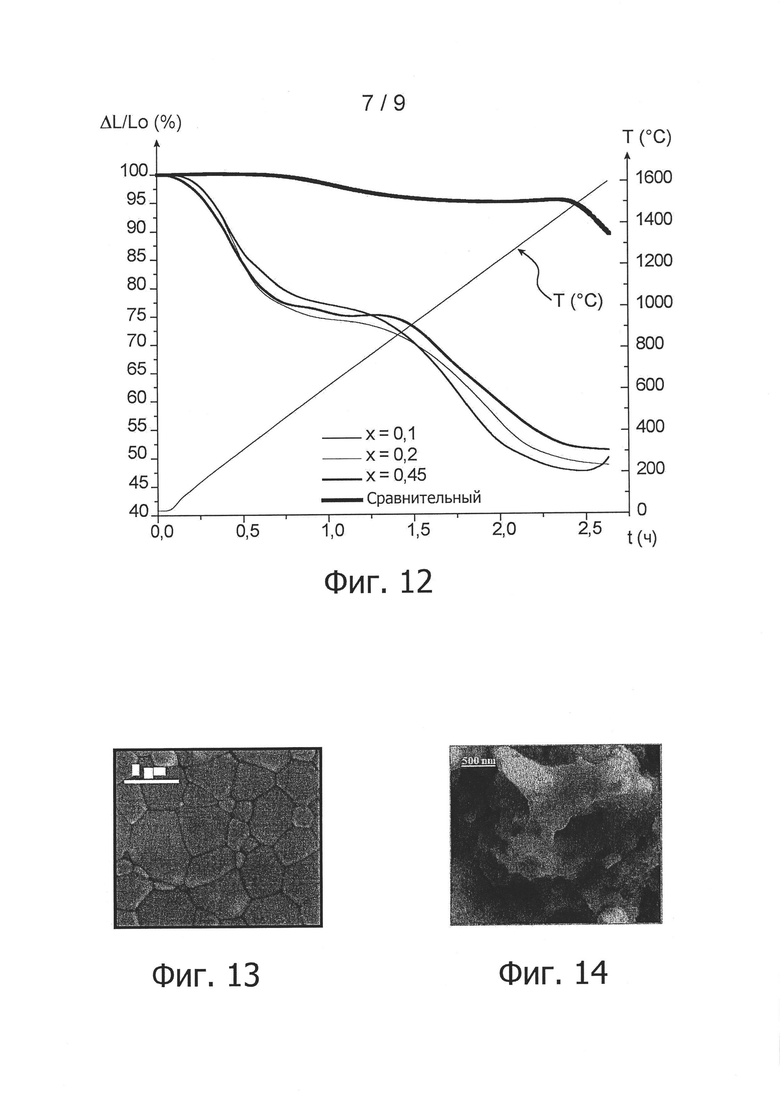
фиг. 10А, 10В и 10С - снимки, полученные сканирующим электронным микроскопом для порошков согласно изобретению в примере 4, приготовленных из смесей А, В и С, соответственно в присутствии этанола в качестве растворителя;
фиг. 10D - снимок, полученный сканирующим электронным микроскопом для сравнительного порошка в примере 4, приготовленного из смеси А в присутствии воды в качестве растворителя;
фиг. 11 - дифрактограммы, полученные методом рентгеновской дифрактометрии и отображающие изменение интенсивности дифрагированных рентгеновских лучей (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка рентгеновских лучей (обозначенного 2θ и выраженного в °), для порошков, приготовленных на основе соответственно смесей А, В и С в примере 4;
фиг. 12 - кривые, отображающие протекание относительной линейной усадки (обозначенной ΔL/L0 и выраженной в %) сравнительной таблетки, также каждой из таблеток, полученных после прессования порошков смешанных оксидов урана (IV) и церия (IV) согласно изобретению в примере 4 в зависимости от применяемой температуры (обозначенной Т и выраженной в °С) и от времени (обозначенного t и выраженного в часах);
фиг. 13 - снимок, полученный сканирующим электронным микроскопом, таблетки согласно изобретению в примере 4, приготовленной из смеси А (х=0,1) после спекания;
фиг. 14 - снимок порошка согласно изобретению в примере 5, полученный сканирующим электронным микроскопом;
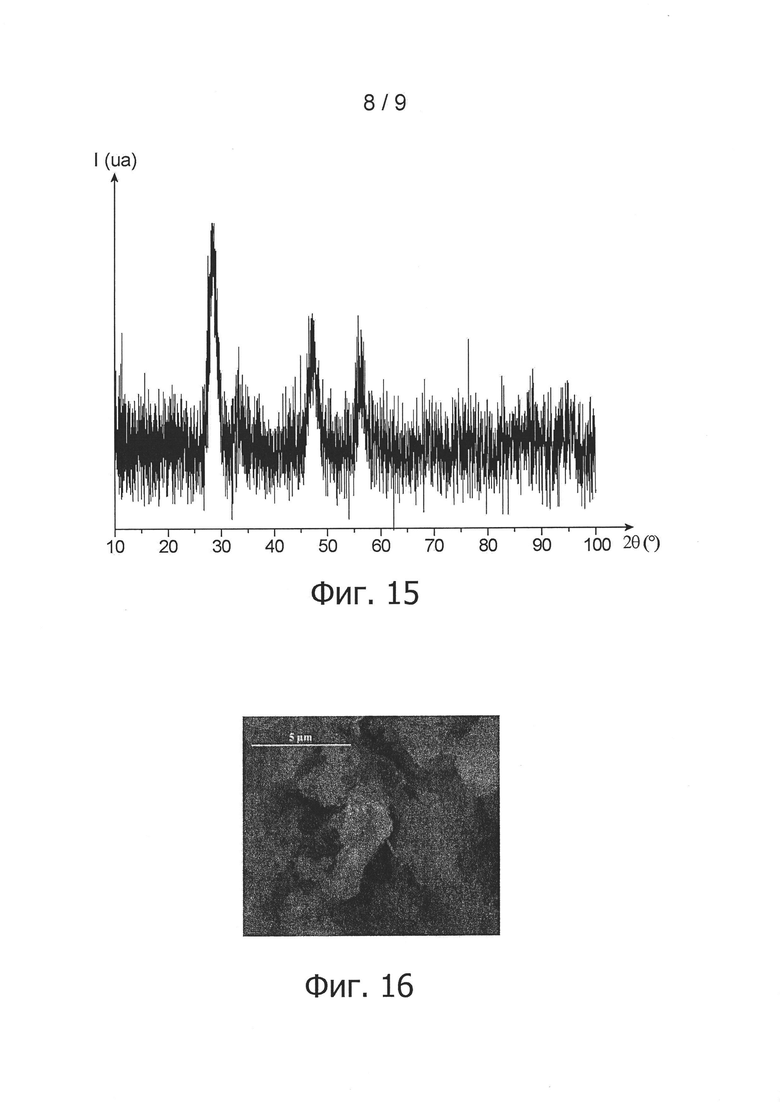
фиг. 15 - дифрактограмма, полученная для порошка согласно изобретению в примере 5 методом рентгеновской дифрактометрии и отображающая обнаруженное изменение интенсивности дифрагированных рентгеновских лучей, (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка рентгеновских лучей (обозначенного 2θ и выраженного в °);
фиг. 16 - снимок, полученный для порошка согласно изобретению в примере 6 с помощью сканирующего электронного микроскопа;
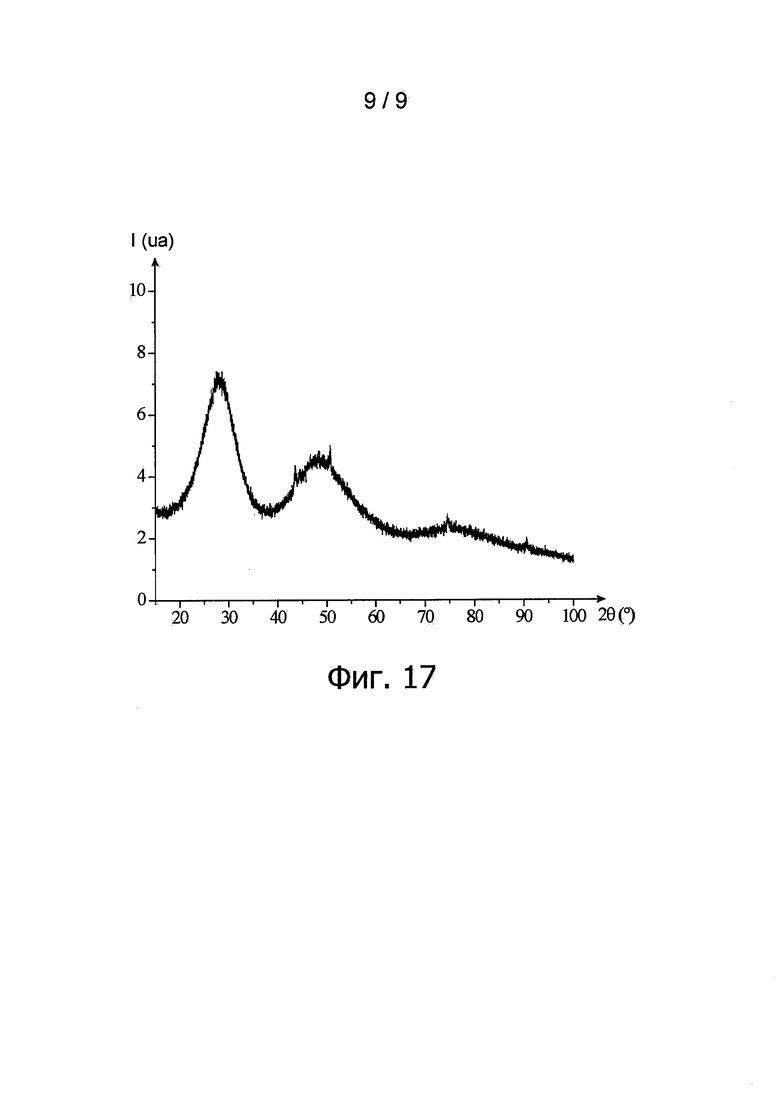
фиг. 17 - дифрактограмма, полученная для порошка согласно изобретению в примере 6 методом рентгеновского структурного анализа и отображающая изменение интенсивности дифрагированных рентгеновских лучей (обозначенной 1 и выраженной условной единицей ua) в зависимости от угла дифракции двойная тета пучка рентгеновских лучей (обозначенного 2θ и выраженного в °).
Осуществление изобретения
Пример 1. Синтез гидратированного и безводного оксидов урана (IV)
Приготовление водного раствора, содержащего хлорид урана (IV)
Для синтеза гидратированного и затем безводного оксидов урана (IV) водный раствор, с содержанием хлорида урана, был приготовлен из металлического урана U0.
Для этого стружки металлического урана очистили с помощью соляной кислоты с молярной концентрацией 2 моль/л от покрывавшего их оксидного слоя. После этой очистки стружек урана U0 их поместили в раствор соляной кислоты с молярной концентрацией 6 моль/л. Для предотвращения бурного протекания реакции стружки вводили постепенно.
