ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к катализаторам гидроочистки, в том числе, гидроочистки остатков, и направлено на производство катализаторов гидроочистки с активной фазой, полученных путем совместного растирания и обладающих структурой и составом, благоприятствующими гидроочистке остатков, особенно гидродеметаллизации. Способ производства, соответствующий изобретению, также позволяет исключить стадию пропитки, обычно проводимую в отношении предварительно формованной подложки.
Изобретение заключается в использовании катализаторов с активной фазой, введенной путем совместного растирания c матрицей из оксида алюминия и содержащей, по меньшей мере, элемент группы VIB, в случае необходимости, по меньшей мере, элемент группы VIII, а также, в случае необходимости, элементарный фосфор. Введение активной фазы этого типа до стадии формования путем совместного растирания с определенным оксидом алюминия, полученным путем обжига особого геля, довольно неожиданно позволяет достичь в процессе гидроочистки, особенно гидроочистки остатков, проводимом в неподвижном слое катализатора, но также и в процессе, проводимом в кипящем слое, существенного повышения активности катализатора в отношении гидрообессеривания, но также и гидродеметаллизации по сравнению с катализаторами, производимыми путем совместного растирания с бомитом, и, одновременно, значительного снижения стоимости производства по сравнению с пропитанными катализаторами предшествующего уровня техники.
УРОВЕНЬ ТЕХНИКИ
Специалистам известно, что каталитическая очистка, осуществляемая путем приведения некоторого углеводородного сырья в контакт с катализатором, свойства которого, а именно, металлы активной фазы и пористость, предварительно тщательно подобраны, позволяет существенно уменьшить содержание в сырье асфальтенов, металлов, серы и других примесей и, одновременно, увеличить отношение водорода к углероду (Н/С) и преобразовать его в той или иной степени в более легкие соединения.
Способы гидроочистки остатков в неподвижном слое катализатора (обычно осуществляемые на установке обессеривания нефтяных остатков, Resid Desulfurization - RDS) обеспечивают высокие параметры очистки: обычно, они позволяют получить фракцию с температурой кипения более 370°С, содержащую менее 0,5% вес. серы и менее 20 частей на миллион металлов, из сырья, содержащего до 5% вес. серы и до 250 частей на миллион металлов (Ni+V). Различные полученные таким образом продукты могут служить основой для производства тяжелого жидкого топлива высокого качества и/или предварительно обработанным сырьем для других процессов, например, каталитического крекинга (Fluid Catalytic Cracking, FCC, флюид-каталитический крекинг). Зато гидроконверсия остатков до более легких соединений, нежели атмосферные остатки (особенно газойль и бензин), как правило, неэффективна, обычно, составляет порядка 10-20% вес. В подобных процессах сырье, предварительно смешанное с водородом, проходит через множество реакторов с неподвижным слоем катализатора, соединенных последовательно и заполненных катализаторами. Общее давление, обычно, составляет от 100 до 200 бар (10-200 МПа), температура составляет от 340 до 420 °С. Отводимые из последнего реактора продукты направляют на стадию фракционирования.
В классическом варианте, способ гидроочистки с неподвижным слоем катализатора состоит, по меньшей мере, из двух этапов (или стадий). Первый этап, именуемый гидродеметаллизацией (HDM) направлен, главным образом, на удаление из сырья большей части металлов с использованием одного или нескольких катализаторов гидродеметаллизации. На этом этапе осуществляют, главным образом, операции по удалению ванадия и никеля и, в меньшей степени, железа.
Второй этап или стадия, именуемый гидрообессеривание (HDS), состоит в пропускании продукта первого этапа над одним или несколькими катализаторами гидрообессеривания, более активными с токи зрения гидрообессеривания и гидрогенизации сырья, но менее устойчивыми к отравлению металлами.
Когда содержание металлов в сырье слишком велико (более 250 частей на миллион) и/или когда целью является значительная степень конверсии (преобразование тяжелой фракции 540°С+ (или 370°С+) в более легкую фракцию 540°С- (или 370°С-), предпочтительными являются способы гидроочистки в кипящем слое катализатора. В процессах данного типа (см. M.S. Rana et al., Fuel 86 (2007), p1216) показатели очистки ниже, чем в способах RDS, но гидроконверсия фракции остатков более эффективна (порядка 45-85% об.). Применяемые высокие температуры, от 415 до 440°С, способствуют повышению эффективности гидроконверсии. Реакции термокрекинга действительно интенсифицируются, хотя катализатор, вообще, не имеет специальной функции в отношении гидроконверсии. Кроме того, образующиеся при таком типе конверсии продукты могут вызывать проблемы, связанные с их устойчивостью (образование осадка).
Таким образом, для гидроочистки остатков необходима разработка эффективных и стабильных поливалентных катализаторов.
В отношении способов, осуществляемых в кипящем слое, в заявке на патент WO 2010/002699 указывается на эффективность использования катализатора, подложка которого характеризуется медианным диаметром пор от 10 до 14 нм с узким распределением. Уточняется, что менее 5% объема пор должны составлять поры с размером боле 21 нм и, точно так же, менее 10% объема пор должны составлять мелкие поры размером менее 9 нм. В патенте US 5968348 подтверждается, что предпочтительно использовать подложку, размер мезопор которой примерно соответствует диапазону от 11 до 13 нм, в некоторых случаях, при наличии макропор, и характеризующуюся увеличенной удельной площадью поверхности по ВЕТ, а именно, по меньшей мере, 175 м2/г.
В отношении способов, осуществляемых в неподвижном слое, в патенте SU 6780817 указывается, что для стабильной работы в неподвижном слое необходимо использовать подложку катализатора, в которой объем макропор составляет, по меньшей мере, 0,32 мл/г. Кроме того, такой катализатор характеризуется медианным диаметром мезопор от 8 до 13 нм и увеличенной удельной площадью поверхности, по меньшей мере, 180 м2/г.
В патенте SU 6919294 также описано использование, так называемой, бимодальной подложки - с мезопорами и макропорами, при этом, объем макропор значителен, а объем мезопор ограничен величиной, самое большее, 0,4 мл/г.
В патентах US 4976848 и US 5089463 описан катализатор гидродеметаллизации и гидрообессеривания тяжелого сырья, содержащий активную фазу гидрогенизации на основании металлов групп VI и VIII и жаропрочную подложку из неорганического оксида, при этом, от 5 до 11% объема пор катализатора составляют макропоры и мезопоры медианным диаметром более 165 нм.
В патенте SU 7169294 описан катализатор гидроконверсии тяжелого сырья, содержащий от 7 до 20% металла группы VI и от 0,5 до 6% металла группы VIII, нанесенных на подложку из оксида алюминия. Катализатор характеризуется удельной площадью поверхности от 100 до 180 м2/г, общим объемом пор, большим или равным 0,55 мл/г, по меньшей мере, 50% общего объема пор заключено в порах размером более 20 нм, по меньшей мере, 5% общего объема пор заключено в порах размером более 100 нм, по меньшей мере, 85% общего объема пор заключено в порах размером от 10 до 120 нм, менее 2% общего объема пор заключено в порах диаметром более 400 нм, менее 1% общего объема пор заключено в порах диаметром более 1000 нм.
Множество усовершенствований направлено именно на оптимизацию распределения пор катализатора или смеси катализаторов с целью оптимизации подложки катализатора.
Так, в патенте SU 6589908, например, описан способ производства оксида алюминия, отличающегося отсутствием макропор: менее 5% общего объема пор составляют поры диаметром более 35 нм, большим объемом пор: более 0,8 мл/г и бимодальным распределением мезопор, в котором два типа пор разделяет от 1 до 20 нм, при этом, исходный тип пор больше, чем медианный диаметр пор. С этой целью описываемый способ производства включает две стадии осаждения прекурсоров оксида алюминия в тщательно регулируемых по температуре, рН и расходу условиях. Первую стадию проводят при температуре от 25 до 60°С, рН от 3 до 10. Затем суспензию нагревают до температуры от 50 до 90°С. В суспензию снова добавляют реагенты, затем ее промывают, сушат, формуют и обжигают с целью создания подложки для катализатора. Затем указанную подложку пропитывают раствором активной фазы с целью получения катализатора гидроочистки; описан катализатор гидроочистки остатков на одномодальной мезопористой подложке с медианными диаметром пор около 20 нм.
В патенте SU 7790652 описаны катализаторы гидроконверсии, которые могут быть получены путем совместного осаждения геля оксида алюминия и последующего введения в полученную подложку металлов любым известным специалистам способом, в том числе, путем пропитки. Получаемый катализатор характеризуется мономодальным распределением мезопор с медианным диаметром мезопор от 11 до 12,6 нм и шириной распределения пор менее 3,3 нм.
Также разработаны альтернативные традиционному введению металлов в подложки из оксида алюминия способы, например, введение в подложку тонкодисперсных катализаторов. Так, в заявке на патент WO 2012/021386 описаны катализаторы гидроочистки, включающие подложку из жаропрочного пористого оксида, сформованную из порошкообразного оксида алюминия, и от 5% до 45% вес. тонкодисперсного катализатора. Подложку, содержащую тонкодисперсные материалы, затем сушат и обжигают. Полученная подложка характеризуется удельной площадью поверхности от 50 м2/г до 450 м2/г, средним диаметром пор от 50 до 200 Å (от 5 до 20 нм) и общим объемом пор, превосходящим 0,55 см3/г. Подложка также содержит металл, введенный в форме металлов, содержащихся в тонкодисперсном катализатора. Готовая подложка может быть обработана хелирующим агентом. Объем пор может быть частично заполнен полярной добавкой, затем пропитываемой раствором металла.
Учитывая предшествующий уровень техники, представляется затруднительным получение катализатора, обладающего одновременно бимодальной пористостью с большим объемом мезопор в сочетании со значительным объемом макропор и увеличенным диаметром мезопор и активной фазой гидро-дегидрогенизации. Кроме того, увеличение пористости часто происходит в ущерб удельной площади поверхности и механической прочности.
Неожиданно, автором настоящей заявки было обнаружено, что катализатор, полученный из оксида алюминия, являющегося продуктом обжига особого геля оксида алюминия, характеризующегося малой степенью диспергируемости, путем совместного растирания активной фазы гидро-дегидрогенизации и обожженного оксида алюминия, обладает пористой структурой, представляющей особый интерес с точки зрения гидроочистки тяжелого сырья, и, одновременно, надлежащим содержанием активной фазы.
Цели изобретения
Изобретение относится к катализатору гидроконверсии/гидроочистки остатков с оптимизированным распределением пор и активной фазой, смешанной с обожженной матрицей из оксида алюминия.
Изобретение также относится к способу производства катализатора, пригодного для гидроконверсии/гидроочистки остатков, путем совместного растирания активной фазы и особого оксида алюминия.
