Область изобретения
Настоящее изобретение относится к новым ингибиторам бета-лактамаз, их фармацевтическим композициям и способам применения. Кроме того, настоящее изобретение относится к терапевтическим способам лечения бактериальных инфекций, включая устранение резистентности бактерий к антибиотикам.
Предшествующий уровень техники
Международное сообщество по микробиологии и инфекционным заболеваниям продолжает выражать серьезную озабоченность в отношении того, что продолжающееся развитие резистентности к антибактериальным средствам может привести к появлению бактериальных штаммов, против которых существующие в настоящее время антибактериальные агенты не будут эффективными. Итогом такого появления могут стать значительные уровни заболеваемости и смертности. В общем, бактериальные патогены можно классифицировать либо как грамположительные патогены, либо как грамотрицательные патогены. Антибиотические соединения с эффективной активностью против грамположительных и грамотрицательных патогенов обычно считаются имеющими широкий спектр действия.
В борьбе против бактериальной инфекции важное место занимают бета-лактамные антибиотики. Бета-лактамы являются представителями широкого класса лекарственных средств, которые в структуре ядра молекулы имеют бета-лактам, и которые обычно проявляют эффективность против широкого спектра грамположительных и грамотрицательных бактерий, ингибируя синтез клеточной стенки бактерии. Поскольку мишень этих лекарственных средств не имеет эукариотического аналога, их токсичность низкая, и они хорошо переносятся. Они остаются в числе самых повсеместно назначаемых, безопасных и эффективных лекарственных средств, доступных для борьбы с бактериальной инфекцией. Однако их эффективность ограничена, так как существуют высокорезистентные инфекционные штаммы, такие как метициллин-резистентный штамм Staphylococcus aureus (MRSA), и резистентные к множеству лекарственных средств (MDR) штаммы Pseudomonas aeruginosa, Acinetobacter baumannii, Escherichia coli, Klebsiella pneumonia и другие Enterobacteriaceae. Такие резистентные бактерии являются основной причиной заболеваемости и смертности пациентов (Helfand, β-lactams Against Emerging 'Superbugs': Progress and Pitfalls, Expert Rev. Clin. Pharmacol. 1(4):559-571 (2008)).
Бета-лактамные антибиотики, одни и в комбинации с ингибиторами бета-лактамаз, продолжают составлять значительную часть антибактериальных агентов, применяемых для борьбы с заболеванием. Резистентность β-лактамов к грамположительным инфекциям стимулируется прежде всего активностью β-лактамаз, и значительная зависимость от β-лактамных антибиотиков привела к расширению разнообразия и увеличению уровня распространения β-лактамаз. Эти β-лактамазы стимулируют резистентность даже к новейшим β-лактамным антибиотикам (Llarrull, et al., The Future of Beta-Lactams, Current Opinion in Microbiology, 13:551-557 (2010)).
Главной опасностью для эффективности этих лекарственных средств является увеличение уровня распространения бета-лактамаз расширенного спектра действия (ESBL). Бета-лактамазы являются ферментами, которые секретируются некоторыми бактериями и которые раскрывают бета-лактамное кольцо бета-лактамного антибиотика, тем самым инактивируя его. В настоящее время существует четыре класса бета-лактамаз, обозначенные Класс А, Класс В, Класс С и Класс D. Бета-лактамазы Класса А, Класса С и Класса D являются сериновыми бета-лактамазами, а бета-лактамазы Класса В являются металло-бета-лактамазами (MBL) (Bush & Jacoby, Updated Functional Classification of β-Lactamases, Antimicrobial Agents and Chemotherapy, 54(3):969-976 (Mar. 2010)).
С целью повышения эффективности бета-лактамных антибиотиков были разработаны некоторые ингибиторы бета-лактамаз. Однако доступные в настоящее время ингибиторы β-лактамаз во многих случаях недостаточно эффективны, чтобы противостоять постоянному увеличению разнообразия β-лактамаз. Тремя самыми известными агентами против сериновых бета-лактамаз, применяемыми в настоящее время, являются клавулановая кислота, тазобактам и сульбактам, которые обладают активностью только против некоторых ферментов Класса А, что существенно ограничивает их применение. Кроме того, проходящие в настоящее время клинические испытания ингибиторы бета-лактамаз, такие как авибактам и MK7655, действуют в первую очередь на ферменты Класса А и Класса С и имеют минимальную эффективность против бета-лактамаз Класса D (Bebrone, et al., Current Challenges in Antimicrobial Chemotherapy: Focus on β-Lactamase Inhibition, Drugs, 70(6):651-679 (2010)). Несмотря на то, что эти агенты обеспечивают значительное улучшение по сравнению с доступными в настоящее время ингибиторами бета-лактамаз, для борьбы со значительной резистентностью к бета-лактамам, наблюдаемой сегодня, нужны агенты, которые эффективно поражают все три сериновые бета-лактамазы. В настоящее время отсутствуют разрешенные к применению ингибиторы β-лактамаз, которые эффективны против β-лактамаз Класса D, и уровни резистентности к традиционным антибиотикам продолжают расти.
Следовательно, существует потребность в новых ингибиторах β-лактамаз, которые эффективны против β-лактамаз по меньшей мере Класса D. Существует очевидная потребность в новых ингибиторах β-лактамаз, которые эффективны против β-лактамаз более чем одного Класса А, С и/или D.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединениям, которые являются ингибиторами бета-лактамаз. Соединения и их фармацевтически приемлемые соли полезны в комбинации с бета-лактамными антибиотиками или сами по себе для лечения бактериальных инфекций, включая инфекции, вызываемые резистентными к лекарственным средствам микроорганизмами, в том числе микроорганизмами с резистентностью к множеству лекарственных средств. Более конкретно, изобретение относится к соединениям формулы (Ia):
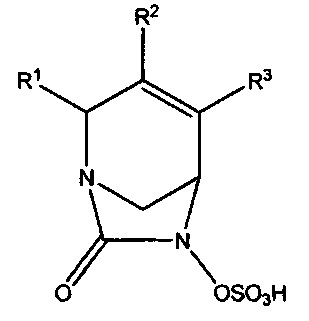
или их фармацевтически приемлемым солям, где R1 представляет собой -CONR'R'', -CN или C1-С3алкил, где каждый алкил возможно замещен C1-С3алкокси, -ОН, -CN, -NR'R'' или -CONR'R''; R2 и R3 независимо выбраны из Н, галогена, -CN, C1-С6алкила, С2-С6алкенила, С2-С6алкинила, С3-С6циклоалкила, C1-С6алкокси, -CONR'R'' или C(O)2R'; где алкил, алкенил, циклоалкил и алкокси в определении R2 или R3 независимо и возможно замещены одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоал килом, C1-С3алкокси, С1-С3галогеналкокси, -NR'R'', 5-7-членным гетероциклом, -C(O)NR'R'' или -NR'C(O)R''; и каждый из R' и R'' независимо выбран из водорода, С1-С6алкила, С3-С6циклоалкила, фенила, 5-6-членного гетероциклила или 5-6-членного гетероарила; где каждый алкил, циклоалкил, фенил, гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоалкилом, C1-С3алкокси, С1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, -NH(C1-C3aлкил), -N(С1-С3алкил)2, 5-7-членным гетероциклилом или 5-7-членным гетероарилом; при условии, что R2 и R3 оба не являются водородом; и когда R1 представляет собой -C(O)NR'R'', тогда ни один из R2 или R3 не представляет собой -C(O)NR'R''.
Подробное описание изобретения
В одном аспекте изобретения предложено являющееся ингибитором бета-лактамаз соединение формулы (I):
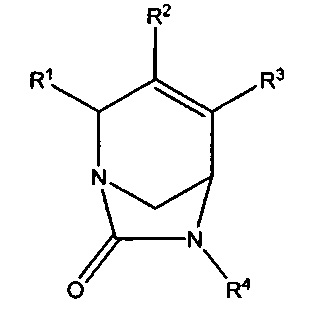
или его фармацевтически приемлемая соль, где R1 представляет собой -CONR'R'', -CN, С1-С3алкил или С1-С2алкокси, где каждый алкил и алкокси независимо и возможно замещен C1-С3алкокси, -ОН, -CN, -NR'R'', -CONR'R'' или 5-7-членным гетероциклом; R2 и R3 независимо выбраны из Н, галогена, -CN, C1-С6алкила, С2-С6алкенила, С2-С6алкинила, С3-С6циклоалкила, С1-С6алкокси, -CONR'R'', C(O)2R', фенила, 5-6-членного гетероциклила или 5-6-членного гетероарила; где алкил, алкенил, циклоалкил, алкокси, фенил, гетероциклил и гетероарил в определении R2 или R3 независимо и возможно замещены одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогенал килом, С3-С6циклоалкилом, C1-С3алкокси, C1-С3галогеналкокси, -NR'R'', 5-7-членным гетероциклилом, -C(O)NR'R'' или -NR'C(O)R''; R4 представляет собой -OS(O)2OH, -S(O)2OH, -ОР(O)2ОН, -Р(O)2ОН, -C(O)NHS(O)2R5, -OCHFCO2H, -OCF2CO2H, или -OCH2CO2H; R5 представляет собой NR'R'', фенил, 5-6-членный гетероциклил или а 5-6-членный гетероарил; и каждый из R' и R'' независимо выбран из водорода, C1-С6алкила, С3-С6циклоалкила, фенила, 5-7-членного гетероциклила или 5-6-членного гетероарила; где каждый алкил, циклоалкил, фенил, гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоалкилом, C1-С3алкокси, C1-С3галогеналкокси, -С(O)(С1-С6алкил), -C(O)(C1-C6алкокси), -NH2, -NH(C1-C3алкил), -N(C1-C3алкил)2, 5-6-членным гетероциклилом или 5-6-членным гетероарилом; или R' и R'' вместе образуют 5-6-членный гетероциклил или гетероарил, где каждый гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоалкилом, C1-С3алкокси, C1-С3галогеналкокси, -NH2, -NH(С1-С3алкил) или -N(C1-C3алкил)2; при условии, что R2 и R3 оба не являются водородом; и когда R1 представляет собой -C(O)NR'R'', тогда ни R2, ни R3 не представляет собой -C(O)NR'R''.
В другом аспекте изобретения предложено соединение формулы (II):
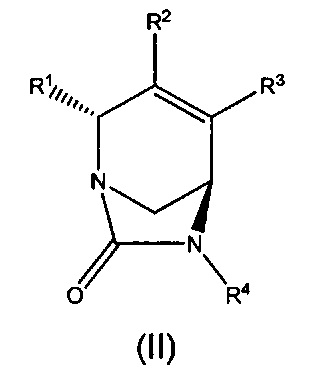
или его фармацевтически приемлемая соль, где переменные R1, R2, R3 и R4 такие, как определено для формулы (I) выше.
В одном аспекте изобретения предложено соединение формулы (III):
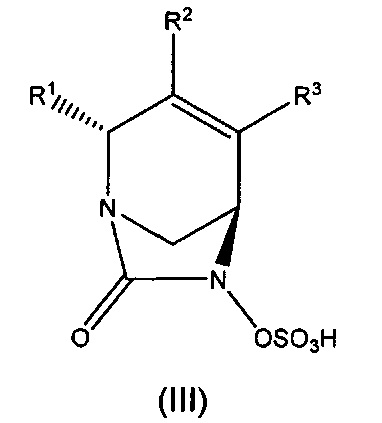
или его фармацевтически приемлемая соль, где переменные R1, R2 и R3 такие, как определено для формулы (Ia) выше.
В одном аспекте изобретения предложено соединение формулы (IV):
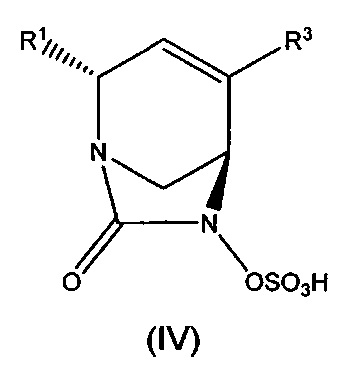
или его фармацевтически приемлемая соль, где переменные R1 и R3 такие, как определено для формулы (Ia).
В одном аспекте изобретения предложено соединение формулы (V):
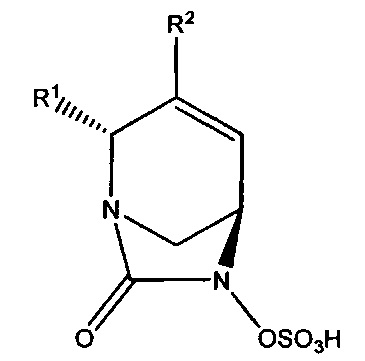
или его фармацевтически приемлемая соль, где переменные R1 и R2 такие, как определено для формулы (Ia).
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONR'R'', -CN или C1-С3алкил, где каждый алкил возможно замещен C1-С3алкокси или -ОН; и R' и R'' в R1 независимо выбраны из группы, состоящей из Н, С1-С3алкила или 5-7-членного гетероциклила, где каждый алкил и гетероциклил в R' и R'' возможно и независимо замещен одним или более чем одним -ОН, С1-С3алкилом, C1-С3алкокси, -ΝΗ2, -NH(С1-С3алкил), -N(С1-С3алкил)2 или 5-7-членным гетероциклилом. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСНз, -CONH(СН2)-сидерофор, -CONH2,  или
или  ; и
; и  обозначает точку присоединения к мостиковому бициклическому ядру. В одном аспекте изобретения в любой одной из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3 или -CONH2.
обозначает точку присоединения к мостиковому бициклическому ядру. В одном аспекте изобретения в любой одной из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3 или -CONH2.
В одном аспекте изобретения в любой из формул (I), (II), (III) или (IV) R1 представляет собой -C(O)NH2, -CN или C1-С3алкил, возможно замещенный одним или более чем одним -ОН, C1-С3алкокси, галогеном, -OC(O)NR'R'', сидерофором или -С(O)NH(сидерофор), где R' и R'' такие, как определено для любой из формул (I), (Ia), (II), (III), (IV) или (V). В одном аспекте изобретения в любой из формул (I), (II), (III) или (IV) R1 представляет собой -C(O)NH2, -CN или С1-С2алкил, возможно замещенный метокси, -ОН или -CN. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONR'R'', -СН2ОСН3 или -CN; и R' и R'' в R1 независимо представляют собой -Н, C1-С3алкил или 5-7-членный гетероциклил, где каждый алкил и гетероциклил в R' и R'' возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, C1-С3алкокси, C1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, -NH(C1-С3алкил), -N(С1-С3алкил)2 или 5-7-членным гетероциклилом. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONH2, -CONH(CH2)nNH2, 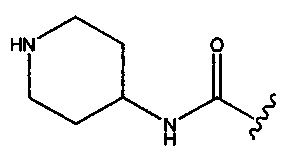 или
или 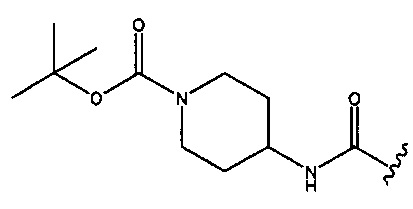 ; n означает целое число от 1 до 3; и
; n означает целое число от 1 до 3; и  обозначает точку присоединения к мостиковому бициклическому ядру. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONH2,
обозначает точку присоединения к мостиковому бициклическому ядру. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONH2, 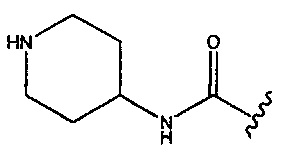 или
или 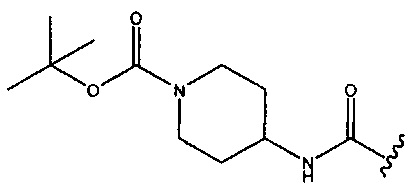 . В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONH2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CN. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОН.
. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CONH2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -CN. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОН.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 выбран из группы, состоящей из Н, C1-С3алкила, С3-С6циклоалкила и -CONR'R'', где алкил и циклоалкил в определении R2 и/или R3 независимо и возможно замещены одной или более чем одной группой, выбранной из галогена, -CN, -ОН, C1-С3алкила, C1-С3галогеналкила, C1-С3алкокси, C1-С3галогеналкокси, -NR'R'', сидерофора, -C(O)NR'R'' и -NR'C(O)R''. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой метил, этил, изопропил или циклопропил, где каждый R2 возможно и независимо замещен одной или более чем одной группой, выбранной из -ОН и C1-С3алкокси. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой метил.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой -Н, -CN, C1-С3алкил, С3-С6циклоалкил, -CO2R', -CONR'R'' или 5-6-членный гетероциклил, где каждый алкил, циклоалкил, гетероциклил, R' и R'' в определении R2 возможно и независимо замещен одной или более чем одной группой, выбранной из галогена, -CN, -ОН, C1-С3алкила, C1-С3галогеналкила, C1-С3алкокси, C1-С3галогеналкокси, -NR'R'', морфолинила, пирролидинила, пиперидинила, пиперазинила, сидерофора, -C(O)NR'R'' и -NR'C(O)R''. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой Н, -CN, метил, этил, изопропил, циклопропил, -CO2(С1-С3алкил), -CONH2, -CONH(C1-C3алкил), -CONC1-С3алкил)2, морфолинил или тиазолил, причем когда R2 не является водородом или циано, тогда каждый R2 возможно и независимо замещен одной или более чем одной группой, выбранной из галогена, -CN, -ОН, С1-С3алкила, C1-С3галогеналкила, С1-С3алкокси, C1-С3галогеналкокси, -NR'R'', морфолинила, пирролидинила, пиперидинила, пиперазинила, сидерофора, -C(O)NR'R'' и -NR'C(O)R''. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой Н, -CN, метил, этил, пропил, изопропил, тиазолил, -CONR'R'' или -СО2СН3, причем когда R2 не является водородом или циано, тогда каждый R2 возможно и независимо замещен одним или более чем одним фтором, хлором, бромом, C1-С3алкилом, C1-С3галогеналкилом, C1-С3алкокси или -NR'R''; и R' и R'', когда они присутствуют в R2, независимо выбраны из Η и метила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (V) R2 представляет собой Н, -CN, метил, изопропил, -CONHCH3, -CONH(CH2)2NH2, -CO2CH3, -(CH2)NH2, -(CH2)2NH2 или тиазолил. В одном аспекте изобретения в любой из формул (I), (Ia), (II) или (III) R2 представляет собой водород.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 выбран из группы, состоящей из Н, C1-С3алкила, С3-С6циклоалкила и -CONR'R'', где алкил и циклоалкил в определении R2 и/или R3 независимо и возможно замещены одной или более чем одной группой, выбранной из галогена, -CN, -ОН, C1-С3алкила, C1-С3галогеналкила, C1-С3алкокси, C1-С3галогеналкокси, -NR'R'', сидерофора, -C(O)NR'R'' и -NR'C(O)R''. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой C1-С3алкил, С2-С6алкенил, С3-С6циклоалкил или -CONR'R'', каждый из которых возможно и независимо замещен одним или более чем одним заместителем, выбранным из группы, состоящей из галогена, -CN, -ОН, C1-С3алкила, циклопропила, C1-С3галогеналкила, C1-С3алкокси, С1-С3галогеналкокси, -NR'R'', сидерофора, -C(O)NR'R'' и -NR'C(O)R''; и каждый из R' и R'' независимо выбран из Η и C1-С3алкила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой метил, этил, изопропил, циклопропил, -CONH2, -CONH(C1-C3aлкил) или -CONH(С1-С3алкил)2, каждый из которых возможно и независимо замещен одной или более чем одной группой, выбранной из -ОН, C1-С3алкила, C1-С3алкокси, -NR'R'', C(O)NR'R'' и -NR'C(O)R''; и каждый из R' и R'' независимо выбран из Η и C1-С3алкила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой C1-С3алкил, циклопропил, -CONR'R'', где каждый алкил и циклопропил возможно и независимо замещен одним или более чем одним -ОН, C1-С3алкокси, -NH2 или NHC(O)(С1-С3алкил); и каждый из R' и R'' независимо выбран из Н, C1-С3алкила и 5-6-членного гетероциклила, где каждый алкил и гетероциклил в определении R' или R'' возможно и независимо замещен одним или более чем одним -ОН, C1-С3алкилом или C1-С3алкокси. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой метил, -СН2ОСН3 или -CONH2.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой Н, C1-С3алкил, С3-С6циклоалкил, -CO2R', -CONR'R'' или 5-6-членный гетероциклил, каждый из которых возможно и независимо замещен одной или более чем одной группой, выбранной из галогена, -CN, -ОН, C1-С3алкила, циклопропила, C1-С3галогеналкила, C1-С3алкокси, С1-С3галогеналкокси, -NR'R'', морфолинила, пирролидинила, пиперидинила, пиперазинила, -C(O)NR'R'' и -NR'C(O)R''; и где каждый из R' и R'', когда они присутствует в R3, независимо выбран из Η и C1-С3алкила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой Н, метил, этил, изопропил, циклопропил, -СО2(С1-С3алкил), -CONH2, -CONH(C1-С3алкил), -CON(С1-С3алкил)2, морфолинил или тиазолил, каждый из которых возможно и независимо замещен одной или более чем одной группой, выбранной из галогена, -CN, -ОН, C1-С3алкила, С1-С3 галогеналкила, циклопропила, C1-С3алкокси, С1-С3галогеналкокси, -NR'R'', морфолинила, пирролидинила, пиперидинила, пиперазинила, -C(O)NR'R'' и -NR'C(O)R''; и где каждый из R' и R'', когда они присутствуют в R3, независимо выбран из Η и C1-С3алкила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой Н, C1-С3алкил, С2-C4алкенил, С3-С6циклоалкил, -CONR'R'' или гетероциклил, где каждый алкил, алкенил и гетероциклил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, C1-С3алкокси, С1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, NH(С1-С3алкил) или -N(C1-С3алкил)2; и каждый из R' и R'', когда они присутствуют в R3, возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, циклопропилом, C1-С3алкокси, С1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, -NH(C1-С3алкил) или -N(С1-С3алкил)2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой -CONR'R''; и один из R' и R'' представляет собой Н, а другой представляет собой C1-С3алкил, возможно замещенный одним или более чем одним галогеном, -CN, -ОН, -CF3, C1-С3алкокси, C1-С3галогеналкокси, циклопропилом, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, -NH(C1-C3aлкил) или -N(С1-С3алкил)2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой -CONH(CH2)nNHR'; R' представляет собой Н, метил, этил, пропил, изопропил или циклопропил; и n означает целое число от 1 до 3. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой -CONH(CH2)2NH2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой метил, изопропил, изопропенил, -CONH2 или -CON(CH3)2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III) или (IV) R3 представляет собой -СН2ОСН3. В одном аспекте изобретения в любой из формул (I), (Ia), (II) или (III) R3 представляет собой водород.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из водорода, C1-С6алкила, С3-С6циклоалкила, фенила, 5-6-членного гетероциклила или 5-6-членного гетероарила; где каждый алкил, циклоалкил, фенил, гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоалкилом, C1-С3алкокси, C1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -NH2, -NH(C1-С3алкил), -N(С1-С3алкил)2, 5-7-членным гетероциклилом или 5-7-членным гетероарилом. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из Н, C1-С3алкила и 5-6-членного гетероциклила, где каждый алкил и гетероциклил в определении R' или R'' возможно и независимо замещен одним или более чем одним -ОН, С1-С3алкилом или C1-С3алкокси. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из Η и С1-С3алкила.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из водорода, C1-С6алкила, С3-С6циклоалкила, фенила, 5-7-членного гетероциклила или 5-6-членного гетероарила; где каждый алкил, циклоалкил, фенил, гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, С1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоалкилом, C1-С3алкокси, C1-С3галогеналкокси, -С(O)(С1-С6алкил), -С(O)(С1-С6алкокси), -ΝΗ2, -NH(C1-С3алкил), -N(С1-С3алкил)2, 5-6-членным гетероциклилом или 5-6-членным гетероарилом; или R' и R'' вместе образуют 5-6-членный гетероциклил или гетероарил, где каждый гетероциклил и гетероарил возможно и независимо замещен одним или более чем одним галогеном, -CN, -ОН, C1-С3алкилом, C1-С3галогеналкилом, С3-С6циклоал килом, C1-С3алкокси, C1-С3галогеналкокси, -NH2, -NH(С1-С3алкил) или -N(С1-С3алкил)2. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из водорода и C1-С6алкила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из водорода, метила, этила, пропила и изопропила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из водорода и метила. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) каждый из R' и R'' независимо выбран из C1-С3алкила, возможно замещенного одним или более чем одним метокси, этокси, -ОН, -NH2, NH(СН3), -N(СН3)2 или сидерофором. В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) один из R' и R'' представляет собой водород, а другой выбран из любых возможных значений, перечисленных выше.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3, -CONH2 или 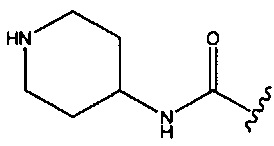 ; R2 представляет собой -Н или -СН3; и R3 представляет собой -Н, -СН3 или -CONH2; при условии, что R2 и R3 оба не представляют собой Н; и когда R1 представляет собой -CONH2 или
; R2 представляет собой -Н или -СН3; и R3 представляет собой -Н, -СН3 или -CONH2; при условии, что R2 и R3 оба не представляют собой Н; и когда R1 представляет собой -CONH2 или 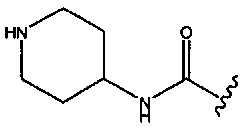 , тогда R3 не представляет собой -CONH2.
, тогда R3 не представляет собой -CONH2.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3, -CONH2, 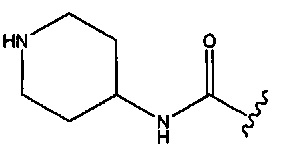 или
или 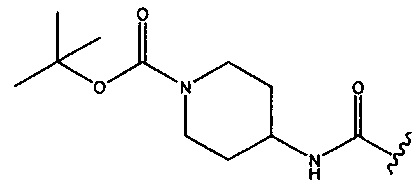 ; R2 представляет собой -Η или метил; R3 представляет собой -Н, -СН3 или -CONH2; и R4 представляет собой -OSO2OH.
; R2 представляет собой -Η или метил; R3 представляет собой -Н, -СН3 или -CONH2; и R4 представляет собой -OSO2OH.
В одном аспекте изобретения в любой из формул (I), (Ia), (II), (III), (IV) или (V) R1 представляет собой -СН2ОСН3, -CONR'R'', 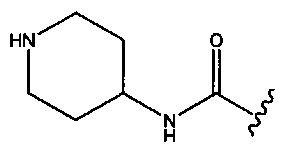 или
или 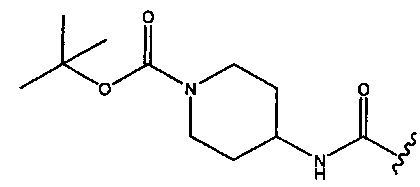 ; R2 представляет собой -Н, C1-С3алкил или С3-С6циклоалкил; R3 представляет собой -Н, C1-С3алкил, С3-С6циклоалкил или -CONR'R''; R4 представляет собой -OSO2OH; и каждый из R' и R'' независимо представляет собой -Н или C1-С3алкил.
; R2 представляет собой -Н, C1-С3алкил или С3-С6циклоалкил; R3 представляет собой -Н, C1-С3алкил, С3-С6циклоалкил или -CONR'R''; R4 представляет собой -OSO2OH; и каждый из R' и R'' независимо представляет собой -Н или C1-С3алкил.
В любом из двух вышеуказанных аспектах изобретения соединение является таким, как определено, при условии, что R2 и R3 оба не представляют собой Н; и когда R1 представляет собой -CONH2, 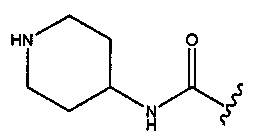 или
или 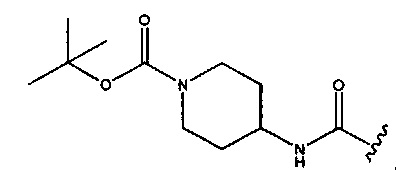 , тогда R3 не представляет собой -CONH2.
, тогда R3 не представляет собой -CONH2.
В одном аспекте изобретения предложено соединение:
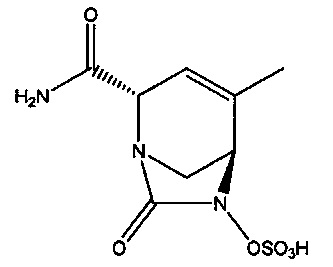
или его фармацевтически приемлемая соль.
В одном аспекте изобретения предложено соединение:
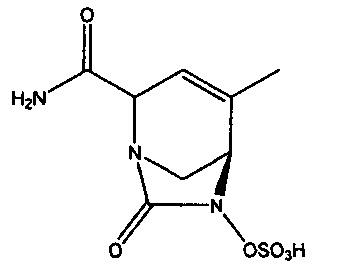
или его фармацевтически приемлемая соль.
В одном аспекте изобретения предложено соединение:
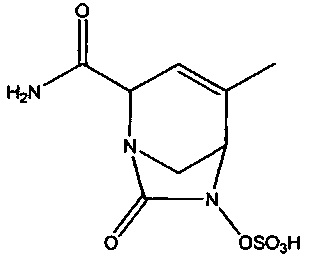
или его фармацевтически приемлемая соль.
В другом аспекте настоящего изобретения предложено соединение:
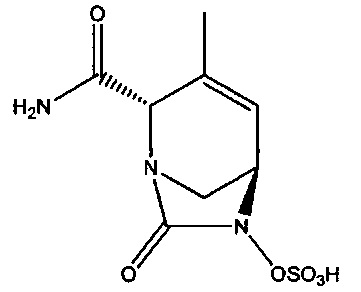
или его фармацевтически приемлемая соль.
В одном аспекте изобретения предложено соединение:
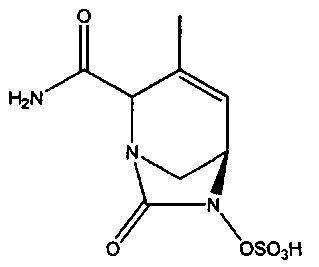
или его фармацевтически приемлемая соль.
В одном аспекте изобретения предложено соединение:
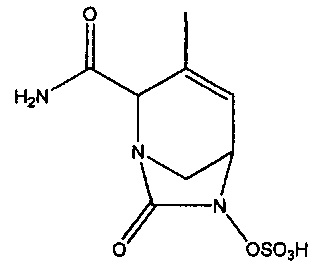
или его фармацевтически приемлемая соль.
Любое воплощение, описанное здесь, может быть объединено с любым другим подходящим воплощением, описанным здесь, для создания дополнительных воплощений. Например, если одно воплощение индивидуально или в совокупности описывает возможные группы для R1, и отдельное воплощение описывает возможные группы для R2, то понятно, что эти воплощения могут быть объединены с получением дополнительного воплощения с использованием возможных групп для R1 с возможными группами для R2. Аналогично, изобретение охватывает любые воплощения, представленные индивидуально для R1, R2, R3, R4, R5, R' и R'', в комбинации с любыми конкретными воплощениями, представленными для каждой из остальных переменных.
Соединения формул (I), (Ia), (II), (III), (IV) и (V) обладают благотворными действенными, метаболическими, токсикологическими и/или фармакодинамическими свойствами.
В одном аспекте изобретения соединение формулы (Ia) выбрано из группы, состоящей из
натриевой соли (2S,5R)-2-карбамоил-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-циано-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-4-метил-7-оксо-2-(пиперидиний-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-карбамоил-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-циано-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-2-(2-аминоэтилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-(метоксиметил)-7-оксо-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-((5-гидрокси-4-оксо-1,4-дигидропиридин-2-ил)метил карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
мононатриевой соли (2S,5R)-4-карбамоил-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2,4-бис(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-(1-(трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-4-(диметилкарбамоил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-(гидроксиметил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-3-метил-7-оксо-2-(пиперидин-1-ий-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-4-(2-амино-2-оксоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-4-карбамоил-2-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-4-(2-аминоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-4-(2-ацетамидоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата;
натриевой соли (2S,5R)-2-(метоксиметил)-4-(метилкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-2-карбамоил-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
натриевой соли (2S,5R)-3-(2-метоксиэтил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата; и
натриевой соли (2S,5R)-2-(((1,5-дигидрокси-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
или их фармацевтически приемлемых солей.
Алкил - Использованный в данном документе термин "алкил" относится к насыщенным углеводородным радикалам с прямой или разветвленной цепью, имеющим конкретно указанное количество атомов углерода. Упоминание индивидуальных алкильных групп, таких как "пропил", относится конкретно только к прямоцепочному варианту, и упоминание индивидуальных алкильных групп с разветвленной цепью, таких как "изопропил", относится конкретно только к варианту с разветвленной цепью. В одном аспекте "алкил" представляет собой метил.
Алкенил - Использованный в данном документе термин "алкенил" относится к углеводородным радикалам с прямой или разветвленной цепью, имеющим конкретно указанное количество атомов углерода и содержащим по меньшей мере одну двойную углерод-углеродную связь. Например, "С2-6алкенил" включает в себя такие группы, как С2-5алкенил, С2-4алкенил, этенил, 2-пропенил, 2-метил-2-пропенил, 3-бутенил, 4-пентенил, 5-гексенил, 2-гептенил и 2-метил-1-гептенил.
Алкинил - Использованный в данном документе термин "алкинил" относится к углеводородным радикалам с прямой или разветвленной цепью, имеющим конкретно указанное количество атомов углерода и содержащим по меньшей мере одну тройную углерод-углеродную связь. Например, "С2-8алкинил" включает в себя такие группы, как С2-6алкинил, С2-4алкинил, этинил, 2-пропинил, 2-метил-2-пропинил, 3-бутинил, 4-пентинил, 5-гексинил, 2-гептинил и 4-метил-5-гептинил.
Галоген - Использованный в данном документе термин "галоген" охватывает фтор, хлор, бром и йод. В одном аспекте "галоген" может относиться к фтору, хлору и брому. В другом аспекте "галоген" может относиться к фтору и хлору. В еще одном аспекте "галоген" может относиться к фтору. В еще одном аспекте "галоген" может относиться к хлору.
Циклоалкил - В одном аспекте "циклоалкил" относится к насыщенному или частично насыщенному моноциклическому углеродному кольцу, в котором одна или более групп -СН2- возможно могут быть заменены соответствующим количеством групп -С(О)-. Иллюстративные примеры "циклоалкила" включают циклопропил, циклобутил, циклопентил и циклопентенил. В одном аспекте "3-5-членный карбоциклил" может представлять собой циклопропил.
5-7-Членный гетероциклил - Термин "5-7-членный гетероциклил" относится к насыщенному или частично насыщенному неароматическому моноциклическому кольцу, содержащему 5-7 кольцевых атомов, из которых по меньшей мере один кольцевой атом выбран из атомов азота, серы и кислорода, и в котором группа -СН2- возможно может быть заменена группой -С(О)-. Аналогично, "5-6-членный гетероциклил" относится к насыщенному или частично насыщенному неароматическому моноциклическому кольцу, содержащему 5-6 кольцевых атомов, из которых по меньшей мере один кольцевой атом выбран из атомов азота, серы и кислорода, и в котором группа -СН2- возможно может быть заменена группой -С(О)-. Если конкретно не указано иное, группы "5-7-членный гетероциклил" и "5-6-членный гетероциклил" могут быть связаны через атом углерода или азота. Кольцевые атомы азота возможно могут быть окислены с образованием Ν-оксида. Кольцевые атомы серы возможно могут быть окислены с образованием S-оксидов или сульфонов. Иллюстративные примеры "5-7-членного гетероциклила" и "5-6-членного гетероциклила" включают, без ограничения, азетидинил, диоксидотетрагидротиофенил, 2,4-диоксоимидазолидинил, 3,5-диоксопиперидинил, фуранил, имидазолил, изотиазолил, изоксазолил, морфолинил, оксазолил, оксетанил, оксоимидазолидинил, 3-оксо-1-пиперазинил, 2-оксопирролидинил, 2-оксотетрагидрофуранил, оксо-1,3-тиазолидинил, пиперазинил, пиперидил, 2Н-пиранил, пиразолил, пиридинил, пирролил, пирролидинил, пиримидинил, пиразинил, пиразолил, пиридазинил, 4-пиридонил, тетрагидрофуранил, тетрагидропиранил, тиазолил, 1,3,4-тиадиазолил, тиазолидинил, тиоморфолинил, тиофенил, 4H-1,2,4-триазолил, пиридин-N-оксидил, тетразолил, оксадиазолил, триазолил, пиразинил, триазинил и гомопиперидинил. В одном воплощении термины "5-7 членный гетероциклил" и "5-6-членный гетероциклил" охватывают сидерофоры из 5-7 или 5-6 членов, которые содержат по меньшей мере один гетероатом.
5- или 6-Членный гетероарил - Термин "5-6-членный гетероарил" относится к моноциклическому ароматическому гетероциклическому кольцу, содержащему 5 или 6 кольцевых атомов, из которых по меньшей мере один кольцевой атом выбран из азота, серы и кислорода. Если конкретно не указано иное, группы "5-6-членный гетероарил" могут быть связаны через атом углерода или азота. Кольцевые атомы азота возможно могут быть окислены с образованием Ν-оксида. Кольцевые атомы серы возможно могут быть окислены с образованием S-оксидов или сульфонов. Иллюстративные примеры "5-6-членного гетероарила" включают фуранил, имидазолил, изотиазолил, изоксазол, оксазолил, пиразинил, пиразолил, пиридазинил, пиримидинил, пиридинил, пирролил, тетразолил, тиадиазолил, тиазолил, тиофенил и триазолил.
6-Членный гетероарил - В одном аспекте "гетероциклил", "5- или 6-членный гетероциклил", "6-членный гетероциклил" и "5- или 6-членный гетероарил" может представлять собой "6-членный гетероарил". Термин "6-членный гетероарил" относится к моноциклическому ароматическому гетероциклическому кольцу, содержащему 6 кольцевых атомов. Кольцевые атомы азота возможно могут быть окислены с образованием Ν-оксида. Кольцевые атомы серы возможно могут быть окислены с образованием S-оксидов или сульфонов. Иллюстративные примеры "6-членного гетероарила" включают пиразинил, пиридазинил, пиримидинил и пиридинил.
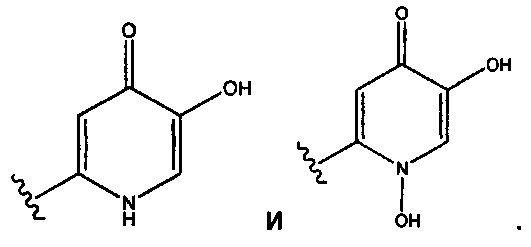
Сидерофор - В одном аспекте "сидерофор" представляет собой низкомолекулярную группировку, которая может связывать ион железа. Сразу после связывания эти "носители железа" могут облегчать транспорт молекулы в бактериальную клетку. Термин "сидерофор" включает, без ограничения, следующие гетероциклилы:
Возможно замещенный - Использованная в данном документе фраза "возможно замещенный" указывает на то, что замещение является возможным и, следовательно, возможно, что указанная группа является либо замещенной, либо незамещенной. В случае если замещение является желательным, соответствующее количество атомов водорода на указанной группе может быть замещено указанными заместителями при условии, что нормальная валентность атомов на конкретном заместителе не превышена, и что в результате замещения образуется стабильное соединение.
В одном аспекте, когда конкретная группа указана как возможно замещенная одним или более заместителями, тогда эта конкретная группа может быть незамещенной. В другом аспекте конкретная группа может нести на себе один заместитель. В другом аспекте конкретный заместитель может нести на себе два заместителя. В еще одном аспекте конкретная группа может нести на себе три заместителя. В еще одном аспекте конкретная группа может нести на себе четыре заместителя. В еще одном аспекте конкретная группа может нести на себе один или два заместителя. В еще одном аспекте конкретная группа может быть незамещенной или может нести на себе один или два заместителя.
Фармацевтически приемлемые - Использованная в данном документе фраза "фармацевтически приемлемые" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в соответствии с обоснованным медицинским суждением пригодны для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений при разумном соотношении польза/риск.
Эффективное количество - Использованная в данном документе фраза "эффективное количество" означает количество соединения или композиции, которая является достаточной для значительного и позитивного модифицирования симптомов и/или состояний, которые лечат (например, обеспечивает положительный клинический ответ). Эффективное количество активного ингредиента для использования в фармацевтической композиции будет варьировать в зависимости от конкретного состояния, которое лечат, тяжести состояния, продолжительности лечения, характера параллельной терапии, конкретного(ых) используемого(ых) активного(ых) ингредиента(ов), конкретного(ых) фармацевтически приемлемого(ых) эксципиента(ов)/носителя(ей) и подобных факторов в объеме знаний и компетенции лечащего врача.
Уходящая группа - Использованная в данном документе фраза "уходящая группа" относится к группам, легко замещаемым нуклеофилом, таким как аминный нуклеофил, спиртовый нуклеофил или тиольный нуклеофил. Примеры подходящих уходящих групп включают галоген, такой как фтор, хлор, бром, и сульфонилоксигруппу, такую как метансульфонилокси и толуол-4-сульфонилокси.
Защитная группа - Использованный в данном документе термин "защитная группа" относится к тем группам, которые используют для защиты реакционноспособных групп (таких как карбокси, амино, гидрокси и меркапто группы) от нежелательных реакций. Иллюстративные примеры подходящих защитных групп для гидрокси группы включают ацильные группы; алканоильные группы, такие как ацетил; ароильные группы, такие как бензоил; силильные группы, такие как три метил сил ил; и арилметильные группы, такие как бензил. Условия удаления вышеуказанных защитных групп для гидрокси группы обязательно будут варьировать в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, может быть удалена, например, в результате гидролиза подходящим основанием, таким как гидроксид щелочного металла, например гидроксид лития или гидроксид натрия. Альтернативно, силильная группа, такая как триметилсилил, может быть удалена, например, фторидом или водной кислотой; или арилметильная группа, такая как бензильная группа, может быть удалена, например, в результате гидрирования в присутствии катализатора, такого как палладий на углероде. Иллюстративные примеры подходящих защитных групп для аминогруппы включают ацильные группы; алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как метоксикарбонил, этоксикарбонил и трет-бутоксикарбонил; арилметоксикарбонильные группы, такие как бензилоксикарбонил; и ароильные группы, такие как бензоил. Условия удаления вышеуказанных защитных групп для аминогруппы обязательно будут варьировать в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа или ароильная группа может быть удалена например, в результате гидролиза подходящим основанием, таким как гидроксид щелочного металла, например гидроксид лития или гидроксид натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки подходящей кислотой, такой как соляная, серная, фосфорная кислота или трифторуксусная кислота, и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий на углероде, или путем обработки кислотой Льюиса, например трихлоридом бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена путем обработки алкиламином, например диметиламинопропиламином или 2-гидроксиэтиламином, или гидразином. Еще одной подходящей защитной группой для аминогруппы является, например, циклическая эфирная группа, такая как тетрагидрофуран, которая может быть удалена путем обработки подходящей кислотой, такой как трифторуксусная кислота. Защитные группы могут быть удалены на любой удобной стадии синтеза с использованием общепринятых методов, известных в химической области, или они могут быть удалены во время более поздней стадии или во время обработки.
Соединения формул (I), (Ia), (II), (III), (IV) или (V) могут образовывать стабильные фармацевтически приемлемые соли присоединения кислоты или основания, и в таких случаях введение соединения в виде соли может быть целесообразным. Примеры солей присоединения кислоты включают соли ацетат, адипат, аскорбат, бензоат, бензолсульфонат, бикарбонат, бисульфат, бутират, камфорат, камфорсульфонат, холин, цитрат, циклогексилсульфамат, диэтилендиамин, этансульфонат, фумарат, глутамат, глюконат, гемисульфат, 2-гидроксиэтил-сульфонат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, гидроксималеат, лактат, малат, малеат, метансульфонат, меглумин, 2-нафталинсульфонат, нитрат, оксалат, памоат, персульфат, фенилацетат, фосфат, дифосфат, пикрат, пивалат, пропионат, хинат, салицилат, стеарат, сукцинат, сульфамат, сульфанилат, сульфат, тартрат, тозилат (пара-толуолсульфонат), трифторацетат и ундеканоат. Примеры солей с основаниями включают аммониевые соли; соли щелочных металлов, такие как натриевые, литиевые и калиевые соли; соли щелочно-земельных металлов, такие как алюминиевые, кальциевые и магниевые соли; соли с органическими основаниями, такие как соли с дициклогексиламином и Ν-метил-D-глюкамином; и соли с аминокислотами, такими как аргинин, лизин, орнитин и т.д. Кроме того, основные азотсодержащие группы могут быть кватернизированы такими агентами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутил-галоген иды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил-сульфаты; диамилсульфаты; длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарил-галогениды; арилалкилгалогениды, такие как бензилбромид и другие. Нетоксичные физиологически приемлемые соли являются предпочтительными, хотя другие соли могут быть полезны, например, для выделения или очистки продукта.
Соли могут быть образованы обычными способами, такими как взаимодействие продукта в форме свободного основания с одним или более эквивалентами соответствующей кислоты в растворителе или в среде, в которой соль является нерастворимой, или в растворителе, таком как вода, который удаляют в вакууме, или посредством сублимационной сушки, или посредством обмена анионов существующей соли на другой анион на подходящей ионообменной смоле.
Соединения формулы (I), (Ia), (II), (III), (IV) или (V) имеют один или более хиральных центров, и понятно, что изобретение охватывает все стереоизомеры, включая энантиомеры и диастереоизомеры. Таким образом, понятно, что поскольку некоторые соединения формул (I), (Ia), (II), (III), (IV) или (V) могут существовать в оптически активных или рацемических формах вследствие наличия одного или более асимметрических атомов углерода, изобретение охватывает в его определении любую такую оптически активную или рацемическую форму, которая обладает вышеупомянутой активностью. Настоящее изобретение охватывает все такие стереоизомеры, обладающие активностью, которая определена в данном документе.
Синтез оптически активных форм может быть осуществлен стандартными способами органической химии, известными в данной области техники, например в результате синтеза из оптически активных исходных веществ или в результате разделения рацемической формы. Рацематы могут быть разделены на индивидуальные энантиомеры известными способами (смотри, например, Advanced Organic Chemistry: 3rd Edition: author J March, p104-107). Подходящий способ включает образование диастереомерных производных в результате реакции рацемического вещества с хиральным вспомогательным веществом с последующим разделением диастереомеров, например хроматографией, и затем отщеплением вспомогательного вещества. Также вышеупомянутая активность может быть оценена стандартными лабораторными методами, описанными здесь ниже.
Таким образом, везде в описании изобретения, где упоминается соединение формулы (I), (Ia), (II), (III), (IV) или (V), термин соединение охватывает изомеры, смеси изомеров и стереоизомеры, которые являются ингибиторами β-лактамаз.
Стереоизомеры могут быть разделены с использованием стандартных методов, например хроматографией или фракционной кристаллизацией. Энантиомеры могут быть выделены путем разделения рацемата, например методом фракционной кристаллизации, перерастворения или ВЭЖХ. Диастереоизомеры могут быть выделены путем разделения на основе разных физических свойств диастереоизомеров, например фракционной кристаллизацией, ВЭЖХ или флэш-хроматографией. Альтернативно, конкретные стереоизомеры могут быть получены в результате хирального синтеза из хиральных исходных веществ в условиях, которые не будут вызывать рацемизацию или эпимеризацию, или в результате дериватизации хиральным реагентом.
Когда обеспечивают получение конкретного стереоизомера (путем разделения, путем хирального синтеза или другими методами), его предпочтительно получают по существу выделенным из других стереоизомеров того же соединения. В одном аспекте смесь, содержащая конкретный стереоизомер соединения формулы (I), (Ia), (II), (III), (IV) или (V), может содержать менее 30%, в частности менее 20% и более конкретно менее 10% (масс.) других стереоизомеров одного и того же соединения. В другом аспекте смесь, содержащая конкретный стереоизомер соединения формулы (I), (Ia), (II), (III), (IV) или (V) может содержать менее 6%, в частности менее 3% и более конкретно менее 2% масс, других стереоизомеров соединения. В другом аспекте смесь, содержащая конкретный стереоизомер соединения формулы (I), (Ia), (II), (III), (IV) или (V) может содержать менее 1%, в частности менее 0,5% и более конкретно менее 0,3%, и еще более конкретно менее 0,1% масс, других стереоизомеров соединения.
Следует иметь в виду, что поскольку некоторые соединения формул (I), (Ia), (II), (III), (IV) или (V), охарактеризованные выше, могут существовать в таутомерных формах, изобретение охватывает любую такую таутомерную форму, которая обладает вышеупомянутой активностью. Таким образом, изобретение относится ко всем таутомерным формам соединений формул (I), (Ia), (II), (III), (IV) или (V), описаны ли они подробно в описании изобретения или нет.
Следует также иметь в виду, что некоторые соединения формул (I), (Ia), (II), (III), (IV) или (V) и их фармацевтически приемлемые соли могут существовать в несольватированной форме и в сольватированных формах, таких как, например, гидратированные формы. Следует иметь в виду, что изобретение охватывает все такие сольватированные формы. Во избежание неясности, это относится к сольватированным (например гидратированным) формам свободной формы соединения, а также к сольватированным (например гидратированным) формам соли соединения.
Для полной ясности, следует иметь в виду, что атомы соединений формул (I), (Ia), (II), (III), (IV) или (V) и любых примеров или воплощений соединений, раскрытых в данном описании, включают все изотопы этих атомов. Например, Η (или водород) включает любую изотопную форму водорода, в том числе 1Н, 2Н (D) и 3Н (Т); С включает любую изотопную форму углерода, в том числе 12С, 13С и 14С; О включает любую изотопную форму кислорода, в том числе 16O, 17O и 18O; N включает любую изотопную форму азота, в том числе 13Ν, 14Ν и 15Ν; Ρ включает любую изотопную форму фосфора, в том числе 31Р и 32Р; S включает любую изотопную форму серы, в том числе 32S и 35S; F включает любую изотопную форму фтора, в том числе 19F и 18F; Cl включает любую изотопную форму хлора, в том числе 35Cl, 37Cl и 36Cl; и т.п. В одном аспекте соединения формул (I), (Ia), (II), (III), (IV) или (V) включают изотопы атомов, охваченные в данном документе, в количествах, соответствующих их относительному содержанию в природе. Однако в некоторых случаях может быть желательным обогащение одного или более чем одного атома в конкретном изотопе, который обычно присутствует в более низком относительном содержании. Например, 1Н может обычно присутствовать в количестве более 99,98%; однако в одном аспекте соединение по изобретению может быть обогащено 2Н или 3Н в одном или более положениях, где находится Η. В другом аспекте, когда соединение по изобретению обогащено радиоактивным изотопом, например 3Н и 14С, соединение может быть полезным в анализах по распределению лекарственного средства и/или субстрата в тканях. Следует понимать, что изобретение охватывает все такие изотопные формы, которые полезны для лечения бактериальных инфекций.
В одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к гинекологической инфекции. В другом аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к инфекции дыхательных путей (RTI). В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к заболеванию, передающемуся половым путем. В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к инфекции мочевыводящих путей (UTI). В дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к острому приступу хронического бронхита (АСЕВ). В еще одном дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к острому среднему отиту. В одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к острому синуситу. В другом аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к инфекции, вызываемой резистентными к лекарственному средству бактериями. В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к сепсису вследствие катетеризации. В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к шанкроиду. В дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к хламидиям. В еще одном дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к внебольничной пневмонии (САР). В еще одном дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к осложненной инфекции кожи и структур кожи. В одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к неосложненной инфекции кожи и структур кожи. В другом аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к эндокардиту. В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к фебрильной нейтропении. В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к гонококковому цервициту. В дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к гонококковому уретриту. В еще одном дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к больничной пневмонии (НАР). В еще одном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к остеомиелиту. В дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к сепсису. В еще одном дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к сифилису. В дополнительном аспекте термины "инфекция" и "бактериальная инфекция" могут относиться к интраабдоминальной инфекции (ΙΑΙ).
В одном воплощении изобретения термины "инфекция" и "бактериальная инфекция" относятся к инфекции, вызываемой грамотрицательными бактериями, также называемой "грамотрицательной инфекцией". В одном аспекте этого воплощения грамотрицательная инфекция представляет собой инфекцию, резистентную к одному или более антибиотикам. В одном аспекте этого воплощения грамотрицательная инфекция представляет собой инфекцию, резистентную к множеству лекарственных средств.
Все вышеупомянутые инфекции могут быть вызваны различными бактериями, которые потенциально могут быть вылечены заявленными агентами в комбинации с ингибиторами пенициллинсвязывающего белка или самими этими агентами. В одном воплощении изобретения предложен способ лечения одной или более инфекций, перечисленных выше, включающий введение субъекту, страдающему бактериальной инфекцией, эффективного количества соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли в комбинации с дополнительным агентом-антибиотиком. В одном аспекте этого воплощения дополнительный агент-антибиотик представляет собой β-лактамный антибиотик. В одном аспекте этого воплощения дополнительным агентом-антибиотиком является ингибитор пенициллинсвязывающего белка.
В одном аспекте предложено применение соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для продуцирования эффекта ингибирования бактериальных пептидогликанов, либо одного(ой), либо в комбинации с ингибитором пенициллинсвязывающего белка, у теплокровного животного, такого как человек.
В другом аспекте предложено применение соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения бактериальной инфекции у теплокровного животного, такого как человек. В одном аспекте соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят в комбинации с дополнительным агентом-антибиотиком, таким как β-лактамный антибиотик. В одном аспекте этого воплощения дополнительным агентом-антибиотиком является ингибитор пенициллинсвязывающего белка.
В еще одном аспекте предложено применение соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения инфекций мочевыводящих путей, пневмонии, простатита, инфекций кожи и мягких тканей, сепсиса и внутрибрюшинных инфекций, у теплокровного животного, такого как человек. В одном аспекте этого воплощения соединение формулы (I), (Ia), (II), (III), (IV) или (V) вводят в комбинации с дополнительным агентом-антибиотиком. В одном аспекте этого воплощения дополнительным агентом-антибиотиком является ингибитор пенициллинсвязывающего белка.
В другом аспекте предложен способ продуцирования эффекта ингибирования бактериальных пептидогликанов у теплокровного животного такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли, либо одного(ой), либо в комбинации с ингибитором пенициллинсвязывающего белка.
В дополнительном аспекте предложен способ лечения бактериальной инфекции у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли. В одном аспекте этого воплощения соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят в комбинации с дополнительным агентом-антибиотиком. В одном аспекте этого воплощения дополнительным агентом-антибиотиком является ингибитор пенициллинсвязывающего белка. В одном аспекте дополнительным агентом антибиотиком является β-лактамный антибиотик.
В еще одном дополнительном аспекте предложен способ лечения инфекций мочевыводящих путей, пневмонии, простатита, инфекций кожи и мягких тканей, сепсиса и внутрибрюшинных инфекций у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли. В одном аспекте этого воплощения соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят в комбинации с дополнительным агентом-антибиотиком. В одном аспекте этого воплощения дополнительным агентом-антибиотиком является ингибитор пенициллинсвязывающего белка. В одном аспекте дополнительным агентом-антибиотиком является β-лактамный антибиотик.
В еще одном дополнительном аспекте предложено соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемая соль для применения в продуцировании эффекта ингибирования бактериальных пептидогликанов, либо одно, либо комбинации с ингибитором пенициллинсвязывающего белка, у теплокровного животного, такого как человек. В одном аспекте предложено соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемая соль для применения в лечении грамотрицательных бактериальных инфекций, либо одно, либо комбинации с бета-лактамным антибиотиком.
В одном аспекте изобретения предложен способ ингибирования одного или более бета-лактамазного фермента, включающий введение соединения формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли животному, нуждающемуся в этом. В дополнительном аспекте одним или более бета-лактамазным ферментом является сериновая бета-лактамаза. В дополнительном аспекте один или более бета-лактамазный фермент выбран из группы, состоящей из бета-лактамаз Класса А, Класса С и Класса D. В дополнительном аспекте один или более бета-лактамазный фермент представляет собой фермент Класса А. В дополнительном аспекте один или более бета-лактамазный фермент представляет собой фермент Класса С. В дополнительном аспекте один или более бета-лактамазный фермент представляет собой фермент Класса D. В дополнительном аспекте один или более бета-лактамазный фермент представляет собой фермент Класса D фермент и один или более ферментов Классов А и С.
Ингибиторы бета-лактамаз формул (I), (Ia), (II), (III), (IV) или (V) можно вводить в комбинации с любым β-лактамным антибиотиком, который относится, без ограничения, к классам клавамов, карбапенемов, монобактамов, пенициллинов и/или цефалоспоринов, или с любым другим соединением, чувствительным к сериновым β-лактамазам. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с одним или более из следующих: пенициллин, метициллин, оксациллин, нафциллин, клоксациллин, диклоксациллин, флуклоксациллин, темоциллин, амоксициллин, ампициллин, ко-амоксиклав, азлоциллин, карбенициллин, трикарциллин, мезлоциллин, пиперациллин, цефалексин, цефалотин, СХА-101, цефазолин, цефаклор, цефуроксим, цефамандол, цефотетан, цефокситин, цефтриаксон, цефотаксим, цефподоксим, цефиксим, цефтазидим, цефтобипрола медокарил, цефепим, цефпиром, цефтаролин, имипенем, меропенем, эртапенем, фаропенем, сулопенем, дорипенем, PZ-601 (Protez Pharmaceuticals), МЕ1036 (Forest Labs), BAL30072, MC-1, томопенем, тебипенем, азтреонам, тигемонам, нокардицин А или табтоксинин-β-лактам. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с меропенемом, азтреонамом или цефтазидимом. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с меропенемом. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с азтреонамом. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с цефтазидимом. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) комбинируют с цефтаролина фосамилом.
В другом аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) вводят в комбинации с β-лактамным антибиотиком и дополнительным антибиотиком и/или дополнительным ингибитором β-лактамазы. В одном аспекте изобретения дополнительный агент-антибиотик выбран из антибиотиков классов аминогликозидов, стрептомицинов, макролидов, кетолидов, стрептограминов, оксазолидинонов, тетрациклинов, фторхинолонов, кумариновых антибиотиков, гликопептидов, липогликопептидов, нитроимидазолов, ансамицинов, фениколов, мупироцина, фосфомицина, тобрамицина, линезолида, даптомицина, ванкомицина и классов, упомянутых в ANTIMICROBIAL AGENTS (ASM Press, Ed: A. Bryskier (2005)).
В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) вводят в комбинации с β-лактамным антибиотиком и вторым агентом, который предназначен для решения проблемы с резистентностью к β-лактамам. В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) вводят в комбинации с β-лактамным антибиотиком и вторым ингибитором сериновых бета-лактамаз. В одном аспекте изобретения второй ингибитор бета-лактамаз выбран из сульбактама, тазобактама, авибактама, клавулановой кислоты, LK-157, LK-176, SA-1-204, SA-2-13, BLI-489 (Pfizer/Wyeth), BAL0029880 и MK7655. В другом аспекте изобретения второй агент, предназначенный для решения проблемы с резистентностью к β-лактамам, может представлять собой ингибитор металло-бета-лактамаз (MBL), также известный как ингибитор β-лактамаз Класса В.
В одном аспекте предложено соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемая соль для применения в лечении бактериальной инфекции у теплокровного животного, такого как человек.
В другом аспекте предложено соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемая соль для применения в лечении инфекций мочевыводящих путей, пневмонии, простатита, инфекций кожи и мягких тканей, сепсиса и внутрибрюшинных инфекций у теплокровного животного такого как человек.
В еще одном аспекте предложена фармацевтическая композиция, содержащая соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
Композиции по изобретению могут быть в форме, подходящей для перорального применения (например, в форме таблеток, пастилок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для наружного применения (например, в форме кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения ингаляцией (например, в форме тонкоизмельченного порошка или жидкого аэрозоля), для введения инсуффляцией (например, в форме тонкоизмельченного порошка) или для парентерального введения (например, в форме стерильного водного или масляного раствора для внутривенного, подкожного или внутримышечного введения дозы или в форме суппозитория для ректального введения дозы). В одном аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят внутривенно. В другом аспекте изобретения соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят внутривенно в комбинации с одним или более другим антибактериальным агентом. В одном аспекте этого воплощения соединение формулы (I), (Ia), (II), (III), (IV) или (V) или его фармацевтически приемлемую соль вводят одновременно с одним или более другими антибактериальными агентами.
Композиции по изобретению могут быть получены общепринятыми способами с использованием традиционных фармацевтических эксципиентов, известных в данной области. Так, композиции, предназначенные для перорального применения, могут содержать, например, один или более красителей, подсластителей, корригентов и/или консервантов.
Подходящие фармацевтически приемлемые эксципиенты для таблеточной формы включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция; гранулирующие и разрыхляющие агенты, такие как кукурузный крахмал или альгиновая кислота; связывающие агенты, такие как крахмал; смазывающие агенты, такие как стеарат магния, стеариновая кислота или тальк; консерванты, такие как этил- или пропил-лара-гидроксибензоат; и антиоксиданты, такие как аскорбиновая кислота. Таблетки могут не иметь покрытия или могут иметь покрытие для модифицирования их разрыхления и последующего всасывания активного ингредиента в желудочно-кишечном тракте или для улучшения их стабильности и/или внешнего вида, в любом случае с использованием традиционных покрытий и методик, известных в данной области.
Композиции для перорального применения могут быть в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в форме мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом, таким как арахисовое масло, вазелиновое масло или оливковое масло.
Водные суспензии обычно содержат активный ингредиент в форме тонкодисперсного порошка или в форме нано- или микрочастиц вместе с одним или более суспендирующими агентами, такими как натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующими или увлажняющими агентами, такими как лецитин или продукты конденсации алкиленоксида с жирными кислотами (например полиоксиэтиленстеарат) или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, образуемыми жирными кислотами и гекситолом, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, образуемыми жирными кислотами и гекситолом, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с частичными сложными эфирами, образуемыми жирными кислотами и ангидридами гекситола, например полиэтиленмоноолеат сорбитана. Водные суспензии могут также содержать один или более консервантов, таких как этил- или пропил-пара-гидроксибензоат; антиоксиданты, такие как аскорбиновая кислота; красители; корригенты; и/или подсластители, такие как сахар, сахарин или аспартам.
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как вазелиновое масло. Масляные суспензии могут также содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие как те, которые указаны выше, и корригенты могут быть добавлены для придания приятного вкуса пероральному препарату. В эти композиции в качестве консерванта может быть добавлен антиоксидант, такой как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантами. Примеры подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов приведены выше. Также могут присутствовать дополнительные эксципиенты, такие как подсластители, корригенты и красители.
Фармацевтические композиции по изобретению могут быть также в форме эмульсии типа масло-в-воде. Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как, например, вазелиновое масло, или их смесь. Подходящими эмульгаторами могут быть, например, природные камеди, такие как аравийская камедь или трагакантовая камедь, природные фосфатиды, такие как соевые фосфатиды, лецитин, сложные эфиры или частичные сложные эфиры, получаемые из жирных кислот и гекситоловых ангидридов (например, моноолеат сорбитана), и продукты конденсации указанных частичных сложных эфиров с этиленоксидом, такие как полиоксиэтилен-сорбитана моноолеат. Эмульсии могут также содержать подсластители, корригенты и консерванты.
Сиропы и эликсиры могут быть приготовлены с подсластителями, такими как глицерин, пропиленгликоль, сорбит, аспартам или сахар, и могут также содержать средство, уменьшающее раздражение, консервант, корригент и/или краситель.
Фармацевтические композиции могут быть также в форме стерильной инъекционной водной или масляной суспензии, которая может быть приготовлена по известным методикам с использованием одного или более подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов, которые упомянуты выше. Стерильный инъекционный препарат может представлять собой также стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле.
Композиции для введения ингаляцией могут быть в форме традиционного аэрозоля под давлением, пригодного для диспергирования активного ингредиента либо в виде аэрозоля, содержащего тонкодисперсное твердое вещество, либо в виде жидких капель. Могут быть использованы стандартные аэрозольные пропелленты, такие как летучие фторированные углеводороды или углеводороды, и аэрозольное устройство обычно сконструировано с возможностью диспергировать отмеренное количество активного ингредиента.
Дополнительную информацию по технологии приготовления лекарственных средств можно найти в главе 25.2 в томе 5 книги Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
Количество активного ингредиента, которое объединяют с одним или более эксципиентами с целью получения стандартной лекарственной формы обязательно будет варьировать в зависимости от хозяина, которого лечат, и конкретного пути введения. Например, лекарственная форма, предназначенная для перорального введения людям, обычно будет содержать, например, от 0,5 мг до 4 г активного агента, смешанного с подходящим и удобным количеством эксципиентов, которое может составлять от примерно 5 до примерно 98 массовых процентов от общей массы композиции. Стандартные лекарственные формы обычно будут содержать от примерно 1 мг до примерно 1000 мг активного ингредиента. Дополнительную информацию по путям введения и режимам дозировки можно найти в главе 25.3 в томе 5 книги Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
Помимо соединений по настоящему изобретению фармацевтическая композиция по данному изобретению может также содержать одно или более известных лекарственных средств, или ее можно совместно вводить (одновременно, последовательно или раздельно) с одним или более известными лекарственными средствами, выбранными из других клинически полезных классов антибактериальных агентов (например, с макролидами, хинолонами, β-лактамами или аминогликозидами), и/или другими противоинфекционными агентами (например, с противогрибковым триазолом или амфотерицином). Они могут содержать карбапенемы, например меропенем или имипенем, для расширения терапевтической эффективности. Соединения по данному изобретению могут также содержать бактерицидные/повышающие проницаемость белковыми (BPI) продукты или ингибиторы эффлюксного насоса для повышения активности против грамотрицательных бактерий и бактерий, резистентных к антимикробным агентам, или их можно вводить совместно с ними.
Как указано выше, величина дозы, необходимой для терапевтического или профилактического лечения конкретного болезненного состояния непременно будет варьировать в зависимости от хозяина, которого лечат, пути введения и тяжести болезни, которую лечат. Предпочтительно использовать суточную дозу в диапазоне 1-50 мг/кг. Подходящая оптимальная дозировка может быть определена практикующим врачом, который лечит конкретного пациента.
