ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента посредством перорального введения терапевтически эффективного количества соединения селективного по альфа-изоформе ингибитора фосфатидилинозитол-3-киназы формулы (I) или его фармацевтически приемлемой соли пациенту по меньшей мере в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около двух дней до около трех дней между указанными последовательными пятидневными циклами; к применению указанного соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного средства для лечения или предотвращения пролиферативного заболевания, вводимого в соответствии с указанной схемой приема; к схеме лечения, включающей в себя введение указанного соединения формулы (I) или его фармацевтически приемлемой соли в соответствии с указанной схемой приема; и к его соответствующим фармацевтическим композициям и упаковкам.
УРОВЕНЬ ТЕХНИКИ
Фосфатидилинозитол-3-киназа («киназа PI-3» или «PI3K») включает семейство липидкиназ, катализирующих перенос фосфата в D-3' положение инозитсодержащих липидов с продуцированием фосфоинозитол-3-фосфата («PIP»), фосфоинозитол-3,4-дифосфата («PIP2») и фосфоинозитол-3,4,5-трифосфата («PIP3»), которые, в свою очередь, действуют как вторичные мессенджеры в сигнальных каскадах посредством стыковки белков, содержащих гомологичные плекстрину, FYVE, Phox и другие фосфолипид-связывающие домены с множеством сигнальных комплексов, часто в плазматической мембране (Vanhaesebroeck и соавт., Annu. Rev. Biochem 70:535 (2001); Katso и соавт., Annu. Rev. Cell Dev. Biol. 17:615 (2001)). Человеческие клетки содержат три гена (PIK3CA, PIK3CB и PIK3CD) кодирующие каталитические субъединицы p110 (α,β,δ-изоформы) ферментов класса IA PI3K. Эти каталитические субъединицы p110α, p110β и p110δ конститутивно связаны с регуляторной субъединицей, которой может являться p85α, p55α, p50α, p85β или p55γ. p110α и p110β экспрессируются в большинстве тканей. Класс 1B PI3K имеет одного члена семейства, гетеродимер, состоящий из каталитической субъединицы p110γ, связанной с одной из двух регуляторных субъединиц, p101 или p84 (Fruman и соавт., Annu Rev. Biochem. 67:481 (1998); Suire и соавт., Curr. Biol. 15:566 (2005)). Модульные домены p85/55/50 субъединиц включают Src-гомологичные домены (SH2), связывающие фосфотирозиновые остатки в определенном контексте последовательности на активированном рецепторе и цитоплазматической тирозинкиназе, что приводит к активации и локализации класса 1A PI3K. Класс 1B, а также p110β при некоторых обстоятельствах, активируется непосредственно соединенными с G-белком рецепторами, которые связывают разнообразный репертуар пептидных и непептидных лигандов (Stephens и соавт., Cell 89:105 (1997)); Katso и соавт., Annu. Rev. Cell Dev. Biol. 17:615-675 (2001)). Таким образом, получаемые в результате фосфолипидные продукты класса I PI3K соединяют расположенные выше по ходу сигнала рецепторы с расположенными ниже по ходу сигнала клеточными активностями, включающими пролиферацию, выживание, хемотаксис, направленную миграцию клеток, подвижность, метаболизм, воспалительные и аллергические реакции, транскрипцию и трансляцию (Cantley и соавт., Cell 64:281 (1991); Escobedo and Williams, Nature 335:85 (1988); Fantl и соавт., Cell 69:413 (1992)).
Нарушенная регуляция PI3K, которая часто повышает выживаемость через активацию Akt, является одним из самых распространенных событий при раке у человека и, как было показано, возникает на множестве уровней. Ген-супрессор опухоли PTEN, дефосфорилирующий фосфоинозитиды в 3'-положениях кольца инозитола, при этом являясь антагонистом активности PI3K, функционально делетируется во множестве опухолей. В других опухолях гены для изоформы p110α, PIK3CA, и для Akt амплифицируются, и повышенная белковая экспрессия продуктов их генов была продемонстрирована для нескольких типов рака человека. Кроме того, мутации и транслокация p85α, которые служат для повышения регуляции комплекса p85-p110, была описана при различных типах рака человека. Наконец, соматические миссенс-мутации в PIK3CA, которые активируют нижележащие сигнальные пути, были описаны со значимой частотой в широком разнообразии типов рака человека, включая 32% рака ободочной и прямой кишки, 27% глиобластом, 25% рака желудка, 36% гепатоцеллюлярных карцином и 18-40% рака груди. (Samuels и соавт., Cell Cycle 3(10):1221 (2004); Hartmann et al, Acta Neuropathol., 109(6):639 (June 2005); Li et al, BMC Cancer 5:29 (March 2005) ; Lee et al, Oncogene, 24(8):1477 (2005); Backman et al, Cancer Biol. Ther. 3(8):772-775 (2004); Campbell и соавт., Cancer Research, 64(21):7678-7681 (2004); Levine и соавт., Clin. Cancer Res., 11(8):2875-2878 (2005); and Wu et al, Breast Cancer Res., 7(5):R609-R616 (2005)). Нарушение регуляции PI3K, включая α-изоформу, является одним из наиболее распространенных нарушений регуляции, связанных с раком и пролиферативными заболеваниями человека (Parsons и соавт., Nature 436:792 (2005); Hennessey и соавт., Nature Rev. Drug Disc. 4:988-1004 (2005)).
(S)-пирролидин-1,2-дикарбоксильной кислоты 2-амид ({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) является специфичным производным соединением 2-карбоксамид циклоаминомочевины, которое сильно и селективно нацелено на альфа(α-изоформу класса IA PI3K. Данное соединение имеет следующую химическую структуру:
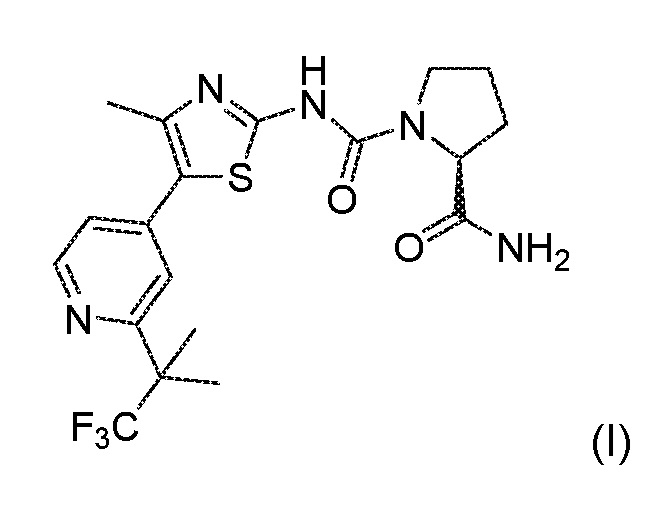
(ниже в настоящем описании «соединение формулы (I)» или «соединение А»). Соединение формулы (I) и его фармацевтически приемлемые соли, подходящие составы и способ приготовления описаны в PCT заявке WO2010/029082.
В клиническом испытании фазы I данное соединение селективного по альфа-изоформе ингибитора PI3K ((S)-пирролидин-1,2-дикарбоксильной кислоты 2-амид ({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид) продемонстрировало клиническую эффективность при монотерапии пациентов, имевших солидные злокачественные образования, несущие альтерацию гена PIK3CA. В фазе подъема дозы пациентам было перорально введено данное соединение (a) в дозировке в пределах от 30 мг до 450 мг один раз в сутки (q.d.) по непрерывному ежедневному графику в течение 28 дней или (b) в дозировке в пределах от 120 мг до 200 мг дважды в сутки (b.i.d.) по непрерывному ежедневному графику в течение 28 дней, руководствуясь байесовской регрессионной логистической моделью с контролем передозировки. После определения максимальной переносимой дозы (MTD) фаза подъема дозы проводилась для дополнительного лечения пациентов, имевших рак головы и шеи с альтерацией PIK3CA, пациентов, имевших солидные опухоли с альтерацией PIK3CA, и пациентов, имевших рак груди PIK3CA дикого типа ER+/HER2-. Клиническая эффективность данного соединения была продемонстрирована предварительно. На 15 февраля 2013 подтвержденный частичный ответ наблюдался у нескольких пациентов, подвергавшихся лечению в дозировке >270 мг/день, включая пациентов, страдавших раком груди (1 пациент, подтвержден), раком ободочной и прямой кишки (1 пациент подтвержден), раком эндометрия (1 пациент, подтвержден) и раком шейки матки (1 пациент подтвержден). (Gonzalez-Angulo и соавт., ʺSafety, pharmacokinetics, and preliminary activity of the α-specific PI3K inhibitor BYL719: results from the first-in-human studyʺ, Presentation at the 2013 ASCO Annual Meeting, held May 31-June 4, 2013 in Chicago, IL.)
Несмотря на клиническую эффективность данного соединения в этом клиническом испытании фазы I, некоторые пациенты, которым вводилось данное соединение один раз в сутки или дважды в сутки по непрерывному ежедневному графику, продемонстрировали по меньшей мере один побочный эффект или нежелательное явление, включая, но не ограничиваясь указанным, гипергликемию (49% пациентов), тошноту (43% пациентов), снижение аппетита (34% пациентов), диарею (35% пациентов), сыпь и аллергию (34% пациентов), астению/усталость (34% пациентов), рвоту, стоматит, дисгевзию и/или диспепсию. (Gonzalez-Angulo и соавт., Presentation at the 2013 ASCO Annual Meeting, held May 31-June 4, 2013 in Chicago, IL.)
В настоящее время существует неудовлетворенная потребность в высокоактивном селективном по альфа(α-изоформе ингибитору PI3K, который может быть введен пациентам в дозировке или режиме дозировки, который является клинически эффективным для лечения пролиферативных болезней, в частности, рака, но также ослабляет, уменьшает или частично снимает любые известные и неизвестные побочные эффекты (например, по тяжести, встречаемости или частоте) лекарственного препарата. Считается, что это не было достигнуто ни для какого селективного по альфа-изоформе ингибитора PI3K до настоящего изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I):

или его фармацевтически приемлемой соли такому пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту сроком в течение от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания, включающему в себя, во-первых, введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в количестве от около 100 мг до около 450 мг ежедневно по непрерывному ежедневному графику через пероральное введение, во-вторых, определение того, что для указанного пациента имеется побочный эффект, выбранный из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени (например, повреждение печени или заболевание печени, повышение уровня аспартатаминотрансферазы, повышение уровня аланинаминотрансферазы, и т.д.), сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи (например, макулопапулезной, угревидной, и т.д.) и гиперчувствительности (например, повышенная чувствительность к ушибам), фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии после введения указанного соединения формулы (I) или его фармацевтически приемлемой соли указанному пациенту, и, в-третьих, уменьшение введения указанного соединения формулы (I) или его фармацевтически приемлемой соли до суточной дозы от около 100 мг до около 450 мг через пероральное введение, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу ослабления по меньшей мере одного побочного эффекта, выбранного из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени (например, повреждение печени или заболевание печени, повышение уровня аспартатаминотрансферазы, повышение уровня аланинаминотрансферазы, и т.д.), сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи (например, макулопапулезной, угревидной, и т.д.) и гиперчувствительности (например, повышенная чувствительность к ушибам), фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии, возникшего после предшествующего лечения соединением формулы (I) или его фармацевтически приемлемой солью, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту в суточной дозе от около 100 мг до около 450 мг, предпочтительно от около 200 мг до около 400 мг или, более предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится нуждающемуся в нем пациенту в суточной дозе от около 100 мг до около 450 мг указанного соединения формулы (I) или его фармацевтически приемлемой соли по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное лекарственное средство не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения или предотвращения пролиферативного заболевания, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции для применения в лечении или профилактике пролиферативного заболевания у нуждающегося в этом пациента, содержащее в количестве от около 100 мг до около 450 мг соединение формулы (I) или его фармацевтически приемлемую соль вместе с одним или более фармацевтически приемлемых эксципиентов, при этом фармацевтическая композиция вводится перорально пациенту, по меньшей мере, в течение двух последовательных пятидневных циклов, и не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к схеме лечения, включающей пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к упаковке, содержащей фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль в суточной дозе от около 100 мг до около 450 мг вместе с одним или более фармацевтически приемлемых эксципиентов в комбинации с инструкциями по пероральному введению указанной фармацевтической композиции, по меньшей мере, в течение двух последовательных пятидневных циклов, и по отсутствию введения указанной композиции в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
ПОДРОБНОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показаны профили концентрации-времени после перорального приема соединения A в дозировке 12,5, 25 и 50 мг/кг в сутки у безтимусных мышей (A) и в 12,5, 25, 40 и 80 мг/кг в сутки у безтимусных крыс (B).
На фигуре 2 показано сравнение наблюдаемых и предсказанных концентраций в плазме после перорального приема соединения A в дозировке 50 мг/кг в сутки у безтимусных мышей (A) и 40 мг/кг в сутки у безтимусных крыс (B).
На фигуре 3 показано сравнение наблюдаемых и предсказанных концентраций в плазме после перорального приема соединения A в дозировке 6,25, 12,5, 25 и 50 мг/кг в сутки у безтимусных мышей (A) и в дозировке 6,25, 12,5, 25, 40, 50 и 80 мг/кг в сутки у безтимусных крыс (B) по непрерывному ежедневному графику.
На фигурах 4А и 4B показано сравнение наблюдаемых и предсказанных концентраций в плазме после перорального приема соединения A в дозировке 40 мг/кг 2 раза в сутки по непрерывному ежедневному графику у безтимусных мышей в модельном исследовании PK (A) и более позднем повторном подтверждающем модельном исследовании PK (B).
На фигуре 5 показано соотношение между концентрацией в ткани опухоли и процентом ингибирования S473P-Akt, измеренными параллельно в опухолях Rat1-myr P110α в различные моменты времени после лечения соединением A.
На фигуре 6 показано соотношение между экспозицией, в соответствии с измеренным в естественных условиях IC80 для ингибирования S473P-Akt, и противоопухолевой эффективностью в опухолях Rat1-myr P110α, подвергавшихся лечению соединением A в дозировке 50 мг/кг в сутки.
На фигуре 7 показано соотношение между реакцией опухолевого маркера PD (pAkt) и противоопухолевой эффективностью, наблюдаемой у мышей и крыс, подвергавшихся пероральном лечению один раз в сутки различными дозами соединения A.
На фигуре 8 показано наблюдаемое по сравнению с предсказанным ингибированием роста опухоли после перорального приема соединения A от 6,25 до 70 мг/кг по непрерывному ежедневному графику в различном режиме у безтимусных мышей и крыс.
На фигуре 9 показано соотношение между концентрациями соединения А в плазме и уровнями инсулина в плазме (A) или уровнями глюкозы в крови (B), измеренными в том же самом исследовании после лечения соединением A безтимусных мышей.
На фигуре 10 показано соотношение между концентрациями соединения А в плазме и уровнями инсулина в плазме (A) или уровнями глюкозы в крови (B), измеренными в том же самом исследовании после лечения соединением A безтимусных крыс.
На фигуре 11 показана корреляция, наблюдаемая между долей времени выше порога гипергликемии в плазме между двумя последовательными дозировками и потерей массы тела у безтимусных мышей и крыс.
На фигуре 12 показана смоделированная кривая эффективности, в соответствии с определенным посредством доли времени выше порога IC80 для S473P-Akt, и кривая переносимости, в соответствии с определенным посредством продолжительностью воздействия выше порога гипергликемии соединения A у безтимусных мышей, подвергавшихся пероральному лечению один раз в сутки или 2 раза в сутки по непрерывному ежедневному графику с увеличивающимися дозами соединения A.
На фигуре 13 показана смоделированная кривая эффективности, в соответствии с определенным посредством доли времени выше порога IC80 для S473P-Akt, и кривая переносимости, в соответствии с определенным посредством продолжительностью воздействия выше порога гипергликемии соединения A у безтимусных крыс, подвергавшихся пероральному лечению один раз в сутки или 2 раза в сутки по непрерывному ежедневному графику с увеличивающимися дозами соединения A.
На фигуре 14 показана смоделированная эффективность для имеющих опухоль Rat1-myr P110α безтимусных крыс, подвергавшихся пероральному лечению соединением A в дозировке 20 мг/кг в альтернативной схеме 1 (A) или в дозировке 14 мг/кг в сутки по непрерывному ежедневному графику (B).
На фигуре 15 показаны смоделированные профили PK в плазме у безтимусных крыс, подвергавшихся пероральному лечению соединением A в дозировке 20 мг/кг в альтернативной схеме 1 (A) или в дозировке 14 мг/кг в сутки по непрерывному ежедневному графику (B)
Подробное описание изобретения
Настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I), в соответствии с определенным в настоящем описании, или его фармацевтически приемлемой соли пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около двух (2) дней до около трех (3) дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Общие термины, используемые в настоящем описании, определены со следующими значениями, если явно не указано иное:
Термины, «содержащий» и «включающий» используются в настоящем описании в их открытом и неограничивающем смысле, если не указано иное.