После растворения стружек в растворе соляной кислоты получили водный раствор, содержащий хлорид урана (IV), от которого отделили центрифугированием мелкие фракции.
Полученный при этом водный раствор, содержащий хлорид урана (IV) титровали посредством колориметрического анализа, а также атомного эмиссионного спектрального анализа с индуктивно связанной плазмой (Inductively Coupled Plasma-Atomic Emission Spectrometry: ICP-AES).
Синтез гидратированного оксида урана (IV)
Синтез гидратированного оксида урана (IV) проводился путем добавки в водный раствор хлорида урана (IV), такой, как приготовленный выше, гидроксида аммония с молярным содержанием 400% относительно молярного содержания хлорида урана в водном растворе. Добавка вносилась при комнатной температуре при перемешивании со скоростью 500 об./мин. Перемешивание длилось в течение часа.
В конце этого часа был получен осадок. После фильтрации осадка анализ фильтрата, проведенный методом ICP-AES, показал количественное осаждение урана при эффективности осаждения более или равном 99,9%.
После нескольких промывок деионизированной водой и затем этанолом, обеспечивающих удаление любых следов остаточной кислоты, осадок отделили от жидкой фазы центрифугированием при скорости 4000 об./мин. Полученный при этом осадок разделили на две фракции.
Первую фракцию осадка сушили на воздухе в течение одних суток при комнатной температуре и атмосферном давлении. Порошок, полученный на этой стадии воздушной сушки, анализировали.
Удельная площадь поверхности, измеренная методом BET (метод Брунауэра-Эммета-Теллера) посредством адсорбции азота при температуре кипения жидкого азота (-196°С), составила около 30 м2/г.
Морфологический анализ этого порошка проводился также методом сканирующей электронной микроскопии. На соответствующем снимке, приведенном на фиг. 1, видно, что образующие порошок частицы сильно агломерированы.
Вторую фракцию осадка поместили в баллон с этанолом.
Для быстрого выпаривания содержащегося в осадке этанола при низкой температуре баллон поместили в динамический вакуум, составлявший менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Одновременно осадок перемешивали при скорости 500 об./мин. и температуре 40°С.
По окончании стадии выпаривания этанола получили порошок. Порошок перемешивали в течение 5 минут, затем вакуум нарушили введением диазота N2 в баллон с помощью вакуумного распределительного устройства с целью предотвращения окисления урана (IV).
Провели анализ порошка, полученного после вакуумной сушки и являвшегося второй фракцией осадка.
Удельная площадь поверхности, которую определяли методом BET путем адсорбции азота при температуре кипения жидкого азота (-196°С), составила около 150 м2/г. Этот показатель, соответствующий большой удельной площади поверхности, свидетельствует о большой реакционной способности полученного порошка, существенно превышающей реакционную способность порошка, полученного с воздушной сушкой.