Помимо применения в терапевтической медицине, соединение формулы (I), (II), (III) или (IV) и его фармацевтически приемлемые соли полезны также в качестве фармакологических средств в разработке и стандартизации in vitro и in vivo тест-систем для оценки эффектов ингибиторов ДНК-гиразы на лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, в рамках исследований новых терапевтических агентов.
Соединения формул (I), (Ia), (II), (III), (IV) или (V) могут быть получены различными способами. Способы, показанные ниже, иллюстрируют способ синтеза соединений формулы (Ia) (где R1, R2 и R3, если не указано иное, такие, как определено выше). Реакции проводят в растворителях, подходящих для используемых реагентов и веществ и подходящих для превращений, которые осуществляют. Кроме того, следует иметь в виду, что в описании способов синтеза, описанных ниже, все предложенные реакционные условия, включая выбранный растворитель, атмосферу, в которой проводят реакцию, реакционную температуру, продолжительность эксперимента и операции обработки, являются условиями, стандартными для данной реакции, которые известны специалисту в данной области. Специалисту в области органического синтеза будет понятно, что функциональные группы, присутствующие в различных частях молекулы, должны быть совместимыми с предлагаемыми реагентами и реакциями. Такие ограничения до заместителей, которые совместимы с реакционными условиями, очевидны для специалиста в данной области, и тогда должны быть использованы альтернативные способы. Представленные Схемы и Способы не отражают исчерпывающий список способов получения соединений формул (I), (Ia), (II), (III), (IV) или (V); напротив, дополнительные способы, о которых знает специалист-химик, также могут быть использованы для синтеза соединений. Притязания не ограничены структурами, показанными на Схемах и в Способах.
Должно быть понятно, что в некоторых реакциях, показанных на Схемах и в Способах, рассмотренных в данном описании, необходимо/желательно защищать чувствительные группы в соединениях. Случаи, когда защита необходима или желательна, известны специалистам в данной области, как и подходящие способы такой защиты. Традиционные защитные группы могут быть использованы в соответствии со стандартной практикой (смотри T.W. Greene, Protective Groups in Organic Synthesis, published by John Wiley и Sons, (1991)) и как описано выше.
Специалист-химик способен использовать и адаптировать информацию, содержащуюся и приведенную в указанных выше источниках информации и в Примерах, приведенных в них, а также в Примерах и на Схемах, приведенных в данном описании, чтобы получить необходимые исходные вещества и продукты.
Если исходные вещества для осуществления способов, описанных в данном документе, коммерчески недоступны, то они могут быть получены способами, выбранными из стандартных способов органической химии, способов, которые аналогичны способам синтеза известных структурных аналогов соединений, или способов, которые аналогичны описанной методике или методикам, описанным в Примерах.
Необходимо отметить, что многие исходные вещества для способов синтеза, которые описаны в данном документе, коммерчески доступны и/или широко освещены в научной литературе, или же они могут быть получены из коммерчески доступных соединений способами, о которых сообщается в научной литературе. Общее руководство по реакционным условиям и реагентам смотри в Advanced Organic Chemistry, 5th Edition, by Jerry March and Michael Smith, published by John Wiley & Sons (2001).
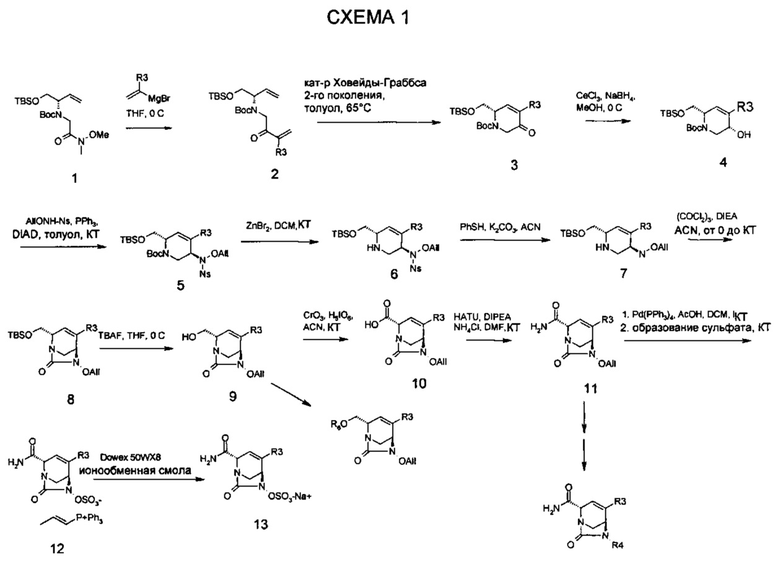
В одном аспекте соединения формулы (I), (Ia), (II), (III), (IV) или (V) или их фармацевтически приемлемые соли могут быть получены способом, представленным на Схеме 1. Из амида Вайнреба (Weinreb), соединение 1, заместители в положение R3 соединений формулы (I), (Ia), (II), (III), (IV) или (V) могут быть введены путем проведения реакции Гриньяра с последующим проведением остальных стадий синтеза, показанных выше, до получения конечных соединений. Соединения с разными заместителями в положении R4 могут быть синтезированы из соединения 11 путем N-O восстановления и деаллилирования с последующим взаимодействием с амином, таким как алкилирование или взаимодействием с замещенным сульфоном или замещенным изоцианатом. Аналогично, соединения с R1=CH2OR могут быть получены из промежуточного соединения 9, используя стандартные методы алкилирования.
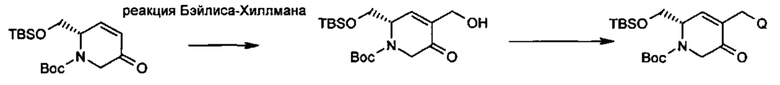
В альтернативном способе синтеза соединений с заместителями R3 используется продукт реакции Бэйлиса-Хиллмана (Baylis-Hillman) енона с последующими стандартными преобразованиями функциональной группы, показанными ниже, где гидроксильная группа может быть превращена в уходящую группу, Q, которая затем может быть замещена подходящим нуклеофилом.
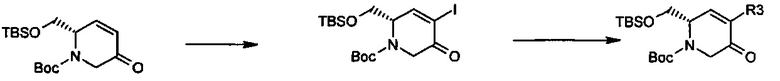
Другие аналоги R3 могут быть получены через перекрестное связывание соответствующего галогенида енона, как показано ниже.
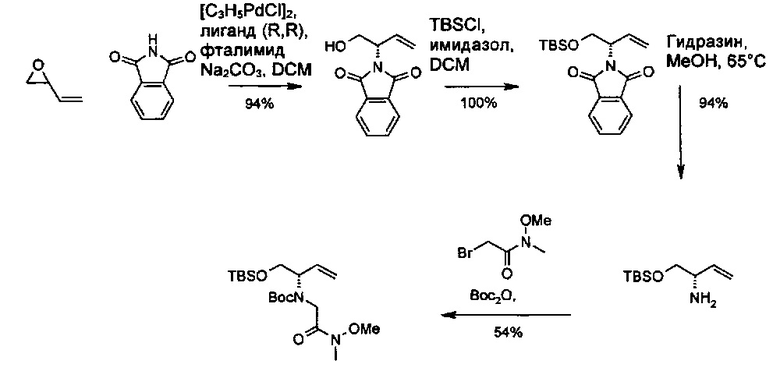
Амид Вайнреба, соединение 1, легко может быть получен из соответствующего амина в результате алкилирования, как показано ниже.
Соединения с замещением по R2 могут быть получены в результате реакции присоединения Михаэля согласно Схеме 2, приведенной ниже.
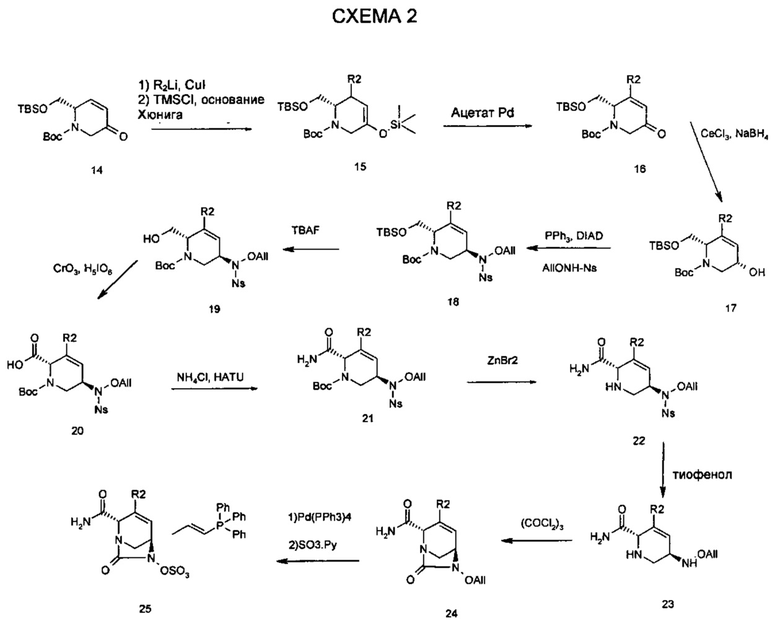
В любых вышеупомянутых фармацевтических композициях, способах, методах, применениях, лекарственных средствах и технологических характеристиках по настоящему изобретению также применимы любые альтернативные воплощения соединений по изобретению, описанные в данном документе.
Примеры
Изобретение далее дополнительно описано со ссылкой на приведенные ниже иллюстративные примеры, в которых, если конкретно не указано иное:
(1) температуры даны в градусах Цельсия (°С); операции проводили при комнатной температуре или при температуре окружающей среды, то есть в диапазоне 18-25°C;
(2) органические растворы сушили над безводным сульфатом магния; выпаривание органического растворителя проводили на роторном испарителе при пониженном давлении (4,5-30 мм рт. ст.) с температурой бани вплоть до 60°C;
(3) хроматография означает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) выполняли на силикагелевых пластинах;
(4) как правило, ход реакций контролировали методом ТСХ или жидкостной хроматографии/масс-спектрометрии (ЖХ/МС), и времена реакций приведены только для иллюстрации;
(5) конечные продукты имеют удовлетворительные спектры протонного ядерного магнитного резонанса (ЯМР) и/или масс-спектральные данные;
(6) выходы приведены только для иллюстрации, и они необязательно те, которые могут быть достигнуты в результате усердной технологической разработки; получения повторяли, если требовалось больше вещества;
(7) ЯМР-данные приведены в форме значений дельта для основных диагностических протонов в миллионных долях (м.д.) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта и определены при 300 МГц в DMSO-d6, если не указано иное;
(8) химические символы имеют их обычные значения;
(9) соотношение растворителей приведено в единицах объем/объем (об./об.);
(10) ISCO Combiflash относится к флэш-хроматографии на силикагеле с использованием системы разделения Isco Combiflash®: нормально-фазовая флэш-колонка RediSep, скорость потока 30-40 мл/мин;
(11) использованы следующие сокращения:

ПРИМЕР 1
Натриевая соль (2S,5R)-2-карбамоил-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
Ионообменную смолу Dowex(R) 50WX8-100 (39 г) кондиционировали путем перемешивания в течение в течение 3 часов в 2 н. гидроксиде натрия (95 мл). Смолу затем загружали в картридж и промывали водой до рН 7. Затем смолу промывали смесью (1/1) ацетон/вода, затем снова водой. (2S,5R)-2-Карбамоил-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 17, 0,2997 г, 0,52 ммоль) переносили в ацетон и разбавляли водой. Этот раствор наносили на смолу и элюировали водой. Фракции, содержащие целевой продукт, объединяли и лиофилизировали. Целевой продукт был получен в виде светло-желтого твердого вещества (140 м г, 90%).
Оптическое вращение: (0,1 г/дл, МеОН)=-219
МС: 278 ЭРИ+ (C8H11N3O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.78 (m, 3Н); 3.20 (m, 2Н); 3.96 (m, 1Н); 4.11 (m, 1Н); 5.42 (m, 1Н); 7.25 (bs, 1Н); 7.51 (bs, 1Н).
Путь 1
Промежуточное соединение 1: (S)-2-(1-гидроксибут-3-ен-2-ил)-изоиндолин-1,3-дион
Реакционную колбу на 2 л с мешалкой, содержащую карбонат натрия (1,981 г, 18,69 ммоль), помещали в глубокий вакуум и сушили струей горячего воздуха в течение десяти минут. После охлаждения колбу заполняли азотом. В нее добавляли хлорид аллилпалладия, димер (0,553 г, 1,53 ммоль), (1R,2R)-(+)-1,2-диаминоциклогексан-N,N'-бис(2-дифенилфосфино-1-нафтоил) (CAS 174810-09-4) (3,36 г, 4,25 ммоль) и фталимид (50 г, 339,83 ммоль). Колбу затем продували азотом в течение десяти минут. Затем добавляли 1,4 л метиленхлорида, предварительно дегазированного азотом в течение десяти минут. Эту суспензию помещали в атмосферу азота; альтернативно, суспензию перемешивали и обрабатывали ультразвуком на протяжении периода времени десять минут, чтобы способствовать растворению. Тогда она представляла собой желтый или светло-оранжевый раствор, содержащий белое твердое вещество. В эту смесь добавляли 2-винилоксиран (24,06 г, 343,23 ммоль). Полученную смесь перемешивали в атмосфере азота при температуре окружающей среды в течение приблизительно 48 часов. Анализ в течение этого времени методами ЖХ/МС и ТСХ (1:1 гексаны:этилацетат) подтверждал развитие реакции, и конечные анализы этими методами подтвердили полное превращение исходного вещества в один основной продукт. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Вязкую желтую жидкость впрыскивали на силикагелевую (330 г) колонку, используя для разжижения неочищенного вещества минимальный объем метиленхлорида. Посредством хроматографии на силикагеле (15-75% этилацетата в гексанах, 40 минут, колонка 330 г) был выделен целевой продукт в виде вязкой желтой жидкости, которая после нескольких часов при пониженном давлении превратилась в бледное желтовато-белое твердое вещество (69,6 г, 94%).
Оптическое вращение: (2,02 г/100 мл, метиленхлорид) литературное значение=-72,2, полученное значение=-71.
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.66 (ddd, J=11.00, 6.47, 5.76 Гц, 1Η) 3.97 (ddd, J=10.95, 9.63, 5.67 Гц, 1Η) 4.69-4.79 (m, 1Η) 5.01 (dd, J=6.52, 5.76 Гц, 1Η) 5.18 (dt, J=2.79, 1.35 Гц, 1Η) 5.23 (dt, J=9.44, 1.42 Гц, 1Η) 6.07 (ddd, J=17.28, 10.67, 6.42 Гц, 1Η) 7.86 (q, J=1.83 Гц, 4Η)
Промежуточное соединение 2: (S)-2-(1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил)изоиндолин-1,3-дион
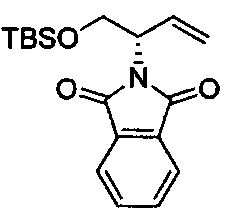
В перемешиваемый раствор (S)-2-(1-гидроксибут-3-ен-2-ил)изоиндолин-1,3-диона (Промежуточное соединение 1, 69,4 г, 319,49 ммоль) и имидазола (26,1 г, 383,39 ммоль) в метиленхлориде (160 мл) при температуре окружающей среды в атмосфере азота добавляли трет-бутилдиметилхлорсилан (55,4 г, 367,41 ммоль) в виде твердого вещества. Это добавление выполняли в течение приблизительно десяти минут. Во время этого добавления наблюдалось нагревание смеси. После перемешивания в течение двух часов раствор вливали в насыщенный водный раствор бикарбоната натрия (приблизительно 150 мл); эту двухфазную смесь встряхивали, и органический слой отделяли. Водный слой экстрагировали три раза по 200 мл метиленхлорида каждый раз. Органические слои объединяли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После сушки в течение ночи в глубоком вакууме целевой продукт был получен в виде бледно-желтого твердого вещества (107 г, 101%).
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.13 (s, 3Η) -0.04 (s, 3Η) 0.67 (s, 9Η) 3.85 (dd, J=10.20, 5.85 Гц, 1Η) 4.08 (t, J=10.01 Гц, 1Η) 4.76-4.86 (m, 1Η) 5.20-5.33 (m, 1Η) 5.25 (dt, J=2.79, 1.35 Гц, 1Η) 6.08 (ddd, J=17.23, 10.53, 6.42 Гц, 1Η) 7.86 (dq, J=4.51,2.21 Гц, 4Η).
Промежуточное соединение 3: (S)-1-(трет-бутилдиметилсилил-окси)бут-3-ен-2-амин
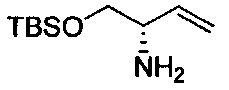
В перемешиваемый раствор (S)-2-(1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил)изоиндолин-1,3-диона (Промежуточное соединение 2, 108,28 г, 326,65 ммоль) в метаноле (1000 мл) при температуре окружающей среды в атмосфере азота добавляли гидразин (35,9 мл, 1143,29 ммоль). Желтый раствор нагревали до 65°С. В пределах 30 минут от достижения реакционной температуры в реакционной смеси наблюдался белый осадок; это твердое вещество быстро становилось объемной смесью, и тогда в реакционную смесь добавляли воду (примерно 150 мл). Реакционную смесь продолжали перемешивать без перерыва и за несколько минут твердое вещество растворялось. После окончания превращения, которое показал анализ методом ЖХ/МС (исходное вещество и продукт оба дают сильные УФ сигналы и легко идентифицируются методом ЖХ/МС), нагрев прекращали и снова добавляли воду (общее содержание воды 600 мл). Смесь оставляли стоять до достижения температуры окружающей среды. Метанол удаляли в вакууме при 35°С (умеренно пониженное давление); вакуумирование убирали, и водную фазу нагревали до примерно 50°С и затем экстрагировали 4×200 мл метиленхлорида. Этот подход может приводить к затруднению отделения водной фазы от органической фазы, поэтому на последней стадии обработки следует использовать большое количество рассола. Органические экстракты объединяли, промывали насыщенным раствором бикарбоната натрия (водн.), промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме при температуре не выше 30°С. Целевой продукт был получен в виде желтой жидкости (58,5 г, 94%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.86 (s, 9Η) 1.51 (br. s., 2Η) 3.22-3.30 (m, 1Η) 3.33-3.48 (m, 2Η) 4.98-5.05 (m, 1Η) 5.17 (dt, J=17.28, 1.84 Гц, 1Η) 5.79 (ddd, J=17.37, 10.39, 5.67 Гц, 1Η).
Промежуточное соединение 4: 2-бром-N-метокси-N-метилацетамид
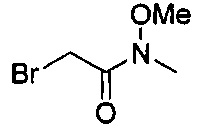
Перемешиваемый раствор карбоната калия (343 г, 2,48 моль) в воде (примерно 800 мл) охлаждали в ледяной бане в течение 15 минут под азотом. Добавляли гидрохлорид Ο,Ν-диметилгидроксиламина (110 г, 1,13 моль) и диэтиловый эфир (примерно 800 мл). В эту смесь затем через воронку добавляли бромацетилбромид (273 г, 1,35 моль) в течение двадцати минут. Ледяную баню удаляли, и смесь перемешивали под азотом в течение двух часов. Слои разделяли, и водный слой экстрагировали диэтиловым эфиром (примерно 350 мл). Органические слои объединяли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Целевой продукт был получен в виде желтой жидкости (143 г, 70%).
1Н ЯМР (300 МГц, CDCl3) δ: 3.24 (s, 3Н); 3.80 (s, 3Н); 4.01 (s, 2Н).
Промежуточное соединение 5: (S)-трет-бутил-1-трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат
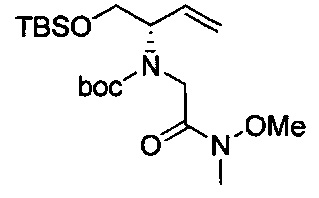
Готовили суспензию (S)-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-амина (Промежуточное соединение 3, 60,4 г, 300 ммоль) и карбоната цезия (103 г, 315 ммоль) в ацетонитриле (примерно 700 мл) и воде (примерно 120 мл) и перемешивали ее в ледяной бане под азотом в течение 5 минут. Смесь была двухфазной и оставалась такой в ходе реакции. В эту смесь затем через воронку добавляли 2-бром-N-метокси-N-метилацетамид (Промежуточное соединение 4, 57,0 г, 285 ммоль) в течение 10 минут. Смесь перемешивали в течение двух суток, поддерживая температуру около 0°С. Смесь хранили в холодильнике в течение ночи. Анализ методом ТСХ (этилацетат, краситель перманганат калия, Rf исходного амина примерно 0,25) показал высокую, но не полную конверсию исходного амина. Добавляли еще 0,05 экв. электрофила. По результатам ТСХ исходный амин полностью не исчезал.
В смесь добавляли ди-трет-бутил-дикарбонат (165 мл, 2М раствор в THF). Смесь перемешивали до тех пор, пока анализ методом ТСХ (этилацетат, краситель перманганат калия) не показал расходование промежуточного соединения. Органический слой отделяли от водного (ТСХ показала, что в водном слое не осталось продукта), и органический слой концентрировали в вакууме. После хроматографии на силикагеле (5-55% этилацетата в гексанах), с разделением на 3 партии, получили целевой продукт в виде бледно-желтого масла (80 г, 66%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (d, J=5.10 Гц, 6Η) 0.84 (s, 9Η) 1.33 (s, 6Η) 1.38 (s, 3Η) 3.02-3.15 (m, 3Η) 3.61-3.68 (m, 3Η) 3.70-3.86 (m, 2Η) 3.95-4.12 (m, 2Η) 4.23-4.68 (m, 1Η) 5.08-5.31 (m, 2Η) 5.75-5.96 (m, 1Η).
Промежуточное соединение 6: (S)-трет-бутил-1-трет-бутилдиметилсилилокси)бут-3-ен-2-ил(3-метил-2-оксобут-3-енил)карбамат
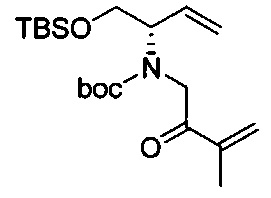
В раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 5, 30,79 г, 76,48 ммоль) в THF (200 мл) при 0°С добавляли бромид проп-1-ен-2-илмагния (0,5М в THF) (300 мл, 149,90 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь гасили 200 мл 10%-ной лимонной кислоты, разбавляли дополнительно 100 мл воды и экстрагировали диэтиловым эфиром. Органические фазы концентрировали, и полученное масло растворяли в диэтиловом эфире и промывали водой и рассолом. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт (26,2 г, 89%) в виде бесцветного масла.
МС: 384 ЭРИ+ (электораспылительная ионизация с регистрацией положительных ионов) (C20H37NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (d, 6Н); 0.83 (s, 9Н); 1.27-1.38 (m, 9Н); 1.80 (m, 3Н); 3.71 (m, 2Н); 4.34 (m, 2Н); 4.61 (m, 1Н); 5.17 (m, 2Н); 5.77 (m, 1Н); 5.85 (m, 1Н); 6.03 (m, 1Н).
Промежуточное соединение 7: (3)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-метил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
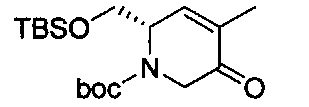
Раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(3-метил-2-оксобут-3-енил)карбамата (Промежуточное соединение 6, 26,18 г, 68,25 ммоль) в толуоле (600 мл) продували азотом в течение 15 минут. Затем добавляли (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенилметилен)рутений (0,987 г, 1,57 ммоль). Реакционную смесь нагревали при 65°С в течение 1,5 часов. Реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили целевой продукт (21,18 г, 87%) в виде бесцветного масла.
МС: 356 ЭРИ+ (C18H33NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (d, 6Н); 0.81 (s, 9Н); 1.42 (s, 9Н); 1.75 (m, 3Н); 3.74-3.89 (m, 3Н); 4.04-4.32 (m, 1Н); 4.67 (m, 1Н); 6.88 (m, 1Н).
Промежуточное соединение 8: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-метил-5,6-дигидропиридин-1 (2Н)-карбоксилат
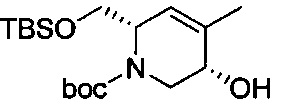
В раствор хлорида церия(III) (14,68 г, 59,57 ммоль) и (8)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-метил-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 7, 21,18 г, 59,57 ммоль) в метаноле (300 мл) при 0°С порциями добавляли боргидрид натрия (2,254 г, 59,57 ммоль). Через 15 минут реакционную смесь разбавляли насыщенным хлоридом аммония (100 мл) и водой (100 мл), затем экстрагировали дважды диэтиловым эфиром. Органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт (19,45 г, 91%) в виде бесцветного масла.
МС: 358 ЭРИ+ (C18H35NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Н); 0.86 (s, 9Н); 1.39 (s, 9Н); 1.69 (m, 3Н); 2.63-2.72 (m, 1Н); 3.59 (m, 2Н); 3.82 (m, 1Н); 4.03 (m, 1Н); 4.21 (m, 1Н); 5.04 (d, 1Н); 5.38 (m, 1Н).
Промежуточное соединение 9: N-(аллилокси)-2-нитробензол-сульфонамид
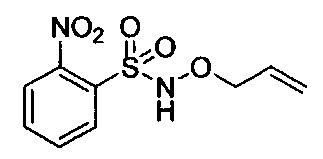
В перемешиваемый раствор гидрохлорида О-аллилгидроксиламина (147,05 г, 1341,59 ммоль) в DCM (2,5 л) при 0°С добавляли пиридин (318 мл, 3948 ммоль), затем порциями добавляли 2-нитробензол-1-сульфонилхлорид (250 г, 1128,05 ммоль) в виде твердого вещества. Реакционную смесь затем перемешивали при той же температуре в течение 1 ч. Завершение реакции контролировали методом ТСХ. Реакционную смесь гасили 1,5 н. HCl (1 л). Органический слой отделяли, промывали водой (250 мл), рассолом (250 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением остатка. Это неочищенное вещество очищали кристаллизацией, используя смесь EtOAc: петролейный эфир (1:3) (800 мл), и получили 202 г указанного в заголовке соединения в виде светло-коричневого твердого вещества. Маточную жидкость концентрировали и очищали колоночной хроматографией на силикагеле (60-120 меш), используя смесь петролейный эфир. EtOAc (7:3), с получением еще 19,1 г указанного в заголовке соединения в виде желтого твердого вещества. Суммарный выход 76%.
УВЭЖХ (ультравысокоэффективная жидкостная хроматография): 257 (М-1) для C9H10N2O5S
1НЯМР (400 МГц, DMSO-d6): δ 4.36-4.38 (m, 2Н), 5.22-5.32 (m, 2Н), 5.84-5.91 (m, 1Н), 7.92-7.96 (m, 2Н), 8.02-8.05 (m, 2Н), 11.07 (s, 1Н).
Промежуточное соединение 10: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
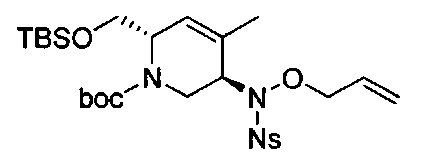
В раствор (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 8, 19,45 г, 54,40 ммоль) в толуоле (300 мл) при комнатной температуре добавляли трифенилфосфин (17,06 г, 65,28 ммоль), Ν-(аллилокси)-2-нитробензолсульфонамид (Промежуточное соединение 9, 14,05 г, 54,40 ммоль) и диизопропил-азодикарбоксилат (12,85 мл, 65,28 ммоль). Через 2 часа реакционную смесь концентрировали на силикагеле и очищали. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили целевой продукт (25,2 г, 78%) в виде желтого масла.
МС: 598 ЭРИ+ (C27H43N3O8SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.00 (s, 6Н); 0.83 (s, 9Н); 1.31 (m, 9Н); 1.34 (m, 3Н); 3.10-3.25 (m, 1Н); 3.59 (m, 2Н); 3.99-4.41 (m, 5Н); 5.17 (m, 2Н); 5.72 (m, 2Н); 7.93-8.16 (m, 4Н).
Промежуточное соединение 11: N-(аллилокси)-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-метил-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамид
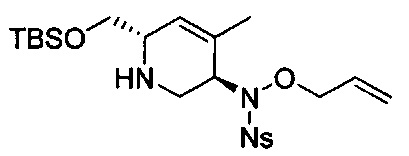
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенил-сульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 10, 13,38 г, 22,38 ммоль) в DCM (200 мл) при комнатной температуре добавляли бромид цинка (3,36 мл, 67,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли DCM и промывали насыщенным раствором бикарбоната натрия и рассолом. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта в виде оранжевого масла (11,14 г, 100%).
МС: 498 ЭРИ+ (C22H35N3O6SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (s, 6Н); 0.85 (s, 9Н); 1.61 (m, 3Н); 2.67 (m, 1H); 2.81 (m, 1H); 3.20 (m, 1H); 3.44 (m, 2H); 4.05 (m, 1H); 4.30 (m, 1H); 4.40 (m, 1H); 5.22 (m, 2H); 5.82 (m, 2H); 7.90-8.15 (m, 4H).
Промежуточное соединение 12: O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-метил-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламин
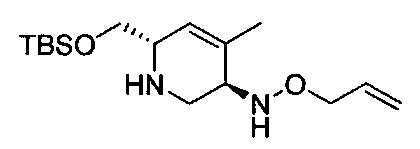
В раствор N-(аллилокси)-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)-метил)-4-метил-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамида (Промежуточное соединение 11, 11,58 г, 23,27 ммоль) и карбоната калия (16,08 г, 116,34 ммоль) в ацетонитриле (200 мл) при комнатной температуре добавляли бензолтиол (11,95 мл, 116,34 ммоль). Через 3 часа реакционную смесь концентрировали досуха и добавляли DCM. Полученное твердое вещество удаляли фильтрованием. Фильтрат концентрировали на силикагеле. После хроматографии на силикагеле (0%-100% этилацетата/гексаны) получили целевой продукт (5,06 г, 69,6%) в виде оранжевого масла.
МС: 313 ЭРИ+ (C16H32N2O2Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Н); 0.86 (s, 9Н); 1.71 (m, 3Н); 2.17 (m, 1Н); 2.81 (m, 2Н); 3.11 (m, 2Н); 3.43 (m, 2Н); 4.09 (m, 2Н); 5.10-5.25 (m, 2Н); 5.48 (m, 1Н); 5.87-5.96 (m, 1Н); 6.35 (d, 1Н).
Промежуточное соединение 13: (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
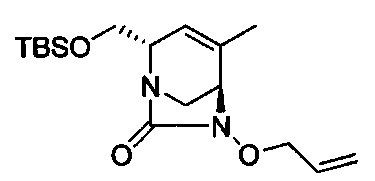
В раствор O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-метил-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламина (Промежуточное соединение 12, 2 г, 6,40 ммоль) и Ν,Ν-диизопропилэтиламина (4,46 мл, 25,60 ммоль) в ацетонитриле (555 мл) при 0°С добавляли трифосген (0,760 г, 2,56 ммоль) в виде раствора в ацетонитриле (45 мл). Раствор трифосгена добавляли посредством шприцевого насоса со скоростью 0,1 мл/мин. Сразу после окончания добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали досуха, разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Было обнаружено, что водные промывочные жидкости содержат некоторое количество продукта, и их экстрагировали дважды этилацетатом. Объединенные органические фазы сушили над сульфатом магния, фильтровали и объединяли с предыдущими экстрактами. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт (1,980 г, 91%) в виде светло-оранжевого масла.
МС: 339 ЭРИ+ (C17H30N2O3Si)
1Н ЯМР (300 МГц, CDCl3) δ: 0.07 (s, 6Н); 0.89 (s, 9Н); 1.87 (m, 3Н); 3.24 (m, 1Н); 3.41 (m, 1Н); 3.61 (m, 1Н); 3.86 (m, 3Н); 4.43 (m, 2Н); 5.33 (m, 3Н); 6.01 (m, 1Н).
Промежуточное соединение 14: (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-4-метил-1.6-диазабицикло[3.2.1]окт-3-ен-7-он
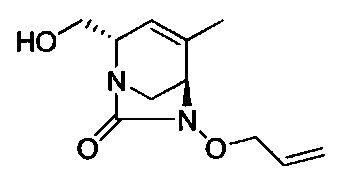
В раствор (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)-метил)-4-метил-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 13, 1,76 г, 5,20 ммоль) в THF (20 мл) при 0°С добавляли фторид тетрабутиламмония (1М в THF) (6,76 мл, 6,76 ммоль). Смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (50%-100% этилацетата/гексаны) получили целевой продукт (1,070 г, 92%) в виде бесцветного масла.
МС: 225 ЭРИ+ (C11Η16Ν2Ο3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.77 (m, 3Н); 3.03 (m, 1Н); 3.23 (m, 1Н); 3.49-3.60 (m, 3Н); 3.78 (m, 1Н); 4.36 (m, 2Н); 4.84 (m, 1Н); 5.24-5.39 (m, 3Н); 5.90-6.01 (m, 1Н).
Промежуточное соединение 15: (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновая кислота
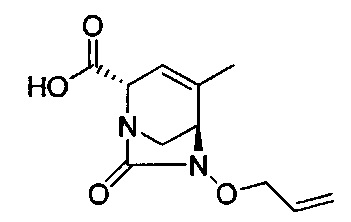
В раствор перйодной кислоты (2 г, 10,42 ммоль) во влажном ацетонитриле (20 мл) (0,75% воды по объему) при комнатной температуре добавляли оксид хрома(VI) (4 мг, 0,04 ммоль). Смесь перемешивали до полного растворения. Этот раствор (5,47 мл, 3 экв.) добавляли по каплям при 0°С в раствор (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-4-метил-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 14, 0,212 г, 0,95 ммоль) во влажном ацетонитриле (10 мл) (0,75% по объему). Реакционная смесь меняла цвет от прозрачного оранжевого до мутного коричневатого и превращалась в мутную зеленую суспензию. Через 30 минут ЖХ/МС показала массу целевого продукта и остаточное исходное вещество. Добавляли еще эквивалент раствора окислителя (1,82 мл). Через 30 минут реакционную смесь разбавляли этилацетатом и промывали смесью 1/1 рассол/вода. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением зеленой пены (0,193 г, 86%).
МС: 239 ЭРИ+ (C11H14N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.80 (m, 3Н); 3.19 (m, 2Н); 3.85 (m, 1Н); 4.27 (m, 1Н); 3.37 (m, 2Н); 5.28-5.43 (m, 3Н); 5.89-6.00 (m, 1Н).
Промежуточное соединение 16: (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
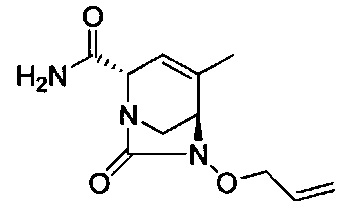
В раствор (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 15, 0,193 г, 0,81 ммоль) в DMF (4 мл) при комнатной температуре добавляли хлорид аммония (0,130 г, 2,43 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,462 г, 1,22 ммоль) и Ν,Ν-диизопропилэтиламин (0,564 мл, 3,24 ммоль). Через 10 минут реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия и рассолом. Объединенные водные промывочные жидкости экстрагировали однократно этилацетатом. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-100% этилацетата/гексаны) получили коричневое масло. Это масло переносили в этилацетат и промывали дважды смесью 1/1 рассол/вода для удаления DMF. Органический слой сушили над сульфатом магния, фильтровали и концентрировали с получением светло-желто-коричневой пены (0,048 г, 25%).
МС: 238 ЭРИ+ (C11H15N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.79 (m, 3Н); 3.19 (m, 2Н); 3.81 (m, 1Н); 4.12 (m, 1Н); 4.36 (m, 2Н); 5.24-5.45 (m, 3Н); 5.89-6.00 (m, 1Н); 7.28 (bs, 1Н); 7.49 (bs, 1Н).
Промежуточное соединение 17: (2S,5R)-2-карбамоил-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
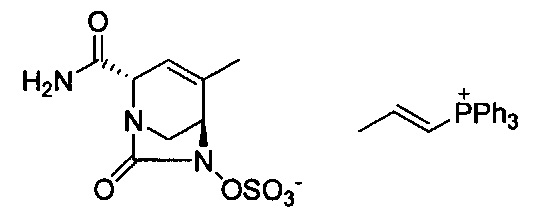
В раствор (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 16, 196,9 мг, 0,83 ммоль) и уксусной кислоты (0,095 мл, 1,66 ммоль) (высушенной над сульфатом натрия) в DCM (9 мл) при комнатной температуре добавляли тетракис(трифенилфосфин)палладий(0) (959 мг, 0,83 ммоль). Раствор перемешивали при комнатной температуре в течение примерно 45 минут, и он становился темно-оранжевым. В реакционную смесь добавляли пиридин (9,00 мл) и комплекс триоксид серы-пиридин (793 мг, 4,98 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре. Суспензию упаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат концентрировали с получением оранжевого масла. После хроматографии на силикагеле (0%-100% ацетона/DCM) получили целевой продукт (300 мг, 62,3%) в виде желтой пены.
МС: 278 ЭРИ+, 303 ЭРИ+ (C8H10N3O6S, C21H20P)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.78 (m, 3Н); 2.17 (m, 3Н); 3.20 (m, 2Н); 3.96 (m, 1H); 4.11 (m, 1H); 5.42 (m, 1H); 6.57-6.74 (m, 1H); 7.22-7.30 (m, 2H); 7.50 (m, 1H); 7.68-7.92 (m, 15H).
Путь 2
Промежуточное соединение 18: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
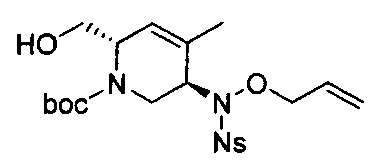
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 10, 1 г, 1,67 ммоль) в THF (11 мл) при 0°С добавляли фторид тетрабутиламмония (1M в THF) (2,175 мл, 2,17 ммоль). Через 90 минут реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (0%-70% этилацетата/гексаны) получили целевой продукт (0,732 г, 90%) в виде желто-коричневой пены.
МС: 484 ЭРИ+ (C21H29N3O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.31 (m, 9Н); 1.35 (m, 3Н); 3.20 (m, 1Н); 3.41 (m, 2Н); 3.96-4.37 (m, 5Н); 4.76 (m, 1Н); 5.19 (m, 2Н); 5.66-5.84 (m, 2Н); 7.94-8.18 (m, 4Н).
Промежуточное соединение 19: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
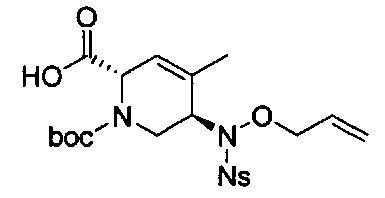
В раствор перйодной кислоты (6 г, 31,26 ммоль) во влажном ацетонитриле (60 мл) (0,75% воды по объему) при комнатной температуре добавляли оксид хрома(VI) (10 мг, 0,10 ммоль). Смесь перемешивали до полного растворения. В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-метил-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 18, 5 г, 10,34 ммоль) во влажном ацетонитриле (60 мл) (0,75% по объему) при 0°С по каплям добавляли ранее образованный раствор перйодной кислоты/оксида хрома (60 мл, 3 экв.). Через 30 минут по результатам ЖХ/МС реакция закончилась. Реакционную смесь разбавляли диэтиловым эфиром и промывали 10%-ной лимонной кислотой, насыщенным бикарбонатом натрия и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением оранжевой пены (4,16 г, 81%).
МС: 498 ЭРИ+ (C21H27N3O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.26 (m, 9Н); 1.31 (m, 3Н); 3.02-3.25 (m, 1Н); 3.90 (m, 1Н); 4.17 (m, 3Н); 4.65-4.77 (m, 1Н); 5.12-5.21 (m, 2Н); 5.68 (m, 1Н); 5.88 (m, 1Н); 7.92-8.17 (m, 4Н).
Промежуточное соединение 20: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
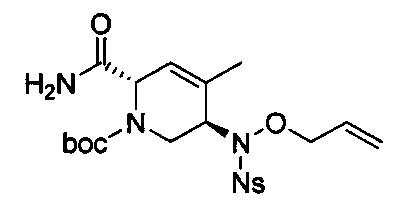
В раствор (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 19, 4,16 г, 8,36 ммоль) в DMF (35 мл) при комнатной температуре добавляли хлорид аммония (0,895 г, 16,72 ммоль), HATU (4,77 г, 12,54 ммоль) и DIEA (5,84 мл, 33,45 ммоль). Через 15 минут реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и дважды смесью 1:1 рассол: вода. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. Хроматографию на силикагеле (0%-80% этилацетата/гексаны) выполняли дважды с получением целевого продукта (2,16 г, 52%) в виде желтой пены.
МС: 497 ЭРИ+ (C21H28N4O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.26 (m, 9Н); 1.37 (m, 3Н); 3.12-3.35 (m, 1Н); 3.80 (m, 1Н); 4.18 (m, 3Н); 4.64-4.79 (m, 1Н); 5.13-5.22 (m, 2Н); 5.68 (m, 1Н); 5.88 (m, 1Н); 7.04 (m, 1Н); 7.45 (bs, 1Н); 7.90-8.18 (m, 4Н).
Промежуточное соединение 21: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамид
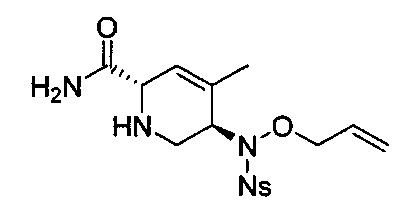
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфон-амидо)-2-карбамоил-4-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 20, 2,16 г, 4,35 ммоль) в DCM (20 мл) при комнатной температуре добавляли бромид цинка (0,700 мл, 13,05 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта (1,450 г, 84%) в виде желтой пены.
МС: 397 ЭРИ+ (C16H20N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.65 (m, 3Н); 2.71 (m, 3Н); 3.76 (m, 1Н); 3.95 (m, 1Н); 4.18-4.42 (m, 2Н); 5.23 (m, 2Н); 5.82 (m, 1Н); 6.02 (m, 1Н); 7.05 (bs, 1Н); 7.30 (bs, 1Н); 7.93-8.18 (m, 4Н).
Промежуточное соединение 22: (2S,5R)-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамид и (2R,5R)-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамид
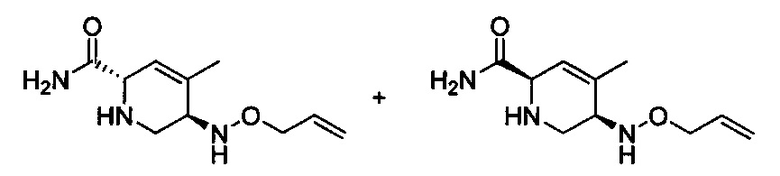
В раствор (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 21, 1,4 г, 3,53 ммоль) и карбоната цезия (9,21 г, 28,25 ммоль) в THF (100 мл) при комнатной температуре добавляли PS-тиофенол (3-(3-меркаптофенил)пропанамидометилполистирол) (1,55 ммоль/г) (9,12 г, 14,13 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтровали через воронку с фильтром из пористого стекла, и смолу промывали дважды DCM. Фильтрат концентрировали с получением желтого масла. После хроматографии на силикагеле (0%-5% метанола/дихлорметан) получили смесь 3:1 транс и цис изомеров (0,473 г, 63,4%) в виде светло-желтого масла. Эту смесь оставляли без разделения.
МС: 212 ЭРИ+ (C10H17N3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.73 (m, 3Н); 2.63 (m, 1Н); 2.97 (m, 1Н); 3.01 (m, 1Н); 3.60 (m, 1Н); 4.12 (m, 2Н); 5.11-5.26 (m, 2Н); 5.92 (m, 1Н); 6.45 (m, 1Н); 7.00 (m, 1Н); 7.33 (bs, 1Н).
Промежуточное соединение 16: (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
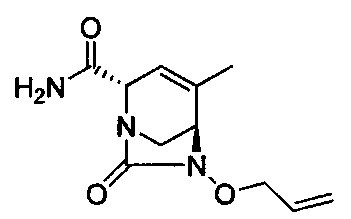
В раствор (2S,5R)-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамида и (2R,5R)-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 22, 0,429 г, 2,03 ммоль) и Ν,Ν-диизопропилэтиламина (1,415 мл, 8,12 ммоль) в ацетонитриле (170 мл) при 0°С добавляли трифосген (0,241 г, 0,81 ммоль) в виде раствора в ацетонитриле (1,5 мл). Раствор трифосгена добавляли со скоростью 0,1 мл/мин. Сразу после окончания добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение выходных дней. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид (0,312 г, 64,8%) в виде светло-желтого масла.
МС: 238 ЭРИ+ (C11H15N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.79 (m, 3Н); 3.19 (m, 2Н); 3.81 (m, 1Н); 4.12 (m, 1Н); 4.36 (m, 2Н); 5.24-5.45 (m, 3Н); 5.89-6.00 (m, 1Н); 7.28 (bs, 1Н); 7.49 (bs, 1Н).
Две конечные стадии Пути 2 получения соединения Примера 1 эквивалентны стадиям, описанным выше для Пути 1.
ПРИМЕР 2
Натриевая соль (2S,5R)-2-циано-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
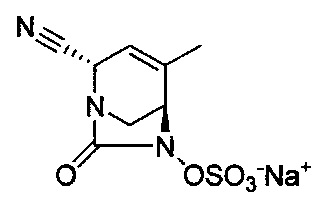
Указанное в заголовке соединение получали из (2S,5R)-2-циано-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 24, 0,1167 г, 0,21 ммоль), по методике, описанной выше для соединения Примера 1. Целевой продукт был получен в виде белого твердого вещества (53,6 мг, 91%).
Оптическое вращение: (0,1 г/дл, DMSO)=-262
МС: 258 ЭРИ- (электрораспылительная ионизация с регистрацией отрицательных ионов) (C8H9N3O5S)
1Н ЯМР (600 МГц, DMSO-d6) δ: 1.82 (s, 3Н); 3.26 (m, 1Н); 3.44 (m, 1Н); 4.08 (m, 1Н); 4.95 (m, 1Н); 5.33 (m, 1Н).
Промежуточное соединение 23: (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрил
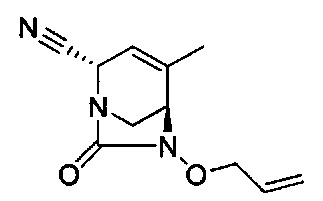
В раствор (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 16, 146 мг, 0,62 ммоль) в DCM (6 мл) под азотом при комнатной температуре порциями добавляли гидроксид (метоксикарбонилсульфамоил)триэтил-аммония, внутреннюю соль (реагент Бургесса (Burgess)) (660 мг, 2,77 ммоль) в течение 2 часов. Реакционную смесь перемешивали при комнатной температуре в течение еще 30 минут. Реакционную смесь промывали смесью 1:1 рассол:вода. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-25% этилацетата/гексаны) получили (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрил (112 мг, 83%) в виде бесцветного масла.
МС: 220 ЭРИ+ (C11H13N3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.83 (m, 3Н); 3.27 (m, 1Н); 3.40 (m, 1Н); 3.97 (m, 1Н); 4.38 (m, 2Н); 4.97 (m, 1Н); 5.26-5.39 (m, 3Н); 5.88-5.99 (m, 1Н).
Промежуточное соединение 24: (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрил
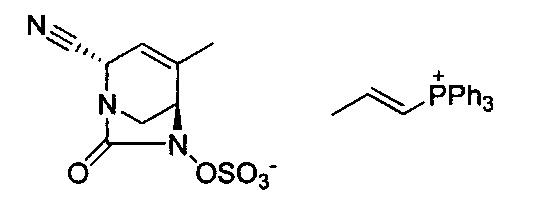
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрила (Промежуточное соединение 23, 111,8 мг, 0,51 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде не совсем белой пены (117 мг, 40,8%).
МС: 258 ЭРИ-, 303 ЭРИ+ (C8H8N3O5S, C21H20P)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.83 (m, 3Н); 2.17 (m, 3Н); 3.26 (m, 1Н); 3.45 (m, 1Н); 4.08 (m, 1Н); 4.94 (m, 1Н); 5.33 (m, 1Н); 6.59-6.72 (m, 1Н); 7.22-7.39 (m, 1Н); 7.68-7.92 (m, 15Н).
ПРИМЕР 3
(2S,5R)-4-метил-7-оксо-2-(пиперидиний-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат
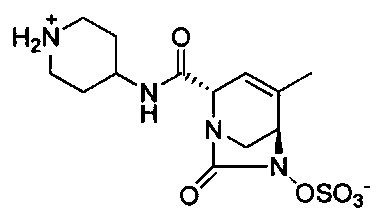
В раствор (2S,5R)-2-(1-(трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 26, 0,1521 г, 0,20 ммоль) в DCM (3 мл) при 0°С добавляли трифторуксусную кислоту (0,031 мл, 0,40 ммоль). Через 30 минут снова добавляли трифторуксусную кислоту (0,031 мл, 0,40 ммоль). Еще через 30 минут добавляли еще 6 эквивалентов TFA при 0°С. Реакционной смеси давали возможность нагреться до комнатной температуры. Через 1 час реакция не была завершена. Реакционную смесь хранили в морозильной камере в течение ночи. Утром добавляли еще 5 экв. TFA при 0°С. Реакционной смеси давали возможность нагреться до комнатной температуры. Через 4 часа реакционную смесь концентрировали и совыпаривали с DCM три раза. Липкое масло затем растирали с диэтиловым эфиром и концентрировали с получением желтого твердого вещества. Это твердое вещество растворяли в воде и промывали дважды DCM. Водную фазу лиофилизировали. Очистку выполняли методом обращенно-фазовой ВЭЖХ (0-10% метанола в воде, YMC Carotenoid С30, 19 мм × 150 мм, 5 мкм) с получением указанного в заголовке соединения в виде белого твердого вещества (3 мг, 4,3%).
МС: 361 ЭРИ+ (C13H20N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.53 (m, 3Н); 1.73 (m, 3Н); 1.78 (m, 3Н); 2.82 (m, 3Н); 3.16 (m, 2Н); 3.77 (m, 1Н); 3.97 (m, 1Н); 4.14 (m, 1Н); 5.37 (m, 1Н); 6.49 (m, 1Н); 8.13 (m, 1Н).
Путь 1
Промежуточное соединение 25: трет-бутил-4-((2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)пиперидин-1-карбоксилат
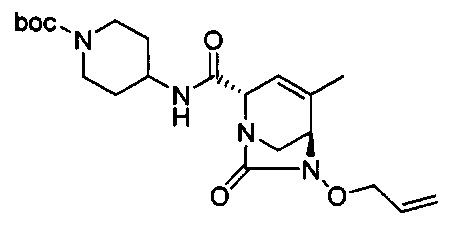
В раствор (2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 15, 0,407 г, 1,71 ммоль) в DMF (7 мл) при комнатной температуре добавляли гидрохлорид 1-Вос-4-амино-пиперидина (0,809 г, 3,42 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,299 г, 3,42 ммоль) и Ν,Ν-диизопропилэтиламин (1,190 мл, 6,83 ммоль). Через 30 минут реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия, рассолом и смесью 1/1 рассол/вода. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-60% этилацетата/гексаны) получили целевой продукт (0,502 г, 69,9%) в виде не совсем белой пены.
МС: 421 ЭРИ+ (C21H32N4O5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.32 (m, 2Н); 1.39 (s, 9Н); 1.66 (m, 2Н); 1.79 (m, 3Н); 2.81 (m, 2Н); 3.14 (m, 1Н); 3.32 (m, 1Н); 3.73 (m, 1Н); 3.84 (m, 3Н); 4.14 (m, 1Н); 4.36 (m, 2Н); 5.24-5.42 (m, 3Н); 5.88-6.02 (m, 1Н); 8.00 (m, 1Н).
Промежуточное соединение 26: (2S,5R)-2-(1-(трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
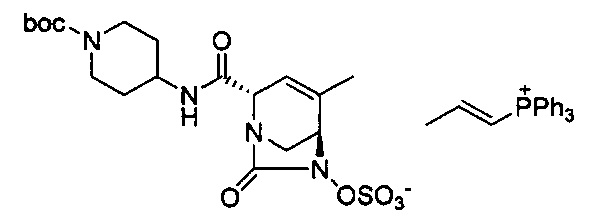
В раствор трет-бутил-4-((2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)пиперидин-1-карбоксилата (Промежуточное соединение 25, 0,502 г, 1,19 ммоль) и уксусной кислоты (0,137 мл, 2,39 ммоль) (высушенной над сульфатом натрия) в DCM (15 мл) при комнатной температуре добавляли тетракис(трифенилфосфин)палладий(0) (0,690 г, 0,60 ммоль). Раствор перемешивали при комнатной температуре в течение 30 минут, и он из желтого становился оранжевым. В реакционную смесь добавляли пиридин (15 мл) и комплекс триоксид серы-пиридин (1,520 г, 9,55 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре. Суспензию выпаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат концентрировали с получением оранжевого масла. После хроматографии на силикагеле (0%-50% ацетона/DCM) получили целевой продукт (0,152 г, 16,7%) в виде белой пены.
МС: 459 ЭРИ-, 303 ЭРИ+ (C18H27N4O8S, C21H20P)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.36 (m, 2Н); 1.41 (s, 9Н); 1.68 (m, 2Н); 1.78 (m, 3Н); 2.11 (m, 2Н); 2.18 (m, 2Н); 2.83 (m, 2Н); 3.20 (m, 1Н); 3.31 (m, 1Н); 3.76 (m, 1Н); 3.87 (m, 2Н); 3.98 (m, 1Н); 4.14 (m, 1Н); 5.40 (m, 1Н); 6.59-6.74 (m, 1Н); 7.24-7.33 (m, 1Н); 7.69-8.01 (m, 15Н).
Путь 2
Промежуточное соединение 27: (2S,5R)-1-трет-бутил-2-метил 5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-5,6-дигидропиридин-1,2(2Н)-дикарбоксилат
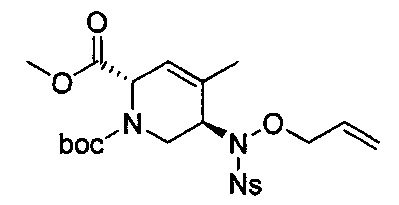
В раствор (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 19, 3 г, 6,03 ммоль) и карбоната калия (3,33 г, 24,12 ммоль) в DMF (30 мл) при комнатной температуре добавляли метилйодид (0,454 мл, 7,24 ммоль). Через 1 час реакционную смесь разбавляли этилацетатом и промывали три раза водой. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-30% этилацетата/гексаны) получили целевой продукт (2,060 г, 66,8%) в виде не совсем белой пены.
МС: 512 ЭРИ+ (C22H29N3O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.27 (m, 9Н); 1.71 (m, 3Н); 2.98-3.19 (m, 1Н); 3.65 (m, 3Н); 3.91 (m, 1Н); 4.17 (m, 3Н); 4.81-4.96 (m, 1Н); 5.12-5.21 (m, 2Н); 5.67 (m, 1Н); 5.86 (m, 1Н); 7.94-8.17 (m, 4Н).
Промежуточное соединение 28: (2S,5R)-метил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилат
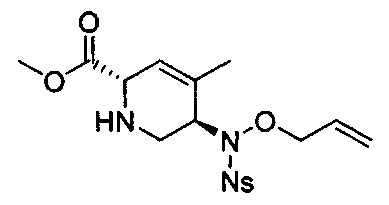
В раствор (2S,5R)-1-трет-бутил-2-метил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-5,6-дигидропиридин-1,2(2Н)-дикарбоксилата (Промежуточное соединение 27, 2,06 г, 4,03 ммоль) в DCM (20 мл) при комнатной температуре добавляли бромид цинка (0,648 мл, 12,08 ммоль). После перемешивания в течение ночи реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта (1,650 г, 100%) в виде светло-желтой пены.
МС: 412 ЭРИ+ (C17H21N3O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.56 (m, 3Н); 2.68 (m, 1Н); 2.91 (m, 1Н); 3.61 (s, 3Н); 4.01 (m, 2Н); 4.28 (m, 1Н); 4.48 (m, 1Н); 5.18-5.28 (m, 2Н); 5.85 (m, 2Н); 7.91-8.17 (m, 4Н).
Промежуточное соединение 29: (2S,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилат и (2R,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилат
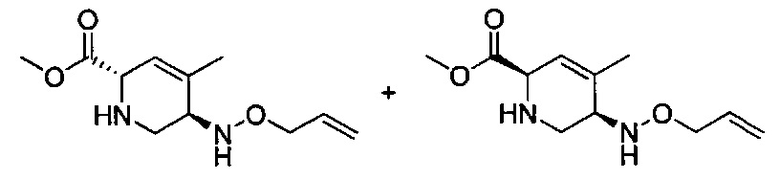
В раствор (2S,5R)-метил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилата (Промежуточное соединение 28, 1,65 г, 4,01 ммоль) и карбоната цезия (7,84 г, 24,06 ммоль) в THF (100 мл) при комнатной температуре добавляли PS-тиофенол (3-(3-меркаптофенил)пропанамидометилполистирол) (1,55 ммоль/г) (7,76 г, 12,03 ммоль). Через 3 часа реакционную смесь фильтровали, и смолу промывали DCM. Фильтрат концентрировали с получением желтого масла. После хроматографии на силикагеле (0%-50% метанол/DCM) получили смесь 1:1 (2S,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилата и (2R,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилата (0,680 г, 74,9%) в виде оранжевого масла. Эту смесь использовали далее без разделения.
МС: 227 ЭРИ+ (C11Η18Ν2O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.74 (m, 6Н); 2.70 (m, 3Н); 3.03 (m, 4Н); 3.61 (s, 3Н); 3.64 (s, 3Н); 3.90 (m, 2Н); 4.10 (m, 4Н); 5.11-5.25 (m, 5Н); 5.60 (m, 2Н); 5.91 (m, 2Н); 6.25 (m, 1Н); 6.41 (m, 1Н).
Промежуточное соединение 30 и Промежуточное соединение 31: (2S,5R)-метил-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксилат и (2R,5R)-метил-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксилат
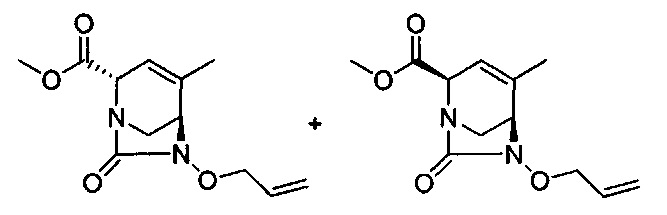
В раствор (2S,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилата и (2R,5R)-метил-5-(аллилоксиамино)-4-метил-1,2,5,6-тетрагидропиридин-2-карбоксилата (смесь 1:1) (Промежуточное соединение 29, 0,68 г, 3,01 ммоль) и Ν,Ν-диизопропилэтиламина (2,094 мл, 12,02 ммоль) в ацетонитриле (250 мл) при 0°С добавляли трифосген (0,357 г, 1,20 ммоль) в виде раствора в ацетонитриле (3 мл). Раствор трифосгена добавляли посредством шприцевого насоса со скоростью 1 мл/ч. Сразу после окончания добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-60% этилацетата/гексаны) получили целевой транс продукт (Промежуточное соединение 30, 0,292 г, 38%) и нежелательный цис продукт (Промежуточное соединение 31, 0,191 г, 25%). цис-Изомер может быть превращен в транс-изомер путем перемешивания в ацетонитриле с 3 эквивалентами триэтиламина в течение 1 часа с последующей обработкой, такой же, как для реакционной смеси.
МС: 253 ЭРИ+ (C12H16N2O4) и для цис и для транс
1Н ЯМР (300 МГц, DMSO-d6) транс Промежуточное соединение 30 δ: 1.80 (m, 3Н); 3.12 (m, 1Н); 3.22 (m, 1Н); 3.69 (s, 3Н); 3.87 (m, 1Н); 4.38 (m, 3Н); 5.24-5.42 (m, 3Н); 5.94 (m, 1Н).
1Н ЯМР (300 МГц, DMSO-d6) цис Промежуточное соединение 31 δ: 1.82 (m, 3Н); 3.19 (m, 1Н); 3.34 (m, 1Н); 3.64 (s, 3Н); 3.89 (m, 1Н); 4.34 (m, 2Н); 4.61 (m, 1Н), 5.23-5.37 (m, 2Н); 5.50 (m, 1Н); 5.92 (m, 1Н).
Промежуточное соединение 32: (2R,5R)-в-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновая кислота
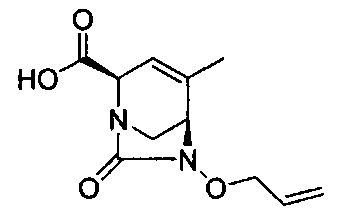
В раствор (2S,5R)-метил-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксилата (Промежуточное соединение 30, 0,479 г, 1,90 ммоль) в THF (10 мл) и воде (5 мл) при 0°С добавляли гидроксид лития (0,045 г, 1,90 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 часов. Добавляли еще 0,5 экв. гидроксида лития. Через 2 часа реакционную смесь нейтрализовали осторожно 1 н. HCl при 0°С, и THF выпаривали. Водную фазу замораживали и лиофилизировали с получением светло-оранжевого твердого вещества (0,464 г, 103% неочищенного).
МС: 239 ЭРИ+ (C11H14N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.72 (m, 3Н); 2.38 (m, 1Н); 3.00 (m, 1Н); 3.47 (m, 1Н); 3.68 (m, 1Н); 3.83 (m, 1Н); 4.33 (m, 1Н); 5.15-55 (m, 4Н); 5.94 (m, 1Н).
Промежуточное соединение 25: трет-бутил-4-((2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)пиперидин-1-карбоксилат
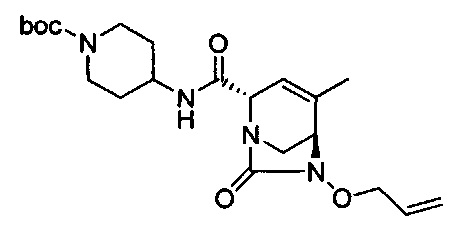
Указанное в заголовке соединение получали из (2R,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 32) по методике, описанной в разделе Путь 1 для Промежуточного соединения 25. Смотри Путь 1, 2 конечные стадии.
ПРИМЕР 4
(2S,5R)-2-карбамоил-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат натриевая соль
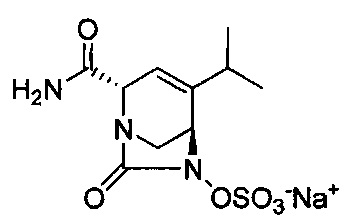
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 44, 71,1 мг, 0,12 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде светло-желтого твердого вещества (31,9 мг, 83%).
Оптическое вращение: (0,1 г/дл, МеОН)=-212
МС: 306 ЭРИ+ (C10H15N3O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.99 (m, 6Н); 2.27 (m, 1Н); 3.15 (m, 1Н); 3.26 (m, 1Н); 4.12 (m, 2Н); 5.38 (m, 1Н); 7.26 (bs, 1Н); 7.52 (bs, 1Н).
Промежуточное соединение 33: (Е)-2,4,6-триизопропил-N'-(3-метилбутан-2-илиден)6ензол-сульфоногидразид
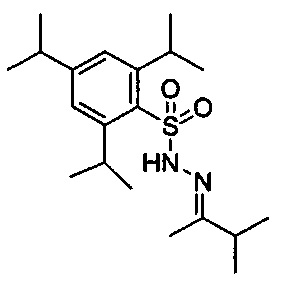
В суспензию 2,4,6-триизопропилбензолсульфонил-гидразида (5,06 г, 16,95 ммоль) и 3-метил-бутан-2-она (1,814 мл, 16,95 ммоль) в этаноле (20 мл) добавляли 2 капли концентрированной соляной кислоты. Суспензия становилась раствором и спустя минуту или две белое твердое вещество начинало выпадать в осадок. Реакционную смесь помещали в холодильник на 2 часа. Белый осадок собирали фильтрованием с получением целевого продукта (4,44 г, 71%).
МС: 367 ЭРИ+ (C20H34N2O2S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.87 (d, 6Н); 1.17 (m, 18Н); 1.75 (s, 3Н); 2.31 (m, 1H); 2.90 (m, 1H); 4.24 (m, 2H); 7.19 (s, 2H); 9.96 (s, 1H).
Промежуточное соединение 34: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(4-метил-3-метилен-2-оксопентил)карбамат
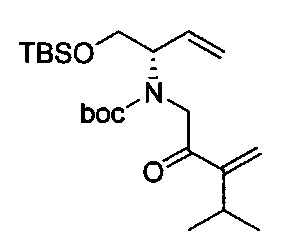
В суспензию (Е)-2,4,6-триизопропил-N'-(3-метилбутан-2-илиден)бензолсульфоногидразида (Промежуточное соединение 33, 8 г, 21,82 ммоль) в гексане (65 мл) и TMEDA (6,50 мл) при -78°С по каплям добавляли н-бутиллитий (1,6М в гексанах) (34,1 мл, 54,56 ммоль). Реакционная смесь становилась оранжевой, и ее перемешивали в течение 30 минут при -78°С, затем нагревали до 0°С и сразу начинали барботирование. Суспензия превращалась в желтый раствор. Через примерно 15 минут барботирование останавливали и добавляли (S)-трет-бутил-1-(трет-бутилдиметилсилил-окси)бут-3-ен-2-ил(2-(метокси(метил)-амино)-2-оксоэтил)карбамат (Промежуточное соединение 5, 4,39 г, 10,91 ммоль) в виде раствора в гексане (2 мл). Через примерно 15 минут ЖХ/МС показала целевой продукт и отсутствие остаточного исходного вещества амида Вайнреба. Реакционную смесь гасили насыщенным бикарбонатом натрия и экстрагировали диэтиловым эфиром дважды. Эфирные экстракты сушили над сульфатом магния, фильтровали и концентрировали с получением желтого масла. После хроматографии на силикагеле (0%-10% этилацетата/гексаны) получили целевой продукт (2,219 г, 49,4%) в виде светло-желтого масла.