Форма единственного числа в контексте описания изобретения (особенно в контексте прилагаемой формулы изобретения) должна быть истолкована как охватывающая и единственное, и множественное число, если иное не указано в настоящем написании или если это явно не противоречит контексту. При использовании формы множественного числа для соединений, солей, и т.п., она считается также обозначающей единственное соединение, соль, и т.п.
Термин «ингибитор фосфатидилинозитол-3-киназы», или «ингибитор PI3K», определяются в настоящем описании как обозначающие соединение, которое нацеливает, снижает или подавляет активность фосфатидилинозитол-3-киназы.
Термин «фармацевтически приемлемый» определяется в настоящем описании как обозначающий соединения, материалы, композиции и/или формы дозировки, которые являются, по результатам тщательной медицинской оценки, подходящими для контакта с тканями пациента без избыточной токсичности, аллергической реакции с раздражением и других создающих проблемы сложностей, в соответствии с целесообразным отношением выгода/риск.
Термин «лечить», «лечащий» или «лечение» при использовании в настоящем описании включает лечение или схему лечения, смягчающие, сокращающие или облегчающие по меньшей мере один симптом у пациента или осуществляющие задержку развития пролиферативного нарушения. Например, лечение может заключаться в уменьшении одного или нескольких симптомов нарушения или полном устранении нарушения, такого как рак. В пределах значений по настоящему изобретению, термин «лечение» также обозначает приостановку, задержку начала (т.е., период до клинического проявления нарушения) и/или снижение риска развития или ухудшения нарушения.
Термин «предотвращать», «предотвращающий» или «предотвращение» при использовании в настоящем описании включает профилактику по меньшей мере одного симптома, связанного или вызванного состоянием, заболеванием или нарушением, которое предотвращается.
Термины «клинически эффективный» или «терапевтически эффективный» означает заметное улучшение по сравнению с исходным уровнем клинически наблюдаемых признаков и симптомов состояния, заболевания или нарушения, которые подвергались лечению терапевтическим средством.
Термин «терапевтически эффективное количество» представляет собой количество, достаточное для обеспечения заметного улучшения по сравнению с исходным уровнем клинически наблюдаемых признаков и симптомов состояния, заболевания или нарушения, которые подвергались лечению терапевтическим средством.
Термин «фармацевтическая композиция» определяется в настоящем описании как означающий смесь или раствор, содержащий по меньшей мере одно терапевтическое средство, которое должно быть введено пациенту в целях предотвращения или лечения определенного заболевания или патологического состояния, поражающего пациента.
Фраза «цикл из пяти последовательных дней» при использовании в настоящем описании означает, что указанное терапевтическое средство вводится пациенту в течение каждого дня в течение пяти дней подряд, и затем не вводится в течение некоторого периода времени перед тем, как то же самое терапевтическое средство будет в следующий раз вводиться пациенту. Следует понимать, что терапевтическое средство может вводиться каждый день в одной единице дозирования или множестве единиц дозирования и/или вводиться каждый день в единственной дозе (один раз в сутки, q.d.) или в разделенных дозах (несколько раз в сутки, например, дважды в сутки, b.i.d.).
Фраза «непрерывный ежедневный график» при использовании в настоящем описании означает, что терапевтическое средство вводится пациенту в течение каждого дня в течение, по меньшей мере, семи дней или в течение неуказанного промежутка времени, или столько, сколько лечение необходимо. Следует понимать, что терапевтическое средство может вводиться каждый день в одной единице дозирования или множестве единиц дозирования и/или вводиться каждый день в единственной дозе (один раз в сутки, q.d.) или в разделенных дозах (несколько раз в сутки, например, дважды в сутки, b.i.d.).
Термины «день» и «сутки» при использовании в настоящем описании относятся или к одному календарному дню, или к одному 24-часовому периоду.
Термин «комбинация» используется в настоящем описании для обозначения фиксированной комбинации в форме одной единицы дозирования, нефиксированной комбинации, или набора частей для комбинированного введения, где соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное терапевтическое средство могут вводиться одновременно, независимо в одно и то же время или по отдельности в пределах временных интервалов, которые позволяют участникам комбинации демонстрировать совместный, например, синергичный, эффект. Термин «фиксированная комбинация» означает, что терапевтические средства, например, соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное терапевтическое средство вводятся пациенту одновременно в форме единого объекта или единицы дозирования. Термин «нефиксированная комбинация» или «набор частей» означает, что терапевтические средства, например, соединение формулы (I) или его фармацевтически приемлемая соль и по меньшей мере одно дополнительное терапевтическое средство вводятся пациенту как отдельные объекты или единицы дозирования, или одновременно, или параллельно, или последовательно без конкретных ограничений по времени, в течение которого такое введение обеспечивает терапевтически эффективные уровни двух терапевтических средств в организме пациента. Последнее также относится к «коктейльной» терапии, например, введению трех или более терапевтических средств.
Термин «комбинированное введение» при использовании в настоящем описании определяется как охватывающий введение выбранных терапевтических средств одному пациенту, и предполагается, что он включает схемы лечения, в которых средства не обязательно вводятся одним и тем же путем введения или в одно и то же время.
Термины «пациент», «субъект» или «теплокровное животное» предназначены для включения животных. Примеры субъектов включают млекопитающих, например, людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных не являющихся человеком животных. В определенных вариантах осуществления субъектом является человек, например, человек, страдающий, или имеющий риск стать страдающим, или потенциально способный стать страдающим опухолью головного мозга. Особенно предпочтительно, чтобы пациент или теплокровное животное являлись человеком.
Термины «около» или «приблизительно» обычно означают нахождение в пределах 10%, более предпочтительно, в пределах 5% от заданного значения или диапазона.
WO2010/029082 описывает конкретные производные 2-карбоксамид цикломочевины, которые, как было обнаружено, обладали высоко селективной ингибирующей активностью в отношении альфа-изоформы фосфатидилинозитол-3-киназы (PI3K). Селективный по альфа-изоформе ингибитор PI3K, подходящий для настоящего изобретения, представляет собой соединение, имеющее следующую формулу (I):

(ниже в настоящем описании называемый «соединение формулы (I)» или «соединение A») или его фармацевтически приемлемые соли. Соединение формулы (I) также известно как химическое соединение (S)-пирролидин-1,2-дикарбоксильной кислоты 2-амид ({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)-пиридин-4-ил]-тиазол-2-ил}-амид). Соединение формулы (I), ее фармацевтически приемлемые соли и подходящие рецептуры описаны в заявке на патент № WO2010/029082, которая включена в настоящее описание посредством ссылки во всей ее полноте, и способы их приготовления были описаны, например, в приведенном там примере 15.
При использовании в настоящем описании, термин «соли» (включая «или его соли» или «или его соль») может присутствовать один или в смеси со свободным соединением формулы (I), и предпочтительно, охватывает фармацевтически приемлемые соли. Такие соли формируются, например, как кислотно-аддитивные соли, предпочтительно, с органическими или неорганическими кислотами, из соединения формулы (I) с основным атомом азота, особенно фармацевтически приемлемые соли. Подходящими неорганическими кислотами являются, например, галогенные кислоты, такие как соляная кислота, серная кислота или фосфорная кислота. Подходящие органические кислоты представляют собой, например, карбоновые кислоты или сульфокислоты, такие как фумаровая кислота или метансульфоновая кислота. В целях выделения или очистки также возможно использование фармацевтически неприемлемых солей, например пикратов или перхлоратов. Для терапевтического применения используются только фармацевтически приемлемые соли или свободное соединение (когда это применимо в форме фармацевтических препаратов), и они, таким образом, являются предпочтительными. Ввиду тесной связи между соединением формулы (I) в свободной форме и в форме его солей, любую ссылку на свободное соединение выше и ниже в настоящем описании следует понимать как относящуюся также к соответствующим солям, если это является подходящим и целесообразным. Соли соединения формулы (I) предпочтительно являются фармацевтически приемлемыми солями; подходящие противоионы, формирующие фармацевтически приемлемые соли, известны в данной области техники.
Ранее было продемонстрировано, что соединение формулы (I) сильно и селективно ингибирует альфа-изоформу PI3K, включая, например, примеры A и C PCT-заявки на патент WO2010/029082. В отличие от ранее известных ингибиторов PI3K, соединение формулы (I) ингибирует альфа-изоформу PI3K (IC50 составляет 0,008 мкМ/л) более сильно, чем бета-изоформу (IC50 составляет 1,212 мкМ/л), дельта-изоформу (IC50 составляет 0,077 мкМ/л) и гамма-изоформу (IC50 составляет 1,097 мкМ/л) в клеточных анализах и не имеет ингибирующей активности в отношении Vps34, mTOR, DNA-PK и ATR. Кроме того, соединение формулы (I) демонстрирует ингибирующую активность против альфа-изоформы PI3K дикого типа, мутантной альфа-изоформы PI3K E545K и мутантной альфа-изоформы PI3K H1047R.
Соединение формулы (I) или его фармацевтически приемлемая соль может вводиться перорально в дозировке от около 100 мг до около 450 мг в сутки нуждающемуся в нем пациенту-человеку. Термин «суточная доза» относится к величине суммарной дозы терапевтического средства, введенного определенному пациенту в течение любого дня. В дальнейших вариантах осуществления соединение формулы (I) может вводиться пациенту в суточной дозе от около 200 до около 400 мг в сутки, или от около 240 мг до около 400 мг в сутки, или от около 300 мг до около 400 мг в сутки, или от около 350 мг до около 400 мг в сутки. В предпочтительном варианте осуществления соединение формулы (I) вводится пациенту-человеку в суточной дозе от около 350 мг до около 400 мг в сутки.
Суточная доза может быть введена пациенту в одной дозе (один раз в сутки, q.d.) или в разделенных дозах (несколько раз в сутки, например, дважды в сутки, b.i.d.). В одном из вариантов осуществления суточная доза вводится один раз в сутки (q.d.) В еще одном варианте осуществления суточная доза вводится дважды в сутки (b.i.d.)
Суточная доза может быть введена пациенту в одной единице дозировки или в объемах множества единиц дозировки, составляющих суточную дозу.
В соответствии со схемой приема по настоящему изобретению, соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней. Предпочтительно, состав или его фармацевтически приемлемая соль не вводятся в течение около 2 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В одном из вариантов осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Следует понимать, что схема приема по настоящему изобретению может быть альтернативно определена в отношении выбора моментов времени фактических введений соединения формулы (I) или его фармацевтически приемлемой соли.
В одном из вариантов осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода около 3 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
В еще одном варианте осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся в течение периода около 2,5 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
Пролиферативными заболеваниями, которые могут подвергаться лечению или предотвращению посредством введения соединения формулы (I) или его фармацевтически приемлемой соли в соответствии со схемой приема по настоящему изобретению, являются, в частности, заболевания, опосредованные альфа-изоформой PI3K. Следует понимать, что один из вариантов осуществления настоящего изобретения включает в себя лечение пролиферативного заболевания, и что еще один вариант осуществления настоящего изобретения включает в себя предотвращение пролиферативного заболевания.
Примеры пролиферативных заболеваний, которые могут подвергаться лечению или предотвращению в соответствии с настоящим изобретением, включают рак, истинную полицитемию, идиопатическую тромбоцитемию, миелофиброз с миелоидной метаплазией, астму, COPD, ARDS, синдром Лоффлера, эозинофильную пневмонию, паразитную инвазию (в частности, многоклеточными организмами) (включая тропическую эозинофилию), бронхолегочный аспергиллез, узелковый полиартериит (включая синдром Хург-Штрауса), эозинофильную гранулему, связанные с эозинофилом нарушения, оказывающие влияние на дыхательные пути, вызванные реакцией на лекарственный препарат, псориаз, контактный дерматит, аллергический дерматит, гнездную алопецию, мультиформную эритему, герпетиформный дерматит, склеродермию, витилиго, аллергические васкулиты, крапивницу, буллезный пемфигоид, красную волчанку, пемфигус, приобретенный буллезный эпидермоз, аутоиммунные гематологические нарушения (например, гемолитическая анемия, апластическая анемия, чистая эритроцитарная анемия и идиопатическая тромбоцитопения), системная красная волчанка, полихондрию, склеродермию, гранулематоз Вегенера, дерматомиозит, хронический активный гепатит, тяжелую миастениию, синдром Стивена-Джонсона, идиопатический синдром мальабсорбции, аутоиммунное воспалительное заболевание кишечника (например, неспецифический язвенный колит и болезнь Крона), эндокринную офтальмопатию, базедову болезнь, саркоидоз, альвеолит, хронический гиперчувствительный пневмонит, рассеянный склероз, первичный желчный цирроз, увеит (передний и задний), внутритканевой фиброз легкого, псориатический артрит, гломерулонефрит, сердечно-сосудистые заболевания, атеросклероз, артериальную гипертензию, тромбоз глубоких вен, инсульт, инфаркт миокарда, нестабильную стенокардию, тромбоэмболию, легочную эмболию, тромболитические болезни, острую артериальную ишемию, периферические тромботические окклюзии, и заболевание коронарной артерии, реперфузионные раны, ретинопатию, такую как диабетическая ретинопатия или гипербарическая вызванная кислородом ретинопатия и патологические состояния, характеризуемые повышенным внутриглазным давлением или секрецией глазной водянистой влаги, такие как глаукома.
Предпочтительно, пролиферативная болезнь является раком. Термин «рак» относится к опухолям и/или росту раковых клеток, предпочтительно, опосредованных альфа-изоформой PI3K. В частности, соединения полезны в лечении рака, включая, например, саркому, рак легкого, бронх, предстательной железы, груди (включая спорадический рак груди и страдающих болезнью Каудена), поджелудочной железы, желудочно-кишечный рак, рак толстой кишки, прямой кишки, карциному толстой кишки, колоректальную аденому, рак щитовидной железы, печени, внутрипеченочных желчных протоков, гепатоцеллюлярный, надпочечников, желудка, гастральный, глиому, глиобластому, рак эндометрия, меланому, рак почки, почечной лоханки, мочевого пузыря, матки, шейки матки, влагалища, яичника, множественную миелому, рак пищевода, лейкоз, острый миелогенный лейкоз, хронический миелогенный лейкоз, лимфолейкоз, миелоидный лейкоз, рак мозга, ротовой полости и глотки, гортань, тонкой кишки, неходжкинскую лимфому, меланому, ворсинчатую аденому толстой кишки, неоплазию, неоплазию эпителиального характера, лимфомы, карциному молочных желез, базально-клеточную карциному, плоскоклеточную карциному, актинический кератоз, рак головы и шеи, истинную полицитемиию, идиопатическую тромбоцитемию, миелофиброз с миелоидной метаплазией и болезнь Вальденстрема.
Пролиферативные заболевания, опосредованные альфа-субъединицей PI3K, могут включать заболевания, демонстрирующие повышенную экспрессию или амплификацию альфы PI3K, соматическую мутацию PIK3CA, или мутацию зародышевой линии или соматическую мутацию PTEN, или мутации и транслокацию p85α, которые служат для повышенной регуляции комплекса p85-p110. В предпочтительном варианте осуществления, рак представляет собой опухоль и/или злокачественный рост, опосредованный альфа-изоформой PI3K.
В одном из вариантов осуществления пролиферативная болезнь представляет собой рак, выбранный из рака легкого, бронха, предстательной железы, груди (включая спорадический рак груди и страдающих болезнью Каудена), толстой кишки, прямой кишки, карциномы толстой кишки, колоректальной аденомы, рака поджелудочной железы, желудочно-кишечного рака, гепатоцеллюлярного рака, рака желудка, гастрального рака, рака яичника, плоскоклеточной карциномы и рака головы и шеи.
В еще одном варианте осуществления пролиферативная болезнь представляет собой рак, выбранный из рака груди, толстой кишки, прямой кишки, карциномы толстой кишки, колоректальной аденомы, рак эндометрия и рака шейки матки.
В еще одном варианте осуществления пролиферативная болезнь представляет собой рак, выбранный из рака легкого, груди (включая спорадический рак груди и страдающих болезнью Каудена), гастральный рак, рак яичника и рак головы и шеи.
В еще одном варианте осуществления настоящее изобретение относится к лечению рака посредством введения соединения формулы (I) или его фармацевтически приемлемой соли в соответствии со схемой приема по настоящему изобретению.
Считается, что снижение дозировки данного соединения сильного селективного по альфа-изоформе ингибитора PI3K формулы (I) или его фармацевтически приемлемой соли с перорального введения в (a) суточной дозе от около 100 мг до около 450 мг ежедневно по непрерывному ежедневному графику до (b) суточной дозы от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение не вводится в течение периода от около 2 дней до около 3 дней между указанными последовательными пятидневными циклами, является эффективным в лечении или предотвращении пролиферативного заболевания, при этом ослабляя, уменьшая или частично снимая тяжесть, встречаемость или частоту любых побочных эффектов. Это, в частности, применимо к лечению или профилактике рака.
