Изобретение относится к области новых способов количественного химического анализа и может быть использовано в области масс-спектрометрического анализа, в частности для контроля остаточного содержания, для клинической диагностики объектов биогенного происхождения, а также в областях науки и техники, связанных с необходимостью применения количественного определения пиперазина. Сущность способа заключается в добавлении перед началом подготовки образцов изотопно меченного пиперазина (в качестве внутренного стандарта); дальнейшем извлечение пиперазина водным раствором муравьиной кислоты, очистку твердофазной экстракцией, дериватизацией очищенного экстракта и анализом методом ВЭЖХ-МС/МС. Данный способ позволяет определять остаточное содержание пиперазина в анализируемых образцах в соответствии с требованиями, предъявляемыми метрологической оценкой к показателям точности, повторяемости и воспроизводимости.
Пиперазин - противогельминтное средство, способ действия которого заключается в параличе паразитов, применяется для лечения аскаридоза и энтеробиоза. По химической природе относится к алифатическим циклическим аминам (гексагидропиразин, диэтилендиамин), C4H10N2.
В промышленности пиперазин применяется как ингибитор коррозии, ускоритель полимеризации хлоропрена, а также в качестве сополимера для получения высокоплавких полиамидов.
Побочными эффектами потребления продуктов с остатками пиперазина являются проблемы с сердечно-сосудистой системой, с ЖКТ (тошнота, боли) и с ЦНС (головная боль, тремор, ухудшение координации). Многие страны тихоокеанского региона, а также страны южной Америки устанавливают максимально допустимые уровни (МДУ) на содержание пиперазина в различных объектах продукции животноводства, или в иных случаях, при условии отсутствия установленного значения нормы, нередко МДУ принимают по умолчанию (для малоисследованных объектов, в отсутствие научно-обоснованных данных, или когда имеют цели снижение рисков попадания продукции, потенциально содержащий пиперазин, к конечному потребителю).
В РФ зарегистрировано несколько лекарственных препаратов, содержащих пиперазина адипинат в качестве действующего вещества, например, Пиперазин таблетки, 0,5 г, №10 производитель Фармстандарт-Лексредства (Россия).
Обращение на рынке препаратов, содержащих пиперазин, а также требование стран, торговых партнеров РФ, актуализирует ситуацию разработки способа количественного определения пиперазина, удовлетворяющего требованиям метрологической оценки, в соответствии с критериями (по валидационным параметрам) общепринятыми в международной практике.
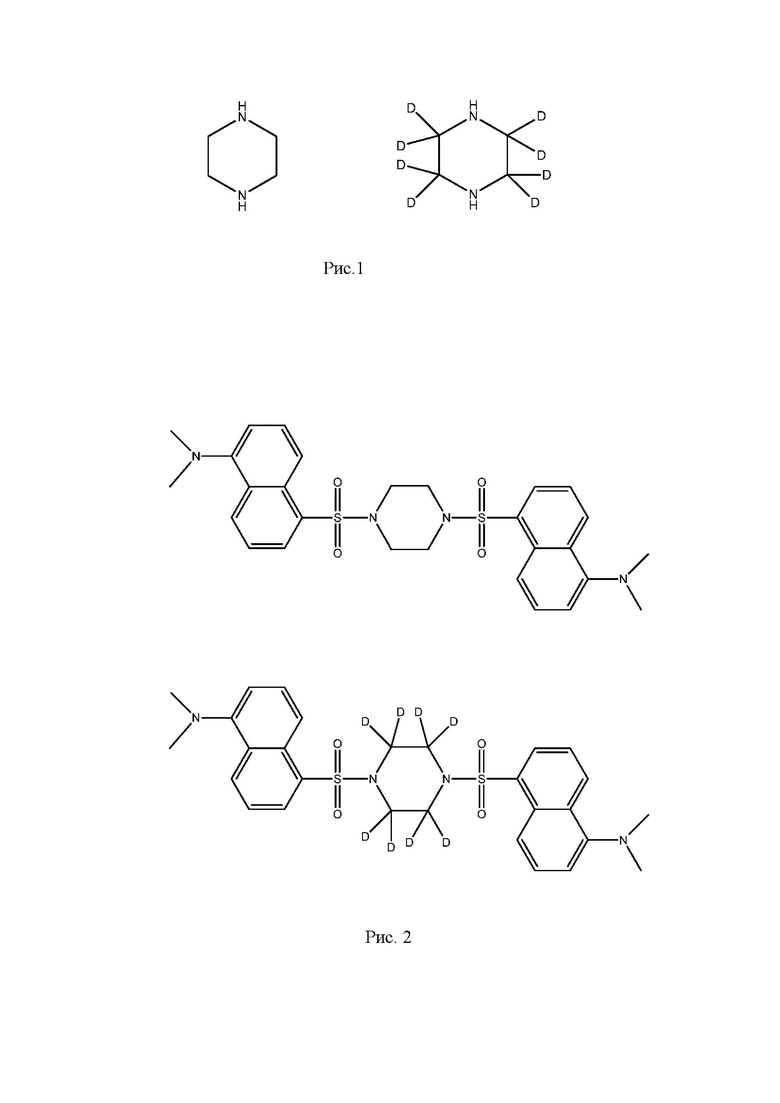
Формулы пиперазина и изотпно меченного пиперазина представлены на рис. 1.
Из уровня техники известен способ определения остаточного содержания пиперазина в вортексентине [1] - Пиперазин является одним из реагентов при синтезе указанного соединения. В предлагаемом способе пиперазин определяют в форме дериватизированных дансилхлоридом моно- и би- производных. Недостатки способа состоят в том, что при аналитической процедуре не используют внутренний стандарт, аналитическая процедура не распространена на матрицы биогенного происхождения, регистрация маркерных молекул проводится в режиме мониторинга единичных реакций (SIM).
Из уровня техники известен способ определения остаточного содержания пиперазина в плазме человека [2]. Подготовка образцов включает дериватизацию дансилхлоридом и жидкостно-жидкостную экстракцию. Анализ проводят методом ВЭЖХ МС/М. Недостатки способа состоят в том, что при аналитической процедуре используют внутренний стандарт не вступающий в реакцию с дериватизирующим агентом (дансилхлоридом), аналитическая процедура не распространена на матрицы продукции животноводства, недостаточное количество точек идентификации (один массовый переход), количественное определение проводят только для монодансилпроизводного пиперазина, т.е результаты анализа основаны на допущении неполной конверсии, вопреки высокой реакционной способности дансилхлорида и концентрации превышающей концентрацию пиперазина более чем в 100 раз.
Наиболее близким к заявляемому предложен способ определения остаточного содержания пиперазина в мышечной ткани курицы [3] - прототип 1. Подготовка образцов включала экстракцию из образов и далее очистку экстракта ТФЭ, с последующим ВЭЖХ-МС/МС анализом в условиях нормальнофазной хроматографии. Недостатки прототипа 1 состоят в том, что при аналитической процедуре не используют внутренний стандарт, аналитическая процедура распространена исключительно на матрицу мышечной ткани курицы. Преимущество данного способа - прямое определение пиперазина, но малая молекулярная масса (87,1 г/моль) пиперазина в MRM-режиме образует фрагменты (44,1/70,1), не все модели масс-спектрометров имеют рабочий диапазон, включающий регистрацию представленных масс, также отмечают, в диапазоне фрагментов малой массы можно наблюдать большое число интерферирующих пиков и высокий уровень шума. Кроме того, молекула пиперазина сильно полярна, удерживание в условиях обращенно-фазной хроматографии практически отсутствует. Хроматографическая колонка и условия нормальнофазной хроматографии, используемые авторами способа прототипа, по-видимому, позволяют решить проблему формирования хроматографического пика, удовлетворяющего условиям регистрации. Авторами, заявляемого патента, был применен аналог хроматографический колонки (другой производитель), авторам не удалось добиться удовлетворительно сформированного хроматографического пика в регистрации образцов матричных растворов, в противоположность стандартным рабочим растворам. Таким образом аппаратурное оформление прототипа 1 в целом представляет затруднение, сравнимое с преимуществом прямого определения пиперазина; не позволяет распространить способ для широкого применения.
