Область техники
Настоящее изобретение относится к способу получения вирусоподобных частиц (VLP; от англ.: virus-like particles) на основе белка L1 папилломавируса человека (HPV; от англ.: human papillomavirus).
Предшествующий уровень техники
Известно более 80 типов папилломавирусов человека (HPV), и более 30 типов вызывают цервикальную инфекцию при половом контакте, причем известно, что половина этих типов связана с раком шейки матки. Кроме того, заболеваемость раком у женщин, инфицированных HPV 16 и 18 типов, в 500 раз выше, чем у неинфицированных женщин, и известно, что HPV 16 и 18 типов инфицируют эпителиальные клетки полового органа, вызывая рак шейки матки вследствие злокачественной трансформации (Hausen, Biochemica. Biophysica. Acta, 1288: 55-78, 1996).
Существующие в настоящее время вакцины для профилактики рака шейки матки разработаны с использованием вирусоподобных частиц (VLP) HPV высокого онкогенного риска. VLP состоят из основного капсидного белка (белка L1 с молекулярной массой, примерно равной 55 кДа) HPV и обладают способностью к самосборке с образованием VLP без продукта гена L1 или продуктов других вирусных генов, что очень сходно со свойствами природного вириона HPV. Кроме того, VLP вызывают долговременный иммунитет, и можно ожидать, что они обеспечат высокую эффективность в качестве экспериментальной вакцины вследствие специфичности типа гена.
Были произведены попытки получения VLP HPV с использованием дрожжевых систем экспрессии. Известны способы очистки VLP HPV, полученных из Saccharomyces cerevisiae, до высокой степени чистоты, описанные ниже. В зарегистрированном корейском патенте №10-0959145 описан способ очистки L1 белка HPV посредством добавления сульфата аммония к лизату дрожжей, который получают посредством культивирования трансформированных дрожжей, экспрессирующих белок L1 HPV, и их лизиса, для осаждения белка и последующего выполнения эксклюзионной хроматографии и катионообменной хроматографии белковых преципитатов.
В тексте публикации процитированы многочисленные статьи и патентные документы и приведены библиографические ссылки на них. Содержание процитированных статей и патентных документов полностью включено в данную публикацию посредством ссылки, а область техники, к которой относится настоящее изобретение, и детали настоящего изобретения разъяснены более подробно.
Сведения, подтверждающие возможность осуществления изобретения
Техническая проблема
Авторы настоящего изобретения выполнили исследования условий очистки с целью повышения выхода продукции при получении и очистке белка L1 HPV в промышленном масштабе с использованием дрожжевой системы экспрессии. В результате, авторы настоящего изобретения подтвердили, что эффективность очистки белка L1 HPV может быть значительно повышена за счет обработки преципитата, полученного при осаждении сульфатом аммония дрожжевого продукта, содержащего белок L1, раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, для удаления загрязнений и последующей хроматографии, и таким образом осуществили настоящее изобретение.
Поэтому первым аспектом настоящего изобретения является обеспечение способа получения вирусоподобных частиц на основе белка L1 HPV.
Другим аспектом настоящего изобретения является обеспечение вирусоподобных частиц на основе белка L1 HPV, полученных способом по настоящему изобретению.
Другие задачи и преимущества настоящего изобретения станут более очевидными из приведенного ниже подробного описания настоящего изобретения, формулы изобретения и графических материалов.
Техническое решение
Согласно первому аспекту настоящего изобретения обеспечен способ получения вирусоподобных частиц (VPL) на основе белка L1 папилломавируса человека (HPV), который включает:
(a) культивирование трансформированных дрожжей, экспрессирующих белок L1 HPV;
(b) выполнение осаждения сульфатом аммония в продукте трансформированных дрожжей, культивированных на стадии (а), с получением преципитата;
(c) обработку преципитата, полученного посредством осаждения сульфатом аммония, раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, с последующей инкубацией и последующее удаление образовавшихся нерастворимых белков; и
(в) выполнение хроматографии продукта, полученного на стадии (с) с получением белка L1.
Авторы настоящего изобретения выполнили исследования условий очистки с целью повышения выхода продукции при получении и очистке белка L1 HPV в промышленном масштабе с использованием дрожжевой системы экспрессии. В результате авторы настоящего изобретения подтвердили, что эффективность очистки белка L1 HPV может быть значительно повышена за счет обработки преципитата, полученного при осаждении сульфатом аммония дрожжевого продукта, содержащего белок L1, раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, для удаления загрязнений и последующей хроматографии.
Далее соответствующие стадии настоящего изобретения будут описаны более подробно.
(а) Культивирование трансформированных дрожжей, экспрессирующих белок L1 HPV
На стадии (а) настоящего изобретения культивируют трансформированные дрожжи, экспрессирующие белок L1 HPV.
В данной публикации клетки, пригодные для экспрессии белка L1 HPV, являются дрожжами; например, могут быть использованы Saccharomyces cerevisiae, Saccharomyces pastorianus, Saccharomyces sp., Schizosaccharomyces pombe, пекарские дрожжи и т.п.
Согласно варианту осуществления настоящего изобретения трансформированными дрожжами по настоящему изобретению являются Saccharomyces cerevisiae.
Термин «трансформированные дрожжи, экспрессирующие белок L1 HPV» относится к дрожжевым клеткам, трансформированным вектором экспрессии, успешно экспрессирующим белок L1 HPV. Вектор экспрессии может содержать факторы, регулирующие транскрипцию или трансляцию, и другие маркерные гены, известные в данной области техники.
Дрожжи, трансформированные для экспрессии белка L1 HPV по настоящему изобретению, можно легко получить с использованием способов, известных в данной области техники, и эти способы раскрыты в публикациях US 7250170, US 6613557, US 5888516, US 5871998, US 5618536 и US 5437951, содержание которых полностью включено в данную работу посредством ссылки.
Трансформированные дрожжи можно культивировать способами, используемыми в данной области техники для получения белка L1 HPV с использованием дрожжей. Например, трансформированные дрожжи можно культивировать в среде с добавлением одного или более источников углерода - глюкозы и галактозы, и примером среды, используемой для культивирования трансформированных дрожжей, является среда YPDG (содержащая экстракт дрожжей, пептон, глюкозу и галактозу). Кроме того, трансформированные дрожжи можно культивировать в течение периода, составляющего менее 96 часов (например, 95 часов или менее, от 10 часов до 95 часов, от 20 часов до 95 часов или от 24 часов до 95 часов).
Согласно варианту осуществления настоящего изобретения, белок L1 HPV выбран из белков HPV 6а типа, HPV 6b типа, HPV 11 типа, HPV 16 типа, HPV 18 типа, HPV 31 типа, HPV 33 типа, HPV 35 типа, HPV 39 типа, HPV 45 типа, HPV 51 типа, HPV 52 типа, HPV 56 типа, HPV 58 типа и HPV 68 типа. Согласно более конкретному варианту осуществления настоящего изобретения, белок L1 HPV является белком HPV 16 типа или HPV 18 типа.
(b) Получение преципитата с использованием сульфата аммония
На стадии (b) настоящего изобретения получают преципитат посредством обработки продукта (содержащего белок L1) трансформированных дрожжей, культивированных на стадии (а), сульфатом аммония. Например, на стадии (b) трансформированные дрожжи, культивированные на стадии (а), лизируют и обрабатывают лизат сульфатом аммония с получением преципитата.
Культивированные дрожжевые клетки можно лизировать с использованием способов получения лизата клеток, известных в данной области техники, например (но не ограничиваясь этим) - посредством обработки ультразвуком, разрушения стеклянными шариками и т.п.
После этого экспрессированные белки осаждают посредством обработки дрожжевого продукта сульфатом аммония (посредством добавления сульфата аммония) и удаляют загрязнения посредством центрифугирования или сходным способом.
Согласно варианту осуществления настоящего изобретения, концентрация сульфата аммония для обработки на стадии (b) лежит в диапазоне от 20 масс. % до 60 масс. %. Согласно более конкретному варианту осуществления настоящего изобретения, концентрация сульфата аммония для обработки лежит в диапазоне от 40 масс. % до 50 масс. % или от 42 масс. % до 48 масс. %.
Согласно варианту осуществления настоящего изобретения, преципитат, полученный посредством осаждения сульфатом аммония на стадии (b), инкубируют в растворе, содержащем соль (например, NaCl).
Согласно конкретному варианту осуществления настоящего изобретения, преципитат, полученный посредством осаждения сульфатом аммония, разбавляют до концентрации, лежащей в диапазоне от 25 мг/мл до 65 мг/мл, посредством добавления буферного раствора, смешанного с 0,5 М или более (например, от 0,5 М до 0,9 М, от 0,6 М до 0,9 М или от 0,7 М до 0,9 М) раствором NaCl, перемешивают в течение предварительно заданного времени и затем инкубируют в течение 12 часов или более (например, в течение периода от 18 часов до 20 часов). Температура инкубации может лежать в диапазоне от 3°C до 5°C (например, она может быть равной 4°C).
(с) Удаление загрязнений посредством обработки раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М
На стадии (с) способа по настоящему изобретению преципитат, полученный посредством обработки сульфатом аммония, обрабатывают раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, и полученный материал инкубируют для формирования загрязнений в форме нерастворимых белков. После этого сформированные нерастворимые белки удаляют, тем самым значительно повышая эффективность очистки белка L1 HPV.
Как описано в примерах, приведенных ниже, способ обработки раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, является стадией, которая способствует значительному повышению эффективности очистки белка L1 HPV (см. Пример 3). То есть, в тех случаях, когда нерастворимые белки, образовавшиеся на этой стадии, удаляют посредством центрифугирования, степень чистоты VLP, элюируемых в процессе хроматографии на следующей стадии, может быть значительно повышена.
Согласно варианту осуществления настоящего изобретения, раствор, содержащий преципитат, полученный посредством осаждения сульфатом аммония, который был инкубирован в течение предварительно заданного времени в присутствии указанной соли (например, NaCl) на стадии (с), диализируют в буферном растворе, содержащем соль в концентрации, лежащей в диапазоне от 0,35 М до 0,60 М, и затем инкубируют, тем самым удаляя загрязнения, находящиеся в нерастворимом состоянии. Буферный раствор, содержащий соль в концентрации, лежащей в диапазоне от 0,35 М до 0,60 М, может содержать неионогенное поверхностно-активное вещество. Например, неионогенное поверхностно-активное вещество может содержаться в концентрации, лежащей в диапазоне от 0,001% до 0,1%.
Согласно варианту осуществления настоящего изобретения, концентрация преципитата, полученного посредством осаждения сульфатом аммония, который подлежит обработке раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М, лежит в диапазоне от 25 мг/мл до 65 мг/мл. Эта концентрация преципитата, полученного посредством осаждения сульфатом аммония, может быть отрегулирована посредством смешивания преципитата, полученного посредством осаждения сульфатом аммония, с буферным раствором.
Согласно более конкретному варианту осуществления настоящего изобретения, концентрация преципитата, полученного посредством осаждения сульфатом аммония, лежит в диапазоне от 28 мг/мл до 62 мг/мл, от 28 мг/мл до 60 мг/мл, от 28 мг/мл до 55 мг/мл, от 28 мг/мл до 50 мг/мл, от 30 мг/мл до 62 мг/мл, от 30 мг/мл до 60 мг/мл, от 30 мг/мл до 55 мг/мл или от 30 мг/мл до 50 мг/мл.
Согласно варианту осуществления настоящего изобретения, солью является NaCl, KCl или Na2CO3.
Согласно варианту осуществления настоящего изобретения, концентрация соли для обработки на стадии (с) лежит в диапазоне от 0,35 М до 0,60 М, от 0,35 М до 0,55 М, от 0,35 М до 0,50 М или от 0,35 М до 0,45 М; от 0,40 М до 0,60 М, от 0,40 М до 0,55 М, от 0,40 М до 0,50 М или от 0,40 М до 0,45 М; от 0,45 М до 0,60 М, от 0,45 М до 0,55 М или от 0,45 М до 0,50 М; или от 0,50 М до 0,60 М, или от 0,50 М до 0,55 М.
Согласно варианту осуществления настоящего изобретения, солью для обработки на стадии (с) является NaCl, и концентрация NaCl для обработки лежит в диапазоне от 0,35 М до 0,60 М или от 0,35 М до 0,55 М; от 0,40 М до 0,60 М или от 0,40 М до 0,55 М; или от 0,45 М до 0,60 М или от 0,45 М до 0,55 М.