Примеры таких побочных эффектов, которые могут быть ослаблены, уменьшены или частично сняты посредством схемы приема по настоящему изобретению, включают, но не ограничены указанным, нейтропению, повышенный билирубин, кардиальную токсичность, нестабильную стенокардию, инфаркт миокарда, стабильную артериальную гипертензию, периферическую сенсорную или моторную невропатию/боль, дисфункцию печени (например, повреждение печени или заболевание печени, повышение уровня аспартатаминотрансферазы, повышение уровня аланинаминотрансферазы, и т.д.), сниженное количество эритроцитов и/или количество лейкоцитов, гипергликемию, тошноту, снижение аппетита, диарею, сыпь (например, макулопапулезную, угревидную, и т.д.) и гиперчувствительность (например, повышенную чувствительность к ушибам), фоточувствительность, астению/усталость, рвоту, стоматит, мукозит слизистой оболочки полости рта, панкреатит, дисгевзию и диспепсию. Специалистам в данной области техники будет понятно, каким образом оценить такие побочные эффекты у пациента, страдающего пролиферативными заболеваниями, с применением своего опыта и ранее полученных знаний и/или путем обращения к стандартным критериям оценки побочных эффектов, например, путем оценки такого пациента с применением общей терминологии критериев оценки побочных эффектов NCI, версия 4.03 (веб-сайт, расположенный по адресу: http://evs.nci.nih.gov/ftp1/CTCAE/About.html), который включен в настоящее описание посредством ссылки во всей его полноте.
В предпочтительном варианте осуществления побочный эффект, ослабленный, уменьшенный или частично снятый посредством схемы приема по настоящему изобретению, является патологическим состоянием, выбранным из гипергликемии, тошноты, сниженного аппетита, диареи, сыпи (например, макулопапулезной, угревидной, и т.д.) и гиперчувствительности (например, увеличенной чувствительности к ушибу), фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, дисгевзии и диспепсии. Более предпочтительно, побочный эффект, ослабленный, уменьшенный или частично снятый посредством схемы приема по настоящему изобретению, является гипергликемией.
С помощью установленных тестовых моделей может быть показано, что схема приема по настоящему изобретению обеспечивает положительные эффекты, описанные выше в настоящем описании. Специалист в данной области техники обладает достаточными способностями для выбора подходящей тестовой модели с целью подтверждения таких положительных эффектов. Фармакологическая активность соединения формулы (I) или его фармацевтически приемлемой соли может, например, быть продемонстрирована в клиническом исследовании, исследовании на животных или в процедуре тестирования, описанной по существу ниже в настоящем описании.
Подходящими клиническими исследованиями являются, в частности, например, исследования с увеличением дозы без контроля плацебо у пациентов с пролиферативным заболеванием, включая, например, заболевание с опухолью, например, рак груди, в которых пациентам вводится перорально соединение формулы (I) в соответствии со схемой приема по настоящему изобретению. Предпочтительно, пациентов распределяют по различным группам, при этом по меньшей мере одной группе вводится соединение формулы (I) по непрерывному ежедневному графику, и по меньшей мере одной группе вводится соединение формулы (I) в соответствии со схемой приема по настоящему изобретению. Такие исследования доказывают, в частности, эффективность терапевтического средства и его влияние на существующие или потенциальные побочные эффекты. Положительные эффекты для пролиферативного заболевания могут быть определены непосредственно через результаты этих исследований, в соответствии с известным специалистам в данной области техники. Такие исследования могут являться, в частности, подходящими для сравнения результатов непрерывного ежедневного графика с применением терапевтических средств и схемы приема по настоящему изобретению. Каждый пациент может получать дозы соединения формулы (I) или его фармацевтически приемлемой соли или один раз в сутки, или несколько раз (например, дважды) в сутки. Эффективность лечения может быть определена в таких исследованиях, например, после 12, 18 или 24 недели посредством оценки симптома и/или измерений размера опухоли каждые 6 недель.
В соответствии с настоящим изобретением, соединение формулы (I) или его фармацевтически приемлемая соль предпочтительно используется или вводится в форме фармацевтических композиций, содержащих терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли вместе с одним или более фармацевтически приемлемых эксципиентов, подходящих для перорального приема. Фармацевтическая композиция может содержать в количестве от около 100 мг до около 450 мг соединения формулы (I) или его фармацевтически приемлемой соли, для введения в одной единице дозирования. Альтернативно, фармацевтическая композиция может содержать количество соединения формулы (I) или его фармацевтически приемлемой соли, разделенное на множество единиц дозирования, и может вводиться в ежедневной дозировке от около 100 мг до около 450 мг соединения формулы (I) или его фармацевтически приемлемой соли.
Фармацевтические композиции, применяемые в соответствии с настоящим изобретением, могут быть приготовлены способом, известным, по сути, в качестве подходящего для перорального введения млекопитающим (теплокровным животным), включая людей. Фармацевтические композиции для перорального введения могут включать, например, композиции в таких формах единиц дозировки, как таблетки в сахарной оболочке, таблетки, капсулы, саше и, кроме того, ампулы. Если не указано иное, они приготовляются способом, известным самим по себе, например, посредством стандартных процессов смешивания, гранулирования, покрытия сахаром, растворения или лиофилизации. Следует понимать, что количество активного ингредиента, содержащееся в индивидуальной дозе или единице дозировки, не должно само по себе составлять терапевтически эффективное количество, так как необходимое эффективное количество может быть достигнуто посредством введения множества единиц дозировки.
Новая фармацевтическая композиция может содержать, например, от около 10% до около 100%, предпочтительно, от около 20% до около 60% активного ингредиента.
При подготовке композиций для пероральной формы единицы дозировки может использоваться любой из обычных фармацевтически приемлемых эксципиентов, таких как, например, вода, гликоли, масла, спирты, ароматизирующие вещества, консерванты, красители; или такие эксципиенты, как крахмалы, сахар, микрокристаллическая целлюлоза, разбавители, средства гранулирования, смазки, связующие вещества, вещества для улучшения разложения и т.п. в случае пероральных твердых составов, таких как, например, порошки, капсулы и таблетки, при этом твердые пероральные составы являются более предпочтительными по сравнению с жидкими составами. Вследствие простоты их введения таблетки и капсулы представляют самую выгодную пероральную форму единицы дозировки, в которой, очевидно, используются твердые фармацевтические носители.
Специалист в данной области техники может выбрать один или больше указанных выше эксципиентов в отношении конкретных требуемых свойств формы единицы дозировки посредством обычного экспериментирования и без какой-либо чрезмерной нагрузки. Количество каждого используемого эксципиента может изменяться в диапазонах, являющихся стандартными в технике. Следующие ссылки, которые включены в настоящее описание посредством ссылок, раскрывают методики и эксципиенты, применяемые для создания рецептур пероральных форм дозировки. (см. The Handbook of Pharmaceutical Excipients, 4th edition, Rowe и соавт., Eds., American Pharmaceuticals Association (2003); and Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003)).
Примеры фармацевтически приемлемых веществ для улучшения разложения включают, но не ограничены указанным, крахмалы; глины; целлюлозы; альгинаты; камеди; поперечно сшитые полимеры, например, поперечно сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL компании International Specialty Products (Уэйн, Нью-Джерси); поперечно сшитую натриевую карбоксиметилцеллюлозу или натриевую кроскармеллозу, например, AC-DI-SOL компании FMC; и поперечно сшитую кальциевую карбоксиметилцеллюлозу; полисахариды сои; и гуаровую камедь. Вещество для улучшения разложения может присутствовать в количестве от около 0% до около 10% по весу композиции. В одном из вариантов осуществления вещество для улучшения разложения присутствует в количестве от около от 0,1% до около 5% по весу композиции.
Примеры фармацевтически приемлемых связующих веществ включают, но не ограничены указанным, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, AVICEL PH компании FMC (Филадельфия, Пенсильвания), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидкросипропилметилцеллюлозу METHOCEL компании Dow Chemical Corp. (Мидленд, Мичиган); сахарозу; декстрозу; кукурузную патоку; полисахариды; и желатин. Связующее вещество может присутствовать в количестве от около 0% до около 50%, например, 2-20% по весу композиции.
Примеры фармацевтически приемлемых смазок и фармацевтически приемлемых регуляторов сыпучести включают, но не ограничены указанным, коллоидный кварц, трисиликат магния, крахмалы, тальк, трехосновный фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, целлюлозу в порошке и микрокристаллическую целлюлозу. Смазка может присутствовать в количестве от около 0% до около 10% по весу композиции. В одном из вариантов осуществления смазка может присутствовать в количестве от около 0,1% до около 1,5% по весу композиции. Регулятор сыпучести может присутствовать в количестве от около 0,1% до около 10% по весу.
Примеры фармацевтически приемлемых наполнителей и фармацевтически приемлемых разбавителей включают, но не ограничены указанным, сахарную глазурь, сахар кусковой, декстраты, декстрин, декстрозу, лактозу, маннитол, микрокристаллическую целлюлозу, целлюлозу в порошке, сорбитол, сахарозу и тальк. Наполнитель и/или разбавитель, например, могут присутствовать в количестве от около 0% до около 80% по весу композиции.
Форма единицы дозировки, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, может быть в форме микротаблеток, заключенных внутри капсулы, например, желатиновой капсулы. Для этого может использоваться желатиновая капсула, такая как используется в фармацевтических составах, такая как твердая желатиновая капсула, известная как CAPSUGEL, поставляемая Pfizer.
Примеры фармацевтически приемлемых веществ для улучшения разложения включают, но не ограничены указанным, крахмалы; глины; целлюлозы; альгинаты; камеди; поперечно сшитые полимеры, например, поперечно сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL компании International Specialty Products (Уэйн, Нью-Джерси); поперечно сшитую натриевую карбоксиметилцеллюлозу или натриевую кроскармеллозу, например, AC-DI-SOL компании FMC; и поперечно сшитую кальциевую карбоксиметилцеллюлозу; полисахариды сои; и гуаровую камедь. Вещество для улучшения разложения может присутствовать в количестве от около 0% до около 10% по весу композиции. В одном из вариантов осуществления вещество для улучшения разложения присутствует в количестве от около от 0,1% до около 5% по весу композиции.
Примеры фармацевтически приемлемых связующих веществ включают, но не ограничены указанным, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, AVICEL PH компании FMC (Филадельфия, Пенсильвания), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидкросипропилметилцеллюлозу METHOCEL компании Dow Chemical Corp. (Мидленд, Мичиган); сахарозу; декстрозу; кукурузную патоку; полисахариды; и желатин. Связующее вещество может присутствовать в количестве от около 0% до около 50%, например, 2-20% по весу композиции.
Примеры фармацевтически приемлемых смазок и фармацевтически приемлемых регуляторов сыпучести включают, но не ограничены указанным, коллоидный кварц, трисиликат магния, крахмалы, тальк, трехосновный фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, целлюлозу в порошке и микрокристаллическую целлюлозу. Смазка может присутствовать в количестве от около 0% до около 10% по весу композиции. В одном из вариантов осуществления смазка может присутствовать в количестве от около 0,1% до около 1,5% по весу композиции. Регулятор сыпучести может присутствовать в количестве от около 0,1% до около 10% по весу.
Примеры фармацевтически приемлемых наполнителей и фармацевтически приемлемых разбавителей включают, но не ограничены указанным, сахарную глазурь, сахар кусковой, декстраты, декстрин, декстрозу, лактозу, маннитол, микрокристаллическую целлюлозу, целлюлозу в порошке, сорбитол, сахарозу и тальк. Наполнитель и/или разбавитель, например, могут присутствовать в количестве от около 0% до около 80% по весу композиции.
В одном из вариантов осуществления настоящее изобретение относится к фармацевтической композиции для применения в лечении или предотвращении пролиферативного заболевания у нуждающегося в этом пациента, содержащей в количестве от около 100 мг до около 450 мг соединения формулы (I) или его фармацевтически приемлемой соли вместе с одним или более фармацевтически приемлемых эксципиентов, при этом фармацевтическая композиция вводится перорально пациенту по меньшей мере, в течение двух последовательных пятидневных циклов, и не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В одном из вариантов осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту в суточной дозе от около 100 мг до около 450 мг, предпочтительно от около 200 мг до около 400 мг, или, более предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней. Предпочтительно, состав или его фармацевтически приемлемая соль не вводятся в течение приблизительно 2 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода около 3 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания у нуждающегося в этом пациента, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся в течение периода около 2,5 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания в соответствии со схемой приема по настоящему описанию, в котором соединение формулы (I) или его фармацевтически приемлемая соль вводятся в течение двух или более указанных последовательных пятидневных циклов до ослабления, уменьшения или частичного снятия тяжести, встречаемости или частоты по меньшей мере одного побочного эффекта у указанного пациента.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания в соответствии со схемой приема по настоящему описанию, в котором соединение формулы (I) или ее его фармацевтически приемлемая соль вводится в течение двух или более указанных последовательных пятидневных циклов до начала прогрессирования заболевания.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания, включающему в себя, во-первых, введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в количестве от около 100 мг до около 450 мг ежедневно по непрерывному ежедневному графику через пероральное введение, во-вторых, определение того, что для указанного пациента имеется побочный эффект, выбранный из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени (например, повреждение печени или заболевание печени, повышение уровня аспартатаминотрансферазы, повышение уровня аланинаминотрансферазы, и т.д.), сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи (например, макулопапулезной, угревидной, и т.д.) и гиперчувствительности (например, повышенная чувствительность к ушибам), фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии после введения указанного соединения формулы (I) или его фармацевтически приемлемой соли указанному пациенту, и, в-третьих, уменьшение введения указанного соединения формулы (I) или его фармацевтически приемлемой соли до суточной дозы от около 100 мг до около 450 мг через пероральное введение, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к способу ослабления по меньшей мере одного побочного эффекта, выбранного из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени (например, повреждение печени или заболевание печени, повышение уровня аспартатаминотрансферазы, повышение уровня аланинаминотрансферазы, и т.д.), сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи (например, макулопапулезной, угревидной, и т.д.) и гиперчувствительности (например, повышенная чувствительность к ушибам), фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии, возникшего после предшествующего лечения соединением формулы (I) или его фармацевтически приемлемой солью, включающему в себя пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту в суточной дозе от около 100 мг до около 450 мг, предпочтительно от около 200 мг до около 400 мг или, более предпочтительно, от около 350 мг до около 400 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Кроме того, настоящее изобретение включает в себя способ лечения или предотвращения пролиферативного нарушения в соответствии с любым другим вариантом осуществления, раскрытым выше для настоящего изобретения.
В одном из вариантов осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится нуждающемуся в нем пациенту в суточной дозе от около 100 мг до около 450 мг, предпочтительно от около 200 мг до около 400 мг или, более предпочтительно, от около 350 мг до около 400 мг, указанного соединения формулы (I) или его фармацевтически приемлемой соли, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное лекарственное средство не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится нуждающемуся в нем пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится нуждающемуся в нем пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средствот перорально вводится нуждающемуся в нем пациенту один раз в сутки (q.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода около 3 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится нуждающемуся в нем пациенту дважды в сутки (b.i.d.) в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся в течение периода около 2,5 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится в соответствии со схемой приема по настоящему описанию, при этом соединение формулы (I) или его фармацевтически приемлемая соль вводится в двух или более указанных последовательных пятидневных циклах до ослабления, уменьшения или частичного снятия тяжести, встречаемости или частоты по меньшей мере одного побочного эффекта у указанного пациента.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство перорально вводится в соответствии со схемой приема по настоящему описанию, при этом соединение формулы (I) или ее его фармацевтически приемлемая соль вводится в двух или более указанных последовательных пятидневных циклах до начала прогрессирования заболевания.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания, при этом указанное лекарственное средство сначала перорально вводится в количестве от около 100 мг до около 450 мг в суточной дозе по непрерывному ежедневному графику, после чего производится снижение до вводимого количества от около 100 мг до около 450 мг в суточной дозе, по меньшей мере, в течение двух последовательных пятидневных циклов через пероральное введение, при этом указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Кроме того, настоящее изобретение включает в себя любое применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания в соответствии со способами лечения или любым вариантом осуществления, раскрытым выше для настоящего изобретения.
В одном из вариантов осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения или предотвращения пролиферативного заболевания, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль вводится перорально нуждающемуся в этом пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Кроме того, настоящее изобретение включает любое применение соединения формулы (I) или его фармацевтически приемлемой соли в соответствии со способами лечения или любым вариантом осуществления, раскрытым выше для настоящего изобретения.