Настоящее изобретение направлено на устранение недостатков перечисленных способов, а именно:
1. применение внутреннего стандарта, изотопно меченного пиперазина (пиперазин d-8), позволяющего адекватно проводить оценку потерь анализируемого соединения на этапах ТФЭ и дериватизации, т.е аргументировано применять количественный метод обсчета через внутренний стандарт в сочетании с общей калибровкой метода;
2. определение биогенных объектов продукции животноводства (мясо, мясо птицы, субпродукты мяса и птицы, яйца и яичные продукты, молоко и молочные продукты, рыба и продукты из рыбы);
3. требуемое количество идентификационных точек, регистрацию маркерных молекул проводят в режиме мониторинга множественных реакций (MRM) по двум массовым переходам;
4. количественное определение проводят только бисдансилпроизводного пиперазина, не допуская неполноты конверсии. Напротив, высокая реакционная способность дансилхлорида и концентрация превышающая концентрацию аналита в не менее чем 200 раз для верхнего значения определяемого диапазона утверждают конверсию близкую к 100%;
5. определение бисдансилпроизводного, во-первых, позволяет регистрировать массовые переходы с относительно высокой массой, это приводит к значительному снижению числа интерферирующих пиков, практическому отсутствию влияния фонового шума, во-вторых, позволяет использовать обращенно-фазную хроматографию, т.е решить проблему формирования хроматографического пика, использовать базовые хроматорафические колонки, находящиеся в широком применении и имеющие широкую коммерческую доступность.
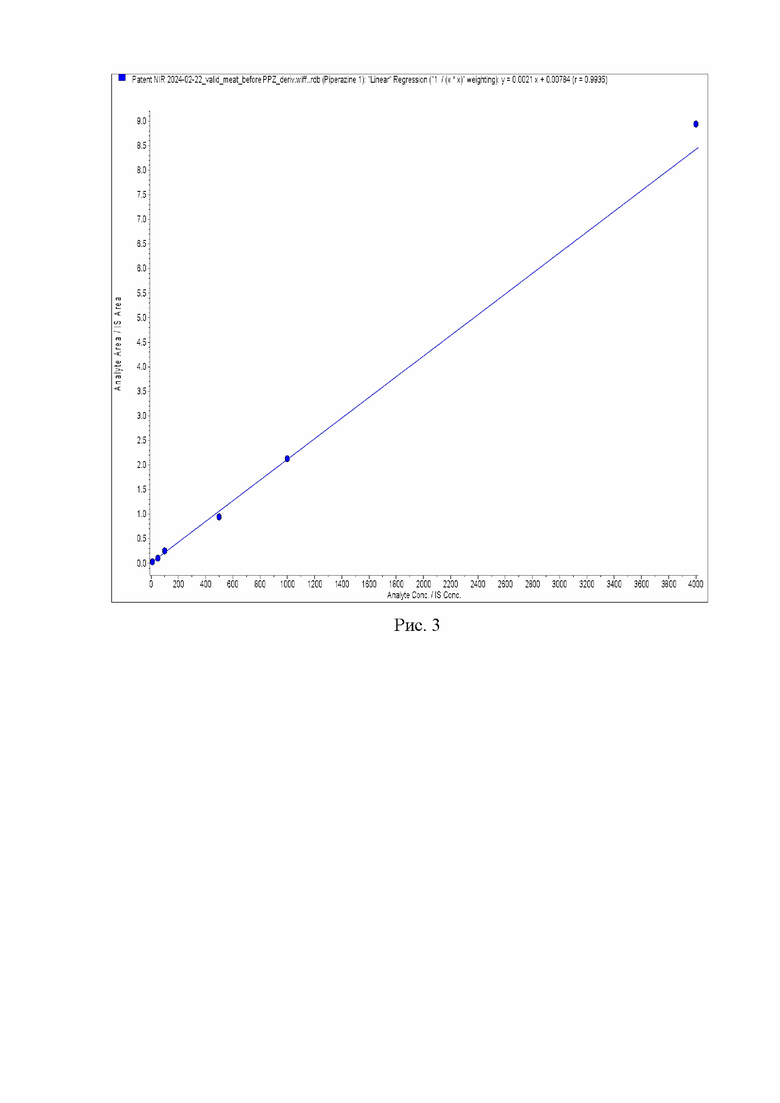
Формулы бисдансильных производных пиперазина и изотопно меченного пиперазина представлены на рис. 2.
Патентуемый способ определения остаточных количеств пиперазина в объектах биогенного происхождения включает: извлечение анализируемого соединения 2% водным раствором муравьиной кислоты, очистку экстракта твердофазной экстракцией, концентрирование очищенного экстракта, дериватизацию концентрата, экстракцию деривата этилацетатом, финальное концентрирование экстракта, растворение остатка в 1 мл деионизованной воды с 0,5% муравьиной кислоты и анализ методом ВЭЖХ-МС/МС
Технический результат выражен в разработке нового способа определения остаточных количеств пиперазина с применением изотопномеченого структурного аналога (пиперезин d-8), позволяющего компенсировать потери анализируемого соединения на этапе ТФЭ, а также компенсировать степень конверсии на этапе дериватизации. Разработанный способ позволяет проводить исследования с предписанной точностью в отношении следующих объектов: кишки и кишечное сырье, мясо и мясная продукция, молоко и молочная продукция, мясо птицы и продукция из мяса птицы, субпродукты, рыба, яйцо и яичная продукция (таблица 5).
Технический результат изобретения достигается за счет применения изотопномеченого пиперазина в качестве внутреннего стандарта, извлечения анализируемого соединения 2% водным раствором муравьиной кислоты, очистки экстракта на обращенно-фазном сорбенте, модифицированном катионобменными функциональными группами, а также концентрирования очищенного экстракта, дериватизации концентрата с дансилхлоридом до бисдансильного производного пиперазина и изотопномеченого пиперазина, экстракции бисдансильного производного пиперазина этилацетатом, финального концентрирования и растворения остатка в деионизованной воде с 0,5% муравьиной кислоты.
Авторам не известны технические решения, в которых осуществляют реализацию полного набора условий патентуемого способа. Патентуемый способ позволяет создать арбитражную методику определения остаточного содержания пиперазина в продукции животноводства методом ВЭЖХ-МС/МС в соответствии с требованиями метрологической аттестации методики.
Полный протокол способа представлен ниже.
Навеску (1,00±0,05) г. помещают в полипропиленовую пробирку вместимостью 15 см3, дозатором вносят 0,05 см3 (С=1000 нг/мл) раствора внутреннего стандарта и помещают в вортекс на 5 с для перемешивания. Добавляют 5 см3 2% раствора муравьиной кислоты и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 20 мин при температуре 4°С. Переносят надосадок экстракт в новую полипропиленовую пробирку вместимостью 15 см3, процедуру экстракции повторяют. Полученные экстракты объединяют и очищают методом твердофазной экстракции.