Согласно другому варианту осуществления настоящего изобретения, солью для обработки на стадии (с) является KCl, и концентрация KCl для обработки лежит в диапазоне от 0,35 М до 0,60 М или от 0,40 М до 0,60 М; от 0,35 М до 0,55 М; или от 0,55 М до 0,60 М.
Согласно еще одному варианту осуществления настоящего изобретения, солью для обработки на стадии (с) является Na2CO3, и концентрация Na2CO3 для обработки лежит в диапазоне от 0,35 М до 0,60 М или от 0,40 М до 0,60 М; от 0,35 М до 0,55 М или от 0,40 М до 0,55 М; от 0,35 М до 0,50 М или от 0,40 М до 0,50 М; или от 0,35 М до 0,45 М.
Согласно варианту осуществления настоящего изобретения, температура для инкубации на стадии (с) лежит в диапазоне от 25°C до 34°C.
Согласно конкретному варианту осуществления настоящего изобретения, температура для инкубации лежит в диапазоне от 26°C до 34°C, от 27°C до 34°C, от 28°C до 34°C, от 29°C до 34°C или от 29,5°C до 34°C, и согласно другому варианту осуществления настоящего изобретения, температура для инкубации лежит в диапазоне от 25°C до 33,5°C, от 26°C до 33,5°C, от 27°C до 33,5°C, от 28°C до 33,5°C, от 29°C до 33,5°C, от 29,5°C до 33,5°C или от 33°C до 33°C.
Согласно варианту осуществления настоящего изобретения, время для инкубации на стадии (с) лежит в диапазоне от 21 часа до 27 часов. Согласно конкретному варианту осуществления настоящего изобретения, время для инкубации лежит в диапазоне от 21 часа до 26 часов или от 22 часов до 26 часов.
(d) Хроматография
На стадии (d) настоящего изобретения выполняют хроматографию раствора, из которого удалены загрязнения, для получения белка L1. В тех случаях, когда раствор, из которого удалены загрязнения, очищают посредством хроматографии, можно очень эффективно удалить остаточные загрязнения.
Согласно варианту осуществления настоящего изобретения, хроматография является аффинной хроматографией или катионообменной хроматографией. Согласно конкретному варианту осуществления настоящего изобретения, аффинная хроматография является хроматографией на гепарин-сефарозе.
Например, в тех случаях, когда выполняют хроматографию на гепарин-сефарозе, гепарин-сефарозную смолу вначале инкубируют с подходящим связывающим буфером перед нанесением материала, полученного на стадии (с), на колонку с гепарин-сефарозной смолой. Связывающий буфер предпочтительно содержит NaCl и неионогенное поверхностно-активное вещество (например, Твин 80), и концентрация NaCl предпочтительно является низкой (от 0,1 М до 0,4 М). Затем наносят пробу для хроматографии на гепарин-сефарозе, после чего элюируют белок из смолы. Для элюирования предпочтителен линейный градиент NaCl, и более предпочтительно загрязнения элюируют в линейном градиенте NaCl в диапазоне от 0,33 М до 0,66 М, а затем желаемый белок L1 элюируют в линейном градиенте NaCl в диапазоне от 0,66 М до 2 М.
В качестве другого примера - в случаях, когда выполняют катионообменную хроматографию, смолу вначале инкубируют с подходящим связывающим буфером перед нанесением на колонку материала, полученного после удаления загрязнений посредством обработки раствором соли с концентрацией, лежащей в диапазоне от 0,35 М до 0,60 М. Связывающий буфер предпочтительно содержит NaCl и неионогенное поверхностно-активное вещество (например, Твин 80), и концентрация NaCl предпочтительно является низкой (от 0,3 М до 0,6 М). Затем на колонку для катионообменной хроматографии наносят пробу, после чего элюируют белки из смолы. Для элюирования предпочтителен ступенчатый градиент NaCl, и, более предпочтительно, желаемый белок L1 элюируют с использованием элюирующего буфера, содержащего NaCl в концентрациях, равных 0,6 М, 0,7 М, 0,8 М и 1 М.
Катионообменную хроматографию можно выполнить с использованием различных смол, и катионообменную хроматографию можно выполнить с использованием, предпочтительно, катионообменного материала, с которым связываются сульфогруппы, сульфоалкильные (например, сульфометильные, сульфоэтильные и сульфопропильные), фосфатные или фосфаталкильные функциональные группы, и наиболее предпочтительно - катионообменного материала, с которым связываются фосфатные функциональные группы.
Фракции белка L1, полученные с использованием процедуры хроматографии, можно сконцентрировать с использованием мембранного фильтра. Например, хроматографические фракции можно сконцентрировать с использованием мембраны, которая способна отсекать вещества с молекулярной массой, лежащей в диапазоне от 50 кДа до 100 кДа. Поскольку VLP, имеющие средний размер около 50 нм, крупнее остальных белков, VLP не могут пройти через мембрану и поэтому концентрируются, тогда как большая часть остаточных загрязнений удаляется за счет проникновения через мембрану.
Согласно варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является хроматографией на гепарин-сефарозе, солью для обработки на стадии (с) является NaCl, и концентрация NaCl для обработки лежит в диапазоне от 0,35 М до 0,60 М, от 0,40 М до 0,60 М, от 0,45 М до 0,60 М или от 0,45 М до 0,55 М.
Согласно другому варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является хроматографией на гепарин-сефарозе, солью для обработки на стадии (с) является KCl, и концентрация KCl для обработки лежит в диапазоне от 0,35 М до 0,60 М, от 0,40 М до 0,60 М, от 0,45 М до 0,60 М, от 0,50 М до 0,60 М или от 0,55 М до 0,60 M.
Согласно еще одному варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является хроматографией на гепарин-сефарозе, солью для обработки на стадии (с) является Na2CO3, и концентрация Na2CO3 для обработки лежит в диапазоне от 0,35 М до 0,60 М или от 0,40 М до 0,60 М; от 0,35 М до 0,45 М; или от 0,55 М до 0,60 М.
Согласно еще одному варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является катионообменной хроматографией, солью для обработки на стадии (с) является NaCl, и концентрация NaCl для обработки лежит в диапазоне от 0,35 М до 0,60 М, от 0,40 М до 0,60 М или от 0,45 М до 0,55 М.
Согласно еще одному варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является катионообменной хроматографией, солью для обработки на стадии (с) является KCl, и концентрация KCl для обработки лежит в диапазоне от 0,35 М до 0,60 М, от 0,35 М до 0,55 М, от 0,35 М до 0,50 М или от 0,35 М до 0,45 М.
Согласно еще одному варианту осуществления настоящего изобретения, в тех случаях, когда хроматография на стадии (d) является катионообменной хроматографией, солью для обработки на стадии (с) является Na2CO3, и концентрация Na2CO3 для обработки лежит в диапазоне от 0,35 М до 0,60 М, от 0,35 М до 0,55 М, от 0,35 М до 0,50 М или от 0,35 М до 0,45 М.
Полезные эффекты
Отличительные признаки и преимущества настоящего изобретения кратко изложены ниже:
(i) Настоящее изобретение обеспечивает способ получения вирусоподобных частиц (VLP) на основе белка L1 HPV.
(ii) Настоящее изобретение может обеспечить белок L1 HPV с высокой степенью чистоты с использованием одного хроматографического процесса за счет способа, в котором загрязнения вначале удаляют посредством осаждения сульфатом аммония, а затем дополнительно удаляют из раствора, содержащего соль в концентрации от 0,35 М до 0,60 М, посредством диализа.
(iii) Антигены HPV, полученные способом по настоящему изобретению, обладают превосходной стабильностью и поэтому пригодны для использования в качестве сырьевого материала для диагностического средства, выявляющего антитела к HPV, требующего высокой стабильности, а также для вакцин.
Краткое описание графических материалов
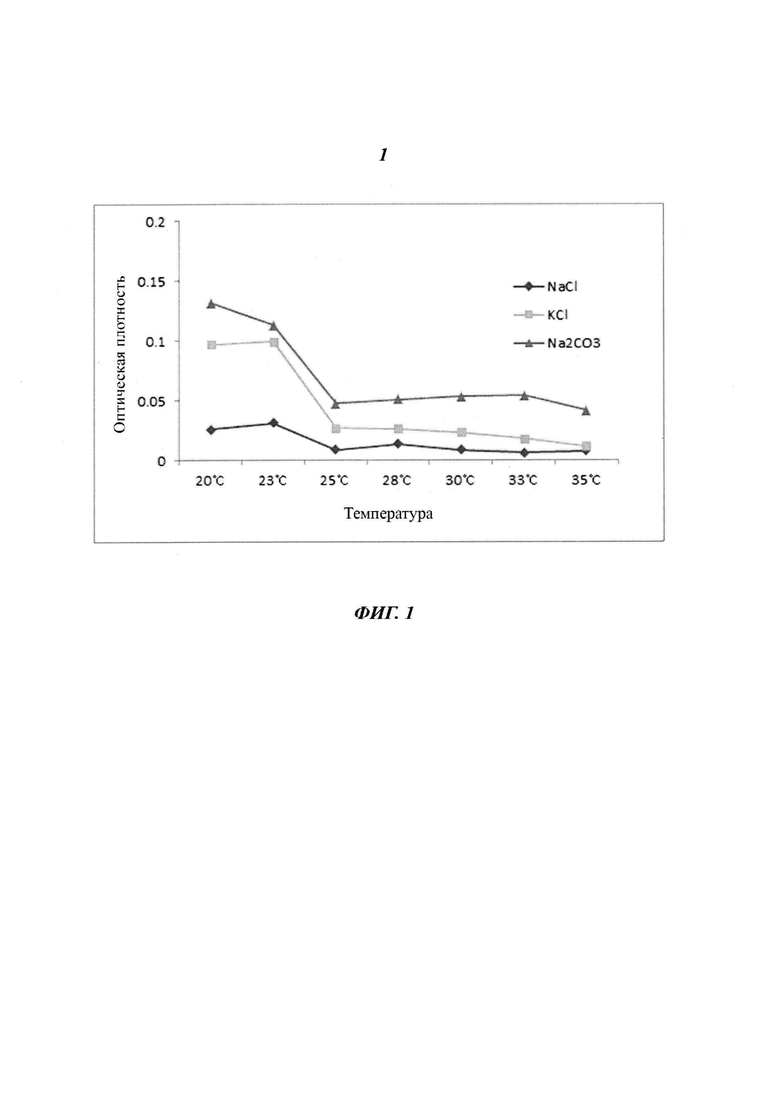
Фиг. 1 является диаграммой, относящейся к Примеру 1, и демонстрирует зависимость эффективности удаления нерастворимых белков от вида буферного раствора, используемого для фракционирования, и температуры инкубации при очистке белка L1 HPV 16 типа.
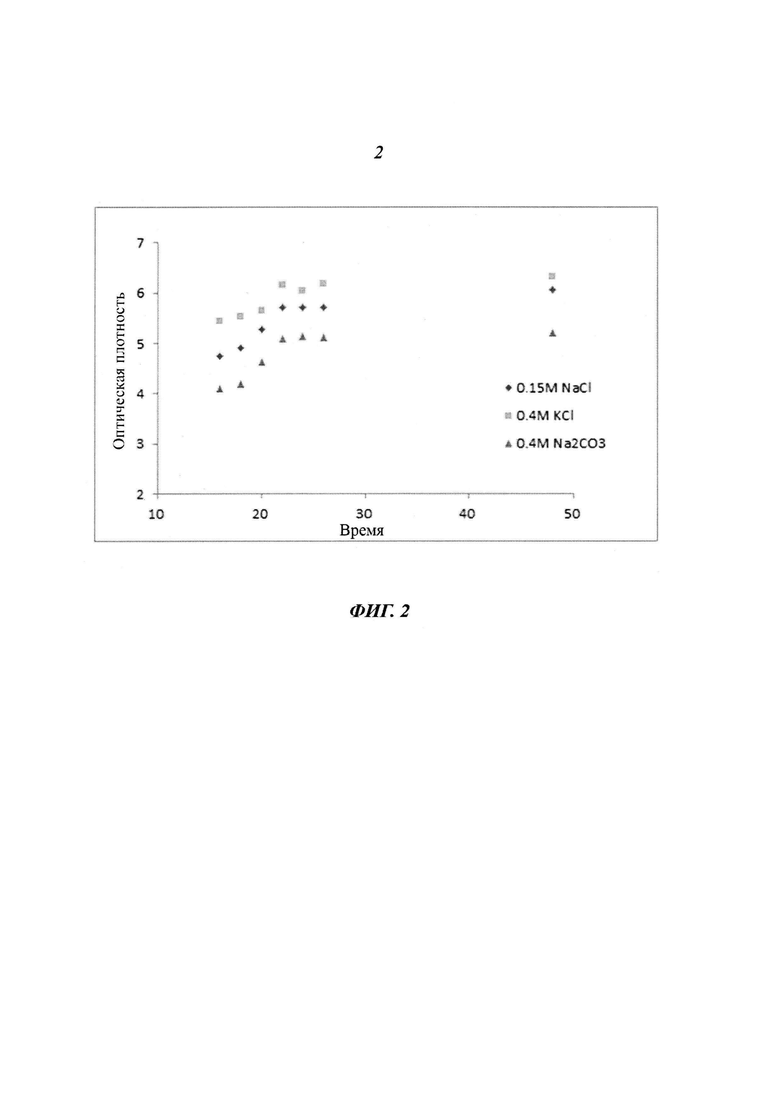
Фиг. 2 является диаграммой, относящейся к Примеру 1, и демонстрирует зависимость эффективности удаления нерастворимых белков от вида буферного раствора, используемого для фракционирования, и времени инкубации при очистке белка L1 HPV 16 типа.