Наконец, изобретение относится к использованию катализатора в способах гидроочистки, в том числе, гидроочистки тяжелого сырья.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к способу производства катализатора с активной фазой, введенной путем совместного растирания, содержащего, по меньшей мере, один металл группы VIB Периодической системы элементов, в некоторых случаях, по меньшей мере, один металл группы VIII Периодической системы элементов, в некоторых случаях, фосфор и оксидную матрицу, в основном, из оксида алюминия, включающему следующие стадии:
а) первая стадия осаждения в водной реакционной среде, по меньшей мере, одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и, по меньшей мере, одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, на которой, по меньшей мере, один из прекурсоров - основный или кислый - содержит алюминий, относительный расход кислого и основного прекурсоров выбран так, чтобы обеспечить рН реакционной среды от 8,5 до 10,5, и расход прекурсора или прекурсоров - кислого и основного, содержащих алюминий, выбран так, чтобы доля выхода указанной первой стадии составляла от 5 до 13%, при этом, доля выхода определяется как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной первой стадии осаждения, к общему количеству оксида алюминия, в эквиваленте Al2O3, образовавшемуся по окончании стадии с) способа производства, при этом, указанную первую стадию осаждения осуществляют при температуре, лежащей в диапазоне от 20 до 90°С, в течение периода времени от 2 минут до 30 минут;
b) стадия нагревания суспензии до температуры, лежащей в диапазоне от 40 до 90°С, на период времени от 7 минут до 45 минут;
с) вторая стадия осаждения суспензии, полученной по окончании стадии b) нагревания, путем добавления в указанную суспензию, по меньшей мере, одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и, по меньшей мере, одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, на которой, по меньшей мере, один из прекурсоров - основный или кислый, содержит алюминий, относительный расход кислого и основного прекурсоров выбирают так, чтобы получить рН реакционной среды от 8,5 до 10,5, расход кислого и/или основного прекурсоров, содержащих алюминий, регулируют так, чтобы достичь доли выхода второй стадии от 87 до 95%, при этом, долю выхода определяют как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной второй стадии осаждения к общему количеству оксида алюминия, в эквиваленте Al2O3, образовавшемуся по окончании стадии с) способа производства, при этом, указанную стадию проводят при температуре, лежащей в диапазоне от 40 до 90°С в течение периода времени от 2 минут до 50 минут;
d) стадия фильтрации суспензии, полученной по окончании второй стадии с) осаждения, с целью получения геля оксида алюминия;
е) стадия сушки указанного геля оксида алюминия, полученного на стадии d), с целью получения порошка;
f) стадия тепловой обработки порошка, полученного по окончании стадии е), при температуре от 500 до 1000°С в течение периода времени от 2 до 10 часов, в присутствии или в отсутствие потока воздуха, содержащего до 60% об. воды, с целью получения обожженного пористого оксида алюминия;
g) стадия совместного растирания полученного обожженного пористого оксида алюминия и раствора, по меньшей мере, одного прекурсора металла активной фазы с целью получения пасты;
h) стадия формования полученной пасты;
i) стадия сушки формованной пасты при температуре, меньшей или равной 200°С, с целью получения высушенного катализатора;
j) возможная стадия тепловой обработки высушенного катализатора при температуре, лежащей в диапазоне от 200 до 1000°С, в присутствии или в отсутствие воды.
Доля выхода первой стадии а) осаждения, преимущественно, составляет от 6 до 12%.
Доля выхода первой стадии а) осаждения, предпочтительно, составляет от 7 до 11%.
Кислый прекурсор, преимущественно, выбирают из сульфата алюминия, хлорида алюминия и нитрата алюминия, предпочтительно, это сульфат алюминия.
Основный прекурсор, преимущественно, выбирают из алюмината натрия и алюмината калия, предпочтительно, этот алюминат натрия.
Предпочтительно, на стадиях а), b), с) водная реакционная среда представляет собой воду, и указанные стадии проводят при перемешивании в отсутствие органических добавок.
Изобретение также относится к мезопористому и макропористому катализатору гидроконверсии, содержащему:
- матрицу из оксида, главным образом, обожженного оксида алюминия;
- активную фазу гидро-дегидрогенизации, включающую, по меньшей мере, один металл группы VIB Периодической системы элементов, в некоторых случаях, по меньшей мере, один металл группы VIII Периодической системы элементов, в некоторых случаях, фосфор,
при этом, указанная активная фаза, по меньшей мере частично, смешана путем растирания с указанной матрицей, главным образом, из обожженного оксида алюминия,
при этом, указанный катализатор характеризуется удельной площадью поверхности SВЕТ более 100 м2/г, медианным по объему диаметром мезопор от 12 до 25 нм, включая границы диапазона, медианным по объему диаметром макропор от 50 до 250 нм, включая границы диапазона, объемом мезопор, измеренным методом ртутной порометрии, большим или равным 0,65 мл/г, и общим объемом пор, измеренным методом ртутной порометрии, большим или равным 0,75 мл/г.
Предпочтительно, указанный катализатор характеризуется медианным по объему диаметром мезопор, измеренным методом ртутной порометрии, от 13 до 17, включая границы диапазона.
Предпочтительно, указанный катализатор характеризуется объемом макропор от 15 до 35% общего объема пор.
Предпочтительно, объем мезопор составляет от 0,65 до 0,75 мл/г.
Предпочтительно, катализатор не содержит микропор.
Предпочтительно, содержание металла группы VIB составляет от 2 до 10% вес. триоксида, по меньшей мере, одного металла группы VIB относительно общей массы катализатора, содержание металла группы VIII составляет от 0,0 до 3,6% вес. оксида, по меньшей мере, одного металла группы VIII относительно общей массы катализатора, содержание элементарного фосфора составляет от 0 до 5% вес. пентоксида фосфора относительно общей массы катализатора.
Активная фаза гидро-дегидрогенизации может быть образована молибденом (Мо) или никелем и молибденом (NiMo) или кобальтом и молибденом (СоМо).
Активная фаза гидро-дегидрогенизации, предпочтительно, также содержит фосфор.
Преимущественно, активная фаза гидро-дегидрогенизации полностью введена путем совместного растирания.
В одном из вариантов осуществления изобретения, часть активной фазы гидро-дегидрогенизации может быть введена путем пропитки оксидной матрицы, главным образом, оксида алюминия.
Изобретение также относится к способу гидроочистки тяжелого углеводородного сырья, выбранного из атмосферных остатков, остатков прямой вакуумной перегонки, деасфальтированной нефти, остатков осуществления способов конверсии, например, образующихся при коксовании нефти, гидроконверсии в неподвижном слое, кипящем слое или подвижном слое, взятых отдельно или в смеси, при этом, указанный способ гидроочистки включает приведение указанного сырья в контакт с водородом и катализатором, допускающим производство согласно изобретению, или катализатором, описанным выше.
Способ может быть реализован, отчасти, в кипящем слое при температуре от 320 до 450°С, парциальном давлении водорода от 3 МПа до 30 МПа, пространственной скорости от 0,1 до 10 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья от 100 до 3000 м3 при н.у. на м3.
Способ может быть реализован, отчасти, в неподвижном слое при температуре от 320 до 450°С, парциальном давлении водорода от 3 МПа до 30 МПа, пространственной скорости от 0,05 до 5 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья от 200 до 5000 м3 при н.у. на м3.
Указанный способ может представлять собой способ гидроочистки тяжелого углеводородного сырья типа остатков в неподвижном слое, включающий, по меньшей мере:
а) стадию гидродеметаллизации
b) стадию гидрообессеривания
и указанный катализатор используют, по меньшей мере, на одной из указанных стадий а) и b).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Автором настоящей заявки обнаружено, что совместное растирание оксида алюминия, полученного из особого геля, изготовленного согласно способу производства, описываемому далее, с металлической композицией, содержащей, по меньшей мере, один элемент группы VIB, в некоторых случаях, по меньшей мере, один элемент группы VIII и, в некоторых случаях, элементарный фосфор, позволяет получить катализатор, характеризующийся одновременно увеличенным общим объемом пор (большим или равным 0,75 мл/г), увеличенными объемом мезопор (большим или равным 0,65 мл/г), увеличенным медианным диаметром мезопор (от 12 до 25 нм), медианным диаметром макропор от 50 до 250 нм, а также параметрами активной фазы, благоприятными для гидроочистки.
С другой стороны, кроме уменьшения количества стадий и, следовательно, стоимости производства, значение совместного растирания по сравнению с пропиткой заключается в том, что исключается любой риск частичного закупоривания пор подложки во время введения активной фазы и, следовательно, появления проблемы ограничений.
Помимо возможности производства при меньших затратах, подобный катализатор обладает значительным преимуществом в отношении гидродеметаллизации по сравнению с другими катализаторами, полученными совместным растиранием, предшествующего уровня техники, таким образом, для достижения той же степени конверсии соединений металлов требуется более низкая рабочая температура. В том числе, при применении указанного катализатора, соответствующего изобретению, в начале технологической цепочки с неподвижным слоем катализатора, либо установки гидродеметаллизации (HDM), либо, затем, установки гидрообессеривания (HDS), общие показатели работы технологической цепочки улучшаются.
Терминология и методы определения параметров
Во всем настоящем документе степень диспергируемости определяется как весовое процентное содержание пептизированного твердого или гелеобразного оксида алюминия, который может быть диспергирован путем центрифугирования в трубке из полипропилена при 3600G (перегрузка) за 3 мин.
Катализатор настоящего изобретения характеризуется определенным распределением пор по размерам, в котором объемы макропор и мезопор измерены по проникновению ртути, а объем микропор измерен по адсорбции азота.
Под «макропорами» понимаются поры с отверстием более 50 нм.
Под «мезопорами» понимаются поры с отверстием в диапазоне от 2 нм до 50 нм, включая границы диапазона.
Под «микропорами» понимаются поры с отверстием строго менее 2 нм.
В последующем описании изобретения под удельной площадью поверхности понимается величина удельной поверхности, определенная методом ВЕТ по адсорбции азота в соответствии со стандартом ASTM D 3663-78, основанном на методе Брунауэра-Эмметта-Теллера, описанном в журнале «The Journal of American Society», 60, 309, (1938).
В нижеследующем описании изобретения под общим объемом пор оксида алюминия или матрицы, преимущественно, из оксида алюминия или катализатора понимается объем, измеренный по проникновению ртути при помощи ртутного порозиметра согласно стандарту ASTM D4284-83 при максимальном давлении 4000 бар (400 МПа), поверхностном натяжении 484 дин/см и угле смачивания 140°. Угол смачивания выбран равным 140° в соответствии с рекомендациями работы авторов Jean Charpin и Bernar Rasneur «Techniques de lʹingénieur, traité analyse et caractérisation» Р 1050-5.
Для достижения большей точности, величина общего порового объема в мл/г, приводимая в последующем тексте, соответствует величине общего объема ртути (общий объем пор, измеренный по проникновению ртути при помощи порозиметра) в мл/г, полученной для одного образца, минус величина общего объема ртути в мл/г, полученная для того же образца при давлении, соответствующем 30 psi (примерно, 0,2 МПа).
Объем макропор и мезопор измеряется при помощи порозиметра по проникновению ртути согласно стандарту ASTM D4284-83 при максимальном давлении 4000 бар (400 МПа), поверхностном натяжении 484 дин/см и угле смачивания 140°.
Исходную величину, начиная с которой ртуть заполняет все межзерновые пустоты, устанавливают равной 0,2 МПа и считают, что затем ртуть проникает во все поры образца.
Объем макропор катализатора определяется как общий объем ртути, введенный под давлением от 0,2 МПа до 30 МПа, соответствующий объему, удерживаемому в порах диаметром, явно превышающим 50 нм.
Объем мезопор катализатора определяется как общий объем ртути, введенный под давлением от 30 МПа до 400 МПа, соответствующий объему, удерживаемому в порах диаметром от 2 до 50 нм.
Объем микропор измеряют по адсорбции азота. Количественный анализ микропористости осуществляют в соответствии с методом «t» (метод Lippens-De Boer, 1965), который предусматривает преобразование исходной изотермы адсорбции, как описано в работе F.Rouquérol, J.Rouquérol, K.Sing «Adsorption by powders and porous solids. Principles, methodology and applications», Academic Press, 1999.
Точно так же, медианный диаметр мезопор определяется как диаметр, для которого все поры, размер которых меньше этого диаметра, составляют 50% общего объема мезопор, измеренного методом ртутной порометрии.
Точно так же, медианный диаметр макропор определяется как диаметр, для которого все поры, размер которых меньше этого диаметра, составляют 50% общего объема макропор, измеренного методом ртутной порометрии.
Далее группы химических элементов указаны в соответствии с классификацией CAS (CRC Handbook of Chemistry and Physics, éditeur CRC press, rédacteur en chef D.R. Lide, 81éme édition, 2000-2001). Например, группа VIII согласно классификации CAS соответствует металлам групп 8, 9 и 10 по новой классификации IUPAC.
Общее описание катализатора
Изобретение направлено на катализатор гидроконверсии с активной фазой, введенной путем совместного растирания, содержащий, по меньшей мере, один металл группы VIB Периодической системы элементов, в некоторых случаях, по меньшей мере, один металл группы VIII Периодической системы элементов, в некоторых случаях, фосфор и оксидную матрицу, в основном, из оксида алюминия, на способ его производства и на его использование в процессе гидроочистки тяжелого углеводородного сырья, такого как нефтяные остатки (атмосферные или вакуумные).
Соответствующий изобретению катализатор представляет собой матрицу, по большей части образованную обожженным пористым жаропрочным оксидом, внутри которой распределены металлы активной фазы.
Изобретение также направлено на способ производства катализатора, который осуществляют путем совместного растирания особого оксида алюминия и раствора металлов, композиция которого согласуется с целевым содержанием металлов в готовом катализаторе.
