ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к получению N-защищенных 6-(пиперидин-4-илкарбамоил)пиперидин-3-илсульфонатов. Эти сульфонаты подходят для применения в качестве промежуточных продуктов, которые посредством серии дополнительных стадий процесса приводят к синтезу 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидов и сложных эфиров. Изобретение также относится к окончательной стадии процесса получения ингибитора бета-лактамазы, (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида.
УРОВЕНЬ ТЕХНИКИ
Некоторые 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамиды представляют собой ингибиторы β-лактомазы и, при использовании их в сочетании с β-лактамными антибиотиками, могут быть эффективны для лечения бактериальных инфекций. См., например, публикацию международной патентной заявки WO2009/091856, где описаны 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамиды и их синтез из промежуточного продукта илида кетосульфоксония, содержащего амидную боковую цепь, где илидный промежуточный продукт циклизуется до 5-оксо-пиперидин-2-карбоксамида при использовании катализатора Ir, Rh или Ru. Аналогично, в документе Baldwin et al. описана трансформация илидов β-кетосульфоксония, полученных из лактона, в β-оксоазот гетероциклы в присутствии родиевого катализатора. См., документ Baldwin et al., 1993, J. Chem. S С., Chem. Commun. 18:1434-1435. В документе Mangion et al. описаны вставки X-H (например, вставки N-H), катализированные иридием, илидов сульфоксония. См., документ Mangion et al., 2009, Org. Lett., 11:3566-3569, и документ Mangion et al., 2011, Org. Lett. 13:5480-5483.
В публикации патентной заявки США US2003/0199541 описаны способы получения азабициклических соединений, которые могут использоваться в качестве лекарственных средств, в частности, в качестве антибактериальных средств. В международной патентной публикации WO2008/039420 описаны способы получения некоторых 7-оксо-2,6-диазабицикло[3.2.0]гептан-2-карбоксамидов, которые могут использоваться в качестве ингибиторов β -лактамазы.
В публикации международной заявки WO2010/126820 описано получение алкиловых эфиров N-защищенных оксоазациклоалкилкарбоновых кислот. Эти сложные эфиры могут использоваться в качестве промежуточных веществ при синтезе 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидов и сложных эфиров.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения соединения формулы III:
 (III),
(III),
который включает:
(B) приведение в контакт лактона формулы II:
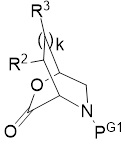 (II)
(II)
с азациклоалкиламином формулы II-Am:
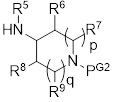 (II-Am)
(II-Am)
после чего происходит приведение в контакт с сульфонилгалогенидом формулы II-Su:
R4-SO2W (II-Su)
в присутствии основания третичного амина с получением соединения формулы III, где:
PG1 представляет собой защитную группу первичного амина, которая образует с азотом амина, к которому она присоединена, карбамат, бензиламин или сульфонамид;
PG2 представляет собой защитную группу вторичного амина, выбранную из (i) карбаматов и (ii) бензиламинов;
k представляет собой целое число, равное 0, 1 или 2;
R2 и R3 определены следующим образом:
(а) R2 представляет собой H, C1-6 алкил, -O-C1-6 алкил, -O-Si(-C1-6 алкил)3 или -O-Si(-C1-6 алкил)(-фенил)2,
и каждый R3 независимо представляет собой H или C1-6 алкил; или
(b) альтернативно и при условии, что k равно 1 или 2, R2 и R3, расположенный рядом с R2, вместе с атомами углерода, к которым каждый присоединен, образуют C5-7 циклоалкил, каждый необязательно замещен от 1 до 3 заместителями, каждый независимо представляет собой C1-6 алкил, -O-C1-6 алкил, -O-Si(-C1-6 алкил)3 или -O-Si(-C1-6 алкил)(-фенил)2; и любой другой R3 представляет собой Н или C1-6 алкил;
R4 представляет собой:
(1) фенил, необязательно замещенный от 1 до 3 заместителями, каждый из которых независимо представляет собой C1-4 алкил, C1-4 галогеналкил, -O-C1-4 алкил, -O-C1-4 галогеналкил, Cl, Br, F или NO2;
(2) C1-4 алкил; или
(3) C1-4 галогеналкил;
R5 представляет собой Н или C1-3 алкил;
R6 и R8 независимо представляют собой H, C1-3 алкил, -O-C1-3 алкил или -N(-C1-3 алкил)2;
каждый R7 и R9 независимо представляет собой H или C1-6 алкил;
W представляет собой галоген;
p равно 0, 1 или 2;
q равно 0, 1 или 2; и
p+q=0, 1, 2 или 3.
Соединение III может использоваться в качестве промежуточного продукта, который в комбинации с серией дополнительных стадий (описанных ниже), обеспечивает конвергентный синтез 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидов и 2-карбоновых эфиров, которые могут использоваться в качестве ингибиторов β -лактамазы (BLI). Применение соединения III также обеспечивает большую гибкость, заключающуюся в представлении более прямого пути к 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновым эфирам, подходящим для применения в качестве BLI. Число всех стадий синтеза снижается, как снижается и цена, и интенсивность расхода продуктов в способе по сравнению со способом, описанным в Mangion et al., 2011, Org Lett 13:5480-5483. Число токсических, дорогих и трудных в обращении реагентов вытесняется при использовании этого способа по сравнению со способом документа Mangion et al., 2011, Org Lett 13:5480-5483. Более того, применение Соединения III позволяет вводить боковые амидные цепи, которые могут быть химически нестабильны в условиях реакции, требуемых на ранних стадиях синтеза. В дополнение, улучшенный способ удаления Вос-защитной группы обеспечивает повышение выхода и снижает интенсивность расхода продуктов.
Другие варианты осуществления, аспекты и признаки настоящего изобретения либо подробно описаны ниже, либо будут понятны из описания, примеров и прилагаемой формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение (альтернативно называемое в настоящем документе как «Способ P») включает способ получения соединения формулы III, который включает Стадию B, как указано выше в сущности изобретения. Стадия B включает непосредственное нуклеофильное раскрытые лактона с амином без применения агентов присоединения. В Стадии B не используются альтернативные методики, такие как получение диазокетона или илида кетосульфоксония для осуществления циклизаций, при которых требуются дорогостоящие переходные металлы или взрывоопасные агенты, такие как диазометан. В некоторых вариантах осуществления Стадия B также может обеспечивать высокий выход; то есть, выход 95% или больше. Соединение формулы III может использоваться в качестве промежуточного продукта в этом способе синтеза ингибитора бета-лактамазы, (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида. В целом процесс синтеза (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида укорочен отчасти за счет того, что он начинается с другого вещества.
Выбор различных защитных групп позволяет осуществлять замену для получения большего выхода, избегая многостадийные реакции защита/снятие защиты и образование большого числа побочных продуктов. Правильный и подходящий выбор защитных групп и активирующих агентов позволяет применять условия селективных реакций для максимального снижения побочных продуктов в течение такой замены (Стадия C). Эти новые защитные группы обеспечивают проведение гидрирования без трудоемкого контроля за реагентами и образования побочных продуктов, так как больше не требуется смена защитных групп (Стадия E). Наконец, новый набор условий конечной стадии снятия защиты обеспечивает высокий выход и чистую реакцию, избегая получения побочных продуктов (Стадия G).
В целом, в некоторых вариантах осуществления настоящее изобретение обеспечивает высокий выход (общий выход ~42%), высокую эффективность и низкую стоимость. В способе также исключено несколько стадий в общем синтезе по сравнению с существующим путем (7 отдельных стадий в новом изобретении по сравнению с 12 в старом пути). Новое изобретение также обеспечивает лучшую экологичность, благодаря замене реакции, катализируемой иридием на промежуточный продукт, который может быть получен путем биокатализа.