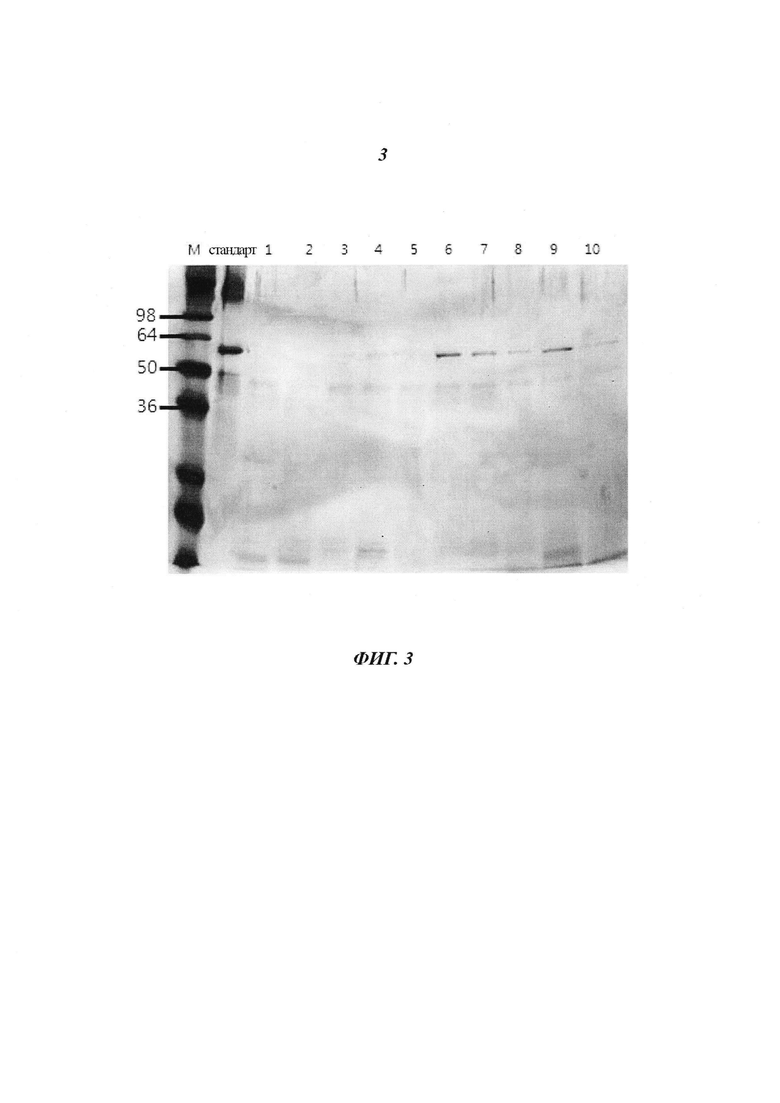
Фиг. 3 является диаграммой, относящейся к Примеру 2, и демонстрирует профили очистки соответствующих фракций в случае, когда инкубацию проводили при 35°C на стадии удаления нерастворимых белков при очистке белка L1 HPV 16 типа.
Фиг. 4 является диаграммой, относящейся к Примеру 2, и демонстрирует изменение выхода белка L1 HPV 16 типа после очистки в зависимости от концентрации преципитата, полученного посредством осаждения сульфатом аммония.
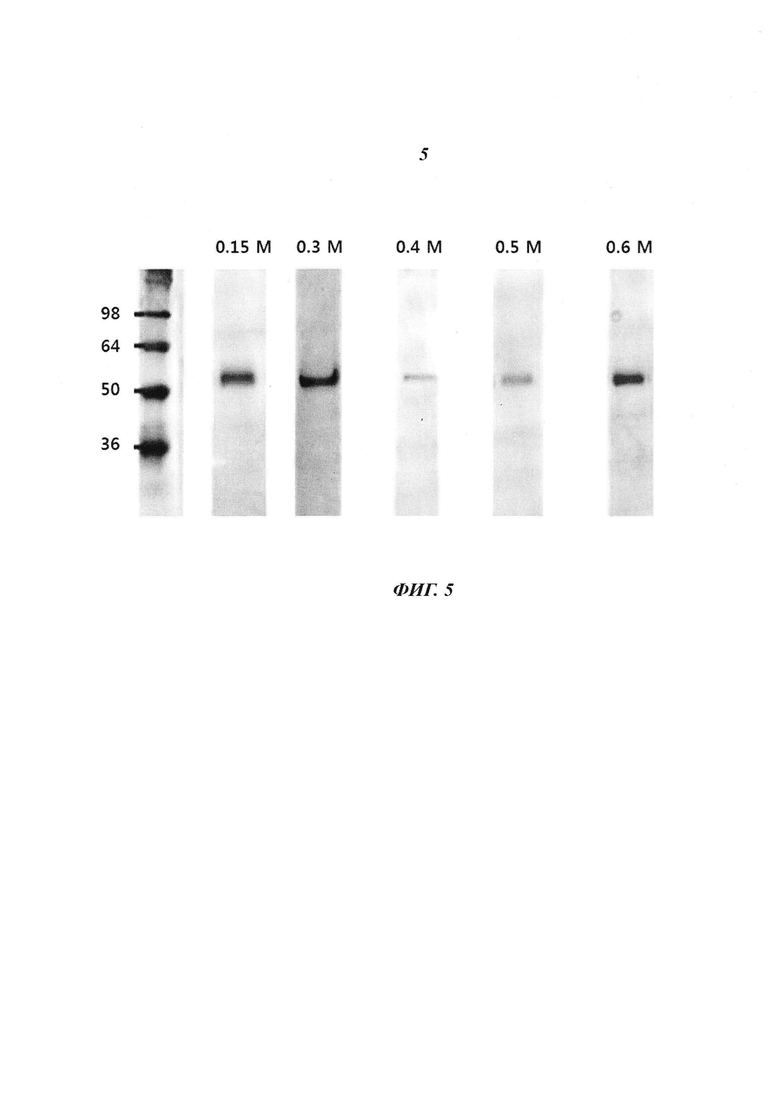
Фиг. 5 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством катионообменной хроматографии после стадии фракционирования с использованием NaCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
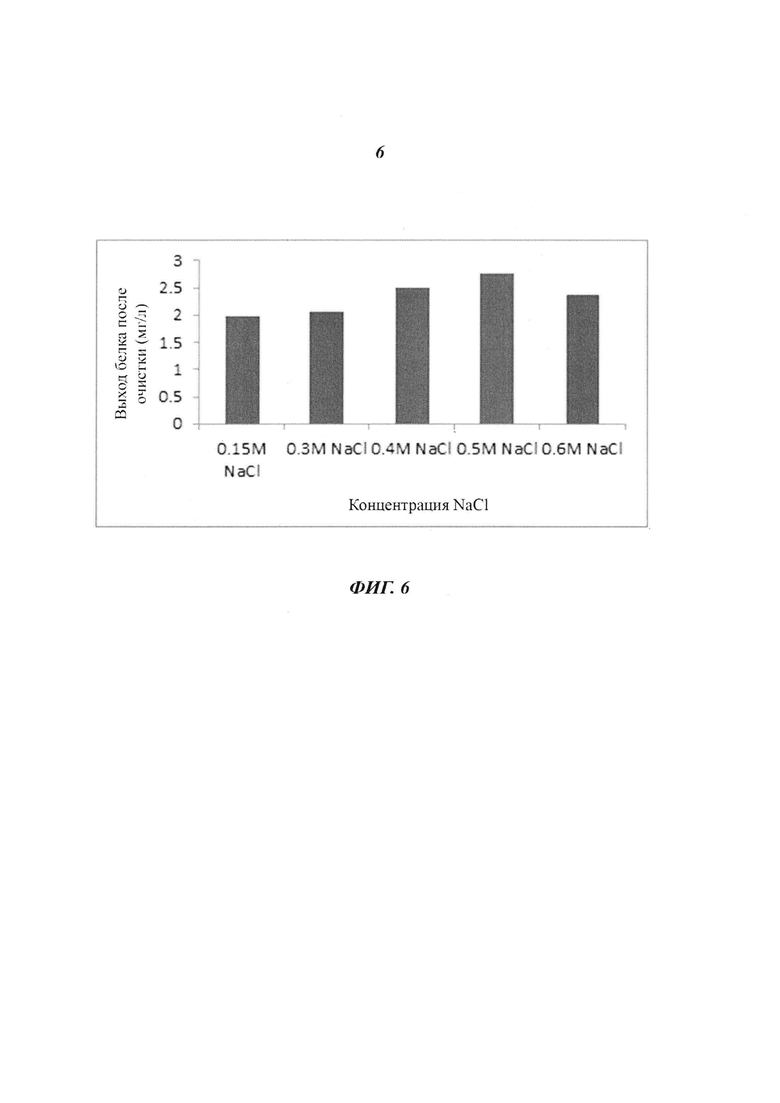
Фиг. 6 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством катионообменной хроматографии после стадии фракционирования с использованием NaCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
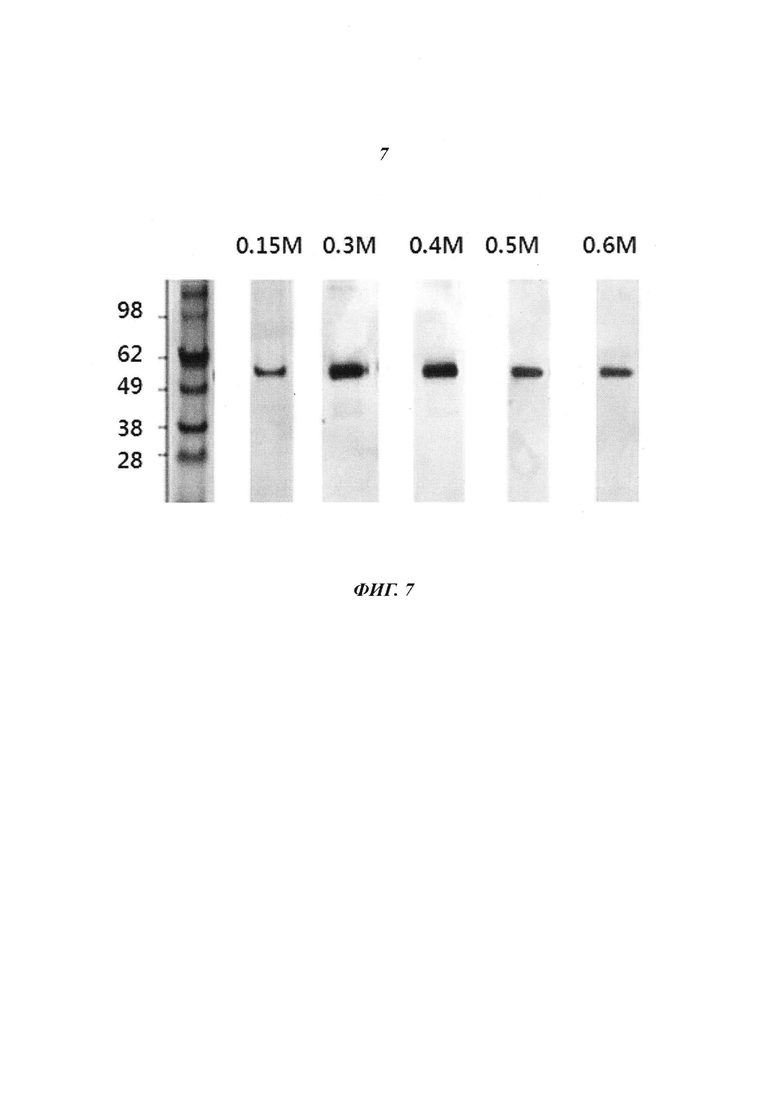
Фиг. 7 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством катионообменной хроматографии после стадии фракционирования с использованием KCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
