Область техники, к которой относится изобретение
Настоящее раскрытие относится к ингибиторам β-лактамаз и их фармацевтическим композициям и применению ингибиторов β-лактамаз для лечения бактериальных инфекций.
Предшествующий уровень техники
Злоупотребление, неправильное применение и сельскохозяйственное применение антибиотиков привело к появлению резистентных бактерий, которые устойчивы к уничтожению с помощью обычных противоинфекционных средств, таких как средства на основе β-лактамов или фторхинолиновых структур. Вызывает беспокойство, что многие из этих резистентных бактерий ответственны за распространенные инфекции, включая, например, пневмонию, сепсис и т.д.
Развитие устойчивости к широко используемым β-лактамным противоинфекционным средствам связано с экспрессией β-лактамаз целевыми бактериями. β-лактамазы обычно гидролизуют β-лактамное кольцо, делая таким образом антибиотик неэффективным против бактерий. Соответственно, ингибирование β-лактамаз с помощью подходящего субстрата может предотвратить разрушение β-лактамного антибиотика, следовательно повышая эффективность вводимого антибиотика и уменьшая появление устойчивости.
Авибактам является известным ингибитором β-лактамазы, который в настоящее время продается в комбинации с цефтазидимом для лечения грамотрицательных бактериальных инфекций. Авибактам следует вводить внутривенно, что ограничивает применение дорогими клиническими условиями.
Сущность Изобретения
Согласно настоящему изобретению соединения имеют структуру формулы (1):
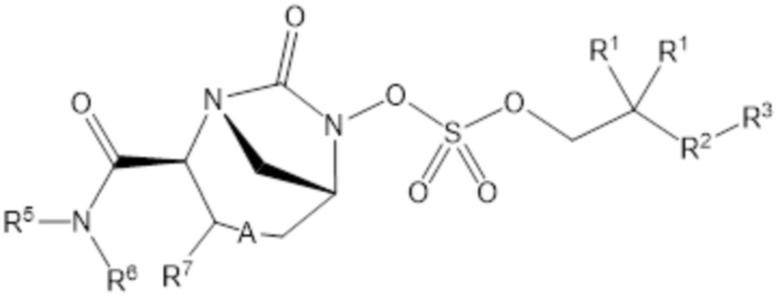 (1)
(1)
или его фармацевтически приемлемой соли, при этом
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила;
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2; при этом
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
Согласно настоящему изобретению фармацевтические композиции содержат соединение согласно настоящему изобретению и фармацевтически приемлемую основу.
Согласно настоящему изобретению способы лечения бактериальной инфекции у пациента включают введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения согласно настоящему изобретению.
Согласно настоящему изобретению способы лечения бактериальной инфекции у пациента включают введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества фармацевтической композиции согласно настоящему изобретению.
Согласно настоящему изобретению способы ингибирования β-лактамазы у пациента включают введение пациенту эффективного количества соединения согласно настоящему изобретению.
Согласно настоящему изобретению способы ингибирования β-лактамазы у пациента включают введение пациенту эффективного количества фармацевтической композиции согласно настоящему изобретению.
Далее сделана ссылка на некоторые соединения и способы. Раскрытые варианты осуществления не предназначены для ограничения формулы изобретения. Наоборот, формула изобретения предназначена для охвата всех альтернативных вариантов, модификаций и эквивалентов.
Подробное описание
Тире («-»), которое находится не между двумя буквами или символами, используют для обозначения точки присоединения для фрагмента или заместителя. Например, -CONH2 присоединяют через атом углерода.
«Алкил» относится к насыщенному или ненасыщенному моновалентному углеводородному радикалу, разветвленному или с прямой цепью, полученному путем удаления одного атома водорода из одного атома углерода исходного алкана, алкена или алкина. Примеры алкильных групп включают метил; этилы, такие как этанил, этенил и этинил; пропилы, такие как пропан-1-ил, пропан-2-ил, проп-1-ен-1-ил, проп-1-ен-2-ил, проп-2-ен-1-ил (аллил), проп-1-ин-1-ил, проп-2-ин-1-ил и т.д.; бутилы, такие как бутан-1-ил, бутан-2-ил, 2-метил-пропан-1-ил, 2-метил-пропан-2-ил, бут-1-ен-1-ил, бут-1-ен-2-ил, 2-метил-проп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил, бут-1-ин-1-ил, бут-1-ин-3-ил, бут-3-ин-1-ил и т.д.; и тому подобное. Термин «алкил» предназначен конкретно для включения групп, имеющих любую степень или уровень насыщения, т.е. групп, имеющих исключительно углерод-углеродные одинарные связи, групп, имеющих одну или более углерод-углеродных двойных связей, групп, имеющих одну или более углерод-углеродных тройных связей, и групп, имеющих комбинации углерод-углеродных одинарных, двойных и тройных связей. Когда предполагается конкретный уровень насыщения, используют термины алканил, алкенил и алкинил. Алкильной группой может быть C1-6 алкил, C1-5 алкил, C1-4 алкил, C1-3 алкил, этил или метил.
«Алкокси» относится к радикалу -OR, где R представляет собой алкил согласно определению настоящего документа. Примеры алкокси групп включают метокси, этокси, пропокси и бутокси. Алкокси группой может быть C1-6 алкокси, C1-5 алкокси, C1-4 алкокси, C1-3 алкокси, этокси или метокси.
«Арил» сам по себе или как часть другого заместителя относится к моновалентному ароматическому углеводородному радикалу, полученному за счет удаления одного атома водорода из одного атома углерода исходной ароматической кольцевой системы. Арил охватывает 5- и 6-членные карбоциклические ароматические кольца, например, бензол; бициклические кольцевые системы, в которых по меньшей мере одно кольцо является карбоциклическим и ароматическим, например, нафталин, индан и тетралин; и трициклические кольцевые системы, в которых по меньшей мере одно кольцо является карбоциклическим и ароматическим, например, флуорен. Арил охватывает множество кольцевых систем, имеющих по меньшей мере одно карбоциклическое ароматическое кольцо, слитое по меньшей мере с одним карбоциклическим ароматическим кольцом, циклоалкильным кольцом или гетероциклоалкильным кольцом. Например, арил содержит фенильное кольцо, слитое с 5-7-членным гетероциклоалкильным кольцом, содержащим один или более гетероатомов, выбранных из N, O и S. Для подобных слитых, бициклических кольцевых систем, в которых только одним из колец является карбоциклическое ароматическое кольцо, атом углерода радикала может находиться в карбоциклическом ароматическом кольце или в гетероциклоалкильном кольце. Примеры арильных групп включают группы, полученные из ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, коронена, флуорантена, флуорена, гексацена, гексафена, гексалена, ассим-индацена, симм-индацена, индана, индена, нафталина, октацена, октафена, окталена, овалена, пентацена, пенталина, пентафена, перилена, феналена, фенантрена, пицена, плеядена, пирена, пирантрена, рубицена, трифенилена, тринафталина и тому подобное. Арильной группой может быть C6-10 арил, C6-9 арил, C6-8 арил или фенил. Однако, арил никоим образом не охватывает или не пересекается с гетероарилом, определенном в настоящем документе отдельно.
«Арилалкил» относится к ациклическому алкильному радикалу, в котором один из атомов водорода, связанных с атомом углерода, заменен арильной группой. Примеры арилалкильных групп включают бензил, 2-фенилэтан-1-ил, 2-фенилэтен-1-ил, нафтилметил, 2-нафтилэтан-1-ил, 2-нафтилэтен-1-ил, нафтобензил и 2-нафтофенилэтан-1-ил. Когда предполагаются конкретные алкильные фрагменты, используют терминологию арилалканил, арилалкенил или арилалкинил. Арилалкильной группой может быть C7-16 арилалкил, например, алканильный, алкенильный или алкинильный фрагмент арилалкильной группы представляет собой C1-6, а арильный фрагмент представляет собой C6-10. Арилалкильной группой может быть C7-16 арилалкил, например, алканильный, алкенильный или алкинильный фрагмент арилалкильной группы представляет собой C1-6, а арильный фрагмент представляет собой C6-10. Арилалкильной группой может быть C7-9 арилалкил, в котором алкильный фрагмент представляет собой C1-3 алкил, а арильный фрагмент представляет собой фенил. Арилалкильной группой может быть C7-16 арилалкил, C7-14 арилалкил, C7-12 арилалкил, C7-10 арилалкил, C7-8 арилалкил или бензил.
«Биодоступность» относится к норме и количеству лекарственного средства, которое достигает системного кровообращения пациента после введения лекарственного средства или его пролекарства пациенту, и может быть определена путем оценки, например, профиля концентрация-время в плазме или крови для лекарственного средства. Параметры, используемые для получения характеристики кривой концентрация-время в плазме или крови, включают площадь под кривой (AUC), время до максимальной концентрации (Tmax) и максимальная концентрация лекарственного средства (Cmax), где Cmax является максимальная концентрация лекарственного средства в плазме или крови пациента после введения пациенту дозы лекарственного средства или формы лекарственного средства, а Tmax является время до максимальной концентрации (Cmax) лекарственного средства в плазме или крови пациента после введения пациенту дозы лекарственного средства или формы лекарственного средства.
«Пероральная биодоступность» (F%) относится к доле введенного перорального лекарственного средства, которая достигает системного кровообращения. Пероральная биодоступность представляет собой произведение поглощенной доли, доли, избежавшей элиминации на стенках кишечника и доли, избежавшей, печеночной элиминации; а факторы, которые влияют на биодоступность, можно разделить на физиологические, физикохимические и биофармацевтические факторы.
«Соединения» и фрагменты, раскрытые в настоящем документе, включают любые конкретные соединения в рамках этих формул. Соединения можно идентифицировать либо по их химической структуре и/или по химическому названию. Соединения называют, используя программу терминологии/структуры ChemBioDraw Ultra Version 14.0.0.117 (CambridgeSoft, Cambridge, MA). При конфликте химической структуры и химического названия определяющим для идентичности соединения является химическая структура. Соединения, описанные в настоящем документе, могут содержать один или более стереогенных центров и/или двойных связей и, следовательно, могут существовать в виде стереоизомеров, таких как изомеры с двойными связями (т.е. геометрические изомеры), энантиомеры, диастереомеры или атропоизомеры. Соответственно, любые химические структуры в рамках объема приведенного описания, в целом или частично, с относительной конфигурацией охватывают все возможные энантиомеры и стереоизомеры проиллюстрированных соединений, включая стереоизомерно чистую форму (например, геометрически чистую, энантиомерно чистую или диастереомерно чистую) и энантиомерные и стереоизомерные смеси. Энантиомерные и стереоизомерные смеси можно разделить на составляющие их энантиомеры или стереоизомеры с использованием методов разделения или методов хирального синтеза, хорошо известных специалисту.
Раскрытые в настоящем документе соединения и фрагменты включают оптические изомеры соединений и фрагментов, их рацематы и другие их смеси. В таких вариантах осуществления отдельные энантиомеры или диастереомеры можно получать посредством асимметричного синтеза или посредством разделения рацематов. Разделение рацематов можно выполнять, например, с помощью обычных способов, таких как кристаллизация в присутствии разделяющего средства или хроматография с использованием, например, колонки для хиральной жидкостной хроматографии высокого давления (HPLC) с хиральными неподвижными фазами. Кроме того, соединения включают формы (Z) и (E) (или формы цис и транс) соединений с двойными связями либо в виде отдельных геометрических изомеров, либо их смесей.
Соединения и фрагменты также могут существовать в нескольких таутомерных формах, включая енольную форму, кетоформу и их смеси. Соответственно, химические структуры, изображенные в настоящем документе, охватывают все возможные таутомерные формы проиллюстрированных соединений. Соединения могут существовать в несольватированных формах, а также сольватированных формах, включая гидратированные формы. Некоторые соединения могут существовать во многих кристаллических, сокристаллических или аморфных формах. Соединения включают их фармацевтически приемлемые соли или фармацевтически приемлемые сольваты в форме свободной кислоты любого из указанного выше, а также кристаллические формы любого из указанного выше.
«Циклоалкил» относится к насыщенному или частично насыщенному циклическому алкильному радикалу. Циклоалкильной группой может быть C3-6 циклоалкил, C3-5 циклоалкил, C5-6 циклоалкил, циклопропил, циклопентил или циклогексил. Циклоалкил можно выбирать из циклопропила, циклобутила, циклопентила и циклогексила.
«Циклоалкилалкил» относится к ациклическому алкильному радикалу, в котором один из атомов водорода, связанных с атомом углерода, заменен циклоалкильной группой согласно определению настоящего документа. Когда предполагаются конкретные алкильные фрагменты, используют терминологию циклоалкилалкил, циклоалкилалкенил или циклоалкилалкинил. Циклоалкилалкильной группой может быть, например, C4-30 циклоалкилалкил, алканильный, алкенильный или алкинильный фрагмент циклоалкилалкильной группы представляет собой C1-10, а циклоалкильный фрагмент циклоалкилалкильного фрагмента представляет собой C3-20. Циклоалкилалкильной группой может быть C4-20 циклоалкилалкил, например, алканильный, алкенильный или алкинильный фрагмент циклоалкилалкильной группы представляет собой C1-8, а циклоалкильный фрагмент циклоалкилалкильной группы представляет собой C3-12. Циклоалкилалкилом может быть C4-9 циклоалкилалкил, в котором алкильный фрагмент циклоалкилалкильной группы представляет собой C1-3 алкил, а циклоалкильный фрагмент циклоалкилалкильной группы представляет собой C3-6 циклоалкил. Циклоалкилалкильной группой может быть C4-12 циклоалкилалкил, C4-10 циклоалкилалкил, C4-8 циклоалкилалкил и C4-6 циклоалкилалкил. Циклоалкилалкильной группой может быть циклопропилметил (-CH2-цикло-C3H5), циклопентилметил (-CH2-цикло-C5H9) или циклогексилметил (-CH2-цикло-C6H11). Циклоалкилалкильной группой может быть циклопропилэтенил (-CH=CH-цикло-C3H5) или циклопентилэтинил (-C≡C-цикло-C5H9).
«Циклоалкилгетероалкил» сам по себе или как часть другого заместителя относится к гетероалкильной группе, в которой один или более атомов углерода (и некоторые ассоциированные атомы водорода) алкильной группы независимо заменены одной и той же или разными гетероатомной группой или группами и в которой один из атомов водорода, связанных с атомом углерода, заменен циклоалкильной группой. Когда предполагаются конкретные алкильные фрагменты, используют терминологию циклоалкилгетероалканил, циклоалкилгетероалкенил и циклоалкилгетероалкинил. В циклоалкилгетероалкиле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -SO- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомная группа представляет собой -O- или -NH-.
«Циклоалкилокси» относится к радикалу -OR, где R представляет собой циклоалкил согласно определению настоящего документа. Примеры циклоалкилокси групп включают циклопропилокси, циклобутилокси, циклопентилокси и циклогексилокси. Циклоалкилокси группой может быть C3-6 циклоалкилокси, C3-5 циклоалкилокси, C5-6 циклоалкилокси, циклопропилокси, циклобутилокси, циклопентилокси или циклогексилокси.
«Заболевание» относится к заболеванию, расстройству, состоянию или симптому любого из указанного выше.
«Фторалкил» относится к алкильной группе согласно определению настоящего документа, в которой один или более атомов водорода заменен фтором. Фторалкильной группой может быть C1-6 фторалкил, C1-5 фторалкил, C1-4 фторалкил или C1-3 фторалкил. Фторалкильной группой может быть пентафторэтил (-CF2CF3) или трифторметил (-CF3).
«Фторалкокси» относится к алкокси группе согласно определению настоящего документа, в которой один или более атомов водорода заменен фтором. Фторалкокси группой может быть C1-6 фторалкокси, C1-5 фторалкокси, C1-4 фторалкокси C1-3, фторалкокси, -OCF2CF3 или -OCF3.
«Галоген» относится к группе фтора, хлора, брома или иода.
«Гетероалкокси» относится к алкокси группе, в которой один или более атомов углерода заменены гетероатом. Гетероалкокси группой может быть C1-6 гетероалкокси, C1-5 гетероалкокси, C1-4 гетероалкокси или C1-3 гетероалкокси. В гетероалкокси гетероатомную группу можно выбирать из -O-, -S-, -NH-, -NR-, -SO2- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомная группа представляет собой -O- и -NH-. Гетероалкокси группой может быть C1-6 гетероалкокси, C1-5 гетероалкокси, C1-4 гетероалкокси или C1-3 гетероалкокси.
«Гетероалкил» сам по себе или как часть другого заместителя относятся к алкильной группе, в которой один или более атомов углерода (и некоторые ассоциированные атомы водорода) независимо заменены одной и той же или разными гетероатомной группой или группами. Примеры гетероатомных групп включают -O-, -S-, -NH-, -NR-, -O-O-, -S-S-, =N-N=, -N=N-, -N=N-NR-, -PR-, -P(O)OR-, -P(O)R-, -POR-, -SO-, -SO2-, -Sn(R)2- и тому подобное, причем каждый R независимо выбирают из водорода, C1-6 алкила, замещенного C1-6 алкила, C6-12 арила, замещенного C6-12 арила, C7-18 арилалкила, замещенного C7-18 арилалкила, C3-7 циклоалкила, замещенного C3-7 циклоалкила, C3-7 гетероциклоалкила, замещенного C3-7 гетероциклоалкила, C1-6 гетероалкила, замещенного C1-6 гетероалкила, C6-12 гетероарила, замещенного C6-12 гетероарила, C7-18 гетероарилалкила и замещенного C7-18 гетероарилалкила. Каждый R можно независимо выбирать из водорода и C1-3 алкила. Ссылка, например, на C1-6 гетероалкил, означает C1-6 алкильную группу, в которой по меньшей мере один из атомов углерода (и некоторые ассоциированные атомы водорода) заменен гетероатом. Например, C1-6 гетероалкил включает группы, имеющие пять атомов углерода и один гетероатом, группы, имеющие четыре атома углерода и два гетероатома и т.д. В гетероалкиле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -SO- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомной группой может быть -O- или -NH-. Гетероалкильной группой может быть C1-6 гетероалкил, C1-5 гетероалкил или C1-4 гетероалкил или C1-3 гетероалкил.
«Гетероарил» сам по себе или как часть другого заместителя относится к моновалентному гетероароматическому радикалу, полученному за счет удаления одного атома водорода из одного атома исходной гетероароматической кольцевой системы. Гетероарил охватывает множество кольцевых систем, имеющих по меньшей мере одно гетероароматическое кольцо, слитое по меньшей мере с одним другим кольцом, которое может быть ароматическим или неароматическим. Например, гетероарил охватывает бициклические кольца, в которых одно кольцо является гетероароматическим, а второе кольцо является гетероциклоалкильным кольцом. Для подобных слитых, бициклических гетероарильных кольцевых систем, в которых только одно из колец содержит один или более гетероатомов, углерод радикала может быть на ароматическом кольце или на гетероциклоалкильном кольце. Когда общее число атомов N, S и O в гетероарильной группе превышает один, гетероатомы могут быть рядом или не рядом друг с другом. Общее число гетероатомов в гетероарильной группе составляет не более чем два. В гетероариле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -S(O)- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомной группой может быть -O- или -NH-. Гетероарильную группу можно выбирать из C5-10 гетероарила, C5-9 гетероарила, C5-8 гетероарила, C5-7 гетероарила, C5-6 гетероарила, C5 гетероарила или C6 гетероарила.
Примеры подходящих гетероарильных групп включают группы, полученные из акридина, арсиндола, карбазола, α-карболина, хромана, хромена, циннолина, фурана, имидазола, индазола, индола, индолина, индолизина, изобензофурана, изохромена, изоиндола, изоиндолина, изохинолина, изотиазола, изоксазола, нафтиридина, оксадиазола, оксазола, перимидина, фенантридина, фенантролина, феназина, фталазина, птеридина, пурина, пирана, пиразина, пиразола, пиридазина, пиридина, пиримидина, пиррола, пирролидина, квиназолина, хинолина, хинолизина, хиноксалина, тетразола, тиадиазола, тиазола, тиофена, триазола, ксантена, тиазолидина, оксазолидина и тому подобное. Гетероарильную группу можно получить из тиофена, пиррола, бензотиофена, бензофурана, индола, пиридина, хинолина, имидазола, оксазола или пиразина. Например, гетероарилом может быть C5 гетероарил, и его можно выбирать из фурила, тиенила, пирролила, имидазолила, пиразолила, изотиазолила или изоксазолила. Гетероарилом может быть C6 гетероарил, и его можно выбирать из пиридинила, пиразинила, пиримидинила и пиридазинила.
«Гетероарилалкил» относится к арилалкильной группе, в которой один из атомов углерода (и некоторых ассоциированных атомов водорода) заменен гетероатом. Гетероарилалкильной группой может быть C6-16 гетероарилалкил, C6-14 гетероарилалкил, C6-12 гетероарилалкил, C6-10 гетероарилалкил, C6-8 гетероарилалкил или C7 гетероарилалкил или C6 гетероарилалкил. В гетероарилалкиле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -SO- и -SO2-, или гетероатомную группу можно выбирать из -O-и -NH-, или гетероатомной группой может быть -O- или -NH-.
«Гетероциклоалкил» сам по себе или как часть другого заместителя относится к насыщенному или ненасыщенному циклическому алкильному радикалу, в котором один или более атомов углерода (и некоторых ассоциированных атомов водорода) независимо заменены тем же самым или другим гетероатом; или к исходной ароматической кольцевой системе, в которой один или более атомов углерода (и некоторых ассоциированных атомов водорода) независимо заменены тем же самым или другим гетероатом таким образом, чтобы кольцевая система нарушала правило Хюккеля. Примеры гетероатомов для замены атома (атомов) углерода включают N, P, O, S и Si. Примеры гетероциклоалкильных групп включают группы, полученные из эпоксидов, азиринов, тииранов, имидазолидина, морфолина, пиперазина, пиперидина, пиразолидина, пирролидина и хинуклидина. Гетероциклоалкилом может быть C5 гетероциклоалкил, и его выбирают из пирролидинила, тетрагидрофуранила, тетрагидротиофенила, имидазолидинила, оксазолидинила, тиазолидинила, доксоланила и дитиоланила. Гетероциклоалкилом может быть C6 гетероциклоалкил, и его можно выбирать из пиперидинила, тетрагидропиранила, пиперизинила, оксазинила, дитианила и диоксанила. Гетероциклоалкильной группой может быть C3-6 гетероциклоалкил, C3-5 гетероциклоалкил, C5-6 гетероциклоалкил, C5 гетероциклоалкил или C6 гетероциклоалкил. В гетероциклоалкиле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -SO- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомной группой может быть -O- или -NH-.
«Гетероциклоалкилалкил» относится к циклоалкилалкильной группе, в которой один или более атомов углерода (и некоторых ассоциированных атомов водорода) циклоалкильного кольца независимо заменены тем же самым или другим гетероатом. Гетероциклоалкилалкилом может быть C4-12 гетероциклоалкилалкил, C4-10 гетероциклоалкилалкил, C4-8 гетероциклоалкилалкил, C4-6 гетероциклоалкилалкил, C6-7 гетероциклоалкилалкил или C6 гетероциклоалкилалкил или C7 гетероциклоалкилалкил. В гетероциклоалкилалкиле гетероатомную группу можно выбирать из -O-, -S-, -NH-, -N(-CH3)-, -SO- и -SO2-, или гетероатомную группу можно выбирать из -O- и -NH-, или гетероатомной группой может быть -O- или -NH-.
«Исходная ароматическая кольцевая система» относится к ненасыщенной циклической или полициклической кольцевой системе, имеющей циклическую сопряженную π (пи) электронную систему с 4n+2 электронами (правило Хюккеля). В определение «исходной ароматической кольцевой системы» включены слитые кольцевые системы, в которых одно или более колец являются ароматическими, а одно или более колец являются насыщенными или ненасыщенными, такими как, например, флуорен, индан, инден, фенален и т.д. Примеры исходных ароматических кольцевых систем включают ацеантрилен, аценафтилен, ацефенантрилен, антрацен, азулен, бензол, хризен, коронен, флуорантен, флуорен, гексацен, гексафен, гексален, ассим-индацен, симм-индацен, индан, инден, нафталин, октацен, октафен, октален, овален, пентацен, пенталин, пентафен, перилен, фенален, фенантрен, пицен, плеяден, пирен, пирантрен, рубицен, трифенилен, тринафталин и тому подобное.
«Гидраты» относится к включению воды в кристаллическую решетку соединения, описанного в настоящем документе, в стехиометрических пропорциях, что приводит к образованию аддукта. Способы получения гидратов включают, но без ограничения хранение в атмосфере, содержащей водяной пар, лекарственные формы, которые содержат воду, или стадии обычной фармацевтической обработки, такие как, например, кристаллизация (т.е. из воды или смешанных водных растворителей), лиофилизация, влажное гранулирование, покрытие водной пленкой или распылительная сушка. В определенных обстоятельствах гидраты также могут образовываться из кристаллических сольватов при воздействии водяного пара или при суспендировании безводного материала в воде. Гидраты также могут кристаллизоваться более чем в одной форме, что приводит к полиморфизму гидратов.
«Метаболический промежуточный продукт» относится к соединению, которое образуется in vivo в результате метаболизма исходного соединения и которое дополнительно подвергается реакции in vivo с высвобождением активного средства. Соединения формулы (1) представляют собой защищенные сульфонатные нуклеофильные пролекарства не β-лактамных ингибиторов β-лактамаз, которые метаболизируются in vivo с образованием соответствующих метаболических промежуточных продуктов, таких как авибактам ([2S,5R]-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфат). Метаболические промежуточные продукты подвергаются нуклеофильной циклизации с высвобождением не β-лактамного ингибитора β-лактамазы, такого как авибактам и одного или более продуктов реакции. Нужно, чтобы продукты реакции или их метаболиты были нетоксичными.
«Неопентил» относится к радикалу, в котором метиленовый углерод связан с атомом углерода, который связан с тремя неводородными заместителями. Примеры неводородных заместителей включают углерод, кислород, азот и серу. Каждым из трех неводородных заместителей может быть углерод. Двумя из трех неводородных заместителей может быть углерод, а третий неводородный заместитель можно выбрать из кислорода и азота. Неопентильная группа может иметь структуру:
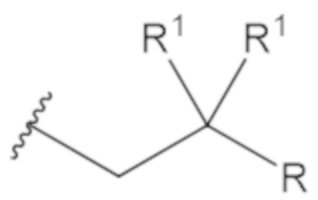
где каждый R1 определяют как для формулы (1).
«Исходная Ароматическая Кольцевая система» относится к ненасыщенной циклической или полициклической кольцевой системе, имеющей сопряженную π электронную систему. В определение «исходной ароматической кольцевой системы» конкретно включены слитые кольцевые системы, в которых одно или более колец являются ароматическими, а одно или более колец являются насыщенными или ненасыщенными, такие как, например, флуорен, индан, инден, фенален и т.д. Примеры исходных ароматических кольцевых систем включают ацеантрилен, аценафтилен, ацефенантрилен, антрацен, азулен, бензол, хризен, коронен, флуорантен, флуорен, гексацен, гексафен, гексален, ассим-индацен, симм-индацен, индан, инден, нафталин, октацен, октафен, октален, овален, пента-2,4-диен, пентацен, пенталин, пентафен, перилен, фенален, фенантрен, пицен, плеяден, пирен, пирантрен, рубицен, трифенилен и тринафталин.
«Исходная гетероароматическая кольцевая система» относится к ароматической кольцевой системе, в которой один или более атомов углерода (и любых ассоциированных атомов водорода) независимо заменены тем же самым или другим гетероатом таким образом, чтобы поддерживать постоянную характеристику π-электронной системы ароматических систем и число π-электронов, соответствующих правилу Хюккеля (4n +2). Примеры гетероатомов для замены атомов углерода включают N, P, O, S и Si и т.д. В определение «исходных гетероароматических кольцевых систем» конкретно включены слитые кольцевые системы, в которых одно или более колец являются ароматическими, а одно или более колец являются насыщенными или ненасыщенными, такие как, например, арсиндол, бензодиоксан, бензофуран, хроман, хромен, индол, индолин и ксантен. Примеры исходных гетероароматических кольцевых систем включают арсиндол, карбазол, β-карболин, хроман, хромен, циннолин, фуран, имидазол, индазол, индол, индолин, индолизин, изобензофуран, изохромен, изоиндол, изоиндолин, изохинолин, изотиазол, изоксазол, нафтиридин, оксадиазол, оксазол, перимидин, фенантридин, фенантролин, феназин, фталазин, птеридин, пурин, пиран, пиразин, пиразол, пиридазин, пиридин, пиримидин, пиррол, пирролидин, квиназолин, хинолин, хинолизин, хиноксалин, тетразол, тиадиазол, тиазол, тиофен, триазол, ксантен, тиазолидин и оксазолидин.
«Пациент» относится к млекопитающему, например, человеку. Термин «пациент» используют взаимозаменяемо с «субъектом».
«Фармацевтически приемлемый» относится к одобренному или находящемуся на утверждении регулирующего органа федерального правительства или правительства штата или указанному в фармакопее США или другой общепризнанной фармакопее для применения на животных и, в частности, на людях.
«Фармацевтически приемлемая соль» относится к соли соединения, которая обладает нужной фармакологической активностью исходного соединения. Подобные соли включают соли присоединения кислоты, образованные с неорганическими кислотами и одной или более протонируемыми функциональными группами, такими как первичный, вторичный или третичный амины в исходном соединении. Примеры неорганических кислот включают соляную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и тому подобное. Соль может быть образована с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, ядлочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил) бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтоевая кислота, салициловая кислота, стеариновая кислота, муконовая кислота и тому подобное. Соль может образовываться, когда один или более протонов кислоты, присутствующих в исходном соединени, заменяют металлическим ионом, таким как ион щелочного металла, ион щелочноземельного металла или ион аллюминия или их комбинации; или координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин и N-метилглюкамин. Фармацевтически приемлемой солью может быть гидрохлоридная соль. Фармацевтически приемлемой солью может быть натриевая соль. В соединениях, имеющих две или более ионизируемых групп, фармацевтически приемлемая соль может содержать один или более противоионов, таких как би-соль, например, дигидрохлоридная соль.
Термин «фармацевтически приемлемая соль» включает гидраты и другие сольваты, а также соли в кристаллической или некристаллической форме. При раскрытии конкретной фармацевтически приемлемой соли подразумевается, что конкретная соль, такая как гидрохлоридная соль, является примером соли, и что с использованием технологий, известных специалисту в данной области, можно получить другие соли. Кроме того, с использованием общеизвестных в данной области технологий специалист в данной области будет в состоянии преобразовать фармацевтически приемлемую соль в соответствующее соединение, свободное основание и/или свободную кислоту.
«Фармацевтически приемлемая основа» относится к фармацевтически приемлемому разбавителю, фармацевтически приемлемому адъюванту, фармацевтически приемлемому вспомогательному средству, фармацевтически приемлемому носителю или комбинации любого из указанного выше, с которой представленное в настоящем раскрытии соединение можно вводить пациенту и которая не нарушает его фармакологическую активность и которая является нетоксичной при введении в дозах, достаточных для обеспечения терапевтически эффективного количества соединения.
«Фармацевтическая композиция» относится к соединению формулы (1) или его фармацевтически приемлемой соли и по меньшей мере одной фармацевтически приемлемой основе, с которой соединение формулы (1) или его фармацевтически приемлемую соль вводят пациенту. Фармацевтически приемлемые основы известны в данной области.
«Предупреждение» или «предотвращение» относится к снижению риска приобретения заболевания или расстройства (то есть к тому, что по меньшей мере один из клинических симптомов заболевания не развивается у пациента, который может быть подвержен или предрасположен к заболеванию, но пока не испытывает и не проявляет симптомов заболевания). В некоторых вариантах осуществления «предупреждение» или «предотвращение» относится к уменьшению симптомов заболевания путем приема соединения профилактическим способом. Применение терапевтического средства для предупреждения или предотвращения заболевания или расстройства, известно как профилактика. Соединения, предоставленные в настоящем раскрытии, могут обеспечить превосходную профилактику из-за более низких долгосрочных побочных эффектов в течение длительных периодов времени.
«Пролекарство» относится к производному молекулы лекарственного средства, которое требует трансформации в организме с высвобождением активного лекарственного средства. Пролекарства часто, хотя и необязательно, являются фармакологически неактивными до преобразования в исходное лекарственное средство.
«Профрагмент» относится к группе, связанной с лекарственным средством, обычно с функциональной группой лекарственного средства, через связь (связи), которые могут расщепляться в определенных условиях использования. Связь (связи) между лекарственным средством и профрагментом могут расщепляться ферментативным или неферментативным способами. В условиях использования, например, после введения пациенту, связь (связи) между лекарственным средством и профрагментом могут расщепляться с высвобождением исходного лекарственного средства. Расщепление профрагмента может происходить спонтанно, например, посредством реакции гидролиза, или его можно катализировать или индуцировать с помощью другого средства, например, с помощью фермента, с помощью света, с помощью кислоты или с помощью изменения или воздействия на физический параметр или параметр окружающей среды, такой как изменение температуры, pH и т.д. Средство может быть эндогенным для условий использования, например, фермент, присутствующий в системном кровообращении пациента, которому вводят пролекарство, или кислая среда желудка, или средство можно подавать экзогенно. Например, для соединения формулы (1), профрагмент может иметь структуру:
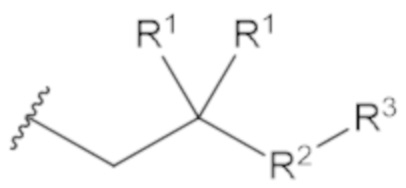
где R1, R2 и R3 соответствуют определению для формулы (1).
«Одинарная связь», как в выражении «R2 выбирают из одинарной связи», относится к фрагменту, в котором R2 представляет собой одинарную связь. Например, во фрагменте, имеющем структуру -C(R1)2-R2-R3, где R2 представляет собой одинарную связь, -R2- соответствует одинарной связи, «-», а фрагмент имеет структуру -C(R1)2-R3.
«Сольват» относится к молекулярному комплексу соединения с одной или несколькими молекулами растворителя в стехиометрическом или нестехиометрическом количестве. Такими молекулами растворителя являются молекулы, которые обычно используют в фармацевтике, которые, как известно, безвредны для пациента, такие как вода, этанол и тому подобное. Молекулярный комплекс соединения или фрагмента соединения и растворителя может быть стабилизирован нековалентными внутримолекулярными силами, такими как, например, электростатические силы, силы Ван-дер-Вальса или водородные связи. Термин «гидрат» относится к сольвату, в котором одна или несколько молекул растворителя представляет собой воду.
«Сольваты» относится к включению растворителей в кристаллическую решетку соединения, описанного в настоящем документе, в стехиометрических пропорциях, что приводит к образованию аддукта. Способы получения сольватов включают, но без ограничения хранение в атмосфере, содержащей растворитель, лекарственные формы, которые включают растворитель, или обычные стадии фармацевтической обработки, такие как, например, кристаллизация (то есть из растворителя или смешанных растворителей) диффузии паров. Сольваты также могут образовываться при определенных обстоятельствах из других кристаллических сольватов или гидратов при воздействии растворителя или при суспендировании материала в растворителе. Сольваты могут кристаллизоваться в более чем одной форме, что приводит к полиморфизму сольватов.
«Замещенный» относится к группе, в которой один или более атомов водорода независимо заменены одним и тем же или другим заместителем (заместителями). Каждый заместитель можно независимо выбирать из дейтерия, галогена, -OH, -CN, -CF3, -OCF3, =O, -NO2, C1-6 алкокси, C1-6 алкила, -COOR, -NR2 и -CONR2; причем каждый R независимо выбирают из водорода и C1-6 алкила. Каждый заместитель можно независимо выбирать из дейтерия, галогена, -NH2, -OH, C1-3 алкокси и C1-3 алкила, трифторметокси, и трифторметила. Каждый заместитель можно независимо выбирать из дейтерия, -OH, метила, этила, трифторметила, метокси, этокси и трифторметокси. Каждый заместитель можно выбирать из дейтерия, C1-3 алкила, =O, C1-3 алкила, C1-3 алкокси и фенила. Каждый заместитель можно выбирать из дейтерия, -OH, -NH2, C1-3 алкила и C1-3 алкокси.
«Терапия» или «лечение» заболевания относится к купированию или ослаблению заболевания или по меньшей мере одного из клинических симптомов заболевания или расстройства, снижению риска получения заболевания или по меньшей мере одного из клинических симптомов заболевания, уменьшению развитя заболевания или по меньшей мере одного из клинических симптомов заболевания или снижению риска развития заболевания или по меньшей мере одного из клинических симптомов заболевания. «Терапия» или «лечение» также относится к ингибированию заболевания, либо физически (например, стабилизация различимого симптома), физиологически (например, стабилизация физического параметра) или и к тому и к другому, и к ингибированию по меньшей мере одного физического параметра или проявления, которое может быть заметно или не заметно пациенту. «Терапия» или «лечение» также относится к задержке наступления заболевания или по меньшей мере одного или более его симптомов у пациента, который может быть подвержен или предрасположен к заболеванию или расстройству, даже если этот пациент еще не испытывает или не проявляет симптомы заболевания.
«Терапевтически эффективное количество» относится к количеству соединения, которое при введении субъекту для лечения заболевания или по меньшей мере одного из клинических симптомов заболевания является достаточным для воздействия на такое лечение заболевания или его симптома. «Терапевтически эффективное количество» может варьировать в зависимости, например, от соединения, заболевания и/или симптомов заболевания, тяжести заболевания и/или симптомов заболевания или расстройства, возраста, веса и/или состояния здоровья пациента, подлежащего лечению, и решения назначающего врача. Подходящее количество в любом конкретном случае может быть установлено специалистами в данной области техники или может быть определено путем обычных экспериментов.
«Терапевтически эффективная доза» относится к дозе, которая обеспечивает эффективное лечение заболевания или расстройства у пациента. Терапевтически эффективная доза может варьировать от соединения к соединению и от пациента к пациенту, и может зависеть от таких факторов, как состояние пациента и путь доставки. Терапевтически эффективную дозу можно определить в соответствии с обычными фармакологическими методиками, известными специалистам в данной области.
«Терапевтически эффективное количество» означает количество соединения, которое при введении пациенту для лечения заболевания является достаточным для воздействия на такое лечение заболевания. «Терапевтически эффективное количество» будет варьировать, например, в зависимости от соединения, заболевания и его тяжести, и от возраста, веса, адсорбции, распределения, метаболизма и экскреции подлежащего лечению пациента.
«Основа» относится к разбавителю, вспомогательному средству или носителю, с которым соединение вводят субъекту. В некоторых вариантах осуществления основа является фармацевтически приемлемой.
Далее сделана подробная ссылка на некоторые варианты осуществления соединений, композиций и способов. Раскрытые варианты осуществления не предназначены для ограничения формулы изобретения. Наоборот, формула изобретения предназначена для охвата всех альтернативных вариантов, модификаций и эквивалентов.
Соединения, представленные в настоящем раскрытии, представляют собой пролекарства в виде сложных эфиров сульфоновой кислоты не β-лактамных ингибиторов β-лактамаз. В пролекарствах не β-лактамного ингибитора β-лактамазы нуклеофильный фрагмент расположен рядом с гидросульфатной группой. In vivo, нуклеофильный фрагмент вступает в реакцию с высвобождением не β-лактамного ингибитора β-лактамазы. Примеры не β-лактамных ингибиторов β-лактамаз включают авибактам ([2S,5R]-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфат), релебактам ((1R,2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфат) и накубактам (1R,2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфат и производные и аналоги любого из указанного выше. Эти соединения являются ингибиторами β-лактамаз класса A, класса C и некоторых класса D, и они полезны в лечении бактериальных инфекций при использовании в сочетании с β-лактамными антибиотиками.
Соединения, представленные в настоящем раскрытии, включают соединения формулы (1):
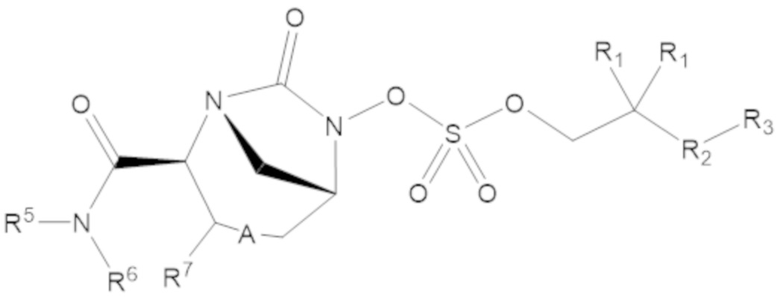 (1)
(1)
или их фармацевтически приемлемую соль, при этом
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила;
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2, в котором
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
В соединениях формулы (1) каждый заместитель можно независимо выбирать из дейтерия, -OH, -CN, -CF3, -OCF3, =O, -NO2, C1-6 алкокси, C1-6 алкила, -COOR, -NR2 и -CONR2; причем каждый R независимо выбирают из водорода и C1-6 алкила, такого как метил, этил, n-пропил, изопропил, n-бутил, трет-бутил или изо-бутил.
В соединениях формулы (1) замещающей группой может быть нуклеофильная группа. Нуклеофильные группы представляют собой функциональную группу, имеющую реакционноспособную пару электронов и обладающую способностью образования химической связи за счет донорства электронов. Примеры подходящих нуклеофильных групп включают сложные эфиры, карбоксилаты, сульфонаты, замещенные или незамещенный амины, спирты (гидроксил), тиолы, сульфиды, гидроксиламины и имины. Другие примеры подходящих нуклеофильных групп включают -OH, -CF3, -O-CF3, -NO2, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), где каждый R4 независимо выбирают из водорода, C1-6 алкила, C1-6 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C6-8 гетероарила, C5-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-6 алкила, замещенного C1-6 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C6-8 гетероарила, замещенного C5-10 арилалкила и замещенного C5-10 гетероарилалкила.
В соединениях формулы (1) каждый заместитель можно независимо выбрать из -OH, -CF3, -O-CF3, -NO2,-O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), где каждый R4 выбирают из водорода, C1-8 алкила и C1-8 гетероалкила.
В соединениях формулы (1) А может быть одинарная связь (-), а R7 может быть водород.
В соединениях формулы (1) А может быть двойная связь (=), а R7 может быть C1-3 алкил, такой как метил, этил, n-пропил или изопропил.
В соединениях формулы (1) каждым из R5, а R6 может быть водород.
В соединениях формулы (1) А может быть одинарная связь (-); R7 может быть водород; и R5 может быть водород.
В соединениях формулы (1) А может быть двойная связь (=), а R7 может быть C1-3 алкил, такой как метил, этил, n-пропил или изопропил; и каждым из R5, а R6 может быть водород.
В соединениях формулы (1) соединение может иметь структуру формулы (2):
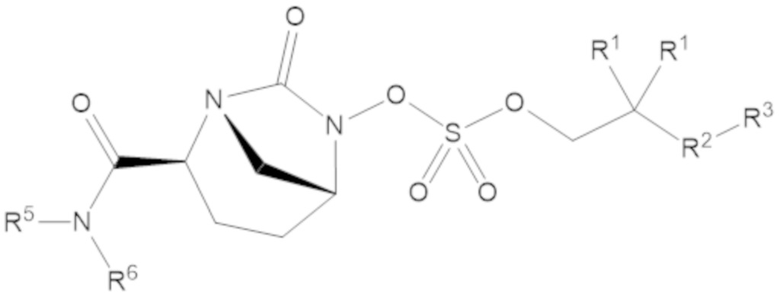 (2)
(2)
В соединениях формулы (1) соединение может иметь структуру формулы (2a):
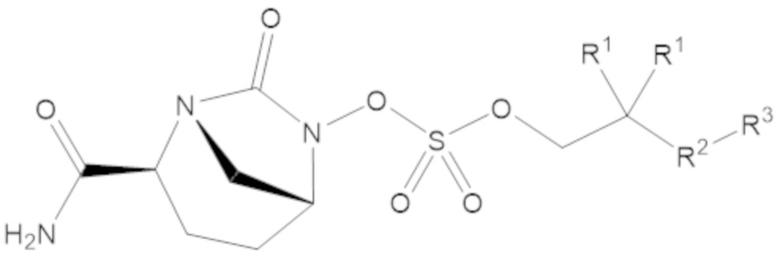 (2a)
(2a)
В соединениях формулы (1) соединение может иметь структуру формулы (3):
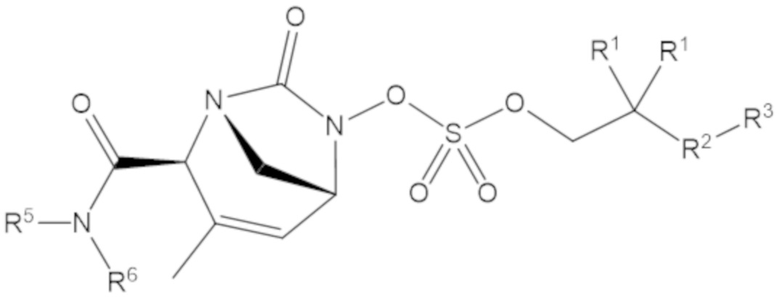 (3)
(3)
В соединениях формулы (1) соединение может иметь структуру формулы (3a):
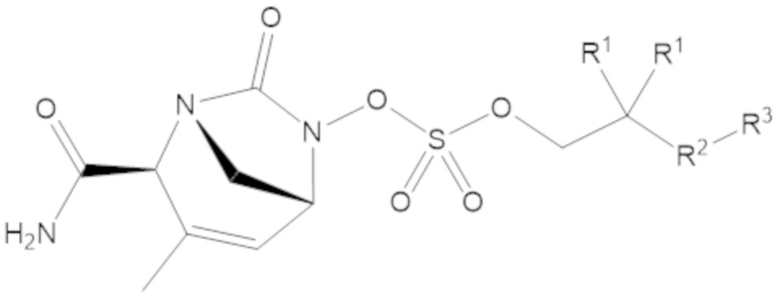 (3a)
(3a)
В соединениях формулы (1) соединение может иметь структуру формулы (4):
 (4)
(4)
В соединениях формулы (1) соединение может иметь структуру формулы (5):
 (5)
(5)
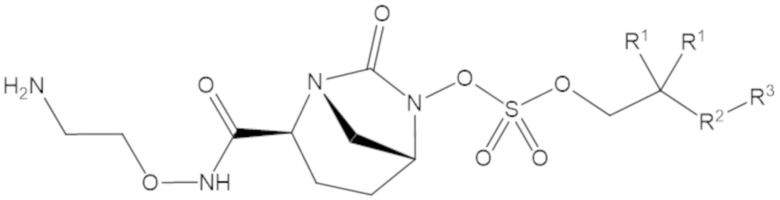
В соединениях формулы (1) R5 может быть C2-6 гетероалкил, содержащий концевую аминогруппу, а R6 может быть водород. Например, R5 может быть -O-(CH2)2-NH2, -CH2-O-CH2-NH2, -(CH2)2-O-CH2-NH2 или -CH2-O-(CH2)2-NH2.
В соединениях формулы (1) А может быть одинарная связь (-), а R7 может быть водород, R5 может быть -O-(CH2)2-NH2, а R6 может быть водород.
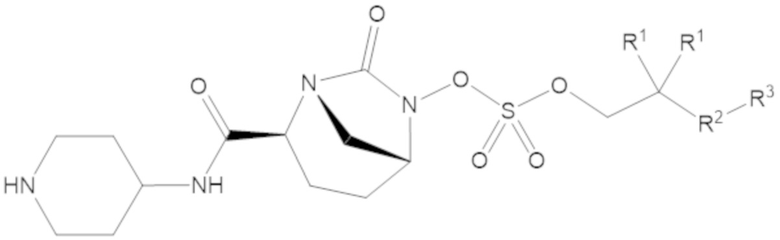
В соединениях формулы (1) R5 может быть C4-6 гетероциклоалкил, содержащий по меньшей мере один -NH- фрагмент, а R6 может быть водород. Например, R5 может быть 2-ил-пиперидин, 3-ил-пиперидин или 4-ил-пиперидин.
В соединениях формулы (1) А может быть одинарная связь (-), а R7 может быть водород, R5 может быть 4-ил-пиперидин, а R6 может быть водород.
В соединениях формулы (1)-(5) каждым R1 независимо может быть C1-6 алкил.
В соединениях формулы (1)-(5) каждым R1 независимо может быть метил, этил или n-пропил.
В соединениях формулы (1)-(5) каждым R1 может быть одинаковым и представляет собой метил, этил или n-пропил.
В соединениях формулы (1)-(5) каждый R1 представляет собой метил.
В соединениях формулы (1)-(5) каждый R1 вместе с геминальным атомом углерода, с которым они связаны, могут образовать C3-6 циклоалкильное кольцо или замещенное C3-6 циклоалкильное кольцо.
В соединениях формулы (1)-(5) каждый R1 вместе с геминальным атомом углерода, с которым они связаны, могут образовать C3-6 циклоалкильное кольцо. Например, каждый R1 вместе с геминальным атомом углерода, с которым они связаны, могут образовать циклопропильное кольцо, циклобутильное кольцо, циклопентильное кольцо или циклогексильное кольцо.
В соединениях формулы (1)-(5) каждый R1 вместе с геминальным атомом углерода, с которым они связаны, могут образовать C3-6 гетероциклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо.
В соединениях формулы (1)-(5) R2 можно выбирать из одинарной связи, C1-2 алкандиила и замещенного C1-2 алкандиила.
В соединениях формулы (1)-(5) R2 может быть одинарная связь.
В соединениях формулы (1)-(5) R2 может быть одинарная связь; и R3 может быть C1-6 алкил.
В соединениях формулы (1)-(5) R2 можно выбирать из C1-2 алкандиила и замещенного C1-2 алкандиила.
В соединениях формулы (1)-(5) R2 может быть метандиил, этандиил, замещенный метандиил или замещенный этандиил.
В соединениях формулы (1)-(5) R2 может быть замещенный C1-2 алкандиил, где замещенную группу можно выбирать из -OH, -CN, -CF3, -OCF3, =O, -NO2, C1-6 алкокси, C1-6 алкила, -COOR, -NR2 и -CONR2; причем каждый R независимо выбирают из водорода и C1-6 алкила.
В соединениях формулы (1)-(5) R2 может быть замещенный C1-2 алкандиил, где замещающей группой может быть нуклеофильная группа. Например, R2 может быть замещенный C1-2 алкандиил, где заместитель группу можно выбирать из -OH, -CF3, -O-CF3, -NO2,-O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), где каждый R4 определяют как для формулы (1), или каждый R4 выбирают из водорода и C1-8 алкила.
В соединениях формулы (1)-(5) R2 может быть замещенный C1-2 алкандиил, где замещающую группу выбирают из -OH, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), замещенного C5-6 арила, -NHR4, -CH(-NH2)(-R4); и R4 определяют как для формулы (1), или каждый R4 выбирают из водорода и C1-8 алкила.
В соединениях формулы (1)-(5), где R2 представляет собой замещенный C1-6 алкандиил, замещенный C1-6 гетероалкандиил или замещенный C5-6 арендиил, стереохимией атома углерода, с которым связана замещающая группа, может быть (S) конфигурация.
В соединениях формулы (1)-(5), где R2 представляет собой замещенный C1-6 алкандиил, замещенный C1-6 гетероалкандиил или замещенный C5-6 арендиил, стереохимией атома углерода, с которым связана замещающая группа, может быть (R) конфигурация.
В соединениях формулы (1)-(5) R2 выбирают из C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C5-6 арендиила и C5-6 гетероциклоалкандиила.
В соединениях формулы (1)-(5) R2 может быть циклопента-1,3-диен-диил, замещенный циклопента-1,3-диен-диил, бензол-диил или замещенный бензол-диил. Например, R2 может быть 1,2-бензол-диил или замещенный 1,2-бензол-диил.
В соединениях формулы (1)-(5) R3 можно выбирать из -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4); где R4 определяют как для формулы (1), или каждый R4 выбирают из водорода и C1-8 алкила.
В соединениях формулы (1)-(5) R3 можно выбирать из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NH-R4 и -CH(-NH2)(-R4); где R4 определяют как для формулы (1), или каждый R4 выбирают из водорода и C1-8 алкила.
В соединениях формулы (1)-(5) R3 представляет собой -C(O)-O-R4); где R4 определяют как для формулы (1), или каждый R4 выбирают из водорода и C1-8 алкила.
В соединениях формулы (1)-(5) R4 можно выбирать из водорода, C1-3 алкила, C5-6 циклоалкила, C5-6 гетероциклоалкила, C5-6 арила, замещенного C1-3 алкила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила и замещенного C5-6 арила.
В соединениях формулы (1)-(5) R4 можно выбирать из метила, этила, фенила и бензила.
В соединениях формулы (1)-(5) R4 можно выбирать из водорода и C1-8 алкила.
В соединениях формулы (1)-(5) R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила, C5-7 гетероциклоалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C7-9 арилалкила и замещенного C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5) R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5) R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
В соединениях формулы (1)-(5) R3 может быть -C(O)-O-R4; и R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C5-7 циклоалкила, C5-7 гетероциклоалкила, C6 арила, C7-9 арилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C6 арила и C7-9 арилалкила,
В соединениях формулы (1)-(5) R3 может быть -C(O)-O-R4; и R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила, C5-7 гетероциклоалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C7-9 арилалкила и замещенного C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5) R3 может быть -C(O)-O-R4; и R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5) R3 можно выбирать из -O-C(O)-CH3, -O-C(O)-CH2-CH3, -O-C(O)-фенила, -O-C(O)-CH2-фенила, -S-C(O)-CH3, -S-C(O)-CH2-CH3, -S-C(O)-фенила, -S-C(O)-CH2-фенила, -NH-C(O)-CH3, -NH-C(O)-CH2-CH3, -NH-C(O)-фенила, -NH-C(O)-CH2-фенила, -O-C(O)-O-CH3, -O-C(O)-O-CH2-CH3, -O-C(O)-O-фенила, -O-C(O)-O-CH2-фенила, -S-C(O)-O-CH3, -S-C(O)-O-CH2-CH3, -S-C(O)-O-фенила, -S-C(O)-O-CH2-фенила, -NH-C(O)-O-CH3, -NH-C(O)-O-CH2-CH3, -NH-C(O)-O-фенила, -NH-C(O)-O-CH2-фенила, -C(O)-O-CH3,-C(O)-O-CH2-CH3, -C(O)-O-фенила, -C(O)-O-CH2-фенила, -C(O)-S-CH3,-C(O)-S-CH2-CH3, -C(O)-S-фенила, -C(O)-S-CH2-фенила, -C(O)-NH-CH3, -C(O)-NH-CH2-CH3, -C(O)-NH-фенила, -C(O)-NH-CH2-фенила, -O-C(O)-O-CH3, -O-C(O)-O-CH2-CH3, -O-C(O)-O-фенила, -O-C(O)-O-CH2-фенила, -O-C(O)-S-CH3, -O-C(O)-S-CH2-CH3, -O-C(O)-S-фенила, -O-C(O)-S-CH2-фенила, -O-C(O)-NH-CH3, -O-C(O)-NH-CH2-CH3, -O-C(O)-NH-фенила, -O-C(O)-NH-CH2-фенила, -S-SH, -S-S-CH3, -S-S-CH2-CH3, -S-S-фенила, -S-S-CH2-фенила, -SH, -S-CH3, -S-CH2-CH3, -S-фенила, -S-CH2-фенила, -NH2, -NH-CH3, -NH-CH2-CH3, -NH-фенила, -NH-CH2-фенила, -CH(-NH2)(-CH3), -CH(-NH2)(-CH2-CH3), -CH(-NH2)(-фенила) и -CH(-NH2)(-CH2-фенила).
В соединениях формулы (1)-(5) R3 можно выбирать из C5-6 циклоалкила, C5-6 гетероциклоалкила, C5-6 арила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила и замещенного C5-6 гетероарила, содержащего по меньшей мере одну нуклеофильную группу. Например, R3 может иметь структуру формулы (4a) или формулы (4b):
(4a) (4b)
В соединениях формулы (1)-(5) R4 можно выбирать из C1-3 алкила, C5-6 циклоалкила, C5-6 гетероциклоалкила, C5-6 арила, замещенного C1-3 алкила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила и замещенного C5-6 арила.
В соединениях формулы (1)-(5) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C4-6 гетероциклоалкильное кольцо, содержащее два соседних S атома, или замещенное C4-6 гетероциклоалкильное кольцо, содержащее по меньшей мере один гетероатом, выбранный из O и S, и карбонильную (=O) замещающую группу, связанную с атомом углерода, примыкающим по меньшей мере к одному гетероатому.
В соединениях формулы (1)-(5) R2 может быть любая связь; R3 может быть C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, образуют C4-6 гетероциклоалкильное кольцо, содержащее два соседних S атома, или замещенное C4-6 гетероциклоалкильное кольцо, содержащее по меньшей мере один гетероатом, выбранный из O и S, и =O замещающую группу, связанную с атомом углерода, примыкающим к гетероатому.
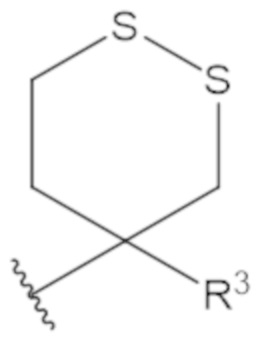
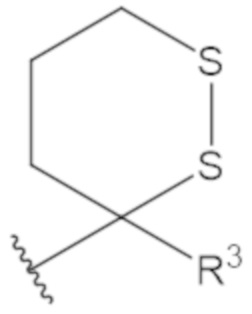
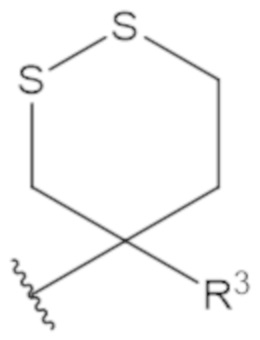
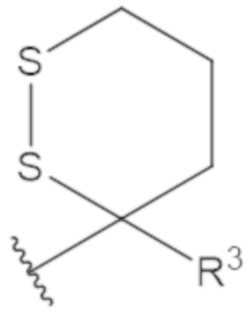
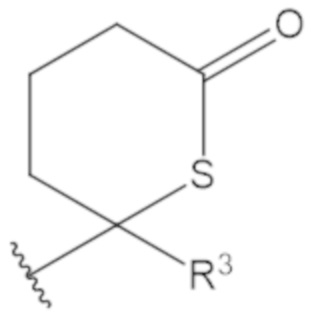
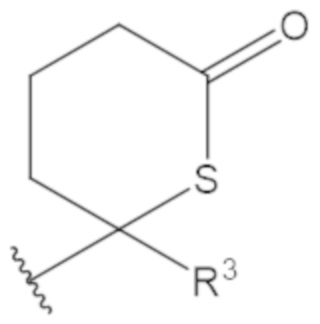
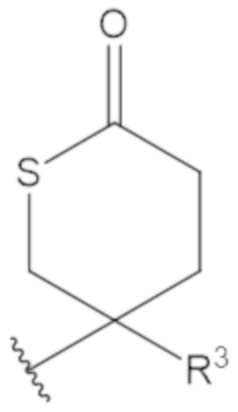
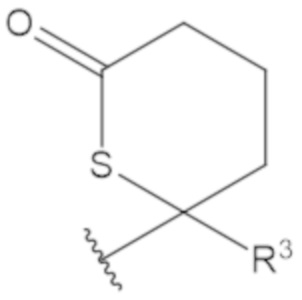
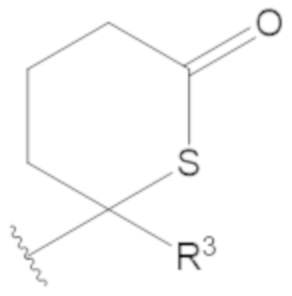
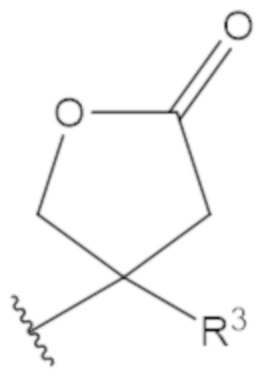
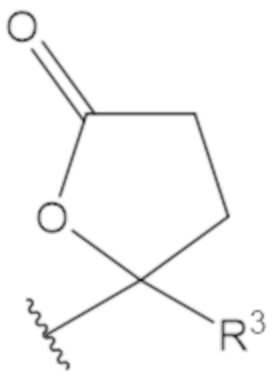
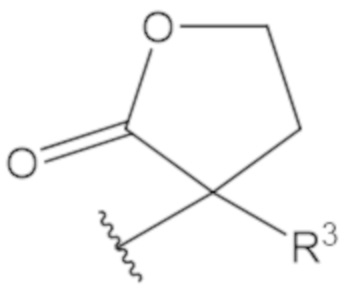
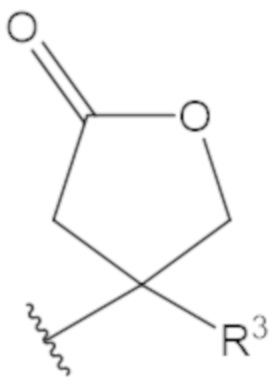
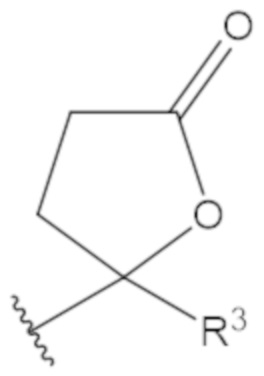
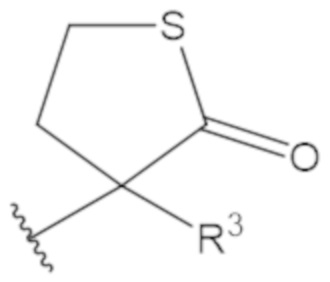
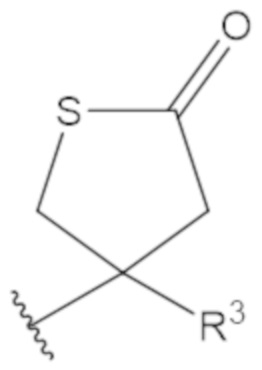
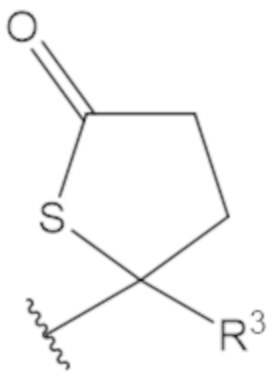
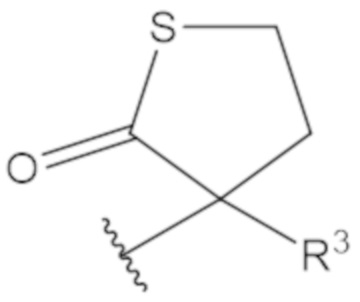
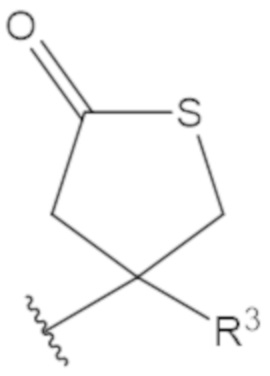
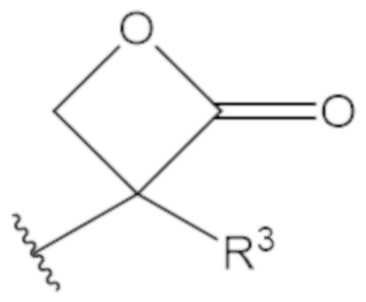
В соединениях формулы (1)-(5) профрагмент -CH2-C(R1)2-R3-R4 может иметь любую из следующих структур, где R3 может быть C1-6 алкил, такой как C1-4 алкил, такой как метил или этил:
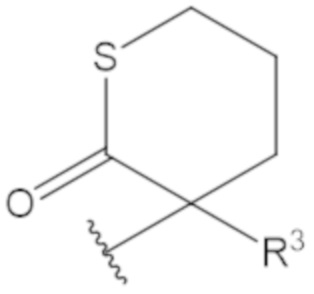
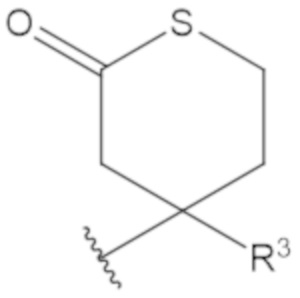
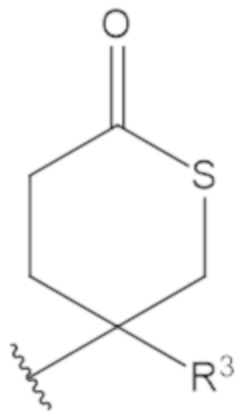
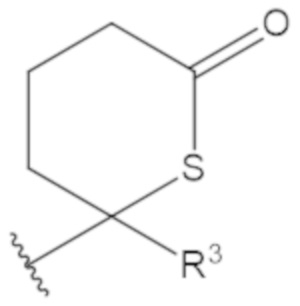

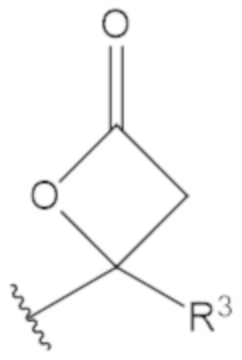
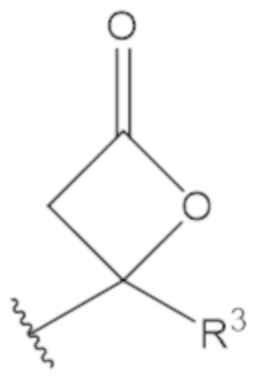
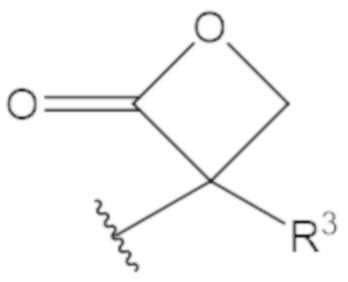
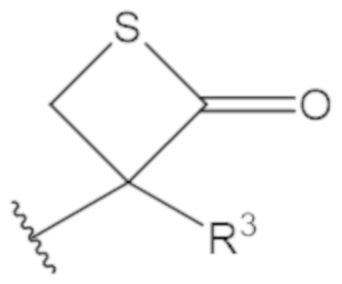
В соединениях формулы (1)-(5) R2 может быть одинарная связь; R3 может быть C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, могут образовать C4-6 гетероциклоалкильное кольцо или замещенное C4-6 гетероциклоалкильное кольцо.
В соединениях формулы (1)-(5) R2 может быть одинарная связь; R3 может быть C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, могут образовать C4-6 гетероциклоалкильное кольцо, содержащее два соседних S атома, или замещенное C4-6 гетероциклоалкильное кольцо, содержащее по меньшей мере один гетероатом, выбранный из O и S, и карбонильную (=O) замещающую группу, связанную с атомом углерода, примыкающим к гетероатому.
В соединениях формулы (1)-(5) R2 может быть одинарная связь; R3 может быть C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, могут образовать 1,2-дитиолан, 1,2-дитановое кольцо, тиэтан-2-оновое кольцо, дигидротиофен-2(3H)-оновое кольцо, тетрагидро-2H-типиран-2-оновое кольцо, оксетан-2-оновое кольцо, дигидрофуран-2(3H)-оновое кольцо или тетрагидро-2H-пиран-2-оновое кольцо.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 можно выбирать из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 можно выбирать из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4), где R4 можно выбирать из водорода, метила, этила, циклопентила, циклогексила, фенила, бензила и 2-пирролидинила.
В соединениях формулы (1)-(5)
каждый R1 и геминальный углерод, с которым они связаны, могут образовать C3-6 циклоалкильное кольцо;
R2 можно выбирать из любой связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 можно выбирать из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4), где R4 можно выбирать из водорода, метила, этила, циклопентила, циклогексила, фенила, бензила и 2-пирролидинила.
В соединениях формулы (1)-(5)
R2 может быть любая связь;
R3 может быть C1-3 алкил; и
каждый R1 вместе с атомом углерода, с которым они связаны, могут образовать 1,2-дитиолан, 1,2-дитановое кольцо, тиэтан-2-оновое кольцо, дигидротиофен-2(3H)-оновое кольцо, тетрагидро-2H-типиран-2-оновое кольцо, оксетан-2-оновое кольцо дигидрофуран-2(3H)-оновое кольцо или тетрагидро-2H-пиран-2-оновое кольцо.
В соединениях формулы (1)-(5) каждым R1 может быть метил;
R2 можно выбирать из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 можно выбирать из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4);
при этом R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 можно выбирать из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 может быть -C(O)-O-R4;
при этом R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 можно выбирать из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 можно выбирать из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4);
при этом R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 можно выбирать из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и
R3 может быть -C(O)-O-R4;
при этом R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4;
при этом R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C7-10 алкиларена и C5-10 гетероалкилциклоалкила.
В соединениях формулы (1)-(5)
каждым R1 может быть метил;
R2 может быть одинарная связь;
R3 может быть -C(O)-O-R4;
при этом R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C7-10 алкиларена и C5-10 гетероалкилциклоалкила;
каждым из R5, R6 и R7 может быть водород; и
А представляет собой одинарную связь.
В соединениях формулы (1)-(5) А может быть одинарная связь, а каждым из R5, R6 и R7 может быть водород.
В соединениях формулы (1)-(5) А может быть одинарная связь; каждым R1 может быть независимо C1-3 алкил; каждым R2 может быть одинарная связь; и каждым из R5, R6 и R7 может быть водород;
В соединениях формулы (1) соединение можно выбирать из:
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил бензоата (2);
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3);
бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил бензоата (6);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил пропионата (7);
бензил (4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) адипата (8);
6-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутокси)-6-оксогексановой кислоты (9);
метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10);
изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11);
гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12);
гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13);
трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14);
2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15);
оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил 1H-имидазол-1-сульфоната (34);
этил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (35);
гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36);
гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37);
2-метоксиэтил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (38);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил пивалата (43);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоата (44);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил бензоата (46);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил пропионата (47);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил) сульфата (48);
2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилацетата (49);
2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилпивалата (50);
S-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) этантиоата (51);
S-(5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил) этантиоата (52);
S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил адамантан-1-карбоксилата (55);
диэтил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилмалоната (56);
пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57);
бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутилпивалата (60);
этил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-этилбутаноата (61);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил бензоата (65);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2-метилбензоата (68);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоата (69);
2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоата (70);
2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил диацетата (71);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72);
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилбутаноата (73);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил) сульфата (74);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
В соединениях формулы (1) соединение можно выбирать из:
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3);
бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4);
метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10);
изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11);
гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12);
гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13);
трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14);
2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15);
оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36);
гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42);
S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53);
пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57);
бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
В соединениях формулы (1) соединение можно выбирать из:
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3);
бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4);
метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10);
изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11);
гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12);
гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13);
трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14);
2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15);
оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
В соединениях формулы (1) соединение можно выбирать из:
гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36);
гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42);
S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53);
пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57);
бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с геминальным атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 может быть одинарная связь;
R3 может быть -C(O)-O-R4; и
R4 можно выбирать из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила, C5-7 гетероциклоалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C7-9 арилалкила и замещенного C5-7 гетероциклоалкила.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 можно выбирать из одинарной связи, метан-диила и этан-диила; и
R3 можно выбирать из -C(O)-O-R4 и -S-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4, где R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть -(CH2)2-; и
R3 может быть -C(O)-O-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединении формулы (2a)
каждый R1 можно выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть -CH2-; и
R3 может быть -S-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила, замещенного C4-10 гетероциклоалкилалкила.
В соединении формулы (2a)
каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, C3-6 циклоалкильное кольцо или C3-6 гетероциклоалкильное кольцо;
R2 может быть одинарная связь; и
R3 может быть C1-3 алкил.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из одинарной связи и метандиила; и
R3 можно выбирать из -O-C(O)-R4 и -C(O)-O-R4, в котором R4 можно выбирать из C1-10 алкила и замещенного фенила.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 может быть одинарная связь;
R3 может быть -CH=C(R4)2, где каждым R4 может быть -C(O)-O-R8, или каждый R4 вместе с атомом углерода, с которым они связаны, образуют замещенное гетероциклогексильное кольцо; и
каждым R8 может быть C1-4 алкил.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из одинарной связи и метандиила; и
R3 может быть замещенный фенил, в котором один или более заместителей можно независимо выбирать из -CH2-O-C(O)-R4 и -O-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила и фенила.
В соединении формулы (2a)
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из -C(R8)2- и -CH2-C(R8)2-, где каждый R8 можно независимо выбирать из C1-3 алкила; и
R3 можно выбирать из -C(O)-O-R4 и -O-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, замещенного C1-10 алкила, замещенного C1-10 гетероалкила и 4(ил-метил)-5-метил-1,3-диоксол-2-она.
В соединении формулы (2a)
каждый R1 вместе с атомом углерода, с которым они связаны, образуют замещенное C5-6 гетероциклическое кольцо;
R2 может быть одинарная связь; и
R3 может быть C1-3 алкил.
Соединением формулы (1) может быть соединение подгруппы (1A) или его фармацевтически приемлемая соль, при этом
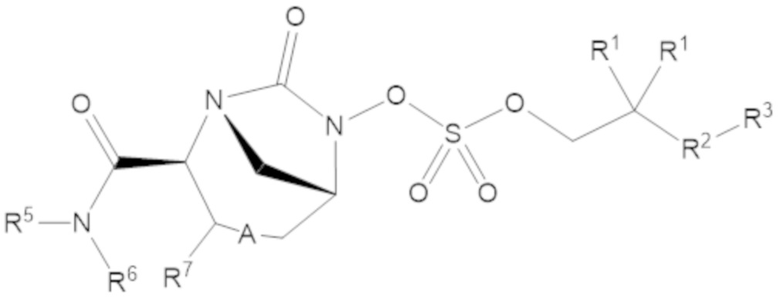 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 можно выбирать из одинарной связи, метан-диила и этан-диила; и
R3 можно выбирать из -C(O)-O-R4 и -S-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединениях подгруппы (1A) каждый R1 можно независимо выбирать из C1-3 алкила.
В соединениях подгруппы (1A) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
В соединениях подгруппы (1A) R2 является одинарная связь.
В соединениях подгруппы (1A) R2 может быть метан-диил.
В соединениях подгруппы (1A) R2 может быть этан-диил.
В соединениях подгруппы (1A) R3 может быть -C(O)-O-R4.
В соединениях подгруппы (1A) R3 может быть -S-C(O)-R4.
В соединениях подгруппы (1A) R4 может быть C1-10 алкил.
В соединениях подгруппы (1A) R4 может быть C1-10 гетероалкил.
В соединениях подгруппы (1A) R4 может быть C5-10 арилалкил.
В соединениях подгруппы (1A) R4 может быть C3-6 гетероциклоалкил.
В соединениях подгруппы (1A) R4 может быть замещенный C4-10 гетероциклоалкилалкил.
Соединением формулы (1) может быть соединение подгруппы (1B) или его фармацевтически приемлемая соль, при этом
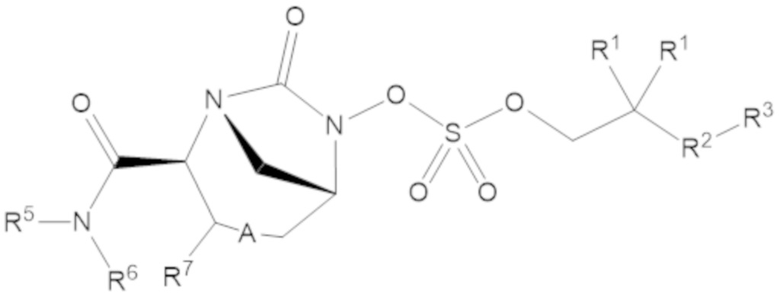 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4, где R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединениях подгруппы (1B) каждый R1 можно независимо выбирать из C1-3 алкила.
В соединениях подгруппы (1B) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
В соединениях подгруппы (1B) R4 можно выбирать из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами может быть кислород, и -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
В соединениях подгруппы (1B) в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами может быть кислород, а один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1B) каждым R1 может быть метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо.
В соединениях подгруппы (1B) R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, бензила, 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
В соединениях подгруппы (1B)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждым R1 может быть метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4, в котором R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, -CH2-фенил (бензила), 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
Соединением формулы (1) может быть соединение подгруппы (1C) или его фармацевтически приемлемая соль, при этом
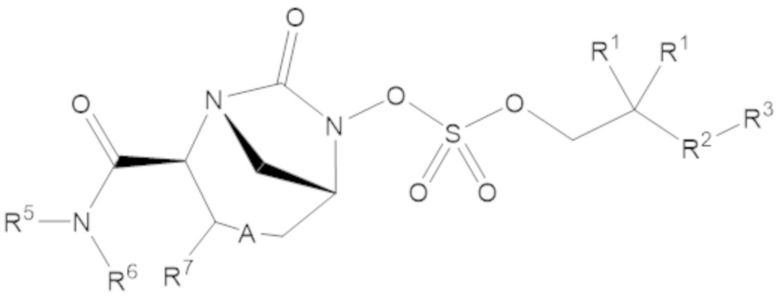 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть -(CH2)2-; и
R3 может быть -C(O)-O-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединениях подгруппы (1C) каждый R1 можно независимо выбирать из C1-3 алкила.
В соединениях подгруппы (1C) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
В соединениях подгруппы (1C) R4 можно выбирать из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
В соединениях подгруппы (1C) в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами может быть кислород, а один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1C) R4 может быть C1-10 алкил.
В соединениях подгруппы (1C)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждым R1 может быть метил;
R2 может быть -(CH2)2-; и
R3 может быть -C(O)-O-R4, в котором R4 можно выбирать из n-гексила и n-гептила.
Соединением формулы (1) может быть соединение подгруппы (1D) или его фармацевтически приемлемая соль, при этом
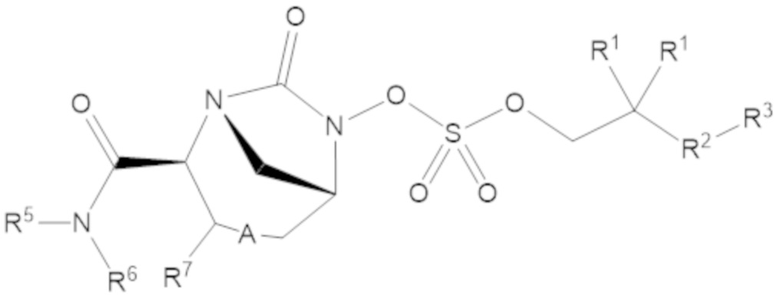 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть -CH2-; и
R3 может быть -S-C(O)-R4, в котором R4 можно выбирать из C1-10C1-1 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила, замещенного C4-10 гетероциклоалкилалкила.
В соединениях подгруппы (1D) каждый R1 можно независимо выбирать из C1-3 алкила.
В соединениях подгруппы (1D) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
В соединениях подгруппы (1D) R4 можно выбирать из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C3-6 замещенного гетероциклоалкила, -(CH2)2-C3-6 замещенного гетероциклоалкила.
В соединениях подгруппы (1D) в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами может быть кислород, а один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1D) R4 может быть C1-10 алкил.
В соединениях подгруппы (1D)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждым R1 может быть метил;
R2 может быть -CH2-; и
R3 может быть -S-C(O)-R4, в котором R4 может быть метил.
Соединением формулы (1) может быть соединение подгруппы (1E) или его фармацевтически приемлемая соль, при этом
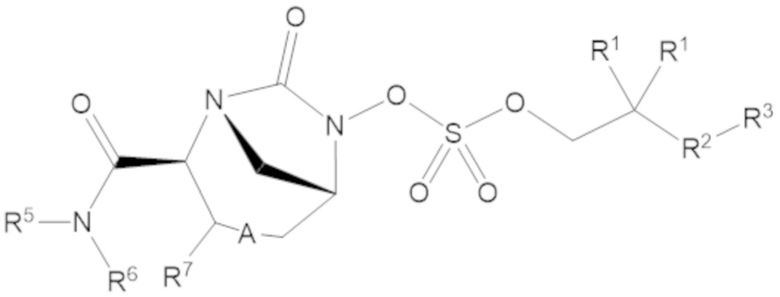 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, C3-6 циклоалкильное кольцо или C3-6 гетероциклоалкильное кольцо;
R2 может быть одинарная связь; и
R3 может быть C1-3 алкил.
