Настоящее изобретение относится к конъюгату антитела с лекарственным средством, способному к связыванию с IGF-1R. В одном аспекте изобретение относится к конъюгату антитела с лекарственным средством, содержащему антитело, способное к связыванию с IGF-1R, где антитело конъюгировано с по меньшей мере одним лекарственным средством, выбранным из производных доластатина 10 и ауристатинов. Изобретение также включает в себя способ лечения и применение указанного конъюгата антитела с лекарственным средством для лечения рака.
Предшествующий уровень техники
Рецептор инсулиноподобного фактора роста 1, называемый IGF-1R (или иногда IGF1R или IGF-IR) представляет собой рецептор, обладающий активностью тирозинкиназы, имеющий 70% гомологию с рецептором инсулина IR. IGF-1R представляет собой гликопротеин с молекулярной массой приблизительно 350000. Он представляет собой гетеродимерный рецептор, каждая половина которого, связанная с другой дисульфидными мостиками, состоит из внеклеточной α-субъединицы и трансмембранной β-субъединицы. IGF-1R связывает IGF1 и IGF2 с очень высоким сродством (Kd #1 нМ), но в равной степени способен к связыванию с инсулином со сродством в 100-1000 раз ниже. Наоборот, IR связывает инсулин с очень высоким сродством, хотя IGF связываются с рецептором инсулина со сродством в 100 раз ниже. Тирозинкиназный домен IGF-1R и IR обладают очень высокой гомологией последовательности, хотя зоны более слабой гомологии соответственно касаются богатого цистеином участка, расположенного на α-субъединице и С-концевой части β-субъединице. Различия последовательности, наблюдаемые в α-субъединице, расположены в зоне связывания лигандов и, следовательно являются источником относительных значений сродства IGF-1R и IR к факторам IGF и инсулину соответственно. Результатом различий в С-концевом участке β-субъединицы является расхождение биохимических путей передачи сигнала двух рецепторов; IGF-1R опосредует митогенные эффекты, дифференцировку и антиапоптические эффекты, тогда как в активацию IR, главным образом, вовлечены эффекты на уровне метаболических биохимических путей.
Цитоплазматические тирозинкиназные белки активируются связыванием лиганда с внеклеточным доменом рецептора. В активацию киназ, в свою очередь, вовлечена стимуляция различных внутриклеточных субстратов, включающих IRS-1, IRS-2, Shc и Grb 10. Два основных субстрата IGF-1R представляют собой IRS и Shc, которые опосредуют за счет активации многочисленных последующих эффекторов большинство действий на рост и дифференцировку, связанных с присоединением IGF к этому рецептору. Доступность субстратов может, следовательно, определять конечное биологическое действие, связанное с активацией IGF-1R. В случае преобладания IRS-1 клетки склонны к пролиферации и к трансформации. В случае преобладания Shc клетки склонны к дифференцировке. По-видимому, в действия по защите против апоптоза, главным образом, вовлечен путь, представляющий собой путь фосфатидилинозит-3-киназ (PI 3-киназ).
За последние десять лет роль системы IGF в канцерогенезе стала предметом интенсивного исследования. За этим интересом последовало открытие того факта, что в дополнение к его митогенным и антиапоптическим свойствам IGF-1R, по-видимому, требуется для установления и поддержания трансформированного фенотипа. В действительности четко установлено, что гиперэкспрессия или конститутивная активация IGF-1R в широком ряде клеток ведет к росту клеток независимо от подпитки средой без фетальной сыворотки теленка и к образованию опухолей у бестимусных мышей. Это свойство само по себе не является уникальным свойством, поскольку к трансформации клеток может вести широкое разнообразие продуктов гиперэкспрессируемых генов, включающих значительное число рецепторов факторов роста. Однако критическим открытием, четко продемонстрировавшим ведущую роль, которую играет IGF-1R в трансформации, стала демонстрация того, что клетки IGR-1R-, в которых инактивирован ген, кодирующий IGF-1R, полностью устойчивы к трансформации различными агентами, которые обычно способны трансформировать клетки, такими как белок Е5 вируса бычьей папилломы, гиперэкспрессия EGFR или PDGFR, антиген Т SV40, активированный ras или комбинация этих двух последних факторов.
IGF-1R экспрессируется в широком ряде опухолей и опухолевых линий, и IGF амплифицируют опухолевый рост посредством их присоединения к IGF-1R. Другие аргументы в пользу роли IGF-1R в канцерогенезе получены из исследований с использованием мышиных моноклональных антител, направленных против рецептора, или с использованием отрицательных доминантов IGF-1R. Действительно, мышиные моноклональные антитела, направленные против IGF-1R, ингибируют пролиферацию различных линий клеток в культуре и рост опухолевых клеток in vivo. Было показано, что отрицательный доминант IGF-1R, вероятно, способен ингибировать пролиферацию опухоли.
Начато большое число проектов по разработке изолированных антител к IGF-1R для лечения форм рака. Тем не менее, к этой дате ни один из этих проектов не имел успеха, и ни одно антитело к IGF-1R не имеется в продаже.
Кроме того, серия клинических исследований, в которых были задействованы антитела к IGF-1R в комбинации с антителами к рецептору эпидермального фактора роста (EGFR) для нацеливания на обе мишени EGFR и IGF-1R, не имела успеха, поскольку ни одно из этих антител не было способно к лечению пациентов с мутацией гомолога вирусного онкогена крысиной саркомы Кирстена (KRAS).
Вследствие этого IGF-1R в настоящее время не считают значительной мишенью, и при исследовании потенциальных терапевтических антител IGF-1R больше не считают представляющим интерес.
Кроме того,также необходимо отметить, что стремления к созданию антител к IGF-1R были сосредоточены на изолированных антителах, т.е. антителах, полезных за счет своих собственных свойств. В этом смысле IGF-1R считают неподходящим в качестве мишени для создания ADC, т.е. конъюгата антитела с лекарственным средством (называемым «ADC»), поскольку IGF-1R описан как мишень, также широко экспрессируемую нормальными клетками, включая кровеносные сосуды. В этом смысле можно отметить, что наиболее недавно полученное антитело к IGF-1R, т.е. AVE1642, разрабатывается в виде изолированного антитела, не нагруженного лекарственным средством. То же верно для других антител IGF-1R, в настоящее время находящихся в разработке, и для всех антител, не имевших успеха в клинических исследованиях.
В данном контексте изобретение относится к ADC или к конъюгату и к его применению для лечения рака, и более конкретно для лечения форм рака, экспрессирующих IGF-1R.
Конъюгаты ADC сочетают связывающую специфичность с активностью лекарственных средств, таких как, например, цитотоксические агенты. В технологии, связанной с разработкой моноклональных антител, применением более эффективных лекарственных средств и конструированием химических линкеров для ковалентного связывания этих компонентов, в последние годы произошел быстрый прогресс.
Применение ADC дает возможность для локальной доставки лекарственных средств, введение которых в виде неконъюгированных лекарственных средств может приводить в результате к неприемлемым уровням токсичности для нормальных клеток.
Иными словами, их применение, таким образом, подразумевает максимальную эффективность при минимальной токсичности. Попытки конструирования и очистки ADC сосредоточены как на селективности антитела, так и на механизме действия лекарственного средства, свойствах связывания лекарственного средства, отношения лекарственное средство/антитело (нагрузки лекарственным средством или drug/antibody ratio, DAR) и высвобождения лекарственного средства. Группировки лекарственного средства могут передавать свои цитотоксические и цитостатические воздействия посредством механизмов, включающих связывание тубулина, связывание ДНК, протеасом, нарушение функции рибосом, синтеза белка и/или ингибирование топоизомераз. Некоторые цитотоксические лекарственные средства склонны к инактивации или к меньшей активности при конъюгации с антителом большого размера.
Необходимо отдельно охарактеризовать каждое антитело, сконструировать подходящий линкер и идентифицировать подходящий цитотоксический агент, который сохраняет свою активность при доставке в опухолевые клетки. Необходимо учитывать антигенную плотность на раковой мишени, а также учитывать, экспрессируют ли нормальные ткани целевой антиген. Другие соображения включают: происходит ли интернализация ADC при связывании мишени; предпочтительно ли цитостатическое или цитотоксическое лекарственное средство при рассмотрении возможного воздействия на нормальную ткань и/или типа и стадии рака, подлежащего лечению; и представляет ли собой линкер, соединяющий антитело с полезной нагрузкой лекарственным средством, расщепляемую или нерасщепляемую связь. Кроме того, в отношении процесса будущей разработки соединения отношение конъюгации антитела с группировкой лекарственного средства должно быть достаточным, и при этом должен отсутствовать риск для связывающей активности антитела и/или активности лекарственного средства, а физико-химические свойства ADC не должны претерпевать модификаций, приводящих в результате к агрегации.
ADC представляет собой комплексную биологическую молекулу, и трудности, связанные с разработкой эффективного ADC, остаются существенным вопросом.
Сущность изобретения
Настоящее изобретение направлено на разрешение данного вопроса и относится к ADC следующей формулы (I):
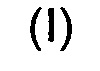
или его фармацевтически приемлемой соли,
где
Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные к связыванию IGF-1R человека, выбранные из:
i) антитело, содержащее три участка, определяющих комплементарность (CDR, от англ. complementarity-determining region), тяжелой цепи, имеющие последовательности SEQ ID NO 1, 2 и 3, и три CDR легкой цепи, имеющие последовательности SEQ ID NO 4, 5 и 6;
ii) антитело, конкурирующее за связывание с IGF-1R с антителом i); и
iii) антитело, связывающееся с тем же эпитопом, что и антитело i) к IGF-1R;
L представляет собой линкер;
D представляет собой группировку лекарственного средства следующей формулы (II):
где:
R2 представляет собой СООН, СООСН3 или тиазолил;
R3 представляет собой Н или (С1-С6)алкил;
R9 представляет собой Н или (С1-С6)алкил;
m представляет собой целое число, составляющее от 1 до 8;
волнистой линией показана точка присоединения к L; и
n равно от 1 до 12.
Воплощение изобретения относится к ADC, где Ab выбрано из:
a) антитела, содержащего три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 10, 5 и 11;
c) антитела, содержащего три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12; и
d) антитела, содержащего три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 8, 2 и 3, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11.
Воплощение изобретения относится к ADC, где Ab выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 13, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 14, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 10, 5 и 11;
c) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 15, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12;
d) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 16, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11; и
e) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 17, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12.
Воплощение изобретения относится к ADC, где Ab выбрано из:
a) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 18, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 19, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
c) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 20, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
d) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 21, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 8, 2 и 3; и
e) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 22, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3.
В воплощении изобретение относится к ADC, где Ab выбрано из:
i) антител 208F2, 212А11, 214F8, 219D6 и 213В10;
ii) антител, конкурирующих за связывание с IGF-1R с антителами i); и
iii) антител, связывающихся с тем же эпитопом IGF-1R, что и антитела i).
Воплощение изобретения относится к ADC, где Ab представляет собой гуманизированное антитело.
Воплощение изобретения относится к ADC, где Ab выбрано из: антитела, содержащего:
a) тяжелую цепь, имеющую CDR-H1, CDR-H2 и CDR-H3 последовательностей SEQ ID NO 7, 2 и 3 соответственно, и FR1, FR2 и FR3, выделенные из IGHV1-46*01 (SEQ ID NO 46) зародышевой линии человека, и FR4, выделенный из IGHJ4*01 зародышевой линии человека (SEQ ID NO 48); и
b) легкую цепь, имеющую CDR-L1, CDR-L2 и CDR-L3 последовательностей SEQ ID NO 9, 5 и 11 соответственно, и FR1, FR2 и FR3, выделенные из IGHV1-39*01 (SEQ ID NO 47) зародышевой линии человека, и FR4, выделенный из IGKJ4*01 зародышевой линии человека (SEQ ID NO 49).
В воплощении изобретения Ab выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 33 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 33, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11; и
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 34 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 34, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11.
В воплощении изобретения Ab выбрано из:
a) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 35 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 35, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3; и
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 36 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 36, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3.
В воплощении изобретения Ab выбрано из:
a) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 37 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 37, и легкую цепь последовательности SEQ ID NO 39 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO
39, или состоящего из этих цепей; и
b) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 38 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 38, и легкую цепь последовательности SEQ ID NO 40 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 40, или состоящего из этих цепей.
В воплощении изобретения Ab выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 или 80; и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность, выбранную из SEQ ID NO 57 и 60 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 57 и 60; и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3; и
c) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 или 80; и три CDR легкой цепи, имеющих последовательности SEQ ID NO 57 или 60 или любую последовательность, по меньшей мере на 80% идентичную SEQ ID NO 57 или 60;
В воплощении изобретения Ab выбрано из:
a) тяжелой цепи, имеющей последовательность, выбранную из SEQ ID NO 58, 63, 65, 67, 69, 71, 73, 75, 77, 79 и 81 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 58, 63, 65, 67, 69, 71, 73, 75, 77, 79 или 81; и
b) легкой цепи, имеющей последовательность, выбранную из SEQ ID NO 59 и 61 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 59 или 61.
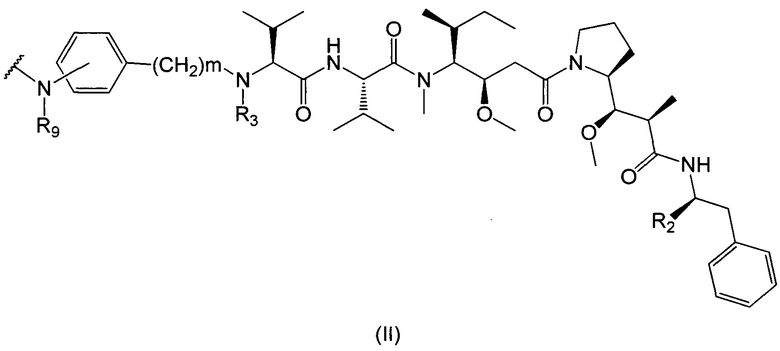
В воплощении изобретение относится к ADC, где L представляет собой линкер следующей формулы (III):
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил или (С2-С6)алкил-карбонил;
W представляет собой аминокислотное звено; w представляет собой целое число, составляющее от 0 до 5;
Y представляет собой РАВ-карбонил, где РАВ представляет собой 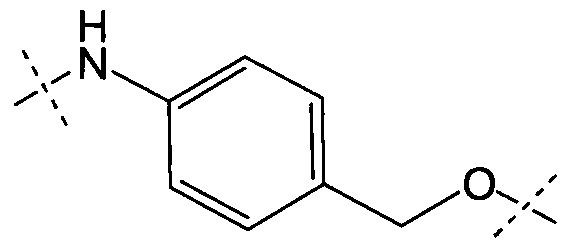 ; у равно 0 или 1;
; у равно 0 или 1;
звездочкой указана точка присоединения к D; и
волнистой линией указана точка присоединения к Ab.
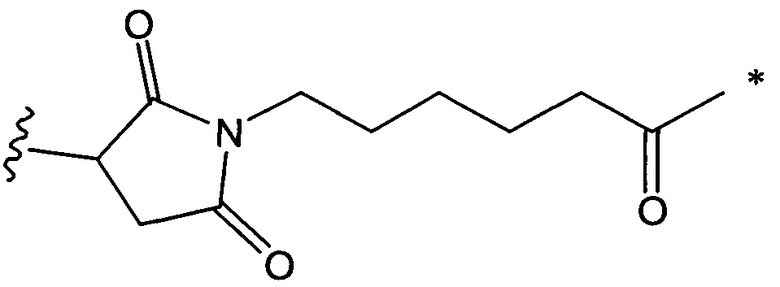
Воплощение изобретения относится к ADC, где L2 имеет следующую формулу (III):
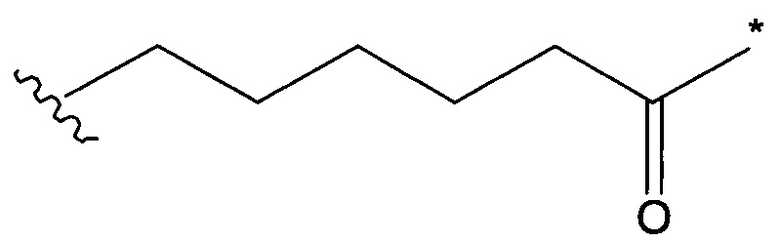
где
звездочкой указана точка присоединения к (W)w; и
волнистой линией указана точка присоединения к атому азота малеимидной группировки формулы:
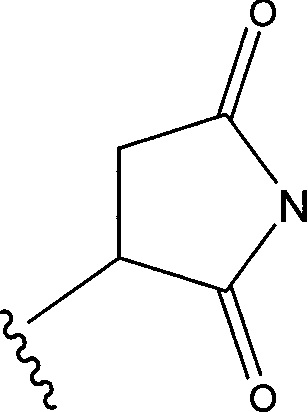
В воплощении изобретения w равно 0, либо w равно 2, и тогда (W)w выбрана из:
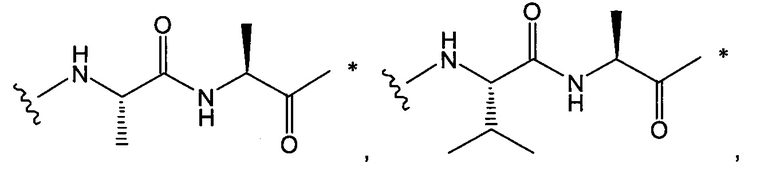
и 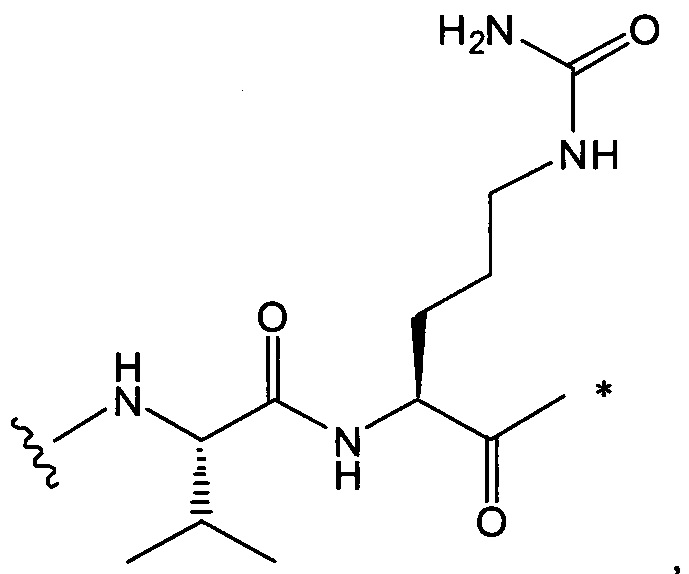
где
звездочкой указана точка присоединения к (Y)y; и
волнистой линией указана точка присоединения к L2.
Воплощение изобретения относится к ADC, где L выбран из:
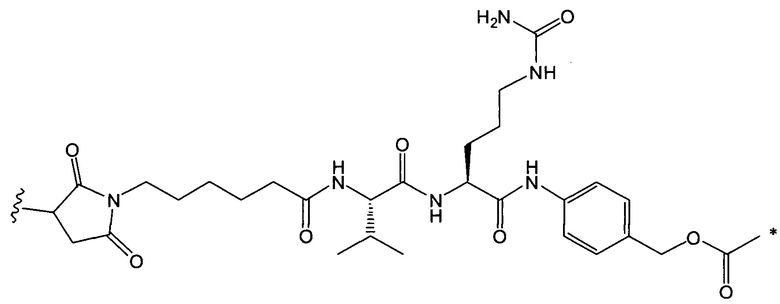 ,
,
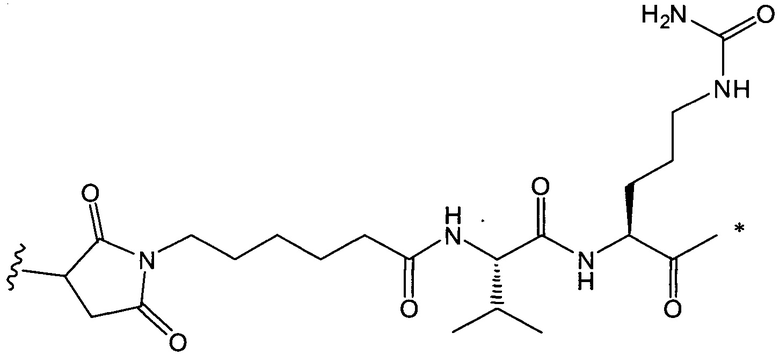 , и
, и
где звездочкой указана точка присоединения к D, а волнистой линией указана точка присоединения к Ab.
Воплощение изобретения относится к ADC, где (L-D) выбрано из:
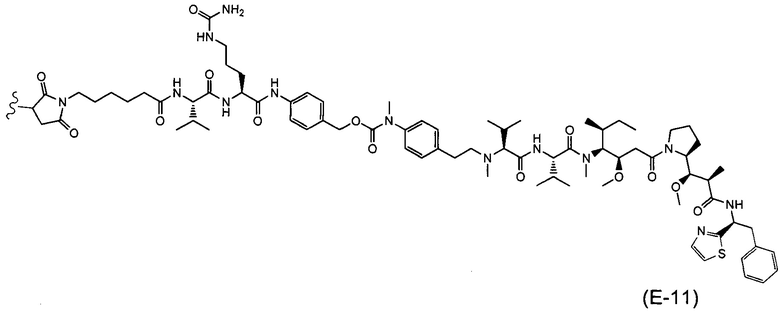
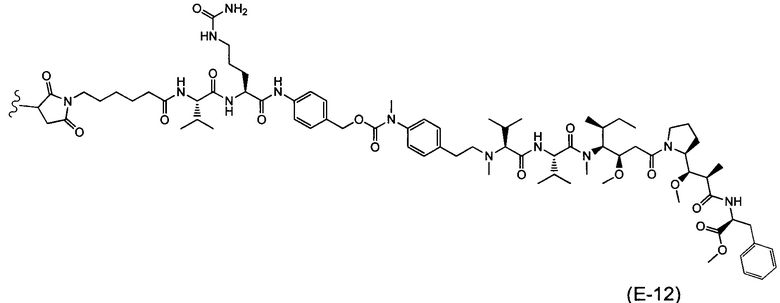
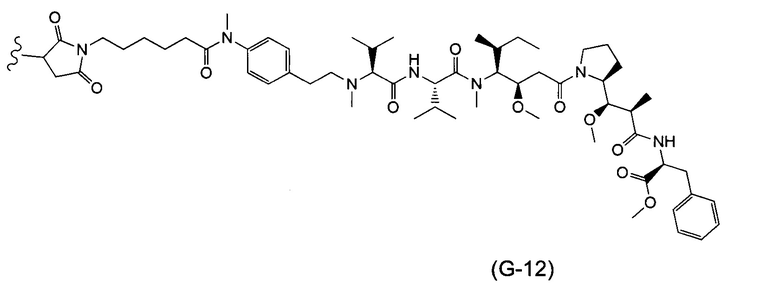
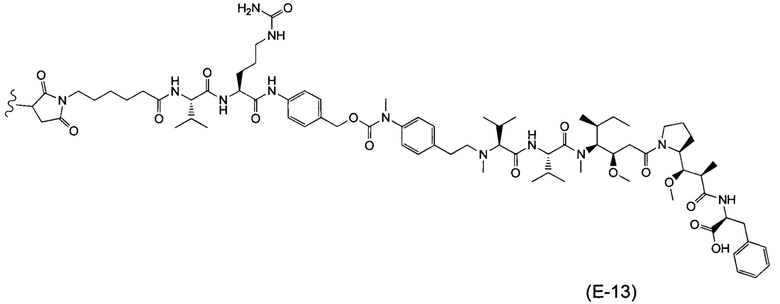
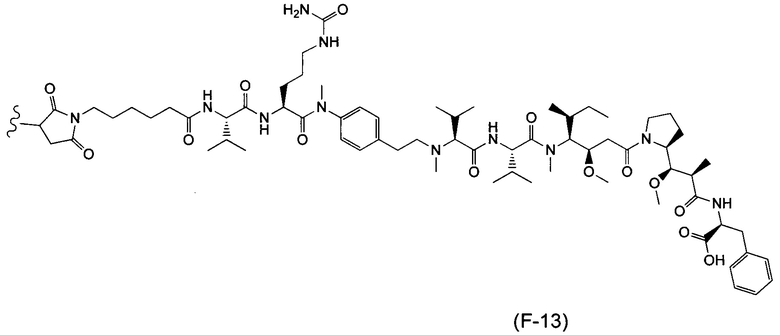
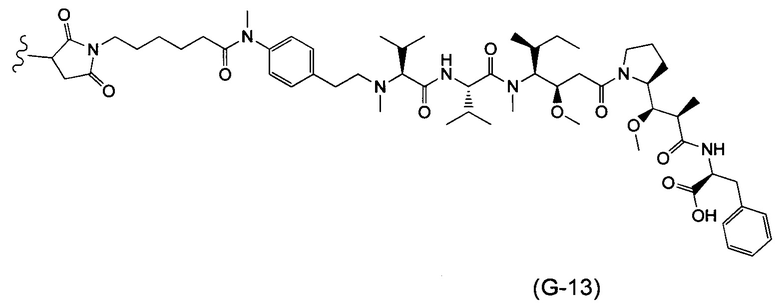
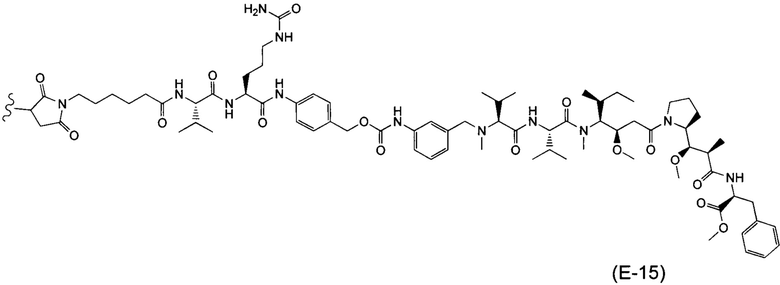
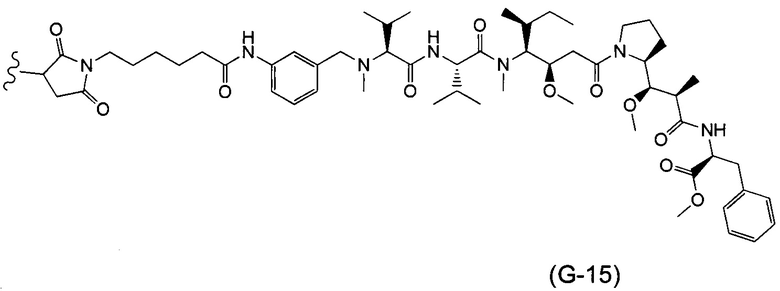
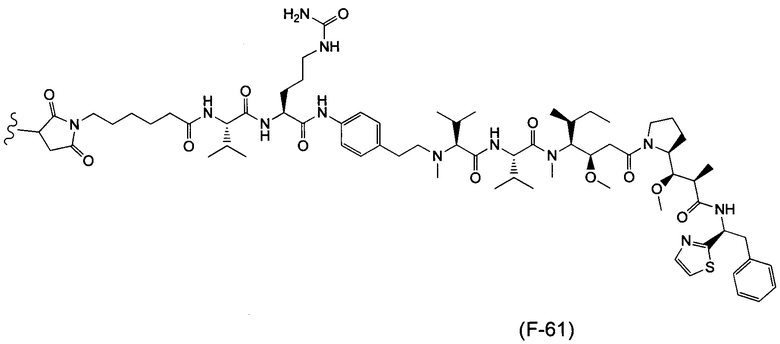
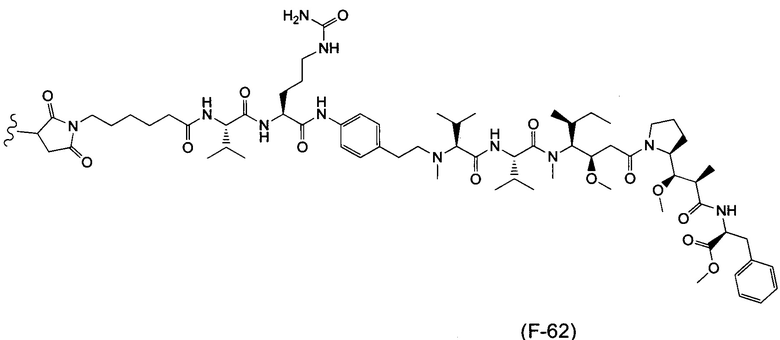
и
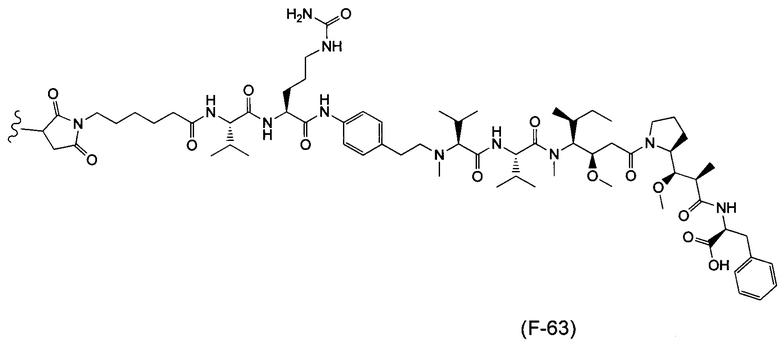
где волнистой линией указана точка присоединения к Аb.
Воплощение изобретения относится к ADC, имеющему формулу, выбранную из:
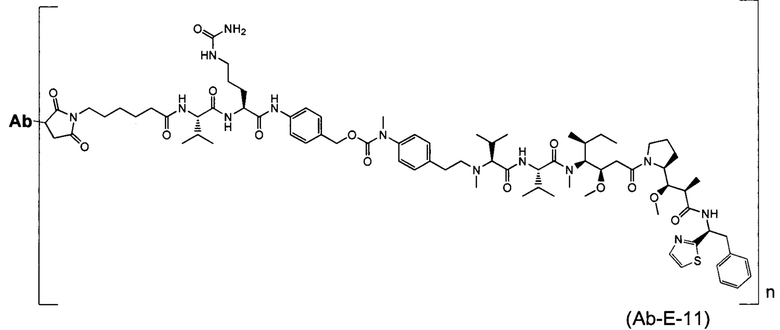
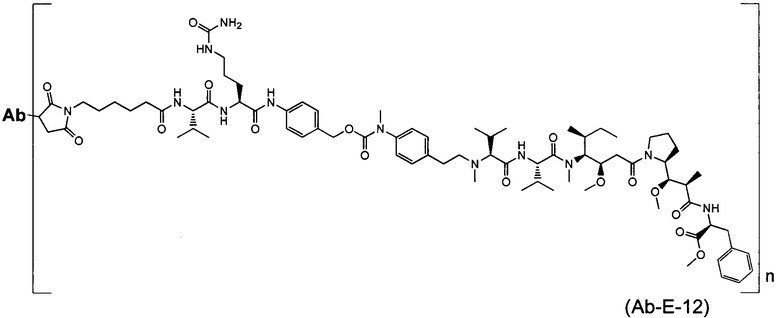
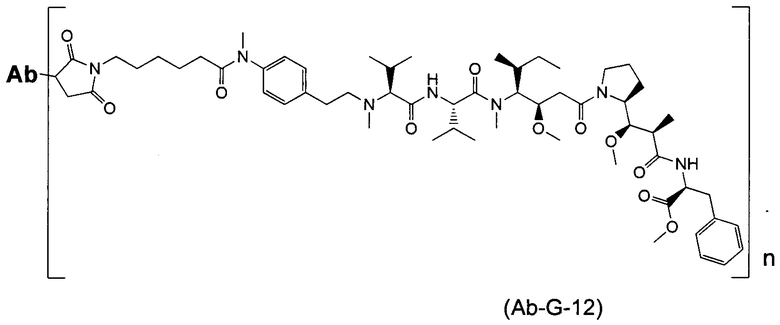
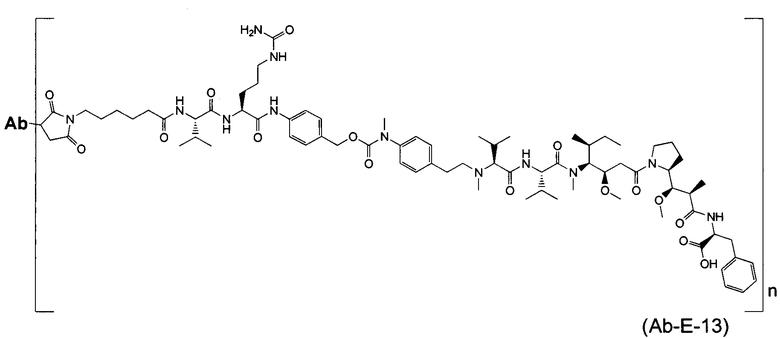
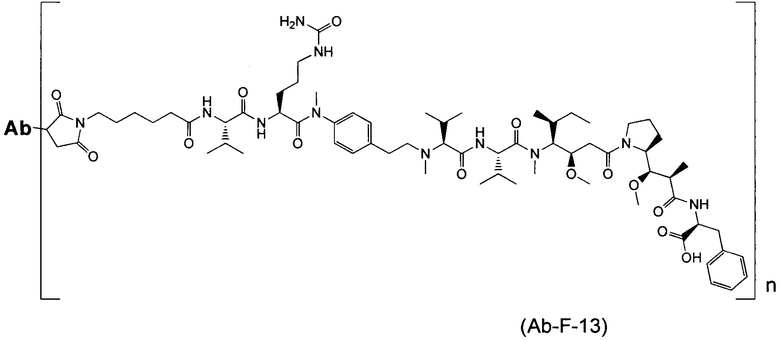
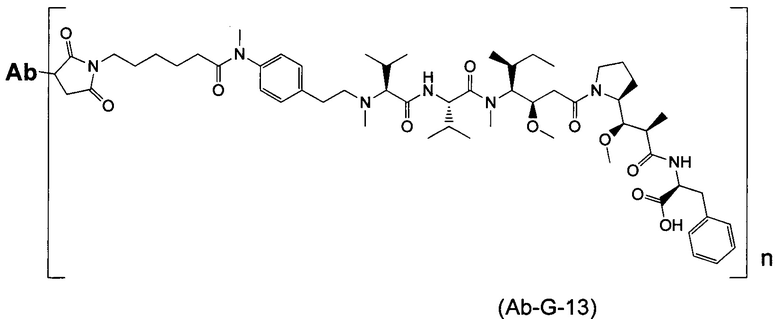
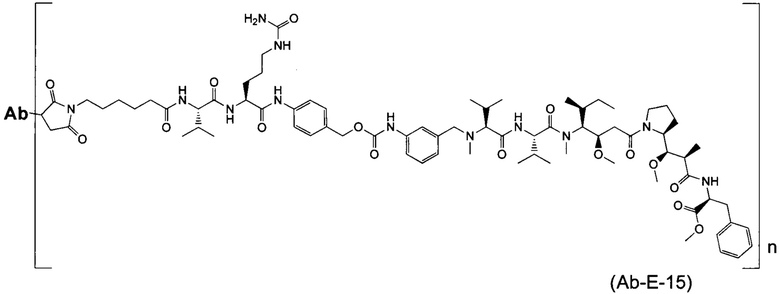
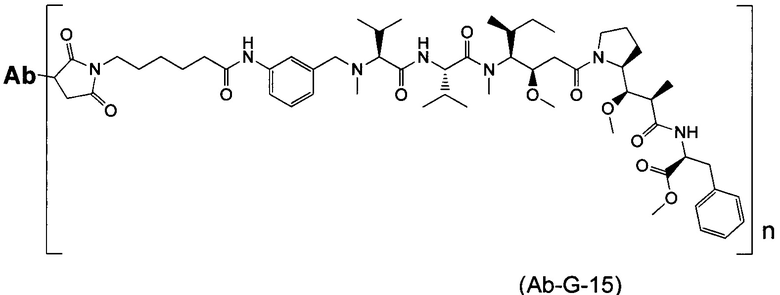
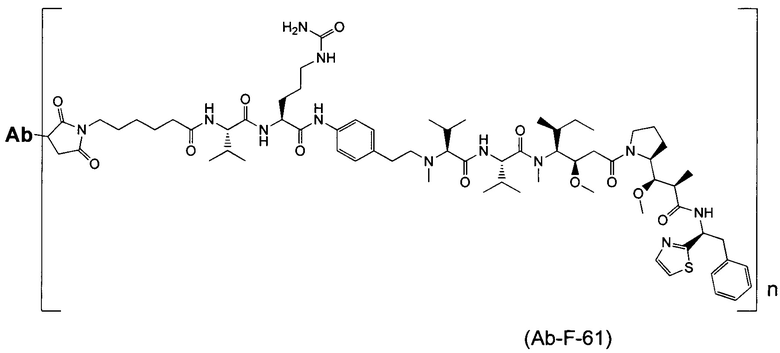
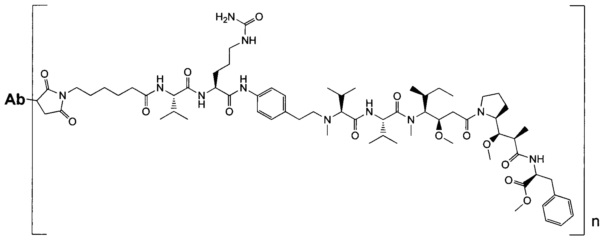
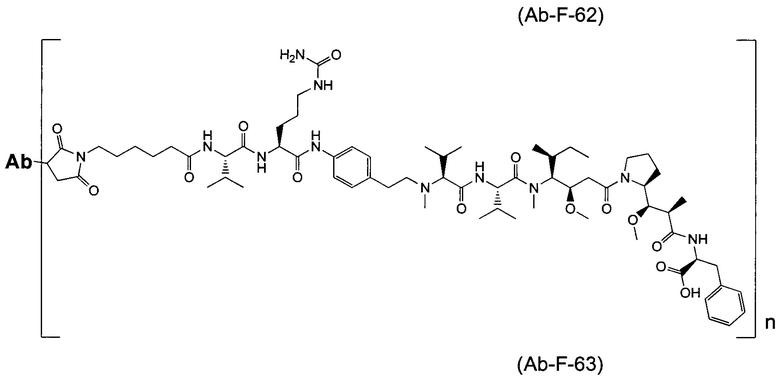
и их фармацевтически приемлемых солей,
где Ab выбрано из группы, состоящей из:
i) антител 208F2, 212А11, 214F8, 219D6 и 213В10;
ii) антител, конкурирующих за связывание с IGF-1R с антителами i); и
iii) антител, связывающихся с тем же эпитопом IGF-1R, что и антитела i).
Воплощение изобретения относится к ADC, где n равно 2.
Воплощение изобретения относится к ADC, где n равно 4.
Воплощение изобретения относится к ADC для применения в качестве лекарственного средства.
Воплощение изобретения относится к композиции, содержащей ADC, как описано выше.
Воплощение изобретения относится к композиции, дополнительно содержащей фармацевтически приемлемый носитель.
Воплощение изобретения относится к композиции для применения при лечении IGF-1R-экспрессирующего рака или форм рака, обусловленных IGF-1R.
IGF-1R-экспрессирующий рак или формы рака, обусловленные IGF-1R, включают опухолевые клетки, экспрессирующие или обладающие гиперэкспрессией полноразмерного IGF-1R или его участка на их поверхности.
Воплощение изобретения относится к композиции, где IGF-1R-экспрессирующий рак представляет собой рак, выбранный из карциномы молочной железы, ободочной кишки, пищевода, печеночно-клеточного рака, рака желудка, глиомы, рака легкого, меланомы, остеосаркомы, рака яичника, предстательной железы, рабдомиосаркомы, рака почки, щитовидной железы, эндометрия матки, мезотелиомы, плоскоклеточной карциномы полости рта и любого лекарственно-устойчивого рака.
Воплощение изобретения относится к способу лечения IGF-1R-экспрессирующего рака у субъекта, нуждающегося в этом, включающему введение субъекту эффективного количества по меньшей мере одного конъюгата антитела с лекарственным средством или композиции согласно изобретению.
Воплощение изобретения относится к набору, содержащему по меньшей мере i) конъюгат антитела с лекарственным средством и/или композицию, как описано выше, и ii) шприц, флакон или ампулу, в которые помещают конъюгат антитела с лекарственным средством и/или композицию.
Подробное описание изобретения
I - Антитело (Ab)
Термины «антитело», «антитела», «ab», «Ab», «MAb» или «иммуноглобулин» используются взаимозаменяемо в самом широком смысле и включают моноклональные антитела, изолированные, сконструированные методами генной инженерии или рекомбинантные антитела (например, полноразмерные или интактные моноклональные антитела), поликлональные антитела, поливалентные антитела или мультиспецифические антитела (например, биспецифические антитела), а также их фрагмент антитела при условии проявления ими желаемой биологической активности.
В одном воплощении изобретения антитело ADC по изобретению состоит из рекомбинантного антитела. Термин «рекомбинантное антитело» относится к антителу, полученному в результате экспрессии рекомбинантной ДНК внутри живых клеток. Рекомбинантное антитело ADC по изобретению получают путем использования лабораторных методик генетической рекомбинации, хорошо известных специалистам в данной области техники, позволяющих создать последовательности ДНК, которые не обнаруживаются в живых организмах.
В другом воплощении изобретения антитело ADC по изобретению состоит из химически синтезированного антитела.
Более конкретно такая молекула состоит из гликопротеина, содержащего по меньшей мере две тяжелые (Н) цепи и две легкие (L) цепи, соединенные друг с другом дисульфидными связями. Каждая тяжелая цепь содержит вариабельную область (или домен) (в данном документе сокращенную как HCVR или VH) и константную область тяжелой цепи. Константная область тяжелой цепи содержит три домена, СН1, СН2 и СН3. Каждая легкая цепь содержит вариабельную область (в данном документе сокращенную как LCVR или VL) и константную область легкой цепи. Константная область легкой цепи содержит один домен, CL. Области VH и VL могут быть дополнительно подразделены на области гипервариабельности, называемые участками, определяющими комплементарность (complementarity determining regions, CDR), расположенные между более консервативными областями, называемые каркасными областями (framework regions, FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат связывающий домен, взаимодействующий с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями или факторами хозяина, включая различные клетки иммунной системы (например, эффекторные клетки) и первый компонент (CIq) классической системы комплемента.
Под «антигенсвязывающим фрагментом» или «IGF-IR-связывающим фрагментом» антитела ADC согласно изобретению подразумевают указание на любой пептид, полипептид или белок, сохраняющий способность к связыванию с мишенью (также в целом называемой антигеном) антитела.
В воплощении изобретения такие «антигенсвязывающие фрагменты» выбраны из группы, состоящей из фрагментов или диател Fv, scFv (sc для одной цепи), Fab, F(ab')2, Fab', scFv-Fc или любого фрагмента, период полувыведения которого увеличен в результате химической модификации, такой как присоединение поли(алкилен)гликоля, такого как поли(этилен)гликоль («ПЭГилирование») (пэгилированные фрагменты, называемые Fv-PEG, scFv-PEG, Fab-PEG, F(ab')2-PEG или Fab'-PEG) («PEG» обозначает поли(этилен)гликоль), или в результате включения в липосому, при этом данные фрагменты имеют по меньшей мере один из участков CDR, характерных для антитела согласно изобретению. Предпочтительные «антигенсвязывающие фрагменты» состоят из частичной последовательности тяжелой или легкой вариабельной цепи антитела, из которого они выделены, или содержат эту последовательность, где данная частичная последовательность достаточна для сохранения такой же специфичности связывания, как у антитела, из которого она происходит, и достаточного сродства по отношению к мишени, предпочтительно по меньшей мере равного 1/100, более предпочтительно по меньшей мере 1/10, от сродства антитела, от которого она происходит. Более предпочтительно «антигенсвязывающие фрагменты» состоят по меньшей мере из трех CDR CDR-H1, CDR-H2 и CDR-H3 тяжелой вариабельной цепи и трех CDR CDR-L1, CDR-L2 и CDR-L3 легкой вариабельной цепи антитела, от которого они выделены, или содержат эти участки.
Термины «связывающий», «связывает» и тому подобное подразумевают, что антитело или любой его антигенсвязывающий фрагмент образует комплекс с антигеном, который является относительно стабильным в физиологических условиях. Специфичное связывание может характеризоваться равновесной константой диссоциации по меньшей мере приблизительно 1×10-6 М. Способы определения, связываются ли две молекулы, хорошо известны в данной области техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс, количественные анализы с радиоактивной меткой и тому подобное. Во избежание сомнений это не означает, что антитело не может связываться или взаимодействовать на низком уровне с другим антигеном. Тем не менее, в качестве воплощения, данное антитело связывается только с данным антигеном.
Как используют в настоящем описании, выражение «антитело к IGF-1R» следует интерпретировать как аналогичное выражению «антитело против IGF-1R» и означает антитело, способное к связыванию с IGF-1R.
В одном воплощении настоящего изобретения эпитоп антитела предпочтительно локализован во внеклеточном домене IGF-1R человека (также называемом IGF-1R ECD).
В конкретном воплощении изобретения антитело или любой его антигенсвязывающий фрагмент способен к связыванию с IGF-1R с ЕС50, составляющей от 10×10-10 до 1×10-10, и более предпочтительно от 8×10-10 до 2×10-10.
Термин «половинная максимальная эффективная концентрация» (ЕС50) соответствует концентрации лекарственного средства, антитела или токсического агента, которая индуцирует ответ, находящийся между исходным уровнем и максимумом, после некоторого заданного времени воздействия. Ее часто используют в качестве меры активности лекарственного средства. ЕС50 возрастающей кривой доза-ответ, таким образом, представляет собой концентрацию соединения, где наблюдается 50% его максимального эффекта. ЕС50 квантовой кривой доза-ответ представляет собой концентрацию соединения, где 50% популяции проявляет ответ после заданной продолжительности воздействия. Меры концентрации в характерном случае следуют сигмоидальной кривой с быстрым возрастанием на протяжении относительно незначительного изменения концентрации. Это может быть определено математически путем построения наилучшей эмпирической кривой.
В качестве предпочтительного воплощения ЕС50, определенная в настоящем изобретении, характеризует активность связывания антитела с IGF-1R ECD, выявленным на поверхности опухолевых клеток человека. Параметр ЕС50 определяют с использованием анализа сортировки флуоресцентно-активированных клеток (FACS). Параметр ЕС50 отражает концентрацию антитела, для которой получают 50% максимального связывания на IGF-1R человека, экспрессируемого на опухолевых клетках человека. Каждое значение ЕС50 было вычислено как средняя точка кривой доза-ответ с использованием программы соответствия четырехпараметрической кривой регрессии (программное обеспечение Prism). Этот параметр выбран в качестве репрезентативного параметра физиологических/патологических условий.
Термин «эпитоп» представляет собой участок антигена, связываемый антителом. Эпитопы могут быть определены как структурные или функциональные. Функциональные эпитопы в целом представляют собой подгруппу структурных эпитопов и имеют остатки, которые вносят непосредственный вклад в сродство взаимодействия. Эпитопы могут быть также конформационными, то есть состоящими из нелинейных аминокислот.В некоторых воплощениях изобретения эпитопы могут включать детерминанты, представляющие собой химически активные поверхностные группировки молекул, таких как аминокислоты, боковые цепи Сахаров, фосфорильные группы или сульфонильные группы, и в некоторых воплощениях могут обладать специфичными трехмерными структурными характеристиками и/или специфичными характеристиками заряда.
Конкуренция за связывание с IGF-1R может быть определена любыми способами или методами, известными специалистам в данной области техники, такими как, без ограничений, радиоактивность, Biacore, твердофазный иммуноферментный анализ (ИФА), проточная цитометрия и т.д. Под «конкурирующим за связывание с IGF-1R» подразумевают конкуренцию по меньшей мере 20%, предпочтительно по меньшей мере 50% и более предпочтительно по меньшей мере 70%.
Определение связывания с тем же эпитопом можно осуществлять любыми способами или методами, известными специалистам в данной области техники, такими как, без ограничений, радиоактивность, Biacore, ИФА, проточная цитометрия и т.д. Под «связывающимся с тем же эпитопом IGF-1R» подразумевают конкуренцию по меньшей мере 20%, предпочтительно по меньшей мере 50% и более предпочтительно по меньшей мере 70%.
Как упомянуто выше, и в противоположность общим сведениям, настоящее изобретение сосредоточено на специфичных антителах к IGF-1R, представляющих высокую способность к интернализации после связывания IGF-1R. При использовании в настоящем документе антитело, которое «интернализуется» или «интернализовано» (эти два выражения аналогичны), представляет собой антитело, которое поглощается клеткой (что означает «входит в клетку») после связывания с IGF-1R на клетке млекопитающего. Такое антитело представляет интерес в составе ADC, поскольку оно адресует или направляет связанный с ним цитотоксический агент в раковые клетки-мишени. Сразу после интернализации цитотоксический агент инициирует гибель раковой клетки.
Неожиданно обнаружено, что все антитела согласно изобретению презентируют одни и те же последовательности CDR-H2, CDR-H3 и CDR-L2, а другие 3 CDR различаются. Это наблюдение, по-видимому, является органичным, поскольку составляет часть общедоступных сведений о том, что в отношении связывающей специфичности антитела CDR-H3 описан как важнейший участок, в максимальной степени задействованный в распознавании эпитопа.
Важными аспектами успеха терапии ADC считают специфичность к антигену-мишени и интернализацию комплексов антиген-антитело в раковые клетки. Очевидно, что не интернализующие антигены менее эффективны для доставки цитотоксических агентов, чем интернализующие антигены. Процессы интернализации различаются среди антигенов и зависят от множества параметров, на которые могут влиять антитела.
В ADC цитотоксический агент придает цитотоксическую активность, а используемое антитело ответственно за специфичность к раковым клеткам, а также используется в качестве вектора внутри клеток для верного направления цитотоксичности. Таким образом, для усовершенствования ADC антитело может проявлять высокую способность к интернализации в раковые клетки-мишени. Эффективность опосредованной антителом интернализации значительно различается в зависимости от эпитопа-мишени. Для выбора антител, активно интернализующих IGF-1R, необходимы различные экспериментальные данные исследований не только понижающей регуляции IGF-1R, но также последующей интернализации антитела к IGF-1R в клетки.
В одном воплощении изобретения интернализацию антитела ADC согласно изобретению можно оценивать с помощью иммунофлуоресценции или FACS (проточной цитометрии) (примеры которой приведены далее в настоящем документе), либо любого способа или процесса, известного специалистам в данной области техники, специфичного для механизма интернализации. В предпочтительном воплощении изобретения антитело ADC согласно изобретению может индуцировать интернализацию после связывания с IGF-1R по меньшей мере на 30%, предпочтительно 50% и более предпочтительно 80%.
Комплекс IGF-IR/антитело интернализуется после связывания антитела с ECD IGF-1R и индуцирует уменьшение количества IGF-1R на поверхности клеток. Это уменьшение можно определить количественно любым способом, известным специалистам в данной области техники, неограничивающими примерами которых являются вестерн-блоттинг, FACS и иммунофлуоресценция.
В одном воплощении изобретения это уменьшение, отражающее, таким образом, интернализацию, можно предпочтительно измерять с помощью FACS и выражать в виде разности или дельта между средней интенсивностью флуоресценции (Mean Fluorescence Intensity, MFI), измеренной при 4°С, и MFI, измеренной при 37°С, после 4 часов инкубации с антителом.
В качестве неограничивающего примера эту разность дельта определяют на основании MFI, полученных на необработанных клетках и на клетках, обработанных антителом, используя i) клетки MCF7 рака молочной железы после 4-часового периода инкубации с антителом, описанный в настоящем документе, и ii) второе антитело, меченое красителем Alexa488. Данный параметр определяют путем вычисления по следующей формуле: Δ(MFI4°C-MFI37°C).
Эта разность между MFI отражает понижающую регуляцию IGF-1R, поскольку MFI пропорциональны IGF-1R, экспрессируемым на клеточной поверхности.
В преимущественном аспекте антитела состоят из антител, инициирующих Д (MFI4°C-MFI37°C) на MCF-7 по меньшей мере 280, предпочтительно по меньшей мере 400.
Более подробно, вышеупомянутую разность дельта можно измерять следующим способом, который должен рассматриваться как иллюстративный и неограничивающий пример:
a) Обработка и инкубация представляющих интерес опухолевых клеток с антителом по изобретению либо в холодной (4°С), либо в теплой (37°С) полной культуральной среде;
b) Обработка обработанных клеток стадии а) и параллельно - необработанных клеток вторым антителом;
c) Измерение MFI (репрезентативной для количества IGF-1R, присутствующего на поверхности) для обработанных и необработанных клеток с помощью второго меченого антитела, способного к связыванию с антителом по изобретению; и
d) Вычисление разности дельта путем вычитания MFI, полученной с обработанными клетками, из MFI, полученной с необработанными клетками.
На основании данного значения дельта MFI можно определить процент интернализации как:
100×(MFI4°C-MFI37°C)/MFI4°C.
Процент интернализации антител ADC согласно изобретению, присутствующих предпочтительно на клетках MCF7, составлял от 50% до 99%, от 70% до 90%, предпочтительно от 75% до 87%.
Особое преимущество антител, описанных в настоящем документе, основано на скорости их интернализации.
Общеизвестно, что для ADC желательно, чтобы используемые антитела проявляли высокую скорость интернализации, предпочтительно в пределах 24 часов от введения антитела и более предпочтительно в пределах 12 часов, и даже более предпочтительно в пределах 6 часов.
В настоящем изобретении скорость интернализации, также называемой уменьшением количества антител, связанных с клеточной поверхностью, или распадом антител клеточной поверхности, выражена в виде t1/2 (время полужизни) и соответствует времени, необходимому для получения уменьшения MFI на 50% Δ(данный аспект будет четко понятен при рассмотрении приведенных ниже примеров).
Особое преимущество состоит в том, что антитела ADC по изобретению имеют t1/2, составляющее от 5 до 25 минут и предпочтительно от 10 и 20 минут.
Конкретное воплощение изобретения относится к ADC, где антитело Ab содержит три участка CDR тяжелой цепи, из которых CDR-H2 имеет последовательность SEQ ID NO 2 и CDR-H3 - последовательность SEQ ID NO 3, и три участка CDR легкой цепи, из которых CDR-L2 имеет последовательность SEQ ID NO 5.
Конкретное воплощение изобретения относится к ADC, где антитело Ab содержит три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 1, 2 и 3, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 4, 5 и 6.
Одно воплощение ADC включает антитело, содержащее три CDR тяжелой цепи, содержащих последовательности SEQ ID NO 1, 2 и 3 или любую последовательность, проявляющую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 1, 2 или 3, или состоящих из этих последовательностей; и три CDR легкой цепи, содержащих последовательности SEQ ID NO 4, 5 и 6 или любую последовательность, проявляющую по меньшей мере 80%), предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 4, 5 или 6, или состоящих из этих последовательностей.
В другом воплощении изобретения антитело или любой его антигенсвязывающий фрагмент содержит три CDR тяжелой цепи, содержащих последовательности SEQ ID NO 1, 2 и 3, и три CDR легкой цепи, содержащих последовательности SEQ ID NO 4, 5 и 6.
Для сравнения вариабельных доменов независимо от рецептора антигена, типа цепи или вида определена уникальная нумерация IMGT [Lefranc М.-Р., Immunology Today 18, 509 (1997) / Lefranc M.-P., The Immunologist, 7, 132-136 (1999)/ Lefranc, M.-P.,  , C., Ruiz, M., Giudicelli, V., Foulquier, E., Truong, L., Thouvenin-Contet, V. and Lefranc, Dev. Comp. Immunol., 27, 55-77 (2003)]. В этой уникальной нумерации IMGT консервативные аминокислоты всегда имеют одно и то же положение, например цистеин 23 (1st-CYS), триптофан 41 (CONSERVED-TRP), гидрофобная аминокислота 89, цистеин 104 (2nd-CYS), фенилаланин или триптофан 118 (J-PHE или J-TRP). Уникальная нумерация IMGT обеспечивает стандартизованное разграничение каркасных областей (FR1-IMGT: положения с 1 по 26, FR2-IMGT: с 39 по 55, FR3-IMGT: с 66 по 104 и FR4-IMGT: с 118 по 128) и участков, определяющих комплементарность: CDR1-IMGT: с 27 по 38, CDR2-IMGT: с 56 по 65 и CDR3-IMGT: с 105 по 117. Поскольку гэпы представляют собой незанятые положения, длины CDR-IMGT (показанные между скобками и разделенные точками, например, [8.8.13]) становятся критической информацией. Уникальную нумерацию IMGT используют в графических изображениях 2D, обозначенных как IMGT Colliers de Perles [Ruiz, M. and Lefranc, M.-P., Immunogenetics, 53, 857-883 (2002) / Kaas, Q. and Lefranc, M.-P., Current Bioinformatics, 2, 21-30 (2007)], и в структурах 3D в IMGT/3Dstructure-DB [Kaas, Q., Ruiz, M. and Lefranc, M.-P., T cell receptor and MHC structural data. Nucl. Acids. Res., 32, D208-D210 (2004)].
, C., Ruiz, M., Giudicelli, V., Foulquier, E., Truong, L., Thouvenin-Contet, V. and Lefranc, Dev. Comp. Immunol., 27, 55-77 (2003)]. В этой уникальной нумерации IMGT консервативные аминокислоты всегда имеют одно и то же положение, например цистеин 23 (1st-CYS), триптофан 41 (CONSERVED-TRP), гидрофобная аминокислота 89, цистеин 104 (2nd-CYS), фенилаланин или триптофан 118 (J-PHE или J-TRP). Уникальная нумерация IMGT обеспечивает стандартизованное разграничение каркасных областей (FR1-IMGT: положения с 1 по 26, FR2-IMGT: с 39 по 55, FR3-IMGT: с 66 по 104 и FR4-IMGT: с 118 по 128) и участков, определяющих комплементарность: CDR1-IMGT: с 27 по 38, CDR2-IMGT: с 56 по 65 и CDR3-IMGT: с 105 по 117. Поскольку гэпы представляют собой незанятые положения, длины CDR-IMGT (показанные между скобками и разделенные точками, например, [8.8.13]) становятся критической информацией. Уникальную нумерацию IMGT используют в графических изображениях 2D, обозначенных как IMGT Colliers de Perles [Ruiz, M. and Lefranc, M.-P., Immunogenetics, 53, 857-883 (2002) / Kaas, Q. and Lefranc, M.-P., Current Bioinformatics, 2, 21-30 (2007)], и в структурах 3D в IMGT/3Dstructure-DB [Kaas, Q., Ruiz, M. and Lefranc, M.-P., T cell receptor and MHC structural data. Nucl. Acids. Res., 32, D208-D210 (2004)].
Должно быть понятно, что в отсутствие противоречащих указаний в настоящем описании участки, определяющие комплементарность, или CDR, означают гипервариабельные участки тяжелых и легких цепей иммуноглобулинов, как определено в соответствии с системой нумерации IMGT.
Тем не менее, CDR также можно определить в соответствии с системой нумерации Кэбота (Kabat et al., Sequences of proteins of immunological interest, 5th Ed., U.S. Department of Health and Human Services, NIH, 1991, и более поздние издания). Существует три участка CDR тяжелой цепи и три участка CDR легкой цепи. В настоящем документе термины «участок CDR» и «участки CDR» используют для указания в зависимости от случая одного, нескольких или даже всех участков, содержащих большинство аминокислотных остатков, ответственных за связывающее сродство антитела к распознаваемому им антигену или эпитопу. В целях упрощения чтения настоящего документа CDR по Кэботу не определены. Тем не менее, специалисту в данной области техники должно быть очевидно использование определения CDR в соответствии с IMGT для определения CDR по Кэботу.
В смысле, относящемся к настоящему изобретению, «идентичность» или «процент идентичности» между двумя последовательностями нуклеиновых кислот или аминокислот означает процент идентичных нуклеотидов или аминокислотных остатков между двумя сравниваемыми последовательностями, полученный после оптимального выравнивания, при этом данный процент является исключительно статистическим, и различия между двумя последовательностями распределены случайным образом по всей их длине. Сравнение двух нуклеиново-кислотных или аминокислотных последовательностей традиционно проводят путем сравнения последовательностей после того, как они оптимально выровнены, при этом сравнение можно проводить посегментно или с использованием «окна выравнивания». Оптимальное выравнивание последовательностей для сравнения можно проводить, кроме сравнения вручную, с помощью алгоритма локальной гомологии Smith and Waterman (1981) [Ad. App. Math. 2:482], с помощью алгоритма локальной гомологии Neddleman and Wunsch (1970) [J. Mol. Biol. 48:443], методом поиска подобия Pearson and Lipman (1988) [Proc. Natl. Acad. Sci. USA 85:2444] или с помощью компьютерных программ, использующих эти алгоритмы (GAP, BESTFIT, FASTA и TFASTA в пакете программ Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, Wl, либо с помощью программ сравнения BLAST NR или BLAST Р).
Процент идентичности вычисляют путем определения числа положений, в которых аминокислота, нуклеотид или остаток идентичны между двумя последовательностями, предпочтительно между двумя полноразмерными последовательностями, деления числа идентичных положений на общее число положений в окне выравнивания и умножения результата на 100 в получением процента идентичности между двумя последовательностями.
Например, можно использовать программу BLAST "BLAST 2 sequences" (Tatusova et al., "Blast 2 sequences - a new tool for comparing protein and nucleotide sequences", FEMS Microbiol., 1999, Lett. 174:247-250), доступную на сайте http://www.ncbi.nlm.nih.gov/gorf/bl2.html, с параметрами по умолчанию (конкретно для параметров: «штраф на открытие гэпа»: 5 и «штраф на удлинение гэпа»: 2; выбранная матрица представляет собой, например, матрицу «BLOSUM 62», предложенную программой); процент идентичности между двумя сравниваемыми последовательностями вычисляется непосредственно программой.
Для аминокислотной последовательности, проявляющей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность эталонной аминокислотной последовательности, предпочтительные примеры включают содержащие эталонную последовательность, некоторые модификации, в частности, делецию, добавление или замену по меньшей мере одной аминокислоты, укорочение или удлинение. В случае замены одной или более последовательных или непоследовательных аминокислот предпочтительны замены, в которых заменяемые аминокислоты замещаются «эквивалентными» аминокислотами. В данном случае под выражением «эквивалентные аминокислоты» подразумевают указание на любые аминокислоты, с большей вероятностью способные к замене одной из структурных аминокислот, при этом, тем не менее, не модифицируя биологические активности соответствующих антител, и конкретные примеры таких аминокислот приведены ниже.
Эквивалентные аминокислоты можно определить либо по их структурной гомологии с аминокислотами, которые они заменяют, либо по результатам сравнительных тестов на биологическую активность между различными антителами, которые, вероятно, будут созданы.
В качестве неограничивающего примера, в таблице 1 ниже приведена сводная информация о возможных заменах, которые, вероятно, будут произведены, не приводя в результате к значительной модификации биологической активности соответствующего модифицированного антитела; обратные замены возможны естественным путем в тех же условиях.
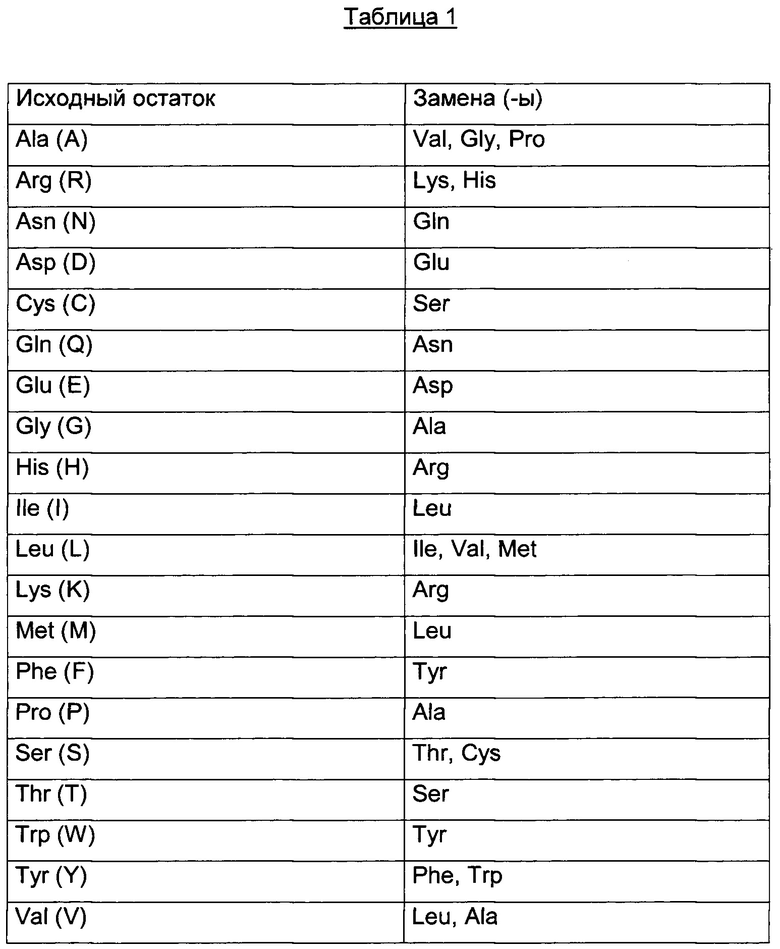
Конкретный аспект изобретения состоит в том, что антитело ADC не связывается с рецептором инсулина (IR). Данный аспект представляет интерес, поскольку антитело, описанное в настоящем документе, не окажет никакого отрицательного влияния на IR, т.е. на метаболизм инсулина.
В другом воплощении изобретения еще один другой предпочтительный аспект антитела ADC по изобретению состоит в том, что оно способно связываться не только с IGF-1R человека, но также с IGF-1R обезьяны и более конкретно с IGF-1R яванского макака. Данный аспект также представляет интерес, поскольку способствует оценке токсичности, требующейся для клинических исследований.
Еще в одном другом воплощении изобретения антитело ADC по изобретению состоит из моноклонального антитела.
Термин «моноклональное антитело» или «Mab», как используют в настоящем документе, относится к антителу, полученному из популяции по существу однородных антител, т.е. отдельные антитела популяции идентичны за исключением возможных встречающихся в природе мутаций, которые могут присутствовать в незначительных количествах. Моноклональные антитела обладают высокой специфичностью, поскольку направлены на единственный эпитоп.Такое моноклональное антитело может продуцироваться единственным клоном В клеток или гибридомы. Моноклональные антитела могут быть также рекомбинантными, т.е. могут быть получены методами белковой инженерии или химического синтеза. Моноклональные антитела могут быть также выделены из фаговых библиотек антител. Кроме того, в противоположность препаратам поликлональных антител, которые в характерном случае включают различные антитела, направленные против различных детерминант или эпитопов, каждое моноклональное антитело направлено против единственного эпитопа антигена.
Моноклональное антитело в настоящем документе включает мышиное, химерное и гуманизированное антитело, такое как описано ниже.