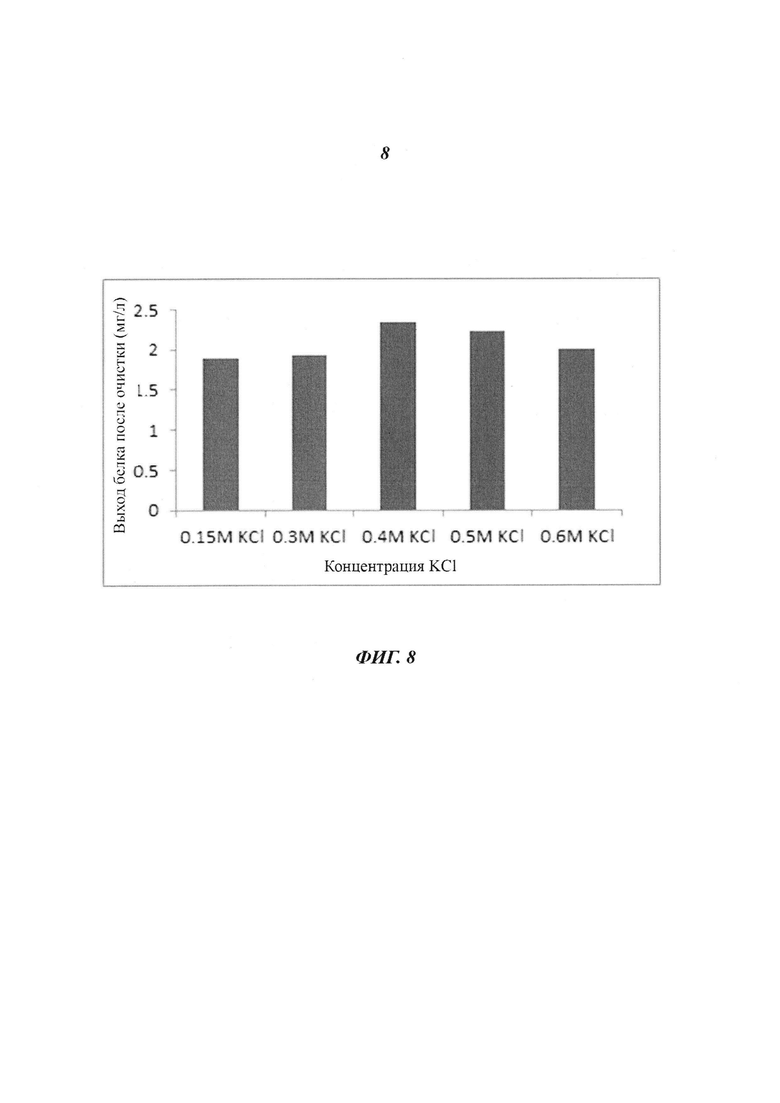
Фиг. 8 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством катионообменной хроматографии после стадии фракционирования с использованием KCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
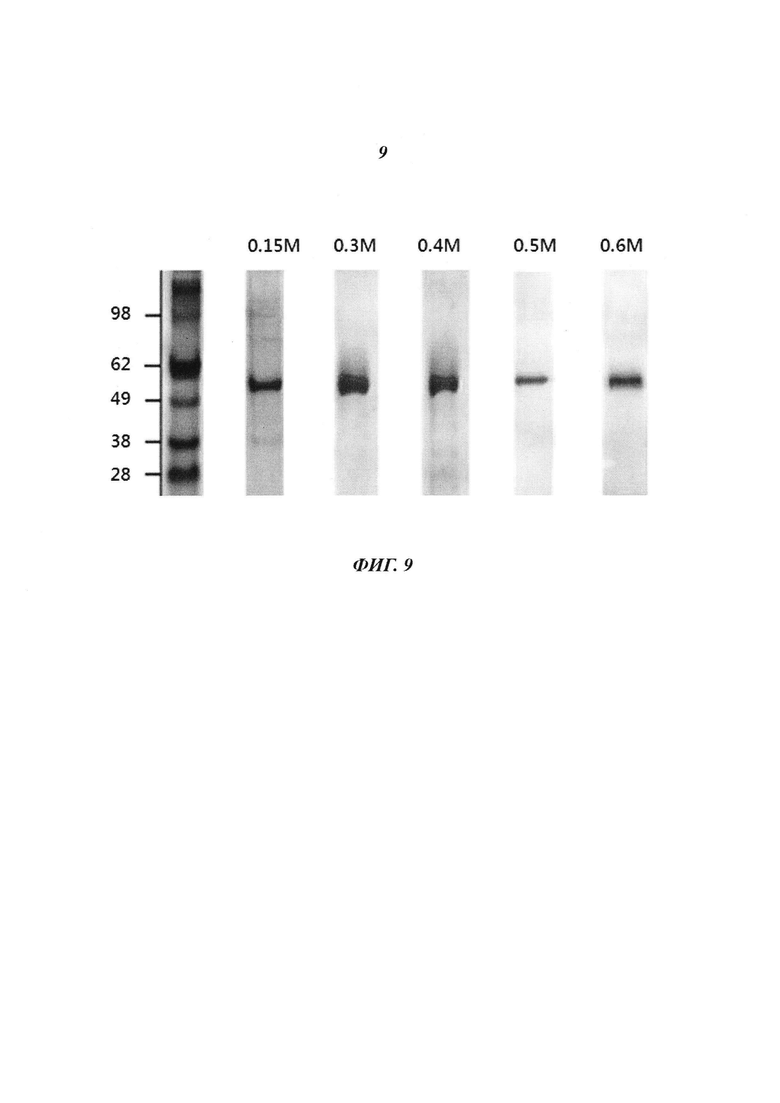
Фиг. 9 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством катионообменной хроматографии после стадии фракционирования с использованием Na2CO3 в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
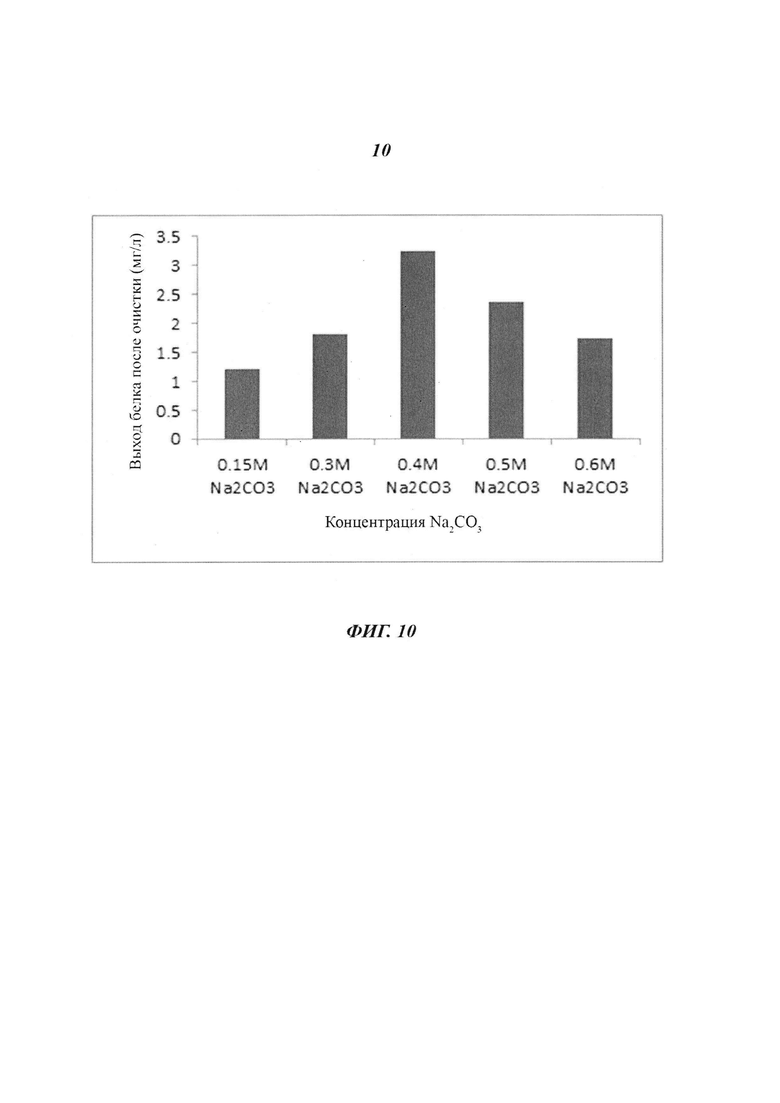
Фиг. 10 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством катионообменной хроматографии после стадии фракционирования с использованием Na2CO3 в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
Фиг. 11 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством аффинной хроматографии после стадии фракционирования с использованием NaCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
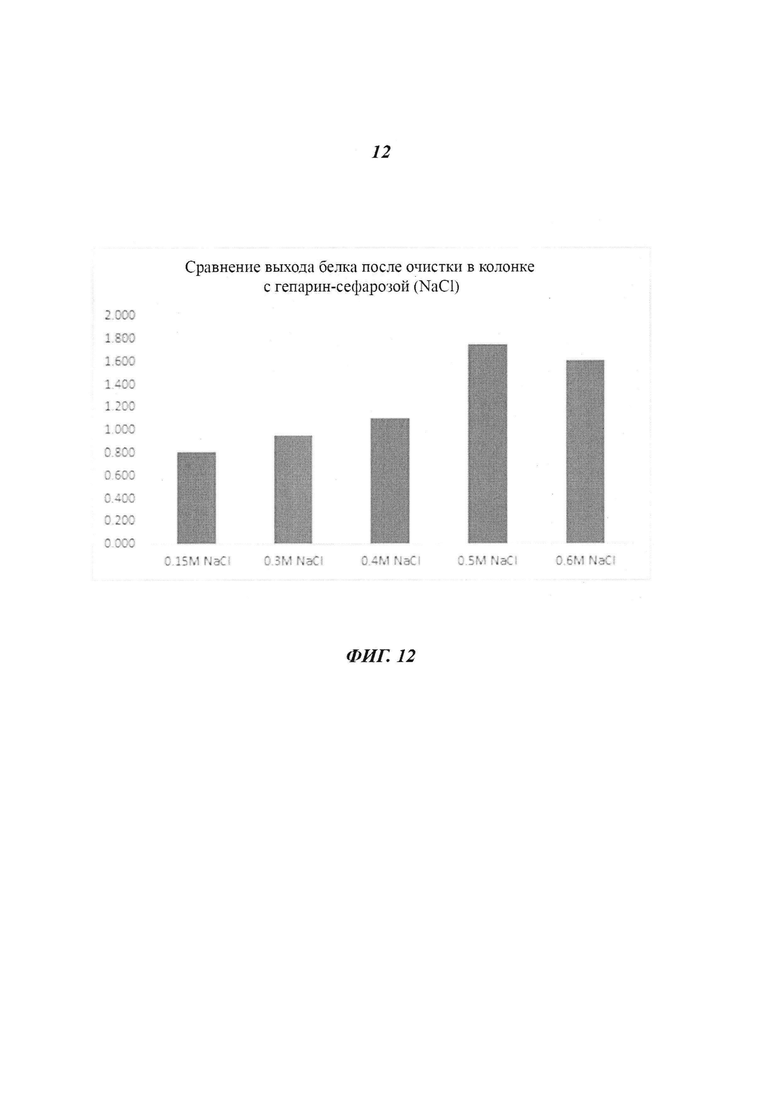
Фиг. 12 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством аффинной хроматографии после стадии фракционирования с использованием NaCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
