Область техники
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается терапии ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же ряда других заболеваний посредством применения соединения, являющегося ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB.
Уровень техники
Функциональные расстройств желудочно-кишечного тракта включают группу гетерогенных клинических состояний, которые проявляются симптомами со стороны среднего и нижнего отделов желудочно-кишечного тракта. Синдром раздраженного кишечника (СРК), колики, вздутие, запор и диарея являются наиболее часто встречающимися расстройствами желудочно-кишечного тракта. Согласно данным Всемирной Гастроэнтерологической Организации, распространенность СРК в Европе и Северной Америке оценивается в 10-15%. Во всем мире этим заболеванием страдает около 11.2% населения (Nat Rev Dis Primers, 2016, 2:16014). В общей структуре гастроэнтерологической патологии СРК занимает в США первое место, на его долю приходится 28% всех случаев обращения к гастроэнтерологам. Другим крайне распространенным расстройством желудочно-кишечного тракта является диарея. Золотым стандартом лечения диареи в настоящее время является лоперамид, представляющий собой агонист мю-опиоидных рецепторов периферического действия. Однако в ряде случаев, в том числе при диарее, вызванной приемом химиотерапевтических препаратов, применение лоперамида оказывается неэффективным. Детские колики, проявляющиеся у младенцев в возрасте от 6 недель до 6 месяцев, затрагивают 10-30% детей, и сопровождаются беспричинным сильным плачем, продолжающимся более 3 часов в день (Zhonghua Er Ke Za Zhi. 2017 Apr 2;55(4):314-317). Причина данного явления в настоящее время неизвестна, поэтому эффективного и безопасного способа купирования приступов колик до сих пор не разработано. В 1980 годах были предприняты попытки использования холиноблокаторов, однако они были прекращены из-за высокой опасности возникновения побочных эффектов. Единственный препарат, одобренный для применения при коликах, симетикон, по результатам недавно проведенного мета-исследования оказался неэффективен. Таким образом, образом, можно утверждать, что существует в создании и внедрении в клинику новых эффективных лекарственных средств для терапии расстройств желудочно-кишечного тракта.
Тахикининовые (нейрокининовые) рецепторы являются перспективной группой терапевтических мишеней инновационных лекарственных средств для терапии заболеваний желудочно-кишечного тракта. Существует три типа тахикининовых рецепторов: NK1, NK2 и NK3. Эти рецепторы широко распространены как в центральной нервной системе, так и в периферических тканях. В желудочно-кишечном тракте (ЖКТ) они экспрессируются в нейронах и эффекторных клетках и оказывают влияние на моторику кишечника, секреторную и иммунную активности, висцеральную чувствительность и ноцицепцию (Holzer P. Tachykinins. In Handbook of Biologically Active Peptides (Second Edition); Kastin A. J., Ed.; Elsevier, 2013; pp. 1330-1337). В связи с этим тахикининовые рецепторы зарекомендовали себя как потенциальные мишени для лечения функциональных заболеваний ЖКТ, одной из разновидностей которых является синдром раздраженного кишечника (СРК). Наиболее изученными и широко исследуемыми рецепторами в связи с лечением функциональных заболеваний кишечника являются NK2-рецепторы (Br J Pharmacol, 2004, 141, 1249-63). Тахикининовые рецепторы NK2 в желудочно-кишечном тракте экспрессируются в клетках мышечного слоя, мышечной пластинки слизистой оболочки, в энтероцитах и иммунных клетках, а также в возбуждающих и тормозящих нейронах подслизистой оболочки и мышечного сплетения (J Comp Neurol, 2007, 503, 381-91). Экспрессия рецепторов NK2 увеличивается в воспалительных клетках собственной пластинки слизистой оболочки и активированных эозинофилах вокруг крипт слизистой оболочки (Naunyn Schmiedebergs Arch Pharmacol, 2003, 367, 104-8). Терапевтические показания к антагонистам NK1 и NK2-рецепторов включают синдром раздраженного кишечника (Neurogastroenterol Motil. 2015 Oct; 27(10):1354-70, Br J Pharmacol. 2004 Apr;141(8):1249-63), язвенный колит (Inflamm Res. 2014 May;63(5):399-409), болезнь Крона (Neurogastroenterol Motil. 2011 May; 23(5):475-83, e179-80), диарею (Br J Pharmacol. 1997 Jun;121(3):375-80), детские колики (Neuropeptides. 2010 Jun;44(3):269-72) послеоперационную непроходимость кишечника, тошноту и рвоту (Am J Health Syst Pharm. 2017 Apr 10), кашель (Pulm Pharmacol Ther. 2004;17(1):11-8), астму (Allergy. 2013 Jan;68(1):48-54, BMC Pulm Med. 2011 Aug 2;11:41), ревматоидный артрит (Neuropeptides. 1998 Jun;32(3):215-23) и псориаз (Pathobiology. 1999;67(1):51-4).
