Область техники, к которой относится изобретение
Настоящее изобретение относится к области гидрообработки углеводородного сырья типа газойля.
Оно, главным образом, относится к применению катализатора на основе особого оксида алюминия в способах, позволяющих осуществлять гидрообработку углеводородного сырья типа газойля. Настоящее изобретение предпочтительно относится к применению в способе гидрообработки по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C, катализатора, содержащего по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, обладающий очень высокой связностью структуры, а также предпочтительно специфическим распределением пор по сравнению с оксидом алюминия предшествующего уровня техники. Аморфный мезопористый оксид алюминия предпочтительно формуют исходя из геля оксида алюминия, обладающего высокой дисперсностью, причем собственно гель оксида алюминия получают осаждением по меньшей мере одной соли алюминия особым способом.
Более предпочтительно настоящее изобретение относится к применению в способе гидрообработки углеводородного сырья катализатора, содержащего носитель, содержащий аморфный мезопористый оксид алюминия, сформованный из геля оксида алюминия, причем гель оксида алюминия получают способом особого осаждения, позволяющим получать по меньшей мере 40 масс.% оксида алюминия в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после одной или нескольких стадий осаждения и в более общем случае после стадий получения геля оксида алюминия, начиная с первой стадии осаждения, причем количество оксида алюминия, образующегося после первой стадии осаждения, может достигать 100%.
Предшествующий уровень техники
Ужесточение норм по загрязнению окружающей среды автомобилями в 2009 году в Европейском Сообществе вынуждает специалистов по нефтепереработке очень сильно уменьшить содержание серы в дизельном топливе и бензине: не более 10 массовых частей на миллион (млн-1) серы в дизельном топливе по состоянию на 1 января 2009 года против 50 млн-1 по состоянию на 1 января 2005 года (при измерении способом ASTM D-4294). Эти ограничения трансформируются в необходимость сооружения новых установок переработки или в значительное повышение активности при равном объеме катализаторов гидрообработки. Новые ограничения обуславливают также повышенную потребность нефтеперерабатывающего предприятия в водороде, необходимом для реакций гидрообессеривания, гидродеазотирования и гидродеароматизации. С другой стороны, новые нормы сопровождаются также ограничениями в отношении качества продукции. Так, например, дизельное топливо должно иметь хорошее цетановое число. Реакции гидрообработки газойлевых фракций затрагивают также гидрирование ароматических ядер, содержащихся в газойлевых фракциях, что, как следствие, улучшает цетановое число конечной газойлевой фракции.
Улучшение эксплуатационных характеристик гидрообработки газойлевых фракций может быть частично обеспечено выбором способа, но во всех случаях использование более активной по своей сути каталитической системы очень часто является ключевым фактором. Таким образом, имеется потребность в разработке новых способов получения катализаторов гидрообработки для дальнейшего улучшения эксплуатационных характеристик этих катализаторов и удовлетворения будущим нормативам.
В общем случае полагают, что катализатор гидрообработки, обладающий сильным каталитическим потенциалом, характеризуется оптимизированной гидрирующей функцией, то есть активной фазой, которая полностью диспергирована на поверхности носителя и в которой содержание металла является высоким. В идеальном случае, независимо от природы обрабатываемого углеводородного сырья катализатор должен обеспечивать возможность доступа к активным центрам со стороны реагентов и продуктов реакции, обладая при этом большой активной поверхностью, что ведет к специфическим требованиям в отношении собственной структуры и текстуры оксидного носителя, составляющего указанные катализаторы.
Состав и применение традиционных катализаторов гидрообработки углеводородного сырья хорошо описаны в издании "J. Scherzer, A.J. Gruia, Marcel Dekker Inc. Hydrocracking Science and Technology, 1996" и в статье B.S. Clausen, H.T. Topsøe, F.E. Massoth, опубликованной в издании "Catalysis Science and Technology, 1996, volume 11, Springer-Verlag". Таким образом, эти катализаторы характеризуются в общем случае наличием активной фазы на основе по меньшей мере одного металла из группы VIB и/или по меньшей мере одного металла из группы VIII Периодической системы элементов. Наиболее традиционные композиции представляют собой композиции типа "кобальт-молибден" (CoMo), "никель-молибден" (NiMo) и "никель-вольфрам" (NiW). Эти катализаторы могут находиться в консистентной форме или в состоянии нанесенного слоя, в случае которого применяют пористые твердые вещества различной природы. В последнем случае пористый носитель, как правило, представляет собой аморфный или нерегулярно кристаллизованный оксид, такой как, например, оксид алюминия, или алюмосиликат, в случае необходимости необязательно сочетаемый с цеолитовым материалом. После получения один или несколько металлов из группы VIB и/или металлов из группы VIII, составляющих указанные катализаторы, часто находятся в форме оксидов. Поскольку активная и стабильная форма указанных катализаторов для процессов гидрообработки представляет собой сульфидированную форму, эти катализаторы необходимо подвергать обработке на стадии сульфидирования. Эта обработка может быть осуществлена на установке, связанной со способом, при этом речь идет о сульфидировании in situ, или перед загрузкой катализатора в установку, при этом речь идет о сульфидировании ex situ.
Традиционные способы, ведущие к образованию активной фазы катализаторов гидрообработки, состоят в нанесении одного или нескольких соединений-предшественников по меньшей мере одного металла из группы VIB и/или по меньшей мере одного металла из группы VIII на оксидный носитель по технологии, называемой "пропитка в сухом состоянии", с последующим осуществлением стадий созревания, сушки и в случае необходимости прокаливания, ведущих к образованию оксидной формы одного или нескольких применяемых металлов. Затем осуществляют конечную стадию сульфидирования, на которой образуется указанная ранее активная фаза.
В частности, в US 7790652 описан новый алюмооксидный носитель, который обладает весьма специфическим распределением и может быть использован в качестве носителя катализатора в способе гидроконверсии тяжелого углеводородного сырья. Указанный носитель, содержащий оксид алюминия, имеет поры со средним диаметром в интервале от 100 до 140Å, ширина интервала распределения которых по размеру составляет меньше 33Å, причем объем пор составляет по меньшей мере 0,75 мл/г, при этом меньше 5% объема пор в этом носителе составляют поры с диаметром, превышающим 210Å.
Указанный носитель, используемый в комбинации с гидрирующей активной фазой, позволяет получать неожиданные каталитические характеристики в случае, когда его применяют при гидроконверсии тяжелого углеводородного сырья, преобладающую часть которого предпочтительно составляют компоненты, кипящие при температуре выше 343°C. В частности, способ гидроконверсии тяжелого сырья согласно US 7790652 позволяет достигать конверсии углеводородных соединений, кипящих при температуре выше 524°C, которая является значительно улучшенной по сравнению с конверсией, достигаемой с традиционными катализаторами предшествующего уровня техники.
Указанный алюмооксидный носитель получают способом, включающим первую стадию образования дисперсии оксида алюминия смешиванием в контролируемом режиме первого водного щелочного раствора и первого водного кислотного раствора, причем по меньшей мере один из указанных кислотных и щелочных растворов или оба раствора содержат соединение алюминия. Кислотный и щелочной раствор смешивают в таких пропорциях, чтобы значение pH получающейся дисперсии находилось в интервале от 8 до 11. Кислотный и щелочной растворы смешивают также в количествах, позволяющих получать дисперсию, содержащую требуемое количество оксида алюминия, причем первая стадия предпочтительно позволяет получать от 25 до 35 масс.% оксида алюминия по отношению к общему количеству оксида алюминия, образующегося после двух стадий осаждения. Первую стадию осуществляют при температуре в интервале от 20 до 40°C. После образования требуемого количества оксида алюминия температуру суспензии повышают до температуры в интервале от 45 до 70°C, а затем нагретую суспензию направляют на вторую стадию осаждения приведением указанной суспензии в контакт со вторым водным щелочным раствором и вторым водным кислотным раствором, причем по меньшей мере один из двух растворов или оба раствора содержат соединение алюминия. Аналогичным образом, значение pH регулируют в интервале от 8 до 10,5 за счет соотношения прибавляемых кислотного и щелочного растворов, а остаточное количество оксида алюминия, образуемого на второй стадии, обеспечивают за счет количества прибавляемых вторых кислотных и щелочных растворов. Вторую стадию осуществляют при температуре в интервале от 20 до 40°C. Полученный таким образом гель оксида алюминия содержит по меньшей мере 95% бемита. Дисперсность геля оксида алюминия, полученного таким образом, не указана. Затем гель оксида алюминия отделяют фильтрованием, промывают и в случае необходимости сушат способами, известными специалистам в данной области техники, без осуществления предварительной стадии термической обработки, для получения порошка оксида алюминия, который далее формуют способами, известными специалистам в данной области техники, и далее прокаливают для получения конечного алюмооксидного носителя.
Первая стадия осаждения способа получения согласно US 7790652 ограничивается получением оксида алюминия в небольшом количестве, т.е. в интервале от 25 до 35 масс.%, поскольку получение оксида алюминия после первой стадии в большем количестве не позволяет осуществлять оптимальное фильтрование полученного геля. В то же время, увеличение количества оксида алюминия, получаемого на первой стадии согласно US 7790652, не позволит формовать полученный таким образом гель.
Заявителем найдено, что катализатор, содержащий по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, обладающий специфическим распределением пор, а также очень высокой связностью структуры, показал каталитическую активность, улучшенную по сравнению с традиционными катализаторами гидрообработки газойлевых фракций, причем указанный катализатор сульфидируют и затем используют в способе гидрообработки по меньшей мере газойлевой фракции согласно настоящему изобретению.
В частности, способ по настоящему изобретению обеспечивает лучшую активность при гидрообессеривании, то есть более низкую рабочую температуру, чем катализаторы предшествующего уровня техники, при заданном содержании серы в уходящем потоке.
На практике, использование катализатора по настоящему изобретению может также позволять специалистам по нефтепереработке осуществлять обессеривание большей доли газойля, поступающего из процессов конверсии ("light cycle oil" (легкого рециклового газойля) или LCO согласно терминологии на английском языке, "coker" (газойля коксования) и т.п.).
Целью настоящего изобретения является разработка способа гидрообработки по меньшей мере газойлевой фракции, в котором применяют катализатор, обладающий улучшенными каталитическими характеристиками предпочтительно в отношении гидрообессеривания и гидродеазотирования обрабатываемых фракций.
Другой целью настоящего изобретения является разработка способа гидрообработки по меньшей мере газойлевой фракции, в котором применяют катализатор, обладающий улучшенными каталитическими характеристиками, причем указанный способ обеспечивает повышенную гидрообессеривающую активность.
Краткое описание и преимущества изобретения
Целью настоящего изобретения является способ гидрообработки по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C, с применением катализатора, содержащего по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, обладающий связностью структуры (Z), значение которой превышает 2,7, причем связность структуры определяют исходя из изотерм адсорбции/десорбции азота, при этом указанный способ гидрообработки осуществляют при температуре в интервале от 250 до 400°C, при общем давлении в интервале от 2 до 10 МПа, при соотношении объема водорода и объема углеводородного сырья в интервале от 100 до 800 литров на литр и при часовой объемной скорости (VVH), определенной как отношение объемного расхода жидкого углеводородного сырья к объему катализатора, загруженного в реактор, в интервале от 1 до 10 ч-1.
Одно из преимуществ настоящего изобретения представляет собой применение в способе гидрообработки по меньшей мере газойлевой фракции катализатора на основе носителя, содержащего оксид алюминия, пористость которого характеризуется очень хорошей связностью структуры, то есть характеризуется очень большим числом пор, являющихся смежными. Повышенная связность структуры дает важное преимущество для диффузии молекул обрабатываемой газойлевой фракции при осуществлении способа гидрообработки согласно настоящему изобретению, в котором используют этот материал, и во время получения катализатора, в частности в случае пропитки растворами соединений металлов, имеющих высокие концентрации металлов.