МС: 412 ЭРИ+ (C22H41NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (m, 6Н); 0.82 (m, 9Н); 0.98 (m, 6Н); 1.28-1.38 (m, 9Н); 2.77 (m, 1Н); 3.71 (m, 2Н); 4.33 (m, 2Н); 4.62 (m, 1Н); 5.16 (m, 2Н); 5.77 (m, 2Н); 6.06 (m, 1Н).
Промежуточное соединение 35: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)-метил)-4-изопропил-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилат
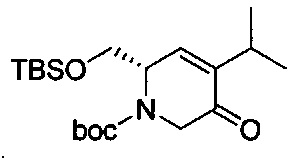
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(4-метил-3-метилен-2-оксопентил)-карбамата (Промежуточное соединение 34, 0,839 г, 2,04 ммоль) по методике, описанной для Промежуточного соединения 7, используя 0,32 экв. (1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенилметилен)рутения. Целевой продукт был получен в виде бесцветного масла (0,667 г, 85%).
МС: 384 ЭРИ+ (C20H37NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.00 (m, 6Н); 0.80 (s, 9Н); 1.00 (m, 6Н); 1.42 (s, 9Н); 2.77 (m, 1Н); 3.85 (m, 3Н); 4.26 (m, 1Н); 4.69 (m, 1Н); 6.80 (m, 1Н).
Промежуточное соединение 36: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
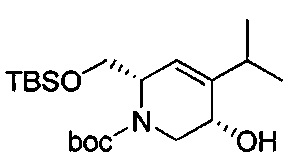
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-изопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 35, 0,667 г, 1,74 ммоль) по методике, описанной для Промежуточного соединения 8. Целевой продукт был получен в виде бесцветного масла (0,464 г, 69%).
МС: 386 ЭРИ+ (C20H39NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Н); 0.86 (s, 9Н); 0.98 (s, 6Н); 1.39 (s, 9Н); 2.64 (m, 2Н); 3.59 (m, 2Н); 3.99 (m, 2Н); 4.21 (m, 1Н); 5.04 (d, 1Н); 5.36 (m, 1Н).
Промежуточное соединение 37: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
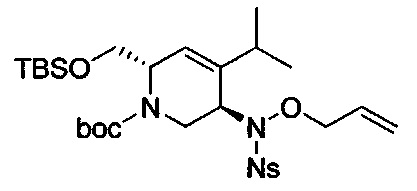
Указанное в заголовке соединение получали из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 36, 1,2 г, 3,11 ммоль) по методике, описанной для Промежуточного соединения 10, используя по 2,4 эквивалента трифенилфосфина и диизопропилазодикарбоксилата. Целевой продукт был получен в виде желтой пены (1,22 г, 62%).
МС: 626 ЭРИ+ (C29H47N3O8SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.00 (s, 6Н); 0.83 (s, 9Н); 1.00 (m, 6Н); 1.34 (m, 9Н); 3.18 (m, 1Н); 3.59 (m, 2Н); 4.22 (m, 4Н); 4.46 (m, 1Н); 5.18 (m, 2Н); 5.73 (m, 2Н); 7.91-8.18 (m, 4H).
Промежуточное соединение 38: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
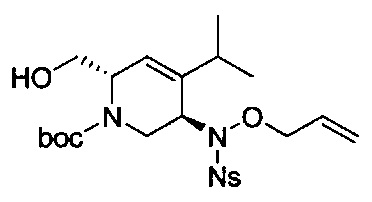
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 37, 0,361 г, 0,58 ммоль) по методике, описанной для Промежуточного соединения 18. Целевой продукт был получен в виде желто-коричневой пены (0,257 г, 87%).
МС: 512 ЭРИ+ (C23H33N3O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.93 (m, 6Н); 1.35 (m, 9Н); 3.13 (m, 1Н); 3.40 (m, 2Н); 4.18 (m, 3Н); 4.41 (m, 1Н); 4.75 (m, 1Н); 5.20 (m, 2Н); 5.74 (m, 2Н); 7.92-8.19 (m, 4H).
Промежуточное соединение 39: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
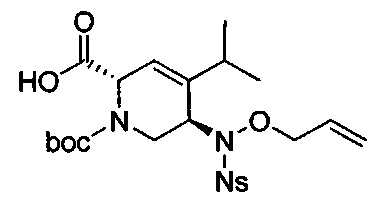
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 38, 0,727 г, 1,42 ммоль) по методике, описанной для Промежуточного соединения 19. Целевой продукт был получен в виде не совсем белой пены (0,65 г, 87%).
МС: 526 ЭРИ+ (C23H31N3O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.00 (m, 6Н); 1.26 (m, 9Н); 2.95-3.13 (m, 1Н); 3.95 (m, 1Н); 4.10-4.34 (m, 3Н); 4.77-4.91 (m, 1Н); 5.19 (m, 2Н); 5.67-5.87 (m, 2Н); 7.92-8.20 (m, 4Н).
Промежуточное соединение 40: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-изопропил-5.6-дигидропиридин-1 (2Н)-карбоксилат
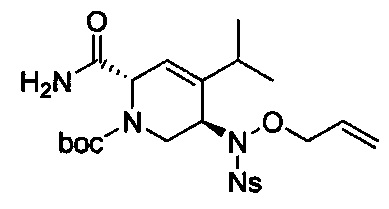
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 39, 0,65 г, 1,24 ммоль) по методике, описанной для Промежуточного соединения 20. Целевой продукт был получен в виде не совсем белого твердого вещества (0,322 г, 51%).
МС: 525 ЭРИ+ (C23H32N4O8S)
1Н ЯМР (300 МГц, CDCl3) δ: 1.00 (m, 6Н); 1.31 (m, 9Н); 3.20 (m, 1Н); 4.20 (m, 3Н); 4.70-4.87 (m, 1Н); 5.19 (m, 2Н); 5.69 (m, 1Н); 5.86 (m, 1Н); 7.03 (m, 1Н); 7.47 (m, 1Н); 7.94-8.21 (m, 4Н).
Промежуточное соединение 41: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
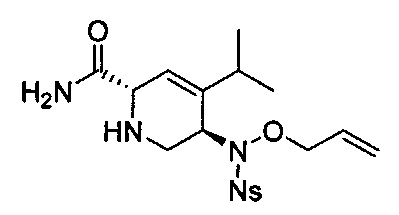
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 40, 0,427 г, 0,81 ммоль) по методике, описанной для Промежуточного соединения 21. Целевой продукт был получен в виде желтой пены (0,247 г, 71%).
МС: 425 ЭРИ+ (C18H24N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.95 (m, 6Н); 2.25 (m, 1Н); 2.67 (m, 2Н); 3.81 (m, 1Н); 4.07 (m, 1Н); 4.30 (m, 2Н); 5.24 (m, 2Н); 5.83 (m, 1Н); 6.03 (m, 1Н); 7.03 (m, 1Н); 7.32 (m, 1Н); 7.93-8.18 (m, 4Н).
Промежуточное соединение 42: (2S,5R)-5-(аллилоксиамино)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
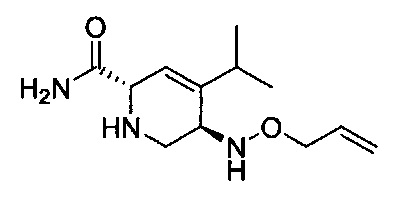
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 41, 0,247 г, 0,58 ммоль) по методике, описанной для Промежуточного соединения 22. Целевой продукт был получен в виде светло-желтого твердого вещества (98 мг, 71%).
МС: 240 ЭРИ+ (C12H21N3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.99 (m, 6Н); 2.37 (m, 1Н); 3.04 (m, 1Н); 3.17 (m, 1Н); 3.60 (m, 1Н); 4.11 (m, 2Н); 5.11-5.26 (m, 2Н); 5.78 (m, 1Н); 5.86-6.00 (m, 1Н); 6.31 (m, 1Н); 7.01 (bs, 1Н); 7.36 (bs, 1Н).
Промежуточное соединение 43: (2S,5R)-6-(аллилокси)-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
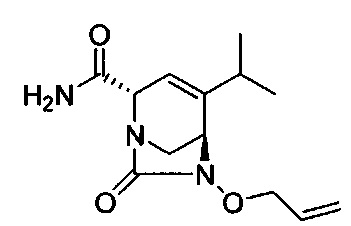
Указанное в заголовке соединение получали из (2S,5R)-5-(аллилоксиамино)-4-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (0,0981 г, 0,41 ммоль) и Ν,Ν-диизопропилэтиламина (Промежуточное соединение 42, 0,286 мл, 1,64 ммоль) по методике, описанной для Промежуточного соединения 16. Целевой продукт был получен в виде светло-желтого масла (69 мг, 63%).
МС: 266 ЭРИ+ (C13H19N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.00 (m, 6Н); 2.28 (m, 1Н); 3.19 (m, 2Н); 4.02 (m, 1Н); 4.14 (m, 1Н); 4.37 (m, 2Н); 5.28-5.42 (m, 3Н); 5.90-6.01 (m, 1Н); 7.28 (bs, 1Н); 7.51 (bs, 1Н).
Промежуточное соединение 44: (2S,5R)-2-карбамоил-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил (проп-1-енил)фосфония
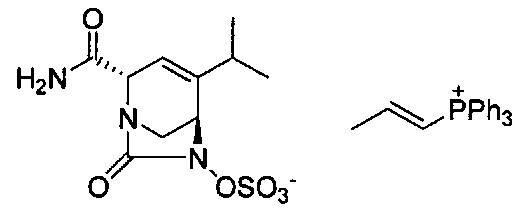
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 43, 68,7 мг, 0,26 ммоль) по методике, описанной для Промежуточного соединения 17, используя 1 эквивалент тетракис(трифенилфосфин)-палладия. Целевой продукт был получен в виде желтого масла (71 мг, 45%).
МС: 306 ЭРИ+, 303 ЭРИ+ (C10H15N3O6S, C21H20P)
ПРИМЕР 5
Натриевая соль (2S,5R)-2-циано-4-изопропил-7-оксо-1,6-диаза6ицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
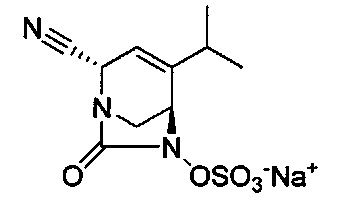
Указанное в заголовке соединение получали из (2S,5R)-2-циано-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 46, 47,2 мг, 0,08 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде белого твердого вещества (18,2 мг, 73%).
Оптическое вращение: (0,1 г/дл, DMSO)=-168
МС: 286 ЭРИ- (C10H13N3O5S)
1Н ЯМР (600 МГц, DMSO-d6) δ: 1.00 (m, 6Н); 2.30 (m, 1Н); 3.20 (m, 1Н); 3.50 (m, 1Н); 4.24 (m, 1Н); 4.97 (m, 1Н); 5.25 (m, 1Н).
Промежуточное соединение 45: (2S,5R)-6-(аллилокси)-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрил
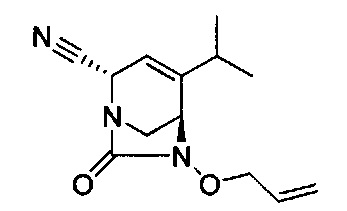
Указанное в заголовке соединение получали из 2S,5R)-6-(аллилокси)-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 43, 143,9 мг, 0,54 ммоль) по методике, описанной для Промежуточного соединения 23. Целевой продукт был получен в виде бесцветного масла (114 мг, 85%).
МС: 248 ЭРИ+ (C13H17N3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.00 (m, 6Н); 2.32 (m, 1Н); 3.21 (m, 1Н); 3.45 (m, 1Н); 4.18 (m, 1Н); 4.38 (m, 2Н); 4.98 (m, 1Н); 5.25-5.39 (m, 3Н); 5.86-6.00 (m, 1Н).
Промежуточное соединение 46: (2S,5R)-2-циано-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)-фосфония
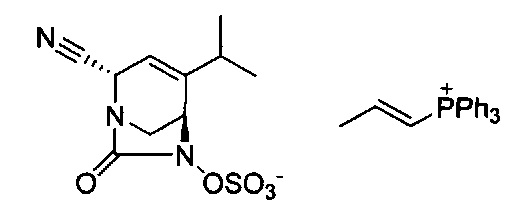
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбонитрила (Промежуточное соединение 45, 114,1 мг, 0,46 ммоль) по методике, описанной для Промежуточного соединения 17, используя 0,75 эквивалента тетракис(трифенил-фосфин)палладия. Целевой продукт был получен в виде светло-желтого масла (47,2 мг, 17%).
МС: 286 ЭРИ-, 303 ЭРИ+ (C10H12N3O5S, C21H20P)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.01 (m, 6Н); 2.17 (m, 3Н); 3.21 (m, 1Н); 3.50 (m, 1Н); 4.24 (m, 1Н); 4.98 (m, 1Н); 5.26 (m, 1Н); 6.64-6.73 (m, 1Н); 7.23-7.37 (m, 1Н); 7.51-7.65 (m, 1Н); 7.68-7.95 (m, 15Н).
ПРИМЕР 6
(2S,5R)-2-(2-аминоэтилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат
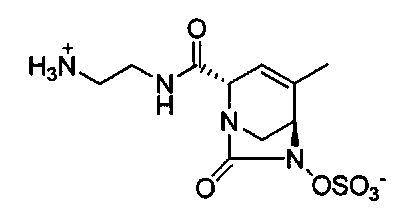
В раствор (2S,5R)-2-(2-(трет-бутоксикарбониламино)этилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил-(проп-1-енил)фосфония (Промежуточное соединение 48, 0,238 г, 0,33 ммоль) в DCM (2 мл) при 0°С добавляли TFA (2 мл). Через 30 минут реакционную смесь концентрировали с получением оранжевого масла. Это масло растирали с диэтиловым эфиром три раза и с этилацетатом три раза с получением оранжевого твердого вещества. Очистку выполняли методом обращенно-фазовой ВЭЖХ (0-10% ацетонитрила в воде, YMC Carotenoid С30, 19 мм × 150 мм, 5 мкм) с получением указанного в заголовке соединения в виде белого твердого вещества (25,4 мг, 13%).
Оптическое вращение: (0,1 г/дл, DMSO)=-120.
МС: 321 ЭРИ+ (C10H16N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.80 (m, 3Н); 2.88 (m, 2Н); 3.17 (m, 1Н); 3.24 (m, 1Н); 3.39 (m, 2Н); 3.99 (m, 1Н); 4.21 (m, 1Н); 4.48 (m, 1Н); 7.43 (m, 2Н); 8.25 (m, 1Н).
Промежуточное соединение 47: трет-бутил-2-((2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)этилкарбамат
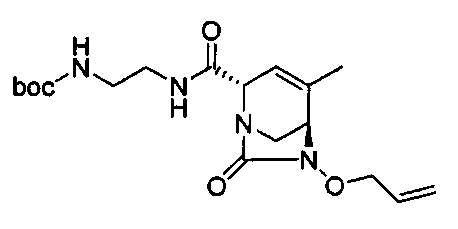
Указанное в заголовке соединение получали из (2R,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 15, 0,348 г, 1,46 ммоль) и гидрохлорида N-Boc-этилендиамина (0,287 г, 1,46 ммоль) по методике, описанной для Промежуточного соединения 25. Целевой продукт был получен в виде светло-розовой пены (230 мг, 41%).
МС: 381 ЭРИ+ (C18H28N4O5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.37 (s, 9Н); 1.79 (m, 3Н); 3.00 (m, 2Н); 3.16 (m, 4Н); 3.82 (m, 1Н); 4.14 (m, 1Н); 4.37 (m, 2Н); 5.24-5.47 (m, 3Н); 5.90-6.00 (m, 1Н); 6.81 (m, 1Н); 8.07 (m, 1Н).
Промежуточное соединение 48: (2S,5R)-2-(2-(трет-бутокси-карбониламино)этилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)-фосфония
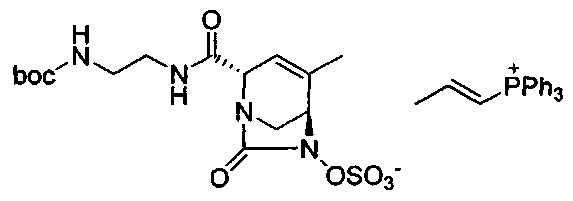
В раствор трет-бутил-2-((2S,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)этилкарбамата (Промежуточное соединение 47, 230 мг, 0,60 ммоль) и уксусной кислоты (0,069 мл, 1,21 ммоль) (высушенной над сульфатом натрия) в DCM (9 мл) при комнатной температуре добавляли тетракис(трифенилфосфин)палладий(0) (349 мг, 0,30 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли пиридин (9,00 мл) и комплекс триоксид серы-пиридин (577 мг, 3,63 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре. Суспензию упаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат концентрировали с получением оранжевого масла. Это масло снова переносили в DCM и фильтровали через фильтр 0,45 мкм. Фильтрат концентрировали с получением оранжевого масла. Это неочищенное вещество переносили на следующую стадию без очистки.
МС: 419 ЭРИ-, 303 ЭРИ+ (C15H23N4O8S, C21H20P)
ПРИМЕР 7
Натриевая соль (2S,5R)-2-(метоксиметил)-7-оксо-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
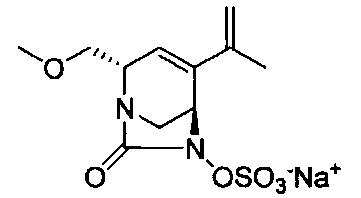
Указанное в заголовке соединение получали из (2S,5R)-2-(метоксиметил)-7-оксо-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 60, 0,283 г, 0,47 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен после очистки методом обращенно-фазовой ВЭЖХ (2-10% ацетонитрила в воде, Synergi Hydro RP, 19 мм × 150 мм, 5 мкм) в виде белого твердого вещества (40 мг, 26%).
Оптическое вращение: (0,1 г/дл, МеОН)=-170.
МС: 305 ЭРИ+ (C11H16N2O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.80 (s, 3Н); 3.24 (m, 5Н); 3.57 (m, 2Н); 3.82 (m, 1Н); 4.48 (m, 1Н); 5.02 (m, 1Н); 5.43 (m, 1Н); 5.57 (m, 1Н).
Промежуточное соединение 49: (S)-трет-бутил-1-(трет-6утилдиметилсилилокси)бут-3-ен-2-ил(2-оксопент-3-енил)карбамат
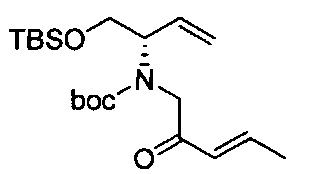
В раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 5, 32,5 г, 80,73 ммоль) в THF (400 мл) под азотом при 0°С по каплям добавляли бромид проп-1-енилмагния (323 мл, 161,45 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа, затем гасили 400 мл 10%-ной лимонной кислотой, затем разбавляли 100 мл воды и экстрагировали диэтиловым эфиром. Органические фазы концентрировали, и полученное масло растворяли в диэтиловом эфире и промывали водой и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (5%-20% этилацетата/гексаны) получили целевой продукт в виде бесцветного масла (27 г, 87%).
МС: 384 ЭРИ+ (C20H37NO4Si)
1Н ЯМР (300 МГц, CDCl3) δ: 0.05 (2, 6Н); 0.88 (s, 9Н); 1.39-1.47 (m, 9Н); 1.90 (m, 3Н); 3.80 (m, 2Н); 4.05-4.18 (m, 2Н); 4.43-4.76 (m, 1Н); 5.22 (m, 2Н); 5.86 (m, 1Н); 6.21 (m, 1Н); 6.91 (m, 1Н).
Промежуточное соединение 50: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
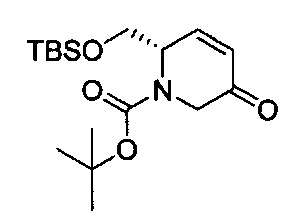
(S)-трет-Бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-оксопент-3-енил)карбамат (Промежуточное соединение 49, 27,0 г, 70,39 ммоль) растворяли в толуоле (650 мл). Раствор продували азотом в течение 15 минут, после чего добавляли катализатор Ховейды-Граббса (Hoveyda-Grubbs) 2-го поколения (0,885 г, 1,41 ммоль). Реакционную смесь нагревали под азотом при 65°С. Через 2 часа ЖХ/МС показала полное образование продукта. Реакционную смесь концентрировали при пониженном давлении. После хроматографии на силикагеле (10%-35% этилацетата/гексаны) получили целевой продукт в виде твердого вещества (17,0 г, 70%).
Оптическое вращение: 0,1 г/дл, метиленхлорид=-175
МС: 342 ЭРИ+ (C17H31NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (s, 6Н); 0.82 (s, 9Н); 1.43 (s, 9Н); 3.78-3.93 (m, 3Н); 4.29 (m, 1Н); 4.70 (m, 1Н); 6.19 (dd, 1Н); 7.15 (m, 1Н).
Промежуточное соединение 51: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-йод-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
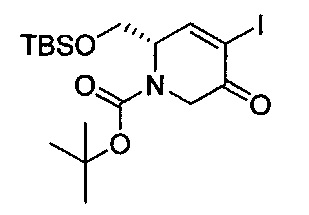
В раствор (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 50, 10 г, 29,28 ммоль) и 4-диметиламинопиридина (0,894 г, 7,32 ммоль) в смеси THF (100 мл)/вода (100 мл) при комнатной температуре добавляли карбонат калия (3,24 г, 23,42 ммоль) и йод (8,92 г, 35,14 ммоль). После перемешивания в течение 15 минут реакционную смесь разбавляли диэтиловым эфиром и промывали насыщенным тиосульфатом натрия дважды, затем 5%-ной лимонной кислотой и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили целевой продукт в виде желто-коричневого масла (11,6 г, 85%).
МС: 468 ЭРИ+ (C17H30INO4Si)
1Н ЯМР (300 МГц, CDCl3) δ: 0.04 (s, 6Н); 0.86 (s, 9Н); 1.49 (s, 9Н); 3.81 (m, 1Н); 3.95 (m, 1Н); 4.17 (m, 1Н); 4.79 (m, 2Н); 7.67 (m, 1Н).
Промежуточное соединение 52: (2S,5R)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-йод-5,6-дигидропиридин-1 (2Н)-карбоксилат
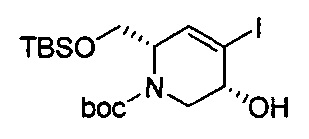
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-йод-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 51, 10 г, 21,39 ммоль) по методике, описанной для Промежуточного соединения 8. Целевой продукт был получен в виде бесцветного масла (8,87 г, 88%).
МС: 470 ЭРИ+ (C17H32INO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Н); 0.86 (s, 9Н); 1.40 (s, 9Н); 2.87 (m, 1Н); 3.65 (m, 2Н); 3.79 (m, 1Н); 4.21 (m, 2Н); 5.74 (d, 1Н); 6.44 (m, 1Н).
Промежуточное соединение 53: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(проп-1-ен-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат
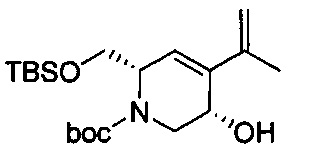
Раствор (2S,5R)-трел7-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-йод-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 52, 8,57 г, 18,26 ммоль), трифтор(проп-1-ен-2-ил)бората калия (5,40 г, 36,51 ммоль), карбоната калия (3,11 мл, 54,77 ммоль) и дихлор[1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (1,190 г, 1,83 ммоль) в диоксане (200 мл) и воде (66,7 мл) при комнатной температуре продували аргоном в течение 5 минут, затем нагревали при 70°С. Реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили смесь 2:1 целевого продукта и исходного вещества в виде коричневого масла (5,36 г, 77%).
МС: 384 ЭРИ+ (C20H37NO4Si)2
Промежуточное соединение 54: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-5,6-дигидропиридин-1 (2Н)-карбоксилат
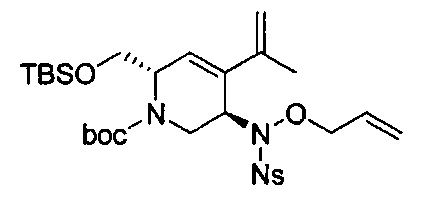
Указанное в заголовке соединение получали из (2S,58S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(проп-1-ен-2-ил)-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 53, 3,86 г, 10,06 ммоль) по методике, описанной для Промежуточного соединения 10. Целевой продукт был получен в виде светло-желтой пены (3,25 г, 52%).
МС: 624 ЭРИ+ (C29H45N3O8SSi)
Промежуточное соединение 55: N-(аллилокси)-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамид
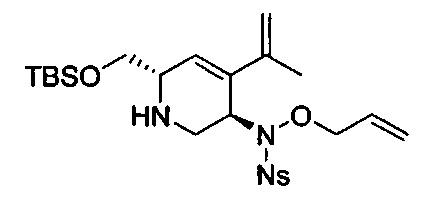
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 54, 3,04 г, 4,87 ммоль) по методике, описанной для Промежуточного соединения 21. Целевой продукт был получен в виде оранжевого масла (2,53 г, 99%).
МС: 524 ЭРИ+ (C24H37N3O6SSi)
Промежуточное соединение 56: O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламин
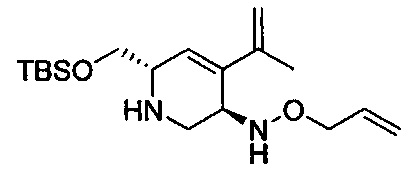
Целевой продукт получали из N-(аллилокси)-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамида (Промежуточное соединение 55, 2,45 г, 4,68 ммоль) по методике, описанной для Промежуточного соединения 22. Целевой продукт был получен в виде желтого масла (1,19 г, 75%).
МС: 339 ЭРИ+ (C18H34N2O2Si)
Промежуточное соединение 57: (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
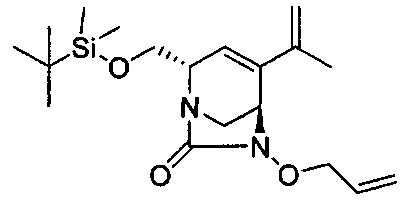
Указанное в заголовке соединение получали из O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)-метил)-4-(проп-1-ен-2-ил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламина (Промежуточное соединение 56, 1.22 г, 3.60 ммоль) по методике, описанной для Промежуточного соединения 16. Целевой продукт был получен в виде светло-желтого масла (1,16 г, 88%).
МС: 365 ЭРИ+ (C18H32N2O3Si)
Промежуточное соединение 58: (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
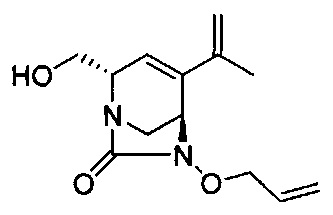
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)-метил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 57, 1,16 г, 3,18 ммоль) по методике, описанной для Промежуточного соединения 14. Целевой продукт был получен в виде бесцветного масла (741 мг, 93%).
МС: 251 ЭРИ+ (C13H18N2O3)
Промежуточное соединение 59: (2S,5R)-6-(аллилокси)-2-(метоксиметил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
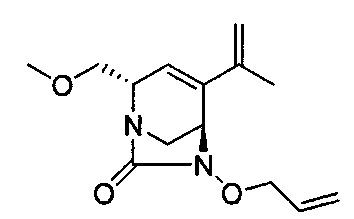
В раствор (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 58, 0,289 г, 1,15 ммоль) в DMF (10 мл) при 0°С добавляли метилйодид (0,435 мл, 6,93 ммоль), затем добавляли гидрид натрия (60% в минеральном масле) (0,051 г, 1,27 ммоль). Реакционную смесь перемешивали в течение 1,5 часов при 0°С. Реакционную смесь разбавляли этилацетатом и промывали дважды водой. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили целевой продукт в виде бледно-желтого масла (233 мг, 76%).
МС: 265 ЭРИ+ (C14H20N2O3)
Промежуточное соединение 60: (2S,5R)-2-(метоксиметил)-7-оксо-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
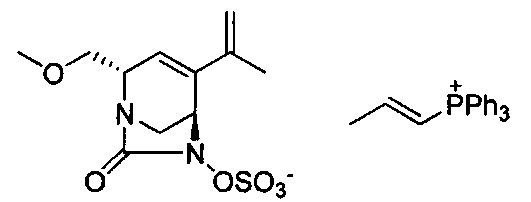
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-2-(метоксиметил)-4-(проп-1-ен-2-ил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 59, 233 мг, 0.88 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде не совсем белой пены (283 мг, 53%).
МС: 305 ЭРИ+, 303 ЭРИ+ (C11Η16Ν2O6S, C21H20P)
ПРИМЕР 8
Натриевая соль (2S,5R)-2-((5-гидрокси-4-оксо-1,4-дигидропиридин-2-ил)метилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
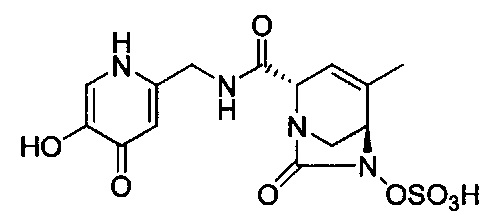
В раствор (2S,5R)-2-((4,5-бис(4-метоксибензилокси)пиридин-2-ил)метилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата (Промежуточное соединение 68, 58 мг, 0,09 ммоль) в DCM (2 мл) при комнатной температуре добавляли трифторуксусную кислоту (0,505 мл, 6,55 ммоль). Реакционную смесь перемешивали в течение 10 минут и затем концентрировали. Полученный остаток растворяли в DCM и концентрировали более чем два раза. Продукт очищали дважды на колонке С18 RediSepRf Gold (15,5 г), элюируя водой вручную с использованием шприца. Целевой продукт был получен после лиофилизации в виде белого твердого вещества (3,4 мг, 9%).
МС: 401 ЭРИ+ (C14H16N4O8S)
1Н ЯМР (300 МГц, D2O) δ: 1.93 (m, 3Н); 3.25 (m, 1Н); 3.58 (m, 1Н); 4.20 (m, 1Н); 4.50 (m, 2Н); 4.59 (m, 1Н); 5.67 (m, 1Н); 6.76 (s, 1Н); 7.77 (s, 1Н).
Промежуточное соединение 61: 2-(гидроксиметил)-5-(4-метоксибензилокси)-4Н-пиран-4-он
В раствор 5-гидрокси-2-(гидроксиметил)-4Н-пиран-4-она (Alfa Aesar, 5,11 г, 35,96 ммоль) в DMF (70 мл) при комнатной температуре по каплям добавляли карбонат калия (9,94 г, 71,92 ммоль) и 1-(хлорметил)-4-метоксибензол (5,86 мл, 43,15 ммоль). Реакционную смесь нагревали при 80°С в течение 1 часа, затем концентрировали. В полученную суспензию добавляли ледяную воду. Осадок собирали фильтрованием, затем растирали с этилацетатом и снова фильтровали. Указанное в заголовке соединение было получено в виде желто-коричневого твердого вещества (6,44 г, 68%).
МС: 263 ЭРИ+ (С14Н14О5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.76 (s, 3Н); 4.28 (s, 2Н); 4.86 (s, 2Н); 5.75 (m, 1Н); 6.31 (s, 1Н); 6.94 (m, 2Н); 7.33 (m, 2Н); 8.13 (s, 1Н).
Промежуточное соединение 62: 2-(гидроксиметил)-5-(4-метоксибензилокси)пиридин-4(1Н)-он
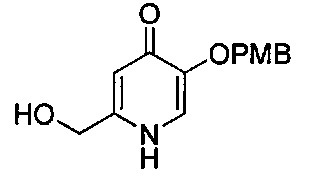
2-(Гидроксиметил)-5-(4-метоксибензилокси)-4Н-пиран-4-он (Промежуточное соединение 63, 6,44 г, 24,56 ммоль) и аммиак (7 н. в МеОН) (59,6 мл, 417,45 ммоль) объединяли реакционном сосуде для работы под давлением и нагревали при 90°С в течение 5 часов. Реакционную смесь охлаждали в течение ночи, затем концентрировали. Твердое вещество суспендировали в воде, затем собирали фильтрованием. Указанное в заголовке соединение было получено в виде коричневого твердого вещества (3,48 г, 54%).
МС: 262 ЭРИ+ (C14H15NO4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.75 (s, 3Н); 4.32 (s, 2Н); 4.93 (s, 2Н); 5.53 (m, 1Н); 6.07 (m, 1Н); 6.92 (m, 2Н); 7.26 (m, 1Н); 7.32 (m, 2Н); 11.02 (m, 1Н).
Промежуточное соединение 63: (4,5-бис(4-метоксибензилокси)-пиридин-2-ил)метанол
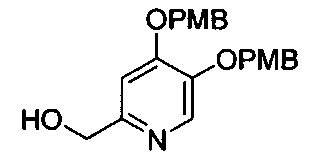
В раствор 2-(гидроксиметил)-5-(4-метоксибензилокси)пиридин-4(1Н)-она (Промежуточное соединение 62, 3,48 г, 13,32 ммоль) в DMF (100 мл) при комнатной температуре добавляли 4-метоксибензилхлорид (1,987 мл, 14,65 ммоль), затем добавляли карбонат калия (2,273 мл, 39,96 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, затем нагревали при 80°С в течение 1,5 часов. Реакционную смесь охлаждали до комнатной температуры, вливали в воду и экстрагировали дважды этилацетатом. Объединенные экстракты промывали водой и рассолом. Этилацетатный слой концентрировали с получением масла. В это масло добавляли 1 н. HCl, при этом образовалось светло-коричневое твердое вещество. Это вещество собирали фильтрованием. Твердое вещество переносили в этилацетат и промывали насыщенным раствором бикарбоната натрия. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения в виде коричневого твердого вещества (2,62 г, 52%).
МС: 382 ЭРИ+ (C22H23NO5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.74 (s, 3Н); 3.76 (s, 3Н); 4.42 (m, 2Н); 5.05 (s, 2Н); 5.12 (s, 2Н); 5.27 (m, 1Н); 6.93 (m, 4Н); 7.17 (s, 1Н); 7.32 (m, 4Н); 8.07 (s, 1Н).
Промежуточное соединение 64: 2-((4,5-бис(4-метоксибензилокси)-пиридин-2-ил)метил)изоиндолин-1,3-дион
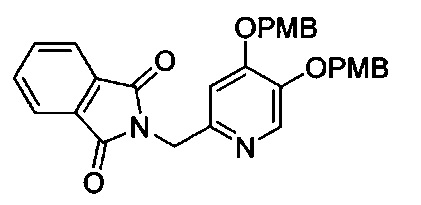
В раствор (4,5-бис(4-метоксибензилокси)пиридин-2-ил)метанола (Промежуточное соединение 63, 1,5 г, 3,93 ммоль), фталимида (0,579 г, 3,93 ммоль) и трифенилфосфина (1,028 г, 3,93 ммоль) в THF (15 мл) при комнатной температуре добавляли диизопропилазодикарбоксилат (2,091 мл, 10,62 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали на силикагеле. После хроматографии на силикагеле (0%-70% этилацетат) получили указанное в заголовке соединение в виде светло-коричневого твердого вещества (1,1 г, 55%).
МС: 511 ЭРИ+ (C30H26N2O6)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.73 (s, 3Н); 3.74 (s, 3Н); 4.78 (s, 2Н); 5.01 (s, 2Н); 5.10 (s, 2Н); 6.89 (m, 4Н); 7.16 (s, 1Н); 7.32 (m, 4Н); 7.89 (m, 4Н); 8.01 (s, 1Н).
Промежуточное соединение 65: (4,5-бис(4-метоксибензилокси)-пиридин-2-ил)метанамин
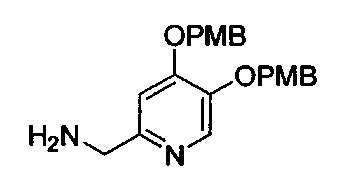
В раствор 2-((4,5-бис(4-метоксибензилокси)пиридин-2-ил)метил)-изоиндолин-1,3-диона (Промежуточное соединение 64, 1,1 г, 2,15 ммоль) в хлороформе (20 мл) и метаноле (10 мл) при комнатной температуре добавляли гидразин гидрат (0,328 мл, 4,31 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли еще 1 эквивалент гидразингидрата. Через 4 часа реакционную смесь фильтровали для удаления твердого вещества. Фильтрат концентрировали с получением оранжевого масла. Это масло растворяли в метаноле и добавляли диэтиловый эфир, чтобы разрушить больше твердого вещества. Это повторяли до тех пор, пока побочного продукта 2,3-дигидрофталазин-1,4-диона не осталось. Указанное в заголовке соединение было получено в виде оранжевой пены (0,82 г, 100%).
МС: 381 ЭРИ+ (C22H24N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.66 (s, 2Н); 3.74 (s, 3Н); 3.76 (s, 3Н); 5.03 (s, 2Н); 5.11 (s, 2Н); 6.92 (m, 4Н); 7.20 (s, 1Н); 7.35 (m, 4Н); 8.05 (s, 1Н).
Промежуточное соединение 66: (2S,5R)-6-(аллилокси)-N-((4,5-бис(4-метоксибензилокси)пиридин-2-ил)метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
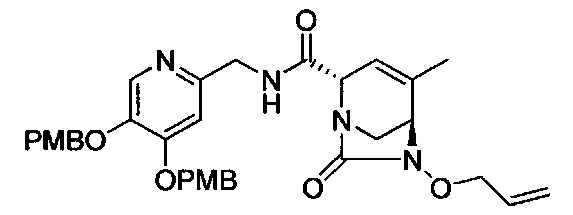
В раствор (R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 32, 0,469 г, 1,97 ммоль) в DMF (10 мл) при комнатной температуре добавляли (4,5-бис(4-метоксибензилокси)пиридин-2-ил)метанамин (Промежуточное соединение 65, 0,824 г, 2,17 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,497 г, 3,94 ммоль) и Ν,Ν-диизопропилэтиламин (1,372 мл, 7,87 ммоль). Через 30 минут реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия, рассолом и смесью 1/1 рассол/вода дважды. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили указанное в заголовке соединение в виде светло-розовой пены (0,427 г, 36%).
МС: 601 ЭРИ+ (C33H36N4O7)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.80 (m, 3Н); 3.20 (m, 2Н); 3.74 (s, 3Н); 3.76 (s, 3Н); 3.80 (m, 1Н); 4.28 (m, 3Н); 4.38 (m, 2Н); 5.05 (s, 2Н); 5.08 (s, 2Н); 5.26 (m, 2Н); 5.49 (m, 1Н); 5.95 (m, 1Н); 6.93 (m, 4Н); 7.03 (s, 1Н); 7.35 (m, 4Н); 8.09 (s, 1Н); 8.53 (m, 1Н).
Промежуточное соединение 67: (2S,5R)-2-((4,5-бис(4-метокси-бензилокси)пиридин-2-ил)метилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил (проп-1-енил)-фосфония
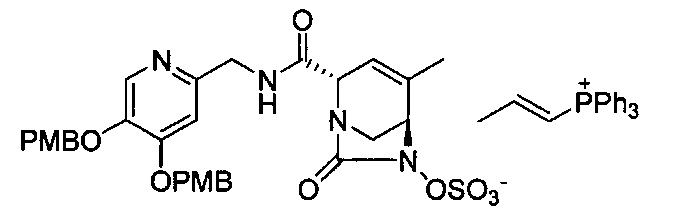
В раствор (2S,5R)-6-(аллилокси)-N-((4,5-бис(4-метоксибензилокси)-пиридин-2-ил)метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 66, 0,427 г, 0,71 ммоль) и уксусной кислоты (0,081 мл, 1,42 ммоль) (высушенной над сульфатом натрия) в DCM (10 мл) при комнатной температуре добавляли тетракис(трифенилфосфин)-палладий(0) (0,821 г, 0,71 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли пиридин (10,00 мл) и комплекс триоксид серы-пиридин (0,679 г, 4,27 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре. Суспензию выпаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат концентрировали и наносили на силикагелевую (24 г) колонку RediSep через фильтр Nalgene 0,45 мкм. После хроматографии на силикагеле (0%-100% ацетона/DCM) получили указанное в заголовке соединение в виде желтой пены (0,393 г, 59%).
МС: 639 ЭРИ-, 303 ЭРИ+ (C30H32N4O10S, С21Н20Р)
Промежуточное соединение 68: натриевая соль (2S,5R)-2-((4,5-бис(4-метоксибензилокси)пиридин-2-ил)метилкарбамоил)-4-метил-7-оксо-1.6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
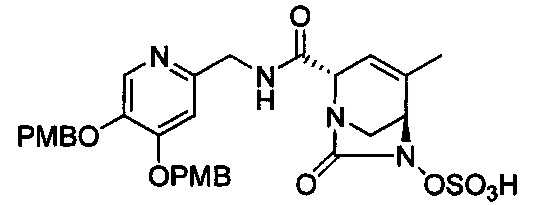
Ионообменную смолу Dowex(R) 50WX8-100 (35 г) кондиционировали путем перемешивания в течение 3 часов в 2 н. гидроксиде натрия (80 мл). Смолу затем загружали в стеклянную колонку (2×12 дюймов (5,08×30,48 см)) и промывали водой до рН 7. Затем ее промывали смесью (1/1) ацетон/вода (примерно 500 мл), затем водой (примерно 500 мл). (2S,5R)-2-((4,5-Бис(4-метоксибензилокси)пиридин-2-ил)метилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 67, 0,393 г, 0,42 ммоль) переносили в ацетон (примерно 2 мл) и разбавляли водой (примерно 4 мл). Желтый раствор наносили на смолу и элюировали водой. Указанное в заголовке соединение было получено после лиофилизации в виде не совсем белого твердого вещества (124 мг, 45%).
МС: 641 ЭРИ+ (C30H32N4O10S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.79 (m, 3Н); 3.23 (m, 2Н); 3.75 (m, 6Н); 3.98 (m, 1Н); 4.27 (m, 3Н); 5.07 (m, 4Н); 5.47 (m, 1Н); 6.93 (m, 4Н); 7.03 (s, 1Н); 7.34 (m,4H); 8.10 (s, 1H).
ПРИМЕР 9
Натриевая соль (2S,5R)-2-карбамоил-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
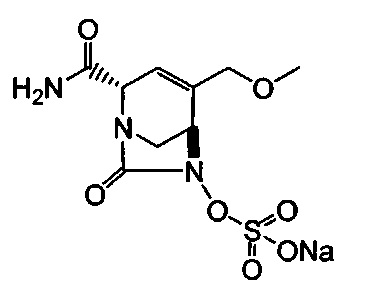
Ионообменную смолу Dowex(R) 50WX8-100 (10 г) кондиционировали путем перемешивания в течение 3 часов в 2 н. гидроксиде натрия (30 мл). Смолу затем загружали в картридж и промывали водой до рН 7. Затем ее промывали смесью (1/1) ацетон/вода (примерно 100 мл), затем водой (примерно 100 мл). (2S,5R)-2-Карбамоил-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 76, 131 мг, ммоль) переносили в воду (примерно 1 мл) и минимум ацетонитрила. Желтый раствор наносили на смолу и промывали водой. Указанное в заголовке соединение было получено после лиофилизации в виде не совсем белого твердого вещества (38 мг, 50%).
МС: 308 ЭРИ+ (C9H12N3NaO7S)
1Н ЯМР (300 МГц, D2O) δ: 3.43 (d, 1Н); 3.53 (s, 3Н); 3.59 (m, 1Н); 3.80 (m, 1Н); 4.24 (q, 2Н); 4.45 (m, 1Н); 6.11 (m, 1Н).
Промежуточное соединение 69: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
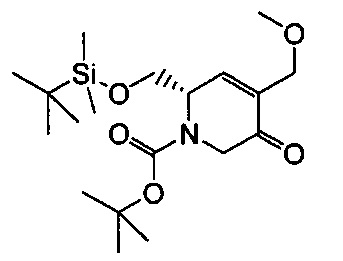
В перемешиваемый раствор (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(гидроксиметил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (17,37 г, 46,75 ммоль) в DCM (480 мл) добавляли N1,N1,N8,N8-тетраметилнафталин-1,8-диамин (60,1 г, 280,51 ммоль). Затем его охлаждали до 0°С и добавляли тетрафторборат триметилоксония (20,74 г, 140,25 ммоль). Его затем перемешивали при комнатной температуре в течение 6 ч. Затем его концентрировали при пониженном давлении. Остаток затем переносили в 100 мл Et2O, фильтровали и промывали 400 мл Et2O. Органический слой затем промывали 10%-ной лимонной кислотой, водным NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали с получением целевого продукта (16,73 г, 93%) в виде масла.
МС: 386 ЭРИ+ (C19H35NO5Si)
1Н ЯМР (300 МГц, CDCl3) δ: 0.01 (s, 6Н); 0.81 (s, 9Н); 1.42 (s, 9Н); 3.28 (s, 3Н); 3.91 (m, 5Н); 4.33 (d, 1Н); 4.78 (m, 1Н); 7.01 (s, 1Н).
Промежуточное соединение 70: (2S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(метоксиметил)-5.6-дигидропиридин-1(2Н)-карбоксилат
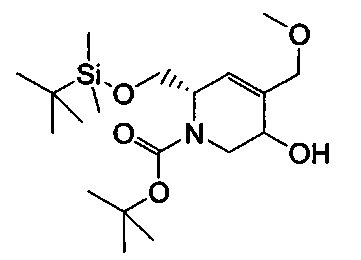
(S)-трет-Бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 69, неочищенное, 16,73 г, 43,39 ммоль) растворяли в МеОН (100 мл), охлаждали до 0°С и добавляли CeCl3 (10,69 г, 43,39 ммоль) с получением раствора. Затем медленно добавляли NaBH4 (1,642 г, 43,39 ммоль) в виде твердого вещества, и эту смесь перемешивали при температуре от 0°С до комнатной температуры в течение 30 мин. Летучий растворитель удаляли. Белое твердое вещество растворяли в 200 мл EtOAc и промывали насыщенным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (0-100% ЕА/Нех) с получением 13,78 г, 82% в виде бесцветного масла.
МС: 410 ЭРИ+ (C19H37NO5Si+Na+)
1Н ЯМР (300 МГц, CDCl3) δ: 0.04 (s, 6Н); 0.89 (s, 9Н); 1.46 (s, 9Н); 3.10 (m, 1Н); 3.36 (s, 3Н); 3.68 (m, 1Н); 3.72 (m, 1Н); 4.11 (m, 2Н); 4.24 (m, 2Н); 4.39 (m, 1Н); 5.75 (m, 1Н).
Промежуточное соединение 71: (2S,5R)-трет-бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5,6-дигидропиридин-1 (2Н)-карбоксилат
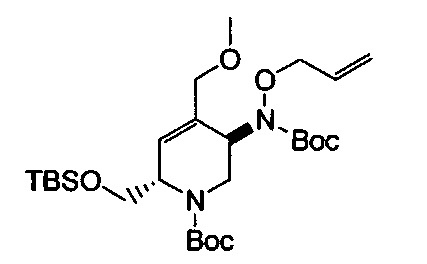
В перемешиваемый раствор (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 70, 13,78 г, 35,55 ммоль) в DCM (200 мл) при 0°С добавляли пиридин (14,38 мл, 177,77 ммоль) и N,N-диметилпиридин-4-амин (217 мг, 1,78 ммоль). Затем добавляли метансульфоновый ангидрид (9,29 г, 53,33 ммоль). Смесь затем перемешивали при температуре от 0°С до комнатной температуры в течение 2 часов. Смесь разбавляли DCM (200 мл) и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5-(метилсульфонилокси)-5,6-дигидропиридин-1(2Н)-карбоксилата (неочищенного, 16,9 г) в виде бледно-желтого масла. Его напрямую использовали на следующей стадии.
В перемешиваемый раствор трет-бутил-аллилоксикарбамата (7,39 г, 42,66 ммоль) в DMF (150 мл) при комнатной температуре добавляли 2-метилпропан-2-олат калия (42,66 мл, 42,66 ммоль) и получили пурпурный раствор. После 30 минут при комнатной температуре смесь охлаждали до 0°С и добавлял и (2S, 5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5-(метилсульфонилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат (неочищенный, 16,55 г, 35,55 ммоль) в 50,0 мл DMF. Смесь затем нагревали до комнатной температуры в течение 1 ч. Затем ее разбавляли этилацетатом (200 мл) и промывали насыщенным водным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали с получением остатка, который содержит некоторое количество исходного вещества. Это неочищенное вещество очищали на силикагелевой колонке с получением бесцветного масла (19,3 г). 1Н ЯМР-анализ показал все еще смесь (с исходным гидроксиамином). Ее напрямую использовали в процессе удаления защитной группы TBS.
МС: 565 ЭРИ+ (C27H50N2O7Si+Na+)
1Н ЯМР (300 МГц, CDCl3) δ: 0.04 (s, 6Н); 0.89 (s, 9Н); 1.47 (s, 9Н); 1.52 (s, 9Н); 3.14 (m, 1Н); 3.31 (s, 3Н); 3.72 (m, 3Н); 4.15 (m, 3Н); 4.44 (m, 3Н); 5.28 (m, 2Н); 5.97 (m, 2Н).
Промежуточное соединение 72: (2S,5R)-трет-бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-(гидроксиметил)-4-(метоксиметил)-5.6-дигидропиридин-1(2Н)-карбоксилат
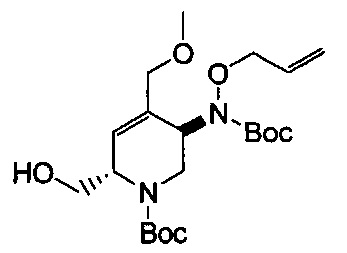
В перемешиваемый раствор (2S,5R)-трет-бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 71, 35,55 ммоль) в THF (100 мл) при 0°С добавляли TBAF (39,11 мл, 39,11 ммоль). После 1 ч при 0°С раствор концентрировали с получением остатка, который очищали на силикагелевой колонке (0-100% ЕА/Нех) с получением целевого продукта (8,82 г, 57,9% за 3 стадии) в виде бесцветного масла.
МС: 451 ЭРИ+ (C21H36N2O7+Na+)
1Н ЯМР (300 МГц, CDCl3) δ: 1.47 (s, 9Н); 1.52 (s, 9Н); 3.14 (dd, 1Н); 3.34 (s, 3Н); 3.71 (m, 3Н); 3.98 (d, 1Н), 4.18 (m, 2H); 4.40 (m, 2H); 4.67 (m, 1H); 5.20 (m, 2H); 5.79 (m, 1H); 5.97 (s, 1H).
Промежуточное соединение 73: (2S,5R)-трет-бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-карбамоил-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
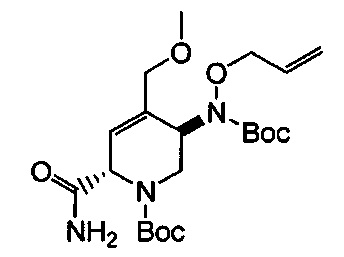
Исходный раствор для окисления: примерно 480 мг конц. HNO3 и примерно 160 мг Na2Cr2O7-2Н2О растворяли в 32 мл Н2О при комнатной температуре.
В перемешиваемый раствор (2S,5R)-трет-бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-(гидроксиметил)-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 72, 8,82 г, 20,58 ммоль) в 100 мл MeCN при 0°С добавляли перйодат натрия (19,37 г, 90,56 ммоль). Затем добавляли 12 мл исходного раствора для окисления. Суспензию затем перемешивали при комнатной температуре в течение 2 суток. Добавляли еще 16 мл вышеуказанного исходного раствора и перемешивали в течение более чем 2 суток. Смесь разбавляли 150 мл этилацетата, 50 мл 1М буфера, рН 7, и 50 мл 2М NaHSO3. Водную фазу экстрагировали 20 мл этилацетата. Этилацетатный слой затем промывали рассолом. Водный слой проверяли методом ЖХ/МС и обнаружили, что он содержит целевую карбоновую кислоту. Водный слой затем экстрагировали 50 мл этилацетата и промывали рассолом. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением желтой сухой пленки (неочищенный продукт, 7,11 г, 78%), которую использовали без дополнительной очистки.
В перемешиваемый раствор (2S,5R)-5-(аллилокси(трет-бутоксикарбонил)амино)-1-(трет-бутоксикарбонил)-4-(метоксиметил)-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (неочищенной, 7,11 г, 16,07 ммоль), хлорида аммония (1,719 г, 32,14 ммоль), HATU (12,22 г, 32,14 ммоль) в DMF (50,0 мл) при комнатной температуре добавляли DIPEA (8,31 г, 64,27 ммоль). После перемешивания при комнатной температуре в течение 1 ч добавляли 100 мл EtOAc. Органический слой промывали водой, рассолом. Остаток очищали на силикагелевой колонке (0-100% Нех/ЕА) с получением целевого продукта (3,07 г, 43,3%) в виде не совсем белого твердого вещества.
МС: 442 ЭРИ+ (C21H35N3O7)
1Н ЯМР (300 МГц, CD3OD) δ: 1.47 (s, 9Н); 1.52 (s, 9Н); 3.17 (d, 1Н); 3.34 (s, 3Н); 4.04 (m, 1Н); 4.12 (m, 4Н); 4.44 (m, 2Н); 5.21 (m, 2Н); 5.77 (m, 1Н); 6.22 (s, 1Н).
Промежуточное соединение 74: (2S,5R)-5-(аллилоксиамино)-4-(метоксиметил)-1,2,5,6-тетрагидропиридин-2-карбоксамид
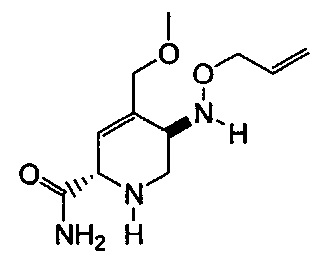
(2S,5R)-трет-Бутил-5-(аллилокси(трет-бутоксикарбонил)амино)-2-карбамоил-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 73, 3,07 г, 6,95 ммоль) растворяли в 20 мл DCM. Затем по каплям добавляли 5,36 мл TFA при 0°С. Смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли в вакууме и совыпаривали дважды с 5 мл МеОН. Остаток растворяли в МеОН (10 мл) и добавляли гидроксид аммония (30% в воде) до тех пор, пока смесь не стала основной. Смесь упаривали на роторном испарителе при комнатной температуре, и остаток подвергали сублимационной сушке в течение ночи с получением твердого вещества. Его растворяли в DCM и очищали на силикагеле, элюируя смесью 0-100% МеОН/DCM, с получением не совсем белого твердого вещества (1,12 г, 66,8%).
МС: 242 ЭРИ+ (C11H19N3O3)
1Н ЯМР (300 МГц, CD3OD) δ: 3.31 (s, 3Н); 3.55 (m, 3Н); 4.05 (m, 4Н); 4.58 (m, 1Н); 5.20 (m, 2Н); 5.96 (m, 2Н).
Промежуточное соединение 75: (2S,5R)-6-(аллилокси)-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
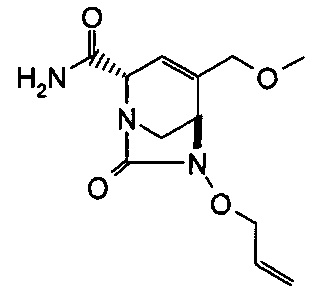
В перемешиваемый раствор (2S,5R)-5-(аллилоксиамино)-4-(метоксиметил)-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 74, 1,12 г, 4,64 ммоль) в ацетонитриле (100 мл) добавляли Ν-этил-N-изопропилпропан-2-амин (4,04 мл, 3,00 г, 23,21 ммоль), затем медленно добавляли трифосген (551 мг, 1,86 ммоль) в 20 мл ацетонитрила посредством шприцевого насоса в течение 4 ч при 0°С. Раствор перемешивали при температуре от 0°С до комнатной температуры в течение ночи. Раствор концентрировали с получением остатка, который переносили в 50 мл EtOAc и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, с получением желтого масла (420 мг, 34%).
МС: 268 ЭРИ+ (C12H17N3O4)
1Н ЯМР (300 МГц, CD2Cl2) δ: 3.03 (d, 1Н); 3.28 (s, 3Н); 3.40 (m, 2Н); 3.95 (m, 3Н); 4.38 (m, 2Н); 5.30 (m, 2Н); 5.98 (m, 2Н); 6.83 (bs, 2Н).
Промежуточное соединение 76: (2S,5R)-2-карбамоил-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
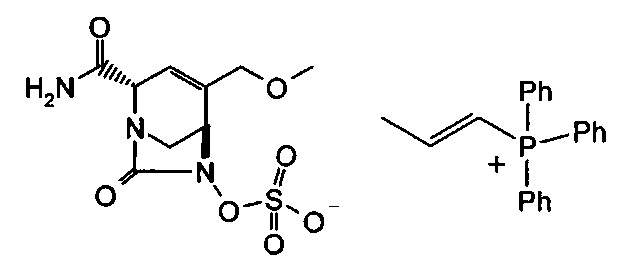
В раствор (2S,5R)-6-(аллилокси)-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 75, 120 мг, 0,45 ммоль) и уксусной кислоты (0,051 мл, 0,90 ммоль) (высушенной над сульфатом натрия) в DCM (4,0 мл) при комнатной температуре добавляли тетракис(трифенилфосфин)палладий(0) (519 мг, 0,45 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли пиридин (2,0 мл) и комплекс триоксид серы-пиридин (429 мг, 2,69 ммоль). Суспензию перемешивали в течение ночи при комнатной температуре. Суспензию выпаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат концентрировали и наносили на силикагелевую (24 г) колонку RediSep через фильтр Nalgene 0,45 мкм. После хроматографии на силикагеле (0%-100% ацетона/DCM) получили указанное в заголовке соединение в виде желтой пены (0,131 г, 48%).
МС: 304 ЭРИ+ (C30H32N3O7PS, C21H20P)
ПРИМЕР 10
Натриевая соль (2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
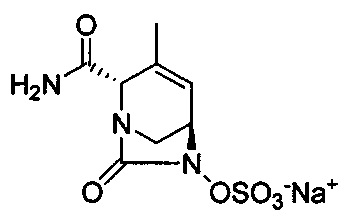
Ионообменную смолу Dowex(R) 50WX8-100 (25 г, 0,33 ммоль) кондиционировали путем перемешивания в течение 3 часов в 2 н. гидроксиде натрия (61 мл, 0,33 ммоль). Смолу затем загружали в картридж и промывали водой до рН 7. Затем ее промывали смесью (1/1) ацетон/вода, затем водой. (2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 87, 68 мг, 0,12 ммоль) переносили в воду. По каплям добавляли ацетон до тех пор, пока все вещество не перешло в раствор. Желтый раствор наносили на смолу и промывали водой. Фракции, содержащие целевой продукт, объединяли и лиофилизировали (25 мг, 79%) с получением белого твердого вещества.
Оптическое вращение: (0,1 г/дл, МеОН)=-287.
МС: 276 ЭРИ- (C8H10N3O6SNa)
1Н ЯМР (300 МГц, ОКСИД ДЕЙТЕРИЯ) δ: 1.76 (dd, J=1.41, 0.85 Гц, 3Η) 3.40-3.49 (m, 1Η) 3.50-3.58 (m, 1Η) 4.27 (dd, J=5.09, 2.45 Гц, 1Η) 4.44 (s, 1Η) 6.23-6.31 (m, 1Η).
Промежуточное соединение 77: (6S)-трет-бутил-6-((трет-бутилдиметилсилилокси)метил)-5-метил-3-(триметилсилилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат
Метиллитий в Et2O (73,2 мл, 117,12 ммоль) по каплям добавляли в течение 20 минут в суспензию йодида меди(I) (11,15 г, 58,56 ммоль) в Et2O (160 мл) и перемешивали при 0°С под азотом. Через 45 минут по каплям добавляли (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат (10 г, 29,28 ммоль) в Et2O (20 мл), и перемешивание продолжали в течение 45 мин. Добавляли TMs-Cl в THF (58,6 мл, 58,56 ммоль), затем добавляли триэтиламин (8,16 мл, 58,56 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч, разбавляли этилацетатом, промывали ледяным насыщенным раствором NaHCO3 (3х) и рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением целевого продукта в виде неочищенного желтого масла (примерно 12,58 г, 29,27 ммоль).
МС: 330 ЭРИ+ (C21H43NO4Si2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.05-0.08 (m, 6Η) 0.21 (br. s., 9Η) 0.89 (br. s., 9H) 1.04 (d, J=6.40 Гц, 2Η) 1.48 (s, 9Η) 2.34 (br. s., 1H) 3.42 (br. s., 2H) 3.54 (dd, J=7.44, 3.67 Гц, 1Η) 3.91-4.04 (m, 1Η) 4.07-4.20 (m, 1Η) 4.87 (br. s., 1H)
Промежуточное соединение 78: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-метил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
Неочищенный (6S)-трет-бутил-6-((трет-бутилдиметилсилилокси)-метил)-5-метил-3-(триметилсилилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 77, 12,58 г, 29,27 ммоль) в 8 мл ACN перемешивали при комнатной температуре с Pd(OAc)2 (6,57 г, 29,27 ммоль) в течение 2 суток. Смесь разбавляли 160 мл EtOAc, фильтровали через целит, концентрировали в вакууме и подвергали флэш-хроматографии (220 г, 0-30% ЕА/Нех) с получением целевого продукта (6,07 г, 58,3%) (за две стадии) в виде бежевого твердого вещества.
МС: 256 ЭРИ+ (C18H33NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.02-0.07 (m, 6Η) 0.80-0.91 (m, 9Η) 1.49 (s, 9Η) 2.04 (d, J=1.13 Гц, 3Η) 3.69-4.06 (m, 3H) 4.32-4.73 (m, 2Η) 6.08 (s, 1Η)
Промежуточное соединение 79: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3-метил-5,6-дигидропиридин-1 (2Н)-карбоксилат
В перемешиваемый раствор гептагидрата хлорида церия(III) (6,36 г, 17,07 ммоль) и (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-метил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 78, 6,07 г, 17,07 ммоль) в МеОН (100 мл) при 0°С добавляли тетрагидроборат натрия (0,646 г, 17,07 ммоль) в виде твердого вещества. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали и разбавляли NH4Cl (водн.), Н2О и экстрагировали диэтиловым эфиром. Эфирный слой отделяли и промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением целевого продукта (5,48 г, 90%) в виде желтого масла.
МС: 258 ЭРИ+ (C18H35NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06 (s, 6Η) 0.78-0.94 (m, 9Η) 1.38-1.50 (m, 9Η) 1.56 (br. s., 1Η) 1.78 (s, 3Η) 2.90-3.39 (m, 1Η) 3.67-4.28 (m, 5Η) 5.80 (br. s., 1H).
Промежуточное соединение 80: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
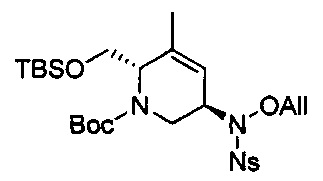
В перемешиваемую суспензию (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 79, 5,48 г, 15,33 ммоль), Ν-(аллилокси)-2-нитробензолсульфонамида (7,92 г, 30,65 ммоль) и трифенилфосфина (12,06 г, 45,98 ммоль) в толуоле (20 мл), охлажденную в ледяной бане по каплям добавляли (Е)-диизопропил-диазен-1,2-дикарбоксилат (8,91 мл, 45,98 ммоль). Реакционной смеси давали нагреться до комнатной температуры, и перемешивание продолжали при комнатной температуре в течение 2 ч. Реакционную смесь упаривали, и неочищенный продукт наносили на силикагель и очищали флэш-хроматографией (750 г, 0-50%) с получением целевого продукта (7,05 г, 77%) в виде желтого масла.
МС: 598 ЭРИ+ (C27H43N3O8SSi)
Промежуточное соединение 81: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
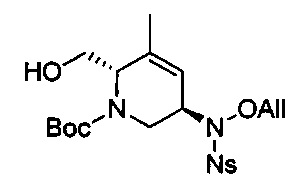
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 80, 7,05 г, 11,79 ммоль) в THF (100 мл) насыщали азотом и охлаждали в ледяной бане. В этот раствор добавляли фторид тетрабутиламмония в THF (14,15 мл, 14,15 ммоль), и смесь перемешивали при комнатной температуре. Реакционную смесь упаривали, и неочищенный продукт наносили на силикагель и очищали флэш-хроматографией (30-100% ЕА/гексаны, 40 г колонка) с получением целевого продукта (4,52 г, 79%) в виде бледно-желтой пены.
МС: 484 ЭРИ+ (C21H29N3O8S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.43 (br. s., 9Η) 1.80 (br. s., 3Η) 3.35 (br. s., 1Η) 3.73 (d, J=6.22 Гц, 1Η) 3.88 (br. s., 1H) 4.35 (br. s., 5H) 5.15-5.29 (m, 2H) 5.45 (s, 1H) 5.77 (br. s., 1H) 7.61 (d, J=7.35 Гц, 1Η) 7.76 (dd, J=12.62, 7.54 Гц, 2Η) 8.13 (d, J=7.72 Гц, 1Η)
Промежуточное соединение 82: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
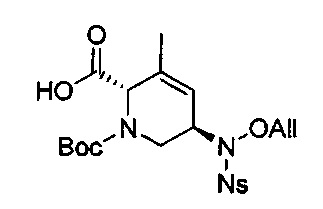
В раствор перйодной кислоты (4,10 г, 17,99 ммоль) во влажном ацетонитриле (25 мл) (0,75% воды по объему) при комнатной температуре добавляли оксид хрома(VI) (0,490 г, 4,90 ммоль). Смесь перемешивали до полного растворения. В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 81, 4,52 г, 9,35 ммоль) во влажном ацетонитриле (25 мл) (0,75% по объему) при 0°С по каплям добавляли ранее образованный раствор перйодной кислоты/оксида хрома. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли CHCl3 и промывали смесью конц. лимонная кислота/вода и затем рассолом (2х). Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта (3,98 г, 86%) в виде бежевой пены.