Настоящее изобретение также относится к схеме лечения, включающей пероральное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в нем пациенту в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли, вводимой в комбинации по меньшей мере с одним дополнительным терапевтическим средством для лечения или профилактики пролиферативного заболевания, при этом соединение формулы (I) или его фармацевтически приемлемая соль вводится в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Подходящие терапевтические средства для применения в соответствии с настоящим изобретением включают, но не ограничены указанным, ингибиторы киназы, антиэстрогены, анти-андрогены, другие ингибиторы, химиотерапевтические противораковые препараты, алкилирующие агенты, хелирующие агенты, модификаторы биологического отклика, вакцины против рака, вещества для антисмысловой терапии. Примеры изложены ниже:
A. Ингибиторы киназ, включая ингибиторы киназ рецептора эпидермального фактора роста (EGFR), такие как низкомолекулярные хиназолины, например, гефитиниб (патент США 5457105, патент США 5616582 и патент США 5770599), ZD-6474 (WO 01/32651), эрлотиниб (тарцева®, патент США 5747498 и WO 96/30347) и лапатиниб (патент США 6727256 и WO 02/02552), и цетуксимаб; ингибиторы киназ рецептора сосудистого эндотелиального фактора роста (VEGFR), включая SU-11248 (WO 01/60814), SU 5416 (патент США 5883113 и WO 99/61422), SU 6668 (патент США 5883113 и WO 99/61422), CHIR-258 (патент США 6605617 и патент США 6774237), ваталаниб или PTK-787 (патент США 6258812), VEGF-Trap (WO 02/57423), B43-генистеин (WO-09606116), фенретинид (ретиноевой кислоты p-гидроксифениламин) (патент США 4323581), IM-862 (WO 02/62826), бевацизумаб или авастин® (WO 94/10202), KRN-951, 3-[5-(метилсульфонилпиперадин метил)-индолил]-хинолон, AG-13736 и AG-13925, пирроло[2,1-f][1,2,4]триазины, ZK-304709, веглин®, VMDA-3601, EG-004, CEP-701 (патент США 5,621,100), Cand5 (WO 04/09769); ингибиторы тирозинкиназы Erb2, такие как пертузумаб (WO 01/00245), трастузумаб и ритуксимаб; ингибиторы протеинкиназы Akt, такие как RX-0201; ингибиторы протеинкиназы C (PKC), такие как LY-317615 (WO 95/17182), и перифозин (патент США 2003171303); ингибиторы киназы Raf/Map/MEK/Ras, включая сорафениб (BAY 43-9006), ARQ-350RP, LErafAON, BMS-354825 AMG-548, MEK162 и другие, раскрытые в WO 03/82272; ингибиторы киназы рецептора фактора роста фибробластов (FGFR); ингибиторы циклин-зависимой киназы (CDK), включая CYC-202 или росковитин (WO 97/20842 и WO 99/02162); ингибиторы киназы рецептора фактора роста тромбоцитов (PDGFR), такие как CHIR-258, 3G3 mAb, AG-13736, SU-11248 и SU6668; и ингибиторы киназы Bcr-Abl и слитые белки, такие как STI-571 или гливек® (иматиниб).
B. Антиэстрогены: нацеленные на эстроген средства включают селективные модуляторы рецепторов эстрогена (SERM), включая тамоксифен, торемифен, ралоксифен; ингибиторы ароматазы, включая аримидекс® или анастрозол; негативные регуляторы рецепторов эстрогена (ERD), включая фаслодекс® или фулвестрант.
C. Антиандрогены: нацеленные на андрогены средства, включая флутамид, бикалутамид, финастерид, аминофинастерид, аминоглутетимид, кетоконазол и кортикостероиды.
D. Другие ингибиторы, включая ингибиторы белка фарнезилтрансферазы, включая типифарниб или R-115777 (патент США 2003134846 и WO 97/21701), BMS-214662, AZD-3409 и FTI-277; ингибиторы топоизомеразы, включая мербарон и дифломотекан (BN- 80915); ингибиторы митотического белка веретена деления кинезина (KSP), включая SB-743921 и MKI-833; модуляторы протеасомы, такие как бортезомиб или велкейд® (патент США 5780454), XL-784; ингибиторы циклооксигеназы 2 (COX-2), включая нестероидные противовоспалительные препараты I (NSAID); летрозол; эксеместан; и эрибулин.
E. Противораковые химиотерапевтические препараты, включая анастрозол (армидекс®), бикалутамид (казодекс®), блеомицина сульфат (бленоксан®), бисульфан (милеран®), бисульфан для инъекций (бисульфекс®), капецитабин (кселода®), N4 пентоксикарбонил-5-дезокси-5-фторцитидин, карбоплатин (параплатин®), кармустин (BiCNU®), хлорамбуцил (лейкеран®), цисплатин (Platinol®), кладрибин (лейстатин®), циклофосфамид (цитоксан® или неозар®), цитарабин, цитозиновый арабинозид (Cytosar-U®), липосомную инъекцию цитарабина (DepoCyt®), дакарбазин (DTIC-Dome®), дактиномицин (актиномицин D, космеган), даунорубицина гидрохлорид (керубидин®), липосомную инъекцию даунорубицина цитрата (DaunoXome®), дексаметазон, доцетаксел (таксотер®), доксорубицина гидрохлорид (адриамицин®, рубекс®), этопозид (вепезид®), флударабина фосфат (флудара®), 5-фторурацил (адруцил®, эфудекс®), флутамид (эйлексин®), тезацитибин, гемцитабин (дифтордезоксицитидин), гидроксимочевину (Hydrea®), идарубицин (идамицин®), ифосфамид (IFEX®), иринотекан (камптозар®), L-аспарагиназа (ELSPAR®), лейковорин кальция, мелфалан (алкеран®), 6-меркаптопурин (пуринетол®), метотрексат (фолекс®), митоксантрон (новантрон®), милотарг, паклитаксел (таксол®), финикс (иттрий90/MX-DTPA), пентостатин, полифепрозан 20 с имплантатом кармустина (глиадел®), цитрат тамоксифена (нолвадекс®), тенипозид (вумон®), 6-тиогуанин, тиотепа, тирапазамин (тиразон®), топотекана гидрохлорид для инъекций (гикамптин®), винбластин (велбан®), винкристин (онковин®) и винорелбин (навелбин®).
F. Алкилирующие средства, включая VNP-40101M или клоретизин, оксалиплатин (патент США 4169846, WO 03/24978 и WO 03/04505), глуфосфабид, мафосфамид, этопофос (патент США 5041424), преднимустин; треосульфан; бисульфан; ирофлувен (ацилфульвен); пенкломедин; пиразолоакридин (PD-115934); O6-бензилгуанин; децитабин (5-аза-2-дезоксицитидин); бросталлицин; митомицин C (MitoExtra); TLK-286 (телцита®); темозоломид; трабектедин (патент США 5478932); AP-5280 (платиновокислый состав цисплатина); пофриромицин; и клеаразид (меклоретамин).
G. Хелирующие средства, включая тетратиомолибдат (WO 01/60814); RP 697; химерик T84.66 (cT84.66); гадофосвесет (вазовист®); дефероксамин; и блеомицин, необязательно, в сочетании с электропорацией (EPT).
H. Модификаторы биологического отклика, такие как иммуномодуляторы, включая стауроцоприн и его макроциклические аналоги, включая UCN-01, CEP 701 и мидостаурин (см. WO 02/30941, WO 97/07081, WO 89/07105, патент США 5621100, WO 93/07153, WO 01/04125, WO 02/30941, WO 93/08809, WO 94/06799, WO 00/27422, WO 96/13506 и WO 88/07045); скваламин (WO 01/79255); DA-9601 (WO 98/04541 и США 6,025,387); алемтузумаб; интерфероны (например, IFN-a, IFN-b и т.д.) ; интерлейкины, предпочтительно, IL-2 или алдеслейкин, а также IL-1, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12 и их активные биологические варианты, имеющие аминокислотные последовательности, составляющие более 70% от нативной последовательности человека; алтрематин (гексален®); SU 101 или лефлумонид (WO 04/06834 и патент США США 6331555); имидазохолины, такие как резиквимод и имихимоод (патенты США 4689338 5389640 5268376 4929624 5266575 5352784 5494916 5482936 5346905 5395937 5238944 и 5525612); и SMIP, включая бензалолы, антрахинон, тиосемикарбазоны и трипантрины (WO 04/87153, WO 04/64759 и WO 04/60308).
I. Противораковые вакцины: противораковые вакцины включая авицин® (Tetrahedron Lett. 26:2269-70 (1974)); ореговомаб (OvaRex®); тератоп® (STn-KLH); вакцины против меланомы; серия GI-4000 (GI-4014, GI-4015 и GI-4016), нацеленные на пять мутаций в белке Ras; GlioVax-1; MelaVax; адвексин® или INGN-201 (WO 95/12660); Sig/E7/LAMP-1, кодирующий HPV-16 E7; вакцина MAGE-3 или M3TK (WO 94/05304); HER-2VAX; ACTIVE, стимулирующий Т-клетки, специфичные для опухолей; противораковую вакцину GM-CSF; и вакцины на основе моноцитогенов Listeria.
J. Средства антисмысловой терапии: противораковые средства, включающие антисмысловые композиции, такие как AEG-35156 (GEM 640); AP-12009 и AP-11014 (TGF-бета-2-специфичные антисмысловые олигонуклеотиды); AVI-4126; AVI-4557; AVI-4472; облимерсен (генасенсе®); JFS2; апринокарсен (WO 97/29780); GTI-2040 (антисмысловой олигонуклеотид, воздействующий на R2 рибонуклеотид-редуктазы мРНК) (WO 98/05769); GTI-2501 (WO 98/05769); капсулированные в липосомах антисмысловые олигодезоксинуклеотиды c-Raf (LErafAON) (WO 98/43095); и Sirna-027 (основанное на интерферирующей РНК терапевтическое нацеливание на мРНК VEGFR-1).
В одном из вариантов осуществления дополнительное терапевтическое средство выбирают из гефитиниба, эрлотиниба, бевацизумаба или авастина®, пертузумаба, трастузумаба, MEK162, тамоксифена, фулвестранта, капецитабина, цисплатина, карбоплатина, цетуксимаба, паклитаксела, темозоломида, летрозола или эксеместана.
Структуру лекарственных веществ, идентифицированных по кодовым номерам, непатентованным наименованиям или торговым маркам, можно получить из Интернета, актуального выпуска стандартного справочника «Индекс Мерка» или из баз данных, например, Patents International, например, IMS World Publications, или из упомянутых выше и ниже публикаций. Соответствующее содержание указанных документов включено в настоящее описание посредством ссылок.
Соединение формулы (I) и дополнительное терапевтическое средство могут быть введены вместе в одной фармацевтической композиции, отдельно в двух или более отдельных формах единиц дозирования, или последовательно. Фармацевтическая композиция или форма единицы дозировки, содержащие дополнительное терапевтическое средство, может быть приготовлено способом, известным самим по себе, и являются подходящими для энтерального, такого как оральное или ректальное, местного и парентерального введения субъектам, включая млекопитающих (теплокровные животные), таких как люди.
В частности терапевтически эффективное количество каждого из терапевтических средств может быть введено одновременно или последовательно, и в произвольном порядке, и компоненты могут быть введены отдельно или как фиксированная комбинация. Например, комбинация по настоящему изобретению может включать: (i) введение первого терапевтического средства (a) в свободной форме или форме фармацевтически приемлемой соли; и (ii) введение терапевтического средства (b) в свободной форме или форме фармацевтически приемлемой соли, одновременно или последовательно, в произвольном порядке, в суммарно терапевтически эффективных количествах, предпочтительно в синергически эффективных количествах, например, в ежедневных или периодических дозировках, соответствующих количествам, описанным в настоящем описании. Отдельные терапевтические средства комбинации могут быть введены отдельно в разные моменты времени в течение курса терапии, или параллельно в разделенной форме или форме единой комбинации.
«Синергия» или «синергический» относится к действию двух терапевтических средств, таких как, например, (a) соединения формулы (I) или его фармацевтически приемлемой соли и (b) ингибитора ароматазы, обеспечивающему такой результат, как, например, замедление симптоматического прогрессирования ракового заболевания или нарушения, в частности, рака, или его симптомов, которое является большим, чем при простом сложении результатов каждого из терапевтических средств, введенных по отдельности. Синергический эффект может быть вычислен, например, с помощью подходящих способов, таких как сигмоидальная Emax-формула (Holford, N. H. G. and Scheiner, L. B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Леве (Loewe, S. and Muischnek, H., Arch. Exp. Pathol Pharmacol. 114: 313-326 (1926)) и уравнение эффекта медианы (Chou, T. C. and Talalay, P., Adv. Enzyme Reg мкл. 22: 27-55 (1984)). Каждое уравнение, упомянутое выше, может быть применено к экспериментальным данным для создания соответствующего графика для помощи в оценке результатов комбинации терапевтических средств. Соответствующие графики, ассоциированные с уравнениями, упомянутыми выше, являются кривыми концентрации-результата, кривой изоболограммы и кривой показателя аддитивности, соответственно. Синергия может быть также показана путем вычисления значения синергии для комбинации в соответствии со способами, известными специалистам.
Эффективная дозировка каждого средства из терапевтического средства (a) или терапевтического средства (b), применяемых в комбинации, может изменяться в зависимости от конкретного используемого компонента или фармацевтической композиции, способа введения, подвергающегося лечению патологического состояния, серьезности подвергающегося лечению патологического состояния. Таким образом, режим дозирования комбинации выбирают в соответствии со множеством факторов, включая тип, вид, возраст, пол и медицинское состояние пациента; серьезности подвергающегося лечению патологического состояния; маршрут введения; функционирование почек и печени у пациента; и конкретное используемое соединение. Обычный врач, врач-клиницист или ветеринар могут легко определить и предписать эффективное количество терапевтического средства, требуемого для предотвращения, противодействия или остановки прогрессирования патологического состояния. Для оптимальной точности в достижении концентрации терапевтического средства в пределах диапазона, обеспечивающего эффективность, требуется режим, основанный на кинетике доступности терапевтического средства в целевых местах. Это включает рассмотрение распределения, равновесия и выведения терапевтического средства.
Примеры пролиферативных заболеваний, которые могут подвергаться лечению комбинацией соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного дополнительного терапевтического вещества, включают изложенные выше примеры, но не ограничены указанным.
С помощью установленных тестовых моделей может быть показано, что комбинация по настоящему изобретению обеспечивает положительные эффекты, описанные выше в настоящем описании. Специалист в данной области техники обладает достаточными способностями для выбора подходящей тестовой модели с целью подтверждения таких положительных эффектов. Фармакологическая активность комбинации по настоящему изобретению может, например, быть продемонстрирована в клиническом исследовании или в процедуре тестирования, описанной по существу ниже в настоящем описании.
Подходящими клиническими исследованиями являются, в частности, например, исследования с увеличением дозы без контроля плацебо у пациентов с пролиферативным заболеванием, включая, например, заболевание с опухолью, например, рак груди. Такие исследования доказывают, в частности, синергизм терапевтических средств комбинации по настоящему изобретению. Положительные эффекты для пролиферативного заболевания могут быть определены непосредственно через результаты этих исследований, что известно специалисту в данной области техники. Такие исследования могут являться, в частности, подходящими для сравнения результатов монотерапии с применением терапевтических средств и комбинации по настоящему изобретению. В одном из вариантов осуществления доза соединения селективного по альфа-изоформе ингибитора PI3K формулы (I) или его фармацевтически приемлемой соли повышается до достижения максимальной переносимой дозы, и партнер по комбинации вводится в фиксированной дозе. Альтернативно, соединение формулы (I) или его фармацевтически приемлемая соль может вводиться в фиксированной дозе, и доза партнера по комбинации может повышаться. Каждый пациент может получить дозы соединения формулы (I) или его фармацевтически приемлемой соли один раз в сутки или несколько раз (например, дважды) в сутки. Эффективность лечения может быть определена в таких исследованиях, например, после 12, 18 или 24 недели посредством проведения оценок симптомов каждые 6 недель.
В комбинации по настоящему изобретению соединение формулы (I) или его фармацевтически приемлемая соль вводится в суточной дозе от около 100 мг до около 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль не вводится пациенту в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
В одном из вариантов осуществления настоящее изобретение относится к способу лечения или предотвращения пролиферативного заболевания посредством введения в соответствии со схемой приема по настоящему изобретению, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль вводится в комбинации по меньшей мере с одним дополнительным терапевтическим средством.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предотвращения пролиферативного заболевания в соответствии со схемой приема по настоящему изобретению, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль вводится в комбинации по меньшей мере с одним дополнительным терапевтическим средством.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения или предотвращения пролиферативного заболевания в соответствии со схемой приема по настоящему изобретению, при этом указанное соединение формулы (I) или его фармацевтически приемлемая соль вводится в комбинации по меньшей мере с одним дополнительным терапевтическим средством.
