Данное изобретение относится к конъюгату антитела и лекарственного средства. В одном из аспектов изобретение относится к конъюгату "антитело - лекарственное средство", содержащему антитело, способное связываться с мишенью, при этом указанное антитело конъюгировано по меньшей мере с одним лекарственным средством, выбранным среди производных доластатина 10 и ауристатинов. Данное изобретение также включает способ лечения и применения указанного конъюгата "антитело - лекарственное средство" для лечения рака.
Уровень техники
Данное изобретение относится к конъюгату "антитело - лекарственное средство" (antibody-drug-conjugate, ADC), или конъюгату, и к его применению для лечения рака.
ADC сочетают специфичность связывания антитела с активностью лекарственных средств, таких как, например, цитотоксические агенты. Методика, связанная с разработкой моноклональных антител, применение более эффективных лекарственных средств и проектирование химических линкеров для ковалентного связывания этих компонентов быстро прогрессировали в последние годы.
Применение ADC позволяет локально доставлять лекарственные средства, которые при введении в виде неконъюгированных лекарственных средств могут привести к неприемлемым уровням токсичности в отношении нормальных клеток.
Другими словами, таким образом достигается максимальная эффективность при минимальной токсичности. Усилия по разработке и усовершенствованию ADC были сосредоточены на селективности антитела, а также на механизме действия лекарственных средств, на связывании лекарственных средств, на соотношении лекарственного средства/антитела (загрузка или DAR, от англ. drug/antibody ratio) и на свойствах высвобождения лекарственных средств. Лекарственные группировки могут осуществлять их цитотоксические и цитостатические эффекты за счет механизмов, включающих связывание тубулина, связывание ДНК, протеасому, нарушение рибосомальной функции, синтез белка и/или ингибирование топоизомеразы. Некоторые цитотоксические лекарственные средства, как правило, являются неактивными или менее активными, когда они конъюгированы с большим антителом.
Каждое антитело должно быть охарактеризовано по отдельности, должен быть разработан соответствующий линкер и выявлен подходящий цитотоксический агент, который сохраняет свою активность при доставке к опухолевым клеткам. Необходимо учитывать плотность антигена на раковой мишени и тот факт, экспрессируют ли нормальные ткани антиген-мишень. Другие соображения включают следующие вопросы: интернализуется ли после связывания мишени весь ADC; является ли предпочтительным цитостатическое или цитотоксическое лекарственное средство, когда рассматривается возможное воздействие на нормальную ткань и/или тип и стадию рака, который лечат; и является ли линкер, связывающий антитело с полезной нагрузкой лекарственного средства, расщепляемой или нерасщепляемой связью. Кроме того, соотношение в конъюгате молекул антитела и лекарственной группировки должно быть достаточным, не ставя под угрозу активность связывания антитела и/или эффективность лекарственного средства и не изменяя физико-химические свойства ADC, приводящие к агрегации или вредным свойствам, касающимся процесса будущей разработки соединения.
ADC представляет собой сложную биологическую молекулу, и проблемы в разработке эффективного ADC остаются серьезным вопросом.
Сущность изобретения
В первом аспекте данное изобретение направлено на конъюгат "антитело - лекарственное средство" следующей формулы (I):
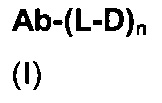
или его фармацевтически приемлемую соль,
где
Ab представляет собой антитело против [мишени] или антитело против антигена или его [мишень]-связывающий фрагмент или его антигенсвязывающий фрагмент;
L представляет собой линкер;
D представляет собой лекарственную группировку следующей формулы (II):
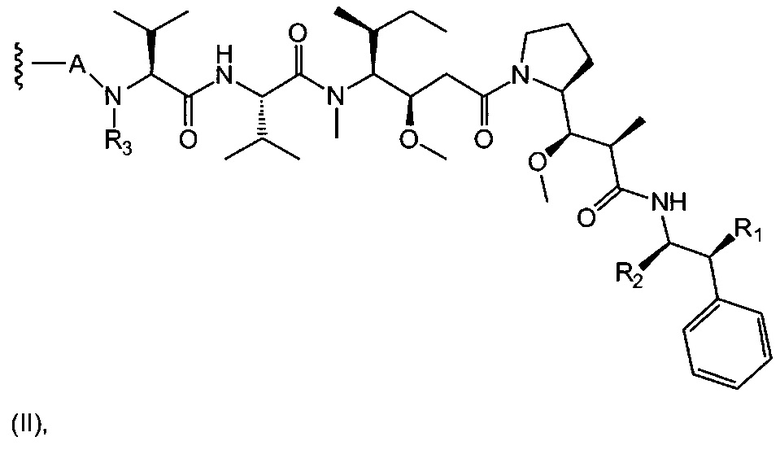
где:
R1 представляет собой Н или ОН;
R2 представляет собой группу: (С1-С6)алкил, СООН, СОО-((С1-С6)алкил) или тиазолил;
R3 представляет собой Н или группу (С1-С6)алкил;
А представляет собой:
- группу формулы -Het-Alk-, где Alk представляет собой (C1-C8)алкандиил и связан с NR3, a Het представляет собой гетероцикл, возможно, замещенный группой (C1-C6)алкил и содержащий по меньшей мере один атом азота, при этом указанный атом азота связан с L, или
- группу формулы -Aa-Ab-, где Aa связан с L и представляет собой О или NR9, при этом R9 представляет собой Н или (С1-С6)алкил, а Ab связан с NR3 и представляет собой:
 (С1-С8)алкандиил,
(С1-С8)алкандиил,
(CH2CH2X1)a1(CH2CH2X2)a2(CH2CH2X3)a3(CH2CH2X4)a4 СН2СН2, где каждый из X1, Х2, Х3 и Х4 независимо от других представляет собой О или NR8; каждый из а1, а2, а3 и а4 независимо от других представляет собой 0 или 1; a R8 представляет собой Н или (С1-С6)алкил,
группу арил-(С1-С8)алкандиил или группу гетероцикл-(С1-С8)алкандиил, где указанная группа, возможно, замещена (C1-C6)алкил, при этом арильная или гетероциклическая группировка связана с Aa, а (С1-С8)алкандиил-группировка связана с NR3;
волнистая линия указывает на точку присоединения к L; и
n составляет от 1 до 12.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где [мишень] или антиген указанного антитела против [мишени] или антитела против антигена согласно изобретению или их [мишень]- или антигенсвязывающих фрагментов выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R, Axl и их внеклеточных мембранных фрагментов (ECD, от англ. extracellular domains).
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где [мишень] или антиген указанного антитела против [мишени] или антитела против антигена согласно изобретению или их [мишень]- или антигенсвязывающих фрагментов выбраны среди HER2, IGF-1R и белка Axl, предпочтительно, человеческого HER2, человеческого IGF-1R и человеческого белка Axl, и их внеклеточных мембранных фрагментов (ECD).
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим IGF-1R, выбранные среди:
i) антител 208F2, 212А11, 214F8, 219D6 и 213В10;
ii) антител, которые конкурируют за связывание с IGF-1R с антителами из i); и
iii) антител, которые связываются с тем же эпитом IGF-1R, что и антитела из i).
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим IGF-1R, выбранные среди:
i) антитела, которое содержит три CDR участка (от англ. complementarity-determining region - участок, определяющий комплементарность) тяжелой цепи последовательностей SEQ ID NO 1, 2 и 3 и три CDR легкой цепи последовательностей SEQ ID NO 4, 5 и 6;
ii) антитела, которое конкурирует за связывание IGF-1R с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом IGF-1R, что и антитело из i).
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где указанное Ab представляет собой антитело, которое содержит:
a) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95; и
b) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим белком Axl, выбранные среди:
i) антитела, которое содержит три CDR тяжелой цепи последовательностей SEQ ID NO 59, 60 и 61 и три CDR легкой цепи последовательностей SEQ ID NO 56, 57 и 58;
ii) антитела, которое конкурирует за связывание Axl с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом Axl, что и антитело из i).
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим HER2, предпочтительно состоящие из трастузумаба.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где L представляет собой линкер следующей формулы (III):
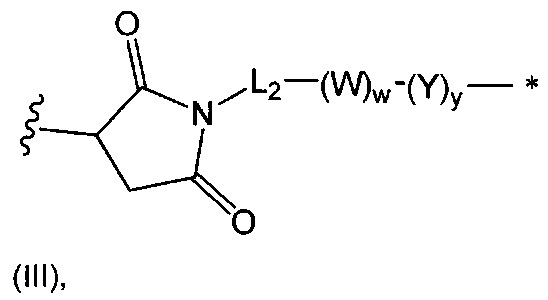
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил, (С2-С6)алкил-карбонил,
W представляет собой аминокислотный блок; w представляет собой целое число от 0 до 5;
Y представляет собой РАВ-карбонил, при этом РАВ представляет собой  у составляет 0 или 1;
у составляет 0 или 1;
звездочка указывает на точку присоединения к D; и
волнистая линия указывает на точку присоединения к Ab.
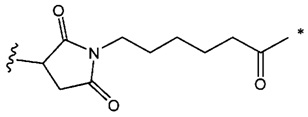
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где L2 имеет следующую формулу:

где
звездочка указывает на точку присоединения к (W)w; и
волнистая линия указывает на точку присоединения к атому азота малеимидной группировки.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где w равно 0 или w равно 2, а (W)w выбран среди:
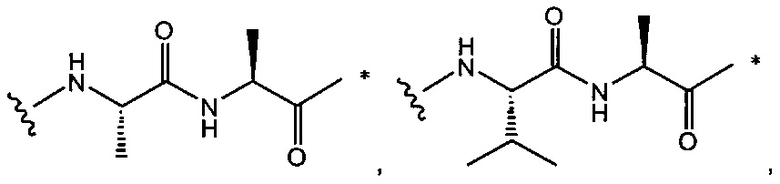
и 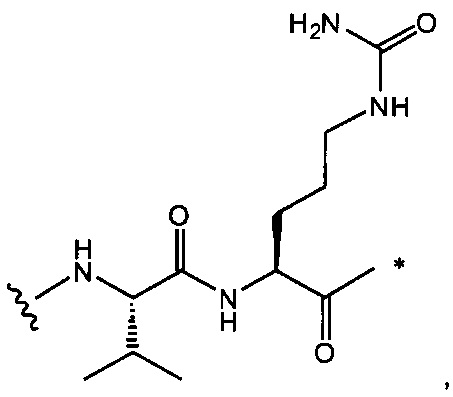
где
звездочка указывает на точку присоединения к (Y)y; и
волнистая линия указывает на точку присоединения к L2.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где L выбран среди:
 ,
,
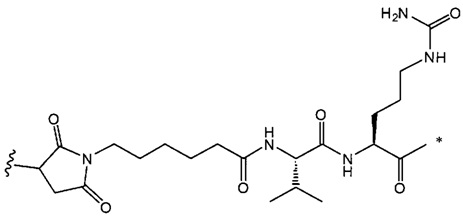 и
и
 ,
,
где звездочка указывает на точку присоединения к D, а волнистая линия указывает на точку присоединения к Ab.
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где А представляет собой группу формулы -Aa-Ab-, в которой Aa является таким, как определено в п. 1, а Ab представляет собой группу:
- фенил-(С1-С2)алкандиил или
- гетероцикл-(С1-С2)алкандиил, возможно, замещенную (С1-С6)алкил (особенно незамещенную), при этом гетероцикл представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 членами, содержащее 1 или 2 атома азота, выбранное, в частности, среди пиридина, пиперидина и имидазола, и предпочтительно представляющее собой пиридин. В одном воплощении данное изобретение относится к конъюгату "антитело -
лекарственное средство" согласно изобретению, где А представляет собой группу
следующей формулы:
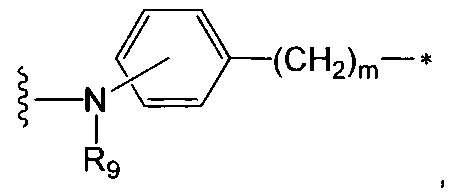
где:
R9 является таким, как определено в п. 1, a m представляет собой целое число от 1 до 8, и предпочтительно R9 представляет собой Н или Me, a m равно 1 или 2,
волнистая линия указывает на точку присоединения к L, а
звездочка указывает на точку присоединения к NR3.
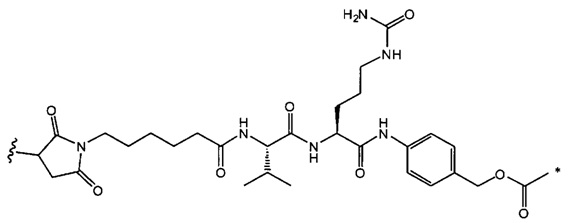
В одном воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, где (L-D) выбран среди:
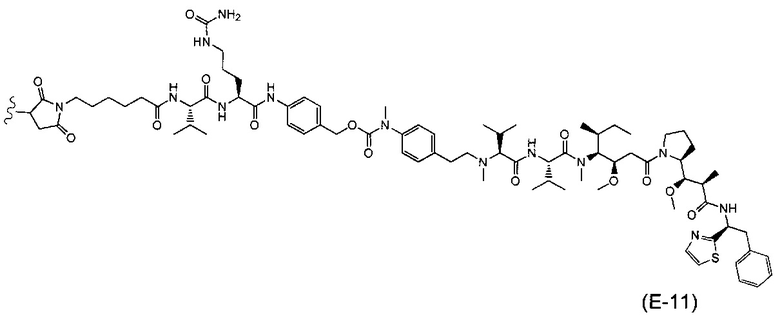
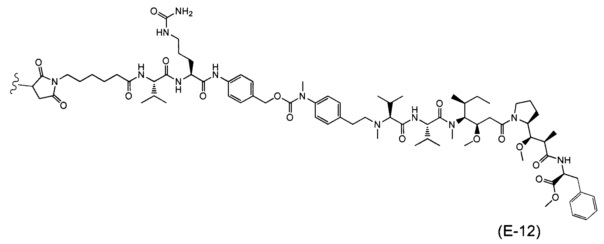
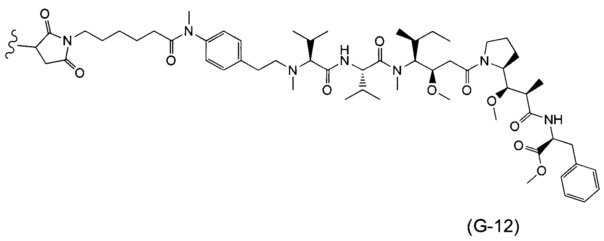
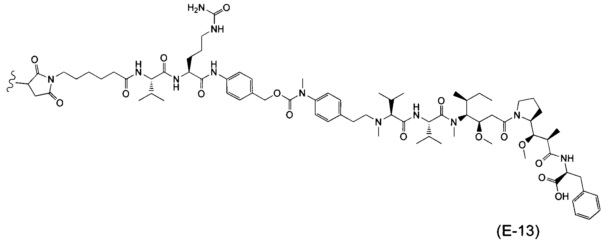
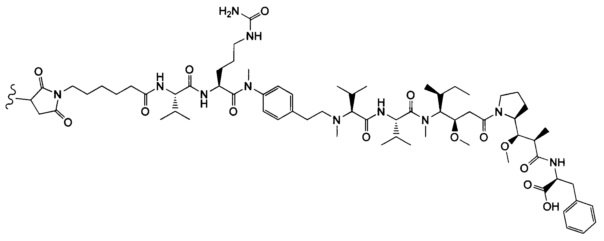

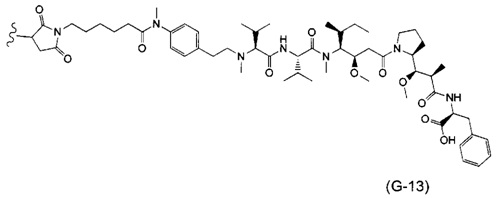
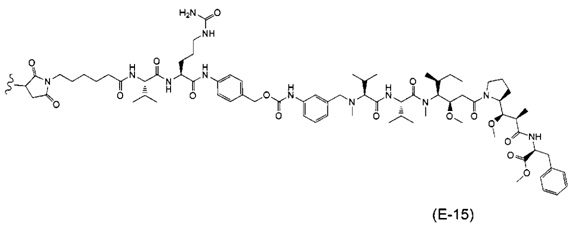
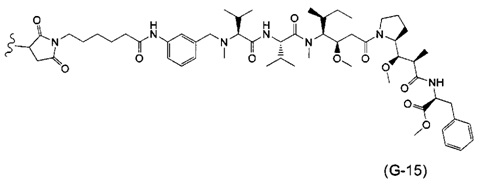
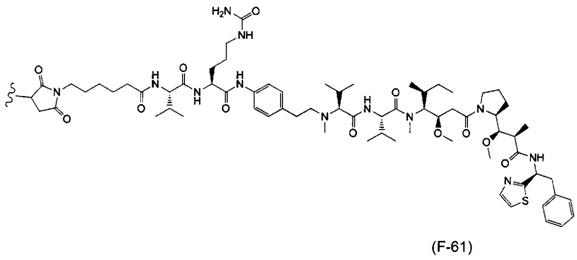
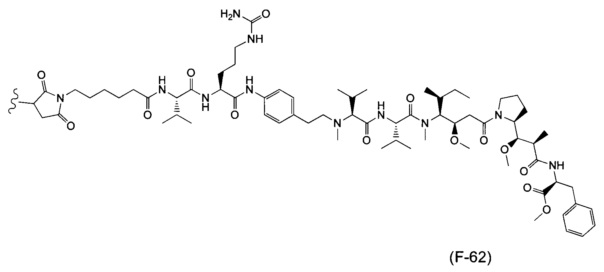
и
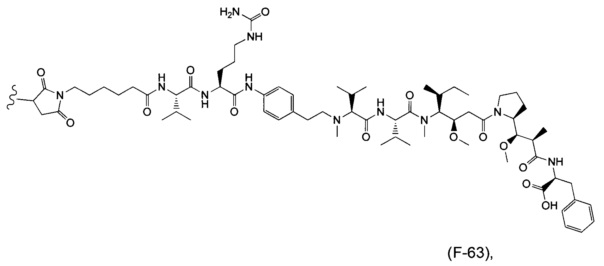
где волнистая линия указывает на точку присоединения к Ab.
В другом воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно изобретению, имеющему формулу, выбранную среди:
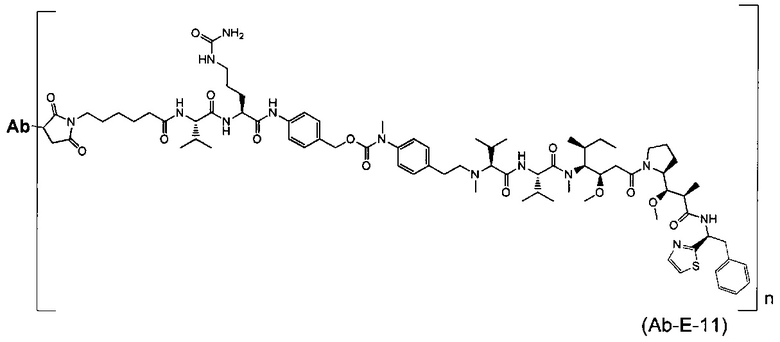
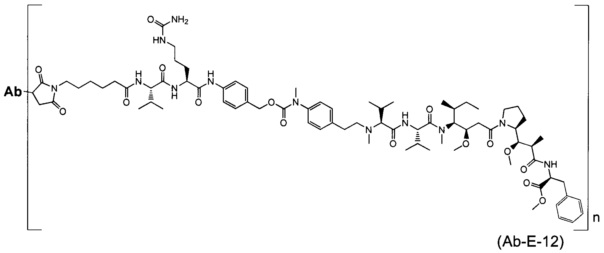
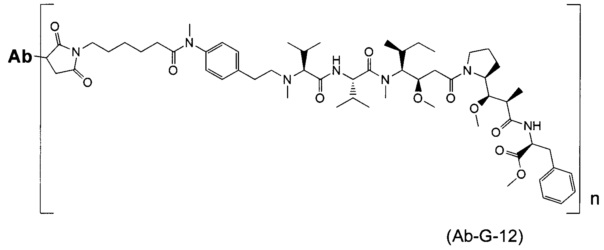
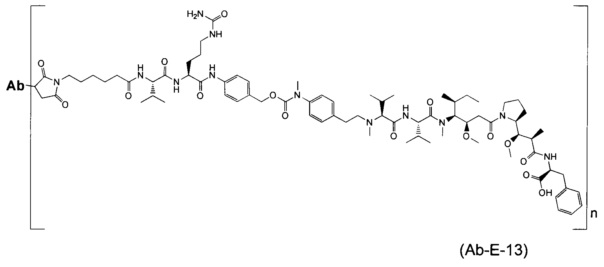
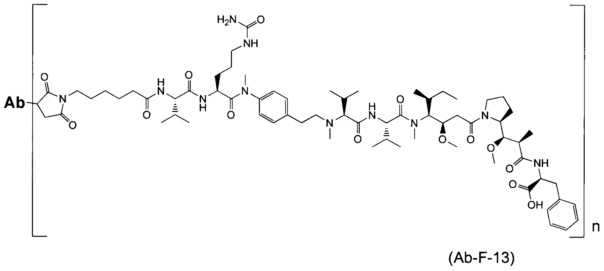
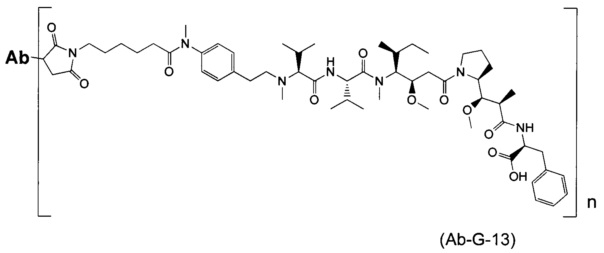
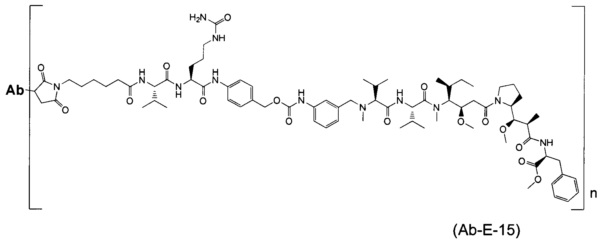
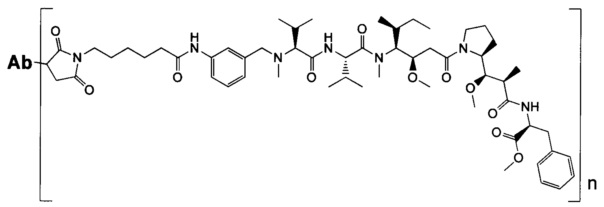

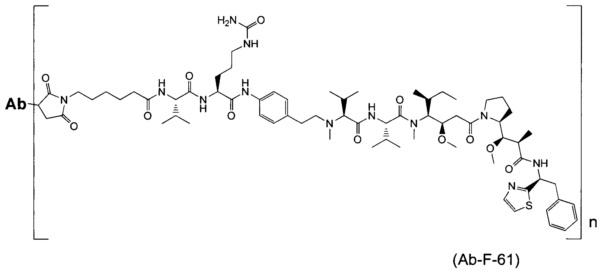
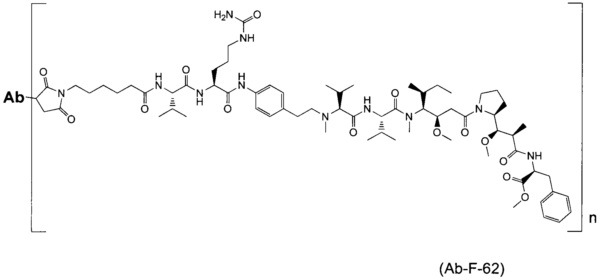
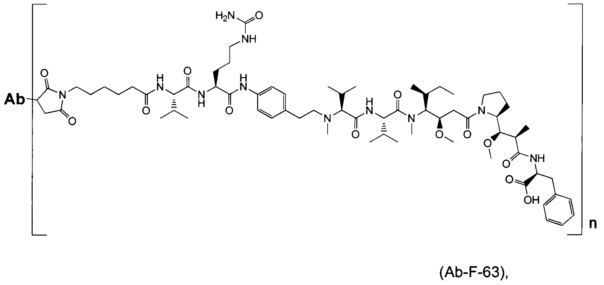
и его фармацевтически приемлемым солям,
где Ab представляет собой антитело против [мишени] или антитело против антигена, или его [мишень]-связывающий фрагмент или его антигенсвязывающий фрагмент.
В одном воплощении и согласно изобретению указанное Ab представляет собой антитело против [мишени] или антитело против антигена, или его [мишень]-связывающий фрагмент или его антигенсвязывающий фрагмент, где указанная [мишень] или антиген выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R, Axl и их внеклеточных мембранных фрагментов (ECD).
В одном воплощении и согласно изобретению указанное Ab представляет собой антитело против [мишени] или антитело против антигена, или его [мишень]-или антигенсвязывающий фрагмент, где указанная [мишень] или антиген выбраны среди HER2, IGF-1R и белка Axl, предпочтительно человеческого HER2, человеческого IGF-1R и человеческого белка Axl, и их внеклеточных мембранных фрагментов (ECD).
В одном воплощении и согласно изобретению указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент и выбрано среди:
a) Ab или его антигенсвязывающего фрагмента, способных связываться с человеческим IGF-1R, выбранных среди:
i) антител 208F2, 212А11, 214F8, 219D6 и 213В10;
ii) антител, которые конкурируют за связывание с IGF-1R с антителами из i); и
iii) антител, которые связываются с тем же эпитом IGF-1R, что и антитела i).
b) Ab или его антигенсвязывающего фрагмента, способных связываться с человеческим IGF-1R, выбранных среди:
i) антитела, которое содержит три CDR тяжелой цепи последовательностей SEQ ID NO 1, 2 и 3 и три CDR легкой цепи последовательностей SEQ ID NO 4, 5 и 6;
ii) антитела, которое конкурирует за связывание IGF-1R с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом IGF-1R, что и антитело из i),
c) Ab, которое содержит:
i) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95; и
ii) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87,
d) Ab или его антигенсвязывающего фрагмента, способных связываться с человеческим белком Axl, выбранных среди:
i) антитела, которое содержит три CDR тяжелой цепи последовательности SEQ ID NO 59, 60 и 61 и три CDR легкой цепи последовательности SEQ ID NO 56, 57 и 58;
ii) антитела, которое конкурирует за связывание Axl с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом Axl, что и антитело из i), и
e) Ab или его антигенсвязывающего фрагмента, способных связываться с человеческим HER2, предпочтительно содержащих трастузумаб или пертузумаб.
В одном воплощении и согласно изобретению n составляет 2 или n составляет 4.
В другом воплощении данное изобретение относится к конъюгату "антитело - лекарственное средство" согласно данному изобретению для применения в качестве лекарственного средства.
В другом воплощении данное изобретение относится к композиции, содержащей по меньшей мере один конъюгат "антитело - лекарственное средство" согласно данному изобретению.
В одном воплощении композиция согласно данному изобретению также содержит фармацевтически приемлемый носитель.
В другом воплощении данное изобретение направлено на композицию согласно данному изобретению для применения в лечении [мишень]- или антиген-экспрессирующего рака, где указанная [мишень] или антиген, предпочтительно, выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R, Axl и их внеклеточных мембранных (ECD) фрагментов, более предпочтительно среди HER2, IGF-1R и белка Axl, и более предпочтительно среди человеческого HER2, человеческого IGF-1R и человеческого белка Axl, а также их внеклеточных мембранных (ECD) фрагментов.
В одном воплощении указанный [мишень]- или антиген-экспрессирующий рак представляет собой рак, выбранный среди рака молочной железы, толстой кишки, карциномы пищевода, гепатоцеллюлярного рака, рака желудка, глиомы, рака легких, меланомы, остеосаркомы, рака яичников, предстательной железы, рабдомиосаркомы, рака почек, щитовидной железы, эндометрия матки, мезотелиомы, плоскоклеточной карциномы ротовой полости, саркомы Капоши, острой лейкемии, колоректальной карциномы, меланомы, протоковой аденокарциномы поджелудочной железы и любого рака с лекарственной устойчивостью.
В одном из воплощений данное изобретение относится к способу лечения [мишень]- или антиген-экспрессирующего рака у субъекта, нуждающегося в этом, включающему введение субъекту эффективного количества по меньшей мере одного конъюгата "антитело - лекарственное средство" согласно данному изобретению или композиции согласно данному изобретению.
Подробное описание изобретения
I - Антитело (Ab)
Термины "антитело", "антитела", "AT" ("ab", от англ. antibody), "МКА (моноклональное антитело)" ("MAb", от англ. monoclonal antibody) или "иммуноглобулин" используются взаимозаменяемо в самом широком смысле и включают моноклональные антитела, предпочтительно выделенные, инженерные или рекомбинантные антитела (например, полноразмерные или интактные моноклональные антитела), поликлональные антитела, поливалентные антитела или мультиспецифические антитела (например, биспецифические антитела), а также фрагменты антител до тех пор, пока они проявляют нужную биологическую активность.
В одном из воплощений антитело согласно данному изобретению состоит из рекомбинантного антитела. Термин "рекомбинантное антитело" относится к антителу, которое является результатом экспрессии рекомбинантной ДНК в живых клетках. Рекомбинантное антитело согласно данному изобретению получают с использованием лабораторных способов генетической рекомбинации, хорошо известных специалистам в данной области, создающих ДНК-последовательности, которые не будут обнаружены в биологических организмах.
В другом воплощении антитело согласно изобретению состоит из химически синтезированного антитела.
Более конкретно, такая молекула состоит из гликопротеина, содержащего по меньшей мере две тяжелые (Н) цепи и две легкие (L) цепи, соединенные дисульфидными связями. Каждая тяжелая цепь содержит вариабельную область (или домен) тяжелой цепи (в данном документе сокращена как HCVR (heavy chain variable region) или VH) и константную область тяжелой цепи. Константная область тяжелой цепи содержит три домена, СН1, СН2 и СН3. Каждая легкая цепь содержит вариабельную область легкой цепи (в данном документе сокращена как LCVR (light chain variable region) или VL) и константную область легкой цепи. Константная область легкой цепи содержит один домен, CL. Области VH и VL могут быть дополнительно подразделены на области гипервариабельности, называемые областями, определяющими комплементарность (CDR), которые перемежаются с областями, которые являются более консервативными, называемыми каркасными областями (framework region, FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат связывающий домен, который взаимодействует с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями или факторами хозяина, включая различные клетки иммунной системы (например, эффекторные клетки), а также первый компонент (C1q) классической системы комплемента.
Под выражением "антигенсвязывающий фрагмент" или "мишень-связывающий фрагмент" антитела согласно данному изобретению понимают любой пептид, полипептид или белок, сохраняющий способность связываться с мишенью (также обычно известной как антиген) указанного антитела и содержащий аминокислотную последовательность по меньшей мере из 10 смежных аминокислотных остатков, по меньшей мере 15 смежных аминокислотных остатков, по меньшей мере 20 смежных аминокислотных остатков, по меньшей мере 25 смежных аминокислотных остатков, по меньшей мере 40 смежных аминокислотных остатков, по меньшей мере 50 смежных аминокислотных остатков, по меньшей мере 60 смежных аминокислотных остатков, по меньшей мере 70 смежных аминокислотных остатков, по меньшей мере 80 смежных аминокислотных остатков, по меньшей мере 90 смежных аминокислотных остатков, по меньшей мере 100 смежных аминокислотных остатков, по меньшей мере 125 смежных аминокислотных остатков, по меньшей мере 150 смежных аминокислотных остатков, по меньшей мере 175 смежных аминокислотных остатков или по меньшей мере 200 смежных аминокислотных остатков, по меньшей мере 250 смежных аминокислотных остатков из аминокислотной последовательности антитела.
В одном воплощении такие "антигенсвязывающие фрагменты" выбраны из группы, которая включает, но не ограничиваясь ими, Fv-, scFv- ("sc" означает "одноцепочечный" - от англ. single chein), Fab-, F(ab')2-, Fab'-, scFv-Fc-фрагменты или димерные антитела или любые фрагменты, период полужизни которых увеличен путем химической модификации, такой как добавление полиалкиленгликоля, такого как полиэтиленгликоль ("ПЭГилирование") (пэгилированные фрагменты, называемые Fv-ПЭГ, scFv-ПЭГ, Fab-ПЭГ, F(ab')2-ПЭГ или Fab'-ПЭГ) ("ПЭГ" означает полиэтиленгликоль), или путем включения в липосому, при этом указанные фрагменты имеют по меньшей мере одну из характерных CDR антитела согласно данному изобретению. Предпочтительно, указанные "антигенсвязывающие фрагменты" будут состоять из или будут содержать частичную последовательность тяжелой или легкой вариабельной цепи антитела, из которого они получены, где указанная частичная последовательность должна быть достаточной для сохранения такой же специфичности связывания, как у антитела, из которого она получена, и достаточной аффинности по отношению к мишени, составляющей предпочтительно по меньшей мере 1/100, в более предпочтительном варианте по меньшей мере 1/10 от аффинности антитела, из которого она получена.
Под термином "связывание", "связывает" и т.п. подразумевается, что антитело или любой его антигенсвязывающий фрагмент образует с антигеном комплекс, который относительно стабилен в физиологических условиях. Специфическое связывание можно охарактеризовать равновесной константой диссоциации, которая составляет по меньшей мере примерно 1×10-6 М. Способы определения того, связываются ли две молекулы, хорошо известны в данной области и включают, например, равновесный диализ, поверхностный плазмонный резонанс, анализы с радиоактивной меткой и т.п. Во избежание сомнений, это не означает, что указанное антитело не может связывать или мешать, на низком уровне, другому антигену. Тем не менее, в качестве предпочтительного воплощения указанное антитело связывается только с указанным антигеном.
Используемое в данном описании выражение "анти-[мишень]-антитело" следует интерпретировать как аналогичное выражению "антитело против [мишени]", и оно означает антитело, способное связываться с [мишенью].
Выражение "мишень" или [мишень] следует интерпретировать как любую молекулу, присутствующую на поверхности клеток, предпочтительно опухолевых клеток, более предпочтительно клеток млекопитающих и человека, которая может быть использована для доставки лекарственного средства. Предпочтительно, мишень специфически экспрессируется или сверхэкспрессируется на поверхности опухолевых клеток по сравнению с нормальными клетками.
Более конкретно, мишень может быть выбрана среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R или Axl (B.A. Teicher, Current Cancer Drug Targets, 2009, 9, 982-1004).
В качестве предпочтительной мишени ею могут быть упоминаемые HER2, IGF-1R и Axl, предпочтительно человеческие HER2, IGF-1R и Axl.
В качестве не ограничивающего примера выражения "HER2-антитело", "IGF-1R-антитело" или "Axl-антитело" следует интерпретировать как аналогичные выражениям "анти-HER2-антитело", "анти-IGF-1R-антитело" или "анти-Axl-антитело", и они означают, что эти антитела способны связываться с HER2, IGF-1R или Axl, соответственно.
В данной заявке эпитоп антитела преимущественно локализуется во внеклеточном домене мишени.
В конкретном воплощении указанное антитело или любой его антигенсвязывающий фрагмент способен связываться с мишенью с ЕС50, находящейся в диапазоне от 10×10-10 до 1×10-10, и более предпочтительно в диапазоне от 8×10-10 до 2×10-10.
Термин "половина максимальной эффективной концентрации" (ЕС50) соответствует концентрации лекарственного средства, антитела или токсиканта, индуцирующей реакцию, находящуюся на середине между базальным и максимальным уровнем после некоторого заданного времени экспозиции. Она широко используется в качестве меры активности лекарственного средства. Таким образом, ЕС50 градуированной кривой "доза - ответ" представляет собой концентрацию соединения, при которой наблюдается 50% от его максимального эффекта. ЕС50 в квантовой кривой "доза - ответ" представляет собой концентрацию соединения, при которой 50% популяции демонстрирует ответ после экспозиции в течение указанного времени. Значения концентрации, как правило, следуют сигмоидальной кривой, быстро увеличиваясь при сравнительно небольшом изменении концентрации. Их можно определить математически путем выведения линии наилучшего соответствия.
В предпочтительном воплощении ЕС50, определенная в данном изобретении, характеризуется силой связывания антитела с ECD мишени, экспонированным на опухолевых клетках человека. Параметр ЕС50 определяют с помощью анализа FACS (от англ. Fluorescence-activated cell sorting - сортировка флуоресцентно-активированных клеток). Параметр ЕС50 отражает концентрацию антитела, при которой достигается 50% от максимального связывания человеческой мишени, экспрессированной на опухолевых клетках. Каждое значение ЕС50 рассчитывали как среднюю точку кривой "доза - ответ" с помощью программы четырехпараметрической подгонки кривой регрессии (Prism Software). Этот параметр был выбран в качестве репрезентативного для физиологических/патологических состояний.
Термин "эпитоп" представляет собой область антигена, которая связывается антителом. Эпитопы могут быть определены как структурные или функциональные. Функциональные эпитопы в целом представляют собой подгруппу структурных эпитопов и имеют те остатки, которые непосредственно способствуют аффинности взаимодействия. Эпитопы также могут быть конформационными, то есть такими, которые состоят из нелинейных аминокислот. В некоторых воплощениях эпитопы могут включать детерминанты, которые являются химически активными поверхностными группировками молекул, такими как аминокислоты, боковые сахарные цепи, фосфорильные группы или сульфонильные группы, а также, в некоторых воплощениях, они могут иметь специфические трехмерные структурные характеристики и/или специфические характеристики заряда.
Конкуренцию за связывание с мишенью можно определить любыми способами или методиками, известными специалистам в данной области, такими как, но не ограничиваясь ими, радиоактивность, Biacore, ELISA (от англ. enzyme-linked immunosorbent assay - твердофазный иммуноферментный анализ (ИФА)), проточная цитометрия и т.д. Под выражением "которое конкурирует за связывание с мишенью" подразумевают конкуренцию, составляющую по меньшей мере 20%, предпочтительно по меньшей мере 50% и более предпочтительно по меньшей мере 70%.
Связывание с тем же эпитопом можно определить любыми способами или методиками, известными специалистам в данной области, такими как, но не ограничиваясь ими, радиоактивность, Biacore, ELISA (ИФА), проточная цитометрия. Под выражением "которое связывается с тем же эпитопом мишени" подразумевают конкуренцию, составляющую по меньшей мере 20%, предпочтительно по меньшей мере 50% и более предпочтительно по меньшей мере 70%.
Как упоминалось выше и в отличие от общих знаний, данное изобретение сфокусировано на специфических антителах, демонстрирующих хорошую способность быть интернализированными после связывания с мишенью. Используемое в данном документе антитело, которое "интернализировано" или "интернализуется" (эти два выражения являются аналогичными), представляет собой такое антитело, которое захватывается клеткой (т.е. оно "входит" в клетку) после связывания с мишенью на клетке млекопитающего. Такое антитело представляет интерес в качестве части ADC, так что оно направляет связанный цитотоксический агент в раковые клетки-мишени. Будучи интернализированным, цитотоксический агент вызывает гибель раковых клеток.
Важными ключами к успеху терапии ADC считается специфичность в отношении антигена-мишени и интернализация комплексов "антиген - антитело" раковыми клетками. Очевидно, что при доставке цитостатических агентов неинтернализующиеся антигены являются менее эффективными, чем интернализующиеся антигены. Процессы интернализации отличаются среди антигенов и зависят от множества параметров, которые могут находиться под влиянием антител.
В ADC цитотоксический агент несет цитотоксическую активность, а используемое антитело ответственно за специфичность в отношении раковых клеток, как и вектор для входа в клетки для правильной адресации цитотоксического агента. Таким образом, чтобы улучшить ADC, антитело должно демонстрировать высокую способность к интернализации раковыми клетками-мишенями. Эффективность, с которой антитело опосредует интернализацию, значительно отличается в зависимости от целевого эпитопа. Выбор мощных интернализирующихся антител требует различных экспериментальных данных, изучающих не только понижающую регуляцию мишени, но и последующее проникновение антитела в клетки.
В предпочтительном воплощении интернализацию антитела согласно изобретению можно оценить с помощью иммунофлуоресценции или FACS (проточной цитометрии) (как проиллюстрировано далее в данной заявке) или с помощью любого способа или процесса, известного специалисту в данной области, специфического для данного механизма интернализации. В предпочтительном воплощении антитело согласно данному изобретению может индуцировать интернализацию после связывания с мишенью по меньшей мере на 30%, предпочтительно на 50% и более предпочтительно на 80%.
Комплекс мишень/антитело интернализируется после связывания антитела с ECD указанной мишени, и это вызывает снижение количества мишени на поверхности клеток. Это снижение можно количественно оценить любым способом, известным специалистам в данной области (вестерн-блотом, FACS, иммунофлуоресценцией и т.п).
В одном из воплощений это снижение, отражающее таким образом интернализацию, можно предпочтительно измерить с помощью FACS и выразить как разность или дельту между средней интенсивностью флуоресценции (Mean Fluorescence Intensity, MFI), измеренной при 4°C, и MFI, измеренной при 37°C, после 4 часов инкубации с антителом.
В качестве неограничивающего примера эта дельта определяется на основе MFI, полученных с необработанными клетками и с клетками, обработанными антителом, с использованием i) раковых клеток, таких как, например, MCF7, после 4-часового периода инкубации с антителом, описанным в данном документе и ii) вторичного антитела, меченного Alexa 488. Этот параметр рассчитывается по следующей формуле: Δ(MFI4°C-MFI37°C). Другие воплощения будут детально описаны в последующих примерах.
Эта разница между значениями MFI отражает понижающую регуляцию мишени, так как значения MFI пропорциональны количеству мишени, экспрессированной на поверхности клетки.
В предпочтительном аспекте антитела состоят из антител, дающих, например, Δ(MFI4°C-MFI37°C) на раковых клетках, таких как MCF7, по меньшей мере 280, предпочтительно по меньшей мере 400.
Более подробно, упомянутая выше дельта может быть измерена в соответствии со следующим процессом, который должен рассматриваться в качестве иллюстративного и неограничивающего примера:
a) обработка и инкубация опухолевых клеток, представляющих интерес, антителом согласно изобретению либо в холодной (4°C), либо в теплой (37°C) полной культуральной среде;
b) параллельная обработка вторичным антителом клеток, обработанных на этапе а), и необработанных клеток,
c) измерение MFI (представляет количество IGF-1R, присутствующего на поверхности) для обработанных и необработанных клеток с помощью вторичного меченого антитела, способного связываться с антителом согласно данному изобретению, и
d) расчет дельты путем вычитания MFI, полученной с обработанными клетками, из MFI, полученной с необработанными клетками.
Из этой дельты MFI процент интернализации может быть определен как:
100×(MFI4°C-MFI37°C)/MFI4°C
Антитела согласно данному изобретению предпочтительно присутствуют с процентом интернализации, который составляет от 50% до 99%, от 70% до 90%, предпочтительно от 75% до 87%.
Особое преимущество антител, описанных в данном документе, основано на скорости их интернализации.
Общеизвестно, что для ADC желательно, чтобы используемые антитела демонстрировали высокую скорость интернализации, предпочтительно в течение 24 часов с момента введения антитела, и более предпочтительно в течение 12 часов, еще более предпочтительно в течение 6 часов.
В данном изобретении скорость интернализации, также определяемая как уменьшение количества антитела, связанного с поверхностью клетки, или как распад антитела на клеточной поверхности, выражается как t1/2 (период полужизни) и соответствует времени, которое необходимо для снижения ΔMFI на 50% (этот аспект будет четко понятен в связи с последующими примерами).
Особым преимуществом является то, что антитела согласно изобретению имеют t1/2 от 5 до 25 минут, и предпочтительно от 10 до 20 минут.
В последующем описании будут проиллюстрированы две предпочтительные мишени.
Для этих двух предпочтительных мишеней под выражением "антитело согласно данному изобретению" или "антитело данного изобретения" следует понимать: "антитело в ADC согласно данному изобретению" или "антитело в ADC данного изобретения", соответственно.
I.1: IGF-1R-антитела
Конкретное воплощение данного изобретения относится к ADC, где антитело Ab содержит три CDR тяжелой цепи с CDR-H2 последовательности SEQ ID NO 2 и CDR-H3 последовательности SEQ ID NO 3, и три CDR легкой цепи с CDR-L2 последовательности SEQ ID NO 5.
Конкретное воплощение данного изобретения относится к ADC, где антитело Ab содержит три CDR тяжелой цепи последовательностей SEQ ID NO 1, 2 и 3 и три CDR легкой цепи последовательностей SEQ ID NO 4, 5 и 6.
Одно из воплощений ADC содержит антитело, содержащее три CDR тяжелой цепи, содержащие последовательности SEQ ID NO 1, 2 и 3 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 1, 2 и 3; и три CDR легкой цепи, содержащие последовательности SEQ ID NO 4, 5 и 6 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 4, 5 и 6.
В другом воплощении антитело или любой его антигенсвязывающий фрагмент содержит три CDR легкой цепи, содержащие последовательности SEQ ID NO 1, 2 и 3; и три CDR тяжелой цепи, содержащие последовательности SEQ ID NO 4, 5 и 6.
Уникальная нумерация IMGT была создана для сравнения вариабельных доменов независимо от антигенного рецептора, типа цепи или вида [Fefranc М.-Р., Immunology Today 18, 509 (1997) / Fefranc М.-Р., The Immunologist, 7, 132-136 (1999) / Fefranc, M.-P., Pommie, C., Ruiz, M., Giudicelli, V., Foulquier, E., Truong, F., Thouvenin-Contet, V. and Fefranc, Dev. Comp. Immunol., 27, 55-77 (2003)]. В уникальной нумерации IMGT консервативные аминокислоты всегда имеют одну и туже позицию, например, цистеин 23 (1st-CYS), триптофан 41 (CONSERVED-TRP), гидрофобная аминокислота 89, цистеин 104 (2nd-CYS), фенилаланин или триптофан 118 (J-PHE или J-TRP). Уникальная нумерация IMGT предлагает стандартизированное разграничение каркасных областей (FR1-IMGT: позиции с 1 по 26, FR2-IMGT: с 39 по 55, FR3-IMGT: с 66 по 104, и FR4-IMGT: с 118 по 128) и областей, определяющих комплементарность: CDR1-IMGT: с 27 по 38, CDR2-IMGT: с 56 по 65, и CDR3-IMGT: со 105 по 117. Поскольку промежутки представляют незанятые позиции, длины CDR-IMGT (показаны в скобках и разделены точками, например, [8,8,13]) становятся важной информацией. Уникальная нумерация IMGT используется в 20-графических представлениях, как описано в Colliers de Perles [Ruiz, M. and Fefranc, M.-P., Immunogenetics, 53, 857-883 (2002) / Kaas, Q. and Fefranc, M.-P., Current Bioinformatics, 2, 21-30 (2007)], и в 3D-структурах в IMGT/3Dstructure-DB [Kaas, Q., Ruiz, M. and Lefranc, M.-P., T cell receptor and MHC structural data. Nucl. Acids. Res., 32, D208-D210 (2004)].
Необходимо понимать, что, без противоречия описанию в данном документе, области, определяющие комплементарность, или CDR, обозначают гипервариабельные области тяжелых и легких цепей иммуноглобулинов, как они определены согласно системе нумерации IMGT.
Тем не менее, CDR также могут быть определены согласно системе нумерации Кабат (Kabat et al., Sequences of proteins of immunological interest, 5th Ed., U.S. Department of Health and Human Services, NIH, 1991, и более поздние издания). Существует три CDR тяжелой цепи и три CDR легкой цепи. В данном документе термин "CDR" в единственном или множественном числе применяется для обозначения, в зависимости от случая, одной или более или даже всех этих областей, которые содержат большинство аминокислотных остатков, ответственных за аффинность связывания антитела с антигеном или эпитопом, который оно распознает. Для того чтобы упростить чтение данной заявки, CDR согласно Kabat не определены. Тем не менее, для специалиста в данной области будет очевидным определение CDR в соответствии с Kabat с использованием определения CDR согласно IMGT.
В контексте данного изобретения понятие "идентичности" или "процента идентичности" двух последовательностей нуклеиновых кислот или аминокислот означает процент идентичных нуклеотидов или аминокислотных остатков в двух сравниваемых последовательностях, полученный после оптимального выравнивания, при этом данный процент является чисто статистическим, и различия между этими двумя последовательностями распределены случайным образом по их длине. Сравнение двух нуклеиновокислотных или аминокислотных последовательностей традиционно проводится путем сравнения последовательностей после их оптимального выравнивания, при этом указанное сравнение можно проводить по сегментам или с помощью "окна выравнивания". Оптимальное выравнивание последовательностей для сравнения может быть осуществлено, помимо сравнения вручную, с помощью алгоритма локальной гомологии Смита и Вотермана (1981) [Ad. Арр. Math. 2:482], с помощью алгоритма локальной гомологии Нидлмана и Вунша (1970) [J. Mol. Biol. 48:443], с помощью способа поиска сходства Пирсона и Липмана (1988) [Proc. Natl. Acad. Sci. USA 85:2444] или с помощью компьютерного программного обеспечения с использованием этих алгоритмов (GAP, BESTFIT, FASTA и TFASTA в Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Мэдисон, Висконсин, или с помощью программного обеспечения для сравнения BLAST NR или BLAST Р).
Процент идентичности рассчитывают путем определения числа позиций, в которых аминокислоты, нуклеотиды или остатки идентичны в двух последовательностях, предпочтительно в двух полных последовательностях, деления числа идентичных позиций на общее число позиций в окне выравнивания и умножения результата на 100 для получения процентной идентичности двух последовательностей.
Например, программа BLAST "BLAST 2 sequences" (Tatusova et al, "Blast 2 sequences - a new tool for comparing protein and nucleotide sequences", FEMS Microbiol, 1999, Lett. 174: 247-250), доступная на сайте http://www.ncbi.nlm.nih.gov/gorf/bl2.html, может быть использована с параметрами по умолчанию (в частности, с параметрами "штраф за открытие делеции": 5 и "штраф за удлинение делеции": 2; выбранной матрицей будет, например, матрица "BLOSUM 62", предложенная в программе); процентная идентичность двух последовательностей для сравнения рассчитывается непосредственно программой.
Для аминокислотной последовательности, демонстрирующей идентичность с референсной аминокислотной последовательностью по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98%, предпочтительные примеры включают те, которые содержат референсную последовательность, определенные модификации, в частности делеции, добавления или замены по меньшей мере одной аминокислоты, усечения или удлинения. В случае замены одной или более последовательных или непоследовательных аминокислот предпочтительными являются те замены, при которых замещаемые аминокислоты заменяются "эквивалентными аминокислотами". В данном случае выражение "эквивалентные аминокислоты" применяется для обозначения любых аминокислот, которые могут заменить одну из структурных аминокислот без изменения биологической активности соответствующих антител, а также тех конкретных примеров, которые приведены ниже.
Эквивалентные аминокислоты могут быть определены либо по их структурной гомологии с аминокислотами, которые они будут замещать, либо по результатам сравнительных анализов биологической активности различных антител, которые могут быть получены.
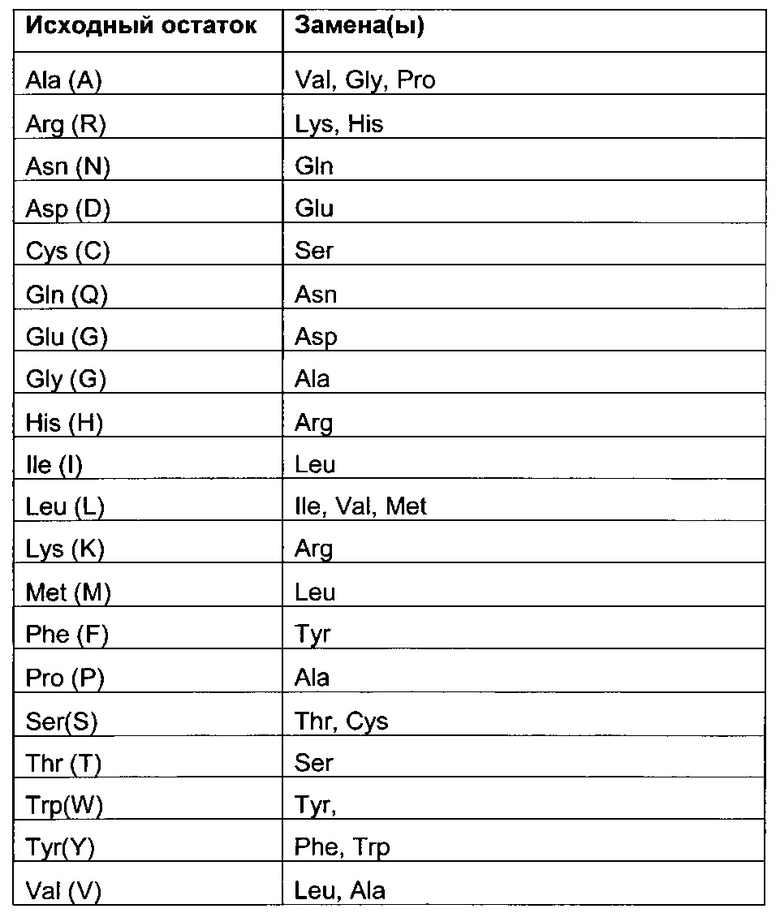
В качестве неограничивающего примера, в таблице 1 ниже приведены возможные замены, которые могут быть проведены без значительного изменения биологической активности соответствующего модифицированного антитела; обратные замены, естественно, возможны при тех же условиях.

Конкретным аспектом данного изобретения является то, что данное антитело не связывается с рецептором инсулина (IR, insulin receptor). Этот аспект представляет интерес, поскольку антитело, описанное в данном документе, не будет иметь никакого негативного влияния на IR, т.е. на метаболизм инсулина.
Другим предпочтительным аспектом антитела в ADC данного изобретения является то, что оно способно связываться не только с человеческим IGF-1R, но также с IGF-1R обезьяны и, более конкретно, с IGF-1R яванского макака. Этот аспект также представляет интерес, поскольку он будет облегчать испытания токсичности, необходимые для клинических испытаний.
В другом воплощении антитело в ADC данного изобретения состоит из моноклонального антитела.
Термин "моноклональное антитело" или "МКА" ("Mab"), используемый в данном документе, относится к антителу, полученному из популяции по существу гомогенных антител, т.е. отдельные антитела данной популяции являются идентичными за исключением возможных встречающихся в природе мутаций, которые могут присутствовать в незначительных количествах. Моноклональные антитела являются высоко специфичными, будучи направленными против одного эпитопа. Такое моноклональное антитело может быть получено с помощью одного клона В-клеток или с помощью гибридомы. Моноклональные антитела также могут быть рекомбинантными, т.е. полученными путем белковой инженерии или химического синтеза. Моноклональные антитела также могут быть выделены из фаговых библиотек антител. Кроме того, в отличие от препаратов поликлональных антител, которые обычно включают различные антитела, направленные против различных детерминант, или эпитопов, каждое моноклональное антитело направлено против одного эпитопа антигена.
Моноклональное антитело в данном документе включает мышиное, химерное и гуманизированное антитело, такое как описано ниже.
Антитело предпочтительно получено из гибридомы мышиного происхождения, сохраненной во Французской коллекции культур микроорганизмов (CNCM, Институт Пастера, 25 Rue du Docteur Roux, 75724, Paris Cedex 15, Франция), при этом указанная гибридома получена путем слияния спленоцитов/лимфоцитов от иммунизированных мышей BALB/C и клеток миеломной клеточной линии Sp2/O-Ag14.
В одном из воплощений антитело против IGF-1R в ADC согласно данному изобретению состоит из мышиного антитела, далее обозначаемого как m[название антитела].
В одном из воплощений антитело против IGF-1R состоит из химерного антитела, далее обозначаемого как с[название антитела].
В одном из воплощений антитело против IGF-1R состоит из гуманизированного антитела, далее обозначаемого как hz[название антитела].
Во избежание сомнений, в последующем описании выражения "IGF-1R-антитело" и "[название антитела]" являются аналогичными и включают (без противоречия описанию) мышиные, химерные и гуманизированные варианты указанного IGF-1R-антитела или указанного "[название антитела]"-антитела. При необходимости используется префикс m - (мышиное), с - (химерное) или hz - (гуманизированное).
Для большей ясности следующая таблица 2 иллюстрирует последовательности CDR, определенные согласно IMGT, для предпочтительных антител.
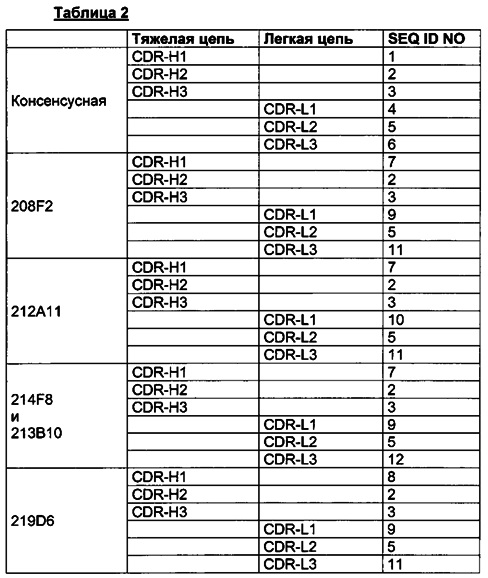
Специалисту в данной области будет очевидно, что любую комбинацию шести CDR, описанных выше, следует рассматривать как часть данного изобретения.
Как можно видеть в этой таблице 2, все антитела, описанные в данном документе, имеют одни и те же последовательности для CDR-H2, CDR-H3 и CDR-L2, причем данное свойство представляет особый интерес, как описано выше.
Конкретный аспект относится к мышиному антителу, которое характеризуется тем, что указанное антитело также содержит константные области легкой цепи и тяжелой цепи, полученные из антитела вида, гетерологичного по отношению к мыши, в частности, из человеческого антитела.
Другой конкретный аспект относится к химерному (с) антителу, которое характеризуется тем, что указанное антитело также содержит константные области легкой цепи и тяжелой цепи, полученные из антитела вида, гетерологичного по отношению к мыши, в частности, из человеческого антитела.
Химерное антитело представляет собой антитело, содержащее природную вариабельную область (легкой цепи и тяжелой цепи), полученную из антитела данного вида, в сочетании с константными областями легкой цепи и тяжелой цепи из антитела другого вида, гетерологичного по отношению к указанному данному виду.
Химерные антитела могут быть получены с использованием методик рекомбинантной генетики. Например, химерное антитело может быть получено путем клонирования рекомбинантной ДНК, содержащей промотор и последовательность, кодирующую вариабельную область нечеловеческого моноклонального антитела в ADC согласно данному изобретению, в частности мышиного, и последовательность, кодирующую константную область антитела гетерологичного вида, предпочтительно, человека. Химерное антитело в ADC в соответствии с данным изобретением, кодируемое одним из таких рекомбинантных генов, может представлять собой, например, химеру "мышь-человек", при этом специфичность этого антитела определяется вариабельной областью, полученной из мышиной ДНК, а его изотип определяется константной областью, полученной из человеческой ДНК.
В предпочтительном, но не ограничивающем воплощении антитело в ADC согласно данному изобретению выбрано среди следующих:
a) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 13 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 13, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 11;
b) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 14 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 14, и три CDR легкой цепи последовательностей SEQ ID NO 10, 5 и 11;
c) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 15 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 15, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 12;
d) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 16 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 16, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 11; и
е) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 17 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 17, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 12.
Под "любой последовательностью, демонстрирующей по меньшей мере 80%, 85%, 90%, 95% и 98% идентичность с SEQ ID NO 13-17" понимается последовательность, демонстрирующая три CDR тяжелой цепи SEQ ID NO 1, 2 и 3 и, кроме того, демонстрирующая по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 13-17 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 1, 2 и 3).
В другом предпочтительном, но не ограничивающем воплощении антитело в ADC согласно данному изобретению выбрано среди следующих:
a) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 18 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 18, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3;
b) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 19 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 19, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3;
c) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 20 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 20, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3;
d) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 21 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 21, и три CDR тяжелой цепи последовательностей SEQ ID NO 8, 2 и 3; и
e) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 22 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 22, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3.
Под "любой последовательностью, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 18-22" понимаются последовательности, демонстрирующие три CDR легкой цепи SEQ ID NO 4, 5 и 6 и, кроме того, демонстрирующие по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 18-22 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 4, 5 и 6).
Одно из воплощений согласно данному изобретению относится к ADC, где Ab представляет собой антитело, выбранное среди следующих:
a) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 13 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 13, и вариабельный домен легкой цепи последовательности SEQ ID NO 18 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ IDNO 18;
b) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 14 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 14, и вариабельный домен легкой цепи последовательности SEQ ID NO 19 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 19;
c) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 15 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 15, и вариабельный домен легкой цепи последовательности SEQ ID NO 20 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 20;
d) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 16 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 16, и вариабельный домен легкой цепи последовательности SEQ ID NO 21 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 21; и
e) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 17 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 17, и вариабельный домен легкой цепи последовательности SEQ ID NO 22 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 22.
Химерные антитела, описанные в данном документе, также могут характеризоваться константным доменом, и, более конкретно, указанные химерные антитела могут быть выбраны или сконструированы как IgG1, IgG2, IgG3, IgM, IgA, IgD или IgE, но не ограничиваясь ими. Более предпочтительно, в контексте данного изобретения указанные химерные антитела представляют собой IgG1 или IgG4.
Одно из воплощений данного изобретения относится к ADC, где Ab представляет собой химерное антитело, содержащее вариабельные домены VH и VL, описанные выше, в формате IgG1. Более предпочтительно, указанное химерное антитело содержит константный домен для VH последовательности SEQ ID NO 43 и каппа-домен для VL последовательности SEQ ID NO 45.
Одно из воплощений данного изобретения относится к ADC, где Ab представляет собой химерное антитело, содержащее вариабельные домены VH и VL, описанные выше, в формате IgG4. Более предпочтительно, указанное химерное антитело содержит константный домен для VH последовательности SEQ ID NO 44 и каппа-домен для VL последовательности SEQ ID NO 45.
В другом предпочтительном, но не ограничивающем воплощении антитело в ADC данного изобретения выбрано среди следующих:
a) антитело, содержащее (или состоящее из) тяжелую цепь последовательности SEQ ID NO 23 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 23, и легкую цепь последовательности SEQ ID NO 28 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 28;
b) антитело, содержащее (или состоящее из) тяжелую цепь последовательности SEQ ID NO 24 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 24, и легкую цепь последовательности SEQ ID NO 29 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 29;
c) антитело, содержащее (или состоящее из) тяжелую цепь последовательности SEQ ID NO 25 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 25, и легкую цепь последовательности SEQ ID NO 30 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 30;
d) антитело, содержащее (или состоящее из) тяжелую цепь последовательности SEQ ID NO 26 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 26, и легкую цепь последовательности SEQ ID NO 31 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 31; и
e) антитело, содержащее (или состоящее из) тяжелую цепь последовательности SEQ ID NO 27 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 27, и легкую цепь последовательности SEQ ID NO 32 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 32.
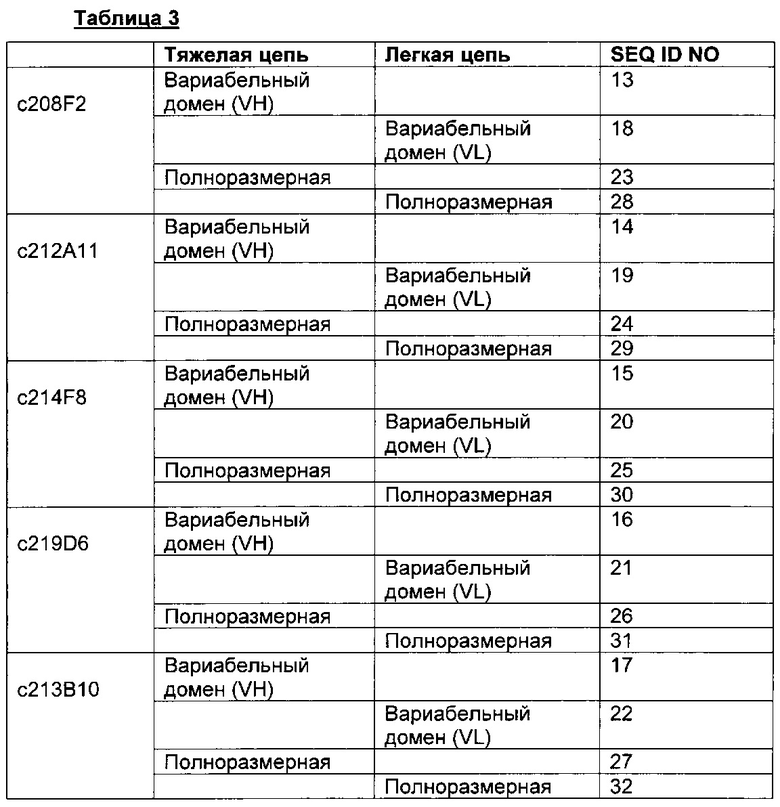
Для большей ясности следующая таблица 3 иллюстрирует последовательности VH и VL, соответственно, для предпочтительных химерных антител.
Другой конкретный аспект данного изобретения относится к ADC, где Ab представляет собой гуманизированное антитело, характеризующееся тем, что константные области легкой цепи и тяжелой цепи, полученные из человеческого антитела, являются, соответственно, областями лямбда или каппа и гамма-1, гамма-2 или гамма-4.
Под "гуманизированными антителами" понимаются антитела, которые содержат CDR-области, полученные из антитела нечеловеческого происхождения, при этом другие части молекулы антитела получены из одного (или нескольких) человеческих антител. Кроме того, некоторые остатки сегментов скелета (называемые FR) могут быть изменены в целях сохранения аффинности связывания.
Гуманизированные антитела или их фрагменты могут быть получены с помощью методик, известных специалистам в данной области. Такие гуманизированные антитела являются предпочтительными для их применения в способах, используемых в диагностике in vitro или в профилактическом и/или терапевтическом лечении in vivo. Также можно отметить другие методики гуманизации, также известные специалистам в данной области, такие как, например, методика "CDR-прививки", описанная PDL в патентах ЕР 0451261, ЕР 0682040, ЕР 09127, ЕР 0566647 или US 5530101, US 6180370, US 5585089 и US 5693761, патентах US 5639641 или US 6054297, US 5886152 и US 5877293.
В качестве конкретного воплощения данного изобретения, и как будет пояснено более подробно в примерах далее, в данном документе описано антитело, состоящее из hz208F2. Такая гуманизация также может быть применена к частям других антител согласно данному изобретению.
В предпочтительном воплощении антитело в ADC согласно данному изобретению содержит вариабельный домен тяжелой цепи (VH), имеющий:
i) CDR-H1, CDR-H2 и CDR-H3 последовательностей SEQ ID NO 7, 2 и 3, соответственно, и
ii) FR1, FR2 и FR3, полученные из человеческой зародышевой последовательности IGHV1-46*01 (SEQ ID NO 46), и
iii) FR4, полученный из человеческой зародышевой последовательности IGHJ4*01 (SEQ ID NO 48).
В предпочтительном воплощении антитело в ADC согласно данному изобретению содержит вариабельный домен легкой цепи (VL), имеющий:
i) CDR-L1, CDR-L2 и CDR-L3 последовательностей SEQ ID NO 9, 5 и 11, соответственно, и
ii) FR1, FR2 и FR3, полученные из человеческой зародышевой последовательности IGKV1-39*01 (SEQ ID NO 47), и
iii) FR4, полученный из человеческой зародышевой последовательности IGKJ4*01 (SEQ ID NO 49).
В предпочтительном, но не ограничивающем воплощении согласно данному изобретению антитело в ADC содержит:
а) тяжелую цепь, имеющую CDR-H1, CDR-H2 и CDR-H3 последовательностей SEQ ID NO 7, 2 и 3, соответственно, и FR1, FR2 и FR3, полученные из человеческой зародышевой последовательности IGHV1-46*01 (SEQ ID NO 46), и FR4, полученную из человеческой зародышевой последовательности IGHJ4*01 (SEQ ID NO 48); и
b) легкую цепь, имеющую CDR-L1, CDR-L2 и CDR-L3 последовательностей SEQ ID NO 9, 5 и 11, соответственно, и FR1, FR2 и FR3, полученные из человеческой зародышевой последовательности IGKV1-39*01 (SEQ ID NO 47), и FR4, полученную из человеческой зародышевой последовательности IGKJ4*01 (SEQ ID NO 49).
В одном воплощении антитело в ADC согласно данному изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33 и вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35. Указанное гуманизированное антитело будет называться далее hz208F2 ("вариант 1" или "вар. 1").
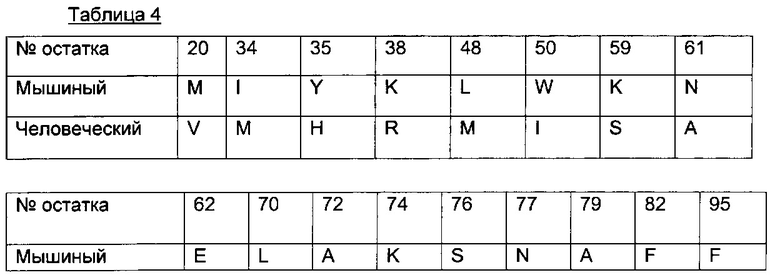
В другом воплощении антитело в ADC согласно данному изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95.
Под выражением "обратная мутация" подразумевается мутация или замена человеческого остатка, присутствующего в зародышевой последовательности, соответствующим остатком, первоначально присутствовавшим в мышиной последовательности.
В другом воплощении антитело в ADC согласно данному изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 или 17 обратных мутаций, выбранных среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95.
Для большей ясности в следующей таблице 4 показаны предпочтительные обратные мутации.

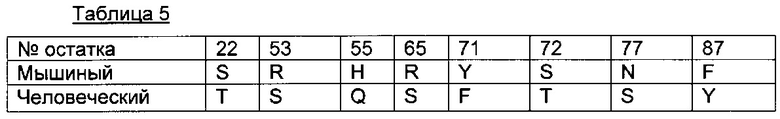
В одном из воплощений антитело в ADC согласно данному изобретению содержит вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87.
В одном из воплощений антитело в ADC согласно данному изобретению содержит вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит 2, 3, 4, 5, 6, 7 или 8 обратных мутаций, выбранных среди остатков 22, 53, 55, 65, 71, 72, 77 или 87.
В другом воплощении антитело в ADC согласно данному изобретению содержит:
a) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95; и
b) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87.
Для большей ясности следующая таблица 5 иллюстрирует предпочтительные обратные мутации.
В таком воплощении антитело в ADC согласно данному изобретению содержит все обратные мутации, указанные выше, и соответствует антителу, содержащему вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 34 и вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 36. Указанное гуманизированное антитело далее будет называться hz208F2 ("вариант 3" или "вар. 3").
В другом воплощении все гуманизированные формы, заключенные между вариантом 1 и вариантом 3, также охватываются данным изобретением. Другими словами, антитело в ADC согласно данному изобретению соответствует антителу, содержащему вариабельный домен тяжелой цепи (VH) "консенсусной" последовательности SEQ ID NO 41 и вариабельный домен легкой цепи (VL) "консенсусной" последовательности SEQ ID NO 42. Указанное гуманизированное антитело целиком будет далее называться hz208F2 ("вариант 2" или "вар. 2").
В предпочтительном, но не ограничивающем воплощении антитело в ADC согласно данному изобретению выбрано среди следующих:
a) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 33 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 33, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 11; или
b) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 34 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 34, и три CDR легкой цепи последовательностей SEQ ID NO 9, 5 и 11.
Под "любой последовательностью, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 33 или 34" понимаются последовательности, демонстрирующие три CDR тяжелой цепи SEQ ID NO 1, 2 и 3 и, кроме того, демонстрирующие по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 33 или 34 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 1, 2 и 3).
В предпочтительном, но не ограничивающем воплощении антитело в ADC согласно данному изобретению выбрано среди следующих:
a) антитело, содержащее вариабельный домен легкой цепи последовательности SEQ ID NO 35 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 35, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3; и
b) антитело, содержащее вариабельный домен тяжелой цепи последовательности SEQ ID NO 36 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 36, и три CDR тяжелой цепи последовательностей SEQ ID NO 7, 2 и 3.
Под "любой последовательностью, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% или 98% идентичность с SEQ ID NO 35 или 36" понимаются последовательности, демонстрирующие три CDR легкой цепи SEQ ID NO 4, 5 и 6 и, кроме того, демонстрирующие по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 35 и 36 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 4, 5 и 6).
Гуманизированные антитела, описанные в данном документе, также могут характеризоваться константным доменом, и, более конкретно, указанные гуманизированные антитела могут быть выбраны или сконструированы как IgG1, IgG2, IgG3, IgM, IgA, IgD или IgE, но не ограничиваясь ими. Более предпочтительно в контексте данного изобретения указанные гуманизированные антитела представляют собой IgG1 или IgG4.
Одно из воплощений согласно данному изобретению относится к ADC, в котором Ab представляет собой гуманизированное антитело, содержащее вариабельные домены VH и VL, описанные выше, в формате IgG1. Более предпочтительно, указанное гуманизированное антитело содержит константный домен для VH последовательности SEQ ID NO 43 и каппа-домен для VL последовательности SEQ ID NO 45.
Одно из воплощений согласно данному изобретению относится к ADC, в котором Ab представляет собой гуманизированное антитело, содержащее вариабельные домены VH и VL, описанные выше, в формате IgG4. Более предпочтительно, указанное гуманизированное антитело содержит константный домен для VH последовательности SEQ ID NO 44 и каппа-домен для VL последовательности SEQ ID NO 45.
Еще одно воплощение данного изобретения относится к ADC, в котором Ab представляет собой антитело, выбранное среди следующих:
a) антитело, содержащее тяжелую цепь последовательности SEQ ID NO 37 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 37, и легкую цепь последовательности SEQ ID NO 39 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 39; и
b) антитело, содержащее тяжелую цепь последовательности SEQ ID NO 38 или любой последовательности, демонстрирующей по меньшей мере 80% идентичности с SEQ ID NO 38, и легкую цепь последовательности SEQ ID NO 40 или любой последовательности, демонстрирующей по меньшей мере 80% идентичность с SEQ ID NO 40.
Для большей ясности следующая таблица 6 иллюстрирует неограничивающие примеры последовательностей VH и VL для варианта 1 (вар. 1) и варианта 3 (вар. 3) гуманизированного антитела hz208F2. Она также содержит консенсусную последовательность для варианта 2 (вар. 2).