В соединениях подгруппы (1E) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 гетероциклоалкильное кольцо или C3-6 гетероциклоалкильное кольцо;
В соединениях подгруппы (1E) одним или более гетероатомами может быть кислород, а одним или более заместителями может быть =O.
В соединениях подгруппы (1E)
каждый R1 вместе с атомом углерода, с которым они связаны, образуют дигидрофуран-2(3H)-оновое кольцо;
R2 может быть одинарная связь; и
R3 может быть метил.
Соединением формулы (1) может быть соединение подгруппы (1F) или его фармацевтически приемлемая соль, при этом
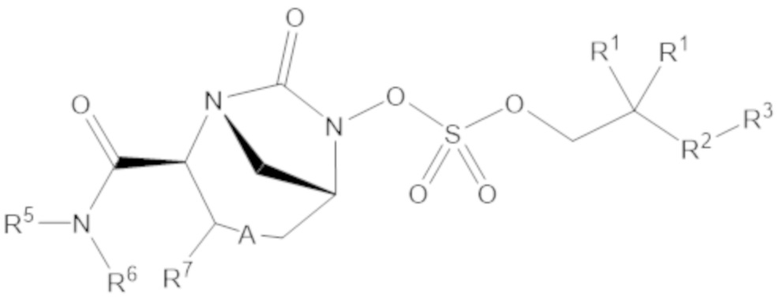 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из одинарной связи и метандиила; и
R3 можно выбирать из -O-C(O)-R4 и -C(O)-O-R4, в котором R4 можно выбирать из C1-10 алкила и замещенного фенила.
В соединениях подгруппы (1F) R2 может быть одинарная связь.
В соединениях подгруппы (1F) R2 может быть метандиил.
В соединениях подгруппы (1F) R3 может быть -O-C(O)-R4.
В соединениях подгруппы (1F) R2 может быть метандиил; и R3 может быть -O-C(O)-R4.
В соединениях подгруппы (1F) R3 может быть -C(O)-O-R4.
В соединениях подгруппы (1F) R2 может быть одинарная связь; и R3 может быть -C(O)-O-R4.
В соединениях подгруппы (1E) R2 может быть одинарная связь; R3 может быть -C(O)-O-R4; и R4 может быть C1-3 алкил.
В соединениях подгруппы (1F) R4 может быть C1-10 алкил.
В соединениях подгруппы (1F) R4 может быть C1-4 алкил.
В соединениях подгруппы (1F) R4 может быть замещенный фенил.
В соединениях подгруппы (1F) R2 может быть метандиил; R3 может быть -O-C(O)-R4; и R4 может быть замещенный фенил.
В соединениях подгруппы (1F) один или более заместителей можно независимо выбирать из галогена, C1-3 алкила и C1-3 алкокси.
В соединениях подгруппы (1F) замещенным фенилом может быть 2,6-замещенный фенил.
В соединениях подгруппы (1F) каждый из заместителей можно выбирать из C1-3 алкила и C1-3 алкокси.
В соединениях подгруппы (1F) замещенным фенилом может быть 2,5,6-замещенный фенил.
В соединениях подгруппы (1F) каждый из заместителей в позициях 2 и 6 можно независимо выбирать из C1-3 алкила и C1-3 алкокси; и заместителем в 5 позиции может быть галоген.
Соединением формулы (1) может быть соединение подгруппы (1G) или его фармацевтически приемлемая соль, при этом
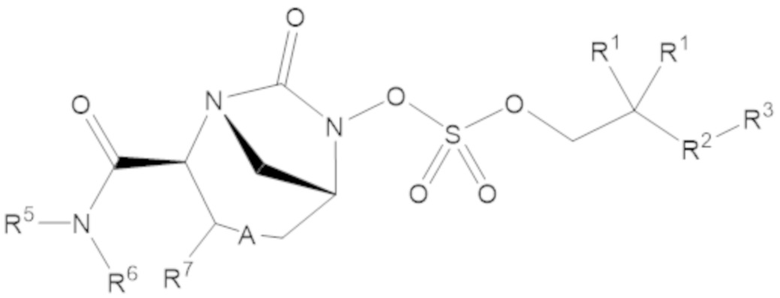 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 может быть одинарная связь; и
R3 может быть -CH=C(R4)2, где каждым R4 может быть -C(O)-O-R8, или каждый R4 вместе с атомом углерода, с которым они связаны, образуют замещенное гетероциклогексильное кольцо; и
каждым R8 может быть C1-4 алкил.
В соединениях подгруппы (1G) каждым R4 может быть -C(O)-O-R8.
В соединениях подгруппы (1G) каждым R4 может быть -C(O)-O-R8, или каждый R4 вместе с атомом углерода, с которым они связаны, образуют замещенное гетероциклогексильное кольцо.
В соединениях подгруппы (1G) в замещенном гетероциклогексильном кольце одним или более гетероатомами может быть кислород.
В соединениях подгруппы (1G) в замещенном гетероциклогексильном кольце один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1G) замещенным гетероциклоалкильным кольцом может быть 2,2-диметил-5-ил-1,3-диоксан-4,6-дион.
Соединением формулы (1) может быть соединение подгруппы (1H) или его фармацевтически приемлемая соль, при этом
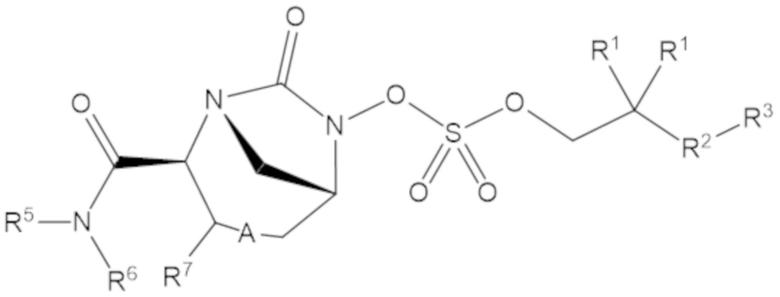 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из одинарной связи и метандиила; и
R3 может быть замещенный фенил, в котором один или более заместителей можно независимо выбирать из -CH2-O-C(O)-R4 и -O-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила и фенила.
В соединениях подгруппы (1H) R2 может быть одинарная связь.
В соединениях подгруппы (1H) R2 может быть 2-замещенный фенил.
В соединениях подгруппы (1H) одним или более заместителями может быть -CH2-O-C(O)-R4.
В соединениях подгруппы (1H) одним или более заместителями может быть -O-C(O)-R4.
В соединениях подгруппы (1H) R4 может быть C1-10 алкил.
В соединениях подгруппы (1H) R4 можно выбирать из метила, этила, изопропила, пивалолила и фенила.
Соединением формулы (1) может быть соединение подгруппы (1I) или его фармацевтически приемлемая соль, при этом
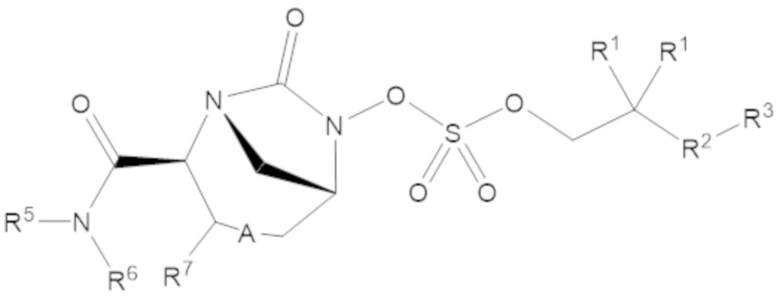 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из -C(R8)2- и -CH2-C(R8)2-, где каждый R8 можно независимо выбирать из C1-3 алкила; и
R3 можно выбирать из -C(O)-O-R4 и -O-C(O)-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, замещенного C1-10 алкила, замещенного C1-10 гетероалкила и 4(ил-метил)-5-метил-1,3-диоксол-2-она.
В соединениях подгруппы (1I) каждым R1 может быть метил.
В соединениях подгруппы (1I) R2 может быть -C(R8)2-.
В соединениях подгруппы (1I) R2 может быть -CH2-C(R8)2-.
В соединениях подгруппы (1I) каждым R8 может быть метил.
В соединениях подгруппы (1I) каждым R1 может быть метил; и каждым R8 может быть метил.
В соединениях подгруппы (1I) R3 может быть -C(O)-O-R4.
В соединениях подгруппы (1I) R3 может быть -O-C(O)-R4.
Соединением формулы (1) может быть соединение подгруппы (1J) или его фармацевтически приемлемая соль, при этом
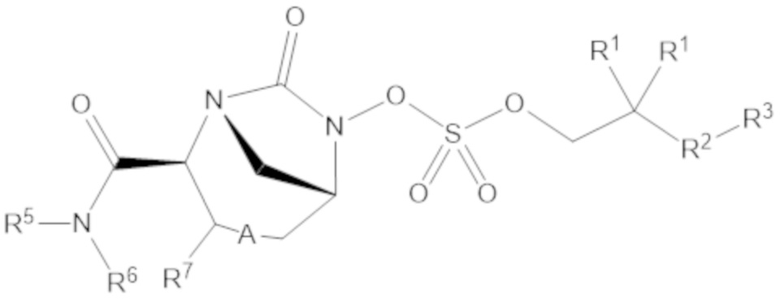 (1)
(1)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 вместе с атомом углерода, с которым они связаны, образуют замещенное C5-6 гетероциклическое кольцо;
R2 может быть одинарная связь; и
R3 может быть C1-3 алкил.
В соединениях подгруппы (1J) в замещенном C5-6 гетероциклическом кольце одним или более гетероатомами может быть кислород; и один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1J) каждый R1 вместе с атомом углерода, с которым они связаны, образуют тетрагидро-2H-пиран-2-оновое кольцо.
В соединениях подгруппы (1J)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила;
R2 можно выбирать из C2-4 алкандиила; и
R3 может быть замещенный C5-6 гетероциклоалкил, в котором один или более гетероатомов можно независимо выбирать из N и O; и один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (1J) R3 может иметь структуру формулы (6):
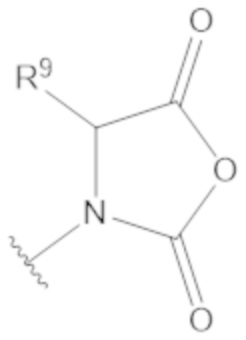 (6)
(6)
в которой R9 можно выбирать из водорода, C1-6 алкила, C4-6 циклоалкила, C1-6 гетероалкила, C4-6 гетероциклоалкила, замещенного C1-6 алкила, замещенного C4-6 циклоалкила, замещенного C1-6 гетероалкила и замещенного C4-6 гетероциклоалкила.
В соединениях подгруппы (1J) R9 можно выбирать из водорода и C1-6 алкила, такого как C1-4 алкил, такой как метил или этил.
Соединением формулы (4) может быть соединение подгруппы (4A) или его фармацевтически приемлемая соль, при этом
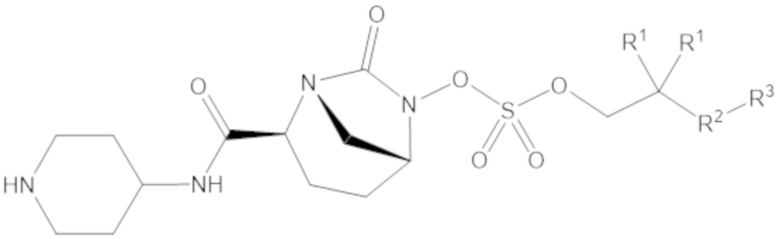 (4)
(4)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
каждый R1 можно независимо выбирать из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4, в котором R4 можно выбирать из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
В соединениях подгруппы (4A) каждый R1 можно независимо выбирать из C1-3 алкила.
В соединениях подгруппы (4A) каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
В соединениях подгруппы (4A) R4 можно выбирать из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами может быть кислород, -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
В соединениях подгруппы (4A) в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами может быть кислород, а один или более заместителей можно независимо выбирать из C1-3 алкила и =O.
В соединениях подгруппы (4A) каждым R1 может быть метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо.
В соединениях подгруппы (4A) R4 можно выбирать из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, бензила, 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
В соединениях подгруппы (4A)
каждым из R5, R6 и R7 может быть водород;
А может быть одинарная связь;
R2 может быть одинарная связь; и
R3 может быть -C(O)-O-R4, в котором R4 может быть C1-10 алкил.
Соединение формулы (4) можно выбирать из:
этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (20);
2-метоксиэтил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (21);
4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22);
4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23);
4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (24);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (25);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Соединение формулы (4) можно выбирать из:
этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (20);
4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22);
4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (25);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Соединение формулы (5) можно выбирать из:
этил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27);
2-метоксиэтил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28);
соли TFA (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29);
соли TFA гексил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30);
соли TFA гептил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (31);
соли TFA этил-1-((((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (32);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Соединением формулы (1)-(5) может быть сольват, фармацевтически приемлемая соль или их комбинация.
В соединениях формулы (1)-(5) фармацевтически приемлемой солью может быть гидрохлоридная соль.
В соединениях формулы (1)-(5) фармацевтически приемлемой солью может быть дигидрохлоридная соль.
Соединением формулы (1)-(5) может быть фармацевтически приемлемая соль соединения формулы (1)-(5), ее гидрат или сольват любого из указанного выше.
Описанное в настоящем документе соединение можно синтезировать с использованием способов, известных в данной области. Синтез различных диазабицикло[3.2.1]октановых структур, описанных в настоящем документе, является традиционным и хорошо известнен специалистам в данной области (Tandiparthi et al. Публикация международной заявки PCT № WO 2016/116788; Lampilas et al. Патент США № 7112592; King et al., ACS Chemical Biology 2016; 11, 864; и Bush et al. Cold Spring Harb Perspect Med 2016; 6:a025247). Образование сложных эфиров серной кислоты также хорошо известно в данной области (Simpson et al. J. Am. Chem. Soc. 2006, 128, 1605; Li et al. Публикация заявки США № 2009/0099253; Jandeleit et al. Публикация международной заявки PCT № WO 2009/033054; Jandeleit et al. Публикация международной заявки PCT № WO 2009/033079; и Jandeleit et al. Публикация международной заявки PCT № WO 2011/150380).
Аналоги сложных моноэфиров серной кислоты сульфатсодержащих соединений можно получать с помощью реакции гидроксилзамещенного сульфатсодержащего соединения со сложными моноэфирами хлорсульфоновой кислоты с образованием соответствующего аналога сложного моноэфира серной кислоты. Способы можно использовать при получении пролекарств сульфатсодержащих фармацевтических соединений.
Пролекарства представляют собой дериватизированные формы лекарственных средств, которые после введения превращаются или метаболизируются в активную форму исходного лекарственного средства in vivo. Пролекарства используют для модификации одного или нескольких аспектов фармакокинетики лекарственного средства таким образом, чтобы повысить терапевтическую эффективность исходного лекарственного средства. Например, пролекарства часто используют для повышения пероральной биодоступности лекарственного средства. Для терапевтической эффективности лекарственных средств, проявляющих плохую пероральную биодоступность, могут потребоваться частое введение доз, большие вводимые дозы, или может быть необходимо введение, отличное от перорального пути, например, внутривенно. В частности, многие лекарственные средства с сульфатными группами проявляют плохую пероральную биодоступность.
Для изменения фармакокинетики лекарственных средств использовались стратегии пролекарств с внутримолекулярной циклизацией. Стратегии пролекарств с высвобождением в результате внутримолекулярной циклизации применяли к лекарственным средствам, содержащим функциональные группы сульфоновой кислоты. Например, в патенте США № 7994218 и патенте США № 8168617 описаны, например, пролекарства, содержащие замещенное производное неопентилового эфира сульфоновой кислоты, в котором неопентильную группу удаляют in vivo, маскируя нуклеофильный гетероатом, связанный с замещенным неопентильным фрагментом, с последующей внутримолекулярной циклизацией для получения исходного лекарственного средства в форме сульфоновой кислоты или соли сульфоновой кислоты. В таких пролекарствах нуклеофильный гетероатом может представлять собой азот или кислород, а нуклеофильный азот или кислород может быть замаскирован защитной группой амина или спирта, соответственно, с которой можно снять защиту in vivo.
Аналоги сложных моноэфиров серной кислоты сульфатсодержащего соединения можно получать с помощью реакции гидроксилзамещенного аналога сульфатсодержащего соединения со сложным моноэфиром хлорсульфоновой кислоты в щелочных условиях с образованием соответствующего аналога сложного моноэфира серной кислоты. Сложные моноэфиры хлорсульфоновой кислоты можно получать с помощью реакции сульфурилхлорида со спиртом, имеющим нужный профрагмент. Неопентиловые спирты, имеющие неопентиловые профрагменты, можно получать с помощью стандартных методов синтеза, таких как методы, описанные в патентах США №№ 7994218 и 8168617.
Например, аналоги сложных моноэфиров серной кислоты авибактама, представленные в настоящем раскрытии, можно синтезировать с помощью реакции (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида со сложными моноэфирами хлорсульфоновой кислоты, имеющими нужный профрагмент, с образованием соответствующих (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сложных моноэфиров серной кислоты.
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид можно получать путем гидрирования (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида с применением способов, описанных, например, в патентах США №№ 8772490; 9035062; и 9284273.
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид можно вводить в реакцию со сложными моноэфирами хлорсульфоновой кислоты в присутствии основания с образованием соответствующего аналога сложного моноэфира серной кислоты авибактама. Подходящие способы раскрыты, например, в J. Am. Chem. Soc. 2006, 128, 1605-1610.
Аналогичные способы можно приспособить для получения аналогов сложных моноэфиров серной кислоты релебактама и накубактама. Например, защищенный трет-бутил карбоксилатом 6-гидроксильный аналог релебактама можно вводить в реакцию со сложными моноэфирами хлорсульфоновой кислоты в присутствии основания с образованием соответствующего защищенного трет-бутил карбоксилатом аналога сложного моноэфира серной кислоты релебактама. Затем с соединения можно снять защиту в присутствии кислоты с получением аналога сложного моноэфира серной кислоты релебактама. Для получения аналогов сложных моноэфиров серной кислоты накубактама можно использовать способы, аналогичные способам, используемым для получения аналогов сложных моноэфиров серной кислоты релебактама.
Например, аналог сложного моноэфира серной кислоты формулы (80a) можно синтезировать с помощью реакции циклической гидроксамовой кислоты формулы (80b) со сложным моноэфиром хлорсульфоната формулы (80c) в щелочных условиях:
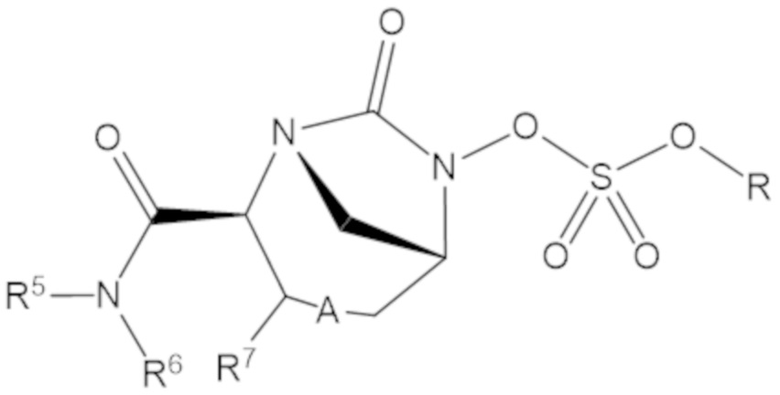 (80a)
(80a) 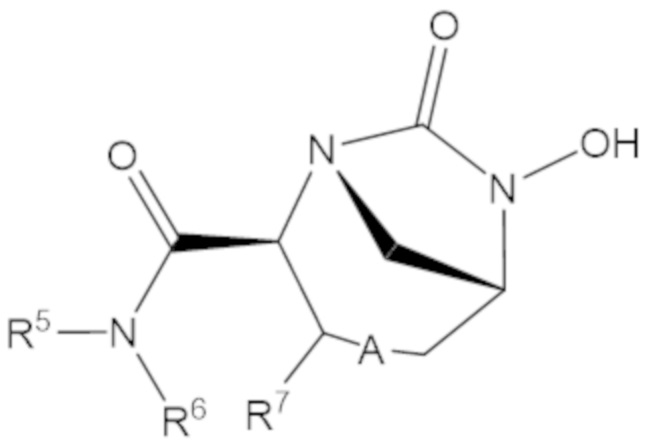 (80b)
(80b)
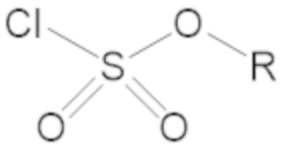 (80c)
(80c)
где
R выбирают из C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
Сложные моноэфиры хлорсульфоновой кислоты могут содержать неопентиловый сложный эфир хлорсульфоновой кислоты, такой как неопентиловый сложный эфир хлорсульфоновой кислоты формулы (81):
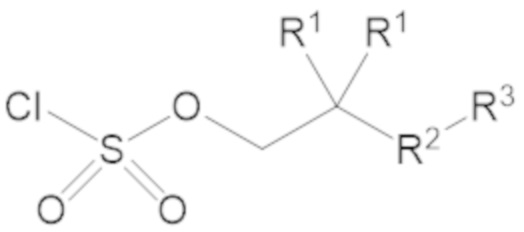 (81)
(81)
в котором
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила; и
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2, в котором
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила.
Сложные моноэфиры хлорсульфоновой кислоты можно синтезировать с помощью реакции спирта, такого как неопентиловый спирт, с сульфурилхлоридом.
Способ можно использовать для связывания любого подходящего сложного эфира хлорсульфоновой кислоты с циклической гидроксамовой кислотой, такого как, например, сложный эфир хлорсульфоновой кислоты формулы (82) и циклической гидроксамовой кислоты формулы (83) с образованием соответствующего аналога сложного моноэфира серной кислоты формулы (84):
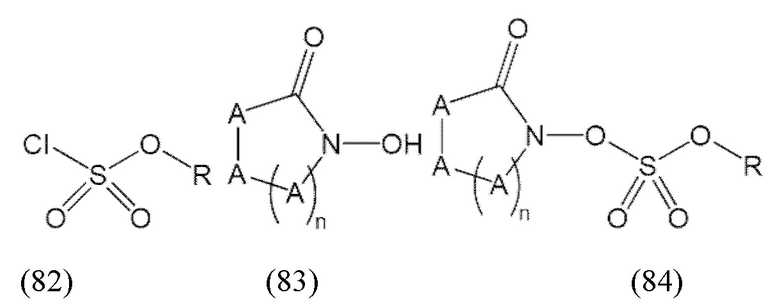
R можно выбирать из C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
n может быть целое число от 1 до 6;
каждый A можно независимо выбирать из -(CH2)-, -(CHR)-, -(CR2)-, -NH-, -NR-, O и S, где R независимо выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила; или один A связан с другим A посредством группы -L-, где L выбирают из C1-8 алкила, C1-8 гетероалкила, замещенного C1-8 алкила и замещенного C1-8 гетероалкила.
R может дополнительно содержать любой из профрагментов, раскрытых в настоящем документе, таких как профрагмент, имеющий структуру:
где R1, R2 и R3 соответствуют определению формулы (1).
Композиции, представленные в настоящем документе, содержат терапевтически эффективные количества одного или более соединений, представленных в настоящем документе, которые полезны для предотвращения, лечения или улучшения одного или более симптомов заболеваний или расстройств, описанных в настоящем документе, и основу. Основы, подходящие для введения соединений, представленных в настоящем документе, включают любые подобные носители, известные специалистам в данной области, подходящих для конкретного режима введения. Кроме того, соединения можно составлять в виде единственного активного ингредиента в композиции или можно комбинировать с другими активными ингредиентами.
Композиции содержат одно или более соединений, представленных в настоящем документе. В некоторых вариантах осуществления соединения готовят в виде подходящих препаратов, таких как растворы, суспензии, таблетки, диспергируемые таблетки, пилюли, капсулы, порошки, готовые формы замедленного высвобождения или элексиры, для перорального введения или в стерильных растворах или суспензиях для парентерального введения, а также местного введения, трансдермального введения и пероральной ингаляции через небулайзеры, дозированные аэрозольные ингаляторы и порошковые ингаляторы. В некоторых вариантах осуществления описанные выше соединения составляют в композиции с применением технологий и методик, хорошо известных в данной области (см. Например, Ansel, Introduction to Pharmaceutical Dosage Forms, Seventh Edition (1999)).
В композициях эффективные концентрации одного или более соединений или их производных смешивают с подходящей основой. Соединения можно дериватизировать в виде соответствующих солей, сложных эфиров, енольных эфиров или сложных эфиров, ацеталей, кеталей, ортоэфиров, полуацеталей, полукеталей, кислот, оснований, сольватов, пар ионов, гидратов или пролекарств перед получением готовых форм, которые описаны выше. Концентрации соединений в композициях являются эффективными для доставки при введении некоторого количества, которое лечит, ведет к предотвращению или улучшению одного или более симптомов заболеваний или расстройств, описанных в настоящем документе. В некоторых вариантах осуществления композиции составляют для введения одной дозировки. Для составления композиции массовую долю соединения растворяют, суспендируют, диспергируют или иным образом перемешивают в выбранной основе с эффективной концентрацией таким образом, чтобы облегчить, предотвратить подлежащее лечению состояние, или улучшить один или более симптомов.
Активное соединение включают в основу в количестве, достаточном для оказания терапевтически полезного действия в отсутствие нежелательных побочных действий на получающего лечение пациента. Терапевтически эффективную концентрацию можно эмпирически спрогнозировать путем тестирования соединений в системах in vitro и in vivo, хорошо известных специалистам в данной области, а затем экстраполировать из них дозировки для людей. Затем дозы для людей обычно точно подбирают в клинических испытаниях и тритруют до ответа.
Концентрация активного соединения в композиции будет зависеть от скорости всасывания, инактивации и выведения активного соединения, физико-химических характеристик соединения, схемы дозировки и вводимого количества, а также других факторов, известных специалистам в данной области. Например, доставляемое количество является достаточным для ослабления одного или нескольких симптомов заболеваний или расстройств, которые описаны в настоящем документе.
В случаях, когда соединения проявляют недостаточную растворимость, можно использовать способы солюбилизации соединений, такие как использование липосом, пролекарств, комплексообразования/хелатирования, наночастиц или эмульсий или третичных шаблонов. Такие способы известны специалистам в данной области и включают, но без ограничения, использование сорастворителей, таких как диметилсульфоксид (ДМСО), использование поверхностно-активных веществ или модификаторов поверхности, таких как TWEEN®, комплексообразующих средств, таких как циклодекстрин или растворение при усиленной ионизации (т.е. растворение в водном бикарбонате натрия). При составлении эффективных композиций также можно использовать производные соединений, такие как пролекарства соединений.
При смешивании или добавлении соединения (соединений) полученная смесь может представлять собой раствор, суспензию, эмульсию или тому подобное. Форма полученной смеси зависит от ряда факторов, включая предполагаемый режим введения и растворимость соединения в выбранной основе. Эффективная концентрация достаточна для ослабления симптомов подлежащего лечению заболевания, расстройства или состояния, и может быть определена эмпирически.
Композиции предназначены для введения людям и животным в показанных соответствующих лекарственных формах, таких как порошковые ингаляторы (DPI), дозированные аэрозольные ингаляторы (pMDI), небулайзеры, таблетки, капсулы, пилюли, сублингвальные ленты/биоразрушаемые стрипсы, таблетки или капсулы, порошки, гранулы, пастилки, лосьоны, мази, суппозитории, быстрые расплавы, трансдермальные пластыри или другие устройства/препараты для трансдермального применения, стерильные парентеральные растворы или суспензии и пероральные растворы или суспензии, а также масляно-водные эмульсии, содержащие подходящие количества соединений или их производных. В некоторых вариантах осуществления терапевтически активные соединения и их производные составляют и вводят в единичных лекарственных формах или множественных лекарственных формах. В рамках настоящего изобретения единичные лекарственные формы относятся к физически дискретным единицам, подходящим для людей и животных и упакованным индивидуально, как известно в данной области. Каждая стандартная доза содержит заранее определенное количество терапевтически активного соединения, достаточное для получения желаемого терапевтического эффекта, в сочетании с необходимой основой. Примеры единичных лекарственных форм включают ампулы и шприцы и индивидуально упакованные таблетки или капсулы. Единичные лекарственные формы можно вводить долями или кратно им. Множественная лекарственная форма представляет собой множество идентичных единичных лекарственных форм, упакованных в один контейнер, для введения в виде раздельных единичных лекарственных форм. Примеры множественных лекарственных форм включают флаконы, бутылки с таблетками или капсулами или бутылки с измерением в пинтах или галлонах. Следовательно, множественная лекарственная форма представляет собой множество стандартных доз, которые не разделены в упаковке.
Жидкие композиции можно получать, например, путем растворения, диспергирования или иного смешивания активного соединения, которое определено выше, и необязательных вспомогательных веществ в основе, такой как, например, вода, физиологический раствор, водная декстроза, глицерин, гликоли, этанол и тому подобное, с образованием таким образом раствора или суспензии, коллоидной дисперсии, эмульсии или липосомальной готовой формы. При желании композиция для введения может также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие средства, эмульгирующие средства, солюбилизирующие средства, буферные средства для поддержания рН и тому подобное, например, ацетат, цитрат натрия, производные циклодекстрина, сорбитанмонолаурат, триэтаноламин ацетат натрия, триэтаноламин олеат и другие подобные средства.
Современные способы приготовления таких лекарственных форм известны или будут очевидны специалистам в данной области; например, см. Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 15th Edition, 1975 или более поздние издания.
Можно получать лекарственные формы или композиции, содержащие активный ингредиент в диапазоне от 0,005% до 100% с остатком, состоящим из основы или носителя. Способы получения этих композиций известны специалистам в данной области. Рассматриваемые композиции могут содержать 0,001%-100% активного ингредиента, в одном варианте осуществления 0,1-95%, в другом варианте осуществления 0,4-10%.
Композиции могут представлять собой композиции без лактозы, содержащие вспомогательные средства, которые хорошо известны в данной области и перечислены, например, в Фармакопее США (USP) 25 NF20 (2002). Как правило, композиции без лактозы содержат активные ингредиенты, связующее вещество/наполнитель и смазывающее вещество в совместимых количествах. Конкретные лекарственные формы без лактозы содержат активные ингредиенты, микрокристаллическую целлюлозу, предварительно желатинизированный крахмал и стеарат магния.
Кроме того, предоставлены безводные композиции и лекарственные формы, содержащие активные ингредиенты, поскольку вода может способствовать разложению некоторых соединений. Например, добавление воды (например, 5%) широко признано в качестве средства имитации длительного хранения, чтобы определить такие характеристики, как срок годности или стабильность готовых форм во времени. См., например, Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80. Фактически вода и тепло ускоряют разложение некоторых соединений. Таким образом, влияние воды на готовую форму может иметь большое значение, так как при изготовлении, обработке, упаковке, хранении, транспортировке и использовании готовых форм обычно присутствует влага и/или влажность.
Безводные композиции и лекарственные формы, представленные в настоящем документе, можно получать с использованием безводных или содержащих низкую влажность ингредиентов и условий с низким содержанием влаги или низкой влажностью.
Безводную композицию следует получать и хранить таким образом, чтобы сохранить ее безводную природу. Соответственно, безводные композиции обычно упаковывают с использованием материалов, которые, как известно, предотвращают воздействие воды, так чтобы их можно было включать в подходящие рецептурные наборы. Примеры подходящей упаковки включают, но без ограничения герметически закрытую фольгу, пластмассы, контейнеры с единичной дозой (например, флаконы), блистерные упаковки и ленточные упаковки.
Пероральные лекарственные формы являются либо твердыми, гелевыми или жидкими. Твердые лекарственные формы представляют собой таблетки, капсулы, гранулы и сыпучие порошки. Типы пероральных таблеток включают прессованные жевательные пастилки и таблетки, которые могут быть покрыты энтеросолюбильной, сахарной или пленочной оболочкой. Капсулы могут быть твердыми или мягкими желатиновыми капсулами, в то время как гранулы и порошки могут быть предоставлены в не шипучей или шипучей форме в сочетании с другими ингредиентами, известными специалистам в данной области.
Готовые формы могут представлять собой твердые лекарственные формы, такие как, например, капсулы или таблетки. Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать один или несколько из следующих ингредиентов или соединений аналогичной природы: связующее вещество; смазывающее вещество; разбавитель; глидант; дезинтегрирующее средство; красящее средство; подсластитель; ароматизирующее средство; смачивающее средство; энтеросолюбильное покрытие; покрывающее пленкой средство и модифицирующее высвобождение средство. Примеры связующих веществ включают микрокристаллическую целлюлозу, метилпарабен, полиалкиленоксиды, трагакантовую камедь, раствор глюкозы, экстракт слизи акации, желатиновый раствор, мелассу, поливинилпирролидин, повидон, кросповидоны, сахарозу и крахмал и производные крахмала. Смазывающие вещества включают тальк, крахмал, стеарат магния/кальция, ликоподий и стеариновую кислоту. Разбавители включают, например, лактозу, сахарозу, трегалозу, лизин, лейцин, лецитин, крахмал, каолин, соль, маннит и дикальцийфосфат. Глиданты включают, но без ограничения, коллоидный диоксид кремния. Разрыхлители включают кроскармеллозу натрия, натрий крахмал гликолят, альгиновую кислоту, кукурузный крахмал, картофельный крахмал, бентонит, метилцеллюлозу, агар и карбоксиметилцеллюлозу. Красящие средства включают, например, любой из утвержденных сертифицированных водорастворимых красителей FD и C, их смеси; и нерастворимые в воде красители FD и C, суспендированные в гидрате оксида алюминия, и улучшенные красящие или предотвращающие подделку красящие/опалесцирующие добавки, известные специалистам в данной области. Подсластители включают сахарозу, лактозу, маннит и искусственные подсластители, такие как сахарин и любое количество высушенных распылением ароматизаторов. Ароматизирующие средства включают натуральные ароматизаторы, экстрагируемые из растений, такие как фрукты и синтетические смеси соединений, которые вызывают приятное ощущение или маскируют неприятный вкус, такие как, но без ограничения, мята перечная и метилсалицилат. Смачивающие средства включают моностеарат пропиленгликоля, моноолеат сорбитана, монолаурат диэтиленгликоля и полиоксиэтиленлауриловый эфир. Энтеросолюбильные покрытия включают жирные кислоты, жиры, воски, шеллак, аммонизированный шеллак и фталаты ацетата целлюлозы. Пленочные покрытия включают гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу натрия, полиэтиленгликоль 4000 и ацетатфталат целлюлозы. Модифицирующие высвобождение средства включают полимеры, такие как серия Eudragit®, и сложные эфиры целлюлозы.
Соединение или его производное может быть предоставлено в композиции, которая защищает его от кислой среды желудка. Например, композиция может быть составлена в виде энтеросолюбильной оболочки, которая сохраняет свою целостность в желудке и высвобождает активное соединение в кишечнике. Композиция также может быть составлена в комбинации с антацидом или другим таким ингредиентом.
Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, помимо материала вышеуказанного типа, жидкий носитель, такой как жирное масло. Кроме того, стандартные лекарственные формы могут содержать различные другие материалы, которые модифицируют физическую форму стандартной дозы, например, покрытия из сахара и других энтеросолюбильных агентов. Соединения также можно вводить в качестве компонента эликсира, суспензии, сиропа, вафли, обсыпки, жевательной резинки или тому подобное. В дополнение к активным соединениям сироп может содержать сахарозу в качестве подсластителя и некоторые консерванты, красители, красящие средства и ароматизаторы.
Активные материалы также можно смешивать с другими активными материалами, которые не ухудшают желаемое действие, или с материалами, которые дополняют желаемое действие, такими как антациды, блокаторы H2 и диуретики. Активный ингредиент представляет собой соединение или его производное, которое описано в настоящем документе. Можно включать более высокие концентрации, примерно до 98 мас.% активного ингредиента.
Во всех вариантах осуществления готовые формы в виде таблеток и капсул могут быть покрыты оболочкой, как известно специалистам в данной области, для модификации или поддержания растворения активного ингредиента. Так, например, они могут быть покрыты обычным растворимым в кишечнике покрытием, таким как фенилсалицилат, воски и фталат ацетата целлюлозы.
Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, восстанавливаемые из нешипучих гранул, и шипучие препараты, восстанавливаемые из шипучих гранул. Водные растворы включают, например, эликсиры и сиропы. Эмульсии представляют собой масло в воде или воду в масле.
Эликсиры представляют собой прозрачные, подслащенные, водноспиртовые препараты. Основы, используемые в эликсирах, включают растворители. Сиропы представляют собой концентрированные водные растворы сахара, например сахарозы, и могут содержать консервант. Эмульсия представляет собой двухфазную систему, в которой одна жидкость диспергирована в форме маленьких шариков в другой жидкости. Носителями, используемыми в эмульсиях, являются неводные жидкости, эмульгирующие средства и консерванты. В суспензиях используют суспендирующие средства и консерванты. Приемлемые вещества, используемые в нешипучих гранулах, подлежащих восстановлению в жидкую пероральную лекарственную форму, включают разбавители, подсластители и смачивающие средства. Приемлемые вещества, используемые в шипучих гранулах, подлежащих восстановлению в жидкую пероральную лекарственную форму, включают органические кислоты и источник углекислого газа. Красящие и ароматизирующие средства используются во всех вышеперечисленных лекарственных формах.
Растворители включают глицерин, сорбит, этиловый спирт и сироп. Примеры консервантов включают глицерин, метил и пропилпарабен, бензойную кислоту, бензоат натрия и спирт. Примеры неводных жидкостей, используемых в эмульсиях, включают минеральное масло и хлопковое масло. Примеры эмульгирующих средств включают желатин, акацию, трагакант, бентонит и поверхностно-активные вещества, такие как полиоксиэтилен сорбитанмоноолеат. Суспендирующие средства включают натрий карбоксиметилцеллюлозу, пектин, трагакант, вигум и акацию. Подсластители включают сахарозу, сиропы, глицерин и искусственные подсластители, такие как сахарин. Смачивающие средства включают моностеарат пропиленгликоля, моноолеат сорбитана, монолаурат диэтиленгликоля и полиоксиэтиленлауриловый эфир. Органические кислоты включают лимонную и винную кислоты. Источники углекислого газа включают бикарбонат натрия и карбонат натрия. Красящие вещества включают любой из утвержденных сертифицированных водорастворимых красителей FD и C и их смеси. Ароматизирующие средства включают натуральные ароматизаторы, экстрагируемые из растений, таких как фрукты, и синтетические смеси соединений, которые вызывают приятные вкусовые ощущения.
Для твердой лекарственной формы раствор или суспензия, например, в пропиленкарбонате, растительных маслах или триглицеридах, в некоторых вариантах осуществления заключены в желатиновую капсулу. Такие растворы, а также их получение и капсулирование раскрыты в патентах США №№ 4328245; 4409239; и 4410545. Для жидкой лекарственной формы раствор, например, в полиэтиленгликоле, можно разбавлять достаточным количеством жидкой основы, например воды, чтобы его можно было легко измерить для введения.
Альтернативно, жидкие или полутвердые пероральные готовые формы можно получать путем растворения или диспергирования активного соединения или соли в растительных маслах, гликолях, триглицеридах, сложных эфирах пропиленгликоля (например, пропиленкарбонате) и других таких носителях и инкапсулирования этих растворов или суспензий в твердые или мягкие желатиновые капсульные оболочки. Другие используемые готовые формы включают готовые формы, изложенные в патентах США № RE28819 и 4358603. Вкратце, такие готовые формы включают, но без ограничения, готовые формы, которые содержат соединение, представленное в настоящем документе, диалкилированный моно- или полиалкиленгликоль, включая, но без ограничения, 1,2-диметоксиэтан, диглим, триглим, тетраглим, диметиловый эфир полиэтиленгликоля 350, диметиловый эфир полиэтиленгликоля 550, диметиловый эфир полиэтиленгликоля 750, где 350, 550 и 750 относятся к приблизительной средней молекулярной массе полиэтиленгликоля, и один или несколько антиоксидантов, таких как бутилированный гидрокситолуол (ВНТ), бутилированный гидроксианизол (ВНА), пропил галлат, витамин Е, гидрохинон, гидроксикумарины, этаноламин, лецитин, цефалин, аскорбиновая кислота, яблочная кислота, сорбит, фосфорная кислота, тиодипропионовая кислота и ее сложные эфиры и дитиокарбаматы.
Другие готовые формы включают, но без ограничения, водно-спиртовые растворы, включая ацеталь. Спирты, используемые в этих готовых формах, представляют собой любые смешивающиеся с водой растворители, имеющие одну или несколько гидроксильных групп, включая, но без ограничения, пропиленгликоль и этанол. Ацетали включают, но без ограничения, ди (низший алкил) ацетали низших алкильных альдегидов, таких как ацетальдегид диэтилацеталь.
В некоторых вариантах осуществления, характеризующихся подкожной, внутримышечной или внутривенной инъекцией, также в настоящем документе рассматривается парентеральное введение. Инъецируемые препараты можно получать в обычных формах, либо в виде жидких растворов или суспензий, твердых форм, подходящих для раствора или суспензии в жидкости перед инъекцией, либо в виде эмульсий. Инъецируемые препараты, растворы и эмульсии также содержат один или несколько вспомогательных средств. Подходящими эксципиентами являются, например, вода, физиологический раствор, декстроза, глицерин или этанол. Кроме того, если необходимо, композиции для введения также могут содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие или эмульгирующие средства, буферные средства для поддержания рН, стабилизаторы, усилители растворимости и другие подобные средства, такие как, например, ацетат натрия, сорбитанмонолаурат, триэтаноламин олеат и циклодекстрины.
Также в настоящем документе предусмотрено внедрение системы с медленным высвобождением или с замедленным высвобождением, так чтобы поддерживать постоянный уровень дозировки (см., например, патент США № 3710795). Вкратце, представленное в настоящем документе соединение диспергировано в твердой внутренней матрице, например, в полиметилметакрилате, полибутилметакрилате, пластифицированном или непластифицированном поливинилхлориде, пластифицированном нейлоне, пластифицированном полиэтилентерефталате, натуральном каучуке, полиизопрене, полиизобутилене, полибутадиене, полиэтилене, сополимерах этилена и винилацетата, силиконовых каучуках, полидиметилсилоксанах, силикон-карбонатных сополимерах, гидрофильных полимерах, таких как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, сшитый поливиниловый спирт и сшитый частично гидролизованный поливинилацетат, который окружен внешней полимерной мембраной, такой как полиэтилен, полипропилен, этилен/пропиленовые сополимеры, этилен/этилакрилатные сополимеры, этилен/винилацетатные сополимеры, силиконовые каучуки, полидиметилсилоксаны, неопреновый каучук, хлорированный полиэтилен, поливинилхлорид, винилхлоридные сополимеры с винилацетатом, винилиденхлорид, этилен и пропилен, иономер полиэтилентерефталат, бутилкаучуковые эпихлоргидриновые каучуки, сополимер этилен/винилового спирта, терполимер этилен/винилацетат/винилового спирта и сополимер этилен/винилоксиэтанола, который нерастворим в жидкостях организма. Соединение диффундирует через внешнюю полимерную мембрану на стадии регулирования скорости высвобождения. Процентное содержание активного соединения, содержащегося в таких парентеральных композициях, сильно зависит от его конкретной природы, а также от активности соединения и потребностей пациента.
Парентеральное введение композиций включает внутривенное, подкожное и внутримышечное введения. Препараты для парентерального введения включают стерильные растворы, готовые для инъекций, стерильные сухие растворимые продукты, такие как лиофилизированные порошки, готовые для соединения с растворителем непосредственно перед применением, включая таблетки для подкожных инъекций, стерильные суспензии, готовые для инъекций, стерильные сухие нерастворимые продукты, готовые для соединения с основой непосредственно перед применением, и стерильные эмульсии. Растворы могут быть водными или неводными.
При внутривенном введении подходящие носители включают физиологический солевой раствор или физиологический раствор с фосфатным буфером (PBS) и растворы, содержащие загущающие и солюбилизирующие средства, такие как глюкоза, полиэтиленгликоль и полипропиленгликоль, и их смеси.
Основы, используемые в парентеральных препаратах, включают водные основы, неводные основы, антимикробные средства, изотонические средства, буферы, антиоксиданты, местные анестетики, суспендирующие и диспергирующие средства, эмульгирующие средства, изолирующие или хелатирующие средства и другие вещества.
Примеры водных основ включают раствор натрия хлорида для инъекций, раствор Рингера для инъекций, раствор изотонической декстрозы для инъекций, стерильную воду для инъекций, раствор декстрозы и лактатный раствор Рингера для инъекций. Неводные парентеральные основы включают жирные масла растительного происхождения, хлопковое масло, кукурузное масло, кунжутное масло и арахисовое масло. К парентеральным препаратам, упакованным в контейнеры для многократного применения, необходимо добавлять противомикробные средства в бактериостатических или фунгистатических концентрациях, которые включают фенолы или крезолы, ртуть, бензиловый спирт, хлорбутанол, метиловые и пропиловые эфиры пара гидроксибензойной кислоты, тимеросал, хлорид бензалкония и хлорид бензетония. Изотонические средства включают хлорид натрия и декстрозу. Буферы включают фосфат и цитрат. Антиоксиданты включают бисульфат натрия. Местные анестетики включают гидрохлорид прокаина. Суспендирующие и диспергирующие средства включают натрий карбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу и поливинилпирролидон. Эмульгирующие средства включают полисорбат 80 (Tween® 80). Секвестрирующее или хелатообразующее средство ионов металлов включает EDTA. Носители также включают этиловый спирт, полиэтиленгликоль и пропиленгликоль для смешиваемых с водой основ; и гидроксид натрия, соляную кислоту, лимонную кислоту или молочную кислоту для регулирования рН.
Концентрацию соединения регулируют таким образом, чтобы инъекция обеспечивала эффективное количество для получения желаемого фармакологического эффекта. Как известно в данной области, точная доза зависит от возраста, веса, площади поверхности тела и состояния пациента или животного.
Единичную дозу парентеральных препаратов упаковывают в ампулу, флакон или шприц с иглой. Все препараты для парентерального введения должны быть стерильными, как это известно и практикуется в данной области.
Иллюстративно, внутривенная или внутриартериальная инфузия стерильного водного раствора, содержащего активное соединение, представляет собой эффективный способ введения. Другим вариантом осуществления является стерильный водный или масляный раствор или суспензия, содержащая активный материал, вводимый по мере необходимости для получения желаемого фармакологического эффекта.
Инъекционные препараты предназначены для местного и системного применения. В некоторых вариантах осуществления составляют терапевтически эффективную дозировку, чтобы она содержала концентрацию по меньшей мере от 0,01 мас.% до 90 мас.% или более, например, более 0,1 мас.% активного соединения на обработанную ткань (ткани).
Соединение можно суспендировать в микронизированной или другой подходящей форме или можно дериватизировать для получения более растворимого активного продукта или для получения пролекарства. Форма получающейся в результате смеси зависит от ряда факторов, включая предполагаемый способ введения и растворимость соединения в выбранном носителе или основе. Эффективная концентрация достаточна для ослабления симптомов состояния и может быть определена эмпирически.
Активные ингредиенты, представленные в настоящем документе, можно вводить с помощью средства регулируемого высвобождения или с помощью устройств доставки, которые хорошо известны рядовым специалистам в данной области. Примеры включают, но без ограничения примеры, описанные в патентах США №№: 3845770; 3916899; 3536809; 3598123; 4008719; 5674533; 5059595; 5591767; 5120548; 5073543; 5639476; 5354556; 5639480; 5733566; 5739108; 5891474; 5922356; 5972891; 5980945; 5993855; 6045830; 6087324; 6113943; 6197350; 6248363; 6264970; 6267981; 6376461; 6419961; 6589548; 6613358; 6699500 и 6740634. Подобные лекарственные формы можно использовать для обеспечения медленного или регулируемого высвобождения одного или более активных ингредиентов с использованием, например, гидроксипропилметилцеллюлозы, других полимерных матриц, гелей, проницаемых мембран, осмотических систем, многослойных покрытий, микрочастиц, липосом, микросфер или их комбинаций с получением нужного профиля высвобождения в различных пропорциях. Подходящие готовые формы регулируемого высвобождения, известные рядовым специалистам в данной области, включая готовые формы, описанные в настоящем документе, можно легко выбрать для использования с активными ингредиентами, представленными в настоящем документе.
Все продукты с регулируемым высвобождением имеют общую цель улучшения лекарственной терапии по сравнению с тем, что достигается их не регулируемыми аналогами. В идеале, использование в лечении оптимально разработанного препарата с регулируемым высвобождением отличается минимальным количеством лекарственного вещества, используемого для лечения или управления состоянием за минимальное количество времени. Преимущества готовых форм с регулируемым высвобождением включают расширенную активность лекарственного средства, сниженную частоту введения дозы и повышенную приверженность пациента. Кроме того, композиции с регулируемым высвобождением можно использовать для влияния на время начала действия или других характеристик, таких как уровни лекарственного средства в крови, и, таким образом, они могут влиять на возникновение побочных (например, неблагоприятных) эффектов.
Большинство готовых форм с регулируемым высвобождением предназначены для первоначального высвобождения некоторого количества лекарственного средства (активного ингредиента), которое быстро производит нужный терапевтический эффект, и постепенного и непрерывного высвобождения других количеств лекарственного средства для поддержания этого уровня терапевтического или профилактического эффекта в течение продолжительного периода времени. Чтобы поддерживать этот постоянный уровень лекарственного средства в организме, лекарственное средство должно высвобождаться из лекарственной формы со скоростью, которая будет замещать количество лекарственного средства, которое метаболизируется и выводится из организма. Регулируемое высвобождение активного ингредиента можно стимулировать с помощью различных условий, включая, но без ограничения, pH, температуру, ферменты, воду или другие физиологические условия или соединения.
Средство можно вводить с использованием внутривенной инфузии, имплантируемого осмотического насоса, трансдермального пластыря, липосом или других способов введения. В некоторых вариантах осуществления можно использовать насос (см. Sefton, CRC Crit. Ref. Biomed. Eng. 14: 201 (1987); Buchwald et al., Surgery 88: 507 (1980); Saudek et al., N. Engl. J. Med. 321: 574 (1989)). В других вариантах можно использовать полимерные материалы. В других вариантах осуществления систему с регулируемым высвобождением можно размещать в непосредственной близости от терапевтической мишени, то есть, таким образом, требуя лишь доли системной дозы (см., например, Goodson, Medical Applications of Controlled Release, vol. 2, pp. 115138 (1984)). В некоторых вариантах осуществления устройство с регулируемым высвобождением вводят пациенту вблизи участка ненужной иммунной активации или опухоли. Другие системы с регулируемым высвобождением обсуждаются в обзоре Langer (Science 249: 1527 1533 (1990)). Активный ингредиент можно диспергировать в твердой внутренней матрице, например, полиметилметакрилате, полибутилметакрилате, пластифицированном или непластифицированном поливинилхлориде, пластифицированном нейлоне, пластифицированном полиэтилентерефталате, натуральном каучуке, полиизопрене, полиизобутилене, полибутадиене, полиэтилене, сополимерах этилена и винилацетата, силиконовых каучуках, полидиметилсилоксанах, силикон-карбонатных сополимерах, гидрофильных полимерах, таких как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, сшитый поливиниловый спирт и сшитый частично гидролизованный поливинилацетат, который окружен внешней полимерной мембраной, такой как полиэтилен, полипропилен, этилен/пропиленовые сополимеры, этилен/этилакрилатные сополимеры, этилен/винилацетатные сополимеры, силиконовые каучуки, полидиметилсилоксаны, неопреновые каучуки, хлорированный полиэтилен, поливинилхлорид, винилхлоридные сополимеры с винилацетатом, винилиденхлорид, этилен и пропилен, иономер полиэтилентерефталат, бутилкаучуковые эпихлоргидриновые каучуки, сополимер этилена/винилового спирта, терполимер этилена/винилацетата/винилового спирта и сополимер этилена/винилоксиэтанола, который нерастворим в жидкостях организма. Затем активный ингредиент диффундирует через внешнюю полимерную мембрану на стадии регулирования скорости высвобождения. Процентное содержание активного ингредиента, содержащегося в таких парентеральных композициях, сильно зависит от его конкретной природы, а также от потребностей пациента.
В данном случае представляют интерес также лиофилизированные порошки, которые можно восстанавливать для введения в виде растворов, эмульсий и других смесей. Их также можно восстанавливать и составлять в виде твердых веществ или гелей.
Стерильный лиофилизированный порошок получают путем растворения представленного в настоящем документе соединения, или его производного в подходящем растворителе. Растворитель может содержать вспомогательное средство, которое улучшает стабильность, или другой фармакологический компонент порошка или полученного из порошка восстановленного раствора. Вспомогательные вещества, которые можно использовать, включают, но без ограничения, антиоксидант, буфер и объемообразующее средство. В некоторых вариантах осуществления вспомогательное средство выбирают из декстрозы, сорбита, фруктозы, кукурузного сиропа, ксилита, глицерина, глюкозы, сахарозы и других подходящих средств. Растворитель может содержать буфер, такой как цитрат, фосфат натрия или калия, или другой такой буфер, известный специалистам в данной области, с нейтральным pH. Нужную готовую форму дает последующая стерильная фильтрация раствора с последующей лиофилизацией в стандартных условиях, известных специалистам в данной области. В некоторых вариантах осуществления полученный раствор следует распределять во флаконы для лиофилизации. Каждый флакон будет содержать одну дозу или несколько доз соединения. Лиофилизированный порошок можно хранить в соответствующих условиях, например при температуре от 4°С до комнатной температуры.
Восстановление этого лиофилизированного порошка водой для инъекций обеспечивает готовую форму для использования при парентеральном введении. Для восстановления лиофилизированный порошок добавляют в стерильную воду или другой подходящий носитель. Точное количество зависит от выбранного соединения. Такое количество может быть определено опытным путем.
Смеси для местного применения получают, как описано для местного и системного введения. Полученная смесь может представлять собой раствор, суспензию, эмульсии или тому подобное, и ее составляют в виде кремов, гелей, мазей, эмульсий, растворов, эликсиров, лосьонов, суспензий, настоек, паст, пен, аэрозолей, ирригаций, спреев, суппозиториев, повязок, кожных пластырей или любых других готовых форм, подходящих для местного применения.
Соединения или их производные можно составлять в виде аэрозолей для местного применения, например, путем ингаляции (см., например, патенты США №№ 4044126, 4414209 и 4364923, в которых описаны аэрозоли для доставки стероида, применяемого для лечения воспалительных заболеваний, в частности астмы). Эти готовые формы для введения в дыхательные пути могут быть в виде аэрозоля или раствора для небулайзера или в виде мелкодисперсного порошка для инсуффляции, отдельно или в комбинации с инертным носителем, таким как лактоза. В таком случае в некоторых вариантах осуществления частицы готовой формы будут иметь среднемассовые геометрические диаметры менее 5 микрон, в других вариантах осуществления менее 10 микрон.
Готовые формы для пероральной ингаляции соединений или производных, пригодных для ингаляции, включают ингаляторы с отмеренной дозой, порошковые ингаляторы и жидкие препараты для введения из небулайзера или системы дозирования отмеренной жидкости. Как для ингаляторов с дозированной дозой, так и для порошковых ингаляторов кристаллическая форма соединений или производных является предпочтительной физической формой лекарственного средства для обеспечения большей стабильности продукта.
В дополнение к методам уменьшения размера частиц, известным специалистам в данной области, кристаллические частицы соединений или производных можно получать с использованием обработки сверхкритической средой, что дает значительные преимущества при получении таких частиц для ингаляционной доставки путем получения пригодных для вдыхания частиц нужного размера в одну стадию (например, международная публикация PCT № WO 2005/025506). Регулируемый размер частиц для микрокристаллов можно выбрать так, чтобы гарантировать, что значительная часть соединений или производных задерживается в легких. В некоторых вариантах осуществления эти частицы имеют среднемассовый аэродинамический диаметр от 0,1 микрона до 10 микрон, в других вариантах осуществления 1 микрона до 5 микрон, и в других вариантах от 1,2 микрона до 3 микрон.
Инертные и невоспламеняющиеся пропелленты HFA выбирают из HFA 134a (1,1,1,2 тетрафторэтан) и HFA 227e (1,1,1,2,3,3,3 гептафторпропан) и предоставляют по отдельности или в соотношении, соответствующем плотности кристаллических частиц соединений или производных. Соотношение также выбирают, чтобы обеспечить предотвращение вредного осаждения суспензии продукта или крема (которые могут вызвать необратимую агломерацию) и вместо этого способствовать образованию слабо флокулированной системы, которая легко диспергируется при встряхивании. Полагают, что слабо колеблющиеся системы обеспечивают оптимальную стабильность для коробок pMDI. В результате свойств готовой формы готовая форма не содержал этанола и поверхностно-активных веществ/стабилизирующих средств.
Соединения можно составлять для локального или местного применения, такого как местное нанесение на кожу и слизистые оболочки, например, в глазу, в форме гелей, кремов и лосьонов и для нанесения на глаз или для внутрицистернального или интраспинального применения. Местное введение предназначено для трансдермальной доставки, а также для введения в глаза или слизистую оболочку или для ингаляционной терапии. Также можно вводить носовые растворы активного соединения отдельно или в комбинации с другими наполнителями.
Для назального введения препарат может содержать этерифицированное фосфонатное соединение, растворенное или суспендированное в жидком носителе, в частности, в водном носителе, для аэрозольного применения. Носитель может содержать солюбилизирующие или суспендирующие средства, такие как пропиленгликоль, поверхностно-активные вещества, усилители абсорбции, такие как лецитин или циклодекстрин, или консерванты.
Растворы, особенно растворы, предназначенные для офтальмологического применения, можно составлять в виде изотонических растворов 0,01-10%, рН 5-7,4, с соответствующими солями.
В настоящем документе также предусмотрены другие пути введения, такие как трансдермальные пластыри, включая ионофоретические и электрофоретические устройства и ректальное введение.
Чрескожные пластыри, включая ионофоретические и электрофоретические устройства, хорошо известны специалистам в данной области. Например, такие пластыри раскрыты в патентах США №№ 6267983, 6261595, 6256533, 6167301, 6024975, 6010715, 5985317, 5983134, 5948433 и 5860957.
Например, лекарственные формы для ректального введения представляют собой ректальные суппозитории, капсулы и таблетки для системного действия. Ректальные суппозитории, используемые в настоящем документе, означают твердые тела для введения в прямую кишку, которые плавятся или размягчаются при температуре тела, высвобождая один или несколько фармакологически или терапевтически активных ингредиентов. Вещества, используемые в ректальных суппозиториях, представляют собой основы или носители и агенты для повышения температуры плавления. Примеры основ включают масло какао (масло растений рода теоброма), желатин-глицерин, карбовакс (полиоксиэтиленгликоль) и соответствующие смеси моно-, ди- и триглицеридов жирных кислот. Можно использовать комбинации различных оснований. Средства для повышения температуры плавления суппозиториев включают спермацет и воск. Ректальные суппозитории можно получать прессованным способом или формованием. В одном варианте осуществления масса ректального суппозитория составляет от 2 г. До 3 г. Таблетки и капсулы для ректального введения изготавливаются с использованием того же вещества и теми же способами, что и для составов для перорального введения.
Соединения, представленные в настоящем документе, или их производные также можно составлять с нацеливанием на конкретную ткань, рецептор или другую область организма подлежащего лечению субъекта. Специалистам в данной области хорошо известны многие подобные способы нацеливания. В настоящем документе предусмотрены все подобные способы нацеливания для применения в настоящих композициях. Для неограничивающих примеров способов нацеливания, см., например, патенты США №№ 6316652, 6274552, 6271359, 6253872, 6139865, 6131570, 6120751, 6071495, 6060082, 6048736, 6039975, 6004534, 5985307, 5972366, 5900252, 5840674, 5759542 и 5709874.
В некоторых вариантах осуществления в качестве носителей также могут подойти липосомные суспензии, включая липосомы, нацеленные на ткани, такие как липосомы, нацеленные на опухоль. Их можно получать в соответствии со способами, известными специалистам в данной области. Например, липосомные готовые формы можно получать, как описано в патенте США № 4522811. Вкратце, липосомы, такие как многослойные везикулы (MLV), можно получать путем высушивания фосфатидилхолина и фосфатидилсерина (молярное соотношение 7:3) внутри колбы. Добавляют раствор соединения, представленного в настоящем документе, в фосфатно-солевом буфере, в котором отсутствуют двухвалентные катионы (PBS), и колбу встряхивают до диспергирования липидной пленки. Полученные везикулы промывают для удаления неинкапсулированного соединения, осаждают центрифугированием, а затем повторно суспендируют в PBS.
Соединения или производные можно упаковать в виде изделий, содержащих упаковочный материал, соединение или его производное, представленные в настоящем документе, которые эффективны для лечения, предотвращения или ослабления одного или нескольких симптомов заболеваний или нарушений, выше, в упаковочном материале, и этикетку, которая указывает, что соединение или композиция или его производное используется для лечения, профилактики или ослабления одного или нескольких симптомов заболеваний или расстройств, выше.
Представленные в настоящем документе готовые изделия содержат упаковочные материалы. Упаковочные материалы для использования в упаковочных продуктах хорошо известны специалистам в данной области. См., например, патенты США №№ 5323907, 5052558 и 5033252. Примеры упаковочных материалов включают, но без ограничения, блистерные упаковки, бутылки, пробирки, ингаляторы, насосы, пакеты, флаконы, контейнеры, шприцы, бутылки и любой упаковочный материал, подходящий для выбранной готовой формы и предполагаемого способа введения и обработки. Предусмотрен широкий спектр готовых форм соединений и композиций, представленных в настоящем документе, а также различные способы лечения любого описанного в настоящем документе заболевания или расстройства.
Для применения для лечения или профилактики инфекционного заболевания описанные в настоящем документе соединения или композиции или их фармацевтические композиции можно вводить или применять в терапевтически эффективном количестве. При лечении человека врач определяет схему лечения, которая наиболее подходит в соответствии с профилактическим или терапевтическим лечением и в соответствии с возрастом, весом, стадией заболевания и другими факторами, специфичными для подлежащего лечению субъекта. Количество активного ингредиента в представленных в настоящем документе готовых формах, которое будет эффективным для профилактики или лечения инфекционного заболевания, будет варьировать в зависимости от природы и тяжести заболевания или состояния и пути введения активного ингредиента. Частота и дозировка также будут варьировать в зависимости от факторов, специфичных для каждого субъекта, в зависимости от назначенной конкретной терапии (например, терапевтических или профилактических средств), тяжести инфекции, пути введения, а также возраста, массы тела, массы тела, реакции и прежней истории болезней субъекта.
Иллюстративные дозы препарата включают количество активного соединения в миллиграммах или микрограммах на килограмм субъекта (например, от 1 микрограмма на килограмм до 50 миллиграммов на килограмм, от 10 микрограммов на килограмм до 30 миллиграммов на килограмм, от 100 микрограммов на килограмм до 10 миллиграммов на килограмм или от 100 микрограммов на килограмм до 5 миллиграммов на килограмм).
В некоторых вариантах осуществления терапевтически эффективная дозировка должна давать концентрацию активного ингредиента в сыворотке от 0,001 нг/мл до 50 мкг/мл до 200 мкг/мл. В других вариантах осуществления композиции должны обеспечивать дозировку от 0,0001 до 70 мг соединения на килограмм массы тела в день. Стандартные лекарственные формы готовят для обеспечения от 0,01 до 0,1 мг, от 1 до 500 мг или от 1000 до 5000 мг, а в некоторых вариантах осуществления от 10 до 500 мг активного ингредиента или комбинации основных ингредиентов на стандартную лекарственную форму.
Активный ингредиент можно вводить сразу или можно разделить на несколько меньших доз, вводимых через определенные промежутки времени. Понятно, что точная дозировка и продолжительность лечения являются функцией подлежащего лечению заболевания и могут быть определены эмпирически с использованием известных протоколов тестирования или путем экстраполяции из данных испытаний in vivo или in vitro или последующего клинического тестирования. Следует отметить, что концентрации и значения дозировки также могут варьировать в зависимости от тяжести подлежащего облегчению состояния. Кроме того, следует понимать, что для любого конкретного субъекта конкретные схемы лечения необходимо корректировать с течением времени в соответствии с индивидуальной потребностью и профессиональным суждением лица, управляющего или контролирующего введение композиций, и что изложенные в данном документе диапазоны концентраций являются всего лишь иллюстративными и не предназначены для ограничения объема или практического использования заявленных композиций.
Как будет очевидно для специалистов в данной области, в некоторых случаях может быть необходимо использовать дозы активного ингредиента, выходящие за пределы, раскрытые в данном документе. Кроме того, следует отметить, что врач или лечащий врач должен знать, как и когда прервать, скорректировать или прекратить терапию в связи с ответом субъекта.
Для системного введения терапевтически эффективную дозу первоначально можно оценить из анализов in vitro. Например, дозу можно сформулировать в животных моделях для достижения диапазона концентраций в кровотоке, который включает IC50, как определено в клеточной культуре (то есть концентрацию тестируемого соединения, которая летальна для 50% культуры клеток), MIC, как определено в клеточной культуре (то есть минимальную ингибирующую концентрацию для роста) или IC100, как определено в клеточной культуре (т.е. концентрацию производного антимикробного сульфонамида, которая летальна для 100% клеточной культуры). Такая информация может быть использована для более точного определения подходящих доз у людей.
Начальные дозы также можно оценить по данным in vivo (например, на животных моделях) с использованием методик, которые хорошо известны в данной области. Специалист в данной области может легко оптимизировать введение людям на основании данных животных.
Альтернативно, начальные дозы можно определить из вводимых дозировок известных антимикробных средств путем сравнения IC50, MIC и/или I100 конкретного соединения, раскрытого в настоящем документе, с дозировкой известного антимикробного средства, и соответствующей корректировки начальных доз. Оптимальная дозировка может быть получена из этих начальных значений путем обычной оптимизации.
В случаях местного введения или селективного поглощения используемое соединение с эффективной локальной концентрацией может не быть связано с концентрацией в плазме. Специалист в данной области сможет оптимизировать терапевтически эффективные местные дозы без чрезмерных экспериментов.
В идеале терапевтически эффективная доза описанных в настоящем документе соединений будет обеспечивать терапевтическую пользу, не вызывая существенной токсичности. Токсичность соединений можно определять с использованием стандартных фармацевтических процедур для клеточных культур или экспериментальных животных, например, путем определения LD50 (доза, летальная для 50% популяции) или LD100 (доза, летальная для 100% популяции). Соотношение доз между токсическим и терапевтическим эффектом является терапевтическим показателем. Соединения, которые проявляют высокие терапевтические показатели, являются предпочтительными. Данные, полученные из этих анализов клеточных культур и исследований на животных, можно использовать при составлении диапазона доз, который не токсичен для применения у субъектов. Дозировка описанных в настоящем документе соединений предпочтительно находится в диапазоне концентраций в кровотоке, которые включают эффективную дозу с небольшой токсичностью или без нее. Дозировка может варьировать в этом диапазоне в зависимости от используемой лекарственной формы и используемого пути введения. Точную готовую форму, способ введения и дозировку может выбирать отдельный врач с учетом состояния пациента (см., например, Fingl et al., 1975, In: The Pharmacological Basis of Therapeutics, Ch.1, p.1).
Лечение можно периодически повторять, пока обнаруживаются инфекции, или даже когда они не обнаруживаются. Введение той же самой представленной в настоящем документе композиции можно повторять, и введения можно разделить по меньшей мере на 1 день, 2 дня, 3 дня, 5 дней, 10 дней, 15 дней, 30 дней, 45 дней, 2 месяца, 75 дней, 3 месяцев или 6 месяцев.
Соединение формулы (1) и/или его фармацевтическую композицию обычно можно использовать в количестве, эффективном для достижения намеченной цели. Для применения для лечения заболевания, такого как бактериальная инфекция, соединение формулы (1) и/или его фармацевтические композиции можно вводить или применять в терапевтически эффективном количестве.
Количество соединения формулы (1) и/или его фармацевтической композиции, которое будет эффективным при лечении конкретного раскрытого в данном описании расстройства или состояния, будет частично зависеть от природы расстройства или состояния и может быть определено с помощью стандартных клинических методик, известных в данной области. Кроме того, для помощи в определении оптимальных диапазонов доз можно использовать анализы in vitro или in vivo. Количество вводимого соединения формулы (1) и/или его фармацевтической композиции будет зависеть, среди прочих факторов, от субъекта, которого лечат, от веса пациента, тяжести заболевания, способа введения и оценки назначающего врача.
Соединение формулы (1) можно анализировать in vitro и in vivo на предмет желаемой терапевтической активности перед применением у людей. Например, анализы in vitro можно использовать для определения того, является ли предпочтительным введение конкретного соединения или комбинации соединений. Можно также продемонстрировать, что соединения эффективны и безопасны с использованием систем животных моделей.
Терапевтически эффективная доза соединения формулы (1) и/или его фармацевтической композиции будет обеспечивать терапевтическую пользу, не вызывая существенной токсичности. Токсичность соединений формулы (1) и/или их фармацевтических композиций можно определять с использованием стандартных фармацевтических методик, и она может быть легко установлена специалистом в данной области. Соотношение доз между токсическим и терапевтическим эффектом является терапевтическим показателем. Соединение формулы (1) и/или его фармацевтическая композиция проявляют особенно высокий терапевтический индекс при лечении заболевания и расстройств. Доза соединения формулы (1) и/или его фармацевтической композиции будет находиться в диапазоне концентраций в кровотоке, которые включают эффективную дозу с минимальной токсичностью.
Соединение формулы (1), его фармацевтически приемлемая соль или фармацевтическая композиция любого из вышеперечисленного можно включить в набор, который можно использовать для введения соединения пациенту в терапевтических целях. Набор может включать фармацевтическую композицию, содержащую соединение формулы (1), подходящее для введения пациенту, и инструкции по введению фармацевтической композиции пациенту. Набор для применения при лечении бактериальной инфекции у пациента содержит соединение формулы (1) или его фармацевтически приемлемую соль, фармацевтически приемлемую основу для введения соединения и инструкции по введению соединения пациенту. Инструкции, прилагаемые к набору, можно напечатать и/или предоставлять, например, в виде электронно-читаемого носителя, видеокассеты, аудиокассеты, устройства флэш-памяти или можно опубликовать на веб-сайте в Интернете или распространять среди пациентов и/или поставщиков медицинских услуг в виде электронного сообщения.
Количество соединения формулы (1), которое будет эффективным при лечении бактериальной инфекции, будет зависеть, по меньшей мере частично, от природы заболевания и может быть определено стандартными клиническими методами, известными в данной области. Кроме того, для помощи в определении оптимальных диапазонов дозировок можно использовать анализы in vitro или in vivo. Схемы лечения и интервалы дозировок также можно определять с помощью способов, известных специалистам в данной области. Количество вводимого соединения формулы (1) может зависеть, помимо других факторов, от субъекта, которого лечат, веса субъекта, тяжести заболевания, пути введения и решения назначающего врача.
Для системного введения терапевтически эффективную дозу можно первоначально оценить из анализов in vitro. Начальные дозы также можно оценить по данным in vivo, например, на животных моделях с использованием методик, известных в данной области. Такую информацию можно использовать для более точного определения применяемых доз для человека. Специалист в данной области может оптимизировать введение людям на основе данных о животных.
для поддержания устойчивой терапевтически эффективной концентрации соединения формулы (1) в крови пациента и в некоторых вариантах осуществления без превышения минимальной неблагоприятной концентрации можно выбирать дозу соединения формулы (1) и соответствующие интервалы получения лекарства.
Фармацевтические композиции, содержащие соединение формулы (1), можно вводить один раз в день, два раза в день или с интервалами более одного раза в день. Прием лекарства можно обеспечивать отдельно или в комбинации с другими лекарственными средствами, и он может продолжаться столько времени, сколько требуется для эффективного лечения заболевания. Прием лекарства также можно осуществлять с использованием непрерывного или полунепрерывного введения в течение определенного периода времени. Прием лекарства включает введение фармацевтической композиции млекопитающему, такому как человек, в сытом состоянии или натощак.
Фармацевтическую композицию можно вводить в форме разовой дозы или в многодозовой форме или в виде непрерывной или накапливаемой дозы в течение некоторого периода времени. При использовании многодозовой формы количество соединения формулы (1), содержащееся в каждой из многодозовых форм, может быть одинаковым или различным.
Подходящие диапазоны суточной дозировки для введения могут составлять от 2 мкг до 20 мг соединения формулы (1) на килограмм массы тела.
Подходящие диапазоны суточной дозировки для введения могут составлять от 1 мкг до 50 мг соединения формулы (1) на квадратный метр (м2) поверхности тела.
Соединение формулы (1) можно вводить для лечения бактериальной инфекции у пациента в количестве от 1 мг до 2000 мг в день, от 100 мкг до 1500 мг в день, от 20 мкг до 1000 мг в день или с любой другой подходящей суточной дозой.
Фармацевтическую композицию, содержащую соединение формулы (1), можно вводить для лечения бактериальной инфекции у субъекта, чтобы обеспечить терапевтически эффективную концентрацию соединения формулы (1) в крови или плазме субъекта. Терапевтически эффективная концентрация соединения формулы (1) в крови или плазме субъекта составляет от 1 мкг/мл до 60 мкг/мл, от 2 мкг/мл до 50 мкг/мл, от 5 мкг/мл до 40 мкг/мл, от 5 мкг/мл до 20 мкг/мл или от 5 мкг/мл до 10 мкг/мл. Терапевтически эффективная концентрация соединения формулы (1) в крови или плазме субъекта составляет по меньшей мере 2 мкг/мл, по меньшей мере 5 мкг/мл, по меньшей мере 10 мкг/мл, по меньшей мере 15 мкг/мл, по меньшей мере 25 мкг/мл или по меньшей мере 30 мкг/мл. Терапевтически эффективная концентрация соединения формулы (1) в крови или плазме субъекта меньше, чем количество, которое вызывает неприемлемые неблагоприятные эффекты, включая неблагоприятные эффекты для гомеостаза. Терапевтически эффективная концентрация соединения формулы (1) в крови или плазме субъекта представляет собой количество, достаточное для восстановления и/или поддержания гомеостаза у субъекта.
Фармацевтическую композицию, содержащую соединение формулы (1), можно вводить для лечения бактериальной инфекции у пациента, чтобы обеспечить терапевтически эффективную концентрацию соединения формулы (1) в крови или плазме субъекта в течение длительного периода времени, например, в течение по меньшей мере 4 часов, в течение по меньшей мере 6 часов, в течение по меньшей мере 8 часов, в течение по меньшей мере 10 часов или в течение по меньшей мере 12 часов.
Количество вводимого соединения формулы (1) может варьировать в течение схемы лечения.
В дополнение к соединению формулы (1) фармацевтические композиции, представленные в настоящем описании, могут дополнительно содержать одно или несколько фармацевтически активных соединений. Такие соединения можно обеспечить для лечения бактериальной инфекции, которую лечат соединением формулы (1), или для лечения заболевания, расстройства или состояния, отличного от бактериальной инфекции, которую лечат соединением формулы (1).
Соединения и композиции, описанные в настоящем документе, можно использовать для широкого спектра вариантов применения для лечения инфекционных заболеваний у субъекта. Способы обычно включают введение субъекту терапевтически эффективного количества соединения формулы (1) или его фармацевтической композиции или введение субъекту терапевтически эффективного количества соединения формулы (1) и антибиотика или его фармацевтической композиции.
Соединения, представленные в настоящем описании, являются пролекарствами ингибиторов β-лактамазы. Соединения и композиции, представленные в настоящем раскрытии, можно использовать для лечения заболевания, при котором этиология заболевания связана с экспрессией β-лактамаз. Например, некоторые бактериальные инфекции устойчивы к β-лактамазным антибиотикам, потому что β-лактамазы, продуцируемые бактериями, гидролизуют β-лактамное кольцо β-лактамного антибиотика.
Соединения и композиции, предусмотренные настоящим раскрытием, можно использовать для лечения бактериального заболевания у пациента.
Соединения и композиции, представленные в настоящем раскрытии, можно использовать для лечения бактериальной инфекции. Например, соединения и композиция, предусмотренные настоящим раскрытием, можно использовать для лечения бактериальной инфекции, связанной с такими бактериями, как облигатные аэробные бактерии, облигатные анаэробные бактерии, факультативные анаэробные бактерии и микроаэрофильные бактерии.
Примеры облигатных аэробных бактерий включают грамотрицательные кокки, такие как Moraxella catarrhalis, Neisseria gonorrhoeae и N. meningitidi; грамположительные бациллы, такие как Corynebacterium jeikeium; кислотоустойчивые бациллы, такие как Mycobacterium avium complex, M. kansasii, M. leprae, M. tuberculosis и вид Nocardia; неферментативные, не энтеробактериальные, такие как Acinetobacter calcoaceticus, Elizabethkingia meningoseptica (ранее Flavobacterium meningosepticum), Pseudomonas aeruginosa, P. alcaligenes, другие виды Pseudomonas и Stenotrophomonas maltophilia; грамотрицательные коккобациллы и бациллы со сложными питательными потребностями, такие как виды Brucella, Bordetella, Francisella и Legionella; и treponemataceae (спиральные бактерии), такие как вид Leptospira.
Примеры облигатных анаэробных бактерий включают грамотрицательные бактерии, такие как Bacteroides fragilis, другие виды Bacteroides, и виды Fusobacterium, виды Prevotella; грамотрицательные кокки, такие как виды Veillonella; грамположительные кокки, такие как Peptococcus niger и виды Peptostreptococcus; не образующие спор грамположительные бациллы, такие как Clostridium botulinum, C. perfringens, C. tetani, другие виды Clostridium; и эндоспорообразующие грамположительные бациллы, такие как Clostridium botulinum, C. perfringens, C. tetani и другие виды Clostridium.
Примеры факультативных анаэробных бактерий включают грамположительные кокки, каталазаположительные, такие как Staphylococcus aureus (коагулазаположительный), S. epidermidis (коагулазаотрицательный) и другие коагулазаотрицательные стафилококки; грамположительные кокки, каталазаотрицательные, такие как Enterococcus faecalis, E. faecium, Streptococcus agalactiae (стрептококки группы B), S. bovis, S. pneumoniae, S. pyogenes (стрептококки группы A), стрептококки группы viridans (S. mutans, S. mitis, S. salivarius, S. sanguis), группы S. anginosus (S. anginosus, S. milleri, S. constellatus) и Gemella morbillorum; грамположительные бациллы, такие как Bacillus anthracis, Erysipelothrix rhusiopathiae и Gardnerella vaginalis (грамвариабельные); грамотрицательные бациллы, такие как Enterobacteriaceae (вид Citrobacter, Enterobacter aerogenes, Escherichia coli, вид Klebsiella, Morganella morganii, вид Proteus, Plesiomonas shigelloides, Providencia rettgeri, Salmonella typhi, другие виды Salmonella, Serratia marcescens и вид Shigella, Yersinia enterocolitica, Y. pestis); ферментативные, не являющиеся Enterobacteriaceae, такие как Aeromonas hydrophila, Chromobacterium violaceum и Pasteurella multocida; грамотрицательные коккобациллы и бациллы со сложными питательными потребностями, такие как Actinobacillus actinomycetemcomitans, Bartonella bacilliformis, B. henselae, B. quintana, Eikenella corrodens, Haemophilus influenzae и другие виды Haemophilus; микоплазму, такую как Mycoplasma pneumoniae; и Treponemataceae (спиральные бактерии), такие как Borrelia burgdorferi и Treponema pallidum.
Примеры микроаэрофильных бактерий включают изогнутые бактерии, такие как Campylobacter jejuni, Helicobacter pylori, Vibrio cholerae и V. vulnificus; облигатные внутриклеточные паразитарные; chlamydiaceae, такие как Chlamydia trachomatis, Chlamydophila pneumoniae и C. psittaci; coxiellaceae, такие как Coxiella burnetii; и rickettsiales, такие как Rickettsia prowazekii, R. rickettsii, R. typhi, R. tsutsugamushi, Ehrlichia chaffeensis и Anaplasma phagocytophilum.
Соединения и композиции, представленное в настоящем раскрытии, можно использовать для лечения бактериального заболевания, в котором бактерии вырабатывают β-лактамазу. Примеры бактерий, которые вырабатывают β-лактамазу, включают Mycobacterium tuberculosis, метициллин-резистентный Staphylococcus aureus, Staphyloccus, Enterobacteriaceae, Pseudomonas aeruginosa, Haemophilus influenzae, Klebsiella pneumoniae, Citrobacter и Morganella.