Настоящее изобретение также относится к упаковке, содержащей фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль в суточной дозе от около 100 мг до около 450 мг вместе с одним или более фармацевтически приемлемых эксципиентов в комбинации с инструкциями по пероральному введению указанной фармацевтической композиции, по меньшей мере, в течение двух последовательных пятидневных циклов, и по отсутствию введения указанной композиции в течение периода от около 2 дней до около 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
Полезность схемы приема соединений формулы (I) по настоящему изобретению может быть продемонстрирована in vitro, в методах тестирования на животных, а также в клинических исследованиях. Например, полезность соединений формулы (I) в соответствии с настоящим изобретением может быть продемонстрирована в соответствии со способами, описанным ниже в настоящем описании:
Пример 1:
Материалы и методы
Животные и условия содержания: Эксперименты выполнялись на самках Hsd: безтимусные мыши Nude-nu CPB (Харлан Винкелман, Германия). Животные имели возраст от 12 до 14 недель в начале лечения и были размещены при оптимизированных гигиенических условиях (OHC) в клетках Makrolon типа III (максимально 5 животных на клетку) со свободным доступом к еде и воде. Кроме того, эксперименты также выполнялись на самках безтимусных крыс Rowett Hsd: RH-Fox1rnu (Харлан (Нидерланды). Животные имели возраст 6-9 недель во время введения соединения. Животные размещались при оптимизированных гигиенических условиях (OHC) в клетках Makrolon типа III (максимально 2 животных на клетку) со свободным доступом к еде и воде. Допускалась их адаптация в течение по меньшей мере 6 дней до начала эксперимента.
Клеточная линия и клеточная культура: Клетки Rat1-Myr-p110α были выращены в минимальной эссенциальной среде Игла, модифицированной по способу Дульбекко (DMEM), содержащей 4,5 г/л глюкозы с добавлением 10% термоинактивированной фетальной телячьей сыворотки (FCS), 2 мМ L-глютамина, 1мМ пирувата натрия, и инкубировались при 37°C в увлажненной атмосфере с 5% CO2. Клетки были отобраны с помощью трипсина-EDTA, ресуспендированы в питательной среде (с добавками) и подсчитаны с помощью системы Casy®. Наконец, клетки были центрифугированы, суспендированы в ледяном сбалансированном солевом растворе Хенкса (HBSS) в концентрации 3×107 клеток/мл. Реактивы клеточной культуры были закуплены у BioConcept (Allschwil, Швейцария).
Клетки Rat1-Myr-p110α были созданы посредством способа, описанного в работе Maira и соавт., Molecular Cancer Therapeutics, 11:317-328 (2012), которая включена в настоящее описание посредством ссылки во всей ее полноте. Кратко, клетки Rat1 были трансфецированы для стабильной экспрессии конститутивно активной формы каталитических α-изоформ p110 PI3K класса I посредством добавления сигнала миристилирования к N-концу.
Имплантация ксенотрансплантатов опухоли в естественных условиях: опухоли Rat1-Myr-p110α были имплантированы посредством подкожной инъекции 5x106 клеток в 100 мкл HBSS (Sigma #H8264) в правый бок безтимусных мышей или безтимусных крыс. Для эффективности экспериментов, лечение начиналось, когда средние объемы опухоли составляли приблизительно 300 мм3 (от 14 до 15 дней после инъекции опухолевых клеток). Для экспериментов с единственной дозой PK/PD животные подвергались лечению один раз перорально соединением A, когда опухоли достигали размера приблизительно 400-500 мм3 (от 21 до 23 дней после инъекции опухолевых клеток).
Получение соединения и лечение животных: соединение A было приготовлено для дозирования в форме гомогенных суспензий 1% карбоксиметилцеллюлозы: 0,5% Tween® 80: 98,5% деионизированной воды. Свежие суспензии готовились раз в 4 дня и хранились при 4°C. Соединение A или наполнитель вводились перорально в объеме 10 мл/кг.
Оценка анти-опухолевой активности: объемы опухоли были измерены с помощью кронциркуля и определены согласно формуле: длина×диаметр2×π/6. В дополнение к представлению изменений объемов опухоли в течение курса лечения, анти-опухолевая активность выражается в %T/C (среднее изменение объема опухоли подвергавшихся лечению/среднее изменение объема опухоли контрольных животных)×100. Регрессии (%) были вычислены согласно формуле ((медианный объем опухоли в конце лечения - медианный объем опухоли в начале лечения)/медианный объем опухоли в начале лечения)×100. Масса тела и объемы опухоли записывались от двух до трех раз в неделю.
Взятие образцов: образцы крови были собраны в различные моменты времени после последнего лечения (1 час, 2 часа, 4 часа, 8 часов, 12 часов, 16 часов и 24 часа, n=2-3 за момент времени) в пробирки, покрытые K-EDTA. Образцы крови центрифугировались, и собранная плазма немедленно замораживалась на -80°C до заключительной обработки. Опухоли были собраны при умерщвлении в 7 временных точках (1 час, 2 часа, 4 часа, 8 часов, 12 часов, 16 часов и 24 часа, n=2-3 за момент времени), подвергнуты быстрому замораживанию, и хранились при -80°C до заключительной обработки.
Фармакокинетические/фармакодинамические исследования
A. Пульверизация ткани животных: после рассечения опухоли были подвергнуты быстрому замораживанию в жидком азоте и хранились при -80°C. Замороженные опухоли подвергались пульверизации с помощью шаровой миксерной дробилки MM20 Retsch (Арлесхайм, Швейцария) с металлическими цилиндрами, предварительно охлажденными до -80°C в морозильнике. Порошок был снят с металлических цилиндров на сухом льду и перенесен в предварительно охлажденные пробирки Эппендорфа на 1,5 мл с предотвращением оттаивания.
B. Биоаналитика (LC/MS-MS) для квантификации соединения A: концентрации соединения A в плазме и опухоли были определены в отдельном анализе с помощью жидкостной хроматографии сверхвысокого давления/тандемной масс-спектрометрии (UPLC/MS-MS). После добавления 25 мкл внутреннего стандарта (1 мкг/мл) к аналитическим аликвотам (25 мкл) плазмы или (20 мг) порошка опухоли, белки осаждались посредством добавления 200 мкл ацетонитрила. Супернатант был перенесен в новый сосуд. После испарения до сухости образцы были повторно растворены в 60 мкл ацетонитрила/воды (1/1 объем/объем). Аликвота (5 мкл) этого раствора была отделена на колонке ACQUITY UPLC BEH C18 (Waters™, размер частиц 1,7 мкм, 2,1×50 мм) с подвижной фазой, состоящей из смеси 0,1% муравьиной кислоты в воде (растворитель A) и 0,1% муравьиной кислоты в ацетонитриле (растворитель B). Применялось программирование градиента со скоростью потока 600 мкл/мин. После уравновешивания 95%-м растворителем A, было инъецировано 5 мкл образца. После периода ожидания 0,25 мин образец был элюирован с линейным градиентом 5-100% растворителя B в течение 0,65 минут, после чего удерживался в течение 0,35 минут. Колонка была подготовлена к следующему образцу путем повторного уравновешивания в течение более 0,25 минут до исходных условий. Растворитель для элюирования колонки был непосредственно введен в источник ионов тройного квадрупольного масс-спектрометра TQD™ (Waters Corporation, Милфорд, Массачусетс, США), управляемого программным обеспечением Masslynx™ 4.1. Мониторинг нескольких реакций положительной ионизации электрораспылением (ESI+) применялся для обнаружения MS/MS анализируемого вещества. Переносы ионов от предшественника к продукту для соединения A и соответствующего внутреннего стандарта суммированы в приведенной ниже таблице:
Предел квантификации (LOQ) для соединения A был установлен на 2,5 нг/мл (CV и общее отклонение менее 30%). Регрессионный анализ и дальнейшие вычисления выполнялись с помощью QuanLynx™ 4.1 (Micromass) и Excel™ 2007 (Microsoft). Концентрации неизвестных образцов были вычислены посредством обратных вычислений на основании отношений площадей пиков анализируемого вещества/IS по калибровочной кривой, построенной с помощью калибровочных образцов, маркированных в чистой плазме, или опухоли, полученной из животных, которые подвергались лечению наполнителем.
C. Квантификация Ser473 P-Akt и Akt с помощью методики обращенного скрининга белков (RPPA).
Приблизительно 20 мг порошка замороженной ткани были отвешены и лизированы в 100 мкл буферной смеси для лизиса белков NP40 (исходный раствор буфера для лизиса (4°C): 2,5 мл Трис HCL 2M pH 7,8 КТ, 1 мл NP40 (100%) КТ, 2,4 мл NaCl 5M КТ, 2,5 мл NaF 1M КТ, 4 мл 1M бета-глицерофосфат динатриевая соль пентагидрат -20°C и вода до 100 мл; Раствор буфера для лизиса (4°C): 10 мл исходного раствора буфера для лизиса; 10 мкл Na3VO3 100 мМ 4°C, 10 мкл DTT 1M -20°C, 10 мкл PMSF 100 мМ 4°C, 10 мкл бензамидина 1M -20°C, и 10 мкл микроцистина -20°C).
Каждый образец был перемешан вихревым способом и центрифугирован в течение 10 минут на 10000 об/мин. Цикл замораживания-оттаивания выполнялся при -80°C в течение 30 минут. Образцы хранились, после дополнительного шага центрифугирования на 10000 об/мин в течение 10 минут, при -80°C для последующего анализа. Белковые концентрации были квантифицированы с помощью Coomassie Plus Kit (#23236 Thermo Scientific, Рокфорд, Иллинойс, США) согласно протоколу производителя. Разбавленные образцы были перенесены в 96-луночный планшет (#269620, NUNC), и поглощающая способность была измерена при 595 нм с помощью спектрофотометра для прочтения планшетов SpectraMAX Plus производства Molecular Devices. Количество белка было вычислено с помощью программного обеспечения Softmax Pro 5.0 (Molecular Devices, США) и затем нормализовано на 1 мг/мл с использованием соответствующего буфера для лизиса. Нормализованные образцы были дополнительно разбавлены 1:10 с использованием буфера для нанесения CSBL1 CeLyA (Zeptosens, cat. № 9020) с добавлением 1 мМ ортованадата натрия (Sigma, cat No. S-6508). Лизат был перенесен на 96-луночный планшет с V-образным дном (Fisher Scientific, cat. № 6067Y), после чего проводилась стадия центрифугирования (5 минут, 1500 об/мин при 19°C в центрифуге Eppendorf 5810R) для не подвергшегося лизису клеточного детрита.
Автоматизированная рабочая станция для пипетирования MATRIX 2x2 (Thermo Fisher Scientific, Великобритания) использовалось для переформатирования лизатов из 96-луночного планшета с V-образным дном в 384-луночные планшеты (Greiner, cat. № 781201). Для получения желаемой структуры для нанесения каждый образец был разбавлен до 4 различных концентраций (d1=100%, d2=75%, d3=50%, d4=25%) путем разведения лизата клеток соответствующим объемом смеси буферов для лизиса и нанесения (10% буфера для лизиса; 90% буфера для нанесения CSBL1 с добавлением 1 мМ ортованадата натрия).
Нанесение образцов проводилось на микрочипах для исследования белков ZeptoMARK® PWG (Zeptosens, Witterswil, Швейцария) с помощью основанного на пьезоэлектрическом микродозаторе бесконтактного прибора Nano-Plotter 2.1 (GeSiM, Гросзеркманнсдорф, Германия). Каждый образец был нанесен в 4 различных концентрациях (d1=100%, d2=75%, d3=50%, d4=25%) путем разведения лизата клеток соответствующим объемом смеси буферов для лизиса и нанесения. После нанесения на микрочипы для исследования белков ZeptoMARK® чипы инкубировались в течение 1 часа при 37°C. Для обеспечения однородных результатов блокирования с помощью ультразвукового распылителя вводился блокирующий буфер CeLyA BB1 (Zeptosens, cat. № 9040). После блокирования в течение 20 мин чипы тщательно промывались деионизированной водой (качества Milli-Q, 18 МОм×см) и высушивались в токе смеси азота с воздухом.
Затем чипы ZeptoMARK® были перенесены в ZeptoCARRIER (Zeptosens, cat. № 1100) и дважды промыты 200 мкл буфера для анализа CAB1 CeLyA (Zeptosens, cat. № 9032). Затем проводилось отсасывание буфера для анализа, и каждая секция инкубировалась со 100 мкл первичных антител-мишеней (pAkt Ser473 (партия № 9) (Cell Signaling Technology, каталожный № 4060); Akt1 pan (партия № E0401) (Epitomics, каталожный № 1085-1); Zenon® Alexa Fluor 647 кролика (Invitrogen, каталожный № Z25308)) при комнатной температуре (RT) в течение ночи. После инкубации первичное антитело было удалено, ряды были дважды промыты буфером CAB1, и проводилось дальнейшее инкубирование со 100 мкл меченых с помощью Alexa fluor 647 фрагментов кроличьих антител к IgG Fab (Invitrogen; #Z25305) в течение одного часа при КТ в темноте. После инкубации ряды были дважды промыты 200 мкл буфера CAB1. Флуоресценция связанных с мишенью фрагментов Fab считывалась с помощью прибора ZeptoReader (Zeptosens, Witterswil, Швейцария) с использованием лазера (длина волны возбуждения 635 нм) и камеры на основе устройства с зарядовой связью. Сигнал флуоресценции регистрировался при длительности экспонирования, равной 1, 3, 5 и 10 секунд, в зависимости от интенсивности сигнала. Изображения флуоресценции для каждого множества были проанализированы с помощью программного обеспечения ZeptoVIEW Pro 2.0 (Zeptosens, Witterswil, Швейцария), и была вычислена RFI (относительная интенсивность флуоресценции) для каждого сигнала.
D. Моделирование PK-PD: Phoenix WinNonlin 6.3 (Pharsight) применялся для моделирования временных профилей медианной концентрации в плазме после многократного дозирования с применением методики некомпартментной непараметрической суперпозиции данных, сгенерированных в результате исследования эффективности для мышей или крыс. Прогнозы основаны на норме накопления, вычисленной по концевому наклону (Лямбда Z), что обеспечивает возможность прогнозирования на основании простых или сложных схем приема.
Статистический анализ: Абсолютные значения для роста первичной опухоли и массы тела использовались для проведения статистических сравнений между группами (односторонний дисперсионный анализ с последующим тестом Даннетта для нормально распределенных данных; ранговый дисперсионный анализ для не распределенных нормально данных с последующим тестом Даннетта для группы равного размера или Данна для групп неравного размера). Уровень значимости был выбран как p<0,05. Площади под кривой (AUC), полученные для 24 ч после последнего лечения, были определены с помощью метода правила трапеций. Все статистические вычисления были выполнены с помощью SigmaStat.
Результаты
Преклиническая модель PK-PD-эффективности-переносимости для соединения A была построена с применением способов, изложенных выше. Для этой преклинической модели PK-PD-эффективности-переносимости для соединения A:
Фармакокинетические исследования и PK-моделирование для соединения A: фармакокинетика соединения A была линейной по диапазону протестированных доз (фиг. 1A: 12,5, 25 и 50 мг/кг в сутки у безтимусных мышей; фиг. 1B: 12,5, 25, 40 и 80 мг/кг в сутки у безтимусных крыс) и была ассоциирована с аналогичным изменением AUC между 12,5 и 50 мг/кг у безтимусных мышей. Аналогичная взаимосвязь наблюдалось у безтимусных крыс для доз до 80 мг/кг. На фигурах 2A и 2B предоставлена непараметрическая суперпозиционная модель для демонстрации соотношения наблюдаемых по сравнению с предсказанными концентрациями в плазме после перорального приема соединения A в дозировке 50 мг/кг в сутки у безтимусных мышей и 40 мг/кг в сутки у безтимусных крыс. (Фиг. 2 A и B). Фиг. 3A и B предоставляют сравнение наблюдаемых концентраций в плазме и предсказаний модели, и показывают, что данная используемая модель PK является хорошо предсказывающей у безтимусных мышей (R2=0,99, n=25, p<0,001) в дозах ниже 150 мг/кг в сутки и безтимусных крыс (R2=0,89, n=31, p<0,01) в дозах ниже 100 мг/кг в сутки. Кроме того, фигура 4A показывает, что эта используемая модель PK является также предсказывающей для моделирования PK-профилей соединения A, вводимого два раза в сутки (2qd) безтимусным мышам.