Другим возможным направлением терапии расстройств желудочно-кишечного тракта является применение агонистов каннабиноидных рецепторов CB1R. Агонисты CB1R рецепторов замедляют ионный транспорт в слизистой кишечника, снижая аккумуляцию воды. Вероятно, этот эффект опосредован скорее взаимодействием с нервными проводниками, нежели прямым действием на эпителий кишечника. Способность агонистов СВ1-рецепторов ослаблять перистальтику кишечника, понижать секрецию и чувствительность нервных окончаний может быть использована для терапии пациентов с синдромом раздраженного кишечника (J Pharmacol Exp Ther. 2014 Jul;350(1):69-78) и диареи (Drug News Perspect. 2009 Sep;22(7):383-92), включая диарею индуцированную химиотерапией (Curr Gastroenterol Rep. 2015 Feb;17(2):429). Кроме того, агонисты каннабиноидных рецепторов CB1R могут использоваться для лечения нейродегенеративных заболеваний (в том числе болезни Паркинсона, болезнь Альцгеймера, болезнь Хантигтона (Handb Exp Pharmacol. 2015;231:233-59), рассеянного склероза (J Med Chem. 2016 Jul 28;59(14):6753-71) и энцефаломиелита (Mult Scler Relat Disord. 2015 Nov;4(6):505-11).
Антагонисты 5-гидрокситриптаминовых рецепторов 5-HT3 применяются для терапии боли ассоциированной с синдромом раздраженного кишечника и другими расстройствами желудочно-кишечного тракта (Aliment Pharmacol Ther 1997; 11: 3-15). В то же время, антагонисты 5-гидрокситриптаминовых рецепторов 5-HT2B по-видимому могут оказаться полезными не только для уменьшения боли, ассоциированной с развитием паталогии, но так же иметь прямое действие на патогенез заболевания (Mini Rev Med Chem. 2004 Mar;4(3):325-30). Кроме того, антагонисты 5-гидрокситриптаминовых рецепторов 5-HT2B могут применяться для лечения мигрени (Expert Opin Investig Drugs. 2017 Mar; 26(3):269-277).
Сравнительно новым и относительно малоизученным направлением, является применение агонистов прокинетициновых рецепторов первого и второго типа для терапии расстройств желудочно-кишечного тракта и в частности синдрома раздраженного кишечника (Neurogastroenterol Motil. 2012 Jan;24(1):65-75). Для терапии болезни Крона, язвенного колита (Br J Pharmacol. 2013 Jan;168(2):389-402) и ряда других заболеваний (ишемии (Stroke. 2009 Jan;40(1):285-93), аллергической астмы (Pharmacol Res. 2016 Feb;104:132-9) и гипергликемии (J Cardiovasc Pharmacol. 2012 Jul;60(1):61-9)) могут применяться агонисты брадикининового рецептора BRDKB1. Необходимо отметить, что функциональный ответ, вызванный активацией ряда рецепторов, связан с сигнальным путем транскрипционого фактора NF-kB. Ингибиторы сигнального пути NF-kB могут применяться для лечения псориаза (Int Immunopharmacol. 2015 Feb;24(2):392-9), рассеянного склероза (J Neuroinflammation. 2015 Sep 30;12:184), язвенного колита (Mol Cell Biochem. 2016 Aug;419(1-2):65-74), болезни Крона (J Steroid Biochem Mol Biol. 2007 Jan;103(1):51-60) и ряда других заболеваний.