Соединения и композиции, представленные в настоящем раскрытии, можно использовать для лечения бактериального заболевания, в котором ингибитор β-лактамазы является эффективным при лечении такого бактериального заболевания, как бактериальная инфекция.
Инфекционным заболеванием может быть бактериальная инфекция. Бактериальной инфекцией может быть инфекция грамположительных бактерий. Бактериальной инфекцией может быть инфекция грамотрицательных бактерий. Примеры грамотрицательных бактерий включают Acinetobacter, Aeromonas, Bacteroides, Burkholderia, Citrobacter, Enterobacter, Escherichia, Fusobacterium, Haemophilus, Klebsiella, Moraxella, Morganella, Mycoplasma, Neisseria, Pantoea, Pasteurella, Plesiomonas, Porphyromonas, Prevotella, Proteus, Providencia, Pseudomonas, Salmonella, Serratia, Shigella, Spirillum, Stenotrophomonas, Streptobacillus, Treponema или Yersinia. Примеры грамотрицательных бактерий включают Acinetobacter baumannii, Aeromonas hydrophila, Arizona hinshawii, Bacteroides fragilis, Branhamella catarrhalis, Burkholderia cepacia, Citrobacter diversus, Citrobacter freundii, Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Fusobacterium nucleatum, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella oxytoca, Klebsiella pneumoniae, Moraxella catarrhalis, Morganella morganii, Neisseria gonorrhoeae, Neisseria meningitidis, Pantoea agglomerans, Pasteurella multocida, Plesiomonas shigelloides, Prevotella melaninogenica, Proteus mirabilis, Proteus rettgeri, Proteus vulgaris, Pseudomonas aeruginosa, Pseudomonas diminuta, Pseudomonas fluorescens, Pseudomonas stutzeri, Salmonella enterica, Salmonella enteritidis, Salmonella typhi, Serratia marcescens, Spirillum minus, Stenotrophomonas maltophilia, Streptobacillus moniliformis, Treponema pallidum или Yersinia enterocolitica.
Развитие устойчивости к антибиотикам продолжает расти, как проблема, с которой сталкиваются пациенты и врачи. Соответственно, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США выявило следующие потенциально опасные для здоровья населения следующие патогены: виды Acinetobacter, виды Aspergillus, комплекс Burkholderia cepacia, виды Campylobacter, виды Candida, Clostridium difficile, виды Coccidioides, виды Cryptococcus, Enterobacteriaceae (например, Klebsiella pneumoniae), виды Enterococcus, Helicobacter pylori, комплекс Mycobacterium tuberculosis, Neisseria gonorrhoeae, N. meningitidis, нетуберкулезные виды микобактерий, виды Pseudomonas, Staphylococcus aureus, Streptococcus agalactiae, S. pneumoniae, S. pyogenes и Vibrio cholerae. Управление по контролю за продуктами и лекарствами определило эти организмы как «квалифицированные патогены» для целей Закона «о создании условий для инвестиции в антибиотики сейчас» (GAIN), призванного стимулировать разработку новых антибактериальных и противогрибковых препаратов для лечения серьезных или опасных для жизни инфекций. Другие типы бактерий можно добавлять или вычитать из списка «квалифицированных патогенов», а способы, предусмотренные настоящим раскрытием, охватывают любые вновь добавленные бактерии. Соединения, композиции, способы и наборы, раскрытые в данном документе, полезны для лечения заболеваний, инфекций и т.д., вызываемых также многими из этих организмов.
Соединения и композиции, описанные в настоящем документе, можно использовать для лечения или профилактики различных заболеваний, вызванных вышеуказанными бактериями. Они включают, но без ограничения, венерические заболевания, пневмонию, осложненные инфекции мочевыводящих путей, инфекции мочевыводящих путей, осложненные внутрибрюшные инфекции и внутрибрюшные инфекции.
Способы, предусмотренные настоящим раскрытием, также можно использовать для ингибирования β-лактамазы у пациента. Соединения и композиции, предлагаемые в настоящем изобретении, можно вводить пациенту для ингибирования любого подходящего типа β-лактамазы. Примеры типов β-лактамаз включают β-лактамазы расширенного спектра, такие как β-лактамазы TEM (класс A), β-лактамазы SHV (класс A), β-лактамазы CTX-M (класс A), β-лактамазы OXA (класс D) и другие бета-лактамазы расширенного спектра, такие как бета-лактамазы PER, VEB, GES и IBC; устойчивые к ингибитору β-лактамазы; β-лактамазы AmpC-типа (класс С); карбапенемазы, такие как карбапенемазы IMP-типа (металло-β-лактамазы) (класс B), VIM (интегронокодированную металло-β-лактамазу из Вероны (класс B), OXA (оксциллиназу) группу β-лактамаз (класс D), KPC (K. pneumoniae карбапенемазу) (класс A), CMY (класс C), SME, IMI, NMC и CcrA и NDM-1 (металло-β-лактамазу из нью-дели) (класс B).
Примеры типов β-лактамаз включают цефалоспориназы, пенициллиназы, цефалоспориназы, β-лактамазы широкого спектра, β-лактамазы расширенного спектра, устойчивые к ингибитору β-лактамазы, карбенициллиназу, клоксициллиназы, оксациллиназы, карбапенемазы и металлоферменты.
Типы β-лактамаз включают в себя β-лактамазы класса A, класса B, класса C и класса D.
Соединения и композиции, предусмотренные настоящим раскрытием, можно вводить перорально.
Соединения, представленные в настоящем изобретении, при пероральном введении обеспечивают повышенную пероральную биодоступность ингибитора β-лактамазы по сравнению с пероральной биодоступностью исходного ингибитора β-лактамазы. Например, соединения формулы (1) могут проявлять пероральную биодоступность (% F) по меньшей мере 10%, по меньшей мере 20%, по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50% или по меньшей мере 60%.
Фармацевтические композиции, представленные в настоящем описании, в дополнение к соединению формулы (1) могут дополнительно включать одно или несколько фармацевтически активных соединений. Такие соединения можно обеспечивать для лечения бактериальной инфекции, которую лечат соединением формулы (1), или для лечения заболевания, расстройства или состояния, отличного от бактериальной инфекции, которую лечат соединением формулы (1).
Соединение формулы (1) можно использовать в комбинации по меньшей мере с одним другим терапевтическим средством. Соединение формулы (1) можно вводить пациенту вместе с другим соединением для лечения бактериальной инфекции у пациента. По меньшей мере одно другое терапевтическое средство может представлять собой другое соединение формулы (1). Соединение формулы (1) и по меньшей мере одно другое терапевтическое средство могут действовать аддитивно или синергически. По меньшей мере одно дополнительное терапевтическое средство может быть включено в ту же самую фармацевтическую композицию или основу, содержащую соединение формулы (1), или может быть в отдельной фармацевтической композиции или основе. Соответственно, способы, предоставленные в настоящем раскрытии, дополнительно включают, помимо введения соединения формулы (1), введение одного или нескольких терапевтических средств, эффективных для лечения бактериальной инфекции или другого заболевания, расстройства или состояния, чем бактериальная инфекция. Способы, предусмотренные настоящим раскрытием, включают введение соединения формулы (1) и одного или нескольких других терапевтических средств при условии, что комбинированное введение не ингибирует терапевтическую эффективность соединения формулы (1) и/или не приводит к неблагоприятной комбинации эффектов.
Фармацевтические композиции, содержащие соединение формулы (1), можно вводить одновременно с введением другого терапевтического средства, которое может являться частью той же фармацевтической композиции, что и фармацевтическая композиция, отличная от той, которая содержит соединение формулы (1). Соединение формулы (1) можно вводить до или после введения другого терапевтического средства. В некоторых вариантах осуществления комбинированной терапии комбинированная терапия может включать чередование введения соединения формулы (1) и композиции, содержащей другое терапевтическое средство, например, чтобы минимизировать неблагоприятные действия лекарственного средства, связанные с конкретным лекарственным средством. Когда соединение формулы (1) вводят одновременно с другим терапевтическим средством, которое потенциально может оказывать неблагоприятное лекарственное воздействие, включая, например, токсичность, другое терапевтическое средство можно вводить в дозе, которая падает ниже порогового значения, при котором возникает вредная реакция на лекарственное средство.
Фармацевтические композиции, содержащие соединение формулы (1), можно вводить с одним или несколькими веществами для усиления, модуляции и/или регулирования высвобождения, биодоступности, терапевтической эффективности, терапевтической эффективности, стабильности и тому подобное соединения формулы (1). Например, чтобы повысить терапевтическую эффективность соединения формулы (1), соединение формулы (1) или фармацевтическую композицию, содержащую соединение формулы (1), можно вводить совместно с одним или несколькими активными средствами для увеличения абсорбции или диффузии соединения формулы (1) из желудочно-кишечного тракта в системный кровоток или для ингибирования распада соединения формулы (1) в крови субъекта. Фармацевтическую композицию, содержащую соединение формулы (1), можно вводить совместно с активным средством, обладающим фармакологическим действием, которое повышает терапевтическую эффективность соединения формулы (1).
Соединение формулы (1) можно вводить вместе с другим терапевтическим соединением, где соединение формулы (1) усиливает эффективность другого терапевтического соединения. Например, другое терапевтическое соединение может представлять собой антибиотик, такой как β-лактамный антибиотик, и соединение формулы (1), которое обеспечивает системный ингибитор β-лактамазы, может усиливать эффективность β-лактамного антибиотика путем ингибирования гидролиза β-лактамного кольца β-лактамазами.
Соединения и композиции, представленные в настоящем изобретении, можно вводить в комбинации с антибиотиком, таким как β-лактамный антибиотик.
Антибиотики включают, например, аминогликозиды, такие как амикацин, гентамицин, неомицин, стрептомицин и тобрамицин; β-лактамы (цефалоспорины первого поколения), такие как цефадроксил, цефазолин, цефалексин; β-лактамы (цефалоспорины второго поколения), такие как цефаклор, цефотетан, цефокситин, цефпрозил и цефуроксим; β-лактамы (цефалоспорины третьего поколения), такие как цефотаксим, цефподоксим, цефтазидим, цефтибутен и цефтриаксон; β-лактамы (цефалоспорины шестого поколения), такие как цефепим; β-лактамы (цефалоспорины пятого поколения), такие как цефтаролин; β-лактамы (пенициллины), такие как амоксициллин, ампициллин, диклоксациллин, нафциллин и оксациллин, пенициллин G, пенициллин G бензатин, пенициллин G прокаин, пиперациллин и тикарциллин; β-лактамные монобактамы, такие как азтреонам; β-лактамные карбапенемы, такие как эртапенем, имипенем, меропенем и дорипенем; фторхиниолоны, такие как ципрофлоксацин, гемифлоксацин, левофлоксацин, моксифлоксацин, норфлоксацин и офлоксацин; макролиды, такие как азитромицин, кларитромицин, эритромицин, фидаксомицин, лактобионат, глюцептат и телитромицин; сульфонамиды, такие как сульфизоксазол, сульфаметизол, сульфаметоксазол и триметоприм; тетрациклины, такие как доксициклин, миноциклин, тетрациклин и тигециклин; и другие антибиотики, такие как клиндамицин, хлорамфеникол, колистин (полимиксин Е), далбаванцин, даптомицин, фосфомицин, линезолид, метронидазол, нитрофурантоин, оритаванцин, хинупристин, дальфопризин, рифампин, рифапентин, тедизолид, телаванцин и ванкомицин. Антибиотиком может быть цефтазидим.
Другие примеры антибиотиков включают пенициллины, такие как аминопенициллины, включая амоксициллин и ампициллин, антипсевдомонадные пенициллины, включая карбенициллин, пеперациллин и тикарциллин, ингибиторы β-лактамаз, включая амоксициллин, ампициллин, пиперациллин и клавуланат, природные пенициллины, включая пенициллин g бензатин, пенициллин v калия и прокаин пенициллин, и устойчивый к пенициллиназе пенициллин, включая оксациллин, диклоксациллин и нафциллин; тетрациклины; цефалоспорины, такие как авибактам, тазобактам, цефадроксил, дефазолин, цефалексин и цефазолин; хинолоны, такие как ломефлоксацин, офлоксацин, норфлоксацин, гатифлоксацин, ципрофлоксацин, моксифлоксацин, левофлоксацин, гемифлоксацин, делафоксацин, циноксацин, налидиксовая кислота, тровафлоксацин и спарфлоксацин; линкомицины, такие как линкомицин и клиндамицин; макролиды, такие как детолиды, включая телитромицин, и макролиды, такие как эритромицин, азитромицин, кларитромицин и фидаксомицин; сульфонамиды, такие как сульфаметоксазол/триметоприм, сульфизоксазол; гликопептиды; аминогликозиды, такие как паромомицин, тобрамицин, гентамицин, амикацин, канамицин и неомицин; и карбапенемы, такие как дорипенем, меропенем, эртапенем и циластатин/имипенем. Примеры подходящих β-лактамных антибиотиков включают в себя пенамы, такие как чувствительные к β-лактамазе пенамы, такие как бензатин пенициллин, бензилпенициллин, феноксиметил пенциллин и прокаинпенициллин; устойчивые к β-лактамазе пенамы, такие как клоксациллин, диклоксациллин, флуклоксациллин, метициллин, нафциллин, оксациллин и темоциллин; пенамы широкого спектра, такие как амоксициллин и ампициллин; пенамы с расширенным спектром, такие как мецилланам; карбоксипенициллины, такие как карбенициллин и тикарциллин, и уреидопенициллины, такие как азлоциллин, мезлоциллин и пеперациллин.
Примеры подходящих β-лактамных антибиотиков включают цефамы, такие как цефамы первого поколения, включая цефазолин, цефалексин, цефалоспорин С, цефалотин; цефамы второго поколения, такие как цефаклор, цефамоандол, цефуроксим, цефотетан и цефокситин; цефамы третьего поколения, такие как цефиксим, цефотаксим, цефподоксим, цефлазидим и цефтриаксон; цефамы четвертого поколения, такие как цефипим и цефпиром; и цефамы пятого поколения, такие как цефтаролин.
Примеры подходящих β-лактамных антибиотиков включают карбапенемы и пенемы, такие как биапенем, дорипенем, эртапенем, фаропенем, имипенем, меропенем, панипернем, разупенем, тебипенем и тиенамицин.
Примеры подходящих β-лактамных антибиотиков включают монобактамы, такие как азтреонам, тигемонам, нокардицин А и β-лактам табтоксинина.
Соединения и фармацевтические композиции, представленные в настоящем изобретении, можно вводить с ингибиторами β-лактамазы и/или ингибиторами карбапенемазы или их фармацевтическими композициями. Примеры подходящих ингибиторов β-лактамазы и/или ингибиторов карбапенемазы включают клавулановую кислоту, сульбактам, авибактам, тазобактам, релебактам, ваборбактам, ETX 2514, RG6068 (т.е. OP0565) (Livermore et al., J AntiMicrob Chemother 2015, 70: 3032) и RPX7009 (Hecker et al., J Med Chem 2015 58: 3682 3692).
Соединения и композиции, предусмотренные настоящим раскрытием, следует использовать в сочетании с одним или несколькими другими активными ингредиентами. Соединение можно вводить в комбинации или последовательно с другим терапевтическим средством. Такие другие терапевтические средства включают средства, известные для лечения, профилактики или ослабления инфекционного заболевания.
Следует понимать, что любая подходящая комбинация соединений и фармацевтических композиций, представленных в настоящем документе, с одним или несколькими из указанных выше терапевтических средств и, необязательно, одним или несколькими другими фармакологически активными веществами, считается включенной в объем настоящего изобретения. В некоторых вариантах осуществления соединения и фармацевтические композиции, представленные в настоящем раскрытии, вводят до или после введения одного или нескольких дополнительных активных ингредиентов.
АСПЕКТЫ ИЗОБРЕТЕНИЯ
Аспект 1. Соединение формулы (1):
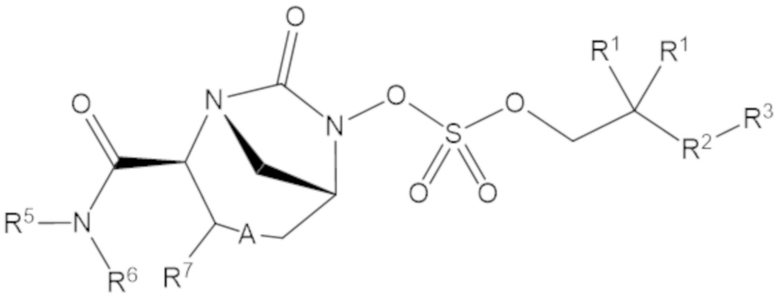 (1)
(1)
в котором
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила;
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2, в котором
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
Аспект 2. Соединение согласно аспекту 1, где каждый заместитель независимо выбирают из -OH, -CN, -CF3, -OCF3, =O, -NO2, C1-6 алкокси, C1-6 алкила, -COOR, -NR2 и -CONR2; причем каждый R независимо выбирают из водорода и C1-6 алкила.
Аспект 3. Соединение согласно любому из аспектов 1-2, где каждый заместитель независимо выбирают из -OH, -CF3, -O-CF3, -NO2,-O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), где каждый R4 выбирают из водорода, C1-8 алкила и C1-8 гетероалкила.
Аспект 4. Соединение согласно любому из аспектов 1-3, в котором А представляет собой одинарную связь (-), а R7 представляет собой водород.
Аспект 5. Соединение согласно любому из аспектов 1-4, в котором А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
Аспект 6. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (2):
(2)
Аспект 7. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (2a):
(2a)
Аспект 8. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (3):
(3)
Аспект 9. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (3a):
(3a)
Аспект 10. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (4):
(4)
Аспект 11. Соединение согласно любому из аспектов 1-5, причем соединение имеет структуру формулы (5):
(5)
Аспект 12. Соединение согласно любому из аспектов 1-11, в котором, R5 представляет собой водород; и R6 представляет собой водород.
Аспект 13. Соединение согласно любому из аспектов 1-12, в котором, R5 представляет собой C2-6 гетероалкил, содержащий концевую аминогруппу; и R6 представляет собой водород.
Аспект 14. Соединение согласно любому из аспектов 1-13, в котором, R5 представляет собой -O-(CH2)2-NH2; и R6 представляет собой водород.
Аспект 15. Соединение согласно любому из аспектов 1-14, в котором, R5 представляет собой C4-6 гетероциклоалкил, содержащий по меньшей мере один -NH- фрагмент; и R6 представляет собой водород.
Аспект 16. Соединение согласно любому из аспектов 1-15, в котором, R5 представляет собой 4-ил-пиперидин; и R6 представляет собой водород.
Аспект 17. Соединение согласно любому из аспектов 1-16, в котором А представляет собой одинарную связь; R5 выбирают из водорода, -O-(CH2)2-NH2 и 4-ил-пиперидина; R6 представляет собой водород; и R7 представляет собой водород.
Аспект 18. Соединение согласно любому из аспектов 1-17, где каждый R1 независимо представляет собой C1-6 алкил.
Аспект 19. Соединение согласно любому из аспектов 1-18, где каждый R1 представляет собой метил.
Аспект 20. Соединение согласно любому из аспектов 1-19, где каждый R1 вместе с геминальным атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо или замещенное C3-6 циклоалкильное кольцо.
Аспект 21. Соединение согласно любому из аспектов 1-20, где каждый R1 вместе с геминальным атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 22. Соединение согласно любому из аспектов 1-21, где каждый R1 вместе с геминальным атомом углерода, с которым они связаны, образуют циклопропильное кольцо, циклобутильное кольцо, циклопентильное кольцо или циклогексильное кольцо.
Аспект 23. Соединение согласно любому из аспектов 1-22, в котором каждый R1 вместе с геминальным атомом углерода, с которым они связаны, образуют C3-6 гетероциклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо.
Аспект 24. Соединение согласно любому из аспектов 1-23, в котором R2 представляет собой одинарную связь.
Аспект 25. Соединение согласно любому из аспектов 1-24, в котором R2 представляет собой одинарную связь; и R3 представляет собой C1-6 алкил.
Аспект 26. Соединение согласно любому из аспектов 1-25, при этом R2 выбирают из C1-2 алкандиила и замещенного C1-2 алкандиила.
Аспект 27. Соединение согласно аспекту 26, в котором замещающую группу выбирают из -OH, -CN, -CF3, -OCF3, =O, -NO2, C1-6 алкокси, C1-6 алкила, -COOR, -NR2 и -CONR2; причем каждый R независимо выбирают из водорода и C1-6 алкила.
Аспект 28. Соединение согласно аспекту 26, в котором замещающую группу выбирают из -OH, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4) и -CH(-NH2)(-R4); и R4 выбирают из водорода и C1-6 алкила.
Аспект 29. Соединение согласно любому из аспектов 26-28, в котором, R2 представляет собой замещенный C1-2 алкандиил; и стереохимия атома углерода, с которым связана замещающая группа, представляет собой (S) конфигурацию.
Аспект 30. Соединение согласно любому из аспектов 26-28, в котором, R2 представляет собой замещенный C1-2 алкандиил; и стереохимия атома углерода, с которым связана замещающая группа, представляет собой (R) конфигурацию.
Аспект 31. Соединение согласно любому из аспектов 1-30, в котором R2 выбирают из C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила и C5-6 гетероциклоалкандиила.
Аспект 32. Соединение согласно любому из аспектов 1-31, при этом R3 выбирают из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NH-R4 и -CH(-NH2)(-R4).
Аспект 33. Соединение согласно любому из аспектов 1-32, при этом R3 представляет собой -C(O)-O-R4.
Аспект 34. Соединение согласно любому из аспектов 1-33, при этом R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C5-7 циклоалкила, C5-7 гетероциклоалкила, C6 арила, C7-9 арилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C6 арила и C7-9 арилалкила.
Аспект 35. Соединение согласно любому из аспектов 1-34, при этом R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила, C5-7 гетероциклоалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C7-9 арилалкила и замещенного C5-7 гетероциклоалкила.
Аспект 36. Соединение согласно любому из аспектов 1-35, при этом R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
Аспект 37. Соединение согласно любому из аспектов 1-36, при этом R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
Аспект 38. Соединение согласно любому из аспектов 1-37, в котором, R3 представляет собой -C(O)-O-R4; и R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C5-7 циклоалкила, C5-7 гетероциклоалкила, C6 арила, C7-9 арилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C6 арила и C7-9 арилалкила.
Аспект 39. Соединение согласно любому из аспектов 1-38, в котором, R3 представляет собой -C(O)-O-R4; и R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила, C5-7 гетероциклоалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C7-9 арилалкила и замещенного C5-7 гетероциклоалкила.
Аспект 40. Соединение согласно любому из аспектов 1-39, в котором, R3 представляет собой -C(O)-O-R4; и R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
Аспект 41. Соединение согласно любому из аспектов 1-40, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C4-6 гетероциклоалкильное кольцо, содержащее два соседних S атома, или замещенное C4-6 гетероциклоалкильное кольцо, содержащее по меньшей мере один гетероатом, выбранный из O и S, и =O замещающую группу, связанную с атомом углерода, примыкающим по меньшей мере к одному гетероатому.
Аспект 42. Соединение согласно любому из аспектов 1-41, в котором, R2 представляет собой одинарную связь; R3 представляет собой C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, образуют C4-6 гетероциклоалкильное кольцо или замещенное C4-6 гетероциклоалкильное кольцо.
Аспект 43. Соединение согласно любому из аспектов 1-42, в котором, R2 представляет собой одинарную связь; R3 представляет собой C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, образуют C4-6 гетероциклоалкильное кольцо, содержащее два соседних S атома, или замещенное C4-6 гетероциклоалкильное кольцо, содержащее по меньшей мере один гетероатом, выбранный из O и S, и =O замещающую группу, связанную с атомом углерода, примыкающим к гетероатому.
Аспект 44. Соединение согласно любому из аспектов 1-43, в котором, R2 представляет собой одинарную связь; R3 представляет собой C1-3 алкил; и каждый R1 вместе с атомом углерода, с которым они связаны, образуют 1,2-дитиолан, 1,2-дитановое кольцо, тиэтан-2-оновое кольцо, дигидротиофен-2(3H)-оновое кольцо, тетрагидро-2H-типиран-2-оновое кольцо, оксетан-2-оновое кольцо дигидрофуран-2(3H)-оновое кольцо или тетрагидро-2H-пиран-2-оновое кольцо.
Аспект 45. Соединение согласно любому из аспектов 1-44, в котором, каждый R1 представляет собой метил; R2 выбирают из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и R3 выбирают из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4); причем R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
Аспект 46. Соединение согласно любому из аспектов 1-45, в котором, каждый R1 представляет собой метил; R2 выбирают из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и R3 выбирают из -C(O)-O-R4; причем R4 выбирают из C1-8 алкила, C1-8 гетероалкила, C7-9 арилалкила и C5-7 гетероциклоалкила.
Аспект 47. Соединение согласно любому из аспектов 1-46, в котором, каждый R1 представляет собой метил; R2 выбирают из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и R3 выбирают из -O-C(O)-R4, -C(O)-O-R4, -S-C(O)-R4, -C(O)-S-R4, -S-S-R4, -NHR4 и -CH(-NH2)(-R4); причем R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
Аспект 48. Соединение согласно любому из аспектов 1-47, в котором, каждый R1 представляет собой метил; R2 выбирают из одинарной связи, метандиила, этандиила, -CH(-OH)-, -CH(-O-C(O)-CH2CH3)- и 1,2-бензол-диила; и R3 выбирают из -C(O)-O-R4; причем R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, втор-бутил изобутила, трет-бутила, 2-метоксиэтила, метилбензола, оксетан-3-окси-ила, циклопентила, циклогексила и 2-пирролидинила.
Аспект 49. Соединение согласно любому из аспектов 1-48, в котором А представляет собой одинарную связь, и каждый из R5, R6 и R7 представляет собой водород.
Аспект 50. Соединение согласно любому из аспектов 1-49, в котором А представляет собой одинарную связь; каждый R1 независимо представляет собой C1-3 алкил; каждый R2 представляет собой одинарную связь; и каждый из R5, R6 и R7 представляет собой водород.
Аспект 51. Соединение согласно любому из аспектов 1-50, в котором, каждый R1 представляет собой метил; R2 представляет собой одинарную связь; и R3 может быть -C(O)-O-R4; причем R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C7-10 алкиларена и C5-10 гетероалкилциклоалкила.
Аспект 52. Соединение согласно любому из аспектов 1-51, в котором, каждый R1 представляет собой метил; R2 представляет собой одинарную связь; R3 может быть -C(O)-O-R4; причем R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C7-10 алкиларена и C5-10 гетероалкилциклоалкила; каждый из R5, R6 и R7 представляет собой водород; и представляет собой одинарную связь.
Аспект 53. Соединение согласно аспекту 1, причем соединение выбирают из:
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил бензоата (2);
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3);
бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил бензоата (6);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил пропионата (7);
бензил (4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) адипата (8);
6-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутокси)-6-оксогексановой кислоты (9);
метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10);
изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11);
гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12);
гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13);
трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14);
2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15);
оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил 1H-имидазол-1-сульфоната (34);
этил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (35);
гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36);
гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37);
2-метоксиэтил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (38);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил пивалата (43);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоата (44);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил бензоата (46);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил пропионата (47);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил) сульфата (48);
2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилацетата (49);
2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилпивалата (50);
S-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) этантиоата (51);
S-(5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил) этантиоата (52);
S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54);
3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил адамантан-1-карбоксилата (55);
диэтил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилмалоната (56);
пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57);
бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутилпивалата (60);
этил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-этилбутаноата (61);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил бензоата (65);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2-метилбензоата (68);
4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоата (69);
2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоата (70);
2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил диацетата (71);
5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72);
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилбутаноата (73);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил) сульфата (74); фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Аспект 54. Фармацевтическая композиция, содержащая соединение согласно любому из аспектов 1-53 и фармацевтически приемлемую основу.
Аспект 55. Фармацевтическая композиция согласно аспекту 54, дополнительно содержащая антибиотик.
Аспект 56. Фармацевтическая композиция согласно аспекту 55, в которой антибиотик представляет собой β-лактамный антибиотик.
Аспект 57. Фармацевтическая композиция согласно любому из аспектов 54-56, в котором фармацевтическая композиция имеет готовую форму с дозировкой для перорального применения.
Аспект 58. Фармацевтическая композиция согласно любому из аспектов 54-57, в котором фармацевтическая композиция имеет пероральную лекарственную форму.
Аспект 59. Фармацевтическая композиция согласно любому из аспектов 54-58, содержащая количество соединения по п. 1, эффективное для лечения бактериальной инфекции у пациента.
Аспект 60. Способ лечения бактериальной инфекции у пациента, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения согласно любому из аспектов 1-53.
Аспект 61. Способ согласно аспекту 60, в котором введение включает в себя пероральное введение.
Аспект 62. Способ согласно любому из аспектов 60-61, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 63. Способ согласно любому из аспектов 60-62, дополнительно включающий введение антибиотика пациенту.
Аспект 64. Способ согласно любому из аспектов 60-63, в которой антибиотик представляет собой β-лактамный антибиотик.
Аспект 65. Способ лечения бактериальной инфекции у пациента, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества фармацевтической композиции согласно любому из аспектов 54-59.
Аспект 66. Способ согласно аспекту 65, в котором введение включает в себя пероральное введение.
Аспект 67. Способ согласно любому из аспектов 65-66, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 68. Способ согласно любому из аспектов 65-67, дополнительно включающий введение антибиотика пациенту.
Аспект 69. Способ согласно аспекту 66, в котором антибиотик представляет собой β-лактамный антибиотик.
Аспект 70. Способ ингибирования β-лактамазы у пациента включающий введение пациенту эффективного количества соединения согласно любому из аспектов 1-53.
Аспект 71. Способ согласно аспекту 70, в котором введение включает в себя пероральное введение.
Аспект 72. Согласно любому из аспектов 70 to 71, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 73. Способ ингибирования β-лактамазы у пациента, включающий введение пациенту эффективного количества фармацевтической композиции согласно любому из аспектов 54-59.
Аспект 74. Способ согласно аспекту 73, в котором введение включает в себя пероральное введение.
Аспект 75. Способ согласно любому из аспектов 73 to 74, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 76. Соединение формулы (1):
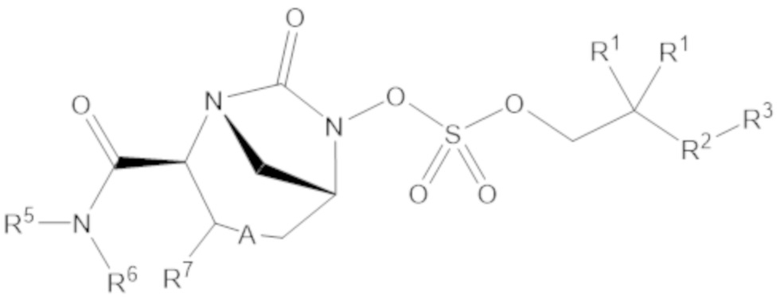 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 выбирают из одинарной связи, метан-диила и этан-диила; и
R3 выбирают из -C(O)-O-R4 и -S-C(O)-R4, в котором R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
Аспект 77. Соединение согласно аспекту 76, где каждый R1 независимо выбирают из C1-3 алкила.
Аспект 78. Соединение согласно аспекту 76, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 79. Соединение согласно аспекту 76, при этом R2 является одинарная связь.
Аспект 80. Соединение согласно любому из аспектов 76-79, при этом R2 представляет собой метан-диил.
Аспект 81. Соединение согласно любому из аспектов 76-79, при этом R2 представляет собой этан-диил.
Аспект 82. Соединение согласно любому из аспектов 76-81, при этом R3 представляет собой -C(O)-O-R4.
Аспект 83. Соединение согласно любому из аспектов 76-81, при этом R3 представляет собой -S-C(O)-R4.
Аспект 84. Соединение согласно любому из аспектов 76-83, при этом R4 представляет собой C1-10 алкил.
Аспект 85. Соединение согласно любому из аспектов 76-83, при этом R4 представляет собой C1-10 гетероалкил.
Аспект 86. Соединение согласно любому из аспектов 76-83, при этом R4 представляет собой C5-10 арилалкил.
Аспект 87. Соединение согласно любому из аспектов 76-83, при этом R4 представляет собой C3-6 гетероциклоалкил.
Аспект 88. Соединение согласно любому из аспектов 76-83, при этом R4 представляет собой замещенный C4-10 гетероциклоалкилалкил.
Аспект 89. Соединение формулы (1):
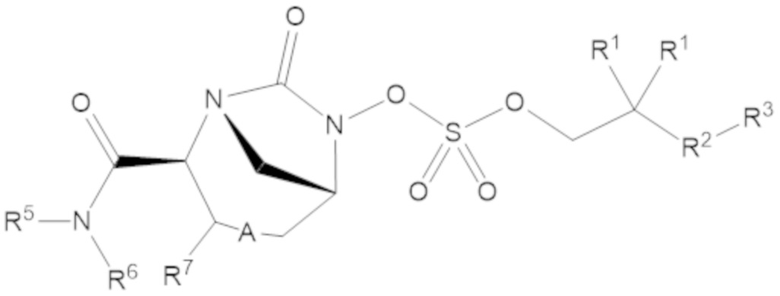 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой -C(O)-O-R4, где R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
Аспект 90. Соединение согласно аспекту 89, где каждый R1 независимо выбирают из C1-3 алкила.
Аспект 91. Соединение согласно аспекту 89, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 92. Соединение согласно любому из аспектов 89-91, при этом R4 выбирают из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами является кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами является кислород, и -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
Аспект 93. Соединение согласно аспекту 92, в котором в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами является кислород, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 94. Соединение согласно любому из аспектов 89-94, где каждый R1 представляет собой метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо.
Аспект 95. Соединение согласно любому из аспектов 89-95, при этом R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, бензила, 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
Аспект 96. Соединение согласно аспекту 89, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 представляет собой метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой -C(O)-O-R4, в котором R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, -CH2-фенил (бензила), 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
Аспект 97. Соединение формулы (1):
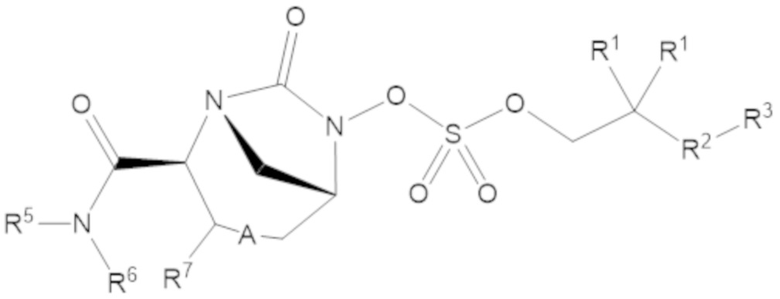 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 представляет собой -(CH2)2-; и
R3 представляет собой -C(O)-O-R4, в котором R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
Аспект 98. Соединение согласно аспекту 97, где каждый R1 независимо выбирают из C1-3 алкила.
Аспект 99. Соединение согласно аспекту 97, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 100. Соединение согласно любому из аспектов 97-99, при этом R4 выбирают из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами является кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами является кислород, -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
Аспект 101. Соединение согласно аспекту 100, в котором в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами является кислород, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 102. Соединение согласно любому из аспектов 97-99, при этом R4 представляет собой C1-10 алкил.
Аспект 103. Соединение согласно аспекту 97, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 представляет собой метил;
R2 представляет собой -(CH2)2-; и
R3 представляет собой -C(O)-O-R4, где R4 выбирают из n-гексила и n-гептила.
Аспект 104. Соединение формулы (1):
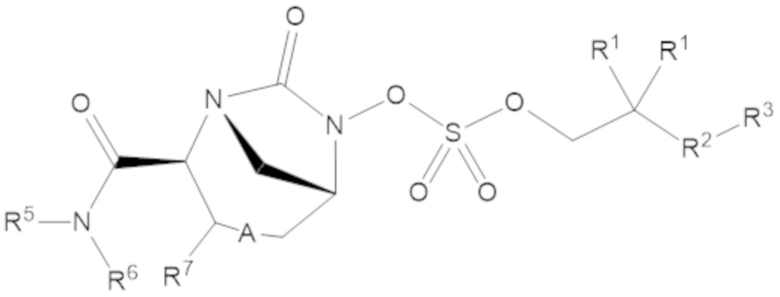 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 выбирают из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 представляет собой -CH2-; и
R3 представляет собой -S-C(O)-R4, в котором R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила, замещенного C4-10 гетероциклоалкилалкила.
Аспект 105. Соединение согласно аспекту 104, где каждый R1 независимо выбирают из C1-3 алкила.
Аспект 106. Соединение согласно аспекту 104, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 107. Соединение согласно любому из аспектов 104-106, при этом R4 выбирают из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами является кислород, -CH2CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами является кислород, -CH2-C3-6 замещенного гетероциклоалкила, -(CH2)2-C3-6 замещенного гетероциклоалкила.
Аспект 108. Соединение согласно аспекту 107, в котором, в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами является кислород, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 109. Соединение согласно любому из аспектов 104-106, при этом R4 представляет собой C1-10 алкил.
Аспект 110. Соединение согласно аспекту 104, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 представляет собой метил;
R2 представляет собой -CH2CH2-; и
R3 представляет собой -S-C(O)-R4, в котором R4 представляет собой метил
Аспект 111. Соединение формулы (1):
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, C3-6 циклоалкильное кольцо или C3-6 гетероциклоалкильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой C1-3 алкил.
Аспект 112. Соединение согласно аспекту 111, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 гетероциклоалкильное кольцо или C3-6 гетероциклоалкильное кольцо;
Аспект 113. Соединение согласно аспекту 112, в котором одним или более гетероатомами является кислород, а одним или более заместителями является =O.
Аспект 114. Соединение согласно аспекту 111, при этом
каждый R1 вместе с атомом углерода, с которым они связаны, образуют дигидрофуран-2(3H)-оновое кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой метил.
Аспект 115. Соединение формулы (1):
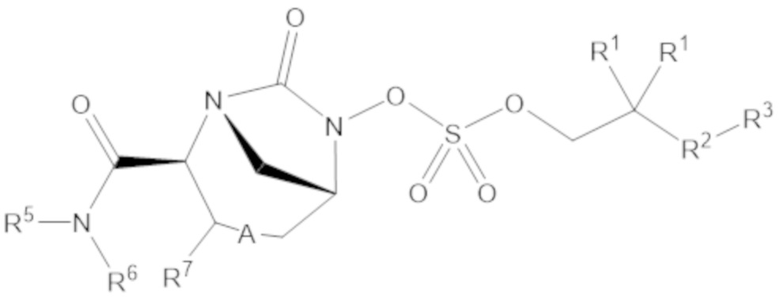 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила;
R2 выбирают из одинарной связи и метандиила; и
R3 выбирают из -O-C(O)-R4 и -C(O)-O-R4, в котором R4 выбирают из C1-10 алкила и замещенного фенила.
Аспект 116. Соединение согласно аспекту 115, при этом R2 представляет собой одинарную связь.
Аспект 117. Соединение согласно аспекту 115, при этом R2 представляет собой метандиил.
Аспект 118. Соединение согласно любому из аспектов 115-117, при этом R3 представляет собой -O-C(O)-R4.
Аспект 119. Соединение согласно любому из аспектов 115-117, при этом R2 представляет собой метандиил; и R3 представляет собой -O-C(O)-R4.
Аспект 120. Соединение согласно любому из аспектов 115-117, при этом R3 представляет собой -C(O)-O-R4.
Аспект 121. Соединение согласно любому из аспектов 115-117, при этом R2 представляет собой одинарную связь; и R3 представляет собой -C(O)-O-R4.
Аспект 122. Соединение согласно аспекту 115, при этом R2 представляет собой одинарную связь; R3 представляет собой -C(O)-O-R4; и R4 представляет собой C1-3 алкил.
Аспект 123. Соединение согласно любому из аспектов 115-122, при этом R4 представляет собой C1-10 алкил.
Аспект 124. Соединение согласно любому из аспектов 115-122, при этом R4 представляет собой C1-4 алкил.
Аспект 125. Соединение согласно любому из аспектов 115-122, при этом R4 представляет собой замещенный фенил.
Аспект 126. Соединение согласно аспекту 115, при этом R2 представляет собой метандиил; R3 представляет собой -O-C(O)-R4; и R4 представляет собой замещенный фенил.
Аспект 127. Соединение согласно аспекту 126, в котором один или более заместителей независимо выбирают из галогена, C1-3 алкила и C1-3 алкокси.
Аспект 128. Соединение согласно аспекту 126, в котором замещенный фенил представляет собой 2,6-замещенный фенил.
Аспект 129. Соединение согласно аспекту 128, где каждый из заместителей выбирают из C1-3 алкила и C1-3 алкокси.
Аспект 130. Соединение согласно аспекту 126, в котором замещенный фенил представляет собой 2,5,6-замещенный фенил.
Аспект 131. Соединение согласно аспекту 130, где каждый из заместителей в позициях 2 и 6 независимо выбирают из C1-3 алкила и C1-3 алкокси; и заместителем а 5 позиция является галоген.
Аспект 132. Соединение формулы (1):
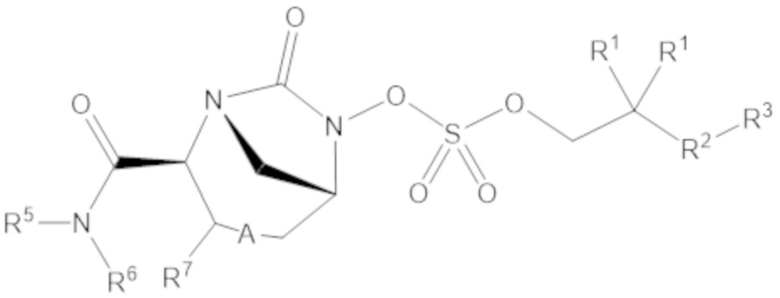 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила;
R2 представляет собой одинарную связь; и
R3 представляет собой -CH=C(R4)2, где каждый R4 представляет собой -C(O)-O-R8, или каждый R4 вместе с атомом углерода, с которым они связаны, образуют замещенное гетероциклогексильное кольцо; и
каждый R8 представляет собой C1-4 алкил.
Аспект 133. Соединение согласно аспекту 132, каждый R4 представляет собой -C(O)-O-R8.
Аспект 134. Соединение согласно аспекту 132, каждый R4 представляет собой -C(O)-O-R8, или каждый R4 вместе с атомом углерода, с которым они связаны, образуют замещенное гетероциклогексильное кольцо.
Аспект 135. Соединение согласно аспекту 133, в котором в замещенном гетероциклогексильном кольце одним или более гетероатомами является кислород.
Аспект 136. Соединение согласно любому из аспектов 134-135, в котором в замещенном гетероциклогексильном кольце один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 137. Соединение согласно аспекту 134, в котором замещенным гетероциклоалкильным кольцом является 2,2-диметил-5-ил-1,3-диоксан-4,6-дион.
Аспект 138. Соединение формулы (1):
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила;
R2 выбирают из одинарной связи и метандиила; и
R3 представляет собой замещенный фенил, в котором один или более заместителей независимо выбирают из -CH2-O-C(O)-R4 и -O-C(O)-R4, в котором R4 выбирают из C1-10 алкила и фенила.
Аспект 139. Соединение согласно аспекту 138, при этом R2 представляет собой одинарную связь.
Аспект 140. Соединение согласно аспекту 138, при этом R2 представляет собой метандиил;
Аспект 141. Соединение согласно аспекту 138, при этом R2 представляет собой 2-замещенный фенил.
Аспект 142. Соединение согласно любому из аспектов 138-141, в котором один или более заместителей представляет собой -CH2-O-C(O)-R4.
Аспект 143. Соединение согласно любому из аспектов 138-141, в котором один или более заместителей представляет собой -O-C(O)-R4.
Аспект 144. Соединение согласно любому из аспектов 138-143, при этом R4 представляет собой C1-10 алкил.
Аспект 145. Соединение согласно любому из аспектов 138-143, при этом R4 выбирают из метила, этила, изопропила, пивалолила и фенила.
Аспект 146. Соединение формулы (1):
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила;
R2 выбирают из -C(R8)2- и -CH2-C(R8)2-, где каждый R8 независимо выбирают из C1-3 алкила; и
R3 выбирают из -C(O)-O-R4 и -O-C(O)-R4, в котором R4 выбирают из C1-10 алкила, C1-10 гетероалкила, замещенного C1-10 алкила, замещенного C1-10 гетероалкила и 4(ил-метил)-5-метил-1,3-диоксол-2-она.
Аспект 147. Соединение согласно аспекту 146, где каждый R1 представляет собой метил.
Аспект 148. Соединение согласно любому из аспектов 146-147, при этом R2 представляет собой -C(R8)2-.
Аспект 149. Соединение согласно любому из аспектов 146-147, при этом R2 представляет собой -CH2-C(R8)2-.
Аспект 150. Соединение согласно любому из аспектов 146-149, где каждый R8 представляет собой метил.
Аспект 151. Соединение согласно любому из аспектов 146-149, где каждый R1 представляет собой метил; и каждый R8 представляет собой метил.
Аспект 152. Соединение согласно любому из аспектов 146-151, при этом R3 представляет собой -C(O)-O-R4.
Аспект 153. Соединение согласно любому из аспектов 146-151, при этом R3 представляет собой -O-C(O)-R4.
Аспект 154. Соединение формулы (1):
(1)
или его фармацевтически приемлемая соль, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 вместе с атомом углерода, с которым они связаны, образуют замещенное C5-6 гетероциклическое кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой C1-3 алкил.
Аспект 155. Соединение согласно аспекту 155, в котором в замещенном C5-6 гетероциклическом кольце одним или более гетероатомами является кислород;, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 156. Соединение согласно аспекту 155, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют тетрагидро-2H-пиран-2-оновое кольцо.
Аспект 157. Соединение согласно аспекту 155, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
каждый R1 независимо выбирают из C1-3 алкила;
R2 выбирают из C2-4 алкандиила; и
R3 представляет собой замещенный C5-6 гетероциклоалкил, в котором один или более гетероатомов независимо выбирают из N и O;, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 158. Соединение согласно аспекту 157, при этом R4 имеет структуру формулы (6):
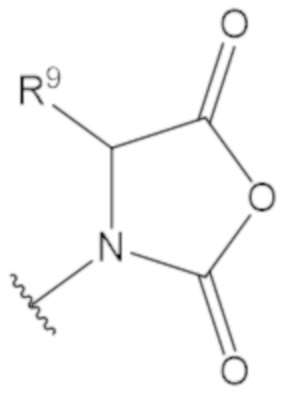 (6)
(6)
в котором R9 выбирают из водорода, C1-6 алкила, C4-6 циклоалкила, C1-6 гетероалкила, C4-6 гетероциклоалкила, замещенного C1-6 алкила, замещенного C4-6 циклоалкила, замещенного C1-6 гетероалкила и замещенного C4-6 гетероциклоалкила.
Аспект 159. Соединение согласно аспекту 158, при этом R9 выбирают из водорода и C1-6 алкила.
Аспект 160. Соединение согласно аспекту 1, причем соединение выбирают из:
этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3);
бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4);
метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10);
изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11);
гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12);
гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13);
трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14);
2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15);
оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36);
гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37);
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42);
S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53);
пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57);
бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Аспект 161. Соединение формулы (4):
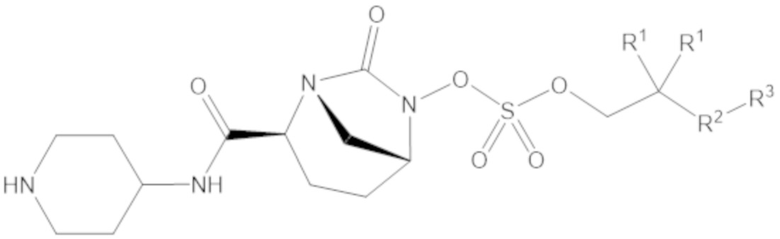 (4)
(4)
или его фармацевтически приемлемая соль, при этом
каждый R1 независимо выбирают из C1-3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой -C(O)-O-R4, в котором R4 выбирают из C1-10 алкила, C1-10 гетероалкила, C5-10 арилалкила, C3-6 гетероциклоалкила и замещенного C4-10 гетероциклоалкилалкила.
Аспект 162. Соединение согласно аспекту 161, где каждый R1 независимо выбирают из C1-3 алкила.
Аспект 163. Соединение согласно аспекту 161, где каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3-6 циклоалкильное кольцо.
Аспект 164. Соединение согласно любому из аспектов 161-163, при этом R4 выбирают из C1-7 алкила, C1-10 гетероалкила, в котором одним или более гетероатомами является кислород, -CH2-C4-6 циклоалкила, -(CH2)2-C4-6 циклоалкила, C3-6 гетероциклоалкила, в котором одним или более гетероатомами является кислород, -CH2-C3-6 замещенного гетероциклоалкила и -(CH2)2-C3-6 замещенного гетероциклоалкила.
Аспект 165. Соединение согласно аспекту 163, в котором в замещенном C3-6 гетероциклоалкиле одним или более гетероатомами является кислород, а один или более заместителей независимо выбирают из C1-3 алкила и =O.
Аспект 166. Соединение согласно аспекту 161, где каждый R1 представляет собой метил, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют циклогексильное кольцо или циклопентильное кольцо.
Аспект 167. Соединение согласно любому из аспектов 161-166, при этом R4 выбирают из метила, этила, n-пропила, изопропила, n-бутила, n-гексила, n-гептила, -CH2-CH2-O-CH3, бензила, 3-оксетанила и метил-5-метил-1,3-диоксол-2-она.
Аспект 168. Соединение согласно аспекту 161, при этом
каждый из R5, R6 и R7 представляет собой водород;
А представляет собой одинарную связь;
R2 представляет собой одинарную связь; и
R3 представляет собой -C(O)-O-R4, в котором R4 представляет собой C1-10 алкил.
Аспект 169. Соединение формулы (4) выбирают из:
этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (20);
2-метоксиэтил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (21);
4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22);
4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23);
4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (24);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (25);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Аспект 170. Соединение формулы (4) выбирают из:
этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (20);
4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22);
4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23);
(5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (25);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Аспект 171. Соединение формулы (5) выбирают из:
этил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27);
2-метоксиэтил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28);
соли TFA (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29);
соли TFA гексил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30);
соли TFA гептил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (31);
соли TFA этил-1-((((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (32);
фармацевтически приемлемой соли любого из перечисленного выше; и
комбинации любых из перечисленных выше.
Аспект 172. Фармацевтическая композиция, содержащая соединение согласно любому из аспектов 76-171 и фармацевтически приемлемая основа.
Аспект 173. Фармацевтическая композиция согласно аспекту 172, дополнительно содержащая антибиотик.
Аспект 174. Фармацевтическая композиция согласно аспекту 173, в которой антибиотик представляет собой β-лактамный антибиотик.
Аспект 175. Фармацевтическая композиция согласно любому из аспектов 172-174, в котором фармацевтическая композиция имеет готовую форму с дозировкой для перорального применения.
Аспект 176. Фармацевтическая композиция согласно любому из аспектов 172-175, в котором фармацевтическая композиция имеет пероральную лекарственную форму.
Аспект 177. Фармацевтическая композиция согласно любому из аспектов 172-176, включающий количество соединения согласно любому из аспектов 76-171, эффективное для лечения бактериальной инфекции у пациента.
Аспект 178. Способ лечения бактериальной инфекции у пациента, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения согласно любому из аспектов 76-171.
Аспект 179. Способ согласно аспекту 178, в котором введение включает в себя пероральное введение.
Аспект 180. Способ согласно любому из аспектов 178-179, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 181. Способ согласно любому из аспектов 178-180, дополнительно включающий введение антибиотика пациенту.
Аспект 182. Способ согласно любому из аспектов 178-181, в которой антибиотик представляет собой β-лактамный антибиотик.
Аспект 183. Способ лечения бактериальной инфекции у пациента, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества фармацевтической композиции согласно любому из аспектов 172-177.
Аспект 184. Способ согласно аспекту 183, в котором введение включает в себя пероральное введение.
Аспект 185. Способ согласно любому из аспектов 183-184, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 186. Способ согласно любому из аспектов 183-185, дополнительно включающий введение антибиотика пациенту.
Аспект 187. Способ согласно аспекту 184, в которой антибиотик представляет собой β-лактамный антибиотик.
Аспект 188. Способ ингибирования β-лактамазы у пациента включающий введение пациенту эффективного количества соединения согласно любому из аспектов 76-171.
Аспект 189. Способ согласно аспекту 188, в котором введение включает в себя пероральное введение.
Аспект 190. Согласно любому из аспектов 188-189, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 191 A способ ингибирования β-лактамазы у пациента включающий введение пациенту эффективного количества фармацевтической композиции согласно любому из аспектов 172-177.
Аспект 192. Способ согласно аспекту 191, в котором введение включает в себя пероральное введение.
Аспект 193. Способ согласно любому из аспектов 191-192, в котором введение включает в себя введение пероральной лекарственной формы.
Аспект 194. Способ получения аналога сложного моноэфира серной кислоты циклической гидроксамовой кислоты, включающий реакцию циклической гидроксамовой кислоты со сложным эфиром хлорсульфоновой кислоты в присутствии основания с получением аналога сложного моноэфира серной кислоты.
Аспект 195. Способ согласно аспекту 194, в котором циклическая гидроксамовая кислота имеет структуру формулы (82), а сложный эфир хлорсульфоновой кислоты имеет структуру формулы (83):
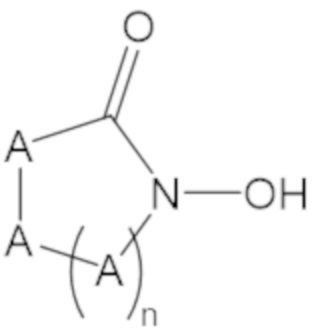
(82) (83)
где
R выбирают из C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
n представляет собой целое число от 1 до 6;
каждый A независимо выбирают из -(CH2)-, -(CHR)-, -(CR2)-, -NH-, -NR-, O и S, где R независимо выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила; или один A связан с другим A посредством группы -L-, где L выбирают из C1-8 алкила, C1-8 гетероалкила, замещенного C1-8 алкила и замещенного C1-8 гетероалкила.
Аспект 196. Способ согласно аспекту 194, в котором аналог сложного моноэфира серной кислоты содержит соединение формулы (80a), а циклическая гидроксамовая кислота содержит соединение формулы (80b):
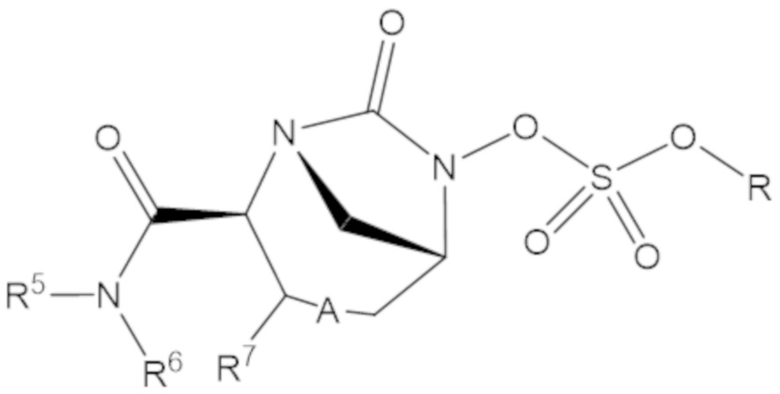
(80a) (80b)
в котором
R выбирают из C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
Аспект 197. Способ согласно любому из аспектов 194-196, в котором сложный моноэфир хлорсульфоновой кислоты представляет собой неопентиловый сложный эфир хлорсульфоновой кислоты.
Аспект 198. Способ согласно любому из аспектов 194-197, в котором неопентиловый сложный эфир хлорсульфоновой кислоты имеет структуру формулы (81):
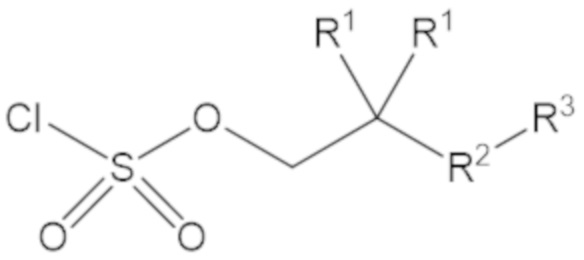 (81)
(81)
в котором
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила; и
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2, в котором
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила.
Аспект 199. Способ согласно любому из аспектов 194-198, в котором сложные моноэфиры хлорсульфоновой кислоты получают с помощью реакции спирта с сульфурилхлоридом.
Аспект 200. Способ согласно аспекту 199, в котором спирт представляет собой неопентиловый спирт.
Аспект 201. Способ согласно аспекту 194, в котором аналог сложного моноэфира серной кислоты, имеет структуру формулы (1):
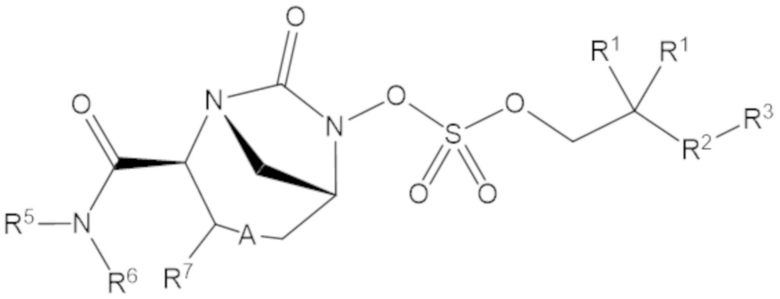 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый R1 независимо выбирают из C1-6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3-6 циклоалкильное кольцо, C3-6 гетероциклоалкильное кольцо, замещенное C3-6 циклоалкильное кольцо или замещенное C3-6 гетероциклоалкильное кольцо;
R2 выбирают из одинарной связи, C1-6 алкандиила, C1-6 гетероалкандиила, C5-6 циклоалкандиила, C5-6 гетероциклоалкандиила, C6 арендиила, C5-6 гетероарендиила, замещенного C1-6 алкандиила, замещенного C1-6 гетероалкандиила, замещенного C5-6 циклоалкандиила, замещенного C5-6 гетероциклоалкандиила, замещенного C6 арендиила и замещенного C5-6 гетероарендиила;
R3 выбирают из C1-6 алкила, -O-C(O)-R4, -S-C(O)-R4, -NH-C(O)-R4, -O-C(O)-O-R4, -S-C(O)-O-R4, -NH-C(O)-O-R4, -C(O)-O-R4, -C(O)-S-R4, -C(O)-NH-R4, -O-C(O)-O-R4, -O-C(O)-S-R4, -O-C(O)-NH-R4, -S-S-R4, -S-R4, -NH-R4, -CH(-NH2)(-R4), C5-6 гетероциклоалкила, C5-6 гетероарила, замещенного C5-6 циклоалкила, замещенного C5-6 гетероциклоалкила, замещенного C5-6 арила, замещенного C5-6 гетероарила и -CH=C(R4)2, в котором
R4 выбирают из водорода, C1-8 алкила, C1-8 гетероалкила, C5-8 циклоалкила, C5-8 гетероциклоалкила, C5-10 циклоалкилалкила, C5-10 гетероциклоалкилалкила, C6-8 арила, C5-8 гетероарила, C7-10 арилалкила, C5-10 гетероарилалкила, замещенного C1-8 алкила, замещенного C1-8 гетероалкила, замещенного C5-8 циклоалкила, замещенного C5-8 гетероциклоалкила, замещенного C5-10 циклоалкилалкила, замещенного C5-10 гетероциклоалкилалкила, замещенного C6-8 арила, замещенного C5-8 гетероарила, замещенного C7-10 арилалкила и замещенного C5-10 гетероарилалкила;
R5 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила;
R6 выбирают из водорода, C1-6 алкила, C5-8 циклоалкила, C6-12 циклоалкилалкила, C2-6 гетероалкила, C5-8 гетероциклоалкила, C6-12 гетероциклоалкилалкила, замещенного C1-6 алкила, замещенного C5-8 циклоалкила, замещенного C6-12 циклоалкилалкила, замещенного C2-6 гетероалкила, замещенного C5-8 гетероциклоалкила и замещенного C6-12 гетероциклоалкилалкила; и
А представляет собой одинарную связь (-), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1-3 алкил.
ПРИМЕРЫ
В следующих примерах подробно описан синтез соединений формулы (1), характеристика соединений формулы (1) и применение соединений формулы (1). Специалистам в данной области будет понятно, что на практике можно реализовать множество модификаций как материалов, так и способов без выхода за рамки объема раскрытия.
Пример 1
Синтез (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1)
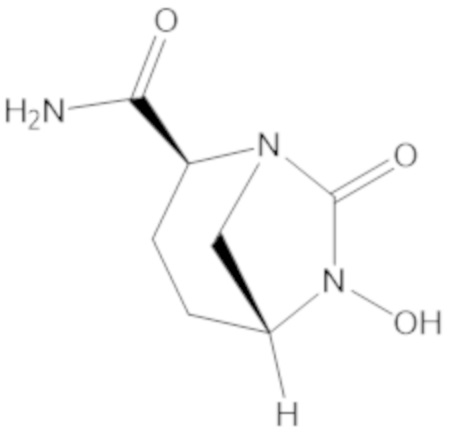
Сделана ссылка на публикацию международной заявки № WO 2012/086241 и международной заявки № PCT/2012/016553 вместе с соответствующими методиками из патентной литературы. Встряхиваемую смесь (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (550 мг, 2,0 ммоль), палладия на углероде (10% по массе; 340 мг, 0,3 ммоль) и MeOH (18 мл) гидратировали при 1 атм. (баллон) до анализа с помощью тонкослойной хроматографии (TLC) не показал завершение реакции (приблизительно, 30 мин; реакцию контролировали с помощью TLC с использованием MeOH/CH2Cl2 5:95 в качестве элюента). Смесь фильтровали через слой Celite®, и слой тщательно промывали MeOH (около 20 мл). Фильтрат концентрировали в вакууме (температура водяной бани не выше 25°C) с получением продукта в виде прозрачного и бесцветного масла. Масло сушили в вакууме в течение 1 ч, и остаток сразу использовали на следующей стадии без дополнительной очистки. Выход принимали по количеству. LC-MS: м/з=186,0 [M+H]+.
Пример 2
Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил бензоата (2)
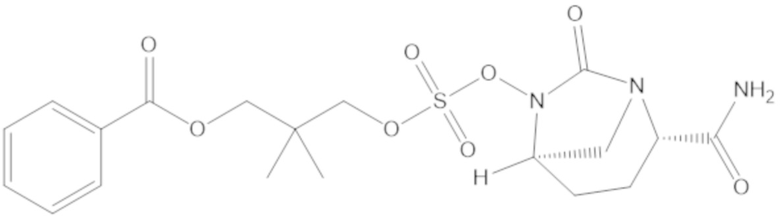
Стадия 1: Синтез 3-гидрокси-2,2-диметилпропил бензоата (2a).
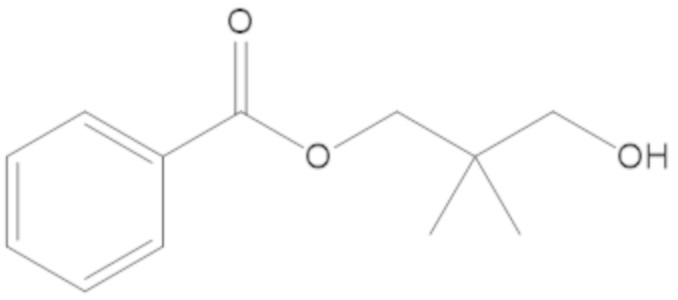
Бензоилхлорид (4,0 мл, 34,5 ммоль) по каплям добавляли во встряхиваемый раствор 2,2-диметилпропан-1,3-диола (10,8 г, 103,4 ммоль), пиридина (5,8 мл, 71,6 ммоль) и N, N-4-диметиламинопиридина (840 мг, 6,9 ммоль) в дихлорметане (207 мл) приблизительно при 0°C. Смесь встряхивали в течение ночи с постепенным нагревом до комнатной температуры, гасили путем добавления 1 н. HCl (100 мл) при 0°C и два раза экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным водным NaHCO3 (100 мл), рассолом (100 мл), сушили (Na2SO4), фильтровали, и растворитель концентрировали в вакууме с получением неочищенного остатка. Остаток разделяли на две партии и очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:4) с получением продукта (5,95 г, 99%) в виде бесцветного масла (примечание: масло сушили в вакууме в течение 2 дней). LC-MS: м/з=209,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): 8,05 (m, 2H), 7,58 (m, 1H), 7,45 (m, 2H), 4,19 (s, 2H), 3,38 (d, J=6,3 Гц, 2H), 2,29 (t, J=6,3 Гц, 1H), 1,02 (s, 6H).
Стадия 2: Синтез 3-((хлорсульфонил)окси)-2,2-диметилпропил бензоата (2b).
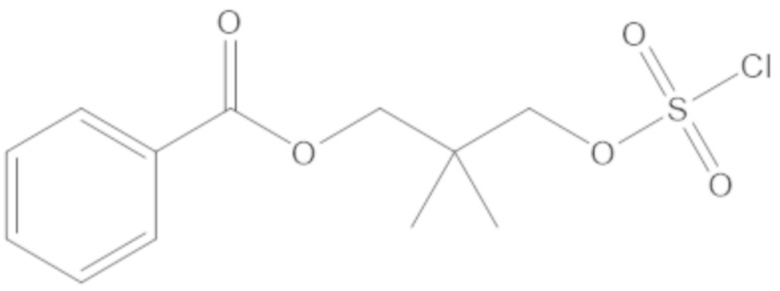
Сделана ссылка на J. Am. Chem. Soc. 2006, 128, 1605-1610. Раствор дистиллированного сульфурилхлорида (1,2 мл, 15,8 ммоль) в Et2O (15 мл) охлаждали до -78°C в атмосфере аргона. Затем с помощью шприца по каплям добавляли раствор 3-гидрокси-2,2-диметилпропил бензоата (2a) (3,0 г, 14,4 ммоль) и пиридина (1,2 мл, 14,4 ммоль) в Et2O (3,0 мл) в течение 1 ч. Шприц промывали Et2O (3×1 мл), причем каждый смыв добавляли в реакционную смесь. Ванну ацетон/CO2 удаляли, и обеспечивали нагревание смеси до комнатной температуры, затем встряхивали при комнатной температуре в течение 4 ч. анализ TLC (EtOAc/гексаны; 3:7) не показал завершение реакции, поэтому повторно охлаждали до -78°C, и еще добавляли SO2Cl2 (0,1 мл), затем обеспечивали нагревание до комнатной температуры и встряхивали в течение дальнейших 2 ч. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением продукта (3,97 г, 89%) в виде масла. 1H-NMR (300 МГц, CDCl3): 8,03 (m, 2H), 7,61-7,57 (m, 1H), 7,49-7,44 (m, 2H), 4,41 (s, 2H), 4,18 (s, 2H), 1,16 (s, 6H).
Стадия 3: Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил бензоата (2).
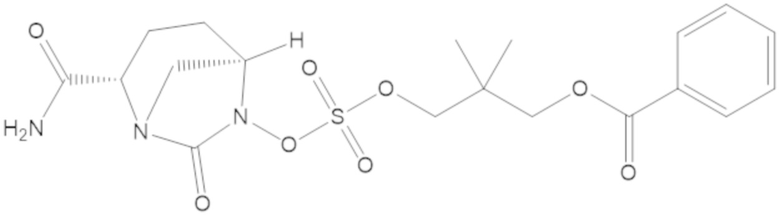
Сделана ссылка на J. Am. Chem. Soc. 2006, 128, 1605-1610. (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (7,0 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3,0 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. По каплям добавляли раствор NaHMDS в THF (1M; 2,2 мл, 2,2 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь быстро добавляли 3-((хлорсульфонил)окси)-2,2-диметилпропил бензоат (2b) (674 мг, 2,2 ммоль). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали при комнатной температуре до завершения оценки посредством LC-MS и анализа TLC. Добавляли EtOAc (20 мл) и насыщенный водный NaHCO3 (20 мл), и разделяли органические и водные слои. Органический слой промывали водой (3×20 мл), рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (400 мг, 43%) в виде твердого вещества. После очистки с помощью колоночной хроматографии продукт, по-видимому, до некоторой степени разлагался после сушки соединения в вакууме в течение выходных - одним продуктом разложения предположительно был авибактам по данным LC-MS, м/з=529,0 [2M-H]-. Часть материала впоследствии повторно очищали с помощью колоночной хроматографии на силикагеле с применением системы элюентов, подробно описанной выше. Затем сразу после выделения продукт сохраняли при -20°C. LC-MS: м/з=456,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,05 (d, J=6,9 Гц, 2H), 7,59-7,54 (m, 1H), 7,47-7,42 (m, 2H), 6,49 (s, 1H), 5,91 (s, 1H), 4,69 (d, J=9,3 Гц, 1H), 4,44 (d, J=9,3 Гц, 1H), 4,16-4,14 (m, 3H), 4,00 (d, J=7,5 Гц, 1H), 3,24-3,20 (m, 1H), 2,96 (d, J=11,7 Гц, 1H), 2,43-2,36 (m, 1H), 2,16-2,09 (m, 1H), 1,97-1,80 (m, 2H), 1,13 (s, 3H), 1,12 (s, 3H). 13C-NMR (300 МГц, CDCl3): δ 171,1, 167,1, 166,4, 133,3, 130,0, 129,8, 128,6, 80,7, 69,1, 62,0, 60,2, 47,1, 35,7, 21,5, 20,8, 17,6.
Пример 3
Синтез этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3)
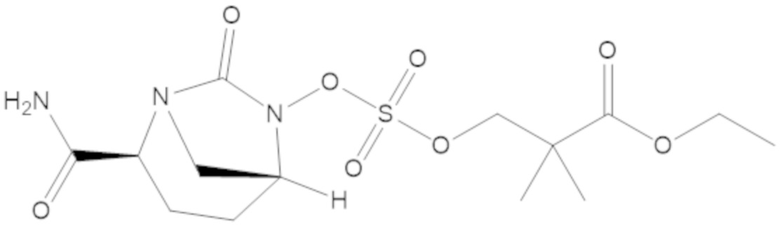
Стадия 1: Синтез этил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (3a).
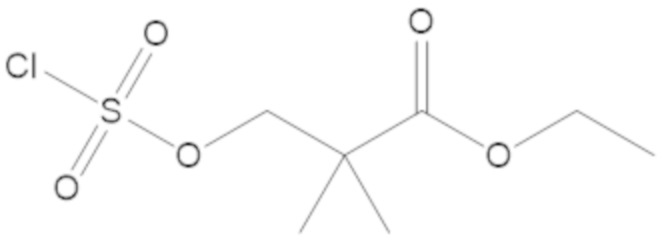
Раствор дистиллированного сульфурилхлорида (0,55 мл, 7,5 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере аргона. Затем в течение 1 ч с помощью шприца по каплям добавляли раствор этил 3-гидрокси-2,2-диметилпропаноата (2a) (1,0 г, 6,8 ммоль) и пиридина (0,55 мл, 6,8 ммоль) в Et2O (1,0 мл). Шприц промывали Et2O (3×1 мл), причем каждый смыв добавляли в реакционную смесь. Ванну ацетон/CO2 удаляли, и обеспечивали нагревание смеси до комнатной температуры, затем встряхивали при комнатной температуре в течение 4 ч. анализ TLC (EtOAc/гексаны; 3:7) не показал, что реакция завершалась. Смесь повторно охлаждали до -78°C, и еще добавляли SO2Cl2 (0,11 мл), и обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение дальнейших 2 ч. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением продукта (выход принимали по количеству). 1H-NMR (300 МГц, CDCl3): δ 4,50 (s, 2H), 4,19 (q, J=6,9 Гц, 2H), 1,31 (s, 6H), 1,28 (t, J=6,9 Гц, 3H).
Стадия 2: Синтез этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (3).
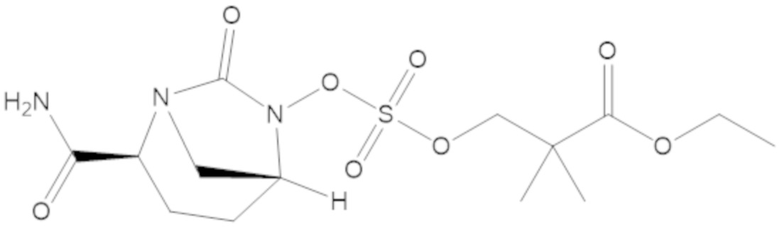
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (7,0 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3,0 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. По каплям добавляли раствор NaHMDS в THF (1M; 2,2 мл, 2,2 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца быстро добавляли раствор этил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (3a) (538 мг, 2,2 ммоль) в THF (1 мл). Шприц промывали THF (3×0,5 мл), причем каждый смыв добавляли в реакционную смесь. Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали при комнатной температуре до завершения оценки посредством LC-MS и анализа TLC (около 2 h). Добавляли EtOAc (20 мл) и насыщенный водный NaHCO3 (20 мл), и разделяли органические и водные слои. Органический слой промывали насыщенным NaHCO3 (20 мл), водой (3×20 мл), рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (318 мг, 39%) в виде твердого вещества. LC-MS: м/з=394,1 [M+H]+ . 1H-NMR (CDCl3, 300 МГц): δ 6,50 (s, 1H), 5,78 (s, 1H), 4,71 (d, J=8,7 Гц, 1H), 4,59 (d, J=8,7 Гц, 1H), 4,22-4,12 (m, 3H), 4,05 (d, J=6,9 Гц, 1H), 3,34-3,30 (m, 1H), 3,01 (d, J=12,3 Гц, 1H), 2,46-2,40 (m, 1H), 2,18-2,12 (m, 1H), 2,00-1,79 (m, 2H), 1,28-1,24 (m, 9H). 13C-NMR (300 МГц, CDCl3): δ 174,2, 171,2, 167,1, 80,5, 61,9, 61,4, 60,2, 47,2, 42,8, 22,2, 21,7, 20,8, 17,5, 14,2.
Пример 4
Синтез бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4)
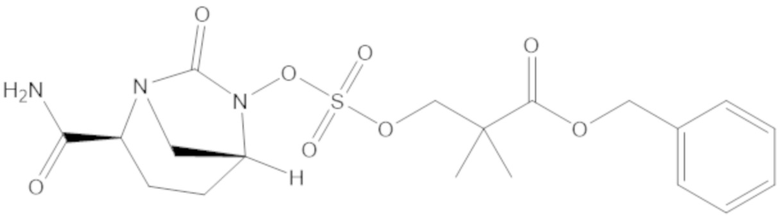
Стадия 1: Синтез бензил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (4a).
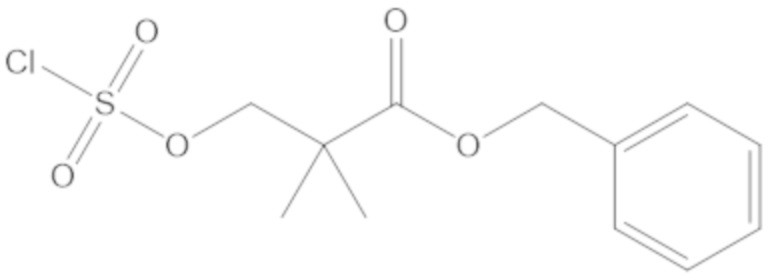
Раствор дистиллированного сульфурилхлорида (0,77 мл, 10,6 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере аргона. Затем в течение 1 ч с помощью шприца по каплям добавляли раствор этил 3-гидрокси-2,2-диметилпропаноата (2a) (Sigma-Aldrich; 2,0 г, 9,6 ммоль) и пиридина (0,85 мл, 10,6 ммоль) в Et2O (2,0 мл). Шприц промывали Et2O, причем каждый смыв добавляли в реакционную смесь. Ванну ацетон/CO2 удаляли и обеспечивали нагревание смеси до комнатной температуры, затем встряхивали при комнатной температуре в течение 30 минут анализ TLC (EtOAc/гексаны; 3:7) не показал завершение реакции, поэтому повторно охлаждали до -78°C, и еще добавляли SO2Cl2 (0,07 мл), затем обеспечивали нагревание до комнатной температуры и встряхивали в течение дальнейшего 1 ч. Добавляли Et2O (5 мл), и смесь встряхивали в течение нескольких минут, затем фильтровали, и фильтрат концентрировали в вакууме с получением продукта (2,19 г, 75%). 1H-NMR (300 МГц, CDCl3): δ 7,41-7,32 (m, 4H), 5,18 (s, 2H), 4,52 (s, 2H), 1,34 (s, 6H).
Стадия 2: Синтез бензил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (4).
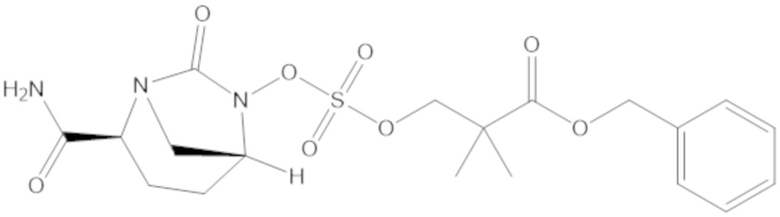
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (7,0 мл), и добавляли 1,3-диметилтетрагидропиримидин-2(1H)-он (2,0 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. По каплям добавляли раствор NaHMDS в THF (1M; 2,2 мл, 2,2 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца быстро добавляли раствор этил бензил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (4a) (674 мг, 2,2 ммоль) в THF (1 мл). Шприц промывали THF (3×0,5 мл), причем каждый смыв добавляли в реакционную смесь. Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали при комнатной температуре до завершения оценки посредством LC-MS и анализа TLC. Добавляли EtOAc (20 мл) и насыщенный водный NaHCO3 (20 мл), и разделяли органические и водные слои. Органический слой промывали насыщенным NaHCO3 (20 мл), водой (3×20 мл), рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (244 мг, 26%) в виде твердого вещества. LC-MS: м/з=456,2 [M+H]+. 1H-NMR (CDCl3, 300 МГц): δ 7,39-7,28 (m, 4H), 6,49 (s, 1H), 5,84 (s, 1H), 5,20-5,11 (m, 2H), 4,74 (d, J=9,0 Гц, 1H), 4,61 (d, J=9,0 Гц, 1H), 4,15-4,14 (m, 1H), 4,04 (d, J=6,9 Гц, 1H), 3,29-3,25 (m, 1H), 2,99 (d, J=11,7 Гц, 1H), 2,45-2,38 (m, 1H), 2,17-2,10 (m, 1H), 1,99-1,78 (m, 2H), 1,30 (s, 3H), 1,29 (s, 3H). 13C-NMR (CDCl3, 75 МГц): δ 174,1, 171,1, 167,1, 135,7, 128,7, 128,4, 128,0, 80,3, 67,0, 62,0, 60,2, 47,2, 43,0, 22,2, 21,7, 20,8, 17,5.
Пример 5
Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил фенил сульфата (5)
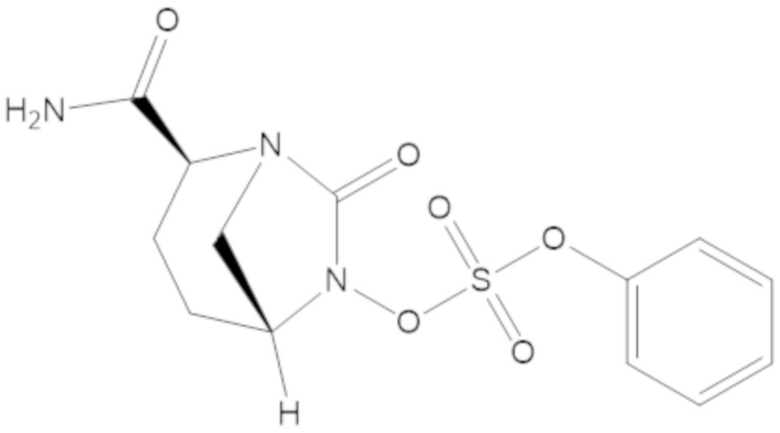
Стадия 1: Синтез фенил сульфохлорида (5a).
