ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение в основном относится к новым соединениям, селективно модулирующим ДНК-зависимую протеинкиназу (DNA-PK), и к их фармацевтически приемлемым солям. Настоящее изобретение также относится к фармацевтическим композициям, содержащим одно или более чем одно такое соединение в качестве активного ингредиента, и к применению этих соединений в лечении заболевания, ассоциированного с DNA-PK, включая рак.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
DNA-PK представляет собой ядерный серин/треониновый протеинкиназный комплекс, состоящий из каталитической субъединицы DNA-PKcs и гетеродимера белков Ku (Ku70/Ku80). Функционально DNA-PK является важнейшим компонентом в репарации двунитевых разрывов ДНК (DSB), способствуя поддержанию целостности генома, и в процессе V(D)J-рекомбинации, приводя к крайне разнообразному репертуару антител/иммуноглобулинов и Т-клеточных рецепторов, обнаруживаемых на В- и Т-клетках соответственно. Кроме того, DNA-PK и ее компоненты вовлечены во множество других физиологических процессов, включая модуляцию структуры хроматина, сохранение теломер, регуляцию транскрипции и ответ на репликативный стресс (Smith and Jackson, 1999; Goodwin and Knudsen, 2014).
Геном человека в виде ДНК постоянно подвергается атаке реакционноспособных форм кислорода (ROS), которые преимущественно являются побочными продуктами окислительного метаболизма. ROS способны вызывать повреждение ДНК в виде однонитевых разрывов. DSB могут возникать, если предыдущие однонитевые разрывы произошли в непосредственной близости. Кроме того, одно- и одно-двунитевые разрывы возникают, когда репликационная вилка ДНК встречает поврежденные группы оснований. Кроме того, внешние воздействия, такие как ионизирующее излучение (например, гамма-излучение или излучение частиц) и некоторые противоопухолевые препараты (например блеомицин) способны вызывать DSB. DSB также могут возникать как промежуточные звенья соматической рекомбинации, процесса, важного для формирования функциональной иммунной системы всех позвоночных.
Если DSB не устранены или устранены некорректно, возникают мутации и/или хромосомные аберрации, которые могут привести к клеточной смерти. Чтобы справиться с серьезными угрозами, создаваемыми DSB, эукариотические клетки выработали несколько механизмов (например, негомологичное соединение концов ДНК (NHEJ) и гомологичная рекомбинация (HR)), чтобы опосредовать их репарацию, в которых DNA-PK играет ключевую роль. Биохимические исследования показали, что DNA-PK наиболее эффективно активируется при появлении DSB ДНК. Клеточные линии, компоненты DNA-PK которых мутированы и нефункциональны, оказались чувствительными к радиации (Smith and Jackson, 1999). Ингибиторы DNA-PK также могут быть эффективными в качестве монотерапии при опухолях с высокими уровнями эндогенных повреждений ДНК. Было продемонстрировано, что ингибиторы DNA-PK, используемые в онкологии, могут включать нацеливание на опухоли с высоким уровнем репликативного стресса (Lin et al., 2014; Ashley et al., 2014; Buisson et al., 2015) либо в качестве монотерапии, либо в комбинации с другими агентами при раке предстательной железы (Goodwin et al., 2013) и раке молочной железы (Medunjanin et al., 2010).
Следовательно, соединения, которые ингибируют DNA-PK, необходимы в качестве фармакологических средств и представляют значительный интерес в качестве лекарственных средств для лечения расстройств, ассоциированных с DNA-PK, таких как рак.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предложено соединение формулы (I):
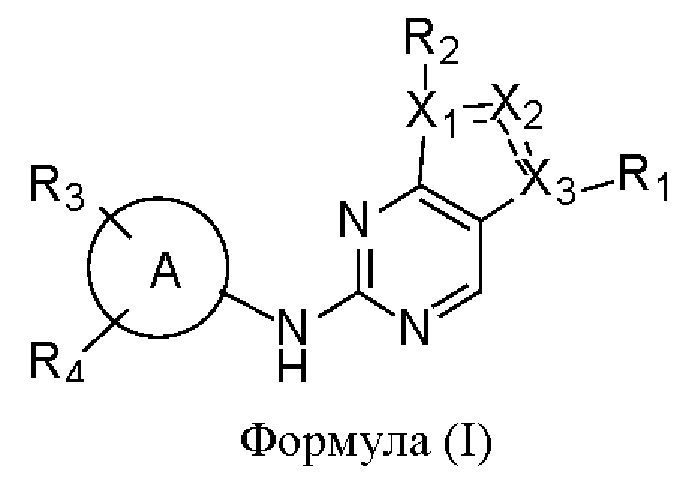
или его фармацевтически приемлемая соль, где X1, Х2, Х3, R1, R2, R3, R4 и кольцо А являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложено соединение формулы (Ia):
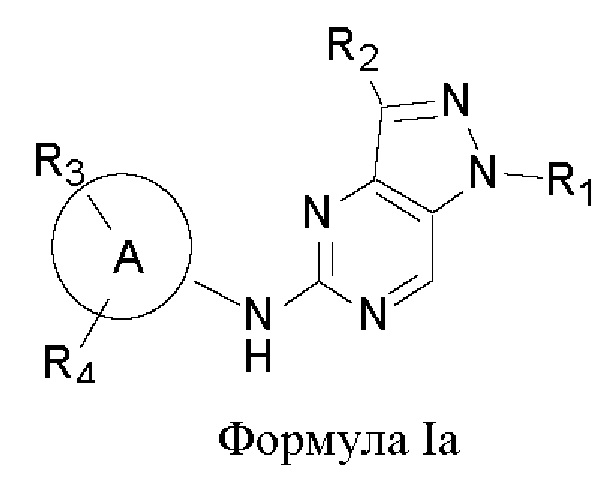
или его фармацевтически приемлемая соль, где R1, R2, R3, R4, n и кольцо А являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложено соединение формулы (Ib):
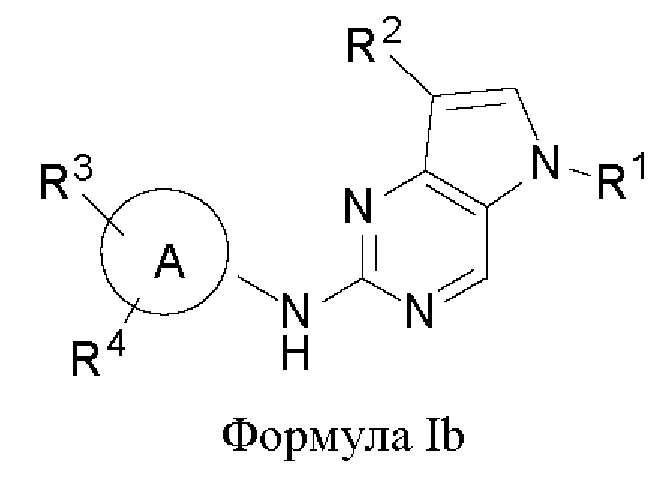
или его фармацевтически приемлемая соль, где R1, R2, R3, R4 и кольцо А являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложено соединение формулы (Ic):
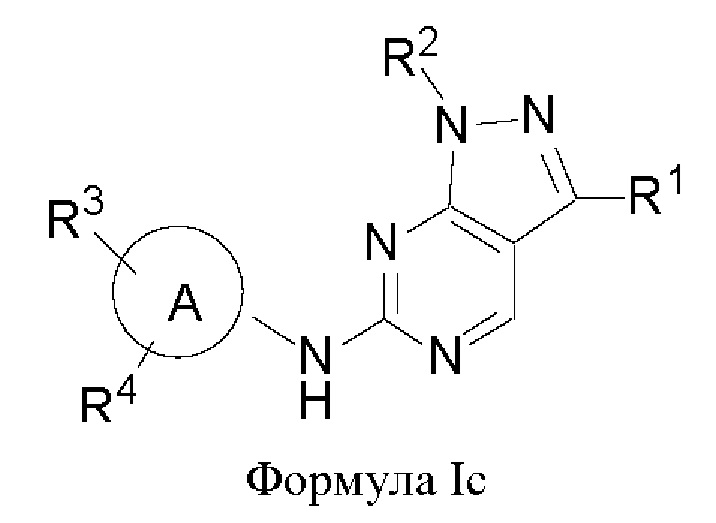
или его фармацевтически приемлемая соль, где R1, R2, R3, R4 и кольцо А являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложено соединение формулы (Id):
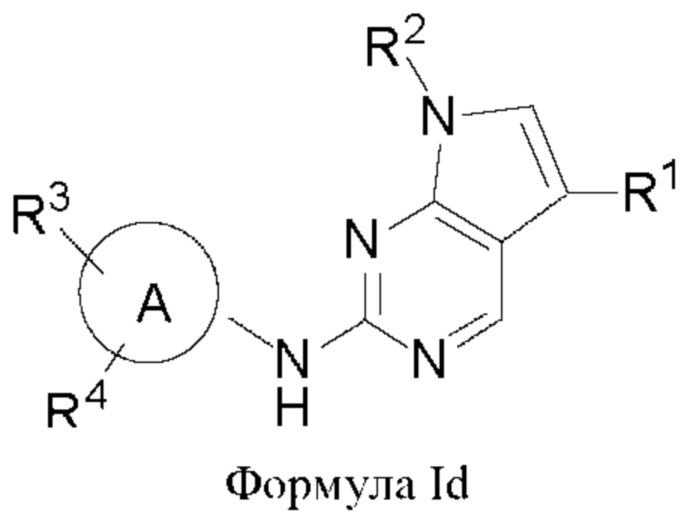
или его фармацевтически приемлемая соль, где R1, R2, R3, R4 и кольцо А являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложено соединение формулы (Ie):
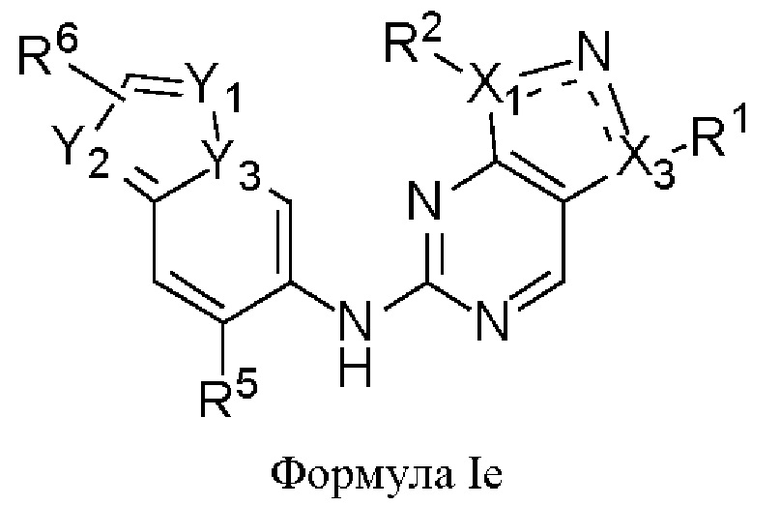
или его фармацевтически приемлемая соль, где X1, Х3, Y1, Y2, Y3, R1, R2, R5 и R6 являются такими, как определено здесь.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая одно или более чем одно соединение формулы (I), формулы (Ia), формулы (Ib), формулы (Ic), формулы (Id), формулы (Ie) или его фармацевтически приемлемую соль в качестве активного ингредиента.
В другом аспекте настоящего изобретения дополнительно предложено соединение формулы (I), или его фармацевтически приемлемая соль, или фармацевтическая композиция одного или более чем одного из вышеперечисленного для применения в ингибировании киназы DNA-PK.
В другом аспекте настоящего изобретения предложено применение соединений формулы (I), или их фармацевтически приемлемых солей, или фармацевтической композиции одного или более чем одного из вышеперечисленных в изготовлении лекарственного средства для ингибирования киназы DNA-PK у субъекта.
В другом аспекте настоящего изобретения предложен способ ингибирования киназы DNA-PK путем применения одного или более чем одного соединения формулы (I), или его фармацевтически приемлемой соли, или фармацевтической композиции одного или более чем одного из вышеперечисленного.
В другом аспекте настоящего изобретения предложен способ лечения расстройства, ассоциированного с DNA-PK (например, рака) путем применения соединения формулы (I), или их фармацевтически приемлемых солей, или фармацевтической композиции одного или более чем одного из вышеперечисленного. В дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль в комбинации со вторым терапевтическим агентом, предпочтительно противоопухолевым агентом.
В другом аспекте настоящего изобретения предложено комбинированное применение соединения формулы (I) или его фармацевтически приемлемой соли и второго терапевтического агента, предпочтительно противоопухолевого агента.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
В одном аспекте настоящего изобретения предложены соединения формулы (I):
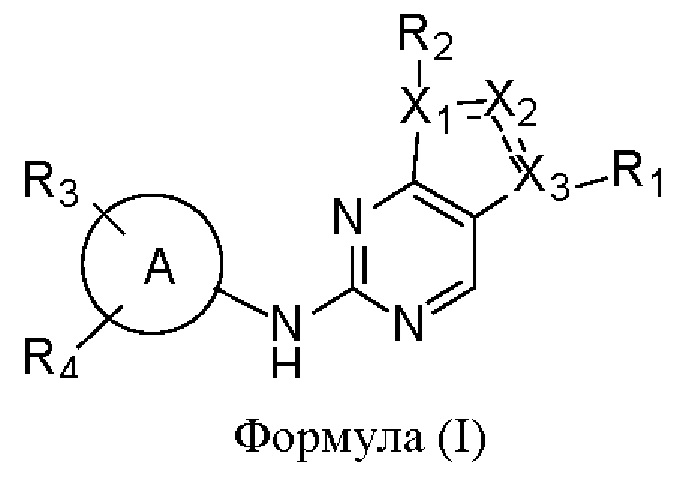
или их фармацевтически приемлемая соль,
где,
X1, Х2 и Х3 каждый независимо представляет собой С или N, при условии, что по меньшей мере один из X1, Х2 и Х3 представляет собой N, и по меньшей мере один из X1, Х2 и Х3 представляет собой С;
пунктирная линия  означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связь, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь;
означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связь, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь;
R1 отсутствует, представляет собой галоген или C1-6алкил, где указанный С1-6алкил возможно может быть моно- или независимо полизамещен гидроксилом, галогеном или дейтерием;
каждый из R2, R3 и R4 отсутствует или независимо выбран из галогена, гидроксила, циано, С1-6алкила, С1-6алкоксила, -(CH2)n-Q, которые возможно могут быть моно- или независимо полизамещены дейтерием, гидроксилом, амино, циано, галогеном, C1-6алкилом, C1-6галогеналкилом, (С≡N)-С1-6алкилом, C1-6алкоксилом, C1-6галогеналкоксилом, С3-8циклоалкилом, С3-8циклоалкоксилом, 3-8-членным арилом или 3-8-членным гетероциклилом,
где n равен 0, 1 или 2, Q представляет собой 3-8-членный насыщенный или ненасыщенный карбоциклил или 3-8-членный насыщенный или ненасыщенный гетероциклил;
кольцо А представляет собой 5-12-членный арил, 5-12-членный гетероарил, имеющий 1-5 кольцевых гетероатомов, выбранных из кислорода, серы и азота, 8-10-членное бициклическое кольцо, имеющее 0-5 кольцевых гетероатомов, выбранных из кислорода, серы и азота, где кольцо А не является фенилом.
В некоторых воплощениях R2 выбран из метила, этила, пропила, бутила, циклопропила, циклобутила, оксетанила, циклопентанила, тетрагидрофурила, циклогексанила, тетрагидропиранила, циклогептанила, пиперидинила, фенила, пиридинила, пиридонила, оксоканила, тетрагидропиранила, дигидропиранила, спиро[3.3]гептанила, спиро[2.5]октанила, бицикло[1.1.1]пентанила, бицикло[3.2.1]октанила, 8-оксабицикло[3.2.1]октан-3-ила, которые возможно могут быть моно- или независимо полизамещены гидроксилом, циано, галогеном, C1-6алкилом, C1-6галогеналкилом, C1-6алкоксилом, C1-6галогеналкоксилом, С3-8циклоалкилом, С3-8циклоалкоксилом, 3-8-членным арилом или 3-8-членным гетероциклилом, которые дополнительно возможно могут быть моно- или независимо полизамещены галогеном, дейтерием, гидроксилом, амино, циано, C1-6алкилом, C1-6галогеналкилом, С1-6алкоксилом или С1-6галогеналкоксилом.
В некоторых воплощениях R2 выбран из:



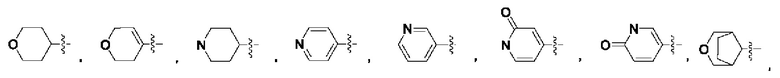

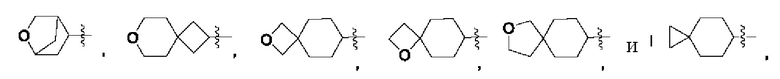
которые возможно могут быть моно- или независимо полизамещены гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом.
В некоторых воплощениях R2 представляет собой циклогексанил или тетрагидропиранил, которые возможно могут быть моно- или независимо полизамещены галогеном, C1-6алкилом или С1-6алкоксилом.
В некоторых воплощениях R1 представляет собой метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил или изобутил, которые возможно могут быть моно- или независимо полизамещены гидроксилом, галогеном или дейтерием.
В некоторых воплощениях R1 представляет собой метил, этил, трифторметил или тридейтерийметил.
В некоторых воплощениях кольцо А представляет собой 6-членный гетероарил, имеющий 1 кольцевой гетероатом азота, 9-членное бициклическое кольцо, имеющее 2-3 кольцевых гетероатома, выбранных из кислорода, серы и азота, возможно 9-членное бициклическое кольцо представляет собой фенил- или пиридинил-конденсированное бициклическое кольцо, возможно кольцо А выбрано из
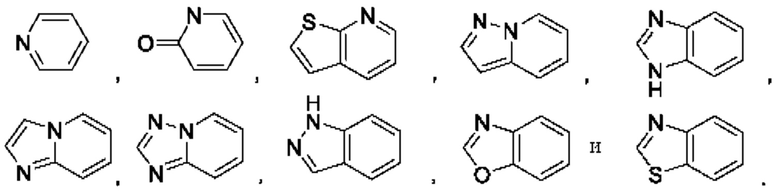
В некоторых воплощениях каждый R3 и R4 отсутствует или независимо выбран из галогена, гидроксила, циано, C1-6алкила, CN-C1-6алкила, С1-6галогеналкила, C1-6алкоксила, C1-6галогеналкоксила, 3-8-членного насыщенного или ненасыщенного гетероциклила, где указанный гетероциклил возможно может быть дополнительно моно- или независимо полизамещен C1-3алкилом.
В некоторых воплощениях кольцо А представляет собой  или
или  каждый из R3 и R4 отсутствует или независимо выбран из метила, циано, метоксила, хлора, цианометила, пиразолила, оксазолила, где указанные пиразолил или оксазолил возможно могут быть дополнительно моно- или независимо полизамещены C1-3алкилом.
каждый из R3 и R4 отсутствует или независимо выбран из метила, циано, метоксила, хлора, цианометила, пиразолила, оксазолила, где указанные пиразолил или оксазолил возможно могут быть дополнительно моно- или независимо полизамещены C1-3алкилом.
В некоторых воплощениях соединения, предложенные здесь, имеют структуру формулы Ia
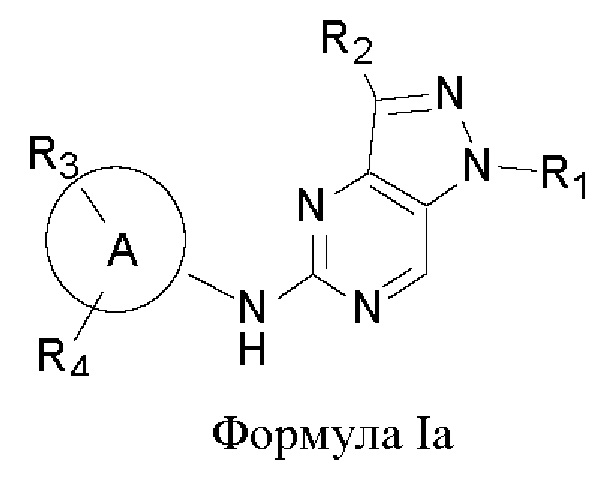
и их фармацевтически приемлемая соль, где R1, R2, R3, R4, кольцо А являются такими, как определено здесь.
В некоторых воплощениях соединения, предложенные здесь, имеют структуру формулы Ib
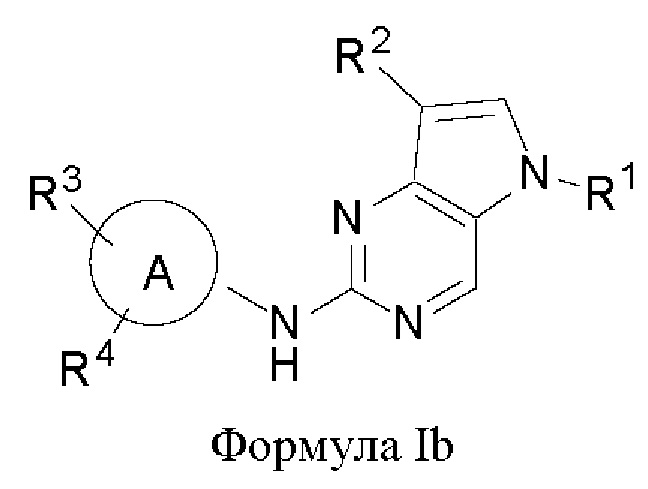
и их фармацевтически приемлемая соль, где R1, R2, R3, R4, кольцо А являются такими, как определено здесь.
В некоторых воплощениях соединения, предложенные здесь, имеют структуру формулы Ic
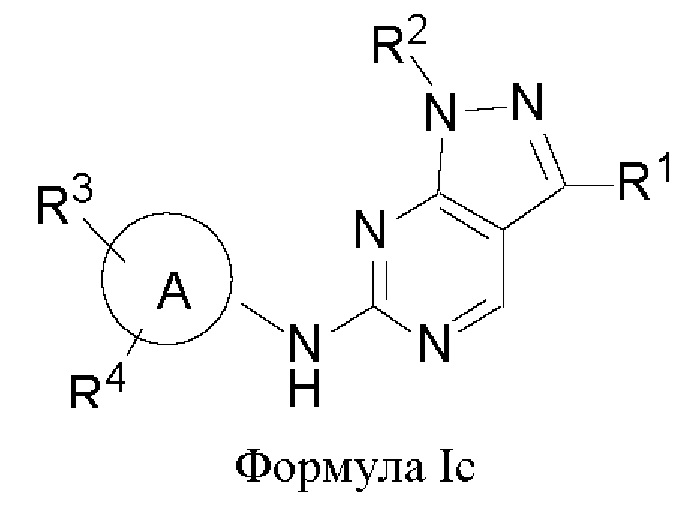
и их фармацевтически приемлемая соль, где R1, R2, R3, R4, кольцо А являются такими, как определено здесь.
В некоторых воплощениях соединения, предложенные здесь, имеют структуру формулы Id
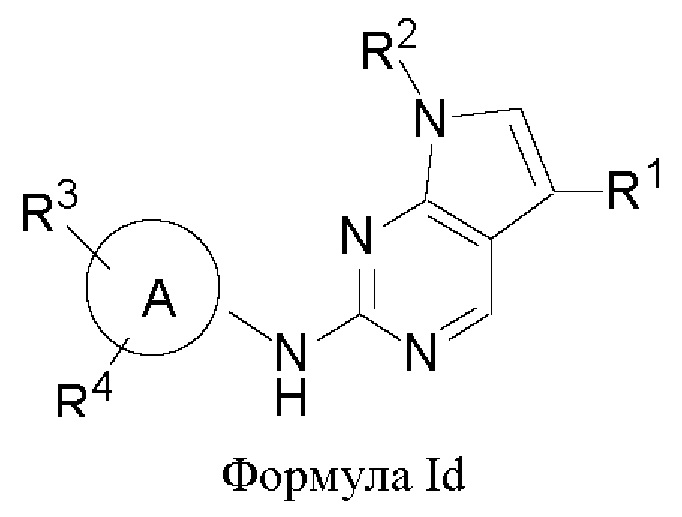
и их фармацевтически приемлемая соль, где R1, R2, R3, R4, кольцо А являются такими, как определено здесь.
В некоторых воплощениях соединения, предложенные здесь, имеют структуру формулы 1е
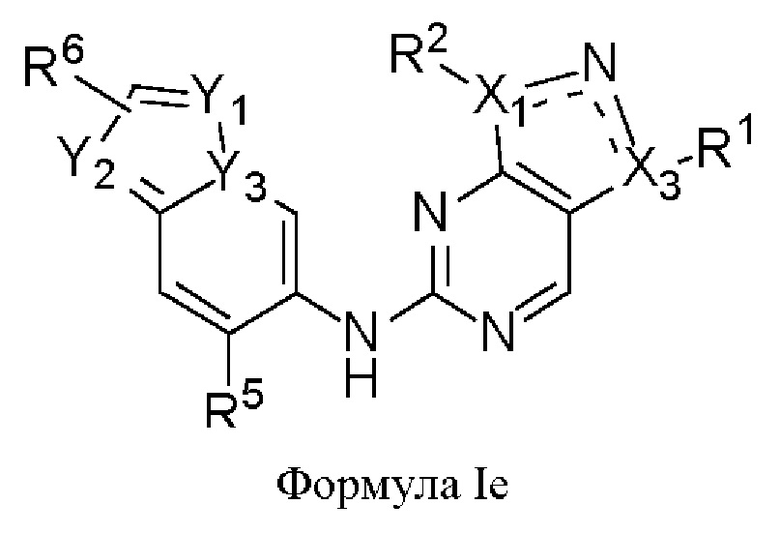
и их фармацевтически приемлемая соль,
где
один из X1 и Х3 представляет собой N, и другой представляет собой С, пунктирная линия  означает, что связь между X1 и N и между N и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и N и между N и Х3 представляет собой простую связь;
означает, что связь между X1 и N и между N и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и N и между N и Х3 представляет собой простую связь;
R1 представляет собой С1-3алкил,
R2 представляет собой циклопентил, циклогексанил, тетрагидропиранил или 8-оксабицикло[3.2.1]октан-3-ил, которые возможно могут быть моно- или независимо полизамещены галогеном или С1-3алкоксилом,
Y1, Y2 и Y3 каждый независимо представляет собой С или N, при условии, что по меньшей мере один из Y1, Y2 и Y3 представляет собой N;
R5 представляет собой галоген или С1-3алкил,
R6 представляет собой С1-3алкил.
В некоторых воплощениях R2 в формуле Ie представляет собой незамещенный циклопентил, циклогексанил, тетрагидропиранил или 8-оксабицикло[3.2.1]октан-3-ил, которые возможно могут быть моно- или независимо полизамещены галогеном или С1-3алкоксилом.
В некоторых воплощениях Y3 в формуле Ie представляет собой N, и по меньшей мере один из Y1 и Y2 представляет собой N.
В некоторых воплощениях R5 в формуле Ie представляет собой метил.
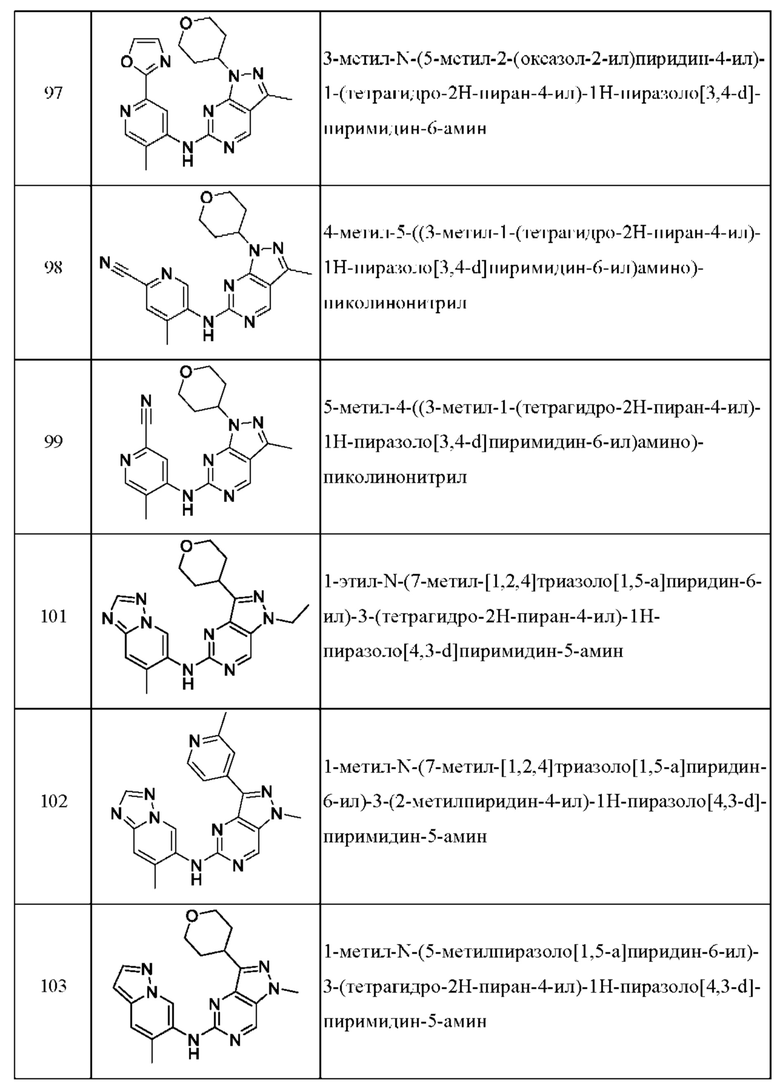
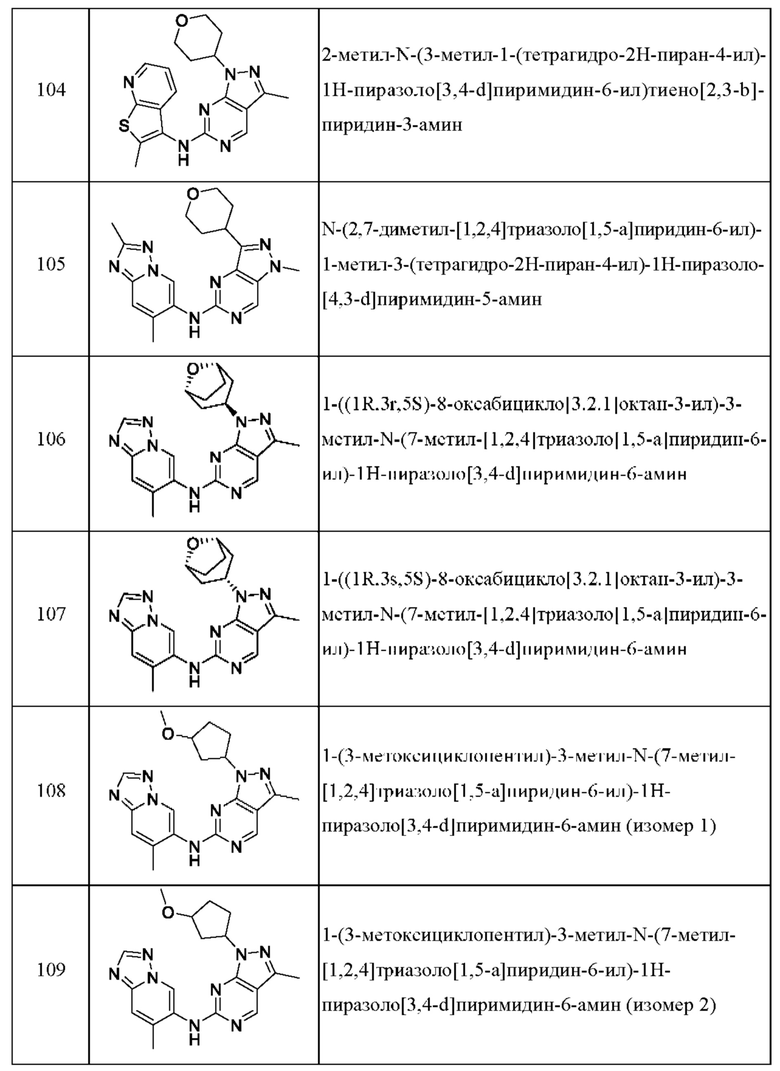
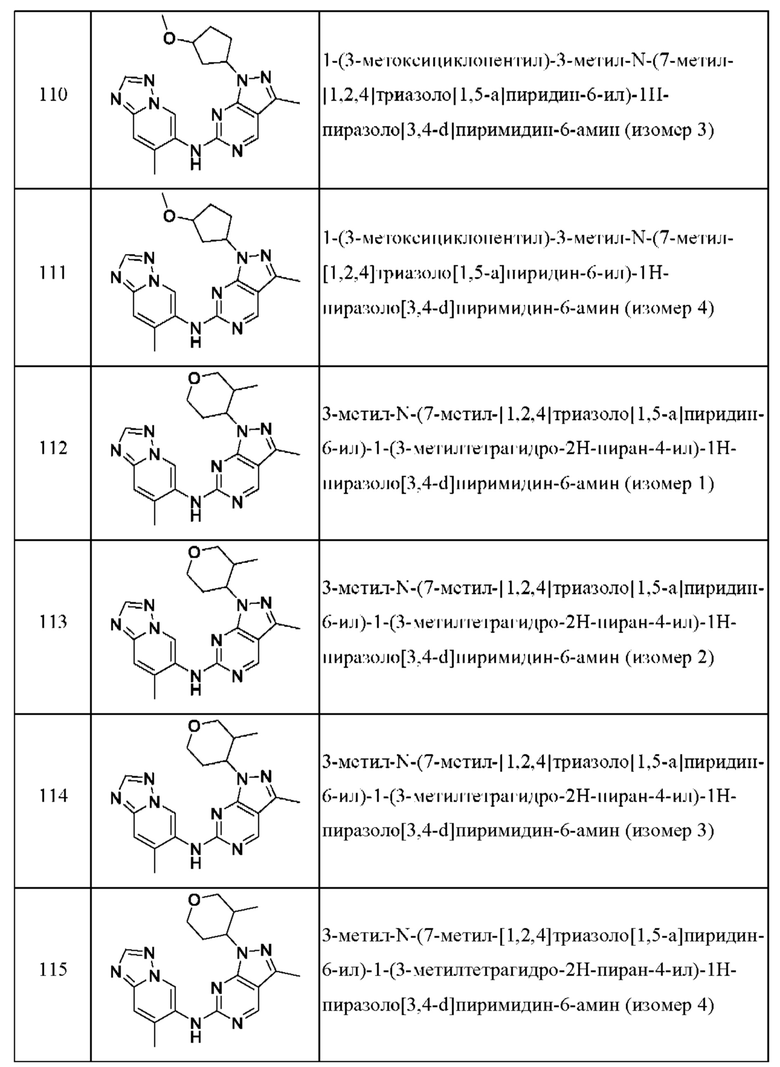
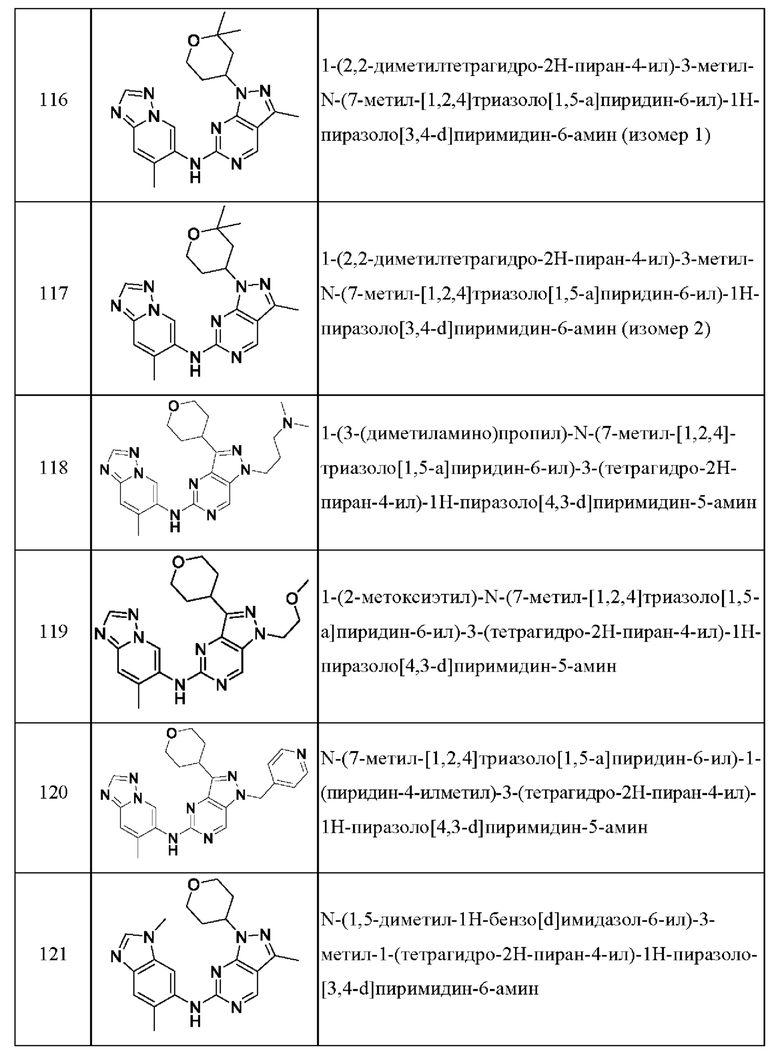
Соединения формулы (I) по примерам 1-149 представлены в таблице 1 ниже.