Таким образом, на сегодняшний день известно множество терапевтических подходов к терапии расстройств желудочно-кишечного тракта. Однако до сих пор нет ни одного зарегистрированного лекарственного препарата действующего по нескольким из перечисленных механизмов. Поэтому сохраняется потребность в создании и внедрении в клинику новых эффективных лекарственных средств для терапии расстройств желудочно-кишечного тракта и других заболеваний.
Данное изобретение касается применения соединения бензил (2S)-2-[2-(4-гидроксифенил) ацетамидо]-3-фенила пропаноата или его аддукта, гидрата, сольвата, являющегося ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB, в терапии ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же ряда других заболеваний.
Раскрытие изобретения
Задачей настоящего изобретения является разработка нового лекарственного средства, являющегося ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB и эффективного для лечения ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты.
Техническим результатом данного изобретения является разработка и получение эффективного ингибитора катепсина S, агониста каннабиноидных рецепторов первого типа, антагониста тахикининовых рецепторов первого и второго типа, антагониста прокинетициновых рецепторов первого и второго типа, антагониста брадикининового рецептора первого типа, антагониста меланокортиновых рецепторов MC4R, и ингибитора сигнального пути NB-kB характеризующегося высокой активностью и фармакокинетическими характеристиками, позволяющими использовать данное соединение при топическом применении, для терапии ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же прочих заболеваний связанных с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и сигнального пути NB-kB.
Указанный технический результат достигается путем применения соединения бензил (2S)-2-[2-(4-гидроксифенил) ацетамидо]-3-фенила пропаноата (Соединение 1)
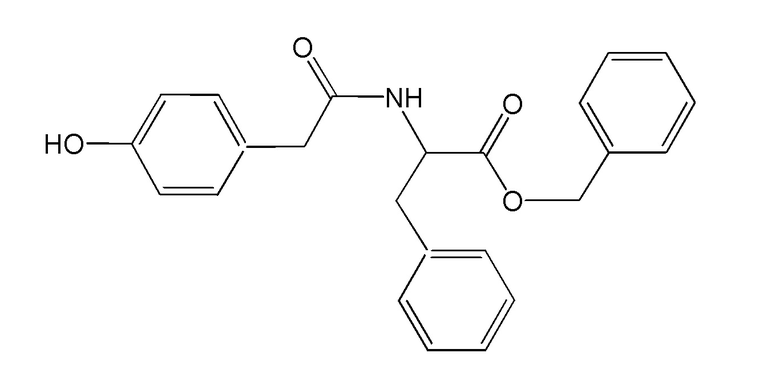
или его аддукта, гидрата, сольвата в качестве ингибитора катепсина S, агониста каннабиноидных рецепторов первого типа, антагониста тахикининовых рецепторов первого и второго типа, антагониста прокинетициновых рецепторов первого и второго типа, антагониста брадикининового рецептора первого типа, антагониста меланокортиновых рецепторов MC4R, антагониста серотониновых рецепторов 5-HT2B и ингибитора сигнального пути NB-kB.
Соединение бензил (2S)-2- [2-(4-гидроксифенил) ацетамидо]-3-фенила пропаноат раскрыто и описано в международной заявке WO 2006101422.
Также настоящее изобретение относится к применению соединения бензил (2S)-2-[2-(4-гидроксифенил) ацетамидо]-3-фенила пропаноата или его аддукта, гидрата, сольвата для получения фармацевтической композиции для предупреждения и/или лечения ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же прочих расстройств связанных с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, серотониновых рецепторов 5-HT2B и сигнального пути NB-kB.
Кроме того, изобретение относится к фармацевтической композиции для предупреждения и/или лечения ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же прочих расстройств связанных с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, серотониновых рецепторов 5-HT2B и сигнального пути NB-kB, содержащей эффективное количество Соединения 1 по изобретению и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах воплощениях изобретения вспомогательное вещество представляет собой фармацевтически приемлемый носитель и/или эксципиент.
Изобретение также включает способ предупреждения и/или лечения расстройства, связанного с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, серотониновых рецепторов 5-HT2B и сигнального пути NB-kB в организме, включающий введение в указанный организм фармацевтической композиции по изобретению. В некоторых неограничивающих вариантах воплощения изобретения заболевание представляет собой ожирение, псориаз, болезнь Крона, колит, синдром раздраженного кишечника, диарею, тошноту и рвоту. В частных случаях воплощения изобретения организм представляет собой организм человека или животного.
