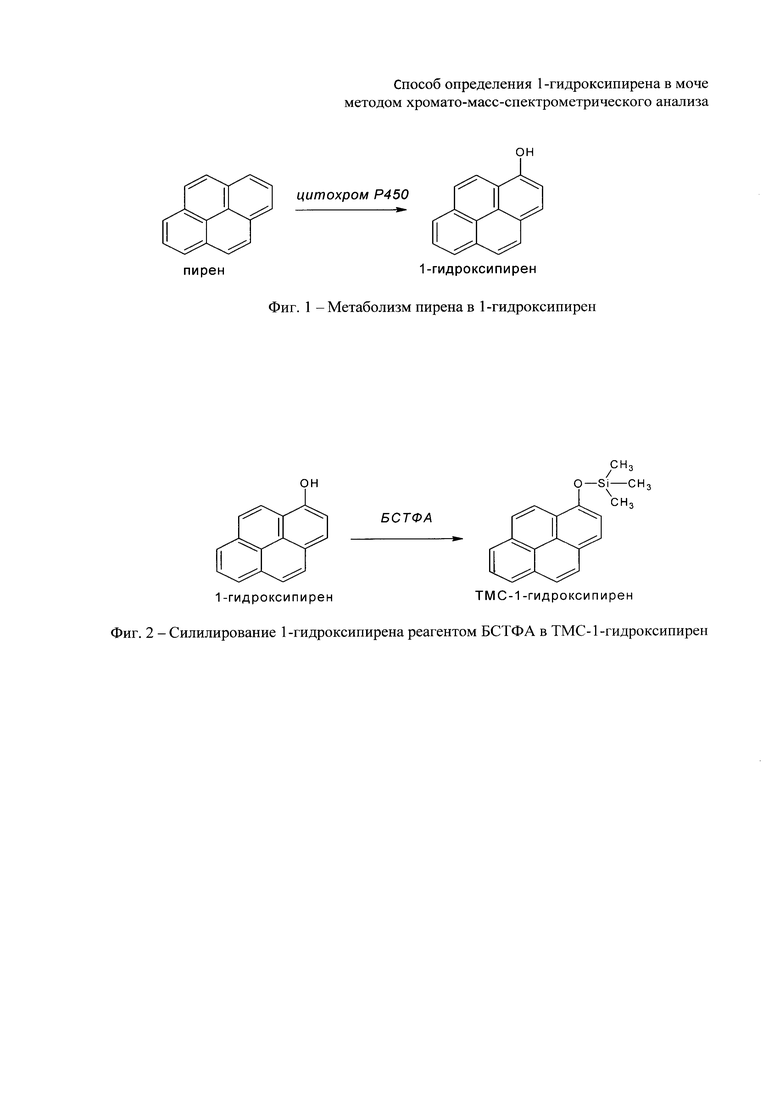
Изобретение относится к области аналитической токсикологии и биологического мониторинга, и может быть использовано при оценке воздействия полициклических ароматических углеводородов (ПАУ) на организм человека. В подавляющем случае ПАУ присутствуют в виде смесей, одним из основных компонентов по уровню концентрации является пирен. Метаболит пирена - 1-гидроксипирен (Фиг. 1) является стабильным соединением, относительно легко и надежно определяемым в моче.
Известный способ определения 1-гидроксипирена в моче методом высокоэффективной жидкостной хроматографии (ВЭЖХ) включает в себя предварительный ферментативный гидролиз β-глюкуронидазой при 37°С 16 ч, твердофазную экстракцию на картридже С18, анализ метанольного экстракта методом ВЭЖХ с флуориметрическим детектором. Предел обнаружения 0,1 нг/см3, нижний предел количественного определения 2 нг/см3 при объеме пробы 10 см3, относительное стандартное отклонение воспроизводимости (RSD) 12,6%, коэффициент корреляции градуировочного графика 0,990, правильность (R) 88±9%. [Jongeneelen F.J., Anzion R.B.M., Henderson Р.Т. Determination of hydroxylated metabolites of polycyclic aromatic hydrocarbons in urine // J. of Chromatography. 1987. V. 413. P. 227-232]. К недостаткам данного способа можно отнести большой объем пробы, длительность процедуры ферментативного гидролиза (16 ч) и ВЭЖХ анализа (50 мин), недостаточная чувствительность для определения концентрации 1-гидроксипирена в моче у населения, а также невысокая точность (RSD 12% и R=88%).