Антитело предпочтительно выделяют из гибридомы мышиного происхождения, зарегистрированной во французской коллекции культур микроорганизмов (CNCM, Институт Пастера, 25 рю дю Доктор Руа, 75724 Париж, Седекс 15, Франция), где гибридому получают путем слияния спленоцитов/лимфоцитов иммунизированных мышей Balb/C и клеток линии клеток миеломы Sp 2/O-Ag 14.
В одном воплощении изобретения антитело к IGF-1R ADC по изобретению состоит из мышиного антитела, далее обозначенного как m[название антитела].
В одном воплощении изобретения антитело к IGF-1R состоит из химерного антитела, далее обозначенного как с[название антитела].
В одном воплощении изобретения антитело к IGF-1R состоит из гуманизрованного антитела, далее обозначенного как hz[название антитела].
Во избежание сомнений в дальнейшем описании выражения «антитело к IGF-1R» и «[название антитела]» аналогичны и включают (в отсутствие противоположного описания) мышиный, химерный и гуманизированный варианты антитела к IGF-1R или «[название антитела]». При необходимости используют префикс m-(мышиное), с-(химерное) или hz-(гуманизированное).
Для большей ясности в следующей таблице 2 проиллюстрированы последовательности CDR, определенные в соответствии с IMGT, для предпочтительных антител.
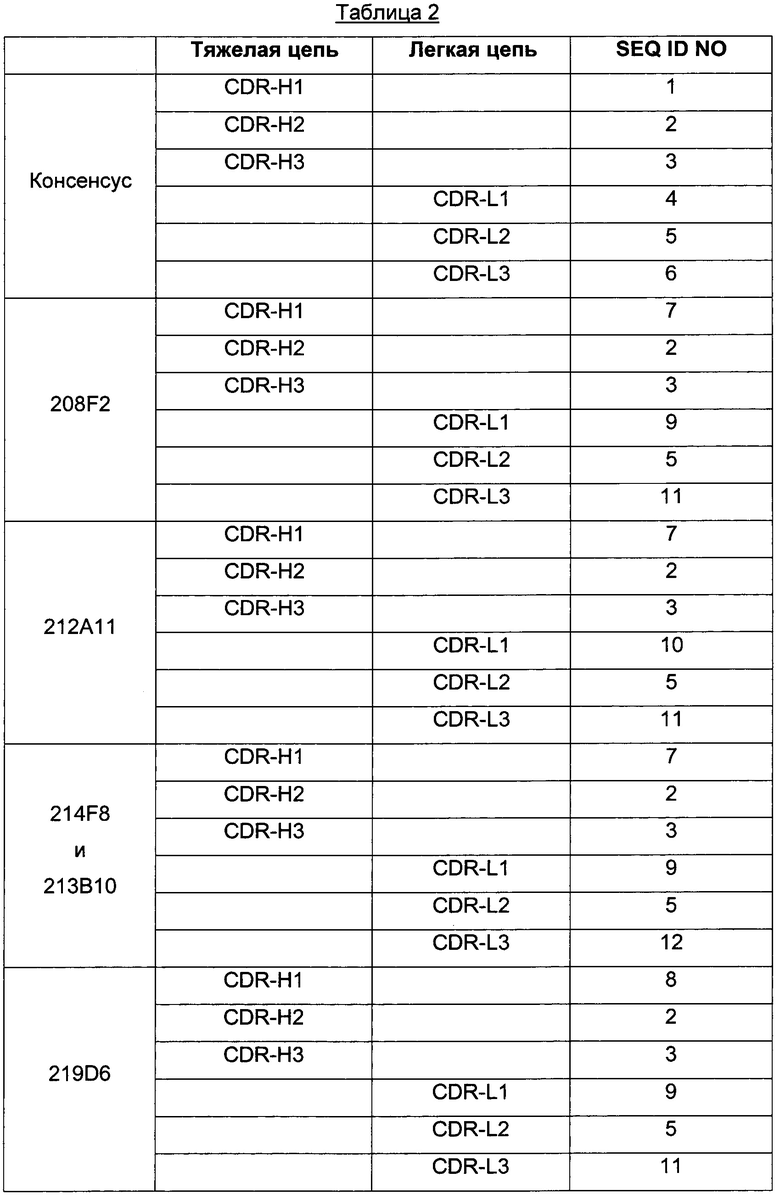
Специалисту в данной области техники очевидно, что любую комбинацию 6 CDR, как описано выше, следует рассматривать в составе настоящего изобретения.
Как можно наблюдать из данной таблицы 2, все антитела, описанные в настоящем документе, имеют одинаковые последовательности CDR-H2, CDR-H3 и CDR-L2, и это свойство представляет особый интерес, как описано выше.
Конкретный аспект относится к ADC, где антитело представляет собой мышиное антитело, отличающееся тем, что антитело также содержит константные области легкой цепи и тяжелой цепи, выделенные из антитела вида, гетерологичного для мыши, в частности, человека.
Другой конкретный аспект относится к ADC, где антитело представляет собой химерное (с) антитело, отличающееся тем, что антитело также содержит константные области легкой цепи и тяжелой цепи, выделенные из антитела вида, гетерологичного для мыши, в частности, человека.
Химерное антитело представляет собой антитело, содержащее природную вариабельную область (легкой цепи и тяжелой цепи), выделенную из антитела данного вида в комбинации с константными областями легкой цепи и тяжелой цепи антитела вида, гетерологичного для данного вида.
Химерные антитела могут быть получены путем использования методов рекомбинантной генетики. Например, химерное антитело может быть получено путем клонирования рекомбинантной ДНК, содержащей промотор и последовательность, кодирующую вариабельную область моноклонального антитела, отличающегося от человеческого, в частности, мышиного, и последовательность, кодирующую константную область антитела гетерологичного вида, предпочтительно человека. Химерное антитело ADC согласно изобретению, кодируемое одним таким рекомбинантным геном, может представлять собой, например, химерное антитело мыши-человека, при этом специфичность этого антитела определяется вариабельной областью, выделенной из мышиной ДНК, а его изотип определяется константной областью, выделенной из ДНК человека.
В предпочтительном, но не ограничивающем воплощении изобретения антитело ADC по изобретению выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 13 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 13, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 14 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 14, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 10, 5 и 11;
c) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 15 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 15, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12;
d) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 16 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 16, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11; и
e) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 17 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 17, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12.
Под «любой последовательностью, проявляющей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO с 13 по 17» подразумевают обозначение последовательностей, содержащих три CDR тяжелой цепи SEQ ID NO 1, 2 и 3, и, кроме того, проявляющих по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полноразмерной последовательностью SEQ ID NO с 13 по 17 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 1, 2 и 3).
В другом предпочтительном, но не ограничивающем воплощении изобретения антитело ADC по изобретению выбрано из:
a) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 18 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 18, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 19 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 19, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
c) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 20 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 20, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
d) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 21 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 21, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 8, 2 и 3; и
e) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 22 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 22, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3.
Под «любой последовательностью, проявляющей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO с 18 по 22» подразумевают обозначение последовательностей, содержащих три CDR тяжелой цепи SEQ ID NO 4, 5 и 6, и, кроме того, проявляющих по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полноразмерной последовательностью SEQ ID NO с 18 по 22 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 4, 5 и 6).
Воплощение изобретения относится к ADC, где Ab представляет собой антитело, выбранное из:
a) антитела, содержащего вариабельный домен тяжелой цепи последовательности SEQ ID NO 13 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 13, и вариабельный домен легкой цепи последовательности SEQ ID NO 18 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 18;
b) антитела, содержащего вариабельный домен тяжелой цепи последовательности SEQ ID NO 14 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 14, и вариабельный домен легкой цепи последовательности SEQ ID NO 19 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 19;
c) антитела, содержащего вариабельный домен тяжелой цепи последовательности SEQ ID NO 15 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 15, и вариабельный домен легкой цепи последовательности SEQ ID NO 20 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 20;
d) антитела, содержащего вариабельный домен тяжелой цепи последовательности SEQ ID NO 16 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 16, и вариабельный домен легкой цепи последовательности SEQ ID NO 21 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 21; и
е) антитела, содержащего вариабельный домен тяжелой цепи последовательности SEQ ID NO 17 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 17, и вариабельный домен легкой цепи последовательности SEQ ID NO 22 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 22.
Химерные антитела, описанные в настоящем документе, могут также отличаться константным доменом, и более конкретно химерные антитела могут быть выбраны или обозначены как, например, без ограничения IgG1, IgG2, IgG3, IgM, IgA, IgD или IgE. Более предпочтительно в контексте настоящего изобретения химерные антитела представляют собой IgG1 или IgG4.
Одно воплощение изобретения относится к ADC, где Ab представляет собой химерное антитело, содержащее вариабельные домены VH и VL, как описано выше, в формате IgG1. Более предпочтительно это химерное антитело содержит константный домен для VH последовательности SEQ ID NO 43 и домен каппа для VL последовательности SEQ ID NO 45.
Одно воплощение изобретения относится к ADC, где Ab представляет собой химерное антитело, содержащее вариабельные домены VH и VL, как описано выше, в формате IgG4. Более предпочтительно это химерное антитело содержит константный домен для VH последовательности SEQ ID NO 44 и домен каппа для VL последовательности SEQ ID NO 45.
В другом предпочтительном, но не ограничивающем воплощении изобретения антитело ADC по изобретению выбрано из:
a) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 23 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 23, и легкую цепь последовательности SEQ ID NO 28 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 28, или состоящего из этих цепей;
b) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 24 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 24, и легкую цепь последовательности SEQ ID NO 29 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 29, или состоящего из этих цепей;
c) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 25 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 25, и легкую цепь последовательности SEQ ID NO 30 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 30, или состоящего из этих цепей;
d) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 26 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 26, и легкую цепь последовательности SEQ ID NO 31 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 31, или состоящего из этих цепей; и
e) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 27 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 27, и легкую цепь последовательности SEQ ID NO 32 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 32, или состоящего из этих цепей.
Для большей ясности в следующей таблице 3 проиллюстрированы последовательности VH и VL соответственно для предпочтительных химерных антител.
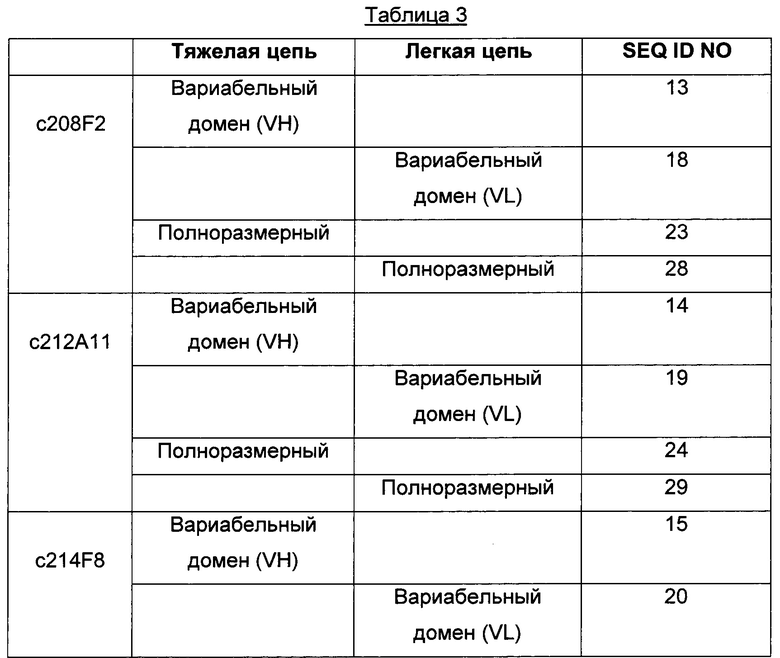
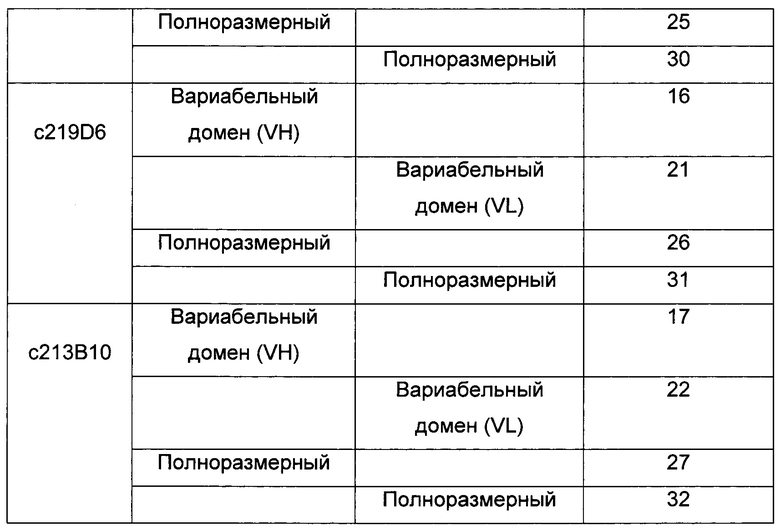
Еще один другой конкретный аспект настоящего изобретения относится к ADC, где «Ab» представляет собой гуманизированное антитело, отличающееся тем, что константные области легкой цепи и тяжелой цепи, выделенные из антитела человека, представляют собой соответственно лямбда- или каппа-область и область гамма-1, гамма-2 или гамма-4.
«Гуманизированные антитела» означают антитело, которое содержит участки CDR, выделенные из антитела происхождения, отличающегося от человеческого, где другие части молекулы антитела выделены из одного (или нескольких) антитела человека. Кроме того, некоторые из остатков каркасного сегмента (называемого FR) могут быть модифицированы для сохранения связывающего сродства.
Гуманизированные антитела или их фрагменты могут быть получены методами, известными специалистам в данной области техники. Такие гуманизированные антитела предпочтительны для применения в способах, включающих диагностику in vitro или превентивное и/или терапевтическое лечение in vivo. Другие методы гуманизации, также известные специалистам в данной области техники, такие как, например, метод «прививания CDR», описанный PDL в патентах ЕР 0 451 216, ЕР 0 682 040, ЕР 0 939 127, ЕР 0 566 647 или US 5,530,101, US 6,180,370, US 5,585,089 и US 5,693,761. Можно также процитировать патенты US 5,639,641 или 6,054,297, 5,886,152 и 5,877,293.
В качестве конкретного воплощения изобретения, и как будет более подробно объяснено ниже в примерах, в настоящем документе описано антитело, состоящее из hz208F2. Такую гуманизацию можно также применять к другим антителам, составляющим часть настоящего изобретения.
В предпочтительном воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен тяжелой цепи (VH), имеющий:
i) CDR-H1, CDR-H2 и CDR-H3 последовательностей SEQ ID NO 7, 2 и 3 соответственно и
ii) FR1, FR2 и FR3, выделенные из IGHV1-46*01 (SEQ ID NO 46) зародышевой линии человека, и
iii) FR4, выделенный из IGHJ4*01 (SEQ ID NO 48) зародышевой линии человека.
В предпочтительном воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен легкой цепи (VL), имеющий:
i) CDR-L1, CDR-L2 и CDR-L3 последовательностей SEQ ID NO 9, 5 и 11 соответственно и
ii) FR1, FR2 и FR3, выделенные из IGHV1-39*01 (SEQ ID NO 47) зародышевой линии человека, и
iii) FR4, выделенный из IGHJ4*01 (SEQ ID NO 49) зародышевой линии человека.
В предпочтительном, но не ограничивающем воплощении изобретения антитело содержит:
a) тяжелую цепь, имеющую CDR-H1, CDR-H2 и CDR-H3 последовательностей SEQ ID NO 7, 2 и 3 соответственно, и FR1, FR2 и FR3, выделенные из IGHV1-46*01 (SEQ ID NO 46) зародышевой линии человека, и FR4, выделенный из IGHJ4*01 зародышевой линии человека (SEQ ID NO 48); и
b) легкую цепь, имеющую CDR-L1, CDR-L2 и CDR-L3 последовательностей SEQ ID NO 9, 5 и 11 соответственно, и FR1, FR2 и FR3, выделенные из IGHV1-39*01 (SEQ ID NO 47) зародышевой линии человека, и FR4, выделенный из IGKJ4*01 зародышевой линии человека (SEQ ID NO 49).
В одном воплощении изобретения антитело ADC согласно изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33 и вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35. Данное гуманизированное антитело далее в настоящем документе называют hz208F2 («вариант 1» или «вар. 1»).
В другом воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где данная последовательность SEQ ID NO 33 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 и 95.
Под выражением «обратная мутация» подразумевают мутацию или замену остатка, присутствующего в зародышевой линии человека, соответствующим остатком, исходно присутствующим в мышиной последовательности.
В другом воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где данная последовательность SEQ ID NO 33 содержит 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 или 17 обратных мутаций, выбранных из остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 и 95.
Для большей ясности в следующей таблице 4 проиллюстрированы предпочтительные обратные мутации.
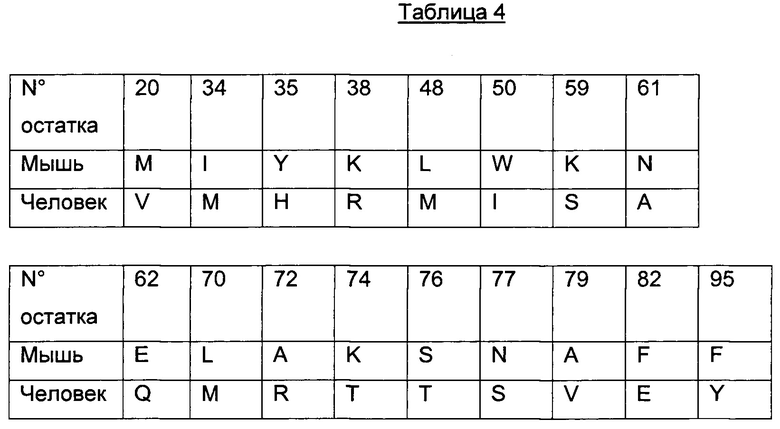
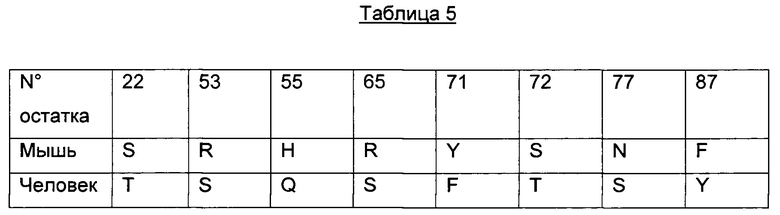
В одном воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где данная последовательность SEQ ID NO 35 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 22, 53, 55, 65, 71, 72, 77 и 87.
В одном воплощении изобретения антитело ADC согласно настоящему изобретению содержит вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где данная последовательность SEQ ID NO 35 содержит 2, 3, 4, 5, 6, 7 или 8 обратных мутаций, выбранных из остатков 22, 53, 55, 65, 71, 72, 77 или 87.
В другом воплощении изобретения антитело ADC согласно настоящему изобретению содержит:
a) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где данная последовательность SEQ ID NO 33 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 и 95; и
b) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где данная последовательность SEQ ID NO 35 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 22, 53, 55, 65, 71, 72, 77 и 87.
Для большей ясности в следующей таблице 5 проиллюстрированы предпочтительные обратные мутации.
В таком воплощении изобретения антитело ADC согласно изобретению содержит все упомянутые выше обратные мутации и соответствует антителу, содержащему вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 34 и вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 36. Данное гуманизированное антитело далее в настоящем документе называют hz208F2 («вариант 3» или «вар. 3»).
В другом воплощении изобретения все гуманизированные формы, содержащиеся между вариантом 1 и вариантом 3, также охвачены настоящим изобретением. Иными словами, антитело согласно изобретению соответствует антителу, содержащему вариабельный домен тяжелой цепи (VH) «консенсус» последовательности SEQ ID NO 41 и вариабельный домен легкой цепи (VL) «консенсус» последовательности SEQ ID NO 42. Данное гуманизированное антитело в целом далее в настоящем документе будет называться hz208F2 («вариант 2» или «вар. 2»).
В предпочтительном, но не ограничивающем воплощении изобретения антитело ADC по изобретению выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 33 или любую последовательность, проявляющую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 33, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11; и
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 34 или любую последовательность, проявляющую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 34, и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11.
Под «любой последовательностью, проявляющей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 33 или 34» подразумевают обозначение последовательностей, содержащих три CDR тяжелой цепи SEQ ID NO 1, 2 и 3, и, кроме того, проявляющих по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полноразмерной последовательностью SEQ ID NO 33 или 34 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 1, 2 и 3).
Если это не указано в соответствующих абзацах, в настоящем описании под любой последовательностью или под последовательностью, проявляющей по меньшей мере 80% идентичность конкретной последовательности, должно быть понятно, что эта последовательность проявляет по меньшей мере 80% и предпочтительно 85%, 90%, 95% и 98% идентичность эталонной последовательности. Если эти последовательности содержат последовательности CDR, это подразумевает, что последовательности, проявляющие идентичность по меньшей мере этих участков CDR участкам CDR эталонной последовательности 80%, предпочтительно 85%, 90%, 95% и 98%, идентичность полноразмерной последовательности, которую следует вычислить для остальной последовательности, расположена вне последовательностей, соответствующих этим участкам CDR.
В предпочтительном, но не ограничивающем воплощении изобретения антитело по изобретению выбрано из:
а) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 35 или любую последовательность, проявляющую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 35, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3; и
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 36 или любую последовательность, проявляющую по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 36, и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3.
Под «любой последовательностью, проявляющей по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность SEQ ID NO 35 или 36» подразумевают обозначение последовательностей, содержащих три CDR легкой цепи SEQ ID NO 4, 5 и 6, и, кроме того, проявляющих по меньшей мере 80%, предпочтительно 85%, 90%, 95% и 98% идентичность с полноразмерной последовательностью SEQ ID NO 35 или 36 вне последовательностей, соответствующих CDR (т.е. SEQ ID NO 4, 5 и 6).
Гуманизированные антитела, описанные в настоящем документе, могут также отличаться константным доменом, и более конкретно гуманизированные антитела могут быть выбраны или обозначены как, например, без ограничения IgG1, IgG2, IgG3, IgM, IgA, IgD или IgE. Более предпочтительно в контексте настоящего изобретения гуманизированные антитела представляют собой IgG1 или IgG4.
Одно воплощение изобретения относится к ADC, где «Аb» представляет собой гуманизированное антитело, содержащее вариабельные домены VH и VL, как описано выше, в формате IgG1. Более предпочтительно это гуманизированное антитело содержит константный домен для VH последовательности SEQ ID NO 43 и домен каппа для VL последовательности SEQ ID NO 45.
Одно воплощение изобретения относится к ADC, где «Аb» представляет собой гуманизированное антитело, содержащее вариабельные домены VH и VL, как описано выше, в формате IgG4. Более предпочтительно это гуманизированное антитело содержит константный домен для VH последовательности SEQ ID NO 44 и домен каппа для VL последовательности SEQ ID NO 45.
Еще одно другое воплощение изобретения относится к ADC, где «Ab» представляет собой антитело, выбранное из:
a) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 37 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 37, и легкую цепь последовательности SEQ ID NO 39 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 39, или состоящего из этих цепей; и
b) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 38 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 38, и легкую цепь последовательности SEQ ID NO 40 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 40, или состоящего из этих цепей.
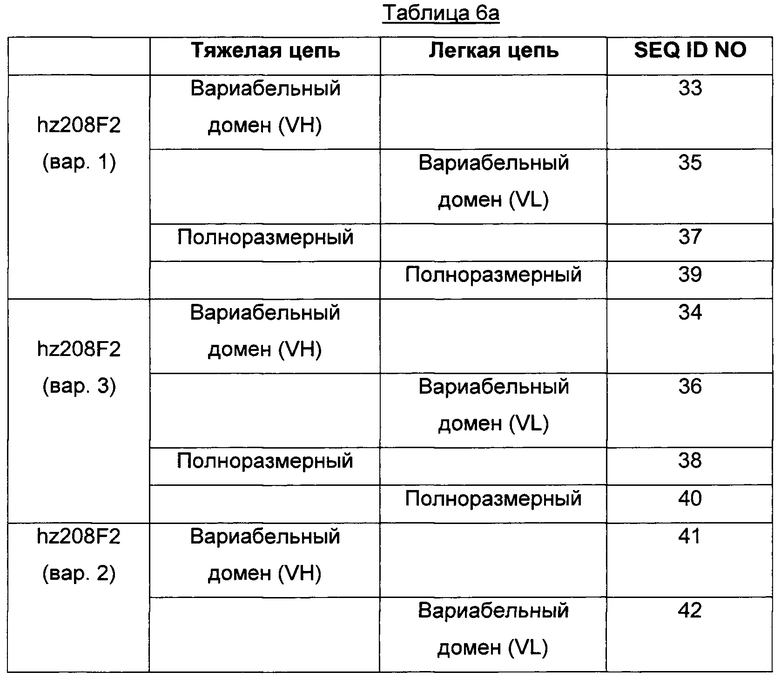
Для большей ясности в следующей таблице 6а проиллюстрированы неограничивающие примеры последовательностей VH и VL для варианта 1 (вар. 1) и варианта 3 (вар. 3) гуманизированного антитела hz208F2. Она также содержит консенсус-последовательность для варианта 2 (вар. 2).
В другом предпочтительном, но не ограничивающем воплощении изобретения антитело ADC по изобретению выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80 или любой последовательности, по меньшей мере на 80%, предпочтительно на 85%, 90%, 95% и 98% идентичной SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80; и три CDR легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 1;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность, выбранную из SEQ ID NO 57 или 60 или любой последовательности, по меньшей мере на 80%, предпочтительно на 85%, 90%, 95% и 98% идентичной SEQ ID NO 57 или 60; и три CDR тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3; и
с) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80 или любой последовательности, по меньшей мере на 80%, предпочтительно на 85%, 90%, 95% и 98% идентичной SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80; и вариабельный домен легкой цепи, имеющий последовательность, выбранную из SEQ ID NO 57 или 60, или любую последовательность, по меньшей мере на 80%, предпочтительно на 85%, 90%, 95% и 98% идентичную SEQ ID NO 57 или 60.
Еще одно другое воплощение изобретения относится к ADC, где «Ab» представляет собой антитело, выбранное из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80 или любую последовательность, по меньшей мере на 80% идентичную SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 или 80; и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 57; и
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 56, 64, 68 и 78 или любую последовательность, проявляющую по меньшей мере 80% идентичность с SEQ ID NO 56, 64, 68 или 78; и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 60 или любую последовательность, проявляющую по меньшей мере 80% идентичность SEQ ID NO 60.
Еще одно другое воплощение изобретения относится к ADC, где Ab представляет собой антитело, выбранное из:
a) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 58 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 58, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
b) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 58 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 58, и легкую цепь последовательности SEQ ID NO 61 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 61, или состоящего из этих цепей;
с) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 63 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 63, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
d) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 65 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 65, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
e) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 65 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 65, и легкую цепь последовательности SEQ ID NO 61 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 61, или состоящего из этих цепей;
f) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 67 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 67, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
g) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 69 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 69, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
h) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 69 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 69, и легкую цепь последовательности SEQ ID NO 61 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 61, или состоящего из этих цепей;
i) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 71 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 71, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
j) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 73 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 73, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
k) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 75 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 75, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
l) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 77 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 77, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
m) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 79 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 79, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей;
n) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 79 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 79, и легкую цепь последовательности SEQ ID NO 61 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 61, или состоящего из этих цепей; и
о) антитела, содержащего тяжелую цепь последовательности SEQ ID NO 81 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 81, и легкую цепь последовательности SEQ ID NO 59 или любой последовательности, проявляющей по меньшей мере 80% идентичность SEQ ID NO 59, или состоящего из этих цепей.
Иными словами, изобретение относится к ADC, где Ab представляет собой антитело, содержащее:
а) тяжелую цепь, имеющую последовательность, выбранную из SEQ ID NO 58, 63, 65, 67, 69, 71, 73, 75, 77, 79 и 81 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 58, 63, 65, 67, 69, 71, 73, 75, 77, 79 или 81; и
b) легкую цепь, имеющую последовательность, выбранную из SEQ ID NO 59 и 61 или любой последовательности, по меньшей мере на 80% идентичной SEQ ID NO 59 или 61.
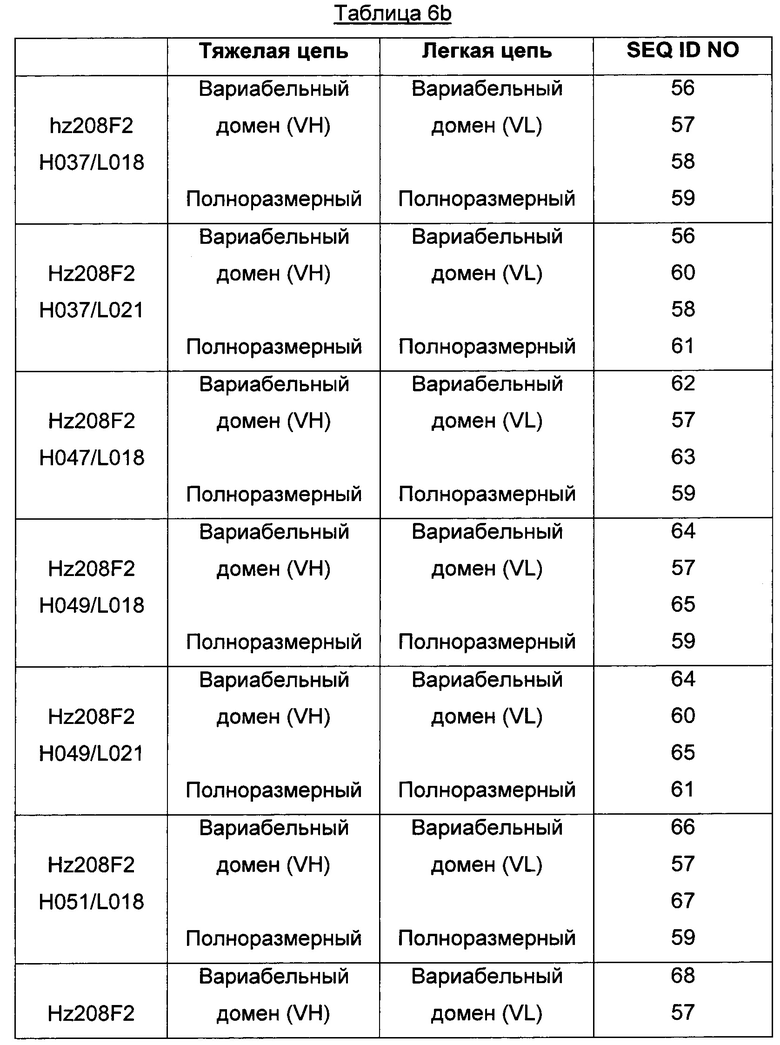
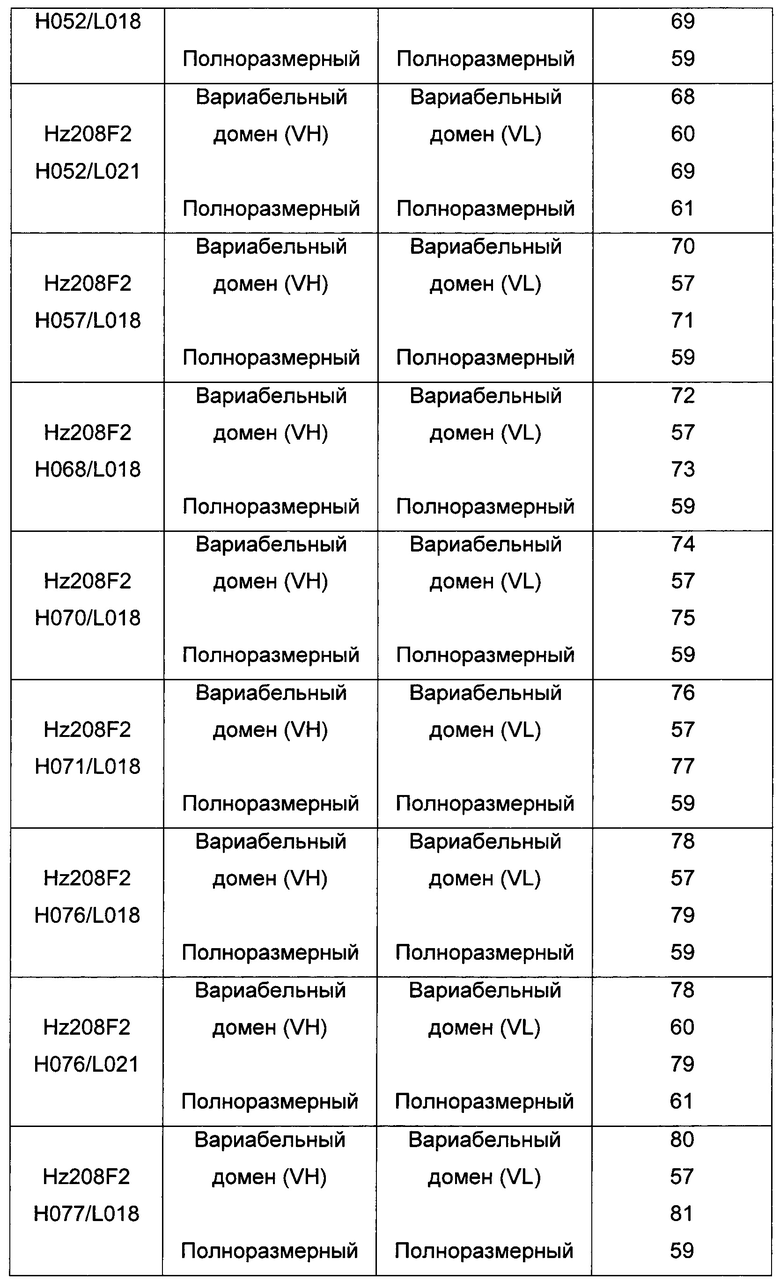
Для большей ясности в следующей таблице 6b проиллюстрированы неограничивающие примеры последовательностей VH и VL (вариабельного домена и полноразмерной) для различных вариантов гуманизированного антитела hz208F2.
Другой аспект настоящего изобретения представляет собой ADC, в котором Ab представляет собой антитело, выбранное из i) антитела, продуцируемого гибридомой 1-4757, 1-4773, 1-4775, 1-4736 или 1-4774, депонированной в CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года соответственно, или ii) антитела, которое конкурирует за связывание с IGF-1R с антителом i); или iii) антитела, которое связывается с тем же эпитопом IGF-1R, что и антитело i).
Действительно, в настоящем документе описана мышиная гибридома, выбранная из гибридомы I-4757, I-4773, I-4775, I-4736 и I-4774, которые депонированы в CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года соответственно.
Также описана изолированная нуклеиновая кислота, кодирующая антитело или его антигенсвязывающий фрагмент согласно изобретению.
Термины «нуклеиновая кислота», «последовательность нуклеиновой кислоты», «нуклеиново-кислотная последовательность», «полинуклеотид», «олигонуклеотид», «полинуклеотидная последовательность» и «нуклеотидная последовательность», используемые в настоящем документе взаимозаменяемо, означают точную последовательность нуклеотидов, модифицированную или не модифицированную, определяющую фрагмент или участок нуклеиновой кислоты, содержащую или не содержащую неприродные нуклеотиды и представляющую собой либо двунитевую ДНК, либо однонитевую ДНК, либо продукты транскрипции этих ДНК.
Эти последовательности изолированы и/или очищены, т.е. отобраны прямым или непрямым путем, например путем копирования, из их окружения, по меньшей мере частично модифицированного. В настоящем документе следует также упомянуть изолированные нуклеиновые кислоты, полученные методами рекомбинантной генетики, например, посредством клеток-хозяев, или полученные путем химического синтеза.
Также описан вектор, содержащий нуклеиновую кислоту, кодирующую антитело или его антигенсвязывающий фрагмент ADC согласно изобретению, в частности, клонирующие и/или экспрессионные векторы, которые содержат такую нуклеотидную последовательность.
Векторы предпочтительно содержат элементы, которые дают возможность для экспрессии и/или секреции нуклеотидных последовательностей в данной клетке-хозяине. Таким образом, вектор может содержать промотор, сигналы инициации и терминации трансляции, а также подходящие регуляторные участки транскрипции. Он должен обладать способностью к стабильному сохранению в клетке-хозяине и может необязательно содержать специфичные сигналы, которые задают секрецию транслированного белка. Эти различные элементы выбирует и оптимизирует специалист в данной области техники в соответствии с используемой клеткой-хозяином. Для этой цели нуклеотидные последовательности могут быть встроены в векторы, автономно реплицирующиеся внутри выбранной клетки-хозяина, или в интегративные векторы выбранной клетки-хозяина.
Векторы представляют собой, например, векторы плазмидного или вирусного происхождения. Их используют для трансформации клеток-хозяев с целью клонирования или экспрессии нуклеотидных последовательностей по изобретению.
Такие векторы получают способами, обычно используемыми специалистами в данной области техники, и полученные в результате клоны можно вводить в подходящую клетку-хозяина стандартными способами, такими как липофекция, электропорация, конъюгация, тепловой шок или химические способы.
Эти изолированные клетки-хозяева трансформированы описанным выше вектором или содержат этот вектор.
Клетка-хозяин может быть выбрана из прокариотических или эукариотических систем, таких как, например, бактериальные клетки, а также дрожжевые клетки или клетки животных, в частности, клетки млекопитающих (за исключением человека). Можно также использовать клетки насекомых или растений.
Также раскрыт способ получения антитела ADC согласно изобретению или его антигенсвязывающего фрагмента, включающий следующие стадии:
a) культивирование в среде в условиях культивирования, подходящих для описанной выше клетки-хозяина; и
b) выделение полученного таким путем антитела из культуральной среды или из культивируемых клеток.
Трансформированные клетки используют в способах получения рекомбинантных антител ADC согласно изобретению. В настоящее описание также включены способы получения антител в рекомбинантной форме с использованием вектора и/или клетки, трансформированной вектором, как раскрыто выше. Предпочтительно клетку, трансформированную вектором, как описано выше, культивируют в условиях, дающих возможность экспрессии описанного выше антитела и выделения этого антитела.
Как уже упоминалось, клетка-хозяин может быть выбрана из прокариотических или эукариотических систем. В частности, возможно идентифицировать нуклеотидные последовательности, способствующие секреции в такой прокариотической или эукариотической системе. Вектор, как раскрыто выше, несущий такую последовательность, можно, следовательно, преимущественно использовать для получения секретируемых рекомбинантных белков. Действительно, очистка этих представляющих интерес рекомбинантных белков облегчается за счет факта их более вероятного присутствия в супернатанте, чем внутри клеток-хозяев.
Антитело ADC по настоящему изобретению может быть также получено путем химического синтеза. Один такой способ получения также составляет объект изобретения. Специалисту в данной области техники известны способы химического синтеза, такие как твердофазные методы или частичные твердофазные методы, путем конденсации фрагментов или путем традиционного синтеза в растворе. Можно также указать полипептиды, полученные путем химического синтеза и способные к содержанию соответствующих неприродных аминокислот.
В настоящее изобретение также включено антитело, которое может быть получено описанным выше способом.
Согласно конкретному аспекту изобретение относится к ADC, в котором АВ представляет собой антитело или его антигенсвязывающий фрагмент, как описано выше, для применения в качестве направляющего носителя для доставки цитотоксического агента в сайт-мишень хозяина, где сайт-мишень хозяина состоит из эпитопа, локализованного в IGF-1R, предпочтительно во внеклеточном домене IGF-1R, более предпочтительно IGF-1R человека (SEQ ID NO 50) и еще более предпочтительно во внеклеточном домене IGF-1R человека (SEQ ID NO 51) и еще более предпочтительно в N-концевом участке внеклеточного домена IGF-1R человека (SEQ ID NO 52) в любом природном варианте этой последовательности.
В предпочтительном воплощении изобретения сайт-мишень хозяина представляет собой сайт-мишень клетки млекопитающего, более предпочтительно клетки человека, более предпочтительно клетки, которые естественно или путем генетической рекомбинации экспрессируют IGF-1R.
В более предпочтительном воплощении изобретения сайт-мишень хозяина представляет собой сайт-мишень клетки пациента, предпочтительно человека, страдающего раком, предпочтительно IGF-IR-экспрессирующим раком или раком, обусловленным IGF-1R.
IGF-1R-экспрессирующие формы рака или формы рака, обусловленные IGF-1R, включают, в частности, формы рака, где опухолевые клетки экспрессируют или характеризуются гиперэкспрессией полноразмерного IGF-1R или его участка на их поверхности.
II - Лекарственное средство (D)
Группировка лекарственного средства согласно изобретению имеет следующую формулу (II):
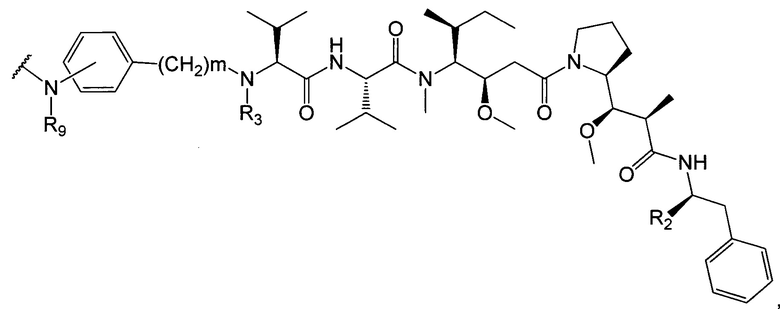
где:
- R2 представляет собой СООН, СООСН3 или тиазолил (такой как тиазол-2-ил),
- R3 представляет собой Н или (С1-С6)алкил (такой как метил), в частности, (С1-С6)алкильную группу,
- R9 представляет собой Н или (С1-С6)алкил (такой как метил),
- m представляет собой целое число, составляющее от 1 до 8, и
- волнистой линией указана точка присоединения к L.
Под «алкилом» в настоящем изобретении подразумевают прямоцепочечную или разветвленную насыщенную углеводородную цепь. Например, можно упомянуть метильные, этильные, пропильные, изопропильные, бутильные, изобутильные, втор-бутильные, трет-бутильные, пентильные или гексильные группы.
Под «(Сх-Су)алкилом» в настоящем изобретении подразумевают алкильную цепь, такую как определено выше, содержащую от×до у атомов углерода. Таким образом, (С1-С6)алкильная группа представляет собой алкильную цепь, имеющую от 1 до 6 атомов углерода.
(С1-С6)алкил преимущественно представляет собой (С1-С4)алкил, предпочтительно (С1-С2)алкил.
Среди соединений по изобретению одному из особенно приемлемых классов группировок лекарственного средства соответствуют группировки лекарственного средства формулы (II), в которых R2 представляет собой группу СООН.
Другому особенно приемлемому классу группировок соответствуют группировки формулы (II), в которых R2 представляет собой тиазол (в частности, группу тиазол-2-ил).
Другому классу особенно приемлемых группировок соответствуют группировки формулы (II), в которых R2 представляет собой СООМе.
Согласно одному конкретному воплощению настоящего изобретения R2 более предпочтительно представляет собой группу СООН, СООМе или тиазол-2-ил.
Согласно первому предпочтительному воплощению R2 представляет собой СООН.
Согласно второму предпочтительному воплощению R2 представляет собой СООМе.
R3, в частности, представляет собой (С1-С6)алкил, преимущественно метильную группу.
m представляет собой целое число, составляющее от 1 до 8, в частности, от 1 до 6, преимущественно от 1 до 4, предпочтительно равно 1 или 2.
В предпочтительном воплощении изобретения R2 представляет собой СООН, R3 представляет собой метильную группу, и m равно 1 или 2.
Среди группировок лекарственных средств по изобретению одному из особенно приемлемых классов группировок лекарственного средства соответствуют группировки лекарственного средства формулы (II), в которых R9 представляет собой метильную группу или атом водорода.
В предпочтительном воплощении изобретения:
- R2 представляет собой СООН, R3 представляет собой метильную группу, R9 представляет собой метильную группу, и m равно 1 или 2, или
- R2 представляет собой СООН, R3 представляет собой метильную группу, R9 представляет собой атом водорода, и m равно 1 или 2.
Согласно предпочтительному воплощению изобретения группа NR9 расположена на фенильном кольце в пара-положении по отношению к группе (СН2)m.
Преимущественно группировка лекарственного средства выбрана из следующих группировок:
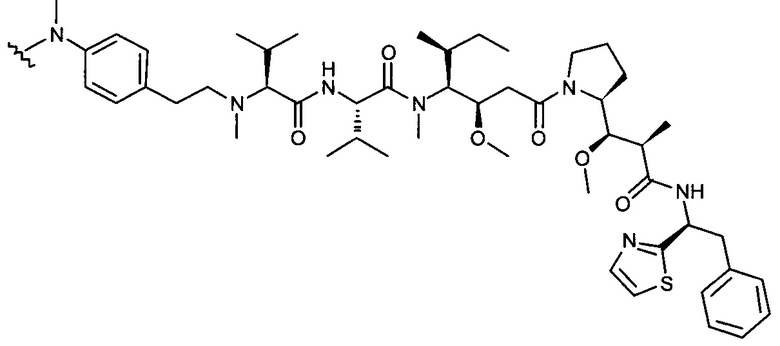
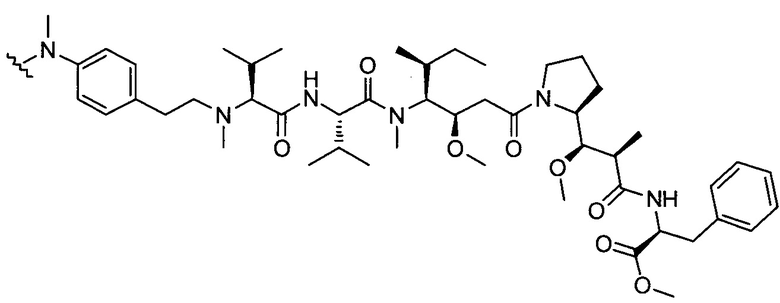
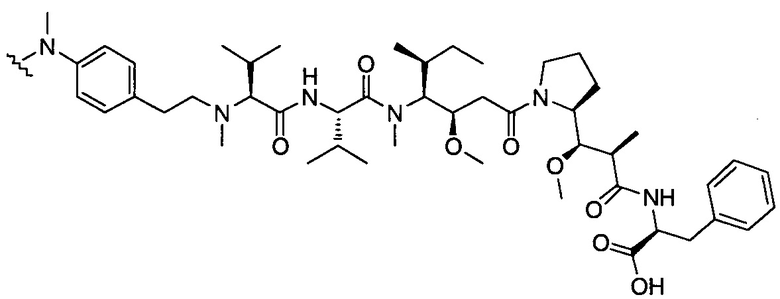
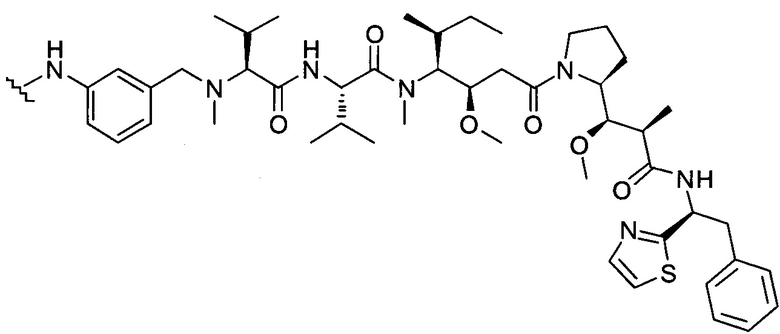
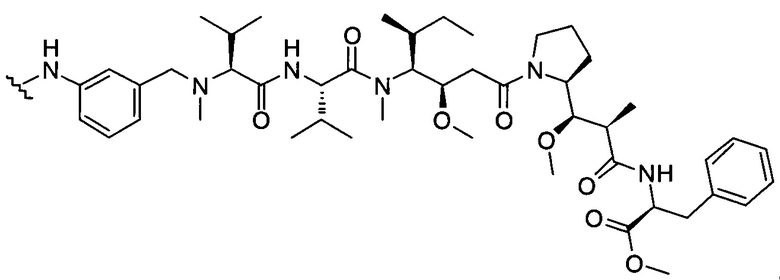
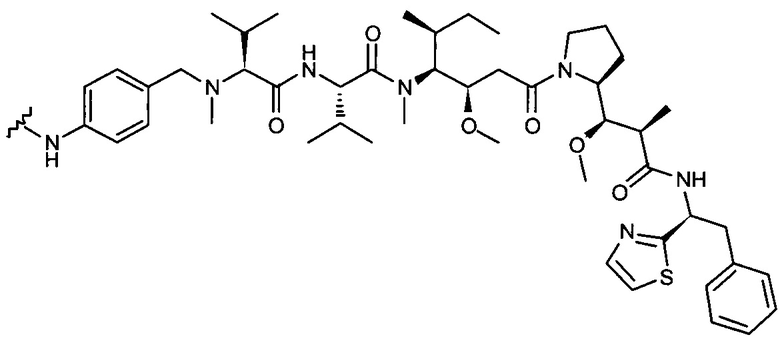 ,
,
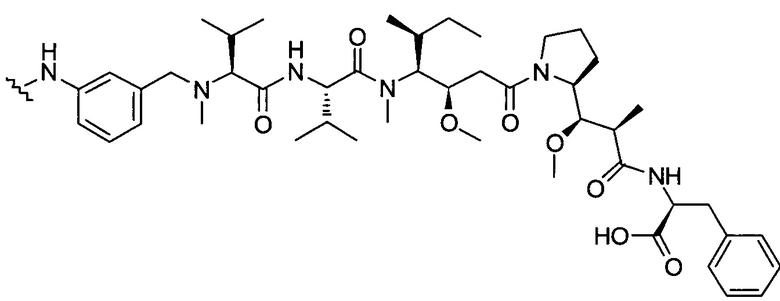 ,
,
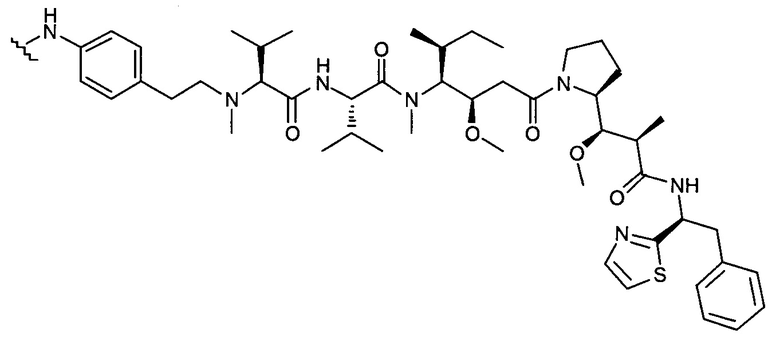 ,
,
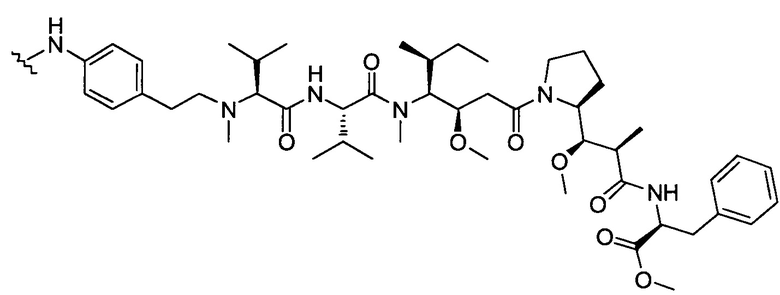 , и
, и
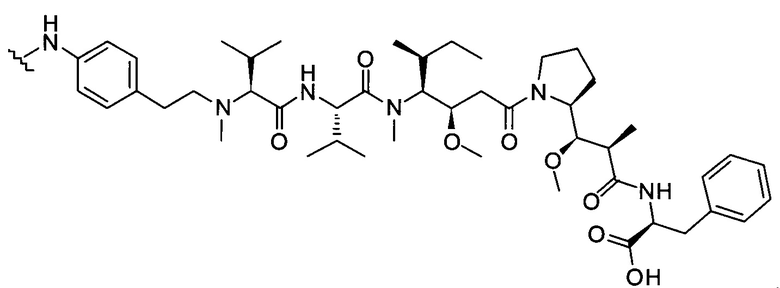 .
.
Получение лекарственного средства (Формулы РН):
Лекарственное средство может быть получено общими способами, описанными на следующих ниже схемах синтеза, необязательно дополненных при необходимости любой стандартной операцией, описанной в литературе или хорошо известной специалистам в данной области техники, либо описанной в примерах в экспериментальном разделе настоящего документа.
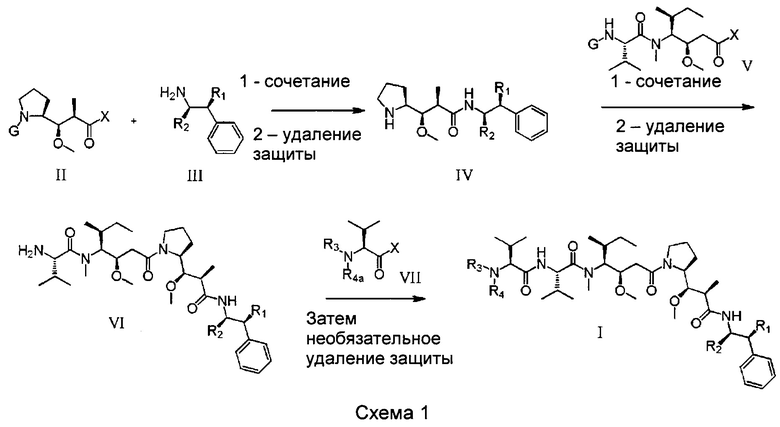
На схеме 1 проиллюстрирован первый общий способ, который можно использовать для получения лекарственного средства. В приведенных выше общих формулах R1-Н, R2 и R3 являются такими, как определено выше для формулы II, R4 представляет собой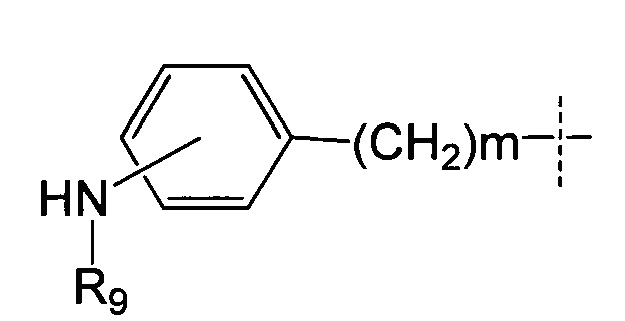 , R4a представляет собой группу R4, как определено выше, необязательно в защищенной форме, и G представляет собой защитную группу.
, R4a представляет собой группу R4, как определено выше, необязательно в защищенной форме, и G представляет собой защитную группу.
Первая стадия состоит в конденсации соединения (II), защищенного на его аминной функциональной группе защитной группой G, с соединением (III). X может представлять собой уходящую группу, такую как атом хлора. В этом случае первая стадия состоит во взаимодействии между хлорангидридом и амином. Это взаимодействие можно проводить, используя способы и методы, хорошо известные специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие двух химических соединений в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3, K2CO3 в растворителе, таком как ТГФ, дихлорметан, ДМФ, ДМСО, при температуре, в частности, от -20°С до 100°С. X может также представлять собой гидроксил (ОН). В этом случае первая стадия представляет собой реакцию конденсации между карбоновой кислотой (II) и амином (III). Это взаимодействие можно проводить, следуя способам и методам, хорошо известным специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии агента сочетания, такого как 1-(3-диметиламинопропил)-3-этил-карбодиимид (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-он, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре, в частности, от -15°С до 40°С. При другом особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии диэтилфосфороцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре, в частности, от -15°С до 40°С. Другой особенно приемлемый способ состоит в том, что вызывают взаимодействие двух химических соединений в присутствии O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата (HATU), а третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре от -15°С до 100°С.
После удаления защиты промежуточного соединения с использованием методов, хорошо известных специалистам в данной области техники («Protective Groups in Organic Synthesis», T.W. Greene, John Wiley & Sons, 2006 и «Protecting Groups», P.J. Kocienski, Thieme Verlag, 1994), соединение (IV) можно конденсировать с соединением (V), следуя способам и методам, описанным выше, ведущим к соединению (VI) после стадии удаления защиты. Затем это соединение после конденсации с промежуточным соединением (VII) и необязательного удаления защиты ведет к образованию лекарственного средства. Соединение (VI) можно также подвергать сочетанию с соединением (VII'), в котором R'3 представляет собой предшественник R3, в частности, группу R3, защищенную защитной группой. Сочетание, следующее за удалением защиты группы R'3 с получением R3 можно осуществлять, следуя таким же методикам, как описано выше.
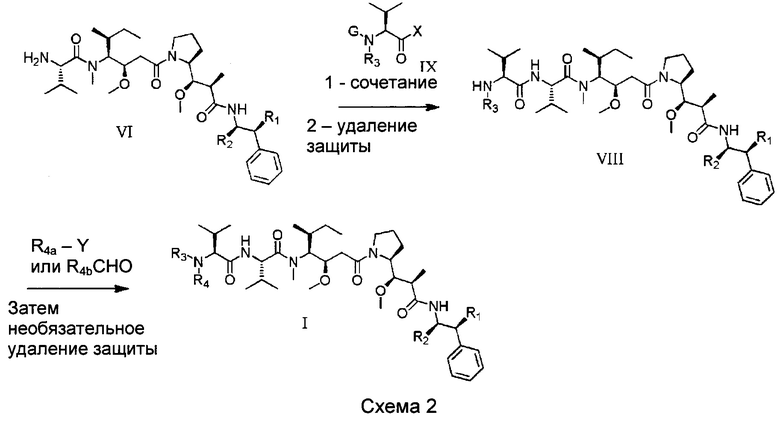
На схеме 2 проиллюстрирован второй общий способ, который можно использовать для получения лекарственного средства. В приведенных выше общих формулах G представляет собой защитную группу, R1-Н, R2, R3 и R4a являются такими, как определено выше, и R4b представляет собой 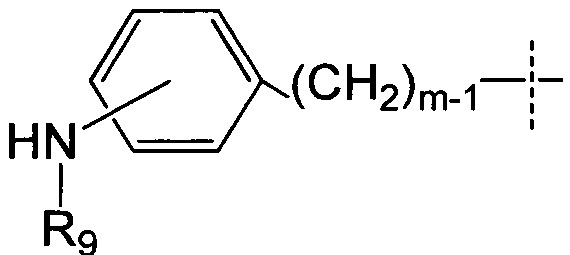 .
.
На первой стадии соединение (IX), защищенное на его аминной функциональной группе защитной группой G, конденсируют с соединением (VI). X может представлять собой уходящую группу, например, атом хлора. В этом случае первая стадия состоит во взаимодействии между хлорангидридом и амином. Это взаимодействие можно проводить, используя способы и методы, хорошо известные специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие двух химических соединений в присутствии органического или неорганического основания, например Et3N, iPr2NEt, пиридина, NaH, Cs2CO3, K2CO3 в растворителе, таком как ТГФ, дихлорметан, ДМФ, ДМСО, при температуре, в частности, от -20°С до 100°С. X может также представлять собой гидроксил. В этом случае первая стадия представляет собой реакцию конденсации между карбоновой кислотой (IX) и амином (VI). Это взаимодействие можно проводить, следуя способам и методам, хорошо известным специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие двух химических соединений в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимид (EDC), 3-гидрокси-1,2,3-бензотриазин- 4(3Н)-он, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре, в частности, от -15°С до 40°С. При другом особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии диэтилфосфороцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре, в частности, от -15°С до 40°С.
После удаления защиты промежуточного соединения, используя методы, хорошо известные специалистам в данной области техники, полученное соединение (VIII) может приводить к получению лекарственного средства после взаимодействия с R4Y. В этом случае Y представляет собой уходящую группу, такую как Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил. Реакцию проводят в присутствии органического или неорганического основания, такого как Et3N, iPr2NEt, NaH, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как дихлорметан, ТГФ, ДМФ, ДМСО, при температуре, в частности, от -20° до 100°С. При другом особенно приемлемом способе вызывают взаимодействие соединения (VIII) с альдегидом формулы R4b-СНО, где R4b соответствует предшественнику R4. В этом случае реакция представляет собой восстановительное аминирование в присутствие восстанавливающего агента, такого как NaBH4, NaBH3CN, NaBH(OAc)3, в полярном растворителе, таком как 1,2-дихлорэтан, дихлорметан, ТГФ, ДМФ, МеОН, необязательно в присутствии изопропоксида титана (IV), при рН, который можно регулировать добавлением кислоты, такой как уксусная кислота, при температуре от-20°С до 100°С.
На приведенных выше схемах синтеза лекарственное средство может привести к получению другого лекарственного средства после дополнительной стадии взаимодействия, такого как омыление, например, используя способы, хорошо известные специалистам в данной области техники, посредством которых группа R2, представляющая собой сложный эфир (СООМе), изменяется на группу R2, представляющую собой карбоновую кислоту (СООН).
Если желательно выделить лекарственное средство, содержащее по меньшей мере одну функциональную группу основания в состоянии соли присоединения кислоты, это возможно путем обработки лекарственного средства в форме свободного основания (содержащего по меньшей мере одну функциональную группу основания) подходящей кислотой, предпочтительно в эквивалентном количестве. Подходящая кислота может, в частности, представлять собой трифторуксусную кислоту.
III - Линкер (L)
«Линкер», «линкерное звено», «L» или «связь» означают в настоящем изобретении химическую группировку, содержащую ковалентную связь или цепь атомов, которая ковалентно присоединяет антитело по меньшей мере к одному лекарственному средству.
Линкеры могут быть получены с использованием разнообразных бифункциональных агентов сочетания белков, таких как N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP), сукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилат (SMCC), иминотиолан (IT), бифункциональных производных сложных имидоэфиров (таких как диметиладипимидат HCI), активных сложных эфиров (таких как дисукцинимидилсуберат), альдегидов (таких как глутаральдегид), бис-азидосоединений (таких как бис(пара-азидобензоил)гександиамин), бис-диазониевых производных (таких как бис-(пара-диазонийбензоил)-этилендиамин), диизоцианатов (таких как толуол-2,6-диизоцианат) и бис-активных соединений фтора (таких как 1,5-дифтор-2,4-динитробензол). Углерод-14-меченая 1-изотиоцианатобензил-3-метилдиэтилентриаминпентауксусная кислота (MX-DTPA) является иллюстративным хелатирующим агентом для конъюгации цитотоксических агентов с направляющей системой. Другие перекрестно-сшивающие агенты могут представлять собой BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, МРВН, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, сульфо-EMCS, сульфо-GMBS, сульфо-KMUS, сульфо-MBS, сульфо-SIAB, сульфо-SMCC и сульфо-SMPB и SVSB (сукцинимидил-(4-винилсульфон)бензоат), которые имеются в продаже (например, от Pierce Biotechnology, Inc., Рокфорд, III., США).
Линкер может быть «нерасщепляемым» или «расщепляемым».
В предпочтительном воплощении изобретения линкер представляет собой «расщепляемый линкер», способствующий высвобождению лекарственного средства в клетке. Например, можно использовать кислотолабильный линкер, пептидазочувствительный линкер, фотолабильный линкер, диметильный линкер или дисульфид-содержащий линкер. В предпочтительном воплощении изобретения линкер является расщепляемым во внутриклеточных условиях, так что в результате расщепления линкера лекарственное средство высвобождается от антитела во внутриклеточную среду.
Например, в некоторых воплощениях изобретения линкер является расщепляемым расщепляющим агентом, присутствующим во внутриклеточной среде (например, внутри лизосомы, или эндосомы, или кавеолы). Линкер может представлять собой, например, пептидильный линкер, который расщепляется внутриклеточными ферментами пептидазой или протеазой, включая без ограничений лизосомальную или эндосомальную протеазу. Как правило, пептидильный линкер содержит по меньшей мере две последовательные аминокислоты или по меньшей мере три последовательные аминокислоты, или имеет длину по меньшей мере две аминокислоты или длину длину по меньшей мере три аминокислоты. Расщепляющие агенты могут включать катепсины В и D и плазмин, при этом известно, что все эти агенты гидролизуют дипептидные производные лекарственных средств, приводя в результате к высвобождению активного лекарственного средства внутри клеток-мишеней. Например, можно использовать пептидильный линкер, расщепляемый тиолзависимой протеазой катепсином-В, обладающей высоким уровнем экспрессии в раковой ткани (например, линкер, содержащий или представляющий собой Phe-Leu или Gly-Phe-Leu-Gly). В конкретных воплощениях изобретения пептидильный линкер, расщепляемый внутриклеточной протеазой, содержит или представляет собой Val-Cit или Phe-Lys. Одно из преимуществ использования внутриклеточного протеолитического высвобождения лекарственного средства состоит в том, что при конъюгации лекарственное средство, как правило, аттенуировано, и стабильности конъюгатов в сыворотке, как правило, высоки.
В других воплощениях изобретения расщепляемый линкер является рН-чувствительным, т.е. чувствительным к гидролизу при некоторых значениях рН. В характерном случае рН-чувствительный линкер является гидролизуемым в кислых условиях. Например, можно использовать кислотолабильный линкер, который подвергается гидролизу в лизосоме (например, гидразон, семикарбазон, тиосемикарбазон, цис-аконитовый амид, сложный ортоэфир, ацеталь, кеталь или тому подобное). Такие линкеры относительно стабильны в нейтральных условиях рН, например, в крови, но нестабильны при рН менее 5,5 или 5,0, приблизительном рН лизосомы. В некоторых воплощениях изобретения гидролизуемый линкер представляет собой простой тиоэфирный линкер (такой как, например, простой тиоэфир, присоединенный к лекарственному средству посредством ацилгидразоновой связи).
Еще в других воплощениях линкер является расщепляемым в восстанавливающих условиях (например, дисульфидный линкер). В данной области техники известен ряд дисульфидных линкеров, включающих, например, линкеры, которые могут быть образованы с использованием SATA (М-сукцинимидил-S-ацетилтиоацетат), SPDP (N-сукцинимидил-3-(2-пиридилдитио)пропионат), SPDB (N- сукцинимидил-3-(2-пиридилтио)бутират) и SMPT (N-сукцинимидил-оксикарбонил-альфа-метил-альфа-(2-пиридил-дитио)толуол).
В некоторых предпочтительных воплощениях изобретения линкерное звено может иметь следующую общую формулу:
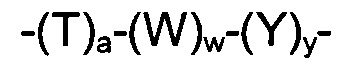
где:
Т представляет собой удлиняющее звено;
а равно 0 или 1;
W представляет собой аминокислотное звено;
w представляет собой целое число в диапазоне от 0 до 12;
Y представляет собой спейсерное звено;
y равно 0, 1 или 2.