Изобретение относится к способу предупреждения и/или лечения расстройства, связанного с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, серотониновых рецепторов 5-HT2B и сигнального пути NB-kB, у субъекта, нуждающегося в таком лечении, включающему введение терапевтически эффективного количества Соединения 1 указанному субъекту.
Также, изобретение относится к способу предупреждения и/или лечения ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества Соединения 1 указанному субъекту.
Изобретение относится также к применению Соединения 1 для получения лекарственного средства.
Также, настоящее изобретение относится к комбинации, содержащей Соединение 1 в сочетании с одним или несколькими другими дополнительными терапевтическими агентами.
Подробное раскрытие изобретения
Получение Соединения 1, являющегося предметом настоящего изобретения, описано в международной заявке WO 2006101422. В указанном заявке раскрываются фенилсодержащие N-ацильные производные биогенных аминов и аминокислот, обладающие способностью к ингибированию циклооксигеназ. И, в свою очередь, обладающие анальгетическими и противовоспалительными свойствами, без побочных эффектов, в частности ульцерогенности и проспазматического действия, способностью потенцировать действие других анальгетиков, обладающих, кроме того, антигипоксическим, антидепрессантным и противопаркинсоническим действием.
В ходе проведения масштабного скрининга фармакологических мишеней Соединения 1 неожиданно оказалось, что Соединение 1 является ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB.
В соответствии со спектром экспериментально определенных терапевтических мишеней Соединения 1 были определены показания, в которых применение Соединения 1 представлялось наиболее перспективным. Однако в ходе исследования фармакокиненики Соединения 1 неожиданно оказалось, что Соединение 1 обладает крайне низкой стабильностью в плазме крови животных и человека. Это неожиданное свойство Соединения 1 позволяет данному соединению оказывать исключительно местное действие. Таким образом, применение Соединения 1 будет безопасно, т.к. не будут возникать ни какие системные побочные эффекты связанные с мультитаргентым действием данного лекарственного препарата.
Таким образом, Соединение 1 является новым ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB, который может применяться для терапии ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же других заболеваний, связанных с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и сигнального пути NB-kB. Фармакокинетические параметры, позволяющие использовать данное соединение при топическом применении, обеспечивают высокую безопасности и отсутствие системных эффектов при применении Соединения 1.
Термины и определения
Термин «Соединение 1» относится к бензил (2S)-2-[2-(4-гидроксифенил) ацетамидо]-3-фенила пропаноату, также представленному структурной формулой:
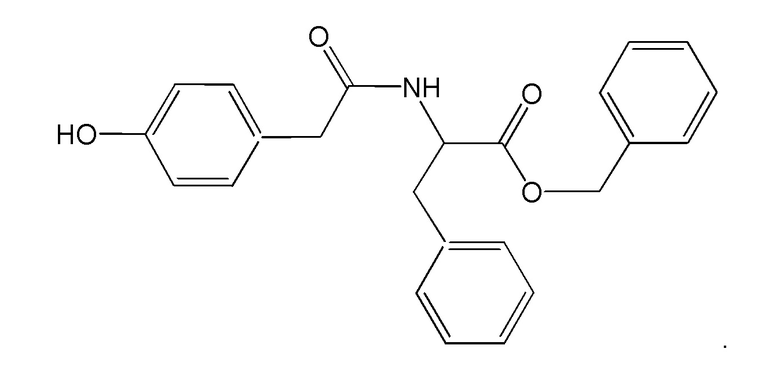
Термин «С», когда он используется со ссылкой на температуру, означает стоградусную шкалу или температурную шкалу Цельсия.
Термин «IC50» означает концентрацию тестируемого соединения, при которой достигается полумаксимальное ингибирование фермента или агонистическое или анагонистическое действие.