Картриджи для твердофазной экстракции предварительно кондиционируют, последовательно пропуская 3 см3 метанола и 3 см3 2% раствора муравьиной кислоты. Затем пропускают через картридж полученный экстракт, последовательно промывают картридж 3 см3 2% раствора муравьиной кислоты и 3 см3 метанола, просушивают. Элюируют в новую полипропиленовую пробирку вместимостью 15 см3 4 см3 5% раствора аммиака в метаноле. Упаривают элюат до 0,1-0,2 см3 в токе азота при температуре не выше 40°С. Полученный остаток дериватизируют, добавляя 1 см3 свежеприготовленного раствора дансилхлорида (С=1 мг/см3), 0,2 см3 карбонатного буферного раствора (смесь 0,1 М раствора карбоната натрия и 0,1 М раствор бикарбоната натрия в объёмном соотношении 5/4), 0,1 см3 концентрированного раствора аммиака (не менее 25%), встряхивают на вортексе и помещают в ультразвуковую баню на 1 мин. Выдерживают при температуре 50°C в течение 30 мин, предохраняя от прямого солнечного света. После дериватизации пробу оставляют на 5 мин при комнатной температуре в темном месте. Приливают 4 см3 этилацетата и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 10 мин при температуре 4°С. Переносят верхний слой в новую полипропиленовую пробирку вместимостью 15 см3, помещают её на нагревательный модуль и упаривают в токе азота при температуре не выше 50°С досуха. К полученному остатку приливают 1,0 см3 подвижной фазы А, перемешивают и помещают в ультразвуковую баню на 1 мин. Переливают экстракт в микроцентрифужную полипропиленовую пробирку вместимостью 1,5 см3 и центрифугируют при 10000 об/мин при температуре 4°С в течение 20 мин. Полученный раствор (не фильтруя) переносят дозатором в виалу для жидкостной хроматографии из тёмного стекла и используют для ВЭЖХ-МС/МС измерения.
Градуировочную характеристику определяют в каждой новой серии измерений. Для нахождения градуировочной характеристики используют не менее четырех градуировочных растворов. Для приготовления проводят обработку "чистых" проб (бланков), не содержащих действующих веществ, к которым перед обработкой добавляют раствор внутреннего стандарта и растворы анализируемого соединения в количестве, соответствующем уровню градуировки. Приготовление матричных градуировочных растворов представлено в таблице 2.
Градуировочную зависимость строят в координатах «отношение площади пика определяемого вещества к площади пика внутреннего стандарта» - «концентрация определяемого вещества в градуировочном растворе к концентрации внутреннего стандарта».
При построении градуировочной зависимости используют линейную регрессию, квадрат коэффициента корреляции не менее 0,98.
Основное оборудование и реагенты: Масс-спектрометр, оснащенный источником ионизации с электрораспылением, квадрупольными анализаторами, с диапазоном измерений от 50 до 1050 атомных единиц массы (а.е.м.), и относительным среднеквадратическим отклонением выходного сигнала по площади пика не более 10%, с режимом получения и анализа фрагментных ионов (режим МС/МС); Система высокоэффективной жидкостной хроматографии; Колонка хроматографическая, обращенно-фазная С18, длиной не менее 100 мм с диаметром частиц сорбента не более 5,0 мкм; Пиперазин, доля основного вещества не менее 80%, CAS 110-85-0; Пиперазин-Д8 гидрохлорид, доля основного вещества не менее 80%, CAS 849482219.
Рекомендуемые параметры настройки жидкостного хроматографа температура колонки 40°С; скорость потока подвижной фазы 0,35 см3/мин; объём вводимой пробы 20 мм3; Фаза А 0,5% водный раствор муравьиной кислоты; Фаза Б 0,5% раствор муравьиной кислоты в метаноле.
Градиентное элюирование проводят согласно таблице 4.
Режим градиентного элюирования представлен в таблице 3.
Значения фрагментных ионов в положительном режиме MRM в условиях электрораспыления представлены в таблице 4
Метрологические характеристики методики определения представлены в таблице 5.