Удлиняющее звено (Т) при его наличии связывает антитело с аминокислотным звеном (W) при его наличии, или со спейсерным звеном при его наличии, или непосредственно с лекарственным средством. Полезные функциональные группы, которые могут присутствовать на антителе, либо естественным путем, либо посредством химической манипуляции, включают сульфгидрил, амино, гидроксил, аномерную гидроксильную группу углевода и карбоксил. Подходящими функциональными группами являются сульфгидрил и амино. Сульфгидрильные группы могут быть образованы путем восстановления внутримолекулярных дисульфидных связей антитела при их наличии. Альтернативно сульфгидрильные группы могут быть образованы путем взаимодействия аминогруппы лизиновой группировки антитела с 2-иминотиоланом или с другими реагентами, образующими сульфгидрил. В конкретных воплощениях изобретения антитело сконструировано таким образом, что оно несет один или более лизинов. Более предпочтительно антитело может быть сконструировано таким образом, что оно несет один или более цистеинов (см. ThioMab).
В некоторых конкретных воплощениях изобретения удлиняющее звено образует связь с атомом серы антитела. Атом серы может иметь происхождение от сульфгидрильной (-SH) группы восстановленного антитела.
В некоторых других конкретных воплощениях изобретения удлиняющее звено связано с антителом посредством дисульфидной связи между атомом серы антитела и атомом серы удлиняющего звена.
В других конкретных воплощениях изобретения активная группа удлиняющего звена содержит активный центр, который может взаимодействовать с аминогруппой антитела. Аминогруппа может представлять собой аминогруппу аргинина или лизина. Подходящие активные центры, взаимодействующие с аминами, включают без ограничений активированные сложные эфиры (такие как сукцинимидные сложные эфиры, 4-нитрофенильные сложные эфиры, пентафторфенильные сложные эфиры), ангидриды, хлорангидриды, сульфонилхлориды, изоцианаты и изотиоцианаты.
Еще в одном другом аспекте изобретения реакционная функциональная группа удлиняющего звена содержит активный центр, который взаимодействует с модифицированной углеводной группой, которая может присутствовать на антителе. В конкретном воплощении изобретения антитело гликозилировано ферментативным путем с получением углеводной группировки или гликозилировано естественным путем. Углевод можно подвергать мягкому окислению реагентом, таким как периодат натрия, и полученное в результате карбонильное звено окисленного углевода может быть конденсировано с удлиняющим звеном, которое содержит функциональную группу, такую как гидразид, оксим, активный амин, гидразин, тиосемикарбазид, гидразинкарбоксилат или арилгидразид.
Согласно конкретному воплощению изобретения удлиняющее звено имеет следующую формулу:
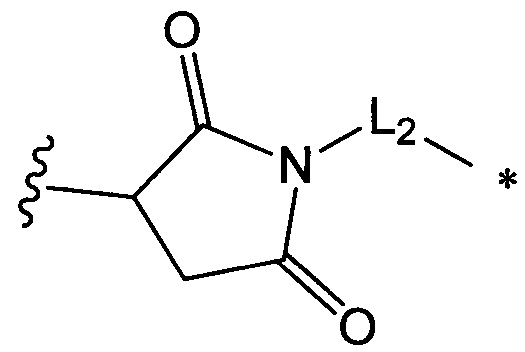
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил или (С2-С6)алкил-карбонил (циклоалкильные или алкильные группировки связаны с атомом азота малеимидной группировки),
звездочкой указана точка присоединения к аминокислотному звену при его наличии, к спейсерному звену при его наличии или к лекарственному средству D, и
волнистой линией указана точка присоединения к антителу Ab.
Под «(С4-С10)циклоалкилом» в настоящем изобретении подразумевают углеводородный цикл, имеющий от 4 до 10 атомов углерода, включающий без ограничений циклопентил, циклогексил и тому подобное.
L2 может преимущественно представлять собой (С2-С6)алкил-карбонил, такой как пентил-карбонил, следующей формулы:
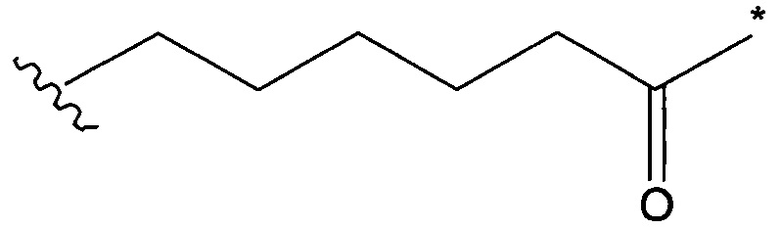
где
звездочкой указана точка присоединения к аминокислотному звену при его наличии, к спейсерному звену при его наличии или к лекарственному средству D; и
волнистой линией указана точка присоединения к атому азота малеимидной группировки.
Аминокислотное звено (W) при его наличии связывает удлиняющее звено (Т) при его наличии или в противном случае антитело со спейсерным звеном (Y) при наличии спейсерного звена или с лекарственным средством в отсутствие спейсерного звена.
Как упомянуто выше, (W)w отсутствует (w равно 0) или может представлять собой дипептидное, трипептидное, тетрапептидное, пентапептидное, гексапептидное, гептапептидное, октапептидное, нонапептидное, декапептидное, ундекапептидное или додекапептидное звено, где аминокислоты, образующие пептиды, могут отличаться друг от друга.
Так, (W)w может быть представлен следующей формулой: (W1)w1(W2)w2(W3)w3(W4)w4(W5)w5, где каждый из W1-W5 представляет собой независимо друг от друга аминокислотное звено, и каждое w1-w5 равно 0 или 1.
В некоторых воплощениях изобретения аминокислотное звено (W)w может содержать аминокислотные остатки, такие как встречающиеся в природе, а также минорные аминокислоты и не встречающиеся в природе аналоги аминокислот, такие как цитруллин.
Аминокислотные остатки аминокислотного звена (W)w включают без ограничения аланин, валин, лейцин, изолейцин, метионин, фенилаланин, триптофан, пролин, лизин, незащищенный или защищенный ацетилом или формилом, аргинин, аргинин, незащищенный или защищенный тозильными группами или нитрогруппами, гистидин, орнитин, орнитин, защищенный ацетилом или формилом, и цитруллин. Иллюстративные компоненты аминокислотного линкера включают предпочтительно дипептид, трипептид, тетрапептид или пентапептид, в частности, дипептид или трипептид.
Иллюстративные дипептиды включают: Val-Cit, Ala-Val, Ala-Ala, Val-Ala, Lys-Lys, Cit-Cit, Val-Lys, Ala-Phe, Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, lle-Cit, Trp-Cit, Phe-Ala, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg.
Иллюстративные трипептиды включают: Val-Ala-Val, Ala-Asn-Val, Val-Leu-Lys, Ala-Ala-Asn, Phe-Phe-Lys, Gly-Gly-Gly, D-Phe-Phe-Lys, Gly-Phe-Lys.
Иллюстративные тетрапептиды включают: Gly-Phe-Leu-Gly (SEQ ID NO 53), Ala-Leu-Ala-Leu (SEQ ID NO 54).
Иллюстративный пентапептид включает: Pro-Val-Gly-Val-Val (SEQ ID NO 55).
Согласно конкретному воплощению изобретения (W)w может представлять собой дипептид (т.е. w равно 2), такой как Val-Cit, либо в линкере отсутствует аминокислотное звено (w равно 0). При отсутствии в линкере аминокислотного звена предпочтительно в нем также отсутствует спейсерное звено.
Согласно предпочтительному воплощению изобретения w равно 0 (т.е. (W)w представляет собой простую связь) или w равно 2 (т.е. (W)w представляет собой дипептид) и (W)w может быть, таким образом, выбран из:
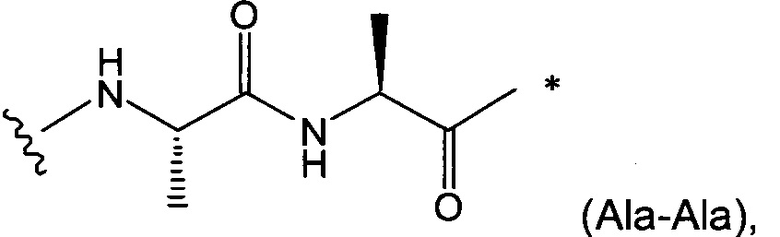
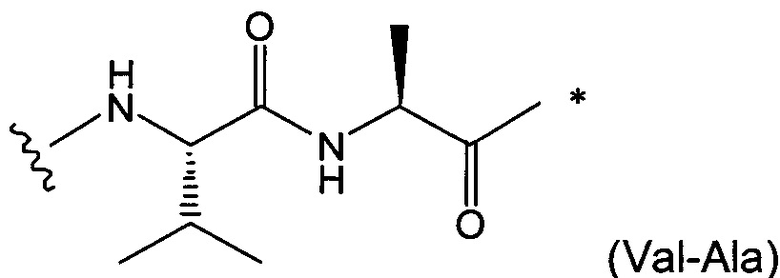 и
и
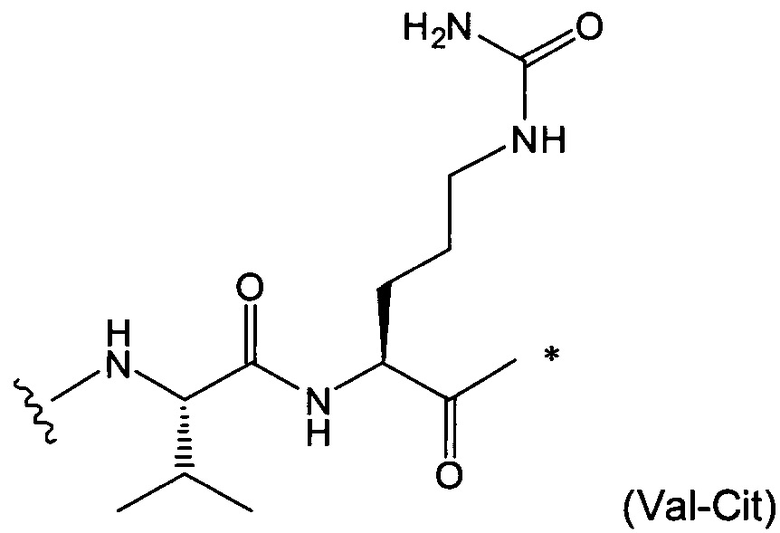
и, в частности, представляет собой Val-Cit,
где
звездочкой указана точка присоединения к спейсерному звену при его наличии или к лекарственному средству D; и
волнистой линией указана точка присоединения к L2.
Компоненты аминокислотного линкера могут быть сконструированы и оптимизированы по их селективности к ферментативному расщеплению конкретным ферментом, например, опухолеассоциированной протеазой, катепсином В, С и D или плазминовой протеазой.
Аминокислотное звено линкера может ферментативно расщепляться ферментом, включающим без ограничения опухолеассоциированную протеазу, с высвобождением лекарственного средства.
Аминокислотное звено может быть сконструировано и оптимизировано по его селективности к ферментативному расщеплению конкретной опухолеассоциированной протеазой. Подходящие звенья представляют собой звенья, расщепление которых катализируется протеазами, катепсином В, С и D и плазмином.
Спейсерное звено (Y) при его наличии связывает аминокислотное звено при его наличии или удлиняющее звено при его наличии или в противном случае антитело с лекарственным средством. Спейсерные звенья принадлежат к двум различным типам: саморасщепляющиеся и не саморасщепляющиеся. Не саморасщепляющееся спейсерное звено представляет собой звено, в котором все спейсерное звено или его часть остается связанным с лекарственным средством после ферментативного отщепления аминокислотного звена от конъюгата антитела с лекарственным средством. Примеры не саморасщепляющегося спейсерного звена включают без ограничений спейсерное звено (глицин-глицин) и глициновое спейсерное звено. Для высвобождения лекарственного средства внутри клетки-мишени должна происходить независимая реакция гидролиза для расщепления связи глицин-лекарственное звено.
В конкретном воплощении изобретения не саморасщепляющееся спейсерное звено (Y) представляет собой Gly.
Альтернативно конъюгат антитела с лекарственным средством, содержащий саморасщепляющееся спейсерное звено, может высвобождать лекарственное средство без необходимости в отдельной стадии гидролиза. В этих воплощениях изобретения (Y) представляет собой остаток звена пара-аминобензилового спирта (РАВ), который связан с (W)w посредством атома азота группы РАВ и соединен непосредственно с лекарственным средством сложноэфирной, карбонатной, карбаматной или простой эфирной группой.
Другие примеры саморасщепляющихся спейсеров включают без ограничений ароматические соединения, электронно эквивалентные группе РАВ, такие как остатки производных 2-аминоимидазол-5-метанола и орто- или пара-аминобензилацеталей. Можно использовать спейсеры, легко претерпевающие циклизацию после гидролиза амидной связи, такие как замещенные и незамещенные амиды 4-аминомасляной кислоты, замещенные соответствующим образом бицикло[2.2.1] и бицикло[2.2.2] кольцевые системы и амиды 2-аминофенилпропионовой кислоты.
В альтернативном воплощении изобретения спейсерное звено представляет собой разветвленное бис(гидроксиметил)стирольное (BHMS) звено, которое можно использовать для включения дополнительных лекарственных средств.
В конкретном воплощении изобретения спейсерное звено (Y) представляет собой РАВ-карбонил, где РАВ представляет собой 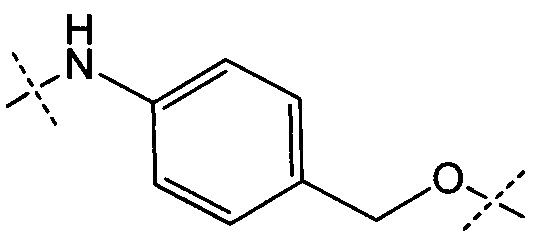 (где атом кислорода звена РАВ связан с карбонилом), и y равно 1, или в линкере отсутствует спейсерное звено (y равно 0).
(где атом кислорода звена РАВ связан с карбонилом), и y равно 1, или в линкере отсутствует спейсерное звено (y равно 0).
В конкретном воплощении изобретения линкер имеет следующую формулу (III):
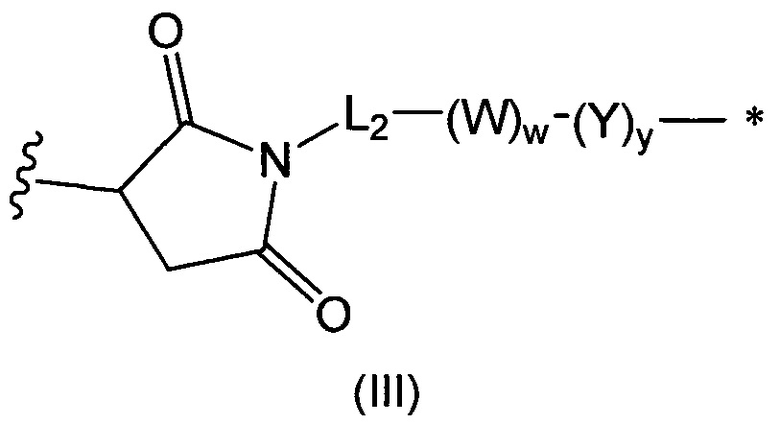
где
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил или (С2-С6)алкил-карбонил (где карбонил этих группировок при его наличии связан с (W)w),
W представляет собой аминокислотное звено, где w представляет собой целое число, составляющее от 0 до 5,
Y представляет собой РАВ-карбонил, где РАВ представляет собой
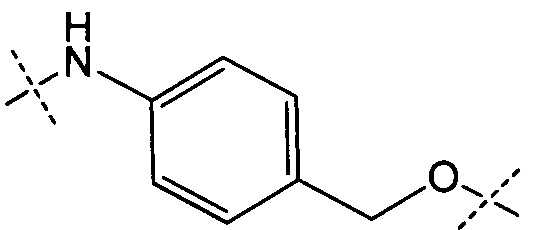 (атом кислорода звена РАВ связан с карбонилом), и y равно 0 или 1 (предпочтительно y равно 0, когда w равно 0, и y равно 0 или 1, когда w составляет от 1 до 5),
(атом кислорода звена РАВ связан с карбонилом), и y равно 0 или 1 (предпочтительно y равно 0, когда w равно 0, и y равно 0 или 1, когда w составляет от 1 до 5),
звездочкой указана точка присоединения к лекарственному средству D, и
волнистой линией указана точка присоединения к антителу Ab.
Преимущественно L2 представляет собой (С2-С6)алкил-карбонил, такой как пентил-карбонил следующей формулы:
где
звездочкой указана точка присоединения к (W)w; и
волнистой линией указана точка присоединения к атому азота малеимидной группировки.
Согласно предпочтительному воплощению изобретения линкер L выбран из:
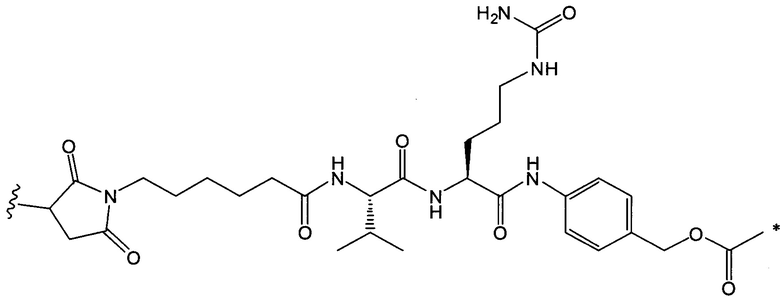
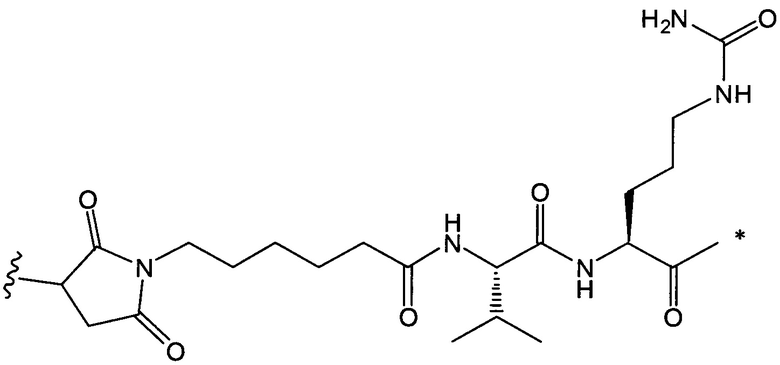
и
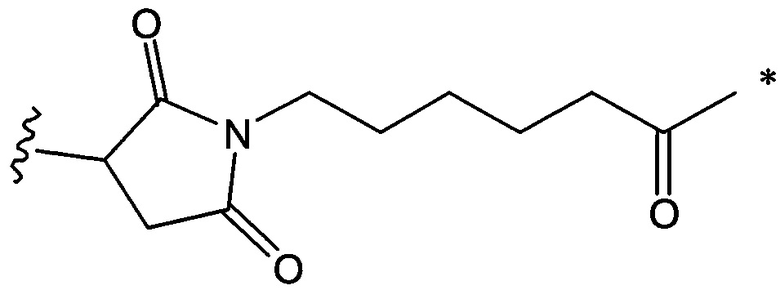
где звездочкой указана точка присоединения к лекарственному средству D, а волнистой линией указана точка присоединения к антителу Аb.
IV - Конъюгат антитела с лекарственным средством (ADC)
В предпочтительном воплощении изобретения конъюгат антитела с лекарственным средством по изобретению может быть получен любым способом, известным специалистам в данной области техники, таким как, без ограничений, i) взаимодействие нуклеофильной группы антитела с двухвалентным линкерным реагентом с последующим взаимодействием с нуклеофильной группой лекарственного средства или ii) взаимодействие нуклеофильной группы лекарственного средства с двухвалентным линкерным реагентом с последующим взаимодействием с нуклеофильной группой антитела.
Нуклеофильные группы на антителе включают без ограничений N-концевые аминные группы, аминные группы боковой цепи (например, лизина), тиольные группы боковой цепи и гидроксильные или аминогруппы сахара в случае гликозилированного антитела.
Нуклеофильные группы на лекарственном средстве включают без ограничений аминные, тиольные и гидроксильные группы, и предпочтительно аминные группы.
Аминные, тиольные и гидроксильные группы являются нуклеофильными и способны взаимодействовать с образованием ковалентных связей с электрофильными группами на линкерных группировках и линкерных реагентах, включающих без ограничений активные сложные эфиры, такие как сложные эфиры N-гидроксисукцинимиды (NHS), сложные эфиры N-гидроксибензотриазолы (HOBt), галогенформиаты и галогенангидриды; алкил- и бензилгалогениды, такие как галогенацетамиды; альдегиды; кетоны; карбоксил и малеимидные группы. Антитело может иметь восстанавливаемые межцепные дисульфидные связи, т.е. цистеиновые мостики. Антитело можно сделать активным для конъюгации с линкерными реагентами путем обработки восстанавливающим агентом, таким как ДТТ (дитиотрейтол). Каждый цистеиновый мостик, таким образом, теоретически образует две активные тиольные нуклеофильные группы. Дополнительные нуклеофильные группы можно вводить в антитело посредством любой реакции, известной специалистам в данной области техники. В качестве неограничивающего примера активные тиольные группы можно вводить в антитело путем введения одного или более остатков цистеина.
Конъюгаты антитела с лекарственным средством могут быть также получены путем модификации антитела с введением электрофильных группировок, которые могут взаимодействовать с нуклеофильными заместителями на линкерном реагенте. Сахара гликозилированного антитела могут быть окислены с образованием альдегидных или кетонных групп, которые могут взаимодействовать с аминной группой линкерных реагентов или лекарственного средства. Полученные в результате иминогруппы, представляющие собой Шиффовы основания, могут образовать стабильную связь или могут быть восстановлены с образованием стабильных аминных связей. В одном воплощении изобретения в результате взаимодействия углеводной части гликозилированного антитела либо с галактозооксидазой, либо с мета-периодатом натрия можно получить карбонильные (альдегидные или кетонные) группы в белке, которые могут взаимодействовать с соответствующими группами на лекарственном средстве. В другом воплощении изобретения белки, содержащие N-концевые остатки серина или треонина, могут взаимодействовать с мета-периодатом натрия с получением в результате альдегида вместо первой аминокислоты.
В предпочтительном воплощении изобретения конъюгат антитела с лекарственным средством по изобретению получают путем подготовки группировки лекарственное средство-линкер с последующим сочетанием между нуклеофильной группой антитела (например, группой SH цистеиновой группировки) и электрофильной группой группировки лекарственное средство-линкер (например, малеимида).
1. Лекарственное средство-линкер
Группировка лекарственное средство-линкер может быть получена путем сочетания:
- линкера с лекарственным средством,
- части линкера с лекарственным средством до завершения синтеза линкера,
- линкера с частью или предшественником лекарственного средства до завершения синтеза лекарственного средства, или
- части линкера с частью или предшественником лекарственного средства до завершения синтеза лекарственного средства.
Реакции сочетания представляют собой хорошо известные реакции для специалиста в данной области техники между нуклеофильной группой и электрофильной группой.
Нуклеофильная группа может представлять собой, в частности, аминную, тиольную или гидроксильную группу. В предпочтительном воплощении изобретения она представляет собой первичную или вторичную аминную группу.
Электрофильная группа может представлять собой карбоново-кислотную группу (СООН), необязательно в активированной форме, или активированную карбонатную сложноэфирную группировку.
Под «активированной формой» карбоновой кислоты подразумевают карбоновую кислоту, в которой группировка ОН функциональной группы СООН замещена активированной уходящей группой (LG), обеспечивающей сочетание активированной карбоново-кислотной группы с аминогруппой с целью образования амидной связи и высвобождения соединения LG-H. Активированные формы могут представлять собой активированные сложные эфиры, активированные амиды, ангидриды или галогенангидриды, такие как хлорангидриды. Активированные сложные эфиры включают производные, образованные путем взаимодействия карбоново-кислотной группы с N-гидроксибензотриазолом или N-гидроксисукцинимидом.
Под «активированным карбонатным сложным эфиром» подразумевают карбонатный сложный эфир, содержащий группировку -OC(O)OR, в которой OR представляет собой хорошую уходящую группу, обеспечивающую сочетание активированного карбонатного сложного эфира с аминогруппой с целью образования карбаматной группировки и высвобождения соединения ROH. Группа R активированного карбонатного сложного эфира включает без ограничений пара-нитрофенильную, пентафторфенильную, 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ильную и бензильную группы, предпочтительно пара-нитрофенильную и пентафторфенильную группы.
Когда линкер имеет следующую формулу (III):
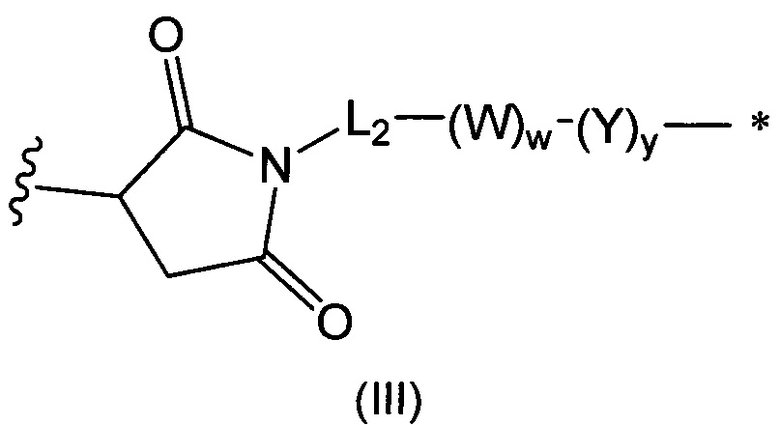
группировка лекарственное средство-линкер имеет следующую формулу (III):
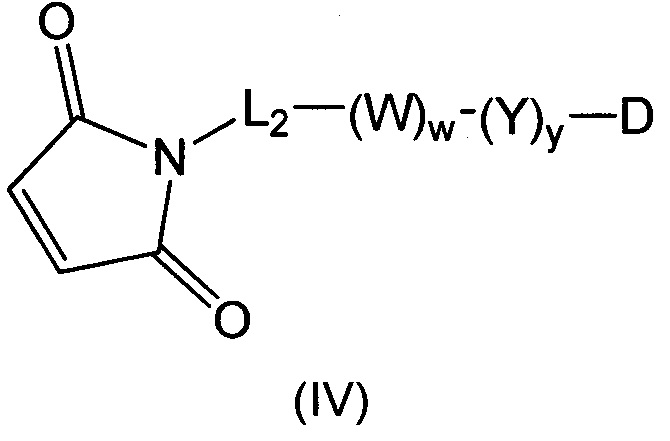
и последняя стадия синтеза группировки лекарственное средство-линкер в целом представляет собой сочетание соединения следующей формулы (V):
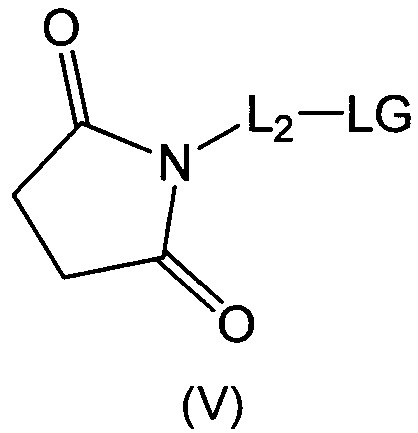
где L2 является таким, как определено выше, и LG представляет собой уходящую группу, в частности, галогенид, такой как хлорид или группу, полученную из N-гидроксисукцинимида,
и соединения следующей формулы (VI):
Когда y равно 1 и Y - РАВ-карбонил, соединение формулы (VI) может быть получено путем сочетания между лекарственным средством (DH) и соединением следующей формулы (VII), предпочтительно его защищенной формы:
где W и w являются такими, как определено выше, и R является таким, как определено в определении «активированного карбонатного сложного эфира», и G представляет собой Н или защитную группу.
Когда соединение формулы (VII) находится в защищенной форме, необходима конечная стадия удаления защиты.
Когда y равен 0, соединение (VI) имеет формулу H-(W)W-D, где (W)w и предпочтительно D состоят из аминокислотных звеньев. Следовательно, соединение (VI) может быть получено в данном случае способом традиционного пептидного синтеза, хорошо известного специалистам в данной области техники.
2. Ab-линкер-лекарственное средство
Предпочтительное воплощение согласно изобретению состоит в сочетании между цистеином, присутствующим на антителе, и электрофильной группой группировки лекарственное средство-линкер, предпочтительно с малеимидной группировкой, присутствующей на группировке лекарственное средство-линкер.
Сочетание малеимид-цистеин можно выполнять способами, хорошо известными специалистам в данной области техники.
В целом антитела не содержат большое количество или совсем не содержат свободных и активных тиольных групп цистеина, которые можно сшивать с группировкой лекарственного средства. Большинство тиольных групп цистеиновых остатков в антителах существует в виде дисульфидных мостиков и должны быть восстановлены восстанавливающим агентом, таким как дитиотрейтол (ДТТ) или трис-2-карбоксиэтилфосфин (ТСЕР), в условиях частичного или полного восстановления. Нагрузку (отношение лекарственное средство/антитело) ADC можно регулировать несколькими различными путями, включающими: (i) ограничение молярного избытка промежуточного соединения лекарственное средство-линкер (D-L) или линкерного реагента относительно антитела, (ii) ограничение времени или температуры реакции конъюгации, и (iii) условия частичного или ограниченного восстановления для модификации тиола цистеина.
Структура дисульфидной связи IgG человека в настоящее время хорошо установлена (обзор в Liu and May, mAbs 4 (2012): 17-23). В действительности имеется множество подобий и некоторые различия, касающиеся структур дисульфидной связи 4 подклассов IgG человека, а именно IgG1, IgG2, IgG3 и IgG4. Все подклассы IgG содержат 12 инвариантных внутрицепных дисульфидных мостиков, и различия заключаются в их межцепных дисульфидных связях, образованных между тяжелой и легкой цепями. Каждая внутрицепная дисульфидная связь ассоциирована с отдельным доменом IgG, т.е. вариабельными (VL и VH) и константными (CL, СН1, СН2 и СН3) доменами. 2 тяжелые цепи сшиты в их шарнирной области вариабельным числом дисульфидных мостиков: 2 для IgG1 и IgG4, 4 для IgG2 и 11 для IgG3. Тяжелые и легкие цепи IgG1 соединены дисульфидной связью между последним остатком цистеина легкой цепи и пятым остатком тяжелой цепи, тогда как для других подклассов, IgG2, IgG3 и IgG4, легкая цепь сшита с тяжелой цепью дисульфидной связью между последним остатком цистеина легкой цепи и третьим остатком цистеина тяжелой цепи, который расположен на границе доменов VH и СН1. Структуры дисульфидных связей, отличающиеся от этих классических структур, описаны для IgG2 и IgG4 (обзор в Liu and May, mAbs 4 (2012): 17-23). Межцепные дисульфидные связи в высокой степени подвергаются воздействию растворителя и, следовательно, значительно более активны, чем внутрицепные дисульфидные связи, которые углублены в антипараллельных структурах бета-слоя внутри каждого домена и не подвергаются воздействию растворителя. По этим причинам независимо от изотипа антитела сочетание будет происходить на межцепных открытых остатках цистеина после восстановления в мягких условиях. Таким образом, каждый межцепной дисульфидный мостик может теоретически образовать два центра конъюгации.
Дополнительные нуклеофильные группы можно вводить в антитела посредством взаимодействия лизинов с 2-иминотиоланом (реагентом Трота), приводящего в результате к преобразованию амина в тиол. Активные тиольные группы также можно вводить в антитело (или его фрагмент) путем конструирования одного, двух, трех, четырех или более остатков цистеина (например, получения мутантных антител, содержащих один или более ненативных аминокислотных остатков цистеина). В патенте US 7521541 заявлено конструирование антител путем введения активных цистеиновых аминокислот.
Цистеиновые аминокислоты могут быть сконструированы в активных центрах в антителе, и которые не образуют внутрицепные или межмолекулярные дисульфидные сшивки (Junutula, et al., 2008b Nature Biotech., 26(8): 925-932; Dornan et al (2009) Blood 114(13): 2721-2729; US 7521541; US 7723485; WO 2009/052249). Сконструированные тиолы цистеина могут взаимодействовать с линкерными реагентами или с реагентами лекарственное средство-линкер по настоящему изобретению, которые имеют взаимодействующие с тиолом, электрофильные группы, такие как малеимид, или альфа-галогенамиды с образованием ADC с антителами со сконструированными цистеинами и группировками лекарственного средства. Таким образом, положение группировки лекарственного средства может быть сконструировано, отрегулировано и известно. Нагрузку лекарственным средством можно регулировать, поскольку сконструированные тиольные группы цистеина в характерном случае взаимодействуют с линкерными реагентами, взаимодействующими с тиолом, или реагентами лекарственное средство-линкер с высоким выходом. В результате конструирования IgG антитела с введением аминокислоты цистеина путем замены в одном сайте на тяжелой или легкой цепи получают два новых цистеина на симметричном антителе. Нагрузка лекарственным средством около 2 может быть достигнута почти с однородностью продукта конъюгации ADC.
Где более чем одна нуклеофильная или электрофильная группа антитела взаимодействует с промежуточным соединением лекарственное средство-линкер или линкерным реагентом с последующим взаимодействием с реагентом группировки лекарственного средства, полученный в результате продукт представляет собой смесь соединений ADC с распределением группировок лекарственного средства, присоединенных к антителу, например, 1, 2, 3 и т.д. Способами жидкостной хроматографии, такими как полимерная обращенная фаза (ПЛОФ) и гидрофобная интерактивная хроматография (ГИХ), можно разделять соединения в смеси по величине нагрузки лекарственным средством. Можно выделить препараты ADC с одной величиной нагрузки лекарственным средством (р), однако, эти ADC с одной величиной нагрузки лекарственным средством могут все же представлять собой гетерогенные смеси, поскольку группировки лекарственного средства могут быть присоединены посредством линкера в различных сайтах на антителе.
Для некоторых конъюгатов антитело-лекарственное средство долю лекарственного средства можно ограничить числом сайтов присоединения на антителе. Высокая нагрузка лекарственным средством, например, доля лекарственного средства более 5, может вызвать агрегацию, нерастворимость, токсичность или потерю клеточной проницаемости некоторых конъюгатов антитело-лекарственное средство. Как правило, в ходе реакции конъюгации с антителом конъюгируют меньшее число группировок лекарственного средства, чем теоретический максимум.
Нагрузка лекарственным средством, также называемая отношением лекарственного средства к антителу (DAR), представляет собой среднее число группировок лекарственного средства на клеточно-связывающий агент.
В случае изотипов антитела IgG1 и IgG4, где лекарственные средства связывают с цистеинами после частичного восстановления антитела, нагрузка лекарственным средством может находиться в диапазоне от 1 до 8 группировок лекарственного средства (D) на антитело, т.е. где 1, 2, 3, 4, 5, 6, 7 и 8 группировок лекарственного средства ковалентно присоединены к антителу.
В случае изотипа антитела IgG2, где лекарственные средства связывают с цистеинами после частичного восстановления антитела, нагрузка лекарственным средством может находиться в диапазоне от 1 до 12 группировок лекарственного средства (D) на антитело, т.е. где 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 и 12 группировок лекарственного средства ковалентно присоединены к антителу.
Композиции ADC включают коллекции клеточно-связывающих агентов, например, антител, конъюгированных с лекарственными средствами в количестве, находящемся в диапазоне от 1 до 8 или от 1 до 12.
Среднее число группировок лекарственного средства на антитело в препаратах ADC из реакций конъюгации можно охарактеризовать традиционными методами, такими как УФ-спектрометрия, высокоэффективная жидкостная хроматография (ВЭЖХ) с обращенной фазой, ГИХ, масс-спектрометрия, количественный анализ ИФА и электрофорез.
В качестве неограничивающего воплощения изобретения в настоящем документе представлена конъюгация с антителом C208F2. В этом случае лекарственное средство подвергают сочетанию по меньшей мере с одним цистеином, выбранным из i) остатка Cys в положении 214 легкой цепи последовательности SEQ ID NO 28 и ii) остатков Cys в положении 223, 229 и 232 тяжелой цепи последовательности SEQ ID NO 23.
В качестве неограничивающего воплощения изобретения в настоящем документе представлена конъюгация с антителом C208F2. В этом случае лекарственное средство подвергают сочетанию с двумя, тремя или четырьмя цистеинами, выбранными из i) остатка Cys в положении 214 легкой цепи последовательности SEQ ID NO 28 и ii) остатков Cys в положении 223, 229 и 232 тяжелой цепи последовательности SEQ ID NO 23.
В качестве неограничивающего воплощения изобретения в настоящем документе представлена конъюгация с антителом hz208F2. (вар. 1). В этом случае лекарственное средство подвергают сочетанию по меньшей мере с одним цистеином, выбранным из i) остатка Cys в положении 214 легкой цепи последовательности SEQ ID NO 39 и ii) остатков Cys в положении 223, 229 и 232 тяжелой цепи последовательности SEQ ID NO 37.
В качестве неограничивающего воплощения изобретения в настоящем документе представлена конъюгация с антителом hz208F2 (вар. 3). В этом случае лекарственное средство подвергают сочетанию с двумя, тремя или четырьмя цистеинами, выбранными из i) остатка Cys в положении 214 легкой цепи последовательности SEQ ID NO 40 и ii) остатков Cys в положении 223, 229 и 232 тяжелой цепи последовательности SEQ ID NO 38.
Альтернатива состоит в сочетании лизина. Антитело может содержать, например, многие остатки лизина, которые не взаимодействуют с промежуточным соединением лекарственное средство-линкер (D-L) или с линкерным реагентом. Только наиболее активные группы лизина могут взаимодействовать с линкерным реагентом, реагирующим с амином. Также только наиболее активные тиольные группы цистеина могут взаимодействовать с линкерным реагентом, реагирующим с тиолом.
В случае связывания соединений по изобретению с лизинами нагрузка лекарственным средством может находиться в диапазоне от 1 до 80 группировок лекарственного средства (D) на клеточное антитело, хотя может быть предпочтителен верхний предел 40, 20, 10 или 8. Композиции ADC включают коллекции клеточно-связывающих агентов, например, антител, конъюгированных с лекарственными средствами в количестве, находящемся в диапазоне от 1 до 10 или от 1 до 8.
ADC формулы (I) согласно изобретению может находиться в форме фармацевтически приемлемой соли.
В настоящем изобретении под «фармацевтически приемлемым» подразумевают то, что можно применять при получении фармацевтической композиции, в целом безопасной, нетоксичной и ни биологически, ни иначе нежелательной, и приемлемой как для ветеринарного применения, так и для фармацевтического применения для человека.
Под «фармацевтически приемлемой солью» соединения подразумевают соль, которая является фармацевтически приемлемой, как определено в настоящем документе, и которая обладает желаемой фармакологической активностью исходного соединения.
Фармацевтически приемлемые соли, в частности, включают:
(1) соли присоединения фармацевтически приемлемой кислоты, образованные с фармацевтически приемлемыми неорганическими кислотами, такими как хлористоводородная, бромистоводородная, фосфорная, серная и подобные кислоты; или образованные с фармацевтически приемлемыми органическими кислотами, такими как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая, молочная и подобные кислоты; и
(2) соли присоединения фармацевтически приемлемого основания, образованные в случае, когда протон кислоты, присутствующий в исходном соединении, либо замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координирован с фармацевтически приемлемым органическим основанием, таким как лизин, аргинин и тому подобное; либо с фармацевтически приемлемым неорганическим основанием, таким как гидроксид натрия, поташ, гидроксид кальция и тому подобное.
Эти соли могут быть получены из соединений по изобретению, содержащих основную или кислотную функциональную группу, и соответствующих кислот или оснований с использованием традиционных химических методов.
V - Лечение
Наконец, изобретение относится к ADC, как описано выше, для применения в качестве лекарственного средства, в частности, при лечении рака.
Следующим объектом настоящего изобретения является соединение формулы (I), такое как определено выше, для применения в качестве лекарственного препарата, в частности, для лечения рака.
Настоящее изобретение также относится к применению соединения формулы (I), такого как определено выше, для получения лекарственного препарата, в частности, предназначенного для лечения рака.
Настоящее изобретение также относится к способу лечения рака, включающему введение человеку, нуждающемуся в этом, эффективного количества соединения формулы (I), такого как определено выше.
Формы рака могут быть предпочтительно выбраны из IGF-IR-обусловленных форм рака, включающих опухолевые клетки, экспрессирующие или обладающие гиперэкспрессией полноразмерного IGF-1R или его участка на их поверхности.
Более конкретно эти формы рака представляют собой рак молочной железы, рак ободочной кишки, карциному пищевода, печеночно-клеточный рак, рак желудка, глиому, рак легкого, меланому, остеосаркому, рак яичника, рак предстателньой железы, рабдомиосаркому, рак почки, рак щитовидной железы, рак эндометрия матки, шванному, нейробластому, плоскоклеточный рак полости рта, мезотелиому, лейомиосаркому и любые лекарственно-устойчивые явления или формы рака.
Во избежание сомнений под лекарственно-устойчивыми IGF-1R-экспрессирующими формами рака необходимо понимать не только устойчивые раки, которые исходно экспрессируют IGF-1R, но также формы рака, которые исходно не проявляют экспрессии или гиперэкспрессии IGF-1R, но начинают экспрессировать IGF-1R, как только становятся устойчивыми к предшествующей терапии.
Другим объектом изобретения является фармацевтическая композиция, содержащая ADC, как раскрыто в описании.
Более конкретно изобретение относится к фармацевтической композиции, содержащей ADC по изобретению, по меньшей мере с эксципиентом и/или фармацевтически приемлемым носителем.
В настоящем описании выражение «фармацевтически приемлемый носитель» или «эксципиент» предназначен для обозначения соединения или комбинации соединений, входящих в фармацевтическую композицию, не вызывающих вторичных реакций, и которые дают возможность, например, облегчить введение активного (-ых) соединения (-ий), увеличить продолжительность его (их) пребывания и/или эффективности в организме, увеличить его (их) растворимость в растворе или, кроме того, улучшить его (их) сохранность. Эти фармацевтически приемлемые носители и эксципиенты хорошо известны и приспособлены специалистом в данной области техники в зависимости от их природы и выбранного способа введения активного (-ых) соединения (-ий).
Активный ингредиент можно вводить единичных формах введения, в смеси с традиционными фармацевтическими носителями, животным или людям. Подходящие единичные формы введения включают формы посредством перорального пути и формы для введения посредством парентерального пути (подкожного, внутрикожного, внутримышечного или внутривенного).
В качестве твердых композиций для перорального введения можно применять таблетки, пилюли, порошки (твердые или мягкие желатиновые капсулы) или гранулы. В этих композициях активный ингредиент по изобретению смешивают с одним или более инертных разбавителей, таких как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния, в потоке аргона. Эти композиции могут также содержать вещества, отличающиеся от разбавителей, например одно или более смазывающих веществ, таких как стеарат магния или тальк, красящее вещество, покрытие (таблетки с покрытием) или глазурь.
Стерильные композиции для парентерального введения могут предпочтительно представлять собой водные или неводные растворы, суспензии или эмульсии. В качестве растворителя или носителя можно применять воду, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое масло, инъекционные органические сложные эфиры, например, этилолеат, или другие подходящие органические растворители. Эти композиции могут также содержать адъюванты, в частности, увлажняющие, изотонические, эмульгирующие, диспергирующие и стабилизирующие средства. Стерилизацию можно выполнять несколькими путями, например, путем стерилизационного фильтрования, путем включения стерилизующих агентов в композицию, путем облучения или путем нагревания. Их можно также готовить в форме твердых стерильных композиций, которые можно растворять во время применения в стерильной воде или любой другой инъекционной стерильной среде.
Предпочтительно эти ADC будут вводить системным путем, в частности, внутривенным путем, внутримышечным, внутрикожным, интраперитонеальным или подкожным путем, либо пероральным путем. Более предпочтительно композицию, содержащую ADC согласно изобретению, будут вводить несколько раз последовательно.
Изобретение, таким образом, также относится к набору, содержащему по меньшей мере i) конъюгат антитела с лекарственным средством и/или фармацевтическую композицию согласно изобретению и ii) шприц, или флакон, или ампулу, в которые помещают конъюгат антитела с лекарственным средством и/или фармацевтическую композицию.
Способы ее введения, дозировки и оптимальные фармацевтические формы могут быть определены в соответствии с критериями, обычно принимаемыми во внимание при установлении терапии, адаптированной для пациента, такими как, например, возраст или масса тела пациента, серьезность его/ее общего состояния, переносимость терапии и отмеченных вторичных эффектов.
Другие характеристики и преимущества изобретения очевидны при продолжении описания примерами и графическими материалами, подписи к которым приведены ниже.
Подписи к графическим материалам
Фигуры 1А-1С: Связывание антитела с нативным IGF-1R человека на основании анализов FACS. На Фиг. 1А представлена кривая титрования на линии клеток MCF-7. MFI представляет собой среднюю интенсивность флуоресценции. На Фиг. 1 В представлена ЕС50 обоих антител к IGF-1R, мышиного и химерного, на линии клеток MCF-7. На Фиг. 1С представлена Bmax химерных антител к IGF-1R на линии клеток MCF-7.
Фигуры 2А-2В: Оценка распознавания hIGF-1R с использованием трансфицированных клеток по сравнению с нетрансфицированными. На фиг. 2А) представлены кривые титрования одного химерного Ab к IGF-1R на линии клеток IGF-1R+. MFI представляет собой среднюю интенсивность флуоресценции. На фиг. 2В представлено связывание химерных Ab к IGF-1R на линии клеток IGF-1R- человека.
Фигуры 3А-3В: Оценка специфичности Ab к IGF-1R по сравнению с hIR с использованием трансфицированных клеток. На фиг. 3А представлено связывание мышиного Ab к IGF-1R на линии трансфицированных клеток hIR+. На фиг. 3В представлено связывание химерного Ab к IGF-1R на линии клеток IR+. MFI представляет собой среднюю интенсивность флуоресценции. Mab GRO5 к hIR (Calbiochem) было введено в качестве положительного контроля.
Фигура 4: Связывание мышиного Ab к IGF-1R на линии клеток IM-9. MFI представляет собой среднюю интенсивность флуоресценции. Mab GRO5 к hIR было введено в качестве положительного контроля.
Фигуры 5А-5С: Оценка распознавания IGF-1R обезьяны. На фиг. 5А представлены кривые титрования химерного Ab к IGF-1R на линии клеток COS-7. MFI представляет собой среднюю интенсивность флуоресценции. На Фиг. 5В представлена ЕС50 обоих антител к IGF-1R, мышиного и химерного, на линии клеток COS-7. На Фиг. 5С представлена ЕС50 химерных антител к IGF-1R на обеих линиях клеток - трансфицированных клетках NIH 3Т3 hIGF-1R+ и клетках COS-7.
Фигура 6: Сенсограммы, полученные с помощью Biacore Х100 на основе технологии поверхностного плазмонного резонанса (SPR), используя сенсорный чип СМ5, активированный более 11000 резонансных единиц (РЕ) мышиного антитела к гистидиновой метке, привитого химическим путем на карбоксиметилдекстрановом матриксе. Эксперимент проводили при скорости тока 30 мкл/мин при 25°С, используя HBS-EP+ в качестве буфера для хроматографии и для разведения проб. На фигуре показано совмещение 4 независимых сенсограмм, выровненных по х-оси в начале первого ввода проб аналитов и по y-оси по исходному уровню, определенному непосредственно перед этим первым вводом пробы. Сенсограммы, полученные с захватом последовательности на основе последовательности рекомбинантного растворимого IGF1R человека, отмечены ромбиками. Сенсограммы, полученные с захватом последовательности на основе последовательности рекомбинантного растворимого IGF1R яванского макака, отмечены треугольниками. Белые символы соответствуют циклам холостой пробы (5 вводов пробы буфера для хроматографии), а черные символы соответствуют вводам ряда возрастающих концентраций C208F2 (5, 10, 20, 40 и 80 нМ).
Фигура 7: Оценка собственного действия антител к hIGF-1R фосфорилирование рецептора по сравнению с IGF1.
Фигура 8: Ингибирование фосфорилирования IGF-1R в ответ на IGF-1 мышиным антителом к hIGF-1R.
Фигура 9: Интенсивность сигнала клеточной поверхности антител к IGF-1R претерпевает понижающую регуляцию после инкубации клеток при 37°С. Клетки MCF-7 инкубировали при 4°С или 37°С в течение 4 ч с 10 мкг/мл Ab. На фигуре представлена ΔMFI.
Фигуры 10А-10В: Распад антител клеточной поверхности. Связанное с клеточной поверхностью антитело оценивали после 10, 20, 30, 60 и 120 мин при 37°С. На Фиг. 10А представлен % остаточного IGF-1R в сравнении с интенсивностью сигнала, измеренной при 4°С. На Фиг. 10В представлено вычисление периода полувыведения с использованием программного обеспечения Prims и с использованием совпадения с экспоненциальной кривой распада.
Фигура 11: Ab к hIGF-1R интернализуются. Клетки инкубировали с 10 мкг/мл мышиных Ab в течение 0, 30 или 60 мин при 37°С. Клетки подвергали или не подвергали пермеабилизации и инкубировали со вторым антимышиным IgG-Alexa 488. Мембрана соответствует пермеабилизации масс/об. интенсивности сигнала. Общий сигнал соответствует интенсивности сигнала после пермеабилизации клеток, а цитоплазматический соответствует интернализованному Ab. Название каждого оцениваемого антитела изображено сверху каждого графика.
Фигуры 12А-12В: Визуализация интернализации Ab. Фигура 12А: Клетки MCF-7 инкубировали с m208F2 в течение 20 мин при 4°С и промывали, после чего инкубировали (W) при 37°С в течение 15 (X), 30 (Y) и 60 (Z) мин. Клетки фиксировали и подвергали пермеабилизации. Ab m208F2 выявляли с использованием антимышиного IgG Alexa488 и Lamp-1, используя кроличье антитело к Lamp-1 и второй антикроличий IgG Alexa 555. Фигура 12В: Клетки MCF-7 инкубировали в течение 30 минут при 37°С с мышиными антителами к hIGF-1R и коаршивали, как описано выше. Колокализацию идентифицировали, используя маркерный плагин колокализации программы ImageJ.
Фигура 13: Вовлечение лизосомального биохимического пути в разложение антитела.
Фигура 14: Кислый рН уменьшает связывающую способность пяти мышиных антител к IGF-1R.
Фигуры 15A-15D: Связывание, характеристическое для первой гуманизированной формы Mab C208F2. Связывающие свойства hz208F2 VH3/VL3 mAb оценивали на линии клеток человека MCF-7 (А), на линии клеток обезьяны COS-7 (В) и на трансфицированной линии клеток мыши, экспрессирующей рецептор инсулина человека (С). Связывание обоих mAb, мышиного и химерного 208F2, оценивали параллельно. Антитело к hIR, клон GRO5, использовали для подтверждения экспрессии hIR на трансфицированной линии клеток (D).
Фигура 16: Распад поверхностного антитела hz208F2 VH3/VL3
Фигура 17: Совмещение сенсограмм, полученных с помощью устройства Biacore Х100 на основе SPR при температуре 25°С с сенсорным чипом СМ5, активированным на обоих проточных кюветах приблизительно 12000 РЕ мышиных моноклональных антител к гистидиновой метке, привитых химическим путем на карбоксиметилдекстрановом матриксе, используя HBS-EP+ в качестве буфера для хроматографии при скорости тока 30 мкл/мин. Каждая сенсограмма (первая отмечена треугольниками, а вторая отмечена ромбиками) соответствует полному циклу:
1 - Ввод в течение одной минуты пробы раствора рекомбинантного h-IGF-1R (10 мкг/мл) во вторую проточную кювету.
2 - Для первой сенсограммы 5 вводов пробы буфера для хроматографии в течение 90 с каждый.
Для второй сенсограммы: пять вводов пробы в ряде возрастающих концентраций растворов антитела C208F2 к IGF-1R в течение 90 с каждый.
3 - Отсрочка 300 с для определения кинетики скорости диссоциации.
4 - Регенерация поверхности путем ввода в течение 45 с 10 мМ буфера глицин, HCl рН 1,5.
Фигура 18: Сенсограмма, соответствующая вычитанию холостой сенсограммы (5 вводов HBS-EP+) из сенсограммы, полученной для ряда возрастающих концентраций растворов антител C208F2 к IGF-1R, представлена серым цветом. Теоретическая сенсограмма, соответствующая модели 1:1 со следующими параметрами: kon=(1,206±0,036)×106 М-1⋅с-1, koff=(7,81±0,18)×10-5 с-1, Rmax=307,6±0,3 РЕ представлена тонкой черной линией. Вычисленные концентрации C208F2 приведены на графике: только самую высокую концентрацию (24 нМ) рассматривают в качестве константы).
Фигура 19: Константы диссоциации, соответствующие среднему из четырех экспериментов, проведенных для каждого антитела, и соответствующие отношению: koff/kon×1012, выраженному в единицах пМ. Усы диаграммы соответствуют стандартной ошибке (n равно 4).
Фигура 20: Периоды полужизни соответствуют среднему из четырех экспериментов, проведенных для каждого антитела, и соответствуют отношению: Ln(2)/koff/3600, выраженному в единицах ч. Усы диаграммы соответствуют стандартной ошибке (n равно 4).
Фигура 21: Клеточная цитотоксичность антитела к IGF-1R при сочетании с тремя различными соединениями. Пять химерных антител к IGF-1R подвергали сочетанию с Е-13, G-13 или F-63. Нерелевантное антитело c9G4 также подвергали сочетанию с теми же соединениями.
Фигуры 22А-22С: Оценка in vivo C208F2-E-13 (Фиг. 22А), C208F2-G-13 (Фиг. 22В) и C208F2-F-63 (Фиг. 22С) в модели ксенотрансплантата MCF-7.
Фигуры 23А-23В: Оценка in vivo обоих антител C208F2-E-13 (Фиг. 23А) и C208F2-G-13 (Фиг. 23В) в сравнении с контролем ADC (C9G4-E13 и C9G4-G-13) в модели ксенотрансплантата MCF-7.
Фигуры 24А и В: Кислый рН уменьшает специфичность связывания гуманизированных антител к IGF-1R hz208F2 H076/L024 (А) и hz208F2 (H077/L018 (В). Фигура 25: Оценка цитотоксичности C208F2-G-13 на нормальных клетках.
Фигура 26: Клеточная цитотоксичность гуманизированных вариантов hz208F2 в сочетании с G-13. Нерелевантное антитело c9G4 также подвергали сочетанию с тем же соединением.
Фигура 27: Оценка in vivo гуманизированных форм 208F2-G-13 по сравнению с C208F2-G-13 в модели ксенотрансплантата MCF-7.
Фигуры 28А и 28В: Оценка in vivo либо C208F2-G-13 (28А), либо hz208F2-4-G-13 (28 В), введенных 4 раза, по сравнению с одним вводом в модели ксенотрансплантата MCF-7.
Фигуры 29А и 29В: Оценка in vivo C208F2-E-13 (29А) и C208F2-G-13 (29В) в модели ксенотрансплантата CaOV-З.
ПРИМЕРЫ
Пример 1: Получение мышиных антител, индуцированных против IGF-1RECD
Для получения мышиных моноклональных антител (Mab) против человеческого внеклеточного домена (ECD) рецептора IGF-1 человека (hIGF-1R) 5 мышей BALB/c иммунизировали 3 раза подкожно (s.c.) 10 мкг белка rhIGF-1R (R&D Systems, № по каталогу 391-GR). В качестве альтернативы у некоторых животных было проведено три дополнительных иммунизации 10 мкг мышиного внеклеточного домена (ECD) IGF-1R (R&D Systems, № по каталогу 6630-GR/Fc). Первая иммунизация была выполнена в присутствии полного адъюванта Фрейнда (Sigma, Сент-Луис, штат Мэриленд, США). Для последующих иммунизаций добавляли неполный адъювант Фрейнда (Sigma). Через три дня после слияния иммунизированным мышам проводили бустер-инъекцию 10 мкг белка rhIGF-1R. Спленоциты и лимфоциты получали путем перфузии селезенки и путем измельчения проксимальных лимфатических узлов соответственно, собранных от одной из 5 иммунизированных мышей (отобранной после титрования сыворотки всех мышей) и проводили слияние с клетками миеломы SP2/0-Ag14 (АТСС, Роквилл, штат Мэриленд, США). Протокол слияния описан в статье Kohler and Milstein (Nature, 256: 495-497, 1975). Затем слитые клетки подвергали отбору на среде с гипоксантином, аминоптерином и тимидином (HAT). Как правило, для получения моноклональных антител или их функциональных фрагментов, особенно мышиного происхождения, можно сделать ссылку на методы, которые описаны, в частности, в руководстве "Antibodies" (Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor NY, pp. 726, 1988). Приблизительно через 10 дней после слияния гибридные клетки подвергали скринингу. Для первичного скрининга супернатанты гидридом оценивали на секрецию Mab, индуцированных против белка IGF-1R ECD, с помощью анализа FACS с использованием опухолевых клеток MCF7 рака молочной железы человека (АТСС) и/или клеток обезьяны COS7 (трансформированных SV40 клеток почек африканской зеленой мартышки), экспрессирующих IGF-1R на их клеточной поверхности. Точнее, для отбора с помощью проточной цитометрии 105 клеток (либо MCF7, либо COS7) высевали в каждую лунку 96-луночного планшета в фосфатно-солевом буфере (ФСБ), содержащем 1% бычий сывороточный альбумин (БСА) и 0,01% азид натрия (буфер FACS) при 4°С. После 2 мин центрифугирования при 2000 об/мин буфер удаляли и добавляли тестируемые супернатанты гибридомы. После 20 мин инкубации при 4°С клетки дважды промывали и добавляли Alexa 488-конъюгированное козье антимышиное антитело 1/500°, разведенное в буфере FACS (#А11017, Molecular Probes Inc., Юджин, США) и инкубировали в течение 20 мин при 4°С. После окончательной отмывки буфером FACS клетки анализировали с помощью FACS (Facscalibur, Becton-Dickinson) после добавления в каждую пробирку пропидия йодида при конечной концентрации 40 мкг/мл. В качестве отрицательного контроля включали лунки, содержащие только клетки и клетки, проинкубированные со вторым Alexa 488-конъюгированным антителом. В каждом эксперименте использовали контроль изотипа (Sigma, ref M90351MG). Для вычисления среднего значения интенсивности флуоресценции (MFI) оценивали 5000 клеток.
С целью отбора только интернализующих антител дополнительно проводили анализ интернализации. Для этого анализа линию опухолевых клеток MCF7 культивировали в среде RMPI 1640 без фенолового красного с 1% L-глутамином и 10% FACS в течение 3 дней до эксперимента. Затем клетки отсоединяли, используя трипсин, и 100 мкл клеточной суспензии высевали при 4⋅105 клеток/мл в 96-луночные планшеты в среде RPMI1640 без фенолового красного с 1% L-глутамином и 5% фетальной бычьей сывороткой (ФБС). После 2 мин центрифугирования при 2000 об/мин клетки ресуспендировали в 50 мкл либо супернатантов гибридомы, либо растворов контрольного антитела (положительный контроль и контроль изотипа 1 мкг/мл). После 20 мин периода инкубации при 4°С клетки центрифугировали в течение 2 мин при 2000 об/мин и ресуспендировали либо в холодной (4°С), либо в теплой (37°С) полной культуральной среде. Затем клетки инкубировали в течение 2 часов либо при 37°С, либо при 4°С. Затем клетки промывали три раза буфером FACS. Alexa 488-меченое антитело козий антимышиный IgG инкубировали в течение 20 минут, и клетки промывали три раза, после чего проводили анализ FACS на популяции клеток, не окрашивающихся пропидия йодидом.
После анализа FACS определяли два параметра: (i) разность сигнала флуоресценции, определяемого на поверхности клеток, проинкубированных при 4°С, и полученного с клетками, проинкубированными при 37°С с одним супернатантом гибридомы, и (ii) процент остаточного IGF-1R на клеточной поверхности.
Процент остаточного hIGF-1R вычисляют следующим образом: % остаточного IGF-1R=(MFIAb37°C/MFIAb4°C)×100.
Кроме того, было проведено три твердофазных иммуноферментных анализа (ИФА) (либо до, либо после клонирования) для исследования связывания антител на рекомбинантных белках человека (hIGF-1R) и мыши (mlGF-1R) и на рекомбинантном белке рецептора инсулина человека (hIR). Сохраняли гибридому, секретирующую антитело, проявляющее связывание на rh- и/или rm-IGF-1R и отсутствие связывания на rhIR. Кратко, в 96-луночные планшеты ELISA (Costar 3690, Корнинг, штат Нью-Йорк, США) наносили 100 мкл/лунка либо белка rhIGF-1R (R&D Systems, № по каталогу 391-GR) концентрации 0,6 мкг/мл, либо белка rmIGF-1R (R&D Systems, № по каталогу 6630-GR/Fc) при 1 мкг/мл, либо белка rhIR (R&D Systems, № по каталогу 1544-IR/CF) концентрации 1 мкг/мл в ФСБ в течение ночи при 4°С. Затем планшеты блокировали ФСБ, содержащим 0,5% желатин (№22151, Serva Electrophoresis GmbH, г. Гейдельберг, Германия) и прочно связывали в течение 2 ч при 37°С. После удаления буфера насыщения путем постукивания перевернутых планшетов в каждую лунку добавляли 100 мкл каждого разведения супернатанта (либо неразведенного супернатанта гибридомы, либо серийных разведений супернатанта) и инкубировали в течение 1 ч при 37°С. После трех промывок добавляли 100 мкл конъюгированного с пероксидазой хрена поликлонального козьего антимышиного IgG (№115-035-164, Jackson Immuno-Research Laboratories, Inc., Уэст-Гров, штат Пенсильвания, США) добавляли при разведении 1/5000 в ФСБ, содержащем 0,5% желатин и 0,05% Твин 20 (масс/масс.) в течение 1 ч при 37°С. Затем планшеты для ИФА промывали 3 раза и добавляли тетраметилбензидиновый субстрат (ТМБ) (№ UP 664782, Uptima, Interchim, Франция). После 10 мин периода инкубации при комнатной температуре реакцию останавливали, используя 1 М серную кислоту, и измеряли оптическую плотность при 450 нм.
Гибридому, секретирующую представляющее интерес антитело, размножали и клонировали путем предельного разведения. После изотипирования по одному клону под каждым кодом размножали и замораживали. Каждое представляющее интерес антитело получали в системах продуцирования in vitro, называемых CellLine (Integra Biosciences), для дальнейшей характеризации.
Дополнительные количественные определения для анализов направленной связывающей специфичности FACS проводили на клетках IM9 (В-лимфобластах человека, экспрессирующих IR), а также на клетках, трансфицированных hIGF-1R, по сравнению с нетрансфицированными клетками.
Все данные, соответствующие отобранным антителам, суммированы в таблице 7, и показано, что пять отобранных антител характеризуются сильным распознаванием нативного человеческого IGF-1R, экспрессируемого либо на клетках MCF-7 рака молочной железы, либо на трансфицированных клетках. Они также распознают IGF-1R обезьяны на клетках COS-7. Эти антитела перекрестно не реагируют с рецептором инсулина человека, экспрессируемого на клетках IM9 в высокой степени. Следует отметить, что эти антитела слабо распознают белок rhIGF-1R ECD при непосредственном нанесении на планшеты ИФА.
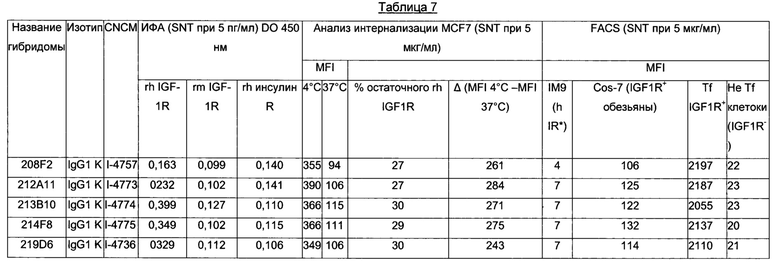
Пример 2: Связывание антитела с нативным IGF-1R человека на основании анализов FACS
Пять мышиных антител к IGF-1R были химеризованы. Связывающие свойства обоих антител к IGF-1R, мышиного и химерного, оценивали с помощью анализов FACS на линии клеток MCF-7 аденокарциномы молочной железы человека (АТСС#НТВ-22), используя возрастающие концентрации антитела. С этой целью клетки (1×106 клеток/мл) инкубировали с антителами к IGF-1R в течение 20 мин при 4°С в буфере FACS (ФСБ, 0,1% БСА, 0,01% NaN3). Затем их промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488, в течение 20 дополнительных минут при 4°С в темноте, после чего промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). Максимальная интенсивность сигнала, полученная с каждым антителом, была обозначена как Bmax и выражена как средняя интенсивность флуоресценции (MFI). ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0).
Кривая титрования каждого мышиного или химерного Ab продемонстрировала, что все полученные антитела способны распознавать нативную форму IGF-1R с типичным профилем насыщения (Фиг. 1А). С целью ранжирования антител и сравнения связывающих свойств обоих Ab, мышиного и химерного, ЕС50 связывания каждого соединения определяли с использованием анализа нелинейной регрессии. Сравнение ЕС50 каждого мышиного Ab с его соответствующей химерной формой показало, что 2 формы проявляли одинаковые связывающие свойства, демонстрируя, что химеризация Ab не влияет на распознавание IGF-1R (Фиг. 1В-С). Значения ЕС50 и Bmax химерных антител суммированы в таблице 8.
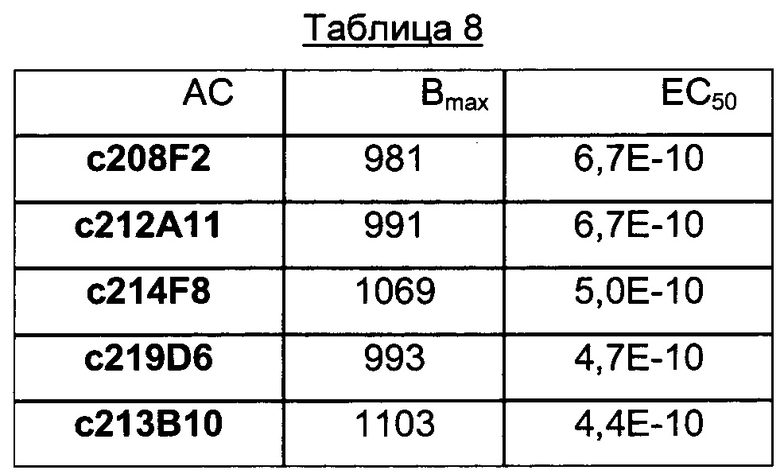
Пример 3: Подтверждение специфичности антитела путем использования клеток, трансфицированных либо IGF-1R, либо IR, или клеток IM9. экспрессирующих значительные уровни IR
Чтобы подтвердить специфичность полученных антител к IGF-1R по сравнению с IR, стабильные трансформанты, экспрессирующие либо hIGF-1R, либо hIR, оценивали с помощью анализов FACS. Кратко, химерные mAb в возрастающих концентрациях инкубировали с клетками в течение 20 мин при 4°С в буфере FACS (ФСБ, 0,1% БСА, 0,01% NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488 перед инкубацией в течение 20 дополнительных минут при 4°С в темноте, а затем промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0).