При использовании метода ВЭЖХ возникают проблемы из-за ошибок при идентификации зарегистрированных компонентов. Количественное определение их также может быть затруднено вследствие наложения или неполного разделения хроматографических пиков.
Другим подходящим способом (прототип) является определение 1-гидроксипирена в моче газовой хроматографией с масс-селективным детектированием (ГХ-МС) в режиме электронного удара.
Способ включает в себя следующие стадии: ферментативный гидролиз β-глюкуронидазой при 37°С 17-18 ч, твердофазную экстракцию, упаривание экстракта в токе азота, дериватизацию сухого остатка силилирующим реагентом N,O-бис-(триметилсилил) трифторацетамид (БСТФА) при температуре 60°С 40 мин, ГХ-МС анализ в течение 40 мин. Предел обнаружения 0,5 нг/см3, нижний предел количественного определения 1 нг/см3 при объеме пробы 3 см3, относительное стандартное отклонение воспроизводимости (RSD) 6,7-13,1%, коэффициент корреляции градуировочного графика 0,995, правильность (R) 88,7%. [С.Schummer, О. Delhomme, B.M.R. Appenzeller. Comparison of MTBSTFA and BSTFA in derivatization reaction of polar compounds prior to GC/MS analysis / Talanta 2009. - V. 77. P. 1473-1482].
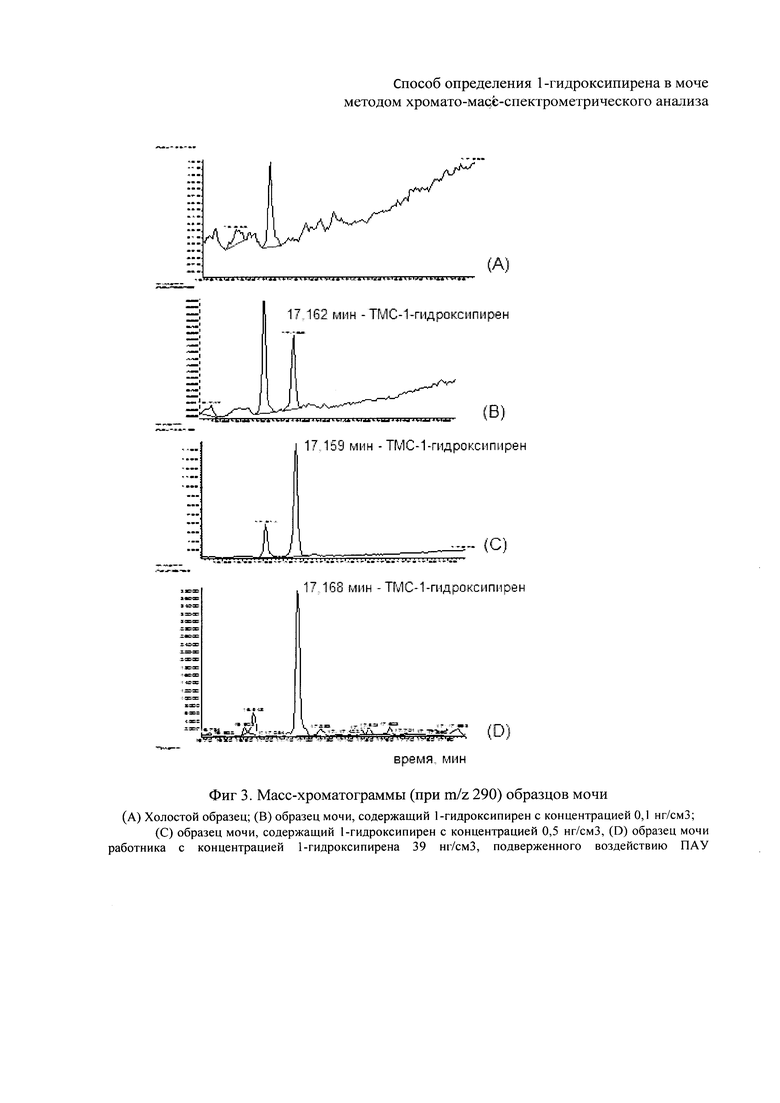
Технической задачей предлагаемого способа является повышение точности определения методом ГХ-МС за счет использования изотопно-меченного стандарта 1-гидроксипирена-d9, повышение чувствительности определения за счет увеличения степени экстракции из биологической матрицы, а также значительное сокращение времени пробоподготовки за счет уменьшения продолжительности ее следующих стадий: ферментативного гидролиза, дериватизации 1-гидроксипирена силилирующим реагентом N,O-бис-(триметилсилил) трифторацетамид (БСТФА) (Фиг. 2), ГХ-МС анализа.