МС: 498 ЭРИ+ (C21H27N3O9S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.43 (s, 9Η) 1.88 (br. s., 3Η) 3.43 (d, J=15.07 Гц, 1Η) 4.09-4.51 (m, 5Η) 4.60-4.92 (m, 1Η) 5.14-5.28 (m, 2Η) 5.46 (br. s., 1H) 5.74 (br. s., 1H) 7.63 (d, J=6.03 Гц, 1Η) 7.69-7.85 (m, 2Η) 8.12 (d, J=7.72 Гц, 1Η).
Промежуточное соединение 83: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилат
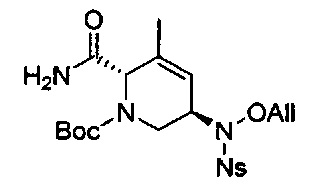
В раствор (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 82, 3,98 г, 8,00 ммоль) в DMF (20 мл) при комнатной температуре добавляли хлорид аммония (0,856 г, 16,00 ммоль), гексафторфосфат(V) 2-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (4,56 г, 12,00 ммоль) и N-этил-N-изопропилпропан-2-амин (5,57 мл, 32,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия и три раза смесью 1:1 рассол:вода. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (220 г, 10%-80% этилацетата/гексаны) с получением целевого продукта (2,79 г, 70,2%) в виде желтой пены.
МС: 497 ЭРИ+ (C21H28N4O8S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.40-1.48 (m, 9Η) 1.81 (s, 3Η) 3.34 (d, J=12.43 Гц, 1Η) 4.19-4.53 (m, 4Η) 4.84 (br. s., 1Η) 5.14-5.38 (m, 3Η) 5.52 (br. s., 1H) 5.74 (br. s., 1H) 6.45 (br. s., 1H) 7.58-7.64 (m, 1H) 7.76 (dtd, J=14.01, 7.69, 7.69, 6.22 Гц, 2Η) 8.11 (d, J=7.72 Гц, 1Η)
Промежуточное соединение 84: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоксамид
К (2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-метил-5,6-дигидропиридин-1(2Н)-карбоксилату (Промежуточное соединение 83, 2,785 г, 5,61 ммоль) и бромиду цинка(II) (2,53 г, 11,22 ммоль) добавляли DCM (10 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта (2,015 г, 91%) в виде бежевой пены.
МС: 397 ЭРИ+ (C16H20N4O6S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.97 (s, 3Η) 2.96 (br. s., 2Η) 3.77 (br. s., 1H) 4.29 (br. s., 1H) 4.34-4.52 (m, 2H) 5.21-5.44 (m, 4H) 5.55 (br. s., 1H) 5.75-5.94 (m, 1H) 7.13 (br. s., 1H) 7.63 (dd, J=7.82, 1.41 Гц, 1Η) 7.70-7.88 (m, 2Η) 8.15 (dd, J=7.72, 1.51 Гц, 1Η)
Промежуточное соединение 85: (2S,5R)-5-(аллилоксиамино)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоксамид
К (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоксамиду (Промежуточное соединение 84, 2,01 г, 5,07 ммоль) и карбонату калия (2,172 г, 15,72 ммоль) в ацетонитриле (10 мл) добавляли бензолтиол (1,557 мл, 15,21 ммоль), и смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали при 30°С, растворяли в DCM и небольшой порции МеОН, фильтровали и наносили на силикагель при 30°С. Неочищенную смесь очищали флэш-хроматографией (колонка 220 г, 0-20% MeOH/DCM), фракции концентрировали при 35°С и сушили в глубоком вакууме с получением целевого продукта (0,759 г, 70,9%) в виде не совсем белого твердого вещества.
МС: 212 ЭРИ+ (C10H17N3O2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.94 (s, 3Η) 2.93-3.09 (m, 2Η) 3.40 (br. s., 1Η) 3.78 (s, 1Η) 4.17-4.23 (m, 2Η) 5.19-5.25 (m, 1Η) 5.25-5.33 (m, 1Η) 5.37 (br. s., 1H) 5.53 (br. s., 1H) 5.94 (ddt, J=17.00, 10.60, 5.98, 5.98 Гц, 1Η) 7.14 (br. s., 1H)
Промежуточное соединение 86: (2S,5R)-6-(аллилокси)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
Раствор (2S,5R)-5-(аллилоксиамино)-3-метил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 85, 0,758 г, 3,59 ммоль) и N-этил-N-изопропилпропан-2-амина (2,507 мл, 14,35 ммоль) в ацетонитриле (350 мл) охлаждали до температуры ниже 0°С в бане лед-соль, и в него добавляли раствор бис(трихлорметил)карбоната (0,426 г, 1,44 ммоль) в ACN (15 мл) со скоростью 0,1 мл/мин. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворители выпаривали при 30°С, и неочищенную смесь растворяли в EtOAc. Неочищенный органический раствор промывали насыщенным раствором NaHCO3, рассолом, сушили над MgSO4 и фильтровали. Органические фазы упаривали, и неочищенный продукт наносили на силикагель при 30°С и очищали флэш-хроматографией (80 г, 0-100% EtOAc/гексаны) с получением целевого продукта (0,700 г, 82%) в виде бледно-желтого масла.
МС: 238 ЭРИ+ (C11H15N3O3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.92 (d, J=0.75 Гц, 3Η) 3.12-3.20 (m, 1Η) 3.26-3.34 (m, 1Η) 3.82 (dd, J=4.99, 2.73 Гц, 1Η) 4.30 (s, 1Η) 4.34-4.50 (m, 2Η) 5.28-5.41 (m, 2Η) 5.44 (br. s., 1Η) 5.94-6.09 (m, 1Η) 6.09-6.15 (m, 1Η) 6.68 (br. s., 1Η)
Промежуточное соединение 87: (2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)-фосфония
В раствор (2S,5R)-6-(аллилокси)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 86, 200 мг, 0,84 ммоль) и АсОН (0,097 мл, 1,69 ммоль) (высушенного над сульфатом натрия) в CH2Cl2 (7 мл) при комнатной температуре добавляли Pd(Ph3P)4 (450 мг, 0,39 ммоль). Раствор перемешивали при комнатной температуре в течение 45 мин. В эту реакционную смесь добавляли пиридин (7,00 мл) и комплекс триоксида серы с пиридином (496 мг, 3,12 ммоль), и перемешивание продолжали в атмосфере N2 при комнатной температуре в течение ночи. Суспензию выпаривали досуха при 39°С и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,45 мкм. Фильтрат наносили на колонку. После хроматографии на силикагеле (80 г колонка, 0%-100% ацетона/DCM) получили целевой продукт (68,0 мг, 13,92%) в виде не совсем белой пены.
МС: 276 ЭРИ- (C29H30N3O6PS)
ПРИМЕР 11
Натриевая соль (2S,5R)-2-карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
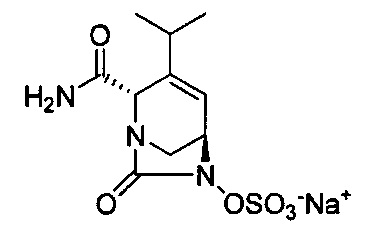
(2S,5R)-2-Карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат (4,02 мг, 89%) был получен способом, аналогичным способу, описанному в Примере 10, в виде белого твердого вещества, используя (2S,5R)-2-карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 98, 9 мг, 0,12 ммоль).
МС: 304 ЭРИ- (C10H14N3O6SNa)
1Н ЯМР (600 МГц, ОКСИД ДЕЙТЕРИЯ) δ: 0.95 (d, J=7.15 Гц, 3Η) 1.01 (d, J=6.78 Гц, 3Η) 2.13 (spt, J=6.78 Гц, 1Η) 3.38 (dd, J=11.29, 2.26 Гц, 1Η) 3.54 (d, J=11.29 Гц, 1Η) 4.27 (dd, J=5.27, 2.64 Гц, 1Η) 4.49 (s, 1Η) 6.26 (d, J=5.27 Гц, 1Η)
Промежуточное соединение 88: (6S)-трет-бутил-6-((трет-бутилдиметилсилилокси)метил)-5-изопропил-3-(триметилсилилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат
(2S,5R)-2-Карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат (теоретически 6,7 г) был получен способом, аналогичным способу, описанному для Промежуточного соединения 77, в виде желтого масла, используя (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 50, 5 г, 14,64 ммоль) в качестве исходного вещества.
МС: 330 ЭРИ+ (C23H47NO4Si2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.05-0.08 (m, 6Η) 0.21 (br. s., 9Η) 0.89 (br. s., 9H) 1.04 (d, J=6.40 Гц, 2Η) 1.48 (s, 9Η) 2.34 (br. s., 1H) 3.42 (br. s., 2H) 3.54 (dd, J=7.44, 3.67 Гц, 1Η) 3.91-4.04 (m, 1Η) 4.07-4.20 (m, 1Η) 4.87 (br. s., 1H)
Промежуточное соединение 89: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-изопропил-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилат
(S)-трет-Бутил-2-((трет-бутилдиметилсилилокси)метил)-3-изопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат (804 мг, 14,32%) (за две стадии) был получен, как описано для Промежуточного соединения 78, в виде желтого масла, используя (6S)-трет-бутил-6-((трет-бутилдиметилсилилокси)метил)-5-изопропил-3-(триметилсилилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 88, теоретически 6,7 г, 14,64 ммоль).
МС: 384 ЭРИ+ (C20H37NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.01 (d, J=3.96 Гц, 6Η) 0.84 (s, 9Η) 1.14-1.23 (m, 6Η) 1.49 (s, 9Η) 2.35-2.50 (m, 1Η) 3.77-3.94 (m, 2H) 3.96-4.05 (m, 1Η) 4.32-4.85 (m, 2Η) 6.05-6.14 (m, 1Η)
Промежуточное соединение 90: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
(2S,5S)-трет-Бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат (0,732 г, 91%), был получен способом, аналогичным способу, описанному для Промежуточного соединения 79, в виде бесцветного масла, используя (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)-метил)-3-изопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 89, 804 мг, 2,10 ммоль) и N-(аллилокси)-2-нитробензолсульфонамид (0,979 г, 3,79 ммоль) в качестве исходных веществ.
МС: 286 ЭРИ+ (C20H39NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06 (d, J=1.51 Гц, 6Η) 0.89 (s, 9Η) 1.09 (dd, J=10.55, 6.78 Гц, 6Η) 1.48 (s, 9Η) 2.13-2.26 (m, 1Η) 2.86 (br. s., 1Η) 3.36-3.52 (m, 1Η) 3.60-3.78 (m, 2Η) 3.82-4.42 (m, 3Η) 5.73-5.91 (m, 1Η)
Промежуточное соединение 91: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-изопропил-5,6-дигидропиридин-1 (2Н)-карбоксилат
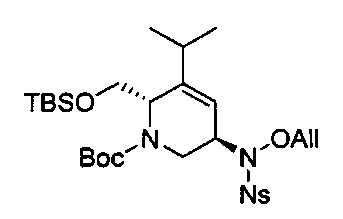
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-изопропил-5,6-дигидропиридин-1 (2Н)-карбоксилат (0,928 г, 78%) был получен, как описано для Промежуточного соединения 80, в виде бледно-желтого масла, используя (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3-изопропил-5,6-дигидропиридин-1 (2Н)-карбоксилат (Промежуточное соединение 90, 0,731 г, 1,90 ммоль) в качестве исходного вещества.
МС: 626 ЭРИ+ (C29H47N3O8SSi)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.01 (s, 6Η) 0.86 (s, 9Η) 0.95-1.09 (m, 6Η) 1.42 (br. s., 9Η) 2.14-2.37 (m, 1Η) 3.50 (dd, J=14.60, 4.43 Гц, 1Η) 3.64-3.89 (m, 2Η) 4.21-4.61 (m, 5Η) 5.12-5.53 (m, 3Η) 5.66-5.88 (m, 1Η) 7.61 (d, J=7.72 Гц, 1Η) 7.68-7.83 (m, 2Η) 8.13 (d, J=8.10 Гц, 1Η)
Промежуточное соединение 92: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
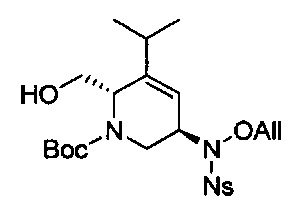
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3-изопропил-5,6-дигидропиридин-1 (2Н)-карбоксилат (660 мг, 87%) был получен способом, аналогичным способу, описанному для Промежуточного соединения 81, в виде желтого масла, используя (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 91, 0,928 г, 1,48 ммоль) в качестве исходного вещества.
МС: 512 ЭРИ+ (C23H33N3O8S)
Промежуточное соединение 93: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
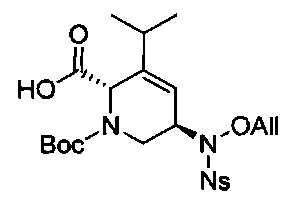
((2S,5R)-5-(N-(Аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота (0,595 г, 87,76%) была получена способом, аналогичным способу, описанному для Промежуточного соединения 82, в виде желтого масла, используя (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 92, 660 мг, 1,29 ммоль) в качестве исходного вещества.
МС: 526 ЭРИ+ (C23H31N3O9S)
1Н ЯМР (300 МГц, DMSO-d) δ: 0.62-1.02 (m, 6Η) 1.36 (br. s, 9Η) 2.78-3.26 (m, 1Η) 3.34-3.62 (m, 1Η) 3.65-4.10 (m, 1Η) 4.10-4.45 (m, 3Η) 4.46-4.84 (m, 1Η) 5.01-5.64 (m, 3Η) 5.64-5.99 (m, 1Η) 7.87-8.14 (m, 4Η) 13.03 (br. s, 1Η)
Промежуточное соединение 94: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
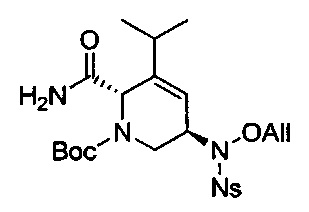
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат (0,260 г, 43,7%) был получен способом, аналогичным способу, описанному для Промежуточного соединения 83, в виде бледно-желтого масла, используя (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоновую кислоту (Промежуточное соединение 93, 0,595 г, 1,13 ммоль) в качестве исходного вещества.
МС: 525 ЭРИ+ (C23H32N4O8S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.02 (d, J=6.97 Гц, 6Η) 1.44 (br. s., 9Η) 2.24 (s, 1Η) 3.29-3.40 (m, 1Η) 4.18-4.50 (m, 4Η) 5.02 (br. s., 1H) 5.15-5.28 (m, 3H) 5.73 (s, 2H) 6.44 (s, 1H) 7.60-7.64 (m, 1H) 7.76 (dqd, J=14.76, 7.54, 7.54, 7.54, 1.60 Гц, 2Η) 8.11 (dd, J=7.91, 1.32 Гц, 1Η)
Промежуточное соединение 95: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
(2S,5R)-5-(N-(Аллилокси)-2-нитрофенилсульфонамидо)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид (160 мг, 86%) был получен способом, аналогичным способу, описанному для Промежуточного соединения 84, в виде бежевого твердого вещества, используя (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-изопропил-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 94, 260 мг, 0,5 ммоль) в качестве исходного вещества.
МС: 425 ЭРИ+ (C18H24N4IO6S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.01-1.12 (m, 6Η) 2.64-2.77 (m, 1Η) 2.82-3.06 (m, 2Η) 3.93 (s, 1Η) 4.33 (br. s., 1Η) 4.35-4.55 (m, 2Η) 5.21-5.31 (m, 3Η) 5.59 (br. s., 1H) 5.76-5.93 (m, 1H) 7.04 (br. s., 1H) 7.62 (dd, J=7.54, 1.51 Гц, 1Η) 7.71-7.85 (m, 2Η) 8.14(dd, J=7.82, 1.41 Гц, 1Η)
Промежуточное соединение 96: (2S,5R)-5-(аллилоксиамино)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
(2S,5R)-5-(Аллилоксиамино)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид (55,0 мг, 61,0%) был получен способом, аналогичным способу, описанному для Промежуточного соединения 85, в виде не совсем белого твердого вещества, используя (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфон-амидо)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид (Промежуточное соединение 95, 160 мг, 0,38 ммоль) в качестве исходного вещества.
МС: 340 ЭРИ+ (C12H21N3O2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.05 (d, J=6.97 Гц, 3Η) 1.11 (d, J=6.78 Гц, 3Η) 2.54-2.68 (m, 1Η) 2.94-3.11 (m, 2Η) 3.45 (br. s, 1Η) 3.95 (br. s., 1H) 4.20 (d, J=5.84 Гц, 2Η) 5.22 (d, J=10.36 Гц, 1Η) 5.29 (dd, J=17.14, 1.51 Гц, 1Η) 5.38 (br. s., 1Η) 5.58 (d, J=3.58 Гц, 1H) 5.87-6.02 (m, 1H) 7.00 (br. s., 1H)
Промежуточное соединение 97: (2S,5R)-6-(аллилокси)-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
(2S,5R)-6-(Аллилокси)-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид (0,46 мг, 77%) был получен способом, аналогичным способу, описанному для Промежуточного соединения 86, в виде бледно-желтого масла, используя (2S,5R)-5-(аллилоксиамино)-3-изопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид (Промежуточное соединение 96, 0,54 мг, 0,23 ммоль) в качестве исходного вещества.
МС: 266 ЭРИ+ (C13H19N3O3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.02 (d, J=6.78 Гц, 3Η) 1.10 (d, J=6.78 Гц, 3Η) 2.60 (quin, J=6.69 Гц, 1Η) 3.14-3.22 (m, 1Η) 3.24-3.32 (m, 1Η) 3.86 (dd, J=5.27, 2.83 Гц, 1Η) 4.33-4.50 (m, 3Η) 5.26-5.52 (m, 3Η) 5.94-6.15 (m, 2Η) 6.63 (br. s., 1Η)
Промежуточное соединение 98: (2S,5R)-2-карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
(2S,5R)-2-Карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (9,00 мг, 8,73%) был получен, способом, аналогичным способу, описанному для Промежуточного соединения 87, в виде бесцветного масла, используя (2S,5R)-6-(аллилокси)-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид (Промежуточное соединение 97, 45 мг, 0,17 ммоль) в качестве исходного вещества.
МС: 304 ЭРИ- (C31H34N3O6PS)
ПРИМЕР 12
Мононатриевая соль (2S,5R)-4-карбамоил-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
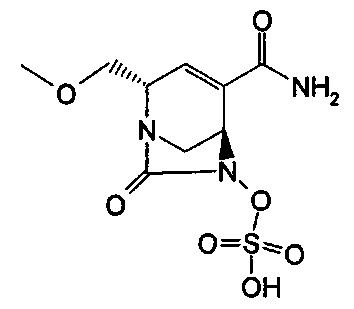
Готовили раствор (2S,5R)-6-гидрокси-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамида (Промежуточное соединение 116, 21 мг, 0,09 ммоль) в пиридине (2 мл) при перемешивании и помещали его в атмосферу азота. В него добавляли комплекс триоксид серы-пиридин (6 экв., 88 мг, 0,55 ммоль). Эту смесь перемешивали в течение ночи при температуре окружающей среды; смесь затем концентрировали при пониженном давлении, и остаток растворяли в небольшом объеме воды. Колонку Dowex подготавливали следующим образом: Dowex(R) 50WX8-100, ионообменную смолу (16 г) кондиционировали путем перемешивания в течение 3 часов в 2 н. гидроксиде натрия (34 мл). Смолу затем загружали в картридж и промывали водой до рН 7. Затем ее промывали смесью (1/1) ацетон/вода, затем снова водой. Водный раствор неочищенного продукта наносили на эту колонку; целевой продукт элюировали водой. Фракции, содержащие целевой продукт, объединяли и лиофилизировали. Полученное вещество растирали с метанолом, и фильтрат концентрировали и снова подвергали сублимационной сушке. Целевой продукт был получен в виде белого твердого вещества (19 мг, 62%).
МС: 308 ЭРИ+ (C9H13N3O7S)
1Н ЯМР (300 МГц, D2O) δ: 3.37-3.50 (m, 4Η) 3.51-3.63 (m, 2Η) 3.72-3.88 (m, 3Η) 4.26 (ddd, J=7.79, 4.67, 3.21 Гц, 1Η) 4.77 (d, J=1.51 Гц, 1Η) 6.61 (d, J=2.83 Гц, 1Η)
Промежуточное соединение 99: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-оксоэтил)карбамат
При перемешивании готовили раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 5, 28,36 г, 70,44 ммоль) в метиленхлориде (247 мл), охлаждали его в бане сухой лед/ацетон и помещали в атмосферу азота. В него добавляли гидрид диизобутилалюминия (1,0 Μ в метиленхлориде) (106 мл, 105,66 ммоль), и эту смесь выдерживали при этой температуре в течение примерно 2,5 часов. Реакцию контролировали методом ТСХ (25% этилацетата в гексанах). После полного превращения исходного вещества реакцию гасили метанолом. Полученную смесь разбавляли метиленхлоридом (примерно 400 мл) и промывали 10%-ным водным раствором (масс/масс.) соли Рошелля (Rochelle). Органический слой отделяли, и водный слой реэкстрагировали метиленхлоридом (примерно 250 мл). Два органических слоя объединяли, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Целевой продукт выделяли нормально-фазовой хроматографией (колонка 330 г, 45 минут, 5-45% этилацетата в гексанах). Полученный продукт очищали второй раз нормально-фазовой хроматографией (0-20% ацетона в гексанах, колонка 120 г) с получением желтого масла, 16,1 г (66%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.84 (s, 9Η) 1.30-1.46 (m, 9Η) 3.65-3.91 (m, 4Η) 4.43-4.73 (m, 1Η) 5.17 (dt, J=8.12, 1.61 Гц, 1Η) 5.21 (dd, J=1.70, 0.76 Гц, 1Η) 5.69-5.87 (m, 1Η) 9.38-9.50 (m, 1Η)
Промежуточное соединение 100: метил-4-(трет-бутоксикарбонил((S)-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил)амино)-3-гидрокси-2-метиленбутаноат
В перемешиваемый раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-оксоэтил)карбамата (Промежуточное соединение 99, 16,1 г, 46,8 ммоль), метилакрилата (6,3 мл, 70 ммоль) и метанола (примерно 2 мл) под азотом при температуре окружающей среды добавляли хинуклидин (2,1 г, 19 ммоль) в виде твердого вещества. Смесь затем перемешивали в течение 1 суток; анализ методом ТСХ (15% этилацетата в гексанах) показал неполное превращение исходного вещества. Добавляли еще 0,8 экв. хинуклидина каждые 24 часа; добавляли еще 0,75 экв. метилакрилата и 1 мл метанола каждые двое суток. Через 6 суток реакционную смесь разбавляли водой и экстрагировали этилацетатом. ТСХ показала, что только одна экстракция примерно 250 мл этилацетата была необходима для переноса всего неочищенного продукта в органический слой, который затем промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. После нормально-фазовой хроматографии (5-30% этилацетата в гексанах, колонка 220 г, 35 минут) получили целевое соединение (16 г, 80%, бледное масло) в виде смеси диастереомеров, в которой по результатам протонного ЯМР-анализа присутствует примерно 10 мол. % исходного альдегида. Это вещество использовали в этом состоянии.
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.85 (s, 9Η) 1.37 (s, 9Η) 3.19-3.47 (m, 2Η) 3.68 (s, 3Η) 3.73-3.92 (m, 2Η) 3.95-4.19 (m, 1Η) 4.47-4.61 (m, 1Η) 5.03-5.25 (m, 3Η) 5.78-5.95 (m, 2Η) 6.16 (br. s, 1Η)
Промежуточное соединение 101: (2S,5S)-1-трет-бутил-4-метил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1,4(2Н)-дикарбоксилат
Перемешиваемый раствор метил-4-(трет-бутоксикарбонил((S)-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил)амино)-3-гидрокси-2-метиленбутаноата (Промежуточное соединение 100, 16 г, 37 ммоль) в толуоле (примерно 300 мл) при температуре окружающей среды в высушенной нагреванием колбе тщательно дегазировали аргоном и помещали в атмосферу азота. В этот раствор добавляли катализатор Ховейды-Граббса 2-го поколения (460 мг, 0,74 ммоль). Анализ смеси 3,5 часа спустя методами ЖХ/МС и ТСХ (15% этилацетата в гексанах) показал очень высокую степень превращения исходного вещества в два основных продукта. Реакционную смесь концентрировали в вакууме в присутствии достаточного количества силикагеля с получением сыпучего порошка после сушки. Нормально-фазовую хроматографию (0-100% этилацетата в гексанах, колонка 330 г, 45 минут) использовали для выделения двух соединений; диастереомер, показанный выше (6,4 г, 43%, темное масло) элюировался первым.
МС: 402 ЭРИ+ (C19H35NO6Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (s, 6Η) 0.83 (s, 9Η) 1.41 (s, 9Η) 2.81-3.10 (m, 1Η) 3.68-3.78 (m, 5Η) 3.93-4.15 (m, 1Η) 4.34 (br. s., 1Η) 4.42-4.64 (m, 1Η) 4.99 (d, J=5.48 Гц, 1Η) 6.94 (d, J=6.80 Гц, 1Η)
Промежуточное соединение 102: (2S,5R)-1-трет-бутил-4-метил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1,4(2Н)-дикарбоксилат
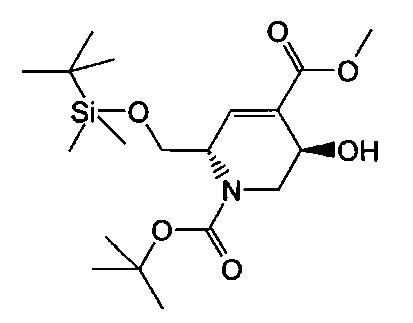
Этот продукт (6,6 г, 44%, темное масло) был получен в результате такой же реакции, как и Промежуточное соединение 101, и элюировался вторым при хроматографической очистке, описанной в этой методике.
МС: 402 ЭРИ+ (C19H35NO6Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Η) 0.86 (s, 9Η) 1.40 (s, 9Η) 2.80 (br. s., 1Η) 3.68 (s, 3Η) 3.76 (d, J=5.67 Гц, 2Η) 4.03 (m, J=7.20 Гц, 1Η) 4.20-4.33 (m, 1Η) 4.42 (br. s., 1Η) 5.24 (d, J=5.85 Гц, 1Η) 6.64 (br. d, J=1.00 Гц, 1Η)
Промежуточное соединение 103: (3S,6S)-1-(трет-бутоксикарбонил)-6-((трет-бутилдиметилсилилокси)метил)-3-гидрокси-1,2,3,6-тетрагидропиридин-4-карбоновая кислота
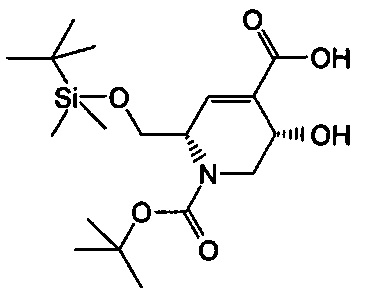
Перемешиваемый раствор (2S,5S)-1-трет-бутил-4-метил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1,4(2Н)-дикарбоксилата (Промежуточное соединение 101, 5,9 г, 15 ммоль) в THF (55 мл) и воде (18 мл) получали в условиях окружающей среды. В него добавляли гидроксид лития (700 мг, 29 ммоль) в виде твердого вещества. Смесь перемешивали в атмосфере азота при температуре окружающей среды. Твердое вещество растворялось за несколько минут. Анализ смеси методом ТСХ (25% этилацетата в гексанах) через 30 минут показал полное расходование исходного вещества. Примерно 20 мл 1 н. водного раствора HCl добавляли в эту смесь при перемешивании. Для доведения pH до значения в диапазоне 4-5 использовали 10%-ный водный раствор лимонной кислоты. Смесь разбавляли примерно 60 мл воды и экстрагировали дважды этилацетатом. Органические экстракты объединяли, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Темно-коричневый смолистый остаток (5,5 г, 96%, темное масло) анализировали методами протонного ЯМР и ЖХ/МС как целевой продукт, и его использовали без дополнительной очистки.
МС: 386 ЭРИ- (C18H33NO6Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Η) 0.86 (s, 9Η) 1.40 (s, 9Η) 2.82 (br. s., 1Η) 3.30 (br. s., 2Η) 3.75 (d, J=6.04 Гц, 2Η) 3.96-4.11 (m, 1Η) 4.25 (t, J=7.55 Гц, 1Η) 4.41 (br. s., 1H) 6.65 (d, J=3.59 Гц, 1Η)
Промежуточное соединение 104: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5-гидрокси-5.6-дигидропиридин-1(2Н)-карбоксилат
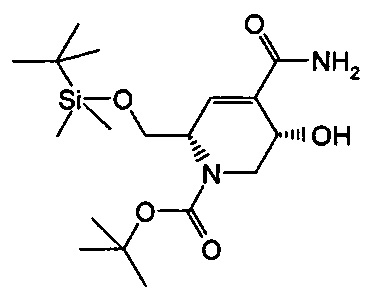
Перемешиваемый раствор (3S,6S)-1-(трет-бутоксикарбонил)-6-((трет-бутилдиметилсилилокси)метил)-3-гидрокси-1,2,3,6-тетрагидропиридин-4-карбоновой кислоты (Промежуточное соединение 103, 5,5 г, 14 ммоль) и Ν,Ν-диизопропилэтиламина (7,4 мл, 42 ммоль) в DMF (примерно 50 мл) получали в условиях окружающей среды. В него добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (8,0 г, 21 ммоль). Реакционный сосуд продували аргоном в течение одной минуты, и темную, прозрачную смесь перемешивали под азотом в течение 5 минут при температуре окружающей среды. В смесь затем добавляли хлорид аммония (1,5 г, 22 ммоль). Смесь перемешивали под азотом при температуре окружающей среды в течение ночи. Утром анализ методом ЖХ/МС показал расходование исходного вещества и присутствие одного основного продукта с более длительным временем удерживания и желаемой массой. Водная обработка с использованием диэтилового эфира привела к отделению неочищенного продукта от водорастворимых веществ. Органические фазы объединяли, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. После нормально-фазовой хроматографии (1-6% метанола в метиленхлориде, 220 г, примерно 50 мин) получили целевой продукт (2,4 г, 44%, желто-коричневое твердое вещество).
МС: 387 ЭРИ+ (C18H34N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Η) 0.86 (s, 9Η) 1.40 (s, 9Η) 2.74 (br. s., 1Η) 3.72 (d, J=5.48 Гц, 2Η) 4.09 (br. s., 1Η) 4.18-4.32 (m, 1Η) 4.40 (br. s., 1H) 5.59 (d, J=5.48 Гц, 1Η) 6.54 (d, J=3.02 Гц, 1Η) 7.15 (br. s., 1H) 7.36 (br. s., 1H)
Промежуточное соединение 105: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2,4-динитрофенилсульфонамидо)-2-((трет-бутилдиметил-силилокси)метил)-4-карбамоил-5,6-дигидропиридин-1 (2Н)-карбоксилат
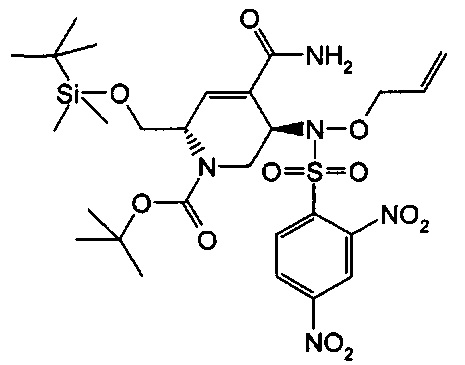
Раствор (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 104, 1,4 г, 3,6 ммоль) в толуоле (35 мл) готовили и перемешивали под азотом при температуре окружающей среды. В него добавляли трифенилфосфин (1,1 г, 4,3 ммоль) и N-(аллилокси)-2,4-динитробензолсульфонамид (1,1 г, 3,6 ммоль). После растворения этих веществ смесь перемешивали в течение еще десяти минут. В смесь затем шприцем добавляли диизопропилазодикарбоксилат (840 мкл, 4,3 ммоль). Поверхность смеси обдували аргоном, и смесь затем помещали в атмосферу азота и перемешивали в течение ночи. Анализ методом ЖХ/МС утром, а также методом ТСХ (5% метанола в метиленхлориде) показал расходование исходного спирт. Реакционную смесь адсорбировали на силикагель, и после нормально-фазовой хроматографии (15-75% этилацетата в гексанах, колонка 220 г) получили целевой продукт (оранжевое твердое вещество; выход примерно 100%).
МС: 673 ЭРИ+ (C27H41N5O11SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Η) 0.84 (s, 9Η) 1.29-1.49 (m, 9Η) 3.62-3.78 (m, 2Η) 4.00 (s, 1Η) 4.15-4.87 (m, 4Η) 5.16-5.36 (m, 2Η) 5.67-5.90 (m, 1Η) 6.76-6.88 (m, 1Η) 6.99-7.80 (m, 2Η) 8.24-8.38 (m, 1Η) 8.62 (ddd, J=8.64, 6.18, 2.36 Гц, 1Η) 9.04 (d, J=2.27 Гц, 1Η)
Промежуточное соединение 106: (3R,6S)-3-(N-(аллилокси)-2,4-динитрофенилсульфонамидо)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-4-карбоксамид
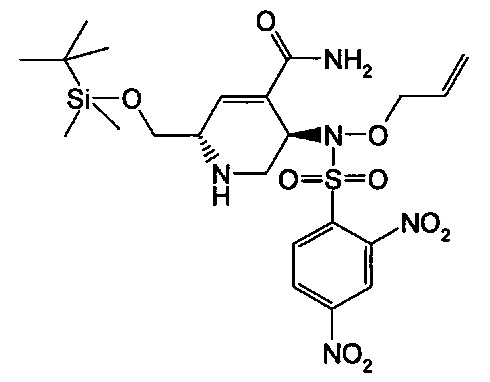
Перемешиваемый раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2,4-динитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 105, 1,8 г, 2,6 ммоль) в метиленхлориде (примерно 25 мл) получали в условиях окружающей среды; раствор обдували аргоном в течение 30 секунд, и раствор затем перемешивали при температуре окружающей среды. В него добавляли бромид цинка (4,1 г, 18 ммоль). Смесь перемешивали в течение ночи. Утром твердое, смолистое желтое вещество покрывало дно колбы, и анализ методом ЖХ/МС показал неполное превращение. Добавляли небольшое количество THF (примерно 2 мл). Смесь становилась гомогенной в течение следующих 25 секунд. Анализ через следующие несколько часов показал увеличение скорости реакции с образованием небольшого количества или без увеличения образования побочного продукта. Добавляли дополнительное количество бромида цинка (еще 5 г) частями. Если после добавления бромида цинка происходило осаждение, опять добавляли небольшой объем THF. К концу суток расходование исходного вещества все еще было неполным, но высоким. Реакционную смесь осторожно переносили в насыщенный раствор бикарбоната натрия. Наблюдалось интенсивное газовыделение. Неочищенный продукт экстрагировали этилацетатом до тех пор, пока ТСХ (5% метанола в метиленхлориде) не показала отсутствие УФ-активного вещества в водной фазе. Органические экстракты объединяли, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток растворяли в метиленхлориде и подвергали нормально-фазовой хроматографии (0-4% метанола в метиленхлориде за 35 минут, колонка 80 г) с получением целевого соединения (1,1 г, 72%, бледно-желтое твердое вещество).
МС: 573 ЭРИ+ (C22H33N5O9SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.01-0.10 (m, 6Η) 0.78-0.91 (m, 9Η) 2.85 (br. s., 2Η) 3.38-3.66 (m, 3Η) 4.31-4.49 (m, 2Η) 4.60 (br. s., 1Η) 5.17-5.39 (m, 3Η) 5.78-5.97 (m, 1Η) 6.73-6.91 (m, 2Η) 7.36 (br. s., 1H) 8.32 (d, J=8.69 Гц, 1Η) 8.62 (dd, J=8.88, 2.27 Гц, 1Η) 9.01 (d, J=2.27 Гц, 1Η)
Промежуточное соединение 107: (3R,6S)-3-(аллилоксиамино)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-4-карбоксамид
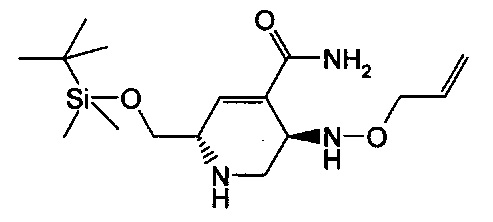
В красновато-коричневую перемешиваемую суспензию (3R,6S)-3-(N-(аллилокси)-2,4-динитрофенил-сульфонамидо)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 106, 1,1 г, 1,9 ммоль) и карбоната цезия (3,1 г, 9,5 ммоль) в THF (примерно 45 мл) при температуре окружающей среды под аргоном добавляли бензолтиол (полимер-связанный реагент, 1,55 ммоль/г) (4,5 г, 7,0 ммоль). Эту смесь перемешивали в течение ночи при температуре окружающей среды. Утром анализы методами ЖХ/МС и ТСХ (5% метанола в метиленхлориде) показали полное расходование исходного вещества. Смесь фильтровали, и отфильтрованное вещество промывали THF до тех пор, пока не осталось вещество, реагирующее на УФ или I2 краситель на силикагелевой пластине. Полученный красно-оранжевый раствор концентрировали и растворяли в метиленхлориде. После нормально-фазовой хроматографии (0-6% метанола в метиленхлориде, 80 г, 25 минут) получили целевое соединение (510 мг, 80%, оранжево-коричневое твердое вещество).
МС: 343 ЭРИ+ (C16H31N3O3Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Η) 0.87 (s, 9Η) 2.39 (br. s., 1Η) 2.74 (dd, J=12.84, 3.40 Гц, 1Η) 2.94 (dd, J=12.84, 3.40 Гц, 1Η) 3.31-3.37 (m, 1Η) 3.49-3.57 (m, 2Η) 3.57-3.66 (m, 1Η) 4.11 (dt, J=5.62, 1.16 Гц, 2Η) 5.13 (ddt, J=10.50, 2.10, 1.16, 1.16 Гц, 1Η) 5.22 (dq, J=17.37, 1.70 Гц, 1Η) 5.92 (ddt, J=17.37, 10.39, 5.67, 5.67 Гц, 1Η) 6.41 (d, J=8.50 Гц, 1Η) 6.68 (dd, J=3.21, 0.57 Гц, 1Η) 6.98 (br. s., 1Η) 7.36 (br. s., 1H)
Промежуточное соединение 108: (2S,5R)-6-(аллилокси)-2-((трет-6утилдиметилсилилокси)метил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
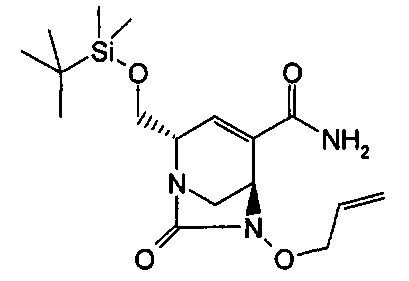
Готовили раствор (3R,6S)-3-(аллилоксиамино)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 107, 505 мг, 1,48 ммоль) и Ν,Ν-диизопропилэтиламина (1,03 мл, 5,91 ммоль) в ацетонитриле (150 мл). Над ним продували аргон в течение одной минуты. Раствор затем помещали в атмосферу аргона и охлаждали в ледяной бане в течение 15 минут. В него добавляли раствор трифосгена (180 мг, 0,59 ммоль) в ацетонитриле (10 мл) посредством шприцевого насоса, установив подачу 0,1 мл/мин. В ходе добавления поддерживали температуру 0°С или около 0°С. После добавления всего трифосгена оранжевый раствор перемешивали при 0°С в течение еще 30 минут. Ледяную баню затем удаляли, и смесь перемешивали в течение еще 30 минут при температуре окружающей среды. Анализ после этого методом ЖХ/МС показал превращение в целевой продукт. Реакционную смесь концентрировали в вакууме, и остаток растворяли в метиленхлориде. Нормально-фазовую хроматографию (15-65% этилацетата в метиленхлориде, колонка 40 г, 25 минут) использовали для выделения целевого продукта (454 мг, 84%, белое твердое вещество).
МС: 349 ЭРИ+ (C17H29N3O4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.06 (s, 6Η) 0.87 (s, 9Η) 3.11-3.20 (m, 1Η) 3.25-3.35 (m, 1Η) 3.71-3.79 (m, 1Η) 3.79-3.95 (m, 2Η) 4.34 (dt, J=5.90, 1.20 Гц, 2Η) 4.51 (d, J=2.83 Гц, 1Η) 5.23 (ddt, J=10.43, 1.89, 1.01, 1.01 Гц, 1Η) 5.33 (dq, J=17.30, 1.60 Гц, 1Η) 5.80-6.00 (m, 1Η) 6.49 (dd, J=2.83, 0.94 Гц, 1Η) 7.08 (br. s., 1Η) 7.51 (br. s., 1Η)
Промежуточное соединение 109: (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
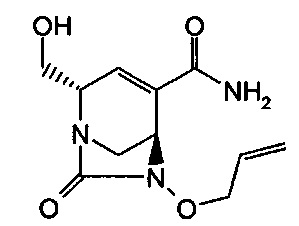
(2S,5R)-6-(Аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид (Промежуточное соединение 108, 450 мг, 1,2 ммоль) растворяли в THF (примерно 7 мл). Атмосферу из реакционного сосуда откачивали, реакционный сосуд заполняли аргоном, и раствор перемешивали в ледяной бане в течение десяти минут. В него затем по каплям добавляли фторид тетрабутиламмония (1M в THF) (1,5 мл, 1,5 ммоль). Через один час анализ смеси методом ЖХ/МС показал расходование исходного вещества и образование одного основного продукта. Смесь концентрировали и растворяли в метиленхлориде. Нормально-фазовую хроматографию (0-10% метанола в метиленхлориде, колонка 25 г, 25 минут) использовали для выделения целевого продукта (284 мг, 92%, белое твердое вещество).
МС: 254 ЭРИ+ (C11Η15Ν3Ο4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.04-3.28 (m, 2Η) 3.54-3.80 (m, 3Η) 4.34 (dt, J=6.00, 1.16 Гц, 2Η) 4.50 (d, J=2.83 Гц, 1Η) 5.01-5.10 (m, 1Η) 5.18-5.27 (m, 1Η) 5.33 (dq, J=17.28, 1.54 Гц, 1Η) 5.78-6.02 (m, 1Η) 6.52 (d, J=1.70 Гц, 1Η) 7.06 (br. s., 1Η) 7.49 (br. s., 1Η)
Промежуточное соединение 110: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилат
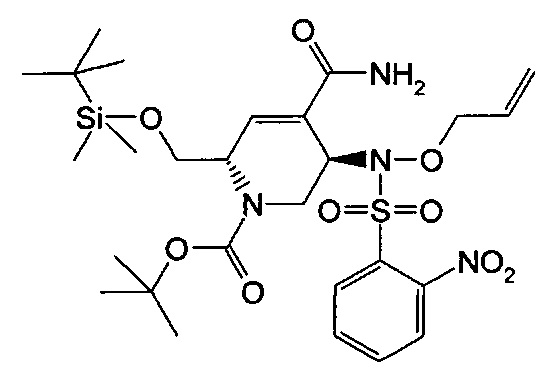
Указанное в заголовке соединение (4,0 г, 74%) было получено из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 104, 3,36 г, 8,69 ммоль) по методике для получения Промежуточного соединения 105 из Промежуточного соединения 104, но используя N-(аллилокси)-2-нитробензолсульфонамид (Промежуточное соединение 9, 2,24 г, 8,69 ммоль) в качестве реагента вместо N-(аллилокси)-2,4-динитробензолсульфонамида.
МС: 627 ЭРИ+ (C27H42N4O9SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Η) 0.84 (s, 9Η) 1.36 (br. s., 9Η) 2.84-3.22 (m, 1Η) 3.59-3.77 (m, 2Η) 4.10-4.46 (m, 3Η) 4.64 (br. s., 1Η) 4.82 (d, J=13.22 Гц, 1Η) 5.08-5.42 (m, 2Η) 5.63-5.93 (m, 1Η) 6.72-6.83 (m, 1Η) 7.37 (br. s., 1H) 7.49-7.71 (m, 1H) 7.85 (dt, J=8.21, 4.01 Гц, 1Η) 7.97 (br. s., 2H) 8.00-8.11 (m, 1H)
Промежуточное соединение 111: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-карбамоил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
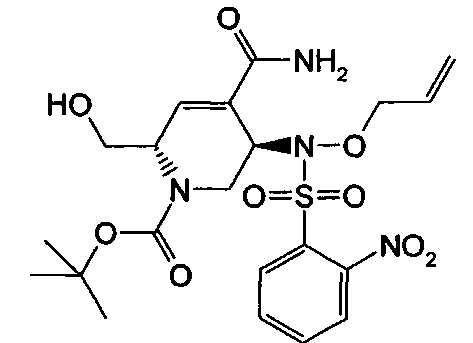
Указанное в заголовке соединение (617 мг, 67%) было получено из (2S,5R)-трет-бутил 5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 110, 1,2 г, 1,80 ммоль) по методике для получения Промежуточного соединения 109 из Промежуточного соединения 108.
МС: 513 ЭРИ+ (C21H28N4O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.19-1.56 (m, 9Η) 2.86-3.20 (m, 1Η) 3.44-3.55 (m, 2Η) 3.89-4.65 (m, 4Η) 4.82 (d, J=11.14 Гц, 1Η) 4.94-5.07 (m, 1Η) 5.12-5.36 (m, 2Η) 5.57-5.92 (m, 1Η) 6.70-7.10 (m, 2Η) 7.36 (br. s., 1Η) 7.78-7.89 (m, 1Η) 7.96 (d, J=2.83 Гц, 2Η) 8.00-8.14 (m, 1Η)
Промежуточное соединение 112: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-карбамоил-2-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
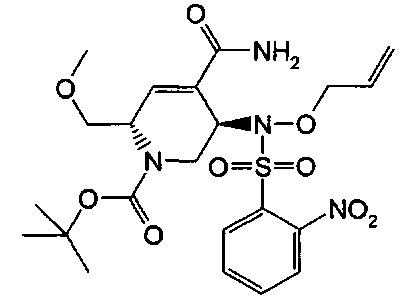
Раствор Промежуточного соединения 111 (427 мг, 0,83 ммоль) и метилйодида (6 экв.) (311 мкл, 5,00 ммоль) в ацетонитриле (примерно 4 мл) готовили в условиях окружающей среды и помещали в атмосферу азота. В этой смеси суспендировали оксид серебра (1,1 экв.) (212 мг, 0,92 ммоль). Смесь защищали от света, используя алюминиевую фольгу, и перемешивали до тех пор, пока анализы методами ТСХ (1:1 ацетонитрил: DCM) и ЖХ/МС не показали полное расходование исходного вещества, около 3 суток. После фильтрования реакционной смеси через фильтр 0,45 мкм получили бронзовый раствор. Его концентрировали в вакууме и растворяли в метиленхлориде. Неочищенную смесь очищали нормально-фазовой хроматографией (10-50% ацетонитрила в DCM) с получением целевого продукта в виде бесцветного остатка (109 мг, 25%). Выделенное вещество лиофилизировали (белый порошок) для облегчения определения характеристик.
МС: 527 ЭРИ+ (C22H30N4O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.38 (d, J=6.99 Гц, 9Η) 2.82-3.15 (m, 1Η) 3.26 (s, 3Η) 3.44 (d, J=3.02 Гц, 2H) 3.90-4.39 (m, 3Η) 4.46-4.94 (m, 2Η) 5.13-5.33 (m, 2Η) 5.64-5.89 (m, 1Η) 6.71-6.84 (m, 1Η) 6.86-7.15 (m, 1Η) 7.35 (br. s., 1Η) 7.85 (t, J=8.03 Гц, 1Η) 7.90-8.15 (m, 3Η)
Промежуточное соединение 113: (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-1,2,3,6-тетрагидропиридин-4-карбоксамид
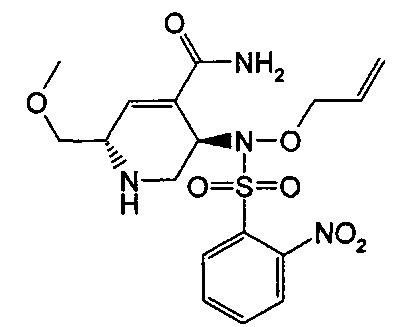
В раствор Промежуточного соединения 112 (729 мг, 1,38 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (2133 мкл, 27,69 ммоль). Этот раствор после добавления стал немного темнее. Реакционную смесь помещали в атмосферу азота и перемешивали до тех пор, пока анализ методом ЖХ/МС не показал расходование исходного вещества (примерно 45 минут). После этого растворитель удаляли в вакууме. Остаток переносили в толуол и обрабатывали ультразвуком, и затем толуол удаляли в вакууме. Эту процедуру повторяли дважды с получением оранжевого твердого вещества высокой чистоты по результатам анализа методом протонного ЯМР. Продукт использовали далее без дополнительной очистки.
МС: 427 ЭРИ+ (C17H22N4O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.24-3.38 (m, 5Η) 3.53-3.71 (m, 2Η) 4.28 (m, J=11.10, 6.40 Гц, 2Η) 4.34-4.44 (m, 1Η) 5.11 (br. s., 1Η) 5.20-5.35 (m, 2Η) 5.84 (ddt, J=17.14, 10.43, 6.42, 6.42 Гц, 1Η) 6.61 (d, J=1.70 Гц, 1Η) 7.28 (br. s., 1Η) 7.65 (br. s., 1H) 7.88-7.99 (m, 1H) 8.01-8.15 (m, 3H) 8.85-9.45 (m, 1H)
Промежуточное соединение 114: (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-1,2,3,6-тетрагидро-пиридин-4-карбоксамид
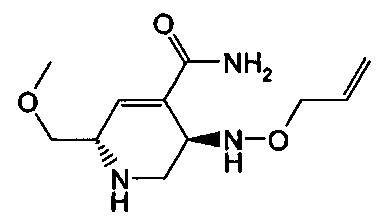
Это промежуточное соединение было получено из (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 113, 734 мг, 1,36 ммоль) по той же методике и с использованием того же метода выделения, которые описаны для получения Промежуточного соединения 107 из Промежуточного соединения 106. Указанное в заголовке соединение было получено в виде оранжевого масла (173 мг, 53%).
МС: 242 ЭРИ+ (C11H19N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.70-2.79 (m, 1Η) 2.70-2.78 (m, 1Η) 2.89-2.99 (m, 1Η) 2.94 (dd, J=12.94, 3.30 Гц, 1Η) 3.22-3.53 (m, 4Η) 3.26 (s, 3Η) 3.61 (br. s., 1Η) 4.11 (dt, J=5.62, 1.25 Гц, 2Η) 5.09-5.29 (m, 2Η) 5.92 (ddt, J=17.33, 10.48, 5.74, 5.74 Гц, 1Η) 6.34-6.49 (m, 1Η) 6.65 (dd, J=3.21, 0.76 Гц, 1Η) 7.00 (br. s., 1Η) 7.39 (br. s., 1H)
Промежуточное соединение 115: (2S,5R)-6-(аллилокси)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
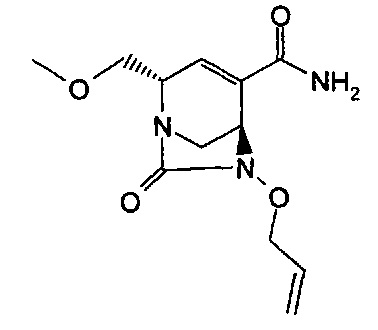
Готовили раствор (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-1,2,3,6-тетрагидро-пиридин-4-карбоксамида (Промежуточное соединение 114, 125 мг, 0,52 ммоль) и триэтиламина (210 мг, 2,07 ммоль) в ацетонитриле (45 мл). Над этим раствором продували аргон в течение одной минуты. Раствор затем помещали в атмосферу аргона и охлаждали в ледяной бане в течение 15 минут. В него медленно добавляли раствор дифосгена (56 мг, 0,28 ммоль) в 5 мл ацетонитрила посредством шприцевого насоса со скоростью 0,1 мл/мин. Сразу после окончания добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем концентрировали в вакууме. Остаток растворяли в метиленхлориде и подвергали нормально-фазовой хроматографии (0-7% метанола в DCM) с получением 60 мг (43%) бледно-желтой пены после приложения глубокого вакуума.
МС: 268 ЭРИ+ (C12H17N3O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.27-3.42 (m, 5Η) 3.59-3.77 (m, 2Η) 4.07-4.15 (m, 1Η) 4.36-4.55 (m, 3Η) 5.27-5.31 (m, 1Η) 5.32-5.36 (m, 1Η) 5.38-5.70 (m, 2Η) 5.92-6.13 (m, 1Η) 6.39 (dd, J=3.02, 1.13 Гц, 1Η)
Промежуточное соединение 116: (2S,5R)-6-гидрокси-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
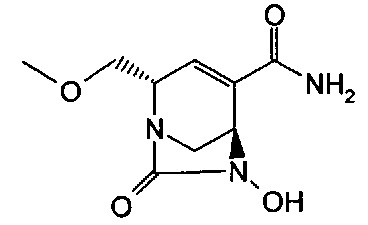
Готовили раствор Промежуточного соединения 115 (60 мг, 0,22 ммоль) в дихлорметане (2 мл) и помещали его в атмосферу азота. В него добавляли уксусную кислоту (25,7 мкл, 0,45 ммоль), затем добавляли тетракис(трифенилфосфин)палладий(0) (259 мг, 0,22 ммоль). Смесь перемешивали под азотом при температуре окружающей среды в течение примерно 60 минут. После полного расходования исходного вещества, что было показано результатами ЖХ/МС-анализа, растворитель удаляли в вакууме. Добавление MeCN приводило к осаждению желто-коричневого твердого вещества, которое отфильтровывали и отбрасывали. Фильтрат адсорбировали на Celite, и целевой продукт (21 мг, 41%, белое твердое вещество) выделяли обращенно-фазовой хроматографией (100%-ная вода) с последующей сублимационной сушкой.
МС: 228 ЭРИ+ (C9H13N3O4)
1Н ЯМР (300 МГц, МЕТАНОЛ-d4) δ: 3.38 (dt, J=3.26, 1.68 Гц, 2Η) 3.46 (s, 3Η) 3.68-3.83 (m, 2Η) 4.05 (td, J=5.76, 3.02 Гц, 1Η) 4.38-4.44 (m, 1Η) 6.58 (dd, J=2.93, 1.04 Гц, 1Η)
ПРИМЕР 13
Натриевая соль (2S,5R)-2,4-бис(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
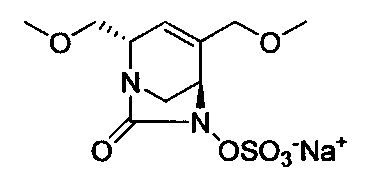
В раствор Промежуточного соединения 124 (100 мг, 0,37 ммоль) в DCM (3 мл) добавляли уксусную кислоту (0,043 мл, 0,75 ммоль) (высушенную над Na2SO4) и Pd(PPh3)4 (215 мг, 0,19 ммоль) под аргоном при комнатной температуре. Желтый раствор перемешивали при комнатной температуре в течение 1 ч. ТСХ и ЖХ/МС показали полное превращение исходного вещества в продукт. Реакционную смесь концентрировали и сушили в вакууме. Затем ее разбавляли 5 мл 10% CH3CN в воде, наносили на колонку 30 г ISCO gold С18 и элюировали 0% CH3CN в течение 5 минут, затем 0-50% CH3CN/H2O в течение 15 минут. Первые несколько фракций собирали и концентрировали с получением желтого масла (126 мг). Это масло растворяли в пиридине (3 мл) при комнатной температуре. Затем добавляли SO3-Pyr (353 мг, 2,22 ммоль) в виде твердого вещества. Смесь затем перемешивали при комнатной температуре в течение ночи. ЖХ/МС показала завершение реакции. Реакционный раствор концентрировали досуха и подвергали азеотропной перегонке с 2×5 мл толуола. Неочищенный раствор затем наносили на примерно 30 г предварительно обработанной смолы Dowex (200 мл 2 н. NaOH в течение 2 ч, затем упаковывали и промывали H2O до нейтрального pH) и очищали на самотечной колонке с использованием H2O, собирая каждые 10 мл. Фракции, содержащие целевое соединение, объединяли и лиофилизировали с получением белого твердого вещества, которое затем очищали методом ОФ-ВЭЖХ с получением примерно 20 мг белого твердого вещества.
МС: 309 ЭРИ+ (C10H16N2O7S)
1Н ЯМР (300 МГц, D2O) δ: 3.31 (s, 3Η) 3.40 (s, 3Η) 3.44 (s, 2Η) 3.57-3.77 (m, 2Η) 3.96-4.12 (m, 3Η) 4.24 (s, 1Η) 5.62 (br. s., 1Η)
Промежуточное соединение 117: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(гидроксиметил)-5-оксо-5.6-дигидропиридин-1(2Н)-карбоксилат
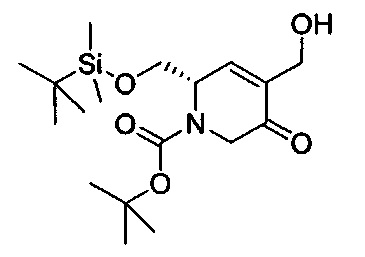
В раствор (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 7, 18 г, 52,71 ммоль) в смеси DCM (900 мл) и МеОН (350 мл) добавляли последовательно формальдегид (42,8 мл, 527,06 ммоль) и трибутилфосфин (0,692 мл, 3,16 ммоль) при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 2-3 ч до тех пор, пока ТСХ (4:1 Нех/ЕА) не показала, что реакция завершена. Раствор концентрировали при пониженном давлении и очищали на силикагелевой (120 г) колонке (0-50% Нех/ЕА) с получением указанного в заголовке соединения (20,00 г, 102%) в виде масла.
МС: 372 ЭРИ+ (C18H33NO5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.13-0.08 (m, 6Η) 0.73-0.97 (m, 9Η) 1.43 (s, 9Η) 3.87 (m, 3Η) 4.11 (m, 2Η) 4.22-4.39 (m, 1Η) 4.69-4.86 (m, 1Η) 5.01 (m, 1Η) 6.92-7.07 (m, 1Η).
Промежуточное соединение 118: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
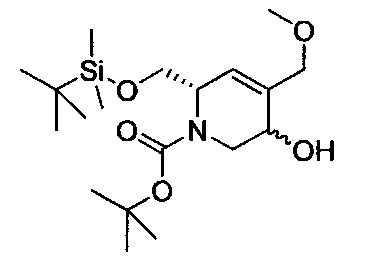
В перемешиваемый раствор Промежуточного соединения 117 (7,3 г, 19,65 ммоль) в DCM (180 мл) добавляли N1,N1,N8,N8-тетраметилнафталин-1,8-диамин (25,3 г, 117,89 ммоль). Затем раствор охлаждали до 0°С и добавляли тетрафторборат триметилоксония (8,72 г, 58,94 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение нескольких часов до тех пор, пока ЖХ/МС не показала, что все исходное вещество израсходовано. Затем ее концентрировали при пониженном давлении. Остаток переносили в 100 мл Et2O и фильтровали и промывали 100 мл Et2O. Органический слой затем промывали 10%-ной лимонной кислотой, водным NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток переносили в МеОН (180 мл), охлаждали до 0°С и затем добавляли хлорид церия(III) (7,32 г, 19,65 ммоль) с получением прозрачного раствора. Затем добавляли NaBH4 (0,743 г, 19,65 ммоль) в виде твердого вещества, и смесь перемешивали при температуре от 0°C до комнатной температуры в течение 1 ч. Реакционную смесь концентрировали в вакууме. Белое твердое вещество растворяли в 200 мл Et2O и промывали 10%-ной лимонной кислотой, NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (20-80% Нех/ЕА, 80 г) с получением указанного в заголовке соединения (6,20 г, 81%) в виде бесцветного масла (смесь 2 диастереомеров в соотношении примерно 3,8:1).
МС: 388 ЭРИ+ (C19H37NO5Si)
Промежуточное соединение 119: (S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
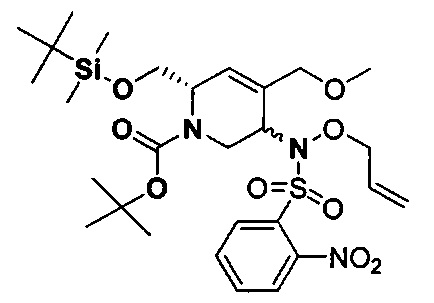
В перемешиваемый раствор Промежуточного соединения 118 (6,2 г, 16,00 ммоль), N-(аллилокси)-2-нитробензолсульфонамида (4,34 г, 16,80 ммоль) и трифенилфосфина (5,03 г, 19,20 ммоль) в толуоле (140 мл) добавляли (Е)-диизопропил-диазен-1,2-дикарбоксилат (3,67 г, 17,60 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение выходных дней. Смесь затем концентрировали, и остаток очищали на силикагелевой колонке (120 г, 0-100% Нех/ЕА) с получением указанного в заголовке соединения (8,80 г, 88%) в виде масла
МС: 628 ЭРИ+ (C28H45N3O9SSi)
Промежуточное соединение 120: (S)-N-(аллилокси)-N-(6-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамид
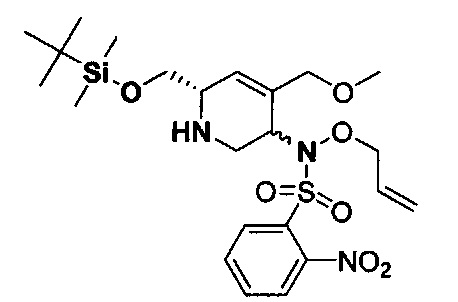
Раствор Промежуточного соединения 119 (8,8 г, 14,02 ммоль) в DCM (120 мл) при комнатной температуре добавляли к бромиду цинка (9,47 г, 42,05 ммоль). Реакционную смесь перемешивали под N2 при комнатной температуре в течение ночи. ЖХ/МС показала, что реакция завершена с образованием вторичного амина. Смесь фильтровали и промывали DCM. DCM-слой промывали водным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали с получением остатка, который адсорбировали на силикагель и очищали на силикагелевой колонке (40 г, 20-80% hex/EA) с получением указанного в заголовке соединения (5,00 г, 67,6%) в виде масла.
МС: 528 ЭРИ+ (C23H37N3O7SSi)
Промежуточное соединение 121: (S)-O-аллил-N-(6-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламин
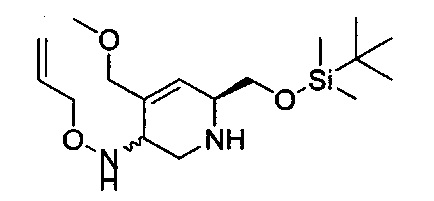
В перемешиваемую суспензию Промежуточного соединения 120 (5 г, 9,47 ммоль) и K2CO3 (7,86 г, 56,85 ммоль) в ацетонитриле (100 мл) добавляли PhSH (3,90 мл, 37,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Ее фильтровали и концентрировали, затем разбавляли DCM и фильтровали через одноразовый фильтр. Фильтрат концентрировали и очищали на силикагелевой колонке (0-100% Нех/ЕА, 80 г ISCO) с получением указанного в заголовке соединения (2,04 г, 63%) в виде масла.
МС: 343 ЭРИ+ (C17H34N2O3Si)
Промежуточное соединение 122: (S)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-4-(метоксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
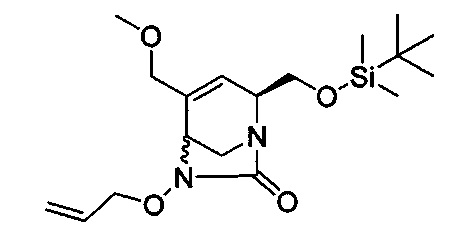
В перемешиваемый раствор Промежуточного соединения 121 (2,04 г, 5,96 ммоль) в ацетонитриле (500 мл) добавляли N-этил-N-изопропилпропан-2-амин (4,15 мл, 23,82 ммоль) и трифосген (0,721 г, 2,38 ммоль) в виде раствора в 20 мл CH3CN посредством шприцевого насоса (0,1 мл/мин) при 0°С. Реакционную смесь перемешивали и оставляли нагреваться от 0°С до комнатной температуры в течение ночи. ЖХ/МС показала полное превращение в продукт. Раствор концентрировали с получением остатка, который затем переносили в 150 мл EtOAc и промывали 5%-ной лимонной кислотой, NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (40 г, 0-60% Нех/ЕА) с получением указанного в заголовке соединения (1,700 г, 77%) в виде масла.
МС: 369 ЭРИ+ (C18H32N2O4Si)
Промежуточное соединение 123: (S)-6-(аллилокси)-2-(гидроксиметил)-4-(метоксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
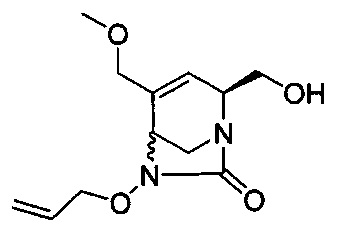
В перемешиваемый раствор Промежуточного соединения 122 (1,7 г, 4,61 ммоль) в THF (40 мл) добавляли TBAF (6,92 мл, 6,92 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч. ЖХ/МС показала исчезновение исходного вещества и образование целевого продукта. Смесь концентрировали и очищали на силикагелевой колонке (40 г, 50-100% Нех/ЕА) с получением указанного в заголовке соединения (1,050 г, 90%) в виде белого твердого вещества.