Кривые титрования, полученные на линии клеток, трансфицированных hIGF-1R (Фиг. 2А), по сравнению с нетрансфицированными клетками (Фиг. 2В) подтвердили специфичность связывания химерных Ab с IGF-1R человека. Значения ЕС50 и Bmax химерных антител суммированы в таблице 9.
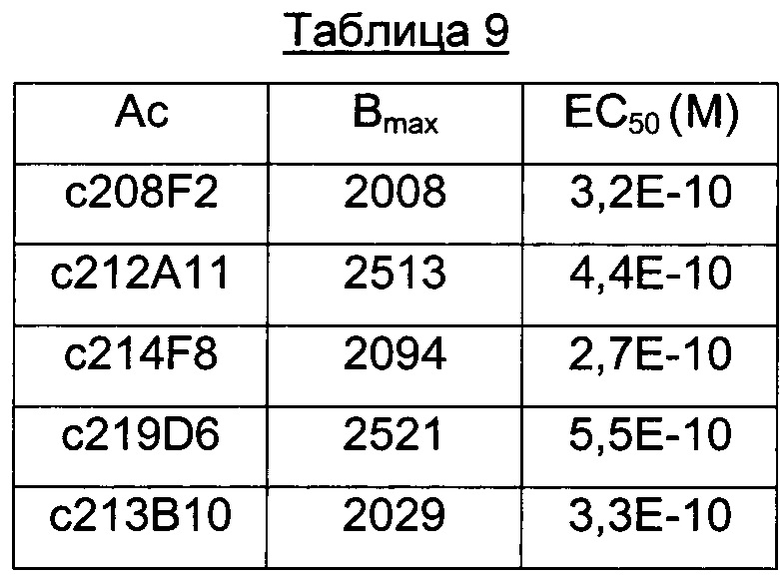
С целью проверки отсутствия связывания обоих антител, мышиного и химерного, на hIR, использовали стабильную линию клеток, экспрессирующую IR человека (hIR). Распознавание hIR человека на клеточной поверхности обоими Аb, мышиным и химерным, исследовали с помощью анализов FACS. Либо мышиные, либо химерные mAb в возрастающих концентрациях инкубировали на линии трансфицированных клеток hlR+ в течение 20 минут при 4°С в буфере FACS (ФСБ, 0,1%) БСА, 0,01%) NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488 перед инкубацией в течение 20 дополнительных минут при 4°С в темноте, а затем промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0). В качестве положительного контроля использовали антитело к hIR, клон GRO5. Мышиное и химерное антитела к 9G4 вводили в качестве нерелевантных антител.
Высокий уровень экспрессии hIR на клеточной поверхности трансфицированных клеток подтверждали с использованием коммерческого антитела к hIR GRO5 (Фиг. 3А и 3В). Даже при использовании высоких концентраций либо мышиного (Фиг. 3А), либо химерного (Фиг. 3В) Ab к hIGF-1R связывание на клеточной поверхности клеток, трансфицированных hIR+, не наблюдали. Эти результаты продемонстрировали, что ни мышиные, ни химерные Ab к hIGF-1R не распознавали NR.
Эта специфичность распознавания hIGF-1R по сравнению с IR также продемонстрирована с помощью анализов FACS с использованием клеток IM9 линии клеток В-лимфомы, экспрессирующих hIR (Фиг. 4). Для этих анализов FACS протокол был таким же, как описано выше, и мышиные антитела использовали с целью предотвращения перекрестной реактивности второго Ab против человека (клетки IM9 экспрессируют Ig человека на их клеточной поверхности). Результаты, представленные на Фиг. 4, еще раз продемонстрировали, что ожидаемый сигнал наблюдали при использовании антитела GRO5 к hIR, при этом ни одно из оцениваемых мышиных антител не проявляло какого-либо значительного сигнала связывания на данной линии клеток.
Пример 4: Связывание антитела с нативным IGF-1R обезьяны на основании анализов FACS и Biacore
Одной из первых предпосылок для нормативных токсикологических исследований является нахождение релевантного вида животного для оценки выбранного соединения. Поскольку серия антител, описанных в настоящем документе, не способна распознавать мышиный IGF-1R, наиболее вероятным видом для токсикологической оценки является нечеловекообразный примат (NHP).
С целью оценки связывания антител к IGF-1R на IGF-1R обезьяны сначала оценивали связывание обоих антител к hIGF-1R, мышиного и химерного, с помощью анализов FACS на линии клеток COS-7, используя возрастающие концентрации антитела. Клетки (1×106 клеток/мл) инкубировали с антителами к IGF-1R в течение 20 мин при 4°С в буфере FACS (ФСБ, 0,1% БСА, 0,01% NaN3). Затем клетки промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488 перед инкубацией в течение 20 дополнительных минут при 4°С в темноте и, наконец, промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу оценивали на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0).
Кривые титрования, полученные на линии клеток COS-7 обезьяны, показали, что Ab к hIGF-1R специфично распознавали IGF-1R, экспрессируемый на поверхности в линии клеток обезьян (Фиг. 5А). Определение ЕС50 для каждого мышиного и химерного Ab показало, что 2 формы в высокой степени сравнимы по их связывающим свойствам с IGF-1R обезьяны (Фиг. 5В). Эти результаты показали, что все полученные антитела к hIGF-1R распознавали IGF-1R обезьяны.
Сравнение ЕС50 связывания на клетках COS-7 со связыванием на трансфицированных клетках IGF-1R проводили с целью проверки величины распознавания химерным антителом IGF-1R человека по сравнению с IGF-1R обезьяны. Результаты, показанные на Фиг. 5С, продемонстрировали сходное распознавание IGF-1R человека и обезьяны всеми антителами.
С целью подтверждения распознавания на другом типе обезьян клетки трансфицировали IGF-1R яванского макакадля продуцирования растворимого IGF-1R ECD обезьяны и проводили эксперименты с помощью устройства Biacore с одним из химерных антител (C208F2) с целью сравнения их свойств связывания либо hIGF-1R, либо IGF-1R яванского макака.
Эксперименты по распознаванию проводили на устройстве Biacore ×100, используя сенсорный чип СМ5, активированный антителом к гистидиновой метке (набор для антитела захвата His (capture kit) GE Healthcare, номер по каталогу 28-9950-56). Более 11000 РЕ антител прививают химическим путем на карбоксиметилдекстрановом матриксе, используя набор для аминной химии. Эксперименты проводили при 25°С при скорости тока 30 мкл/мин, используя буфер HBS-EP (GE Healthcare) в качестве буфера для хроматографии и разведения образца. Для определения кинетических параметров связывания химерной формы антитела 208F2 (C208F2) на hIGF-1R по сравнению с IGF-1R макаки использовали кинетическую схему одного цикла.
Раствор растворимого рекомбинантного варианта гетеротетрамера IGF-1R, состоящего из 2α цепей и внеклеточных доменов из 2β цепей, экспрессируемого с дополнительной С-концевой меткой 10-His, на основе последовательности либо человека (R&D Systems номер по каталогу 305-GR-50), либо последовательности яванского макака (продуцируемого в лаборатории), вводили в течение 1 минуты во вторую проточную кювету при разведении, определенном для захвата около 160 РЕ антигена. После фазы захвата либо вводили буфер для хроматографии 5 раз (каждый ввод пробы 90 с), либо вводили ряд возрастающих 5 концентраций C208F2 (каждый ввод пробы 90 с) в обе проточные ячейки. В конце пятого ввода пробы буфер для хроматографии пропускали с целью определения скорости диссоциации.
Затем поверхность регенерировали вводом буфера 10 мМ глицин, HCI рН 1,5 в течение 30 с.
Вычисленный с помощью компьютерной программы сигнал соответствует разности между ответом проточной ячейки 2 (с иммобилизованным IGF-1R) и ответом проточной ячейки 1 (без каких-либо молекул IGF-1R) (Фиг. 6).
Для каждой молекулы IGF-1R (человека или яванского макака) сигнал за счет ввода ряда возрастающих концентраций C208F2 корректировали путем вычитания сигнала, полученного в результате 5 вводов буфера (двойной контроль). Полученные в результате сенсограммы анализировали с использованием программного обеспечения Biaevaluation в модели 1:1. Кинетические коэффициенты оценивали либо независимо (2 кинетических коэффициента C208F2 на каждом IGF-1R), либо совместно (одинаковые кинетические коэффициенты связывания C208F2 на IGF-1R человека и яванского макака). Качество соответствия оценивали на основании отношения Chi2/Rmax ниже 0,05 РЕ.
Кинетические коэффициенты связывания (см. таблицу 10), определенные по отдельности для каждого IGF-1R, близки, и качество соответствия обеих сенсограмм с одинаковыми кинетическими коэффициентами является высоким.
Антитело C208F2 распознает как рекомбинантный человеческий IGF-1R, так и IGF-1R яванского макака с константой диссоциации (KD) приблизительно 0,2 нМ. Значения сродства, определенные в данном исследовании, соответствуют значениям функционального сродства (авидности) антител для уровня захвата IGF-1R человека и яванского макака около 160 РЕ.
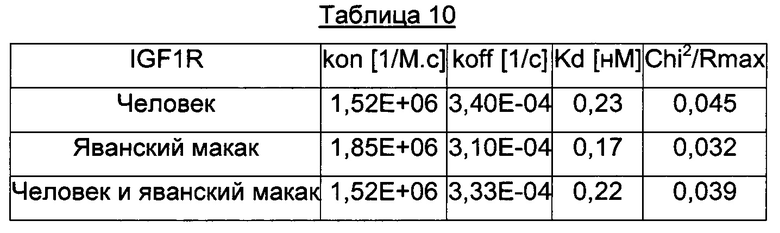
Пример 5: Агонистическое действие полученных антител на фосфорилирование IGF-1R
Хорошо известно, что антитела могут индуцировать агонистическое действие при их связывании с тирозинкиназными рецепторами. Поскольку авторы изобретения не хотели бы выбрать такие агонистические антитела, была проведена оценка фосфорилирования hIGF-1R с использованием химерных антител.
Для этой цели клетки MCF-7 инкубировали в среде без сыворотки в течение ночи. Затем добавляли либо IGF-1 (100 нМ), либо тестируемые Ab (10 мкг/мл) в течение 10 минут при 37°С. Среду удаляли, и клетки соскребали в буфер для лизиса (рН 7,5), содержащий 10 мМ буфер Трис HCl (рН 7,5), 15% NaCl (1 М), 10% смесь детергентов (10 мМ Tris-HCl, 10% буфер для лизиса Igepal) (Sigma Chemical Co.), 5% дезоксихолат натрия (Sigma Chemical Co.), 1 таблетку TM полной смеси ингибиторов протеаз (Roche), 1% смесь ингибиторов фосфатаз Cocktail Set II (Calbiochem) и инкубировали в течение 90 мин при 4°С. Лизаты осветляли центрифугированием при 4°С, нагревали в течение 5 мин при 100°С и выдерживали при -20°С или непосредственно наносили на 4-12% полиакриламидные гели с додецилсульфатом натрия для электрофореза (ДСН-ПААГ). Инкубацию с первым антителом проводили в течение 2 ч при комнатной температуре, а затем инкубировали с HRP-связанными вторыми антителами в течение 1 ч при комнатной температуре. Мембраны промывали в буфере TBST, после чего белки визуализировали методом электрохемилюминесцентного определения (ЭХЛ). Количественное определение блоттингов проводят с помощью программного обеспечения Image J. Значения фосфорилирования белков нормализовали по глицеральдегид-3-фосфатдегидрогеназе (GAPDH). Фосфорилирование hIGF-1R в ответ на IGF-1 считали за 100% стимуляцию. Действие Ab к hIGF-1R на фосфорилирование hIGF-1R определяли как % фосфорилирования, индуцированного IGF-1.
Результаты, описанные на Фиг. 7, представляют собой среднее значение % plGF-1R в ответ на химерные Ab к IGF-1R 3 независимых экспериментов +/- S.D. по сравнению с IGF-1. Как проиллюстрировано, ни значительного, ни незначительного (менее 10%) фосфорилирования hIGF-1R не было обнаружено при инкубации клеток MCF-7 с 10 мкг Ab к IGF-1R.
Пример 6: Ингибирование фосфорилирования IGF-1R в ответ на IGF-1 мышиными антителами к hIGF-1R
Чтобы охарактеризовать отобранные антитела, исследовали их способность к ингибированию IGF1-индуцированного фосфорилирования. Для этой цели клетки MCF-7 инкубировали в среде без сыворотки в течение ночи. Затем клетки инкубировали в течение 5 минут с мышиными Ab к hIGF, после чего добавляли IGF-1 в течение 2 минут при 37°С. Среду удаляли, и клетки соскребали в буфер для лизиса (рН 7,5), содержащий 10 мМ буфер Трис HCl (рН 7,5), 15% NaCl (1 М), 10% смесь детергентов (10 мМ трис-HCl, 10% буфер для лизиса Igepal) (Sigma Chemical Co.), 5% дезоксихолат натрия (Sigma Chemical Co.), 1 таблетку TM полной смеси ингибиторов протеаз (Roche), 1% смесь ингибиторов фосфатаз Cocktail Set II (Calbiochem) и инкубировали в течение 90 мин при 4°С. Лизаты осветляли центрифугированием при 4°С, нагревали в течение 5 мин при 100°С и выдерживали при -20°С или непосредственно наносили на 4-12% полиакриламидные гели с додецилсульфатом натрия для электрофореза (ДСН-ПААГ). Инкубацию с первым антителом проводили в течение 2 ч при комнатной температуре, а затем инкубировали с HRP-связанными вторыми антителами в течение 1 ч при комнатной температуре. Мембраны промывали в буфере TBST, после чего белки визуализировали методом электрохемилюминесцентного определения (ЭХЛ). Количественное определение блоттингов проводят с помощью программного обеспечения Image J. Значения фосфорилирования белков нормализовали по глицеральдегид-3-фосфатдегидрогеназе (GAPDH). Фосфорилирование hIGF-1R в ответ на IGF-1 считали за 100% стимуляцию. Действие Ab к hIGF-1R на фосфорилирование hIGF-1R определяли как % фосфорилирования, индуцированного IGF-1.
Все Ab к IGF-1R сильно ингибировали фосфорилирование hIGF-1R в ответ на IGF-1 (уменьшение >80%) (Фиг. 8). Лучшими ингибиторами IGF1-индуцированного фосфорилирования hIGF-1R являются Mab m208F2, m212A11 и m214F8.
Пример 7: Исследование интернализации IGF-1R после связывания полученных антител к IGF-1R с помощью анализов FACS
Клетки MCF-7 инкубировали с 10 мкг/мл химерных антител при 4°С в течение 20 мин. Затем клетки промывали и инкубировали при 4°С или 37°С в течение 4 ч. Количество связанного с клеточной поверхностью антитела определяли с использованием второго антитела. ΔMFI определяли как разность между MFI, измеренной при 4°С, и MFI, измеренной при 37°С, после 4-часового периода инкубации, в соответствии с количеством интернализованного Ab. ΔMFI была представлена на Фиг. 9 и в таблице 11. Процент интернализации при 10 мкг/мл Ab вычисляли, следуя формуле 100*(MFI при 4°С - MFI при 37°C)/MFI при 4°С, и результаты представлены в таблице 11.
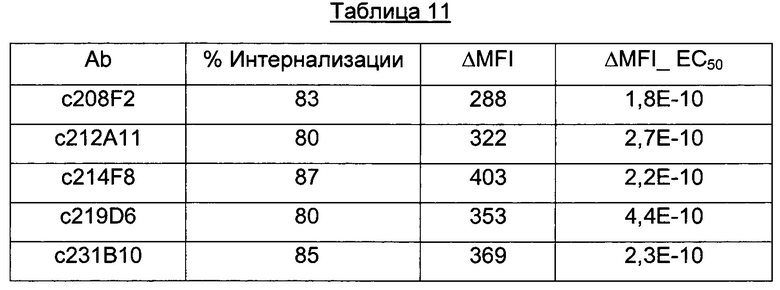
Чтобы определить, способны ли антитела, которые также распознают IGF-1R обезьяны, к интернализации данного рецептора, был проведен такой же эксперимент по интернализации. Результаты, суммированные в таблице 12, продемонстрировали, что все протестированные антитела способны к опосредованию интернализации IGF-1R.
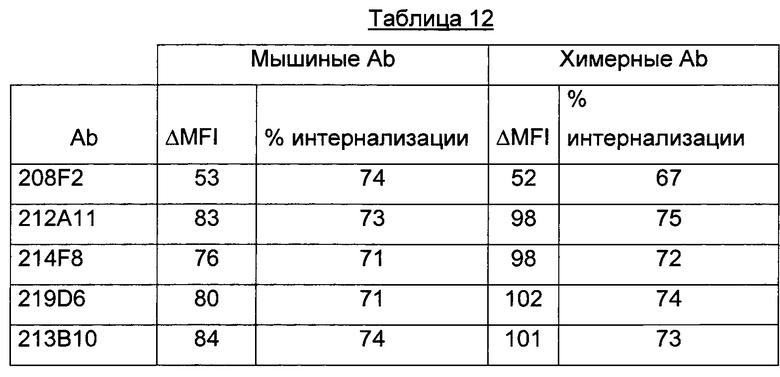
Далее оценивали кинетику уменьшения количества антитела, связанного с клеточной поверхностью. С этой целью клетки MCF-7 высевали в 96-луночные планшеты и инкубировали с 10 мкг/мл мышиного антитела в течение 20 мин при 4°С. Затем клетки промывали для удаления несвязанного антитела и помещали в среду при 37°С на 10, 20, 30, 60 или 120 мин. В каждый момент времени клетки центрифугировали, а затем метили клеточную поверхность на льду вторым антимышиным антителом IgG-Alexa488, чтобы определить количество антитела, оставшегося на клеточной поверхности. Интенсивность флуоресценции для каждого мышиного Ab и для каждого момента времени нормализовали по сигналу при 4°С (% оставшегося IGF-1R) и приводили в соответствие с экспоненциальным распадом, чтобы определить время полужизни (t1/2). t1/2 считали временем, в течение которого получали 50% снижение сигнала. Как проиллюстрировано на Фиг. 10, уровень всех мышиных Ab на поверхности быстро падал в течение первых 30 мин, и снижение было почти максимальным после 60 мин инкубации (Фиг. 10А). Вычисленное время полужизни составляло от 10 до 18 мин в соответствии с мышиным Ab (Фиг. 10В).
С целью валидации, что уменьшение сигнала клеточной поверхности является следствием интернализации Ab и не связана с шеддингом рецептора клетки инкубировали с мышиными Ab в течение 0, 30 и 60 мин при 37°С (Фиг. 11). Затем клетки фиксировали и либо пермеабилизировали, либо не пермеабилизировали, чтобы определить антитело, связанное с клеточной поверхностью (в отсутствие пермеабилизации), и суммарный сигнал антитела, соответствующий связанному с клеточной поверхностью+интернализованному Ab (с пермеабилизацией). Количество интернализованного Ab (цитоплазматического) определяли следующим образом: MFI после пермеабилизации - MFI без пермеабилизации. Этот эксперимент показал, что снижение количества связанного с клеточной поверхностью Ab происходило вследствие возрастания количества цитоплазматических Аb, что демонстрирует, что Ab были интернализованы (Фиг. 11). Кроме того, расщепление Ab начиналось после 1 ч инкубации, что показано на основании снижения сигнала после пермеабилизации (суммарного).
Пример 8: Исследование интернализации IGF-1R после связывания полученных антител к IGF-1R с помощью анализов FACS
Для дополнительного подтверждения интернализации антител проводили конфокальную микроскопию, чтобы оценить субклеточное распределение антител после внутриклеточного транспорта. Клетки инкубировали с Ab к hIGF-1R 37°С, фиксировали и пермеабилизировали. Таким образом, клетки окрашивали с использованием второго антитела Alexa-488 и кроличьего антитела к Lamp-1, которое было выявлено с использованием второго антикроличьего IgG, меченого Alexa 555. Перед инкубацией при 37°С мышиное Ab 208F2 было локализовано на мембране клеток MCF-7 (Фиг. 12А). Отсутствие колокализации с лизосомным маркером lamp-1 было отмечено с использованием маркерный плагин колокализации программы Image J. Количество связанного с клеточной поверхностью антитела резко уменьшалось после 15 мин инкубации при 37°С. Одновременно с уменьшением связанного с клеточной поверхностью антитела внутриклеточное антитело обнаруживалось в везикулах. Можно было наблюдать редкую колокализацию с lamp-1. После 30 мин инкубации связанное с клеточной поверхностью антитело с трудом обнаруживалось. Тем не менее колокализация Ab в лизосоме возрастала. После 1 ч инкубации уменьшалось как окрашивание внутриклеточного Аb, так и число колокализаций с lamp-1. Кинетика связанного с клеточной поверхностью антитела и его внутриклеточной аккумуляции коррелировала с кинетикой распада поверхностного антитела, измеренной с помощью FACS. Кроме того, как уже описано для исследований FACS, на основании конфокальной микроскопии расщепление мышиных Ab начиналось после 1 ч инкубации.
Также оценивали интернализацию всех остальных мышиных антител к hIGF-1R и их колокализацию с Lamp-1 (Фиг. 12В). После 30 мин инкубации при 37°С обнаруживалось внутриклеточное антитело, и можно было наблюдать колокализацию с lamp-1, указывающую на то, что все отобранные антитела к IGF-1R эффективно интернализировались в лизосомы.
Пример 9: Ингибирование расщепления Ab с использованием ингибитора лизосом бафиломицина А1
Чтобы подтвердить, что антитела, достигшие лизосомы, расщепляются, клетки обрабатывали бафиломицином А1, представляющим собой активный ингибитор функции лизосом, или оставляют без обработки. Затем клетки инкубировали с 10 мкг/мл тестируемого Ab при 4°С, промывали и инкубировали в течение 2 ч при 37°С. Интернализованное Ab определяли после пермеабилизации клеток, используя второе антимышиное Ab IgG-Alexa 488. Добавление бафиломицина А1 предотвращало расщепление внутриклеточного Ab (Фиг. 13), что указывает на то, что Ab были эффективно интернализованы и расщеплялись в лизосомах.
Пример 10: Действие рН на связывание антитело-IGF-IR
Поскольку антитела были отобраны на основании их потенциала к интернализации, и, как показано выше, колокализуются с ранними эндосомами перед вхождением в лизосомальный компартмент, интересный подход состоял в отборе антител, для которых происходит модулирование стабильности связывания Ab/hIGF-1R в зависимости от рН окружающей среды, и преимущественно антител, диссоциирующих от IGF-1R, когда рН окружающей среды становится кислым. Действительно, основное различие между ранними эндосомами и лизосомами состоит в рН их просвета: в эндосомальном компартменте рН составляет приблизительно 6, тогда как в лизосомальном компартменте рН составляет около 4,5.
Хорошо известно, что сразу после интернализации после связывания лиганда (IGF1) hIGF-1R возвращается к клеточной поверхности посредством биохимического пути рециклинга.
Без связи с какой-либо теорией гипотеза, описанная в настоящем документе, состоит в том, что антитела, более склонные к раннему высвобождению от их мишени при кислом рН, вероятно, будут способствовать рециклингу мишени на мембране и, следовательно, могут считаться лучшими кандидатами для подходов ADC.
Чтобы изучить, проявляют ли некоторые из антител такое свойство, и исследовать корреляцию этого свойства с цитотоксической активностью, связывание мышиных Mab к hIGF-1R на линии клеток MCF-7 проводили в буферах при различных значениях рН. Возрастающие концентрации мышиных mAb инкубировали на линии клеток MCF-7 в течение 20 мин при 4°С при различных значениях рН в диапазоне от 5 до 8. Затем клетки промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488, в буфере FACS. Клетки инкубировали дополнительно в течение 20 минут при 4°С в темноте, а затем промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид, который окрашивает мертвые клетки. ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0). Все отобранные мышиные антитела к IGF-1R проявляли сниженную связывающую способность при кислом рН, как проиллюстрировано на Фиг. 14.
Связывание гуманизированных Mab к IGF-1R на линии клеток MCF-7 проводили в буферах при различных рН. Возрастающие концентрации мышиных mAb инкубировали на линии клеток MCF-7 в течение 20 мин при 4°С при различных значениях рН в диапазоне от 5 до 8. Затем клетки промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488, в буфере FACS. Клетки инкубировали дополнительно в течение 20 минут при 4°С в темноте, а затем промывали 3 раза в буфере FACS. Связывание гуманизированных антител к IGF-1 R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид, который окрашивает мертвые клетки. ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0). Гуманизированные антитела к IGFR проявляли сниженную связывающую способность при кислом рН, как проиллюстрировано на Фиг. 24.
Пример 12: Оценка гуманизированной формы 208F2 Mab
12.1 Оценка связывания и интернализации первой гуманизированной формы hz208F2 VH3/VL3 (также называемой hz208F2 H026/L024)
Связывание первой гуманизированной формы c208F2 mAb оценивали на линиях клеток MCF-7, COS-7 и NIH 3Т3 IR+. К каждой клеточной линии добавляли возрастающие концентрации m208F2, c208F2 или hz208F2 VH3VL3 на 20 мин при 4°С. Затем клетки промывали, и связывание тестируемых mAb выявляли с использованием соответствующего второго антитела. С целью валидации экспрессии IR человека на трансфицированной линии клеток использовали клон GRO5 коммерческого антитела к hIR и использовали в качестве примера профиля распознавания, приеденного на Фиг. 15D.
Сравнение гуманизированной формы с мышиными или химерными формами на клетках MCF-7 (Фиг. 15А) или COS-7 обезьяны (Фиг. 15В) показало близкие профили для 3 тестируемых форм. Процесс гуманизации не модифицировал специфичность распознавания антитела, которая является точно сравнимой с мышиной и химерной формами по отсутствию перекрестной реактивности на рецепторе инсулина человека (Фиг. 15С).
Вычисленные значения ЕС50 первой гуманизированной формы 208F2 на линии клеток MCF-7 человека и линии клеток COS-7 обезьяны были сходными со значением, определенным либо для мышиной, либо для химерной формы mAb 208F2.
Способность mAb hz208F2 VH3/VL3 к интернализации оценивали с помощью проточной цитометрии. Клетки MCF-7 инкубировали с 10 мкг/мл антител при 4°С в течение 20 мин. Затем клетки промывали и инкубировали при 4°С или 37°С в течение 4 ч. Количество связанного с клеточной поверхностью антитела определяли с использованием второго антитела. ΔMFI, определяемая как разность между MFI, измеренной при 4°С, и MFI, измеренной при 37°С, после 4-часового периода инкубации соответствует количеству интернализованного Ab. ΔMFI представлена на Фиг. 16 и в таблице 13. Процент интернализации при 10 мкг/мл Ab вычисляли, следуя формуле 100*(MFI при 4°С - MFI при 37°C)/MFI при 4°С, и результаты представлены в таблице 13. Таким образом, гуманизированное антитело hz208F2 VH3/VL3 обладало свойствами связывания и интернализации, подобными свойствам, измеренным для соответствующего мышиного и химерного антител 208F2.

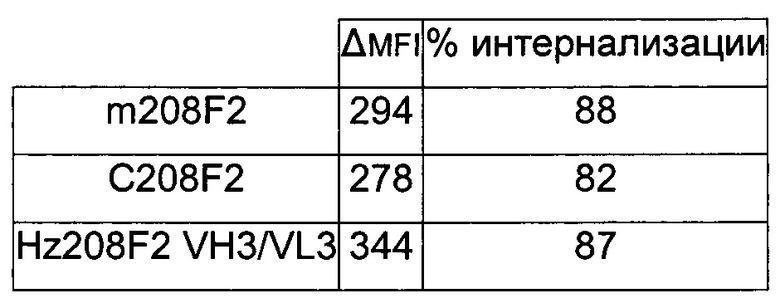
12.2 Оценка связывания дополнительных гуманизированных форм hz208F2
mAb 208F2 подвергали гуманизации и оценивали связывающие свойства шестнадцати гуманизированных вариантов (включая первую форму, описанную в 12.1). Связывающие свойства гуманизированных вариантов оценивали с помощью анализов FACS на линии клеток MCF-7 аденокарциномы молочной железы человека и линии клеток Cos-7 обезьяны с использованием возрастающих концентраций антитела. С этой целью клетки (1×106 клеток/мл) инкубировали с антителами к IGF-1R в течение 20 мин при 4°С в буфере FACS (ФСБ, 0,1% БСА, 0,01% NaN3). Затем их промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488, в течение 20 дополнительных минут при 4°С в темноте, после чего промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). ЕС50 связывания, выраженную в молярности (М), вычисляли, используя анализ нелинейной регрессии (GraphPad Prims 4.0).
Значения ЕС50 гуманизированных вариантов показали, что все гуманизированные варианты проявляют эквивалентные связывающие свойства на обеих линиях клеток человека и обезьяны.
Значения ЕС50 гуманизированных антител были суммированы в таблице 13b.
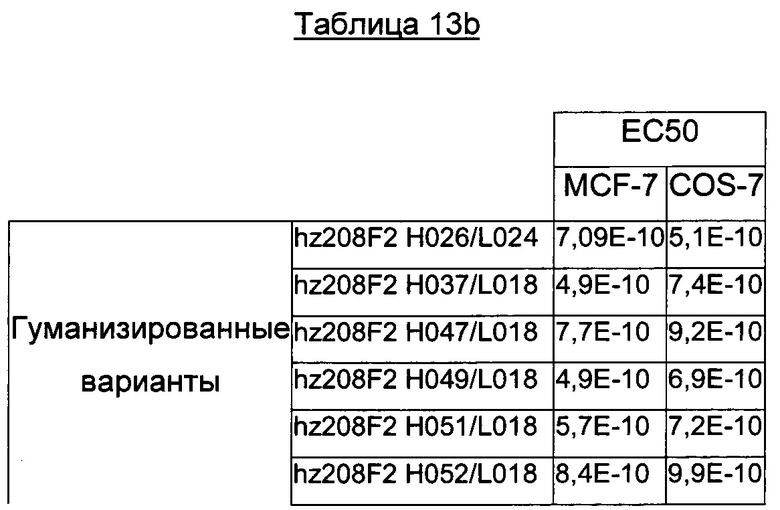
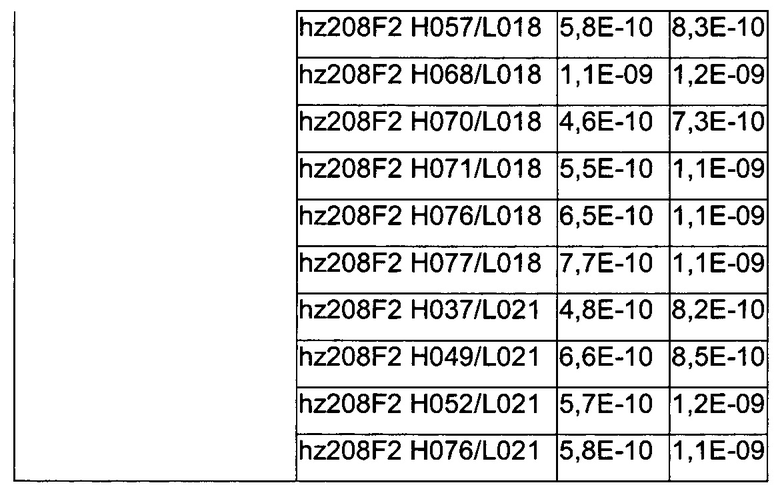
12.3 Оценка интернализации другой гуманизированной формы hz208F2
Клетки MCF-7 инкубировали с 10 мкг/мл гуманизированных антител при 4°С в течение 20 мин. Затем клетки промывали и инкубировали при 4°С или 37°С в течение 4 ч. Количество связанного с клеточной поверхностью антитела определяли с использованием второго антитела на проточном цитометре FacsCalibur Flow (Becton Dickinson). ΔMFI, определяемая как разность между MFI, измеренной при 4°С, и MFI, измеренной при 37°С, после 4-часового периода инкубации соответствует количеству интернализованного Ab. ΔMFI представлена в таблице 13 с.Процент интернализации при 10 мкг/мл Ab вычисляли по следующей формуле: 100*(MFI при 4°С - MFI при 37°C)/MFI при 4°С. Гуманизированное антитело hz208F2 H077/L018 способно индуцировать значимую интернализацию IGF-1R.

Пример 13: Определение константы диссоциации (Кп) связывания пяти химерных антител к IGF-1R (c208F2, с213В10, С212А11, C214F8 и C219D6) и гуманизированного варианта (VH3/VL3) антитела 208F2 на растворимом рекомбинантном IGF-1R человека
Константы диссоциации (KD) связывания антител на рекомбинантном растворимом IGF-1R человека определяли по отношению между скоростью диссоциации (koff) и скоростью ассоциации (kon). Эксперименты по кинетике проводили на устройстве Biacore ×100, используя сенсорный чип СМ5, активированный мышиным моноклональным антителом к гистидиновой метке. Около 12 000 РЕ антител прививают химическим путем на карбоксиметилдекстрановом матриксе, используя набор для аминной химии.
Эксперименты проводили при 25°С при скорости тока 30 мкл/мин, используя буфер HBS-EP (GE Healthcare) в качестве буфера для хроматографии и разведения образца.
Для определения кинетических параметров связывания антител к IGF-1R на растворимом рекомбинантном IGF-1R человека, захваченном его двумя С-концевыми 10-гистидиновыми метками, использовали кинетическую схему одного цикла.
1 - Растворение растворимого рекомбинантного варианта гетеротетрамера IGF-1R человека: 2α цепи и внеклеточные домены 2р цепей, экспрессируемые с дополнительной С-концевой меткой 10-His (R&D Systems номер по каталогу 305-GR-50), вводили в течение одной минуты во вторую проточную ячейку при концентрации 10 мкг/мл. В среднем в каждом из 24 циклов, выполненных для данного исследования, было захвачено 587 РЕ (со стандартным отклонением 24 РЕ) растворимого рецептора.
2 - После фазы захвата либо вводили буфер для хроматографии 5 раз (каждый ввод пробы 90 с), либо вводили ряд возрастающих 5 концентраций одного из шести антител (каждый ввод пробы 90 с) в обе проточные ячейки. В конце пятого ввода пробы буфер для хроматографии пропускали в течение 5 минут с целью определения скорости диссоциации.
3 - Затем поверхность регенерировали вводом буфера 10 мМ глицин, HCl рН 1,5 в течение 45 с.
Вычисленный с помощью компьютерной программы сигнал соответствует разности между ответом проточной ячейки 2 (с иммобилизованным IGF-1R) и ответом проточной ячейки 1 (без каких-либо молекул IGF-1R).
Для каждого IGF-1R сигнал за счет ввода ряда возрастающих концентраций одного антитела корректировали путем вычитания сигнала, полученного в результате 5 вводов буфера (двойной контроль), см. Фиг. 17.
Полученные в результате сенсограммы анализировали с использованием программного обеспечения Biaevaluation в модели 1:1.
Для каждого антитела проводили четыре эксперимента, используя два различных ряда концентраций: 40, 20, 10, 5 и 2,5 нМ для первых двух экспериментов и 24, 12, 6, 3 и 1,5 нМ для двух последних экспериментов, проведенных с каждым антителом.
Для 6 антител, тестируемых в данном эксперименте, экспериментальные данные соответствовали модели 1:1 со значимыми значениями koff, где более высокую концентрацию определяли в качестве константы, и вычисляли четыре другие концентрации (см. Фиг. 18).
Константы диссоциации (KD), вычисленные как отношение: koff/kon, а время полужизни комплексов, вычисленное как отношение: Ln(2)/koff, представлены на Фиг. 19 и 20. Они соответствуют среднему значению четырех независимых экспериментов, проведенных для каждого антитела. Усы диаграммы соответствуют стандартной ошибке (n равно 4) значений.
Константы диссоциации находятся в диапазоне от 10 до 100 пМ. Антитело C208F2 обладает более слабым сродством (более высоким значением константы диссоциации) к h-IGF-1R (при KD около 75 пМ), а его гуманизированный вариант обладает по меньшей мере таким же хорошим сродством, как химерный вариант (при KD около 60 рМ). Четыре других химерных антитела к IGF-1R обладают в достаточной степени подобным сродством к hIGF1-R (при KD около 30 пМ). Различие значений сродства, в основном, связано со скоростью диссоциации полученного в результате времени полужизни комплексов. С антителом 208F2 время полужизни комплекса составляет от 2 до 3 часов для химерного и гуманизированного (VH3/VL3) вариантов. Для четырех других химерных антител средние значения времени полужизни составляют от 7,0 до 9,4 ч.
Эти очень медленные значения кинетики диссоциации явным образом связаны с двухвалентной структурой антител, которые способны к одновременному связыванию обоими плечами Fab с двумя соседними молекулами hIGF-1R. В этом случае уровень захваченных молекул IGF-1R может влиять на скорость диссоциации. Значения сродства, определенные в данном исследовании, соответствуют значениям функционального сродства (или авидности) антител для уровня захвата h-IGF-1R около 600 РЕ. 3-кратное различие KD, наблюдаемое между представленными выше данными (таблица 10), и значения, приведенные в Примере 13, связаны с изменением уровня захвата hIGF-1R (600 РЕ по сравнению с 160 РЕ в Примере 4).
Пример 14: Синтез лекарственных средств по изобретению
В приведенных ниже Примерах использованы следующие сокращения:
aq. водный раствор
эи энантиомерный избыток
экв эквивалент
ИЭР Ионизация электрораспылением
ЖХ/МС Жидкостная хроматография, сопряженная с масс-спектрометрией
ВЭЖХ Высокоэффективная жидкостная хроматография
ЯМР Ядерный магнитный резонанс
нас. насыщенный
УФ ультрафиолетовое излучение
Эталонное соединение 1
(S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, бис-трифторуксусная кислота
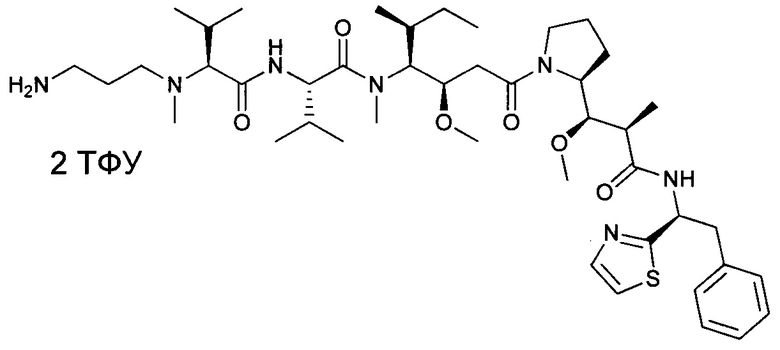
Соединение 1А: (4R, 5S)-4-метил-5-фенил-3-пропаноил-1,3-оксазолидин-2-он
(4R, 5S)-4-метил-5-фенил-1,3-оксазолидин-2-он (5,8 г, 32,7 ммоль, 1,00 экв) растворяли в тетрагидрофуране (ТГФ, 120 мл) в инертной атмосфере. Смесь охлаждали до -78°С и добавляли по каплям н-бутиллитий (14,4 мл). После перемешивания в течение 30 минут при -78°С добавляли пропаноилхлорид (5,7 мл). Перемешивание продолжали в течение 30 минут при -78°С, затем в течение ночи при температуре окружающей среды. Реакционную смесь концентрировали, затем повторно растворяли в 200 мл воды. рН раствора доводили до 7 насыщенным водным раствором бикарбоната натрия. Водную фазу экстрагировали 3 раза 100 мл этилацетата (EtOAc). Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 6,8 г (89%) соединения 1А в форме желтого масла.
Соединение 1В: трет-Бутил-(2S)-2-[(1R,2R)-1-гидрокси-2-метил-3-[(4R,5S)-4-метил-2-оксо-5-фенил-1,3-оксазолидин-3-ил]-3-оксопропил]пирролидин-1-карбоксилат
Соединение 1А (17,6 г, 75,45 ммоль, 1,00 экв) растворяли в дихлорметане (ДХМ, 286 мл) в инертной атмосфере. Этот раствор охлаждали в ледяной бане. Добавляли по каплям триэтиламин (ТЭА, 12,1 мл, 1,15 экв) и Bu2BOTf (78,3 мл, 1,04 экв), при этом поддерживая температуру реакционной смеси ниже 2°С. Перемешивание продолжали при 0°С в течение 45 минут, после чего реакционную смесь охлаждали до -78°С. Добавляли по каплям раствор трет-бутил (2S)-2-формилпирролидин-1-карбоксилат (8,5 г, 42,66 ммоль, 0,57 экв) в ДХМ (42 мл). Перемешивание продолжали в течение 2 часов при -78°С, затем в течение 1 часа при 0°С и, наконец, 1 часа при температуре окружающей среды. Реакционную смесь нейтрализовали 72 мл фосфатного буфера (рН составляет от 7,2 до 7,4) и 214 мл метанола и охлаждали до 0°С. Добавляли по каплям раствор 30% пероксида водорода в метаноле (257 мл), поддерживая при этом температуру ниже 10°С. Перемешивание продолжали в течение 1 часа при 0°С. Реакционную смесь нейтрализовали 142 мл воды, затем концентрировали при пониженном давлении. Полученный в результате водный раствор экстрагировали 3 раза 200 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и петролейного эфира (EtOAc:РЕ составляет 1:8) с получением 13, 16 г (40%) соединения 1В в форме бесцветного масла.
Соединение 1С: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-гидрокси-2-метилпропановая кислота
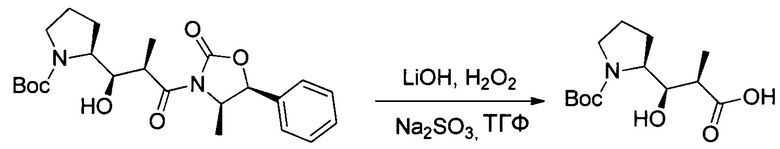
Соединение 1В (13,16 г, 30,43 ммоль, 1,00 экв) растворяли в ТГФ (460 мл) в присутствии пероксида водорода (30% в воде, 15,7 мл), затем охлаждали в ледяной бане. Добавляли по каплям водный раствор гидроксида лития (0,4 моль/л, 152,1 мл), поддерживая при этом температуру реакционной смеси ниже 4°С. Реакционную смесь перемешивали в течение 2,5 часов при 0°С. Добавляли по каплям водный раствор Na2SO3 (1 моль/л, 167,3 мл), поддерживая при этом температуру реакционной смеси при 0°С. Реакционную смесь перемешивали в течение 14 часов при температуре окружающей среды, затем нейтрализовали 150 мл холодного насыщенного раствора бикарбоната натрия и промывали 3 раза 50 мл ДХМ. рН водного раствора доводили до 2-3 1М водным раствором KHSO4. Водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, промывали один раз насыщенным раствором NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 7,31 г (88%) соединения 1С в форме желтого масла.
Соединение 1D: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-метокси-2-метилпропановая кислота
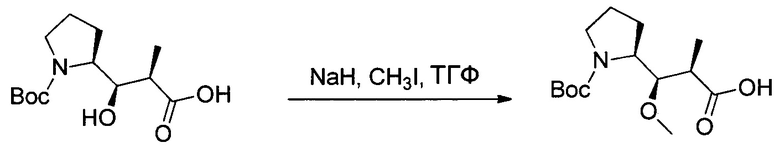
Соединение 1С (7,31 г, 26,74 ммоль, 1,00 экв) растворяли в инертной атмосфере в ТГФ (135 мл) в присутствии йодметана (25,3 мл). Реакционную смесь охлаждали в ледяной бане, после чего добавляли порциями NaH (60% в масле, 4,28 г). Реакционную смесь оставляли при перемешивании на 3 дня при 0°С, а затем нейтрализовали 100 мл насыщенного водного раствора бикарбоната натрия и промывали 3 раза 50 мл эфира. рН водного раствора доводили до 3 1М водным раствором KHSO4. Водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, промывали один раз Na2S2O3 (5% в воде), один раз насыщенным раствором NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали с получением 5,5 г (72%) соединения 1D в форме бесцветного масла.
Соединение 1Е: N-метокси-N-метил-2-фенилацетамид
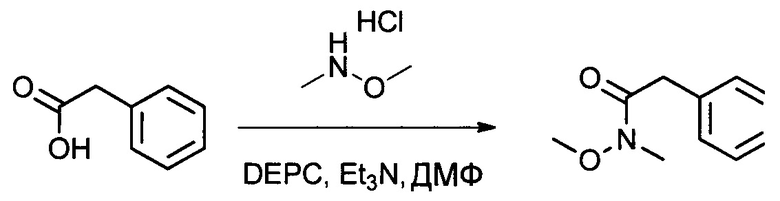
2-Фенилуксусную кислоту (16,2 г, 118,99 ммоль, 1,00 экв) растворяли в диметилформамиде (ДМФ, 130 мл), затем охлаждали до -10°С. Добавляли диэтилфосфороцианидат (DEPC, 19,2 мл), гидрохлорид метокси(метил)амина (12,92 г, 133,20 ммоль, 1,12 экв) и триэтиламин (33,6 мл). Реакционную смесь перемешивали в течение 30 минут при -10°С, затем 2,5 часов при температуре окружающей среды. Затем ее дважды экстрагировали 1 литром EtOAc. Органические фазы объединяли, дважды промывали 500 мл NaHCO3 (нас.), один раз 400 мл воды, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:3) с получением 20,2 г (95%) соединения 1Е в форме желтого масла.
Соединение 1F: 2-Фенил-1-(1,3-тиазол-2-ил)этан-1-он
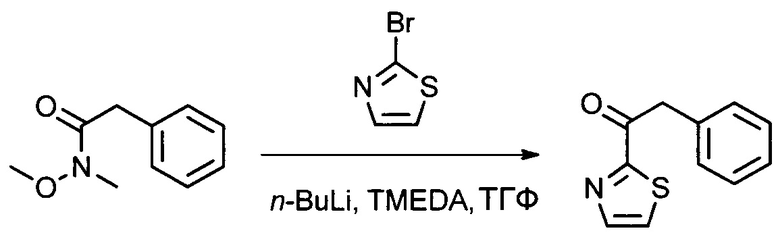
Тетраметилэтилендиамин (TMEDA, 27,2 мл) растворяли в ТГФ (300 мл) в инертной атмосфере, затем охлаждали до -78°С, после чего добавляли по каплям n-BuLi (67,6 мл, 2,5 М). Добавляли по каплям 2-бром-1,3-тиазол (15,2 мл), и перемешивание продолжали в течение 30 минут при -78°С. Добавляли по каплям соединение 1Е (25 г, 139,50 ммоль, 1,00 экв), растворенное в ТГФ (100 мл) Перемешивание продолжали в течение 30 минут при -78°С, затем 2 часов при -10°С. Реакционную смесь нейтрализовали 500 мл KHSO4 (нас.), затем экстрагировали 3 раза 1 литром EtOAc. Органические фазы объединяли, дважды промывали 400 мл воды и дважды 700 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:10) с получением 25 г (88%) соединения 1F в форме желтого масла.
Соединение 1G: (1R)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-ол
В инертной атмосфере раствор соединения 1F (15 г, 73,8 ммоль, 1,00 экв.) в эфире (300 мл) добавляли по каплям к (+)-В-хлордиизопинокамфеилборана ((+)-Ipc2BCl, 110,8 мл). Реакционную смесь перемешивали в течение 24 часов при 0°С, затем нейтрализовали 300 мл смеси (1:1) NaOH (10% в воде) и H2O2 (30% в воде) и, наконец, экстрагировали три раза 500 мл EtOAc. Органические фазы объединяли, дважды промывали 300 мл K2CO3 (нас.) и один раз 500 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:20 до 1:2) с получением 6,3 г (42%) соединения 1G в форме белого твердого вещества.
Соединение 1Н: 2-[(1S)-1-азидо-2-фенилэтил]-1,3-тиазол
Соединение 1G (6 г, 29,23 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (150 мл) в присутствии трифенилфосфина (13 г, 49,56 ммоль, 1,70 экв.), затем охлаждали до 0°С. Добавляли по каплям диэтилазодикарбоксилат (DEAD, 7,6 мл) с последующим добавлением дифенилфосфорилазида (DPPA, 11 мл), затем охлаждающую баню удаляли, и раствор оставляли при перемешивании на 48 часов при температуре окружающей среды. Смесь концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:30) с получением 8 г частично очищенного соединения 1Н в форме желтого масла. Соединение 1Н использовали в таком виде в следующей стадии.
Соединение 11: трет-Бутил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамат
Соединение 1Н (6,5 г, 28,2 ммоль, 1,00 экв) растворяли в инертной атмосфере в ТГФ (100 мл) в присутствии трифенилфосфина (6,5 г, 33,9 ммоль, 1,20 экв.) и нагревали до 50°С в течение 2 часов. Затем добавляли аммиак (70 мл), и нагревание продолжали в течение 3 часов. Реакционную смесь охлаждали, нейтрализовали 500 мл воды, затем экстрагировали 3 раза 500 мл EtOAc. Органические фазы объединяли и дважды экстрагировали 500 мл 1 н. HCl. Водные фазы объединяли, доводили до рН 8-9 добавлением раствора гидроксида натрия (10%) в воде), затем экстрагировали 3 раза 500 мл ДХМ. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 4,8 г (83%) (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина в форме желтого масла. Затем это соединение защищали группой Boc ((трет-бутокси)карбонил), чтобы его можно было очистить. Его растворяли в инертной атмосфере в 1,4-диоксане (40 мл), затем охлаждали до 0°С. Добавляли по каплям (Boc)2O (10,26 г, 47,01 ммоль, 2,00 экв), разведенный в 20 мл 1,4-диоксана. Охлаждающую баню удаляли, и раствор оставляли при перемешивании на ночь при температуре окружающей среды, после чего нейтрализовали 300 мл воды и дважды экстрагировали 500 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:20, эи равно 93%). Затем смесь перекристаллизовали в смеси гексан/ацетон (~5-10/1,1 г/10 мл) с получением 6 г (84%) соединения 1I в форме белого твердого вещества (эи >99%).
Соединение 1J: трет-Бутил (2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-карбоксилат
Соединение 1I (3 г, 9,86 ммоль, 1,00 экв) растворяли в инертной атмосфере в 10 мл ДХМ. Добавляли трифторуксусную кислоту (ТФУ, 10 мл), и раствор оставляли при перемешивании на ночь при температуре окружающей среды, затем концентрировали при пониженном давлении с получением 2,0 г (64%) (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина; трифторуксусная кислота в форме желтого масла. Это промежуточное соединение повторно растворяли в 20 мл ДХМ, после чего добавляли соединение 1D (1,8 г, 6,26 ммоль, 1,05 экв), DEPC (1,1 г, 6,75 ммоль, 1,13 экв) и диизопропилэтиламин (DIEA, 1,64 г, 12,71 ммоль, 2,13 экв). Реакционную смесь оставляли при перемешивании на ночь при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:3) с получением 2,3 г (81%) соединения 1J в форме бледно-желтого твердого вещества.
Соединение 1K: (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамид; трифторуксусная кислота
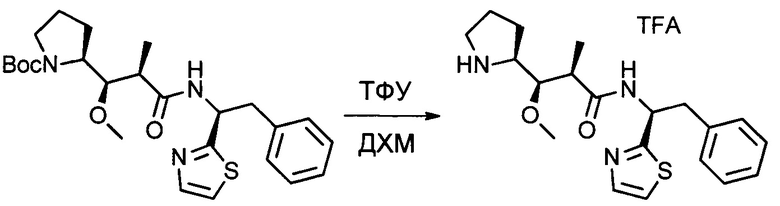
Соединение 1J (2,25 г, 4,75 ммоль, 1,00 экв) растворяли в инертной атмосфере в 10 мл ДХМ. Добавляли ТФУ (10 мл), и раствор оставляли при перемешивании на ночь при температуре окружающей среды, затем концентрировали при пониженном давлении с получением 2,18 г (94%) соединения 1K в форме желтого масла.
Соединение 1L: (2S,3S)-2-(бензиламино)-3-метилпентановая кислота
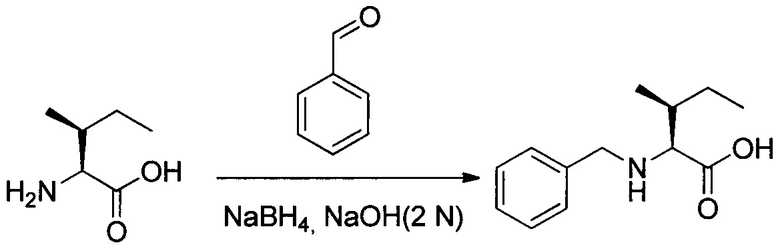
(2S,3S)-2-амино-3-метилпентановую кислоту (98,4 г, 750 ммоль, 1,00 экв) добавляли при температуре окружающей среды и порциями к 2 н. раствору гидроксида натрия (375 мл). Быстро добавляли бензальдегид (79,7 г, 751,02 ммоль, 1,00 экв), и полученный в результате раствор перемешивали в течение 30 минут.Добавляли небольшими порциями боргидрид натрия (10,9 г, 288,17 ммоль, 0,38 экв), поддерживая при этом температуру от 5 до 15°С. Перемешивание продолжали в течение 4 часов при температуре окружающей среды. Реакционную смесь разбавляли 200 мл воды, затем дважды промывали 200 мл EtOAc. рН водного раствора доводили до 7 2 н. раствором соляной кислоты. Образовавшийся осадок собирали фильтрованием и получили 149,2 г (90%) соединения 1L в форме белого твердого вещества.
Соединение 1М: (2S,3S)-2-[бензил(метил)амино]-3-метилпентановая кислота
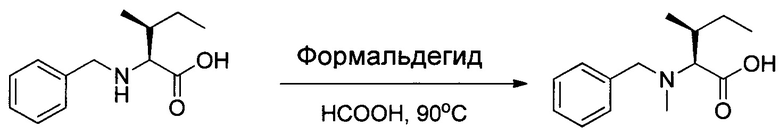
Соединение 1L (25 г, 112,97 ммоль, 1,00 экв) растворяли в инертной атмосфере в муравьиной кислоте (31,2 г) в присутствии формальдегида (36,5% в воде, 22,3 г). Раствор перемешивали в течение 3 часов при 90°С, затем концентрировали при пониженном давлении. Остаток растирали в 250 мл ацетона, затем концентрировали. Операцию растирания/выпаривания повторяли дважды с 500 мл ацетона с получением 21,6 г (81%) соединения 1M в форме белого твердого вещества.
Соединение 1N: (2S,3S)-2-[бензил(метил)амино]-3-метилпентан-1-ол
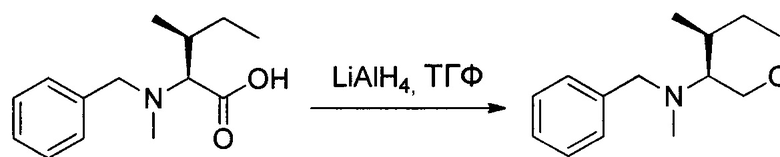
LiAIH4 (0,36 г) суспендировали в 10 мл ТГФ в инертной атмосфере при 0°С. Добавляли небольшими порциями соединение 1М (1,5 г, 6,37 ммоль, 1,00 экв), поддерживая при этом температуру от 0 до 10°С. Реакционную смесь перемешивали в течение 2 часов при 65°С и снова охлаждали до 0°С, после чего нейтрализовали последовательными добавлениями 360 мкл воды, 1 мл 15% гидроксида натрия и 360 мкл воды. Осаждающиеся соли алюминия отделяли фильтрованием. Фильтрат высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:50) с получением 820 мг (58%) соединения 1N в форме бледно-желтого масла.
Соединение 1O: (2S,3S)-2-[бензил(метил)амино]-3-метилпентаналь
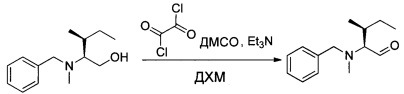
Оксалилхлорид (0,4 мл) растворяли в ДХМ (15 мл) в инертной атмосфере. Раствор охлаждали до -70°С и добавляли по каплям раствор диметилсульфоксида (ДМСО (0,5 мл) в ДХМ (10 мл)) в течение 15 минут. Реакционную смесь перемешивали 30 минут, после чего добавляли по каплям раствор соединения 1N (820 мг, 3,70 ммоль, 1,00 экв) в ДХМ (10 мл) в течение 15 минут. Реакционную смесь перемешивали дополнительно в течение 30 минут при низкой температуре, затем медленно добавляли триэтиламин (2,5 мл). Реакционную смесь перемешивали 1 час при -50°С, затем охлаждающую баню удаляли, и реакционную смесь нейтрализовали 25 мл воды, при этом давая температуре смеси вернуться к обычной. Раствор дважды промывали 30 мл насыщенного раствора NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:200) с получением 0,42 г (52%) соединения 1O в форме желтого масла.
Соединение 1Р: (2S,3S)-N-бензил-1,1-диметокси-N,3-диметилпентан-2-амин
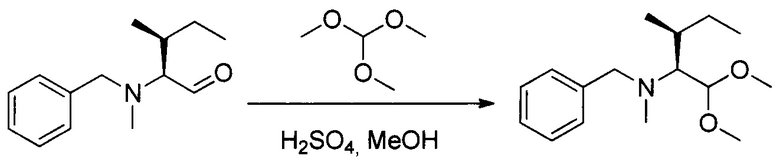
Соединение 1O (4,7 г, 21,43 ммоль, 1,00 экв) растворяли в 20 мл метанола при 0°С. Добавляли по каплям концентрированную серную кислоту (4,3 мл), и перемешивание продолжали в течение 30 минут при 0°С. Добавляли триметилортоформиат (21,4 мл), охлаждающую баню удаляли, и реакционную смесь оставляли при перемешивании на 3 часа при температуре окружающей среды. Реакционную смесь разбавляли 200 мл EtOAc, последовательно промывали 100 мл 10% Na2CO3 и 200 мл насыщенного раствора NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,4 г (60%) соединения 1Р в форме бледно-желтого масла.
Соединение 1Q: [[1-(трет-бутокси)этенил]окси](трет-бутил)диметилсилан
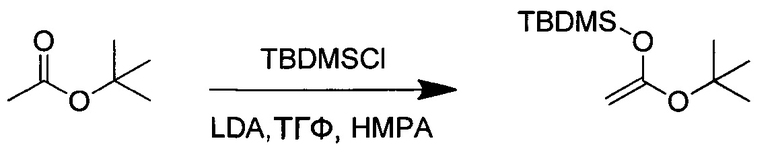
Диизопропиламин (20 г, 186,71 ммоль, 1,08 экв) растворяли в 170 мл ТГФ в инертной атмосфере и охлаждали до -78°С. Добавляли по каплям nBuLi (2,4 М, 78,8 мл), и раствор перемешивали в течение 30 минут при низкой температуре (с получением LDA - диизопропиламида лития), после чего добавляли трет-бутилацетат (20 г, 172, 18 ммоль, 1,00 экв). Реакционную смесь перемешивали 20 минут при -78°С, после чего добавляли гексаметилфосфорамид (НМРА, 25,8 мл) и раствор трет-бутилдиметилхлорсилана (TBDMSCI, 28 г, 185,80 ммоль, 1,08 экв) в 35 мл ТГФ. Перемешивание продолжали дополнительно в течение 20 минут при низкой температуре, а затем охлаждающую баню удаляли. Раствор концентрировали при пониженном давлении. Остаток повторно растворяли в 100 мл воды, затем экстрагировали 3 раза 100 мл РЕ. Органические фазы объединяли, промывали один раз 500 мл насыщенного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали перегонкой с получением 16,6 г (83%) соединения 1Q в форме бесцветного масла.
Соединение 1R: трет-Бутил (3R,4S,5S)-4-[бензил(метил)амино]-3-метокси-5-метилгептаноат
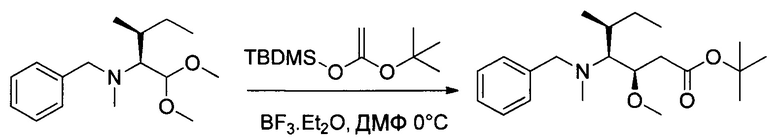
Соединение 1Р (2,0 г, 7,54 ммоль, 1,00 экв) и соединение 1Q (2,6 г, 11,28 ммоль, 1,50 экв) растворяли в 33 мл ДХМ в инертной атмосфере. Раствор охлаждали до 0°С. Добавляли по каплям ДМФ (1,2 г) вместе с раствором BF3⋅Et2O (2,1 г) в 7,5 мл ДХМ. Перемешивание продолжали в течение 24 часов при 0°С. Реакционную смесь промывали один раз 30 мл карбоната натрия (10%) и дважды 50 мл насыщенного водного раствора NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:100) с получением 1,82 г (91%) соединения 1R в форме желтого масла.
Соединение 1S: Гидрохлорид (3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноата
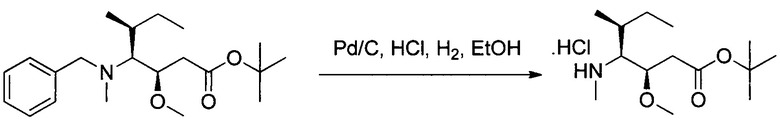
Соединение 1R (2,4 г, 6,87 ммоль, 1,00 экв) растворяли в инертной атмосфере в 35 мл этанола в присутствии Pd/C (0,12 г) и концентрированной соляной кислоты (0,63 мл). Атмосферу азота вытесняли атмосферой водорода, и реакционную смесь оставляли при перемешивании на 18 часов при температуре окружающей среды. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток растирали в 50 мл гексана, и супернатант удаляли, в результате после высушивания при пониженном давлении получили 1,66 г (82%) соединения 1S в форме белого твердого вещества.
Соединение 1T: трет-Бутил (3R,4S,5S)-4-[(2S)-2-[[(бензилокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
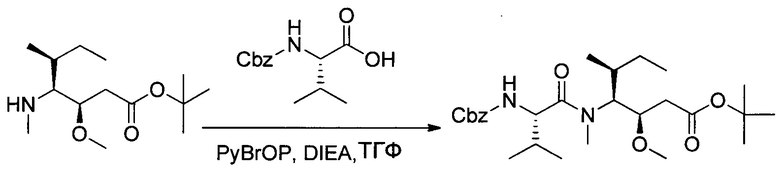
(2S)-2-[[(бензилокси)карбонил]амино]-3-метилбутановую кислоту (15 г, 0,40 ммоль, 1,00 экв) растворяли в 300 мл ДХМ в присутствии DIEA (38,3 мл) и бромтрипирролидинфосфония гексафторфосфата (РуВгОР, 32,3 г). Раствор перемешивали 30 минут при температуре окружающей среды, после чего добавляли соединение 1S (15,99 г, 0,42 ммоль, 1,07 экв). Реакционную смесь перемешивали в течение 2 часов, а затем концентрировали. Остаток очищали на колонке с обращенной фазой (С18) смесью ацетонитрила (ACN) и воды (от 30:70 до 100:0 за 40 минут) с получением 17 г (58%) соединения 1Т в форме бесцветного масла.
Соединение 1U: трет-Бутил-(3R,4S,5S)-4-[(2S)-2-амино-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
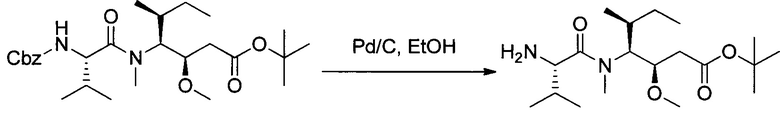
Соединение 1Т (76 мг, 0,15 ммоль, 1,00 экв) растворяли в инертной атмосфере в 10 мл этанола в присутствии Pd/C (0,05 г). Атмосферу азота вытесняли атмосферой водорода, и реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 64 мг соединения 1U в форме бесцветного масла.
Соединение 1V: (3R,4S,5S)-4-[(2S)-2-[[(9H-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
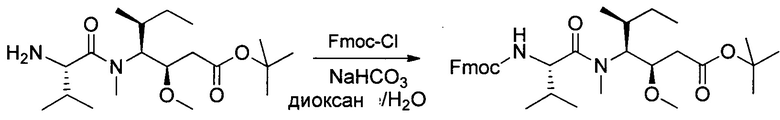
Соединение 1U (18,19 г, 50,74 ммоль, 1,00 экв) растворяли в 400 мл смеси 1,4-диоксан/вода (1:1) в присутствии бикарбоната натрия (12,78 г, 152 ммоль, 3,00 экв) и 9Н-флуорен-9-илметилхлорформиат (Fmoc-CI, 19,69 г, 76 ммоль, 1,50 экв), затем перемешивали в течение 2 часов при температуре окружающей среды. Затем реакционную смесь разбавляли 500 мл воды и экстрагировали 3 раза 200 мл EtOAc. Органические фазы объединяли, промывали один раз 200 мл насыщенного водного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 40 г частично очищенного соединения 1V в форме бледно-желтого масла.
Соединение 1W: (3R,4S,5S)-4-[(2S)-2-[[(9H-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептановая кислота
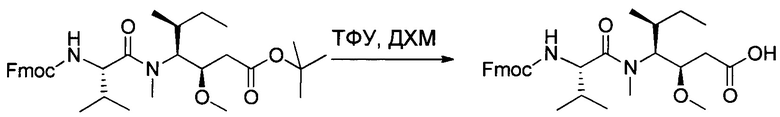
Соединение 1V (40 г, 68,88 ммоль, 1,00 экв) растворяли в нейтральной атмосфере в 600 мл ДХМ. Добавляли ТФУ (300 мл). Раствор перемешивали 2 часа при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (1:10) с получением 23,6 г (65%) соединения 1W в форме бесцветного масла.
Соединение 1Х: 9Н-флуорен-9-илметил N-[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамат
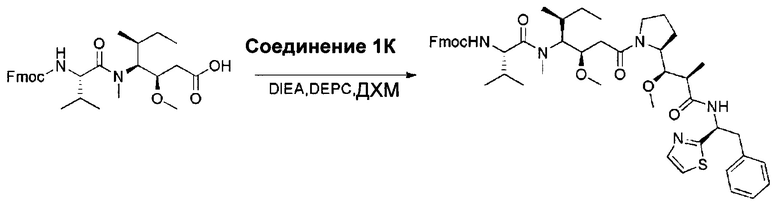
Соединение 1W (2,53 г, 4,82 ммоль, 1,08 экв) растворяли в 20 мл ДХМ в присутствии соединения 1K (2,18 г, 4,47 ммоль, 1,00 экв), DEPC (875 мг, 5,37 ммоль, 1,20 экв) и DIEA (1,25 г, 9,67 ммоль, 2,16 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, затем последовательно промывали 50 мл насыщенного раствора KHSO4 и 100 мл воды, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (от 1:200 до 1:40) с получением 2,8 г (71%) соединения 1Х в форме бледно-желтого твердого вещества.
