Область техники, к которой относится изобретение
Данное изобретение относится к области переработки отработавших ядерных топлив.
Более конкретно, данное изобретение относится к способу, позволяющему получать порошок, содержащий однородную смесь частиц октаоксида триурана U3O8 и частиц диоксида плутония PuO2, который дополнительно может содержать частицы диоксида четырёхвалентного актинида, выбранного из тория и нептуния, при использовании водных потоков, образующихся в результате гидрометаллургической обработки отработавших ядерных топлив.
Полученный порошок может находить применение в особенности для получения свежих ядерных топлив типа MOX (смешанное оксидное топливо), которые можно облучать, например, в легководных реакторах (реакторы LWR) или реакторах на быстрых нейтронах (реакторы FNR).
Предшествующий уровень техники
В настоящее время обработка/переработка отработавших ядерных топлив для повторного использования содержащегося в них плутония и получения топлива MOX, включает в себя, в частности, следующие стадии:
1. растворение отработавших ядерных топлив в азотной кислоте для переведения в растворимое состояние различных актинидов и продуктов деления, присутствующих в указанных топливах;
2. разделение/очистка урана и плутония для получения двух водных потоков, одного, содержащего очищенный уран (VI), и другого, содержащего очищенный плутоний (IV);
3. превращение урана и плутония, присутствующих в упомянутых водных потоках, в две отдельные твёрдые фазы диоксидного типа, UO2 и PuO2; и
4. изготовление таблеток топлива MOX способом, последовательно включающим в себя стадии смешивания/прессования/спекания диоксидов UO2 и PuO2 вместе со смесью, которая может необязательно дополняться шамотом (отходы производства таблеток).
Один из предусматриваемых путей усиления противостояния цикла ядерного топлива риску присвоения плутония в незаконных целях заключается в максимально возможном ограничении количества стадий с использованием только одного очищенного плутония, в частности, на стадии превращения водных потоков, содержащих уран (VI) и плутоний (IV) в форме нитратов, в диоксиды, как описано в международной заявке PCT WO 2007/13517, далее в настоящем документе ссылка [1].
Было предложено несколько способов совместной переработки урана и плутония, конкретно на основе получения стабильного водного раствора урана (VI) и плутония (IV) до стадии превращения. Превращение урана (VI) и плутония (IV), присутствующих в указанном водном растворе, предназначено для получения смешанного оксидного порошка (U,Pu)O2 или чаще порошка, образованного однородно смешанными частицами UO2 и PuO2, в качестве конечного продукта. Главное различие между разными способами превращения, которые разработаны в пилотном или промышленном масштабе, сопряжено со стадией превращения актинидов, находящихся в растворе, в твёрдую фазу. Данная стадия основывается либо на способе типа термического деазотирования, либо на способе типа осаждения/прокалки.
В отношении способов термического деазотирования для совместной переработки урана/плутония можно упомянуть способы, описанные авторами Numao et al. (материалы конференции GLOBAL 2007: Advanced Nuclear Fuel Cycles and Systems, Бойсе, США, 9-13 сентября 2007 г., далее в настоящем документе ссылка [2]) и авторами Felker et al. (материалы конференции ATALANTE 2008: Nuclear Fuel Cycles for a Sustainable Future, Монпелье, Франция, 19-23 мая 2008 г., далее в настоящем документе ссылка [3]).
Указанные способы являются в целом относительно компактными, но требуют предварительного интенсивного концентрирования водных растворов актинидов. В дополнение к этому, трудно контролировать характеристики оксидных порошков, получаемых для непосредственного применения, и необходимо прибегать к многочисленным обработкам (размалывание, гранулирование, …) перед формованием топлива.
В аспекте способов с использованием осаждения/прокалки для совместной переработки урана/плутония встречаются три типа:
- способы, прибегающие к соосаждению урана и плутония в форме двойных карбонатов аммония;
- способы, прибегающие к аммиачному соосаждению;
- способы, прибегающие к оксалатному соосаждению.
Соосаждение урана и плутония в форме двойных карбонатов аммония можно применять только в случае шестивалентных актинидов. Ввиду указанной причины это требует осуществления трудной стадии доведения плутония (IV) по валентности до плутония (VI). В дополнение к этому, реакция соосаждения не является количественной вследствие довольно заметной растворимости двойных карбонатов аммония в водной среде, что приводит к низким выходам.
Аммиачное соосаждение обладает большим недостатком, заключающимся в том, что обращение с потоками нитрата аммония является проблематичным.
Оксалатное соосаждение основывается либо на соосаждении актинидов только в степени окисления IV, как описано в международной заявке PCT WO 02/28778, далее в настоящем документе ссылка [4], либо на соосаждении актинидов в степенях окисления III и IV, как описано в международной заявке PCT WO 2005/119699, далее в настоящем документе ссылка [5].
В случае оксалатного соосаждения актинидов только в степени окисления IV проблема заключается в совместном удерживании урана и плутония в степени окисления IV, хотя уран IV является мощным восстановителем плутония (IV) в водной среде. Следовательно, необходимо добавлять один или несколько сильных комплексообразователей, способных стабилизировать катионы двух упомянутых металлов в степени окисления IV до оксалатного соосаждения. Указанная стабилизация, хотя и является эффективной, ограничивает рамки рабочих условий способа, в частности, в отношении контролирования уровня pH. В дополнение к этому, данный способ влечёт за собой добавление больших количеств комплексообразователя (комплексообразователей) в водный раствор, с сопутствующим добавлением основания, например, гидроксида натрия или аммиака. Это приводит к осложнению управления образующимися потоками.
В случае оксалатного соосаждения актинидов в степенях окисления III и IV, оно позволяет количественно осаждать данные актиниды и приводит к образованию смешанного оксида типа твёрдого раствора (ссылка [5]). Однако в такое соосаждение вовлечена потенциально проблематичная стадия восстановления плутония (IV) до плутония (III) и стабилизации последнего. В дополнение к этому, максимально достижимое отношение Pu/(U+Pu) ограничивается значением около 50 атомных % с учётом фазовой диаграммы полученного смешанного оксалата гексагональной формы.
В рамках других вариантов применения оксалатное осаждение урана (VI) и плутония (IV) в водном растворе описано авторами Delegard et al. (PNNL-13934, 2002, далее в настоящем документе ссылка [6]) для отделения урана от плутония за счёт использования высокой растворимости оксалата урана (VI) в воде по сравнению с низкой растворимостью плутония (IV) в воде.
И наконец, можно отметить применение на основе оксалатного соосаждения урана (IV) и тория (IV), для приготовления порошка смешанного оксида (U,Th)O2 (Atlas et al., Journal of Nuclear Materials, 2001, 294, 344-348, далее в настоящем документе ссылка [7]).
Следовательно, учитывая вышеизложенное, авторы настоящего изобретения ставили целью разработку способа превращения урана и плутония, который позволяет и расширять рамки рабочих условий для указанного превращения, и упрощать его воплощение.
Как часть своих исследований, авторы настоящего изобретения вопреки всем ожиданиям выявили, что можно получать водную суспензию, которая, хотя и содержит частицы и оксалата урана (IV), и оксалата плутония (IV), является стабильной и однородной, преодолевая за счёт этого определённое число рабочих ограничений, с которыми сталкиваются до сих пор в случае совместного превращения урана и плутония.
Данное изобретение основывается на обнаружении упомянутых неожиданных экспериментальных фактов.