МС: 255 ЭРИ+ (C12H18N2O4)
Промежуточное соединение 124: (S)-6-(аллилокси)-2,4-бис(метоксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
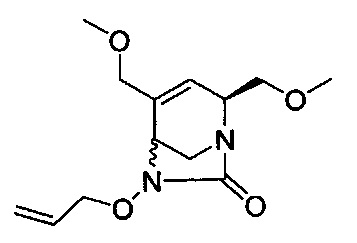
В перемешиваемый раствор Промежуточного соединения 123 (0,3 г, 1,18 ммоль) в ACN (12 мл) добавляли йодметан (0,734 мл, 11,80 ммоль) и оксид серебра (1,094 г, 4,72 ммоль). Реакционную смесь в сосуде, обернутом алюминиевой фольгой, перемешивали при комнатной температуре в течение ночи. ЖХ/МС показала продукт. Смесь фильтровали и концентрировали. Остаток затем очищали на силикагелевой колонке (40 г, 0-100% Нех/ЕА) с получением указанного в заголовке соединения (0,100 г, 31,6%) в виде масла.
МС: 269 ЭРИ+ (C13H20N2O4)
ПРИМЕР 14
Натриевая соль (2S,5R)-2-(1-(трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
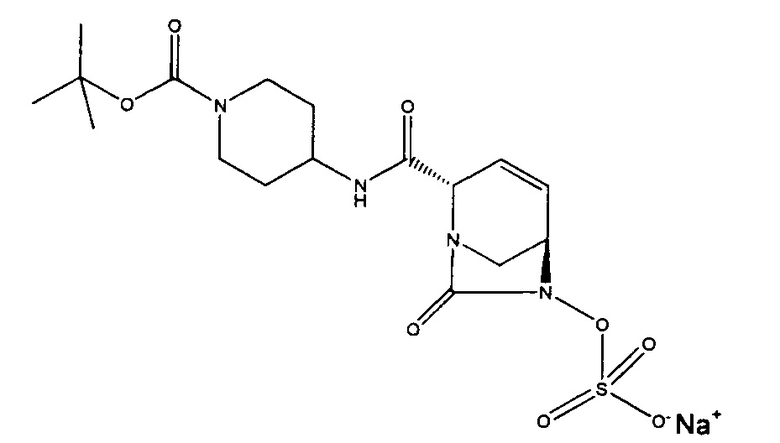
В лабораторном стакане смолу DOWEX 50WX8 (15 г) кондиционировали путем перемешивания ее в течение 20 минут в 2 н. гидроксиде натрия (39,3 мл, 78,69 ммоль), загружали в картридж, промывали водой до рН 7 и затем промывали смесью ацетон/вода (1/1), затем снова водой. Наносили фосфониевую соль (Промежуточное соединение 132, 140 мг), используя воду и минимальное количество ацетона, и элюировали водой. Фракции замораживали и лиофилизировали с получением целевой натриевой соли в виде белого твердого вещества (39 мг).
МС: 445 (М-Н) (C17H26N4O8S⋅[Na+])
1Н ЯМР (300 МГц, DMSO-d) δ: 1.31-1.40 (m, 2Н) 1.40-1.45 (s, 9Н) 1.70 (m., 2Н) 2.79-2.86 (m, 2Н) 3.21 (d, 1Н) 3.85 (dd,, 2Н) 3.75 (m, 1Н) 3.80-3.93 (m, 2Н) 4.01-4.12 (m, 1Н) 4.25 (s, 1Н) 5.82 (d, 1Н) 6.30-6.36 (m, 1Н)
Промежуточное соединение 125: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
В перемешиваемый раствор хлорида церия (4,36 г, 11,71 ммоль) и (S)-трет-бутил-2-(трет-бутилдиметилсилилокси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 50, 4,00 г, 11,71 ммоль) в 50 мл МеОН при 0°С добавляли NaBH4 (0,443 г, 11,71 ммоль) в виде твердого вещества. Смесь перемешивали при температуре окружающей среды в течение 15 минут. Смесь концентрировали и разбавляли 10%-ной лимонной кислотой (водн.), Н2О и этилацетатом. Органический слой отделяли и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением остатка, который очищали на силикагелевой колонке (25 г, 20-40% Нех/ЕА) с получением целевого продукта (2,6 г) в виде бесцветного масла.
МС: 344 ЭРИ+ (C17H33NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.10-0.07 (m, 6Η) 0.65-0.88 (m, 9Η) 1.25 (br. s., 6Η) 3.60 (d, J=5.67 Гц, 3Η) 5.08 (d, J=5.10 Гц, 1Η) 5.67 (br. s., 1H) 5.72-5.86 (m, 1H)
Промежуточное соединение 126: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2,4-динитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат
В перемешиваемый раствор (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 125, 17,50 г, 50,94 ммоль), трифенилфосфора (14,70 г, 56,04 ммоль), N-(аллилокси)-2,4-динитробензолсульфонамида (16,22 г, 53,49 ммоль) в толуоле (380 мл) добавляли диизопропилазодикарбоксилат (11,03 мл, 56,04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, и остаток очищали на силикагелевой колонке (220 г, 0-40% Нех/ЕА) с получением целевого продукта (30,8 г) в виде желтой смолы.
МС: 429 ЭРИ+ (C26H40N4O10SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.10-0.07 (m, 6Η) 0.65-0.88 (m, 9Η) 1.25 (br. s., 6Η) 1.32 (s, 4Η) 5.14 (d, J=10.01 Гц, 2Η) 8.01 (dd, J=9.25, 1.89 Гц, 4Η) 8.10 (s, 1Η)
Промежуточное соединение 127: N-(аллилокси)-N-((3R,6S)-6-((третбутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-3-ил)-2,4-динитробензолсульфонамид
В перемешиваемый раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2,4-динитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 126, 19,5 г, 31,01 ммоль) в DCM (250 мл) под азотом при комнатной температуре добавляли бромид цинка (20,95 г, 93,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч (ЖХ/МС показала, что реакционная смесь представляла собой чистую реакционную смесь) и разбавляли 50 мл DCM и промывали насыщенным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали с получением целевого продукта в виде оранжевого масла (17,5 г).
МС: 529 ЭРИ+ (C21H32N4O8SSi)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.17-0.11 (m, 6Η) 0.65-0.87 (m, 9Η) 3.1 (m, 2H), 3.75(m, 3Н) 4.6(m 3Н) 5.26 (d, J=8.69 Гц, 2Η) 5.86 (m, J=8.69 Гц, 3Η) 8.17-8.57 (m, 3Н)
Промежуточное соединение 128: O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламин
В перемешиваемую суспензию N-(аллилокси)-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-3-ил)-2,4-динитробензолсульфонамида (Промежуточное соединение 127, 17,50 г, 33,10 ммоль) и карбоната калия (13,73 г, 99,31 ммоль) в ацетонитриле (700 мл) добавляли бензолтиол (5,10 мл, 49,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов. ЖХ/МС показала завершение реакции. Реакционную смесь концентрировали, разбавляли EtOAc и фильтровали через одноразовый фильтр. Фильтрат концентрировали, и неочищенный продукт очищали на силикагелевой колонке (0-4% МеОН в DCM) с получением целевого продукта в виде желтого масла (5,6 г).
МС: 299 ЭРИ+ (C15H30N2O2Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.08-0.09 (m, 6Η) 0.75-0.92 (m, 9Η) 2.53-2.62 (m, 1Η) 3.03 (dd,.J=11.71, 4.72 Гц, 1Η) 3.15-3.25 (m, 1Η) 3.29 (s, 1Η) 3.37 (td, J=4.58, 2.36 Гц, 1Η) 3.41-3.49 (m, 2Η) 4.08 (dt, J=5.57, 1.37 Гц, 2Η) 5.04-5.27 (m, 2Η) 5.61-5.80 (m, 2Η) 5.80-6.00 (m, 1Η) 6.40 (d, J=7.37 Гц, 1Η)
Промежуточное соединение 129: (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
В перемешиваемый раствор O-аллил-N-((3R,6S)-6-((трет-бутилдиметилсилилокси)метил)-1,2,3,6-тетрагидропиридин-3-ил)-гидроксиламина (Промежуточное соединение 128, 2,0 г, 6,70 ммоль) в ацетонитриле (560 мл) добавляли N-этил-N-изопропилпропан-2-амин (4,67 мл, 26,80 ммоль). Бис(трихлорметил)карбонат (0,795 г, 2,68 ммоль), растворенный в 40 мл CH3CN добавляли посредством шприцевого насоса (со скорость 0,1 мл/мин) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч. Ее затем перемешивали при комнатной температуре в течение ночи. Смесь затем концентрировали и разбавляли EtOAc (50 мл). Органический слой промывали водой, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке (12 г, 0-25% Нех/ЕА) с получением целевого продукта в виде бесцветного масла (1,64 г).
МС: 325 ЭРИ+ (C16H28N2O3Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.05-0.07 (m, 6Η) 0.67-0.87 (m, 9Η) 2.92-3.08 (m, 1Η) 3.32 (d, J=11.14 Гц, 1Η) 3.48-3.62 (m, 1Η) 3.68-3.82 (m, 2Η) 3.87 (dd, J=5.00, 2.74 Гц, 1Η) 4.29 (dt, J=6.00, 1.25 Гц, 2Η) 5.09-5.39 (m, 2Η) 5.50-6.02 (m, 2H) 6.11-6.39 (m, 1Η)
Промежуточное соединение 130: (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
В перемешиваемый раствор (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 129, 1,64 г, 5,05 ммоль) в THF (40 мл) добавляли фторид тетрабутиламмония (6,06 мл, 6,06 ммоль) при 0°C. Реакционную смесь перемешивали при 0°С в течение 1 ч. Смесь концентрировали и очищали на силикагелевой колонке (40 ч, 50-100% Нех/ЕА) с получением целевого продукта (1,03 г) в виде желтого масла.
МС: 211 ЭРИ+ (C10H14N2O3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 3.31-3.37 (m, 2Η) 3.69-3.81 (m, 2Η) 3.88 (dd, J=4.34, 1.51 Гц, 1Η) 3.97-4.11 (m, 1Η) 4.46 (ddt, J=7.86, 6.63, 1.06, 1.06 Гц, 2Η) 5.24-5.45 (m, 2Η) 5.56-5.72 (m, 1Η) 5.92-6.18 (m, 1Η) 6.30-6.49 (m, 1Η)
Промежуточное соединение 131: трет-бутил-4-((2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)пиперидин-1-карбоксилат
В раствор перйодной кислоты (6 г, 31,26 ммоль) во влажном ацетонитриле (60 мл) (0,75% воды по объему) при комнатной температуре добавляли оксид хрома(VI) (10 мг, 0,10 ммоль). Смесь перемешивали до полного растворения. Этот раствор ортоперйодной кислоты использовали на следующей стадии.
В перемешиваемый раствор (2S,5R)-6-(аллилокси)-2-(гидроксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 130, 216 мг, 1,03 ммоль) во влажном CH3CN (10 мл) (0,75% воды по объему) при комнатной температуре по каплям добавляли 9 мл ранее образованного раствора ортоперйодной кислоты. Смесь затем перемешивали при комнатной температуре в течение 50 минут при добавлении 50 мл EtOAc. Органический раствор промывали небольшим количеством воды и рассолом, затем сушили над MgSO4, фильтровали и концентрировали с получением неочищенной кислоты в виде светло-желтого масла (200 мг). В раствор неочищенной кислоты в DMF (5 мл) при 0°С добавляли трет-бутил-4-аминопиперидин-1-карбоксилат (214 мг, 1,07 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (509 мг, 1,34 ммоль) и TEA (497 мкл, 3,57 ммоль). Реакционную смесь перемешивали при 0°C в течение 35 минут, затем смесь разбавляли этилацетатом и промывали рассолом и водой. Объединенные водные промывочные жидкости экстрагировали однократно этилацетатом. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (12 г колонка, 50%-100% EtOAc) получили целевой продукт в виде бесцветного масла (110 мг).
МС: 407 3PM+ (C20H30N4O5)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.31-1.43 (m, 2Η) 1.42-1.51 (m, 9Η) 1.90 (br. s., 2Η) 2.79-2.96 (m, 2Η) 3.01 (d, J=10.39 Гц, 1Η) 3.38 (dd, J=10.48, 1.61 Гц, 1Η) 3.85 (dd, J=5.00, 3.12 Гц, 1H) 3.89-3.98 (m, 1Η) 4.01-4.12 (m, 2Η) 4.35-4.52 (m, 3Η) 5.22-5.45 (m, 2Η) 5.93-6.08 (m, 1Η) 6.15 (dd, J=9.35, 3.30 Гц, 1Η) 6.32-6.50 (m, 1Η) 6.84 (d, J=7.55 Гц, 1Η)
Промежуточное соединение 132: (2S,5R)-2-(1-трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил (проп-1-енил)-фосфония
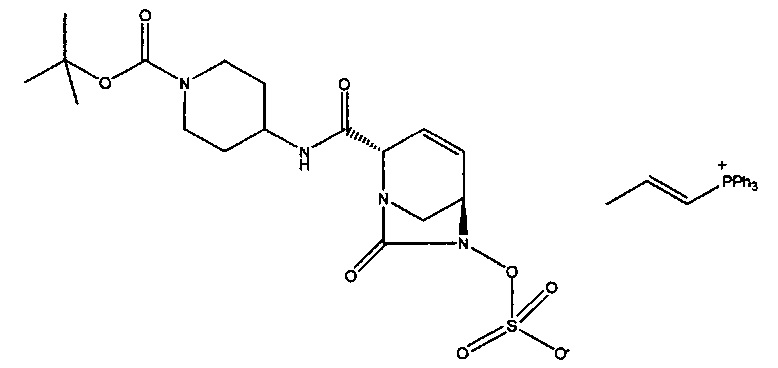
В раствор трет-бутил-4-((2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-енкарбоксамидо)пиперидин-1-карбоксилата (Промежуточное соединение 131, 110 мг, 0,52 ммоль) в DCM (5 мл) добавляли АсОН (59,2 мкл, 1,03 ммоль) (высушенный над Na2SO4) и тетракис(трифенил-фосфин)палладий(0) (597 мг, 0,52 ммоль) под азотом при комнатной температуре. Желтый раствор перемешивали при комнатной температуре в течение 1 ч. В смесь добавляли пиридин (10 мл), затем комплекс пиридин/триоксид серы (493 мг, 3,10 ммоль). Суспензию перемешивали при комнатной температуре в течение ночи под азотом. Реакционную смесь концентрировали досуха (≤ 30°С) и затем очищали на силикагелевой колонке (ацетон в гексане (от 5 до 100%)) с получением не совсем белого твердого вещества (140 мг).
ПРИМЕР 15
Натриевая соль (2S,5R)-4-(диметилкарбамоил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
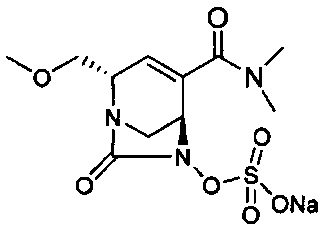
Ионообменную смолу Dowex(R) 50WV8, 100-200 меш, (0,63 г, 9,41 мкмоль) кондиционировали путем перемешивания в течение 3 часов в 2 н. NaOH (1,6 мл, 3,20 ммоль). Смолу затем загружали в картридж и промывали водой до pH 7. Затем ее промывали смесью (1/1) ацетон/вода, затем водой. (2S,5R)-4-(диметилкарбамоил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 144, 6 мг, 9,41 мкмоль) переносили в воду. Желтый раствор наносили на смолу и промывали водой. Продукт собирали и сушили в лиофилизаторе с получением целевого продукта (3,3 мг, 98%) в виде не совсем белого твердого вещества.
МС: 334 ЭРИ- (C11H16N3O7SNa)
1Н ЯМР (300 МГц, D2Ο) δ: 2.99 (s, 7Η) 3.13 (s, 7Η) 3.44 (s, 6Η) 3.56 (d, J=1.70 Гц, 4Η) 3.65-3.79 (m, 5Η) 4.24 (ddd, J=8.34, 4.47, 3.01 Гц, 2Η) 4.47 (s, 2Η) 5.97 (d, J=1.88 Гц, 2Η)
Промежуточное соединение 133: (2S,5R)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
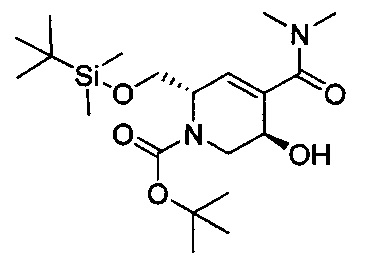
В сухую двухгорлую круглодонную колбу, оснащенную дефлегматором, снабженным в его верхней части входным отверстием для подачи азота, резиновой мембраной и магнитной мешалкой, загружали толуол (7,5 мл) и быстро промывали азотом, после чего через мембрану в колбу впрыскивали триметилалюминий в гексанах (3,5 мл, 7,0 ммоль). Раствор перемешивали и охлаждали в бане лед-соль при температуре от -10° до -15°, и медленно шприцем добавляли диметиламин в THF (3,36 мл, 6,72 ммоль). Через двадцать минут после окончания добавления охлаждающую баню удаляли, содержимое колбы перемешивали и оставляли медленно нагреваться до комнатной температуры в течение периода времени 45 минут. Готовили раствор (2S,5R)-1-трет-бутил-4-метил 2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1,4(2Н)-дикарбоксилата (Промежуточное соединение 102, 2,25 г, 5,60 ммоль) в толуоле (3 мл) и впрыскивали его в двухгорлую колбу через мембрану под азотом, при этом сразу наблюдалось образование пузырей. Полученный раствор перемешивали в течение ночи при комнатной температуре. ЖХ/МС показала, что осталось очень малое количество исходного вещества. Еще одну партию Me2AlNMe2 получали с использованием 0,6 мл амина и 0,6 мл AlMe3 в 1 мл толуола и переносили ее в реакционную смесь. Реакционную смесь перемешивали в течение более чем 3 часов. Реакционную смесь затем подвергали гидролизу, медленно осторожно добавляя 7,82 мл (7,82 ммоль) 1М HCl. Смесь затем перемешивали в течение 30 минут, чтобы гарантировать полный гидролиз. Верхний органический слой отделяли, и водный слой экстрагировали тремя порциями по 50 мл EtOAc. Органические экстракты объединяли, промывали рассолом, сушили над безводным Na2SO4 и упаривали при пониженном давлении с получением целевого продукта (2,194 г, 94%) в виде прозрачного коричневого масла.
МС: 415 ЭРИ+ (C20H38N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (s, 6Η) 0.83-0.86 (m, 9Η) 1.40-1.44 (m, 9Η) 2.91 (br. s., 7Η) 3.66-3.73 (m, 2Η) 4.04 (m, J=7.00 Гц, 1Η) 4.15-4.22 (m, 1Η) 4.33-4.51 (m, 1Η) 4.98 (d, J=6.22 Гц, 1Η) 5.88 (d, J=3.58 Гц, 1Η)
Промежуточное соединение 134: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
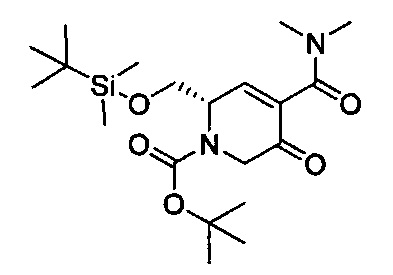
(2S,5R)-трет-Бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 133, 2,194 г, 5,29 ммоль) в DCM (65 мл) охлаждали в ледяной бане, затем добавляли перйодинан Десс-Мартина в DCM (26,5 мл, 7,94 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Органический раствор промывали насыщенным раствором Na2S2O3, насыщенным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и упаривали. Неочищенный продукт очищали флэш-хроматографией (10-75% ЕА/EtOAc) с получением целевого продукта (1,050 г, 48,1%).
МС: 413 ЭРИ+ (C20H36N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Η) 0.83 (s, 9Η) 1.44 (s, 9Η) 2.81 (s, 3Η) 2.89 (s, 3Η) 3.79-4.02 (m, 3Η) 4.30-4.48 (m, 1Η) 4.72-4.87 (m, 1Η) 7.10-7.22 (m, 1Η)
Промежуточное соединение 135: (2S,5S)-трет-бутил-2-(трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
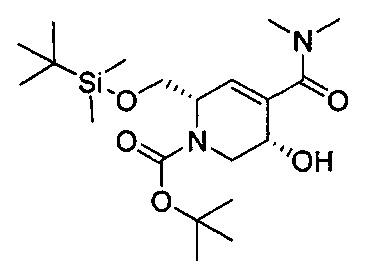
В перемешиваемый раствор гептагидрата хлорида церия(III) (0,948 г, 2,54 ммоль) и (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 134, 1,05 г, 2,54 ммоль) в 1 мл МеОН при 0°С добавляли натрия тетрагидроборат (0,096 г, 2,54 ммоль) в виде твердого вещества. Смесь перемешивали при температуре окружающей среды в течение 15 минут. Смесь концентрировали и разбавляли NH4Cl (водн.), H2O и диэтиловым эфиром. Эфирный слой отделяли и промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением целевого продукта (1,034 г, 98%) в виде бежевого твердого вещества.
МС: 415 ЭРИ+ (C20H38N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.76-0.92 (m, 9Η) 1.30-1.48 (m, 9Η) 2.71-3.05 (m, 7Η) 3.74 (d, J=4.71 Гц, 2Η) 4.18 (br. s., 3Η) 5.34 (d, J=5.46 Гц, 1Η) 5.66 (br. s., 1Η)
Промежуточное соединение 136: (3S,6S)-1-(трет-бутоксикарбонил)-6-((трет-бутилдиметилсилилокси)метил)-3-гидрокси-1,2,3,6-тетрагидропиридин-4-карбоновая кислота
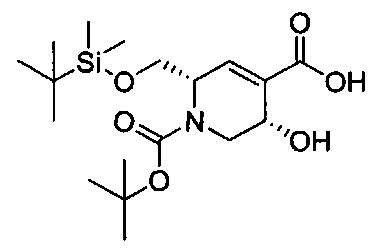
(2S,5S)-1-трет-Бутил-4-метил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-5,6-дигидропиридин-1,4(2Н)-дикарбоксилат (Промежуточное соединение 101, 4,22 г, 10,51 ммоль) растворяли в THF (64,0 мл) и воде (32 мл) и затем добавляли LiOH (0,277 г, 11,56 ммоль), и эту смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли 1 н. HCl, экстрагировали EtOAc, промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением целевого продукта (4,07 г, 100%) в виде прозрачного масла.
МС: 386 ЭРИ- (C18H33NO6Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Η) 0.84-0.90 (m, 9Η) 1.40 (s, 9Η) 2.73-2.88 (m, 1Η) 3.75 (d, J=5.46 Гц, 2Η) 4.25 (t, J=7.16 Гц, 1Η) 4.41 (br. s., 1Η) 5.15 (br. s., 1Η) 6.65 (d, J=3.96 Гц, 1Η) 12.46 (br. s., 1H)
Промежуточное соединение 137: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
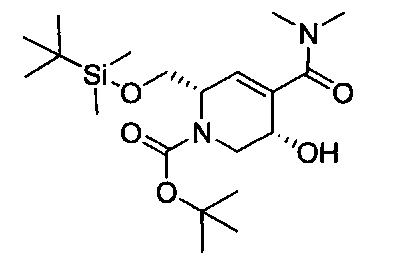
В раствор (3S,6S)-1-(трет-бутоксикарбонил)-6-((трет-бутилдиметилсилилокси)метил)-3-гидрокси-1,2,3,6-тетрагидропиридин-4-карбоновой кислоты (Промежуточное соединение 136, 4,07 г, 10,50 ммоль) в DMF (20 мл) при комнатной температуре добавляли 2М диметиламин в THF (5,25 мл, 10,50 ммоль), HATU (5,99 г, 15,75 ммоль) и затем DIEA (5,50 мл, 31,51 ммоль). Реакционную смесь перемешивали при комнатной температуре. Реакцию проводили до полного расходования исходного вещества, затем ее гасили водой и экстрагировали EtOAc. Органические растворы затем промывали рассолом и очищали флэш-хроматографией (35-100% ЕА/Нех) с получением целевого продукта (3,05 г, 70,0%) в виде розового стеклообразного твердого вещества.
МС: 415 ЭРИ+ (C20H38N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.76-0.92 (m, 9Η) 1.30-1.48 (m, 9Η) 2.71-3.05 (m, 7Η) 3.74 (d, J=4.71 Гц, 2Η) 4.18 (br. s., 3Η) 5.34 (d, J=5.46 Гц, 1Η) 5.66 (br. s., 1Η)
Промежуточное соединение 138: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилат
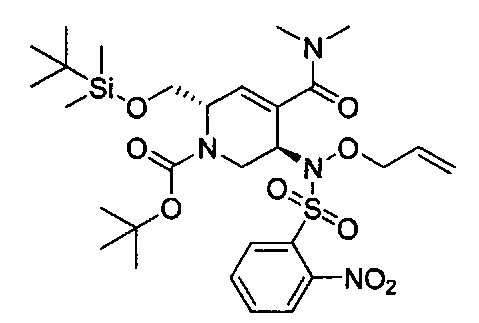
Перемешиваемую суспензию (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(диметилкарбамоил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 137, 3,09 г, 7,45 ммоль), N-(аллилокси)-2-нитробензолсульфонамида (2,348 г, 9,09 ммоль) и трифенилфосфина (2,150 г, 8,20 ммоль) в толуоле (65 мл) охлаждали в бане соль-лед и затем по каплям добавляли (Е)-диизопропил-диазен-1,2-дикарбоксилат (1,588 мл, 8,20 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение еще 2 ч. Растворитель удаляли, и неочищенный продукт наносили на силикагель и очищали флэш-хроматографией (25-75% ЕА/Нех) с получением целевого продукта (4,0 г, 82%) в виде не совсем белого твердого вещества.
МС: 655 ЭРИ+ (C29H46N4O9SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.01 (s, 6Η) 0.82 (s, 9Η) 1.37 (d, J=12.24 Гц, 9Η) 2.56-3.06 (m, 6Η) 3.06-3.25 (m, 1Η) 3.60-3.81 (m, 2Η) 4.07-4.41 (m, 3Η) 4.41-4.68 (m, 1Η) 4.73 (br. s., 1Η) 5.14-5.33 (m, 2Η) 5.70-5.92 (m, 1Η) 6.18-6.31 (m, 1Η) 7.83-8.09 (m, 4Η)
Промежуточное соединение 139: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(диметилкарбамоил)-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
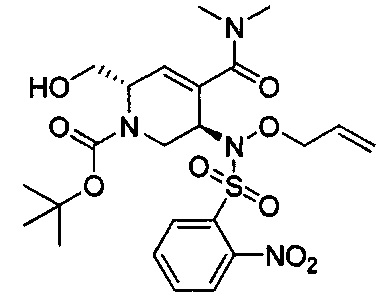
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметил-силилокси)метил)-4-(диметилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 138, 4 г, 6,11 ммоль) в THF (20 мл) насыщали азотом и охлаждали в ледяной бане. В этот раствор по каплям добавляли 1М TBAF в THF (7,33 мл, 7,33 ммоль), и смесь перемешивали в течение 1 ч. Растворитель затем удаляли, и неочищенный продукт наносили на силикагель и очищали флэш-хроматографией (0-20%MeOH-DCM) с получением целевого продукта (2,98 г, 90%) в виде бежевого твердого вещества.
МС: 541 ЭРИ+ (C23H32N4O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.38 (d, J=6.03 Гц, 9Η) 2.85 (br. s., 6Η) 3.04-3.27 (m, 1Η) 3.51 (br. s., 2Η) 4.27 (br. s., 3H) 4.39-4.64 (m, 1H) 4.75 (br. s., 1H) 4.87-4.99 (m, 1H) 5.24 (t, J=3.96 Гц, 2Η) 5.73-5.91 (m, 1Η) 6.18-6.35 (m, 1Η) 8.00 (br. s., 4H)
Промежуточное соединение 140: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(диметилкарбамоил)-2-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
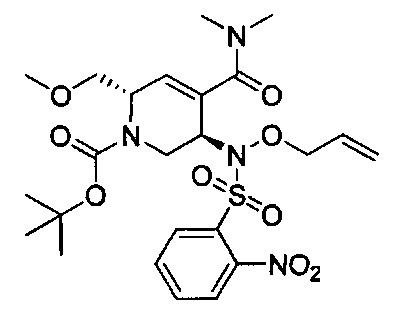
(2S,5R)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(диметилкарбамоил)-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 139, 1,64 г, 3,03 ммоль) и оксид серебра (2,81 г, 12,13 ммоль) растворяли в ацетонитриле (40 мл). В этот раствор добавляли йодметан (1,897 мл, 30,34 ммоль) под N2, и защищенный от света раствор перемешивали в течение 5 суток. Реакционную смесь затем фильтровали через Celite, промывали EtOAc, и неочищенный продукт наносили на силикагель. Продукт очищали флэш-хроматографией (0-20% MeOH/DCM) с получением целевого продукта (1,643 г, 98%) в виде не совсем белого твердого вещества.
МС: 555 ЭРИ+ (C24H34N4O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.38 (d, J=7.35 Гц, 9Η) 2.57-3.20 (m, 7Η) 3.22 (s, 3Η) 3.44 (d, J=3.58 Гц, 2Η) 4.08-4.35 (m, 3Η) 4.57-4.82 (m, 2Η) 5.17-5.31 (m, 2Η) 5.73-5.90 (m, 1Η) 6.16-6.32 (m, 1Η) 7.82-7.92 (m, 1Η) 7.99 (d, J=6.22 Гц, 3Н)
Промежуточное соединение 141: (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-N,N-диметил-1.2,3,6-тетрагидропиридин-4-карбоксамид
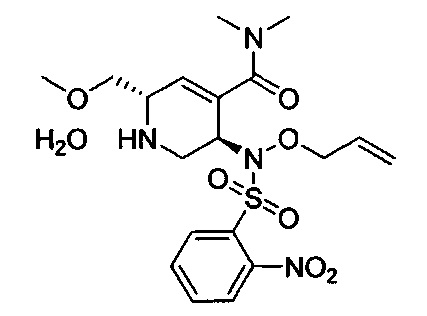
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(диметилкарбамоил)-2-(метоксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 140, 1,642 г, 2,96 ммоль) в DCM (8 мл) добавляли трифторуксусную кислоту (4,33 мл, 56,25 ммоль), и эту смесь перемешивали при комнатной температуре в течение 30 минут. По окончании добавления цвет раствора сразу становился розовым. Затем растворители выпаривали, и неочищенный продукт растворяли в DCM и промывали 0,5 н. NaOH, рассолом, фильтровали и концентрировали с получением целевого продукта (1,346 г, 100%) в виде бежевой пены.
МС: 455 ЭРИ+ (C19H26N4O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.62-3.03 (m, 9Η) 3.20-3.28 (m, 5Η) 3.55 (br. s., 1Η) 4.31-4.44 (m, 1Η) 4.48 (d, J=6.97 Гц, 1Η) 4.65 (br. s., 1Η) 5.19-5.33 (m, 2Η) 5.81-5.97 (m, 1Η) 6.12 (s, 1Η) 7.86-7.92 (m, 1Η) 7.95-8.09 (m, 2Η)
Промежуточное соединение 142: (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-N,N-диметил-1,2,3,6-тетрагидропиридин-4-карбоксамид
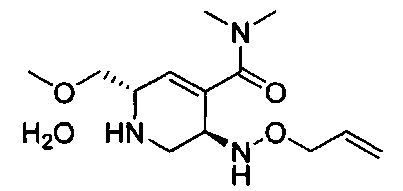
В раствор (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-N,N-диметил-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 141, 1,346 г, 2,96 ммоль) и карбоната калия (2,456 г, 17,77 ммоль) в ацетонитриле (50 мл) добавляли бензолтиол (1,825 мл, 17,77 ммоль), и эту смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, затем неочищенную смесь растворяли в DCM и небольшом количестве МеОН, фильтровали, наносили на силикагель. Неочищенный продукт очищали флэш-хроматографией (0-20% MeOH/DCM) и сушили в глубоком вакууме с получением целевого продукта (0,725 г, 91%) в виде бледно-желтого масла.
МС: 270 ЭРИ+ (C13H23N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.78-3.00 (m, 9Η) 3.20-3.28 (m, 5Η) 3.46 (br. s., 1Η) 3.58 (d, J=8.10 Гц, 1Η) 4.02 (dt, J=5.51, 1.39 Гц, 2Η) 5.08-5.23 (m, 2Η) 5.79-5.92 (m, 2Η) 6.36-6.41 (m, 1Η)
Промежуточное соединение 143: (2S,5R)-6-(аллилокси)-2-(метоксиметил)-N,N-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
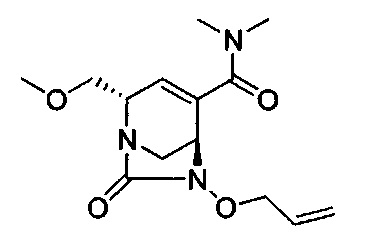
Раствор (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-N,N-диметил-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 142, 725 мг, 2,69 ммоль) и основания Хюнига (1,880 мл, 10,77 ммоль) в ацетонитриле (350 мл) охлаждали до температуры ниже 0°С в бане лед-соль и затем добавляли раствор трифосгена (320 мг, 1,08 ммоль) в ACN (22 мл) со скоростью 0,1 мл/мин. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции растворители удаляли, и неочищенную смесь растворяли EtOAc, промывали насыщенным раствором NaHCO3 и рассолом, сушили над MgSO4, фильтровали и концентрировали с получением целевого продукта (658 мг, 83%) в виде бледно-желтого масла.
МС: 296 ЭРИ+ (C14H21N3O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.92 (br. s., 6Η) 3.16-3.22 (m, 1Η) 3.28 (s, 3Η) 3.32-3.38 (m, 1Η) 3.51-3.65 (m, 2Η) 3.92 (ddd, J=6.40, 4.99, 2.92 Гц, 1Η) 4.16 (d, J=2.64 Гц, 1Η) 4.34 (dd, J=5.84, 1.32 Гц, 2Η) 5.20-5.35 (m, 2Η) 5.80 (dd, J=2.92, 1.04 Гц, 1Η) 5.81-5.95 (m, 1Η)
Промежуточное соединение 144: (2S,5R)-4-(диметилкарбамоил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония 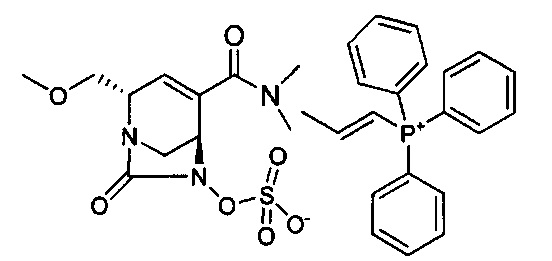
В раствор (2S,5R)-6-(аллилокси)-2-(метоксиметил)-N,N-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамида (Промежуточное соединение 143, 115 мг, 0,39 ммоль) и АсОН (0,045 мл, 0,78 ммоль) (высушенной над сульфатом натрия) в CH2Cl2 (3 мл) при комнатной температуре добавляли Pd(Ph3P)4 (450 мг, 0,39 ммоль) в атмосфере азота. Раствор перемешивали при комнатной температуре в течение 1 ч. В эту реакционную смесь добавляли пиридин (3,00 мл) и комплекс триоксида серы с пиридином (496 мг, 3,12 ммоль), и перемешивание продолжали под N2 при комнатной температуре в течение ночи. По результатам ЖХ/МС в реакционной смеси наблюдалась желаемая масса. Суспензию выпаривали досуха и затем ресуспендировали в DCM. Твердое вещество отфильтровывали через фильтр Nalgene 0,2 мкм. Фильтрат концентрировали с получением желтого масла. 80 мг неочищенного вещества очищали препаративной ВЭЖХ с использованием смеси вода/муравьиная кислота. Продукт затем лиофилизировали с получением 6 мг слегка загрязненного примесями продукта, который хранили в инертной атмосфере до использования на следующей стадии.
ПРИМЕР 16
Натриевая соль (2S,5R)-2-(гидроксиметил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
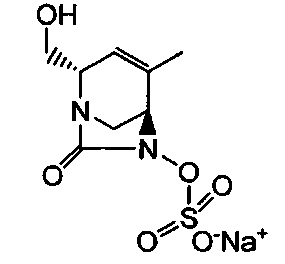
В лабораторном стакане на 150 мл смолу DOWEX 50WX8 (50 г) кондиционировали путем перемешивания ее в течение 20 минут в 2 н. гидроксиде натрия (40 мл), загружали ее в картридж, промывали водой до pH 7, затем промывали смесью ацетон/вода (1/1) и снова водой. Наносили (2S,5R)-2-((трет-бутилдиметилсилилокси)-метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 145, 190 мг), используя воду и минимальное количество ацетона, и элюировали водой. Фракции замораживали и лиофилизировали с получением соединения в виде белого твердого вещества (130 мг), которое очищали обращенно-фазовой хроматографией (MeCN в воде: 0-4%) с получением белого твердого вещества в качестве целевого продукта (31 мг).
МС: 263 ЭРИ- (C8H12N2O6S[Na+])
1Н ЯМР (300 МГц. DMSO-d6) δ: 1.76 (t, J=1.70 Гц, 3Η) 3.02-3.14 (m, 1Η) 3.15-3.25 (m, 1Η) 3.41-3.52 (m, 1Η) 3.52-3.62 (m, 2Η) 3.92 (d, J=2.83 Гц, 1Η) 4.72-4.89 (m, 1Η) 5.27 (s, 1Η)
Промежуточное соединение 145: (2S.5R)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
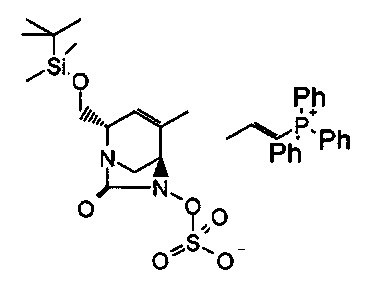
В раствор (2S,5R)-6-(аллилокси)-2-((трет-бутилдиметилсилилокси)метил)-4-метил-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 13, 0,40 г, 1,18 ммоль) в DCM (20 мл) добавляли высушенную уксусную кислоту (0,135 мл, 2,36 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,365 г, 1,18 ммоль) под азотом при комнатной температуре. Желтый раствор перемешивали при комнатной температуре в течение 4 часов. ЖХ/МС показала, что исходного вещества не осталось. Затем в смесь добавляли пиридин (20 мл), затем комплекс пиридин-триоксид серы (1,128 г, 7,09 ммоль). Суспензию перемешивали при комнатной температуре в течение ночи в атмосфере азота. Смесь концентрировали досуха(≤30°С), растворяли в DCM и фильтровали. Неочищенный продукт очищали на силикагелевой колонке (ацетон в DCM: 10-40%) с получением целевого продукта в виде белого твердого вещества (190 мг).
МС: 377 ЭРИ- (C14H25N2O6SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: м.д. -0.11-0.09 (m, 6Η) 0.73-0.88 (m, 9Η) 1.21-1.22 (m, 0Η) 1.71 (t, J=1.89 Гц, 3Η) 2.06-2.19 (m, 2Η) 3.03 (dd, J=11.05, 2.93 Гц, 1Η) 3.42-3.57 (m, 1Η) 3.60-3.79 (m, 2Η) 3.88 (d, J=3.02 Гц, 1Η) 5.20 (dt, J=2.74, 1.46 Гц, 1Η) 6.44-6.73 (m, 1Η) 7.08-7.36 (m, 1Η) 7.51-7.79 (m, 10Η) 7.80-7.98 (m, 3Η)
Пример 17
(2S,5R)-3-метил-7-оксо-2-(пиперидин-1-ий-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат
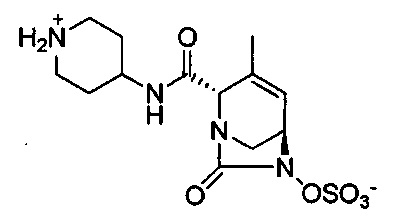
В раствор (2S,5R)-2-((1-(трет-бутоксикарбонил)пиперидин-4-ил)карбамоил)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-ен-1-ил)фосфония (Промежуточное соединение 152, 0,2 г, 0,26 ммоль) в DCM (3 мл) при 0°С добавляли TFA (0,5 мл). Реакционную смесь перемешивали в течение 30 минут и концентрировали с получением желтого масла. Остаток растворяли в DCM и экстрагировали водой. Водную фазу лиофилизировали. Очистку выполняли методом обращенно-фазовой ВЭЖХ (0%-20% ацетонитрила в воде, колонка Synergi Polar RP 21,2 мм × 100 мм, 4 мкм, спаренная с YMC С30, 20 мм × 150 мм, 5 мкм) с получением указанного в заголовке соединения в виде белого твердого вещества (18,7 мг, 20%).
Оптическое вращение: (0,16 г/дл, DMSO)=-291
МС: 359 ЭРИ- (C13H20N4O6S)
1Н ЯМР (300 МГц, D2O) δ: 1.74 (m, 3Н); 1.82 (m, 2Н); 2.19 (m, 2Н); 3.15 (m, 2Н); 3.47 (m, 3Н); 3.61 (m, 1Н); 4.05 (m, 1Н); 4.29 (m, 1Н); 4.39 (m, 1Н); 6.30 (m, 1Н).
Промежуточное соединение 146: N-[(3R,6S)-6-[[(трет-бутилдиметилсилил)окси]метил]-5-метил-1,2,3,6-тетрагидропиридин-3-ил]-2-нитро-N-(проп-2-ен-1-илокси)бензол-1-сульфонамид
В круглодонную колбу на 250 мл, которую продували азотом и поддерживали в инертной атмосфере азота, помещали раствор трет-бутил-(3R,6S)-6-[[(трет-бутилдиметилсилил)окси]метил]-5-метил-3-[N-(проп-2-ен-1-илокси)(2-нитробензол)сульфонамидо]-1,2,3,6-тетрагидропиридин-1-карбоксилата (Промежуточное соединение 80, 13,6 г, 22,75 ммоль, 1,00 экв.) в дихлорметане (100 мл). После этого несколькими порциями добавляли ZnBr2 (10,2 г, 45,29 ммоль, 2,00 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Полученный раствор разбавляли 500 мл дихлорметана. Полученную смесь промывали 2×200 мл бикарбоната натрия (водн.) и 2×200 мл NH4Cl (водн.). Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. В результате получили 12 г (неочищенный продукт) указанного в заголовке соединения в виде желтого масла.
МС: 498 ЭРИ+ (C22H35N3O6SSi)
Промежуточное соединение 147: (3R,6S)-6-[[(трет-бутилдиметилсилил)окси]метил]-5-метил-N-(проп-2-ен-1-илокси)-1,2,3,6-тетрагидропиридин-3-амин
В круглодонную колбу на 250 мл, которую продували азотом и поддерживали в инертной атмосфере азота, помещали раствор N-[(3R,6S)-6-[[(трет-бутилдиметилсилил)окси]метил]-5-метил-1,2,3,6-тетрагидропиридин-3-ил]-2-нитро-N-(проп-2-ен-1-илокси)бензол-1-сульфонамида (Промежуточное соединение 146, 12 г, 24,11 ммоль, 1,00 экв.) в Ν,Ν-диметилформамиде (100 мл) и 2-сульфанилуксусную кислоту (4,4 г, 47,77 ммоль, 2,00 экв.). После этого порциями добавляли LiOH (5,8 г, 242,17 ммоль, 10,00 экв.). Полученный раствор перемешивали в течение 2 ч при комнатной температуре. Полученный раствор разбавляли 500 мл воды. Полученный раствор экстрагировали 5×200 мл этилацетата, и органические слои объединяли. Полученную смесь промывали 3×200 мл рассола и 2×200 мл бикарбоната натрия (водн.). Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. В результате получили 8,4 г (неочищенный продукт) указанного в заголовке соединения в виде желтого масла.
МС: 313 ЭРИ+ (C16H32N2O2Si)
Промежуточное соединение 148: (2S,5R)-2-[[(трет-6утилдиметилсилил)окси]метил]-3-метил-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
В 3-хгорлую круглодонную колбу на 2000 мл, которую продували азотом и поддерживали в инертной атмосфере азота, помещали раствор (3R,6S)-6-[[(трет-бутилдиметилсилил)окси]метил]-5-метил-N-(проп-2-ен-1-илокси)-1,2,3,6-тетрагидропиридин-3-амина (Промежуточное соединение 147, 8,4 г, 26,88 ммоль, 1,00 экв.) в ацетонитриле (1600 мл) и Ν,Ν-диизопропилэтиламин (14,2 г, 109,87 ммоль, 4,00 экв.). После этого по каплям добавляли раствор дитрихлорметилкарбонат (2,9 г, 9,77 ммоль, 0,40 экв.) в ацетонитриле (100 мл) при перемешивании при -15°С через 3 ч. Полученный раствор перемешивали в течение ночи при комнатной температуре. Полученную смесь концентрировали в вакууме. Полученный раствор разбавляли 500 мл этилацетата. Полученную смесь промывали 2×400 мл NH4Cl (водн.) и 2×400 мл рассола. Полученную смесь концентрировали в вакууме. Остаток наносили на силикагелевую колонку и элюировали смесью этилацетат/петролейный эфир (1:10). В результате получили 3,9 г (43%) указанного в заголовке соединения в виде желтого масла.
МС: 339 ЭРИ+ (C17H30N2O3Si)
Промежуточное соединение 149: (2S,5R)-2-(гидроксиметил)-3-метил-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
В круглодонную колбу на 100 мл помещали тетрагидрофуран (30 мл) и (2S,5R)-2-[[(трет-бутилдиметилсилил)окси]метил]-3-метил-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он (Промежуточное соединение 148, 3,2 г, 9,45 ммоль, 1,00 экв.), и эту смесь охлаждали до 0°С, затем по каплям добавляли TBAF (14,2 мл 1 н. в THF, 1,50 экв.). Полученный раствор перемешивали в течение 1 ч при 0°С в бане вода/лед. Полученную смесь концентрировали в вакууме. Остаток наносили на силикагелевую колонку и элюировали смесью этилацетат/петролейный эфир (1:5-1:2). В результате получили 1,6 г (75%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
МС: 225 ЭРИ+ (C11H16N2O3)
1Н ЯМР (300 МГц, CDCl3) δ: 1.63 (3Н, d), 3.20 (2Н, d), 3.62-3.84 (2Н, m), 3.85-3.90 (2Н, m), 4.35-4.48 (2Н, m), 5.28-5.39 (2Н, m), 5.95-6.08 (2Н, m).
Промежуточное соединение 150: (2S,5R)-3-метил-7-оксо-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновая кислота
В раствор H5IO6(12,93 г,56,71 ммоль) во влажном CH3CN (150 мл, 0,75% H2O об./об.) при комнатной температуре добавляли CrO3 (128 мг, 1,28 ммоль). Эту смесь перемешивали до полного растворения. В круглодонную колбу на 100 мл помещали влажный ацетонитрил (35 мл) и (2S,5R)-2-(гидроксиметил)-3-метил-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он (Промежуточное соединение 149, 740 мг, 3,30 ммоль, 1,00 экв.), и эту смесь охлаждали до 0°С, затем по каплям добавляли приготовленный выше раствор для окисления (35 мл, 3,00 экв.) в течение 30 минут при 0°С. Полученный раствор перемешивали в течение 1 ч при 0°С в бане вода/лед, и методом ТСХ было определено, что исходное вещество полностью израсходовано. Затем смесь разбавляли 200 мл хлороформа и 50 мл раствора лимонной кислоты (25%). Органический слой отделяли, промывали 3×50 мл рассола, сушили над безводным сульфатом натрия и концентрировали в вакууме. В результате получили 0,700 г неочищенного указанного в заголовке соединения в виде желтого масла.
МС: 239 ЭРИ+ (C11H16N2O4)
Промежуточное соединение 151: трет-бутил-4-[(2S,5R)-3-метил-7-оксо-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-2-амидо]пиперидин-1-карбоксилат
В круглодонную колбу на 100 мл помещали Ν,Ν-диметилформамид (20 мл), неочищенную (2S,5R)-3-метил-7-оксо-6-(проп-2-ен-1-илокси)-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновую кислоту (Промежуточное соединение 150, 700 мг, 2,94 ммоль, 1,00 экв.), трет-бутил-4-аминопиперидин-1-карбоксилат (1,17 г, 5,84 ммоль, 1,99 экв.), HATU (1,67 г, 4,39 ммоль, 1,49 экв.) и DIEA (1,51 г, 11,68 ммоль, 3,98 экв.) при 0°С. Полученный раствор перемешивали в течение 12 ч при 25°С. Полученный раствор разбавляли 150 мл этилацетата и промывали 3×100 мл рассола, сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток наносили на силикагелевую колонку и элюировали смесью этилацетат/петролейный эфир (1:5-1:2). В результате получили 0,500 г (40%) указанного в заголовке соединения в виде белого твердого вещества.
МС: 421 ЭРИ+ (C21H32N4O5)
1Н ЯМР (300 МГц, CDCl3) δ: 1.36 (2Н, m), 1.46 (9Н, s), 1.88-2.05 (5Н, m), 2.90 (2Н, m), 3.07 (1Н, m), 3.24 (1Н, m), 3.79-3.92 (2Н, m), 4.06 (2Н, m), 4.23 (1Н, s), 4.44 (2Н, m), 5.29-5.39 (2Н, m), 5.96-6.10 (2Н, m), 6.73 (1Н, d).
Промежуточное соединение 152: (2S,5R)-2-((1-(трет-бутоксикарбонил)пиперидин-4-ил)карбамоил)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-ен-1-ил)-фосфония
Указанное в заголовке соединение получали из трет-бутил-4-((2S,5R)-6-(аллилокси)-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамидо)-пиперидин-1-карбоксилата (Промежуточное соединение 151, 0,763 г, 1,81 ммоль) по методике, описанной для Промежуточного соединения 17. После хроматографии на силикагеле (0%-5% метанола/дихлорметан) получили целевой продукт в виде желтой пены (1,22 г, 88%).
МС: 459 ЭРИ-, 303 ЭРИ+ (C18H27N4O8S, C21H20P)
Пример 18
Натриевая соль (2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
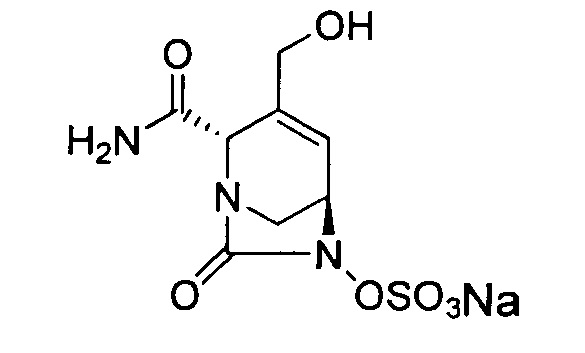
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата (Промежуточное соединение 167, раствор из ионообменной колонки) по методике, описанной в Примере 1. Целевой продукт был получен в виде не совсем белого рыхлого твердого вещества, 5,6 мг.
Путь, описанный в этом примере, может быть использован для синтеза других соединений по изобретению, таких как соединение, описанное в Примере 10.
МС: 292 ЭРИ- (C8H10N3O7S)
1Н ЯМР (300 МГц, ОКСИД ДЕЙТЕРИЯ-d) δ: 3.44-3.58 (m, 2Н) 4.14 (s, 2Н) 4.37 (d, J=5.09 Гц, 1Н) 4.63 (s, 1Н) 6.52 (d, J=4.90 Гц, 1Н).
Промежуточные соединения для получения соединения Примера 18 получали следующим образом:
Промежуточное соединение 153: трет-бутил-бензилоксикарбамат
В раствор гидрохлорида О-бензилгидроксиламина (225 г, 1,44 моль) в дихлорметане (300 мл) добавляли раствор бикарбоната натрия (261 г, 3,11 моль, 300 мл). Через 1 час добавляли ди-трет-бутил-дикарбонат (375 г, 1,72 моль) при 0°С. Полученный раствор перемешивали в течение 60 минут при 0°С в бане вода/лед, и затем реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Реакцию затем гасили добавлением 300 мл водного раствора бикарбоната натрия. Водную фазу экстрагировали 3×500 мл дихлорметана, и органические слои объединяли и сушили над безводным сульфатом натрия и концентрировали в вакууме. После флэш-хроматографии на силикагеле (РЕ:ЕА=10:1) получили 220 г (69%) указанного в заголовке соединения в виде желтого масла.
1Н ЯМР (300МГц, CDCl3) δ: 1.43 (9Н, s), 4.85 (2Н, s), 7.19-7.21 (1Н, brs), 7.30-7.40 (5Н, m).
Промежуточное соединение 154: (R)-трет-бутил-бензилокси(1-гидроксибут-3-ен-2-ил)карбамат
В колбе RBF (круглодонная колба) на 3 литра через раствор трет-бутил-бензилоксикарбамата (Промежуточное соединение 153, 86,9 г, 389,22 ммоль), N,N(1S,2S)-циклогексан-1,2-диил)бис(2-(дифенилфосфино)-1-нафтамида) (18,5 г, 23,39 ммоль), Pd2(dba)3-CHCl3 (8,05 г, 7,78 ммоль) и ТВАВ (150,5 г, 466,81 ммоль) в ацетонитриле (1,8 л) барботировали N2 в течение 30 минут, и колбу закрывали резиновой пробкой. Раствор затем держали в морозильнике при -20°С в течение 2 ч. Добавляли 2-винилоксиран (30 г, 428,02 ммоль, 1,10 экв.), и смесь перемешивали в морозильнике (-20-25°С) в течение 3 суток. Смесь фильтровали, и фильтрат упаривали. Остаток очищали на силикагелевой колонке, элюируя смесью EtOAc/гексан (0-40%), с получением указанного в заголовке соединения (106 г, 93%) в виде бледно-желтого масла.
МС: 194 ЭРИ+ (C16H23NO4)
Промежуточное соединение 155: (R)-трет-бутил-бензилокси(1-(1,3-диоксоизоиндолин-2-ил)бут-3-ен-2-ил)карбамат
Суспензию (R)-трет-бутил-бензилокси(1-гидроксибут-3-ен-2-ил)-карбамата (Промежуточное соединение 154, 74 г, 252,25 ммоль), фталимида (66,8 г, 454,02 ммоль) и трифенилфосфина (119 г, 453,70 ммоль) в смеси толуол/THF (200/200 мл) охлаждали под азотом с использованием льда. По каплям добавляли DIAD (91,8 г, 453,98 ммоль) в течение 20 минут, и смесь перемешивали при 0°С в течение 20 минут без охлаждения в течение 2 ч. Смесь фильтровали, и фильтрат выпаривали. Остаток переносили в диэтиловый эфир и фильтровали. Фильтрат выпаривали, и остаток очищали на силикагелевой колонке, элюируя смесью EtOAc/гексан (0-50%), с получением указанного в заголовке соединения (74 г, 79%) в виде бесцветного масла.
МС: 323 ЭРИ+ (C24H26N2O5)
Промежуточное соединение 156: (R)-трет-бутил-1-аминобут-3-ен-2-ил(бензилокси)карбамат
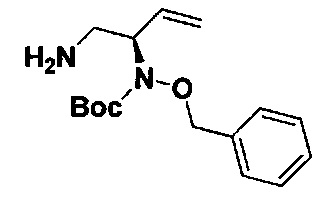
Раствор (R)-трет-бутил-бензилокси(1-(1,3-диоксоизоиндолин-2-ил)бут-3-ен-2-ил)карбамата (Промежуточное соединение 155, 80 г, 189,36 ммоль) и гидразингидрата (47,4 г, 946,86 ммоль) в МеОН (800 мл) перемешивали при комнатной температуре в течение ночи. Смесь разбавляли диэтиловым эфиром и фильтровали. Фильтрат выпаривали, и остаток очищали на силикагелевой колонке, элюируя смесью MeOH/DCM (0-10%), с получением указанного в заголовке соединения (44,3 г, 80%) в виде бесцветного масла.
1Н ЯМР (300 МГц, МЕТАНОЛ-d) δ: 7.36-7.40 (m, 5Н), 5.76-5.88 (m, 1Н), 5.14-5.21 (m, 2Н), 4.83 (q, 2Н), 4.27-4.30 (m, 1Н), 2.81-2.88 (m, 1Н), 2.67-2.73 (m, 1Н), 1.45 (s, 9Н).
Промежуточное соединение 157: 2-(((трет-бутилдиметилсилил)-окси)метил)проп-2-ен-1-ол
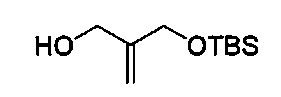
В сухую колбу, содержащую гидрид натрия (2,270 г, 56,75 ммоль) в THF (150 мл), под N2 при 0°С медленно добавляли 2-метиленпропан-1,3-диол (5 г, 56,75 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали 45 минут. Одной порцией добавляли TBS-Cl (8,55 г, 56,75 ммоль), и перемешивание продолжали в течение еще 45 минут до окончания реакции по результатам ТСХ. В реакционную смесь добавляли воду, и смесь экстрагировали три раза EtOAc и промывали рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали примерно при 10-15°С. После хроматографии на силикагеле (20%-50% EtOAc/Нех) получили указанное в заголовке соединение (11,36 г, 99%) в виде прозрачного масла.
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (s, 6Η) 0.88 (s, 10Η) 3.91 (d, J=5.65 Гц, 2Η) 4.10 (s, 2Η) 4.74 (t, J=5.46 Гц, 1Η) 5.00 (t, J=1.51 Гц, 2Η).
Промежуточное соединение 158: 2-(((трет-бутилдиметилсилил)-окси)метил)акрилальдегид
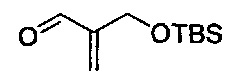
В раствор 2-(((трет-бутилдиметилсилил)окси)метил)проп-2-ен-1-ола (Промежуточное соединение 157, 11,35 г, 56,09 ммоль) в DCM (150 мл) добавляли диоксид марганца (активированный, 5 мкм) (28,7 г, 280,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение трех суток. Снова добавляли MnO2, и перемешивание продолжали до тех пор, пока ТСХ не показала минимум исходного вещества. Реакционную смесь фильтровали через слой диоксида кремния, упаривали при примерно 15°С и очищали на силикагелевой колонке (5-20%, ЕА/Нех) с получением указанного в заголовке соединения (6,70 г, 59,6%) в виде бесцветного масла.
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06-0.13 (m, 6Η) 0.89-0.97 (m, 9Η) 4.41 (t, J=2.07 Гц, 2Η) 6.11 (q, J=1.88 Гц, 1Η) 6.50-6.56 (m, 1Η) 9.63 (s, 1Η).
Промежуточное соединение 159: трет-бутил-((2R)-1-((((9Н-флуорен-9-ил)метокси)карбонил)(2-(((трет-бутилдиметилсилил)окси)метил)-1-цианоаллил)амино)бут-3-ен-2-ил)(бензилокси)карбамат
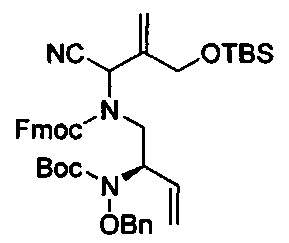
2-(((трет-Бутилдиметилсилил)окси)метил)акрилальдегид (Промежуточное соединение 158, 4,76 г, 23,76 ммоль) добавляли в раствор (R)-трет-бутил-(1-аминобут-3-ен-2-ил)(бензилокси)карбамата (Промежуточное соединение 156, 5,79 г, 19,80 ммоль) в THF (100 мл), затем добавляли триметилсилилцианид (10,62 мл, 79,19 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Снова добавляли TMS-CN, и перемешивание продолжали до тех пор, пока методом ЖХ/МС не определили меньшее количество исходного вещества. В реакционную смесь добавляли MgSO4 и перемешивали в течение 1 ч. Добавляли бикарбонат натрия (3,33 г, 39,58 ммоль), затем (9Н-флуорен-9-ил)метилкарбонохлоридат (7,68 г, 29,69 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли на 0,3 экв. больше FmocCl и перемешивали более 5 ч. Смесь фильтровали, и фильтрат выпаривали. Остаток очищали на силикагелевой колонке (0-22%, EtOAc/гексан) с получением указанного в заголовке соединения (3,32 г, 23,18%) в виде бледно-желтого густого масла.
МС: 724 ЭРИ+ (C42H53N3O6Si)
Промежуточное соединение 160: (5R)-(9Н-флуорен-9-ил)метил-5-((бензилокси)(трет-бутоксикарбонил)амино)-3-(((трет-бутилдиметил-силил)окси)метил)-2-циано-5.6-дигидропиридин-1(2Н)-карбоксилат
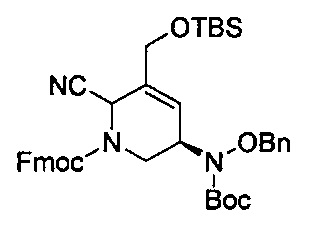
Через раствор трет-бутил-((2R)-1-((((9Н-флуорен-9-ил)метокси)-карбонил)(2-(((трет-бутилдиметилсилил)окси)метил)-1-цианоаллил)амино)бут-3-ен-2-ил)(бензилокси)карбамата (Промежуточное соединение 159, 3,32 г, 4,59 ммоль) в толуоле (150 мл) барботировали азот в течение 15 минут. Добавляли катализатор Ховейды-Граббса 2-го поколения (0,288 г, 0,46 ммоль), и через смесь барботировали азот в течение еще 15 минут. Полученный раствор нагревали под азотом при 90°С в течение 2 суток. Реакционную смесь выпаривали. Остаток очищали на силикагелевой колонке (0-30% EtOAc/гексан) с получением указанного в заголовке соединения (1,300 г, 40,7%) в виде светло-коричневой пленки.
МС: 596 ЭРИ+ (C40H49N3O6Si)
Промежуточное соединение 161: (5R)-(9Н-флуорен-9-ил)метил 5-((бензилокси)(трет-бутоксикарбонил)амино)-3-(((трет-бутилдиметил-силил)окси)метил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилат
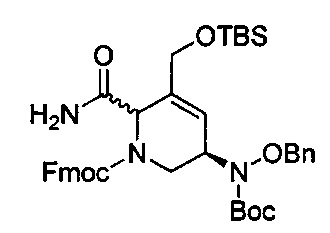
Приготовление хлорида меди(II) на молекулярных ситах Å: молекулярные сита 4 Å (2 г, 2,00 ммоль) добавляли в раствор дигидрата хлорида меди(II) (0,341 г, 2,00 ммоль) в воде (200 мл), и суспензию перемешивали в течение ночи. Твердое вещество отфильтровывали, промывали водой и ацетоном и сушили в термостате при 140°С в течение 1 ч с получением 2,104 г синего твердого вещества. Это вещество активировали при температуре 140°С в течение пары часов и охлаждали до комнатной температуры в эксикаторе до использования.
Смесь (5R)-(9Н-флуорен-9-ил)метил-5-((бензилокси)(трет-бутоксикарбонил)амино)-3-(((трет-бутилдиметилсилил)окси)метил)-2-циано-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 160, 1,3 г, 1,87 ммоль), ацетальдегидоксима (0,569 мл, 9,34 ммоль) и активированного хлорида меди(II) на молекулярных ситах 4 Å (187 мг, 0,15 ммоль) (использовали 100 мг CuCl2/моль сит на 1 ммоль субстрата) в МеОН (5 мл) перемешивали под азотом при 65°С в течение 6 ч. Смесь фильтровали и упаривали. Остаток очищали на силикагелевой колонке (0-60%, EtOAc/гексаны). Два диастереомера объединяли вместе с получением указанного в заголовке соединения (1,190 г, 89%) в виде бледно-желтого пенистого твердого вещества.
МС: 714 ЭРИ+ (C40H51N3O7Si)
Промежуточное соединение 162: (513)-(9Н-флуорен-9-ил)метил-5-((бензилокси)амино)-3-(((трет-бутилдиметилсилил)окси)метил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилат
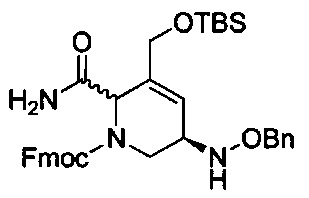
В раствор (5R)-(9Н-флуорен-9-ил)метил-5-((бензилокси)(трет-бутоксикарбонил)амино)-3-(((трет-бутилдиметилсилил)окси)метил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 161, 1,19 г, 1,67 ммоль) в DCM (20 мл) при комнатной температуре добавляли бромид цинка (1,502 г, 6,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия. Водную промывочную жидкость экстрагировали более трех-четырех раз DCM. Органические фазы промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения (0,970 г, 95%) в виде не совсем белой пены.
МС: 614 ЭРИ+ (C35H43N3O5Si)
Промежуточное соединение 163: (5R)-5-((бензилокси)амино)-3-(((трет-бутилдиметилсилил)окси)метил)-1,2,5,6-тетрагидропиридин-2-карбоксамид
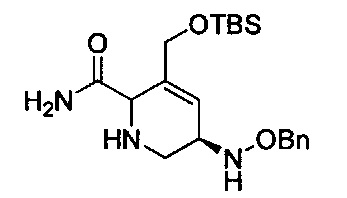
Диэтиламин (0,824 мл, 7,89 ммоль) добавляли в раствор (5R)-(9H-флуорен-9-ил)метил-5-((бензилокси)амино)-3-(((трет-бутилдиметилсилил)окси)метил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 162, 0,968 г, 1,58 ммоль) в DCM (20 мл), и полученный раствор перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли выпариванием, и остаток очищали на силикагеле (0-12%, MeOH/DCM, УФ 220 нм) с получением указанного в заголовке соединения (0,564 г, 91%) в виде желтой пленки.
МС: 392 ЭРИ+ (C20H33N3O3Si)
Промежуточное соединение 164: (2S,5R)-6-(бензилокси)-3-(((трет-бутилдиметилсилил)окси)метил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
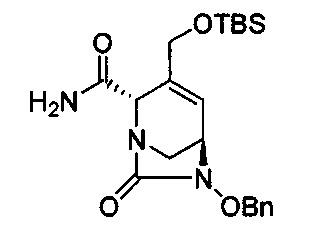
В раствор (5R)-5-((бензилокси)амино)-3-(((трет-бутилдиметилсилил)-окси)метил)-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 163, 0,562 г, 1,44 ммоль) и диизопропилэтиламина (1,000 мл, 5,74 ммоль) в ацетонитриле (200 мл) при 0°С добавляли трифосген (0,145 г, 0,49 ммоль) в виде раствора в ацетонитриле (6 мл). Раствор трифосгена добавляли со скоростью 4 мл/час. Сразу после окончания добавления реакционную смесь перемешивали при 0°С в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия. Водный слой снова экстрагировали EtOAc и затем DCM. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-100, этилацетата/гексаны) получили указанное в заголовке соединение (0,225 г, 37,5%) в виде бесцветной пленки.
МС: 418 ЭРИ+ (C21H31N3O4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.08 (s, 6Η) 0.76 (s, 9Η) 2.89-3.00 (m, 1Η) 3.04-3.13 (m, 1Η) 3.32 (dd, J=5.09, 2.45 Гц, 1Η) 4.01-4.09 (m, 1Η) 4.19-4.28 (m, 1Η) 4.30 (s, 1 Η) 4.67-4.76 (m, 1Η) 4.83-4.93 (m, 1Η) 5.51 (br. s., 1Η) 6.09-6.21 (m, 1Η) 6.63 (br. s., 1H) 7.19-7.36 (m, 5H).
Промежуточное соединение 165: (2S,5R)-3-(((трет-бутилдиметилсилил)окси)метил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат триметиламмония
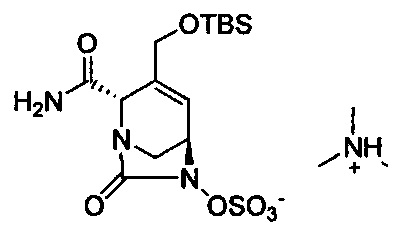
В раствор (2S,5R)-6-(бензилокси)-3-(((трет-бутилдиметилсилил)окси)метил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 164, 30 мг, 0,07 ммоль) в EtOAc (1,33 мл), воде (1,995 мл) и EtOH (0,660 мл) добавляли Pd/C (влажный, типа Degussa Е101 NE/W) (7,65 мг, 7,18 мкмоль), SO3. TMA (12,00 мг, 0,09 ммоль) и TEA (2,003 мкл, 0,01 ммоль) в атмосфере N2. Реакционную смесь дегазировали и заполняли Н2, используя баллон. Реакционную смесь перемешивали при комнатной температуре при подключенном баллоне с Н2 в течение 5 часов. Баллон с Н2 удаляли, и реакционную смесь перемешивали в течение ночи. Реакционную смесь фильтровали, и органические фазы упаривали. Оставшуюся водную смесь лиофилизировали, и полученный остаток очищали препаративной ВЭЖХ (колонка Synergi Polar RP 21,2 мм × 100 мм 4 мкм, спаренная с YMC С30 20 мм × 100 мм 5 мкм) с получением указанного в заголовке соединения в виде белого твердого вещества, 5 мг, 15%.
МС: 406 ЭРИ- (C14H24N3O7SSi)
Промежуточное соединение 166: натриевая соль (2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
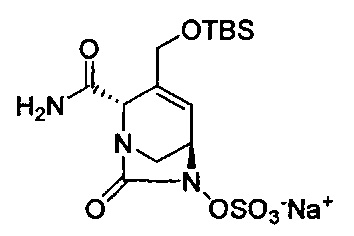
Смолу Dowex 50WX8, 100-200 меш, (0,75 г, 0,01 ммоль) кондиционировали путем перемешивания в течение 3 часов в 2 н. NaOH (1,8 мл, 3,60 ммоль). Смолу затем загружали в картридж и промывали водой до рН 7. Затем ее промывали смесью (1/1) ацетон/вода, затем водой. (2S,5R)-3-(((трет-Бутилдиметилсилил)окси)метил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Промежуточное соединение 165, 5 мг, 0,01 ммоль) в воде пропускали через смолу 5 раз и затем лиофилизировали с получением указанного в заголовке соединения (3,00 мг, 56,7%).
МС: 406 ЭРИ+ (C14H24N3O7SSi)
Промежуточное соединение 167: (2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат
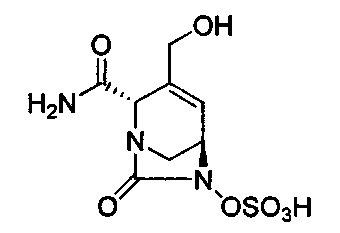
Смолу Dowex 50WX8, 100-200 меш, (0,48 г, 6,43 мкмоль) в 2 мл воды загружали в картридж, и воде давали стечь. Натриевую соль (2S,5R)-3-(((трет-бутилдиметилсилил)окси)метил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Промежуточное соединение 166, 3 мг, 6,43 мкмоль) в воде пропускали через смолу 2 раза (рН примерно 3). Раствор направляли в конечную ионообменную колонку (смотри Пример 18).
Пример 19
Натриевая соль (2S,5R)-4-(2-амино-2-оксоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
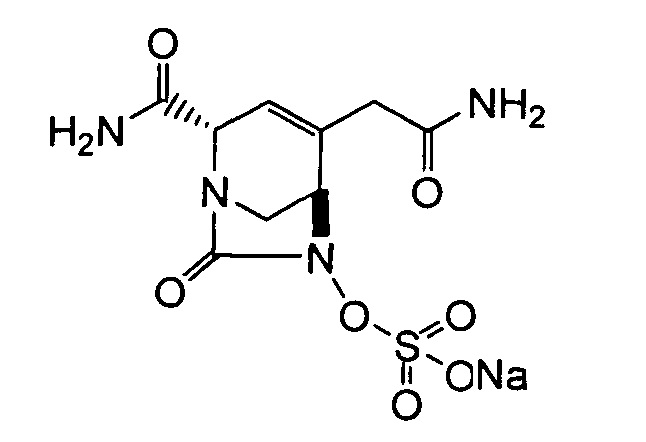
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-4-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Промежуточное соединение 179, 34 мг, 0,04 ммоль) по методике, описанной для Примера 1. Затем его очищали обращенно-фазовой ВЭЖХ на колонке Synergi Polar RP (21,2 мм × 100 мм 4 мкм), спаренной с YMC С30 (20×150 мм 5 мкм) (подвижная фаза А: 100% H2O, подвижная фаза В: 100% ацетонитрил), с получением бледно-желтого твердого вещества, 1,6 мг, 3%.
МС: 343 ЭРИ+ (C9H12N4NaO7S)
1Н ЯМР (300 МГц, D2O) δ: 3.17 (s, 2Н); 3.37 (d, 1Н); 3.70 (dd, 1Н); 4.23 (d, 1Н); 4.64 (d, 1Н); 5.93 (d, 1Н).
Промежуточные соединения для получения соединения Примера 19 получали следующим образом:
Промежуточное соединение 168: (S)-трет-бутил-2-(((трет-бутилдиметилсилил)окси)метил)-4-(((4-метоксибензил)-окси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
В раствор (2S)-трет-бутил-5-гидрокси-4-(гидроксиметил)-2-(изопропилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 117, 13,0 г, 34,99 ммоль) в толуоле (150 мл) добавляли 4-метоксибензил 2,2,2-трихлорацетимидат (7,65 мл, 10,42 г, 36,74 ммоль) и La(OTf)3 (205 мг, 0,35 ммоль). Смесь перемешивали при 50°С в течение 4 ч. Водную фазу обрабатывали этилацетатом, и органический слой сушили над MgSO4 с получением неочищенного продукта.
МС: 492 ЭРИ+ (C26H41NO6Si)
Промежуточное соединение 169: (2S)-трет-бутил-2-(((трет-бутилдиметилсилил)окси)метил)-5-гидрокси-4-(((4-метоксибензил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат
Указанное в заголовке соединение получали из (S)-трет-бутил-2-(((трет-бутилдиметилсилил)окси)метил)-4-(((4-метоксибензил)окси)метил)-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 168, 34,99 ммоль) по методике, описанной для Промежуточного соединения 8. Реакционную смесь концентрировали, и белое твердое вещество растворяли в 200 мл EtOAc и промывали насыщенным раствором NaHCO3, рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, с получением бесцветного масла, 17,10 г, 99% за 2 стадии.
МС: 494 ЭРИ+ (C26H43NO6Si)
1Н ЯМР (300 МГц, CDCl3) δ: 0.01 (s, 6Н); 0.83 (s, 9Н); 1.41 (s, 9Н); 3.62 (m, 1Н); 3.75 (s, 3Н); 3.75 (m, 1Н); 4.14 (m,4H); 4.34 (m, 1H); 4.41 (s, 2H); 5.70 (d, 1H); 6.83 (d, 2H); 7.21 (d, 2H).
Промежуточное соединение 170: (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)-окси)метил)-4-(((4-метоксибензил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат
Указанное в заголовке соединение получали из (S)-трет-бутил-2-(((трет-бутилдиметилсилил)окси)метил)-4-(((4-метоксибензил)-окси)метил)-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 169, 17,10 г, 34,78 ммоль) и N-(аллилокси)-2-нитробензолсульфонамида (Промежуточное соединение 9, 9,88 г, 38,26 ммоль) по методике, описанной для Промежуточного соединения 10. Целевой продукт был получен в виде бледно-желтого масла, 18,70 г, 74%.
МС: 734 ЭРИ+ (C35H51N3O10SSi)
1Н ЯМР (300 МГц, CDCl3) δ: 0.05 (s, 6Н); 0.87 (s, 9Н); 1.26 (s, 9Н); 3.71 (m, 2Н); 3.81 (s, 3Н); 4.45 (m, 4Н); 4.57 (d, 2Н); 5.28 (m, 2Н); 5.32 (m, 2Н); 5.71 (m, 1Н); 5.93 (m, 2Н); 6.12 (d, 1Н); 6.86 (m,2H);, 7.70 (m, 5H); 8.13 (m, 1H).
Промежуточное соединение 171: (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
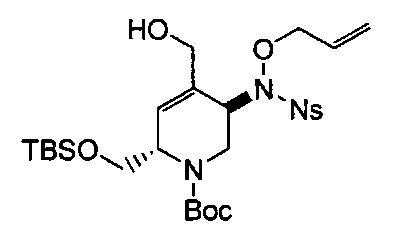
В перемешиваемый раствор (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(((4-метоксибензил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 170, 18,70 г, 25,48 ммоль) в смеси DCM/вода (180 мл/20 мл) добавляли DDQ (6,94 г, 30,57 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали. Остаток очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, с получением бледно-желтого масла, 12,38 г, 79%.
МС: 614 ЭРИ+ (C27H43N3O9SSi)
1Н ЯМР (300 МГц, CDCl3) δ: 0.05 (s, 6Н); 0.87 (s, 9Н); 1.40 (s, 9Н); 1.81 (bs, 1Н); 3.63 (d, 1Н); 3.70 (m, 2Н); 4.27 (m, 6Н); 4.60 (m, 1Н); 5.18 (m, 2Н); 5.71 (m, 1Н); 6.09 (m, 1Н); 7.60 (t, 1Н); 7.77 (m, 2Н); 8.14 (t, 1Н).
Промежуточное соединение 172: (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(цианометил)-5,6-дигидропиридин-1 (2Н)-карбоксилат
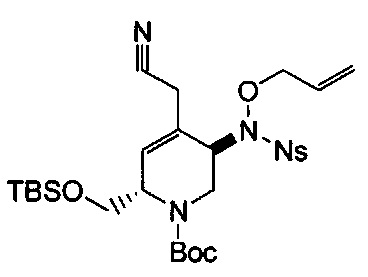
В перемешиваемый раствор (2S)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 171, 12,38 г, 20,17 ммоль) в DCM (200 мл) при 0°С добавляли пиридин (2,39 г, 30,25 ммоль) и N,N-диметилпиридин-4-амин (123 мг, 1,01 ммоль). Затем добавляли метансульфоновый ангидрид (4,22 г, 24,20 ммоль). Смесь затем перемешивали при температуре от 0°С до комнатной температуры в течение 2 часов. Смесь разбавляли DCM (100 мл) и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением неочищенного вещества в виде бледно-желтого масла. Его напрямую использовали на следующей стадии.
В перемешиваемый раствор цианида натрия (4,94 г, 100,85 ммоль) в воде (20,0 мл) добавляли (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(((метилсульфонил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат (неочищенный, 20,17 ммоль) в DMF (100 мл). Смесь затем перемешивали при комнатной температуре в течение 15 ч. Она становилась темно-оранжевым раствором. Этот раствор затем разбавляли насыщенным раствором NaHCO3 и водой, экстрагировали этилацетатом и сушили над MgSO4. Неочищенное вещество очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, с получением бледно-желтого масла, 5,92 г, 47,1%.
МС: 623 ЭРИ+ (C28H42N4O8SSi)
1Н ЯМР (300 МГц, CDCl3) δ: 0.05 (s, 6Н); 0.88 (s, 9Н); 1.41 (s, 9Н); 3.12 (m, 1Н); 3.32 (m, 1Н); 3.74 (m, 2Н); 4.21 (m, 5Н); 4.65 (s, 1Н); 5.18 (m, 2Н); 5.69 (m, 1Н); 6.31 (bs, 1Н); 7.65 (m, 1Н); 7.81 (m, 2Н); 8.15 (d, 1Н).
Промежуточное соединение 173: (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(2-амино-2-оксоэтил)-2-(((трет-бутилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат
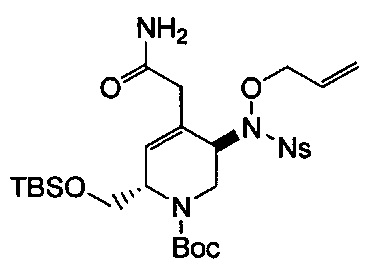
Приготовление катализатора: молекулярные сита 4 Å (MS-4Å) импрегнировали соответствующей солью металла (CuCl2-2H2O) следующим образом: 1 ммоль соли растворяли в 100 мл деионизированной H2O и перемешивали с 1 г MS-4Å при комнатной температуре в течение 6 ч. Твердое вещество отфильтровывали, промывали деионизированной H2O и ацетоном и сушили в термостате при 150°С в течение 1 ч.
(2S)-трет-Бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(((трет-бутилдиметилсилил)окси)метил)-4-(цианометил)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 172, 3,60 г, 5,78 ммоль), катализатор Cu(II)Cl2-4Å (200 мг), ацетальоксим (1,762 мл, 28,90 ммоль, 5,0 экв.) и МеОН (40,0 мл) перемешивали при 60°С в течение 15 ч. Твердое вещество отфильтровывали, промывали МеОН, и фильтрат упаривали с получением неочищенного продукта.
МС: 641 ЭРИ+ (C28H42N4O8SSi)
Промежуточное соединение 174: (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(2-амино-2-оксоэтил)-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
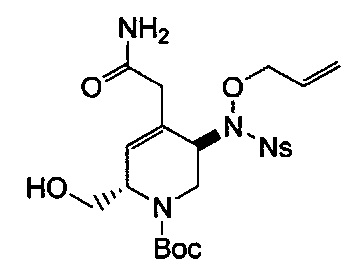
Указанное в заголовке соединение получали из (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(2-амино-2-оксоэтил)-2-(((трет-бутилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 173, неочищенное, 5,78 ммоль) по методике, описанной для Промежуточного соединения 18. После очистки на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, получили бледно-желтое твердое вещество, 2,61 г, 86%.
МС: 527 ЭРИ+ (C22H30N4O9S)
1Н ЯМР (300 МГц, CDCl3) δ: 1.39 (s, 9Н); 3.03 (m, 2Н); 3.33 (m, 2Н); 3.70 (m, 2Н); 4.20 (m, 1Н); 4.35 (m, 2Н); 4.68 (bs, 1Н); 5.17 (m, 2Н); 5.72 (m, 2Н); 6.00 (s, 1Н); 6.08 (bs, 1Н); 6.32 (bs, 1Н); 7.65 (d, 1Н); 7.80 (m, 2Н); 8.15 (d, 1Н).
Промежуточное соединение 175: (2S)-трет-бутил 5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(2-амино-2-оксоэтил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилат
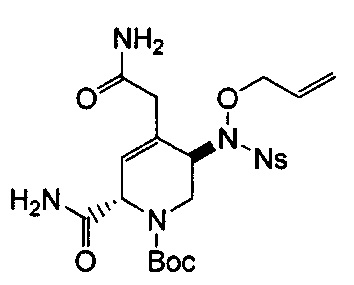
Исходный раствор: 240 мг конц. ΗΝΟ3 и 80 мг Na2Cr2O7-2Н2О растворяли в 16 мл Н2О при комнатной температуре.
В раствор (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфон-амидо)-4-(2-амино-2-оксоэтил)-2-(((трет-бутилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 174, 2,61 г, 4,96 ммоль) в ацетонитриле (50 мл) при комнатной температуре добавляли перйодат натрия (4,66 г, 21,81 ммоль) и каталитический раствор NaCr2O7/HNO3 (6,0 мл), и смесь перемешивали в течение 15 ч. Смесь разбавляли 100 мл этилацетата, 25 мл 1М буфера с рН 7, 25 мл 2М NaHSO3. Водный раствор экстрагировали 50 мл этилацетата. Этилацетатный слой затем промывали рассолом и сушили над MgSO4, фильтровали и концентрировали с получением (2S)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-(2-амино-2-оксоэтил)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты, которую напрямую использовали на следующей стадии без дополнительной очистки. Бледно-желтое твердое вещество, неочищенное, 2,79 г, 104%.
МС: 541 ЭРИ+ (C22H28N4O10S)
В перемешиваемый раствор (2S)-5-(N-(аллилокси)-2-нитрофенил-сульфонамидо)-4-(2-амино-2-оксоэтил)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (неочищенной, 2,79 г, 5,16 ммоль), хлорида аммония (552 мг, 10,32 ммоль), HATU (3,93 г, 10,32 ммоль) в DMF (20 мл) при 0°С добавляли DIPEA (2,67 г, 20,65 ммоль). После перемешивания при комнатной температуре в течение 1 ч добавляли 50 мл EtOAc и 50 мл воды. Органический слой промывали водой, рассолом, сушили над MgSO4, фильтровали и концентрировали с получением остатка, который очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны, с получением не совсем белого твердого вещества, 1,36 г, 49%.
МС: 540 ЭРИ+ (C22H29N5O9S)
1Н ЯМР (300 МГц, CDCl3) δ: 1.38 (s, 9Н); 3.08 (m, 2Н); 4.24 (m, 1Н); 4.40 (m, 2Н); 5.17 (m, 2Н); 5.68 (m, 1Н); 6.02 (m, 2Н); 6.20 (m, 2Н); 6.40 (bs, 1Н); 6.51 (bs, 1H);7.64 (d, 1Н); 7.78 (m, 2Н); 8.14 (d, 1Н).
Промежуточное соединение 176: (2S)-трет-бутил-5-((аллилокси)-амино)-4-(2-амино-2-оксоэтил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилат
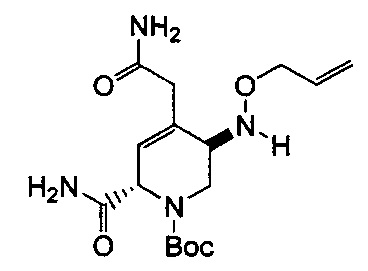
В раствор (2S)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфон-амидо)-4-(2-амино-2-оксоэтил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 175, 1,36 г, 2,52 ммоль) и карбоната калия (1,742 г, 12,60 ммоль) в ацетонитриле (20,0 мл) при 0°С добавляли бензолтиол (1,389 г, 12,60 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь концентрировали, и полученный остаток растирали с DCM. Твердое вещество удаляли фильтрованием. Фильтрат концентрировали до бледно-желтой пленки, которую очищали на силикагелевой колонке, элюируя смесью 0-50% MeOH/DCM, с получением бледно-желтого твердого вещества, 330 мг, 36,9%.
МС: 355 ЭРИ+ (C16H26N4O6)
1Н ЯМР (300 МГц, CDCl3) δ: 1.50 (s, 9Н); 3.05 (m, 2Н); 3.20 (m,1H); 3.45 (bs, 1Н); 4.23 (d, 2Н); 4.500 (m, 1Н); 5.03 (m, 1Н); 5.24 (m, 2Н); 5.88 (m, 2Н); 5.94 (m, 2Н); 6.57 (bs, 1Н); 6.76 (bs, 1Н).
Промежуточное соединение 177: (R)-5-(аллилоксиамино)-4-(2-амино-2-оксоэтил)-1,2,5,6-тетрагидропиридин-2-карбоксамид
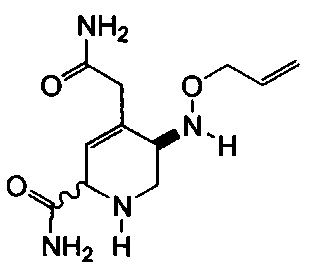
В раствор (2S)-трет-бутил 5-((аллилокси)амино)-4-(2-амино-2-оксоэтил)-2-карбамоил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 176, неочищенное, 330 мг, 0,93 ммоль) в DCM (5,0 мл) при 0°С добавляли HCl (4,0М в диоксане, 0,233 мл, 0,93 ммоль). Смесь перемешивали при температуре от 0°С до комнатной температуры в течение 2 ч. Затем в смесь добавляли еще 0,233 мл HCl и перемешивали в течение 2 ч. Смесь нейтрализовали добавлением K2CO3 и TEA, фильтровали, и растворитель удаляли в вакууме. Неочищенное вещество очищали на силикагелевой колонке, элюируя смесью 0-50% MeOH/DCM, с получением целевых диастереомерных продуктов в виде белых твердых веществ: 100 мг диастереомера 1, 32 мг диастереомера 2, выход 56%.
Диастереомер 1:
МС: 255 ЭРИ+ (C11H18N4O3)
1Н ЯМР (300 МГц, CD3OD) δ: 3.22 (m, 1Н); 3.38 (m, 2Н); 3.61 (m, 2Н); 3.82 (m, 1Н); 4.27 (d, 2Н); 4.75 (m, 1Н); 5.30 (m, 2Н); 6.03 (m, 1Н); 6.15 (d, 1Н).
Диастереомер 2:
МС: 255 ЭРИ+ (C11H18N4O3)
1Н ЯМР (300 МГц, CD3OD) δ: 3.16 (m, 2Н); 3.33 (m, 2Н); 3.58 (m, 2Н); 4.16 (m, 2Н); 4.38 (bs, 1Н); 5.19 (m, 2Н); 5.89 (d, 1Н); 5.95 (m, 1Н).
Промежуточное соединение 178: (2S,5R)-6-(аллилокси)-4-(2-амино-2-оксоэтил)-7-оксо-1.6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
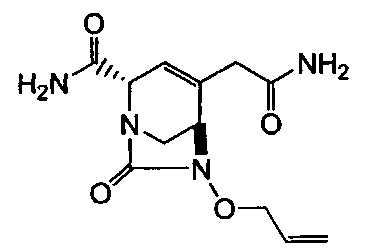
Указанное в заголовке соединение получали из (R)-5-(аллилоксиамино)-4-(2-амино-2-оксоэтил)-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 177, 132 мг, 0,52 ммоль) по методике, описанной для Промежуточного соединения 16. Реакционную смесь перемешивали при 0°С в течение 2 ч. Летучие вещества удаляли в вакууме, и неочищенное вещество очищали на силикагелевой колонке, элюируя смесью 0-100% этилацетата/гексаны и затем 95% этилацетата/5% МеОН, с получением бледно-желтого твердого вещества, 62 мг, 43%.
МС: 281 ЭРИ+ (C12H16N4O4)
1Н ЯМР (300 МГц, CD3Cl) δ: 3.13 (m, 3Н); 3.43 (dd, 1Н); 4.10 (d, 1Н); 4.43 (m, 3Н); 5.35 (m, 2Н); 5.57 (bs, 1Н); 5.80 (m, 2Н); 5.90 (d, 1Н); 6.01 (m, 1Н); 6.86 (bs, 1Н).
Промежуточное соединение 179: (2S,5R)-4-(2-амино-2-оксоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат
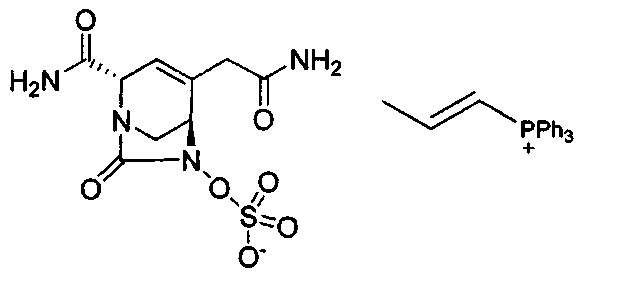
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-4-(2-амино-2-оксоэтил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 178, 62 мг, 0,22 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде бледно-желтого масла, 34 мг, 48%.
МС: 304; 320 (C21H20P; C9H11N4O7S).
1Н ЯМР (300 МГц, CD3Cl) δ: 2.31 (m, 1H); 2.64 (s, 3Н); 3.15 (m, 4Н); 3.63 (dd, 1Н); 4.22 (d, 1Н); 4.45 (bs, 1Н); 5.62 (bs, 1Н); 5.96 (d, 1Н); 6.01 (bs, 1Н); 6.64 (bs, 1Н); 6.96 (bs, 1Н); 7.68 (m, 15Н); 8.60 (bs, 1H).
Пример 20
Натриевая соль (2S,5R)-4-карбамоил-2-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
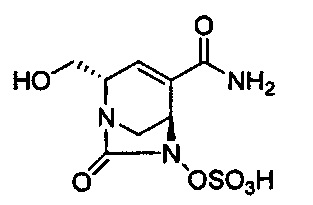
Смолу DOWEX 50WX8 (50 г) кондиционировали путем перемешивания в течение 20 минут в 2 н. гидроксиде натрия (40 мл). DOWEX загружали в картридж, промывали водой до рН=7, затем промывали смесью ацетон/вода (1/1) и снова водой. Соединение (Промежуточное соединение 180, 0,31 г, 0,76 ммоль) наносили, используя воду и минимальное количество ацетона, и элюировали водой. Фракции снова наносили на картридж и промывали снова водой. Этот процесс нанесения и промывки повторяли пять раз, затем конечные фракции сразу замораживали и лиофилизировали с получением белого твердого вещества (130 мг). После обращенно-фазовой хроматографии (0%-4% MeCN в воде) получили указанное в заголовке соединение в виде белого твердого вещества (31 мг).
МС: 292 ЭРИ- (C6H10N3O7S)
1Н ЯМР (600 МГц, ОКСИД ДЕЙТЕРИЯ) δ: 3.56 (d, J=11.67 Гц, 1Н); 3.64 (br. s., 1Н); 3.69-3.81 (m, 1Н); 3.97 (d, J=6.40 Гц, 2H); 4.18 (br. s., 1H); 4.84 (br. s., 1H); 6.73 (br. s., 1H).
Промежуточные соединения для получения соединения Примера 20 получали следующим образом:
Промежуточное соединение 180: (2S,5R)-2-(((трет-бутилдиметил-силил)окси)метил)-4-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-ен-1-ил)-фосфония
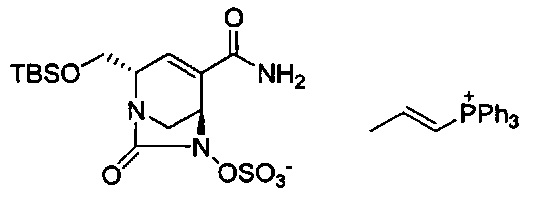
В раствор (2S,5R)-6-(аллилокси)-2-(((трет-бутилдиметилсилил)окси)-метил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамида (Промежуточное соединение 108, 0,32 г, 0,87 ммоль) в DCM (15 мл) добавляли уксусную кислоту (высушенную над сульфатом натрия) (0,100 мл, 1,74 ммоль) и тетракис(трифенилфосфин)палладии(0) (1,006 г, 0.87 ммоль) под азотом при комнатной температуре. Желтый раствор перемешивали при комнатной температуре в течение 3,5 часов. В смесь добавляли пиридин (10 мл), затем комплекс триоксида серы с пиридином (0,832 г, 5,22 ммоль). Суспензию перемешивали при комнатной температуре в течение ночи под азотом. Реакционную смесь концентрировали досуха (≤30°С), суспендировали в DCM, фильтровали, концентрировали и очищали на силикагелевой колонке (0%-100% МеОН в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (0,31 г).
МС: 406 ЭРИ- (C14H24N3O7SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.07-0.05 (m, 6Н); 0.71-0.86 (m, 9Н); 2.10 (dt, J=6.28, 2.05 Гц, 3H); 3.03-3.18 (m, 1Н); 3.22-3.32 (m, 1Н); 3.60-3.73 (m, 1Н); 3.79 (d, J=6.42 Гц, 2H); 4.48 (d, J=3.02 Гц, 1H); 6.40 (dd, J=2.93, 1.04 Гц, 1H); 6.43-6.70 (m, 1Н); 7.17 (br. s., 2H); 7.56-7.96 (m, 16Η).
Пример 21
Натриевая соль (2S,5R)-2-карбамоил-3.4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
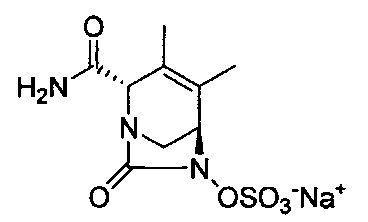
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 199, 0,22 г, 0,37 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде белого твердого вещества (99,6 мг, 86%).
Оптическое вращение: (0,2 г/дл, МеОН)=-228
МС: 290.0 ЭРИ- (C9H13N3O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.51 (s, 3Н); 1.75 (s, 3Н); 3.03 (dd, J=10.76, 3.02 Гц, 1H); 3.70 (d, J=10.76 Гц, 1H); 3.88-4.06 (m, 2Н); 7.23 (br. s., 1H); 7.76 (br. s., 1Η).
Промежуточные соединения для получения соединения Примера 21 получали следующим образом:
Промежуточное соединение 181: (R)-3-трет-бутил-4-метил-2,2-диметилоксазолидин-3,4-дикарбоксилат
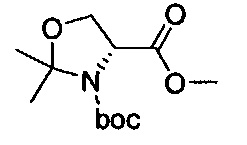
Раствор (R)-метил-2-(трет-бутоксикарбониламино)-3-гидрокси-пропаноата (Aldrich, 9,26 мл, 45,61 ммоль) в дихлорметане (18,10 мл) охлаждали до 0°С в бане лед-вода. Через 20 минут добавляли 2,2-диметоксипропан (Aldrich, 28,6 мл, 232,63 ммоль) и моногидрат пара-толуолсульфоновой кислоты (1,301 г, 6,84 ммоль) при 0°С и перемешивали в течение ночи при 25°С. Через 24 часа исходное вещество все еще присутствовало, поэтому добавляли еще 270 мг моногидрата пара-толуолсульфоновой кислоты, и реакционную смесь перемешивали при 25°С в течение 3 часов. Реакционную смесь вливали в насыщенный водный раствор NaHCO3, и полученный раствор экстрагировали диэтиловым эфиром. Объединенные органические слои промывали раствором бикарбоната натрия и рассолом. Полученные органические слои сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-70% этилацетата/гексаны) получили бесцветное маслянистое твердое вещество (9,53 г, 81%).
МС: 260,1 ЭРИ+ (C12H21NO5)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.37-1.57 (m, 12Н); 1.62-1.71 (m, 3Н); 3.70-3.81 (m, 3Н); 3.98-4.22 (m, 2Н); 4.33-4.54 (m, 1Η).
Промежуточное соединение 182: (R)-трет-бутил-4-формил-2,2-диметилоксазолидин-3-карбоксилат
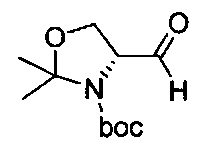
В раствор (R)-3-трет-бутил-4-метил 2,2-диметилоксазолидин-3,4-дикарбоксилата (Промежуточное соединение 181, 18,80 г, 72,50 ммоль) в толуоле (153 мл) при -78°С по каплям (через капельную воронку) добавляли DIBAL-H (1,0 Μ в толуоле) (116 мл, 116,01 ммоль) в течение 1,5 часа. Реакционную смесь перемешивали при -78°С в течение 2 часов в атмосфере азота. Холодную реакционную смесь обрабатывали 38 мл МеОН (примерно 1,91 М) и затем в реакционную смесь вливали 750 мл холодного 1 н. раствора HCl (примерно 0,09М). Реакционную смесь удаляли из бани -78°С, переносили в делительную воронку и экстрагировали этилацетатом. Органические слои сушили над сульфатом магния, фильтровали и концентрировали. В связи с трудностью разделения проводили две хроматографии на силикагелевых колонках (0%-50% этилацетата/гексаны) с получением указанного в заголовке соединения в виде бесцветного маслянистого твердого вещества (10,2 г, 61%).
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.36-1.74 (m, 15Н); 3.95-4.49 (m, 3Н); 9.45-9.68 (m, 1Η).
Промежуточное соединение 183: (R)-трет-бутил-4-(1-гидроксиэтил)-2,2-диметилоксазолидин-3-карбоксилат
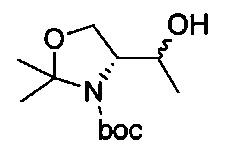
В раствор (R)-трет-бутил-4-формил-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 182, 10,18 г, 44,40 ммоль) в THF (188 мл) при -78°С по каплям добавляли бромид метилмагния (16,28 мл, 48,84 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи в атмосфере азота. Снова охлаждали реакционную смесь до -78°С и по каплям добавляли еще 13 мл (0,9 экв., доводя в сумме до 2 экв.) бромида метилмагния. Реакционную смесь перемешивали при -78°С в течение 3 часов и нагревали до комнатной температуры. Реакционную смесь гасили водой и разбавляли этилацетатом насыщенным хлоридом натрия (рассолом). Полученную эмульсию фильтровали через целит, и слои разделяли. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение в виде бесцветного маслянистого твердого вещества (8,42 г, 77%).
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.09-1.22 (m, 3Н); 1.36-1.64 (m, 15Н); 3.70-4.27 (m, 4Η)
Промежуточное соединение 184: (R)-трет-бутил-4-ацетил-2,2-диметилоксазолидин-3-карбоксилат
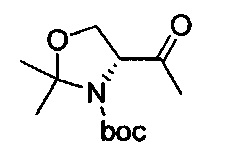
Указанное в заголовке соединение получали из (R)-трет-бутил-4-(1-гидроксиэтил)-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 183, 8,42 г, 34,32 ммоль) по методике, описанной для Промежуточного соединения 231. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили целевой продукт в виде бесцветного маслянистого твердого вещества (8,16 г, 98%).
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.37-1.57 (m, 12Н); 1.61-1.77 (m, 3Н); 2.20 (br. s., 3Н); 3.89-4.03 (m, 1Н); 4.07-4.21 (m, 1Н); 4.24-4.47 (m, 1Η).
Промежуточное соединение 185: (S)-трет-бутил-2,2-диметил-4-(проп-1-ен-2-ил)оксазолидин-3-карбоксилат
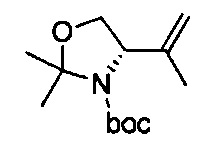
Указанное в заголовке соединение получали из (R)-трет-бутил-4-ацетил-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 184, 8,16 г, 33,54 ммоль) по методике для JC75. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт в виде бесцветного маслянистого твердого вещества (7,25 г, 90%).
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.37-1.54 (m, 12Н); 1.66 (br. s., 3Н); 1.74 (s, 3Н); 3.75 (dd, J=9.06, 3.02 Гц, 1H); 4.02-4.19 (m, 1Н); 4.20-4.44 (m, 1Н); 4.86 (br. s., 2Η).
Промежуточное соединение 186: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-илкарбамат
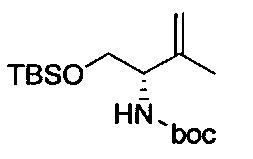
Указанное в заголовке соединение получали из (S)-трет-бутил-2,2-диметил-4-(проп-1-ен-2-ил)оксазолидин-3-карбоксилата (Промежуточное соединение 185, 6,92 г, 28,67 ммоль) по методике для JC76. После хроматографии на силикагеле (0%-10% этилацетата/гексаны) получили целевой продукт в виде бесцветного масла (8,28 г, 92%).
МС: 316.3 ЭРИ+ (C16H33NO3Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.05 (d, J=2.27 Гц, 6Н); 0.84-0.93 (m, 9Н); 1.45 (s, 9Н); 1.76 (s, 3Н); 3.59-3.81 (m, 2Н); 4.06 (d, J=6.99 Гц, 1Н); 4.81-5.05 (m, 2Η).
Промежуточное соединение 187: (S)-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-амин
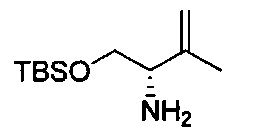
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-илкарбамата (Промежуточное соединение 186, 8,28 г, 26,24 ммоль) по методике, описанной для Промежуточного соединения 234. Органические слои сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (5,57 г, 99%).
МС: 216,2 ЭРИ+ (C11H25NOSi)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06-0.15 (m, 6Н); 0.88-0.96 (m, 9Н); 1.78-1.88 (m, 3Н); 3.00 (br. s., 2Н); 3.53-3.64 (m, 1Н); 3.66-3.79 (m, 1Н); 3.91 (dd, J=10.29, 3.30 Гц, 1H); 4.97-5.11 (m, 2Η).
Промежуточное соединение 188: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат
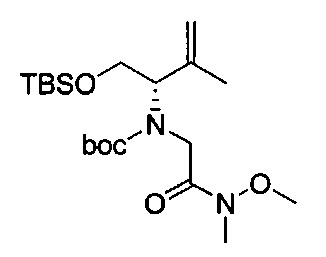
Указанное в заголовке соединение получали из (S)-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-амина (Промежуточное соединение 187, 5,57 г, 25,86 ммоль) и 2-бром-N-метокси-N-метилацетамида (Промежуточное соединение 4, 4,28 г, 23,53 ммоль) по методике, описанной для Промежуточного соединения 5. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт в виде бесцветного масла (4,24 г, 39%).
МС: 417,3 ЭРИ+ (C20H40N2O5Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.03-0.15 (m, 6Н); 0.81-0.97 (m, 9Н); 1.41-1.63 (m, 9Н); 1.72-1.82 (m, 3Н); 3.55-3.79 (m, 2Н); 3.92-4.23 (m, 1Н); 4.85-5.02 (m, 2Η).
Промежуточное соединение 189: (S)-трет-бутил-1-(трет-бутил-диметилсилилокси)-3-метилбут-3-ен-2-ил(3-метил-2-оксобут-3-енил)карбамат
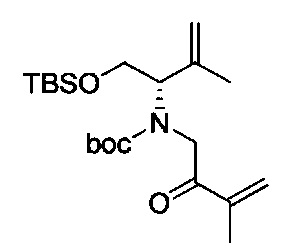
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 188, 4,24 г, 10,18 ммоль) по методике, описанной для Промежуточного соединения 6, с заменой на релевантный бромид изопренилмагния (0,5 Μ в THF) (204 мл, 101,77 ммоль). После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт в виде светло-желтого масла (3,72 г, 92%).
МС: 398,3 ЭРИ+ (C21H39NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.02-0.08 (m, 6Н); 0.82-0.90 (m, 9Н); 1.35-1.51 (m, 9Н); 1.78 (s, 3Н); 1.90 (s, 3Н); 3.68-3.99 (m, 2Н); 4.23-4.75 (m, 3Н); 4.84-5.01 (m, 2Н); 5.84-5.98 (m, 2Η).
Промежуточное соединение 190: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3,4-диметил-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилат
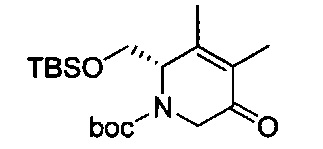
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-метилбут-3-ен-2-ил(3-метил-2-оксобут-3-енил)карбамата (Промежуточное соединение 189, 3,72 г, 9,36 ммоль) по методике, описанной для Промежуточного соединения 7, за исключением того, что реакционную смесь нагревали при 110-120°С в течение 48 часов, и загрузку катализатора удваивали (0,6 эквивалентов). После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили целевой продукт в виде светлого желто-коричневого маслянистого твердого вещества (2,39 г, 69%).
Оптическое вращение: (1 г/дл, МеОН)=-40
МС: 370,3 ЭРИ+ (C19H35NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.00 (d, J=7.93 Гц, 6Н); 0.82 (s, 9Н); 1.48 (s, 9Н); 1.79-1.87 (m, 3Н); 1.98 (s, 3Н); 3.79-4.06 (m, 4Н); 4.36-4.58 (m, 1Н).
Промежуточное соединение 191: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3,4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилат
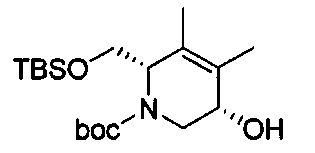
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3,4-диметил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 190, 2,39 г, 6,47 ммоль) по методике, описанной для Промежуточного соединения 8. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт в виде бесцветного маслянистого твердого вещества (1,73 г, 72%).
МС: 372,3 ЭРИ+ (C19H37NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.01-0.12 (m, 6Н); 0.83-0.94 (m, 9Н); 1.48 (s, 9Н); 1.69 (s, 3Н); 1.84 (s, 3Н); 3.34 (d, J=12.84 Гц, 1Н); 3.68 (d, J=8.12 Гц, 2Н); 3.87 (br. s., 1Н); 4.00-4.29 (m, 2Η).
Промежуточное соединение 192: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилил-окси)метил)-3.4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилат
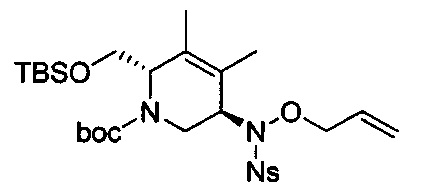
Указанное в заголовке соединение получали из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-3,4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 191, 1,73 г, 4,66 ммоль) и N-(аллилокси)-2-нитробензолсульфонамида (Промежуточное соединение 9, 1,26 г, 4,89 ммоль) по методике, описанной для Промежуточного соединения 10. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили целевой продукт в виде не совсем белого твердого вещества (3,15 г, 100%).
МС: 612,3 ЭРИ+ (C28H45N3O8SSi)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.05-0.08 (m, 6Н); 0.78-0.91 (m, 9Н); 1.31-1.89 (m, 15Н); 3.17-3.86 (m, 4Н); 4.17-4.31 (m, 1Н); 4.39 (br. s., 1Н); 4.57 (dt, J=6.23, 1.23 Гц, 2H); 5.05-5.43 (m, 2Н); 5.94 (ddt, J=17.09, 10.48, 6.33, 6.33 Гц, 1H); 7.50-7.64 (m, 1Н); 7.66-8.00 (m, 1Н); 8.02-8.32 (m, 2Η).
Промежуточное соединение 193: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3.4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилат
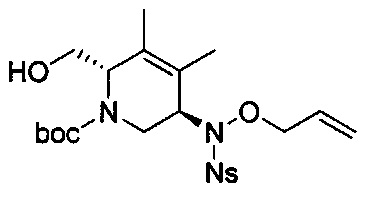
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)-метил)-3,4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 192, 3,15 г, 5,15 ммоль) по методике, описанной для Промежуточного соединения 18. После проведения двух хроматографии на силикагеле (0%-100% этилацетата/гексаны) получили целевой продукт в виде белого пенистого твердого вещества (1,68 г, 65,6%).
МС: 498,2 ЭРИ+ (C22H31N3O8S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.15-1.92 (m, 15Н); 2.98-3.46 (m, 1Н); 3.55-3.96 (m, 3Н); 4.01-4.35 (m, 3Н); 4.57 (d, J=6.80 Гц, 1Н); 5.09-5.35 (m, 2Н); 5.54-5.91 (m, 1Н); 7.61 (d, J=7.37 Гц, 1Н); 7.67-7.85 (m, 2Н); 8.08-8.22 (m, 1Η).
Промежуточное соединение 194: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3.4-диметил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
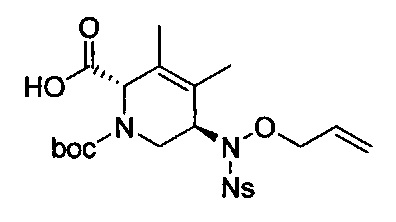
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-3,4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 193, 1,68 г, 3,38 ммоль) по методике, описанной для Промежуточного соединения 19. Полученную после обработки органическую фазу сушили над сульфатом магния, фильтровали и концентрировали с получением неочищенного вещества в виде светло-оранжевой пены (1,48 г, 86%).
МС: 512,2 ЭРИ+ (C22H29N3O9S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.22-1.61 (m, 12Н); 1.86 (s, 3Н); 3.15-3.42 (m, 1Н); 3.82-4.07 (m, 1Н); 4.09-4.34 (m, 2Н); 4.92 (s, 1Н); 5.09-5.24 (m, 2Н); 5.31 (s, 1Н); 5.56-5.84 (m, 1Н); 7.52-7.67 (m, 1Н); 7.69-7.91 (m, 2Н); 8.13 (dd, J=7.84, 1.42 Гц, 1Η).
Промежуточное соединение 195: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3,4-диметил-5,6-дигидропиридин-1 (2Н)-карбоксилат
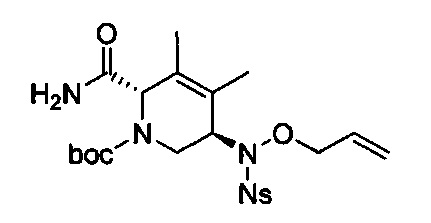
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3,4-диметил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 194, 1,48 г, 2,89 ммоль) по методике, описанной для Промежуточного соединения 20. После хроматографии на силикагеле (0%-80% этилацетата/гексаны) получили целевой продукт в виде светло-желтого пенистого твердого вещества (1,41 г, 95%).
Оптическое вращение: (0,4 г/дл, МеОН)=-51
МС: 511,2 ЭРИ+ (C22H30N4O8S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.15-1.48 (m, 9Н); 1.51-1.85 (m, 6Н); 2.93-3.36 (m, 2Н); 4.02-4.34 (m, 2Н); 4.83 (br. s., 1Н); 5.04-5.40 (m, 3Н); 5.51-5.77 (m, 1Н); 7.60 (d, J=7.36 Гц, 1H); 7.65-7.85 (m, 2Н); 7.99-8.18 (m, 1Η).
Промежуточное соединение 196: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3,4-диметил-1,2,5,6-тетрагидропиридин-2-карбоксамид
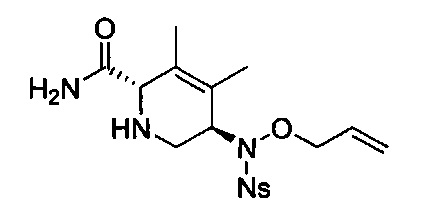
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3,4-диметил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 195, 1,36 г, 2,66 ммоль) по методике, описанной для Промежуточного соединения 21. Полученный после обработки органический слой сушили над сульфатом магния, фильтровали и концентрировали с получением целевого продукта в виде светло-розового твердого вещества (0,82 г, 75%).
МС: 411,1 ЭРИ+ (C17H22N4O6S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.87 (s, 3Н); 1.98 (s, 3Н); 2.67-2.89 (m, 2Н); 3.66 (s, 1Н); 3.92-4.31 (m, 2Н); 4.51 (dd, J=11.33, 6.04 Гц, 1Н); 5.15-5.34 (m, 2Н); 5.67-5.93 (m, 1Н); 7.61 (dd, J=7.84, 1.42 Гц, 1Н); 7.68-7.94 (m, 2Н); 8.05-8.18 (m, 1Η).
Промежуточное соединение 197: (2S,5R)-5-(аллилоксиамино)-3,4-диметил-1,2,5,6-тетрагидропиридин-2-карбоксамид
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3,4-диметил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 196, 0,82 г, 2,00 ммоль) по методике, описанной для Промежуточного соединения 12. После хроматографии на силикагеле (0%-10% метанол/дихлорметан) получили целевой продукт в виде белого твердого вещества (0,31 г, 68,9%).
МС: 226,2 ЭРИ+ (C11H19N3O2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.80 (s, 3Н); 1.86 (s, 3Н); 2.83-2.97 (m, 1Н); 3.12 (br. s., 1Н); 3.22 (d, J=13.79 Гц, 1H); 3.76 (br. s., 1H); 4.20 (dt, J=6.00, 1.25 Гц, 2H); 5.16-5.35 (m, 2H); 5.42 (br. s., 1H); 5.95 (ddt, J=17.28, 10.34, 6.02, 6.02 Гц, 1H); 7.06-7.20 (m, 1H).
Промежуточное соединение 198: (2S,5R)-6-(аллилокси)-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
Указанное в заголовке соединение получали из (2S,5R)-5-(аллилоксиамино)-3,4-диметил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 197, 0,31 г, 1,38 ммоль) по методике, описанной для Промежуточного соединения 13. После хроматографии на силикагеле (0%-70% этилацетата/гексаны) получили целевой продукт в виде белого твердого вещества (0,23 г, 66,5%).
МС: 252,2 ЭРИ+ (C12H17N3O3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.76-1.82 (m, 3Н); 1.83-1.90 (m, 3Н); 3.10-3.33 (m, 1Н); 3.66 (d, J=2.64 Гц, 1Н); 4.23 (s, 1Н); 4.42 (qdt, J=12.20, 12.20, 12.20, 6.35, 1.09, 1.09 Гц, 2Н); 5.24-5.51 (m, 3Н); 6.02 (ddt, J=17.00, 10.53, 6.35, 6.35 Гц, 1Н); 6.61 (br. s., 2Η).
Промежуточное соединение 199: (2S,5R)-2-карбамоил-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 198, 0,227 г, 0,9 ммоль) по методике, описанной для Промежуточного соединения 17. После хроматографии на силикагеле (0%-100% ацетона/дихлорметан) получили целевой продукт в виде светло-желтого маслянистого твердого вещества (0,5 г, 93%).
МС: 290 ЭРИ-, 303 ЭРИ+ (C9H12N3O6S, C21H20P)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.77 (s, 3Н); 1.86-1.95 (m, 3Н); 2.28-2.39 (m, 3Н); 3.14 (d, J=10.76 Гц, 1Н); 3.46 (dd, J=10.76, 3.02 Гц, 1Н); 4.29 (d, J=2.83 Гц, 1Н); 5.38 (br. s., 1Н); 6.51-6.80 (m, 1Н); 7.20-7.54 (m, 6Н); 7.58-7.91 (m, 10Н).
Пример 22
Натриевая соль (2S,5R)-2-карбамоил-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
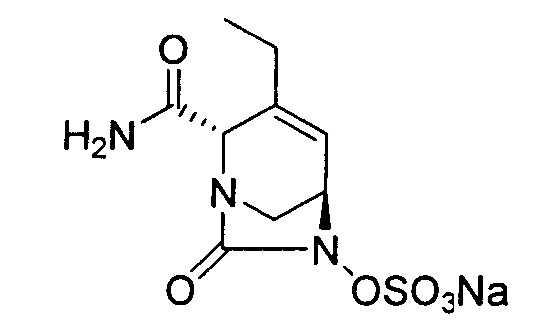
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-ен-1-ил)фосфония (Промежуточное соединение 216, 0,77 г, 1,30 ммоль) по методике, описанной в Примере 1. Фракции 5-7 объединяли и лиофилизировали отдельно от фракций 8-17, которые были желтыми. Указанное в заголовке соединение было получено из фракций 5-7 (0,275 г, 67,5%) в виде не совсем белого твердого вещества.
МС: 292 ЭРИ+ (C9H13N3O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.94 (t, J=7.54 Гц, 3Н) 1.59-2.19 (m, 2Н) 3.07 (dd, J=10.93, 1.88 Гц, 1Н) 3.73 (d, J=10.55 Гц, 1Н) 4.06 (dd, J=4.52, 2.26 Гц, 1Н) 4.15 (s, 1Н) 6.03 (d, J=4.52 Гц, 1Н) 7.29 (br. s., 1Н) 7.83 (br. s., 1Н).
Промежуточные соединения для получения соединения Примера 22 получали следующим образом:
Промежуточное соединение 200: (R)-трет-бутил-4-(3-гидрокси-пентан-3-ил)-2,2-диметилоксазолидин-3-карбоксилат
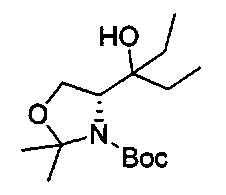
Суспензию хлорида церия (III) (119 г, 482,07 ммоль) в THF (350 мл) при комнатной температуре энергично перемешивали в течение 2 часов. Суспензию охлаждали до -78°С и по каплям добавляли бромид этилмагния (482 мл, 482,07 ммоль). Смесь перемешивали при -78°С в течение 1,5 часов. Затем по каплям добавляли (R)-3-трет-бутил-4-метил-2,2-диметилоксазолидин-3,4-дикарбоксилат (Aldrich, 25 г, 96,41 ммоль) в THF (100 мл) при -78°С. Реакционную смесь перемешивали при -78°С в течение 30 минут и затем нагревали до 0°С в течение 15 минут. Реакционную смесь гасили насыщенным NH4Cl, разбавляли дополнительно водой и экстрагировали дважды диэтиловым эфиром. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение (26,4 г, 95%) в виде бесцветного масла.
МС: 288 ЭРИ+ (C15H29NO4)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.77-0.98 (m, 6Η) 1.24-1.81 (m, 19Η) 3.86 (d, J=8.29 Гц, 1Η) 3.95-4.07 (m, 1Η) 4.17 (d, J=7.35 Гц, 1Η) 4.97 (br. s., 1Η).
Промежуточное соединение 201: (S)-трет-бутил-2.2-диметил-4-(пент-2-ен-3-ил)оксазолидин-3-карбоксилат
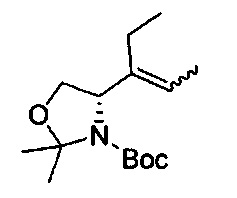
В раствор (R)-трет-бутил-4-(3-гидроксипентан-3-ил)-2,2-диметил-оксазолидин-3-карбоксилата (Промежуточное соединение 200, 26,4 г, 91,86 ммоль) в дихлорметане (350 мл) при 0°С добавляли триэтиламин (128 мл, 918,60 ммоль) и затем медленно добавляли метансульфонилхлорид (35,8 мл, 459,30 ммоль). Желтую реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи в течение примерно 16 ч. Реакционную смесь разбавляли диэтиловым эфиром и вливали в воду. Органический слой промывали 10%-ной лимонной кислотой, насыщенным раствором бикарбоната натрия, насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением коричневого масла. После очистки колоночной флэш-хроматографией (0-20% этилацетата/гексаны) получили указанное в заголовке соединение (17,70 г, 71,5%) в виде очень бледного желтого масла.
МС: 270 ЭРИ+ (C15H27NO3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.02 (q, J=7.47 Гц, 3Η) 1.38-1.54 (m, 12Η) 1.61-1.73 (m, 6Η) 1.83-2.29 (m, 2Η) 3.59-3.75 (m, 1Η) 4.03-4.16 (m, 1Η) 4.18-5.01 (m, 1Η) 5.32 (br. s., 1Η).
Промежуточное соединение 202: (S)-трет-бутил-3-этил-1-гидрокси-пент-3-ен-2-илкарбамат
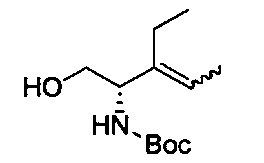
Раствор (S)-трет-бутил-2,2-диметил-4-(pent-2-en-3-ил)оксазолидин-3-карбоксилата (Промежуточное соединение 201, 15,815 г, 58,71 ммоль) и моногидрата пара-толуолсульфоновой кислоты (3,35 г, 17,61 ммоль) в метаноле (100 мл) нагревали до 85°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли триэтиламин (8,18 мл, 58,71 ммоль) и ди-трет-бутил-дикарбонат (6,82 мл, 29,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении с получением желтого масла. После очистки колоночной флэш-хроматографией (0-100% этилацетата/гексаны) получили указанное в заголовке соединение (10,12 г, 75%) в виде белого твердого вещества.
МС: 230 ЭРИ+ (C12H23NO3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.99-1.08 (m, 3Η) 1.46 (q, J=2.07 Гц, 9Η) 1.64-1.75 (m, 3Η) 1.92-2.22 (m, 3Η) 3.54-3.76 (m, 2Η) 4.15-4.89 (m, 2Η) 5.43 (q, J=6.84 Гц, 1Η).
Промежуточное соединение 203: (S,E)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-илкарбамат
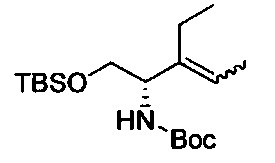
В раствор (S)-трет-бутил-3-этил-1-гидроксипент-3-ен-2-илкарбамата (Промежуточное соединение 202, 11,855 г, 51,70 ммоль) в DCM (120 мл) добавляли имидазол (5,28 г, 77,55 ммоль), DMAP (1,263 г, 10,34 ммоль) и TBDMS-Cl (9,35 г, 62,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли диэтиловым эфиром и промывали рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После очистки колоночной флэш-хроматографией (0-50% этилацетата/гексаны) получили указанное в заголовке соединение (17,20 г, 97%) в виде прозрачного масла.
МС: 344 ЭРИ+ (C18H37NO3Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06 (d, J=1.13 Гц, 6Η) 0.89 (dd, J=2.83, 1.32 Гц, 9Η) 0.96-1.08 (m, 3Η) 1.45 (s, 9Η) 1.54-1.76 (m, 3Η) 1.89-2.20 (m, 2Η) 3.50-3.76 (m, 2Η) 3.93-4.71 (m, 1Η) 4.87 (br. s., 1Η) 5.27-5.46 (m, 1Η).
Промежуточное соединение 204: (S)-1-(трет-бутилдиметил-силилокси)-3-этилпент-3-ен-2-амин
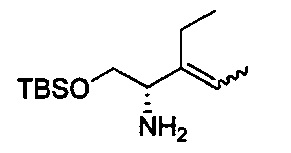
В раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-илкарбамата (Промежуточное соединение 203, 17,2 г, 50,06 ммоль) в DCM (300 мл) при комнатной температуре добавляли бромид цинка (45,1 г, 200,25 ммоль). Реакционную смесь перемешивали в течение 2 суток. Реакционную смесь фильтровали через воронку с фильтром из пористого стекла и промывали насыщенным раствором бикарбоната натрия. Водный слой экстрагировали более двух раз DCM, и органические фазы объединяли, сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения (12,19 г, 100%) в виде желтого масла.
МС: 244 ЭРИ+ (C13H29NOSi)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.02-0.12 (m, 6Η) 0.86-0.94 (m, 9Η) 0.95-1.05 (m, 3Η) 1.60-1.72 (m, 3Η) 1.87-2.22 (m, 2Η) 3.29-3.57 (m, 2Η) 3.58-4.00 (m, 1Η) 5.24-5.51 (m, 1Η).
Промежуточное соединение 205: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат
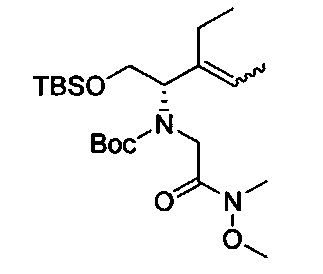
Смесь (S)-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-амина (Промежуточное соединение 204, 14,14 г, 58,08 ммоль) и карбоната цезия (18,92 г, 58,08 ммоль) в DMF (140 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли 2-бром-N-метокси-N-метилацетамид (Промежуточное соединение 4, 9,61 г, 52,80 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли ди-трет-бутил-дикарбонат (12,74 мл, 55,44 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и фильтровали для удаления неорганических солей. Фильтрат концентрировали, и остаток переносили в диэтиловый эфир и промывали насыщенным водным раствором бикарбоната натрия, водой, водной 5%-ной лимонной кислотой, водой и рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-30% этилацетата/гексаны) получили указанное в заголовке соединение (13,71 г, 58,4%) в виде светло-желтого масла.
МС: 445 ЭРИ+ (C22H44N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: -0.05-0.05 (m, 6Η) 0.83 (s, 9Η) 0.87-0.98 (m, 3Η) 1.27-1.43 (m, 9Η) 1.59 (dd, J=8.57, 7.06 Гц, 3Η) 1.84-2.15 (m, 2Η) 3.01-3.11 (m, 3Η) 3.63 (d, J=6.03 Гц, 3Η) 3.69-3.92 (m, 3Η) 3.98-4.22 (m, 1Η) 4.43-4.91 (m, 1Η) 5.27-5.42 (m, 1Η).
Промежуточное соединение 206: (S)-трет-бутил-1-(трет-бутил-диметилсилилокси)-3-этилпент-3-ен-2-ил(2-оксопент-3-енил)карбамат
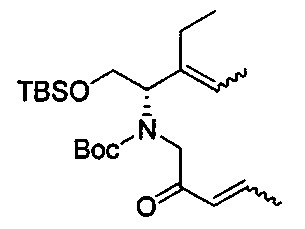
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-ил (2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 205, 13,7 г, 30,81 ммоль) по методике, описанной для Промежуточного соединения 236. Реакцию гасили насыщенным раствором NH4Cl, смесь разбавляли дополнительно водой и экстрагировали дважды диэтиловым эфиром. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-45% этилацетата/гексаны) получили указанное в заголовке соединение (10,95 г, 83%) в виде бесцветного масла.
МС: 426 ЭРИ+ (C23H43NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.02-0.07 (m, 6Η) 0.83-0.91 (m, 9Η) 1.00 (t, J=7.35 Гц, 3Η) 1.33-1.51 (m, 9Η) 1.57-1.74 (m, 3Η) 1.82-1.92 (m, 3Η) 1.92-2.21 (m, 2Η) 3.65-3.97 (m, 3Η) 3.97-4.34 (m, 1Η) 4.54-5.08 (m, 1Η) 5.25-5.44 (m, 1Η) 6.12-6.35 (m, 1Η) 6.79-7.00 (m, 1Η).
Промежуточное соединение 207: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-этил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
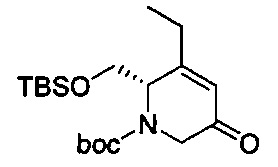
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-этилпент-3-ен-2-ил(2-оксопент-3-енил)карбамата (Промежуточное соединение 206, 10,95 г, 25,72 ммоль) по методике, описанной для Промежуточного соединения 7, за исключением того, что реакционную смесь нагревали при 86°С в течение 6 суток. После хроматографии на силикагеле (0-10% этилацетата/гексаны) получили указанное в заголовке соединение (5,15 г, 54,1%) в виде светло-желтого масла.
МС: 370 ЭРИ+ (C19H35NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: -0.18-0.09 (m, 6Η) 0.70-0.94 (m, 9Η) 1.17 (t, J=7.35 Гц, 3Η) 1.48 (s, 9Η) 2.19-2.43 (m, 2Η) 3.68-4.06 (m, 3Η) 4.28-4.78 (m, 2Η) 6.08 (s, 1Η).
Промежуточное соединение 208: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-этил-5-гидрокси-5,6-дигидропиридин-1 (2Н)-карбоксилат
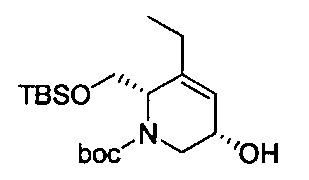
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-этил-5-оксо-5,6-дигидропиридин-1 (2Н)-карбоксилата (Промежуточное соединение 207, 5,146 г, 13,92 ммоль) по методике, описанной для Промежуточного соединения 8, за исключением того, что смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали и разбавляли водным раствором NH4Cl, водой и EtOAc. Органический слой отделяли и промывали три раза рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (5,19 г, 100%) в виде мутного масла.
МС: 372 ЭРИ+ (C19H37NO4Si)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.06 (s, 6Η) 0.80-0.93 (m, 9Η) 1.05-1.14 (m, 3Η) 1.48 (s, 9Η) 1.98-2.17 (m, 2Η) 2.83-3.53 (m, 2Η) 3.74 (d, J=11.30 Гц, 2Η) 4.06-4.35 (m, 3Η) 5.82 (br. s., 1Η).
Промежуточное соединение 209: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-этил-5,6-дигидропиридин-1(2Н)-карбоксилат
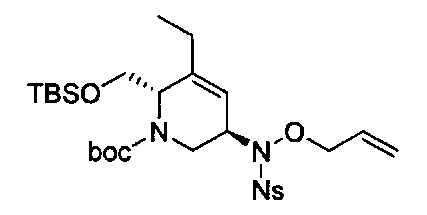
В перемешиваемую суспензию (2S,5R)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-этил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 208, 6,36 г, 17,12 ммоль), Ν-(аллилокси)-2-нитробензолсульфонамида (Промежуточное соединение 9) (8,84 г, 34,23 ммоль) и трифенилфосфина (13,47 г, 51,35 ммоль) в толуоле (25 мл) при 0°С по каплям добавляли (Е)-диизопропил-диазен-1,2-дикарбоксилат (9,95 мл, 51,35 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. После хроматографии на силикагеле (0-40% этилацетата/гексаны) получили указанное в заголовке соединение (7,00 г, 66,8%) в виде желтого масла.
МС: 612 ЭРИ+ (C28H45N3O8SSi)
Промежуточное соединение 210: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-этил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат

Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)-метил)-3-этил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 209, 7 г, 11,44 ммоль) по методике, описанной для Промежуточного соединения 18. После хроматографии на силикагеле (30%-90% этилацетата/гексаны) получили указанное в заголовке соединение (4,93 г, 87%) в виде желто-коричневой пены.
МС: 498 ЭРИ+ (C22H31N3O8S)
Промежуточное соединение 211: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-этил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
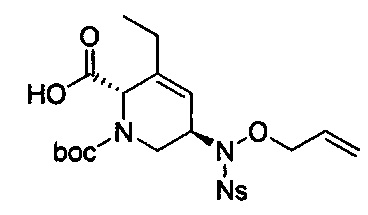
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-этил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 210, 2,35 г, 4,72 ммоль) по методике, описанной для Промежуточного соединения 19. Указанное в заголовке соединение было получено в виде желто-коричневой пены (1,89 г, 78%).
МС: 512 ЭРИ+ (C22H29N3O9S)
Промежуточное соединение 212: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-этил-5,6-дигидропиридин-1 (2Н)-карбоксилат
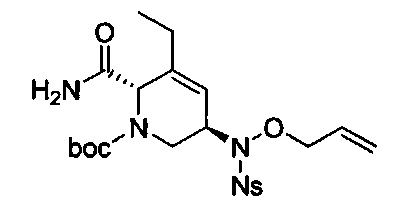
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-этил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 211, 1,89 г, 3,69 ммоль) по методике, описанной для Промежуточного соединения 20. После хроматографии на силикагеле (0%-80% этилацетата/гексаны) получили указанное в заголовке соединение (1,445 г, 77%) в виде бежевой пены.
МС: 511 ЭРИ+ (C22H29N3O9S)
Промежуточное соединение 213: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-этил-1,2,5,6-тетрагидропиридин-2-карбоксамид
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-этил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 212, 1,44 г, 2,82 ммоль) по методике, описанной для Промежуточного соединения 21, за исключением того, что реакционную смесь перемешивали в течение выходных дней при комнатной температуре. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия. Образовалась эмульсия, и водную фазу экстрагировали боле трех-четырех раз DCM. Органические фазы промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения (0,870 г, 75%) в виде желтой пены.
МС: 411 ЭРИ+ (C17H22N4O6S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.04 (t, J=7.54 Гц, 3Η) 2.17-2.35 (m, 1Η) 2.35-2.52 (m, 1Η) 2.94 (d, J=5.27 Гц, 2Η) 3.79 (s, 1Η) 4.29 (d, J=3.01 Гц, 1Η) 4.35-4.56 (m, 2Η) 5.18-5.39 (m, 3Η) 5.55 (br. s., 1Η) 5.74-5.94 (m, 1Η) 7.04 (br. s., 1H) 7.55-7.67 (m, 1H) 7.69-7.86 (m, 2H) 8.06-8.23 (m, 1H).
Промежуточное соединение 214: (R)-5-(аллилоксиамино)-3-зтил-1,2,5,6-тетрагидропиридин-2-карбоксамид
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-этил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 213, 0.87 г, 2.12 ммоль) по методике, описанной для Промежуточного соединения 12. После хроматографии на силикагеле (0%-8% метанола/дихлорметан) получили указанное в заголовке соединение (0,355 г, 74,3%) в виде не совсем белого твердого вещества после сушки в глубоком вакууме.
МС: 226 ЭРИ+ (C11H19N3O2)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.07 (t, J=7.54 Гц, 3Η) 2.10-2.48 (m, 3Η) 2.90-3.08 (m, 2Η) 3.43 (d, J=2.26 Гц, 1Η) 3.84 (s, 1Η) 4.21 (dd, J=6.78, 1.51 Гц, 2Η) 5.16-5.67 (m, 5Η) 5.95 (ddt, J=17.14, 10.74, 6.03, 6.03 Гц, 1Η) 7.08 (br. s., 1Η).
Промежуточное соединение 215: (2S,5R)-6-(аллилокси)-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
В раствор (R)-5-(аллилоксиамино)-3-этил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 214, 0,35 г, 1,55 ммоль) и диизопропилэтиламина (1,082 мл, 6,21 ммоль) в ацетонитриле (110 мл) при 0°С добавляли трифосген (0,157 г, 0,53 ммоль) в виде раствора в ацетонитриле (4 мл). Раствор трифосгена добавляли со скоростью 4 мл/час. Сразу после окончания добавления реакционную смесь перемешивали при 0°С в течение примерно 1 часа. Реакционную смесь разбавляли этилацетатом, затем промывали насыщенным раствором бикарбоната натрия и рассолом. Водные промывочные жидкости экстрагировали один раз дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-100% этилацетата/гексаны) получили указанное в заголовке соединение (0,340 г, 87%) в виде бесцветного масла.
МС: 252 ЭРИ+ (C12H17N3O3)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 1.05 (t, J=7.16 Гц, 3Η) 2.08-2.47 (m, 2Η) 3.15-3.34 (m, 2Η) 3.85 (dd, J=5.27, 3.01 Гц, 1Η) 4.31-4.50 (m, 3Η) 5.27-5.51 (m, 3Η) 5.93-6.15 (m, 2Η) 6.65 (br. s., 1Η).
Промежуточное соединение 216: (2S,5R)-2-карбамоил-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 215, 340 мг, 1,35 ммоль) по методике, описанной для Промежуточного соединения 17. После хроматографии на силикагеле (0%-100% ацетона/DCM) получили указанное в заголовке соединение (770 мг, 96%) в виде желтой пены.
МС: 303 ЭРИ+ (С21Н20Р) и 290 ЭРИ- (C9H12N3O6S)
1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ: 0.99 (t, J=7.16 Гц, 3Η) 2.05-2.16 (m, 1Η) 2.07-2.16 (m, 1Η) 2.34 (td, J=4.33, 1.88 Гц, 3Η) 2.36-2.47 (m, 1Η) 3.13 (d, J=10.55 Гц, 1Η) 3.45-3.55 (m, 1Η) 4.31 (s, 1Η) 4.42 (dd, J=5.27, 3.01 Гц, 1Η) 5.40 (br. s., 1Η) 6.18 (d, J=5.27 Гц, 1Η) 6.53-6.81 (m, 2Η) 7.35-7.47 (m, 2Η) 7.62-7.87 (m, 13Η).
Пример 23
(2S,5R)-4-(2-аминоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат
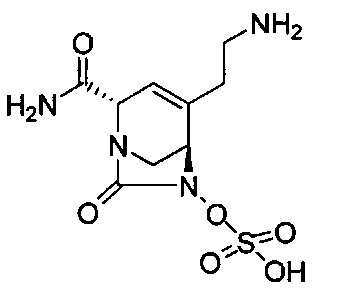
В суспензию (2S,5R)-4-(2-(трет-бутоксикарбониламино)этил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата (Промежуточное соединение 229, 0,060 г, 0,15 ммоль) в дихлорметане (2 мл) при 0°С добавляли трифторуксусную кислоту (0,100 мл). Через 15 минут растворитель удаляли в вакууме, и остаток сушили в глубоком вакууме. После очистки обращенно-фазовой хроматографией с использованием смеси ацетонитрил/вода получили указанное в заголовке соединение в виде белого твердого вещества после лиофилизации (0,006 г, 13%).
МС: 305 ЭРИ- (C9H14N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.19-2.33 (m, 2Η) 2.71-2.87 (m, 1Η) 2.93 (d, J=9.80 Гц, 1Η) 3.13 (br. s., 2Η) 3.89-4.14 (m, 2Η) 5.49 (br. s., 1Η) 7.27 (br. s., 1H) 7.52 (d, J=11.30 Гц, 3Η).
Промежуточные соединения для получения соединения Примера 23 получали следующим образом:
Промежуточное соединение 217: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
Указанное в заголовке соединение было получено в виде бесцветного масла (13,7 г, 88%) по методике, описанной для Промежуточного соединения 79, из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-(гидроксиметил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 117).
МС: 374 ЭРИ+ (C18H35NO5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.00 (s, 6Η) 0.75-0.89 (m, 9Η) 1.37 (s, 9Η) 3.59 (d, J=5.27 Гц, 2Η) 3.88-4.00 (m, 3Η) 4.15-4.36 (m, 1Η) 4.57 (t, J=5.65 Гц, 1Η) 5.00 (d, J=5.27 Гц, 1Η) 5.62 (d, J=2.26 Гц, 1Η).
Промежуточное соединение 218: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-((этоксикарбонилокси)метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
В раствор (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 217, 13,7 г, 36,67 ммоль) в дихлорметане (20 мл) при 0°С добавляли пиридин (5,93 мл, 73,35 ммоль), затем по каплям добавляли этилхлороформиат (3,87 мл, 40,34 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры в течение 3 часов. Смесь разбавляли дихлорметаном и промывали насыщенным раствором NH4Cl, экстрагировали дихлорметаном три раза, объединяли и сушили над Na2SO4. Растворитель удаляли с получением указанного в заголовке соединения в виде желтого масла (14,5 г, 89%).
МС: 446 ЭРИ+ (C21H39NO7Si)
Промежуточное соединение 219: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(2-нитроэтил)-5.6-дигидропиридин-1(2Н)-карбоксилат
(2S,5S)-трет-Бутил-2-((трет-бутилдиметилсилилокси)метил)-4-((этоксикарбонилокси)метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 218, 14,5 г, 32,54 ммоль) переносили в нитрометан (100 мл, 1854,52 ммоль) и дегазировали в течение 5 минут. Добавляли Pd2(dba)3 (1,49 г, 1,63 ммоль), и реакционную смесь перемешивали под азотом при комнатной температуре в течение 18 ч. Реакционную смесь сушили на диоксиде кремния и очищали флэш-хроматографией, используя смесь 0-50% EtOAc/гексаны. Указанное в заголовке соединение было получено в виде желтого масла (6 г, 44,3%).
МС: 417 ЭРИ+ (C19H36N2O6Si)
Промежуточное соединение 220: (2S,5R)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилил-окси)метил)-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилат
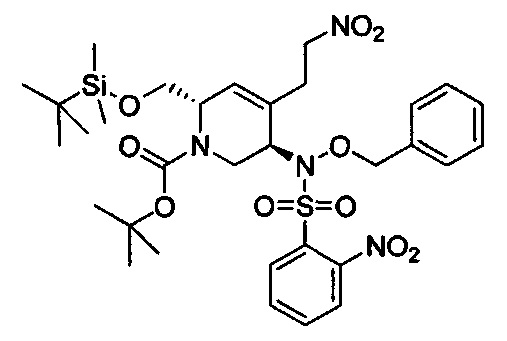
Указанное в заголовке соединение было получено в виде бледно-желтого масла (5 г, 30%) по методике, описанной для Промежуточного соединения 80, из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-5-гидрокси-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 219, 6,73 г, 16,3 ммоль) и N-(бензилокси)-2-нитробензол-сульфонамида (7,40 г, 24,00 ммоль).
МС: 707 ЭРИ+ (C32H46N4O10SSi)
Промежуточное соединение 221: (2S,5R)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилат
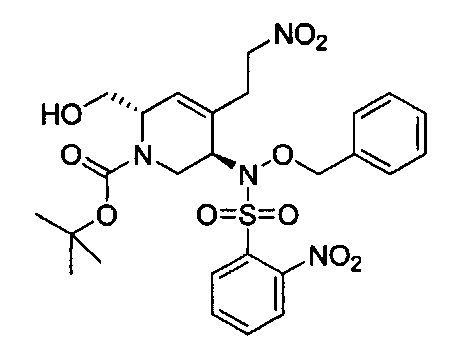
(2S,5R)-трет-Бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилат (Промежуточное соединение 220, 5 г, 7,07 ммоль) переносили в метанол (150 мл), охлажденный до 0°С, и добавляли ацетилхлорид (0,075 мл, 1,06 ммоль). Реакционную смесь перемешивали в течение 3 часов при 0°С. Растворитель удаляли в вакууме, и остаток переносили в EtOAc, промывали насыщенным раствором бикарбоната натрия, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде масла (4,1 г, 98%).
МС: 593 ЭРИ+ (C26H32N4O10S)
Промежуточное соединение 222: (2S,5R)-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-(2-нитроэтил)-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
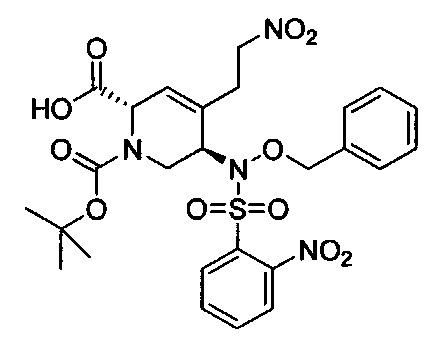
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-(гидроксиметил)-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 221, 4,1 г, 6,92 ммоль) по методике, описанной для Промежуточного соединения 19. Целевой продукт был получен в виде не совсем белой пены (4,0 г, 98%).
МС: 606 ЭРИ+ (C26H30N4O11S)
Промежуточное соединение 223: (2S,5R)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-(2-нитроэтил)-5,6-дигидропиридин-1(2H)-карбоксилат

Указанное в заголовке соединение получали из (2S,5R)-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-(2-нитроэтил)-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 222, 4,0 г, 6,59 ммоль) по методике, описанной для Промежуточного соединения 20. Целевой продукт был получен в виде не совсем белой пены (2 г, 50%).
МС: 606 ЭРИ+ (C26H31N5O10S)
Промежуточное соединение 224: (2S,5R)-трет-бутил-5-(бензилоксиамино)-2-карбамоил-4-(2-нитроэтил)-5,6-дигидропиридин-1 (2Н)-карбоксилат
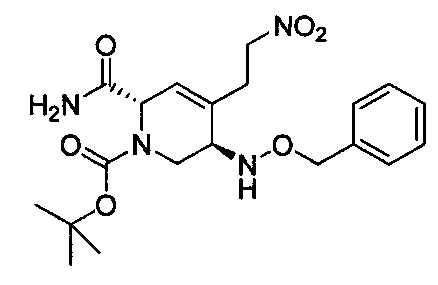
Целевой продукт получали из (2S,5R)-трет-бутил-5-(N-(бензилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 223, 2 г, 3,30 ммоль) по методике, описанной для Промежуточного соединения 22. Целевой продукт был получен в виде желтого масла (0,8 г, 68%).
МС: 421 ЭРИ+ (C20H28N4O6)
Промежуточное соединение 225: (2S,5R)-5-(бензилоксиамино)-4-(2-нитроэтил)-1,2,5,6-тетрагидропиридин-2-карбоксамид
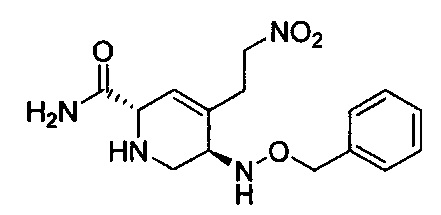
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(бензилоксиамино)-2-карбамоил-4-(2-нитроэтил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 224, 0,800 г, 3,30 ммоль) по методике, описанной для Промежуточного соединения 21. Целевой продукт был получен в виде желтого масла (0,6 г, 98%).
МС: 321 ЭРИ+ (C15H2oN4O4)
Промежуточное соединение 226: (2S,5R)-6-(бензилокси)-4-(2-нитроэтил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
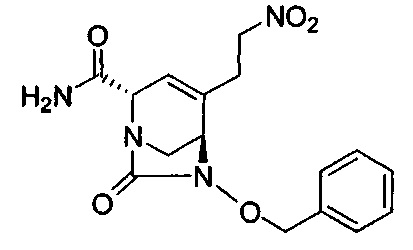
Указанное в заголовке соединение получали из (2S,5R)-5-(бензилоксиамино)-4-(2-нитроэтил)-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 225, 0,600 г, 1,87 ммоль) по методике, описанной для Промежуточного соединения 16. Целевой продукт был получен в виде светло-желтого масла (0,510 г, 79%).
МС: 347 ЭРИ+ (C16H18N4O5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.69 (t, J=6.78 Гц, 2Η) 3.17 (s, 2 Η) 3.88 (s, 1Η) 4.09-4.18 (m, 1Η) 4.55-4.70 (m, 2Η) 4.91 (d, J=3.01 Гц, 2Η) 5.56 (br. s., 1Η) 7.30 (br. s., 1Η) 7.35-7.46 (m, 5Η) 7.50 (br. s., 1H).
Промежуточное соединение 227: трет-бутил-2-((2S,5R)-6-(6ензилокси)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-ил)этилкарбамат
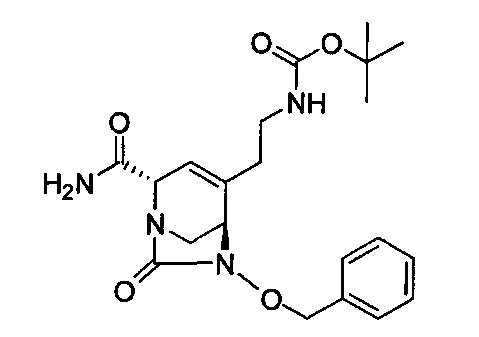
(2S,5R)-6-(Бензилокси)-4-(2-нитроэтил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид (Промежуточное соединение 226, 0,420 г, 1,21 ммоль) переносили в этанол (абсолютный, 99,5%) (20 мл) и добавляли цинковую пыль (1,983 г, 30,32 ммоль), затем добавляли уксусную кислоту (2,78 мл, 48,51 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Добавляли DIPEA (2,118 мл, 12,13 ммоль), затем ВОС-ангидрид (0,845 мл, 3,64 ммоль). Реакционную смесь перемешивали в течение 2 часов, затем разбавляли DCM, промывали насыщенным бикарбонатом и концентрировали досуха. После флэш-хроматографии с использованием смеси 0-10% MeOH/DCM получили указанное в заголовке соединение в виде прозрачного масла (0,31 г, 61%).
МС: 417 ЭРИ+ (C21H28N4O5)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.10-1.26 (m, 3Η) 1.28-1.46 (m, 9Η) 2.17 (d, J=6.03 Гц, 1Η) 2.79-3.16 (m, 2Η) 3.18-3.26 (m, 1Η) 3.55 (br. s., 1Η) 4.21 (br. s., 1Η) 4.71 (s, 3Η) 4.76-4.95 (m, 2Η) 5.53 (d, J=2.26 Гц, 1Η) 7.23-7.46 (m, 4Η).
Промежуточное соединение 228: трет-бутил-2-((2S,5R)-2-карбамоил-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-ил)этилкарбамат
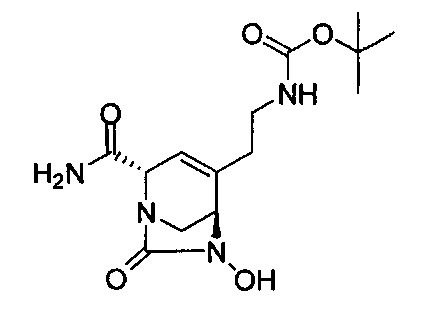
В раствор трет-бутил-2-((2S,5R)-6-(бензилокси)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-ил)этилкарбамата (Промежуточное соединение 227, 0,300 г, 0,72 ммоль) в EtOAc (5 мл) добавляли 10% Pd/C (0,077 г, 0,72 ммоль). Реакционную смесь перемешивали в атмосфере водорода в течение 30 минут. Реакционную смесь фильтровали через целит и промывали EtOAc. Растворитель удаляли в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (0,23 г, 98%).
МС: 327 ЭРИ+ (C14H22N4O5)
Промежуточное соединение 229: (2S,5R)-4-(2-(трет-бутоксикарбониламино)этил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфат
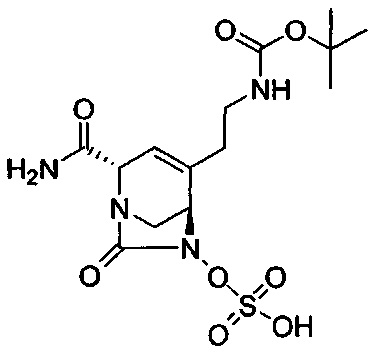
В раствор трет-бутил-2-((2S,5R)-2-карбамоил-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-ил)этилкарбамата (Промежуточное соединение 228, 0,230 г, 0,70 ммоль) в пиридине (5 мл) добавляли комплекс пиридин/триоксид серы (0,673 г, 4,23 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в вакууме, и остаток очищали обращенно-фазовой хроматографией с использованием смеси ацетонитрил/вода. Желаемые фракции объединяли и лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (0,23 г, 80%) в виде белого твердого вещества.
МС: 405 ЭРИ- (C14H22N4O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.36 (s, 9Н) 2.17 (t, J=6.40 Гц, 2Н) 2.90-3.27 (m, 4Н) 4.08 (d, J=18.84 Гц, 2Н) 5.43 (d, J=2.26 Гц, 1Н) 6.61 (br. s., 1Н) 7.25 (br. s., 1H) 7.47 (br. s., 1H) 7.80-7.95 (m, 1H) 8.38 (t, J=7.54 Гц, 1H) 8.83 (d, J=5.27 Гц, 1H).
Пример 24
Натриевая соль (2S,5R)-2-карбамоил-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
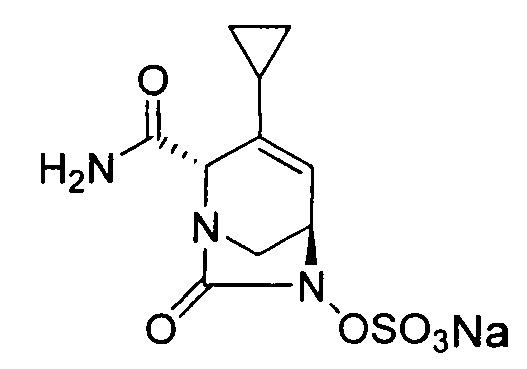
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 246, 0,39 г, 0,64 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде не совсем белого твердого вещества (158 мг, 75%).
Оптическое вращение: (0,1 г/дл, МеОН)=-50
МС: 302 ЭРИ- (C10H13N3O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.38 (m, 2Н); 0.59 (m, 2Н); 1.19 (m, 1Н); 3.03 (m, 1Н); 3.78 (m, 1Н); 4.02 (m, 1Н); 4.20 (m, 1Н); 5.91 (m, 1Н); 7.28 (bs, 1Н); 7.85 (bs, 1Н).
Промежуточные соединения для получения соединения Примера 24 получали следующим образом:
Промежуточное соединение 230: (R)-трет-бутил-4-(циклопропил-(гидрокси)метил)-2,2-диметилоксазолидин-3-карбоксилат
В раствор (R)-трет-бутил-4-формил-2,2-диметилоксазолидин-3-карбоксилата (Aldrich, 12,44 г, 54,26 ммоль) в THF (150 мл) при -78°С по каплям добавляли циклопропилмагния бромид (217 мл, 108,52 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры, и его перемешивали в течение ночи. Реакционную смесь гасили водой и разбавляли этилацетатом и рассолом. Полученную эмульсию фильтровали через целит, и слои разделяли. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение в виде светло-желтого масла (12,47 г, 85%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.16 (m, 2Н); 0.37 (m, 2Н); 0.82 (m, 1Н); 1.45 (m, 15Н); 2.87 (m, 1H); 3.86 (m, 2H); 3.97 (m, 1H); 4.74 (m, 1H).
Промежуточное соединение 231: (R)-трет-бутил-4-(циклопропанкарбонил)-2,2-диметилоксазолидин-3-карбоксилат
В раствор (R)-трет-бутил-4-(циклопропил(гидрокси)метил)-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 230, 12,47 г, 45,95 ммоль) в DCM (300 мл) при комнатной температуре добавляли перйодинан Десс-Мартина (29,2 г, 68,93 ммоль). Реакционную смесь перемешивали в течение ночи, затем разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия. Образовавшуюся эмульсию фильтровали через целит. Слои разделяли, и органический слой промывали рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение в виде бесцветного масла (11,15 г, 90%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.90 (m, 4Н); 1.38 (m, 12Н); 1.54 (m, 3Н); 2.12 (m, 1H); 3.94 (m, 1H); 4.18 (m, 1H); 4.56 (m, 1H).
Промежуточное соединение 232: (S)-трет-бутил-4-(1-циклопропилvinyl)-2,2-диметилоксазолидин-3-карбоксилат
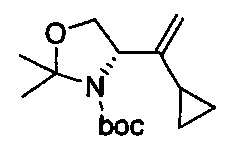
В суспензию трет-бутоксида калия (9,29 г, 82,80 ммоль) в диэтиловом эфире (250 мл) при комнатной температуре добавляли бромид метилтрифенилфосфония (29,6 г, 82,80 ммоль). Смесь становилась темно-желтой, и ее нагревали до 40°С в течение 1 часа. Смесь охлаждали до комнатной температуры, добавляли раствор (R)-трет-бутил-4-(циклопропанкарбонил)-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 231, 11,15 г, 41,40 ммоль) в диэтиловом эфире (30 мл), и реакционную смесь перемешивали в течение 2 часов. Реакцию гасили водой (10 мл), и слои разделяли. Водный слой экстрагировали один раз диэтиловым эфиром. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили указанное в заголовке соединение в виде бесцветного масла (9,84 г, 89%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.42 (m, 2Н); 0.65 (m, 2Н); 1.43 (m, 16Н); 3.76 (m, 1H); 4.09 (m, 1H); 4.27 (m, 1H); 4.66 (m, 2H).
Промежуточное соединение 233: (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-илкарбамат
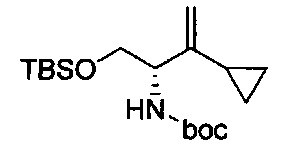
В раствор (S)-трет-бутил-4-(1-циклопропилвинил)-2,2-диметилоксазолидин-3-карбоксилата (Промежуточное соединение 232, 8,25 г, 30,86 ммоль) в метаноле (100 мл) при комнатной температуре добавляли моногидрат пара-толуолсульфоновой кислоты (1,174 г, 6,17 ммоль). Реакционную смесь нагревали до 80°С в течение ночи. Добавляли еще 0,2 экв. моногидрата пара-толуолсульфоновой кислоты. Нагревание продолжали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. Добавляли триэтиламин (4,29 мл, 30,86 ммоль) и ди-трет-бутил-дикарбонат (3,37 г, 15,43 ммоль). Реакционную смесь перемешивали в течение выходных дней, затем концентрировали. Остаток растворяли в этилацетате и промывали один раз насыщенным раствором бикарбоната натрия. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. Полученное масло растворяли в DCM (100 мл). Добавляли имидазол (2,73 г, 40,11 ммоль), 4-диметиламинопиридин (0,754 г, 6,17 ммоль) и трет-бутилдиметилсилилхлорид (4,65 г, 30,86 ммоль), и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали для удаления твердого вещества и промывали рассолом дважды. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-10% этилацетата/гексаны) получили указанное в заголовке соединение в виде бесцветного масла (6,77 г, 64%).
МС: 342 ЭРИ+ (C18H35NO3Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Н); 0.39 (m, 2Н); 0.63 (m, 2Н); 0.85 (s, 9Н); 1.32 (m, 1Н); 1.37 (m, 9Н); 3.55 (m, 1Н); 3.67 (m, 1Н); 3.99 (m, 1Н); 4.63 (s, 1Н);4.78 (s, 1Н); 6.80 (m, 1Н).
Промежуточное соединение 234: (S)-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-амин
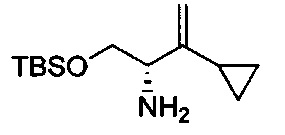
В раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-илкарбамата (Промежуточное соединение 233, 6,77 г, 19,82 ммоль) в DCM (100 мл) при комнатной температуре добавляли бромид цинка (17,86 г, 79,28 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли еще 1 экв. бромида цинка. Через несколько часов реакционную смесь фильтровали и промывали насыщенным раствором бикарбоната натрия. Полученную эмульсию фильтровали через нейлоновый фильтр, и слои разделяли. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения в виде желтого масла (4,61 г, 96%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (s, 6Н); 0.39 (m, 2Н); 0.63 (m, 2Н); 0.87 (s, 9Н); 1.35 (m, 1Н); 1.81 (m, 2Н); 3.33 (m, 1Н); 3.45 (m, 1Н); 3.67 (m, 1Н); 4.59 (s, 1Н); 4.83 (m, 1Н).
Промежуточное соединение 235: (S)-трет-бутил-1-(трет-бутил-диметилсилилокси)-3-циклопропилбут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат
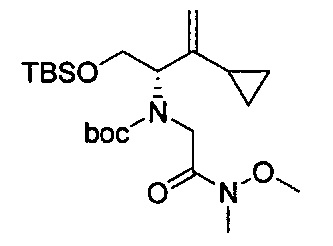
Указанное в заголовке соединение получали из (S)-1-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-амина (Промежуточное соединение 234, 4,61 г, 19,09 ммоль) и 2-бром-N-метокси-N-метилацетамида (Промежуточное соединение 4, 3,16 г, 17,36 ммоль) по методике, описанной для Промежуточного соединения 5. Целевой продукт был получен в виде светло-желтого масла (4,94 г, 64%).
МС: 443 ЭРИ+ (C22H42N2O5Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (m, 6Н); 0.35 (m, 1Н); 0.48 (m, 1Н); 0.61 (m, 2Н); 0.83 (m, 9Н); 1.35 (m, 9Н); 3.07 (m, 3Н); 3.65 (m, 3Н); 3.84 (m, 2Н); 4.02 (m, 2Н); 4.54 (m, 1Н); 4.83 (m, 2Н).
Промежуточное соединение 236: (S)-трет-бутил-1-(трет-бутил-диметилсилилокси)-3-циклопропилбут-3-ен-2-ил(2-оксопент-3-енил)-карбамат
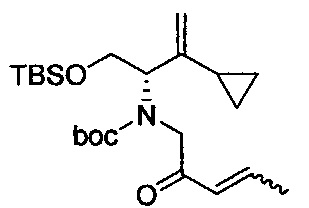
Суспензию хлорида церия (III) (27,8 г, 112,95 ммоль) в THF (100 мл) при комнатной температуре энергично перемешивали в течение 2 часов. Суспензию охлаждали до -78°С и по каплям добавляли бромид (Е)-проп-1-енилмагния (0,5 Μ в THF) (226 мл, 112,95 ммоль). Смесь перемешивали при -78°С в течение 1,5 часов. Затем по каплям добавляли (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат (Промежуточное соединение 235, 5 г, 11,30 ммоль) в THF (20 мл) при -78°С. Реакционную смесь перемешивали при -78°С в течение 30 минут и затем нагревали до 0°С в течение 15 минут. Реакционную смесь гасили 10%-ной лимонной кислотой, разбавляли дополнительно водой и экстрагировали дважды диэтиловым эфиром. Органический слой промывали один раз рассолом, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение в виде светло-желтого масла (4,0 г, 84%).
МС: 424 ЭРИ+ (C23H41NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (m, 6Н); 0.43 (m, 2Н); 0.61 (m, 2Н); 0.83 (m, 9Н); 1.34 (m, 10Н); 1.84 (m, 2Н); 2.04 (m, 1Н); 3.74 (m, 1Н); 3.84 (m, 2Н); 4.03 (m, 1Н); 4.57 (m, 1Н); 4.79 (m, 2Н); 6.28 (m, 1Н); 6.84 (m, 1Н).
Промежуточное соединение 237: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
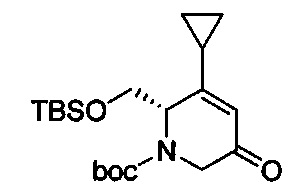
Указанное в заголовке соединение получали из (S,Е)-трет-бутил-1-(трет-бутилдиметилсилилокси)-3-циклопропилбут-3-ен-2-ил(2-оксопент-3-енил)карбамата (Промежуточное соединение 236, 4 г, 9,44 ммоль) по методике, описанной для Промежуточного соединения 7, за исключением того, что реакционную смесь нагревали при 110°С в течение ночи. Целевой продукт был получен в виде светло-коричневого масла (2,97 г, 82%).
МС: 382 ЭРИ+ (C20H35NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (m, 6Н); 0.62 (m, 1Н); 0.80 (s, 9Н); 1.00 (m, 3Н); 1.42 (s, 9Н); 1.61 (m, 1Н); 3.80 (m, 1Н); 3.95 (m, 2Н); 4.19 (m, 1Н); 4.75 (m, 1Н); 5.72 (s, 1Н).
Промежуточное соединение 238: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
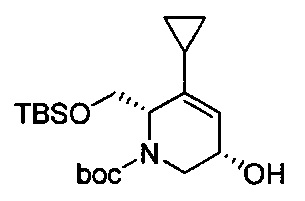
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 237, 2,97 г, 7,78 ммоль) по методике, описанной для Промежуточного соединения 8. Целевой продукт был получен в виде желто-коричневого масла (2,74 г, 92%).
МС: 384 ЭРИ+ (C20H37NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (m, 6Н); 0.34 (m, 1Н); 0.47 (m, 1Н); 0.64 (m, 2Н); 0.85 (m, 9Н); 1.26 (m, 1Н); 1.39 (s, 9Н); 2.65 (m, 1Н); 3.89 (m, 3Н); 4.05 (m, 1Н); 4.95 (m, 1Н); 5.34 (m, 1Н).
Промежуточное соединение 239: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5,6-дигидропиридин-1 (2Н)-карбоксилат
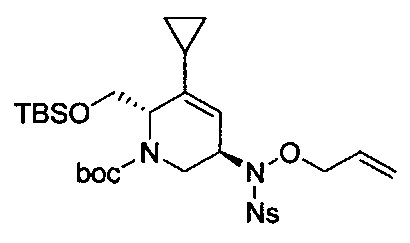
Указанное в заголовке соединение получали из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 238, 2,74 г, 7,14 ммоль) и N-(аллилокси)-2-нитробензолсульфонамида (Промежуточное соединение 9, 1,85 г, 7,14 ммоль) по методике, описанной для Промежуточного соединения 10. Целевой продукт был получен в виде светло-желтого масла (3,19 г, 71%).
МС: 624 ЭРИ+ (C29H45N3O8SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.00 (m, 6Н); 0.34 (m, 1Н); 0.63 (m, 2Н); 0.83 (m, 9Н); 1.37 (m, 9Н); 3.30 (m, 1Н); 3.84 (m, 2Н); 4.30 (m, 4Н); 5.18 (m, 2Н); 5.75 (m, 1Н); 8.04 (m, 4Н).
Промежуточное соединение 240: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-циклопропил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
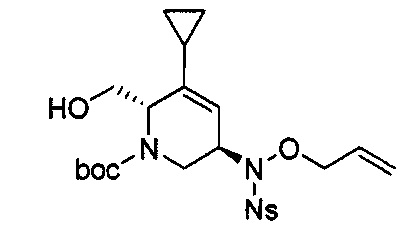
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-3-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 239, 3,19 г, 5,11 ммоль) по методике, описанной для Промежуточного соединения 18. Целевой продукт был получен в виде желто-коричневой пены (2,35 г, 90%).
МС: 510 ЭРИ+ (C23H31N3O8S)
1H ЯМР (300 МГц, DMSO-d6) δ: 0.32 (m, 2Н); 0.62 (m, 2Н); 1.35 (m, 9Н); 3.30 (m, 1Н); 3.67 (m, 2Н); 4.27 (m, 4Н); 4.71 (m, 1Н); 5.19 (m, 2Н); 5.71 (m, 1Н); 8.04 (m, 4Н).
Промежуточное соединение 241: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
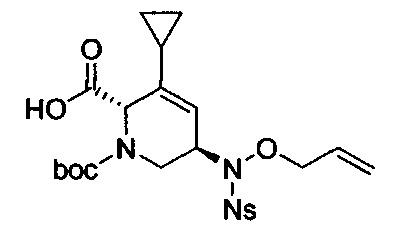
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-циклопропил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 240, 2,35 г, 4,61 ммоль) по методике, описанной для Промежуточного соединения 19. Целевой продукт был получен в виде оранжевой пены (2,28 г, 94%).
МС: 524 ЭРИ+ (C23H29N3O9S)
Промежуточное соединение 242: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
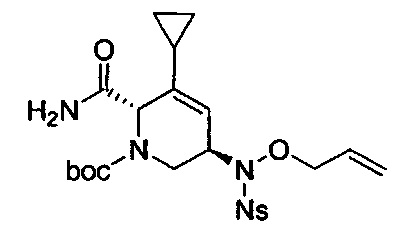
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-3-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 241, 2,28 г, 4,35 ммоль) по методике, описанной для Промежуточного соединения 20. Целевой продукт был получен в виде оранжевой пены (1,07 г, 47%).
МС: 523 ЭРИ+ (C23H30N4O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.23 (m, 2Н); 0.59 (m, 2Н); 1.35 (m, 9Н); 3.58 (m, 1Н); 4.23 (m, 3Н); 4.72 (m, 1Н); 5.19 (m, 2Н); 5.71 (m, 1Н); 7.18 (m, 1Н); 7.59 (m, 1Н); 8.04 (m, 4Н).
Промежуточное соединение 243: (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
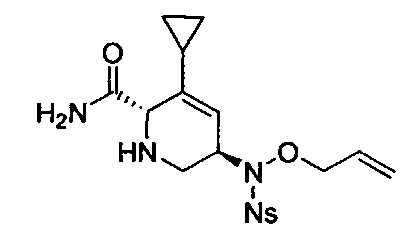
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-3-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 242, 0,932 г, 1,78 ммоль) по методике, описанной для Промежуточного соединения 21. Целевой продукт был получен в виде оранжевой пены (0,518 г, 68%).
МС: 423 ЭРИ+ (C18H22N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.18 (m, 2Н); 0.53 (m, 2Н); 1.29 (m, 1Н); 2.30 (m, 1Н); 2.58 (m, 1Н); 2.95 (m, 1Н); 3.72 (m, 1Н); 4.22 (m, 1Н); 4.36 (m, 2Н); 4.96 (m, 1Н); 5.24 (m, 2Н); 5.80 (m, 1Н); 7.07 (bs, 1Н); 7.39 (bs, 1Н); 8.04 (m, 4Н).
Промежуточное соединение 244: (R)-5-(аллилоксиамино)-3-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 243, 0,518 г, 1,23 ммоль) по методике, описанной для Промежуточного соединения 12. Целевой продукт был получен в виде светло-желтого масла (0,171 г, 59%). Продукт представляет собой смесь диастереомеров.
МС: 238 ЭРИ+ (C12H19N3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.28 (m, 2Н); 0.41 (m, 2Н); 0.54 (m, 2Н); 1.33 (m, 1Н); 2.49 (m, 1Н); 2.64 (m, 1Н); 2.93 (m, 1Н); 3.23 (m, 1Н); 3.65 (m, 1Н); 4.07 (m, 2Н); 5.19 (m, 3Н); 5.89 (m, 1Н); 6.26 (m, 1Н); 6.97 (bs, 1Н); 7.34 (bs, 1Н).
Промежуточное соединение 245: (2S,5R)-6-(аллилокси)-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
Указанное в заголовке соединение получали из (R)-5-(аллилоксиамино)-3-циклолропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 244, 0,316 г, 1,33 ммоль) по методике, описанной для Промежуточного соединения 16. Целевой продукт был получен в виде бесцветного масла (0,261 г, 74%).
МС: 264 ЭРИ+ (C13H17N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.37 (m, 2Н); 0.60 (m, 2Н); 1.20 (m, 1Н); 2.98 (m, 1Н); 3.79 (m, 1Н); 3.92 (m, 1Н); 4.20 (m, 1Н); 4.33 (m, 2Н); 5.28 (m, 2Н); 5.93 (m, 2Н); 7.30 (bs, 1Н); 7.86 (bs, 1Н).
Промежуточное соединение 246: (2S,5R)-2-карбамоил-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 245, 0,261 г, 0,99 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде желтой пены (0,39 г, 65%).
МС: 302 ЭРИ-, 303 ЭРИ+ (C10H12N3O6S-⋅C21H20P+)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.39 (m, 2Н); 0.58 (m, 2Н); 1.18 (m, 2Н); 2.16 (m, 3Н); 3.03 (m, 1H); 3.78 (m, 1H); 4.01 (m, 1H); 4.20 (m, 1H); 5.91 (m, 1H); 6.65 (m, 1H); 7.38 (m, 2H); 7.78 (m, 15H).
Пример 25
Натриевая соль (2S,5R)-4-(2-ацетамидоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата
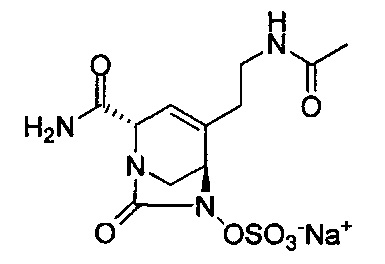
Указанное в заголовке соединение получали из пиридин-(2S,5R)-4-(2-ацетамидоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Промежуточное соединение 248, 0,007 г, 0,020 ммоль) по методике, описанной для Примера 1. Целевой продукт был получен в виде белого твердого вещества (3 мг, 58%).
МС: 347 ЭРИ- (C11H16N4O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.85 (d, J=6.78 Гц, 1Η) 1.70-1.84 (m, 3Η) 2.18 (t, J=6.78 Гц, 1Η) 3.12-3.21 (m, 2Η) 3.97-4.17 (m, 2Η) 5.42 (br. s., 1Η) 7.27 (br. s., 1Η) 7.49 (s, 1Η) 7.71 (s, 1Η).
Промежуточные соединения для получения соединения Примера 25 получали следующим образом:
Промежуточное соединение 247: (2S,5R)-4-(2-ацетамидоэтил)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
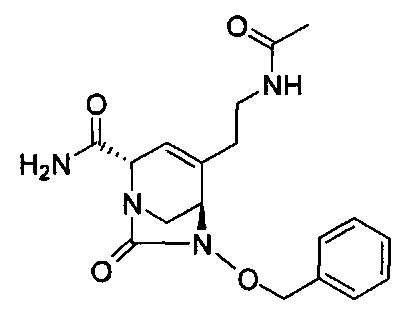
В раствор (2S,5R)-6-(бензилокси)-4-(2-нитроэтил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 226, 0,300 г, 0,87 ммоль) в этаноле (10 мл) добавляли цинковую пыль (1,416 г, 21,66 ммоль) и уксусную кислоту (1,984 мл, 34,65 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Затем в реакционную смесь добавляли DIPEA (2,118 мл, 12,13 ммоль), затем добавляли уксусный ангидрид (0,245 мл, 2,60 ммоль). Реакционную смесь перемешивали в течение еще 2 часов, затем разбавляли DCM, промывали насыщенным бикарбонатом и концентрировали досуха. После флэш-хроматографии (0%-10% MeOH/DCM) получили указанное в заголовке соединение в виде прозрачного масла (0,31 г, 61%).
МС: 359 ЭРИ+ (C18H22N4O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.75 (s, 3Η) 2.08-2.21 (m, 2Η) 2.92-3.05 (m, 1Η) 3.09-3.24 (m, 3Η) 3.72 (s, 1Η) 4.12 (br. s., 1Η) 4.89 (s, 2Η) 5.44 (br. s., 1Η) 7.29 (br. s., 1H) 7.34-7.51 (m, 6H) 7.70 (br. s., 1H).
Промежуточное соединение 248: (2S,5R)-4-(2-ацетамидоэтил)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
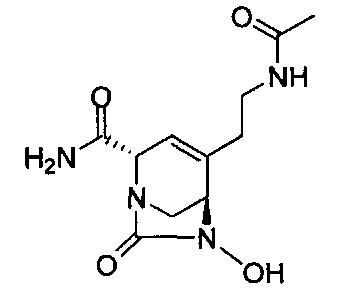
В раствор 4-(2-ацетамидоэтил)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 247, 0,120 г, 0,33 ммоль) в EtOAc (20 мл) и этаноле (6,67 мл) добавляли Pd/C (0,036 г, 0,33 ммоль). Реакционную смесь перемешивали под водородом при давлении 1 атм в течение 1 часа, затем фильтровали через целит и концентрировали досуха с получением указанного в заголовке соединения в виде белого твердого вещества (0,09 г, 100%).
Промежуточное соединение 249: пиридин-(2S,5R)-4-(2-ацетамидоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат
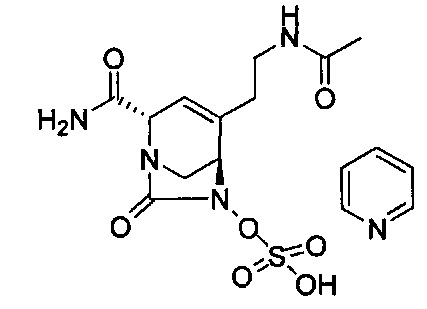
В раствор (2S,5R)-4-(2-ацетамидоэтил)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 248, 0,090 г, 0,34 ммоль) в пиридине (5 мл) добавляли комплекс пиридин/триоксид серы (0,320 г, 2,01 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в вакууме, и остаток очищали обращенно-фазовой ВЭЖХ. Указанное в заголовке соединение было получено в виде белого твердого вещества после лиофилизации (0,007 г, 6%).
МС: 347 ЭРИ- (C11H16N4O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.85 (d, J=6.78 Гц, 1Η) 1.68-1.82 (m, 3Η) 2.18 (t, J=7.16 Гц, 2Η) 3.06 (dt, J=12.81, 6.40 Гц, 1Η) 3.47 (d, J=6.78 Гц, 1Η) 3.99-4.17 (m, 3Η) 5.42 (d, J=1.51 Гц, 1Η) 7.28 (br. s., 1Η) 7.49 (br. s., 1Η) 7.72 (t, J=5.27 Гц, 1Η) 7.91-8.02 (m, 1Η) 8.48 (t, J=7.91 Гц, 1Η) 8.89 (br. s., 1H).
Пример 26
Натриевая соль (2S,5R)-2-(метоксиметил)-4-(метилкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
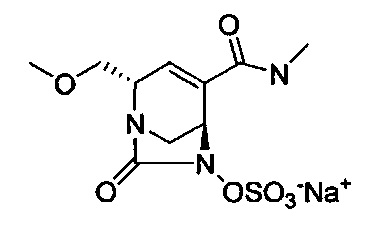
Указанное в заголовке соединение (0,08 г, 44%) получали по методике, описанной в Примере 1, начиная с (2S,5R)-2-(метоксиметил)-4-(метилкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 260, 0,33 г).
Оптическое вращение: (0,1 г/дл, МеОН)=-136
МС: 320 ЭРИ- (C10H15N3O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.65 (d, J=4.52 Гц, 3Η) 3.19-3.29 (m, 6Η) 3.53-3.69 (m, 2Η) 3.89 (td, J=5.75, 3.01 Гц, 1Η) 4.50-4.61 (m, 1Η) 6.38 (dd, J=3.01, 0.94 Гц, 1Η) 7.76 (d, J=4.52 Гц, 1Η).
Промежуточные соединения для получения соединения Примера 26 получали следующим образом:
Промежуточное соединение 250: (S)-трет-бутил-1-гидроксибут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамат
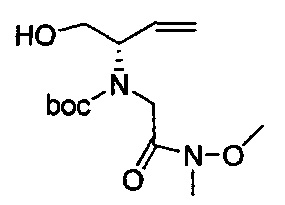
В раствор (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 5, 10,7 г, 26,58 ммоль) в THF (25 мл) добавляли TBAF (31,9 мл, 31,89 ммоль) при 0°С. Через 1 час реакционную смесь концентрировали и очищали хроматографией на силикагеле (гексаны/этилацетат) с получением продукта в виде светло-желтого масла (5,9 г, 77%).
МС: 289 ЭРИ+ (C13H24N2O5)
1Н ЯМР (300 МГц, CDCl3) δ: 1.36-1.61 (m, 10Η) 3.25 (s, 3Η) 3.35-3.52 (m, 1Η) 3.57-3.72 (m, 2Η) 3.72-3.82 (m, 3Η) 4.26-4.56 (m, 1Η) 4.83 (ddd, J=8.24, 4.19, 2.07 Гц, 2Η) 5.10-5.34 (m, 2Η) 5.57-5.84 (m, 1Η)
Промежуточное соединение 251: (S)-трет-бутил-2-(метокси(метил)-амино)-2-оксоэтил(1-метоксибут-3-ен-2-ил)карбамат
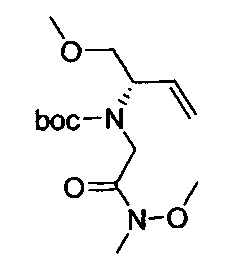
В раствор (S)-трет-бутил-1-гидроксибут-3-ен-2-ил(2-(метокси(метил)-амино)-2-оксоэтил)карбамата (Промежуточное соединение 250, 7 г, 24,28 ммоль) в THF (75 мл) при 0°С добавляли диметилсульфат (2,436 мл, 25,49 ммоль), затем добавляли LiHMDS (26,7 мл, 26,70 ммоль). Реакционную смесь оставляли медленно нагреваться до комнатной температуры и перемешивали ее в течение двух часов. Реакционную смесь затем распределяли между этилацетатом и водой. Слои разделяли, и органический слой промывали насыщенным раствором бикарбоната натрия, водой и рассолом. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (5%-30% этилацетата/гексаны) получили указанное в заголовке соединение в виде прозрачного масла (6,89 г, 91%).
МС: 303 ЭРИ+ (C14H264N2O5)
1Н ЯМР (300 МГц, CDCl3 δ: 1.18-1.42 (m, 9Η) 3.02 (s, 3Η) 3.14 (d, J=6.03 Гц, 3Η) 3.41-3.64 (m, 5Η) 3.76-4.13 (m, 2Η) 4.57-4.82 (m, 1Η) 4.93-5.13 (m, 2Η) 5.60-5.90 (m, 1Η).
Промежуточное соединение 252: (S)-трет-бутил-1-метоксибут-3-ен-2-ил(2-оксоэтил)карбамат
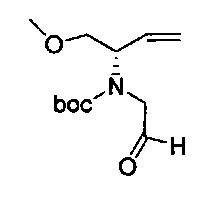
В раствор (S)-трет-бутил-2-(метокси(метил)амино)-2-оксоэтил-(1-метоксибут-3-ен-2-ил)карбамата (Промежуточное соединение 251, 3,35 г, 11,08 ммоль) в дихлорметане (60 мл) при -78°С под азотом добавляли гидрид диизобутилалюминия (1,5 экв., 1.0М в дихлорметане) (16,62 мл, 16,62 ммоль). Реакционную смесь перемешивали при этой температуре в течение двух часов. Реакционную смесь гасили медленным добавлением метанола при низкой температуре и медленно нагревали до температуры окружающей среды. Реакционную смесь разбавляли дихлорметаном и промывали 10%-ным водным раствором тартрата калия-натрия дважды, затем водой и рассолом. Органические фазы сушили над сульфатом натрия, фильтровали и концентрировали. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили указанное в заголовке соединение в виде прозрачного масла (1,8 г, 68%).
1Н ЯМР (300 МГц, CDCl3 δ: 1.46 (s, 9Η) 3.31 (s, 3Η) 3.46-3.65 (m, 2Η) 3.65-3.80 (m, 1Η) 3.80-4.05 (m, 1Η) 4.65-5.01 (m, 1Η) 5.13-5.36 (m, 2Η) 5.64-5.90 (m, 1Η) 9.49 (s, 1Η).
Промежуточное соединение 253: метил-4-(трет-бутоксикарбонил-((S)-1-метоксибут-3-ен-2-ил)амино)-3-гидрокси-2-метиленбутаноат
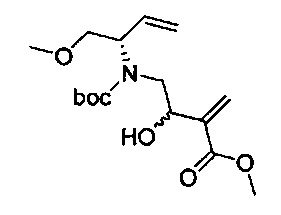
В раствор (S)-трет-бутил-1-метоксибут-3-ен-2-ил(2-оксоэтил)карбамата (Промежуточное соединение 252, 1,475 г, 6,06 ммоль) и метилакрилата (0,82 мл, 9,09 ммоль) в МеОН (0,2 мл) под азотом при комнатной температуре добавляли хинуклидин (0,337 г, 3,03 ммоль). Реакционную смесь перемешивали при этой температуре в течение ночи. Добавляли еще 100 мг хинуклидина, и реакционную смесь перемешивали в течение еще 24 часов. Реакционную смесь затем распределяли между водой и этилацетатом. Слои разделяли, и органический слой промывали водой и рассолом, затем сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-30% этилацетата/гексаны) получили указанное в заголовке соединение в виде прозрачного масла (1,28 г, 65%). Продукт представляет собой смесь двух диастереомеров.
МС: 330 ЭРИ+ (C16H27NO6)
Промежуточное соединение 254: (2S,5S)-1-трет-бутил-4-метил-5-гидрокси-2-(метоксиметил)-5,6-дигидропиридин-1,4(2Н)-дикарбоксилат
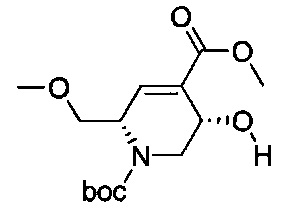
Указанное в заголовке соединение (0,45 г, 40%) было получено по методике, описанной для Промежуточного соединения 101, начиная с метил-4-(трет-бутоксикарбонил((S)-1-метоксибут-3-ен-2-ил)амино)-3-гидрокси-2-метиленбутаноата (Промежуточное соединение 253, 1,22 г).
МС: 302 ЭРИ+ (C14H23NO6)
1Н ЯМР (300 МГц, CDCl3) δ: 1.49 (s, 9Η) 2.96-3.18 (m, 1Η) 3.39 (s, 3Η) 3.52-3.73 (m, 2Η) 3.88 (s, 3Η) 3.99-4.19 (m, 1Η) 4.19-4.38 (m, 1Η) 4.52-4.59 (m, 1Η) 4.59-4.81 (m, 1Η) 7.01 (d, J=4.14 Гц, 1Η).
Промежуточное соединение 255: (2S,5S)-трет-бутил-5-гидрокси-2-(метоксиметил)-4-(метилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилат
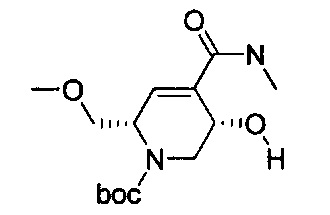
Сухую 2-горлую круглодонную колбу снабжали магнитной мешалкой и продували азотом дважды. Затем добавляли триметилалюминий (0,191 мл, 0,38 ммоль) и затем толуол (0,5 мл). Раствор охлаждали до -10°С и медленно добавляли метанамин (0,183 мл, 0,37 ммоль). Реакционную смесь перемешивали при этой температуре в течение 20 минут. Медленно добавляли раствор (2S,5S)-1-трет-бутил-4-метил-5-гидрокси-2-(метоксиметил)-5,6-дигидропиридин-1,4(2Н)-дикарбоксилата (Промежуточное соединение 254, 100 мг, 0,33 ммоль) в толуоле (0,5 мл) при комнатной температуре. После перемешивания в течение ночи реакционную смесь гасили 1М HCl и перемешивали в течение 15 минут до завершения гидролиза. Реакционную смесь экстрагировали этилацетатом. Органические слои промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (MeOH/DCM) получили указанное в заголовке соединение в виде прозрачного масла (53 мг).
МС: 302 ЭРИ+ (C14H24N2O5)
1Н ЯМР (300 МГц, CDCl3) δ: 1.48 (s, 9Η) 1.69 (br. s., 2Η) 2.57-2.94 (m, 3Η) 3.08 (dd, J=13.00, 9.04 Гц, 1Η) 3.36 (s, 3Η) 3.40-3.56 (m, 1Η) 3.51-3.72 (m, 1Η) 4.13 (dd, J=13.00, 4.71 Гц, 1Η) 4.47 (t, J=6.78 Гц, 1Η) 4.60-4.66 (m, 1Η) 6.60 (d, J=4.14 Гц, 1Η).
Промежуточное соединение 256: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-(метоксиметил)-4-(метилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилат
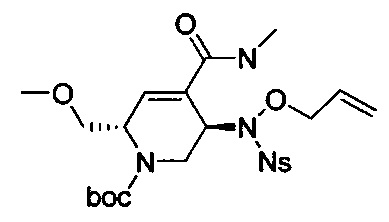
Указанное в заголовке соединение в виде не совсем белого твердого вещества (1 г, 63%) было получено по методике для Промежуточного соединения 10, начиная с (2S,5S)-трет-бутил 5-гидрокси-2-(метоксиметил)-4-(метилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 255, 0,87 г).
МС: 541 ЭРИ+ (C23H32N4O9S)
Промежуточное соединение 257: (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-N-метил-1,2,3,6-тетрагидропиридин-4-карбоксамид
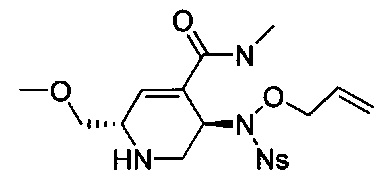
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенил-сульфонамидо)-2-(метоксиметил)-4-(метилкарбамоил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 256, 1 г, 1,85 ммоль) в диоксане (10 мл) при комнатной температуре добавляли 4М HCl в диоксане (2 мл, 57,16 ммоль). Реакционную смесь перемешивали при этой температуре в течение 4-5 часов. Реакционную смесь концентрировали, и неочищенное вещество переносили в 2М NaOH и экстрагировали дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. После хроматографии на силикагеле (с увеличением процента метанола в дихлорметане) получили указанное в заголовке соединение в виде густого масла (0,56 г, 69%).
МС: 441 ЭРИ+ (C18H24N4O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.58 (d, J=3.96 Гц, 3Н); 2.77 (br. s., 1H); 3.14-3.26 (m, 2H); 3.24 (s, 3Н) 3.48 (d, J=4.52 Гц, 1Η) 4.16-4.42 (m, 2Η) 4.66 (br. s., 1Η) 5.12-5.33 (m, 2Η) 5.83 (dd, J=17.24, 10.46 Гц, 1Η) 6.63 (d, J=2.26 Гц, 1Η) 7.77-7.94 (m, 2Η) 7.94-8.02 (m, 2Η) 8.08 (d, J=7.91 Гц, 1Η) (2Η пики, скрытые под пиком воды).
Промежуточное соединение 258: (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-N-метил-1,2,3,6-тетрагидропиридин-4-карбоксамид
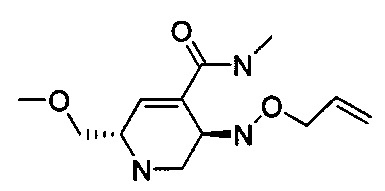
В смесь (3R,6S)-3-(N-(аллилокси)-2-нитрофенилсульфонамидо)-6-(метоксиметил)-N-метил-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 257, 561 мг, 1,27 ммоль) и карбоната калия (880 мг, 6,37 ммоль) в ацетонитриле (15 мл) при комнатной температуре добавляли бензолтиол (0,654 мл, 6,37 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (0%-20% метанол/дихлорметан) получили указанное в заголовке соединение в виде не совсем белого твердого вещества (0,26 г, 81%).
МС: 256 ЭРИ+ (C12H21N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.64 (d, J=4.71 Гц, 3Η) 2.73 (d, J=9.04 Гц, 1Η) 2.94 (dd, J=12.90, 3.11 Гц, 1Η) 3.20-3.28 (m, 2Η) 3.26 (s, 3Η) 3.37-3.52 (m, 1Η) 3.54-3.68 (m, 1Η) 4.12 (dt, J=5.70, 1.39 Гц, 2Η) 5.00-5.30 (m, 2Η) 5.79-6.04 (m, 1Η) 6.43 (d, J=8.48 Гц, 1Η) 6.53-6.65 (m, 1Η) 7.86 (d, J=4.52 Гц, 1Η) (1Η peak buried underneath вода peak).
Промежуточное соединение 259: (2S,5R)-6-(аллилокси)-2-(метоксиметил)-N-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамид
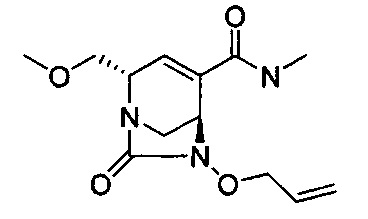
Указанное в заголовке соединение (0,28 г, 68%) было получено по методике, описанной для Промежуточного соединения 13, начиная с (3R,6S)-3-(аллилоксиамино)-6-(метоксиметил)-N-метил-1,2,3,6-тетрагидропиридин-4-карбоксамида (Промежуточное соединение 258, 0,52 г)
МС: 282 ЭРИ+ (C13H19N3O4)
1Н ЯМР (300 МГц, CDCl3) δ: 2.88 (d, J=4.90 Гц, 3Η) 3.27-3.38 (m, 2Η) 3.40 (s, 3Η) 3.49-3.79 (m, 2Η) 4.11 (td, J=5.79, 2.92 Гц, 1Η) 4.36-4.41 (m, 1Η) 4.41-4.56 (m, 2Η) 5.14-5.46 (m, 2Η) 5.94 (br. s., 1Η) 5.96-6.14 (m, 1Η) 6.26 (d, J=2.26 Гц, 1Η).
Промежуточное соединение 260: (2S,5R)-2-(метоксиметил)-4-(метилкарбамоил)-7-оксо-1.6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
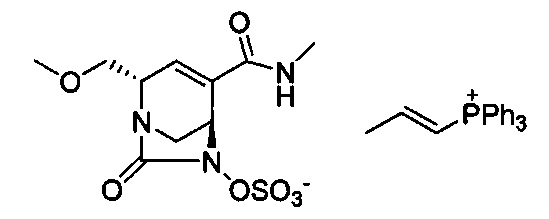
Указанное в заголовке соединение (0,33 г, 79%) было получено по методике, описанной для Промежуточного соединения 17, начиная с (2S,5R)-6-(аллилокси)-2-(метоксиметил)-N-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-4-карбоксамида (Промежуточное соединение 259, 0,19 г).
МС: 319 ЭРИ-, 303 ЭРИ+ (C10H15N3O7S, C21H20P)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.17 (dt, J=6.36, 2.00 Гц, 4Н) 2.65 (d, J=4.71 Гц, 3Н) 3.17-3.28 (m, 4Н) 3.51-3.69 (m, 2Н) 3.89 (td, J=5.79, 3.11 Гц, 1Н) 4.57 (d, J=1.13 Гц, 1Н) 6.38 (dd, J=2.92, 1.04 Гц, 1Н) 6.54-6.78 (m, 1Н) 7.18-7.41 (m, 1Н) 7.63-7.84 (m, 14Н) 7.85-8.00 (m, 3Н).
Пример 27
Натриевая соль (2S,5R)-2-карбамоил-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
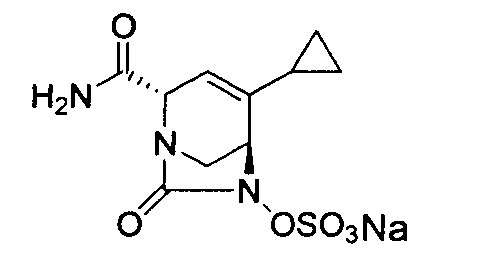
Указанное в заголовке соединение получали из (2S,5R)-2-карбамоил-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-енил)фосфония (Промежуточное соединение 272, 0,305 г, 0,50 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде белого твердого вещества (66,4 мг, 40%) после очистки методом ВЭЖХ (Synergi Polar RP 19 мм × 100 мм 5 мкм).
Оптическое вращение: (0,1 г/дл, МеОН)=-229
МС: 302 ЭРИ- (C10H13N3O6S)
1Н ЯМР (300 МГц, D2O) δ: 0.56 (m, 1Н); 0.78 (m, 3Н); 1.56 (m, 1Н); 3.28 (d, 1Н); 3.59 (m, 1Н); 4.07 (m, 1Н); 4.51 (m, 1Н); 5.63 (m, 1Н).
Промежуточные соединения для получения соединения Примера 27 получали следующим образом:
Промежуточное соединение 261: (Е)-N'-(1-циклопропилэтилиден)-2,4,6-триизопропилбензолсульфоногидразид
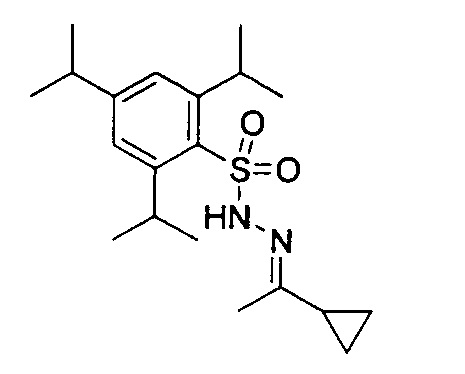
Указанное в заголовке соединение получали из 2,4,6-триизопропилбензолсульфонилгидразида (Aldrich, 20 г, 67,01 ммоль) и 1-циклопропилэтанона (Aldrich, 6,28 мл, 67,01 ммоль) по методике, описанной для Промежуточного соединения 33. Целевой продукт был получен в виде белого твердого вещества (15 г, 61%).
МС: 365 ЭРИ+ (C20H32N2O2S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.56 (m, 3Н); 0.75 (m, 1Н); 1.17 (m, 18Н); 1.47 (m, 1H); 1.61 (s, 3Н); 2.91 (m, 1H); 4.25 (m, 2H); 7.20 (s, 2H); 9.97 (s, 1H).
Промежуточное соединение 262: (S)-трет-бутил-1 -(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(3-циклопропил-2-оксобут-3-енил)карбамат
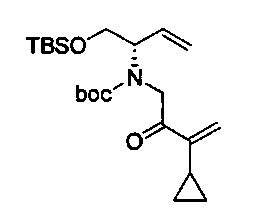
Указанное в заголовке соединение получали из (E)-N'-(1-циклопропилэтилиден)-2,4,6-триизопропилбензолсульфоногидразида (Промежуточное соединение 261, 17,58 г, 48,22 ммоль) и (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(2-(метокси(метил)амино)-2-оксоэтил)карбамата (Промежуточное соединение 5, 9,71 г, 24,11 ммоль) по методике, описанной для Промежуточного соединения 34. Целевой продукт был получен в виде светло-желтого масла (4,86 г, 49%).
МС: 410 ЭРИ+ (C22H39NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (m, 6Н); 0.45 (m, 2Н); 0.75 (m, 2Н); 0.83 (m, 10Н); 1.32 (m, 9Н); 0.70 (m, 1Н); 3.71 (m, 2Н); 4.35 (m, 2Н); 5.18 (m, 2Н); 5.46 (m, 1Н); 5.78 (m, 1Н); 5.88 (m, 1Н).
Промежуточное соединение 263: (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
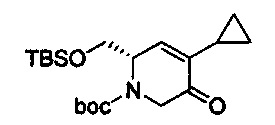
Указанное в заголовке соединение получали из (S)-трет-бутил-1-(трет-бутилдиметилсилилокси)бут-3-ен-2-ил(3-циклопропил-2-оксобут-3-енил)карбамата (Промежуточное соединение 262, 5,16 г, 12,60 ммоль) по методике, описанной для Промежуточного соединения 7, за исключением того, что реакционную смесь нагревали при 85°С в течение ночи. Целевой продукт был получен в виде светло-желтого масла (3,82 г, 79%).