Термин «фармацевтически приемлемые аддукты» или «аддукты» включает продукт прямого присоединения молекул друг к другу, которые получены с помощью относительно нетоксичных соединений. Примерами фармацевтически приемлемых нетоксичных аддуктов могут служить аддукты образованные нетоксичными нитропроизводными или мочевиной. К другим фармацевтически приемлемым аддуктам относятся аддукты неионных тензидов, циклодекстринов и другие, а так же комплексы с переносом заряда (π-аддукты). Необходимо отметить, что термин «аддукты» включает так же аддукты нестехиометрического состава.
Термин «сольват» используется для описания молекулярного комплекса, содержащего соединение по изобретению и одну или более молекул фармацевтически приемлемого растворителя, например этанола. Термин «гидрат» используется, когда указанным растворителем является вода.
Термин «вспомогательное вещество» означает любое фармацевтически приемлемое вещество неорганического или органического происхождения, входящее в состав лекарственного препарата или используемое в процессе производства, изготовления лекарственного препарата для придания ему необходимых физико-химических свойств.
Термин «AUC» (area under the curve) означает фармакокинетический параметр, характеризующий суммарную концентрацию лекарственного препарата в плазме крови в течение всего времени наблюдения. Математически определяется как интеграл от 0 до ∞ функции концентрации препарата (фармакокинетической кривой) в плазме крови от времени и равен площади фигуры, ограниченной фармакокинетической кривой и осями координат.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) снижение, б) блокирование (приостановку) течения заболевания, в) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания, г) реверсирование заболевания или состояния, к которому данный термин применяется, или одного или более симптомов данного заболевания или состояния.
Термин «профилактика», «предотвращение» охватывает устранение факторов риска, а также профилактическое лечение субклинических стадий заболевания у млекопитающих, предпочтительно у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. Пациенты для профилактической терапии отбираются на основе факторов, которые, на основании известных данных, влекут увеличение риска возникновения клинических стадий заболевания по сравнению с общим населением. К профилактической терапии относится а) первичная профилактика и б) вторичная профилактика. Первичная профилактика определяется как профилактическое лечение у пациентов, клиническая стадия заболевания у которых еще не наступила. Вторичная профилактика - это предотвращение повторного наступления того же или близкого клинического состояния заболевания.
Применение Соединения 1, являющееся предметом данного изобретения, может использоваться для лечения ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, а так же прочих заболеваний связанных с активностью катепсина S, каннабиноидных рецепторов первого типа, тахикининовых рецепторов первого и второго типа, прокинетициновых рецепторов первого и второго типа, брадикининового рецептора первого типа, меланокортинового рецепторов MC4R, серотониновых рецепторов 5-HT2B и сигнального пути NB-kB.
Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества Соединения 1 по изобретению. Под терапевтически эффективным количеством подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Соединение по изобретению или фармацевтическая композиция, содержащая соединение, может быть введено в организм пациента в любом количестве и любым путем введения, эффективным для лечения или профилактики заболевания. Предпочтительно, суточная доза действующего вещества составляет до 5 г на пациента в сутки, наиболее предпочтительно, суточная доза составляет 5-500 мг/сутки. Предпочтительно, Соединение 1 вводят перорально или путем местного нанесения.
После смешения Соединения 1 с подходящим фармацевтически приемлемом носителем в желаемой дозировке, фармацевтические композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, топически, ингаляционно и т.п.
Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Кроме того, Соединение 1 может вводиться в организм пациента ежедневно в течение определенного периода дней, например, 2-10 дней, а затем следует период без приема вещества, например, 1-30 дней.
В случае, когда Соединение 1 используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции (Лекарственные средства)
Изобретение также относится к фармацевтической композиции, которая содержит Соединение 1 по изобретению или его аддукт, гидрат, сольват, и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не влияют на фармакологическую активность этого соединения, и являются нетоксичными при введении в дозах, достаточных для доставки терапевтического количества соединения. Фармацевтические композиции, заявляемые в данном изобретении, содержат Соединение 1 данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, скользящие материалы и т.д., подходящие для конкретной формы дозирования.
Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахаридами, а также их производными; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы.
Также, в составе композиции могут быть другие нетоксичные совместимые скользящие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, топически, ингаляционно, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом, методами микрокапсулирования, методами приготовления наноформ препарата, или другими методами, известными в фармацевтике.
Фармацевтические композиции настоящего изобретения могут быть получены путем смешения Соединения 1 с фармацевтически приемлемым носителем.