Амино-защитная группа PG1 в Стадии B в сочетании азотом амина, к которому она присоединена, может быть карбаматом или бензиламином или сульфонамидом. Подходящие защитные группы карбамат, бензиламин и сульфонамид и способы их получения и отщепления описаны в Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973 и в T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 3ее издание, 1999, и 2ое издание, 1991. В одном из вариантов осуществления PG1 представляет собой (1)°C(=O)-O-(CH2)0-1-CH=CH2, (2) -C(=O)-O-CH2-AryB, где AryB представляет собой фенил, каждый необязательно замещен от 1 до 3 заместителями, каждый из которых независимо представляет собой галоген, -NO2, -C1-4 алкил или -O-C1-4 алкил, (3) -C(=O)-O-C1-4 алкил или (4) -CH2-AryC, в котором AryC представляет собой фенил, каждый необязательно замещен от 1 до 3 заместителями, каждый из которых независимо представляет собой галоген, -NO2, -C1-4 алкил или -O-C1-4 алкил. В еще одном варианте осуществления PG1 представляет собой трет-бутилоксикарбонил (Boc), аллилоксикарбонил (Alloс), бензилоксикарбонил (Cbz), p-метоксибензилоксикарбонил, p-нитробензилоксикарбонил, p-бромбензилоксикарбонил, p-хлорбензилоксикарбонил, 2,4-дихлорбензилоксикарбонил или бензил. В еще одном варианте осуществления PG1 представляет собой Bос. В другом варианте осуществления PG1 представляет собой сульфонильную группу, полученную из сульфонилгалогенидов, таких как хлорид метансульфонила, хлорид хлорметансульфонила, хлорид дихлорметансульфонила, хлорид бензолсульфонил, хлорид p-трифторметилбензолсульфонила, хлорид p-толуолсульфонила, хлорид p-бромбензолсульфонила, хлорид p-фторбензолсульфонила, хлорид p-метоксибензолсульфонила, хлорид 2-нитробензолсульфонила, хлорид 4-нитробензолсульфонила и хлорид 2,4-дихлорбензолсульфонила, хлорид хлорметансульфонила, хлорид p-трифторетилбензолсульфонила, хлорид p-бромбензолсульфонила и хлорид 2,4-дихлорбензолсульфонила, хлорид хлорметансульфонила, хлорид p-трифторметилбензолсульфонила и хлорид p-бромбензолсульфонила. Предпочтительный сульфонилгалогенид представляет собой хлорид 2-нитробензолсульфонила.
PG2 представляет собой амино-защитную группу, которая является стабильной в условиях, при которых группа PG1 является нестабильной. Другими словами, PG2 представляет собой группу, которая не отщепляется в условиях, подходящих для удаления PG1. PG2, в сочетании с азотом амино, к которому она присоединена, предпочтительно представляет собой алкил карбамат, арил карбамат, винил карбамат, аллил карбамат, ацетамид (включая ттрифторацетамид) или бензиламин. Подходящие группы PG2 включают Bос, Cbz, Allос, p-метокси-бензил и бензил. Предпочтительный PG2 представляет собой Bос.
Амины формулы II-Am могут быть получены, например, путем восстановительного аминирования соответствующего кетона или путем восстановления водородом соответствующего имина. Дополнительное описание способов, подходящих для получения аминов формулы II-Am, может быть найдено в Richard Larосk, Comprehensive Organic Transformations, 2ое издание, Wiley-VCH Publishers Inc, 1999, pp.753-879.
Стадию B проводят в органическом растворителе. Подходящие растворители включают DCE, THF, DMF, NMP, DMSO, 1,4-диоксан, диметилацетамид, толуол, ксилены, хлорбензол, трифтортолуол, 2-метил-THF и ацетонитрил. Предпочтительные растворители представляют собой THF и ацетонитрил.
Добавление амина формулы II-Am в Стадию B можно проводить при температуре от приблизительно 25°С до приблизительно 100°С в течение от 2 до 72 часов или от 10 до 72 часов и обычно его проводят при температуре в диапазоне от приблизительно 60°С до приблизительно 85°С в течение от 8 до 24 часов или от 15 до 24 часов. Как используется в настоящем описании термин “приблизительно” может относится к отклонению ±5%, ±10% или ±15%.
Добавление сульфонилгалогенида формулы II-Su в Стадии B можно подходящим образом осуществлять в присутствии основания третичного амина в течение времени от 10 минут до 10 часов, предпочтительно, от 30 минут до 90 минут.
Класс подходящих третичных аминов включает TEA, DIPEA, 4-NMM и 4-диметиламинопиридин. 4-Диметиламинопиридин предпочтительно представляет собой основание. Основание обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 3 эквивалентов на эквивалент Соединения III, и чаще используют в количестве в диапазоне от приблизительно 1,1 до приблизительно 2 эквивалентов (например, приблизительно 1,6 эквивалентов).
Примеры сульфонилгалогенидов, подходящих для применения в Стадии B, включают хлорид метансульфонила, хлорид хлорметансульфонила, хлорид дихлорметансульфонила, хлорид бензолсульфонила, хлорид p-трифторметилбензолсульфонила, хлорид p-толуолсульфонила, хлорид p-бромбензолсульфонила, хлорид p-фторензолсульфонила, хлорид p-метоксибензолсульфонила, хлорид 2-нитробензолсульфонила, хлорид 4-нитробензолсульфонила и хлорид 2,4-дихлорбензолсульфонила. Класс подходящих сульфонилгалогенидов состоит из хлорида хлорметансульфонила, p-трифторметилбензолсульфонила, хлорида p-бромбензолсульфонила и хлорида 2,4-дихлорбензолсульфонил. Еще один класс подходящих сульфонилгалогенидов состоит из хлорида хлорметансульфонила, хлорида p-трифторметилбензолсульфонила, хлорида 2-нитробензолсульфонила и хлорида p-бромбензолсульфонила. Предпочтительный сульфонилгалогенид представляет собой хлорид p-трифторметилбензолсульфонила. Еще одним предпочтительным сульфонилгалогенидом является хлорид 2-нитробензолсульфонила. Сульфонилгалогенид обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 2 эквивалентов на эквивалент Соединения III, и чаще используют в количестве в диапазоне от приблизительно 1 до приблизительно 1,5 эквивалентов (например, приблизительно 1,3 эквивалента).
Добавление сульфонилгалогенида формулы II-Su в Стадии B можно осуществлять при температуре от приблизительно 0°С до приблизительно 50°С и предпочтительно осуществляют при температуре в диапазоне от приблизительно 10°С до приблизительно 30°С в течение времени от 30 минут до 90 минут.
Другие варианты осуществления Соединения III и Стадии B включают следующее:
(1a) k равно 0 или 1;
(1b) k равно 0;
(1c) k равно 1;
(2a) R2 представляет собой Н, C1-4 алкил, -O-C1-4 алкил, -O-Si(-C1-4 алкил)3 или -O-Si(-C1-4 алкил)(фенил)2, и каждый R3 представляет собой Н или C1-4 алкил;
(2b) R2 представляет собой Н, CH3, -ОСH3, -O-триметилсилил (TMS), -O-трет-бутилдифенилсилил (TBDPS), -O-трет-бутилдиметилсилил (TBS) или -O-триизопропилсилил (TIPS), и каждый R3 представляет собой Н или CH3;
(2c) R2 представляет собой Н или CH3, и каждый R3 представляет собой Н или CH3;
(2d) R2 представляет собой Н, и каждый R3 представляет собой Н;
(2e) при условии, что k равно 1 или 2, R2 и R3, расположенный рядом с R2, вместе с атомами углерода, к которым каждый присоединен, образуют C5-6 циклоалкил; и любой другой R3 представляет собой Н.
Один или несколько этих вариантов осуществления от (1) до (2) может быть объединен с каждым другим, где каждая такая комбинация представляет собой отдельный вариант осуществления Соединения III и Стадии B. Другими словами, любой вариант осуществления из группы 1 (1a, 1b или 1c) может быть объединен с любым вариантом осуществления из группы 2 (2a, 2b, 2c, 2d или 2e).
Один из вариантов способа P включает Стадию B, как только что описано выше, и дополнительно включает:
(А) Приведение в контакт соединения формулы I:
 (I)
(I)
с PG1-продуцирующим агентом в присутствии водного основания, после чего следует добавление третичного основания с получением Соединения II. В одном из вариантов осуществления приведение в контакт проходит в присутствии органического или неорганического основания с получением Соединения II.
Стадию A проводят в органическом растворителе и воде. Подходящие растворители включают ацетон, толуол, дихлорметан, DCE, DMF, DMAC, DMSO, THF, хлорбензол, 1,2-дихлорбензол, циклопентилметиловый эфир, ацетонитрил, EtOAc, IPAc, MeOAc, нитрометан, трифторметилбензол, метил этил кетон, DME и 2-MeTHF. Предпочтительные растворители представляют собой ацетон и этилацетат.
Реакцию в Стадии A можно подходящим образом подходящим образом проводить при температуре в диапазоне от приблизительно 0°С до приблизительно 30°С в течение времени от 10 минут до 10 часов и ее предпочтительно осуществляют при температуре в диапазоне от приблизительно 10°С до приблизительно 25°С в течение времени от 30 минут до 60 минут.
Подходящие PG1-продуцирующе агенты включают хлорид 2-нитробензол-1-сульфонила, хлорид 4-нитробензол-1-сульфонила и (BОС)2O. PG1-продуцирующий агент обычно используют в количестве в диапазоне от приблизительно 1 эквивалента до приблизительно 5 эквивалентов и чаще используют в количестве в диапазоне от приблизительно 1,3 до приблизительно 2 эквивалентов.