Другой аспект данного изобретения представляет собой ADC, в котором Ab представляет собой антитело, выбранное среди следующих: i) антитело, продуцируемое гибридомой I-4757, I-4773, I-4775, I-4736 или I-4774, депонированной в CNCM, Институт Пастера, 30 мая 2013 года, 26 июня 2013, 26 июня 2013, 24 апреля 2013 и 26 июня 2013 года, соответственно, ii) антитело, которое конкурирует за связывание IGF-1R с антителом из i); или iii) антитело, которое связывается с тем же эпитопом IGF-1R, что и антитело из i).
В данном документе описана мышиная гибридома, выбранная среди гибридом I-4757, I-4773, I-4775, I-4736 и I-4774, депонированных в CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013, 26 июня 2013, 24 апреля 2013 и 26 июня 2013 года, соответственно.
Также описана выделенная нуклеиновая кислота, кодирующая антитело или его антигенсвязывающий фрагмент согласно данному изобретению.
Термины "нуклеиновая кислота", "нуклеиновая последовательность", "нуклеиновокислотная последовательность", "полинуклеотид", "олигонуклеотид", "полинуклеотидная последовательность" и "нуклеотидная последовательность", взаимозаменяемо используемые в данном описании, означают точную последовательность нуклеотидов, модифицированных или нет, определяющую фрагмент или область нуклеиновой кислоты, содержащей или не содержащей неприродные нуклеотиды и являющейся либо двухцепочечной ДНК, либо одноцепочечной ДНК, либо транскрипционными продуктами указанных ДНК.
Нуклеиновые последовательности антител в ADC данного изобретения были выделены и/или очищены, т.е. они были отобраны прямо или косвенно, например, путем копирования, и их окружающая среда по меньшей мере частично была изменена. Также здесь следует упомянуть выделенные нуклеиновые кислоты, полученные путем рекомбинантной генетики, например, с помощью клеток-хозяев, или путем химического синтеза.
Также здесь описан вектор, содержащий нуклеиновую кислоту, кодирующую антитело в ADC согласно данному изобретению или его антигенсвязывающий фрагмент.
В частности, раскрыты клонирующие векторы и/или экспрессионные векторы, которые содержат такую нуклеотидную последовательность.
Векторы предпочтительно содержат элементы, которые позволяют экспрессировать и/или секретировать нуклеотидные последовательности в данной клетке-хозяине. Вектор, таким образом, может содержать промотор, сигналы инициации и терминации трансляции, а также подходящие области регуляции транскрипции. Он должен иметь способность стабильно сохраняться в клетке-хозяине и может дополнительно иметь специфические сигналы, которые определяют секрецию транслированного белка. Эти различные элементы отбираются и оптимизируются специалистами в данной области в соответствии с используемой клеткой-хозяином. Для этой цели нуклеотидные последовательности могут быть вставлены в самореплицирующихся векторах в выбранного хозяина или могут быть интегративными векторами выбранного хозяина.
Векторы являются, например, векторами плазмидного или вирусного происхождения. Они используются для трансформации клеток-хозяев для клонирования или экспрессии нуклеотидных последовательностей согласно изобретению.
Такие векторы изготавливают, как правило, с помощью способов, использующихся специалистами в данной области, и полученные в результате клоны могут быть введены в подходящего хозяина с помощью стандартных способов, таких как липофекция, электропорация, конъюгация, тепловой шок или химические способы.
Также раскрыты выделенные клетки-хозяева, трансформированные вектором, описанным выше, или содержащие его.
Клетка-хозяин может быть выбрана среди прокариотических или эукариотических систем, таких как, например, бактериальные клетки, а также дрожжевые клетки или клетки животных, в частности клетки млекопитающих (за исключением человека). Также могут быть использованы клетки насекомых или растений.
Также раскрыт способ получения антитела согласно данному изобретению или его антигенсвязывающего фрагмента, при этом указанный способ характеризуется тем, что включает следующие этапы:
a) культивирование клеток-хозяев, описанных выше, в среде в подходящих культуральных условиях; и
b) извлечение антитела, полученного таким образом, из культуральной среды или из указанных культивируемых клеток.
Трансформированные клетки используются в способах получения рекомбинантных антител. В данное описание также входят способы получения антител в рекомбинантной форме с помощью вектора и/или клетки, трансформированной вектором. Предпочтительно, клетку, трансформированную вектором, описанным выше, культивируют в условиях, которые делают возможной экспрессию указанного выше полипептида и извлечение указанного антитела.
Как уже упоминалось, клетка-хозяин может быть выбрана среди прокариотических или эукариотических систем. В частности, можно определить нуклеотидные последовательности, которые облегчают секрецию в такой прокариотической или эукариотической системе. Вектор согласно данному изобретению, несущий такую последовательность, может быть выгодно использован для продукции секретируемых рекомбинантных белков. Действительно, очистке этих рекомбинантных белков, представляющих интерес, будет способствовать тот факт, что они присутствуют в супернатанте клеточной культуры, а не внутри клеток-хозяев.
Антитело в ADC согласно данному изобретению также может быть получено путем химического синтеза. Один такой способ получения также является объектом изобретения. Специалистам в данной области известны способы химического синтеза, такие как твердофазные методики или частично твердофазные методики, путем конденсации фрагментов или путем обычного синтеза в растворе. В изобретение также входят полипептиды, которые получены путем химического синтеза и могут содержать соответствующие неприродные аминокислоты.
В данное изобретение также входит ADC, содержащий антитело, вероятно, полученное описанным выше способом.
В соответствии с конкретным аспектом данное изобретение относится к ADC, в котором Ab представляет собой антитело или его антигенсвязывающий фрагмент, описанный выше, для применения в качестве адресующего носителя для доставки цитотоксического агента в место-мишень хозяина, при этом указанное место-мишень хозяина состоит из эпитопа, локализованного в IGF-1R, предпочтительно во внеклеточном домене IGF-1R, более предпочтительно в человеческом IGF-1R (SEQ ID NO 50), и еще более предпочтительно во внеклеточном домене человеческого IGF-1R (SEQ ID NO 51), и еще более предпочтительно на N-конце внеклеточного домена человеческого IGF-1R (SEQ ID NO 52), или последовательности любого их природного варианта.
В предпочтительном воплощении указанное место-мишень хозяина представляет собой место-мишень клетки млекопитающего, более предпочтительно клетки человека, более предпочтительно клетки, которая естественным образом или путем генетической рекомбинации экспрессирует IGF-1R.
I.2: Axl-антитела
Без какого-либо противоречивого указания определения или выражения, используемые и определенные в параграфе "I.1, IGF-1R-антитела", являются такими же в данном параграфе и могут быть применены ко всем ADC согласно данному изобретению.
Антитело против Axl способно связываться с человеческим белком Axl. Более конкретно, указанная мишень представляет собой эпитоп, расположенный во внеклеточном домене Axl (называемом как Axl ECD, что является сокращением для "Axl Extra Cellular Domain").
Axl ECD представляет собой фрагмент из 451 аминокислоты, соответствующий аминокислотам 1-451 последовательности SEQ ID NO 103, последовательность которого представлена в списке последовательностей как SEQ ID NO 105. Аминокислоты 1-25 соответствуют сигнальному пептиду, ECD человеческого белка Axl без сигнального пептида соответствует аминокислотам 26-451 последовательности SEQ ID NO 104, представленным последовательностью SEQ ID NO 106.
Axl-антитело в ADC согласно данному изобретению включает три CDR легкой цепи, содержащие последовательности SEQ ID NO 56, 57 и 58 или любую последовательность, демонстрирующую по меньшей мере 90%, предпочтительно 95% и 98% идентичность с SEQ ID NO 56, 57 и 58; и три CDR тяжелой цепи, содержащие последовательности SEQ ID NO 59, 60 и 61 или любую последовательность, демонстрирующую по меньшей мере 90%, предпочтительно 95% и 98% идентичность с SEQ ID NO 59, 60 и 61.
В одном из воплощений Axl-антитело содержит три CDR легкой цепи, содержащие, соответственно, последовательности SEQ ID NO 56, 57 и 58, и три CDR тяжелой цепи, содержащие, соответственно, последовательности SEQ ID NO 59, 60 и 61.
В одном воплощении Axl-антитело состоит из m1613F12, содержащего i) вариабельный домен легкой цепи последовательности SEQ ID NO 62 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 62; и/или ii) вариабельный домен тяжелой цепи последовательности SEQ ID NO 63 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 63.
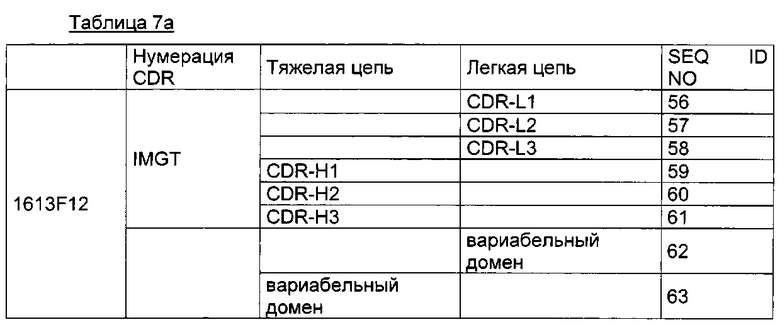
Для большей ясности в таблице 7а ниже приведены различные аминокислотные последовательности, соответствующие Axl-антителу.
В одном воплощении Axl-антитело состоит из c1613F12, содержащего три CDR легкой цепи, содержащие последовательности SEQ ID NO 56, 57 и 58 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 56, 57 и 58; и три CDR тяжелой цепи, содержащие последовательности SEQ ID NO 59, 60 и 61 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ №№ №59, 60 и 61.
В одном воплощении c1613F12 содержит три CDR легкой цепи, содержащие, соответственно, последовательности SEQ ID NO 56, 57 и 58; и три CDR тяжелой цепи, содержащие, соответственно, последовательности SEQ ID NO 59, 60 и 61.
В одном воплощении Axl-антитело состоит из c1613F12, содержащего i) вариабельный домен легкой цепи последовательности SEQ ID NO 62 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 62; и/или ii) вариабельный домен тяжелой цепи последовательности SEQ ID NO 63 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 63.
В одном воплощении Axl-антитело состоит из hz1613F12, содержащего три CDR легкой цепи, содержащие последовательности SEQ ID NO 56, 57 и 58 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 56, 57 и 58; и три CDR тяжелой цепи, содержащие последовательности SEQ ID NO 59, 60 и 61 или любую последовательность, демонстрирующую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 59, 60 и 61.
В одном воплощении hz1613F12 содержит три CDR легкой цепи, содержащие, соответственно, последовательности SEQ ID NO 56, 57 и 58; и три CDR тяжелой цепи, содержащие, соответственно, последовательности SEQ ID NO 59, 60 и 61.
В одном воплощении hz1613F12 содержит вариабельный домен легкой цепи, состоящий из последовательности SEQ ID NO 70 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 70; и три CDR тяжелой цепи, состоящие из последовательностей SEQ ID NO 59, 60 и 61.
В другом воплощении данного изобретения hz1613F12 содержит вариабельный домен легкой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 71-81, или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 71-81; и три CDR тяжелой цепи, состоящие из SEQ ID NO 59, 60 и 61.
Чтобы проиллюстрировать процент идентичности, определенный выше, под "любой последовательностью, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 70-81" понимаются последовательности, демонстрирующие три CDR легкой цепи SEQ ID NO 56, 57 и 58 и, кроме того, демонстрирующие по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 70-81 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 56, 57 и 58).
Для большей ясности в таблице 7b ниже приведены различные аминокислотные последовательности, соответствующие легкой цепи (VL) гуманизированного Axl-антитела в ADC данного изобретения (где hz. - гуманизированное).
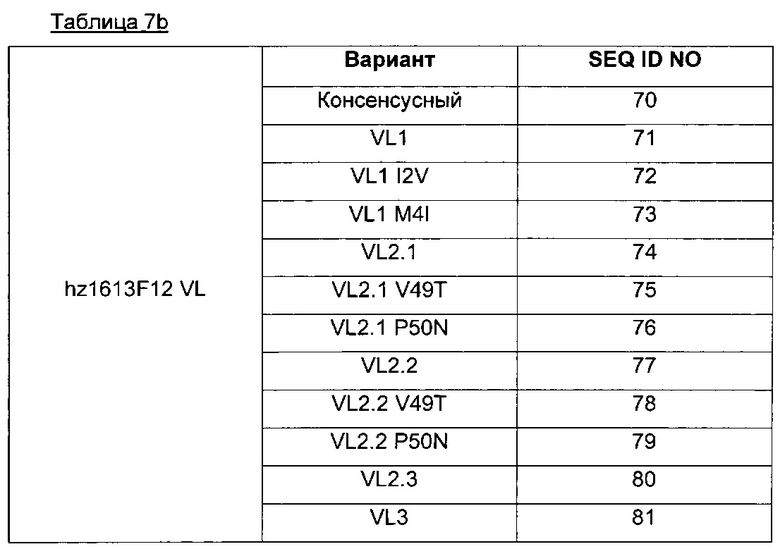
В одном воплощении hz1613F12 содержит вариабельный домен тяжелой цепи, состоящий из последовательности SEQ ID NO 82 или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 82; и три CDR легкой цепи, состоящие из последовательностей SEQ ID NO 56, 57 и 58.
В другом воплощении hz1613F12 содержит вариабельный домен тяжелой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 83-102, или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 83-102; и три CDR легкой цепи, состоящие из SEQ ID NO 56, 57 и 58.
Чтобы проиллюстрировать процент идентичности, определенный выше, под "любой последовательностью, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 82-102", понимаются последовательности, демонстрирующие три CDR тяжелой цепи SEQ ID NO 59, 60 и 61 и, кроме того, демонстрирующие по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полной последовательностью SEQ ID NO 82-102 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 59, 60 и 61).
Для большей ясности в таблице 7с ниже приведены различные аминокислотные последовательности, соответствующие тяжелой цепи (VH) гуманизированноого антигенсвязывающего белка согласно данному изобретению (где hz - гуманизированное)
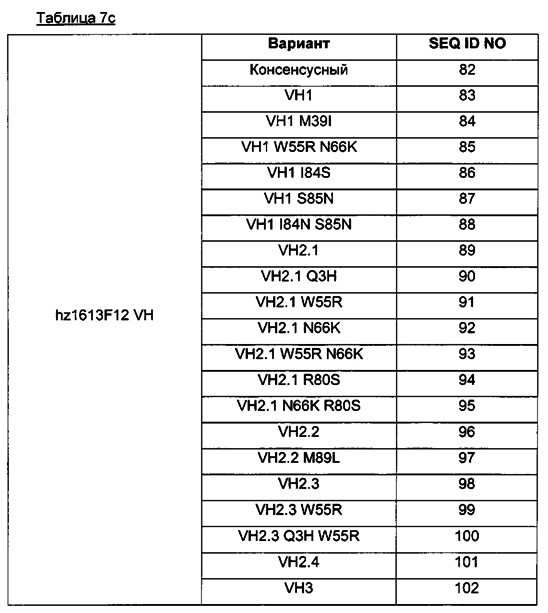
В одном воплощении hz1613F12 содержит вариабельный домен легкой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 70, и любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%), 95% и 98% идентичность с SEQ ID NO 70; и вариабельный домен тяжелой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 82, и любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 82.
В одном воплощении hz1613F12 содержит вариабельный домен легкой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 71-81, или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 71-81; и вариабельный домен тяжелой цепи последовательности, выбранной из группы, состоящей из SEQ ID NO 83-102, или любой последовательности, демонстрирующей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с SEQ ID NO 83-102.
Другой аспект данного изобретения представляет собой ADC, где Ab представляет собой антитело, выбранное среди следующих: i) антитело, продуцируемое гибридомой I-4505, депонированной в CNCM, Институт Пастера, 28 июля 2011, ii) антитело, которое конкурирует за связывание Axl с антителом из i); или iii) антитело, которое связывается с тем же эпитопом Axl, что и антитело из i).
Согласно другому аспекту описана мышиная гибридома, выбранная среди гибридомы I-4505.
В таблице 8 ниже приведены нуклеотидные последовательности, касающиеся CDR 1613F12.
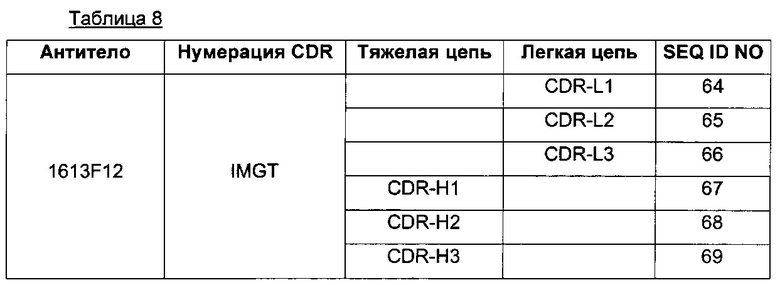
В одном воплощении нуклеиновокислотные последовательности CDR могут быть выбраны среди последовательностей SEQ ID NO 64-69.
В качестве неограничивающего воплощения в данном документе описана выделенная нуклеиновая кислота или их комбинация, кодирующая антитело, характеризующееся тем, что указанная нуклеиновая кислота или их комбинация содержит шесть CDR-последовательностей SEQ ID NO 64-69.
I.3: HER2-антитела
НЕР2-антитела могут состоять из любого антитела, способного связываться с HER2, такого как, но не ограничиваясь ими, TrasGEX (гликотоп), трастузумаб, пертузумаб или любой их биоаналог.
В предпочтительном воплощении антитело против HER2 представляет собой трастузумаб (Herceptin®; 4D5; Genentech, Сан-Франциско, Калифорния).
II - Лекарственное средство (D)
Лекарственная группировка согласно данному изобретению имеет следующую формулу (II)
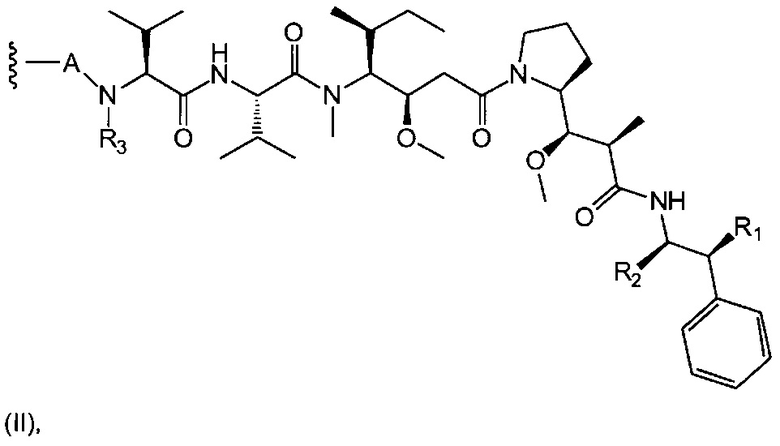
где:
R1 представляет собой Н или ОН;
R2 представляет собой группу: (С1-С6)алкил (например, метил), СООН, СОО-((С1-С6)алкил) (такую как СООМе) или тиазолил (такую как тиазол-2-ил);
R3 представляет собой Н или (С1-С6)алкил (такую как метил), в частности, (С1-С6)алкил,
А представляет собой:
- группу формулы -Het-Alk-, где Alk представляет собой (C1-C8)алкандиил и связан с NR3, a Het представляет собой гетероцикл, возможно, замещенный (С1-С6)алкил и содержащий по меньшей мере один атом азота, при этом указанный атом азота связан с L, или
- группу формулы -Aa-Ab-, где Aa связан с L и представляет собой О или NR9, при этом R9 представляет собой Н или (С1-С6)алкил (такой как метил), а Ab связан с NR3 и представляет собой:
(С1-С8)алкандиил (такую как -(СН2)m-, где m составляет от 1 до 8),
-(СН2СН2Х1)а1(СН2СН2Х2)a2(CH2CH2X3)a3(СН2СН2Х4)a4 СН2СН2, где каждый из Х1, Х2, Х3 и Х4 независимо от других представляет собой О или NR8; каждый из а1, а2, а3 и а4 независимо от других представляет собой 0 или 1 (в частности, где а1+а2+а3+а4=1 или 2, в частности, 1); a R8 представляет собой Н или (С1-С6)алкил (такую как метил),
арил-(С1-С8)алкандиил- или гетероцикл-(С1-С8)алкандиил, где указанная группа, возможно, замещена (С1-С6)алкил, при этом арильная или гетероциклическая группировка связана с Aa, а группировка (C1-С8)алкандиил связана с NR3;
волнистая линия указывает на точку присоединения к L.
Радикалы R2, R3 и А могут представлять собой хиральные группы и могут находиться в форме их различных стереоизомеров и, возможно, в форме смеси стереоизомеров.
Под "стереоизомером" в значении данного изобретения понимается геометрический изомер или оптический изомер.
Геометрические изомеры являются результатом различного расположения заместителей на двойной связи, следовательно, могут иметь Z- или Е-конфигурацию.
Оптические изомеры являются результатом, в частности, различного расположения в пространстве заместителей на атоме углерода, содержащем 4 различных заместителя. Этот атом углерода затем образует хиральный или асимметричный центр. Оптические изомеры содержат диастереоизомеры и энантиомеры. Оптические изомеры, которые являются зеркальным отражением друг друга, но которые не могут быть наложены друг на друга, называются "энантиомерами". Оптические изомеры, которые не являются зеркальным отражением друг друга, называются "диастереоизомерами".
Смесь, содержащую равные количества двух отдельных энантиомерных форм с противоположной хиральностью, называют "рацемической смесью".
Под "алкилом" в данном изобретении понимается моновалентная, линейная или разветвленная, насыщенная углеводородная цепь. Например, можно упомянуть группы: метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил или гексил.
Под "(Сх-Су)алкилом" в значении данного изобретения понимается алкильная цепь, такая как определено выше, содержащая от х до у атомов углерода. Кроме того, (С1-С6)алкил представляет собой алкильную цепь, содержащую от 1 до 6 атомов углерода. (С1-С6)алкил преимущественно представляет собой (С1-С4)алкил, предпочтительно (С1-С2)алкил.
Под "алкандиилом" в данном изобретении понимается двухвалентная, линейная или разветвленная, насыщенная углеводородная цепь. Например, можно упомянуть метандиил, этандиил, пропандиил, бутандиил, пентандиил, гександиил и т.п.
Под "(Сх-Су)алкандиилом" в значении данного изобретения понимается алкандиил-цепь, такая как определено выше, содержащая от х до у атомов углерода. Кроме того, (С1-С8)алкандиил представляет собой алкандиил-цепь, содержащую от 1 до 8 атомов углерода.
Под "арилом" в значении данного изобретения понимается ароматическая углеводородная группа, предпочтительно содержащая от 6 до 10 атомов углерода и способная содержать от одного до двух конденсированных колец. Например, можно упомянуть фенил или нафтил. Преимущественно, она представляет собой фенил.
Под "гетероциклом" в значении данного изобретения понимается насыщенная, ненасыщенная или ароматическая углеводородная группа, содержащая 1 или 2 конденсированных кольца, в которой один или более, преимущественно от 1 до 4, более преимущественно 1 или 2 атома углерода заменены гетероатомом, выбранным среди кислорода, азота и серы. Преимущественно гетероцикл содержит от 5 до 10 атомов углерода и гетероатомов. Например, можно упомянуть фуран, пиррол, тиофен, тиазол, изотиазол, оксадиазол, имидазол, оксазол, изоксазол, пиридин, пиримидин, пиперазин, пиперидин, хиназолин, хинолин, хиноксалин, бензофуран, бензотиофен, индолин, индолизин, бензотиазол, бензотиофен, бензопиран, бензоксазол, бензо[1,3]диоксол, бензоизоксазол, бензимидазол, хроман, хромен, дигидробензофуран, дигидробензотиофен, дигидроизоксазол, изохинолин, дигидробензо[1,4]диоксин, имидазо[1,2-а]пиридин, фуро[2,3-с]пиридин, 2,3-дигидро-1Н-инден, [1,3]диоксоло[4,5-с]пиридин, пирроло[1,2-с]пиримидин, пирроло[1,2-а]пиримидин, тетрагидронафтален и бензо[b][1,4]оксазин.
В данном изобретении гетероцикл более предпочтительно является насыщенным, ненасыщенным или ароматическим кольцом с 5 или 6 членами, содержащим 1 или 2 атома азота. Например, можно упомянуть пиррольное, имидазольное, пиридиновое, пиримидиновое, пиперазиновое и пиперидиновое кольца. Предпочтительно оно представляет собой пиридин, пиперидин или имидазол.
Среди лекарственных группировок согласно данному изобретению один особо оцененный класс лекарственных группировок соответствует лекарственным группировкам формулы (II), в которой R1 представляет собой ОН, a R2 представляет собой (С1-С6)алкил, такую как метил.
Другой особо оцененный класс лекарственных группировок соответствует лекарственным группировкам формулы (II), в которой R1 представляет собой водород, a R2 представляет собой тиазолил (в частности, тиазол-2-ил).
Другой особо оцененный класс лекарственных группировок соответствует лекарственным группировкам формулы (II), в которой R1 представляет собой водород, a R2 представляет собой СОО-((С1-С6)алкил), такую как СООМе.
Другой класс особо оцененных лекарственных группировок соответствует лекарственным группировкам формулы (II), в которой R1 представляет собой водород, a R2 представляет собой СООН.
Согласно одному конкретному воплощению данного изобретения R2 представляет собой метил-, СООН-, СООМе- или тиазол-2-ил.
Кроме того, лекарственные группировки согласно данному изобретению преимущественно являются лекарственными группировками формулы (II), в которой:
- R1 представляет собой ОН, a R2 представляет собой Me (метил), или
- R1 представляет собой Н, a R2 представляет собой СООН, СООМе или тиазол-2-ил.
Согласно предпочтительному воплощению R1 представляет собой Н, a R2 представляет собой СООН, СООМе или тиазол-2-ил, предпочтительно R2 представляет собой СООН или СООМе, и более предпочтительно R2 представляет собой СООН.
R3, в частности, представляет собой Н или метил, преимущественно метил.
В предпочтительном воплощении R1 представляет собой Н, R2 представляет собой СООН, a R3 представляет собой метил.
В определении А:
- (С1-C8)алкандиил преимущественно представляет собой (С1-С6)алкандиил, предпочтительно (С1-С2)алкандиил и более предпочтительно (С1-С2)алкандиил, и, в частности, она представляет собой прямую цепь с формулой -(СН2)m-, где m представляет собой целое число от 1 до 8, преимущественно от 1 до 6, предпочтительно от 1 до 4, более предпочтительно m составляет 1 или 2,
- арильная группа преимущественно представляет собой фенильную группу, и
- гетероцикл преимущественно представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 членами, имеющее 1 или 2 атома азота; например, пиррольное, имидазольное, пиридиновое, пиримидиновое, пиперазиновое или пиперидиновое кольцо; предпочтительно пиридин, пиперидин или имидазол; более предпочтительно пиридин.
Когда А представляет собой группу формулы -Het-Alk-, Het более предпочтительно является гетероциклом, выбранным среди пиперидина и имидазола.
Когда А представляет собой группу формулы -Aa-Ab-, где Ab представляет собой группу гетероцикл-(С1-С8)алкандиил, возможно, замещенную (С1-С6)алкилом, гетероцикл более предпочтительно является пиридином.
Согласно предпочтительному воплощению А представляет собой группу формулы -Aa-Ab-, определенную выше.
Aa представляет собой О или NR9, и предпочтительно NR9, при этом R9 является таким, как определено выше, и предпочтительно R9 представляет собой Н или Me.
Согласно конкретному воплощению данного изобретения Ab представляет собой группу:
- (С1-С6)алкандиил, в частности, (С2-С6)алкандиил (такой как -(СН2)m-, где m составляет от 1 до 6, предпочтительно от 2 до 6),
- -(СН2СН2Х1)а1(СН2СН2Х2)a2CH2CH2-, где а1+а2 преимущественно представляет собой 1 или 2, в частности 1,
- арил-(С1-С6)алкандиил, или
- гетероцикл-(C1-С6)алкандиил, возможно, замещенную (C1-С6)алкилом (особенно незамещенную).
Согласно другому конкретному воплощению данного изобретения Ab представляет собой группу:
- (С1-С4)алкандиил, в частности, (С2-С4)алкандиил (такую как -(СН2)m-, где m составляет от 1 до 4, и предпочтительно составляет 2, 3 или 4),
- -(CH2CH2X1)CH2CH2- или -(CH2CH2X1)(CH2CH2X2)CH2CH2-,
- арил-(C1-С4)алкандиил, или
- гетероцикл-(С1-С4)алкандиил, возможно, замещенную (C1-С6)алкилом (особенно незамещенную).
Согласно другому конкретному воплощению данного изобретения Ab представляет собой группу:
- (С1-С4)алкандиил, в частности, (С2-С4)алкандиил (такую как -(СН2)m-, где m составляет от 1 до 4, и предпочтительно составляет 2, 3 или 4),
- -(CH2CH2X1)CH2CH2-,
- арил-(C1-С2)алкандиил, или
- гетероцикл-(C1-С2)алкандиил, возможно, замещенную (C1-С6)алкилом (особенно незамещенную).
Согласно другому конкретному воплощению Ab представляет собой:
арил-(С1-С8)алкандиил; или гетероцикл-(C1-С8)алкандиил, возможно, замещенную (С1-С6)алкилом (особенно незамещенную);
особенно арил-(С1-С6)алкандиил; или гетероцикл-(С1-С6)алкандиил, возможно, замещенную (С1-С6)алкилом (особенно незамещенную);
преимущественно арил-(C1-С4)алкандиил; или гетероцикл-(С1-С4)алкандиил, возможно, замещенную (С1-С6)алкилом (особенно незамещенную);
и предпочтительно арил-(С1-С2)алкандиил; или гетероцикл-(C1-С2)алкандиил, возможно, замещенную (С1-С6)алкилом (особенно незамещенную).
Согласно еще одному конкретному воплощению Ab представляет собой группу арил-(С1-С8)алкандиил; особенно арил-(С1-С6)алкандиил; преимущественно арил-(C1-С4)алкандиил; и предпочтительно арил-(C1-С2)алкандиил.
В приведенных выше конкретных воплощениях для Ab арильная группа преимущественно представляет собой фенильную группу.
В приведенных выше конкретных воплощениях для Ab гетероцикл преимущественно представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 членами, содержащее 1 или 2 атома азота. Например, можно упомянуть пиррольное, имидазольное, пиридиновое, пиримидиновое, пиперазиновое или пиперидиновое кольцо. Предпочтительно оно представляет собой пиридин, пиперидин или имидазол, и более предпочтительно, оно представляет собой пиридин.
Преимущественно, Ab представляет собой группу:
- фенил-(С1-С2)алкандиил, или
- гетероцикл-(С1-С2)алкандиил, возможно, замещенную (C1-С6)алкилом (особенно незамещенную), при этом гетероцикл представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 членами, содержащее 1 или 2 атома азота, выбранное, в частности, среди пиридина, пиперидина и имидазола и предпочтительно представляющее собой пиридин.
В предпочтительном воплощении Ab представляет собой фенил-(C1-С2)алкандиил.
Согласно предпочтительному воплощению А имеет следующую формулу:
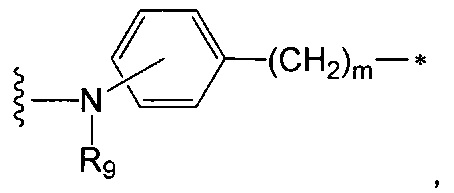
и предпочтительно следующую формулу:
где:
R9 и m являются такими, как определено ранее, и где предпочтительно R9 представляет собой Н или Me, а m равно 1 или 2,
волнистая линия указывает на точку присоединения к L, и
звездочка указывает на точку присоединения к NR3.
Преимущественно лекарственная группировка выбрана среди следующих группировок:
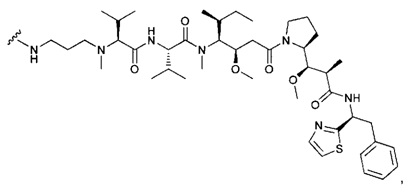
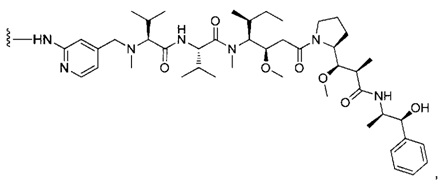

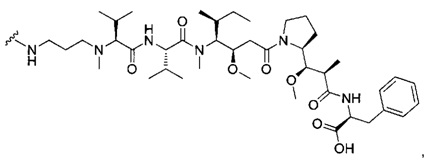
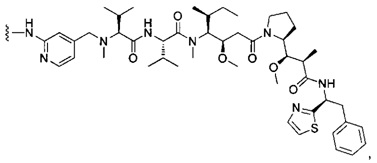
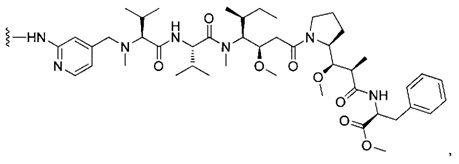
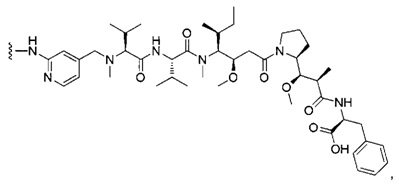
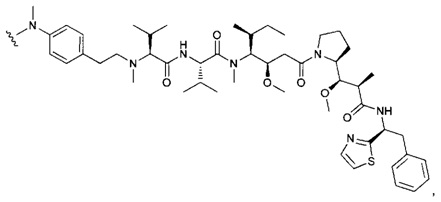
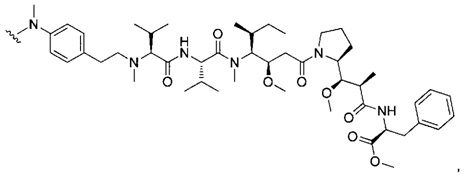
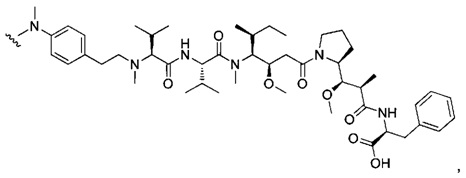
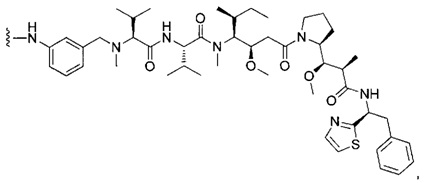
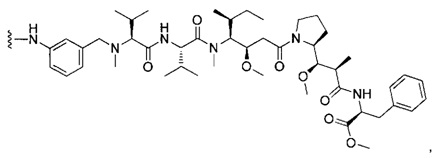
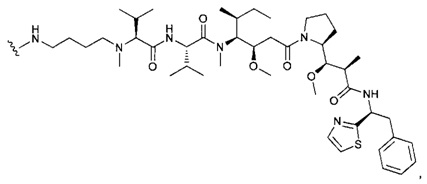
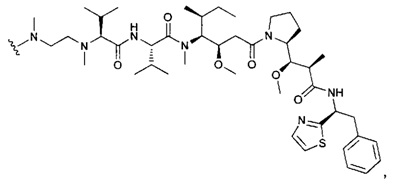
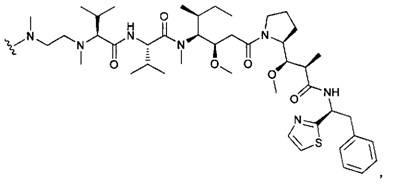

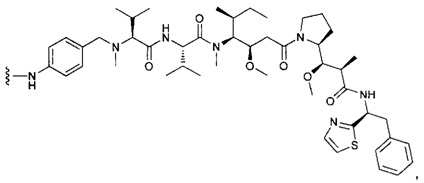
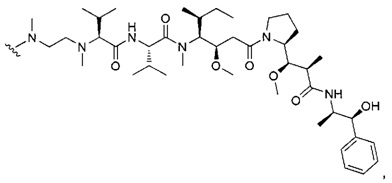
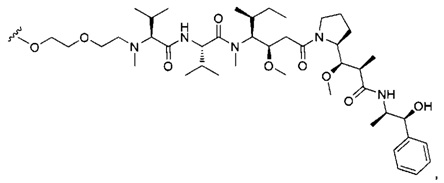

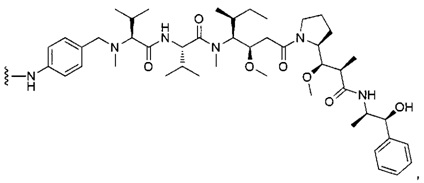
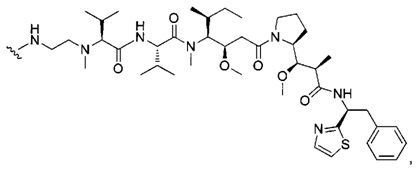
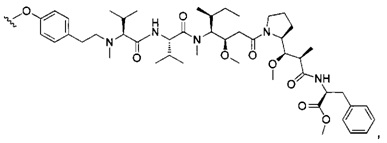
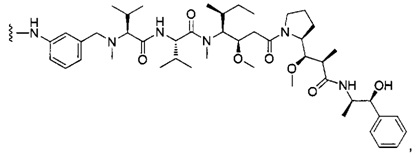
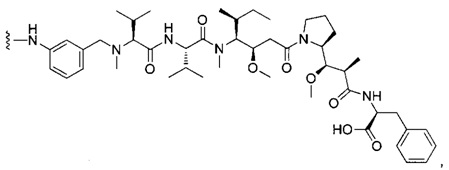
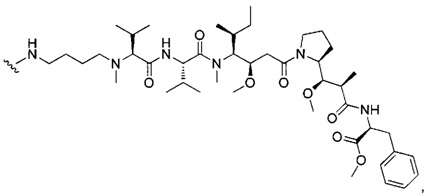
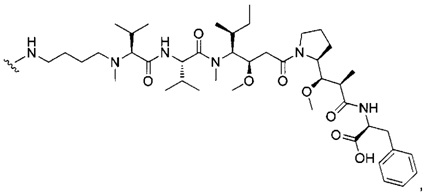
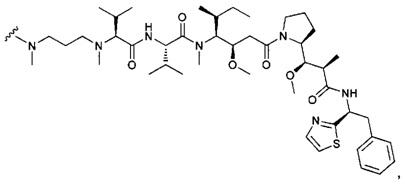
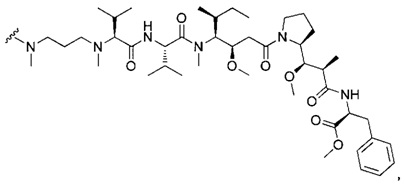
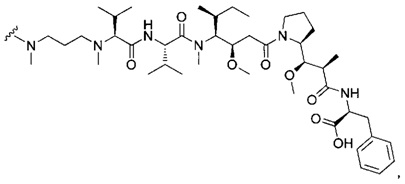
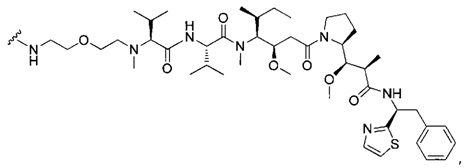
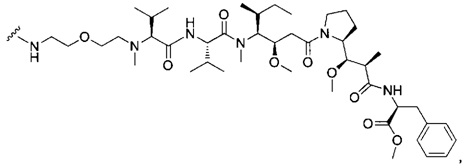
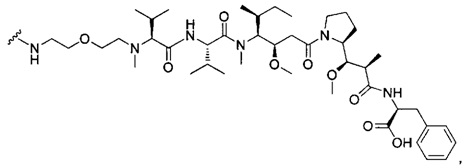
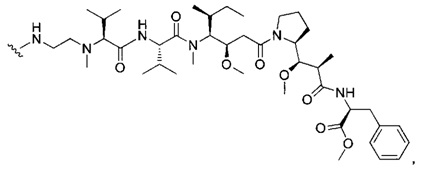
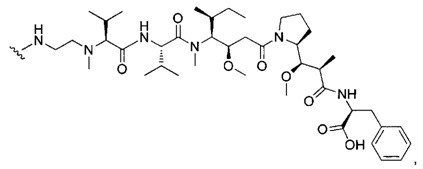
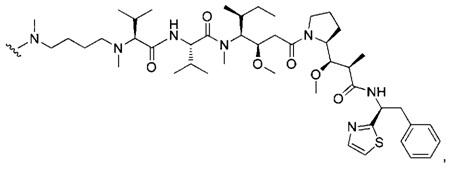
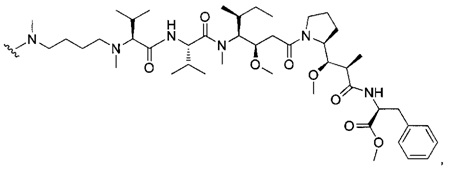
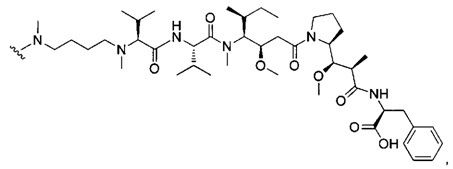
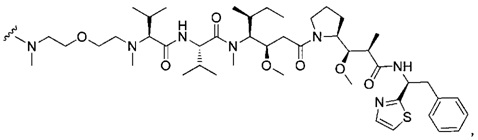
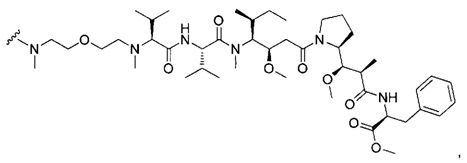
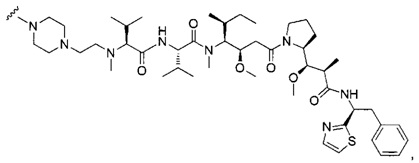
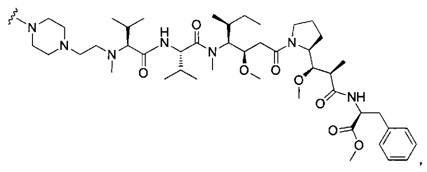

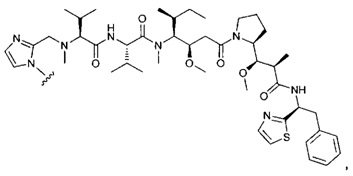
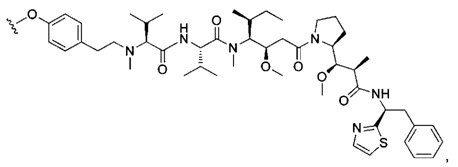
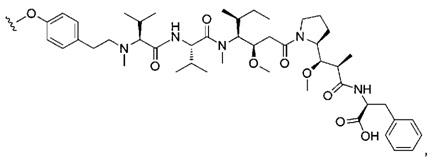