Раскрытие изобретения
С учётом вышесказанного, объектом изобретения является способ получения порошка, содержащего частицы октаоксида триурана U3O8 и частицы диоксида плутония PuO2, который включает в себя:
a) получение водной суспензии S1 частиц оксалата урана (IV) и водной суспензии S2 частиц оксалата плутония (IV) при помощи реакций оксалатного осаждения;
b) смешивание водной суспензии S1 с водной суспензией S2 для получения водной суспензии S1+2, содержащей частицы оксалата урана (IV) и частицы оксалата плутония (IV);
c) разделение водной суспензии S1+2 на водную фазу и твёрдую фазу, содержащую частицы оксалата урана (IV) и частицы оксалата плутония (IV); и
d) прокаливание твёрдой фазы для превращения (1) частиц оксалата урана (IV) в частицы октаоксида триурана и (2) частиц оксалата плутония (IV) в частицы диоксида плутония, в результате чего образуется порошок;
и при этом стадии b) и c) осуществляют одновременно или последовательно.
Согласно данному изобретению стадия a) предпочтительно включает в себя:
- приведение водного раствора A1, содержащего азотную кислоту и нитрат урана (IV), или урановый нитрат, в контакт с водным раствором A2, содержащим осадитель, выбранный из щавелевой кислоты, её солей (например, оксалата аммония) и алкилпроизводных (например, диметилоксалата), для образования реакционной среды, в которой уран (IV) осаждается в форме оксалата урана (IV); и
- приведение водного раствора A’1, содержащего азотную кислоту и нитрат плутония (IV), в контакт с водным раствором A’2, содержащим осадитель, выбранный из щавелевой кислоты, её солей и алкилпроизводных, для образования реакционной среды, в которой плутоний (IV) осаждается в форме оксалата плутония (IV).
Водные растворы A1 и A’1 предпочтительно содержат от 0,5 моль/л до 5 моль/л азотной кислоты.
В рамках данного изобретения концентрация урана (IV) в водном растворе A1 и концентрация плутония (IV) в водном растворе A’1 могут изменяться в широких пределах, но предпочтительно указанные концентрации составляют от 0,001 моль/л до 1 моль/л.
Концентрация осадителя в водных растворах A2 и A’2 обычно составляет от 0,05 моль/л до 1 моль/л.
Указанная концентрация предпочтительно выбрана так, что с учётом объёмных соотношений водных растворов A1 и A’1, соответственно, к водным растворам A2 и A’2, соответственно, которые используются для приведения в контакт с указанными растворами, осадитель присутствует в реакционных средах в избытке относительно стехиометрических условий реакций оксалатного осаждения урана (IV) и плутония (IV).
Как правило, упомянутый избыток определяется так, чтобы в конце реакций оксалатного осаждения в реакционных средах достигалась остаточная концентрация оксалат-ионов, находящаяся в диапазоне от 0,01 моль/л до 0,5 моль/л.
На стадии a) реакционные среды предпочтительно поддерживают при температуре, находящейся в диапазоне от 10°C до 60°C, на протяжении всего периода выполнения процедур оксалатного осаждения.
В данном изобретении водный раствор A1 может дополнительно содержать соединение, способное стабилизировать уран в степени окисления IV и, следовательно, предотвращать его окисление в уран (VI) некоторыми соединениями, присутствующими в реакционной среде (азотная кислота, азотистая кислота, …), или окружающей атмосферой (например, кислородом окружающего воздуха).
Стабилизирующее соединение предпочтительно представляет собой соединение, которое высвобождает в водном растворе однозарядный катион, содержащий только атомы углерода, водорода, кислорода и/или азота (катион CHON). Например, упомянутое соединение является антиазотистым реагентом, таким как соль гидразиния или алкилгидразиния, которую используют предпочтительно в концентрации от 0,05 моль/л до 0,2 моль/л.
К тому же, водный раствор A’1 дополнительно может содержать нитрат урана (VI) или уранилнитрат, в таком случае концентрация данного нитрата в растворе A’1 обычно составляет от 0,001 моль/л до 0,05 моль/л.
Для обеспечения стабильности условий химического осаждения в отношении содержания свободной азотной кислоты и избытка оксалата, и посредством этого стабильного однородного распределения по размерам частиц оксалата урана (IV) и частиц оксалата плутония (IV), стадия a) предпочтительно включает в себя:
- добавление водных растворов A1 и A2 к третьему водному раствору, содержащему азотную кислоту и осадитель, идентичный осадителю водного раствора A2; и
- добавление водных растворов A’1 и A’2 к четвёртому водному раствору, содержащему азотную кислоту и осадитель, идентичный осадителю водного раствора A’2.
В данном случае, если в растворе A1 присутствует реагент, стабилизирующий уран (IV), то указанный стабилизирующий реагент предпочтительно также присутствует в третьем водном растворе. Аналогичным образом, если в водном растворе A’1 присутствует нитрат урана (VI), то нитрат урана (VI) предпочтительно также присутствует в четвёртом водном растворе.
Приготовление водных суспензий S1 и S2 можно проводить в реакторе любого типа, подходящем для осаждения актинидов, например, реакторе вихревого типа, используемом в ядерной промышленности, или реакторе с псевдоожиженным слоем, таком, как описан в международной заявке PCT WO 2010/070064, далее в настоящем документе ссылка [8].
Согласно данному изобретению, стадию b), соответствующую смешиванию водной суспензии S1 с водной суспензией S2, можно осуществлять с использованием любой процедуры, позволяющей плотно контактировать указанным водным суспензиям, а следовательно, частицам оксалата, содержащимся в них, и аналогичным образом, стадию c), соответствующую разделению водной суспензии S1+2, полученной на стадии b), на водную фазу и твёрдую фазу, можно осуществлять с использованием любой методики разделения твёрдое тело-жидкость, например, путём фильтрования, в частности, фильтрования под действием вакуума или давления, или путём центрифугирования.
Стадии b) и c) предпочтительно выполняют одновременно и, если это другой случай, то стадию c) предпочтительно осуществляют максимум в пределах 10 часов после стадии b).
В любом случае, вследствие сходности морфологических и структурных характеристик оксалата урана (IV) и оксалата плутония (IV), смешивание частиц данных оксалатов происходит легко и однородно. Указанная однородность далее обнаруживается в распределении частиц оксалата урана (IV) и частиц оксалата плутония (IV) в твёрдой фазе, получаемой в конце стадии c). В результате, любое последующее разделение твёрдых частиц урана и плутония становится практически невозможным.
Прокалку твёрдой фазы, полученной по завершении стадии c), проводят предпочтительно при температуре, по меньшей мере, 550°C и предпочтительно не выше 1 250°C и в окислительной атмосфере, например, воздухе или смеси кислорода и азота.
Таким путём получается порошок, содержащий равномерную, однородную смесь частиц октаоксида триурана и частиц диоксида плутония, при этом гомогенность, достигаемая на стадиях b) и c), сохраняется в смеси оксидов урана и плутония, получаемой на стадии d).
Как указано ранее, способ настоящего изобретения позволяет также получать порошок, содержащий частицы диоксида актинида (IV), выбранного из тория и нептуния, в дополнение к частицам октаоксида триурана и диоксида плутония.
Для воплощения этого существуют два варианта порядка выполнения процедур:
- либо приготовляют водную суспензию S3 частиц актинида (IV), которую затем смешивают с водными суспензиями S1 и S2;
- либо нитрат актинида (IV) добавляют к водному раствору A1, содержащему нитрат урана (IV), в этом случае получают водную суспензию S1, содержащую двойной оксалат урана (IV) и актинида (IV), который затем смешивают с водной суспензией S2.