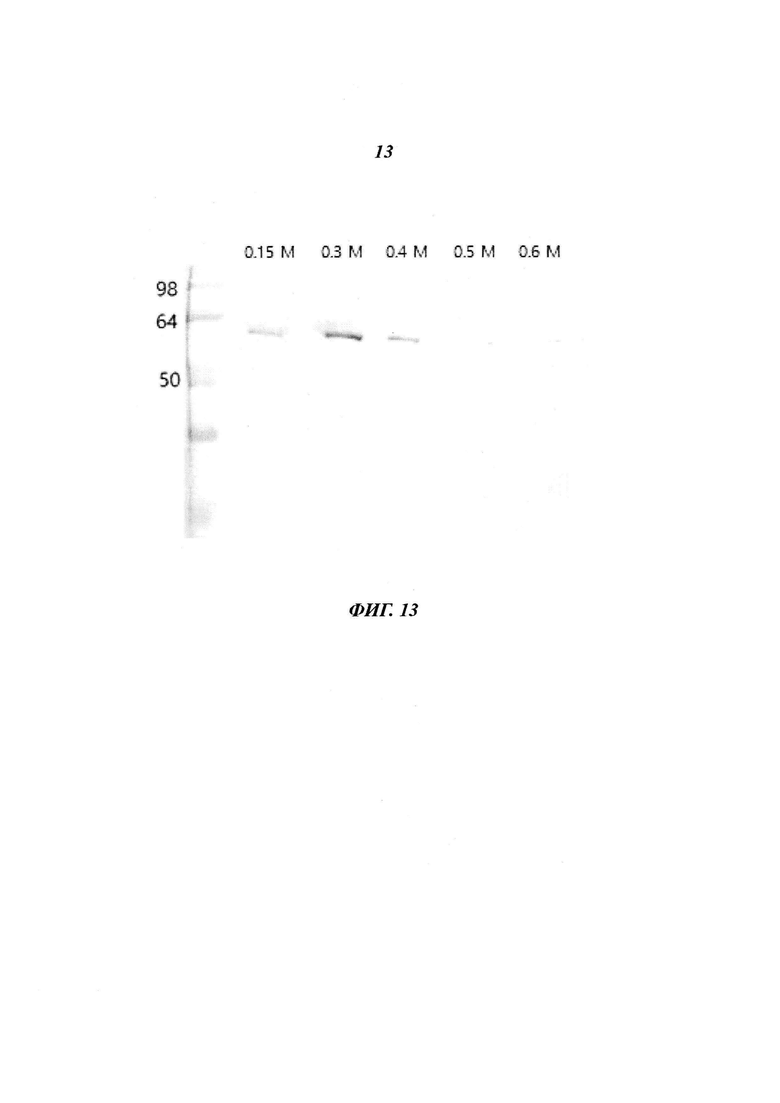
Фиг. 13 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством аффинной хроматографии после стадии фракционирования с использованием KCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
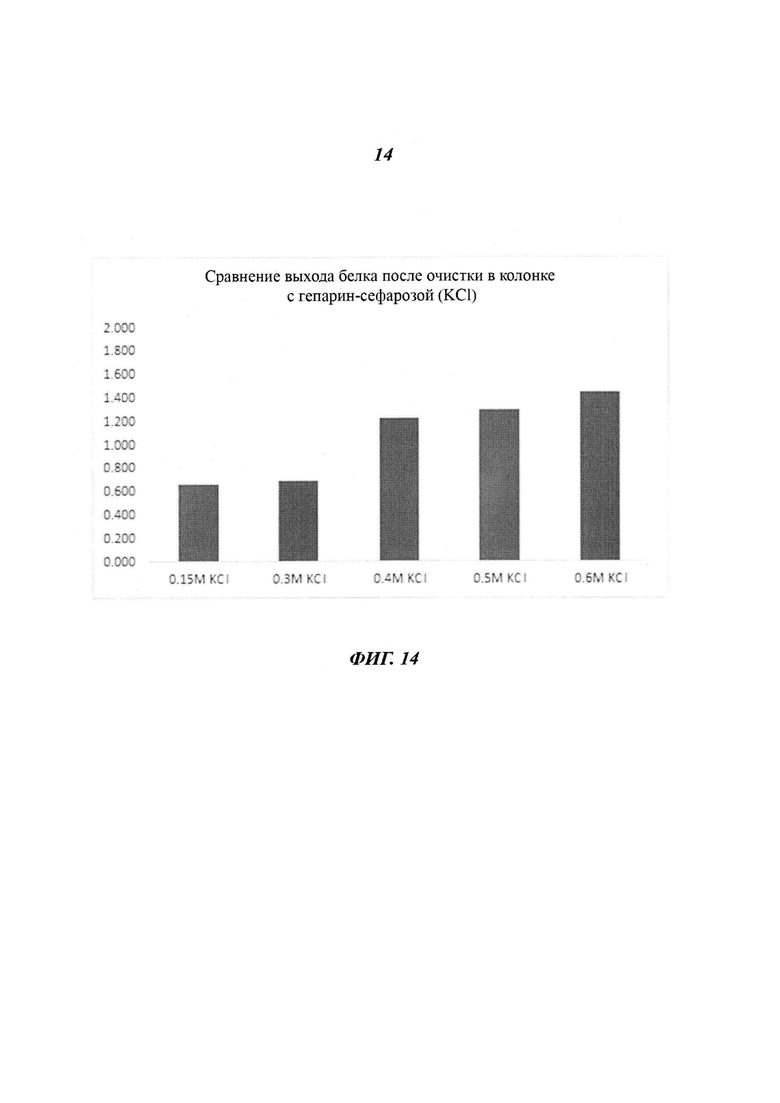
Фиг. 14 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством аффинной хроматографии после стадии фракционирования с использованием KCl в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
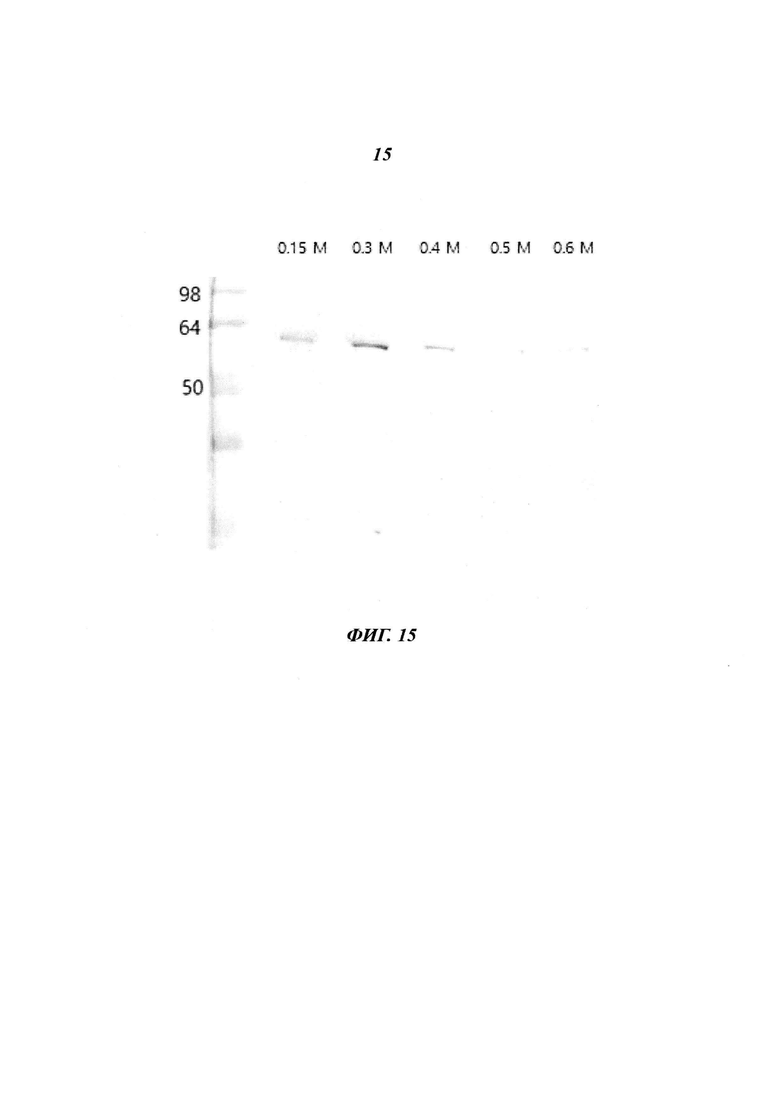
Фиг. 15 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством аффинной хроматографии после стадии фракционирования с использованием Na2CO3 в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
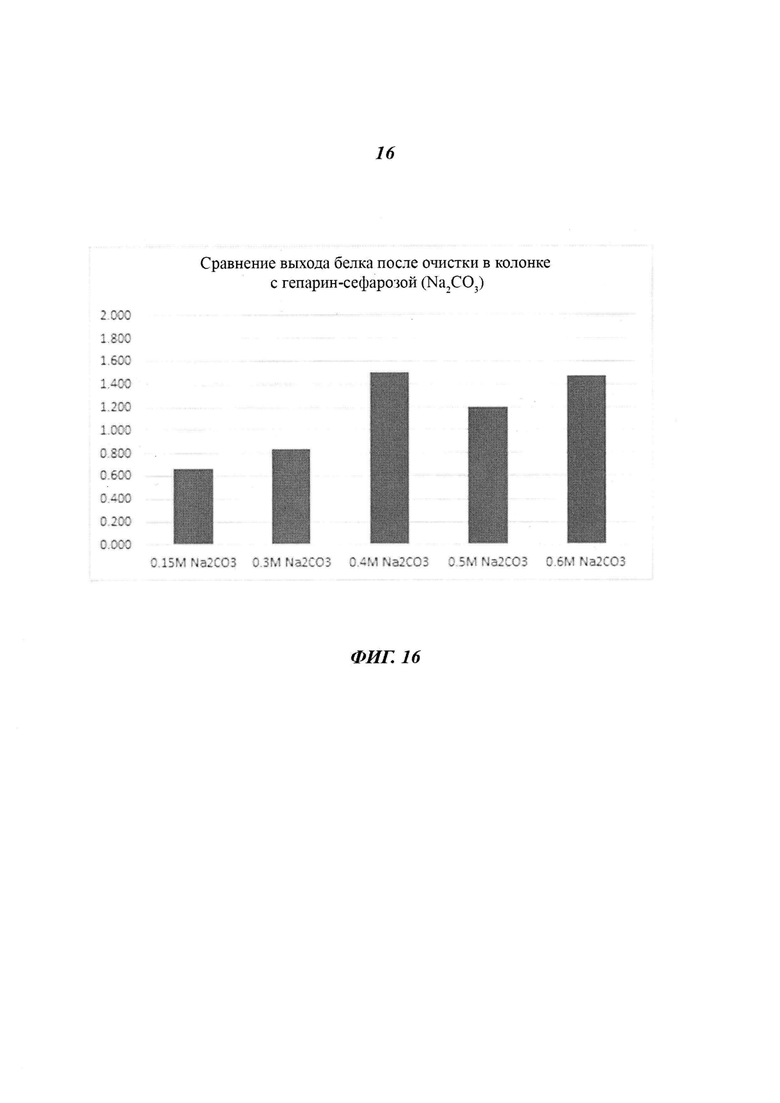
Фиг. 16 является диаграммой, относящейся к Примеру 3, и демонстрирует выходы белка после очистки посредством аффинной хроматографии после стадии фракционирования с использованием Na2CO3 в концентрации, лежащей в диапазоне от 0,15 М до 0,6 М.
Фиг. 17 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством катионообменной хроматографии белков L1 HPV 16 и 18 типов. Дорожка 1: 0,5 М NaCl, Дорожка 2: 0,4 М KCl, Дорожка 3: 0,4 М Na2CO3.