Соединение 1Y: (2S)-2-амино-N-[(3R,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил]-N,3-диметилбутанамид
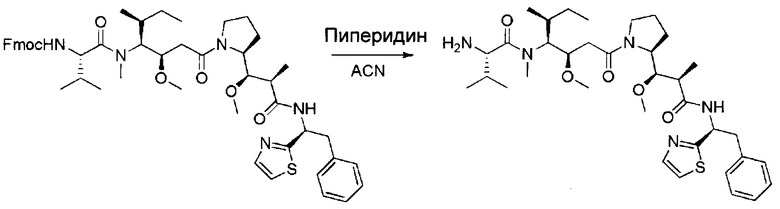
Соединение 1Х (2,8 г, 3,18 ммоль, 1,00 экв) растворяли в ацетонитриле (ACN, 12 мл) в присутствии пиперидина (3 мл) и оставляли при перемешивании на 18 часов при температуре окружающей среды. Реакционную смесь нейтрализовали 50 мл воды, затем дважды экстрагировали 100 мл ДХМ. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (от 1:100 до 1:40) с получением 1,2 г (57%) соединения 1Y в форме желтого твердого вещества.
Соединение 1ZA: (2S)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутановая кислота
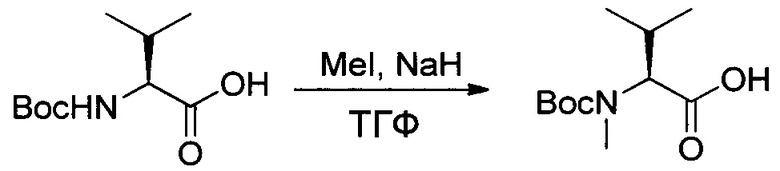
(2S)-2-[[(трет-бутокси)карбонил]амино]-3-метилбутановую кислоту (63 г, 289,97 ммоль, 1,00 экв) растворяли в инертной атмосфере в ТГФ (1000 мл) в присутствии йодметана (181 мл). Раствор охлаждали до 0°С, после чего добавляли гидрид натрия (116 г, 4,83 моль, 16,67 экв) небольшими порциями. Реакционную смесь перемешивали в течение 1,5 часов при 0°С, затем охлаждающую баню удаляли, и перемешивание продолжали в течение 18 часов. Реакционную смесь нейтрализовали 200 мл воды, а затем концентрировали при пониженном давлении. Остаточную водную фазу разбавляли 4 литрами воды, промывали один раз 200 мл EtOAc и доводили рН до значения между 3 и 4 1 н. раствором соляной кислоты. Полученную смесь экстрагировали 3 раза 1,2 л EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 60 г (89%) соединения 1ZA в форме желтого масла.
Соединение 1ZB: бензил-(28)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутаноат
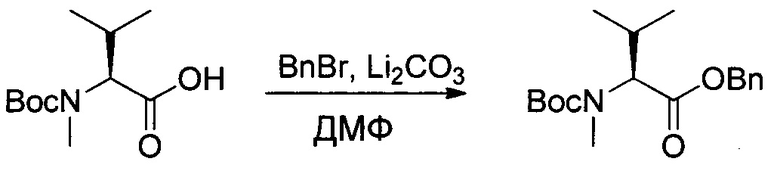
Соединение 1ZA (47 г, 203,21 ммоль, 1,00 экв) растворяли в ДМФ (600 мл) в присутствии Li2CO3 (15,8 г, 213,83 ммоль, 1,05 экв). Раствор охлаждали до 0°С, затем добавляли по каплям бензилбромид (BnBr 57,9 г, 338,53 ммоль, 1,67 экв). Реакционную смесь оставляли на ночь при перемешивании, после чего нейтрализовали 400 мл воды и фильтровали. Полученный раствор дважды экстрагировали 500 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (от 1:100 до 1:20) с получением 22,5 г (34%) соединения 1ZB в форме желтого масла.
Соединение 1ZC: гидрохлорид бензил-(2S)-3-метил-2-(метиламино)бутаноата
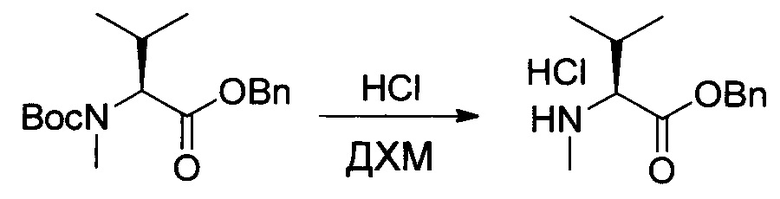
Соединение 1ZB (22,5 г, 70,00 ммоль, 1,00 экв) растворяли в 150 мл ДХМ. Барботировали газообразную соляную кислоту. Реакционную смесь перемешивали 1 час при температуре окружающей среды, а затем концентрировали при пониженном давлении с получением 17 г (94%) соединения 1ZC в форме желтого твердого вещества.
Соединение 1ZD: трет-Бутил-N-(3,3-диэтоксипропил)карбамат
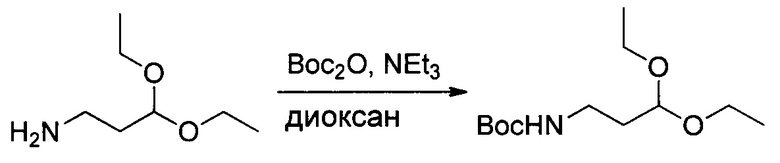
3,3-Диэтоксипропан-1-амин (6 г, 40,76 ммоль, 1,00 экв) растворяли в 1,4-диоксане (30 мл) в присутствии TEA (4,45 г, 43,98 ммоль, 1,08 экв), затем охлаждали до 0°С. Добавляли по каплям (Вос)2O (9,6 г, 43,99 ммоль, 1,08 экв), разведенный в 20 мл 1,4-диоксана. Раствор перемешивали 2 часа при 0°С, затем в течение ночи при температуре окружающей среды, после чего нейтрализовали 10 мл воды. рН доводили до 5 HCl (1%). Раствор экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 8,21 г (81%) соединения 1ZD в форме бледно-желтого масла.
Соединение 1ZE: трет-Бутил-N-(3-оксопропил)карбамат
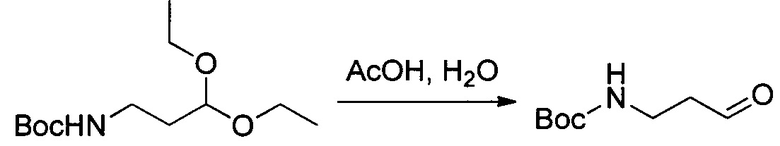
Соединение 1ZD (8,20 г, 33,15 ммоль, 1,00 экв) растворяли в 18,75 мл уксусной кислоты и оставляли на ночь при перемешивании при температуре окружающей среды. Затем реакционную смесь экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, промывали 3 раза 30 мл насыщенного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 5 г (87%) соединения 1ZE в форме темно-красного масла.
Соединение 1ZF: (2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метилбутановая кислота
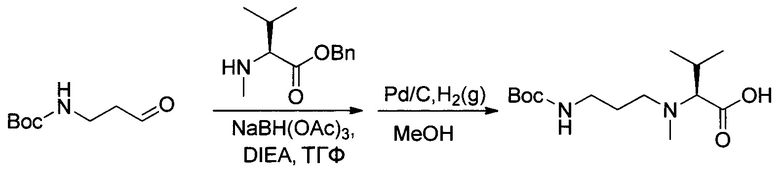
Соединение 1ZE (2,4 г, 13,86 ммоль, 1,00 экв) растворяли в 50 мл ТГФ в присутствии соединения 1ZC (3,56 г, 13,81 ммоль, 1,00 экв) и DIEA (9,16 мл, 4,00 экв). Реакционную смесь перемешивали 30 минут при температуре окружающей среды, после чего добавляли триацетоксиборгидрид натрия (5,87 г, 27,70 ммоль, 2,00 экв). Перемешивание продолжали в течение ночи, затем реакционную смесь нейтрализовали 100 мл воды и экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток частично очищали на колонке силикагеля смесью EtOAc и РЕ (1:4). Полученный неочищенный продукт повторно растворяли в 20 мл метанола в присутствии Pd/C (1,2 г) и гидрогенизировали в течение 20 минут при обычной температуре и давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 200 мг (5%) соединения 1ZF в форме белого твердого вещества.
Соединение 1ZG: трет-бутил-N-(3-[[(1S)-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]тиил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]пропил)карбамат
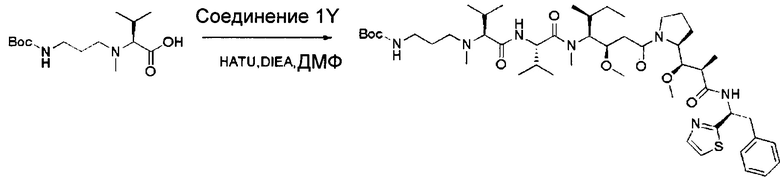
Соединение 1Y (50 мг, 0,08 ммоль, 1,00 экв) растворяли в 2 мл ДМФ в присутствии соединения 1ZF (26,2 мг, 0,09 ммоль, 1,20 экв), DIEA (37,7 мл) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU, 43,3 мг, 0,11 ммоль, 1,50 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, затем разбавляли 10 мл воды и экстрагировали 3 раза 5 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 100 мг соединения 1ZG в форме частично очищенного бесцветного масла.
Соединение 1ZG (90 мг, 0,10 ммоль, 1,00 экв) растворяли в нейтральной атмосфере в 2 мл ДХМ, и раствор охлаждали в ледяной бане. ТФУ (1 мл) добавляли, и реакционную смесь перемешивали 2 часа при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 18% до 31% ACN в течение 7 минут, затем от 31% до 100% ACN в течение 2 минут; Waters 2489, УФ детектор при 254 нм и 220 нм). Соединение 1 было получено при выходе 25% (23 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Atlantis Т3, 3 мкм, 4,6×100 мм; 35°С; 1 мл / мин, от 30% до 60% ACN в воде (20 мМ ацетат аммония в течение 6 минут); ИЭР (C44H73N7O6S, точная масса 827,53) m/z: 829 (МН+), 5,84 мин (93,7%, 254 нм).
1Н ЯМР (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.69-7.66 (m, 1Н), 7.40-7.10 (m, 5Н), 5.80-5.63 (m, 1Н), 4.80-4.65 (m, 2Н), 4.22-4.00 (m, 1Н), 3.89-0.74 (m, 58Н).
Эталонное соединение 2
(S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N/,3-диметилбутанамид, трифторуксусная кислота
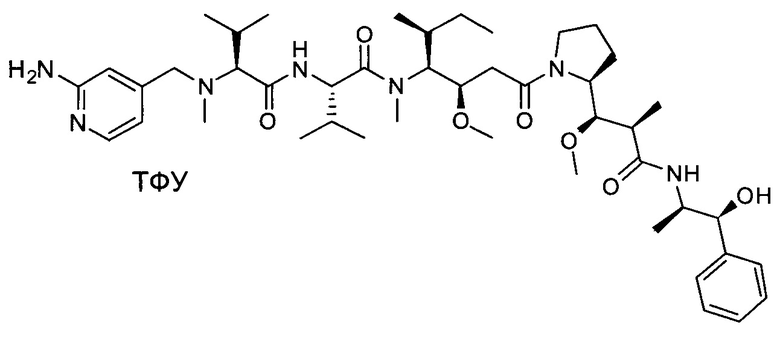
Соединение 2А: трет-бутил-(S)-2-((1R,2R)-3-((1S,2R)-1-гтдрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-карбоксилат
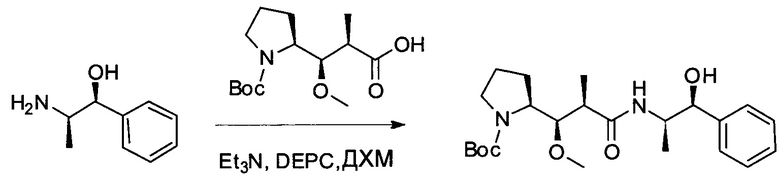
Соединение 1D (2,5 г, 8,70 ммоль, 1,00 экв) и (1S,2R)-2-амино-1-фенилпропан-1-ол (1,315 г, 8,70 ммоль, 1,00 экв) растворяли в инертной атмосфере в ДМФ (35 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,39 мл) и TEA (1,82 мл). Реакционную смесь перемешивали 2 часа при 0°С, затем 4 часа при температуре окружающей среды. Реакционную смесь разбавляли 200 мл воды и экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHCO3 (нас.), один раз 50 мл NaCI (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,6 г (98%) соединения 2А в форме желтого твердого вещества.
Соединение 2В: (2R,3R)-N-((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамид 2,2,2-трифторацетат
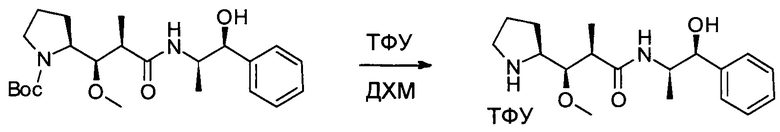
Соединение 2А (2,7 г, 6,42 ммоль, 1,00 экв) растворяли в инертной атмосфере в ДХМ (40 мл), затем охлаждали до 0°С. Добавляли ТФУ (25 мл), и раствор перемешивали в течение 2 часов при 0°С. Реакционную смесь концентрировали при пониженном давлении с получением 4,4 г соединения 2В в форме желтого масла.
Соединение 2С: (9Н-флуорен-9-ил)метил ((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)(метил)амино)-3-метил-1-оксобутан-2-ил)карбамат
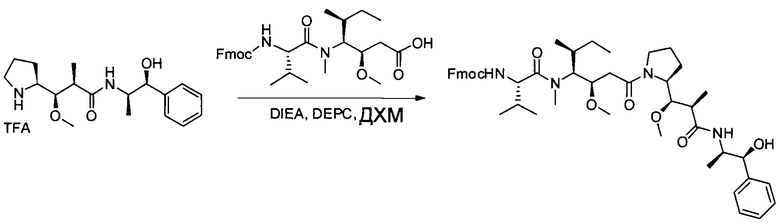
Соединения 2В (4,4 г, 10,13 ммоль, 1,00 экв) и 1W (5,31 г, 10,12 ммоль, 1,00 экв) растворяли в инертной атмосфере в ДХМ (45 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,62 мл) и DIEA (8,4 мл). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 200 мл воды и экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHCO3 (нас.), один раз 50 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,3 г (39%) соединения 2С в форме желтого твердого вещества.
Соединение 2D: (S)-2-амино-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
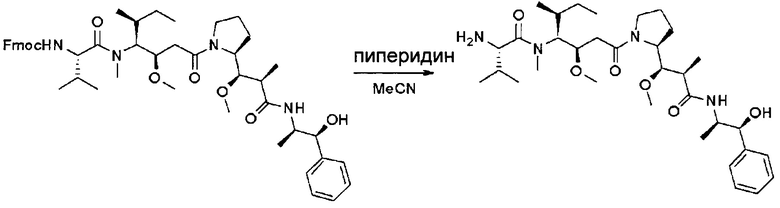
Соединение 2С (300 мг, 0,36 ммоль, 1,00 экв) растворяли в инертной атмосфере в ACN (2 мл) и пиперидине (0,5 мл). Раствор оставляли при перемешивании на ночь при температуре окружающей среды, затем выпаривали до сухого состояния при пониженном давлении. Остаток очищали на колонке силикагеля смесью ДХМ и МеОН (1:100) с получением 150 мг (68%) соединения 2D в форме белого твердого вещества.
Соединение 2Е: метил 2-((трет-бутоксикарбонил)амино)изоникотинат
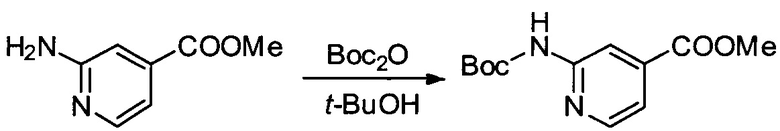
Метил-2-аминопиридин-4-карбоксилат (2 г, 13,14 ммоль, 1,00 экв) растворяли в трет-бутаноле (20 мл), после чего добавляли ди-трет-бутилдикаронат (4,02 г, 18,42 ммоль, 1,40 экв). Реакционную смесь перемешивали при 60°С в течение ночи, затем реакцию останавливали посредством добавления водного раствора 1М NaHCO3 (50 мл). Твердое вещество выделяли фильтрованием, промывали 50 мл EtOH, затем высушивали в вакууме с получением 2,5 г (75%) соединения 2Е в форме белого твердого вещества.
Соединение 2F: трет-бутил (4-(гидроксиметил)пиридин-2-ил)карбамат
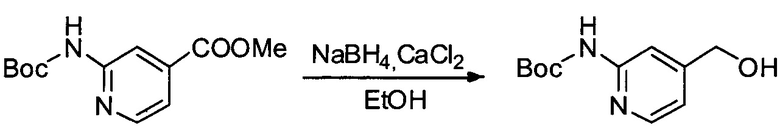
Соединение 2Е (2,5 г, 9,91 ммоль, 1,00 экв) и CaCl2 (1,65 г) растворяли в EtOH (30 мл). Раствор охлаждали до 0°С, затем постепенно добавляли NaBH4 (1,13 г, 29,87 ммоль, 3,01 экв). Раствор оставляли при перемешивании на ночь при температуре окружающей среды, затем реакцию останавливали добавлением воды (50 мл). Смесь экстрагировали три раза 20 мл EtOAc. Органические фазы объединяли, промывали дважды 20 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 2,0 г (90%) соединения 2F в форме бесцветного твердого вещества.
Соединение 2G: трет-бутил (4-формилпиридин-2-ил)карбамат
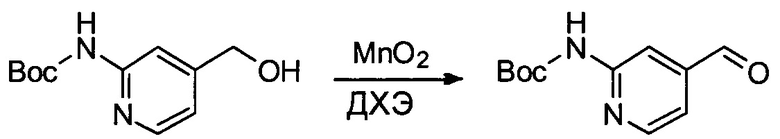
Соединение 2F (2,5 г, 11,15 ммоль, 1,00 экв) растворяли в ДХЭ (25 мл), затем добавляли 19,4 г (223,14 ммоль, 20,02 экв) MnO2. Смесь оставляли при перемешивании на ночь при 70°С, затем твердые вещества удаляли фильтрованием. Фильтрат выпаривали до сухого состояния с получением 1,4 г (57%) соединения 2G в форме белого твердого вещества.
Соединение 2Н: бензил-(S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутаноат
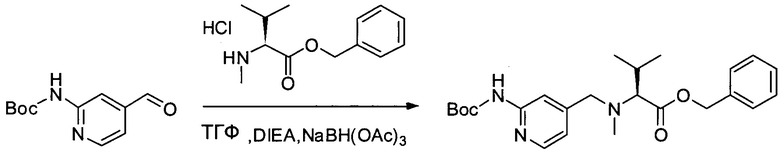
Соединение 2G (2,3 г, 10,35 ммоль, 1,00 экв) растворяли в 25 мл ТГФ в присутствии соединения 1ZC (2,93 г, 11,37 ммоль, 1,10 экв), DIEA (5,39 г, 41,71 ммоль, 4,03 экв) и NaBH(OAc)3 (4,39 г, 20,71 ммоль, 2,00 экв). Реакционную смесь перемешивали в течение 6 часов при температуре окружающей среды, затем нейтрализовали 60 мл NaHCO3 (нас.) и экстрагировали 3 раза 20 мл AcOEt. Органические фазы объединяли, промывали дважды 20 мл NaCl (нас.), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:15) с получением 2,7 г (61%) соединения 2Н в форме белого твердого вещества.
Соединение 2I: (S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутановая кислота
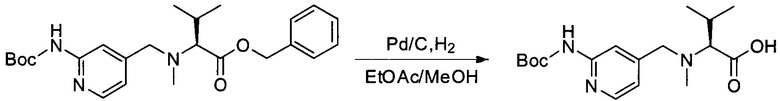
Соединение 2Н (500 мг, 1,17 ммоль, 1,00 экв) растворяли в 10 мл AcOEt и 2 метанола в присутствии Pd/C (250 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 254 мг (64%) соединения 21 в форме бесцветного твердого вещества.
Соединение 2J: трет-бутил (4-((3S,6S,9S,10R)-9-((S)-втор-бутил)-10-(2-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-3,6-диизопропил-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазидодецил)пиридин-2-ил)карбамат
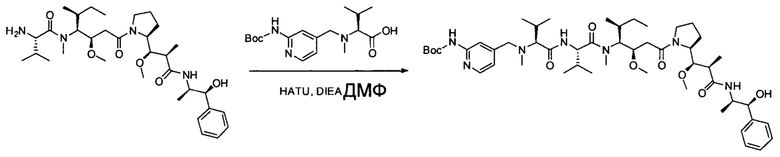
Соединение 2J было получено аналогично соединению 1ZG из амина 2D (85,2 мг, 0,14 ммоль, 1,50 экв), кислоты 21 (31,7 мг, 0,09 ммоль, 1,00 экв), HATU (42,9 мг, 0,11 ммоль, 1,20 экв) и DIEA (36,7 мг, 0,28 ммоль, 3,02 экв) в ДМФ (3 мл). После выпаривания до сухого состояния было получено 100 мг неочищенного продукта в форме белого твердого вещества.
Соединение 2J (100 мг, 0,11 ммоль, 1,00 экв) растворяли в 2 мл ДХМ и 1 мл ТФУ. Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм). Соединение 2 было получено при выходе 6% (6,3 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,8 мл / мин, от 10% до 95% ACN в воде (0,05% ТФУ) в течение 6 минут; ИЭР (C45H73N7O7, точная масса 823,56) m/z: 824,5 (МН+) и 412,9 (М.2Н+/2, 100%), 3,21 мин (99,2%, 210 нм)
1Н ЯМР (400 МГц, CD3OD,ppm): δ (присутствие ротамеров) 7.81-7.79 (m, 1Н); 7.39-7.29 (m, 5Н); 6.61-6.59 (m, 2Н); 4.84-4.52 (m, 1Н); 4.32-4.02 (m, 1Н); 3.90-2.98 (m, 10Н); 2.90-2.78 (m, 1Н); 2.55-0.81 (m, 39Н).
Эталонное соединение 3
метил ((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
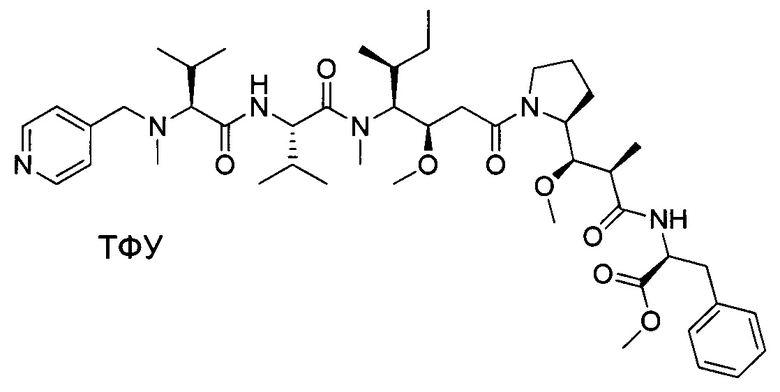
Соединение 3А: трет-бутил (S)-2-((1R,2R)-1-метокси-3-(((S)-1-метокси-1-оксо-3-фенилпропан-2-ил)амино)-2-метил-3-оксопропил)пирролидин-1-карбоксилат
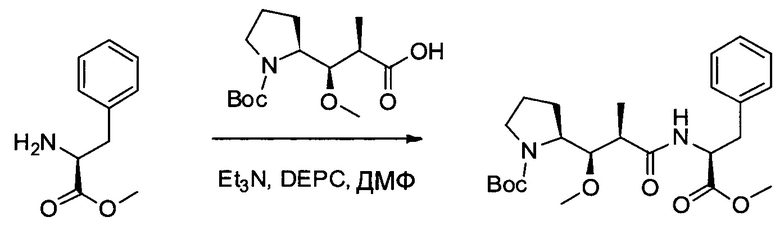
Соединение 1D (3 г, 10,44 ммоль, 1,00 экв) и метил-(S)-2-амино-3-фенилпропаноат (2,25 г, 12,55 ммоль, 1,20 экв) растворяли в инертной атмосфере в ДМФ (40 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,67 мл, 1,05 экв) и TEA (3,64 мл, 2,50 экв). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 100 мл воды и экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, промывали один раз 100 мл KHSO4 (1 моль/л), один раз 100 мл NaHCO3 (нас.), один раз 100 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 4 г (85%) соединения 3А в форме бесцветного масла.
Соединение 3В: 2,2,2-трифторацетат метил (S)-2-((2R,3R)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамидо)-3-фенилпропаноат
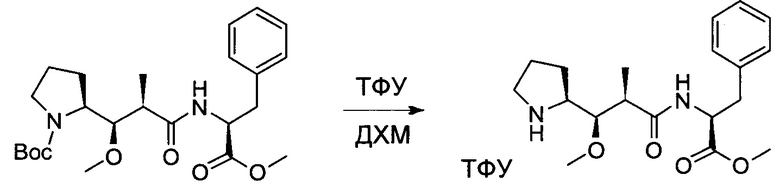
Соединение 3А (5 г, 11,15 ммоль, 1,00 экв) растворяли в инертной атмосфере в ДХМ (40 мл). Добавляли ТФУ (25 мл), и раствор перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением 8 г соединения 3В в форме желтого масла.
Соединение 3С: метил (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
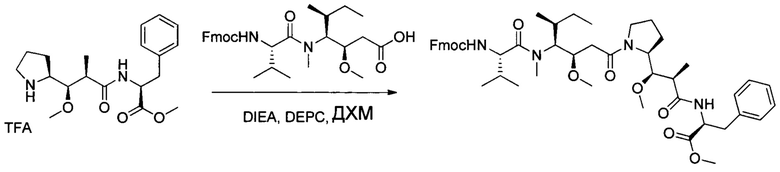
Соединения 3В (8,03 г, 17,36 ммоль, 1,00 экв) и 1W (9,1 г, 17,34 ммоль, 1,00 экв) растворяли в инертной атмосфере в ДХМ (80 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (2,8 мл) и DIEA (12 мл). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 200 мл воды и экстрагировали три раза 50 мл ДХМ. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHCO3(нас.), один раз 50 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 5 г (34%) соединения ЗС в форме желтого твердого вещества.
Соединение 3D: метил (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-амино-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3- фенилпропаноат
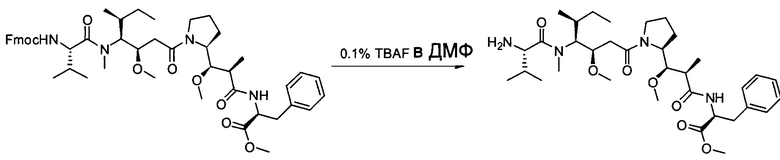
Соединение 3С (5,5 г, 6,43 ммоль, 1,00 экв) растворяли в инертной атмосфере в растворе фторида тетрабутиламмония (TBAF, 2,61 г, 9,98 ммоль, 1,55 экв) в ДМФ (100 мл). Раствор перемешивали при температуре окружающей среды в течение 2 часов, затем разбавляли 100 мл воды и экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,3 г (81%) соединения 3D в форме желтого твердого вещества.
Соединение 3Е: бензил-(S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутаноат
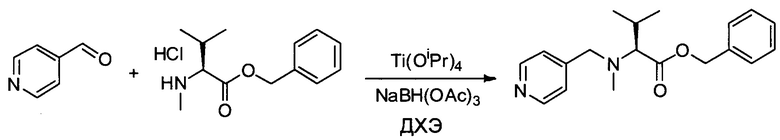
Пиридин-4-карбальдегид (1 г, 9,34 ммоль, 1,00 экв) растворяли в 10 мл 1,2-дихлорэтана (ДХЭ) в присутствии соединения 1ZC (2,9 г, 11,25 ммоль, 1,21 экв) и изопропоксида титана (IV) (4,19 мл, 1,40 экв). Смесь перемешивали при температуре окружающей среды в течение 30 минут, затем добавляли 2,77 г NaBH(OAc)3 (13,07 ммоль, 1,40 экв). Реакционную смесь оставляли на ночь при перемешивании, затем нейтрализовали 100 мл воды, и смесь экстрагировали 3 раза 50 мл AcOEt. Органические фазы объединяли и выпаривали до сухого состояния. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:20) с получением 1,3 г (45%) соединения 3Е в форме бесцветного масла.
Соединение 3F: (3)-3-метил-2-(метил(пиридин-4-илметил)амино)бутановая кислота
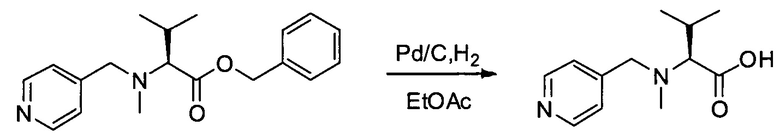
Соединение 3Е (800 мг, 2,56 ммоль, 1,00 экв) растворяли в 30 мл AcOEt в присутствии Pd/C (300 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью ДХМ и МеОН (от 100:1 до 5:1) с получением 100 мг (18%) соединения 3F в форме белого твердого вещества.
Соединения 3D (50 мг, 0,08 ммоль, 1,00 экв) и 3F (26,34 мг, 0,12 ммоль, 1,50 экв) растворяли в 3 мл ДХМ. Раствор охлаждали до 0°С, затем добавляли по каплям 0,018 мл DEPC и 0,0392 мл DIEA. Реакционную смесь перемешивали при 0°С в течение 2 часов, затем при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали при пониженном давлении, и остаток (70 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2545, детектор УФ при 254 нм и 220 нм). Соединение 3 было получено при выходе 27% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл / мин, от 10% до 95% ACN в воде (0,05% ТФУ) в течение 8 минут; ИЭР (C46H72N6O8, точная масса 836,5) m/z: 837,5 (МН+) и 419,4 (M.2HV2 (100%)), 7,04 мин (90,0%, 210 нм)
1Н ЯМР (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.76-8.74 (m, 2Н); 8.53-8.48 (m, 0.4Н, NHCO неполный обмен); 8.29-8.15 (m, 0.8Н, NHCO неполный обмен); 8.01 (s, 2Н), 7.31-7.22 (m, 5Н), 4.88-4.68 (m, 3Н); 4.31-4.07 (m, 2Н); 3.94-2.90 (m, 18Н); 2.55-0.86 (m, 38Н).
Эталонное соединение 4
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
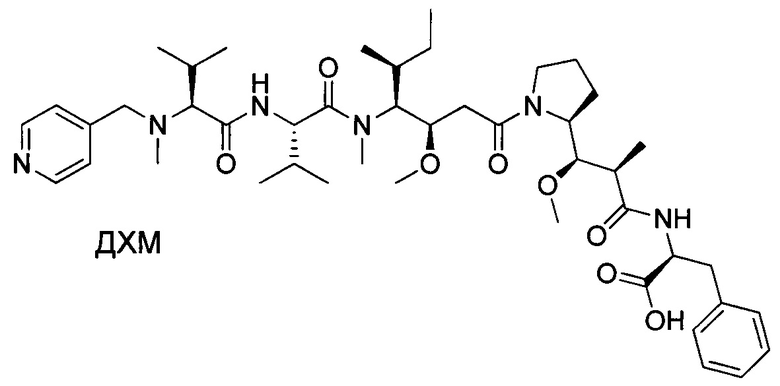
Соединение 3 (100 мг, 0,11 ммоль, 1,00 экв) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли при перемешивании на ночь, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2545, детектор УФ при 254 нм и 220 нм).
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл / мин, от 10% до 95% ACN в воде (0,05% ТФУ) в течение 8 минут; ИЭР (C45H70N6O8, точная масса 822,5) m/z: 823,5 (МН+) и 412,4 (М.2Н+/2, 100%,), 6,84 мин (89,1%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.79-8.78 (m, 2Н); 8.09 (m, 2Н); 7.30-7.21 (m, 5Н); 4.80-4.80 (m, 1Н), 4.36-0.87 (m, 58Н).
Эталонное соединение 6
метил (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-трифторуксусная кислота
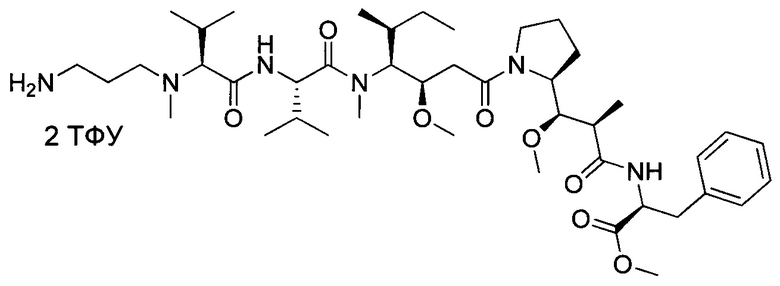
Соединение 6А: метил (2S)-2-[(2R)-2-[(R)-[(2S)-1-[(3R,4S,5S)-4-[(2S)-2-[(2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метил бутанамидо]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноил]пирролидин-2-ил](метокси)метил]пропанамидо]-3-фенилпропаноат
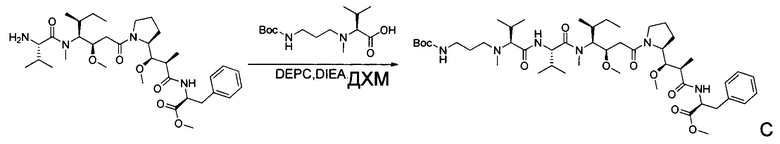
соединение 3D (157,5 мг, 0,25 ммоль, 1,00 экв) растворяли при 0°С в инертной атмосфере в 3 мл ДХМ в присутствии карбоновой кислоты 1ZF (78,7 мг, 0,27 ммоль, 1,10 экв), DEPC (46 мкл) и DIEA (124 мкл). Реакционную смесь перемешивали в течение 2 часов при низкой температуре, а затем охлаждающую баню удаляли, и перемешивание продолжали в течение 4 часов. Затем ее концентрировали при пониженном давлении с получением 200 мг соединения 6А в форме неочищенного желтого масла. Смесь использовали в таком виде в следующей стадии.
Соединение 6А (200 мг, 0,22 ммоль, 1,00 экв) растворяли в инертной атмосфере при 0°С в 2 мл ДХМ. Добавляли по каплям ТФУ (1 мл), и охлаждающую баню удаляли. Реакционную смесь перемешивали 1 час при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм) с получением 60 мг (26%, выход за 2 стадии) соединения 6 в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Zorbax Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл / мин, 40°С; от 30 до 80% метанола в воде (0,1% H3PO4) в течение 18 минут; ИЭР (C43H74N6O8, точная масса 802,56) m/z: 804 (МН+); 11,50 мин (91,5%, 210 нм).
1Н ЯМР (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.52 (d, 0.3Н, NHCO неполный обмен); 8.25 (d, 0.5Н, NHCO неполный обмен); 7.30-7.22 (m, 5Н); 4.9-4.6 (m, 3Н); 4.2-4.0 (m, 1Н); 4.0-0.86 (m, 61Н).
Эталонное соединение 7
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-Т,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-трифторуксусная кислота
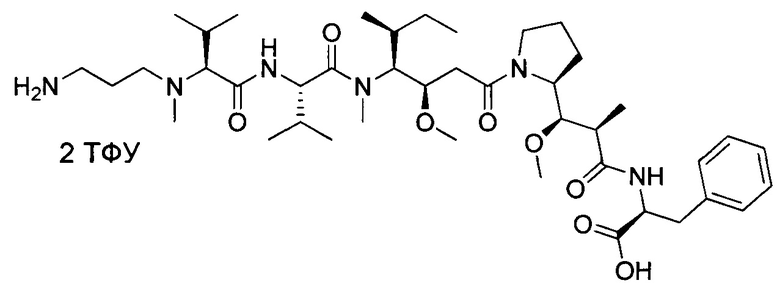
Соединение 6 (70 мг, 0,08 ммоль, 1,00 экв) растворяли в смеси воды (5 мл), ACN (2,5 мл) и пиперидин (5 мл). Реакционную смесь оставляли при перемешивании на ночь при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep C18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм) с получением 14,6 мг (21%) соединения 7 в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл / мин, 40°С; от 0 до 80% метанола в воде (0,05% ТФУ) в течение 8 минут; ИЭР (C42H72N6O8, точная масса 788,54) m/z: 790 (МН+), 5,71 мин (96,83%, 210 нм).
1Н ЯМР (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.42 (d, 0.3Н, NHCO неполный обмен); 8.15 (d, 0.2Н, NHCO неполный обмен); 7.31-7.21 (m, 5Н); 4.9-4.6 (m, 3Н); 4.25-4.0 (m, 1Н); 4.0-0.86 (m, 59Н).
Соединение 11
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((8)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамид, трифторуксусная кислота
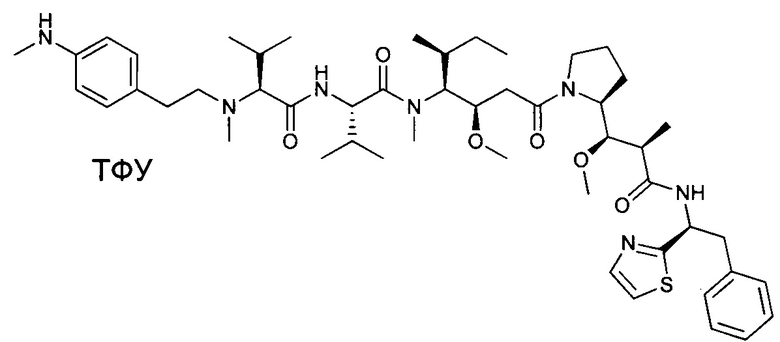
Соединение 11А: трет-бутил-N-[4-(2-гидроксиэтил)фенил]карбамат
Ди-трет-бутилдикарбонат (16,7 г, 77 ммоль, 1,05 экв) добавляли к раствору 2-(4-аминофенил)этанола (10 г, 72,9 ммоль, 1 экв) в ТГФ (200 мл), и реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Смесь разбавляли EtOAc (200 мл), промывали водой (200 мл), затем HCl 1М (100 мл), затем насыщенным водным раствором NaHCO3 (100 мл), затем соляным раствором (100 мл). Органическую фазу высушивали над MgSO4, затем выпаривали до сухого состояния при пониженном давлении. Неочищенный продукт дважды растирали с гептаном (150 мл) и высушивали в вакууме с получением соединения 11А в виде белого твердого вещества (14,7 г, 84%).
Соединение 11 В: трет-бутил-N-[4-(2-оксоэтил)фенил]карбамат
Соединение 11А (2,5 г, 10,5 ммоль, 1,00 экв) растворяли в 25 мл ДХМ, затем охлаждали до -78°С. Добавляли по каплям раствор периодинана Десса-Мартина (DMP, 6,71 г, 15,8 ммоль, 1,5 экв) в ДХМ (10 мл) Охлаждающую баню удаляли, и перемешивание продолжали в течение 1 часа при температуре окружающей среды. Реакционную смесь нейтрализовали 60 мл смеси 50/50 насыщенного водного раствора бикарбоната натрия и насыщенного водного раствора Na2S2O3. Полученный в результате раствор экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, дважды промывали насыщенным водным раствором NaCI, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (EtOAc/РЕ 1/15) с получением 1,0 г (40%) соединения 11В в форме бледно-желтого твердого вещества.
Соединение 11С: бензил-(2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил) этил](метил)амино]-3-метилбутаноат
Соединение 1ZC (3,5 г, 13,6 ммоль, 1,1 экв) растворяли в ТГФ (30 мл) в присутствии DIEA (6,4 г, 49,7 ммоль, 4,0 экв), альдегида 11 В (2,9 г, 12,3 ммоль, 1,0 экв) и триацетоксиборгидрида натрия (5,23 г, 49,7 ммоль, 2,0 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, затем нейтрализовали 60 мл насыщенного раствора бикарбоната натрия. Полученный в результате раствор экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, дважды промывали насыщенным водным раствором NaCI, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (EtOAc/РЕ 1:20) с получением 3,7 г (68%) соединения 11С в форме желтого масла.
Соединение 11D: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил)этил](метил)амино]-3-метилбутановая кислота
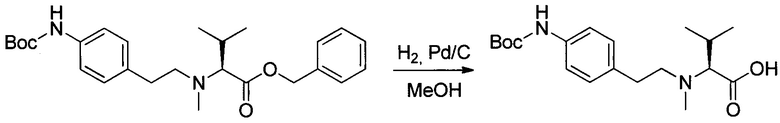
Соединение 11С (2 г, 4,5 ммоль, 1 экв) растворяли в 10 мл метанола в присутствии Pd/C (2 г) и гидрогенизировали в течение 2 часов при обычной температуре и давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 1,2 г (75%) соединения 11D в форме желтого масла.
Соединение 11Е: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил](метил)амино]фенил)этил](метил)амино]-3-метилбутановая кислота
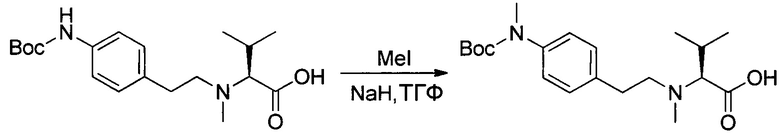
Соединение 11D (1,2 г, 3,4 ммоль, 1,00 экв) растворяли в инертной атмосфере в ТГФ (20 мл). Реакционную смесь охлаждали в ледяной бане, после чего добавляли порциями NaH (60% в масле, 549 мг, 13,7 ммоль, 4,0 экв) с последующим добавлением йодметана (4,9 г, 34 ммоль, 10 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, затем нейтрализовали водой и промывали 100 мл EtOAc. рН водного раствора доводили до 6-7 1 н. раствором HCl. Водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 800 мг (64%) соединения 11Е в форме желтого твердого вещества.
Соединение 11F: трет-бутил-N-[4-(2-[[(1S)-1-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]этил)фенил]-N-метилкарбамат
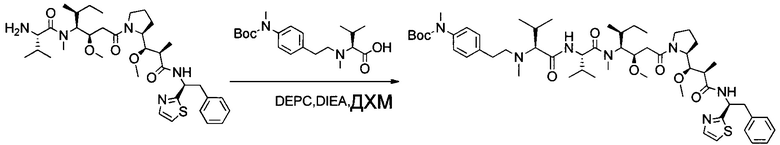
Соединение 11F было получено аналогично соединению 6А из амина 1Y (150 мг, 0,22 ммоль, 1,2 экв) и кислоты 11Е (70 мг, 0,19 ммоль, 1,0 экв). После очистки на силикагеле (EtOAc/РЕ 1:1) получили 100 мг (52%) желаемого продукта в форме бледно-желтого твердого вещества.
Соединение 11 было получено также, как и соединение 1, из промежуточного соединения 11F (100 мг, 0,1 ммоль). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм). Соединение 11 было получено при выходе 39% (39,7 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл / мин, 40°С; от 50 до 95% метанола в воде (0,05% ТФУ) в течение 18 минут); ИЭР (C50H77N7O6S, точная масса 903,57) m/z: 904,5 (МН+), 7,53 мин (93,68%, 254 нм).
1Н ЯМР (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.84 (d, 0.5Н, NHCO неполный обмен); 8.7-8.5 (m, 0.9Н, NHCO неполный обмен); 7.76-7.73 (m, 1Н); 7.55-7.4 (m, 1Н); 7.28-7.22 (m, 7Н); 7.08-7.05 (m, 2Н); 5.51-5.72 (m, 1Н); 4.9-4.80 (m, 2Н); 4.3-0.7 (m, 60Н).
Соединение 12
метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
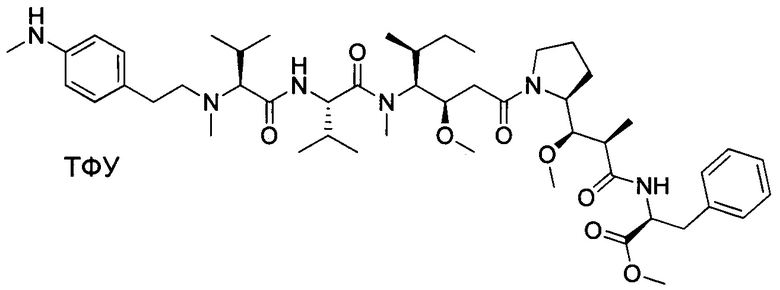
Так же, как и для конечных фаз синтеза соединения 1, соединение 12 было получено за две стадии из амина 3D (118 мг, 0,19 ммоль) и кислоты 11Е (82 мг, 0,22 ммоль). Конечный остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм). Соединение 12 было получено при выходе 7% (13,7 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл / мин, 40°С; от 40 до 95% метанола в воде (0,05% ТФУ) в течение 18 минут); ИЭР (C49H78N9O8, точная масса 878,59) m/z: 879,7 (МН+), 10,07 мин (90,6%, 254 нм).
1Н ЯМР (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.40 (se, 2Н); 7.38-7.22 (m, 7Н); 4.95-4.7 (m, 3Н); 4.2-4.0 (m, 1Н); 3.9-0.86 (m, 62Н).
Соединение 13
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамидо)-3-метокси-5-метил гептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
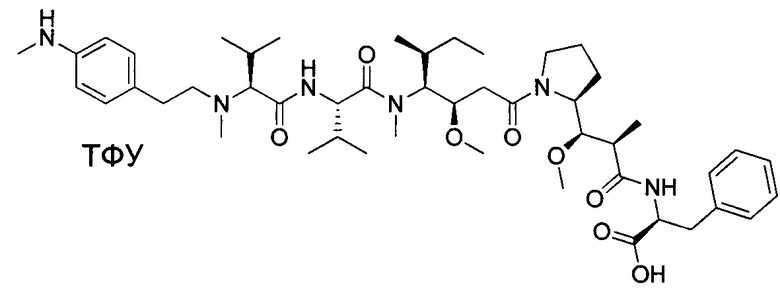
Соединение 13 было получено так же, как и соединение 7, из соединения 12 (100 мг, 0,10 ммоль). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489, детектор УФ при 254 нм и 220 нм). Соединение 13 было получено при выходе 20% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл / мин, 40°С; от 10 до 95% метанола в воде (0,05% ТФУ) в течение 8 минут; ИЭР (C48H76N6O8, точная масса 864,57) m/z: 865,6 (МН+), 6,05 мин (90,9%, 210 нм).
1Н ЯМР: (300 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.32-7.19 (m, 9Н); 4.9-4.65 (m, 3Н); 4.2-4.0 (m, 1Н); 3.9-0.86 (m, 59Н).
Соединение 14
(S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота
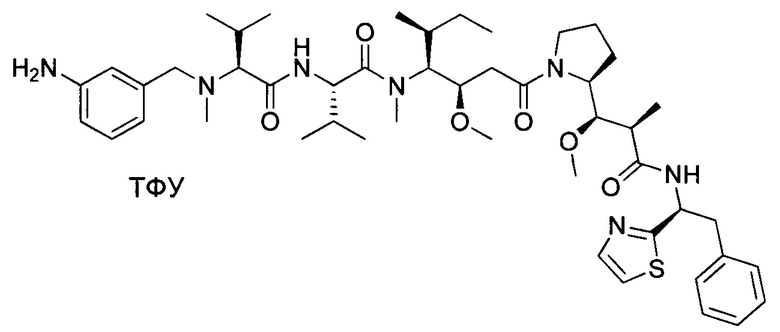
Соединение 14А: трет-бутил (3-(гидроксиметил)фенил)карбамат
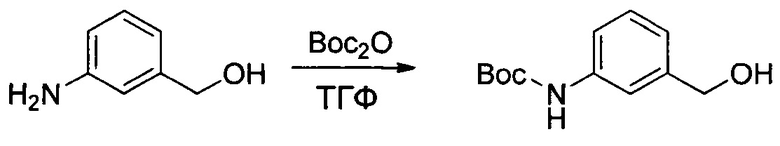
(3-Аминофенил)метанол (3 г, 24,36 ммоль, 1,00 экв) растворяли в ТГФ (60 мл), после чего добавляли ди-трет-бутилдикарбонат (6,38 г, 29,23 ммоль, 1,20 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, а затем реакционную смесь разбавляли добавлением 200 мл воды. Продукт экстрагировали 3 раза 100 мл AcOEt, а затем органические фазы повторно объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта (13,85 г соединения 14А) в форме желтого масла.
Соединение 14В: трет-бутил(3-формилфенил)карбамат
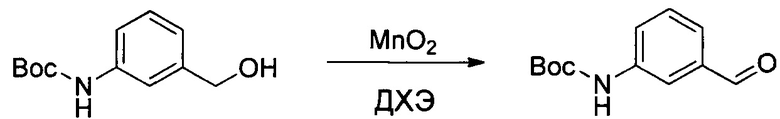
Соединение 14А (13,8 г, 61,81 ммоль, 1,00 экв) растворяли в ДХЭ (400 мл), а затем добавляли MnO2 (54 г, 621,14 ммоль, 10,05 экв). Смесь оставляли при перемешивании при температуре окружающей среды на 3 дня, после чего твердые вещества удаляли фильтрованием. Фильтрат выпаривали до сухого состояния, и остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:30) с получением 3 г (22%) соединения 14В в форме белого твердого вещества.
Соединение 14С: бензил (S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутаноат
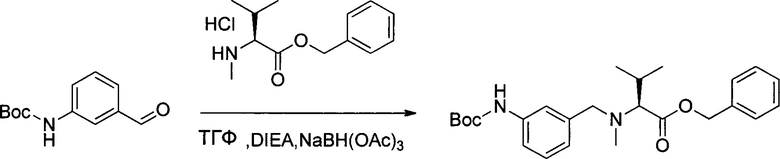
Соединение 14В (1 г, 4,52 ммоль, 1,00 экв) растворяли в 20 мл ТГФ в присутствии соединения 1ZC (1,16 г, 4,50 ммоль, 1,00 экв), DIEA (3 мл) и NaBH(OAc)3 (1,92 г, 9,06 ммоль, 2,01 экв). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, а затем нейтрализовали 100 мл воды и экстрагировали 3 раза 50 мл AcOEt. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и РЕ (1:50) с получением 1,9 г (99%) соединения 14С в форме белого твердого вещества.
Соединение 14D: (S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутановая кислота
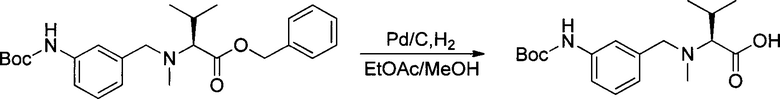
Соединение 14С (1 г, 2,34 ммоль, 1,00 экв) растворяли в 30 мл AcOEt и 4 мл метанола в присутствии Pd/C (400 мг) и гидрогенизировали в течение 1 часа при температуре окружающей среды и атмосферном давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 680 мг (86%) соединения 14D в форме белого твердого вещества.
Соединение 14Е: трет-бутил (3-((3S,6S,9S,10R)-9-((S)-втор-бутил)-3,6-диизопропил-10-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)фенил)карбамат
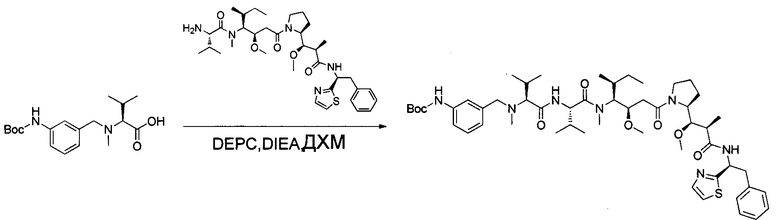
Соединение 14Е синтезировали так же, как для соединения 3, из амина 1Y (100 мг, 0,15 ммоль, 1,00 экв), кислоты 14D (102,27 мг, 0,30 ммоль, 2,00 экв), DEPC (0,053 мл) и DIEA (0,046 мл) в ДХМ (3 мл). Неочищенный продукт (80 г) очищали на колонке силикагеля смесью EtOAc и РЕ (1:1) с получением 100 мг (67%) соединения 14Е в форме бледно-желтого масла.
Соединение 14 синтезировали так же, как соединение 2, из промежуточного соединения 14Е (100 мг, 0,10 ммоль, 1,00 экв). Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05%) ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2545, детектор УФ при 254 нм и 220 нм). Соединение 14 было получено при выходе 10% (10 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse plus С8, 3,5 мкм, 4,6×150 мм; 40°С; 1,0 мл / мин, от 40% до 95% МеОН в воде (0,05% ТФУ) в течение 18 минут); ИЭР (C48H73N7O6S, точная масса 875,5) m/z: 876,5 (МН+) и 438,9 (М.2Н+/2, 100%), 11,35 мин (95,6%, 210 нм).
1Н ЯМР (400МГц, CD3OD, ppm): δ (присутствие ротамеров) 8.92-8.86 (m, 0.4Н, NH неполный обмен); 8.70-8.54 (m, 0.6Н, NH неполный обмен); 7.88-7.78 (m, 1Н); 7.60-7.50 (m, 1Н); 7.45-6.97 (m, 9Н); 5.80-5.65 (m, 1Н); 4.85-4.70 (m, 1Н); 4.40-0.80 (m, 56Н).
Соединение 15
метил (S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
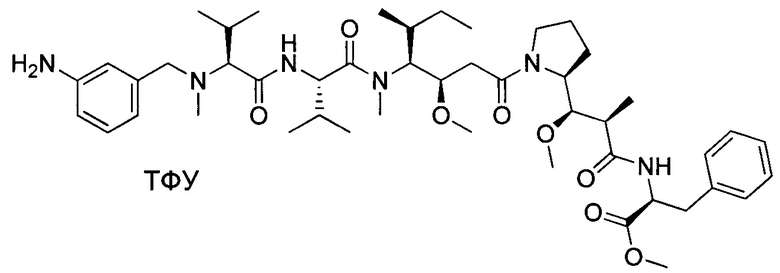
Соединение 15А: Метил-(S)-2-((2R,3R)-3-((S)-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-Т,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
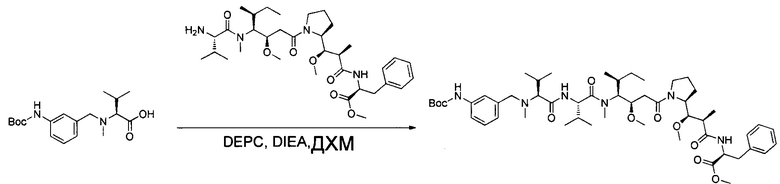
Соединение 15А синтезировали так же, как для соединения 3, из амина 3D (200 мг, 0,32 ммоль, 1,00 экв), кислоты 14D (212,6 мг, 0,63 ммоль, 2,00 экв), DEPC (0,1103 мл) и DIEA (0,157 мл, 3,00 экв) в ДХМ (5 мл). Неочищенный продукт очищали на колонке силикагеля смесью EtOAc и РЕ (1:1) с получением 200 мг (67%) соединения 15А в форме желтого твердого вещества.
Соединение 15: Соединение 15 синтезировали так же, как соединение 2, из промежуточного соединения 15А (200 мг, 0,21 ммоль, 1,00 экв). Неочищенный продукт очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2545, детектор УФ при 254 нм и 220 нм). Соединение 15 было получено при выходе 19% (38,6 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл / мин, от 10% до 95% МеОН в воде (0,05% ТФУ) в течение 8 минут; ИЭР (C47H74N6O8, точная масса 850,5) m/z: 851,5 (МН+) и 426,4 (М.2Н+/2, 100%), 6,61 min (91,1%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.53-7.42 (m, 1Н); 7.35-7.18 (m, 8Н); 4.88-4.79 (m, 2Н); 4.42-4.00 (m, 3Н); 3.93-2.71 (m, 22Н); 2.61-0.81 (m, 33Н).
Соединение 20
(S)-2-((S)-2-((4-аминобензил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота
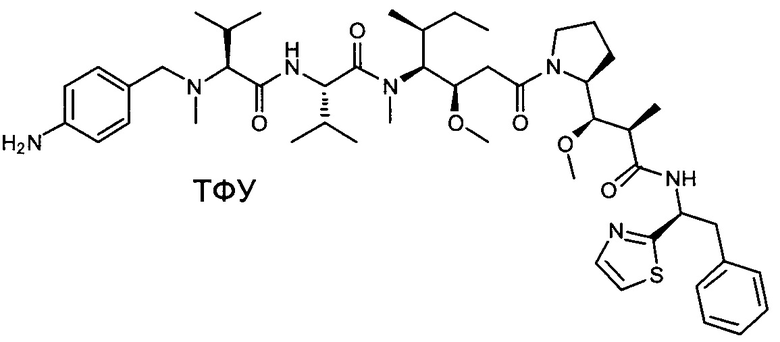
Соединение 20 было получено так же, как и соединение 1, из амина 1ZC и соответствующего альдегида.
4-Нитробензальдегид, вовлеченный в получение соединения 20, приобретен коммерческим путем.
Синтез соединения 20 выполняли путем восстановления нитрогруппы. Синтез проводили следующим образом: (2S)-N-[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-2-[[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]карбамоил]-1-метокси-2-метилэтил]пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил]-N,3-диметил-2-[(2S)-3-метил-2-[метил[(4-нитрофенил)метил]амино]бутанамидо]бутанамид (40 мг, 0,05 ммоль, 1,0 экв) растворяли в 15 мл этанола. Добавляли дигидрат хлорида олова (II) (317 мг, 1,4 ммоль, 30 экв), и раствор оставляли при перемешивании на 3 дня при температуре окружающей среды. Реакционную смесь нейтрализовали 50 мл воды, затем экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 20 в неочищенном состоянии (чистота: 93,2%; количество: 21,6 мг).
Соединение очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2489 детектор УФ при 254 нм и 220 нм).
1Н ЯМР: (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.6-7.5 (m, 1Н); 7.4-7.15 (m, 5Н); 7.1-7.05 (m, 2Н); 6.73-6.70 (m, 2Н); 5.8-5.55 (m, 1Н); 5.0-4.7 (m, 2Н); 4.25-4.05 (m, 1Н); 4.0-0.8 (m, 54Н). ЖХ/МС/УФ ИЭР: (C48H73N7O7S, точная масса 875,53) m/z 876 (МН+), 439 [75%, (М.2Н+)/2]; УФ: время удерживания (RT) равно 4,83 мин (96,8%, 254 нм). 1Н ЯМР (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.6-7.5 (m, 1Н); 7.4-7.1 (m, 7Н); 6.76-6.72 (m, 2Н); 5.8-5.55 (m, 1Н); 4.9-4.65 (m, 2Н); 4.25-4.05 (m, 1Н); 4.0-0.8 (m, 54Н).
Соединение 29
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
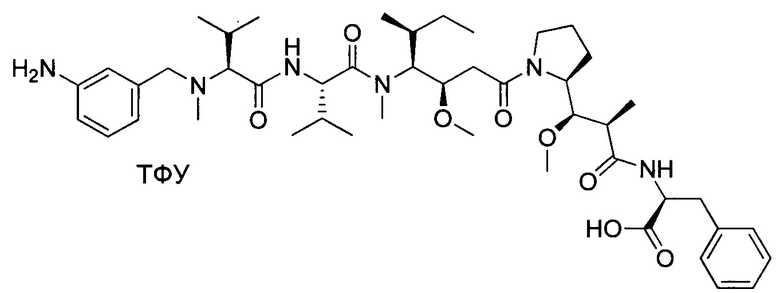
Соединение 15 (100 мг, 0,10 ммоль, 1,00 экв) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли на ночь при перемешивании при температуре окружающей среды, а затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода / ACN, забуференная 0,05% ТФУ; градиент от 20% до 40% ACN в течение 10 минут, затем от 40% до 100% ACN в течение 2 минут; Waters 2545 детектор УФ при 254 нм и 220 нм).
ЖХ/МС/УФ (колонка Eclipse plus С8, 3,5 мкм, 4,6×150 мм; 40°С; 1,0 мл / мин, от 40% до 95% МеОН в воде (0,05% ТФУ) в течение 18 минут); ИЭР (C46H72N6O8, точная масса 836,54) m/z: 837,5 (МН+) и 419,4 (М.2Н+/2, 100%), 10,61 мин (92,5%, 210 нм).
1Н ЯМР: (400 МГц, CD3OD, ppm): δ (присутствие ротамеров) 7.38-7.15 (m, 6Н); 7.00-6.99 (m, 3Н); 4.85-4.68 (m, 2Н); 4.37-3.38 (m, 11Н); 3.31-2.70 (m, 8Н); 2.60-0.82 (m, 35Н).
Соединение 61
(S)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N-(3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2- ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
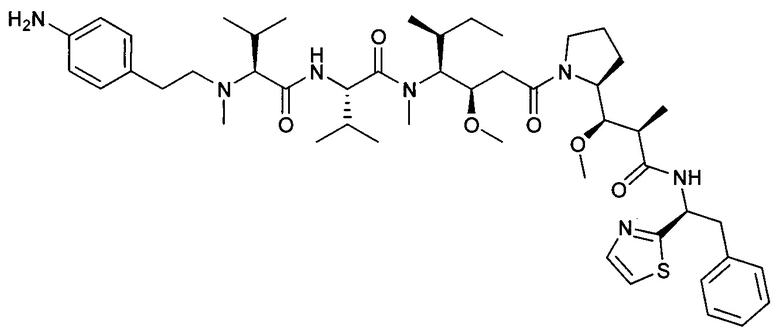
Соединение 61А: Дигидрохлорид N-(4-аминофенетил)-N-метил-L-валина
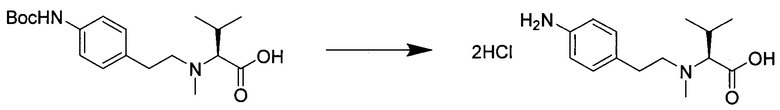
Соединение 11D (962 мг, 2,75 ммоль) растворяли в 10 мл имеющегося в продаже раствора HCl в пропан-2-оле (5-6 М) и перемешивали при комнатной температуре в течение 2 часов. Анализ ТСХ показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и полученное в результате желтое твердое вещество растирали с Et2O (2×10 мл). Продукт высушивали в вакууме с получением соединения 61А в виде желтого твердого вещества (322 мг, 47%).
Соединение 61: Карбоновую кислоту 61А (73 мг, 0,23 ммоль, 1 экв) и амин 1Y (150 мг, 0,23 ммоль, 1 экв) растворяли в сухом ДМФ (2 мл). Добавляли DIEA (158 мкл, 0,90 ммоль, 4 экв) и DECP (также называемый DEPC) (51 мкл, 0,34 ммоль, 1,5 экв), и реакционную смесь перемешивали в течение 4 часов при комнатной температуре. Анализ с помощью ЖХ-МС показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле (ДХМ/МеОН) с получением соединения 61 в виде светло-желтого твердого вещества (83 мг, 40%).
1Н ЯМР: (500 МГц, ДМСО-d6, ppm): δ (присутствие ротамеров), 8.86 (d, 0.5Н, NHCO); 8.65 (d, 0.5Н, NHCO), 8.11-8.05 (m, 1H, NHCO), 7.80 (d, 0.5H, тиазол), 7.78 (d, 0.5H, тиазол), 7.65 (d, 0.5H, тиазол), 7.63 (d, 0.5H, тиазол), 7.32-7.12 (m, 5H), 6.83 (d, J равно 8.3 Гц, 2H), 6.45 (d, J равно 8.3 Гц, 2H), 5.56-5.49 (m, 0.5 H), 5.42-5.35 (m, 0.5H), 4.78 (s, 2H, NH2), 4.74-4.46 (m, 2H), 4.01-0.66 (m, 57H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°С, от 0 до 95% MeCN в воде (0,1% ТФУ) в 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr составляет 1,31 мин (96,5%, 220 нм).
m/z (Q-TOF ИЭР+) 890,5558 (2%, МН+, C49H76N7O6S требуется 890,5572), 445,7834 (100%, (МН2)2+, C49H77N7O6S требуется 445,7823).
Соединение 62
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат
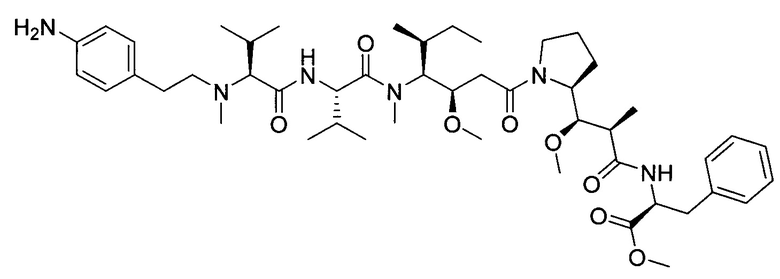
Соединение 62 было получено так же, как и соединение 61, с использованием карбоновой кислоты 61А (69 мг, 0,21 ммоль, 1 экв), амина 3D (135 мг, 0,21 ммоль, 1 экв), DIEA (75 мкл, 0,43 ммоль, 2 экв) и DECP (49 мкл, 0,32 ммоль, 1,5 экв). Неочищенный продукт очищали флэш-хроматографией на силикагеле (ДХМ/МеОН) с получением соединения 62 в виде желтоватого твердого вещества (82 мг, 45%).
1Н ЯМР: (500 МГц, ДМСО-d6, ppm): δ (присутствие ротамеров), 8.50 (d, J равно 8.3, 0.5Н, NHCO); 8.27 (d, J равно 8.0, 0.5H, NHCO), 8.15-8.04 (m, 1H, NHCO), 7.27-7.13 (m, 5H), 6.86-6.79 (m, 2H), 6.48-6.42 (m, 2H), 4.78 (s, 2H, NH2), 4.74-4.44 (m, 3H), 4.01-3.72 (m, 1.5H), 3.66 (s, 1.5H, CO2Me), 3.63 (s, 1.5H, CO2Me), 3.57-0.65 (m, 55.5H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) в течение 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr равно 1,29 мин (95,3%, 220 нм).
m/z (Q-TOF ИЭР+) 865,5800 (2%, МН+, C48H77N6O8 требуется 865,5797), 433,2937 (100%, (МН2)2+, C48H78N6O8 требуется 433,2935).
Соединение 63
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((3)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
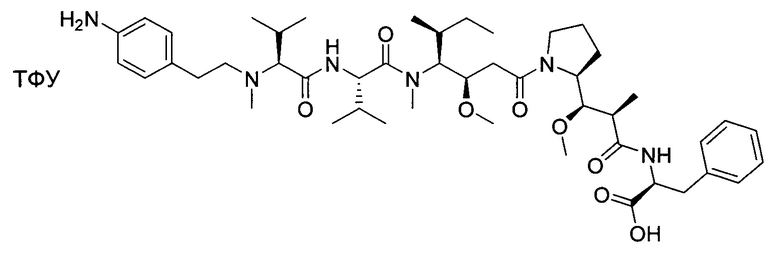
Соединение 62 (23 мг, 0,03 ммоль) растворяли в смеси воды (1 мл) и ацетонитрила (1 мл). Добавляли пиперидин (0,75 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Анализ ТСХ показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода/MeCN, забуференная 0,1% ТФУ; градиент от 20%) до 40%) MeCN в течение 10 минут, затем от 40% до 100% MeCN в течение 2 минут; Waters 2545, детектор УФ при 254 нм и 220 нм). Соединение 63 было получено в виде белого твердого вещества (14 мг, 66%).
1Н ЯМР: (500 МГц, ДМСО-d6, ppm): δ (присутствие ротамеров), 12.7 (s(br), 1Н, CO2H), 9.58 (m(br), 1Н); 9.04-8.89 (m, 1Н), 8.41 (d, 0.6Н, NHCO), 8.15 (d, 0.4Н, NHCO), 7.27-7.13 (m, 5H), 7.13-6.99 (m(br), 2H), 6.90-6.64 (s(br), 2H), 4.77-3.40 (m, 10H), 3.34-2.75 (m, 20H), 2.34-1.94 (m, 4H), 1.90-0.7 (m, 25H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) в течение 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr равно 1,24 мин (100%, 220 нм).
m/z (Q-TOF ИЭР+) 851,5641 (6%, МН+, C47H75N6O8 требуется 851,5641), 426,2854 (100%, (МН2)2+, C47H76N6O8 требуется 426,2857).
Пример 15: Антипролиферативная активность лекарственных средств
Методика:
Культура клеток Клетки А549 (немелкоклеточный рак легкого - АТСС CCL-185) и MDA-MB-231 (аденокарцинома молочной железы - АТСС НТВ-26) культивировали в минимальной питательной среде Игла (MEM) с 5% фетальной телячьей сывороткой (ФСТ) и модифицированной Дульбекко среде Игла (DMEM) с 10% ФСТ соответственно. Клетки MCF7 (дуктальная карцинома молочной железы - АТСС НТВ-22) и клетки SN-12C (карцинома почки - АТСС) поддерживали в среде RPMI1640 (без фенолового красного для клеток MCF7), содержащей 10% ФСТ. Во все среды добавляли фунгизон (1,25 мкг/мл) и пенициллин-стрептомицин (100 ед./ 100 мкг/мл). Клетки культивировали в стандартных условиях в инкубаторе при 37°С, 5% CO2 и 95% атмосферной влажности.
Антипролиферативная активность на 4 линиях опухолевых клеток Выбранные лекарственные средства исследовали на их антипролиферативную активность с использованием количественного определения пролиферации ATPIite (Perkin Elmer, Villebon sur Yvette, Франция) на объемной панели из 4 линий клеток. Клетки высевали в 96-луночные планшеты (103 клеток/лунка для А549, 2.103 для MCF7, MDA-MB-231 и SN12C) в день 0 при концентрации, обеспечивающей сохранение клеток в логарифмической фазе роста на протяжении 72 ч периода обработки лекарственным средством. После 24 ч периода инкубации все клетки инкубировали с серийными разведениями тестируемых соединений (11 мкл 10× раствора в 1% ДМСО - 6 клеток/условие). Во избежание прилипания соединений к наконечникам пипеток наконечники меняли между двумя последовательными разведениями. Затем клетки помещали в инкубатор при 37°С, 5% CO2. На день 4 жизнеспособность клеток оценивали путем определения дозы АТФ, высвобождаемого жизнеспособными клетками. Проводили анализ числа жизнеспособных клеток по сравнению с числом клеток, инкубируемых с растворителем. Значения ЕС50 определяли с помощью анализа аппроксимации кривой (модели нелинейной регрессии с сигмоидной кривой доза-ответ, переменный коэффициент наклона Хилла), проведенного с помощью алгоритма, обеспечиваемого программой GraphPad (GraphPad Software Inc., штат Калифорния, США).
Результаты:
Различные лекарственные средства:
Различные лекарственные средства исследовали с определением их антипролиферативной активности на линии клеток MDA-MB-231, следуя описанной выше методике. При измерении активностей получили значения ЕС50 менее 0,1 мкМ.
В нескольких приведенных ниже Примерах, выбранных из приведенных выше иллюстративных лекарственных средств, показаны их полностью значимые антипролиферативные свойства:
Пример 12: ЕС50=5,80×10-10 М; Пример 13: ЕС50=7,95×10-8 М; Пример 15: ЕС50=1,70×10-10 М; Пример 27: ЕС50=1,20×10-10М.
Различные линии клеток:
Соединение 15 исследовали на различных линиях клеток (А549, MDA-MB-231, MCF-7, SN12C), следуя описанной выше методике. При измерении активностей на всех исследованных линиях клеток получили значения ЕС50 менее 0,1 мкМ.
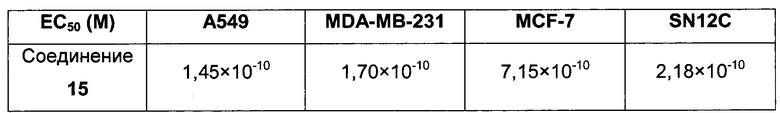
Сравнительные примеры:
Замещение на фенильном кольце (амино по сравнению с карбоксилом) было исследовано в приведенных ниже сравнительных примерах и показало улучшенную антипролиферативную активность лекарственных средств согласно изобретению, содержащих амино-заместитель.
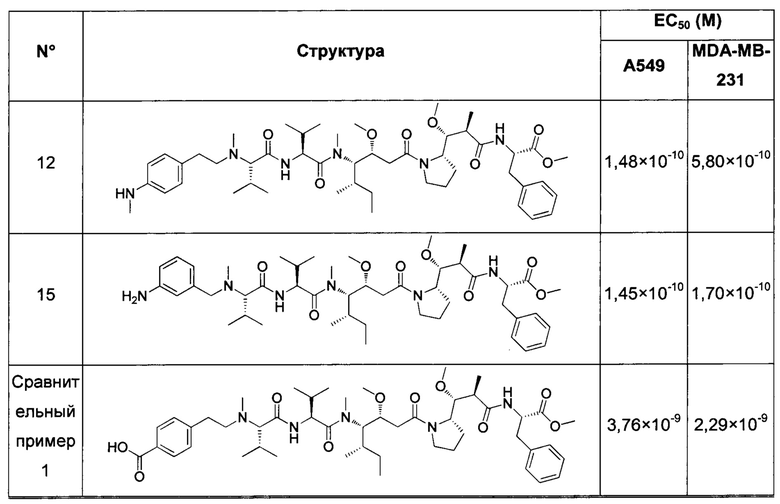
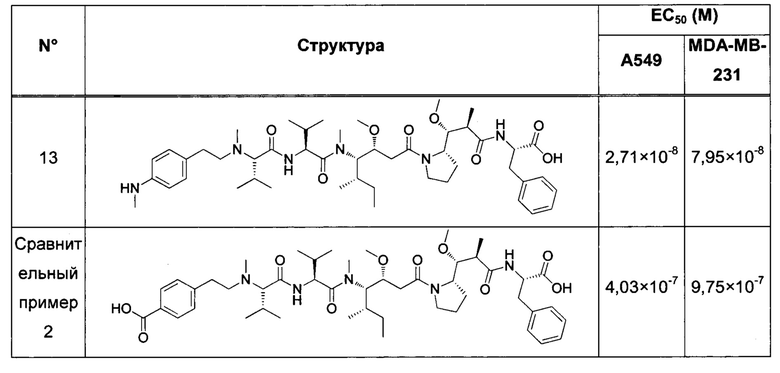
Пример 16: Синтез группировки лекарственное средство-линкер
Соединение Е-11
4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил(4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)(метил)карбамата 2,2,2-трифторацетат
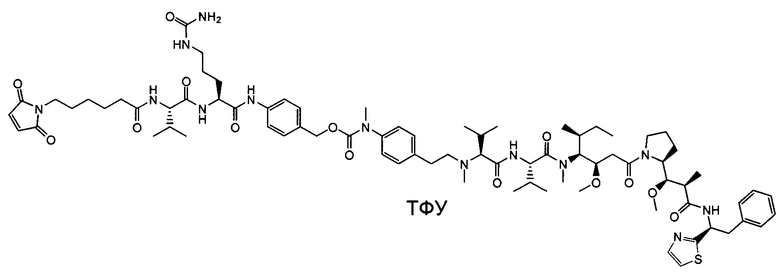
Соединение Е-11-1: гидрохлорид метил-(S)-2-амино-5-уреидопентаноата
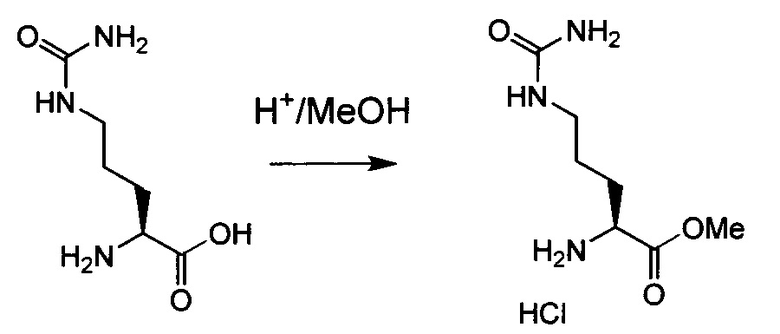
Ацетилхлорид (10 мл) добавляли по каплям к МеОН (120 мл) при 0°С при перемешивании. Через 20 минут добавляли L-цитруллин (10 г, 57 ммоль, 1,00 экв), и смесь нагревали с обратным холодильником в течение ночи. Растворитель выпаривали при пониженном давлении с получением 15 г (116%) соединение Е-11-1 в виде белого твердого вещества. Продукт использовали в следующей стадии без дополнительной сушки.
Соединение Е-11-2: метил-(S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентаноат
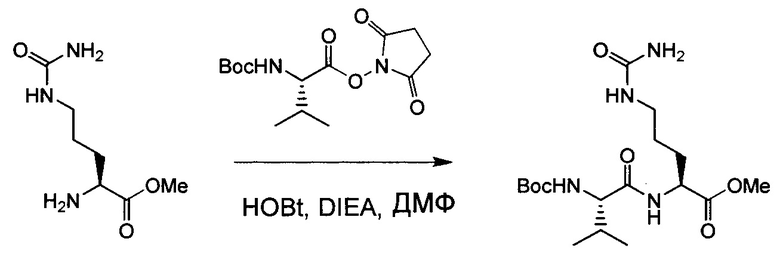
Соединение Е-11-1 (13 г, 57,6 ммоль, 1,1 экв) растворяли в ДМФ (140 мл) при 0°С в инертной атмосфере. Добавляли DIEA (30 мл, 173 ммоль, 3,0 экв), гидроксибензотриазол (HOBt - 10,59 г, 69,1 ммоль, 1,2 экв) и сложный эфир гидроксисукцинимид Boc-L-валина (Boc-Val-OSu - 18,1 г, 57,6 ммоль, 1,0 экв). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды, затем растворитель выпаривали при пониженном давлении. Остаток растворяли в воде (100 мл) и дважды экстрагировали ДХМ (150 мл). Органические фазы объединяли, высушивали над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали на силикагеле (ДХМ/МеОН) с получением 18,8 г (84%) соединения Е-11-2 в виде белого твердого вещества.
Соединение Е-11-3: (S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентановая кислота
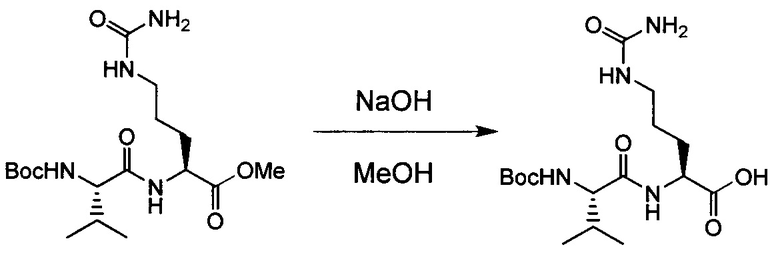
Соединение Е-11-2 (18,8 г, 48,4 ммоль, 1 экв) растворяли в МеОН (200 мл) при 0°С. Добавляли раствор NaOH 1 М (72 мл, 72 ммоль, 1,5 экв), и смесь перемешивали в течение 2 часов при комнатной температуре. МеОН удаляли при пониженном давлении, и остаточный водный раствор подкисляли HCl 1 М. Водную фазу выпаривали до сухого состояния, и остаток очищали на силикагеле (ДХМ/МеОН) с получением 18 г (99%) соединения Е-11-3 в виде белого твердого вещества.
Соединение Е-11-4: трет-бутил-((S)-1-(((S)-1-((4-(гидроксиметил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-3-метил-1-оксобутан-2-ил)карбамат
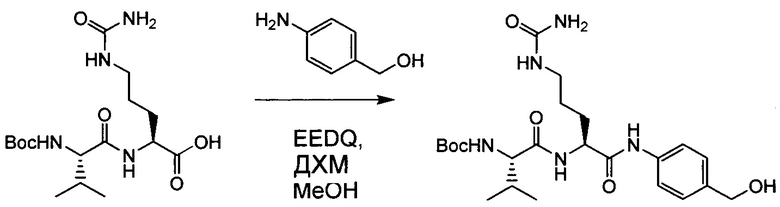
Соединение Е-11-3 (5 г, 13,4 ммоль, 1 экв) растворяли в смеси безводного ДХМ (65 мл) и безводного МеОН (35 мл). Добавляли (4-аминофенил)метанол (1,81 г, 14,7 ммоль, 1,1 экв) и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ - 6,60 г, 26,7 ммоль, 2 экв), и смесь перемешивали в темноте в течение ночи. Растворители выпаривали при пониженном давлении, и остаток очищали на силикагеле (ДХМ/МеОН) с получением 5,2 г (73%) соединения Е-11-4 в виде беловатого твердого вещества.
Соединение Е-11-5: трет-бутил-((S)-3-метил-1-(((S)-1-((4-((((4-нитрофенокси)карбонил)окси)метил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-1-оксобутан-2-ил)карбамат
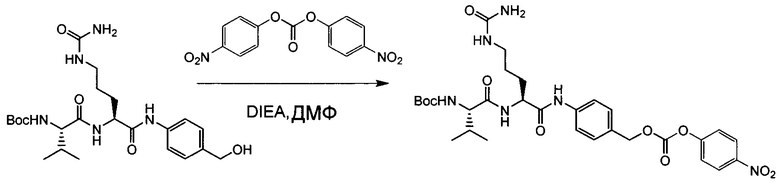
Соединение Е-11-4 (1,1 г, 2,29 ммоль, 1 экв) растворяли в безводном ДМФ (5 мл) при температуре окружающей среды в инертной атмосфере. Добавляли бис(4-нитрофенил)карбонат (1,40 г, 4,59 ммоль, 2 экв) с последующим добавлением DIEA (600 мкл, 3,44 ммоль, 1,5 экв.), и полученный в результате желтый раствор перемешивали в течение ночи. ДМФ выпаривали при пониженном давлении, и остаток очищали на силикагеле (ДХМ/МеОН) с получением 1,27 г (84%) соединения Е-11-5 в виде беловатого твердого вещества.
Соединение Е-11-6: 4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил (4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11- диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)(метил)карбамата 2,2,2-трифторацетат
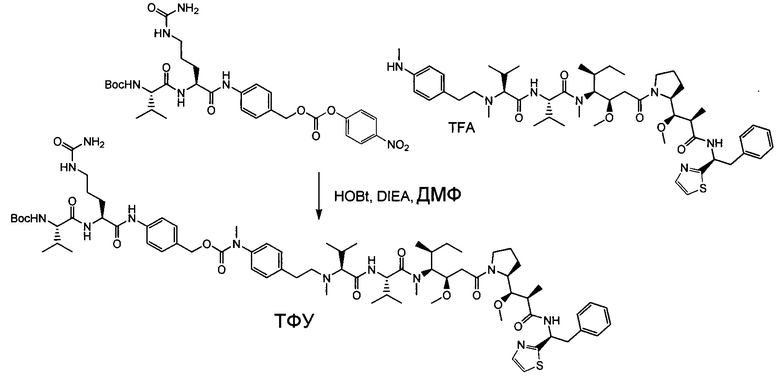
Карбонат Е-11-5 (114 мг, 0,177 ммоль, 1,2 экв) и анилин 11F (150 мг, 0,147 ммоль, 1 экв) растворяли в безводном ДМФ (4 мл). Добавляли HOBt (38 мг, 0,295 ммоль, 2 экв) и DIEA (54 мкл, 0,295 ммоль, 2 экв), и смесь перемешивали в течение выходных дней при комнатной температуре. ДМФ выпаривали при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле, элюируя ДХМ. Продукт подвергали переочистке препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединение Е-11-6 в виде белого твердого вещества (89 мг, 39%).
Соединение Е-11:
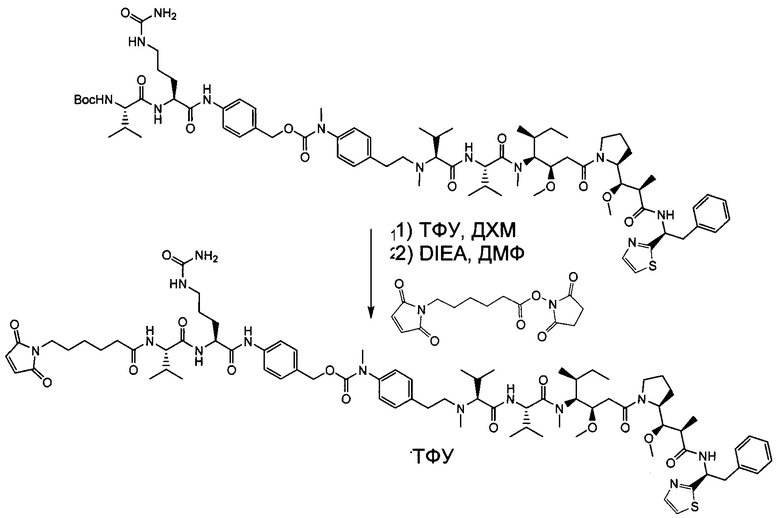
Соединение Е-11-6 (21 мг, 0,014 ммоль, 1,0 экв) растворяли в ДХМ (0,25 мл) и добавляли ТФУ (40 мкл). Раствор перемешивали в течение 2 часов при комнатной температуре, после чего анализ ЖХ-МС показал полное расходование исходного вещества. Смесь быстро охлаждали (баня с жидким азотом), при этом одновременно добавляли ДМФ (0,5 мл), затем DIEA (100 мкл), чтобы нейтрализовать ТФУ. Затем охлаждающую баню удаляли и добавляли 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1 -ил)гексаноат (4 мг, 0,012 ммоль, 1 экв). Смесь перемешивали при комнатной температуре в течение 48 часов, и продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487 детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения Е-11 в виде белого твердого вещества (11 мг, 54%). m/z (Q-TOF МС ИЭР+) 1524,8282 (2%, MNa+, C79H115N13NaO14S требуется 1524,8299), 751,9283 (100%, (МН2)2+ C79H117N13O14S требуется 751,9276).
Соединение Е-12
метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5- метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
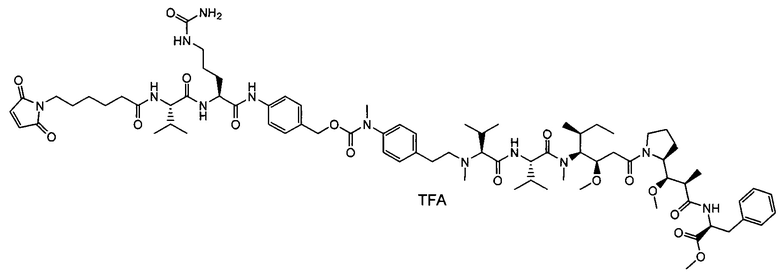
Соединение Е-12-1: трет-бутил-((S)-3-метил-1-оксо-1-(((S)-1-оксо-1-((4-((((перфторфенокси)карбонил)окси)метил)фенил)амино)-5-уреидопентан-2-ил)амино)бутан-2-ил)карбамат
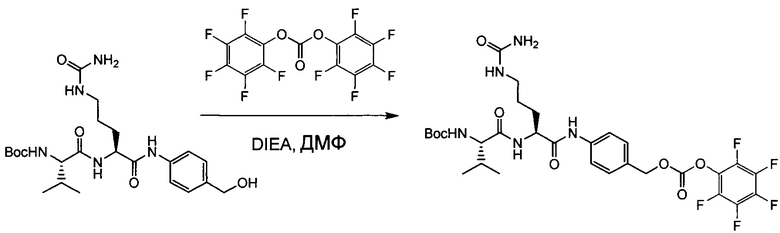
Соединение Е-11-4 (670 мг, 1,26 ммоль, 1 экв) растворяли в безводном ДМФ (6 мл) при 0°С в инертной атмосфере. Добавляли бис(перфторфенил)карбонат (991 мг, 2,51 ммоль, 2 экв) с последующим добавлением DIEA (329 мкл, 1,89 ммоль, 1,5 экв), и полученный в результате бесцветный раствор перемешивали в течение 30 минут при комнатной температуре. ДМФ выпаривали при пониженном давлении, и остаток очищали на силикагеле (ДХМ/МеОН) с получением 836 мг (96%) соединения Е-12-1 в виде беловатого твердого вещества.
Соединение Е-12-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
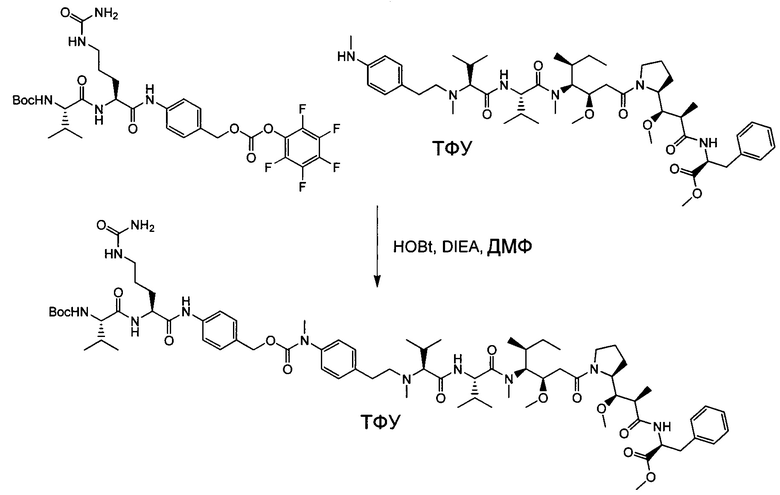
Анилин 12 (165 мг, 0,189 ммоль, 1,0 экв) растворяли в ДМФ (5 мл) при 0°С в инертной атмосфере. Добавляли карбонат Е-12-1 (194 мг, 0,282 ммоль, 1,5 экв), HOBt (51 мг, 0,375 ммоль, 2 экв) и DIEA (66 мкл, 0,375 ммоль, 2 экв), и смесь перемешивали при комнатной температуре в течение 8 часов. Растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487 детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения Е-12-7 в виде белого твердого вещества (247 мг, 77%).
Соединение Е-12-3: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
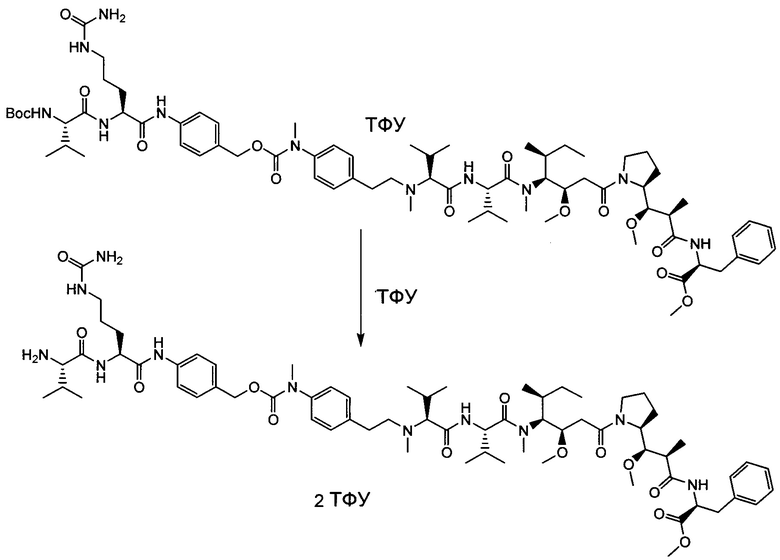
Соединение Е-12-2 (5,6 мг, 4,04 мкмоль, 1,0 экв) растворяли ТФУ (100 мкл). Через 5 минут добавляли 2 мл воды, и смесь лиофилизировали в течение ночи с получением соединения Е-12-3 в виде беловатого твердого вещества (5,6 мг, 98%).
Соединение Е-12:
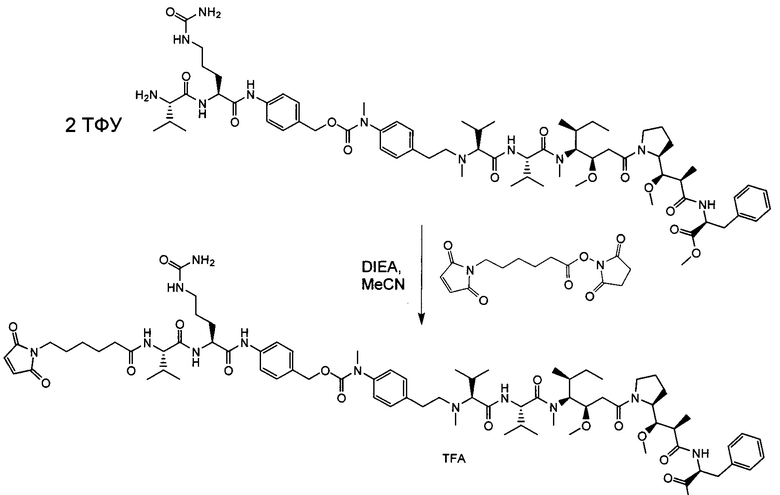
Соединение Е-12-3 (5,6 мг, 4 мкмоль, 1,0 экв) растворяли в ацетонитриле (0,5 мл) и добавляли DIEA (5 мкл, 7 экв) с последующим добавлением 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноата (2,5 мг, 8 мкмоль, 2 экв). Смесь перемешивали в течение 6 часов при комнатной температуре. После проверки реакции по ЖХ-МС добавляли 200 мкл воды, и полученный в результате раствор очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения Е-12 в виде белого твердого вещества (4,6 мг, 70%).
m/z (Q-TOF МС ИЭР+) 739,4389 (100%, (МН2)2+, C78H118N12Ol6 требуется 739,4389).
Соединение Е-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
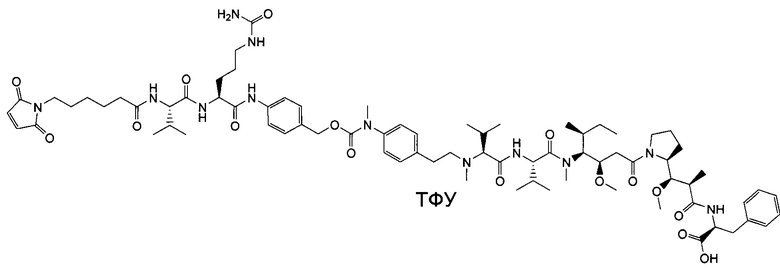
Соединение Е-13-1: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин
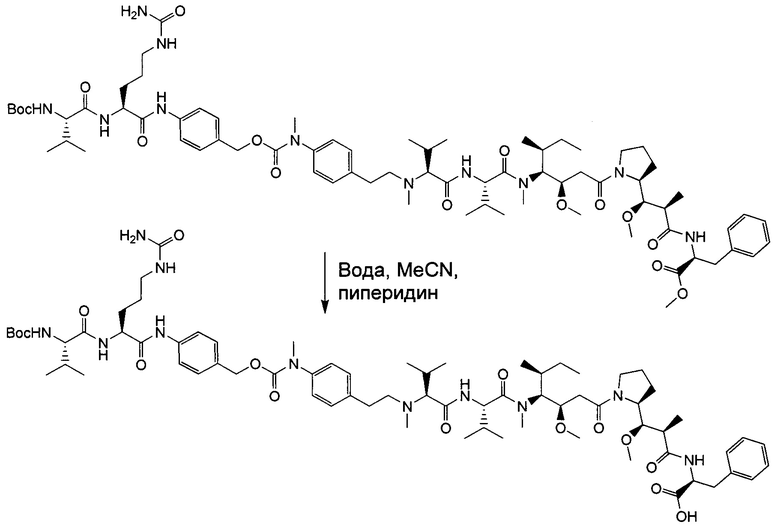
Соединение Е-12-2 (185 мг, 0,123 ммоль, 1,0 экв) растворяли в смеси воды (5 мл) и ацетонитрила (5 мл) при комнатной температуре. Добавляли пиперидин (3,67 мл, 300 экв), и смесь перемешивали в течение 6 часов при комнатной температуре. Растворители выпаривали до сухого состояния при пониженном давлении, и остаток растирали с Et2O (60 мл). Твердое вещество дважды промывали Et2O (20 мл) и высушивали в вакууме с получением соединения Е-13-1 в виде беловатого твердого вещества (175 мг, 95%).
Соединение Е-13-2: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина бис-(2,2,2-трифторацетат)
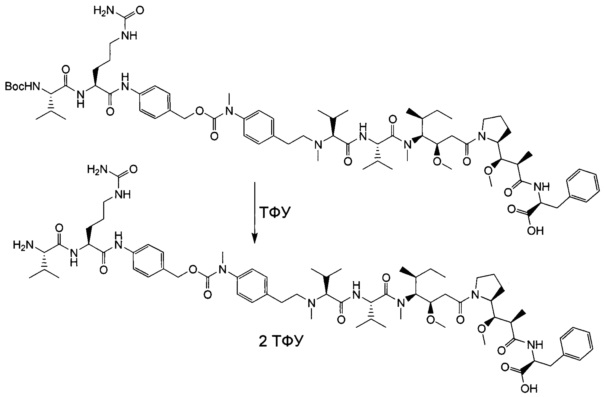
Соединение Е-13-1 (175 мг, 0,128 ммоль, 1,0 экв) растворяли ТФУ (200 мкл). Через 5 минут добавляли воду (1 мл) и ацетонитрил (1 мл), и раствор лиофилизировали в течение ночи с получением соединения Е-12-2 в виде беловатого твердого вещества (180 мг, 87%).
Соединение Е-13: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)(метил)амино)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
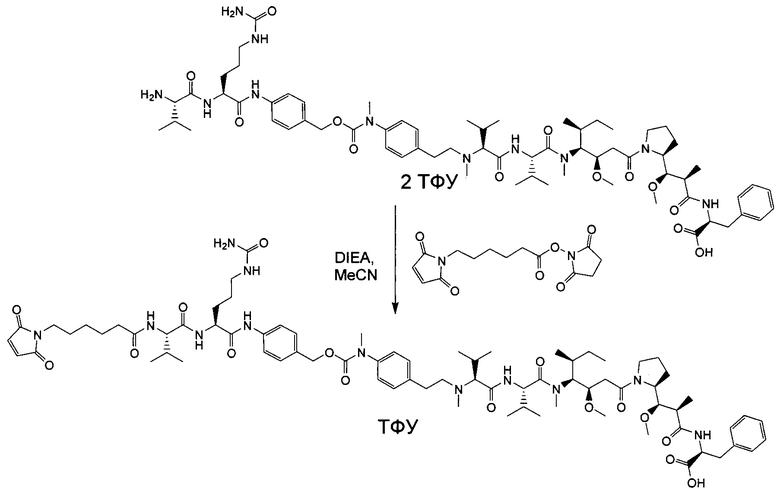
Соединение Е-13-2 (80 мг, 0,058 ммоль, 1,0 экв) растворяли в смеси ацетонитрила (1,5 мл) и ДМФ (0,4 мл). Добавляли DIEA (50 мкл, 0,289 ммоль, 5 экв) с последующим добавлением 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноата (36 мг, 0,116 ммоль, 2 экв). Смесь перемешивали в течение 3 часов при комнатной температуре. После проверки реакции по ЖХ-МС растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения Е-13 в виде белого твердого вещества (32 мг, 35%).
m/z (Q-TOF МС ИЭР-) 1461,8336 (100%, (M-H)-, C77H113N12O16 требуется 1461,8403).
m/z (Q-TOF МС ИЭР+) 1463,8565 (2%, МН+, C77H115Nl2O16 требуется 1463,8549), 732,4317 (100%, (МН2)2+, C77H116N12O16 требуется 732,4311).
Соединение Е-15
метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5- метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
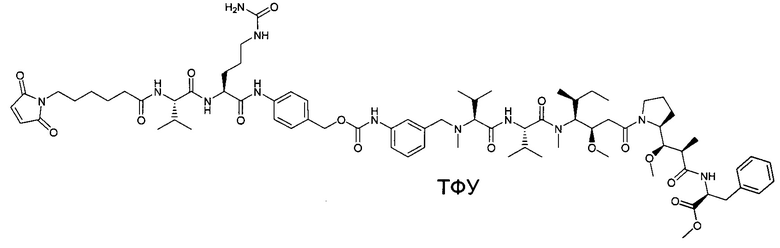
Соединение Е-15-1: метил-((2R,3R)-3-((S)-1 -((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси)карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
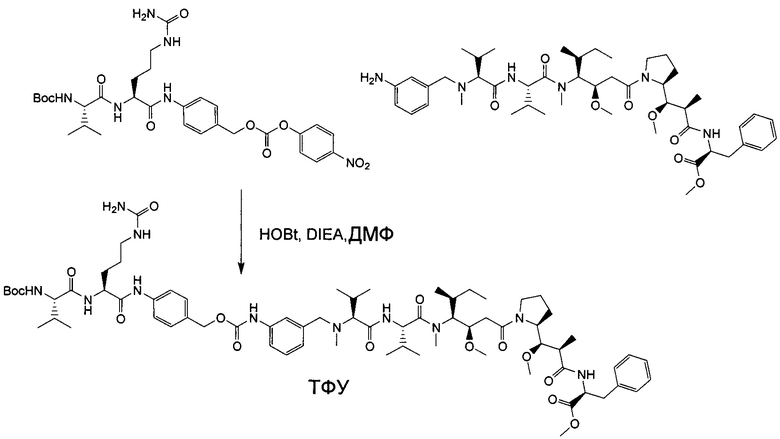
Соединение Е-15-1 было получено в соответствии с такой же методикой, как для соединения Е-11-6, с использованием карбоната Е-11-5 (28 мг, 0,044 ммоль, 1 экв), анилина 15 (42 мг, 0,044 ммоль, 1 экв), HOBt (3 мг, 0,022 ммоль, 0,5 экв) и DIEA (15 мкл, 0,087 ммоль, 2 экв) в ДМФ (2 мл). Соединение Е-15-1 выделили в виде твердого вещества (8,2 мг, 13%).
Соединение Е-15-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)бензил)окси) карбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
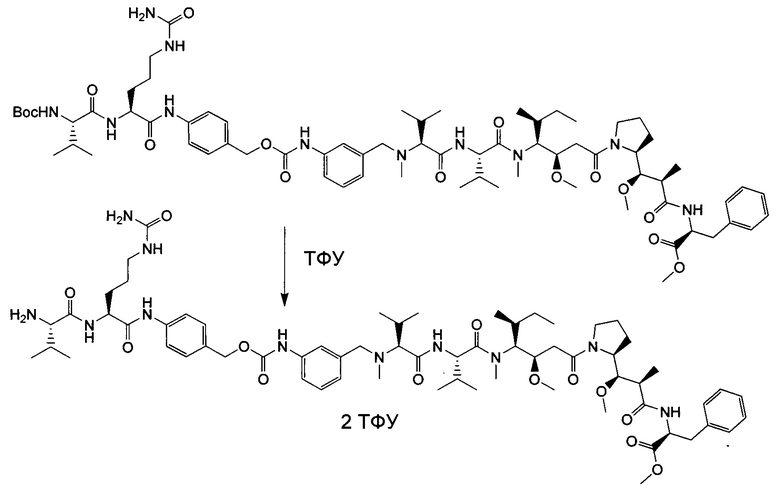
Соединение Е-15-1 (8,2 мг, 5,58 мкмоль, 1,0 экв) растворяли в ТФУ (200 мкл). Через 5 минут добавляли воду (1 мл), и раствор лиофилизировали в течение ночи с получением соединения Е-15-8 в виде белого твердого вещества (7,6 мг, 99%).
Соединение Е-15:
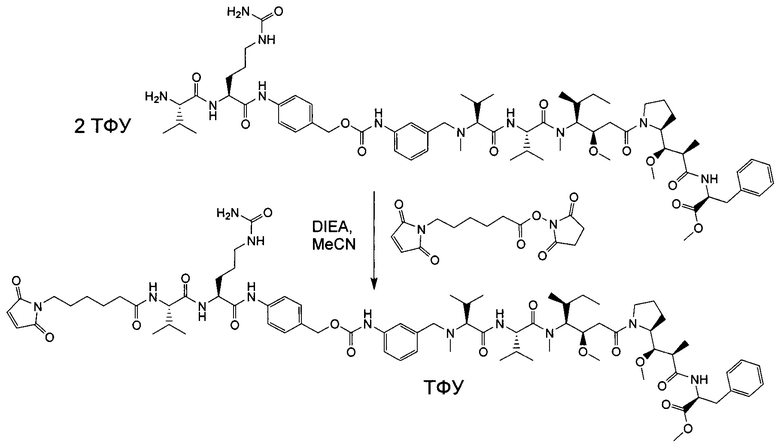
Соединение Е-15 было получено в соответствии с такой же методикой, как для соединения Е-12, с использованием амина Е-15-2 (7,6 мг, 5,55 мкмоль, 1 экв), 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноата (2 мг, 6,65 мкмоль, 1.2 экв) и DIEA (5 мкл, 0,028 ммоль, 5 экв) в ацетонитриле (0,5 мл). Соединение Е-15 выделили в виде белого твердого вещества (4,2 мг, 48%). m/z (Q-TOF МС ИЭР+) 1471,8169 (2%, MNa+, C76H112N12NaO16 требуется 1471,8211), 725,4223 (100%, (МН2)2+, C76H114N12O16 требуется 725,4232), 483,9482 (10%, (МН3)3+, C76H115N12O16 требуется 483,9513).
Соединение F-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
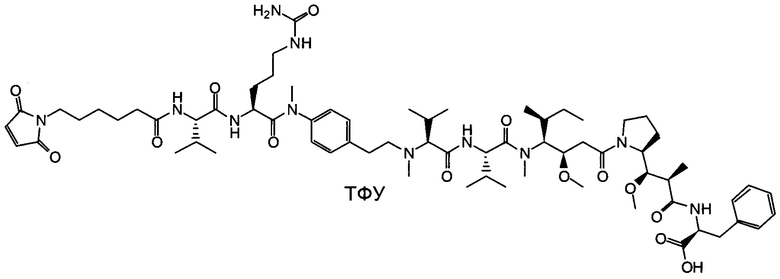
Соединение F-13-1: бензил-N-(4-((трет-бутоксикарбонил)(метил)амино)фенетил)-N-метил-L-валинат
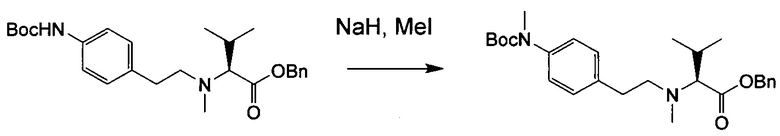
Соединение 11С (250 мг, 0,567 ммоль, 1 экв) растворяли в ТГФ (10 мл) с последующим добавлением NaH (60% суспензия в минеральном масле, 68 мг, 1,702 ммоль, 3 экв). Смесь перемешивали в течение 5 минут, затем добавляли йодметан (106 мкл, 1,702 ммоль, 3 экв). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, после чего гасили водой и разделяли между EtOAc (100 мл) и водой (50 мл). Органическую фазу высушивали над MgSO4 и выпаривали до сухого состояния с получением соединения F-13-1 в виде желтого масла (250 мг, 97%), которое использовали без дополнительной очистки.
Соединение F-13-2: бензил-N-метил-N-(4-(метиламино)фенетил)-L-валинат
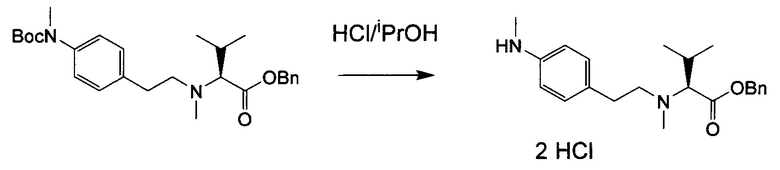
Вос-защищенный анилин F-13-1 (250 мг, 0,550 ммоль, 1 экв) растворяли в МеОН (5 мл) с последующим добавлением 1 мл имеющегося в продаже раствора HCl в iРrОН (5-6 М). Раствор перемешивали при комнатной температуре в течение 2 часов, после чего выпаривали до сухого состояния при пониженном давлении. Полученное в результате желтое масло растирали с Et2O с получением соединения F-13-2 в виде желтого твердого вещества (202 мг, 94%).
Соединение F-13-3: бензил-N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенетил)-N-метил-L-валинат
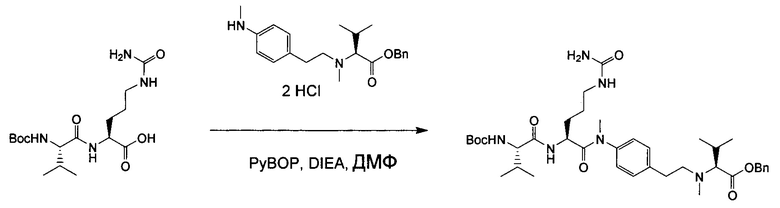
Кислоту Е-11-3 (190 мг, 0,508 ммоль, 1,5 экв) растворяли в безводном ДМФ (1 мл) с последующим добавлением DIEA (118 мкл, 0,677 ммоль, 2 экв), бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата (РуВОР - 264 мг, 0,508 ммоль, 1,5 экв) и анилина F-13-2 (120 мг, 0,339 ммоль, 1 экв). Смесь перемешивали при комнатной температуре в течение ночи, и растворители выпаривали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-13-3 в виде белого твердого вещества (140 мг, 45%).
Соединение F-13-4: N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенетил)-N-метил-L-валин
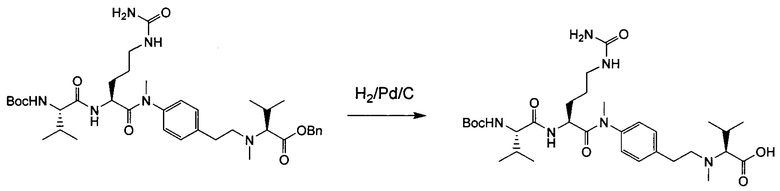
Соединение F-13-3 (116 мг, 0,163 ммоль, 1 экв) растворяли в МеОН (5 мл) в присутствии Pd/C 10%) (30 мг) и гидрогенизировали в течение 2 часов при температуре окружающей среды и атмосферном давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 110 мг (99%) соединения F-13-4 в виде бежевого твердого вещества.
Соединение F-13-5: метил-((2R3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((3)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3- диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
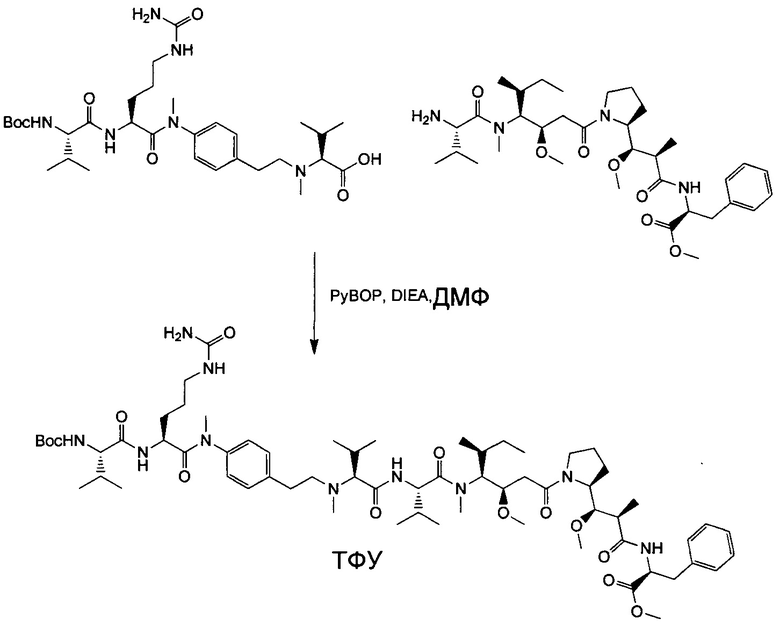
Амин 3D (89 мг, 0,140 ммоль, 1 экв) и кислоту F-13-4 (145 мг, 0,210 ммоль, 1,5 экв) растворяли в безводном ДМФ (4 мл) и добавляли РуВОР (109 мг, 0,210 ммоль, 1,5 экв) и DIEA (73 мкл, 0,420 ммоль, 3 экв). Смесь перемешивали в течение 1 часа при комнатной температуре, и растворитель выпаривали. Остаток разделяли между EtOAc и водой, и органическую фазу высушивали над MgS04, фильтровали и выпаривали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-13-5 в виде белого твердого вещества (140 мг, 73%).
Соединение F-13-6: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-N-метил-5-уреидопентанамидо) фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
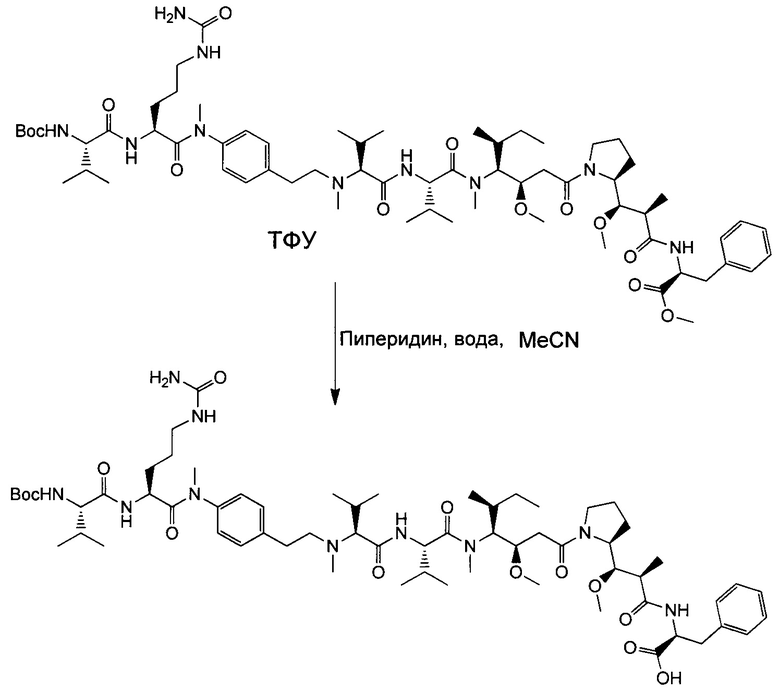
Соединение F-13-5 (140 мг, 0,104 ммоль, 1 экв) растворяли в смеси воды (4 мл), ацетонитрила (4 мл) и пиперидина (2 мл) и перемешивали при комнатной температуре в течение 4 часов. Растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487 детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-13-6 в виде белого твердого вещества (115 мг, 83%).
Соединение F-13:
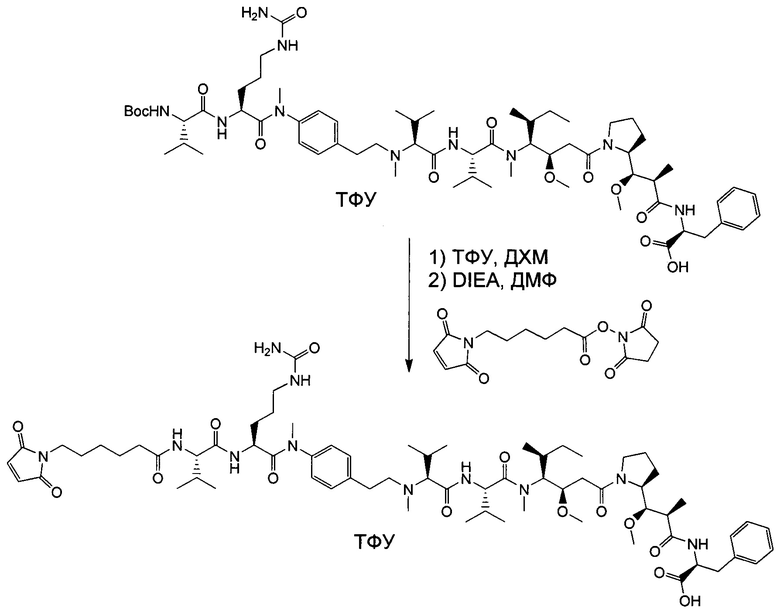
Соединение F-13 было получено в соответствии с такой же методикой, как для соединения Е-11, с использованием Вос-защищенного амина F-13-6 (55 мг, 0,041 ммоль, 1,0 экв) в ДХМ (0,5 мл) и ТФУ (100 мкл, 30 экв) с последующим разбавлением ДМФ (1 мл), гашением DIEA (320 мкл, 45 экв), затем взаимодействием с 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноатом (15 мг, 0,049 ммоль, 1,2 экв). После очистки препаративной ВЭЖХ и лиофилизации соединение F-13 было получено в виде белого твердого вещества (14 мг, 24%).
m/z (Q-TOF МС ИЭР+) 1314,8067 (2%, МН+, C69H108N11O14 требуется 1314,8072), 657,9067 (100%, (МН2)2+, C69H109N11O14 требуется 657,9072).
Соединение F-61
N((S)-1-(((S)-1-((4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-Диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)амино)-1-оксо-5-уреидопентан-2- ил)амино)-3-метил-1-оксобутан-2-ил)-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамида 2,2,2-трифторацетат
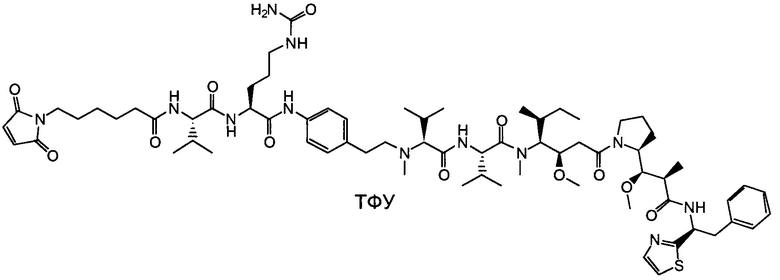
Соединение F-61-1: дигидрохлорид бензил-N-(4-аминофенетил)-N-метил-L-валинат
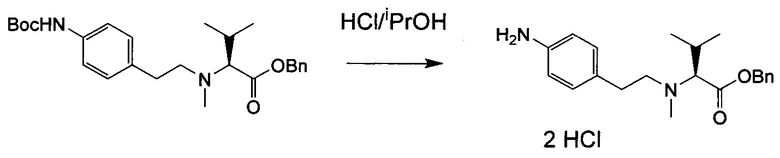
Соединение 11С (1,0 г, 2,27 ммоль, 1 экв) растворяли в 8 мл имеющегося в продаже раствора HCl в iPrOH (5-6 М). Смесь перемешивали в течение 2 часов при комнатной температуре, после чего выпаривали до сухого состояния при пониженном давлении. Остаток дважды растирали с Et2O (30 мл) и высушивали в вакууме с получением соединения F-61-1 в виде белого твердого вещества (916 мг, 98%).
Соединение F-61-2: бензил-N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)-N-метил-L-валинат
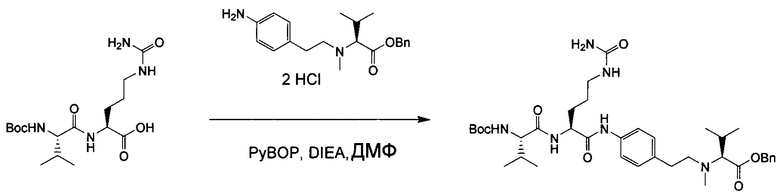
Кислоту Е-11-3 (769 мг, 2,05 ммоль, 1,5 экв) растворяли в безводном ДМФ (2,5 мл) с последующим добавлением DIEA (957 мкл, 5,48 ммоль, 4 экв) и РуВОР (1,07 г, 2,05 ммоль, 1,5 экв). Добавляли анилин F-61-1 (566 мг, 1,369 ммоль, 1 экв), и смесь перемешивали при комнатной температуре в течение ночи. Растворители выпаривали при пониженном давлении, и остаток очищали на силикагеле (ДХМ/МеОН) с получением 969 мг (102%) соединения F-61-2 в виде белого твердого вещества.
Соединение F-61-3: N-(4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)-N-метил-L-валин
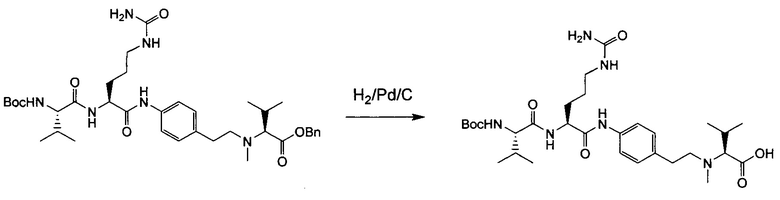
Соединение F-61-2 (969 мг, 1,28 ммоль, 1 экв) растворяли в МеОН (20 мл) в присутствии Pd/C 10% (270 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную смесь фильтровали и концентрировали при пониженном давлении, и остаток очищали на силикагеле (ДХМ/МеОН/АсОН) с получением 520 мг (67%) соединения F-61-3 в виде белого твердого вещества.
Соединение F-61-4: трет-бутил-((S)-1-(((S)-1-((4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-Диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)амино)-1-оксо-5-уреидопентан-2-ил)амино)-3-метил-1-оксобутан-2-ил)карбамата 2,2,2-трифторацетат
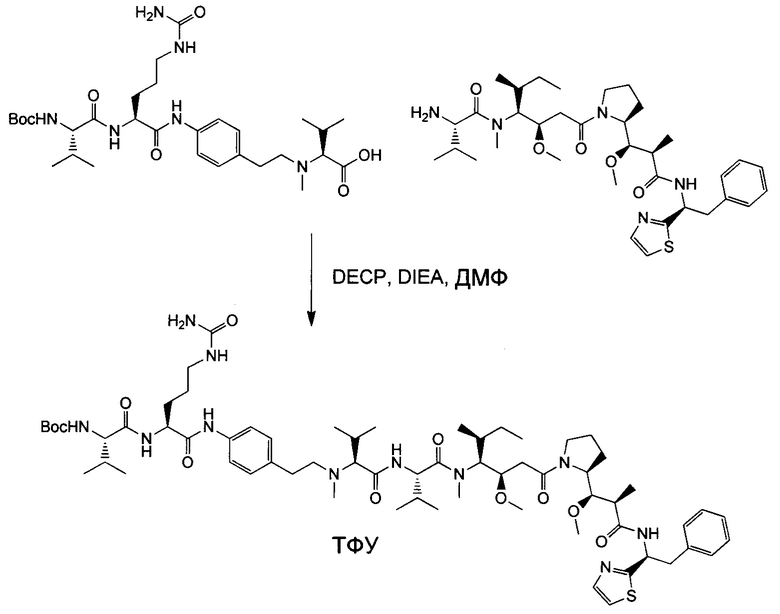
Кислоту F-61-3 (67,5 мг, 0,111 ммоль, 1,5 экв) растворяли в безводном ДМФ (2 мл) и добавляли DECP (17 мкл, 0,111 ммоль, 1,5 экв) и DIEA (39 мкл, 0,223 ммоль, 3 экв). После перемешивания в течение 15 минут при комнатной температуре добавляли амин 1Y (50 мг, 0,074 ммоль, 1 экв), и раствор перемешивали в течение ночи. Растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-61-4 в виде белого твердого вещества (28 мг, 28%).
Соединение F-61-5: (S)-2-((S)-2-амино-3-метилбутанамидо)-N-(4-((3R,4S,7S,10S)-4-((S)-втор-бутил)-7,10-Диизопропил-3-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазатридекан-13-ил)фенил)-5-уреидопентанамида бис(2,2,2-трифторацетат)
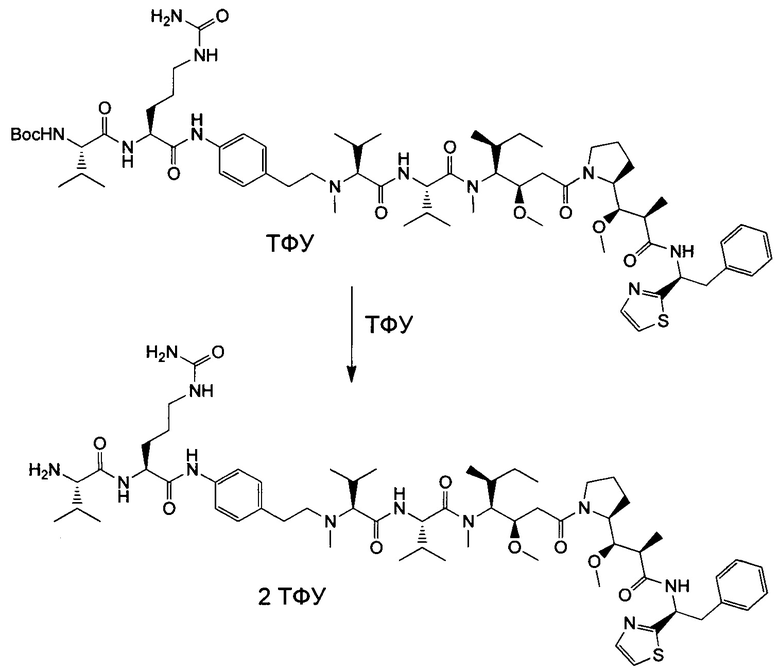
Соединение F-61-4 (28 мг, 0,021 ммоль, 1,0 экв) растворяли в ТФУ (200 мкл). Через 5 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл), и раствор лиофилизировали в течение ночи с получением соединения F-61-5 в виде бесцветного масла (38 мг, 134%).
Соединение F-61:
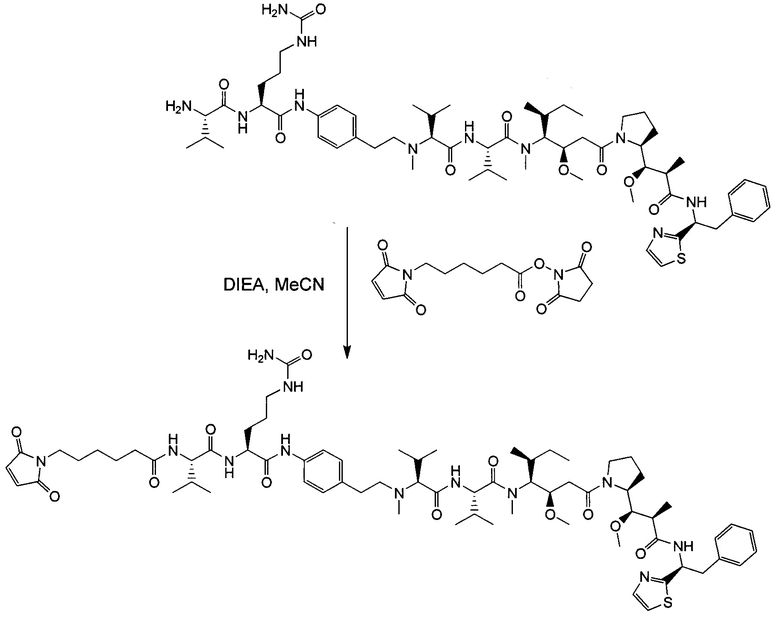
Соединение F-61-5 (28,3 мг, 0,020 ммоль, 1 экв) растворяли в ацетонитриле (0,5 мл) с последующим добавлением 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноата (9 мг, 0,029 мкмоль, 1,4 экв) и DIEA (25 мкл, 0,143 ммоль, 7 экв). Смесь перемешивали в течение 4,5 часов, после чего анализ ВЭЖХ показал присутствие исходного вещества, но полное расходование сукцинимида. Поэтому добавляли дополнительное количество 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноата (3 мг, 0,01 мкмоль, 0,5 экв), и реакционную смесь перемешивали в течение 1,5 часов. Анализ ВЭЖХ показал полное расходование исходного вещества. Растворитель выпаривали до сухого состояния, и остаток дважды растирали со смесью EtOAc/Et2O (80/20) с получением соединения F-61 в виде беловатого твердого вещества (19,4 мг, 70%).
m/z (Q-TOF МС ИЭР+) 1361,7725 (2%, MNa+, C70H106N12NaO12S требуется 1361,7666), 670,3961 (100%, (МН2)2+, C70H108N12O12S требуется 670,3960).
Соединение F-62:
метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
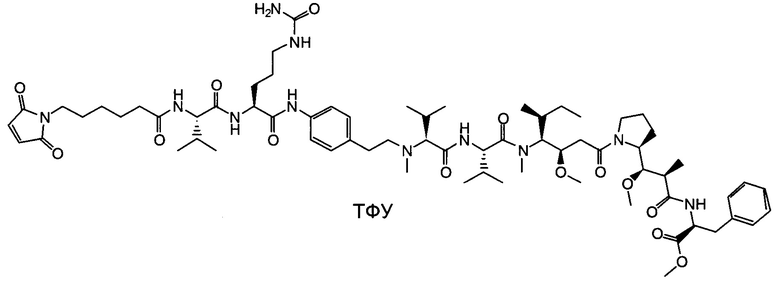
Соединение F-62-1: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
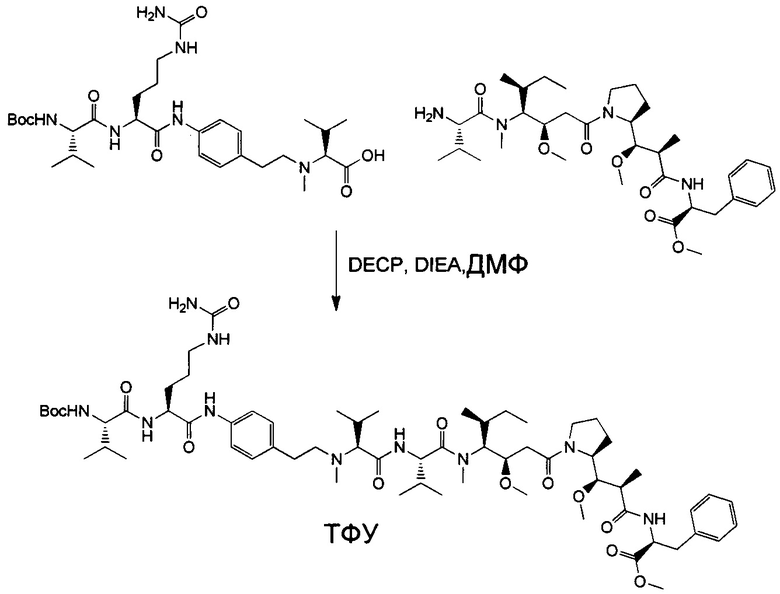
Соединение F-62-1 было получено аналогично соединению F-61-4 из амина 3D (100 мг, 0,158 ммоль, 0,9 экв), кислоты F-61-3 (108 мг, 0,178 ммоль, 1 экв), DECP (41 мкл, 0,267 ммоль, 1,5 экв) и DIEA (93 мкл, 0,534 ммоль, 3 экв) в ДМФ (2 мл). После очистки препаративной ВЭЖХ соединение F-62-1 было получено в виде белого твердого вещества (93 мг, 39%).
Соединение F-62-2: метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил) пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината бис(2,2,2-трифторацетат)
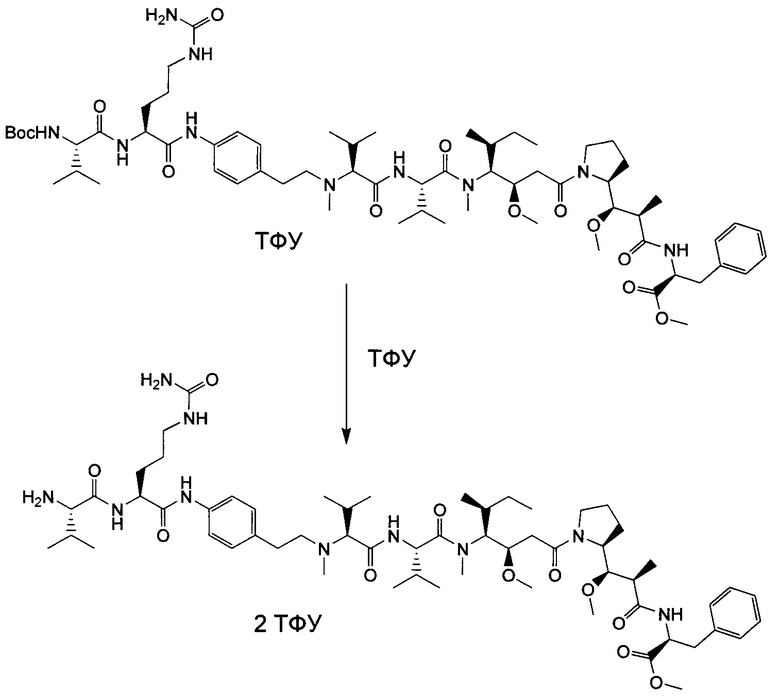
Соединение F-62-1 (35 мг, 0,026 ммоль, 1,0 экв) растворяли в ТФУ (200 мкл). Через 10 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл), и раствор лиофилизировали в течение ночи с получением соединения F-62-2 в виде белого твердого вещества (34 мг, 105%).
Соединение F-62:
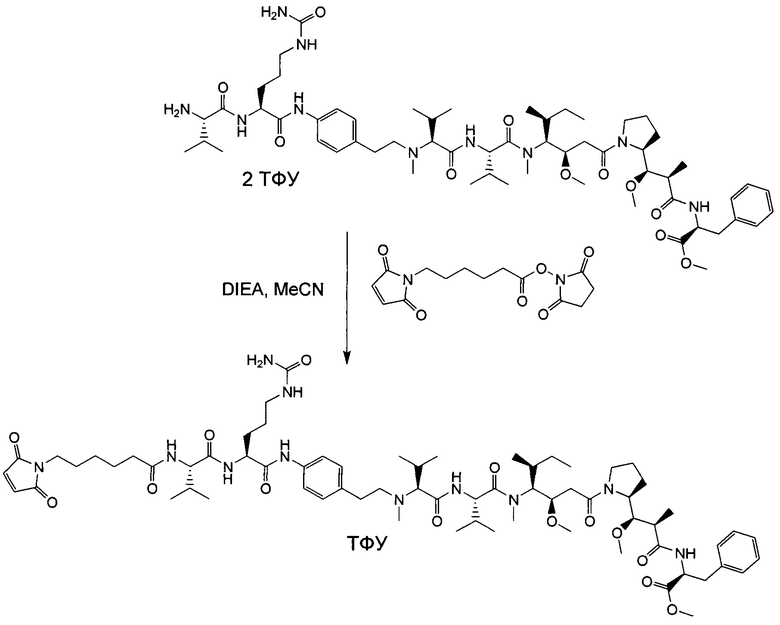
Амин F-62-2 (34 мг, 5,55 мкмоль, 1 экв) растворяли в ацетонитриле (3 мл). Добавляли DIEA (5 мкл, 0,028 ммоль, 5 экв) и 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноат (2 мг, 6,65 мкмоль, 1,2 экв). Анализ ВЭЖХ показал полное расходование исходного вещества. Растворитель выпаривали до сухого состояния, и остаток растирали со смесью EtOAc/Et2O (80/20). Неочищенный продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-62 в виде белого твердого вещества (5,5 мг, 13%). m/z (Q-TOF МС ИЭР+) 1336,7859 (2%, MNa+, C69H107N11NaO14 требуется 1336,7891), 657,9073 (100%, (МН2)2+, C69H109N11O14 требуется 657,9072).