Так, при получении композиции, например, в форме таблетки, Соединение 1 смешивают с одним или несколькими фармацевтическими эксципиентами, такими как, желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки могут быть покрыты сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая Соединение 1с фармацевтически приемлемым носителем (не понятно какими другими веществами). и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например, пропиленгликоль или бутиленгликоль.
Примеры фармацевтических композиций
Вещество, описанное в данном изобретении, может быть использовано для профилактики и/или лечения болезней человека, или животных в виде следующих составов (под «Веществом» понимается активный ингредиент - Соединение 1):
Таблетка I мг/таблетка
Вещество 3,0
Микрокристаллическая целлюлоза 64,0
Карбоксиметилкрахмал натрия 2,3
Магния стеарат 0,7
Таблетка II мг/таблетка
Вещество 30,0
Микрокристаллическая целлюлоза 640,0
Карбоксиметилкрахмал натрия 23,0
Магния стеарат 7,0
Таблетка III мг/таблетка
Вещество 3,0
Микрокристаллическая целлюлоза 64,0
Карбоксиметилкрахмал натрия 2,3
Магния стеарат 0,7
Оболочка кишечнорастворимая Acryl-EZE® MP 2,0
Таблетка IV мг/таблетка
Вещество 30,0
Микрокристаллическая целлюлоза 640,0
Карбоксиметилкрахмал натрия 23,0
Магния стеарат 7,0
Оболочка кишечнорастворимая Acryl-EZE® MP 20,0
Таблетка V мг/таблетка
Вещество 200,0
Лактоза Ph. Eur 182,75
Кроскармеллоза натрия 12,0
Кукурузный крахмал (5% w/v паста) 2,25
Стеарат магния 3,0
Капсула мг/капсула
Вещество 10,0
Лактоза Ph. Eur 488,5
Магнезия 1,5
Состав для инъекций I мг/100мл
Вещество 310,0
Полиэтиленгликоль-400 44,4
Динатрия эдетат 5,0
Вода для инъекций до 100 мл
Мазь I г/100г
Вещество 0,103
Токоферол 0,100
Ланетт SX 10,900
Масло касторовое 11,000
Полиэтиленоксид 1500 31,906
Полисорбат 80 4,491
1,2-Пропандиол 41,500
Мазь II г/100г
Вещество 0,103
Бутилгидрокситолуол (ионол) 0,100
Ланетт SX 10,900
Масло касторовое 11,000
Полиэтиленоксид 1500 31,906
Полисорбат 80 4,491
1,2-Пропандиол 41,500
Данные составы могут быть приготовлены в соответствии со стандартными фармацевтическими методиками.
Применение Соединения 1 в комбинированной терапии
Несмотря на то, что Соединение 1 по данному изобретению может вводиться в качестве индивидуального активного фармацевтического средства, его также можно использовать в сочетании с одним или несколькими другими дополнительными терапевтическими агентами, в частности, другой агент может представлять собой антибиотик, НПВС или другое противовоспалительное средство, антитело, обезболивающее средство, цитостатическое средство, и т.д. При совместном приеме внутрь терапевтические агенты могут представлять собой разные лекарственные формы, которые вводятся одновременно или последовательно в разное время, либо терапевтические агенты могут быть объединены в одну лекарственную форму.
Фраза «комбинированная терапия» в отношении Соединения 1 данного изобретения в сочетании с другими терапевтическими агентами, означает одновременный или последовательный прием всех агентов, который так или иначе обеспечит благоприятное воздействие сочетания лекарственных средств. Совместное введение подразумевает, в частности, совместную доставку, например, в одной таблетке, капсуле, инъекции или в другой форме, имеющий фиксированное соотношение активных веществ, также как и одновременную доставку в нескольких, отдельных лекарственных формах для каждого соединения соответственно.
Таким образом, введение соединений данного изобретения может быть осуществлено в сочетании с дополнительными методами лечения, известными специалистам в области профилактики и лечения соответствующих заболеваний, включающими применение антибактериальных, цитостатических и цитотоксических препаратов, препаратов для подавления симптомов или побочных эффектов одного из лекарств.