Параметры геля, используемого при получении оксида алюминия, а также достигаемые структурные свойства и состав активной фазы, придают катализатору, соответствующему изобретению, его особые свойства.
Металлы группы VIB, преимущественно, выбраны из молибдена и вольфрама, предпочтительно, указанный металл группы VIB представляет собой молибден.
Металлы группы VIII, преимущественно, выбраны из железа, никеля или кобальта, предпочтительны никель или кобальт или сочетание двух этих металлов.
Соответствующие количества металла группы VIB и металла группы VIII, преимущественно, таковы, что атомное отношение металла(ов) группы VIII к металлу(ам) группы VIB (VIII:VIB) лежит в диапазоне от 0,0:1 до 0,7:1, предпочтительно, от 0,1:1 до 0,6:1, более предпочтительно, от 0,2:1 до 0,5:1. Это отношение может быть более точно подобрано в соответствии с типом сырья и используемым процессом.
Соответствующие количества металла группы VIB и фосфора таковы, что атомное отношение фосфора к металлу(ам) группы VIB (Р/VIB) лежит в диапазоне от 0,2:1 до 1,0:1, предпочтительно, от 0,4:1 до 0,9:1, более предпочтительно, от 0,5:1,0 до 0,85:1.
Содержание металла группы VIB, преимущественно, составляет от 2 до 10% вес. триоксида, по меньшей мере, одного металла группы VIB относительно общей массы катализатора, предпочтительно, от 3 до 8%, более предпочтительно, от 4 до 7% вес.
Содержание металла группы VIII, преимущественно, составляет от 0,0 до 3,6% вес. оксида, по меньшей мере, одного металла группы VIII относительно общей массы катализатора, предпочтительно, от 0,4 до 2,5%, более предпочтительно, от 0,7 до 1,8% вес.
Содержание элементарного фосфора, преимущественно, составляет от 0,0 до 5% вес. пентоксида фосфора относительно общей массы катализатора, предпочтительно, от 0,6 до 3,5% вес., более предпочтительно, от 1,0 до 3,0% вес.
Матрица, преимущественно, из обожженного оксида алюминия указанного катализатора, соответствующего изобретению, содержит оксид алюминия в количестве, большем или равном 90%, и оксид кремния в количестве, самое большее, 10% вес. в эквиваленте SiO2 относительно массы матрицы, предпочтительно, содержание оксида кремния составляет менее 5% вес., более предпочтительно, менее 2% вес.
Оксид кремния может быть введен любым способом, известным специалистам, во время синтеза геля оксида алюминия или во время совместного растирания.
Еще более предпочтительно, матрица из оксида алюминия не содержит ничего другого, кроме оксида алюминия.
Указанный катализатор с активной фазой, введенной путем совместного растирания, соответствующий изобретению, вообще, имеет любую форму, известную специалистам. Предпочтительно, он образован экструдатами диаметром, вообще, от 0,5 до 10 мм, предпочтительно, от 0,8 до 3,2 мм, более предпочтительно, от 1,0 до 2,5 мм. Он может, преимущественно, иметь форму цилиндрических, трехдольчатых или четырехдольчатых экструдатов. Предпочтительно, его форма трехдольчатая или четырехдольчатая. Форма долей может быть выбрана в соответствии со всеми способами, известными из предшествующего уровня техники.
Полученный путем совместного растирания катализатор, соответствующий изобретению, обладает особыми структурными свойствами. Катализатор, соответствующий изобретению, характеризуется общим объемом пор (VPT), по меньшей мере, 0,75 мл/г, предпочтительно, по меньшей мере, 0,80 мл/г. В предпочтительном варианте осуществления изобретения, катализатор характеризуется общим объемом пор от 0,80 до 1,05 мл/г.
Катализатор, используемый согласно изобретению, преимущественно, характеризуется объемом макропор, Vmacro или V50нм, определяемым как объем пор диаметром более 50 нм, от 15 до 35% общего объема пор, предпочтительно, от 15 до 30% общего объема пор. В предпочтительном варианте осуществления изобретения, объем макропор составляет от 20 до 30% общего объема пор.
Объем мезопор (Vméso) катализатора составляет, по меньшей мере, 0,65 мл/г, предпочтительно, от 0,65 до 0,80 мл/г. В предпочтительном варианте осуществления изобретения, объем мезопор катализатора составляет от 0,65 до 0,75 мл/г.
Медианный диаметр мезопор (Dpméso) составляет от 12 нм до 25 нм, включая границы диапазона, предпочтительно, от 12 нм до 25 нм, включая границы диапазона. Особенно предпочтительно, медианный диаметр мезопор составляет от 13 нм до 17 нм, включая границы диапазона.
Преимущественно, медианный диаметр макропор (Dpmacro) катализатора составляет от 50 до 250 нм, предпочтительно, от 80 до 200 нм, более предпочтительно, от 80 до 150 нм. Особенно предпочтительно, медианный диаметр макропор составляет от 90 до 130 нм.
Катализатор, соответствующий изобретению, характеризуется удельной площадью поверхности по ВЕТ (SВЕТ), по меньшей мере, 100 м2/г, предпочтительно, по меньшей мере, 120 м2/г, более предпочтительно, от 150 до 250 м2/г.
Предпочтительно, катализатор характеризуется небольшой микропористостью, особенно предпочтительно, при помощи азотного порозиметра никакая микропористость не обнаруживается.
Если нужно, можно увеличить содержание металла посредством введения второй части активной фазы путем пропитки катализатора, уже прошедшего стадию совместного растирания с первой частью активной фазы.
Важно отметить, что катализатор, соответствующий изобретению, структурно отличается от катализатора, полученного путем простой пропитки прекурсором металла подложки из оксида алюминия, в которой оксид алюминия образует подложку, а активная фаза введена в поры подложки. Без связи с какой-либо определенной теорией полагают, что способ производства катализатора, соответствующего изобретению, путем совместного растирания особого пористого оксида алюминия с одним или несколькими прекурсорами металлов позволяет получить композиционный материал, в котором металлы и оксид алюминия тщательно перемешаны и образуют, таким образом, саму структуру катализатора с некоторой пористостью и содержанием активной фазы для требуемых реакций.
Способ производства катализатора согласно изобретению
Основные стадии
Катализатор, соответствующий изобретению, произведен путем совместного растирания обожженного пористого оксида алюминия, полученного из особого геля оксида алюминия, и прекурсора или прекурсоров металлов.
Способ производства катализатора, соответствующего изобретению, включает следующие стадии:
а)- е) Синтез геля-прекурсора пористого оксида;
f) Тепловая обработка порошка, полученного по окончании стадии е);
g) Совместное растирание полученного пористого оксида и, по меньшей мере, одного прекурсора активной фазы;
h) Формование пасты, полученной путем растирания, например, путем экструзии;
i) Сушка полученной формованной пасты;
j) Необязательная тепловая обработка (предпочтительно, в сухом воздухе).
Твердый материал, полученный по окончании стадий а)-f), подвергают обработке на стадии g) совместного растирания. Затем его формуют на стадии h), после чего он может быть просто высушен при температуре, меньшей или равной 200°С, (стадия i) или высушен, а затем снова подвергнут тепловой обработке путем обжига на необязательной стадии j).
Перед использованием в процессе гидроочистки, катализатор обычно подвергают обработке на последней стадии сульфирования. Эта стадия заключается в активации катализатора путем преобразования, по меньшей мере частично, оксидной фазы в восстановительную серосодержащую среду. Такая активация путем сульфирования известна специалистам и может быть осуществлена любым способом, уже известным и описанным в литературе. Один из классических способов сульфирования, хорошо известный специалистам, состоит в нагревании смеси твердых материалов в потоке, состоящем из смеси водорода и сероводорода, или в потоке, состоящем из смеси водорода и углеводородов, содержащих соединения серы, при температуре от 150 до 800°С, предпочтительно, от 250 до 600°С, обычно, в реакционной зоне с поперечным слоем.
Подробное описание способа производства
Соответствующий изобретению катализатор с активной фазой, полученный путем совместного растирания, получают из особого геля оксида алюминия, который перед совместным растиранием с активной фазой сушат и обжигают, затем катализатор формуют.
Стадии производства геля оксида алюминия, осуществляемые в ходе производства катализатора, соответствующего изобретению, подробно описаны далее.
В соответствии с изобретением, указанный способ производства геля оксида алюминия включает первую стадию а) осаждения, стадию b) нагревания, вторую стадию c) осаждения, стадию d) фильтрации, стадию е) сушки.
Доля выхода каждой из стадий осаждения определяется как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной первой или второй стадии осаждения, к общему количеству оксида алюминия, в эквиваленте Al2O3, образовавшемуся по окончании двух стадий осаждения и, вообще, по окончании стадий производства геля оксида алюминия, в частности, по окончании стадии с) способа производства, соответствующего изобретению.
Стадия а) - первая стадия осаждения
Данная стадия состоит в приведении в контакт в водной реакционной среде, по меньшей мере, одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и, по меньшей мере, одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, при этом, по меньшей мере, один из прекурсоров - основный или кислый - содержит алюминий, относительный расход кислого и основного прекурсоров выбран так, чтобы обеспечить рН реакционной среды от 8,5 до 10,5, и расход прекурсора или прекурсоров - кислого и основного, содержащих алюминий, выбран так, чтобы доля выхода первой стадии составляла от 5 до 13%, при этом, доля выхода определяется как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной стадии а) осаждения, к общему количеству оксида алюминия, в эквиваленте Al2O3, образовавшемуся по окончании стадии с), при этом, указанную стадию осуществляют при температуре, лежащей в диапазоне от 20 до 90°С, в течение периода времени от 2 минут до 30 минут.
Необходимо, чтобы в смеси в водной реакционной среде, по меньшей мере, одного основного прекурсора и, по меньшей мере, одного кислого прекурсора, по меньшей мере, один из прекурсоров - кислый или основной, содержал алюминий. Также возможно, чтобы, по меньшей мере, два прекурсора - основный и кислый - содержали алюминий.
Основными прекурсорами, содержащими алюминий, являются алюминат натрия и алюминат калия. Предпочтительным основным прекурсором является алюминат натрия.
Кислыми прекурсорами, содержащими алюминий, являются сульфат алюминия, хлорид алюминия и нитрат алюминия. Предпочтительным кислым прекурсором является сульфат алюминия.
Предпочтительно, водной реакционной средой является вода.
Предпочтительно, указанную стадию а) проводят при перемешивании.
Предпочтительно, указанную стадию а) осуществляют в отсутствие органических добавок.
Кислый и основный прекурсоры, которые содержат или не содержат алюминий, смешивают, предпочтительно в растворе, в водной реакционной среде в таком соотношении, чтобы рН получаемой суспензии лежал в диапазоне от 8,5 до 10,5.
В соответствии с изобретением, кислые прекурсоры оксида алюминия и основные прекурсоры оксида алюминия могут быть использованы на стадии осаждения по отдельности или в смеси.
В соответствии с изобретением относительный расход кислого и основного прекурсоров, которые содержат или не содержат алюминий, подбирают так, чтобы рН водной реакционной среды составлял от 8,5 до 10,5.
В предпочтительном случае, в котором основный и кислый прекурсоры представляют собой, соответственно, алюминат натрия и сульфат алюминия, массовое отношение указанного основного прекурсора к указанному кислому прекурсору, преимущественно, лежит в диапазоне от 1,60 до 2,05.
Для других основных и кислых прекурсоров, которые содержат или не содержат алюминий, массовое отношение основание/кислота устанавливается по кривой нейтрализации основания кислотой. Такая кривая без труда может быть получена специалистом.
Предпочтительно, указанную стадию а) осаждения проводят при рН в диапазоне от 8,5 до 10,0, наиболее предпочтительно, от 8,7 до 9,9.
В соответствии с изобретением, указанную стадию а) осаждения проводят при температуре, лежащей в диапазоне от 20 до 90°С, предпочтительно, от 20 до 70°С, более предпочтительно, от 30 до 50°С.
В соответствии с изобретением, указанную стадию а) осаждения проводят с течение периода времени от 2 до 30 минут, предпочтительно, от 5 до 20 минут, более предпочтительно, от 5 до 15 минут.