Данное исследование по моделированию PK было повторено с целью подтверждения предшествующих данных о том, что данная модель PK является предсказательной для моделирования профилей PK соединения A, вводимого два раза в сутки (2qd) безтимусным мышам. Результаты этого повторного исследования предоставлены на фигуре 4B и подтверждают, что данная модель PK является предсказательной для моделирования профилей PK для соединения A, вводимого два раза в сутки (2qd) безтимусным мышам. Эти данные с фигуры 4B предоставлены в настоящем описании исключительно для демонстрации дополнительного подтверждения исследования по моделированию PK у безтимусных мышей.
Моделирование PK-PD-эффективности
A. Модуляция фосфорилирования Akt: концентрации в опухоли, дающие 50% (IC50 в естественных условиях) и 80% (IC80 в естественных условиях) ингибирования S473P-Akt (0,6 и 4 мкМ/л, соответственно) по сравнению с контрольными группами были определены путем измерения уровня фосфорилирования Akt с применением RPPA и специфичной концентрации противоопухолевого лекарственного препарата в сопоставленных образцах у множества животных (безтимусные мыши и крысы) и для множества временных точек после лечения соединением A (фиг. 5). После коррекции в отношении связывания белка плазмы крови соединения A у мышей (PPB=91,2%), значения IC50 (53 нмоль/л) и IC80 (на 352 нмоль/л) в естественных условиях грубо аппроксимируют значения in vitro для клеток IC50 и IC80, составляющие 74 нмоль/л и 301 нмоль/л соответственно.
B. Противоопухолевая активность соединения A в модели опухоли Rat1-myr-p110α: соединение A было введено перорально имеющим опухоль Rat1-myr-p110α мышам и крысам в различных дозах. Результаты ингибирования роста опухоли подытожены ниже:
(наблюда-емое)
(наблюда-емый)
(наблюда-емый)
(наблюда-емый)
Ингибирование, как оказалось, было зависящим от дозы. Регресс опухоли наблюдался при суточных дозах выше 25 мг/кг у безтимусных мышей и крыс, и при вводимых дважды в сутки дозах выше 12,5 мг/кг у безтимусных мышей.
C. Взаимосвязь PK/PD/эффективность: на фигуре 6 предоставлена взаимосвязь между экспозицией (как измерено по времени превышения IC80 в естественных условиях) и противоопухолевой эффективностью. Далее, близкая к линейной взаимосвязь идентифицирована между величиной противоопухолевой эффективности и продолжительностью экспозиции лекарственного препарата (как измерено по времени превышения IC80 в естественных условиях) по IC80 (R2=0,89; фиг. 7). Из этой взаимосвязи было определено, что 80%-е ингибирование фосфорилирования Akt, по меньшей мере, для 25% интервала дозирования требуется для того, чтобы соединение А индуцировало остановку роста опухоли, и уровень ингибирования пути должен поддерживаться, по меньшей мере, для 45% интервала дозирования для обеспечения 30%-го регресса опухоли.
На фигуре 8 предоставлено сравнение наблюдаемого ингибирования роста опухоли и предсказанного по модели ингибирования роста опухоли после перорального введения соединения А в дозировке от 6,25 до 70 мг/кг в сутки и 2qd. Таким образом, данная модель взаимосвязи PK/PD является предсказательной для противоопухолевой эффективности альтернативных схем приема у мышей и крыс, подвергавшихся пероральному лечению различными дозами соединения А (R2=0,93, n=12, p<0,001).
Моделирование PK-PD-переносимости
A. Модуляция уровней глюкозы и инсулина: Для того, чтобы оценить, нарушает ли соединение A гомеостаз глюкозы, были измерены уровни инсулина в плазме и глюкозы в крови, и они сравнивались с концентрациями лекарственного препарата в плазме в сопоставленных образцах множества животных и в множество моментов времени. В этом анализе уровни инсулина в плазме возрастали пропорционально концентрациям соединения А в плазме, в то время как уровни глюкозы в крови сохранялись близко к нормальным вплоть до 20 мкМ/л соединения А у безтимусных мышей (фиг. 9 A и B) и до 15 мкМ/л соединения А у безтимусных крыс (фиг. 10 A и B). Однако выше 20 мкМ/л у безтимусных мышей и 15 мкМ/л у безтимусных крыс, зависящее от концентрации соединения повышение уровня глюкозы, которое приводило к гипергликемии, наблюдалось несмотря на повышение уровня инсулина в плазме. Таким образом, связанный с соединением А гипергликемический порог был определен как составляющий 20 мкМ/л и 15 мкМ/л у мышей и крыс, соответственно.
B. Взаимосвязь PK/PD/переносимости: Кроме того, близкая к линейной взаимосвязь наблюдалась между величиной потери массы тела и продолжительностью экспозиции соединения А выше порога гипергликемии (20 мкМ/л для безтимусных мышей и 15 мкМ/л для безтимусных крыс; R2=0,98, фиг. 11). Из этой взаимосвязи понятно, что уровни экспозиции соединения должны поддерживаться не более чем в 35% интервала дозирования выше порога отсечения гипергликемии для поддержания потери массы тела ниже 5% у мышей и крыс.
Моделирование PK-PD-эффективности
Смоделированные кривые эффективности (как определено долей времени выше порога IC80 для S473P-Akt) и кривые переносимости (как определено продолжительностью экспозиции выше порога гипергликемии соединения А (20 мкМ/л)) у мышей, подвергавшихся пероральному лечению один раз в сутки возрастающими дозами соединения A, показаны на графике на фигуре 12. Моделирование предполагает, что при дозе 70 мг/кг в сутки (меньше чем 5% потеря BW), 80% ингибирование pAkt будет достигнуто в течение 65% времени между двумя последовательными лечениями, обеспечивая 55%-й регресс опухоли (фиг. 12, фиг. 7). Если доза 70 мг/кг в сутки вводится как 35 мг/кг два раза в сутки (2qd), модель показывает, что 80% ингибирование pAkt будет достигнуто в течение 100% времени между двумя последовательным лечениями, обеспечивая регресс опухоли. У безтимусных крыс, для которых порог гипергликемии был установлен на 15 мкМ/л, доза 30 мг/кг в сутки (отсутствие потери BW) приводила к 80% ингибированию pAkt в течение 83% времени между двумя последовательными лечениями, обеспечивая 80%-й регресс опухоли (фиг. 13, фиг. 7).
Пример применения: 20 мг/кг в сутки в режиме дозирования «альтернативная схема 1» у безтимусных крыс
На основании предшествующего анализа, преклиническое моделирование PK-PD-эффективности-переносимости для соединения А, описанное выше, является ценным инструментом для предсказания эффективности и переносимости следующей схемы приема соединения A: пероральное введение соединения А один раз в сутки (q.d.) или дважды в сутки (b.i.d.) в течение пяти последовательных дней, после чего соединение А не вводится в течение двух дней (цикл 1), и затем повторение той же схемы приема [т.е., пероральный прием соединения А один раз в сутки (q.d.) или дважды в сутки (b.i.d.) в течение пяти последовательных дней, после чего соединение А не вводится в течение двух дней] в одном или более последовательных циклах. Эта альтернативная схема приема называется «альтернативной схемой 1». Как описано в настоящем описании, эта модель в настоящем описании применяется для исследования и направления создания схемы приема в клинических исследованиях.
На фигуре 14 предоставлены графики, показывающие смоделированную эффективность соединения А у имевших опухоль Rat1-myr P110α безтимусных крыс, которым перорально вводилось соединение А в дозировке 20 мг/кг в альтернативной схеме 1 (A), по сравнению с 14 мг/кг в сутки в непрерывном ежедневном графике (т.е. без прерывания введения лекарственного препарата) (B). На фигуре 15 предоставлен смоделированный PK-профиль в плазме у имевших опухоль Rat1-myr P110α безтимусных крыс, которым перорально вводилось соединение А в дозировке 20 мг/кг по альтернативной схеме 1 (A), по сравнению с 14 мг/кг в сутки в непрерывном ежедневном графике (т.е. без прерывания введения лекарственного препарата).
На основании нашего моделирования (фиг. 7), альтернативная схема 1 для соединения A может (a) достигать аналогичной или повышенной противоопухолевой эффективности, наблюдаемой у безтимусных крыс, которым перорально вводилось соединение А один раз в сутки (q.d.) по непрерывному ежедневному графику и (b) достижение, по меньшей мере, частичного регресса (30%-й регресс опухоли) за весь период лечения, если концентрация соединения А в плазме выше IC80 для pAkt в течение 45% времени между двумя периодами лечения. На основании эквивалентного AUC, доза соединения А для человека, составляющая 300-350 мг/день перорально (Cmax: 3500 нг/мл=8 мкмоль/л; AUC: 35000 h.нг/мл=80 h.мкмоль/л), соответствует 20 мг/кг пероральной дозы в альтернативной схеме 1 у безтимусных крыс. Соответствующая суммарная пероральная доза в сутки при непрерывном ежедневном графике составила бы 14 мг/кг.
Согласно этой модели у безтимусных крыс (фиг. 13) соединение А в дозировке 20 мг/кг приведет приблизительно к 60%-й регрессу опухоли. Таким образом, предсказанная эффективность для 20 мг/кг в альтернативной схеме 1 у безтимусных крыс представлена на фиг. 14A. Макс. эффективность составляет 60%-й регресс с восстановлением до 30% регресса в конце 2 дней отсутствия введения лекарственного препарата. Предсказанная эффективность для ежедневной дозировки 14 мг/кг составляет 30% регресс опухоли (фиг. 14B).
Предсказанные уровни в плазме соединения А после перорального лечения у мышей и крыс в дозировке 14 мг в сутки в непрерывном ежедневном графике или 20 мг в альтернативной схеме 1 не превысит 15 мкмоль (порог гипергликемии) (фиг. 15).
Если предположить, что взаимосвязь между PD и эффективностью у людей и ксенотрансплантатов является аналогичной, то данная модель и анализ могут быть полезными для предсказания ответа опухоли у людей на альтернативную схему 1.
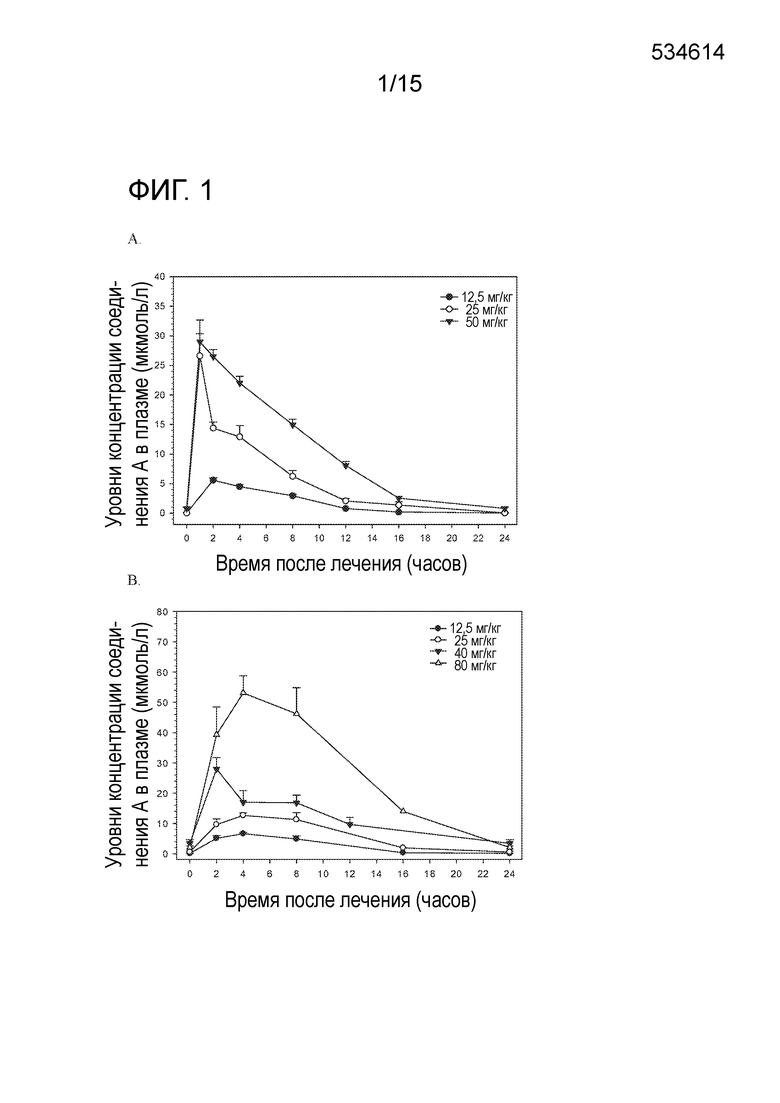
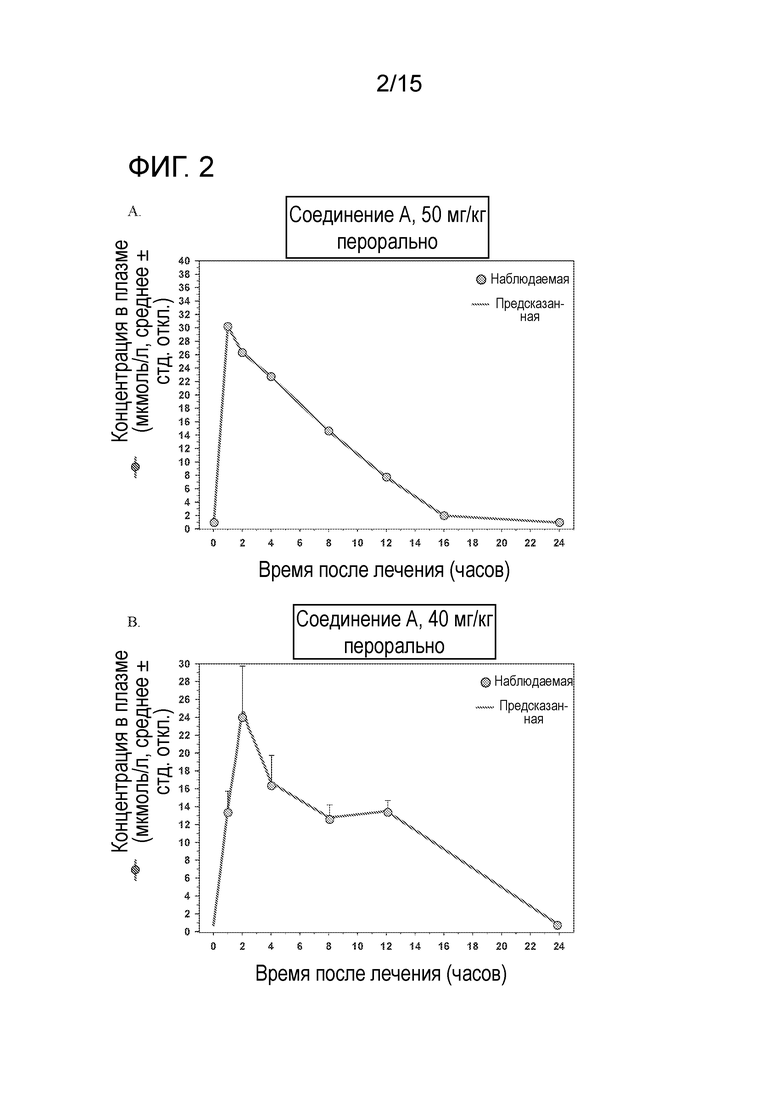
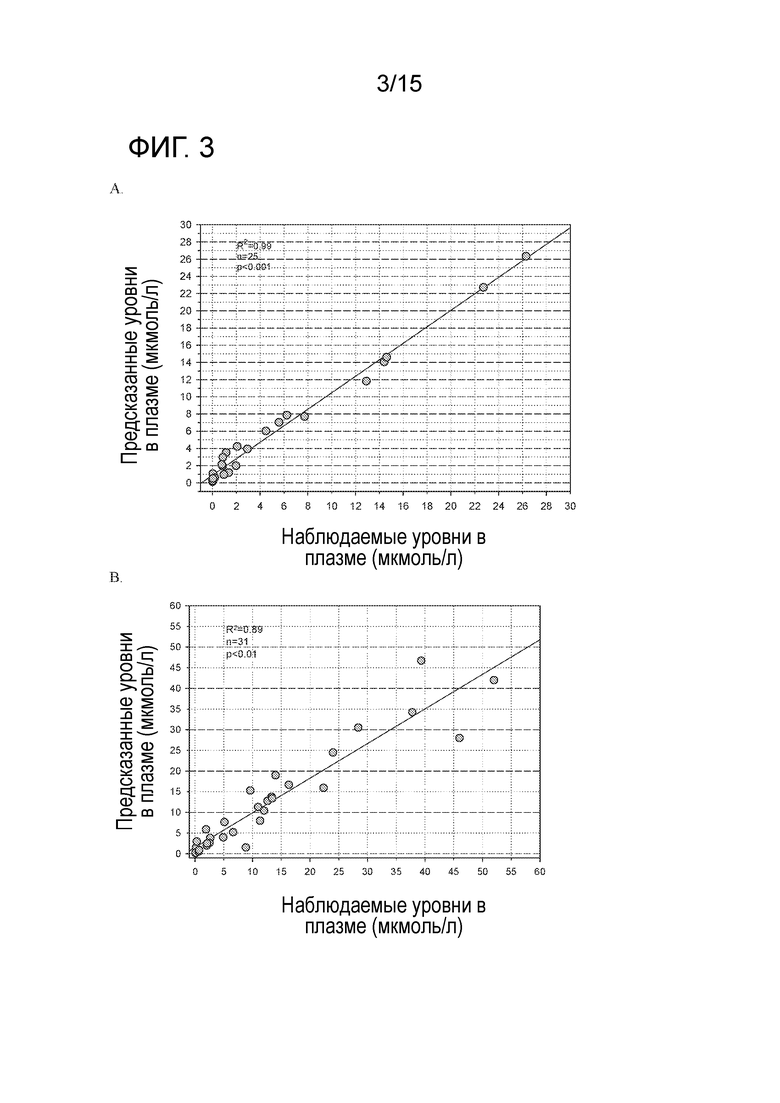
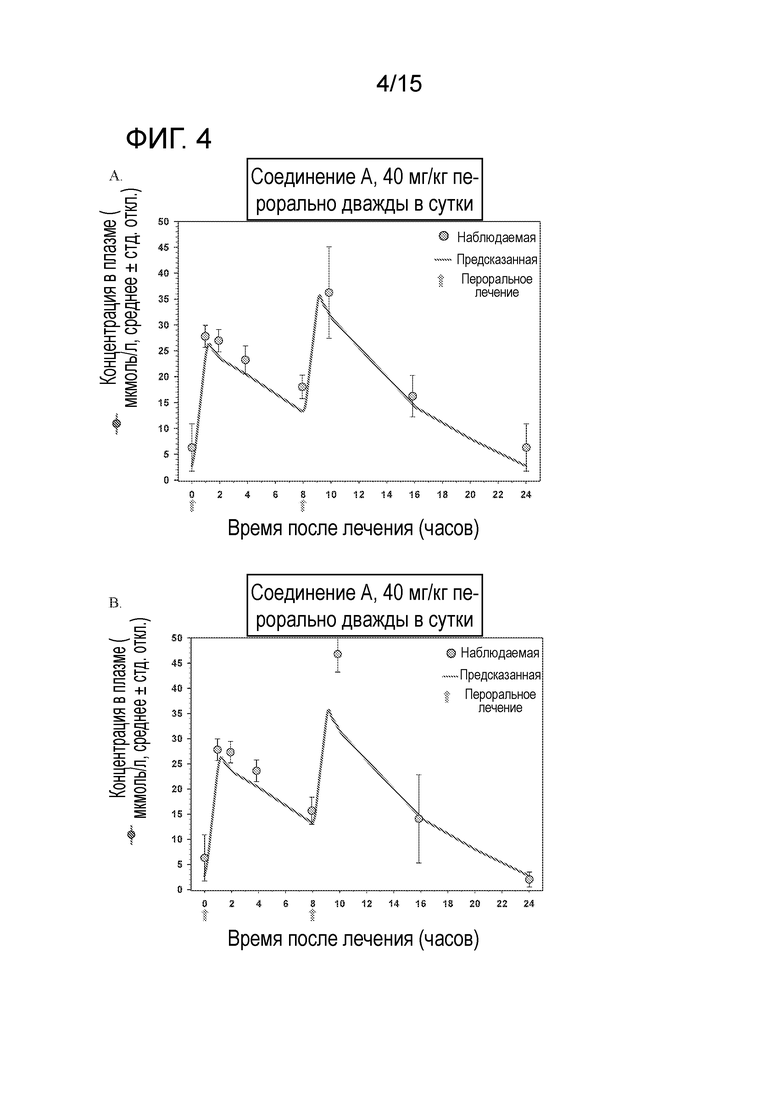
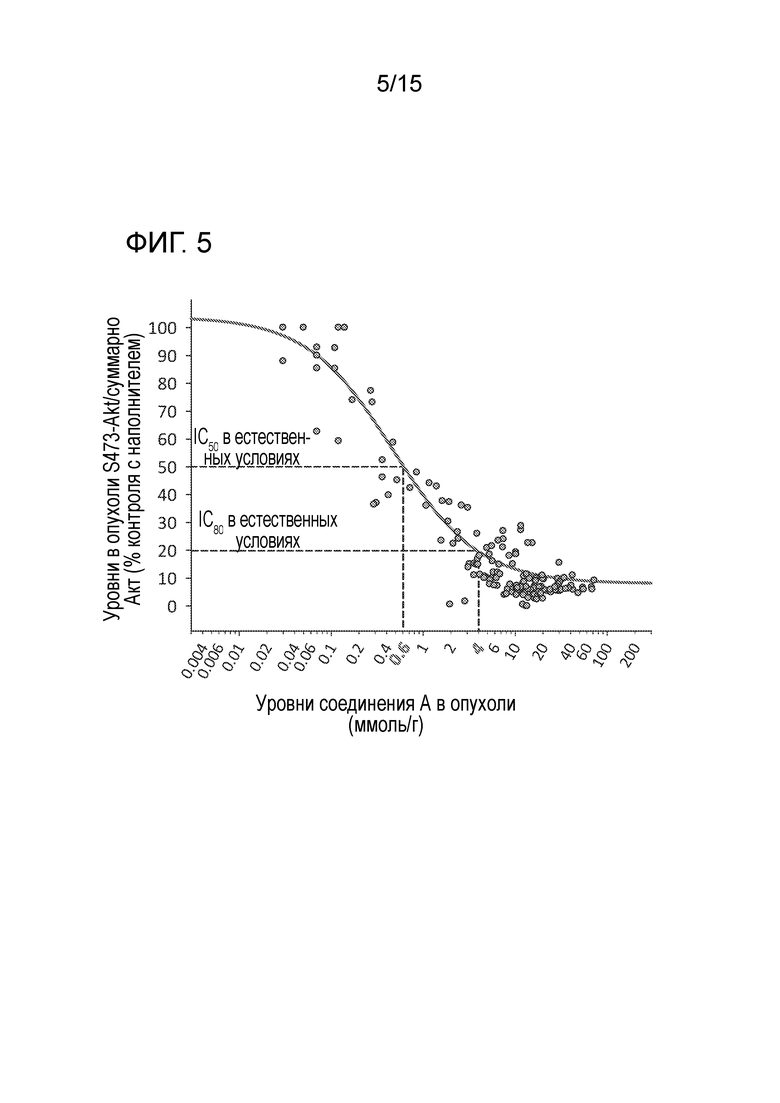
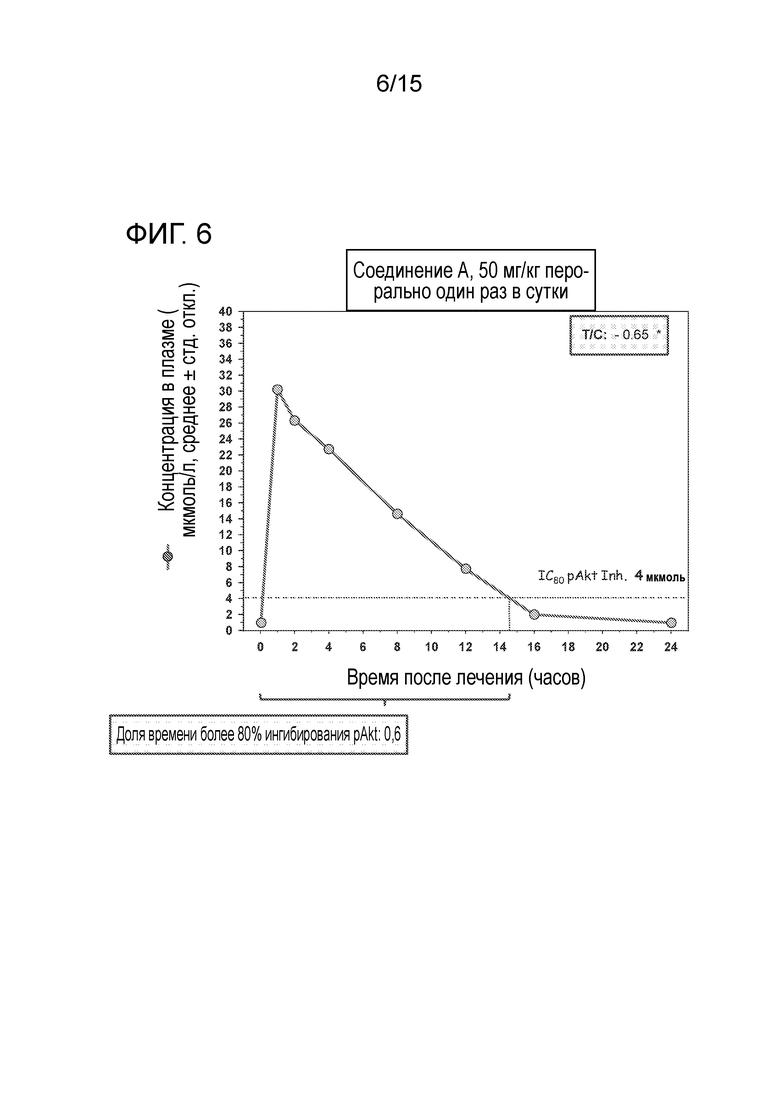
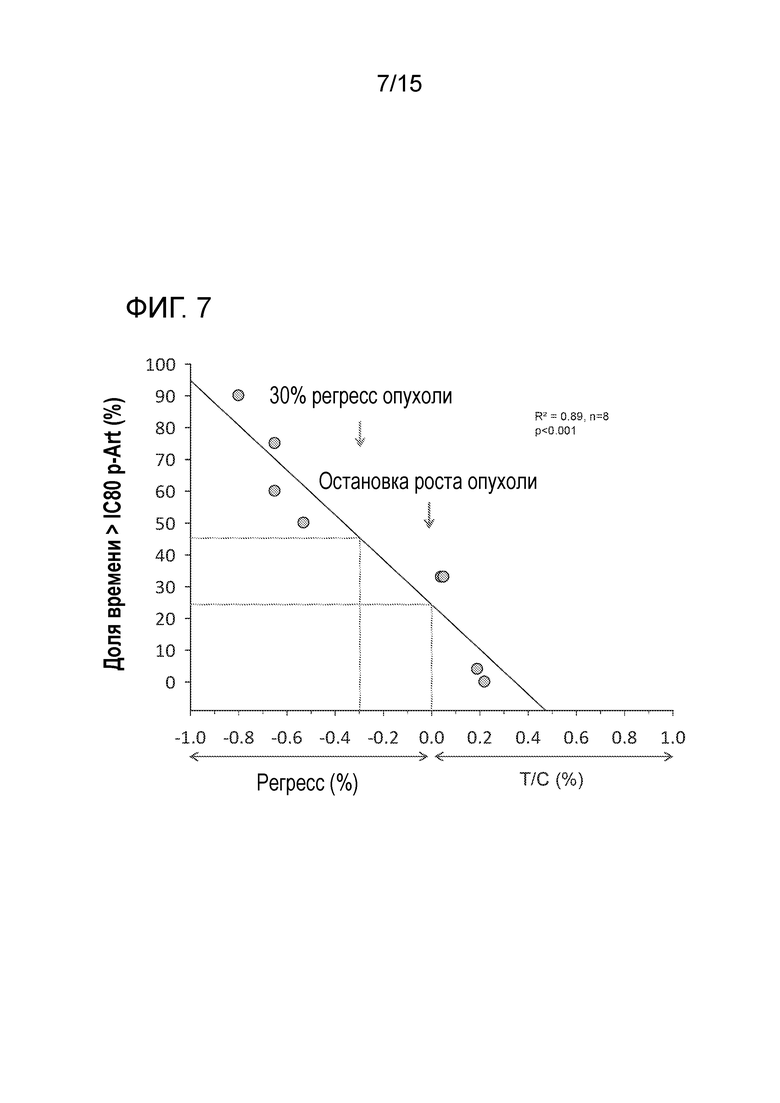
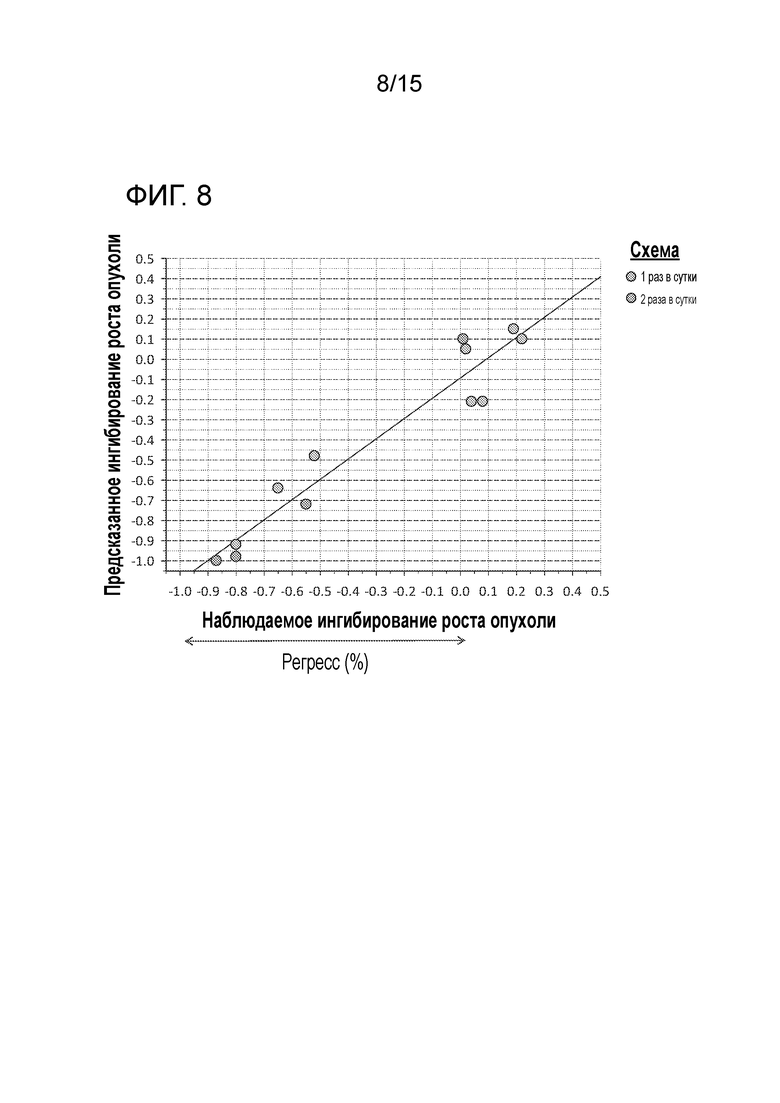
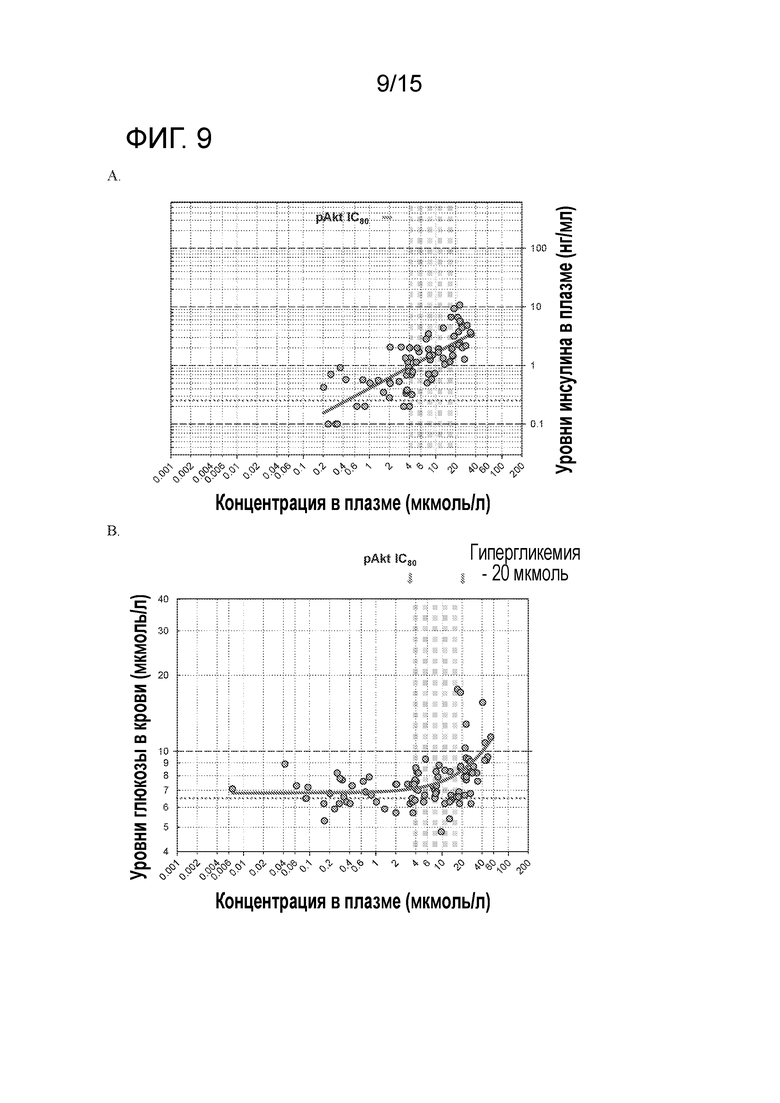
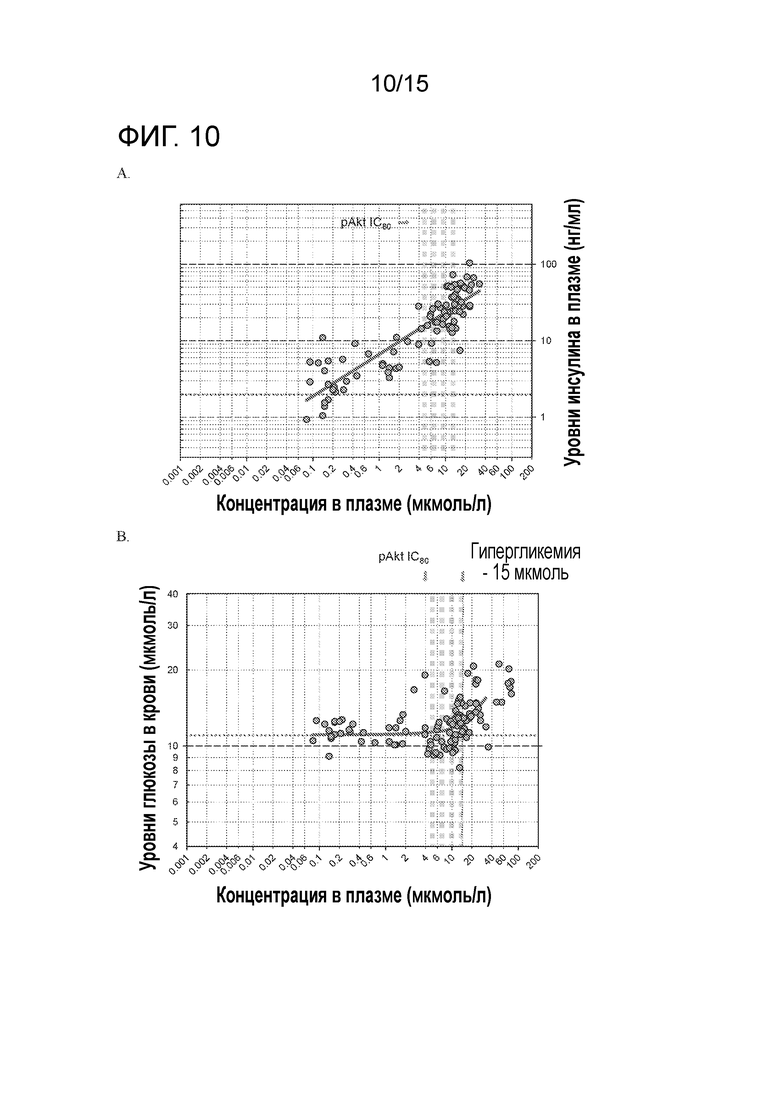

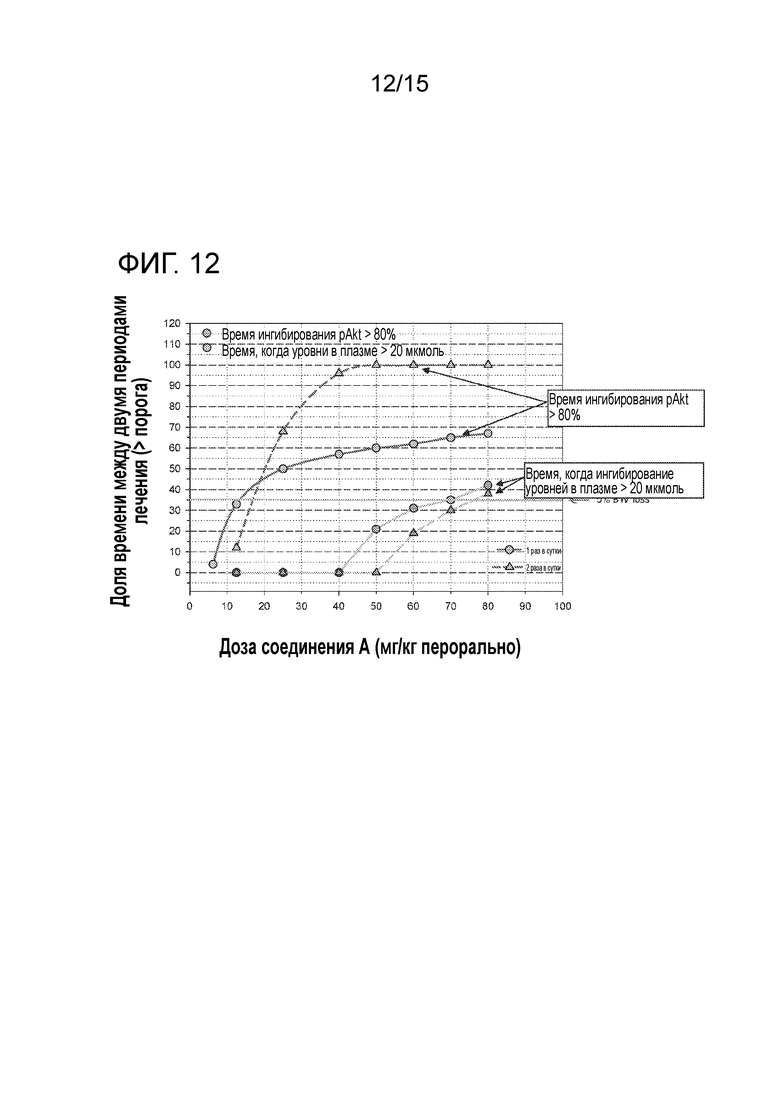

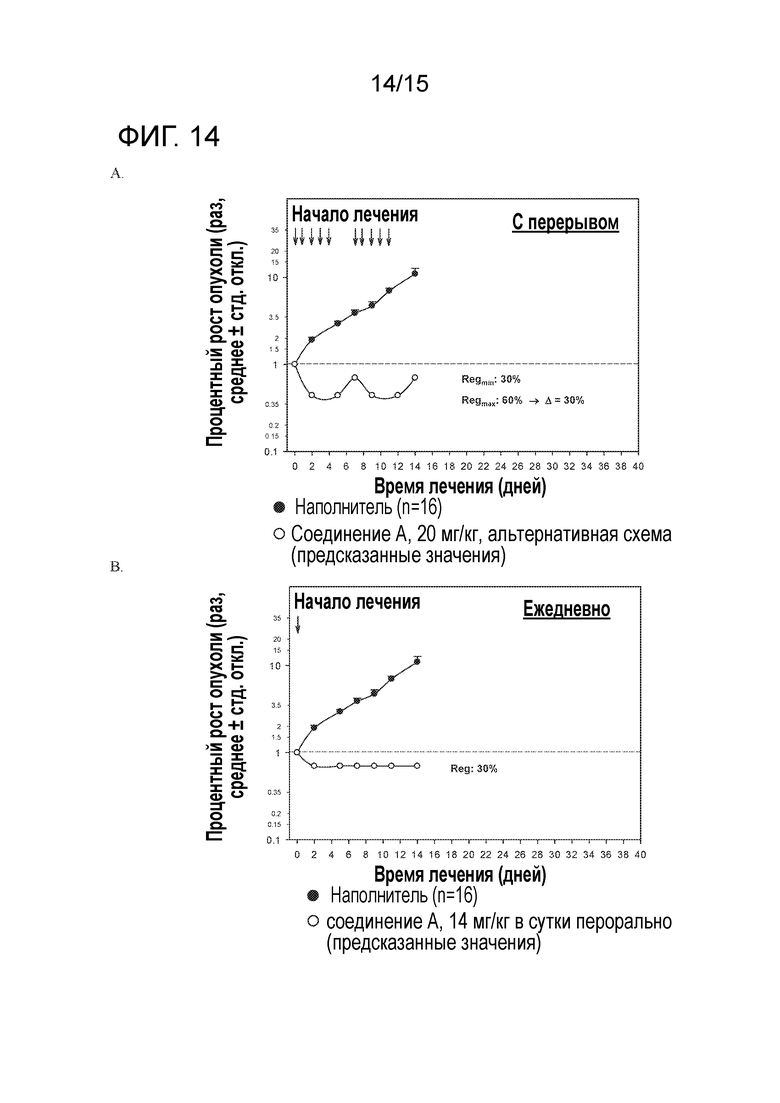
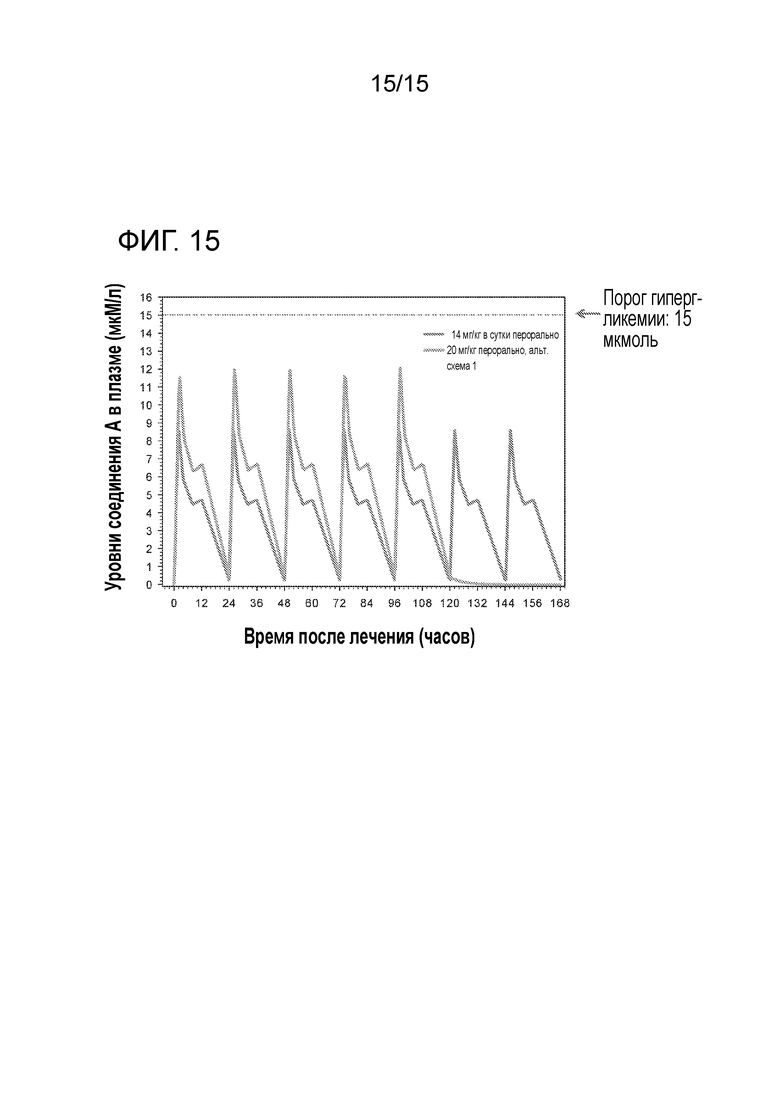
Группа изобретений относится к медицине, а именно к онкологии, и может быть использована для лечения рака у нуждающегося в этом пациента. Для этого пациенту перорально вводят терапевтически эффективное количество соединения селективного по альфа-изоформе ингибитора фосфатидилинозитол-3-киназы формулы (I) в суточной дозе от 100 мг до 450 мг, по меньшей мере, в течение двух последовательных пятидневных циклов, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от 2 дней до 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней. Также предложен способ лечения рака, в случае, если у указанного пациента имеется побочный эффект после введения указанного соединения формулы (I). Группа изобретений обеспечивает лечение рака у пациента. 2 н. и 10 з.п. ф-лы, 15 ил., 2 табл., 1 пр.
1. Способ лечения рака у нуждающегося в этом пациента, включающий пероральное введение терапевтически эффективного количества соединения формулы (I)

или его фармацевтически приемлемой соли пациенту в суточной дозе от 100 мг до 450 мг по меньшей мере в течение двух последовательных пятидневных циклов, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от 2 дней до 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
2. Способ лечения рака, включающий, во-первых, введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в количестве от 100 мг до 450 мг ежедневно по непрерывному ежедневному графику через пероральное введение, во-вторых, определение того, что для указанного пациента имеется побочный эффект, выбранный из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени, сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи и гиперчувствительности, фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии после введения указанного соединения формулы (I) или его фармацевтически приемлемой соли указанному пациенту, и, в-третьих, уменьшение введения указанного соединения формулы (I) или его фармацевтически приемлемой соли до суточной дозы от 100 мг до 450 мг через пероральное введение по меньшей мере в течение двух последовательных пятидневных циклов, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода от 2 дней до 3 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
3. Способ по п. 1, где суточная доза соединения формулы (I) или его фармацевтически приемлемой соли составляет от 200 мг до 400 мг.
4. Способ по п. 1, где соединение формулы (I) или фармацевтически приемлемой соли вводится перорально один раз в сутки (q.d.) в суточной дозе от 100 мг до 450 мг по меньшей мере в течение двух последовательных пятидневных циклов.
5. Способ по п. 1, где соединение формулы (I) или фармацевтически приемлемой соли вводится перорально дважды в сутки (b.i.d.) в суточной дозе от 100 мг до 450 мг по меньшей мере в течение двух последовательных пятидневных циклов.
6. Способ по любому из пп. 1 и 3-5, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода 2 дней между одним циклом из пяти последовательных дней и следующим за ним циклом из пяти последовательных дней.
7. Способ по п. 4, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода 3 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
8. Способ по п. 5, где указанное соединение или его фармацевтически приемлемая соль не вводятся пациенту в течение периода 2,5 дней между последним введением указанного соединения или его фармацевтически приемлемой соли в одном цикле из пяти последовательных дней и первым введением указанного соединения или его фармацевтически приемлемой соли в следующем цикле из пяти последовательных дней.
9. Способ по п. 1, где соединение формулы (I) или его фармацевтически приемлемая соль вводятся в двух или более указанных последовательных пятидневных циклах до ослабления, уменьшения или частичного снятия тяжести, встречаемости или частоты по меньшей мере одного побочного эффекта у указанного пациента.
10. Способ по п. 9, где побочный эффект является патологическим состоянием, выбранным из нейтропении, повышенного билирубина, кардиальной токсичности, нестабильной стенокардии, инфаркта миокарда, стабильной артериальной гипертензии, периферической сенсорной или моторной невропатии/боли, дисфункции печени, сниженного количества эритроцитов и/или количества лейкоцитов, гипергликемии, тошноты, снижения аппетита, диареи, сыпи и гиперчувствительности, фоточувствительности, астении/усталости, рвоты, стоматита, мукозита слизистой оболочки полости рта, панкреатита, дисгевзии и диспепсии.
11. Способ по п. 1, где пролиферативная болезнь является раком, выбранным из рака легкого, бронха, простаты, груди, толстой кишки, прямой кишки, карциномы толстой кишки, колоректальной аденомы, рака поджелудочной железы, желудочно-кишечного рака, печеночно-клеточного рака, рака желудка, гастрального рака, рака яичника, плоскоклеточной карциномы и рака головы и шеи.
12. Способ по п. 1, где соединение формулы (I) или его фармацевтически приемлемая соль вводится в комбинации по меньшей мере с одним дополнительным терапевтическим средством.
| JURIC D | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Published By American Association for Cancer Research | |||
| Термосно-паровая кухня | 1921 |
|
SU72A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| КОМБИНАЦИЯ И СПОСОБЫ ВВЕДЕНИЯ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ И КОМБИНИРОВАННОЙ ТЕРАПИИ | 2006 |
|
RU2452482C2 |
| WO 2012062694 А1, 18.05.2012 | |||
| WO 2010029082 A1, 18.03.2010 | |||
| FURET PASCAL ET AL | |||
| ТЕПЛОВОЙ ДВИГАТЕЛЬ | 1923 |
|
SU719A1 |
| BIOORGANIC MEDICINAL CHEMISTRY LETTERS, PERGAMON, AMSTERDAM, NL, vol | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Насос | 1917 |
|
SU13A1 |