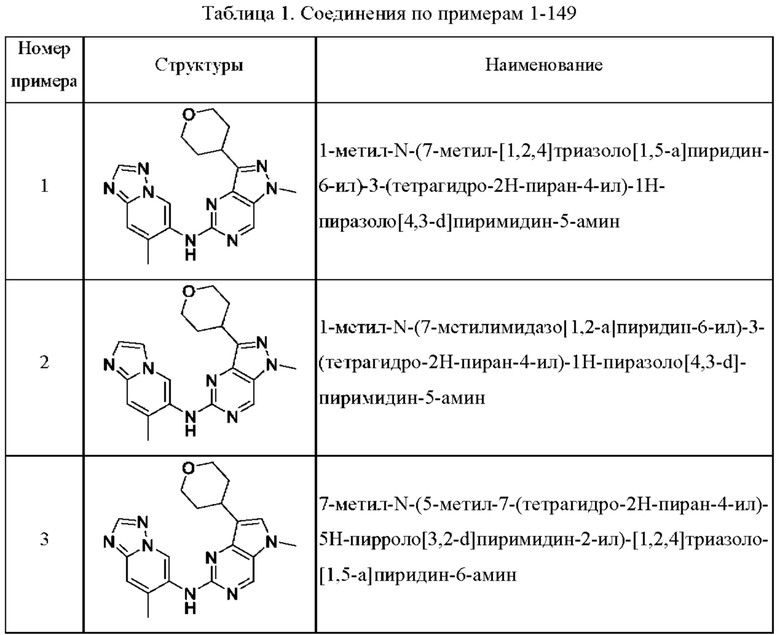
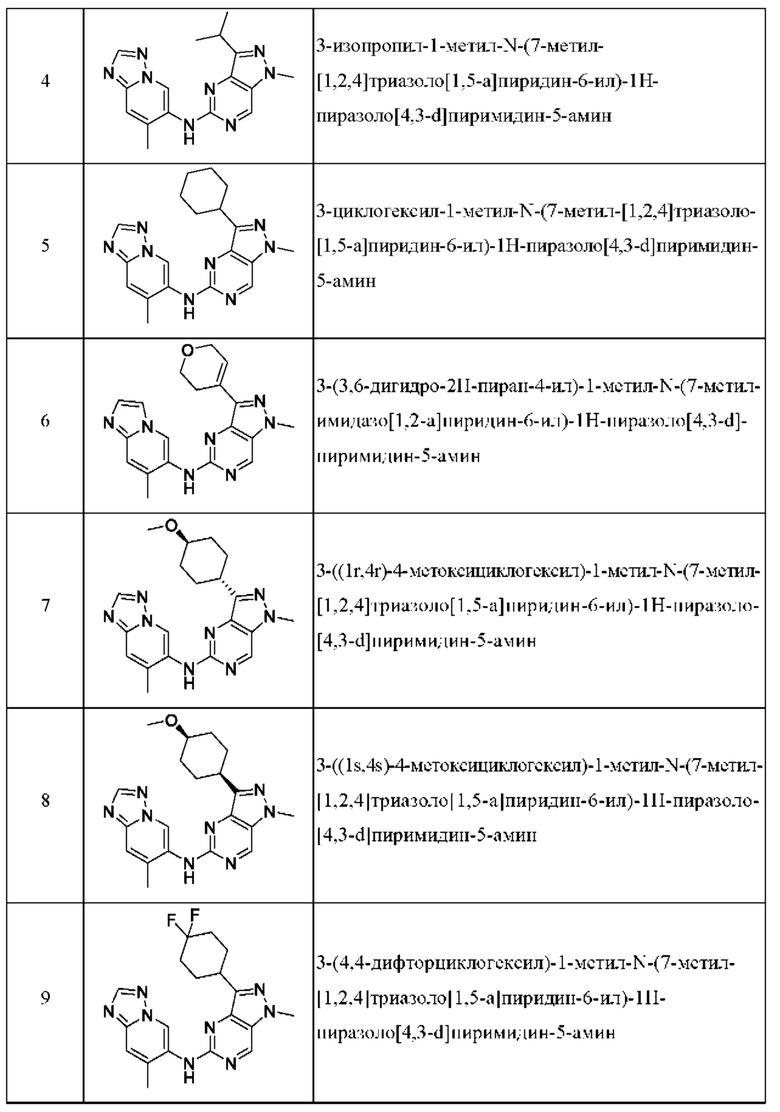
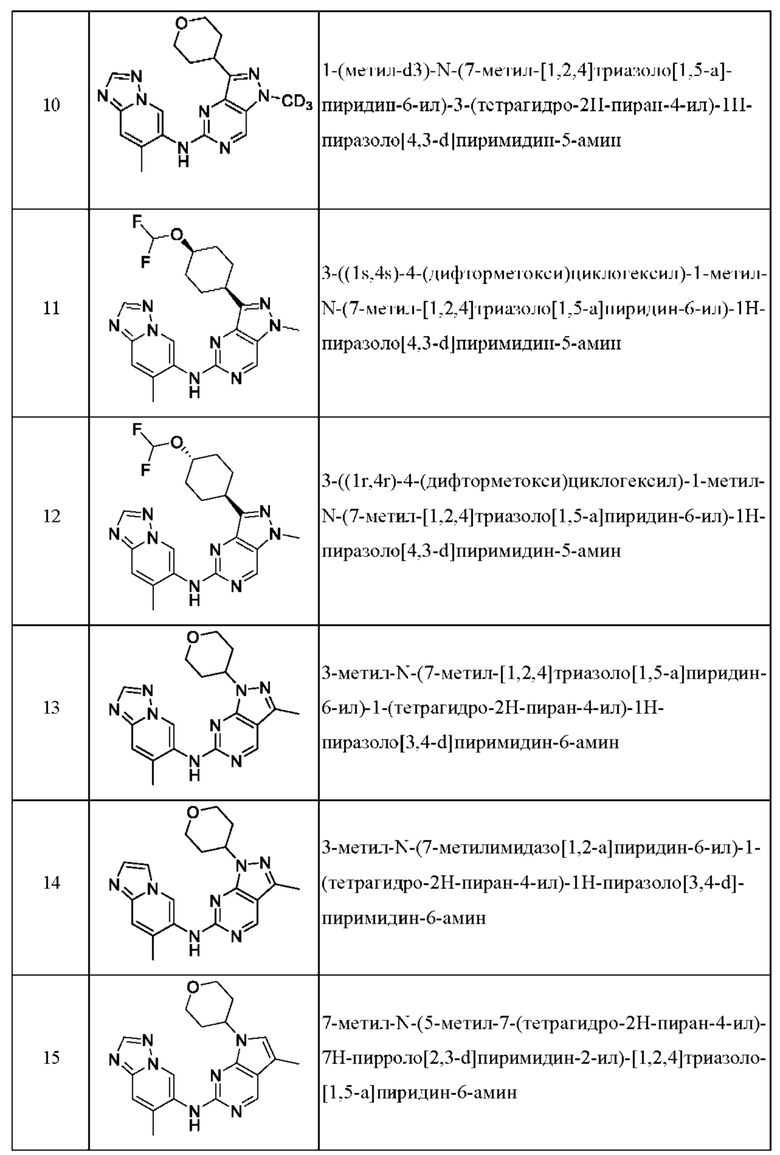
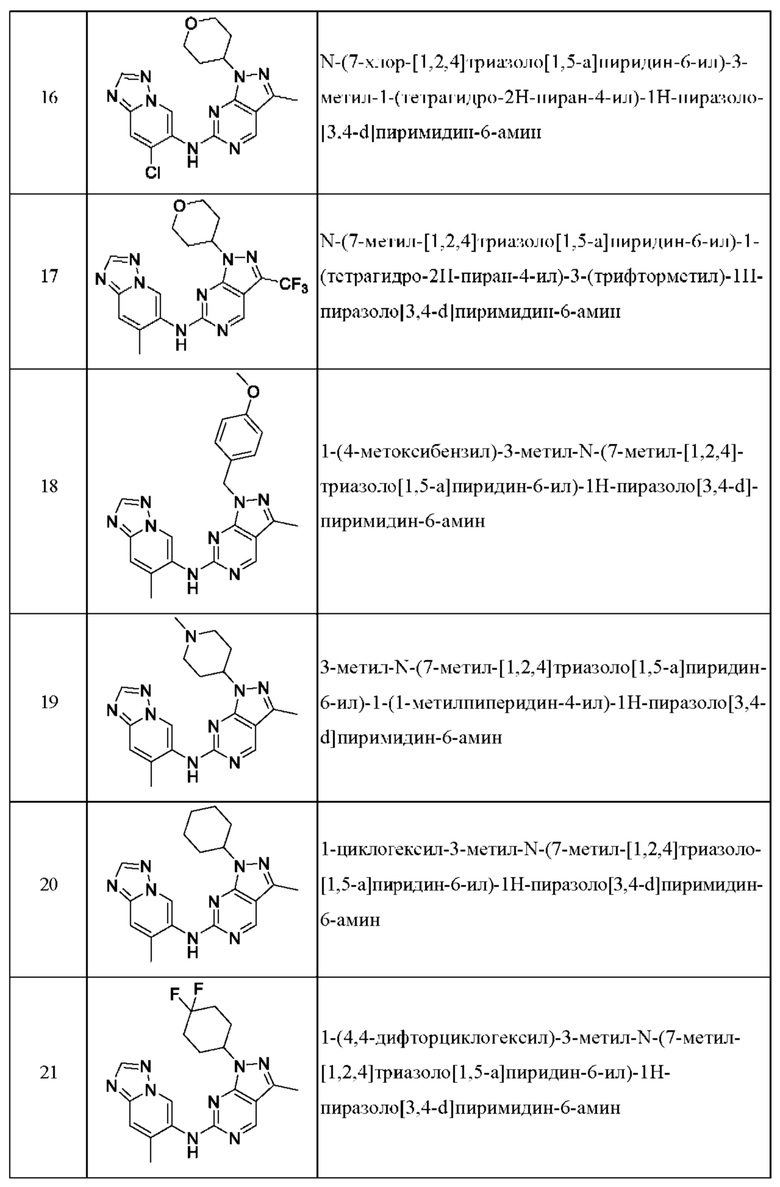
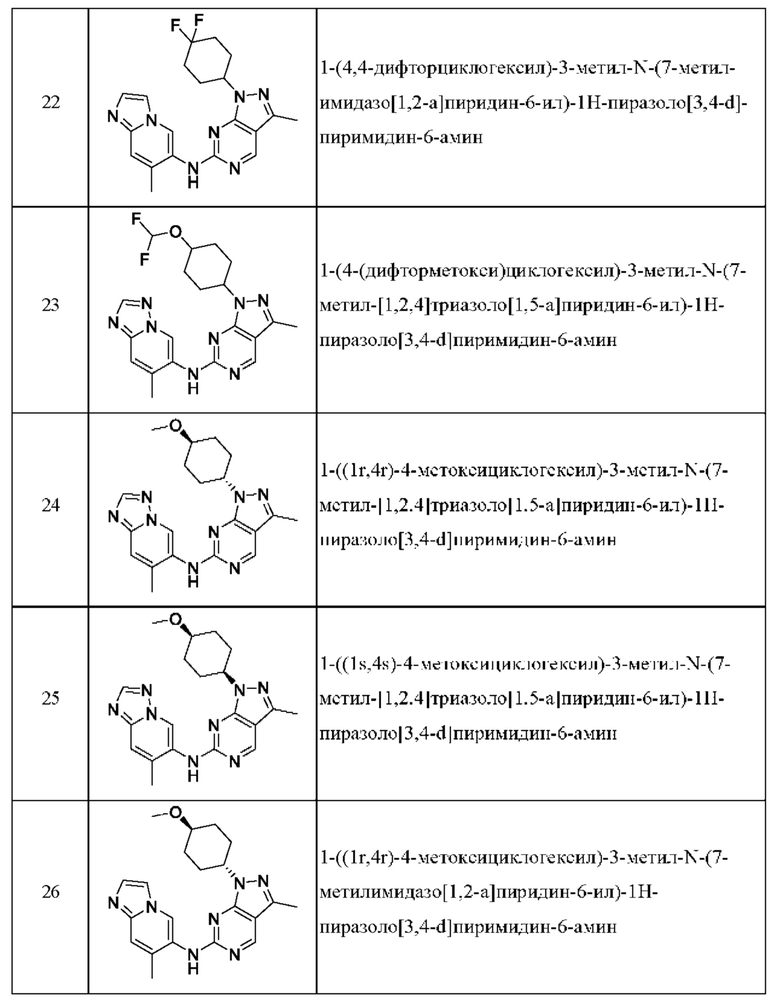
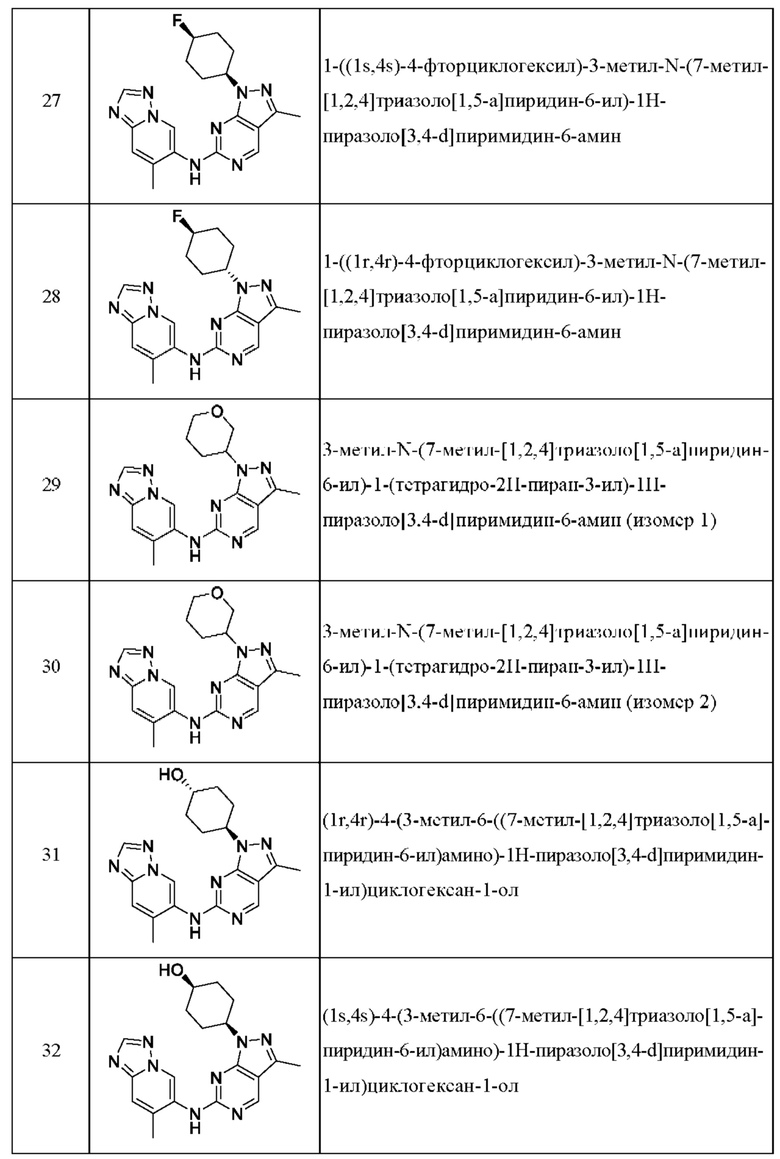
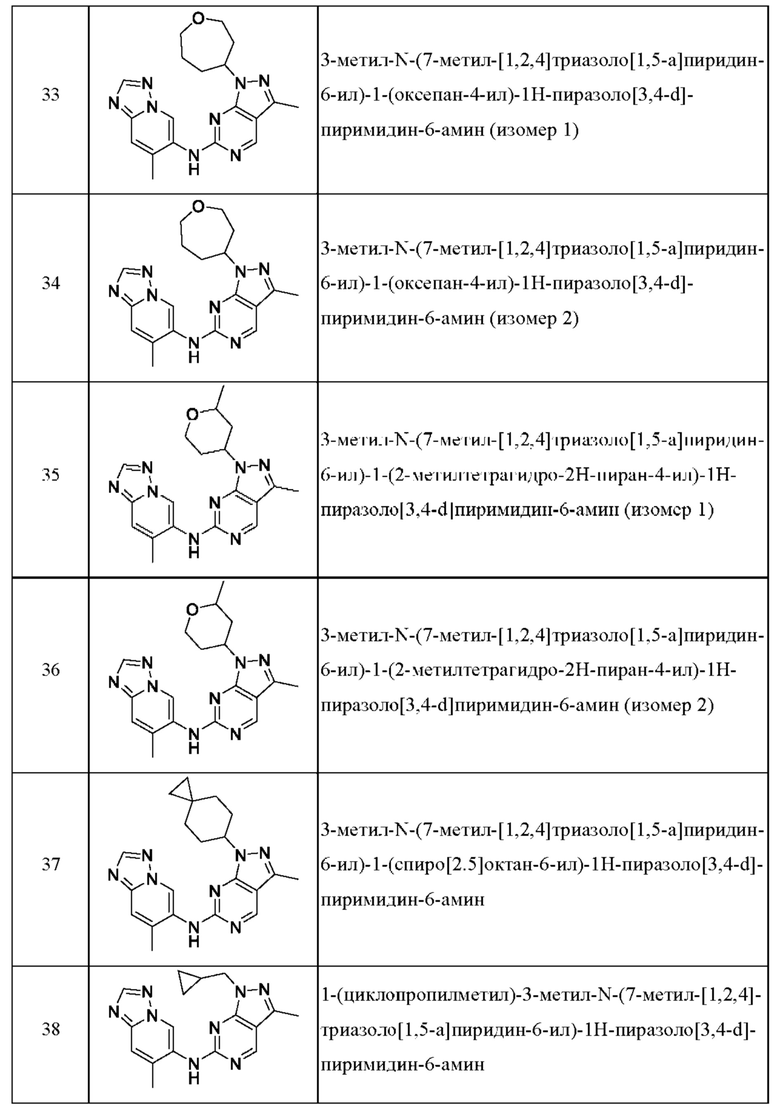
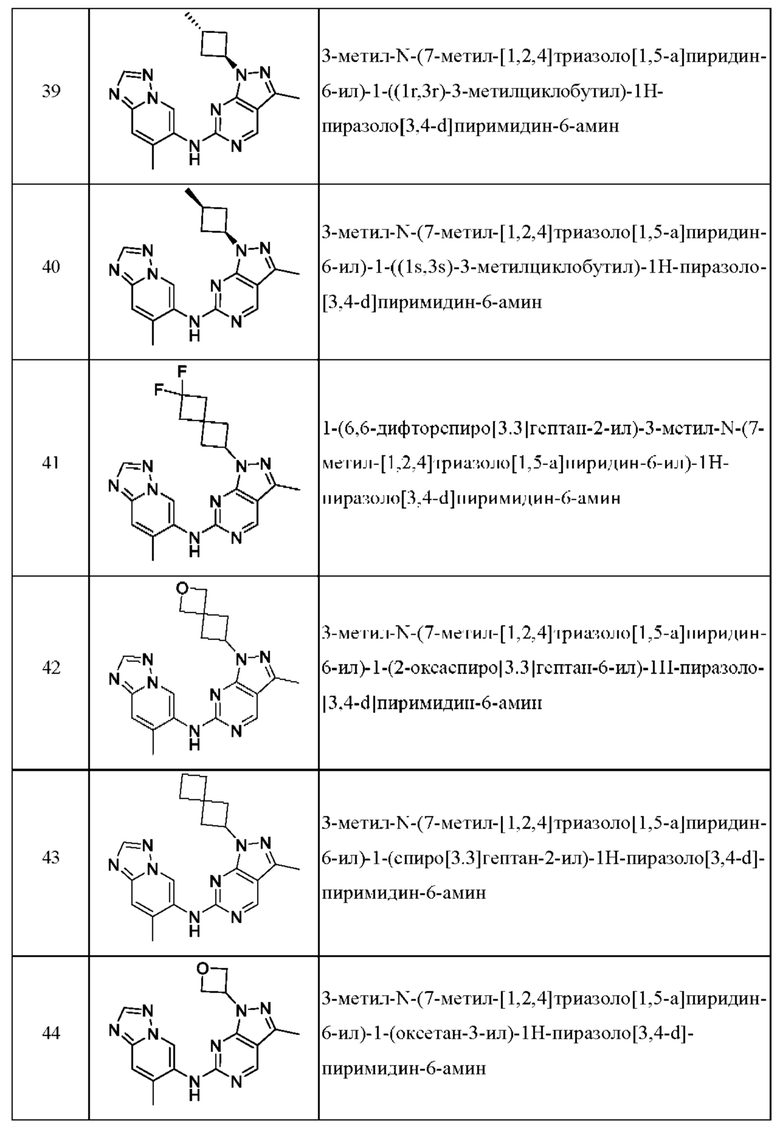
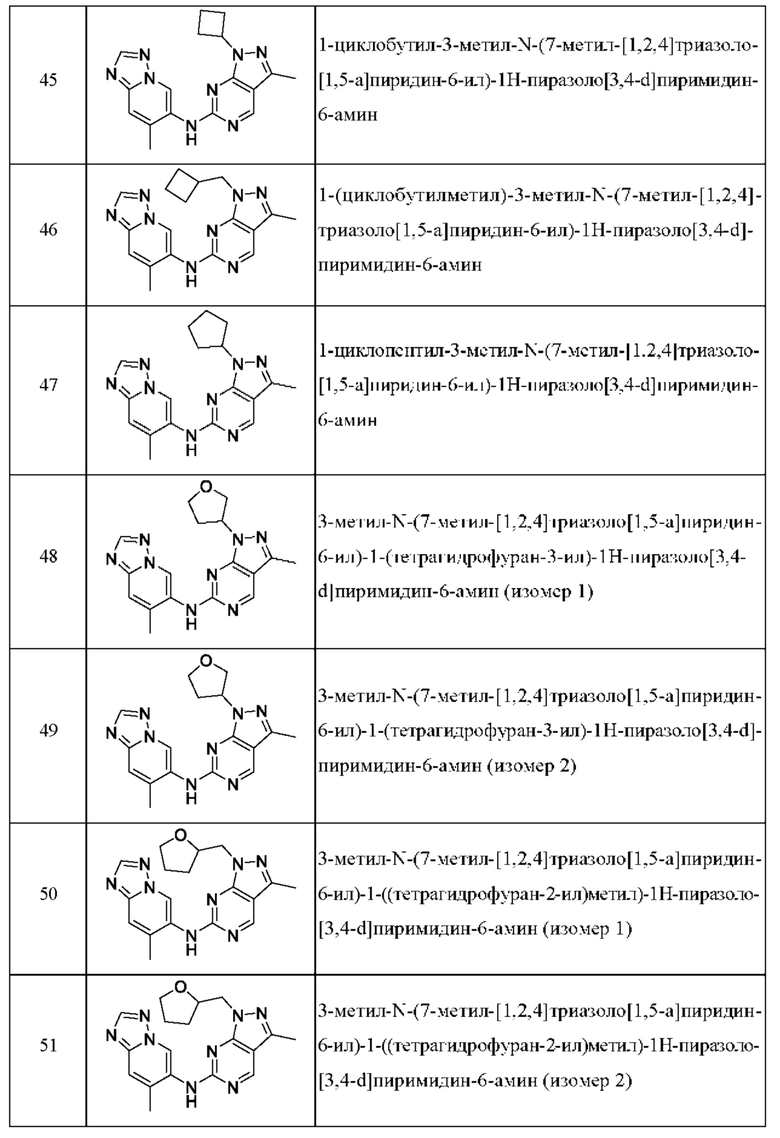
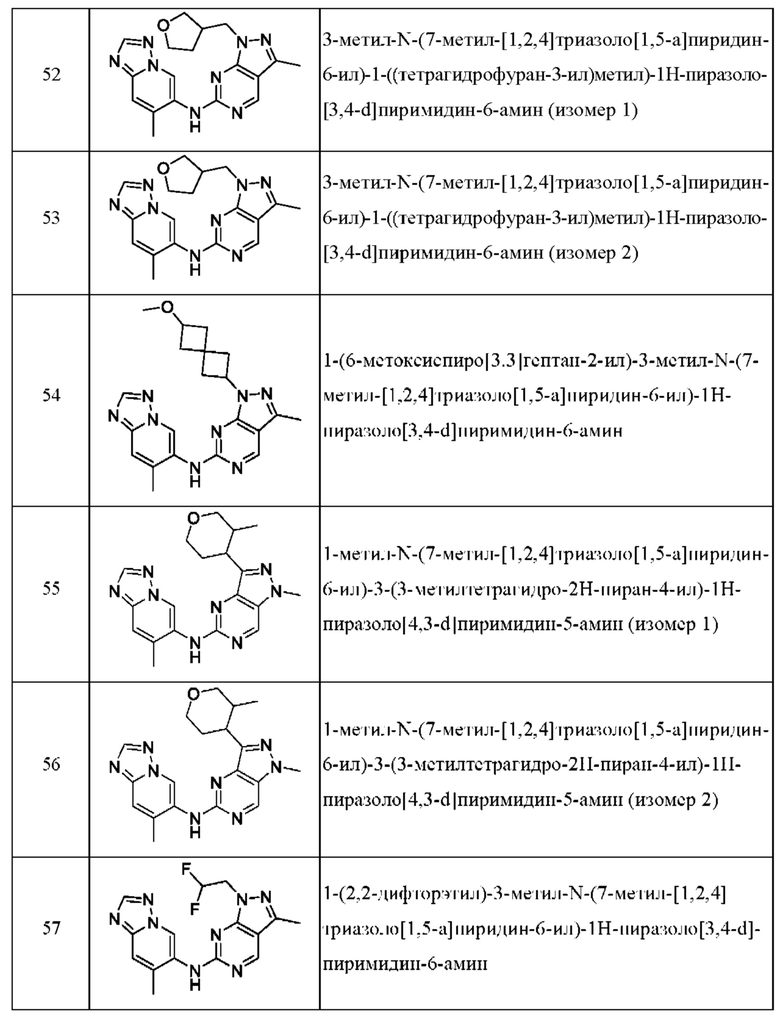
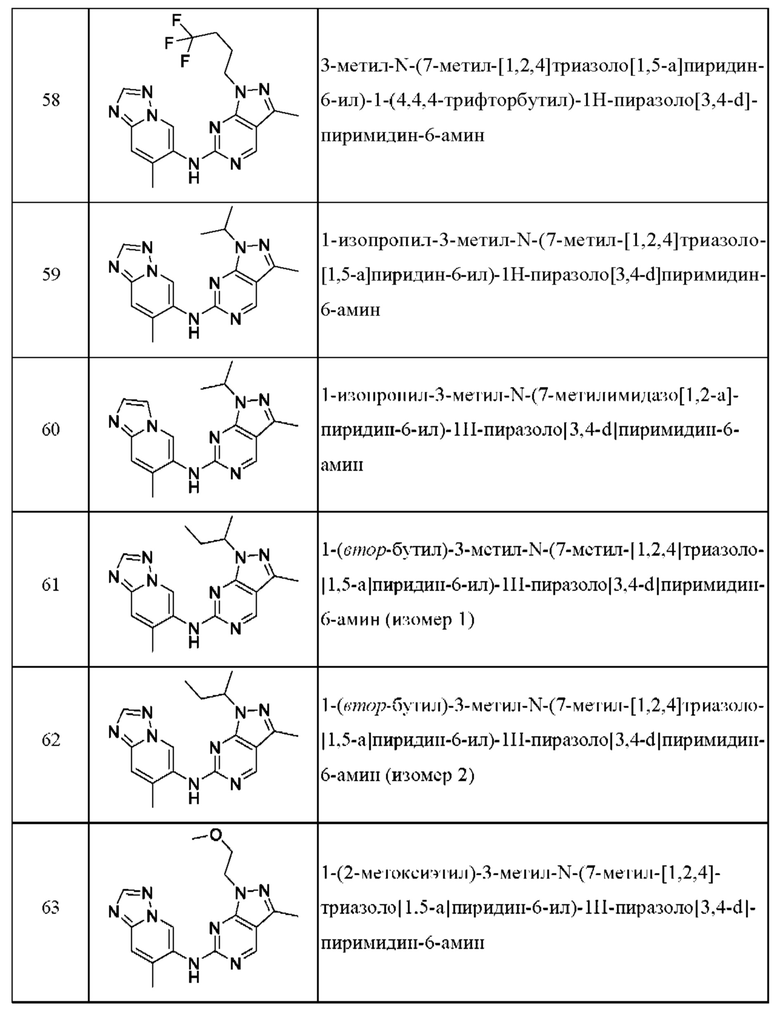
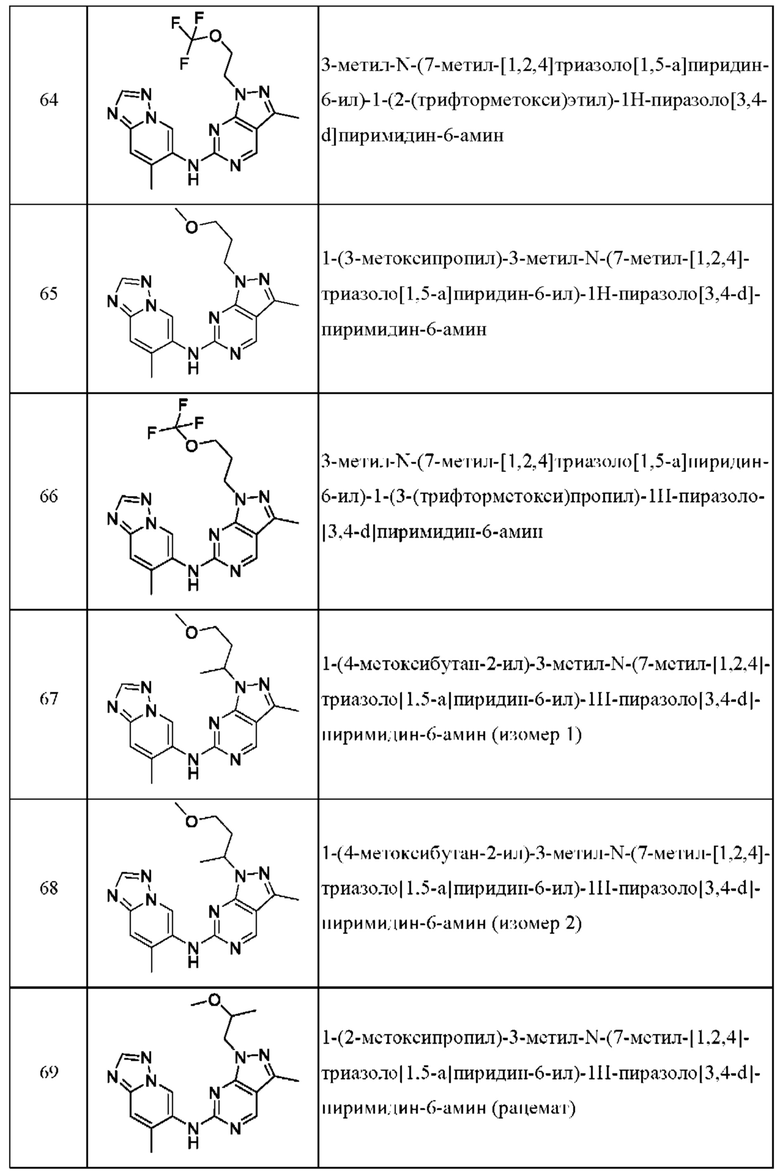
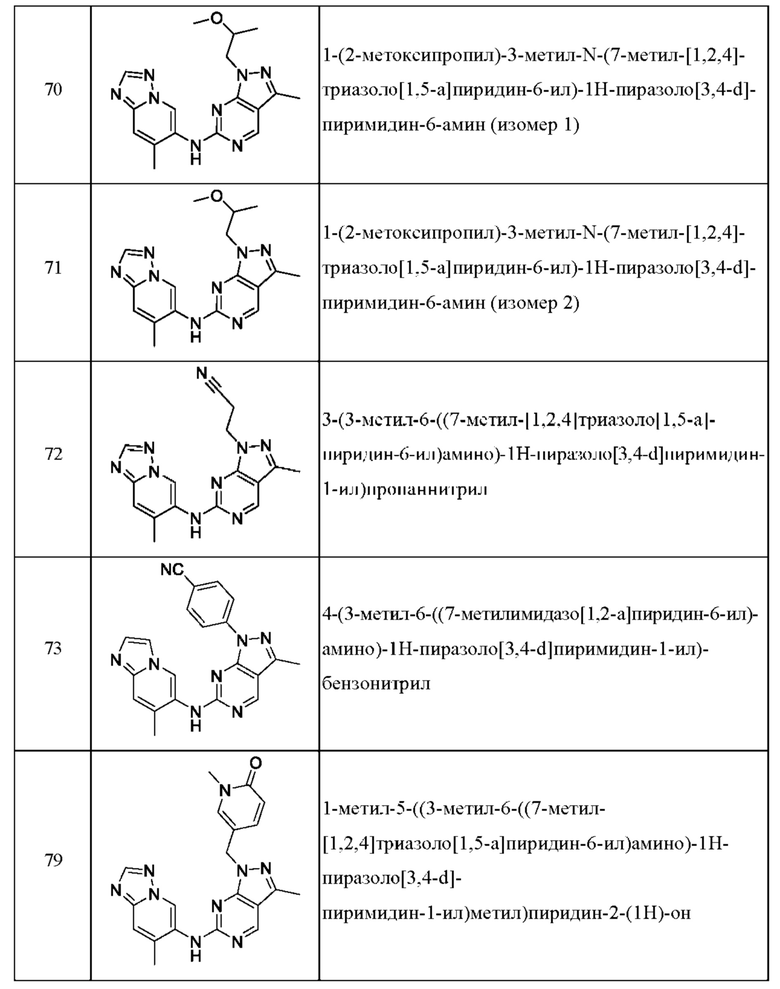
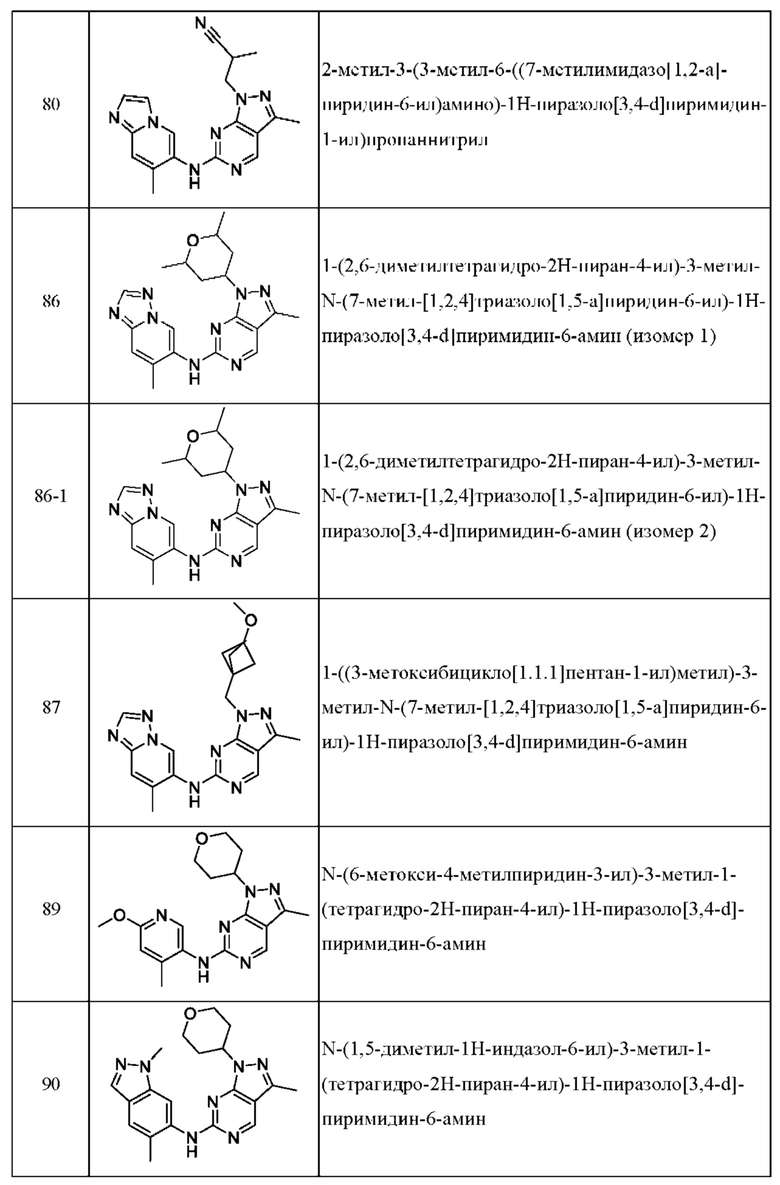
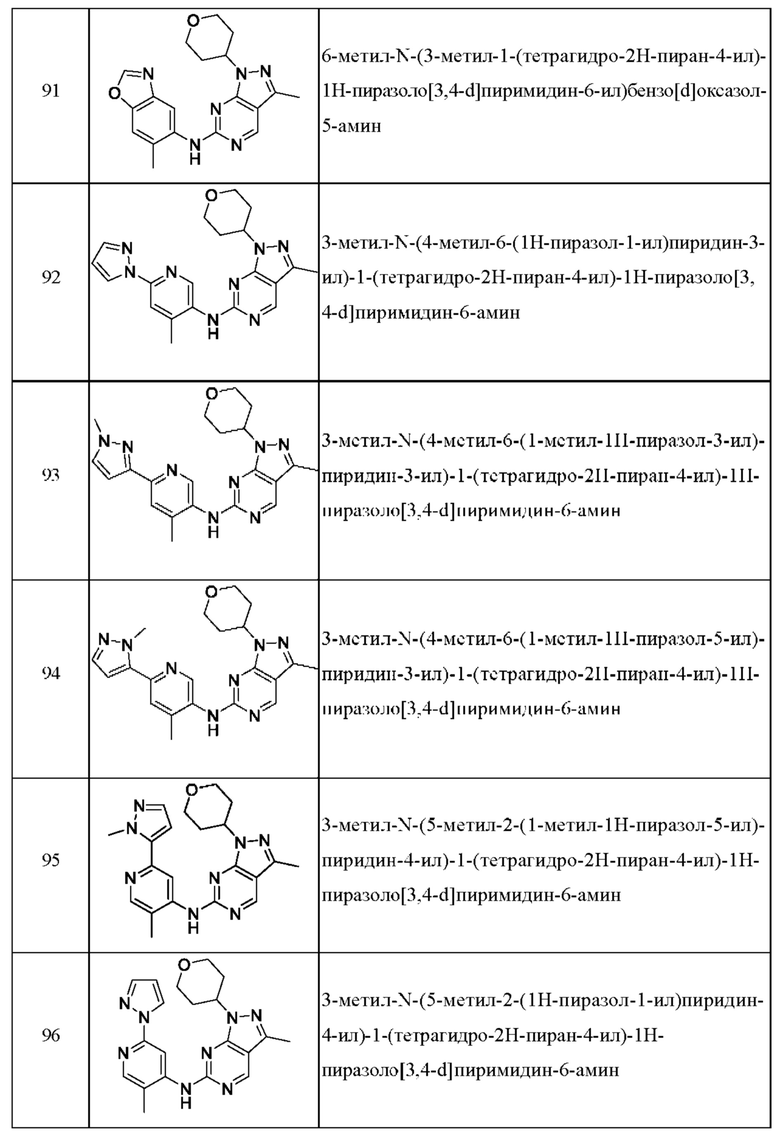
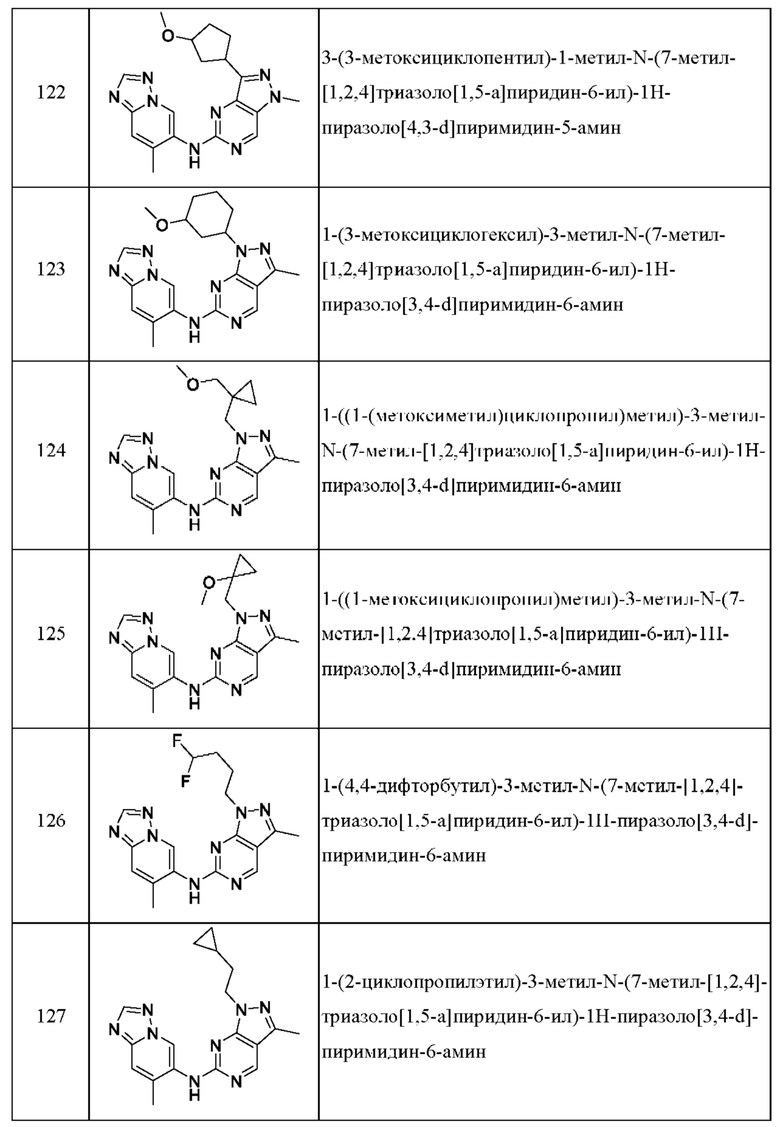
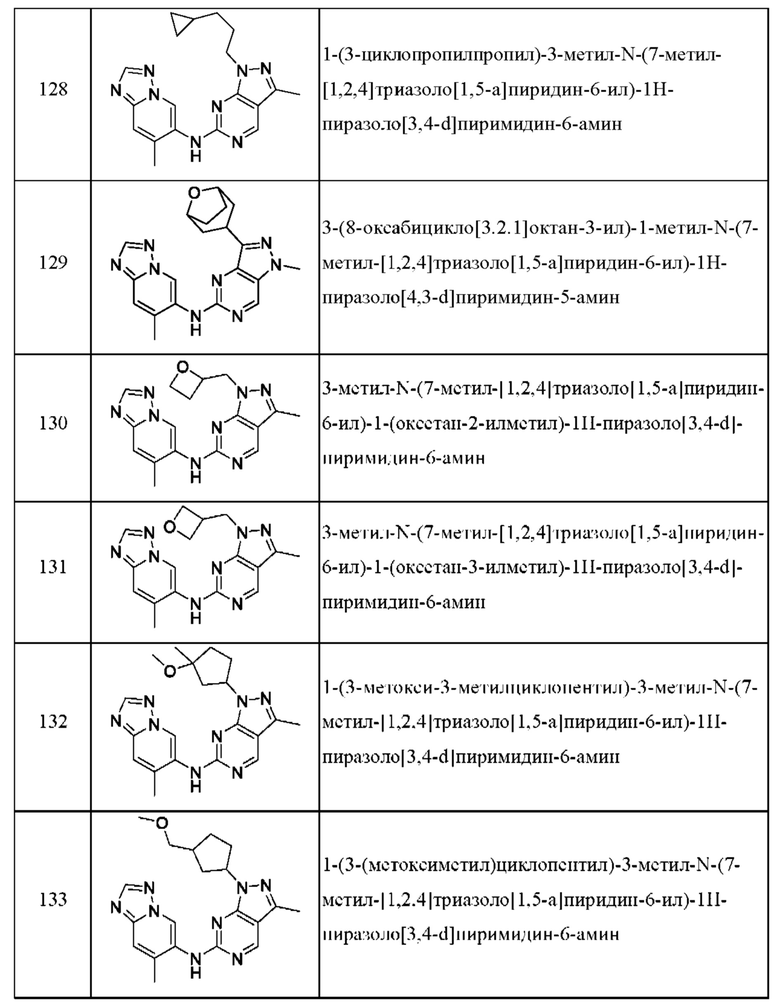
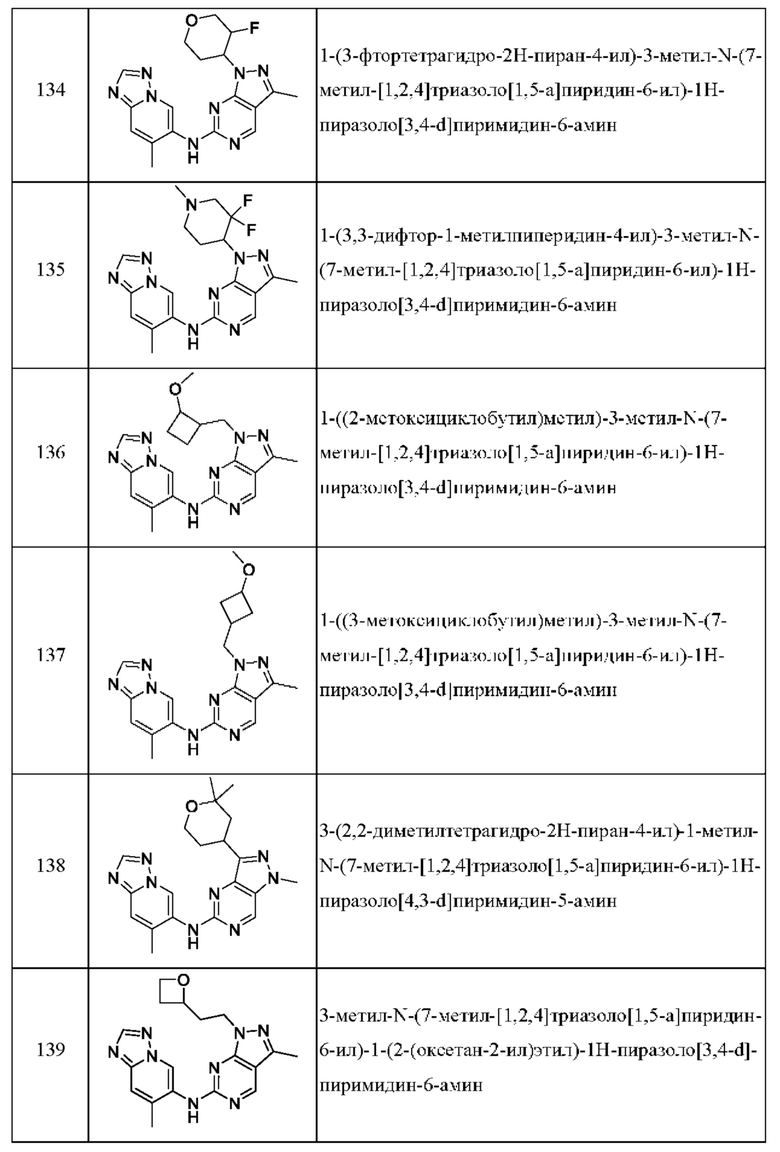
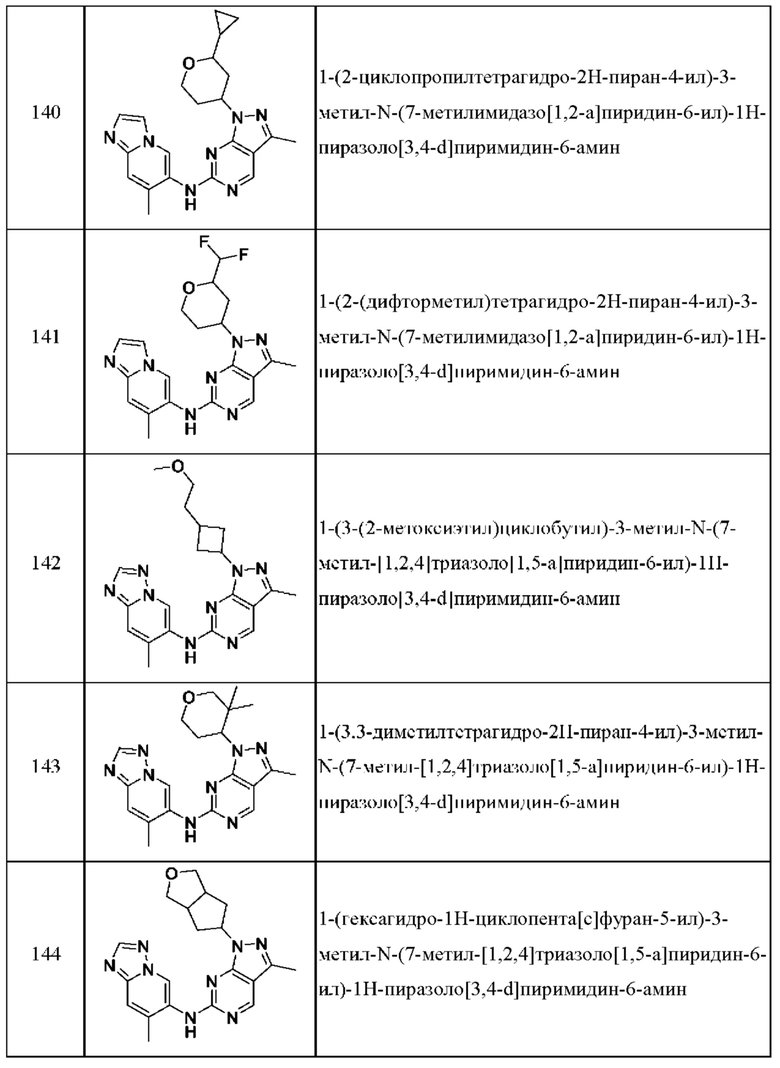
Следует отметить, что некоторые признаки настоящего изобретения, которые для лучшего понимания описаны в контексте отдельных воплощений, также могут быть представлены в комбинации в одном воплощении. И наоборот, различные признаки настоящего изобретения, которые для краткости описаны в контексте одного воплощения, также могут быть представлены отдельно или в любой подходящей подкомбинации.