Подходящие органические или неорганические основания включают LiOH, NaOH, KOH, Cs(OH)2, Li2CO3, Na2CO3, K2CO3, CsCO3, LiHCO3, NaHCO3, KHCO3, Li3PO4, Na3PO4, K3PO4, Li2HPO4, NaHPO4, KHPO4, диизопропиламин, триэтиламин, диизопропилэтиламин, морфолин, 4-метилморфолин, DABCO, DBU, пиридин, лютидин, коллидин и тому подобное.
Подходящие основания включают NaOH, KOH, LiOH, K2CO3, Cs2CO3, Na2CO3, K3PO4 и Na3PO4. Водное основание обычно используют в количестве в диапазоне от приблизительно 1 эквивалентов до приблизительно 10 эквивалентов и чаще используют в количестве в диапазоне от приблизительно 1,5 до приблизительно 2,0 эквивалентов.
Подходящие третичные основания включают TEA, DIPEA, 4-NMM и диэтилизопропиламин. Третичное основание обычно используют в количестве в диапазоне от приблизительно 2,0 эквивалентов до приблизительно 10,0 эквивалентов и чаще используют в количестве в диапазоне от приблизительно 3,0 до приблизительно 5,0 эквивалентов.
Основание третичного амина в Стадии A обычно представляет собой три-C1-4 алкиламин. Класс подходящих третичных аминов включает TEA, DIPEA, 4-NMM и диэтилизопропиламин. TEA предпочтительно представляет собой основание. Основание обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 3 эквивалентов на эквивалент Соединения I, и чаще используют в количестве в диапазоне от приблизительно 1,1 до приблизительно 2 эквивалентов (например, приблизительно 1,8 эквивалентов).
Один из вариантов способа P включает Стадию B, как только что было описано выше, или Стадии A и B, как только что описано, и дополнительно включает:
(C) обработку соединения формулы III N-4-нитробензолсульфонил-O-бензилгидроксиламином в присутствии основания, а затем обработку нуклеофильным реагентом, таким как тиол, с получением соединения формулы IV, или его фармацевтически приемлемой соли:
 (IV)
(IV)
Стадию C проводят в органическом растворителе. Подходящие растворители включают DMAC, DMF, NMP, THF, метанол и DME. Предпочтительный растворитель представляет собой DMAC и метанол.
Подходящие основания в Стадии C включают Li t-бутоксид, Na трет-бутоксид, K трет-бутоксид, карбонат цезия, карбонат натрия, KHMDS и NaHMDS. Класс подходящих оснований состоит из Li трет-бутоксида, Na трет-бутоксида, K трет-бутоксида, карбоната натрия и карбоната цезия. Предпочтительные основания представляют собой t-бутоксид и карбонат натрия. Основание обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 2 эквивалентов на эквивалент соединения формулы III, и чаще используют в количестве в диапазоне от приблизительно 1 до приблизительно 1,5 эквивалентов (например, приблизительно 1,2 эквивалентов).
N-4-нитробензолсульфонил-O-бензилгидроксиламин обычно используют в диапазоне от приблизительно 1 до приблизительно 2 эквивалентов на эквивалент Соединения III, и чаще используют в количестве в диапазоне от приблизительно 1 до приблизительно 1,5 эквивалентов (например, приблизительно 1,2 эквивалентов).
Реакцию в Стадии C можно подходящим образом проводить при температуре в диапазоне от приблизительно 30°C до приблизительно 90°C и ее предпочтительно осуществляют при температуре в диапазоне от приблизительно 45°C до приблизительно 70°C в течение времени от 18 до 30 часов.
Подходящие тиолы Стадии C включают тиофенол или 2-меркаптоуксусную кислоту. Предпочтительный нуклеофил представляет собой 2-меркаптоуксусную кислоту. Кислоту обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 10 эквивалентов на эквивалент Соединения III, и чаще используют в количестве в диапазоне от приблизительно 3 до приблизительно 6 эквивалентов.
Другие варианты осуществления Стадии C включают следующие признаки IV:
(3a) R5 представляет собой Н или CH3;
(3b) R5 представляет собой Н;
(4a) R6 представляет собой Н или C1-3 алкил;
(4b) R6 представляет собой Н или CH3;
(4c) R6 представляет собой Н;
(5a) p равно 1, и q равно 1;
(5b) p равно 1, и q равно 0.
Один или несколько из этих вариантов осуществления, от (3) до (5), могут быть осуществлены каждый с другим и/или с вариантами осуществления, описанными выше, где каждая такая комбинация, представляет собой отдельный вариант осуществления соединения, используемого в Стадии C.
Еще один вариант осуществления способа P включает Стадии от B до C, как описано выше, или Стадии от A до C, как описано выше, и дополнительно включает:
(D) приведение в контакт соединения (IV) с фосгеном, дифосгеном, трифосгеном или эквивалентом фосгена, таким как карбодимидазол или галогенформиат, в присутствии третичного амина, и затем добавление водного раствора кислоты с получением соединения формулы V:
 (V)
(V)
В одном из аспектов этого варианта осуществления приведение в контакт можно осуществлять в присутствии амино-основания и обрабатывать водой с получением соединения формулы V.
Стадию D проводят в органическом растворителе. Подходящие растворители включают DCM и ацетонитрил. Предпочтительный растворитель представляет собой DCM и ацетонитрил.
Подходящие кислоты в Стадии D включают хлористоводородную кислоту, серную кислоту, трифторуксусную кислоту и фосфорную кислоту. Предпочтительной кислотой является фосфорная кислота. Кислота обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 6 эквивалентов на эквивалент Соединения IV, и чаще используют в количестве в диапазоне от приблизительно 3 до приблизительно 5 эквивалентов (например, приблизительно 3,2 эквивалента).
Третичный амин в Стадии D предпочтительно представляет собой три-C1-4 алкиламин. Класс подходящих аминов состоит из TEA, DIPEA и диэтилизопропиламина. DIPEA предпочтительно представляет собой амин. Амин обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 6 эквивалентов на эквивалент Соединения IV, и чаще используют в количестве в диапазоне от приблизительно 3 до приблизительно 5 эквивалентов (например, приблизительно 3,2 эквивалента).
Трифосген, дифосген или фосген или эквивалент фосгена, такой как карбодимидазол или галогенформиат обычно используют в Стадии D в количестве в диапазоне от приблизительно от 0,5 до 1 эквивалента на эквивалент Соединения IV, и чаще используют в количестве в диапазоне от приблизительно 0,7 до приблизительно 1 эквивалента (например, приблизительно 0,8 эквивалентов). Трифосген является более предпочтительным, чем дифосген и фосген.
Приведение в контакт Соединения IV с трифосгеном, дифосгеном или фосгеном или эквивалентом фосгена, таким как карбодимидазол или галогенформиат, в Стадии D может подходящим образом быть проведено при температуре в диапазоне от приблизительно -15°C до приблизительно 100°C или -15°C до приблизительно 40°C и его предпочтительно осуществляют при температуре в диапазоне от приблизительно -5°C до приблизительно 80°C или -5°C до приблизительно 25°C. Последующее добавление и реакция с кислотой или, альтернативно, с обработкой водой, может подходящим образом быть проведено при температуре в диапазоне от приблизительно 0°C до приблизительно 40, или от 0°C до приблизительно 25°C, в течение времени от 5 до 72 часов, особенно, от 10 до 30 часов.
Соединение формулы V может быть последовательно обработано, как описано в международной патентной заявке WO2010/126820, с получением ингибитора бета-лактамазы.
Таким образом, в еще одном варианте осуществления способ P включает Стадии от B до D, как описано выше, или Стадии от A до D, как описано выше, и дополнительно включает:
(E) приведение в контакт соединения V с источником водорода в присутствии катализатора гидрогенолиза с получением соединения формулы VI:
 (VI);
(VI);
(F) приведение в контакт соединения VI с агентом сульфатирования в присутствии органического основания с получением соединения формулы VII, или его фармацевтически приемлемой соли:
 (VII);
(VII);
(G) обработку соединение VII кислотой с получением соединения формулы VIII:
 (VIII),
(VIII),
или его фармацевтически приемлемой соли.
Стадию E проводят в органическом растворителе. Подходящие растворители включают этилацетат, DMAC, NMP, DMF, трет-бутанол, триэтиламин и THF. Предпочтительный растворитель представляет собой THF.
Источник водорода в Стадии E обычно представляет собой газообразный водород, необязательно, в смеси с газом-носителем, который является химически инертным в условиях реакции, используемых в Стадии E (например, азот или благородный газ, такой как гелий или аргон). Давление не является критическим аспектом в Стадии E, хотя атмосферное и суператмосферное является более подходящим. Источник водорода может альтернативно быть водород-переносящей молекулой, такой как формиат аммония, циклогексен или циклогексадиен.
Поглощение водорода не является критическим параметром способа, хотя обычно используют по меньшей мере стехиометрическое количество газообразного водорода или другого источника водорода.