Таким образом, преимущество настоящего изобретения состоит в разработке способа, в котором применяют катализатор, содержащий специфический носитель, обеспечивающий достижение улучшенной активности при гидрообессеривании по сравнению с катализаторами предшествующего уровня техники.
Другой целью настоящего изобретения является также способ гидрообработки по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C, причем способ осуществляют при температуре в интервале от 250 до 400°C, при общем давлении в интервале от 2 до 10 МПа, при соотношении объема водорода и объема углеводородного сырья в интервале от 100 до 800 литров на литр и при часовой объемной скорости (VVH), определенной как отношение объемного расхода жидкого углеводородного сырья к объему катализатора, загруженного в реактор, в интервале от 1 до 10 ч-1, причем в способе применяют по меньшей мере один катализатор, содержащий по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, причем указанный оксид алюминия получают, осуществляя по меньшей мере следующие стадии:
a) по меньшей мере одну первую стадию осаждения оксида алюминия в водной реакционной смеси исходя по меньшей мере из одного основного предшественника, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере из одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, причем по меньшей мере один из основных или кислотных предшественников содержит алюминий, относительный расход кислотных и основных предшественников выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5, расход одного или нескольких кислотных и основных предшественников, содержащих алюминий, регулируют с целью достижения выхода на первой стадии в интервале от 40 до 100%, при этом выход определяют как отношение количества оксида алюминия, образующегося во время указанной стадии a) осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после одной или нескольких стадий осаждения и в более общем случае после стадий получения геля оксида алюминия, причем стадию a) осуществляют при температуре в интервале от 10 до 50°C в течение промежутка времени от 2 до 30 минут;
b) стадию термической обработки суспензии, полученной после стадии a), при температуре в интервале от 50 до 200°C в течение промежутка времени от 30 минут до 5 часов, что обеспечивает получение геля оксида алюминия;
c) стадию фильтрования суспензии, полученной после стадии b) термической обработки, с последующим осуществлением по меньшей мере одной стадии промывки полученного геля;
d) стадию сушки геля оксида алюминия, полученного после стадии c), для получения порошка;
e) стадию формования порошка, полученного после стадии d), для получения сырого материала;
f) стадию термической обработки сырого материала, полученного после стадии e), при температуре в интервале от 500 до 1000°C, необязательно в токе воздуха, содержащего до 60 об.% воды.
Преимущество настоящего изобретения состоит в разработке нового способа гидрообработки, в котором применяют катализатор, алюмооксидный носитель которого получают способом особого осаждения, позволяющим формовать сильно диспергированный гель оксида алюминия, в свою очередь полученный на стадии осаждения, так что по меньшей мере 40 масс.% оксида алюминия в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образовавшегося после завершения получения геля, образуются на первой стадии осаждения. Этот способ реализуют благодаря осуществлению стадии термической обработки и, в частности, стадии созревания, обеспечивающих получение геля оксида алюминия, обладающего улучшенной фильтруемостью, и облегчающих его формование.
Определения и способы измерения
В последующем тексте индекс дисперсности понимают как массовую процентную долю пептизированного геля оксида алюминия, который можно отделить центрифугированием в полипропиленовой пробирке при 3600 об/мин в течение 10 мин.
Дисперсность определяют, отделяя 10% бемита или геля оксида алюминия в водной суспензии, содержащей также 10% азотной кислоты в расчете на массу бемита. Затем суспензию центрифугируют при 3600 об/мин в течение 10 мин. Собранные осадки сушат при 100°C в течение ночи и затем взвешивают.
Индекс дисперсности получают, рассчитывая ID по следующей формуле: ID (%)=100% - масса сухих осадков (%).
В последующем тексте связность структуры оксида алюминия, применяемого в качестве носителя катализатора, или катализатора, применяемого в способе по настоящему изобретению, понимают как число пор, являющихся смежными с отдельной порой. Связность структуры согласно настоящему изобретению определяется исходя из изотерм адсорбции/десорбции азота и характеризуется всей совокупностью пор оксида алюминия или катализатора и, в частности, всей совокупностью мезопор оксида алюминия, то есть всей совокупностью пор, имеющих средний диаметр в интервале от 2 до 50 нм.
Связность структуры представляет собой относительную величину, измеряемую согласно процедуре, описанной в публикации Ситона (Liu H., Zhang L., Seaton N.A., Chemical Engineering Science, 47, 17-18, pp. 4393-4404, 1992). Речь идет о моделировании по методу Монте-Карло, исходя из изотерм адсорбции/десорбции азота. Эти параметры связности структуры основаны на теории перколяции. Связность структуры коррелирует с числом смежных пор, при этом повышенная связность структуры дает преимущество в отношении диффузии обрабатываемых молекул во время каталитических реакций.
Оксид алюминия и катализатор, применяемые в способе по настоящему изобретению, обладают также специфическим распределением пор, в случае которого объем макропор и мезопор определяют по интрузии ртути, а объем микропор определяют по адсорбции азота.
Под "макропорами" понимают поры, размер устьев которых превышает 50 нм.
Под "мезопорами" понимают поры, размер устьев которых находится в интервале от 2 до 50 нм, включая граничные значения.
Под "микропорами" понимают поры, устье которых меньше 2 нм.
В последующем тексте описания настоящего изобретения распределение пор, определяемое ртутной порометрией, определяют по интрузии ртутным порометром по стандарту ASTM D4284-83 при максимальном давлении 4000 бар (400 МПа), при поверхностном натяжении 484 дин/см и краевом угле 140°. Угол смачивания был принят равным 140° по рекомендации из книги Jean Charpin и Bernard Rasneur "Techniques de l'ingenieur, traite analyse et caracterisation" на стр. 1050-5.
При 0,2 МПа фиксируют значение, начиная с которого ртуть находится во всех межгранульных пустотах, и считают, что затем ртуть проникает в поры образца.
Для получения лучшей точности значение общего объема пор соотносят со значением общего объема пор, определенного по интрузии ртутным порометром и измеренного в образце, за вычетом значения общего объема пор, определенного по интрузии ртутным порометром и измеренного в том же образце при давлении, соответствующем 30 пси (около 0,2 МПа).
Объем макропор катализатора определяют как суммарный объем ртути, введенной при давлении в интервале от 0,2 до 30 МПа, что соответствует объему, образованному порами с кажущимся диаметром, превышающим 50 нм.
Объем мезопор катализатора определяют как суммарный объем ртути, введенной при давлении в интервале от 30 до 400 МПа, что соответствует объему, образованному порами с кажущимся диаметром в интервале от 2 до 50 нм.
Объем микропор определяют азотной порометрией. Количественное определение микропористости осуществляют, исходя из способа "t" (способ Липпенса-Де Бура, 1965 г.), который соответствует трансформации изотермы исходной адсорбции соответственно описанию в книге F. Rouquerol, J. Rouquerol и K. Sing "Adsorption by powders and porous solids. Principles, methodology and applications. Academic Press, 1999".
Также определяют медианный диаметр мезопор (Dp, нм) как диаметр, такой что все поры, размер которых меньше этого диаметра, составляют 50% от объема мезопор, определенного ртутным порометром.
Распределение пор, определяемое по адсорбции азота, определяли по модели Барретта-Джойнера-Халенды (BJH). Изотерма адсорбции-десорбции азота согласно модели BJH описана E.P. Barrett, L.G. Joyner и P.P. Halenda в "The Journal of American Society, 73, 373, (1951)". В последующем тексте описания настоящего изобретения под объемом адсорбции азота понимают объем, определенный для P/P0=0,99, т.е. при давлении, в случае которого полагают, что азот заполняет все поры.
В последующем тексте описания настоящего изобретения под удельной поверхностью понимают удельную поверхность БЭТ, определенную по адсорбции азота соответственно стандарту ASTM D 3663-78, разработанному на основе способа БРУНАУЭРА-ЭММЕТТА-ТЕЛЛЕРА, описанного в "The Journal of American Society, 60, 309, (1938)".
В приведенном далее тексте группы химических элементов указаны согласно классификации CAS ("CRC Handbook of Chemistry and Physics", издательство "CRC press", под редакцией D.R. Lide, 81-е издание, 2000-2001 гг.) Например, группа VIII согласно классификации CAS соответствует металлам столбцов 8, 9 и 10 согласно новой классификации ИЮПАК.
Описание изобретения
Настоящее изобретение относится к способу гидрообработки и предпочтительно гидрообессеривания по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C.
TMP определяют, исходя из температуры, при которой отгоняются 5, 50 и 70% объема исходной смеси, по следующей формуле: TMP=(T5%+2T50%+4T70%)/7. TMP рассчитывают, исходя из значений, полученных при модельной перегонке. Интервал перегонки обрабатываемого углеводородного сырья в общем случае соответствует интервалу от 150 до 500°C и предпочтительно от 180 до 450°C.
В последующем тексте это сырье традиционно обозначается как газойль, но это обозначение не имеет никакого ограничительного характера. Любое углеводородное сырье, содержащее серу и азотистые соединения, ингибирующие гидрообработку, и имеющее TMP, соответствующую газойлевой фракции, может быть обработано способом, являющимся целью настоящего изобретения. Углеводородное сырье может быть любой химической природы, то есть характеризоваться любым распределением химических соединений различных групп, в частности, парафинов, олефинов, нафтенов и ароматических соединений.
Сырье
Сырье, используемое в способе гидрообработки согласно настоящему изобретению, представляет собой газойлевую фракцию, содержащую серу.
Содержание серы в сырье в общем случае составляет меньше 5 масс.%, предпочтительно от 0,2 до 4 масс.% и более предпочтительно от 0,25 до 3 масс.%.
Содержание общего азота (нейтрального и основного) в сырье превышает или равно 50 млн-1, предпочтительно оно находится в интервале от 200 до 6000 млн-1, более предпочтительно от 300 до 4000 млн-1 и наиболее предпочтительно от 400 до 4000 млн-1. Содержание основного азота составляет по меньшей мере третью часть от содержания общего азота.
Содержание основного азота в общем случае превышает или равно 10 млн-1 и составляет более предпочтительно от 65 до 2000 млн-1 и наиболее предпочтительно от 100 до 2000 млн-1.
Обрабатываемое сырье в общем случае содержит очень мало смол, в общем случае содержание смол составляет меньше 1 масс.%.
Газойлевые фракции, используемые в способе по настоящему изобретению, преимущественно выбирают из газойлевых фракций, получаемых прямой перегонкой (или "gazole straight run" (прямогонный газойль) согласно терминологии на английском языке) и используемых индивидуально или в смеси по меньшей мере с одной фракцией, получаемой на установке коксования ("coking" (коксование) согласно терминологии на английском языке), или по меньшей мере с одной фракцией, получаемой каталитическим крекингом ("Fluid Catalytic Cracking" (крекинг с псевдоожиженным катализатором) согласно терминологии на английском языке), или также по меньшей мере с одной газойлевой фракцией, получаемой другими способами конверсии, такими как мягкий гидрокрекинг или гидрообработка нефтяных остатков. Газойлевые фракции, используемые в способе по настоящему изобретению, представляют собой фракции, в которых по меньшей мере 90% соединений предпочтительно имеют температуру кипения в интервале от 250 до 400°C.