Сделана ссылка на J. Am. Chem. Soc. 2013, 135, 10638-10641. Раствор дистиллированного сульфурилхлорида (2,6 мл, 35,1 ммоль) в Et2O (30 мл) охлаждали до -78°C в атмосфере аргона. Затем одновременно, но из разных шприцов, по каплям в течение 1 ч добавляли раствор фенола (3,0 г, 31,9 ммоль) в Et2O (3,0 мл) и пиридина (2,6 мл, 31,9 ммоль). Каждый шприц промывали Et2O, и каждый смыв добавляли в реакционную смесь. Обеспечивали медленное нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением продукта (4,65 г), загрязненного другими продуктами и исходным фенольным материалом. Продукт фенил сульфохлорид дополнительно не очищали и использовали непосредственно на следующей стадии.
Стадия 2: Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил фенил сульфата (5).
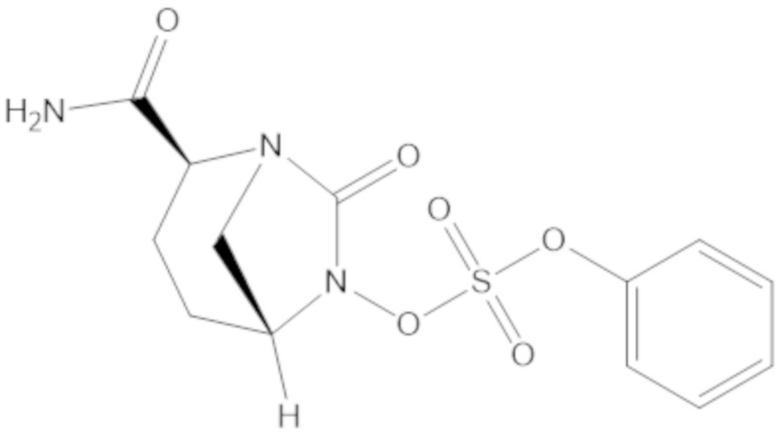
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (7,0 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2,0 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. По каплям добавляли раствор NaHMDS в THF (1M; 2,2 мл, 2,2 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца быстро добавляли неразбавленный фенил сульфохлорид (5a) (423 мг, 2,2 ммоль). Шприц промывали THF (3×0,5 мл), причем каждый смыв добавляли в реакционную смесь. Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали при комнатной температуре до завершения оценки посредством LC-MS и анализа TLC (около 1 ч). Добавляли EtOAc (20 мл) и насыщенный водный NaHCO3 (20 мл), и разделяли органические и водные слои. Органический слой промывали насыщенным NaHCO3 (20 мл), водой (3×20 мл), рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (126 мг, 18%) в виде твердого вещества. LC-MS: м/з=342,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,54-7,51 (m, 2H), 7,47-7,42 (m, 2H), 7,39-7,33 (m, 1H), 6,53 (s, 1H), 5,88 (s, 1H), 4,24 (fd, J=2,4 Гц, 1H), 4,09 (d, J=6,9 Гц, 1H), 3,34 (d, J=11,7 Гц, 1H), 3,05 (d, J=12,3 Гц, 1H), 2,46-2,39 (m, 1H), 2,19-2,11 (m, 1H), 2,02-1,81 (m, 2H). 13C-NMR (75 МГц, CDCl3): δ 171,2, 166,8, 150,9, 130,2, 128,1, 121,3, 62,1, 60,6, 47,0, 20,8, 17,6.
Пример 6
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил бензоата (6)
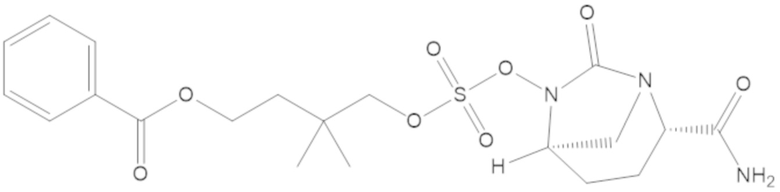
Стадия 1: Синтез 2,2-диметилбутан-1,4-диола (6a).
В суспензию аллюмогидрида лития (8,3 г, 219,0 ммоль) в THF (80 мл) при 0°C (ледяная ванна) по каплям добавляли раствор 2,2-диметилянтарной кислоты (10,0 г, 68,4 ммоль) в THF (150 мл). Смесь нагревали до комнатной температуры в течение 20 мин, а затем нагревали с обратным холодильником в течение 1,5 ч. По завершении (реакцию контролировали с помощью TLC с использованием MeOH/CH2Cl2 5:95 в качестве элюента) реакцию гасили очень осторожно и путем добавления воды по каплям (10 мл), 3 M NaOH (15 мл) и воды (20 мл). Смесь встряхивали при комнатной температуре в течение 20 мин, и твердые вещества фильтровали через слой Celite®. Фильтрационный осадок тщательно промывали THF. Фильтрат концентрировали в вакууме с получением смеси титульного соединения и неидентифицированных побочных продуктов в виде неочищенного масла. Масло очищали с помощью колоночной хроматографии на силикагеле с использованием MeOH/CH2Cl2 (0:1-1:9) в качестве элюента с получением продукта (4,649 г, 57%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 4,11 (s, 2H), 3,66 (t, J=5,9 Гц, 2H), 3,30 (s, 2H), 1,52 (t, J=5,6 Гц, 2H), 0,89 (s, 6H).
Стадия 2: Синтез 4-гидрокси-3,3-диметилбутил бензоата (6b).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (6a) (0,30 г, 2,5 ммоль) в безводном дихлорметане (9 мл) добавляли бензоилхлорид (0,30 мл, 2,5 ммоль), Et3N (0,71 мл, 5,1 ммоль) и каталитическое количество N, N-4-диметиламинопиридина при 0°C (ледяная ванна). Смесь постепенно нагревали до комнатной температуры и встряхивали в течение ночи. После полного расходования исходного материала (реакцию контролировали с помощью TLC с использованием EtOAc/гексанов 2:8 в качестве элюента) реакцию гасили путем добавления 1 н. HCl (20 мл) при 0°C (ледяная ванна), и смесь два раза экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным водным NaHCO3, рассолом, сушили (Na2SO4), фильтровали, и растворитель концентрировали с получением смеси по меньшей мере двух продуктов в виде прозрачного и бесцветного масла. Масло очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (0:1-4:6) в качестве элюента с получением продукта (0,29 г, 51%) в виде масла (которое сушили при большом разрежении в течение 2 д). 1H-NMR (300 МГц, CDCl3): δ 8,04-8,01 (m, 2H), 7,58-7,53 (m, 1H), 7,46-7,41 (m, 2H), 4,41 (t, J=7,4 Гц, 2H), 3,41 (s, 2H), 1,78 (t, J=7,4 Гц, 2H), 1,70 (s, 1H), 0,99 (s, 6H).
Стадия 3: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил бензоата (6c).
Раствор свежедистиллированного сульфурилхлорида (0,11 мл, 1,5 ммоль) в Et2O (3 мл) охлаждали до -78°C в атмосфере аргона. В охлажденный раствор (в течение 1 ч) по каплям добавляли раствор 4-гидрокси-3,3-диметилбутил бензоата (6b) (0,28 г, 1,3 ммоль) и пиридина (0,10 мл, 1,3 ммоль) в Et2O (2 мл). Смесь нагревали до комнатной температуры и встряхивали в течение 30 минут. (реакцию контролировали с помощью TLC с использованием EtOAc/гексанов 2:8 в качестве элюента). Смесь повторно охлаждали до -78°C, и добавляли сульфурилхлорид (0,02 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут. Добавляли Et2O (5 мл), и смесь встряхивали в течение нескольких минут. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением продукта (6c) (0,305 г, 75%). 1H-NMR (300 МГц, CDCl3): δ 8,03 (d, J=8,1 Гц, 2H), 7,60-7,54 (m, 1H), 7,47-7,42 (m, 2H), 4,44-4,38 (m, 2H), 4,29 (s, 2H), 1,89-1,85 (m, 2H), 1,13 (s, 6H).
Стадия 4: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил бензоата (6).
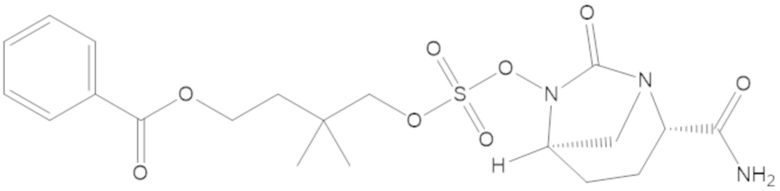
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (176 мг, 1,0 ммоль) растворяли в THF (4 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли NaHMDS (1,0 M в THF; 1,05 мл, 1,05 ммоль), и смесь встряхивали при -78°C в течение 10 мин. В смесь быстро добавляли 4-((хлорсульфонил)окси)-3,3-диметилбутил бензоат (6c) (305 мг, 1,0 ммоль) в THF (0,5 мл). Шприц промывали THF (3×0,5 мл), и также добавляли в смесь. Спустя 10 мин смесь нагревали до комнатной температуры и встряхивали до завершения оценки с помощью анализ TLC. В смесь добавляли EtOAc (10 мл), и насыщенный водный NaHCO3 (10 мл), и разделяли органические и водные слои. Органический слой промывали насыщенным водным NaHCO3 (10 мл), водой (6×10 мл), рассолом (10 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. В результате очистки с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) получали продукт (6) (215 мг, 18%) в виде твердого вещества. LC-MS: 470,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,04-8,01 (m, 2H), 7,58-7,53 (m, 1H), 7,46-7,41 (m, 2H), 6,48 (s, 1H), 5,80 (s, 1H), 4,60 (d, J=9,3 Гц, 1H), 4,40 (t, J=6,9 Гц, 2H), 4,29 (d, J=9,3 Гц, 1H), 4,17-4,16 (m, 1H), 4,02 (d, J=7,8 Гц, 1H), 3,34-3,30 (m, 1H), 3,00 (d, J=12,3 Гц, 1H), 2,45-2,35 (m, 1H), 2,17-2,11 (m, 1H), 1,98-1,78 (m, 4H), 1,10 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 167,1, 166,6, 133,1, 130,2, 129,7, 128,5, 83,7, 61,9, 61,5, 60,2, 47,2, 36,9, 34,1, 24,1, 23,8, 20,8, 17,5.
Пример 7
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил пропионата (7)
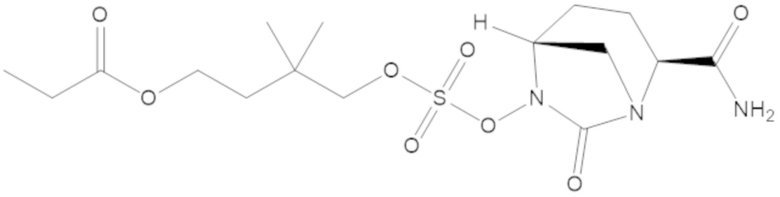
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутил пропионата (7a).
Раствор пропионилхлорида (0,74 мл, 8,5 ммоль) в безводном дихлорметане (5 мл) добавляли во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (6a) (1,00 г, 8,5 ммоль), Et3N (2,4 мл, 16,9 ммоль) и 4-N, N-диметиламинопиридина (52 мг) в безводном дихлорметане (20 мл) при -78°C в атмосфере аргона. Смесь встряхивали в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры, встряхивали при комнатной температуре в течение 1 ч, затем повторно охлаждали до -78°C и обеспечивали медленное нагревание до комнатной температуры, позволяя смеси оставаться в холодной ванне и обеспечивая возгонку сухого льда (после добавления всех реагентов рекомендовано обеспечивать нагрев до комнатной температуры от -78°C). После полного расходования исходного материала (TLC 50% EtOAc/гексаны) реакцию гасили путем добавления 0,5 N HCl (10 мл) при 0°C. Разделяли органические и водные слои, и водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои промывали насыщенным водным NaHCO3 (20 мл), рассолом (20 мл), затем сушили (Na2SO4), фильтровали, и растворитель концентрировали в вакууме, получая неочищенное масло. Масло очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (0:1-4:1) в качестве элюента с получением продукта (7a) (463 мг, 22%) в виде масла, загрязненного значительными остатками растворителя EtOAc. 1H-NMR (300 МГц, CDCl3): δ 4,14 (t, J=7,4 Гц, 2H), 3,32 (s, 2H), 2,30 (q, J=7,6 Гц, 2H), 1,88 (s, 1H), 1,61 (t, J=7,7 Гц, 2H), 1,13 (t, J=7,5 Гц, 3H), 0,91 (fd, J=1,2 Гц, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил пропионата (7b).
Раствор свежедистиллированного сульфурилхлорида (0,15 мл, 2,0 ммоль) в Et2O (3,5 мл) охлаждали до -78°C в атмосфере аргона. Раствор 4-гидрокси-3,3-диметилбутил пропионата (7a) (чистота 73%, остальное EtOAc; 441 мг, 1,8 ммоль) и пиридина (0,15 мл, 1,8 ммоль) в Et2O (2,5 мл) в охлажденный раствор в течение 1 ч по каплям добавляли. Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут (контролировали с помощью TLC, 30% EtOAc/гексаны), повторно охлаждали до -78°C, и добавляли сульфурилхлорид (0,03 мл) и пиридин (0,03 мл), нагревали до комнатной температуры и встряхивали в течение 30 минут. Смесь снова повторно охлаждали до -78°C, и добавляли еще одну часть сульфурилхлорида (0,15 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут. Добавляли Et2O (5 мл), и смесь встряхивали в течение нескольких минут. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением продукта (7b) (401 мг, 79%). 1H-NMR: (300 МГц, CDCl3): 4,22 (s, 2H), 4,14 (t, J=6,8 Гц, 2H), 2,30 (q, J=7,6 Гц, 2H), 1,70 (t, J=6,8 Гц, 2H), 1,11 (t, J=7,7 Гц, 3H), 1,05 (s, 6H).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил пропионата (7).
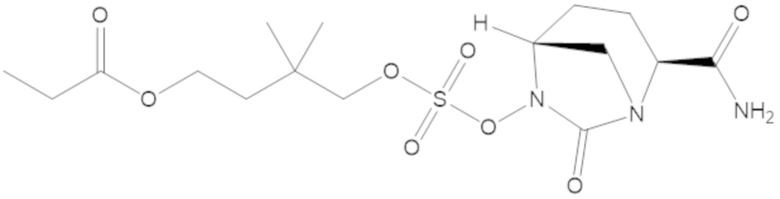
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (185 мг, 1,0 ммоль) растворяли в THF (4 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1 мл) в атмосфере аргона. Полученный в результате раствор охлаждали до -78°C. В охлажденный раствор по каплям добавляли NaHMDS (1,0 M в THF; 1,1 мл, 1,1 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли 4-((хлорсульфонил)окси)-3,3-диметилбутил пропионат (7b) (272 мг, 1,0 ммоль) в THF (1 мл). Шприц промывали THF (3×0,5 мл), и также добавляли в смесь. В смесь добавляли еще THF (3 мл), обеспечивая реакцию с эффективным перемешиванием. Спустя 10 мин обеспечивали нагревание смеси до комнатной температуры. По завершении (1 ч; TLC, 70% EtOAc/гексаны) в смесь добавляли EtOAc (10 мл) и насыщенный водный NaHCO3 (10 мл). Водные и органические слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (10 мл), водой (6×10 мл), рассолом (10 мл), затем сушили (Na2SO4), фильтровали, и растворитель концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (1:0-1:0) в качестве элюента с получением продукта (7) (93 мг, 22%) в виде твердого вещества. LC-MS: 422,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,51 (s, 1H), 5,76 (s, 1H), 4,54 (d, J=8,7 Гц, 1H), 4,25 (d, J=8,7 Гц, 1H), 4,17-4,12 (m, 3H), 4,04 (d, J=6,9 Гц, 1H), 3,36-3,32 (m, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,47-2,40 (m, 1H), 2,32 (q, J=7,6 Гц, 2H), 2,18-2,15 (m, 1H), 2,00-1,79 (m, 2H), 1,75-1,61 (m, 2H), 1,13 (t, J=7,7 Гц, 3H), 1,03 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 174,5, 171,0, 167,1, 83,6, 62,0, 60,8, 60,2, 47,2, 36,8, 34,0, 27,7, 24,1, 23,7, 20,8, 17,6, 9,2.
Пример 8
Синтез бензил (4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) адипата (8)
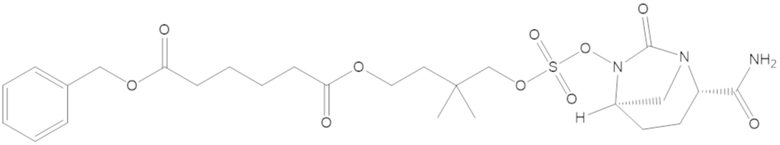
Стадия 1: Синтез бензил (перфторфенил) адипата (8a).
Во встряхиваемый раствор сложного монобензилового эфира адипиновой кислоты (1,03 г, 4,3 ммоль) и пентафторфенола (0,87 г, 4,7 ммоль) в EtOAc (18,7 мл) при 0°C добавляли N, N'-дициклогексилкарбодиимид (0,97 г, 4,7 ммоль). Обеспечивали нагревание смеси до комнатной температуры, а затем встряхивали в течение ночи. Полученное в результате твердое вещество удаляли посредством вакуумной фильтрации через слой Celite®. Фильтрационный осадок промывали EtOAc. Фильтрат загружали на силикагель методом сухого ввода и очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (0:1-4:6) в качестве элюента с получением продукта (8a) (1,59 г, 93%) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 7,37-7,35 (m, 5 H), 5,13 (s, 2H), 2,68 (t, J=6,8 Гц, 2H), 2,44 (t, J=6,5 Гц, 2H), 1,82-1,78 (m, 4H).
Стадия 2: Синтез бензил (4-гидрокси-3,3-диметилбутил) адипата (8b).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (6a) (0,22 г, 1,8 ммоль) в безводном дихлорметане (4 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли бензил (перфторфенил) адипат (8a) (0,36 г, 0,9 ммоль), Et3N (0,25 мл, 1,8 ммоль) и каталитическое количество 4-N, N-диметиламинопиридина (небольшое невзвешиваемое количество). Смесь постепенно нагревали до комнатной температуры, а затем при комнатной температуре в течение ночи. Смесь загружали на силикагель методом сухого ввода и очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением продукта, загрязненного региоизомерным продуктом. Эту смесь повторно очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением чистого продукта (8b) (113 мг 38%). 1H-NMR (300 МГц, CDCl3): 7,36-7,34 (m, 5H), 5,11 (s, 2H), 4,14 (t, J=7,2 Гц, 2H), 3,34 (d, J=5,7 Гц, 2H), 2,38-2,31 (m, 4H), 1,68-1,59 (m, 6H), 0,92 (s, 6H). Для получения большего количества материала реакцию можно повторить.
Стадия 3: Синтез бензил (4-((хлорсульфонил)окси)-3,3-диметилбутил) адипата (8c).
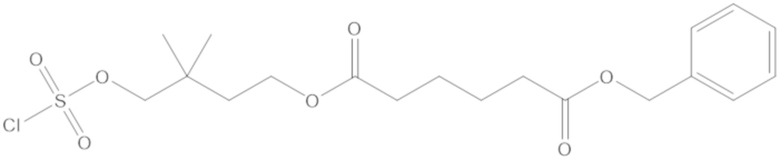
Раствор свежедистиллированного сульфурилхлорида (0,12 мл, 1,6 ммоль) в Et2O (5 мл) охлаждали до -78°C в атмосфере аргона. В охлажденный раствор в течение 1 ч по каплям добавляли раствор бензил (4-гидрокси-3,3-диметилбутил) адипата (8b) (446 мг, 1,3 ммоль) и пиридина (0,11 мл, 1,3 ммоль) в Et2O (3,5 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут (контролировали с помощью TLC, 30% EA/гекс). Реакцию не завершили, поэтому смесь снова охлаждали до -78°C, затем добавляли сульфурилхлорид (0,05 мл) и пиридин (0,05 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут. Добавляли Et2O (5 мл), и смесь встряхивали в течение нескольких минут. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением продукта (8c) (446 мг, 77%). 1H-NMR (300 МГц, CDCl3): δ 7,39-7,29 (m, 5H), 5,11 (s, 2H), 4,22 (s, 2H), 4,15 (t, J=6,8 Гц, 2H), 2,40-2,29 (m, 4H), 1,73-1,59 (m, 6H), 1,06 (s, 6H).
Стадия 4: Синтез бензил (4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) адипата (8).
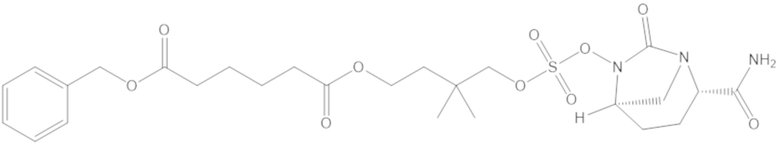
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (185 мг, 1,0 ммоль) растворяли в THF (9 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли NaHMDS (1,0 M раствор в THF; 1,1 мл, 1,1 ммоль), и встряхивали в течение 10 мин. В смесь быстро добавляли бензил (4-((хлорсульфонил)окси)-3,3-диметилбутил) адипат (8c) (435 мг, 1,0 ммоль) в THF (1 мл). Шприц промывали THF (3×0,5 мл), и также добавляли в смесь. Спустя 10 мин обеспечивали нагревание смеси до комнатной температуры. По завершении (30 мин; TLC, 70% EtOAc/гексаны) добавляли EtOAc (10 мл) и насыщенный водный NaHCO3 (10 мл). Водные и органические слои разделяли, и органический слой промывали насыщенным водным NaHCO3 (10 мл), водой (3×10 мл), рассолом (10 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением твердого вещества. Твердое вещество растирали в порошок с Et2O (2×2 мл) с получением продукта (8) (43 мг, 7%) в виде твердого вещества [вместе с менее чистым материалом (99 мг, чистота 93%), которое использовали на следующей стадии без дополнительной очистки]. Данные для 43 мг чистого продукта подробно изложены ниже. LC-MS: 584,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,39-7,30 (m, 5H), 6,52 (s, 1H), 5,65 (s, 1H), 5,11 (s, 2H), 4,54 (d, J=9,6 Гц, 1H), 4,24 (d, J=8,7 Гц, 1H), 4,16-4,08 (m, 3H), 4,04 (d, J=7,2 Гц, 1H), 3,35-3,31 (m, 1H), 3,01 (d, J=12,3 Гц, 1H), 2,47-2,30 (m, 5H), 2,18-2,12 (m, 1H), 1,99-1,77 (m, 2H), 1,74-1,65 (m, 6H), 1,03 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 173,34, 173,31, 171,0, 167,1, 136,1, 128,7, 128,4, 128,3, 83,5, 66,4, 62,0, 60,9, 60,2, 47,2, 36,8, 34,0, 24,5, 24,4, 24,2, 23,8, 20,8, 17,6.
Пример 9
Синтез 6-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутокси)-6-оксогексановой кислоты (9)
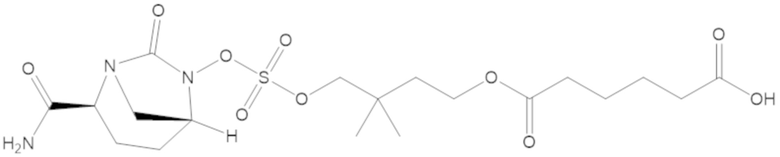
В колбу Парра, загруженную бензил (4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) адипатом (8) (чистота 93%; 50 мг, 0,1 ммоль) в MeOH (14 мл), добавляли палладий на углероде (10% по массе; 13 мг). Смесь гидратировали при 1 атмосфере H2 (баллон), при комнатной температуре в течение 30 минут (контролировали с помощью TLC, 100% EtOAc; окрашивание PMA; LC-MS: комнатная температура продукта=4,66 мин и м/з=494,2 [M+H]+, комнатная температура исходного материала=5,48 мин и м/з=584,3 [M+H]+). Смесь фильтровали через слой Celite®, и фильтрационный осадок промывали MeOH (около 20 мл). Фильтрат концентрировали в вакууме, затем очищали с помощью колоночной хроматографии на силикагеле с использованием MeOH/CH2Cl2 (0:1-4:96) в качестве элюента с получением продукта (9) (12 мг) в виде твердого вещества (чистота около 73% по данным LC/MS). LC-MS: 494,2 [M+H]+.
Пример 10
Синтез метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10)
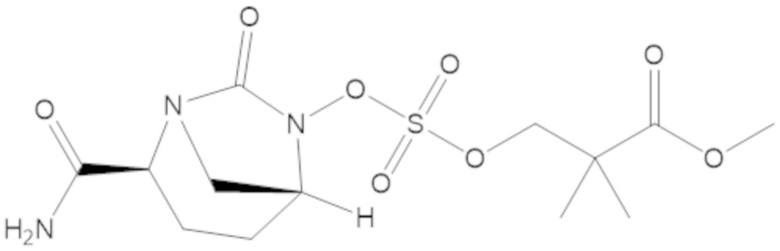
Стадия 1: Реакция получения метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (10a).
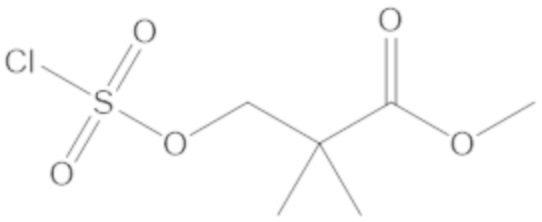
Раствор свежедистиллированного сульфурилхлорида (3,3 мл, 45,4 ммоль) в Et2O (45 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 30 мин по каплям добавляли раствор метил 2,2-диметил-3-гидроксипропионата (3,0 г, 22,7 ммоль) и пиридина (2,2 мл, 27,2 ммоль) в Et2O (20 мл). Колбу промывали Et2O (3×5 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения (контролировали с помощью TLC, 30% EA/гекс, 30 мин). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (10a) (5,6 г, выход 70%). Смесь хранили при -78°C и сразу использовали для следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3) δ 4,50 (s, 2H), 3,74 (s, 3H), 1,31 (s, 6H).
Стадия 2: Реакция получения метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (10).
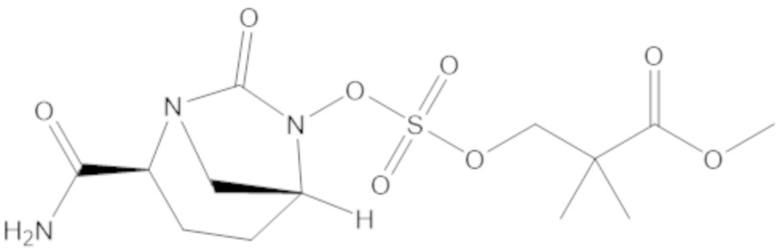
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (673 мг, 3,6 ммоль) растворяли в THF (35 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (4,0 мл, 4,0 ммоль). После завершения добавления смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли неразбавленный метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (10a) (1,27 г, 3,6 ммоль, 1,0 эквив., чистота 66%). Шприц промывали THF (3×2,5 мл), а смыв в смесь добавляли. Спустя 10 мин обеспечивали нагревание реакционной смеси до 23°C. По завершении с помощью TLC (30 мин; 70% EtOAc/гексаны) добавляли в реакционную смесь EtOAc (50 мл) и насыщенный водный NaHCO3 (50 мл). Слои разделяли и органический слой промывали насыщенным водным NaHCO3 (50 мл), водой (3×50 мл), рассолом (50 мл), затем сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (10) (98 мг, 7% выход). LC-MS: 380,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,47 (s, 1H), 5,61 (s, 1H), 4,72 (d, J=9,3 Гц, 1H), 4,58 (d, J=9,3 Гц, 1H), 4,18 (m, 1H), 4,05 (d, J=6,9 Гц, 1H), 3,72 (s, 3H), 3,35-3,31 (m, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,47-2,41 (m, 1H), 2,19-2,13 (m, 1H), 2,01-1,79 (m, 2H), 1,29 (s, 3H), 1,28 (s, 3H), 13C-NMR (75 МГц, CDCl3): δ 174,7, 171,1, 167,1, 80,4, 62,0, 60,2, 52,6, 47,2, 43,0, 22,2, 21,8, 20,9, 17,6
Пример 11
Синтез изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11)
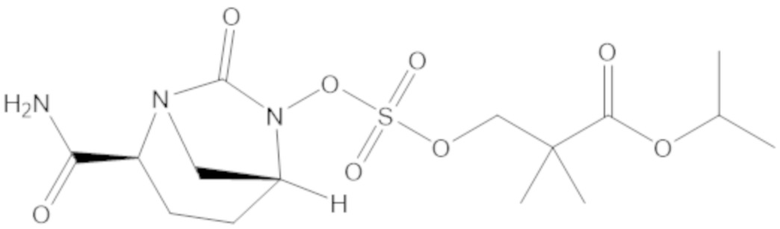
Стадия 1: Реакция получения изопропил 3-гидрокси-2,2-диметилпропаноата (11a).
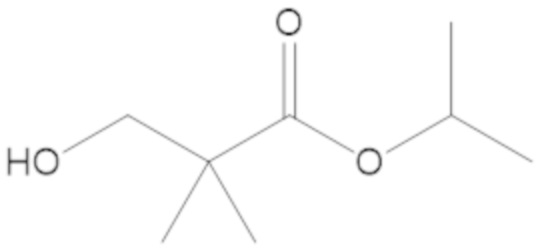
Сделана ссылка на публикацию заявки Германии № DE3045373. Смесь 3-гидрокси-2,2-диметилпропионовой кислоты (4,7 г, 40 ммоль), изопропанола (70 мл) и концентрированной серной кислоты (или дымящейся серной кислоты; 1 мл) нагревали с обратным холодильником и встряхивали в течение ночи. После обеспечения охлаждения смесь концентрировали в вакууме, а остаток разделяли на EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Водную смесь промывали H2O (50 мл), насыщенным NaHCO3 (50 мл) и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта в виде масла. Продукт (11a) использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 5,08-4,95 (m, 1H), 3,53 (fd, J=1,8 Гц, 2H), 2,49 (s, 1H), 1,25 (fd, J=2,4 Гц, 3H), 1,22 (fd, J=2,4 Гц, 3H), 1,17 (s, 3H), 1,16 (s, 3H).
Стадия 2: Реакция получения изопропил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (11b).
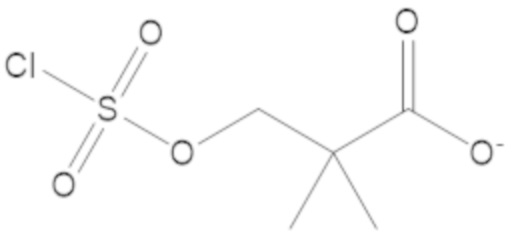
Раствор сульфурилхлорида (2,7 мл, 37,5 ммоль) в Et2O (45 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 30 мин по каплям добавляли раствор изопропил 3-гидрокси-2,2-диметилпропаноата (11a) (3,0 г, 18,7 ммоль) и пиридина (1,82 мл, 22,5 ммоль) в Et2O (20 мл). Колбу промывали Et2O (3×5 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения с помощью TLC (30 мин; 30% EA/гекс). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением изопропил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (11b) (4,1 г, 85% выход). Смесь хранили при -78°C и сразу использовали для следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 5,10-4,98 (m, 1H), 4,49 (s, 2H), 1,29 (s, 6H), 1,26 (s, 3H), 1,24 (s, 3H).
Стадия 3: Реакция получения изопропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (11).
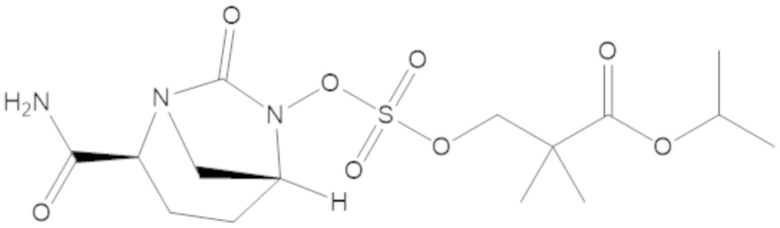
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (673 мг, 3,6 ммоль) растворяли в THF (35 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (4,0 мл, 4,0 ммоль), и встряхивали в течение 20 мин. В реакционную смесь быстро добавляли неразбавленный изопропил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (11b) (0,94 г, 3,6 ммоль). Шприц промывали THF (3×3 мл), а смыв также добавляли в смесь. Спустя 20 мин обеспечивали нагревание смеси до комнатной температуры. По завершении реакции с помощью TLC (30 мин; 70% EtOAc/гексаны) добавляли в реакционную смесь EtOAc (50 мл) и насыщенный водный NaHCO3 (50 мл). Водные и органические слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (50 мл), водой (3×50 мл), рассолом (50 мл), затем сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии (колонка ISCO 40 г) на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (11) (63 мг, 4%) в виде твердого вещества. LC-MS: 408,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,50 (s, 1H), 5,74 (s, 1H), 5,02 (quint, J=6,3 Гц, 1H), 4,70 (d, J=8,7 Гц, 1H), 4,60 (d, J=9,3 Гц, 1H), 4,17 (m, 1H), 4,05 (d, J=7,2 Гц, 1H), 3,34-3,30 (m, 1H), 3,02 (d, J=11,7 Гц, 1H), 2,47-2,40 (m, 1H), 2,19-2,12 (m, 1H), 2,04-1,66 (m, 2H), 1,26-1,23 (m, 12H). 13C-NMR (75 МГц, CDCl3): δ 173,7, 171,1, 167,0, 80,6, 68,8, 62,0, 60,2, 47,2, 42,9, 22,2, 21,7, 21,6, 20,9, 17,5.
Пример 12
Синтез гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12)
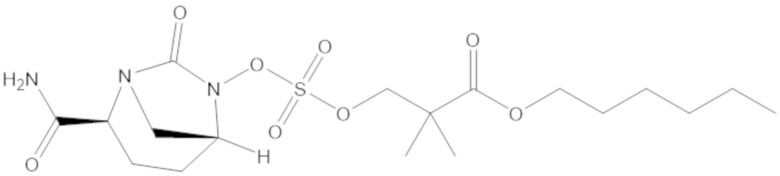
Стадия 1: Реакция получения гексил 3-гидрокси-2,2-диметилпропаноата (12a).
Сделана ссылка на публикацию заявки Германии № DE3045373. Смесь 3-гидрокси-2,2-диметилпропионовой кислоты (4,7 г, 40 ммоль), 1-гексанола (70 мл) и концентрированной серной кислоты (или дымящейся серной кислоты; 1 мл) нагревали до 80°C и встряхивали в течение ночи. После обеспечения охлаждения смесь концентрировали в вакууме (необходим высоковакуумный насос), а остаток разделяли на EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Водную смесь промывали H2O (50 мл), насыщенным NaHCO3 (50 мл) и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта (12a) в виде масла. Продукт использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,04-3,98 (m, 2H), 3,47-3,45 (m, 2H), 2,26 (s, 1H), 1,58-1,32 (m, 2H), 1,32-1,23 (m, 6H), 1,12 (s, 3H), 1,11 (s, 3H).
Стадия 2: Реакция получения гексил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (12b).
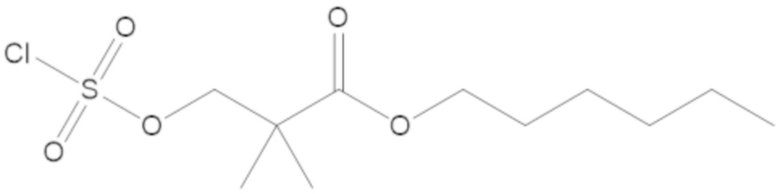
Раствор сульфурилхлорида (2,1 мл, 29,7 ммоль) в Et2O (40 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 30 мин по каплям добавляли раствор гексил 3-гидрокси-2,2-диметилпропаноата (12a) (3,0 г, 14,8 ммоль) и пиридина (1,4 мл, 17,8 ммоль) в Et2O (15 мл). Колбу промывали Et2O (3×5 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения с помощью TLC (30 мин; 30% EA/гекс). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением продукта (12b) (3,7 г, 83% выход). Смесь хранили при -78°C и сразу использовали для следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): 4,50 (s, 2H), 4,13 (t, J=6,8 Гц, 2H), 1,69-1,60 (m, 2H), 1,40-1,27 (m, 12H), 0,91-0,87 (m, 3H).
Стадия 3: Реакция получения гексил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (12).
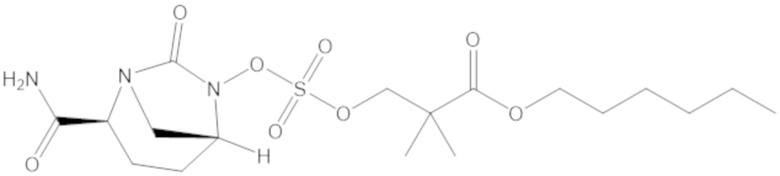
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (673 мг, 3,6 ммоль) растворяли в THF (35 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (4,0 мл, 4,0 ммоль), и встряхивали в течение 20 мин. В реакционную смесь быстро добавляли неразбавленный гексил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (12b) (1,1 г, 3,6 ммоль). Шприц промывали THF (3×3 мл), а смыв также добавляли в смесь. Спустя 10 мин реакционную смесь нагревали до 23°C и встряхивали до завершения реакции, что определяли с помощью TLC и LC-MS. В смесь добавляли EtOAc (50 мл) и насыщенный водный NaHCO3 (50 мл). Слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (50 мл), водой (3×50 мл), рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с последующей высокоэффективной жидкостной хроматографией с получением продукта (12) (44 мг, 3%) в виде твердого вещества. LC-MS: 450,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,49 (s, 1H), 5,71 (s, 1H), 4,71 (d, J=8,7 Гц, 1H), 4,60 (d, J=9,3 Гц, 1H), 4,17-4,04 (m, 4H), 3,34-3,30 (m, 1H), 3,04-3,00 (d, J=12,6 Гц, 1H), 2,47-2,40 (m, 1H), 2,18-2,13 (m, 1H), 2,01-1,79 (m, 2H), 1,66-1,59 (m, 2H), 1,37-1,27 (m, 12H), 0,91-0,86 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 174,3, 171,1, 167,0, 80,5, 65,6, 62,0, 60,2, 47,2, 43,0, 31,5, 28,6, 25,6, 22,6, 22,3, 21,8, 20,9, 17,6, 14,1.
Пример 13
Синтез гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13)
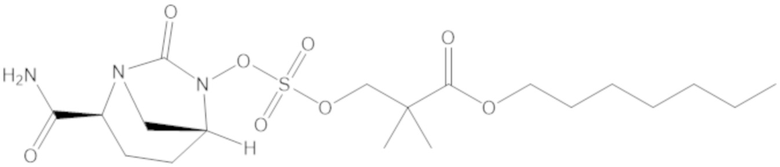
Стадия 1: Реакция получения гептил 3-гидрокси-2,2-диметилпропаноата (13a).
Сделана ссылка на публикацию заявки Германии № DE3045373. Смесь 3-гидрокси-2,2-диметилпропионовой кислоты (4,7 г, 40 ммоль), 1-гептанола (70 мл) и концентрированной серной кислоты (или дымящейся серной кислоты; 1 мл) нагревали до 80°C и встряхивали в течение ночи. После обеспечения охлаждения смеси смесь концентрировали в вакууме (необходим высоковакуумный насос), а остаток разделяли на EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Водный раствор промывали H2O (50 мл), насыщенным NaHCO3 (50 мл) и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта (13a) в виде масла. Продукт использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,31 (t, J=6,5 Гц, 2H), 3,77 (s, 2H), 1,87-1,81 (m, 2H), 1,53-1,50 (m, 8H), 1,41 (s, 6H), 1,12-1,08 (m, 3H).
Стадия 2: Реакция получения гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (13b).
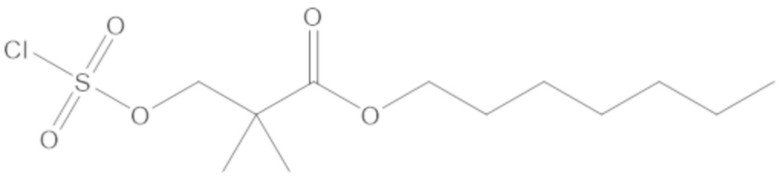
Раствор сульфурилхлорида (2,0 мл, 27,7 ммоль) в Et2O (40 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 30 мин по каплям добавляли раствор гептил 3-гидрокси-2,2-диметилпропаноата (13a) (3,0 г, 13,9 ммоль) и пиридина (1,4 мл, 16,6 ммоль) в Et2O (15 мл). Колбу промывали Et2O (3×5 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения, что контролировали с помощью TLC (30 мин; 30% EA/гекс). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (13b) (3,3 г, 75%). Смесь хранили при -78°C и сразу использовали для следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,46 (s, 2H), 4,11-4,00 (m, 2H), 1,64-1,55 (m, 2H), 1,26-1,24 (m, 8H), 0,85-0,81 (m, 3H).
Стадия 3: Реакция получения гептил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (13).
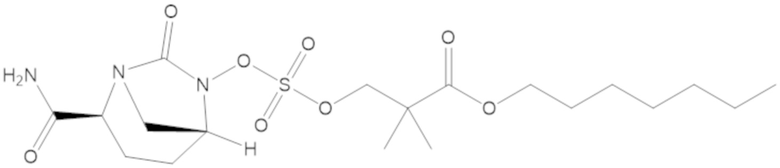
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (673 мг) растворяли в THF (35 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (4,0 мл, 4,0 ммоль), и встряхивали в течение 20 мин. В реакционную смесь быстро добавляли неразбавленный гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (13b) (1,3 г, 4,0 ммоль). Шприц промывали THF (3×3 мл), а смыв также добавляли в смесь. Спустя 10 мин реакционную смесь нагревали до 23°C и встряхивали до завершения, что определяли с помощью TLC и LC-MS. В смесь добавляли EtOAc (50 мл) и насыщенный водный NaHCO3 (50 мл). Водные и органические слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (50 мл), водой (3×50 мл), рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с последующей очисткой с применением высокоэффективной жидкостной хроматографии с получением продукта (13) (65 мг, 4%) в виде твердого вещества. LC-MS: 464,3 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (s, 1H), 5,71 (s, 1H), 4,71 (d, J=9,6 Гц, 1H), 4,60 (d, J=9,3 Гц, 1H), 4,18-4,04 (m, 4H), 3,34-3,29 (m, 1H), 3,02 (d, J=11,7 Гц, 1H), 2,47-2,40 (m, 1H), 2,19-2,11 (m, 1H), 2,01-1,79 (m, 2H), 1,66-1,59 (m, 2H), 1,37-1,26 (m, 14H), 0,90-0,86 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 174,3, 171,1, 167,0, 80,5, 65,6, 62,0, 60,2, 47,2, 43,0, 31,8, 29,0, 28,6, 25,9, 22,7, 22,2, 21,8, 20,9, 17,6, 14,2.
Пример 14
Синтез трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14)
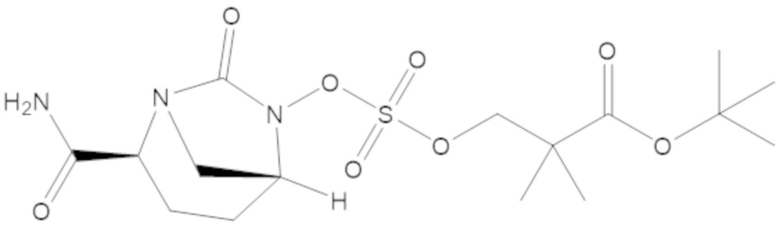
Стадия 1 и Стадия 2: Реакция получения трет-бутил 3-гидрокси-2,2-диметилпропаноата (14a).
соединение синтезировали в соответствии с публикацией международной заявки PCT № WO 2007116922. В охлажденный раствор трет-бутил метил малоната (4 г) в THF (100 мл) при 0°C в атмосфере Ar добавляли натрий гидрид (60% в минеральном масле; 2,0 г). Смесь встряхивали при 0°C в течение 10 мин. В смесь добавляли MeI (3,2 мл), и встряхивание продолжали в течение 3 ч (к этому времени смесь была при комнатной температуре). В смесь добавляли рассол и EtOAc, и органический слой разделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта (около 4,5 г), который использовали непосредственно на следующей стадии.
В течение 15 мин в раствор трет-бутил метил 2,2-диметил-малоната (2,2 г) в THF (100 мл) в атмосфере Ar порциями добавляли твердый три-трет-бутоксиалюмогидрид лития (7,1 г, 28 ммоль). Затем смесь нагревали с обратным холодильником и встряхивали в течение ночи. После охлаждения до комнатной температуры добавляли насыщенный раствор NH4Cl и EtOAc, и разделяли водные и органические слои. Органический слой промывали H2O и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением продукта (14a) (900 мг) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,50 (d, J=5,1 Гц, 2H), 2,53 (t, J=6,5 Гц, 1H), 1,45 (s, 9 H), 1,14 (s, 6H)
Стадия 3: Реакция получения трет-бутил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (14b).
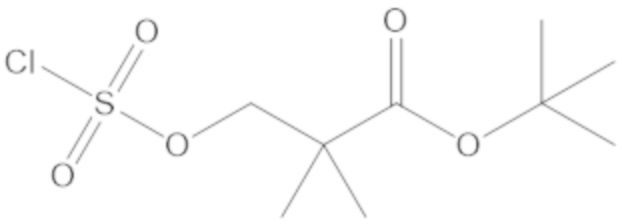
Раствор сульфурилхлорида (0,31 мл, 4,2 ммоль) в Et2O (6 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор трет-бутил 3-гидрокси-2,2-диметилпропаноата (14a) (0,49 г, 2,8 ммоль) и пиридина (0,25 мл, 3,1 ммоль) в Et2O (6 мл). После выявления с помощью TLC, что реакция не завершилась (10% EtOAc/гексаны), смесь встряхивали при -78°C в течение 90 мин и обеспечивали нагревание до 23°C. Смесь повторно охлаждали до -78°C, и добавляли дополнительный 1 эквивaлент сульфурилхлорида, встряхивали в течение 10 мин, и обеспечивали нагревание смеси до 23°C (примечание: после добавления и во время периода нагрева обеспечивали встряхивание смеси в общей сложности в течение 1 ч). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением трет-бутил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (14b) (961 мг, выход принимали по количеству) в виде прозрачного масла. 1H-NMR (300 МГц, CDCl3): δ 4,46 (fd, J=1,5 Гц, 2H), 1,47 (fd, J=1,2 Гц, 9H), 1,27 (s, 6H).
Стадия 4: Реакция получения трет-бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (14).
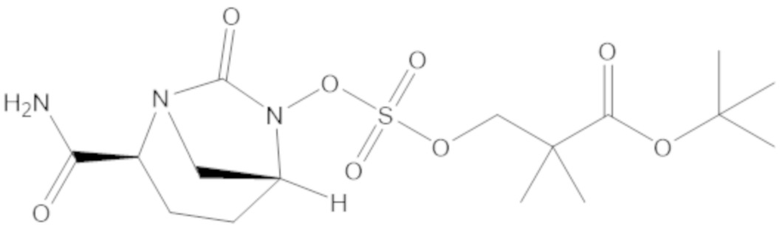
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (463 мг, 2,5 ммоль) растворяли в THF (25 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1,5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (2,8 мл, 2,8 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли неразбавленный трет-бутил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (14b) (0,75 г, 2,8 ммоль). Шприц промывали THF (3×3 мл), и эти смывы также быстро добавляли в смесь. Спустя 20 мин обеспечивали нагревание реакционной смеси до 23°C. После встряхивания в течение 70 мин реакция завершалась, что определяли с помощью TLC (70% EA/гексаны). Смесь охлаждали до 0°C, разбавляли EtOAc (50 мл) и гасили насыщенным водным NaHCO3 (50 мл). Водные и органические слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (50 мл), водой (3×50 мл) и рассолом (50 мл), затем сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (14) (368 мг, выход 35%) в виде твердого вещества. LC-MS: 422,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,50 (s, 1H), 5,85 (s, 1H), 4,66 (d, J=9 Гц, 1H), 4,56 (d, J=8,7 Гц, 1H), 4,17 (s, 1H), 4,04 (d, J=6,9 Гц, 1H), 3,33-3,29 (m, 1H), 3,02 (d, J=12 Гц, 1H), 2,46-2,39 (m, 1H), 2,17-2,12 (m, 1H), 2,00-1,79 (m, 2H), 1,45 (s, 9H), 1,23 (s, 3H), 1,21 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 173,3, 171,1, 167,0, 81,6, 80,9, 62,0, 60,2, 47,2, 43,4, 28,0, 22,2, 21,6, 20,8, 17,6.
Пример 15
Синтез 2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15)
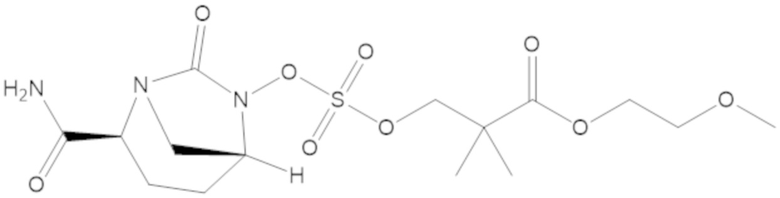
Стадия 1: Реакция получения 2-метоксиэтил 3-гидрокси-2,2-диметилпропаноата (15a).
3-гидрокси-2,2-диметилпропановую кислоту (1,2 г, 10,3 ммоль) и Cs2CO3 (3,4 г, 10,4 ммоль) суспендировали в DMF (25 мл) при 23°C, затем добавляли 2-бромэтилметиловый эфир (1,0 мл, 10,4 ммоль). Полученную в результате смесь встряхивали при 70°C в течение ночи. После охлаждения смесь фильтровали через слой Celite®. Фильтрат разбавляли EtOAc (150 мл), и смесь промывали водой (3×100 мл) и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (1:4-4:1) в качестве элюента с получением продукта (15a) (неочищенная масса 1,3 г) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 4,28 (t, J=4,8 Гц, 2H), 3,62-3,55 (m, 4H), 3,38 (s, 3H), 2,65 (t, J=6,0 Гц, 1H), 1,21 (s, 6H).
Стадия 2: Реакция получения 2-метоксиэтил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (15b).
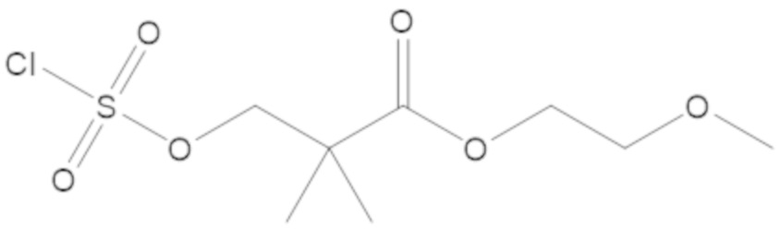
Раствор свежедистиллированного сульфурилхлорида (0,2 мл, 2,8 ммоль) в Et2O (7,0 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 11 мин по каплям добавляли раствор 2-метоксиэтил 3-гидрокси-2,2-диметилпропаноата (15a) (0,48 г, 2,7 ммоль) и пиридина (0,24 мл, 3,0 ммоль) в Et2O (1 мл). Колбу промывали Et2O (3×1 мл), который также добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения (контролировали с помощью TLC, 30% EtOAc/гекс, 30 мин). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением продукта (15b) (0,5 г, 67%) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки [Примечание: 1HNMR показал нужный продукт с остатком пиридина и вместе с исходным материалом].
Стадия 3: Реакция получения 2-метоксиэтил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (15).
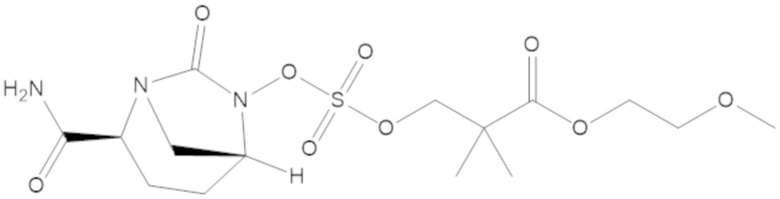
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (162 мг, 0,9 ммоль) растворяли в THF (2,5 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,3 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (1,0 мл, 1,0 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли 2-метоксиэтил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (15b) (0,3 г, 1,1 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание смеси до 23°C, и встряхивали в течение 30 минут. Смесь разбавляли EtOAc (40 мл) и водой. Водные и органические слои разделяли, а органический слой промывали водой (3×20 мл), и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (колонка 4 г) с использованием EtOAc/гексанов (3:7-1:0) в качестве элюента с получением твердого вещества с примесями. Продукт растворяли в Et2O (20 мл) с помощью обработки ультразвуком и осаждали гексанами. Полученное в результате твердое вещество фильтровали и сушили в вакууме с получением продукта (15) (72 мг, 19,4%) в виде твердого вещества. LCMS: м/з=424,3 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (br. s, 1H), 5,56 (br. s, 1H), 4,62 (dd, J=28,8, 8,7 Гц, 2H), 4,33-4,22 (m, 2H), 4,17 (br. s, 1H), 4,05 (d, J=6,9 Гц, 1H), 3,60 (t, J=4,6 Гц, 2H), 3,38 (s, 3H), 3,33 (d, J=11,1 Гц, 1H), 3,02 (d, J=12,0 Гц, 1H), 2,46-2,41 (m, 1H), 2,18-2,13 (m, 1H), 1,98-1,84 (m, 2H), 1,31 (s, 3H), 1,29 (s, 3H). 13C-NMR (75 МГц, CDCl3), δ 174,1, 170,8, 166,9, 80,2, 70,2, 64,1, 61,8, 60,0, 59,0, 47,1, 42,9, 22,1, 21,6, 20,7, 17,4.
Пример 16
Синтез оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16)
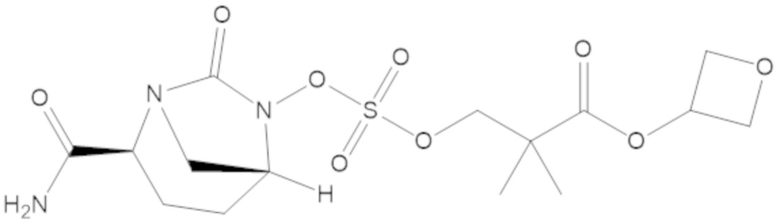
Стадия 1: Реакция получения оксетан-3-ил 3-гидрокси-2,2-диметилпропаноата (16a).
3-гидрокси-2,2-диметилпропановую кислоту (4,7 г, 40 ммоль) и Cs2CO3 (13,0 г, 40 ммоль) суспендировали в DMF (100 мл) при 23°C, затем добавляли 3-иодоксетан (7,4 г, 40 ммоль). Полученную в результате смесь встряхивали при 70°C в течение ночи. После охлаждения смесь разбавляли EtOAc (150 мл), и смесь промывали водой (3×100 мл) и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов с получением продукта (16a) (3,6 г, 51%) в виде масла.
Стадия 2: Реакция получения оксетан-3-ил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (16b).
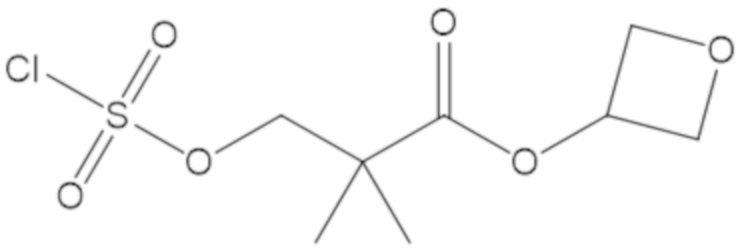
Раствор свежедистиллированного сульфурилхлорида (0,2 мл, 2,7 ммоль) в Et2O (3 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 11 мин по каплям добавляли раствор оксетан-3-ил 3-гидрокси-2,2-диметилпропаноата (16a) (0,46 г, 2,6 ммоль) и пиридина (0,2 мл, 2,7 ммоль) в Et2O (2 мл). Колбу промывали Et2O (3×1 мл), который также добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения (контролировали с помощью TLC, 30% EtOAc/гекс, 30 мин). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением продукта (16b) (0,5 г, 69%) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 5,50-5,46 (m, 1H), 4,94-4,89 (m, 2H), 4,65-4,60 (m, 2H), 4,52 (s, 2H), 1,72 (br. s, 1H), 1,36 (s, 6H).
Стадия 3: Реакция получения оксетан-3-ил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (16).
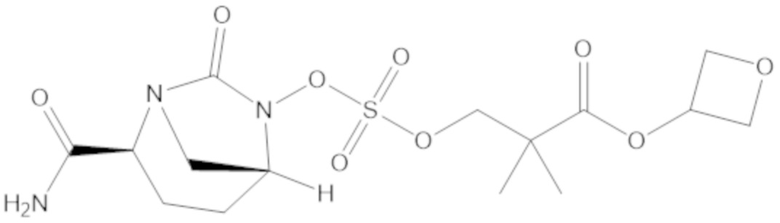
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (200 мг, 1,1 ммоль) растворяли в THF (3 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2 мл), добавляли DMPU, а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (1,2 мл, 1,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор оксетан-3-ил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (16b) (0,37 г, 1,4 ммоль) в THF (2 мл). После встряхивания при -78oC в течение 10 мин обеспечивали нагревание смеси до 23°C, и встряхивали в общей сложности в течение 1 ч. Смесь разбавляли EtOAc (40 мл) и H2O. Водные и органические слои разделяли, и органический слой промывали H2O (3×20 мл), рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (колонка 4 г) с использованием EtOAc/гексанов (3:7-1:0) в качестве элюента с получением масла. Масло растирали в порошок с Et2O с помощью обработки ультразвуком, и фильтрационный осадок промывали Et2O с получением продукта (16) (170 мг, 37%) в виде твердого вещества. LCMS: м/з=422,3 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (br. s, 1H), 5,54 (br. s, 1H), 5,48-5,44 (m, 1H), 4,89 (t, J=7,4 Гц, 2H), 4,79 (d, J=8,7 Гц, 1H), 4,66- 4,58 (m, 3H), 4,18 (br. s, 1H), 4,05 (d, J=6,9 Гц, 1H), 3,33 (d, J=12,3 Гц, 1H), 2,44-2,42 (m, 1H), 2,20-2,16 (m, 1H), 2,00-1,80 (m, 2H), 1,32 (s, 3H), 1,31 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 173,4, 170,8, 167,0, 80,0, 68,6, 76,6, 61,9, 60,2, 47,1, 42,7, 21,9, 21,6, 20,7, 17,4.
Пример 17
Синтез этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17)
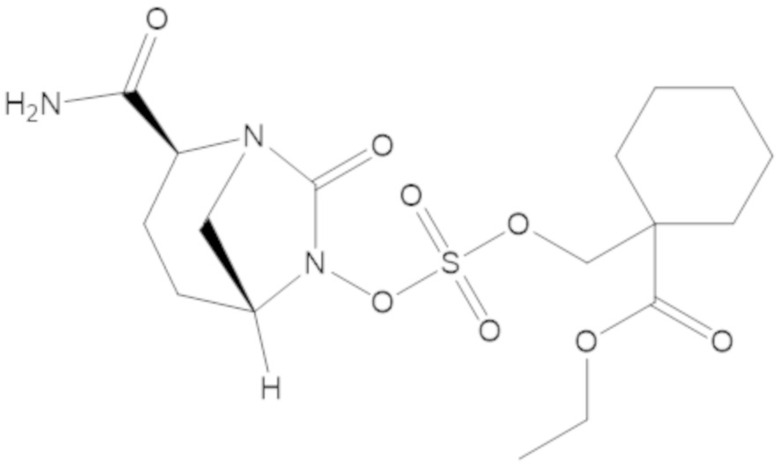
Стадия 1: Реакция получения этил-1-(((хлорсульфонил)окси)метил)циклогексанкарбоксилата (17a).
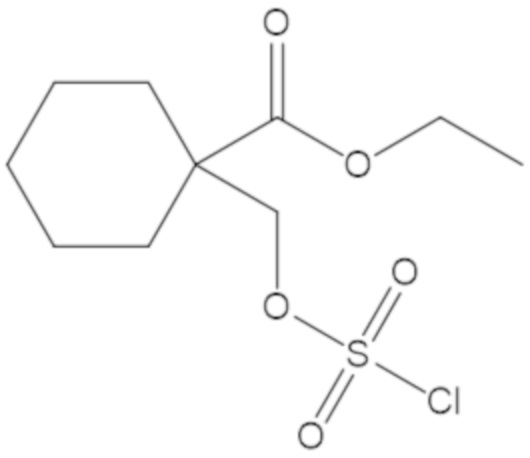
Раствор свежедистиллированного сульфурилхлорида (77 мкл, 1,1 ммоль) в Et2O (3 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 11 мин по каплям добавляли раствор этил-1-(гидроксиметил)циклогексанкарбоксилата (0,2 г, 1,0 ммоль) и пиридина (85 мкл, 1,1 ммоль) в Et2O (2 мл). Колбу промывали Et2O (3×1 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения (около 30 мин; контролировали с помощью TLC, 30% EtOAc/гекс). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения в виде масла, которое использовали непосредственно на следующей стадии без очистки. Вторая партия с использованием 476 мг исходного спирта давала 600 мг продукта (17a) (чистота приблизительно 85% по данным 1H-NMR).
Стадия 2: Реакция получения этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17).
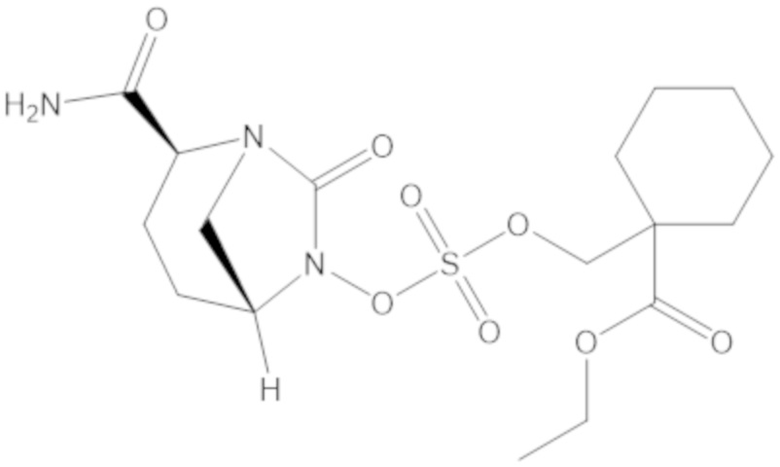
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,33 г, 1,8 ммоль) растворяли в THF (7 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3,5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В течение 20 мин по каплям добавляли 1,0 M раствора NaHMDS в THF (1,8 мл, 1,8 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли этил-1-(((хлорсульфонил)окси)метил)циклогексанкарбоксилат (17a) (0,51 г, 1,8 ммоль) в THF (2 мл). Спустя 10 мин встряхивания при -78°C обеспечивали нагревание смеси до 23°C и встряхивали в общей сложности в течение 1 ч. Реакционную смесь разбавляли EtOAc (40 мл) и H2O при -60°C. Водные и органические слои разделяли, а органический слой промывали H2O (3×20 мл) и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка (330 мг). Масло очищали с помощью колоночной хроматографии на силикагеле (колонка 4 г) с использованием EtOAc/гексанов (3:7-1:0) в качестве элюента с последующей очисткой с использованием препаративной HPLC (10-90% MeCN/H2O в течение 20 мин с использованием УФ-детектирования при 254/220 нМ) с получением продукта (17) (223 мг, 34%) в виде твердого вещества. LCMS: м/з=434,3 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,50 (br. s, 1H), 5,80 (br.s, 1H), 4,66 (dd, J=48,6, 12,8 Гц, 2H), 4,21-4,15 (m, 3H), 4,04 (d, J=6,9 Гц, 1H), 3,31 (d, J=3,0 Гц, 1H), 3,01 (d, J=11,7 Гц, 1H), 2,44-2,39 (m, 1H), 2,16-1,78 (m, 5H), 1,57-1,39 (m, 8H), 1,27 (t, J=7,1 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ 173,2, 171,0, 167,0, 80,2, 61,8, 61,1, 60,1, 47,1, 30,4, 30,0, 25,4, 22,2, 22,0, 20,7, 17,4, 14,1.
Пример 18
Синтез этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопентан-1-карбоксилата (18)
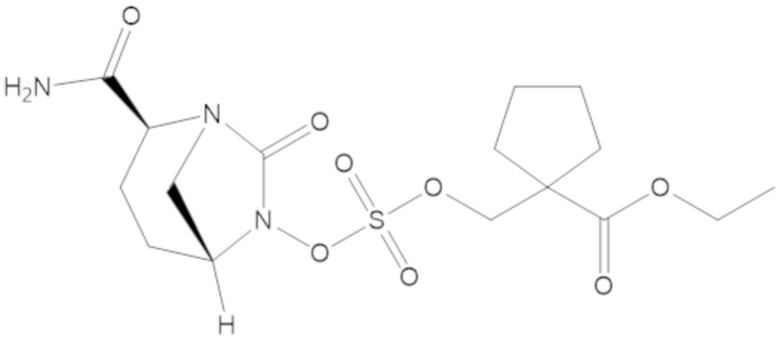
Стадия 1: Реакция получения этил-1-(((хлорсульфонил)окси)метил)циклопентан-1-карбоксилата (18a).
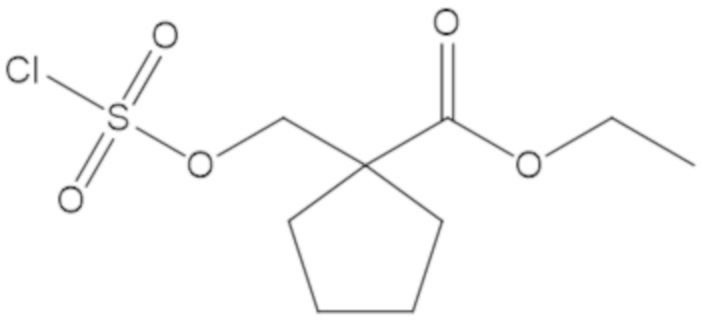
Раствор свежедистиллированного сульфурилхлорида (200 мкл, 2,7 ммоль) в Et2O (3 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 7 мин по каплям добавляли раствор этил-1-(гидроксиметил)циклопентанкарбоксилата (0,48 г, 2,7 ммоль) и пиридина (222 мкл, 2,7 ммоль) в Et2O (2 мл). Колбу промывали Et2O (2×1 мл), и оба смыва добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 1,5 ч. Осадок фильтровали, и фильтрационный осадок промывали Et2O (4 мл). Фильтрат концентрировали в вакууме с получением титульного соединения (18a) в виде масла, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: Реакция получения этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклопентан-1-карбоксилата (18).
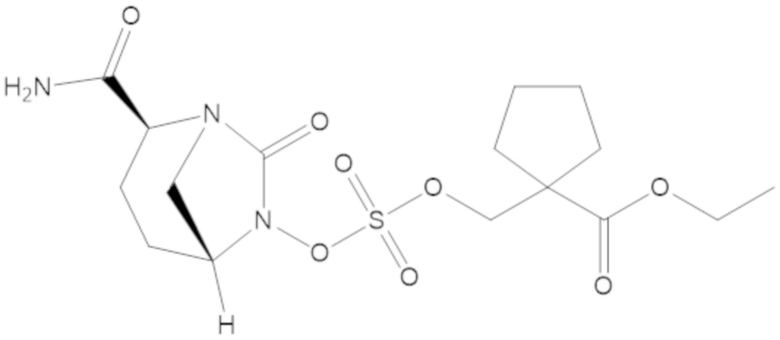
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (200 мг, 1,1 ммоль) растворяли в THF (5 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере Ar. В течение 10 мин по каплям добавляли 1,0 M раствора NaHMDS в THF (1,3 мл, 1,3 ммоль. Примечание: поскольку сульфонил хлорид содержит приблизительно 20% исходный спирт, добавляли дополнительно 0,2 экв. NaHMDS). Примечание: реакционную смесь погружали и доставали из охлаждающей ванны с получением раствора для встряхивания в противном случае это был гель. В реакционную смесь быстро добавляли этил-1-(((хлорсульфонил)окси)метил)циклопентанкарбоксилат (18a) (0,47 г, 1,7 ммоль) в THF (2×1 мл). Спустя 10 мин обеспечивали нагревание смеси до 23°C, и встряхивали в общей сложности в течение 2 ч. Реакционную смесь разбавляли EtOAc (40 мл) и рассолом при -60°C. Водные и органические слои разделяли, и органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (3:7-1:0) в качестве элюента, с последующей препаративной HPLC с обращенной фазой с получением титульного соединения (18) (62 мг, 14%) в виде твердого вещества. LCMS: м/з=420,3 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,55 (br. s, 1H), 6,15 (br. s, 1H), 4,69 (dd, J=20,9, 9,3 Гц, 2H), 4,20-4,10 (m, 3H), 4,02 (d, J=6,6 Гц, 1H), 3,29 (d, J=12,3 Гц, 1H), 3,01 (d, J=11,7, 1H), 2,40-2,36 (m, 1H), 2,14-1,66 (m, 11 ч), 1,24 (t, J=7,4 Гц, 3H). 13C-NMR (75 Гц, CDCl3): δ 174,4, 171,2, 167,0, 78,9, 61,8, 61,2, 60,1, 53,0, 47,0, 33,9, 32,9, 25,6, 25,5, 20,7, 17,5, 14,1.
Пример 19
Синтез этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19)
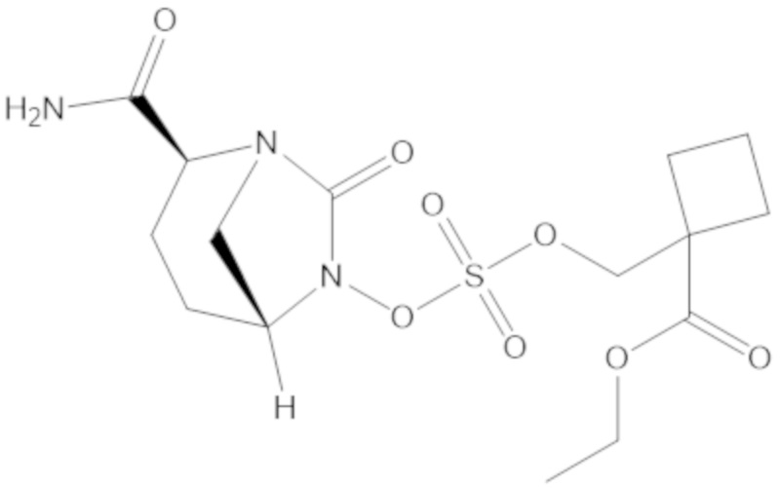
Стадия 1: Реакция получения этил-1-(((хлорсульфонил)окси)метил)циклобутанкарбоксилата (19a).
Раствор свежедистиллированного сульфурилхлорида (451 мкл, 6,2 ммоль) в Et2O (5 мл) охлаждали до -78°C в атмосфере Ar. В раствор сульфурилхлорида в течение 11 мин по каплям добавляли раствор этил-1-(гидроксиметил)циклобутанкарбоксилата (1,0 г, 6,1 ммоль) и пиридина (500 мкл, 6,2 ммоль) в Et2O (10 мл). Колбу промывали Et2O (3×1 мл), который также добавляли в реакционную смесь. Смесь встряхивали при -78°C, затем обеспечивали нагревание до окружающей температуры в течение 4 ч. Осадок фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (1,2 г, 76%) в виде масла, которое использовали непосредственно на следующей стадии без дополнительной очистки. Примечание: 1HNMR показал нужный продукт (19a) вместе с исходным материалом.
Стадия 2: Реакция получения этил-1-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19).
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,33 г, 1,8 ммоль) растворяли в THF (8 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В течение 10 мин по каплям добавляли 1,0 M раствора NaHMDS в THF (2 мл, 2,1 ммоль. Примечание: поскольку сульфонил хлорид содержит приблизительно 30% исходный спирт, дополнительно добавляли 0,3 экв. NaHMDS). В реакционную смесь быстро добавляли этил-1-(((хлорсульфонил)окси)метил)циклобутанкарбоксилат (19a) (1,0 г, 3,9 ммоль) в THF (2×1 мл). Спустя 10 мин обеспечивали нагревание смеси до 23°C, и встряхивали в течение 1 ч. Смесь разбавляли EtOAc (40 мл) и H2O при -60°C. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Масло очищали с помощью препаративной HPLC с получением твердого вещества (303 мг). Твердое вещество растворяли в DCM и фильтровали через фильтровальный шприц с получением продукта (19) (273 мг, 35%) в виде твердого вещества. LCMS: м/з=406,1 [M+H]+, 1H-NMR (300 МГц, CDCl3): δ 6,57 (br. s, 1H), 6,28 (br. s, 1H), 4,85 (dd, J=63,9, 9,3 Гц, 2H), 4,30-4,13 (m, 3H), 4,01 (d, J=7,2 Гц, 1H), 3,27 (d, J=10,8 Гц, 1H), 3,01 (d, J=11,7 Гц, 1H), 2,50-2,35 (m, 3H), 2,13-1,76 (m, 8H), 1,24 (t, J=7,3 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ173,1, 171,3, 167,2, 77,9, 61,8, 61,2, 60,7, 60,1, 47,0, 46,3, 27,1, 26,1, 20,7, 17,5, 15,6, 14,0.
Пример 20
Синтез этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (20)
Стадия 1: Синтез (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (20a).
Раствор дистиллированного сульфурилхлорида (0,61 мл, 7,5 ммоль) в Et2O (10 мл) охлаждали в азоте до -78°C. Затем в течение 1 ч с помощью шприца по каплям добавляли раствор этил 3-гидрокси-2,2-диметилпропаноата (2a) (1,0 г, 6,8 ммоль) и пиридина (0,55 мл, 6,8 ммоль) в Et2O (2,0 мл). Реакционную смесь в течение 1 ч встряхивали при -78°C, и обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение дальнейших 2 ч. После фильтрования смеси фильтрат концентрировали в вакууме с получением продукта (20a) в виде бесцветной жидкости (1,46 г, выход 87%). 1H-NMR (300 МГц, CDCl3): δ 4,50 (s, 2H), 4,19 (q, J=6,9 Гц, 2H), 1,31 (s, 6H), 1,28 (t, J=6,9 Гц, 3H).
Стадия 2: Синтез трет-бутил 4-((2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20b).
В смесь (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (20a) (10 г, 36,2 ммоль), трет-бутил 4-аминопиперидин-1-карбоксилата (7,26 г, 36,2 ммоль) в DCM (200 мл) добавляли HATU (13,76 г, 36,2 ммоль) и DIPEA (6,31 мл, 36,2 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение ночи. Смесь промывали насыщенным NH4Cl раствором, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1) в качестве элюента с получением продукта (20b) (10,3 г, выход 62%) в виде белого твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 7,36-7,44 (m, 5H), 6,55 (d, 1H, J=8,1 Гц), 5,05 (d, 1H, J=11,7 Гц), 4,90 (d, 1H, J=11,1 Гц), 4,02 (br, s, 1H), 3,87-3,99 (m, 2H), 3,29 (s, 1H), 3,01 (d, 1H), 2,85 (t, 2H), 2,64 (d, 1H), 2,37 (dd, 1H), 1,84-2,05 (m, 4H), 1,55-1,67 (m, 2H), 1,45 (s, 9H), 1,23-1,36 (m, 2H). MS (ESI) C24H34N4O5=459,1 (M+1)+.
Стадия 3: Синтез трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20c).
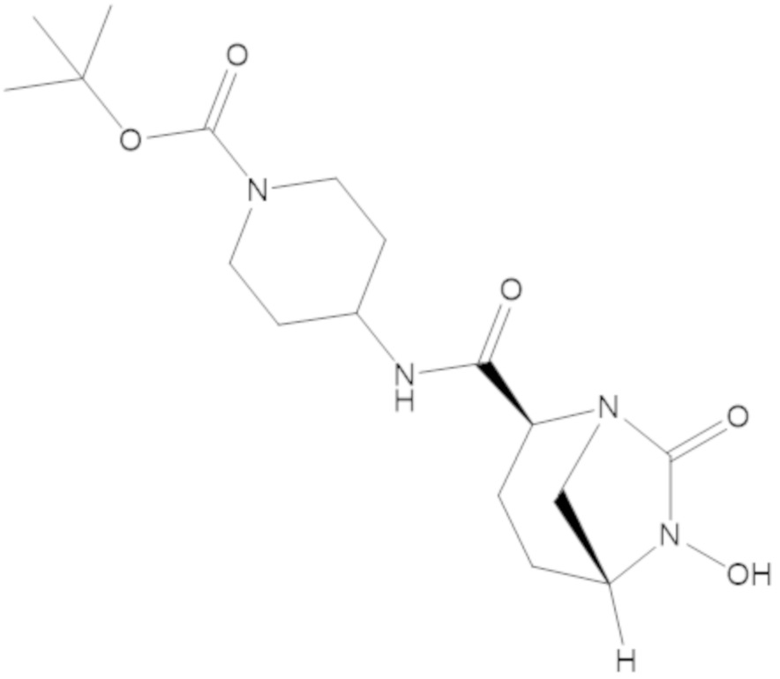
В раствор трет-бутил 4-((2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20b) (0,6 г, 1,31 ммоль) в MeOH (6 мл) добавляли 10% палладий на углероде (0,2 г). Реакционную смесь в течение 1 ч встряхивали при давлении водорода 1 атм. После фильтрования смеси через слой Celite® фильтрат концентрировали в вакууме с получением неочищенного продукта (20c) (0,48 г, выход 100%) который использовали непосредственно для следующей стадии. 1H-NMR (300 МГц, CDCl3): δ 6,62 (d, 1H, J=7,8 Гц), 3,86-4,01 (m, 4H), 3,75 (s, 1H), 3,17 (d, 1H), 2,91 (t, 2H), 2,81 (d, 1H), 2,42 (m, 1H), 2,13 (m, 1H), 1,88 (m, 4H), 1,74 (m, 1H), 1,45 s, 9H), 1,31 (m, 2H).
Стадия 4: Синтез трет-бутил 4-((2S,5R)-6-(((3-этокси-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20d).
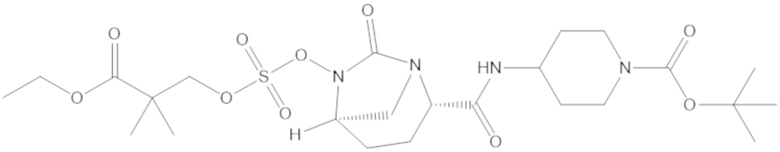
Трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (20c) (1,31 ммоль) растворяли в THF (7,0 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 1,31 мл, 1,31 ммоль), и смесь в течение 10 мин встряхивали при -78°C. Затем в реакционную смесь с помощью шприца добавляли раствор этил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (3a) (352 мг, 1,44 ммоль) в THF (1 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали насыщенным NaHCO3, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1) в качестве элюента с получением продукта (20d) (330 мг, выход 44%) в виде белой пены. 1H-NMR (300 МГц, CDCl3): δ 6,44 (d, 1H, J=8,1 Гц), 4,59-4,73 (dd, 2H, J=8,7 Гц), 3,89-4,23 (m, 7H), 3,28 (d, 1H), 2,83-2,92 (m, 3H), 2,42-2,49 (m, 1H), 2,14-2,17 (m, 1H), 1,80-1,97 (m, 4H), 1,46 (s, 9H), 1,58-1,23 (m, 11H). MS (ESI) C24H40N4O10S=577 (M+1)+.
Стадия 5: Синтез этил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (TFA соль) (20).