Катализатор гидрогенолиза включает переходные металлы на подложке или без нее, или соединение, соль или комплекс переходного металла, на подложке или без нее. Катализатор, обычно используемый на Стадии E, представляет собой металл Pd и Pt на подложке или без нее, или соединение, соль или комплекс Pd и Pt на подложке или без. Подложка для подходящего катализатора включает углерод, оксид кремния, алюминий, карбид кремния, фторид алюминия и фторид кальция. Класс подходящих катализаторов состоит из черного Pd (то есть, мелкие частицы металлического палладия), Pd(OH)2, Pd/C (то есть, палладий на угле), PtO2 и Pt/C. Pd/C предпочтительно представляет собой катализатор гидрогенолиза. Катализатор обычно используют в количестве в диапазоне от приблизительно 5 до приблизительно 20 масс.% относительно количества соединения V, и чаще используют в количестве в диапазоне от приблизительно 5 до приблизительно 15 масс.% (например, приблизительно 10 масс.%).
Реакцию на Стадии E можно подходящим образом проводить при температуре в диапазоне от приблизительно 10°C до приблизительно 50°C и ее предпочтительно осуществляют при температуре в диапазоне от приблизительно 15°C до приблизительно 30°C.
Сульфатирующий агент на Стадии F предпочтительно представляет собой комплекс триоксида серы и амин, где амин предпочтительно представляет собой третичный амин, включающий, например, ациклические амины (например, триметиламин, TEA, DIPEA, диметилфениламин и диметилбензиламин), циклические амины (например, 1-метилпирролидин и 1-метилпиперидин) и ароматические амины, имеющие один или более атомов N как часть ароматического кольца (например, 1-метилимидазол, пиридин и пиримидин). Галогенсульфоновые кислоты (например, хлорсульфоновая кислота) и комплексы третичного амида SO3 (например, DMF-SO3) также являются подходящими сульфатирующими агентом. Класс подходящих сульфатирующих агентов состоит из комплексов каждого следующего амина с триоксидом серы: пиридин, триметиламин и триэтиламин. Еще один класс подходящих сульфатирующих агентов состоит из комплекса пиридин-SO3, комплекса DMF-SO3 и хлорсульфоновой кислоты. Сульфатирующий реагент обычно используют в количестве в диапазоне от приблизительно 1,5 до приблизительно 7,0 эквивалентов на эквивалент соединения VI, и чаще используют в количестве в диапазоне от приблизительно 3,0 до приблизительно 4,5 эквивалентов.
Органическое основание предпочтительно представляет собой пиридин или третичный амин, такой как 2-пиколин, 2,6-лютидин, отдельный триметилпиридин, или смесь двух или более триметилпиридинов. Класс подходящих оснований состоит из пиколина (например, 2-пиколин, 2,6-лютидин и 2,4,6-триметилпиридин. В предпочтительном варианте осуществления основание представляет собой 2-пиколин или пиридин. Основание обычно используют в количестве в диапазоне от приблизительно 1 до приблизительно 3 эквивалентов на эквивалент соединения VI, и чаще используют в количестве в диапазоне от приблизительно 1,7 до приблизительно 2,2 эквивалентов.
Стадию F проводят в органическом растворителе. Подходящие растворители включают дихлорметан, ацетонитрил, THF, DMF или пиридин. Предпочтительный растворитель представляет собой THF.
Реакция в Стадии F может подходящим образом быть проведена при температуре в диапазоне от приблизительно 0°C до приблизительно 40°C и предпочтительно осуществляют при температуре в диапазоне от приблизительно 10°C до приблизительно 28°C.
Обработка кислотой на Стадии G удаляет Вос-защитную группу. Кислота предпочтительно представляет собой неорганическую кислоту, кислоту Льюиса или органическую кислоту. Подходящие неорганические кислоты включают галогенводороды (HCl, HBr и HF, в виде газа или водного раствора), серную кислоту, тетрафторборную кислоту и азотную кислоту. Подходящие органические кислоты включают карбоновые кислоты, алкилсульфоновые кислоты и арилсульфоновые кислоты. Примеры органических кислот включают трифторуксусную кислоту (TFA), толуолсульфоновую кислоту, бензолсульфоновоую кислоту, метансульфоновую кислоту и трифторметансульфоновую кислоту. Подходящие кислоты Льюиса включают BF3·Et2O, SnCl4, ZnBR2, Me3SiI, Me3SiCl, Me3SiBr, Me3SiOTf и AlCl3. Класс подходящих кислот состоит из Me3SiOTf, TFA и тетрафторборной кислоты. Кислоту обычно используют в количестве в диапазоне от приблизительно 1,0 до приблизительно 2,0 эквивалентов на эквивалент Соединения V, и чаще используют в количестве в диапазоне от приблизительно 1,2 до приблизительно 1,5 эквивалентов. Обработку предпочтительно проводят при температуре в диапазоне от приблизительно -10°C до приблизительно 25°C и предпочтительно осуществляют при температуре в диапазоне от приблизительно 0°C до приблизительно 10°C. Предпочтительная кислота для удаления Вос-защитной группы на Стадии G представляет собой триметилсилил йодид (TMSI), необязательно, в присутствии силилирующего реагента, например, N,O-бис(триметилсилил)ацетамида (BSA) или N,O-Bis(триметилсилил)трифторацетамида (TFBSA). Реакция может обеспечить полное преобразование TMSI в диапазоне от 0,2 эквивалентов до 1,5 эквивалентов или 0,9 эквивалентов до 1,5 эквивалентов. Предпочтительное количество TMSI равно 0,2-1,4 эквивалентов или 1,2-1,4 эквивалентов, которое добавляют к реакционной смеси в течение по меньшей мере 1 часа. Предпочтительные растворители для этой реакции включают MeCN и DCM при приблизительно от 6 объемов до 12 объемов. Обработку предпочтительно проводят при температуре в диапазоне от -20°С до комнатной температуры и предпочтительно осуществляют при температуре в диапазоне от -10 до 25°С или 0-5°С до первоначального образования карбамата TMS как промежуточного продукта.
Промежуточный продукт гасили путем добавляя воду и разнообразные спирты (например, первичные спирты, такие как MeOH, EtOH, n-PrOH, вторичные спирты, такие как iPA, и третичные спирты, такие как трет-BuOH). H2O предпочтительно представляет собой реагент для гашения реакции с получением моногидрата непосредственно в виде кристаллического твердого продукта. pH реакционной смеси необязательно доводят до нейтрального, добавляя органическое основание или кислоту, которая является растворимой в органическом растворителе, таком как триэтиламин, основание Хунига, диизопропиламин и диэтилизопропиламин, ацетат тетра-n-бутиламмония, гидроксид тетра-n-бутиламмония, уксусная кислота, пропионовая кислота, муравьиная кислота, до выделения моногидратного продукта. Количество H2O, используемого для гашения, имеет минимальное влияние на параметры реакции, однако, большая потеря маточного раствора наблюдалась при использовании большого количества H2O. Оптимальное количество H2O составляет 3-6 эквивалента.
При использовании TMSI сырой продукт в виде моногидрата может быть непосредственно выделено путем фильтрации после реакции, а затем провки водным MeCN, с получением не совсем твердого продукта. Сырой продукт может быть перекристаллизован из системы MeCN/H2O/1-бутанол, если необходимо.
Соединения, попадающие в объем Формулы VIII, могут демонстрировать ингибирование β-лактамазы и, таким образом, могут использоваться в качестве ингибиторов β-лактамазы в сочетании с β-лактамными антибиотиками, например, имипенем, цефтазидим и пиперациллин) для лечения бактериальных инфекций, вызванных микроорганизмами, обычно резистентыми β-лактамным антибиотикам в виду присутствия β-лактамаз. Соединения, которые представляют особенный интерес, представляют собой соединения Формулы VIII, в которых R2=R3=H и k=1.
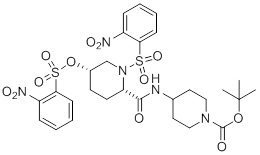
Подвариант осуществления способа P представляет собой способ получения Соединение 3:
 (3),
(3),
который включает:
(B) приведение в контакт лактона 2:
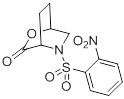 (2)
(2)
с азациклоалкиламином 2-Am:
 (2-Am)
(2-Am)
с последующим приведением в контакт с сульфонилгалогенидом 2-Su:
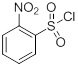 (2-Su)
(2-Su)
в присутствии 4-диметиламинопиридина.
Еще один подвариант осуществления способа P включает Стадию B, как это только что было описано выше, и дополнительно включает:
(A) приведение в контакт соединения 1:
 (1)
(1)
с хлоридом 2-нитробензол-1-сульфонила в присутствии водного основания, а затем добавление TEA, DIPEA или диэтилизопропиламина с получением соединения 2. В одном из вариантов осуществления приведение в контакт проходит в присутствии органического или неорганического основания с получением соединения 2.