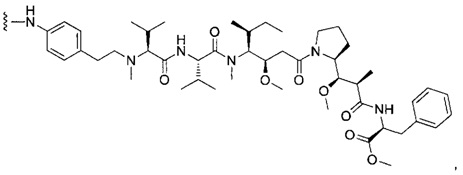
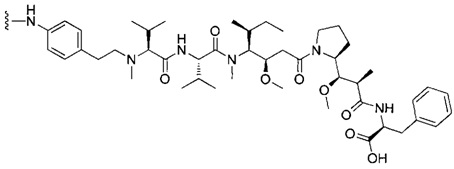 и
и
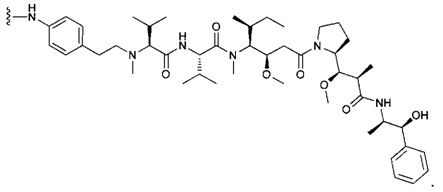
Получение лекарственного средства (Формулы DH):
Лекарственное средство может быть получено с использованием общих способов, описанных в последующих схемах синтеза, возможно дополненных любой стандартной операцией, когда это необходимо, которые описаны в литературе или хорошо известны специалистам в данной области или описаны в примерах, приведенных в экспериментальной части.
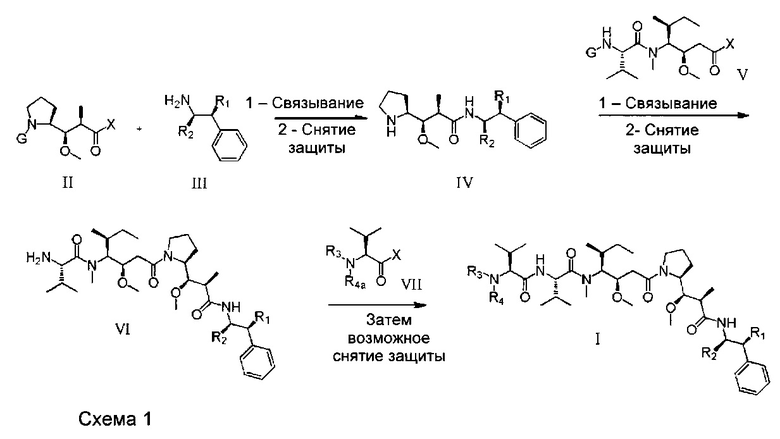
Схема 1 иллюстрирует первый общий способ, который можно использовать для изготовления лекарственного средства. В приведенных выше общих формулах R1, R2 и R3 являются такими, как определено выше, R4 представляет собой -АН, а R4a представляет собой такую группу R4, как указано выше, возможно, в защищенной форме, a G представляет собой защитную группу.
Первый этап состоит из конденсации соединения (II), защищенного на его аминной функциональной группе защитной группой G, с соединением (III). X может представлять собой уходящую группу, такую как хлор. В этом случае первый этап состоит из реакции между хлорангидридом и амином. Эта реакция может быть проведена с использованием способов и методик, хорошо известных специалистам в данной области. В одном особо оцененном способе эти две группировки вступают в реакцию в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3, K2CO3, в растворителе, таком как ТГФ (тетрагидрофуран), дихлорметан, ДМФА (N,N-диметилформамид), ДМСО (диметилсульфоксид), при температуре, в частности, от -20°C до 100°C. X также может представлять собой гидроксил (ОН). В этом случае первый этап представляет собой реакцию конденсации между карбоновой кислотой (II) и амином (III). Эта реакция может быть проведена в соответствии со способами и методиками, хорошо известными специалистам в данной области. В одном особо оцененном способе эти две группировки вступают в реакцию в присутствии связывающего агента, такого как 1-(3-диметиламинопропил)-3-этил-карбодиимид (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-он, третичный амин, такой как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре, в частности, от -15°C до 40°C. В другом особо оцененном способе эти две группировки вступают в реакцию в присутствии диэтилфосфороцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре от -15°C до 40°C. Другой особо оцененный способ заключается в введении в реакцию этих двух группировок в присутствии O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата (HATU), третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре от -15°C до 100°C.
После удаления защитной группы с промежуточного продукта с использованием методик, хорошо известных специалистам в данной области («Protective Groups in Organic Synthesi», T.W. Greene, John Wiley & Sons, 2006 и «Protecting Groups», P.J. Kocienski, Thieme Verlag, 1994), соединение (IV) можно конденсировать с соединением (V) в соответствии со способами и методиками, описанными выше, получая соединение (VI) после этапа удаления защитной группы. Это соединение может затем, после конденсации с промежуточным продуктом (VII) и возможным удалением защитной группы, привести к образованию лекарственного средства. Соединение (VI) также может быть соединено с соединением (VII'), в котором R'3 является предшественником R3, в частности, группой R3, защищенной защитной группой. Связывание с последующим удалением защитной группы R'3, которое образует R3, можно выполнить с помощью тех же процедур, что описаны ранее.
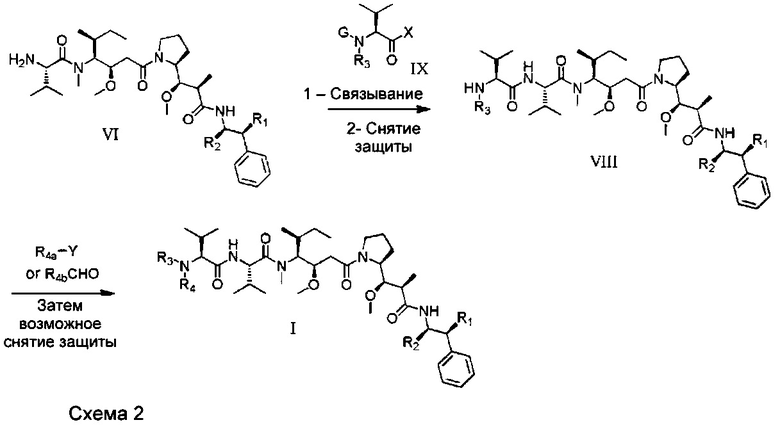
Схема 2 иллюстрирует второй общий способ, который может быть использован для получения лекарственного средства. В приведенных выше общих формулах г представляет собой защитную группу, R1, R2, R3 и R4a являются такими, как определено ранее, a R4b представляет -Ас-AaH, где Aa является таким, как определено ранее, а Ас представляет собой:
(С1-С7)алкандиил,
группу -(СН2СН2Х1)а1(СН2СН2Х2)а2(СН2СН2Х3)а3(СН2СН2Х4)а4СН2*, где группа СН2, отмеченная звездочкой, связана с группой СНО R4bCHO,
арильную или гетероциклическую группу,
группу арил-(С1-С7)алкандиил или группу гетероцикл-(С1-С7)алкандиил, где указанная группа, возможно, замещена группой (C1-C6)алкил, при этом группировка (С1-С7)алкандиил связана с группой СНО R4bCHO.
На первом этапе соединение (IX), защищенное на его аминной функциональной группе защитной группой G, конденсируют с соединением (VI). X может представлять собой уходящую группу, например, хлор. В этом случае первый этап состоит из реакции между хлорангидридом и амином. Эта реакция может быть проведена с использованием способов и методик, хорошо известных специалистам в данной области. В одном особо оцененном способе эти две группировки вступают в реакцию в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3, K2CO3, в растворителе, таком как ТГФ, дихлорметан, ДМФА, ДМСО, при температуре, в частности, от -20°C до 100°C. X также может представлять собой гидроксил. В этом случае первый этап представляет собой реакцию конденсации между карбоновой кислотой (IX) и амином (VI). Эта реакция может быть проведена в соответствии со способами и методиками, хорошо известными специалистам в данной области. В одном особо оцененном способе эти две группировки вступают в реакцию в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре, в частности, от -15°C до 40°C. В другом особо оцененном способе эти две группировки вступают в реакцию в присутствии диэтилфосфороцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре от -15°C до 40°C.
После удаления защитной группы с промежуточного продукта с использованием методик, хорошо известных специалистам в данной области, полученное соединение (VIII) может привести к образованию лекарственного средства после реакции с R4Y. В этом случае Y представляет собой уходящую группу, такую как Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил. Реакцию проводят в присутствии органического или неорганического основания, такого как Et3N, iPr2NEt, NaH, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как дихлорметан, ТГФ, ДМФА, ДМСО, при температуре, в частности, от -20°C до 100°C. В другом особо оцененном способе соединение (VIII) подвергают взаимодействию с альдегидом формулы R4b-CHO, где R4b соответствует предшественнику R4. В этом случае реакция представляет собой восстановительное аминирование в присутствии восстанавливающего агента, такого как NaBH4, NaBH3CN, NaBH(OAc)3, в полярном растворителе, таком как 1,2-дихлорэтан, дихлорметан, ТГФ, ДМФА, МеОН, в возможном присутствии изопропоксида титана (IV) при pH, который можно контролировать путем добавления кислоты, такой как уксусная кислота, при температуре, в частности, от -20°C до 100°C.
В приведенных выше схемах синтеза лекарственное средство может привести к образованию другого лекарственного средства после дополнительного реакционного этапа, такого как омыление, например, с использованием способов, хорошо известных специалистам в данной области, в ходе которых группа R2, представляющая собой сложный эфир (СООМе), изменяется на группу R2, представляющую собой карбоновую кислоту (СООН).
Если желательно выделить лекарственное средство, содержащее по меньшей мере одну основную функциональную группу в форме соли присоединения кислоты, это возможно путем обработки свободного основания средства (содержащего по меньшей мере одну основную функциональную группу) подходящей кислотой, предпочтительно, в эквивалентном количестве. Подходящая кислота может быть, в частности, трифторуксусной кислотой.
III - линкер (L)
Понятия "линкер", "линкерный блок" или "L" обозначают в данном изобретении химическую группировку, содержащую ковалентную связь или цепь атомов, которая ковалентно присоединяет антитело по меньшей мере к одному лекарственному средству.
Линкеры могут быть получены с использованием множества бифункциональных агентов, связывающих белки, таких как N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP), сукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилат (SMCC), иминотиолан (IT), бифункциональные производные имидоэфиров (такие как диметиладипимидат HCl), активные сложные эфиры (такие как дисукцинимидилсуберат), альдегиды (такие как глутаральдегид), бис-азидо-соединения (такие как бис(пара-азидобензоил)гександиамин), бис-диазония производные (такие как бис(пара-диазониябензоил)этилендиамин), диизоцианаты (такие как толуол-2,6-диизоцианат) и бис-активные соединения фтора (такие как 1,5-дифтор-2,4-динитробензол). Меченная углеродом-14 1-изотиоцианатобензил-3-метилдиэтилентриаминпентауксусная кислота (MX-DTPA) является примером хелатирующего агента для конъюгации цитотоксических агентов с адресующей системой. Другие связывающие реагенты могут представлять собой BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, МРВН, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, сульфо-EMCS, сульфо-GMBS, сульфо-KMUS, сульфо-MBS, сульфо-SIAB, сульфо-SMCC и сульфо-SMPB и SVSB (сукцинимидил-(4-винилсульфон)бензоат), которые являются коммерчески доступными (например, от Pierce Biotechnology, Inc., Рокфорд, Иллинойс, США).
Линкер может быть "нерасщепляемым" или "расщепляемым".
В предпочтительном воплощении он состоит из "расщепляемого линкера", облегчающего высвобождение лекарственного средства в клетке. Например, может быть использован кислотно-лабильный линкер, чувствительный к пептидазе линкер, фотолабильный линкер, диметиловый линкер или линкер, содержащий дисульфид. В предпочтительном воплощении линкер расщепляется во внутриклеточных условиях таким образом, что расщепление линкера высвобождает лекарственное средство из антитела во внутриклеточную среду.
Например, в некоторых воплощениях линкер расщепляется расщепляющим агентом, который присутствует во внутриклеточной среде (например, в лизосоме, эндосоме или кавеоле). Линкер может представлять собой, например, пептидильный линкер, который расщепляется внутриклеточным ферментом пептидазой или протеазой, в том числе, но не ограничиваясь ими, лизосомальной или эндосомальной протеазой. Как правило, пептидильный линкер содержит по меньшей мере две последовательные аминокислоты или по меньшей мере три последовательные аминокислоты, или имеет по меньшей мере две аминокислоты в длину или по меньшей мере три аминокислоты в длину. Расщепляющие агенты могут включать катепсины В и D и плазмин, которые, как известно, гидролизуют дипептидные производные лекарственных средств, приводя к высвобождению активного лекарственного средства внутри клетки-мишени. Например, может быть использован пептидильный линкер, который расщепляется тиол-зависимой протеазой катепсином В, которая экспрессируется на высоком уровне в раковой ткани (например, линкер, содержащий или представляющий собой Phe-Leu или Gly-Phe-Leu-Gly). В конкретных воплощениях пептидильный линкер, расщепляемый внутриклеточной протеазой, содержит или представляет собой Val-Cit или Phe-Lys. Одно из преимуществ использования внутриклеточного протеолитического высвобождения лекарственного средства заключается в том, что лекарственное средство, как правило, ослабляется при конъюгации, и сывороточная стабильность конъюгатов, как правило, является высокой.
В других воплощениях расщепляемый линкер является pH-чувствительным, т.е. чувствительным к гидролизу при определенных значениях pH. Как правило, pH-чувствительный линкер гидролизуется в кислых условиях. Например, может быть использован кислотно-лабильный линкер, который гидролизуется в лизосомах (например, гидразон, семикарбазон, тиосемикарбазон, цис-аконитовый амид, ортоэфир, ацеталь, кеталь и т.п.). Такие линкеры относительно стабильны в нейтральных условиях pH, таких как условия в крови, но нестабильны при pH ниже 5,5 или 5,0, приближенном к pH лизосом. В некоторых воплощениях гидролизуемый линкер представляет собой тиоэфирный линкер (такой как, например, тиоэфир, прикрепленный к лекарственному средству через ацилгидразоновую связь).
В других воплощениях линкер расщепляется в восстанавливающих условиях (например, дисульфидный линкер). В данной области известно большое число дисульфидных линкеров, включая, например, те, которые могут быть образованы с использованием SATA (N-сукцинимидил-S-ацетилтиоацетата), SPDP (N-сукцинимидил-3-(2-пиридилдитио)пропионата), SPDB (N-сукцинимидил-3-(2-пиридилдитио)бутирата) и SMPT (N-сукцинимидил-алкилоксикарбонил-альфа-метил-альфа-(2-пиридил-дитио)толуола).
В некоторых предпочтительных воплощениях линкерный блок может иметь следующую общую формулу:
-(T)a-(W)w-(Y)y-,
в которой:
Т представляет собой растяжку (растягивающий блок);
а равен 0 или 1;
W представляет собой аминокислотный блок;
w представляет собой целое число от 0 до 12;
Y является спейсерным блоком;
у равен 0, 1 или 2.
Растягивающий блок (Т), если он присутствует, связывает антитело с аминокислотным блоком (W), если он присутствует, или со спейсерным блоком, если он присутствует, или напрямую с лекарственным средством. Используемые функциональные группы, которые могут присутствовать на антителе либо от природы, либо за счет химических манипуляций, включают сульфгидрильную, аминную, гидроксильную, аномерную гидроксильную группу углеводорода и карбоксильную группу. Подходящими функциональными группами являются сульфгидрильные и аминогруппы. Сульфгидрильные группы могут быть получены при восстановлении внутримолекулярных дисульфидных связей антитела, если они присутствуют. В качестве альтернативы, сульфгидрильные группы могут быть получены в результате реакции аминогруппы лизиновой группировки антитела с 2-иминотиоланом или другими сульфгидрил-образующими реагентами. В конкретных воплощениях антитело сконструировано таким образом, чтобы нести один или более чем один лизин. Более предпочтительно, антитело может быть сконструировано таким образом, чтобы нести один или более чем один цистеин (см. ThioMab).
В некоторых конкретных воплощениях растягивающий блок образует связь с атомом серы антитела. Атом серы может быть получен из сульфгидрильной (-SH) группы восстановленного антитела.
В некоторых других конкретных воплощениях растягивающий блок связан с антителом через дисульфидную связь между атомом серы антитела и атомом серы растягивающего блока.
В других конкретных воплощениях реакционноспособная группа растяжки содержит реакционноспособный сайт, который может реагировать с аминогруппой антитела. Аминогруппа может быть таковой группой аргинина или лизина. Подходящие сайты, способные реагировать с аминогруппой, включают, но не ограничиваясь ими, активированные сложные эфиры (такие как сукцинимидные сложные эфиры, 4-нитрофениловые эфиры, пентафторфениловые эфиры), ангидриды, кислые хлориды, сульфонилхлориды, изоцианаты и изотиоцианаты.
В еще одном аспекте реакционноспособная функциональная группа растяжки содержит реакционноспособный сайт, который может вступать в реакцию с модифицированной углеводородной группой, которая может присутствовать на антителе. В конкретном воплощении антитело гликозилируется ферментативно, образуя углеводородную группировку, или является природно гликозилированным. Углеводород может быть слегка окислен таким реагентом как периодат натрия, и полученный в результате карбонильный блок окисленного углеводорода может быть конденсирован с растяжкой, которая содержит функциональную группу, такую как гидразид, оксим, реакционноспособный амин, гидразин, тиосемикарбазид, гидразинкарбоксилат или арилгидразид.
Согласно конкретному воплощению растягивающий блок имеет следующую формулу:
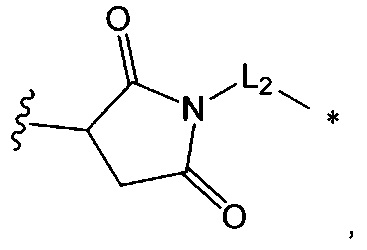
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил или (С2-С6)алкил-карбонил (при этом циклоалкильные или алкильные группировки связаны с атомом азота малеимидной группировки),
звездочка указывает на точку присоединения к аминокислотному блоку, если он присутствует, к спейсерному блоку, если он присутствует, или к лекарственному средству D, и
волнистая линия указывает на точку присоединения к антителу Ab.
Под "(С4-С10)циклоалкилом" в данном изобретении понимается углеводородный цикл, содержащий от 4 до 10 атомов углерода, включая, но не ограничиваясь ими, циклопентил, циклогексил и т.п.
L2 преимущественно может быть (С2-С6)алкил-карбонилом, таким как пентил-карбонил следующей формулы:
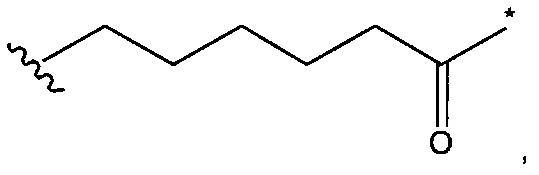
где
звездочка указывает на точку присоединения к аминокислотному блоку, если он присутствует, к спейсерному блоку, если он присутствует, или к лекарственному средству D; и
волнистая линия указывает на точку присоединения к атому азота малеимидной группировки.
Аминокислотный блок (W), если он присутствует, связывает растягивающий блок (Т), если он присутствует, или, напротив, антитело со спейсерным блоком (Y), если спейсерный блок присутствует, и с лекарственным средством, если спейсерный блок отсутствует.
Как упоминалось выше, (W)w отсутствует (w равно 0) или может представлять собой дипептидный, трипептидный, тетрапептидный, пентапептидный, гексапептидный, гептапептидный, октапептидный, нонапептидный, декапептидный, ундекапептидный или додекапептидный блок, где аминокислоты, формирующие указанные пептиды, могут отличаться друг от друга.
Таким образом, (W)w может быть представлен следующей формулой: (W1)w1(W2)w2(W3)W3(W4)W4(W5)w5, где каждый из W1-W5 представляет независимо друг от друга аминокислотный блок, и каждый из w1-w5 составляет 0 или 1.
В некоторых воплощениях аминокислотный блок (W)w может содержат аминокислотные остатки, такие как те, которые являются природными, а также минорные аминокислоты и неприродные аминокислотные аналоги, такие как цитруллин.
Аминокислотные остатки аминокислотного блока (W)w включают, но не ограничиваясь ими, аланин, валин, лейцин, изолейцин, метионин, фенилаланин, триптофан, пролин, защищенный ацетилом или формилом или незащищенный лизин, аргинин, защищенный тозил- или нитро-группами или незащищенный аргинин, гистидин, орнитин, защищенный ацетилом или формилом орнитин и цитруллин. Иллюстративные аминокислотные линкерные компоненты предпочтительно включают дипептид, трипептид, тетрапептид или пентапептид, в частности, дипептид или трипептид.
Примеры дипептидов включают: Val-Cit, Ala-Val, Ala-Ala, Val-Ala, Lys-Lys, Cit-Cit, Val-Lys, Ala-Phe, Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Ala, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg.
Примеры трипептидов включают: Val-Ala-Val, Ala-Asn-Val, Val-Leu-Lys, Ala-Ala-Asn, Phe-Phe-Lys, Gly-Gly-Gly, D-Phe-Phe-Lys, Gly-Phe-Lys.
Примеры тетрапептидов включают: Gly-Phe-Leu-Gly (SEQ ID NO 53), Ala-Leu-Ala-Leu (SEQ ID NO 54).
Примеры пентапептидов включают: Pro-Val-Gly-Val-Val (SEQ ID NO 55).
Согласно конкретному воплощению (W)w может представлять собой дипептид (т.е. w равно 2), такой как Val-Cit, или линкер теряет аминокислотный блок (w равно 0). Когда линкер теряет аминокислотный блок, предпочтительно он также теряет спейсерный блок.
Согласно предпочтительному воплощению w равно 0 (т.е. (W)w представляет собой одиночную связь) или w равно 2 (т.е. (W)w представляет собой дипептид), и (W)w может, таким образом, быть выбран среди:
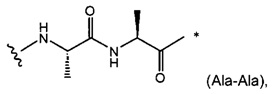
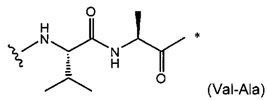 и
и
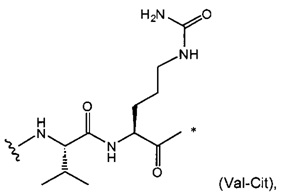
и, в частности, представляет собой Val-Cit,
где
звездочка указывает на точку присоединения к спейсерному блоку, если он присутствует, или к лекарственному средству D; и
волнистая линия указывает на точку присоединения к L2.
Аминокислотные линкерные компоненты могут быть разработаны и оптимизированы по их селективности в отношении ферментативного расщепления конкретным ферментом, например, ассоциированной с опухолью протеазой, катепсином В, С и D и протеазой плазмином.
Для высвобождения лекарственного средства аминокислотный блок линкера может быть ферментативно отщеплен с помощью фермента, включая ассоциированную с опухолью протеазу, но не ограничиваясь ею.
Аминокислотный блок может быть разработан и оптимизирован по его селективности в отношении ферментативного расщепления конкретной ассоциированной с опухолью протеазой. Подходящими блоками являются те блоки, расщепление которых катализируется протеазами, катепсином В, С и D и плазмином.
Спейсерный блок (Y), если он присутствует, связывает аминокислотный блок, если он присутствует, с лекарственным средством. Спейсерные блоки бывают двух основных типов: саморазрушающиеся и несаморазрушающиеся. Несаморазрушающийся спейсерный блок является таким, в котором часть или весь спейсерный блок остается связанным с лекарственным средством после ферментативного отщепления аминокислотного блока из конъюгата "антитело - лекарственное средство". Примеры несаморазрушающегося спейсерного блока включают, но не ограничиваясь ими, спейсерный блок "глицин - глицин" и спейсерный блок "глицин". Для того чтобы высвободить лекарственное средство, внутри клетки-мишени должна произойти независимая реакция гидролиза, расщепляющая связь "глицин - лекарственное средство".
В конкретном воплощении несаморазрушающийся спейсерный блок (Y) представляет собой Gly.
В качестве альтернативы, конъюгат "антитело - лекарственное средство", содержащий саморазрушающийся спейсерный блок, может высвободить лекарственное средство без необходимости отдельного этапа гидролиза. В этих воплощениях (Y) представляет собой остаток пара-аминобензилового спиртового (РАВ) блока, который связан с (W)w через атом азота группы РАВ и соединен непосредственно с лекарственным средством через сложноэфирную, карбонатную, карбаматную или эфирную группу.
Другие примеры саморазрушающихся спейсеров включают, но не ограничиваясь ими, ароматические соединения, которые электронно эквивалентны группе РАВ, такие как остатки производных 2-аминоимидазол-5-метанола и орто- или пара-аминобензилацетали. Могут быть использованы спейсеры, которые легко подвергаются циклизации после гидролиза амидной связи, такие как замещенные и незамещенные амиды 4-аминомасляной кислоты, соответствующим образом замещенные бицикло[2.2.1]- и бицикло[2.2.2]-кольцевые системы и амиды 2-аминофенилпропионовой кислоты.
В альтернативном воплощении спейсерный блок представляет собой разветвленный бис(гидроксиметил)стироловый (BHMS) блок, который может быть использован для включения дополнительных лекарственных средств.
В конкретном воплощении спейсерный блок (Y) представляет собой РАВ-карбонил, при этом РАВ представляет собой 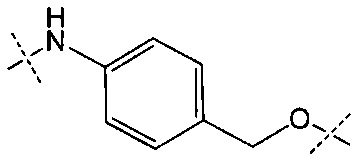 (кислород блока РАВ связан с карбонилом), и у равен 1, или линкер теряет спейсерный блок (у равен 0).
(кислород блока РАВ связан с карбонилом), и у равен 1, или линкер теряет спейсерный блок (у равен 0).
В конкретном воплощении линкер имеет следующую формулу (III):
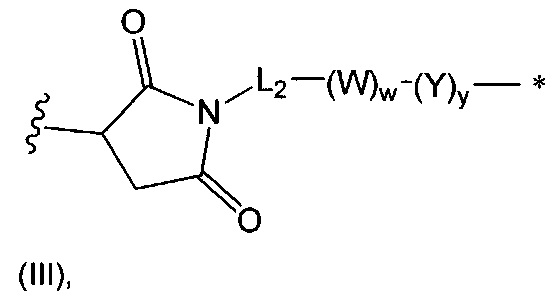
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил или (С2-С6)алкил-карбонил (карбонил этих группировок, если он присутствует, связан с (W)w),
W представляет аминокислотный блок, где w представляет собой целое число в диапазоне от 0 до 5,
Y представляет собой РАВ-карбонил, при этом РАВ представляет собой  (кислород блока РАВ связан с карбонилом), а у составляет 0 или 1 (предпочтительно у составляет 0, когда w составляет 0, и Y составляет 0 или 1, когда w составляет от 1 до 5),
(кислород блока РАВ связан с карбонилом), а у составляет 0 или 1 (предпочтительно у составляет 0, когда w составляет 0, и Y составляет 0 или 1, когда w составляет от 1 до 5),
звездочка указывает на точку присоединения к лекарственному средству D, и
волнистая линия указывает на точку присоединения к антителу Ab.
Преимущественно L2 представляет собой (С2-С6)алкил-карбонил, такой как пентил-карбонил следующей формулы:
где
звездочка указывает на точку присоединения к (W)w; и
волнистая линия указывает на точку присоединения к атому азота малеимидной группировки.
Согласно предпочтительному воплощению линкер L выбран среди:
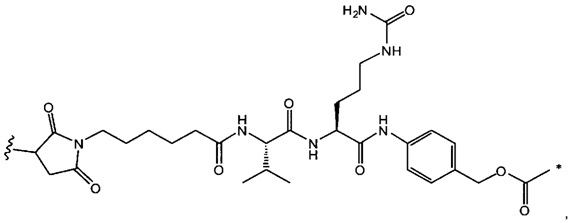
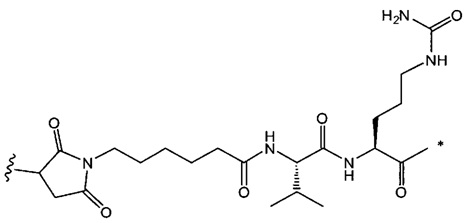 и
и
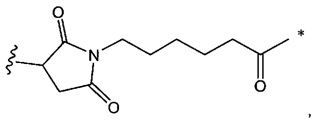
где звездочка указывает на точку присоединения к лекарственному средству D, а волнистая линия указывает на точку присоединения к антителу Ab.
IV - Конъюгат "антитело - лекарственное средство" (ADC)
В предпочтительном воплощении конъюгат "антитело - лекарственное средство" согласно данному изобретению могут быть получены любым способом, известным специалисту в данной области, таким как, но не ограничиваясь ими, i) реакция нуклеофильной группы антитела с двухвалентным линкерным реагентом с последующей реакцией с нуклеофильной группой лекарственного средства или ii) реакция нуклеофильной группы лекарственного средства с двухвалентным линкерным реагентом с последующей реакцией с нуклеофильной группой антитела.
Нуклеофильные группы на антителе включают, но не ограничиваясь ими, N-концевые аминогруппы, аминогруппы боковой цепи (например, лизина), тиоловые группы боковой цепи и гидроксильные или аминогруппы сахаров, когда антитело гликозилировано.
Нуклеофильные группы на лекарственном средстве включают, но не ограничиваясь ими, аминные, тиоловые и гидроксильные группы, предпочтительно аминные группы.
Аминные, тиоловые и гидроксильные группы являются нуклеофильными и способны вступать в реакцию с образованием ковалентных связей с электрофильными группами на линкерных группировках и линкерных реагентах, включая, но не ограничиваясь ими, активные сложные эфиры, такие как сложные эфиры NHS, сложные эфиры HOBt, галогенформиаты и галогениды кислот; алкил- и бензилгалогениды, такие как галогенацетамиды; альдегиды; кетоны; карбоксильные и малеимидные группы. Антитело может иметь восстанавливаемые межцепочечные дисульфиды, т.е. цистеиновые мостики. Антитело можно сделать реакционноспособным для конъюгации с линкерными реагентами путем обработки восстанавливающим агентом, таким как DTT (дитиотреитол). Каждый цистеиновый мостик, таким образом, теоретически будет формировать два реактивных тиоловых нуклеофила. Дополнительные нуклеофильные группы могут быть введены в антитело посредством любой реакции, известной специалистам в данной области. В качестве неограничивающего примера, реакционноспособные тиоловые группы могут быть введены в антитело путем введения одного или более чем одного остатка цистеина.
Конъюгат "антитело - лекарственное средство" также могут быть получены путем модификации антитела введением электрофильных группировок, которые могут вступать в реакцию с нуклеофильными заместителями на линкерном реагенте. Сахара гликозилированного антитела могут быть окислены с образованием альдегидных или кетоновых групп, которые могут вступать в реакцию с аминогруппой линкерных реагентов или лекарственного средства. Полученные в результате иминовые группы оснований Шиффа могут формировать стабильную связь или могут быть восстановлены с образованием стабильных аминных связей. В одном из воплощений реакция углеводородной части гликозилированного антитела либо с галактозооксидазой, либо с метапериодатом натрия может образовывать в белке карбонильные (альдегидные и кетоновые) группы, которые могут взаимодействовать с соответствующими группами на лекарственном средстве. В другом воплощении белки, содержащие N-концевые остатки серина или треонина, могут вступать в реакцию с метапериодатом натрия, что приводит к образованию альдегида на месте первой аминокислоты.
В предпочтительном воплощении конъюгат "антитело - лекарственное средство" согласно данному изобретению получают путем изготовления группировки "лекарственное средство - линкер" с последующим соединением нуклеофильной группы антитела (например, SH-группы цистеиновой группировки) и электрофильной группы группировки "лекарственное средство - линкер" (например, малеимида).
1. Лекарственное средство - линкер
Группировка "лекарственное средство - линкер" может быть получена путем связывания:
- линкера с лекарственным средством,
- части линкера с лекарственным средством до завершения синтеза линкера,
- линкера с частью или предшественником лекарственного средства до завершения синтеза лекарственного средства, или
- части линкера с частью или предшественником лекарственного средства до завершения синтеза линкера и лекарственного средства.
Специалистам в данной области хорошо известны реакции связывания (сочетания) между нуклеофильной группой и электрофильной группой.
Нуклеофильная группа, в частности, может представлять собой аминную, тиольную или гидроксильную группу и, в частности, аминную или гидроксильную группу. В предпочтительном воплощении она представляет собой первичную или вторичную аминогруппу.
Электрофильная группа может представлять собой карбоксильную группу (СООН), возможно, в активированной форме или в форме активированной группировки сложного эфира угольной кислоты.
Под "активированной формой" карбоновой кислоты подразумевают карбоновую кислоту, в которой ОН-группировка функциональной СООН-группы была заменена активированной уходящей группой (leaving group, LG), которая делает возможным связывание активированной карбоксильной группы с аминогруппой с целью формирования амидной связи и высвобождения соединения LG-H. Активированные формы могут представлять собой активированные сложные эфиры, активированные амиды, ангидриды или ацилгалогениды, такие как ацилхлориды. Активированные сложные эфиры включают производные, образуемые взаимодействием карбоксильной группы с N-гидроксибензотриазолом или N-гидроксисукцинимидом.
Под "активированным сложным эфиром угольной кислоты" подразумевают сложный эфир угольной кислоты, содержащий группировку -OC(O)OR, в которой OR представляет собой хорошую уходящую группу, которая делает возможным связывание активированного сложного эфира угольной кислоты с аминогруппой с целью формирования карбаматной группировки и высвобождения соединения ROH. R-группа активированного сложного эфира угольной кислоты включает группы, но не ограничиваясь ими: пара-нитро-фенил, пентафторфенил, 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил и бензил, предпочтительно пара-нитрофенил и пентафторфенил.
Когда линкер имеет следующую формулу (III):
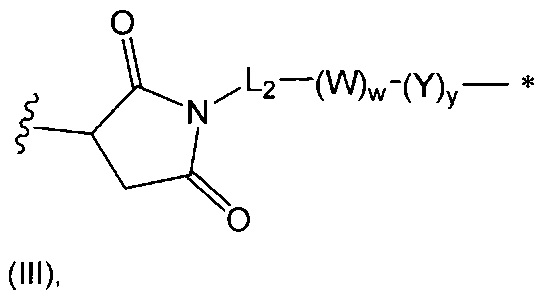
группировка "лекарственное средство - линкер" имеет следующую формулу (IV):
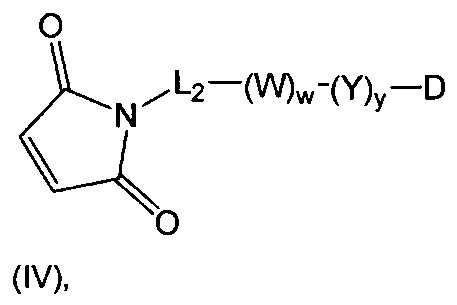
и последний этап синтеза группировки "лекарственное средство - линкер", как правило, представляет собой связывание соединения следующей формулы (V):
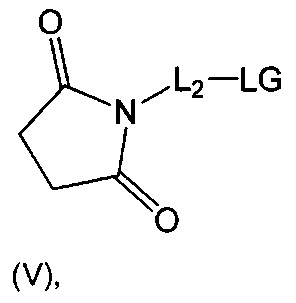
где L2 является таким, как определено ранее, a LG представляет собой уходящую группу, в частности, такую как галогенид, такой как хлорид, или группу, полученную из N-гидроксисукцинимида,
и соединения следующей формулы (VI):
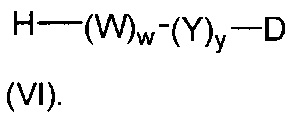
Когда у равен 1 и Y равен РАВ-карбонил, соединение формулы (VI) может быть получено путем связывания лекарственного средства (DH) и соединения следующей формулы (VII) или его защищенной формы:
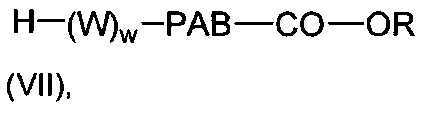
где W и w являются такими, как определено ранее, a R является таким, как описано в определении "активированного сложного эфира угольной кислоты".
Когда соединение формулы (VII) находится в защищенной форме, необходим финальный этап снятия защиты.
Когда у равен 0, соединение (VI) имеет формулу H-(W)w-D, где (W)w и D состоят из аминокислотных блоков. Следовательно, соединение (VI) в этом случае может быть получено путем стандартного пептидного синтеза, хорошо известного специалистам в данной области.
2. Антитело - линкер - лекарственное средство
Предпочтительное воплощение согласно данному изобретению состоит в связывании цистеина, присутствующего на антителе, с электрофильной группой группировки "лекарственное средство - линкер", предпочтительно с малеимидной группировкой, присутствующей на группировке "лекарственное средство - линкер".
Сязывание малеимида и цистеина может быть выполнено способами, хорошо известными специалистам в данной области.
Как правило, антитела не содержат много, если таковые вообще имеются, свободных и реакционноспособных тиольных групп цистеинов, которые могут быть связаны с лекарственной группировкой. Большинство тиольных групп цистеиновых остатков в антителах существуют в виде дисульфидных мостиков и должны быть восстановлены восстанавливающим агентом, таким как дитиотреитол (DTT) или ТСЕР, в частично или полной восстанавливающих условиях. Загрузку (соотношение лекарственное средство/антитело) в ADC можно контролировать несколькими различными способами, включая: (i) ограничение молярного избытка промежуточного продукта "лекарственное средство - линкер" (D-L) или линкерного реагента по отношению к антителу, (ii) ограничение времени реакции конъюгации или температуры, и (iii) частично или ограниченно восстанавливающие условия для модификации тиольных групп цистеинов.
Структура дисульфидных связей человеческих IgG в настоящее время хорошо известна (обзор в Liu and May, mAbs 4 (2012): 17-23). На самом деле существует много общего и некоторые различия, касающиеся структур дисульфидных связей в четырех подклассах человеческих IgG, а именно IgG1, IgG2, IgG3 и IgG4. Все подклассы IgG неизменно содержат 12 внутрицепочечных дисульфидных мостиков, а различия располагаются в их межцепочечных дисульфидных связях, образованных между тяжелыми и легкими цепями. Каждая внутрицепочечная дисульфидная связь ассоциирована с отдельным доменом IgG, т.е. вариабельным (VL и VH) и константным (CL, СН1, СН2 и СН3) доменами. Эти две тяжелые цепи связаны между собой в их шарнирной области переменным числом дисульфидных мостиков: 2 для IgG1 и IgG4, 4 для IgG2 и 11 для IgG3. Тяжелые и легкие цепи IgG1 связаны дисульфидной связью между последним остатком цистеина легкой цепи и пятым остатком тяжелой цепи, в то время как в других подклассах, IgG2, IgG3 и IgG4, легкая цепь связана с тяжелой цепью дисульфидной связью между последним остатком цистеина легкой цепи и третьим остатком цистеина тяжелой цепи, который расположен на стыке доменов VH и СН1. Помимо этих классических структур, для IgG2 и IgG4 были описаны другие структуры дисульфидных связей (обзор в Liu and May, mAbs 4 (2012): 17-23). Внутрицепочечные дисульфидные связи хорошо подвергаются действию растворителя и, следовательно, намного более реакционноспособны, чем внутрицепочечные дисульфидные связи, которые скрыты в антипараллельных бета-складчатых структурах внутри каждого домена и не подвергаются действию растворителя. По этим причинам независимо от изотипа антитела связывание будет происходить на межцепочечных экспонированных остатках цистеина после мягкого восстановления. Таким образом, каждый межцепочечный дисульфидный мостик теоретически может формировать два сайта конъюгации.
Дополнительные нуклеофильные группы можно вводить в антитела посредством реакции лизинов с 2-иминотиоланом (реагент Траута), что приводит к превращению амина в тиол. Реакционноспособные тиольные группы также можно ввести в антитело (или его фрагмент) путем инженерии одного, двух, трех, четырех или более остатков цистеина (например, получая мутантные антитела, содержащие один или более чем один ненативный остаток аминокислоты цистеина). В US 7521541 описана инженерия антител путем введения реакционноспособных аминокислот цистеинов.
Аминокислоты цистеины могут быть сконструированы на реакционноспособных сайтах в антителе, и могут быть такими, которые не образуют внутрицепочечные или межмолекулярные дисульфидные связи (Junutula, et al., 2008b Nature Biotech., 26(8): 925-932; Dornan et al (2009) Blood 114(13): 2721-2729; US 7521541; US 7723485; WO 2009/052249). Инженерные тиольные группы цистеина могут вступать в реакцию с линкерными реагентами или реагентами "лекарственное средство - линкер" согласно данному изобретению, которые имеют тиольно-реакционноспособные электрофильные группы, такие как малеимид или альфа-галогенамиды, для формирования ADC с цистеин-инженерными антителами и лекарственными группировками. Расположение лекарственной группировки, таким образом, можно разработать, контролировать, и оно может быть известно. Лекарственную загрузку можно контролировать, поскольку сконструированные тиольные группы цистеинов, как правило, вступают в реакцию с тиольно-реакционноспособными линкерными реагентами или реагентами "лекарственное средство - линкер" с высоким выходом. Инженерия антитела IgG с введением аминокислоты цистеина путем замещения в одном сайте на тяжелой или легкой цепи дает два новых цистеина на симметричном антителе. Лекарственная загрузка с примерным значением 2 может быть достигнута с прктически гомогенным продуктом конъюгации ADC.
Если более чем одна нуклеофильная или электрофильная группа антитела реагирует с промежуточным продуктом "лекарственное средство - линкер" или с линкерным реагентом, а затем с лекарственной группировкой, то полученный продукт представляет собой смесь соединений ADC с распределением лекарственных группировок, присоединенных к антителу, например 1, 2, 3 и т.д. Способы жидкостной хроматографии, такие как полимерная с обращенной фазой (polymeric reverse phase, PLRP) и хроматография гидрофобного взаимодействия (hydrophobic interaction, HIC), могут разделить соединения в смеси по значению лекарственной загрузки. Можно выделить препараты ADC с одним значением лекарственной загрузки (р), тем не менее, эти ADC с одним значением загрузки по-прежнему могут представлять собой гетерогенные смеси, поскольку лекарственные группировки могут быть присоединены через линкер к различным сайтам на антителе.
Для некоторых конъюгатов соотношение антитела и лекарственного средства может быть ограничено числом участков присоединения на антителе. Высокая лекарственная загрузка, например, соотношение лекарственного средства более 5, может вызвать агрегацию, нерастворимость, токсичность или потерю клеточной проницаемости для определенных конъюгатов "антитело - лекарственное средство". Как правило, в ходе реакции конъюгации с антителом конъюгируется меньшее число молекул лекарственного средства, чем теоретический максимум.
Лекарственная загрузка, которую также называют соотношением "лекарственное средство - антитело" (Drug-Antibody ratio, DAR), представляет собой среднее число молекул лекарственного средства на одну молекулу агента, связывающего клетку.
В случае антитела изотипов IgG1 и IgG4, когда лекарственные средства связываются с остатками цистеина после частичного восстановления антитела, лекарственная загрузка может находиться в диапазоне от 1 до 8 молекул лекарственного средства (D) на молекулу антитела, т.е. 1, 2, 3, 4, 5, 6, 7 и 8 молекул лекарственного средства ковалентно присоединены к антителу.
В случае антитела изотипа IgG2, когда лекарственные средства связываются с цистеинами после частичного восстановления антитела, лекарственная загрузка может находиться в диапазоне от 1 до 12 молекул лекарственного средства (D) на молекулу антитела, т.е. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 и 12 молекул лекарственного средства ковалентно присоединены к антителу.
Композиции ADC включают коллекции агентов, связывающих клетку, например, антител, конъюгированных с диапазоном молекул лекарственного средства от 1 до 8 или от 1 до 12.
Среднее число молекул лекарственного средства на молекулу антитела в препаратах ADC из реакций конъюгации можно охарактеризовать стандартными способами, такими как УФ, обращенно-фазовая HPLC, HIC, масс-спектрометрия, ELISA-анализ и электрофорез.
В качестве неограничивающего воплощения в данном документе представлена конъюгация с IGF-1R-антителом c208F2. В этом случае лекарственное средство связано по меньшей мере с одним цистеином, выбранным среди i) остатка цистеина в позиции 214 для легкой цепи последовательности SEQ ID NO 28, и ii) остатков Cys в позициях 223, 229 и 232 для тяжелой цепи последовательности SEQ ID NO 23.
В качестве неограничивающего воплощения в данном документе представлена конъюгация с IGF-1R-антителом c208F2. В этом случае лекарственное средство связано с двумя, тремя или четырьмя цистеинами, выбранными среди i) остатка цистеина в позиции 214 для легкой цепи последовательности SEQ ID NO 28, и ii) остатков Cys в позициях 223, 229 и 232 для тяжелой цепи последовательности SEQ ID NO 23.
В качестве неограничивающего воплощения в данном документе представлена конъюгация с IGF-1R-антителом hz208F2 (вар. 1). В этом случае лекарственное средство связано по меньшей мере с одним цистеином, выбранным среди i) остатка цистеина в позиции 214 для легкой цепи последовательности SEQ ID NO 39, и ii) остатков Cys в позициях 223, 229 и 232 для тяжелой цепи последовательности SEQ ID NO 37.
В качестве неограничивающего воплощения в данном документе представлена конъюгация с IGF-1R-антителом hz208F2 (вар. 3). В этом случае лекарственное средство связано с двумя, тремя или четырьмя цистеинами, выбранными среди i) остатка цистеина в позиции 214 для легкой цепи последовательности SEQ ID NO 40, и ii) остатков Cys в позициях 223, 229 и 232 для тяжелой цепи последовательности SEQ ID NO 38.
Такой же подход может быть легко адаптирован для других антител, таких как, например, Axl-антитела.
Альтернатива состоит в связывании лизина. Антитело может содержать, например, много остатков лизина, которые не вступают в реакцию с промежуточным продуктом "лекарственное средство - линкер" (D-L) или линкерным реагентом. Только наиболее реакционноспособные группы лизина могут вступать в реакцию с амино-реакционноспособным линкерным реагентом. Кроме того, только наиболее реакционноспособные тиольные группы цистеина могут вступать в реакцию с тиол-реакционноспособным линкерным реагентом.
Когда соединения согласно данному изобретению связаны с лизинами, то лекарственная загрузка может находиться в диапазоне от 1 до 80 молекул лекарственного средства (D) на молекулу антитела, связывающего клетку, хотя предпочтительным может быть верхний предел 40, 20, 10 или 8. Композиции ADC включают коллекции агентов, связывающих клетку, например, антител, конъюгированных с диапазоном молекул лекарственного средства от 1 до 80, от 1 до 40, от 1 до 20, от 1 до 10 или от 1 до 8.
ADC формулы (I) согласно данному изобретению могут находиться в форме фармацевтически приемлемой соли.
В данном изобретении "фармацевтически приемлемый" означает такой, который может быть использован в изготовлении фармацевтической композиции, которая, как правило, является безопасной, нетоксичной и ни биологически, ни иным образом нежелательной, и который является приемлемым для применения в ветеринарии, а также для фармацевтического применения у людей.
Под термином "фармацевтически приемлемая соль" соединения подразумевают соль, которая является фармацевтически приемлемой, как определено в данном документе, и которая обладает необходимой фармакологической активностью исходного соединения.
Фармацевтически приемлемые соли, в частности, включают следующие:
(1) соли присоединения фармацевтически приемлемой кислоты, образованные с помощью фармацевтически приемлемых неорганических кислот, такими как соляная, бромистоводородная, фосфорная, серная и другие подобные кислоты; или образованные с помощью фармацевтически приемлемых органических кислот, таких как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая, молочная и другие подобные кислоты; а также
(2) соли присоединения фармацевтически приемлемого основания, образованные, когда кислотный протон, присутствующий в исходном соединении, либо заменяется ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координируется с фармацевтически приемлемым органическим основанием, таким как лизин, аргинин и т.п.; или образованные с помощью фармацевтически приемлемого неорганического основания, такого как гидроксид натрия, гидроксид калия, гидроксид кальция и т.п.
Эти соли могут быть получены из соединений согласно данному изобретению, содержащих основные или кислотные функциональные группы, а также соответствующих кислот или оснований, с применением стандартных химических способов.
V - Лечение
Наконец, данное изобретение относится к ADC, описанному выше, для применения в качестве лекарственного средства, в частности, для лечения рака.
Еще одним объектом данного изобретения является соединение формулы (I), такое как определено выше, для применения в качестве фармацевтического продукта, в частности, для лечения рака.
Данное изобретение также относится к применению соединения формулы (I), такого как определено выше, для получения фармацевтического продукта, в частности, предназначенного для лечения рака.
Данное изобретение также относится к способу лечения рака, включающему введение человеку, нуждающемуся в этом, эффективного количества соединения формулы (I), такого как определено выше.
Рак предпочтительно может быть выбран среди раков, связанных с мишенью, включающих опухолевые клетки, экспрессирующие или сверхэкспресирующие на своей поверхности мишень целиком или ее часть.
Более конкретно, указанные виды рака представляют собой рак молочной железы, рак толстой кишки, карциному пищевода, гепатоцеллюлярный рак, рак желудка, глиому, рак легких, меланому, остеосаркому, рак яичников, рак предстательной железы, рабдомиосаркому, рак почки, рак щитовидной железы, рак эндометрия матки, шванному, нейробластому, плоскоклеточную карциному ротовой полости, мезотелиому, леймиосаркому, саркому Капоши, острую лейкемию, колоректальную карциному, меланому, протоковую аденокарциному поджелудочной железы и любой рак с лекарственной устойчивостью.
Во избежание сомнений, виды рака с лекарственной устойчивостью, экспрессирующие мишень, следует понимать не только как резистентные виды рака, которые изначально экспрессируют мишень, но и такие виды рака, которые изначально не экспрессировали или не сверхэкспрессировали мишень, но которые начинали экспрессировать мишень, когда они стали устойчивыми к предыдущему лечению.
Другим объектом данного изобретения является фармацевтическая композиция, содержащая ADC, описанный в данном документе.
Более конкретно, данное изобретение относится к фармацевтической композиции, содержащей ADC согласно изобретению по меньшей мере с одним эксципиентом и/или его фармацевтически приемлемым носителем.
В данном описании выражение "фармацевтически приемлемый носитель" или "эксципиент" обозначает соединение или комбинацию соединений, входящих в фармацевтическую композицию, которые не провоцируют побочные реакции и которые, например, облегчают введение активного соединения (соединений), увеличивают продолжительность его жизни и/или его эффективность в организме, увеличивают его растворимость в растворе или также улучшают его сохранность. Эти фармацевтически приемлемые носители и эксципиенты хорошо известны и будут адаптированы специалистами в данной области в зависимости от природы и способа введения выбранного активного соединения (соединений).
Активный ингредиент можно вводить в виде единичных форм введения, в смеси с обычными фармацевтическими носителями, животным или человеку. Подходящие единичные формы введения включают формы для введения пероральным путем и формы для введения парентеральным путем (подкожным, внутрикожным, внутримышечным или внутривенным).
В качестве твердых композиций для перорального введения можно применять таблетки, пилюли, порошки (твердые или мягкие желатиновые капсулы) или гранулы. В этих композициях активный ингредиент согласно изобретению смешивают с одним или более чем одним инертным разбавителем, таким как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния, в потоке аргона. Эти композиции помимо разбавителей также могут содержать другие вещества, например, один или более чем один смазывающий агент, такой как стеарат магния или тальк, краситель, покрывающий агент (таблетки с покрытием) или лак.
Стерильные композиции для парентерального введения предпочтительно могут быть водными или неводными растворами, суспензиями или эмульсиями. В качестве растворителя или носителя может быть использована вода, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое масло, инъекционные органические сложные эфиры, например этилолеат, или другие подходящие органические растворители. Эти композиции также могут содержать адъюванты, в частности, смачивающие, изотонические, эмульгирующие, диспергирующие и стабилизирующие агенты. Стерилизацию можно выполнить несколькими способами, например, путем стерилизующей фильтрации, путем включения стерилизующих агентов в композиции, путем облучения или нагревания. Они также могут быть получены в виде твердых стерильных композиций, которые могут быть растворены в момент применения в стерильной воде или любой другой пригодной для инъекций стерильной среде.
Предпочтительно эти ADC будут вводиться системным путем, в частности, внутривенно, внутримышечно, внутрикожно, внутрибрюшинно или подкожно, либо пероральным путем. В более предпочтительном способе композиция, содержащая ADC согласно изобретению, будет вводиться несколько раз в последовательном порядке.
Таким образом, изобретение также относится к набору, содержащему по меньшей мере i) конъюгат "лекарственное средство - антитело" согласно данному изобретению и/или фармацевтическую композицию согласно данному изобретению, и ii) шприц или флакон или ампулу, в которую помещен указанный конъюгат "лекарственное средство - антитело" и/или фармацевтическая композиция.
Способы введения, дозировки и оптимальные фармацевтические формы могут быть определены в соответствии с критериями, которые обычно принимаются во внимание при разработке лечения, адаптированного для пациента, такими как, например, возраст или вес тела пациента, тяжесть его/ее общего состояния, устойчивость к лечению и отмеченные побочные эффекты.
Другие отличительные признаки и преимущества данного изобретения станут ясны в продолжение описания с примерами и графическими материалами, легенды к которым представлены ниже.
Легенды к графическим материалам
Фиг. 1А-1С: Связывание антитела с человеческим нативным IGF-1R в анализах FACS. Фиг. 1А представляет кривую титрования на клеточной линии MCF-7. MFI представляет собой среднюю величину интенсивности флуоресценции. Фиг. 1 В представляет ЕС50 как мышиных, так и химерных анти-IGF-1R-антител на клеточной линии MCF-7. Фиг. 1С представляет Bmax химерных анти-IGF-1R-антител на клеточной линии MCF-7.
Фиг. 2А-2В: Оценка распознавания hIGF-IR с помощью трансфицированных и нетрансфицированных клеток. Фиг. 2А представляет кривые титрования одного из химерных анти-IGF-1R-антител на клеточной линии IGF-1R+. MFI представляет собой среднюю величину интенсивности флуоресценции. Фиг. 2В представляет связывание химерных анти-IGF-1R-антител на человеческой клеточной линии IGF-1R-.
Фиг. 3А-3В: Оценка специфичности антител против hIGF-1 R и hIR с использованием трансфицированных клеток. Фиг. 3А представляет связывание мышиного анти-IGF-1R-антитела на hIR+-трансфицированной клеточной линии. Фиг. 3В представляет связывание химерного анти-IGF-1R-антитела на IR+ клеточной линии. MFI представляет собой среднюю величину интенсивности флуоресценции. Анти-hIR-MKA GRO5 (Calbiochem) было введено в качестве положительного контроля.
Фиг. 4: Связывание мышиного анти-IGF-1R-антитела на клеточной линии IM-9. MFI представляет собой среднюю величину интенсивности флуоресценции. Анти-hIR-MKA GRO5 было введено в качестве положительного контроля.
Фиг. 5А-5С: Оценка распознавания обезьяньего IGF-1R. Фиг. 5А представляет кривые титрования химерного анти-IGF-1R-антитела на клеточной линии COS-7. MFI представляет собой среднюю величину интенсивности флуоресценции. Фиг. 5В представляет ЕС50 как мышиных, так и химерных анти-IGF-1R-антител на клеточной линии COS-7. Фиг. 5С представляет ЕС50 химерных анти-IGF-1R-антител как на hIGF-1R+-трансфицированных клетках NIH 3Т3, так и на клеточных линиях COS-7.
Фиг. 6: Сенсограммы, полученные по методике SPR на основе Biacore Х100 с использованием сенсорного чипа СМ5, активированного более чем 11000 RU мышиного анти-Tag-His-антитела, химически привитого к карбоксиметилдекстрановой матрице. Эксперимент проводили со скоростью потока 30 мкл/мин при 25°C с использованием HBS-EP+ в качестве рабочего буфера и буфера для разведения образцов. На этой фиг. представлено наложение четырех независимых сенсограмм, выровненных на оси х по началу первого введения анализируемых веществ и на оси у по базальному уровню, определенному непосредственно перед этим первым введением. Сенсограммы, полученные при захвате последовательности на основе человеческого рекомбинантного растворимого IGF-1R, отмечены ромбами. Сенсограммы, полученные при захвате последовательности на основе рекомбинантного растворимого IGF-1R яванского макака, отмечены треугольниками. Белые символы соответствуют пустым циклам (5 введений рабочего буфера), а черные символы соответствуют введениям возрастающего диапазона концентраций c208F2 (5, 10, 20, 40 и 80 нМ).
Фиг. 7: Оценка собственного влияния анти-hIGF-1R-антител на фосфорилирование рецептора по сравнению с IGF1.
Фиг. 8: Ингибирование фосфорилирования IGF-1R в ответ на IGF-1 мышиным анти-hIGF-1R.
Фиг. 9: Интенсивность сигнала связывания анти-IGF-1R-антител на клеточной поверхности снижается после инкубации клеток при температуре 37°C. Клетки MCF-7 инкубировали при 4°C или при 37°C в течение 4 ч с 10 мкг/мл антител. Фигура представляет ΔMFI.
Фиг. 10А-10В: Распад антитела на поверхности. Антитело, связанное с поверхностью клетки, оценивали через 10, 20, 30, 60 и 120 мин при 37°C. На фиг. 10А представлен % от оставшегося IGF-1R в сравнении с интенсивностью сигнала, измеренного при температуре 4°C. Фиг. 10В представляет вычисление периода полужизни с помощью программного обеспечения Prims Software и с использованием настроек экспоненциального распада.
Фиг. 11: Анти-hIGF-1R-антитела интернализированы. Клетки инкубировали с 10 мкг/мл мышиных антител в течение 0, 30 или 60 мин при 37°C. Клетки пермеабилизовали или не пермеабилизовали и инкубировали со вторичным противомышиным антителом IgG, конъюгированным с Alexa 488. Мембранное значение соответствует интенсивности сигнала без пермеабилизации. Общее значение соответствует интенсивности сигнала после клеточной пермеабилизации, а цитоплазматическое значение соответствует интернализированному антителу. Название каждого оцениваемого антитела приведено в верхней части каждого графика.
Фиг. 12А-12В: Визуализация интернализации антитела. Фиг. 12А: клетки MCF-7 инкубировали с m208F2 в течение 20 мин при 4°C и промывали перед инкубацией (W) при 37°C в течение 15 (X), 30 (Y) и 60 (Z) минут. Клетки фиксировали и пермеабилизовали. Антитело m208F2 выявляли с помощью противомышиного IgG-антитела, конъюгированного с Alexa488, a Lamp-1 выявляли с помощью кроличьего анти-Lamp-1-антитела и вторичного противокроличьего IgG, конъюгированного с Alexa 555. Фиг. 12В: клетки MCF-7 инкубировали в течение 30 минут при 37°C с мышиными анти-hIGF-1R-антителами, а затем окрашивали, как описано выше. Колокализацию идентифицировали с помощью плагина для выделения колокализации в программном обеспечении Image J.
Фиг. 13: Участие лизосомального пути в деградации антител.
Фиг. 14: Кислый pH снижает связывающую способность пяти мышиных анти-IGF-1R-антител.
Фиг. 15A-15D: Связывающая характеристика первой гуманизированной формы МКА c208F2. Связывающие свойства МКА hz208F2 VH3A/L3 оценивали на человеческой клеточной линии MCF-7 (А), на обезьяньей клеточной линии COS-7 (В) и на трансфицированной мышиной клеточной линии, экспрессирующей рецептор человеческого инсулина (С). Связывание как мышиных, так и химерных МКА 208F2 оценивали в параллели. Клон анти-hIR-антитела GRO5 использовали для проверки экспрессии hIR на трансфицированной клеточной линии (D).
Фиг. 16: Распад антитела hz208F2 VH3/VL3 на поверхности.
Фиг. 17: Совмещение сенсограмм, полученных с помощью устройства Biacore Х100 на основе SPR при температуре 25°C с сенсорным чипом СМ5, активированным на обеих проточных ячейках с помощью примерно 12000 RU мышиных моноклональных анти-His-Tag-антител, химически привитых к карбоксиметилдекстрановой матрице, с использованием HBS-EP+ в качестве рабочего буфера со скоростью потока 30 мкл/мин. Каждая сенсограмма (первая отмечена треугольниками, а вторая отмечена ромбами) соответствует полному циклу:
1 - введение в течение одной минуты раствора рекомбинантного h-IGF-1R (10 мкг/мл) во вторую проточную ячейку.
2 - для первой сенсограммы: 5 введений рабочего буфера, каждое по 90 с.
- для второй сенсограммы: пять введений растворов анти-IGF-1R-антитела c208F2 с возрастающим диапазоном концентраций, каждое по 90 с.
3 - задержка 300 с для определения кинетических скоростей диссоциации.
4 - регенерация поверхности с помощью введения буфера с 10 мМ глицином, HCl pH 1,5, в течение 45 с.
Фиг. 18: Сенсограмма, соответствующая вычитанию фоновой сенсограммы (5 введений HBS-EP+) из сенсограммы, полученной с растворами анти-IGF-1R-антитела c208F2 с возрастающим диапазоном концентраций, представлена в сером цвете. Теоретическая сенсограмма, соответствующая модели 1:1 со следующими параметрами: kon=(1,206±0,036)×106 М-1.с-1, koff=(7,81±0,18)×10-5 с-1, Rmax=307,6±0,3 RU, представлена тонкой черной линией. Рассчитанные концентрации c208F2 представлены на графике: только самая высокая концентрация (24 нМ) рассматривается как константа.
Фиг. 19: Константы диссоциации соответствуют среднему значению от четырех экспериментов для каждого антитела и соответствуют соотношению: koff/kon×1012, будучи выраженными в единицах "пМ". Столбцы ошибок соответствуют стандартной ошибке (n равен 4).
Фиг. 20: Периоды полужизни соответствуют среднему значению от четырех экспериментов для каждого антитела и соответствуют соотношению: Ln(2)/koff/3600, будучи выраженными в единицах "час". Столбцы ошибок соответствуют стандартной ошибке (n равен 4).
Фиг. 21: Клеточная цитотоксичность анти-IGF-1R, соединенных с тремя различными соединениями. Пять химерных анти-IGF-1R-антител были соединены с Е-13, G-13 или F-63. Нерелевантное антитело c9G4 также соединяли с этими же соединениями.
Фиг. 22А-22С: Оценка in vivo c208F2-E-13 (фиг. 22А), c208F2-G-13 (фиг. 22В) и c208F2-F-63 (фиг. 22С) на модели с ксенотрансплантатом MCF-7.
Фиг. 23А-23В: Оценка in vivo c208F2-E-13 (фиг. 23A) и c208F2-G-13 (фиг. 23В) по сравнению с контрольными ADC (c9G4-E13 и c9G4-G-13) на модели с ксенотрансплантатом MCF-7.
Фиг. 24А, 24В и 24С: Специфичность связывания 1613F12 на иммобилизованном белке rhAxl-Fc (24А), rhDtk-Fc (24В) или rhMer-Fc (24С) в анализе ELISA.
Фиг. 25: FACS-анализ связывания 1613F12 на опухолевых клетках человека.
Фиг. 26: Эксперименты ELISA по изучению связывания белка rhAxl-Fc с m1613F12 и hz1613F12.
Фиг. 27А, 27В и 27С: Иммунофлуоресцентная микроскопия клеток SN12C после инкубации с 1613F12. Фиг. 27А - Фотоснимки условий изотипического контроля mIgG1 как для мембранного, так и для внутриклеточного окрашивания. Фиг. 27В - Мембранное окрашивание. Фиг. 27С - Внутриклеточное окрашивание как рецептора Axl с использованием 1613F12, так и раннего эндосомального маркера ЕЕА1. Ниже представлено наложение изображений, и визуализированная колокализация обозначена стрелками.
Фиг. 28А и 28В: Фиг. 28А представляет цитотоксическую активность hz1613F12-E-13 на обеих клеточных линиях - SN12c (Axl+) и MCF-7 (Axl-). Фиг. 28В представляет цитотоксическую активность hz1613F12-G-13 на обеих клеточных линиях - SN12c (Axl+) и MCF-7 (Axl-).
Фиг. 29А и В: Активность in vivo антитела трастузумаба, конъюгированного с соединениями Е-13 (29А) или G-13 (29В) на модели с ксенотрансплантатом Calu-3.
Фиг. 30А-30С: Активность in vivo антитела трастузумаба, конъюгированного с соединениями Е-13 (фиг. 30А) или G-13 (фиг. 30В) или в одиночку (фиг. 30С) на модели с ксенотрансплантатом JIMT-1.
Примеры
Пример 1: Создание мышиных антител, направленных против ECD IGF-1R
Для создания мышиных моноклональных антител (МКА) против внеклеточного домена (ECD) человеческого рецептора IGF-1 (hIGF-1R) пять мышей BALB/c иммунизировали 3 раза путем подкожного введения 10 мкг белка rhIGF-1R (R&D Systems, кат. №391-GR). В качестве альтернативы, некоторым животным выполняли три дополнительных иммунизации с использованием 10 мкг мышиного внеклеточного домена (ECD) IGF-1R (R&D Systems, кат. №6630-GR/Fc). Первую иммунизацию проводили в присутствии полного адъюванта Фрейнда (Sigma, Сент-Луис, Мэриленд, США). К последующим иммунизациям добавляли неполный адъювант Фрейнда (Sigma). За три дня до слияния иммунизированных мышей бустировали с использованием 10 мкг белка rhIGF-1R. Затем получали спленоциты и лимфоциты путем перфузии селезенки и измельчения проксимальных лимфатических узлов, соответственно, собранных от 1 из 5 иммунизированных мышей (выбранной после титрования сывороток всех мышей), и сливали их с клетками миеломы SP2/0-Ag14 (АТСС, Роквилл, Мэриленд, США). Протокол слияния описан Kohler и Milstein (Nature, 256: 495-497, 1975). Затем гибридные клетки подвергали НАТ-селекции (селекции с использованием гипоксантина, аминоптерина и тимидина). Как правило, для получения моноклональных антител или их функциональных фрагментов, особенно мышиного происхождения, можно обратиться к методикам, которые описаны, в частности, в руководстве "Antibodies" (Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor NY, pp. 726, 1988). Примерно через 10 дней после слияния колонии гибридных клеток подвергали скринингу. Для первичного скрининга супернатанты гибридом оценивали на предмет секреции МКА, выработанных против белка rhIGF-1R ECD, с помощью FACS-анализа с использованием клеток человеческой опухоли молочной железы MCF7 (АТСС) и/или клеток обезьяны COS7 (клетки почки африканский зеленой мартышки, трансформированные SV40), которые экспрессируют обезьяний IGF-1R на своей клеточной поверхности. Более точно, для отбора с помощью проточной цитометрии 105 клеток (MCF7 или COS7) высевали в каждую лунку 96-луночного планшета в PBS, содержащем 1% BSA и 0,01% азид натрия (FACS-буфер), при 4°C. После центрифугирования в течение 2 минут со скоростью 2000 об/мин буфер удаляли и добавляли гибридомные супернатанты, которые нужно было проанализировать. Через инкубации в течение 20 минут при температуре 4°C клетки дважды промывали и добавляли Alexa 488-конъюгированное козлиное противомышиное антитело, разведенное 1/500 в FACS-буфере (№ А11017, Molecular Probes Inc., Юджин, США), и инкубировали в течение 20 минут при температуре 4°C. После финальной промывки FACS-буфером клетки анализировали с помощью FACS (FACSCalibur, Becton-Dickinson) после добавления пропидия йодида в каждую пробирку до конечной концентрации 40 мкг/мл. Лунки, содержащие только клетки, и лунки, содержащие клетки, инкубированные с вторичным Alexa 488-конъюгированным антителом, были включены в качестве отрицательных контролей. В каждом эксперименте использовали изотипические контроли (Sigma, № M90351MG). Для вычисления среднего значения интенсивности флуоресценции (MFI) оценивали по меньшей мере 5000 клеток.
Кроме того, проводили анализ интернализации для того, чтобы выбрать только интернализированные антитела. Для этого анализа линию опухолевых клеток MCF7 культивировали в RMPI 1640 без фенола красного, с 1% L-глутамином и 10% FACS, в течение 3 дней до начала эксперимента. Затем клетки отделяли с помощью трипсина, и 100 мкл клеточной суспензии с плотностью 4×105 клеток/мл высевали в 96-луночные планшеты в RPMI 1640 без фенола красного, с 1% L-глутамином и 5% FBS. После центрифугирования в течение 2 минут со скоростью 2000 об/мин клетки ресуспендировали в 50 мкл либо гибридомных супернатантов, либо растворов контрольных антител (положительные и изотипические контроли в концентрации 1 мкг/мл). После инкубации в течение 20 минут при 4°C клетки центрифугировали в течение 2 минут со скоростью 2000 об/мин и ресуспендировали либо в холодной (4°C), либо в теплой (37°C) полной культуральной среде. Затем клетки инкубировали в течение 2 часов либо при 37°C, либо при 4°C. Затем клетки промывали три раза FACS-буфером. Alexa 488-меченное козлиное противомышиное IgG антитело инкубировали в течение 20 минут и клетки промывали три раза перед FACS-анализом популяции клеток, отрицательных при окрашивании пропидия йодидом.
После FACS-анализа определяли два параметра: (i) разность флуоресцентного сигнала, обнаруженного на поверхности клеток, инкубированных при 4°C, и сигнала, полученного с клетками, инкубированными при 37°C, с супернатантом одной гибридомы и (и) процент оставшегося IGF-1R на поверхности клетки.
Процент оставшегося hIGF-1R рассчитывается следующим образом:
% оставшегося IGF-1R=(MFIAT при 37°С/MFIAT при 4°C)×100.
Помимо этого, проводили три ELISA (либо до, либо после клонирования) для изучения связывания антител с рекомбинантным человеческим (hIGF-1R) и мышиным (mIGF-IR) белками, а также с белком рекомбинантного человеческого рецептора инсулина (hIR). Сохраняли гибридомы, секретирующие антитела, которые демонстрировали связывание с rh- и/или rm-IGF-1R и не связывались с rhIR. Вкратце, 96-луночные планшеты для ELISA (Costar 3690, Corning, Нью-Йорк, США) покрывали 100 мкл/лунка либо раствора белка rhIGF-1R (R&D Systems, кат. №391-GR) с концентрацией 0,6 мкг/мл, либо раствора белка rmIGF-IR (R&D Systems, кат. №6630-GR/Fc) с концентрацией 1 мкг/мл, либо раствора белка rhIR (R&D Systems, кат. №1544-IR/CF) с концентрацией 1 мкг/мл в PBS в течение ночи при температуре 4°C. Затем планшеты блокировали с помощью PBS, содержащего 0,5% желатин (№22151, Serva Electrophoresis GmbH, Гейдельберг, Германия) в течение 2 ч при 37°C. Сразу после удаления буфера насыщения путем стряхивания планшетов в каждую лунку добавляли по 100 мкл каждого разведения супернатанта (либо неразведенного супернатанта от гибридомы, либо серийных разведений супернатантов) и инкубировали в течение 1 ч при 37°C. После трех промываний добавляли 100 мкл конъюгированного с пероксидазой хрена поликлонального козлиного противомышиного антитела IgG (№115-035-164, Jackson Immuno-Research Laboratories, Inc., Вест-Гров, Пенсильвания, США) в разведении 1/5000 в PBS, содержащем 0,1% желатин и 0,05% Tween 20 (по массе), на 1 ч при температуре 37°C. Затем ELISA-планшеты промывали 3 раза и добавляли субстрат ТМВ (№ UP664782, Uptima Interchim, Франция). После 10-минутного периода инкубации при комнатной температуре реакцию останавливали с помощью 1 М серной кислоты и измеряли оптическую плотность при длине волны 450 нм.
Гибридому, секретирующую антитело, представляющее интерес, размножали и клонировали путем серийных разведений. После изотипирования один клон каждого кода размножали и замораживали. Каждое антитело, представляющее интерес, получали в системах продукции in vitro под названием CellLine (Integra Biosciences) для дальнейшей характеризации.
Дополнительные анализы для определения специфичности связывания проводили с помощью FACS-анализов на клетках IM9 (человеческие IR-экспрессирующие В-лимфобласты), а также на hIGF-IR-трансфицированных клетках в сравнении с нетрансфицированными клетками.
Все данные, соответствующие выбранным антителам, приведены в таблице 9, и они показывают, что пять отобранных антител хорошо распознают нативный человеческий IGF-1R, экспрессированный либо на клетках рака молочной железы MCF-7, либо на трансфицированных клетках. Они также распознают обезьяний IGF-1R на клетках COS-7. Эти антитела не вступают в перекрестную реакцию с рецептором инсулина человека, высоко экспрессированного на клетках IM9. Следует заметить, что эти антитела плохо распознают белок rhIGF-1Р ECD, когда ELISA-планшет непосредственно им покрыт.
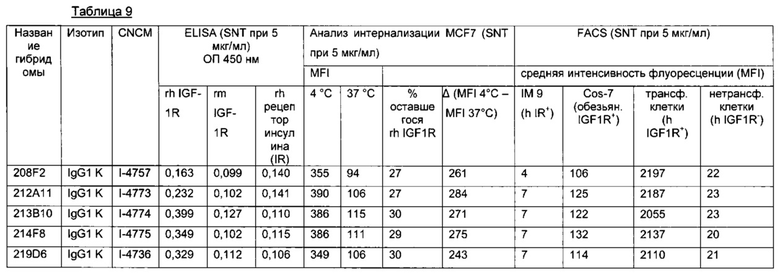
rh - от англ. recombinant human - рекомбинантный человеческий
rm - от англ. recombinant murine - рекомбинантный мышиный
h - от англ. human - человеческий
ОП - оптическая плотность
SNT - от англ. serum neutralization test - реакция сывороточной нейтрализации
Пример 2: Связывание антител с человеческим нативным IGF-1R в FACS-анализах
Пять мышиных IGF-1R-антител были химеризированными. Связывающие свойства и мышиных, и химерных IGF-1R-антител оценивали с помощью FACS-анализов на клеточной линии человеческой аденокарциномы молочной железы MCF-7 (АТСС №НТВ-22) с использованием возрастающих концентраций антитела. Для этой цели клетки (1×106 клеток/мл) инкубировали с анти-IGF-1R-антителами в течение 20 мин при 4°C в FACS-буфере (PBS, 0,1% BSA, 0,01% NaN3). Затем их промывали 3 раза и инкубировали с соответствующим вторичным антителом, конъюгированным с Alexa 488, в течение 20 дополнительных минут при 4°C в темноте, а затем промывали 3 раза FACS-буфером. Связывание анти-IGF-1R-антител немедленно проводили на жизнеспособных клетках, которые идентифицировали с помощью пропидия йодида (который окрашивает мертвые клетки). Максимум интенсивности сигнала, полученный с каждым антителом, обозначали как Bmax и выражали в средней интенсивности флуоресценции (MFI). ЕС50 связывания, выраженную в молярности (М), рассчитывали с помощью нелинейного регрессионного анализа (GraphPad Prims 4.0).
Кривые титрования каждого мышиного или химерного антитела показали, что все созданные антитела способны распознавать нативную форму IGF-1R с типичным профилем насыщения (фиг. 1А). Для того чтобы ранжировать антитела и сравнить связывающие свойства мышиных и химерных антител, для каждого соединения определяли ЕС50 связывания с использованием нелинейного регрессионного анализа. Сравнение ЕС50 каждого мышиного антитела с ЕС50 его соответствующей химерной формы показало, что две формы демонстрировали одни и те же связывающие свойства, показывая, что химеризация антитела не влияла на распознавание IGF-1R (фиг. 1В-С). Значения ЕС50 и Bmax химерных антител приведены в таблице 10.
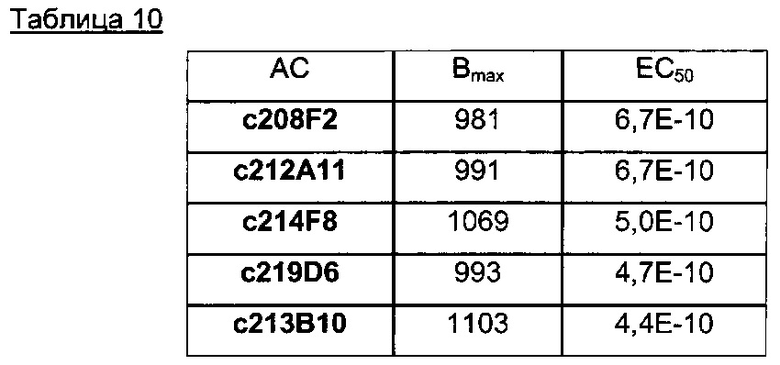
Пример 3: Подтверждение специфичности антитела с использованием IGF-1R- или IR-трансфицированных клеток или клеток IM9, которые экспрессируют существенные уровни IR
Для того чтобы подтвердить специфичность созданных антител в отношении IGF-1R по сравнению с IR, с помощью FACS-анализов оценивали стабильные трансфектанты, экспрессирующие либо hIGF-1R, либо hIR. Вкратце, возрастающие концентрации химерных МКА инкубировали с клетками в течение 20 минут при 4°C в FACS-буфере (PBS, 0,1% BSA, 0,01% NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторичным антителом, конъюгированным с Alexa 488, и инкубировали в течение 20 дополнительных минут при 4°C в темноте, а затем промывали 3 раза FACS-буфером. Связывание анти-IGF-1R-антител немедленно проводили на жизнеспособных клетках, которые идентифицировали с помощью пропидия йодида (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), рассчитывали с помощью нелинейного регрессионного анализа (GraphPad Prims 4,0).
Кривые титрования, полученные на hIGF-1R-трансфицированной клеточной линии (фиг. 2А) по сравнению с нетрансфицированными клетками (фиг. 2В) подтвердили специфичность связывания химерных антител с человеческим IGF-1R. Значения ЕС50 и Bmax химерных антител приведены в таблице 11.
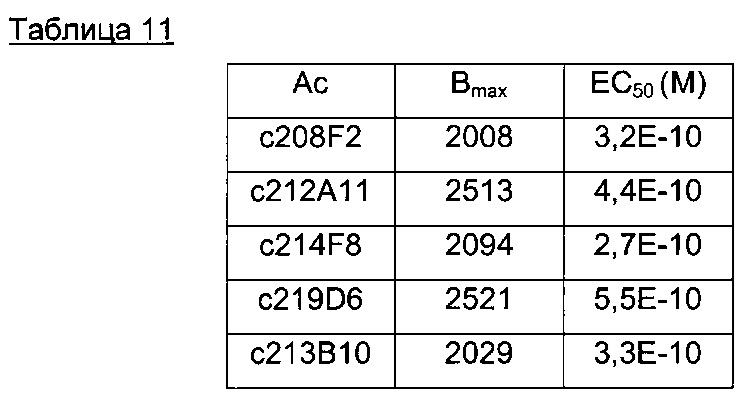
Для того чтобы подтвердить отсутствие связывания мышиных и химерных антител с hIR, использовали клеточную линию, стабильно экспрессирующую человеческий IR (hIR). Распознавание человеческого hIR на клеточной поверхности как мышиным, так и химерным антителом проводили путем FACS-анализов. Возрастающие концентрации мышиных или химерных МКА инкубировали с hIR+-трансфицированной клеточной линией в течение 20 минут при 4°C в FACS-буфере (PBS, 0,1% BSA, 0,01% NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторичным антителом, конъюгированным с Alexa 488, и инкубировали в течение 20 дополнительных минут при 4°C в темноте, а затем промывали 3 раза в FACS-буфере. Связывание анти-IGF-1R-антител немедленно проводили на жизнеспособных клетках, которые идентифицировали с помощью пропидия йодида (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), рассчитывали с помощью нелинейного регрессионного анализа (GraphPad Prims 4.0). Анти-hIR-антител о, клон GRO5, использовали в качестве положительного контроля. Мышиные и химерные антитела c9G4 вводили в качестве нерелевантных антител.
Высокий уровень экспрессии hIR на клеточной поверхности трансфицированных клеток подтверждали с использованием коммерческого анти-hIR-антитела GRO5 (фиг. 3А и 3В). Даже с использованием высоких концентраций либо мышиного (фиг. 3А), либо химерного (фиг. 3В) анти-hIGF-IR-антител никакого связывания на клеточной поверхности hIR+-трансфицированных клеток не наблюдалось. Эти результаты показали, что ни мышиное, ни химерное анти-hIGF-IR-антитела не распознавали hIR.
Эта специфичность распознавания hIGF-1R в сравнении с IR также была продемонстрирована в FACS-анализах с использованием клеток IM9, клеточной линии В-лимфомы, которая экспрессирует hIR (фиг. 4). Для этих FACS-анализов протокол был таким же, как описанный выше, а мышиные антитела использовали для того, чтобы предотвратить перекрестную реактивность вторичного противочеловеческого антитела (клетки IM9 экспрессируют человеческий иммуноглобулин на своей клеточной поверхности). Результаты, представленные на фиг. 4, еще раз показали, что ожидаемый сигнал наблюдался при использовании анти-hIR-антитела GRO5, в то время как ни одно из оцениваемых мышиных антител не демонстрировало никакого существенного сигнала связывания на этой клеточной линии.
Пример 4: Связывание антитела с нативным обезьяньим IGF-1R в анализах FACS и Biacore
Одной из первых предпосылок для контрольных токсикологических исследований является выявление релевантного вида животного для оценки выбранного соединения. Поскольку серия антител, описанных в данном документе, не способна распознавать мышиный IGF-1R, то, скорее всего, видом для токсикологической оценки будет примат, не являющийся человеком (nоn human primate, NHP).
Для того чтобы оценить связывание анти-IGF-1R-антител с обезьяньим IGF-1R, оценивали связывание как мышиных, так и химерных анти-hIGF-IR-антител с помощью FACS-анализов на линии клеток COS-7 с использованием возрастающих концентраций антител. Клетки (1×106 клеток/мл) инкубировали с анти-IGF-1R-антителами в течение 20 минут при 4°C в FACS-буфере (PBS, 0,1% BSA, 0,01% NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторичным антителом, конъюгированным с Alexa 488, а затем инкубировали в течение 20 дополнительных минут при 4°C в темноте, и затем промывали 3 раза в FACS-буфере. Связывание анти-IGF-1R-антител немедленно проводили на жизнеспособных клетках, которые идентифицировали с помощью пропидия йодида (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), рассчитывали с помощью нелинейного регрессионного анализа (GraphPad Prims 4.0).
Кривые титрования, полученные на линии клеток обезьян COS-7, показали, что все анти-hIGF-IR-антител а специфически распознавали IGF-1R, экспрессированный на поверхности линии клеток обезьян (фиг. 5А). Определение ЕС50 для каждого из мышиных и химерных антител показало, что две формы были сравнительно хорошими касательно их свойств связывания с обезьяньим IGF-1R (фиг. 5В). Эти результаты показали, что все созданные анти-hIGF-IR-антитела распознают обезьяний IGF-1R.
Сравнение ЕС50 связывания на клетках COS-7 и IGF-1R-трансфицированных клетках проводили с целью проверки величины распознавания химерным антителом человеческого IGF-1R по сравнению с обезьяньим IGF-1R. Результаты, показанные на фиг. 5С, продемонстрировали аналогичное распознавание человеческого и обезьяньего IGF-1R всеми антителами.
Для того чтобы подтвердить распознавание на другом типе обезьян, клетки трансфицировали IGF-1R от яванского макака, получая растворимый обезьяний IGF-1R ECD, и выполняли эксперименты Biacore с одним из химерных антител (c208F2), чтобы сравнить его связывающие свойства либо в отношении hIGF-IR, либо в отношении IGF-1R яванского макака.
Эксперименты по распознаванию проводили на устройстве Biacore X с использованием сенсорного чипа СМ5, активированного анти-His-Tag-антителом (набор His Capture Kit от GE Healthcare, кат. номер 28-9950-56). Более 11000 RU антител химически присоединяли к карбоксиметилдекстрановой матрице с использованием набора для аминной химической реакции. Эксперименты проводили при 25°C со скоростью потока 30 мкл/мин с использованием буфера HBS-EP (GE Healthcare) в качестве рабочего буфера и буфера для разведения образцов. Кинетическую схему с одним циклом использовали для определенных кинетических параметров связывания химерной формы анти-IGF-1R-антитела 208F2 (c208F2) на hIGF-1R в сравнении с IGF-1R макаки.
Раствор растворимого рекомбинантного варианта гетеротетрамера hIGF-IR, состоящего из цепей 2α и внеклеточных доменов цепей 2β, экспрессированных с дополнительной C-концевой 10-His-меткой, основанного либо на человеческой последовательности (R&D Systems, кат. номер 305-GR-50), либо на последовательности яванского макака (получен самостоятельно), вводили в течение 1 минуты во вторую проточную ячейку в разведении, определенном для захвата примерно 160 RU антигена. После фазы захвата 5 раз вводили либо рабочий буфер (каждое введение по 90 с), либо возрастающий диапазон пяти концентраций c208F2 (каждое введение по 90 с) в обе проточные ячейки. В конце пятого введения прогоняли рабочий буфер для определения скорости диссоциации.
Затем поверхность подвергали регенерации путем введения буфера с 10 мМ глицином, HCl, pH 1,5, в течение 30 с.
Вычисленный сигнал соответствует разности между ответом из проточной ячейки 2 (с захваченным IGF-1R) и ответом из проточной ячейки 1 (без каких-либо молекул IGF-1R) (фиг. 6).
Для каждой молекулы IGF-1R (человека или обезьяны) сигнал от введения возрастающего диапазона концентраций c208F2 корректировали путем вычитания сигнала, полученного с 5 введениями буфера (двойной референс). Полученные сенсограммы анализировали с использованием программного обеспечения Biaevaluation с моделью 1:1. Кинетические показатели оценивали либо независимо (2 кинетические скорости связывания c208F2 на каждом IGF-1R), либо вместе (одни и те же кинетические скорости связывания c208F2 на IGF-1R человека и яванского макака). Качество подгонки оценивали по соотношению Chi2/Rmax ниже 0,05 RU.
Кинетические скорости связывания (см. таблицу 12), определенные по отдельности для каждого IGF-1R, были близкими, и подгонка обеих сенсограмм с одинаковыми кинетическими скоростями имеет хорошее качество.
Антитело c208F2 распознает рекомбинантные IGF-1R человека и яванского макака с константой диссоциации (KD) примерно 0,2 нМ. Аффинности, определенные в данном исследовании, соответствуют функциональным аффинностям (авидностям) антител с уровнем захваченного IGF-1R человека и яванского макака примерно 160 RU.
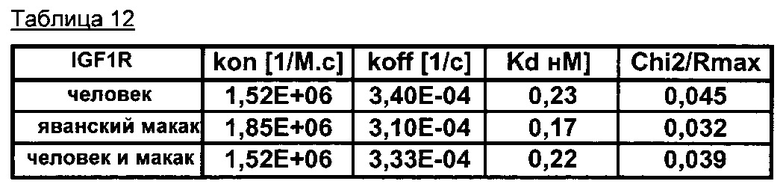
Пример 5: Собственное влияние созданных антител на фосфорилирование IGF-1R
Хорошо известно, что антитела могут индуцировать агонистическое действие, когда они связываются с тирозинкиназными рецепторами. Поскольку мы не хотели выбирать такие антитела-агонисты, мы проводили оценку фосфорилирования hIGF-IR с использованием химерных антител.
Для этой цели клетки MCF-7 инкубировали в бессывороточной среде в течение ночи. Затем добавляли либо IGF-1 (100 нМ), либо анализируемые антитела (10 мкг/мл) на 10 минут при 37°C. Среду отбрасывали и клетки соскабливали в лизирующий буфер (pH 7,5), содержащий 10 мМ буфер Tris-HCl (pH 7,5), 15% раствор NaCl (1 М), 10% смесь детергентов (10 мМ Tris-HCl, 10% лизирующий буфер Igepal) (Sigma Chemical Co.), 5% дезоксихолат натрия (Sigma Chemical Co.), 1 таблетку смеси ингибиторов протеаз ТМ (Roche), 1% смесь ингибиторов фосфатаз Cocktail Set II (Calbiochem), на 90 минут при температуре 4°C. Лизаты осветляли центрифугированием при 4°C, нагревали в течение 5 минут при 100°C и хранили при -20°C или сразу загружали в 4-12% SDS-PAGE-гели. Инкубацию с первичным антителом проводили в течение 2 ч при комнатной температуре, а затем проводили инкубацию с HRP-связанными вторичными антителами в течение 1 ч при комнатной температуре. Мембраны промывали в TBST, а затем белки визуализировали с помощью ECL. Блоты оценивали количественно, используя программное обеспечение Image J. Значения "фосфор - белок" нормировали по GAPDH. Фосфорилирование hIGF-1R в ответ на IGF-1 рассматривали как 100% стимуляции. Влияние анти-hIGF-IR-антител на фосфорилирование hIGF-1R определяли как % фосфорилирования, индуцированный IGF-1.
Результаты, описанные на фиг. 7, представляют собой среднее значение от % pIGF-1R в ответ на химерные анти-IGF-1R-антитела из трех независимых экспериментов плюс/минус S.D. (от англ. standard deviation - среднеквадратическое отклонение) в сравнении с IGF-1. Как было показано, при инкубации клеток MCF-7 с 10 мкг анти-IGF-1R-антител не было обнаружено никакого существенного или минорного (менее 20%) фосфорилирования hIGF-1R.
Пример 6: Ингибирование фосфорилирования IGF-1R в ответ на IGF-1 мышиными анти-hIGF-IR-антителами
Чтобы охарактеризовать выбранные антитела, исследовали их способность ингибировать IGF1-индуцированное фосфорилирование. Для этой цели клетки MCF-7 инкубировали в бессывороточной среде в течение ночи. Затем клетки инкубировали в течение 5 минут с мышинымИ анти-hIGF-IR-антителами, а затем добавляли IGF-1 на 2 минуты при 37°C. Среду отбрасывали и клетки соскабливали в лизирующий буфер (pH 7,5), содержащий 10 мМ буфер Tris-HCl (pH 7,5), 15% раствор NaCl (1 М), 10% смесь детергентов (10 мМ Tris-HCl, 10% лизирующий буфер Igepal) (Sigma Chemical Co.), 5% дезоксихолат натрия (Sigma Chemical Co.), 1 таблетку смеси ингибиторов протеаз ТМ (Roche), 1% смесь ингибиторов фосфатаз Cocktail Set II (Calbiochem), на 90 минут при температуре 4°C. Лизаты осветляли центрифугированием при 4°C, нагревали в течение 5 минут при 100°C и хранили при -20°C или сразу загружали в 4-12% SDS-PAGE-гели. Инкубацию с первичным антителом проводили в течение 2 ч при комнатной температуре, а затем проводили инкубацию с HRP-связанными вторичными антителами в течение 1 ч при комнатной температуре. Мембраны промывали в TBST, а затем белки визуализировали с помощью ECL. Блоты оценивали количественно, используя программное обеспечение Image J. Значения "фосфор-белок" нормировали по GAPDH. Фосфорилирование hIGF-1R в ответ на IGF-1 рассматривали как 100% стимуляции. Влияние анти-hIGF-IR-антител на фосфорилирование hlGF-1R определяли как % фосфорилирования, индуцированный IGF-1.
Все анти-IGF-1R-антитела сильно ингибировали фосфорилирование hIGF-1R в ответ на IGF-1 (снижение более 80%) (фиг. 8). Лучшими ингибиторами IGF1-индуцированного фосфорилирования hlGF-1R являются МКА m208F2, m212A11 и m214F8.
Пример 7: Исследование интернализации IGF-1R после связывания с созданными анти-IGF-1R-антителами с помощью FACS-анализов
Клетки MCF-7 инкубировали с 10 мкг/мл химерных антител при 4°C в течение 20 минут. Затем клетки промывали и инкубировали при 4°C или 37°C в течение 4 ч. Количество антител, связанных с клеточной поверхностью, определяли с помощью вторичного антитела. Значение ΔMFI, которое определяли как разность между MFI, измеренной при 4°C, и MFI, измеренной при 37°C, по истечении периода инкубации 4 часа, соответствовало количеству интернализированного антитела. Значения ΔMFI представлены на фиг. 9 и в таблице 11. Процент интернализации при 10 мкг/мл антитела рассчитан как 100*(MFI при 4°C - MFI при 37°C)/MFI при 4°C и представлен в таблице 13.
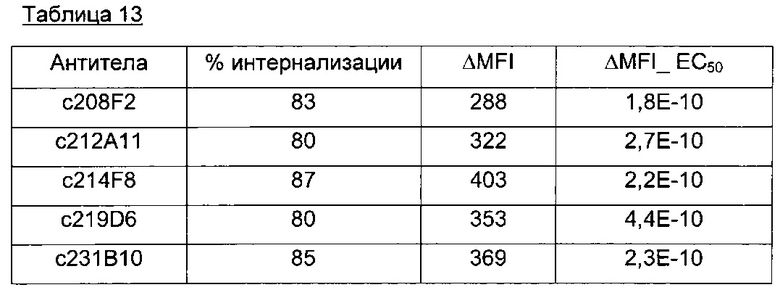
Чтобы определить, могут ли антитела, которые также распознают обезьяний IGF-1R, интернализовать этот рецептор, проводили такой же эксперимент по интернализации. Результаты, приведенные в таблице 14, показывают, что все проанализированные антитела способны опосредовать интернализацию обезьяньего IGF-1R.
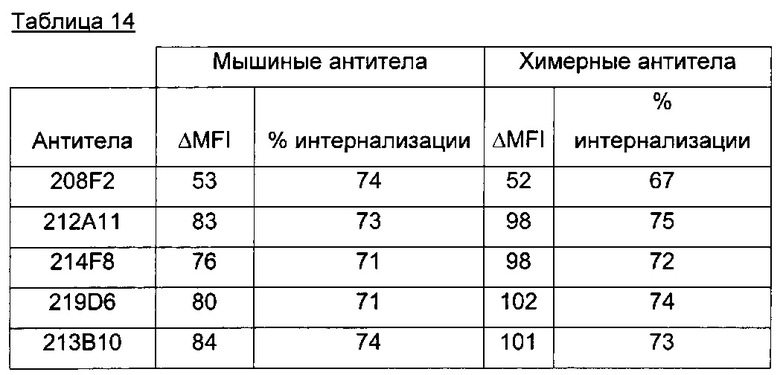
Также оценивали кинетику уменьшения количества антитела, связанного с клеточной поверхностью. С этой целью клетки MCF-7 высевали в 96-луночные планшеты и инкубировали с 10 мкг/мл мышиного антитела в течение 20 минут при температуре 4°C. Затем клетки промывали для удаления несвязанного антитела и в средах при температуре 37°C в течение 10, 20, 30, 60 или 120 мин. В каждый момент времени клетки центрифугировали, а затем на льду метили поверхность вторичным противомышиным антителом IgG-Alexa488 для определения количества антитела, оставшегося на поверхности клетки. Интенсивность флуоресценции для каждого мышиного антитела и для каждой временной точки нормировали по сигналу, полученному при 4°C (% оставшегося IGF-1R), и подводили к экспоненциальному затуханию, чтобы определить период полужизни (t1/2). t1/2 рассматривали как время, необходимое для уменьшения сигнала на 50%. Как показано на фиг. 10, поверхностный уровень всех мышиных антител быстро падал в течение первых 30 минут, и снижение было почти максимальным через 60 минут инкубации (фиг. 10А). Рассчитанный период полужизни находился в диапазоне от 10 до 18 мин касательно мышиного антитела (фиг. 10В).
Для того чтобы подтвердить, что уменьшение сигнала с клеточной поверхности обусловлено интернализацией антитела, а не слущиванием рецептора, клетки инкубировали с мышиными антителами в течение 0, 30 и 60 мин при 37°C (фиг. 11). Затем клетки фиксировали и пермеабилизовали или не пермеабилизовали для того, чтобы определить сигнал от антитела, связанного с клеточной поверхностью (без пермеабилизации), и общий сигнал от антитела, который соответствует антителу, связанному с клеточной поверхностью плюс интернализированному антителу (с пермеабилизацией). Количество интернализированного антитела (цитоплазматический сигнал) определяли следующим образом: MFI после пермеабилизации - MFI без пермеабилизации. Этот эксперимент показал, что уменьшение антитела, связанного с клеточной поверхностью, было связано с увеличением цитоплазматических антител, доказывая, что антитела были интернализованы (фиг. 11). Кроме того, деградация антител начиналась через 1 ч инкубации, что показано уменьшением сигнала после пермеабилизации (общий сигнал).
Пример 8: Исследование интернализации IGF-1R после связывания созданных анти-IGF-1R-антител путем конфокальных анализов
Для дальнейшего подтверждения интернализации антител проводили конфокальную микроскопию, чтобы оценить субклеточное распределение антител после клеточного перемещения. Клетки инкубировали с анти-hIGF-IR-антителами при 37°C, фиксировали и пермеабилизировали. Затем клетки окрашивали с помощью вторичного антитела, конъюгированного с Alexa-488, и кроличьего анти-lamp-1-антитела, которое обнаруживали с помощью вторичного противокроличьего антитела IgG, конъюгированного с Alexa 555. Перед инкубацией при 37°C мышиное антитело 208F2 локализовали на мембране клеток MCF-7 (фиг. 12А). При использовании плагина для выделения колокализации в программном обеспечении Image J с лизосомным маркером, lamp-1, колокализация не была выявлена. Количество связанных с поверхностью клетки антител резко уменьшалось после 15-минутной инкубации при 37°C. Одновременно с уменьшением количества антитела, связанного с клеточной поверхностью, внутриклеточное антитело определялось в везикулах. Можно было наблюдать редкую колокализацию с lamp-1. После 30-минутной инкубации антитело, связанное с клеточной поверхностью, почти не обнаруживалось. Тем не менее, колокализация антитела в лизосомах увеличивалась. Через 1 ч инкубации внутриклеточное окрашивание антитела уменьшалось, как и уровень колокализации с lamp-1. Кинетика антитела, связанного с клеточной поверхностью, и его внутриклеточное накопление коррелировали с кинетикой распада антитела на поверхности, измеренной путем FACS. Кроме того, как уже было описано в FACS-исследованиях, деградация мышиных антител начиналась через 1 ч инкубации по результатам конфокальной микроскопии.
Также оценивали интернализацию всех других мышиных hIGF-IR-антител и их колокализацию с lamp-1 (фиг. 12В). После 30-минутной инкубации при 37°C внутриклеточное антитело обнаруживалось, и можно было наблюдать колокализацию с lamp-1, что указывало на то, что все выбранные анти-IGF-1R-антитела были эффективно интернализованы в лизосомы.
Пример 9: Ингибирование деградации антител с помощью лизосомального ингибитора, бафиломицина А1
Для того чтобы подтвердить, что антитела, которые достигли лизосомы, были деградированы, клетки обрабатывали или не обрабатывали бафиломицином А1, сильным ингибитором лизосомфальных функций. Затем клетки инкубировали с 10 мкг/мл анализируемого антитела при 4°C, промывали и инкубировали в течение 2 ч при 37°C. Интернализированное антитело обнаруживали после клеточной пермеабилизации с использованием вторичного противомышиного антитела IgG? конъюгированного с Alexa 488. Добавление бафиломицина А1 предотвращало деградацию внутриклеточного антитела (фиг. 13), что указывало на то, что антитела были эффективно интернализованы и деградировали в лизосомах.
Пример 10: Влияние pH на связывание IGF-1R-антитела
Поскольку антитела выбирали на основании их потенциала интернализации, и ранее была показана колокализация с ранними эндосомами перед входом в лизосомальный компартмент, интересующий подход заключался в выборе антител, для которых стабильность связывания антитело/hIGF-1R модулировалась относительно pH среды, и предпочтительными являются такие антитела, которые предпочтительно диссоциируют от IGF-1R тогда, когда pH среды становится кислым. В самом деле, основное различие между ранними эндосомами и лизосомами заключается в их полостном pH: в эндосомальном компартменте pH составляет примерно 6, в то время как в лизосомальном компартменте pH составляет примерно 4,5.
Хорошо известно, что hIGF-IR, однажды интернализированный после связывания лиганда (IGF1), возвращается обратно на поверхность клетки через путь рециркуляции.
Без связи с какой-либо теорией, описанная в данном документе гипотеза заключается в том, что антитела, более склонные к раннему освобождению от их мишеней при кислом pH, вероятно, будут благоприятствовать рециркуляции мишени к мембране, и, следовательно, их можно рассматривать в качестве более подходящих кандидатов для подходов с использованием ADC. Для того чтобы исследовать, демонстрируют ли какие-либо из наших антител такое свойство, и чтобы соотнести это свойство с цитотоксической активностью, было проведено связывание мышиных анти-hIGF-IR-МКА на клеточной линии MCF-7 в буферах с различными значениями pH. Возрастающие концентрации мышиных МКА инкубировали на клеточной линии MCF-7 в течение 20 минут при 4°C и при различных pH в диапазоне от 5 до 8. Затем эти клетки промывали 3 раза и инкубировали с соответствующим вторичным антителом, конъюгированным с Alexa 488, в FACS-буфере. Клетки инкубировали в течение 20 дополнительных минут при 4°C в темноте, а затем промывали 3 раза FACS-буфером. Связывание анти-hIGF-1R-антител немедленно проводили на жизнеспособных клетках, которые идентифицировали с помощью пропидия йодида, который окрашивает мертвые клетки. ЕС50 связывания, выраженную в молярности (М), рассчитывали с помощью нелинейного регрессионного анализа (GraphPad Prims 4,0). Все выбранные мышиные анти-IGF-1R-антитела показали более низкую связывающую способность при кислом pH, что показано на фиг. 14.
Пример 11: Оценка гуманизированной формы МКА 208F2
Связывание первой гуманизированной формы МКА c208F2 оценивали на IR+ клеточных линиях MCF-7, COS-7 и NIH 3Т3. Возрастающие концентрации m208F2, c208F2 или hz208F2 VH3VL3 добавляли к каждой клеточной линии на 20 минут при 4°C. Затем клетки промывали и связывание анализируемого МКА обнаруживали с помощью соответствующего вторичного антитела. Для валидации экспрессии человеческого IR на трансфицированной клеточной линии использовали коммерческое анти-hIR-антитело, клон GRO5, и его профиль распознавания, проиллюстрированный на фиг. 15D.
Сравнение гуманизированной формы либо с мышиной, либо с химерной формой на MCF-7 (фиг. 15А) или на обезьяньих клетках COS-7 (фиг. 15В) показало близкие профили для трех проанализированных форм. Процесс гуманизации не изменял специфичность распознавания антитела, которая была отлично сравнима с мышиной и химерной формами касательно отсутствия перекрестной реактивности с рецептором человеческого инсулина (фиг. 15С).
Рассчитанные ЕС50 первой гуманизированной формы 208F2 на линии клеток человека MCF-7 и линии клеток обезьяны COS-7 были похожи на те, которые были определены для мышиной или химерной формы МКА 208F2.
Способность МКА hz208F2 VH3/VL3 быть интернализированным оценивали с помощью проточной цитометрии. Клетки MCF-7 инкубировали с 10 мкг/мл антител при 4°C в течение 20 мин. Затем клетки промывали и инкубировали при 4°C или 37°C в течение 4 ч. Количество антител, связанных с клеточной поверхностью, определяли с помощью вторичного антитела. Значение ΔMFI, которое определяли как разность между MFI, измеренной при 4°C, и MFI, измеренной при 37°C, по истечении периода инкубации 4 часа, соответствовало количеству интернализированного антитела. Значения ΔMFI представлены на фиг. 16 и в таблице 13. Процент интернализации при 10 мкг/мл антитела рассчитан как 100*(MFI при 4°C - MFI при 37°C)/MFI при 4°C и представлен в таблице 15. Таким образом, гуманизированное hz208F2 VH3/VL3 имело свойства связывания и интернализации, сходные с измеренными у соответствующего мышиного и химерного антител 208F2.
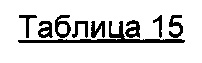
Пример 12: Определение константы диссоциации (KD) связывания пяти химерных анти-IGF-1R-антител (c208F2, с213В10, c212A11, c214F8 и c219D6) и гуманизированной версии (VH3/VL3) антитела 208F2 на растворимом рекомбинантном человеческом IGF-1R
Константы диссоциации (KD) связывания антител на рекомбинантном растворимом человеческом IGF-1R определяли по соотношению между скоростью диссоциации (koff) и скоростью ассоциации (kon). Кинетические эксперименты проводили на устройстве Biacore Х100 с использованием сенсорного чипа СМ5, активированного мышиным моноклинальным анти-His-Tag-антителом. Примерно 12000 RU антител химически присоединяли к карбоксиметилдекстрановой матрице с использованием набора для аминной химической реакции.
Эксперименты проводили при 25°C со скоростью потока 30 мкл/мин с использованием буфера HBS-EP+(GE Healthcare) в качестве рабочего буфера и буфера для разведения образцов.
Кинетическую схему с одним циклом использовали для определения кинетических параметров связывания анти-IGF-1R-антител на растворимом рекомбинантном человеческом IGF-1R, захваченном с помощью двух его C-концевых 10-His-меток.
1 - Раствор растворимого рекомбинантного варианта человеческого гетеротетрамера hIGF-IR: цепи 2α и внеклеточные домены цепей 2β, экспрессированные с дополнительной C-концевой 10-His-меткой (R&D Systems, кат. номер 305-GR-50), вводили в течение 1 минуты во вторую проточную ячейку в концентрации 10 мкг/мл. В среднем 587 RU (со стандартным отклонением 24 RU) растворимого рецептора было захвачено в каждом из 24 циклов, реализованных в данном исследовании.
2 - После фазы захвата в обе проточные ячейки вводили либо рабочий буфер 5 раз (каждое введение по 90 с), либо возрастающий диапазон из пяти концентраций одного из шести антител (каждое введение по 90 с). В конце пятого введения рабочий буфер прогоняли в течение 5 минут для определения скорости диссоциации.
3 - Затем поверхность подвергали регенерации введением буфера с 10 мМ глицином, HCl, pH 1,5, в течение 45 с.
Вычисленный сигнал соответствует разности между ответом из проточной ячейки 2 (с захваченным IGF-1R) и ответом из проточной ячейки 1 (без каких-либо молекул IGF-1R).
Для каждого IGF-1R сигнал от введения возрастающего диапазона концентраций одного антитела корректировали путем вычитания сигнала, полученного с 5 введениями буфера (двойной референс), см. фиг. 17.
Полученные сенсограммы анализировали с использованием программного обеспечения BIAevaluation с моделью 1:1.
Для каждого антитела было проведено четыре эксперимента с использованием двух различных диапазонов концентраций: 40, 20, 10, 5 и 2,5 нМ для двух первых экспериментов и 24, 12, 6, 3 и 1,5 нМ для двух последних экспериментов для каждого антитела.
В этом эксперименте для 6 анализируемых антител экспериментальные данные подводили в соответствии с моделью 1:1 со значимыми значениями koff, когда более высокую концентрацию определяли как константу, а остальные четыре концентрации вычисляли (см. фиг. 18).
Константы диссоциации (KD), рассчитанные как отношение: koff/kon, и периоды полужизни комплексов, рассчитанные как отношение Ln(2)/koff, представлены на фиг. 19 и 20. Они соответствуют среднему значению от четырех независимых экспериментов, проведенных для каждого из антител. Столбцы ошибок соответствуют стандартным ошибкам (n равен 4) значений.
Константы диссоциации находятся в диапазоне от 10 до 100 пМ. Антитело c208F2 показывает более слабую аффинность (более высокое значение константы диссоциации) в отношении MGF-1R (с KD примерно 75 пМ), а его гуманизированный вариант является по меньшей мере таким же хорошим, как химерный вариант (с KD примерно 60 пМ). Четыре других химерных анти-IGF-1R-антитела показывают весьма похожую аффинность в отношении hIGF1-R (с KD примерно 30 пМ). Разница в аффинности, главным образом, связана со скоростью диссоциации или результирующим периодом полужизни комплексов. У 208F2 период полужизни комплекса составляет от 2 до 3 ч для химерной и гуманизированной (VH3/VL3) версий. Для четырех других химерных антител средние периоды полужизни составляют от 7,0 до 9,4 ч.
Эти очень низкие значения кинетики диссоциации однозначно связаны с двухвалентной структурой антител, которые способны связываться одновременно обеими своими Fab-ветвями с двумя соседними молекулами h-IGF-1R. В этом случае уровень захваченных молекул IGF-1R может оказать влияние на скорость диссоциации. Аффинности, определенные в данном исследовании, соответствуют функциональным аффинностям (или авидностям) антител на уровне захваченного h-IGF-1R примерно 600 RU. 3-кратная разница KD, которая наблюдалась между данными, приведенными выше (таблица 10), и значениями, представленными в примере 13, связана с изменением уровня захвата hIGF-1R (600 RU по сравнению с 160 RU в примере 4).
Пример 13: Создание 1613F12
Для создания мышиных моноклональных антител (МКА) против внеклеточного домена (ECD) рецептора Axl пять мышей BALB/c иммунизировали 5 раз путем подкожного введения 15-20×106 клеток CHO-Axl и 2 раза 20 мкг rh Axl ECD. Первую иммунизацию проводили в присутствии полного адъюванта Фрейнда (Sigma, Сент-Луис, Мэриленд, США). К последующим иммунизациям добавляли неполный адъювант Фрейнда (Sigma).
За три дня до слияния иммунизированных мышей бустировали с использованием 20×106 клеток CHO-Axl и 20 мкг rhAxl ECD с неполным адъювантом Фрейнда.
Для создания гибридом получали спленоциты и лимфоциты путем перфузии селезенки и измельчения проксимальных лимфатических узлов, соответственно, собранных от 1 из 5 иммунизированных мышей (выбранной после титрования сывороток всех мышей), и сливали их с клетками миеломы SP2/0-Ag14 (АТСС, Роквилл, Мэриленд, США). Протокол слияния описан Kohler и Milstein (Nature, 256: 495-497, 1975). Затем гибридные клетки подвергали НАТ-селекции. Как правило, для получения моноклональных антител или их функциональных фрагментов, особенно мышиного происхождения, можно обратиться к методикам, которые описаны, в частности, в руководстве "Antibodies" (Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor NY, pp. 726, 1988).
Примерно через 10 дней после слияния колонии гибридных клеток подвергали скринингу. Для первичного скрининга супернатанты гибридом оценивали на предмет секреции МКА, выработанных против белка Axl ECD, с помощью ELISA. Параллельно для выбора МКА, способных связываться с клеточной формой Axl, присутствующей на клеточной поверхности, проводили FACS-анализ с использованием клеток СНО дикого типа и клеток СНО, экспрессирующих Axl.
Как можно скорее выбранные гибридомы клонировали путем серийных разведений, а затем подвергали скринингу на их реакционную способность в отношении белка Axl ECD. Затем клонированные МКА изотипировали с использованием набора Isotyping kit (кат. №5300.05, Southern Biotech, Бирмингем, Алабама, США). Один клон, полученный из каждой гибридомы, выбирали и размножали.
Анализы ELISA проводили, как описано далее, либо с использованием чистого гибридомного супернатанта, либо, когда определяли содержание IgG в супернатантах, титрование осуществляли, начиная с 5 мкг/мл. Затем проводили  серийное разведение в следующих 11 рядах. Вкратце, 96-луночные ELISA-планшеты (Costar 3690, Corning, Нью-Йорк, США) покрывали белком rhAxl-Fc в концентрации 50 мкл/лунка (R and D Systems, кат. №154-AL) или rhAxl ECD в концентрации 2 мкг/мл в PBS в течение ночи при температуре 4°C. Затем планшеты блокировали с помощью PBS, содержащего 0,5% желатин (№22151, Serva Electrophoresis GmbH, Гейдельберг, Германия), в течение 2 ч при 37°C. Сразу после удаления буфера насыщения путем стряхивания планшетов в ELISA-планшеты добавляли по 50 мкл чистых гибридомных клеточных супернатантов или 50 мкл раствора с концентрацией 5 мкг/мл и инкубировали в течение 1 ч при 37°C. После трех промываний добавляли 50 мкл конъюгированного с пероксидазой хрена поликлонального козлиного противомышиного антитела IgG (№115-035-164, Jackson Immuno-Research Laboratories, Inc., Вест-Гров, Пенсильвания, США) в разведении 1/5000 в PBS, содержащем 0,1% желатин и 0,05% Tween 20 (по массе), на 1 ч при температуре 37°C. Затем ELISA-планшеты промывали 3 раза и добавляли субстрат ТМВ (№ UP664782, Uptima Interchim, Франция). После 10-минутного периода инкубации при комнатной температуре реакцию останавливали с помощью 1 М серной кислоты и измеряли оптическую плотность при длине волны 450 нм.
серийное разведение в следующих 11 рядах. Вкратце, 96-луночные ELISA-планшеты (Costar 3690, Corning, Нью-Йорк, США) покрывали белком rhAxl-Fc в концентрации 50 мкл/лунка (R and D Systems, кат. №154-AL) или rhAxl ECD в концентрации 2 мкг/мл в PBS в течение ночи при температуре 4°C. Затем планшеты блокировали с помощью PBS, содержащего 0,5% желатин (№22151, Serva Electrophoresis GmbH, Гейдельберг, Германия), в течение 2 ч при 37°C. Сразу после удаления буфера насыщения путем стряхивания планшетов в ELISA-планшеты добавляли по 50 мкл чистых гибридомных клеточных супернатантов или 50 мкл раствора с концентрацией 5 мкг/мл и инкубировали в течение 1 ч при 37°C. После трех промываний добавляли 50 мкл конъюгированного с пероксидазой хрена поликлонального козлиного противомышиного антитела IgG (№115-035-164, Jackson Immuno-Research Laboratories, Inc., Вест-Гров, Пенсильвания, США) в разведении 1/5000 в PBS, содержащем 0,1% желатин и 0,05% Tween 20 (по массе), на 1 ч при температуре 37°C. Затем ELISA-планшеты промывали 3 раза и добавляли субстрат ТМВ (№ UP664782, Uptima Interchim, Франция). После 10-минутного периода инкубации при комнатной температуре реакцию останавливали с помощью 1 М серной кислоты и измеряли оптическую плотность при длине волны 450 нм.
Для отбора с помощью проточной цитометрии 105 клеток (СНО дикого типа или CHO-Axl) высевали в каждую лунку 96-луночного планшета в PBS, содержащем 1% BSA и 0,01% азид натрия (FACS-буфер), при 4°C. После центрифугирования в течение 2 минут со скоростью 2000 об/мин буфер удаляли и добавляли гибридомные супернатанты или очищенные МКА (1 мкг/мл), которые нужно было проанализировать. После 20-минутной инкубации при температуре 4°C клетки дважды промывали и добавляли Alexa 488-конъюгированное козлиное противомышиное антитело, разведенное 1/500 в FACS-буфере (№ А11017, Molecular Probes Inc., Юджин, США), и инкубировали в течение 20 минут при температуре 4°C. После финального промывания FACS-буфером клетки анализировали с помощью FACS (Facscalibur, Becton-Dickinson) после добавления пропидия йодида в каждую пробирку в конечной концентрации 40 мкг/мл. Лунки, содержащие только клетки, и лунки, содержащие клетки, инкубированные с вторичным Alexa 488-конъюгированным антителом, были включены в качестве отрицательных контролей. Изотипические контроли использовали в каждом эксперименте (Sigma, № M90351MG). Для вычисления среднего значения интенсивности флуоресценции (MFI) оценивали по меньшей мере 5000 клеток.
Гибридома, продуцирующая 1613F12, была выбрана в качестве кандидата.
Пример 14: Гуманизация 1613F12
Использование мышиных антител (МКА) для терапевтического применения у людей обычно приводит к значительным побочным эффектам, у пациентов вырабатывается человеческое противомышиное антитело (human anti-mouse antibody, НАМА), что снижает эффективность лечения и предотвращает дальнейшее введение препарата. Один из подходов для преодоления этой проблемы заключается в гуманизации мышиных МКА путем замены мышиных последовательностей их человеческими аналогами, но без изменения антигенсвязывающей активности. Это может быть достигнуто двумя основными способами: (i) путем конструирования химерных мышиных/человеческих антител, в которых вариабельные мышиные области соединены с человеческими константными областями (Boulianne et al., 1984) и (ii) путем прививания областей, определяющих комплементарность (CDR), из мышиных вариабельных областей в тщательно отобранные человеческие вариабельные области, а затем соединения этих "преобразованных человеческих" вариабельных областей с человеческими константными областями (Riechmann et al., 1988).
14.1 Гуманизация вариабельного домена легкой цепи VL
В качестве предварительного этапа нуклеотидную последовательность 1613F12 VL сравнивали с частью последовательностей мышиных зародышевых генов из базы данных IMGT (http://www.imgt.org). Были определены мышиные зародышевые гены IGKV16-104*01 и IGKJ5*01. Для того чтобы выявить лучшего человеческого кандидата для CDR-прививки, был найден человеческий зародышевый ген, демонстрирующий лучшую идентичность с мышиной последовательностью 1613F12 VL. С помощью инструментов анализа базы данных IMGT были идентифицированы возможные акцепторные человеческие V-области для мышиных 1613F12 VL CDR: IGKV1-27*01 и IGKJ4*02. Для того чтобы выполнить гуманизацию, каждому остатку вариабельного домена легкой цепи, который отличался в человеческой и мышиной последовательностях, был дан ранг приоритета. Эти приоритеты (1-4) были использованы для создания 11 различных гуманизированных вариантов вариабельной области легкой цепи, содержащих до 14 обратных мутаций.