В различных частях настоящего изобретения описаны связывающие заместители. В случаях, когда структура явно требует наличия связывающей группы, тогда под переменными Маркуша, перечисленными для этой группы, понимают связывающие группы. Например, если для структуры требуется связывающая группа и в определении группы Маркуша для этой переменной перечислен «алкил», то подразумевается, что «алкил» представляет собой связывающую алкиленовую группу.
Использованный здесь термин «замещенный», когда он относится к химической группе, означает, что эта химическая группа имеет один или более чем один атом водорода, который(е) удален(ы) и заменен(ы) заместителями. Использованный здесь термин «заместитель» имеет обычное значение, известное в данной области техники, и относится к химической группировке, которая ковалентно присоединена к родительской группе или, если целесообразно, конденсирована с ней. Использованный здесь термин «возможно замещенный» или «возможно… замещенный» означает, что химическая группа может не иметь заместителей (то есть быть незамещенной) или может иметь один или более чем один заместитель (то есть быть замещенной). Следует понимать, что замещение по данному атому ограничено валентностью.
Использованный здесь термин «Ci-j» обозначает диапазон числа атомов углерода, где i и j являются целыми числами, и диапазон числа атомов углерода включает конечные числа (то есть i и j) и каждое целое число между ними, и где j больше i. Например, С1-6 обозначает диапазон от одного до шести атомов углерода, включая один атом углерода, два атома углерода, три атома углерода, четыре атома углерода, пять атомов углерода и шесть атомов углерода. В некоторых воплощениях термин «С1-12» означает от 1 до 12, включая от 1 до 10, от 1 до 8, от 1 до 6, от 1 до 5, от 1 до 4, от 1 до 3 или от 1 до 2 атомов углерода.
Использованный здесь термин «алкил», независимо от того, используется ли он как часть другого термина или сам по себе, относится к насыщенной или ненасыщенной углеводородной цепи, при этом последняя может быть дополнительно подразделена на углеводородную цепь, имеющую по меньшей мере одну двойную или тройную связь (алкенил или алкинил). В некоторых воплощениях алкил относится к насыщенной углеводородной цепи. Упомянутая выше углеводородная цепь может быть линейной или разветвленной. Термин «Ci-jалкил» относится к алкилу, имеющему от i до j атомов углерода. Примеры насыщенной алкильной группы включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил высшие гомологи, такие как 2-метил-1-бутил, н-пентил, 3-пентил, н-гексил, 1,2,2-триметилпропил и тому подобное, но не ограничиваются ими. Примеры ненасыщенных алкильных групп включают этенил, н-пропенил, изопропенил, н-бутенил, втор-бутенил, этинил, пропин-1-ил, пропин-2-ил и тому подобное, но не ограничиваются ими. Примеры группы «С1-6алкил» включают метил, этил, пропил, изопропил, н-бутил, изобутил и трет-бутил, но не ограничиваются ими. Примеры группы «C1-3алкил» включают метил, этил, пропил и изопропил, но не ограничиваются ими.
Когда «алкил» представляет собой связывающую алкиленовую группу, примеры алкиленовых групп включают метилен, 1,1-этилен, 1,2-этилен, 1,1-пропилен, 1,2-пропилен, 1,3-пропилен, 2,2-пропилен, трет-бутанилен и тому подобное, но не ограничиваются ими.
Использованный здесь термин «амино» относится к группе формулы «-NH2».
Использованный здесь термин «карбамоил» относится к аминокарбонильной группе (то есть NH2-C(=O)-).
Использованный здесь термин «циано» относится к группе формулы «-C≡N».
Использованный здесь термин «галоген» относится к группам фтор, хлор, бром или йод.
Использованный здесь термин «гидроксил» относится к группе формулы «-ОН».
Использованный здесь термин «алкокси», независимо от того, используется ли он как часть другого термина или сам по себе, относится к группе формулы -О-алкил.
Термин «Ci-jалкокси» означает, что алкильная группировка алкоксигруппы имеет от i до j атомов углерода. Примеры алкоксигрупп включают метоксил, этоксил, пропоксил (например н-пропокси и изопропокси), трет-бутокси и тому подобное, но не ограничиваются ими. Примеры группы «C1-12алкоксил» представляют собой метоксил, этоксил и пропоксил.
Использованный здесь термин «гидроксиС1-12алкил» относится к группе формулы «-С1-12алкил-ОН», где алкильная группировка группы имеет от 1 до 12 атомов углерода, и одна или более чем одна гидроксильная группа может быть связана с любыми атомами углерода в алкильной группировке. В некоторых воплощениях «Ci-jалкил-ОН» имеет одну гидроксильную группу. Примерами группы «С1-12алкил-ОН» являются гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил и 1-гидроксиизопропил.
Использованный здесь термин «Ci-jгалогеналкил» относится к галогензамещенной (моно- или полизамещенной) Ci-j-алкильной группе. Примерами группы «С1-12галогеналкил» являются фторметил, дифторметил, трифторметил, фторэтил, дифторэтил, трифторэтил, хлорэтил и бромизопропил. Примером группы «дифторэтил» является 1,1-дифторэтил. Примерами группы «трифторэтил» являются 2,2,2-трифторэтил и 1,2,2-трифторэтил.
Примерами группы «Ci-jгалогеналкоксил» являются фторметоксил, дифторметоксил или трифторметоксил. Примерами группы «трифторэтокси» являются 2,2,2-трифторэтокси и 1,2,2-трифторэтокси.
Использованный здесь термин «арил» или «ароматический», независимо от того, используется ли он как часть другого термина или сам по себе, относится к кольцевой системе с чередующимися двойными и простыми связями между атомами, образующими кольца. В настоящем изобретении подразумевается, что термин «арил» или «ароматический» также включает псевдоароматический. Термин «псевдоароматический» относится к кольцевой системе, которая не является строго ароматической, но которая стабилизирована посредством делокализации электронов и ведет себя аналогично ароматическим кольцам. Арил или ароматическая группа может иметь моно- или поликольцо(а). Примеры арильных групп включают фенил, нафтил, тетрагидронафтил, инданил и тому подобное, но не ограничиваются ими.
Использованный здесь термин «гетероарил» относится к арилу, который содержит по меньшей мере один гетероатом, образующий кольцо, выбранный из О, S, N, Р и тому подобного. Гетероарил включает фурил, тиенил, пиридинил, триазинил, пиридил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, индолизинил, индолил, изоиндолил, индолинил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,4-оксадиазол-5-он, 1,2,3-триазолил, 1,3,4-тиадиазолил, пиридазинил, пиримидинил, пиразинил, хиназолинил, изохиназолинил, 1,3,5-триазинил, 1Н-тиено[2,3-с]пиразолил, тиено[2,3-b]фурил, 3Н-индолил, бензо[b]фуранил, бензо[b]тиофенил, 1H-индазолил, бензимидазолил, тетразолил, уридинил и цитозинил, но не ограничивается ими.
Использованный здесь термин «карбоциклил», независимо от того, используется ли он как часть другого термина или сам по себе, относится к любому кольцу, включая моно- или полициклическое кольцо(а) (например имеющее 2 или 3 конденсированных, мостиковых или спирокольца), в котором все атомы кольца представляют собой углерод и которое содержит по меньшей мере три атома углерода, образующих кольцо. Использованный здесь термин «спиро» кольца относится к кольцевым системам, в которых два кольца соединены одним общим атомом; термин «конденсированные» кольца относится к кольцевым системам, имеющим два кольца, имеющих два общих смежных атома; и термин «мостиковые» кольца относится к кольцевым системам с двумя кольцами, имеющими три или более общих атомов.
В некоторых воплощениях карбоциклил может содержать от 3 до 12 атомов углерода, образующих кольцо (то есть 3-12 атомов углерода), от 3 до 10 атомов углерода, образующих кольцо, от 3 до 9 атомов углерода, образующих кольцо, или от 3 до 8 атомов углерода, образующих кольцо. Карбоциклильные группы могут быть насыщенными, частично ненасыщенными или полностью ненасыщенными. В некоторых воплощениях карбоциклильная группа может быть насыщенной циклической алкильной группой. В некоторых воплощениях карбоциклильная группа может быть ненасыщенной циклической алкильной группой, которая содержит по меньшей мере одну двойную связь в своей кольцевой системе. В некоторых воплощениях ненасыщенная карбоциклильная группа может содержать одно или более чем одно ароматическое кольцо. В некоторых воплощениях одна или более чем одна образующая кольцо группа -СН2- насыщенного или ненасыщенного карбоциклила может быть заменена группой -С(O)-.
В некоторых воплощениях карбоциклильная группа представляет собой моноциклическую алкильную группу. В некоторых воплощениях карбоциклическая группа представляет собой насыщенную моноциклическую алкильную группу. Примеры насыщенных моноциклических алкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил и тому подобное, но не ограничиваются ими.
«3-8-членный насыщенный или ненасыщенный карбоциклил» представляет собой насыщенную, частично ненасыщенную или полностью ненасыщенную моно- или полициклическую кольцевую систему, содержащую от 3 до 8, от 3 до 6 или от 5 до 8 атомов углерода, образующих кольцо, соответственно, где одна или более чем одна образующая кольцо группа -СН2- возможно может быть заменена группой -С(O)-.
Примерами группы «3-8-членный насыщенный или ненасыщенный карбоциклил» являются С3-6циклоалкил, циклогексил, циклогексенил, циклопентил, фенил, нафтил и бицикло[1.1.1]пентан-1-ил. Примерами группы «С3-8циклоалкил» являются циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Термин «С3-8циклоалкоксил» относится к группе формулы «С3-8циклоалкил-О-».
Использованный здесь термин «гетероциклил» относится к карбоциклической группе, в которой один или более чем один (например 1, 2 или 3) кольцевой атом заменен гетероатомами, которые включают О, S, N, Р и тому подобное, но не ограничиваются ими. В некоторых воплощениях гетероциклил представляет собой насыщенный гетероциклил. В некоторых воплощениях гетероциклил представляет собой ненасыщенный гетероциклил, имеющий одну или более чем одну двойную связь в кольцевой системе. В некоторых воплощениях гетероциклил представляет собой частично ненасыщенный гетероциклил. В некоторых воплощениях гетероциклил представляет собой полностью ненасыщенный гетероциклил. В некоторых воплощениях ненасыщенная гетероциклильная группа может содержать одно или более чем одно ароматическое кольцо. В некоторых воплощениях одна или более чем одна образующая кольцо группа -CH2- гетероциклила возможно может быть заменена группой -С(O)-, -S-, -S(O)- или -S(O)2. В некоторых воплощениях, где гетероциклил содержит серу в своей кольцевой системе, указанный атом серы, образующий кольцо, возможно может быть окислен с образованием S-оксидов. В некоторых воплощениях гетероциклил связан с другой частью соединения посредством образующего его кольцо атома углерода. В некоторых воплощениях гетероциклил связан с другой частью соединения посредством образующего его кольцо атома азота.
В некоторых воплощениях 3-8-членный насыщенный или ненасыщенный моно-или полициклический гетероциклил содержит 1, 2 или 3 гетероатома, выбранных из N, О или S.
«3-8-членный насыщенный или ненасыщенный гетероциклил» представляет собой насыщенную, частично ненасыщенную или полностью ненасыщенную моно- или полициклическую кольцевую (например имеющую 2 или 3 конденсированных, мостиковых или спирокольца) систему, содержащую от 3 до 8 образующих кольцо атомов соответственно, из которых по меньшей мере один образующий кольцо атом выбран из азота, серы или кислорода, которая, если не оговорено особо, может быть связана с другой частью соединения посредством образующего ее кольцо атома углерода или азота, где одна или более чем одна образующая кольцо группа -СН2-насыщенного или ненасыщенного карбоциклила может быть заменена группой -С(O)-, -S-, -S(O)- или -S(O)2-, и где в случае, когда гетероциклил содержит серу в своей кольцевой системе, указанный кольцевой атом серы возможно может быть окислен с образованием S-оксидов.
Примеры моноциклических гетероциклильных групп включают оксетанил, пиранил, 1,1-диоксотиетанилпирролидил, тетрагидрофурил, тетрагидротиенил, пирролил, фуранил, тиенил, пиразолил, имидазолил, триазолил, оксазолил, тиазолил, пиперидил, пиперидил, пиперазинил, морфолинил, пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил, пиридонил, пиримидонил, пиразинонил, пиримидонил, пиридазонил, триазинонил и тому подобное, но не ограничиваются ими.
Примеры спирогетероциклила включают спиропиранил, спирооксазинил и тому подобное, но не ограничиваются ими. Примеры конденсированного гетероциклила включают конденсированное с фенилом кольцо или конденсированное с пиридинилом кольцо, такое как хинолинильная, изохинолинильная, хиноксалинильная, хинолизинильная, хиназолинильная, азаиндолизинильная, птеридинильная, хроменильная, изохроменильная, индолильная, изоиндолильная, индолизинильная, индазолильная, пуринильная, бензофуранильная, изобензофуранильная, бензимидазолильная, бензотиенильная, бензотиазолильная, карбазолильная, феназинильная, фенотиазинильная, фенантридинильная, имидазо[1,2-а]пиридинильная, [1,2,4]триазоло[4,3-а]пиридинильная, [1,2,3]триазоло[4,3-а]пиридинильная группы и тому подобное, но не ограничиваются ими. Примеры мостикового гетероциклила включают морфанил, гексаметилентетраминил, 8-аза-бицикло[3.2.1]октан, 1-аза-бицикло[2.2.2]октан, 1,4-диазабицикло[2.2.2]октан (DABCO) и тому подобное, но не ограничиваются ими.
Подразумевается, что «соединение» по настоящему изобретению включает все стереоизомеры, геометрические изомеры и таутомеры изображенных структур, если не оговорено особо.
Термин «стереоизомер» относится к любой из различных стереоизомерных конфигураций (например к энантиомерам, диастереомерам и рацематам) асимметрического соединения (например к тем, которые имеют один или более чем один асимметрически замещенный атом углерода или «асимметрический центр»). Соединения по настоящему изобретению, которые содержат асимметрические центры, могут быть выделены в оптически активных (энантиомеры или диастереомеры) или оптически неактивных (рацемических) формах. Термин «энантиомер» включает пары стереоизомеров, которые являются неналагающимися зеркальными изображениями Друг Друга. Смесь пары энантиомеров в соотношении 1:1 представляет собой «рацемическую смесь». Термины «диастереомеры» или «диастереоизомеры» включают стереоизомеры, которые имеют по меньшей мере два асимметрических атома, но не являются зеркальными отражениями друг друга. Некоторые соединения, содержащие один или более чем один асимметрический центр, могут иметь энантиомеры, диастереомеры или другие стереоизомерные формы, которые могут быть определены на основании абсолютной конфигурации как (R)- или (S)- в каждом асимметрическом центре согласно R-S системе Кана-Ингольда-Прелога. Разделенные соединения, абсолютная конфигурация которых неизвестна, могут быть обозначены с использованием термина «или» по асимметрическому центру. Способы получения оптически активных форм из рацемических смесей, такие как разделение посредством ВЭЖХ (высокоэффективная жидкостная хроматография) или стереоселективный синтез, известны в данной области техники.
Термины «геометрические изомеры» или «цис- и транс-изомеры» относятся к соединениям с одинаковой формулой, функциональные группы которых повернуты в разной ориентации в трехмерном пространстве.
Термин «таутомеры» включает прототропные таутомеры, которые представляют собой изомерные состояния протонирования соединений, имеющих одну и ту же формулу, и общий заряд. Примеры прототропных таутомеров включают пары кетон-енол, пары амид-имидовая кислота, пары лактам-лактим, пары енамин-имин и кольцевые формы, где протон может занимать два или более положений гетероциклической системы, например 1Н- и 3Н-имидазол, 1Н-, 2Н- и 4Н- 1,2,4-триазол, 1Н- и 2Н-изоиндол и 1Н- и 2Н-пиразол, но не ограничиваются ими. Таутомеры могут находиться в равновесии или быть стерически заблокированными в одной форме посредством соответствующего замещения. Подразумевается, что соединения по настоящему изобретению, идентифицированные по названию или структуре как одна конкретная таутомерная форма, включают другие таутомерные формы, если не оговорено особо.
Подразумевается также, что «соединение» по настоящему изобретению также включает все изотопы атомов в соединениях. Изотопы атома включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, подразумевается, если не оговорено особо, что водород, углерод, азот, кислород, фосфор, сера, фтор, хлор, бром или йод в «соединении» по настоящему изобретению также включают изотопы, такие как: 1H, 2H, 3H, 11C, 12С, 13С, 14С, 14N, 15N, 16О, 17О, 18О, 31Р, 32Р, 32S, 33S, 34S, 36S, 17F, 19F, 35Cl, 37Cl, 79Br, 81Br, 127I и 131I, но не ограничиваются ими. В некоторых воплощениях водород включает протий, дейтерий и тритий. В некоторых воплощениях термин «замещенный дейтерием» или «дейтерий-замещенный» означает замену другой изоформы водорода (например протия) в химической группе дейтерием. В некоторых воплощениях углерод включает 12С и 13С. В некоторых воплощениях «соединение» по настоящему изобретению включает только изотопы водорода в соединении. В некоторых воплощениях «соединение» по настоящему изобретению включает только распространенные в природе изотопы атомов.
Также следует понимать, что «соединение» по настоящему изобретению может существовать в сольватированных, а также в несольватированных формах, таких как, например, гидратированные формы, твердые формы, и подразумевается, что настоящее изобретение включает все такие сольватированные и несольватированные формы.
Кроме того, следует понимать, что «соединение» по настоящему изобретению может существовать в виде фармацевтически приемлемых солей.
Использованный здесь термин «фармацевтически приемлемый» относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримы с разумным соотношением польза/риск. В некоторых воплощениях соединения, вещества, композиции и/или лекарственные формы, которые являются фармацевтически приемлемыми, относятся к тем, которые одобрены регулирующим органом (таким как Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов Китая или Европейское агентство лекарственных средств) или перечислены в общепризнанной фармакопее (такой как Фармакопея США, Китайская фармакопея или Европейская фармакопея) для применения у животных и, более конкретно, у людей.
Использованный здесь термин «фармацевтически приемлемые соли» относится к производным соединений по настоящему изобретению, в которых исходное соединение модифицировано путем превращения существующей кислотной группировки (например карбоксила и тому подобного) или основной группировки (например амина, щелочи и тому подобного) в его солевую форму. Во многих случаях соединения по настоящему изобретению способны образовывать кислые и/или основные соли благодаря присутствию амино- и/или карбоксильных групп или подобных им групп. Фармацевтически приемлемые соли представляют собой соли кислот и/или оснований, которые сохраняют биологическую эффективность и свойства исходного соединения, которые обычно не являются нежелательными с биологической или иной точки зрения. Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению включают, например, соль присоединения кислоты, которая может быть образована, например, из неорганической кислоты (например соляной, бромистоводородной, серной, азотной, фосфорной кислоты и тому подобных) или органической кислоты (например муравьиной, уксусной, пропионовой, гликолевой, щавелевой, малеиновой, малоновой, янтарной, фумаровой, винной, тримезиновой, лимонной, молочной, фенилуксусной, бензойной, миндальной, метансульфоновой, нафталиндисульфоновой, этансульфоновой, толуолсульфоновой, трифторуксусной, салициловой, сульфосалициловой кислот и тому подобных). В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль муравьиной кислоты. В некоторых воплощениях фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой соль TFA (трифторуксусной кислоты).
Подходящие фармацевтически приемлемые соли соединения по настоящему изобретению также включают, например, соль присоединения основания, которая может быть образована, например, из неорганических оснований (например солей и гидроксида натрия, калия, аммония, и карбонатов, бикарбонатов металлов I - XII групп периодической таблицы, таких как кальций, магний, железо, серебро, цинк, медь и тому подобных) или органических оснований (например первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов, основных ионообменных смол и тому подобных). Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин, но не ограничиваются ими. Специалистам в данной области техники понятно, что также возможно добавление кислот или оснований для образования солей присоединения кислот/оснований, отличных от показанных в примерах. Перечни дополнительных подходящих солей можно найти, например в "Remington's Pharmaceutical Sciences", 20th ed.. Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). В некоторых воплощениях подходящие фармацевтически приемлемые соли соединения по настоящему изобретению представляют собой соль неорганических оснований.
Настоящее изобретение также включает активные промежуточные соединения, активные метаболиты и пролекарства соединений по настоящему изобретению. Использованный здесь термин «активное промежуточное соединение» относится к промежуточному соединению в процессе синтеза, которое проявляет такую же или по существу такую же биологическую активность, что и конечное синтезированное соединение.
Использованный здесь термин «активный метаболит» относится к продукту разложения или к конечному продукту соединения по настоящему изобретению, его соли или пролекарству, полученному посредством метаболизма или биотрансформации в организме животного или человека, который проявляет такую же или по существу такую же биологическую активность, что и указанное соединение. Такие метаболиты могут быть результатом, например, окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного введенного соединения, или соли, или пролекарства.
Использованный здесь термин «пролекарства» относится к любым соединениям или конъюгатам, которые высвобождают активное исходное лекарственное средство при введении животному или человеку. Пролекарства могут быть получены путем модификации функциональных групп, присутствующих в соединениях, таким образом, что эти модификации могут быть отщеплены от исходных соединений либо посредством обычной обработки, либо in vivo. Пролекарства включают соединения, в которых гидроксильная, амино, сульфгидрильная или карбоксильная группа связана с любой группой, которая при введении млекопитающему расщепляется с образованием свободной гидроксильной, амино, сульфгидрильной или карбоксильной группы соответственно. Примеры пролекарств включают ацетатные, формиатные и бензоатные производные спиртовых и аминных функциональных групп в соединениях по настоящему изобретению, но не ограничиваются ими. Приготовление и применение пролекарств обсуждается в THiguchi and V. Stella, "Pro-drugs as Novel Delivery Systems", Vol.14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, оба из которых включены в данное описание изобретения посредством ссылки во всей их полноте.
Здесь раскрыты новые соединения или фармацевтически приемлемые соли, которые могут селективно ингибировать DNA-PK. Соединения по настоящему изобретению или их фармацевтически приемлемые соли по сравнению с другими клинически доступными ингибиторами DNA-PK демонстрируют некоторые улучшенные свойства, например более высокое проникновение через ВВВ (гематоэнцефалический барьер) (что делает их потенциально полезными для лечения раковых заболеваний, которые метастазировали в ЦНС (центральную нервную систему), в частности метастазов в головной мозг и лептоменингеальных метастазов), лучшую активность и так далее. Они также могут обладать благоприятными профилями токсичности и/или благоприятными метаболическими или фармакокинетическими профилями по сравнению с известными ингибиторами DNA-PK.
Поэтому такие соединения или их фармацевтически приемлемые соли могут быть особенно полезными в лечении рака, особенно с метастазами в головной мозг.
Способ синтеза
Синтез предложенных здесь соединений, включая их соли, сложные эфиры, гидраты, или сольваты, или стереоизомеры, проиллюстрирован на схемах синтеза в примерах. Предложенные здесь соединения могут быть получены с использованием любых известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, и, таким образом, эти схемы являются только иллюстративными и не предназначены для ограничения других возможных способов, которые могут быть использованы для получения предложенных здесь соединений. Кроме того, стадии в схемах предназначены для лучшей иллюстрации и могут быть изменены, как целесообразно. Воплощения соединений в примерах были синтезированы в Китае для целей исследования и возможно для представления в регулирующие органы.
Реакции получения соединений по изобретению могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по существу химически инертными в отношении исходных веществ (реагентов), промежуточных соединений или продуктов при температурах, при которых проводят взаимодействия, например при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данное взаимодействие может быть проведено в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии взаимодействия квалифицированный специалист может выбрать подходящие растворители для конкретной стадии взаимодействия.
Получение соединений по изобретению может включать защиту и снятие защиты с различных химических групп. Необходимость защиты и снятия защиты и выбор соответствующих защитных групп могут быть легко определены специалистом в данной области техники. Химию защитных групп можно найти, например, в TW Greene and PGM Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), которая включена в описание изобретения посредством ссылки во всей своей полноте.
Ход взаимодействий можно контролировать посредством любого подходящего способа, известного в данной области техники. Например, образование продукта можно контролировать с помощью спектроскопии, такой как спектроскопия ядерного магнитного резонанса (например 1H или 13С), инфракрасная спектроскопия, спектрофотометрия (например в УФ (ультрафиолетовой) и видимой областях спектра), масс-спектрометрия, или с помощью хроматографических методов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография-масс-спектроскопия (ЖХМС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистами в данной области техники различными способами, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) («Preparative LC-MS Purification: Improved Compound Specific Method Optimization» Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, которая включена в описание изобретения посредством ссылки во всей своей полноте) и нормально-фазовую хроматографию на диоксиде кремния.
Использованные здесь сокращения определены следующим образом: «1×» или «×1» означает однократно, «2×» или «×2» означает двукратно, «3×» или «×3» означает трехкратно, «4×» или «×4» означает четырехкратно, «5×» или «×5» означает пятикратно, «°С» означает градусы Цельсия, «экв» или «эквив.» означает эквивалент или эквиваленты, «г» означает грамм или граммы, «мг» означает миллиграмм или миллиграммы, «л» означает литр или литры, «мл» означает миллилитр или миллилитры, «мкл» означает микролитр или микролитры, «н.» означает нормальный, «М» означает молярный, «ммоль» означает миллимоль или миллимоли, «мин» означает минуту или минуты, «ч» означает час или часы, «К.Т.» или «КТ» означает комнатную температуру, «атм» означает атмосферу, «ф/кв. дюйм» означает фунты на квадратный дюйм, «конц.» означает концентрированный, «нас.» или «насыщ.» означает насыщенный, «МС» означает масс-спектрометрию, «ESI» означает ионизацию электрораспылением при масс-спектроскопии, «ЖХМС» означает жидкостную хроматографию-масс-спектрометрию, «ВЭЖХ» означает высокоэффективную жидкостную хроматографию, «ОФ» означает обращенную фазу, «ТСХ» означает тонкослойную хроматографию, «ИВ» означает исходное вещество, «ЯМР» означает спектроскопию ядерного магнитного резонанса, «1H» означает протон, «δ» означает дельта, «s» означает синглет, «d» означает дублет, «t» означает триплет, «q» означает квартет, «m» означает мультиплет, «br» означает уширенный, и «Гц» означает Герц. «α», «β», «R», «S», «Е» и «Z» представляют собой стереохимические обозначения, знакомые специалисту в данной области техники.
Фармацевтическая композиция
Согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере одно соединение по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит более одного соединения по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция содержит одно или более чем одно соединение по настоящему изобретению и фармацевтически приемлемый носитель.
Фармацевтически приемлемые носители представляют собой обычные в данной области техники носители для лекарственных средств, которые могут быть получены посредством способов, хорошо известных в области фармацевтики. В некоторых воплощениях соединения по настоящему изобретению могут быть смешаны с фармацевтически приемлемым носителем для приготовления фармацевтической композиции.
Использованный здесь термин «фармацевтически приемлемый носитель» относится к фармацевтически приемлемому веществу, композиции или носителю, такому как жидкий или твердый наполнитель, разбавитель, эксципиент, растворитель или инкапсулирующее вещество, участвующему в переносе или транспортировке предложенного здесь соединения из одной области, жидкости организма, ткани, органа (внутреннего или внешнего) или части тела в другую область, жидкость организма, ткань, орган или часть тела. Фармацевтически приемлемые носители могут представлять собой носители, разбавители, эксципиенты или другие вещества, которые могут быть использованы для контакта с тканями животного без чрезмерной токсичности или побочных эффектов. Примеры фармацевтически приемлемых носителей включают сахара, крахмал, целлюлозы, мальтозу, трагакант, желатин, раствор Рингера, альгиновую кислоту, изотонический раствор, буферные агенты и тому подобное. Фармацевтически приемлемый носитель, который может быть использован в настоящем изобретении, включает носители, общеизвестные в данной области техники, такие как раскрытые в «Remington Pharmaceutical Sciences» Mack Pub. Co., New Jersey (1991), который включен в данное описание изобретения посредством ссылки.
Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) мальтозу; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) спирт, такой как этиловый спирт и пропиловый спирт; (20) фосфатные буферные растворы; и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях, такие как ацетон.
Фармацевтические композиции могут содержать фармацевтически приемлемые вспомогательные вещества, необходимые для приближения к физиологическим условиям, такие как регуляторы рН и буферные агенты, агенты, регулирующие токсичность, и тому подобное, например ацетат натрия, хлорид натрия, хлорид калия, хлорид кальция, лактат натрия и тому подобное.
Форма фармацевтических композиций зависит от ряда критериев, включая путь введения, степень заболевания или дозу, которую необходимо ввести, без ограничения ими. Фармацевтические композиции могут быть приготовлены в виде препаратов для перорального, назального, ректального, чрескожного, внутривенного или внутримышечного введения. Например, лекарственные формы для назального введения могут быть для удобства приготовлены в виде аэрозолей, растворов, капель, гелей или сухих порошков; лекарственные формы для интраназального введения могут быть приготовлены в виде жидкой композиции. В соответствии с нужным путем введения фармацевтические композиции могут быть приготовлены в виде таблеток, капсул, пилюль, драже, порошка, гранул, саше, облаток, лепешек, суспензий, эмульсий, растворов, сиропов, аэрозолей (твердых частиц или в жидкой среде), спрея, мази, пасты, крема, лосьона, геля, пластыря, ингалятора или суппозитория.
Фармацевтические композиции также могут быть приготовлены в виде препаратов для обеспечения немедленного, замедленного или отсроченного высвобождения активного ингредиента после введения пациенту с применением способов, известных в данной области техники. В некоторых воплощениях фармацевтическую композицию готовят в виде формы с замедленным высвобождением. Использованный здесь термин «форма с замедленным высвобождением» относится к высвобождению активного агента из фармацевтической композиции, так что он становится доступным для биоабсорбции у субъекта, в первую очередь в желудочно-кишечном тракте субъекта, в течение длительного периода времени (пролонгированное высвобождение) или в определенной области (контролируемое высвобождение). В некоторых воплощениях длительный период времени может составлять от 1 часа до 24 часов, от 2 часов до 12 часов, от 3 часов до 8 часов, от 4 часов до 6 часов, от 1 до 2 суток или более. В некоторых воплощениях длительный период времени составляет по меньшей мере примерно 4 часа, по меньшей мере примерно 8 часов, по меньшей мере примерно 12 часов или по меньшей мере примерно 24 часа. Фармацевтическая композиция может быть приготовлена в виде таблетки. Например, скорость высвобождения активного агента может контролироваться не только с помощью растворения активного агента в желудочно-кишечной жидкости и последующей диффузии из таблетки или пилюль независимо от значения рН, но также может зависеть от физических процессов разрушения и размывания таблетки. В некоторых воплощениях полимерные вещества, раскрытые в «Medical Applications of Controlled Release,» Langer and Wise (eds.), CRC Pres., Boca Raton, Florida (1974); «Controlled Drug Bioavailability,» Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J MacromolSci. Rev. Macromol Chem. 23:61; смотри также Levy et al., 1985, Science 228:190; During et al., 1989, Ann. Neurol. 25:351; Howard et al., 1989, J. Neurosurg. 71:105, могут быть использованы для замедленного высвобождения. Вышеупомянутые ссылочные документы включены в данное описание изобретения посредством ссылки во всей своей полноте.
В некоторых воплощениях фармацевтические композиции содержат от примерно 0,0001 мг до примерно 100 мг соединений по настоящему изобретению (например от примерно 0,0001 мг до примерно 10 мг, от примерно 0,001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 0,1 мг до примерно 5 мг, от примерно 0,1 мг до примерно 4 мг, от примерно 0,1 мг до примерно 3 мг, от примерно 0,1 мг до примерно 2 мг, от примерно 0,1 мг до примерно 1 мг, от примерно 0,1 мг до примерно 0,5 мг, от примерно 1 мг до примерно 10 мг, от примерно 1 мг до примерно 5 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг). Подходящие дозировки на субъекта в сутки могут составлять от примерно 0,1 мг до примерно 10 мг, предпочтительно от примерно 0,1 мг до примерно 5 мг, от примерно 5 мг до примерно 10 мг или от примерно 1 мг до примерно 5 мг.
В некоторых воплощениях фармацевтические композиции могут быть приготовлены в виде стандартной лекарственной формы, причем каждая доза содержит от примерно 0,0001 мг до примерно 10 мг, от примерно 0,001 мг до примерно 10 мг, от примерно 0,01 мг до примерно 10 мг, от примерно 0,1 мг до примерно 10 мг, от примерно 0,1 мг до примерно 5 мг, от примерно 0,1 мг до примерно 4 мг, от примерно 0,1 мг до примерно 3 мг, от примерно 0,1 мг до примерно 2 мг, от примерно 0,1 мг до примерно 1 мг, от примерно 0,1 мг до примерно 0,5 мг, от примерно 1 мг до примерно 10 мг, от примерно 5 мг до примерно 10 мг, от примерно 5 мг до примерно 20 мг, от примерно 5 мг до примерно 30 мг, от примерно 5 мг до примерно 40 мг, от примерно 5 мг до примерно 50 мг, от примерно 10 мг до примерно 100 мг, от примерно 20 мг до примерно 100 мг, от примерно 30 мг до примерно 100 мг, от примерно 40 мг до примерно 100 мг, от примерно 50 мг до примерно 100 мг соединений по настоящему изобретению. Термин «стандартные лекарственные формы» относится к физически дискретным единицам, подходящим в качестве стандартных дозировок для людей и других млекопитающих, где каждая единица содержит заранее определенное количество активного вещества, рассчитанное на получение требуемого терапевтического эффекта, в сочетании с подходящим фармацевтическим носителем.
В некоторых воплощениях фармацевтические композиции содержат одно или более чем одно соединение по настоящему изобретению в качестве первого активного ингредиента и, кроме того, содержат второй активный ингредиент. Второй активный ингредиент может представлять собой любой иммуномодулятор или противоопухолевый агент, известный в данной области техники, включая химиотерапевтические средства, иммунотерапевтические средства, ингибиторы сигнальной трансдукции клеток, алкилирующие агенты, ингибиторы топоизомеразы, ингибиторы митоза, антигормональные агенты и так далее, без ограничения ими. Примерами таких иммуно модуляторов или противоопухолевых агентов являются химиотерапевтические средства на основе платины (например цисплатин (DDP), карбоплатин (СВР), сульфато-1,2-диаминоциклогексанплатина (SHP), недаплатин, оксалиплатин (ОХА), лабоплатин), доцетаксел, паклитаксел, доксорубицин, этопозид, митоксантрон, ингибиторы CTLA-4 (антиген цитотоксических Т-лимфоцитов 4 класса), анти-CTLA-4 антитела, ингибиторы PD-1 (рецептор программируемой клеточной смерти), ингибиторы PD-L1 (1 лиганд белка PD-1), анти-PD-1/PD-L1 антитела, ингибиторы CD39, анти-С039 антитела, ингибиторы CD73, анти-CD73 антитела, ингибиторы CCR2 (СС-рецептор хемокина типа 2), анти-CCR2 антитела, ингибиторы EGFR (рецептор эпидермального фактора роста), ингибиторы CDK 4/6 (циклинзависимые киназы 4 и 6), ингибиторы MELK (киназа материнской эмбриональной лейциновой молнии), агонисты OX40 (4 член суперсемейства рецепторов фактора некроза опухоли), антиандрогенные ингибиторы, антитела изотипа IgG4 (иммуноглобулин-04), ингибиторы тирозинкиназы, ингибиторы ДНК-метилтрансферазы, ингибиторы Hsp90 (белок теплового шока 90), ингибиторы FGFR (рецептор фактора роста фибробластов), ингибиторы mTOR (мишень рапамицина млекопитающих), ингибиторы ароматазы, ингибиторы VEGF (фактор роста эндотелия сосудов), антагонисты LHRH (гормон высвобождения лютеинизированного гормона), ингибиторы PI3K (фосфоинозитид-3-киназы), ингибиторы АКТ (протеинкиназа В альфа), ингибиторы киназы Aurora, ингибиторы МЕК (киназа митоген-активируемой протеинкиназы), ингибиторы HDAC (деацетилаза гистонов), ингибиторы BET (бромодомен и экстра-терминальный домен), ингибиторы PIK3CA, ингибиторы протеасом, другие SERD (селективные деструкторы эстрогеновых рецепторов), ингибиторы фарнезилтрансферазы, антитела к VEGF-A, антитела к мембранному белку ErbB3 (Her3) (рецептор эпидермального фактора роста 3 типа), ингибиторы протеасом, ингибиторы протеинкиназы Сβ, анти-IGF-1R антитела (рецептор инсулино подобного фактора роста 1 типа), анти-HER2 (рецептор эпидермального фактора роста 2 типа) антитела, SERM (селективные модуляторы эстрогеновых рецепторов), ингибиторы IGF, анти-IgG антитела и тому подобное. Типичные примеры противоопухолевых средств для лечения различных видов рака или опухолей могут включать цисплатин, карбоплатин, SHP, недаплатин, оксалиплатин, лабоплатин, доцетаксел, паклитаксел, доксорубицин, этопозид, митоксантрон, винкристин, винбластин, гемцитабин, циклофосфамид, хлорамбуцил, кармустин, метотрексат, фторурацил, актиномицин, эпирубицин, антрациклин, блеомицин, митомицин-С, иринотекан, топотекан, тенипозид, интерлейкин, интерферон, тремелимумаб, ипилимумаб, пембролизумаб, ниволумаб, авелумаб, дурвалумаб, атезолизумаб, IPH 52, IPH 53, CPI-006, плозализумаб, MLN1202, цетуксимаб, лапатиниб, эрлотиниб, гефитиниб, нератиниб, трастузумаб, адо-трастузумаб эмтанзин, пертузумаб, MCLA-128, анастрозол, ралоксифен, G1T38, тамоксифен, гозерелин, энзалутамид, вориностат, энтиностат, сунитиниб, пазопаниб, бевацизумаб, ранибизумаб, пегаптаниб, цедираниб, дазатиниб, GDC-0980, гедатолисиб, алпелисиб, ВКМ120, копанлисиб, AZD8835, GDC-0941, таселисиб, темсиролимус, эверолимус, сапанисертиб, AZD5363, МК2206, панитумумаб, пембролизумаб, сорафениб, палбоциклиб, абемациклиб, рибоциклиб, кризотиниб, довитиниб, руксолитиниб, азацитидин, СС-486, HSP90 ганетеспиб, Debio 1347, эрдафитиниб, вистусертиб, алисертиб, селуметиниб, GS-5829, GSK525762, MLN9708, GDC-0810, AFP464, типифарниб, серибантумаб, бортезомиб, энзастаурин, AVE1642, ксентузумаб, далотузумаб, AMG 479 и тому подобное, но не ограничиваются ими.
Примеры такого противоопухолевого средства также можно найти в Cancer Principles and Practice of Oncology. V. T. Devita and S. Hellman (editors), 6th edition (Feb. 15, 2001), Lippincott Williams & Wilkins Publishers. Специалист в данной области техники также сможет установить, какие комбинации средств будут полезны, на основе конкретных характеристик лекарственных средств и имеющегося рака.
В соответствии с этим аспектом настоящего изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), как определено здесь ранее, или его фармацевтически приемлемую соль и любой из иммуномодуляторов или противоопухолевых средств, перечисленных выше.
Поэтому в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с иммуномодулятором или химиотерапевтическими средствами, выбранными из одного из перечисленных выше.
Следует понимать, что использованный здесь термин «комбинация» относится к одновременному, раздельному или последовательному введению. В некоторых воплощениях «комбинация» относится к одновременному введению. В другом аспекте настоящего изобретения «комбинация» относится к раздельному введению. В еще одном аспекте настоящего изобретения «комбинация» относится к последовательному введению. Если введение является последовательным или раздельным, отсрочка введения второго компонента не должна быть такой, чтобы положительный эффект комбинации был потерян.
В соответствии с еще одним аспектом настоящего изобретения предложена фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с иммуномодулятором или противоопухолевым средством, выбранным из перечисленных выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с еще одним аспектом настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с иммуномодулятором или противоопухолевым средством, выбранным из перечисленных выше, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в получении иммуномодулирующего или противоопухолевого эффекта.
В соответствии с еще одним аспектом настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с иммуномодулятором или противоопухолевым средством, выбранным из перечисленных выше, в сочетании с фармацевтически приемлемым разбавителем или носителем для применения в лечении расстройств, ассоциированных с DNA-PK, например NSCLC (немелкоклеточный рак легкого), RCC (почечно-клеточный рак), рака предстательной железы или рака молочной железы и так далее.
В соответствии с еще одним аспектом настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с иммуномодулятором или противоопухолевым средством, выбранным из перечисленных выше.
Согласно еще одному аспекту настоящего изобретения, предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) иммуномодулятор или противоопухолевое средство, выбранное из перечисленных выше, во второй стандартной лекарственной форме; и
с) контейнер для размещения указанных первой и второй лекарственных форм.
Помимо применения в терапевтического медицине соединения формулы (I) или их фармацевтически приемлемая соль также полезны в качестве фармакологических инструментов в разработке и стандартизации in vitro и in vivo тест-систем для оценки активности или экспрессии DNA-PK у лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, в рамках поиска новых терапевтических агентов.
В другой фармацевтической композиции, процессе, способе, применении и особенностях получения лекарственного средства, упомянутых выше, также применимы альтернативные и предпочтительные воплощения соединений по настоящему изобретению, описанные здесь.