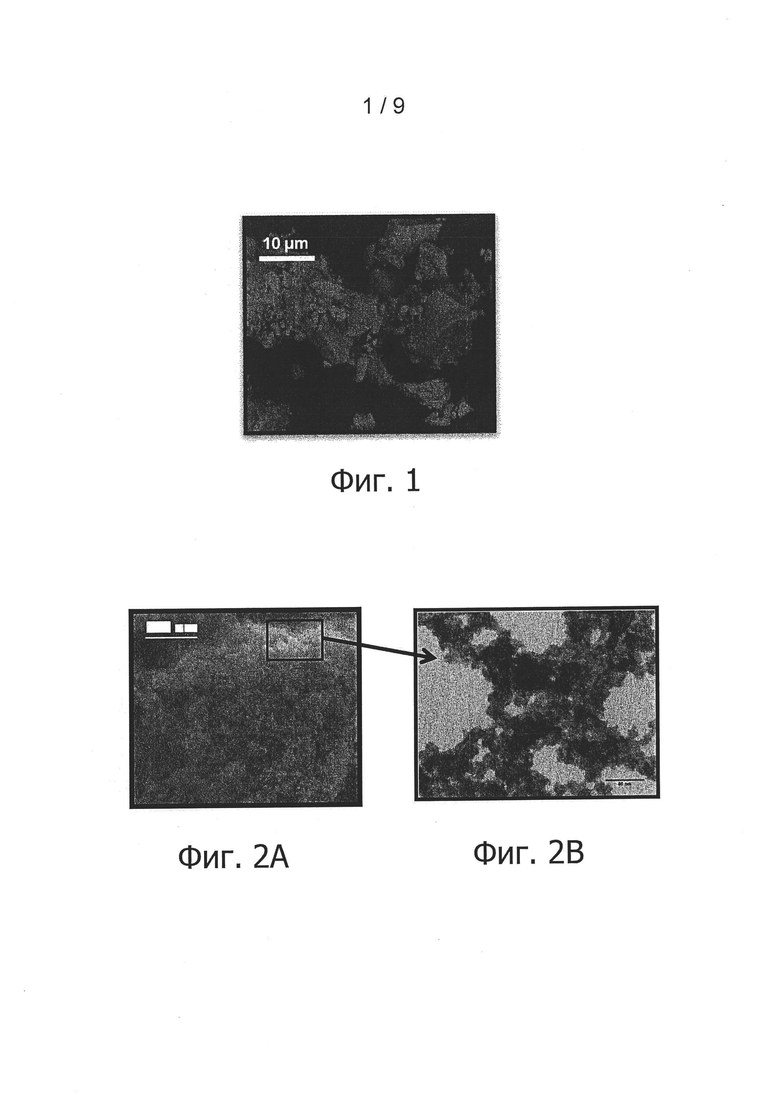
Морфологический анализ высушенного в вакууме порошка также проводился методом сканирующей электронной микроскопии. Соответствующий снимок на фиг. 2А показывает, что этот порошок не агломерирует и что он образован из частиц нанометрического размера. Этот факт подтверждается впрочем снимком, выполненным методом просвечивающей электронной микроскопии и приведенным на фиг.2 В. Действительно, снимок на фиг. 2В свидетельствует о присутствии частиц размером с десяток нанометров.
По окончании стадии вакуумной 0 сушки второй фракции осадка получили порошок, который не образует агрегатов и обладает существенно возросшей реакционной способностью по сравнению с порошком, полученным способом с воздушной сушкой этого же осадка.
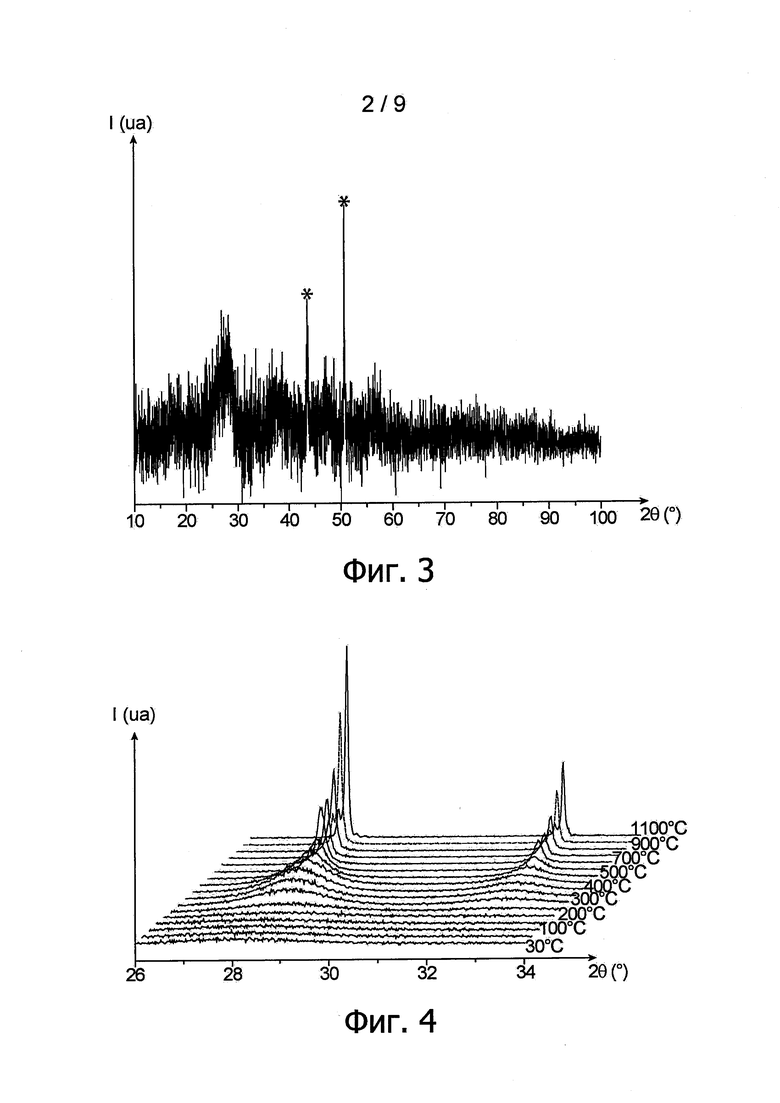
Для определения структуры частиц высушенного в вакууме порошка провели анализ методом рентгеновской дифрактометрии. Соответствующая дифрактограмма приведена на фиг. 3, при этом следует уточнить, что два наиболее высоких пика, соответствующих значениям угла 2θ 43,5° и 50,5° и отмеченных звездочкой (*), соответствуют интенсивности рентгеновских лучей, дифрактированных носителем пробы порошка.
Можно видеть, что полученная при этом дифрактограмма указывает на кристаллографическую кубическую гранецентрированную структуру типа флюорита пространственной группы Fm-3m, которая характерна для диоксидов актинидов. Эта дифрактограмма свидетельствует также о низкой степени кристаллизации частиц, образующих порошок.
Дополнительный анализ методом спектрометрии комбинационного рассеяния (Рамановская) выявил отсутствие характерной полосы вибрации групп ОН и подтвердил присутствие гидратированного оксида. Такие анализы методом рентгеновской дифрактометрии и спектрометрии комбинационного рассеяния позволяют объяснить явления, происходящие во время реакции хлорида урана с гидроксидом аммония.