В соответствии с изобретением, доля выхода указанной первой стадии а) осаждения составляет от 5 до 13%, предпочтительно, от 6 до 12%, более предпочтительно, от 7 до 11%. Следовательно, кислый и основный прекурсоры, содержащие алюминий, должны быть поданы в количествах, позволяющих получить суспензию, содержащую необходимое количество оксида алюминия, в зависимости от требуемой конечной концентрации оксида алюминия. В частности, указанная стадия а) позволяет получить от 5 до 13% вес. оксида алюминия относительно общего количества оксида алюминия, в эквиваленте Al2O3, образовавшегося по окончании стадии с) данного способа производства.
Стадия b) - нагревание
В соответствии с изобретением, указанный способ производства включает стадию b) нагревания суспензии, полученной по окончании первой стадии а) осаждения.
Согласно изобретению, до осуществления второй стадии осаждения, между двумя стадиями осаждения проводят стадию нагревания суспензии, полученной по окончании стадии а) осаждения.
Указанную стадию нагревания суспензии, полученной по окончании стадии а), осуществляемую между указанной первой стадией а) осаждения и второй стадией с) осаждения, проводят при температуре, лежащей в диапазоне от 40 до 90°С, предпочтительно, от 40 до 80°С, более предпочтительно, от 40 до 70°С, наиболее предпочтительно, от 40 до 65°С.
Указанную стадию нагревания осуществляют в течение периода времени от 7 до 45 минут, предпочтительно, от 7 до 35 минут.
Указанную стадию нагревания, преимущественно, осуществляют любым способом нагревания, известным специалистам.
Стадия с) - вторая стадия осаждения
Согласно изобретению, указанный способ производства включает вторую стадию осаждения суспензии, полученной по окончании стадии b) нагревания, при этом, указанную вторую стадию проводят путем добавления в указанную суспензию, по меньшей мере, одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и, по меньшей мере, одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, при этом, по меньшей мере, один из прекурсоров - основный или кислый, содержит алюминий, относительный расход кислого и основного прекурсоров выбирают так, чтобы получить рН реакционной среды от 8,5 до 10,5, расход кислого и/или основного прекурсоров, содержащих алюминий, регулируют так, чтобы достичь доли выхода второй стадии от 87 до 95%, при этом, долю выхода определяют как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной второй стадии осаждения, к общему количеству оксида алюминия, в эквиваленте Al2O3, образовавшемуся по окончании стадии с) способа производства, при этом, указанную стадию проводят при температуре, лежащей в диапазоне от 40 до 90°С в течение периода времени от 2 минут до 50 минут.
Основный(ые) и кислый(ые) прекурсор(ы) добавляют на указанной второй стадии осаждения в водном растворе.
Как и на первой стадии а) осаждения, при добавлении в нагретую суспензию, по меньшей мере, основного прекурсора и, по меньшей мере, кислого прекурсора, требуется, чтобы, по меньшей мере, один из прекурсоров - основный или кислый - содержал алюминий. Также возможно, чтобы, по меньшей мере, два прекурсора, основный и кислый, содержали алюминий.
Основными прекурсорами, содержащими алюминий, являются алюминат натрия и алюминат калия. Предпочтительным основным прекурсором является алюминат натрия.
Кислыми прекурсорами, содержащими алюминий, являются сульфат алюминия, хлорид алюминия и нитрат алюминия. Предпочтительным кислым прекурсором является сульфат алюминия.
Предпочтительно, указанную вторую стадию осаждения проводят при перемешивании.
Предпочтительно, указанную вторую стадию осуществляют в отсутствие органических добавок.
Кислый и основный прекурсоры, которые содержат или не содержат алюминий, смешивают, предпочтительно в растворе, с суспензией в таком соотношении, чтобы рН получаемой суспензии лежал в диапазоне от 8,5 до 10,5.
Как и на стадии а) осаждения, относительный расход кислого и основного прекурсоров, которые содержат или не содержат алюминий, подбирают так, чтобы рН получаемой реакционной среды лежал в диапазоне от 8,5 до 10,5, предпочтительно, от 8,5 до 10, более предпочтительно, от 8,7 до 9,9.
В предпочтительном случае, в котором основный и кислый прекурсоры представляют собой, соответственно, алюминат натрия и сульфат алюминия, массовое отношение указанного основного прекурсора к указанному кислому прекурсору, преимущественно, лежит в диапазоне от 1,60 до 2,05.
Для других основных и кислых прекурсоров, которые содержат или не содержат алюминий, массовое отношение основание/кислота устанавливается по кривой нейтрализации основания кислотой. Такая кривая без труда может быть получена специалистом.
Прекурсоры оксида алюминия равномерно смешивают в количествах, позволяющих получить суспензию, содержащую необходимое количество оксида алюминия, в зависимости от заданной конечной концентрации оксида алюминия. В частности, указанная вторая стадия осаждения позволяет получать от 87 до 95% вес. оксида алюминия, в эквиваленте Al2O3, относительно общего количества оксида алюминия, образующегося по окончании двух стадий осаждения.
Как и на стадии а) осаждения, расход прекурсора или прекурсоров - кислого и основного, содержащих алюминий, регулируют так, чтобы получит долю выхода второй стадии от 87 до 95%, предпочтительно, от 88 до 94%, более предпочтительно, от 89 до 93%, при этом, доля выхода определяется как процентная доля оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной второй стадии осаждения, относительно общего количества алюминия, в эквиваленте Al2O3, образовавшегося по окончании стадии с) способа производства.
Таким образом, в зависимости от концентрации оксида алюминия, которой нужно достичь по завершении стадий осаждения, предпочтительно, от 20 до 100 г/л, рассчитывают количества алюминия, которые должны быть внесены кислым и/или основным прекурсорами, и расход прекурсоров регулируют в зависимости от концентрации алюминия в указанных добавляемых прекурсорах, от количества воды, добавляемой в реакционную среду, и от надлежащей доли выхода на каждой стадии осаждения.
Как и на стадии а) осаждения, расход кислого и/или основного прекурсоров, содержащих алюминий, зависит от размеров используемого реактора, а также от количества воды, добавляемой в реакционную среду.
Например, если используется реактор объемом 3 л, и если нужно получить 1 л суспензии оксида алюминия с конечной концентраций Al2O3 50 г/л, целевая доля выхода на первой стадии осаждения составляет 10%, 10% общего количества оксида алюминия должно быть внесено на стадии а) осаждения. Прекурсорами оксида алюминия являются алюминат натрия в концентрации 155 г/л в эквиваленте Al2O3, и сульфат алюминия в концентрации 102 г/л в эквиваленте Al2O3. рН осаждения на первой стадии устанавливают равным 9,5, на второй - 9. Количество воды, добавляемое в реактор составляет 620 мл.
На первой стадии а) осаждения, проводимой при 30°С в течение 8 минут, расход сульфата алюминия должен составлять 2,1 мл/мин, расход алюмината натрия - 2,6 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, следовательно, равно 1,91.
На второй стадии осаждения, проводимой при 70°С в течение 30 минут, расход сульфата алюминия должен составлять 5,2 мл/мин, расход алюмината натрия - 6,3 мл/мин. Таким образом, массовое отношение алюмината натрия к сульфату алюминия равно 1,84.
Предпочтительно, вторую стадию осаждения проводят при температуре, лежащей в диапазоне от 40 до 80°С, предпочтительно, от 45 до 70°С, наиболее предпочтительно, от 50 до 70°С.
Предпочтительно, вторую стадию осаждения осуществляют в течение периода времени от 5 до 45 минут, предпочтительно, от 7 до 40 минут.
Вообще, вторая стадия осаждения позволяет получить суспензию оксида алюминия с концентрацией Al2O3, лежащей в диапазоне от 20 до 100 г/л, предпочтительно, от 20 до 80 г/л, более предпочтительно, от 20 до 50 г/л.
Стадия d) - фильтрация
Способ производства оксида алюминия, соответствующий изобретению, также включает стадию фильтрации суспензии, полученной по окончании второй стадии с) осаждения. Указанную стадию фильтрации осуществляют согласно способам, известным специалистам.
Фильтруемость суспензии, полученной по окончании двух стадий осаждения, улучшена благодаря низкой диспергируемости получаемого геля оксида алюминия, поэтому возможно повышение производительности способа, соответствующего изобретению, а также выведение способа на промышленный уровень.
За указанной стадией фильтрации, преимущественно, следует, по меньшей мере, одна стадия промывки, предпочтительно, водой, предпочтительно, от одной до трех стадий промывки количеством воды, равным количеству отфильтрованного осадка.
Последовательность стадий а) первого осаждения, b) нагревания и с) второго осаждения и стадии d) фильтрации позволяет получить особый гель оксида алюминия, обладающий степенью диспергируемости менее 15%, предпочтительно, от 5 до 15%, более предпочтительно, от 6 до 14%, еще более предпочтительно, от 7 до 13%, наиболее предпочтительно, от 7 до 10% и размером кристаллитов от 1 до 35 нм, предпочтительно, от 2 до 35 нм.
Получаемый таким образом гель оксида алюминия также, преимущественно, характеризуется содержанием серы, измеренным рентгенофлуоресцентным методом, от 0,001% до 2% вес., предпочтительно, от 0,01% до 0,2% вес. и содержанием натрия, измеренным методом спектрометрии индуктивно связанной плазмы (IPC), от 0,001% до 2% вес., предпочтительно, от 0,01% до 0,1% вес., процентное содержание выражено относительно общей массы геля оксида алюминия.
В частности, гель оксида алюминия или бомит в порошкообразной форме, соответствующий изобретению, образован кристаллитами, размер которых, полученный по формуле Шеррера дифракции рентгеновского излучения по кристаллографическим осям (020) и (120), составляет, соответственно, от 2 до 20 нм и от 2 до 35 нм.
Предпочтительно, гель оксида алюминия, соответствующий изобретению, характеризуется размером кристаллитов по кристаллографической оси (020) от 1 до 15 нм и размером кристаллитов по кристаллографической оси (120) от 1 до 35 нм.
Рентгенодифракционный анализ геля оксида алюминия или бомита был проведен с использованием классического порошкового метода при помощи дифрактометра.
Формула Шеррера является формулой, используемой в рентгенодифракционном анализе порошков или поликристаллических образцов, которая устанавливает связь между шириной на середине высоты пиков дифракции и размером кристаллитов. Подробно она описана в следующем ссылочном документе: Appl. Cryst. (1978). 11, 102-113 Scherrer after sixty years: A survey and some new results in the determination of crystallite size, J. I. Langford and A. J. C. Wilson.
Низкая степень диспергируемости полученного таким образом геля позволяет упростить стадию формования указанного геля любым способом, известным специалистам, в частности способом экструзионного смешивания, способом гранулирования, так называемым, способом капельной коагуляции (oil drop согласно англо-американской терминологии).
Стадия е) -сушка геля оксида алюминия
В соответствии с изобретением, гель оксида алюминия, полученный по окончании второй стадии с) осаждения, сопровождаемой стадией d) фильтрации, сушат на стадии е) сушки с целью получения порошка, при этом, указанную стадию сушки осуществляют посредством сушки, например, сушки при температуре от 20 до 200°С, в течение периода времени от 8 ч до 15 ч или путем распылительной сушки или любым другим способом сушки, известным специалистам.
В том случае, когда указанную стадию е) сушки осуществляют путем распылительной сушки, массу, полученную по окончании второй стадии осаждения, сопровождаемой стадией фильтрации, снова суспендируют. Затем указанную суспензию распыляют мелкими каплями в вертикальной цилиндрической камере в контакте с потоком теплого воздуха с целью испарения воды согласно принципу, хорошо известному специалистам. Полученный порошок увлекается потоком теплого воздуха в циклон или в рукавный фильтр, где происходит отделение порошка от воздуха.
Предпочтительно, в том случае, когда указанная стадия е) сушки проводится способом распылительной сушки, распылительную сушку осуществляют в соответствии с протоколом, описанным в публикации Asep Bayu Dani Nandiyanto, Kikuo Okuyama, Advanced Powder Technology, 22, 1-19, 2011.
Стадия f) -тепловая обработка порошка, полученного по окончании стадии е)
В соответствии с изобретением, порошок, полученный по окончании стадии е) сушки, подвергают тепловой обработке на стадии f) при температуре, лежащей в диапазоне от 500 до 1000°С в течение периода времени от 2 до 10 ч в присутствии или в отсутствие потока воздуха, содержащего до 60% об. воды.