Согласно настоящему изобретению гидрообработку или гидрообессеривание по меньшей мере газойлевой фракции по настоящему изобретению осуществляют при температуре в интервале от 250 до 400°C, предпочтительно от 320 до 380°C при общем давлении в интервале от 2 до 10 МПа и предпочтительно от 3 до 9 МПа, при соотношении объема водорода и объема углеводородного сырья в интервале от 100 до 800 литров на литр и предпочтительно от 200 до 400 литров на литр и при часовой объемной скорости (VVH), определенной как отношение объемного расхода жидкого углеводородного сырья к объему катализатора, загруженного в реактор, в интервале от 1 до 10 ч-1 и предпочтительно от 2 до 8 ч-1.
Согласно настоящему изобретению катализатор, применяемый в способе гидрообработки или гидрообессеривания, содержит по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий и предпочтительно представляющий собой аморфный мезопористый оксид алюминия, обладающий связностью структуры (Z), значение которой превышает 2,7, причем связность структуры указанного оксида алюминия определяют исходя из изотерм адсорбции/десорбции азота.
Катализатор, применяемый по настоящему изобретению, имеет также связность структуры (Z), значение которой преимущественно превышает 2,7 и предпочтительно находится в интервале от 2,7 до 10, предпочтительно от 2,8 до 10, более предпочтительно от 3 до 9, еще более предпочтительно от 3 до 8 и наиболее предпочтительно от 3 до 7, причем связность структуры катализатора определяют исходя из изотерм адсорбции/десорбции азота.
Элементы из группы VIII предпочтительно выбирают из благородных и неблагородных металлов группы VIII и предпочтительно из железа, кобальта, никеля, рутения, родия, палладия, осмия, иридия или платины, применяемых индивидуально или в смеси.
В случае, когда элементы из группы VIII выбирают из неблагородных металлов группы VIII, элементы из группы VIII предпочтительно выбирают из кобальта, никеля, железа и предпочтительно из кобальта и никеля, применяемых индивидуально или в смеси.
Элементы из группы VIB предпочтительно выбирают из вольфрама и молибдена, применяемых индивидуально или в смеси. В случае, когда функциональная группа с гидрирующей способностью содержит элементы из группы VIII и элементы из группы VIB, предпочтительными являются следующие сочетания металлов: никель-молибден, кобальт-молибден, железо-молибден, железо-вольфрам, никель-вольфрам, кобальт-вольфрам, и более предпочтительными являются: никель-молибден, кобальт-молибден, никель-вольфрам. Можно также использовать сочетания трех металлов, такие как, например, никель-кобальт-молибден. Если используют комбинацию металлов из группы VI и из группы VIII, то катализатор предпочтительно применяют в сульфидированной форме.
Содержание определяемых металлов выражают в расчете на их эквивалентную оксидную форму в случае, когда определение не осуществляли по рентгеновской флуоресценции в твердом веществе, которое было прокалено, хотя катализатор необязательно может быть прокален перед сульфидированием.
В случае, когда катализатор содержит по меньшей мере один металл из группы VIB в комбинации по меньшей мере с одним неблагородным металлом из группы VIII, содержание металла из группы VIB предпочтительно находится в интервале от 10 до 35 масс.%, более предпочтительно от 15 до 30 масс.% и наиболее предпочтительно от 18 до 25 масс.% в расчете на оксид по отношению к общей массе катализатора, а содержание неблагородного металла из группы VIII предпочтительно находится в интервале от 1 до 10 масс.%, более предпочтительно от 1,5 до 8 масс.% и наиболее предпочтительно от 2 до 6 масс.% в расчете на оксид по отношению к общей массе катализатора.
Молярное соотношение одного или нескольких металлов из группы VIII и одного или нескольких металлов из группы VIB в оксидном катализаторе предпочтительно находится в интервале от 0,1:1,0 до 0,8:1,0, более предпочтительно от 0,2:1,0 до 0,6:1,0 и наиболее предпочтительно от 0,3:1,0 до 0,5:1,0.
Катализатор, применяемый в способе по настоящему изобретению, предпочтительно может содержать по меньшей мере один легирующий элемент, выбранный из фосфора, бора, фтора или кремния, применяемых индивидуально или в смеси. Легирующий элемент предпочтительно представляет собой фосфор или бор.
Если катализатор содержит фосфор, то содержание фосфора в оксидном катализаторе предпочтительно составляет от 0,5 до 15 масс.%, более предпочтительно от 1 до 10 масс.% и наиболее предпочтительно от 2 до 8 масс.% в расчете на P2O5. Содержание фосфора предпочтительно выбирают также так, чтобы молярное соотношение фосфора и молибдена находилось в интервале от 0,1:1,0 до 0,8:1,0 и более предпочтительно в интервале от 0,2:1,0 до 0,6:1,0.
Если катализатор содержит бор, то содержание бора в оксидном катализаторе предпочтительно составляет от 0,2 до 8 масс.%, более предпочтительно от 0,5 до 5 масс.% и наиболее предпочтительно от 1 до 4 масс.% в расчете на B2O3. Содержание бора предпочтительно выбирают также так, чтобы молярное соотношение бора и молибдена находилось в интервале от 0,1:1,0 до 0,8:1,0 и предпочтительно в интервале от 0,2:1,0 до 0,6:1,0.
Если катализатор содержит кремний, то содержание кремния в оксидном катализаторе предпочтительно составляет от 0,5 до 30 масс.% и более предпочтительно от 3 до 10 масс.% в расчете на SiO2.
Если катализатор содержит фтор, то содержание фтора в оксидном катализаторе предпочтительно составляет от 0,5 до 5 масс.% и более предпочтительно от 1 до 3 масс.%.
Катализатор, применяемый в способе по настоящему изобретению, предпочтительно содержит носитель, содержащий и предпочтительно представляющий собой аморфный мезопористый оксид алюминия, причем указанный оксид алюминия получают согласно настоящему изобретению.
Носитель катализатора, применяемый по настоящему изобретению, предпочтительно содержит и предпочтительно представляет собой аморфный мезопористый оксид алюминия, обладающий связностью структуры (Z), значение которой находится в интервале от 2,7 до 10, предпочтительно от 2,8 до 10, более предпочтительно от 3 до 9, еще более предпочтительно от 3 до 8 и наиболее предпочтительно от 3 до 7.
Оксид алюминия, применяемый в качестве носителя в катализаторе, применяемом согласно настоящему изобретению, представляет собой мезопористый оксид алюминия с контролируемой мезопористостью, обладающий хорошей термической и химической стабильностью, имеющий единообразное и контролируемое центральное распределение мезопор по размеру.
Оксид алюминия и носитель, содержащий указанный оксид алюминия, имеют удельную поверхность и распределение пор, нормализованные и адаптированные для применения в способе гидрообработки по меньшей мере газойлевой фракции.
Оксид алюминия, применяемый в качестве носителя в катализаторе, применяемом в способе по настоящему изобретению, и носитель катализатора предпочтительно обладают специфическим распределением пор.
Мезопористый оксид алюминия предпочтительно не имеет микропор.
Мезопористый носитель, содержащий указанный оксид алюминия, предпочтительно не имеет микропор.
Оксид алюминия и носитель, содержащий указанный оксид алюминия, предпочтительно характеризуются следующим распределением пор, определенным ртутной порометрией:
- процентная доля объема, соответствующего порам с размерами в интервале от 2 до 6 нм, составляет от 1 до 25% по отношению к общему объему пор;
- процентная доля объема, соответствующего порам с размерами, превышающими 6 нм и меньшими 15 нм, составляет от 60 до 95% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 15 до 50 нм, составляет от 0 до 8% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 50 до 7000 нм и относящегося к объему макропор, составляет от 0 до 5% и предпочтительно от 0 до 2% от общего объема пор.
Медианный диаметр мезопор (Dp, нм), определенный по объему ртутной порометрией в оксиде алюминия и в носителе, предпочтительно находится в интервале от 7 до 12,5 нм.
Общий объем пор оксида алюминия и носителя, определенный ртутной порометрией, предпочтительно находится в интервале от 0,5 до 0,85 мл/г.
Объем мезопор оксида алюминия и носителя, определенный ртутной порометрией, предпочтительно находится в интервале от 0,5 до 0,8 мл/г, предпочтительно от 0,55 до 0,75 мл/г и более предпочтительно от 0,60 до 0,75 мл/г.
Общий объем макропор оксида алюминия и носителя, определенный ртутной порометрией, предпочтительно находится в интервале от 0 до 0,04 мл/г и более предпочтительно от 0 до 0,02 мл/г.
Оксид алюминия и носитель предпочтительно имеют удельную поверхность, превышающую 180 м2/г и предпочтительно превышающую 220 м2/г.
Оксид алюминия, применяемый по настоящему изобретению, и носитель, содержащий указанный оксид алюминия, предпочтительно не являются мезоструктурными.
Оксид алюминия, используемый в качестве носителя катализатора, применяемого в способе по настоящему изобретению, предпочтительно получают способом получения, который включает по меньшей мере следующие стадии:
a) по меньшей мере одну первую стадию осаждения оксида алюминия в водной реакционной смеси исходя по меньшей мере из одного основного предшественника, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере из одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, причем по меньшей мере один из основных или кислотных предшественников содержит алюминий, относительный расход кислотных и основных предшественников выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5, расход одного или нескольких кислотных и основных предшественников, содержащих алюминий, регулируют с целью достижения выхода на первой стадии в интервале от 40 до 100%, при этом выход определяют как отношение количества оксида алюминия, образующегося во время первой стадии осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после одной или нескольких стадий осаждения, причем первую стадию осаждения осуществляют при температуре в интервале от 10 до 50°C в течение промежутка времени от 2 до 30 минут;
b) стадию термической обработки суспензии, нагретой до температуры в интервале от 50 до 200°C, в течение промежутка времени от 30 минут до 5 часов, что обеспечивает получение геля оксида алюминия;
c) стадию фильтрования суспензии, полученной после стадии b) термической обработки, с последующим осуществлением по меньшей мере одной стадии промывки полученного геля;
d) стадию сушки геля оксида алюминия, полученного после стадии c), для получения порошка;
e) стадию формования порошка, полученного после стадии d), для получения сырого материала;
f) стадию термической обработки сырого материала, полученного после стадии e), при температуре в интервале от 500 до 1000°C, необязательно в токе воздуха, содержащего до 60 об.% воды.
В общем случае под "выходом" на n-ной стадии осаждения понимают процентную долю оксида алюминия, образующегося на n-ной стадии, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после совокупности стадий осаждения и в более общем случае после стадий получения геля оксида алюминия.
В случае, когда выход на стадии a) осаждения, составляет 100%, стадия a) осаждения в общем случае позволяет получать суспензию оксида алюминия с концентрацией Al2O3 в интервале от 20 до 100 г/л, предпочтительно от 20 до 80 г/л и более предпочтительно от 20 до 50 г/л.
Стадия a) осаждения
При смешивании в водной реакционной среде по меньшей мере одного основного предшественника и по меньшей мере одного кислотного предшественника требуется, чтобы по меньшей мере один основный или кислотный предшественник содержал алюминий или чтобы оба предшественника, основный и кислотный, содержали алюминий.
Основные предшественники, содержащие алюминий, представляют собой алюминат натрия и алюминат калия. Предпочтительный основный предшественник представляет собой алюминат натрия.
Кислотные предшественники, содержащие алюминий, представляют собой сульфат алюминия, хлорид алюминия и нитрат алюминия. Предпочтительный кислотный предшественник представляет собой сульфат алюминия.
Один или несколько основных и кислотных предшественников предпочтительно прибавляют на первой стадии осаждения a) в виде водного раствора, содержащего один или несколько указанных предшественников.
Один или несколько основных и кислотных предшественников предпочтительно прибавляют на первой стадии осаждения a) в виде водных растворов.
Водная реакционная среда предпочтительно представляет собой воду.
Стадию a) предпочтительно осуществляют при перемешивании.