14.2 Гуманизация вариабельного домена тяжелой цепи VH
Для того чтобы выявить лучшего человеческого кандидата для CDR-прививки, были найдены мышиный и человеческий зародышевые гены, демонстрирующие лучшую идентичность с 1613F12 VH. Нуклеотидную последовательность 1613F12 VH совмещали с последовательностями мышиного и человеческого зародышевых генов с помощью программного обеспечения для выравнивания последовательностей "IMGT/V-QUEST", которое является частью базы данных IMGT. Для проверки результатов выравнивания нуклеотидных последовательностей также выполняли выравнивания аминокислотных последовательностей с помощью программного обеспечения "Align X" пакета VectorNTI. Выравнивания с мышиными зародышевыми генами показали, что мышиные зародышевые V-ген IGHV14-3*02 и J-ген IGHJ2*01 являются наиболее гомологичными мышиными зародышевыми генами. С помощью базы данных IMGT мышиный зародышевый D-ген IGHD1-1*01 был идентифицирован в качестве гомологичной последовательности. Для того чтобы выбрать соответствующий человеческий зародышевый ген для прививания CDR, был идентифицирован человеческий зародышевый ген, обладающий самой высокой гомологией с мышиной последовательностью 1613F12 VH. С помощью баз данных и инструментов IMGT человеческий зародышевый ген IGHV1-2*02 и человеческий зародышевый ген IGHJ5*01 J были выбраны в качестве человеческих акцепторных последовательностей для мышиных 1613F12 VH CDR. Для того чтобы выполнить гуманизацию, каждому остатку вариабельного домена тяжелой цепи, который отличался в человеческой и мышиной последовательностях, был дан ранг приоритета (1-4). Эти приоритеты были использованы для создания 20 различных гуманизированных вариантов вариабельной области тяжелой цепи, содержащих до 18 обратных мутаций.
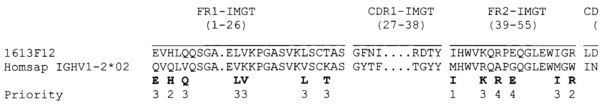
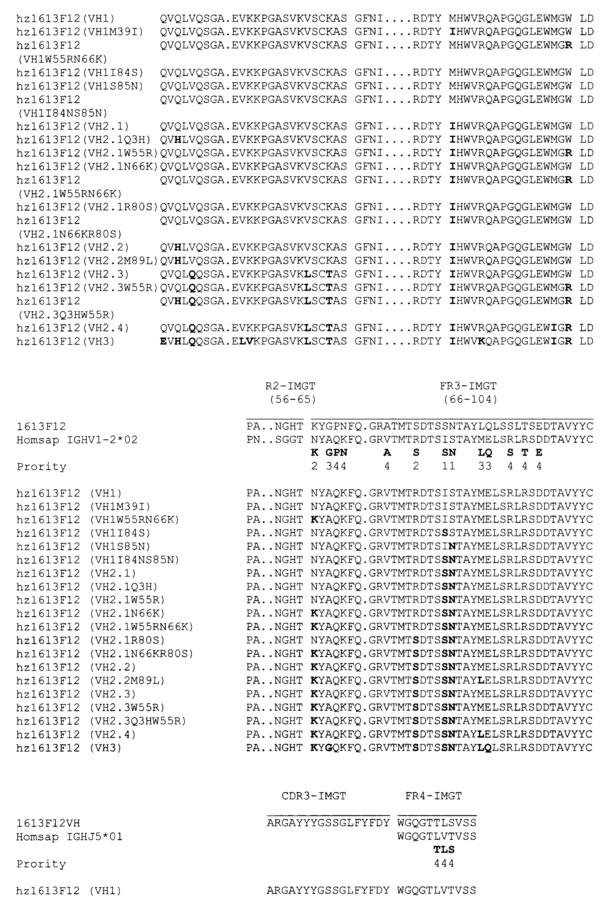

Пример 15: Специфичность связывания Axl
В этом примере сначала было изучено связывание 1613F12 с белком rhAxl-Fc. Затем было изучено его связывание с двумя другими членами ТАМ-семейства, rhDtk-Fc и rhMer-Fc.
Вкратце, 96-луночные планшеты Immulon II покрывали рекомбинантными человеческими белками Axl-Fc (R and D Systems, кат. №154AL/CF), rhDtk (R and D Systems, кат. №859-DK) или rhMer-Fc (R and D Systems, кат. №891-MR) в течение ночи при 4°C, проводили этап блокировки в течение 1 ч с использованием 0,5% раствора желатина, и затем добавляли 1613F12 еще на 1 ч при 37°C в исходной концентрации 5 мкг/мл (3,33×10-8 М). Затем были сделаны  серийные разведения в 12 столбцах. Планшеты промывали и добавляли козлиное противомышиное специфическое IgG-антитело, конъюгированное с HRP (Jackson), на 1 ч при 37°C. Развитие реакции проводили с использованием раствора субстрата ТМВ. Параллельно использовали изотипическое контрольное антитело mIgG1 и коммерческое анти-Axl-MKA 154. Контроли покрытия выполняли в присутствии козлиной противочеловеческой IgG Fc поликлональной сыворотки, меченной HRP (Jackson, №109-035-098), и/или в присутствии HRP-связанного анти-гистидин-антитела (R and D Systems, № МАВ050Н).
серийные разведения в 12 столбцах. Планшеты промывали и добавляли козлиное противомышиное специфическое IgG-антитело, конъюгированное с HRP (Jackson), на 1 ч при 37°C. Развитие реакции проводили с использованием раствора субстрата ТМВ. Параллельно использовали изотипическое контрольное антитело mIgG1 и коммерческое анти-Axl-MKA 154. Контроли покрытия выполняли в присутствии козлиной противочеловеческой IgG Fc поликлональной сыворотки, меченной HRP (Jackson, №109-035-098), и/или в присутствии HRP-связанного анти-гистидин-антитела (R and D Systems, № МАВ050Н).
Результаты представлены на фиг. 24А, 24В и 24С, соответственно.
Этот пример показывает, что 1613F12 связывается только с белком rhAxl-Fc и не связывается с двумя другими членами ТАМ-семейства, rhDtk или rhMer. Отсутствие перекрестной специфичности связывания 1613F12 не наблюдается между членами ТАМ-семейства. В отсутствие первичного антитела (разбавитель) неспецифическое связывание не наблюдалось. В присутствии контрольного изотипического антитела связывание не наблюдалось.
Пример 16: 1613F12 распознает клеточную Форму Axl, экспрессированную на человеческих опухолевых клетках
Уровень экспрессии Axl на клеточной поверхности человеческих опухолевых клеток сначала определяли с использованием коммерческого Axl-антитела (R and D Systems, № МАВ154) в параллели с калибровочными гранулами, чтобы количественно оценить уровень экспрессии Axl. Затем связывание Axl клеточной поверхности исследовали с использованием 1613F12.
Для исследований связывания на клеточной поверхности готовили двукратные серийные разведения 10 мкг/мл (6,66×10-8 М) раствора первичного антитела (1613F12, МАВ154 или изотипического контрольного МКА mIgG1 9G4) и наносили на 2×105 клеток на 20 минут при 4°C. После 3 промываний фосфатно-буферным солевым раствором (PBS) с добавлением 1% BSA и 0,01% NaN3 клетки инкубировали с вторичным козлиным противомышиным антителом, конъюгированным с Alexa 488 (разведение 1/500), в течение 20 минут при 4°C. После 3 дополнительных промываний в PBS с добавлением 1% BSA и 0,1% NaN3 клетки анализировали с помощью FACS (Facscalibur, Becton-Dickinson). Для вычисления среднего значения интенсивности флуоресценции оценивали по меньшей мере 5000 клеток.
Для количественной оценки ABC с использованием МАВ154 использовали калибровочные гранулы QIFIKIT®. Затем, параллельно с гранулами QIFIKIT®, клетки инкубировали с поликлональными козлиными противомышиными иммуноглобулинами/FITC, козлиным F(ab')2, в насыщающей концентрации. Затем на клетках в образце определяли число антигенных сайтов путем интерполяции калибровочной кривой (интенсивность флуоресценции отдельных популяций гранул относительно числа молекул МКА на гранулах).
16.1 Количественная оценка уровня экспрессии Axl на клеточной поверхности
Уровень экспрессии Axl на поверхности человеческих опухолевых клеток определяли с помощью проточной цитометрии с использованием непрямой реакции иммунофлюоресценции (способ QIFIKIT® (Dako, Дания), набора для количественной проточной цитометрии для оценки антигенов на поверхности клеток. Сравнение средней интенсивности флуоресценции (MFI) от известных уровней антигена на гранулах через калибровочный график позволяет определить связывающую способность антитела (antibody binding capacity, ABC) в отношении указанных клеточных линий.
В таблице 16 представлены уровни экспрессии Axl, обнаруженные на поверхности различных опухолевых клеточных линий человека (SN12C, Calu-1, MDA-MB435S, MDA-MB231, NCI-H125, MCF7, Panc1), определенные с использованием QIFIKIT® и МАВ154 (R and D Systems). Значения приведены в виде антигенсвязывающего комплекса (antigen binding complex, ABC).
Результаты, полученные с МАВ154, показали, что рецептор Axl экспрессируется на различных уровнях в зависимости от рассматриваемой человеческой опухолевой клетки.
16.2. Обнаружение Axl с помощью 1613F12 на человеческих опухолевых клетках
Более конкретно связывание Axl изучали с помощью 1613F12.
Были получены кривые "доза - ответ" для 1613F12. Затем с помощью программного обеспечения Prism анализировали MFI, полученные с использованием различных человеческих опухолевых клеток. Данные представлены на фиг. 25.
Данные показывают, что 1613F12 специфически связывается с мембранным рецептором Axl, о чем свидетельствуют профили кривой насыщения. Тем не менее, наблюдались различные интенсивности мечения, указывая на различные уровни рецепторов Axl на клеточной поверхности человеческих опухолевых клеток. При использовании клеточной линии человеческой опухоли молочной железы MCF7 связывание рецептора Axl не наблюдалось.
Пример 17: Валидация hz1613F12 в сравнении с m1613F12
Для того чтобы установить, сравнимо ли антитело hz1613F12 с его мышиной формой, проводили эксперименты связывания с помощью ELISA с использованием белковых анализов rhAxl-Fc.
В этом анализе 96-луночные планшеты (Immulon II, Thermo Fisher) покрывали раствором 1613F12 с концентрацией 5 мкг/мл в 1×PBS в течение ночи при 4°C. После этапа насыщения диапазон белка Axl-Fc (R and D Systems, №154-AL) инкубировали в течение 1 часа при температуре 37°C в покрытых планшетах. На следующем этапе добавляли биотинилированное Axl-антитело (собственный продукт) в концентрации 0,85 мкг/мл на 1 ч при 37°C. Это Axl-антитело относится к отдельной группе эпитопов. Затем в лунки добавляли раствор конъюгата авидина и пероксидазы хрена в разведении 1/2000 в буфере для разведения. Затем на 5 минут добавляли раствор субстрата ТМВ. После добавления стоп-раствора с помощью микропланшетного ридера измеряли оптическую плотность при 405 нм.
На фиг. 26 показано, что мышиная и гуманизированная версии 1613F12 аналогично связываются с белком rhAxl-Fc.
Пример 18: Исследование интернализации 1613F12 с использованием флуоресцентного иммуноцитохимического мечения
Дополнительные результаты по интернализации получали путем конфокальной микроскопии с использованием способа непрямого флуоресцентного мечения.
Вкратце, линию опухолевых клеток SN12C культивировали в RPMI 1640 с 1% L-глутамином и 10% FCS в течение 3 дней до начала эксперимента. Затем клетки отделяли с помощью трипсина и высевали в 6-луночный планшет, содержащий покровное стекло, в RPMI 1640 с 1% L-глутамином и 5% FCS. На следующий день добавляли 1613F12 в концентрации 10 мкг/мл. Также были включены клетки, обработанные нерелевантным антителом. Затем клетки инкубировали в течение 1 ч и 2 ч при температуре 37°C, 5% CO2. Для получения точки Т0 клетки инкубировали в течение 30 минут при 4°C до определения связывания антитела на поверхности клетки. Клетки промывали PBS и фиксировали параформальдегидом в течение 15 минут. Клетки промывали и инкубировали с козлиным противомышиным антителом IgG, конъюгированным с Alexa 488, в течение 60 минут при 4°C, чтобы определить оставшееся антитело на клеточной поверхности. Чтобы проследить за проникновением антител в клетки, клетки фиксировали и пермеабилизовали сапонином. Козлиное противомышиное антитело IgG, конъюгированное с Alexa 488 (Invitrogen), использовали для окрашивания как мембранного, так и внутриклеточного антитела. Ранние эндосомы идентифицировали с помощью кроличьего поликлонального антитела против ЕЕА1, которое открывали козлиным противокроличьим антителом IgG, конъюгированным с Alexa 555 (Invitrogen). Клетки промывали три раза и окрашивали ядра с помощью Draq5. После окрашивания клетки заливали в гистологическую среду Prolong Gold (Invitrogen) и анализировали с помощью конфокального микроскопа Zeiss LSM 510.
Фотографии представлены на фиг. 27А-4С.
Изображения были получены путем конфокальной микроскопии. В присутствии изотипического контроля mIgG1 (9G4) ни мембранного окрашивания, ни внутриклеточного мечения не наблюдалось (фиг. 27А). Прогрессирующая потеря мечения мембранного анти-Axl наблюдалась сразу после 1 часа инкубации клеток SN12C с 1613F12 (фиг. 27В). Внутриклеточное накопление антитела 1613F12 отчетливо наблюдалось через 1 ч и 2 ч (фиг. 27С). Внутриклеточное антитело колокализовалось с ЕЕА1, ранним эндосомальным маркером. Эти фотографии подтверждают интернализацию 1613F12 в клетки SN12C.
Пример 19: Синтез лекарственных средств данного изобретения
В следующих далее примерах используются следующие сокращения:
водн. водный
ее энантиомерный избыток
экв. эквивалент
ESI ионизация электрораспылением
LC/MS жидкостная хроматография с масс-спектрометрией
HPLC высокоэффективная жидкостная хроматография
NMR ядерно-магнитный резонанс
насыщ. насыщенный
UV ультрафиолет
Соединение 1
(S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, бис-соль трифторуксусной кислоты
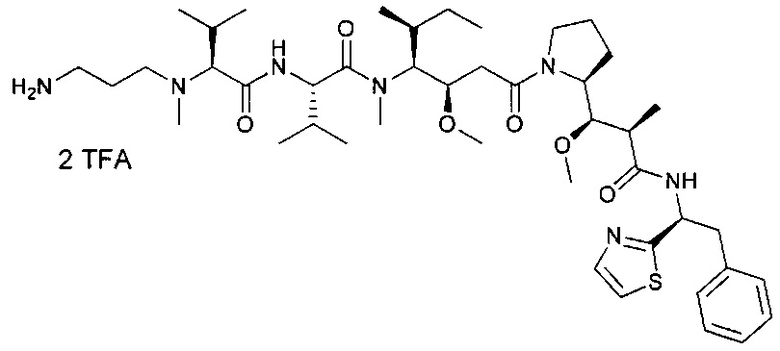
Соединение 1А: (4R,5S)-4-метил-5-фенил-3-пропаноил-1,3-оксазолидин-2-он
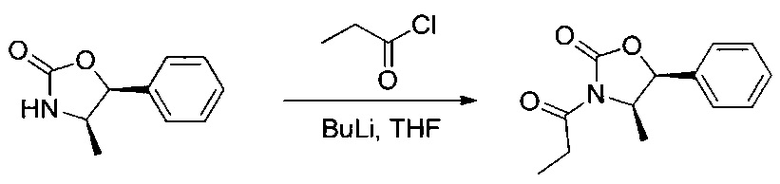
(4R, 5S)-4-метил-5-фенил-1,3-оксазолидин-2-он (5,8 г, 32,7 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (ТГФ, 120 мл) в инертной атмосфере. Смесь охлаждали до -78°C и по каплям добавляли н-бутиллитий (14,4 мл). После встряхивания в течение 30 минут при -78°C добавляли пропаноил хлорид (5,7 мл). Встряхивание продолжали в течение 30 минут при -78°C, а затем в течение ночи при температуре окружающей среды. Реакционную смесь концентрировали, затем повторно растворяли в 200 мл воды. pH раствора подводили до 7 насыщенным водным раствором натрия бикарбоната. Эту водную фазу экстрагировали 3 раза с помощью 100 мл этилацетата (EtOAc). Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 6,8 г (89%) соединения 1А в форме желтого масла.
Соединение 1В: трет-бутил-(2S)-2-[(1R,2R)-1-гидрокси-2-метил-3-[(4R,5S)-4-метил-2-оксо-5-фенил-1,3-оксазолидин-3-ил]-3-оксопропил]пирролидин-1-карбоксилат
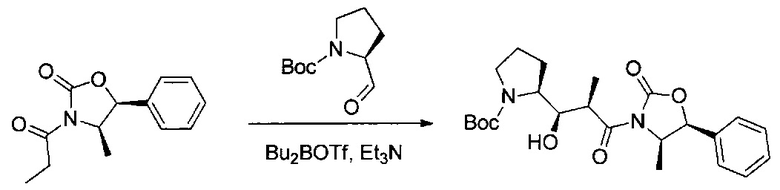
Соединение 1А (17,6 г, 75,45 ммоль, 1,00 экв.) растворяли в дихлорметане (DCM, 286 мл) в инертной атмосфере. Этот раствор охлаждали на ледяной бане. По каплям добавляли триэтиламин (TEA, 12,1 мл, 1,15 экв.) и Bu2BOTf (78,3 мл, 1,04 экв.), поддерживая температуру реакционной смеси ниже 2°C. Встряхивание продолжали при 0°C в течение 45 мин, после чего реакционную смесь охлаждали до -78°C. По каплям добавляли раствор трет-бутил(2S)-2-формилпирролидин-1-карбоксилата (8,5 г, 42,66 ммоль, 0,57 экв.) в DCM (42 мл). Встряхивание продолжали в течение 2 ч при -78°C, затем в течение 1 ч при 0°C и, наконец, в течение 1 ч при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 72 мл фосфатного буфера (pH равен 7,2-7,4) и 214 мл метанола и охлаждали до 0°C. По каплям добавляли раствор 30% пероксида водорода в метаноле (257 мл), поддерживая температуру ниже 10°C. Встряхивание продолжали в течение 1 ч при 0°C. Реакционную смесь нейтрализовали с помощью 142 мл воды, затем концентрировали при пониженном давлении. Полученный водный раствор экстрагировали 3 раза с помощью 200 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и петролейного эфира (EtOAc:РЕ=1:8), получая 13,16 г (40%) соединения 1В в форме бесцветного масла.
Соединение 1С: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-гидрокси-2-метилпропановая кислота
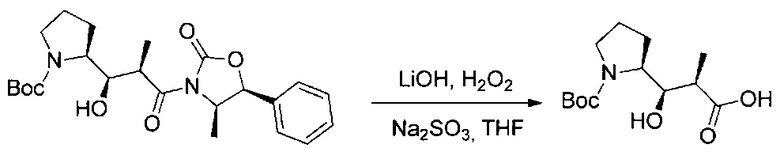
Соединение 1В (13,16 г, 30,43 ммоль, 1,00 экв.) растворяли в ТГФ (460 мл) в присутствии пероксида водорода (30% в воде, 15,7 мл), затем охлаждали на ледяной бане. По каплям добавляли водный раствор лития гидроксида (0,4 моль/л, 152,1 мл), поддерживая реакционную смесь при температуре ниже 4°C. Реакционную смесь встряхивали в течение 2,5 ч при 0°C. По каплям добавляли водный раствор Na2SO3 (1 моль/л, 167,3 мл), поддерживая температуру при 0°C. Реакционную смесь встряхивали в течение 14 ч при температуре окружающей среды, затем нейтрализовали с помощью 150 мл холодного насыщенного раствора натрия бикарбоната и промывали 3 раза с помощью 50 мл DCM. pH водного раствора подводили до 2-3 с помощью 1М водного раствора KHSO4. Этот водный раствор экстрагировали 3 раза с помощью 100 мл EtOAc. Органические фазы объединяли, промывали один раз насыщенным раствором NaCl, сушили над сульфатом натрия, фильтровали и концентрировали, получая 7,31 г (88%) соединения 1С в форме бесцветного масла.
Соединение 1D: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-метокси-2-метилпропановая кислота

Соединение 1С (7,31 г, 26,74 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (135 мл) в присутствии йодометана (25,3 мл). Реакционную среду охлаждали на ледяной бане, после чего порциями добавляли NaH (60% в масле, 4,28 г). Реакционную смесь оставляли встряхиваться в течение 3 дней при 0°C, а затем нейтрализовали с помощью 100 мл насыщенного водного раствора натрия бикарбоната и промывали 3 раза с помощью 50 мл эфира. pH водного раствора подводили до 3 с помощью 1 М водного раствора KHSO4. Этот водный раствор экстрагировали 3 раза с помощью 100 мл EtOAc. Органические фазы объединяли, промывали один раз с помощью 100 мл Na2S2O3 (5% в воде), один раз насыщенным раствором NaCl, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая 5,5 г (72%) соединения 1D в форме бесцветного масла.
Соединение 1Е: N-метокси-N-метил-2-фенилацетамид
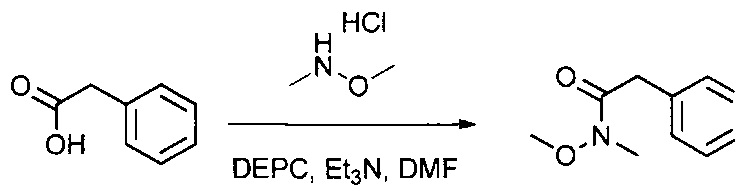
2-фенилуксусную кислоту (16,2 г, 118,99 ммоль, 1,00 экв.) растворяли в диметилформамиде (ДМФА, 130 мл), затем охлаждали до -10°C. Добавляли диэтилфосфороцианидат (DEPC, 19,2 мл), метокси(метил)амина гидрохлорид (12,92 г, 133,20 ммоль, 1,12 экв.) и триэтиламин (33,6 мл). Реакционную смесь встряхивали в течение 30 мин при -10°C, а затем в течение 2,5 ч при температуре окружающей среды. Затем ее дважды экстрагировали с помощью 1 л EtOAc. Органические фазы объединяли, промывали дважды с помощью 500 мл NaHCO3 (насыщ.), один раз с помощью 400 мл воды, затем сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем с помощью смеси EtOAc и РЕ (от 1:100 до 1:3), получая 20,2 г (95%) соединения 1Е в форме желтого масла.
Соединение 1F: 2-фенил-1-(1,3-тиазол-2-ил)этан-1-он
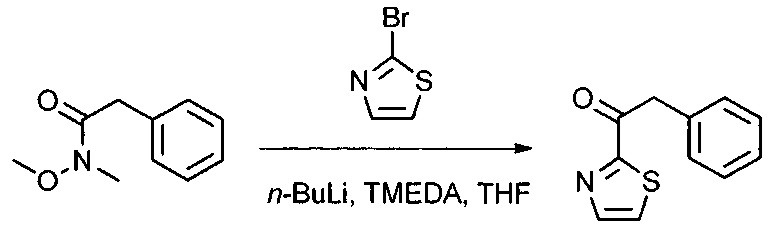
Тетраметилэтилендиамин (TMEDA, 27,2 мл) растворяли в ТГФ (300 мл) в инертной атмосфере, затем охлаждали до -78°C, затем по каплям добавляли н-BuLi (67,6 мл, 2,5 М). По каплям добавляли 2-бром-1,3-тиазол (15,2 мл) и продолжали встряхивание в течение 30 мин при -78°C. По каплям добавляли соединение 1Е (25 г, 139,50 ммоль, 1,00 экв.), растворенное в ТГФ (100 мл). Встряхивание продолжали в течение 30 минут при -78°C, а затем в течение 2 ч при -10°C. Реакционную смесь нейтрализовали с помощью 500 мл KHSO4 (насыщ.), затем экстрагировали 3 раза с помощью 1 л EtOAc. Органические фазы объединяли, промывали дважды с помощью 400 мл воды и дважды с помощью 700 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:100 до 1:10), получая 25 г (88%) соединения 1F в форме желтого масла.
Соединение 1G: (1R)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-ол
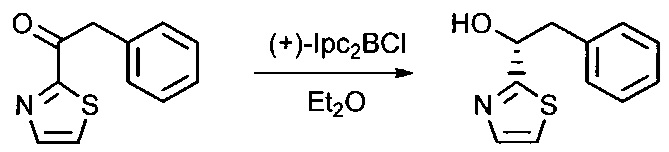
В инертной атмосфере по каплям добавляли раствор соединения 1F (15 г, 73,8 ммоль, 1,00 экв.) в эфире (300 мл) к (+)-В-хлордиизопинокамфеилборану ((+)-Ipc2BCl, 110,8 мл). Реакционную смесь встряхивали в течение 24 ч при 0°C, затем нейтрализовали с помощью 300 мл (1:1) смеси NaOH (10% в воде) и H2O2 (30% в воде) и, наконец, экстрагировали три раза с помощью 500 мл EtOAc. Органические фазы объединяли, промывали дважды с помощью 300 мл K2CO3 (насыщ.) и один раз с помощью 500 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:20 до 1:2), получая 6,3 г (42%) соединения 1G в форме белого твердого вещества.
Соединение 1Н: 2-[(1S)-1-азидо-2-фенилэтил]-1,3-тиазол

Соединение 1G (6 г, 29,23 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (150 мл) в присутствии трифенилфосфина (13 г, 49,56 ммоль, 1,70 экв.), затем охлаждали до 0°C. По каплям добавляли диэтилазодикарбоксилат (DEAD, 7,6 мл), затем дифенилфосфорилазид (DPPA, 11 мл), затем ледяную баню удаляли и раствор оставляли встряхиваться в течение 48 ч при температуре окружающей среды. Среду концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:100 до 1:30), получая 8 г частично очищенного соединения 1Н в форме желтого масла. Соединение 1Н использовали как таковое на следующем этапе.
Соединение 1I: трет-бутил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамат.
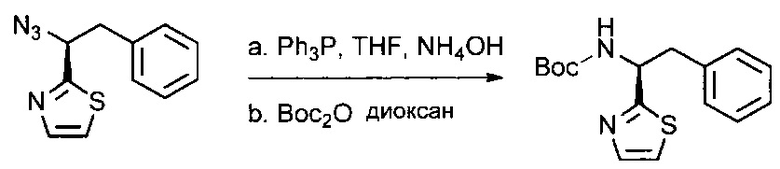
Соединение 1Н (6,5 г, 28,2 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (100 мл) в присутствии трифенилфосфина (6,5 г, 33,9 ммоль, 1,20 экв.) и нагревали до 50°C в течение 2 ч. Затем добавляли амииак (70 мл) и продолжали нагревание в течение 3 ч. Реакционную смесь охлаждали, нейтрализовали с помощью 500 мл воды, затем экстрагировали 3 раза с помощью 500 мл EtOAc. Органические фазы объединяли и дважды экстрагировали с помощью 500 мл 1 н HCl. Водные фазы объединяли, доводили до pH 8-9 добавлением раствора натрия гидроксида (10% в воде), затем экстрагировали 3 раза с помощью 500 мл DCM. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 4,8 г (83%) (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина в форме желтого масла. Затем это соединение защищали Вос-группой ((трет-бутокси)карбонил) так, чтобы его можно было очистить. Его растворяли в инертной атмосфере в 1,4-диоксане (40 мл), затем охлаждали до 0°C. По каплям добавляли (Вос)2O (10,26 г, 47,01 ммоль, 2,00 экв.), разведенный в 20 мл 1,4-диоксана. Ледяную баню удаляли и раствор оставляли встряхиваться в течение ночи при температуре окружающей среды, а затем нейтрализовали с помощью 300 мл воды и дважды экстрагировали с помощью 500 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:100 до 1:20, ее = 93%). Затем его рекристаллизовывали в смеси гексана/ацетона (примерно 5-10/1,1 г/10 мл), получая 6 г (84%) соединения 1I в форме белого твердого вещества (ее более 99%).
Соединение 1J: трет-бутил-(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-карбоксилат
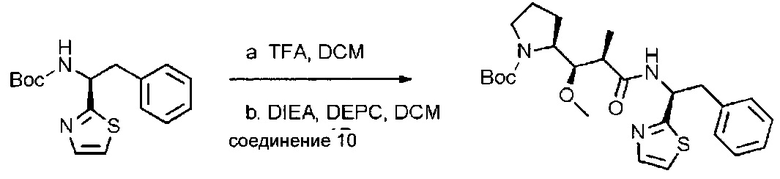
Соединение 1I (3 г, 9,86 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл DCM. Добавляли трифторуксусную кислоту (TFA, 10 мл) и раствор оставляли встряхиваться в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении, получая 2,0 г (64%) соли трифторуксусной кислоты (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина в форме желтого масла. Этот промежуточный продукт повторно растворяли в 20 мл DCM, после чего добавляли соединение 1D (1,8 г, 6,26 ммоль, 1,05 экв.), DEPC (1,1 г, 6,75 ммоль, 1,13 экв.) и диизопропилэтиламин (DIEA, 1,64 г, 12,71 ммоль, 2,13 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:100 до 1:3), получая 2,3 г (81%) соединения 1J в форме бледно-желтого твердого вещества.
Соединение 1K: (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамид; соль трифторуксусной кислоты
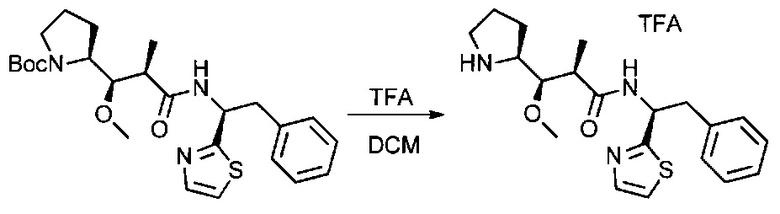
Соединение 1J (2,25 г, 4,75 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл DCM. Добавляли TFA (10 мл) и раствор оставляли встряхиваться в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении, получая 2,18 г (94%) соединения 1K в форме желтого масла.
Соединение 1L: (2S,3S)-2-(бензиламино)-3-метилпентановая кислота
(2S,3S)-2-амино-3-метилпентановую кислоту (98,4 г, 750 ммоль, 1,00 экв.) добавляли при температуре окружающей среды и порциями к 2 н раствору натрия гидроксида (375 мл). Быстро добавляли бензальдегид (79,7 г, 751,02 ммоль, 1,00 экв.) и полученный раствор встряхивали в течение 30 мин. Маленькими частями добавляли натрия борогидрид (10,9 г, 288,17 ммоль, 0,38 экв.), поддерживая температуру между 5 и 15°C. Встряхивание продолжали в течение 4 ч при температуре окружающей среды. Реакционную смесь разводили 200 мл воды, затем промывали дважды с помощью 200 мл EtOAc. pH водного раствора подводили до 7 с помощью 2 н раствора соляной кислоты. Образованный осадок собирали путем фильтрации и получали 149,2 г (90%) соединения 1L в форме белого твердого вещества.
Соединение 1М: (2S,3S)-2-[бензил(метил)амино]-3-метилпентановая кислота
Соединение 1L (25 г, 112,97 ммоль, 1,00 экв.) растворяли в инертной атмосфере в муравьиной кислоте (31,2 г) в присутствии формальдегида (36,5% в воде, 22,3 г). Раствор встряхивали в течение 3 ч при 90°C, затем концентрировали при пониженном давлении. Осадок растирали в 250 мл ацетона, затем концентрировали. Эту процедуру растирания/выпаривания повторяли дважды с помощью 500 мл ацетона, получая 21,6 г (81%) соединения 1М в форме белого твердого вещества.
Соединение 1N: (2S,3S)-2-[бензил(метил)амино]-3-метилпентан-1-ол
LiAlH4 (0,36 г) суспендировали в 10 мл ТГФ в инертной атмосфере при 0°C. Маленькими частями добавляли соединение 1М (1,5 г, 6,37 ммоль, 1,00 экв.), поддерживая температуру между 0 и 10°C. Реакционную смесь встряхивали в течение 2 ч при 65°C, затем снова охлаждали до 0°C, а затем нейтрализовали с помощью избыточных количеств 360 мкл воды, 1 мл 15% натрия гидроксида и 360 мкл воды. Соли алюминия, которые выпадали в осадок, удаляли путем фильтрации. Фильтрат сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:50), получая 820 мг (58%) соединения 1N в форме бледно-желтого масла.
Соединение 1O: (2S,3S)-2-[бензил(метил)амино]-3-метилпентанал
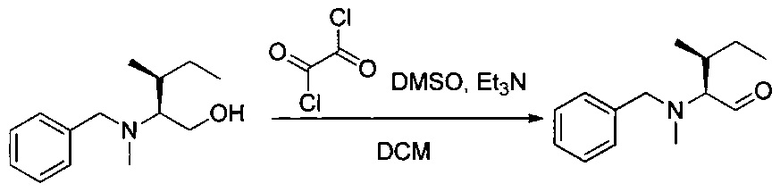
Оксалилхлорид (0,4 мл) растворяли в DCM (15 мл) в инертной атмосфере. Раствор охлаждали до -70°C и по каплям добавляли раствор диметилсульфоксида (ДМСО (0,5 мл) в DCM (10 мл) в течение 15 минут. Реакционную смесь встряхивали в течение 30 минут, после чего по каплям добавляли раствор соединения 1N (820 мг, 3,70 ммоль, 1,00 экв.) в DCM (10 мл) в течение 15 минут. Реакционную смесь встряхивали еще в течение 30 минут при низкой температуре, затем медленно добавляли триэтиламин (2,5 мл). Реакционную смесь встряхивали в течение 1 ч при -50°C, затем ледяную баню удаляли и реакционную смесь нейтрализовали с помощью 25 мл воды, позволяя температуре вернуться к нормальной. Раствор промывали один раз с помощью 30 мл насыщенного водного раствора NaCl, затем сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:200), получая 0,42 г (52%) соединения 1O в форме желтого масла.
Соединение 1Р: (2S,3S)-N-бензил-1,1-диметокси-N,3-диметилпентан-2-амин
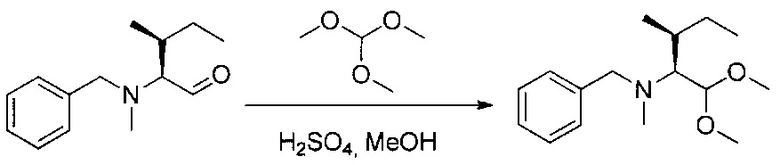
Соединение 1O (4,7 г, 21,43 ммоль, 1,00 экв.) растворяли в 20 мл метанола при 0°C. По каплям добавляли концентрированную серную кислоту (4,3 мл) и продолжали встряхивание в течение 30 минут при 0°C. Добавляли триметилортоформиат (21,4 мл), ледяную баню удаляли и реакционную среду оставляли встряхиваться в течение 3 ч при температуре окружающей среды. Реакционную среду разводили 200 мл EtOAc, тщательно промывали 100 мл 10% раствора Na2CO3 и 200 мл насыщенного NaCl, затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 3,4 г (60%) соединения 1Р в форме бледно-желтого масла.
Соединение 1Q: [[1-(трет-бутокси)этенил]окси](трет-бутил)диметилсилан
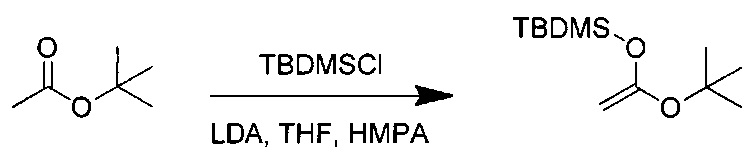
Диизопропиламин (20 г, 186,71 ммоль, 1,08 экв.) растворяли в 170 мл ТГФ в инертной атмосфере и охлаждали до -78°C. По каплям добавляли H-BuLi (2,4 М, 78,8 мл) и раствор встряхивали в течение 30 минут при низкой температуре (получая LDA-лития диизопропиламид), а затем добавляли трет-бутилацетат (20 г, 172,18 ммоль, 1,00 экв.). Реакционную смесь встряхивали в течение 20 мин при -78°C, а затем добавляли гексаметилфосфорамид (НМРА, 25,8 мл) и раствор трет-бутилдиметилхлорсилана (TBDMSCI, 28 г, 185,80 ммоль, 1,08 экв.) в 35 мл ТГФ. Встряхивание продолжали в течение 20 дополнительных минут при низкой температуре, и затем ледяную баню удаляли. Раствор концентрировали при пониженном давлении. Осадок повторно растворяли в 100 мл воды и экстрагировали 3 раза с помощью 100 мл РЕ. Органические фазы объединяли, промывали один раз с помощью 500 мл насыщенного водного раствора NaCl, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали путем дистилляции, получая 16,6 г (83%) соединения 1Q в форме бесцветного масла.
Соединение 1R: трет-бутил(3R,4S,5S)-4-[бензил(метил)амино]-3-метокси-5-метилгептаноат
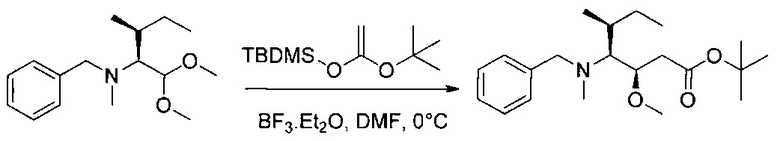
Соединение 1Р (2,0 г, 7,54 ммоль, 1,00 экв.) и соединение 1Q (2,6 г, 11,28 ммоль, 1,50 экв.) растворяли в 33 мл DCM в инертной атмосфере. Раствор охлаждали до 0°C. По каплям добавляли ДМФА (1,2 г) вместе с раствором BF3⋅Et2O (2,1 г) в 7,5 мл DCM. Встряхивание продолжали в течение 24 ч при 0°C. Реакционную среду промывали один раз с помощью 30 мл натрия карбоната (10%) и дважды с помощью 50 мл насыщенного водного раствора NaCl, затем сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:100), получая 1,82 г (91%) соединения 1R в форме желтого масла.
Соединение 1S: (3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноата гидрохлорид
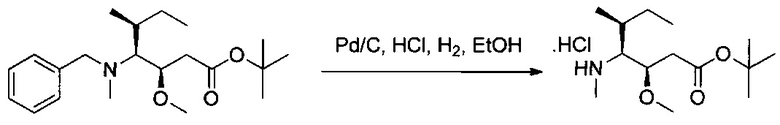
Соединение 1R (2,4 г, 6,87 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 35 мл этанола в присутствии Pd/C (0,12 г) и концентрированной соляной кислоты (0,63 мл). Атмосферу азота заменяли атмосферой водорода и реакционную среду оставляли встряхиваться в течение 18 ч при температуре окружающей среды. Реакционную среду фильтровали и концентрировали при пониженном давлении. Осадок растирали в 50 мл гексана и удаляли супернатант, после высушивания при пониженном давлении получая 1,66 г (82%) соединения 1S в форме белого твердого вещества.
Соединение 1T: трет-бутил(3R,4S,5S)-4-[(2S)-2-[[(бензилокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
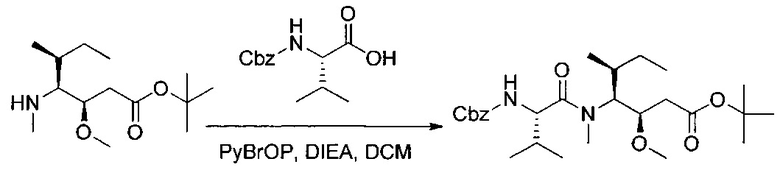
(2S)-2-[[(бензилокси)карбонил]амино]-3-метилбутановую кислоту (15 г, 0,40 ммоль, 1,00 экв.) растворяли в 300 мл DCM в присутствии DIEA (38,3 мл) и бромтрипирролидинофосфора гексафторфосфата (PyBrOP, 32,3 г). Раствор встряхивали в течение 30 минут при температуре окружающей среды, а затем добавляли соединение 1S (15,99g, 0,42 ммоль, 1,07 экв.). Реакционную среду встряхивали в течение 2 ч, а затем концентрировали. Осадок очищали на обращенной фазе (С18) смесью ацетонитрила (ACN) и воды (от 30:70 до 100:0 в течение 40 мин), получая 17 г (58%) соединения 1Т в форме бесцветного масла.
Соединение 1U: трет-бутил(3R,4S,5S)-4-[(2S)-2-амино-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
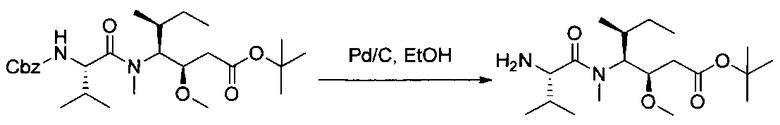
Соединение 1Т (76 мг, 0,15 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл этанола в присутствии Pd/C (0,05 г). Атмосферу азота заменяли атмосферой водорода и реакционную смесь встряхивали в течение 2 ч при температуре окружающей среды. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 64 мг соединения 1U в форме бесцветного масла.
Соединение 1V: (3R,4S,5S)-4-[(2S)-2-[[(9H-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
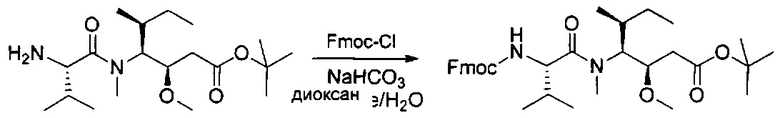
Соединение 1U (18,19 г, 50,74 ммоль, 1,00 экв.) растворяли в 400 мл смеси 1,4-диоксана/воды (1:1) в присутствии натрия бикарбоната (12,78 г, 152 ммоль, 3,00 экв.) и 9Н-флуорен-9-илметил хлорформиата (Fmoc-Cl, 19,69 г, 76 ммоль, 1,50 экв.), затем встряхивали в течение 2 ч при температуре окружающей среды. Реакционную среду затем разводили 500 мл воды и экстрагировали 3 раза с помощью 200 мл EtOAc. Органические фазы объединяли, промывали один раз с помощью 200 мл насыщенного водного раствора NaCl, сушили над сульфатом натрия, фильтровали и концентрировали, получая 40 г частично очищенного соединения 1V в форме бледно-желтого масла.
Соединение 1W: (3R,4S,5S)-4-[(2S)-2-[[(9H-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептановая кислота

Соединение 1V (40 г, 68,88 ммоль, 1,00 экв.) растворяли в нейтральной атмосфере в 600 мл DCM. Добавляли TFA (300 мл). Раствор встряхивали в течение 2 ч при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью метанола и DCM (1:10), получая 23,6 г (65%) соединения 1W в форме бесцветного масла.
Соединение 1Х: 9Н-флуорен-9-илметил-N-[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамат
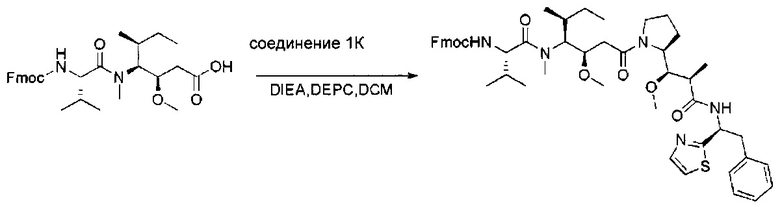
Соединение 1W (2,53 г, 4,82 ммоль, 1,08 экв.) растворяли в 20 мл DCM в присутствии соединения 1K (2,18 г, 4,47 ммоль, 1,00 экв.), DEPC (875 мг, 5,37 ммоль, 1,20 экв.) и DIEA (1,25 г, 9,67 ммоль, 2,16 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем тщательно промывали с помощью 50 мл насыщенного KHSO4 и 100 мл воды, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью метанола и DCM (от 1:200 до 1:40), получая 2,8 г (71%) соединения 1Х в форме бледно-желтого твердого вещества.
Соединение 1Y: (2S)-2-амино-N-[(3R,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил]-N,3-диметилбутанамид
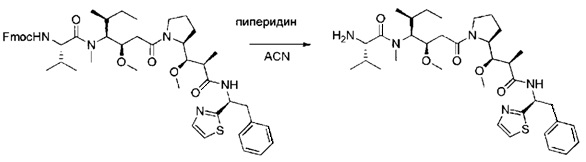
Соединение 1Х (2,8 г, 3,18 ммоль, 1,00 экв.) растворяли в ацетонитриле (ACN, 12 мл) в присутствии пиперидина (3 мл) и оставляли встряхиваться в течение 18 ч при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 50 мл воды, затем дважды экстрагировали с помощью 100 мл DCM. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью метанола и DCM (от 1:100 до 1:40), получая 1,2 г (57%) соединения 1Y в форме желтого твердого вещества.
Соединение 1ZA: (2S)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутановая кислота
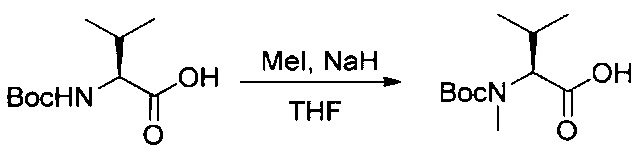
(2S)-2-[[(трет-бутокси)карбонил]амино]-3-метилбутановую кислоту (63 г, соединение 289,97 ммоль, 1,00 экв.) растворяли в  сфере в ТГФ (1000 мл) в присутствии йодометана (181 мл). Раствор охлаждали до 0°C, а затем маленькими частями добавляли натрия гидрид (116 г, 4,83 mol, 16,67 экв.). Реакционную смесь встряхивали в течение 1,5 ч при 0°C, затем ледяную баню удаляли и продолжали встряхивание в течение 18 ч. Реакционную смесь нейтрализовали с помощью 200 мл воды, а затем концентрировали при пониженном давлении. Оставшуюся водную фазу разводили 4 л воды, промывали один раз с помощью 200 мл EtOAc и подводили ее pH до 3-4 с помощью 1 н раствора соляной кислоты. Полученную смесь экстрагировали 3 раза с помощью 1,2 л EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 60 г (89%) соединения 1ZA в форме желтого масла.
сфере в ТГФ (1000 мл) в присутствии йодометана (181 мл). Раствор охлаждали до 0°C, а затем маленькими частями добавляли натрия гидрид (116 г, 4,83 mol, 16,67 экв.). Реакционную смесь встряхивали в течение 1,5 ч при 0°C, затем ледяную баню удаляли и продолжали встряхивание в течение 18 ч. Реакционную смесь нейтрализовали с помощью 200 мл воды, а затем концентрировали при пониженном давлении. Оставшуюся водную фазу разводили 4 л воды, промывали один раз с помощью 200 мл EtOAc и подводили ее pH до 3-4 с помощью 1 н раствора соляной кислоты. Полученную смесь экстрагировали 3 раза с помощью 1,2 л EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 60 г (89%) соединения 1ZA в форме желтого масла.
Соединение 1ZB: бензил(2S)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутаноат

Соединение 1ZA (47 г, 203,21 ммоль, 1,00 экв.) растворяли в ДМФА (600 мл) в присутствии Li2CO3 (15,8 г, 213,83 ммоль, 1,05 экв.). Раствор охлаждали до 0°C, затем по каплям добавляли бензилбромид (BnBr 57,9 г, 338,53 ммоль, 1,67 экв.). Реакционную смесь оставляли встряхиваться в течение ночи, а затем нейтрализовали с помощью 400 мл воды и фильтровали. Полученный раствор дважды экстрагировали с помощью 500 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (от 1:100 до 1:20), получая 22,5 г (34%) соединения 1ZB в форме желтого масла.
Соединение 1ZC: бензил(2S)-3-метил-2-(метиламино)бутаноата гидрохлорид
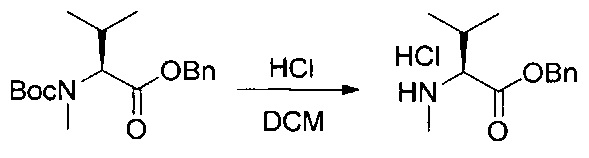
Соединение 1ZB (22,5 г, 70,00 ммоль, 1,00 экв.) растворяли в 150 мл DCM. Пропускали газообразную хлороводородную кислоту. Реакционную смесь встряхивали в течение 1 ч при температуре окружающей среды, а затем концентрировали при пониженном давлении, получая 17 г (94%) соединения 1ZC в форме желтого твердого вещества.
Соединение 1ZD: трет-бутил N-(3,3-диэтоксипропил)карбамат
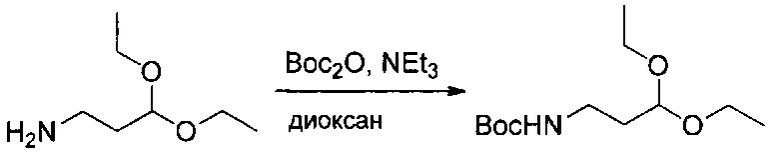
3,3-диэтоксипропан-1-амин (6 г, 40,76 ммоль, 1,00 экв.) растворяли в 1,4-диоксане (30 мл) в присутствии TEA (4,45 г, 43,98 ммоль, 1,08 экв.), затем охлаждали до 0°C. По каплям добавляли (Вос)2O (9,6 г, 43,99 ммоль, 1,08 экв.), разведенный в 20 мл 1,4-диоксана. Раствор встряхивали в течение 2 ч при 0°C, затем в течение ночи при температуре окружающей среды, а затем нейтрализовали с помощью 10 мл воды. pH подводили до 5 с помощью HCl (1%). Раствор экстрагировали 3 раза с помощью 50 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 8,21 г (81%) соединения 1ZD в форме бледно-желтого масла.
Соединение 1ZE: трет-бутил N-(3-оксопропил)карбамат

Соединение 1ZD (8,20 г, 33,15 ммоль, 1,00 экв.) растворяли в 18,75 мл уксусной кислоты и оставляли встряхиваться в течение ночи при температуре окружающей среды. Затем реакционную среду экстрагировали 3 раза с помощью 30 мл EtOAc. Органические фазы объединяли, промывали 3 раза с помощью 30 мл насыщенного раствора NaCl, сушили над сульфатом натрия, фильтровали и концентрировали, получая 5 г (87%) соединения 1ZE в форме темно-красного масла.
Соединение 1ZF: (2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метилбутановая кислота
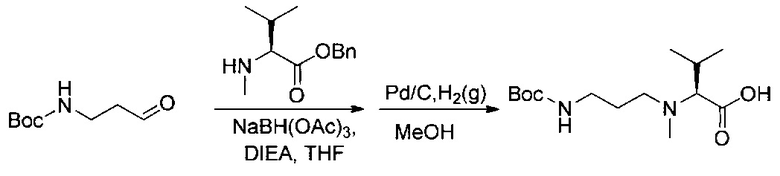
Соединение 1ZE (2,4 г, 13,86 ммоль, 1,00 экв.) растворяли в 50 мл ТГФ в присутствии соединения 1ZC (3,56 г, 13,81 ммоль, 1,00 экв.) и DIEA (9,16 мл, 4,00 экв.). Реакционную смесь встряхивали в течение 30 мин при температуре окружающей среды, а затем добавляли натрия триацетоксиборогидрид (5,87 г, 27,70 ммоль, 2,00 экв.). Встряхивание продолжали в течение ночи, затем реакционную смесь нейтрализовали с помощью 100 мл воды и экстрагировали 3 раза с помощью 50 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок частично очищали на колонке с силикагелем смесью EtOAc и РЕ (1:4). Полученный неочищенный продукт повторно растворяли в 20 мл метанола в присутствии Pd/C (1,2 г) и гидрогенировали в течение 20 минут при нормальной температуре и давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 200 мг (5%) соединения 1ZF в форме белого твердого вещества.
Соединение 1ZG: трет-бутил-N-(3-[[(1S)-1-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-phnyl-1-(1,3-тиазол-2-ил)этил]карбамоил]thy]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]пропил) карбамат
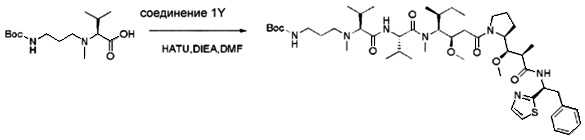
Соединение 1Y (50 мг, 0,08 ммоль, 1,00 экв.) растворяли в 2 мл ДМФА в присутствии соединения 1ZF (26,2 мг, 0,09 ммоль, 1,20 экв.), DIEA (37,7 мл) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU, 43,3 мг, 0,11 ммоль, 1,50 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем разводили 10 мл воды и экстрагировали 3 раза с помощью 5 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 100 мг соединения 1ZG в форме частично очищенного бесцветного масла.
Соединение 1ZG (90 мг, 0,10 ммоль, 1,00 экв.) растворяли в нейтральной атмосфере в 2 мл DCM и раствор охлаждали на ледяной бане. Добавляли TFA (1 мл) и реакционную смесь встряхивали в течение 2 ч при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 18% до 31% ACN в течение 7 мин, затем от 31% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 1 получали с выходом 25% (23 мг) в форме белого твердого вещества.
LC/MS/UV (Atlantis Т3 колонка, 3 мкм, 4,6×100 мм; 35°C; 1 мл/мин, от 30% до 60% ACN в воде (20 мМ аммония ацетат в течение 6 мин); ESI (C44H73N7O6S, точная масса 827,53) m/z: 829 (МН+), 5,84 мин (93,7%, 254 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,85-7,80 (m, 1Н); 7,69-7,66 (m, 1Н), 7,40-7,10 (m, 5Н), 5,80-5,63 (m, 1Н), 4,80-4,65 (m, 2Н), 4,22-4,00 (m, 1Н), 3,89-0,74 (m, 58Н).
Соединение 2
(S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты
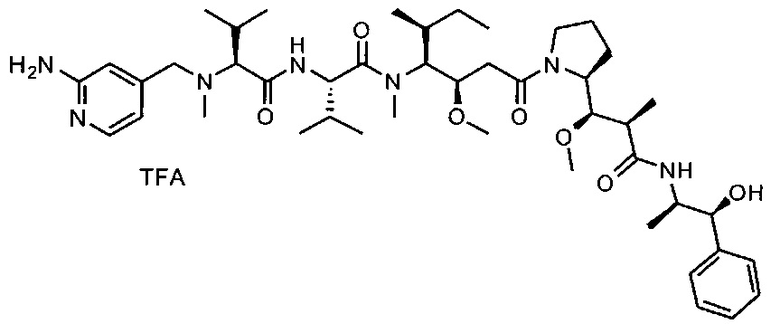
Соединение 2А: трет-бутил(S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-карбоксилат
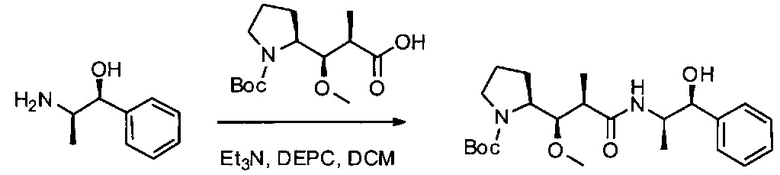
Соединение 1D (2,5 г, 8,70 ммоль, 1,00 экв.) и (1S,2R)-2-амино-1-фенилпропан-1-ол (1,315 г, 8,70 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДМФА (35 мл). Раствор охлаждали до 0°C, затем по каплям добавляли DEPC (1,39 мл) и TEA (1,82 мл). Реакционную смесь встряхивали в течение 2 ч при 0°C, затем 4 ч при температуре окружающей среды. Реакционную смесь разводили в 200 мл воды и экстрагировали три раза с помощью 50 мл EtOAc. Органические фазы объединяли, промывали один раз с помощью 50 мл KHSO4 (1 моль/л), один раз с помощью 50 мл NaHCO3 (насыщ.), один раз с помощью 50 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 3,6 г (98%) соединения 2А в форме желтого твердого вещества.
Соединение 2В: (2R,3R)-N-((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамид2,2,2-трифторацетат
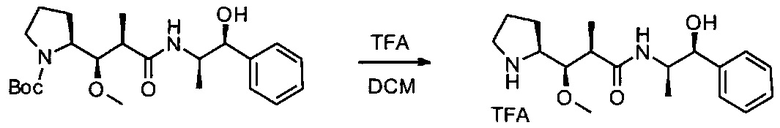
Соединение 2А (2,7 г, 6,42 ммоль, 1,00 экв.) растворяли в инертной атмосфере в DCM (40 мл), затем охлаждали до 0°C. Добавляли TFA (25 мл) и раствор встряхивали в течение 2 ч при 0°C. Реакционную смесь концентрировали при пониженном давлении, получая 4,4 г соединения 2В в форме желтого масла.
Соединение 2С: (9Н-флуорен-9-ил)метил((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)(метил)амино)-3-метил-1-оксобутан-2-ил)карбамат
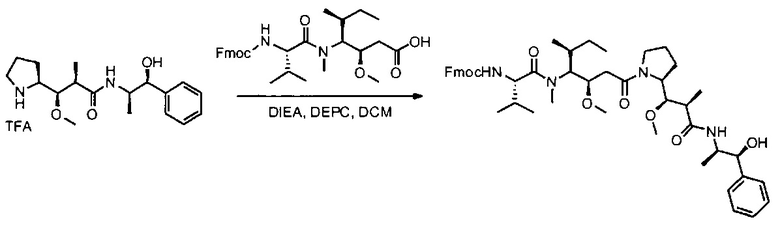
Соединения 2В (4,4 г, 10,13 ммоль, 1,00 экв.) и 1W (5,31 г, 10,12 ммоль, 1,00 экв.) растворяли в инертной атмосфере в DCM (45 мл). Раствор охлаждали до 0°C, затем по каплям добавляли DEPC (1,62 мл) и DIEA (8,4 мл). Реакционную смесь встряхивали в течение 2 ч при 0°C, затем при температуре окружающей среды в течение ночи. Реакционную смесь разводили 100 мл воды и экстрагировали три раза с помощью 50 мл DCM. Органические фазы объединяли, промывали один раз с помощью 50 мл KHSO4 (1 моль/л), один раз с помощью 50 мл NaHCO3 (насыщ.), один раз с помощью 50 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали под давлением, получая 3,3 г (39%) соединения 2С в форме желтого твердого вещества.
Соединение 2D: (S)-2-амино-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
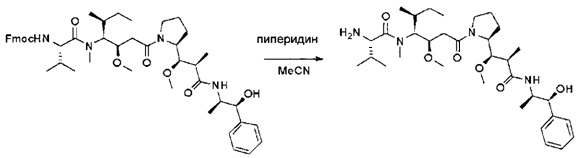
Соединение 2С (300 мг, 0,36 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ACN (2 мл) и пиперидине (0,5 мл). Раствор оставляли встряхиваться при температуре окружающей среды в течение ночи, затем выпаривали досуха при пониженном давлении. Осадок очищали на колонке с силикагелем, элюировали смесью DCM и МеОН (1:100), получая 150 мг (68%) соединения 2D в форме белого твердого вещества.
Соединение 2Е: метил-2-((трет-бутоксикарбонил)амино)изоникотинат
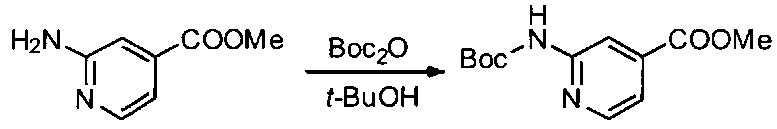
Метил-2-аминопиридин-4-карбоксилат (2 г, 13,14 ммоль, 1,00 экв.) растворяли в трет-бутаноле (20 мл), после чего добавляли ди-трет-бутилдикарбонат (4,02 г, 18,42 ммоль, 1,40 экв.). Реакционную смесь встряхивали при 60°C в течение ночи, затем реакцию останавливали путем добавления водного 1 М NaHCO3 раствора (50 мл). Твердое вещество извлекали путем фильтрации, промывали 50 мл EtOH, затем сушили in vacuo, получая 2,5 г (75%) соединения 2Е в форме белого твердого вещества.
Соединение 2F: трет-бутил-(4-(гидроксиметил)пиридин-2-ил)карбамат
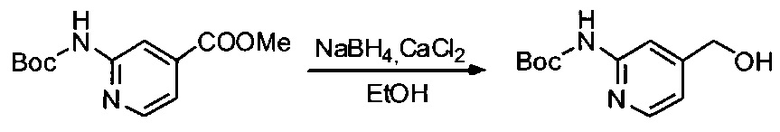
Соединение 2Е (2,5 г, 9,91 ммоль, 1,00 экв.) и CaCl2 (1,65 г) растворяли в EtOH (30 мл). Раствор охлаждали до 0°C, затем постепенно добавляли NaBH4 (1,13 г, 29,87 ммоль, 3,01 экв.). Раствор оставляли встряхиваться в течение ночи при температуре окружающей среды, затем реакцию останавливали добавлением воды (50 мл). Смесь экстрагировали три раза с помощью 20 мл EtOAc. Органические фазы объединяли, промывали дважды с помощью 20 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 2,0 г (90%) соединения 2F в форме бесцветного твердого вещества.
Соединение 2G: трет-бутил-(4-формилпиридин-2-ил)карбамат
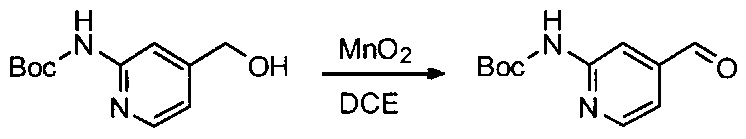
Соединение 2F (2,5 г, 11,15 ммоль, 1,00 экв.) растворяли в DCE (25 мл), затем добавляли 19,4 г (223,14 ммоль, 20,02 экв.) MnO2. Смесь оставляли встряхиваться в течение ночи при 70°C, затем твердые вещества удаляли путем фильтрации. Фильтрат выпаривали досуха, получая 1,4 г (57%) соединения 2G в форме белого твердого вещества.
Соединение 2Н: бензил-(S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил) (метил)амино)-3-метилбутаноат
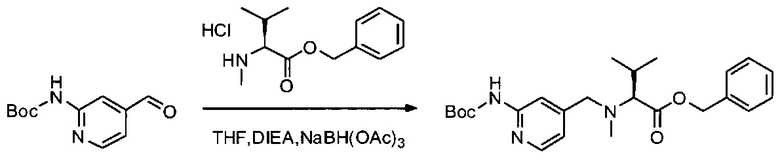
Соединение 2G (2,3 г, 10,35 ммоль, 1,00 экв.) растворяли в 25 мл ТГФ в присутствии соединения 1ZC (2,93 г, 11,37 ммоль, 1,10 экв.), DIEA (5,39 г, 41,71 ммоль, 4,03 экв.) и NaBH(OAc)3 (4,39 г, 20,71 ммоль, 2,00 экв.). Реакционную смесь встряхивали в течение 6 ч при температуре окружающей среды, затем нейтрализовали с помощью 60 мл NaHCO3 (насыщ.) и экстрагировали 3 раза с помощью 20 мл AcOEt. Органические фазы объединяли, промывали дважды с помощью 20 мл NaCl (насыщ.), сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:15), получая 2,7 г (61%) соединения 2Н в форме белого твердого вещества.
Соединение 2I: (S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутановая кислота
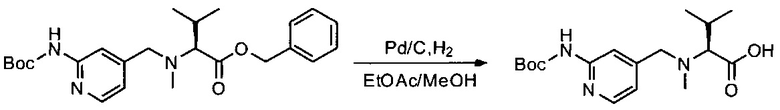
Соединение 2Н (500 мг, 1,17 ммоль, 1,00 экв.) растворяли в 10 мл AcOEt и 2 мл метанола в присутствии Pd/C (250 мг) и гидрогенировали в течение 3 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 254 мг (64%) соединения 2I в форме бесцветного твердого вещества
Соединение 2J: трет-бутил-(4-((3S,6S,9S,10R)-9-((S)-втор-бутил)-10-(2-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-3,6-диизопропил-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)пиридин-2-ил)карбамат
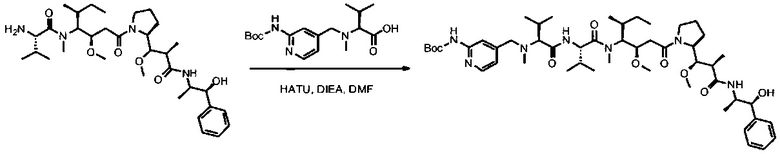
Соединение 2J получали таким же образом, как соединение 1ZG, из амина 2D (85,2 мг, 0,14 ммоль, 1,50 экв.), кислоты 2I (31,7 мг, 0,09 ммоль, 1,00 экв.), HATU (42,9 мг, 0,11 ммоль, 1,20 экв.) и DIEA (36,7 мг, 0,28 ммоль, 3,02 экв.) в ДМФА (3 мл). После выпаривания досуха получали 100 мг неочищенного продукта в форме белого твердого вещества.
Соединение 2J (100 мг, 0,11 ммоль, 1,00 экв.) растворяли в 2 мл DCM и 1 мл TFA. Реакционную смесь встряхивали в течение 1 ч при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок (80 мг) очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 2 получали с выходом 6% (6,3 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,8 мл/мин, от 10% до 95% ACN в воде (0,05% TFA) в течение 6 мин); ESI (C45H73N7O7, точная масса 823,56) m/z: 824,5 (МН+) и 412,9 (М, 2Н+/2, 100%), 3,21 мин (99,2%, 210 нм)
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,81-7,79 (m, 1Н); 7,39-7,29 (m, 5Н); 6,61-6,59 (m, 2Н); 4,84-4,52 (m, 1Н); 4,32-4,02 (m, 1Н); 3,90-2,98 (m, 10Н); 2,90-2,78 (m, 1Н); 2,55-0,81 (m, 39Н).
Референсное соединение 3
Метил-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты
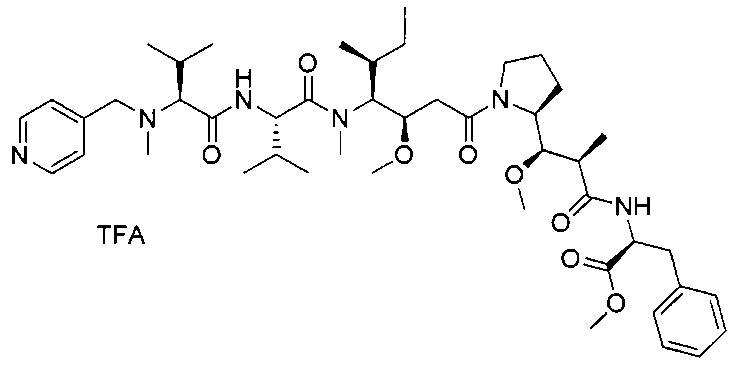
Соединение 3А: трет-бутил-(S)-2-((1R,2R)-1-метокси-3-(((S)-1-метокси-1-оксо-3-фенилпропан-2-ил)амино)-2-метил-3-оксопропил)пирролидин-1-карбоксилат
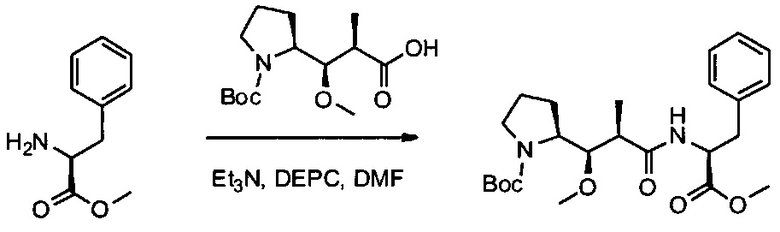
Соединение 1D (3 г, 10,44 ммоль, 1,00 экв.) и метил-(S)-2-амино-3-фенилпропаноат (2,25 г, 12,55 ммоль, 1,20 экв.) растворяли в инертной атмосфере в ДМФА (40 мл). Раствор охлаждали до 0°C, затем по каплям добавляли DEPC (1,67 мл, 1,05 экв.) и TEA (3,64 мл, 2,50 экв.). Реакционную смесь встряхивали в течение 2 ч при 0°C, затем при температуре окружающей среды в течение ночи. Реакционную смесь разводили 100 мл воды и экстрагировали три раза с помощью 50 мл EtOAc. Органические фазы объединяли, промывали один раз с помощью 100 мл KHSO4 (1 моль/л), один раз с помощью 100 мл NaHCO3 (насыщ.), один раз с помощью 100 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали под давлением, получая 4 г (85%) соединения 3А в форме бесцветного масла.
Соединение 3В: 2,2,2-трифторацетатметил-(S)-2-((2R,3R)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамидо)-3-фенилпропаноат
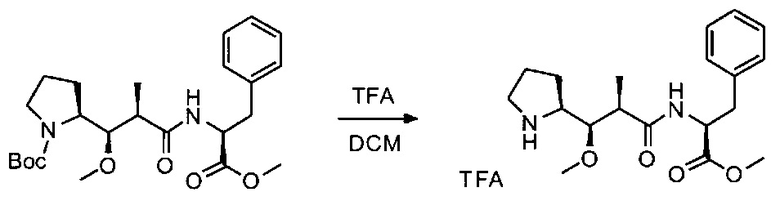
Соединение 3А (5 г, 11,15 ммоль, 1,00 экв.) растворяли в инертной атмосфере в DCM (40 мл). Добавляли TFA (25 мл) и раствор встряхивали в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, получая 8 г соединения 3В в форме желтого масла.
Соединение 3С: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
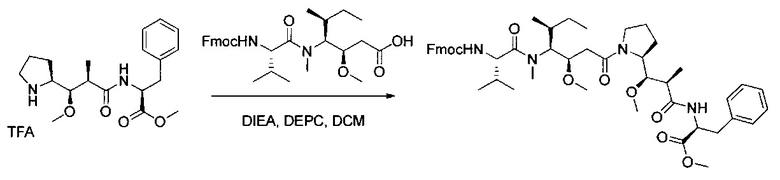
Соединения 3В (8,03 г, 17,36 ммоль, 1,00 экв.) и 1W (9,1 г, 17,34 ммоль, 1,00 экв.) растворяли в инертной атмосфере в DCM (80 мл). Раствор охлаждали до 0°C, затем по каплям добавляли DEPC (2,8 мл) и DIEA (12 мл). Реакционную смесь встряхивали в течение 2 ч при 0°C, затем при температуре окружающей среды в течение ночи. Реакционную смесь разводили в 200 мл воды и экстрагировали три раза с помощью 50 мл DCM. Органические фазы объединяли, промывали один раз с помощью 50 мл KHSO4 (1 моль/л), один раз с помощью 50 мл NaHCO3 (насыщ.), один раз с помощью 50 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 5 г (34%) соединения 3С в форме желтого твердого вещества.
Соединение 3D: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-амино-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
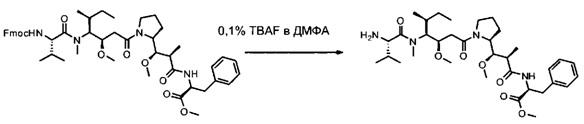
Соединение 3С (5,5 г, 6,43 ммоль, 1,00 экв.) растворяли в инертной атмосфере в растворе тетрабутиламмония фторида (TBAF, 2,61 г, 9,98 ммоль, 1,55 экв.) в ДМФА (100 мл). Раствор встряхивали при температуре окружающей среды в течение 2 ч, затем разводили 100 мл воды и экстрагировали три раза с помощью 50 мл EtOAc. Органические фазы объединяли, затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 3,3 г (81%) соединения 3D в форме желтого твердого вещества.
Соединение 3Е: бензил-(S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутаноат
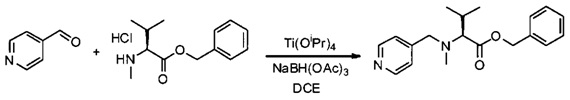
Пиридин-4-карбальдегид (1 г, 9,34 ммоль, 1,00 экв.) растворяли в 10 мл 1,2-дихлорэтана (DCE) в присутствии соединения 1ZC (2,9 г, 11,25 ммоль, 1,21 экв.) и титана изопропоксида (IV) (4,19 мл, 1,40 экв.). Смесь встряхивали при температуре окружающей среды в течение 30 минут, затем добавляли 2,77 г NaBH(OAc)3 (13,07 ммоль, 1,40 экв.). Реакционную среду оставляли встряхиваться в течение ночи, затем нейтрализовали с помощью 100 мл воды и смесь экстрагировали 3 раза с помощью 50 мл AcOEt. Органические фазы объединяли и выпаривали досуха. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:20), получая 1,3 г (45%) соединения 3Е в форме бесцветного масла.
Соединение 3F: (S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутановая кислота