Один из вариантов способа P включает Стадию B, как это только что было описано выше, или Стадию A и B, как только что описано, и дополнительно включает:
(C) обработку соединения 3 N-4-нитробензолсульфонил-O-бензилгидроксиламином в присутствии основания, а затем обработку нуклеофильным реагентом, таким как тиол, с получением соединения 4 или его фармацевтически приемлемой соли:
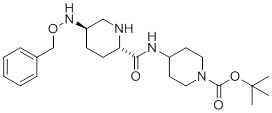 (4).
(4).
Еще одни вариант осуществления способа P включает Стадии от B до С, как описано выше, или Стадию от A до C, как описано выше, и дополнительно включает:
(D) приведение в контакт соединения 4 с фосгеном, дифосгеном или трифосгеном, или эквивалентом фосгена, таким как карбодимидазол или галогенформиат, в присутствии три-C1-4 алкиламина, и затем добавление водного раствора кислоты с получением соединения 5:
 (5).
(5).
Еще один вариант осуществления способа P включает Стадии от B до D, как описано выше, или Стадию от A до D, как описано выше, и дополнительно включает:
(E) приведение в контакт соединения 5 с галогеном в присутствии катализатора Pd и, необязательно, Bос-продуцирующего агента, выбранного из группы, состоящей из ди-t-бутилкарбоната и Вос-ON с получением соединения 6:
 (6);
(6);
(F) приведение в контакт соединения 6 с сульфатирующим агентом, выбранным из группы, состоящей из комплекса пиридин-SO3, хлорсульфоновой кислоты и комплекса DMF-SO3, в присутствии 2-пиколина с получением соединения 7, или его фармацевтически приемлемой соли:
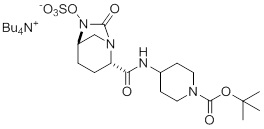 (7); и
(7); и
(G) обработку Соединения 7 кислотой с получением Соединения 8:
 (8),
(8),
или его фармацевтически приемлемой соли. В альтернативном варианте осуществления стадию (F) осуществляют в присутствии пиридина вместо 2-пиколина.
Примерная схема

Растворители, агенты, катализаторы, реакционные количества температура реакции, и тому подобное, как описано выше, для Стадии от A до G в способе P, что дает соединение VIII, применимы для Стадии от A до G, как указано в предыдущих подвариантах осуществления, что дает соединение 8, кроме случаев, когда выраженные ограничения накладываются на одну или несколько этих переменных в подвариантах осуществления.
Следует понимать, что растворители, агенты, катализаторы, количество реакций, температура реакции и тому подобное, как описано выше, в отношении способа P и его вариантов осуществления и субвариантов осуществления, предназначены только для иллюстрации, а не для ограничения объема способа. Например, растворитель, используемый на любой Стадии от А до G, может быть любым органическим веществом, которое в условиях реакции, используемых на интересующей стадии в жидкой фазе, является химически инертным, и будет растворять, суспендировать и/или диспергировать реагенты и любые реагенты, так чтобы привести реагенты и реактивы в контакт и обеспечивать прохождение реакцию. Аналогичные соображения применимы к выбору оснований, катализаторов, реагентов и других реагентов, используемых в стадиях способа. Кроме того, каждую стадию можно проводить при любой температуре, при которой реакция, при которой образуется желаемый продукт, может быть осуществлена. Реагирующие вещества, катализаторы и реагенты в данной стадии могу быть использованы в любых количествах, которые приводят к образованию по крайней мере некоторых из желаемых продуктов. Конечно, высокая степень превращения (например, по меньшей мере приблизительно 60% и предпочтительно выше) исходных веществ в комбинации с высоким выходом (например, по меньшей мере приблизительно 50% и предпочтительно выше) желаемых продуктов обычно является целью каждой стадии, и предпочтителен выбор растворителей, агентов, катализаторов, реакционных количеств, температуры и тому подобное, которые могут обеспечить удовлетворительное превращение и выход продукта, и более предпочтителен выбор, который может обеспечить оптимальное превращение и выход. Конкретные растворители, агенты, катализаторы, количество реакций, температура реакции и тому подобное описаны выше со ссылкой на Способ Р и его варианты осуществления и подварианты осуществлени, могут обеспечить от удовлетворительных до оптимальных превращений и выход.
Время реакции для стадий способа, как описано выше, зависит от таких факторов как (i) выбор и относительные пропорции исходного вещества и других реагентов, (ii) выбор растворителя, (iii) выбор комнатной температуры, и (iv) уровень требуемого превращения. Реакции обычно проводят в течение времени, достаточного для достижения 100% или около 100% превращения (например, 99,5%, 99,0%, 98,0%, 97,0% или 95%).
Прогресс любой стадии реакции, указанной в настоящем описании, может сопровождаться контролем за расходом реагента и/или за появлением желаемого продукта, используя такие аналитическое методики как ТСХ, ВЭЖХ, ИК, ЯМР или GC.
Если не указано иного, то все диапазоны, указанные в настоящем документе, являются включительными; то есть диапазон включает значения верхнего и нижнего предела этого диапазона, а также все значения между ними. Например, под фенильным кольцом, описанным как необязательно замещенное «от 1 до 3 заместителями», понимают, что оно включает в качестве аспектов кольцо, замещенное от 1 до 3 заместителей, от 2 до 3 заместителей, 3 заместителями, от 1 до 2 заместителей, 2 заместителями и 1 заместителем. В качестве еще одного примера, диапазон температур, диапазон эквивалентов и тому подобное, как описано в настоящем документе, включает верхний и нижний предел этого диапазона и любое значение в его пределах.
Термин “алкил” относится к моновалентной прямой или разветвленной цепи, насыщенному алифатическому углеводородному радикалу, имеющему определенное число атомов углерода. Таким образом, например, “C1-6 алкил” (или C1-C6 алкил”) относится к любому алкильному изомеру, гексилу и пентилу, а также к n-, изо-, втор- и трет-бутилу, n- и изо-пропилу, этилу и метилу. В качестве еще одного примера, “C1-4 алкил” относится к n-, изо-, втор- и трет-бутил, n- и изопропилу, этилу и метилу. В качестве еще одного примера “C1-3 алкил” относится к n-пропилу, изопропилу, этилу и метилу.
Термин «разветвленный алкил» относится к алкильной группе, как определено выше, кроме алкильных групп с прямой цепью в определенном диапазоне, которые исключаются. Как определено в настоящем документе, разветвленный алкил включает алкильные группы, в котором алкил присоединен к остальной части соединения через вторичный или третичный углерод; например, изопропил представляет собой разветвленную алкильную группу.
Термин «галоген» относится к атому фтора, хлора, брома и йода (альтернативно называется фтор, хлор, бром и йод).
Термин «галогеналкил» относится к алкильной группе, как определено выше, в котором один или несколько атомов водорода заменены галогеном (то есть, F, Cl, Br и/или I). Таким образом, например, “C1-4 галогеналкил” (или “C1-C4 галогеналкил”) относится к С1-C4 линейной или разветвленной алкильной группе, как определено выше, с одним или несколькими галогеновыми заместителями. Термин “фторалкил” имеет аналогичное значение, за исключение того, что галогеновые заместители ограничены фтором. Подходящие фторалкилы включают серию (CH2)0-4CF3 (то есть, трифторметил, 2,2,2-трифторэтил, 3,3,3-трифтор-n-пропил, и тому подобное).
Настоящее изобретение также относится к соединению:
 (III)
(III)
где:
PG1 представляет собой защитную группу первичного амина, которая образует вместе с азотом амино, к которому она присоединена карбамат, бензиламин или сульфонамид;
PG2 представляет собой защитную группу вторичного амина, выбранную из (i) карбаматов и (ii) бензиламинов,
k представляют собой целое число, равное 0, 1 или 2;
R2 и R3 определены следующим образом:
(а) R2 представляет собой Н, C1-6 алкил, -O-C1-6 алкил, -O-Si(-C1-6 алкил)3 или -O-Si(-C1-6 алкил)(-фенил)2,
и каждый R3 представляет собой Н или C1-6 алкил; или
(b) альтернативно и при условии, что k равно 1 или 2, R2 и R3, расположенный рядом с R2, вместе с атомами углерода, к которым каждый присоединен, образуют C5-7 циклоалкил, каждый необязательно замещен от 1 до 3 заместителями, каждый из которых независимо представляет собой C1-6 алкил, -O-C1-6 алкил, -O-Si(-C1-6 алкил)3 или -O-Si(-C1-6 алкил)(-фенил)2; и любой другой R3 представляет собой Н или C1-6 алкил;
R4 представляют собой:
(1) фенил, необязательно замещенный от 1 до 3 заместителями, каждый из которых независимо представляет собой C1-4 алкил, C1-4 галогеналкил, -O-C1-4 алкил, -O-C1-4 галогеналкил, Cl, Br, F или NO2;
(2) C1-4 алкил; или
(3) C1-4 галогеналкил;
R5 представляет собой Н или C1-3 алкил;
R6 и R8 независимо представляют собой H, C1-3 алкил, -O-C1-3 алкил или -N(-C1-3 алкил)2;
каждый R7 и R9 независимо представляет собой H или C1-6 алкил;
W представляет собой галоген;
p равно 0, 1 или 2;
q равно 0, 1 или 2; и
p + q = 0, 1, 2 или 3.