Действительно, при контакте с гидроксидом аммония катионы U4+, присутствующие в водном растворе хлорида урана, выпадают в осадок в виде гидроксида урана (IV) в соответствии со следующей химической реакцией (1):
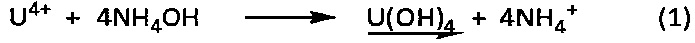
Однако этот осадок гидроксида урана (IV) является очень реакционноспособным соединением, способным самопроизвольно образовывать гидратированный оксид урана (IV) в соответствии со следующей химической реакцией (2):
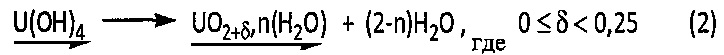
Термообработка гидратированного оксида урана (IV)
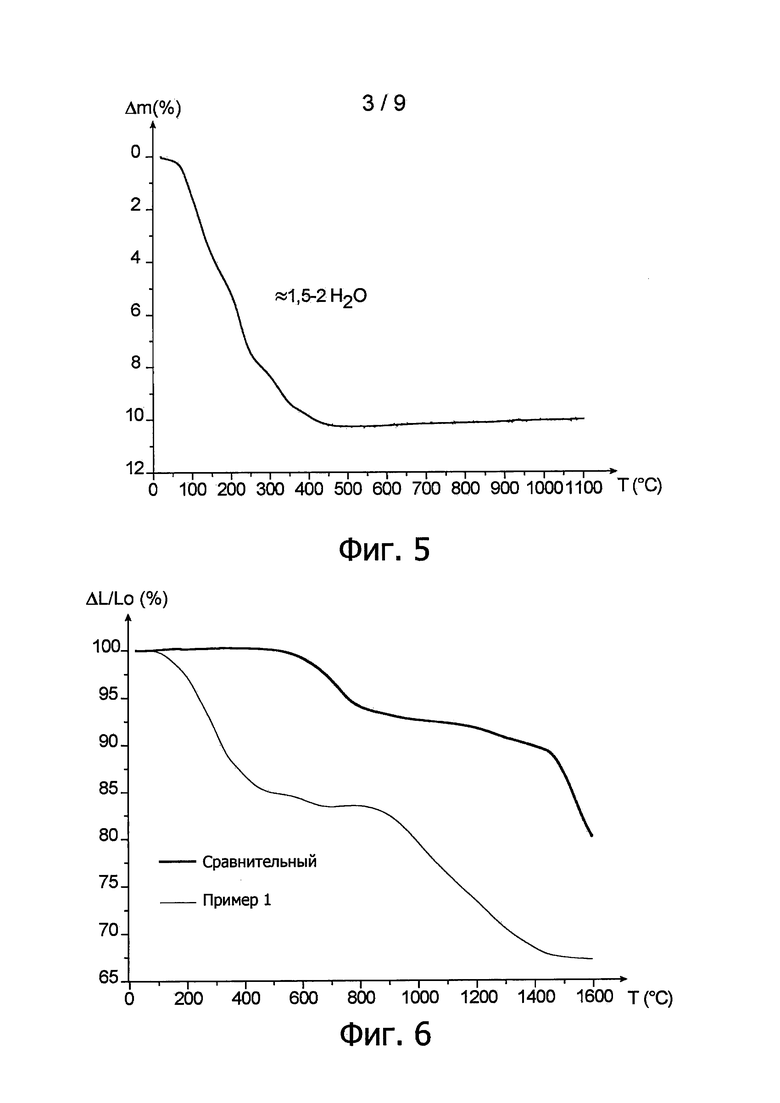
* При первой термообработке проводился контроль за изменением кристалличности порошка гидратированного оксида урана (IV) в зависимости от температуры. Эта термообработка проходила in situ в инертной атмосфере в присутствии диазота с применением температурного градиента, возраставшего от 30 до 1100°С.
Полученные при этом и представленные на фиг. 4 дифрактограммы показывают, что кристаллографическая гранецентрированная кубическая структура типа флюорита сохранялась на протяжении первой термообработки и что кристалличность порошка резко возросла, начиная с температуры 600°С.
* При второй термообработке проводилось полная дегидратация порошка гидратированного оксида урана (IV) в инертной атмосфере, а именно аргона.
После этой дегидратации проводился термогравиметрический анализ с целью наблюдения за изменением массы пробы порошка гидратированного оксида урана (IV) в зависимости от применяемой температуры. Это показано соответствующей кривой на фиг. 5.
Обратившись к фиг. 5, можно видеть, что дегидратация порошка происходит за одну стадию, завершающуюся при температуре около 450°С. Начиная с этой температуры, достигается максимальная величина относительной потери массы. Эта величина, составляющая около 10%, соответствует полной дегидратации гидратированного оксида урана (IV) с образованием безводного оксида урана (IV). Такая относительная потеря массы, показанная кривой на фиг. 5, соответствует потере от 1,5 до 2 молекул воды.
Прессование гидратированного оксида урана (IV)
Порошок гидратированного оксида урана (IV), полученный после вакуумной сушки, прессовали при одноосном прессовании при давлении 500 МПа. Такое прессование, называемое также таблетированием, позволило получить сырую таблетку с плотностью от 40 до 45%.
Уплотнение гидратированного оксида урана (IV)
Контроль за линейной усадкой спрессованной таблетки производился методом дилатометрии с учетом температуры. Полученная при этом соответствующая кривая показана на фиг. 6.
Эта кривая на фиг. 6 состоит из двух частей, из которых первая часть соответствует температуре от комнатной до около 800°С, т.е. это - температурный интервал, в котором происходит дегидратация гидратированного оксида урана (IV) с получением безводного оксида урана (IV), а вторая часть, соответствует температуре от 800 до около 1600°С, т.е. это - температурный интервал, в котором происходит спекание частиц безводного оксида урана (IV) с образованием таблетки.
Для сравнения на той же фиг. 6 построена кривая линейной усадки сравнительной таблетки, спрессованной при тех же условиях, что и предыдущая таблетка, но изготовленная из порошка, полученного способом, описанным в публикации N. Hingant и др. ("Preparation, sintering and leaching of optimized uranium thorium dioxides", Journal of Nuclear materials, 385 (2009), 400-406), [5].
Следует уточнить, что полученный способом из источника информации [5] порошок содержит частицы размером около 1 мкм и что эти частицы, полученные этим способом, агломерируют в виде квадратных пластинок со стороной от около 5 до 10 мкм. Это явление агломерации происходит как до сушки, так и после, сушка проводится на воздухе или даже в соответствии с признаками стадий с) и d) способа согласно изобретению.
Следовательно было отмечено, что скорость максимальной линейной усадки достигается при температуре выше или равной 1600°С для сравнительной таблетки, в то время как эта скорость достигается при температуре около 1200°С для таблетки, полученной способом согласно изобретению, т.е. выигрыш около 400°С.
Геометрические измерения позволили охарактеризовать таблетку, полученную способом согласно изобретению, в конце анализа методом дилатометрии. Эти геометрические измерения указывают на то, что полученный материал обладает плотностью 95%.
Этот факт подтверждается микрографическим снимком той же таблетки, полученной после термообработки, фиг. 7. На этом снимке четко видно, что материал, полученный после спекания таблетки из безводного оксида, обладал особой плотностью.
Пример 2. Синтез гидратированного оксида церия (IV)
Приготовление водного раствора, содержащего сульфат церия (IV)
Для синтеза гидратированного оксида церия (IV) приготовили водный раствор, содержащий сульфат церия (IV), растворением гидратированного сульфата церия в деионизированной воде с последующим разбавлением также деионизированной водой для доведения до должной концентрации.
Для сведения к минимуму ошибок в связи с гигроскопическим характером сульфата церия (IV) этот водный раствор, содержащий сульфат церия (IV), титровали методом атомной эмиссионной спектрометрии с индуктивно связанной плазмой (ICP-AES).
Синтез гидратированного оксида церия (IV)
Синтез гидратированного оксида церия (IV) проводили путем добавки в водный раствор сульфата церия, такой, как приготовленный выше, гидроксида аммония при молярном содержании 400% по отношению к молярному содержанию сульфата церия (IV) в водном растворе. Добавку производили при комнатной температуре и при перемешивании со скоростью 500 об./мин. Перемешивание длилось в течение одного часа.
В конце этого часа получили осадок. После фильтрации осадка анализ методом ICP-AES показал, что осаждение церия в количественном отношении произошло при эффективности осаждения более или равной 99,9%.
После нескольких промывок деионизированной водой и затем этанолом, что позволило удалить все следы остаточной кислоты, осадок отделили от жидкой фазы центрифугированием при скорости 4000 об./мин.
Полученный при этом осадок поместили в баллон с этанолом. Баллон, содержащий осадок и этанол, расположили в динамическом вакууме, составлявшем менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Осадок перемешивали при скорости 500 об./мин. и температуре 40°С для выпаривания этанола.
По окончании стадии выпаривания этанола получили порошок. Перемешивание этого порошка проводилось в течение 5 минут, затем вакуум нарушили.
Провели анализ порошка, полученного после вакуумной сушки осадка.