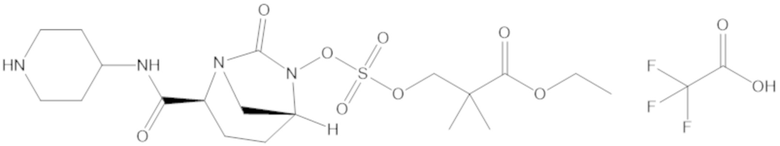
В смесь трет-бутил 4-((2S,5R)-6-(((3-этокси-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20d) (240 мг, 0,42 ммоль) в DCM (1,4 мл) добавляли трифторуксусную кислоту (1,4 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 30 минут. Анализ LC/MS показал, что исходный материал полностью израсходован. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием MeCN/H2O, содержащей 0,1% TFA (5-100%) с получением титульного соединения (20) (103 мг, выход 42%) в виде порошка грязно-белого цвета. 1H-NMR (300 МГц, CDCl3): δ 9,42 (br s, 1H), 9,06 (br s, 1H), 6,71 (d, 1H, J=7,8 Гц), 4,57-4,73 (dd, 2H, J=9,0 Гц), 3,99-4,19 (m, 5H), 3,48 (d, 2H), 3,26 (d, 1H), 3,00 (m, 2H), 2,88 (d, 1H), 1,82-2,39 (m, 7H), 1,23-1,30 (m, 9H). 13C-NMR (75 МГц, CDCl3): δ 174,5, 168,9, 167,3, 80,8, 62,0, 61,6, 60,4, 46,8, 44,9, 43,6, 43,3, 28,7, 22,3, 21,9, 20,9, 18,0, 14,4. 19F NMR (282 МГц, CDCl3): δ -75,8. MS (ESI) C19H32N4O8S=477 (M+1)+.
Аналитическую HPLC проводили на системе Agilent 1200 с использованием колонки C18 Phenomenex® (150×4,6 мм в/д). Подвижной фазой были MeCN и вода с линейным градиентом (0,1% TFA, от 5% MeCN до 100% MeCN за 15 мин). Скорость потока поддерживали 1 мл/мин, а элюент контролировали с помощью УФ-детектора при 220 и 254 нМ. Время удерживания HPLC: 7,31 мин.
Пример 21
Синтез 2-метоксиэтил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (21).
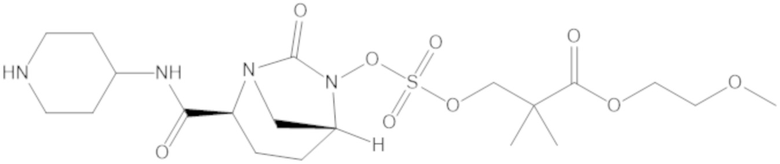
Стадия 1: Синтез 2-метоксиэтил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (21a).
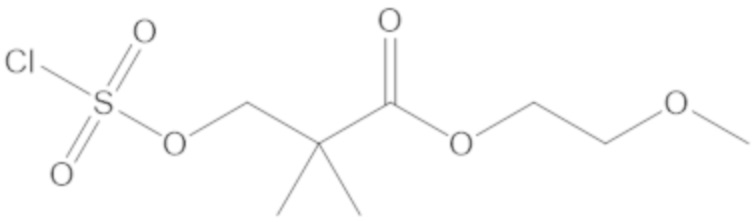
Раствор дистиллированного сульфурилхлорида (0,51 мл, 6,2 ммоль) в Et2O (10 мл) охлаждали в азоте до -78°C. Затем в течение 1 ч с помощью шприца по каплям добавляли раствор 2-метоксиэтил 3-гидрокси-2,2-диметилпропаноата (15a) (1,0 г, 5,68 ммоль) и пиридина (0,46 мл, 5,68 ммоль) в Et2O (2,0 мл). Реакционную смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 2 ч. После фильтрования смеси фильтрат концентрировали в вакууме с получением продукта (21a) в виде бесцветной жидкости (1,5 г, выход 96%). 1H-NMR (300 МГц, CDCl3): δ 4,40 (s, 2H), 4,29 (t, 3H), 3,59 (t, 3H), 3,37 (s, 3H), 1,32 (s, 6H).
Стадия 2: Синтез трет-бутил 4-((2S,5R)-6-(((3-(2-метоксиэтокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (21b).
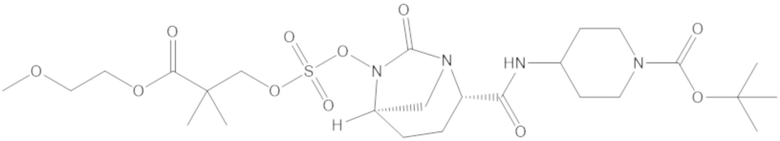
трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (20c) (3,26 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (6 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 3,59 мл, 3,59 ммоль), и смесь в течение 10 мин встряхивали при -78°C. Затем в реакционную смесь с помощью шприца добавляли раствор 2-метоксиэтил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (21a) (878 мг, 3,59 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали насыщенным NaHCO3, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1) в качестве элюента с получением продукта (21b) (0,96 г, выход 48%) в виде белой пены. MS (ESI) C25H42N4O11S=607,0 (M+1)+.
Стадия 3: Синтез 2-метоксиэтил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (TFA соль) (21).
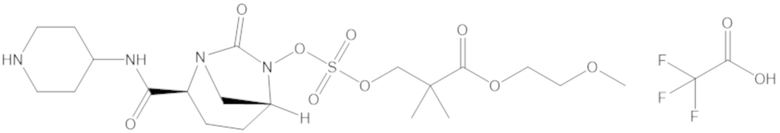
В смесь трет-бутил 4-((2S,5R)-6-(((3-(2-метоксиэтокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (21b) (0,86 г, 1,42 ммоль) в DCM (4,3 мл) добавляли трифторуксусную кислоту (4,3 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 30 минут. Анализ LC/MS показал, что исходный материал полностью израсходован. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием MeCN/H2O, содержащей 0,1% TFA (5-75%) с получением титульного соединения (21) (513 мг, выход 58%) в виде желтого порошка. 1H-NMR (300 МГц, CDCl3): δ 9,09 (br s, 1H), 8,75 (br s, 1H), 6,83 (d, 1H, J=7,8 Гц), 4,59-4,71 (dd, 2H, J=9,3 Гц), 3,99-4,36 (m, 5H), 3,60 (m, 2H), 3,50 (d, 2H), 3,39 (s, 3H), 3,30 (d, 1H), 3,02 (m, 2H), 2,89 (d, 1H), 1,87-2,40 (m, 7H), 1,25-1,30 (m, 9H). 13C-NMR (75 МГц, CDCl3): δ 174,4, 168,9, 167,4, 80,6, 70,6, 64,5, 62,0, 60,4, 59,3, 46,8, 44,9, 43,6, 43,2, 28,7, 22,4, 21,8, 20,9, 18,0. 19F NMR (282 МГц, CDCl3): δ -75,8. MS (ESI) C20H34N4O9S=507 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 6,75 мин.
Пример 22
Синтез 4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22).
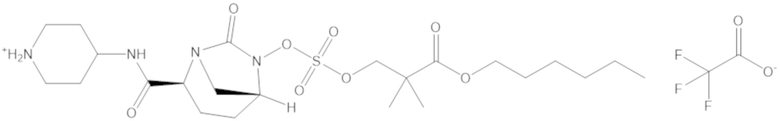
Стадия 1: Синтез гексил 3-гидрокси-2,2-диметилпропаноата (22a).
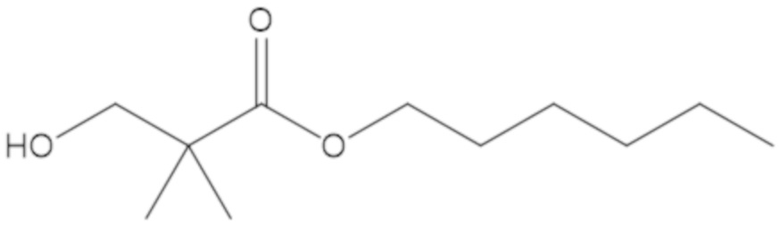
Смесь 3-гидрокси-2,2-диметилпропионовой кислоты (4,7 г, 40 ммоль),-1-гексанол (70 мл) и концентрированной серной кислоты (или дымящейся серной кислоты, 1 мл) нагревали до 80°C и встряхивали в течение ночи. После обеспечения охлаждения смесь концентрировали в вакууме (необходим высоковакуумный насос), а затем остаток разделяли на EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Водную фазу промывали H2O (50 мл), насыщенным NaHCO3 (50 мл) и рассолом (50 мл), а затем сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением продукта в виде масла. Продукт было трудно очищать с использованием хроматографии на силикагеле; и вследствие этого продукт дистиллировали при большом разрежении при 47°C с получением 4,92 г чистого продукта сложного эфира (22a) (выход 61%). 1H-NMR (300 МГц, CDCl3) δ 4,10 (td, J=6,7, 1,3 Гц, 2H), 3,55 (d, J=5,1 Гц, 2H), 2,42 (s, 1H), 1,64 (s, 1H), 1,72-1,56 (m, 1H), 1,35 (s, 1H), 1,31 (s, 6H), 1,27-1,11 (m, 6H), 0,95-0,84 (m, 3H). MS (ESI) C11H22O3=203 (M+1)+.
Стадия 2: Синтез гексил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (22b).
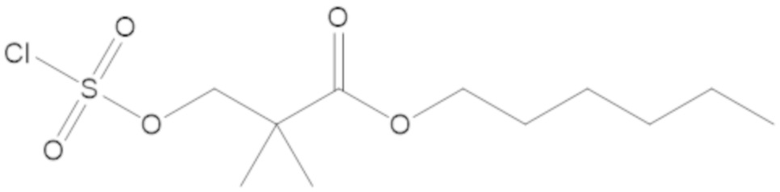
Раствор свежедистиллированного сульфурилхлорида (0,60 мл, 7,4 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере N2. В раствор сульфурилхлорида в течение 20 мин по каплям добавляли раствор гексил 3-гидрокси-2,2-диметилпропаноата (22a) (1,0 г, 4,94 ммоль) и пиридина (0,48 мл, 5,93 ммоль) в Et2O (5 мл). Колбу промывали Et2O (3×1 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения с помощью TLC (30 мин; 30% EA/гексан). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением неочищенного продукта (22b) в виде сплошной пены и использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,50 (s, 2H), 4,13 (t, J=6,8 Гц, 2H), 1,69-1,60 (m, 2H), 1,40-1,27 (m, 12H), 0,91-0,87 (m, 3H).
Стадия 3: Синтез трет-бутил 4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (22d).
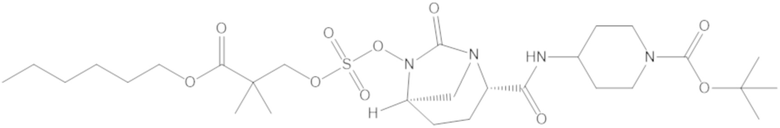
Гидроксамовую кислоту (2,39 ммоль) растворяли в THF (12 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3,4 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере N2. По каплям добавляли раствор NaHMDS в THF (2,4 мл, 1,0 M, 2,4 ммоль), и смесь встряхивали в течение 20 мин. В реакционную смесь быстро добавляли неразбавленный гексил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (22b) (0,973 г, 2,64 ммоль). Шприц промывали THF (3×4 мл), а смыв также добавляли в смесь. Спустя 10 мин реакционную смесь нагревали до комнатной температуры и встряхивали до завершения, что определяли с помощью TLC и LC-MS. В смесь добавляли EtOAc (30 мл) и насыщенный водный NaHCO3 (30 мл). Слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (30 мл), водой (3×20 мл) и рассолом (30 мл), а затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:9-1:0) в качестве элюента с последующей высокоэффективной жидкостной хроматографией с получением продукта (22d) (740 мг, выход 49% в течение 3 стадий) в виде сплошной пены. 1H-NMR (300 МГц, CDCl3) δ 6,43 (d, J=8,2 Гц, 1H), 4,76-4,64 (m, 1H), 4,60 (d, J=9,0 Гц, 1H), 4,19-4,03 (m, 5H), 3,98 (d, J=7,5 Гц, 2H), 3,28 (d, J=12,0 Гц, 1H), 2,90 (d, J=12,0 Гц, 2H), 2,45 (dd, J=14,8, 6,2 Гц, 1H), 2,14 (s, 1H), 1,97-1,84 (m, 3H), 1,62 (q, J=7,0 Гц, 11H), 1,46 (s, 9H), 1,34-1,19 (m, 14H), 0,88 (d, J=7,0 Гц, 3H). MS (ESI) C28H48N4O10S=633 (M+1)+.
Стадия 4: Синтез 4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (22).
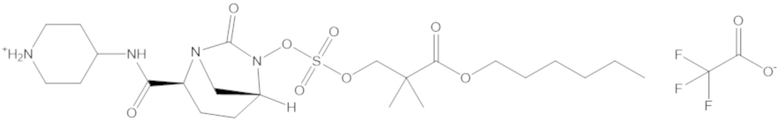
Трет-бутил 4-((2S,5R)-6-(((3-(гексилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (22d) (300 мг, 0,474 ммоль) растворяли в DCM (2 мл) и охлаждали до -10 oC. В раствор по каплям добавляли TFA (2 мл). Реакцию контролировали с помощью LCMS или TLC до завершения (около 10 мин). Растворитель удаляли in vacuo, а остаток очищали с использованием препаративной HPLC с MeCN/H2O, содержащей 0,1% TFA (20-100%), в качестве элюента с получением после лиофилизации титульного соединения (22) (212,4 мг, выход 84%) в виде пены. 1H-NMR (300 МГц, CDCl3) δ 9,08 (s, 1H), 8,74 (s, 1H), 6,88 (d, J=7,9 Гц, 1H), 4,70-4,51 (m, 2H), 4,23-3,92 (m, 6H), 3,47 (d, J=12,6 Гц, 2H), 3,31-3,19 (m, 1H), 2,95 (dd, J=19,6, 11,0 Гц, 3H), 2,34 (dd, J=15,0, 6,2 Гц, 1H), 2,10 (s, 2H), 1,91 (ddd, J=15,8, 12,6, 8,0 Гц, 1H), 1,61 (ddd, J=12,5, 8,1, 6,3 Гц, 3H), 1,40-1,16 (m, 14H), 0,91-0,80 (m, 3H). 13C-NMR (75 МГц, CDCl3) δ 174,2, 168,6, 167,1, 80,4, 65,5, 61,7, 60,1, 46,6, 44,7, 43,3, 42,9, 42,9, 31,4, 31,4, 28,5, 25,6, 25,5, 22,2, 22,2, 21,6, 20,7, 17,8, 14,0. 19F NMR (282 МГц, CDCl3) δ -75,6. MS (ESI) C23H40N4O8S=533 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 8,18 мин.
Пример 23
Синтез 4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23)
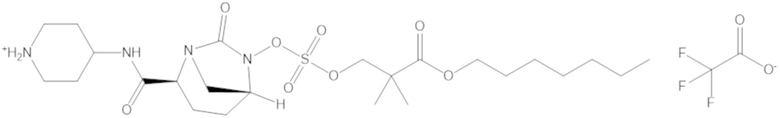
Стадия 1: Синтез гептил 3-гидрокси-2,2-диметилпропаноата (23a).
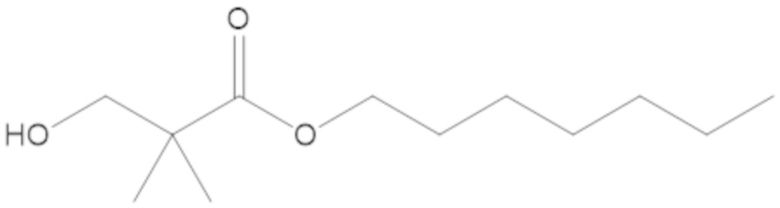
Смесь 3-гидрокси-2,2-диметилпропионовой кислоты (4,7 г, 40 ммоль), 1-гептанола (70 мл) и концентрированной серной кислоты (1 мл) нагревали до 80°C и встряхивали в течение ночи. После обеспечения охлаждения смесь концентрировали в вакууме (необходим высоковакуумный насос), а остаток разделяли на EtOAc (100 мл) и насыщенный водный NaHCO3 (100 мл). Водную фазу промывали H2O (50 мл), насыщенным NaHCO3 (50 мл) и рассолом (50 мл), а затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением продукта в виде масла. Продукт дистиллировали при большом разрежении при 65°C с получением титульного соединения (23a) в виде масла (6,7 г, 77% выход). 1H-NMR (300 МГц, CDCl3) δ 4,09 (td, J=6,7, 0,9 Гц, 2H), 3,55 (d, J=6,1 Гц, 2H), 2,43 (t, J=6,7 Гц, 1H), 1,60 (d, J=22,8 Гц, 4H), 1,3-1,58 (m, 6H), 1,27-1,14 (m, 6H), 0,92-0,83 (m, 3H).
Стадия 2: Синтез гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (23b)
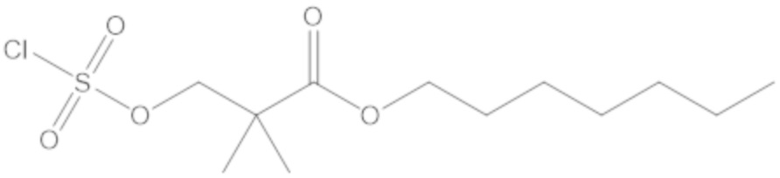
Раствор сульфурилхлорида (0,6 мл, 7,4 ммоль) в Et2O (15 мл) охлаждали до -78°C в атмосфере N2. В раствор сульфурилхлорида в течение 30 мин по каплям добавляли раствор гептил 3-гидрокси-2,2-диметилпропаноата (23a) (1,0 г, 4,94 ммоль) и пиридина (479 мкл, 5,93 ммоль) в Et2O (1 мл). Колбу промывали Et2O (3×1 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения, что контролировали с помощью TLC (30 мин; 30% EA/гексан). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (23b) (1,37 г, выход 92%). Смесь хранили при -78°C и использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3) δ 4,50 (s, 2H), 4,20-4,02 (m, 2H), 1,68 (m, 2H), 1,31 (d, J=3,1 Гц, 13H), 1,23 (s, 1H), 0,95-0,83 (m, 3H).
Стадия 3: Синтез трет-бутил 4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (23c).
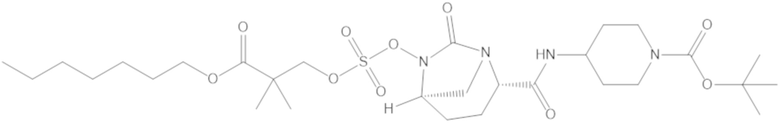
Гидроксамовую кислоту (1) (2,399 ммоль, в результате гидрогенизации без дополнительной очистки) растворяли в THF (12 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (2,4 мл, 2,4 ммоль), и встряхивали в течение 20 мин. В реакционную смесь быстро добавляли гептил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (23b) (0,79 г, 2,63 ммоль) в THF (5 мл). Шприц промывали THF (3×2 мл), а смыв также добавляли в смесь. Спустя 10 мин реакционную смесь нагревали до комнатной температуры и встряхивали до завершения, что определяли с помощью TLC и LC-MS. В смесь добавляли EtOAc (50 мл) и насыщенный водный NaHCO3 (50 мл). Водные и органические слои разделяли, а органический слой промывали насыщенным водным NaHCO3 (10 мл), водой (3×10 мл), рассолом (20 мл), а затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (5% to 95%) в качестве элюента, с получением 740,0 мг (49% выход) продукта (23c).
Стадия 4: Синтез 4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (23).
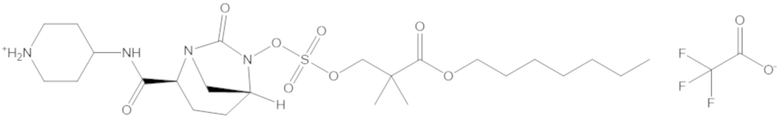
Охлаждали до -10°C трет-бутил 4-((2S,5R)-6-(((3-(гептилокси)-2,2-диметил-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (23c) (472,1 мг, 0,73 ммоль), растворенный в DCM (5 мл), в который по каплям добавляли TFA (5 мл). После завершения растворитель испаряли in vacuo, а остаток очищали с помощью препаративной HPLC с использованием MeCN/H2O, содержащей 0,1% TFA (20-100%), с получением титульного соединения (23) (390 мг, выход 81%) в виде сплошной пены. 1H-NMR (300 МГц, CDCl3) δ 8,93 (d, J=10,4 Гц, 1H), 8,62 (s, 1H), 6,90 (d, J=7,8 Гц, 1H), 4,70-4,51 (m, 2H), 4,18-3,92 (m, 6H), 3,48 (d, J=12,2 Гц, 2H), 3,26 (d, J=11,5 Гц, 1H), 2,98 (dt, J=24,4, 11,7 Гц, 3H), 2,34 (dd, J=15,1, 6,3 Гц, 1H), 2,10 (s, 2H), 1,60 (h, J=6,6 Гц, 3H), 1,24 (q, J=11,1, 9,8 Гц, 18H), 0,90-0,79 (m, 4H). 13C-NMR (75 МГц, CDCl3) δ 174,2, 168,6, 167,1, 80,3, 73,9, 65,4, 61,6, 60,1, 46,5, 44,6, 43,4, 42,8, 42,8, 31,6, 28,8, 28,4, 28,3, 25,8, 25,7, 22,5, 22,1, 22,1, 21,5, 20,6, 17,8, 14,0. 19F NMR (282 МГц, CDCl3) δ -75,7. MS (ESI) C24H42N4O8S=547 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 9,59 мин.
Пример 24
Синтез 4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетата (24)
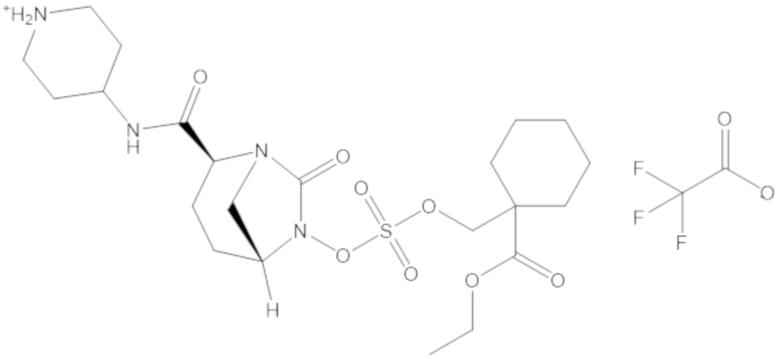
Стадия 1: Синтез этил-1-(гидроксиметил)циклогексанкарбоксилата (24a).

В THF (50 мл) растворяли диэтил циклогексан-1,1-дикарбоксилат (2,12 г, 9,29 ммоль), в который частями добавляли LiAl(OtBu)3 (5,9 г, 23,2 ммоль). Реакционную смесь встряхивали с обратным холодильником в течение ночи. Реакционную смесь охлаждали в ледяной ванне и осторожно обрабатывали 10% водным раствором KHSO4 (30 мл) со встряхиванием в течение 10 мин. Образовавшийся осадок фильтровали через слой Celite®. Фильтрат экстрагировали EtOAc (3×40 мл), а органическую фазу объединяли и промывали рассолом (50 мл), сушили над NaSO4, фильтровали и концентрировали in vacuo. Остаток очищали с помощью CombiFlash (SiO2) в 0-5% MeOH/DCM с получением нужного продукта (24) в виде масла (1,23 г, выход 71%). 1H-NMR (300 МГц, CDCl3) δ 4,19 (qd, J=7,1, 0,8 Гц, 2H), 3,62 (d, J=6,4 Гц, 2H), 3,46 (s, 1H), 2,00 (dt, J=11,5, 6,4 Гц, 4H), 1,57-1,22 (m, 9H).
Стадия 2: Синтез этил-1-(((хлорсульфонил)окси)метил)циклогексанкарбоксилата (24b).
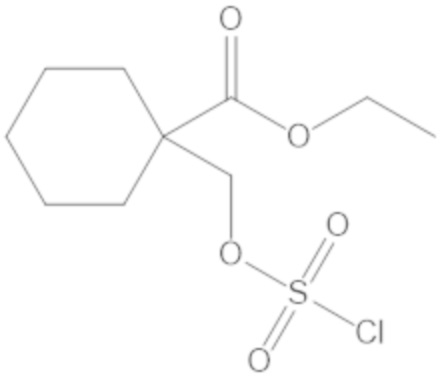
Раствор свежедистиллированного сульфурилхлорида (294 мкл, 3,63 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере азота. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор этил-1-(гидроксиметил)циклогексанкарбоксилата (24a) (0,615 г, 3,3 ммоль) и пиридина (294 мкл, 3,63 ммоль) в Et2O (6 мл). Колбу промывали Et2O (3×1 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C до завершения (около 30 мин; контролировали с помощью TLC, 30% EtOAc/гексан). Осадок фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (24b) в виде масла, 0,94 г в количественном выходе, которое использовали непосредственно на следующей стадии без очистки. 1H-NMR (300 МГц, CDCl3): δ 4,52 (s, 2H), 4,21 (q, J=7,1 Гц, 2H), 2,04 (s, 2H), 1,53-1,39 (m, 8H), 1,39-1,21 (m, 3H).
Стадия 3: Синтез трет-бутил 4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (24c).
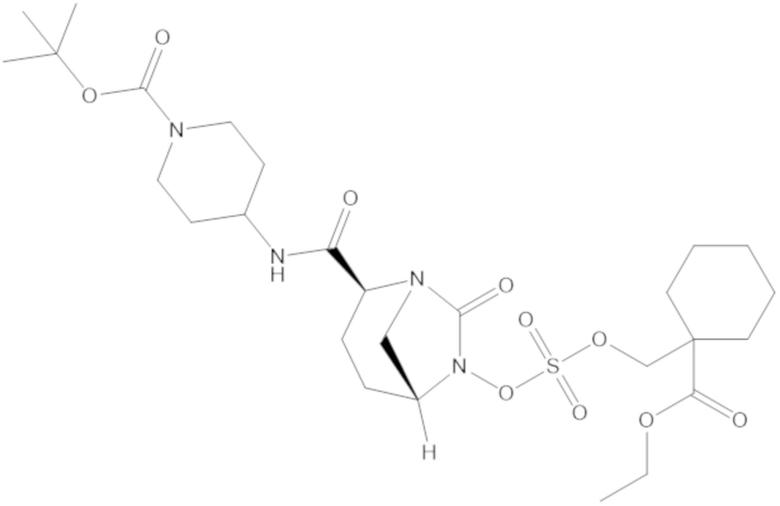
Гидроксамовую кислоту (2,73 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (7 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. В течение 20 мин по каплям добавляли 1,0 M раствора NaHMDS в THF (2,73 мл, 2,73 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли этил-1-(((хлорсульфонил)окси)метил)циклогексанкарбоксилат (24b) (0,94 г, 3,3 ммоль) в THF (2 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Реакционную смесь разбавляли EtOAc (60 мл) и H2O при -60°C. Водные и органические слои разделяли, а органический слой промывали H2O (3×30 мл) и рассолом (50 мл), затем сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением неочищенного остатка (330 мг). Масло очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (3:7-1:0) в качестве элюента с получением продукта (24c) (0,98 г, выход 59%) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 6,43 (d, J=8,2 Гц, 1H), 4,75 (d, J=9,2 Гц, 1H), 4,59 (d, J=9,1 Гц, 1H), 4,28-4,05 (m, 5H), 4,04-3,90 (m, 3H), 2,87 (t, J=12,4 Гц, 3H), 2,45 (dd, J=15,0, 5,7 Гц, 1H), 2,08-1,84 (m, 4H), 1,56 (d, J=10,6 Гц, 3H), 1,46 (s, 9H), 1,46-1,34 (m, 5H), 1,37-1,20 (m, 8H). MS (ESI) C27H44N4O10S: 617 (M+H)+.
Стадия 4: 4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-иум 2,2,2-трифторацетат (24).
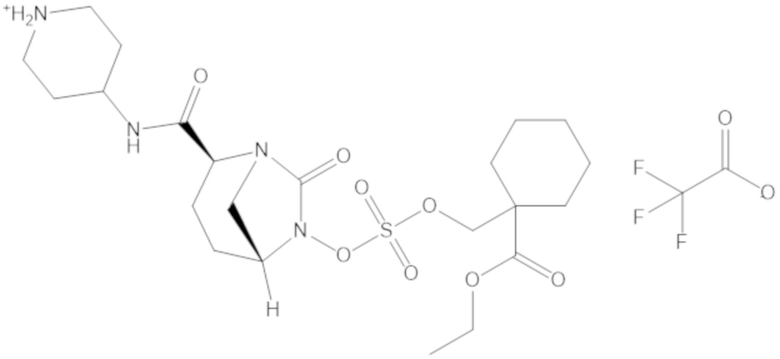
Охлаждали до -10°C (солевая ледяная ванна) трет-бутил 4-((2S,5R)-6-((((1-(этоксикарбонил)циклогексил)метокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат (24c) (403,3 мг, 0,65 ммоль), растворенный в DCM (4 мл), в который по каплям добавляли TFA (4 мл). Реакцию контролировали с помощью LCMS. Спустя 30 минут она завершалась. Растворитель удаляли in vacuo, а остаток очищали с помощью препаративной HPLC в MeCN/H2O, содержащей 0,1% TFA (20-100%), с получением титульного соединения (24) (263,7 мг, выход 78%) в виде сплошной пены. 1H-NMR (300 МГц, CDCl3) δ 9,02 (d, J=10,6 Гц, 1H), 8,66 (s, 1H), 6,87 (d, J=7,8 Гц, 1H), 4,70 (d, J=9,1 Гц, 1H), 4,54 (d, J=9,0 Гц, 1H), 4,16 (dtd, J=12,9, 6,7, 6,3, 3,1 Гц, 4H), 4,00 (q, J=8,7, 7,2 Гц, 3H), 3,47 (d, J=12,3 Гц, 2H), 3,26 (d, J=11,5 Гц, 1H), 2,95 (dd, J=25,8, 11,9 Гц, 3H), 2,35 (dd, J=15,3, 6,3 Гц, 1H), 2,11 (t, J=10,3 Гц, 4H), 1,99 (s, 2H), 2,08-1,87 (m, 2H), 1,89-1,72 (m, 4H), 1,54 (d, J=8,0 Гц, 5H), 1,25 (dd, J=14,4, 3,8 Гц, 2H). 13C-NMR (75 МГц, CDCl3) δ 173,2, 168,5, 167,0, 80,2, 61,7, 61,1, 60,0, 47,0, 46,6, 44,6, 43,3, 30,4, 29,9, 28,3, 25,3, 22,4, 22,2, 22,0, 20,6, 17,7, 14,1. 19F NMR (282 МГц, CDCl3) δ -75,7. MS (ESI) C22H36N4O8S=517 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 8,15 мин.
Пример 25
Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (25)
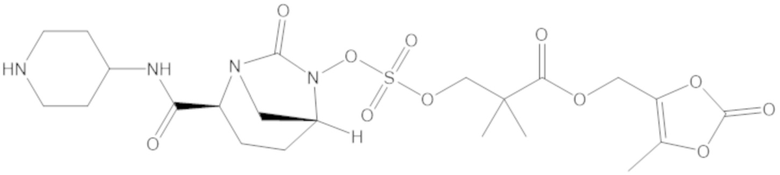
Стадия 1: Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-гидрокси-2,2-диметилпропаноата (25a).
Во встряхиваемый раствор 3-гидрокси-2,2-диметилпропановой кислоты (4,0 г, 33,9 ммоль) и карбоната калия (4,68 г, 33,9 ммоль) в DMF (45 мл) при 0°C по каплям в течение 1 ч добавляли 4-(гидроксиметил)-5-метил-1,3-диоксол-2-он (5,03 г, 33,9 ммоль) в DMF (5 мл). Реакционную смесь встряхивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtOAc и промывали водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:4-2:3) в качестве элюента с получением продукта (25a) в виде желтой жидкости (1,6 г, выход 21%). 1H-NMR (300 МГц, CDCl3): δ 4,86 (s, 2H), 3,58 (s, 2H), 2,18 (s, 3H), 1,20 (s, 6H).
Стадия 2: Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (25b).
Раствор дистиллированного сульфурилхлорида (0,61 мл, 7,53 ммоль) в Et2O (15 мл) охлаждали в азоте до -78°C. Добавляли раствор (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-гидрокси-2,2-диметилпропаноата (25a) (1,48 г, 6,43 ммоль) в Et2O (1 мл). После этого в течение периода 1 ч добавляли раствор пиридина (0,55 мл, 6,86 ммоль) в Et2O (1 мл). Реакционную смесь встряхивали при -78°C в течение 1 ч. После фильтрования смеси фильтрат концентрировали в вакууме с получением продукта (25b) в виде желтого масла (1,6 г, выход 76%). 1H-NMR (300 МГц, CDCl3): δ 4,90 (s, 2H), 4,49 (s, 2H), 2,19 (s, 3H), 1,33 (s, 6H).
Стадия 3: Синтез трет-бутил 4-((2S,5R)-6-(((2,2-диметил-3-((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (25c).
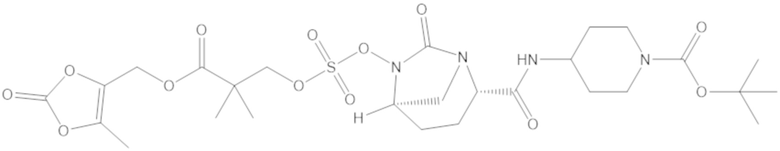
трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилат(20c) (2,18 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (6 мл), а полученный в результате раствор охлаждали в азоте до -78°C. По каплям добавляли раствор NaHMDS в THF (1M, 2,62 мл, 2,62 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца добавляли раствор (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (25b) (106b) (0,86 г, 2,62 ммоль) в THF (1 мл). После встряхивания в течение 1 ч при -78°C реакционную смесь разбавляли EtOAc и промывали насыщенным NaHCO3, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:3-1:1) в качестве элюента с получением продукта (25c) в виде желтой пасты (0,44 г, выход 31%). 1H-NMR (300 МГц, CDCl3): δ 6,73 (d, 1H, J=8,1 Гц), 4,78-4,98 (m, 3H), 4,47 (d, 1H, J=8,7 Гц), 3,93-4,15 (m, 5H), 3,27 (d, 1H), 2,83-2,92 (m, 3H), 2,41-2,45 (m, 1H), 2,18 (s, 3H), 2,15 (m, 1H), 1,78-1,92 (m, 4H), 1,45 (s, 9H), 1,23-1,58 (m, 8H). MS (ESI) C27H40N4O13S=661 (M+1)+.
Стадия 5: Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 2,2-диметил-3-(((((2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)пропаноата (TFA соль) (25).
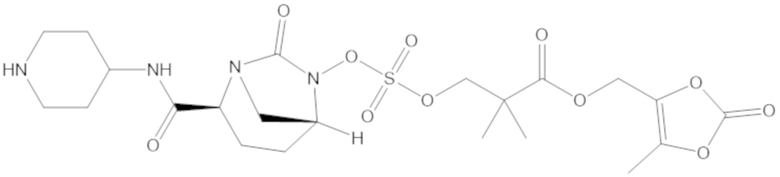
В раствор трет-бутил 4-((2S,5R)-6-(((2,2-диметил-3-((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)-3-оксопропокси)сульфонил)окси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (25c) (100 мг, 0,15 ммоль) в DCM (3 мл) добавляли трифторуксусную кислоту (0,4 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 1 ч. Анализ LC/MS показал, что расходования исходного материала. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием с использованием MeCN/H2O, содержащей 0,1% TFA (5-80%), с получением титульного соединения (25) в виде порошка грязно-белого цвета (55,2 мг, выход 55%). 1H-NMR (300 МГц, CDCl3): δ 9,43 (br s, 1H), 9,05 (br s, 1H), 7,14 (d, 1H, J=6,9 Гц), 4,99 (d, 1H, J=13,5 Гц), 4,95 (d, 1H, J=8,1 Гц), 4,78 (d, 1H, J=14,1 Гц), 4,41 (d, 1H, J=9,3 Гц), 4,14 (s, 1H), 4,06 (m, 1H), 3,98 (d, 1H, J=6,3 Гц), 3,47 (d, 2H), 3,29 (d, 1H), 3,04 (m, 2H), 2,86 (d, 1H), 1,82-2,40 (m, 11H), 1,29-1,33 (ds, 6H). 13C-NMR (75 МГц, CDCl3): δ 173,9, 169,1, 167,3, 152,9, 141,0, 133,7, 80,3, 62,0, 60,4, 54,8, 46,8, 44,7, 43,4, 43,3, 28,5, 22,3, 22,0, 20,8, 18,0, 9,6. 19F NMR (282 МГц, CDCl3): δ -75,9. MS (ESI) C22H32N4O11S=561 (M+1)+.
Аналитическую HPLC проводили на системе Agilent 1200 с использованием колонки C18 Phenomenex® (150×4,6 мм в/д). Подвижной фазой были MeCN и вода с линейным градиентом (0,1% TFA, от 5% MeCN до 100% MeCN за 15 мин). Скорость потока поддерживали 1 мл/мин, а элюент контролировали с помощью УФ-детектора при 220 нМ и 254 нМ. Время удерживания HPLC: 7,25 мин.
Пример 26
Синтез (2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфата (26)
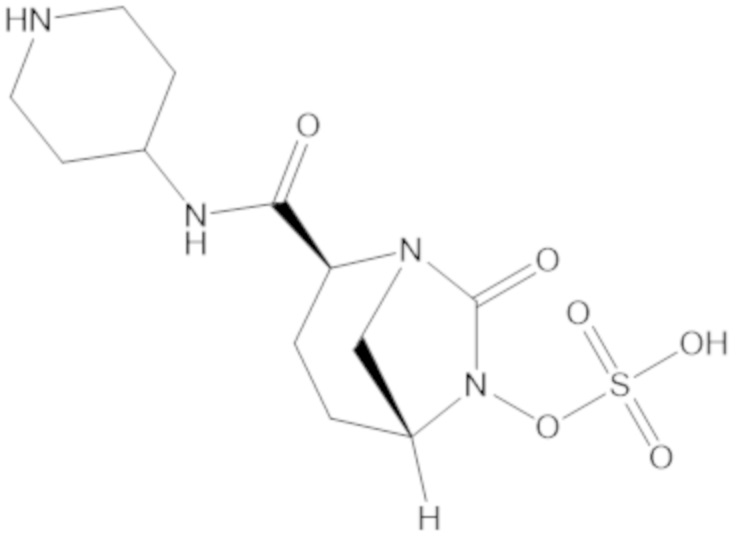
Стадия 1: Синтез тетрабутиламмоний (2S,5R)-2-((1-(трет-бутоксикарбонил)пиперидин-4-ил)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сульфата (26a).
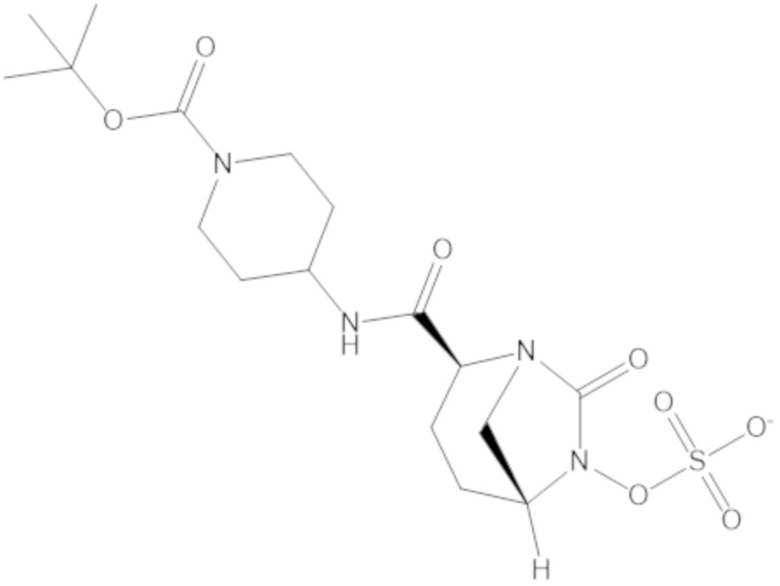
В раствор трет-бутил 4-((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)пиперидин-1-карбоксилата (20c) (1,92 г, 5,21 ммоль) в DCM (30 мл) добавляли триэтиламин (3 мл) и комплекс триоксида серы и пиридина (3,34 г, 21,0 ммоль). Реакционную смесь встряхивали при 35°C в течение ночи. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток встряхивали с 0,5 N водным раствором дигидрофосфата калия (30 мл) в течение 1 ч. Полученный в результате раствор экстрагировали три раза DCM. Объединенный органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением триэтиламиновой соли (2S,5R)-2-((1-(трет-бутоксикарбонил)пиперидин-4-ил)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сульфата (26a) (2,3 г, выход 80%).
В раствор приведенного выше продукта (26a) (2,2 г, 4,0 ммоль) в DCM (30 мл) добавляли 0,5 N водный гидрофосфат калия (12,4 мл) при 0°C. После встряхивания при 0°C в течение 10 мин добавляли тетрабутил аммоний гидросульфат (1,49 г, 4,4 ммоль). Полученный в результате раствор встряхивали при комнатной температуре в течение 30 минут. После разделения органического слоя водный слой экстрагировали три раза DCM. Объединенный органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента 10% MeOH в DCM с получением продукта (26a) (1,35 г, выход 49%) в виде белого твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 6,55 (d, 1H, J=8,1 Гц), 4,33 (br s, 1H), 4,03 (m, 2H), 3,91-3,95 (m, 1H), 3,86 (d, 1H), 3,48 (m, 1H), 3,25-3,31 (m, 10H), 2,85 (m, 2H), 2,73 (d, 1H), 2,39 (dd, 1H), 2,13 (m, 1H), 1,81-1,92 (m, 4H), 1,60-1,71 (m, 11H), 1,29-1,50 (m, 14H), 1,00 (t, 12H). MS (ESI) C17H28N4O8S=446,9 (M-1)+.
Стадия 2: Синтез (2S,5R)-7-оксо-2-(пиперидин-4-илкарбамоил)-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфата (26).
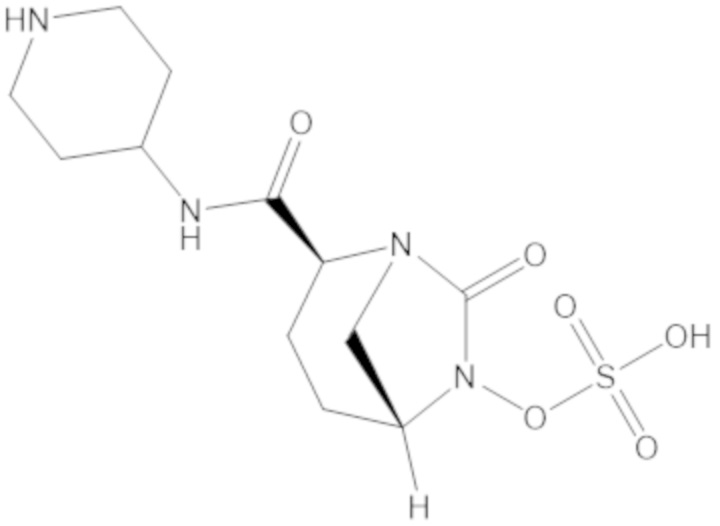
В раствор тетрабутиламмоний (2S,5R)-2-((1-(трет-бутоксикарбонил)пиперидин-4-ил)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сульфата (26a) (1,35 г, 1,96 ммоль) в DCM (15 мл) при -10°C добавляли TFA (15 мл). После расходования исходного материала по данным LC/MS смесь концентрировали в вакууме с получением неочищенного остатка. Остаток встряхивали с диэтиловым эфиром с получением осадка. Твердое вещество фильтровали и промывали два раза ацетоном с получением титульного соединения (26) в виде грязно-белого твердого вещества (0,56 г, выход 82%). 1H-NMR (300 МГц, DMSO-d6): δ 8,30 (br s, 2H), 8,21 (d, 1H, J=7,5 Гц), 3,97 (s, 1H), 3,86 (m, 1H), 3,71 (d, 1H), 3,25 (m, 2H), 2,97 (m, 4H), 2,06 (m, 1H), 1,83 (m, 3H), 1,64 (m, 4H). 13C-NMR (75 МГц, DMSO-d6): 169,5, 166,7, 59,6, 58,0, 46,7, 43,9, 42,5, 28,0, 20,6, 18,6. MS (ESI) C12H20N4O6S=346,9 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 1,60 мин.
Пример 27
Синтез этил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27).
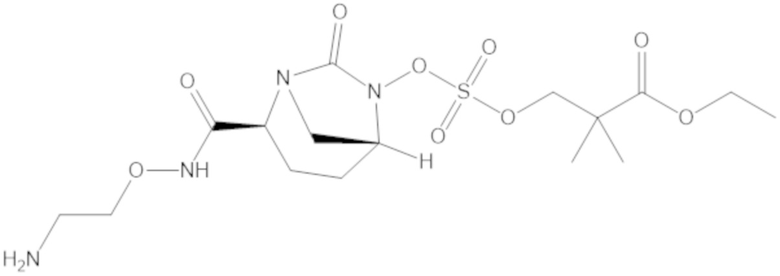
Стадия 1: Синтез N-(2-трет-Бок-аминоэтокси)фталимида (27a).
В смесь трет-бутил (2-бромэтил)карбамата (5,0 г, 22,3 ммоль) и N-гидроксифталимида (3,64 г, 22,3 ммоль) в ацетонитриле (80 мл) при комнатной температуре добавляли триэтиламин (7,46 мл, 53,5 ммоль). Реакционную смесь встряхивали при 70°C в течение 20 ч, а затем концентрировали. Смесь разбавляли этилацетатом и обильно промывали 1 N HCl, насыщенным NaHCO3 и водой. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного продукта (27a) в виде грязно-белого твердого вещества (3,8 г, выход 56%). 1H-NMR (300 МГц, CDCl3): δ 7,76-7,87-4,90 (m, 4H), 5,65 (m, 1H), 4,25 (t, 3H), 3,44 (t, 3H), 1,46 (s, 9H).
Стадия 2: Синтез трет-бутил (2-(аминоокси)этил)карбамата (27b).
В раствор N-(2-трет-Бок-аминоэтокси)фталимида (27a) (3,8 г, 12,4 ммоль) в EtOH (38 мл) при комнатной температуре добавляли гидразин моногидрат (0,63 мл, 13,0 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение 2 ч. Смесь фильтровали и промывали этилацетатом. Фильтрат концентрировали, и образовывалось белое твердое вещество. Белое твердое вещество удаляли путем фильтрации и промывали этилацетатом. Этот процесс повторяли еще три раза. Затем объединенный фильтрат концентрировали с получением продукта (27b) в виде желтой пасты (2,16 г, выход 99%). 1H-NMR (300 МГц, CDCl3): δ 5,46 (br s, 2H), 4,91 (br s, 1H), 3,70 (m, 2H), 3,35 (m, 2H), 1,44 (s, 9H).
Стадия 3: Синтез трет-бутил (2-(((2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамата (27c).
В смесь (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (20a) (3,19 г, 11,6 ммоль), трет-бутил (2-(аминоокси)этил)карбамата (27b) (2,06 г, 11,7 ммоль) в DCM (20 мл) добавляли HATU (4,39 г, 11,6 ммоль) и DIPEA (2,02 мл, 11,6 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение ночи. Смесь промывали насыщенным NH4Cl раствором, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1-3:1) в качестве элюента с получением продукта (27c) в виде белой пасты (4,0 г, выход 79%). 1H-NMR (300 МГц, CDCl3): δ 9,46 (br, s, 1H), 7,26-7,43 (m, 5H), 5,46 (t, 1H), 4,80-5,10 (dd, 2H, J=11,1 Гц), 2,75-3,97 (m, 8H), 1,61-2,33 (m. 4H), 1,43 (t, 9H). MS (ESI) C21H30N4O6=435 (M+1)+.
Стадия 4: Синтез трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамата (27d).
В раствор трет-бутил (2-(((2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамата (27c) (1,0 г, 2,30 ммоль) в MeOH (10 мл) добавляли 10% палладий на углероде (0,3 г). Реакционную смесь в течение 1 ч встряхивали при давлении водорода 1 атм. После фильтрования смеси через слой Celite® фильтрат концентрировали в вакууме с получением неочищенного продукта (27d) (0,79 г, выход 100%), который использовали непосредственно на следующей стадии.
Стадия 5: Синтез этил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27e).
Трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамат (27d) (0,79 г, 2,30 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (6 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 3,45 мл, 3,45 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца добавляли раствор этил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (3a) (675 мг, 2,76 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали насыщенным NaHCO3, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1-3:1) в качестве элюента с получением продукта (27e) в виде грязно-белой пены (0,68 г, выход 54%). 1H-NMR (300 МГц, CDCl3): δ 9,60 (br, s, 1H), 5,27 (br, t, 1H), 4,58-4,73 (dd, 2H, J=9,3 Гц), 3,02-4,22 (m, 10H), 1,62-2,40 (m. 4H), 1,44 (t, 9H), 1,26-1,28 (m, 9H). MS (ESI) C21H36N4O11S=553 (M+1)+.
Стадия 6: Синтез этил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27).
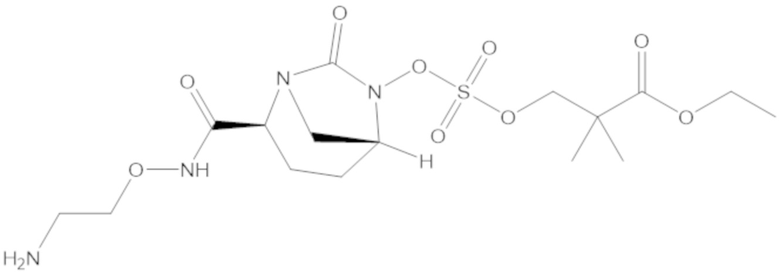
В смесь этил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (27e) (340 мг, 0,62 ммоль) в DCM (4 мл) добавляли трифторуксусную кислоту (4 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 30 минут. Анализ LC/MS показал полное расходование исходного материала. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием MeCN/H2O, содержащей 0,1% TFA (5-80%), с получением титульного соединения (27) в виде грязно-белой пены (243 мг, выход 72%). 1H-NMR (300 МГц, CDCl3): δ 8,10 (br s, 3H), 4,52-4,67 (dd, 2H, J=9,3 Гц), 3,12-4,21 (m, 10H), 1,93-2,23 (m, 4H), 1,19-1,29 (m, 9H). 13C-NMR (75 МГц, CDCl3): δ 174,7, 169,5, 167,6, 80,8, 73,0, 61,7, 60,7, 60,2, 46,5, 43,1, 38,5, 22,3, 21,9, 20,6, 18,5, 14,3. 19F NMR (282 МГц, CDCl3): δ -75,6. MS (ESI) C16H28N4O9S=453 (M+1)+.
Аналитическую HPLC проводили с использованием системы Agilent 1200 с колонкой C18 Phenomenex® (150×4,6 мм в/д). Подвижной фазой были MeCN и вода с линейным градиентом (0,1% TFA, от 5% MeCN до 100% MeCN за 15 мин). Скорость потока поддерживали 1 мл/мин, а элюент контролировали с помощью УФ-детектора при 220 нМ и при 254 нМ. Время удерживания HPLC: 7,10 мин.
Пример 28
Синтез 2-метоксиэтил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28)
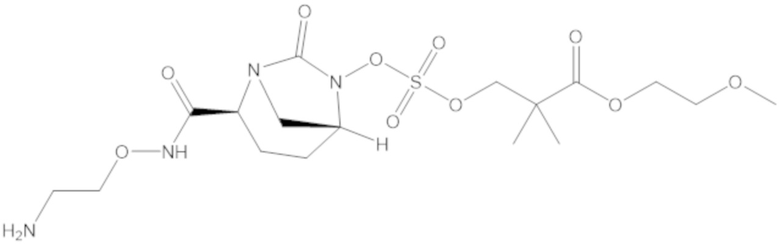
Стадия 1: Синтез 2-метоксиэтил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28a).
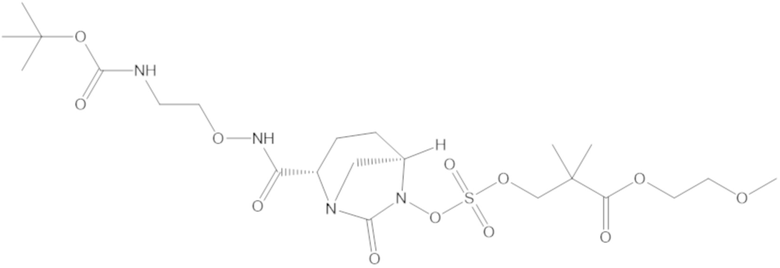
Трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамат (27d) (0,79 г, 2,30 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-он (6 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 2,76 мл, 2,76 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца добавляли раствор 2-метоксиэтил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (15b) (758 мг, 2,76 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали насыщенным NaHCO3, водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексана (1:1-3:1) в качестве элюента с получением продукта (28a) в виде белой пены (0,65 г, выход 49%). 1H-NMR (300 МГц, CDCl3): δ 9,60 (br, s, 1H), 5,30 (br, t, 1H), 4,60-4,72 (dd, 2H, J=8,7 Гц), 3,02-4,22 (m, 15H), 1,62-2,40 (m. 4H), 1,45 (t, 9H), 1,21-1,30 (m, 6H). MS (ESI) C22H38N4O12S=583 (M+1)+.
Стадия 2: Синтез 2-метоксиэтил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28).
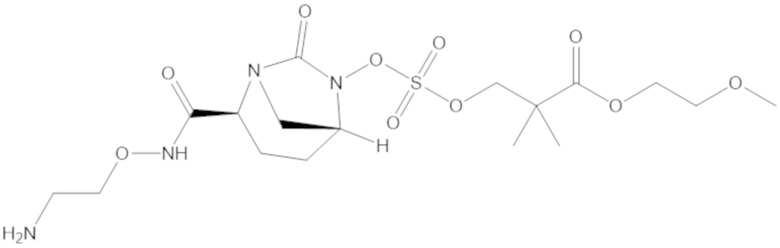
В смесь 2-метоксиэтил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (28a) (0,33 г, 0,57 ммоль) в DCM (4 мл) добавляли трифторуксусную кислоту (4 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 30 минут. Анализ LC/MS показал, что исходный материал полностью израсходован. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием с использованием MeCN/H2O, содержащей 0,1% TFA (5-80%), с получением титульного соединения (28) в виде грязно-белой пены (128 мг, выход 39%). 1H-NMR (300 МГц, CDCl3): δ 8,10 (br s, 3H), 4,53-4,68 (dd, 2H, J=8,7 Гц), 3,11-4,21 (m, 15H), 1,96-2,23 (m, 4H), 1,28 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 174,6, 169,5, 167,6, 80,6, 72,9, 70,6, 64,5, 60,6, 60,1, 59,2, 46,5, 43,2, 38,4, 22,2, 21,9, 20,5, 18,5. 19F NMR (282 МГц, CDCl3): δ 75,6. MS (ESI) C17H30N4O10S=483 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 6,59 мин.
Пример 29
Синтез соли TFA (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29)
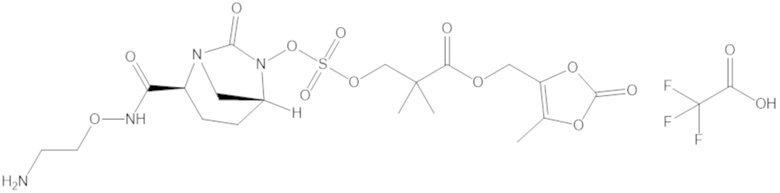
Стадия 1: Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29a).
Трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамат (27d) (0,80 г, 2,32 ммоль) растворяли в THF (23 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (3,3 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 2,32 мл, 2,32 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца добавляли раствор (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (25b) (916 мг, 2,8 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексана (0-99%) с получением продукта (29a) в виде грязно-белой пены (399,8 мг, выход 27%). 1H-NMR (300 МГц, хлороформ-d) δ 9,75 (s, 1H), 5,37 (s, 1H), 5,30 (d, J=0,7 Гц, 2H), 5,00-4,76 (m, 2H), 4,46 (d, J=9,3 Гц, 1H), 4,16-4,00 (m, 2H), 3,94 (s, 4H), 3,42 (s, 2H), 3,38-3,26 (m, 4H), 3,03 (d, J=12,2 Гц, 2H), 2,19 (s, 6H), 2,16 (d, J=5,5 Гц, 2H), 2,02 (d, J=14,0 Гц, 3H), 1,92 (s, 2H), 1,56 (m, 1H), 1,48-1,41 (m, 9H), 1,41-1,20 (m, 6H). MS (ESI) C24H36N4O14S: 637 (M+1)+.
Стадия 2: Синтез соли TFA (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29)
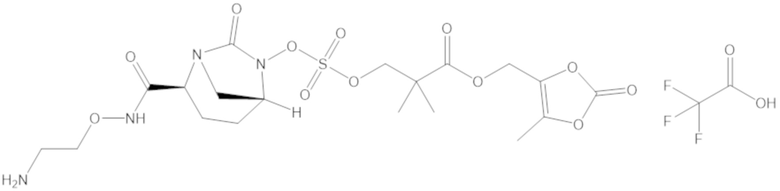
В смесь (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (29a) (0,33 г, 0,52 ммоль) в DCM (3 мл) добавляли трифторуксусную кислоту (0,3 мл) при -10°C. Реакционную смесь встряхивали при -10°C в течение 4 ч. Анализ LC/MS показал, что исходный материал полностью израсходован. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием с использованием MeCN/H2O, содержащей 0,1% TFA (0-80%), с получением титульного соединения (29) в виде грязно-белой пены (9,1 мг, выход 3,2%). 1H-NMR (300 МГц, MeCN-d3): δ 7,61 (s, 1H), 4,97-4,83 (m, 3H), 4,74-4,60 (m, 1H), 4,55 (d, J=9,3 Гц, 1H), 4,24-4,00 (m, 2H), 3,94-3,83 (m, 1H), 3,73-3,62 (m, 2H), 3,29 (d, J=12,3 Гц, 2H), 3,19 (s, 3H), 2,15 (dq, J=1,3, 0,6 Гц, 3H), 1,93 (s, 1H), 1,39-1,07 (m, 8H). 19F NMR (282 МГц, MeCN-d3): δ -76,1, -76,3. MS (ESI) C19H28N4O12S: 537 (M+1)+
Пример 30
Синтез соли TFA гексил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30)
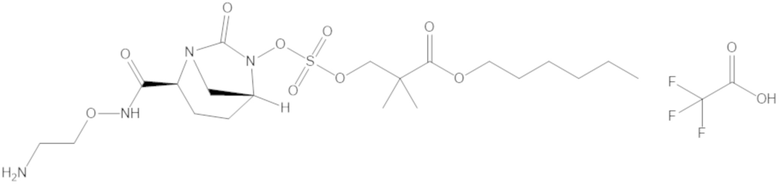
Стадия 1: Синтез гексил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30a).
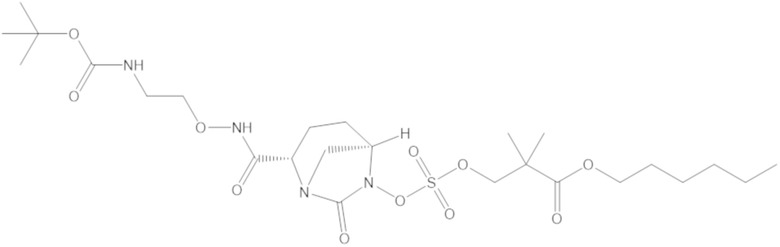
Трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамат (27d) (0,51 г, 1,48 ммоль) растворяли в THF (18 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. По каплям добавляли раствор NaHMDS в THF (1M, 1,9 мл, 1,9 ммоль), и смесь встряхивали при -78°C в течение 10 мин. Затем в реакционную смесь с помощью шприца добавляли раствор гексил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (730 мг, 2,42 ммоль) в THF (2 мл). Спустя 10 мин при -78°C обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Реакционную смесь разбавляли EtOAc и промывали водой и рассолом. Органический слой сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексана (0-99%) с получением продукта (30a) в виде грязно-белой пены (194,3 мг, выход 22%). 1H-NMR (300 МГц, CDCl3): δ 9,54 (s, 1H), 5,26 (s, 1H), 4,60 (d, J=9,0 Гц, 2H), 4,21-4,02 (m, 7H), 3,46 (d, J=16,3 Гц, 1H), 3,31 (s, 2H), 3,03 (d, J=12,0 Гц, 1H), 2,39 (dd, J=14,9, 6,3 Гц, 1H), 2,19 (s, 1H), 2,08-1,89 (m, 2H), 1,65-1,55 (m, 2H), 1,45 (s, 10H), 1,35-1,19 (m, 10H), 0,94-0,79 (m, 3H). MS (ESI) C25H44N4O11S: 607 (M-1)+.
Стадия 2: Синтез соли TFA гексил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30).
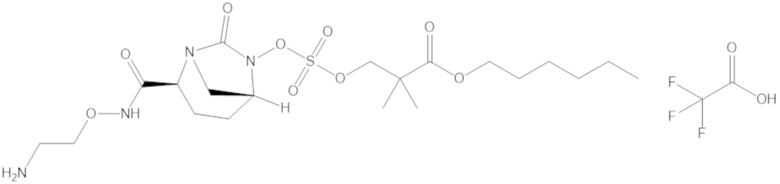
В раствор гексил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (30a) (144,3 мг, 0,237 ммоль) в DCM (3 мл) добавляли трифторуксусную кислоту (0,15 мл) при 0°C. Реакционную смесь встряхивали при 0°C в течение 1 ч. Анализ LC/MS показал, что исходный материал полностью израсходован. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью препаративной HPLC на колонке C18 с элюированием с использованием MeCN/H2O, содержащей 0,1% TFA (0-80%), с получением титульного соединения (30) в виде коричневого масла (68,1 мг, выход 56%). 1H-NMR (300 МГц, CDCl3): δ 7,80-7,73 (m, 2H), 6,94 (s, 1H), 4,57 (dt, J=28,2, 14,1 Гц, 2H), 4,22 (s, 1H), 4,08 (ddt, J=17,3, 13,2, 8,0 Гц, 6H), 3,96 (s, 2H), 3,28 (d, J=10,2 Гц, 2H), 2,10 (s, 1H), 1,93 (s, 1H), 1,62 (m, 2H), 1,28 (ddd, J=12,8, 6,5, 3,7 Гц, 12H), 1,19 (s, 2H), 0,94-0,82 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 176,6, 174,3, 169,2, 167,3, 161,6, 118,2, 114,3, 80,4, 73,7, 72,6, 65,5, 65,3, 60,3, 59,9, 46,3, 42,9, 42,8, 38,4, 31,4, 28,4, 28,3, 25,5, 25,5, 22,5, 22,1, 22,0, 21,6, 20,3, 18,2, 14,0. 19F NMR (282 МГц, CDCl3): δ -75,8. MS (ESI) C20H36N4O9S: 509 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 8,96 мин.
Пример 31
Синтез соли TFA гептил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (31).
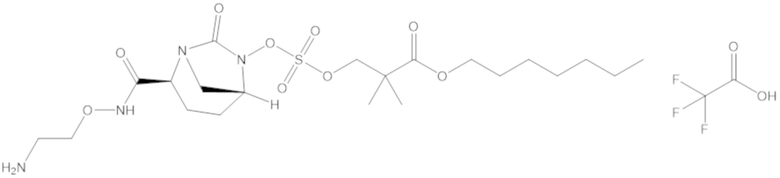
Стадия 1: Синтез гептил 3-(((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (31a).
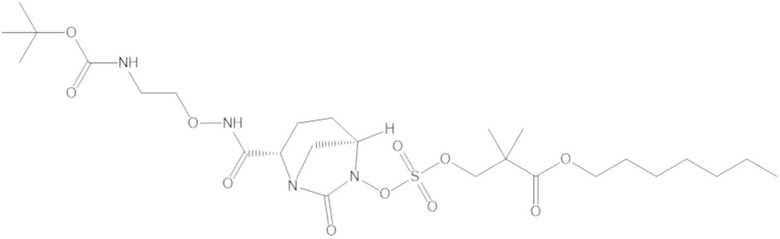
Способ, аналогичный способу стадии 1 примера 30, использовали для получения 274,0 мг титульного соединения (31a) (44% выход) в виде грязно-белого твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 9,75 (s, 1H), 5,37 (t, J=6,2 Гц, 1H), 4,72-4,52 (m, 2H), 4,09 (dddd, J=18,6, 10,4, 3,8, 2,1 Гц, 3H), 3,91 (t, J=4,8 Гц, 2H), 3,51-3,19 (m, 3H), 3,05 (d, J=12,1 Гц, 1H), 2,43-2,29 (m, 1H), 2,21-2,07 (m, 1H), 2,06-1,77 (m, 3H), 1,62 (t, J=6,9 Гц, 2H),1,40 (s, 9H), 1,40-1,17 (m, 16H), 0,92-0,81 (m, 3H). MS (ESI) C26H46N4O11S: 523 (M+1-Бок)+.
Стадия 2: Синтез соли TFA гептил 3-(((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (31).
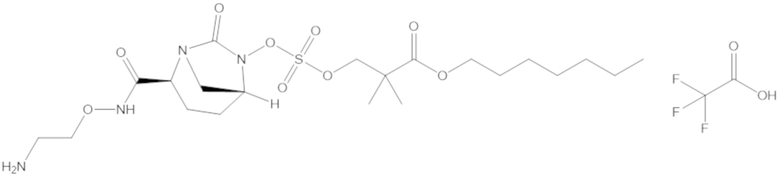
Способ, аналогичный способу стадии 2 примера 30, использовали для получения титульного соединения (31) (214,5 мг, 93%) в виде коричневого масла. 1H-NMR (300 МГц, CDCl3): δ 10,61 (s, 1H), 8,02 (s, 3H), 4,63 (d, J=8,7 Гц, 1H), 4,53 (d, J=8,9 Гц, 1H), 4,17 (d, J=13,3 Гц, 3H), 4,12-3,97 (m, 4H), 3,95 (s, 1H), 3,26 (s, 3H), 3,18-3,08 (m, 1H), 2,22 (s, 1H), 2,10 (s, 1H), 1,61 (q, J=7,0 Гц, 2H), 1,25 (m, 12H), 1,18 (s, 2H), 0,87 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 176,5, 174,3, 169,1, 167,3, 80,3, 73,7, 72,6, 65,5, 65,3, 60,3, 59,9, 46,3, 42,9, 42,8, 38,3, 31,7, 31,7, 28,9, 28,9, 28,4, 25,8, 25,8, 22,6, 22,1, 22,0, 22,0, 21,6, 20,3, 18,3, 14,0. 19F NMR (282 МГц, CDCl3): δ -75,71. MS (ESI) C21H38N4O9S: 523 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 9,37 мин.
Пример 32
Синтез соли TFA этил-1-((((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (32)
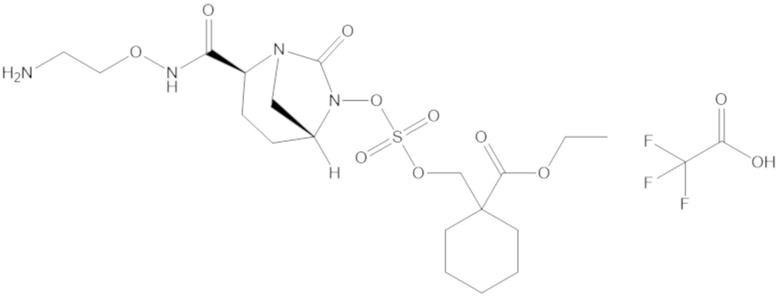
Стадия 1: Синтез этил-1-((((((2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (32a).
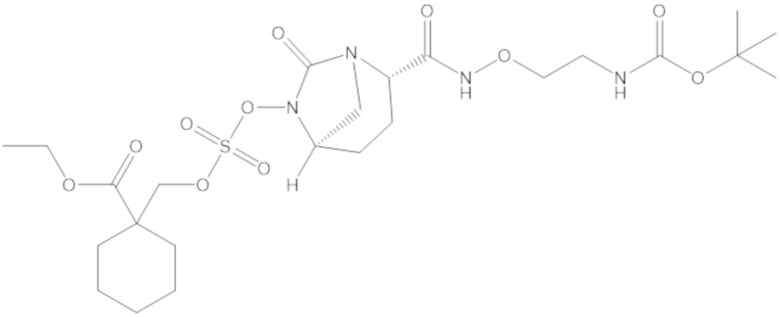
Способ, аналогичный способу стадии 1 примера 30, использовали для соединения между трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбаматом и этил-1-(((хлорсульфонил)окси)метил)циклогексанкарбоксилатом (27d) с получением 122,2 мг титульного соединения (32a) (выход 14% в течение 3 стадий) в виде грязно-белого твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 9,92 (d, J=5,6 Гц, 1H), 5,46 (d, J=5,7 Гц, 1H), 4,68 (d, J=9,2 Гц, 1H), 4,52 (d, J=9,2 Гц, 1H), 4,24-3,98 (m, 5H), 3,88 (t, J=4,8 Гц, 2H), 3,37 (d, J=6,7 Гц, 1H), 3,25 (dt, J=10,1, 5,1 Гц, 2H), 3,04 (d, J=12,1 Гц, 1H), 2,30 (t, J=6,8 Гц, 1H), 2,16-1,80 (m, 6H), 1,52 (d, J=9,3 Гц, 3H), 1,4 (s, 9H), 1,34 (m, 2H), 1,22 (td, J=7,2, 5,0 Гц, 3H). MS (ESI) C24H40N4O11S: 593 (M+1)+.
Стадия 2: Синтез соли TFA этил-1-((((((2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (32).
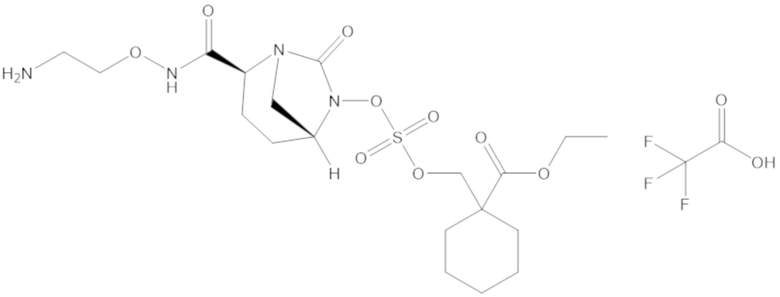
Способ, аналогичный способу стадии 2 примера 30, использовали для получения 91,4 мг титульного соединения (32) (выход 90%) в виде грязно-белого твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 10,74 (s, 1H), 7,98 (s, 3H), 4,65 (d, J=9,2 Гц, 1H), 4,50 (d, J=9,1 Гц, 1H), 4,26-4,04 (m, 3H), 4,13 (s, 3H), 3,95 (s, 1H), 3,26 (d, J=9,9 Гц, 3H), 3,14 (d, J=11,6 Гц, 1H), 2,02 (s, 4H), 1,92 (s, 3H), 1,50-1,22 (m, 8H), 1,22 (d, J=7,5 Гц, 2H). 13C-NMR (75 МГц, CDCl3): δ 175,6, 173,5, 169,2, 167,3, 118,5, 114,6, 80,3, 73,6, 72,6, 61,3, 61,1, 60,3, 60,0, 47,2, 47,1, 46,2, 38,2, 30,6, 30,2, 30,1, 25,5, 25,3, 22,4, 22,1, 22,0, 20,3, 18,3, 14,1. 19F NMR (282 МГц, CDCl3): δ -75,7. MS (ESI) C19H32N4O9S: 493 (M+1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 8,05 мин.
Пример 33
Синтез (2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфата (33)
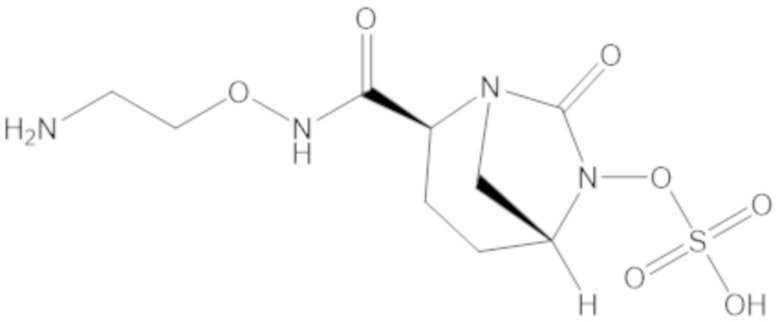
Стадия 1: Синтез тетрабутиламмоний (2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сульфата (33a).
В раствор трет-бутил (2-(((2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамидо)окси)этил)карбамата (27d) (0,79 г, 2,30 ммоль) в пиридине (35 мл) добавляли комплекс триоксида серы и пиридина (1,46 г, 9,2 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение 64 ч. Добавляли еще комплекс триоксида серы и пиридина (1,46 г, 9,2 ммоль), и реакционную смесь встряхивали при 35°C в течение 16 ч. Смесь концентрировали в вакууме с получением неочищенного остатка. В раствор приведенного выше продукта в DCM (30 мл) добавляли 0,5 N водный гидрофосфат калия (7,4 мл) при 0°C. После встряхивания при 0°C в течение 10 мин добавляли тетрабутил аммоний гидросульфат (0,86 г, 2,53 ммоль). Полученный в результате раствор встряхивали при комнатной температуре в течение 30 минут. После разделения органического слоя водный слой экстрагировали три раза DCM. Объединенные экстрагированные органические слои сушили безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента 10% MeOH в DCM с получением продукта (33a) в виде грязно-белого твердого вещества (0,51 г, выход 33%). MS (ESI) C14H24N4O9S=423 (M-1)+.
Стадия 2: Синтез (2S,5R)-2-((2-аминоэтокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил гидросульфата (33).
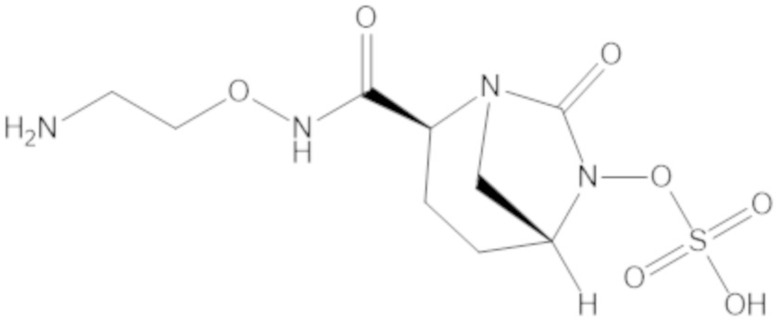
В раствор тетрабутиламмоний (2S,5R)-2-((2-((трет-бутоксикарбонил)амино)этокси)карбамоил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил сульфата (33a) (0,51 г, 0,77 ммоль) в DCM (10 мл) при -10°C добавляли TFA (10 мл). Анализ LC/MS показал, что реакция завершалась спустя 1 ч. Смесь концентрировали в вакууме с получением неочищенного остатка. Остаток встряхивали с диэтиловым эфиром с получением желтого осадка. Твердое вещество фильтровали и промывали ацетоном. Остаток растворяли в воде и ацетонитриле (1:1) и лиофилизировали с получением желтого твердого вещества. Остаток снова промывали ацетоном. Остаток растворяли в воде и ацетонитриле (1:1) и лиофилизировали с получением титульного соединения (33) в виде желтого твердого вещества (68 мг, выход 27%). 1H-NMR (300 МГц, DMSO-d6): δ 7,84 (br s, 3H), 3,82-4,00 (m, 3H), 2,92-3,33 (m, 5H), 2,00 (m, 1H), 1,87 (m, 1H), 1,68 (m, 2H). 13C-NMR (75 МГц, DMSO-d6): 169,1, 166,5, 72,8, 58,6, 58,3, 47,7, 38,2, 21,3, 19,0. MS (ESI) C9H16N4O7S=323 (M-1)+. Время удерживания HPLC (MeCN/H2O в 0,1% TFA): 1,52 мин.
Пример 34
Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил 1H-имидазол-1-сульфоната (34)
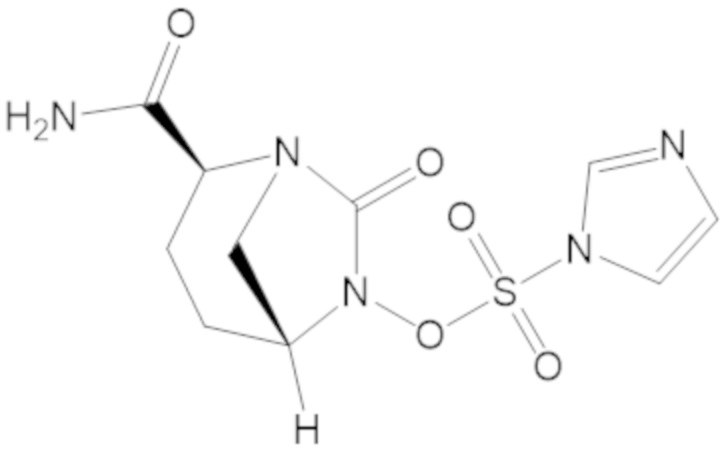
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (741 мг, 4,0 ммоль) растворяли в тетрагидрофуране (36 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2 мл), а полученный в результате раствор охлаждали до -78°C. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (4,4 мл, 4,4 ммоль), и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли 1-((1H-имидазол-1-ил)сульфонил)-3-метил-1H-имидазол-3-иум трифторметансульфонат (полученный согласно Org. Lett. 2013, 15, 18-21 & J. Org. Chem. 2003, 68, 115-119) (2,90 г, 8,0 ммоль). Спустя 10 мин реакционную смесь нагревали до 0°C (реакцию контролировали с помощью TLC с использованием 70% EtOAc/гексаны). Смесь встряхивали в течение 1 ч при комнатной температуре, затем разбавляли EtOAc (50 мл) и гасили насыщенным водным NaHCO3 (50 мл). Разделяли органические и водные слои, и органический слой промывали насыщенным водным NaHCO3 (50 мл), H2O (3×50 мл) и рассолом (50 мл), а затем сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-1:0) с получением продукта (34) (0,393 г, 31%) в виде твердого вещества. LC-MS: м/з=316,0 [M+H]+. 1H-NMR (300 МГц, 1,4-диоксан-d8): δ 8,15 (s, 1H), 7,58 (ft, J=1,5 Гц, 1H), 7,15 (s, 1H), 6,92 (s, 1H), 6,48 (s, 1H), 3,95 (d, J=6,3 Гц, 1H), 3,70 (s, 1H), 3,08 (s, 2H), 2,23-2,17 (m, 1H), 2,04-1,97 (m, 1H), 1,88-1,74 (m, 2H). 13C-NMR (75 МГц, 1,4-диоксан-d8): δ 171,4, 167,6, 139,2, 132,0, 119,9, 62,9, 61,8, 46,9, 21,5, 18,7.
Пример 35
Синтез этил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (35)
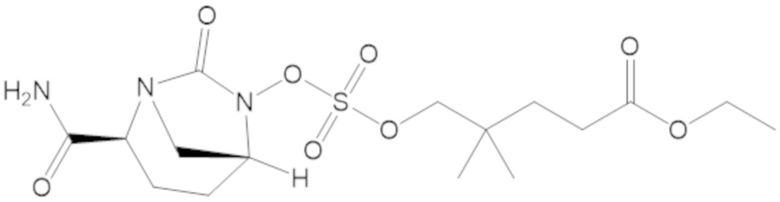
Стадия 1: Синтез этил 5-гидрокси-4,4-диметилпентаноата (35a).
В суспензию натрий 5-этокси-2,2-диметил-5-оксопентаноата (3,77 г, 17,9 ммоль) в смеси тетрагидрофурана (39 мл) и DMF (13 мл) добавляли раствор изопропилового хлорформиата, 1,0 M в толуоле (27,0 мл, 27,0 ммоль) при 0°C. Смесь встряхивали при 0°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 2 ч. Раствор охлаждали до 0°C, и добавляли борогидрид натрия (1,21 г, 35,9 ммоль). Смесь встряхивали в течение 20 мин, затем в раствор добавляли метанол (6,5 мл). После 10 мин встряхивания добавляли этилацетат (25 мл), модифицированный несколькими каплями триэтиламина, и насыщенный водный раствор NH4Cl (25 мл). Слои разделяли, и водный слой экстрагировали EtOAc (2×40 мл). Объединенные органические слои промывали рассолом, сушили (MgSO4), фильтровали, и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов, модифицированных 0,1% TEA (5:95-4:6) с получением продукта (35a) (2,01 г, 64% неочищенного) в виде бесцветного масла. Для подавления лактонизации в продукт добавляли одну каплю триэтиламина.
Стадия 2: Синтез этил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (35b).
Раствор сульфурилхлорида (0,64 мл, 8,7 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере азота. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор этил 5-гидрокси-4,4-диметилпентаноата (35a) (0,76 г, 4,4 ммоль) и пиридина (0,39 мл, 4,8 ммоль) в Et2O (10 мл). Шприц промывали Et2O (3×1 мл), и также добавляли в смесь. Смесь встряхивали при -78°C в течение 1,5 ч, еще добавляли пиридин (0,9 эквив.), и смесь фильтровали через слой Celite®. Фильтрат концентрировали в вакууме с получением продукта (35b) (0,897 г) в виде бесцветного масла. Его использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез этил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (35).