Способы лечения
Согласно настоящему изобретению предложен способ лечения расстройств, ассоциированных с DNA-PK, включающий введение субъекту эффективного количества одного или более чем одного соединения, его фармацевтически приемлемой соли или фармацевтической композиции по настоящему изобретению.
Согласно настоящему изобретению также предложен способ лечения расстройств, ассоциированных с DNA-PK. В некоторых воплощениях способ включает введение субъекту эффективного количества одного или более чем одного соединения, его фармацевтически приемлемой соли или фармацевтической композиции по настоящему изобретению.
Использованный здесь термин «расстройства, связанные с DNA-PK» относится к заболеваниям, начало или развитие которых, или и то, и другое связано с экспрессией или активностью DNA-PK. Примеры включают гиперпролиферативное расстройство (например рак), но не ограничиваются им.
В некоторых воплощениях связанные с DNA-PK расстройства представляют собой рак, предпочтительно рак со сверхэкспрессией DNA-PK. «Рак со сверхэкспрессией DNA-PK» представляет собой рак, который имеет значительно более высокие уровни белка DNA-PK в раковой или опухолевой клетке по сравнению с нераковой клеткой того же типа ткани. Такая сверхэкспрессия может быть вызвана амплификацией гена или повышенной транскрипцией или трансляцией. Сверхэкспрессия DNA-PK может быть определена в диагностическом или прогностическом анализе путем оценки повышенных уровней белков DNA-PK, присутствующих в клетке (например с помощью иммуногистохимического анализа; IHC). Альтернативно или дополнительно, можно измерить уровни нуклеиновой кислоты, кодирующей DNA-PK, в клетке, например, с помощью флуоресцентной гибридизации in situ (FISH; смотри WO 98/45479, опубликованную в октябре 1998 года), методов саузерн-блоттинга или полимеразной цепной реакции (ПЦР), таких как количественная ПЦР в реальном времени (RT-PCR) (Methods 132: 73-80 (1990)). Помимо вышеуказанных способов анализа специалисту в данной области техники доступны различные способы анализа in vivo. Например, можно подвергнуть клетки в организме пациента воздействию антитела, которое возможно помечено детектируемой меткой, например радиоактивным изотопом, и связывание антитела с клетками пациента можно оценить, например посредством внешнего сканирования радиоактивности или посредством анализа биопсии, взятой у пациента, которого ранее подвергли воздействию антитела.
В частности, раковые заболевания включают рак легких (например немелкоклеточный рак легкого (NSCLC), мелкоклеточный рак легкого, аденокарциному легких, крупноклеточный рак легкого, плоскоклеточный рак легкого), почечно-клеточный рак (RCC), рак предстательной железы, рак молочной железы, рак яичников, рак эндометрия, рак шейки матки, рак костей, рак матки, рак толстой кишки, лейкоз, глиобластому, меланому, хондросаркому, рак головного мозга, холангио карциному, остеосаркому, лимфому, аденому, миелому, гепатоцеллюлярную карциному, рак коры надпочечников, рак поджелудочной железы, рак мочевого пузыря, рак печени, рак желудка, колоректальный рак, рак пищевода, рак яичка, рак кожи, различные раковые заболевания почек, мезотелиому, нейробластому, рак щитовидной железы, различные раковые заболевания головы и шеи, различные раковые заболевания пищевода, различные раковые заболевания глаза, рак носоглотки или рак полости рта, но не ограничиваются ими. В некоторых воплощениях рак представляет собой NSCLC, RCC, рак предстательной железы или рак молочной железы. Упомянутый здесь рак может находиться на любой стадии, если не оговорено особо. В некоторых воплощениях рак представляет собой рак на ранней стадии. В некоторых воплощениях рак представляет собой местнораспространенный рак. В некоторых воплощениях рак представляет собой местнораспространенный и/или метастатический рак. В некоторых воплощениях рак представляет собой инвазивный рак. В некоторых воплощениях рак представляет собой рак, устойчивый к существующим методам лечения.
Использованные здесь термины «лечение», «лечить» и «лечащий» относятся к реверсированию, облегчению, задержке начала или ингибированию развития заболевания или расстройства или одного или более чем одного его симптома, как описано здесь. В некоторых воплощениях лечение можно проводить после развития одного или более чем одного симптомов. В других воплощениях лечение можно проводить в отсутствие симптомов. Например, лечение может быть назначено предрасположенному индивидууму до появления симптомов (например с учетом симптомов в анамнезе и/или с учетом генетических или других факторов предрасположенности). Лечение также может быть продолжено после ликвидации симптомов, например, чтобы предупредить или отсрочить их повторное появление.
В некоторых воплощениях одно или более чем одно соединение, его фармацевтически приемлемая соль или фармацевтическая композиция, предложенные здесь, вводят парентеральным путем или непарентеральным путем. В некоторых воплощениях одно или более чем одно соединение, его фармацевтически приемлемую соль, гидрат, сольват или стереоизомер, или фармацевтическую композицию вводят перорально, энтерально, трансбуккально, назально, интраназально, трансмукозально, эпидермально, трансдермально, дермально, офтальмологически, пульмонально, ректально, сублингвально, вагинально, местно, подкожно, внутривенно, внутримышечно, внутриартериально, интратекально, внутрикапсулярно, интраорбитально, внутрисердечно, интрадермально, внутрибрюшинно, транстрахеально, субкутикулярно, интраартикулярно, субкапсулярно, интраспинально, субарахноидально или интрастернально.
Предложенные здесь соединения могут быть введены в чистом виде, в комбинации с другими активными ингредиентами или в виде фармацевтических композиций по настоящему изобретению. В некоторых воплощениях предложенные здесь соединения могут быть введены нуждающемуся субъекту одновременно или последовательно в комбинации с одним или более чем одним противоопухолевым или противовоспалительным средством; известным в данной области техники. Отдельные соединения таких комбинаций могут быть введены либо последовательно, либо одновременно в отдельных или комбинированных фармацевтических композициях. Предпочтительно, отдельные соединения вводят одновременно в комбинированной фармацевтической композиции. Соответствующие дозы известных терапевтических агентов будут легко определены специалистами в данной области техники.
В некоторых воплощениях введение осуществляют один раз в сутки, два раза в сутки, три раза в сутки или один раз в двое суток, один раз в трое суток, один раз в четверо суток, один раз в пятеро суток, один раз в шестеро суток, один раз в неделю.
Терапевтически эффективное количество предложенного здесь соединения или его фармацевтически приемлемых солей будет зависеть от различных факторов, известных в данной области техники, таких как масса тела, возраст, история болезни, применяемые лекарственные препараты, состояние здоровья субъекта и возможность перекрестного взаимодействия, аллергические реакции, чувствительность и побочные эффекты, а также путь введения и степень развития заболевания. Дозировки могут быть пропорционально уменьшены или увеличены специалистом в данной области техники (например врачом или ветеринаром) в соответствии с этими и другими обстоятельствами или требованиями.
В некоторых воплощениях одно или более чем одно предложенное здесь соединение, его фармацевтически приемлемую соль или фармацевтическую композицию вводят перорально. Для перорального введения подходит любая доза, позволяющая достичь желаемых целей. В некоторых воплощениях подходящие суточные дозы составляют примерно 0,001 - 100 мг, предпочтительно от 0,1 мг до 5 г, более предпочтительно от 5 мг до 1 г, более предпочтительно от 10 мг до 500 мг, и введение осуществляют один раз в сутки, два раза в сутки, три раза в сутки, каждые сутки или 3-5 суток в неделю. В некоторых воплощениях доза одного или более чем одного предложенного здесь соединения, их фармацевтически приемлемых солей или фармацевтической композиции находится в диапазоне от примерно 0,0001 мг, предпочтительно 0,001 мг, 0,01 мг, 0,1 мг, 0,2 мг, 0,3 мг, 0,4 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 2 мг, 3 мг, 4 мг, 5 мг, 6 мг, 7 мг, 8 мг, 9 мг, 10 мг в сутки.
Применение соединений
В некоторых воплощениях согласно настоящему изобретению предложено применение соединений, их фармацевтически приемлемых солей или фармацевтической композиции по настоящему изобретению в изготовлении лекарственных средств для лечения расстройств, ассоциированных с DNA-PK. В некоторых воплощениях расстройства, связанные с DNA-PK, включают раковые заболевания.
Соединения и их фармацевтические композиции в настоящем изобретении могут быть использованы в предупреждении или лечении начала или развития любого из расстройств, ассоциированных с DNA-PK (экспрессией или активностью), у млекопитающих, особенно у человека.
В таком случае в настоящем изобретении также предложен способ отбора пациента, подходящего для лечения соединениями или фармацевтической композицией по настоящему изобретению отдельно или в комбинации с другими ингредиентами (например вторым активным ингредиентом, например, противовоспалительным или противоопухолевым агентом). Способ включает секвенирование образцов тканей пациентов и обнаружение накопления DNA-PK у пациента.
ПРИМЕРЫ
Нижеследующее примеры дополнительно объясняет общие способы по настоящему изобретению. Соединения по настоящему изобретению могут быть получены посредством способов, известных в данной области техники. Нижеследующее иллюстрирует подробные способы получения предпочтительных соединений по настоящему изобретению. Однако эти примеры никоим образом не ограничивают способы получения соединений по настоящему изобретению.
ПРИМЕРЫ СИНТЕЗА
Синтез предложенных здесь соединений, включая их фармацевтически приемлемые соли, проиллюстрирован на схемах синтеза в примерах. Предложенные здесь соединения могут быть получены с использованием любых известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, и, таким образом, эти схемы являются только иллюстративными и не предназначены для ограничения других возможных способов, которые могут быть использованы для получения соединений, предложенных здесь. Кроме того, стадии в этих схемах предназначены для лучшей иллюстрации и могут быть изменены, как целесообразно. Воплощения соединений в примерах были синтезированы для целей исследования и возможно для представления в регулирующие органы.
Реакции получения соединений по настоящему изобретению могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по существу инертными в отношении исходных веществ (реагентов), промежуточных соединений или продуктов при температурах, при которых проводят взаимодействия, например при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данное взаимодействие может быть проведено в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии взаимодействия квалифицированный специалист может выбрать подходящие растворители для конкретной стадии взаимодействия.
Получение соединений по настоящему изобретению может включать защиту и снятие защиты с различных химических групп. Необходимость защиты и снятия защиты и выбор подходящих защитных групп могут быть легко определены специалистом в данной области техники. Химию защитных групп можно найти, например, в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), которая включена в описание изобретения посредством ссылки во всей своей полноте.
Взаимодействия можно контролировать посредством любого подходящего метода, известного в данной области техники. Например, образование продукта можно контролировать с помощью методов спектроскопии, таких как спектроскопия ядерного магнитного резонанса (например 1H или 13С), инфракрасная спектроскопия, спектрофотометрия (например в УФ (ультрафиолетовой) и видимой областях спектра), масс-спектрометрия, или с помощью хроматографических методов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ), жидкостная хроматография-масс-спектроскопия (ЖХМС) или тонкослойная хроматография (ТСХ). Соединения могут быть очищены специалистами в данной области техники различными методами, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) («Preparative LC-MS Purification: Improved Compound Specific Method Optimization» Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883, которая включена в описание изобретения посредством ссылки во всей своей полноте) и нормально-фазовую хроматографию на диоксиде кремния.
Структуры соединений в примерах охарактеризованы с помощью ядерного магнитного резонанса (ЯМР) или/и жидкостной хроматографии-масс-спектрометрии (ЖХ-МС). Химический сдвиг ЯМР (δ) приведен в единицах 10-6 (м.д.). Спектры 1H-ЯМР регистрируют в растворителе диметилсульфоксид-d6 (DMSO-d6) или CDCl3, или CD3OD, или D2O, или ацетон-d6, или CD3CN (от Innochem, или Sigma-Aldrich, или Cambridge Isotope Lab., me.) на спектрометре Bruker AVANCE NMR (300 МГц или 400 МГц) с использованием ICON-NMR (под контролем программы TopSpin) с тетраметилсиланом в качестве внутреннего стандарта.
МС-измерение проводят с использованием масс-спектрометра Shimadzu 2020 с источником электрораспыления в режиме положительных и отрицательных ионов.
Измерение посредством высокоэффективной жидкостной хроматографии (ВЭЖХ) проводят на системах Shimadzu LC-20AD, Shimadzu LC-20ADXR или Shimadzu LC-30AD с использованием колонки Shim-pack XR-ODS C18 (3,0×50 мм, 2,2 мкм), или колонки Ascentis Express C18 (2,1×50 мм, 2,7 мкм), или колонки Agilent Poroshell HPH-C18 (3,0×50 мм, 2,7 мкм).
Тонкослойную хроматографию проводят с использованием пластин силикагеля Sinopharm Chemical Reagent Beijing Co., Ltd. и Xinnuo Chemical. Пластины силикагеля, используемые для тонкослойной хроматографии (ТСХ), имеют толщину слоя 175-225 мкм. Пластины силикагеля, используемые для разделения и очистки продуктов посредством ТСХ, имеют толщину слоя 1,0 мм.
В хроматографической колонке для очистки используют в качестве носителя силикагель (100 ~ 200, 200 ~ 300 или 300 ~ 400 меш, производства Rushanshi Shangbang Xincailiao Co., Ltd. или Rushan Taiyang Desiccant Co., Ltd. и так далее) или флэш-колонку (колонка C18 с обращенной фазой 20-45 мкм, производства Agela Technologies) в флэш-системе Agela Technologies. Размер колонок регулируется в зависимости от количества соединений.
Известные исходные вещества по настоящему изобретению могут быть синтезированы с использованием известных в данной области техники способов или в соответствии с ними, либо их можно приобрести у Alfa Aesar, TCI, Sigma-Aldrich, Bepharm, Bide Pharmatech, PharmaBlock, Enamine, Innochem и JW&Y PharmLab и так далее.
Если не оговорено особо, все взаимодействия проводят в атмосфере аргона или азота. Атмосфера аргона или азота означает, что реакционная колба соединена с баллоном с аргоном или азотом объемом около 1 л. Гидрирование обычно проводят под давлением. Если не оговорено особо, температура взаимодействий в примерах соответствует температуре окружающей среды, которая составляет 10°С~30°С. За ходом взаимодействий следят с помощью ТСХ или/и ЖХ-МС. Системы элюентов, используемые для взаимодействий, включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей регулируют в соответствии с разными полярностями соединений.
Система элюирования колоночной хроматографии, используемая для очистки соединений, и система элюентов ТСХ включают систему дихлорметан-метанол и систему петролейный эфир-этилацетат. Объемные соотношения растворителей регулируют в соответствии с разными полярностями соединений. Для корректировки может быть добавлено небольшое количество щелочных или кислотных агентов (0,1%~1%), таких как муравьиная кислота, или уксусная кислота, или TFA, или аммиак.
Сокращения для химических веществ, используемых в синтезе предложенных здесь соединений, указаны ниже:
Пример 1
Получение 1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 1)
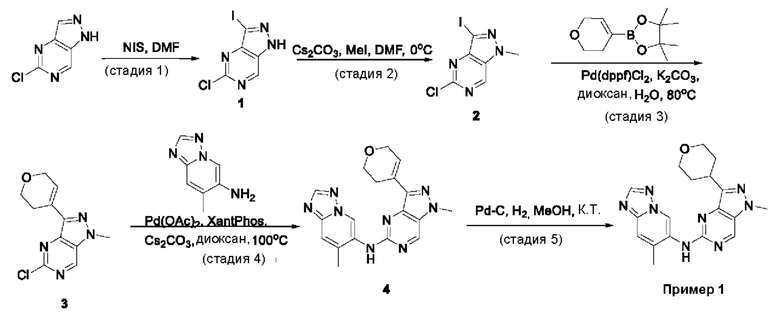
СХЕМА 1
Стадия 1. 5-Хлор-3-иод-1Н-пиразоло[4,3-d]пиримидин
Смесь 5-хлор-1Н-пиразоло[4,3-d]пиримидина (3,00 г; 19,410 ммоль; 1,00 экв.) и NIS (N-йодсукцинимид) (7,86 г; 34,936 ммоль; 1,80 экв.) в DMF (60,00 мл) перемешивали в течение ночи при 0°С в воздушной атмосфере. Полученную смесь экстрагировали EtOAc (3×150 мл). Объединенные органические слои промывали рассолом (3×200 мл). Объединенные органические слои промывали Na2S2Os (3×200 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя смесью РЕ/EtOAc (20:1) с получением 5-хлор-3-иод-1H-пиразоло[4,3-d]пиримидина (2,4 г; 44,09%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 281,0.
Стадия 2. 5-Хлор-3-иод-1-метил-1Н-пиразоло[4,3-d]пиримидин
Смесь CS2CO3 (3,49 г; 10,697 ммоль; 3 экв.), CH3I (2,53 г; 17,828 ммоль; 5,00 экв.) и 5-хлор-3-иод-1Н-пиразоло[4,3-d]пиримидина (1,00 г; 3,566 ммоль; 1,00 экв.) в DMF (20,00 мл) перемешивали в течение 1 часа при 0°С в атмосфере азота. Полученную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3×80 мл). Объединенные органические слои промывали рассолом (3×100 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт перекристаллизовывали из смеси EtOAc/РЕ (1:5; 300 мл) с получением 5-хлор-3-иод-1-метил-1Н-пиразоло[4,3-d]пиримидина (850 мг; 80,95%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 295,0.
Стадия 3. 5-Хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-1Н-пиразоло[4,3-d]пиримидин
K2CO3 (1210,85 мг; 8,761 ммоль; 3 экв.), Pd(dppf)Cl2 CH2Cl2 (476,98 мг; 0,584 ммоль; 0,2 экв.), 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (797,57 мг; 3,797 ммоль; 1,3 экв.) и 5-хлор-3-иод-1-метил-1Н-пиразоло[4,3-d]пиримидин (860,00 мг; 2,920 ммоль; 1,00 экв.) в диоксане (15,00 мл) и H2O (3,00 мл) перемешивали в течение 16 часов при 80°С в атмосфере азота. Полученную смесь разбавляли водой (100 мл). Полученную смесь экстрагировали смесью CH2Cl2/MeOH 12:1 (3×50 мл). Объединенные органические слои промывали рассолом (2×100 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением 5-хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-1Н-пиразоло[4,3-d] пиримидина (380 мг; 51,90%) в виде серого твердого вещества. ЖХМС: m/z (ESI), [М+H]+ составляет 251,2.
Стадия 4. 3-(3,6-Дигидро-2Н-пиран-4-ил)-1-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил]-1Н-пиразоло[4,3-d]пиримидин-5-амин
Смесь CS2CO3 (2599,38 мг; 7,978 ммоль; 2,50 экв.), XantPhos (553,94 мг; 0,957 ммоль; 0,30 экв.), Pd(OAc)2 (143,29 мг; 0,638 ммоль; 0,20 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (567,40 мг; 3,829 ммоль; 1,20 экв.) и 5-хлор-3-(3,6-дигидро-2Н-пиран-4- ил)-1 -метил- 1Н-пиразоло[4,3-d]пиримидина (800,00 мг; 3,191 ммоль; 1,00 экв.) в диоксане (20,00 мл) перемешивали в течение ночи при 100°С в атмосфере азота. Полученную смесь разбавляли водой (200 мл). Полученную смесь экстрагировали смесью CH2Cl2/МеОН (12:1) (3×200 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт перекристаллизовывали из смеси EtOAc/РЕ (1:6; 300 мл) с получением 3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[4,3-d]пиримидин-5-амина (600 мг; 51,88%) в виде коричневого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 363,3.
Стадия 5. 1-Метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-(оксан-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 1)
Смесь Pd/C (47,92 мг; 0,450 ммоль; 1,36 экв.) и 3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[4,3-d]пиримидин-5-амина (120 мг; 0,331 ммоль; 1 экв.) в МеОН (200 мл) и THF (100 мл) перемешивали в течение 2 часов при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3×100 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт (120 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN (ацетонитрил); скорость потока: 60 мл/мин; градиент: от 25 В до 51 В за 7 минут) с получением 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-(оксан-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (55 мг; 45,12%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 365,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.95 (4Н, t), 2.42 (3Н, d), 3.14-3.30 (1H, m), 3.47 (2H, d), 3.93 (2H, d), 4.04 (3Н, s), 7.71 (1H, t), 8.37 (1H, s), 8.84 (1H, s), 9.15 (1H, s), 9.34 (1H, s).
Пример 2
Получение 1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (пример 2) и 3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 6)
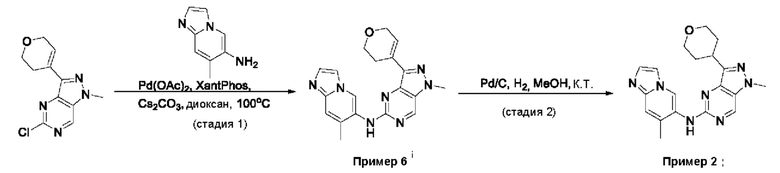
СХЕМА 2
Стадия 1. 3-(3,6-Дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 6)
Смесь CS2CO3 (682,34 мг; 2,094 ммоль; 2,5 экв.), XantPhos (96,94 мг; 0,168 ммоль; 0,2 экв.), Pd(OAc)2 (37,61 мг; 0,168 ммоль; 0,2 экв.), 7-метилимидазо[1,2-а]пиридин-6-амина (147,95 мг; 1,005 ммоль; 1,2 экв.) и 5-хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-метилпиразоло[4,3-d]пиримидина (210,00 мг; 0,838 ммоль; 1,00 экв.) в диоксане (6,00 мл) перемешивали в течение ночи при 100°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь фильтровали, осадок на фильтре промывали DCM (3×50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/MeOH, 12:1) с получением неочищенного продукта. Неочищенный продукт (170 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 26 В до 36 В за 7 минут) с получением 3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (100 мг; 57,65%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 362,2. 1H ЯМР (300 МГц, DMSO-d6) δ 2.30 (3Н, d), 2.58 (2H, s), 3.83 (2H, t), 4.06 (3H, s), 4.25 (2H, d), 7.07-7.12 (1H, m), 7.43 (1H, d), 7.49 (1H, d), 7.84 (1H, t), 8.81 (2H, d), 9.14 (1H, s).
Стадия 2. 1-Метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]-3-(оксан-4-ил)-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 2)
Смесь Pd/C (70,67 мг; 0,664 ммоль; 3,00 экв.) и 3-(3,6-дигидро-2Н-пиран-4-ил)-1-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (80,00 мг; 0,221 ммоль; 1,00 экв.) в МеОН (20,00 мл) перемешивали в течение 3 часов при 40°С в атмосфере водорода. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3×10 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт (50 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 22 В до 33 В за 7 минут; RT1 (время удерживания): 6,63) с получением 1-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]-3-(оксан-4-ил)пиразоло[4,3-d]пиримидин-5-амина (20 мг; 24,61%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 364,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.96 (4Н, d), 2.28 (3Н, d), 3.21 (1H, t), 3.47 (2H, d), 3.93 (2H, d), 4.03 (3Н, s), 7.42 (1H, q), 7.49 (1H, d), 7.81 (1H, t), 8.69 (1H, s), 8.84 (1H.s), 9.09 (1H,s).
Пример 3
Получение 7-метил-N-(5-метил-7-(тетрагидро-2Н-пиран-4-ил)-5Н-пирроло[3,2-d]пиримидин-2-ил)-[1,2,4]триазоло[1,5-a]пиридин-6-амина (соединение по примеру 3)
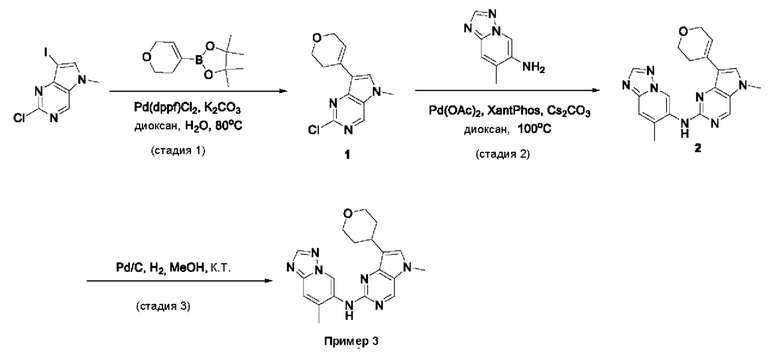
СХЕМА 3
Стадия 1. 2-Хлор-7-(3,6-дигидро-2Н-пиран-4-ил)-5-метил-5Н-пирроло[3,2-d] пиримидин
Смесь 2-хлор-7-иод-5-метилпирроло[3,2-d]пиримидина (300,00 мг; 1,022 ммоль; 1,00 экв.), 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (279,16 мг; 1,329 ммоль; 1,30 экв.), Pd(dppf)Cl2 (149,59 мг; 0,204 ммоль; 0,2 экв.) и K2CO3 (423,81 мг; 3,067 ммоль; 3 экв.) в диоксане (6,00 мл) и H2O (1,20 мл) перемешивали в течение 3 часов при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 1:3) с получением 2-хлор-7-(3,6-дигидро-2Н-пиран-4-ил)-5-метилпирроло[3,2-d]пиримидина (196 мг; 76,79%) в виде коричневого твердого вещества. ЖХМС: m/z (ESI), [M+H]+ составляет 250,2.
Стадия 2. 7-(3,6-Дигидро-2Н-пиран-4-ил)-5-метил-N-[7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил]пирроло[3,2-d]пиримидин-2-амин
Смесь 2-хлор-7-(3,6-дигидро-2Н-пиран-4-ил)-5-метилпирроло[3,2-d]пиримидина (196,00 мг; 0,785 ммоль; 1,00 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (139,56 мг; 0,942 ммоль; 1,20 экв.), Pd(AcO)2 (35,25 мг; 0,157 ммоль; 0,20 экв.), XantPhos (136,25 мг; 0,235 ммоль; 0,30 экв.) и CS2CO3 (639,37 мг; 1,962 ммоль; 2,50 экв.) в диоксане (3,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1) с получением 7-(3,6-дигидро-2Н-пиран-4-ил)-5-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пирроло[3,2-d]пиримидин-2-амина (170 мг; 59,93%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [M+H]+ составляет 362,3.
Стадия 3. 7-Метил-N-(5-метил-7-(тетрагидро-2Н-пиран-4-ил)-5Н-пирроло[3,2-d]пиримидин-2-ил)-[1,2,4]триазоло[1,5-а]пиридин-6-амин (соединение по примеру 3)
Смесь 7-(3,6-дигидро-2Н-пиран-4-ил)-5-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пирроло[3,2-d]пиримидин-2-амина (170,00 мг; 0,470 ммоль; 1,00 экв.) и Pd/C (250,29 мг; 2,352 ммоль; 5,00 экв.) в МеОН (20,00 мл) и THF (50,00 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (5×30 мл). Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN; скорость потока: 25 мл/мин; градиент: от 30 В до 45 В за 7 минут; RT1: 6,02) с получением 5-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-7-(оксан-4-ил)пирроло[3,2-d]пиримидин-2-амина (29 мг; 16,96%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 364,3. 1H ЯМР (300 МГц, DMSO-d6) δ 1.76 (2Н, d), 1.88-2.01 (2Н, m), 2.46 (3H, d), 3.02 (1H, t), 3.47 (2Н, td), 3.80 (3H, s), 3.88-4.05 (2Н, m), 7.51 (1H, s), 7.61-7.75 (1H, m), 8.33 (2Н, d), 8.74 (1H, s), 9.49 (1H, s).
Пример 4
Получение 3-изопропил-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 4)
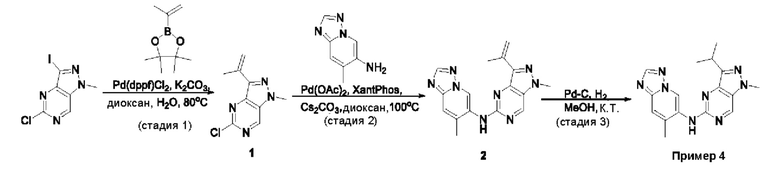
СХЕМА 4
Стадия 1. 5-Хлор-1-метил-3-(проп-1-ен-2-ил)-1Н-пиразоло[4,3-d]пиримидин
Смесь Pd(dppf)Cl2 (124,24 мг; 0,170 ммоль; 0,2 экв.), K2CO3 (293,33 мг; 2,122 ммоль; 2,5 экв.), 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолана (213,99 мг; 1,273 ммоль; 1,5 экв.) и 5-хлор-3-иод-1-метилпиразоло[4,3-d] пиримидина (250,00 мг; 0,849 ммоль; 1,00 экв.) в диоксане (5,00 мл) и Н2О (1,00 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 1:3) с получением 5-хлор-1-метил-3-(проп-1-ен-2-ил)пиразоло[4,3-d]пиримидина (130 мг; 73,39%) в виде розового твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 209,6. 1H ЯМР (300 МГц, DMSO-d6) δ 2.22 (3H. t). 4.18 (3H. s). 5.50 (1H. p). 6.43 (1H. d). 9.45 (1H, s).
Стадия 2. 1-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(проп-1-ен-2-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин
Смесь CS2CO3 (468.47 мг; 1.438 ммоль; 2.5 экв.). XantPhos (66,56 мг; 0,115 ммоль; 0,2 экв.), Pd(AcO)2 (25,82 мг; 0,115 ммоль; 0,2 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (127,82 мг; 0,863 ммоль; 1,50 экв.) и 5-хлор-1-метил-3-(проп-1-ен-2-ил)пиразоло[4,3-d]пиримидина (120,00 мг; 0,575 ммоль; 1,00 экв.) в диоксане (4,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 15:1) с получением 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-(проп-1-ен-2-ил)пиразоло[4,3-d]пиримидин-5-амина (90 мг; 48.85%) в виде желтого твердого вещества. ЖХМС: m/z (ESI). [М+Н]+ составляет 321.3. 1H ЯМР (300 МГц. DMSO-d6) δ 2.18 (3Н. s). 2.45 (3H. d). 4.09 (3H. d). 5.33 (1H, d), 6.35 (1H, d), 7.70-7.78 (1H, m), 8.38 (1H, s), 8.96 (1H, s), 9.22 (1H, s), 9.37 (1H,s).
Стадия 3. 3-Изопропил-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 4)
Смесь Pd/C (89.69 мг; 0.843 ммоль; 3 экв.) и 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-(проп-1-ен-2-ил)пиразоло[4,3-d]пиримидин-5-амина (90,00 мг; 0,281 ммоль; 1,00 экв.) в МеОН (10,00 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водорода. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (4×50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2:МеОН, 12:1) с получением неочищенного продукта. Неочищенный продукт (80 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: МеОН; скорость потока: 25 мл/мин; градиент: от 58 В до 70 В за 7 минут; RT1: 5,57) с получением 3-изопропил-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (50 мг; 54,66%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 323,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.39 (6Н, d), 2.45 (3H, d), 3.18-3.34 (1H, m), 4.04 (3H, s), 7.69-7.75 (1H, m), 8.37 (1H, s), 8.79 (1H, s), 9.16 (1H, s), 9.44 (1H, s).
Пример 5
Получение 3-циклогексил-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 5)
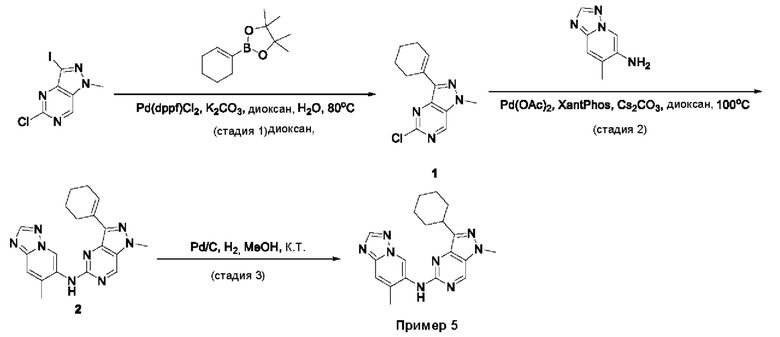
СХЕМА 5
Стадия 1. 5-Хлор-3-(циклогекс-1-ен-1-ил)-1-метил-1Н-пиразоло[4,3-d]пиримидин K2CO3 (293,33 мг; 2,122 ммоль; 2,50 экв.), 2-(циклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (265,01 мг; 1,273 ммоль; 1,50 экв.) и 5-хлор-3-иод-1-метилпиразоло[4,3-d]пиримидин (250,00 мг; 0,849 ммоль; 1,00 экв.) в диоксане (5,00 мл) и H2O (1,00 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 1:2) с получением 5-хлор-3-(циклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]пиримидина (150 мг; 71,04%) в виде розового твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 249,3. 1H ЯМР (300 МГц, DMSO-d6) δ 1.61-1.80 (4Н, m), 2.29 (2H, s), 2.54 (2H, s), 4.14 (3Н, s), 7.17-7.27 (1H, m), 9.40 (1H, s).
Стадия 2. 3-(Циклогекс-1-ен-1-ил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин
Смесь CS2CO3 (458,51 мг; 1,407 ммоль; 2,5 экв.), XantPhos (65,14 мг; 0,113 ммоль; 0,2 экв.), Pd(AcO)2 (25,28 мг; 0,113 ммоль; 0,2 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (125,11 мг; 0,844 ммоль; 1,5 экв.) и 5-хлор-3-(циклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]пиримидина (140,00 мг; 0,563 ммоль; 1,00 экв.) в диоксане (4,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 15:1) с получением 3-(циклогекс-1-ен-1-ил)-1-метил-Н-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (140 мг; 69,00%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 361,3. 1H ЯМР (300 МГц, DMSO-d6) δ 1.61-1.83 (4Н, m), 2.17-2.33 (4H, m), 2.46 (3H, d), 4.06 (3H, s), 7.21 (1H, s), 7.73 (1H, s), 8.38 (1H, s), 8.88 (1H, s), 9.19 (1H, s), 9.46 (1H, s).
Стадия 3. 3-Циклогексил-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 5)
Раствор 3-(никлогекс-1-ен-1-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]-пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (160,00 мг; 0,444 ммоль; 1,00 экв.) и Pd/C (236,21 мг; 2,220 ммоль; 5,00 экв.) в THF (40,00 мл) и МеОН (80,00 мл) перемешивали в течение 3 суток при комнатной температуре в атмосфере водорода. Осажденные твердые вещества собирали посредством фильтрации и промывали МеОН (5х30 мл). Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт (150 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 40 В до 50 В за 7 минут; RT1: 6,55) с получением 3-циклогексил-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (17,41 мг; 10,82%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [M+H]+ составляет 363,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.33 (3H, d), 1.76 (5H, d), 2.00 (2H, d), 2.46 (3H, d), 2.88-3.05 (1H, m), 4.04 (3H, s), 7.72 (1H, t), 8.38 (1H, s), 8.78 (1H, s), 9.15 (1H,s),9.51(1H,s).
Пример 8
Получение 3-((1r,4r)-4-метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 7) и 3-((1s,4s)-4-метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 8)
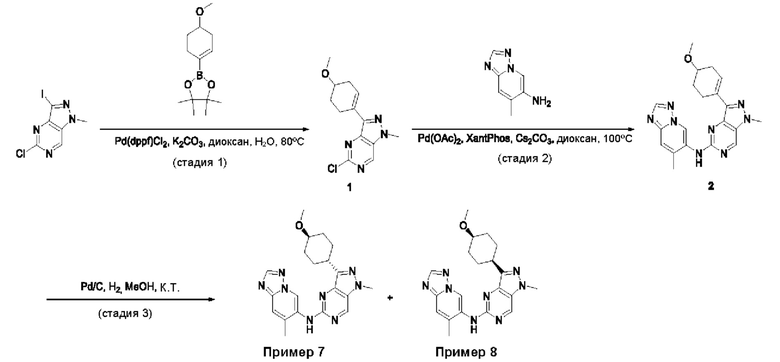
СХЕМА 8
Стадия 1. 5-Хлор-3-(4-метоксициклогекс-1-ен-1-ил)-1-метил-1Н-пиразоло[4,3-d]пиримидин
Смесь K2CO3 (234,66 мг; 1,698 ммоль; 2,5 экв.), Pd(dppf)Cl2 CH2Cl2 (110,93 мг; 0,136 ммоль; 0,2 экв.), 2-(4-метоксициклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (194,07 мг; 0,815 ммоль; 1,2 экв.) и 5-хлор-3-иод-1-метилпиразоло[4,3-d]пиримидина (200,00 мг; 0,679 ммоль; 1,00 экв.) в диоксане (5,00 мл) и H2O (1,00 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 1:2) с получением 5-хлор-3-(4-метоксициклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]пиримидина (175 мг; 92,44%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 279,3. 1H ЯМР (300 МГц, CDCl3) δ 1.89 (2Н, d), 2.11 (1H, m), 2.33 (1H, t), 2.66 (1H, m), 2.88 (1H, d), 3.43 (3H, s), 3.60 (1H, d), 4.12 (3H, s), 7.27 (1H, d), 8.89 (1H, s).
Стадия 2. 3-(4-Метоксициклогекс-1-ен-1-ил)-1-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин
Смесь CS2CO3 (496,78 мг; 1,525 ммоль; 2,5 экв.), XantPhos (70,58 мг; 0,122 ммоль; 0,2 экв.), Pd(AcO)2 (27,39 мг; 0,122 ммоль; 0,2 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (99,40 мг; 0,671 ммоль; 1,1 экв.) и 5-хлор-3-(4-метоксициклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]пиримидина (170,00 мг; 0,610 ммоль; 1,00 экв.) в диоксане (5,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением 3-(4-метоксициклогекс-1-ен-1-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (100 мг; 41,99%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 392,4. 1H ЯМР (300 МГц, DMSO-d6) δ 1.67 (1Н, d), 2.00 (1H, s), 2.27 (1H, m), 2.46 (3H, d), 2.72 (1H, d), 3.17 (2H, d), 3.33 (3H, s), 3.53 (1H, s), 4.07 (3H, s), 7.08 (1H, s), 7.73 (1H, s), 8.39 (1H, s), 8.91 (1H, s), 9.19 (1H, s), 9.43 (1H, s).
Стадия 3. 3-((1r,4r)-4-Метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 7) и 3-((1s,4s)-4-метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 8)
Смесь Pd/C (65,41 мг; 0,615 ммоль; 3 экв.) и 3-(4-метоксициклогекс-1-ен-1-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (80,00 мг; 0,205 ммоль; 1,00 экв.) в МеОН (150,00 мл) и THF (80,00 мл) перемешивали в течение ночи при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3×50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/MeOH, 12:1) с получением неочищенного продукта. Неочищенный продукт (80 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3•H2O), подвижная фаза В: ACN; скорость потока: 25 мл/мин; градиент: от 37 В до 41 В за 7 минут; RT1: 5,30/5,92) с получением 3-((1r,4r)-4-метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 7; 60 мг; 22,73%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 393,3. 1H ЯМР (400 МГц, DMSO-d6) δ 1.28 (2H, q), 1.83 (2H, q), 2.09 (4H, t), 2.43 (3H, d), 2.95 (1H, t), 3.30 (4H,m), 4.04 (3H, s), 7.73 (1H, s), 8.38 (1H, s), 8.81 (1H, s), 9.16 (1H, s), 9.51 (1H, s); 3-((1s,4s)-4-метоксициклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 8; 10 мг; 12,19%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 393,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.57 (2H, t), 1.76 (2H, m), 1.86 (2H, m), 2.04 (2H, q), 2.43 (3H, d), 3.05 (1H, m), 3.20 (3H, s), 3.42 (1H, s), 4.04 (3H, s), 7.72 (1H, s), 8.37 (1H, s), 8.80 (1H, s), 9.14 (1H, s), 9.34 (1H, s).
Пример 9
Получение 3-(4,4-дифторциклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 9)
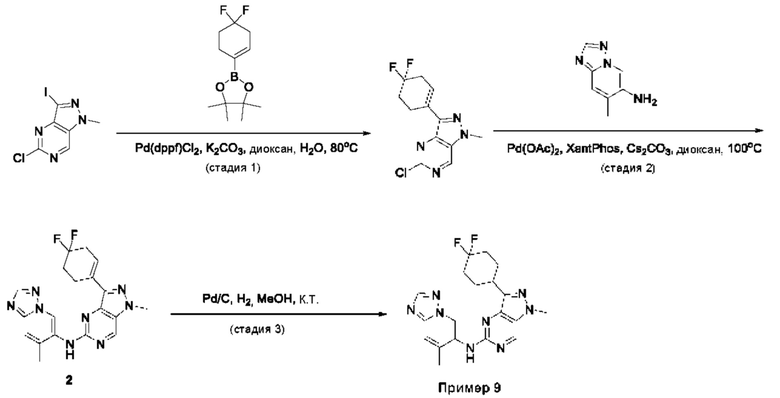
СХЕМА 9
Стадия 1. 5-Хлор-3-(4,4-дифторциклогекс-1-ен-1-ил)-1-метил-1Н-пиразоло[4,3-d]пиримидин
Смесь 5-хлор-3-иод-1-метилпиразоло[4,3-d]пиримидина (200,00 мг; 0,679 ммоль; 1,00 экв.), 2-(4,4-дифторциклогекс-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (198,93 мг; 0,815 ммоль; 1,20 экв.), Pd(dppf)Cl2 (99,39 мг; 0,136 ммоль; 0,2 экв.) и K2CO3 (234,66 мг; 1,698 ммоль; 2,5 экв.) в диоксане (3,00 мл) и H2O (0,60 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 2:3) с получением 5-хлор-3-(4,4-дифторциклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]пиримидина (100 мг; 51,72%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 285,3.
Стадия 2. 3-(4,4-Дифторциклогекс-1-ен-1-ил)-1-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин
Смесь 5-хлор-3-(4,4-дифторциклогекс-1-ен-1-ил)-1-метилпиразоло[4,3-d]-пиримидина (100,00 мг; 0,351 ммоль; 1,00 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (62,45 мг; 0,421 ммоль; 1,20 экв.), Pd(OAc)2 (15,77 мг; 0,070 ммоль; 0,20 экв.), XantPhos (60,97 мг; 0,105 ммоль; 0,30 экв.) и Cs2CO3 (286,12 мг; 0,878 ммоль; 2,50 экв.) в диоксане (2,50 мл) перемешивали в течение 2 часов при 100°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением 3-(4,4-дифторциклогекс-1-ен-1-ил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (110 мг; 79,00%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 397,3.
Стадия 3. 3-(4,4-Дифторциклогексил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 9)
Смесь 3-(4,4-дифторциклогекс-1-ен-1-ил)-1-метил-N-[7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (130,00 мг; 0,328 ммоль; 1,00 экв.) и Pd/C (174,50 мг; 1,640 ммоль; 5,00 экв.) в МеОН (60,00 мл) и THF (30,00 мл) перемешивали в течение 5 часов при 30°С в атмосфере водорода. Полученную смесь фильтровали, осадок на фильтре промывали CH2Cl2 (3×40 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1). Неочищенный продукт (70 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅Н2О), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 34 В до 48 В за 7 минут; RT1: 5,97) с получением 3-(4,4-дифторциклогексил)-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (17 мг; 13,01%) в виде розового твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 399,3. 1H ЯМР (300 МГц, MeOD-d4) δ 1.80-2.22 (8Н, m), 2.49 (3Н, d), 3.22 (1H, s), 4.03 (3Н, s), 7.58 (1H, t), 8.27 (1H, s), 8.98 (1H, s), 9.62 (1H, s).