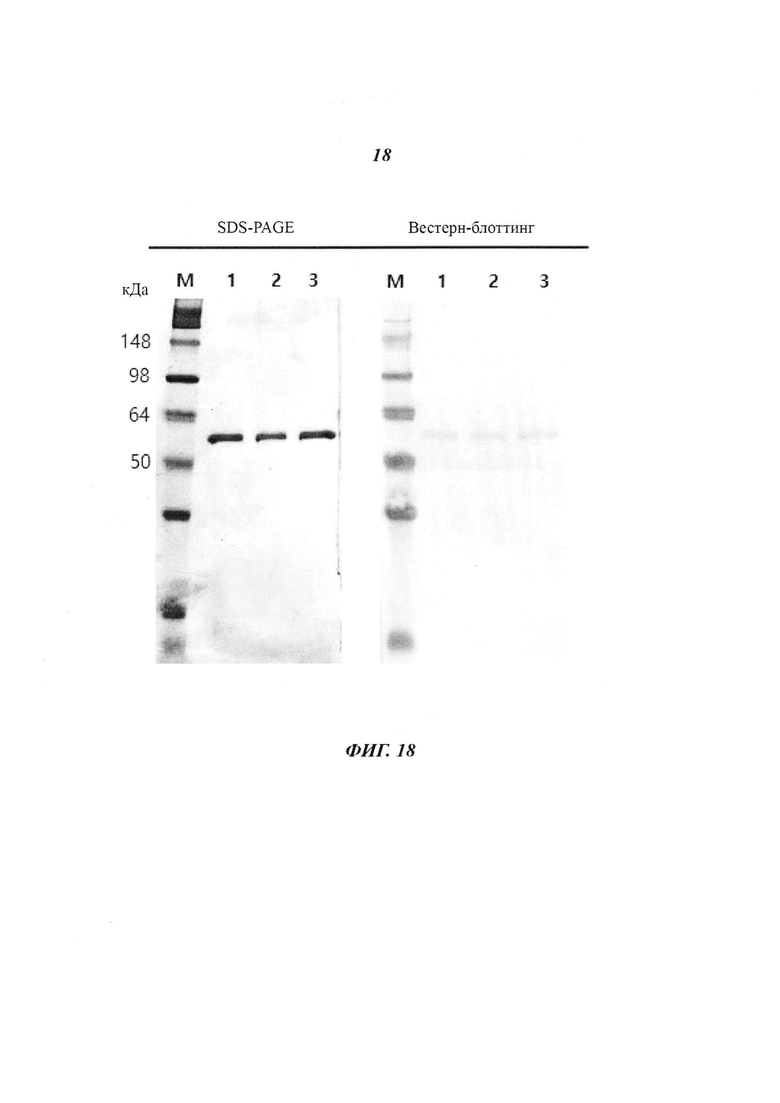
Фиг. 18 является диаграммой, относящейся к Примеру 3, и демонстрирует результаты очистки посредством аффинной хроматографии белка L1 HPV 16 типа. Дорожка 1: 0,5 М NaCl, Дорожка 2: 0,4 М KCl, Дорожка 3: 0,4 М Na2CO3.
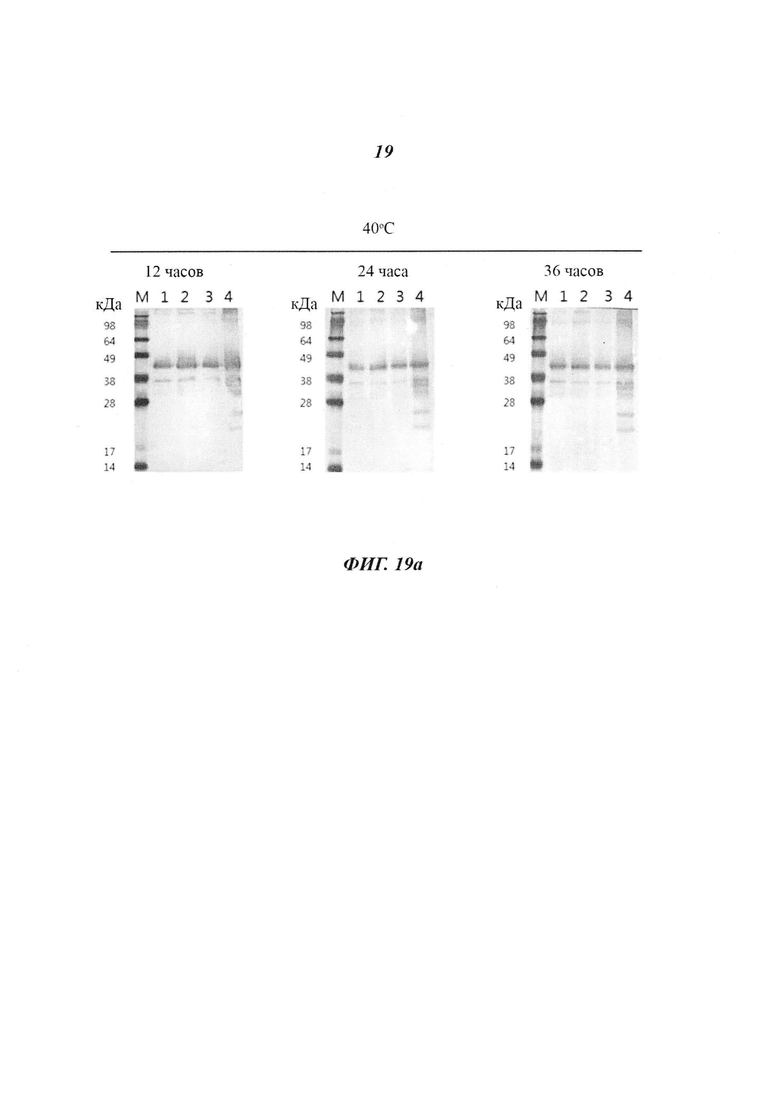
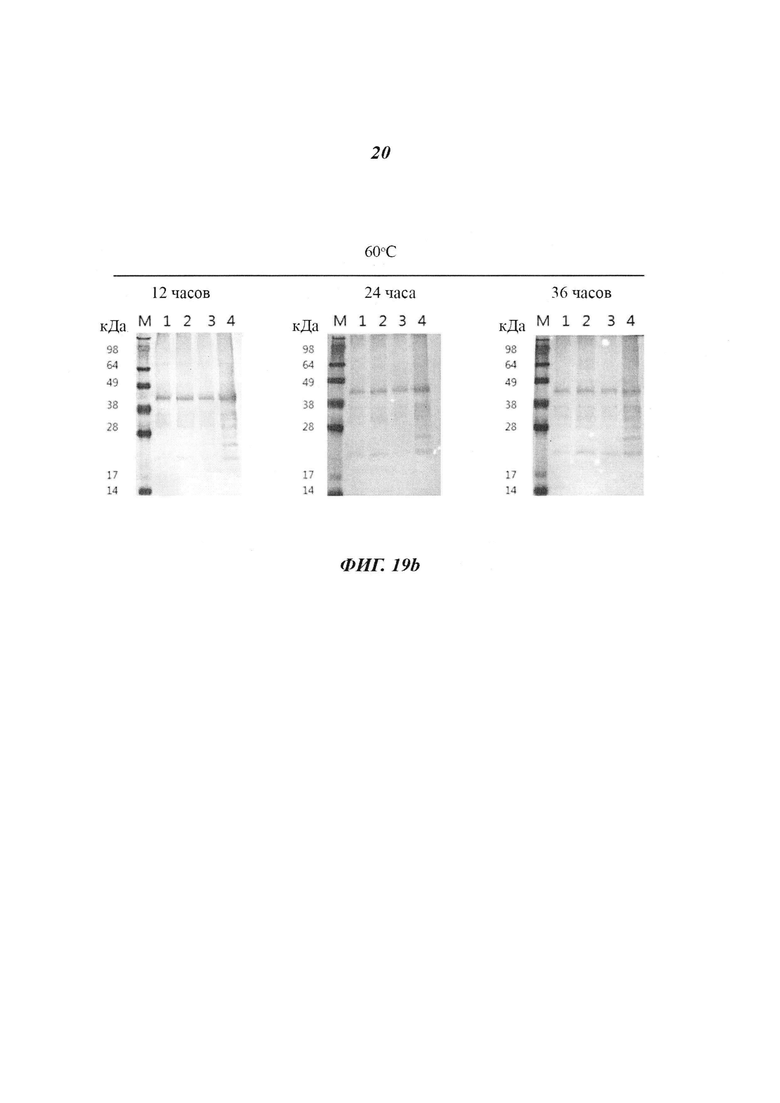
Фиг. 19 является диаграммой, относящейся к Примеру 4, и демонстрирует результаты оценки стабильности антигена HPV в зависимости от вида и концентрации соли и температуры реакции на стадии фракционирования. Дорожка 1: 0,5 М NaCl, Дорожка 2: 0,4 М KCl, Дорожка 3: 0,4 М Na2CO3, Дорожка 4: 0,15 М NaCl.
Описание примеров осуществления изобретения
Далее настоящее изобретение будет описано подробно со ссылкой на примеры его осуществления. Эти примеры предназначены исключительно для более конкретной иллюстрации настоящего изобретения, и специалистам в данной области техники будет очевидно, что объем настоящего изобретения не ограничен этими примерами.
Пример 1
Определение температуры и времени для эффективного удаления нерастворимых белков
1-1. Способы
С целью определения температурных и временных диапазонов для эффективного удаления нерастворимых белков в способе получения белков L1 HPV 16 и 18 типов провели следующее испытание.
Дрожжевые клетки (Saccharomyces cerevisiae; КССМ11036Р и КССМ11037Р, соответственно), которые были трансформированы, соответственно, YEGα-HPV16L1-ROS и YEGα-HPV18L1-ROS, в которые были включены гены рекомбинантных белков L1 HPV 16 типа и HPV 18 типа, инокулировали в синтетические полные культуральные среды без урацила (SD-ura), находившиеся в колбах с ребрами, после чего производили встряхивание культуры при 30°C. Для экспрессии белков L1 HPV 16 типа и HPV 18 типа с промотора GAL10 использовали YPDG среды. Ко всем средам добавили 1% экстракта дрожжей (производства компании Duchefa Biochem, Нидерланды) и 2% пептона (производства компании Duchefa Biochem, Нидерланды), а также глюкозу и галактозу. После инокуляции трансформированного штамма дрожжей в YPDG среду клетки культивировали при 30°C с использованием ферментера. Время культивирования не превышало 96 часов.
Дрожжевые клетки смешали с буферным раствором (20 мМ фосфата натрия, pH 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА (этилендиаминтетрауксусная кислота), 0,01% Твина 80). Затем клетки гомогенизировали гомогенизатором клеток и отделили супернатант посредством центрифугирования гомогената клеток. 45 масс. % сульфата аммония добавили к собранному супернатанту для осаждения, после чего выполнили центрифугирование при высокой скорости (24000 g), чтобы удалить супернатант и собрать только преципитат. Концентрацию белков в полученном преципитате измерили посредством анализа с бицинхониновой кислотой (ВСА; от англ.: bicinchoninic acid).
Преципитат разбавили фосфатным буферным раствором с Твином 20 (1Х PBS-T; от англ.: phosphate buffer solution) до концентрации, равной 30 мг/мл, и к разбавленному раствору добавили NaCl до конечной концентрации, равной 0,8 М. После перемешивания в течение примерно 30 минут раствор инкубировали при 4°C в течение 18 часов, при этом он находился в покое.
После инкубации с NaCl суспензию разделили на четыре части и соответствующие части суспензии поместили в диализные мембраны для проведения диализа с буферами для фракционирования (PBS+0,01% Твина 80) с добавлением (0,15 М NaCl, 0,5 М NaCl, 0,4 М KCl, 0,4 М Na2CO3) трех видов солей (NaCl, KCl, Na2CO3). После завершения диализа добавили соответствующие буферные растворы для фракционирования для разбавления концентрации белка до 10 мг/мл или менее и инкубировали растворы в инкубаторе при температуре, лежавшей в диапазоне от 20°C до 35°C (±2°C) в течение 24 часов, при этом они находились в покое. Через 24 часа образовавшийся материал центрифугировали при высокой скорости (24000 g) для сбора супернатанта и затем измерили мутность собранного супернатанта с использованием спектрометра (определили оптическую плотность при длине волны, равной 600 нм).
Кроме того, пробы преципитата диализировали с буферными растворами для фракционирования, к которым были добавлены соли четырех типов, после чего их оставляли в инкубаторе при температуре, равной 30°C (±2°C), на 16-26 часов (с отбором проб с двухчасовыми интервалами) или на 48 часов для определения промежутка времени, во время которого мутность была минимальной.
1-2. Результаты
Процедуру удаления нерастворимых белков осуществляют посредством осаждения загрязнений при комнатной температуре для повышения степени чистоты во время очистки. Чем ниже значение оптической плотности супернатанта, отделенного посредством центрифугирования после инкубации, тем большее количество загрязнений удалено.