Соединение F-63:
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
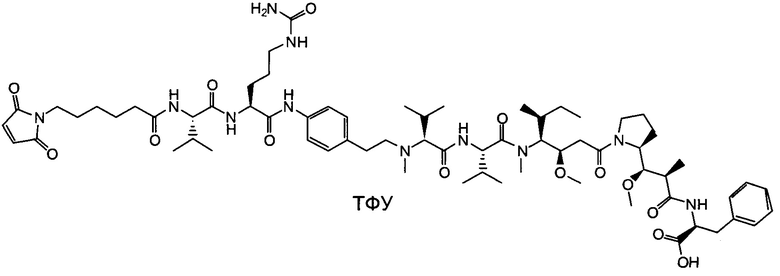
Соединение F-63-1: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-((трет-бутоксикарбонил)амино)-3-метилбутанамидо)-5-уреидопентанамидо)фенетил) (метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин
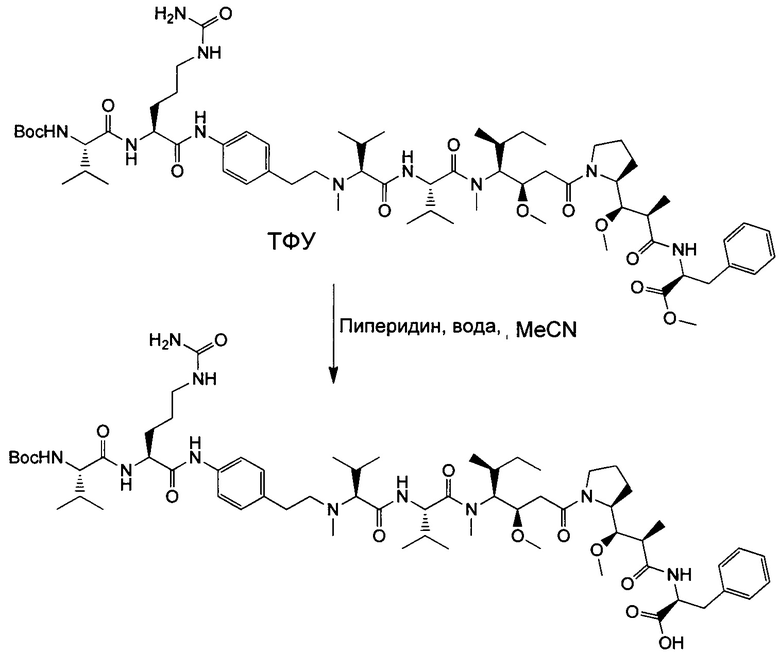
Соединение F-62-1 (157 мг, 0,118 ммоль, 1 экв) растворяли в смеси воды (4,5 мл), ацетонитрила (4,5 мл) и пиперидина (3,5 мл) и перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали при пониженном давлении, и остаток растирали с Et2O (60 мл). Твердое вещество собирали фильтрованием и дважды промывали Et2O (10 мл) с получением соединения F-63-1 в виде беловатого твердого вещества (153 мг, 100%).
Соединение F-63-2: ((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((S)-2-((S)-2-амино-3-метилбутанамидо)-5-уреидопентанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил) пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина бис-2,2,2-трифторацетат
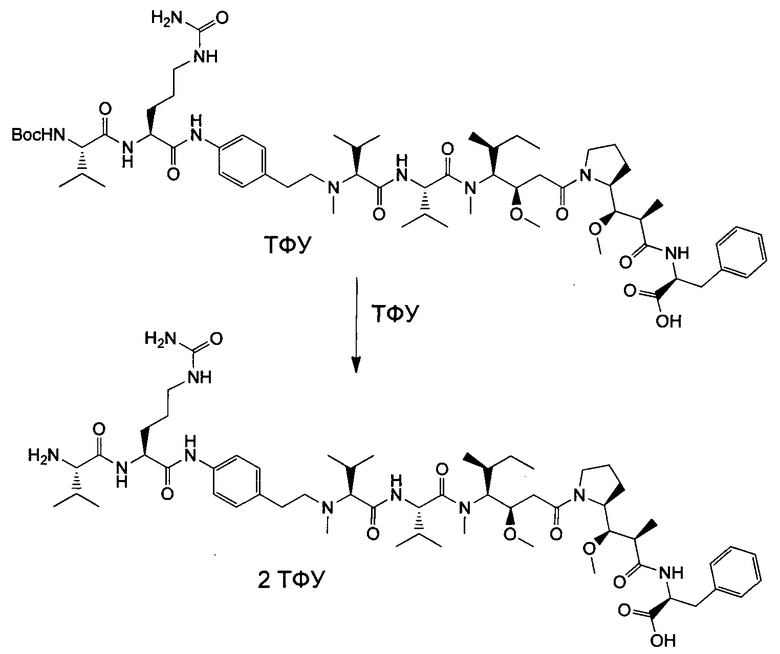
Соединение F-63-1 (153 мг, 0,127 ммоль, 1,0 экв) растворяли в ТФУ (200 мкл). Через 10 минут добавляли воду (2 мл) и ацетонитрил (0,5 мл), и раствор лиофилизировали в течение ночи с получением соединения F-63-2 в виде белого твердого вещества (34 мг, 105%).
Соединение F-63:
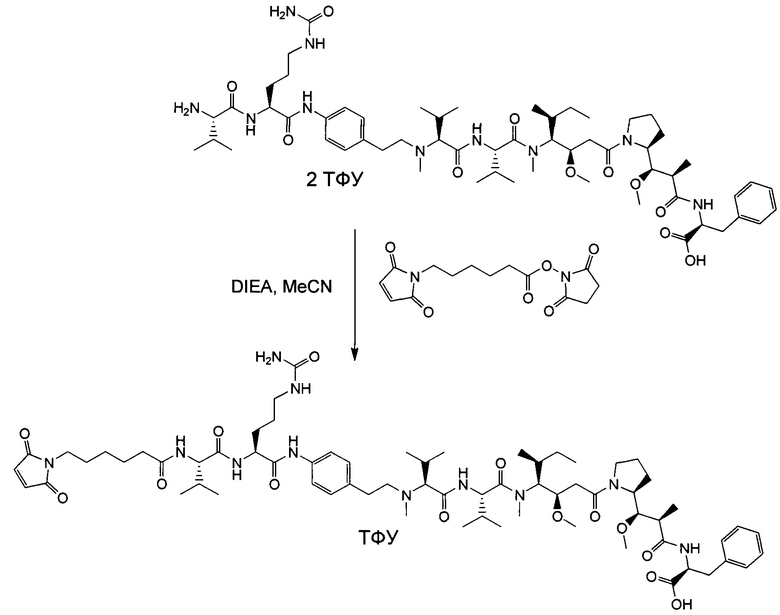
Амин F-63-2 (100 мг, 0,082 ммоль, 1 экв) растворяли в смеси ацетонитрила (2 мл) и ДМФ (0,5 мл) и добавляли 2,5-диоксопирролидин-1-ил-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноат (45 мг, 0,147 ммоль, 1,8 экв) и DIEA (71 мкл, 0,409 ммоль, 5 экв). После перемешивания при комнатной температуре в течение 4,5 часов растворитель выпаривали при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения F-63 в виде белого твердого вещества (42 мг, 36%).
m/z (Q-TOF МС ИЭР+) 1300,7901 (2%, МН+, C67H106N11O14 требуется 1300,7915), 650,8990 (100%, (МН2)2+, C68H107N11O14 требуется 650,8994).
Соединение G-12
метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)-N-метилгексанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
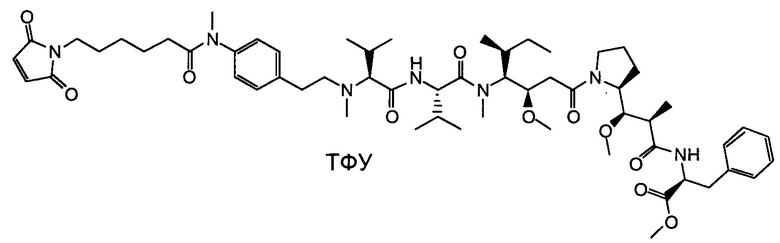
Соединение G-12-1: дигидрохлорид бензил-N-(4-аминофенетил)-N-метил-L-валината
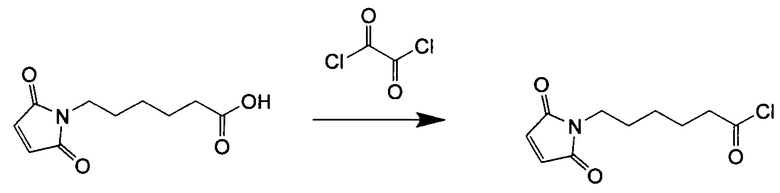
В оксалилхлориде (3 мл) растворяли 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексановую кислоту (200 мг, 0,947 ммоль, 1 экв). Раствор перемешивали при комнатной температуре в течение 5 часов, после чего выпаривали до сухого состояния при пониженном давлении. Соединение G-12-1 было получено в виде бежевого твердого вещества (217 мг, 100%) и использовано в следующей стадии без очистки.
Соединение G-12:
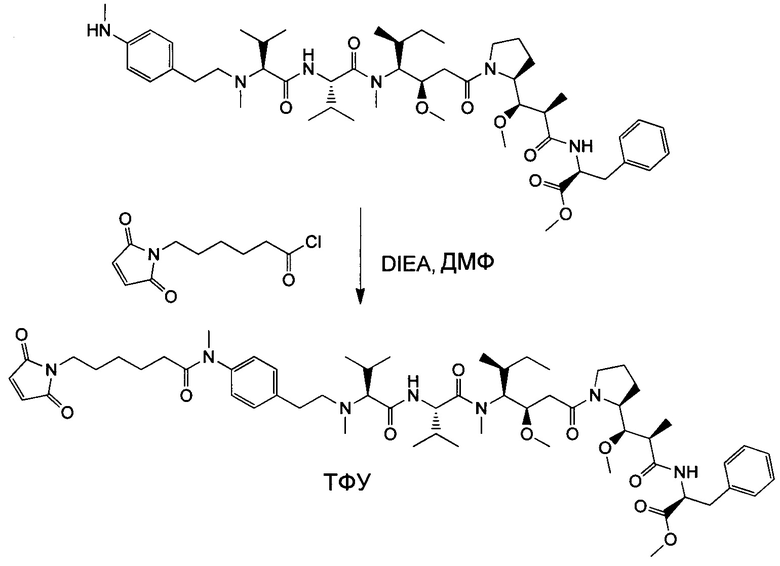
Анилин 12 (40 мг, 0,045 ммоль, 1 экв) растворяли в безводном ДХМ (1 мл) при 0°С и добавляли DIEA (8 мкл, 0,045 ммоль, 1 экв). После перемешивания в течение 30 минут вводили раствор соединения G-12-1 (10 мг, 0,45 ммоль, 1 экв) в безводном ДХМ (1 мл), и реакционную смесь перемешивали в течение 1 часа при 0°С. Смесь разбавляли ДХМ (25 мл) и дважды промывали водой (20 мл), один раз соляным раствором (10 мл). Органическую фазу высушивали над Na2SO4, фильтровали и выпаривали при пониженном давлении с получением неочищенного продукта в виде светло-коричневого твердого вещества (54 мг). Этот продукт очищали флэш-хроматографией на силикагеле (ДХМ/МеОН), а затем препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Выделенный продукт лиофилизировали с получением белого твердого вещества (23 мг), которое подвергали переочистке препаративной ВЭЖХ, и отобранные фракции объединяли и лиофилизировали с получением соединения G-12 в виде белого твердого вещества (9 мг, 16%).
m/z (Q-TOF МС ИЭР+) 1094,6543 (20%, MNa+, C59H89N7NaO11 требуется 1094,6512), 1072,6722 (16%, МН+, C59H90N7OH требуется 1072,6693), 536,8358 (100%, (МН2)2+, C59H91N7O11 требуется 536,8383).
Соединение G-13
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метилгексанамидо)фенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланина 2,2,2-трифторацетат
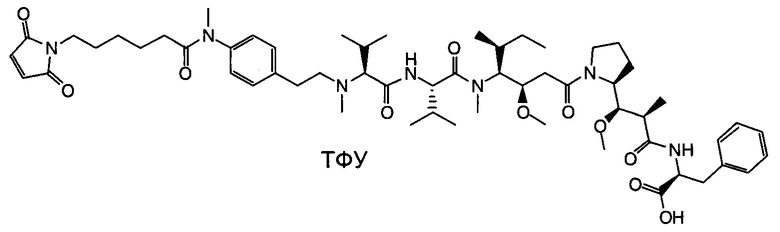
Соединение G-13:
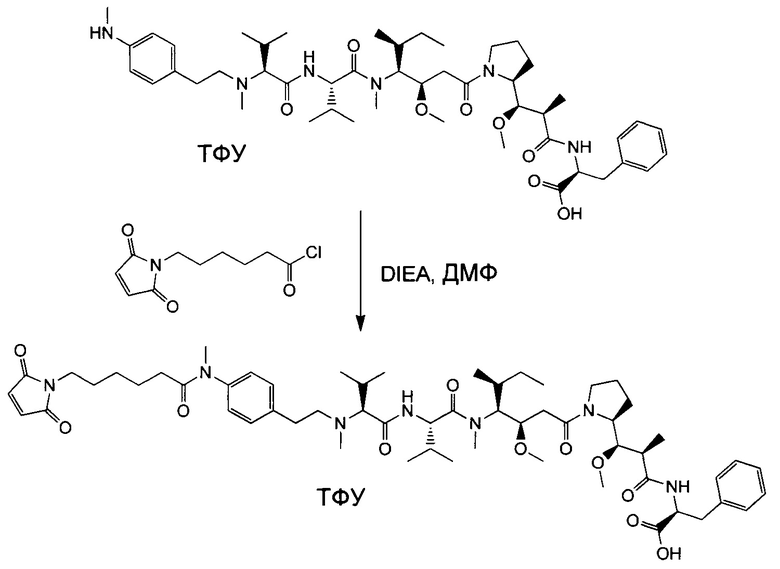
Анилин 13 (15 мг, 0,015 ммоль, 1 экв) растворяли в безводном ДХМ (1,5 мл) при 0°С и добавляли DIEA (8 мкл, 0,046 ммоль, 3 экв). Вводили раствор соединения G-12-1 (3,5 мг, 0,046 ммоль, 1 экв) в безводном ДХМ (0,5 мл), и реакционную смесь перемешивали в течение 1,5 часов при 0°С. Растворитель выпаривали при пониженном давлении, и неочищенный продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100% MeCN в течение 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения G-13 в виде белого твердого вещества (11,4 мг, 62%).
m/z (Q-TOF МС ИЭР+) 1058,6510 (30%, МН+, C58H88N7O11 требуется 1058,6536), 529,8285 (100%, (МН2)2+, C58H89N7O11 требуется 529,8305).
Соединение G-15
метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланината 2,2,2-трифторацетат
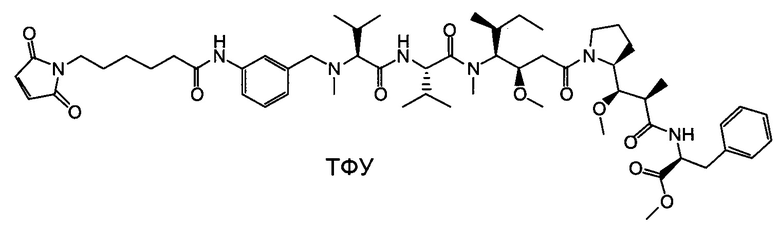
Соединение G-15:
Анилин 15 (40 мг, 0,047 ммоль, 1 экв) растворяли в сухом ДХМ (2 мл) при 0°С и добавляли DIEA (10 мкл, 0,056 ммоль, 1,2 экв). Вводили раствор соединения G-12-1 (108 мг, 0,47 ммоль, 10 экв) в сухом ДХМ (1 мл), и реакционную смесь перемешивали в течение 1,5 часов при 0°С. Смесь разбавляли ДХМ (10 мл) и дважды промывали водой (5 мл). Органическую фазу высушивали над MgSO4, фильтровали и выпаривали при пониженном давлении с получением неочищенного продукта в виде бежевого твердого вещества (54 мг). Этот продукт очищали препаративной ВЭЖХ (Waters 600Е, колонка SunFire Prep С18 OBD, 5 мкм, 19×100 мм; элюирующая фаза: вода / MeCN, забуференная 0,1% ТФУ; градиент от 5% до 100%) MeCN в 15 минут; Waters 2487, детектор УФ при 220 нм). Отобранные фракции объединяли и лиофилизировали с получением соединения G-15 в виде белого твердого вещества (27 мг, 50%).
m/z (Q-TOF МС ИЭР+) 1066,6517 (2%, MNa+, C57H85N7NaO11 требуется 1066,6199), 522,8224 (100%, (МН2)2+, C57H87N7O11 требуется 522,8226).
Пример 17: Синтез, очистка и характеризация ADC
Описанная ниже методика применима к химерным и гуманизированным формам IgG1. Необходимо понимать, что для других форм, таких как IgG2, IgG4 и т.д., специалист в данной области техники способен адаптировать эту методику с использованием общих знаний.
Антитела (1-5 мг/мл) частично восстанавливали гидрохлоридом трис(2-карбоксиэтил)фосфина (ТСЕР) в 10 мм боратном буфере рН 8,4, содержащем 150 мМ NaCl и 2 мМ ЭДТА, в течение 2 ч при 37°С. Как правило, для получения целевых отношений лекарственного средства к антителу (DAR) около 4 соответственно использовали 2,5-3 молярных эквивалента ТСЕР. Частичное восстановление антитела подтверждали анализом в ДСН-ПААГ электрофорезе в невосстанавливающих условиях. Перед сочетанием группировки линкер-лекарственное средство с высвобожденными межцепными остатками цистеина восстановленную смесь оставляли охлаждаться до комнатной температуры. Затем концентрацию антитела доводили до 1 мг/мл 10 мМ боратным буфером рН 8,4, содержащим 150 мМ NaCl и 2 мМ ЭДТА, и добавляли 5-кратный молярный избыток лекарственного средства к антителу из 10 мМ раствора в диметилсульфоксиде (ДМСО). Конечную концентрацию ДМСО доводили до 10% для поддержания растворимости лекарственного средства в водной среде в процессе сочетания. Реакцию проводили в течение 1 ч при комнатной температуре. Избыток лекарственного средства гасили добавлением 1,5 моль N-ацетилцистеина на моль лекарственного средства и инкубации в течение 1 ч при комнатной температуре. После диализа против 25 мМ His буфера рН 6,5, содержащего 150 мМ NaCl, в течение ночи при 4°С конъюгат антитела с лекарственными препаратами очищали путем использования способов, известных специалистам в данной области техники, основанных на коммерческих хроматографических колонках и ультрафильтрационных устройствах. Сначала несвязанное лекарственное средство и агрегаты ADC удаляли посредством эксклюзионной хроматографии (ЭХ) на колонке S200 (GE Life Sciences) или TSK G3000 SW (Tosoh). Затем очищенные мономеры ADC концентрировали до 2-3 мг/мл посредством ультрафильтрации на фильтрационных устройствах с номинальным отсечением по молекулярной массе (НОММ) 30 или 50 кДа или аффинной хроматографии на белке А. Очищенные ADC хранили при 4°С после стерилизации фильтрованием на фильтрах 0,2 мкм. Далее их анализировали путем электрофореза в ДСН-ПААГ в восстанавливающих и в невосстанавливающих условиях для подтверждения конъюгации лекарственного средства и путем хроматографии на ЭХ или аналитических колонках S200 или TSK G3000 SWXL для определения содержания мономеров и агрегированных форм. Концентрации белка определяли путем количественного анализа с использованием бицинхониновой кислоты (БЦК) с IgG в качестве стандарта. DAR оценивали для каждого очищенного ADC с помощью ГИХ и ЖХ-МС. Как правило, содержание агрегированных форм было ниже 5%, и DAR составляло от 3,5 до 5.
Пример 18: Анализ цитотоксичности антител IGF-1R. конъюгированных с различными лекарственными средствами
18.1 Оценка химерных антител на клетках MCF-7
Было показано, что пять антител к IGF-1R претерпевают быструю интернализацию в лизосомах и обладают низкой связывающей способностью в кислых средах. В связи с этим данные Ab обладали всеми свойствами для использования в составе ADC. Таким образом, пять химерных антител к IGF-1R подвергали сочетанию с тремя различными соединениями (G-13; Е-13 и F-63). Отношение лекарственного средства к антителу этих ADC составляло около 4. С целью оценки неспецифической токсичности нерелевантное химерное антитело c9G4 также подвергали сочетанию с этими соединениями при таких же DAR. Клетки MCF-7 инкубировали с возрастающими концентрациями каждого ADC при 37°С в течение 6 дней в полной культуральной среде. Жизнеспособность клеток оценивали, используя количественное определение жизнеспособности клеток с помощью люминесцентного анализа (CellTiter-Glo, Promega). Люминесцентный сигнал считывали с использованием планшет-ридера Mithras (Berthold Technologies). Нерелевантное химерное антитело c9G4, конъюгированное с любым из Е-13, G-13 или F-63, не проявляло или проявляло умеренную цитотоксическую активность на клетках MCF-7 (Фиг. 21). Напротив, при добавлении всех остальных ADC, полученных в результате сочетания антител к IGF-1R с любым из Е-13, G-13 или F-63, жизнеспособность клеток MCF-7 резко снижалась.
18.2 Оценка химерных антител на нормальных клетках Уровни экспрессии IGF-1R оценивали на первичной культуре нормальных клеток (PromoCell GmbH) с использованием c208F2 mAb. С этой целью клетки (0,5×106 клеток/мл) инкубировали с 10 мкг/мл антитела C208F2 в течение 20 мин при 4°С в буфере FACS (ФСБ, 0,1% БСА, 0,01% NaN3). Затем их промывали 3 раза и инкубировали с соответствующим вторым антителом, связанным с Alexa 488, в течение 20 дополнительных минут при 4°С в темноте, после чего промывали 3 раза в буфере FACS. Связывание антител к IGF-1R сразу проводили на жизнеспособных клетках, которые идентифицировали, используя пропидия йодид (который окрашивает мертвые клетки). Уровень экспрессии (Bmax) на нормальных клетках (таблица 14) был низким по сравнению с экспрессией IGF-1R на клетках MCF-7 (см. пример 2, таблица 8).
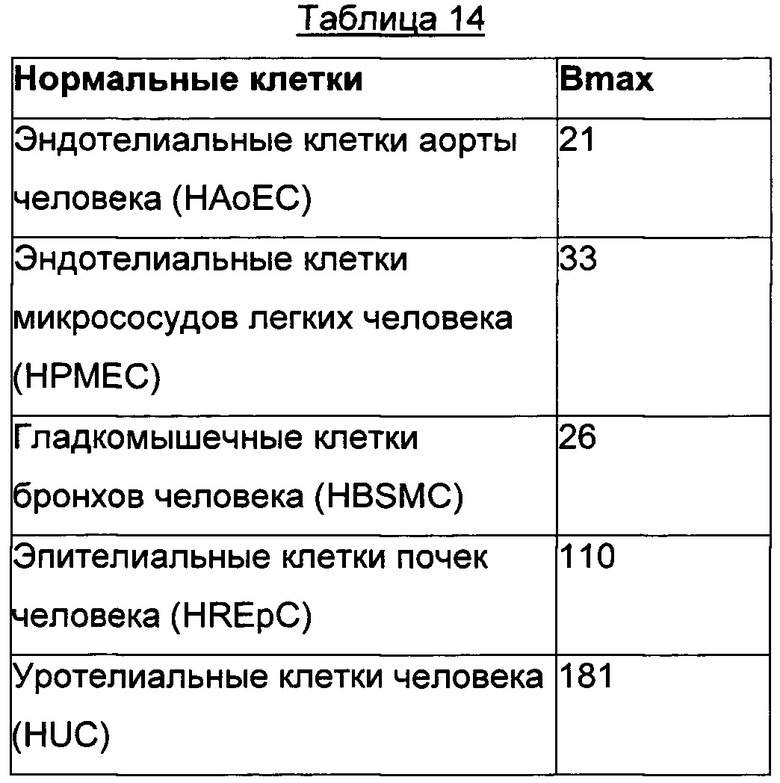
Цитотоксичность ADC C208F2-G-13 оценивали на нормальных клетках. Клетки инкубировали с возрастающими концентрациями C208F2-G-13 при 37°С в течение 6 дней в полной культуральной среде. Жизнеспособность клеток оценивали, используя количественное определение жизнеспособности клеток с помощью люминесцентного анализа (CellTiter-Glo, Promega). Люминесцентный сигнал считывали с использованием планшет-ридера Mithras (Berthold Technologies). На HBSMC, НРМЕС, НАоЕС и HREpC значительной цитотоксичности не наблюдали (Фиг. 25). Незначительная цитотоксичность была измерена только на клетках HUC при высоких концентрациях C208F2-G-13.
18.3 Оценка гуманизированных вариантов hz208F2
Шестнадцать гуманизированных вариантов 208F2 подвергали сочетанию с соединением G-13. Отношение лекарственного средства к антителу этих ADC составляло около 4, С целью оценки неспецифической токсичности нерелевантное химерное антитело c9G4 также подвергали сочетанию с этими соединениями при таких же DAR. Химерное антитело C208F2 также подвергали сочетанию с G-13. Клетки MCF-7 инкубировали с возрастающими концентрациями каждого ADC при 37°С в течение 6 дней в полной культуральной среде. Жизнеспособность клеток оценивали, используя количественное определение жизнеспособности клеток с помощью люминесцентного анализа (CellTiter-Glo, Promega). Люминесцентный сигнал считывали с использованием планшет-ридера Mithras (Berthold Technologies). Нерелевантное химерное антитело c9G4, конъюгированное с G-13, не проявляло или проявляло умеренную цитотоксическую активность на клетках MCF-7 (Фиг. 26). Напротив, при добавлении всех остальных ADC, полученных в результате сочетания антител к IGF-1R с G-13, жизнеспособность клеток MCF-7 резко снижалась. Способность шестнадцати гуманизированных вариантов к индукции клеточной цитотоксичности была по меньшей мере эквивалентной или даже лучшей по сравнению с определенной для химерной формы C208F2-G-13, как показано в таблице 15 и проиллюстрировано для одного гуманизированного варианта на Фиг. 26.


Пример 19: Активность in vivo антитела c208F2, конъюгированного с любым из соединений Е-13. G-13 или F-63 в модели ксенотрансплантата MCF-7
В целях подтверждения, что эффективность C208F2 в сочетании с соединениями G-13, Е-13 или F-63 in vitro может быть транслирована in vivo, их тестировали в модели ксенотрансплантата MCF-7.
Методики работы с животными выполняли в соответствии с руководящими принципами Директивы 2010763/ЕС по защите животных, используемых в научных целях. Протокол был одобрен Комитетом по контролю этических норм обращения с животными института Pierre Fabre. Пять миллионов клеток MCF-7 инъецировали подкожно 7-недельным мышам линии Swiss/Nude. Перед инъекцией клеток в левый бок мышей имплантировали пеллеты эстрогена (Innovative Research of America), необходимого для роста in vivo опухолей MCF-7.
Через двадцать дней после имплантации MCF-7, когда опухоли достигали среднего размера 120-150 мм3, животных делили на группы по 5 мышей в соответствии с размером и аспектом опухоли. Различные препараты вводили путем интраперитонеальных инъекций. Состояние здоровья животных контролировали ежедневно. Объем опухоли измеряли два раза в неделю с помощью электронного циркуля до конца исследования. Объем опухоли вычисляют по следующей формуле: π/6 × длина × ширина × высота. Токсичность оценивали путем наблюдения за массой тела животных три раза в неделю. Статистические анализы проводили для каждого показателя с помощью критерия Уилкоксона-Манна-Уитни. Все соединения инъецировали интраперитонеально (i.p.). В данном Примере противоопухолевая активность C208F2 mAb в сочетании с любым из Е-13, F-13 или F-63 при DAR около 4 оценивали после 2 инъекций дозы 7 мг/кг при D20 и D27 (Фиг. 22А, 22В и 22С). Параллельно блокированные группировки лекарственного средства E-13, F-13 и F-63 инъецировали в эквивалентной дозе, соответствующей дозе 7 мг/кг C208F2-E-13, C208F2-F-13 и C208F2-F-63 DAR около 4.
Инъекция любого из С208-Е-13 (Фиг. 22А), C208F2-G-13 (Фиг. 22 В) или C208F2-F-63 (Фиг. 22С) приводила к значительному ингибированию и даже индукции полной регрессии роста опухоли (р менее 0,05 по сравнению с соответствующим блокированным лекарственным средством). Статистически значимое различие активности С208-Е-13, C208F2-G-13 и C208F2-F-63 не было отмечено. Блокированные лекарственные средства не оказывали воздействия на рост опухоли MCF-7 (р>0,05 по сравнению с контрольной группой).
Вторая серия экспериментов была проведена с C208F2, конъюгированным либо с Е-13, либо с G-13 и с нерелевантным антителом c9G4, конъюгированным либо с Е-13, либо с G-13, в модели ксенотрансплантата MCF-7, как описано выше. Мышам инъецировали i.p. 7 мг/кг каждого ADC при D20 и D27 (Фиг. 23А и 23В).
Инъекция обоих C9G4-E-13 и C9G4-F-13 оказывала умеренное и транзиторное влияние на рост опухолей ксенотрансплантата MCF-7. Тем не менее, этот второй эксперимент подтвердил, что инъекции либо C208-E-13, либо C208F2-G-13 приводят к индукции полной регрессии D43, что показывает высокую противоопухолевую активность ADC.
Пример 20: Активность in vivo антитела hz208F2, конъюгированного с соединением G-13. в модели ксенотрансплантата 3+ MCF-7
Гуманизированные формы 208F2 в сочетании с соединением G-13 оценили in vivo в модели ксенотрансплантата MCF-7.
Методики работы с животными выполняли в соответствии с руководящими принципами Директивы 2010763/ЕС по защите животных, используемых в научных целях. Протокол был одобрен Комитетом по контролю этических норм обращения с животными института Pierre Fabre. Пять миллионов клеток MCF-7 инъецировали подкожно 7-недельным мышам линии Swiss/Nude. Перед инъекцией клеток в левый бок мышей имплантировали пеллеты эстрогена (Innovative Research of America), необходимого для роста in vivo опухолей MCF-7.