Указанная техническая задача достигается путем внесения в анализируемые пробы мочи изотопно-меченного стандарта 1-гидроксипирена-d9, проведения ферментативного гидролиза в течение 1 часа при 55°С, 2-х кратной жидкостно-жидкостной экстракции гексаном, упаривания гексанового экстракта досуха в токе азота, силилирования сухого остатка N,O-бис-(триметилсилил) трифторацетамидом (БСТФА) при температуре 22-25°С в течение 5 мин и проведении газохроматографического анализа на капиллярной колонке с масс-селективным детектированием в течение 20 мин.
Из патентной и научно-технической литературы нам неизвестны способы количественного определения 1-гидроксипирена в моче, содержащие совокупность предложенных нами признаков, что позволяет сделать вывод о новизне заявляемого технического решения.
Кроме того, из существующего уровня техники нам неизвестно использование существенных признаков, характеризующих предлагаемый способ, для достижения описываемого технического результата.
Способ осуществляют следующим образом:
К 2 см3 мочи в центрифужной пробирке вместимостью 10 см3 вносят 20 мм3 раствора 1-гидроксипирена-d9 в ацетонитриле (5 мкг/см3), 1 см3 ацетатно-уксусного буфера с рН=5, 20 мм3 водного раствора β-глюкуронидазы (активность >85000), нагревают 1 ч при 55°С, затем охлаждают 20 мин до температуры 20-25°С, добавляют 0,5 г сульфата магния, 2 см3 гексана и интенсивно встряхивают на мультивортексе 2 мин, после этого разделяют фазы на центрифуге при 4000 об/мин 10 мин, отделяют гексановый экстракт, перенося его во флакон вместимостью 5 см3 с коническим дном, снова вносят 2 см3 гексана, интенсивно встряхивают на мультивортексе 2 мин, разделяют фазы на центрифуге при 4000 об/мин 10 мин, отделяют гексановый экстракт, объединяя с первым экстрактом. Объединенный гексановый экстракт упаривают досуха в небольшом токе азота при температуре водяной бани 60°С, затем к сухому остатку вносят 100 мм3 реагента БСТФА и выдерживают 5 мин.
Основное же преимущество метода ГХ-МС по сравнению с ВЭЖХ - это более лучшее разделение компонентов и возможность применения изотопно-меченного стандарта 1 -гидроксипирен-d9 (дейтерированный стандарт).
Хромато-масс-спектрометрический анализ осуществляют в следующих условиях (табл. 1).

Идентификацию на хроматограммах 1-гидроксипирена и 1-гидроксипирена-d9 в виде производных (триметилсиланов) проводят по времени удерживания и соотношению интенсивностей регистрируемых ионов (таблица 2).

Количественное определение по предлагаемому способу проводят методом внутреннего стандарта по растворам известной концентрации 1-гидроксипирена в моче от 0,1 до 100 нг/см3, предварительно обработанным аналогично анализируемым пробам. При градуировке процедуру ферментативного гидролиза допускается не проводить.
При реакции силилирования 1-гидроксипирена реагентом БСТФА, содержащим 1% триметилхлорсилана, установлено, что температура и время реакции не оказывают значимого влияния на аналитический сигнал, особенно если в качестве аналитического сигнала выступает относительная площадь (высота) пика (табл. 3).
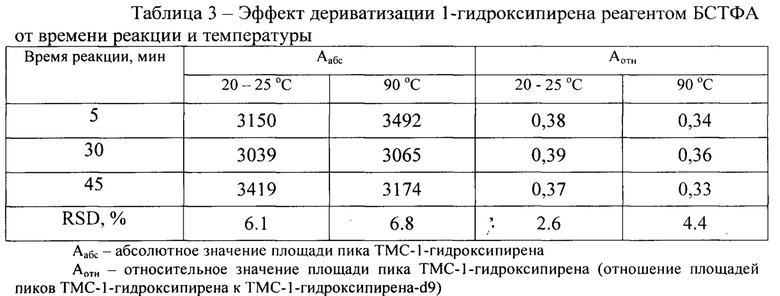
Данные показывают, что силилирование достаточно проводить при комнатной температуре 20-25°С в течение 5 мин и с внутренним стандартом, так как величина RSD самая минимальная.