Пример 10
Получение 1-(метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 10)
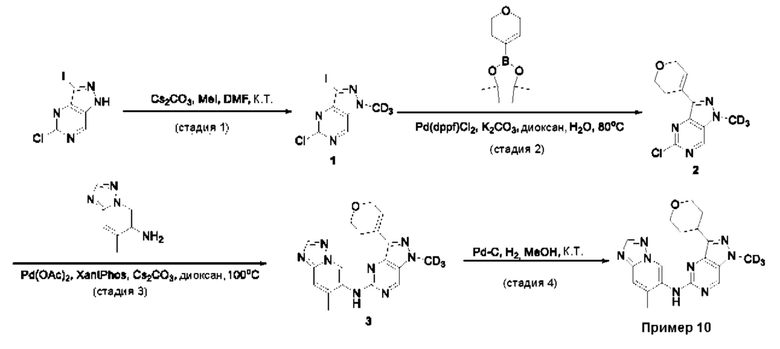
СХЕМА 10
Стадия 1. 5-Хлор-3-иод-1-(метил-d3)-1H-пиразоло[4.3-d]пиримидин
Смесь Cs2CO3 (1045,60 мг; 3,209 ммоль; 3 экв.), CD3I (775,31 мг; 5,349 ммоль; 5 экв.) и 5-хлор-3-иод-1Н-пиразоло[4,3-d]пиримидина (300,00 мг; 1,070 ммоль; 1,00 экв.) в DMF (6,00 мл) перемешивали в течение 1 часа при 0°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь разбавляли водой (50 мл). Полученную смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным рассолом (3×50 мл) и Na2S2O3 (3×50 мл) и сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении с получением 5-хлор-3-иод-1-(метил-d3)-1Н-пиразоло[4,3-d]пиримидина (260 мг; 81,70%) в виде розового твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 298,0. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.45 (1H, s).
Стадия 2. 5-Хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-(метил-d3)-1Н-пиразоло[4,3-d]пиримидин
Смесь K2CO3 (313,58 мг; 2,269 ммоль; 2,5 экв.), Pd(dppf)Cl2 CH2Cl2 (148,23 мг; 0,182 ммоль; 0,2 экв.), 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (228,79 мг; 1,089 ммоль; 1,2 экв.) и 5-хлор-3-иод-1-(метил-d3)-1Н-пиразоло[4,3-d]пиримидина (270,00 мг; 0,908 ммоль; 1,00 экв.) в диоксане (5,00 мл) и воде (1,00 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (гексан/EtOAc, 1:2) с получением 5-хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-(метил-d3)-1Н-пиразоло[4,3-d]пиримидина (100 мг; 43,43%) в виде розового твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 254,2. 1Н ЯМР (400 МГц, DMSO-d6) δ 2,62 (2Н, m), 3,86 (2Н, t), 4,33 (2Н, q), 7.18 (1Н, t), 9.43 (1Н, s).
Стадия 3. 3-(3,6-Дигидро-2Н-пиран-4-ил)-1-(метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4.3-d]пиримидин-5-амин
Смесь Cs2CO3 (385,28 мг; 1,182 ммоль; 3 экв.), XantPhos (45,61 мг; 0,079 ммоль; 0,2 экв.), Pd(ОАс)2 (17,70 мг; 0,079 ммоль; 0,2 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (70,08 мг; 0,473 ммоль; 1,20 экв.) и 5-хлор-3-(3,6-дигидро-2Н-пиран-4-ил)-1-(метил-d3)-1Н-пиразоло[4,3-d]пиримидина (100,00 мг; 0,394 ммоль; 1,00 экв.) в диоксане (3,00 мл) перемешивали в течение 2 часов при 100°C в атмосфере азота. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/MeOH, 12:1) с получением 3-(3,6-дигидро-2Н-пиран-4-ил)-1-(метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (100 мг; 69,43%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 366,3. 1Н ЯМР (400 МГц, DMSO-d6) δ 2.44 (2Н, d), 2.61 (3H, s), 3.17 (1H, d), 3.85 (2Н, t), 4.26 (2Н, q), 7.72 (1H, s), 8.38 (1H, s), 8.94 (1H, s), 9.20 (1H, s), 9.37 (1Н, s).
Стадия 4. 1-(Метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин (соединение по примеру 10)
Смесь Pd/C (87,37 мг; 0,821 ммоль; 3 экв.) и 3-(3,6-дигидро-2Н-пиран-4-ил)-1-(метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[4,3-d]пиримидин-5-амина (100,00 мг; 0,274 ммоль; 1,00 экв.) в МеОН (10,00 мл) и THF (10,00 мл) перемешивали в течение 2 часов при 35°С в атмосфере водорода. Требуемый продукт можно было определить посредством ЖХМС. Полученную смесь фильтровали, осадок на фильтре промывали МеОН (3×30 мл) и DCM (3×30 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением неочищенного продукта. Неочищенный продукт (40 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 15 В до 35 В за 7 минут; RT1: 6,4) с получением 1-(метил-d3)-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амина (10 мг; 9,85%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 368,2. 1H ЯМР (400 МГц, DMSO-d6) δ 1.94 (4Н, d), 2.43 (3H, d), 3.24 (1H, d), 3.48 (2Н, m), 3.93 (2Н, m), 7.71 (1H, s), 8.37 (1H, s), 8.81 (1H, s), 9.15 (1H, s), 9.33 (1H, s).
Пример 11/12
Получение 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1s,4s)-4-(дифторметокси)циклогексил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 11) и 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1r,4r)-4-(дифторметокси)циклогексил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 12)
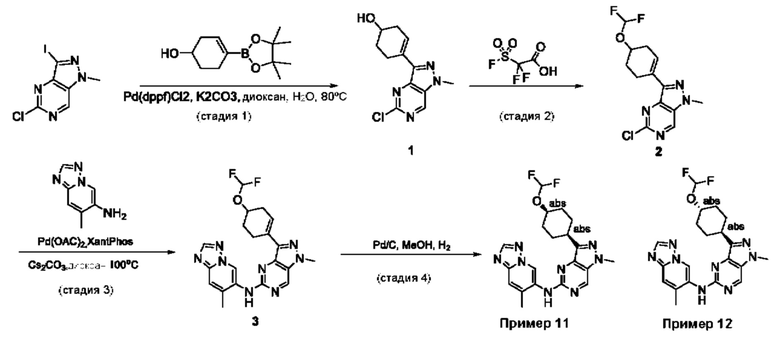
СХЕМА 11/12
Стадия 1. 4-(5-Хлор-1-метил-1H-пиразоло[4,3-d]пиримидин-3-ил)циклогекс-3-ен-1-ол
В сосуд на 40 мл добавляли 5-хлор-3-иод-1-метилпиразоло[4,3-d]пиримидин (500,00 мг; 1,698 ммоль; 1,00 экв.) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-3-ен-1-ол (570,78 мг; 2,547 ммоль; 1,50 экв.), Pd(dppf)Cl2 (248,47 мг; 0,340 ммоль; 0,20 экв.), K2CO3 (938,64 мг; 6,792 ммоль; 4 экв.), диоксан (10,00 мл) и H2O (2,00 мл) при комнатной температуре. Затем смесь перемешивали при 80°С в атмосфере азота в течение 3 часов. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (РЕ:ЕА, 1:4) с получением 4-[5-хлор-1-метилпиразоло[4,3-d]пиримидин-3-ил]циклогекс-3-ен-1-ола (253 мг; 56,29%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 265,2.
Стадия 2. 5-Хлор-3-(4-(дифторметокси)циклогекс-1-енил)-1-метил-1Н-пиразоло-[4,3-d] пиримидин
В сосуд на 40 мл добавляли 4-[5-хлор-1-метилпиразоло[4,3-d]пиримидин-3-ил]циклогекс-3-ен-1-ол (253,00 мг; 0,956 ммоль; 1,00 экв.), CuI (63,71 мг; 0,335 ммоль; 0,35 экв.), MeCN (10,00 мл) при 50°С, и в смесь по каплям добавляли 2,2-дифтор-2-(фторсульфонил)уксусную кислоту (510,61 мг; 2,867 ммоль; 3,00 экв.). Затем смесь перемешивали при 50°С в воздушной атмосфере в течение 3 часов. Полученную смесь разбавляли водой (10 мл). Водный слой экстрагировали DCM (3×30 мл). Полученную смесь разбавляли DCM (5 мл). Полученную смесь фильтровали, осадок на фильтре промывали DCM (10 мл × 3). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 1:1) с получением 5-хлор-3-[4-(дифторметокси)циклогекс-1-ен-1-ил]-1-метилпиразоло-[4,3-d]пиримидина (180 мг; 47,87%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 315,2.
Стадия 3. 3-(4-(Дифторметокси)циклогекс-1-енил)-1-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[4,3-d]пиримидин-5-амин
В сосуд на 40 мл добавляли 5-хлор-3-[4-(дифторметокси)циклогекс-1-ен-1-ил]-1-метилпиразоло[4,3-d]пиримидин (180,00 мг; 0,572 ммоль; 1,00 экв.) и 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амин (101,69 мг; 0,686 ммоль; 1,20 экв.), Pd(OAc)2 (38,52 мг; 0,172 ммоль; 0,30 экв.), XantPhos (99,28 мг; 0,172 ммоль; 0,30 экв.), Cs2CO3 (559,05 мг; 1,716 ммоль; 3,00 экв.) и диоксан (10,00 мл) при комнатной температуре. Затем смесь перемешивали при 70°С в атмосфере азота в течение 3 часов. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (DCM:MeOH, 15:1) с получением 3-[4-(дифторметокси)циклогекс-1-ен-1-ил]-1-метил-N-[7-метил-[1,2,4]триазоло[l,5-a]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (135 мг; 55,35%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 427,1. 1Н ЯМР (400 МГц, CDCl3) δ1.97-2.23 (3Н, m), 2.55 (4Н, s), 2.68-2.87 (2Н, m), 2.98 (1H, d), 4.11 (3H, s), 4.54-4.62 (1Н, m), 6.35 (1H, t), 7,00 (1H, s), 7.61 (1H, s), 8.29 1H, s), 8.82 (1H, s), 9.90 (1H, s).
Стадия 4. Получение 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1s,4s)-4-(дифторметокси)циклогексил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 11) и 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1r,4r)-4-(дифторметокси)циклогексил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 12)
В перемешиваемую смесь 3-[4-(дифторметокси)циклогекс-1-ен-1-ил]-1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[4,3-d]пиримидин-5-амина (135,00 мг; 0,317 ммоль; 1,00 экв.) в МеОН (20 мл) добавляли Pd/C (168,45 мг; 1,583 ммоль; 5,00 экв.) в воздушной атмосфере. Полученную смесь перемешивали в течение 4 часов при 40°С в атмосфере водорода. Полученную смесь фильтровали, и осадок на фильтре промывали DCM (8×100 мл). Фильтрат концентрировали при пониженном давлении с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 34 В до 54 В за 7 минут; RT1: 5,93) с получением твердого вещества. Затем продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK IG, 2×25 см, 5 мкм; подвижная фаза А: гексан:DCM, 3:1, подвижная фаза В: EtOH; скорость потока: 20 мл/мин; градиент: от 10 В до 10 В за 15 минут; RT1: 10; RT2: 11;) с получением 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1s,4s)-4-(дифторметокси)циклогексил]пиразоло[4,3-d]пиримидин-5-амина (соединение по примеру 11; 15 мг; 50,00%) в виде белого твердого вещества и 1-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-[(1r,4r)-4-(дифторметокси)циклогексил]пиразоло-[4,3-d]пиримидин-5-амина (соединение по примеру 12; 5 мг; 16,67%) в виде белого твердого вещества.
(Соединение по примеру 11) ЖХМС: m/z (ESI), [М+Н]+ составляет 429,3. 1H ЯМР (300 МГц, DMSO-d6) δ 1.87 (2Н, d), 1.97 (4Н, d), 2.09 (2Н, d), 2.43 (3H, s), 3.07 (1H, d), 4.04 (3H, s), 4.37 1H, s), 6.69 (1H, t), 7.70 (1H, s), 8.36 (1H, s), 8.79 1H, s), 9.14 (1H, s), 9.33 (1H, s);
(Соединение по примеру 12) ЖХМС: m/z (ESI), [М+Н]+ составляет 429,3. 1Н ЯМР (300 МГц, MeOD-d4) δ 1.59-1.71 (2Н, m), 1.95-2.09 (2Н, m), 2.15-2.20 (4Н, m), 2.55 (3H,s), 3.05-3.14 (1H, m), 4.09 (3H, s), 4.20-4.27 (1H, m), 6.71 (1H, t), 7.64 (1H, s), 8.31 1H, s), 9.03 (1Н, s), 9.85 (1H, s).
Пример 13
Получение 3-метил-Н-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 13)
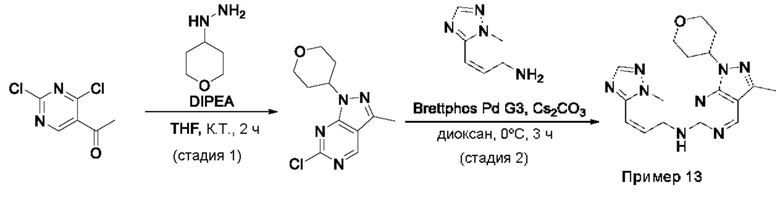
СХЕМА 13
Стадия 1. 6-Хлор-3-метил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d] пиримидин
В перемешиваемую смесь 1-(2,4-дихлорпиримидин-5-ил)этанона (500,00 мг; 2,618 ммоль; 1,00 экв.) и DIPEA (N,N-диизопропилэтиламин) (1353,26 мг; 10,471 ммоль; 4,00 экв.) в THF (5,00 мл) порциями добавляли оксан-4-илгидразин (364,89 мг; 3,141 ммоль; 1,20 экв.) при комнатной температуре в атмосфере азота. И смесь перемешивали в течение 2 часов при комнатной температуре в атмосфере азота. Полученную смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (3х10 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 2:1) с получением 6-хлор-3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидина (350 мг; 52,91%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 253,2.
Стадия 2. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 13)
В перемешиваемую смесь 6-хлор-3-метил-1-(оксан-4-ил)-1Н-пиразоло[3,4-d]пиримидина (200 мг; 0,791 ммоль; 1 экв.), Cs2CO3 (644,68 мг; 1,979 ммоль; 2,5 экв.) и 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (140,72 мг; 0,950 ммоль; 1,2 экв.) в диоксане (20 мл) порциями добавляли BrettPhos Pd G3 (143,49 мг; 0,158 ммоль; 0,2 экв.) при комнатной температуре в атмосфере азота. И смесь перемешивали в течение 3 часов при 100°С в атмосфере азота. Полученную смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали водой (3×50 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: Xselect CSH OBD, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% TFA), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 18% В до 29% В за 7 минут; RT: 6,30 мин) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-(оксан-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (74 мг; 25,66%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 365,3. 1H ЯМР (DMSO-d6, 300 МГц) δ 1.7-1.9 (2Н, m), 2.0-2.2 (2Н, m), 2.4-2.5 (6Н, m), 3.4 (2Н, td), 3.9-4.1 (2Н, m), 4.5-4.7 (1H, m), 7.8 (1H, s), 8.6 (1H, s), 8.9 (1H, s), 9.3 (1H, s), 9.3 (1H, s).
Пример 14
Получение 3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 14)
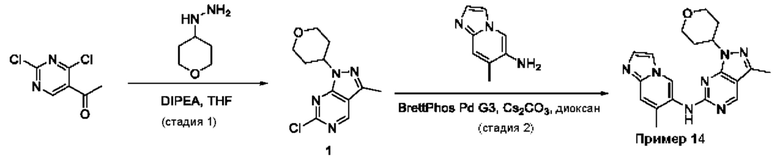
СХЕМА 14
Стадия 1. 6-Хлор-3-метил-1 -(тетрагидро-2Н-пиран-4-ил)- 1Н-пиразоло[3,4-d]пиримидин
Раствор 1-(2,4-дихлорпиримидин-5-ил)этанона (250,00 мг; 1,309 ммоль; 1,00 экв.) и оксан-4-илгидразина (182,45 мг; 1,571 ммоль; 1,20 экв.), DIPEA (338,32 мг; 2,618 ммоль; 2,00 экв.) в THF (10,00 мл) перемешивали в течение 2 часов при 0°С в атмосфере азота. Полученную смесь экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали рассолом (10 мл × 3), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ/EtOAc, 5:1) с получением 6-хлор-3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидина (230 мг; 69,54%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 253,2.
Стадия 2. 3-Метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-1H-пиразоло[3.4-d]пиримидин-6-амин (соединение по примеру 14)
Смесь 7-метилимидазо[1,2-а]пиридин-6-амина (139,78 мг; 0,950 ммоль; 1,50 экв.) и 6-хлор-3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидина (160 мг; 0,633 ммоль; 1,00 экв.), BrettPhos Pd G3 (57,40 мг; 0,063 ммоль; 0,10 экв.), Cs2CO3 (412,59 мг; 1,266 ммоль; 2,00 экв.) в диоксане (5,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Остаток очищали посредством колоночной хроматографии на силикагеле, элюируя смесью CH2Cl2/МеОН (10:1) с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ВЭЖХ с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 17 В до 37 В за 7 минут; RT1: 6,75) с получением 3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]-1-(оксан-4-ил)пиразоло[3,4-d]пиримидин-6-амина (140 мг; 60,84%) в виде не совсем белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 364,1, 1H ЯМР (DMSO-d6, 300 МГц) δ 1.79 (2Н, d), 2.02-2.18 (2Н, m), 2.23 (3H, d), 2.42 (3H, s), 3.44 (2Н, dd), 3.94 (2Н, dd), 4.62 (1H, dd), 7.42 (1H, s), 7.48 (1H, d), 7.84 (1H, s), 8.69 (1H, s), 8.85 (1H, s), 9.04 (1Н, s). Пример 15
Получение 7-метил-N-(5-метил-7-(тетрагидро-2Н-пиран-4-ил)-7Н-пирроло[2,3-d]пиримидин-2-ил)-[1,2,4]триазоло[l,5-a]пиридин-6-амина (соединнеие по примеру 15)
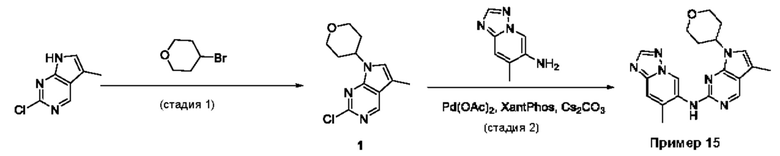
СХЕМА 15
Стадия 1. 2-Хлор-5-метил-7-(тетрагидро-2Н-пиран-4-ил)-7Н-пирроло[2,3-d]пиримидин
Смесь 2-хлор-5-метил-7Н-пирроло[2,3-d]пиримидина (400,00 мг; 2,387 ммоль; 1,00 экв.), 4-бромоксана (3,94 г; 23,874 ммоль; 10,00 экв.) и K2CO3 (824,62 мг; 5,967 ммоль; 2,50 экв.) в DMSO (50,00 мл; 12,922 ммоль) перемешивали в течение ночи при 120°С в атмосфере азота. Полученную смесь разбавляли водой (150 мл). Полученную смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали рассолом (3×30 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1). Неочищенный продукт (70 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 28 В до 48 В за 7 минут; RT1: 5,80) с получением 2-хлор-5-метил-7-(оксан-4-ил)пирроло[2,3-d]пиримидина (40 мг; 6,66%) в виде желтого твердого вещества.
Стадия 2. 7-Метил-N-(5-метил-7-(тетрагидро-2Н-пиран-4-ил)-7Н-пирроло[2,3-d]пиримидин-2-ил)-[1,2,4]триазоло[1,5-а]пиридин-6-амин (соединение по примеру 15)
Смесь 2-хлор-5-метил-7-(оксан-4-ил)пирроло[2,3-d]пиримидина (30,00 мг; 0,119 ммоль; 1,00 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (21,19 мг; 0,143 ммоль; 1,20 экв.), Pd(AcO)2 (5,35 мг; 0,024 ммоль; 0,20 экв.), XantPhos (20,69 мг; 0,036 ммоль; 0,30 экв.) и CS2CO3 (97,08 мг; 0,298 ммоль; 2,50 экв.) в диоксане (1,50 мл) перемешивали в течение 2 часов при 100°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением неочищенного продукта. Неочищенный продукт (40 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 26 В до 46 В за 7 минут; RT1: 6,37) с получением 5-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-7-(оксан-4-ил)пирроло[2,3-d]пиримидин-2-амина (6 мг; 13,85%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 364,3. 1Н ЯМР (300 МГц, MeOD-d4) δ1.79-1.91 (2Н, m), 1.95-2.14 (2Н, m), 2.24 (3H, d), 2.43 (3H, d), 3.47 (2Н, td), 3.93-4.05 (2Н, m), 4.55-4.72 (1H, m), 7.18 (1H, d), 7.58-7.74 (1H, m), 8.37 (1H, s), 8.62 (2Н, d), 9.32 (1H, s).
Пример 16
Получение N-(7-хлор-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-метил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 16)
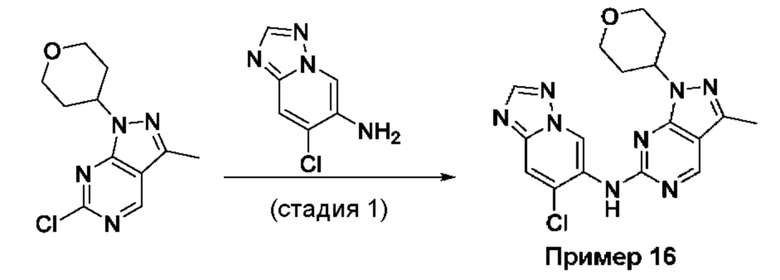
СХЕМА 16
Стадия 1. N-(7-Хлор-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-3-метил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 16)
Смесь 7-хлор-[1,2,4]триазоло[1,5-а]пиридин-6-амина (50,00 мг; 0,297 ммоль; 1,00 экв.), 6-хлор-3-метил-1-(оксан-4-ил)пиразоло[3,4-(1]пиримидина (74,95 мг; 0,297 ммоль; 1,00 экв.), Pd(AcO)2 (13,32 мг; 0,059 ммоль; 0,2 экв.), XantPhos (51,48 мг; 0,089 ммоль; 0,3 экв.) и CS2CO3 (241,59 мг; 0,741 ммоль; 2,5 экв.) в диоксане (4,00 мл) перемешивали в течение 2 часов при 100°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1). Неочищенный продукт (90 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 25 мл/мин; градиент: от 30 В до 40 В за 7 минут; RT1: 6,62) с получением N-[7-хлор-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидин-6-амина (32,77 мг; 28,71%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 385,2. 1Н ЯМР (300 МГц, DMSO-d6) δ1.84 (2Н, d), 2.13 (2Н, dd), 2.46 (3H, s), 3.39-3.57 (2Н, m), 3.97 (2Н, dd), 4.54-4.77 (1Н, m), 8.23 (1H, s), 8.57 (1H, s), 8.93 (1H, s), 9.36 (2Н, d).
Пример 17
Получение N-(7-метил-[1,2,4]триазоло [1,5-а]пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-3-(трифторметил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 17)
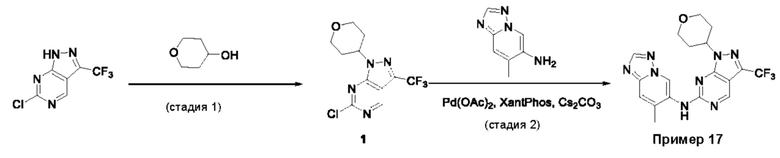
СХЕМА 17
Стадия 1; 6-Хлор-1-(тетрагидро-2Н-пиран-4-ил)-3-(трифторметил)-1Н-пиразоло[3,4-d]пиримидин
В перемешиваемую смесь 6-хлор-3-(трифторметил)-1Н-пиразоло[3,4-d]пиримидина (340,00 мг; 1,528 ммоль; 1,00 экв.), оксан-4-ола (624,11 мг; 6,111 ммоль; 4,00 экв.) и PPh3 (1442,48 мг; 5,500 ммоль; 3,60 экв.) в THF (2,50 мл) по каплям добавляли DIAD (диизопропилазодикарбоксилат) (1112,07 мг; 5,500 ммоль; 3,60 экв.) при 0°С в атмосфере азота. Полученную смесь экстрагировали EtOAc (3×20 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1) с получением 6-хлор-1-(тетрагидро-2Н-пиран-4-ил)-3-(трифторметил)-1Н-пиразоло[3,4-d]пиримидина (370 мг; 78,98%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 307,3.
Стадия 2. N-(7-Метил-[1,2,4]триазоло[1,5-а1пиридин-6-ил)-1-(тетрагидро-2Н-пиран-4-ил)-3-(трифторметил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 17)
Смесь 6-хлор-1-(оксан-4-ил)-3-(трифторметил)пиразоло[3,4-d]пиримидина (70,00 мг; 0,228 ммоль; 1,00 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (33,82 мг; 0,228 ммоль; 1,00 экв.), Pd(AcO)2 (10,25 мг; 0,046 ммоль; 0,20 экв.), XantPhos (39,62 мг; 0,068 ммоль; 0,30 экв.) и Cs2CO3 (185,93 мг; 0,571 ммоль; 2,50 экв.) в диоксане (3,00 мл) перемешивали в течение 3 часов при 70°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1). Неочищенный продукт (90 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 30 В до 50 В за 7 минут; RT1: 6,67) с получением N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-(оксан-4-ил)-3-(трифторметил)пиразоло[3,4-d]пиримидин-6-амина (54,44 мг; 53,86%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 419,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.77-2.01 (2Н, m), 2.01-2.24 (2Н, m), 2.39 (3H, d), 3.40-3.59 (2Н, m), 3.98 (2Н, d), 4.82 (1H, t), 7.78 (1H, d), 8.44 (1H, s), 9.13 (2Н, d), 9.69 (1H, s).
Пример 18
Получение 1-(4-метоксибензил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 18)
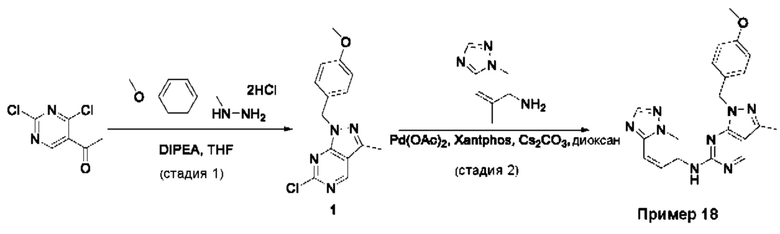
СХЕМА 18
Стадия 1. 6-Хлор-1-(4-метоксибензил)-3-метил-1H-пиразоло[3,4-d]пиримидин
В перемешиваемую смесь 1-(2,4-дихлорпиримидин-5-ил)этанона (640,00 мг; 3,351 ммоль; 1,00 экв.) и DIPEA (433,04 мг; 3,351 ммоль; 1,00 экв.) в THF по каплям добавляли [(4-метоксифенил)метил]гидразин (764,93 мг; 5,026 ммоль; 1,50 экв.) при 0°С в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (РЕ:ЕА, 1:1) с получением 6-хлор-1-[(4-метоксифенил)метил]-3-метилпиразоло[3,4-d]пиримидина (623 мг; 64,40%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 289,2. 1Н ЯМР (300 МГц, CDCl3) δ2.58 (3H, s), 3.77 (3H, s), 5.47 (2Н, s), 6.82-6.87 (2Н, m), 7.30-7.35 (2Н, m), 8.90 (1H, s).
Стадия 2. 1-(4-Метоксибензил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 18)
В перемешиваемую смесь 6-хлор-1-[(4-метоксифенил)метил]-3-метилпиразоло[3,4-d]пиримидина (100,00 мг; 0,346 ммоль; 1,00 экв.) и 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (76,97 мг; 0,519 ммоль; 1,50 экв.) в диоксане (5 мл) по каплям добавляли CS2CO3 (338,53 мг; 1,039 ммоль; 3,00 экв.) и XantPhos (40,08 мг; 0,069 ммоль; 0,20 экв.), Pd(AcO)2 (15,55 мг; 0,069 ммоль; 0.20 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2/Cl2/МеОН, 10:1) с получением желтого твердого вещества. Неочищенный продукт (70 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 26 В до 46 В за 7 минут; RT1: 7,07) с получением 1-[(4-метоксифенил)метил]-3-метил-N-[7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (20 мг; 28,57%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 401,2. 1Н ЯМР (400 МГц, DMSO-d6) δ 2.38 (3H, s), 2.44 (3H, s), 3.71 (3H, s), 5.24 (2Н, s), 6.85-6.88 (2Н, m), 7.18 (2Н, t), 7.77 (1H, s), 8.43 (1H, s), 8.95 (1H, s), 9.22 (2Н, d).
Пример 20
Получение 1-циклогексил-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 20)
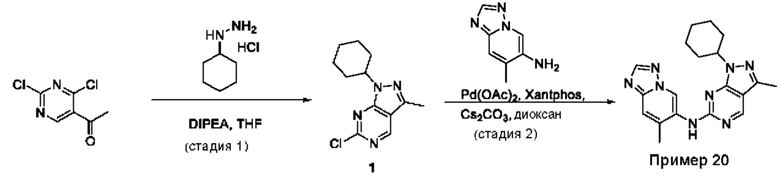
СХЕМА 20
Стадия 1. 6-Хлор-1-циклогексил-3-метил-1Н-пиразоло[3,4-d]пиримидин
В перемешиваемую смесь 1-(2,4-дихлорпиримидин-5-ил)этанона (180,00 мг; 0,942 ммоль; 1,00 экв.) и DIPEA (487,17 мг; 3,769 ммоль; 4,00 экв.) в THF(10 мл) порциями добавляли гидрохлорид циклогексилгидразина (184,56 мг; 1,225 ммоль; 1,30 экв.) при 0°С в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при 25°С в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (РЕ/ЕА, 1:2) с получением 6-хлор-1-циклогексил-3-метилпиразоло[3,4-d]пиримидина (145 мг; 61,37%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 251,2. 1Н ЯМР (300 МГц, CDCl3) δ 1.42-1.48 (2Н, m), 1.51-1.55 (2Н, m), 1.99-2.08 (6Н, m), 2.60 (3H, s), 4.66-4.76 (1H, m), 8.90 (1H, s).
Стадия 2. 1-Циклогексил-3-метил-Н-(7-метил-[1,2.4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло [3,4-d]пиримидин-6-амин (соединение по примеру 20)
В перемешиваемую смесь 6-хлор-1-циклогексил-3-метилпиразоло[3,4-d]пиримидина (120,00 мг; 0,479 ммоль; 1,00 экв.) и 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (106,37 мг; 0,718 ммоль; 1,5 экв.) в диоксане (6 мл) порциями добавляли Cs2CO3 (467,81 мг; 1,436 ммоль; 3 экв.) и XantPhos (55,39 мг; 0,096 ммоль; 0,2 экв.), Pd(AcO)2 (21,49 мг; 0,096 ммоль; 0,2 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2/Cl2/МеОН, 10:1) с получением неочищенного твердого вещества. Неочищенный продукт (80 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: % В; 254; 220 нм; RT1: 6,50) с получением 1-циклогексил-3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (60 мг; 34,59%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [M+H]+ составляет 363,2. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.23-1.38 (3H, m), 1.67 (1H, d), 1.81-1.97 (6Н, m), 2.41-2.44 (6Н, m), 4.36-4.43 (1H, m), 7.74 (1Н, s), 8.41 (1H, s), 8.91 (1H, s), 9.15 (1H, s), 9.22 (1H, s).
Пример 21
Получение 1-(4,4-дифторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 21)
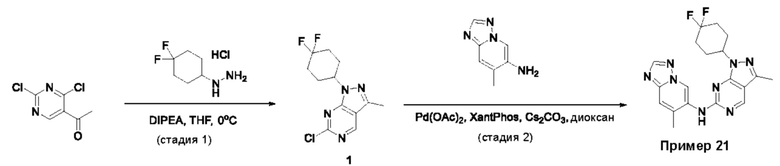
СХЕМА 21
Стадия 1. 6-Хлор-1-(4,4-дифторциклогексил)-3-метил-1Н-пиразоло[3,4-d]пиримидин
Смесь DIPEA (338,32 мг; 2,618 ммоль; 5 экв.), (4,4-дифторциклогексил)гидразина (86,48 мг; 0,576 ммоль; 1,10 экв.) и 1-(2,4-дихлорпиримидин-5-ил)этанона (100,00 мг; 0,524 ммоль; 1,00 экв.) в THF (5,00 мл) перемешивали в течение 3 часов при 0°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (гексан/EtOAc, 1:1) с получением 6-хлор-1-(4,4-дифторциклогексил)-3-метилпиразоло[3,4-d]пиримидина (80 мг; 53,30%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 287,3. 1Н ЯМР (300 МГц, CDCl3) δ 2.03 (4Н, d), 2.35 (4Н, d), 2.60 (3H, s), 4.85 (1H, t), 8.92 (1H, s).
Стадия 2. 1-(4.4-Дифторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 21)
Смесь Cs2CO3 (227,28 мг; 0,698 ммоль; 2,5 экв.), XantPhos (32,29 мг; 0,056 ммоль; 0,2 экв.), Pd(AcO)2 (12,53 мг; 0,056 ммоль; 0,2 экв.), 7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-амина (45,48 мг; 0,307 ммоль; 1,1 экв.) и 6-хлор-1-(4,4-дифторциклогексил)-3-метилпиразоло[3,4-d]пиримидина (80,00 мг; 0,279 ммоль; 1,00 экв.) в диоксане (3,00 мл) перемешивали в течение 2 часов при 100°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением неочищенного продукта. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 31 В до 51 В за 7 минут; RT1: 6,30) с получением 1-(4,4-дифторциклогексил)-3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (80 мг; 71,96%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 399,3. 1H ЯМР (300 МГц, DMSO-d6) δ 1.99 (3H, s), 2.15 (5Н, s), 2.38 (3H, d), 2.45 (3H, s), 4.62 (1H, s), 7.75 (1H, d), 8.42 (1H, s), 8.93 (1H, s), 9.13 (1H, s), 9.22 (1H, s).
Пример 22
Получение 1-(4,4-дифторциклогексил)-3-метил-N-(7-метилимидазо[1,2-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 22)
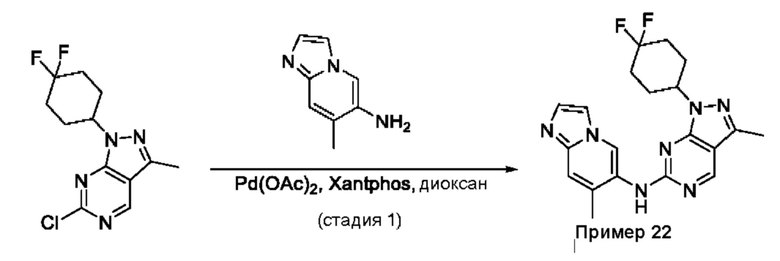
СХЕМА 22
Стадия 1. 1-(4,4-Дифторциклогексил)-3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 22)
Смесь 6-хлор-1-(4,4-дифторциклогексил)-3-метилпиразоло[3,4-d]пиримидина (67,00 мг; 0,234 ммоль; 1,00 экв.), 7-метилимидазо[1,2-а]пиридин-6-амина (68,79 мг; 0,467 ммоль; 2 экв.), Pd(AcO)2 (10,49 мг; 0,047 ммоль; 0,20 экв.), XantPhos (40,56 мг; 0,070 ммоль; 0,30 экв.) и Cs2CO3 (190,35 мг; 0,584 ммоль; 2,50 экв.) в диоксане (5.00 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1). Неочищенный продукт (50 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 30×150 мм, 5 мкм; подвижная фаза А: вода (10 ммоль/л NH4HCO3 плюс 0,1% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 38 В до 50 В за 7 минут; RT1: 5,63) с получением 1-(4,4-дифторциклогексил)-3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (20 мг) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 398,2. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.95 (3H, d), 2.15 (5Н, d), 2.26 (3H, d), 2.44 (3H, s), 4.62 (1H, s), 7.44 (1Н, s), 7.50 (1H, d), 7.86 (1H, t), 8.72 (1H, s), 8.88 (1H, s), 9.02 (1H, s).
Пример 26
Получение 1-((1r,4r)-4-метоксициклогексил)-3-метил-N-(7-метилимидазо[1,2-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 26)
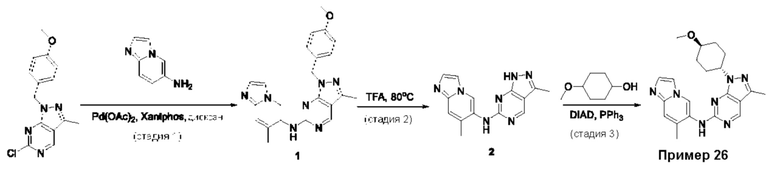
СХЕМА 26
Стадия 1. 1-(4-Метоксибензил)-3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1H-пиразоло [3,4-d]пиримидин-6-амин
Смесь 6-хлор-1-[(4-метоксифенил)метил]-3-метилпиразоло[3,4-d]пиримидина (200,00 мг; 0,693 ммоль; 1,00 экв.), 7-метилимидазо[1,2-а]пиридин-6-амина (152,92 мг; 1,039 ммоль; 1,50 экв.), XantPhos (120,24 мг; 0,208 ммоль; 0,30 экв.), Pd(AcO)2 (31,10 мг; 0,139 ммоль; 0,20 экв.) и Cs2CO3 (564,21 мг; 1,732 ммоль; 2,50 экв.) в диоксане (3 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением 1-[(4-метоксифенил)метил]-3-метил-N-[7-метилимидазо[1,2-a]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (200 мг; 72,28%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 400,3.
Стадия 2. 3-Метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин
Раствор 1-[(4-метоксифенил)метил]-3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (190,00 мг; 0,476 ммоль; 1,00 экв.) и TFA (70,00 мл; 613,910 ммоль; 1981,34 экв.) перемешивали в течение 2 суток при 80°С в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Полученную смесь разбавляли DCM (10 мл). Смесь доводили до значения рН 8 насыщенным NaHCO3 (водн.), фильтровали и промывали DCM (2×3 мл). Осажденные твердые вещества собирали с получением 3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (160 мг; 80,00%), ЖХМС: m/z (ESI), [М+Н]+ составляет 280,2.
Стадия 3. 1-((1r,4r)-4-Метоксициклогексил)-3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 26)
Смесь 3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (150,00 мг; 0,537 ммоль; 1,00 экв.), 4-метоксициклогексан-1-ола (174,79 мг; 1,343 ммоль; 2,50 экв.) и PPh3 (422,58 мг; 1,611 ммоль; 3,00 экв.) в THF (2,50 мл) перемешивали в течение 20 минут при 0°С в атмосфере азота, затем добавляли DIAD (325,78 мг; 1,611 ммоль; 3,00 экв.), и смесь перемешивали в течение 2 часов при 70°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/EtOAc, 12:1) с получением неочищенного твердого вещества. Неочищенный продукт (60,00 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD CI8, 30×150 мм, 5 мкм; подвижная фаза А: вода (10 ммоль/л NH4HCO3 плюс 0,1% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 29 В до 39 В за 9 минут; RT1: 6,22, 7,43) с получением 3-метил-N-[7-метилимидазо[1,2-a]пиридин-6-ил]-1-[(1r,4r)-4-метоксициклогексил]пиразоло[3,4-d]пиримидин-6-амина (16 мг; 26,67%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [M+H]+ составляет 392,2. 1Н ЯМР (300 МГц, MeOD-d4) δ 1.31-1.46 (2Н, m), 2.00 (2Н, s), 2.03-2.14 (2Н, m), 2.21 (2Н, d), 2.39 (3H, d), 2.50 (3H, s), 3.23 (1Н, d), 3.38 (3H, s), 4.51 (1Н, t), 7.46 (1H, s), 7.54 (1H, d), 7.80 (1H, s), 8.82 (2Н, s).
Пример 27
Получение 1-((1s,4s)-4-фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло-[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 27) и 1-((1r,4r)-4-фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 28)
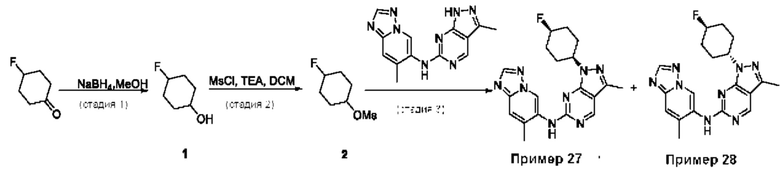
СХЕМА 27
Стадия 1. 4-Фторциклогексан-1-ол
В перемешиваемую смесь 4-фторциклогексан-1-она (1,00 г; 8,611 ммоль; 1,00 экв.) и МеОН (100,00 мл) порциями добавляли NaBH4 (0,98 мг; 0,026 ммоль; 3,0 экв.) при 0°С. Полученную смесь перемешивали в течение 16 часов при комнатной температуре. Реакцию останавливали посредством добавления воды при 0°С. Полученную смесь концентрировали при пониженном давлении. Полученную смесь разбавляли водой (50 мл). Полученную смесь экстрагировали CH2Cl2 (3×50 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении с получением 4-фторциклогексан-1-ола (800 мг; 78,63%) в виде светло-желтого твердого вещества. 1Н ЯМР (300 МГц, DMSO-d6) δ1.52-1.56 (5Н, m), 1.88-1.90 (3H, m), 3.54-3.57 (1H, m), 4.55-4.58 (1H, m), 4.70-4.72 (1H, т).
Стадия 2. 4-Фторциклогексилметансульфонат
В перемешиваемую смесь 4-фторциклогексан-1-ола (400,00 мг; 3,385 ммоль; 1,00 экв.) и TEA (1027,74 мг; 10,156 ммоль; 3,00 экв.) в DCM (10,00 мл) по каплям добавляли метансульфонилхлорид (581,66 мг; 5,078 ммоль; 1,5 экв.) при 0°С. Полученную смесь перемешивали в течение 2 часов при комнатной температуре. Полученную смесь разбавляли водой (50 мл). Полученную смесь экстрагировали CH2Cl2 (3×50 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Полученную смесь концентрировали при пониженном давлении с получением 4-фторциклогексилметансульфоната (650 мг; 97,84%) в виде светло-желтого твердого вещества. Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
1Н ЯМР (300 МГц, DMSO-d6) δ 1.52-1.90 (8Н, m), 3.22 (3H, s), 4.60-4.78 (2Н, m).