Соединение 3Е (800 мг, 2,56 ммоль, 1,00 экв.) растворяли в 30 мл AcOEt в присутствии Pd/C (300 мг) и гидрогенировали в течение 3 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью DCM и МеОН (от 100:1 до 5:1), получая 100 мг (18%) соединения 3F в форме белого твердого вещества.
Соединения 3D (50 мг, 0,08 ммоль, 1,00 экв.) и 3F (26,34 мг, 0,12 ммоль, 1,50 экв.) растворяли в 3 мл DCM. Раствор охлаждали до 0°C, затем добавляли 0,018 мл DEPC и 0,0392 мл DIEA. Реакционную смесь встряхивали при 0°C в течение 2 ч, затем при температуре окружающей среды в течение ночи. Реакционную среду концентрировали при пониженном давлении и осадок (70 мг) очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 3 получали с выходом 27% (20 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% ACN в воде (0,05% TFA) в течение 8 минут); ESI (C46H72N6O8, точная масса 836,5) m/z: 837,5 (МН+) и 419,4 (М, 2Н+/2 (100%)), 7,04 мин (90,0%, 210 нм)
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,76-8,74 (m, 2Н); 8,53-8,48 (m, 0,4Н, NHCO неполный обмен); 8,29-8,15 (m, 0,8Н, NHCO неполный обмен); 8,01 (s, 2Н), 7,31-7,22 (m, 5Н), 4,88-4,68 (m, 3Н); 4,31-4,07 (m, 2Н); 3,94-2,90 (m, 18Н); 2,55-0,86 (m, 38Н).
Референсное соединение 4
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, соль трифторуксусной кислоты

Соединение 3 (100 мг, 0,11 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли встряхиваться в течение ночи, затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 20 мг (20%) соединения 4 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% ACN в воде (0,05% TFA) в течение 8 минут); ESI (C45H70N6O8, точная масса 822,5) m/z: 823,5 (МН+) и 412,4 (М, 2Н+/2, 100%), 6,84 мин (89,1%, 210 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,79-8,78 (m, 2Н); 8,09 (m, 2Н); 7,30-7,21 (m, 5Н); 4,80-4,80 (m, 1Н), 4,36-0,87 (m, 58Н).
Соединение 6
Метил-(S)-2((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
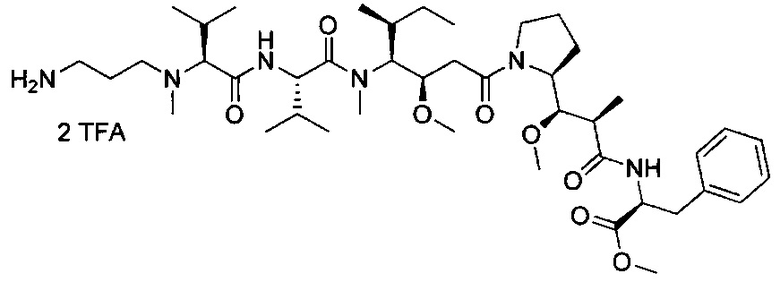
Соединение 6А: метил-(2S)-2-[(2R)-2-[(R)-[(2S)-1-[(3R,4S,5S)-4-[(2S)-2-[(2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метил
бутанамидо]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноил]пирролидин-2-ил](метокси)метил]пропанамидо]-3-фенилпропаноат
Соединение 3D (157,5 мг, 0,25 ммоль, 1,00 экв.) растворяли при 0°C в инертной атмосфере в 3 мл DCM в присутствии карбоновой кислоты 1ZF (78,7 мг, 0,27 ммоль, 1,10 экв.), DEPC (46 мкл) и DIEA (124 мкл). Реакционную смесь встряхивали в течение 2 ч при низкой температуре, затем ледяную баню удаляли и продолжали встряхивание в течение 4 ч. Затем ее концентрировали при пониженном давлении, получая 200 мг соединения 6А в форме неочищенного желтого масла. Его использовали как таковое на следующем этапе.
Соединение 6А (200 мг, 0,22 ммоль, 1,00 экв.) растворяли в инертной атмосфере при 0°C в 2 мл DCM. По каплям добавляли TFA (1 мл) и ледяную баню удаляли. Реакционную смесь встряхивали в течение 1 ч при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм), получая 60 мг (26%, выход в 2 этапа) соединения 6 в форме белого твердого вещества.
LC/MS/UV (Zorbax Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°C, от 30 до 80% метанола в воде (0,1% H3PO4) в течение 18 минут); ESI (C43H74N6O8, точная масса 802,56) m/z: 804 (МН+); 11,50 мин (91,5%, 210 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,52 (d, 0,3H, NHCO неполный обмен); 8,25 (d, 0,5Н, NHCO неполный обмен); 7,30-7,22 (m, 5Н); 4,9-4,6 (m, 3Н); 4,2-4,0 (m, 1Н); 4,0-0,86 (m, 61Н).
Соединение 7
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-соль трифторуксусной кислоты
Соединение 6 (70 мг, 0,08 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (2,5 мл) и пиперидина (5 мл). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм), получая 14,6 мг (21%) соединения 7 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°C, от 0 до 80% метанола в воде (0,05% TFA) в течение 8 минут); ESI (C42H72N6O8, точная масса 788,54) m/z: 790 (МН+), 5,71 мин (96,83%, 210 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,42 (d, 0,3H, NHCO неполный обмен); 8,15 (d, 0,2Н, NHCO неполный обмен); 7,31-7,21 (m, 5Н); 4,9-4,6 (m, 3Н); 4,25-4,0 (m, 1Н); 4,0-0,86 (m, 59Н).
Соединение 8
(S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты
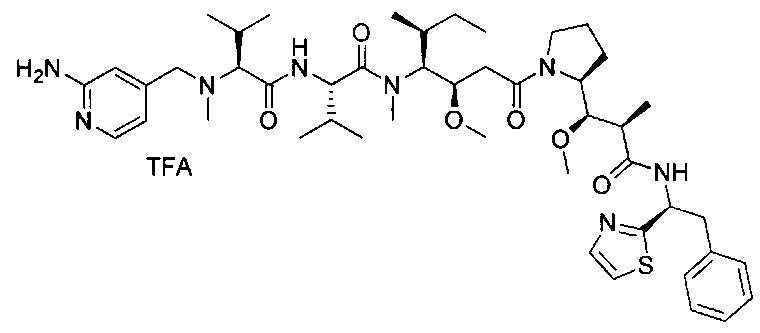
Соединение 8А: трет-бутил-(4-((3S,6S,9S,10R)-9-((S)-втор-бутил)-3,6-диизопропил-10-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)пиридин-2-ил)карбамат
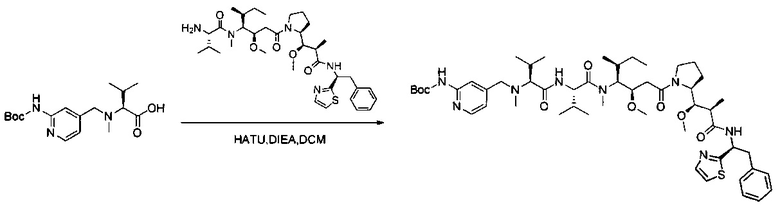
Соединение 8А синтезировали аналогично соединению 2J из амина 1Y (39 мг, 0,06 ммоль, 1,00 экв.), кислоты 2I (20 мг, 0,06 ммоль, 1,00 экв.), HATU (27 мг, 0,07 ммоль, 1,20 экв.) и DIEA (23,2 мг, 0,18 ммоль, 3,01 экв.) в DCM (3 мл). Неочищенный продукт не очищали.
Соединение 8: Соединение 8 синтезировали аналогично соединению 2 из промежуточного продукта 8А (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт (100 мг) очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 18% до 31% ACN в течение 7 мин, затем от 31%) до 100%) ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 8 получали с выходом 8% (8 мг) в форме белого твердого вещества.
LC/MS/UV (Atlantis Т3 колонка, 3 мкм, 4,6×100 мм; 35°C; 1,8 мл/мин, от 25% до 80% ACN в воде (0,05% TFA) в течение 7 мин); ESI (C47H72N8O6S, точная масса 876,5) m/z: 877,5 (МН+) и 439,5 (М, 2Н+/2, 100%), 4,87 мин (95,1%, 254 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,83-7,78 (m, 2Н); 7,56-7,52 (m, 1Н); 7,34-7,17 (m, 5Н); 6,64-6,62 (m, 2Н); 5,77-5,61 (m, 1Н); 4,86-4,68 (m, 2Н); 4,25-4,05 (m, 1Н); 3,87-2,83 (m, 17Н); 2,56-0,84 (m, 37Н).
Соединение 9
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты
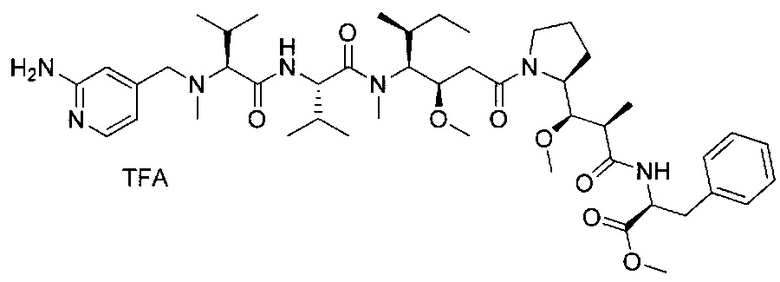
Соединение 9А: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
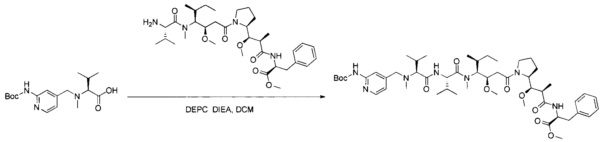
Соединение 9А синтезировали аналогично соединению 3 из амина 3D (170 мг, 0,27 ммоль, 1,00 экв.), кислоты 2I (99,7 мг, 0,30 ммоль, 1,10 экв.), DEPC (0,049 мл, 1,05 экв.) и DIEA (0,133 мл, 3,00 экв.) в DCM (5 мл). Неочищенный продукт очищали на колонке с силикагелем смесью EtOAc и РЕ (1:1), получая 200 мг (78%) соединения 9А в форме бледно-желтого твердого вещества.
Соединение 9: Соединение 9 синтезировали аналогично соединению 2 из промежуточного продукта 9А (200 мг, 0,21 ммоль, 1,00 экв.) в DCM (4 мл) и TFA (2 мл). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 9 получали с выходом 10% (20 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C46H73N7O8, точная масса 851,6) m/z: 852,5 (МН+) и 426,9 (М, 2Н+/2, 100%), 6,92 мин (92,7%, 254 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,51-8,45 (m, 0,5Н, NH неполный обмен); 8,30-8,24 (m, 0,3H, NH неполный обмен); 8,17-8,07 (m, 0,8Н, NH неполный обмен); 7,79-7,77 (m, 1Н); 7,36-7,18 (m, 5Н); 7,21-7,16 (m, 1Н); 6,94- 6,89 (m, 1H); 4,85-4,65 (m, 3Н); 4,20-3,10 (m, 20H); 3,00-2,85 (m, 2H); 2,55-0,80 (m, 36H).
Соединение 10
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, соль трифторуксусной кислоты
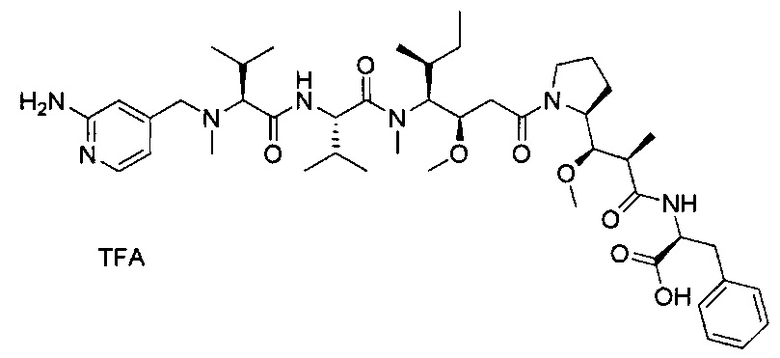
Соединение 10: Соединение 9 (100 мг, 0,11 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, а затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 32,2 мг (33%) соединения 10 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C45H71N7O6, точная масса 837,5) m/z: 838,5 (МН+) и 419,9 (М, 2Н+/2, 100%), 6,81 мин (97,7%, 220 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,41-8,32 (m, 0,3Н, NH неполный обмен); 8,20-8,07 (m, 0,8Н, NH неполный обмен); 7,82-7,75 (m, 1Н); 7,36-7,158 (m, 5Н); 7,12-7,03 (m, 1Н); 6,94-6,88 (m,1Н); 4,85-4,66 (m, 3Н); 4,20-3,10 (m, 16Н); 3,00-2,85 (m, 2Н); 2,57-0,80 (m, 37Н).
Соединение 11
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенэтил)амино)бутанамидо)бутанамид, соль трифторуксусной кислоты
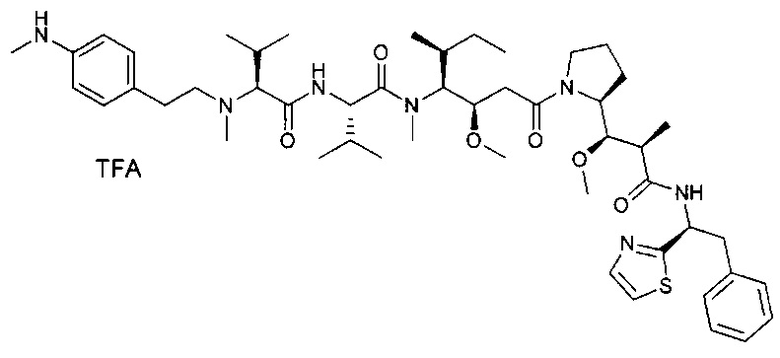
Соединение 11А: трет-бутил-N-[4-(2-гидроксиэтил)фенил]карбамат
Ди-трет-бутилдикарбонат (16,7 г, 77 ммоль, 1,05 экв.) добавляли к раствору 2-(4-аминофенил)этанола (10 г, 72,9 ммоль, 1 экв.) в ТГФ (200 мл), и реакционную смесь встряхивали в течение ночи при температуре окружающей среды. Смесь разводили EtOAc (200 мл), промывали водой (200 мл), затем 1 М HCl (100 мл), затем насыщенным водным раствором NaHCO3 (100 мл), затем насыщенным солевым раствором (100 мл). Органическую фазу сушили над MgSO4, затем выпаривали досуха при пониженном давлении. Неочищенный продукт растирали дважды с гептаном (150 мл) и сушили в вакууме, получая соединение 11А в виде белого твердого вещества (14,7 г, 84%).
Соединение 11В: трет-бутил N-[4-(2-оксоэтил)фенил]карбамат
Соединение 11А (2,5 г, 10,5 ммоль, 1,00 экв.) растворяли в 25 мл DCM, затем охлаждали до -78°C. По каплям добавляли раствор периодинана Десс-Мартина (DMP, 6,71 г, 15,8 ммоль, 1,5 экв.) в DCM (10 мл). Ледяную баню удаляли и продолжали встряхивание в течение 1 ч при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 60 мл 50/50 смеси насыщенного водного раствора натрия бикарбоната и насыщенного водного раствора Na2S2O3. Полученный раствор экстрагировали 3 раза с помощью 30 мл EtOAc. Органические фазы объединяли, промывали дважды насыщенным водным раствором NaCl, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ 1/15), получая 1,0 г (40%) соединения 11В в форме бледно-желтого твердого вещества.
Соединение 11С: бензил-(2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил)этил](метил)амино]-3-метилбутаноат
Соединение 1ZC (3,5 г, 13,6 ммоль, 1,1 экв.) растворяли в ТГФ (30 мл) в присутствии DIEA (6,4 г, 49,7 ммоль, 4,0 экв.), альдегида 11В (2,9 г, 12,3 ммоль, 1,0 экв.) и натрия триацетоксиборогидрида (5,23 г, 49,7 ммоль, 2,0 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем нейтрализовали с помощью 60 мл насыщенного раствора натрия бикарбоната. Полученный раствор экстрагировали 3 раза с помощью 30 мл EtOAc. Органические фазы объединяли, промывали дважды насыщенным водным раствором NaCl, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ 1:20), получая 3,7 г (68%) соединения 11С в форме желтого масла.
Соединение 11D: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил)этил](метил)амино]-3-метилбутановая кислота
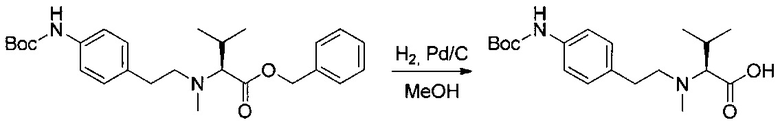
Соединение 11С (2 г, 4,5 ммоль, 1 экв.) растворяли в 10 мл метанола в присутствии Pd/C (2 г) и гидрогенировали в течение 2 ч при нормальной температуре и давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 1,2 г (75%) соединения 11D в форме желтого масла.
Соединение 11Е: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил](метил)амино]фенил)этил](метил)амино]-3-метилбутановая кислота
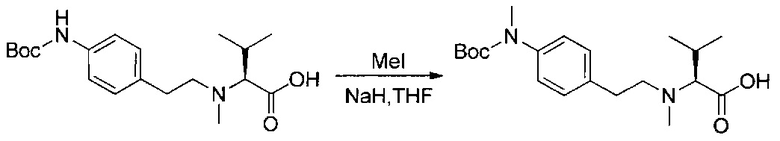
Соединение 11D (1,2 г, 3,4 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (20 мл). Реакционную среду охлаждали на ледяной бане, после чего порциями добавляли NaH (60% в масле, 549 мг, 13,7 ммоль, 4,0 экв.), а затем йодометан (4,9 г, 34 ммоль, 10 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, затем нейтрализовали водой и промывали 100 мл EtOAc. pH водного раствора подводили до 6-7 с помощью 1 н HCl. Этот водный раствор экстрагировали 3 раза с помощью 100 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 800 мг (64%) соединения 11Е в форме желтого твердого вещества.
Соединение 11F: трет-бутил-N-[4-(2-[[(1S)-1-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]этил)фенил]-N-метилкарбамат
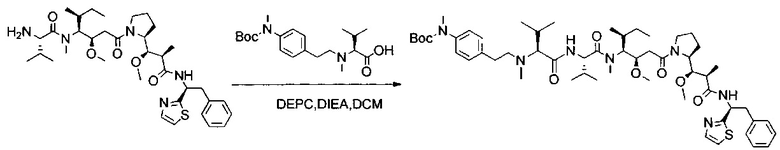
Соединение 11F получали таким же образом, как соединение 6А, из амина 1Y (150 мг, 0,22 ммоль, 1,2 экв.) и кислоты 11Е (70 мг, 0,19 ммоль, 1,0 экв.). После очистки на силикагеле (EtOAc/РЕ 1:1) получали 100 мг (52%) нужного продукта в форме бледно-желтого твердого вещества.
Соединение 11 получали аналогично соединению 1 из промежуточного продукта 11F (100 мг, 0,1 ммоль). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 11 получали с выходом 39% (39,7 мг) в форме белого твердого вещества.
LC/MS/UV (Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°C, 50 до 95% метанол в воде (0,05% TFA) в течение 18 минут); ESI (C50H77N7O6S, точная масса 903,57) m/z: 904,5 (МН+), 7,53 мин (93,68%, 254 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,84 (d, 0,5Н, NHCO неполный обмен); 8,7-8,5 (m, 0,9Н, NHCO неполный обмен); 7,76-7,73 (m, 1Н); 7,55-7,4 (m, 1Н); 7,28-7,22 (m, 7Н); 7,08-7,05 (m, 2Н); 5,51-5,72 (m, 1Н); 4,9-4,80 (m, 2Н); 4,3-0,7 (m, 60Н).
Соединение 12
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенэтил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты
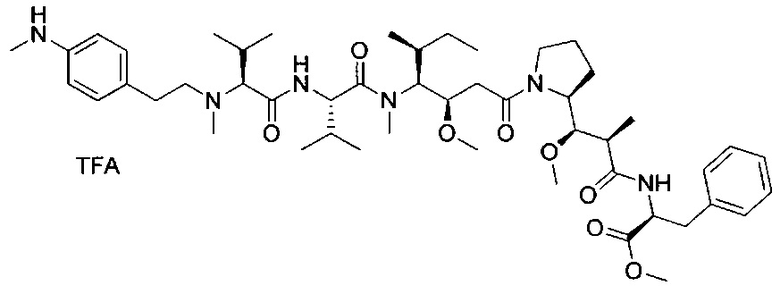
Аналогичным образом, как на последних этапах синтеза соединения 1, соединение 12 получали в два этапа из амина 3D (118 мг, 0,19 ммоль) и кислоты 11Е (82 мг, 0,22 ммоль). Окончательный осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 12 получали с выходом 7% (13,7 мг) в форме белого твердого вещества.
LC/MS/UV (Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°C, 40 до 95% метанол в воде (0,05% TFA) в течение 18 минут); ESI (C49H78N6O8, точная масса 878,59) m/z: 879,7 (МН+), 10,07 мин (90,6%, 254 нм).
1H: NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,40 (se, 2Н); 7,38-7,22 (m, 7Н); 4,95-4,7 (m, 3Н); 4,2-4,0 (m, 1Н); 3,9-0,86 (m, 62Н).
Соединение 13
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенэтил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, соль трифторуксусной кислоты
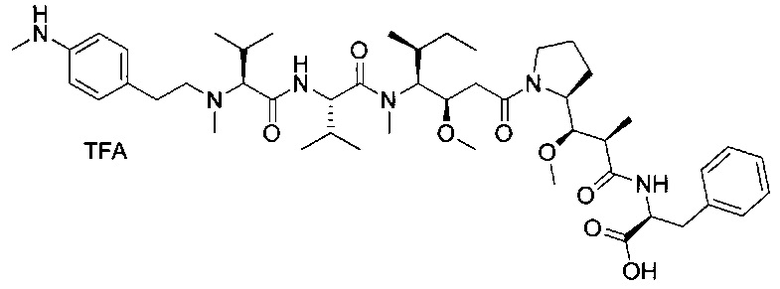
Соединение 13 получали аналогично соединению 7 из соединения 12 (100 мг, 0,10 ммоль). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 13 получали с выходом 20% (20 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°C, от 10 до 95% метанол в воде (0,05% TFA) в течение 8 минут); ESI (C48H76N6O8, точная масса 864,57) m/z: 865,6 (МН+), 6,05 мин (90,9%, 210 нм).
1Н NMR: (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,32-7,19 (m, 9Н); 4,9-4,65 (m, 3Н); 4,2-4,0 (m, 1Н); 3,9-0,86 (m, 59Н).
Соединение 14
(S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты
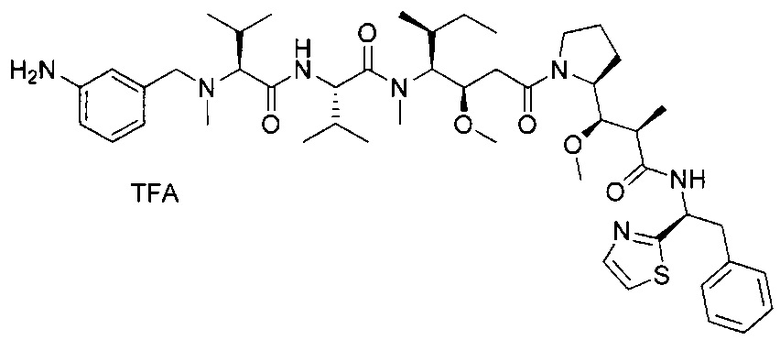
Соединение 14А: трет-бутил-(3-(гидроксиметил)фенил)карбамат
(3-аминофенил)метанол (3 г, 24,36 ммоль, 1,00 экв.) растворяли в ТГФ (60 мл), после чего добавляли ди-трет-бутил дикарбонат (6,38 г, 29,23 ммоль, 1,20 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, и затем реакционную смесь разводили добавлением 200 мл воды. Продукт экстрагировали 3 раза с помощью 100 мл AcOEt и затем органические фазы снова объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая неочищенный продукт (13,85 г соединения 14А) в форме желтого масла.
Соединение 14В: трет-бутил-(3-формилфенил)карбамат
Соединение 14А (13,8 г, 61,81 ммоль, 1,00 экв.) растворяли в DCE (400 мл) и затем добавляли MnO2 (54 г, 621,14 ммоль, 10,05 экв.). Смесь оставляли встряхиваться при температуре окружающей среды в течение 3 дней, после чего твердые вещества удаляли путем фильтрации. Фильтрат выпаривали досуха и осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:30), получая 3 г (22%) соединения 14В в форме белого твердого вещества.
Соединение 14С: бензил-(S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутаноат
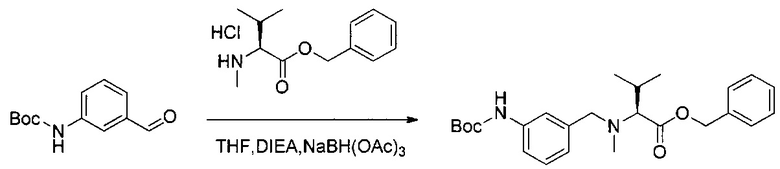
Соединение 14В (1 г, 4,52 ммоль, 1,00 экв.) растворяли в 20 мл ТГФ в присутствии соединения 1ZC (1,16 г, 4,50 ммоль, 1,00 экв.), DIEA (3 мл) и NaBH(OAc)3 (1,92 г, 9,06 ммоль, 2,01 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, а затем нейтрализовали с помощью 100 мл воды и экстрагировали 3 раза с помощью 50 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:50), получая 1,9 г (99%) соединения 14С в форме белого твердого вещества.
Соединение 14D: (S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутановая кислота
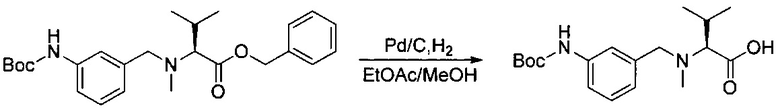
Соединение 14С (1 г, 2,34 ммоль, 1,00 экв.) растворяли в 30 мл AcOEt и 4 мл метанола в присутствии Pd/C (400 мг) и гидрогенировали в течение 1 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 680 мг (86%) соединения 14D в форме белого твердого вещества.
Соединение 14Е: трет-бутил-(3-((3S,6S,9S,10R)-9-((S)-втор-бутил)-3,6-диизопропил-10-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)фенил)карбамат

Соединение 14Е синтезировали аналогично соединению 3 из амина 1Y (100 мг, 0,15 ммоль, 1,00 экв.), кислоты 14D (102,27 мг, 0,30 ммоль, 2,00 экв.), DEPC (0,053 мл) и DIEA (0,046 мл) в DCM (3 мл). Неочищенный продукт (80 мг) очищали на колонке с силикагелем смесью EtOAc и РЕ (1:1), получая 100 мг (67%) соединения 14Е в форме бледно-желтого твердого вещества.
Соединение 14 синтезировали аналогично соединению 2 из промежуточного продукта 14Е (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт (80 мг) очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 14 получали с выходом 10% (10 мг) в форме белого твердого вещества.
LC/MS/UV (Eclipse plus С8 колонка, 3,5 мкм, 4,6×150 мм; 40°C; 1,0 мл/мин, от 40% до 95% МеОН в воде (0,05% TFA) в течение 18 минут); ESI (C48H73N7O6S, точная масса 875,5) m/z: 876,5 (МН+) и 438,9 (М, 2Н+/2, 100%), 11,35 мин (95,6%, 210 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,92-8,86 (m, 0,4Н, NH неполный обмен); 8,70-8,54 (m, 0,6Н, NH неполный обмен); 7,88-7,78 (m, 1Н); 7,60-7,50 (m, 1Н); 7,45-6,97 (m, 9Н); 5,80-5,65 (m, 1Н); 4,85-4,70 (m, 1Н); 4,40-0,80 (m, 56Н).
Соединение 15
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты
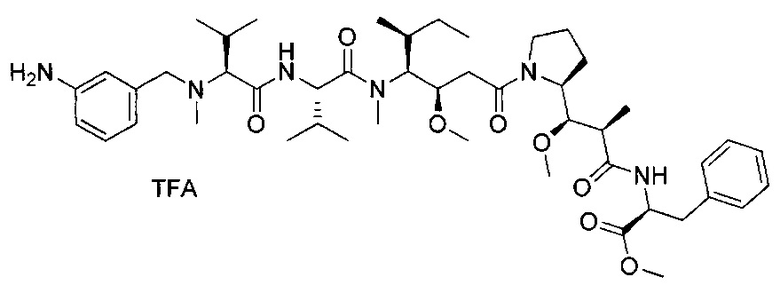
Соединение 15А: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
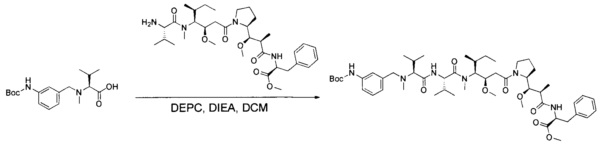
Соединение 15А синтезировали аналогично соединению 3 из амина 3D (200 мг, 0,32 ммоль, 1,00 экв.), кислоты 14D (212,6 мг, 0,63 ммоль, 2,00 экв.), DEPC (0,1103 мл) и DIEA (0,157 мл, 3,00 экв) в DCM (5 мл). Неочищенный продукт очищали на колонке с силикагелем смесью EtOAc и РЕ (11), получая 200 мг (67%) соединения 15А в форме желтого твердого вещества.
Соединение 15 синтезировали аналогично соединению 2 из промежуточного продукта 15А (200 мг, 0,21 ммоль, 1,00 экв) Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм, элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters УФ-детектор 2545 при 254 нм и 220 нм). Соединение 15 получали с выходом 19% (38,6 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C47H74N6O8, точная масса 850,5) m/z: 851,5 (МН+) и 426,4 (М, 2Н+/2, 100%), 6,61 мин (91,1%, 210 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,53-7,42 (m, 1Н); 7,35-7,18 (m, 8Н); 4,88-4,79 (m, 2Н); 4,42-4,00 (m, 3Н); 3,93-2,71 (m, 22Н); 2,61-0,81 (m, 33Н).
Соединения 16-20
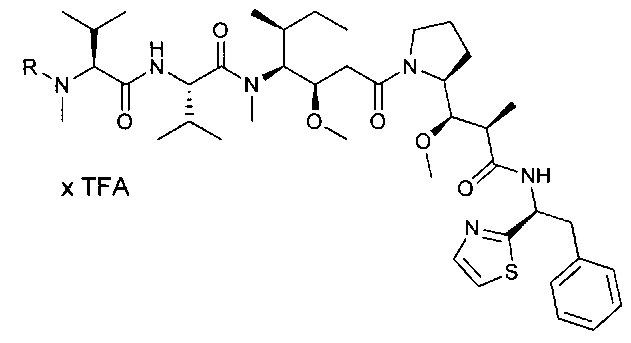
Соединения 16-20 получали аналогично соединению 1 из аминов 1Y и 1ZC и соответствующих альдегидов.
Трет-бутил-(4-оксобутил)карбамат, вовлеченный в получение соединения 16, получали в виде соединения 1ZE в 2 этапа из 4,4-диэтоксибутан-1-амина.
Трет-бутил-N-метил-N-(2-оксоэтил)карбамат, вовлеченный в получение соединения 17, был коммерческим.
2-(2-((трет-бутилдиметилсилил)окси)этокси)ацетальдегид, вовлеченный в получение соединения 18, получали в 2 этапа следующим образом:
2-(2-гидроксиэтокси)этан-1-ол (7 г, 66 ммоль, 9,9 экв.) растворяли в инертной атмосфере в пиридине (10 мл) в присутствии 4-диметиламинопиридина (DMAP, 80 мг, 0,65 ммоль, 0,1 экв.). Раствор охлаждали до 0°C, затем порциями добавляли TBDMSCI (1 г, 6,6 ммоль, 1,0 экв.). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, разводили в 100 мл EtOAc и дважды тщательно промывали с помощью 100 мл 1 н HCl и дважды насыщенным водным раствором NaCl. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 1,3 г (88%) 2-[2-[(трет-бутилдиметилсилил)окси]этокси]этан-1-ола в форме бесцветного масла.
Оксалилхлорид (760 мг, 6 ммоль, 1,3 экв.) растворяли в инертной атмосфере в DCM (40 мл) и охлаждали до -78°C. Добавляли по каплям диметилсульфоксид (ДМСО, 1,07 г, 13,7 ммоль, 3 экв.), разведенный в DCM (5 мл). После встряхивания в течение 30 минут при низкой температуре добавляли 2-[2-[(трет-бутилдиметил силил)окси]этокси]этан-1-ол (1 г, 4,5 ммоль, 1,0 экв.), растворенный в 5 мл DCM. Встряхивание продолжали в течение 1 ч при низкой температуре, а затем добавляли TEA (2,78 г, 27 ммоль, 6 экв.). Реакционную смесь встряхивали в течение 15 мин при -78°C и в течение ночи при температуре окружающей среды, а затем нейтрализовали с помощью 100 мл воды. Затем ее экстрагировали 3 раза с помощью 100 мл DCM. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ 1:20) и получали 0,8 г (80%) 2-[2-[(трет-бутилдиметилсилил)окси]этокси]ацетальдегида в форме бесцветного масла.
Трет-бутил-4-формилфенилкарбонат, вовлеченный в получение соединения 19, получали на одном этапе следующим образом: 4-гидроксибензальдегид (2,5 г, 20,5 ммоль, 1,0 экв.) растворяли в инертной атмосфере в ТГФ (20 мл) в присутствии 18-краун-6 (0,25 г) и калия карбоната (5 г). Реакционную смесь охлаждали до 0°C и затем добавляли ди-трет-бутилдикарбонат (5,8 г, 26,58 ммоль, 1,30 экв.). Встряхивание продолжали в течение 1 ч при низкой температуре, после чего реакционную смесь нейтрализовали с помощью 30 мл воды. Полученный раствор экстрагировали три раза с помощью 200 мл EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ 1:10) и получали 4,2 г (92%) трет-бутил-4-формилфенилкарбоната в форме бледно-желтого твердого вещества.
4-нитробензальдегид, вовлеченный в получение соединения 20, был коммерческим.
Синтез соединения 18 завершали снятием защиты силилированного спирта. Это проводили следующим образом: (S)-N-((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)(метил)амино)-3-метил-1-оксобутан-2-ил)-11-изопропил-2,2,3,3,10-пентаметил-4,7-диокса-10-аза-3-силадодекан-12-амид (40 мг, 0,04 ммоль, 1,0 экв.) растворяли в инертной атмосфере в ТГФ (10 мл) в присутствии TBAF (2 мг, 0,09 ммоль, 2 экв.) и встряхивали в течение 4 ч при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 50 мл воды, затем экстрагировали три раза с помощью 50 мл EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая соединение 18 в неочищенном виде.
Синтез соединения 20 завершали восстановлением нитро-группы. Это проводили следующим образом: (2S)-N-[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-2-[[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]карбамоил]-1-метокси-2-метилэтил]пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил]-N,3-диметил-2-[(2S)-3-метил-2-[метил[(4-нитрофенил)метил]амино]бутанамидо]бутанамид (40 мг, 0,05 ммоль, 1,0 экв.) растворяли в 15 мл этанола. Добавляли дигидратированный хлорид олова (II) (317 мг, 1,4 ммоль, 30 экв.) и раствор оставляли встряхиваться в течение 3 дней при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 50 мл воды, затем экстрагировали три раза с помощью 50 мл EtOAc. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая соединение 20 в неочищенном виде.

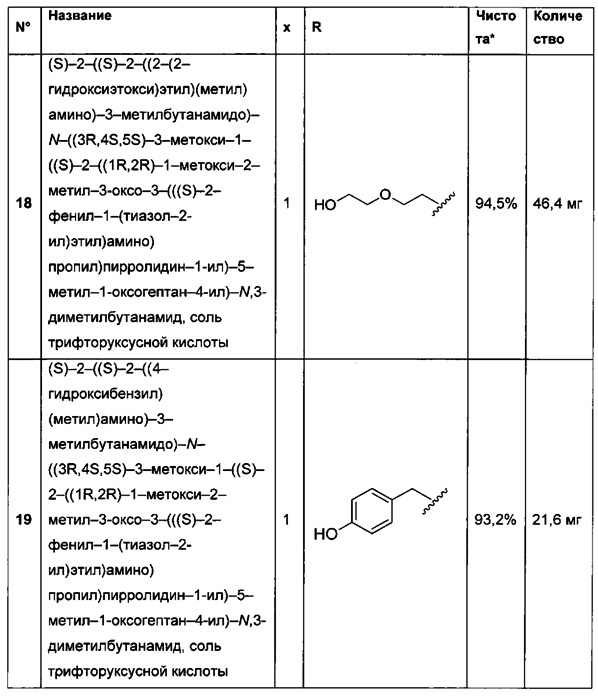
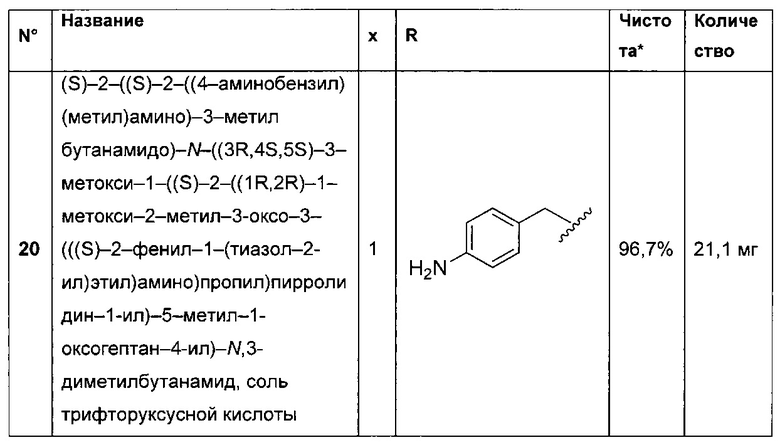
* Соединения очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм), получая соответствующие соли TFA в форме белых твердых веществ.
Характеризация конечных продуктов: Соединение 16 LC/MS/UV (Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°C, от 5 до 95% метанола в воде (0,05% TFA) в течение 18 минут); ESI: (C45H75N7O6S, точная масса 841,55) m/z 842,5 (МН+), 421,9 (100%, (М, 2Н+)/2); UV: 14,02 мин (94,9%, 210 нм). 1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,55-8,2 (m, 0,8Н, NHCO неполный обмен); 8,0 (0,55Н, NHCO неполный обмен); 7,70 (d, 1Н); 7,44 (d, 1Н); 7,21-7,15 (m, 5Н); 5,65-5,45 (m, 1Н); 4,8-4,5 (m, 2Н); 4,15-3,9 (m, 2Н); 3,8-0,6 (m, 59Н). Соединение 17 LC/MS/UV ESI: (C44H73N7O6S, точная масса 827,53) m/z 828 (МН+), 415 [100%, (М, 2Н+)/2]; UV: RT составвляет 6,72 мин (99,6%, 254 нм) 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,82-7,80 (m, 1Н); 7,56-7,54 (m, 1Н); 7,35-7,20 (m, 5Н); 5,8-5,55 (m, 1Н); 4,85-4,6 (m, 1Н); 4,25-4,05 (m, 1Н); 3,95-0,8 (m, 60Н). Соединение 18 LC/MS/UV (Atlantis Т3, 3 мкм, 4,6×100 мм; 1,2 мл/мин, 40°C, от 5 до 95% метанола в воде (0,05% TFA) в течение 7 минут) i; ESI: (C45H74N6O8S, точная масса 858,53) m/z 859 (МН+), 881 (MNa+), 430 (100%, (М, 2Н+)/2); UV: 4,85 мин (96,8%, 220 нм). 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,75-8,55 (m, 0,5Н, NHCO неполный обмен); 7,85-7,80 (m, 1Н); 7,6-7,5 (m, 1Н); 7,40-7,15 (m, 5Н); 5,8-5,6 (m, 1Н); 4,8-4,55 (m, 2Н); 4,15-4,0 (m, 1Н); 4,0-0,8 (m, 60Н). Соединение 19 LC/MS/UV ESI: (C48H72N6O7S, точная масса 876,52) m/z 877 (MH+), 439 [100%, (M, 2H+)/2]; UV: RT составляет 1,76 мин (93,2%, 220 нм). Соединение 20 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,85-7,80 (m, 1Н); 7,6-7,5 (m, 1Н); 7,4-7,15 (m, 5Н); 7,1-7,05 (m, 2Н); 6,73-6,70 (m, 2Н); 5,8-5,55 (m, 1Н); 5,0-4,7 (m, 2Н); 4,25-4,05 (m, 1Н); 4,0-0,8 (m, 54Н). LC/MS/UV ESI: (C48H73N7O7S, точная масса 875,53) m/z 876 (МН+), 439 [75%, (М, 2Н+)/2]; UV: RT составляет 4,83 мин (96,8%, 254 нм). 1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,85-7,80 (m, 1Н); 7,6-7,5 (m, 1Н); 7,4-7,1 (m, 7Н); 6,76-6,72 (m, 2Н); 5,8-5,55 (m, 1Н); 4,9-4,65 (m, 2Н); 4,25-4,05 (m, 1Н); 4,0-0,8 (m, 54Н).
Соединения 21-24
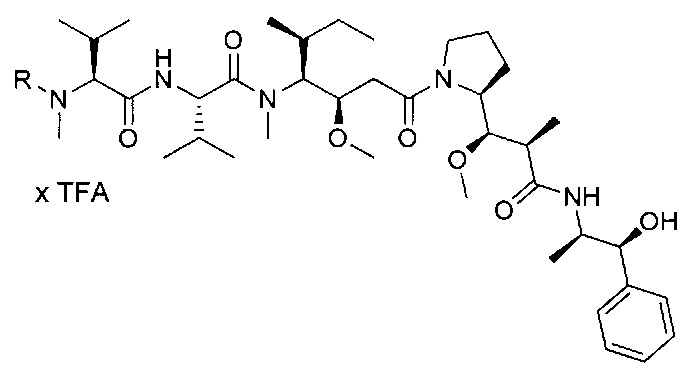
Соединения 21-24 получали аналогично соединениям 17-20, замещая амин 1Y амином 2D.


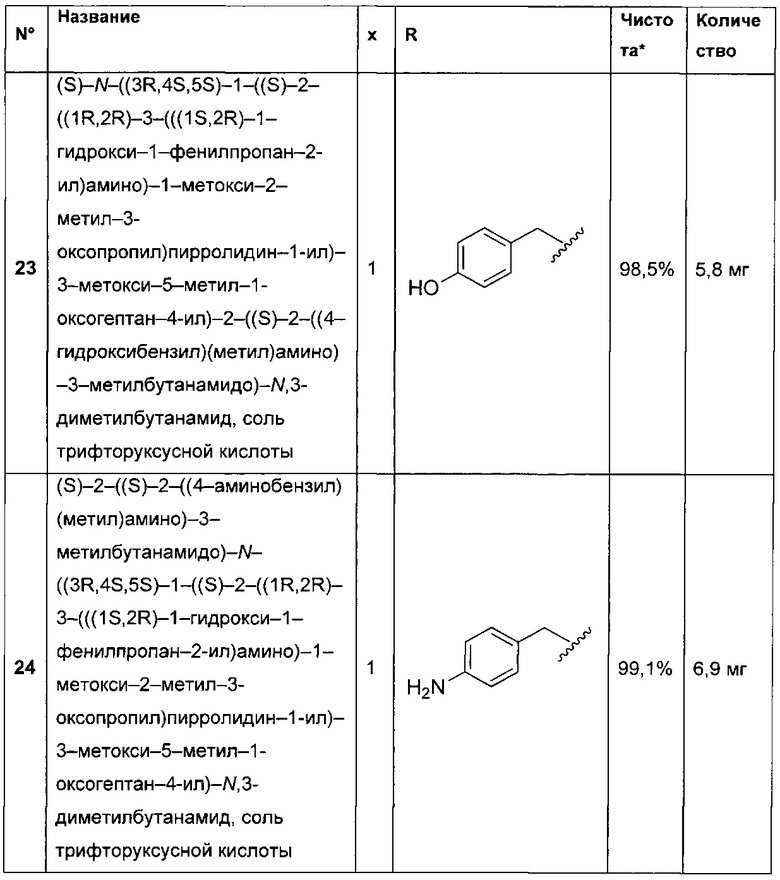
* Соединения очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм), получая соответствующие соли TFA в форме белых твердых веществ.
Характеризация конечных продуктов: Соединение 21 LC/MS/UV (ESI) (C42H74N6O7, точная масса 774,56) m/z 775 (МН+), 797 (MNa+), 388 (100%, (М, 2Н+)/2); UV: 3,14 мин (97,6%, 215 нм). 1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,05-7,7 (m, 0,8Н, NHCO неполный обмен); 7,45-7,15 (m, 5Н); 4,9-4,45 (m, 2Н); 4,35-4,00 (m, 2Н); 3,95-0,8 (m, 61Н). Соединение 22 LC/MS/UV (ESI) (C43H75N5O9, точная масса 805,56) m/z 806 (МН+), 828 (MNa+), 404 (100%, (М, 2Н+)/2); UV: 4,47 мин (95,6%, 215 нм). 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,1-7,7 (m, 0,4Н, NHCO неполный обмен); 7,45-7,15 (m, 5Н); 4,9-4,5 (m, 3Н); 4,4-4,05 (m, 2Н); 4,05-0,8 (m, 61Н). Соединение 23 LC/MS/UV (ESI) (C46H73N5O8, точная масса 823,55) m/z 824 (МН+), 846 (MNa+), 413 (100%, (М, 2Н+)/2); UV: 4,76 мин (98,5%, 215 нм). 1Н NMR (400 MHz, CDCl3, ppm): δ (наличие ротамеров) 7,5-7,2 (m, 5Н); 7,9-7,75 (m, 2Н); 5,5-5,3 (m, 1Н); 4,9-4,6 (m, 2Н); 4,55-4,15 (m, 4Н); 4,0-0,8 (m, 55Н). Соединение 24 LC/MS/UV (ESI) (C46H74N6O7) точная масса 822,56) m/z 823 (МН+), 845 (MNa+), 861 (MK+); UV: 3,68 мин (99,15%, 254 нм). 1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,0-7,7 (m, 0,5Н, NHCO неполный обмен); 7,5-7,0 (m, 7Н); 6,75-6,65 (m, 2Н); 4,85-4,5 (m, 2Н); 4,4-4,05 (m, 2Н); 4,0-0,8 (m, 56Н).
Соединение 26
(S)-2-((S)-2-((2-аминоэтил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, бис-соль трифторуксусной кислоты
Соединение 26А: бензил-(S)-2-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)-3-метилбутаноат

Соединение 26А получали аналогично соединению 2Н из амина 1ZC (1,3 г, 5,04 ммоль, 1,00 экв.), трет-бутил-(2-оксоэтил)карбамата (800 мг, 5,03 ммоль, 1,00 экв.), DIEA (3,52 г, 27,24 ммоль, 5,42 экв.) и NaBH(OAc)3 (2,25 г, 10,62 ммоль, 2,11 экв.) в ТГФ (25 мл). Смесь оставляли встряхиваться в течение ночи и нейтрализовали с помощью 50 мл воды. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (10:1), получая 0,6 г (33%) соединения 26А в форме бесцветного масла.
Соединение 26В: (S)-2-((2-((трет-бутоксикарбонил)амино)этил)(метил)амино)-3-метилбутановая кислота
Соединение 26А (600 мг, 1,65 ммоль, 1,00 экв.) растворяли в 40 мл ТГФ в присутствии Pd/C (300 мг) и гидрогенировали в течение 1 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью EtOAc и МеОН, получая 0,4 г (89%) соединения 26В в форме бесцветного масла.
Соединение 26С: трет-бутил-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)карбамат

Соединение 26С получали аналогично соединению 3 из амина 1Y (70 мг, 0,11 ммоль, 1,00 экв.), кислоты 26В (58,4 мг, 0,21 ммоль, 2,00 экв.), DEPC (0,032 мл) и DIEA (0,053 мл) в DCM (3 мл). После выпаривания досуха получали соединение 26С в форме желтого масла (100 мг).
Соединение 26: Соединение 26 синтезировали аналогично соединению 2 из промежуточного продукта 26С (100 мг, 0,11 ммоль, 1,00 экв.) в DCM (3 мл) и TFA (1,5 мл). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 45% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 26 получали с выходом 38% (38,1 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,0 мл/мин, от 5% до 95% МеОН в воде (0,05% TFA) в течение 18 минут); ESI (C43H71N7O6S, точная масса 813,52) m/z: 814,5 (МН+) и 407,9 (М, 2Н+/2, 100%), 15,78 мин (91,2%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,90-8,82 (m, 0,5Н, NH неполный обмен); 8,71-8,65 (m, 0,3H, NH неполный обмен); (7,85-7,77 (m, 1Н); 7,60-7,49 (m, 1Н); 7,37-7,15 (m, 5Н); 5,78-5,55 (m, 1Н); 4,82-4,62 (m, 1,6Н); 4,32-3,83 (m, 3,6Н); 3,75-3,35 (m, 7,4Н); 3,30-2,60 (m, 13Н); 2,58-0,80 (m, 42Н).
Соединение 27
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксифенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты

Соединение 27: Соединение 27 получали аналогично соединению 3 из амина 3D (70 мг, 0,11 ммоль, 1,00 экв.), кислоты 49С (55,5 мг, 0,22 ммоль, 2,00 экв.), DEPC (0,034 мл, 2,00 экв.) и DIEA (0,055 мл, 3,00 экв.) в DCM (3 мл). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 45% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 27 получали с выходом 3% (2,9 мг) в форме белого твердого вещества.
LC/MS/UV (Eclipse Plus С8 колонка, 3,5 мкм, 4,6×150 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C48H75N5O9, точная масса 866,56) m/z: 866,5 (МН+) и 433,9 (М, 2Н+/2, 100%), 6,61 мин (89,1%, 210 нм).
1Н NMR (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,70-8,49 (m, 0,9Н, NH/OH неполный обмен); 8,30-8,22 (m, 0,3H, NH неполный обмен); 7,36-7,02 (m, 7Н); 6,86-6,62 (m, 2Н); 4,82-4,69 (m, 2Н); 4,20-4,03 (m, 1Н); 3,91-3,33 (m, 12Н); 3,30-2,90 (m, 17Н); 2,55-0,80 (m, 35Н).
Соединение 28
(S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты
Соединение 28А: трет-бутил-(3-((3S,6S,9S,10R)-9-((S)-втор-бутил)-10-(2-((S)-2-((1R,2R)-3-(((1S.2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-3,6-диизопропил-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)фенил)карбамат
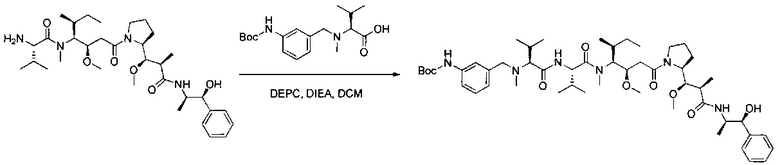
Соединение 28А получали аналогично соединению 3 из амина 2D (100 мг, 0,17 ммоль, 1,00 экв.), кислоты 14D (111,25 мг, 0,33 ммоль, 2,00 экв.), DEPC (0,058 мл) и DIEA (0,05 мл) в DCM (3 мл). Осадок очищали на колонке с силикагелем смесью EtOAc и гексана (1:1), получая 100 мг (66%) соединения 28А в форме белого твердого вещества.
Соединение 28: Соединение 28 синтезировали аналогично соединению 2 из промежуточного продукта 28А (100 мг, 0,11 ммоль, 1,00 экв.). Неочищенный продукт (80 мг) очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 28 получали с выходом 20% (20 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95%) МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C46H74N6O7, точная масса 822,56) m/z: 823,5 (МН+) и 412,4 (М, 2Н+/2, 100%), 12,45 мин (87,2%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,47-7,20 (m, 5Н); 7,10-7,01 (m, 1Н); 6,80-6,56 (m, 3Н); 4,82-4,52 (m, 3Н); 4,33-4,03 (m, 2Н); 3,91-3,82 (m, 0,5Н); 3,75-3,35 (m, 9,5Н); 3,28-3,10 (m, 2Н); 2,79-2,90 (m, 1Н); 2,60-2,40 (m, 2Н); 2,30-0,80 (m, 40Н).
Соединение 29
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, соль трифторуксусной кислоты
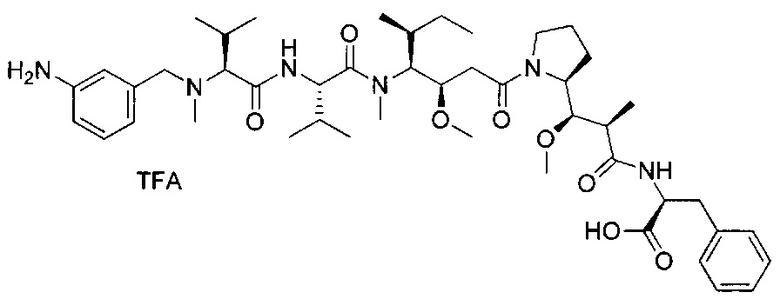
Соединение 15 (100 мг, 0,10 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды, а затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep C18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 20 мг (20%) соединения 29 в форме белого твердого вещества.
LC/MS/UV (Eclipse Plus C8 колонка, 3,5 мкм, 4,6×150 мм; 40°C; 1,0 мл/мин, от 40% до 95% МеОН в воде (0,05% TFA) в течение 18 минут); ESI (C46H72N6O8, точная масса 836,54) m/z: 837,5 (МН+) и 419,4 (М, 2Н+/2, 100%), 10,61 мин (92,5%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,38-7,15 (m, 6Н); 7,00-6,99 (m, 3Н); 4,85-4,68 (m, 2Н); 4,37-3,38 (m, 11Н); 3,31-2,70 (m, 8Н); 2,60-0,82 (m, 35Н).
Соединение 30
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминобутил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
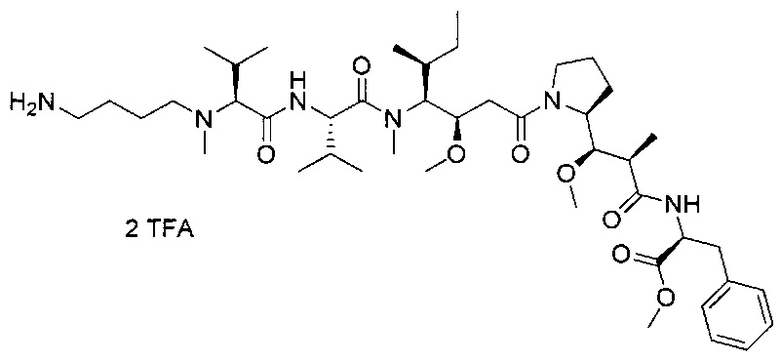
Соединение 30 получали аналогично соединению 16 из амина 3D и соответствующей карбоновой кислоты.
LC/MS/UV (Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°C, от 10 до 95% метанол в воде (0,05% TFA) в течение 15 мин); ESI (C44H76N6O8, точная масса 816,57) m/z: 817,6 (МН+), 409,4 (М, 2Н+/2); 12,0 мин (90%, 210 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,7-8,2 (m, 1Н, NHCOs, неполный обмен); 7,4-7,1 (m, 5Н); 4,95-4,7 (m, 3Н); 4,2-4,0 (m, 1Н); 3,9-0,8 (m, 63Н).
Соединение 31
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминобутил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-соль трифторуксусной кислоты
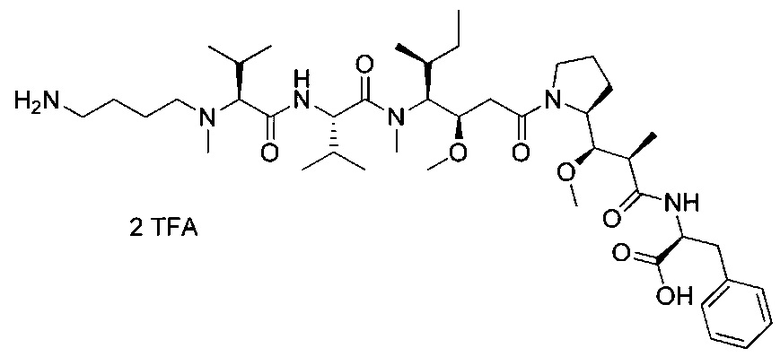
Соединение 31 получали аналогично соединению 4 из метилового эфира 30.
LC/MS/UV (Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°C, от 10 до 95% метанола в воде (0,05% TFA) в течение 18 минут); ESI (C43H74N6O8, точная масса 802,56) m/z: 803,6 (МН+), 402,4 (М, 2Н+/2); 13,68 мин (98,9%, 210 нм).
1Н NMR (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,4-7,1 (m, 5Н); 4,95-4,7 (m, 3Н); 4,2-4,0 (m, 1Н); 3,9-0,8 (m, 61Н).
Соединение 32
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(3-(метиламино)пропил)амино)бутанамидо)бутанамид, бис-соль трифторуксусной кислоты
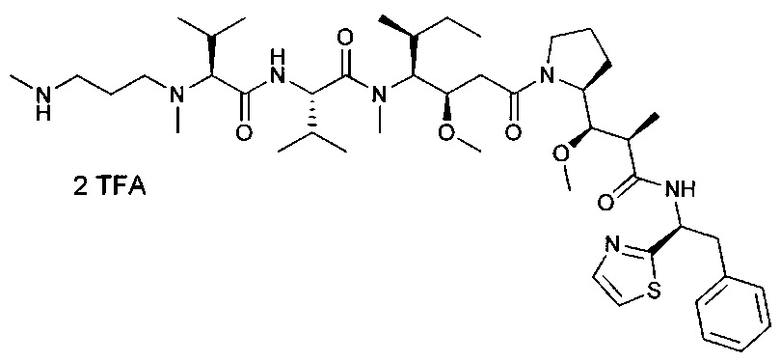
Соединение 32А: трет-бутил-(3,3-диэтоксипропил)(метил)карбамат
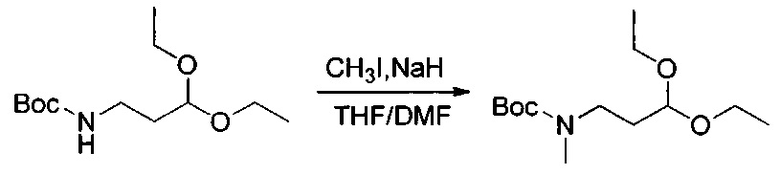
Соединение 1ZD (247 мг, 1 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 30 мл смеси 1:1 ТГФ и ДМФА. Реакционную среду охлаждали на ледяной бане, после чего порциями добавляли NaH (60% в масле, 60 мг, 1,5 экв.), затем по каплям MeI (0,28 мл). Реакционную смесь оставляли встряхиваться в течение 2 дней при температуре окружающей среды, затем нейтрализовали с помощью 5 мл насыщенного водного раствора NH4Cl и дважды экстрагировали с помощью 15 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 200 мг (77%) соединения 32А в форме желтого твердого вещества.
Соединение 32В: (S)-2-((3-((трет-бутоксикарбонил)(метил)амино)пропил)(метил)амино)-3-метилбутановая кислота
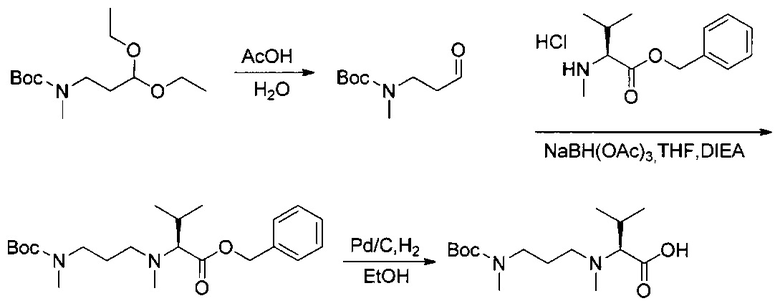
Соединение 32В получали, следуя тому же протоколу, который описан для получения соединения 1ZF, заменяя соединение 1ZD соединением 32А.
Соединение 32: Соединение 32 получали в два этапа, следуя тому же протоколу, который описан для получения соединения 1, из амина 1Y и карбоновой кислоты 32В.
LC/MS/UV (Zorbax SB-Aq, 1,8 мкм, 4,6×100 мм, 40°C, от 10 до 95% метанола в воде (0,05% TFA) в течение 13 минут); ESI (C45H75N7O6S, точная масса 842,19) m/z: 843 (МН+), 421,9 (М, 2Н+/2); 11,91 мин (88%, 210 нм).
1Н NMR: (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,5-9,0 (m, 0,5Н, неполный обмен NHCOs), 7,85-7,80 (m, 1Н); 7,60-7,50 (m, 1Н), 7,35-7,15 (m, 5Н), 5,80-5,63 (m, 1Н), 4,80-4,65 (m, 2Н), 4,30-4,00 (m, 1Н), 3,95-0,80 (m, 61Н).
Соединения 33 и 34
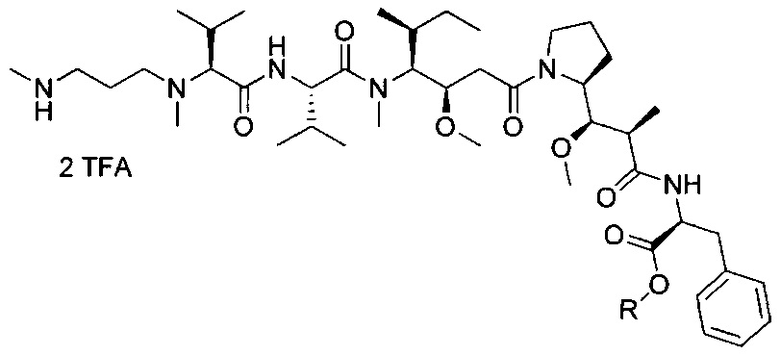
Соединения 33 и 34 получали аналогично соединениям 6 и 7, заменяя карбоновую кислоту 1ZF соединением 32В.
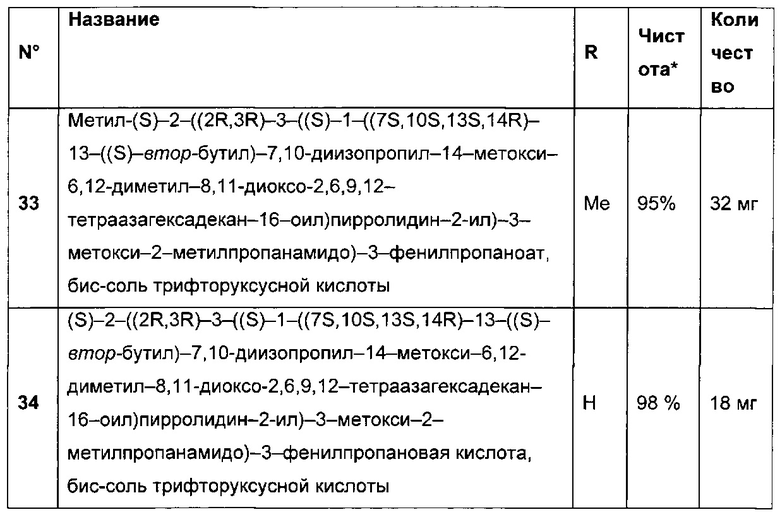
* Соединения очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм), получая соответствующие соли TFA в форме белых твердых веществ.
Характеризация конечных продуктов: Соединение 33 LC/MS/UV (ESI) (C44H76N6O8, точная масса 816,57) m/z 817,6 (МН+), 409,4 (М, 2Н+/2); UV: 5,94 мин (95%, 210 нм). 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) δ 8,6-8,2 (m, 0,8H, NHCO неполный обмен) 7,30-7,22 (m, 5Н), 4,80 (m, 2Н), 4,23-0,86 (m, 66Н). Соединение 34 LC/MS/UV (ESI) (C43H74N6O8, точная масса 802,56) m/z 803,6 (МН+), 402,4 (М, 2Н+/2); UV: 5,94 мин (97,5%, 210 нм). 1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) δ 7,30-7,20 (m, 5Н), 4,80 (m, 2Н), 4,25-0,86 (m, 63Н).
Соединение 35
(S)-2-((S)-2-((2-(2-аминоэтокси)этил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, бис-соль трифторуксусной кислоты
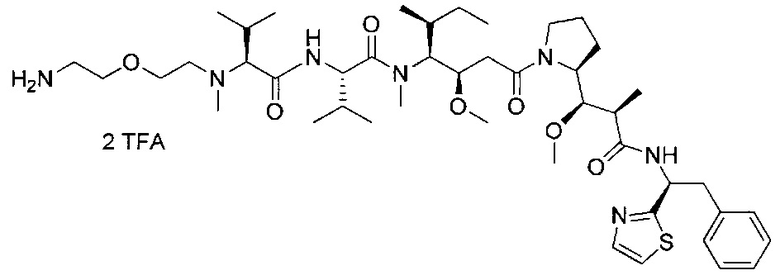
Соединение 35А: трет-бутил-(2-(2-гидроксиэтокси)этил)карбамат
2-(2-аминоэтокси)этанол (5 г, 47,56 ммоль, 1,00 экв.) растворяли в ТГФ (100 мл) при 0°C и затем добавляли натрия гидроксид (2 г, 50,00 ммоль, 1,05 экв.) (раствор в 25 мл воды). По каплям добавляли раствор ди-трет-бутилдикарбоната (10,38 г, 47,56 ммоль, 1,00 экв.) в ТГФ (20 мл) и затем реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды. Реакционную смесь разводили добавлением 50 мл воды и продукт экстрагировали 3 раза с помощью 75 мл AcOEt. Органические фазы объединяли, промывали один раз с помощью 100 мл NaCl (насыщ.), затем сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 9 г (92%) соединения 35А в форме желтого масла.
Соединение 35В: трет-бутил-(2-(2-оксоэтокси)этил)карбамат
Раствор ДМСО (3,46 мл, 5,00 экв.) в DCM (20 мл) добавляли по каплям к раствору оксалилхлорида (1,9 мл, 2,30 экв.) в DCM (20 мл) при -78°C в атмосфере азота. После добавления (30 минут) раствор встряхивали в течение 30 минут и затем добавляли раствор соединения 35А (2 г, 9,74 ммоль, 1,00 экв.) в 20 мл DCM. После добавления TEA (12,2 мл) реакционную смесь встряхивали при -78°C в течение 30 минут, а затем при температуре окружающей среды в течение ночи. Реакционную смесь разводили добавлением 100 мл воды. Продукт экстрагировали 3 раза с помощью 50 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 1,9 г соединения 35В в форме желтого масла.
Соединение 35С: бензил-(S)-12-изопропил-2,2,11-триметил-4-оксо-3,8-диокса-5,11-диазатридекан-13-оат
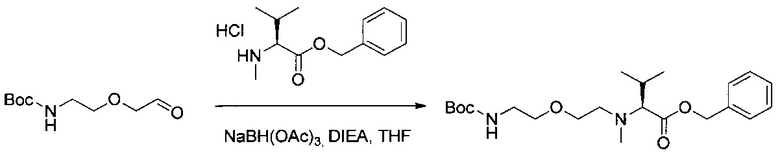
Соединение 35С синтезировали аналогично соединению 14С из амина 1ZC (2,4 г, 9,31 ммоль, 1,00 экв.), альдегида 35В (1,9 г, 9,35 ммоль, 1,00 экв.), NaBH(OAc)3 (3,96 г, 18,68 ммоль, 2,00 экв.) и DIEA (6,2 мл) в ТГФ (40 мл). Реакционную смесь нейтрализовали с помощью 200 мл воды и экстрагировали 3 раза с помощью 100 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали, получая 2,3 г соединения 35С в форме желтого масла.
Соединение 35D: (S)-12-изопропил-2,2,11-триметил-4-оксо-3,8-диокса-5,11-диазатридекан-13-овая кислота
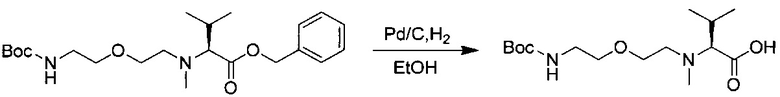
Соединение 35С (200 мг, 0,49 ммоль, 1,00 экв.) растворяли в 10 мл EtOH в присутствии Pd/C (200 мг) и гидрогенировали в течение ночи. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 150 мг (96%) соединения 35D в форме белого твердого вещества.
Соединение 35Е: трет-бутил-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2,14-диокса-5,8,11-триазагексадекан-16-ил)карбамат
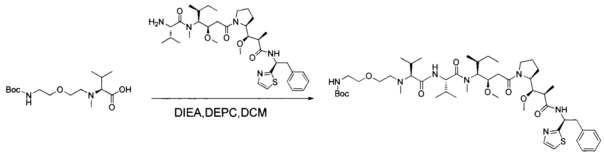
Соединение 35Е синтезировали аналогично соединению 3 из амина 1Y (70 мг, 0,11 ммоль, 1,00экв.), кислоты 35D (40,6 мг, 0,13 ммоль, 1,20 экв.), DEPC (0,0324 мл) и DIEA (0,0527 мл) в DCM (3 мл). Неочищенный продукт (100 мг, 98%) выделяли в форме желтого масла и затем использовали как таковое.
Соединение 35: Соединение 35 синтезировали аналогично соединению 2 из промежуточного продукта 35Е (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-010), SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 35 получали с выходом 23% (22,9 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C45H75N7O7S, точная масса 857,54) m/z: 858,5 (МН+) и 429,9 (М, 2Н+/2, 100%), 5,89 мин (89,7%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) δ 8,9-8,5 (m, 0,5Н, NHCO неполный обмен), 7,8-7,7 (m, 1Н), 7,55-7,45 (m, 1Н), 7,35-7,1 (m, 5Н), 5,45-5,5 (m, 1Н), 4,9-4,6 (m, 1Н), 4,3-0,75 (m, 62Н).
Соединение 36
Метил-(S)-2-((2R,3R)-3-((S)-1-((7S,10S,13S,14R)-1-амино-13-((S)-втор-бутил)-7,10-диизопропил-14-метокси-6,12-диметил-8,11-диоксо-3-окса-6,9,12-триазагексадекан-16-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
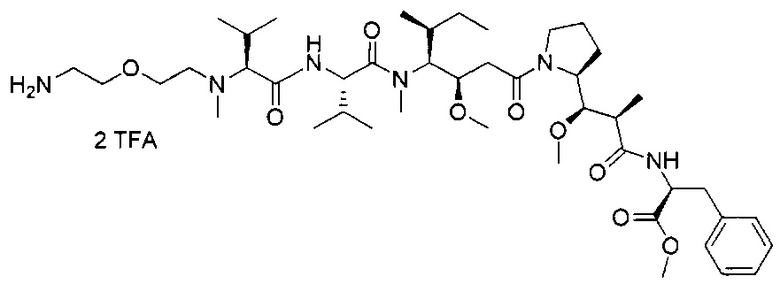
Соединение 36А: Метил-(S)-2-((2R,3R)-3-((S)-1-((12S,15S,18S,19R)-18-((S)-втор-бутил)-12,15-диизопропил-19-метокси-2,2,11,17-тетраметил-4,13,16-триоксо-3,8-диокса-5,11,14,17-тетраазагенэйкозан-21-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
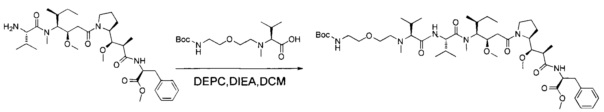
Соединение 36А синтезировали аналогично соединению 3 из амина 3D (50 мг, 0,08 ммоль, 1,00 экв.), кислоты 35D (25 мг, 0,11 ммоль, 1,48 экв.), DEPC (0,0337 мл) и DIEA (0,0548 мл) в DCM (3 мл). Неочищенный продукт (100 мг) выделяли в форме желтого масла и затем использовали как таковое.
Соединение 36: Соединение 36 синтезировали аналогично соединению 2 из промежуточного продукта 36А (100 мг, 0,11 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 36 получали с выходом 13%) (12,7 мг) в форме белого твердого вещества.
LC/MS/UV (Agilent ZORBAX SB-Aq колонка, 1,8 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, 2% МеОН в воде (0,05% TFA) в течение 1 минуты, затем от 2% до 95% МеОН в воде в течение 13 минут, затем 95% МеОН в воде в течение 2 минут); ESI (C44H76N6O9, точная масса 832,57) m/z: 833,5 (MH+) и 417,4 (М, 2Н+/2, 100%), 11,58 мин (98,5%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,1-8,5 (m, 0,8Н, NHCO неполный обмен), 7,30-7,1 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 64Н).
Соединение 37
(S)-2-((2R,3R)-3-((S)-1-((7S,10S,13S,14R)-1-амино-13-((S)-втор-бутил)-7,10-диизопропил-14-метокси-6,12-диметил-8,11-диоксо-3-окса-6,9,12-триазагексадекан-16-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-соль трифторуксусной кислоты

Соединение 37 получали аналогично соединению 4 из соединения 36 (100 мг, 0,11 ммоль, 1,00 экв.). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, Atlantis Prep OBD Т3 колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 18,4 мг (19%) соединения 37 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C43H74N6O9, точная масса 818,6) m/z: 819,6 (МН+) и 410,4 (М, 2Н+/2, 100%), 5,48 мин (96,7%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,35-7,2 (m, 5Н), 5,0-4,65 (m, 3Н), 4,3-0,8 (m, 61Н).
Соединение 38
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((2-аминоэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
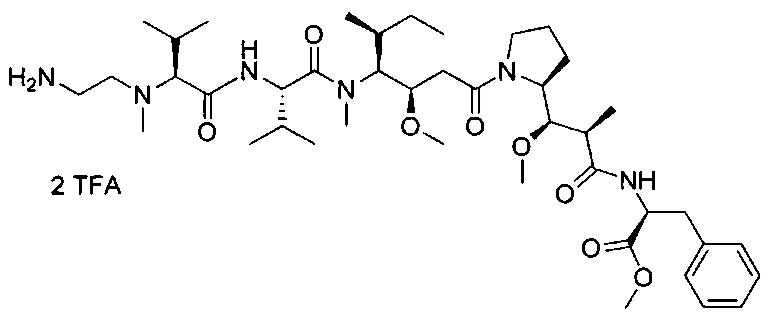
Соединение 38А: метил-(S)-2-((2R,3R)-3-((S)-1-((9S,12S,15S,16R)-15-((S)-втор-бутил)-9,12-диизопропил-16-метокси-2,2,8,14-тетраметил-4,10,13-триоксо-3-окса-5,8,11,14-тетраазаоктадекан-18-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
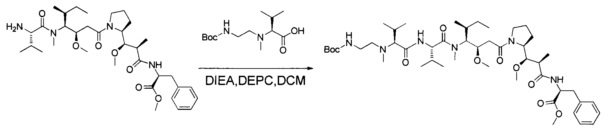
Соединение 38А синтезировали аналогично соединению 3 из амина 3D (70 мг, 0,11 ммоль, 1,00 экв.), кислоты 26В (60,7 мг, 0,22 ммоль, 2,00 экв.), DEPC (0,0337 мл) и DIEA (0,0548 мл) в DCM (3 мл). Неочищенный продукт (100 мг) выделяли в форме желтого масла.
Соединение 38: Соединение 38 синтезировали аналогично соединению 2 из промежуточного продукта 38А (100 мг, 0,11 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLCPre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 38 получали с выходом 10% (10,3 мг) в форме белого твердого вещества.
LC/MS/UV (Agilent ZORBAX SB-Aq колонка, 1,8 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, 2% МеОН в воде (0,05% TFA) в течение 1 минуты, затем от 2% до 95% МеОН в воде в течение 13 минут, затем 95% МеОН в воде в течение 2 минут); ESI (C42H72N6O8, точная масса 788,5) m/z: 789,5 (МН+) и 395,4 (М, 2Н+/2, 100%), 12,97 мин (91,0%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,30-7,1 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 60Н).
Соединение 39
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((2-аминоэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-соль трифторуксусной кислоты
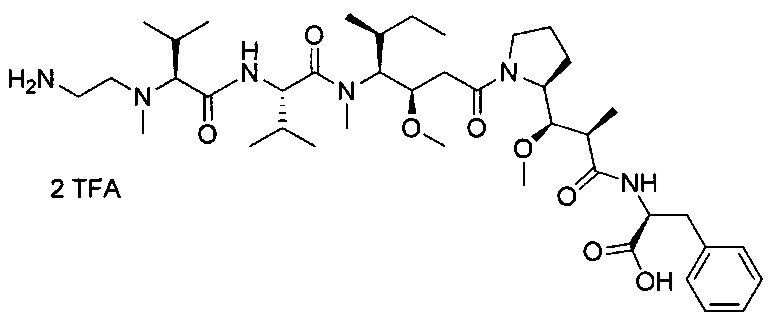
Соединение 39 получали аналогично соединению 4 из соединения 38 (100 мг, 0,11 ммоль, 1,00 экв.). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep C18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 8,2 мг (8%) соединения 39 в форме белого твердого вещества.
LC/MS/UV (Eclipse Plus С18 колонка, 3,5 мкм, 4,6×150 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C41H70N6O8, точная масса 774,5) m/z: 775,5 (МН+) и 388,4 (М, 2Н+/2, 100%), 6,47 мин (93,6%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,35-7,15 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 57Н).
Соединение 40
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)бутил)амино)бутанамидо)бутанамид, бис-соль трифторуксусной кислоты
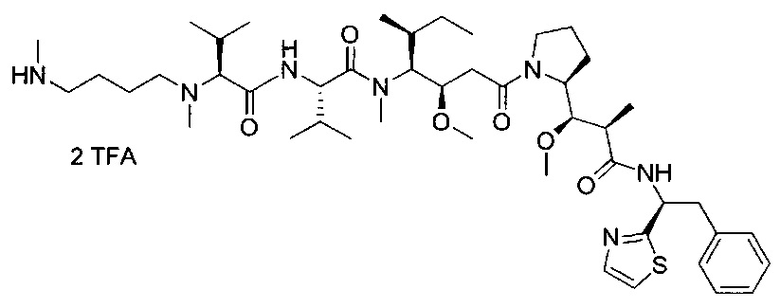
Соединение 40А: трет-бутил-(4,4-диэтоксибутил)(метил)карбамат