В отношении следующих матриц: кишки и кишечное сырье, мясо и мясная продукция, молоко и молочная продукция, мясо птицы и продукция из мяса птицы, субпродукты, рыба, яйца птицы и продукты их переработки представленный протокол поясняет, но не ограничивают настоящее изобретение.
Пример 1
Девять навесок мышечной ткани свинины (1,00±0,05) г. помещают в полипропиленовые пробирки вместимостью 15 см3, дозатором вносят 0,05 см3 (С=1000 нг/мл) раствора внутреннего стандарта и помещают в вортекс на 5 с для перемешивания. В две пробирки делают добавки, раствора пиперазина 0,1 см3 (С=100 нг/мл, приготовленного разведением раствора С=1000 нг/мл; ожидаемое значение массовой доли 10 мкг/кг), в другие шесть пробирок делают добавки, соответствующие градуировочным уровням (таблица 2), последняя пробирка остается как бланковый образец. Добавляют 5 см3 2% раствора муравьиной кислоты и помещают пробирки в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 20 мин при температуре 4°С. Переносят надосадок экстракт в новую полипропиленовую пробирку вместимостью 15 см3, процедуру экстракции повторяют. Полученные экстракты объединяют и очищают методом твердофазной экстракции.
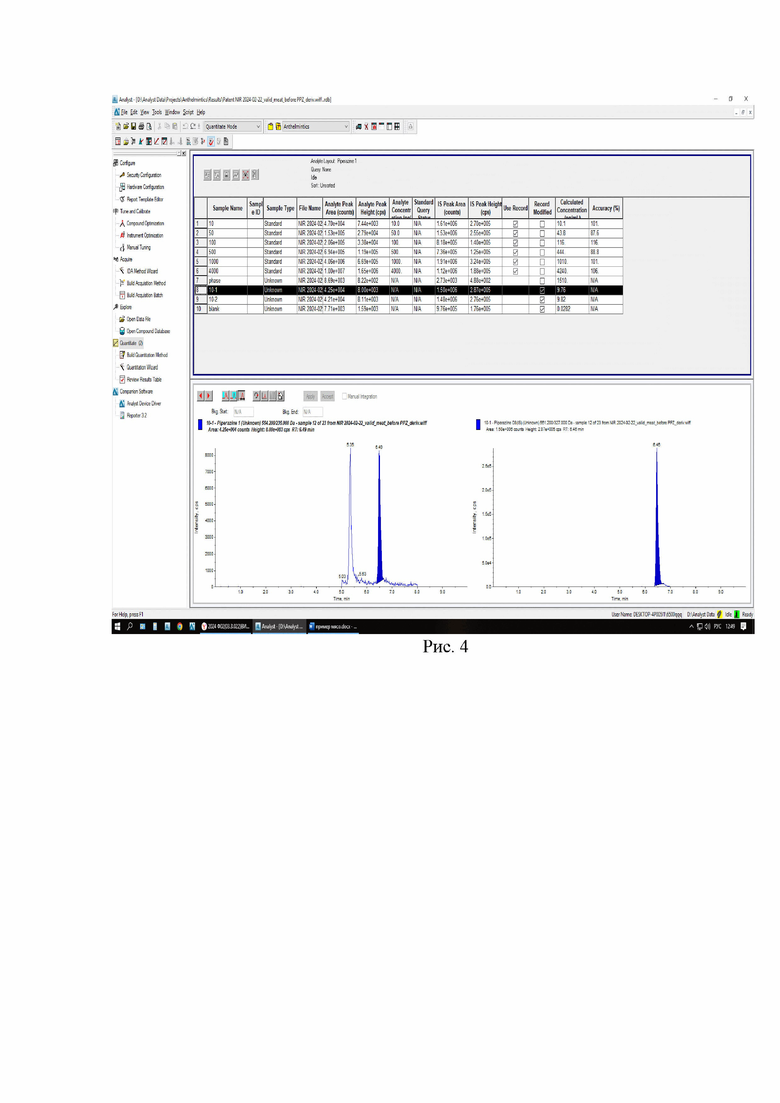
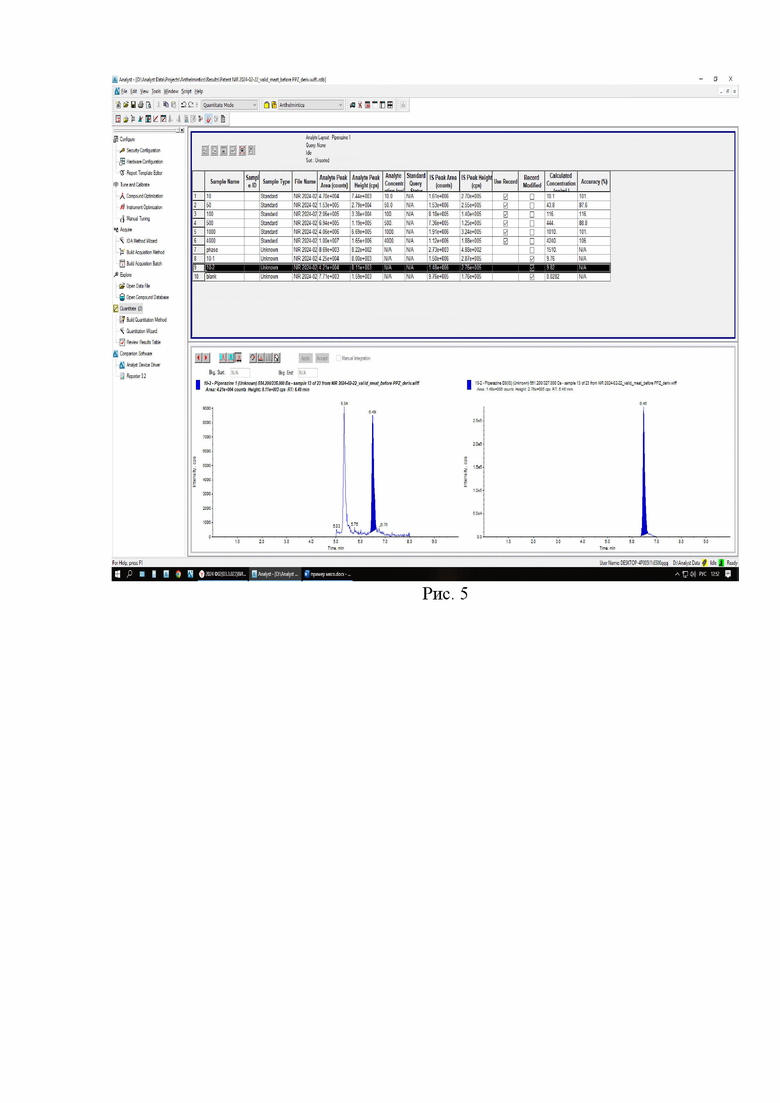
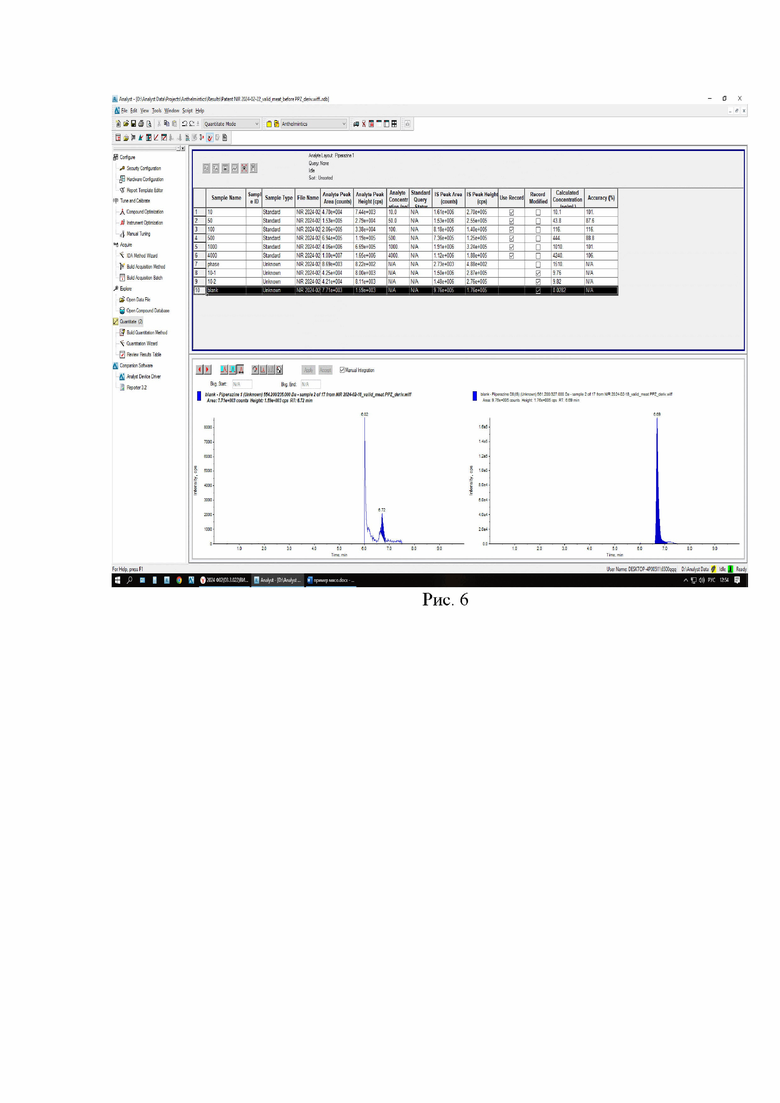
Картриджи для твердофазной экстракции предварительно кондиционируют, последовательно пропуская 3 см3 метанола и 3 см3 2% раствора муравьиной кислоты. Затем пропускают через картридж полученный экстракт, последовательно промывают картридж 3 см3 2% раствора муравьиной кислоты и 3 см3 метанола, просушивают. Элюируют в новую полипропиленовую пробирку вместимостью 15 см3 4 см3 5% раствора аммиака в метаноле. Упаривают элюат до 0,1-0,2 см3 в токе азота при температуре не выше 40°С. Полученный остаток дериватизируют, добавляя 1 см3 свежеприготовленного раствора дансилхлорида (С=1 мг/см3), 0,2 см3 карбонатного буферного раствора (смесь 0,1 М раствора карбоната натрия и 0,1 М раствор бикарбоната натрия в объёмном соотношении 5/4), 0,1 см3 концентрированного раствора аммиака (не менее 25%), встряхивают на вортексе и помещают в ультразвуковую баню на 1 мин. Выдерживают при температуре 50°C в течение 30 мин, предохраняя от прямого солнечного света. После дериватизации пробу оставляют на 5 мин при комнатной температуре в темном месте. Приливают 4 см3 этилацетата и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 10 мин при температуре 4°С. Переносят верхний слой в новую полипропиленовую пробирку вместимостью 15 см3, помещают её на нагревательный модуль и упаривают в токе азота при температуре не выше 50°С досуха. К полученному остатку приливают 1,0 см3 подвижной фазы А, перемешивают и помещают в ультразвуковую баню на 1 мин. Переливают экстракт в микроцентрифужную полипропиленовую пробирку вместимостью 1,5 см3 и центрифугируют при 10000 об/мин при температуре 4°С в течение 20 мин. Полученный раствор (не фильтруя) переносят дозатором в виалу для жидкостной хроматографии из тёмного стекла и используют для ВЭЖХ-МС/МС измерения. Градуировочная прямая и масс-хроматограммы образцов с добавкой и образца бланка представлены на рис 3-6
Пример 2
Девять навесок говяжьей печени (1,00±0,05) г. помещают в полипропиленовые пробирки вместимостью 15 см3, дозатором вносят 0,05 см3 (С=1000 нг/мл) раствора внутреннего стандарта и помещают в вортекс на 5 с для перемешивания. В две пробирки делают добавки, раствора пиперазина 0,05 см3 (С=1000 нг/мл; ожидаемое значение массовой доли 50 мкг/кг), в другие шесть пробирок делают добавки, соответствующие градуировочным уровням (таблица 2), последняя пробирка остается как бланковый образец. Добавляют 5 см3 2% раствора муравьиной кислоты и помещают пробирки в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 20 мин при температуре 4°С. Переносят надосадок экстракт в новую полипропиленовую пробирку вместимостью 15 см3, процедуру экстракции повторяют. Полученные экстракты объединяют и очищают методом твердофазной экстракции.