Через двадцать дней после имплантации MCF-7, когда опухоли достигали среднего размера 120-150 мм3, животных делили на группы по 6 мышей в соответствии с размером и аспектом опухоли. Различные препараты вводили путем интраперитонеальных инъекций по протоколу 4 инъекций: одна инъекция один раз в четыре дня (Q4d4). Состояние здоровья животных контролировали ежедневно. Объем опухоли измеряли два раза в неделю с помощью электронного циркуля до конца исследования. Объем опухоли вычисляют по следующей формуле: π/6 × длина × ширина × высота. Токсичность оценивали путем наблюдения за массой тела животных три раза в неделю. Статистические анализы проводили для каждого показателя с помощью критерия Уилкоксона-Манна-Уитни. Все соединения инъецировали интраперитонеально (i.p.). В данном Примере противоопухолевую активность c208F2 mAb, конъюгированного с соединением G-13, сравнивали с различными гуманизированными формами, также конъюгированными с G-13 (Фиг. 27). Протестированные гуманизированные формы описаны в таблице 16 ниже:

Инъекция любой из гуманизированных форм C208-G-13 или 208F2 приводила к значительному ингибированию и даже индукции полной регрессии роста опухоли (р менее0,05 по сравнению с соответствующим контролем). Статистически 15 значимого различия активности между C208F2-G-13 и протестированными гуманизированными формами не наблюдали.
Вторую серию экспериментов проводили либо с C208F2, либо с hz208F2-4 в сочетании с G-13 в модели ксенотрансплантата MCF-7, как описано выше (Фиг. 28А и 28В соответственно). Мышам инъецировали i.p.3 мг/кг каждого ADC в виде 4 2 0 инъекций раз в четыре дня (Q4d4) или только один раз.
В модели ксенотрансплантата MCF-7 наблюдали такую же сильную противоопухолевую активность при четырехкратном или только однократном введении ADC.
Пример 21: Активность in vivo антитела 208F2. конъюгированного с соединениями G-13 или Е-13 в модели ксенотрансплантата 2+ CaOV-3
Противоопухолевую активность также исследовали в 2+ экспрессирующей опухоли в модели ксенотрансплантата CaOV-3, представляющей собой линию клеток карциномы яичника. С этой целью мышам инъецировали подкожно на день DO 7×106 клеток. Когда опухоли достигали приблизительно 120 мм3 (19 дней после инъекции опухолевых клеток) животных делили на 5 групп по 5 мышей со сравнимым размером опухоли и вводили интраперитонеально C208F2, конъюгированное либо с Е-13, либо с G-13, и нерелевантное антитело c9G4, конъюгированное либо с Е-13, либо с G-13. Мышам инъецировали i.p. 3 мг/кг каждого ADC по 6 инъекций на цикл; одна инъекция раз в четыре дня. У мышей наблюдали рост ксенотрансплантата. Объем опухоли вычисляли по формуле: π/6 × длина × ширина × высота.
По сравнению с C9G4-E-13, которое умеренно или транзиторно индуцировало замедление роста, C9G4-G-13 не влияло на рост ксенотрансплантатов опухоли CaOV-3. Инъекции либо C208F2-E-13, либо C208F2-G-13 приводили к индукции в среднем 95% и 77% ингибированию роста опухоли соответственно на день 50 (Фиг. 29А и 29В).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2685259C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА, ЭКСПРЕССИРУЮЩЕГО IGF-1R | 2016 |
|
RU2728568C2 |
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО (ADC), КОТОРЫЕ СВЯЗЫВАЮТСЯ С БЕЛКАМИ FLT3 | 2016 |
|
RU2739617C2 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |
| АНТИТЕЛО ПРОТИВ IGF-1R И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ АДРЕСУЮЩЕГО ПЕРЕНОСЧИКА ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2698977C2 |
| КОНЪЮГАТЫ АНТИ-РТК7 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2015 |
|
RU2708075C2 |
| НОВЫЕ КОНЪЮГАТЫ АНТИТЕЛ И ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684468C2 |
| ПИРРОЛОБЕНЗОДИАЗЕПИНОВЫЕ КОНЪЮГАТЫ | 2017 |
|
RU2744200C2 |
| Модулирующие стабильность линкеры для использования с конъюгатами антитело-лекарственное средство | 2015 |
|
RU2680238C2 |




























































































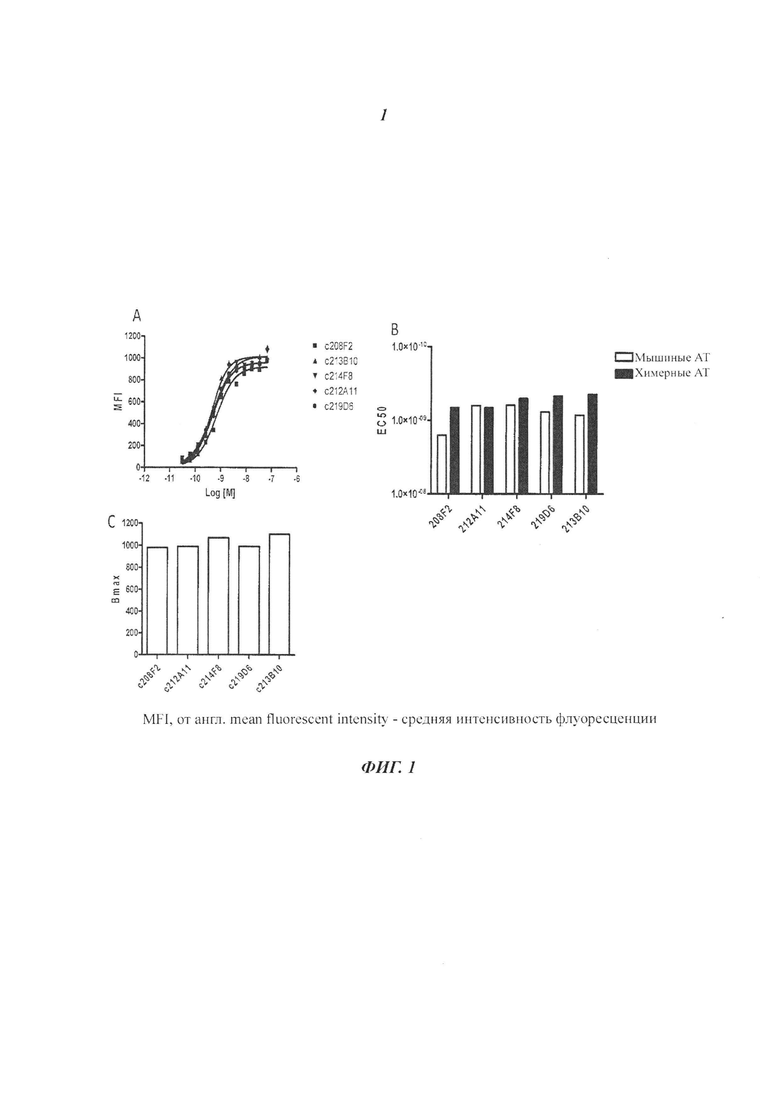
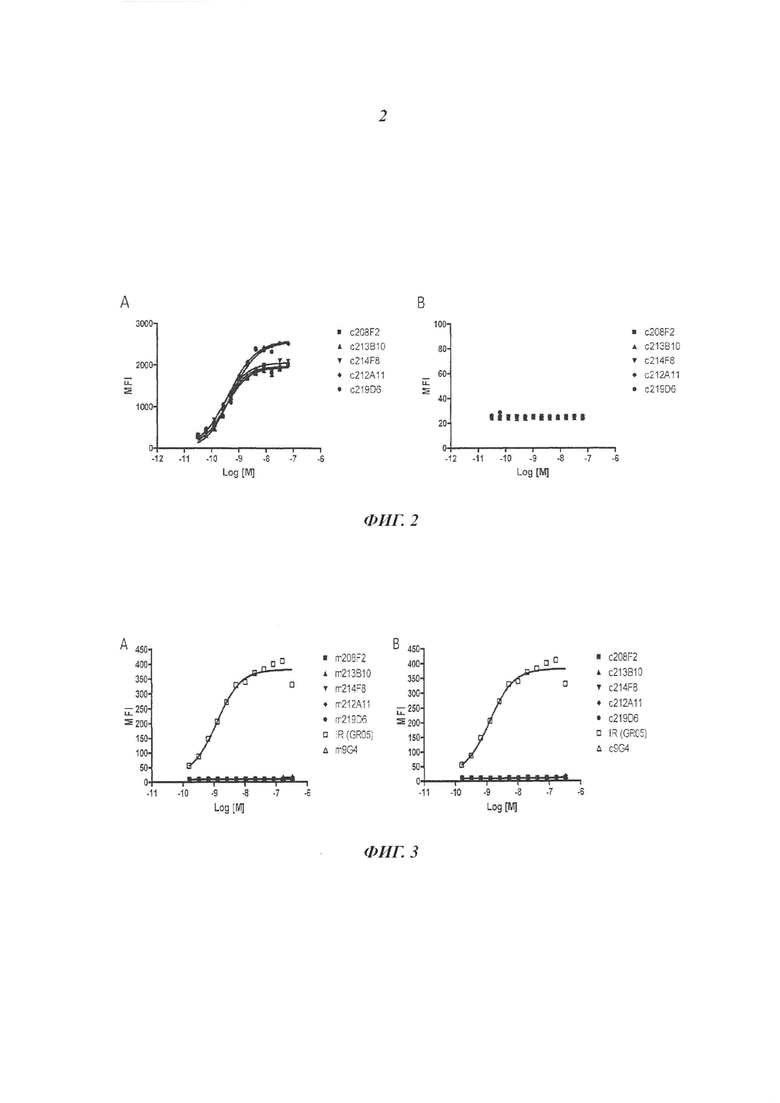
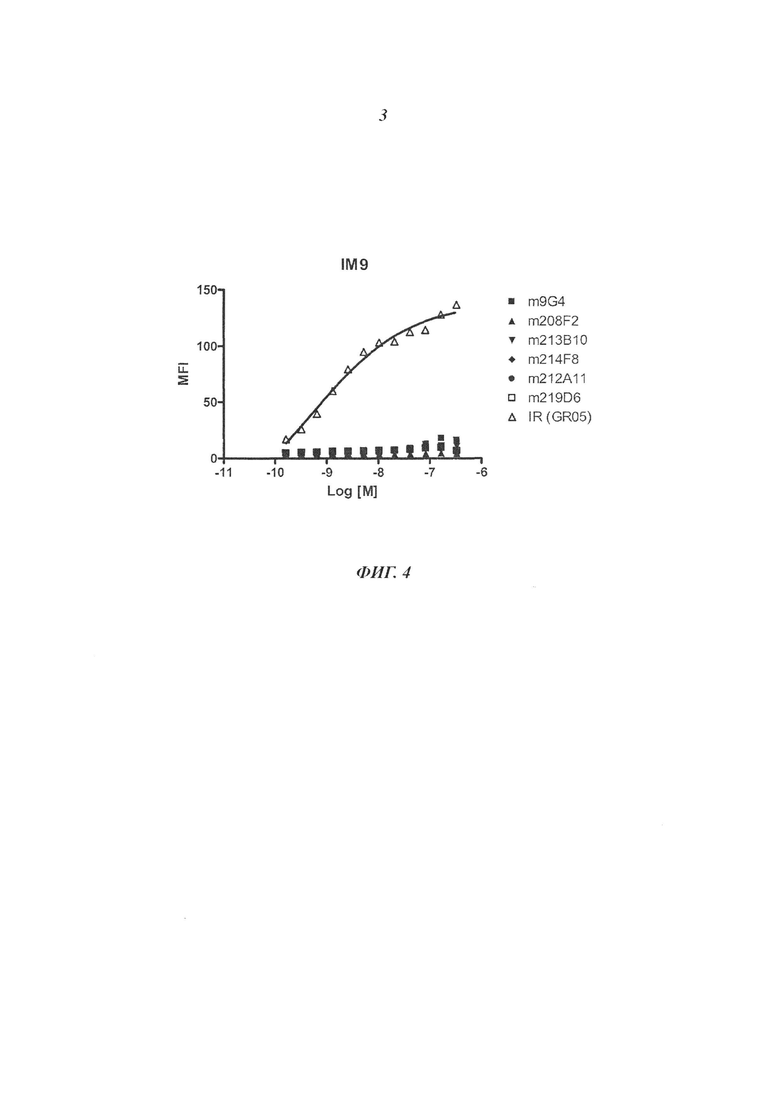
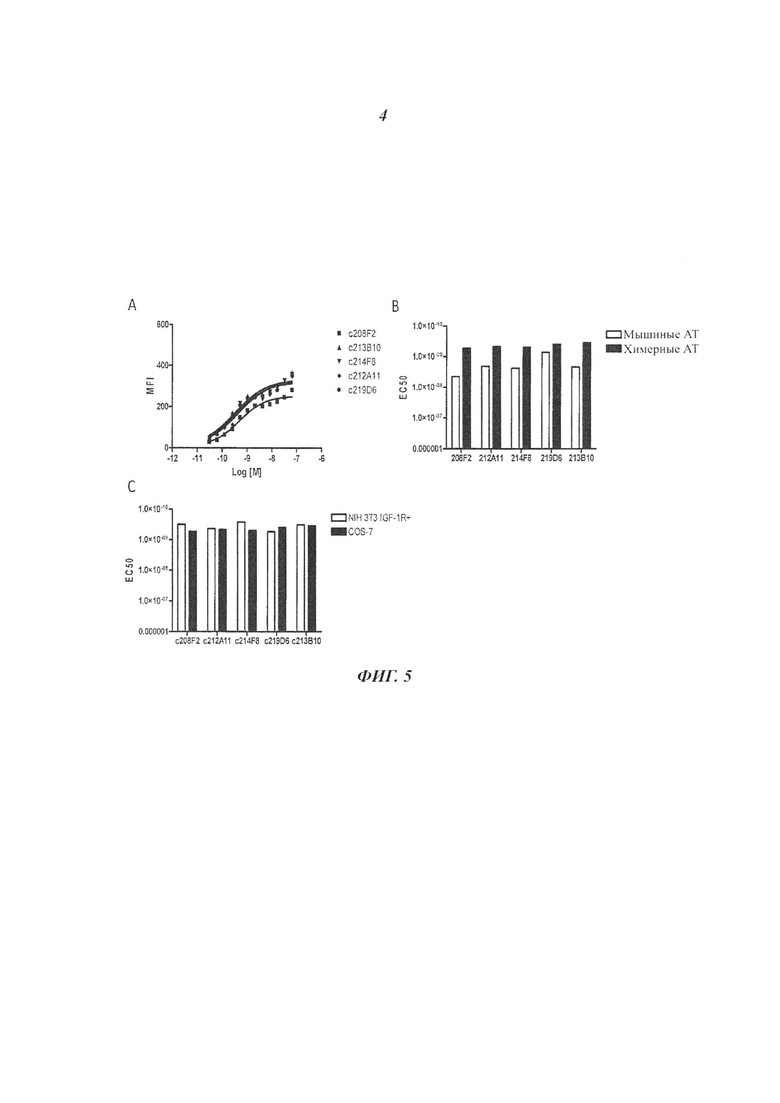
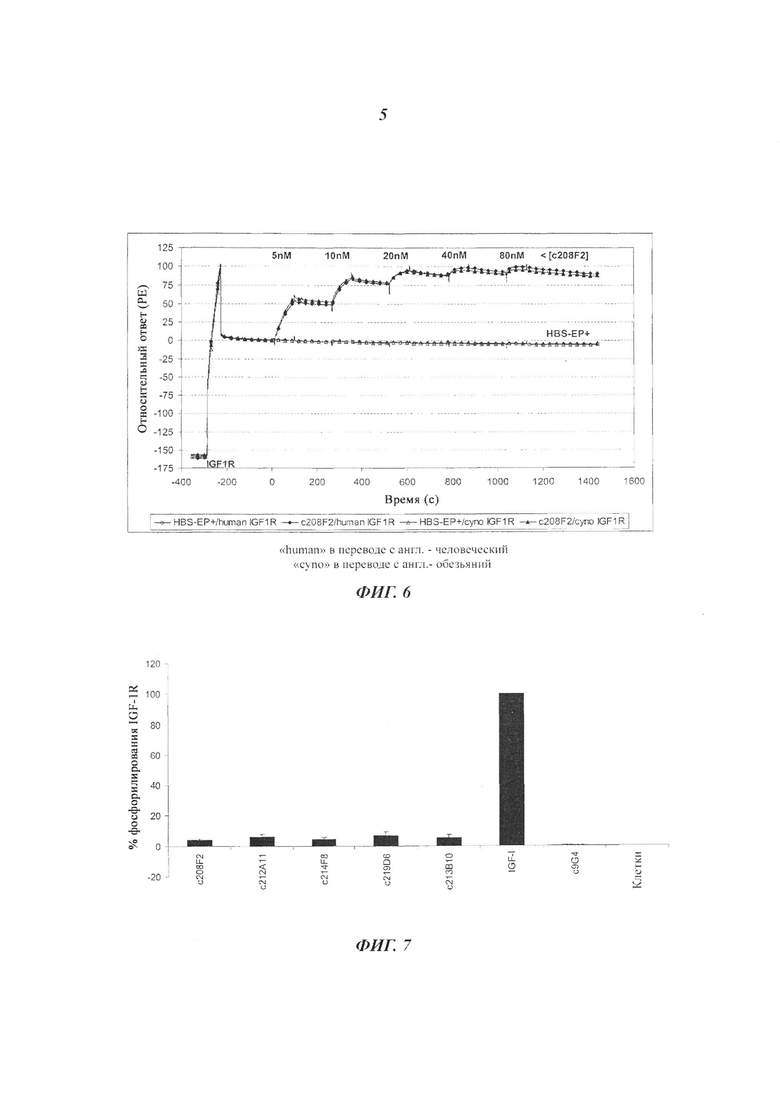
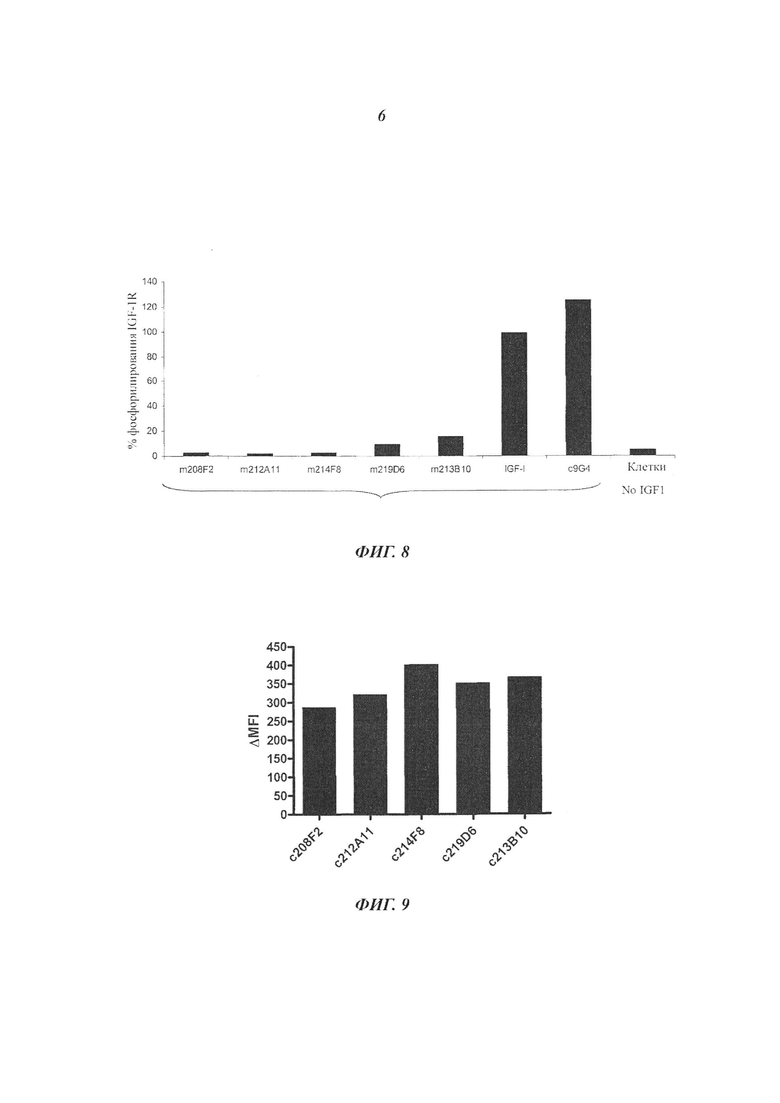
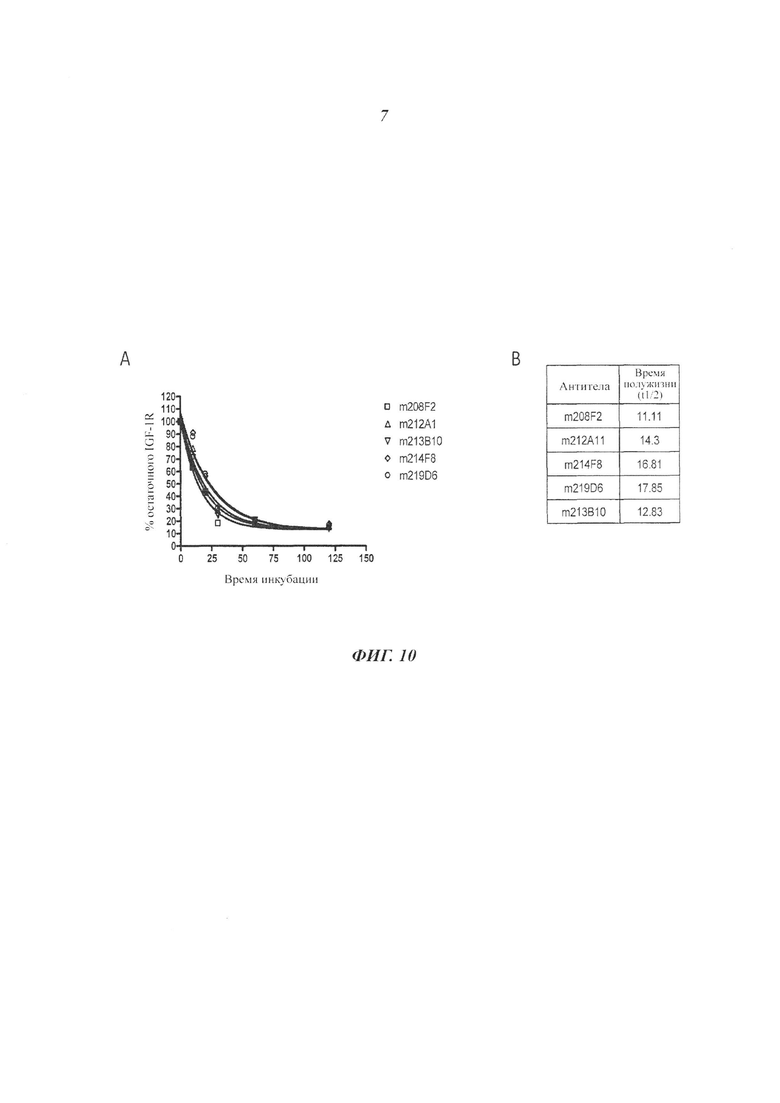
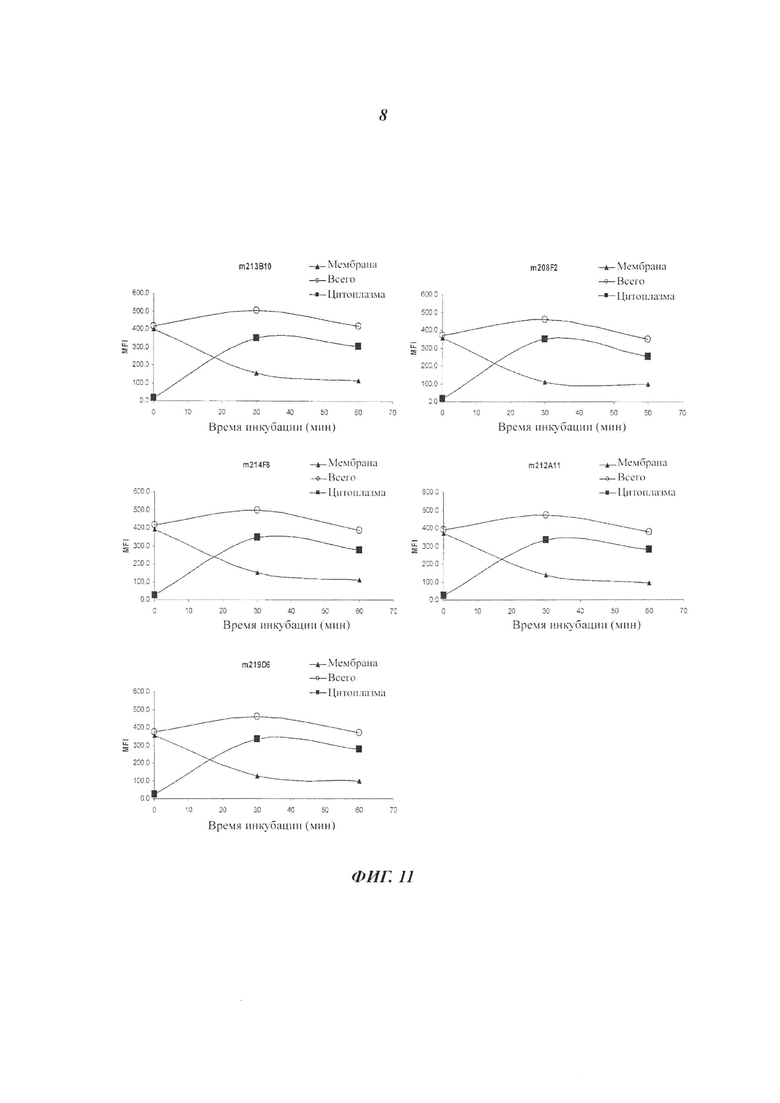
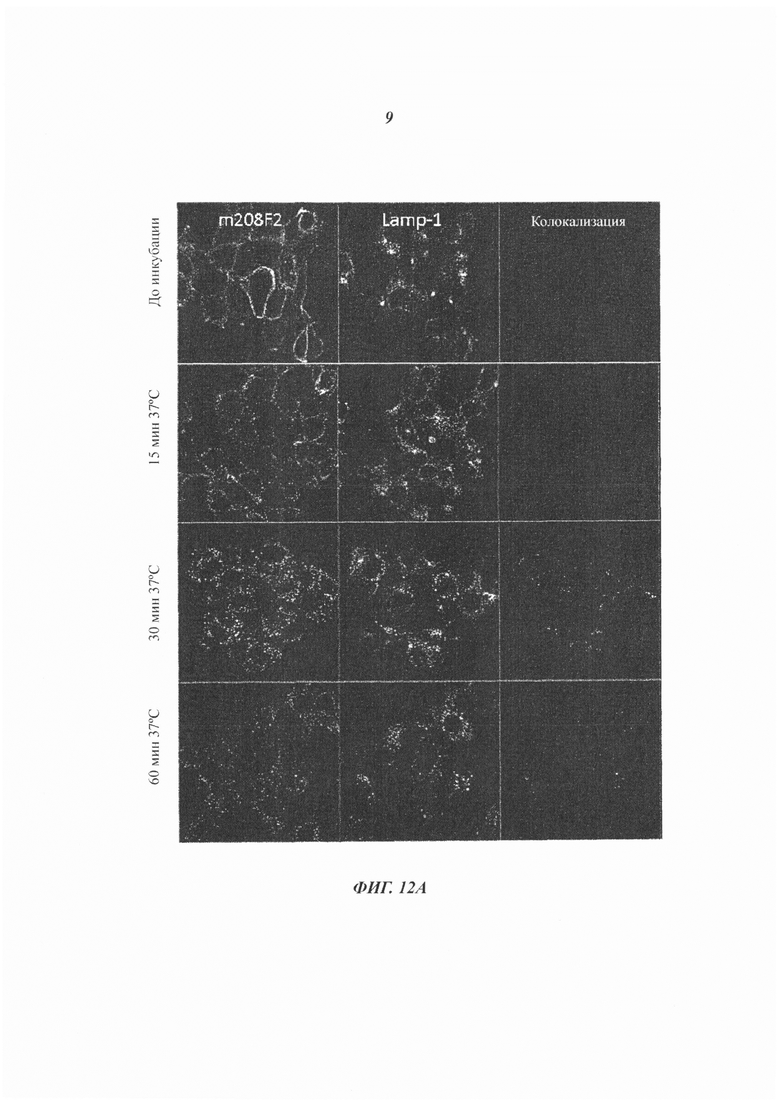
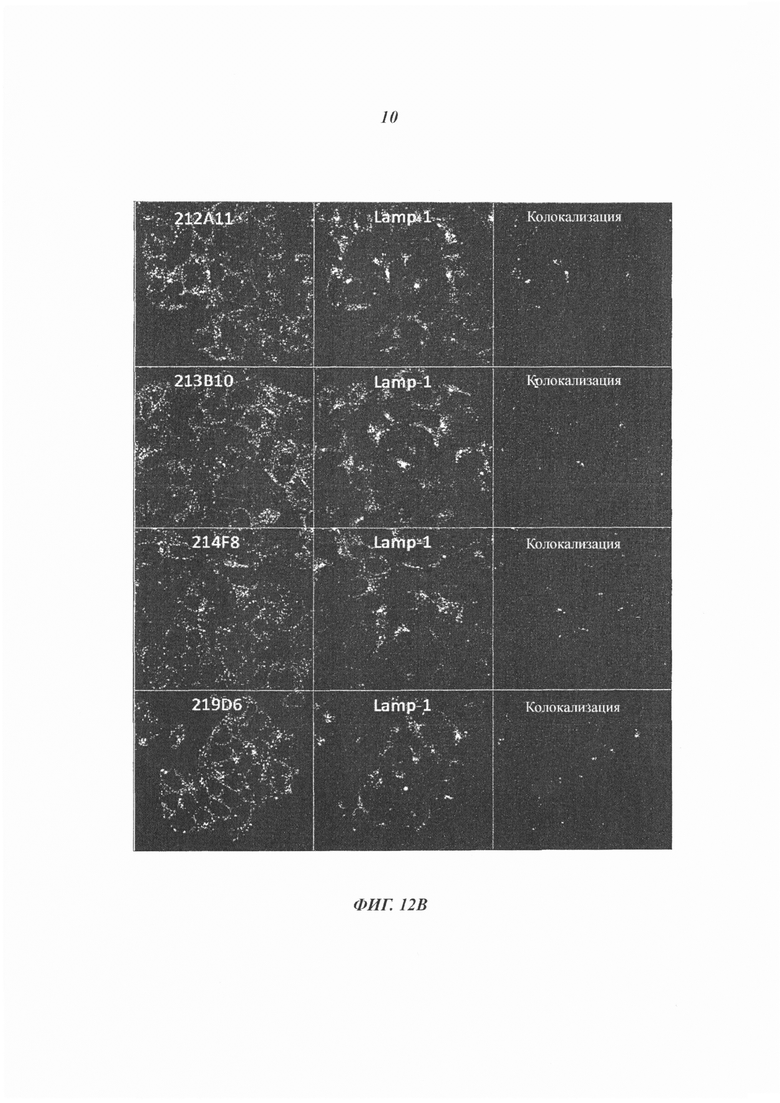
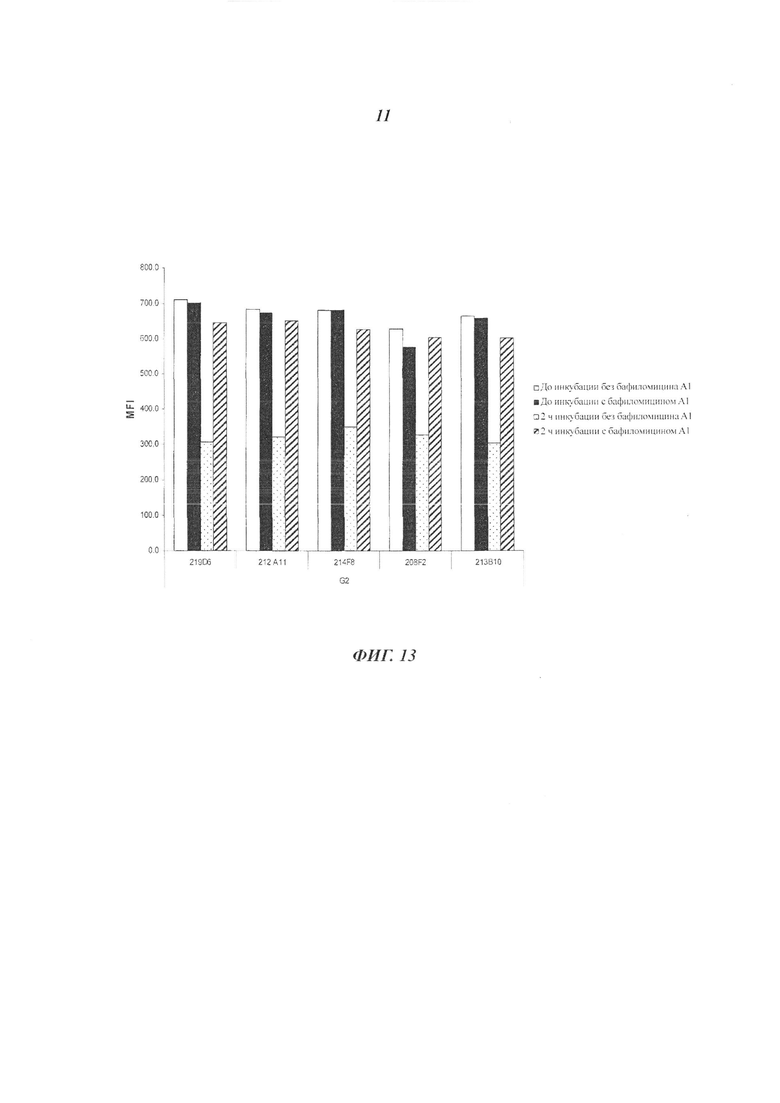
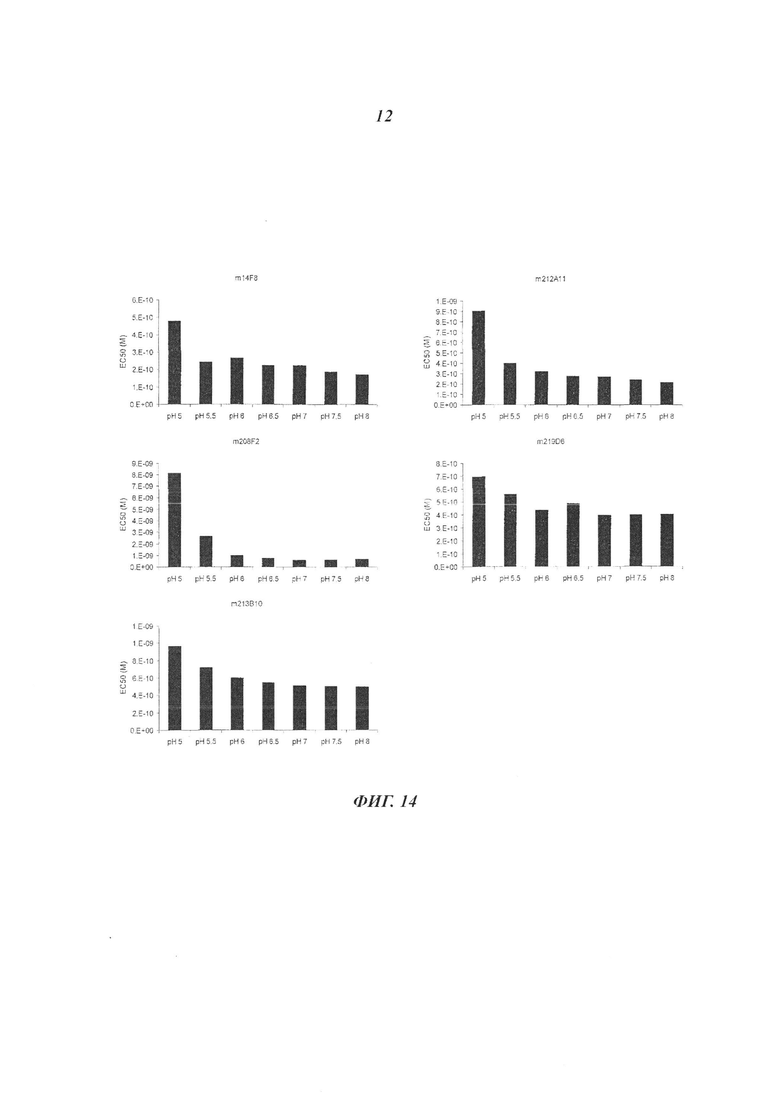
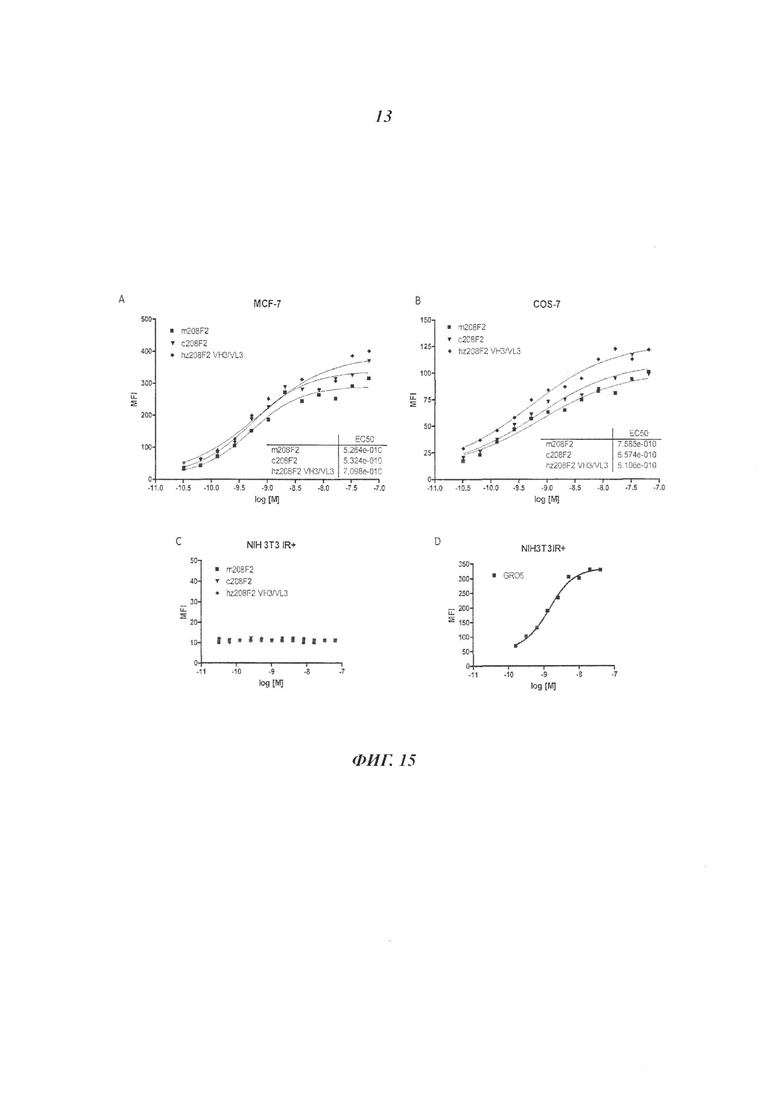
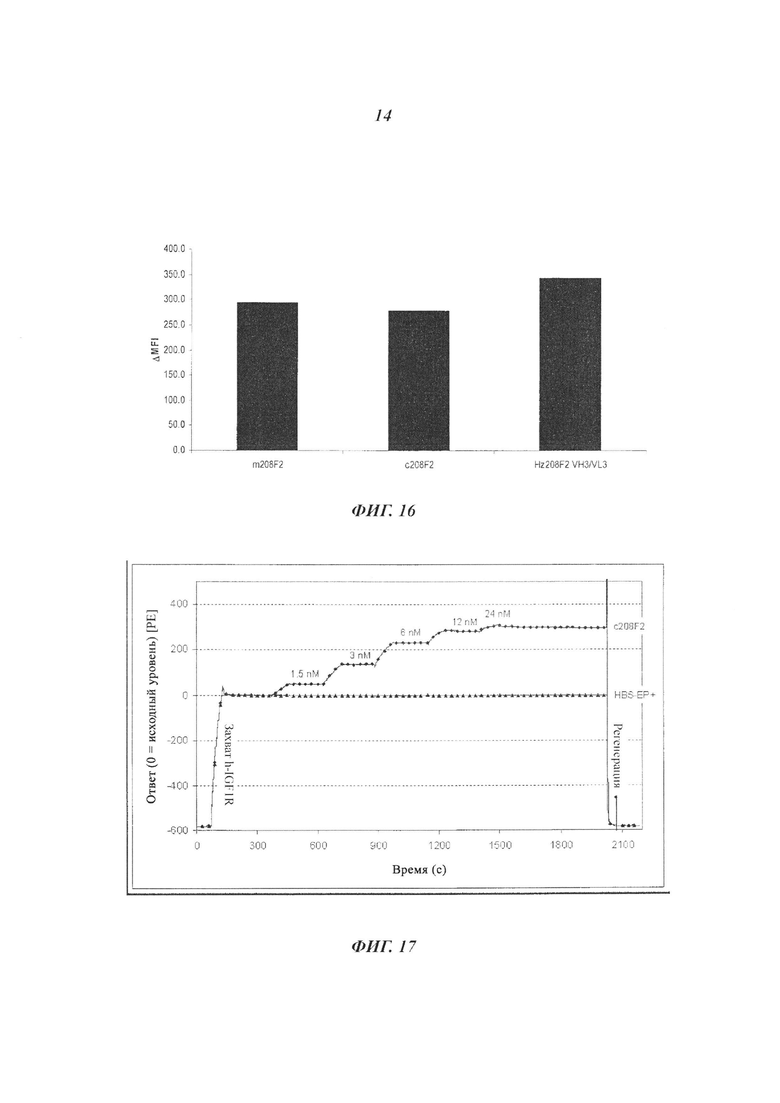
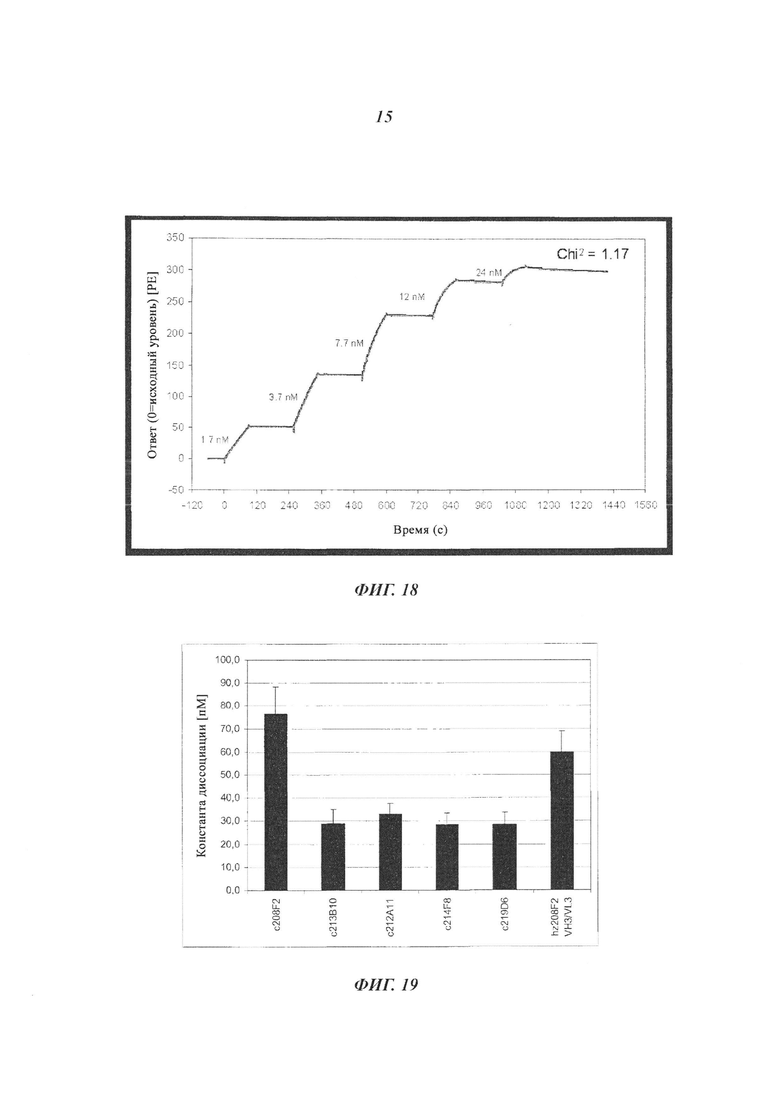
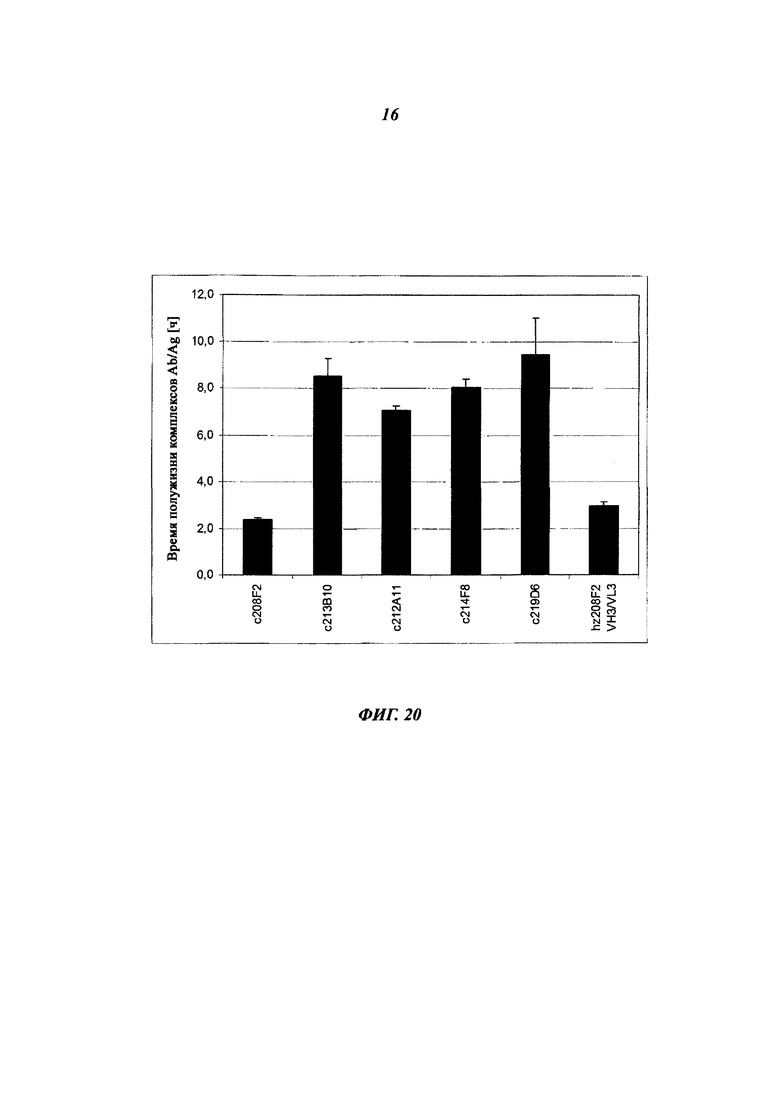
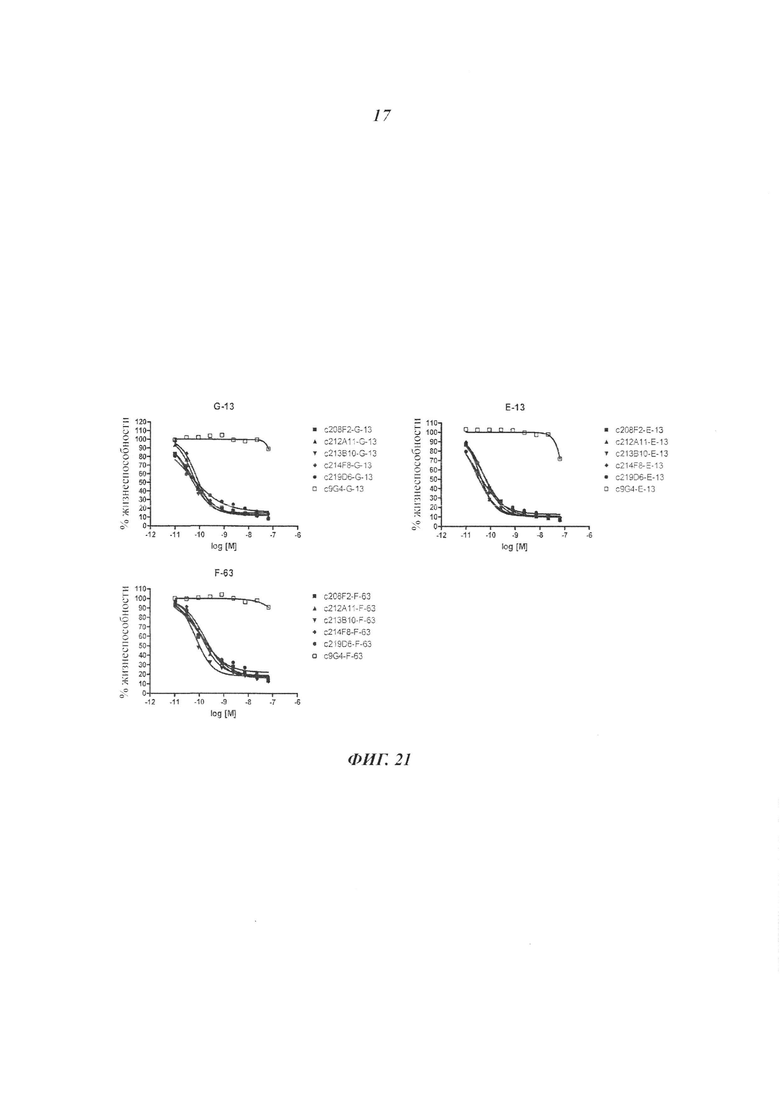
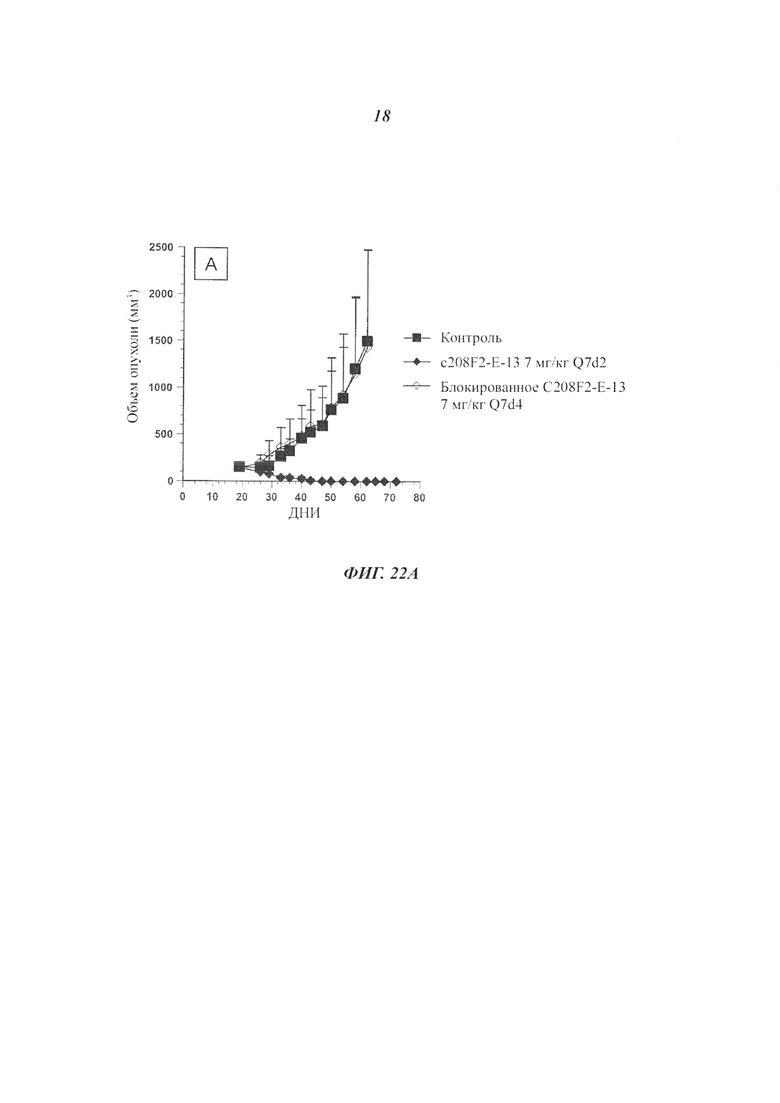
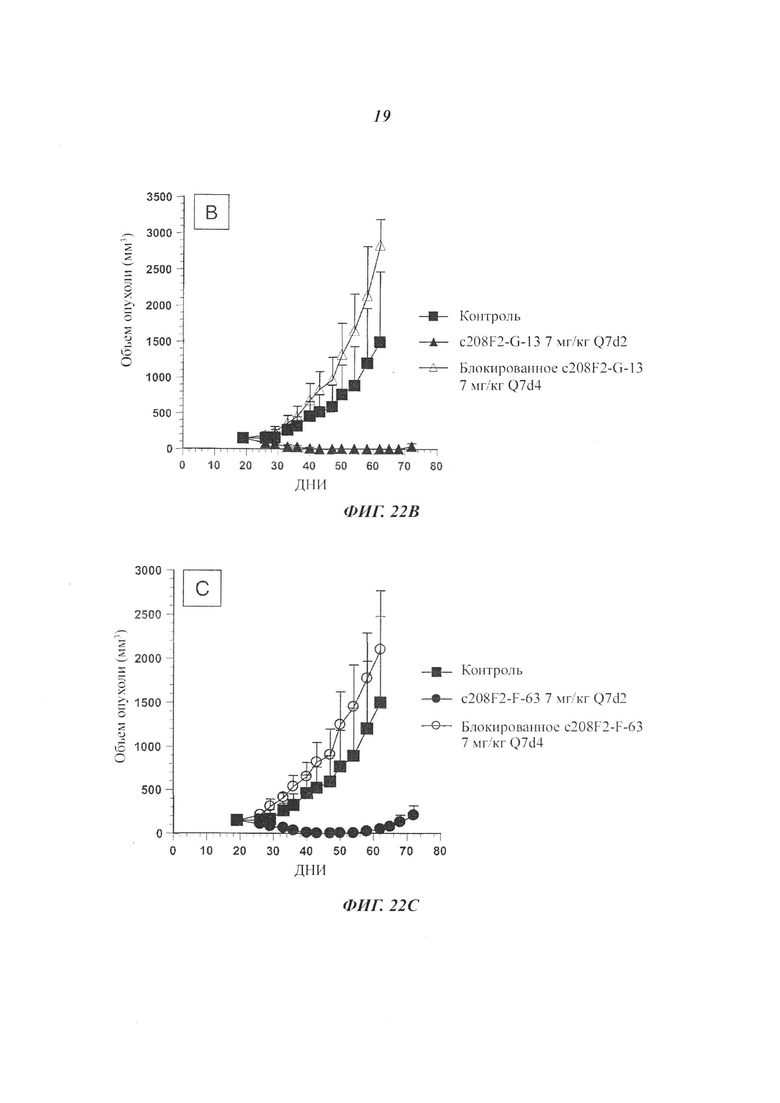
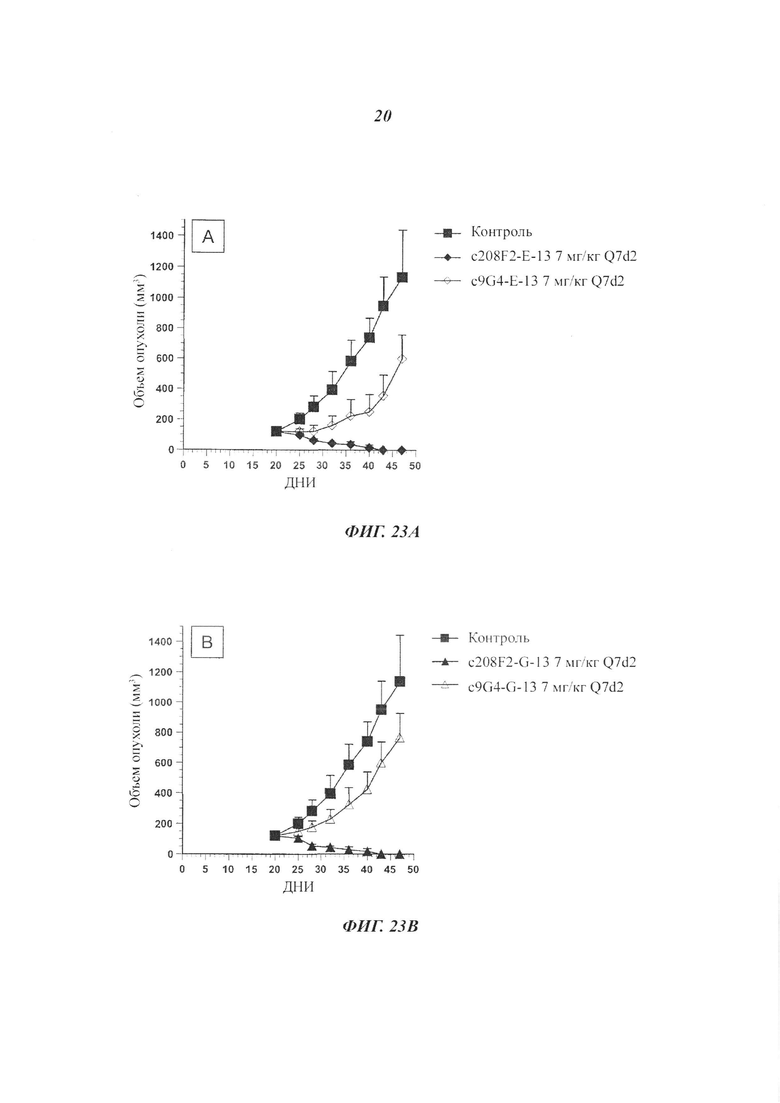
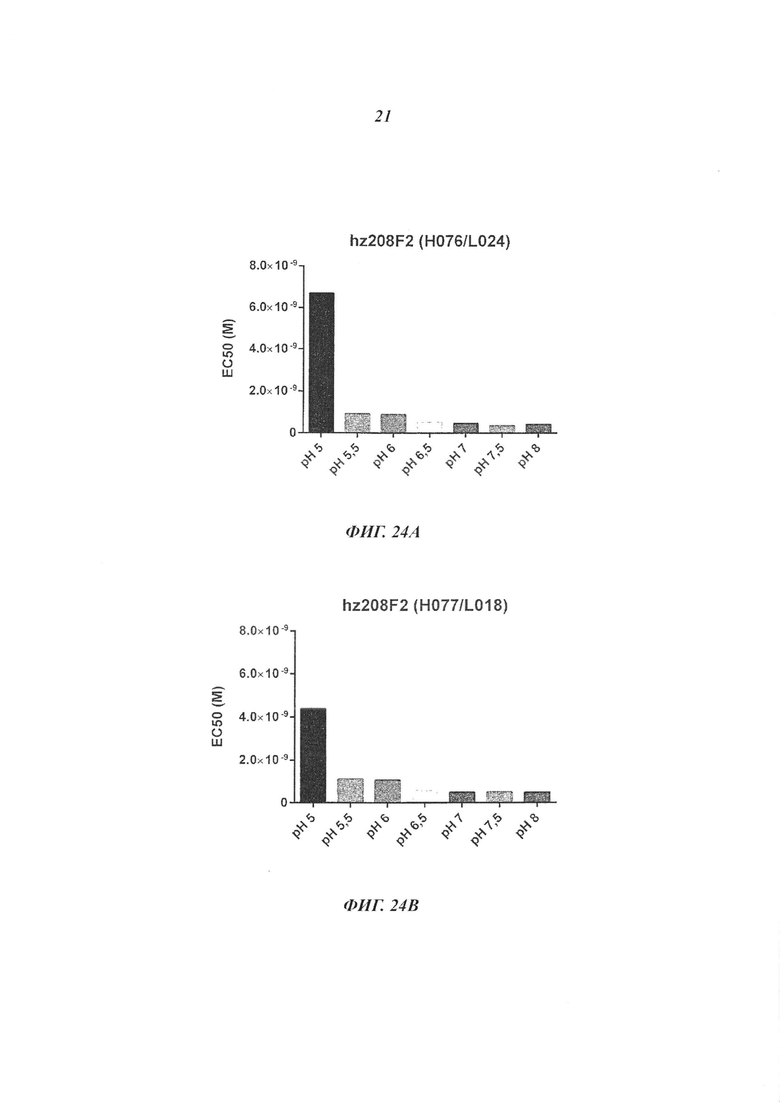
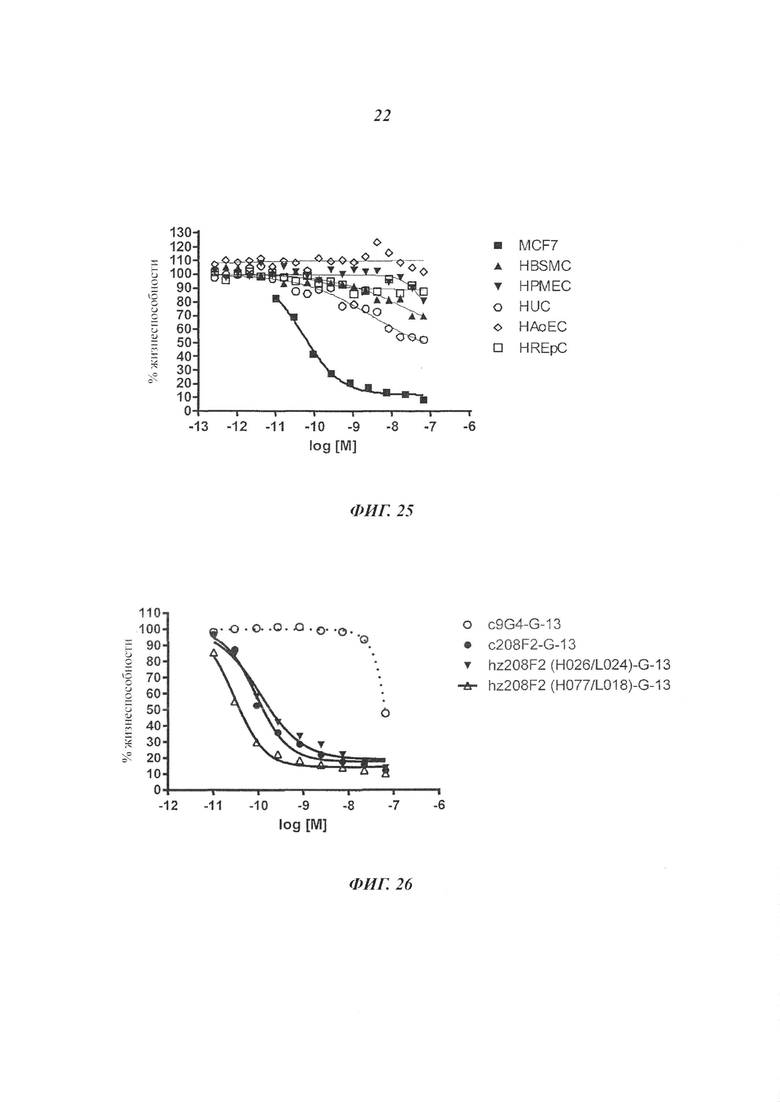
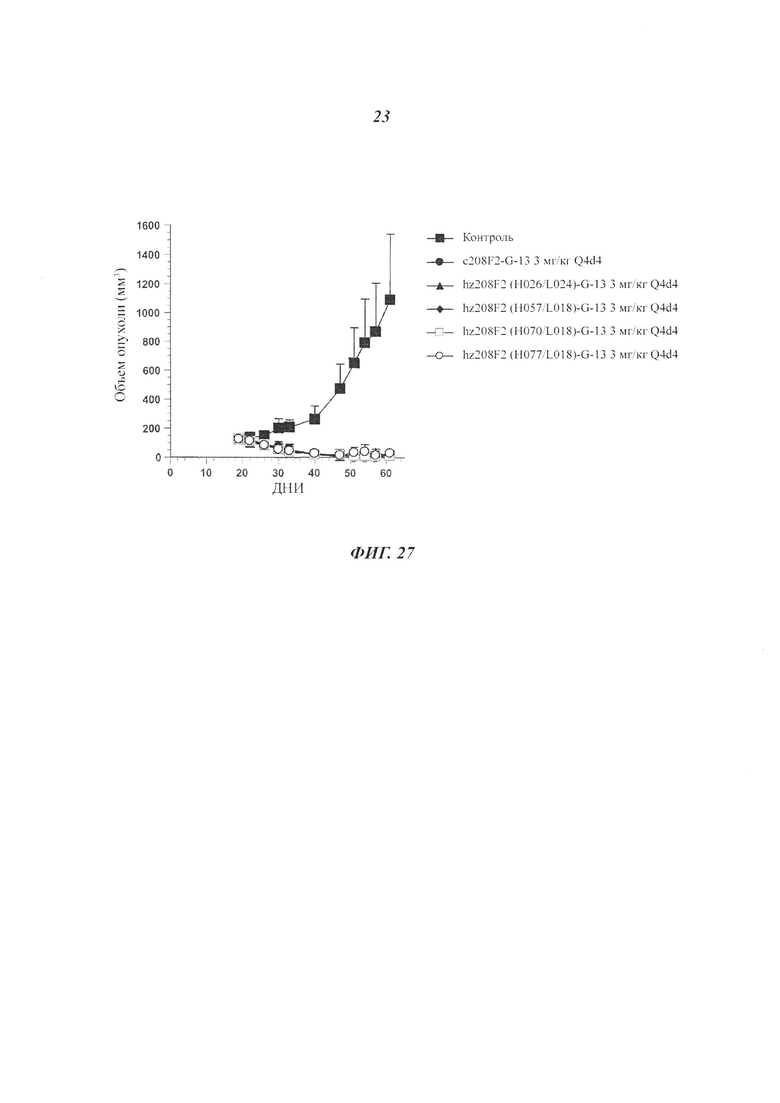
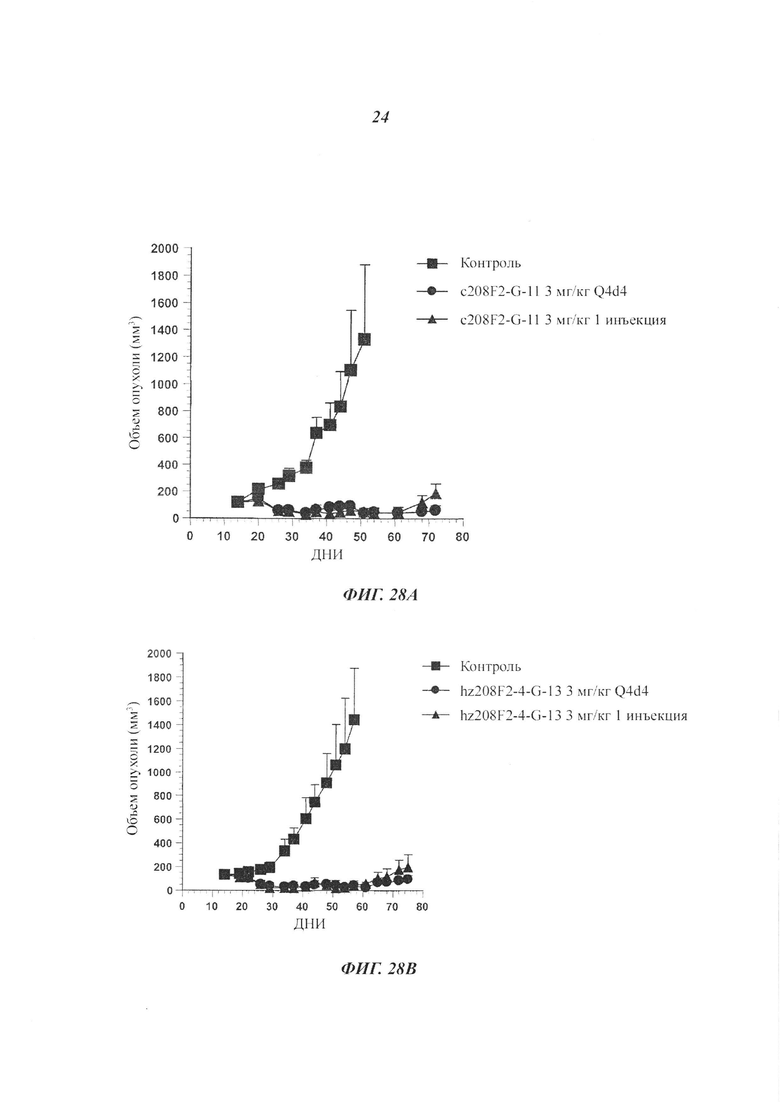
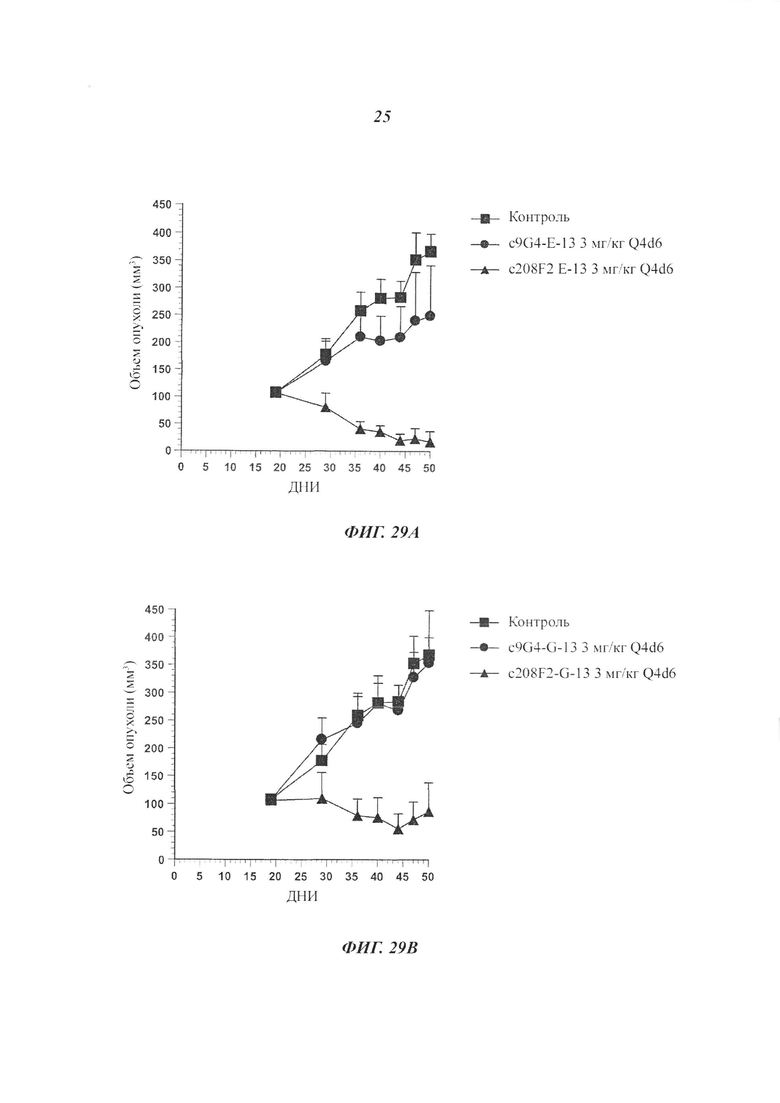
Изобретение относится к области биохимии, в частности к конъюгату антитела с лекарственным средством для лечения IGF-lR-экспрессирующего рака, а также к композиции для лечения IGF-1R-экспрессирующего рака, содержащей эффективное количество, по меньшей мере, одного вышеуказанного конъюгата антитела с лекарственным средством. Также раскрыт способ лечения IGF-1R-экспрессирующего рака у субъекта, нуждающегося в этом, предусматривающий использование вышеуказанного конъюгата. Изобретение позволяет эффективно осуществлять лечение IGF-lR-экспрессирующего рака. 3 н. и 19 з.п. ф-лы, 29 ил., 16 табл., 21 пр.
1. Конъюгат антитела с лекарственным средством для лечения IGF-lR-экспрессирующего рака следующей формулы (I):
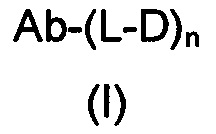
или его фармацевтически приемлемая соль,
где:
Ab представляет собой антитело или его антигенсвязывающий фрагмент, способные к связыванию с IGF-1R человека, причем антитело выбрано из:
a) антитела, содержащего три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 10, 5 и 11;
c) антитела, содержащего три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12; и
d) антитела, содержащего три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 8, 2 и 3, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
L представляет собой линкер следующей формулы (III):
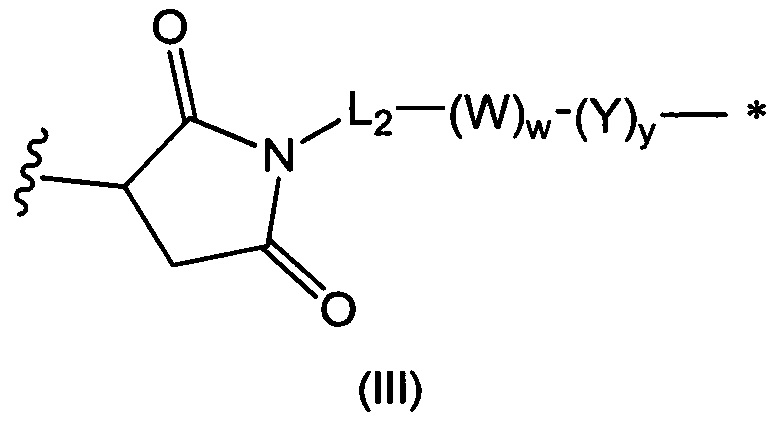
где:
L2 представляет собой (С4-С10)циклоалкил-карбонил, (С2-С6)алкил, (С2-С6)алкил-карбонил,
W представляет собой аминокислотное звено; w представляет собой целое число, составляющее от 0 до 5;
Y представляет собой РАВ-карбонил, где РАВ представляет собой 
у равно 0 или 1;
звездочкой указана точка присоединения к D; и
волнистой линией указана точка присоединения к Ab;
D представляет собой группировку лекарственного средства следующей формулы (II):
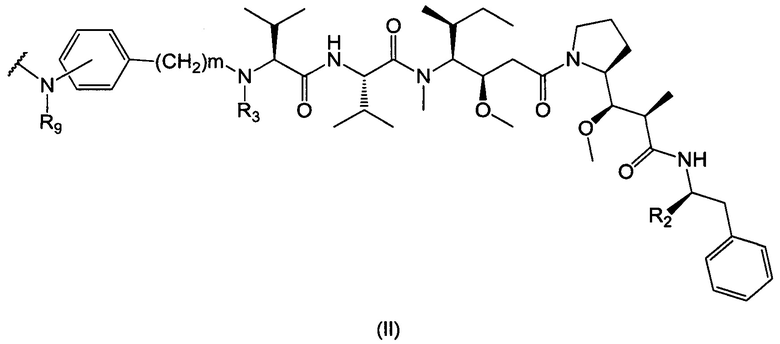
где:
R2 представляет собой СООН, СООСН3 или тиазолил;
R3 представляет собой Н или (С1-С6)алкил;
R9 представляет собой Н или (С1-С6)алкил;
m представляет собой целое число, составляющее от 1 до 8;
волнистой линией показана точка присоединения к L; и
n равно от 1 до 12.
2. Конъюгат антитела с лекарственным средством по п. 1, где Ab выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 13, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 14, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 10, 5 и 11;
c) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 15, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12;
d) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 16, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11; и
e) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 17, и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 12.
3. Конъюгат антитела с лекарственным средством по п. 1, где Ab выбрано из:
a) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 18, и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 19, и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
c) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 20, и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3;
d) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 21, и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 8, 2 и 3; и
e) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 22, и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3.
4. Конъюгат антитела с лекарственным средством по п. 1, где Ab выбрано из антител, продуцируемых гибридомами I-4757, I-4773, I-4775, I-4736 и I-4774, депонированными в Национальной коллекции культур микроорганизмов CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года соответственно.
5. Конъюгат антитела с лекарственным препаратом по п. 1, где Ab содержит: а) вариабельный домен тяжелой цепи (VH) последовательности SEQ ID NO 33, где данная последовательность SEQ ID NO 33 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 20, 34, 35, 38, 48, 50, 59, 61, 62, 70, 72, 74, 76, 77, 79, 82 и 95; и
b) вариабельный домен легкой цепи (VL) последовательности SEQ ID NO 35, где данная последовательность SEQ ID NO 35 содержит по меньшей мере одну обратную мутацию, выбранную из остатков 22, 53, 55, 65, 71, 72, 77 или 87.
6. Конъюгат антитела с лекарственным средством по п. 1, где Ab выбрано из:
a) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80; и три CDR участка легкой цепи, имеющих последовательности SEQ ID NO 9, 5 и 11;
b) антитела, содержащего вариабельный домен легкой цепи, имеющий последовательность, выбранную из SEQ ID NO 57 или 60; и три CDR участка тяжелой цепи, имеющих последовательности SEQ ID NO 7, 2 и 3; и
c) антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность, выбранную из SEQ ID NO 56, 62, 64, 66, 68, 70, 72, 74, 76, 78 и 80; и вариабельный домен легкой цепи, имеющий последовательность, выбранную из SEQ ID NO 57 или 60.
7. Конъюгат антитела с лекарственным средством по п. 1, где Ab содержит:
a) тяжелую цепь, имеющую последовательность, выбранную из SEQ ID NO 58, 63, 65, 67, 69, 71, 73, 75, 77, 79 и 81; и
b) легкую цепь, имеющую последовательность, выбранную из SEQ ID NO 59 и 61.
8. Конъюгат антитела с лекарственным средством по п. 1, где L2 имеет следующую формулу:
где
звездочкой указана точка присоединения к (W)w; и
волнистой линией указана точка присоединения к атому азота малеимидной группировки.
9. Конъюгат антитела с лекарственным средством по п. 1, где (W)w выбран из:
простой связи, 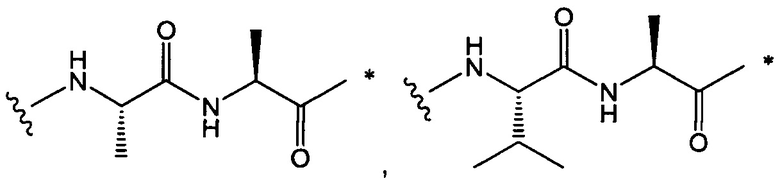
и 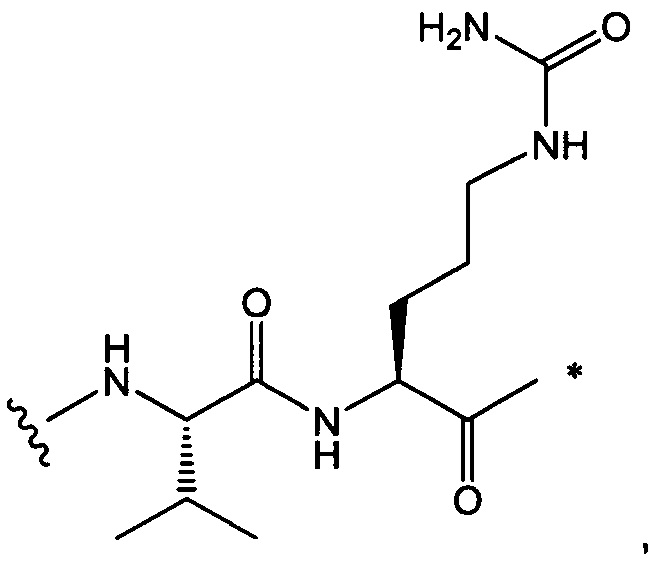
где
звездочкой указана точка присоединения к (Y)y; и
волнистой линией указана точка присоединения к L2.
10. Конъюгат антитела с лекарственным средством по п. 1, где w равно 0; или w равно 2, и (W)w выбран из:
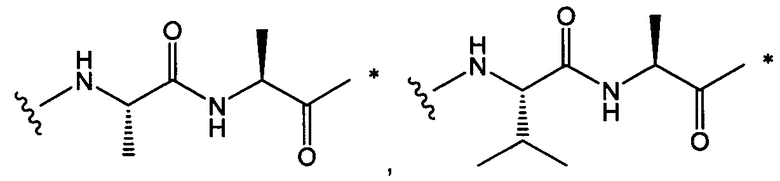
и 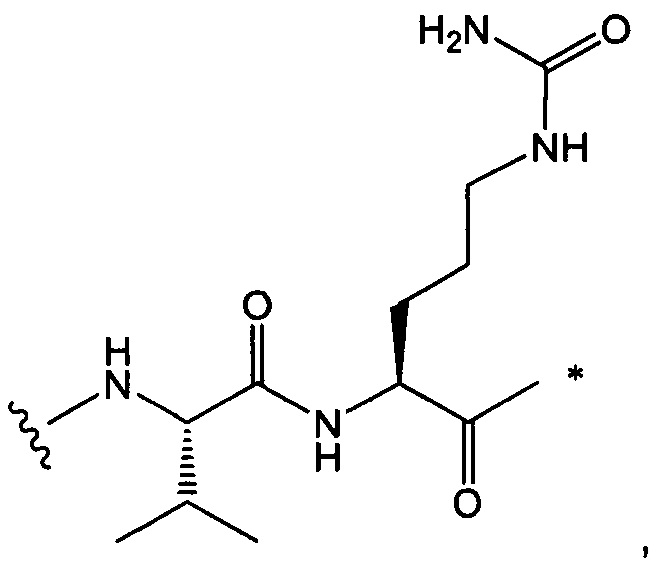
где
звездочкой указана точка присоединения к (Y)y; и
волнистой линией указана точка присоединения к L2.
11. Конъюгат антитела с лекарственным препаратом по п. 1, где (L-D) выбран из:
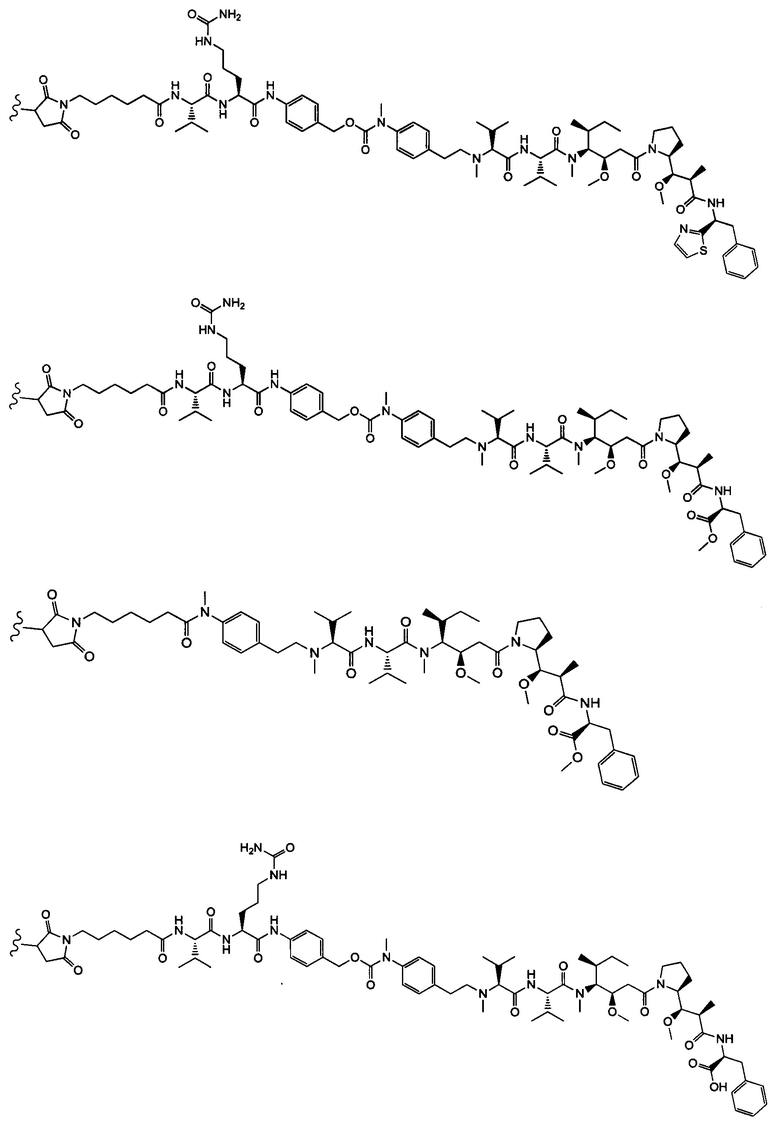
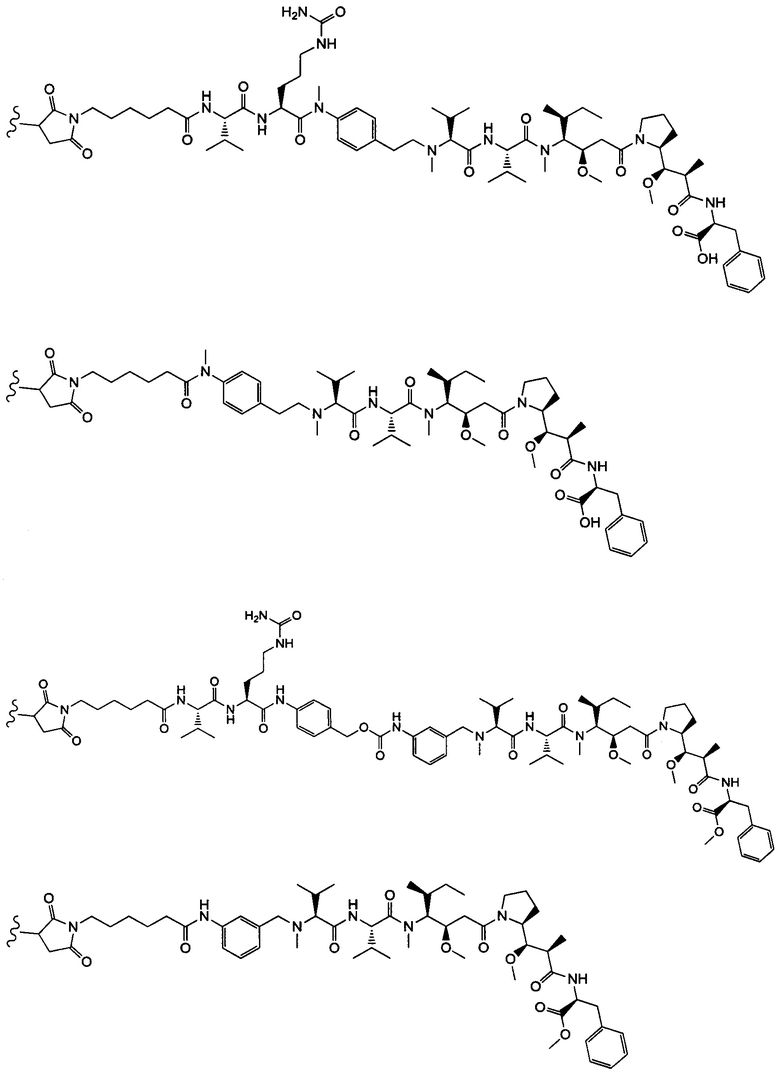
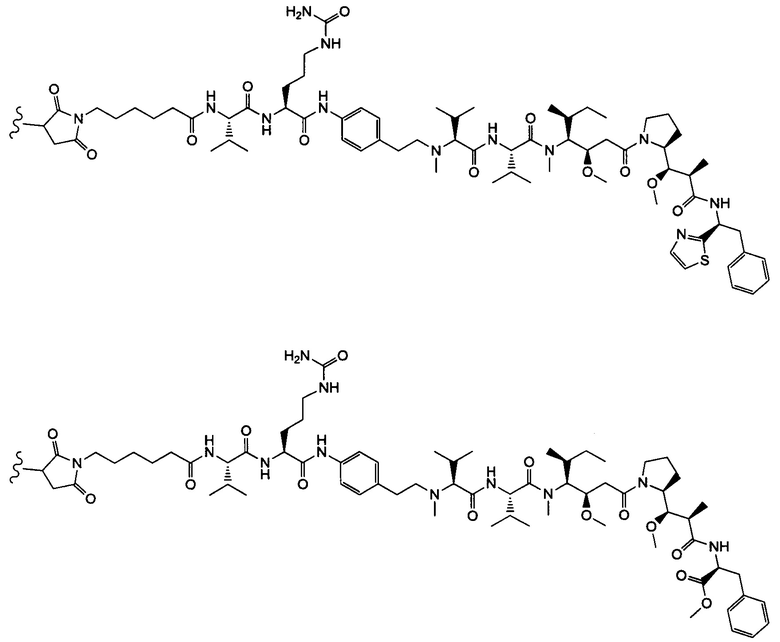
и
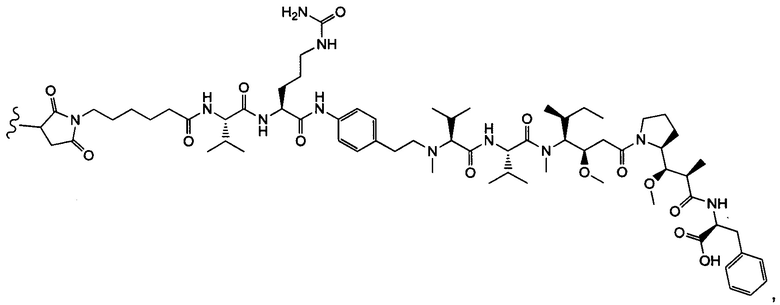
где волнистой линией указана точка присоединения к Ab.
12. Конъюгат антитела с лекарственным средством по п. 1, имеющий формулу, выбранную из:
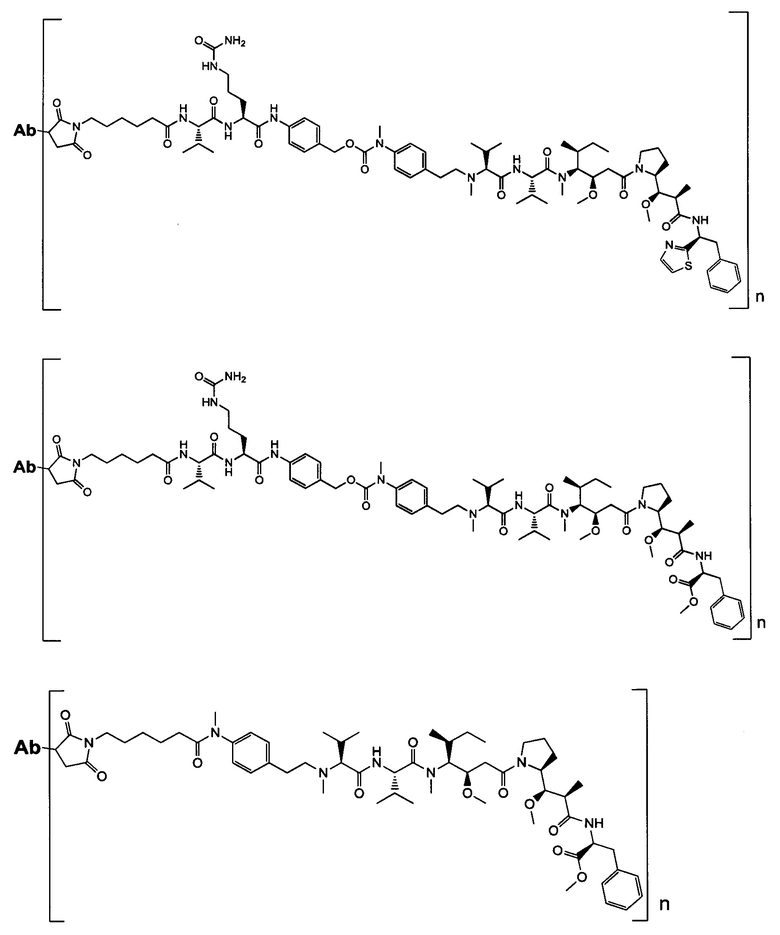
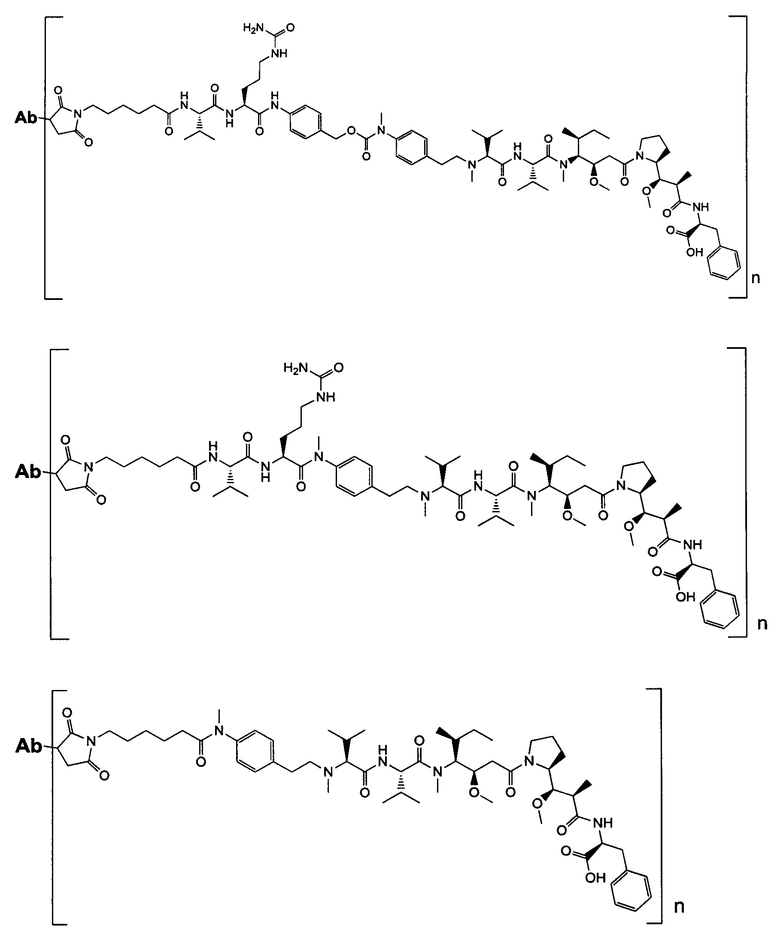
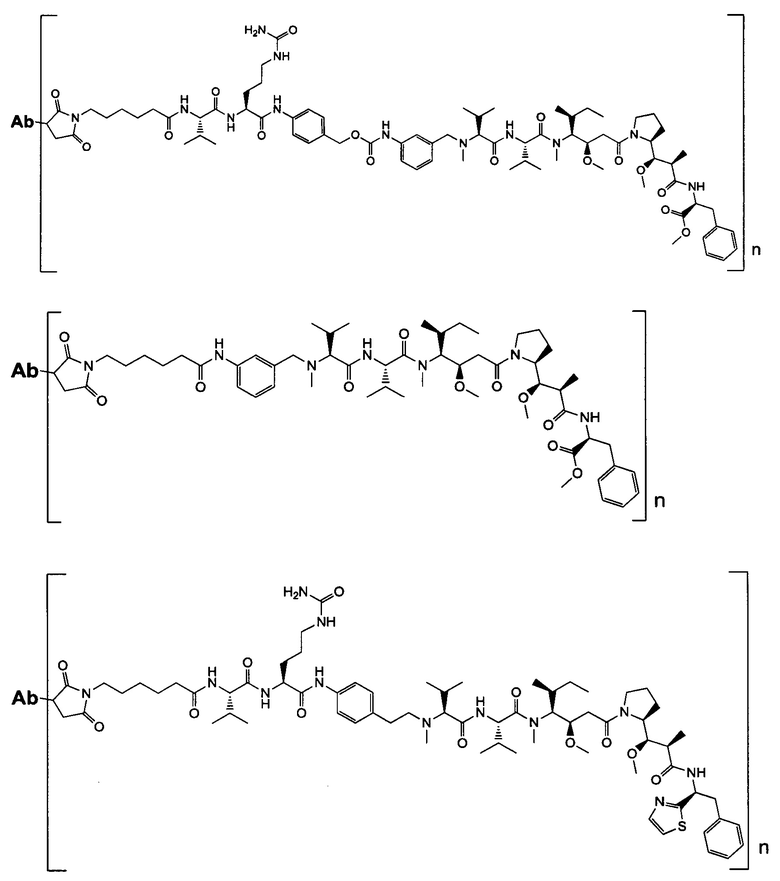
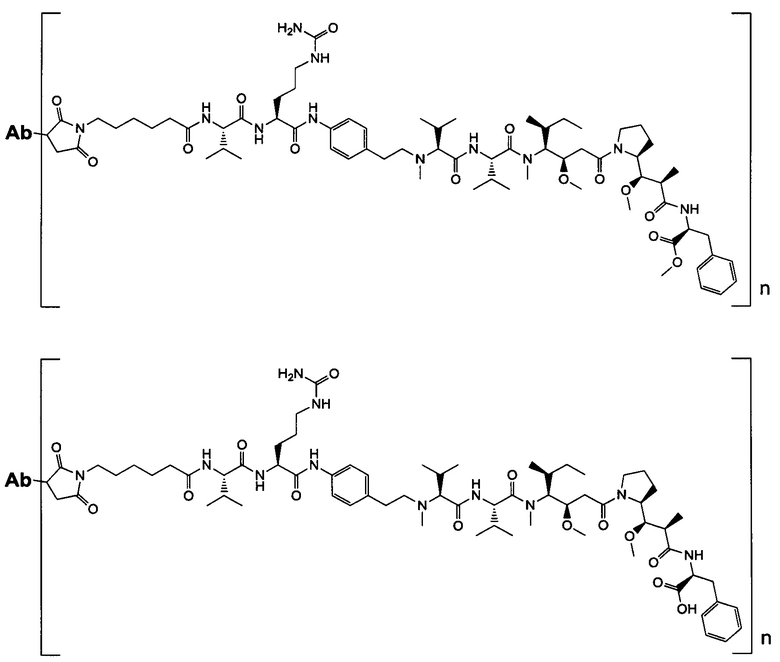
и их фармацевтически приемлемых солей,
где Ab выбрано из группы, состоящей из антител, продуцируемых гибридомами I-4757, I-4773, I-4775, I-4736 и I-4774, депонированными в Национальной коллекции культур микроорганизмов CNCM, Институт Пастера, Франция, 30 мая 2013 года, 26 июня 2013 года, 26 июня 2013 года, 24 апреля 2013 года и 26 июня 2013 года соответственно.
13. Конъюгат антитела с лекарственным средством по п. 11, где Ab выбрано из группы, состоящей из:
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 56, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 56, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 60;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 62, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 64, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 64, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 60;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 66, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 68, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 68, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 60;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 70, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 72, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 74, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 76, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 78, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57;
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 78, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 60; и
- антитела, содержащего вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 80, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57.
14. Конъюгат антитела с лекарственным средством по п. 13, где Ab выбрано из группы, состоящей из:
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 58, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 58, и легкую цепь, имеющую последовательность SEQ ID NO 61;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 63, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 65, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 65, и легкую цепь, имеющую последовательность SEQ ID NO 61;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 67, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 69, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 69, и легкую цепь, имеющую последовательность SEQ ID NO 61;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 71, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 73, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 75, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 77, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 79, и легкую цепь, имеющую последовательность SEQ ID NO 59;
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 79, и легкую цепь, имеющую последовательность SEQ ID NO 61; и
- антитела, содержащего тяжелую цепь, имеющую последовательность SEQ ID NO 81, и легкую цепь, имеющую последовательность SEQ ID NO 59.
15. Конъюгат антитела с лекарственным средством по п. 11, имеющий следующую формулу:
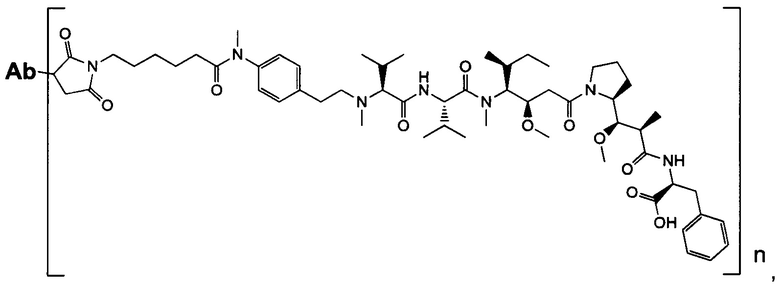
или его фармацевтически приемлемая соль,
где Ab представляет собой антитело, содержащее вариабельный домен тяжелой цепи, имеющий последовательность SEQ ID NO 80, и вариабельный домен легкой цепи, имеющий последовательность SEQ ID NO 57.
16. Конъюгат антитела с лекарственным средством по п. 15, где Ab представляет собой антитело, содержащее тяжелую цепь, имеющую последовательность SEQ ID NO 81, и легкую цепь, имеющую последовательность SEQ ID NO 59.
17. Конъюгат антитела с лекарственным средством по п. 1, где n равно 2.
18. Конъюгат антитела с лекарственным средством по п. 1, где n равно 4.
19. Конъюгат антитела с лекарственным средством по любому из пп. 1-18 для применения в качестве лекарственного препарата.
20. Композиция для лечения IGF-1R-экспрессирующего рака, содержащая эффективное количество по меньшей мере одного конъюгата антитела с лекарственным средством по любому из пп. 1-19 по меньшей мере с эксципиентом и/или фармацевтически приемлемым носителем.
21. Композиция по п. 20, где IGF-1R-экспрессирующий рак представляет собой рак, выбранный из карциномы молочной железы, ободочной кишки, пищевода, печеночно-клеточного рака, рака желудка, глиомы, рака легкого, меланомы, остеосаркомы, рака яичника, предстательной железы, рабдомиосаркомы, рака почки, щитовидной железы, эндометрия матки, мезотелиомы, плоскоклеточной карциномы полости рта и любого лекарственно-устойчивого рака.
22. Способ лечения IGF-1R-экспрессирующего рака у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества по меньшей мере одного конъюгата антитела с лекарственным средством по любому из пп. 1-18 или композиции по п. 20.
| WO2012142164 A1, 18.10.2012 | |||
| SVETLANA O DORONINA et al., Development of potent monoclonal antibody auristatin conjugates for cancer therapy, Nature Biotechnology, 2003, vol.21, pp.778-784 | |||
| RUDIKOFF S | |||
| et al., Single amino acid substitution altering antigen-binding specificity, Proceedings of the National Academy of Sciences, 1982, Т | |||
| Цилиндрический сушильный шкаф с двойными стенками | 0 |
|
SU79A1 |
| 2005 |
|
RU2404810C2 | |