Настоящее изобретение дополнительно относится к соединениям, в том числе:

Аббревиатуры, используемые в настоящем документе, включают следующее:
Следующие примеры служат только для иллюстрации изобретения и его осуществления. Примеры не предназначены для ограничения объема и сущности изобретения.
Пример 1
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфoокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид

Стадия A: Получение (1S,4S)-5-((2-нитрофенил)сульфонил)-2-окса-5-азабицикло[2.2.2]октан-3-она (2)

Реактор (R-1), оснащенный дополнительной воронкой, входом для азота и мешалкой, наполняли (2S,5S)-5-гидроксипиперидин-2-карбоновой кислотой (77,3 масс.%) (50,0 г, 344 ммоль) и водой (150 мл). Начинали перемешивание, значение pH доводили до 10-11, добавляя 10 н NaOH (~46,5 мл), и реактор наполняли ацетоном (50,0 мл).
Отдельный реактор (R-2), оснащенном мешалкой и входом для азота, наполняли хлоридом 2-нитробензол-1-сульфонила (97%) (106,0 г, 478 ммоль) и ацетоном (80 мл). Содержимое R-2 переносили в R-1 при 23-30°C, при этом значение pH раствора поддерживали при 10-11, одновременно добавляя 10 н NaOH. Через 15-30 минут значение pH доводили до приблизительно 6, добавляя 12 н HCl. В раствор подавали EtOAc (500 мл), и значение pH доводили до 3,0, добавляя 12 н HCl. Слои разделяли и водный слой снова экстрагировали EtOAc (150 мл × 2).
Отдельный реактор (R-3) наполняли продуктом 1a в объединенных органических слоях, хлориде 2-нитробензол-1-сульфонила (73,0 г, 329 ммоль) и триэтиламине (130 мл). Партию в R-3 перемешивали при 20-28°С в течение 30 минут. Раствор наполняли водой (100 мл), слои разделяли, и водный слой снова экстрагировали EtOAc (150 мл × 2). Объединенный EtOAc-слой промывали 10% NaHCO3 (100 мл) и насыщенным солевым раствором (100 мл). Органическую фазу концентрировали до 150 мл, при этом образовывалась кристаллическая взвесь. Концентрированный раствор перемешивали при 13-18°С в течение 2-3 часов, а потом отфильтровали кристаллические твердые вещества. Полученный влажный осадок промывали EtOAc (60 мл) и затем сушили в печи под вакуумом при 25-30°С с получением 2 (65,6 г, выход 79%), т.пл. 126,0-126,7°C. 1H ЯМР (CDCl3, 400 МГц) δ: 8,02 (м, 1Н), 7,80-7,71 (м, 2Н), 7,66 (м, 1Н), 4,88 (м, 1Н), 4,55 (дд, J=3,8, 2,7 Гц, 1Н), 3,78 (дт, J=11,2, 3,0 Гц, 1Н), 3,66 (дд, J=11,2, 1,1 Гц, 1Н), 2,44 (м, 1Н), 2,11 (м, 2Н), 1,91 (м, 1Н); 13C ЯМР (CDCl3, 100 MГц) δ: 168,4, 148,3, 134,4, 132,1, 131,0, 130,7, 124,2, 73,5, 51,4, 48,0, 25,1, 23,2
Стадия B: Получение трет-бутил 4-((2S,5S)-1-((2-нитрофенил)сульфонил)-5-(((2-нитрофенил)сульфонил)окси)пиперидин-2-карбоксамидо)пиперидин-1-карбоксилата (3)

Реактор (R-1) наполняли лактоном 2 (65,5 г, 210 ммоль), THF (131 мл) и трет-бутил 4-аминопиперидин-1-карбоксилатом (44,5 г, 222 ммоль). Перемешанный раствор нагревали при кипячении с обратным холодильником (обычно при температуре 72°C) в течение ~18 часов. Реакционную смесь охлаждали до 25-35°C и затем наполняли THF (325 мл) и 4-диметиламинопиридин (40,1 г, 328 ммоль), а затем перемешивали в течение 30 минут.
Отдельный реактор (R-2) наполняли хлоридом 2-нитробензол-1-сульфонила (60,9 г, 275 ммоль) и THF (200 мл). Содержимое R-2 добавляли в R-1 в течение от 45 до 75 минут, поддерживая температуру партии от 20 до 30°С. Партию в R-1 перешивали в течение от 2 до 4 часов при температуре от 20 до 30°С.
Отдельный реактор (R-3) наполняли водой (600 мл) и метанолом (600 мл). Содержимое R-3 загружали в основную партию в течение от 45 до 75 минут при перемешивали, поддерживая температуру от 20 до 30°С. Партию охлаждали до температуры от 5 до -5°С и затем перемешивали при температуре от 5 до -5°С в течение по меньшей мере 4 часов. Твердые продукты фильтровали и затем промывали дважды метанолом (130 мл × 2). Сырой остаток сушили в вакуумной печи при температуре от 40 до 50°С с получением 3 (144,0 г, выход 98%), т.пл. 131,8-133,1°C. 1H ЯМР (CDCl3, 400 MГц) δ: 8,14 (м, 2Н), 7,83-7,74 (м, 6Н), 6,50 (д, J=7,9 Гц, 1Н), 4,69 (м, 1Н), 4,43 (c, 1H), 4,11 (дд, J=13,7, 4,9 Гц, 1H), 3,95 (м, 2H), 3,83 (м, 1H), 3,47 (c, 1H), 3,10 (дд, J=13,7, 11,0 Гц, 1H), 2,81 (м, 2H), 2,51 (м, 1H), 2,12 (м, 1H), 1,85-1,72 (м, 4H), 1,45 (c, 9H), 1,26 (м, 1H); 13C ЯМР (CDCl3, 100 МГц) δ: 166,9, 154,6, 148,2, 147,6, 135,2, 134,8, 132,6, 132,5, 131,9, 131,6, 131,4, 129,7, 124,9, 124,7, 79,8, 76,5, 55,0, 47,1, 46,0, 31,8, 31,5, 28,4, 27,3, 24,4.
Получение N-4-нитробензол сульфонил-O-бензилгидроксиламина

Реактор (R-1) наполняли гидрохлоридом O-бензилгидроксиламина (61,0 г, 382 ммоль) и пиридином (400 мл). Раствор охлаждали до температуры от 5 до -5°С.
Отдельный (R-2) наполняли хлоридом 4-нитробензолсульфонила (89,0 г, 402 ммоль) и пиридином (200 мл). Содержимое R-2 переносили в R-1, поддерживая диапазон температуры от -5 до -5°С. Партию в R-1 перемешивали при температуре от 5 до -5°С в течение от 15 до 45 минут, затем нагревали до температуры от 20 до 30°С в течение от 45 до 75 минут. Затем добавляли воду (250 мл), поддерживая температуру от 20 до 30°С и перемешивая от 5 до 15 минут. Твердые продукты фильтровали и влажный осадок промывали водой (100 мл × 3). Сырой осадок сушили в вакуумной печи при 50°С с получением N-4-нитробензолсульфонил-O-бензилгидроксиламина (113,3 г, выход 96%), т.пл. 128,4-130,0 °C, 1H ЯМР (CDCl3, 400 МГц) δ: 8,36 (д, J=8,9 Гц, 2Н), 8,11 (д, J=8,9 Гц, 2Н), 7,36 (м, 5H), 7,11 (c, 1H), 5,02 (c, 2H); 13C ЯМР (CDCl3, 100 МГц) δ: 151,0, 142,5, 134,9, 130,2, 129,7, 129,3, 128,9, 124,5, 80,2.
Стадия C. Получение трет-бутил 4-((2S,5R)-5-((бензилокси)амино)пиперидин-2-карбоксамидо)пиперидин-1-карбоксилата (4)

Реактор (R-1) наполняли трет-бутил 4-((2R,5R)-1-((2-нитрофенил)сульфонил)-5-(((2-нитрофенил)сульфонил)окси)пиперидин-2-карбоксамидо)пиперидин-1-карбоксилатом (3) (110 г, 158 ммоль), N-4-нитробензол сульфонил-O-бензилгидроксиламином (58 г, 188 ммоль), карбонатом калия (25,9 г, 187 ммоль) и диметилацетамидом (440 мл). Перемешанный раствор нагревали до температуры от 60 до 70°С в течение 24-32 часов. Партию охлаждали до температуры от 20 до 30°С и наполняли толуолом (660 мл). Партию экстрагировали 1 н гидроксидом натрия (3×220 мл), затем промывали водой (220 мл).