Предпочтительно, указанную стадию f) тепловой обработки проводят при температуре от 540 до 850°С.
Предпочтительно, указанную стадию f) тепловой обработки осуществляют в течение периода времени от 2 ч до 10 ч.
Указанная стадия f) тепловой обработки позволяет преобразовать бомит в конечный оксид алюминия.
Стадии тепловой обработки может предшествовать сушка при температуре от 50°С до 120°С, проводимая любым способом, известным специалистам.
В соответствии с изобретением, порошок, полученный по окончании стадии е) сушки, после стадии f) тепловой обработки подвергают совместному растиранию с одним или несколькими прекурсорами металлов активной фазы на стадии g) совместного растирания, позволяющей привести один или несколько растворов, содержащий активную фазу, в контакт с порошком, после чего получаемый материал формуют на стадии h), получая катализатор.
Стадия g) - совместное растирание
На этой стадии обожженный пористый оксид алюминия стадии f) растирают в присутствии активной фазы в форме раствора прекурсоров одного или нескольких металлов, выбранных из элементов группы VIB, в некоторых случаях, элементов группы VIII и, в некоторых случаях, фосфора.
Активная фаза вносится одним или несколькими растворами, содержащими, по меньшей мере, один металл группы VIB, в некоторых случаях, по меньшей мере, один металл группы VIII, в некоторых случаях, элементарный фосфор. Указанный(е) раствор(ы) может(могут) быть водным(и), состоять из органического растворителя или смеси воды и, по меньшей мере, одного органического растворителя (например, этанола или толуола). Предпочтительно, раствор является водно-органическим, еще более предпочтительно, водно-спиртовым. рН этого раствора может быть изменен путем добавления, в некоторых случаях, кислоты.
Среди соединений, которые могут быть введены в раствор в качестве источников элементов группы VIII, фигурируют, преимущественно, цитраты, оксалаты, карбонаты, гидроксикарбонаты, гидроксиды, фосфаты, сульфаты, алюминаты, молибдаты, вольфраматы, оксиды, нитраты, галогениды, например, хлориды, фториды, бромиды, ацетаты или любая смесь указанных соединений.
Что касается источников элементов группы VIB, которые хорошо известны специалистам, среди них, преимущественно, фигурируют, например, для молибдена и вольфрама: оксиды, гидроксиды, молибденовые и вольфрамовые кислоты и их соли, в частности, соли аммония, гептамолибдат аммония, вольфрамат аммония, фосфорномолибденовая кислота, фосфорновольфрамовая кислота и их соли. Предпочтительно, используют оксиды или соли аммония, такие как молибдат аммония, гептамолибдат аммония или вольфрамат аммония.
Предпочтительным источником фосфора является ортофосфорная кислота, а также ее соли и сложные эфиры, такие как щелочные фосфаты, фосфат аммония, фосфат галлия или алкилфосфаты. Преимущественно, могут быть использованы фосфористые кислоты, например, гипофосфорная кислота, фосфорномолибденовая кислота и ее соли, фосфорновольфрамовая кислота и ее соли.
Какая-либо добавка, например, хелирующий агент органической природы, преимущественно, может быть введена в раствор, если специалист сочтет это необходимым.
Любой другой элемент, например, оксид кремния в форме раствора или эмульсии силикатного прекурсора, может быть введен в смесительный резервуар на этой стадии.
Совместное растирание, преимущественно, проводят в смесительном устройстве, например, мешалке типа Brabender, хорошо известной специалистам. Пористый оксид алюминия в форме обожженного порошка, полученный на стадии f), и, в некоторых случаях, один или несколько других элементов помещают в смесительный резервуар. Затем раствор прекурсоров металлов, например, никеля и молибдена, и, в некоторых случаях, пермутированную воду добавляют при помощи шприца или другого средства за несколько минут, обычно, около 2 минут при заданной скорости перемешивания. После получения пасты перемешивание может продолжаться еще несколько минут, например, около 15 минут при 50 об/мин.
Стадия h) - формование
Пасту, полученную по окончании стадии g) совместного растирания, затем формуют любым способом, известным специалистам, например, экструзионного формования, таблетирования, способом капельной коагуляции, способом гранулирования на поворотном столе.
Предпочтительно, указанную подложку, используемую согласно изобретению, формуют путем экструдирования с получением экструдатов диаметром, вообще, от 0,5 до 10 мм, предпочтительно, от 0,8 до 3,2 мм. В предпочтительном варианте осуществления изобретения, она образована трехдольчатыми или четырехдольчатыми экструдатами размером от 1,0 до 2,5 мм в диаметре.
Особенно предпочтительно, указанную стадию g) совместного растирания и указанную стадию h) формования объединяют в одну стадию экструзионного перемешивания. В этом случае, паста, полученная по окончании перемешивания, может быть подана в экструдер с фильерой, имеющей необходимый диаметр, обычно, от 0,5 до 10 мм.
Стадия i) - сушка формованной пасты
В соответствии с изобретением, катализатор, полученный по окончании стадии g) совместного растирания и h) формования подвергают сушке i) при температуре, меньшей или равной 200°С, предпочтительно, менее 150°С согласно любому способу, известному специалистам, в течение периода времени, обычно, от 2 до 12 ч.
Стадия j) - тепловая или гидротермическая обработка
Высушенный таким образом катализатор затем может быть подвергнут дополнительной тепловой или гидротермической обработке на стадии j) при температуре от 200 до 1000°С, предпочтительно, от 300 до 800°С, еще более предпочтительно, от 350 до 550°С в течение периода времени от 2 до 10 ч, в присутствии или в отсутствие потока воздуха, содержащего до 60% об. воды. Может быть осуществлено множество объединенных циклов тепловой или гидротермической обработки.
В том случае, когда катализатор не проходит дополнительную стадию тепловой или гидротермической обработки, катализатор может быть, преимущественно, только высушен на стадии i).
В том случае, когда желательно добавить воду, контакт с паром может происходить при атмосферном давлении (пропаривание) или при повышенном давлении (автоклавная обработка). В случае пропаривания содержание воды, предпочтительно, составляет от 150 до 900 граммов на килограмм сухого воздуха, более предпочтительно, от 250 до 650 граммов на килограмм сухого воздуха.
Согласно изобретению, может предусматриваться введение всех или части перечисленных металлов во время совместного растирания раствора(ов) металлов с обожженным пористым оксидом алюминия.
В одном из вариантов осуществления изобретения, чтобы увеличить общее содержание активной фазы в катализаторе, приготовленном путем совместного растирания, часть металлов может быть введена путем пропитки указанного катализатора по окончании стадии i) или j) любым способом, известным специалистам, чаще всего, это пропитка сухим способом.
В другом варианте осуществления изобретения, всю металлическую фазу вводят во время производства путем совместного растирания пористого оксида алюминия, таким образом, в какой-либо дополнительной стадии пропитки нет необходимости. Предпочтительно, активную фазу катализатора полностью вводят внутрь обожженного пористого оксида алюминия путем совместного растирания.
Описание способа использования катализатора, соответствующего изобретению
Катализатор, соответствующий изобретению, может быть использован в способах гидроочистки, позволяющих обрабатывать тяжелое углеводородное сырье, содержащее серные и металлические примеси. Задача, решаемая путем использования катализаторов настоящего изобретения, состоит в улучшении параметров, в частности, при гидродеметаллизации и гидрообессеривании, при одновременном упрощении их производства по сравнению с катализаторами предшествующего уровня техники. Катализатор, соответствующий изобретению, позволяет улучшить параметры гидродеметаллизации и гидродеасфальтирования по сравнению с обычными катализаторами и, при этом, обладает значительной устойчивостью во времени.
Вообще, способы гидроочистки, позволяющие обрабатывать тяжелое углеводородное сырье, содержащее серные и металлические примеси, осуществляются при температуре от 320 до 450°С, при парциальном давлении водорода от 3 МПа до 30 МПа, пространственной скорости, преимущественно, от 0,05 до 10 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья, преимущественно, от 100 до 5000 м3 при н.у. на м3.
Сырье
Сырье, обрабатываемое способом, соответствующим изобретению, преимущественно, выбрано из атмосферных остатков, вакуумных остатков прямой перегонки, деасфальтированной нефти, остатков осуществления способов конверсии, например, образующихся при коксовании нефти, гидроконверсии в неподвижном слое, кипящем слое или подвижном слое, взятых отдельно или в смеси. Это сырье, преимущественно, может быть использовано без изменения или дополнительно разбавлено углеводородной фракцией или смесью углеводородных фракций, которые могут быть выбраны из продуктов процесса FCC, легкого рециклового масла (light cycle oil, LCO согласно англо-саксонской терминологии), тяжелого рециклового масла (heavy cycle gas oil, HCO согласно англо-саксонской терминологии), отфильтрованного масла (decanted oil, DO согласно англо-саксонской терминологии), суспензии, или могут быть получены при перегонке газойлевых фракций, в том числе, полученных при вакуумной перегонке (vacuum gas oil, VGO согласно англо-саксонской терминологии). Тяжелое сырье, преимущественно, также может содержать фракции, полученные способом ожижения каменного угля, ароматические фракции или любые другие углеводородные фракции.
Указанное тяжелое сырье, как правило, содержит более 1% вес. молекул, характеризующихся температурой кипения более 500°С, Ni+V в количестве более 1 вес. части на миллион, предпочтительно, более 20 вес. частей на миллион, наиболее предпочтительно, более 50 вес. частей на миллион, асфальтены, осаждающиеся в гептане, в количестве более 0,05% вес., предпочтительно, более 1% вес., более предпочтительно, более 2% вес.
Тяжелое сырье, преимущественно, также может быть смешано с каменным углем в порошкообразной форме, такую смесь, как правило, называют суспензией. Такое сырье, преимущественно, может представлять собой побочные продукты конверсии угля, снова смешанные со свежим углем. Содержание угля в тяжелом сырье, как правило и предпочтительно, составляет 1/4 (масло/уголь) и, преимущественно, может изменяться в широком диапазоне от 0,1 до 1. Уголь может содержать лигнит, представлять собой полубитуминозный уголь или битуминозный уголь. Для использования изобретения подходит любой другой тип угля как в реакторах с неподвижным слоем катализатора, так и в реакторах с кипящим слоем.
Использование катализатора, соответствующего изобретению
В соответствии с изобретением, катализатор с активной фазой, введенной путем совместного растирания, предпочтительно, используют в первых слоях катализатора способов, включающих применение последовательно, по меньшей мере, одной стадии гидродеметаллизации и, по меньшей мере, одной стадии гидрообессеривания. Способ, соответствующий изобретению, преимущественно, осуществляют в одном - десяти последовательных реакторах, при этом, катализатор или катализаторы, соответствующие изобретению, преимущественно, могут быть загружены в один или несколько реакторов и/или во все или часть реакторов.
В предпочтительном варианте осуществления изобретения, взаимозаменяемые реакторы, то есть, реакторы, работающие по очереди, в которых, предпочтительно, могут быть использованы катализаторы гидродеметаллизации, соответствующие изобретению, могут быть применены по потоку в начале установки. В этом предпочтительном варианте осуществления изобретения за взаимозаменяемыми реакторами последовательно установлены реакторы, в которых используются катализаторы гидрообессеривания, которые могут быть произведены любым способом, известным специалистам.
В особенно предпочтительном варианте осуществления изобретения, в начале установки используется два взаимозаменяемых реактора, преимущественно, для гидродеметаллизации, в которых размещен один или несколько катализаторов, соответствующих изобретению. За ними могут следовать, преимущественно, от одного до четырех последовательных реакторов, преимущественно, предназначенных для гидрообессеривания.
Способ, соответствующий изобретению, преимущественно, может быть осуществлен в неподвижном слое катализатора с целью удаления металлов и серы и снижения средней температуры кипения углеводородов. В том случае, когда способ, соответствующий изобретению, осуществляют в неподвижном слое катализатора, его осуществляют при температуре, преимущественно, от 320°С до 450°С, предпочтительно, от 350 до 410°С, парциальном давлении водорода от 3 МПа до 30 МПа, предпочтительно, от 10 до 20 МПа, пространственной скорости, преимущественно, от 0,05 до 5 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья, преимущественно, от 200 до 5000 м3 при н.у. на м3, предпочтительно, от 500 до 1500 м3 при н.у. на м3.