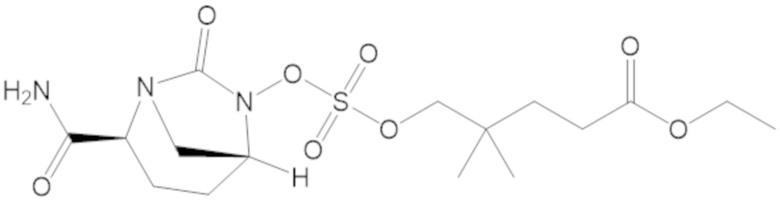
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (278 мг, 1,5 ммоль) растворяли в THF (14 мл) и HMPA (0,6 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. Раствор этил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (35b) (0,45 г, 1,6 ммоль) в THF (3 мл) охлаждали до -78°C, и быстро добавляли в смесь. Колбу, содержащую сульфатирующий реагент, промывали THF (1 мл), при этом температуру колбы поддерживали при -78°C, и быстро добавляли это в реакционную смесь. После встряхивания в течение 15 мин смесь нагревали до комнатной температуры и встряхивали в течение 45 мин. Смесь разбавляли EtOAc (30 мл), а реакцию гасили насыщенным водным NaHCO3 (30 мл). Разделяли органические и водные слои, а органический слой промывали водой (3×30 мл) и рассолом (30 мл), сушили (MgSO4), и растворитель удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов (3:7-4:1) с получением продукта (35) (157 мг, 25%) в виде твердого вещества. LC-MS: м/з=422,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 5,52 (s, 1H), 5,83 (s, 1H), 4,49 (d, J=9,3 Гц, 1H), 4,21-4,08 (m, 4H), 4,03 (d, J=6,9 Гц, 1H), 3,34-3,30 (m, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,43-2,38 (m, 1H), 2,32-2,26 (m, 2H), 2,17-2,11 (m, 1H), 1,99-1,82 (m, 2H), 1,72-1,66 (m, 3H), 1,25 (t, J=7,1 Гц, 3H), 0,98 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ173,5, 171,1, 167,1, 83,4, 62,0, 60,6, 60,3, 47,2, 34,2, 33,3, 29,3, 23,6, 23,3, 20,8, 17,6, 14,3.
Пример 36
Синтез гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36)
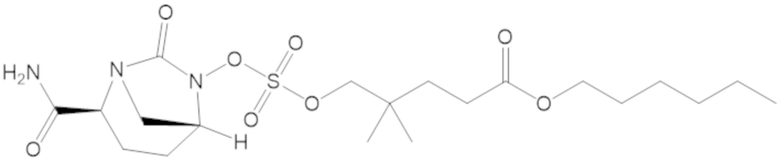
Стадия 1: Синтез натрий-5-(гексилокси)-2,2-диметил-5-оксопентаноата (36a).
В раствор 2,2-диметилглутарового ангидрида (5,0 г, 35,2 ммоль) в 1-гексаноле (50 мл) добавляли раствор натрий гексан-1-олата (5,4 г, 43,5 ммоль) в 1-гексаноле. После 20 ч встряхивания растворитель испаряли, а полученное в результате твердое вещество суспендировали в диэтиловом эфире (80 мл). Смесь фильтровали, а твердое вещество промывали диэтиловым эфиром (2×40 мл). Твердое вещество сушили при большом разрежении с получением продукта (36a) (3,84 г, 41%) в виде твердого вещества. 1H-NMR (300 МГц, D2O): δ 4,14 (t, J=6,5 Гц, 2H), 2,38-2,33 (m, 2H), 1,82-1,77 (m, 2H), 1,75-1,63 (m, 2H), 1,43-1,28 (m, 6H), 1,12 (s, 6H), 0,92-0,88 (m, 3H). Спектр показал, что продукт был загрязнен небольшим количеством неидентифицированного вещества.
Стадия 2: Синтез гексил 5-гидрокси-4,4-диметилпентаноата (36b).
В суспензию натрий 5-(гексилокси)-2,2-диметил-5-оксопентаноата (36a) (3,84 г, 14,4 ммоль) в смеси THF (31 мл) и DMF (10 мл) добавляли изопропил хлорформиат, 1,0 M в толуоле (21,6 мл, 21,6 ммоль) при 0°C, и смесь встряхивали в течение 10 мин. После 3,3 ч встряхивания при комнатной температуре раствор охлаждали до 0°C, и добавляли борогидрид натрия (0,98 г, 28,8 ммоль). Смесь встряхивали в течение 20 мин, и в раствор добавляли MeOH (5,2 мл) (реакцию контролировали с помощью TLC с использованием в качестве элюента 2:8 этилацетата/гексанов). Спустя 15 мин добавляли несколько капель триэтиламина. Еще спустя 15 мин этилацетат (25 мл) встряхивания, и добавляли раствор насыщенного водного NH4Cl (25 мл). Разделяли органические и водные слои, и водный слой экстрагировали EtOAc (2×40 мл). Объединенные органические слои промывали рассолом, сушили (MgSO4) и фильтровали, а фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием EtOAc/гексанов, модифицированных 0,1% Et3N (5:95-3:7) с получением продукта (36b) (2,16 г, 65%) в виде бесцветного масла. Для подавления лактонизации добавляли одну каплю Et3N.
Стадия 3: Синтез гексил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (36c).
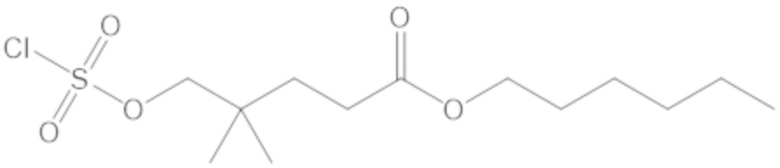
Раствор сульфурилхлорида (0,38 мл, 5,2 ммоль) в Et2O (8,5 мл) охлаждали до -78°C в атмосфере азота. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор гексил 5-гидрокси-4,4-диметилпентаноата (36b) (0,60 г, 2,6 ммоль) и пиридина (0,42 мл, 5,2 ммоль) в Et2O (8,5 мл). Шприц промывали Et2O (3×1 мл), а смыв также добавляли в смесь. Смесь встряхивали в течение 4,5 ч (реакцию контролировали с помощью TLC с использованием в качестве элюента 2:8 EtOAc/гексанов). Твердые вещества фильтровали, и растворитель концентрировали in vacuo с получением продукта (36c) в виде бесцветного масла с количественным выходом. В него добавляли 3 мл THF, и раствор хранили при -78°C. Его использовали на следующей стадии без дополнительной очистки.
Стадия 4: Синтез гексил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (36).
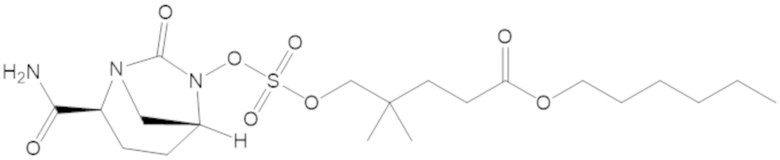
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в тетрагидрофуране (19 мл) и HMPA (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,2 мл, 2,2 ммоль), и встряхивали в течение 10 мин. Раствор гексил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (36c) (0,72 г, 2,2 ммоль) в THF (3 мл) охлаждали до -78°C, и быстро добавляли в реакционную смесь. Колбу, содержащую сульфатирующий реагент, промывали THF (3×1 мл), при этом температуру колбы поддерживали при -78°C, а смыв быстро добавляли в реакционную смесь. После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение ночи. Затем реакцию гасили насыщенным NaHCO3 (40 мл), и экстрагировали EtOAc (40 мл). Органический слой концентрировали, и масляный остаток разделяли на H2O (40 мл) и EtOAc (40 мл). Органический слой промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-8:2) с получением продукта (36) (421 мг, 44%) в виде твердого вещества. LC-MS: м/з=478,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (s, 1H), 5,59 (s, 1H), 4,51 (d, J=8,7 Гц, 1H), 4,22-4,18 (m, 2H), 4,08-4,04 (m, 3H), 3,36-3,32 (m, 1H), 3,02 (d, J=12,6 Гц, 1H), 2,47-2,41 (m, 1H), 2,33-2,28 (m, 2H), 2,18-2,13 (m, 1H), 2,01-1,79 (m, 2H), 1,72-1,59 (m, 4H), 1,35-1,31 (m, 6H), 0,99 (s, 6H), 0,91-0,87 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 173,6, 170,9, 167,1, 83,5, 64,9, 62,0, 60,2, 47,3, 34,3, 33,3, 31,6, 29,3, 28,7, 25,7, 23,6, 23,3, 22,7, 20,9, 17,6, 14,1.
Пример 37
Синтез гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37)
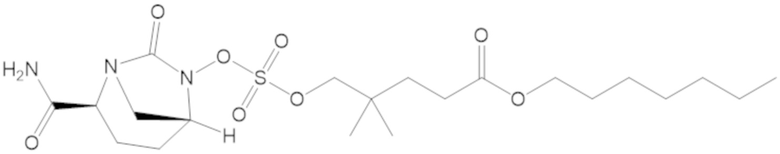
Стадия 1: Синтез натрий 5-(гептилокси)-2,2-диметил-5-оксопентаноата (37a).
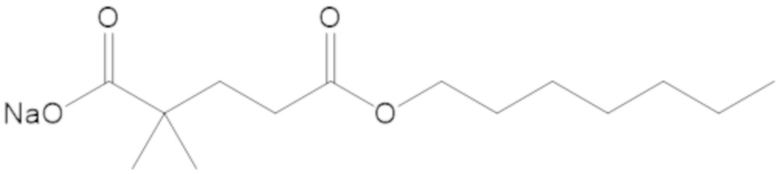
В раствор 2,2-диметилглутарового ангидрида (5,0 г, 35,2 ммоль) в 1-гептаноле (40 мл) добавляли раствор натрий гептан-1-олата (6,01 г, 43,5 ммоль) в 1-гептаноле (30 мл). После встряхивания в течение ночи растворитель испаряли, а полученное в результате твердое вещество суспендировали в Et2O (80 мл). Смесь фильтровали, а твердое вещество промывали Et2O (2×40 мл). Твердое вещество сушили при большом разрежении с получением продукта (37a) (7,89 г, 80%) в виде твердого вещества. 1H-NMR (300 МГц, D2O): δ 4,14 (t, J=6,5 Гц, 2H), 2,36-2,32 (m, 2H), 1,82-1,77 (m, 2H), 1,74-1,63 (m, 2H), 1,40-1,31 (m, 8H), 1,11 (s, 6H), 0,92-0,87 (m, 3H). Спектр показал, что продукт был загрязнен небольшим количеством неидентифицированного вещества.
Стадия 2: Синтез гептил 5-гидрокси-4,4-диметилпентаноата (37b).
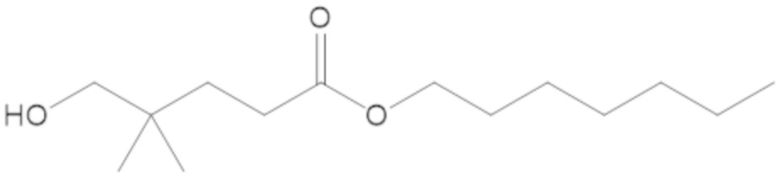
В суспензию натрий 5-(гептилокси)-2,2-диметил-5-оксопентаноата (37a) (7,89 г, 28,1 ммоль) в смеси THF (61 мл) и DMF (20 мл) добавляли изопропил хлорформиат, 1,0 M в толуоле (42,2 мл, 42,2 ммоль) при 0°C, и смесь встряхивали в течение 10 мин. После 4 ч встряхивания при комнатной температуре суспензию охлаждали до 0°C, и добавляли борогидрид натрия (1,9 г, 56,3 ммоль). Смесь встряхивали в течение 20 мин, а затем в раствор добавляли MeOH (10 мл) (реакцию контролировали с помощью TLC с использованием 2:8 этилацетата/гексанов). После 30 мин встряхивания EtOAc (50 мл) добавляли несколько капель Et3N и насыщенный водный раствор NH4Cl (50 мл). Водные и органические слои разделяли, и водный слой экстрагировали EtOAc (2×80 мл). Объединенные органические слои промывали рассолом (80 мл), и фильтрат концентрировали in vacuo. Остаточный раствор промывали H2O (3×100 мл), рассолом (100 мл), сушили (Na2SO4) и концентрировали. Во время всех экстракций для подавления лактонизации в органический слой добавляли несколько капель Et3N. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов, модифицированных 0,1% Et3N (0:1-3:7), с получением продукта (37b) (3,35 г, 49% неочищенный) в виде бесцветного масла.
Стадия 3: Синтез гептил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (37c).
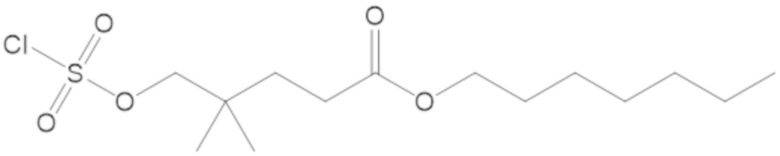
Раствор сульфурилхлорида (0,60 мл, 8,2 ммоль) в Et2O (13 мл) охлаждали до -78°C в атмосфере азота. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор гептил 5-гидрокси-4,4-диметилпентаноата (37b) (1,0 г, 4,1 ммоль) и пиридина (0,66 мл, 8,2 ммоль) в Et2O (13 мл). Шприц промывали диэтиловым эфиром (3×1 мл), и также добавляли в смесь. Смесь встряхивали в течение 4,5 ч (реакцию контролировали с помощью TLC с использованием в качестве элюента этилацетата/гексанов 2:8). Твердые вещества фильтровали, и фильтрат концентрировали in vacuo с получением продукта (37c) (1,13 г) в виде бесцветного масла. В него добавляли 3 мл THF, и раствор сохраняли при -78°C. Его использовали на следующей стадии без дополнительной очистки.
Стадия 4: Синтез гептил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (37).
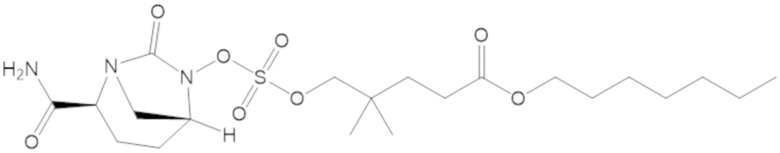
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,56 г, 3,0 ммоль) растворяли в THF (28 мл) и HMPA (1,2 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (3,3 мл, 3,3 ммоль) и встряхивали в течение 10 мин. Раствор гептил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (37c) (1,13 г, 3,3 ммоль) растворяли в THF (3 мл, при температуре, поддерживаемой -78°C) и быстро добавляли в реакционную смесь. Колбу, содержащую сульфатирующий реагент, промывали THF (3×1 мл), при этом температуру колбы поддерживали при -78°C, и также быстро добавляли в реакционную смесь. После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение ночи. Смесь гасили насыщенным водным раствором бикарбоната натрия (60 мл) и экстрагировали EtOAc (60 мл). Органический слой концентрировали, и масляный остаток разбавляли EtOAc (60 мл) и промывали водой (2×60 мл). Органический слой промывали рассолом, сушили (MgSO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле, EtOAc/гексаны (1:9-8:2) в качестве элюента, с получением продукта (37) (473 мг, 32%) в виде твердого вещества. LC-MS: м/з=492,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (s, 1H), 5,62 (s, 1H), 4,51 (d, J=9,3 Гц, 1H), 4,22-4,18 (m, 2H), 4,08-4,04 (m, 3H), 3,36-3,31 (m, 1H), 3,02 (d, J=12,6 Гц, 1H), 2,47-2,41 (m, 1H), 2,33-2,28 (m, 2H), 2,18-2,13 (m, 1H), 2,01-1,80 (m, 2H), 1,72-1,60 (m, 4H), 1,31-1,28 (m, 8H), 0,99 (s, 6H), 0,91-0,86 (m, 3H). 13C-NMR (75 МГц, CDCl3): δ 173,6, 170,9, 167,1, 83,4, 64,9, 62,0, 60,2, 47,2, 34,2, 33,3, 31,8, 29,2, 29,0, 28,7, 26,0, 23,6, 23,3, 22,7, 20,8, 17,6, 14,2.
Пример 38
Синтез 2-метоксиэтил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (38)
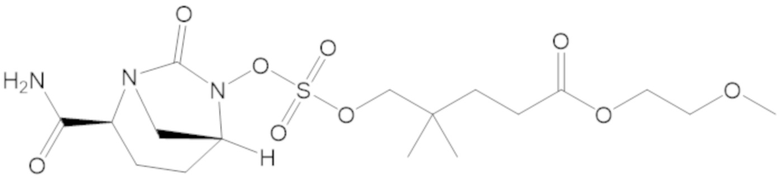
Стадия 1: Синтез натрий 5-(2-метоксиэтокси)-2,2-диметил-5-оксопентаноата (38a).
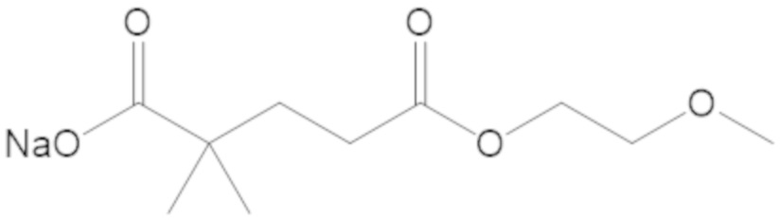
В раствор 2,2-диметилглутарового ангидрида (5,0 г, 35,2 ммоль) в 2-метоксиэтаноле (30 мл) добавляли раствор натрий 2-метоксиэтанолат (4,27 г, 43,5 ммоль) в 2-метоксиэтаноле (30 мл). После 20 ч встряхивания растворитель испаряли, а полученное в результате твердое вещество суспендировали в Et2O (80 мл). Смесь фильтровали, а твердое вещество промывали Et2O (2×40 мл). Твердое вещество сушили при большом разрежении с получением продукта (38a) (6,44 г, 76%) в виде твердого вещества. 1H-NMR (300 МГц, D2O): δ 4,30-4,27 (m, 2H), 3,75-3,72 (m, 2H), 3,42 (s, 3H), 2,41-2,36 (m, 2H), 1,83-1,78 (m, 2H), 1,12 (s, 6H). Спектр показал, что продукт был загрязнен небольшим количеством неидентифицированного вещества.
Стадия 2: Синтез 2-метоксиэтил 5-гидрокси-4,4-диметилпентаноата (38b).
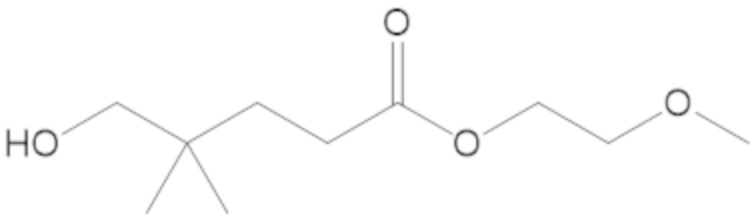
В суспензию натрий 5-(2-метоксиэтокси)-2,2-диметил-5-оксопентаноата (38a) (6,44 г, 26,8 ммоль) в смеси THF (58 мл) и DMF (19 мл) добавляли изопропил хлорформиат, 1,0 M в толуоле (40,2 мл, 40,2 ммоль) при 0°C и встряхивали в течение 10 мин. После 4 ч встряхивания при комнатной температуре смесь хранили при -78°C в течение ночи. Суспензию охлаждали до 0°C, и добавляли борогидрид натрия (1,81 г, 53,6 ммоль). Смесь встряхивали в течение 20 мин, а затем в раствор добавляли MeOH (9,6 мл) (реакцию контролировали с помощью TLC с использованием в качестве элюента EtOAc/гексанов 3:7). После 30 мин встряхивания добавляли EtOAc (50 мл) с несколькими каплями Et3N с последующим насыщенным водным раствором NH4Cl (50 мл). Слои разделяли, и водный слой экстрагировали EtOAc (2×80 мл). Объединенные органические слои промывали рассолом, сушили (MgSO4), фильтровали, и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением продукта (38b) (2,54 г, неочищенный 46%).
Стадия 3: Синтез 2-метоксиэтил 5-((хлорсульфонил)окси)-4,4-диметилпентаноата (38c).
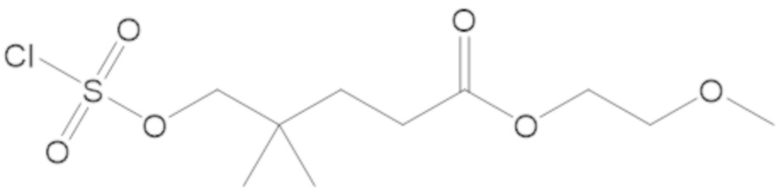
Раствор сульфурилхлорида (0,36 мл, 4,9 ммоль) в Et2O (8 мл) охлаждали до -78°C в атмосфере азота. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор 2-метоксиэтил 5-гидрокси-4,4-диметилпентаноата (38b) (0,50 г, 2,4 ммоль) и пиридина (0,40 мл, 4,9 ммоль) в Et2O (8 мл). Шприц промывали Et2O (3×1 мл), а смыв также добавляли в смесь. Смесь встряхивали в течение 4,5 ч (реакцию контролировали с помощью TLC с использованием в качестве элюента 2:8 EtOAc/гексанов). Твердые вещества фильтровали, и фильтрат концентрировали in vacuo с получением продукта (38c) (0,60 г, 2,0 ммоль) в виде бесцветного масла. В него добавляли 3 мл THF, и раствор хранили при -78°C. Его использовали на следующей стадии без дополнительной очистки.
Стадия 4: Синтез 2-метоксиэтил 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентаноата (38).
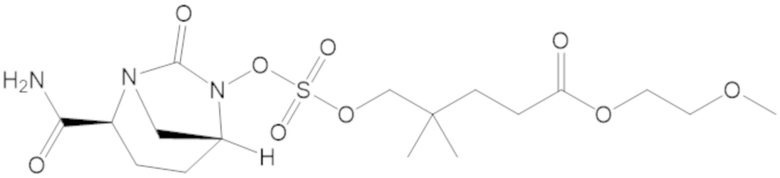
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (19 мл) и HMPA (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере азота. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (2,2 мл, 2,2 ммоль) и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли 2-метоксиэтил 5-((хлорсульфонил)окси)-4,4-диметилпентаноат (38c) (0,60 г, 2,0 ммоль), растворенный в THF (3 мл, его температуру поддерживали -78°C). Колбу, содержащую сульфатирующий реагент, промывали THF (3×1 мл), при этом температуру колбы поддерживали при -78°C, а смыв быстро добавляли в реакционную смесь. После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение ночи. Реакцию гасили насыщенным раствором водного бикарбоната натрия (40 мл) и экстрагировали EtOAc (40 мл). Органический слой концентрировали, и масляный остаток разделяли на H2O (40 мл) и EtOAc (40 мл). Органический слой промывали рассолом, сушили (MgSO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-8:2) с получением продукта (38) (164 мг, 18%) в виде твердого вещества. LC-MS: 452,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,53 (s, 1H), 5,62 (s, 1H), 4,51 (d, J=9,3 Гц, 1H), 4,24-4,18 (m, 4H), 4,05-4,03 (m, 1H), 3,61-3,58 (m, 2H), 3,39-3,32 (m, 4H), 3,01 (d, J=11,7 Гц, 1H), 2,47-2,33 (m, 3H), 2,18-2,13 (m, 1H), 2,00-1,82 (m, 2H), 1,73-1,68 (m, 2H), 0,99 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 173,6, 171,0, 167,1, 83,4, 70,6, 63,7, 62,0, 60,2, 59,1, 47,2, 34,2, 33,2, 29,1, 23,6, 23,3, 20,8, 17,6.
Пример 39
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39)
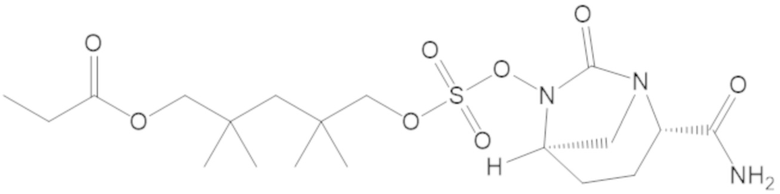
Стадия 1: Синтез 5,5-диметилтетрагидро-2H-пиран-2-она (39a).
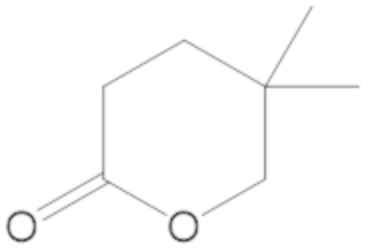
В раствор этил 5-гидрокси-4,4-диметилпентаноата (35a) (26,5 г, 152,1 ммоль) в дихлорметане (683 мл) добавляли трифторуксусную кислоту (1,75 мл, 22,8 ммоль). Смесь встряхивали при комнатной температуре в течение 3 д. Реакцию гасили насыщенным водным раствором бикарбоната натрия (150 мл), быстро встряхивали в течение 30 минут, и слои разделяли. Органический слой промывали рассолом (150 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-45:55) с получением продукта (39a) (8,79 г, 45%) в виде бесцветного масла. Продукт использовали на следующей стадии без дополнительной очистки, и он был загрязнен небольшими количествами неидентифицированных побочных продуктов. 1H-NMR (300 МГц, CDCl3): δ 3,97 (s, 2H), 2,56 (t, J=7,4 Гц, 2H), 1,69 (t, J=7,4 Гц, 2H), 1,05 (s, 6H).
Стадия 2: Синтез 3,3,5,5-тетраметилтетрагидро-2H-пиран-2-она (39b).
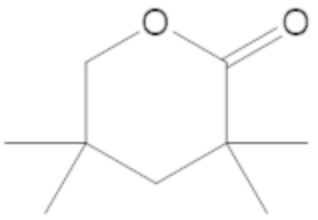
5,5-диметилтетрагидро-2H-пиран-2-он (39a) (8,79 г, 68,6 ммоль) растворяли в безводном DMF (150 мл), а полученный в результате раствор охлаждали до 0°C в инертной атмосфере аргона. В одной части добавляли натрий гидрид, 60% в минеральном масле (8,23 г, 205,7 ммоль), и смесь встряхивали в течение 20 мин. За этим следовало покапельное добавление MeI (17,1 мл, 274,3 ммоль). Полученный в результате раствор встряхивали при 0°C в течение 20 мин, а затем при комнатной температуре в течение 3 д. Смесь разбавляли EtOAc (350 мл), а затем гасили при 0°C с помощью осторожного добавления по каплям насыщенного водного раствора NH4Cl (100 мл). Водные и органические слои разделяли, и водный слой экстрагировали EtOAc (350 мл). Объединенные органические слои промывали H2O (6×300 мл), рассолом (300 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9) с получением продукта (39b) (3,42 г, 32%). Продукт использовали на следующей стадии без дополнительной очистки, и он был загрязнен небольшими количествами различных неидентифицированных побочных продуктов. 1H-NMR (300 МГц, CDCl3): δ 4,01 (s, 2H), 1,62 (s, 2H), 1,30 (s, 6H), 1,02 (s, 6H).
Стадия 3: Синтез 2,2,4,4-тетраметилпентан-1,5-диола (39c).
Колбу с горлышком и круглым дном, содержащую встряхиваемую суспензию 95% LiAlH4 (0,87 г, 21,6 ммоль) в Et2O (126 мл), охлаждали до 0°C в атмосфере аргона. В эту суспензию добавляли раствор 3,3,5,5-тетраметилтетрагидро-2H-пиран-2-она (39b) (2,94 г, 18,8 ммоль) в Et2O (50 мл) в инертной атмосфере аргона. Ее нагревали до комнатной температуры и встряхивали в течение ночи. Смесь аккуратно гасили H2O (80 мл), затем 3 M NaOH (120 мл) и встряхивали в течение 30 минут. Смесь фильтровали через слой Celite®, и слой тщательно промывали Et2O. Водные и органические слои разделяли, и водный слой экстрагировали Et2O (3×100 мл). Объединенные органические слои концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (2:8-6:4) с получением продукта (39c) (2,59 г, 86%) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 3,41 (s, 4H), 2,55 (s, 2H), 1,34 (s, 2H), 0,95 (s, 12H)
Стадия 4: Синтез 5-гидрокси-2,2,4,4-тетраметилпентил пропионата (39d).
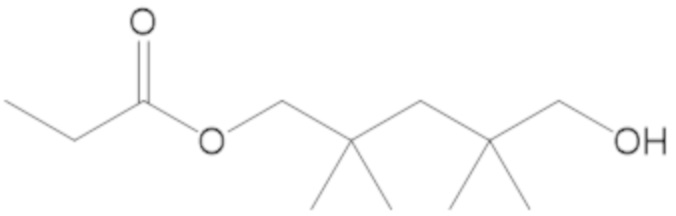
Во встряхиваемый раствор 2,2,4,4-тетраметилпентан-1,5-диола (39c) (0,48 г, 3,0 ммоль) и пиридина (0,24 мл, 3,0 ммоль) в DCM (20 мл) по каплям в течение 30 минут добавляли пропионилхлорид (0,26 мл, 3,0 ммоль) приблизительно при 0°C (ледяная ванна). Реакционную смесь встряхивали в течение ночи при комнатной температуре. Смесь разбавляли H2O (20 мл), и слои разделяли. Водный слой экстрагировали DCM (2×20 мл), и объединенные органические слои промывали рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле, в качестве элюента EtOAc/гексаны (5:95-6:4), с получением продукта (39d) (411 мг, 63%). 1H-NMR (300 МГц, CDCl3): δ 3,85 (s, 2H), 3,32 (s, 2H), 2,37 (q, J=7,7 Гц, 2H), 1,50 (s, 1H), 1,36 (s, 2H), 1,16 (t, J=7,5 Гц, 3H), 1,03 (s, 6H), 0,99 (s 6H).
Стадия 5: Синтез 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39e).
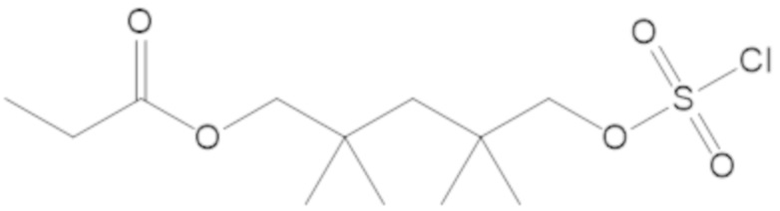
Раствор сульфурилхлорида (0,136 мл, 1,9 ммоль) в Et2O (6,4 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор 5-гидрокси-2,2,4,4-тетраметилпентил пропионата (39d) (404 мг, 1,9 ммоль) и пиридина (0,15 мл, 1,9 ммоль) в Et2O (6,4 мл). Смесь нагревали до комнатной температуры и встряхивали в течение 70 мин. Твердые вещества фильтровали с получением в качестве фильтрата раствора продукта (39e) в Et2O. Выход оценивали количественно, и смесь использовали на следующей стадии без дополнительной очистки.
Стадия 6: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39).
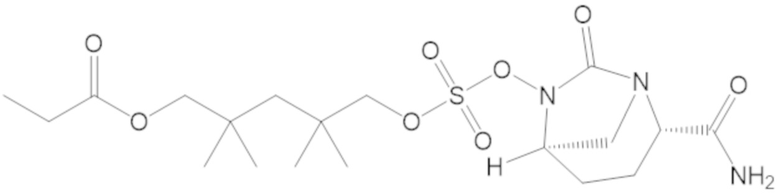
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (346 мг, 1,9 ммоль) растворяли в THF (21,8 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1,0 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,9 мл, 1,9 ммоль), и встряхивали в течение 90 мин. В реакционную смесь (канюля) добавляли раствор 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил пропионата (39e) (0,59 г, 1,9 ммоль) в Et2O (около 20 мл). После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение 4 ч. Затем реакцию гасили насыщенным водным раствором бикарбоната натрия (40 мл) и экстрагировали EtOAc (40 мл). Органический слой промывали H2O (3×40 мл), рассолом (40 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-8:2) с получением продукта (39) (254 мг, 29%) в виде твердого вещества. LC-MS: м/з=464,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,53 (s, 1H), 5,64 (s, 1H), 4,53 (d, J=9 Гц, 1H), 4,23 (d, J=9 Гц, 1H), 4,18 (m, 1H) 4,06-4,04 (m, 1H), 3,80 (s, 2H), 3,36-3,32 (m, 1H), 3,01 (d, J=12,3 Гц, 1H), 2,47-2,33 (m, 3H), 2,19-2,13 (m, 1H), 2,01-1,79 (m, 2H), 1,43 (s, 2H), 1,16 (t, J=7,7 Гц, 3H), 1,09 (s, 6H), 1,03 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 174,6, 171,1, 167,1, 84,9, 73,3, 61,9, 60,2, 47,2, 46,2, 36,0, 35,3, 27,8, 26,6, 26,3, 25,9, 25,3, 20,8, 17,5, 9,3.
Пример 40
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40)
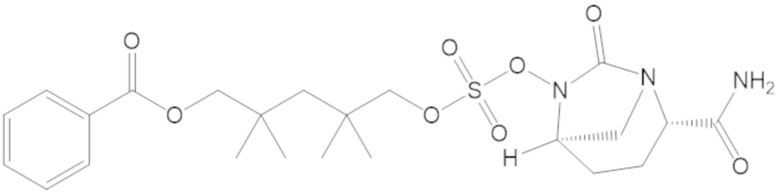
Стадия 1: Синтез 5-гидрокси-2,2,4,4-тетраметилпентил бензоата (40a).
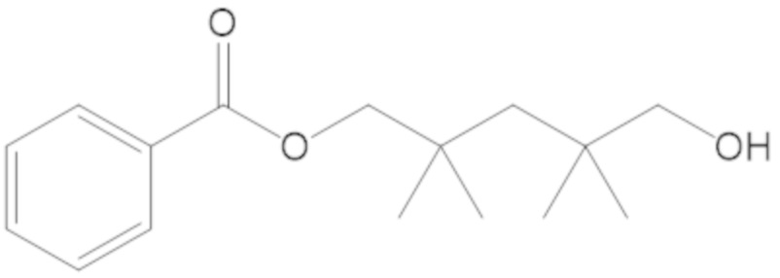
Во встряхиваемый раствор 2,2,4,4-тетраметилпентан-1,5-диола (39c) (0,48 г, 3,0 ммоль) и пиридина (0,24 мл, 3,0 ммоль) в DCM (20 мл) по каплям в течение 30 минут добавляли бензоилхлорид (0,37 мл, 3,0 ммоль) приблизительно при 0°C (ледяная ванна) в атмосфере аргона. Реакционную смесь встряхивали в течение ночи при комнатной температуре. Смесь разбавляли H2O (20 мл), и слои разделяли. Водный слой экстрагировали DCM (2×20 мл), и объединенные органические слои промывали рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (5:95-1:1) с получением продукта (40a) (548 мг, 69%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 8,06 (d, J=8,4 Гц, 2H), 7,59-7,55 (m, 1H), 7,48-7,43 (m, 2H), 4,09 (s, 2H), 3,35 (s, 2H), 1,48 (s, 2H), 1,13 (s, 6H), 1,02 (s, 6H).
Стадия 2: Синтез 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40b).
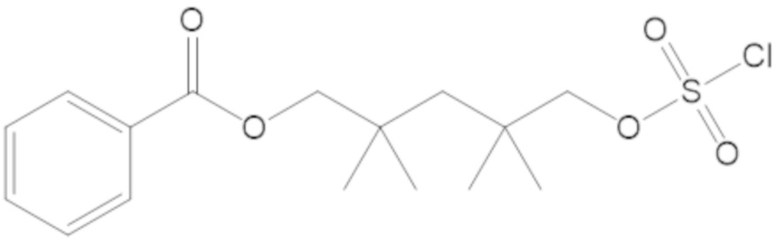
Раствор сульфурилхлорида (0,15 мл, 2,0 ммоль) в Et2O (8,5 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор 5-гидрокси-2,2,4,4-тетраметилпентил бензоата (40a) (541 мг, 2,0 ммоль) и пиридина (0,17 мл, 2,0 ммоль) в Et2O (8,5 мл). Смесь встряхивали при 0°C в течение 20 мин, затем при комнатной температуре в течение 90 мин. Смесь фильтровали, а фильтрат использовали для получения раствора продукта (40b) в Et2O (около 20 мл). Выход оценивали количественно, а продукт использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40).
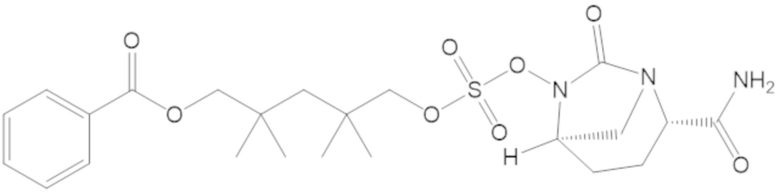
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (370 мг, 2,0 ммоль) растворяли в THF (23 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1,5 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,0 мл, 2,0 ммоль), и встряхивали в течение 90 мин. В реакционную смесь (канюля) добавляли раствор 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил бензоата (40b) (0,73 г, 2,0 ммоль, 1,0 эквив.), растворенный в Et2O (около 20 мл). После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение 4 ч. Смесь гасили насыщенным водным раствором бикарбоната натрия (40 мл) и экстрагировали EtOAc (40 мл). Органический слой промывали H2O (3× 40 мл), рассолом (40 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-8:2) с получением продукта (40) (282 мг, 27%) в виде твердого вещества. LC-MS: м/з=512,15 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,06 (d, J=7,8 Гц, 2H), 7,59-7,55 (m, 1H), 7,49-7,43 (m, 2H), 6,49 (s, 1H), 5,70 (s, 1H), 4,57 (d, J=9 Гц, 1H), 4,26 (d, J=8,7 Гц, 1H), 4,17 (s, 1H), 4,10-4,01 (m, 3H), 3,29-3,25 (m, 1H), 2,98 (d, J=11,7 Гц, 1H), 2,45-2,35 (m, 1H), 2,17-2,11 (m, 1H), 1,99-1,77 (m, 2H), 1,56 (s, 2H), 1,14-1,13 (m, 12H). 13C-NMR (75 МГц, CDCl3): δ 171,0, 167,0, 166,6, 133,1, 130,4, 129,7, 128,6, 85,0, 73,9, 61,9, 60,2, 47,2, 46,2, 36,1, 35,7, 26,7, 26,4, 25,9, 25,4, 20,8, 17,5.
Пример 41
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41)
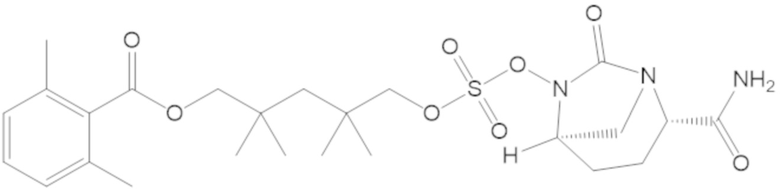
Стадия 1: Синтез 5-гидрокси-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41a).
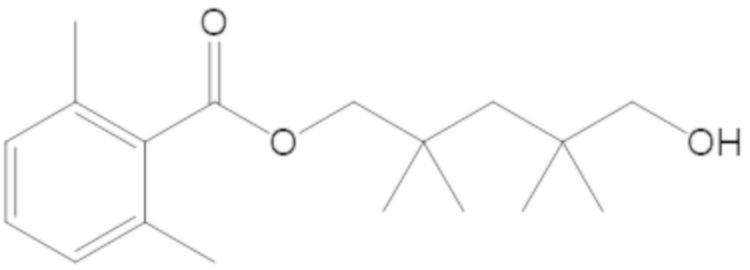
Во встряхиваемый раствор 2,2,4,4-тетраметилпентан-1,5-диола (39c) (0,48 г, 3,0 ммоль) и пиридина (0,24 мл, 3,0 ммоль) в DCM (20 мл) по каплям в течение 30 минут добавляли 2,6-диметилбензоилхлорид (0,45 мл, 3,0 ммоль) при 0°C (ледяная ванна) в атмосфере аргона. Реакционную смесь встряхивали в течение ночи при комнатной температуре. Смесь разбавляли H2O (20 мл), и слои разделяли. Водный слой экстрагировали DCM (2×20 мл), и объединенные органические слои промывали рассолом (20 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (5:95-1:1) с получением продукта (41a) (462 мг, 53%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,22-7,17 (m, 1H), 7,04 (d, J=7,5 Гц, 2H), 4,10 (s, 2H), 3,32 (s, 2H), 2,33 (s, 6H), 1,41 (s, 2H), 1,10 (s, 6H), 1,00 (s, 6H).
Стадия 2: Синтез 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41b).
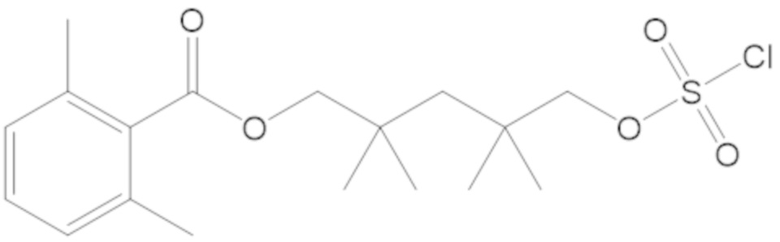
Раствор сульфурилхлорида (0,11 мл, 1,5 ммоль) в Et2O (7 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор 5-гидрокси-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41a) (453 мг, 1,5 ммоль) и пиридина (0,13 мл, 1,5 ммоль) в Et2O (7 мл). Смесь встряхивали в ледяной ванне в течение 20 мин, затем при комнатной температуре в течение 90 мин. Смесь фильтровали, а фильтрат сохраняли для получения раствора продукта (41b) в Et2O (около 20 мл). Выход принимали по количеству. Эту смесь использовали на следующей стадии без дополнительной очистки (небольшое количество концентрировали в вакууме, и проводили NMR остатка). 1H-NMR (300 МГц, CDCl3): δ 7,21 (t, J=7,7 Гц, 1H), 7,05 (d, J=7,2 Гц, 2H), 4,20 (s, 2H), 4,07 (s, 2H), 2,32 (s, 6H), 1,50 (s, 2H), 1,14 (s, 6H), 1,12 (s, 6H).
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41).
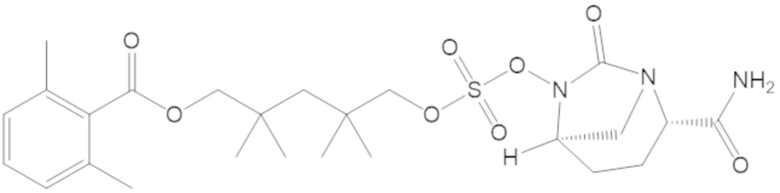
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (286 мг, 1,5 ммоль) растворяли в THF (18 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (2,3 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,5 мл, 1,5 ммоль), и встряхивали в течение 90 мин. В реакционную смесь (канюля) добавляли раствор 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметилбензоата (41b) (0,61 г, 1,5 ммоль) в Et2O (около 20 мл). После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение 4 ч. Смесь гасили насыщенным водным раствором бикарбоната натрия (40 мл) и экстрагировали EtOAc (40 мл). Органический слой промывали H2O (3×40 мл), рассолом (40 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-8:2) с получением продукта (41) (490 мг, 58%) в виде твердого вещества. LC-MS: м/з=540,07 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,20 (t, J=7,7 Гц, 1H), 7,04 (d, J=7,5 Гц, 2H), 6,50 (s, 1H), 5,63 (s, 1H), 4,54 (d, J=8,7 Гц, 1H), 4,23 (d, J=8,7 Гц, 1H), 4,17 (s, 1H), 4,06-4,03 (m, 3H), 3,34-3,29 (m, 1H), 3,00 (d, J=11,7 Гц, 1H), 2,47-2,40 (m, 1H), 2,32 (s, 6H), 2,18-2,14 (m, 1H), 2,00-1,78 (m, 2H), 1,49 (s, 2H), 1,11 (s, 12H). 13C-NMR (75 МГц, CDCl3): δ 170,9, 170,4, 167,0, 135,0, 134,2, 129,4, 127,7, 84,9, 74,5, 62,0, 60,2, 47,2, 46,2, 36,1, 35,4, 26,5, 26,4, 26,0, 25,3, 20,8, 20,0, 17,5.
Пример 42
Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42)
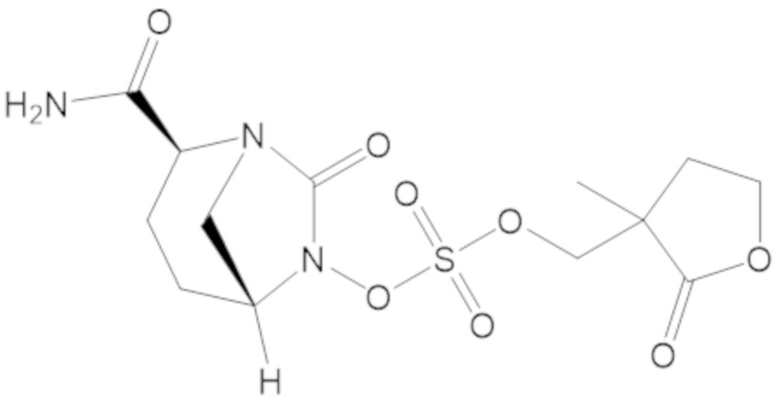
Стадия 1: Синтез (3-метил-2-оксотетрагидрофуран-3-ил)метил сульфохлорида (42a).
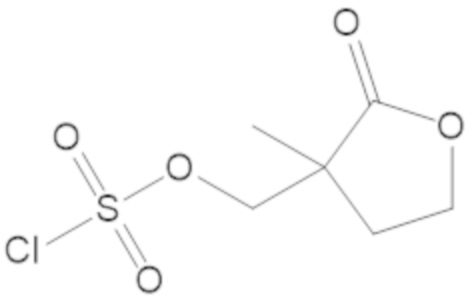
Пиридин (0,28 мл, 3,5 ммоль) добавляли во встряхиваемую смесь 3-(гидроксиметил)-3-метилдигидрофуран-2(3H)-она (полученного согласно Synlett 2010, 2625-2627) (0,30 г, 2,3 ммоль) и Et2O (8 мл) в атмосфере аргона. Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,28 мл, 3,5 ммоль) в Et2O (3 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (42a) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 2: Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфата (42).
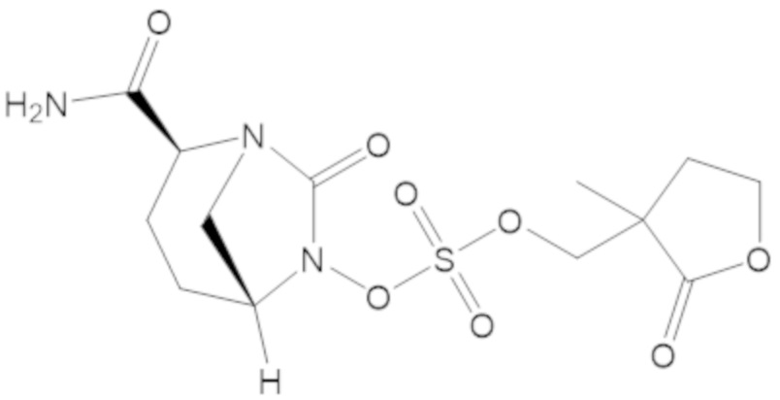
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (0,24 г, 1,3 ммоль) в THF (20 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,4 мл, 1,4 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли (3-метил-2-оксотетрагидрофуран-3-ил)метил сульфохлорид (42a) (0,30 г, 1,3 ммоль) в THF (30 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение ночи. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением твердого вещества (150 мг). NMR показал следовые примеси, которые удаляли посредством растирания с EtOAc с получением продукта (42) (35 мг) в виде твердого вещества. LC/MS: м/з=378,0 [M+H]+. 1H-NMR (300 МГц, d6-DMSO): δ 7,53 (s, 1H), 7,38 (s, 1H), 4,68-4,64 (m, 1H), 4,54 (d, J=9,3 Гц, 1H), 4,32-4,27 (m, 2H), 4,09 (s, 1H), 3,89 (d, J=6,0Hz, 1H), 3,21-3,13(m, 2H), 2,38-2,28 (m, 1H), 2,13-2,00 (m, 2H), 1,91-1,66 (m, 3H), 1,21 (s, 3H). 13C-NMR (75 МГц, d6-DMSO): δ 178,3, 171,0, 168,7, 77,9, 65,6, 61,7, 61,2, 46,3, 43,2, 31,2, 20,8, 19,1, 18,9.
(2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфат (42) разделяли на изомеры (S) и (R), (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил (((S)-3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфат (42(S) и (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил (((R)-3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфат (42(R):
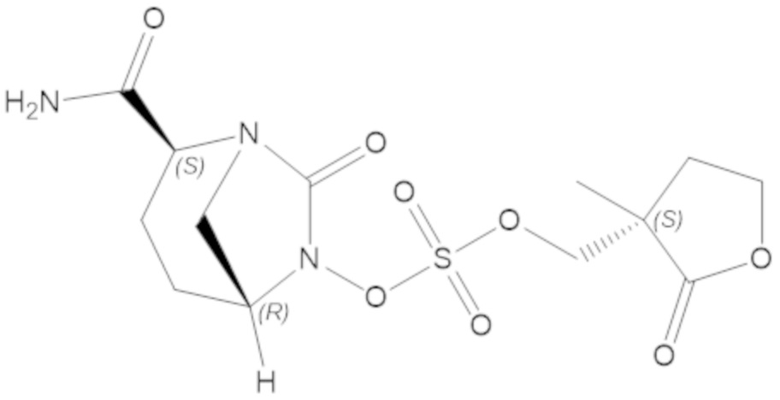
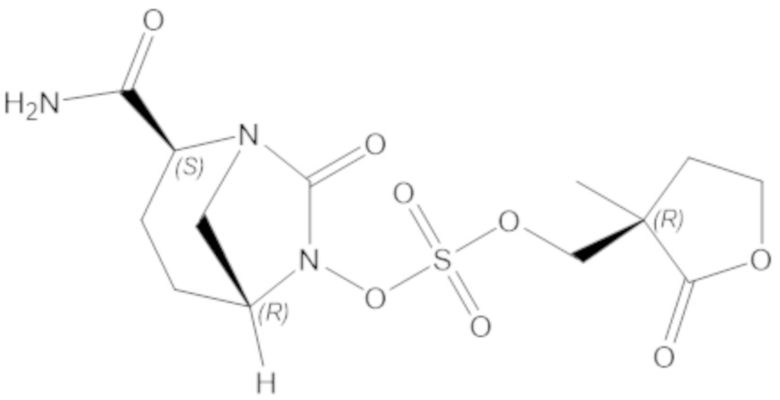
Пример 43
Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил пивалата (43)
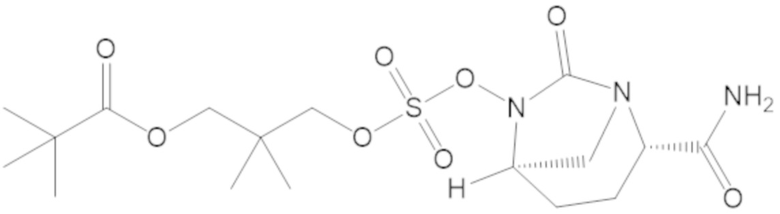
Стадия 1: Синтез 3-гидрокси-2,2-диметилпропил пивалата (43a).
Во встряхиваемый раствор 2,2-диметилпропан-1,3-диола (5,07 г, 48,7 ммоль) в DCM (50 мл) при 0°C в атмосфере аргона добавляли триметилацетилхлорид (2,0 мл, 16,2 ммоль), пиридин (2,63 мл, 32,5 ммоль) и N, N-4-диметиламинопиридин (0,4 г, 3,2 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили добавлением 1 н. HCl (50 мл), затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:5) с получением продукта (43a) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,92 (s, 2H), 3,27 (s, 2H), 1,22 (s, 9H), 0,92 (s, 6H).
Стадия 2: Синтез 3-((хлорсульфонил)окси)-2,2-диметилпропил пивалата (43b).
Пиридин (0,75 мл, 9,3 ммоль) добавляли во встряхиваемую смесь 3-гидрокси-2,2-диметилпропил пивалата (43a) (1,17 г, 6,2 ммоль) и Et2O (20 мл) в атмосфере аргона. Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,75 мл, 9,3 ммоль) в Et2O (8 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (43b) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 3: Реакция получения 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил пивалата (43).
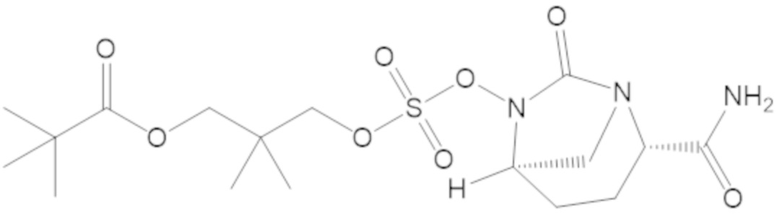
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (1,1 г, 5,9 ммоль) в THF (20 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (6,5 мл, 6,5 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли 3-((хлорсульфонил)окси)-2,2-диметилпропил пивалат (43b) (1,7 г, 5,9 ммоль) в THF (30 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение ночи. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (43) (654 мг) в виде твердого вещества. LC/MS: м/з=436,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (s, 1H), 5,58 (s, 1H), 4,60 (d, J=8,7 Гц, 1H), 4,36 (d, J=9,0 Гц, 1H), 4,17 (s, 1H), 4,04 (d, J=6,3 Гц, 1H), 3,95-3,84 (q, 2H), 3,35-3,31 (m, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,50-2,41 (m, 1H), 2,20-2,05 (m, 1H), 1,99-1,78 (m, 2H), 1,22 (s, 9H), 1,04 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 178,2, 171,1, 167,1, 80,6, 68,4, 62,0, 60,2, 47,2, 39,1, 35,6, 27,3, 21,3, 21,3, 20,8, 17,6.
Пример 44
Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоата (44)
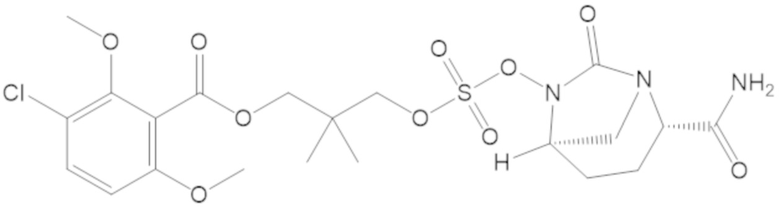
Стадия 1: Синтез 3-гидрокси-2,2-диметилпропил 2,6-диметоксибензоата (44a).
Во встряхиваемый раствор 2,2-диметилпропан-1,3-диола (3,89 г, 37,4 ммоль) в DCM (40 мл) при 0°C в атмосфере аргона добавляли 2,6-диметоксибензоилхлорид (80% чистота; 3,13 г, 12,5 ммоль), пиридин (2,02 мл, 24,9 ммоль) и N, N-4-диметиламинопиридин (0,3 г, 2,5 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили путем добавления 1 н. HCl (50 мл), затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:5) с получением продукта (44a) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,19 (t, J=5,0 Гц, 1H), 6,48 (d, J=8,1 Гц, 2H), 4,09 (s, 2H), 3,71 (s, 6H), 3,33 (s, 2H), 0,89 (s, 6H).
Стадия 2: Синтез 3-((хлорсульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоата (44b).
Пиридин (0,16 мл, 2,0 ммоль) добавляли во встряхиваемую смесь 3-гидрокси-2,2-диметилпропил 2,6-диметоксибензоата (44a) (0,35 г, 1,3 ммоль) и Et2O (10 мл) в атмосфере аргона. Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,16 мл, 2,0 ммоль) в Et2O (8 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (44b) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству). 1H-NMR (300 МГц, CDCl3): δ 7,36 (d, J=8,7 Гц, 1H), 6,66 (d, J=8,7 Гц, 1H), 4,35 (s, 2H), 4,21 (s, 2H), 3,89 (s, 3H), 3,81 (s, 3H), 1,13 (s, 6H).
Стадия 3: Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоата (44).
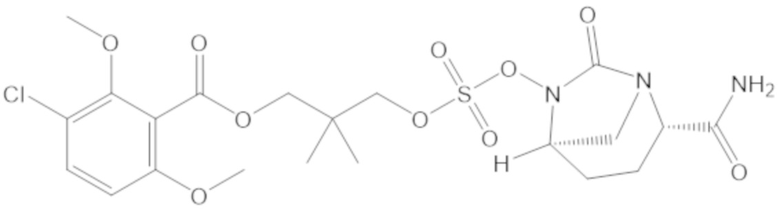
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (237 мг, 1,3 ммоль) в THF (15 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,4 мл, 1,4 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли 3-((хлорсульфонил)окси)-2,2-диметилпропил 3-хлор-2,6-диметоксибензоат (44b) (0,47 г, 1,2 ммоль) в THF (8 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение ночи. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (44) в виде твердого вещества. LC/MS: м/з=550,0 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,36-7,32 (dd, J=1,2 Гц, 1,2 Гц, 1H), 6,65 (d, J=8,1 Гц, 1H), 6,51 (s, 1H), 5,82 (s, 1H), 4,55 (d, J=8,7 Гц, 1H), 4,38 (d, J=9,0 Гц, 1H), 4,25-4,07 (m, 3H), 4,01 (d, J=6,6 Гц, 1H), 3,88 (s, 3H), 3,81 (s, 3H), 3,27 (d, J=11,7 Гц, 1H), 2,98 (d, J=11,7 Гц, 1H), 2,41-2,37 (m, 1H), 2,14-2,10 (m, 1H), 1,94-1,74 (m, 2H), 1,08 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 167,1, 165,2, 156,1, 153,7, 131,7, 119,8, 119,5, 107,9, 80,4, 69,3, 62,2, 62,0, 60,2, 56,3, 47,1, 35,7, 21,3, 21,2, 20,8, 17,5.
Пример 45
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45)
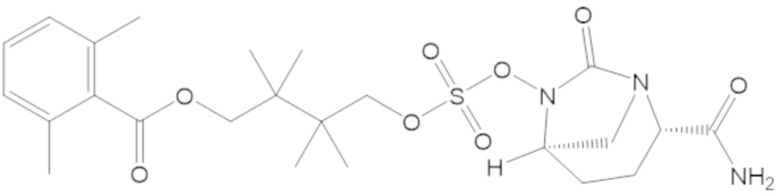
Стадия 1: Синтез 2,2,3,3-тетраметилбутан-1,4-диола (45a).
Во встряхиваемую суспензию LiAlH4 (95%; 0,32 г, 8,1 ммоль) в Et2O (28 мл) добавляли раствор 3,3,4,4-тетраметилдигидрофуран-2(3H)-она (полученного согласно патентам США №№ 3658849) (1,0 г, 7,0 ммоль) в Et2O (28 мл) при 0°C в атмосфере аргона. Смесь нагревали до комнатной температуры и встряхивали в течение ночи. Декагидрат сульфата натрия медленно добавляли до тех пор, пока в колбе не прекращалось выделение пузырьков. Твердое вещество фильтровали через слой Celite®, и слой промывали EtOAc. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (45a) (0,7 г) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 3,41 (s, 4H), 0,88 (s, 12H).
Стадия 2: Синтез 4-гидрокси-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45b).
Во встряхиваемый раствор 2,2,3,3-тетраметилбутан-1,4-диола (45a) (0,71 г, 4,9 ммоль) в DCM (15 мл) при 0°C в атмосфере аргона добавляли 2,6-диметилбензоилхлорид (0,2 мл, 1,6 ммоль), пиридин (0,26 мл, 3,2 ммоль) и N, N-4-диметиламинопиридин (0,04 г, 0,3 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили путем добавления 1 н. HCl (15 мл), затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением продукта (45b) в виде масла (266 мг). 1H-NMR (300 МГц, CDCl3): δ 7,18 (t, J=8,4 Гц, 1H), 7,02 (d, J=6,9 Гц, 2H), 4,25 (s, 2H), 3,51 (s, 2H), 2,31 (s, 6H), 0,98 (s, 6H), 0,93 (s, 6H).
Стадия 3: Синтез 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45c).
Во встряхиваемую смесь 4-гидрокси-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45b) (0,26 г, 0,9 ммоль) и Et2O (10 мл) в атмосфере аргона добавляли пиридин (0,11 мл, 1,3 ммоль). Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,11 мл, 1,3 ммоль) в Et2O (3 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (45c) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоата (45).
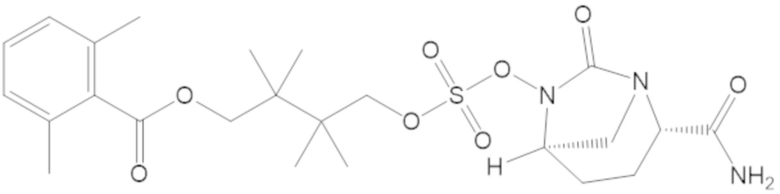
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (255 мг, 1,4 ммоль) в THF (10 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,5 мл, 1,5 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил 2,6-диметилбензоат (45c) (0,52 г, 1,4 ммоль) в THF (5 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (45) в виде твердого вещества. LC/MS: м/з=526,16 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,19 (t, J=7,7 Гц, 1H), 7,03 (d, J=6,9 Гц, 2H), 6,47 (s, 1H), 5,58 (s, 1H), 4,76 (d, J=9,3 Гц, 1H), 4,40 (d, J=9,6 Гц, 1H), 4,20-4,16 (m, 3H), 4,04 (d, J=6,3 Гц, 1H), 3,33 (d, J=12,3 Гц, 1H), 2,99 (d, J=12,3 Гц, 1H), 2,45-2,40 (m, 1H), 2,32 (s, 6H), 2,17-2,13 (m, 1H), 2,04-1,83 (m. 2H), 1,05-1,03 (m, 12H). 13C-NMR (75MHz, CDCl3): δ 170,9, 170,4, 167,1, 134,9, 134,2, 129,4, 127,6, 82,1, 70,8, 62,0, 60,2, 47,3, 39,1, 38,5, 21,0, 20,9, 20,8, 20,5, 20,3, 20,0, 17,5.
Пример 46
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил бензоата (46)
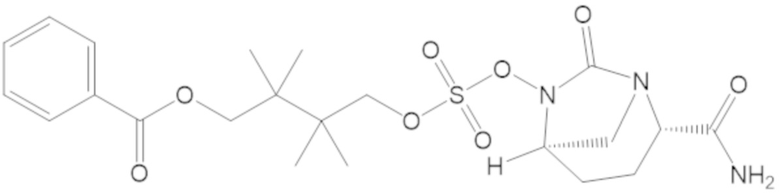
Стадия 1: Синтез 4-гидрокси-2,2,3,3-тетраметилбутил бензоата (46a).
Во встряхиваемый раствор 2,2,3,3-тетраметилбутан-1,4-диола (45a) (0,74 г, 5,0 ммоль) в DCM (15 мл) при 0°C в атмосфере аргона добавляли бензоилхлорид (0,25 мл, 2,0 ммоль), пиридин (0,33 мл, 4,0 ммоль) и N, N-4-диметиламинопиридин (0,05 г, 0,4 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили путем добавления 1 н. HCl (15 мл), затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением продукта (46a) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 8,05 (d, J=7,2 Гц, 2H), 7,58 (t, J=7,4 Гц, 1H), 7,46 (t, J=7,4 Гц, 2H), 4,27 (s, 2H), 3,59 (s, 2H), 1,05 (s, 6H), 0,99 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил бензоата (46b).
Во встряхиваемую смесь 4-гидрокси-2,2,3,3-тетраметилбутил бензоата (46a) (0,70 г, 2,8 ммоль) и Et2O (10 мл) в атмосфере аргона добавляли пиридин (0,29 мл, 3,6 ммоль). Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,29 мл, 3,6 ммоль) в Et2O (3 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (46b) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил бензоата (46).
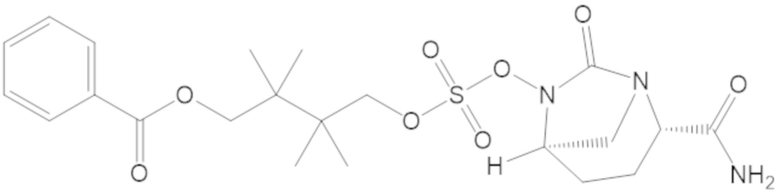
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (0,52 г, 2,8 ммоль) в THF (10 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (3,1 мл, 3,1 ммоль). Смесь встряхивали при -78°C в течение 8 мин, а затем добавляли 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил бензоат (46b) (0,98 г, 2,8 ммоль) в THF (5 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (46) в виде твердого вещества. LC/MS: м/з=498,10 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,05 (d, J=7,2 Гц, 2H), 7,57 (t, J=7,4 Гц, 1H), 7,45 (t, J=7,4 Гц, 2H), 6,47 (s, 1H), 5,61 (s, 1H), 4,87 (d, J=9,0 Гц, 1H), 4,52 (d, J=9,3 Гц, 1H), 4,22-4,17 (m, 3H), 4,01 (d, J=6,3 Гц, 1H), 3,35-3,31 (m, 1H), 2,99 (d, J=14,7 Гц, 1H), 2,43-2,39 (m, 1H), 2,17-2,12 (m, 1H), 1,92-1,81 (m, 2H), 1,10-1,06 (m, 12H). 13C-NMR (75 МГц, CDCl3): δ 170,9, 167,1, 166,6, 133,1, 130,3, 129,7, 128,6, 82,4, 70,5, 62,0, 60,2, 47,3, 39,2, 38,8, 21,1, 21,0, 20,8, 20,5, 20,3, 17,5.
Пример 47
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил пропионата (47)
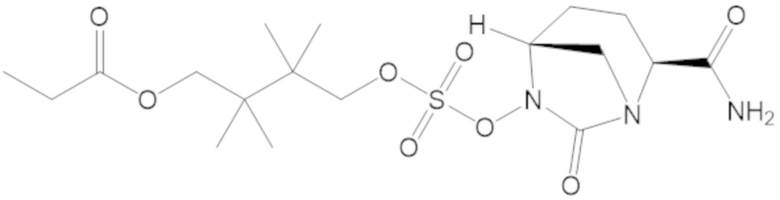
Стадия 1: Синтез 4-гидрокси-2,2,3,3-тетраметилбутил пропионата (47a).
Во встряхиваемый раствор 2,2,3,3-тетраметилбутан-1,4-диола (45a) (0,59 г, 4,0 ммоль) в DCM (15 мл) при 0°C в атмосфере аргона добавляли пропионилхлорид (0,25 мл, 3,1 ммоль), пиридин (0,33 мл, 4,0 ммоль) и N, N-4-диметиламинопиридин (0,05 г, 0,4 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили путем добавления 1 н. HCl (15 мл), а затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением диацилированного материала, с последующим продуктом (47a) (300 мг) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,99 (s, 2H), 3,49 (s, 2H), 2,38-2,31 (q, 2H), 1,15 (t, J=7,8 Гц, 3H), 0,91 (d, J=4,8 Гц, 12H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил пропионата (47b).
Во встряхиваемую смесь 4-гидрокси-2,2,3,3-тетраметилбутил пропионата (47a) (0,30 г, 1,5 ммоль) и Et2O (10 мл) в атмосфере аргона добавляли пиридин (0,16 мл, 1,9 ммоль). Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,16 мл, 1,9 ммоль) в Et2O (3 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением продукта (47b) в виде масла, который использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил пропионата (47).
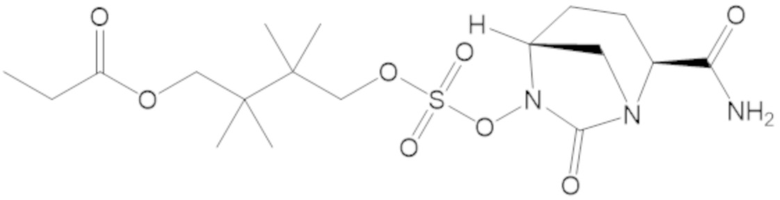
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида 91) (271 мг, 1,5 ммоль) в THF (10 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,6 мл, 1,6 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил пропионат (47b) (0,44 г, 1,5 ммоль) в THF (5 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (47) (300 мг) в виде твердого вещества. LC/MS: м/з=450,09 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,49 (s, 1H), 5,62 (s, 1H), 4,77 (d, J=8,7 Гц, 1H), 4,45 (d, J=9,3 Гц, 1H), 4,19 (s, 1H), 4,05 (d, J=6,3 Гц, 1H), 3,95 (s, 2H), 3,35 (d, J=12,0 Гц, 1H), 3,01 (d, J=12,3 Гц, 1H), 2,46-2,34 (m, 3H), 2,19-2,15 (m, 1H), 1,92-1,83 (m, 2H), 1,16 (t, J=7,7 Гц, 3H), 1,01 (d, J=9,3 Гц, 6H), 0,96 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 174,5, 171,0, 167,1, 82,5, 70,0, 62,0, 60,2, 47,3, 39,1, 38,4, 27,8, 21,0, 20,9, 20,8, 20,4, 20,2, 17,5, 9,3.
Пример 48
Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил) сульфата (48)
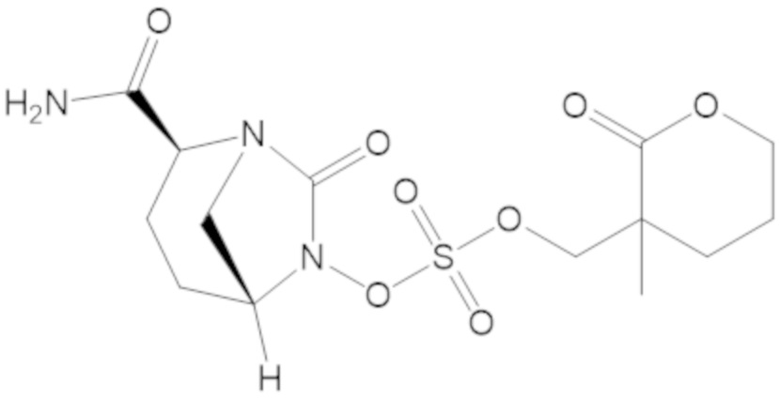
Стадия 1: Синтез 3-((бензилокси)метил)-3-метилтетрагидро-2H-пиран-2-она (48a).
δ-валеролактон (5,23 г, 52,2 ммоль) растворяли в смеси THF (120 мл) и HMPA (9,2 мл) в атмосфере аргона. Реакционную смесь охлаждали до -78°C и встряхивали в течение 10 мин. В течение 5 мин по каплям добавляли раствор диизопропиламида лития, 2,0 M в THF (28,7 мл, 57,5 ммоль). Реакционную смесь встряхивали при -78°C в течение 30 минут, а затем в реакционную смесь в течение 5 мин добавляли неразбавленный MeI (3,3 мл, 52,8 ммоль). Смесь встряхивали при -78°C в течение 30 минут, затем удаляли из охлаждающей ванны и обеспечивали нагревание до 0°C и встряхивали в течение 30 минут. (примечание: в это время смесь постепенно становилась желтой). Смесь повторно охлаждали до -78°C и встряхивали в течение 10 мин, а затем в течение 5 мин добавляли дополнительное количество диизопропиламида лития, 2,0 M в THF (28,7 мл, 57,5 ммоль). Реакционную смесь встряхивали при -78°C в течение 30 минут, затем в течение 5 мин добавляли неразбавленный бензил хлорметиловый эфир (70%; 10,5 мл, 52,8 ммоль). Смесь оставляли нагреваться до комнатной температуры, и встряхивали в течение 16 ч. Затем растворитель удаляли в вакууме, а остаток разделяли на насыщенный хлорид аммония (200 мл) и EtOAc (200 мл). Водный слой экстрагировали EtOAc (2×100 мл), а объединенные органические слои промывали рассолом (2×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме (19 г). Остаток загружали методом сухого ввода на силикагель, и очищали с помощью колоночной хроматографии на силикагеле (картридж 120 г) с использованием в качестве элюента EtOAc/гексанов с получением продукта, загрязненного примесями (6,9 г). Остаток повторно очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента DCM/гексанов (0:1-4:1) с получением продукта (48a) (1,76 г) в виде жидкости. 1H-NMR (300 МГц, CDCl3): δ 7,29-7,37 (m, 5H), 4,61 (dd, J=21,0, 12,3 Гц, 2H), 4,32-4,38 (m, 2H), 3,26-3,81 (dd, J=15,8, 8,1 Гц, 2H), 2,21-2,30 (m, 1H), 1,87-1,94 (m, 2H), 1,59-1,66 (m, 1H), 1,23 (s, 3H).
Стадия 2: Синтез 3-(гидроксиметил)-3-метилтетрагидро-2H-пиран-2-она (48b).
3-((бензилокси)метил)-3-метилтетрагидро-2H-пиран-2-он (48a) (0,52 г, 2,2 ммоль) растворяли в 2-пропаноле (25 мл), и раствор дегазировали и подвергали обратной промывке аргоном. (Примечание: не использовали MeOH в качестве растворителя, так как во время гидрогенизации можно раскрыть кольцо лактона). В смесь добавляли палладий на углероде, 10% (0,26 г, 0,2 ммоль), и систему герметично закрывали. Реакционную смесь дегазировали, подвергали обратной промывке водородом (3 раза) и встряхивали в атмосфере водорода в течение 2 ч. Суспензию фильтровали через слой Celite®, и фильтрационный осадок промывали свежим 2-пропанолом (2×50 мл). Фильтрат концентрировали в вакууме, и продукт (48b) использовали без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,27-4,45 (m, 2H), 3,67 (d, J=11,4 Гц, 1H), 3,52 (d, J=11,1 Гц, 1H), 1,84-2,03 (m, 2H), 1,58-1,64 (m, 1H), 1,29 (s, 3H).
Стадия 3: Синтез (3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил сульфохлорида (48c).
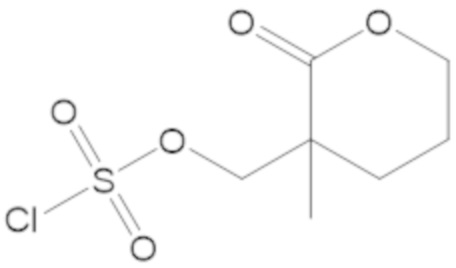
Раствор 3-(гидроксиметил)-3-метилтетрагидро-2H-пиран-2-она (48b) (0,32 г, 2,2 ммоль) и пиридина (0,21 мл, 2,6 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере аргона. В описанный выше раствор с помощью шприца по каплям добавляли неразбавленный сульфурилхлорид (0,21 мл, 2,6 ммоль). Смесь встряхивали при -78°C в течение 10 мин, затем колбу нагревали до комнатной температуры и встряхивали в течение 1 ч (контролировали с помощью TLC EtOAc/гексаны, 3:7). Осадок формовали с получением густой суспензии. Суспензию фильтровали через 0,45 мкМ фильтр Teflon®, и фильтрационный осадок промывали свежим Et2O (2×5 мл). Брали аликвоту (0,5 мл) и концентрировали, и для смеси проводили NMR. Оставшийся раствор, содержащий продукт (48c), использовали непосредственно на следующей стадии. 1H-NMR (300 МГц, CDCl3): δ 4,87 (d, J=9,3 Гц, 1H), 4,25-4,50 (m, 2H), 4,32 (d, J=8,7 Гц, 1H), 2,00-2,20 (m, 2H), 1,75-2,00 (m, 2H), 1,39 (s, 3H).
Стадия 4: Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил) сульфата (48).
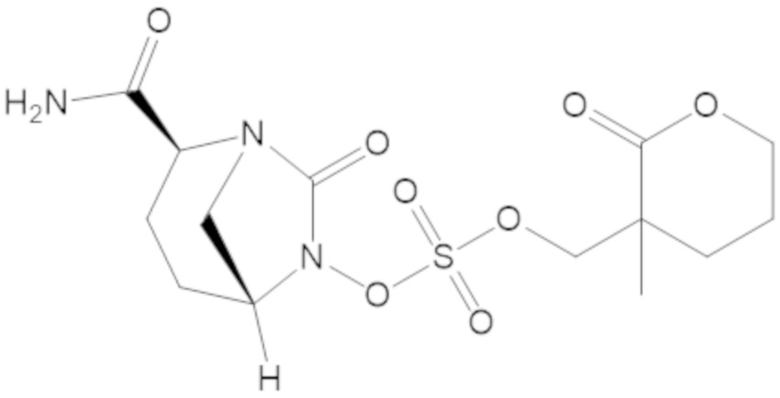
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,39 г, 2,1 ммоль) растворяли в THF (18 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,65 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли NaHMDS, 1,0 M в THF (2,3 мл, 2,3 ммоль) и встряхивали в течение 1 ч. В реакционную смесь быстро добавляли раствор (3-метил-2-оксотетрагидро-2H-пиран-3-ил)метил сульфохлорида (48c) (0,51 г, 2,1 ммоль) в Et2O (из предыдущей реакции). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение ночи. В реакционную смесь добавляли рассол (100 мл) и EtOAc (100 мл), и разделяли водные и органические слои. Водный слой экстрагировали EtOAc (2×100 мл), и объединенные органические слои промывали рассолом (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток загружали методом сухого ввода на силикагель (8 г) и очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:4-1:0) с получением нужного продукта (48) (0,21 г,) в виде твердого вещества. LC-MS: м/з=392 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,55 (br. d, J=9,3 Гц, 1H), 5,77 (br. s, 1H), 4,83-5,03 (m, 1H), 4,56 (m, 0,5H), 4,33-4,45 (m, 2,5H), 4,17 (m, 1H), 4,06 (m, 1H), 3,35 (d, J=9,3 Гц, 1H), 3,04 (m, 1H), 2,38-2,44 (m, 1H), 1,68-2,20 (m, 7H), 1,36 (d, J=7,2 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ 172,8, 172,5, 171,2, 171,0, 167,3, 167,1, 80,4, 79,9, 76,7, 70,8, 70,8, 62,0, 60,3, 60,2, 47,2, 47,1, 43,1, 43,0, 29,8, 29,6, 22,9, 22,9, 20,9 20,8, 20,17. (Примечание: существует несколько сигналов, которые разделены вследствие хиральности в продукте лактона).
Пример 49
Синтез 2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилацетата (49)
Стадия 1: Синтез этил 3-(2-метоксифенил)-2,2-диметилпропаноата (49a).
Встряхиваемый раствор диизопропиламида лития, 2,0 M в THF (26,6 мл, 53,2 ммоль) разбавляли THF (100 мл), охлаждали до -78°C в атмосфере аргона, и встряхивали в течение 5 мин. По каплям добавляли неразбавленный этилизобутират (6,68 мл, 49,7 ммоль) в течение 15 мин, и обеспечивали встряхивание смеси при -78°C в течение 1 ч. По каплям добавляли раствор 1-(бромметил)-2-метоксибензола (полученного согласно J. Am. Chem. Soc. 2013, 135, 11951) (12,0 г, 59,7 ммоль) в THF (100 мл) в течение 30 мин. Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 20 ч. Реакцию гасили рассолом (100 мл), и экстрагировали Et2O (4×100 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле (колонка 120 г) с использованием в качестве элюента EtOAc/гексанов (0:1-5:95) с получением продукта (49a) в виде жидкости (8,06 г, 68%). 1H-NMR (300 МГц, CDCl3): δ 7,18 (dt, J=1,8, 8,1 Гц, 1H), 7,06 (dd, J=1,5, 8,1 Гц, 1H), 6,82-6,87 (m, 2H), 4,12 (q, J=6,9 Гц, 2H), 3,77 (s, 3H), 2,92 (s, 2H), 1,26 (t, J=6,9 Гц, 3H), 1,15 (s, 6H).
Стадия 2: Синтез 3,3-диметилхроман-2-она (49b).