Стадию a) предпочтительно осуществляют в отсутствие органических добавок.
Кислотные и основные предшественники, необязательно содержащие алюминий, смешивают предпочтительно в виде раствора в водной реакционной среде в таких пропорциях, чтобы значение pH образующейся суспензии находилось в интервале от 8,5 до 10,5.
Согласно настоящему изобретению именно относительный расход кислотных и основных предшественников, необязательно содержащих алюминий, выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5.
В предпочтительном случае, в котором основный и кислотный предшественники представляют собой алюминат натрия и сульфат алюминия соответственно, массовое отношение основного предшественника к кислотному предшественнику предпочтительно находится в интервале от 1,6 до 2,05.
Для других основных и кислотных предшественников, необязательно содержащих алюминий, массовое отношение "основание/кислота" определяют по кривой нейтрализации основания кислотой. Такая кривая может быть легко получена специалистами в данной области техники.
Стадию a) осаждения предпочтительно осуществляют при значении pH в интервале от 8,5 до 10 и более предпочтительно от 8,7 до 9,9.
Кислотные и основные предшественники смешивают также в количествах, позволяющих получать суспензию, содержащую требуемое количество оксида алюминия, в зависимости от требуемой конечной концентрации оксида алюминия. В частности, стадия a) позволяет получать от 40 до 100 масс.% оксида алюминия в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после одной или нескольких или стадий осаждения и в более общем случае после стадий получения геля оксида алюминия.
Согласно настоящему изобретению именно расход одного или нескольких кислотных и основных предшественников, содержащих алюминий, регулируют с целью достижения выхода на первой стадии в интервале от 40 до 100%.
Выход на стадии осаждения a) предпочтительно находится в интервале от 40 до 99%, предпочтительно от 45 до 90% и более предпочтительно от 50 до 85%.
В случае, когда выход, достигаемый после стадии a) осаждения, составляет меньше 100%, требуется осуществлять вторую стадию осаждения с целью увеличения количества образующегося оксида алюминия. В этом случае выход определяют как отношение количества оксида алюминия, образующегося на стадии a) осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после двух стадий осаждения способа получения по настоящему изобретению и в более общем случае после стадий получения геля оксида алюминия.
Таким образом, в зависимости от концентрации оксида алюминия, предусматриваемой после одной или нескольких стадий осаждения предпочтительно в интервале от 20 до 100 г/л, рассчитывают количество алюминия, которое должно быть внесено с кислотными и/или основными предшественниками, и расход предшественников регулируют в зависимости от концентрации прибавляемых предшественников алюминия, количества воды, прибавляемой к реакционной смеси, и выхода, требующегося на одной или нескольких стадиях осаждения.
Расходы одного или нескольких кислотных и/или основных предшественников, содержащих алюминий, зависят от размера используемого реактора, а также от количества воды, прибавляемой к реакционной смеси.
Стадию a) осаждения предпочтительно осуществляют при температуре в интервале от 10 до 45°C, предпочтительно от 15 до 45°C, более предпочтительно от 20 до 45°C и наиболее предпочтительно от 20 до 40°C.
Важным обстоятельством является то, что стадию a) осаждения осуществляют при низкой температуре. В случае, когда способ получения по настоящему изобретению включает две стадии осаждения, стадию a) осаждения предпочтительно осуществляют при температуре ниже температуры на второй стадии осаждения.
Стадию a) осаждения предпочтительно осуществляют в течение промежутка времени от 5 до 20 минут и предпочтительно от 5 до 15 минут.
Стадия b) термической обработки
Согласно настоящему изобретению способ получения включает стадию b) термической обработки суспензии, полученной после стадии a) осаждения, причем указанную стадию термической обработки осуществляют при температуре в интервале от 60 до 200°C в течение промежутка времени от 30 минут до 5 часов для получения геля оксида алюминия.
Стадия b) термической обработки предпочтительно представляет собой стадию созревания.
Стадию b) термической обработки предпочтительно осуществляют при температуре в интервале от 65 до 150°C, предпочтительно от 65 до 130°C, более предпочтительно от 70 до 110°C и наиболее предпочтительно от 70 до 95°C.
Стадию b) термической обработки предпочтительно осуществляют в течение промежутка времени от 40 минут до 5 часов, предпочтительно от 40 минут до 3 часов и более предпочтительно от 45 минут до 2 часов.
Вторая стадия осаждения, осуществляемая в случае необходимости
Согласно предпочтительному варианту осуществления в случае, когда выход, достигнутый после стадии a) осаждения, составляет меньше 100%, способ получения после первой стадии осаждения предпочтительно включает вторую стадию осаждения а').
Вторая стадия осаждения позволяет увеличить содержание полученного оксида алюминия.
Вторую стадию осаждения а') предпочтительно осуществляют между первой стадией осаждения a) и стадией b) термической обработки.
В случае, когда осуществляют вторую стадию осаждения, стадию нагревания суспензии, полученной после стадии осаждения a), предпочтительно осуществляют между двумя стадиями осаждения a) и а').
Стадию нагревания полученной после стадии a) суспензии, осуществляемую между стадией a) и второй стадией а') осаждения, предпочтительно осуществляют при температуре в интервале от 20 до 90°C, предпочтительно от 30 до 80°C, более предпочтительно от 30 до 70°C и наиболее предпочтительно от 40 до 65°C.
Стадию нагревания предпочтительно осуществляют в течение промежутка времени от 7 до 45 минут и более предпочтительно от 7 до 35 минут.
Стадию нагревания предпочтительно осуществляют любыми способами нагревания, известными специалистам в данной области техники.
Согласно предпочтительному варианту осуществления способ получения включает вторую стадию осаждения суспензии, полученной после стадии нагревания, причем вторую стадию осуществляют прибавлением к указанной суспензии по меньшей мере одного основного предшественника, выбранный из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, причем по меньшей мере один из основных или кислотных предшественников содержит алюминий, относительный расход кислотных и основных предшественников выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5, а расход одного или нескольких основных и кислотных предшественников, содержащих алюминий, регулируют с целью достижения выхода на второй стадии в интервале от 0 до 60%, при этом выход определяют как отношение количества оксида алюминия, образующегося на второй стадии осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после двух стадий осаждения и в более общем случае после стадий получения геля оксида алюминия и предпочтительно после стадии а') способа получения по настоящему изобретению, причем указанную стадию осуществляют при температуре в интервале от 40 до 90°C и в течение промежутка времени от 2 до 50 минут.
Как и на первой стадии осаждения a), при прибавлении к нагретой суспензии по меньшей мере одного основного предшественника и по меньшей мере одного кислотного предшественника требуется, чтобы по меньшей мере один основный или кислотный предшественник содержал алюминий или чтобы оба предшественника, основный и кислотный, содержали алюминий.
Основные предшественники, содержащие алюминий, представляют собой алюминат натрия и алюминат калия. Предпочтительный основный предшественник представляет собой алюминат натрия.
Кислотные предшественники, содержащие алюминий, представляют собой сульфат алюминия, хлорид алюминия и нитрат алюминия. Предпочтительный кислотный предшественник представляет собой сульфат алюминия.
Один или несколько основных и кислотных предшественников предпочтительно прибавляют на второй стадии осаждения a') в виде водных растворов.
Вторую стадию осаждения предпочтительно осуществляют при перемешивании.
Вторую стадию осаждения предпочтительно осуществляют в отсутствие органических добавок.
Кислотные и основные предшественники, необязательно содержащие алюминий, смешивают предпочтительно в виде раствора в водной реакционной среде в таких пропорциях, чтобы значение pH образующейся суспензии находилось в интервале от 8,5 до 10,5.
Как и на стадии a) осаждения, именно относительный расход кислотных и основных предшественников, необязательно содержащих алюминий, выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5.
В предпочтительном случае, в котором основный и кислотный предшественники представляют собой алюминат натрия и сульфат алюминия соответственно, массовое отношение основного предшественника к кислотному предшественнику предпочтительно находится в интервале от 1,6 до 2,05.
Для других основных и кислотных предшественников, необязательно содержащих алюминий, массовое отношение "основание/кислота" определяют по кривой нейтрализации основания кислотой. Такая кривая может быть легко получена специалистами в данной области техники.
Вторую cтадию осаждения предпочтительно осуществляют при значении pH в интервале от 8,5 до 10 и более предпочтительно от 8,7 до 9,9.
Кислотные и основные предшественники смешивают также в количествах, позволяющих получать суспензию, содержащую требуемое количество оксида алюминия, в зависимости от требуемой конечной концентрации оксида алюминия. В частности, вторая стадия осаждения позволяет получать от 0 до 60 масс.% оксида алюминия в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после двух стадий осаждения.
Как и на стадии a) осаждения, именно расход одного или нескольких основных и кислотных предшественников, содержащих алюминий, регулируют с целью достижения выхода на второй стадии в интервале от 0 до 60%.
Выход на этой стадии осаждения a) предпочтительно находится в интервале от 10 до 55% и более предпочтительно от 15 до 55%.
Таким образом, в зависимости от концентрации оксида алюминия, предусматриваемой после одной или нескольких стадий осаждения предпочтительно в интервале от 20 до 100 г/л, рассчитывают количество алюминия, которое должно быть внесено с кислотными и/или основными предшественниками, и расход предшественников регулируют в зависимости от концентрации прибавляемых предшественников алюминия, количества воды, прибавляемой к реакционной смеси, и выхода, требующегося на каждой из стадий осаждения.
Как и на стадии a) осаждения, расходы одного или нескольких кислотных и/или основных предшественников, содержащих алюминий, зависят от размера используемого реактора, а также от количества воды, прибавляемой к реакционной смеси.
В качестве примера, если работать с реактором вместимостью 3 л и задаться целью получить 1 л суспензии оксида алюминия с конечной концентрацией Al2O3, равной 50 г/л, то намеченный выход на первой стадии осаждения составит 50% в расчете на эквивалент Al2O3. Таким образом, 50% от общего количества оксида алюминия должно быть внесено на стадии a) осаждения. Предшественники оксида алюминия представляют собой алюминат натрия с концентрацией 155 г/л в расчете на Al2O3 и сульфат алюминия с концентрацией 102 г/л в расчете на Al2O3. Значение pH осаждения установлено равным 9,5 на первой стадии, а на второй стадии равным 9. Количество воды, подаваемой в реактор, составляет 622 мл.
При осуществлении первой стадии a) осаждения при 30°C в течение 8 минут расход сульфата алюминия должен составлять 10,5 мл/мин, а расход алюмината натрия должен составлять 13,2 мл/мин. Таким образом, массовое отношение алюмината натрия к сульфату алюминия составляет 1,91.
При осуществлении второй стадии осаждения при 70°C в течение 30 минут расход сульфата алюминия должен составлять 2,9 мл/мин, а расход алюмината натрия должен составлять 3,5 мл/мин. Таким образом, массовое отношение алюмината натрия к сульфату алюминия составляет 1,84.
Вторую стадию осаждения предпочтительно осуществляют при температуре в интервале от 40 до 80°C, более предпочтительно от 45 до 70°C и наиболее предпочтительно от 50 до 70°C.
Вторую стадию осаждения предпочтительно осуществляют в течение промежутка времени от 5 до 45 минут и более предпочтительно от 7 до 40 минут.
Вторая стадия осаждения позволяет в общем случае получать суспензию оксида алюминия с концентрацией Al2O3 в интервале от 20 до 100 г/л, предпочтительно от 20 до 80 г/л и более предпочтительно от 20 до 50 г/л.
В случае, когда осуществляют вторую стадию осаждения, способ получения предпочтительно включает также вторую стадию нагревания суспензии, полученной после второй стадии осаждения, при температуре в интервале от 50 до 95°C и предпочтительно от 60 до 90°C.