Способ, соответствующий изобретению, также, преимущественно, может быть применен, отчасти, в кипящем слое с тем же сырьем. В том случае, когда способ, соответствующий изобретению, осуществляют в кипящем слое, катализатор, преимущественно, используют при температуре от 320°С до 450°С, парциальном давлении водорода, преимущественно, от 3 МПа до 30 МПа, предпочтительно, от 10 до 20 МПа, пространственной скорости, преимущественно, от 0,1 до 10 объемов сырья на объем катализатора в час, предпочтительно, от 0,5 до 2 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья, преимущественно, от 100 до 3000 м3 при н.у. на м3, предпочтительно, 200 до 1200 м3 при н.у. на м3.
Согласно предпочтительному варианту осуществления, способ настоящего изобретения осуществляют в неподвижном слое.
Перед использованием в способе, соответствующем изобретению, катализаторы настоящего изобретения, предпочтительно, подвергают обработке путем сульфирования, позволяющей преобразовать, по меньшей мере частично, металлические соединения в сульфиды до их приведения в контакт с подлежащим обработке сырьем. Такая активация путем сульфирования хорошо известна специалистам и может быть осуществлена любым способом, уже известным и описанным в литературе. Один из классических способов сульфирования, хорошо известный специалистам, состоит в нагревании смеси твердых материалов в потоке, состоящем из смеси водорода и сероводорода, или в потоке, состоящем из смеси водорода и углеводородов, содержащих соединения серы, при температуре от 150 до 800°С, предпочтительно, от 250 до 600°С, обычно, в реакционной зоне с поперечным слоем.
Обработка путем сульфирования может быть осуществлена в другом месте (до загрузки катализатора в реактор гидроочистки/гидроконверсии) или на месте посредством органического серосодержащего прекурсора H2S, например, DMDS (диметилдисульфида).
Приведенные далее примеры поясняют изобретение, тем не менее, никак не ограничивая его объем.
ПРИМЕРЫ
Пример 1. Приготовление растворов металлов А, В
Растворы А и В, используемые для приготовления катализаторов А1, В1, А2, А3, приготовили путем растворения в воде следующих прекурсоров активных фаз: МоО3, Ni(ОН)2, Н3РО4. Все эти прекурсоры получены от Sigma-Aldrich®. Концентрации веществ в различных растворах приведены в нижеследующей таблице.
Таблица 1. Молярная концентрация приготовленных водных растворов (моль/л)
Пример 2. Приготовление катализаторов А1, В1 путем совместного растирания в соответствии с изобретением
Синтез оксида алюминия Al(А1) согласно изобретению проводили в реакторе объемом 5 л за 3 стадии.
Использовали следующие концентрации прекурсоров: сульфата алюминия Al2(SO4)3 102 г/л в эквиваленте Al2O3 и алюмината натрия NaAlOO 155 г/л в эквиваленте Al2O3.
Оксид алюминия Al(А1), соответствующий изобретению, произвели согласно следующим стадиям:
а) Первая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 30°С и рН=9,1 за 8 мин; доля выхода 10%. Доля выхода соответствует доле оксида алюминия, образовавшегося на первой стадии, если конечная концентрация оксида алюминия установлена равной 45 г/л. Если объем реактора равен 5 л, и нужно получить 4 л суспензии оксида алюминия с конечной концентрацией Al2O3 45 г/л при целевой доле выхода первой стадии осаждения 10%, то 10% общего количества оксида алюминия должно быть внесено на стадии а) осаждения. рН на первой стадии осаждения установили равным 9,1, рН на второй стадии осаждения - равным 9,1. Количество воды, изначально присутствовавшее в реакторе, составляло 1330 мл.
Для первой стадии а) осаждения, проводимой при 30°С в течение 8 минут, расход сульфата алюминия должен составлять 7,6 мл/мин, расход алюмината натрия - 9,1 мл/мин, расход воды - 34,6 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,91.
b) Увеличение температуры с 30 до 70°С за 20-30 минут;
с) Вторая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 70°С и рН=9,1 за 30 мин; доля выхода 90%; для второй стадии осаждения, проводимой при 70°С в течение 30 минут, расход сульфата алюминия должен составлять 18,5 мл/мин, расход алюмината натрия - 29 мл/мин, расход воды - 33,8 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,84.
d) Фильтрация путем вытеснения воды в устройстве типа воронки Бюхнера и промывка 3 раза 5 л дистиллированной воды при 70°С;
е) Сушка в течение ночи при 120°С;
f) Обжиг порошка при 750°С.
Синтез оксида алюминия Al(В1) согласно изобретению проводили в реакторе объемом 5 л за 3 стадии.
Использовали следующие концентрации прекурсоров: сульфата алюминия Al2(SO4)3 102 г/л в эквиваленте Al2O3 и алюмината натрия NaAlOO 155 г/л в эквиваленте Al2O3.
Оксид алюминия Al(В1), соответствующий изобретению, произвели согласно следующим стадиям:
а) Первая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 30°С и рН=9,1 за 8 мин; доля выхода 8%. Доля выхода соответствует доле оксида алюминия, образовавшегося на первой стадии, если конечная концентрация оксида алюминия установлена равной 45 г/л. Если объем реактора равен 5 л, и нужно получить 4 л суспензии оксида алюминия с конечной концентрацией Al2O3 45 г/л при целевой доле выхода первой стадии осаждения 8%, то 8% общего количества оксида алюминия должно быть внесено на стадии а) осаждения. рН на первой стадии осаждения установили равным 9,1, рН на второй стадии осаждения - равным 9,1. Количество воды, изначально присутствовавшее в реакторе, составляло 1330 мл.
Для первой стадии а) осаждения, проводимой при 30°С в течение 8 минут, расход сульфата алюминия должен составлять 6,1 мл/мин, расход алюмината натрия - 7,6 мл/мин, расход воды - 69,7 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,91.
b) Увеличение температуры с 30 до 70°С за 20-30 минут;
с) Вторая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 70°С и рН=9,1 за 30 мин; доля выхода 92%; для второй стадии осаждения, проводимой при 70°С в течение 30 минут, расход сульфата алюминия должен составлять 19 мл/мин, расход алюмината натрия - 23 мл/мин, расход воды - 24,7 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,84.
d) Фильтрация путем вытеснения воды в устройстве типа воронки Бюхнера и промывка 3 раза 5 л дистиллированной воды;
е) Сушка в течение ночи при 120°С;
f) Обжиг порошка при 750°С.
Пропиточные растворы А и В были, соответственно, тщательно перемешаны с оксидами алюминия Al(A) и Al(B), приготовленными, как описано выше, с целью получения катализаторов А1 и В1.
Совместное растирание проводили в мешалке типа Brabender с резервуаром объемом 80 см3 со скоростью перемешивания 30 об/мин. Порошкообразный оксид алюминия помещали в смесительный резервуар. Затем при помощи шприца добавляли, примерно, за 2 минуты раствор МоNi(Р) при перемешивании со скоростью 15 об/мин. После получения пасты перемешивание продолжали еще 15 минут со скоростью 50 об/мин. Полученную таким образом пасту подавали в капиллярный реометр MTS через фильеру 2,1 мм со скоростью 10 мм/мин. Полученный таким образом экструдат затем сушили в течение ночи в печи при 80°С, затем обжигали 2 часа в воздухе (1 л/ч/г) в трубчатой печи при 400°С.
Полученные таким образом катализаторы А1 и В1 обладали свойствами, приведенными в таблице 2 ниже.
Таблица 2. Свойства полученных путем совместного растирания катализаторов Е, А1, В1, А2, А3
(% от общего объема)
(30%)
(26%)
(24%)
(54%)
(49%)
Пример 3 (сравнительный). Приготовление катализатора Е путем пропитки сухим способом
Катализатор Е представляет собой катализатор, приготовленный путем экструзионного смешивания бомита, последующих обжига и гидротермической обработки до пропитки сухим способом подложки S(Е) водным раствором так, чтобы содержание металлов было таким же, как и введенное в катализатор А1 путем совместного растирания.
Катализатор Е приготовлен путем пропитки сухим способом подложки S(Е) из оксида алюминия, полученной следующим образом.
Синтез оксида алюминия проводили в реакторе объемом 5 л за три стадии.
Использовали следующие концентрации прекурсоров: сульфата алюминия Al2(SO4)3 102 г/л в эквиваленте Al2O3 и алюмината натрия NaAlOO 155 г/л в эквиваленте Al2O3.
Оксид алюминия произвели согласно следующим стадиям:
а) Первая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 30°С и рН=9,1 за 8 мин; доля выхода 20%. Доля выхода соответствует доле оксида алюминия, образовавшегося на первой стадии, если конечная концентрация оксида алюминия установлена равной 45 г/л. Если объем реактора равен 5 л, и нужно получить 4 л суспензии оксида алюминия с конечной концентрацией Al2O3 45 г/л при целевой доле выхода первой стадии осаждения 20%, то 20% общего количества оксида алюминия должно быть внесено на стадии а) осаждения. рН на первой стадии осаждения установили равным 9,1. Количество воды, изначально присутствовавшее в реакторе, составляло 1330 мл. Для первой стадии а) осаждения, проводимой при 30°С в течение 8 минут, расход сульфата алюминия должен составлять 15,2 мл/мин, расход алюмината натрия - 19 мл/мин, расход воды - 49,2 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,91.
b) Увеличение температуры с 30 до 70°С за 20-30 минут;
с) Вторая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 70°С и рН=9,1 за 30 мин с долей выхода 80%; для второй стадии осаждения, проводимой при 70°С в течение 30 минут, расход сульфата алюминия должен составлять 16,5 мл/мин, расход алюмината натрия - 20 мл/мин, расход воды - 30,1 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,84.
d) Фильтрация путем вытеснения воды в устройстве типа воронки Бюхнера и промывка 3 раза 5 л дистиллированной воды;
е) Сушка в течение ночи при 120°С;
Полученный после фильтрации осадок сушили (на стадии е), минимум, в течение ночи при 120°С. Получили порошок для последующего формования.
Формование проводили в мешалке типа Brabender при содержании кислоты (общем, выраженном относительно сухого оксида алюминия) 1%, степени нейтрализации 20% и потерях при прокаливании кислоты и основания, соответственно, 62 и 64%.
Экструзию проводили в экструдере с трехлодьчатой фильерой диаметром 2,1 мм.
После экструзии экструдат сушили в течение ночи при 80°С и обжигали 2 часа при 800°С в потоке влажного воздуха в трубчатой печи (VVH=1 л/ч/г при 30% влажности). Получили экструдированную подложку S(Е).
Затем подложку S(Е) подвергли пропитке активной фазой NiMoP указанным сухим способом, используя те же прекурсоры, что и в примере 1, а именно, MoO3, Ni(ОН)2, Н3РО4. Концентрацией металлов в растворе устанавливалось их содержание, которые выбирали так, чтобы оно было сравнимым с соответствующими величинами для катализаторов А1 и В1. После пропитки катализатор подвергали обработке на стадии вызревания в течение 24 часов в атмосфере, насыщенной водой, после чего сушили 13 часов при 120°С на воздухе, затем обжигали на воздухе при 400°С в течение 2 часов. Получили катализатор Е. Проверили содержание металлов, полученные величины приведены в таблице 2, размещенной выше.
Пример 4 (сравнительный). Приготовление не соответствующего изобретению катализатора А2 путем совместного растирания
Катализатор А2 приготовили путем совместного растирания активной фазы с обожженным оксидом алюминия Al(А2), полученным из геля оксида алюминия, приготовленного не в соответствии с изобретением (при иной доле выхода первой стадии).
Синтез оксида алюминия Al(А2) провели в соответствии со стадиями осуществления примера 2 (оксид алюминия Al(А1)). Технологические условия были строго идентичными за исключением двух следующих позиций:
- На первой стадии а) осаждения доля выхода составляла 20%.
- На второй стадии с) осаждения доля выхода составляла 80%.
Синтез оксида алюминия, используемого в соответствии с изобретением, провели в реакторе объемом 5 л за 3 стадии.
Использовали следующие концентрации прекурсоров: сульфата алюминия Al2(SO4)3 102 г/л в эквиваленте Al2O3 и алюмината натрия NaAlOO 155 г/л в эквиваленте Al2O3.