Удельная площадь поверхности порошка, измеренная методом BET при адсорбции азота при температуре кипения жидкого азота (-196°С), составила около 120 м2/г, которая соответствует значительной удельной площади поверхности и которая свидетельствует о сильной реакционной способности произведенного порошка.
Морфологический анализ данного порошка был проведен также методом сканирующей электронной микроскопии. На соответствующем снимке, представленном на фиг. 8, можно видеть, что порошок не агломерировался и образован из частиц нанометрического размера.
Для определения структуры частиц порошка, полученного из осадка, провели анализ методом рентгеновской дифрактометрии. Соответствующая дифрактограмма показана на фиг. 9, следует уточнить, что три наиболее высоких пика, соответствующих величинам угла 2θ 43°, 50° и 75°, отмечены звездочкой (*), они соответствуют интенсивности рентгеновских лучей, дифрагированных носителем порошковой пробы.
Отмечается, что полученная дифрактограмма выявляет кристаллографическую кубическую гранецентрированную структуру типа флюорита пространственной группы Fm-3m, которая является характерной для диоксида церия. С другой стороны, данная дифрактограмма также обнаруживает слабую кристалличность частиц, образующих порошок.
Дополнительный анализ методом спектрометрии комбинационного рассеяния (Рамановской) обнаружил отсутствие характерной полосы вибрации групп ОН и подтвердил присутствие гидратированного оксида.
Анализы методами рентгеновской дифрактометрии и спектрометрии комбинационного рассеяния позволяют объяснить явления во время реакции сульфата церия с гидроксидом аммония.
Действительно при контакте с гидроксидом аммония катионы Се4+, присутствующие в водном растворе церия (IV), переходят в осадок в виде гидроксида церия (IV) в соответствии со следующей химической реакцией (3):
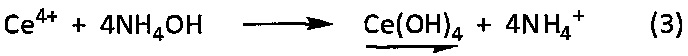
Однако такой осадок гидроксида церия (IV) является очень реакционноспособным соединением, образующим самопроизвольно гидратированный оксид церия (IV) в соответствии со следующей химической реакцией (4):
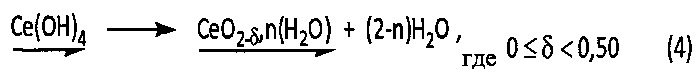
Пример 3. Синтез гидратированного оксида тория (IV)
Приготовление водного раствора, содержащего нитрат тория (IV)
Для синтеза гидратированного оксида тория (IV) водный раствор, содержащий нитрат тория (IV), приготовили путем растворения гидратированного нитрата тория в соляной кислоте с молярной концентрацией 6 моль/л.
Для минимизации ошибок в связи с гигроскопичной природой нитрата тория (IV) этот водный раствор, содержащий нитрат тория (IV), титровали методом атомной эмиссионной спектрометрии с индуктивно связанной плазмой (ICP-AES).
Синтез гидратированного оксида тория (IV)
Синтез гидратированного оксида тория (IV) проводили с добавкой в водный раствор нитрата тория, такого, как приготовленный выше, гидроксида аммония при молярном содержании 400% по отношению к молярному содержанию нитрата тория (IV) в водном растворе. Эта добавка происходила при комнатной температуре при перемешивании со скоростью 500 об./мин. Перемешивание длилось в течение часа.
В конце этого часа получили осадок. После фильтрации осадка анализ методом ICP-AES показал, что осаждение церия в количественном отношении произошло при эффективности осаждения более или равной 99,9%.
После нескольких промывок деионизированной водой и затем этанолом, позволивших удалить все следы остаточной кислоты, осадок отделили от жидкой фазы центрифугированием при скорости 4000 об./мин.
Полученный при этом осадок поместили в баллон с этанолом. Баллон, содержащий осадок и этанол расположили в динамическом вакууме, составившем менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Осадок перемешивали при скорости 500 об./мин. и температуре 40°С для выпаривания этанола.
По окончании стадии выпаривания этанола получили порошок. Перемешивание этого порошка проводилось в течение 5 минут, затем вакуум нарушили.
Провели анализ порошка, полученного после сушки осадка в вакууме.
Удельная площадь поверхности порошка, измеренная методом BET при адсорбции азота при температуре кипения жидкого азота (-196°С), составила более 150 м2/г, которая соответствует значительной удельной площади поверхности и которая свидетельствует о сильной реакционной способности произведенного порошка.
Как и в случае с порошками в примерах 1 и 2, полученными способом согласно изобретению, дополнительные анализы показали, что полученный порошок представляет собой гидратированный оксид тория (IV).
Действительно, при контакте с гидроксидом аммония катионы Th4+, присутствующие в водном растворе нитрата тория (IV), выпадают в осадок в виде гидроксида тория (IV) в соответствии со следующей химической реакцией (5):

Однако этот осадок гидроксида тория (IV) Th(OH)4 представляет собой очень реакционноспособное соединение, которое самопроизвольно образовывает гидратированный оксид тория (IV) в соответствии со следующей химической реакцией (6):
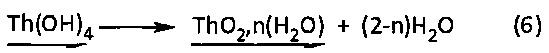
Пример 4. Синтез гидратированных и безводных смешанных оксидов урана (IV) и церия (IV)
Приготовление водных растворов
Приготовили водный раствор, содержащий хлорид урана (IV), путем растворения металлического урана U° в соляной кислоте (6М), как в примере 1.
Затем приготовили водный раствор, содержащий сульфат церия (IV), путем растворения гидратированного сульфата церия в деонизированной воде и разбавили также деонизированной водой для получения требуемой концентрации, как в примере 2.
Как было указано выше, каждый из водных растворов титровали колориметрическим анализом и методом ICP-AES.
Синтез гидратированных смешанных оксидов урана (IV) и церия (IV)
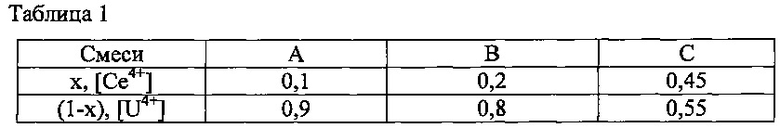
Три смеси, обозначенные, как А, В и С, были приготовлены на основе водных растворов, полученных на предыдущей стадии, при стехиометрии элементов церия и урана, обозначенных соответственно х и (1-х) и приведенных в нижеследующей таблице 1.
Синтез гитратированных смешанных оксидов урана (IV) и церия (IV) проводился с добавкой в каждую из смесей А, В и С, приготовленных, как указано выше, гидроксида аммония с молярным содержанием 400% от суммы молярных содержаний хлорида урана (IV) и сульфата церия (IV) в смесях. В трех опытах добавка гидроксида аммония производилась при комнатной температуре при перемешивании со скоростью 500 об./мин. Перемешивание происходило в течение часа.
В конце этого часа были получены осадки. После фильтрации этих осадков анализы каждого из фильтратов методом ICP-AES показали наличие количественного осадка урана и церия при эффективности осаждения более или равной 99,9%.
После нескольких промывок деонизированной водой и затем этанолом для удаления следов остаточной кислоты каждый из осадков был отделен от жидкой фазы центрифугированием при скорости 4000 об./мин.
Каждый из отделенных осадков приводили в контакт с растворителем, которым являлись либо вода, либо этанол.
Для этого каждый полученный осадок поместили в баллон с указанным растворителем. Баллон с осадком и растворителем разместили в динамическом вакууме, составившем менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Осадок перемешивали при скорости 500 об./мин. и температуре 40°С для выпаривания растворителя.
По окончании этой стадии выпаривания растворителя получили порошок. Перемешивание порошка длилось 5 минут, затем вакуум нарушили введением диазота N2 в баллон с помощью вакуумного распределительного устройства в целях исключения окисления урана (IV).