Как показано на Фиг. 1, значение мутности варьируется в зависимости от вида соли, содержащейся в буфере для фракционирования, однако, в целом, чем выше температура инкубации, тем ниже значение оптической плотности, измеренное при длине волны, равной 600 нм, и на основании этого результата можно сделать вывод о том, что удаление нерастворимых белков легче осуществить при более высокой температуре. Однако было установлено, что при слишком высокой температуре могут возникнуть проблемы со структурой белка. Поэтому при дальнейшем анализе, в ходе которого сравнили выходы белка L1 HPV 16 типа, который был очищен посредством удаления нерастворимых белков после инкубации в течения 24 часов при 30°C и 35°C, определили, что выход белка после очистки при 30°C был равен примерно 2,65 мг/л, тогда как выход белка после очистки при 35°C был равен примерно 1,24 мг/л, и также была продемонстрирована низкая степень чистоты. Эти результаты подтвердили, что удаление нерастворимых белков при превышении подходящего диапазона температур имело низкую эффективность, и температурный диапазон, обеспечивающий эффективную очистку, составлял от примерно 25°C до 34°C.
Что касается времени инкубации нерастворимых белков, то, как показано на Фиг. 2, по результатам измерения мутности в течение периода, лежащего в диапазоне от 16 часов до 26 часов, с 2-часовыми интервалами и через 48 часов при добавлении каждой соли установлено, что мутность суспензии увеличилась из-за осаждения нерастворимых белков и оставалась на определенном уровне в течение периода исследования. Мутность суспензии означает, что степень чистоты белка возрастает вследствие осаждения загрязнений. Тем не менее, поскольку инкубация в течение слишком длительного периода времени может привести к осаждению целевого белка, то определено, что диапазон периода инкубации, подходящего для удаления нерастворимых белков, составляет от 21 часа до 26 часов.
Пример 2
Сравнение зависимости выходов белка после очистки от концентрации преципитата, полученного посредством осаждения сульфатом аммония 2-1.
Способы
Дрожжевые клетки из Примера 1 смешали с буферным раствором (20 мМ фосфата натрия, pH 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА, 0,01% Твина 80). Затем клетки гомогенизировали гомогенизатором клеток и отделили супернатант посредством центрифугирования гомогената клеток при высокой скорости (24000 g). К собранному супернатанту добавили 5 масс. % сульфата аммония для осаждения, после чего выполнили центрифугирование с высокой скоростью (24000 g) для отделения супернатанта и получения только преципитата. Концентрацию белка в полученном преципитате измерили посредством анализа с бицинхониновой кислотой (ВСА), после чего добавили 1Х PBS-T для разбавления концентрации преципитата, полученного посредством осаждения сульфатом аммония, до концентраций, лежавших в диапазоне от 10 мг/мл до 60 мг/мл (с шагом, равным 10 мг/мл). К преципитатам в соответствующих концентрациях добавили NaCl до конечной концентрации, равной 0,8 М, с последующим перемешиванием в течение примерно 30 минут, и инкубировали смеси при 4°C в течение 18 часов, при этом раствор находился в покое. После инкубации с NaCl суспензии с соответствующими концентрациями поместили в мембраны для диализа и диализировали с буферными растворами для фракционирования. После завершения диализа добавили буферные растворы из Примера 1 для разбавления концентрации белка до 10 мг/мл или менее и инкубировали растворы в инкубаторе при температуре, равной 30°C (±2°C), в течение 24 часов, при этом они находились в покое. Через 24 часа образовавшийся материал центрифугировали при высокой скорости (24000 g) для отделения супернатанта. Собранный супернатант поместили в диализную мембрану и диализировали со связывающим буфером (PBS+0,35 М NaCl, pH 7,2, 0,01% Твина 80) при 4°C. Фильтрат очистили посредством катионообменной хроматографии. Более конкретно, колонку Poly-Prep размером 1,6 см × 5 см (производства компании GE Healthcare Life Science, США), заполненную смолой Р-11 на основе фосфата целлюлозы (производства компании Whatman, Соединенное Королевство), уравновесили со связывающим буфером (PBS+0,5 М NaCl), объем которого был в пять раз больше объема смолы. После завершения диализа раствор пропустили через колонку с Р-11 смолой для связывания, после чего промыли колонку связывающим буфером, объем которого был в пять раз больше объема смолы. После промывки белок L1 элюировали посредством пропускания 4-5 мл элюирующих буферов, содержавших 0,6 М, 0,7 М, 0,8 М и 1 М NaCl, через связывающий буфер. Фракции с подтвержденными целевыми полосами объединяли и количественно определяли содержание белка посредством анализа с ВСА, после чего рассчитывали выход белка после очистки.
2-2. Результаты
Как показано на Фиг. 4, по результатам сравнения выхода белка после очистки в зависимости от концентрации преципитата, полученного посредством осаждения солью аммония, выход белка после очистки был наивысшим и равным примерно 2,6 мг/л, если концентрация преципитата, полученного посредством осаждения солью аммония, была равна 30 мг/мл, а следующее значение было равно примерно 2,2 мг/л, если концентрация преципитата, полученного посредством осаждения солью аммония, была равна 40 мг/мл. По этим результатам определили, что диапазон концентраций преципитата, полученного посредством осаждения сульфатом аммония, обеспечивающий высокий выход антигена HPV после очистки, составляет от 25 мг/мл до 65 мг/мл.
Пример 3
Определение вида и концентрации оптимальной соли на стадии фракционирования
3-1. Способы
При получении белка L1 HPV стадию фракционирования выполняют для удаления загрязнений посредством инкубации с использованием соли. Для выбора вида соли, подходящей для очистки антигена VLP HPV, и исследования зависимости степени удаления загрязнений от концентрации соли провели следующее испытание. Основные соли показаны в Таблице 1.
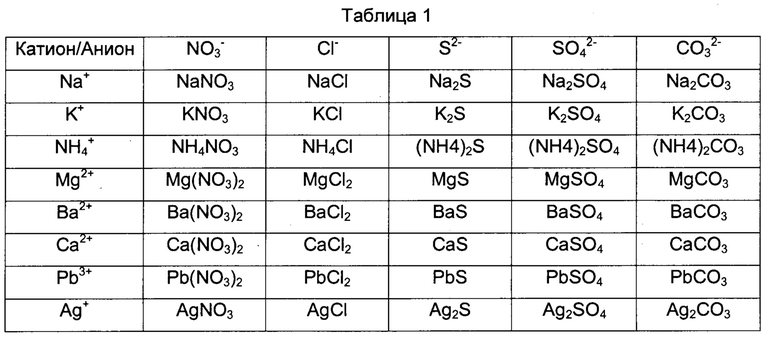
Дрожжевые клетки из Примера 1 смешали с буферным раствором (20 мМ фосфата натрия, pH 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА, 0,01% Твина 80). Затем клетки гомогенизировали гомогенизатором клеток и отделили супернатант посредством центрифугирования гомогената клеток. К собранному супернатанту добавили 45 масс. % сульфата аммония и центрифугировали смесь при высокой скорости (24000 g), чтобы удалить супернатант и оставить только преципитат (в данном случае суспензию получили посредством добавления к преципитату небольшого количества 1Х PBS-T). Концентрацию белка в преципитате измерили посредством анализа с ВСА и добавили 1Х PBS-T для разбавления концентрации преципитата, полученного посредством осаждения сульфатом аммония, до 30 мг/мл или менее. К разбавленному преципитату при медленном перемешивании добавили NaCl до получения конечной концентрации, равной 0,8 М. После перемешивания в течение примерно 30 минут разбавленный раствор инкубировали при 4°C в течение 18 часов, при этом раствор находился в покое. После инкубации с NaCl суспензию поместили в мембрану для диализа и диализировали в буферных растворах для фракционирования с использованием компонентов из Таблицы 2. После завершения диализа добавили соответствующие фракционирующие буферы для разбавления концентрации белка до 10 мг/мл или менее и инкубировали раствор в инкубаторе при температуре, равной 30°C (±2°C), в течение 24 часов, при этом раствор находился в покое.
Через 24 часа полученный материал центрифугировали при высокой скорости (24000 g), чтобы собрать супернатант. Собранный супернатант поместили в диализную мембрану и диализировали со связывающим буферным раствором (PBS+0,35 М NaCl, pH 7,2, 0,01% Твина 80) при 4°C. После диализа со связывающим буферным раствором супернатант профильтровали посредством центрифугирования при высокой скорости (24000 g). Как показано в Примере 2, фильтрат очистили посредством катионообменной хроматографии и затем собрали только фракции высокой степени чистоты для расчета выхода. Кроме того, чтобы исследовать, будут ли получены такие же результаты с другим типом смолы, провели испытание с очисткой посредством аффинной хроматографии с использованием соли, обеспечившей наивысший выход среди соответствующих солей, использованных в катионообменной хроматографии.
Более конкретно, для аффинной хроматографии колонку, заполненную гепарин-сефарозной смолой (производства компании GE Healthcare Life Sciences, США) уравновесили посредством пропускания через нее связывающего буфера (PBS-T, содержавший 0,33 М NaCl) в объеме, в десять раз превышавшем объем смолы. После завершения диализа раствор пропустили через колонку и дали ему возможность связаться с колонкой, заполненной гепарин-сефарозой, после чего промыли содержавшим 2 М NaCl элюирующим буфером, объем которого был в 50 раз больше объема смолы, с линейным градиентом NaCl, лежавшим в диапазоне от 0,33 М NaCl до 0,5 М NaCl. После промывки элюировали белок L1 элюирующим буфером, содержавшим 2 М NaCl, объем которого был в 20 раз больше объема смолы, с линейным градиентом NaCl, лежавшим в диапазоне от 0,5 М NaCl до 1,5 М NaCl. Фракции с подтвержденными целевыми полосами объединили и количественно оценили посредством анализа с ВСА, после чего рассчитали выход белка после очистки. Для аффинной хроматографии была использована колонка объемом 1 мл, заполненная гепарин-сефарозой (производства компании GE, США).
3-2. Результаты
В результате расчета выхода белка после очистки с использованием катионообменной хроматографии с различными солями в различных концентрациях оказалось, что MgCl2 не обеспечивал очистки при всех концентрациях, a NaCl, KCl и Na2CO3 дали результаты, показанные на Фигурах с 5 по 10. Как и в случае катионообменной хроматографии, в случае дополнительно проведенной аффинной хроматографии оказалось, что высокие выходы были получены при высоких концентрациях соли - от 0,3 М и более.
Пробы, полученные с использованием каждой соли, которая обеспечивала наивысший выход, исходя из приведенных выше результатов, количественно оценили посредством анализа с ВСА, после чего 0,5 мкг пробы подвергли электрофорезу в двух 12%-ных акриламидных гелях при 150 В и 150 мА в течение 1,5 часов. Для подтверждения проявленных проб одну из них подвергли окрашиванию серебром, а другую - вестерн-блоттингу с использованием коммерчески доступных моноклональных антител, специфически реагирующих с антигенами HPV 16 и 18 типов.