Соединение 40А получали аналогично соединению 11Е из трет-бутил(4,4-диэтоксибутил)(метил)карбамата (5,5 г, 19,97 ммоль, 1,00 экв.), NaH (60% в масле, 3,2 г, 80,00 ммоль, 4,01 экв.) и йодометана (14 мл) в ТГФ/ДМФА (40/20 мл). Реакционную смесь нейтрализовали с помощью 50 мл NH4Cl (водн.). Соединение 40А выделяли в форме желтого масла, 5,5 г (95%).
Соединение 40 В: трет-бутилметил(4-оксобутил)карбамат
Соединение 40А (3 г, 10,89 ммоль, 1,00 экв.) растворяли в смеси АсОН и воды (15/4 мл) и раствор оставляли встряхиваться в течение ночи. pH доводили до 7-8 с помощью насыщенного водного раствора NaHCO3, а затем дважды экстрагировали с помощью 50 мл EtOAc. Органические фазы объединяли, промывали дважды с помощью 50 мл насыщенного водного раствора NaCl, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Соединение 40В выделяли в форме желтого масла, 2,1 г (96%).
Соединение 40С: бензил-(S)-2-((4-((трет-бутоксикарбонил)(метил)амино)бутил)(метил)амино)-3-метилбутаноат
Соединение 40С синтезировали аналогично соединению 14С из амина 1ZC (2,45 г, 9,53 ммоль, 0,80 экв.), альдегида 40В (2,4 г, 11,92 ммоль, 1,00 экв.), NaBH(OAc)3 (5,06 г, 23,87 ммоль, 2,00 экв.) и DIEA (6,16 г, 47,66 ммоль, 4,00 экв.) в ТГФ (15 мл). Реакционную смесь нейтрализовали с помощью 100 мл воды и дважды экстрагировали с помощью 100 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на силикагеле (EtOAc/РЕ (1:100-1:20)), получая 1,2 г (25%) соединения 40С в форме желтого масла.
Соединение 40D: (S)-2-((4-((трет-бутоксикарбонил)(метил)амино)бутил)(метил)амино)-3-метилбутановая кислота
Соединение 40С (500 мг, 1,23 ммоль, 1,00 экв.) растворяли в 20 мл EtOH в присутствии Pd/C (550 мг) и гидрогенировали в течение 1 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 350 мг (90%) соединения 40D в форме бесцветного масла.
Соединение 40Е: трет-бутил-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-ил)(метил)карбамат
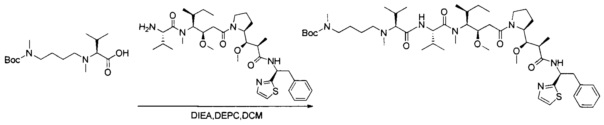
Соединение 40Е синтезировали аналогично соединению 3 из амина 1Y (60 мг, 0,09 ммоль, 1,00 экв.), кислоты 40D (57,8 мг, 0,18 ммоль, 2,00 экв.), DEPC (0,0278 мл) и DIEA (0,0452 мл) в DCM (3 мл). Затем неочищенный продукт (100 мг) использовали как таковой.
Соединение 40: Соединение 40 синтезировали аналогично соединению 2 из промежуточного продукта 40Е (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 40 получали с выходом 24% (24,6 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C46H77N7O6S, точная масса 855,6) m/z: 856,6 (МН+) и 428,8 (М, 2Н+/2, 100%), 5,89 мин (97,0%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,9-8,5 (0,7Н, NHCO неполный обмен), 7,8-7,7 (m, 1Н), 7,55-4,45 (m, 1Н), 7,35-7,1 (m, 5Н), 5,5-5,75 (m, 1Н), 4,9-4,6 (m, 2Н), 4,2-0,8 (m, 64Н).
Соединение 41
Метил-(S)-2-((2R,3R)-3-((S)-1-((8S,11S,14S,15R)-14-((S)-втор-бутил)-8,11-диизопропил-15-метокси-7,13-диметил-9,12-диоксо-2,7,10,13-тетраазагептадекан-17-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
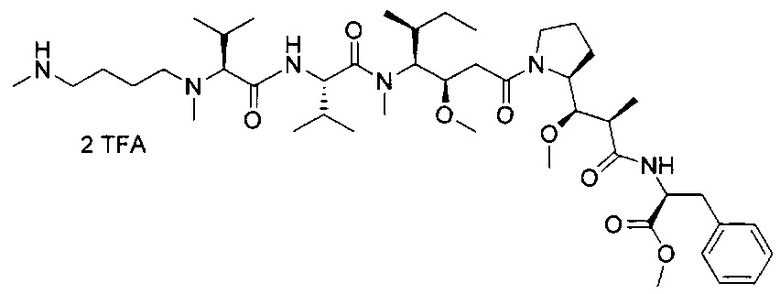
Соединение 41А: метил-(S)-2-((2R,3R)-3-((S)-1-((11S,14S,17S,18R)-17-((S)-втор-бутил)-11,14-диизопропил-18-метокси-2,2,5,10,16-пентаметил-4,12,15-триоксо-3-окса-5,10,13,16-тетраазаэйкозан-20-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
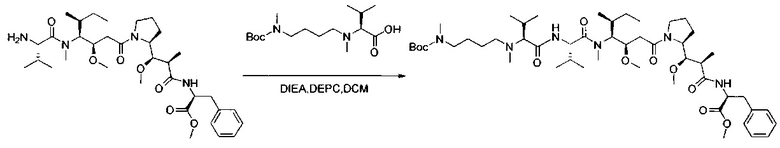
Соединение 41А синтезировали аналогично соединению 3 из амина 3D (170 мг, 0,27 ммоль, 1,00 экв.), кислоты 40D (170 мг, 0,54 ммоль, 2,09 экв.), DEPC (0,0819 мл) и DIEA (0,133 мл) в DCM (5 мл). Затем неочищенный продукт (200 мг) использовали как таковой.
Соединение 41: Соединение 41 синтезировали аналогично соединению 2 из промежуточного продукта 41А (100 мг, 0,11 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 41 получали с выходом 25% (25 мг) в форме белого твердого вещества.
LC/MS/UV (Agilent Zorbax SB-Aq колонка, 1,8 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, 2% МеОН в воде (0,05% TFA) в течение 1 минуты, затем от 2% до 95% МеОН в воде в течение 13 минут, затем 95% МеОН в воде в течение 2 минут); ESI (C45H78N6O8, точная масса 830,6) m/z: 831,6 (МН+) и 416,4 (М, 2Н+/2, 100%), 11,58 мин (97,2%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,55-8,15 (0,75Н, NHCO неполный обмен), 7,30-7,1 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 67Н).
Соединение 42
(S)-2-((2R,3R)-3-((S)-1-((8S,11S,14S,15R)-14-((S)-втор-бутил)-8,11-диизопропил-15-метокси-7,13-диметил-9,12-диоксо-2,7,10,13-тетраазагептадекан-17-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-соль трифторуксусной кислоты
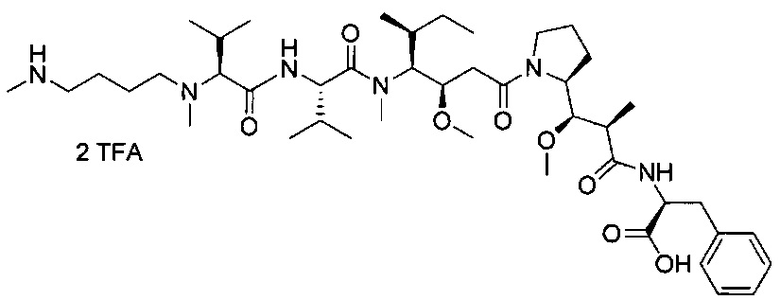
Соединение 42 получали аналогично соединению 4 из соединения 41 (100 мг, 0,11 ммоль, 1,00 экв.). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, Atlantis Prep OBD Т3 колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 30,6 мг (31%) соединения 42 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 0% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C44H76N6O8, точная масса 816,6) m/z: 817,6 (MH+) и 409,4 (М, 2Н+/2, 100%), 5,75 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,5-8,1 (0,3Н, NHCO неполный обмен), 7,30-7,1 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 64Н).
Соединение 43
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((R)-3-метил-2-(метил(2-(2-(метиламино)этокси)этил)амино)бутанамидо)бутанамид, бис-соль трифторуксусной кислоты

Соединение 43А: трет-бутил-(2-(2-((третбутилдиметилсилил)окси)этокси)этил)карбамат

Соединение 35А (трет-бутил-((2-(2-гидроксиэтокси)этил)карбамат) (8,21 г, 40,00 ммоль, 1,00 экв.) и имидазол (6 г, 88,14 ммоль, 2,20 экв.) растворяли в инертной атмосфере в DCM (200 мл). По каплям добавляли третбутилдиметилсиланхлорид (TBDMSCI, 6,62 г, 43,92 ммоль, 1,10 экв.) и реакционную среду оставляли встряхиваться в течение ночи при температуре окружающей среды. Реакционную смесь разводили в 100 мл DCM, затем промывали дважды с помощью 200 мл 0,5 М HCl, дважды с помощью 200 мл NaHCO3 (насыщ.), затем 300 мл NaCl (насыщ.). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ (1:3), получая 10 г (78%) соединения 43А в форме белого твердого вещества.
Соединение 43В: трет-бутил-(2-(2-((трет-бутилдиметилсилил)окси)этокси)этил)(метил)карбамат
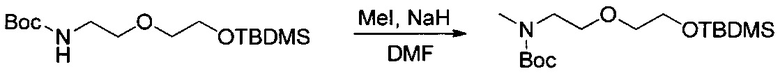
Соединение 43В получали аналогично соединению 11Е из соединения 43А (10 г, 31,30 ммоль, 1,00 экв.), NaH (60% в масле, 5 г, 208,33 ммоль, 4,00 экв.) и йодометана (22 г, 5,00 экв.) в ДМФА (200 мл). Реакционную среду нейтрализовали с помощью 200 мл воды и промывали 3 раза с помощью 100 мл AcOEt, затем 300 мл NaCl (насыщ.). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 10 г (96%) соединения 43В в форме белого твердого вещества.
Соединение 43С: трет-бутил-(2-(2-гидроксиэтокси)этил)(метил)карбамат

Соединение 43В (10 г, 29,89 ммоль, 1,00 экв.) и TBAF, 3H2O (20,8 г, 65,93 ммоль, 2,20 экв.) растворяли в ТГФ (200 мл). Смесь встряхивали при температуре окружающей среды в течение 2 ч, затем экстрагировали 3 раза с помощью 100 мл AcOEt. Органические фазы снова объединяли, промывали дважды с помощью 300 мл воды, затем дважды с помощью 300 мл NaCl (насыщ.), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на силикагеле (EtOAc/РЕ (1:3 - 1:1), получая 6,6 г соединения 43С в форме бесцветного масла.
Соединение 43D: трет-бутилметил(2-(2-оксоэтокси)этил)карбамат
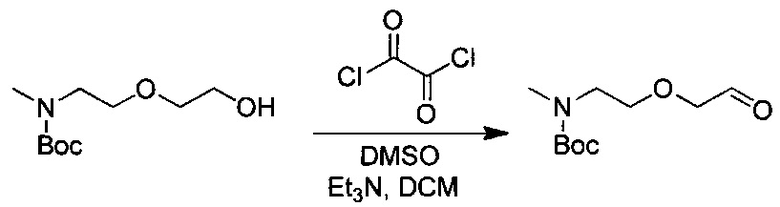
Соединение 43D получали аналогично соединению 35В из соединения 43С (2 г, 9,12 ммоль, 1,00 экв.), оксалилхлорида (1,9 мл), TEA (11,3 мл) и ДМСО (3,3 мл). Соединение 43D (2 г) выделяли в форме желтого масла.
Соединение 43Е: бензил-(S)-12-изопропил-2,2,5,11-тетраметил-4-оксо-3,8-диокса-5,11-диазатридекан-13-оат
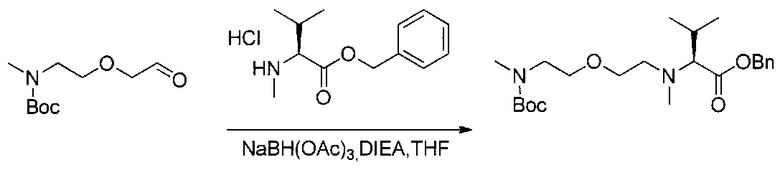
Соединение 43Е синтезировали аналогично соединению 14С из амина 1ZC (2,4 г, 9,31 ммоль, 1,00 экв.), альдегида 43D (2 г, 9,16 ммоль, 1,00 экв.), NaBH(OAc)3 (4 г, 18,87 ммоль, 2,06 экв.) и DIEA (6 мл) в ТГФ (100 мл). Реакционную смесь нейтрализовали с помощью 100 мл воды и экстрагировали 3 раза с помощью 100 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на силикагеле (EtOAc/РЕ (4:1), получая 1 г (37%) соединения 43Е в форме белого твердого вещества.
Соединение 43F: (S)-12-изопропил-2,2,5,11-тетраметил-4-оксо-3,8-диокса-5,11-диазатридекан-13-овая кислота
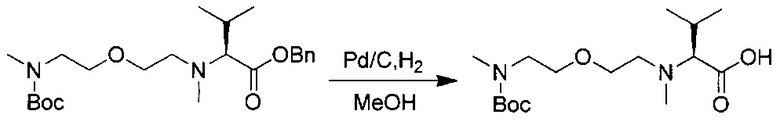
Соединение 43Е (1 г, 2,37 ммоль, 1,00 экв.) растворяли в 40 мл МеОН в присутствии Pd/C (1 г) и гидрогенировали в течение 1 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 600 мг (76%) соединения 43F в форме белого твердого вещества.
Соединение 43G: трет-бутил-((3R,4S,7S,10R)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2,14-диокса-5,8,11-триазагексадекан-16-ил)(метил)карбамат

Соединение 43G синтезировали аналогично соединению 3 из амина 1Y (50 мг, 0,08 ммоль, 1,00 экв.), кислоты 43F (50 мг, 0,08 ммоль, 1,00 экв.), DEPC (24,79 мг, 0,15 ммоль, 2,00 экв.) и DIEA (29,46 мг, 0,23 ммоль, 3,00 экв.) в DCM (1 мл). Затем неочищенный продукт (59 мг) использовали как таковой.
Соединение 43: Соединение 43 синтезировали аналогично соединению 2 из промежуточного продукта 43G (81 мг, 0,08 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 43 получали с выходом 64% (52,6 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% MeCN в воде (0,05% TFA) в течение 8 минут, затем 95% MeCN в воде в течение 2 минут); ESI (C48H77N7O7S, точная масса 871,6) m/z: 872,5 (МН+) и 436,9 (М, 2Н+/2, 100%), 3,90 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,8-7,7 (m, 1Н), 7,55-4,45 (m, 1Н), 7,35-7,1 (m, 5Н), 5,5-5,75 (m, 1Н), 4,9-4,6 (m, 2Н), 4,2-0,8 (m, 64Н).
Соединение 44
Метил-(S)-2-((2R,3R)-3-((S)-1-((9S,12S,15S,16R)-15-((S)-втор-бутил)-9,12-диизопропил-16-метокси-8,14-диметил-10,13-диоксо-5-окса-2,8,11,14-тетраазаоктадекан-18-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-соль трифторуксусной кислоты
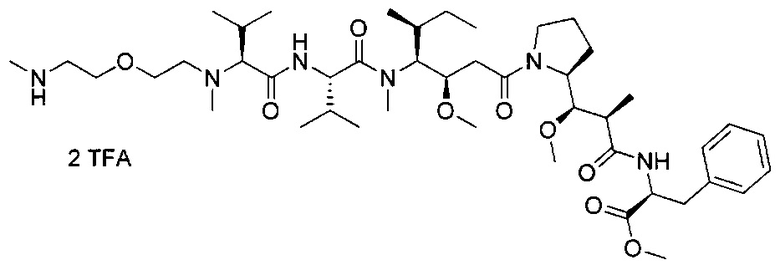
Соединение 44А: метил-(S)-2-((2R,3R)-3-((S)-1-((12S,15S,18S,19R)-18-((S)-втор-бутил)-12,15-диизопропил-19-метокси-2,2,5,11,17-пентаметил-4,13,16-триоксо-3,8-диокса-5,11,14,17-тетраазагенэйкозан-21-оил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат

Соединение 44А синтезировали аналогично соединению 3 из амина 3D (60 мг, 0,09 ммоль, 1,00 экв.), кислоты 43F (47 мг, 0,14 ммоль, 1,50 экв.), DEPC (31 мг, 0,19 ммоль, 2,00 экв.) и DIEA (37 мг, 0,28 ммоль, 3,00 экв.) в DCM (1,5 мл). Затем неочищенный продукт (58 мг) использовали как таковой.
Соединение 44: Соединение 44 синтезировали аналогично соединению 2 из промежуточного продукта 44А (58 мг, 0,06 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 44 получали с выходом 40% (23,7 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% MeCN в воде (0,05% TFA) в течение 8 минут, затем 95% MeCN в воде в течение 2 минут); ESI (C45H78N6O9, точная масса 846,6) m/z: 847,6 (МН+) и 424,4 (М, 2Н+/2, 100%), 3,20 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,3-7,1 (m, 5Н), 4,9-4,6 (m, 3Н), 4,2-0,8 (m, 67Н).
Соединение 45
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(2-(пиперазин-1-ил)этил)амино)бутанамидо)бутанамид, трис-соль трифторуксусной кислоты
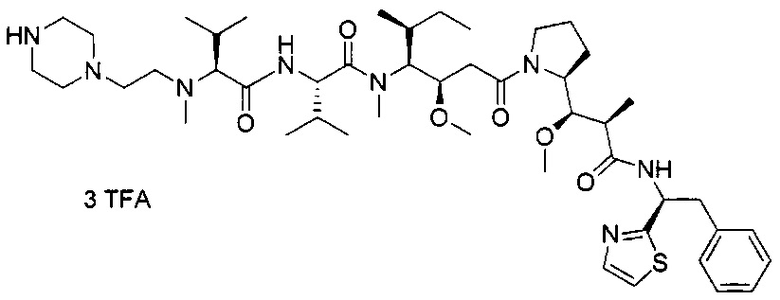
Соединение 45А: трет-бутил-4-(2-гидроксиэтил)пиперазин-1-карбоксилат
2-(пиперазин-1-ил)этан-1-ол (5 г, 38,41 ммоль, 1,00 экв.) растворяли в DCM (100 мл) и по каплям добавляли раствор ди-трет-бутилдикарбоната (8,38 г, 38,40 ммоль, 1,00 экв.) в DCM (20 мл). Реакционную смесь оставляли встряхиваться в течение ночи при температуре окружающей среды. Реакционную смесь выпаривали досуха и осадок растворяли в 200 мл AcOEt, промывали 5 раз с помощью NaCl (насыщ.), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 8,5 г (96%) соединения 45А в форме белого твердого вещества.
Соединение 45В: трет-бутил-4-(2-оксоэтил)пиперазин-1-карбоксилат
Соединение 45В получали аналогично соединению 35В из соединения 45А (1 г, 4,34 ммоль, 1,00 экв.), оксалилхлорида (610 мг, 4,80 ммоль, 1,12 экв.), TEA (2,13 г, 21,09 ммоль, 4,90 экв.) и ДМСО (0,82 г, 2,40 экв.). Соединение 45В (0,8 г, 81%) выделяли в форме бесцветного масла.
Соединение 45С: трет-бутил-(S)-4-(2-((1-(бензилокси)-3-метил-1-оксобутан-2-ил)(метил)амино)этил)пиперазин-1-карбоксилат
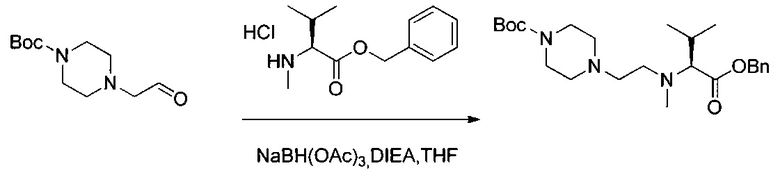
Соединение 45С синтезировали аналогично соединению 14С из амина 1ZC (720 мг, 2,79 ммоль, 0,80 экв.), альдегида 45В (800 мг, 3,50 ммоль, 1,00 экв.), NaBH(OAc)3 (1,6 г, 7,55 ммоль, 2,15 экв.) и DIEA (2,5 мл) в ТГФ (50 мл). Реакционную смесь нейтрализовали с помощью 5 мл воды и экстрагировали 3 раза с помощью 5 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на силикагеле (EtOAc/РЕ (3:1), получая 400 мг (33%) соединения 45С в форме бесцветного масла.
Соединение 45D: (S)-2-((2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)этил)(метил)амино)-3-метилбутановая кислота
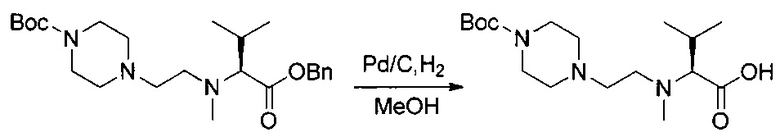
Соединение 45С (400 мг, 0,92 ммоль, 1,00 экв.) растворяли в 30 мл МеОН в присутствии Pd/C (400 мг) и гидрогенировали в течение 1 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 300 мг (95%) соединения 45D в форме белого твердого вещества.
Соединение 45Е: трет-бутил-4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)пиперазин-1-карбоксилат
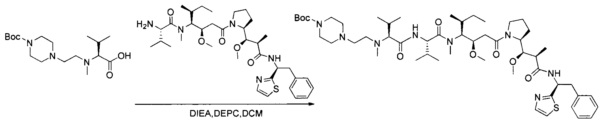
Соединение 45Е синтезировали аналогично соединению 3 из амина 1Y (60 мг, 0,09 ммоль, 1,00 экв.), кислоты 45D (62,7 мг, 0,18 ммоль, 2,00 экв.), DEPC (0,0278 мл) и DIEA (0,0452 мл) в DCM (3 мл). Затем неочищенный продукт (100 мг) использовали как таковой.
Соединение 45: Соединение 45 синтезировали аналогично соединению 2 из промежуточного продукта 45Е (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 45 получали с выходом 19% (19,4 мг) в форме белого твердого вещества.
LC/MS/UV (Agilent ZORBAX SB-Aq колонка, 1,8 мкм, 4,6×100 мм; 40°C; 1,0 мл/мин, 2% МеОН в воде (0,05% TFA) в течение 1 минуты, затем от 2% до 95% МеОН в воде в течение 13 минут, затем 95% МеОН в воде в течение 2 минут); ESI (C47H78N8O6S, точная масса 882,6) m/z: 883,5 (МН+) и 442,4 (М, 2H+/2, 100%), 10,95 мин (98,8%, 210 нм).
1H NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров), 7,80-7,70 (m, 1Н), 7,52-7,43 (m, 1Н), 7,31-7,09 (m, 5Н), 5,70-5,51 (m, 1Н), 4,80-4,60 (m, 1Н), 4,20-0,75 (m, 66Н).
Соединение 46
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(2-(пиперазин-1-ил)этил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трис-соль трифторуксусной кислоты
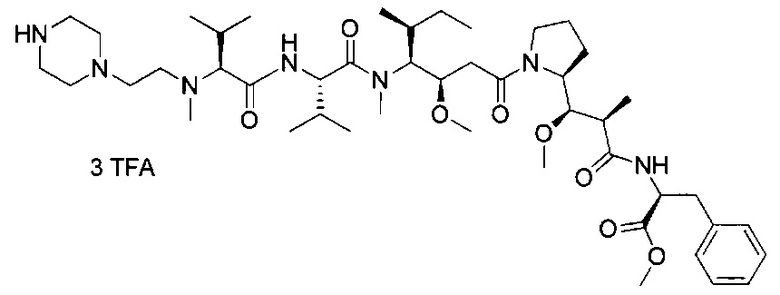
Соединение 46А: трет-бутил-4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-3-(((S)-1-метокси-1-оксо-3-фенилпропан-2-ил)амино)-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)пиперазин-1-карбоксилат
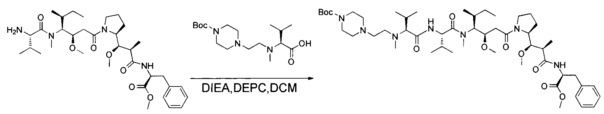
Соединение 46А синтезировали аналогично соединению 3 из амина 3D (170 мг, 0,27 ммоль, 1,00 экв.), кислоты 45D (184,6 мг, 0,54 ммоль, 2,00 экв.), DEPC (0,0819 мл) и DIEA (0,133 мл) в DCM (5 мл). Затем неочищенный продукт (200 мг) использовали как таковой.
Соединение 46: Соединение 46 синтезировали аналогично соединению 2 из промежуточного продукта 46А (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 46 получали с выходом 19% (19,1 мг) в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% MeCN в воде (0,05% TFA) в течение 8 минут, затем 95% MeCN в воде в течение 2 минут); ESI (C46H79N7O8, точная масса 857,6) m/z: 858,6 (МН+) и 429,9 (М, 2Н+/2, 100%), 5,93 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,58-8,50 (m, 0,5 Н, NHCO, неполный обмен), 8,29-8,22 (m, 0,4Н, NHCO, неполный обмен), 7,35-7,15 (m, 5Н), 4,87-4,69 (m, 3Н), 4,22-0,82 (m, 68Н).
Соединение 47
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(2-(пиперазин-1-ил)этил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трис-соль трифторуксусной кислоты
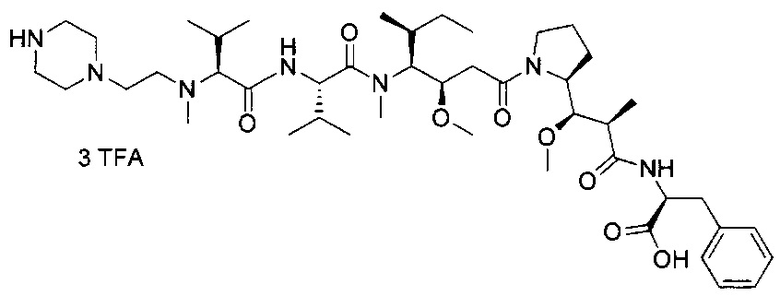
Соединение 47 получали аналогично соединению 4 из соединения 46 (100 мг, 0,10 ммоль, 1,00 экв.). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, Atlantis Prep OBD Т3 колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 32,6 мг (33%) соединения 47 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C46H77N7O8, точная масса 843,6) m/z: 844,6 (МН+) и 422,9 (М, 2Н+/2, 100%), 5,73 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,66-8,57 (m, 0,3 Н, NHCO, неполный обмен), 8,41-8,32 (m, 0,3Н, NHCO, неполный обмен), 8,13-8,06 (m, 0,2H, NHCO, неполный обмен), 7,30-7,10 (m, 5Н), 4,80-4,61 (m, 3Н), 4,19-0,78 (m, 65Н).
Соединение 48
(S)-2-((S)-2-(((1Н-имидазол-2-ил)метил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты
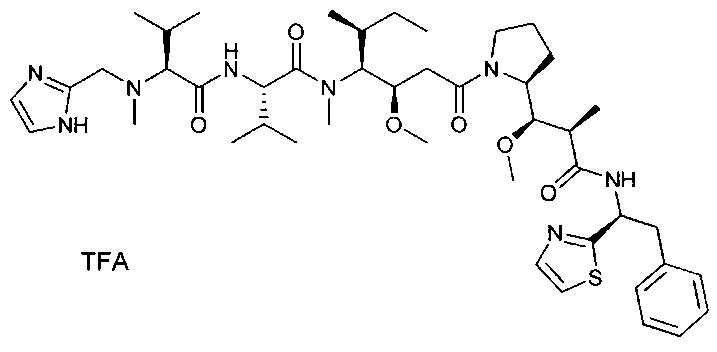
Соединение 48 получали аналогично соединению 1 из аминов 1Y и 1ZC и 1Н-имидазол-2-карбальдегида. Конечный продукт очищали путем препаративной HPLC в следующих условиях: SunFire Prep С18 OBD колонка, 5 мкм, 19×150 мм, подвижные фазы, буферизованные 0,05% TFA, градиент от 15,0 до 30% ACN в воде в течение 10 минут, затем вверх до 95,0% ACN в течение 2 минут, УФ-детекция 220 нм.
LC/MS/UV (Zorbax Eclipse Plus C8, 1,8 мкм, 4,6×100 мм; 1 мл/мин, 40°C, 2% метанол в воде (элюирующие фазы, буферизованные 0,05% TFA) в течение 1 минуты, затем от 2% до 95% метанола в течение 12 минут; ESI (C45H70N8O6S, точная масса 850,51) m/z: 851,2 (МН+), 873,5 (MNa+), 426,3 (М, 2H+/2); 12,75 мин (90,5%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,83-7,81 (m, 1Н), 7,80-7,53 (m, 3Н), 7,53-7,22 (m, 5Н), 5,6-5,8 (m, 1Н), 5,0-4,6 (m, 2Н); 4,6-0,85 (m, 55Н).
Соединение 49
(S)-2-((S)-2-((4-гидроксифенэтил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, соль трифторуксусной кислоты

Соединение 49А: 2-(4-гидроксифенил)ацетальдегид
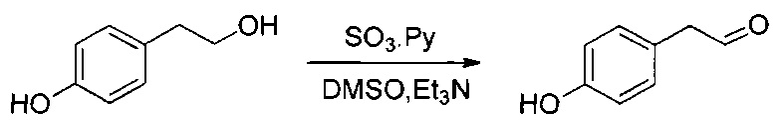
4-(2-гидроксиэтил)фенол (4 г, 28,95 ммоль, 1,00 экв.) растворяли в ДМСО (32 мл) и затем по каплям добавляли TEA (8,8 мл, 2,20 экв.). Добавляли раствор SO3.Py (10 г, 2,20 экв.) в ДМСО (36 мл) и смесь оставляли встряхиваться в течение ночи при температуре окружающей среды. Реакционную смесь нейтрализовали с помощью 250 мл воды и экстрагировали 3 раза с помощью 100 мл AcOEt. Органические фазы объединяли, промывали 5 раз водой (100 мл), затем дважды с помощью 150 мл NaCl (насыщ.), сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на силикагеле (EtOAc/PE (1:10), получая 1 г (25%) соединения 49А в форме бесцветного масла.
Соединение 49В: бензил-(S)-2-((4-гидроксифенэтил)(метил)амино)-3-метилбутаноат
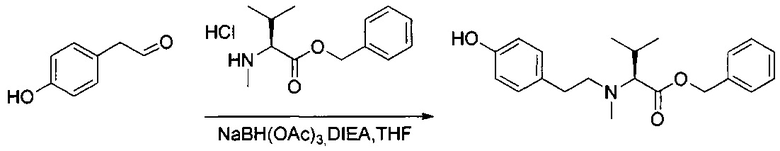
Соединение 49В синтезировали аналогично соединению 14С из амина 1ZC (1,5 г, 5,82 ммоль, 0,99 экв.), альдегида 49А (800 мг, 5,88 ммоль, 1,00 экв.), NaBH(OAc)3 (2,7 г, 12,74 ммоль, 2,17 экв.) и DIEA (4,23 мл) в ТГФ (25 мл). Реакционную смесь нейтрализовали с помощью 50 мл воды и экстрагировали 3 раза с помощью 50 мл AcOEt. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали на силикагеле (EtOAc/РЕ (1:10), получая 600 мг (37%) соединения 49В в форме белого твердого вещества.
Соединение 49С: (S)-2-((4-гидроксифенэтил)(метил)амино)-3-метилбутановая кислота

Соединение 49В (0,5 г, 1,46 ммоль, 1,00 экв.) растворяли в 40 мл МеОН в присутствии Pd/C (250 мг) и гидрогенировали в течение 3 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 0,4 г соединения 49С в форме белого твердого вещества.
Соединение 49: Соединение 49 синтезировали аналогично соединению 3 из амина 1Y (53,4 мг, 0,08 ммоль, 2,00 экв.), кислоты 49С (70 мг, 0,28 ммоль, 1,00 экв.), DEPC (0,032 мл, 2,00 экв.) и DIEA (0,053 мл, 3,00 экв.) в DCM (3 мл). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, Atlantis Prep OBD Т3 колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 45% ACN в течение 10 минут, затем от 45% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 3 мг (1%) соединения 49 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C49H74N6O7S, точная масса 890,5) m/z: 891,5 (МН+) и 446,4 (М, 2H+/2, 100%), 6,69 мин (100%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,92-8,87 (m, 0,5Н, NHCO, неполный обмен), 8,70-8,63 (m, 0,4Н, NHCO, неполный обмен), 8,85-8,77 (m, 1Н), 7,59-7,51 (m, 1Н), 7,35-7,03 (m, 7Н), 6,82-6,71 (m, 2Н), 5,77-5,58 (m, 1Н), 5,81-5,70 (m, 1Н), 4,21-0,80 (m, 58Н).
Соединение 50
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксифенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, соль трифторуксусной кислоты
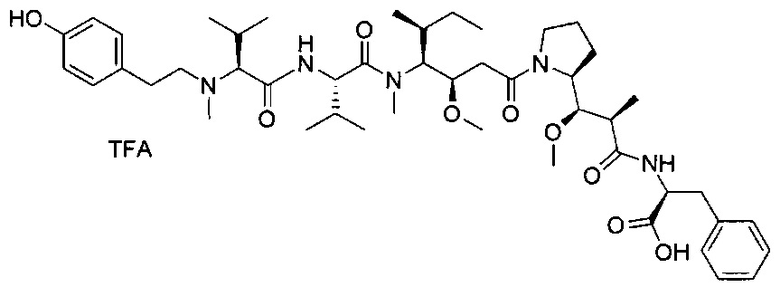
Соединение 50 получали аналогично соединению 4 из соединения 27 (100 мг, 0,10 ммоль, 1,00 экв.). Осадок очищали путем препаративной HPLC (Pre-HPLC-001 SHIMADZU, Atlantis Prep OBD Т3 колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм), получая 10,7 мг (11%) соединения 50 в форме белого твердого вещества.
LC/MS/UV (Ascentis Express С18 колонка, 2,7 мкм, 4,6×100 мм; 40°C; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% TFA) в течение 8 минут); ESI (C47H73N5O9, точная масса 851,5) m/z: 852,5 (МН+) и 426,8 (М, 2H+/2, 100%), 6,46 мин (91,7%, 210 нм).
1Н NMR: (400 MHz, CD3OD, ppm): δ (наличие ротамеров) 7,34-7,15 (m, 5Н); 7,15-7,04 (se, 2Н), 6,82-6,83 (m, 2Н), 4,83-4,70 (m, 1Н), 4,21-4,00 (m, 1Н), 3,90-3,80 (m, 1Н), 3,74-3,62 (m, 1Н), 3,57-2,86 (m, 20Н), 2,56-0,80 (m, 36Н).
Соединение 51
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксибензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, соль трифторуксусной кислоты
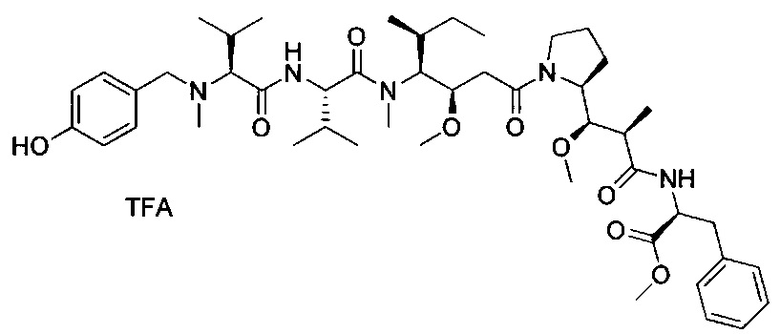
Соединение 51А: трет-бутил-(4-формилфенил)карбонат
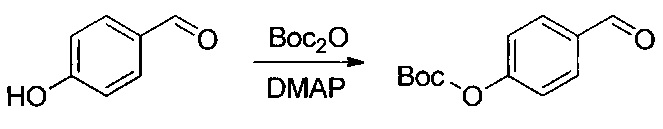
4-гидроксибензальдегид (3,0 г, 24 ммоль) растворяли в 30 мл DCM в присутствии 4-DMAP (300 мг, 2,46 ммоль, 0,1 экв.) и ди-трет-бутилдикарбоната (5,35 г, 24 ммоль, 1,0 экв.) и встряхивали в течение 1 ч при температуре окружающей среды. Затем раствор разводили в 200 мл воды и экстрагировали 3 раза с помощью 100 мл DCM. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 5 г (92%) соединения 51А в форме белого твердого вещества.
Соединение 51В: бензил-(S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутаноат
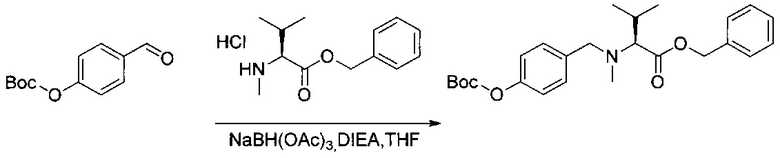
Соединение 51А (220 мг, 0,99 ммоль) растворяли в 5 мл ТГФ в присутствии соединения 1ZC (255 мг, 0,99 ммоль, 1,0 экв.), NaBH(OAc)3 (420 мг, 2 ммоль, 2,0 экв.) и DIEA (654 мкл) и встряхивали в течение ночи при температуре окружающей среды. Раствор затем разводили 100 мл воды и экстрагировали 3 раза с помощью 50 мл EtOAc. Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок очищали на колонке с силикагелем смесью EtOAc и РЕ (1:100), получая 200 мг (47%) соединения 51В в форме белого твердого вещества.
Соединение 51С: (S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутановая кислота
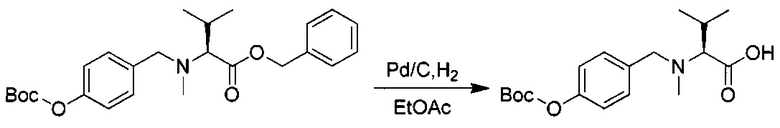
Соединение 51С получали путем гидрирования соединения 51В (200 мг), следуя протоколу, используемому для получения соединения 3F.
Соединение 51D: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
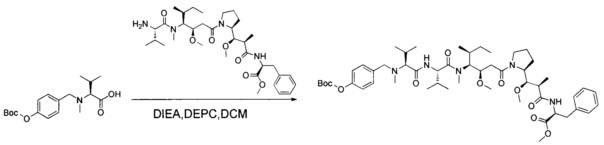
Соединение 51D получали путем связывания соединение 51С с амином 3D, следуя протоколу, используемому для получения соединения 3, получая нужный продукт в форме желтого масла с выходом 60%.
Соединение 51: Соединение 51D (80 мг, 0,08 ммоль) растворяли в 1 мл DCM в присутствии 0,5 мл TFA, встряхивали в течение 2 ч при температуре окружающей среды, а затем концентрировали при пониженном давлении. Осадок очищали путем препаративной HPLC (Pre-HPLC-010, SunFire Prep C18 OBD колонка, 5 мкм, 19×150 мм; элюирующая фаза: вода/ACN, буферизованные 0,05% TFA; градиент от 23% до 40% ACN в течение 10 минут, затем от 40% до 95% ACN в течение 2 минут; УФ-детектор Waters 2489 при 254 нм и 220 нм). Соединение 51 получали с выходом 24% (20 мг) в форме белого твердого вещества.
LC/MS/UV (Zorbax SB-Aq, 1,8 мкм, 4,6×100 мм; 2% МеОН в воде (0,05% TFA) в течение 1 минуты, затем от 2% до 95% МеОН в течение 13 минут); ESI (C47H73N5O9, точная масса 851,54) m/z: 874,5 (MNa+), 426,9 (М, 2Н+/2); 12,48 мин (96%, 210 нм).
1Н NMR: (300 MHz, CD3OD, ppm): δ (наличие ротамеров) 8,1-8,6 (m, 0,9Н, NHCO неполный обмен); 7,29-7,27 (m, 2Н), 7,25-6,86 (m, 5Н), 6,84-6,83 (m, 2Н), 4,83-4,72 (m, 3Н), 4,26-0,82 (m, 58Н).
Соединение 61
(S)-2-((S)-2-((4-аминофензгил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
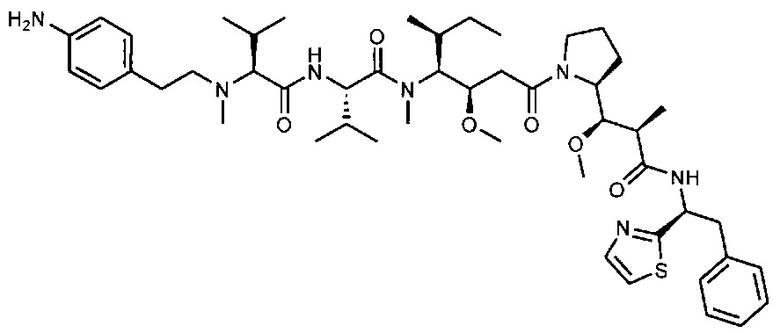
Соединение 61А: N-(4-аминофенэтил)-N-метил-L-валина дигидрохлорид
Соединение 11D (962 мг, 2,75 ммоль) растворяли в 10 мл коммерчески доступного раствора HCl в пропан-2-оле (5-6 М) и встряхивали при комнатной температуре в течение 2 ч. TLC-анализ показывал полное потребление исходного материала. Растворитель выпаривали при пониженном давлении, и полученное желтое твердое вещество растирали с Et2O (2×10 мл). Продукт сушили в вакууме, получая соединение 61А в виде желтого твердого вещества (322 мг, 47%).
Соединение 61: Карбоновую кислоту 61А (73 мг, 0,23 ммоль, 1 экв.) и амин 1Y (150 мг, 0,23 ммоль, 1 экв.) растворяли в сухом ДМФА (2 мл). Добавляли DIEA (158 мкл, 0,90 ммоль, 4 экв.) и DECP (51 мкл, 0,34 ммоль, 1,5 экв.) и реакционную смесь встряхивали в течение 4 ч при комнатной температуре. Анализ путем LC-MS показывал полное потребление исходного материала. Растворитель выпаривали при пониженном давлении, и осадок очищали путем флэш-хроматографии на силикагеле (DCM/MeOH), получая соединение 61 в виде светло-желтого твердого вещества (83 мг, 40%).
1Н NMR: (500 MHz, ДМСО-d6, ppm): δ (наличие ротамеров), 8,86 (d, 0,5Н, NHCO); 8,65 (d, 0,5Н, NHCO), 8,11-8,05 (m, 1Н, NHCO), 7,80 (d, 0,5H, тиазол), 7,78 (d, 0,5Н, тиазол), 7,65 (d, 0,5Н, тиазол), 7,63 (d, 0,5Н, тиазол), 7,32-7,12 (m, 5Н), 6,83 (d, J=8,3 Hz, 2Н), 6,45 (d, J=8,3 Hz, 2H), 5,56-5,49 (m, 0,5 H), 5,42-5,35 (m, 0,5H), 4,78 (s, 2H, NH2), 4,74-4,46 (m, 2H), 4,01-0,66 (m, 57H).
HPLC (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% TFA) в течение 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr составляет 1,31 мин (96,5%, 220 нм).
m/z (Q-TOF ESI+) 890,5558 (2%, MH+, C49H76N7O6S требует 890,5572), 445,7834 (100%, (MH2)2+, C49H77N7O6S требует 445,7823).
Соединение 62
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминофенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат
Соединение 62 получали аналогично соединению 61, используя карбоновую кислоту 61А (69 мг, 0,21 ммоль, 1 экв.), амин 3D (135 мг, 0,21 ммоль, 1 экв.), DIEA (75 мкл, 0,43 ммоль, 2 экв.) и DECP (49 мкл, 0,32 ммоль, 1,5 экв.). Неочищенный продукт очищали путем флэш-хроматографии на силикагеле (DCM/MeOH), получая соединение 62 в виде желтоватого твердого вещества (82 мг, 45%).
1Н NMR: (500 MHz, ДМСО-d6, ppm): δ (наличие ротамеров), 8,50 (d, J=8,3, 0,5Н, NHCO); 8,27 (d, J=8,0, 0,5H, NHCO), 8,15-8,04 (m, 1H, NHCO), 7,27-7,13 (m, 5H), 6,86-6,79 (m, 2H), 6,48-6,42 (m, 2H), 4,78 (s, 2H, NH2), 4,74-4,44 (m, 3H), 4,01-3,72 (m, 1,5H), 3,66 (s, 1.5H, CO2Me), 3,63 (s, 1,5H, CO2Me), 3,57-0,65 (m, 55,5H).
HPLC (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% TFA) в течение 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr составляет 1,29 мин (95,3%, 220 нм).
m/z (Q-TOF ESI+) 865,5800 (2%, МН+, C48H77N6O8 требует 865,5797), 433,2937 (100%, (МН2)2+, C48H78N6O8 требует 433,2935).
Соединение 63
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминофенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат

Соединение 62 (23 мг, 0,03 ммоль) растворяли в смеси воды (1 мл) и ацетонитрила (1 мл). Добавляли пиперидин (0,75 мл) и смесь встряхивали при комнатной температуре в течение 5 ч. TLC-анализ показывал полное потребление исходного материала. Растворитель выпаривали при пониженном давлении и осадок очищали путем препаративной HPLC (SunFire Prep колонка С18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 20% до 40% MeCN в 10 минут, затем от 40% до 100% MeCN в течение 2 минут; УФ-детектор Waters 2545 при 254 нм и 220 нм). Соединение 63 получали в виде белого твердого вещества (14 мг, 66%).
1Н NMR: (500 MHz, ДМСО-d6, ppm): δ (наличие ротамеров), 12,7 (s(br), 1Н, CO2H), 9,58 (m(br), 1Н); 9,04-8,89 (m, 1Н), 8,41 (d, 0,6Н, NHCO), 8,15 (d, 0,4Н, NHCO), 7,27-7,13 (m, 5Н), 7,13-6,99 (m(br), 2Н), 6,90-6,64 (s(br), 2Н), 4,77-3,40 (m, 10Н), 3,34-2,75 (m, 20Н), 2,34-1,94 (m, 4Н), 1,90-0,7 (m, 25Н).
HPLC (Xbridge Shield С18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% TFA) в течение 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr составляет 1,24 мин (100%, 220 нм).
m/z (Q-TOF ESI+) 851,5641 (6%, МН+, C47H75N6O8 требует 851,5641), 426,2854 (100%, (МН2)2+, C47H76N6O8 требует 426,2857).
Пример 20: Антипролиферативная активность лекарственных средств
Метод:
Культура клеток. Клетки А549 (немелкоклеточный рак легких - АТСС CCL-185) и MDA-MB-231 (аденокарциномы молочной железы - АТСС НТВ-26) культивировали в минимальной эссенциальной среде Игла (MEM) с 5% фетальной телячьей сывороткой (FCS) и в среде Игла в модификации Дульбекко (DMEM) с 10% FCS, соответственно. Клетки MCF7 (протоковая карцинома молочной железы - АТСС НТВ-22) и SN-12C (карцинома почки - АТСС) поддерживали в среде RPMI 1640 (без фенола красного для клеток MCF7), содержащей 10% FCS. Все среды были дополнены фунгизоном (1,25 мкг/мл) и пенициллином-стрептомицином (100 Ед/100 мкг/мл). Клетки культивировали в стандартных условиях в инкубаторе при температуре 37°C, 5% CO2 и 95% влажности воздуха.
Антипролиферативная активность на 4 опухолевых клеточных линиях.
Выбранные лекарственные средства исследовали на предмет их антипролиферативной активности с использованием анализа пролиферации ATPLite (Perkin Elmer, Вильбон-сюр-Иветт, Франция) на полной панели 4 клеточных линий. Клетки высевали в 96-луночные планшеты (103 клеток/лунка для А549, 2×103 для MCF7, MDA-MB-231 и SN12C) в 0 день в такой концентрации, чтобы клетки оставались в логарифмической фазе клеточного роста в течение всего 72-часового периода обработки лекарственным средством. После 24-часового инкубационного периода все клетки обрабатывали серийными разведениями анализируемых соединений (11 мкл 10 × раствора в 1% ДМСО - 6 лунок/условие). Для того чтобы избежать прилипания соединения на наконечники, наконечники меняли между двумя последовательными разведениями. Затем клетки помещали в 37°C, 5% CO2-инкубатор. На 4 день жизнеспособность клеток оценивали путем определения доз АТФ, высвобождаемого жизнеспособными клетками. Число жизнеспособных клеток анализировали в сравнении с числом клеток, обработанных растворителем. Значения ЕС50 определяли с помощью анализа подгонки кривой (нелинейная регрессионная модель с сигмоидальной кривой "доза - ответ", переменный коэффициент наклона), выполненного с использованием алгоритма, предоставленного программным обеспечением GraphPad Software (GraphPad Software Inc., Калифорния, США).
Результаты:
Различные лекарственные средства:
Различные лекарственные средства анализировали с целью определения их антипролиферативной активности в отношении клеточной линии MDA-MB-231 согласно описанному выше способу. Измеренные активности давали значения ЕС50 менее 0,1 мкМ.
Несколько следующих примеров, выбранных из числа лекарственных средств, приведенных выше в качестве примеров, иллюстрируют их полностью замечательные антипролиферативные свойства:
Пример 12: ЕС50=5,80×10-10 М; Пример 13: ЕС50=7.95×10-8 М; Пример 15: ЕС50=1,70×10-10 М; Пример 27: ЕС50=1,20×10-10 М.
Различные клеточные линии:
Соединение 15 анализировали на различных клеточных линиях (А549, MDA-МВ-231, MCF-7, SN12C) в соответствии с описанным выше способом. Измеренные активности давали значения ЕС50 менее 0,1 мкМ на всех протестированных клеточных линиях.
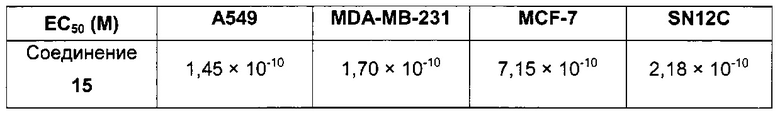
Сравнительные примеры:
В сравнительных примерах, приведенных ниже, было изучено замещение на фенильном кольце (аминный/гидроксильный против карбоксильного заместителя) и показана улучшенная антипролиферативная активность лекарственных средств согласно данному изобретению, содержащих аминный или гидроксильный заместитель.
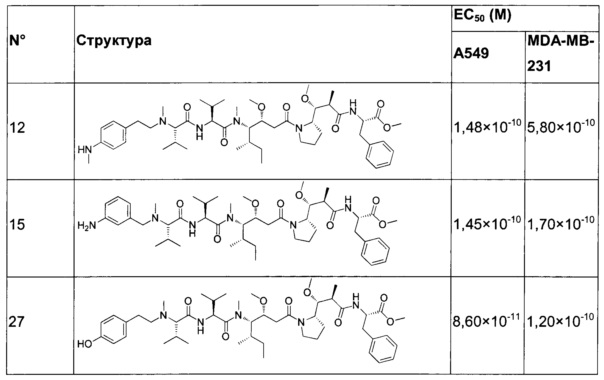
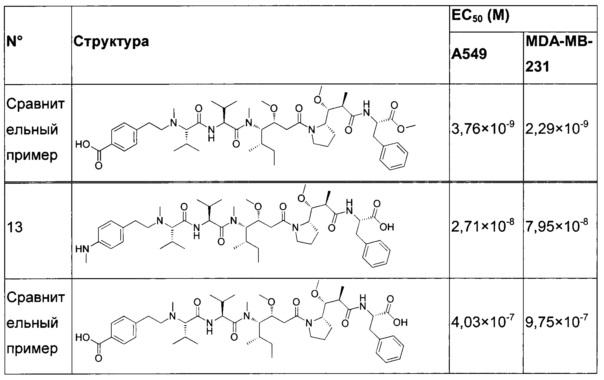
Пример 21: Синтез группировки "лекарственное средство - линкер"
Соединение Е-11
4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил-(4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)(метил)карбамат 2,2,2-трифторацетат
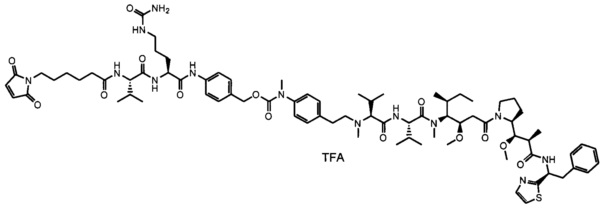
Соединение Е-11-1: метил-(S)-2-амино-5-уреидопентаноата гидрохлорид
К МеОН (120 мл) по каплям добавляли ацетилхлорид (10 мл) при 0°C в условиях встряхивания. Через 20 минут добавляли L-цитруллин (10 г, 57 ммоль, 1,00 экв.) и смесь нагревали с обратным кипячением в течение ночи. Растворитель выпаривали при пониженном давлении, получая 15 г (116%) соединения Е-11-1 в виде белого твердого вещества. Продукт использовали на следующем этапе без дополнительного высушивания.
Соединение Е-11-2: метил-(S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентаноат
Соединение Е-11-1 (13 г, 57,6 ммоль, 1,1 экв.) растворяли в ДМФА (140 мл) при 0°C в инертной атмосфере. Добавляли DIEA (30 мл, 173 ммоль, 3,0 экв.), гидроксибензотриазол (HOBt - 10,59 г, 69,1 ммоль, 1,2 экв.) и Boc-L-валина гидроксисукцинимидный эфир (Boc-Val-OSu - 18,1 г, 57,6 ммоль, 1,0 экв.). Реакционную смесь встряхивали в течение ночи при температуре окружающей среды, затем растворитель выпаривали при пониженном давлении. Осадок растворяли в воде (100 мл) и дважды экстрагировали с помощью DCM (150 мл). Органические фазы объединяли, сушили над Na2SO4 и концентрировали при пониженном давлении. Осадок очищали на силикагеле (DCM/MeOH), получая 18,8 г (84%) соединения Е-11-2 в виде белого твердого вещества.
Соединение Е-11-3: (S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метил бутанамидо)-5-уреидопентановая кислота

Соединение Е-11-2 (18,8 г, 48,4 ммоль, 1 экв.) растворяли в МеОН (200 мл) при 0°C. Добавляли раствор NaOH 1 М (72 мл, 72 ммоль, 1,5 экв.) и смесь встряхивали в течение 2 ч при комнатной температуре. МеОН удаляли при пониженном давлении и оставшийся водный раствор закисляли с помощью 1 М HCl. Водную фазу выпаривали досуха и осадок очищали на силикагеле (DCM/MeOH), получая 18 г (99%) соединения Е-11-3 в виде белого твердого вещества.
Соединение Е-11-4: трет-бутил-((S)-1-(((S)-1-((4-(гидроксиметил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-3-метил-1-оксобутан-2-ил)карбамат
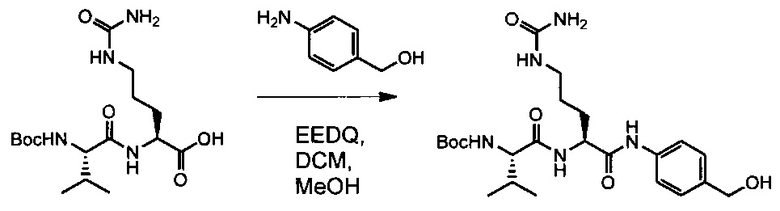
Соединение Е-11-3 (5 г, 13,4 ммоль, 1 экв.) растворяли в смеси сухого DCM (65 мл) и сухого МеОН (35 мл). Добавляли (4-аминофенил)метанол (1,81 г, 14,7 ммоль, 1,1 экв.) и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ - 6,60 г, 26,7 ммоль, 2 экв.) и смесь встряхивали в темноте в течение ночи. Растворители выпаривали при пониженном давлении, и осадок очищали на силикагеле (DCM/MeOH), получая 5,2 г (73%) соединения Е-11-4 в виде твердого вещества белого цвета с желтоватым оттенком.
Соединение Е-11-5: трет-бутил-((S)-3-метил-1-(((S)-1-((4-((((4-нитрофенокси)карбонил)окси)метил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-1-оксобутан-2-ил)карбамат
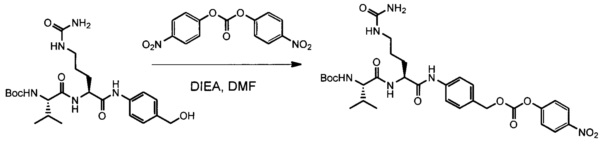
Соединение Е-11-4 (1,1 г, 2,29 ммоль, 1 экв.) растворяли в сухом ДМФА (5 мл) при температуре окружающей среды в инертной атмосфере. Добавляли бис(4-нитрофенил)карбонат (1,40 г, 4,59 ммоль, 2 экв.), затем DIEA (600 мкл, 3,44 ммоль, 1,5 экв.), и полученный желтый раствор встряхивали в течение ночи. ДМФА выпаривали при пониженном давлении и осадок очищали на силикагеле (DCM/MeOH), получая 1,27 г (84%) соединения Е-11-5 в виде твердого вещества белого цвета с желтоватым оттенком.
Соединение Е-11-6: 4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил-(4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)(метил)карбамата 2,2,2-трифторацетат

Карбонат Е-11-5 (114 мг, 0,177 ммоль, 1,2 экв.) и анилин 11F (150 мг, 0,147 ммоль, 1 экв.) растворяли в сухом ДМФА (4 мл). Добавляли HOBt (38 мг, 0,295 ммоль, 2 экв.) и DIEA (54 мкл, 0,295 ммоль, 2 экв.) и смесь встряхивали в течение выходных при комнатной температуре. ДМФА выпаривали при пониженном давлении и осадок очищали путем флэш-хроматографии на силикагеле, элюируя с помощью DCM. Продукт повторно очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение Е-11-6 в виде белого твердого вещества (89 мг, 39%).
Соединение Е-11:
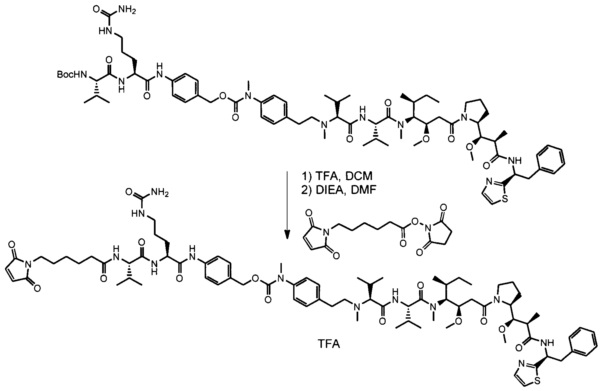
Соединение Е-11-6 (21 мг, 0,014 ммоль, 1,0 экв.) растворяли в DCM (0,25 мл) и добавляли TFA (40 мкл). Раствор встряхивали в течение 2 ч при комнатной температуре, после чего LC-MS-анализ показывал полное потребление исходного материала. Смесь быстро охлаждали (баня с жидким азотом), одновременно добавляя ДМФА (0,5 мл), затем DIEA (100 мкл) для нейтрализации TFA. Затем охлаждающую баню удаляли и добавляли 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (4 мг, 0,012 ммоль, 1 экв.). Смесь встряхивали при комнатной температуре в течение 48 ч и продукт очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение Е-11 в виде белого твердого вещества (11 мг, 54%).
m/z (Q-TOF MS ESI+) 1524,8282 (2%, MNa+, C79H115N13NaO14S требует 1524,8299), 751,9283 (100%, (MH2)2+, C79H117N13O14S требует 751,9276).
Соединение Е-12
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
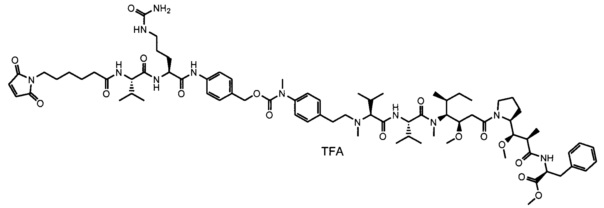
Соединение Е-12-1: трет-бутил-((S)-3-метил-1-оксо-1-(((S)-1-оксо-1-((4-((((перфторфенокси)карбонил)окси)метил)фенил)амино)-5-уреидопентан-2-ил)амино)бутан-2-ил)карбамат
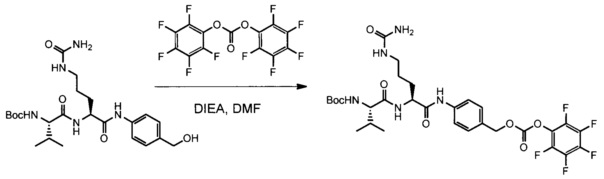
Соединение Е-11-4 (670 мг, 1,26 ммоль, 1 экв.) растворяли в сухом ДМФА (6 мл) при 0°C в инертной атмосфере. Добавляли бис(перфторфенил)карбонат (991 мг, 2,51 ммоль, 2 экв.), затем DIEA (329 мкл, 1,89 ммоль, 1,5 экв.), и полученный бесцветный раствор встряхивали в течение 30 минут при комнатной температуре. ДМФА выпаривали при пониженном давлении, и осадок очищали на силикагеле (DCM/MeOH), получая 836 мг (96%) соединения Е-12-1 в виде твердого вещества белого цвета с желтоватым оттенком.
Соединение Е-12-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
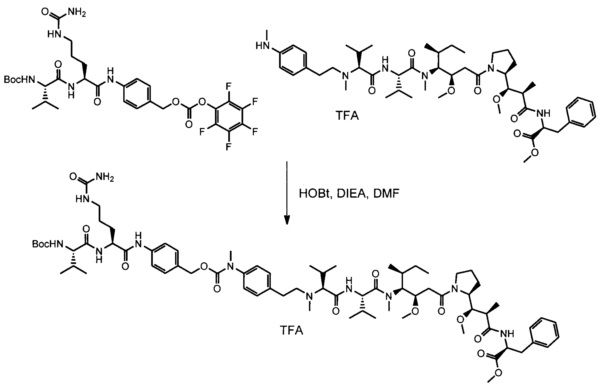
Анилин 12 (165 мг, 0,189 ммоль, 1,0 экв.) растворяли в ДМФА (5 мл) при 0°C в инертной атмосфере. Добавляли карбонат Е-12-1 (194 мг, 0,282 ммоль, 1,5 экв.), HOBt (51 мг, 0,375 ммоль, 2 экв.) и DIEA (66 мкл, 0,375 ммоль, 2 экв.) и смесь встряхивали при комнатной температуре в течение 8 ч. Растворитель выпаривали при пониженном давлении и осадок очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение Е12-7 в виде белого твердого вещества (247 мг, 77%).
Соединение Е-12-3: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
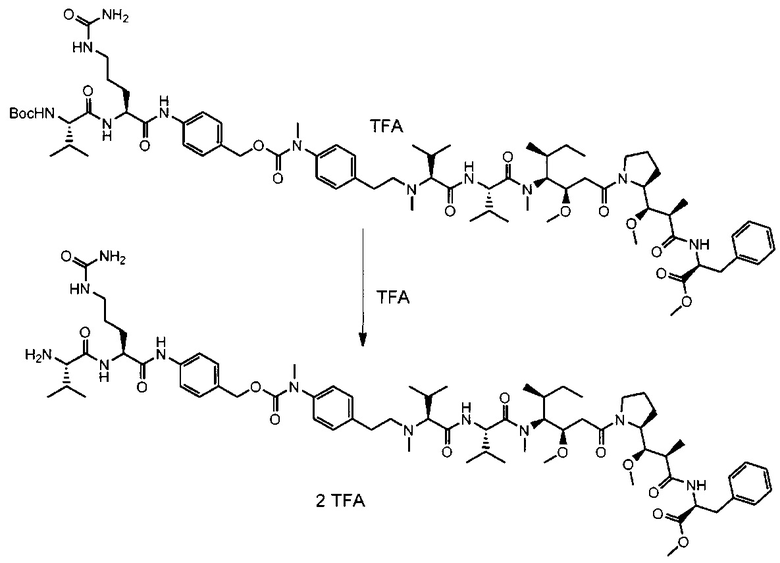
Соединение Е-12-2 (5,6 мг, 4,04 мкмоль, 1,0 экв.) растворяли в TFA (100 мкл). Через 5 минут добавляли 2 мл воды и смесь лиофилизировали в течение ночи, получая соединение Е-12-3 в виде твердого вещества белого цвета с желтоватым оттенком (5,6 мг, 98%).
Соединение Е-12:
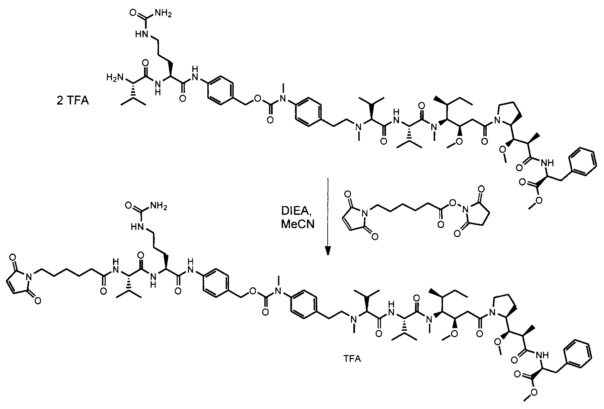
Соединение Е-12-3 (5,6 мг, 4 мкмоль, 1,0 экв.) растворяли в ацетонитриле (0,5 мл), и добавляли DIEA (5 мкл, 7 экв.), затем 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (2,5 мг, 8 мкмоль, 2 экв.). Смесь встряхивали в течение 6 ч при комнатной температуре. После контроля реакционной смеси путем LC-MS добавляли 200 мкл воды и полученный раствор очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение Е-12 в виде белого твердого вещества (4,6 мг, 70%).
m/z (Q-TOF MS ESI+) 739,4389 (100%, (МН2)2+, C78H118N12O16 требует 739,4389).
Соединение Е-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
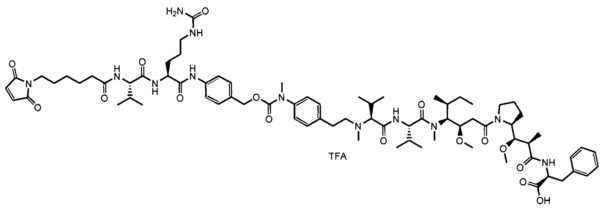
Соединение Е-13-1: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин
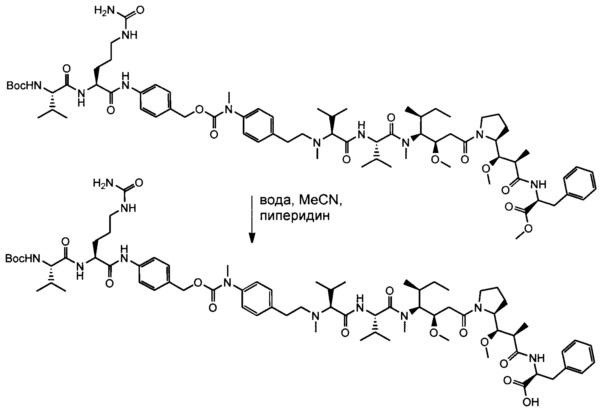
Соединение Е-12-2 (185 мг, 0,123 ммоль, 1,0 экв.) растворяли в смеси воды (5 мл) и ацетонитрила (5 мл) при комнатной температуре. Добавляли пиперидин (3,67 мл, 300 экв.) и смесь встряхивали в течение 6 ч при комнатной температуре. Растворители выпаривали досуха при пониженном давлении, и осадок растирали с Et2O (60 мл). Твердое вещество промывали дважды Et2O (20 мл) и сушили в вакууме, получая соединение Е-13-1 в виде твердого вещества белого цвета с желтоватым оттенком (175 мг, 95%).
Соединение Е-13-2: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина бис(2,2,2-трифторацетат)
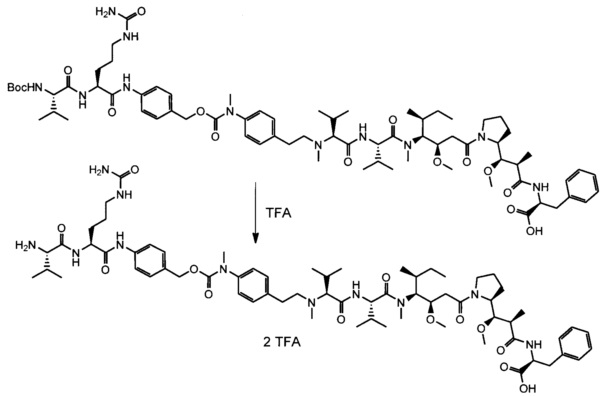
Соединение Е-13-1 (175 мг, 0,128 ммоль, 1,0 экв.) растворяли в TFA (200 мкл). Через 5 минут добавляли воду (1 мл) и ацетонитрил (1 мл) и раствор лиофилизировали в течение ночи, получая соединение Е-13-2 в виде твердого вещества белого цвета с желтоватым оттенком (180 мг, 87%).
Соединение Е-13: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил) пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин, 2,2,2-трифторацетат
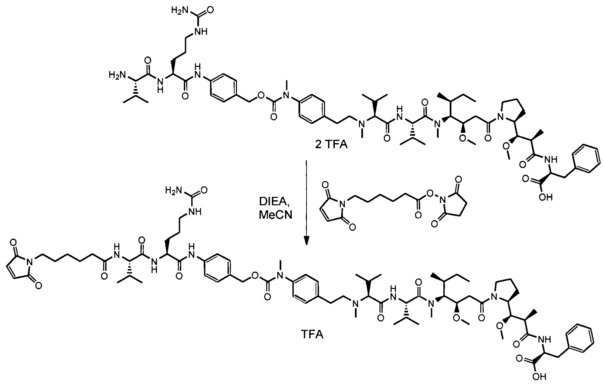
Соединение Е-13-2 (80 мг, 0,058 ммоль, 1,0 экв.) растворяли в смеси ацетонитрила (1,5 мл) и ДМФА (0,4 мл). Добавляли DIEA (50 мкл, 0,289 ммоль, 5 экв.), затем 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (36 мг, 0,116 ммоль, 2 экв.). Смесь встряхивали в течение 3 ч при комнатной температуре. После контроля реакционной смеси путем LC-MS растворитель выпаривали при пониженном давлении и осадок очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение Е-13 в виде белого твердого вещества (32 мг, 35%).
m/z (Q-TOF MS ESI-) 1461,8336 (100%, (M-H-), C77H113N12O16 требует 1461,8403). m/z (Q-TOF MS ESI+) 1463,8565 (2%, MH+, C77H115N12O16 требует 1463,8549), 732,4317 (100%, (МН2)2+, C77H116N12O16 требует 732,4311).
Соединение Е-15
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат 2,2,2-трифторацетат
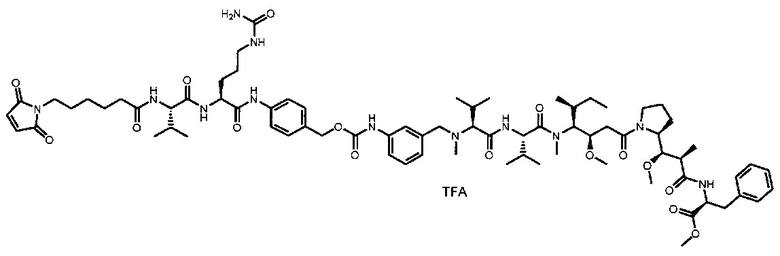
Соединение Е-15-1: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
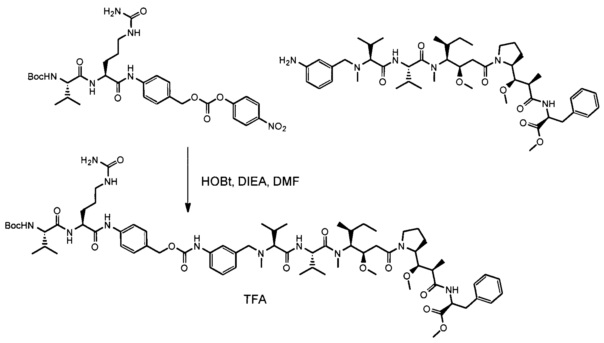
Соединение Е-15-1 получали согласно тому же способу, как для соединения Е-11-6, используя карбонат Е-11-5 (28 мг, 0,044 ммоль, 1 экв.), анилин 15 (42 мг, 0,044 ммоль, 1 экв.), HOBt (3 мг, 0,022 ммоль, 0,5 экв.) и DIEA (15 мкл, 0,087 ммоль, 2 экв.) в ДМФА (2 мл). Соединение Е-15-1 выделяли в виде белого твердого вещества (8,2 мг, 13%).
Соединение Е-15-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси) карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
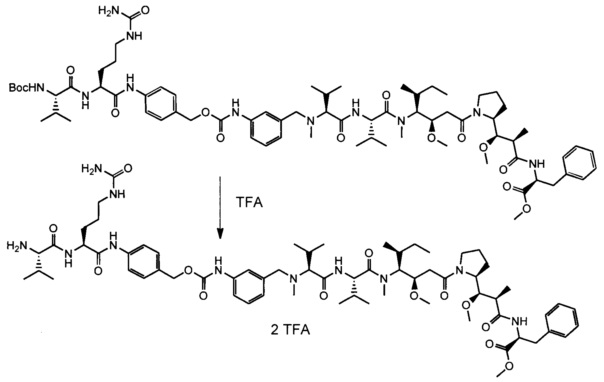
Соединение Е-15-1 (8,2 мг, 5,58 мкмоль, 1,0 экв.) растворяли в TFA (200 мкл). Через 5 минут добавляли воду (1 мл) и раствор лиофилизировали в течение ночи, получая соединение Е-15-8 в виде белого твердого вещества (7,6 мг, 99%).
Соединение Е-15:
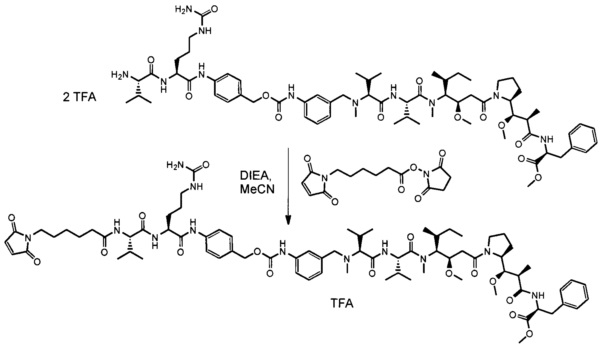
Соединение Е-15 получали согласно тому же способу, как для соединения Е-12, используя амин Е-15-2 (7,6 мг, 5,55 мкмоль, 1 экв.), 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноат (2 мг, 6,65 мкмоль, 1,2 экв.) и DIEA (5 мкл, 0,028 ммоль, 5 экв.) в ацетонитриле (0,5 мл). Соединение Е-15 выделяли в виде белого твердого вещества (4,2 мг, 48%).
m/z (Q-TOF MS ESI+) 1471,8169 (2%, MNa+, C76H112N12NaO16 требует 1471,8211), 725,4223 (100%, (MH2)2+, C76H114N12O16 требует 725,4232), 483,9482 (10%, (MH3)3+, C76H115N12O16 требует 483,9513).
Соединение F-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
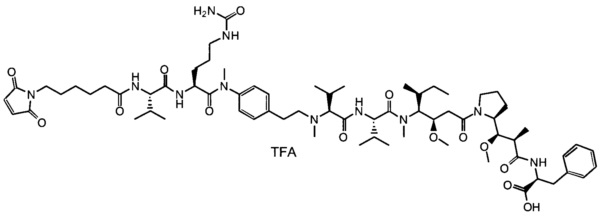
Соединение F-13-1: бензил-N-(4-((трет-бутоксикарбонил)(метил)амино)фенэтил)-N-метил-L-валинат
Соединение 11С (250 мг, 0,567 ммоль, 1 экв.) растворяли в ТГФ (10 мл), затем добавляли NaH (60% суспензия в минеральном масле, 68 мг, 1,702 ммоль, 3 экв.). Смесь встряхивали в течение 5 минут, а затем добавляли йодометан (106 мкл, 1,702 ммоль, 3 экв.). Реакционную смесь встряхивали в течение 2 ч при комнатной температуре, затем гасили водой и разделяли между EtOAc (100 мл) и водой (50 мл). Органическую фазу сушили над MgSO4 и выпаривали досуха, получая соединение F-13-1 в виде желтого масла (250 мг, 97%), которое использовали без дополнительной очистки.
Соединение F-13-2: бензил-N-метил-N-(4-(метиламино)фенэтил)-L-валинат
Вос-защищенный анилин F-13-1 (250 мг, 0,550 ммоль, 1 экв.) растворяли в МеОН (5 мл), затем добавляли 1 мл коммерчески доступного раствора HCl в iPrOH (5-6 М). Раствор встряхивали при комнатной температуре в течение 2 ч, а затем выпаривали досуха при пониженном давлении. Полученное желтое масло растирали с Et2O, получая соединение F-13-2 в виде желтого твердого вещества (202 мг, 94%).
Соединение F-13-3: бензил-N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенэтил)-N-метил-L-валинат