Оксид алюминия Al(А2) произвели согласно следующим стадиям:
а) Первая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 30°С и рН=9,1 за 8 мин; доля выхода 20%. Доля выхода соответствует доле оксида алюминия, образовавшегося на первой стадии, если конечная концентрация оксида алюминия установлена равной 45 г/л. Если объем реактора равен 5 л, и нужно получить 4 л суспензии оксида алюминия с конечной концентрацией Al2O3 45 г/л при целевой доле выхода первой стадии осаждения 20%, то 20% общего количества оксида алюминия должно быть внесено на стадии а) осаждения. рН на первой стадии осаждения установили равным 9,1. Количество воды, изначально присутствовавшее в реакторе, составляло 1330 мл. Для первой стадии а) осаждения, проводимой при 30°С в течение 8 минут, расход сульфата алюминия должен составлять 15,2 мл/мин, расход алюмината натрия - 19 мл/мин, расход воды - 49,2 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,91.
b) Увеличение температуры с 30 до 70°С за 20-30 минут;
с) Вторая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 70°С и рН=9,1 за 30 мин, доля выхода 80%; для второй стадии осаждения, проводимой при 70°С в течение 30 минут, расход сульфата алюминия должен составлять 16,5 мл/мин, расход алюмината натрия - 20 мл/мин, расход воды - 30,1 мл/мин. Массовое отношение алюмината натрия к сульфату алюминия, таким образом, равно 1,84.
d) Фильтрация путем вытеснения воды в устройстве типа воронки Бюхнера и промывка 3 раза 5 л дистиллированной воды при 70°С;
е) Сушка в течение ночи при 120°С;
f) Обжиг порошка при 750°С.
Совместное растирание проводили в мешалке типа Brabender с резервуаром объемом 80 см3 со скоростью перемешивания 30 об/мин. Порошкообразный оксид алюминия помещали в смесительный резервуар. Затем при помощи шприца добавляли, примерно, за 2 минуты раствор А МоNi(Р) при перемешивании со скоростью 15 об/мин. После получения пасты перемешивание продолжали еще 15 минут со скоростью 50 об/мин. Полученную таким образом пасту подавали в капиллярный реометр MTS через фильеру 2,1 мм со скоростью 10 мм/мин. Полученный таким образом экструдат затем сушили в течение ночи в печи при 80°С, затем обжигали 2 часа в воздухе (1 л/ч/г) в трубчатой печи при 400°С.
Получили катализатор А2. Свойства катализатора А2 приведены в таблице 2. В том числе, он обладал слишком большим объемом макропор в ущерб объему мезопор, который остался небольшим, и медианному диаметру мезопор (Dpméso), также небольшому (менее 8 нм).
Пример 5 (сравнительный). Приготовление не соответствующего изобретению катализатора А3 путем совместного растирания
Катализатор А3, не соответствующий изобретению, приготовили путем совместного растирания активной фазы с не обожженным порошкообразным бомитом В(А3).
Синтез бомита провели в реакторе объемом 5 л за 3 стадии.
Использовали следующие концентрации прекурсоров: сульфата алюминия Al2(SO4)3 102 г/л в эквиваленте Al2O3 и алюмината натрия NaAlOO 155 г/л в эквиваленте Al2O3.
Бомит В(А3) произвели согласно следующим стадиям а)-е) в тех же условиях, что и в примере 1, но без стадии f) обжига:
а) Первая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 30°С и рН=9,1 за 8 мин; доля выхода 10%. Доля выхода соответствует доле оксида алюминия, образовавшегося на первой стадии, если конечная концентрация оксида алюминия установлена равной 45 г/л.
b) Увеличение температуры с 30 до 70°С за 20-30 минут;
с) Вторая стадия совместного осаждения сульфата алюминия Al2(SO4)3 и алюмината натрия NaAlOO при 70°С и рН=9,1 за 30 мин, доля выхода 90%;
d) Фильтрация путем вытеснения воды в устройстве типа воронки Бюхнера и промывка 3 раза 5 л дистиллированной воды при 70°С;
е) Сушка в течение ночи при 120°С с получением порошкообразного бомита.
Никакого обжига порошка на данном этапе не было.
Раствор А смешивали с порошкообразным прекурсором оксида алюминия В(А3) (в форме AlООН), полученным на стадии е), без какой-либо его дополнительной тепловой обработки. Таким образом, речь идет о порошкообразном бомите. При этом, используемые условия смешивания были строго такими же, что и описанные ранее.
Совместное растирание проводили в мешалке типа Brabender с резервуаром объемом 80 см3 со скоростью перемешивания 30 об/мин. Порошок помещали в смесительный резервуар. Затем при помощи шприца добавляли, примерно, за 2 минуты раствор МоNi(Р) при перемешивании со скоростью 15 об/мин. После получения пасты перемешивание продолжали еще 15 минут со скоростью 30 об/мин. Полученную таким образом пасту подавали в капиллярный реометр MTS через фильеру 2,1 мм со скоростью 10 мм/мин. Полученный таким образом экструдат затем сушили в течение ночи в печи при 80°С, затем обжигали 2 часа в воздухе (1 л/ч/г) в трубчатой печи при 400°С.
Свойства полученного катализатора А3 приведены в таблице 2. По сравнению с катализатором А2, он обладал меньшим, но все же слишком большим объемом макропор. Кроме того, объем мезопор был слишком мал, медианный диаметр мезопор (Dpméso) по сравнению с катализатором А2 не изменился, то есть, был менее 8 нм.
Пример 6. Оценка катализаторов А1, В1, А2, А3 и Е в ходе модельных испытаний
В таких вариантах применения, как гидроочистка, в частности вакуумных дистиллятов и остатков, функция гидро-дегидрогенизации играет критическую роль, принимая во внимание значительное содержание ароматических соединений в этом сырье. Таким образом, испытание по гидрогенизации толуола использовали для исследования пригодности катализаторов для указанных вариантов применения, в частности для гидроочистки остатков.
Описанные ранее в примерах со 2 до 5 катализаторы сульфировали в динамике, на месте использования в трубчатом реакторе с неподвижным поперечным слоем пилотной установки типа Microcat (изготовитель компания Vinci), где текучие среды циркулировали сверху вниз. Измерение активности в отношении гидрогенизации осуществляли сразу после сульфирования, под давлением, без возвращения на воздух, с углеводородным сырьем, которое использовали для сульфирования катализаторов.
Сырье для сульфирования и проведения испытания состояло из 5,8% из диметилдисульфида (DMDS), 20% толуола и 74,2% циклогексана (по весу).
Сульфирование проводили при температуре от комнатной до 350°С со скоростью нагревания 2°С/мин, VVH=4 ч-1 и Н2/НС=450 л при н.у./л. Каталитические испытания проводили при 350°С, VVH=2 ч-1 и таком же Н2/НС, как и при сульфировании, отбирая, минимум, 4 пробы, которые анализировали методом газовой хроматографии.
Таким образом измеряли стабилизировавшуюся каталитическую активность равных объемов катализаторов в реакции гидрогенизации толуола.
Условия измерения активности:
Пробы жидкого отходящего потока анализировали методом газовой хроматографии. На основании определенных молярных концентраций не преобразованного толуола (Т) и продуктов гидрогенизации (метилциклогексана (МСС6), этилциелогептана (EtСС5) и диметилциклопентанов (DMCC5)) степень гидрогенизации XHYD толуола вычисляли по формуле:
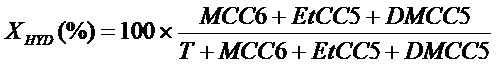
Принимая, что реакция гидрогенизации толуола в условиях испытания является реакцией первого порядка, а реактор ведет себя как реактор идеального вытеснения, активность гидрогенизации AHYD катализаторов вычисляли по формуле:
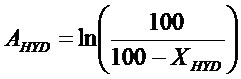
Приведенная ниже таблица позволяет сравнить относительную активность катализаторов в реакции гидрогенизации.
Таблица 3. Сравнение параметров гидрогенизации толуола катализаторов, соответствующих изобретению, (А1, В1) и катализаторов, не соответствующих изобретению А2, А3 и Е
Эти результаты каталитических испытаний демонстрируют наличие определенного влияния совместного растирания раствора металлов с особым оксидом алюминия, описываемым в настоящем документе. Ясно показано, что при осуществлении совместного растирания в соответствии с изобретением, помимо снижения стоимости производства катализатора, наблюдается, практически, сохранение параметров катализаторов, приготовленных сухим способом пропитки (катализатор Е), и значительное улучшение по сравнению с катализаторами, приготовленными путем совместного растирания с использованием обожженного оксида алюминия, полученного из геля оксида алюминия, приготовленного не в соответствии с изобретением (катализатор А2), или бомита (катализатор А3).
Пример 7. Оценка катализаторов А1, В1, А2, А3 и Е в реакторе периодического действия
Катализаторы А1, В1, приготовленные согласно изобретению, а также сравнительные катализаторы А2, А3 и Е подвергли каталитическим испытаниям в реакторе периодического действия с мешалкой с сырьем типа RSV Arabian Light (свойства см. в таблице 4).
Таблица 4. Свойства использованного сырья (RSV Arabian Light)
* ppm - частей на миллион
С этой целью после стадии сульфирования вне реактора путем обеспечения циркуляции газообразной смеси H2S/H2 в течение 2 часов при 350°С, 15 мл катализатора загрузили без доступа воздуха в реактор периодического действия и покрыли 90 мл сырья. Рабочие условия были следующими:
Таблица 5. Рабочие условия в реакторе периодического действия
По окончании испытания реактор охладили и после трехкратной атмосферной перегонки с азотом (10 минут при 1МПа) продукт собрали и проанализировали рентгенофлуоресцентным методом (содержание серы и металлов).
Степень гидрообессеривания (HDS) определили следующим образом:
HDS(%)=((% вес. S)сырье-(% вес. S)продукт)/(% вес. S)сырье×100
Точно так же, степень гидродеметаллизации (HDM):
HDM(%)=((вес. частей на миллион Ni+V)сырье-(вес. частей на миллион Ni+V)продукт)/(вес. частей на миллион Ni+V)сырье×100
Результаты испытаний катализаторов суммированы в таблице 6. Они ясно показывают, что при осуществлении совместного растирания в соответствии с изобретением, помимо снижения стоимости производства катализатора, наблюдается, практически, сохранение эффективности катализаторов, приготовленных сухим способом пропитки, и значительное улучшение по сравнению с катализаторами, приготовленными путем совместного растирания с использованием обожженного оксида алюминия, полученного из геля оксида алюминия, приготовленного не в соответствии с изобретением, или бомита.
Таблица 6. Эффективность катализаторов, соответствующих изобретению (А1, В1), в отношении HDS и HDM и сравнение с катализаторами, не соответствующими изобретению (А2, А3 и Е)
Пример 8. Оценка в ходе гидроочистки в неподвижном слое катализаторов А1 и В1, соответствующих изобретению, в сравнении с каталитической эффективностью катализатора Е
Эффективность катализаторов А1 и В1, приготовленных в соответствии с изобретением, в ходе испытания по гидроочистке нефтяных остатков сравнили с эффективностью катализатора Е. Сырье состояло из смеси атмосферных остатков (RA) происхождением со Среднего Востока (Arabian Мédium) и вакуумных остатков (RSV) происхождением со Среднего Востока (Arabian Light). Сырье отличалось высоким содержанием углерода по Конрадсону (14,4% вес.) и асфальтенов (6,1% вес.) и повышенным количеством никеля (25 вес. частей на миллион), ванадия (79 вес. вес. частей на миллион) и серы (3,90% вес.). Полностью параметры этого сырья приведены в таблице 7.
Таблица 7. Свойства сырья RA AM/RSV AL, использованного для испытания
* ppm - частей на миллион
После стадии сульфирования путем организации циркуляции в реакторе фракции газойля с добавлением DMDS при конечной температуре 350°С, в установку подали нефтяные остатки, описанные выше, в рабочих условиях, приведенных в таблице 8.
Таблица 8. Рабочие условия в реакторе с фиксированным слоем
Сначала подавали смесь сырья RA AM/RSV AL, затем увеличивали температуру испытания. После периода стабилизации 300 часов исследовали параметры гидрообессеривания (HDS) и гидродеметаллизации (HDM).
Полученные результаты (таблица 9) согласуются с результатами примера 7, то есть, подтверждают достаточную эффективность катализаторов, полученных путем совместного растирания в соответствии с изобретением, по сравнению с катализатором сравнения, приготовленным сухим способом пропитки. Потеря активности по сравнению со сравнительным примером незначительная. Таким образом, ясно, что катализаторы, соответствующие изобретению, одновременно с уменьшением стоимости производства, позволяют достичь удовлетворительной активности, почти эквивалентной достигаемой катализатором, приготовленным сухим способом пропитки. Это позволяет подтвердить важность способа производства, соответствующего изобретению, который намного проще реализуем и, следовательно, более дешев для производителя катализатора.
Таблица 9. Эффективность катализаторов А1 и В1 в отношении HDS и HDM по сравнению с катализатором Е
Изобретение относится к мезопористому и макропористому катализатору гидроконверсии с активной фазой, к способу получения такого катализатора, а также к способу гидроочистки тяжелого углеводородного сырья. Катализатор гидроконверсии содержит матрицу из обожженного оксида алюминия и активную фазу гидро-дегидрогенизации, включающую, по меньшей мере, молибден в качестве металла группы VIB Периодической системы элементов, в некоторых случаях, по меньшей мере, никель или кобальт в качестве металла группы VIII Периодической системы элементов, в некоторых случаях фосфор. Активная фаза полностью введена смешиванием путем совместного растирания, с указанной матрицей. Катализатор характеризуется удельной площадью поверхности SBET более 100 м2/г, медианным по объему диаметром мезопор от 12 до 25 нм, включая границы диапазона, медианным по объему диаметром макропор от 50 до 250 нм, включая границы диапазона, объемом мезопор, большим или равным 0,65 мл/г, общим объемом пор, большим или равным 0,75 мл/г, и объемом макропор от 15 до 35% общего объема пор. Способ получения катализатора заключается в том, что вначале проводят первую стадию осаждения в водной реакционной среде при температуре в диапазоне от 20 до 90°С в течение периода времени от 2 минут до 30 минут. Далее осуществляют нагревание суспензии до температуры, лежащей в диапазоне от 40 до 90°С в течение времени от 7 минут до 45 минут. После этого осуществляют вторую стадию осаждения суспензии при температуре, лежащей в диапазоне от 40 до 90°С, в течение периода времени от 2 минут до 50 минут. Затем проводят фильтрацию суспензии с получением геля оксида алюминия. После этого осуществляют стадию сушки геля оксида алюминия с получением порошка. Затем проводят стадию тепловой обработки порошка при температуре от 500 до 1000°С в течение периода времени от 2 до 10 часов, в присутствии или в отсутствие потока воздуха, содержащего до 60% об. воды, с получением обожженного пористого оксида алюминия. После этого проводят стадию совместного растирания обожженного пористого оксида алюминия и раствора по меньшей мере одного прекурсора металла активной фазы с получением пасты. Затем проводят стадию формования полученной пасты. После этого осуществляют стадию сушки формованной пасты при температуре, меньшей или равной 200°С, с получением высушенного катализатора. При необходимости, проводят стадию тепловой обработки высушенного катализатора при температуре, лежащей в диапазоне от 200 до 1000°С, в присутствии или в отсутствие воды. Способ гидроочистки тяжелого углеводородного сырья заключается в том, что углеводородное сырье вводят в контакт с водородом и вышеуказанным катализатором. Изобретение позволяет получить катализатор, обладающий большим объемом мезопор в сочетании со значительным объемом макропор, увеличенным диаметром мезопор и активной фазой гидро-дегидрогенизации. 3 н. и 15 з.п. ф-лы, 9 табл., 8 пр.
1. Способ получения катализатора с активной фазой, полностью введенной смешиванием путем совместного растирания, содержащего, по меньшей мере, молибден в качестве одного металла группы VIB Периодической системы элементов, необязательно, по меньшей мере, никель или кобальт в качестве металла группы VIII Периодической системы элементов, необязательно, фосфор и оксидную матрицу, в основном, из обожженного оксида алюминия, включающий следующие стадии:
а) первая стадия осаждения в водной реакционной среде по меньшей мере одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, на которой по меньшей мере один из прекурсоров - основный или кислый - содержит алюминий, относительный расход кислого и основного прекурсоров выбран так, чтобы обеспечить рН реакционной среды от 8,5 до 10,5, и расход прекурсора или прекурсоров - кислого и основного, содержащих алюминий, выбран так, чтобы доля выхода первой стадии составляла от 5 до 13%, при этом доля выхода определяется как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной первой стадии осаждения, к общему количеству оксида алюминия, образовавшемуся по окончании стадии с) способа производства, при этом указанную стадию осаждения осуществляют при температуре, лежащей в диапазоне от 20 до 90°С, в течение периода времени от 2 минут до 30 минут;
b) стадия нагревания суспензии до температуры, лежащей в диапазоне от 40 до 90°С, на период времени от 7 минут до 45 минут;
с) вторая стадия осаждения суспензии, полученной по окончании стадии b) нагревания, путем добавления в указанную суспензию по меньшей мере одного основного прекурсора, выбранного из алюмината натрия, алюмината калия, аммиака, гидроксида натрия и гидроксида калия, и по меньшей мере одного кислого прекурсора, выбранного из сульфата алюминия, хлорида алюминия, нитрата алюминия, серной кислоты, соляной кислоты и азотной кислоты, на которой, по меньшей мере один из прекурсоров - основный или кислый, содержит алюминий, относительный расход кислого и основного прекурсоров выбирают так, чтобы получить рН реакционной среды от 8,5 до 10,5, и расход прекурсора или прекурсоров - кислого и основного, содержащих алюминий, регулируют так, чтобы достичь доли выхода второй стадии от 87 до 95%, при этом долю выхода определяют как отношение количества оксида алюминия, в эквиваленте Al2O3, образовавшегося на указанной второй стадии осаждения, к общему количеству оксида алюминия, образовавшемуся по окончании стадии с) способа производства, при этом указанную стадию проводят при температуре, лежащей в диапазоне от 40 до 90°С, в течение периода времени от 2 минут до 50 минут;
d) стадия фильтрации суспензии, полученной по окончании второй стадии с) осаждения, с целью получения геля оксида алюминия;
е) стадия сушки указанного геля оксида алюминия, полученного на стадии d), с целью получения порошка;
f) стадия тепловой обработки порошка, полученного по окончании стадии е), при температуре от 500 до 1000°С в течение периода времени от 2 до 10 часов, в присутствии или в отсутствие потока воздуха, содержащего до 60% об. воды, с целью получения обожженного пористого оксида алюминия;
g) стадия совместного растирания полученного обожженного пористого оксида алюминия и раствора по меньшей мере одного прекурсора металла активной фазы с целью получения пасты;
h) стадия формования полученной пасты;
i) стадия сушки формованной пасты при температуре, меньшей или равной 200°С, с целью получения высушенного катализатора;
j) возможная стадия тепловой обработки высушенного катализатора при температуре, лежащей в диапазоне от 200 до 1000°С, в присутствии или в отсутствие воды.
2. Способ по п. 1, в котором доля выхода первой стадии а) осаждения составляет от 6 до 12%.
3. Способ по одному из пп. 1 или 2, в котором доля выхода первой стадии а) осаждения составляет от 7 до 11%.
4. Способ по одному из пп. 1-3, в котором кислый прекурсор выбран из сульфата алюминия, хлорида алюминия и нитрата алюминия, предпочтительно это сульфат алюминия.
5. Способ по одному из пп. 1-4, в котором основный прекурсор выбран из алюмината натрия и алюмината калия, предпочтительно это алюминат натрия.
6. Способ по одному из пп. 1-5, в котором на стадиях а), b), с) водная реакционная среда представляет собой воду, и указанные стадии проводят при перемешивании в отсутствие органических добавок.
7. Мезопористый и макропористый катализатор гидроконверсии, содержащий:
- матрицу из обожженного оксида алюминия;
- активную фазу гидро-дегидрогенизации, включающую, по меньшей мере, молибден в качестве металла группы VIB Периодической системы элементов, в некоторых случаях, по меньшей мере, никель или кобальт в качестве металла группы VIII Периодической системы элементов, в некоторых случаях, фосфор,
при этом указанная активная фаза полностью введена смешиванием путем совместного растирания с указанной матрицей, главным образом, из обожженного оксида алюминия,
при этом указанный катализатор характеризуется удельной площадью поверхности SBET более 100 м2/г, медианным по объему диаметром мезопор от 12 до 25 нм, включая границы диапазона, медианным по объему диаметром макропор от 50 до 250 нм, включая границы диапазона, объемом мезопор, измеренным методом ртутной порометрии, большим или равным 0,65 мл/г, и общим объемом пор, измеренным методом ртутной порометрии, большим или равным 0,75 мл/г, и объемом макропор от 15 до 35% общего объема пор.
8. Катализатор гидроконверсии по п. 7, характеризующийся медианным по объему диаметром мезопор, измеренным методом ртутной порометрии, от 13 до 17, включая границы диапазона.
9. Катализатор гидроконверсии по одному из пп. 7, 8, в котором объем мезопор составляет от 0,65 до 0,75 мл/г.
10. Катализатор гидроконверсии по одному из пп. 7-9, который не содержит микропор.
11. Катализатор гидроконверсии по одному из пп. 7-10, в котором содержание металла группы VIB составляет от 2 до 10% вес. триоксида металла группы VIB относительно общей массы катализатора, содержание металла группы VIII составляет от 0,0 до 3,6% вес. оксида металла группы VIII относительно общей массы катализатора, содержание элементарного фосфора составляет от 0 до 5% вес. пентоксида фосфора относительно общей массы катализатора.
12. Катализатор гидроконверсии по одному из предшествующих пунктов, в котором активная фаза гидро-дегидрогенизации образована молибденом или никелем и молибденом, или кобальтом и молибденом.
13. Катализатор гидроконверсии по п. 12, в котором активная фаза гидро-дегидрогенизации также содержит фосфор.
14. Способ гидроочистки тяжелого углеводородного сырья, выбранного из атмосферных остатков, остатков прямой вакуумной перегонки, деасфальтированной нефти, остатков осуществления способов конверсии, гидроконверсии в неподвижном слое, кипящем слое или подвижном слое, взятых отдельно или в смеси, включающий приведение указанного сырья в контакт с водородом и катализатором, получаемым согласно одному из пп. 1-6, или катализатором по одному из пп. 7-13.
15. Способ гидроочистки по п. 14, где тяжелое углеводородное сырье выбрано из остатков осуществления способов конверсии, образующихся при коксовании нефти.
16. Способ гидроочистки по любому из пп. 14-15, реализованный в кипящем слое при температуре от 320 до 450°С, парциальном давлении водорода от 3 МПа до 30 МПа, объемной скорости, преимущественно, от 0,1 до 10 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья, преимущественно, от 100 до 3000 м3 при н.у. на м3.
17. Способ гидроочистки по любому из пп. 14-16, реализованный в неподвижном слое при температуре от 320°С до 450°С, парциальном давлении водорода от 3 МПа до 30 МПа, объемной скорости от 0,05 до 5 объемов сырья на объем катализатора в час и при соотношении газообразного водорода и жидкого углеводородного сырья от 200 до 5000 м3 при н.у. на м3.
18. Способ гидроочистки тяжелого углеводородного сырья типа остатков в неподвижном слое по п. 16, включающий, по меньшей мере:
а) стадию гидродеметаллизации,
b) стадию гидрообессеривания,
в котором указанный катализатор используют по меньшей мере на одной из указанных стадий а) и b).
| US 5620592 A, 15.04.1997 | |||
| КАТАЛИЗАТОР ПЕРЕРАБОТКИ ТЯЖЕЛЫХ НЕФТЯНЫХ ФРАКЦИЙ | 2012 |
|
RU2506997C1 |
| КАТАЛИЗАТОР, СОДЕРЖАЩИЙ ФОСФОР, И СПОСОБ ГИДРООЧИСТКИ НЕФТЯНОГО СЫРЬЯ С ИСПОЛЬЗОВАНИЕМ ЭТОГО КАТАЛИЗАТОРА | 1997 |
|
RU2192923C2 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |