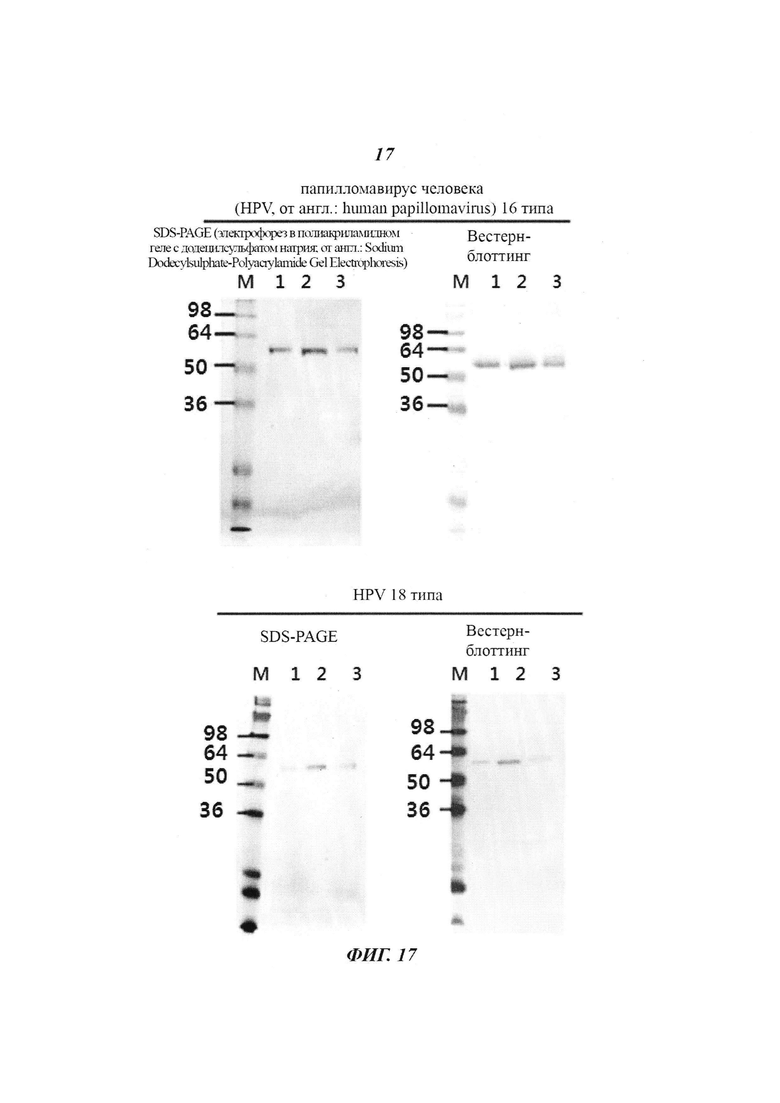
Как показано на Фиг. 17, результаты подтвердили, что все пробы имели высокую степень чистоты и достаточную реакционную способность при взаимодействии с антителами в условиях, когда использовали соответствующие буферные растворы для катионообменной хроматографии (Дорожка 1 : 0,5 М NaCl, Дорожка 2 : 0,4 М KCl, Дорожка 3 : 0,4 М Na2CO3).
На основании приведенных выше результатов было подтверждено, что солями, оказывающими благоприятные эффекты при получении VLP антигенов HPV 16 и 18 типов, были NaCl, KCl и Na2CO3, а оптимальным диапазоном концентраций этих солей для удаления нерастворимых белков был диапазон от 0,35 М до 0,60 М.
Пример 4
Оценка стабильности в зависимости от вида и концентрации соли на стадии фракционирования
4-1. Способы
Стабильность антигена оценивали посредством варьирования температуры реакции и времени хранения антигенов, полученных с использованием различных солей и различных концентраций солей на стадии фракционирования. С этой целью провели процедуру фракционирования в соответствии с видом и концентрацией соли и получили антиген L1 VLP HPV 16 типа (0,15 М NaCl, 0,5 М NaCl, 0,4 М KCl, 0,4 М Na2CO3). Стабильность антигена оценили, задав температуру реакции получения антигена равной 40°C и 60°C, а время реакции равным 12, 24 и 36 часам. По 140 мкл антигена дозировали в новые пробирки в соответствии с условиями, указанными в Таблице 3. Пробы антигена получали через каждый час и подтверждали состояние антигена посредством вестерн-блоттинга. Вестерн-блоттинг выполнили посредством количественной оценки каждой пробы анализом с ВСА и электрофореза 0,6 мкг пробы в 12%-ном акриламидном геле при 150 В и 150 мА в течение 1,5 часов с последующим переносом полученной пробы на нитроцеллюлозную мембрану. С перенесенной пробой выполнили реакцию с использованием моноклонального антитела, доступного на рынке, которое специфически реагирует с антигенами HPV 16 и 18 типов, после чего выполнили проявление.
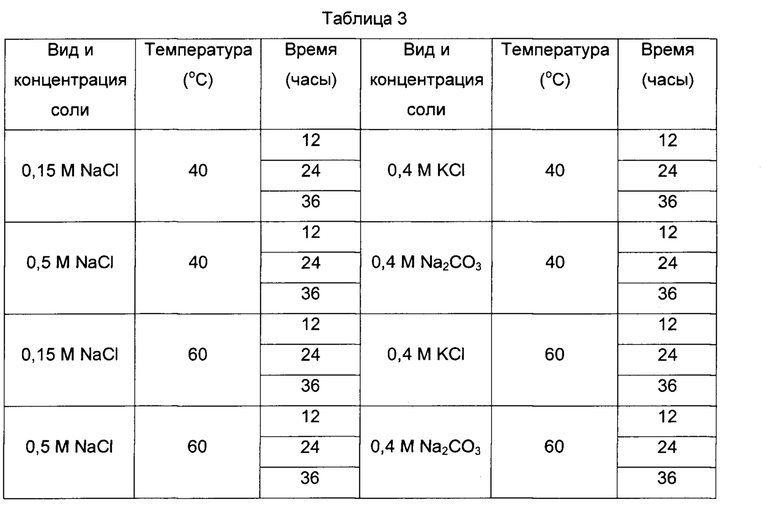
4-2. Результаты
Как показано на Фиг. 19, согласно результатам вестерн-блоттинга проб антигена, полученных при различных температурах, пробы сохраняли стабильность в качестве антигена при 40°C и 60°C. С учетом того, что 40°C и 60°C являются жесткими температурными условиями, приведенные выше результаты подтверждают, что антигены HPV, полученные способом получения по настоящему изобретению, могут сохранять стабильность в качестве антигена при хранении в стандартном холодильнике и при температурах замораживания.
Также, как показано на Фиг. 19, при исследовании зависимости от вида и концентрации соли стабильность в качестве антигена была высокой при использовании 0,4 М KCl, 0,4 М Na2CO3 и 0,5 М NaCl, а не при низких концентрациях соли, таких как 0,15 М NaCl. Эти результаты показывают, что антигены HPV, полученные способом получения по настоящему изобретению, превосходили антигены HPV, полученные с использованием 0,15 М NaCl, в отношении выхода белка после очистки и стабильности, и поэтому они пригодны для применения в качестве сырьевого материала для получения диагностического средства для выявления антител к HPV.
Хотя настоящее изобретение было описано подробно со ссылкой на специфические признаки, специалистам в данной области техники будет очевидно, что это описание относится лишь к предпочтительному варианту осуществления настоящего изобретения и не ограничивает объем настоящего изобретения. Поэтому объем настоящего изобретения по существу определен формулой изобретения и его эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИМЕРНАЯ ЧАСТИЦА HPV | 2012 |
|
RU2642287C2 |
| Способ получения рекомбинантной вакцины для профилактики папилломавирусной инфекции человека, рекомбинантная вакцина | 2018 |
|
RU2681174C1 |
| РЕКОМБИНАНТНЫЙ ШТАММ ДРОЖЖЕЙ Hansenula polymorpha - ПРОДУЦЕНТ ГЛАВНОГО КАПСИДНОГО БЕЛКА L1 ВИРУСА ПАПИЛЛОМЫ ЧЕЛОВЕКА ТИПА 56 | 2014 |
|
RU2546240C1 |
| РЕКОМБИНАНТНЫЙ ШТАММ ДРОЖЖЕЙ Hansenula polymorpha - ПРОДУЦЕНТ ГЛАВНОГО КАПСИДНОГО БЕЛКА L1 ВИРУСА ПАПИЛЛОМЫ ЧЕЛОВЕКА ТИПА 16 | 2014 |
|
RU2546241C1 |
| ПАПИЛЛОМАВИРУСНЫЕ ВАКЦИНЫ | 1995 |
|
RU2206608C2 |
| РЕКОМБИНАНТНАЯ ВАКЦИНА ДЛЯ ПРОФИЛАКТИКИ ПАПИЛЛОМАВИРУСНОЙ ИНФЕКЦИИ ЧЕЛОВЕКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2546243C1 |
| Рекомбинантный штамм дрожжей Hansenula polymorpha - продуцент главного капсидного белка L1 вируса папилломы человека типа 11 | 2018 |
|
RU2676160C1 |
| Рекомбинантный штамм дрожжей Hansenula polymorpha - продуцент главного капсидного белка L1 вируса папилломы человека типа 6 | 2018 |
|
RU2675471C1 |
| ОЧИЩЕННЫЕ БЕЛКИ ВИРУСА ПАПИЛЛОМЫ | 1995 |
|
RU2161651C2 |
| РЕКОМБИНАНТНЫЙ ШТАММ ДРОЖЖЕЙ Hansenula polymorpha - ПРОДУЦЕНТ ГЛАВНОГО КАПСИДНОГО БЕЛКА L1 ВИРУСА ПАПИЛЛОМЫ ЧЕЛОВЕКА ТИПА 18 | 2014 |
|
RU2546242C1 |


Изобретение относится к биохимии. Описан способ получения вирусоподобных частиц (VLP) на основе белка L1 папилломавируса человека (HPV), включающий: (a) культивирование трансформированных дрожжей, экспрессирующих белок L1 HPV; (b) выполнение осаждения сульфатом аммония в продукте трансформированных дрожжей, культивированных на стадии (а), для получения преципитата; (c) обработку преципитата, полученного посредством осаждения сульфатом аммония, NaCl, KCl или Na2CO3 в концентрации, находящейся в диапазоне от 0,35 до 0,60 М, с последующей инкубацией и удаление образовавшихся нерастворимых белков; и (d) выполнение хроматографии продукта, полученного на стадии (с), с получением вирусоподобных частиц (VLP) на основе белка L1 папилломавируса человека (HPV). Изобретение расширяет арсенал способов получения вирусоподобных частиц на основе белка L1 папилломавируса человека. 7 з.п. ф-лы, 19 ил., 3 табл., 4 пр.
1. Способ получения вирусоподобных частиц (VLP, от англ. virus-like particles) на основе белка L1 папилломавируса человека (HPV, от англ. human papillomavirus), включающий:
(a) культивирование трансформированных дрожжей, экспрессирующих белок L1 HPV;
(b) выполнение осаждения сульфатом аммония в продукте трансформированных дрожжей, культивированных на стадии (а), для получения преципитата;
(c) обработку преципитата, полученного посредством осаждения сульфатом аммония, NaCl, KCl или Na2CO3 в концентрации, находящейся в диапазоне от 0,35 М до 0,60 М, с последующей инкубацией и удаление образовавшихся нерастворимых белков; и
(d) выполнение хроматографии продукта, полученного на стадии (с), с получением вирусоподобных частиц (VLP) на основе белка L1 папилломавируса человека (HPV).
2. Способ по п. 1, отличающийся тем, что концентрация сульфата аммония при обработке на стадии (b) находится в диапазоне от 40 мас. % до 50 мас.%.
3. Способ по п. 1, отличающийся тем, что он дополнительно включает между стадией (b) и стадией (с) инкубацию преципитата, полученного посредством осаждения сульфатом аммония, в растворе, содержащем соль.
4. Способ по п. 1, отличающийся тем, что преципитат, полученный посредством осаждения сульфатом аммония на стадии (с), имеет концентрацию, находящуюся в диапазоне от 25 мг/мл до 65 мг/мл.
5. Способ по п. 1, отличающийся тем, что концентрация соли для обработки на стадии (с) находится в диапазоне от 0,35 М до 0,55 М.
6. Способ по п. 1, отличающийся тем, что инкубацию на стадии (с) выполняют при температуре от 25°С до 34°С.
7. Способ по п. 1, отличающийся тем, что инкубацию на стадии (с) выполняют в течение периода от 21 ч. до 27 ч.
8. Способ по п. 1, отличающийся тем, что папилломавирус человека выбран из группы, включающей HPV 6а типа, HPV 6b типа, HPV 11 типа, HPV 16 типа, HPV 18 типа, HPV 31 типа, HPV 33 типа, HPV 35 типа, HPV 39 типа, HPV 45 типа, HPV 51 типа, HPV 52 типа, HPV 56 типа, HPV 58 типа HPV 68 типа.
| УСТРОЙСТВО МОНИТОРИНГА ЗАРЯДКИ | 2008 |
|
RU2444103C2 |
| ПАПИЛЛОМАВИРУСНЫЕ ВАКЦИНЫ | 1995 |
|
RU2206608C2 |
| МОЛЕКУЛА НУКЛЕИНОВОЙ КИСЛОТЫ, КОДИРУЮЩАЯ HPV31 L1, ЭКСПРЕССИРУЮЩИЙ ВЕКТОР, КЛЕТКА-ХОЗЯИН, ВИРУСОПОДОБНАЯ ЧАСТИЦА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ, ВАКЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ С ИХ ИСПОЛЬЗОВАНИЕМ | 2004 |
|
RU2356943C2 |