Этил 3-(2-метоксифенил)-2,2-диметилпропаноат (49a) (8,1 г, 34,2 ммоль) растворяли в DCM (200 мл) и охлаждали до 0°C в атмосфере аргона. В холодный раствор по каплям добавляли раствор BBr3 (3,6 мл, 37,7 ммоль) в DCM (100 мл). Смесь нагревали до комнатной температуры и встряхивали в течение ночи (во время реакции образовалось твердое вещество). Окрашенную суспензию охлаждали в ледяной водяной ванне, и в смесь добавляли воду (150 мл). Разделяли органические и водные слои, и водный слой экстрагировали DCM (3×75 мл). Объединенные органические слои сушили (MgSO4; примечание: раствор становилась более темным), фильтровали и концентрировали в вакууме с получением продукта (49b) (4,85 г, 80%) в виде масла. Этот материал использовали без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,01-7,25 (m, 3H), 2,85 (s, 2H), 1,29 (s, 6H).
Стадия 3: Синтез 2-(3-гидрокси-2,2-диметилпропил)фенола (49c).
LiAlH4 (1,94 г, 51,1 ммоль) суспендировали в Et2O (52,5 мл) в атмосфере аргона, и смесь охлаждали до 0°C в ледяной водяной ванне, и в суспензию по каплям добавляли раствор 3,3-диметилхроман-2-она (49b) (4,85 г, 27,5 ммоль) в Et2O (50 мл) в течение 30 мин. Смесь нагревали до комнатной температуры и встряхивали в течение 20 ч. Смесь охлаждали в ледяной водяной ванне и воде (2 мл), после чего путем медленного добавления добавляли 15% водный гидроксид натрия (2 мл) и воду (6 мл). Смесь нагревали до комнатной температуры и встряхивали в течение 15 мин. В суспензию добавляли безводный MgSO4, и смесь встряхивали в течение 15 мин. Смесь фильтровали, и фильтрационный осадок промывали Et2O (3×50 мл). Фильтрат концентрировали в вакууме с получением продукта (49c) (4,34 г, 88%) в виде твердого вещества. Этот материал использовали без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,15 (dt, J=8,1, 1,5 Гц, 1H), 7,04 (dd, J=7,5, 1,8 Гц, 1H), 6,82-7,01 (m, 2H), 3,22 (s, 2H), 2,61 (s, 2H), 0,98 (s, 6H).
Стадия 4: Синтез 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенола (49d).
Раствор 2-(3-гидрокси-2,2-диметилпропил)фенола (49c) (4,0 г, 22,2 ммоль) и имидазола (3,8 г, 56,0 ммоль) растворяли в DMF (50 мл), и в раствор добавляли трет-бутилдиметилсилил хлорид (4,0 г, 26,6 ммоль) и встряхивали в течение 2 ч. Растворитель удаляли при большом разрежении, а остаток очищали с помощью колоночной хроматографии на силикагеле (картридж 40 г) с гексанами (5:95-2:3) в качестве элюента с получением продукта (49d) в виде масла (7,34 г, >100%). Соединение было чистым приблизительно на 90%, и его использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,11 (dt, J=7,5, 1,8 Гц, 1H), 7,10 (dd, J=7,5, 1,8 Гц, 1H), 6,90 (dd, J=8,1, 1,5 Гц, 1H), 6,79 (dt, J=6,9, 0,9 Гц, 1H), 3,17 (s, 2H), 2,57 (s, 2H), 0,97 (s, 9H), 0,92 (s, 6H), 0,13 (s, 6H).
Стадия 5: Синтез 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенилацетата (49e).
Раствор 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенола (49d) (чистота около 90%; 2,5 г, 7,6 ммоль) и Et3N (2,3 г, 22,9 ммоль) в THF (90 мл) охлаждали до 0°C в ледяной ванне в атмосфере аргона. По каплям в смесь добавляли ацетилхлорид (0,65 мл, 9,2 ммоль), а после завершения добавления ледяную ванну удаляли. Обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение 2 ч. Суспензию фильтровали, и твердое вещество промывали свежим THF (2×20 мл). Фильтрат концентрировали в вакууме, а остаток загружали на силикагель методом сухого ввода, затем очищали с помощью колоночной хроматографии на силикагеле (картридж 40 г) с использованием в качестве элюента 0-8% EtOAc/гексанов (0:1-8:92) с получением продукта (49e) (2,16 г, 84%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,11-7,27 (m, 3H), 7,04 (d, J=7,5 Гц, 1H), 3,25 (s, 2H), 2,51 (s, 2H), 2,30 (s, 3H), 0,93 (s, 9H), 0,81 (s, 6H), 0,06 (s, 6H).
Стадия 6: Синтез 2-(3-гидрокси-2,2-диметилпропил)фенилацетата и 3-(2-гидроксифенил)-2,2-диметилпропил ацетата (49f).
Пиридин гидрофторид (70%, 1,3 мл, 10,4 ммоль) добавляли во встряхиваемый раствор 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенилацетата (49e) (0,70 г, 2,1 ммоль) и пиридина (2,5 мл, 31,2 ммоль) в THF (25 мл) при комнатной температуре в атмосфере аргона, и смесь встряхивали в течение 24 ч. Растворитель удаляли в вакууме (температуру бани устанавливали 25°C), и остаток растворяли в EtOAc (100 мл), промывали рассолом (3×75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением смеси нужного спирта и 3-(2-гидроксифенил)-2,2-диметилпропил ацетата 65:35. анализ NMR показал присутствие обоих сложных эфиров продукта (49f). Этот материал использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3) нужного продукта: δ 6,8-7,26 (m, 4H), 3,79 (s, 2H), 3,27 (s, 2H), 2,62 (s, 2H), 2,53 (s, 2H), 2,33 (s, 3H), 2,13 (s, 3H), 0,974 (s, 6H), 0,90 (s, 6H).
Стадия 7: Синтез 2-(3-((хлорсульфонил)окси)-2,2-диметилпропил)фенилацетата (49g).
Раствор сульфурилхлорида (172 мкл, 2,1 ммоль) в Et2O (6,8 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида через канюлю по каплям добавляли раствор 2-(3-гидрокси-2,2-диметилпропил)фенилацетата (49f) (0,43 г, 1,9 ммоль) и пиридина (172 мкл, 2,1 ммоль) в Et2O (2,0 мл). Смесь встряхивали при -78°C в течение 10 мин, затем колбу нагревали до комнатной температуры и встряхивали в течение 1,5 ч (контролировали с помощью TLC 30% EtOAc/гексаны). Суспензию фильтровали через 0,45 мкм шприцевой фильтр из PTFE, и шприцевой фильтр промывали свежим Et2O (10 мл) с получением продукта (49g). Фильтрат сразу использовали на следующей стадии без дополнительной очистки. Выход оценивали количественно.
Стадия 8: Синтез 2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилацетата (49).
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,44 г, 2,4 ммоль) растворяли в THF (22 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,1 мл, 2,1 ммоль) и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 2-(3-((хлорсульфонил)окси)-2,2-диметилпропил)фенилацетата (49g) (0,62 г, 1,9 ммоль) в Et2O из предыдущей реакции. Спустя 10 мин реакционную смесь нагревали до комнатной температуры и встряхивали в течение ночи. В реакционную смесь добавляли рассол (100 мл), и разделяли водные и органические слои. Водный слой экстрагировали EtOAc (2×200 мл), и объединенные органические слои промывали рассолом (3×75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в DMF (6 мл) и фильтровали через 0,45 мкм фильтр, а затем очищали с помощью препаративной HPLC с 20-90% MeCN/вода (без модификатора) в качестве элюента с получением продукта (49) (0,18 г, 20%) в виде твердого вещества. LC-MS: м/з=470 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,23-7,29 (m, 1H), 7,18-7,20 (m, 2H), 7,08 (d, J=7,5 Гц, 1H), 6,52 (br. s, 1H), 5,78 (br. s, 1H), 4,55 (d, J=7,5 Гц, 1H), 4,19-4,22 (m, 2H), 4,05 (br. d, J=7,2 Гц, 1H), 3,30 (br. d, J=11,7 Гц, 1H), 2,99 (d, J=11,7 Гц, 1H), 2,51-2,62 (m, 2H), 2,39-2,44 (m, 1H), 2,34 (s, 3H), 2,10-2,19 (m, 1H), 1,84-2,00 (m, 2H), 0,98 (d, J=3,0 Гц, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 169,6, 167,1, 149,6, 132,8, 129,1, 128,0, 125,8, 122,7, 83,4, 62,0, 60,3, 47,2, 38,1, 36,3, 23,7, 21,3, 20,8, 17,6.
Пример (50)
Синтез 2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилпивалата (50)
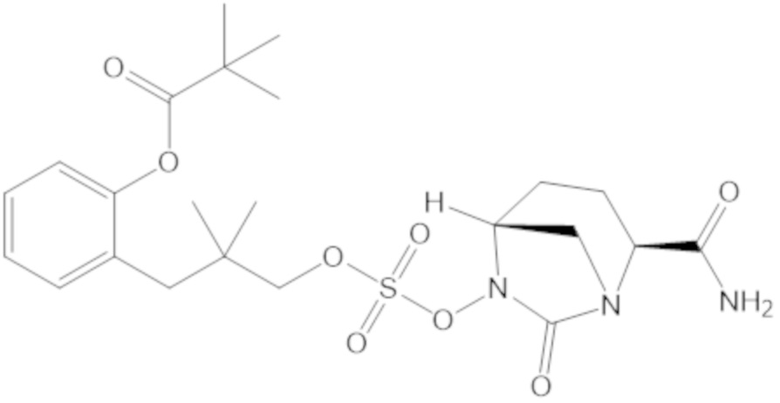
Стадия 1: Синтез 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенилпивалата (50a).
2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенол (0,9 г, 3,1 ммоль) и N, N-4-диметиламинопиридин (0,93 г, 7,6 ммоль) растворяли в THF (50 мл) в атмосфере аргона. По каплям в смесь добавляли триметилацетилхлорид (0,45 мл, 3,7 ммоль) при комнатной температуре с немедленным образованием белого твердого вещества, и добавление продолжали до образования суспензии. Реакционную смесь встряхивали при комнатной температуре в течение 2 ч, а затем фильтровали, и фильтрационный осадок промывали THF (10 мл). Фильтрат загружали на силикагель методом сухого ввода (15 г) и очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-6:94) с получением продукта (50a), загрязненного около 3% исходного материала по данным анализа NMR. Этот материал использовали без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,27 (dd, J=7,2, 2,1 Гц, 1H), 7,21 (dt, J=7,5, 1,8 Гц, 1H), 7,15 (dt, J=7,8, 1,8 Гц, 1H), 6,97 (dd, J=8,1, 1,8 Гц, 1H), 3,25 (s, 2H), 2,49 (s, 2H), 1,38 (s, 9H), 0,92 (s, 9H), 0,82 (s, 6H), 0,05 (s 6H).
Стадия 2: Синтез 2-(3-гидрокси-2,2-диметилпропил)фенилпивалата (50b).
Пиридин гидрофторид (70%, 1,3 мл, 10,4 ммоль) добавляли во встряхиваемый раствор 2-(3-((трет-бутилдиметилсилил)окси)-2,2-диметилпропил)фенилпивалата (50a) (0,70 г, 1,8 ммоль) и пиридина (2,5 мл, 31,2 ммоль) в THF (25 мл) при комнатной температуре в атмосфере аргона, и смесь встряхивали в течение 24 ч. Растворитель удаляли в вакууме (температуру бани устанавливали 25°C), и остаток растворяли в EtOAc (100 мл) и промывали рассолом (3×75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением нужного продукта (50b) в виде масла. Этот материал использовали непосредственно на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,12-7,26 (m, 3H), 6,98 (m, 1H), 3,31 (s, 2H), 2,51 (s, 2H), 1,39 (s, 9H), 0,89 (s, 9H).
Стадия 3: Синтез 2-(3-((хлорсульфонил)окси)-2,2-диметилпропил)фенилпивалата (50c).
Раствор сульфурилхлорида (173 мкл, 2,1 ммоль) в Et2O (7,5 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида через канюлю по каплям добавляли раствор 2-(3-гидрокси-2,2-диметилпропил)фенилпивалата (50b) (0,47 г, 1,8 ммоль) и пиридина (173 мкл, 2,1 ммоль) в Et2O (2,2 мл). Смесь встряхивали при -78°C в течение 10 мин, а затем колбу нагревали до комнатной температуры и встряхивали в течение 1,5 ч (контролировали с помощью TLC 30% EtOAc/гексаны). Суспензию фильтровали через 0,45-мкм шприцевой фильтр из PTFE, и шприцевой фильтр промывали свежим Et2O с получением продукта (50c). Фильтрат сразу использовали на следующей стадии без дополнительной очистки. Выход оценивали количественно.
Стадия 4: Синтез 2-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил)фенилпивалата (50).
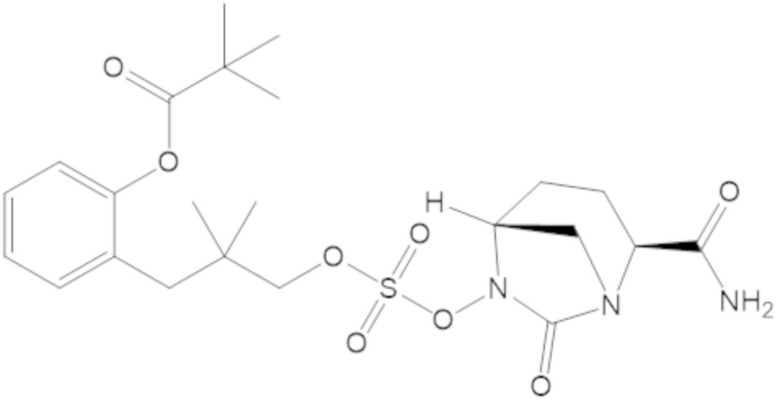
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,41 г, 2,2 ммоль) растворяли в THF (23 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,2 мл, 2,2 ммоль), и раствор встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 2-(3-((хлорсульфонил)окси)-2,2-диметилпропил)фенилпивалата (50c) (0,65 г, 1,8 ммоль) в Et2O из предыдущей реакции. Спустя 10 мин реакционную смесь нагревали до комнатной температуры и встряхивали в течение ночи. В реакционную смесь добавляли рассол (100 мл), и разделяли водные и органические слои. Водный слой экстрагировали EtOAc (2×200 мл), и объединенные органические слои промывали рассолом (3×75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в DMF (6 мл) и фильтровали через 0,45-мкм фильтр, а затем очищали с помощью препаративной HPLC с получением продукта (50) (0,21 г, 23%) в виде твердого вещества. LC-MS: м/з=512 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,12-7,28 (m, 3H), 6,99 (d, J=7,5 Гц, 1H), 6,50 (br. s, 1H), 5,78 (br. s, 1H), 4,57 (d, J=9,3 Гц, 1H), 4,26 (d, J=8,7 Гц, 1H), 4,17 (br. s, 1H), 4,04 (d, J=6,3 Гц, 1H), 3,28 (br. d, J=11,7 Гц, 1H), 3,03 (d, J=11,7 Гц, 1H), 2,52-2,62 (m, 2H), 2,39-2,50 (m, 1H), 2,10-2,20 (m, 1H), 1,78-1,98 (m, 2H), 1,38 (s, 9H), 0,97 (d, J=4,2 Гц, 6H). 13C-NMR (75 МГц, CDCl3): δ 177,2, 171,1, 167,0, 150,1, 132,5, 129,2, 127,9, 125,6, 122,6, 83,7, 61,9, 60,2, 47,2, 39,3, 37,5, 36,2, 27,4, 23,5, 20,8, 17,6.
Пример 51
Синтез S-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) этантиоата (51)
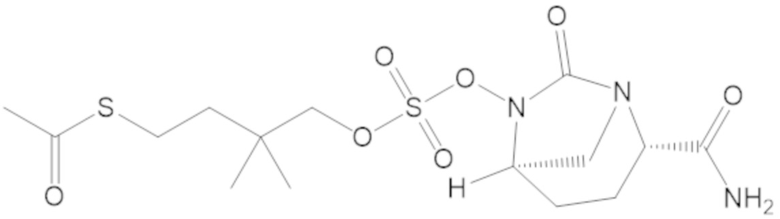
Стадия 1: Синтез S-(4-((хлорсульфонил)окси)-3,3-диметилбутил) этантиоата (51a).
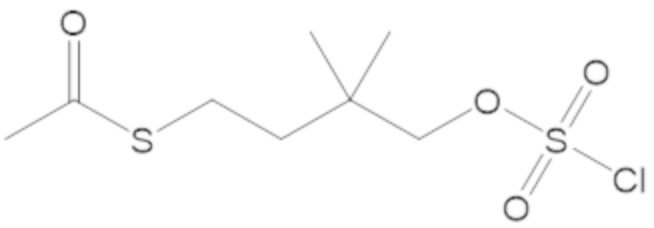
Раствор свежедистиллированного сульфурилхлорида (271 мкл, 3,7 ммоль) в Et2O (5 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор S-(4-гидрокси-3,3-диметилбутил) этантиоата (полученного согласно Chem. Commun. 2011, 47, 2038) (500 мг, 2,8 ммоль) и пиридина (267 мкл, 3,3 ммоль) в Et2O (3 мл). Колбу промывали диэтиловым эфиром (2×5 мл), а смыв также добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 1 ч и обеспечивали нагревание до комнатной температуры и встряхивали при комнатной температуре еще в течение 20 мин. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (51a) в виде масла, которое сразу использовали для следующей стадии без дополнительной очистки.
Стадия 2: Синтез S-(4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил) этантиоата (51).
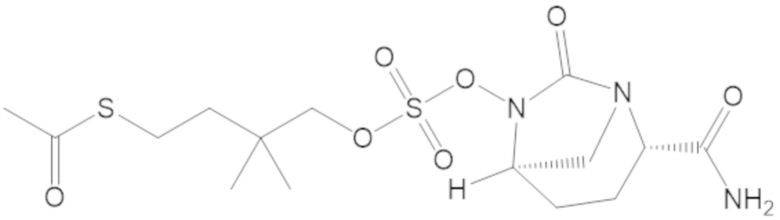
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (430 мг, 2,3 ммоль) растворяли в пиридине (8 мл) и охлаждали до 0°C в атмосфере аргона. В реакционную смесь при 0°C добавляли раствор S-(4-((хлорсульфонил)окси)-3,3-диметилбутил) этантиоата (51a) (0,75 г, 2,7 ммоль) в THF (4 мл). Полученную в результате смесь встряхивали в течение 1 ч, и растворители удаляли при большом разрежении при комнатной температуре. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-9:1) с получением продукта (50) в виде твердого вещества. LCMS: м/з=424,2 [M+1]+. 1H-NMR (300 МГц, CDCl3): δ 6,47 (br. s, 1H), 5,61 (br. s, 1H), 4,53 (d, J=8,7 Гц, 1H), 4,24 (d, J=9,0 Гц, 1H), 4,17 (br. s, 1H), 4,05-4,03 (m, 1H), 3,39-3,30 (m, 1H), 3,01 (d, J=9,0 Гц, 1H), 2,85-2,80 (m, 2H), 2,48-2,40 (m, 1H), 2,32 (s, 3H), 2,20-2,18 (m, 1H), 2,01-1,80 (m, 2H), 1,03 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 195,4, 170,9, 166,9, 83,1, 61,8, 60,0, 47,1, 38,4, 34,8, 30,5, 23,9, 23,4, 23,2, 20,6, 17,4.
Пример 52
Синтез S-(5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил) этантиоата (52)
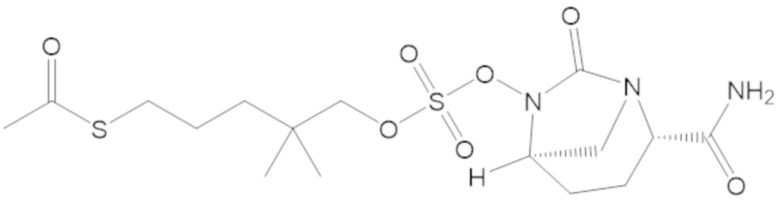
Стадия 1: Синтез 5-бром-2,2-диметилпентан-1-ола (52a).
В LiBH4 (0,66 г, 30,4 ммоль) добавляли DCM (18 мл) с последующим добавлением по каплям безводного MeOH (1,2 мл, 30,4 ммоль) в течение 20 мин в атмосфере аргона. После прекращения выделения газа H2 в течение 20 мин по каплям добавляли раствор этил 5-бром-2,2-диметилпентаноата (полученного согласно публикации заявки PCT № 2011046771) (4,5 г, 19,0 ммоль) в DCM (10 мл). Реакционную смесь нагревали с обратным холодильником в течение 16 ч, охлаждали до комнатной температуры и осторожно гидролизировали насыщенным раствором NH4Cl (30 мл). Суспензию экстрагировали DCM (3×50 мл). Объединенные органические слои промывали 1 н. HCl (26 мл) и рассолом (40 мл), сушили и концентрировали в вакууме с получением продукта (52a) (3,61 г, 97%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,39 (t, J=6,9 Гц, 2H), 3,24 (s, 2 H), 1,90-1,76 (m, 2 H), 1,48 (br. s, 1H), 1,41-1,36 (m, 2H), 0,88 (s, 6H).
Стадия 2: Синтез S-(5-гидрокси-4,4-диметилпентил) этантиоата (52b).
Раствор 5-бром-2,2-диметилпентан-1-ола (52a) (2,0 г, 10,3 ммоль) и тиоацетата калия (2,34 г, 20,5 ммоль) в ацетоне (22 мл) встряхивали в инертной атмосфере при комнатной температуре в течение 23 ч. После удаления растворителей в вакууме при комнатной температуре остаток очищали с помощью колонки колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:3) с получением продукта (52b) (1,2 г, 61%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,31 (s, 2H), 2,85 (t, J=7,8 Гц, 2H), 2,32 (s, 3 H), 1,62-1,48 (m, 2 H), 1,32-1,21 (m, 2H), 0,86 (s, 6H).
Стадия 3: Синтез S-(5-((хлорсульфонил)окси)-4,4-диметилпентил) этантиоата (52c).
Раствор свежедистиллированного сульфурилхлорида (379 мкл, 5,2 ммоль) в Et2O (8 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор S-(5-гидрокси-4,4-диметилпентил) этантиоата (52b) (700 мг, 3,6 ммоль) и пиридина (374 мкл, 4,6 ммоль) в Et2O (4 мл). Смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (10 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (52c) в виде масла, которое сразу использовали для следующей стадии без дополнительной очистки.
Стадия 4: Синтез S-(5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил) этантиоата (52).
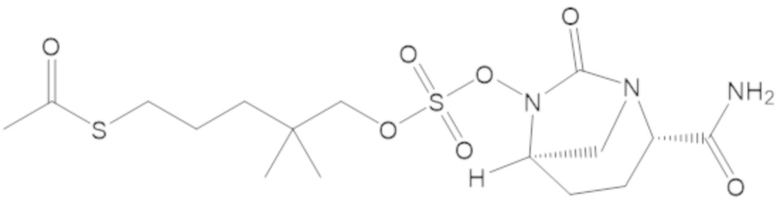
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (600 мг, 3,2 ммоль) растворяли в пиридине (9 мл) и охлаждали до 0°C в атмосфере аргона. В реакционную смесь при 0°C добавляли раствор S-(5-((хлорсульфонил)окси)-4,4-диметилпентил) этантиоата (52c) (857 мг, 3,0 ммоль) в THF (5 мл). Полученную в результате смесь встряхивали в течение 1 ч, и растворители удаляли при большом разрежении при комнатной температуре. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-9:1) с получением продукта (52) (70 мг, 5%) в виде твердого вещества. LC-MS: м/з=438,2 [MH]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (br. s, 1H), 5,56 (br. s, 1H), 4,49 (d, J=9,0 Гц, 1H), 4,19 (d, J=9,6 Гц, 1H), 4,17 (br. s, 1H), 4,05-4,03 (m, 1H), 3,38-3,30 (m, 1H), 3,01 (d, J=9,0 Гц, 1H), 2,85 (t, J=6,9 Гц, 2H), 2,46-2,40 (m, 1H), 2,32 (s, 3H), 2,21-2,18 (m, 1H), 1,98-1,82 (m, 2H), 1,40-1,33 (m, 2H), 0,97 (s, 3H), 0,96 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 175,7, 170,8, 166,9, 83,6, 61,8, 60,0, 47,1, 37,4, 34,3, 30,6, 29,4, 23,9, 23,6, 23,4, 20,6, 17,4.
Пример 53
Синтез S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53)
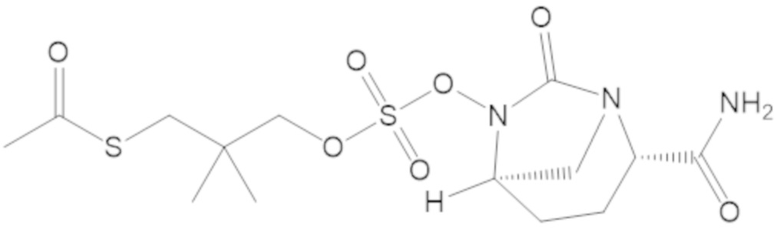
Стадия 1: Синтез S-(3-гидрокси-2,2-диметилпропил) этантиоата (53a).
Калия тиоацетат (4,1 г, 35,8 ммоль) растворяли в DMF (20 мл) в атмосфере аргона. Добавляли 3-гидрокси-2,2-диметилпропил 4-метилбензолсульфонат (полученного согласно публикации заявки PCT № 2012165648) (4,2 г, 16,3 ммоль), и смесь встряхивали при 80°C в течение 2,5 ч. После охлаждения добавляли рассол (100 мл), и смесь экстрагировали Et2O (3×100 мл). Объединенные органические слои промывали рассолом (5×50 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме (остаточный DMF удаляли с помощью большого разрежения). Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-15:85) с получением продукта (53a) (1,06 г, 40%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,23 (br. s, 2H), 2,89 (s, 2H), 2,62 (br. s, 1H), 2,37 (s, 3H), 0,94 (s, 6H).
Стадия 2: Синтез S-(3-((хлорсульфонил)окси)-2,2-диметилпропил) этантиоата (53b).
Раствор свежедистиллированного сульфурилхлорида (283 мкл, 3,9 ммоль) в Et2O (4 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор S-(3-гидрокси-2,2-диметилпропил) этантиоата (53a) (520 мг, 3,1 ммоль) и пиридина (327 мкл, 4,0 ммоль) в Et2O (6 мл). Смесь встряхивали при -78°C в течение 1 ч, затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (10 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (53b) в виде масла, которое сразу использовали для следующей стадии без дополнительной очистки.
Стадия 3: Синтез S-(3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил) этантиоата (53).
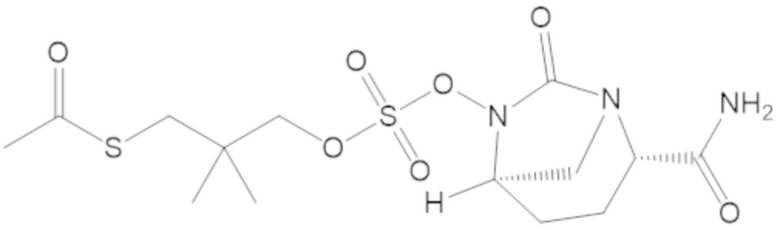
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (600 мг, 3,2 ммоль) растворяли в пиридине (10 мл) и охлаждали до 0°C в атмосфере аргона. В реакционную смесь при 0°C добавляли раствор S-(5-((хлорсульфонил)окси)-4,4-диметилпентил) этантиоата (53b) (800 мг, 3,1 ммоль) в THF (6 мл). Полученную в результате смесь встряхивали в течение 2 ч, и растворители удаляли при большом разрежении при комнатной температуре. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (53) (90 мг, 7%) в виде твердого вещества. LCMS: м/з=410,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,47 (br. s, 1H), 5,56 (br. s, 1H), 4,51 (d, J=9,3 Гц, 1H), 4,25 (d, J=9,6 Гц, 1H), 4,17 (br. s, 1H), 4,06-4,03 (m, 1H), 3,36-3,24 (m, 1H), 3,06-2,88 (m, 2H), 2,51-2,41 (m, 1H), 2,36 (s, 3H), 2,21-2,08 (m, 1H), 1,98-1,82 (m, 2H), 1,36-1,22 (m, 1H), 1,04 (s, 3H), 1,02 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 194,7, 170,8, 166,9, 81,9, 61,8, 60,1, 47,1, 36,7, 35,6, 30,6, 23,2, 22,9, 20,6, 17,4.
Пример 54
Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54)
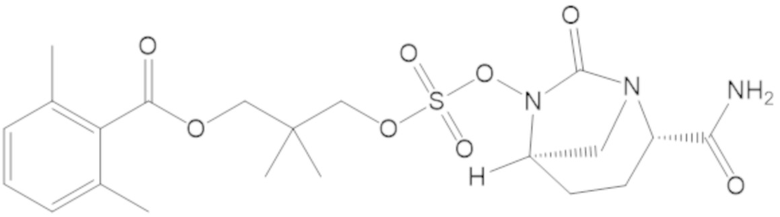
Стадия 1: Синтез 3-гидрокси-2,2-диметилпропил 2,6-диметилбензоата (54a).
Во встряхиваемый раствор 2,2-диметилпропан-1,3-диола (2,5 г, 24,3 ммоль) в DCM (60 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли 2,6-диметилбензоилхлорид (1,2 мл, 8,1 ммоль), пиридин (1,1 мл, 13,7 ммоль) и N, N-4-диметиламинопиридин (99 мг, 0,8 ммоль). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Реакцию гасили путем добавления 1 н. HCl, и смесь экстрагировали DCM (два раза). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 и рассола, сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:9-2:3) с получением продукта (54a) (1,5 г, 78%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,21 (m, 1H), 7,04 (m, 2H), 4,18 (s, 2H), 3,41 (s, 2H), 2,32 (s, 6H), 2,20 (br. s, 1H), 0,99 (s, 6H).
Стадия 2: Синтез 3-((хлорсульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54b).
Раствор свежедистиллированного сульфурилхлорида (0,25 мл, 3,9 ммоль) в Et2O (6 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор 3-гидрокси-2,2-диметилпропил 2,6-диметилбензоата (54a) (500 мг, 2,1 ммоль) и пиридина (0,26 мл, 3,3 ммоль) в Et2O (6 мл). Смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (54b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54).
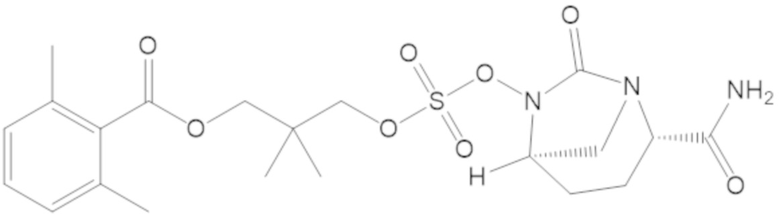
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (420 мг, 2,3 ммоль) растворяли в THF (12 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,7 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,3 мл, 2,3 ммоль), и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 3-((хлорсульфонил)окси)-2,2-диметилпропил 2,6-диметилбензоата (54b) (660 мг, 2,0 ммоль) в THF (8 мл). После встряхивания при -78°C в течение 10 мин обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 2 ч. Добавляли EtOAc (400 мл), насыщенный водный NaHCO3 (40 мл) и воду (40 мл). Органический слой разделяли и промывали насыщенным водным NaHCO3 (60 мл), H2O (3×50 мл), рассолом (60 мл), а затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (180 мг, 19%) в виде твердого вещества. LC-MS: м/з=484,01 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,19 (m, 1H), 7,04 (m, 2H), 6,47 (s, 1H), 5,55 (s, 1H), 4,58 (d, J=9,3 Гц, 1H), 4,39 (d, J=9,0 Гц, 1H), 4,22 (d, J=11,1 Гц, 1H), 4,16 (m, 1H), 4,11 (d, J=11,1 Гц, 1H), 4,04-4,02 (m, 1H), 3,33-3,29 (m, 1H), 3,01-2,98 (m, 1H), 2,45-2,40 (m, 1H), 2,32 (s, 6H), 2,20-2,08 (m, 1H), 1,93-1,76 (m, 2H), 1,10 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 170,8, 169,9, 166,9, 134,9, 133,6, 129,4, 127,5, 80,2, 69,1, 61,8, 60,1, 47,1, 35,2, 21,4, 21,3, 20,7, 19,8, 17,4.
Пример 55
Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил адамантан-1-карбоксилата (55)
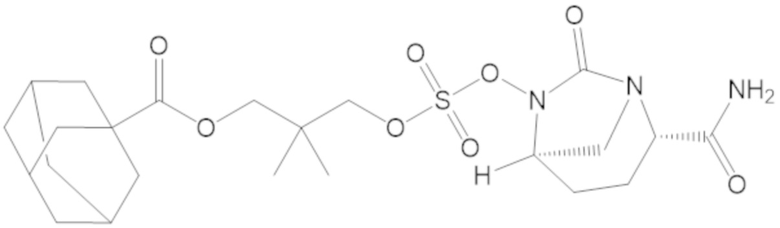
Стадия 1: Синтез 3-гидрокси-2,2-диметилпропил (3r,5r,7r)-адамантан-1-карбоксилата (55a).
Во встряхиваемый раствор 2,2-диметилпропан-1,3-диола (2,5 г, 24,3 ммоль) в DCM (60 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли-1-адамантан-карбонил хлорид (1,36 г, 6,9 ммоль), пиридин (1,1 мл, 13,7 ммоль) и N, N-4-диметиламинопиридин (99 мг, 0,8 ммоль). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Реакцию гасили путем добавления 1 н. HCl, и смесь экстрагировали DCM (два раза). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 и рассола, сушили (MgSO4), фильтровали и концентрировали в вакууме с получением продукта (55a) (1,82 г, 100%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 3,91 (s, 2H), 3,25 (s, 2H), 2,01 (br. s, 3H), 1,89 (br. s, 6H), 1,71 (br. s, 7H), 0,91 (s, 6H).
Стадия 2: Синтез 3-((хлорсульфонил)окси)-2,2-диметилпропил (3r,5r,7r)-адамантан-1-карбоксилата (55b).
Раствор свежедистиллированного сульфурилхлорида (266 мкл, 3,3 ммоль) в Et2O (4 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор 3-гидрокси-2,2-диметилпропил-адамантан-1-карбоксилата (55a) (600 мг, 2,2 ммоль) и пиридина (0,28 мл, 3,5 ммоль) в Et2O (4 мл). Колбу промывали Et2O (5 мл), а смыв также добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (55b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропил адамантан-1-карбоксилата (55).
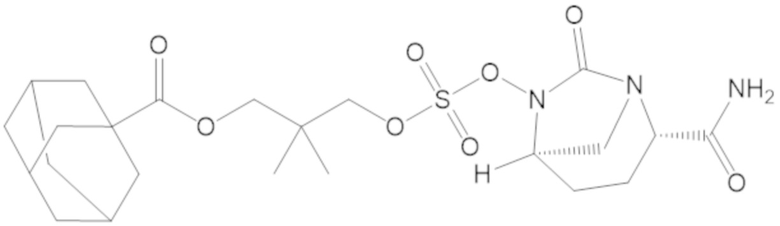
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (442 мг, 2,4 ммоль) растворяли в THF (14 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,6 мл, 2,6 ммоль), и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 3-((хлорсульфонил)окси)-2,2-диметилпропил-адамантан-1-карбоксилата (55b) (750 мг, 2,1 ммоль) в THF (8 мл). После встряхивания при -78°C в течение 10 мин обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 2 ч. Добавляли EtOAc (400 мл) и насыщенный водный NaHCO3 (40 мл) и H2O (40 мл). Органический слой разделяли и промывали насыщенным водным NaHCO3 (60 мл), H2O (3×50 мл), рассолом (60 мл), затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (55) (180 мг, 17%) в виде твердого вещества. LC-MS: м/з=514,12 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,48 (s, 1H), 5,55 (s, 1H), 4,58 (d, J=8,7 Гц, 1H), 4,36 (d, J=9,3 Гц, 1H), 4,17 (m, 1H), 4,05 (d, J=6,9 Гц, 1H), 3,93 (d, J=11,1 Гц, 1H), 3,84 (d, J=11,1 Гц, 1H), 3,34-3,32 (m, 1H), 3,03-2,99 (m, 1H), 2,49-2,41 (m, 1H), 2,20-2,14 (m, 1H), 2,04 (br. s, 3H), 1,91-1,8 (m, 8H), 1,87 (br. s, 6H), 1,03 (s, 7H). 13C-NMR (75 МГц, CDCl3): δ 178,0, 171,7, 167,7, 81,3, 68,7, 62,6, 60,8, 47,8, 41,6, 39,5, 37,2, 36,2, 28,6, 22,0, 21,9, 21,4, 18,1.
Пример 56
Синтез диэтил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилмалоната (56)
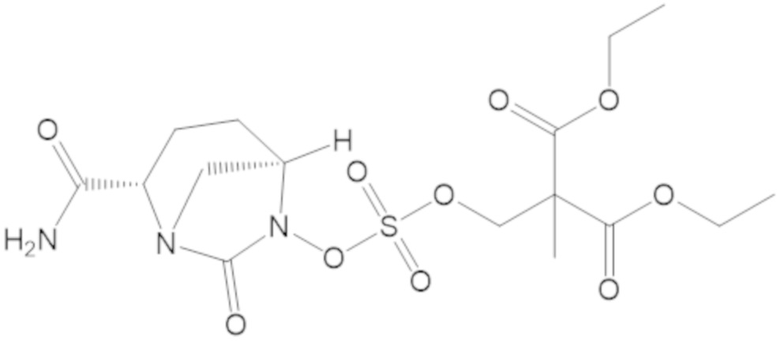
Стадия 1: Синтез диэтил 2-(гидроксиметил)-2-метилмалоната (56a).
В суспензию параформальдегида (1,3 г, 43,3 ммоль) и K2CO3 (11 г, 79 ммоль) в EtOH (150 мл) добавляли диэтил 2-метилмалонат (4,5 мл, 26,3 ммоль). Смесь встряхивали при комнатной температуре в течение 17 ч, затем фильтровали через слой Celite®, и фильтрационный осадок промывали EtOH (2×30 мл). Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением продукта (56a) (4,0 г, 74%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 4,22 (q, J=6,9 Гц, 4H), 3,83 (d, J=6,9 Гц, 2H), 2,90 (t, J=7,8 Гц, 1H), 1,42 (s, 3H), 1,26 (t, J=6,9 Гц, 6H).
Стадия 2: Синтез диэтил 2-(((хлорсульфонил)окси)метил)-2-метилмалоната (56b).
Раствор свежедистиллированного сульфурилхлорида (248 мкл, 3,0 ммоль) в Et2O (8 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор диэтил 2-(гидроксиметил)-2-метилмалоната (56a) (500 мг, 2,4 ммоль) и пиридина (0,26 мл, 3,2 ммоль) в Et2O (4 мл). Колбу промывали Et2O (5 мл), а смыв также добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (56b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез диэтил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилмалоната (56).
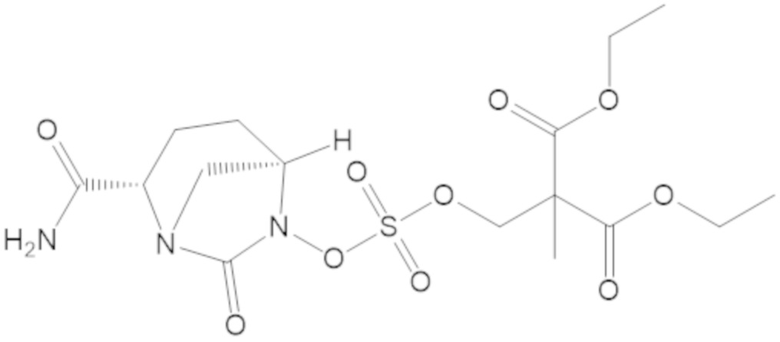
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (390 мг, 2,1 ммоль) растворяли в THF (10 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,2 мл, 2,2 ммоль), и встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор диэтил 2-(((хлорсульфонил)окси)метил)-2-метилмалоната (56b) (638 мг, 2,1 ммоль) в THF (8 мл). После встряхивания при -78°C в течение 10 мин обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 2 ч. Добавляли EtOAc (400 мл) и насыщенный водный NaHCO3 (40 мл) и H2O (40 мл). Органический слой разделяли и промывали насыщенным водным NaHCO3 (60 мл), H2O (3×50 мл), рассолом (60 мл), затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-9:1) с получением продукта (56) (166 мг, 17%) в виде твердого вещества. LC-MS: м/з=452,03 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,49 (s, 1H), 5,58 (s, 1H), 5,02 (d, J=8,7 Гц, 1H), 4,93 (d, J=9,3 Гц, 1H), 4,24 (q, J=7,2 Гц, 4H), 4,17 (m, 1H), 4,05 (d, J=6,9 Гц, 1H), 3,35 (m, 1H), 3,01 (d, J=11,1 Гц, 1H), 2,49-2,41 (m, 1H), 2,20-2,14 (m, 1H), 1,98-1,81 (m, 2H), 1,56 (s, 3H), 1,28 (t, J=7,2 Гц, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 168,6, 168,5, 167,1, 76,5, 62,3, 61,9, 60,2, 53,9, 47,1, 20,8, 17,8, 17,5, 14,0.
Пример 57
Синтез пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57)
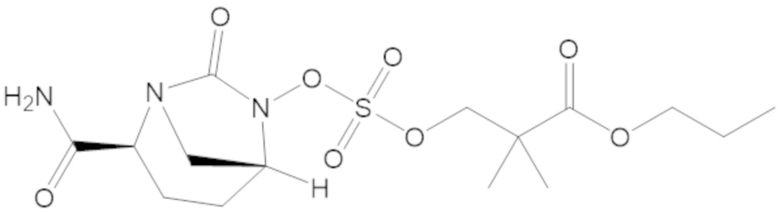
Стадия 1: Синтез пропил 3-гидрокси-2,2-диметилпропаноата (57a).
Загружали смесь 3-гидрокси-2,2-диметилпропановой кислоты (1,15 г, 9,7 ммоль), и 1-пропанол (15 мл) и концентрированную H2SO4 (70 мкл, 1,3 ммоль) в 20 мл сосуде для микроволновой обработки встряхивали при комнатной температуре, а затем нагревали в микроволновом реакторе при 80°C в течение 2 ч, и встряхивали при комнатной температуре в течение ночи. Когда нужный продукт идентифицировали с помощью TLC (EtOAc/гексаны; 3:7), смесь концентрировали в вакууме (40°C) и разбавляли EtOAc (80 мл) и H2O (30 мл). Органический слой промывали H2O (два раза) и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали с получением продукта (57a) (1,18 г, 76%) в виде масла. Материал использовали непосредственно на следующей стадии без очистки. 1H-NMR (300 МГц, CDCl3): δ 4,07 (t, J=6,6 Гц, 2H), 3,55 (s, 2H), 2,42 (br. s, 1H), 1,70-1,61 (m, 2H), 1,19 (s, 6H), 0,95 (t, J=7,5 Гц, 3H).
Стадия 2: Синтез пропил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (57b).
Раствор свежедистиллированного сульфурилхлорида (194 мкл, 2,7 ммоль) в Et2O (1,0 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор пропил 3-гидрокси-2,2-диметилпропаноата (57a) (0,42 г, 2,6 ммоль) и пиридина (215 мкл, 2,7 ммоль) в Et2O. Колбу промывали Et2O (3×1 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 5 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, а фильтрат концентрировали в вакууме с получением титульного соединения (57b) (0,56 г, 83%) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,50 (s, 2H), 4,10 (t, J=6,6 Гц, 2H), 1,72-1,64 (m, 2H), 1,32 (s, 6H), 0,95 (t, J=7,2 Гц, 3H).
Стадия 3: Синтез пропил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (57).
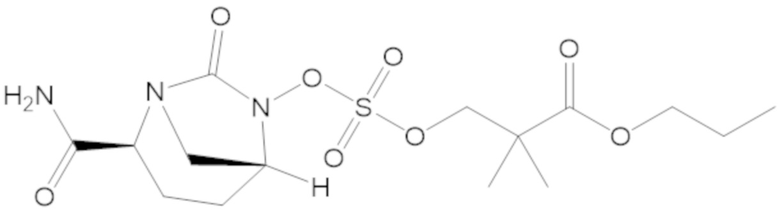
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (5,5 мл) и HMPA (1,0 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,5 мл, 1,5 ммоль), а затем в реакционную смесь быстро добавляли раствор пропил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (57b) (0,40 г, 1,5 ммоль) в THF (2×1 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь гасили H2O и разбавляли EtOAc (40 мл). Водные и органические слои разделяли, и органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (3:7-1:0) с получением нужного продукта (57) (190 мг, 39%) в виде твердого вещества. LCMS: м/з=408,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,47 (br. s, 1H), 5,57 (br. s, 1H), 4,66 (dd, J=9,0, 35,1 Гц, 2H), 4,17-4,04 (m, 4H), 3,32 (d, J=12,3 Гц, 1H), 3,02 (d, J=10,8 Гц, 1H), 2,46-2,41 (m, 1H), 2,14-2,13 (m, 1H), 1,99-1,83 (m, 2H), 1,71-1,66 (m, 2H), 1,29 (s, 3H), 1,28 (s, 3H), 0,95 (t, J=7,6 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ174,1, 170,9, 166,8, 80,3, 66,8, 61,8, 60,1, 47,1, 42,9, 22,1, 21,9, 21,6, 20,7, 17,4, 10,3.
Пример (58)
Синтез бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58)
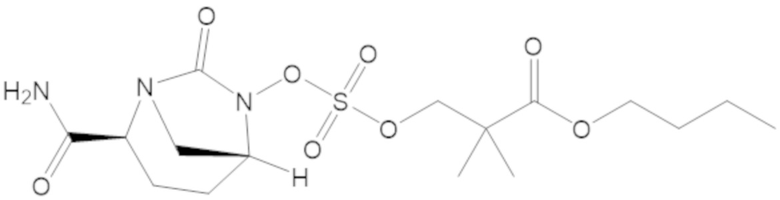
Стадия 1: Синтез бутил 3-гидрокси-2,2-диметилпропаноата (58a).
Загружали смесь 3-гидрокси-2,2-диметилпропановой кислоты (1,15 г, 9,7 ммоль), и 1-бутанол (15 мл) и концентрированную H2SO4 (70 мкл, 1,3 ммоль) в 20 мл сосуде для микроволновой обработки встряхивали при комнатной температуре, затем нагревали в микроволновом реакторе при 80°C в течение 2 ч, затем встряхивали при комнатной температуре в течение ночи. Когда нужный продукт идентифицировали с помощью TLC (EtOAc/гексаны; 3:7), смесь концентрировали в вакууме (40°C; совместно испаряли с толуолом×3) и разбавляли EtOAc (80 мл) и H2O (30 мл). Органический слой промывали H2O (два раза) и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали с получением продукта (58a) (1,24 г, 81%) в виде масла. Материал использовали непосредственно на следующей стадии без очистки. 1H-NMR (300 МГц, CDCl3): δ 4,11 (t, J=6,5 Гц, 2H), 3,55 (s, 2H), 2,42 (br. s, 1H), 1,65-1,58 (m, 2H), 1,43-1,35 (m, 2H), 1,19 (s, 6H), 0,94 (t, J=7,5 Гц, 3H).
Стадия 2: Синтез бутил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (58b).
Раствор свежедистиллированного сульфурилхлорида (198 мкл, 2,7 ммоль) в Et2O (1,0 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор пропил 3-гидрокси-2,2-диметилпропаноата (58a) (0,47 г, 2,7 ммоль) и пиридина (219 мкл, 2,7 ммоль) в Et2O. Колбу промывали Et2O (3×1 мл), который добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 5 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (58b) (0,52 г, 72%) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,50 (s, 2H), 4,14 (t, J=6,8 Гц, 2H), 1,66-1,59 (m, 2H), 1,43-1,35 (m, 2H), 1,32 (s, 6H), 0,94 (t, J=7,4 Гц, 3H).
Стадия 3: Синтез бутил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (58).
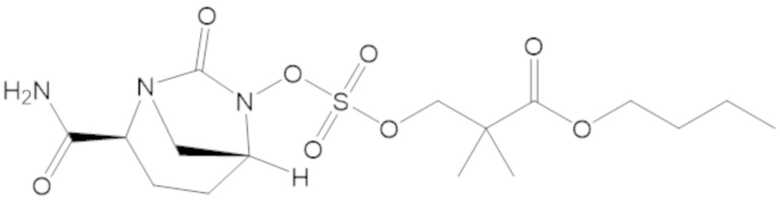
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (5 мл) и HMPA (1 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. Добавляли раствор NaHMDS, 1,0 M в THF (1,5 мл, 1,5 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор бутил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (58b) (0,41 г, 1,5 ммоль) в THF (5×1 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь гасили H2O и разбавляли EtOAc (40 мл). Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток растворяли в DMF и очищали с помощью препаративной HPLC с получением нужного продукта (58) (70 мг, 14%) в виде твердого вещества. LCMS: м/з=422,2 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,47 (br. s, 1H), 5,67 (br. s, 1H), 4,65 (dd, J=34,8, 9,0 Гц, 2H), 4,16-4,04 (m, 4H), 3,32 (d, J=11,7 Гц, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,47-2,40 (m, 1H), 2,18-2,13 (m, 1H), 2,01-1,80 (m, 2H), 1,67-1,58 (m, 3H), 1,45-1,32 (m, 2H), 1,28 (s, 3H), 1,27 (s, 3H), 0,94 (t, J=7,4 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ 174,1, 170,8, 166,8, 80,3, 65,1, 61,8, 60,1, 47,1, 42,8, 30,5, 22,1, 21,6, 20,7, 19,0, 17,4, 13,6.
Пример 59
Синтез (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (59)
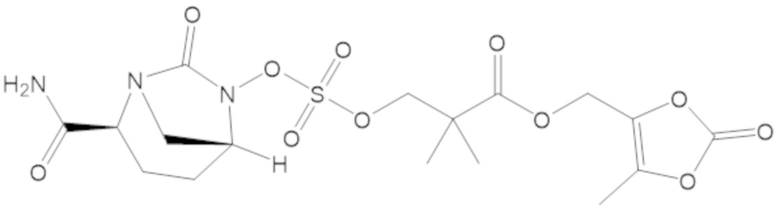
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (10 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,3 мл, 1,3 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор (5-метил-2-оксо-1,3-диоксол-4-ил)метил 3-((хлорсульфонил)окси)-2,2-диметилпропаноата (0,42 г, 1,3 ммоль) в THF (5×1 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc (40 мл). Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью препаративной HPLC с получением нужного продукта (59) (189 мг, 34%) в виде твердого вещества. LCMS: м/з=478,1 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,68 (br. s, 1H), 5,74 (br. s, 1H), 4,95-4,79 (m, 3H), 4,50 (d, J=9,3 Гц, 1H), 4,14 (br. s, 1H), 4,03 (d, J=7,2 Гц, 1H), 3,32 (d, J=12,3 Гц, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,45-2,39 (m, 1H), 2,17-2,09 (m, 4H), 1,98-1,79 (m, 2H), 1,30 (s, 3H), 1,29 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 177,6, 171,0, 167,0, 152,2, 140,5, 133,2, 80,0, 61,8, 60,2, 54,4, 47,0, 43,0, 21,8, 21,7, 20,7, 17,5, 9,3.
Пример 60
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутилпивалата (60)
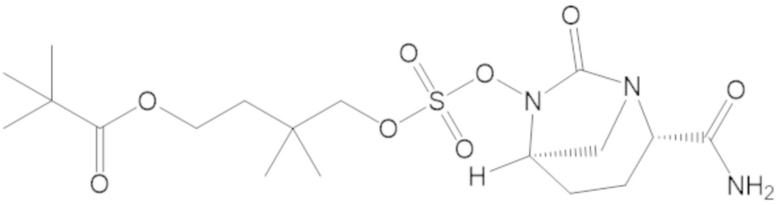
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутилпивалата (60a).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (0,86 г, 7,3 ммоль) в DCM (9 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли триметилацетилхлорид (0,89 мл, 7,3 ммоль), Et3N (1,17 мл, 14,5 ммоль) и N, N-4-диметиламинопиридин (каталитическое количество). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Смесь гасили путем добавления 1 н. HCl (50 мл). Разделяли органические и водные слои, и водный слой экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным NaHCO3 и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением нужного продукта (60a) (0,42 г, 28%). 1H-NMR (300 МГц, CDCl3): δ 4,13 (t, J=7,1 Гц, 2H), 3,35 (s, 2H), 1,61 (q, J=6,9 Гц, 2H), 1,19 (s, 9H), 0,93 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутилпивалата (60b).
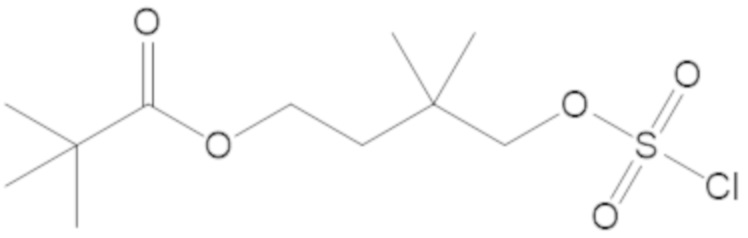
Раствор свежедистиллированного сульфурилхлорида (153 мкл, 2,1 ммоль) в Et2O (4,5 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 60 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилбутилпивалата (60a) (0,42 г, 2,1 ммоль) и пиридина (203 мкл, 2,5 ммоль) в Et2O (3 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (60b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,23 (s, 2H), 4,13 (t, J=6,8 Гц, 2H), 1,71 (t, J=6,6 Гц, 2H), 1,19 (s, 9H), 1,08 (s, 6H).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутилпивалата (60).
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,41 г, 2,2 ммоль) растворяли в THF (18,5 мл) и HMPA (0,9 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (2,2 мл, 2,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 4-((хлорсульфонил)окси)-3,3-диметилбутилпивалата (60b) (0,73 г, 2,4 ммоль) в THF (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 16 ч. Смесь гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (60) (176 мг) в виде твердого вещества. LCMS: м/з=450,15 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,49 (br. s, 1H), 5,59 (br. s, 1H), 4,55 (d, J=9,3 Гц, 1H), 4,26-4,04 (m, 5H), 3,34 (d, J=11,7 Гц, 1H), 3,02 (d, J=12,3 Гц, 1H), 2,45-2,41 (m, 1H), 2,19-2,15 (m, 1H), 2,01-1,85 (m, 2H), 1,69 (t, J=6,8 Гц, 2H), 1,19 (s, 9H), 1,05 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ178,6, 171,0, 167,0, 83,8, 61,9, 60,8, 60,2, 47,2, 38,7, 36,9, 34,0, 27,2, 24,0, 23,7, 20,8, 17,5.
Пример 61
Синтез этил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-этилбутаноата (61)
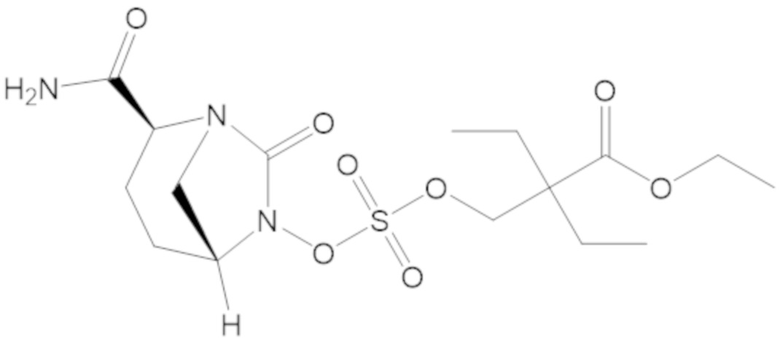
Стадия 1: Синтез этил 2-(((хлорсульфонил)окси)метил)-2-этилбутаноата (61a).
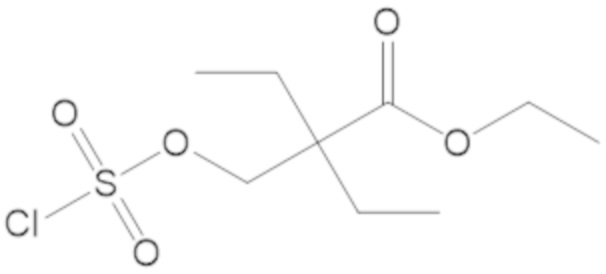
Раствор свежедистиллированного сульфурилхлорида (126 мкл, 1,7 ммоль) в Et2O (3,2 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 60 мин по каплям добавляли раствор этил 2-этил-2-(гидроксиметил)бутаноата (экс енамин) (0,30 г, 1,7 ммоль) и пиридина (153 мкл, 1,9 ммоль) в Et2O (2,1 мл). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 30 минут. Смесь повторно охлаждали до -78°C, добавляли сульфурилхлорид (20 мкл), обеспечивали нагревание реакционной смеси до комнатной температуры и дополнительно встряхивали в течение 30 мин. Добавляли Et2O (5 мл), и смесь встряхивали в течение 5 мин, затем фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (61a), которое сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 4,62 (s, 2H), 4,21 (q, J=7,3 Гц, 2H), 1,78-1,58 (m, 4H), 1,28 (t, J=7,1 Гц, 3H), 0,88 (t, J=7,7 Гц, 6H).
Стадия 2: Синтез этил 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-этилбутаноата (61).
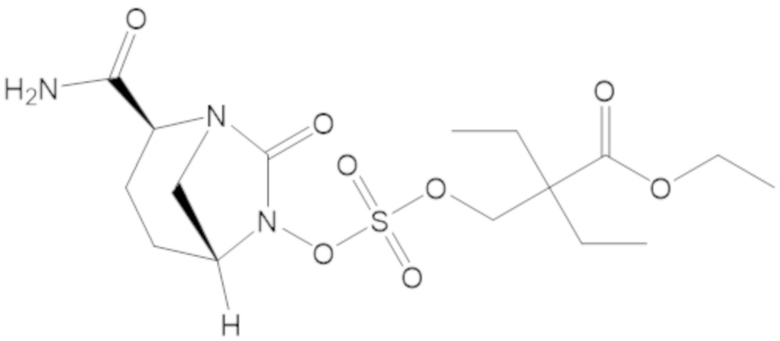
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,25 г, 1,4 ммоль) растворяли в THF (11,2 мл) и HMPA (0,6 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,4 мл, 1,4 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор этил 2-(((хлорсульфонил)окси)метил)-2-этилбутаноата (61a) (0,41 г, 1,5 ммоль) в THF (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 16 ч. Смесь гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением продукта (61) (162 мг, 29%) в виде твердого вещества. LCMS: м/з=422,03 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,50 (br. s, 1H), 5,66 (br. s, 1H), 4,87 (d, J=9,9 Гц, 1H), 4,70 (d, J=9,9 Гц, 1H), 4,22-4,14 (m, 3H), 4,06 (d, J=7,2 Гц, 1H), 3,33 (d, J=11,7 Гц, 1H), 3,02 (d, J=12,3 ч, 1H), 2,47-2,41 (m, 1H), 2,22-2,12 (m, 1H), 2,01-1,60 (m, 6H), 1,27 (t, J=7,1 Гц, 3H), 0,92-0,83 (m, 6H). 13C-NMR (75 МГц, CDCl3): δ 173,3, 171,1, 167,1, 75,2, 62,0, 61,1, 60,2, 50,6, 47,2, 26,2, 25,8, 20,9, 17,5, 14,3, 8,4, 8,3.
Пример 62
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62)
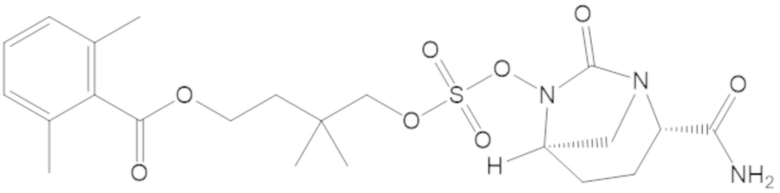
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутил 2,6-диметилбензоата (62a).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (0,84 г, 7,1 ммоль) в DCM (9 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли 2,6-диметилбензоилхлорид (1,0 г, 5,9 ммоль), пиридин (0,96 мл, 11,9 ммоль) и N, N-4-диметиламинопиридин (каталитическое количество). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Смесь гасили путем добавления 1 н. HCl (50 мл). Разделяли органические и водные слои, и водный слой экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным NaHCO3, а затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением нужного продукта (62a) (0,42 г, 28%). 1H-NMR (300 МГц, CDCl3): δ 7,18 (t, J=7,6 Гц, 1H), 7,04-7,01 (m, 2H), 4,41 (t, J=7,6 Гц, 2H), 3,37 (s, 2H), 2,31 (s, 6H), 1,76 (t, J=7,5 Гц, 2H), 0,97 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62b).
Раствор свежедистиллированного сульфурилхлорида (122 мкл, 1,7 ммоль) в Et2O (1,0 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилбутил 2,6-диметилбензоата (62a) (0,42 г, 1,7 ммоль) и пиридина (136 мкл, 1,7 ммоль) в Et2O (1,5 мл). Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (62b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки (не чистая). 1H-NMR (300 МГц, CDCl3): δ 7,19 (t, J=7,7 Гц, 1H), 7,03 (d, J=7,5 Гц, 2H), 4,41 (t, J=7,4 Гц, 2H), 4,23 (s, 2H), 2,31 (s, 6H), 1,84 (t, J=6,9 Гц, 2H), 1,11 (s, 6H).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62).
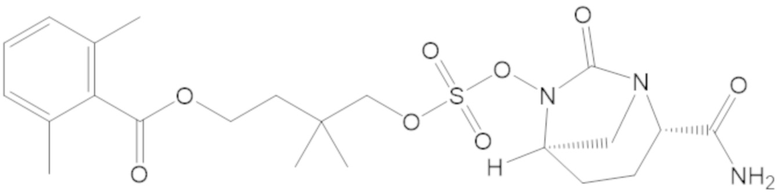
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (10 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,2 мл, 1,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 4-((хлорсульфонил)окси)-3,3-диметилбутил 2,6-диметилбензоата (62b) (0,42 г, 1,2 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (62) (192 мг, 32%) в виде твердого вещества. LCMS: м/з=498,08 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,18 (t, J=7,5 Гц, 1H), 7,02 (d, J=7,5 Гц, 2H), 6,48 (br. s, 1H), 5,63 (br. s, 1H), 4,57 (d, J=9,0 Гц, 1H), 4,40 (t, J=7,5 Гц, 2H), 4,25 (d, J=8,7 Гц, 1H), 4,17 (br. s, 1H), 4,04 (d, J=6,3 Гц, 1H), 3,32 (d, J=12,3 Гц, 1H), 3,00 (d, J=12,3 Гц, 1H), 2,47-2,40 (m, 1H), 2,31 (s, 6H), 2,18-2,14 (m, 1H), 1,97-1,80 (m, 4H), 1,08 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 170,9, 170,0, 167,1, 134,8, 133,8, 129,4, 127,6, 83,4, 61,9, 61,3, 60,1, 47,1, 36,7, 33,9, 23,9, 23,6, 20,7, 19,8, 17,4.
Пример 63
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63)
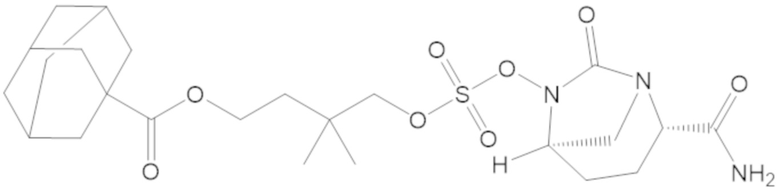
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутил адамантан-1-карбоксилата (63a).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (0,72 г, 6,1 ммоль) в DCM (20 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли-1-адамантан-карбонил хлорид (1,1 г, 10,1 ммоль), пиридин (0,82 мл, 10,1 ммоль) и N, N-4-диметиламинопиридин (0,03 г, 0,3 ммоль). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Смесь гасили путем добавления 1 н. HCl. Разделяли органические и водные слои, и водный слой экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным NaHCO3 и рассолом, а затем сушили (MgSO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:1) с получением нужного продукта (63a) (0,49 г, 35%). 1H-NMR (300 МГц, CDCl3): δ 4,14-4,09 (m, 2H), 3,34 (s, 2H), 2,00 (m, 3H), 1,90-1,86 (m, 6H), 1,75-1,59 (m, 6H), 1,59 (t, J=7,1 Гц, 2H), 0,92 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63b).
Раствор свежедистиллированного сульфурилхлорида (127 мкл, 1,7 ммоль) в Et2O (1,2 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилбутил адамантан-1-карбоксилата (63a) (0,48 г, 1,7 ммоль) и пиридина (141 мкл, 1,7 ммоль) в Et2O (1,7 мл). Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (63b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки (не чистая). 1H-NMR (300 МГц, CDCl3): δ 4,25 (s, 2H), 4,13 (t, J=6,8 Гц, 2H), 2,01 (m, 3H), 1,90-1,85 (m, 6H), 1,73-1,69 (m, 8H), 1,08 (s, 6H).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63).
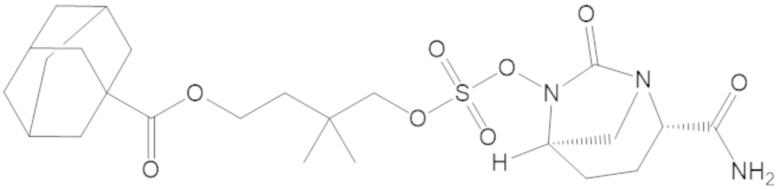
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (10 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,2 мл, 1,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 4-((хлорсульфонил)окси)-3,3-диметилбутил адамантан-1-карбоксилата (63b) (0,66 г, 1,7 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением твердого вещества, которое растирали в порошок с гексанами с получением продукта (63) (230 мг, 36%) в виде твердого вещества. LCMS: м/з=528,17 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,18 (t, J=7,5 Гц, 1H), 7,02 (d, J=7,5 Гц, 2H), 6,48 (br. s, 1H), 5,63 (br. s, 1H), 4,57 (d, J=9,0 Гц, 1H), 4,40 (t, J=7,5 Гц, 2H), 4,25 (d, J=8,7 Гц, 1H), 4,17 (br. s, 1H), 4,04 (d, J=6,3 Гц, 1H), 3,32 (d, J=12,3 Гц, 1H), 3,00 (d, J=12,3 Гц, 1H), 2,47-2,40 (m, 1H), 2,31 (s, 6H), 2,18-2,14 (m, 1H), 1,97-1,80 (m, 4H), 1,08 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 177,8, 171,0, 167,1, 83,9, 62,0, 60,7, 60,2, 47,2, 40,7, 39,0, 39,0, 37,0, 36,6, 34,1, 28,0, 24,0, 23,8, 20,8, 17,5.
Пример 64
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64)
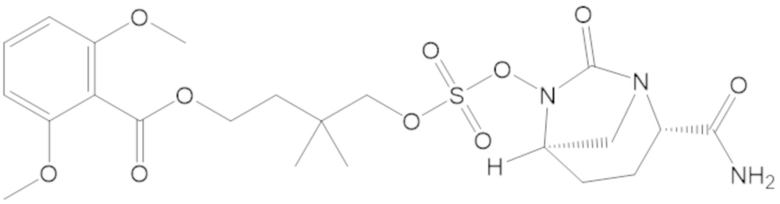
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутил 2,6-диметоксибензоата (64a).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (1,85 г, 15,7 ммоль) в DCM (28 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли 2,6-диметоксибензоилхлорид (80%; 3,93 г, 15,7 ммоль), Et3N (2,5 мл, 31,3 ммоль) и N, N-4-диметиламинопиридин (каталитическое количество). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение ночи. Смесь концентрировали в вакууме и суспендировали в EtOAc, а затем фильтровали, и фильтрационный осадок промывали EtOAc. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:3) с получением нужного продукта (64a) (чистота около 80%; 0,92 г). 1H-NMR (300 МГц, CDCl3): δ 7,29-7,26 (m, 1H), 6,57-6,53 (m, 3H), 4,43-4,39 (m, 2H), 3,83 (s, 6H), 3,36 (s, 2H), 1,74 (t, J=6,5 Гц, 2H), 0,95 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64b).
Раствор свежедистиллированного сульфурилхлорида (0,2 мл, 2,7 ммоль) в Et2O (1,9 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилбутил 2,6-диметоксибензоата (64a) (около 80% чистота; 0,97 г, 2,7 ммоль) и пиридина (222 мкл, 2,7 ммоль) в Et2O (2,7 мл). Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (64b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки (не чистая).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64).
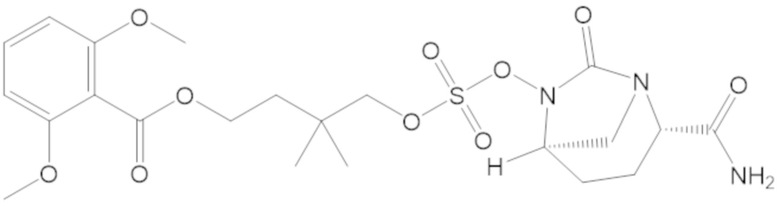
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,21 г, 1,1 ммоль) растворяли в THF (9,4 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,1 мл, 1,1 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 4-((хлорсульфонил)окси)-3,3-диметилбутил 2,6-диметоксибензоата (64b) (0,65 г, 1,7 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (64) (100 мг, 17%) в виде твердого вещества. LCMS: м/з=530,01 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,31-7,25 (m, 1H), 6,54 (d, J=9,0 Гц, 2H), 6,50 (br. s, 1H), 5,58 (br. s, 1H), 4,54 (d, J=8,7 Гц, 1H), 4,40 (t, J=6,8 Гц, 2H), 4,28 (d, J=8,7 Гц, 1H), 4,16 (br. s, 1H), 4,03 (d, J=6,9 Гц, 1H), 3,81 (s, 6H), 3,33 (d, J=11,7 Гц, 1H), 2,99 (d, J=7,2 Гц, 1H), 2,44-2,39 (m, 1H), 2,21-2,13 (m, 1H), 1,93-1,79 (m, 4H), 1,07 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ171,0, 167,1, 166,7, 157,3, 131,2, 113,1, 103,9, 84,0, 62,0, 61,8, 60,2, 56,0, 47,2, 36,6, 34,1, 23,8, 23,6, 20,8, 17,5.
Пример 65
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил бензоата (65)
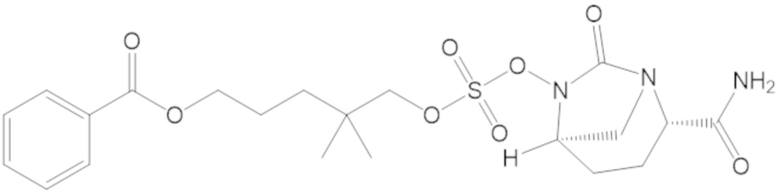
Стадия 1: Синтез 5-гидрокси-4,4-диметилпентил бензоата (65a).
Во встряхиваемый раствор 2,2-диметилпентан-1,5-диола (J. Org. Chem. 2010, 75, 1892-1897; PCT International Publication № WO 2002092606) (1,55 г, 11,7 ммоль) в DCM (20 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона добавляли бензоилхлорид (1,5 мл, 12,9 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение 2,5 ч и концентрировали в вакууме. В остаток добавляли EtOAc, и смесь встряхивали. Фильтрат концентрировали под остатком, очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:4) с получением продукта (65a) (1,38g, 50%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 8,04 (d, J=6,9 Гц, 2H), 7,56 (t, J=7,5 Гц, 1H), 7,44 (t, J=7,5 Гц, 2H), 4,31 (t, J=6,8 Гц, 2H), 3,36 (s, 2H), 1,81-1,71 (m, 2H), 1,42-1,36 (m, 2H), 0,92 (s, 6H).
Стадия 2: Синтез 5-((хлорсульфонил)окси)-4,4-диметилпентил бензоата (65b).
Раствор свежедистиллированного сульфурилхлорида (0,2 мл, 2,7 ммоль) в Et2O (1,9 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилпентил бензоата (65a) (0,76 г, 3,2 ммоль) и пиридина (218 мкл, 2,7 ммоль) в Et2O (2,7 мл). Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (65b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки (не чистая). 1H-NMR (300 МГц, CDCl3): δ 8,04 (d, J=7,5 Гц, 2H), 7,57-7,55 (m, 1H), 7,48-7,33 (m, 1H), 4,35-4,29 (m, 2H), 4,23 (s, 2H), 1,81-1,74 (m, 2H), 1,53-1,21 (m, 2H), 1,06 (s, 6H).
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил бензоата (65).
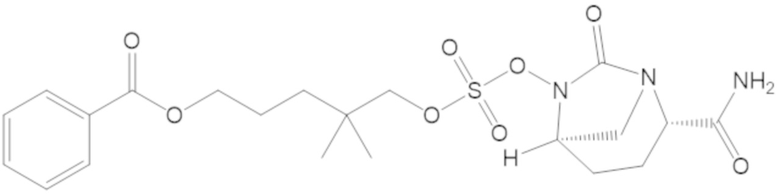
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,21 г, 1,1 ммоль) растворяли в THF (9,4 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,1 мл, 1,1 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 5-((хлорсульфонил)окси)-4,4-диметилпентил бензоата (65b) (0,42 г, 1,2 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (65) (160 мг, 29%) в виде твердого вещества. LCMS: м/з=484,10 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,04 (d, J=7,5 Гц, 2H), 7,57-7,54 (m, 1H), 7,47-7,42 (m, 2H), 6,43 (br. s, 1H), 5,51 (br. s, 1H), 4,56 (d, J=9,0 Гц, 1H), 4,32-4,17 (m, 4H), 4,02 (d, J=7,2 Гц, 1H), 3,28 (d, J=12,3 Гц, 1H), 2,97 (d, J=12,3 Гц, 1H), 2,44-2,40 (m, 1H), 2,20-2,14 (m, 1H), 1,94-1,72 (m, 4H), 1,51-1,45 (m, 2H), 1,02 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,0, 167,1, 166,8, 133,1, 130,4, 129,7, 128,5, 83,6, 65,3, 61,9, 60,2, 47,2, 34,8, 34,3, 23,9, 23,6, 23,3, 20,8, 17,5.
Пример 66
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66)
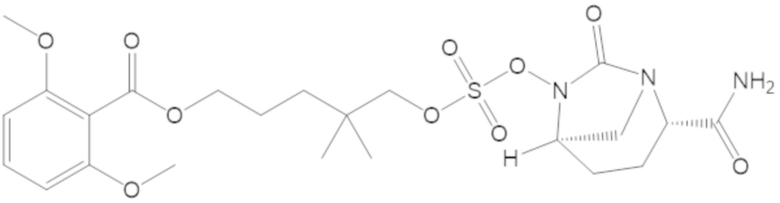
Стадия 1: Синтез 5-гидрокси-4,4-диметилпентил 2,6-диметоксибензоата (66a).
Во встряхиваемый раствор 2,2-диметилпентан-1,5-диола (1,5 г, 11,3 ммоль) в пиридине (8,3 мл) при 0°C в атмосфере аргона добавляли одну часть 2,6-диметоксибензоилхлорида (80%; 1,4 г, 5,6 ммоль). В течение 3 ч обеспечивали нагревание реакционной смеси до комнатной температуры. Реакционную смесь концентрировали до сухого состояния, и добавляли EtOAc. Смесь фильтровали, а фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением продукта (66a) (0,65 г, 39%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,31-7,26 (m, 2H), 6,55 (d, J=8,1 Гц, 2H), 4,33 (t, J=6,2 Гц, 2H), 3,82 (s, 6H), 3,33 (s, 2H), 1,77-1,67 (m, 2H), 1,41-1,35 (m, 2H), 0,92 (s, 6H).
Стадия 2: Синтез 5-((хлорсульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66b).
Раствор свежедистиллированного сульфурилхлорида (0,16 мл, 2,2 ммоль) в Et2O охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 5-гидрокси-4,4-диметилпентил 2,6-диметоксибензоата (66a) (0,65 г, 2,2 ммоль) и пиридина (177 мкл, 2,2 ммоль) в Et2O. Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (66b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки (не чистая). 1H-NMR (300 МГц, CDCl3): δ 7,32-7,26 (m, 1H), 6,56 (d, J=8,7 Гц, 2H), 4,34 (t, J=6,2 Гц, 2H), 4,21 (s, 2H), 3,81 (s, 6H), 1,77-1,71 (m, 2H), 1,52-1,46 (m, 2H), 1,03 (s, 6H).
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66).
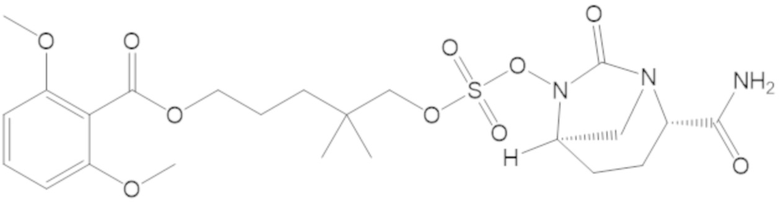
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,21 г, 1,1 ммоль) растворяли в THF (9,4 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,1 мл, 1,1 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 5-((хлорсульфонил)окси)-4,4-диметилпентил 2,6-диметоксибензоата (66b) (0,49 г, 1,2 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (66) (115 мг, 18%) в виде твердого вещества. LCMS: м/з=544,17 [M+H]+ и 589,11 [M+HCOOH]+. 1H-NMR (300 МГц, CDCl3): δ 7,31-7,25 (m, 1H), 6,55 (d, J=8,7 Гц, 2H), 6,51 (br. s, 1H), 5,51 (br. s, 1H), 4,53 (d, J=9,0 Гц, 1H), 4,33 (t, J=6,2 Гц, 2H), 4,23-4,16 (m, 2H), 4,03 (d, J=7,5 Гц, 1H), 3,82 (s, 6H), 3,31 (d, J=12,3 Гц, 1H), 2,98 (d, J=11,7 Гц, 1H), 2,42-2,39 (m, 1H), 2,20-2,18 (m, 1H), 2,00-1,68 (m, 4H), 1,49-1,44 (m, 2H), 1,00 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,0, 167,0, 166,7, 157,4, 131,2, 110,1, 104,1, 83,9, 65,6, 62,0, 60,2, 56,1, 47,2, 34,7, 34,3, 23,8, 23,6, 23,4, 20,8, 17,6.
Пример 67
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67)
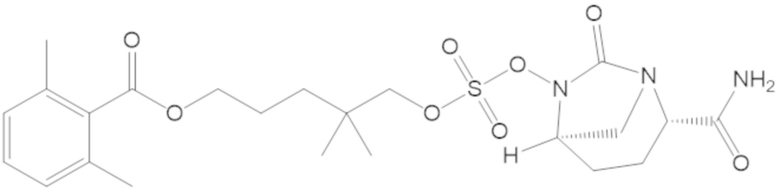
Стадия 1: Синтез 5-гидрокси-3,3-диметилпентил 2,6-диметилбензоата (67a).
Во встряхиваемый раствор 2,2-диметилпентан-1,5-диола (1,1 г, 8,3 ммоль) в пиридине (8,3 мл) при 0°C в атмосфере аргона добавляли одну часть 2,6-диметилбензоилхлорида. Обеспечивали нагревание реакционной смеси до комнатной температуры в течение 3 ч. Реакционную смесь концентрировали до сухого состояния, и добавляли EtOAc. Смесь фильтровали, а фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:4) с получением продукта (67a) (0,44 г, 25%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,18 (t, J=7,7 Гц, 1H), 7,03 (d, J=7,5 Гц, 2H), 4,32 (t, J=6,3 Гц, 2H), 3,34 (s, 2H), 2,32 (s, 6H), 1,78-1,68 (m, 2H), 1,40-1,34 (m, 2H), 0,90 (s, 6H).
Стадия 2: Синтез 5-((хлорсульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67b).
Раствор свежедистиллированного сульфурилхлорида (122 мкл, 1,7 ммоль) в Et2O охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 5-гидрокси-4,4-диметилпентил 2,6-диметилбензоата (67a) (0,44 г, 1,7 ммоль) и пиридина (135 мкл, 1,7 ммоль) в Et2O. Колбу промывали Et2O (2×20 мл), а смыв добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, а затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме с получением титульного соединения (67b), которое сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,19 (t, J=7,5 Гц, 1H), 7,03 (d, J=7,5 Гц, 2H), 4,33 (t, J=6,2 Гц, 2H), 4,20 (s, 2H), 2,32 (s, 6H), 1,81-1,71 (m, 2H), 1,51-1,45 (m, 2H), 1,04 (s,6H).