С учётом вышесказанного, первый вариант порядка выполнения процедур данного способа включает в себя:
a’) приготовление водной суспензии S1 частиц оксалата урана (IV), водной суспензии S2 частиц оксалата плутония (IV) и водной суспензии S3 частиц оксалата актинида (IV) при помощи реакций оксалатного осаждения;
b’) смешивание водных суспензий S1, S2 и S3 друг с другом для получения водной суспензии S1+2+3, содержащей частицы оксалата урана (IV), частицы оксалата плутония (IV) и частицы оксалата актинида (IV);
c’) разделение водной суспензии S1+2+3 на водную фазу и твёрдую фазу, содержащую частицы оксалата урана (IV), частицы оксалата плутония (IV) и частицы оксалата актинида (IV); и
d’) прокаливание твёрдой фазы для превращения частиц оксалата урана (IV) в частицы октаоксида триурана, частиц оксалата плутония (IV) в частицы диоксида плутония и частиц оксалата актинида (IV) в частицы диоксида актинида (IV);
при этом стадии b’) и c’) выполняют одновременно или последовательно.
В указанном случае водную суспензию частиц оксалата актинида (IV) получают в соответствии с теми же методиками, что и описанные ранее для приготовления водных суспензий S1 и S2, но с использованием водного раствора нитрата указанного актинида (IV) взамен и вместо водных растворов A1 и A’1.
К тому же, стадии b’), c’) и d’) осуществляют в соответствии с теми же методами, что и описанные ранее для проведения стадий b), c) и d).
В случае второго варианта порядка выполнения процедур данный способ включает в себя:
a’’) приготовление водной суспензии S1 частиц двойного оксалата урана (IV) и актинида (IV), а также водной суспензии S2 частиц оксалата плутония (IV) при помощи реакций оксалатного осаждения;
b’’) смешивание водной суспензии S1 с водной суспензией S2 для получения водной суспензии S1+2, содержащей частицы двойного оксалата урана (IV) и актинида (IV) и частицы оксалата плутония (IV);
c’’) разделение водной суспензии S1+2 на водную фазу и твёрдую фазу, образованную частицами двойного оксалата урана (IV) и актинида (IV) и частицами оксалата плутония (IV); и
d’’) прокаливание твёрдой фазы для превращения (1) частиц двойного оксалата урана (IV) и актинида (IV) в частицы октаоксида триурана и диоксида актинида (IV), и (2) частиц оксалата плутония (IV) в частицы диоксида плутония, в результате чего получают порошок;
при этом стадии b’’) и c’’) выполняют одновременно или последовательно.
В указанном случае водную суспензию S1 получают в соответствии с теми же методиками, что и описанные ранее для приготовления водной суспензии S1, но с использованием водного раствора, в котором часть нитрата урана (IV) заменена нитратом актинида (IV).
Кроме того, стадии b’’), c’’) и d’’) также осуществляют в соответствии с теми же методами, что и описанные ранее для выполнения стадий b), c) и d).
Помимо уже упомянутых преимуществ, способ изобретения предоставляет следующие преимущества.
Поскольку уран (IV) и плутоний (IV) приводят в контакт друг с другом только при смешивании водных суспензий S1 и S2, риск протекания окислительно-восстановительной реакции между ураном и плутонием исключается. Это предотвращает возникновение риска изменения степени окисления указанных катионов: восстановления плутония (IV) до плутония (III) и окисления урана (IV) до урана (VI), что могло бы сделать невозможным количественное осаждение урана.
К тому же, способ изобретения позволяет получать порошки оксидов урана и плутония с содержанием плутония, которое может изменяться в широком диапазоне: от величины более 10% до величины более 50 атомных %.
Это также позволяет регулировать отношение Pu/(U+Pu) в порошках оксидов урана и плутония, получаемых с желаемым отношением, путём воздействия либо на концентрации урана (IV) и плутония (IV), соответственно, в растворах A1 и A’1, используемых для реакций оксалатного осаждения, либо на соотношения, в которых водные суспензии S1 и S2 смешивают друг с другом. Поскольку растворимости оксалатов актинида (IV) в водной среде, а конкретнее, растворимости оксалатов урана (IV) и плутония (IV) являются близкими и очень низкими в рабочих условиях способа изобретения, реакции оксалатного осаждения протекают количественно (с выходом выше 99%) для всех актинидов (IV). С учётом вышесказанного, степень превращения урана (IV) и плутония (IV), соответственно, присутствующих в водных растворах A1 и A’1, в оксалаты не оказывает никакого влияния на содержание указанных элементов в получаемых порошках оксидов урана и плутония.
С учётом вышесказанного, при использовании способа настоящего изобретения можно получать оксидные порошки, которые изначально имеют содержание плутония, нацеленное на изготовление ядерного топлива, например, от 10 до 30 атомных % в случае MOX топлива, а также получать базовые оксидные порошки, имеющие содержание плутония выше 50 атомных %, которые далее будут разбавляться порошком оксида урана с целью доведения содержания плутония до величины, желаемой для изготовления ядерного топлива.
Способ настоящего изобретения также позволяет легко включать оксид тория или нептуния в порошки получаемых оксидов урана и плутония, опять же с возможностью регулирования содержания данного оксида в указанных порошках. Следовательно, способ позволяет увеличивать количество элементов отработавших ядерных топлив, которые можно возвращать в цикл, и за счёт этого снижать радиотоксичность конечных отходов на основе обработки отработавших ядерных топлив.
Способ изобретения дополнительно предоставляет преимущество возможности его осуществления как после способа обработки отработавших ядерных топлив, который нацелен на раздельное управление очисткой урана и очисткой плутония и который, следовательно, приводит к образованию двух водных потоков, одного, содержащего очищенный уран, и другого, содержащего очищенный плутоний, как в случае способа PUREX; так и после способа обработки отработавших ядерных топлив, в котором обеспечивается совместное обращение с потоками урана и плутония на стадиях очистки.
И наконец, способ настоящего изобретения обладает преимуществом особенно упрощённого управления остаточными водными потоками, образованными в основном азотной кислотой и содержащими умеренное количество осадителя, без какого-либо иного добавления реагента для контроля уровня pH и/или комплексообразователя, что могло бы ухудшить процесс ввиду сложности обращения и окончательного кондиционирования.
Другие характеристики и преимущества данного изобретения станут очевидными при прочтении остальной части описания, представленной ниже, в совокупности с одним примером варианта осуществления изобретения и обращением к прилагаемым фигурам.
Очевидно, что указанный пример приведён только с целью иллюстрирования изобретения, и он не ограничивает его никоим образом.
Краткое описание чертежей
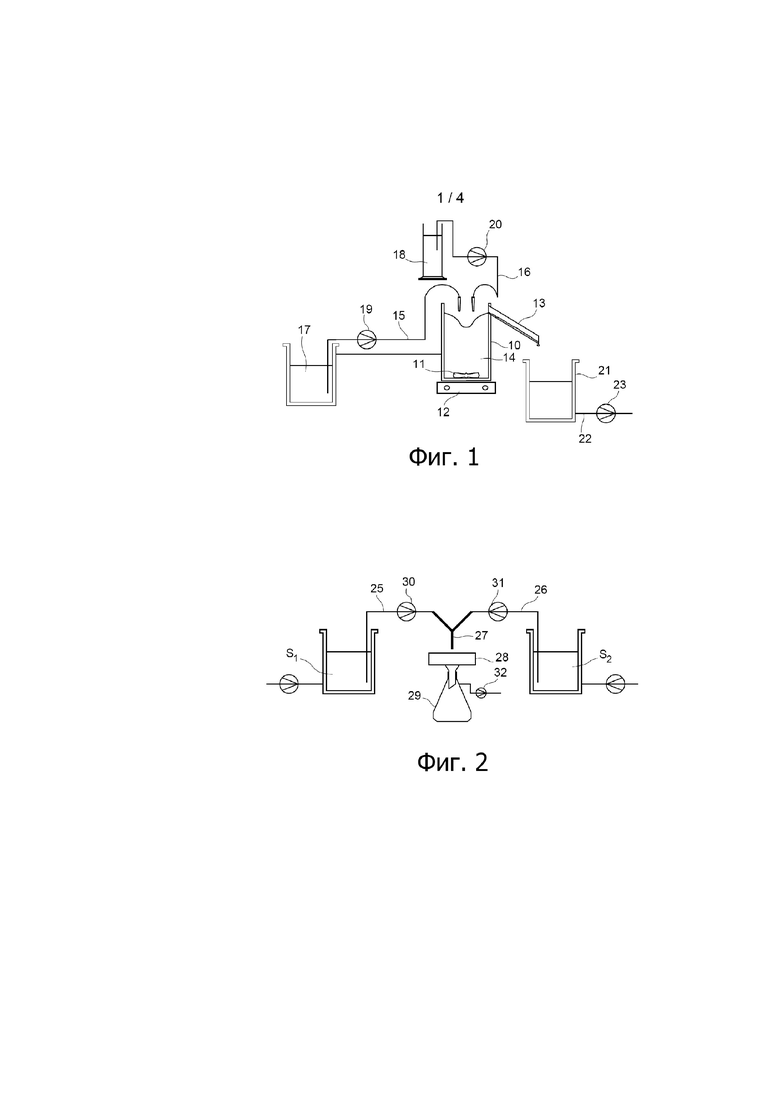
Фиг. 1 представляет собой технологическую схему узла, используемого для приготовления водной суспензии S1 частиц оксалата урана (IV) и водной суспензии S2 частиц оксалата плутония (IV) в примере варианта осуществления способа настоящего изобретения, описанного ниже.
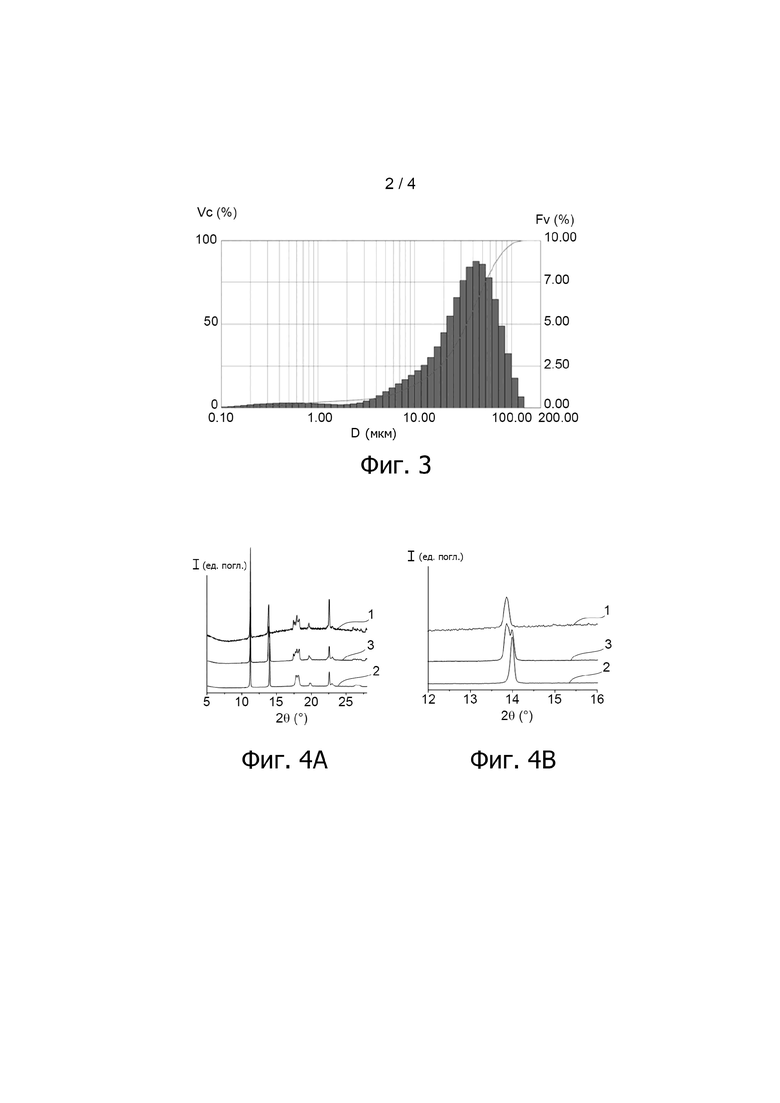
Фиг. 2 представляет собой технологическую схему узла, используемого для смешивания водной суспензии S1 частиц оксалата урана (IV) с водной суспензией S2 частиц оксалата плутония (IV) и для практически одновременной фильтрации суспензии S1+2, получаемой из указанной смеси в примере варианта осуществления способа настоящего изобретения, описанного ниже.
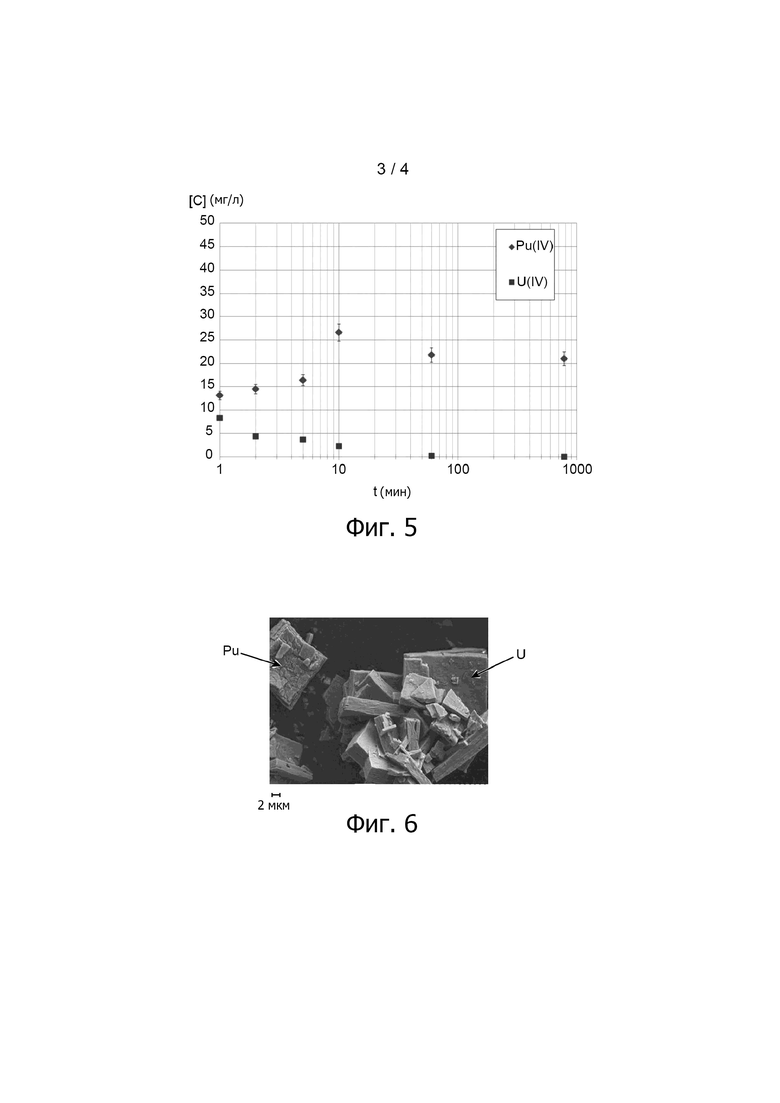
Фиг. 3 иллюстрирует измеренное лазерным методом распределение по размерам частиц оксалата урана (IV) и оксалата плутония (IV) в водной суспензии S1+2, полученной в примере варианта осуществления способа настоящего изобретения, описанного ниже; диаметр частиц, обозначенный D и выраженный в мкм, приведён на оси X: частота нахождения частиц в объёме, обозначенная Fv и выраженная в %, представлена на правой оси Y, тогда как суммарный объём частиц, обозначенный Vc и выраженный в %, представлен на левой оси Y.