Вторую стадию нагревания предпочтительно осуществляют в течение промежутка времени от 7 до 45 минут.
Вторую стадию нагревания предпочтительно осуществляют любыми способами нагревания, известными специалистам в данной области техники.
Вторая стадия нагревания позволяет повысить температуру реакционной смеси перед подачей полученной суспензии на стадию b) термической обработки.
Стадия c) фильтрования
Согласно настоящему изобретению способ получения оксида алюминия по настоящему изобретению включает также стадию c) фильтрования суспензии, полученной после стадии b) термической обработки, с последующим осуществлением по меньшей мере одной стадии промывки полученного геля. Стадию фильтрования осуществляют способами, известными специалистам в данной области техники.
Фильтруемость суспензии, полученной после стадии a) осаждения или двух стадий осаждения, улучшается благодаря наличию стадии b) конечной термической обработки полученной суспензии, причем стадия термической обработки благоприятствует производительности процесса получения, а также внедрению способа в промышленном масштабе.
После стадии фильтрования предпочтительно осуществляют по меньшей мере одну стадию промывки водой и более предпочтительно три стадии промывки, применяя воду в количестве, равном количеству отфильтрованного осадка.
Включение в технологическую цепочку стадий a) b) и c) и в случае необходимости второй стадии осаждения, второй стадии нагревания и в случае необходимости стадии фильтрования позволяет получать особый гель оксида алюминия, который характеризуется индексом дисперсности, превышающим 70%, размером кристаллитов в интервале от 1 до 35 нм, а также содержит серу в количестве от 0,001 до 2 масс.% и натрий в количестве от 0,001 до 2 масс.%, при этом массовые процентные доли выражены по отношению к общей массе геля оксида алюминия.
Гель оксида алюминия, полученный таким образом и называемый также бемитом, характеризуется индексом дисперсности в интервале от 70 до 100%, предпочтительно от 80 до 100%, более предпочтительно от 85 до 100°C и наиболее предпочтительно от 90 до 100%.
Гель оксида алюминия, полученного таким образом, предпочтительно характеризуется размером кристаллитов в интервале от 2 до 35 нм.
Гель оксида алюминия, полученный таким образом, предпочтительно содержит серу в количестве от 0,001 до 1 масс.%, предпочтительно от 0,001 до 0,40 масс.%, более предпочтительно от 0,003 до 0,33 масс.% и наиболее предпочтительно от 0,005 до 0,25 масс.%.
Гель оксида алюминия, полученный таким образом, предпочтительно содержит натрий в количестве от 0,001 до 1 масс.%, предпочтительно от 0,001 до 0,15 масс.%, более предпочтительно от 0,0015 до 0,10 масс.% и от 0,002 до 0,040 масс.%.
В частности, гель оксида алюминия или бемит в форме порошка по настоящему изобретению состоит из кристаллитов, размер которых, полученный по формуле Шеррера по дифракции рентгеновских лучей при кристаллографических направлениях (020) и (120), находится в интервале от 2 до 20 нм и от 2 до 35 нм соответственно.
Гель оксида алюминия по настоящему изобретению предпочтительно характеризуется размером кристаллитов при кристаллографическом направлении (020) в интервале от 2 до 15 нм и размер кристаллитов при кристаллографическом направлении (120) в интервале от 2 до 35 нм.
Дифракцию рентгеновских лучей в гелях оксида алюминия или бемитах исследовали традиционным способом анализа порошков дифрактометром.
Формула Шеррера представляет собой формулу, используемую при дифракции рентгеновских лучей в случае порошков или поликристаллических образцов и связывающую ширину дифракционных пиков на полувысоте с размером кристаллитов. Она подробно описана в статье "J.I. Langford and A.J. Wilson. Scherrer after sixty years: A survey and some new results in the determination of crystallite size. Appl. Cryst. (1978), 11, 102-113".
Гель оксида алюминия, полученный таким образом и имеющий высокую дисперсность, позволяет облегчать стадию формования этого геля любыми способами, известными специалистам в данной области техники, и предпочтительно пластицированием-экструзией, гранулированием и по технологии, называемой "oil drop" (гранулирование разбрызгиванием в масло) согласно терминологии на английском языке.
Стадия d) сушки
Согласно настоящему изобретению гель оксида алюминия, полученный после стадии c) фильтрования, сушат на стадии d) сушки для получения порошка.
Указанную стадию сушки предпочтительно осуществляют при температуре в интервале от 20 до 50°C в течение промежутка времени от 1 суток до 3 недель или распылением.
В случае, когда стадию d) сушки осуществляют при температуре в интервале от 20 до 50°C в течение промежутка времени от 1 суток до 3 недель, указанная стадия d) сушки предпочтительно может быть осуществлена в закрытом вентилируемом сушильном шкафу, при этом стадию сушки предпочтительно осуществляют при температуре в интервале от 25 до 40°C в течение промежутка времени от 3 суток до двух недель.
В случае, когда стадию d) сушки осуществляют распылением, осадок, полученный после стадии термической обработки с последующим осуществлением в случае необходимости стадии фильтрования, суспендируют. Затем полученную суспензию распыляют в виде тонкодисперсных капелек в цилиндрической вертикальной камере в контакте с потоком горячего воздуха для испарения воды согласно принципу, хорошо известному специалистам в данной области техники. Полученный порошок перемещается тепловым потоком в циклон или в рукавный фильтр, в котором происходит разделение воздуха и порошка. В случае, когда стадию d) сушки осуществляют распылением, распыление предпочтительно осуществляют по технологии, описанной в публикации "Asep Bayu Dani Nandiyanto, Kikuo Okuyama. Advanced Powder Technology, 22, 1-19, 2011".
Стадия e) формования
Согласно настоящему изобретению порошок, полученный после стадии d) сушки, формуют на стадии e) для получения сырого материала.
Под сырым материалом понимают сформованный материал, не подвергнувшийся стадии термической обработки.
Стадию e) формования предпочтительно осуществляют пластицированием-экструзией, таблетированием, способом гранулирования разбрызгиванием в масло ("oil drop"), гранулированием на вращающейся тарелке или любым другим способом, известным специалистам в данной области техники.
Более предпочтительно стадию e) формования осуществляют пластицированием-экструзией.
Стадия f) термической обработки
Согласно настоящему изобретению сырой материал, полученный после стадии e) формования, затем на стадии f) подвергают термической обработке при температуре в интервале от 500 до 1000°C в течение промежутка времени от 2 до 10 ч необязательно в токе воздуха, содержащего до 60 об.% воды.
Стадию f) термической обработки предпочтительно осуществляют при температуре в интервале от 540 до 850°C.
Стадию f) термической обработки предпочтительно осуществляют в течение промежутка времени от 2 до 10 ч.
Стадия f) термической обработки обеспечивает переход бемита в конечный оксид алюминия.
Способ получения оксида алюминия, используемого в качестве носителя катализатора, применяемого в способе по настоящему изобретению, позволяет получать аморфный мезопористый оксид алюминия, обладающий связностью структуры, значение которой превышает 2,7, и контролируемой мезопористостью, причем указанный оксид алюминия, обладает хорошей термической и химической стабильностью, имеет единообразное и контролируемое центральное распределение мезопор по размеру и нормализованные удельную поверхность и объем пор и, в частности, мезопор и специфическое распределение пор, описанное далее.
Катализатор, применяемый в способе гидрообработки согласно настоящему изобретению, получают, прибавляя элементы, составляющие активную фазу.
Катализатор по настоящему изобретению может быть получен по любой технологии, известной специалистам в данной области техники, и предпочтительно пропиткой выбранного носителя элементами из групп VIII и/или VIB. Прибавление легирующего элемента, такого как фосфор, может быть осуществлено в то же самое время, что и прибавление солей металлов. Пропитка может быть осуществлена способом, который известен специалистам в данной области техники под названием "пропитка в сухом состоянии" и в случае которого количество требуемых элементов в форме солей, растворимых в выбранном растворителе, например в деминерализованной воде, прямо вводят так, чтобы заполнить поры носителя в возможно более точной мере. Пропитка также может быть осуществлена избытком, если это будет сочтено необходимым специалистами в данной области техники. Носитель, заполненный раствором сообразно одному или другому из указанных ранее способов, предпочтительно сушат. Этой стадия предпочтительно предшествует стадия созревания, продолжительность которой составляет меньше 72 часов и предпочтительно находится в интервале от 0 до 24 часов и более предпочтительно от 1 до 12 часов. Стадию сушки, осуществляемую далее, предпочтительно реализуют в среде воздуха или инертного газа при температуре в интервале от 50 до 200°C, предпочтительно от 65 до 180°C и более предпочтительно от 75 до 160°C. После этой стадии сушки в случае необходимости осуществляют стадию прокаливания в общем случае в интервале от 200 до 550°C и предпочтительно от 300 до 500°C. При этом прокаливание осуществляют в среде воздуха или инертного газа.
В некоторых случаях предпочтительным может быть осуществление пропитки по меньшей мере на двух стадиях. Если это решение является предпочтительным, то при этом возможно осуществление промежуточных стадий созревания и сушки и даже прокаливания.
Предшественники одного или нескольких металлов из группы VIII предпочтительно выбирают из оксидов, цитратов, оксалатов, карбонатов, гидроксикарбонатов, гидроксидов, фосфатов, сульфатов, алюминатов, молибдатов, вольфраматов, нитратов, галогенидов, например из хлоридов, фторидов, бромидов, ацетатов, или из любых смесей указанных здесь соединений. Гидроксикарбонат никеля, нитрат кобальта или никеля, карбонат кобальта или гидроксид кобальта или никеля используют предпочтительно.
Применяемые предшественники молибдена хорошо известны специалистам в данной области техники. Например, в качестве источников молибдена можно использовать оксиды и гидроксиды, молибденовые кислоты и их соли, предпочтительно соли аммония, такие, как молибдат аммония, гептамолибдат аммония, фосфорномолибденовую кислоту (H3PMo12O40) и ее соли и при необходимости кремниймолибденовую кислоту (H4SiMo12O40) и соответствующие соли. Источниками молибдена могут быть также любые гетерополисоединения, например, типа соединений Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина, Доусона, Андерсона, Страндберга. Предпочтительно используют триоксид молибдена и гетерополисоединения типа соединений Страндберга, Кеггина, лакунарной структуры Кеггина или замещенной структуры Кеггина, известные специалистам в данной области техники. Применяемые предшественники вольфрама хорошо известны специалистам в данной области техники. Например, в качестве источников вольфрама можно использовать оксиды и гидроксиды, вольфрамовые кислоты и их соли, предпочтительно соли аммония, такие, как вольфрамат аммония, метавольфрамат аммония, фосфорновольфрамовую кислоту (H3PWo12O40) и ее соли и при необходимости кремнийвольфрамовую кислоту (H4SiW12O40) и ее соли. Источниками вольфрама могут быть также любые гетерополисоединения, например, типа соединений Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина, Доусона. Предпочтительно используют оксиды и соли аммония, такие, как метавольфрамат аммония, или гетерополианионы типа соединений Кеггина, лакунарной структуры Кеггина, замещенной структуры Кеггина, известные специалистам в данной области техники.
Хелатообразующий агент органической природы, предпочтительно может быть введен в виде одного или нескольких растворов, если их применение будет сочтено необходимым специалистами в данной области техники.
Катализатор, необязательно подвергнутый обработке на стадии прокаливания, затем может быть пропитан одним или несколькими органическими агентами, применяемыми индивидуально или в смеси в растворителе органической или водной природы. В этом случае специалисты в данной области техники могут опираться на множество существующих литературных источников.