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67).
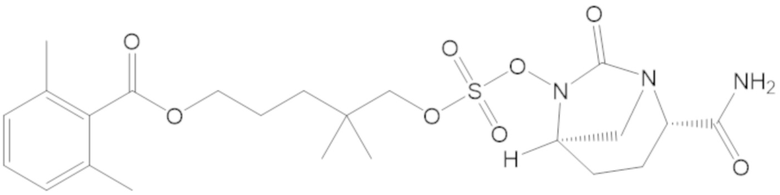
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,21 г, 1,1 ммоль) растворяли в THF (9,4 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,1 мл, 1,1 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 5-((хлорсульфонил)окси)-4,4-диметилпентил 2,6-диметилбензоата (67b) (0,49 г, 1,4 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (67) (200 мг, 32%) в виде твердого вещества. LCMS: м/з=512,18 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,19 (t, J=7,5 Гц, 1H), 7,03 (d, J=7,5 Гц, 2H), 6,45 (br. s, 1H), 5,52 (br. s, 1H), 4,54 (d, J=9,3 Гц, 1H), 4,31 (t, J=7,4 Гц, 2H), 4,23-4,17 (m, 2H), 4,03 (d, J=7,2 Гц, 1H), 3,31 (d, J=11,7 Гц, 1H), 2,99 (d, J=12,3 Гц, 1H), 2,45-2,40 (m, 1H), 2,32 (s, 6H), 2,18-2,14 (m, 1H), 1,98-1,70 (m, 4H), 1,48-1,42 (m, 2H), 1,00 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 170,9, 170,2, 167,1,135,0, 134,1, 129,4, 127,7, 83,6, 65,4, 62,0, 60,2, 47,2, 34,9, 34,3, 23,7, 23,6, 23,3, 20,8, 19,9, 17,5.
Пример 68
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2-метилбензоата (68)
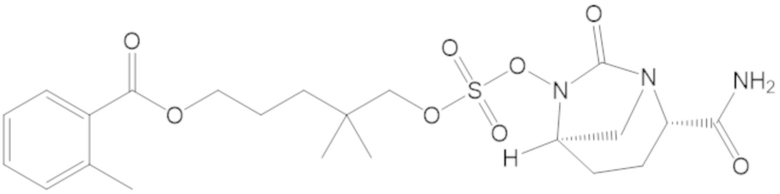
Стадия 1: Синтез 4-гидрокси-3,3-диметилбутил 2-метилбензоата (68a).
Во встряхиваемый раствор 2,2-диметилбутан-1,4-диола (0,80 г, 6,8 ммоль) в пиридине (5 мл) приблизительно при 0°C (ледяная ванна) в атмосфере аргона по каплям добавляли толуоил хлорид (0,89 мл, 6,8 ммоль). Обеспечивали постепенное нагревание реакционной смеси до комнатной температуры, и смесь встряхивали в течение 4 ч. Смесь концентрировали в вакууме и суспендировали в EtOAc, а затем фильтровали, и фильтрационный осадок промывали EtOAc. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:7) с получением нужного продукта (68a) (0,7 г, 44%). 1H-NMR (300 МГц, CDCl3): δ 7,88 (d, J=8,4 Гц, 1H), 7,40 (t, J=7,1 Гц, 1H), 7,26-7,24 (m, 2H), 4,38 (t, J=7,3 Гц, 2H), 3,41 (s, 3H), 2,60 (s, 3H), 1,78 (t, J=7,5 Гц, 2H), 0,98 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-3,3-диметилбутил 2-метилбензоата (68b).

Раствор свежедистиллированного сульфурилхлорида (96 мкл, 1,3 ммоль) в Et2O (0,8 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор 4-гидрокси-3,3-диметилбутил 2-метилбензоата (68a) (0,31 г, 1,3 ммоль) и пиридина (106 мкл, 1,3 ммоль) в Et2O (1,1 мл). Колбу промывали Et2O (2×20 мл), который добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 30 минут. Смесь фильтровали, и продукт (68b) сразу использовали на следующей стадии без дополнительной очистки. 1H-NMR (300 МГц, CDCl3): δ 7,89 (d, J=8,1 Гц, 1H), 7,41-7,39 (m, 1H), 7,26-7,25 (m, 2H), 4,41-4,35 (m, 2H), 4,28 (s, 2H), 2,61 (s, 3H), 1,87 (t, J=7,2 Гц, 2H), 1,13 (s, 6H).
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-4,4-диметилпентил 2-метилбензоата (68).
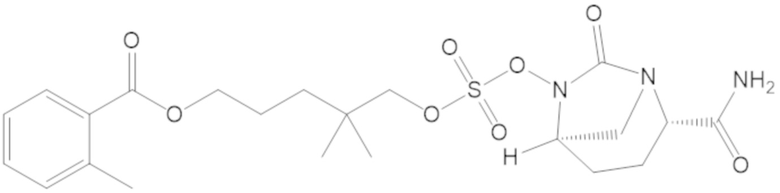
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (10 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,2 мл, 1,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор 4-((хлорсульфонил)окси)-3,3-диметилбутил 2-метилбензоата (68b) (0,42 г, 1,3 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (68) (231 мг, 40%) в виде твердого вещества. LCMS: м/з=484,06 [M+1]+. 1H-NMR (300 МГц, CDCl3): δ 7,90 (d, J=7,5 Гц, 1H),7,40 (t, J=7,5 Гц, 1H), 7,26-7,24 (m, 2H), 6,44 (br. s, 1H), 5,53 (br. s, 1H), 4,60 (d, J=8,7 Гц, 1H), 4,35 (t, J=7,1 Гц, 2H), 4,28 (d, J=9,0 Гц, 1H), 4,17 (br. s, 1H), 4,03 (d, J=7,2 Гц, 1H), 3,33 (d, J=12,3 Гц, 1H), 2,99 (d, J=11,7 Гц, 1H), 2,60 (s, 3H), 2,47-2,40 (m, 1H), 2,18-2,14 (m, 1H), 1,95-1,82 (m, 4H), 1,10 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,0, 167,5, 167,1, 140,5, 132,2, 131,9, 130,7, 129,5, 125,9, 83,7, 62,0, 61,2, 60,2, 47,2, 36,9, 34,0, 24,1, 23,8, 21,9, 20,8, 17,5.
Пример 69
Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоата (69)
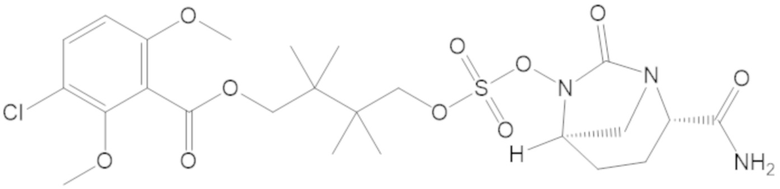
Стадия 1: Синтез 4-гидрокси-2,2,3,3-тетраметилбутил 2,6-диметоксибензоата (69a).
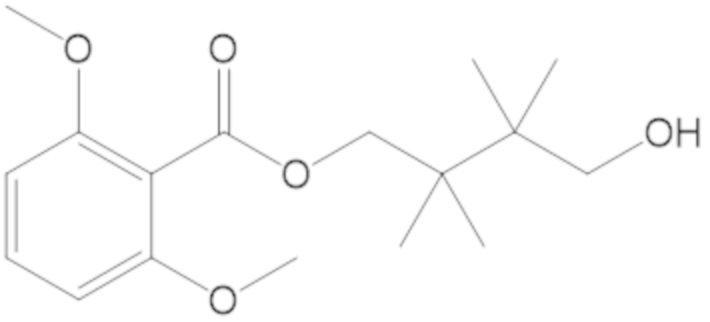
Во встряхиваемый раствор 2,2,3,3-тетраметилбутан-1,4-диола (45a) (0,7 г, 4,8 ммоль) в DCM (20 мл) при 0°C в атмосфере аргона добавляли 2,6-диметоксибензоилхлорид (80%; 0,55 г, 2,2 ммоль), пиридин (0,36 мл, 4,4 ммоль) и N, N-4-диметиламинопиридин (0,05 г, 0,4 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°C, и реакцию гасили путем добавления 1 н. HCl (15 мл), а затем экстрагировали DCM (два раза). Объединенные органические слои промывали насыщенным бикарбонатом натрия и рассолом, затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением продукта (69a) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 7,29 (t, J=8,4 Гц, 1H), 6,56 (d, J=8,1 Гц, 2H), 4,24 (s, 2H), 3,81 (s, 6H), 3,49 (s, 2H), 0,98 (s, 6H), 0,92 (s, 6H).
Стадия 2: Синтез 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоата (69b).
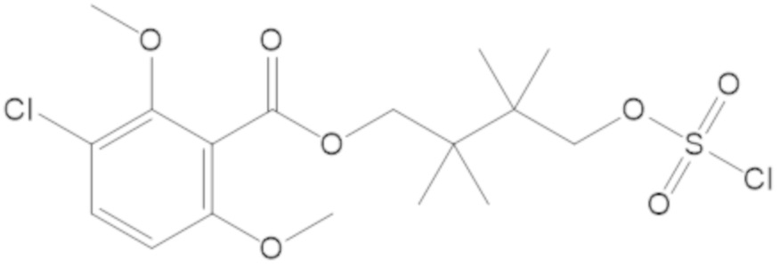
Во встряхиваемую смесь 4-гидрокси-2,2,3,3-тетраметилбутил пропионата (69a) (0,30 г, 1,5 ммоль) и Et2O (10 мл) в атмосфере аргона добавляли пиридин (0,15 мл, 1,8 ммоль). Раствор охлаждали до -78°C, и медленно добавляли сульфурилхлорид (0,15 мл, 1,8 ммоль) в Et2O (3 мл) при -78°C. Смесь встряхивали при -78°C в течение 1 ч, а затем нагревали до комнатной температуры и встряхивали в течение 1 ч. Реакционную смесь фильтровали для удаления пиридиновой соли, и фильтрат концентрировали в вакууме с получением титульного соединения (69b) в виде масла, которое использовали непосредственно на следующей стадии без дополнительной очистки (выход принимали по количеству).
Стадия 3: Синтез 4-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоата (69).
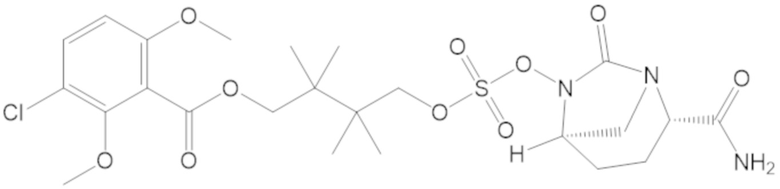
Во встряхиваемую смесь (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (1) (235 мг, 1,3 ммоль) в THF (10 мл) в атмосфере аргона добавляли несколько капель 1,3-диметилтетрагидропиримидин-2(1H)-она. Смесь охлаждали до -78°C и встряхивали в течение 10 мин, затем по каплям добавляли раствор NaHMDS, 1,0 M в THF (1,4 мл, 1,4 ммоль). Смесь встряхивали при -78°C в течение 8 мин, затем добавляли 4-((хлорсульфонил)окси)-2,2,3,3-тетраметилбутил 3-хлор-2,6-диметоксибензоат (69b) (0,52 г, 1,2 ммоль) в THF (5 мл) при -78°C. Смесь встряхивали при -78°C в течение 10 мин, затем обеспечивали нагревание до комнатной температуры и встряхивали в течение 1 ч. Смесь разбавляли EtOAc и насыщенным раствором бикарбоната натрия. Водные и органические слои разделяли, а органический слой промывали водой, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:0) с получением продукта (69) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 7,33 (d, J=9,0 Гц, 1H), 6,65 (d, J=8,7 Гц, 1H), 6,51 (s, 1H), 5,74 (s, 1H), 4,75 (d, J=9,6 Гц, 1H), 4,44 (d, J=8,7 Гц, 1H), 4,22-4,15 (m, 3H), 4,02 (d, J=6,3 Гц, 1H), 3,88 (s, 3H), 3,81 (s, 3H), 3,31 (d, J=11,7 Гц, 1H), 2,99 (d, J=12,3 Гц, 1H), 2,43-2,39 (m, 1H), 2,16-2,12 (m, 1H), 1,91-1,80 (m, 2H), 1,05-1,01 (m, 12H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 167,1, 165,6, 156,0, 153,6, 131,6, 120,1, 119,5, 107,8, 82,4, 71,2, 62,2, 61,9, 60,2, 56,2, 47,2, 39,0, 38,7, 20,8, 20,7, 20,3, 20,2, 17,5.
Пример 70
Синтез 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоата (70)
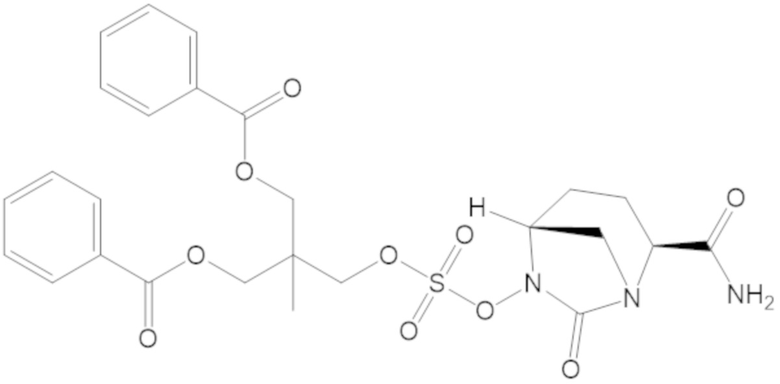
Стадия 1: Реакция получения 2-(гидроксиметил)-2-метилпропан-1,3-диил дибензоата (70a).
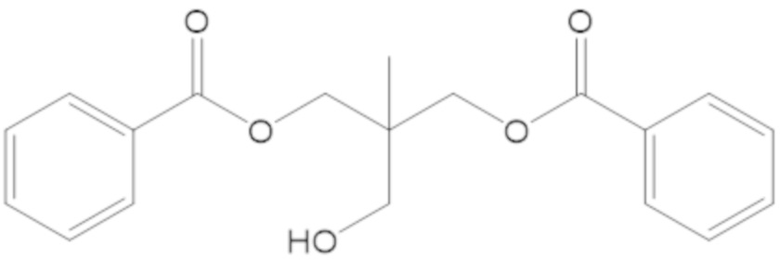
Бензоилхлорид (2,46 мл, 20,0 ммоль) добавляли по каплям в смесь 2-(гидроксиметил)-2-метилпропан-1,3-диола (1,2 г, 10,0 ммоль), пиридина (2,02 мл, 25,0 ммоль) и N, N-4-диметиламинопиридина (0,06 г, 0,4 ммоль) в DCM (30 мл) при комнатной температуре. После встряхивания при комнатной температуре в течение ночи, органическую фазу промывали 1 M HCl, водой и рассолом, сушили (MgSO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:3) с получением продукта (70a) (1,3 г, 40%) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 8,06-8,02 (m, 4H), 7,62-7,56 (m, 2H), 7,49-7,42 (m, 4H), 4,39 (s, 2H), 4,38 (s, 2H), 3,59 (s, 2H), 1,16 (s, 3H).
Стадия 2: Синтез 2-(((хлорсульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоата (70b).
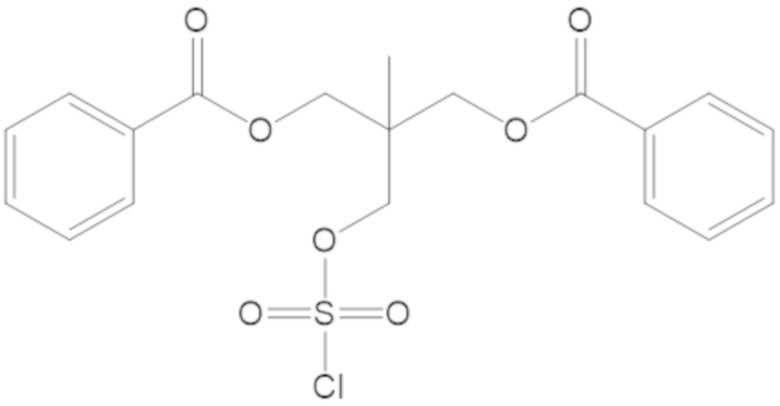
Раствор свежедистиллированного сульфурилхлорида (0,3 мл, 3,7 ммоль) в Et2O (5 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор 2-(гидроксиметил)-2-метилпропан-1,3-диил дибензоата (70a) (800 мг, 2,4 ммоль) и пиридина (0,32 мл, 3,9 ммоль) в Et2O (5 мл). Колбу промывали Et2O (3 мл), который также добавляли в смесь. Смесь встряхивали при -78°C в течение 1 ч, а затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (70b) в виде масла, которое сразу использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоата (70).
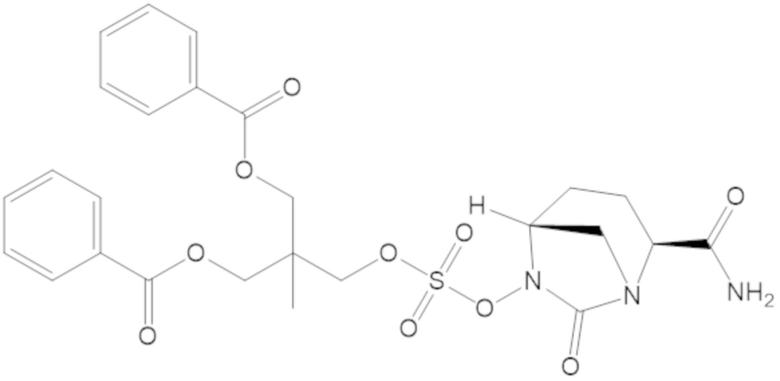
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (400 мг, 2,2 ммоль) растворяли в THF (10 мл) и HMPA (1,1 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли 1,0 M раствора NaHMDS в THF (2,3 мл), и встряхивали в течение 10 мин. 2-(((хлорсульфонил)окси)метил)-2-метилпропан-1,3-диил дибензоат (70b) (922 мг, 2,2 ммоль) растворяли в THF (8 мл) и быстро добавляли в реакционную смесь. После встряхивания при -78°C в течение 10 мин обеспечивали нагревание реакционной смеси до комнатной температуры. После встряхивания при комнатной температуре в течение 2 ч добавляли EtOAc (400 мл) и насыщенный водный NaHCO3 (40 мл) и H2O (40 мл). Водные и органические слои разделяли, и органический слой промывали насыщенным водным NaHCO3 (60 мл), водой (3× 50 мл), рассолом (60 мл), затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:9) с получением продукта (70) (186 мг, 15%) в виде масла. LC-MS: м/з=576 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 8,06-8,03 (m, 4H), 7,60-7,55 (m, 2H), 7,44-7,26 (m, 4H), 6,41 (s, 1H), 5,49 (s, 1H), 4,97 (d, J=9,3 Гц, 1H), 4,68 (d, J=9,3 Гц, 1H), 4,43-4,39 (m, 4H), 4,13 (m, 1H), 4,01-3,99 (m, 1H), 3,13 (m, 1H), 2,95-2,91 (m, 1H), 2,45-2,40 (m, 1H), 2,20-2,08 (m, 1H), 1,93-1,76 (m, 2H), 1,12 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 170,8, 167,1, 166,1, 133,3, 129,8, 129,7, 128,5, 77,2, 66,1, 65,9, 61,8, 60,1, 46,9, 39,4, 20,6, 17,4, 16,9.
Пример 71
Синтез 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил диацетата (71)
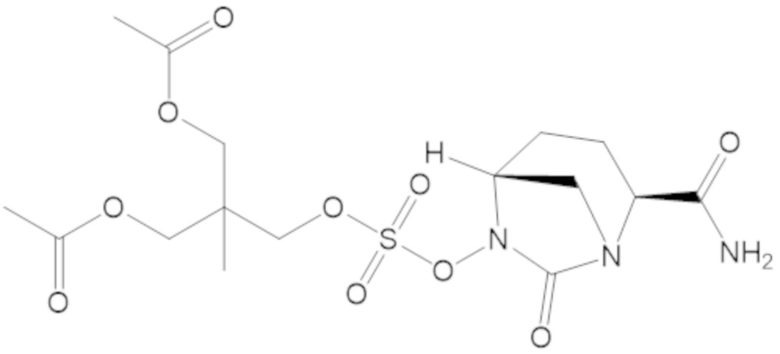
Стадия 1: Синтез 2-(гидроксиметил)-2-метилпропан-1,3-диил диацетата (71a).
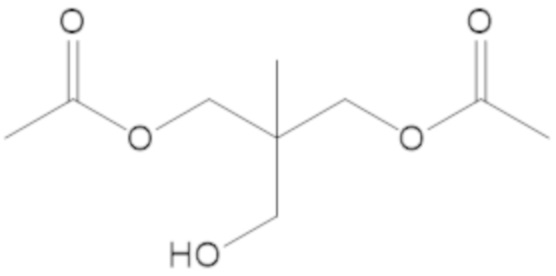
В смесь 2-(гидроксиметил)-2-метилпропан-1,3-диола (2,2 г, 18,0 ммоль), пиридина (12 мл, 25,0 ммоль) и N, N-4-диметиламинопиридина (0,05 г) при комнатной температуре по каплям добавляли уксусный ангидрид (3,46 мл, 36,6 ммоль). После встряхивания при комнатной температуре в течение ночи смесь концентрировали в вакууме. Смесь суспендировали в EtOAc (100 мл), и медленно добавляли H2O (20 мл) при 0°C. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4), затем концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-3:2) с получением продукта (71a) (1,0 г, 26%). 1H-NMR (300 МГц, CDCl3): δ 4,02 (s, 4H), 3,41 (s, 2H), 2,08 (s, 6H), 0,96 (s, 3H).
Стадия 2: Синтез 2-(((хлорсульфонил)окси)метил)-2-метилпропан-1,3-диил диацетата (71b).
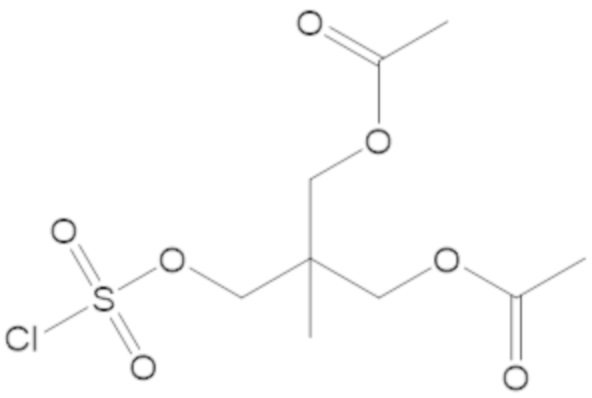
Раствор свежедистиллированного сульфурилхлорида (0,33 мл, 4,0 ммоль) в Et2O (4 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 5 мин по каплям добавляли раствор 2-(гидроксиметил)-2-метилпропан-1,3-диил диацетата (71a) (550 мг, 2,7 ммоль) и пиридина (0,35 мл, 4,3 ммоль) в Et2O (4 мл). Колбу промывали Et2O (5 мл), который также добавляли в смесь. Смесь встряхивали при -78°C в течение 1 ч, затем обеспечивали нагревание до комнатной температуры. Осадок (быстро) фильтровали, и фильтрационный осадок промывали Et2O (12 мл). Фильтрат концентрировали в вакууме при комнатной температуре с получением титульного соединения (71b) в виде масла, которое сразу использовали для следующей стадии без дополнительной очистки.
Стадия 3: Синтез 2-((((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)метил)-2-метилпропан-1,3-диил диацетата (71).
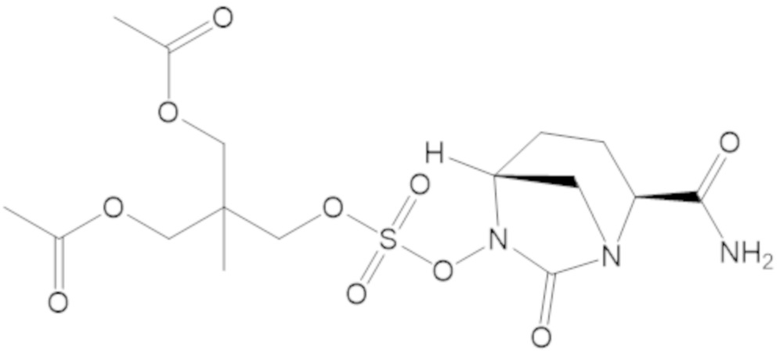
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (430 мг, 2,3 ммоль) растворяли в THF (8 мл) и HMPA (0,8 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли NaHMDS, 1,0 M в THF (2,4 мл, 2,4 ммоль), и встряхивали в течение 10 мин. 2-(((хлорсульфонил)окси)метил)-2-метилпропан-1,3-диил диацетат (71b) (703 мг, 2,3 ммоль) растворяли в THF (8 мл), и быстро добавляли в реакционную смесь. После встряхивания при -78°C в течение 10 мин обеспечивали нагревание реакционной смеси до комнатной температуры. После встряхивания при комнатной температуре в течение 2 ч добавляли EtOAc (400 мл) и насыщенный водный NaHCO3 (40 мл) и H2O (40 мл). Водные и органические слои разделяли, и органический слой промывали насыщенным водным NaHCO3 (60 мл), H2O (3×50 мл), рассолом (60 мл), а затем сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:9) с получением продукта (71) (198 мг, 19%) в виде масла. LC-MS: м/з=452 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 6,50 (s, 1H), 5,68 (s, 1H), 4,72 (d, J=9,3 Гц, 1H), 4,46 (d, J=9,3 Гц, 1H), 4,16 (m, 1H), 4,12-4,08 (m, 4H), 3,36-3,32 (m, 1H), 3,04-3,00 (m, 1H), 2,45-2,40 (m, 1H), 2,20-2,12 (m, 1H), 2,09 (s, 6H), 1,91-1,76 (m, 2H), 1,11 (s, 3H). 13C-NMR (75 МГц, CDCl3): δ 171,1, 170,8, 170,7, 167,1, 76,9, 65,2, 61,9, 60,2, 47,1, 38,8, 20,8, 20,7, 17,5, 16,6.
Пример 72
Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72)
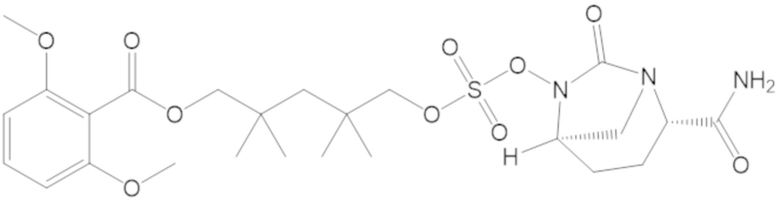
Стадия 1: Синтез 5-гидрокси-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72a).
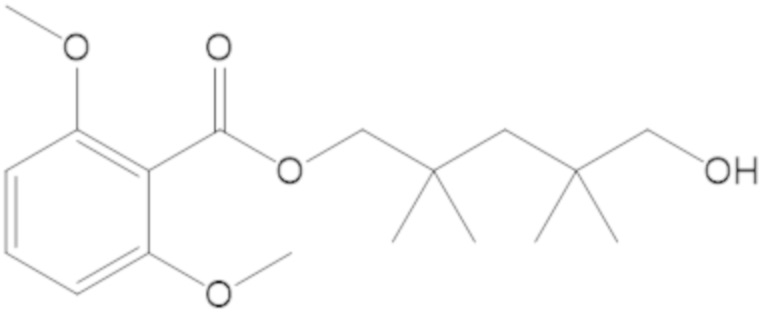
Во встряхиваемый раствор 2,2,4,4-тетраметилпентан-1,5-диола (39c) (0,64 г, 4,0 ммоль) и пиридина (0,32 мл, 4,0 ммоль) в DCM (27 мл) по каплям добавляли 2,6-диметоксибензоилхлорид (80%; 1,0 г, 4,0 ммоль) в DCM (10 мл) в течение 30 минут при 0°C (ледяная ванна) в атмосфере аргона. Обеспечивали нагревание реакционной смеси до комнатной температуры, и встряхивали в течение ночи. Смесь разбавляли H2O (30 мл), и слои разделяли. Водный слой экстрагировали DCM (2×30 мл), и объединенные органические слои промывали рассолом (30 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:98) с получением продукта (72a) (927 мг, 71%) в виде масла. Соединение было загрязнено предположительно диацелированным побочным продуктом. Материал использовали на следующей стадии без дополнительной очистки.
Стадия 2: Синтез 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72b).
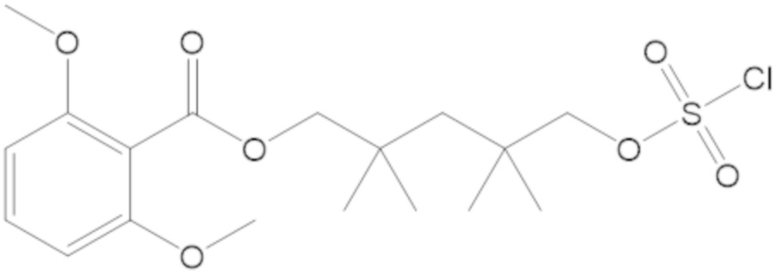
Раствор сульфурилхлорида (0,21 мл, 2,8 ммоль) в Et2O (13 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 10 мин по каплям добавляли раствор 5-гидрокси-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72a) (921 мг, 2,8 ммоль) и пиридина (0,23 мл, 2,8 ммоль) в Et2O (13 мл). Смесь встряхивали при -78°C в течение 5 ч. Смесь фильтровали, а фильтрат сохраняли для получения раствора продукта (72b) в Et2O (около 20 мл). Выход оценивали количественно. Эту смесь использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 5-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72).
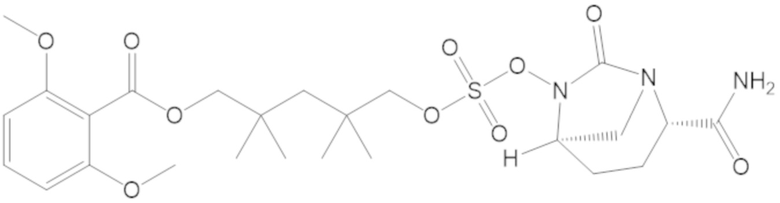
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (525 мг, 2,8 ммоль) растворяли в THF (33 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (4 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли раствор NaHMDS, 1,0 M в THF (2,8 мл, 2,8 ммоль), и встряхивали в течение 90 мин. В реакционную смесь (канюлю) добавляли раствор 5-((хлорсульфонил)окси)-2,2,4,4-тетраметилпентил 2,6-диметоксибензоата (72b) (1,2 г, 2,8 ммоль) в Et2O (около 20 мл). После встряхивания в течение 10 мин смесь нагревали до комнатной температуры и встряхивали в течение 2 ч. Смесь гасили насыщенным водным раствором бикарбоната натрия (40 мл) и экстрагировали EtOAc (40 мл). Органический слой промывали H2O (3×40 мл), рассолом (40 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (2:3-9:1) с получением продукта (72) (726 мг, 43%) в виде твердого вещества. LC-MS: м/з=572,08 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ 7,32-7,26 (m, 1H), 6,56 (d, J=8,1 Гц, 2H), 6,52 (s, 1H), 5,64 (s, 1H), 4,53 (d, J=8,7 Гц, 1H), 4,24 (d, J=9,0 Гц, 1H), 4,17 (s, 1H), 4,07-4,04 (m, 3H), 3,81 (s, 6H), 3,34-3,30 (m, 1H), 3,00 (d, J=12,3 Гц, 1H), 2,47-2,40 (m, 1H), 2,14 (m, 1H), 2,05-1,84 (m, 2H), 1,48 (s, 2H), 1,11 (s, 6H), 1,10 (s, 6H). 13C-NMR (75 МГц, CDCl3): δ 171,0, 167,0, 166,8, 157,5, 131,1, 113,3, 103,9, 85,2, 74,2, 61,9, 60,2, 56,0, 47,2, 46,1, 36,0, 35,7, 26,4, 26,3, 25,9, 25,2, 20,8, 17,5.
Пример 73
Синтез этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилбутаноата (73)
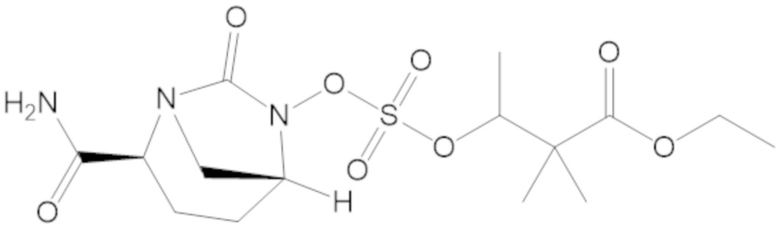
Стадия 1: Синтез R/S-этил 3-((хлорсульфонил)окси)-2,2-диметилбутаноата (73a).
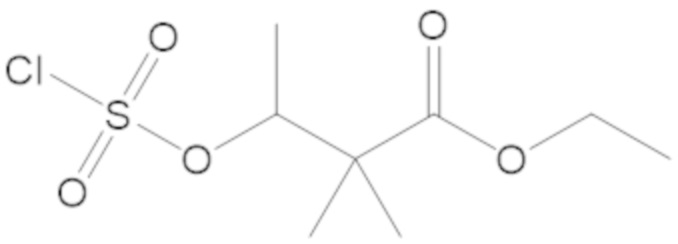
Раствор свежедистиллированного сульфурилхлорида (148 мкл, 2,0 ммоль) в Et2O (0,2 мл) охлаждали до -78°C в атмосфере аргона. В раствор сульфурилхлорида в течение 15 мин по каплям добавляли раствор этил 3-гидрокси-2,2-диметилбутаноата (полученного согласно J. Med. Chem. 1987, 30, 366-374 и Ad. Synth. Catal. 2009, 351, 3128-3132) (324 мг, 2,0 ммоль) и пиридина (164 мкл, 2,0 ммоль) в Et2O (0,2 мл). Колбу промывали Et2O (2×20 мл), который добавляли в реакционную смесь. Смесь встряхивали при -78°C в течение 30 минут. Смесь фильтровали, а продукт (73a) использовали непосредственно на следующей стадии с принятым количественным выходом. 1H-NMR (300 МГц, CDCl3): δ 5,34-5,29 (m, 1H), 4,22-4,14 (m, 2H), 1,55-1,52 (m, 3H), 1,35-1,08 (m, 9H).
Стадия 2: Синтез этил 3-(((((2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-2,2-диметилбутаноата (73).
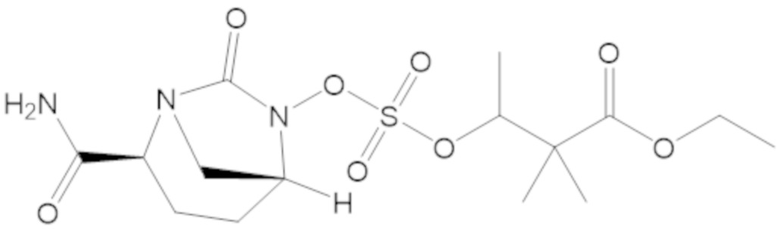
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,22 г, 1,2 ммоль) растворяли в THF (10 мл) и HMPA (0,5 мл), а полученный в результате перемешанный раствор охлаждали до -78°C в атмосфере аргона. В смесь добавляли раствор NaHMDS, 1,0 M в THF (1,2 мл, 1,2 ммоль), и смесь встряхивали в течение 10 мин. В реакционную смесь быстро добавляли раствор этил 3-((хлорсульфонил)окси)-2,2-диметилбутаноата (73a) (0,51 г, 2,0 ммоль) в Et2O (20 мл). После 10 мин встряхивания при -78°C обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 1 ч. Смесь охлаждали до 0°C, гасили H2O и разбавляли EtOAc. Водные и органические слои разделяли, а органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-7:3) с получением продукта (73) (70 мг, 15%) в виде твердого вещества. 1H-NMR (300 МГц, CDCl3): δ 6,51 (br. s, 1H), 5,66 (br. s, 1H), 5,32 (q, J=6,3 Гц,1H), 4,21-4,14 (m, 3H), 4,07 (t, J=6,3 Гц, 1H), 3,35-3,31 (m, 1H), 3,04-2,98 (m, 1H), 2,44-2,39 (m, 1H), 2,17-2,09 (m, 1H), 1,95-1,83 (m, 2H), 1,58-1,48 (m, 3H), 1,31-1,20 (m, 9H). 13C-NMR (75 МГц, CDCl3): δ 174,5, 174,4, 171,2, 171,1, 166,8, 166,6, 91,1, 90,6, 62,0, 62,0, 61,5, 61,5, 60,2, 47,2, 47,2, 47,1, 21,2, 20,9, 20,8, 20,8, 20,6, 20,3, 17,5, 17,5, 15,9, 15,5, 14,2 (Примечание: 13C-NMR показал некоторые дублированные пики из-за смеси диастереомеров).
Пример 74
Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил) сульфата (74)
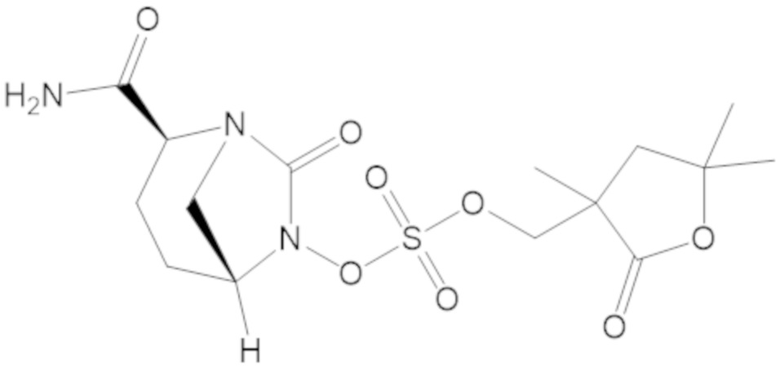
Стадия 1: Синтез 3,5,5-триметилдигидрофуран-2(3H)-она (74a).
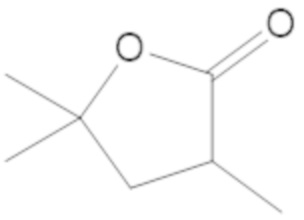
5,5-диметилдигидрофуран-2(3H)-он (4,7 г, 41,2 ммоль) растворяли в THF (94 мл), и смесь охлаждали до -78°C в атмосфере аргона. В раствор диизопропиламида лития, 2,0 M в течение 10 мин по каплям добавляли раствор в THF (22,6 мл, 45,2 ммоль). Реакционную смесь встряхивали при -78°C в течение 2 ч, а затем в реакционную смесь в течение 5 мин добавляли неразбавленный MeI (2,6 мл, 41,6 ммоль). Реакционную смесь встряхивали при -78°C в течение 45 мин, а затем обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 16 ч. Реакцию гасили насыщенным NH4Cl (25 мл), и смесь концентрировали для удаления THF. Водный остаток разбавляли H2O для растворения твердого вещества, а затем экстрагировали этилацетатом (3×40 мл). Объединенный органический слой концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:3) с получением жидкости, которая затвердевала при отстаивании. Это твердое вещество дополнительно очищали посредством дистилляции в Kugelrohr с получением продукта (74a) (3,2 г) в виде масла. 1H-NMR (300 МГц, CDCl3): δ 2,78-2,87 (m, 1H), 2,33 (dd, J=9,3, 12,3 Гц, 1H), 1,71 (t, J=12,3 Гц, 1H), 1,45 (s, 3H), 1,38 (s, 3H), 1,29 (d, J=6,9 Гц, 3H).
Стадия 2: Синтез 3-((бензилокси)метил)-3,5,5-триметилдигидрофуран-2(3H)-она (74b).
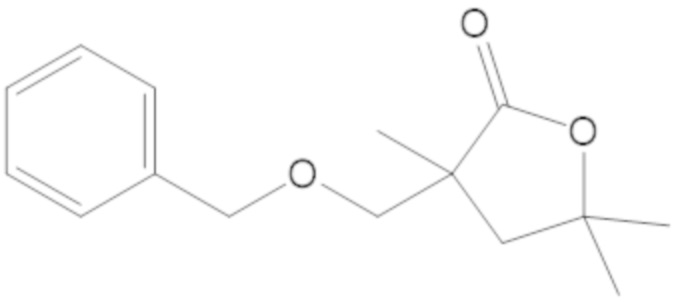
3,5,5-Триметилдигидрофуран-2(3H)-он (74a) (3,2 г, 25,0 ммоль) растворяли в THF (60 мл), и смесь охлаждали до -78°C в атмосфере аргона. В течение 10 мин по каплям добавляли раствор диизопропиламида лития, 2,0 M в THF (13,7 мл, 27,5 ммоль). Смесь встряхивали при -78°C в течение 30 минут, затем в течение 5 мин добавляли неразбавленный бензил хлорметиловый эфир (90%; 4,2 мл, 27,5 ммоль). Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение 16 ч. Добавляли насыщенный NH4Cl (10 мл) и H2O (10 мл), и растворитель удаляли в вакууме. Остаток экстрагировали EtOAc (2×75 мл), и объединенные органические слои промывали рассолом (2×75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме (5,8 г). Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-2:3) с получением продукта (2,27 г) и с фракциями примесей (1,35 г). Фракции примесей повторно очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (0:1-1:4) с получением дополнительного чистого продукта (74b) (1,39 г). Продуктом (3,66 г) было масло. 1H-NMR (300 МГц, CDCl3): δ 7,28-7,34 (m, 5H), 4,62 (dd, J=11,7, 35,1 Гц, 2H), 3,61 (d, J=11,7 Гц, 1H), 3,32 (d, J=11,7 Гц, 1H), 2,48 (d, J=12,9 Гц, 1H), 1,89 (d, J=12,9 Гц, 1H), 1,45 (d, J=6,9 Гц, 6H), 1,26 (s, 3H).
Стадия 3: Синтез 3-(гидроксиметил)-3,5,5-триметилдигидрофуран-2(3H)-она (74c).
3-((бензилокси)метил)-3,5,5-триметилдигидрофуран-2(3H)-он (74b) (1,8 г, 7,2 ммоль) растворяли в 2-пропаноле (60 мл), и раствор дегазировали аргоном. В колбу добавляли твердый 10,0% палладий на углероде (0,31 г, 0,3 ммоль). Колбу герметично закрывали, и проводили вакуумное дегазирование, а затем обратное промывание водородом (3 раза). Реакционную смесь встряхивали в течение 6 ч. Суспензию фильтровали через Celite®, и фильтрационный осадок промывали 2-пропанолом (15 мл). Фильтрат концентрировали в вакууме с получением продукта (74c) в виде неочищенного масла. 1H-NMR (300 МГц, CDCl3): δδ 3,75 (dd, J=6,9, 11,1 Гц, 1H), 3,51 (dd, J=5,7, 11,1 Гц, 1H), 2,33 (d, J=12,9 Гц, 1H), 2,23 (t, J=6 Гц, 1H), 1,94 (d, J=12,9 Гц, 1H), 1,48 (d, J=6,9 Гц, 6H), 1,32 (s, 3H).
Стадия 4: Синтез (3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил сульфохлорида (74d).
Раствор 3-(гидроксиметил)-3,5,5-триметилдигидрофуран-2(3H)-она (74c) (0,50 г, 3,2 ммоль) и пиридина (0,28 мл, 3,5 ммоль) в Et2O (10 мл) охлаждали до -78°C в атмосфере аргона. В указанный выше раствор с помощью шприца по каплям добавляли неразбавленный сульфурилхлорид (0,28 мл, 3,5 ммоль). Смесь встряхивали при -78°C в течение 10 мин, затем колбу нагревали до комнатной температуры и встряхивали в течение 1 ч (контролировали с помощью TLC 30% EA/гексаны). Образовался осадок с получением густой суспензии. Суспензию фильтровали через 0,45-мкм фильтр Teflon®, и фильтрационный осадок промывали свежим Et2O (2×5 мл). Брали аликвоту (0,5 мл) и концентрировали, и для смеси проводили NMR. Оставшийся содержащий продукт (74d) раствор использовали непосредственно на следующей стадии. 1H-NMR (300 МГц, CDCl3): δ 4,60 (d, J=9,3 Гц, 1H), 4,36 (d, J=9,3 Гц, 1H), 2,37 (d, J=14,1 Гц, 1H), 2,09 (d, J=13,5 Гц, 1H), 1,51 (d, J=8,4 Гц, 6H), 1,44 (s, 3H).
Стадия 5: Синтез (2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил) сульфата (74).
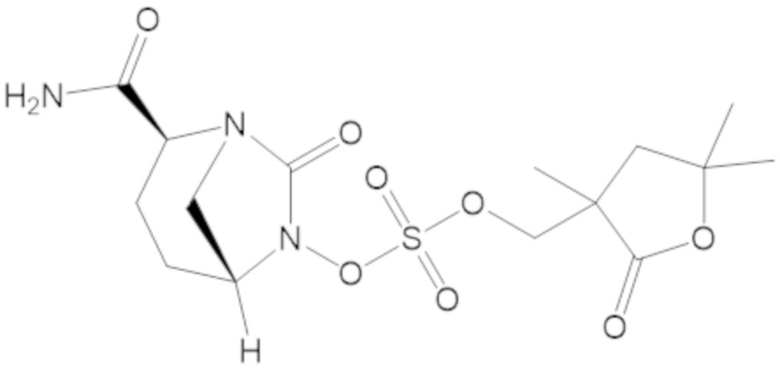
(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1) (0,64 г, 3,5 ммоль) растворяли в THF (30 мл) и 1,3-диметилтетрагидропиримидин-2(1H)-оне (1,4 мл), а полученный в результате раствор охлаждали до -78°C в атмосфере аргона. В охлажденный раствор по каплям добавляли NaHMDS, 1,0 M раствор в THF (3,5 мл, 3,5 ммоль), и смесь встряхивали в течение 1 ч. В реакционную смесь быстро добавляли раствор (3,5,5-триметил-2-оксотетрагидрофуран-3-ил)метил сульфохлорида (74d) (0,81 г, 3,2 ммоль) в Et2O из предыдущей реакции. Обеспечивали нагревание смеси до комнатной температуры, и встряхивали в течение ночи. Добавляли рассол (100 мл) и EtOAc (100 мл), и разделяли водные и органические слои. Водный слой экстрагировали EtOAc (2×100 мл), и объединенные органические слои промывали рассолом (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента EtOAc/гексанов (1:4-1:0) с получением твердого вещества (0,42 г). Твердое вещество растирали в порошок с Et2O (100 мл) в течение 16 ч, фильтровали, и фильтрационный осадок промывали свежим Et2O (3×20 мл) с получением продукта (74) (0,28 г) в виде твердого вещества. LC-MS: м/з=406 [M+H]+. 1H-NMR (300 МГц, CDCl3): δ, 6,55 (br. d, J=44,7 Гц, 1H), 5,82 (br. s, 1H), 4,86 (dd, J=9,3, 63,6 Гц, 1H), 4,55 (dd, J=9,3, 46,2 Гц, 1H), 4,03-4,16 (m, 2H), 3,30-3,35 (m, 1H), 3,06 (dd, J=4,8, 12,3 Гц, 1H), 2,38-2,45 (m, 2H), 2,10-2,20 (m, 1H), 1,8-2,04 (m, 3H), 1,48 (s, 3H), 1,477 (d, J=6,6 Гц, 3H), 1,40 (d, J=4,2 Гц, 3H). 13C-NMR (75 МГц, CDCl3): δ 177,3, 177,0, 170,9, 170,8, 167,3, 167,0, 82,0, 81,9, 78,6, 77,8, 61,9, 60,2, 60,2, 47,1, 46,9, 45,8, 45,7, 43,6, 43,2, 30,1, 29,8, 29,7, 22,5, 20,7, 20,7, 17,4 (Примечание: 13C-NMR показал некоторые дублированные пики, из-за смеси диастереомеров).
Пример 75
Синтез этил 3-(((((2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил)окси)сульфонил)окси)-2,2-диметилпропаноата (75)
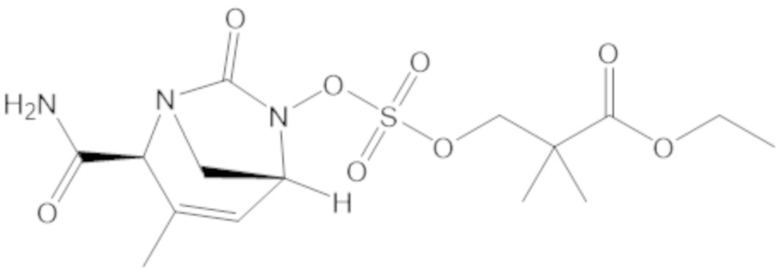
(2S,5R)-6-(Бензилокси)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид (1) (3,6 мг, 0,01 ммоль) растворяли в смешанном растворителе (EtOAc/H2O/EtOH: 0,22/0,34/0,11 мл), в который добавляли Et3N (0,25 мкл, 0,002 ммоль) и Pd/C (сухой, 10%; 1,3 мг, 20 моль%) в N2 при комнатной температуре. Для замены азота на реакционную колбу помещали водородный баллон. Реакционную смесь дегазировали в вакууме и загружали водородом (3 раза). Смесь встряхивали при комнатной температуре в течение 5 ч и контролировали с помощью LCMS. Когда реакция завершалась, смесь разбавляли EtOAc (2 мл) и промывали рассолом. Органическую фазу выделяли и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток (4,3 мг, 0,02 ммоль) растворяли в THF (0,4 мл) и охлаждали до -78°C. По каплям добавляли NaHMDS (1M в THF; 21,3 мкл, 0,02 ммоль). Реакционную смесь встряхивали при -78°C в течение 20 мин, а затем добавляли этил 3-((хлорсульфонил)окси)-2,2-диметилпропаноат (7,8 мг, 0,03 ммоль). Реакционную смесь встряхивали при -78°C в течение 20 мин, а затем медленно нагревали до комнатной температуры и встряхивали в течение ночи (реакцию контролировали с помощью LCMS). После завершения реакции добавляли EtOAc (5 мл), и органический слой промывали насыщенным NaHCO3 с последующим рассолом. Органическую фазу выделяли и сушили (Na2SO4), и продукт (75) концентрировали в вакууме. LC/MS: м/з=406 [M+H]+
Пример 76
Пероральную Биодоступность у Крыс
Фармакокинетическое (PK) исследование проводили на трех самцах крыс Sprague-Dawley (SD) после внутривенного (IV) и перорального (PO) введения авибактама в дозе 2 мг/кг и тестируемых соединений в дозе 10 мг/кг, соответственно, и авибактам измеряли в плазме.
Авибактам, растворяли в забуференном фосфатом солевом растворе (PBS) (pH 7,5) при 0,4 мг/мл для внутривенной (IV) инъекции. Соединения для перорального введения получади в 10% этаноле/40% полиэтиленгликоле (PEG) 400/50% воде для инъекции (WFI) (pH 6,5) при 1 мг/мл. Объемы вводимых доз составляли 5 мл/кг для IV и 10 мл/кг для PO. Все аспекты этой работы, включая содержание, экспериментальную часть и ликвидацию животных проводили в целом в соответствиис «Guide for the Care and Use of Laboratory Animals: Eighth Edition» (National Academies Press, Washington, D.C. 2011); и Suckow et al. Ed. Laboratory Rat. 2nd Edition. Academic Press. New York. 2005. Животные имели доступ к стандартной лабораторной пище и автоклавированной водопроводной воде ad libitum.
Аликвоты крови (от 300 мкл до 400 мкл) брали у крыс с катетеризацией яремной вены в пробирки, покрытые гепарином лития, в различные моменты времени. Пробирки осторожно перемешивали и хранили на льду, а затем центрифугировали при 2500 об/мин в течение 15 минут при 4°С в течение 1 часа после сбора. Для животных в контрольных группах кровь собирали путем пункции сердца, а плазму собирали и хранили замороженной при -70°С до дальнейшего анализа. Beaudoin et al., Bioanalytical method validation for the simultaneous determination of ceftazidime and avibactam in rat plasma. Bioanalysis. 2016 8:111-22.
Образцы плазмы обрабатывали с использованием осаждения ацетонитрилом и анализировали методом LC-MS/MS. Калибровочную кривую плазмы получали с аликвотами плазмы, не содержащей лекарств, с добавлением тестируемого вещества при указанных уровнях концентрации. Образцы плазмы с добавлениями обрабатывали вместе с неизвестными образцами плазмы, используя ту же процедуру. Обработанные образцы плазмы хранили при -70°С до получения анализа LC-MS/MS, в который регистрировали площади пиков по времени, а концентрации тестируемого вещества в неизвестных образцах плазмы определяли с использованием соответствующей калибровочной кривой. Определяли отчетный линейный диапазон анализа, а также нижний предел количественного определения (LLQ). Строили графики зависимости концентрации соединения в плазме от времени. Фармакокинетические параметры соединения после внутривенного и перорального введения (AUClast, AUCINF, T1/2, Tmax, и Cmax) получали из некомпартментального анализа (NCA) данных плазмы с использованием WinNonlin. WinNonlin® Certara L.P. Pharsight, St. Louis, MO.
В этих тестах авибактам показал пероральную биодоступность (%F) 1,2%, а соединения (3), (4), (10), (11), (12), (13), (14), (15), (16), (17), (18) и (19) показали пероральную биодоступность (%F) более 10%. Кроме того, соединения (36), (37), (42), (53), (57), (58) и (59) показали пероральную биодоступность (% F), превышающую 10%.
В этих тестах релабактам проявлял пероральную биодоступность (%F) 1,8%, а соединения (20), (22), (23) и (25) демонстрировали пероральную биодоступность (%F) более 5%.
Пример 77
Минимальная Ингибирующая концентрация
Значения минимальной концентрации ингибитора (MIC) исследуемых монобактамов и ингибиторов β-лактамазы определяли с помощью тестирования на чувствительность к микроразведению в бульоне, проводимого в соответствии с рекомендациями Института клинических и лабораторных стандартов (Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Tenth Edition. CLSI document M07-A10. CLSI, 950 West Valley Road, Suite 2500, Wayne, PA 19087-1898 USA, 2015; CLSI. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Sixth Informational Supplement. CLSI document M100-S26, CLSI, 950 West Valley Road, Suite 2500, Wayne, PA 19087 USA, 2016) относительно панели бактериальных штаммов, экспрессирующих характерные β-лактамазы, которые придают устойчивость к β-лактамам. Zasowski et al., The β-Lactams Strike Back: Ceftazidime-Avibactam. Pharmacotherapy, 2015 35:755-70; Levasseur et al., In vitro antibacterial activity of the ceftazidime-avibactam combination against enterobacteriaceae, including strains with well-characterized β-lactamases. Antimicrob Agents Chemother, 2015 59:1931-4. Соединения хранили в виде сухого порошка и хранили при -20°С до испытания. Эти соединения и препараты сравнения растворяли в соответствующем растворителе в день анализа. Все препараты тестировали с использованием диапазона концентраций препаратов от 0,001 мкг/мл до 64 мкг/мл. Ингибиторы β-лактамазы тестировали в постоянной концентрации 4 мкг/мл. Изоляты наносили на соответствующие среды и инкубировали в течение ночи при 35°С. Значения MIC определяли с использованием бульона Мюллера-Хинтона со стандартизированным содержанием катионов (MHBII; BD, Sparks, MD) в соответствии с рекомендациями CLSI на планшетах 96-луночного формата. MIC регистрировали через 18 ч инкубации при 35°С. MIC считывали и регистрировали в виде самой низкой концентрации препарата, которая подавляла видимый рост организма.
Пример 78
Пероральная Биодоступность у собак
Оценивали у собак пероральную биодоступность некоторых пролекарств авибактама, представленных в настоящем раскрытии.
Дозированные готовые формы получали в день введения. Внутривенные готовые формы получали в асептических уловиях, стерильно фильтровали и доводили до комнатной температуры перед введением. Внутривенная готовая форма содержала авибактам в итоговой концентрации 2,0 мг/мл в PBS при pH 7,5.
Пероральные дозированные готовые формы имели итоговую концентрацию либо авибактама, либо пролекарства авибактама 2 мг/мл в растворе 1 мл этанола, 4 мл PEG400 и 5 мл воды для инъекции с pH, доведенным до 7 с помощью 1 н. NaOH.
Готовые формы вводили вводили самцам собак породы бигль весом от 8 кг до 410 кг. Животных содержали в соответствии с Руководством по уходу и использованию лабораторных животных, National Research Council, National Academies Press, Washington, DC, 2011.
Собаки получали либо IV болюсную дозу 10 мг/кг, либо пероральную дозу 20 мг/кг. Уровни дозы выбирали чтобы преодолеть разрыв между первичными историческими контрольными данными и исследованием NHP (Американская ветеринарная медицинская ассоциация. Рекомендации AVMA по эвтаназии. 2013), чтобы точно предсказать активность пролекарства у людей. Внутривенное введение проводили в головную вену с последующим промыванием 0,5 мл стерильным физиологическим раствором. Пероральное введение через желудок с использованием катетера 18 по французской шкале с последующим промыванием 15 мл деионизированной водой. Для каждой группы исследования использовали двух собак.
Концентрацию авибактама в плазме измеряли с некоторыми интервалами после введения. В течение 2 минут после сбора 100 мкл цельной крови переносили в пробирки K2EDTA, содержащие 300 мкл ацетонитрила. Каждую смесь кровь/ацетонитрил встряхивали в течение приблизительно 30 секунд и сразу же замораживали на сухом льду и хранили замороженной (-55°C - -85°C) до анализа. Концентрацию авибактама определяли с использованием LC/MS/MS.
Площадь под кривыми концентрация/время (AUC) расчитывали с использованием линейного трапециевидного метода с линейной интерполяцией. Процентную пероральную биодоступность (%F) авибактама определяли путем сравнения AUC после перорального введения с AUC после IV введения на основе нормированной дозы.
Соединения (3), (13) и (15) продемонстрировали пероральную биодоступность авибактама у самцов собак породы бигль более чем 50%F.
Пример 79
Пероральная Биодоступность у обезъян
Оценивали пероральную биодоступность некоторых пролекарств авибактама, представленных в настоящем раскрытии, у самцов обезъян Яванского макака.
Дозированные готовые формы получали в день введения. Внутривенные готовые формы получали в асептических уловиях, стерильно фильтровали и доводили до комнатной температуры перед введением. Внутривенная готовая форма содержала авибактам в итоговой концентрации 2,0 мг/мл в PBS при pH 7,5.
Пероральные дозированные готовые формы имели итоговую концентрацию либо авибактама, либо пролекарства авибактама 2 мг/мл в растворе 1 мл этанола, 4 мл PEG400 и 5 мл воды для инъекции с pH, доведенным до 7 с помощью 1 н. NaOH.
Готовые формы вводили самцам обезъян Яванского макака весом от 2 кг до 4 кг. Животных содержали в соответствии с Руководством по уходу и использованию лабораторных животных, National Research Council, National Academies Press, Washington, DC, 2011.
Обезъяны получали либо IV болюсную дозу 10 мг/кг, либо пероральную дозу 20 мг/кг. уровни дозы выбирали для имитации терапевтически эффективных системных концентраций у людей. Внутривенное введение проводили в подкожную вену. Пероральное введение осуществляли посредством интубации через рот через гибкую ротовую трубку. Для каждой группы исследования использовали двух обезъян.
Концентрацию авибактама в плазме измеряли с некоторыми интервалами после введения. В течение 2 минут после сбора 100 мкл цельной крови переносили в пробирки K2EDTA, содержащие 300 мкл ацетонитрила. Каждую смесь кровь/ацетонитрил встряхивали в течение приблизительно 30 секунд и сразу же замораживали на сухом льду и хранили замороженными (от -55°C до -85°C) до анализа. Концентрацию авибактама определяли с использованием LC/MS/MS.
Площадь под кривыми концентрация/время (AUC) расчитывали с использованием линейного трапециевидного метода с линейной интерполяцией. Процентную пероральную биодоступность (%F) авибактама определяли путем сравнения AUC после перорального введения с AUC после IV введения на основе нормированной дозы.
Соединения (3), (13) и (15) продемонстрировали пероральную биодоступность авибактама у обезъян Яванского макака более чем 50%F.
В итоге следует отметить, что существуют альтернативные способы реализации раскрытых в данном документе вариантов осуществления. Соответственно, настоящие варианты осуществления следует рассматривать как иллюстративные, а не ограничивающие, и формулу изобретения не следует ограничивать деталями, приведенными в данном документе, но можно изменять в пределах ее объема и эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2815314C2 |
| ВВОДИМЫЕ ПЕРОРАЛЬНО КОМБИНАЦИИ БЕТА-ЛАКТАМНЫХ АНТИБИОТИКОВ И ПРОИЗВОДНЫХ АВИБАКТАМА ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2020 |
|
RU2825281C1 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2715058C2 |
| АЗОТСОДЕРЖАЩИЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2835672C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| ПРОИЗВОДНЫЕ 7-ОКСО-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-3-ЕНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2645678C2 |
Изобретение относится к соединению формулы (1) или его фармацевтически приемлемой соли, где каждый R1 независимо выбирают из C1–6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3–6 циклоалкильное кольцо или C3–6 гетероциклоалкильное кольцо, содержащее один атом кислорода и замещенное 1-3 группами, независимо выбранными из =О и C1–6 алкила; R2 выбирают из одинарной связи, C1–6 алкандиила, C1–6 гетероалкандиила, содержащего один или два атома кислорода, C1–6 алкандиила, замещенного 1-2 C1–6 алкилами, C1–6 гетероалкандиила, содержащего один или два атома кислорода и замещенного =О; R3 выбирают из C1–6 алкила, –O–C(O)–R4, –S–C(O)–R4, –C(O)–O–R4, C6 арила, замещенного –O-C(O)-R4, в котором R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C6 арила, C7–10 арилалкила, C1–8 алкила, замещенного C1–6 алкокси, C1–8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, метил-5-метил-1,3-диоксол-2-она, C6 арила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси; R5 выбирают из водорода, 4-ил-пиперидина и C2–6 гетероалкила, содержащего атом кислорода и замещенного -NH2; R6 представляет собой водород; и А представляет собой одинарную связь (–), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1–3 алкил; или соединение представляет собой: (2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил 1H–имидазол–1–сульфонат (34); 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил адамантан–1–карбоксилат (55); диэтил 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилмалонат (56); 4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил адамантан–1–карбоксилат (63); 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил дибензоат (70); 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил диацетат (71); этил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилбутаноат (73). Изобретение также относится к фармацевтической композиции для ингибирования бета-лактамазы на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения бактериальных инфекций. 7 н. и 23 з.п. ф-лы, 79 пр.
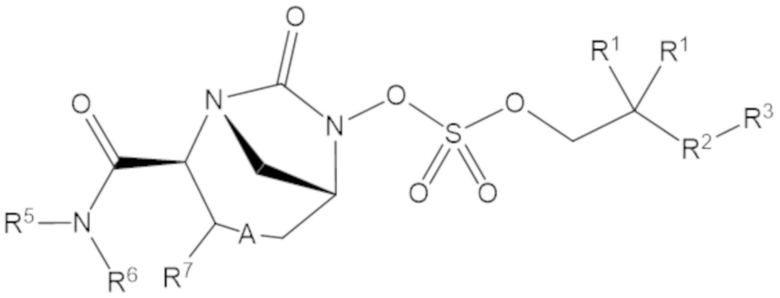 (1)
(1)
1. Соединение формулы (1):
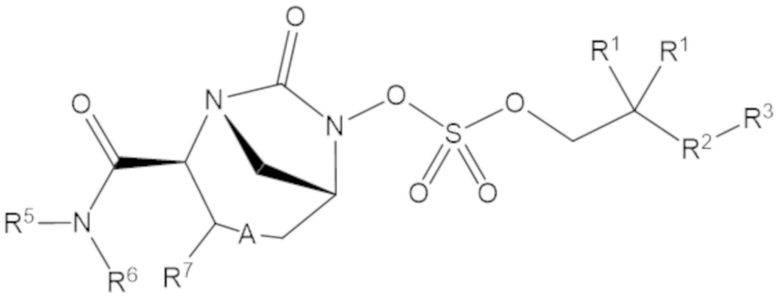 (1)
(1)
или его фармацевтически приемлемая соль, при этом
каждый R1 независимо выбирают из C1–6 алкила, или каждый R1 и геминальный атом углерода, с которым они связаны, образует C3–6 циклоалкильное кольцо или C3–6 гетероциклоалкильное кольцо, содержащее один атом кислорода и замещенное 1-3 группами, независимо выбранными из =О и C1–6 алкила;
R2 выбирают из одинарной связи, C1–6 алкандиила, C1–6 гетероалкандиила, содержащего один или два атома кислорода, C1–6 алкандиила, замещенного 1-2 C1–6 алкилами, C1–6 гетероалкандиила, содержащего один или два атома кислорода и замещенного =О;
R3 выбирают из C1–6 алкила, –O–C(O)–R4, –S–C(O)–R4, –C(O)–O–R4, C6 арила, замещенного –O-C(O)-R4, в котором
R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C6 арила, C7–10 арилалкила, C1–8 алкила, замещенного C1–6 алкокси, C1–8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, метил-5-метил-1,3-диоксол-2-она, C6 арила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси;
R5 выбирают из водорода, 4-ил-пиперидина и C2–6 гетероалкила, содержащего атом кислорода и замещенного -NH2;
R6 представляет собой водород; и
А представляет собой одинарную связь (–), а R7 представляет собой водород, или А представляет собой двойную связь (=), а R7 представляет собой C1–3 алкил;
или
(2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил 1H–имидазол–1–сульфонат (34);
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил адамантан–1–карбоксилат (55);
диэтил 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилмалонат (56);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил адамантан–1–карбоксилат (63);
2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил дибензоат (70);
2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил диацетат (71); или
этил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилбутаноат (73).
2. Соединение по п. 1, причем соединение имеет структуру формулы (2a):
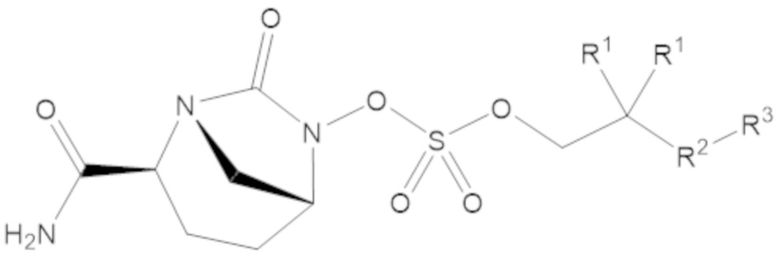 (2a)
(2a)
или его фармацевтически приемлемая соль.
3. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила;
R2 представляет собой одинарную связь;
R3 представляет собой –C(O)–O–R4; и
R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C7–9 арилалкила, C1–8 алкила, замещенного C1–6 алкокси, C1–8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О.
4. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–6 алкила;
R2 представляет собой одинарную связь;
R3 представляет собой –C(O)–O–R4; и
R4 выбирают из C1–6 алкила.
5. Соединение, выбранное из:
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил бензоата (2);
этил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (3);
бензил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (4);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил бензоата (6);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил пропионата (7);
бензил (4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил) адипата (8);
6–(4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутокси)–6–оксогексановой кислоты (9);
метил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (10);
изопропил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (11);
гексил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (12);
гептил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (13);
трет–бутил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (14);
2–метоксиэтил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (15);
оксетан–3–ил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (16);
этил–1–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)циклогексанкарбоксилата (17);
этил–1–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)циклопропанкарбоксилата (18);
этил–1–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)циклобутанкарбоксилата (19);
(2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил 1H–имидазол–1–сульфоната (34);
этил 5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентаноата (35);
гексил 5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентаноата (36);
гептил 5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентаноата (37);
2–метоксиэтил 5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентаноата (38);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,4,4–тетраметилпентил пропионата (39);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,4,4–тетраметилпентил бензоата (40);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,4,4–тетраметилпентил 2,6–диметилбензоата (41);
(2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил ((3–метил–2–оксотетрагидрофуран–3–ил)метил) сульфата (42);
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил пивалата (43);
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил 3–хлор–2,6–диметоксибензоата (44);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,3,3–тетраметилбутил 2,6–диметилбензоата (45);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,3,3–тетраметилбутил бензоата (46);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,3,3–тетраметилбутил пропионата (47);
(2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил ((3–метил–2–оксотетрагидро–2H–пиран–3–ил)метил) сульфата (48);
2–(3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил)фенилацетата (49);
2–(3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил)фенилпивалата (50);
S–(4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил) этантиоата (51);
S–(5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентил) этантиоата (52);
S–(3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил) этантиоата (53);
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил 2,6–диметилбензоата (54);
3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропил адамантан–1–карбоксилата (55);
диэтил 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилмалоната (56);
пропил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (57);
бутил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (58);
(5–метил–2–оксо–1,3–диоксол–4–ил)метил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноата (59);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутилпивалата (60);
этил 2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–этилбутаноата (61);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил 2,6–диметилбензоата (62);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил адамантан–1–карбоксилата (63);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–3,3–диметилбутил 2,6–диметоксибензоата (64);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентил бензоата (65);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентил 2,6–диметоксибензоата (66);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентил 2,6–диметилбензоата (67);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентил 2–метилбензоата (68);
4–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,3,3–тетраметилбутил 3–хлор–2,6–диметоксибензоата (69);
2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил дибензоата (70);
2–((((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)метил)–2–метилпропан–1,3–диил диацетата (71);
5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2,4,4–тетраметилпентил 2,6–диметоксибензоата (72);
этил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилбутаноата (73);
(2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил ((3,5,5–триметил–2–оксотетрагидрофуран–3–ил)метил) сульфата (74);
фармацевтически приемлемой соли любого из перечисленного выше.
6. Соединение по п. 2, где соединение представляет собой этил 5–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–4,4–диметилпентаноат (35):
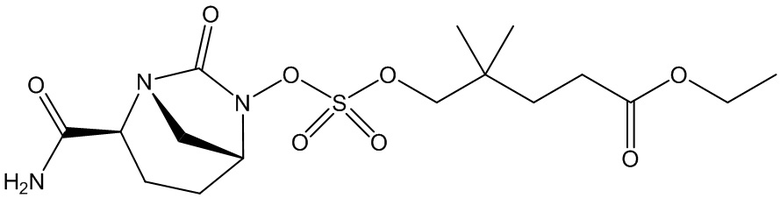
или его фармацевтически приемлемая соль.
7. Соединение по п. 2, где соединение представляет собой 4-(((((1R,2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил)окси)сульфонил)окси)-3,3-диметилбутилпивалат (60):
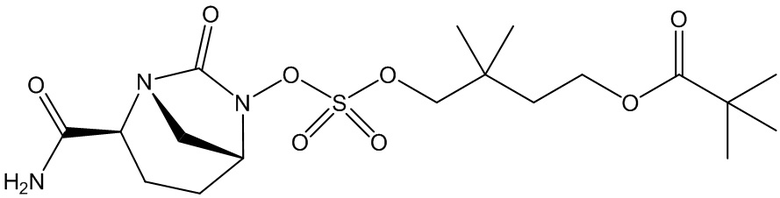
или его фармацевтически приемлемая соль.
8. Соединение по п. 2, где соединение представляет собой (1R,2S,5R)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]октан-6-ил ((3-метил-2-оксотетрагидрофуран-3-ил)метил) сульфат (42):
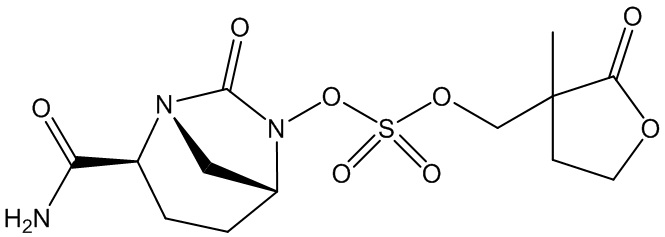
или его фармацевтически приемлемая соль.
9. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо;
R2 выбирают из одинарной связи, метан–диила и этан–диила; и
R3 выбирают из –C(O)–O–R4 и –S–C(O)–R4, в котором R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C6 арила, C7–9 арилалкила, C1-8 алкила, замещенного C1–6 алкокси, C1-8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, C6 арила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси.
10. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой –C(O)–O–R4, где R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C6 арила, C7-9 арилалкила, C1-8 алкила, замещенного C1–6 алкокси, C1-8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, C6 арила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси.
11. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо;
R2 представляет собой –(CH2)2–; и
R3 представляет собой –C(O)–O–R4, в котором R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C6 арила, C7-9 арилалкила, C1-8 алкила, замещенного C1–6 алкокси, C1-8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, C6 арила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси.
12. Соединение по п. 2, в котором
каждый R1 выбирают из C1–3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо;
R2 представляет собой –CH2–; и
R3 представляет собой –S–C(O)–R4, в котором R4 представляет собой C1–8 алкил.
13. Соединение по п. 2, в котором
каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо или C3–6 гетероциклоалкильное кольцо, содержащее один атом кислорода и замещенное 1-3 группами, независимо выбранными из =О и C1–6 алкила;
R2 представляет собой одинарную связь; и
R3 представляет собой C1–3 алкил.
14. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила;
R2 выбирают из одинарной связи и метандиила; и
R3 выбирают из –O–C(O)–R4 и –C(O)–O–R4, в котором R4 выбирают из C1–8 алкила и фенила, замещенного 1-3 заместителями, независимо выбранными из галогена, C1–6 алкила и C1–6 алкокси.
15. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила;
R2 выбирают из одинарной связи и метандиила; и
R3 представляет собой замещенный фенил, в котором заместитель представляет собой –O–C(O)–R4, в котором R4 представляет собой C1–8 алкил.
16. Соединение по п. 2, в котором
каждый R1 независимо выбирают из C1–3 алкила;
R2 выбирают из –C(R8)2– и –CH2–C(R8)2–, где каждый R8 независимо выбирают из C1–3 алкила; и
R3 выбирают из –C(O)–O–R4 и –O–C(O)–R4, в котором R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C1–8 алкила, замещенного C1–6 алкокси, C1–8 гетероалкила, содержащего один атом кислорода и замещенного -СООН и =О, и 4(ил–метил)–5–метил–1,3–диоксол–2–она.
17. Соединение по п. 2, в котором
каждый R1 вместе с атомом углерода, с которым они связаны, образуют C5–6 гетероциклическое кольцо, содержащее один атом кислорода и замещенное 1-3 группами, независимо выбранными из =О и C1–6 алкила;
R2 представляет собой одинарную связь; и
R3 представляет собой C1–3 алкил.
18. Соединение по п. 1, причем соединение имеет структуру формулы (4):
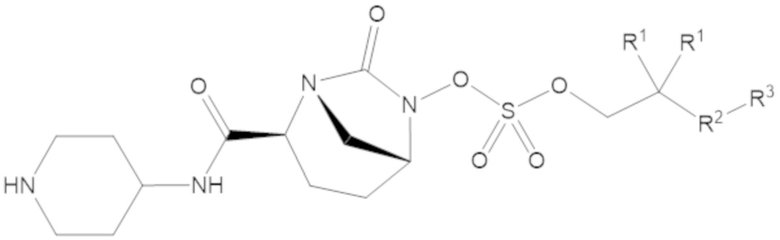 (4)
(4)
или его фармацевтически приемлемой соли.
19. Соединение по п. 18, в котором
каждый R1 независимо выбирают из C1–3 алкила, или каждый R1 вместе с атомом углерода, с которым они связаны, образуют C3–6 циклоалкильное кольцо;
R2 представляет собой одинарную связь; и
R3 представляет собой –C(O)–O–R4, в котором R4 выбирают из C1–8 алкила, C1–8 гетероалкила, содержащего один атом кислорода, C7–10 арилалкила.
20. Соединение по п. 18, причем соединение выбирают из:
этил 2,2–диметил–3–(((((2S,5R)–7–оксо–2–(пиперидин–4–илкарбамоил)–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)пропаноата (20);
2–метоксиэтил 2,2–диметил–3–(((((2S,5R)–7–оксо–2–(пиперидин–4–илкарбамоил)–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)пропаноата (21);
4–((2S,5R)–6–(((3–(гексилокси)–2,2–диметил–3–оксопропокси)сульфонил)окси)–7–оксо–1,6–диазабицикло[3.2.1]октан–2–карбоксамидо)пиперидин–1–иум 2,2,2–трифторацетата (22);
4–((2S,5R)–6–(((3–(гептилокси)–2,2–диметил–3–оксопропокси)сульфонил)окси)–7–оксо–1,6–диазабицикло[3.2.1]октан–2–карбоксамидо)пиперидин–1–иум 2,2,2–трифторацетата (23);
4–((2S,5R)–6–((((1–(этоксикарбонил)циклогексил)метокси)сульфонил)окси)–7–оксо–1,6–диазабицикло[3.2.1]октан–2–карбоксамидо)пиперидин–1–иум 2,2,2–трифторацетата (24);
(5–метил–2–оксо–1,3–диоксол–4–ил)метил 2,2–диметил–3–(((((2S,5R)–7–оксо–2–(пиперидин–4–илкарбамоил)–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)пропаноата (25);
фармацевтически приемлемой соли любого из перечисленного выше.
21. Соединение по п. 1, причем соединение имеет структуру формулы (5):
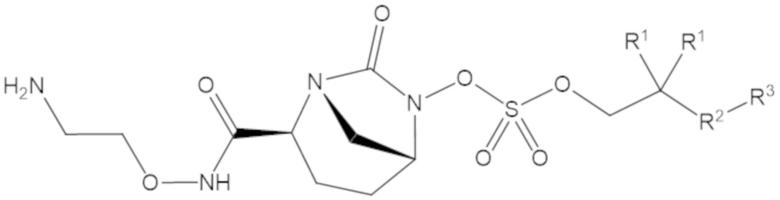 (5)
(5)
или его фармацевтически приемлемой соли.
22. Соединение, представляющее собой этил 3–(((((2S,5R)–2–карбамоил–7–оксо–1,6–диазабицикло[3.2.1]октан–6–ил)окси)сульфонил)окси)–2,2–диметилпропаноат (3):
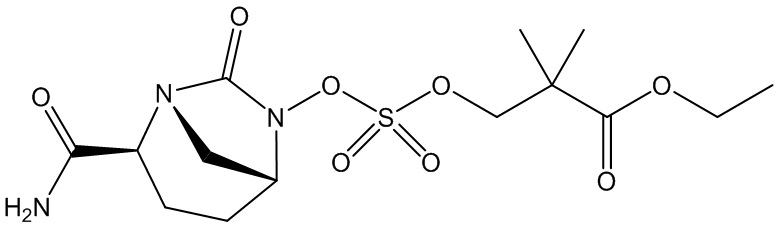
или его фармацевтически приемлемая соль.
23. Фармацевтическая композиция для ингибирования β-лактамазы, содержащая терапевтически эффективное количество соединения по любому из пп. 1-22 и фармацевтически приемлемую основу.
24. Фармацевтическая композиция по п. 23, дополнительно содержащая β-лактамный антибиотик.
25. Фармацевтическая композиция по п. 24, в которой β-лактамный антибиотик включает цефтибутен, перорально биодоступное производное азтреонама, цефподоксима, цефиксима, тебипенема, пенема, или фармацевтически приемлемую соль любого из вышеуказанного, или комбинацию любого из вышеуказанного.
26. Фармацевтическая композиция по п. 24, причем фармацевтическая композиция представляет собой форму с дозировкой для перорального применения.
27. Способ лечения бактериальной инфекции у пациента, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п. 1 или его фармацевтически приемлемой соли и терапевтически эффективного количества β-лактамного антибиотика, где бактерии, вызывающие бактериальную инфекцию, продуцируют β-лактамазу.
28. Способ лечения бактериальной инфекции у пациента, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п. 1 или его фармацевтически приемлемой соли и терапевтически эффективного количества β-лактамного антибиотика,
в котором бактериальная инфекция способна лечиться терапевтически эффективным количеством β-лактамного антибиотика при совместном введении с терапевтически эффективным количеством соединения по п. 1 или его фармацевтически приемлемой соли.
29. Способ ингибирования фермента β-лактамазы у пациента, включающий введение пациенту эффективного количества соединения по п. 1.
30. Способ по любому из пп. 27-29, в котором введение включает пероральное введение.
| WO 2017045510 A1, 23.03.2017 | |||
| WO 2016116788 A1, 28.07.2016 | |||
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |