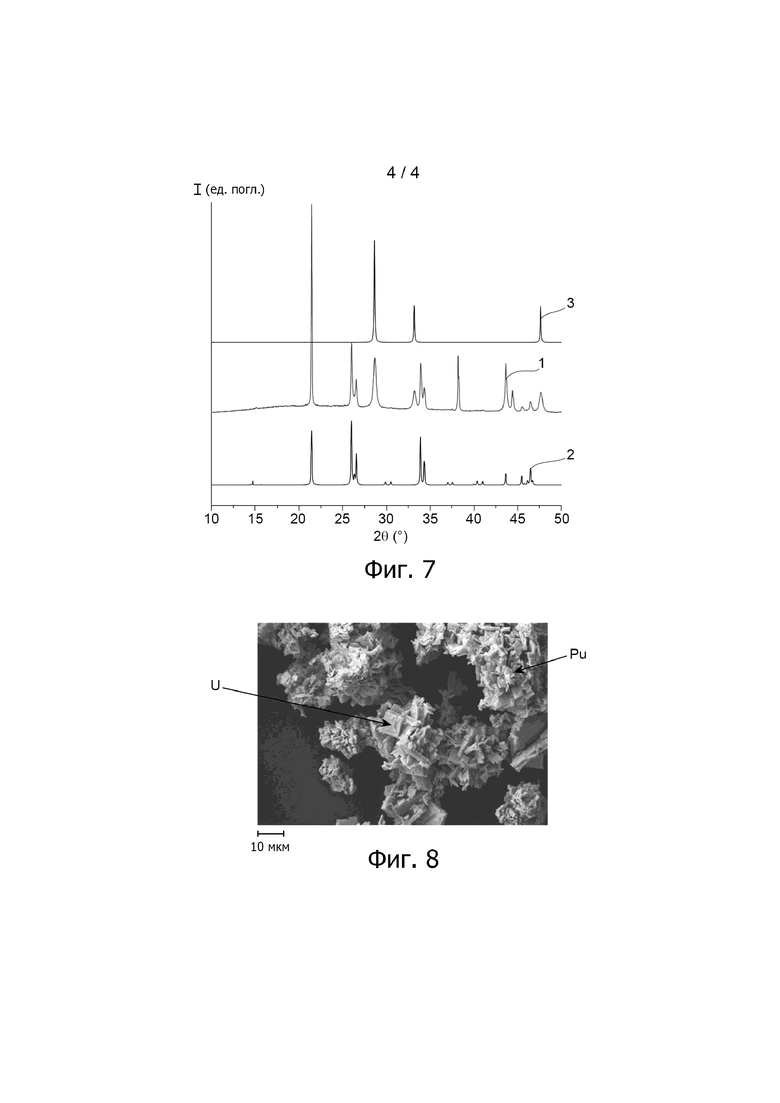
Фиг. 4A и 4B отображают рентгеновские дифрактограммы частиц оксалатов водных суспензий S1, S2 и S1+2, полученных в примере варианта осуществления способа настоящего изобретения, описанного ниже, после фильтрации и обезвоживания указанных суспензий, при этом на фиг. 4B приведено увеличенное изображение фиг. 4A в области пика, расположенного на данной фигуре в точке 2θ = 14°; на каждой из фиг. 4A и 4B дифрактограмма, обозначенная 1, соответствует частицам оксалата урана (IV) в водной суспензии S1, дифрактограмма, обозначенная 2, соответствует частицам оксалата плутония (IV) в водной суспензии S2, тогда как дифрактограмма, обозначенная 3, соответствует частицам оксалата урана (IV) и оксалата плутония (IV) в водной суспензии S1+2.
Фиг. 5 иллюстрирует изменения во времени, обозначенном t и выраженном в минутах на логарифмической шкале, концентраций урана (IV) и плутония (IV), обозначенных [C] и выраженных в мг/л, в пробе водной суспензии S1+2, полученной в примере варианта осуществления способа настоящего изобретения, описанного ниже, которую оставляли для состаривания в течение 15 часов.
На фиг. 6 приведён снимок водной суспензии S1+2, полученной в примере варианта осуществления способа настоящего изобретения, описанного ниже, выполненный при помощи сканирующего электронного микроскопа (СЭМ) после фильтрования и обезвоживания данной суспензии.
На фиг. 7 представлена обозначенная 1 рентгеновская дифрактограмма порошка октаоксида триурана и диоксида плутония, полученного в примере варианта осуществления способа настоящего изобретения, описанного ниже; для сравнения на данной фигуре также приведены расчётная дифрактограмма для частиц октаоксида триурана, обозначенная 2, и расчётная дифрактограмма для частиц диоксида плутония, обозначенная 3.
Фиг. 8 представляет собой выполненный методом СЭМ снимок порошка октаоксида триурана и диоксида плутония, полученного в примере варианта осуществления способа настоящего изобретения, описанного ниже.
Пример варианта осуществления способа изобретения
Данный пример относится к приготовлению порошка, состоящего из смеси частиц U3O8 и частиц PuO2, из водного раствора A1 нитрата урана (IV) и водного раствора A’1 нитрата плутония (IV) и нитрата урана (VI).
Водный раствор A1 содержит 0,15 моль/л нитрата урана (IV) или урановый нитрат формулы U(NO3)4, 2,5 моль/л азотной кислоты и 0,06 моль/л ионов гидразиния N2H5+ (поставляемых в форме нитрата гидразиния N2H5NO3), тогда как водный раствор A’1 содержит 0,15 моль/л нитрата плутония (IV) формулы Pu(NO3)4, 0,038 моль/л нитрата урана (VI) или уранилнитрата формулы UO2(NO3)2 и 2,5 моль/л азотной кислоты.
Концентрация нитрата урана (IV) в водном растворе A1 и концентрация нитрата плутония (IV) в водном растворе A’1 выбраны так, что, с учётом объёмов вовлекаемых водных растворов начальное мольное отношение Pu(IV)/U(IV)+Pu(IV) составляет 0,45.
1. Приготовление порошка:
В данном изобретении приготовление порошка U3O8 и PuO2 последовательно включает в себя:
- приготовление водной суспензии S1 частиц оксалата урана (IV) формулы U(C2O4)2⋅6H2O и водной суспензии S2 частиц оксалата плутония (IV) формулы Pu(C2O4)2⋅6H2O при помощи реакций оксалатного осаждения;
- смешивание водных суспензий S1 и S2 для получения водной суспензии S1+2, содержащей и частицы оксалата урана (IV), и частицы оксалата плутония (IV);
- разделение суспензии S1+2, полученной таким образом, на водную фазу и твёрдую фазу, образованную частицами оксалата урана (IV) и частицами оксалата плутония (IV); и
- прокаливание твёрдой фазы, полученной таким образом, для превращения, с одной стороны, частиц оксалата урана (IV) в частицы U3O8 и, с другой стороны, частиц оксалата плутония (IV) в частицы PuO2.
* Приготовление водных суспензий S1 и S2:
Как проиллюстрировано на фиг. 1, водную суспензию S1 частиц оксалата урана (IV) получают в реакторе 10, который оснащён системой 11, 12 перемешивания и переливной трубой 13 и который первоначально содержит водный раствор 14, содержащий 0,05 моль/л щавелевой кислоты, 0,039 моль/л ионов гидразиния (также поставляемых в форме нитрата гидразиния) и 2 моль/л азотной кислоты.
В реактор 10 загружают водный раствор A1, обозначенный позицией 17 на фиг. 1, и водный раствор A2, обозначенный позицией 18 на фиг. 1, который содержит 0,7 моль/л щавелевой кислоты, через входные патрубки 15 и 16, соответственно.
Скорости добавления водных растворов A1 и A2 в реактор 10 регулируют при помощи насосов 19 и 20, соответственно, при этом каждый из них снабжён измерителем расхода, и скорости составляют 21,7 мл/мин для водного раствора A1 и 11,7 мл/мин для водного раствора A2, что приводит к избытку щавелевой кислоты относительно стехиометрических условий оксалатного осаждения урана (IV).
Добавление водных растворов A1 и A2 в реактор 10 приводит к образованию реакционной среды, в которой уран (IV) осаждается в форме частиц оксалата урана (IV), выгружаемых по переливной трубе 13 в приёмник, расположенный ниже свободного конца указанной переливной трубы. Затем водную суспензию S1, образовавшуюся таким путём, откачивают из приёмника по линии 22, снабжённой насосом 23.
Для приготовления водной суспензии S2 частиц оксалата плутония (IV) используют водный раствор A’2, имеющий состав, идентичный составу водного раствора A2, использованному ранее, и узел, аналогичный изображённому на фиг. 1, за исключением того, что:
- во-первых, водный раствор 14, первоначально содержащийся в реакторе 10, заменяют водным раствором, содержащим 0,05 моль/л щавелевой кислоты, 0,02 моль/л урана (VI) и 2 моль/л азотной кислоты; и
- во-вторых, водный раствор A1 заменяют водным раствором A’1.
Режимы скоростей потоков являются теми же, что описаны ранее для приготовления водной суспензии S1 частиц оксалата урана (IV).
* Смешивание водных суспензий S1 и S2 и разделение суспензии S1+2 на две фазы:
Указанные стадии осуществляют с использованием узла, изображённого на фиг. 2.
Как можно видеть на данной фиг., каждую из водных суспензий S1 и S2 подают по линии, соответственно, 25 и 26, в одну из ветвей Y-образного соединителя 27, при этом указанные водные суспензии объединяются и однородно смешиваются друг с другом для образования водной суспензии S1+2; третья ветвь Y-образного соединителя 27 имеет концевой участок, расположенный чуть выше фильтрационной системы, позволяющей данной суспензии разделяться на водную фазу (или фильтрат) и твёрдую фазу (или лепёшку).
Указанная фильтрационная система состоит из воронки 28 Бюхнера, нижняя часть которой снабжена фильтром (например, стеклянным микроволоконным фильтром типа Whatman™ GF/B filter), на котором удерживаются частицы оксалатов, и вакуумной колбы 29, которая размещена под воронкой и в которой собирается водная фаза суспензии S1+2.
Скорости входных потоков водных суспензий S1 и S2 в соединителе 27 регулируют при помощи насосов, 30 и 31, соответственно, причём каждый из них снабжён измерителем расхода, при этом указанные скорости потоков составляют 48,1 мл/мин для водной суспензии S1 и 39,9 мл/мин для водной суспензии S2.
Фильтрование водной суспензии S1+2 осуществляют без подключения колбы 29 к вакууму, так что частицы оксалатов равномерно распределяются на фильтре. Как только достигается максимальный объём для ёмкости воронки 28 Бюхнера, колбу 29 подключают к вакууму при помощи вакуумного насоса 32 для обезвоживания лепёшки, образованной частицами оксалатов.
* Прокалка твёрдой фазы:
Лепёшку из частиц оксалата, полученную ранее, прокаливают при продувании воздухом.
Для этого лепёшку из частиц помещают в печь, которую нагревают со скоростью 20°C/мин до тех пор, пока её температура не достигает 700°C. Указанную температуру поддерживают в течение 1 часа. Затем прекращают нагревание и оставляют лепёшку из частиц в печи до тех пор, пока температура печи не вернется к температуре окружающей среды. Скорость потока продувочного газа является такой, что объём печи обновляется указанным газом 10 раз на протяжении времени прокалки.
По окончании данной прокалки получают порошок, состоящий из смеси частиц U3O8 и частиц PuO2.
2. Анализы:
*Анализы фильтрата:
Фильтрат, полученный по окончании стадии фильтрования, анализировали с целью определения состава катионов металлов. Анализы показали, что данный фильтрат содержит от 1 мг/л до 10 мг/л урана (IV), от 20 мг/л до 25 мг/л плутония (IV) и от 3 г до 4 г/л урана (VI).
Это подтверждает, что химические условия осаждения, применяемые выше для приготовления водных суспензий S1 и S2, позволяют практически количественно осаждать уран (IV) и плутоний (IV). Следовательно, при контролировании скоростей потоков на стадии смешивания указанных суспензий можно находить мольное отношение Pu(IV)/U(IV)+Pu(IV) в лепёшке оксалатных частиц, которое является аналогичным начальному мольному отношению Pu(IV)/U(IV)+Pu(IV).
Кроме того, начальная концентрация урана (VI) в водном растворе A’1 приводит к обнаружению всей совокупности данного урана в фильтрате.
* Анализы частиц оксалатов:
Водные суспензии S1 и S2, а также водную суспензию S1+2 подвергали лазерному анализу размера частиц (анализатор размера частиц фирмы MALVERN Instruments).
Ниже в таблице приведены значения среднеобъёмного диаметра, обозначенного D [4,3] и выраженного в мкм, полученные для частиц оксалата (оксалатов) указанных суспензий.
Таблица
Указанные значения размера частиц, близкие друг к другу и сосредоточенные вокруг 45 мкм, сравнимы с размерами частиц оксалата плутония (IV), обычно получаемых на промышленных установках превращения плутония (IV) в оксалат.
Кроме того, на фиг. 3 приведено распределение частиц по размерам для водной суспензии S1+2, также полученное лазерным методом анализа размера частиц.
Водные суспензии S1 и S2, а также водную суспензию S1+2 тоже анализировали, но после фильтрования и обезвоживания, методом дифракции рентгеновских лучей (конфигурация θ-2θ дифрактометра BRUKER AXS, оснащённого медным антикатодом, характеризующимся Kα-излучением с длиной волны λ, равным 1,5418 Å, и линейным детектором BRUKER AXS).
Рентгеновские дифрактограммы, полученные таким образом, изображены на фиг. 4A а на фиг. 4B, которая соответствует увеличенному изображению фиг. 4A в области пика, расположенного на фиг. 4A в точке 2θ = 14°.
Как показано на данных фиг., на которых дифрактограмма 1 соответствует водной суспензии S1, дифрактограмма 2 соответствует водной суспензии S2, тогда как дифрактограмма 3 соответствует водной суспензии S1+2, две оксалатные фазы водной суспензии S1+2 кристаллизуются в одной и той же структуре моноклинного типа An(C2O4)2⋅6H2O.
Указанная форма кристаллизации обладает преимуществом удерживать массовую долю воды в фильтровальной лепёшке всего лишь порядка 15%, таким образом придавая едва заметную липкость смеси частиц оксалата урана (IV) и частиц оксалата плутония (IV), образующих данную лепёшку.
На дифрактограммах 2 и 3 отмечается отсутствие каких-либо пиков, которые могли бы соответствовать оксалату урана (VI).
Как показано в вышеизложенном описании, уран (IV) является мощным восстановителем плутония (IV). Путём раздельного приготовления водных суспензий частиц оксалата урана (IV) и частиц оксалата плутония (IV) можно устранить окислительно-восстановительный эффект в водной фазе, когда указанные частицы оксалатов в дальнейшем смешиваются друг с другом. Это демонстрируется на фиг. 5, которая показывает, что измерение концентраций урана (IV) и плутония (IV) в пробе суспензии S1+2, оставленной для состаривания в течение 15 часов, не позволяет обнаруживать никакого явления, которое ставило бы под сомнение химическую стабильность указанных частиц.
В дополнение к этому, как показано на фиг. 6, которая соответствует снимку фильтровальной лепёшки водной суспензии S1+2, выполненному методом СЭМ в режиме вторичных электронов (электронный микроскоп ZEISS с полевой эмиссией, соединённый с детектором ЭДС и детектором ВДС), распределение частиц оксалата урана (IV) и частиц оксалата плутония (IV) в данной лепёшке является однородным.
*Анализ оксидных частиц:
Порошок, полученный по окончании стадии прокалки, анализировали для оценки его удельной площади поверхности по БЭТ, распределения частиц по размерам (методом лазерного анализа размера частиц), состава (методом дифракции рентгеновских лучей) и однородности (методом СЭМ).
Анализы методами лазерного определения размера частиц, дифракции рентгеновских лучей и СЭМ проводили с использованием того же оборудования, что указано ранее.
Данные анализы показали, что порошок:
- имеет удельную площадь поверхности около 3 м2/г;
- имеет среднеобъёмный диаметр D [4,3] около 15 мкм;
- состоит исключительно из U3O8 и PuO2, как проиллюстрировано на фиг. 7, что показывает рентгеновская дифрактограмма упомянутого порошка, обозначенная 1, а также расчётные дифрактограммы для частиц октаоксида триурана и частиц диоксида плутония, соответственно обозначенные 2 и 3; и
- демонстрирует однородность в рамках фаз U3O8 и PuO2, соответствующую однородности, полученной для фаз оксалата урана (IV) и оксалата плутония (IV) в фильтровальной лепёшке водной суспензии S1+2 до прокалок, как отображается снимком СЭМ, приведённым на фиг. 8.
Цитированные ссылки
[1] WO-A-2007/135178
[2] Numao et al., GLOBAL 2007: Advanced Nuclear Fuel Cycles and Systems, Boise, USA, 9-13 September 2007
[3] Felker et al., ATALANTE 2008: Nuclear Fuel Cycles for a Sustainable Future, Montpellier, France, 19-23 May 2008
[4] WO-A-02/28778
[5] WO-A-2005/119699
[6] Delegard et al., PNNL-13934, 2002
[7] Atlas et al., Journal of Nuclear Materials, 2001, 294, 344-348
[8] WO-A-2010/070064
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО ЯДЕРНОГО ТОПЛИВА | 2012 |
|
RU2612659C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА ОКСИДА МЕТАЛЛА, СПОСОБ ИЗГОТОВЛЕНИЯ ТАБЛЕТКИ ИЗ ОКСИДА МЕТАЛЛА, ПОРОШОК И ТАБЛЕТКА, ПОЛУЧЕННЫЕ ЭТИМИ СПОСОБАМИ, И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2675572C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА, ВКЛЮЧАЮЩЕГО ТВЕРДЫЙ РАСТВОР ДИОКСИДА УРАНА И ДИОКСИДА ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ДРУГОГО АКТИНИДА И/ИЛИ ЛАНТАНИДА | 2014 |
|
RU2662526C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО ЯДЕРНОГО ТОПЛИВА НА ОСНОВЕ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО МЛАДШЕГО АКТИНИДА | 2010 |
|
RU2551654C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕШАННЫХ ОКСИДОВ УРАНА И ПЛУТОНИЯ | 2015 |
|
RU2626854C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТВЕРДЫХ РАСТВОРОВ ОКСИДОВ АКТИНИДОВ | 2012 |
|
RU2494479C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТВЕРДОГО РАСТВОРА ДИОКСИДА ПЛУТОНИЯ В МАТРИЦЕ ДИОКСИДА УРАНА | 2010 |
|
RU2446107C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТВЕРДОГО РАСТВОРА ДИОКСИДА ПЛУТОНИЯ В МАТРИЦЕ ДИОКСИДА УРАНА | 2013 |
|
RU2554626C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТВЁРДОГО РАСТВОРА ДИОКСИДА ПЛУТОНИЯ В МАТРИЦЕ ДИОКСИДА УРАНА | 2015 |
|
RU2598943C1 |
| СПОСОБЫ ПРИГОТОВЛЕНИЯ ОКСАЛАТА АКТИНОИДОВ И ПРИГОТОВЛЕНИЯ СОЕДИНЕНИЙ АКТИНОИДОВ | 2009 |
|
RU2505484C2 |
Изобретение относится к области переработки отработавших ядерных топлив. Способ получения порошка, содержащего однородную смесь частиц U3O8 и частиц PuO2, включает в себя: получение водной суспензии S1 частиц оксалата урана (IV) и водной суспензии S2 частиц оксалата плутония (IV) при помощи процедур оксалатного осаждения; смешивание суспензий S1 и S2. Далее проводят разделение водной суспензии S1+2 на водную фазу и твёрдую фазу, содержащую частицы оксалата урана (IV) и частицы оксалата плутония (IV). И потом прокаливание твёрдой фазы для превращения частиц оксалата урана (IV) в частицы октаоксида триурана и частиц оксалата плутония (IV) в частицы диоксида плутония (IV), в результате чего получают порошок. Изобретение позволяет упростить управление остаточными водными потоками. 11 з.п. ф-лы, 1 табл., 9 ил.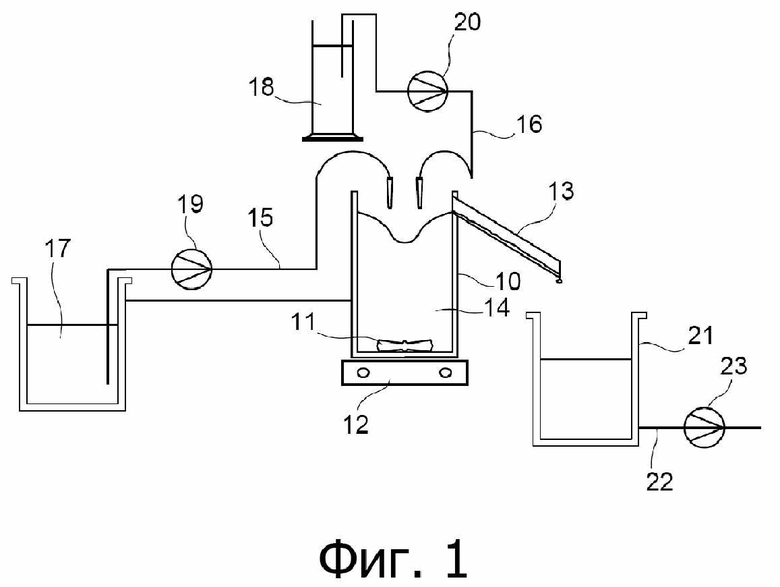
1. Способ получения порошка, содержащего частицы октаоксида триурана и частицы диоксида плутония, отличающийся тем, что он включает в себя следующее:
a) получают водную суспензию S1 частиц оксалата урана (IV) и водную суспензию S2 частиц оксалата плутония (IV) при помощи реакций оксалатного осаждения;
b) смешивают водную суспензию S1 с водной суспензией S2 для получения водной суспензии S1+2, содержащей частицы оксалата урана (IV) и частицы оксалата плутония (IV);
c) разделяют водную суспензию S1+2 на водную фазу и твёрдую фазу, содержащую частицы оксалата урана (IV) и частицы оксалата плутония (IV); и
d) прокаливают твёрдую фазу для превращения (1) частиц оксалата урана (IV) в частицы октаоксида триурана и (2) частиц оксалата плутония (IV) в частицы диоксида плутония (IV), в результате чего образуется порошок;
и тем, что стадии b) и c) осуществляют одновременно или последовательно.
2. Способ по п. 1, отличающийся тем, что стадия a) включает в себя следующее:
- водный раствор A1, содержащий азотную кислоту и нитрат урана (IV), приводят в контакт с водным раствором A2, содержащим осадитель, выбранный из щавелевой кислоты, её солей и алкилпроизводных, для образования реакционной среды, в которой уран (IV) осаждается в форме оксалата урана (IV); и
- водный раствор A’1, содержащий азотную кислоту и нитрат плутония (IV), приводят в контакт с водным раствором A’2, содержащим осадитель, выбранный из щавелевой кислоты, её солей и алкилпроизводных, для образования реакционной среды, в которой плутоний (IV) осаждается в форме оксалата плутония (IV).
3. Способ по п. 2, отличающийся тем, что концентрация азотной кислоты в водных растворах A1 и A’1 составляет от 0,5 моль/л до 5 моль/л.
4. Способ по п. 2 или 3, отличающийся тем, что концентрация нитрата урана (IV) в водном растворе A1 и концентрация нитрата плутония (IV) в водном растворе A’1 составляет от 0,001 моль/л до 1 моль/л.
5. Способ по любому из пп. 2 - 4, отличающийся тем, что концентрация осадителя в водных растворах A2 и A’2 составляет от 0,05 моль/л до 1 моль/л.
6. Способ по любому из пп. 2 - 5, отличающийся тем, что осадитель присутствует в реакционных средах в избытке относительно стехиометрических условий реакций оксалатного осаждения урана (IV) и плутония (IV).
7. Способ по любому из пп. 2 - 6, отличающийся тем, что водный раствор A’1 дополнительно содержит нитрат урана (VI).
8. Способ по любому из пп. 1 - 7, отличающийся тем, что стадия c) включает в себя фильтрование водной суспензии S1+2 под действием вакуума или давления.
9. Способ по любому из пп. 1 - 8, отличающийся тем, что стадии b) и c) выполняют одновременно.
10. Способ по любому из пп. 1 - 9, отличающийся тем, что стадия d) включает в себя обработку твёрдой фазы при температуре, по меньшей мере, 550°C и в окислительной атмосфере.
11. Способ по любому из пп. 1 - 10, отличающийся тем, что порошок дополнительно содержит частицы диоксида актинида (IV), выбранного из тория и нептуния, и тем, что способ включает в себя следующее:
a’) получают водную суспензию S1 частиц оксалата урана (IV), водную суспензию S2 частиц оксалата плутония (IV) и водную суспензию S3 частиц оксалата актинида (IV) при помощи реакций оксалатного осаждения;
b’) смешивают водные суспензии S1, S2 и S3 друг с другом для получения водной суспензии S1+2+3, содержащей частицы оксалата урана (IV), частицы оксалата плутония (IV) и частицы оксалата актинида (IV);
c’) разделяют водную суспензию S1+2+3 на водную фазу и твёрдую фазу, образованную частицами оксалата урана (IV), частицами оксалата плутония (IV) и частицами оксалата актинида (IV); и
d’) прокаливают твёрдую фазу для превращения (1) частиц оксалата урана (IV) в частицы октаоксида триурана, (2) частиц оксалата плутония (IV) в частицы диоксида плутония и (3) частиц оксалата актинида (IV) в частицы диоксида актинида (IV), в результате чего получают порошок;
и тем, что стадии b’) и c’) осуществляют одновременно или последовательно.
12. Способ по любому из пп. 1 - 10, отличающийся тем, что порошок дополнительно содержит частицы диоксида актинида (IV), выбранного из тория и нептуния, и тем, что способ включает в себя следующее:
a’’) получают водную суспензию S1 частиц двойного оксалата урана (IV) и актинида (IV), а также водную суспензию S2 частиц оксалата плутония (IV) при помощи реакций оксалатного осаждения;
b’’) смешивают водную суспензию S1 с водной суспензией S2 для получения водной суспензии S1+2, содержащей частицы двойного оксалата урана (IV) и актинида (IV) и частицы оксалата плутония (IV);
c’’) разделяют водную суспензию S1+2 на водную фазу и твёрдую фазу, образованную частицами двойного оксалата урана (IV) и актинида (IV) и частицами оксалата плутония (IV); и
d’’) прокаливают твёрдую фазу для превращения (1) частиц двойного оксалата урана (IV) и актинида (IV) в частицы октаоксида триурана и диоксида актинида (IV), и (2) частиц оксалата плутония (IV) в частицы диоксида плутония, в результате чего получают порошок;
а также тем, что стадии b’’) и c’’) выполняют одновременно или последовательно.
| GB 978615 A, 23.12.1964 | |||
| СПОСОБЫ ПРИГОТОВЛЕНИЯ ОКСАЛАТА АКТИНОИДОВ И ПРИГОТОВЛЕНИЯ СОЕДИНЕНИЙ АКТИНОИДОВ | 2009 |
|
RU2505484C2 |
| СПОСОБ СООСАЖДЕНИЯ АКТИНОИДОВ С РАЗНОЙ СТЕПЕНЬЮ ОКИСЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ СМЕШАННЫХ СОЕДИНЕНИЙ АКТИНОИДОВ | 2005 |
|
RU2408537C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО ЯДЕРНОГО ТОПЛИВА НА ОСНОВЕ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО МЛАДШЕГО АКТИНИДА | 2010 |
|
RU2551654C2 |
| US 2010301287 A1, 02.12.2010. | |||