Полученный таким образом катализатор, применяемый в способе по настоящему изобретению, предпочтительно характеризуется следующим распределением пор, определенным ртутным порометром:
- процентная доля объема, соответствующего порам с размерами в интервале от 2 до 6 нм, составляет от 1 до 25% по отношению к общему объему пор;
- процентная доля объема, соответствующего порам с размерами, превышающими 6 нм и меньшими 15 нм, составляет от 60 до 95% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 15 до 50 нм, составляет от 0 до 15% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 50 до 7000 нм и относящегося к объему макропор, составляет от 0 до 5% и предпочтительно от 0 до 3% от общего объема пор.
При этом медианный диаметр (Dp, нм) мезопор катализатора, определенный по объему ртутной порометрией, предпочтительно находится в интервале от 8 до 14 нм.
Катализатор предпочтительно имеет удельную поверхность, превышающую 130 м2/г и более предпочтительно превышающую 150 м2/г.
Катализатор предпочтительно имеет также общий объем пор при определении ртутной порометрией, превышающий или равный 0,35 мл/г и предпочтительно превышающий или равный 0,40 мл/г.
Катализатор, применяемый по настоящему изобретению, предпочтительно обладает связностью структуры (Z), значение которой находится в интервале от 2,7 до 10, предпочтительно от 2,8 до 10, более предпочтительно от 3 до 9, еще более предпочтительно от 3 до 8 и наиболее предпочтительно от 3 до 7.
Способ получения катализатора по настоящему изобретению предпочтительно включает по меньшей мере одну стадию сульфидирования для того, чтобы активная фаза катализатора находилась в сульфидированной форме с целью применения указанного катализатора в способе гидрообработки соответственно изложенному в настоящем описании. Такая активирующая обработка сульфидированием хорошо известна специалистам в данной области техники и может быть осуществлена любым способом, известным специалистам в данной области техники. Стадию сульфидирования осуществляют приведением в контакт катализатора, применяемого в способе по настоящему изобретению, по меньшей мере с одним органическим соединением серы, которое может быть разложено с образованием H2S, или приведением указанного катализатора в прямой контакт с газообразным потоком H2S, например, смешанным с водородом. Органическое производное соединение серы предпочтительно выбирают из алкилдисульфидов, таких как диметилдисульфид (DMDS), алкилсульфидов, таких как диметилсульфид, меркаптанов, таких как н-бутилмеркаптан, полисульфидных соединений типа трет-нонилполисульфида, такого как TPS-37 или TPS-54, реализуемые компанией "ARKEMA", или любых других соединений, известных специалистам в данной области техники и позволяющих получить хорошее качество сульфидирования катализатора. Стадия сульфидирования может быть осуществлена in situ (то есть после загрузки катализатора в реакторный блок гидрообработки согласно настоящему изобретению) или ex situ (то есть перед загрузкой катализатора в реакторный блок гидрообработки согласно настоящему изобретению) при температуре в интервале от 200 до 600°C и более предпочтительно от 300 до 500°C. Если сульфидирование осуществляют ex situ, то имеется возможность вводить органическую добавку или растворитель или любые их смеси. Эта альтернатива может обеспечивать лучшую активность или облегчать загрузку и применение катализатора.
Настоящее изобретение поясняется приведенными далее примерами, которые в любом случае не имеют ограничительного характера.
Примеры
Пример 1 (сравнительный). Получение носителя AL-1 (не соответствующего изобретению) согласно US 7790562
Сначала осуществляют синтез геля оксида алюминия, не соответствующего гелю по примеру 1, реализуя способ получения, описанный в US 7790562. В частности, способ получения геля оксида алюминия согласно примеру 1 не включает стадию термической обработки суспензии, полученной после стадий осаждения, а на первой стадии осаждения a) оксид алюминия образуется в количестве, не превышающем 40%, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после второй стадии осаждения.
Синтез осуществляют в реакторе вместимостью 7 л с получением 5 л конечной суспензии на 2 стадиях осаждения. Количество воды, подаваемой в реактор, составляет 3868 мл.
Заданная конечная концентрация оксида алюминия составляет 30 г/л.
Первую стадию соосаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO осуществляют при 30°C и pH=9,3 в течение 8 минут. Используют следующие концентрации предшественников алюминия: Al2(SO4)3 с концентрацией 102 г/л в расчете на Al2O3 и NaAlOO с концентрацией 155 г/л в расчете на Al2O3. Перемешивание осуществляют со скоростью 350 об/мин в течение всего синтеза.
Раствор сульфата алюминия Al2(SO4)3 в непрерывном режиме в течение 8 минут с расходом 19,6 мл/мин прибавляют к раствору алюмината натрия NaAlOO соответственно массовому соотношению "основание/кислота"=1,80, так чтобы установить значение pH, равное 9,3. Температуру реакционной смеси поддерживают равной 30°C.
Таким образом, получают суспензию, содержащую осадок оксида алюминия.
Поскольку заданная конечная концентрация оксида алюминия составляет 30 г/л, то расход содержащих алюминий предшественников, т.е. сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO, вводимых на первой стадии осаждения, составляет 19,6 и 23,3 мл/мин соответственно.
Эти расходы кислотных и основных предшественников, содержащих алюминий, позволяют после первой стадии осаждения получать выход, равный 30%.
Затем полученную суспензию нагревают до температуры от 30 до 57°C.
Далее осуществляют вторую стадию соосаждения полученной суспензии прибавлением сульфата алюминия Al2(SO4)3 с концентрацией 102 г/л в расчете на Al2O3 и алюмината натрия NaAlOO с концентрацией 155 г/л в расчете на Al2O3. При этом раствор сульфата алюминия Al2(SO4)3 прибавляют в непрерывном режиме к нагретой суспензии, полученной после первой стадии осаждения, в течение 30 минут с расходом 12,8 мл/мин к раствору алюмината натрия NaAlOO соответственно массовому соотношению "основание/кислота"=1,68, так чтобы установить значение pH, равное 8,7. Температуру реакционной смеси на второй стадии поддерживают равной 57°C.
Таким образом, получают суспензию, содержащую осадок оксида алюминия.
Поскольку заданная конечная концентрация оксида алюминия составляет 30 г/л, то расход содержащих алюминий предшественников, т.е. сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO, вводимых на второй стадии осаждения, составляет 12,8 и 14,1 мл/мин соответственно.
Эти расходы кислотных и основных предшественников, содержащих алюминий, позволяют после второй стадии осаждения получать выход, равный 70%.
Полученную таким образом суспензию не направляют на стадию термической обработки.
Затем полученную суспензию фильтруют, отделяя воду на устройстве типа воронки Бюхнера с пористой фильтрующей перегородкой, и полученный гель оксида алюминия промывают 3 раза дистиллированной водой объемами по 5 л с температурой 70°C. Продолжительность фильтрования и промывок составляет 4 ч.
Характеристики геля оксида алюминия, полученного таким образом, обобщены в таблице 1.
Таблица 1. Характеристики геля оксида алюминия, полученного согласно примеру 1
Далее гель оксида алюминия сушат распылением при температуре на входе 250°C и на выходе 130°C.
Сухой гель оксида алюминия вводят в смеситель типа "Brabender". Воду, подкисленную азотной кислотой с общим содержанием кислоты, равным 3% и выраженным по массе в расчете на массу сухого геля, введенного в смеситель, прибавляют в течение 5 минут во время пластицирования со скоростью 20 об/мин. Пластицирование с кислотой осуществляют в течение 15 минут. Затем осуществляют стадию нейтрализации прибавлением в смеситель аммиачного раствора до 50%-й степени нейтрализации, выраженной по массе аммиака в расчете на количество азотной кислоты, введенной в смеситель на стадии подкисления. Пластицирование осуществляют в течение 3 минут.
Полученную пасту экструдируют затем через трехдольчатую фильеру размером 2 мм. Полученные экструдированные элементы сушат при 100°C в течение ночи и затем прокаливают в течение 2 ч при 600°C.
Характеристики сформованного оксида алюминия приведены в таблице 2.
Таблица 2. Характеристики оксида алюминия AL-1, полученного согласно примеру 1
Пример 2 (соответственно изобретению). Получение носителей AL-2 и AL-3 (соответствующих изобретению).
Сначала осуществляют синтез двух алюмооксидных носителей AL-2 и AL-3 способом получения согласно настоящему изобретению в реакторе вместимостью 7 л с получением 5 л конечной суспензии на 3 стадиях, т.е. на двух стадиях осаждения с последующей стадией созревания.
Заданная конечная концентрация оксида алюминия составляет 45 г/л. Количество воды, подаваемой в реактор, составляет 3267 мл. Перемешивание осуществляют со скоростью 350 об/мин в течение всего синтеза.
Первую стадию соосаждения в воде сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO осуществляют при 30°C и pH=9,5 в течение 8 минут. Используют следующие концентрации предшественников алюминия: Al2(SO4)3 с концентрацией 102 г/л в расчете на Al2O3 и NaAlOO с концентрацией 155 г/л в расчете на Al2O3.
Раствор сульфата алюминия Al2(SO4)3 в непрерывном режиме в течение 8 минут с расходом 69,6 мл/мин прибавляют к раствору алюмината натрия NaAlOO с расходом 84,5 мл/мин соответственно массовому соотношению "основание/кислота"=1,84, так чтобы установить значение pH, равное 9,5. Температуру реакционной смеси поддерживают равной 30°C.
Таким образом, получают суспензию, содержащую осадок оксида алюминия.
Поскольку заданная конечная концентрация оксида алюминия составляет 45 г/л, то расход содержащих алюминий предшественников, т.е. сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO, вводимых на первой стадии осаждения, составляет 69,6 и 84,5 мл/мин соответственно.
Эти расходы кислотных и основных предшественников, содержащих алюминий, позволяют после первой стадии осаждения получать выход, равный 72%.
Затем полученную суспензию нагревают до температуры от 30 до 68°C.
Далее осуществляют вторую стадию соосаждения полученной суспензии прибавлением сульфата алюминия Al2(SO4)3 с концентрацией 102 г/л в расчете на Al2O3 и алюмината натрия NaAlOO с концентрацией 155 г/л в расчете на Al2O3. При этом раствор сульфата алюминия Al2(SO4)3 прибавляют в непрерывном режиме к нагретой суспензии, полученной после первой стадии осаждения, в течение 30 минут с расходом 7,2 мл/мин к раствору алюмината натрия NaAlOO соответственно массовому соотношению "основание/кислота"=1,86, так чтобы установить значение pH, равное 9. Температуру реакционной смеси на второй стадии поддерживают равной 68°C.
Таким образом, получают суспензию, содержащую осадок оксида алюминия.
Поскольку заданная конечная концентрация оксида алюминия составляет 45 г/л, то расход содержащих алюминий предшественников, т.е. сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO, вводимых на второй стадии осаждения, составляет 7,2 и 8,8 мл/мин соответственно.
Эти расходы кислотных и основных предшественников, содержащих алюминий, позволяют после второй стадии осаждения получать выход, равный 28%.
Затем полученную суспензию нагревают до температуры от 68 до 90°C.
Далее суспензию направляют на стадию термической обработки, на которой ее выдерживают при 90°C в течение 60 минут.
Затем полученную суспензию фильтруют, отделяя воду на устройстве типа воронки Бюхнера с пористой фильтрующей перегородкой, и полученный гель оксида алюминия промывают 3 раза дистиллированной водой объемами по 5 л. Продолжительность фильтрования и промывок составляет 3 ч.
Характеристики геля оксида алюминия, полученного таким образом, обобщены в таблице 3.
Таблица 3. Характеристики геля оксида алюминия, полученного согласно примеру 2
Таким образом, получают гель, имеющий индекс дисперсности, равный 100%.