Экспериментальным путем установлено, что при твердофазной экстракции (ТФЭ) степень экстракции 1-гидроксипирена из мочи составляет 8%, а при жидкостно-жидкостной экстракции (ЖЖЭ) гексаном около 90% (табл. 4).

Несмотря на то, что ТФЭ более современный способ, чем ЖЖЭ, степень извлечения аналита данным способом очень низкая. Это объясняется не тем, что аналит плохо адсорбируется на сорбент C18 и/или плохо элюируется с него метанолом, а тем, что в образце мочи присутствует большое количество полярных соединений, содержащие в своем составе ОН, СООН, NH, NH2 группы, и гораздо в больших концентрациях, чем наш аналит. Эти компоненты оказывают мешающее влияние при силилировании 1-гидроксипирена реагентом БСТФА. Получается, что реакцию силилирования вступает очень малое количество 1-гидроксипирена. При ЖЖЭ гексаном извлекаются в основном соединения неполярной или малополярной природы, которые не оказывают мешающее влияние при дериватизации БСТФА. Поэтому, подходящим способом извлечения 1-гидроксипирена из биологической матрицы является 2-х кратная ЖЖЭ гексаном.
Основными факторами, влияющими на эффективность жидкостно-жидкостной экстракции являются тип экстрагента, продолжительность экстракции, число экстракций, природа и количество высаливающего агента. Опытным путем установлено, что гексан, является более удобным экстрагентом чем, диэтиловый эфир и толуол, так как меньше мешающих влияний при дериватизации. В качестве высаливающего агента подходит сульфат магния, чем сульфат натрия, так как выше процент извлечения аналита. Оптимальные условия проведения ЖЖЭ 1-гидрокиспирена из мочи сводятся к следующим: масса сульфата магния 0,5 г, время экстракции 1-2 мин, кратность экстракции 2.
В таблице 5 приведены результаты анализа реальных образцов мочи работников алюминиевого производства, при двух вариантах ферментативного гидролиза.
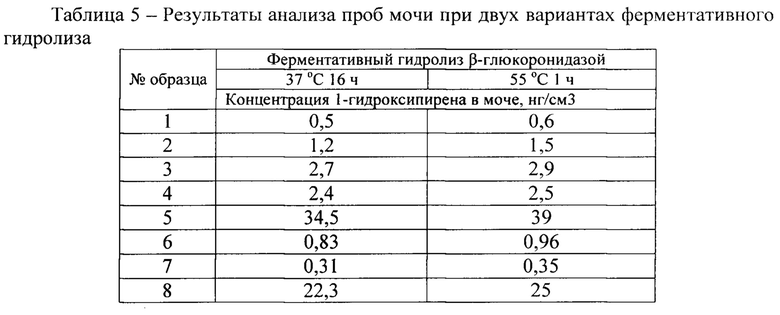
Сравнивая два варианта ферментативного гидролиза, видно, что концентрации 1-гидроксипирена не сильно различаются, при втором варианте концентрации аналита даже выше, поэтому ферментативный гидролиз β-глюкуронидазой лучше проводить при температуре 55°С в течение 60 мин (1 ч).
Установлены валидационные характеристики: предел обнаружения, предел количественного определения, линейный диапазон, повторяемость, внутрилабораторная прецизионность, правильность, матричный эффект, селективность.
Предел обнаружения и предел количественного определения составили 0,02 и 0,1 нг/см3 соответственно. Линейный диапазон установили по 6 модельным образцам мочи с разными концентрациями 1-гидроксипирена (0.1, 2, 10, 20, 40, 100 нг/см3), коэффициент корреляции r>0.999. Приемлемый критерий для r не ниже 0,990. Таким образом, линейный диапазон составил от 0,1 до 100 нг/см3. Прецизионность и точность оценивали для 4 образцов мочи с веденными концентрациями 0,5; 2; 20 и 40 нг/см3, каждый образец анализировали 2 раза 5 дней. По результатам анализа рассчитали относительное стандартное отклонение (RSD, %) повторяемости и внутрилабораторной прецизионности, точность как отношение значения найденной концентрации к значению веденной концентрации (табл. 6).