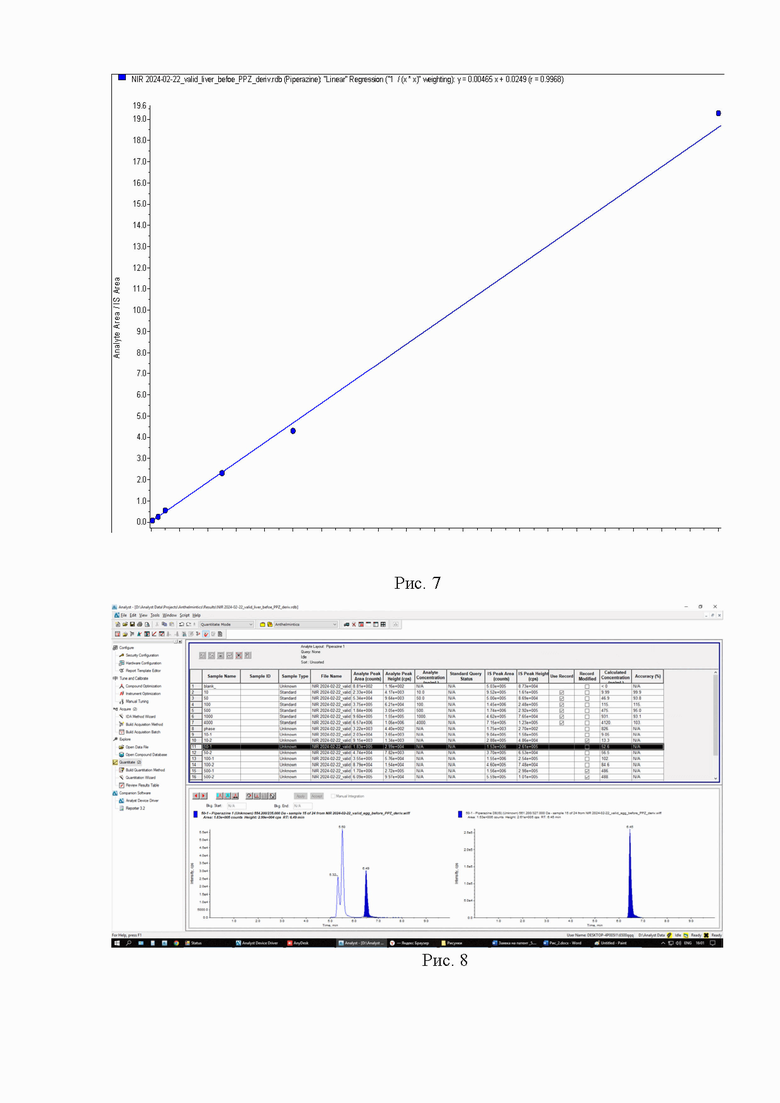
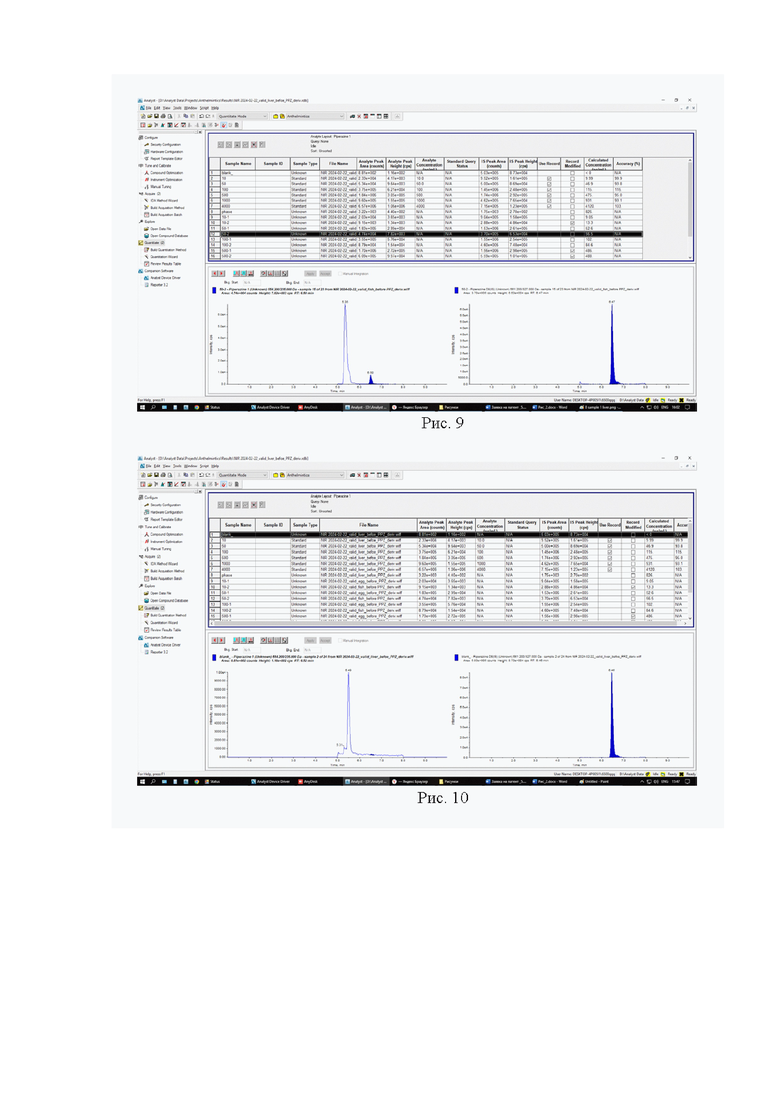
Картриджи для твердофазной экстракции предварительно кондиционируют, последовательно пропуская 3 см3 метанола и 3 см3 2% раствора муравьиной кислоты. Затем пропускают через картридж полученный экстракт, последовательно промывают картридж 3 см3 2% раствора муравьиной кислоты и 3 см3 метанола, просушивают. Элюируют в новую полипропиленовую пробирку вместимостью 15 см3 4 см3 5% раствора аммиака в метаноле. Упаривают элюат до 0,1-0,2 см3 в токе азота при температуре не выше 40°С. Полученный остаток дериватизируют, добавляя 1 см3 свежеприготовленного раствора дансилхлорида (С=1 мг/см3), 0,2 см3 карбонатного буферного раствора (смесь 0,1 М раствора карбоната натрия и 0,1 М раствор бикарбоната натрия в объёмном соотношении 5/4), 0,1 см3 концентрированного раствора аммиака (не менее 25%), встряхивают на вортексе и помещают в ультразвуковую баню на 1 мин. Выдерживают при температуре 50°C в течение 30 мин, предохраняя от прямого солнечного света. После дериватизации пробу оставляют на 5 мин при комнатной температуре в темном месте. Приливают 4 см3 этилацетата и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 10 мин при температуре 4°С. Переносят верхний слой в новую полипропиленовую пробирку вместимостью 15 см3, помещают её на нагревательный модуль и упаривают в токе азота при температуре не выше 50°С досуха. К полученному остатку приливают 1,0 см3 подвижной фазы А, перемешивают и помещают в ультразвуковую баню на 1 мин. Переливают экстракт в микроцентрифужную полипропиленовую пробирку вместимостью 1,5 см3 и центрифугируют при 10000 об/мин при температуре 4°С в течение 20 мин. Полученный раствор (не фильтруя) переносят дозатором в виалу для жидкостной хроматографии из тёмного стекла и используют для ВЭЖХ-МС/МС измерения. Градуировочная прямая и масс-хроматограммы образцов с добавкой и образца бланка представлены на рис 7-10.
Пример 3
Девять навесок яиц птицы (1,00±0,05) г. помещают в полипропиленовые пробирки вместимостью 15 см3, дозатором вносят 0,05 см3 (С=1000 нг/мл) раствора внутреннего стандарта и помещают в вортекс на 5 с для перемешивания. В две пробирки делают добавки, раствора пиперазина 0,1 см3 (С=1000 нг/мл; ожидаемое значение массовой доли 100 мкг/кг), в другие шесть пробирок делают добавки, соответствующие градуировочным уровням (таблица 2), последняя пробирка остается как бланковый образец. Добавляют 5 см3 2% раствора муравьиной кислоты и помещают пробирки в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 20 мин при температуре 4°С. Переносят надосадок экстракт в новую полипропиленовую пробирку вместимостью 15 см3, процедуру экстракции повторяют. Полученные экстракты объединяют и очищают методом твердофазной экстракции.
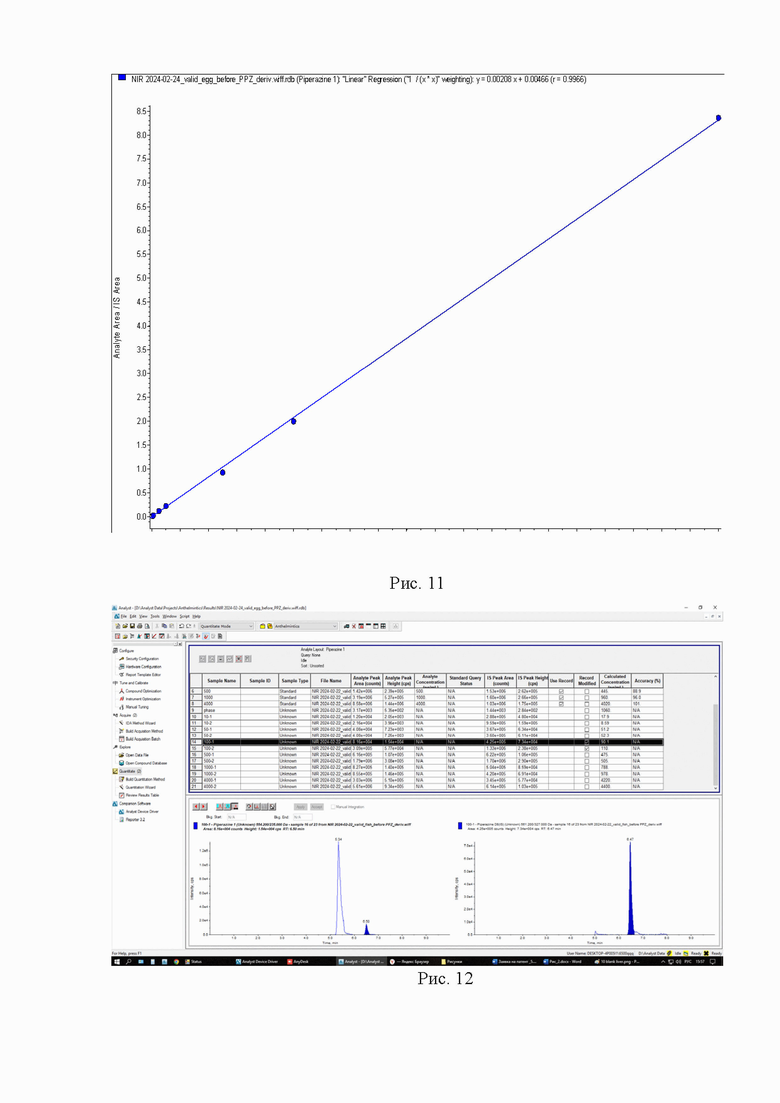
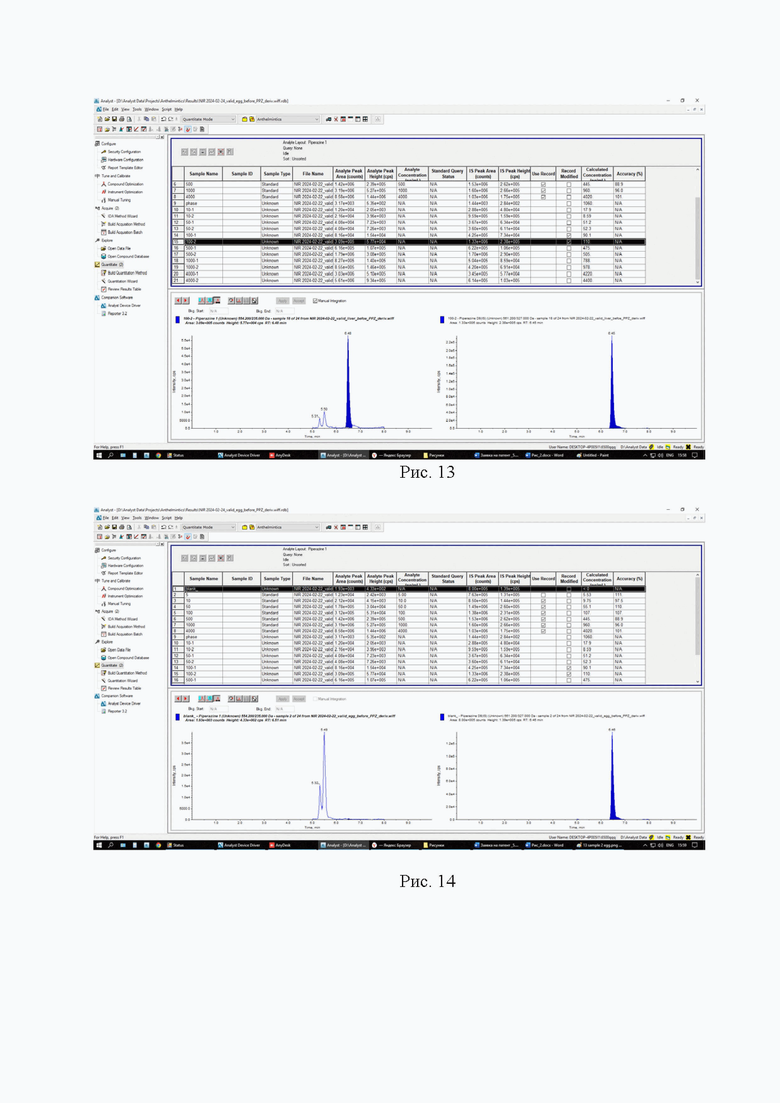
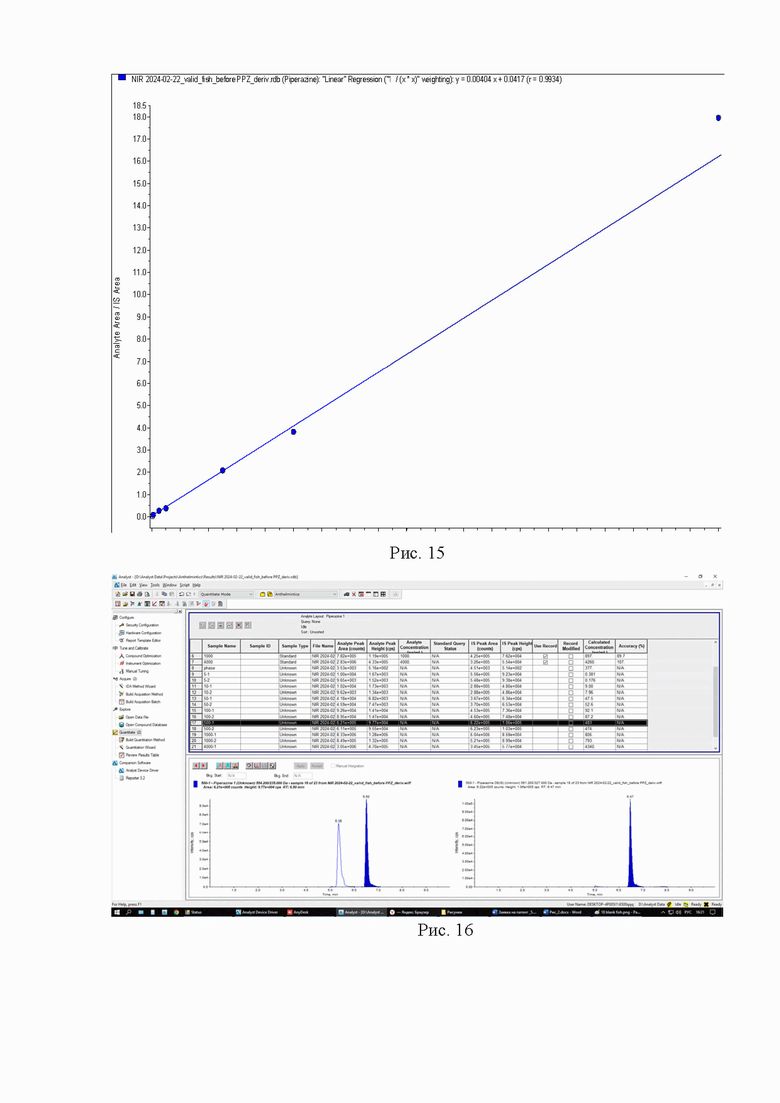
Картриджи для твердофазной экстракции предварительно кондиционируют, последовательно пропуская 3 см3 метанола и 3 см3 2% раствора муравьиной кислоты. Затем пропускают через картридж полученный экстракт, последовательно промывают картридж 3 см3 2% раствора муравьиной кислоты и 3 см3 метанола, просушивают. Элюируют в новую полипропиленовую пробирку вместимостью 15 см3 4 см3 5% раствора аммиака в метаноле. Упаривают элюат до 0,1-0,2 см3 в токе азота при температуре не выше 40°С. Полученный остаток дериватизируют, добавляя 1 см3 свежеприготовленного раствора дансилхлорида (С=1 мг/см3), 0,2 см3 карбонатного буферного раствора (смесь 0,1 М раствора карбоната натрия и 0,1 М раствор бикарбоната натрия в объёмном соотношении 5/4), 0,1 см3 концентрированного раствора аммиака (не менее 25%), встряхивают на вортексе и помещают в ультразвуковую баню на 1 мин. Выдерживают при температуре 50°C в течение 30 мин, предохраняя от прямого солнечного света. После дериватизации пробу оставляют на 5 мин при комнатной температуре в темном месте. Приливают 4 см3 этилацетата и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 10 мин при температуре 4°С. Переносят верхний слой в новую полипропиленовую пробирку вместимостью 15 см3, помещают её на нагревательный модуль и упаривают в токе азота при температуре не выше 50°С досуха. К полученному остатку приливают 1,0 см3 подвижной фазы А, перемешивают и помещают в ультразвуковую баню на 1 мин. Переливают экстракт в микроцентрифужную полипропиленовую пробирку вместимостью 1,5 см3 и центрифугируют при 10000 об/мин при температуре 4°С в течение 20 мин. Полученный раствор (не фильтруя) переносят дозатором в виалу для жидкостной хроматографии из тёмного стекла и используют для ВЭЖХ-МС/МС измерения. Градуировочная прямая и масс-хроматограммы образцов с добавкой и образца бланка представлены на рис. 11-14.
Пример 4
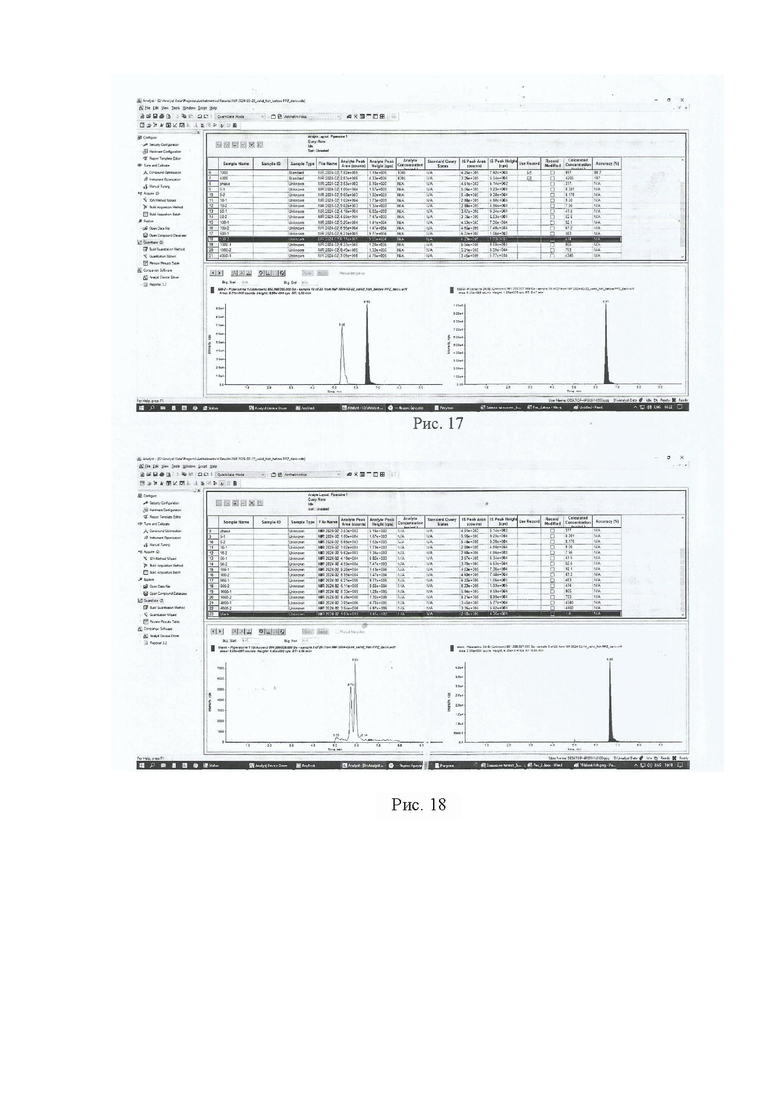
Девять навесок тканей рыбы (1,00±0,05) г. помещают в полипропиленовые пробирки вместимостью 15 см3, дозатором вносят 0,05 см3 (С=1000 нг/мл) раствора внутреннего стандарта и помещают в вортекс на 5 с для перемешивания. В две пробирки делают добавки, раствора пиперазина 0,05 см3 (С=10000 нг/мл; ожидаемое значение массовой доли 500 мкг/кг), в другие шесть пробирок делают добавки, соответствующие градуировочным уровням (таблица 2), последняя пробирка остается как бланковый образец. Добавляют 5 см3 2% раствора муравьиной кислоты и помещают пробирки в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 20 мин при температуре 4°С. Переносят надосадок экстракт в новую полипропиленовую пробирку вместимостью 15 см3, процедуру экстракции повторяют. Полученные экстракты объединяют и очищают методом твердофазной экстракции.
Картриджи для твердофазной экстракции предварительно кондиционируют, последовательно пропуская 3 см3 метанола и 3 см3 2% раствора муравьиной кислоты. Затем пропускают через картридж полученный экстракт, последовательно промывают картридж 3 см3 2% раствора муравьиной кислоты и 3 см3 метанола, просушивают. Элюируют в новую полипропиленовую пробирку вместимостью 15 см3 4 см3 5% раствора аммиака в метаноле. Упаривают элюат до 0,1-0,2 см3 в токе азота при температуре не выше 40°С. Полученный остаток дериватизируют, добавляя 1 см3 свежеприготовленного раствора дансилхлорида (С=1 мг/см3), 0,2 см3 карбонатного буферного раствора (смесь 0,1 М раствора карбоната натрия и 0,1 М раствор бикарбоната натрия в объёмном соотношении 5/4), 0,1 см3 концентрированного раствора аммиака (не менее 25%), встряхивают на вортексе и помещают в ультразвуковую баню на 1 мин. Выдерживают при температуре 50°C в течение 30 мин, предохраняя от прямого солнечного света. После дериватизации пробу оставляют на 5 мин при комнатной температуре в темном месте. Приливают 4 см3 этилацетата и помещают пробирку в шейкер на 10 мин для экстракции, затем центрифугируют при 4000 об/мин в течение 10 мин при температуре 4°С. Переносят верхний слой в новую полипропиленовую пробирку вместимостью 15 см3, помещают её на нагревательный модуль и упаривают в токе азота при температуре не выше 50°С досуха. К полученному остатку приливают 1,0 см3 подвижной фазы А, перемешивают и помещают в ультразвуковую баню на 1 мин. Переливают экстракт в микроцентрифужную полипропиленовую пробирку вместимостью 1,5 см3 и центрифугируют при 10000 об/мин при температуре 4°С в течение 20 мин. Полученный раствор (не фильтруя) переносят дозатором в виалу для жидкостной хроматографии из тёмного стекла и используют для ВЭЖХ-МС/МС измерения. Градуировочная прямая и масс-хроматограммы образцов с добавкой и образца бланка представлены на рис 15-18.
Литературные источники:
1. Shubo Dong, Zhengyu Yan, Hanyue Yang. «A Sensitive Precolumn Derivatization Method for Determination of Piperazine in Vortioxetine Hydrobromide Using a C8 Column and High-Performance Liquid Chromatography-Mass Spectrometry» //Analytical Sciences December 2016, V. 32, pp. 1333-1338
2. Hui Lina, Yuan Tiana, Zunjian Zhanga, Lili Wuc, Yun Chenc. «Quantification of piperazine phosphate in human plasma by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry employing precolumn derivatization with dansyl chloride» // Analytica Chimica Acta 664 (2010) 40-48 //doi:10.1016/j.aca.2010.02.003
3. Kaizhou Xie, Ya'nan Liu, Lirui Sun, Maoda Pang, Xing Xie, Qiang Gao, Bo Wang, Yangyang Zhang, Ran Wang, Genxi Zhang, Guojun Dai1, Jinyu Wang. «Quantification of Piperazine in Chicken Muscle by Ultra-Performance Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry» // Food Anal. Methods // DOI 10.1007/s12161-016-0717-x.
Таблица 1
Таблица 2 - Приготовление матричных градуировочных растворов G1-G6
10000 нг/мл
1000 нг/мл
100 нг/мл
1000 нг/мл
Таблица 3 - Режим градиентного элюирования
Таблица 4 - Значения фрагментных ионов в положительном режиме MRM в условиях электрораспыления
Таблица 5 - Показатели точности методики (значения относительной погрешности (расширенной неопределенности) результатов измерений, относительное среднеквадратическое отклонение повторяемости и внутрилабораторной прецезионности).
n=2)
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ОСТАТОЧНОГО СОДЕРЖАНИЯ СТЕРОИДНЫХ ГОРМОНОВ В РЫБЕ | 2021 |
|
RU2776013C1 |
| Способ определения массовых концентраций фенола и пирокатехина в крови методом высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786509C1 |
| Способ количественного определения окадаиковой кислоты в морепродуктах | 2017 |
|
RU2666247C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙЕССОТОКСИНОВ В МОЛЛЮСКАХ | 2018 |
|
RU2716233C1 |
| Способ подготовки пробы мочи для определения монометилфталата, моноэтилфталата, монобутилфталата, монобензилфталата, моноэтилгексилфталата методом высокоэффективной жидкостной хроматографии/масс-спектрометрии | 2019 |
|
RU2687738C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЛИФОСАТА, ЕГО МЕТАБОЛИТА И ГЛЮФОСИНАТА В ПРОДУКЦИИ ЖИВОТНОВОДСТВА | 2021 |
|
RU2783283C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИМЕТИЛТЕРЕФТАЛАТА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2010 |
|
RU2425380C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ПРОФИЛИРОВАНИЯ СТЕРОИДОВ В СЛЮНЕ ЧЕЛОВЕКА | 2024 |
|
RU2833902C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| Способ определения 1-гидроксипирена в моче методом хромато-масс-спектрометрического анализа | 2018 |
|
RU2687887C1 |
Изобретение относится к области аналитической химии, а именно к новому способу определения остаточного содержания пиперазина в биологических тканях животного происхождения. Способ определения остаточных количеств пиперазина включает применение изотопно меченного пиперазина в качестве внутреннего стандарта, извлечение анализируемого соединения 2%-ным водным раствором муравьиной кислоты, очистку экстракта на обращенно-фазном сорбенте, модифицированном катионобменными функциональными группами, концентрирование очищенного экстракта, дериватизацию концентрата с дансилхлоридом до бисдансильного производного пиперазина и изотопно меченного пиперазина, экстракцию бисдансильного производного пиперазина этилацетатом, финальное концентрирование, растворение остатка в деионизованной воде с 0,5 % муравьиной кислоты и анализ методом ВЭЖХ-МС/МС с использованием обращенно-фазной хроматографии и регистрации по двум массовым переходам в режиме множественных реакций. Техническим результатом является разработка нового способа определения остаточных количеств пиперазина в биологических тканях и объектах животного происхождения. 18 ил., 5 табл. 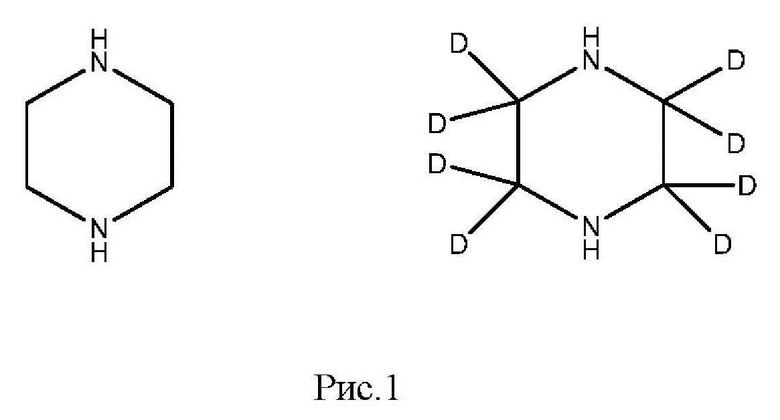
Способ определения остаточных количеств пиперазина, включающий применение изотопно меченного пиперазина в качестве внутреннего стандарта, извлечение анализируемого соединения 2%-ным водным раствором муравьиной кислоты, очистку экстракта на обращенно-фазном сорбенте, модифицированном катионообменными функциональными группами, концентрирование очищенного экстракта, дериватизацию концентрата с дансилхлоридом до бисдансильного производного пиперазина и изотопно меченного пиперазина, экстракцию бисдансильного производного пиперазина этилацетатом, финальное концентрирование, растворение остатка в деионизованной воде с 0,5 % муравьиной кислоты и анализ методом ВЭЖХ-МС/МС, с использованием обращенно-фазной хроматографии и регистрации по двум массовым переходам в режиме множественных реакций.
| KAIZHOU XIE, et al | |||
| QUANTIFICATION OF PIPERAZINE IN CHICKEN MUSCLE BY ULTRA-PERFORMANCE LIQUID CHROMATOGRAPHY-ELECTROSPRAY IONIZATION TANDEM MASS SPECTROMETRY", FOOD ANALYTICAL METHODS, v | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| HUI LINA, et al | |||
| QUANTIFICATION OF PIPERAZINE PHOSPHATE IN HUMAN PLASMA BY HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY-ELECTROSPRAY IONIZATION TANDEM MASS | |||