Стадия 3. 1-((1s,4s)-4-Фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 27) и 1-((1r,4r)-4-фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 28)
Смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло-[3,4-d]пиримидин-6-амина (150,00 мг; 0,535 ммоль; 1,00 экв.), 4-фторциклогексилметансульфоната (1050,18 мг; 5,352 ммоль; 10,00 экв.) и Cs2CO3 (523,09 мг; 1,605 ммоль; 3,00 экв.) в DMF (20,00 мл) перемешивали в течение 16 часов при 110°С в атмосфере азота. Полученную смесь разбавляли водой (20 мл). Полученную смесь экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 39 В до 59 В за 7 минут; RT1: 6,4) с получением:
1-((1s,4s)-4-фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 27; 6,8 мг; 3,34%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 381,2. 1H ЯМР (300 МГц, DMSO-d6) δ 1.66-1.68 (1H, m), 1.75 (3H, d), 2.04-2.06 (2Н, m), 2.18-2.19 (2Н, m), 2.38 (3H, d), 2.45 (3H, s), 4.50-4.53 (1H, m), 4.81-4.93 (1H, s), 7.73 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.15 (2Н, d);
1-((1r,4r)-4-фторциклогексил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 28; 18,8 мг; 9,23%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 381,2. 1Н ЯМР (400 МГц, DMSO-d6) δ 1.63-1.65 (2Н, m), 1.92-1.96 (4Н, m), 2.00-2.04 (2Н, m), 2.14 (3H, s), 2.41 (3H, s), 4.47-4.49 (1H, m), 4.61-4.65 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.19(2Н, d).
Пример 42
Получение 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(2-оксаспиро[3,3]гептан-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 42)
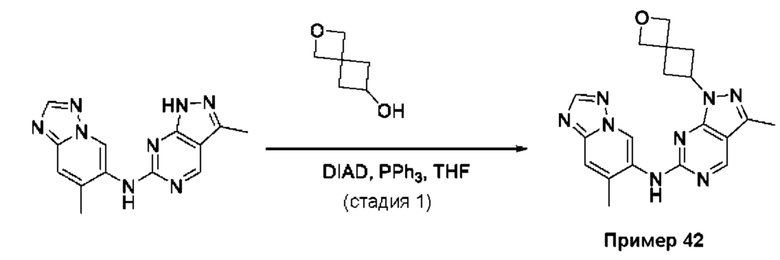
СХЕМА 42
Стадия 1. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(2-оксаспиро[3,3]гептан-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 42)
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (70,00 мг; 0,250 ммоль; 1,00 экв.) и 2-оксаспиро[3,3]гептан-6-ола (57,01 мг; 0,499 ммоль; 2,00 экв.) и PPh3 (196,51 мг; 0,749 ммоль; 3,00 экв.) в THF (10 мл) порциями добавляли DIAD (151,50 мг; 0,749 ммоль; 3,00 экв.) при 0°С в атмосфере N2. Полученную смесь перемешивали в течение 2 часов при 70°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (DCM/MeOH, 15:1) с получением неочищенного твердого вещества. Неочищенный продукт (80 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD C18, 30×150 мм, 5 мкм; подвижная фаза А: вода (10 ммоль/л NH4HCO3 плюс 0,1% NH3⋅Н2О), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 26 В до 36 В за 7 минут; RT1: 5,88) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил]-1-[2-оксаспиро[3,3]гептан-6-ил]пиразоло[3,4-d]пиримидин-6-амина (40 мг; 50,00%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 337,3. 1Н ЯМР (300 МГц, DMSO-d6) δ 2.37 (3H, s), 2.44 (3H, s), 2.65-2.80 (4Н, m), 4.45 (2Н, s), 4.64 (2Н, s), 4.84-4.89 (1H, m), 7.76 (1Н, s), 8.42 (1H, s), 8.92 (1Н, s), 9.13 (1H, s), 9.18 (1H, s).
Следующие соединения в таблице 2 ниже синтезированы способом, аналогичным описанному в примере 42.
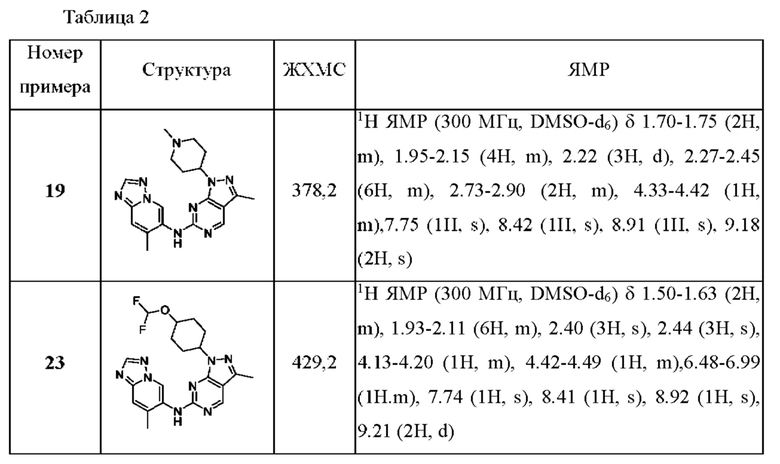
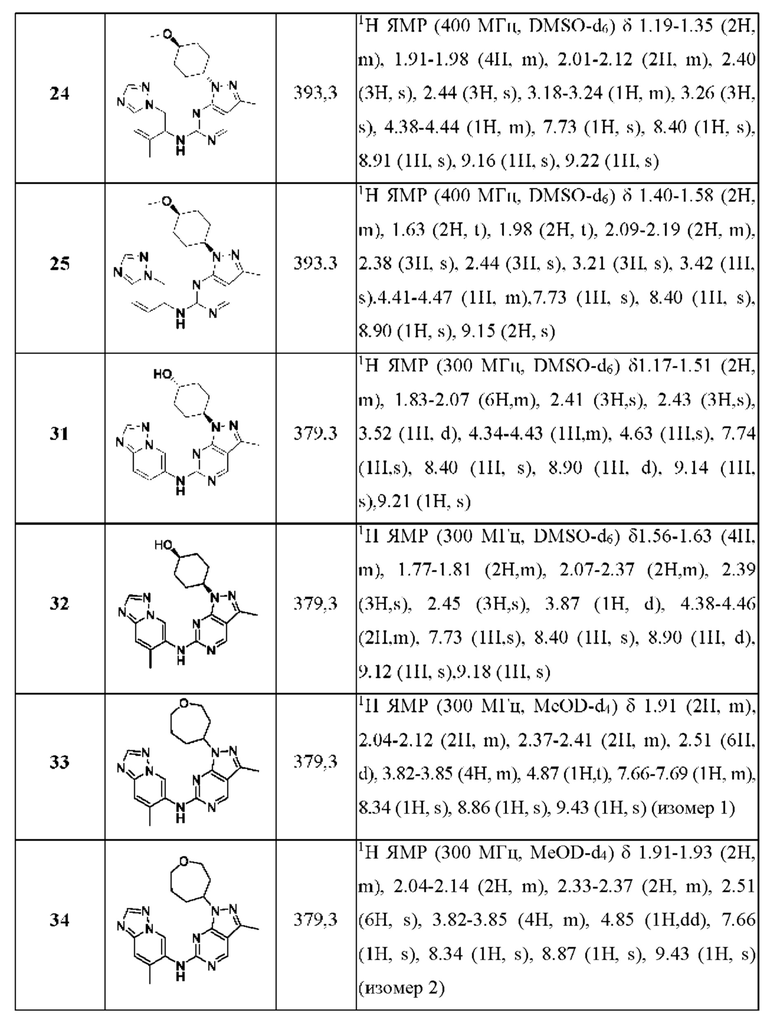
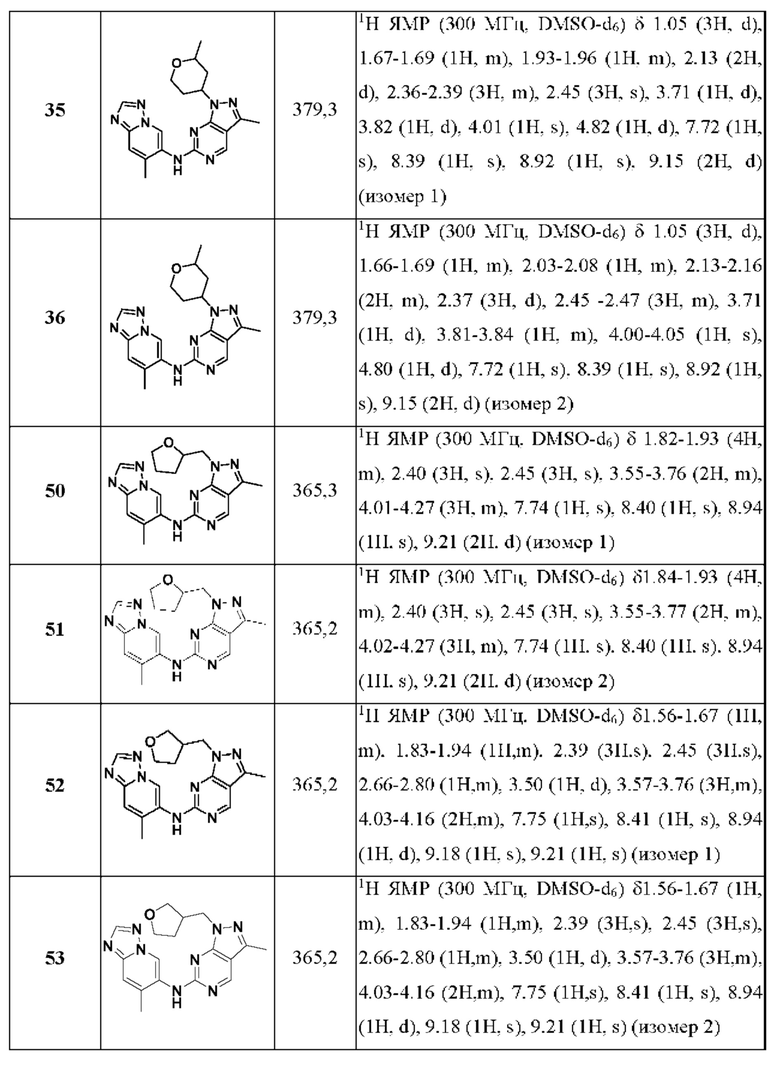
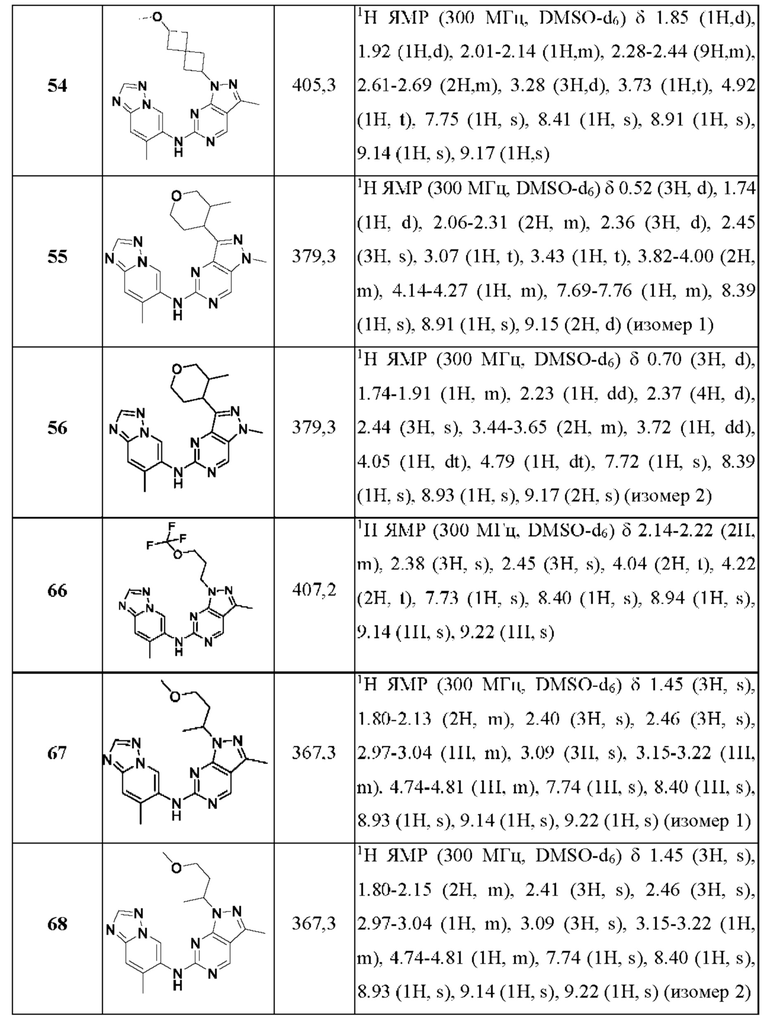
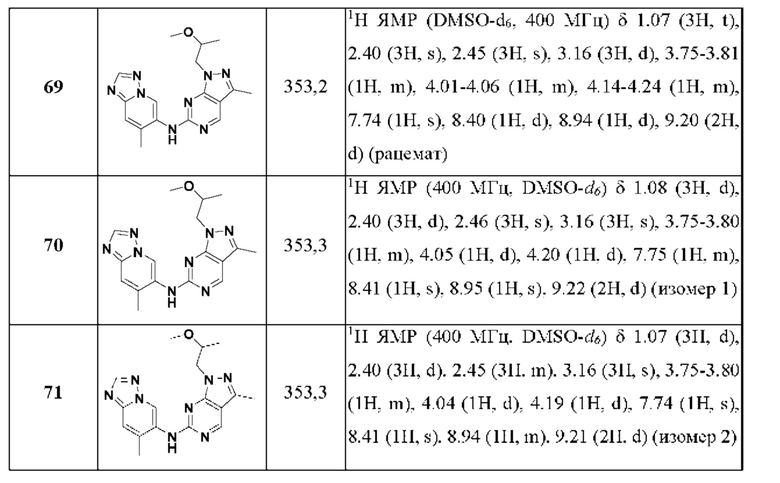
Пример 37
Получение 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(спиро-[2,5]октан-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 37)
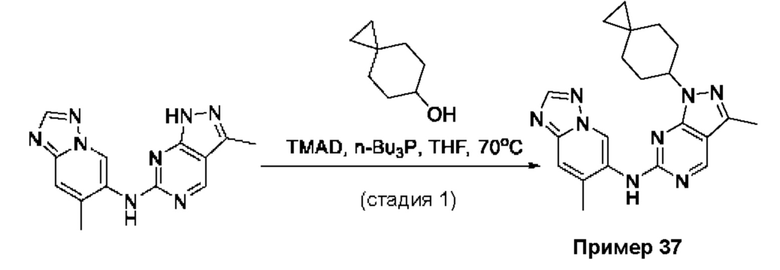
СХЕМА 37
Стадия 1. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(спиро-[2,5]октан-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 37)
Смесь TMAD (тетраметилазодикарбоксамид) (276,43 мг; 1,605 ммоль; 3,00 экв.), н-Bu3P (324,81 мг; 1,605 ммоль; 3,00 экв.), спиро[2,5]октан-6-ола (202,61 мг; 1,605 ммоль; 3,00 экв.) и 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (150,00 мг; 0,535 ммоль; 1,00 экв.) в THF (25,00 мл) перемешивали в течение 2 часов при 70°С в атмосфере азота. Полученную смесь разбавляли водой (100 мл). Полученную смесь экстрагировали CH2Cl2 (3×50 мл), сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт перекристаллизовывали из МеОН (20 мл) с получением неочищенного продукта. Неочищенный продукт (60 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 19×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: МеОН; скорость потока: 25 мл/мин; градиент: от 58 В до 70 В за 7 минут) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[спиро[2,5]октан-6-ил]пиразоло[3,4-d]пиримидин-6-амина (30 мг; 14,29%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 389,3. 1H ЯМР (400 МГц, DMSO-d6) δ 0.20 (2Н, d), 0.33 (2Н, d), 0.98 (2Н, d), 1.84 (4Н, m), 2.08 (2Н, m), 2.39 (3H, d), 2.45 (3H, s), 4.45 (1H, d), 7.74 (1H, s), 8.40 (1H, s), 8.91 (1H, s), 9.16 (2Н, d).
Следующие соединения в таблице 3 ниже синтезированы способом, аналогичным описанному в примере 37.
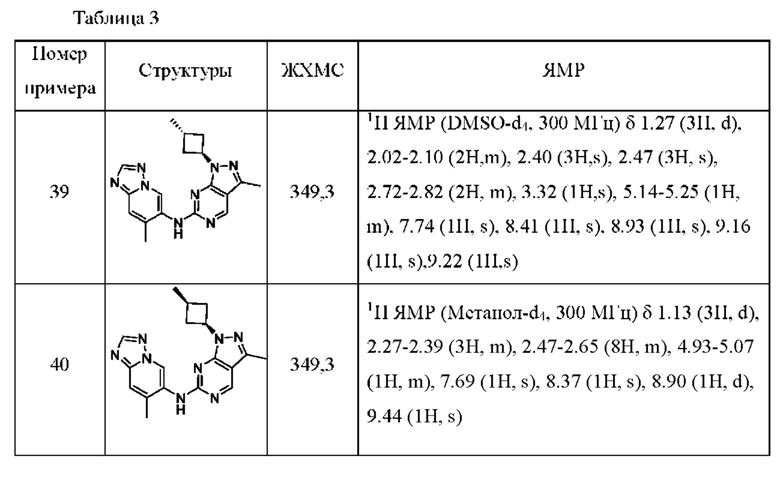
Пример 43
Получение 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(спиро-[3,3]гептан-2-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 43)
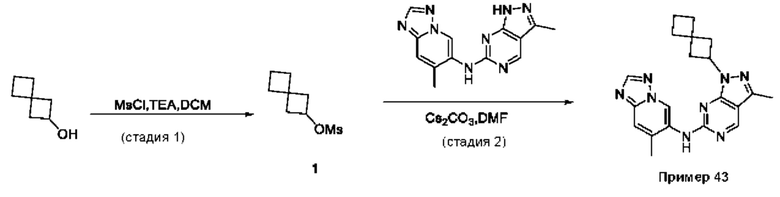
СХЕМА 43
Стадия 1. Спиро[3,3]гептан-2-илметансульфонат
В перемешиваемую смесь спиро[3,3]гептан-2-ола (300,00 мг; 2,674 ммоль; 1,00 экв.) и TEA (811,89 мг; 8,023 ммоль; 3,00 экв.) в DCM (50,00 мл) по каплям добавляли метансульфонилхлорид (459,50 мг; 4,012 ммоль; 1,50 экв.) при 0°С в атмосфере азота. Полученную смесь разбавляли водой (20 мл). Полученную смесь экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. В результате получали спиро[3,3]гептан-2-илметансульфонат (500 мг; 98,26%) в виде светло-желтого масла.
Стадия 2. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(спиро-[3,3]гептан-2-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 43)
Смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло-[3,4-d]пиримидин-6-амина (100,00 мг; 0,357 ммоль; 1,00 экв.), спиро[3,3]гептан-2-илметансульфоната (678,78 мг; 3,568 ммоль; 10,00 экв.) и Cs2CO3 (348,73 мг; 1,070 ммоль; 3,00 экв.) в DMF (20,00 мл) перемешивали в течение 16 часов при 100°С в атмосфере азота. Полученную смесь разбавляли водой (40 мл). Полученную смесь экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали рассолом, сушили над безводным Na2SO4. После фильтрации фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 39 В до 59 В за 7 минут; RT1: 6,4) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[спиро-[3,3]гептан-2-ил]пиразоло[3,4-d]пиримидин-6-амина (31,8 мг; 23,80%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 375,3. 1Н ЯМР (400 МГц, DMSO-d6) δ 1.81-1.95 (2Н, m), 1.89-1.91 (2Н, m), 2.09-2.11 (2Н, m), 2.42 (8Н, d), 2.59-2.63 (2Н, m), 4.89-4.93 (1Н, m), 7.76 (1H, s), 8.42 (1Н, s), 8.92 (1H, s), 9.17 (2Н, d).
Пример 44
Получение 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(оксетан-3-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 44)
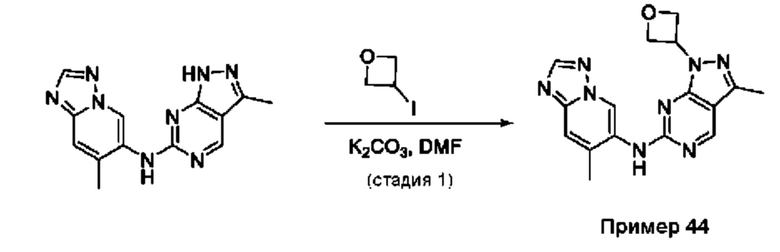
СХЕМА 44
Стадия 1. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(оксетан-3-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 44)
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (80,00 мг; 0,285 ммоль; 1,00 экв.) и 3-иодоксетана (78,76 мг; 0,428 ммоль; 1,50 экв.) в DMF (10 мл) добавляли K2CO3 (118,34 мг; 0,856 ммоль; 3,00 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при 80°С в воздушной атмосфере. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2:МЕОН, 12:1) с получением неочищенного продукта. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅Н2О), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 12 В до 32 В за 7 минут; RT1: 6,62) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил]-1-(оксетан-3-ил)-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 44; 40 мг; 41,67%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 337,2. 1Н ЯМР (300 МГц, DMSO-d6) δ 2.38 (3H, s), 2.49 (3H, s), 4.89-5.02 (4Н, m), 5.72 (1H, t), 7.75 (1H, s), 8.42 (1Н, s), 8.96 (1Н, s), 9.15 (1Н, s), 9.27 (1H, s).
Следующие соединения в таблице 4 ниже синтезированы способом, аналогичным описанному в примере 44.
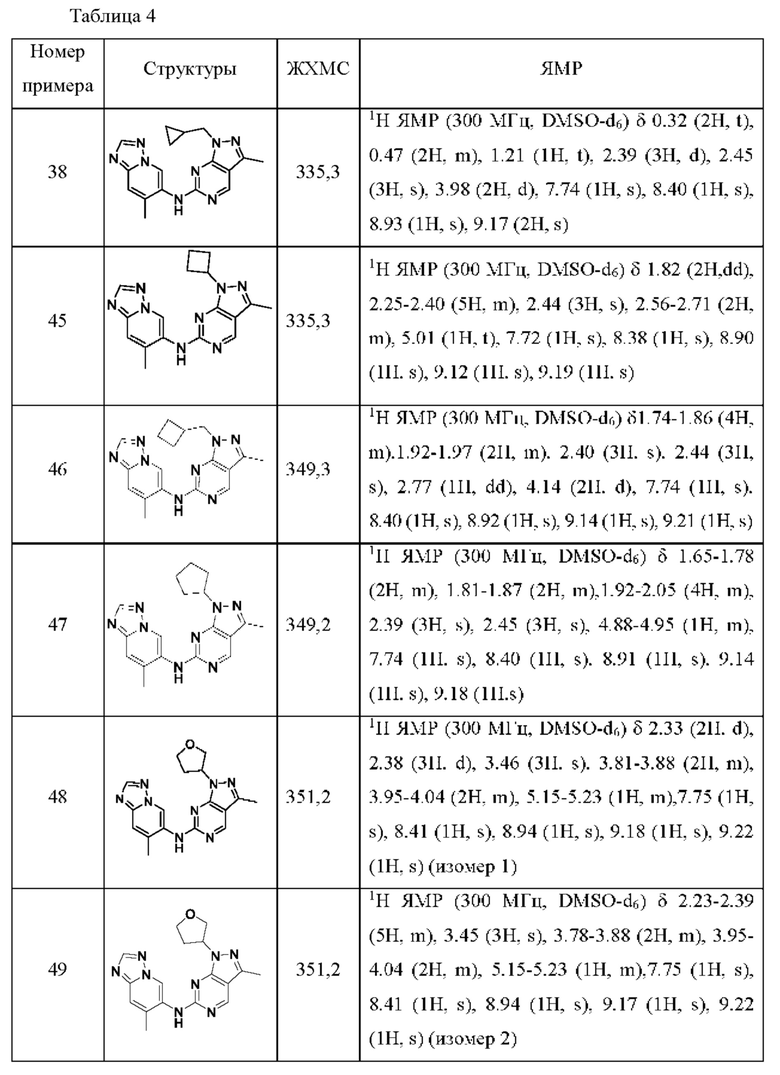
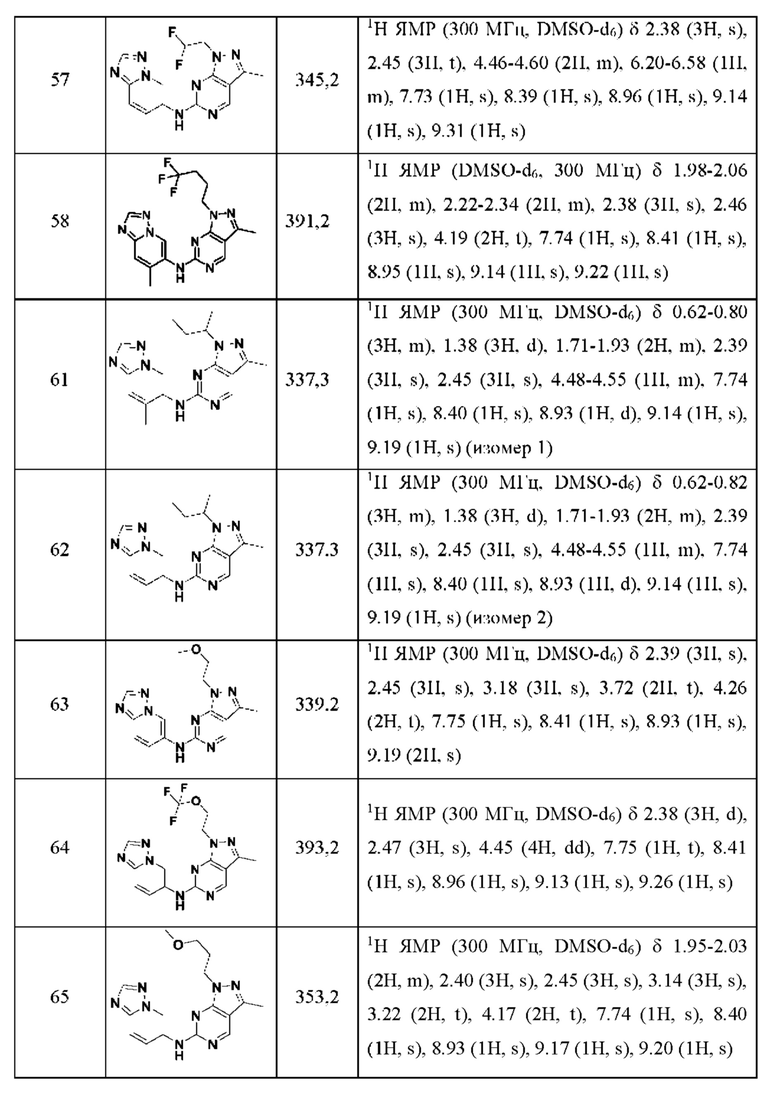
Пример 59
Получение 1-изопропил-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 59)
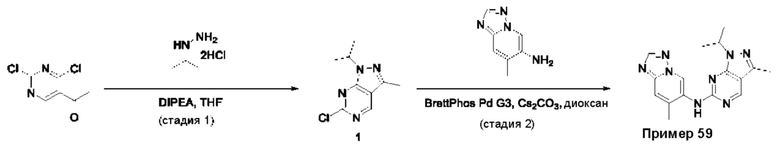
СХЕМА 59
Стадия 1. 6-Хлор-1-изопропил-3-метил-1Н-пиразоло[3,4-d]пиримидин
В перемешиваемую смесь 1-(2,4-дихлорпиримидин-5-ил)этанона (150,00 мг; 0,785 ммоль; 1,00 экв.) и DIPEA (54,13 мг; 0,419 ммоль; 4,00 экв.) в THF по каплям добавляли изопропилгидразин (15,26 мг; 0,136 ммоль; 1,30 экв.) при 0°С в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при комнатной температуре в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (РЕ:ЕА, 1:1) с получением 6-хлор-1-изопропил-3-метилпиразоло[3,4-d]пиримидина (120 мг; 72,53%) в виде желтого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 211,2. 1H ЯМР (300 МГц, CDCl3) δ 1.54 (6Н, d), 2.61 (3H, s), 5.12 (1H, dd), 8.90 (1H, s).
Стадия 2. 1-Изопропил-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 59)
В перемешиваемую смесь 6-хлор-1-изопропил-3-метилпиразоло[3,4-d]пиримидина (120,00 мг; 0,570 ммоль; 1,00 экв.) и 7-метилимидазо[1,2-а]пиридин-6-амина (108,99 мг; 0,740 ммоль; 1,30 экв.) в диоксане (10 мл) добавляли XantPhos (65,92 мг; 0,114 ммоль; 0,20 экв.), Pd(AcO)2 (25,58 мг; 0,114 ммоль; 0,20 экв.) и Cs2CO3 (556,77 мг; 1,709 ммоль; 3,00 экв.) при комнатной температуре в воздушной атмосфере. Полученную смесь перемешивали в течение 2 часов при 60°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1) с получением неочищенного продукта. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; RT1: 6,67) с получением 1-изопропил-3-метил-N-[7-метил-[1,2,4]триазоло-[1,5-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (62 мг; 33,76%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 323,4. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.41 (6Н, d), 2.40 (3H, d), 2.45 (3H, s), 4.73-4.82 (1Н, m), 7.74 (1Н, s), 8.41 (1H, s), 8.92 (1H, s), 9.16 (1H, s), 9.19 (1H, s). Пример 60
Получение 1-изопропил-3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 60)
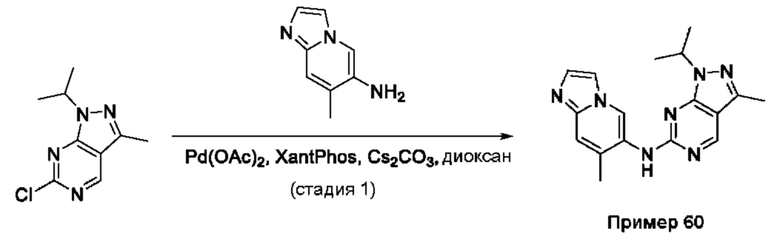
СХЕМА 60
Стадия 1. 1-Изопропил-3-метил-N-(7-метилимидазо[1,2-а]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 60)
Смесь 6-хлор-1-изопропил-3-метилпиразоло[3,4-d]пиримидина (80,00 мг; 0,380 ммоль; 1,00 экв.), 7-метилимидазо[1,2-а]пиридин-6-амина (67,07 мг; 0,456 ммоль; 1,20 экв.), Pd(AcO)2 (17,05 мг; 0,076 ммоль; 0,20 экв.), XantPhos (65,92 мг; 0,114 ммоль; 0,30 экв.) и Cs2CO3 (309,32 мг; 0,949 ммоль; 2,50 экв.) в диоксане (10,00 мл) перемешивали в течение 3 часов при 100°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1). Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅Н2О), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 21 В до 41 В за 7 минут; RT1: 7,02) с получением 1-изопропил-3-метил-N-[7-метилимидазо[1,2-а]пиридин-6-ил]пиразоло-[3,4-d]пиримидин-6-амина (55,77 мг; 45,70%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 322,2. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.38 (6Н, d), 2.25 (3H, d), 2.44 (3H, s), 4.67-4.89 (1Н, m), 7.47 (2Н, dd), 7.87 (1H, t), 8.67 (1H, s), 8.86 (1H, s), 9.03 (1H, s).
Пример 72
Получение 3-(3-метил-6-((7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)амино)-1Н-пиразоло[3,4-d]пиримидин-1-ил)пропаннитрила (соединение по примеру 72)
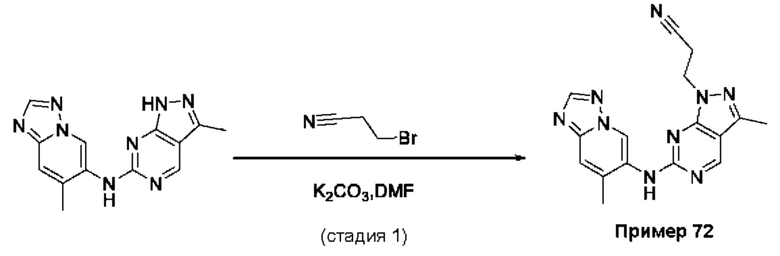
СХЕМА 72
Стадия 1. 3-(3-Метил-6-((7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)амино)-1Н-пиразоло[3,4-d]пиримидин-1-ил)пропаннитрил (соединение по примеру 72)
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (70,00 мг; 0,250 ммоль; 1,00 экв.) и 3-бромпропионитрила (66,92 мг; 0,499 ммоль; 2,00 экв.) в DMF (10 мл) добавляли K2CO3 (103,55 мг; 0,749 ммоль; 3,00 экв.). Полученную смесь перемешивали в течение 2 часов при 80°С в воздушной атмосфере. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (DCM: МеОН, 15:1) с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30x150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 8 В до 28 В за 7 минут; RT1: 7,70) с получением 3-[3-метил-6-([7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]амино)-пиразоло[3,4-d]пиримидин-1-ил]пропаннитрила (40 мг; 48,05%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 334,3. 1Н ЯМР (300 МГц, DMSO-d6) δ 2.39 (3H, s), 2.47 (3H, s), 3.08 (2Н, t), 4.36 (2Н, t), 7.75 (1H, s), 8.41 (1H, s), 8.97 (1Н, s), 9.17 (1H, s), 9.29 (1H, s).
Следующие соединения в таблице 5 ниже синтезированы способом, аналогичным описанному в примере 72.
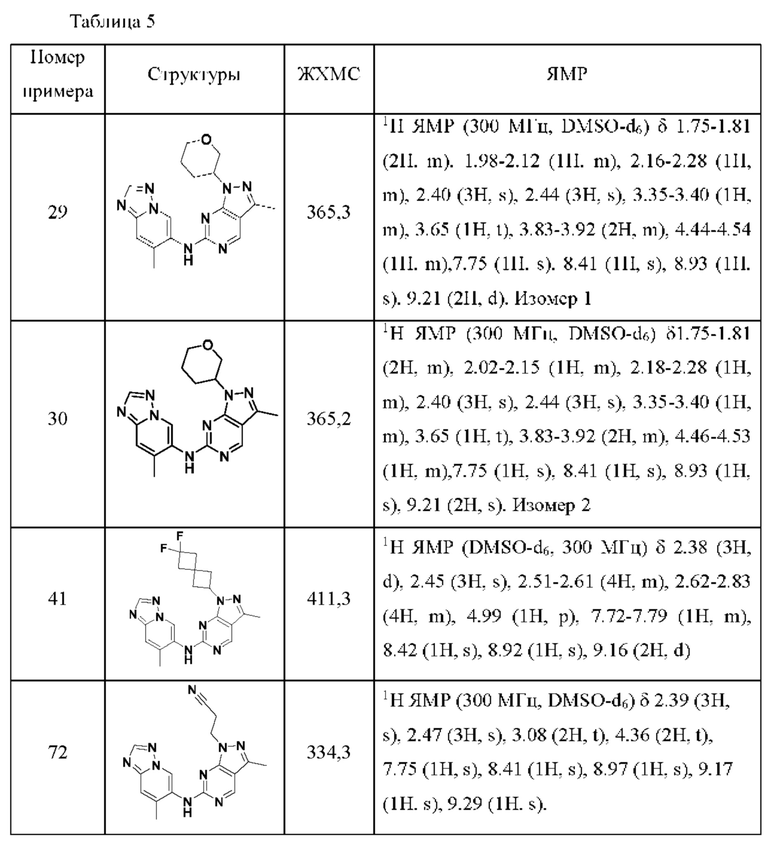
Пример 73
Получение 4-(3-метил-6-(7-метилимидазо[1,2-а]пиридин-6-иламино)-1Н-пиразоло[3,4-d]пиримидин-1-ил)бензонитрила (соединение по примеру 73)
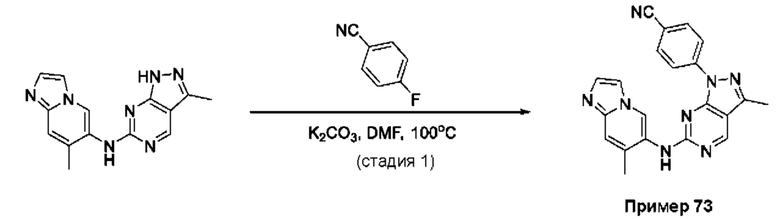
СХЕМА 73
Стадия 1. 4-(3-Метил-6-(7-метилимидазо[1,2-а]пиридин-6-иламино)-1Н-пиразоло-[3,4-d]пиримидин-1-ил)бензонитрил (соединение по примеру 73)
Смесь 7-метил-N-[3-метил-1Н-пиразоло[3,4-d]пиримидин-6-ил]-[1,2,4]триазоло-[1,5-а]пиридин-6-амина (150,00 мг; 0,535 ммоль; 1,00 экв.), 4-фторбензонитрила (97,57 мг; 0,806 ммоль; 1,50 экв.), бензонитрила (97,22 мг; 0,803 ммоль; 1,50 экв.) и K2CO3 (221,88 мг; 1,605 ммоль; 3,0 экв.) в DMF (50,00 мл) перемешивали в течение 16 часов при 100°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1) с получением 4-[3-метил-6-([7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]амино)пиразоло[3,4-d]пиримидин-1-ил]бензонитрила (40 мг; неочищенный) в виде светло-желтого твердого вещества. Неочищенный продукт (40 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Shield RP18 OBD, 19×250 мм, 10 мкм; подвижная фаза А: вода (10 ммоль/л NH4HCO3 плюс 0,1% NH3⋅H2O), подвижная фаза В: ACN; скорость потока: 20 мл/мин; градиент: от 36 В до 46 В за 7 минут; RT1: 5,73) с получением 4-[3-метил-6-([7-метилимидазо[1,2-а]пиридин-6-ил]амино)пиразоло[3,4-d]пиримидин-1-ил]бензонитрила (8,8 мг; 4,31%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 381,3. 1Н ЯМР (300 МГц, CDCl3) δ 2.45 (3H, d), 2.62 (3H, s), 7.08 (1H, s), 7.54 (2Н, s), 7.65 (1H, s), 7.75 (2Н, d), 8.42-8.44 (2Н, m), 8.85 (1H, s), 9.06 (1H, d).
Пример 89
Получение 6-метокси-4-метил-N-[3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидин-6-ил]пиридин-3-амина
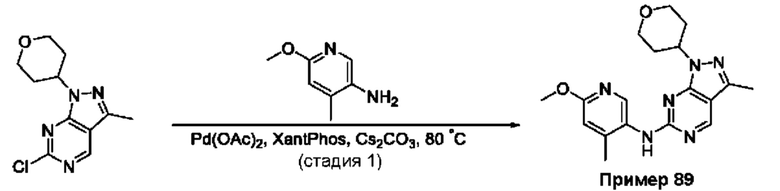
СХЕМА 89
Стадия 1. 6-Метокси-4-метил-N-[3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидин-6-ил]пиридин-3-амин (соединение по примеру 89)
Смесь 6-хлор-3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидина (80,00 мг; 0,317 ммоль; 1,00 экв.), 6-метокси-4-метилпиридин-3-амина (52,49 мг; 0,380 ммоль; 1,20 экв.), Pd(AcO)2 (14,22 мг; 0,063 ммоль; 0,20 экв.), XantPhos (54,95 мг; 0,095 ммоль; 0,30 экв.) и Cs2CO3 (257,87 мг; 0,791 ммоль; 2,50 экв.) в диоксане (2,50 мл) перемешивали в течение 2 часов при 80°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 15:1) с получением неочищенного твердого вещества. Неочищенный продукт (100 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Shield RP18 OBD, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 25 В до 55 В за 7 минут; 254; 220 нм; RT1: 5,20) с получением 6-метокси-4-метил-N-[3-метил-1-(оксан-4-ил)пиразоло[3,4-d]пиримидин-6-ил]пиридин-3-амина (70 мг; 62,39%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 355,2. 1Н-ЯМР (300 МГц, DMSO-d6) δ 1.79 (2Н, d), 1.99-2.20 (5Н, m), 2.42 (3Н, s), 3.43-3.56 (2Н, m), 3.83 (3Н, s), 3.96 (2Н, dd), 4.58 (1Н, t), 6.74 (1Н, s), 8.11 (1Н, s), 8.82 (1Н, s), 8.98 (1Н, s).
Пример 106
Получение 1-((1R,3r,5S)-8-окса-бицикло[3.2.1]октан-3-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина
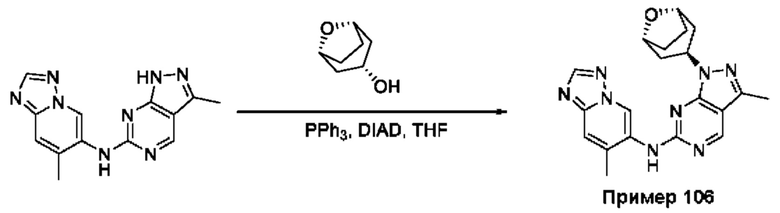
СХЕМА 106
Стадия 1. 1-((1R,3r,5S)-8-Окса-бицикло[3.2.1]октан-3-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (100,00 мг; 0,357 ммоль; 1,00 экв.), (1R,3S,5S)-8-оксабицикло[3.2.1]октан-3-ола (137,18 мг; 1,070 ммоль; 3,00 экв.) и PPh3 (280,72 мг; 1,070 ммоль; 3 экв.) в THF (10,00 мл) по каплям добавляли DIAD (216,42 мг; 1,070 ммоль; 3 экв.) при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 2 часов при 70°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 37 В до 57 В за 7 минут) с получением 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[(1R,3R,5S)-8-оксабицикло[3.2.1]октан-3-ил]пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 106; 50 мг; 50,00%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 391,3. 1Н ЯМР (300 МГц, DMSO-d6) 5 1.74 (4Н, s), 2.22-2.45 (10Н, m), 4.35 (2Н, s), 4.63 (1Н, s), 7.75 (1Н, s), 8.41 (1Н, s), 8.93 (1Н, s), 9.17 (1Н, s), 9.21 (1Н, s).
Пример 107
Получение 1-((1R,3S,5S)-8-окса-бицикло[3.2.1]октан-3-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина
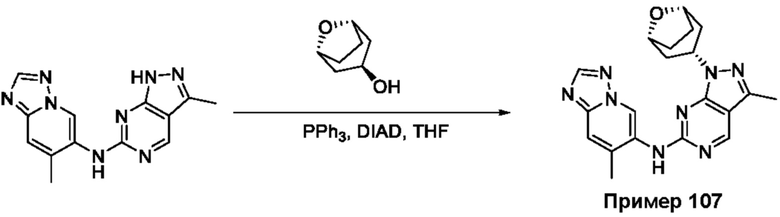
СХЕМА 107
Стадия 1. 1-((1R,3S,5S)-8-Окса-бицикло[3.2.1]октан-3-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (80,00 мг; 0,285 ммоль; 1,00 экв.), (1R,3R,5S)-8-оксабицикло[3.2.1]октан-3-ола (109,75 мг; 0,856 ммоль; 3,00 экв.) и PPh3 (224,58 мг; 0,856 ммоль; 3,00 экв.) в THF (16,00 мл) по каплям добавляли DIAD (173,14 мг; 0,856 ммоль; 3,00 экв.) при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 2 часов при 70°С в атмосфере азота. Полученную смесь концентрировали под вакуумом. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 12:1) с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Prep OBD С18, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 37 В до 50 В за 7 минут) с получением 1-((1R,3s,5S)-8-окса-бицикло[3.2.1]октан-3-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-(1]пиримидин-6-амина (соединение по примеру 107; 45 мг, 45,00%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 391,4. 1Н ЯМР (300 МГц, DMSO-d6) δ 1.78-1.94 (6Н, m), 2.07-2.25 (2Н, m), 2.42 (3H. s), 2.45 (3H, s), 4.44 (2Н, s), 4.88-4.96 (1Н, m), 7.73 (1Н, s), 8.41 (1Н, s), 8.92 (1Н, s), 9.18 (1Н, s), 9.29 (1Н, s).