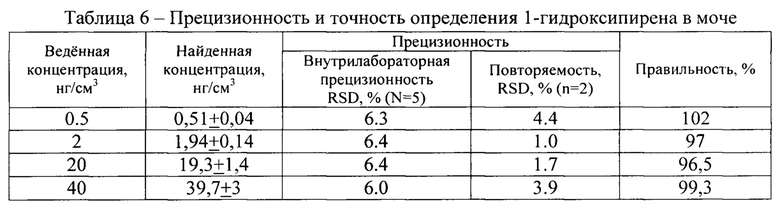
Все значения правильности, лежащие в диапазоне от 96,5 до 102% от номинальной концентрации анализируемого вещества, соответствовали приемлемому критерию (100±7%). Повторяемость (RSD, %) лежит в интервале 1.0-4.4% и внутрилабораторная прецизионность (RSD, %) 6.0-6.4%, также в соответствии с приемлемыми критериями (не выше 12%). Введенные концентрации перекрываются с доверительными интервалами найденных концентраций.
Матричный эффект оценивали в виде относительного смещения ВМЕ, сравнивая относительные сигналы аналита введенного в органический растворитель (As) определенного количества и того же количества веденного в образец мочи (Am) (табл. 10). Расчет осуществляли по следующей формуле (2)


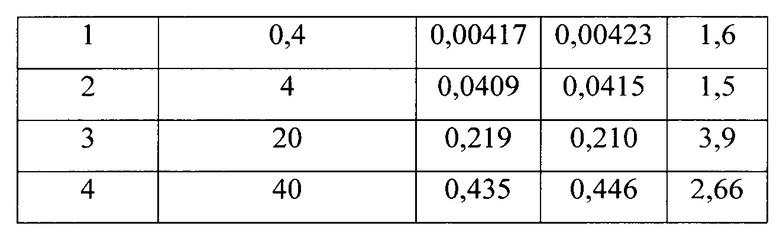
Максимальный матричный эффект составляет 3,9% и не превышает приемлемый критерий 6%.
На рисунке (фиг. 3) приведены масс-хроматограммы холостого образца и 3-х образцов с разными концентрациями 1-гидроксипирена (0.1, 0.5, 39 нг/см3).
Пик ТМС-1-гидроксипирена узкий (полуширина пика 1.5 с), симметричный, мешающие пики отсутствуют. Таким образом, достигается отличная селективность определения. Значение RSD измерения времен удерживания составляет 0,027%.
Данный способ количественного определения 1-гидроксипирена был апробирован на образцах мочи работников (31 человек) производства алюминия. Интервал содержания составил от 0,31 до 263 нг/см3. Также были проанализированы и образцы мочи работников других производств (14 человек), где в производственной воздушной среде отсутствуют ПАУ. Здесь с концентрации лежат от 0,08 до 0,9 нг/см3.
Таким образом, предлагаемым способом с удовлетворительным пределом количественного определения 0.1 нг/см3 можно определять 1-гидроксипирен в моче при более малом объеме анализируемой пробы 2 см3 не только у работников производств алюминия, но и у лиц, не занятых в данном производстве. Применение данного способа позволяет уменьшить продолжительность анализа за счет уменьшения продолжительности ее следующих стадий: ферментативного гидролиза, дериватизации 1-гидроксипирена реагентом БСТФА, ГХ-МС анализа по сравнению с прототипом. Также применение способа позволяет повысить точность определения за счет использования изотопно-меченного стандарта 1-гидроксипирена-d19, повысить чувствительность определения за счет увеличения степени экстракции из биологической матрицы.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения гидроксилированных полициклических ароматических углеводородов в моче | 2023 |
|
RU2814310C1 |
| Способ определения ароматических микробных метаболитов в форме фенилкарбоновых кислот в сыворотке крови | 2017 |
|
RU2663571C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЪЮГИРОВАННЫХ КСЕНОБИОТИКОВ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2011 |
|
RU2451936C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СТЕРОИДНОГО ПРОФИЛЯ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2011 |
|
RU2467331C1 |
| Способ подготовки пробы мочи для определения монометилфталата, моноэтилфталата, монобутилфталата, монобензилфталата, моноэтилгексилфталата методом высокоэффективной жидкостной хроматографии/масс-спектрометрии | 2019 |
|
RU2687738C1 |