Кислоту Е-11-3 (190 мг, 0,508 ммоль, 1,5 экв.) растворяли в сухом ДМФА (1 мл), затем добавляли DIEA (118 мкл, 0,677 ммоль, 2 экв.), бензотриазол-1-ил-окситрипирролидинофосфора гексафторфосфат (РуВОР - 264 мг, 0,508 ммоль, 1,5 экв.) и анилин F-13-2 (120 мг, 0,339 ммоль, 1 экв.). Смесь встряхивали при комнатной температуре в течение ночи и растворители выпаривали при пониженном давлении. Осадок очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F-13-3 в виде белого твердого вещества (140 мг, 45%).
Соединение F-13-4: N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенэтил)-N-метил-L-валин
Соединение F-13-3 (116 мг, 0,163 ммоль, 1 экв.) растворяли в МеОН (5 мл) в присутствии Pd/C 10% (30 мг) и гидрогенировали в течение 2 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, получая 110 мг (99%) соединения F-13-4 в виде бежевого твердого вещества.
Соединение F-13-5: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
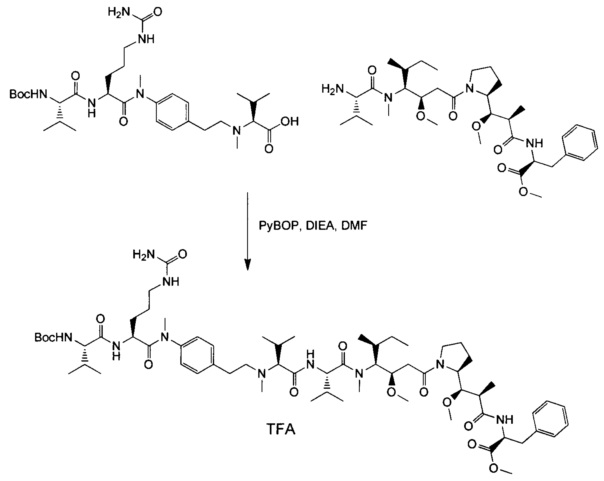
Амин 3D (89 мг, 0,140 ммоль, 1 экв.) и кислоту F-13-4 (145 мг, 0,210 ммоль, 1,5 экв.) растворяли в сухом ДМФА (4 мл), и добавляли РуВОР (109 мг, 0,210 ммоль, 1,5 экв.) и DIEA (73 мкл, 0,420 ммоль, 3 экв.). Смесь встряхивали в течение 1 ч при комнатной температуре и растворитель выпаривали. Осадок разделяли между EtOAc и водой, и органическую фазу сушили над MgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенный продукт очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F-13-5 в виде белого твердого вещества (140 мг, 73%).
Соединение F-13-6: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
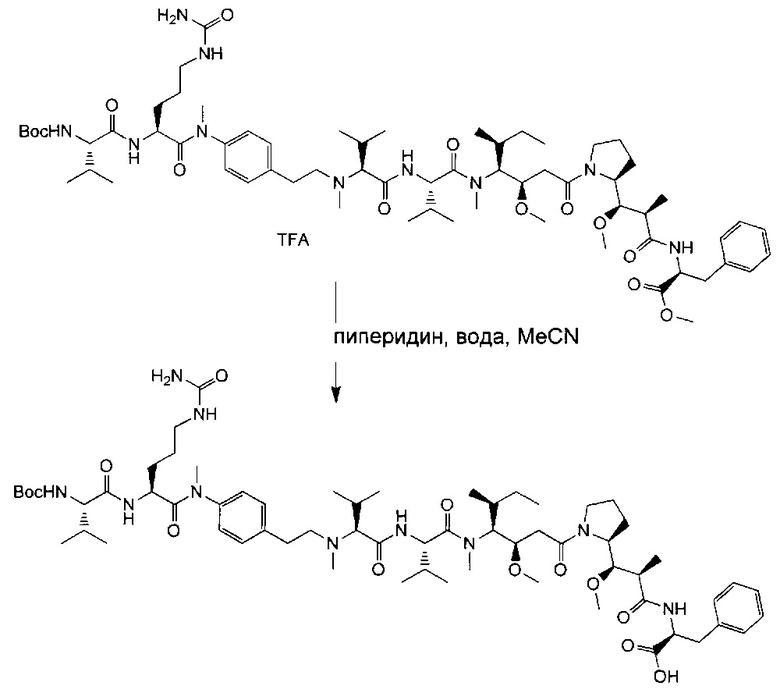
Соединение F-13-5 (140 мг, 0,104 ммоль, 1 экв.) растворяли в смеси воды (4 мл), ацетонитрила (4 мл) и пиперидина (2 мл) и встряхивали при комнатной температуре в течение 4 ч. Растворитель выпаривали при пониженном давлении и осадок очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F-13-6 в виде белого твердого вещества (115 мг, 83%).
Соединение F-13:
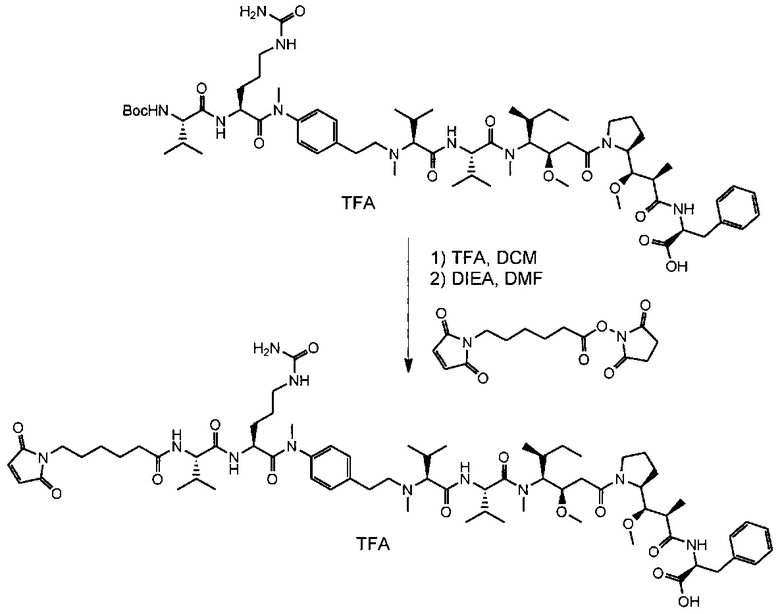
Соединение F-13 получали согласно тому же способу, как для соединения Е-11, используя Вос-защищенный амин F-13-6 (55 мг, 0,041 ммоль, 1,0 экв.) в DCM (0,5 мл) и TFA (100 мкл, 30 экв.), затем разводили ДМФА (1 мл), гасили (DIEA (320 мкл, 45 экв.), затем вводили в реакцию с 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноатом (15 мг, 0,049 ммоль, 1,2 экв.). После очистки путем препаративной HPLC и лиофилизации получали соединение F-13 в виде белого твердого вещества (14 мг, 24%).
m/z (Q-TOF MS ESI+) 1314,8067 (2%, МН+, C69H108N11O14 требует 1314,8072), 657,9067 (100%, (МН2)2+, C69H109N11O14 требует 657,9072).
Соединение F-61
N-((S)-1-(((S)-1-((4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-3-метил-1-оксобутан-2-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 2,2,2-трифторацетат
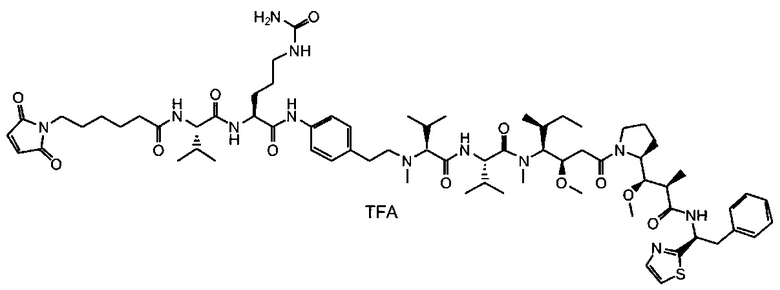
Соединение F-61-1: бензил-N-(4-аминофенэтил)-N-метил-L-валината дигидрохлорид
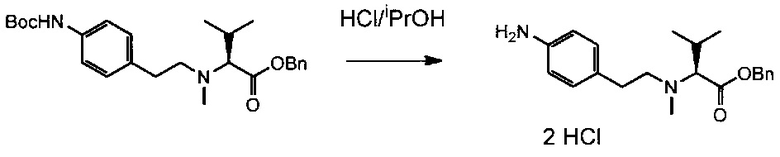
Соединение 11С (1,0 г, 2,27 ммоль, 1 экв.) растворяли в 8 мл коммерчески доступного раствора HCl в iPrOH (5-6 М). Смесь встряхивали в течение 2 ч при комнатной температуре, а затем выпаривали досуха при пониженном давлении. Осадок дважды растирали с Et2O (30 мл) и сушили в вакууме, получая соединение F-61-1 в виде белого твердого вещества (916 мг, 98%).
Соединение F-61-2: бензил-N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)-N-метил-L-валинат
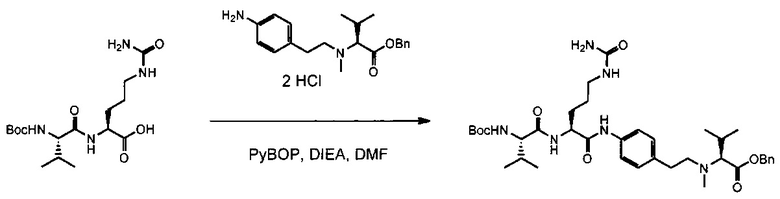
Кислоту Е-11-3 (769 мг, 2,05 ммоль, 1,5 экв.) растворяли в сухом ДМФА (2,5 мл), затем добавляли DIEA (957 мкл, 5,48 ммоль, 4 экв.) и РуВОР (1,07 г, 2,05 ммоль, 1,5 экв.). Добавляли анилин F-61-1 (566 мг, 1,369 ммоль, 1 экв.) и смесь встряхивали при комнатной температуре в течение ночи. Растворители выпаривали при пониженном давлении, и осадок очищали на силикагеле (DCM/MeOH), получая 969 мг (102%) соединения F-61-2 в виде белого твердого вещества.
Соединение F-61-3: N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)-N-метил-L-валин
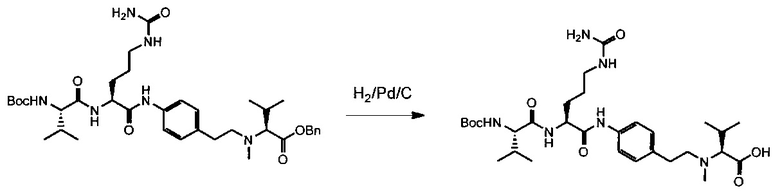
Соединение F-61-2 (969 мг, 1,28 ммоль, 1 экв.) растворяли в МеОН (20 мл) в присутствии Pd/C 10% (270 мг) и гидрогенировали в течение 3 ч при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении, и осадок очищали на силикагеле (DCM/MeOH/AcOH), получая 520 мг (67%) соединения F-61-3 в виде белого твердого вещества.
Соединение F-61-4: трет-бутил-((S)-1-(((S)-1-((4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-3-метил-1-оксобутан-2-ил)карбамата 2,2,2-трифторацетат
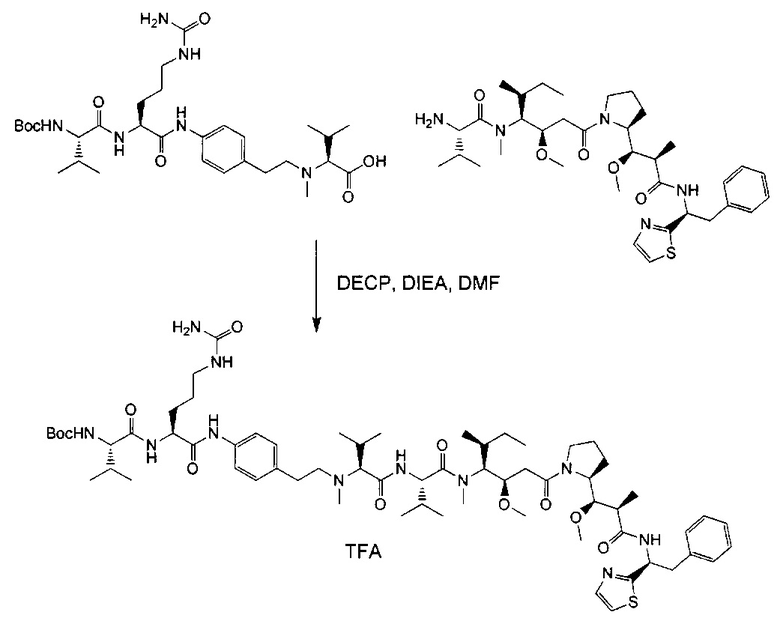
Кислоту F-61-3 (67,5 мг, 0,111 ммоль, 1,5 экв.) растворяли в сухом ДМФА (2 мл) и добавляли DECP (17 мкл, 0,111 ммоль, 1,5 экв.) и DIEA (39 мкл, 0,223 ммоль, 3 экв.). После встряхивания в течение 15 минут при комнатной температуре добавляли амин 1Y (50 мг, 0,074 ммоль, 1 экв.) и раствор встряхивали в течение ночи. Растворитель выпаривали при пониженном давлении, и осадок очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F61-4 в виде белого твердого вещества (28 мг, 28%).
Соединение F-61-5: (S)-2-((S)-2-амино-3-метилбутанамидо)-N-(4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)-5-уреидопентанамид бис(2,2,2-трифторацетат)
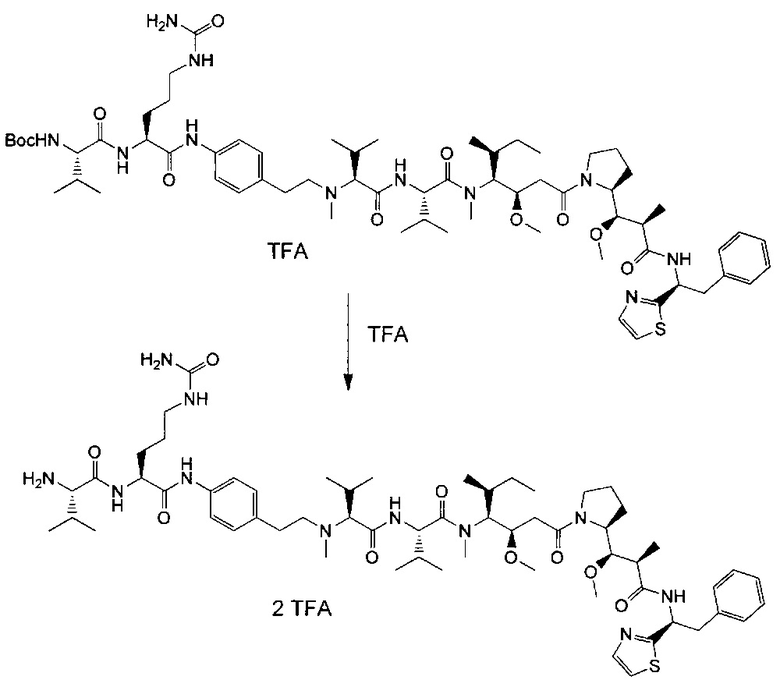
Соединение F-61-4 (28 мг, 0,021 ммоль, 1,0 экв.) растворяли в TFA (200 мкл). Через 5 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл) и раствор лиофилизировали в течение ночи, получая соединение F-61-5 в виде бесцветного масла (38 мг, 134%).
Соединение F-61:
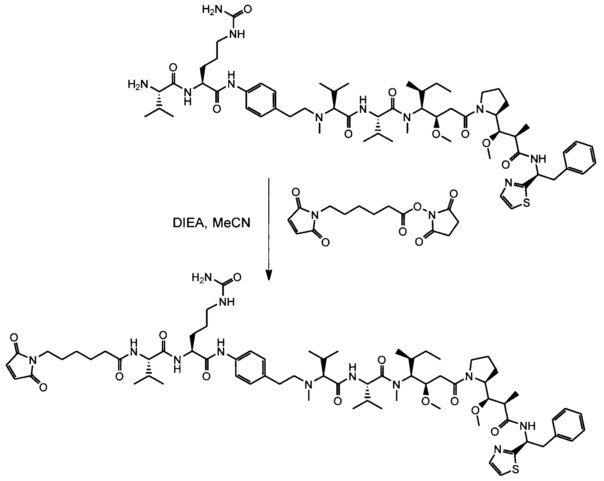
Соединение F-61-5 (28,3 мг, 0,020 ммоль, 1 экв.) растворяли в ацетонитриле (0,5 мл), затем добавляли 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (9 мг, 0,029 мкмоль, 1,4 экв.) и DIEA (25 мкл, 0,143 ммоль, 7 экв.). Смесь встряхивали в течение 4,5 ч, после чего HPLC-анализ показывал присутствие исходного материала, но полное потребление сукцинимида. Также добавляли дополнительный 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (3 мг, 0,01 мкмоль, 0,5 экв.) и реакционную смесь встряхивали в течение 1,5 ч. HPLC-анализ показывал полное потребление исходного материала. Растворитель выпаривали досуха и осадок дважды растирали со смесью EtOAc/Et2O (80/20), получая соединение F-61 в виде твердого вещества белого цвета с желтоватым оттенком (19,4 мг, 70%).
m/z (Q-TOF MS ESI+) 1361,7725 (2%, MNa+, C70H106N12NaO12S требует 1361,7666), 670,3961 (100%, (MH2)2+, C70H108N12O12S требует 670,3960).
Соединение F-62:
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат 2,2,2-трифторацетат
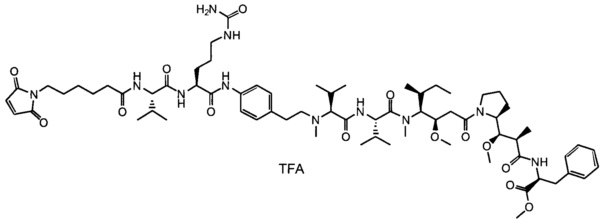
Соединение F-62-1: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат 2,2,2-трифторацетат
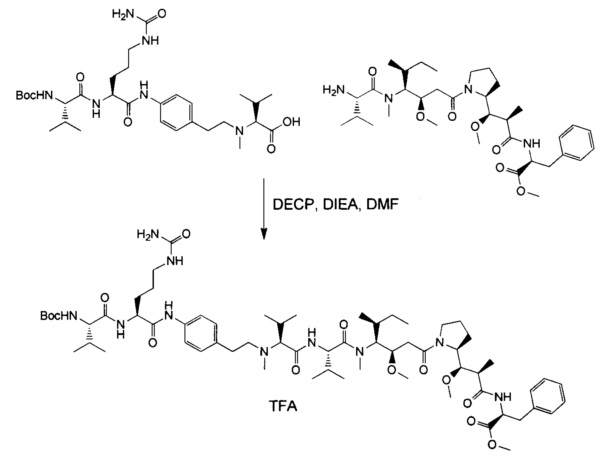
Соединение F-62-1 получали таким же образом, как соединение F-61-4, из амина 3D (100 мг, 0,158 ммоль, 0,9 экв.), кислоты F-61-3 (108 мг, 0,178 ммоль, 1 экв.), DECP (41 мкл, 0,267 ммоль, 1,5 экв.) и DIEA (93 мкл, 0,534 ммоль, 3 экв.) в ДМФА (2 мл). После очистки путем препаративной HPLC получали соединение F-62-1 в виде белого твердого вещества (93 мг, 39%).
Соединение F-62-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
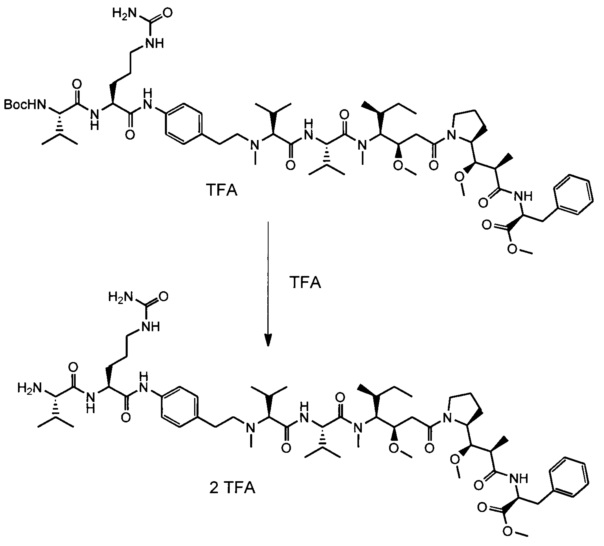
Соединение F-62-1 (35 мг, 0,026 ммоль, 1,0 экв.) растворяли в TFA (200 мкл). Через 10 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл) и раствор лиофилизировали в течение ночи, получая соединение F-62-2 в виде белого твердого вещества (34 мг, 105%).
Соединение F-62:
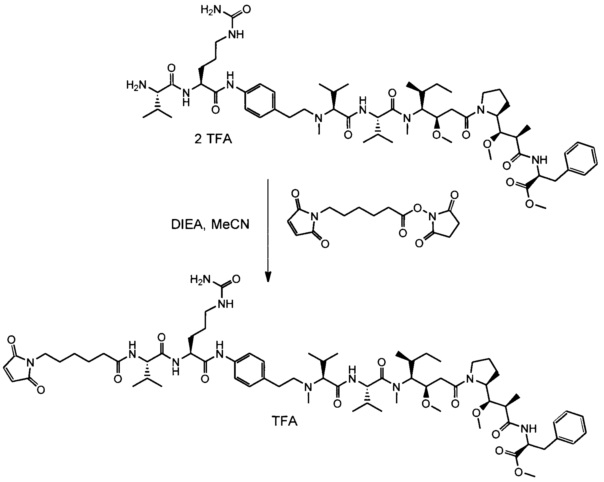
Амин F-62-2 (34 мг, 5,55 мкмоль, 1 экв.) растворяли в ацетонитриле (3 мл). Добавляли DIEA (5 мкл, 0,028 ммоль, 5 экв.) и 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (2 мг, 6,65 мкмоль, 1,2 экв.). HPLC-анализ показывал полное потребление исходного материала. Растворитель выпаривали досуха и осадок растирали смесью EtOAc/Et2O (80/20). Неочищенный продукт очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F-62 в виде белого твердого вещества (5,5 мг, 13%).
m/z (Q-TOF MS ESI+) 1336,7859 (2%, MNa+, C69H107N11NaO14 требует 1336,7891), 657,9073 (100%, (МН2)2+, C69H109N11O14 требует 657,9072).
Соединение F-63:
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
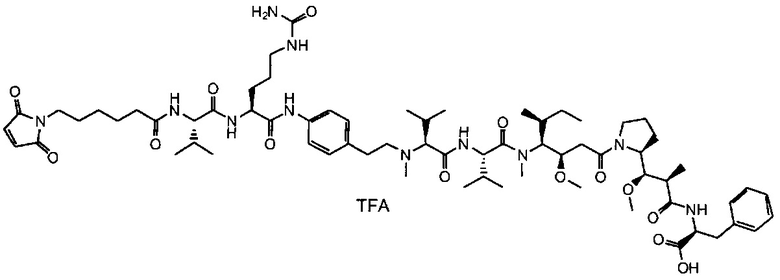
Соединение F-63-1: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин
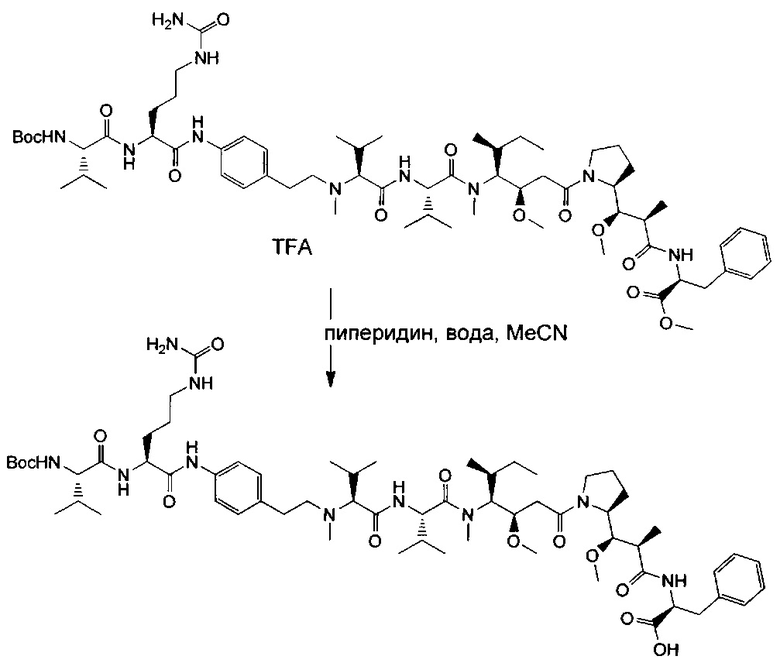
Соединение F-62-1 (157 мг, 0,118 ммоль, 1 экв.) растворяли в смеси воды (4,5 мл), ацетонитрила (4,5 мл) и пиперидина (3,5 мл) и встряхивали при комнатной температуре в течение 5 ч. Растворитель выпаривали при пониженном давлении и осадок растирали с Et2O (60 мл). Твердое вещество собирали путем фильтрации и промывали дважды с помощью Et2O (10 мл), получая соединение F-63-1 в виде твердого вещества белого цвета с желтоватым оттенком (153 мг, 100%).
Соединение F-63-2: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина бис-2,2,2-трифторацетат
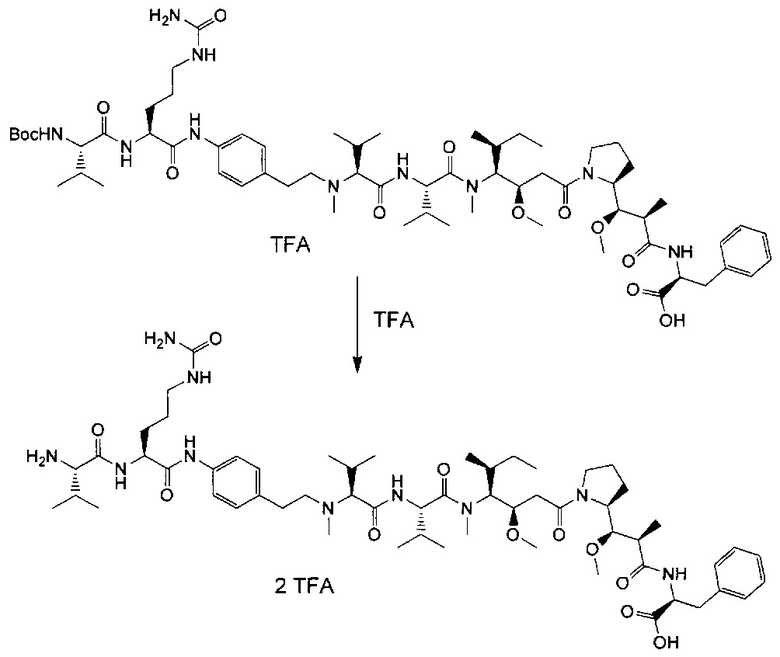
Соединение F-63-1 (153 мг, 0,127 ммоль, 1,0 экв.) растворяли в TFA (200 мкл). Через 10 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл) и раствор лиофилизировали в течение ночи, получая соединение F-63-2 в виде белого твердого вещества (34 мг, 105%).
Соединение F-63:
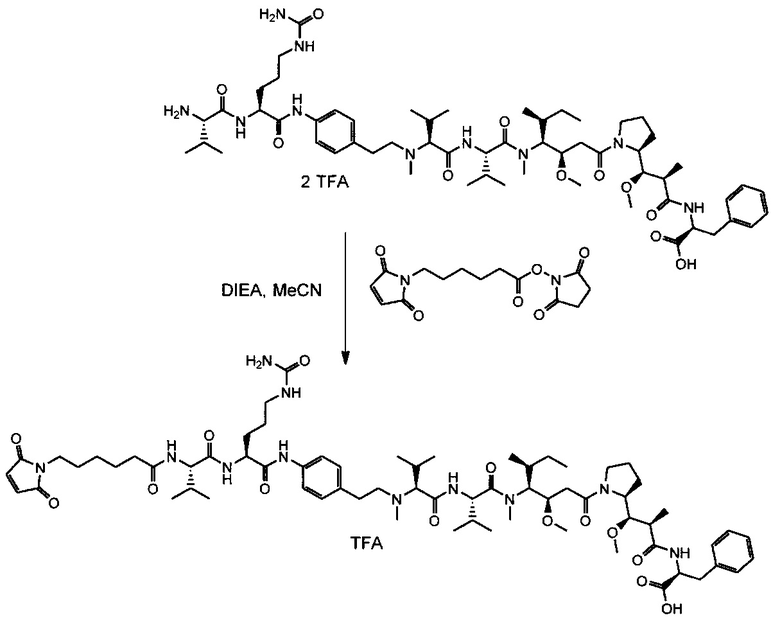
Амин F-63-2 (100 мг, 0,082 ммоль, 1 экв.) растворяли в смеси ацетонитрила (2 мл) и ДМФА (0,5 мл), и добавляли 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноат (45 мг, 0,147 ммоль, 1,8 экв.) и DIEA (71 мкл, 0,409 ммоль, 5 экв.). После встряхивания при комнатной температуре в течение 4,5 ч растворитель выпаривали при пониженном давлении. Неочищенный продукт очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение F-63 в виде белого твердого вещества после (42 мг, 36%).
m/z (Q-TOF MS ESI+) 1300,7901 (2%, МН+, C68H106N11O14 требует 1300,7915), 650,8990 (100%, (МН2)2+, C68H107N11O14 требует 650,8994).
Соединение G-12
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метилгексанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат 2,2,2-трифторацетат
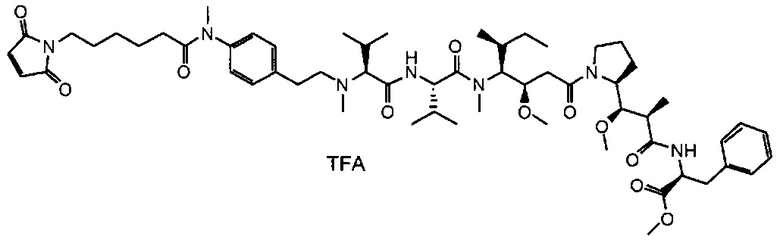
Соединение G-12-1: бензил-N-(4-аминофенэтил)-N-метил-L-валината дигидрохлорид
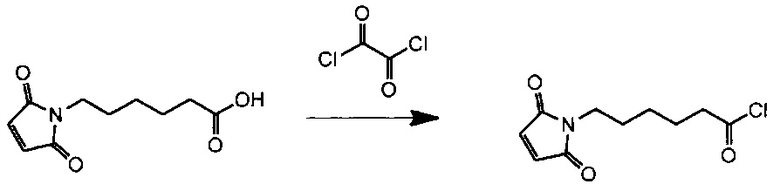
В оксалилхлориде (3 мл) растворяли 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексановую кислоту (200 мг, 0,947 ммоль, 1 экв.). Раствор встряхивали при комнатной температуре в течение 5 ч, а затем выпаривали досуха при пониженном давлении. Соединение G-12-1 получали в виде бежевого твердого вещества (217 мг, 100%) и использовали на следующем этапе без очистки.
Соединение G-12:
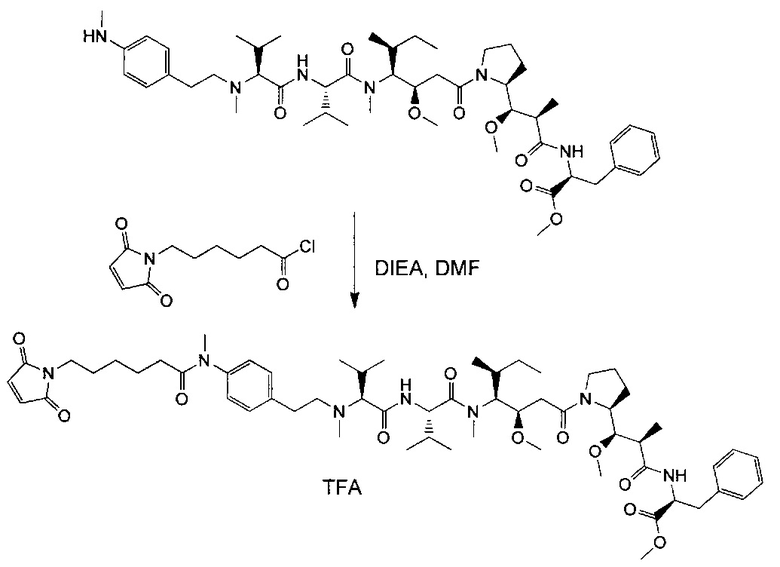
Анилин 12 (40 мг, 0,045 ммоль, 1 экв.) растворяли в сухом DCM (1 мл) при 0°C и добавляли DIEA (8 мкл, 0,045 ммоль, 1 экв.). После встряхивания в течение 30 минут вводили раствор соединения G-12-1 (10 мг, 0,45 ммоль, 1 экв.) в сухом DCM (1 мл) и реакционную смесь встряхивали в течение 1 ч при 0°C. Смесь разводили DCM (25 мл) и промывали дважды водой (20 мл), один раз насыщенным солевым раствором (10 мл). Органическую фазу сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении, получая неочищенный продукт в виде светло-коричневого твердого вещества (54 мг). Его очищали путем флэш-хроматографии на силикагеле (DCM/MeOH), а затем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выделенный продукт лиофилизировали, получая белое твердое вещество (23 мг), которое повторно очищали путем препаративной HPLC, и выбранные фракции объединяли и лиофилизировали, получая соединение G-12 в виде белого твердого вещества (9 мг, 16%).
m/z (Q-TOF MS ESI+) 1094,6543 (20%, MNa+, C59H89N7NaO11 требует 1094,6512), 1072,6722 (16%, MH+, C59H90N7O11 требует 1072,6693), 536,8358 (100%, (МН2)2+, C59H91N7O11 требует 536,8383).
Соединение G-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)-N-метилгексанамидо)фенэтил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
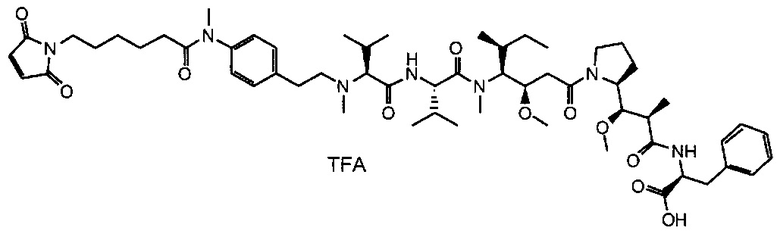
Соединение G-13:
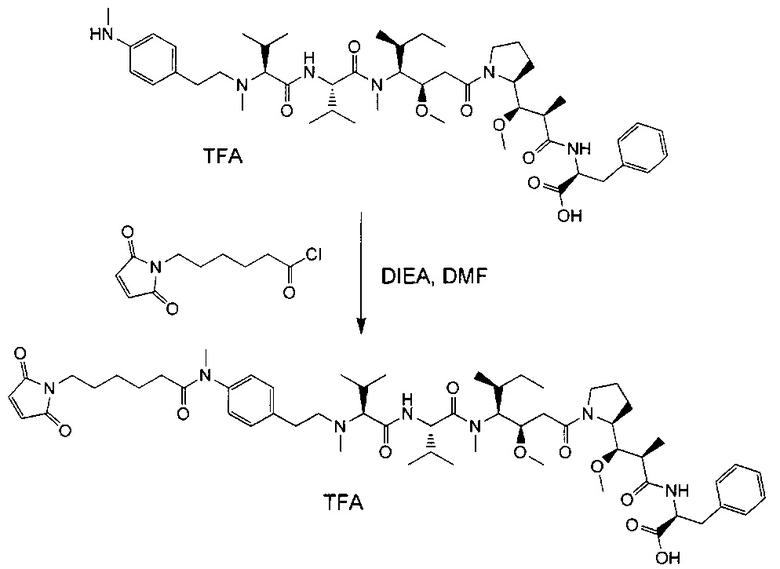
Анилин 13 (15 мг, 0,015 ммоль, 1 экв.) растворяли в сухом DCM (1,5 мл) при 0°C и добавляли DIEA (8 мкл, 0,046 ммоль, 3 экв.). Вводили раствор соединения G-12-1 (3,5 мг, 0,046 ммоль, 1 экв.) в сухом DCM (0,5 мл) и реакционную смесь встряхивали в течение 1,5 ч при 0°C. Растворитель выпаривали при пониженном давлении и неочищенный продукт очищали путем препаративной HPLC (Waters 600Е, SunFire Prep C18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение G-13 в виде белого твердого вещества (11,4 мг, 62%).
m/z (Q-TOF MS ESI+) 1058,6510 (30%, МН+, C58H88N7O11 требует 1058,6536), 529,8285 (100%, (МН2)2+, C58H89N7O11 требует 529,8305).
Соединение G-15
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
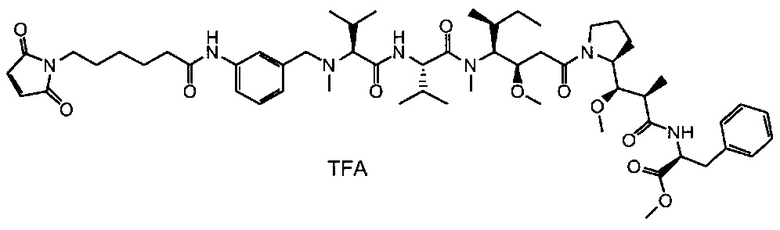
Соединение G-15:
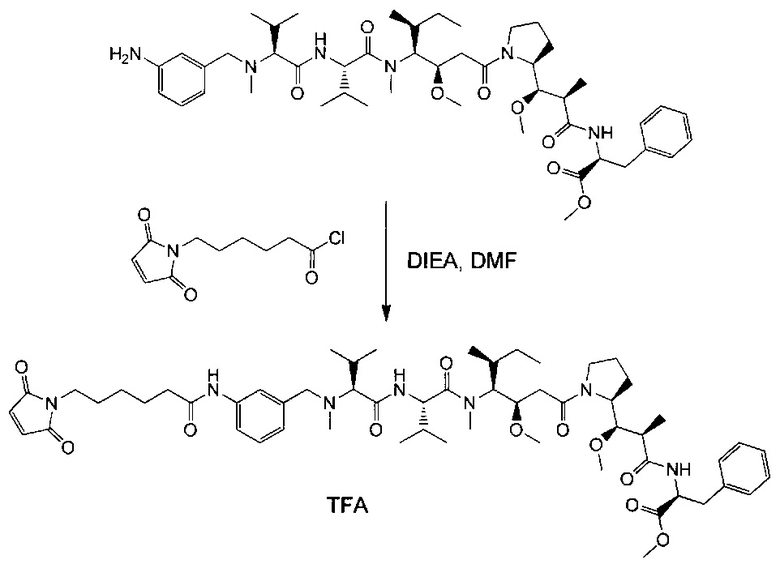
Анилин 15 (40 мг, 0,047 ммоль, 1 экв.) растворяли в сухом DCM (2 мл) при 0°C и добавляли DIEA (10 мкл, 0,056 ммоль, 1,2 экв.). Вводили раствор соединения G-12-1 (108 мг, 0,47 ммоль, 10 экв.) в сухом DCM (1 мл) и реакционную смесь встряхивали в течение 1,5 ч при 0°C. Смесь разводили DCM (10 мл) и промывали дважды водой (5 мл). Органическую фазу сушили над MgSO4, фильтровали и выпаривали при пониженном давлении, получая неочищенный продукт в виде бежевого твердого вещества. Его очищали путем препаративной HPLC (Waters 600Е, SunFire Prep С18 OBD колонка, 5 мкм, 19×100 мм; элюирующая фаза: вода/MeCN, буферизованные 0,1% TFA; градиент от 5% до 100% MeCN в течение 15 минут; УФ-детектор Waters 2487 при 220 нм). Выбранные фракции объединяли и лиофилизировали, получая соединение G15 в виде белого твердого вещества (27 мг, 50%).
m/z (Q-TOF MS ESI+) 1066,6517 (2%, MNa+, C57H85N7NaO11 требует 1066,6199), 522,8224 (100%, (MH2)2+, C57H87N7O11 требует 522,8226).
Пример 22: Синтез, очистка и характеризация ADC
Процедура, описанная ниже, относится к химерным и гуманизированным формам IgG1. Нужно понимать, что для любых других форм, таких как IgG2, IgG4 и т.д., специалист в данной области сможет адаптировать эту процедуру, используя общие знания.
Антитела (1-5 мг/мл) частично восстанавливали с помощью трис(2-карбоксиэтил)фосфингидрохлорида (ТСЕР) в 10 мМ боратном буфере, pH 8,4, содержащем 150 мМ NaCl и 2 мМ EDTA, в течение 2 ч при 37°C. Как правило, 2,5-3 молярных эквивалента ТСЕР использовали для получения соотношений "лекарственное средство - антитело" (DAR) в размере примерно 4, соответственно. Частичное восстановление антитела подтверждали с помощью анализа SDS-PAGE в невосстанавливающих условиях. Перед связыванием группировки "линкер - лекарственное средство" с освобожденными межцепочечными остатками цистеинов восстанавливающей смеси давали остыть до комнатной температуры. Затем концентрацию антитела доводили до 1 мг/мл в 10 мМ боратном буфере с pH 8,4, содержащем 150 мМ NaCl и 2 мМ EDTA, и к антителу добавляли 5-молярный избыток лекарственного средства из 10 мМ раствора в диметилсульфоксиде (ДМСО). Конечную концентрацию ДМСО доводили до 10% для поддержания растворимости лекарственного средства в водной среде в процессе связывания. Реакцию проводили в течение 1 ч при комнатной температуре. Избыточное лекарственное средство останавливали добавлением 1,5 моль N-ацетилцистеина на каждый моль лекарственного средства и инкубацией в течение 1 ч при комнатной температуре. После диализа против 25 мМ His-буфера, pH 6,5, содержащего 150 мМ NaCl, в течение ночи при 4°C конъюгаты "антитело - лекарственное средство" очищали с использованием способов, известных специалистам в данной области, основанных на применении коммерческих хроматографических колонок и ультрафильтрационных блоков. Сначала несвязанное лекарственное средство и агрегаты ADC удаляли путем эксклюзионной хроматографии (SEC) на колонке S200 (GE Life Sciences) или TSK G3000 SW (Tosoh). Затем очищенные мономеры ADC концентрировали до 2-3 мг/мл путем ультрафильтрации на блоках фильтрации с MWCO 30 или 50 кДа или путем аффинной хроматографии на белке А. Очищенные ADC хранили при 4°C после стерильной фильтрации через фильтр с размером пор 0,2 мкм. Далее их анализировали с помощью SDS-PAGE в восстанавливающих и невосстанавливающих условиях, чтобы подтвердить конъюгацию лекарственного средства, и с помощью SEC на аналитических колонках S200 или TSK G3000 SWXL, чтобы определить содержание мономеров и агрегированных форм. Концентрации белка определяли с помощью анализа на основе бицинхониновой кислоты (ВСА) с IgG в качестве стандарта. Для каждого очищенного ADC оценивали DAR путем HIC и LC-MS. Как правило, содержание агрегированных форм было ниже 5%, a DAR находилось в пределах от 3,5 до 5.
Пример 23: Оценка цитотоксичности IGF-1R-антител, связанных с различными лекарственными средствами
Для пяти IGF-1R-антител было показано, что они быстро интернализовались в лизосомы и имели более низкую связывающую способность в кислой среде. В связи с этим данные антитела имели все свойства, которые будут использоваться в ADC. Таким образом, пять химерных анти-IGF-1R-антител связывали с тремя различными соединениями (G-13, Е-13 и F-63). Отношение "лекарственное средство - антитело" в этих ADC составляло примерно 4. Чтобы оценить неспецифическую цитотоксичность, нерелевантное химерное антитело c9G4 также связывали с этими соединениями с таким же DAR. Клетки MCF-7 инкубировали с возрастающими концентрациями каждого ADC при 37°C в течение 6 дней в полной культуральной среде. Жизнеспособность клеток оценивали с использованием люминесцентного анализа жизнеспособности клеток (CellTiter-Glo, Promega). Люминесцентный сигнал считывали с использованием планшетного ридера Mithras (Berthold Technologies). Нерелевантное химерное антитело c9G4, связанное с любым из Е-13, G-13 и F-63, показало отсутствие или скромную цитотоксическую активность на клетках MCF-7 (фиг. 21). Напротив, добавление всех других ADC, полученных после связывания анти-IGF-1R-антитела с любым из R-13, G-13 и F-63, резко снижало жизнеспособность клеток MCF-7.
Пример 24: Активность in vivo антитела c208F2, конъюгированного с соединениями R-13, G-13 или F-63, на модели с ксенотрансплантатом MCF-7
Для того чтобы подтвердить, что эффективность in vitro c208F2, соединенного с соединениями G-13, Е-13 или F-63, может быть переведена на in vivo, они были протестированы на модели с ксенотрансплантатом MCF-7.
Все процедуры на животных проводили в соответствии с руководствами Директивы 2010/63/UE о защите животных, используемых для научных целей. Протокол был одобрен Этическим комитетом в области исследований на животных в Pierre Fabre Institute. Пять миллионов клеток MCF-7 вводили подкожно швейцарским/голым мышам в возрасте 7 недель. Перед введением клеток в левый бок мышей были имплантированы эстрогеновые гранулы (Innovative Research of America), чтобы высвобождать эстрогены, необходимые для роста опухолей MCF-7 in vivo.
Через двадцать дней после имплантации клеток MCF-7, когда опухоли достигали среднего размера 120-150 мм3, животных разделяли на группы по 5 мышей в зависимости от размера и внешнего вида опухоли. Различные подходы к лечению осуществляли путем внутрибрюшинных инъекций. Состояние здоровья животных контролировали ежедневно. Объем опухоли измеряли два раза в неделю электронным штангенциркулем до завершения исследования. Объем опухоли рассчитывали по следующей формуле: π/6 × длина × ширина × высота. Токсичность оценивали три раза в неделю по весу животных. Для каждого измерения проводили статистический анализ с использованием критерия Манна-Уитни. Все соединения вводили внутрибрюшинно (i.p.). В этом примере противоопухолевую активность МКА с208F2, соединенного с Е-13, F-13 или F-63 со значением DAR примерно 4, оценивали после двух инъекций в дозе 7 мг/кг на день 20 и день 27 (фиг. 22А, 22В и 22С). Параллельно вводили кэппированные лекарственные группировки Е-13, F-13 и F-63 в эквивалентной дозе, соответствующей 7 мг/кг с208F2-E-13, с208F2-F-13 и с208F2-F-63 с DAR примерно 4.
Введение с208-Е-13 (фиг. 22А), с208F2-G-13 (фиг. 22В) или с208F2-F-63 (фиг. 22С) значительно ингибировало и даже вызывало полную регрессию роста опухоли (р менее 0,05 по сравнению с соответствующим кэппированным лекарственным средством). Никакой статистической разницы в активности между с208-Е-13, с208F2-G-13 и с208F2-F-63 отмечено не было. Кэппированные лекарственные средства не имели никакого влияния на рост опухоли MCF-7 (р более 0,05 по сравнению с контрольной группой).
Вторую серию экспериментов проводили с с208F2, связанным либо с Е-13, либо с G-13, и с нерелевантным антителом c9G4, связанным либо с Е-13, либо с G-13, на моделях с ксенотрансплантатом MCF-7, которые были описаны ранее. Мышам внутрибрюшинно вводили 7 мг/кг каждого ADC в день 20 и день 27 (фиг. 23А и 23В).
Введение как с9G4-E-13, так и с9G4-F-13 приводило к умеренному и временному росту ксенотрансплантатных опухолей MCF-7. Тем не менее, этот второй эксперимент подтвердил, что введение либо с208-Е-13, либо с208F2-G-13 индуцировало полную регрессию опухоли с 43-го дня, показывая высокую противоопухолевую активность этих ADC.
Пример 25: Мощная цитотоксичность in vitro Axl ADC, связанных с различными лекарственными средствами
Цитотоксическую активность ADC в ингибировании роста опухолевых клеток испытывали в анализе пролиферации клеток с использованием SN12C (человеческая почечная карцинома Axl+) и MCF-7 (человеческая аденокарцинома молочной железы Axl-). Вкратце, клетки высевали в 96-луночные многолуночные планшеты за день до введения лекарственного средства с плотностью 2500 клеток на лунку. ADC и контроли серийно разводили и затем вносили в 96-луночные многолуночные планшеты. Затем клетки инкубировали в течение 6 дней при температуре 37°C и 5% CO2. Жизнеспособность клеток оценивали путем измерения уровня АТФ в лунках с использованием анализа CellTiter-Glo® Luminescent Cell Viability Assay (Promega, кат. № G7571). Процент жизнеспособности клеток рассчитывали, принимая необработанные клетки за 100%. С помощью нелинейного регрессионного анализа (GraphPad Prism 4.0) определяли IC50, т.е. концентрацию соединения, необходимую для получения 50% снижения жизнеспособности по сравнению с необработанными клетками (контроль равен 100%), и выражали ее в молярности (фиг. 28).
Данные на фиг. 28А и 28В показывают, что hz1613F12, конъюгированное либо с Е-13, либо с G-13, дает значения IC50 1,048×10-10 М, 1,413×10-10 М, соответственно. Таким образом, Е-13 и G-13 могут вызывать сильную цитотоксичность, будучи связанными с Axl-антителом, таким как 1613F12. На клетках MCF7 (Axl-) цитотоксичность не наблюдалась.
Пример 26: Активность in vivo антитела трастузумаба, конъюгированного с соединениями Е-13 или G-13, на модели с ксенотрансплантатом Calu-3.
Для того чтобы подтвердить эффективность in vivo антител, связанных с соединениями G-13 или Е-13, их связывали с трастузумабом и анализировали на HER2-чувствительной модели с ксенотрансплантатом Calu-3, известной своей амплификацией и (3+) экспрессией HER2. Антитело трастузумаб было приобретено у Euromedex, 24 Rue des Tuileries 67460, SOUFFELWEYERSHEIM, Франция.
Все процедуры на животных проводили в соответствии с руководствами Директивы 2010/63/UE о защите животных, используемых для научных целей. Протокол был одобрен Этическим комитетом в области исследований на животных в Pierre Fabre Institute. Семь миллионов клеток Calu-3 вводили подкожно мышам SCID в возрасте 7 недель.
Через шесть дней после имплантации клеток Calu-3, когда опухоли достигали среднего размера 250-260 мм3, животных разделяли на группы по 6 мышей в зависимости от размера и внешнего вида опухоли. Различные способы лечения проводили путем внутрибрюшинных инъекций. Состояние здоровья животных контролировали ежедневно. Объем опухоли измеряли два раза в неделю электронным штангенциркулем до завершения исследования. Объем опухоли рассчитывали по следующей формуле: (длина × ширина2)/2. Токсичность оценивали три раза в неделю по весу животных. Для каждого измерения проводили статистический анализ с использованием критерия Манна-Уитни. Все соединения вводили внутрибрюшинно (i.p.). В этом примере противоопухолевую активность МКА трастузумаба, соединенного с Е-13 или G-13 со значением DAR примерно 4, оценивали после 1 инъекции в дозе 3 мг/кг на 6-й день. Параллельно трастузумаб в одиночку вводили в эквивалентной дозе, соответствующей 3 мг/кг голого антитела.
Введение трастузумаба-Е-13 (фиг. 29А) или трастузумаба-G-13 (фиг. 29В) значительно ингибировало рост опухоли и даже индуцировало полную регрессию роста опухоли у всех мышей, получавших лечение (р менее 0,05 по сравнению с соответствующим голым антителом). Между группами, получавшими трастузумаб-Е-13 и трастузумаб-G-13, никакой статистической разницы в активности не наблюдалось. По сравнению с литературными данными (Cretella et al. Molecular Cancer 2014, 13:143) на этой модели с Calu-3 TDM-1 не индуцировал полную регрессию, даже если его дозировка была выше (15 мг/кг каждые 6 дней против 3 мг/кг, одна инъекция, соответственно), чем используемая для трастузумаба-Е-13 или трастузумаба-G-13.
Пример 27: Активность in vivo антитела трастузумаба, конъюгированного с соединениями Е-13 или G-13, на модели с ксенотрансплантатом JIMT-1
Для того чтобы узнать, проявляют ли конъюгаты антитела трастузумаба также активность на модели, известной своей устойчивостью к трастузумабу, оценивали модель с ксенотрансплантатом JIMT-1, которая экспрессирует на высоком уровне HER2, но которая был устойчив к терапии трастузумабом. Все процедуры на животных проводили в соответствии с руководствами Директивы 2010/63/UE о защите животных, используемых для научных целей. Протокол был одобрен Этическим комитетом в области исследований на животных в Pierre Fabre Institute. Семь миллионов клеток JIMT-1 вводили подкожно мышам SCID в возрасте 7 недель.
Через 14 дней после имплантации клеток JIMT-1, когда опухоли достигали среднего размера 220-230 мм3, животных разделяли на группы по 5 мышей в зависимости от размера и внешнего вида опухоли. Различные способы лечения проводили путем внутрибрюшинных инъекций. Состояние здоровья животных контролировали ежедневно. Объем опухоли измеряли два раза в неделю электронным штангенциркулем до завершения исследования. Объем опухоли рассчитывали по следующей формуле: (длина × ширина2)/2. Токсичность оценивали по весу животных три раза в неделю. Для каждого измерения проводили статистический анализ с использованием критерия Манна-Уитни. Все соединения вводили внутрибрюшинно (i.p.). В этом примере противоопухолевую активность МКА трастузумаба, соединенного с Е-13 или G-13 со значением DAR примерно 4, оценивали после 1 инъекции в дозе 3 мг/кг на 6-й день (фиг. 30А и 30В). В первом эксперименте мы показали, что трастузумаб сам по себе не имеет никакого противоопухолевого эффекта (фиг. 30С). Этот результат согласуется с литературными данными.
Введение трастузумаба-Е-13 (фиг. 30А) или трастузумаба-G-13 (фиг. 30В) значительно ингибировало рост опухоли, давая, соответственно, ингибирование роста на 73 и 70% на 34-й день. Между группами, получавшими трастузумаб-Е-13 и трастузумаб-G-13, никакой статистической разницы в активности не наблюдалось. Как уже отмечалось для модели Calu-3 и по сравнению с опубликованными данными (Barok et al. Breast Cancer Research 2011, 13:R46) на этой модели JIMT-1, TDM-1 представляется менее мощным, даже если его дозировка (15 мг/кг каждые 6 дней против 3 мг/кг, одно введение, соответственно) была выше, чем используемая для трастузумаба-Е-13 или трастузумаба-G-13.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛА ПРОТИВ IGF-1R С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2692563C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА, ЭКСПРЕССИРУЮЩЕГО IGF-1R | 2016 |
|
RU2728568C2 |
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |
| Модулирующие стабильность линкеры для использования с конъюгатами антитело-лекарственное средство | 2015 |
|
RU2680238C2 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 И АУРИСТАТИНОВ | 2014 |
|
RU2662951C2 |
| КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО (ADC), КОТОРЫЕ СВЯЗЫВАЮТСЯ С БЕЛКАМИ FLT3 | 2016 |
|
RU2739617C2 |
| Ингибитор ДНК-зависимой протеинкиназы | 2020 |
|
RU2835434C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ ДЛЯ ВНУТРИКЛЕТОЧНОЙ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2629957C2 |



































































































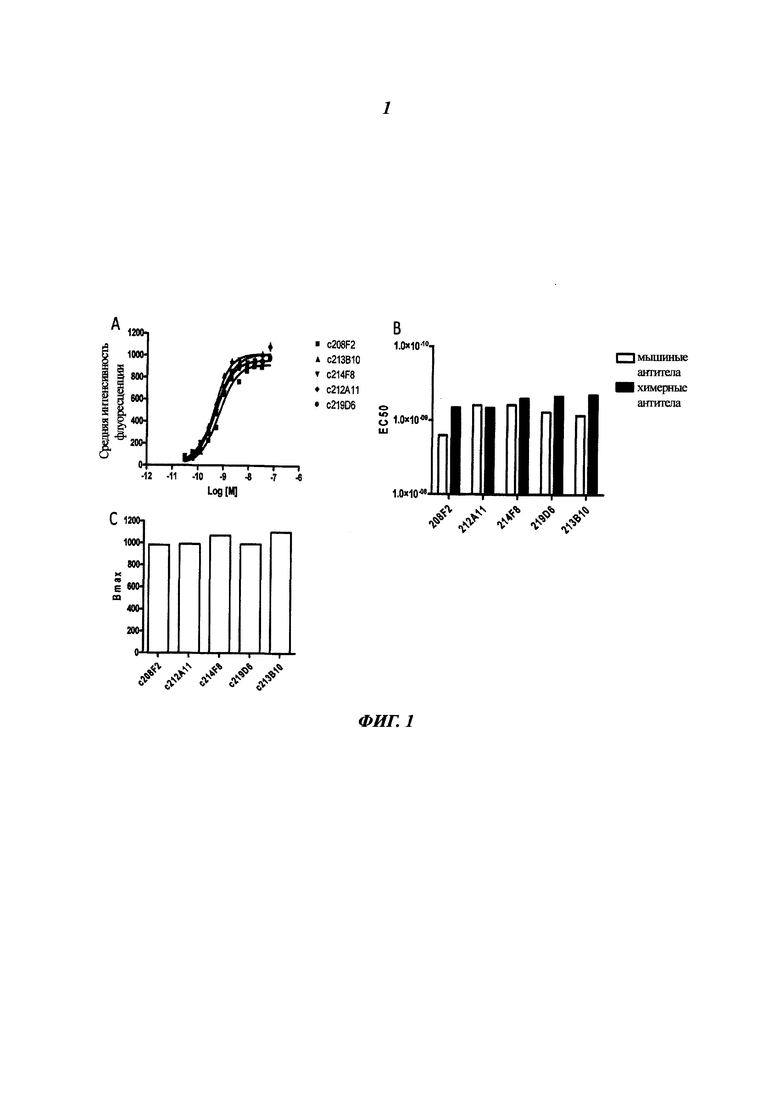
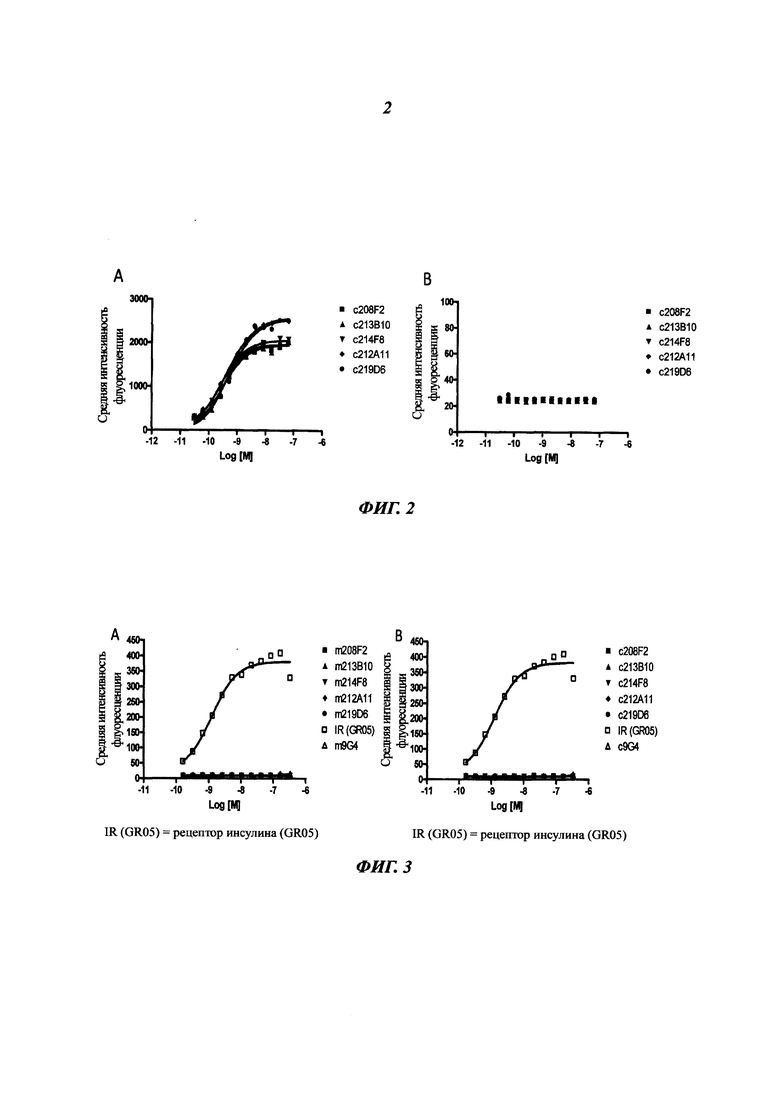
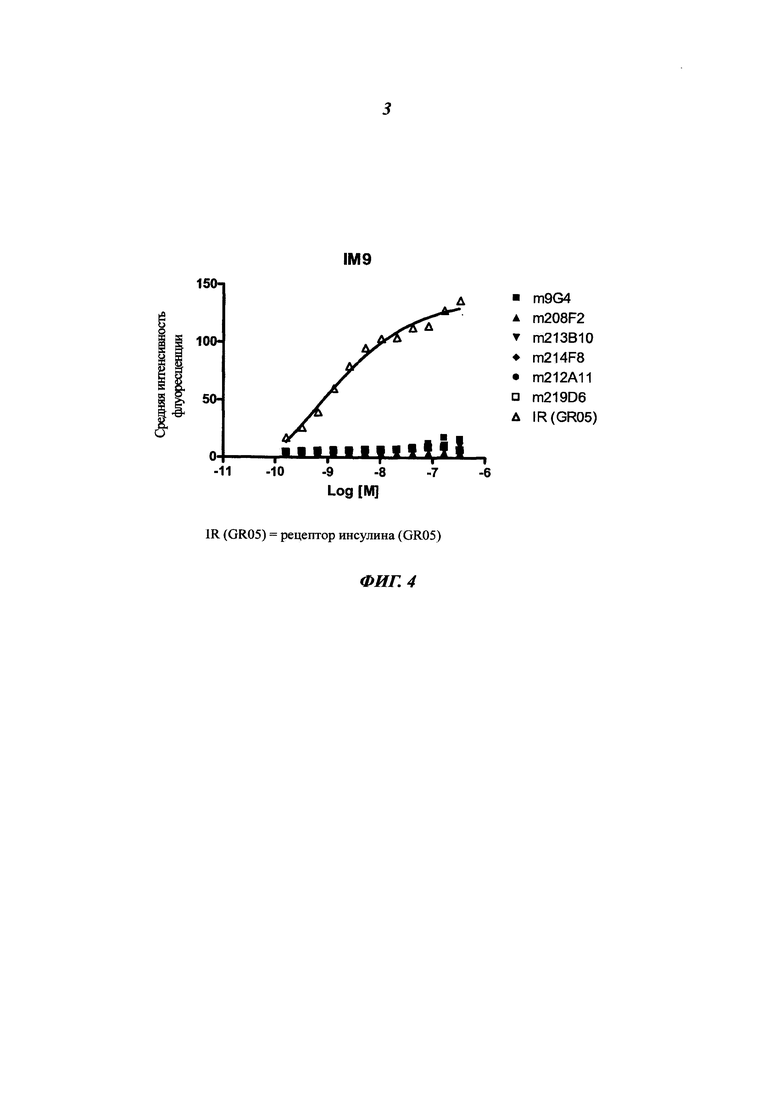
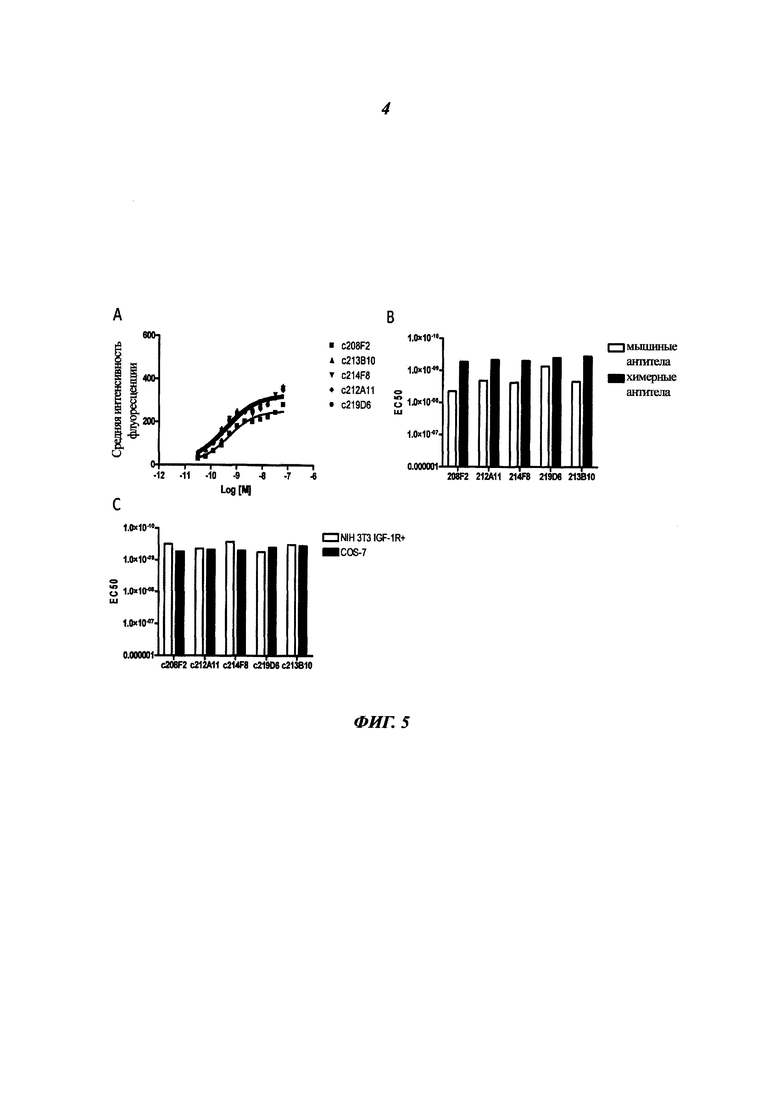

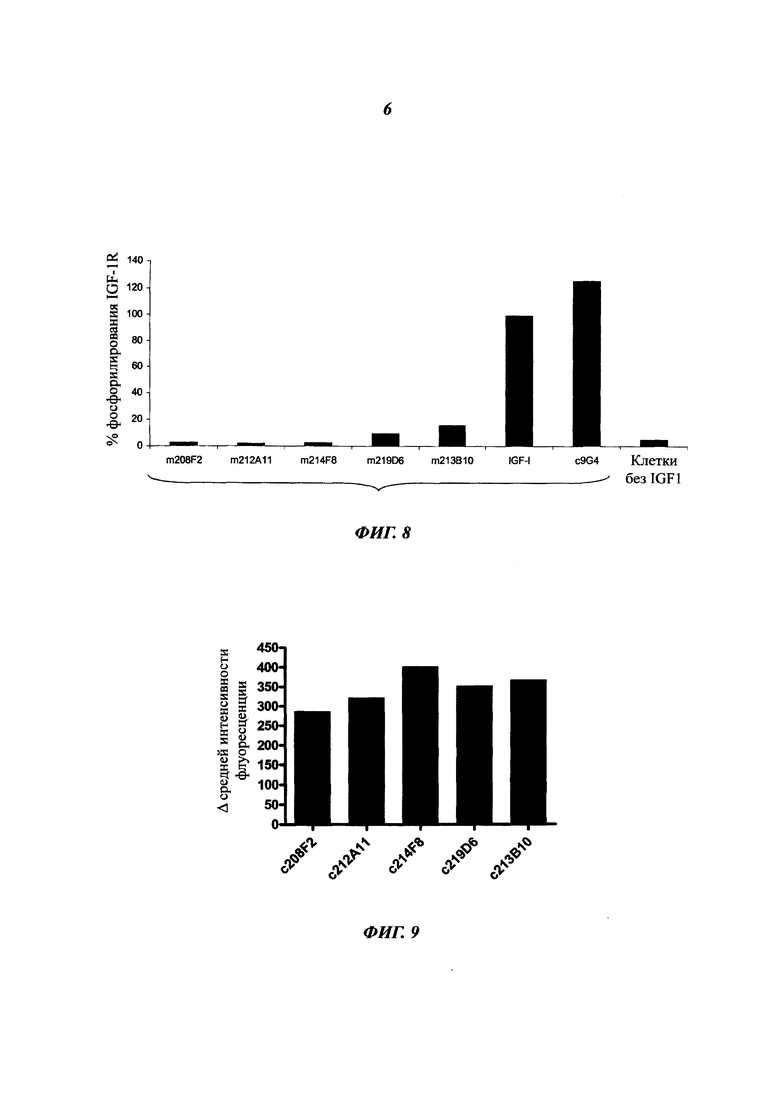
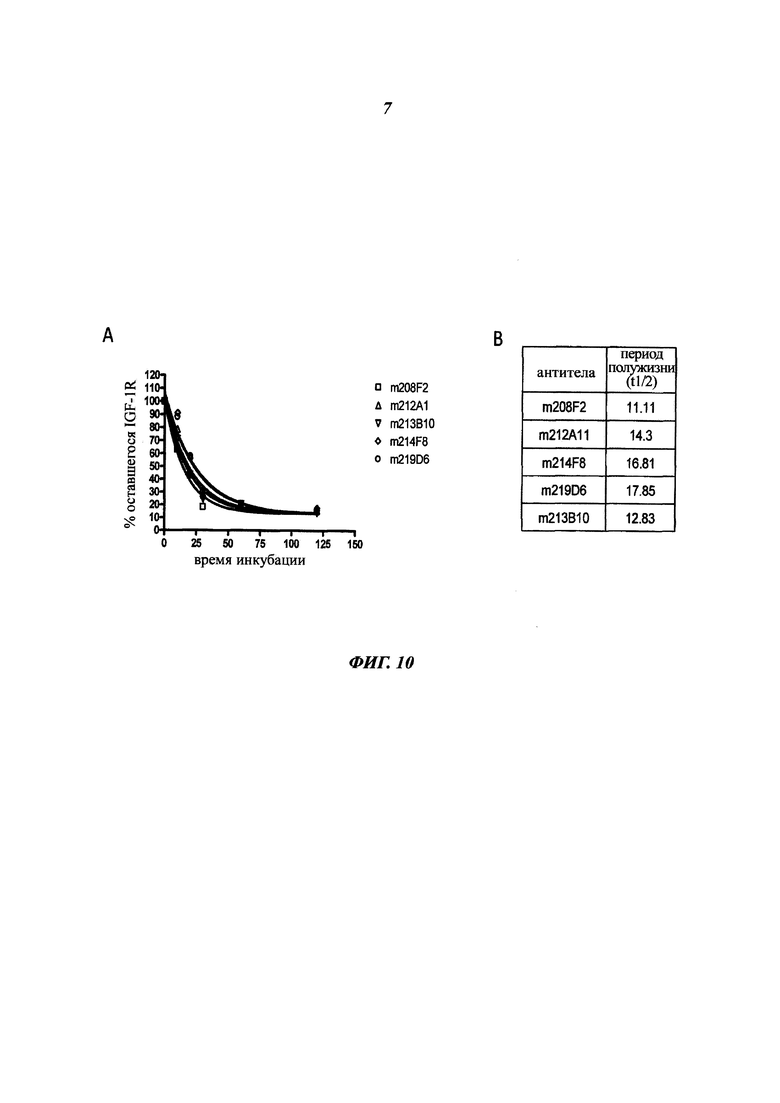
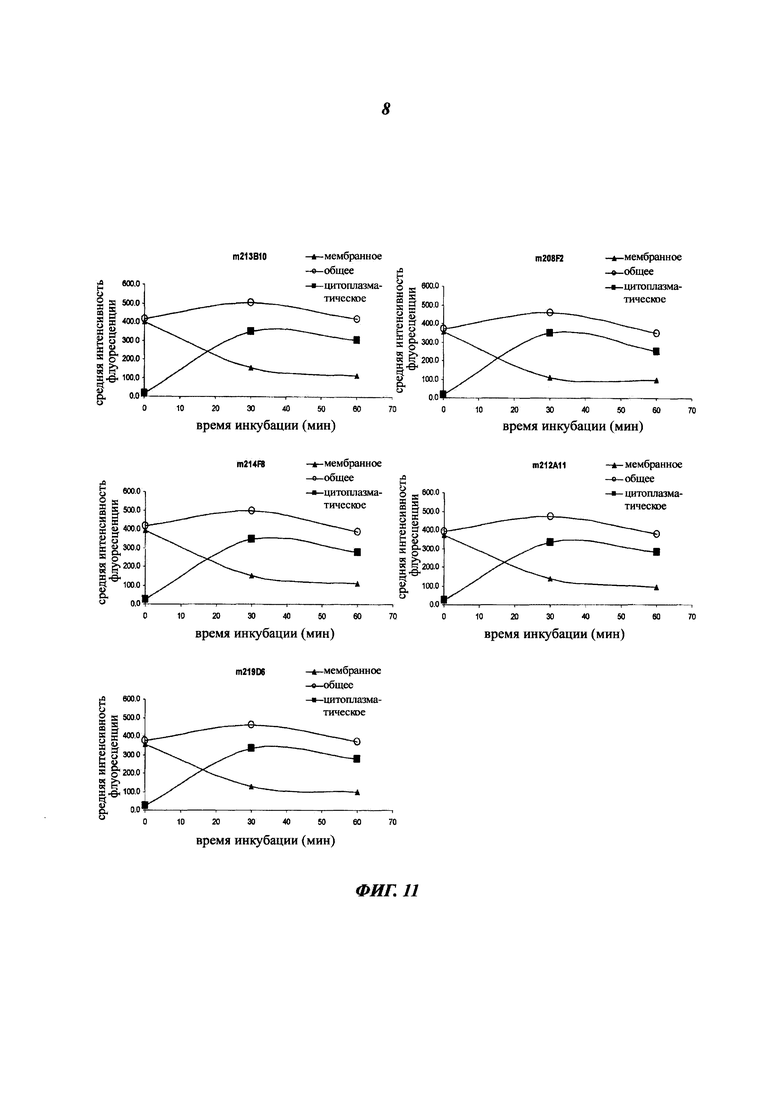