Раствор толуола азотропно дистиллировали при ~50°C до приблизительно 1/3 объема. Растворитель в растворе меняли на MeOH при 45-55°C, доводили до 237 мл.
Партию охлаждали до 20-25°C, наполняли тригликолевой кислотой (57,9 г, 629 ммоль) при 10°C, и затем наполняли безводным K2CO3 (172,0 г, 1225 ммоль). Партию перемешивали при 10-15°C в течение 0,5 часов, нагревали до 20-25°C, перемешивали при 20-25°C в течение 10-15 часов и нагревали при 48-53°C в течение 3-6 часов.
Партию наполняли 10 масс.% хлоридом натрия (1,10 л) и толуолом (880 мл) при приблизительно 40°C. Слои разделяли и водный слой опять экстрагировали толуолом (3×440 мл). Объединенный органический слой промывали 10% NaHCO3 (2×220 мл). Партию концентрировали при 40-50°C до 165 мл, затем охлаждали до 35-40°C. В партию добавляли затравку (50 мг) и перемешивали в течение 1 часа при 35-40°C. Партию наполняли гептаном (110 мл) при 35-40°C в течение 1 часа, затем медленно охлаждали до 15-20°C в течение 1 часа. Партию перемешивали в течение 3 часов, и твердые продукты фильтровали. Сырой осадок промывали толуолом/гептаном (137,5 мл), затем сушили в вакуумной печи при 30°C в течение 3-8 часов с получением 4 (47,3 г, общий выход 70% из 3), т.пл. 117,5-118,0°C, 1H ЯМР (CDCl3, 500 МГц) δ: 7,37-7,29 (м, 5Н), 6,64 (д, J=8,2 Гц, 1Н), 5,36 (ушир.с, 1Н), 4,67 (c, 2Н), 4,00 (м, 2Н), 3,90 (м, 1Н), 3,28 (ддд, J=11,8, 4,0, 1,7 Гц, 1Н), 3,12 (дд, J=10,2, 3,2 Гц, 1Н), 2,95 (м, 1Н), 2,86 (м, 2Н), 2,46 (дд, J=11,8, 9,5 Гц, 1Н), 2,10 (м, 1Н), 1,93-1,83 (м, 3Н), 1,58 (ушир.с, 1Н), 1,45 (c, 9Н), 1,41 (м, 1Н), 1,35-1,23 (м, 3Н); 13C ЯМР (CDCl3, 125 МГц) δ: 172,8, 154,7, 137,7, 128,4 (4C), 127,9, 79,6, 76,9, 59,8, 57,0, 49,2, 46,1, 42,8 (ушир., 2C), 32,0 (2C), 28,4 (3C), 28,3, 27,2.
Стадия D: Получение трет-бутил 4-((1R,2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (5)

Реактор (R-1) наполняли трет-бутил 4-((2S,5R)-5-((бензилокси)амино)пиперидин-2-карбоксамидо)пиперидин-1-карбоксилатом (4) (46,3 г, 107 ммоль), дихлорметаном (463 мл) и основанием Хунига (58,0 мл). Партию охлаждали до -18°C и затем добавляли трифосген четырьмя порциями (всего 25,1 г; 85 ммоль) при <-8°C. Партию перемешивали при температуре от -5 до 0°C в течение 0,5 часов, затем нагружали 11,4 масс.% водным H3PO4 при температуре от -5 до 0°C (347 г, 3541 ммоль). Партию перемешивали при 20-25°C в течение 15-20 часов, затем фазу удаляли. Водный слой снова экстрагировали дихлорметаном (138 мл). Объединенный органический слой промывали 10% NaHCO3 (115 мл), затем водой (115 мл). Органический раствор концентрировали при атмосферном давлении до ~80 мл, затем помещали MTBE (347 мл) при 35-45°C в течение 0,5 часов, затем концентрировали при 35-45°C до 231 мл два раза до образования взвеси.
В взвесь добавляли гептан (139 мл) при 35-45°C в течение 2 часов, затем медленно охлаждали до 15-20°C в течение 1 часа. Партию перемешивали при 15-20°C в течение 6-8 часов. Твердые продукты фильтровали, и сырой остаток промывали MTBE/гептаном (1,4:1,185 мл), затем сушили под вакуумом при 25-30°С в течение 5-10 часов с получением 5 (43,7 г, выход 92%), т.пл. 161,3-161,8°C. 1H ЯМР (CDCl3, 500 МГц) δ: 7,45-7,32 (м, 5Н), 6,55 (д, J=8,2 Гц, 1Н), 5,05 (д, J=11,6 Гц, 1Н), 4,90 (д, J=11,6 Гц, 1Н), 4,02 (м, 2Н), 3,90 (м, 2Н), 3,30 (м, 1Н), 2,99 (дт, J=11,7, 1,1 Гц, 1Н), 2,86 (м, 2Н), 2,64 (д, J=11,7 Гц, 1Н), 2,37 (дд, J=14,6, 6,9 Гц, 1Н), 2,04-1,82 (м, 4Н), 1,58 (м, 1Н), 1,45 (c, 9Н), 1,30 (м, 2Н); 13C ЯМР (CDCl3, 125 МГц) δ: 168,3, 167,5, 154,7, 135,6, 129,2 (2C), 128,8, 128,6 (2 C), 79,7, 78,3, 60,4, 57,8, 47,5, 46,8, 42,5 (ушир., 2 C), 32,0, 31,7, 28,4 (3 C), 20,8, 17,2,
Стадия E: Получение трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата

Трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (9,2 г, 20,1 ммоль) добавляли в стеклянную бутылку, и твердые продукты растворяли в THF (150 мл). Раствор затем помещали в реактор для гидрирования вместе с Pd/Al2O3 (10 масс.%, 1,5 г). Реакционную смесь продували три раза галогеном и затем устанавливали давление водорода, равное 50 фунтов на квадратный дюйм. Температуру реакции доводили до 25°С, и реакционную смесь оставляли перемешиваться в течение 22 часов. После завершения реакции, что определяли с помощью анализом ВЭЖХ, раствор фильтровали через SOLKA-FL°С® (Interational Fiber Corporation, North Tonawanda, NY) для удаления катализатора и отфильтрованный осадок промывали THF. В фильтрате и промывках затем переключали растворитель путем вакуумной дистилляции на iPrOAc до конечного объема 40 мл. Полученную взвесь iPrOAc перемешивали при комнатной температуре в течение 1 часа. Твердые продукты затем фильтровали и промывали iPrOAc (20 мл) и сушили под вакуумом и N2 при 40°С до получения продукта, указанного в заголовке (6,62 g, 17,97 ммоль, выход 90%). Спектральные данные совпали со ссылочным соединением.
Получение (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида

Трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (20 г, 54,3 ммоль), THF (200 мл), 2-пиколин (10,9 мл, 309 ммоль) и комплекс пиридин-SO3 (30,2 г, 190 ммоль) помещали в колбу в атмосфере азота. Гетерогенную смесь оставляли перемешиваться в течение ночи (~15Н). Реакционную смесь охлаждали до -10°С, затем добавляли DCM (200 мл). Добавляли 0,5 M K2HPO4 (168 мл, 84 ммоль) в течение 10 минут. Затем добавляли Bu4NHSO4 (19,4 г, 57 ммоль) в течение 10 минут. Двухфазную смесь перемешивали в течение 30 минут, фазу удаляли и водный слой снова экстрагировали 40 мл DCM. Объединенный раствор DCM промывали водой (120 мл), фазу удаляли, и в растворе с органическим растворителем меняли растворитель на MeCN (320 мл) при вакуумной дистилляции с 3-мя объемами слоя MeCN (всего 1,0 л) и использовали в следующей стадии. Раствор соли Bu4N+ -OSO3 7 в растворе MeCN использовали с предположительным выходом 100% (37,5 г, 54,3 ммоль). Реакционную смесь охлаждали на ледяной бане, и добавляли TMSI (10,26 мл, 70,7 ммоль) с помощью дополнительной воронки в течение 30 минут между 0°C и 5°C. Полученную смесь перемешивали в течение 1-2 часа и затем гасили H2O:MeCN (1:1, 6 мл) с получением взвеси. Взвесь нагревали до комнатной температуры и перемешивали в течение 12 часов и после этого времени значение рН супернатанта составило приблизительно 3,0. Ацетат тeтрабутиламмония (13,6 мл, 13,59 ммоль) медленно добавляли в течение 30 минут. Взвесь перемешивали в течение 1 часа и значение pH супернатанта составило приблизительно 4,0. Твердые продукты собирали путем фильтрации. Твердый продукт промывали 60 мл водным раствором MeCN с получением 19,5 г сырого продукта 8 с выходом 93% соединения 6.
На этой стадии все побочные продукты (включая продукты гидролиза TMS-карбоната) и примеси были растворимы в органической фазе.
Продукт снова растворяли в 140 мл MeCN:H2O (1:2) при комнатной температуре. 1-Бутанол (390 мл) в качестве антирастворителя медленно добавляли в раствор с получением взвеси. Взвесь перемешивали в течение ночи. Белый кристаллический продукт фильтровали и промывали 3:1 IPA:вода (40 мл) и сушили под вакуумом и в атмосфере азота при комнатной температуре с получением продукта, указанного в заголовке в форме кристаллического гидрата. (Выход=16,3 г, 82%). Спектральные данные совпали со ссылочным соединением.
Получение сульфата (2 S ,5 R )-7-оксо-2-(пиперидин-1-ий-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ила (1).
Трет-бутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат 16 (0,54 г, 1,5 ммоль), THF (5,4 мл), 2-пиколин (0,29 мл, 2,9 ммоль) и комплекс пиридин-SO3 (0,70 г, 4,4 ммоль) помещали в пробирку в атмосфере азота. Гетерогенную смесь оставляли перемешиваться в течение ночи (~15 часов). Реакционную смесь охлаждали до -10°С, затем добавляли дихлорметан (5,4 мл). Добавляли 0,5 M K2HPO4 (4,5 мл, 2,3 ммоль) в течение 10 минут. Затем добавляли Bu4NHSO4 (0,53 г, 1,54 ммоль) в течение 10 минут. Двухфазную смесь перемешивали в течение 30 минут, фазу удаляли и водный слой снова экстрагировали 1 мл DCM. Объединенный раствор DCM промывали водой (2,0 мл), фазу удаляли, и в растворе с органическим растворителем меняли растворитель на MeCN (3,2 мл) с помощью вакуумной дистилляции 3-мя объемами слоя MeCN. Продукт использовали в следующей стадии (водное содержимое меньше 1000 ppm).
Раствор соли Bu4N+SO4-- 8 в растворе MeCN использовали с предполагаемым выходом 100% (1,0 г, 1,47 ммоль). Реакционную смесь охлаждали на ледяной бане и добавляли N,O-бис(триметилсилил)трифторацетамид (BSTFA) (0,41 г, 1,59 ммоль) в реакционную смесь и оставляли перемешиваться в течение 10 минут. Добавляли TMSI (0,06 г, 0,27 ммоль) между 0°C и 5°C. Полученную смесь оставляли перемешиваться в течение 2 часов и затем гасили H2O (0,07 г, 4,1 ммоль) и уксусной кислотой (0,08 г, 1,5 ммоль) с получением взвеси. Взвесь нагревали до комнатной температуры и перемешивали в течение 12 часов. В фильтре собирали твердые продукты. Твердые продукты промывали MeCN/вода (94:6, 1 мл × 4) с получением кристаллического продукта 1 (0,38 г) с выходом 75%.
Если наносили N,O-бис(триметилсилил)ацетамид (BSA) (0,32 г, 1,59 ммоль), то реакции требовалось 24 ч для полного превращения.
Несмотря на то, что в вышеприведенном описании описаны принципы настоящего изобретения, с примерами, приведенными с целью иллюстрации, на практике изобретение охватывает все обычные вариации, адаптации и/или модификации, которые находятся в пределах объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| СОЕДИНЕНИЯ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА BRM, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2797832C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2815314C2 |
| ИНГИБИТОРЫ β-ЛАКТАМАЗ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2753401C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| ПРОИЗВОДНЫЕ 7-ОКСО-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-3-ЕНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2645678C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2715058C2 |
| ОПТИЧЕСКИ АКТИВНОЕ ПРОИЗВОДНОЕ ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2591701C2 |
Изобретение относится к способу получения N-защищенных 6-(пиперидин-4-илкарбамоил)пиперидин-3-илсульфонатов формулы III, где соединение формулы III представляет собой соединение 3, который включает приведение в контакт лактона 2 с азациклоалкиламином 2-Am с последующим приведением в контакт с сульфонилгалогенидом 2-Su в присутствии 4-диметиламинопиридина. Изобретение также относится к промежуточным соединениям для получения соединения формулы 3. Технический результат – разработан новый способ получения N-защищенных 6-(пиперидин-4-илкарбамоил)пиперидин-3-илсульфонатов с высоким выходом и чистотой конечного продукта, который является промежуточным для получения 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидов. 2 н. и 11 з.п. ф-лы, 1 пр.
 (3)
(3)  (2)
(2)
 (2-Am)
(2-Am)  (2-Su)
(2-Su)
1. Способ получения соединения формулы III, где соединение формулы III представляет собой соединение 3:
 (3),
(3),
который включает:
(B) приведение в контакт лактона 2:
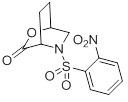 (2)
(2)
с азациклоалкиламином 2-Am:
 (2-Am)
(2-Am)
с последующим приведением в контакт с сульфонилгалогенидом 2-Su:
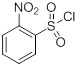 (2-Su)
(2-Su)
в присутствии 4-диметиламинопиридина.
2. Способ по п.1, который дополнительно включает:
(A) приведение в контакт соединения 1:
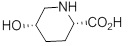 (1)
(1)
с хлоридом 2-нитробензол-1-сульфонила в присутствии органического или неорганического основания с получением соединения 2.
3. Способ по п.1, который дополнительно включает:
(A) приведение в контакт соединения 1:
(1)
с хлоридом 2-нитробензол-1-сульфонила в присутствии водного основания, после чего добавляли TEA, DIPEA или диэтилизопропиламин с получением соединения 2.
4. Способ по п.2 или 3, который дополнительно включает:
(C) обработку соединения 3 N-4-нитробензолсульфонил-O-бензилгидроксиламином в присутствии основания, после чего следует обработка тиолом с получением соединения 4:
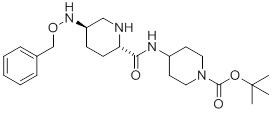 (4).
(4).
5. Способ по п.4, который дополнительно включает:
(D) приведение в контакт соединения 4 с фосгеном, дифосгеном или трифосгеном в присутствии три-C1-4 алкиламина, и затем добавление водного раствора кислоты с получением соединения 5:
 (5).
(5).
6. Способ по п.5, который дополнительно включает:
(E) приведение в контакт соединения 5 с галогеном в присутствии катализатора Pd с получением соединения 6:
 (6).
(6).
7. Способ по п.6, который дополнительно включает:
(F) приведение в контакт соединения 6 с сульфатирующим агентом, выбранным из группы, состоящей из комплекса пиридин-SO3, хлорсульфоновой кислоты и комплекса DMF-SO3, в присутствии 2-пиколина с получением соединения 7:
 (7).
(7).
8. Способ по п.6, который дополнительно включает:
(F) приведение в контакт соединения 6 с сульфатирующим агентом, выбранным из группы, состоящей из комплекса пиридин-SO3, хлорсульфоновой кислоты и комплекса DMF-SO3 в присутствии пиколина с получением соединения 7:
(7).
9. Способ по п.7 или 8, который дополнительно включает:
(G) обработку соединения 7 кислотой с получением соединения 8:
 (8).
(8).
10. Способ по п.9, где кислота представляет собой йодид триметилсилил (TMSI).
11. Способ по п.10, где TMSI используют в присутствии N,O-бис(триметилсилил)ацетамида (BSA) или N,O-бис(триметилсилил)трифторацетамид (BSTFA).
12. Способ по п.9, где соединение 8 непосредственно кристаллизуется как моногидрат из реакционной смеси.
13. Соединение, которое выбрано из группы, состоящей из:
 ,
, 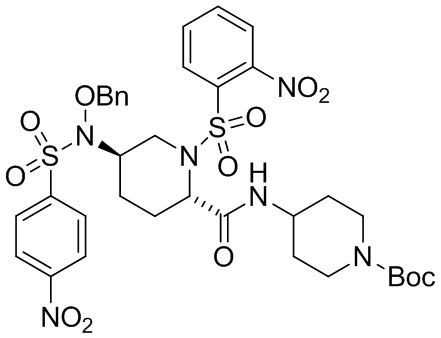 и
и  .
.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| US 5510343 А, 23.04.1996 | |||
| ПРОИЗВОДНЫЕ БЕНЗАМИДА ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ УКАЗАННОГО ПРОИЗВОДНОГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2333198C2 |