| СПОСОБ ВЫЯВЛЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ ПАЦИЕНТОВ, ПРИНИМАВШИХ НАРКОТИЧЕСКИЕ ИЛИ ПСИХОАКТИВНЫЕ ВЕЩЕСТВА | 2009 |
|
RU2419788C2 |
| Способ количественного определения глифосата и N-(фосфонометил)-иминодиуксусной кислоты | 2020 |
|
RU2753453C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ПИПЕРАЗИНА В БИОЛОГИЧЕСКИХ ТКАНЯХ И ОБЪЕКТАХ ЖИВОТНОГО ПРОИСХОЖДЕНИЯ | 2024 |
|
RU2837303C1 |
| Способ определения производных стероидных гормонов в моче | 2021 |
|
RU2764363C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИМЕТИЛТЕРЕФТАЛАТА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2010 |
|
RU2425380C1 |
Изобретение относится к области медицины, в частности к медицинским, токсикологическим исследованиям, и может быть использовано при диагностике экологически обусловленной патологии, вызванной полициклическими ароматическим углеводородами (ПАУ), в лабораториях биохимии, специализированных учреждениях и клинико-диагностических лабораториях медицинских учреждений. Способ определения 1-гидроксипирена в моче включает добавление к пробе изотопно-меченного стандарта 1-гидроксипирена-d9, ферментативный гидролиз β-глюкуронидазой при 55°С в течение 1 ч, двукратную экстракцию гексаном, упаривание полученного экстракта досуха в небольшом токе азота, дериватизацию сухого остатка силилирующим N,O-бис-(триметилсилил)трифторацетамидом при 22-25°С в течение 5 минут с последующим газохроматографическим анализом на капиллярной колонке с масс-селективным детектором в течение 20 минут. Техническим результатом является повышение точности и чувствительности анализа, сокращение времени пробоподготовки. 3 ил., 7 табл.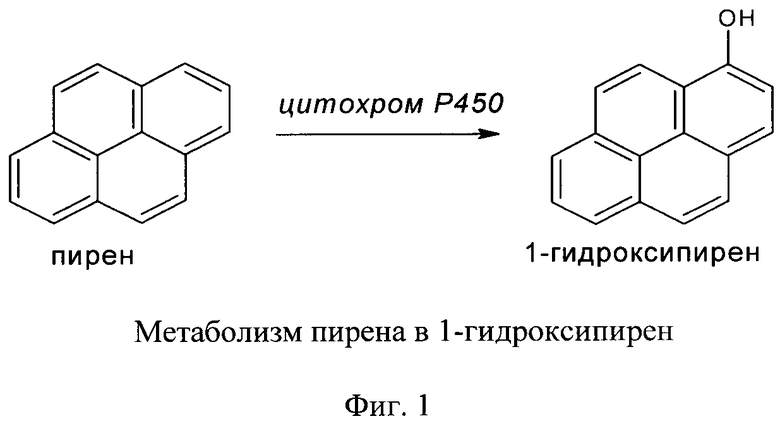
Способ определения 1-гидроксипирена в моче методом хромато-масс-спектрометрического анализа, включающий ферментативный гидролиз β-глюкуронидазой, экстракцию анализируемого вещества, упаривание экстракта в токе азота, дериватизацию сухого остатка силилирующим реагентом N,O-бис-(триметилсилил) трифторацетамид, газохроматографический анализ с масс-селективным детектированием, отличающийся тем, что в пробу добавляют изотопно-меченный стандарт 1-гидроксипирена-d9, проводят ферментативный гидролиз в течение 1 часа при 55°С, двухкратную жидкостно-жидкостную экстракцию гексаном, дериватизацию сухого остатка при 22-25°С в течение 5 минут с последующим газохроматографическим анализом на капиллярной колонке с масс-селективным детектором в течение 20 минут.
| CN 103175920 A, 26.06.2013 | |||
| Зыкова Г | |||
| В., Семёнов С | |||
| Ю., Смирнов В | |||
| Н | |||
| "Определение метаболитов ПАУ в моче человека методом высокоэффективной жидкостной хроматографии" Вестник Российского университета дружбы народов | |||
| Серия: Экология и безопасность жизнедеятельности, no | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Сепаратор-центрофуга с периодическим выпуском продуктов | 1922 |
|
SU128A1 |
| С.Schummer, О | |||
| Delhomme, B.M.R | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Спускная труба при плотине | 0 |
|
SU77A1 |
| Способ разделения смеси галоидных соединений циркония и гафния | 1924 |
|
SU1473A1 |
| Tsai P.J., Shih T.S., Chen H.L | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Technol., 2004, Vol | |||
| Способ сужения чугунных изделий | 1922 |
|
SU38A1 |
| рр | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |