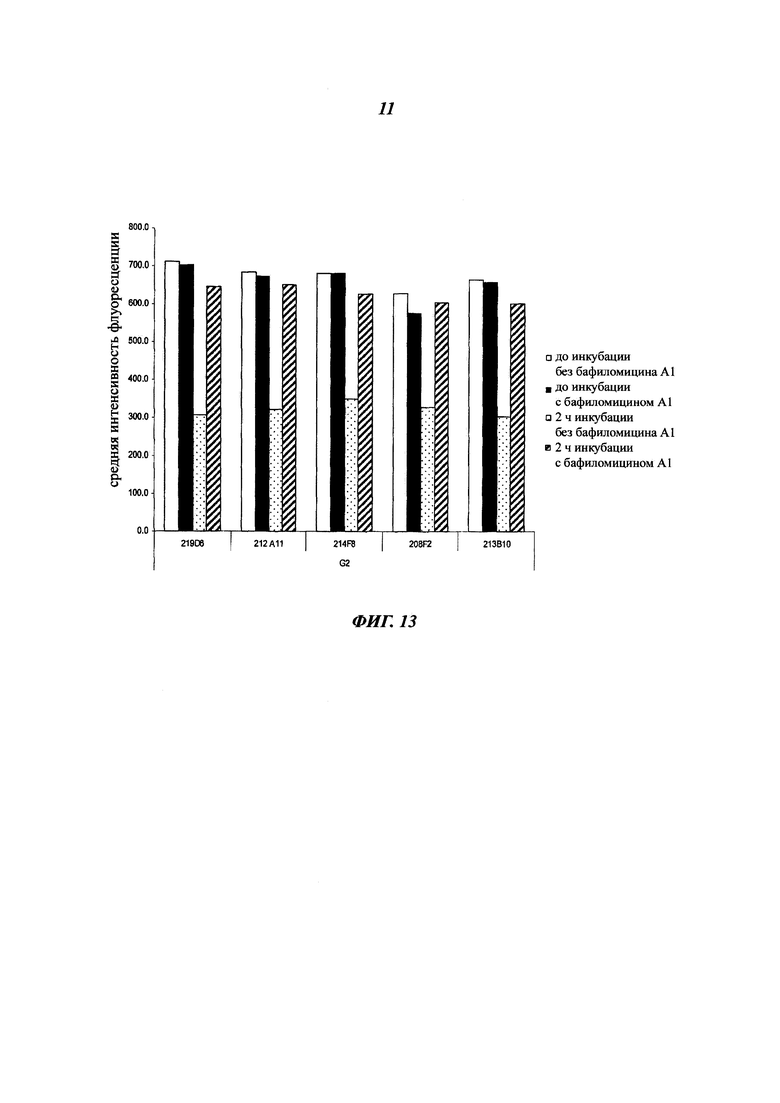
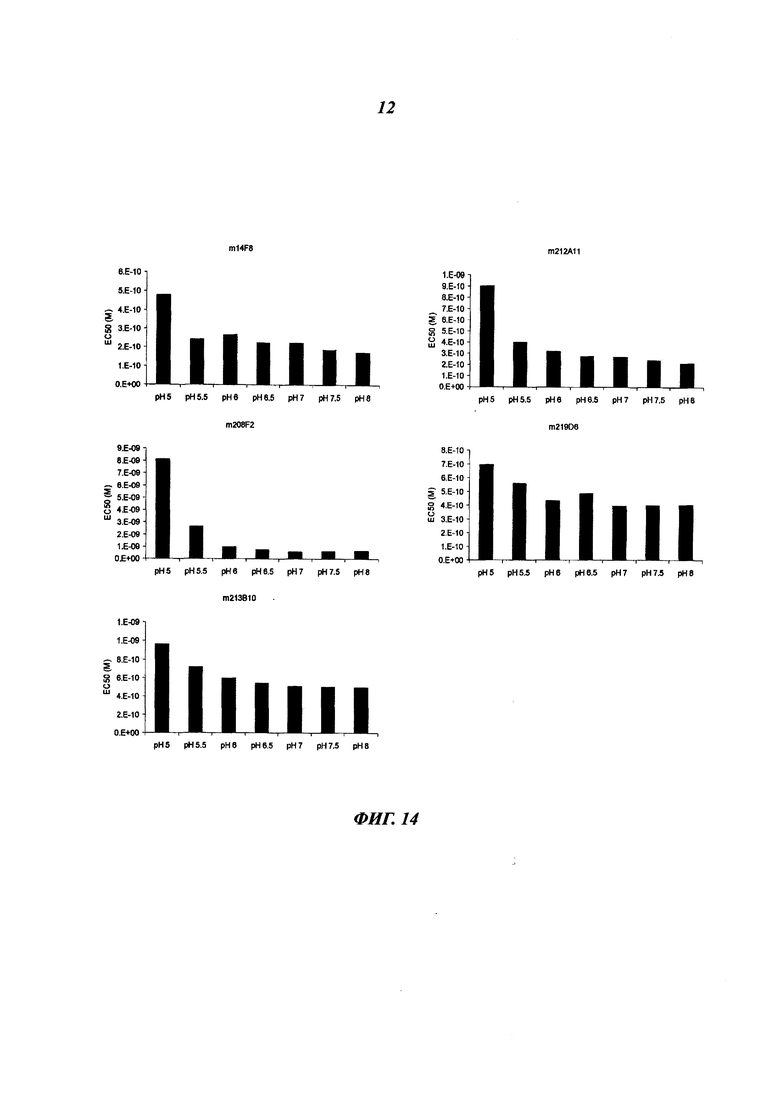
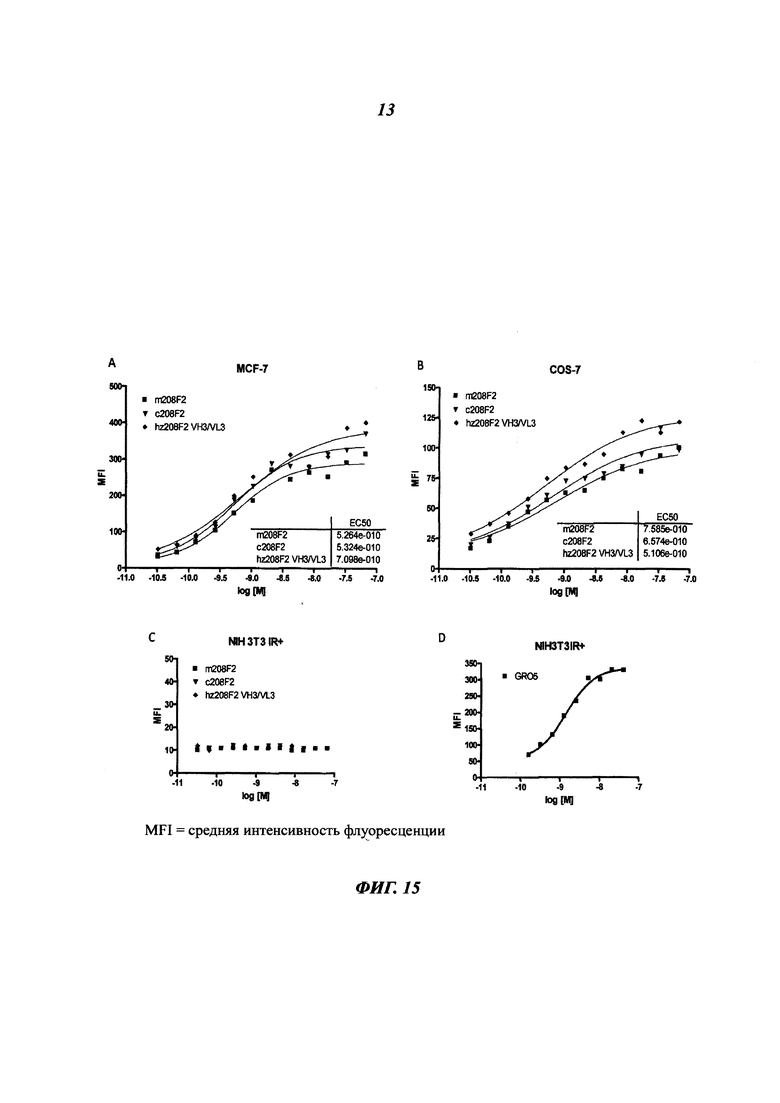
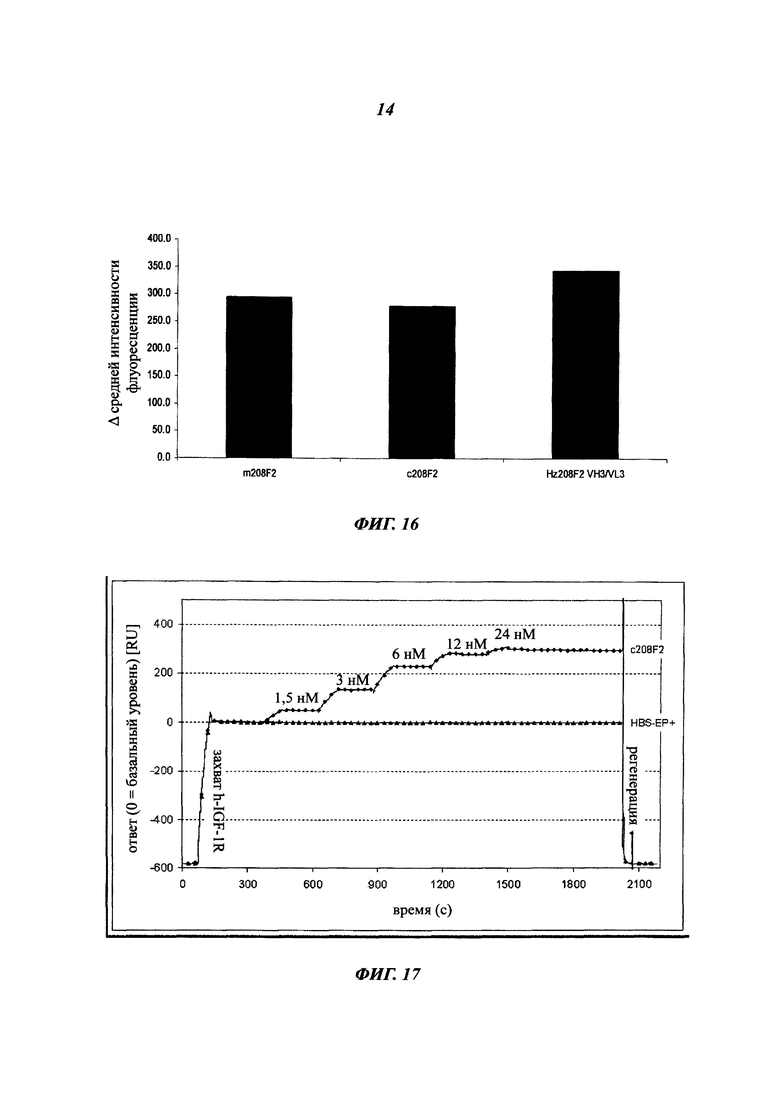
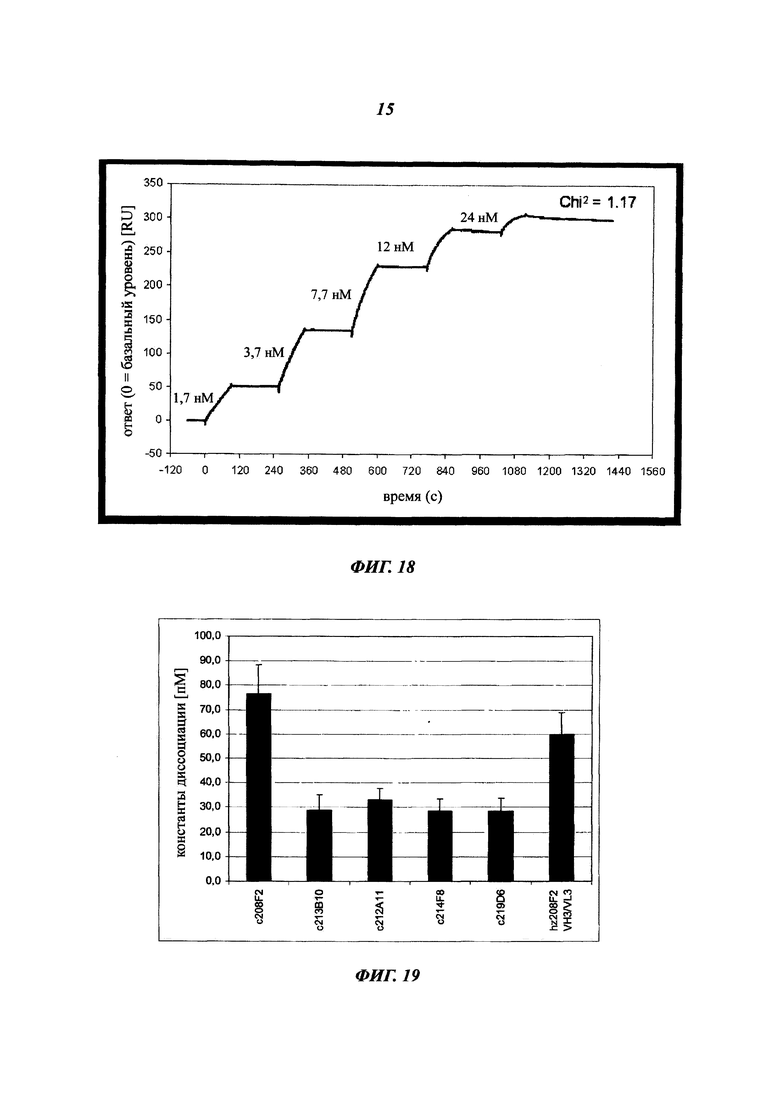
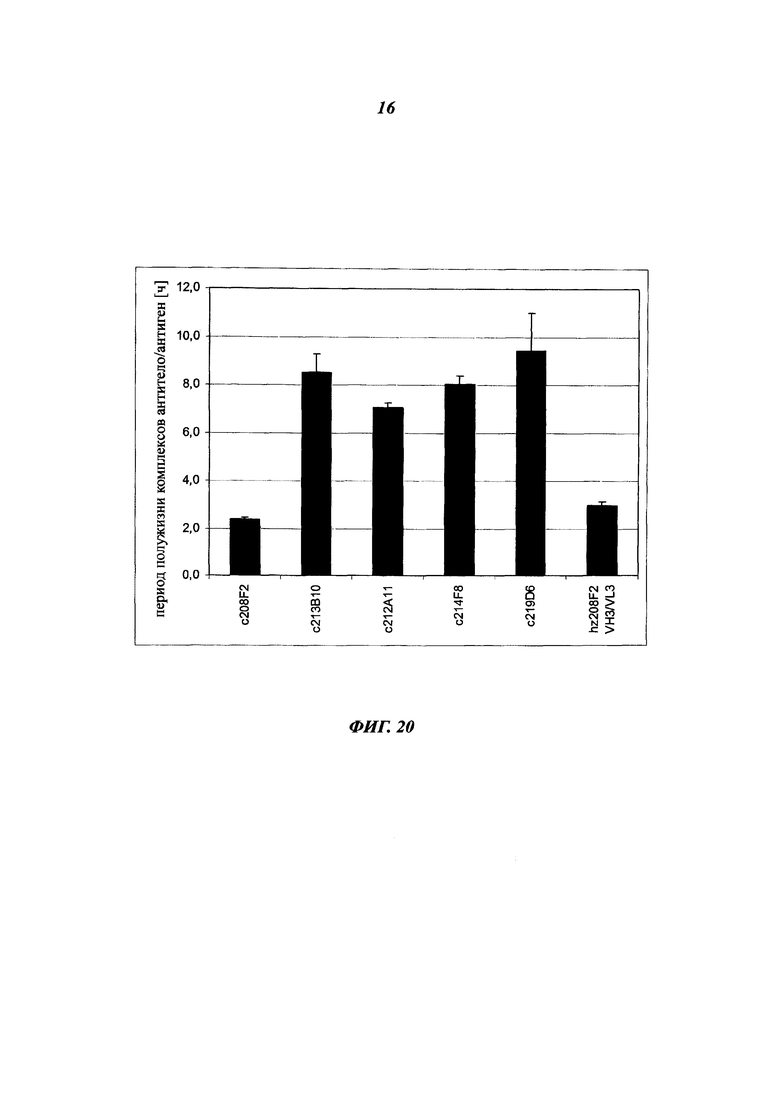

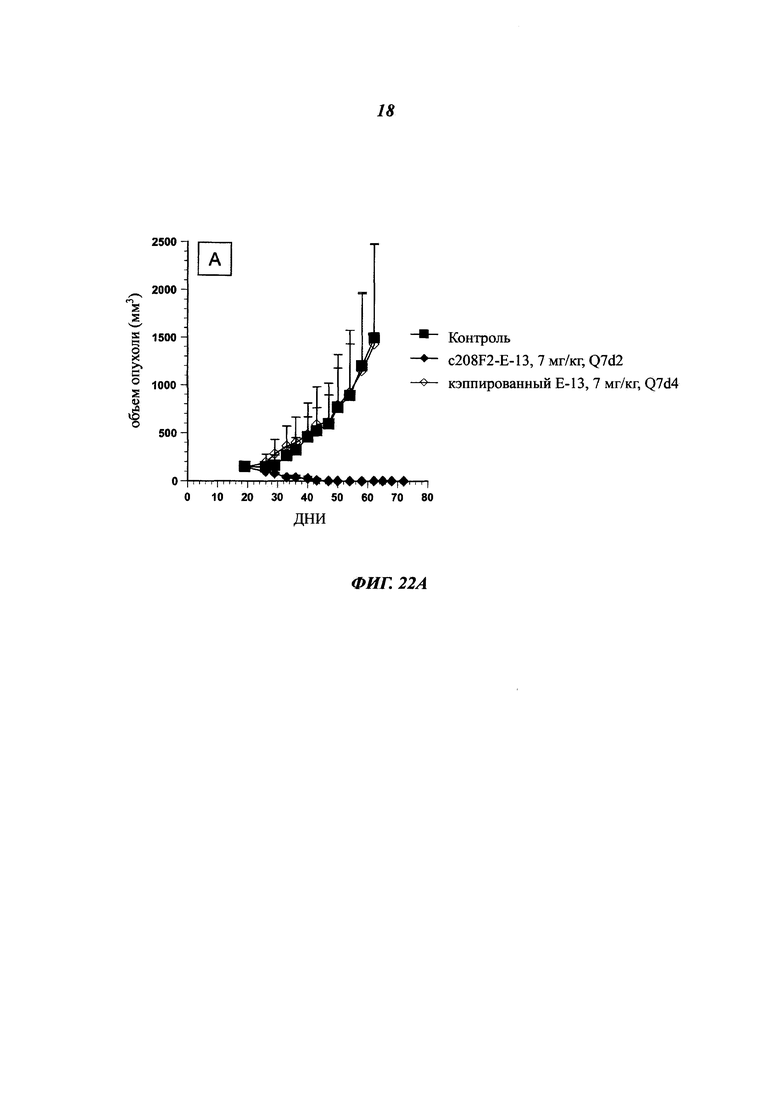
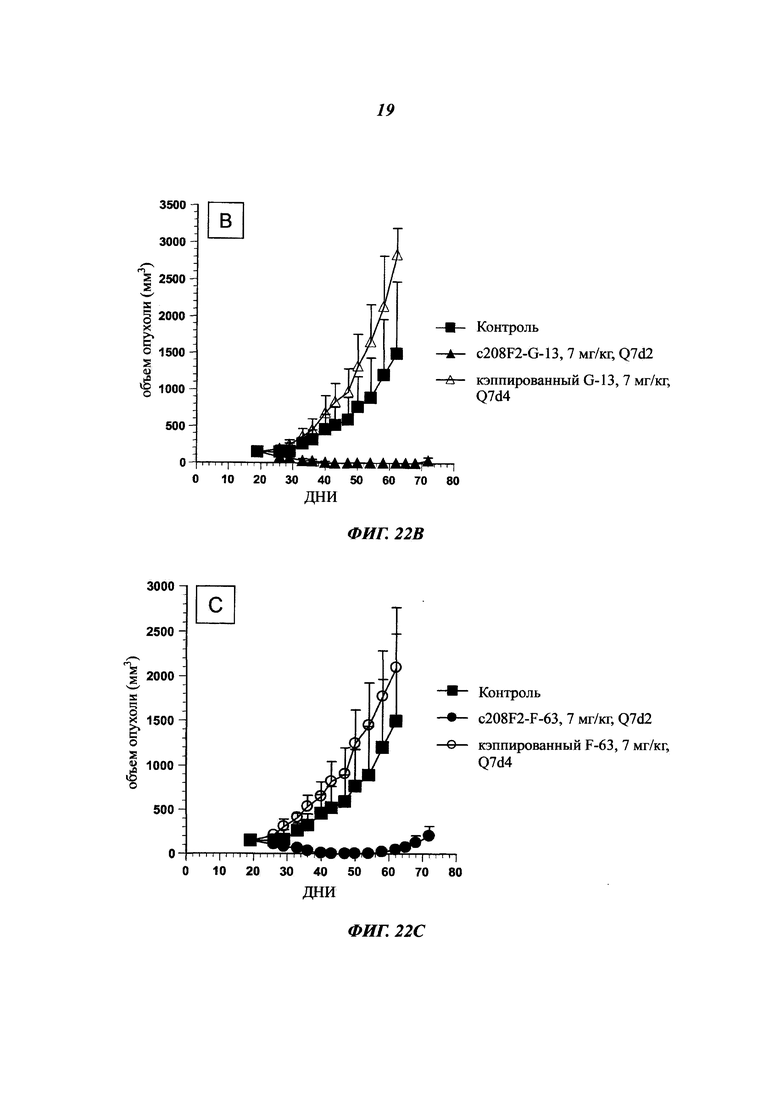
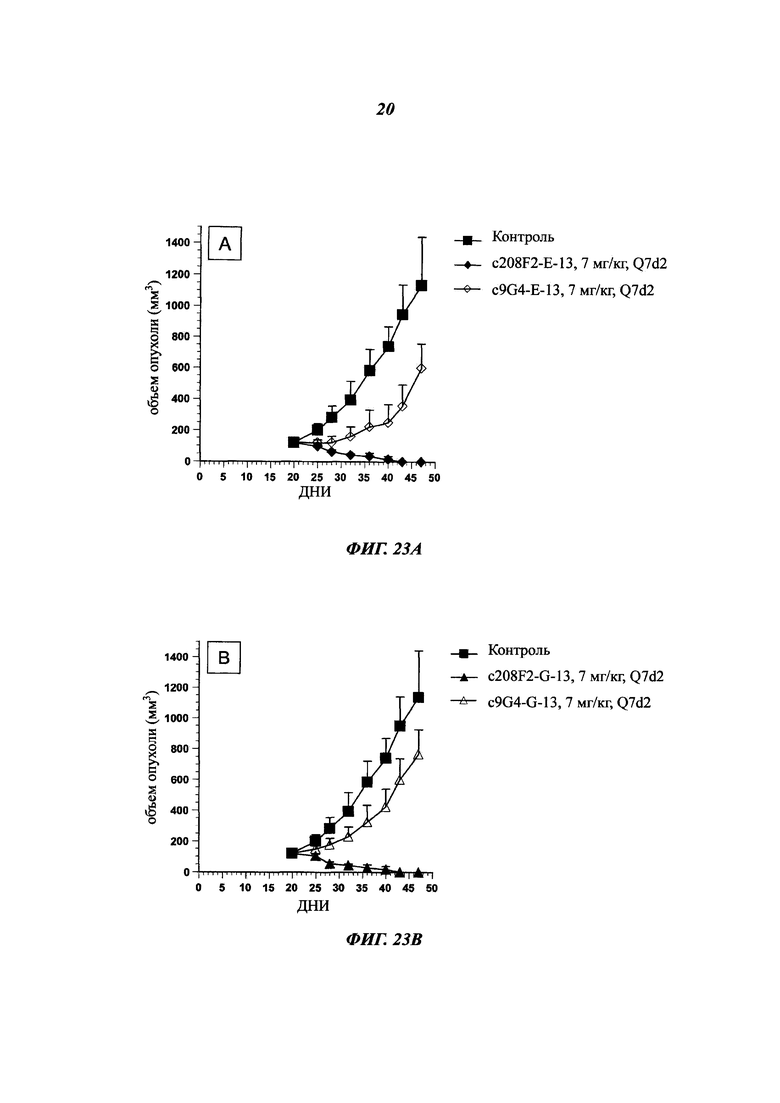
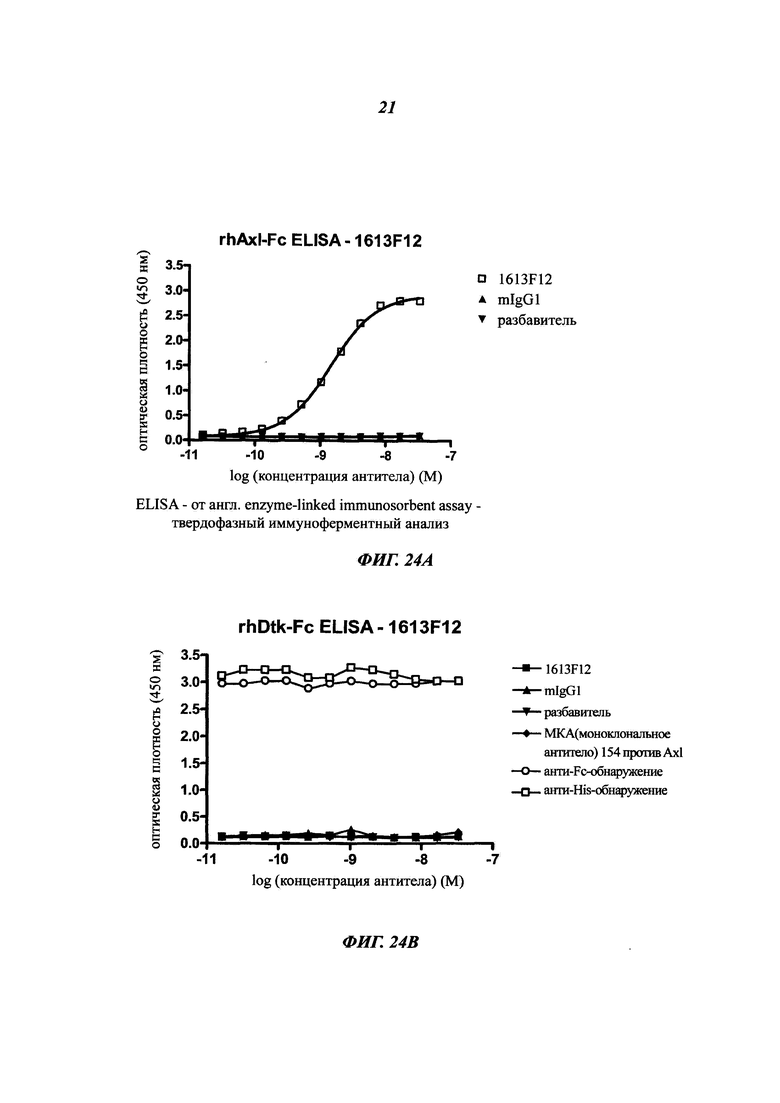
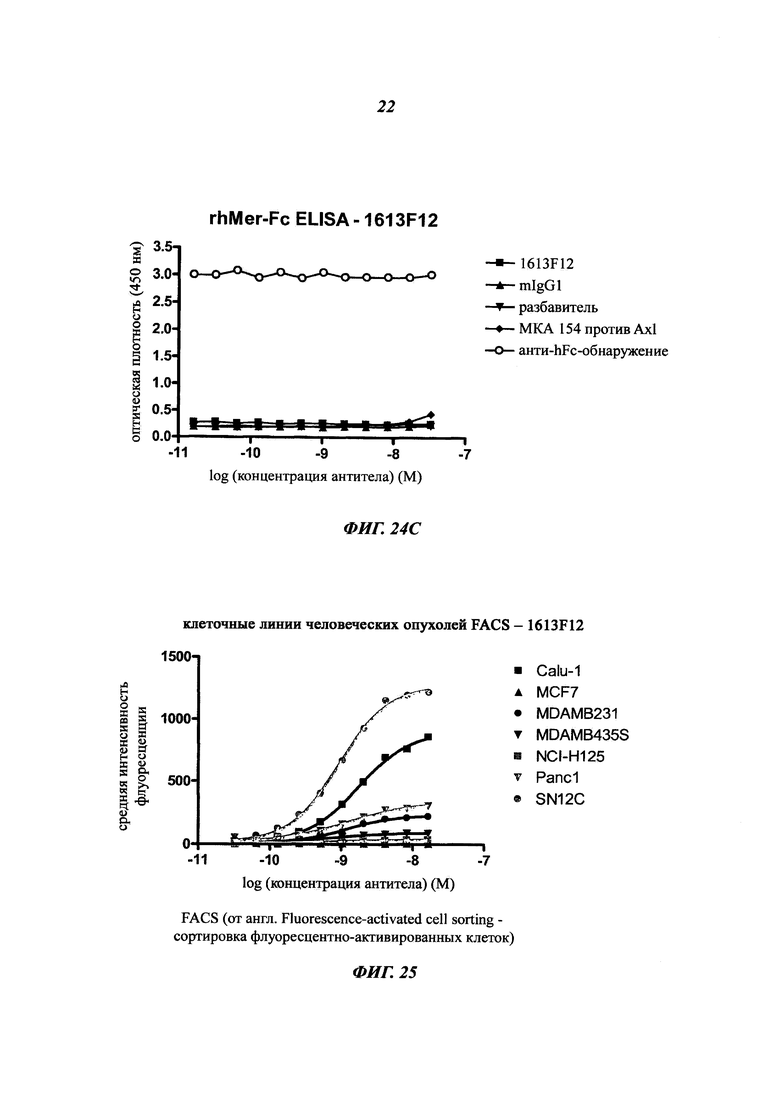
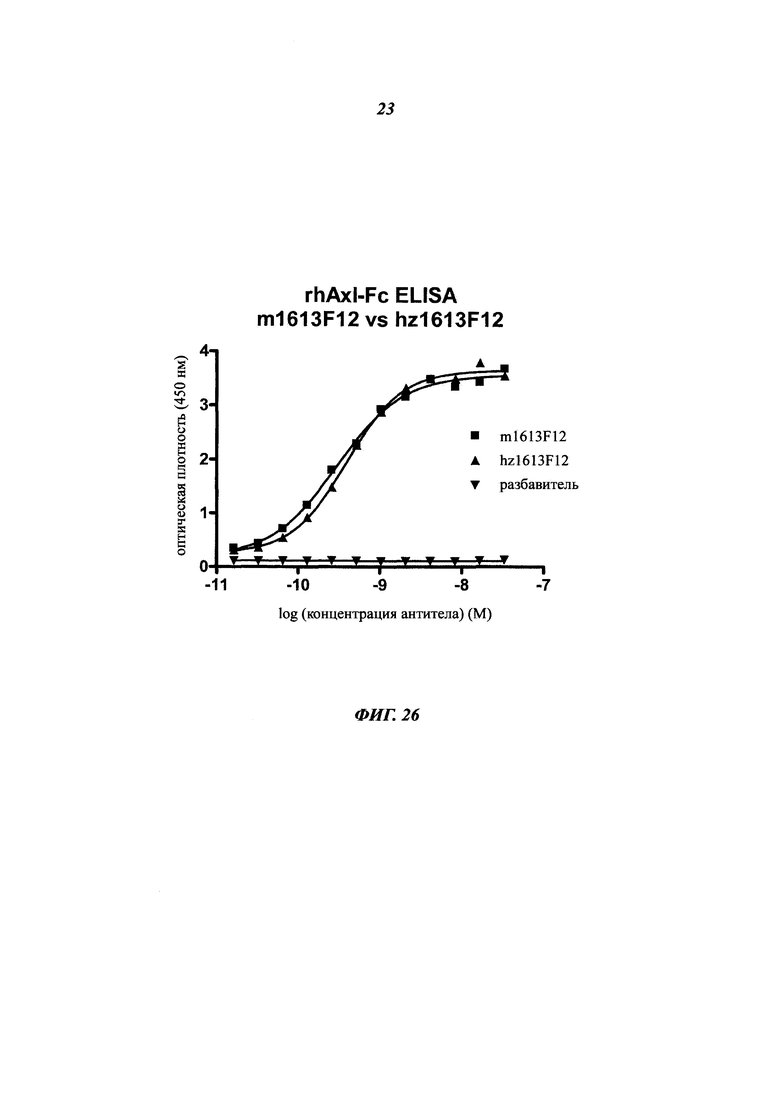

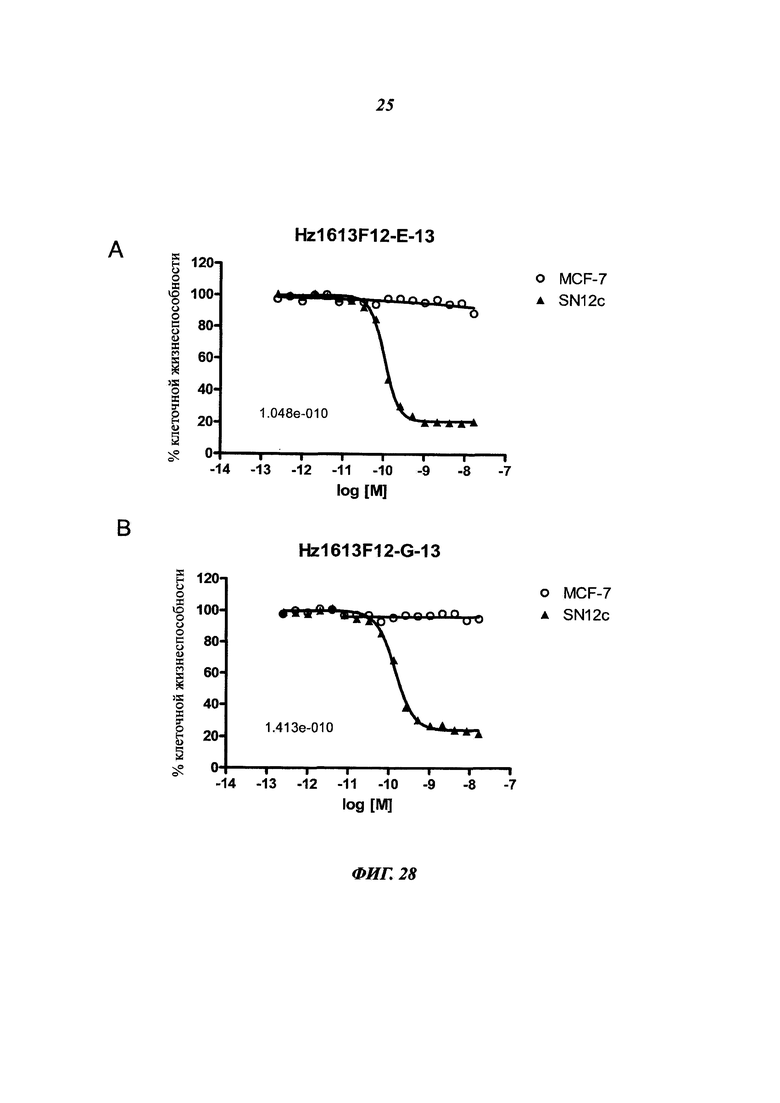
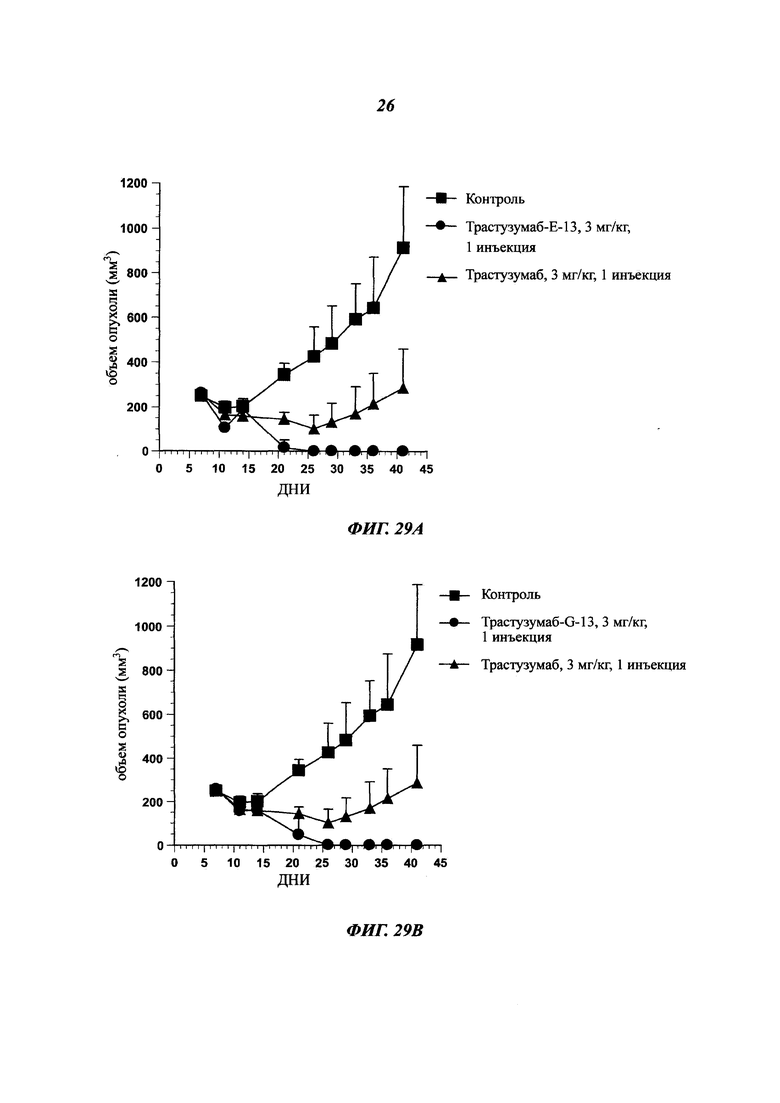
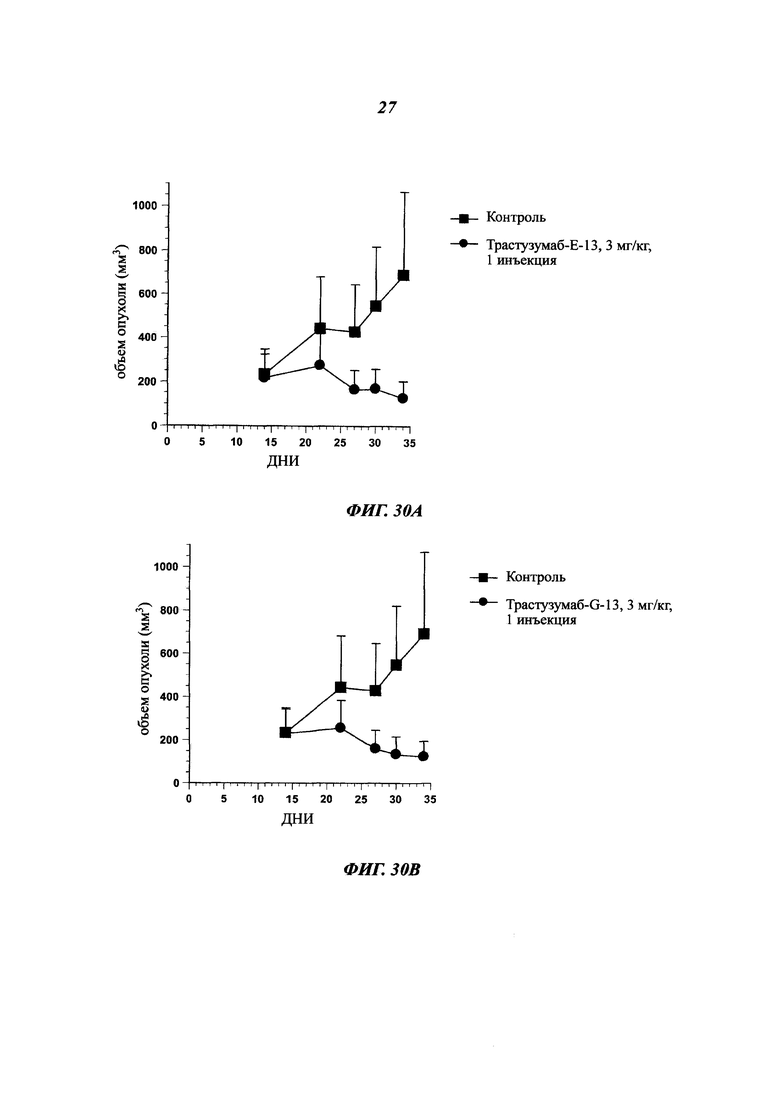
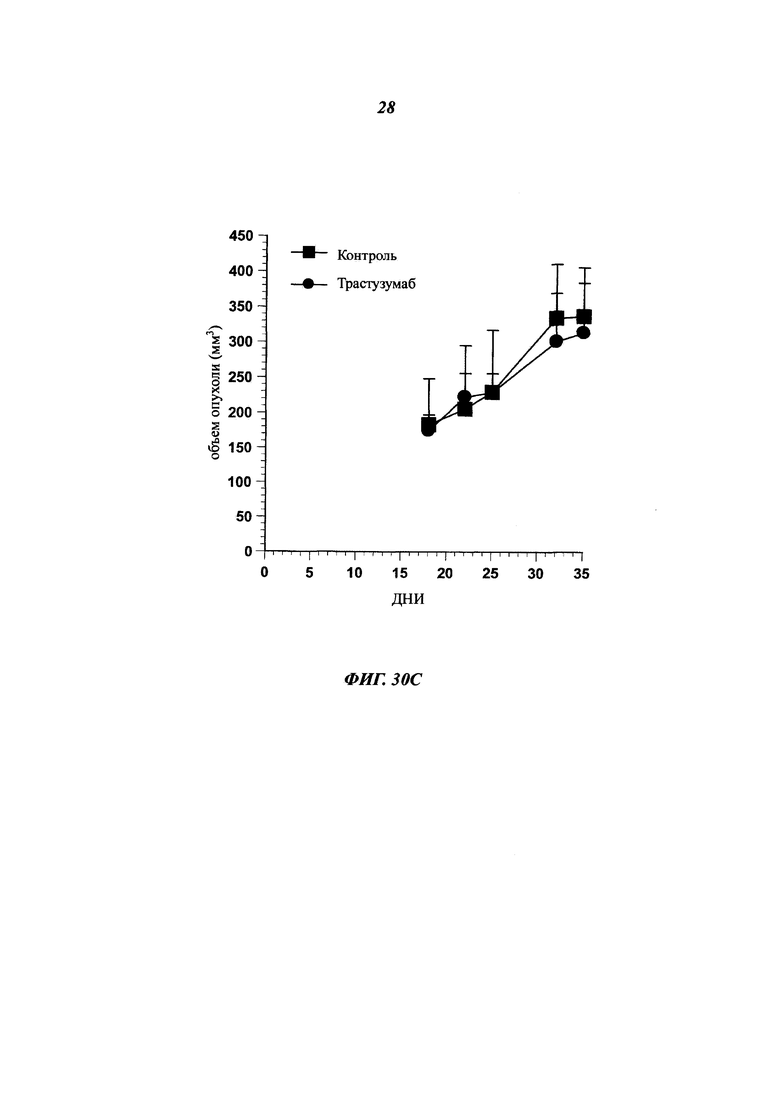
Группа изобретений относится к медицине и касается конъюгата "антитело - лекарственное средство" для лечения рака следующей формулы (I):
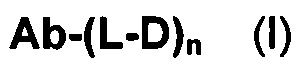
или его фармацевтически приемлемой соли. Где Ab представляет собой антитело против мишени или антитело против антигена, или его мишень-связывающий фрагмент или его антигенсвязывающий фрагмент; L представляет собой линкер следующей формулы (III):
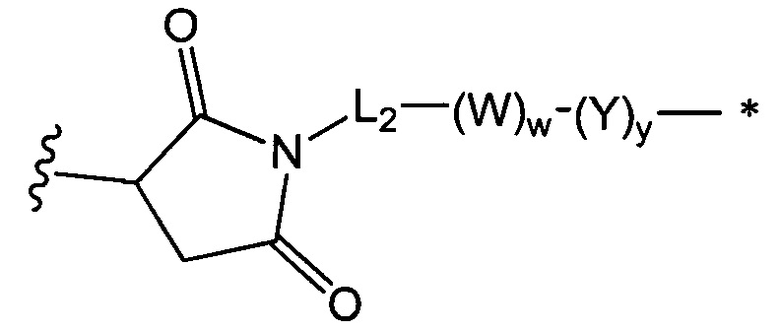 ;
;
D представляет собой лекарственную группировку. Группа изобретений также касается композиции для лечения рака, содержащей по меньшей мере один указанный конъюгат "антитело - лекарственное средство"; способа лечения мишень- или антигенэкспрессирующего рака у субъекта, включающего введение эффективного количества по меньшей мере одного указанного конъюгата "антитело - лекарственное средство". Группа изобретений обеспечивает улучшенную биологическую активность лекарственного средства. 3 н. и 23 з.п. ф-лы, 27 пр., 30 ил., 15 табл.
1. Конъюгат "антитело - лекарственное средство" для лечения рака следующей формулы (I):
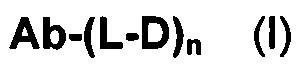
или его фармацевтически приемлемая соль,
где:
Ab представляет собой антитело против мишени или антитело против антигена, или его мишень-связывающий фрагмент или его антигенсвязывающий фрагмент;
L представляет собой линкер следующей формулы (III):
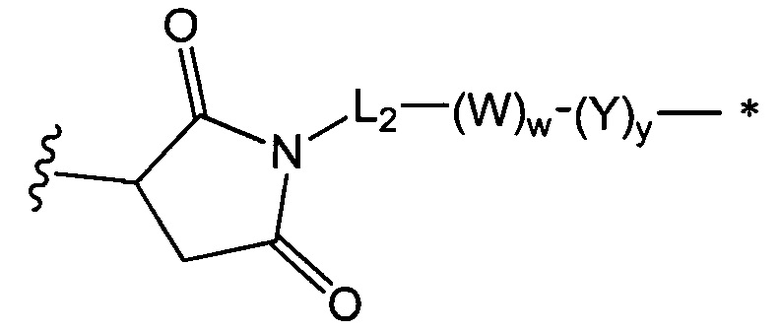 (III),
(III),
где:
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил, (С2-С6)алкил-карбонил,
W представляет собой аминокислотный блок;
w представляет собой целое число от 0 до 5;
Y представляет собой РАВ-карбонил, при этом РАВ представляет собой
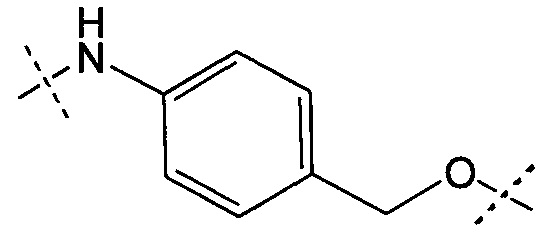 ;
;
у представляет собой 0 или 1;
звездочка указывает на точку присоединения к D; и
волнистая линия указывает на точку присоединения к Ab;
D представляет собой лекарственную группировку следующей формулы (II):
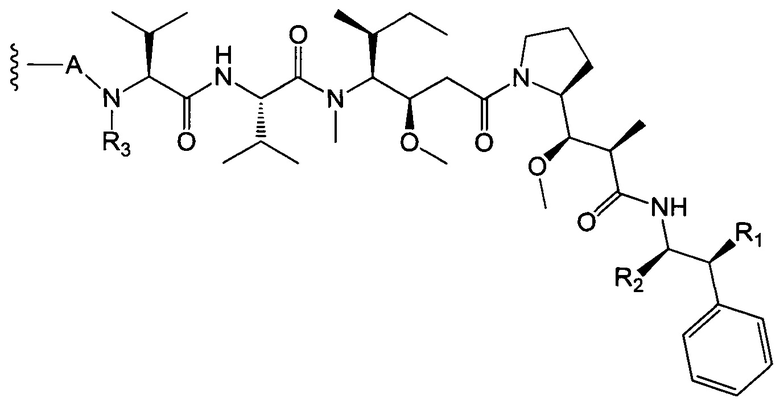 (II),
(II),
где:
R1 представляет собой Н или ОН;
R2 представляет собой группу: (С1-С6)алкил, СООН, СОО-((С1-С6)алкил) или тиазолил;
R3 представляет собой Н или (С1-С6)алкил;
А представляет собой:
группу формулы -Aa-Ab-, где Aa связан с L и представляет собой О или NR9, где R9 представляет собой Н или (С1-С6)алкил, а Ab связан с NR3 и представляет собой:
 группу арил-(С1-С8)алкандиил или группу гетероцикл-(С1-С8)алкандиил, где указанная группа, возможно, замещена (С1-С6)алкилом, при этом арильная или гетероциклическая группировка связана с Aa, а группировка (С1-С8)алкандиил связана cNR3;
группу арил-(С1-С8)алкандиил или группу гетероцикл-(С1-С8)алкандиил, где указанная группа, возможно, замещена (С1-С6)алкилом, при этом арильная или гетероциклическая группировка связана с Aa, а группировка (С1-С8)алкандиил связана cNR3;
волнистая линия указывает на точку присоединения к L; и
n составляет от 1 до 12.
2. Конъюгат "антитело - лекарственное средство" по п. 1, где мишень или антиген указанного антитела против мишени, или антитела против антигена согласно изобретению, или его мишень- или антигенсвязывающего фрагмента выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R, Axl и их внеклеточных мембранных фрагментов (ECD, от англ. extracellular domains).
3. Конъюгат "антитело - лекарственное средство" по п. 1, где указанная мишень или антиген указанного антитела против мишени, или антитела против антигена согласно изобретению, или его мишень- или антигенсвязывающего фрагмента выбраны среди HER2, IGF-1R и белка Axl, предпочтительно человеческого HER2, человеческого IGF-1R и человеческого белка Axl, а также их внеклеточных мембранных фрагментов (ECD).
4. Конъюгат "антитело - лекарственное средство" по п. 1, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим IGF-1R, выбранные среди:
i) антител, продуцируемых гибридомой I-4757, I-4773, I-4775, I-4736 и I-4774, депонированной в CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года, соответственно;
ii) антител, которые конкурируют за связывание с IGF-1R с антителами из i); и
iii) антител, которые связываются с тем же эпитопом IGF-1R, что и антитела i).
5. Конъюгат "антитело - лекарственное средство" по п. 1, где указанное Аb представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим IGF-1R, выбранные среди:
i) антитела, которое содержит три CDR участка тяжелой цепи последовательностей SEQ ID NO 1, 2 и 3 и три CDR участка легкой цепи последовательностей SEQ ID NO 4, 5 и 6;
ii) антитела, которое конкурирует за связывание IGF-1R с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом IGF-1R, что и антитело из i).
6. Конъюгат "антитело - лекарственное средство" по п. 1, где Ab содержит:
а) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95; и
b) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87.
7. Конъюгат "антитело - лекарственное средство" по п. 1, где указанное Аb представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим белком Axl, выбранные среди:
i) антитела, которое содержит три CDR участка тяжелой цепи последовательностей SEQ ID NO 59, 60 и 61 и три CDR участка легкой цепи последовательностей SEQ ID NO 56, 57 и 58;
ii) антитела, которое конкурирует за связывание Axl с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом Axl, что и антитело из i).
8. Конъюгат "антитело - лекарственное средство" по п. 1, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные связываться с человеческим HER2, состоящие из трастузумаба.
9. Конъюгат "антитело - лекарственное средство" по любому из пп. 1-8, где L2 имеет следующую формулу:
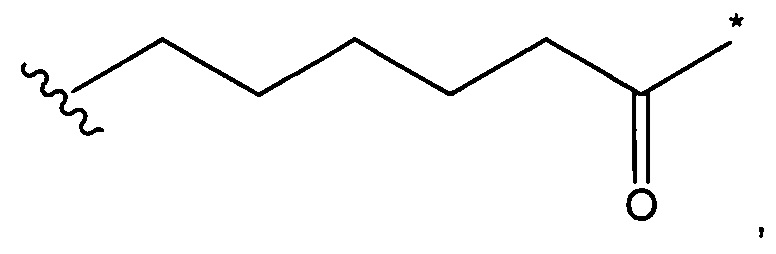
где:
звездочка указывает на точку присоединения к (W)w;
и волнистая линия указывает на точку присоединения к атому азота малеимидной группировки.
10. Конъюгат "антитело - лекарственное средство" по любому из пп. 1-8, где w равно 0; или w равно 2, a (W)w выбран среди:
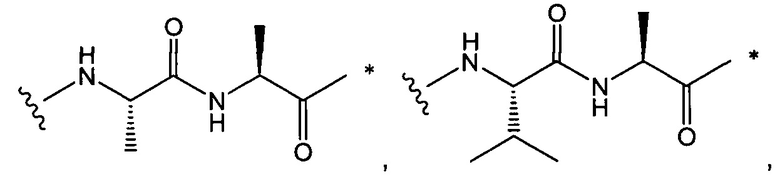
и 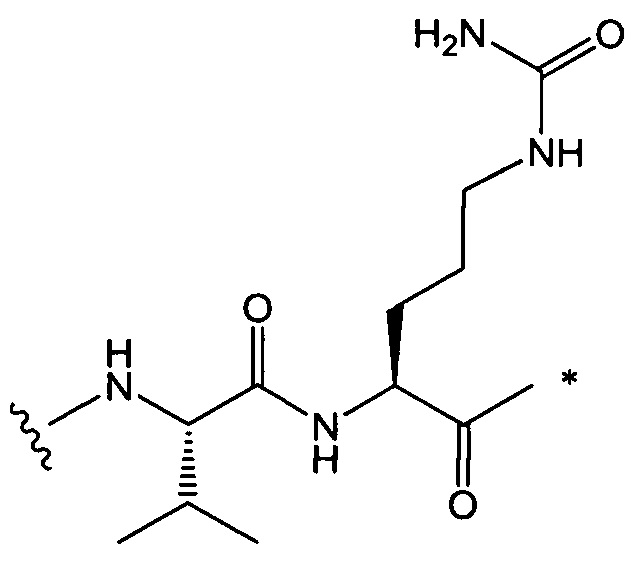
где:
звездочка указывает на точку присоединения к (Y)y; и
волнистая линия указывает на точку присоединения к L2.
11. Конъюгат "антитело - лекарственное средство" по любому из пп. 1-8, где L выбран среди:

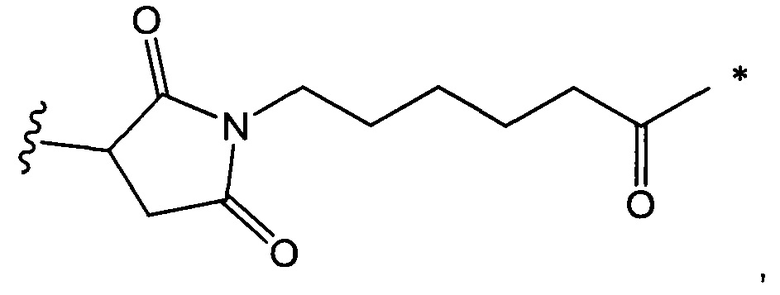
где звездочка указывает на точку присоединения к D, а волнистая линия указывает на точку присоединения к Ab.
12. Конъюгат "антитело - лекарственное средство" по п. 1, где А представляет собой группу формулы -Aa-Ab-, в которой Aa является таким, как определено в п. 1, а Ab представляет собой группу:
- фенил-(С1-С2)алкандиил, или
- гетероцикл-(С1-С2)алкандиил, возможно, замещенную группой (С1-С6)алкил (особенно незамещенную), при этом гетероцикл представляет собой насыщенное, ненасыщенное или ароматическое кольцо с 5 или 6 членами, содержащее 1 или 2 атома азота, выбранное, в частности, среди пиридина, пиперидина и имидазола и предпочтительно представляющее собой пиридин.
13. Конъюгат "антитело - лекарственное средство" по п. 1, где А представляет собой группу следующей формулы:
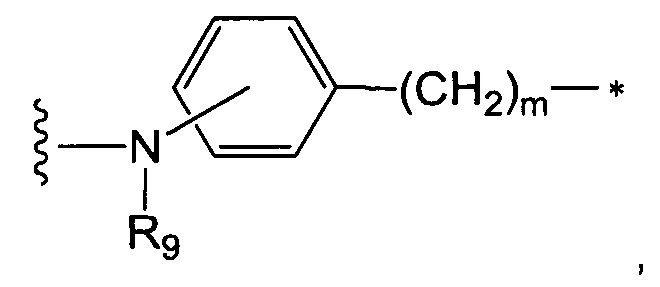
где:
R9 является таким, как определено в п. 1, a m представляет собой целое число от 1 до 8, и где предпочтительно R9 представляет собой Н или Me, a m равен 1 или 2,
волнистая линия указывает на точку присоединения к L, и
звездочка указывает на точку присоединения к NR3.
14. Конъюгат "антитело - лекарственное средство" по п. 1, где (L-D) выбран среди:
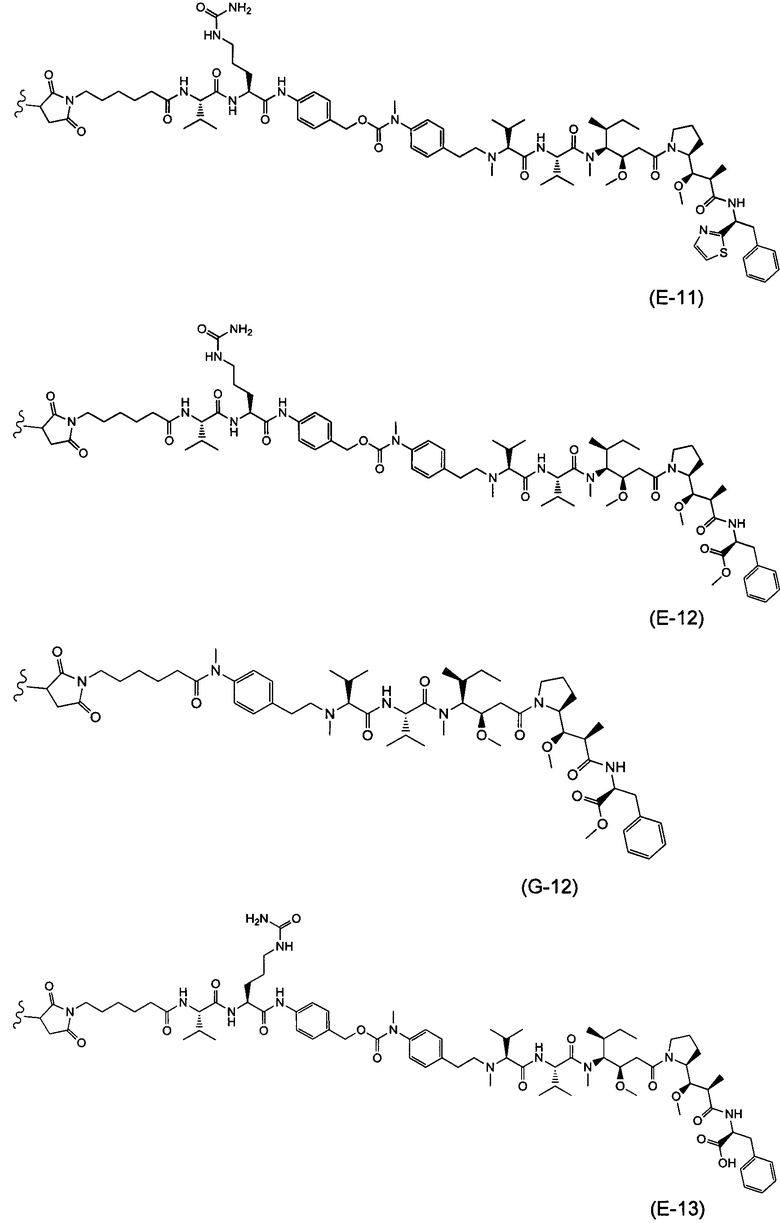
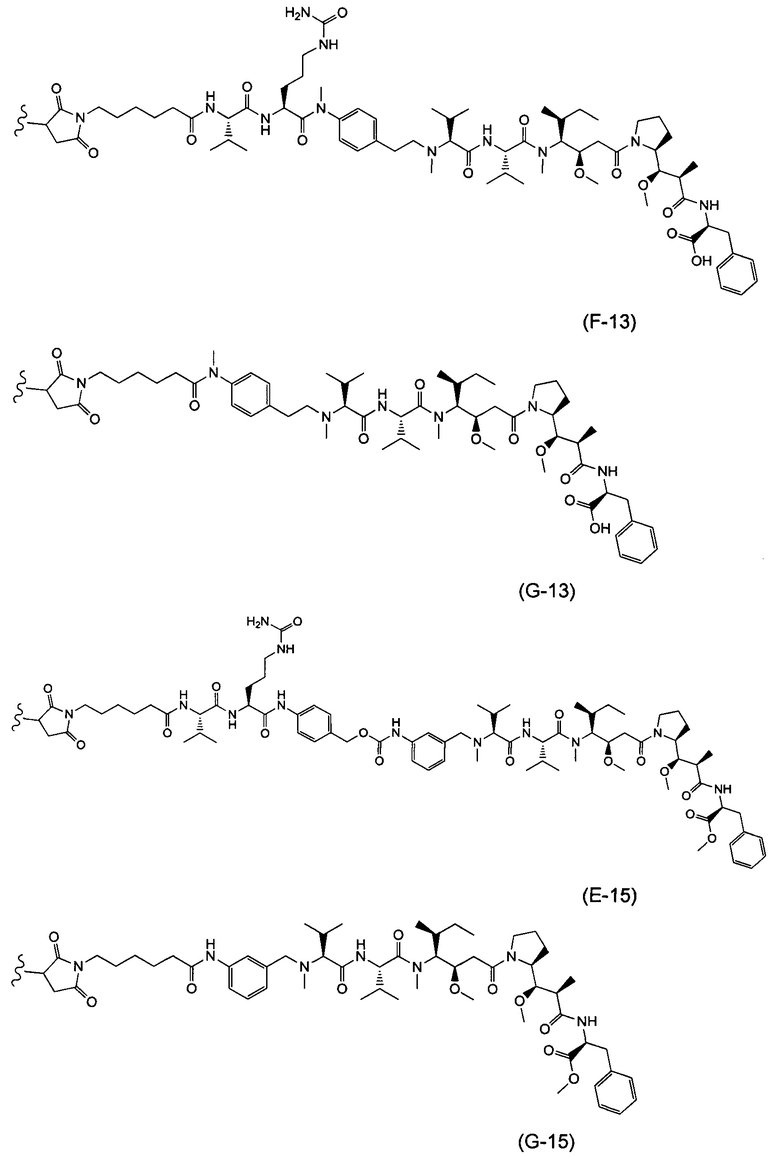

где волнистая линия указывает на точку присоединения к Ab.
15. Конъюгат "антитело - лекарственное средство" по п. 1, имеющий формулу, выбранную среди:
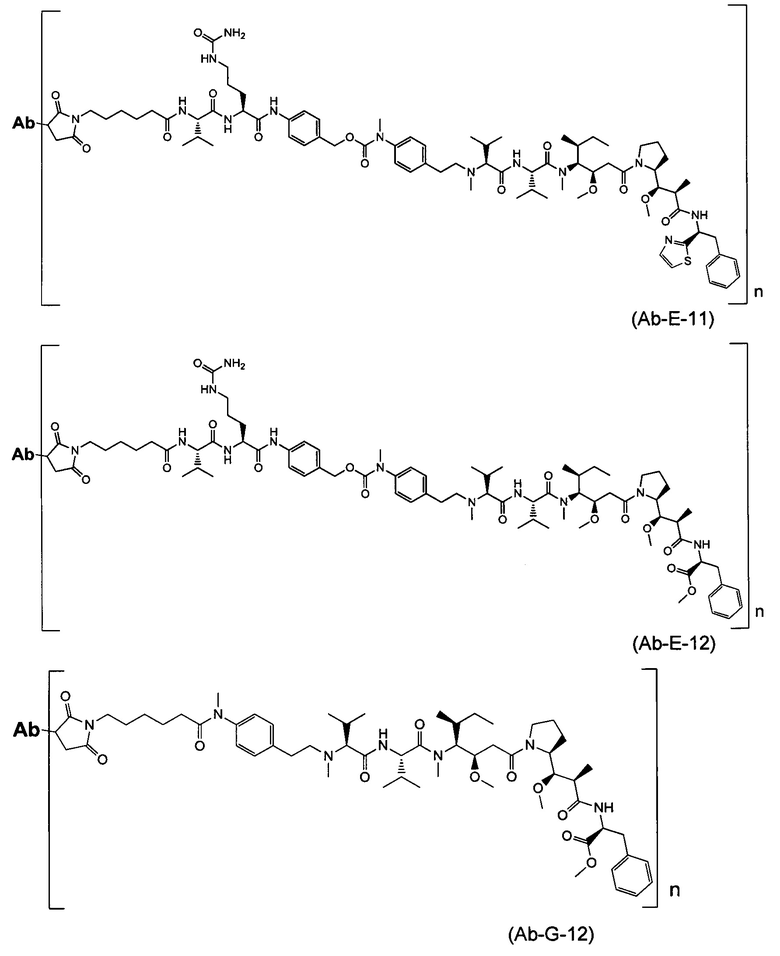
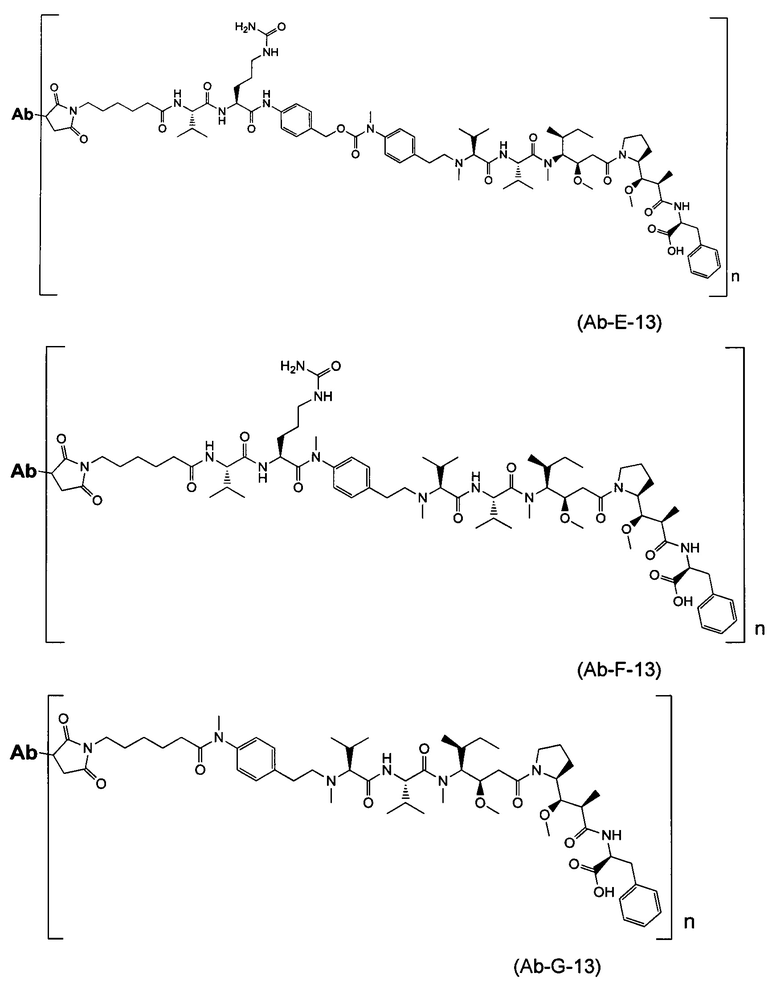
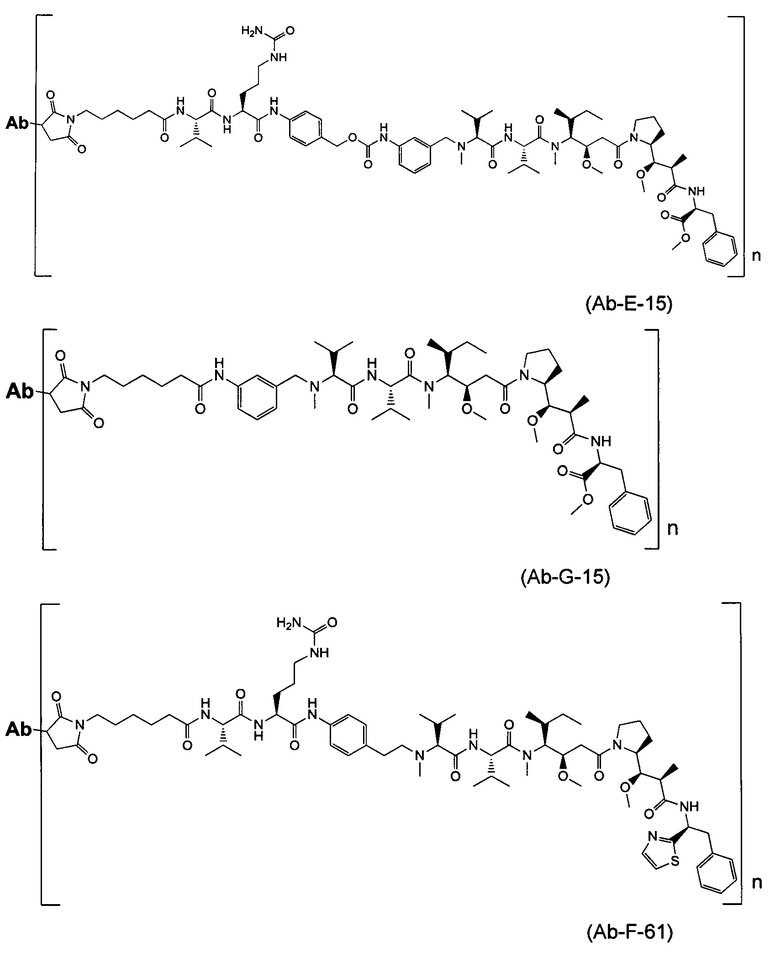
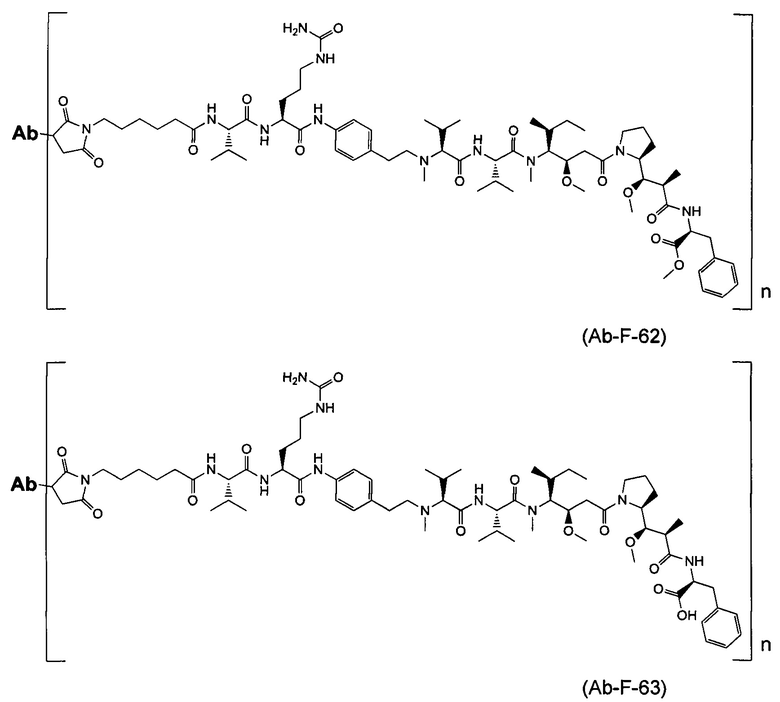
и его фармацевтически приемлемая соль,
где Ab представляет собой антитело против мишени или антитело против антигена, или его мишень-связывающий фрагмент или его антигенсвязывающий фрагмент.
16. Конъюгат "антитело - лекарственное средство" по п. 15, где мишень указанного антитела против мишени или антиген указанного антитела против антигена или его мишень-связывающего фрагмента или его антигенсвязывающего фрагмента выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER1, HER3, HER2, IGF-1R, Axl и их внеклеточных мембранных фрагментов (ECD).
17. Конъюгат "антитело - лекарственное средство" по п. 15, где мишень указанного антитела против мишени или антиген указанного антитела против антигена выбраны среди HER2, IGF-1R и белка Axl, предпочтительно человеческого HER2, человеческого IGF-1R и человеческого белка Axl.
18. Конъюгат "антитело - лекарственное средство" по п. 15, где указанное Ab представляет собой антитело или его антигенсвязывающий фрагмент и выбрано среди:
a) Ab или его антигенсвязывающего фрагмента, способного связываться с человеческим IGF-1R, выбранного среди:
i) антител, продуцируемых гибридомой I-4757, I-4773, I-4775, I-4736 и I-4774, депонированной в CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года, соответственно;
ii) антител, которые конкурируют за связывание с IGF-1R с антителами из i); и
iii) антител, которые связываются с тем же эпитопом IGF-1R, что и антитела из i),
b) Ab или его антигенсвязывающего фрагмента, способного связываться с человеческим IGF-1R, выбранного среди:
i) антитела, которое содержит три CDR участка тяжелой цепи последовательностей SEQ ID NO 1, 2 и 3 и три CDR участка легкой цепи последовательностей SEQ ID NO 4, 5 и 6;
ii) антитела, которое конкурирует за связывание IGF-1R с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом IGF-1R, что и антитело из i),
c) Ab, которое содержит:
i) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где указанная последовательность SEQ ID NO 33 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 или 95; и
ii) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где указанная последовательность SEQ ID NO 35 содержит по меньшей мере 1 обратную мутацию, выбранную среди остатков 22, 53, 55, 65, 71, 72, 77 или 87,
d) Ab или его антигенсвязывающего фрагмента, способного связываться с человеческим белком Axl, выбранного среди:
i) антитела, которое содержит три CDR тяжелой цепи последовательностей SEQ ID NO 59, 60 и 61 и три CDR легкой цепи последовательностей SEQ ID NO 56, 57 и 58;
ii) антитела, которое конкурирует за связывание Axl с антителом из i); и
iii) антитела, которое связывается с тем же эпитопом Axl, что и антитело из i), и
е) Ab или его антигенсвязывающего фрагмента, способного связываться с человеческим HER2, состоящего из трастузумаба или пертузумаба.
19. Конъюгат "антитело - лекарственное средство" по п. 1, где n составляет 2.
20. Конъюгат "антитело - лекарственное средство" по п. 1, где n составляет 4.
21. Композиция для лечения рака, содержащая по меньшей мере один конъюгат "антитело - лекарственное средство" по любому из пп. 1-20.
22. Композиция по п. 21, дополнительно содержащая фармацевтически приемлемый носитель.
23. Композиция по п. 21 или 22 для применения в лечении мишень- или антигенэкспрессирующего рака, где указанная мишень или антиген предпочтительно выбраны среди CD19, CD20, CD22, CD25, CD30, CD33, CD40, CD56, CD64, CD70, CD74, CD79, CD105, CD138, CD174, CD205, CD227, CD326, CD340, MUC16, GPNMB, PSMA, Cripto, ED-B, TMEFF2, EphB2, EphA2, FAP, интегрина av, мезотелина, EGFR, TAG-72, GD2, CAIX, 5T4, HER2, IGF-1R, Axl и их внеклеточных мембранных фрагментов (ECD).
24. Композиция по п. 21 или 22 для применения в лечении HER2-, IGF-1R-или Axl-экспрессирующего рака.
25. Композиция по п. 23, где указанный мишень- или антигенэкспрессирующий рак представляет собой рак, выбранный среди рака молочной железы, толстой кишки, карциномы пищевода, гепатоцеллюлярного рака, рака желудка, глиомы, рака легких, меланомы, остеосаркомы, рака яичников, предстательной железы, рабдомиосаркомы, рака почек, щитовидной железы, эндометрия матки, мезотелиомы, плоскоклеточной карциномы ротовой полости, саркомы Капоши, острой лейкемии, колоректальной карциномы, меланомы, протоковой аденокарциномы поджелудочной железы и любого рака с лекарственной устойчивостью.
26. Способ лечения мишень- или антигенэкспрессирующего рака у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества по меньшей мере одного конъюгата "антитело - лекарственное средство" по любому из пп. 1-20 или композиции по п. 21 или 22.
| DOSIO F., et al., Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components.Toxins (Basel) | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| WO 2014026286 A1, 20.02.2014 | |||
| US 20090035258 A1, 05.02.2009 | |||
| ALLEY SC., et al., Contribution of linker stability to the activities of anticancer immunoconjugates.Bioconjug Chem | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 20080279868 A1, 13.11.2008. | |||