Полученные после стадии вакуумной сушки порошки каждого из осадков анализировали.
Удельные площади поверхности порошков, измеренные методом BET при адсорбции азота при температуре кипения жидкого азота (-196°С), находятся в диапазоне показателей от 100 до 150 м2/г в том случае, когда растворителем, с которым приводился в контакт каждый из осадков, являлся этанол. Такие показатели, свидетельствующие о большой удельной площади поверхности, указывают на большую реакционную способность порошков согласно изобретению, полученных из смесей А, В и С.
Однако отмечается, что в том случае, когда в качестве растворителя используется вода, показатели удельной площади поверхности меньше указанных выше, поскольку они располагаются в интервале от 10 до 30 м2/г.
Морфологический анализ трех порошков согласно изобретению, полученных после их контакта с этанолом в качестве растворителя, был проведен методом сканирующей электронной микроскопии. На соответствующих снимках, приведенных на фигурах 10А, 10В и 10С, можно видеть, что три порошка не агломерируют и содержат частицы нанометрического размера.
Для сравнения, на снимке на фиг. 10D, на котором показан порошок для сравнения, полученный на основе смеси А, но затем приведенный в контакт с водой в качестве растворителя, можно видеть агломерированный порошок, образованный агломератами микрометрического размера.
Для определения структуры частиц трех порошков согласно изобретению провели анализы методом рентгеновской дифрактометрии. Соответствующие дифрактограммы представлены на фиг. 11.
Отмечается, что на всех дифрактограммах на фиг. 11 присутствует твердое тело, имеющее кристаллографическую кубическую гранецентрированную структуру типа флюорита пространственной группы Fm-3m, которая является признаком смешанных диоксидов актинидов. Однако эти же дифрактограммы указывают и на слабую кристалличность частиц трех порошков.
Дополнительными анализами методом комбинационного рассеяния было выявлено отсутствие характерной полосы вибрации групп ОН и подтверждено наличие гидратированного смешанного оксида.
Анализы методами рентгеновской дифрактометрии и спектрометрии комбинационного рассеяния позволяют объяснить явления, происходящие при реакции хлорида урана (IV) и сульфата церия (IV) с гидроксидом аммония.
Действительно, при контакте с гидроксидом аммония катионы U4+ и Се4+, присутствующие в смесях А, В и С, выпадают в осадок в виде смешанного гидроксида урана (IV) и церия (IV) в соответствии со следующей химической реакцией (7):
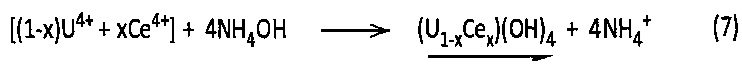
Однако этот осадок смешанного гидроксида урана (IV) и церия (IV) является очень реакционноспособным соединением, которое самопроизвольно образует гидратированный смешанный оксид урана (IV) и церия (IV) в соответствии со следующей химической реакцией (8):
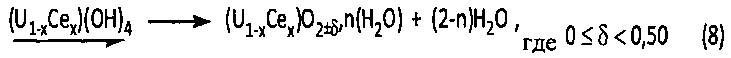
Прессование гидратированных смешанных оксидов урана (IV) и церия (IV) Порошки гидратированных смешанных оксидов урана (IV) и церия (IV) согласно изобретению, полученные из смесей А, В и С, прессовали одноосевым прессованием при давлении 500 МПа. Все полученные при этом сырые таблетки имели плотность от 40 до 45%.
Спекание гидратированных смешанных оксидов урана (IV) и церия (IV)
Контроль за линейной усадкой спрессованных таблеток проводился методом дилатометрии с учетом температуры. Полученные три соответствующие кривые показаны на фиг. 12.
Эти кривые на фиг. 12 состоят из двух частей, при этом первая часть соответствует температуре от комнатной до температуры около 900°С, что образует температурный интервал, в котором происходит дегидратация смешанных оксидов урана (IV) и церия (IV) и их превращение в безводные смешанные оксиды урана (IV) и церия (IV), вторая часть соответствует температуре от около 900°С до температуры около 1600°С, что образует температурный диапазон, в котором происходит спекание частиц безводных смешанных оксидов урана (IV) и церия (IV), каждый из которых образует таблетки.
Для сравнения на той же фиг.12 построена кривая линейной усадки эталонной таблетки, уплотненной при тех же условиях, что и таблетки смешанных оксидов урана (IV) и церия (IV), но изготовленной из порошка, полученного описанным в источнике [5] способом.
Отмечается, что максимальная скорость линейной усадки сравнительной таблетки происходит при температуре около 1600°С, в то время как та же усадка таблетки, изготовленной способом согласно изобретению, происходит при температуре около 1100-1200°С, т.е. выигрыш составляет от 400 до 500°С.
Геометрические измерения позволили охарактеризовать таблетку, полученную из смеси А (х=0,1) способом согласно изобретению, после анализа методом дилатометрии. Такие геометрические замеры показали, что полученный материал обладает плотностью 95%.
Этот вывод подтверждается приведенным на фиг. 13 микрографическим снимком той же таблетки, полученной после термообработки. На этом снимке отчетливо видно, что материал, полученный после спекания таблетки из безводного смешанного оксида урана (IV) и церия (IV) (при х=0,1), обладает особой плотностью.
Пример 5. Синтез гидратированного смешанного оксида церия (IV) и гадолиния (III).
Приготовление водных растворов
Водный раствор, содержащий хлорид церия (III), приготовили растворением гидратированного хлорида церия (III) в деионизированной воде и разбавили также деионизированной водой для получения требуемой концентрации.
Водный раствор, содержащий хлорид гадолиния (III), приготовили растворением гидратированного хлорида гадолиния (III) в деионизированной воде и разбавили также деионизированной водой для получения требуемой концентрации.
Как указано выше, каждый из этих водных растворов титровали методом колориметрического анализа и методом ICP-AES.
Синтез гидратированного смешанного оксида церия (IV) и гадолиния (III)
Смесь D приготовили из водных растворов, таких, как приготовленные на предыдущей стадии, при молярном содержании гадолиния 20%, обозначенном z, при z=0,2.
Синтез гидратированного смешанного оксида церия (IV) и гадолиния (III) проводили с добавкой в смесь D, приготовленную, как указано выше, гидроксида аммония при молярном содержании 400% от суммы молярных содержаний хлорида церия (III) и хлорида гадолиния (III), содержавшихся в смеси D. Добавку гидроксида аммония производили при комнатной температуре при перемешивании со скоростью 500 об./мин. Перемешивание происходило в течение часа.
В конце этого часа получили осадок. После фильтрации осадка анализ фильтрата методом ICP-AES показал количественное осаждение церия и гадолиния при эффективности осаждения более или равной 99,9%.
После нескольких промывок деионизированной водой и затем этанолом, что позволило удалить все следы остаточной кислоты, осадок отделили от жидкой фазы центрифугированием со скоростью 4000 об./мин.
Полученный при этом осадок ввели в баллон с этанолом. Баллон, содержащий осадок и этанол поместили в динамический вакуум, составлявшем менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Осадок перемешивали со скорости 500 об./мин. при температуре 40°С для выпаривания этанола.
В конце стадии выпаривания этанола получили порошок. Перемешивание порошка проводилось в течение 5 минут, затем вакуум нарушили.
Произвели анализ порошка, полученного из осадка после вакуумной сушки. Удельная площадь поверхности, измеренная методом BET при адсорбции азота при температуре кипения жидкого азота (-196°С), находилась в диапазоне значений от 100 до 130 м2/г. Эта величина, соответствующая большой удельной площади поверхности, свидетельствует о сильной реакционной способности полученного порошка.
Микрографический анализ порошка проводился методом сканирующей электронной микроскопии. На соответствующем снимке на фиг. 14 можно видеть, что порошок не агломерирован и образован из частиц нанометрического размера.
Для определения состава частиц данного порошка провели анализ методом рентгеновской дифрактоскопии. Соответствующая дифрактограмма представлена на фиг. 15.
Отмечается, что на дифрактограмме на фиг. 15 прослеживается присутствие твердого тела, имеющего кристаллографическую кубическую гранецентрированную структуру типа флюорита, пространственной группы Fm-3m, что является характерным для диоксида церия. Вместе с тем эта дифрактограмма выявляет также низкую кристалличность частиц порошка.
Дополнительные анализы методом спектрометрии комбинационного рассеяния (Рамановской) показали отсутствие характерной полосы вибрации групп ОН и подтвердили наличие гидратированного смешанного оксида.
Эти анализы, проведенные методами рентгеновской дифрактометрии и спектрометрии комбинационного рассеяния, позволяют объяснить явления во время реакции хлорида церия (III) и хлорида гадолиния (III) с гидроксидом аммония.
Действительно, при контакте с гидроксидом аммония катионы Се3+ и Gd3+, содержащиеся в смеси D, осаждаются в виде смешанного гидроксида церия (III) и гадолиния (III) в соответствии со следующей химической реакцией (9):

Однако этот осадок смешанного гидроксида церия (III) и гадолиния (III) является очень реакционноспособным соединением, способным самопроизвольно образовывать гидратированный смешанный оксид церия (IV) и гадолиния (III) в соответствии со следующей химической реакцией (10):
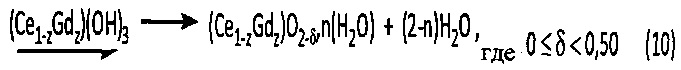
Пример 6. Синтез гидратированного смешанного оксида тория (IV) и урана (IV)
Приготовление водных растворов
Водный раствор, содержащий нитрат урана (IV), приготовили путем растворения урана в виде UO4, 4Н2О в азотной кислоте (6 М).
Водный раствор, содержащий торий (IV), приготовили путем растворения гидратированного нитрата тория в соляной кислоте с молярной концентрацией 6 моль/л.
Для сведения к минимуму ошибок в связи с гигроскопичной природой тория (IV) этот водный раствор, содержащий нитрат тория (IV), титровали методом атомной эмиссионной спектрометрии с индуктивной связанной плазмой (ICP-AES).
Синтез гидратированного смешанного оксида тория (IV) и урана (VI)
Смесь Е получили на основе водных растворов, таких, как приготовленные на предыдущей стадии, с соответствующей стехиометрией элементов торий и уран от 0,8 до 0,2.
Синтез гидратированного смешанного оксида тория (IV) и урана (VI) проводился при добавке в приготовленную выше смесь Е гидроксида аммония с молярным содержанием 400% от суммы молярных содержаний нитрата тория (IV) и нитрата урана (VI), содержавшихся в смеси Е. Эта добавка гидроксида аммония проводилась при комнатной температуре при перемешивании со скоростью 500 об./мин. Перемешивание длилось в течение часа.
В конце этого часа получили осадок. После его фильтрации анализы фильтрата методом ICP-AES показали количественное осаждение урана и тория при эффективности осаждения более или равной 99,9%.
После нескольких промывок деионизированной водой и затем этанолом, что позволило удалить все следы остаточной кислоты, осадок отделили от жидкой фазы центрифугарованием со скоростью 4000 об./мин.
Полученный при этом осадок ввели в баллон с этанолом. Баллон, содержащий осадок и этанол, поместили в динамический вакуум, составивший менее 100 Па (1 мбар), с помощью вакуумного распределительного устройства. Осадок перемешивали со скорости 500 об./мин. при температуре 40°С для выпаривания этанола.
В конце стадии выпаривания этанола получили порошок. Перемешивание порошка проводилось в течение 5 минут, затем вакуум нарушили введением в баллон диазота N2 посредством вакуумного распределительного устройства.
Провели анализ порошка, полученного из осадка после вакуумной сушки.
Удельная площадь поверхности этого порошка, измеренная методом BET при адсорбции азота при температуре кипения жидкого азота (-196°С) составила более 100 м2/г. Эта величина, соответствующая большой удельной площади поверхности, свидетельствует о сильной реакционной способности порошка, полученного из смеси Е.
Морфологический анализ порошка проводился также методом сканирующей электронной микроскопии. На соответствующем снимке на фиг. 16 можно видеть, что порошок не агломерирует и образован из частиц нанометрического размера.
Для определения структуры частиц данного порошка провели анализ методом рентгеновской дифрактоскопии. Соответствующая дифрактограмма представлена на фиг. 17.
Отмечается, что на дифрактограмме на фиг. 17 прослеживается присутствие твердого тела, имеющего кристаллографическую кубическую гранецентрированную структуру типа флюорита, пространственной группы Fm-3m, что является характерным для смешанных диоксидов актинидов. Вместе с тем, эта дифрактограмма выявляет слабую кристалличность образующих порошок частиц.
Библиография:
[1] GB 1,128,838
[2] US 4,314,952
[3] US 4,382,885
[4] US 4,971,734
[5] N. Hingant и др. Journal of Nuclear Materials, 2009, 385, стр. 400-406.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОРОШОК ОКСИДА МЕТАЛЛА, ПОРОШОК ОКСИДА ТИТАНА, СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА ОКСИДА МЕТАЛЛА | 1994 |
|
RU2127221C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА, ВКЛЮЧАЮЩЕГО ТВЕРДЫЙ РАСТВОР ДИОКСИДА УРАНА И ДИОКСИДА ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ДРУГОГО АКТИНИДА И/ИЛИ ЛАНТАНИДА | 2014 |
|
RU2662526C2 |
| ЧАСТИЦЫ ОКСИДА ЦЕРИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2017 |
|
RU2746315C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИТОВ ИЗ ОКСИДА АЛЮМИНИЯ И СМЕШАННЫХ ОКСИДОВ ЦЕРИЯ И ЦИРКОНИЯ | 2012 |
|
RU2590162C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА, СОДЕРЖАЩЕГО ЧАСТИЦЫ ОКТАОКСИДА ТРИУРАНА И ЧАСТИЦЫ ДИОКСИДА ПЛУТОНИЯ | 2018 |
|
RU2767779C2 |
| ПОДЛОЖКА КАТАЛИЗАТОРА ИЗ ОКСИДА АЛЮМИНИЯ, УСТОЙЧИВАЯ К СЕРЕ | 2011 |
|
RU2615991C2 |
| КОМПОЗИЦИЯ НА ОСНОВЕ НАНОМЕТРИЧЕСКОГО ОКСИДА ЦЕРИЯ НА НОСИТЕЛЕ С ПОВЫШЕННОЙ ВОССТАНОВИТЕЛЬНОЙ СПОСОБНОСТЬЮ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ КАТАЛИЗАТОРА | 2007 |
|
RU2411995C2 |
| Способ получения оксида скандия | 2015 |
|
RU2608033C1 |
| ПОРИСТЫЙ НЕОРГАНИЧЕСКИЙ КОМПОЗИТНЫЙ ОКСИД | 2011 |
|
RU2606505C2 |
| СПОСОБ СООСАЖДЕНИЯ АКТИНОИДОВ И СПОСОБ ПОЛУЧЕНИЯ СМЕШАННЫХ ОКСИДОВ АКТИНОИДОВ | 2001 |
|
RU2282590C2 |
Изобретение относится к способу получения порошка оксида по меньшей мере одного металла, при этом степень окисления каждого металла составляет от (III) до (VI). Способ включает в себя последовательно в следующем порядке: а) проведение реакции водного раствора, содержащего, для каждого металла, по меньшей мере одну соль с катионом этого металла, с соединением, содержащим гидроксид, b) отделение полученного осадка, с) приведение в контакт отделенного осадка с органическим протонным полярным растворителем, d) удаление органического протонного полярного растворителя путем вакуумной сушки осадка. Изобретение относится также к способу изготовления таблетки из оксида по меньшей мере одного металла, к порошку и таблетке, получаемым этим способом, а также к их применению. 9 н. и 18 з.п. ф-лы, 1 табл., 17 ил.
1. Способ получения порошка оксида по меньшей мере одного металла, при этом степень окисления каждого металла составляет от (III) до (VI), который включает в себя последовательно в следующем порядке:
а) проведение реакции водного раствора, содержащего, для каждого металла, по меньшей мере одну соль с катионом этого металла, с соединением, содержащим по меньшей мере один анион гидроксида ОН- и по меньшей мере один катион, выбранный из катиона аммония NH4+, катиона гидразиния N2H5+, катиона щелочного металла и катиона щелочноземельного металла, с получением осадка гидратированного оксида указанного по меньшей мере одного металла,
b) отделение полученного осадка,
с) приведение в контакт отделённого осадка со спиртом,
d) удаление спирта вакуумной сушкой осадка с получением порошка гидратированного оксида указанного по меньшей мере одного металла, при этом указанный порошок образован из частиц, средний диаметр которых менее или равен 1 мкм.
2. Способ по п. 1, в котором порошок гидратированного оксида указанного по меньшей мере одного металла образован из частиц, средний диаметр которых менее или равен 100 нм, предпочтительно менее или равен 20 нм, более предпочтительно менее или равен 10 нм.
3. Способ по п. 1 или 2, в котором порошок гидратированного оксида указанного по меньшей мере одного металла имеет удельную площадь поверхности, измеренную методом ВЕТ, превышающую или равную 30 м2/г, предпочтительно превышающую или равную 80 м2/г, более предпочтительно превышающую или равную 100 м2/г.
4. Способ по любому из пп. 1-3, который дополнительно включает после стадии b) и перед стадией с) стадию промывки отделённого осадка, причём указанная промывка, в частности, проводится протонным растворителем при необходимости в смеси с водой, предпочтительно являющейся деионизированной.
5. Способ по любому из пп. 1-4, который дополнительно включает после стадии d):
e) термообработку порошка гидратированного оксида указанного по меньшей мере одного металла с получением порошка безводного оксида указанного по меньшей мере одного металла.
6. Способ по любому из пп. 1-5, в котором каждый металл выбран из актинидов, лантанидов и переходных металлов.
7. Способ по п. 6, в котором металл, являющийся актинидом, выбран из химических элементов группы, состоящей из: U, Th, Pu, Np, Am и Сm.
8. Способ по п. 6, в котором металл, являющийся лантанидом, выбран из химических элементов группы, состоящей из: Ce, Gd, Nd, Sm и Еu.
9. Способ по п. 6, в котором металл, являющийся переходным металлом, выбран из химических элементов группы, состоящей из: Ti, Cr, Zr, Sc, Y и Hf.
10. Способ по любому из пп. 1-9, в котором гидратированный оксид указанного по меньшей мере одного металла является простым оксидом, причём этот оксид предпочтительно выбран из UO2+δ, UO3, U3O8, CeO2-δ, ThO2, PuO2-δ, NpO2+δ, ZrO2 и HfO2.
11. Способ по любому из пп. 1-9, в котором гидратированный оксид указанного по меньшей мере одного металла является смешанным оксидом, предпочтительно выбранным из: (U,Ce)O2±δ, (U,Pu)O2±δ, (U,Am)O2±δ, (U,Th)O2+δ, (Ce,Gd)O2-δ, (U,Gd)O2±δ, (Th,Pu)O2-δ, (Th,Y)O2-δ и (U,Pu,Am)O2±δ.
12. Способ по любому из пп. 1-11, в котором для каждого металла соль с катионом указанного по меньшей мере одного металла выбрана из сульфата, нитрата, галогенида, такого, как хлорид или бромид.
13. Способ по любому из пп. 1-12, в котором соединение, используемое на стадии (а), представляет собой гидроксид аммония или гидроксид гидразиния.
14. Способ по любому из пп. 1-13, в котором молярное содержание соединения, используемого на стадии (а), является избыточным по сравнению с общим молярным содержанием катиона (катионов) указанного по меньшей мере одного металла, причём это молярное содержание указанного соединения предпочтительно составляет от 150 до 600%, предпочтительно от 300 до 500%, от общего молярного содержания катиона (катионов) указанного по меньшей мере одного металла.
15. Способ по любому из пп. 1-14, в котором спирт представляет собой одноатомный спирт или диол.
16. Способ по п. 15, в котором спирт выбран из группы, состоящей из метанола, этанола и этандиола.
17. Способ по любому из пп. 1-16, в котором вакуумная сушка проводится посредством вакуумного распределительного устройства.
18. Порошок гидратированного оксида по меньшей мере одного металла, причём степень окисления каждого металла составляет от (III) до (VI), полученный способом по любому из пп. 1–17, при этом указанный порошок образован из частиц, средний диаметр которых меньше или равен 1 мкм и удельная площадь поверхности которых, измеренная методом ВЕТ, превышает или равна 80 м2/г.
19. Применение порошка по п. 18 в производстве ядерного топлива.
20. Применение порошка по п. 18 в качестве носителя катализатора.
21. Применение порошка по п. 18, в котором металлом является уран, для приготовления октаоксида триурана U3O8.
22. Применение порошка по п. 18 в способе гидрофторирования.
23. Применение порошка по п. 18 для изготовления ионного проводника.
24. Применение порошка по п. 18 для производства керамического изделия.
25. Способ изготовления таблетки из оксида по меньшей мере одного металла, причём степень окисления каждого металла составляет от (III) до (VI), при этом способ включает в себя последовательно в следующем порядке:
1) получение порошка оксида по меньшей мере одного металла, при этом степень окисления каждого металла составляет от (III) до (VI), способом по любому из пп. 1-17,
2) прессование указанного порошка и
3) термообработку спрессованного порошка с получением таблетки из оксида по меньшей мере одного металла.
26. Способ по п. 25, в котором таблетка из оксида по меньшей мере одного металла обладает плотностью по меньшей мере 90%, предпочтительно по меньшей мере 95%.
27. Способ по п. 25 или 26, в котором стадия 3) термообработки проводится с использованием температурного градиента в диапазоне от комнатной температуры до температуры менее или равной 1600°С, предпочтительно менее или равной 1400°С.
| US 4382885 A, 10.05.1983 | |||
| US 3963828 A, 15.06.1976 | |||
| СПОСОБ ПОЛУЧЕНИЯ ФРИТТИРОВАННЫХ ТАБЛЕТОК ОКИСЕЙ | 1989 |
|
RU2012071C1 |
| Устройство для когерентного сложения сигналов с угловой модуляцией | 1972 |
|
SU478442A1 |
| ИНГИБИТОРЫ АСПАРТАТ-ПРОТЕАЗЫ | 2006 |
|
RU2424231C2 |
| FR 1489438 A, 21.07.1967 | |||
| US 3923933 A, 02.12.1975 | |||
| Способ получения производных 2этинилпиперазина | 1973 |
|
SU491635A1 |