Пример 108/109/110/111
1-(3-Метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 108, изомер 1)/ 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 109, изомер 2)/ 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 110, изомер 3)/ 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-(1]пиримидин-6-амин (соединение по примеру 111, изомер 4)
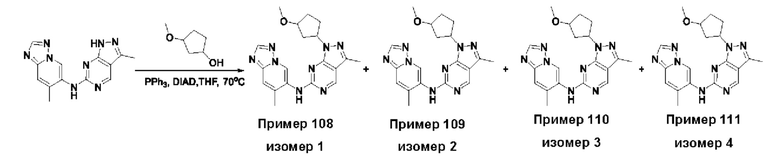
СХЕМА 108-111
Стадия 1. Получение 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-в]пиримидин-6-амина (смесь соединений по примерам 108/109) и получение 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-(11пиримидин-6-амина (смесь соединений по примерам 110/111)
В перемешиваемую смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амина (220,00 мг; 0,785 ммоль; 1,00 экв.), PPh3 (617,59 мг; 2,355 ммоль; 3,00 экв.) и 3-метоксициклопентан-1-ола (273,52 мг; 2,355 ммоль; 3,00 экв.) в THF (20,00 мл) по каплям добавляли DIAD (476,13 мг; 2,355 ммоль; 3,00 экв.) в THF (3 мл) в течение 10 минут при 0°С. Полученную смесь перемешивали в течение 2 часов при 70°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 10:1) с получением 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (150 мг) в виде светло-желтого твердого вещества. Неочищенный продукт (150 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: YMC-Actus Triart С18, 30×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 34 В до 46 В за 8,5 минут) с получением 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (смесь соединений по примерам 108/109; 25 мг; 16,67%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3. 1Н ЯМР (400 МГц, DMSO-d6) δ 1.81-1.85 (2Н, m), 1.98-2.02 (2Н, m), 2.09-2.11 (1Н, m), 2.38-2.41 (4Н, m), 2.45 (3Н, s), 3.18 (3Н, s), 3.83-3.85 (1Н, m), 4.85-4.89 (1Н, m), 7.74 (1Н, s), 8.40 (1Н, s), 8.91 (1Н, s), 9.14 (2Н, s), и 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-(1]пиримидин-6-амина (смесь соединений по примерам 110/111; 80 мг; 53,33%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3. 1Н ЯМР (400 МГц, DMSO-d6) δ 1.61-1.63 (1Н, m), 1.82-1.98 (1Н, m), 2.38-2.41 (4Н, m), 2.39 (3Н, s), 2.45 (3Н, s), 3.18 (3Н, s), 3.93-3.96 (1Н, m), 5.00-5.06 (1Н, m), 7.74 (1Н, s), 8.40 (1Н, s), 8.91 (1Н, s), 9.15 (2Н, d).
Стадия 2. Получение 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-d]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 108, изомер 1)/ 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 109, изомер 2)
Смесь соединений по примерам 108/109 (25 мг) очищали посредством хиральной препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK AD-H, 2,0 см I.D. (внутренний диаметр) × 25 см L (длина); подвижная фаза А: гексан (8 ммоль/л NH3⋅МеОН)-ВЭЖХ, подвижная фаза В: IPA (изопропанол)-ВЭЖХ; скорость потока: 40 мл/мин; градиент: от 20 В до 20 В за 18 минут) с получением 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 108; 4,5 мг; 18,00%) (изомер 1) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 1.81-1.85 (2Н, m), 1.98-2.01 (2Н, m), 2.06-2.09 (1Н, m), 2.38-2.43 (4Н, m), 2.45 (3Н, s), 3.18 (3Н, s), 3.80-3.85 (1Н, m), 4.82-4.87 (1Н, m), 7.74 (1Н, s), 8.40 (1Н, s), 8.91 (1Н, s), 9.14 (2Н, s), и 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 109; 3,8 мг, 12,00%) (изомер 2) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 1.81-1.83 (2Н, m), 1.97-2.01 (2Н, m), 2.10-2.12 (1Н, m), 2.37-2.43 (4Н, m), 2.45 (3Н, s), 3.18 (3Н, s), 3.81-3.85 (1Н, m), 4.80-4.89 (1Н, m), 7.73 (1Н, s), 8.40 (1Н, s), 8.91 (1Н, s), 9.14 (2Н, s).
Стадия 4. 1-(3-Метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 110, изомер 3)/ 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 111. изомер 4)
Смесь соединений по примерам 110/111 (80 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK AD-H, 2,0 см I.D. × 25 см L; подвижная фаза А: гексан (8 ммоль/л NH3⋅МеОН)-ВЭЖХ, подвижная фаза В: IPA-ВЭЖХ; скорость потока: 40 мл/мин; градиент: от 30 В до 30 В за 12 минут) с получением 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4d]пиримидин-6-амина (соединение по примеру 110; 39,1 мг; 48,87%) (изомер 3) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1H ЯМР (400 МГц, DMSO-d6) δ 1.67-1.69 (1Н, m), 1.93-1.99 (1Н, m), 2.10-2.15 (4Н, m), 2.39 (3Н, s), 2.44 (3Н, s), 3.16 (3Н, s), 3.94 (1Н, s), 5.00-5.08 (1Н, m), 7.74 (1Н, s), 8.40 (1Н, s), 8.91 (1Н, s), 9.16 (2Н, d) и 1-(3-метоксициклопентил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 111; 35,2 мг) (изомер 4) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1H ЯМР (400 МГц, DMSO-d6) δ 1.65-1.69 (1Н, m), 1.93-1.97 (1H, m), 2.09-2.14 (4H, m), 2.39 (3H, d), 2.44 (3H, s), 3.15 (3H, s), 3.93-3.96 (1H, m), 5.00-5.08 (1H, m), 7.74 (1H, s), 8.41 (1H, s), 8.91 (1H, s), 9.16 (2H, d).
Пример 112/113/114/115
Получение 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 112, изомер 1)/ 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2H-пиран-4-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 113, изомер 2)/ 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 114, изомер 3)/ 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 115, изомер 4)
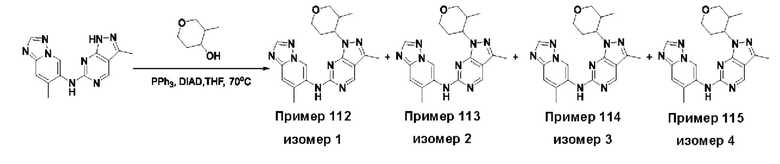
СХЕМА 112-115
Стадия 1. 3-Метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[3-метилоксан-4-ил]пиразоло[3,4-d]пиримидин-6-амин (смесь соединений по примерам 112/113) и 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[3-метилоксан-4-ил]пиразоло[3,4-d]пиримидин-6-амин (смесь соединений по примерам 114/115)
Смесь 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло-[3,4-d]пиримидин-6-амина (200,00 мг; 0,714 ммоль; 1,00 экв.), 3-метилоксан-4-ола (248,65 мг; 2,141 ммоль; 3,00 экв.) и PPh3 (561,45 мг; 2,141 ммоль; 3,00 экв.) в THF (10,00 мл) перемешивали при 0°С и по каплям добавляли DIAD (432,85 мг; 2,141 ммоль; 3,00 экв.) в атмосфере азота при 70°С и перемешивали в течение 2 часов. Полученную смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 15:1) с получением неочищенного твердого вещества. Неочищенный продукт (120 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: XBridge Shield RP18 OBD, 30×150 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 18 В до 48 В за 7 минут) с получением 3 метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1-[3-метилоксан-4-ил]пиразоло[3,4-d]пиримидин-6-амина (смесь соединений по примерам 112/113; 30 мг; 25,00%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 0.52 (3Н, d), 1.74 (2Н, d), 2.06-2.31 (1Н, m), 2.36 (3Н, d), 2.45 (3Н, s), 3.07 (1Н, t), 3.43 (1Н, t), 3.82-4.00 (2Н, m), 4.14-4.27 (1Н, m), 7.69-7.76 (1Н, m), 8.39 (1Н, s), 8.91 (1Н, s), 9.15 (2Н, d) и 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил]-1-[-3-метилоксан-4-ил]пиразоло[3,4-d]пиримидин-6-амина (смесь соединений по примерам 114/115; 70 мг, 57,75%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 0.70 (3Н, d), 1.74-1.91 (1Н, m), 2.23 (1Н, d), 2.37 (4Н, d), 2.44 (3Н, s), 3.44-3.65 (2Н, m), 3.72 (1Н, dd), 4.05 (1Н, dt), 4.79 (1Н, dt), 7.72 (1Н, s), 8.39 (1Н, s), 8.93 (1Н, s), 9.17 (2Н, s).
Стадия 2. 3-Метил-N-(7-метил-[1,24]триазоло[1,5-a]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 112. изомер 1) и 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 113, изомер 2)
Неочищенный продукт 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (смесь соединений по примерам 112/113; 30,00 мг; 0,079 ммоль; 1,00 экв.) очищали посредством хиральной препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK IE-3, 4,6×50 мм, 3 мкм; подвижная фаза А: гексан (0,1% DEA) : EtOH, 50:50) с получением 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 112, изомер 1; 12,22 мг; 40,73%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 0.52 (3Н, d), 1.74 (2Н, d), 2.06-2.31 (1Н, m), 2.36 (3Н, d), 2.45 (3Н, s), 3.07 (1Н, t), 3.43 (1Н, t), 3.82-4.00 (2Н, m), 4.14-4.27 (1Н, m), 7.69-7.76 (1Н, m), 8.39 (1Н, s), 8.91 (1Н, s), 9.15 (2Н, d), и 3-метил-Н-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 113, изомер 2; 8,37 мг; 27,90%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (400 МГц, DMSO-d6) δ 0.52 (3Н, d), 1.74 (2Н, d), 2.06-2.31 (1Н, m), 2.36 (3Н, d), 2.45 (3Н, s), 3.07 (1Н, t), 3.43 (1Н, t), 3.82-4.00 (2Н, m), 4.14-4.27 (1Н, m), 7.69-7.76 (1H, m), 8.39 (1H, s), 8.91 (1H, s), 9.15 (2H, d).
Стадия 3. 3-Метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 114, изомер 3) и 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1H-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 115, изомер 4)
Неочищенный продукт 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин (смесь соединений по примерам 114/115; 70,00 мг; 0,185 ммоль; 1,00 экв.) очищали посредством хиральной препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK IE-3, 4,6×50 мм, 3 мкм; подвижная фаза А: гексан (0,1% DEA):EtOH, 50:50) с получением 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 114, изомер 3; 32,98 мг; 47,11%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1H ЯМР (300 МГц, DMSO-d6) δ 0.70 (3Н, d), 1.74-1.91 (1Н, m), 2.23 (1Н, d), 2.37 (4Н, d), 2.44 (3Н, s), 3.44-3.65 (2Н, m), 3.72 (1Н, d), 4.05 (1Н, t), 4.79 (1Н, t), 7.72 (1Н, s), 8.39 (1Н, s), 8.93 (1Н, s), 9.17 (2Н, s), и 3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1-(3-метилтетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 115, изомер 4; 33,45 мг; 41,39%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 379,3; 1Н ЯМР (300 МГц, DMSO-d6) δ 0.70 (3Н, d), 1.74-1.91 (1Н, m), 2.23 (1Н, d), 2.37 (4Н, d), 2.44 (3Н, s), 3.44-3.65 (2Н, m), 3.72 (1Н, d), 4.05 (1Н, t), 4.79 (1Н, t), 7.72 (1Н, s), 8.39 (1Н, s), 8.93 (1Н, s), 9.17 (2Н, s).
Пример 116/117
Получение 1-(2,2-диметилтетрагидро-2Н-пиран-4-ил)-3-метил-N-(7-метил-[1,2,4]-триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 116, изомер 1) и 1-(2,2-диметилтетрагидро-2Н-пиран-4-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-a]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 117, изомер 2)
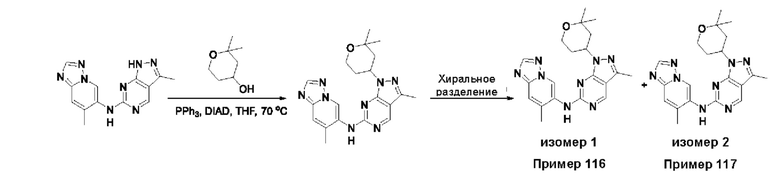
СХЕМА 116
Стадия 1. 1-(2,2-Диметилтетрагидро-2Н-пиран-4-ил)-3-метил-N-(7-метил-[1,2,4]-триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[3,4-d]пиримидин-6-амин
В круглодонную колбу на 250 мл добавляли 2,2-диметилоксан-4-ол (627,03 мг; 4,816 ммоль; 3,00 экв.) и 3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]-1Н-пиразоло[3,4-d]пиримидин-6-амин (450,00 мг; 1,605 ммоль; 1,00 экв.), PPh3 (1263,26 мг; 4,816 ммоль; 3,00 экв.) в THF (60,00 мл) при 0°С. К вышеуказанному раствору добавляли по каплям раствор DIAD (973,90 мг; 4,816 ммоль; 3,00 экв.) в THF (10 мл) при 0°С в атмосфере N2 и перемешивали при комнатной температуре в течение 3 минут. Реакционную смесь продолжали перемешивать при 70°С в течение 2 часов. Полученную смесь концентрировали при пониженном давлении. Полученную смесь разбавляли DCM (20 мл) и фильтровали, осадок на фильтре промывали DCM (2×5 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали посредством препаративной ТСХ (CH2Cl2/МеОН, 15:1) с получением неочищенного продукта. Неочищенный продукт (250 мг) очищали посредством препаративной ВЭЖХ в следующих условиях (колонка: YMC-Actus Triart С18, 30×250 мм, 5 мкм; подвижная фаза А: вода (0,05% NH3H2O), подвижная фаза В: ACN; скорость потока: 60 мл/мин; градиент: от 37 В до 57 В за 7 минут) с получением 1-[2,2-диметилоксан-4-ил]-3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (170 мг; 26,98%) в виде белого твердого вещества. ЖХМС: m/z (ESI), [М+Н]+ составляет 393,2.
Стадия 2. 1-(2,2-Диметилтетрагидро-2Н-пиран-4-ил)-3-метил-N-(7-метил-[1,2,4]-триазоло[1,5-а]пиридин-6-ил)-1H-пиразоло[3,4-d]пиримидин-6-амин (соединение по примеру 116. изомер 1)/ 1-(2,2-диметилтетрагидро-2Н-пиран-4-ил)-3-метил-N-(7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил)-1Н-пиразоло[34-d]пиримидин-6-амин (соединение по примеру 117. изомер 2)
Неочищенный продукт (170 мг) очищали посредством хиральной препаративной ВЭЖХ в следующих условиях (колонка: CHIRALPAK-AD-H-UL001, 20×250 мм, 5 мкм; подвижная фаза А: гексан (8 ммоль/л NH3⋅МеОН)-ВЭЖХ, подвижная фаза В: IPA-ВЭЖХ; скорость потока: 20 мл/мин; градиент: от 25 В до 25 В за 15 минут; RT1: 10,12; RT2: 11,691) с получением rel-1-[(4R)-2,2-диметилоксан-4-ил]-3-метил-N-[7-метил-[1,2,4]триазоло[1,5-а]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 116, изомер 1) (70 мг; 41,18%), ЖХМС: m/z (ESI), [М+Н]+ составляет 393,3; 1Н-ЯМР (400 МГц, DMSO-d6) δ 1.21 (6Н, d), 1.80 (2Н, dd), 1.89 (1Н, t), 2.05 (1Н, qd), 2.35-2.48 (6Н, m), 3.66-3.81 (2Н, m), 4.75-4.95 (1Н, m), 7.74 (1Н, d), 8.40 (1Н, s), 8.92 (1Н, s), 9.20 (2Н, d); и rel-1-[(4R)-2,2-диметилоксан-4-ил]-3-метил-N-[7-метил-[l,2,4]триазоло[1,5-a]пиридин-6-ил]пиразоло[3,4-d]пиримидин-6-амина (соединение по примеру 117, изомер 2) (70 мг; 41,18%) в виде белого твердого вещества, ЖХМС: m/z (ESI), [М+Н]+ составляет 393,3; 1Н-ЯМР (400 МГц, DMSO-d6) δ 1.21 (6Н, d), 1.80 (2Н, dd), 1.89 (1Н, t), 2.05 (1Н, qd), 2.35-2.48 (6Н, m), 3.66-3.81 (2Н, m), 4.75-4.95 (1Н, m), 7.74 (1Н, d), 8.40 (1Н, s), 8.92 (1Н, s), 9.20 (2Н, d).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Типичные соединения по примерам, раскрытые здесь, были охарактеризованы в одном или более чем в одном из следующих биологических анализов.
Биологический пример 1:
Ферментативный анализ
Ингибиторную активность соединений в отношении DNA-PK определяли посредством измерения с помощью метода TR-FRET (резонансный перенос энергии флюоресценции с временным разрешением) превращения флуоресцентно меченого пептидного субстрата в фосфорилированный продукт. Все анализы проводили в черных 384-луночных планшетах Greiner малого объема (Greiner) в общем реакционном объеме 6 мкл и с конечной концентрацией DMSO 0,5% (об./об.). Полноразмерный белок DNA-PK человека, пептидный субстрат флуоресцеин-Р53 (Ser15) (флуоресцеин-EPPLSQEAFADLWKK) и набор Tb-анти-фосфо-р53 [pSer15] антител LanthaScreen™ были приобретены у Thermo Fisher Scientific. Первоначально белок DNA-PK инкубировали с соединением в течение 30 минут при комнатной температуре в реакционном буфере (50 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) рН 7,5, 0,01% Brij-35 (лауриловый эфир полиоксиэтилена), 10 мМ MgCl2, 1 мМ EGTA (этиленгликольтетрауксусная кислота), 1 мМ DTT (дитиотреитол), 10 мкг/мл ДНК тимуса теленка). Затем инициировали реакцию посредством добавления АТФ (аденозинтрифосфорная кислота) и флуоресцеин-Р53 (Ser15) пептидного субстрата. Киназную реакцию (10 мкМ АТФ, 1,6 мкМ пептидного субстрата) гасили через 60 минут путем добавления 6 мкл стоп-буфера, содержащего 20 мМ EDTA (этилендиаминтетрауксусная кислота), 4 нМ Tb-анти-фосфо-р53 [Serl5] антитела. Реакционную смесь инкубировали еще в течение часа, и планшеты считывали на Spark 20М (Tecan). Данные анализировали, и концентрацию соединения, вызывающую 50% ингибирование соответствующей киназы (IC50), рассчитывали с использованием четырехпараметрической логистической аппроксимации в XLfit. Ингибиторные активности соединений по примерам в отношении DNA-PK показаны в таблице 5 ниже. Как можно видеть, соединения по настоящему изобретению продемонстрировали сильную ингибиторную активность в отношении DNA-PK.
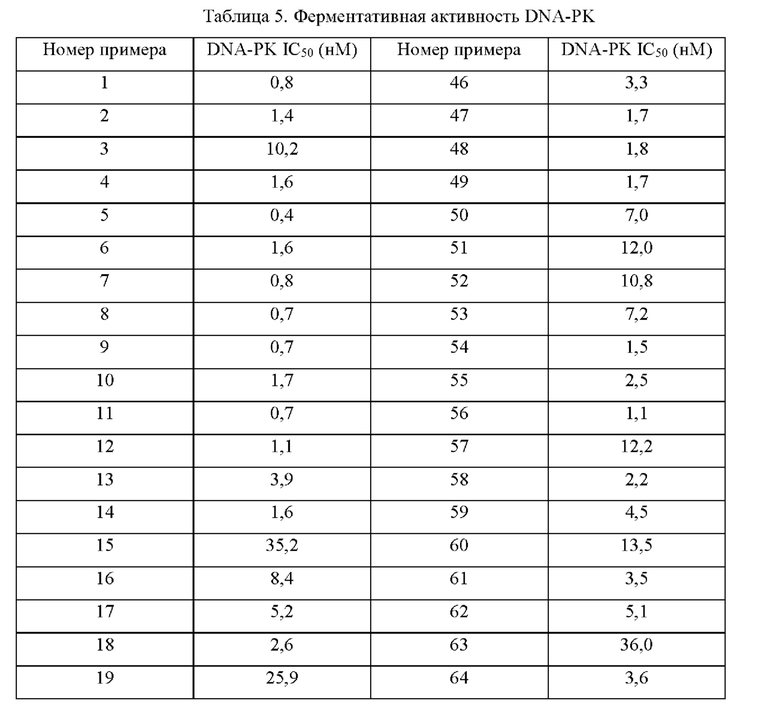
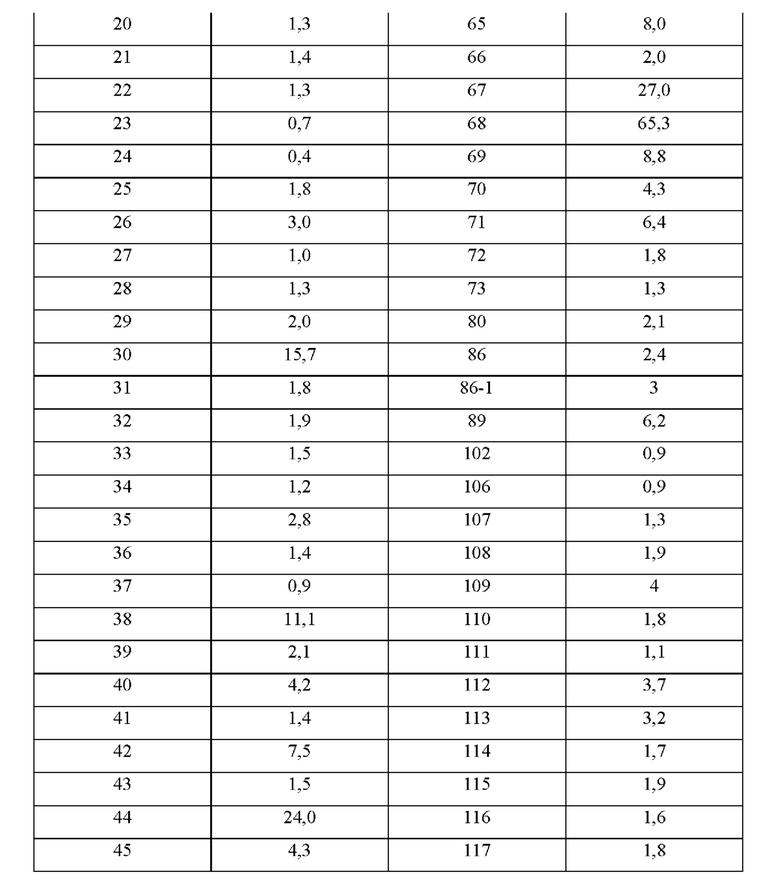
Биологический пример 2:
Анализ метаболической стабильности (Clint (внутренний клиренс) гепатоцитов крысы)
Жизнеспособность криоконсервированных гепатоцитов определяли с использованием трипанового синего, а концентрацию клеток доводили до 106 клеток на мл с помощью буфера. 1 мкМ соединения (в ацетонитриле; 0,01% DMSO) инкубировали с 250 мкл гепатоцитов (1 миллион клеток на мл) в 96-луночном планшете с глубокими лунками. Реакцию останавливали в разных временных точках (0; 0,5; 5; 15; 30; 45; 60; 80; 100 и 120 мин) посредством добавления 3 объемов охлажденного ацетонитрила к 20 мкл реакционной смеси и центрифугировали при 4°С в течение 15 минут.40 мкл надосадочной жидкости разбавляли чистой водой до 200 мкл и анализировали с использованием ЖХ-МС/МС.
Клиренс гепатоцитов in vitro оценивали на основании определения периода полувыведения (Т1/2) соединений, исходя из их исходных концентраций. Рассчитывали отношения площадей пиков каждого соединения (тестируемого или контрольного) к IS (внутреннему стандарту). Константу скорости выведения лекарственного средства к (мин-1), Т1/2 (мин) и внутренний клиренс CLint (мкл/мин/106 клеток) in vitro рассчитывали согласно следующим уравнениям:
k =-угловой коэффициент
Т1/2 = 0,693/k
CLint = k/Chep
где Chep (клетки × мкл-1) представляет собой концентрацию клеток в инкубационной системе.
Данные показаны в таблице 6 ниже.
Анализ метаболической стабильности (Clint в микросомах человека)
1 мкМ соединения инкубировали с 1 мг/мл микросом (объединенные HLM (объединенные микросомы печени человека) с концентрацией белка 20 мг/мл) при 37°С в 250 мкл буфера (100 мМ фосфатный буфер, рН 7,4), содержащего 1 мМ раствор NADPH (восстановленный никотинамидадениндинуклеотидфосфат). 20 мкл инкубационной смеси гасили 5 объемами охлажденного ацетонитрила в разные моменты времени (0; 0,5; 5; 10; 15; 20 и 30 мин) в свежем 96-луночном планшете. Планшет после остановки реакции центрифугировали при 4000 об/мин в течение 15 минут.40 мкл надосадочной жидкости разбавляли чистой водой до 200 мкл и анализировали с использованием ЖХ-МС/МС.
Собственный клиренс лекарственных средств в микросомах CLint (мкл/мин/мг) in vitro рассчитывали аналогично расчету CLint в гепатоцитах. Данные также показаны в таблице 6 ниже.
Анализ эффлюкса через MDCKII-MDR1-BCRP
Апикально-базолатеральный (А-В) и базолатерально-апикальный (В-А) транспорт соединения в HBSS (сбалансированный солевой раствор Хэнка) (25 мМ HEPES, рН 7,4) измеряли через клеточные монослои MDCKII-MDR1-BCRP. Инкубацию проводили при температуре примерно 37°С в течение 120 минут, при этом функциональность тест-системы подтверждали с использованием 5 мкМ дигоксина в качестве субстрата для положительного контроля. Транспорт 5 мкМ соединения и контрольного соединения определяли путем количественного определения концентрации субстрата в инкубационной среде донорского компартмента в начале инкубационного периода, а также и донорского, и принимающего компартментов в конце инкубационного периода. Данные использовали для расчета кажущейся проницаемости (Рарр). Все инкубации проводили в трех повторах, а целостность клеточных монослоев подтверждали с использованием маркера люцифер желтый.
Коэффициент проницаемости Pexact (см/с) рассчитывали с использованием уравнения:
Рарр = (dCr/dt) × Vr / (А×С0)
Pexact = -(Vd×Vr)/(Vd+Vr)/A/t×ln(1-(Vd+Vr)×Cr/(Vd×Cd+Vr×Cr))
Соотношение Pexact рассчитывали по формуле:
Соотношение Pexact или Рарр = Pexact или Рарр(+ингибитор)/Pexact или Рарр(-ингибитор)
Коэффициент эффлюкса рассчитывали по формуле:
Коэффициент эффлюкса = Pexact или Papp(BA)/Pexact или Рарр(АВ),
где dCr/dt представляет собой кумулятивную концентрацию соединения в принимающей камере как функцию времени (мкМ/с); Vr представляет собой объем раствора в принимающей камере (0,1 мл на апикальной стороне, 0,3 мл на базолатеральной стороне); А представляет собой площадь поверхности для переноса, т.е. 0,11 см2 для площади монослоя; С0 представляет собой начальную концентрацию в донорской камере (мкМ). Данные показаны в таблице 6 ниже.
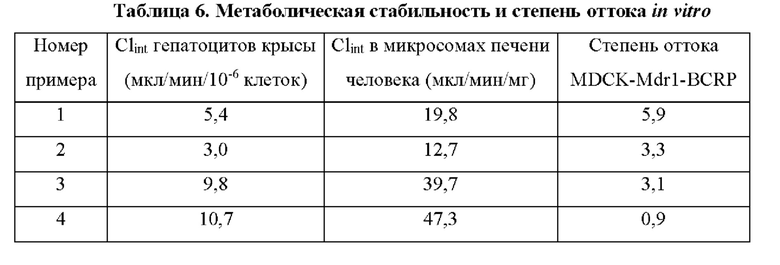
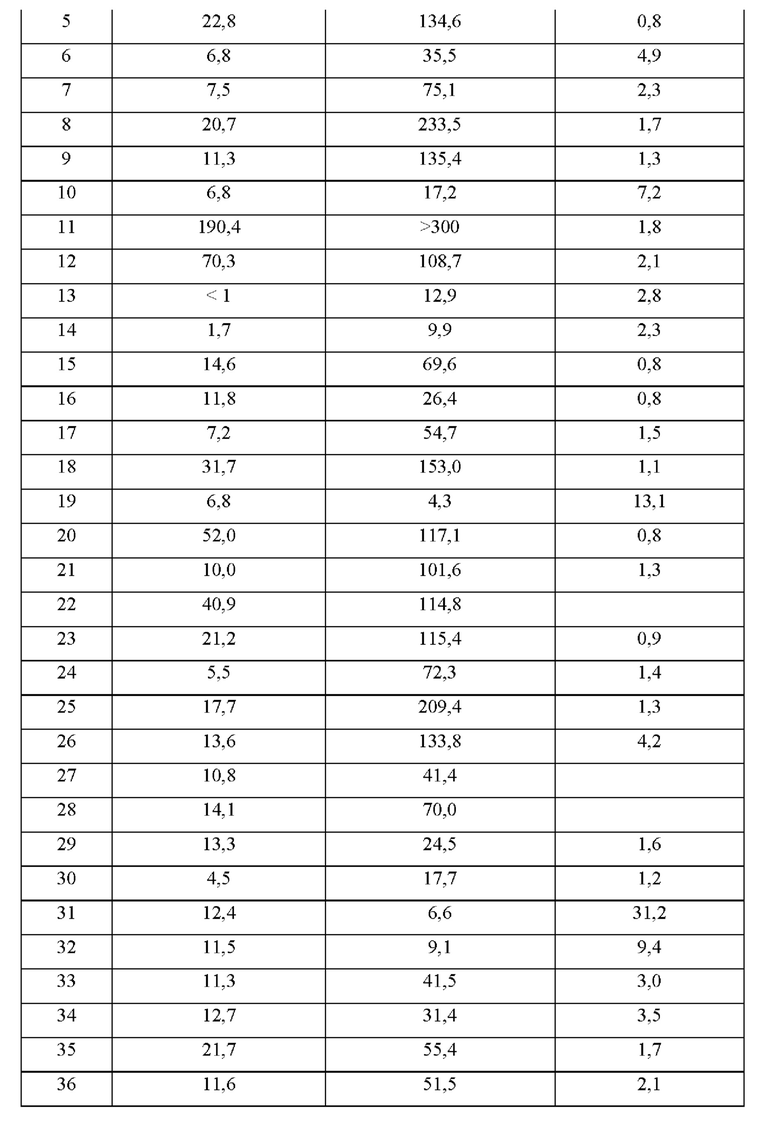
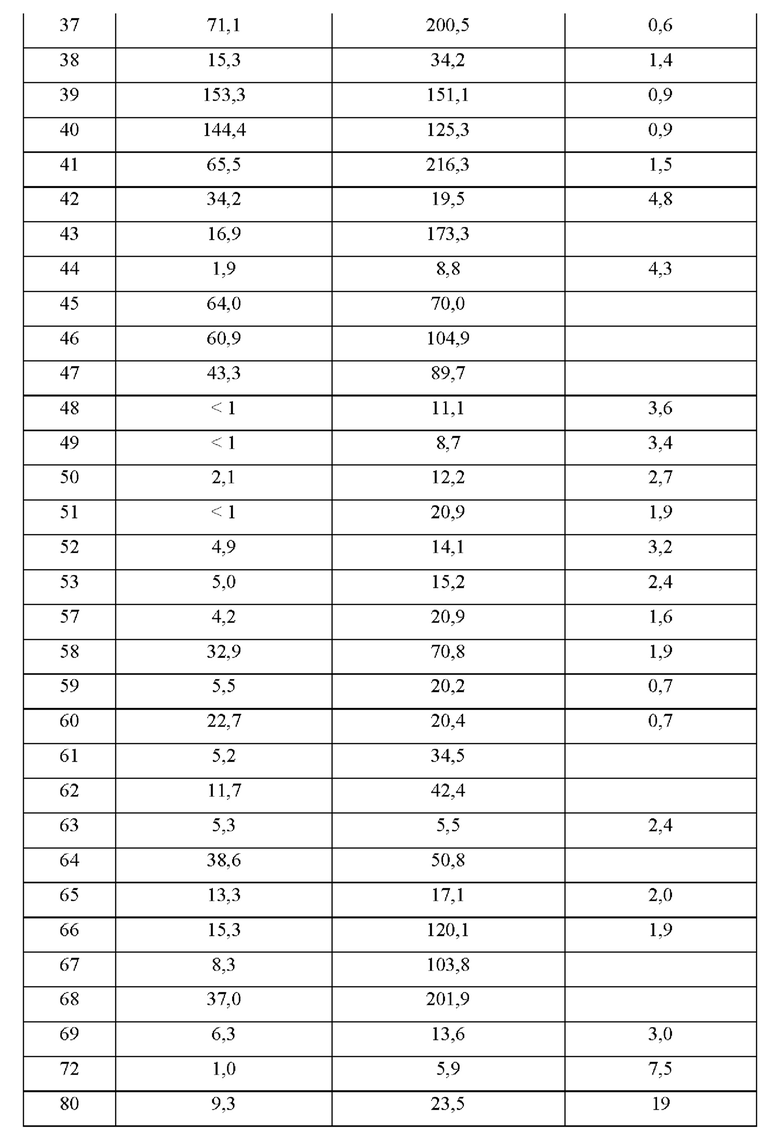
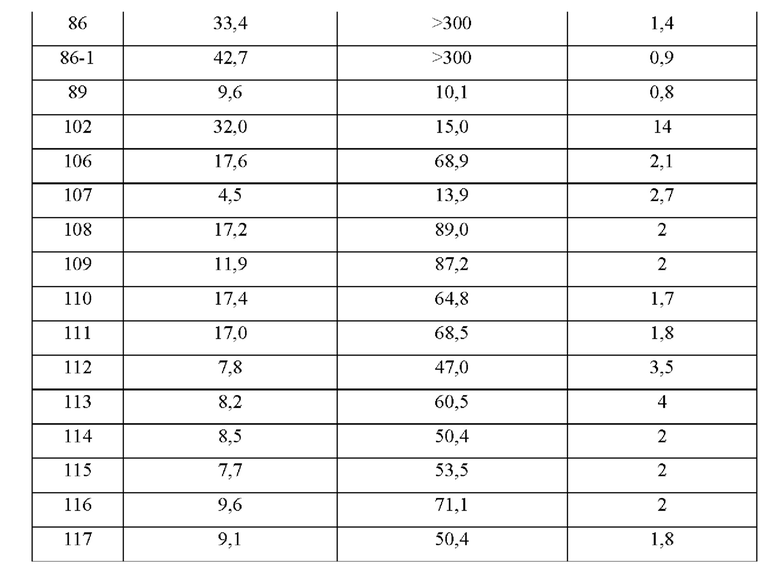
Биологический пример 3:
Анализ проникновения через гемато-энцефалический барьер
Считается, что Kp,uu, соотношение между концентрациями несвязанного лекарственного средства в головном мозге и в плазме, является ключом к прогнозированию действия ЦНС и должно быть основным параметром, измеряемым и оптимизируемым при поиске новых лекарственных средств (Di L et al., Journal of Medicinal Chemistry [2013], 56: 2-12).
Анализ связывания в плазме и головном мозге in vitro проводили с использованием метода равновесного диализа через полупроницаемую мембрану. В плазму и разбавленный гомогенат головного мозга (1:4 DPBS (фосфатно-солевой буферный раствор Дульбекко) рН 7,4) вводили 5 мкМ тестируемого соединения (в трех повторах) и проводили диализ против равного объема 150 мкл 100 мМ буфера PBS (рН 7,4) при 37°С в течение 18 часов в медленно вращающемся планшете. В конце инкубации отбирали аликвоту 50 мкл со стороны приемной камеры и 5 мкл из донорской камеры. Образец объемом 5 мкл дополнительно разбавляли 45 мкл исходной плазмы или гомогената головного мозга. Парные образцы приводили в соответствие с матриксом либо с помощью буфера, либо с помощью исходной плазмы/гомогената мозга и смешивали в течение 2 минут, а затем осаждали 150 мкл холодного ацетонитрила с 100 нг/мл толбутамида в качестве внутреннего стандарта. После центрифугирования при 4000 об/мин в течение 20 минут надосадочную жидкость разбавляли 0,1% водным раствором муравьиной кислоты и анализировали с помощью ЖХ/МС/МС (API 4000, Applied Biosystems, Foster City). Несвязанную фракцию (fu) тестируемого соединения в гомогенате мозга и разбавленной плазме рассчитывали по соотношению реакции со стороны буферного раствора к реакции со стороны гомогената мозга/плазмы, а несвязанные фракции (fu, плазм, и fu, мозг.) тестируемого соединения в неразбавленной плазме и ткани рассчитывали по измеренному fu в гомогенате и плазме с помощью следующего уравнения: fu, плазм, (fu, мозг.) = (1/D)/[(1/fu -1)+1/D)]. D представляет собой коэффициент разбавления.
Модель кратковременного всасывания при пероральном введении (SOA) представляет собой модель скрининга in vivo для определения проникновения соединения в мозг. Шести самцам крыс линии Han Wistar, приобретенным у Beijing Vital River, перорально вводили соединение в дозе 10 мг/кг в 1% метил целлюлозе. Через 0,5, 1, 2, 4, 7 и 16 часов после введения отбирали спинномозговую жидкость (CSF) из большой цистерны мозга. Образцы плазмы обрабатывали для получения плазмы посредством центрифугирования при температуре примерно 4°С и 3000 g в течение получаса после сбора. Образцы плазмы переносили в маркированную пробирку и хранили при -80°С до анализа. Ткань головного мозга собирали и гомогенизировали в 3-кратном объеме 100 мМ фосфатно-солевого буфера (рН 7,4). Все образцы хранили при примерно -70°С до проведения анализа с помощью ЖХ/МС/МС.
Стандарты готовили путем введения исходной плазмы, гомогената головного мозга и искусственной CSF, охватывая диапазон концентраций от 0,5 до 500 нг/мл. Гомогенизированную ткань головного мозга вместе с образцами плазмы осаждали посредством добавления 3-кратного объема холодного ацетонитрила, содержащего внутренний стандарт (40 нг/мл дексаметазона и 40 нг/мл диклофенака), и 10 мкл образцов CSF осаждали с помощью 100 мкл холодного ацетонитрила, содержащего внутренний стандарт. После вихревого перемешивания в течение 2 минут и центрифугирования в течение 5 минут при 14000 об/мин, надосадочную жидкость анализировали с помощью ЖХ/МС/МС (API 4000, Applied Biosystems, Foster City). Два набора стандартных кривых были построены в начале и в конце каждой серии анализа образцов плазмы. Для образцов головного мозга и спинномозговой жидкости была проанализирована одна стандартная кривая наряду с тестируемыми образцами.
Общие уровни в головном мозге, выраженные как соотношение мозг/плазма (Kp), измеряли с помощью ППК (площадь под кривой) мозг./ППК плазм, у грызунов после перорального введения. Свободную фракцию тестируемого соединения в биологическом матриксе определяли с помощью анализа связывания в плазме и головном мозге in vitro. Kp,uu рассчитывали по следующему уравнению: Kp,uu = ППК (мозг.)/ППК (плазм.)×(fu, мозг./fu, плазм.). Данные показаны в таблице 7.
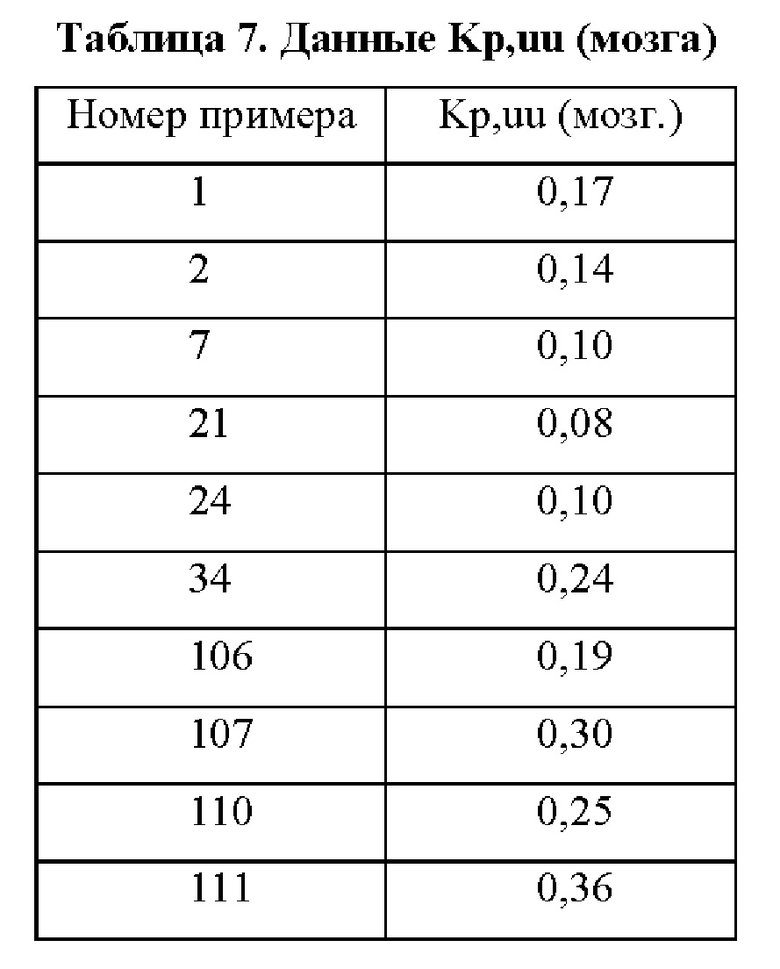
Хотя настоящее изобретение было конкретно проиллюстрировано и описано со ссылкой на конкретные воплощения (некоторые из которых являются предпочтительными воплощениями), специалистам в данной области техники следует понимать, что различные изменения в форме и деталях могут быть сделаны в нем без отступления от сущности и объема настоящего изобретения, раскрытого здесь.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
Группа изобретений относится к области органической химии и включает соединение формулы (I), его фармацевтически приемлемые соли, индивидуальные соединения, представленные в формуле изобретения, фармацевтическую композицию, способ ингибирования DNA-PK и способ лечения расстройства, связанного с DNA-PK. В формуле I X1, Х2 и Х3 каждый независимо представляет собой С, СН или N, при условии, что по меньшей мере один из X1, Х2 и Х3 представляет собой N, и по меньшей мере один из X1, Х2 и Х3 представляет собой С; пунктирная линия  означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь; R1 отсутствует или представляет собой C1-6алкил, где указанный C1-6алкил возможно может быть замещен от 1 до 3 галогеном или дейтерием; каждый R2 и R4 отсутствует или независимо выбран из галогена, C1-6алкила, C1-6алкоксила, -(CH2)n-Q, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, С1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом, C1-6 галогеналкоксилом, С3-8циклоалкилом или С3-8циклоалкоксилом, или R2 выбран из
означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь; R1 отсутствует или представляет собой C1-6алкил, где указанный C1-6алкил возможно может быть замещен от 1 до 3 галогеном или дейтерием; каждый R2 и R4 отсутствует или независимо выбран из галогена, C1-6алкила, C1-6алкоксила, -(CH2)n-Q, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, С1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом, C1-6 галогеналкоксилом, С3-8циклоалкилом или С3-8циклоалкоксилом, или R2 выбран из 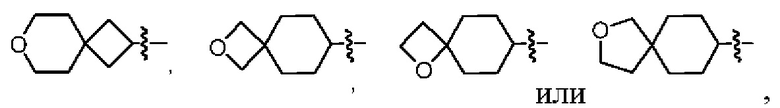 которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом; R3 выбран из галогена, C1-6алкила, C1-6алкоксила, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, C1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом или C1-6 галогеналкоксилом, где n равен 0, 1 или 2, Q представляет собой 3-8-членный насыщенный или ненасыщенный карбоциклил или 3-8-членный насыщенный или ненасыщенный гетероциклил, имеющий 1 или 2 кольцевых гетероатома, независимо выбранных из кислорода и азота, где одна образующая кольцо группа -CH2- гетероциклила может быть заменена -С(О)-; кольцо А представляет собой 6-членный гетероарил, имеющий 1 кольцевой гетероатом азота, или 9-членное бициклическое кольцо, имеющее 2-3 кольцевых гетероатома, выбранных из кислорода, серы и азота. Технический результат - соединение формулы (I) в качестве ингибитора ДНК-зависимой протеинкиназы (DNA-PK). 5 н. и 21 з.п. ф-лы, 7 табл., 120 пр.
которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом; R3 выбран из галогена, C1-6алкила, C1-6алкоксила, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, C1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом или C1-6 галогеналкоксилом, где n равен 0, 1 или 2, Q представляет собой 3-8-членный насыщенный или ненасыщенный карбоциклил или 3-8-членный насыщенный или ненасыщенный гетероциклил, имеющий 1 или 2 кольцевых гетероатома, независимо выбранных из кислорода и азота, где одна образующая кольцо группа -CH2- гетероциклила может быть заменена -С(О)-; кольцо А представляет собой 6-членный гетероарил, имеющий 1 кольцевой гетероатом азота, или 9-членное бициклическое кольцо, имеющее 2-3 кольцевых гетероатома, выбранных из кислорода, серы и азота. Технический результат - соединение формулы (I) в качестве ингибитора ДНК-зависимой протеинкиназы (DNA-PK). 5 н. и 21 з.п. ф-лы, 7 табл., 120 пр.
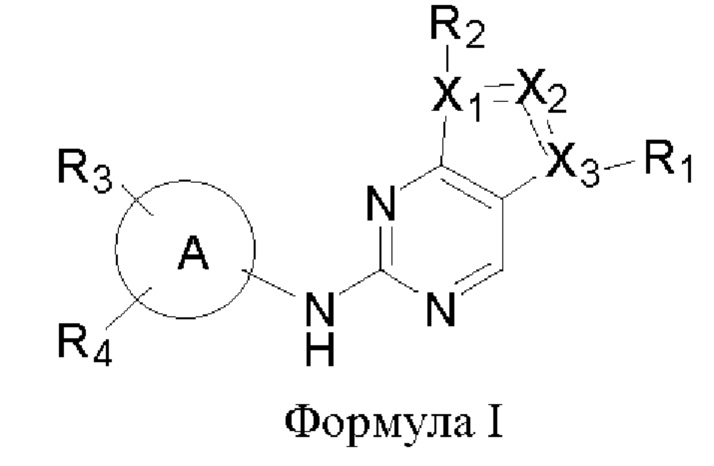
1. Соединение формулы I
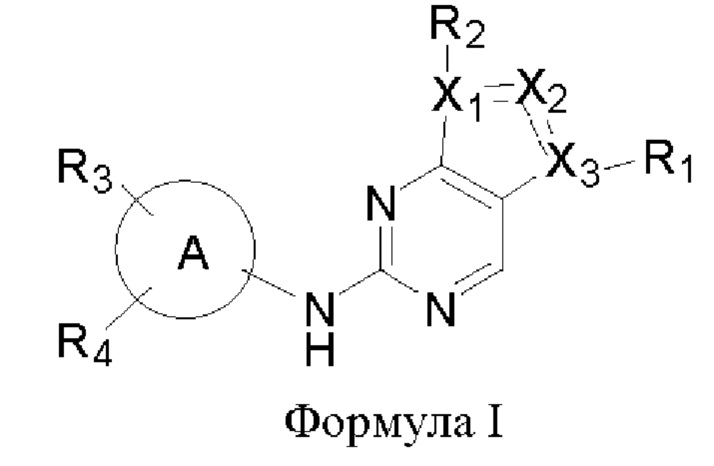
и его фармацевтически приемлемая соль, где
X1, Х2 и Х3 каждый независимо представляет собой С, СН или N, при условии, что по меньшей мере один из X1, Х2 и Х3 представляет собой N, и по меньшей мере один из X1, Х2 и Х3 представляет собой С;
пунктирная линия  означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь;
означает, что связь между X1 и Х2 и между Х2 и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и Х2 и между Х2 и Х3 представляет собой простую связь;
R1 отсутствует или представляет собой C1-6алкил, где указанный C1-6алкил возможно может быть замещен от 1 до 3 галогеном или дейтерием;
каждый R2 и R4 отсутствует или независимо выбран из галогена, C1-6алкила, C1-6алкоксила, -(CH2)n-Q, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, С1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом, C1-6 галогеналкоксилом, С3-8циклоалкилом или С3-8циклоалкоксилом, или
R2 выбран из  которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом;
которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом;
R3 выбран из галогена, C1-6алкила, C1-6алкоксила, которые возможно могут быть независимо замещены от 1 до 3 дейтерием, гидроксилом, циано, галогеном, C1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом или C1-6 галогеналкоксилом,
где n равен 0, 1 или 2, Q представляет собой 3-8-членный насыщенный или ненасыщенный карбоциклил или 3-8-членный насыщенный или ненасыщенный гетероциклил, имеющий 1 или 2 кольцевых гетероатома, независимо выбранных из кислорода и азота, где одна образующая кольцо группа -CH2- гетероциклила может быть заменена -С(О)-;
кольцо А представляет собой 6-членный гетероарил, имеющий 1 кольцевой гетероатом азота, или 9-членное бициклическое кольцо, имеющее 2-3 кольцевых гетероатома, выбранных из кислорода, серы и азота.
2. Соединение по п. 1, имеющее структуру формулы Ia
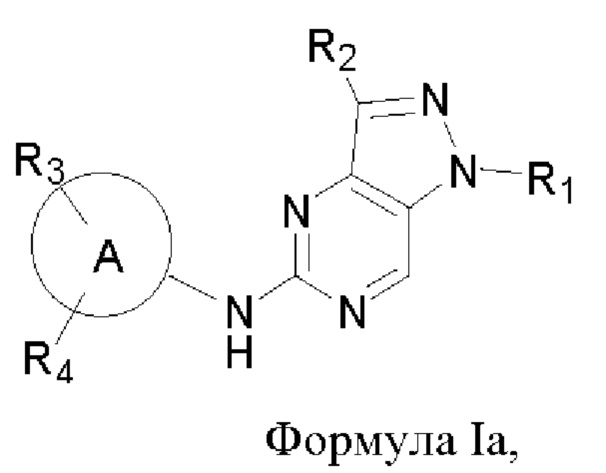
и его фармацевтически приемлемая соль.
3. Соединение по п. 1, имеющее структуру формулы Ib
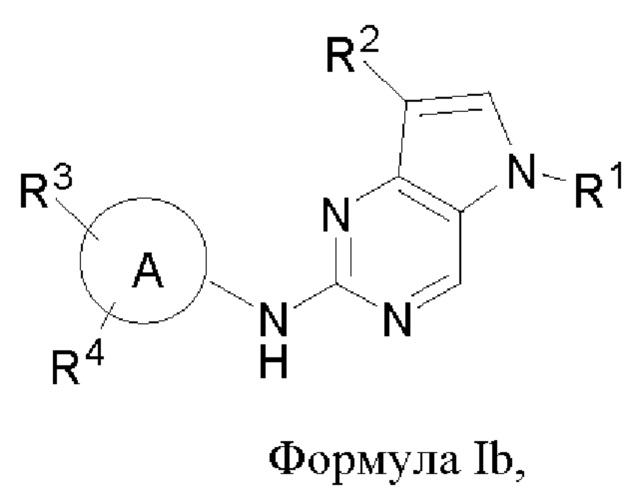
и его фармацевтически приемлемая соль.
4. Соединение по п. 1, имеющее структуру формулы Ic
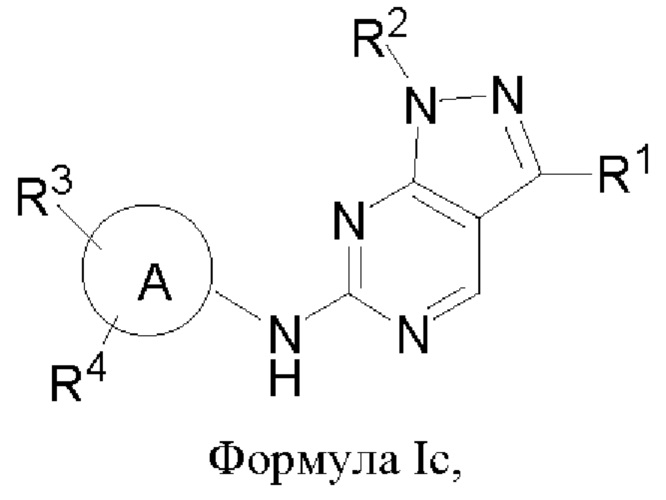
и его фармацевтически приемлемая соль.
5. Соединение по п. 1, имеющее структуру формулы Id
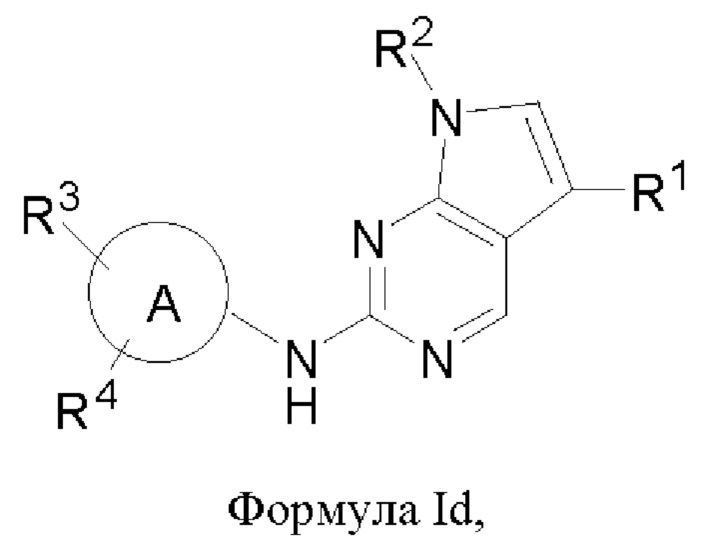
и его фармацевтически приемлемая соль.
6. Соединение по п. 1, где R2 выбран из метила, этила, пропила, бутила, циклопропила, циклобутила, оксетанила, циклопентанила, тетрагидрофурила, циклогексанила, тетрагидропиранила, циклогептанила, пиперидинила, фенила, пиридинила, пиридонила, оксоканила, дигидропиранила, спиро[3.3]гептанила, спиро[2.5]октанила, бицикло[1.1.1]пентанила, бицикло[3.2.1]октанила, 8-оксабицикло[3.2.1]октан-3-ила, которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, галогеном, C1-6алкилом, C1-6 галогеналкилом, C1-6алкоксилом, C1-6 галогеналкоксилом, С3-8циклоалкилом или С3-8циклоалкоксилом.
7. Соединение по п. 1, где R2 выбран из:
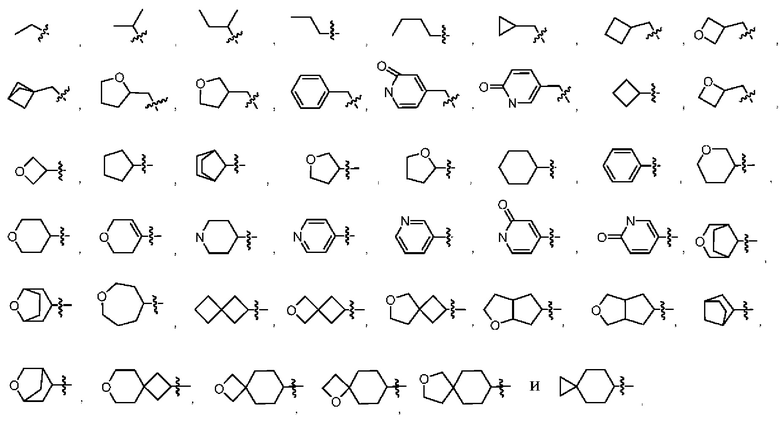
которые возможно могут быть независимо замещены от 1 до 3 гидроксилом, циано, фтором, хлором, бромом, метилом, этилом, метоксилом, дифторметилом, дифторметоксилом или трифторметоксилом.
8. Соединение по п. 1, где R2 представляет собой циклогексанил или тетрагидропиранил, которые возможно могут быть независимо замещены от 1 до 3 галогеном, C1-6алкилом или C1-6алкоксилом.
9. Соединение по п. 1, где R1 представляет собой метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил или изобутил, которые возможно могут быть замещены от 1 до 3 галогеном или дейтерием.
10. Соединение по п. 1, где R1 представляет собой метил, этил, трифторметил или тридейтерийметил.
11. Соединение по п. 1, где кольцо А представляет собой 9-членное бициклическое кольцо, имеющее 2-3 кольцевых гетероатома, выбранных из кислорода, серы и азота.
12. Соединение по п. 11, где кольцо А представляет собой фенил- или пиридинил-конденсированное бициклическое кольцо.
13. Соединение по п. 12, где кольцо А выбрано из
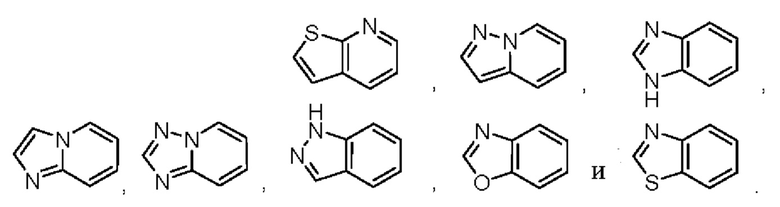
14. Соединение по п. 1, где R4 выбран из галогена, C1-6алкила, С1-6 галогеналкила, С1-6алкоксила, C1-6галогеналкоксила, 3-8-членного насыщенного или ненасыщенного гетероциклила, имеющего 1 кольцевой гетероатом, выбранный из кислорода и азота, где указанный гетероциклил возможно может быть замещен от 1 до 3 С1-3алкилом,
R3 выбран из галогена, C1-6алкила, C1-6галогеналкила, С1-6алкоксила, C1-6галогеналкоксила.
15. Соединение по п. 1, где кольцо А представляет собой 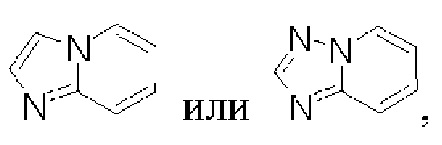
R4 отсутствует или независимо выбран из метила, метоксила, хлора или цианометила,
R3 выбран из метила, метоксила, хлора или цианометила.
16. Соединение по п. 1, имеющее структуру формулы Ie
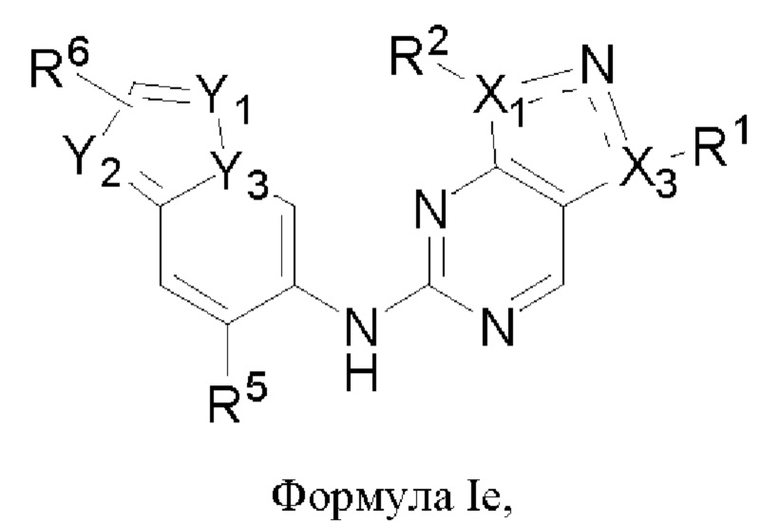
и его фармацевтически приемлемая соль,
где
один из X1 и Х3 представляет собой N, и другой представляет собой С, пунктирная линия  означает, что связь между X1 и N и между N и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и N и между N и Х3 представляет собой простую связь;
означает, что связь между X1 и N и между N и Х3 может представлять собой простую или двойную связи, при условии, что по меньшей мере одна из связей между X1 и N и между N и Х3 представляет собой простую связь;
R1 представляет собой С1-3алкил,
R2 представляет собой циклопентил, циклогексанил, тетрагидропиранил или 8-оксабицикло[3.2.1]октан-3-ил, которые возможно могут быть независимо замещены от 1 до 3 галогеном или C1-3алкоксилом,
Y1, Y2 и Y3 каждый независимо представляет собой С, СН или N, при условии, что по меньшей мере два из Y1, Y2 и Y3 представляют собой N;
R5 представляет собой галоген или C1-3алкил,
R6 представляет собой C1-3алкил.
17. Соединение по п. 16, где R2 представляет собой незамещенный циклопентил, циклогексанил, тетрагидропиранил или 8-оксабицикло[3.2.1]октан-3-ил, которые возможно могут быть независимо замещены от 1 до 3 галогеном или С1-3алкоксилом.
18. Соединение по п. 16, где Y3 представляет собой N и по меньшей мере один из Y1 и Y2 представляет собой N.
19. Соединение по п. 16, где R5 представляет собой метил.
20. Соединение, выбранное из группы, состоящей из: 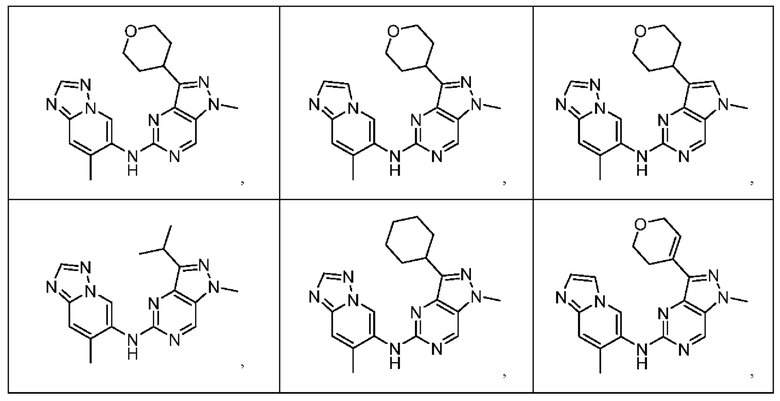
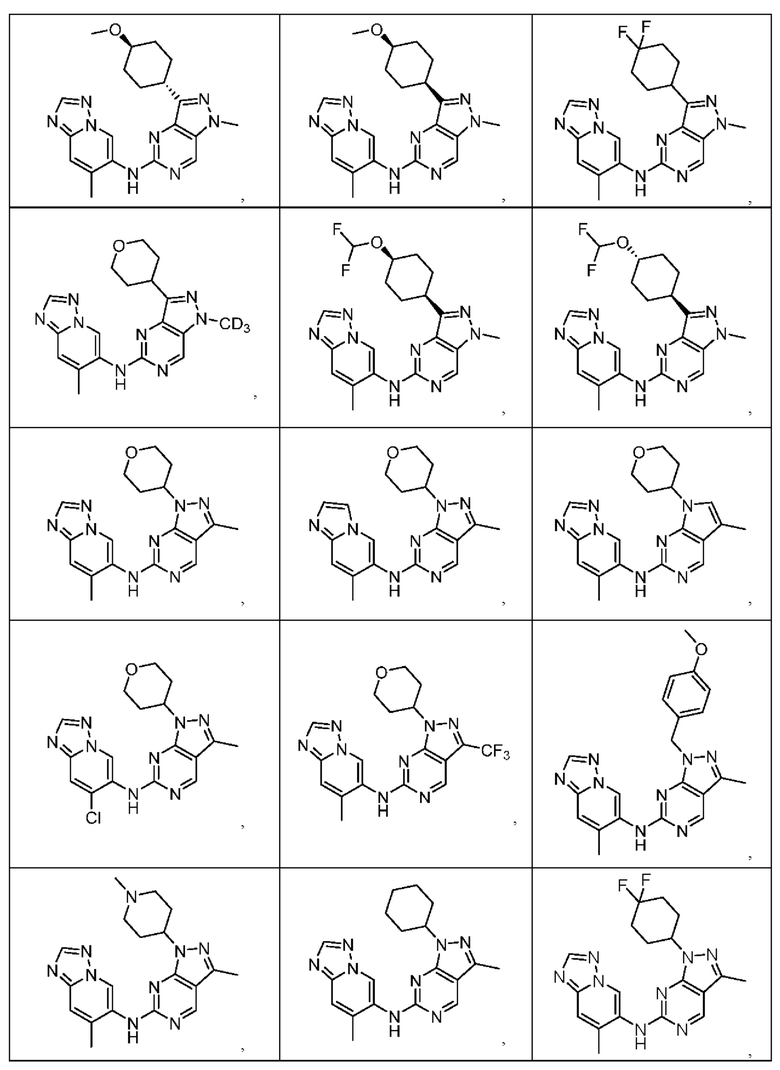
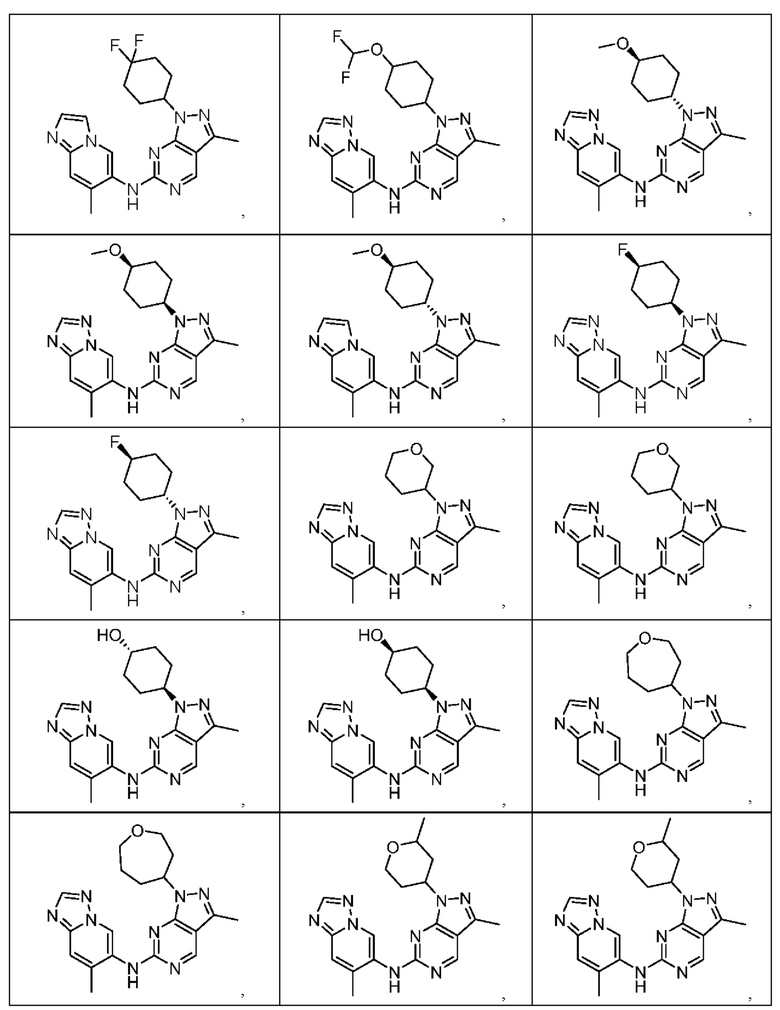
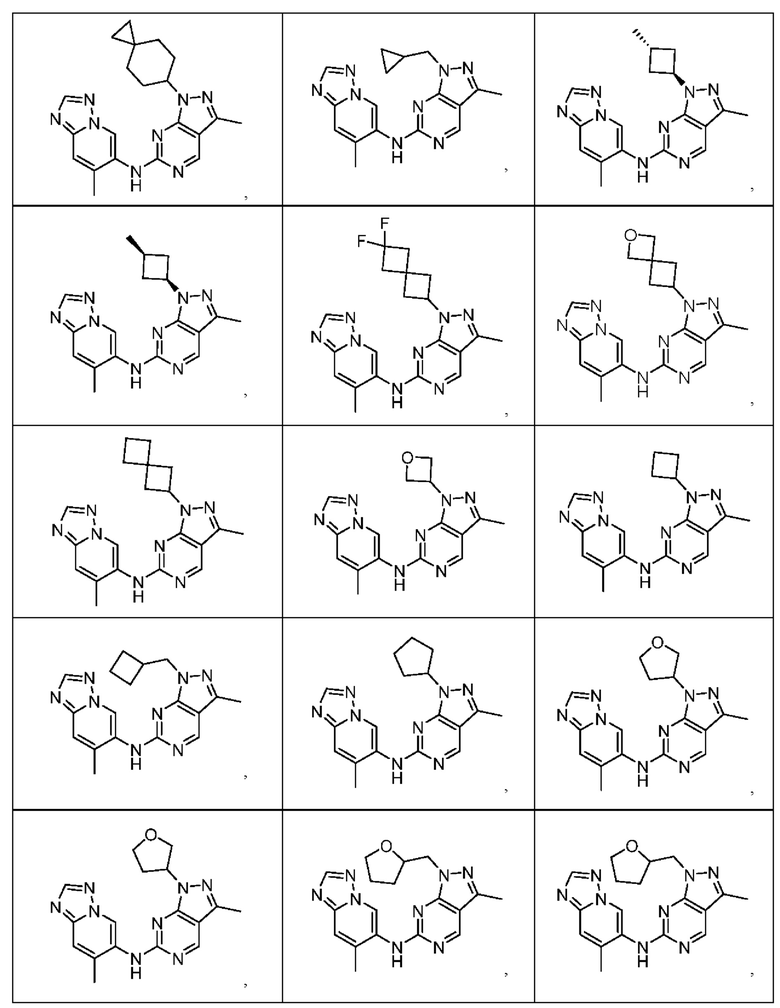
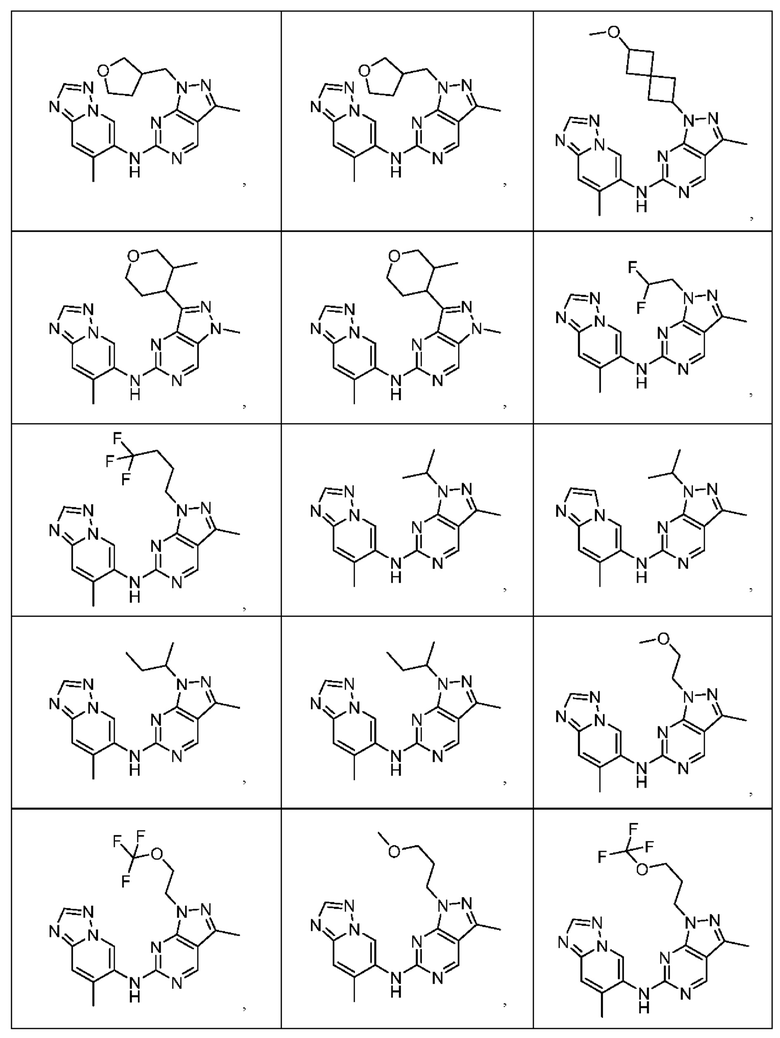
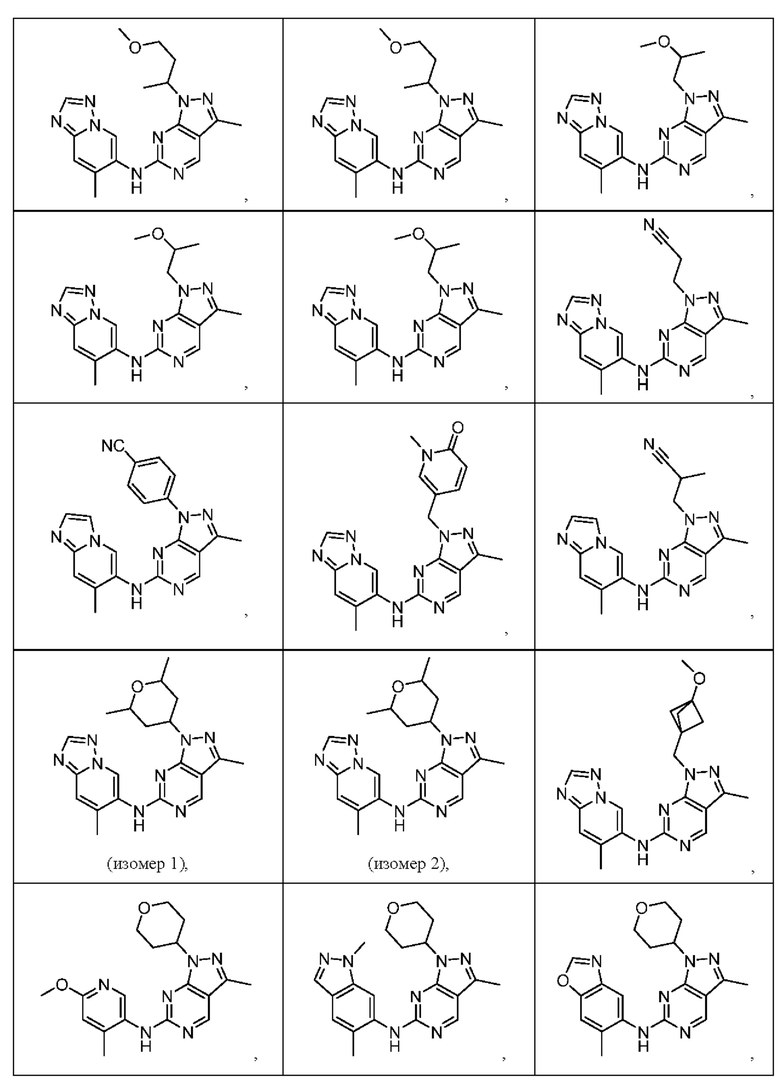
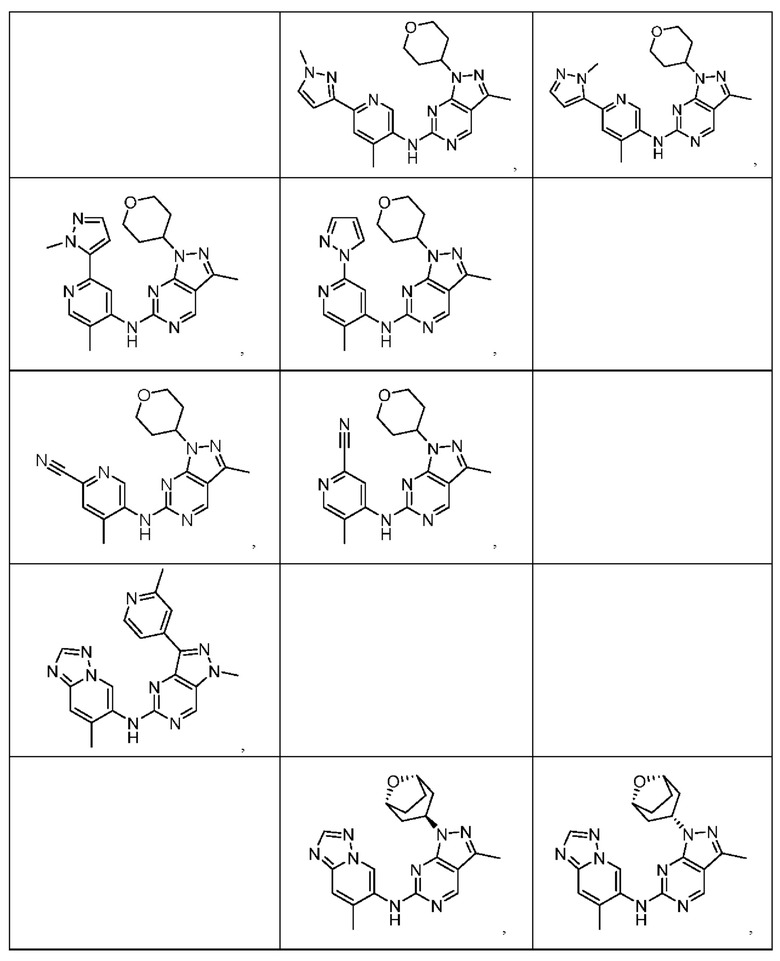
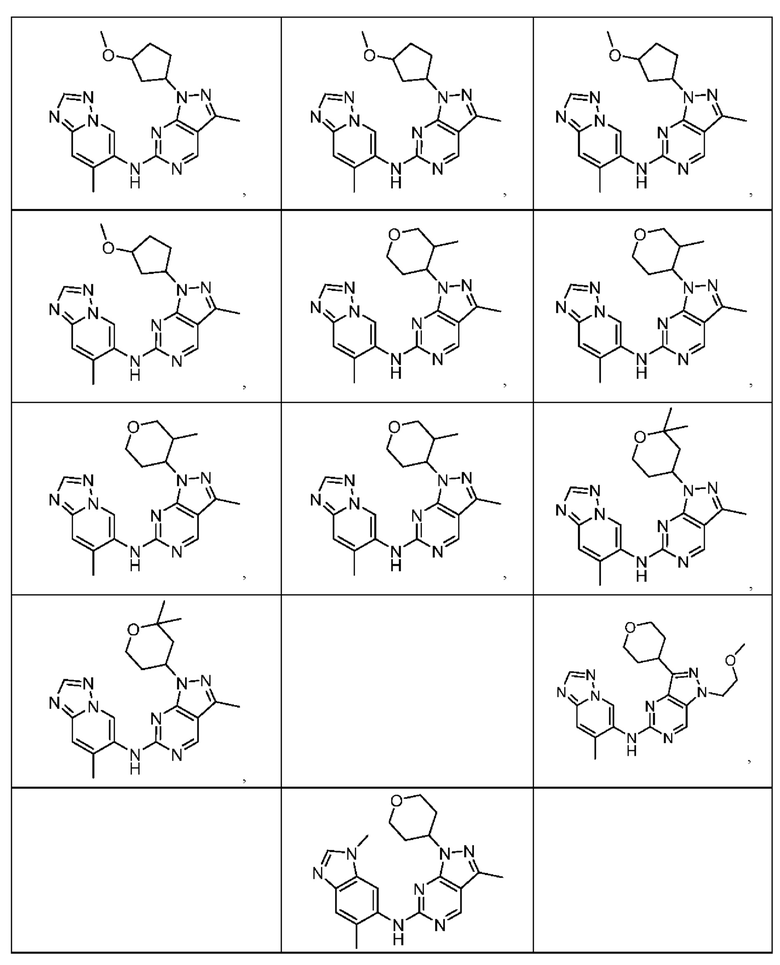
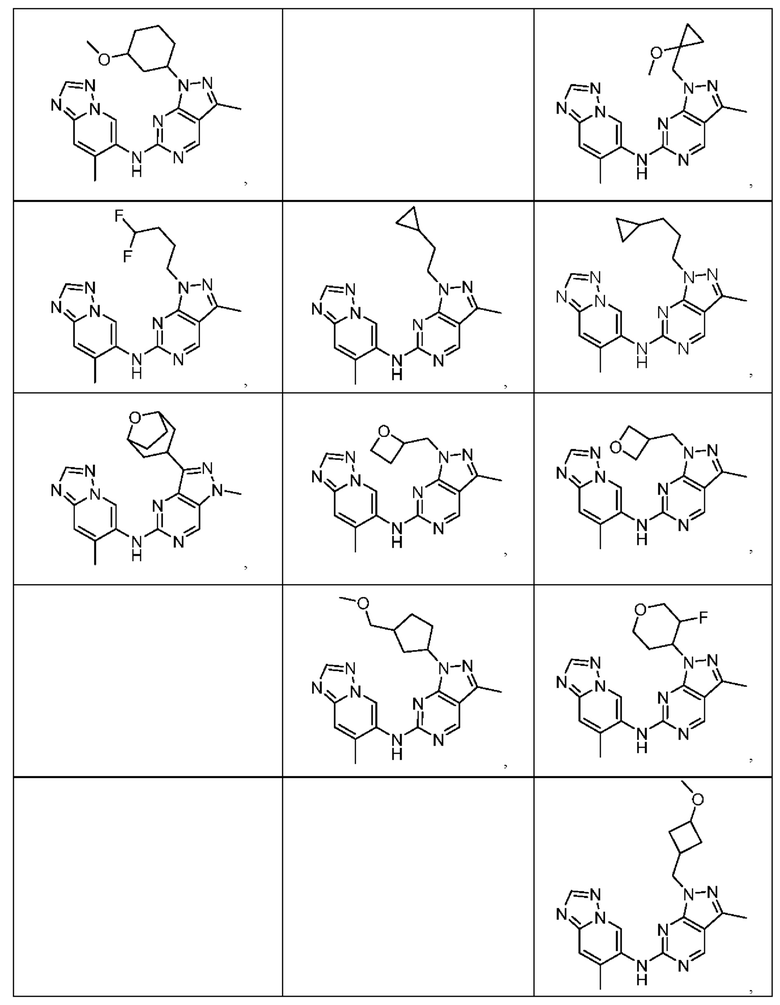
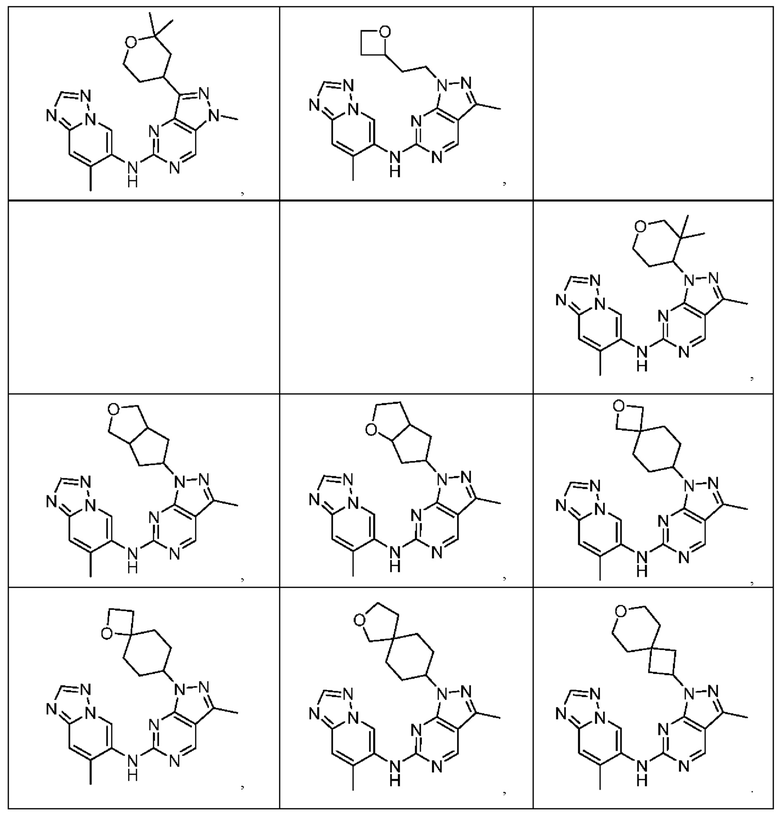
и его фармацевтически приемлемая соль.
21. Фармацевтическая композиция, обладающая активностью ингибиторов ДНК-зависимой протеинкиназы (DNA-PK), которая содержит эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-20 и фармацевтически приемлемый разбавитель, эксципиент или носитель.
22. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-20 или фармацевтическая композиция по п. 21 для ингибирования ДНК-зависимой протеинкиназы.
23. Способ ингибирования DNA-PK путем применения соединения или его фармацевтически приемлемой соли по любому из пп. 1-20 или фармацевтической композиции по п. 21.
24. Способ лечения расстройства, связанного с DNA-PK, у субъекта, включающий введение субъекту эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-20 или фармацевтической композиции по п. 21.
25. Способ по п. 24, где субъект представляет собой теплокровное животное, такое как человек.
26. Способ по п. 24, где расстройство, связанное с DNA-PK, представляет собой рак.
| WO 2005107760 A1, 17.11.2005 | |||
| ЕА 200802332 А1, 30.06.2009 | |||
| JP 2006241089 A, 14.09.2006 | |||
| WO 2006074984 A1, 20.07.2006 | |||
| WO 2006074985 A1, 20.07.2006 | |||
| WO 2018114999 А1, 28.06.2018 | |||
| WO 2014159690 А1, 02.10.2014 | |||
| WO 2013163190 А1, 31.10.2013. |