МС: 382 ЭРИ+ (C20H35NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (m, 6Н); 0.45 (m, 2Н); 0.75 (m, 1Н); 0.83 (m, 10Н); 1.28 (m, 1Н); 1.41 (m, 9Н); 1.71 (m, 1Н); 3.85 (m, 2Н); 4.05 (m, 1Н); 4.66 (m, 1Н); 6.56 (m, 1Н).
Промежуточное соединение 264: (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
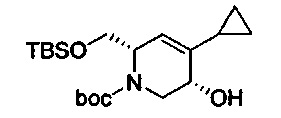
Указанное в заголовке соединение получали из (S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 263, 3,82 г, 10,01 ммоль) по методике, описанной для Промежуточного соединения 8. Целевой продукт был получен в виде бесцветного масла (3 г, 78%).
МС: 384 ЭРИ+ (C20H37NO4Si)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (s, 6Н); 0.22 (m, 1Н); 0.57 (m, 3Н); 0.85 (s, 9Н); 1.38 (s, 9Н); 1.57 (m, 1Н); 2.69 (m, 1Н); 3.58 (m, 2Н); 3.93 (m, 1Н); 4.17 (m, 1Н); 5.11 (d, 1Н); 5.22 (m, 1Н).
Промежуточное соединение 265: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
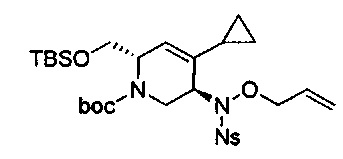
Указанное в заголовке соединение получали из (2S,5S)-трет-бутил-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 264, 3 г, 7.82 ммоль) и N-(аллилокси)-2-нитробензолсульфонамида (Промежуточное соединение 9, 2,020 г, 7,82 ммоль) по методике, описанной для Промежуточного соединения 10. Целевой продукт был получен в виде желтого масла (3,62 г, 74%).
МС: 624 ЭРИ+ (C29H45N3O8SSi)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (s, 6Н); 0.40 (m, 3Н); 0.83 (s, 9Н); 1.31 (m, 9Н); 3.14 (m, 1Н); 3.57 (m, 2Н); 4.10 (m, 4Н); 4.43 (m, 1Н); 5.20 (m, 2Н); 5.70 (m, 2Н); 8.02 (m, 4Н).
Промежуточное соединение 266: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-циклопропил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилат
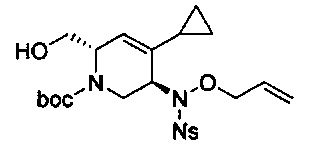
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-((трет-бутилдиметилсилилокси)метил)-4-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 265, 3,62 г, 5,80 ммоль) в THF (30 мл) по методике, описанной для Промежуточного соединения 18. Целевой продукт был получен в виде не совсем белой пены (2,64 г, 89%).
МС: 510 ЭРИ+ (C23H31N3O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.46 (m, 3Н); 1.34 (m, 9Н); 3.07 (m, 1Н); 3.38 (m, 2Н); 4.05 (m, 2Н); 4.29 (m, 3Н); 4.73 (m, 1Н); 5.20 (m, 2Н); 5.69 (m, 2Н); 8.04 (m, 4Н).
Промежуточное соединение 267: (2S,5R)-5-(N-(аллилокси)-2-нитро-фенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоновая кислота
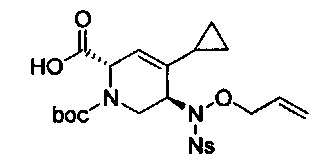
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-циклопропил-2-(гидроксиметил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 266, 2,64 г, 5,18 ммоль) по методике, описанной для Промежуточного соединения 19. Целевой продукт был получен в виде оранжевой пены (2,33 г, 86%).
МС: 524 ЭРИ+ (C23H29N3O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.46 (m, 3Н); 1.28 (m, 9Н); 3.10 (m, 1Н); 3.90 (m, 1Н); 4.29 (m, 3Н); 4.78 (m, 1Н); 5.19 (m, 2Н); 5.69 (m, 2Н); 8.04 (m, 4Н).
Промежуточное соединение 268: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилат
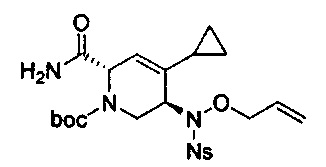
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-1-(трет-бутоксикарбонил)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоновой кислоты (Промежуточное соединение 267, 2,33 г, 4,45 ммоль) по методике, описанной для Промежуточного соединения 20. Целевой продукт был получен в виде желто-коричневой пены (1,47 г, 63%).
МС: 523 ЭРИ+ (C23H30N4O8S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.53 (m, 4Н); 1.28 (m, 9Н); 3.15 (m, 1Н); 3.93 (m, 1Н); 4.27 (m, 3Н); 4.73 (m, 1Н); 5.19 (m, 2Н); 5.73 (m, 2Н); 7.03 (m, 1Н); 7.43 (m, 1Н); 8.05 (m, 4Н).
Промежуточное соединение 269: (2S,5R)-5-(N-(аллилокси)-2-нитро-фенилсульфонамидо)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
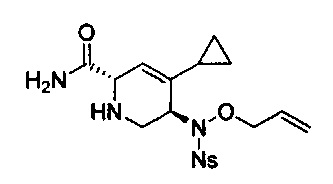
Указанное в заголовке соединение получали из (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-2-карбамоил-4-циклопропил-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 268, 1,47 г, 2,81 ммоль) по методике, описанной для Промежуточного соединения 21. Целевой продукт был получен в виде светло-желтой пены (0,95 г, 80%).
МС: 423 ЭРИ+ (C18H22N4O6S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.30 (m, 1Н); 0.50 (m, 2Н); 0.81 (m, 1Н); 1.28 (m, 1Н); 2.70 (m, 2Н); 3.75 (m, 1Н); 4.07 (m, 1Н); 4.37 (m, 2Н); 5.24 (m, 2Н); 5.84 (m, 2Н); 7.02 (m, 1Н); 7.28 (m, 1Н); 8.04 (m, 4Н).
Промежуточное соединение 270: (R)-5-(аллилоксиамино)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамид
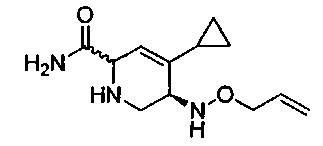
Указанное в заголовке соединение получали из (2S,5R)-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 269, 0,95 г, 2,25 ммоль) по методике, описанной для Промежуточного соединения 22. Целевой продукт был получен в виде светло-желтого масла (0,307 г, 57%).
МС: 238 ЭРИ+ (C12Η19Ν3O2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.52 (m, 4Н); 1.43 (m, 2Н); 2.46 (m, 1Н); 3.04 (m, 2Н); 3.59 (m, 1Н); 4.13 (m, 2Н); 5.20 (m, 2Н); 5.39 (m, 1Н); 5.92 (m, 1Н); 6.39 (m, 1Н); 7.00 (m, 1Н); 7.34 (bs, 1Н).
Промежуточное соединение 271: (2S,5R)-6-(аллилокси)-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
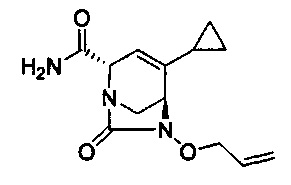
Указанное в заголовке соединение получали из (R)-5-(аллилоксиамино)-4-циклопропил-1,2,5,6-тетрагидропиридин-2-карбоксамида (Промежуточное соединение 270, 0,307 г, 1,29 ммоль) по методике, описанной для Промежуточного соединения 16. Целевой продукт был получен в виде желтого масла (0,168 г, 49%).
МС: 264 ЭРИ+ (C13H17N3O3)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.56 (m, 4Н); 1.43 (m, 1Н); 3.17 (m, 2Н); 3.71 (m, 1Н); 4.11 (m, 1Н); 4.36 (m, 2Н); 5.34 (m, 3Н); 5.94 (m, 1Н); 7.27 (bs, 1Н); 7.49 (bs, 1Н).
Промежуточное соединение 272: (2S,5R)-2-карбамоил-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-енил)фосфония
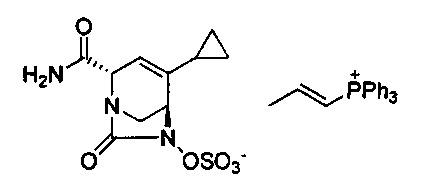
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 271, 0,168 г, 0,64 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде желтой пены (0,305 г, 79%).
МС: 302 ЭРИ-, 303 ЭРИ+ (C10H12N3O6S-⋅C21H20P+)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.56 (m, 3Н); 0.85 (m, 1Н); 1.42 (m, 1Н); 2.16 (m, 3Н); 3.18 (m, 2Н); 3.71 (m, 1Н); 4.10 (m, 1Н); 5.44 (m, 1Н); 6.65 (m, 1Н); 7.25 (m, 2Н); 7.51 (m, 1Н); 7.79 (m, 15Н).
Пример 28
Натриевая соль (2S,5R)-3-(2-метоксиэтил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
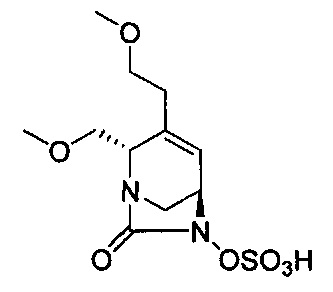
Указанное в заголовке соединение получали из (2S,5R)-3-(2-метоксиэтил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата (Е)-трифенил(проп-1-ен-1-ил)фосфония (Промежуточное соединение 288, 0,328 г, 0,53 ммоль) по методике, описанной для соединения Примера 1. Целевой продукт был получен в виде не совсем белого твердого вещества (146 м г, 81%).
Оптическое вращение: (0,22 г/дл, DMSO)=-263
МС: 321 ЭРИ- (C11H18N2O7S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.13 (m, 2Н); 3.07 (m, 1Н); 3.22 (s, 3Н); 3.28 (s, 3Н); 3.36 (m, 3Н); 3.68 (m, 3Н); 4.00 (m, 1Н); 6.03 (m, 1Н).
Промежуточные соединения для получения соединения Примера 28 получали следующим образом:
Промежуточное соединение 273: ((3-бромбут-3-ен-1-ил)окси)(трет-бутил)диметилсилан
В раствор 3-бромбут-3-ен-1-ола (ACROS, 15,18 г, 100,53 ммоль) в DCM (300 мл) при комнатной температуре добавляли имидазол (8,90 г, 130,69 ммоль), 4-диметиламинопиридин (2,456 г, 20,11 ммоль) и трет-бутилдиметилсилилхлорид (16,67 г, 110,58 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем фильтровали для удаления твердого вещества и промывали рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили указанное в заголовке соединение (24,67 г, 93%) в виде бесцветного масла.
1Н ЯМР (300 МГц, CDCl3) δ: 0.08 (s, 3Н), 0.90 (s, 9Н), 2.63 (t, 2Н), 3.80 (t, 2Н), 5.46 (m, 1Н), 5.64 (m, 1Н).
Промежуточное соединение 274: (S,Е)-N-(2-((трет-бутилдиметил-силил)окси)этилиден)-2-метилпропан-2-сульфинамид
В раствор 2-((трет-бутилдиметилсилил)окси)ацетальдегида (Aldrich, 15 г, 86,05 ммоль) в DCM (200 мл) при комнатной температуре добавляли сульфат меди(II) (41,2 г, 258,16 ммоль) и (S)-2-метилпропан-2-сульфинамид (Aldrich, 15,64 г, 129,08 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем фильтровали через целит, промывали DCM и концентрировали с получением масла. После хроматографии на силикагеле (0%-25% этилацетата/гексаны) получили указанное в заголовке соединение (14,73 г, 61,7%) в виде бесцветного масла.
1Н ЯМР (300 МГц, CDCl3) δ: 0.11 (s, 6Н), 0.93 (s, 9Н), 1.22 (s, 9Н), 4.55 (d, 2Н), 8.07 (t, 1Н).
Промежуточное соединение 275: (S)-2-метил-N-((S)-2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)пропан-2-сульфинамид
В раствор ((3-бромбут-3-ен1-ил)окси)(трет-бутил)диметилсилана (Промежуточное соединение 273, 24,66 г, 92,97 ммоль) в THF (200 мл) при -78°С по каплям через канюлю добавляли трет-бутиллитий (1,7М в пентане) (120 мл, 204,54 ммоль). Реакционную смесь перемешивали в течение 45 минут при -78°С. По каплям добавляли (S,Е)-N-(2-((трет-бутилдиметилсилил)окси)-этилиден)-2-метилпропан-2-сульфинамид (Промежуточное соединение 274, 17,2 г, 61,98 ммоль) в THF (50 мл). Реакционную смесь перемешивали в течение примерно 1,5 часов при -78°С. Реакционную смесь гасили насыщенным раствором бикарбоната натрия и экстрагировали дважды диэтиловым эфиром. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили указанное в заголовке соединение (21,07 г, 73,3%) в виде бесцветного масла.
1Н ЯМР (300 МГц, CDCl3) δ: 0.06 (m, 12Н), 0.90 (m, 18Н), 1.24 (s, 9H), 2.25 (m, 2H), 3.57 (m, 1H); 3.73 (m, 3Н); 3.95 (m, 2H); 5.04 (m, 1H); 5.19 (m, 1H).
Промежуточное соединение 276: гидрохлорид (S)-2-амино-3-метиленпентан-1,5-диола
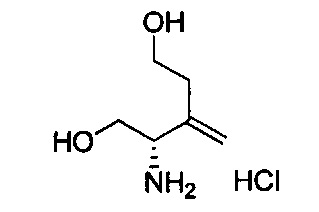
В раствор 2-метил-N-((S)-2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)пропан-2-сульфинамида (Промежуточное соединение 275, 21,07 г, 45,42 ммоль) в метаноле (100 мл) при 0°С добавляли соляную кислоту (4М в диоксане) (22,71 мл, 90,85 ммоль). Реакционную смесь перемешивали при 0°С в течение примерно 20 минут. По результатам ЖХ/МС исходного вещества не осталось. Реакционную смесь концентрировали с получением масла (7,6 г, 100%).
МС: 132 ЭРИ+ (C6H13NO2)
Промежуточное соединение 277: (S)-2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-амин
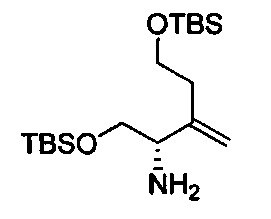
В раствор (S)-2-амино-3-метиленпентан-1,5-диола.HCl (Промежуточное соединение 276, 7,6 г, 45,34 ммоль) в DCM (200 мл) при комнатной температуре добавляли имидазол (12,35 г, 181,35 ммоль), 4-диметиламинопиридин (2,77 г, 22,67 ммоль) и трет-бутилдиметилсилилхлорид (20,50 г, 136,01 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали для удаления твердого вещества и промывали рассолом. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-5% метанола/дихлорметан) получили указанное в заголовке соединение (14,34 г, 88%) в виде желтого масла.
МС: 359 ЭРИ+ (C18H41NO2Si2)
1Н ЯМР (300 МГц, CDCl3) δ: 0.06 (m, 12Н), 0.90 (m, 18Н), 2.30 (m, 2H), 3.41 (m, 2H); 3.72 (m, 3Н); 4.90 (m, 1H); 5.09 (m, 1H).
Промежуточное соединение 278: (S)-трет-бутил-(2-(метокси(метил)амино)-2-оксоэтил)(2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)карбамат
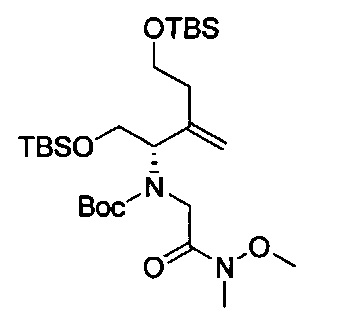
Смесь (S)-2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-амина (14,34 г, 39,87 ммоль) и карбоната калия (Промежуточное соединение 277, 5,51 г, 39,87 ммоль) в DMF (300 мл) перемешивали при комнатной температуре в течение 1 часа. Добавляли 2-бром-N-метокси-N-метилацетамид (Промежуточное соединение 4, 7,26 г, 39,87 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия. Образовавшуюся эмульсию фильтровали для удаления твердого вещества. Слои разделяли, и органический слой промывали смесью 1:1 рассол:вода. Органический слой сушили над сульфатом магния, фильтровали и концентрировали. Полученное желтое масло растворяли в THF (100 мл) и добавляли ди-трет-бутил-дикарбонат (17,40 г, 79,73 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем при 50°С в течение примерно 4 часов, затем при комнатной температуре в течение выходных дней. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия. Слои разделяли, и органический слой промывали дважды смесью 1:1 рассол:вода, сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-30% этилацетата/гексаны) получили указанное в заголовке соединение (13,55 г, 60,6%) в виде светло-желтого масла.
МС: 561 ЭРИ+ (C27H56N2O6S12)
1Н ЯМР (300 МГц, CDCl3) δ: 0.06 (m, 12Н), 0.88 (m, 18Н), 1.45 (m, 9H), 2.32 (m, 2H); 3.16 (s, 3Н); 3.69 (m, 5H); 3.80 (m, 2H); 3.02 (m, 2H); 4.64 (m, 1H); 5.04 (m, 2H).
Промежуточное соединение 279: (S)-трет-бутил-(2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)(2-оксопент-3-ен-1-ил)карбамат
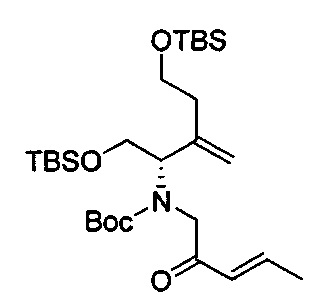
Суспензию хлорида церия (III) (47,6 г, 193,26 ммоль) в THF (200 мл)3 при комнатной температуре энергично перемешивали в течение 2 часов. Суспензию охлаждали до -78°С и по каплям добавляли бромид (Е)-проп-1-енилмагния (0,5 Μ в THF) (387 мл, 193,26 ммоль). Смесь перемешивали при -78°С в течение 1,5 часов. Затем по каплям добавляли (S)-трет-бутил-(2-(метокси(метил)амино)-2-оксоэтил)(2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)карбамат (Промежуточное соединение 278, 13,55 г, 24,16 ммоль) в THF (50 мл) при -78°С. Реакционную смесь перемешивали при -78°С в течение 1 часа и затем нагревали до 0°С в течение 30 минут. Реакционную смесь гасили 10%-ной лимонной кислотой, разбавляли водой и экстрагировали дважды диэтиловым эфиром. Органические слои сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение (10 г, 76%) в виде желтого масла.
МС: 542 ЭРИ+ (C28H55NO5Si2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (m, 12Н), 0.85 (m, 18H), 1.33 (m, 9H), 1.84 (m, 2H); 2.18 (m, 2H); 3.71 (m, 6H); 4.56 (m, 1H); 4.93 (m, 2H); 6.20 (m, 1H); 6.85 (m, 1H).
Промежуточное соединение 280: (S)-трет-бутил-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)-метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилат
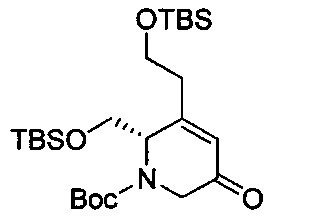
Раствор (S,Е)-трет-бутил-(2,2,3,3,11,11,12,12-октаметил-7-метилен-4,10-диокса-3,11-дисилатридекан-6-ил)(2-оксопент-3-ен-1-ил)карбамата (Промежуточное соединение 279, 10 г, 18,45 ммоль) в толуоле (400 мл) продували азотом в течение 15 минут. Затем добавляли катализатор Ховейды-Граббса 2-го поколения (2,320 г, 3,69 ммоль). Реакционную смесь нагревали при 100°С в течение ночи. Добавляли еще 0,05 экв. катализатора, и реакционную смесь нагревали при 100°С в течение еще 2 часов. Реакционную смесь концентрировали на силикагеле. После хроматографии на силикагеле (0%-15% этилацетата/гексаны) получили указанное в заголовке соединение (8,55 г, 93%) в виде светло-коричневого масла.
МС: 500 ЭРИ+ (C25H49NO5Si2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.01 (m, 12Н), 0.81 (m, 18H), 1.42 (s, 9H), 2.50 (m, 2H); 3.79 (m, 5H); 4.20 (m, 1H); 4.67 (m, 1H); 6.07 (s, 1H).
Промежуточное соединение 281: (2S,5S)-трет-бутил-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)-метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилат
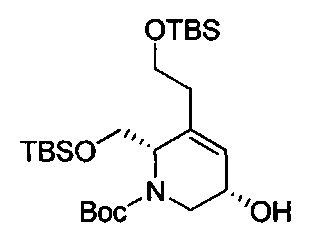
Указанное в заголовке соединение получали из (S)-трет-бутил-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)метил)-5-оксо-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 280, 8,55 г, 17,11 ммоль) по методике, описанной для Промежуточного соединения 8, с получением целевого продукта (7,33 г, 85%) в виде светло-желтого масла.
МС: 502 ЭРИ+ (C25H51NO5Si2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.02 (m, 12Н), 0.85 (m, 18H), 1.39 (s, 9H), 2.21 (m, 2H); 2.68 (m, 1H); 3.70 (m, 4H); 4.02 (m, 2H); 4.21 (m, 1H); 4.99 (m, 1H); 5.55 (s, 1H).
Промежуточное соединение 282: (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилат
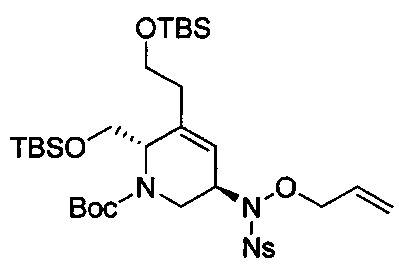
В раствор (2S,5S)-трет-бутил-3-(2-((трет-бутилдиметилсилил)окси)-этил)-2-(((трет-бутилдиметилсилил)окси)метил)-5-гидрокси-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 281, 7,33 г, 14,61 ммоль) в толуоле (100 мл) при комнатной температуре добавляли трифенилфосфин (4,58 г, 17,53 ммоль), N-(аллилокси)-2-нитробензол-сульфонамид (3,77 г, 14,61 ммоль) и диизопропилазодикарбоксилат (3,45 мл, 17,53 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем концентрировали. Полученное масло растирали с гексаном и фильтровали для удаления трифенилфосфиноксида. Фильтрат концентрировали на силикагеле. После хроматографии на силикагеле (0%-20% этилацетата/гексаны) получили указанное в заголовке соединение (7,5 г, 69,2%) в виде светло-желтого масла.
МС: 743 ЭРИ+ (C34H59N3O9SSi2)
Промежуточное соединение 283: N-(аллилокси)-N-((3R,6S)-5-(2-((трет-бутилдиметилсилил)окси)этил)-6-(((трет-бутилдиметилсилил)-окси)метил)-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамид
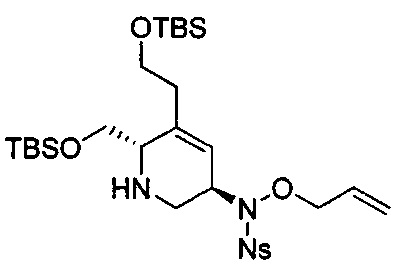
В раствор (2S,5R)-трет-бутил-5-(N-(аллилокси)-2-нитрофенилсульфонамидо)-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)метил)-5,6-дигидропиридин-1(2Н)-карбоксилата (Промежуточное соединение 282, 7,5 г, 10,11 ммоль) в DCM (100 мл) при комнатной температуре добавляли бромид цинка (6,83 г, 30,32 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли еще эквивалент бромида цинка, и реакционную смесь перемешивали в течение еще 24 часов. Реакционную смесь разбавляли DCM и добавляли насыщенный раствор бикарбоната натрия. Двухфазную смесь фильтровали через целит для удаления твердого вещества, и слои разделяли. Органический слой промывали один раз рассолом, сушили над сульфатом магния, фильтровали и концентрировали с получением оранжевого масла (6,49 г, 100%).
МС: 642 ЭРИ+ (C29H51N3O7SSi2)
Промежуточное соединение 284: O-аллил-N-((3R,6S)-5-(2-((трет-бутилдиметилсилил)окси)этил)-6-(((трет-бутилдиметилсилил)окси)-метил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламин
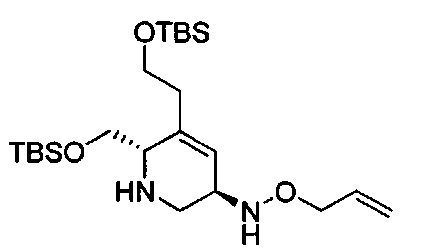
Указанное в заголовке соединение получали из N-(аллилокси)-N-((3R,6S)-5-(2-((трет-бутилдиметилсилил)окси)этил)-6-(((трет-бутилдиметилсилил)окси)метил)-1,2,3,6-тетрагидропиридин-3-ил)-2-нитробензолсульфонамида (Промежуточное соединение 283, 6,49 г, 10,11 ммоль) по методике, описанной для Промежуточного соединения 12. После хроматографии на силикагеле (0%-5% метанола/дихлорметан) получили целевой продукт (3,95 г, 86%) в виде желтого масла.
МС: 457 ЭРИ+ (C23H48N2O3Si2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.03 (m, 12Н), 0.85 (m, 18H), 2.15 (m, 2H), 2.65 (m, 1H); 2.89 (m, 1H); 3.20 (m, 2H); 3.61 (m, 4H); 4.08 (m, 2H); 5.15 (m, 2H); 5.44 (m, 1H); 5.88 (m, 1H); 6.16 (m, 1H).
Промежуточное соединение 285: (2S,5R)-6-(аллилокси)-3-(2-((трет-бутилдиметилсилил)окси)этил)-2-(((трет-бутилдиметилсилил)окси)-метил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
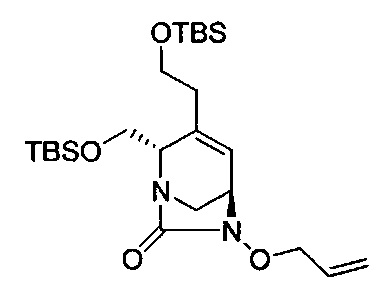
В раствор O-аллил-N-((3R)-5-(2-((трет-бутилдиметилсилил)окси)этил)-6-(((трет-бутилдиметилсилил)окси)метил)-1,2,3,6-тетрагидропиридин-3-ил)гидроксиламина (Промежуточное соединение 284, 3,95 г, 8,65 ммоль) и диизопропилэтиламина (6,02 мл, 34,58 ммоль) в ацетонитриле (600 мл) при 0°С добавляли трифосген (0,871 г, 2,94 ммоль) в виде раствора в ацетонитриле (26 мл). Раствор трифосгена добавляли со скоростью 4 мл/час. Сразу после окончания добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и смесью 1:1 рассол:вода. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-30% этилацетата/гексаны) получили указанное в заголовке соединение в виде желтого масла (3.69 г, 88%). Эту реакцию проводили в двух отдельных 1-литровых колбах из-за большого объема, и реакционные смеси объединяли для обработки и очистки.
МС: 483 ЭРИ+ (C24H46N2O4Si2)
1Н ЯМР (300 МГц, DMSO-d6) δ: 0.04 (m, 12Н), 0.85 (m, 18H), 2.11 (m, 2H), 3.01 (m, 1H); 3.44 (m, 1H); 3.63 (m, 3H); 3.93 (m, 2H); 4.32 (m, 2H); 5.28 (m, 2H); 5.93 (m, 1H);6.05 (m, 1H).
Промежуточное соединение 286: (2S,5R)-6-(аллилокси)-3-(2-гидроксиэтил)-2-(гидроксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
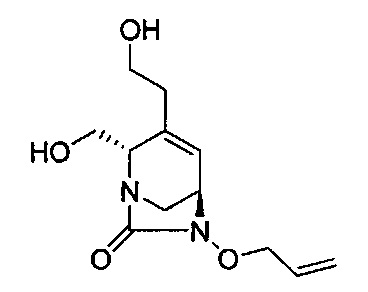
В раствор (2S,5R)-6-(аллилокси)-3-(2-((трет-бутилдиметилсилил)окси)-этил)-2-(((трет-бутилдиметилсилил)окси)метил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 285, 0,96 г, 1,99 ммоль) в THF (6 мл) при 0°С добавляли фторид тетрабутиламмония (1М в THF) (5,97 мл, 5,97 ммоль). Реакционную смесь перемешивали в течение примерно 1 часа и затем концентрировали на силикагеле. После хроматографии на силикагеле (0%-10% метанола/дихлорметан) получили указанное в заголовке соединение (0,446 г, 88%) в виде мутного желтого масла.
МС: 255 ЭРИ+ (C12H18N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.06 (m, 2Н), 2.98 (m, 1Н), 3.42 (m, 3Н), 3.60 (m, 1Н); 3.71 (m, 2Н); 3.88 (m, 1Н); 4.33 (m, 2Н); 4.51 (t, 1Н); 4.84 (t, 1Н); 5.27 (m, 2Н); 5.96 (m, 2Н).
Промежуточное соединение 287: (2S,5R)-6-(аллилокси)-3-(2-метоксиэтил)-2-(метоксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-он
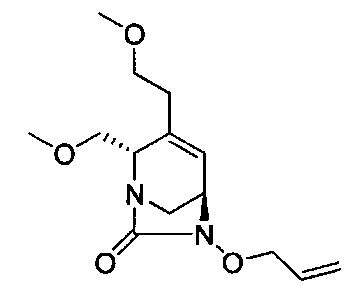
В раствор (2S,5R)-6-(аллилокси)-3-(2-гидроксиэтил)-2-(гидроксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 286, 0,446 г, 1,75 ммоль) и йодметана (0,655 мл, 10,52 ммоль) в DMF (8 мл) при 0°С добавляли гидрид натрия (60% в минеральном масле) (0,210 г, 5,26 ммоль). Реакционную смесь перемешивали в течение 10 минут при 0°С, затем гасили водой и разбавляли этилацетатом. Слои разделяли, и водный слой экстрагировали однократно DCM. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-50% этилацетата/гексаны) получили указанное в заголовке соединение (0,138 г, 28%) в виде желтого масла.
МС: 283 ЭРИ+ (C14H22N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 2.12 (m, 2Н), 3.02 (m, 1Н), 3.21 (s, 3Н), 3.27 (s, 3Н); 3.35 (m, 3Н); 3.66 (m, 3Н); 3.90 (m, 1Н); 4.33 (m, 2Н); 5.27 (m, 2Н); 5.96 (m, 2Н).
Промежуточное соединение 288: (2S,5R)-3-(2-метоксиэтил)-2-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-ен-1-ил)фосфония
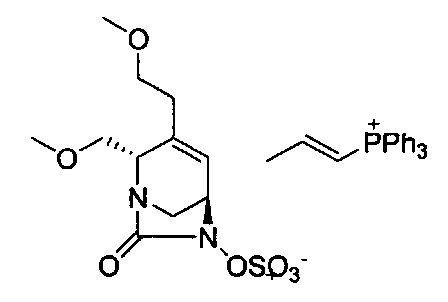
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-3-(2-метоксиэтил)-2-(метоксиметил)-1,6-диазабицикло[3.2.1]окт-3-ен-7-она (Промежуточное соединение 287, 0,177 г, 0,63 ммоль) по методике, описанной для Промежуточного соединения 17. Целевой продукт был получен в виде светло-желтой пены (0,328 г, 84%).
МС: 323, 303 ЭРИ+ (C14H22N2O4, C21H20P)
Пример 29
Натриевая соль (2S,5R)-2-(((1,5-дигидрокси-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
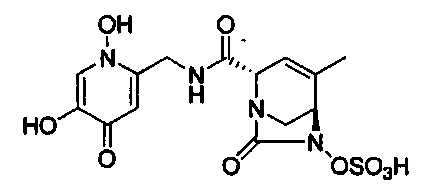
В раствор (2S,5R)-2-(((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата (Промежуточное соединение 296, 58,6 мг, 0,08 ммоль) в анизоле (906 мкл, 8,34 ммоль) при 0°С добавляли трифторуксусную кислоту (321 мкл, 4,17 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2,5 часов. Реакционную смесь разбавляли DCM и водой. Слои разделяли, и водный слой экстрагировали более одного раза DCM. Водный слой напрямую наносили на колонку 5,5 г RediSep Gold С18 и пропускали через нее 100%-ную воду. Фракции 1 и 2 содержали целевой продукт, и их объединяли и лиофилизировали с получением светло-оранжевого твердого вещества. Соединение прогоняли через другую С18 колонку с водой и собирали в пробирки 1 и 2. Их объединяли и лиофилизировали. После очистки методом ВЭЖХ (колонка YMC Carotenoid С30 21,2 мм × 150 мм 5 мкм, спаренная с колонкой Synergi Polar RP 100 мм × 21,2 мм 4 мкм, 0%-15% ацетонитрила/вода) получили указанное в заголовке соединение в виде розового твердого вещества (3,4 мг, 9,3%) после лиофилизации.
МС: 417 ЭРИ+ (C14H16N4O9S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.93 (s, 3Н); 3.31 (m, 1Н); 3.61 (m, 1Н); 4.22 (m, 1Н); 4.49 (m, 2Н); 4.61 (m, 1Н); 5.69 (m, 1Н); 6.49 (s, 1Н); 7.64 (s, 1Н).
Промежуточные соединения для получения соединения Примера 29 получали следующим образом.
Промежуточное соединение 289: 2-(гидроксиметил)-5-[(4-метокси-бензил)окси]-4H-пиран-4-он
В суспензию койевой кислоты (500 г, 3,518 моль) в сухом DMF (5 л) добавляли сухой карбонат калия (972 г, 7,036 моль), затем 4-метоксибензилхлорид (661 г, 4,221 моль) при 0°С. Реакционную смесь нагревали до комнатной температуры и затем нагревали при 80°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь вливали в ледяную воду (15 л) и энергично перемешивали. Осадок собирали фильтрованием и сушили в вакууме с получением указанного в заголовке соединения в виде бледно-коричневого твердого вещества (687 г, 75%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.74 (s, 3Н); 4.27 (s, 2Н); 4.83 (s, 2Н); 5.68 (bs, 1Н); 6.29 (s, 1Н); 6.93 (d, 2Н); 7.33 (d, 2Н); 8.12 (s, 1Н).
Промежуточное соединение 290: 1-гидрокси-2-(гидроксиметил)-5-[(4-метоксибензил)окси]пиридин-4(1Н)-он
В перемешиваемый раствор 2-(гидроксиметил)-5-[(4-метоксибензил)окси]-4Н-пиран-4-она (Промежуточное соединение 289, 101,7 г, 0,388 моль) в пиридине (1,35 л) добавляли гидрохлорид гидроксиламина (134,7 г, 1,94 моль). Реакционную смесь нагревали при 85°С в течение 4 часов, затем упаривали досуха и растирали с водой (700 мл). Осадок собирали фильтрованием и промывали водой (250 мл). Твердое вещество затем перемешивали в изопропаноле (100 мл) в течение 12 часов, собирали фильтрованием и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (41,2 г, 38%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.75 (s, 3Н); 4.55 (s, 2Н); 5.05 (s, 2Н); 6.95 (d, 2Н); 7.06 (s, 1Н); 7.38 (d, 2Н); 7.47 (m, 1Н); 8.25 (s, 1Н); 8.62 (d, 1Н).
Промежуточное соединение 291: 1-(дифенилметокси)-2-(гидроксиметил)-5-[(4-метоксибензил)окси]пиридин-4(1Н)-он
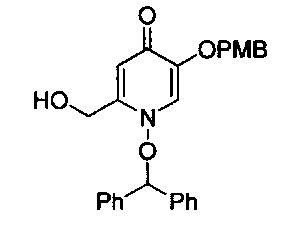
В перемешиваемый охлажденный во льду раствор 1-гидрокси-2-(гидроксиметил)-5-[(4-метоксибензил)окси]пиридин-4(1H)-она (Промежуточное соединение 290, 45 г, 0,162 моль) добавляли трет-бутоксид калия (18,2 г, 0,162 моль), затем бензилхлорид (36,14 г, 0,178 моль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем вливали в ледяную воду (3 л) и перемешивали в течение 1 часа. Осадок собирали фильтрованием и сушили в вакууме с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (70,4 г, 98%).
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.76 (s, 3Н); 4.25 (d, 2Н); 4.65 (s, 2Н); 5.54 (bs, 1Н); 6.05 (s, 1Н); 6.51 (s, 1Н); 6.94 (d, 2Н); 7.25 (d, 2Н); 7.41 (m, 11Η).
Промежуточное соединение 292: 2-((1-(6ензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)изоиндолин-1,3-дион
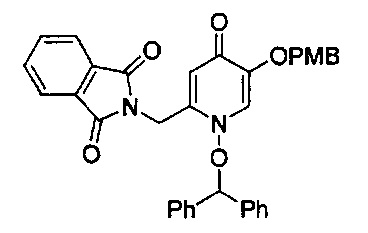
В раствор 1-(бензгидрилокси)-2-(гидроксиметил)-5-((4-метоксибензил)-окси)пиридин-4(1Н)-она (Промежуточное соединение 291, 2 г, 4,51 ммоль), фталимида (0,664 г, 4,51 ммоль) и трифенилфосфина (1,178 г, 4,51 ммоль) в THF (20 мл) при комнатной температуре добавляли диизопропилазодикарбоксилат (2,397 мл, 12,18 ммоль). Реагенты являются нерастворимыми. Добавляли DMF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и концентрировали на силикагеле. Хроматография на силикагеле (0%-4% метанола/дихлорметан) не обеспечила удаление примесей из целевого. Фракции объединяли и снова очищали. После хроматографии на силикагеле (0%-30% ацетона/дихлорметан) получили указанное в заголовке соединение (1,66 г, 64,3%) в виде светло-желтой пены.
МС: 573 ЭРИ+ (C335H28N2O6)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.76 (s, 3Н); 4.51 (s, 2Н); 4.70 (s, 2Н); 5.71 (s, 1Н); 6.63 (s, 1Н); 6.94 (m, 2Н); 7.26 (m, 2Н); 7.45 (m, 10Н); 7.62 (s, 1Н); 7.88 (m, 4Н).
Промежуточное соединение 293: 2-(аминометил)-1-(бензгидрилокси)-5-((4-метоксибензил)окси)пиридин-4(1Н)-он
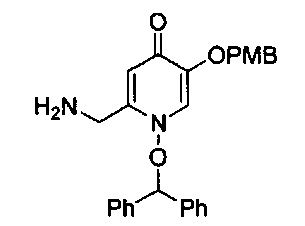
В раствор 2-((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)изоиндолин-1,3-диона (Промежуточное соединение 292, 1,66 г, 2,90 ммоль) в хлороформе (20 мл) и метаноле (10 мл) при комнатной температуре добавляли моногидрат гидразина (0,284 мл, 5,80 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли еще 1 экв. моногидрата гидразина. Через 3 часа исходное вещество все еще присутствовало. Добавляли еще 1 экв. моногидрата гидразина. Через 2 часа реакционную смесь фильтровали для удаления твердого вещества. Фильтрат концентрировали. Остаток растирали с МеОН и диэтиловым эфиром, и твердое вещество отфильтровывали. Это повторяли более двух раз. Полученное твердое вещество растирали один раз с хлороформом и МеОН, и твердое вещество удаляли фильтрованием. Фильтрат концентрировали с получением желтой пены (1,01 г, 79%).
МС: 443 ЭРИ+ (C27H26N2O4)
1Н ЯМР (300 МГц, DMSO-d6) δ: 3.76 (s, 3Н); 4.25 (d, 2Н); 4.65 (s, 2Н); 5.54 (bs, 1Н); 6.05 (s, 1Н); 6.51 (s, 1Н); 6.94 (d, 2Н); 7.25 (d, 2Н); 7.29 (s, 1Н); 7.41 (m, 11Η).
Промежуточное соединение 294: (2S,5R)-6-(аллилокси)-N-((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамид
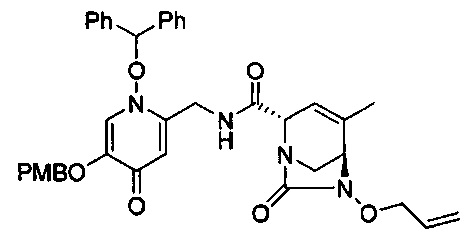
В раствор (2R,5R)-6-(аллилокси)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоновой кислоты (Промежуточное соединение 32, 0,54 г, 2,27 ммоль) в DMF (10 мл) при комнатной температуре добавляли 2-(аминометил)-1-(бензгидрилокси)-5-((4-метоксибензил)окси)-пиридин-4(1Н)-он (Промежуточное соединение 293, 1 г, 2,27 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,Ν,Ν',Ν'-тетраметилурония (1,724 г, 4,53 ммоль) и Ν,Ν-диизопропилэтиламин (1,579 мл, 9,07 ммоль). Через 15 минут реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия, рассолом и смесью 1/1 рассол/вода дважды. Органические фазы сушили над сульфатом магния, фильтровали и концентрировали. После хроматографии на силикагеле (0%-70% ацетон/дихлорметан) получили указанное в заголовке соединение (0,704 г, 47%) в виде светло-оранжевой пены.
МС: 663 ЭРИ+ (C38H38N4O7)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.80 (m, 3Н); 3.18 (m, 2Н); 3.76 (s, 3Н); 3.85 (m, 1Н); 4.06 (m, 2Н); 4.26 (m, 1Н); 4.37 (m, 2Н); 4.68 (s, 2Н); 5.33 (m, 3Н); 5.82 (s, 1Н); 5.95 (m, 1Н); 6.52 (s, 1Н); 6.94 (m, 2Н); 7.24 (m, 2Н); 7.42 (m, 10Н); 7.47 (s, 1Н).
Промежуточное соединение 295: (2S,5R)-2-(((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил-(проп-1-ен-1-ил)фосфония
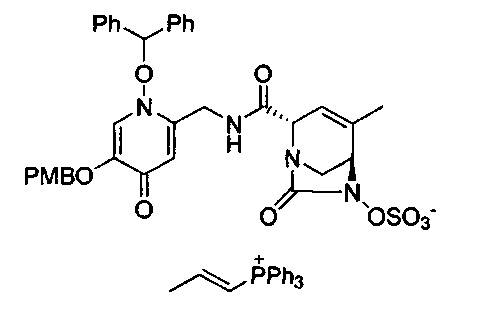
Указанное в заголовке соединение получали из (2S,5R)-6-(аллилокси)-N-((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-2-карбоксамида (Промежуточное соединение 294, 0,704 г, 1,06 ммоль) по методике, описанной для Промежуточного соединения 17. После хроматографии на силикагеле (0%-5% метанола/дихлорметан) получили указанное в заголовке соединение (0,379 г, 35,5%) в виде желтой пены.
МС: 703, 303 ЭРИ+ (C35H33N4O10S, C21H20P)
Промежуточное соединение 296: натриевая соль (2S,5R)-2-(((1-(бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата
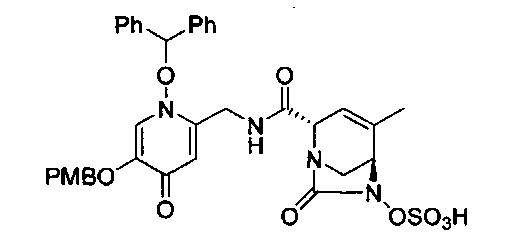
Ионообменную смолу Dowex(R) 50WX8-100 (35 г, 0,38 ммоль) кондиционировали путем перемешивания в течение 2 часов в 2 н. гидроксиде натрия (80 мл, 0,38 ммоль). Смолу затем загружали в колонку и промывали водой до pH 7. Затем ее промывали смесью (1/1) ацетон/вода (примерно 100 мл), затем водой (примерно 100 мл). (2S,2R)-2-(((1-(Бензгидрилокси)-5-((4-метоксибензил)окси)-4-оксо-1,4-дигидропиридин-2-ил)метил)карбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфат (Е)-трифенил(проп-1-ен-1-ил)фосфония (Промежуточное соединение 295, 0,379 г, 0,38 ммоль) переносили в ацетонитрил (примерно 2 мл), наносили на смолу и пропускали через нее воду. Масса целевого продукта наблюдалась в пробирках 1-2 (смешанные фракции), 3-14 (чистые фракции). Две смешанных фракции снова прогоняли через колонку. Пробирки 22-23 (смешанные фракции) и пробирки 24-31 (чистые фракции) лиофилизировали. Целевой продукт был получен в виде не совсем белого твердого вещества (113,4 мг, 43%).
МС: 703 ЭРИ+ (C35H34N4O10S)
1Н ЯМР (300 МГц, DMSO-d6) δ: 1.78 (m, 3Н); 3.19 (m, 2Н); 3.36 (s, 2Н); 3.75 (s, 3Н); 4.00 (m, 2Н); 4.23 (m, 1Н); 4.67 (m, 2Н); 5.40 (m, 1Н); 5.83 (s, 1Н); 6.50 (s, 1Н); 6.93 (m, 2Н); 7.24 (m, 2Н); 7.41 (m, 10Н); 7.45 (s, 1Н).
ПРИМЕР 30: БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Минимальные ингибирующие концентрации (MIC) определяли методом микроразведений в бульоне в соответствии с инструкциями Института клинических и лабораторных стандартов (CLSI) (Clinical Laboratory Standards Institute: Methods for Dilution Antimicrobial Susceptability Tests for Bacteria That Grow Aerobically (8th Ed. (2009)) M07-A8). В краткой форме, суспензии микроорганизмов доводили до стандарта мутности по МакФарланду 0,5 и далее разводили с получением конечного инокулята от 3×105 до 7×105 колониеобразующих единиц (CFU)/Mn. Бактериальные инокуляты готовили в стерильном, уравновешенном по содержанию катионов бульоне Мюллера-Хинтона (Mueller-Hinton) (Beckton Dickinson) либо для Escherichia coli, Klebsiella pneumoniae, либо для Pseudomonas aeruginosa. Бактериальные инокуляты Haemophilus influenzae готовили в стерильном, уравновешенном по содержанию катионов бульоне Мюллера-Хинтона (Beckton Dickinson), содержащем 0,5% дрожжевого экстракта (Beckton Dickinson) плюс 15 мкг/мл бычьего гематина (Sigma) и 15 мкг/мл β-никотинамид-аденин-динуклеотида (Sigma). Бактериальные инокуляты готовили в стерильном, уравновешенном по содержанию катионов бульоне Мюллера-Хинтона (Beckton Dickinson) для Staphylococcus aureus или в стерильном, уравновешенном по содержанию катионов бульоне Мюллера-Хинтона, содержащем 2,5% подвергнутой лизису лошадиной крови (Hema Resource & Supply Inc.) для Streptococcus pneumonia и Streptococcus pyogenes. Инокулят в объеме 100 мкл добавляли в лунки (используя робот Tecan EVO), содержащие 2 мкл DMSO, содержащего 2 кратные последовательные разведения лекарственного средства. Все планшеты с инокулированными микроразведениями инкубировали в атмосфере окружающего воздуха при 35°С в течение 18-24 часов. После инкубирования самую низкую концентрацию лекарственного средства, которая предотвращала видимый рост, который считывали при OD600 нм, регистрировали как MIC. Осуществление анализа контролировали, используя лабораторные красители для контроля качества и коммерчески доступные контрольные соединения с определенными диапазонами MIC, в соответствии с инструкциями CLSI.
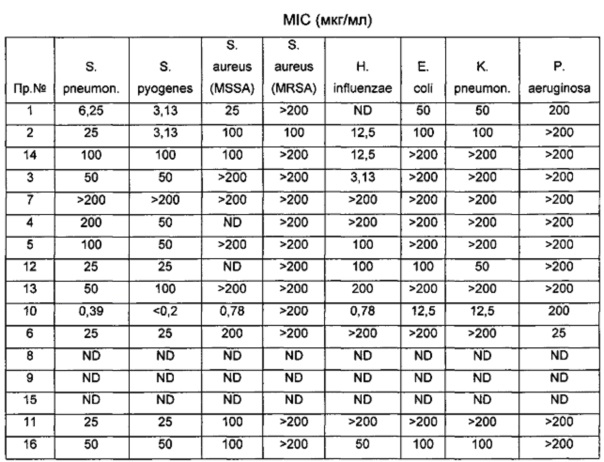
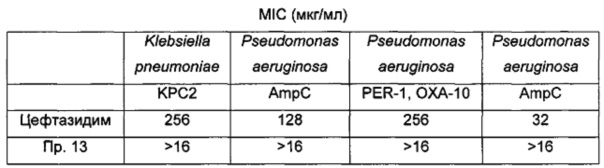
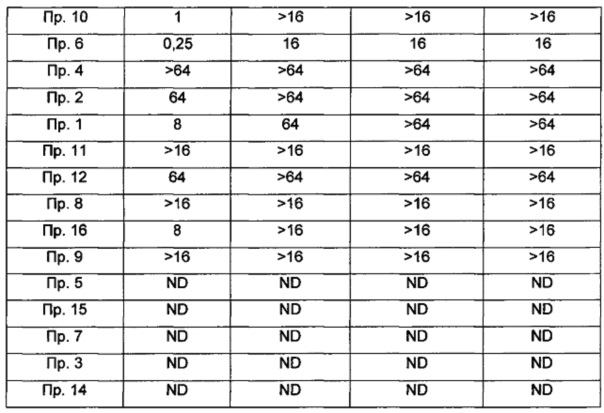
Пример 31
Оценивали синергию с β-лактамами против нескольких микроорганизмов, продуцирующих различные β-лактамазы, относящиеся к разным классам (Bush & Jacoby, Updated Functional Classification of β-Lactamases, Antimicrobial Agents and Chemotherapy, 54:969-76 (2010)). В анализах ингибирующую рост активность (MIC) измеряли с использованием каждых двукратных разведений β-лактама-партнера при фиксированной концентрации соединений формулы (I). Соединения готовили в DMSO, и определения MIC с использованием комбинаций соединение формулы (I)/β-лактам выполняли согласно инструкциям CLSI (Clinical Laboratory Standards Institute: Methods for Dilution Antimicrobial Susceptability Tests for Bacteria That Grow Aerobically (8th Ed. (2009)) M07-A8). Синергию определяли как четырехкратное или выше снижение MIC β-лактама в присутствии соединения формулы (I) по сравнению с одним β-лактамом.
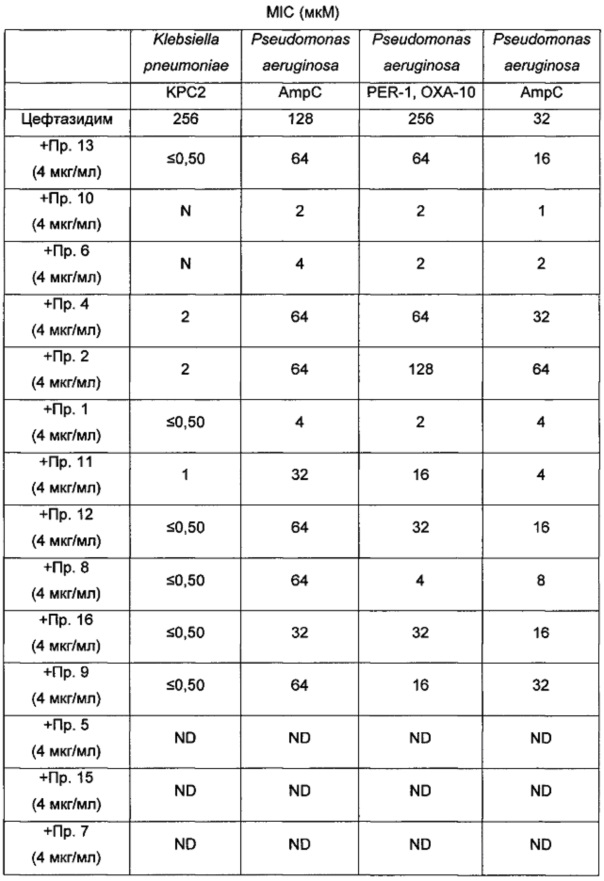
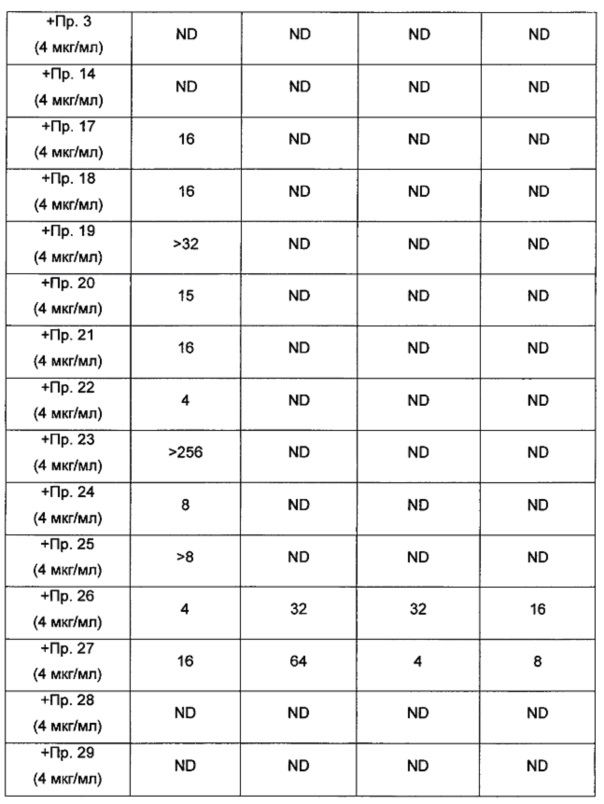
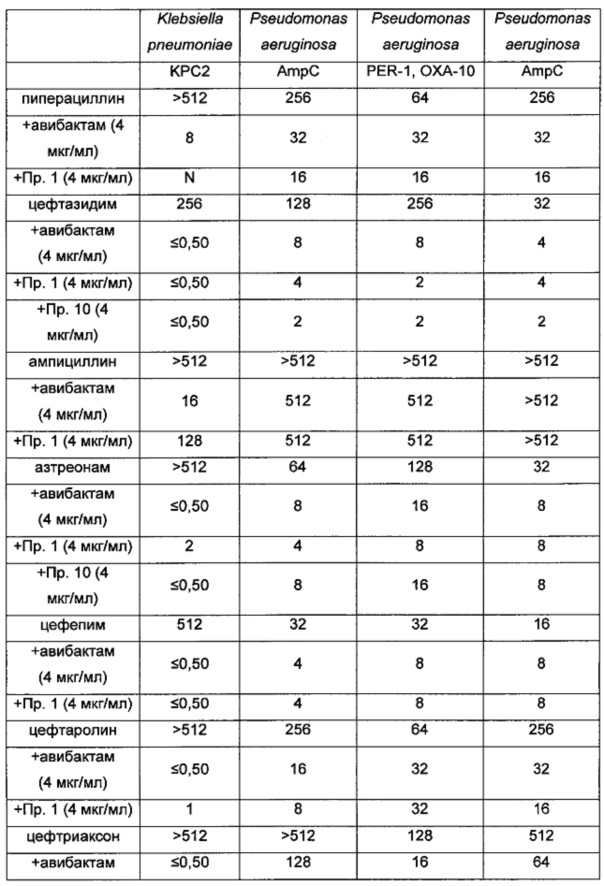
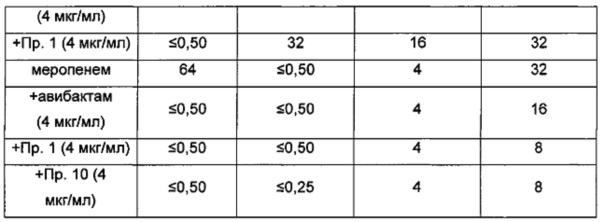
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ИЛИ ПРЕДОТВРАЩЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2741496C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| ОПТИЧЕСКИ АКТИВНОЕ ПРОИЗВОДНОЕ ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2591701C2 |
| СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА | 2012 |
|
RU2622391C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ПРОФИЛАКТИКЕ ИЛИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2783160C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (Ia), или к его стереоизомеру, или к его фармацевтически приемлемой соли, где R1 представляет собой -CONR'R'' или -CN; R2 и R3 независимо выбраны из Н, C1-С6алкила, С3-С6циклоалкила; где алкил в определении R2 или R3 независимо и возможно замещен -ОН, С1-С3алкокси, -NR'R'', -C(O)NR'R'' или -NR'C(O)R''; и каждый из R' и R'' независимо выбран из водорода, С1-С6алкила, 6-членного насыщенного гетероциклила, содержащего один гетероатом N; где каждый алкил и гетероциклил возможно и независимо замещен -С(О)(С1-С6алкокси) или -NH2; при условии, что R2 и R3 оба не являются водородом. Также изобретение относится к конкретным соединениям, фармацевтической композиции на основе соединения формулы (Ia), его применению и способу лечения бактериальной инфекции, основанному на использовании соединения формулы (Ia). Технический результат: получены новые производные диазабициклооктена, полезные при лечении бактериальной инфекции. 6 н. и 9 з.п. ф-лы, 31 пр.
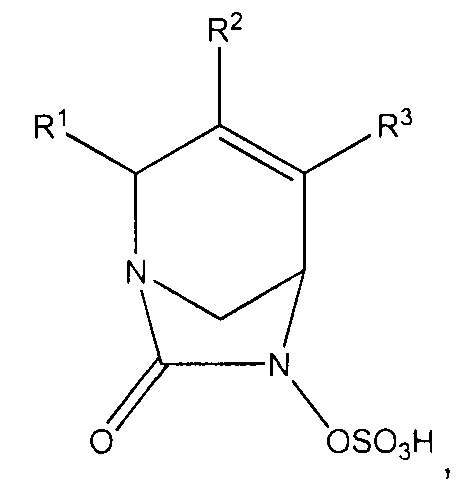
1. Соединение формулы (Ia):
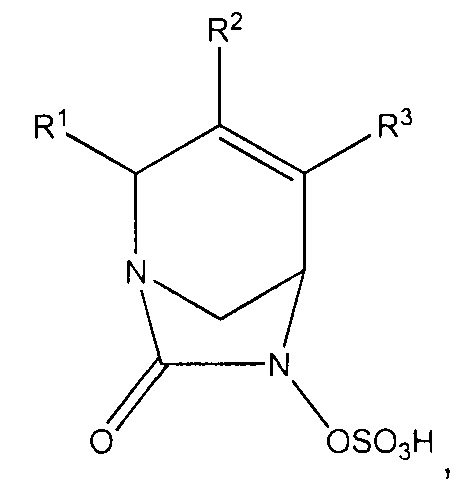
включая его стереоизомер, или его фармацевтически приемлемая соль, где:
R1 представляет собой -CONR'R'' или -CN;
R2 и R3 независимо выбраны из Н, C1-С6алкила, С3-С6циклоалкила; где алкил в определении R2 или R3 независимо и возможно замещен -ОН, С1-С3алкокси, -NR'R'', -C(O)NR'R'' или -NR'C(O)R''; и
каждый из R' и R'' независимо выбран из водорода, С1-С6алкила, 6-членного насыщенного гетероциклила, содержащего один гетероатом N; где каждый алкил и гетероциклил возможно и независимо замещен -С(О)(С1-С6алкокси) или -NH2;
при условии, что R2 и R3 оба не являются водородом.
2. Соединение по п.1 формулы (V):
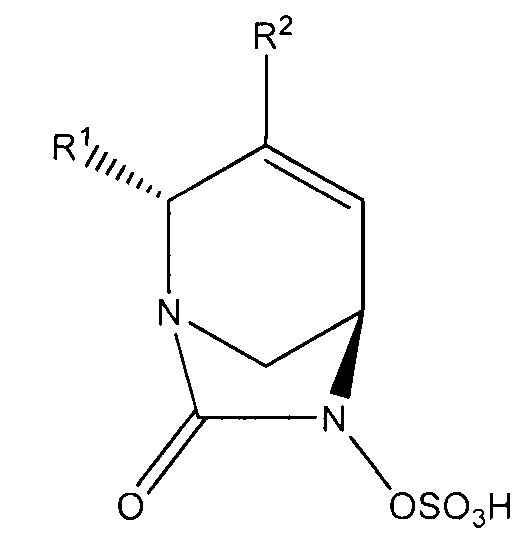
или его фармацевтически приемлемая соль.
3. Соединение по п.2 или его фармацевтически приемлемая соль, где R2 представляет собой метил, этил, изопропил или циклопропил, где алкил возможно и независимо замещен -ОН или С1-С3алкокси.
4. Соединение по п.3 или его фармацевтически приемлемая соль, где R2 представляет собой метил.
5. Соединение по п.1 формулы (IV):
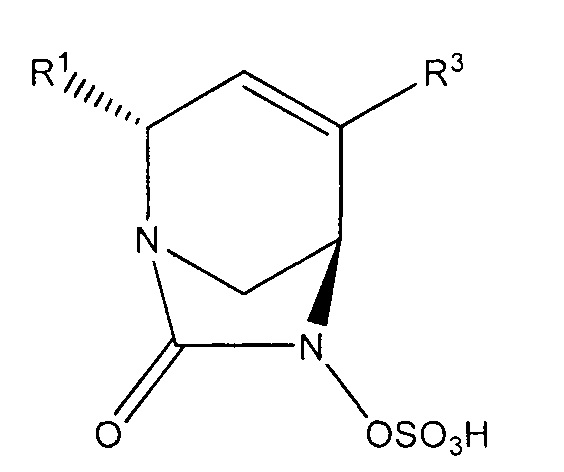
или его фармацевтически приемлемая соль.
6. Соединение по п.5 или его фармацевтически приемлемая соль, где R3 представляет собой метил или -СН2ОСН3.
7. Соединение по п. 1 или его фармацевтически приемлемая соль, где
R1 представляет собой -CONH2,  или
или  и
и
 обозначает точку присоединения к мостиковому бициклическому ядру.
обозначает точку присоединения к мостиковому бициклическому ядру.
8. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой -CONH2.
9. Соединение по п.1 или его фармацевтически приемлемая соль, где:
R1 представляет собой -CONH2 или 
R2 представляет собой -Н или -СН3; и
R3 представляет собой -Н или -СН3;
при условии, что R2 и R3 оба не представляют собой Н.
10. Соединение:
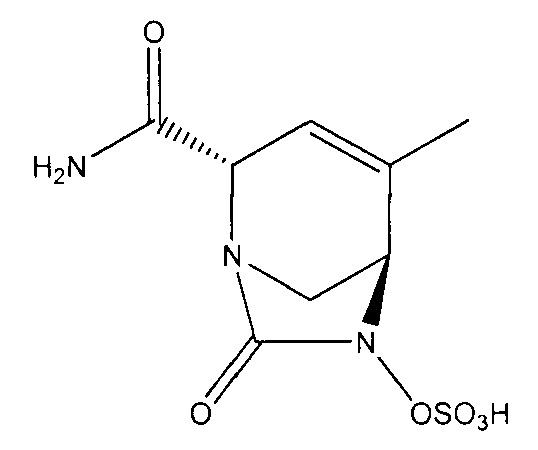
или его фармацевтически приемлемая соль.
11. Соединение:
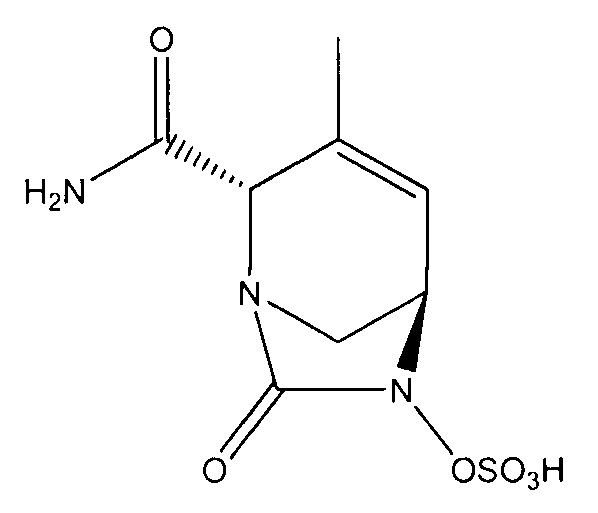
или его фармацевтически приемлемая соль.
12. Соединение по п.1, выбранное из группы, состоящей из
(2S,5R)-2-карбамоил-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-циано-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-4-метил-7-оксо-2-(пиперидиний-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-2-карбамоил-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-циано-4-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-(2-аминоэтилкарбамоил)-4-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-карбамоил-4-(метоксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-карбамоил-3-метил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-карбамоил-3-изопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-(1-(трет-бутоксикарбонил)пиперидин-4-илкарбамоил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-3-метил-7-оксо-2-(пиперидин-1-ий-4-илкарбамоил)-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-2-карбамоил-3-(гидроксиметил)-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-4-(2-амино-2-оксоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-карбамоил-3,4-диметил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-2-карбамоил-3-этил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-4-(2-аминоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-гидросульфата;
(2S,5R)-2-карбамоил-3-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
(2S,5R)-4-(2-ацетамидоэтил)-2-карбамоил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия и
(2S,5R)-2-карбамоил-4-циклопропил-7-оксо-1,6-диазабицикло[3.2.1]окт-3-ен-6-ил-сульфата натрия;
или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция, содержащая соединение по любому из пп.1-12 или его фармацевтически приемлемую соль в эффективном количестве и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент, для использования в качестве лекарственного средства для лечения бактериальной инфекции.
14. Применение соединения по любому из пп.1-12 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения бактериальной инфекции.
15. Способ лечения бактериальной инфекции у теплокровного животного, такого как человек, включающий введение указанному животному эффективного количества соединения по любому из пп.1-12 или его фармацевтически приемлемой соли.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| Устройство для подачи пара в тепловые аккумуляторы типа аккумуляторов Рутса | 1926 |
|
SU4920A1 |
| О.Н.Зефирова и др.: "Об истории возникновения и развития концепции биоизостеризма", Вестник московского университета, серия 2, Химия, т.43, 4, стр.251-256, 2002 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ДИАЗАБИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2124014C1 |