Затем полученный гель оксида алюминия сушат распылением при температуре на входе 250°C и на выходе 130°C. Гель, высушенный распылением, обозначен как гель № 1.
Гель оксида алюминия, полученный согласно примеру 2, также сушили в вентилируемом сушильном шкафу при 35°C в течение 4 суток. Гель, высушенный в шкафу, обозначен как гель № 2.
Далее в смеситель типа "Brabender" вводят сухие гели оксида алюминия № 1 и № 2 соответственно. Воду, подкисленную азотной кислотой с общим содержанием кислоты, равным 3% и выраженным по массе в расчете на массу сухого геля, введенного в смеситель, прибавляют в течение 5 минут во время пластицирования со скоростью 20 об/мин. Пластицирование с кислотой осуществляют в течение 15 минут. Затем осуществляют стадию нейтрализации прибавлением в смеситель аммиачного раствора до 50%-й степени нейтрализации, выраженной по массе аммиака в расчете на количество азотной кислоты, введенной в смеситель на стадии подкисления. Пластицирование осуществляют в течение 3 минут.
Полученную пасту экструдируют затем через трехдольчатую фильеру размером 2 мм. Полученные экструдированные элементы сушат при 100°C в течение ночи и затем прокаливают в течение 2 ч при 600°C.
Характеристики сформованных образцов оксида алюминия AL-2 и AL-3 приведены в таблице 4.
Таблица 4. Характеристики образцов оксида алюминия AL-2 и AL-3, полученных по примеру 2
Пример 3. Получение катализаторов C1 и C2 (не соответствующих изобретению) исходя из алюмооксидного носителя AL-1 и C3 и C4 (соответствующих изобретению) исходя из алюмооксидных носителей AL-2 и AL-3 соответственно
Катализаторы C1 и C2 получают пропиткой в сухом состоянии алюмооксидного носителя AL-1 в форме экструдированных элементов, полученного согласно примеру 1, пропиточным раствором, полученным растворением при нагревании оксида молибдена и гидроксикарбоната кобальта в растворе фосфорной кислоты в виде водного раствора, причем объем пропиточного раствора строго равен объему пор массы алюмооксидного носителя. Концентрации предшественников в пропиточном растворе регулируют так, чтобы нанести на алюмооксидный носитель требуемые массовые количества Mo, Co и P. После стадии пропитки в сухом состоянии экструдированные элементы оставляют созревать в атмосфере, насыщенной водой, в течение 12 ч и затем сушат в течение ночи при 90°C перед прокаливанием при 450°C в течение 2 часов.
При этом катализатор C1 имеет следующий конечный состав, выраженный в пересчете на оксиды и определенный по рентгеновской флуоресценции: MoO3=20,4±0,2 масс.%, CoO=3,6±0,1 масс.% è P2O5=5,9±0,1 масс.%, что соответствует соотношению Co/Mo=0,34 моль/моль и соотношению P/Mo=0,59 моль/моль.
При этом катализатор C2 имеет следующий конечный состав, выраженный в пересчете на оксиды и определенный по рентгеновской флуоресценции: MoO3=25,6±0,2 масс.%, CoO=3,9±0,1 масс.% и P2O5=2,8±0,1 масс.%, что соответствует соотношению Co/Mo=0,29 моль/моль и соотношению P/Mo=0,22 моль/моль.
Для катализаторов C1 и C2 значение Z остается близким к значению в случае оксида алюминия AL-1: 2,4 и 2,3 соответственно.
Катализатор C3 получают аналогично катализатору C1, но исходя из носителя AL-2. Он сохраняет значение Z, превышающее значение в случае катализатора C1 и равное 6,4.
При этом катализатор C3 имеет следующий конечный состав, выраженный в пересчете на оксиды и определенный по рентгеновской флуоресценции: MoO3=20,1±0,2 масс.%, CoO=3,4±0,1 масс.% и P2O5=6,0±0,1 масс.%, что соответствует соотношению Co/Mo=0,33 моль/моль и соотношению P/Mo=0,60 моль/моль.
Катализатор C4 получают аналогично катализатору C1, но исходя из носителя AL-3. Он также сохраняет значение Z, превышающее значение в случае катализатора C2 и равное 6,1.
При этом катализатор C4 имеет следующий конечный состав, выраженный в пересчете на оксиды: MoO3=25,1±0,2 масс.%, CoO=3,6±0,1 масс.% и P2O5=2,9±0,1 масс.%, что соответствует соотношению Co/Mo=0,28 моль/моль и соотношению P/Mo=0,23 моль/моль.
Пример 4. Оценка каталитических характеристик катализаторов C1 и C2 (не соответствующих изобретению) и C3 и C4 (соответствующих изобретению) в испытании, моделирующем гидрирование толуола в циклогексан под давлением и в присутствии H2S
В вариантах применения, таких как гидрообработка, когда для достижения требуемого содержания серы необходимо осуществлять обессеривание преобладающей части устойчивых соединений, таких как 4,6-DMDBT, гидрирующая-дегидрирующая функция играет критическую роль. Таким образом, испытание по гидрированию толуола использовали для изучения эффективности катализаторов, предназначенных для гидрообработки такого сырья.
Описанные ранее катализаторы C1-C4 сульфидируют in situ в динамическом режиме в трубчатом реакторе с неподвижным проницаемым слоем на экспериментальной установке типа "Microcat" (разработчик: компания "Vinci"), причем потоки проходят сверху вниз. Определение гидрирующей активности осуществляют немедленно после сульфидирования под давлением и без выгрузки из реактора на воздух с использованием углеводородного сырья, служившего для сульфидирования катализаторов.
Сульфидирующая и испытуемая смесь состоит из 5,88% диметилдисульфида (DMDS), 20% толуола и 74,12% циклогексана (по массе). DMDS позволяет за счет разложения на метан и H2S поддерживать катализатор в сульфидированной форме.
Сульфидирование осуществляют при температуре от комнатной до 350°C со скоростью подъема температуры 2°C/мин, VVH=4 ч-1 и H2/HC=450 нл/л. Испытание по катализу осуществляют при 350°C, VVH=2 ч-1 и отношении H2/HC, равном отношению при сульфидировании, с анализом в потоке по меньшей мере в 4 пробах/уходящих потоках для определения состава.
Таким образом, определяют стабилизированную каталитическую активность равных объемов катализаторов в реакции гидрирования толуола.
Приняты следующие конкретные условия определения активности:
- общее давление: 6,0 МПа;
- давление толуола: 0,37 МПа;
- давление циклогексана: 1,42 МПа;
- давление метана: 0,22 МПа;
- давление водорода: 3,68 МПа;
- давление H2S: 0,22 МПа.
Вносят катализатор в объеме, равном 4 см3 (в виде экструдированных элементов длиной от 2 до 4 мм) и определенном по навеске исходя из плотности слоя катализатора.
Определение молярных концентраций непревращенного толуола (T) и концентраций продуктов его гидрирования (метилциклогексан (MCC6), этилциклопентан (EtCC5) и диметилциклопентаны (DMCC5)) позволяет рассчитывать степень гидрирования толуола XHYD, определяемую по формуле:
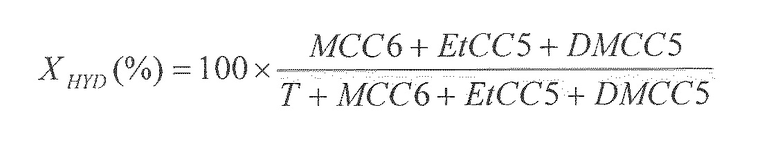
Поскольку реакция гидрирования толуола в условиях осуществляемого испытания имеет 1-й порядок, а реактор соответствует реактору идеального вытеснения, гидрирующую активность AHYD катализаторов рассчитывают, используя формулу:
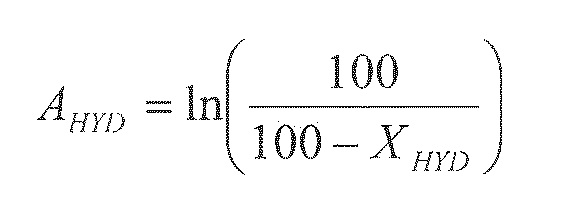
Приведенная далее таблица 5 позволяет сравнить относительные гидрирующие активности катализаторов C1-C4 в зависимости от пропитанной активной фазы и значения связности структуры Z.
Таблица 5. Относительная активность по сравнению с катализатором C1 (не соответствующим изобретению) при гидрировании толуола с катализаторами C3 и C4 (соответствующими изобретению) и с катализатором C2 (не соответствующим изобретению)
Результаты, представленные в таблице 5, показывают, что каталитические характеристики в отношении гидрирующей активности катализатора C3, полученного по настоящему изобретению исходя из носителя AL-2 и характеризующегося значением Z, равным 6,4, значительно превышают характеристики не соответствующего изобретению катализатора C1, полученного исходя из носителя AL-1 и характеризующегося значением Z, равным 2,4.
Это повышение гидрирующей активности является особенно предпочтительным, поскольку оно позволяет, в частности, достигать уровня активности, близкого к уровню катализатора C2, не соответствующего изобретению, но только с 20% MoO3 против 25% MoO3 в случае катализатора C2. В то же время, при этом содержании молибдена катализатор C4, полученный по настоящему изобретению исходя из носителя AL-3, проявляет еще более высокую гидрирующую активность.
В отсутствие диффузионного ограничения для реакционной смеси при испытании, осуществляемом в газовой фазе, этот результат показывает эффективность пропитки металлической фазой носителя с высокой связностью структуры. При этом качество нанесенной активной фазы представляется улучшенным.
Пример 5. Оценка каталитических характеристик катализаторов C1 и C2 (не соответствующих изобретению) и катализаторов C3 и C4 (соответствующих изобретению) при гидрообработке прямогонного газойля
Катализаторы C1-C4 с целью их оценки при гидрообессеривании (HDS) газойля обрабатывают на стадии сульфидирования in situ в реакторе с проницаемым слоем (30 см3 катализатора в форме экструдированных элементов, смешанного с 10 см3 SiC в гранулах размером 0,8 мм). Сульфидирование осуществляют при 30 бар (3 МПа), VVH=2 ч-1, с соотношением (объемным расходом) H2/HC на входе=250 нл/л. Сульфидирующую смесь (газойль с добавкой 2% присадки "DMDS Evolution®" компании "Arkema") вводят в реактор при подаче H2 по достижении 150°C. После выдерживания в течение часа при 150°C температуру повышают со скоростью 25°C/ч до 220°C и затем со скоростью 12°C/ч до достижения плато 350°C, выдерживаемого в течение 12 часов.
После сульфидирования температуру понижают до 330°C и вводят испытуемую исходную смесь (без DMDS). Испытание по катализу осуществляют при общем давлении 3 МПа, со сбросом водорода (без рециркуляции), VVH=2 ч-1, с объемным соотношением H2/HC на входе 250 нл/л (расход H2=24 нл·ч-1, расход исходной смеси=60 см3·ч-1) и при трех разных значениях температуры (330°C, а затем 340 и 350°C), осуществляя в каждой точке выдержку продолжительностью, достаточной для того, чтобы содержание серы стабилизировалось.
Для того чтобы иметь возможность оценивать эксплуатационные характеристики катализаторов при HDS и для освобождения уходящего потока от присутствия H2S, приемный сосуд, содержащий уходящий поток, продувают азотом из расчета 10 л·ч-1.
Используемый в данном случае газойль получен из неочищенной тяжелой арабской нефти. Она содержит 0,89 масс.% серы, 150 млн-1 азота и имеет плотность 0,848 г/см3 и средневзвешенную температуру (TMP), определенную исходя из температуры, при которой отгоняются 5, 50 и 70% объема исходной смеси, по формуле TMP=(T5+2T50+4T95)/7 и равную 324°C.
Эксплуатационные каталитические характеристики катализаторов представлены в таблице 6. Они выражены в относительной активности с учетом того, что активность катализатора C1 принята равной 100, и в расчете, что их значения составляют порядка 1,25 в отношении серы (указана средняя активность, рассчитанная при трех значениях температуры). Другой способ количественного определения повышения активности состоит в повышении температуры, при которой уходящий поток содержит 50 млн-1 серы. Аналогичным образом, катализатор C1 принимают в качестве базы.
Таблица 6. Относительная активность (определенная при равенстве объемов катализаторов) при HDS прямогонного газойля с катализаторами C1-C4
* Более низкая температура указывает на повышение активности.
Результаты, представленные в таблице 6, показывают значительное повышение активности, достигнутой в случае катализатора C3, полученного способом по настоящему изобретению путем пропитки носителя, характеризующегося высоким значением Z (6,4), по сравнению с катализатором C1, полученным с носителем, характеризующимся низким значением Z (2,4).
Эффект подтверждается при более высоком содержании металлов (катализатор C2, полученный способом предшествующего уровня техники, по сравнению с катализатором C4, полученным по настоящему изобретению).
В более общем смысле, полученные результаты показывают, что катализатор, полученный по настоящему изобретению, позволяет снизить приблизительно на 2°C рабочую температуру установки при одном и том же содержании серы в уходящем потоке по сравнению с катализаторами предшествующего уровня техники. Это повышение эффективности может также позволить специалистам по нефтепереработке обрабатывать сырье, которое является более стойким и неприемлемым при рабочих температурах, требуемых катализаторами предшествующего уровня техники, но которое становится приемлемым, если применять катализаторы по настоящему изобретению.
Настоящее изобретение относится к области гидрообработки углеводородного сырья типа газойля. Описан способ гидрообработки по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C, причем способ осуществляют при температуре в интервале от 250 до 400°C, при общем давлении в интервале от 2 до 10 МПа, при соотношении объема водорода и объема углеводородного сырья в интервале от 100 до 800 литров на литр и при часовой объемной скорости (VVH), определенной как отношение объемного расхода жидкого углеводородного сырья к объему катализатора, загруженного в реактор, в интервале от 1 до 10 ч-1, причем в способе применяют по меньшей мере один катализатор, содержащий по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, причем указанный оксид алюминия получают, осуществляя по меньшей мере следующие стадии: a) по меньшей мере одну первую стадию осаждения оксида алюминия в водной реакционной смеси исходя по меньшей мере из одного основного предшественника, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере из одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, cтадию нагревания полученной после стадии a) суспензии, осуществляемую между стадией a) и второй стадией а') осаждения, которую осуществляют при температуре в интервале от 20 до 90°C в течение промежутка времени от 7 до 45 минут; a’) вторую стадию осаждения, которую осуществляют между первой стадией осаждения a) и стадией b) термической обработки, b) стадию термической обработки суспензии, полученной после стадии a), при температуре в интервале от 50 до 200°C в течение промежутка времени от 30 минут до 5 часов, что обеспечивает получение геля оксида алюминия; c) стадию фильтрования суспензии, полученной после стадии b) термической обработки, с последующим осуществлением по меньшей мере одной стадии промывки полученного геля; d) стадию сушки геля оксида алюминия, полученного после стадии c), для получения порошка; e) стадию формования порошка, полученного после стадии d), для получения сырого материала; f) стадию термической обработки сырого материала, полученного после стадии e), при температуре в интервале от 500 до 1000°C, необязательно в токе воздуха, содержащего до 60 об.% воды. Технический результат - разработка способа гидрообработки по меньшей мере газойлевой фракции, в котором применяют катализатор, обладающий улучшенными каталитическими характеристиками, причем указанный способ обеспечивает повышенную гидрообессеривающую активность. 13 з.п. ф-лы, 5 пр., 6 табл.
1. Способ гидрообработки по меньшей мере газойлевой фракции, имеющей средневзвешенную температуру (TMP) в интервале от 240 до 350°C, причем способ осуществляют при температуре в интервале от 250 до 400°C, при общем давлении в интервале от 2 до 10 МПа, при соотношении объема водорода и объема углеводородного сырья в интервале от 100 до 800 литров на литр и при часовой объемной скорости (VVH), определенной как отношение объемного расхода жидкого углеводородного сырья к объему катализатора, загруженного в реактор, в интервале от 1 до 10 ч-1, причем в способе применяют по меньшей мере один катализатор, содержащий по меньшей мере один металл из группы VIB и/или по меньшей мере один металл из группы VIII Периодической системы элементов и носитель, содержащий аморфный мезопористый оксид алюминия, причем указанный оксид алюминия получают, осуществляя по меньшей мере следующие стадии:
a) по меньшей мере одну первую стадию осаждения оксида алюминия в водной реакционной смеси исходя по меньшей мере из одного основного предшественника, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере из одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, причем по меньшей мере один из основных или кислотных предшественников содержит алюминий, относительный расход кислотных и основных предшественников выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5, расход одного или нескольких кислотных и основных предшественников, содержащих алюминий, регулируют с целью достижения выхода на первой стадии в интервале от 45 до 90%, при этом выход определяют как отношение количества оксида алюминия, образующегося во время первой стадии осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после одной или нескольких стадий осаждения, причем первую стадию осаждения осуществляют при температуре в интервале от 20 до 40°C в течение промежутка времени от 2 до 30 минут;
cтадию нагревания полученной после стадии a) суспензии, осуществляемую между стадией a) и второй стадией а') осаждения, которую осуществляют при температуре в интервале от 20 до 90°C в течение промежутка времени от 7 до 45 минут;
a’) вторую стадию осаждения, которую осуществляют между первой стадией осаждения a) и стадией b) термической обработки,
указанная вторая стадия a’) осаждения осуществляется путем добавления к суспензии по меньшей мере одного основного предшественника, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере одного кислотного предшественника, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, причем по меньшей мере один из основных или кислотных предшественников содержит алюминий, относительный расход кислотных и основных предшественников выбирают так, чтобы получить значение pH реакционной смеси в интервале от 8,5 до 10,5, расход одного или нескольких кислотных и основных предшественников, содержащих алюминий, регулируют с целью достижения выхода на первой стадии в интервале от 10 до 55%, при этом выход определяют как отношение количества оксида алюминия, образующегося во время второй стадии осаждения, в расчете на эквивалент Al2O3 по отношению к общему количеству оксида алюминия, образующегося после стадии a’) осаждения, причем указанную стадию осаждения осуществляют при температуре в интервале от 40 до 90°C в течение промежутка времени от 2 до 50 минут;
вторую стадию нагревания суспензии, полученной после второй стадии осаждения, при температуре в интервале от 50 до 95°C,
b) стадию термической обработки суспензии, полученной после стадии a), при температуре в интервале от 50 до 200°C в течение промежутка времени от 30 минут до 5 часов, что обеспечивает получение геля оксида алюминия;
c) стадию фильтрования суспензии, полученной после стадии b) термической обработки, с последующим осуществлением по меньшей мере одной стадии промывки полученного геля;
d) стадию сушки геля оксида алюминия, полученного после стадии c), для получения порошка;
e) стадию формования порошка, полученного после стадии d), для получения сырого материала;
f) стадию термической обработки сырого материала, полученного после стадии e), при температуре в интервале от 500 до 1000°C, необязательно в токе воздуха, содержащего до 60 об.% воды.
2. Способ по п. 1, в котором используемую газойлевую фракцию выбирают из газойлевых фракций, получаемых прямой перегонкой, и используют индивидуально или в смеси по меньшей мере с одной фракцией, получаемой на установке коксования, или по меньшей мере с одной фракцией, получаемой каталитическим крекингом, или также по меньшей мере с одной газойлевой фракцией, получаемой другими способами конверсии, такими как мягкий гидрокрекинг или гидрообработка нефтяных остатков.
3. Способ по п. 1 или 2, в котором элементы из группы VIII выбирают из кобальта и никеля, применяемых индивидуально или в смеси.
4. Способ по любому из пп. 1-3, в котором элементы из группы VIB выбирают из вольфрама и молибдена, применяемых индивидуально или в смеси.
5. Способ по любому из пп. 1-4, в котором, в случае когда катализатор содержит по меньшей мере один металл из группы VIB в комбинации по меньшей мере с одним неблагородным металлом из группы VIII, содержание металла из группы VIB находится в интервале от 10 до 35 мас.% в расчете на оксид по отношению к общей массе катализатора, а содержание неблагородного металла из группы VIII находится в интервале от 1 до 10 мас.% в расчете на оксид по отношению к общей массе катализатора.
6. Способ по любому из пп. 1-5, в котором катализатор содержит по меньшей мере один легирующий элемент, выбранный из фосфора, бора, фтора или кремния, применяемых индивидуально или в смеси.
7. Способ по п. 6, в котором содержание фосфора в катализаторе находится в интервале от 0,5 до 15 мас.% в расчете на P2O5.
8. Способ по любому из пп. 1-7, в котором носитель катализатора содержит аморфный мезопористый оксид алюминия, обладающий связностью структуры (Z), значение которой находится в интервале от 3 до 7.
9. Способ по любому из пп. 1-8, в котором катализатор обладает связностью структуры (Z), значение которой находится в интервале от 3 до 7.
10. Способ по любому из пп. 1-9, в котором носитель катализатора отличается следующим распределением пор, определенным ртутной порометрией:
- процентная доля объема, соответствующего порам с размерами в интервале от 2 до 6 нм, составляет от 1 до 25% по отношению к общему объему пор;
- процентная доля объема, соответствующего порам с размерами, превышающими 6 нм и меньшими 15 нм, составляет от 60 до 95% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 15 до 50 нм, составляет от 0 до 8% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 50 до 7000 нм и относящегося к объему макропор, составляет от 0 до 5%.
11. Способ по любому из пп. 1-10, в котором носитель отличается медианным диаметром мезопор в интервале от 7 до 12,5 нм при определении по объему ртутной порометрией.
12. Способ по любому из пп. 1-11, в котором носитель имеет объем мезопор, определенный ртутной порометрией, в интервале от 0,5 до 0,8 мл/г.
13. Способ по любому из пп. 1-12, в котором катализатор отличается следующим распределением пор, определенным ртутным порометром:
- процентная доля объема, соответствующего порам с размерами в интервале от 2 до 6 нм, составляет от 1 до 25% по отношению к общему объему пор;
- процентная доля объема, соответствующего порам с размерами, превышающими 6 нм и меньшими 15 нм, составляет от 60 до 95% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 15 до 50 нм, составляет от 0 до 15% от общего объема пор;
- процентная доля объема, соответствующего порам с размерами в интервале от 50 до 7000 нм и относящегося к объему макропор, составляет от 0 до 5% от общего объема пор.
14. Способ по любому из пп. 1-13, в котором катализатор имеет общий объем пор, определенный ртутной порометрией и превосходящий или равный 0,35 мл/г.
| WO 2013095856 A1, 27.06.2013 | |||
| СПОСОБ И КАТАЛИЗАТОР ГИДРОКОНВЕРСИИ ТЯЖЕЛОГО УГЛЕВОДОРОДНОГО ИСХОДНОГО СЫРЬЯ | 2004 |
|
RU2376059C2 |
| FR 2972648 A1, 21.09.2012 | |||
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИТИЧЕСКОЙ КОМПОЗИЦИИ СООСАЖДЕНИЕМ, КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ГИДРООБРАБОТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ | 2000 |
|
RU2242283C2 |