Если лекарственная форма представляет собой единичную дозированную форму, такая комбинация использует соединения данного изобретения в приемлемом дозовом диапазоне. Соединение 1 по данному изобретению также может быть введено в организм пациента последовательно с другими агентами, в том случае, когда комбинация этих препаратов невозможна. Изобретение не ограничено последовательностью введения; соединение данного изобретения может быть введено в организм пациента совместно, до или после введения другого препарата.
Примеры
Получение соединения по изобретению
Получение Соединения 1 описано и раскрыто в международной заявке WO 2006101422. В той же заявке описана и раскрыта способность Соединения 1 ингибировать активность циклооксигеназ.
Характеристика биологической активности соединения по изобретению
Биологическая активность Соединения 1, являющегося предметом настоящего изобретения, была изучена в различных in vitro и in vivo экспериментах. В частности, при изучении активности Соединения 1 в различных in vitro и in vivo моделях было показано ингибирующее действие Соединения 1 в модели диареи, индуцированной касторовым маслом у мышей. Данное биологическое действие Соединения 1 не может быть предсказано или объяснено на основе предшествующих знаний о способности Соединения 1 к ингибированию циклооксигеназ.
Исследования биологической активности Соединения 1 in vitro, позволили установить, что Соединение 1 является ингибитором фермента катепсина S, агонистом канабиоидного рецептора первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, и ингибитором сигнального пути NB-kB. По-видимому, активность Соединения 1 в моделях псориаза, а так же в различных моделях расстройств желудочно-кишечного тракта связана с действием на вышеуказанные белки.
Пример 1. Исследование влияния Соединения 1 на ферментативную активность катепсина S
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовался человеческий рекомбинантный катепсин S, экспрессированный в E.coli. Тестируемые соединения преинкубировали 15 минут при 37С с ферментом, активность которого определяли по скорости превращения субстрата Z-Phe-Arg-AMC (6 мкМ) методом флуоресцентной спектроскопии (Protein Sci. 1996 Apr;5(4):789-91).
В результате исследования было установлено, что Соединения 1 является ингибитором катепсина S c IC50=6,7 мкM.
Пример 2. Исследование влияния Соединения 1 на активность тахикининового рецептора первого типа (NK1R)
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки U373, экспрессирующие NK1R, которые после преинкубации с агонистом [Sar9,Met(O2)11]-SP (1 нМ) инкубировали с Соединением 1. Активность рецепторов определяли по внутриклеточной концентрации кальция методом флуоресцентной спектроскопии (Glia. 1992; 6(2):89-95).
В результате исследования было установлено, что Соединение 1 является антагонистом тахикининового рецептора первого типа c IC50=4,1 мкM.
Пример 3. Исследование влияния Соединения 1 на активность тахикининового рецептора второго типа (NK2R)
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки CHO, экспрессирующие NK2R, которые после преинкубации с агонистом [Nleu10]-NKA-(4-10) (10 нМ) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации кальция методом флуоресцентной спектроскопии (Biochem Biophys Res Commun. 1994 May 16;200(3):1512-20).
В результате исследования было установлено, что Соединение 1 является антагонистом тахикининового рецептора второго типа c IC50=8,4 мкM.
Пример 4. Исследование влияния Соединения 1 на активность каннабиноидного рецептора первого типа
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки CHO, экспрессирующие CB1R, которые инкубировали с тестируемым соединением. В качестве контроля использовали соединение CP55940 (30 нМ). Активность рецепторов определяли по внутриклеточной концентрации кальция методом гомогенной время разрешенной флуоресцентной спектроскопии (Mol Pharmacol. 1995 Sep;48(3):443-50).
В результате исследования было установлено, что Соединение 1 является агонистом каннабиноидного рецептора первого типа c IC50=3,3 мкM.
Пример 5. Исследование влияния Соединения 1 на активность брадикининового рецептора BRDKB1
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки CHO, экспрессирующие брадикининовые рецепторы B1, которые после преинкубации с агонистом LysdesArg9-BK (3 nM) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации кальция методом флуоресцентной спектроскопии (Eur J Pharmacol. 2000 Mar 24;392(1-2):1-9).
В результате исследования было установлено, что Соединение 1 является антагонистом брадикининовых рецепторов c IC50=3,7 мкM.
Пример 6. Исследование влияния Соединения 1 на активность прокинетициновых рецепторов первого типа (PK1)
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки HEK-293, экспрессирующие прокинетициновые рецепторы PK1, которые после преинкубации с агонистом PK1 (3 nM) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации кальция методом флуоресцентной спектроскопии (Mol Pharmacol. 2005 Jun;67(6):2070-6).
В результате исследования было установлено, что Соединение 1 является антагонистом прокинетицинового рецептора первого типа (PK1) c IC50=5,7 мкM.
Пример 7. Исследование влияния Соединения 1 на активность прокинетициновых рецепторов второго типа (PK2)
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки HEK-293, экспрессирующие прокинетициновые рецепторы PK2, которые после преинкубации с агонистом PK2 (2 nM) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации кальция методом флуоресцентной спектроскопии (Mol Pharmacol. 2005 Jun;67(6):2070-6).
В результате исследования было установлено, что Соединение 1 является антагонистом прокинетицинового рецептора второго типа (PK2) c IC50=5,4 мкM.
Пример 8. Исследование влияния Соединения 1 на активность меланокортиновых рецепторов MC4R
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки CHO, экспрессирующие меланокортиновый рецептор МС4R, которые после преинкубации с агонистом NDP-alpha-MSH (30 nM) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации циклического аденозинмонофосфата методом гомогенной времяразрешенной флуоресцентной спектроскопии.
В результате исследования было установлено, что Соединение 1 является антагонистом меланокортиновых рецепторов MC4R c IC50=7,6 мкM.
Пример 9. Исследование влияния Соединения 1 на активность серотониновых рецепторов 5-HT2B
Соединение 1 растворяли в ДМСО до концентрации 100 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались клетки CHO, экспрессирующие серотониновые рецепторы 5-HT2B, которые после преинкубации с агонистом серотонином (30 nM) инкубировали с тестируемым соединением. Активность рецепторов определяли по внутриклеточной концентрации фисфатидилинозитола методом гомогенной времяразрешенной флуоресцентной спектроскопии (Br J Pharmacol. 1999 Sep;128(1):13-20).
В результате исследования было установлено, что Соединение 1 является антагонистом серотониновых рецепторов 5-HT2B c IC50=8,9 мкM.
Пример 10. Исследование влияния Соединения 1 на активность сигнального пути NF-kB
Соединение 1 растворяли в ДМСО до концентрации 50 мМ; затем стоковый раствор серийно разбавляли ДМСО. Максимальная стартовая концентрация вещества - 100 μМ. Эффект определяли при 5 концентрациях тестируемых соединений, каждую концентрацию исследовали дважды. В эксперименте использовались человеческие T лимфоциты линии Jurkat, трансфецированные lacZ-опероном, в котором транскрипция β-галактозидазы находилась под контролем NFAT-1 транскрипционного фактора. Тестируемые соединения преинкубировали с клетками. β-галактозидазную активность клеток определяли по скорости превращения субстрата FDG (флуоресцеин-ди-β-D-галактопиранозид) флуоресцентной спектрофотометрией.
В результате исследования было установлено, что Соединение 1 является ингибитором сигнального пути NF-kB c IC50=9,9 мкM.
Пример 11. Исследование активности Соединения 1 на модели имиквимод-индуцированного псориаза на ухе у мышей
Псориаз моделировали у самок мышей линии balb/c путем нанесения крема Алдара (5% имиквимод) на внутреннюю сторону правого уха ежедневно 1 раз в сутки в течение 10 дней (J Immunol. 2009 May 1;182(9):5836-45). Интактным животным наносили вазелин: 20 мг на правое ухо.
Оценку развития патологии проводили на 2, 4, 6, 8 и 10 сутки перед очередным нанесением крема Алдара путем измерения толщины левого и правого уха.
Результаты исследования представлены в таблице 1.
Таблица 1 - Прирост толщины правого (пораженного) уха в определенный день исследования относительно толщины правого (пораженного) уха на 0 день исследования при изучении активности Соединения 1 на модели псориаза уха у мышей, % (M±m, n=10)
4,6
5,6
8,3
плацебо
9,4*
12,9*
16,1*
19,5*
16,0*
2,6
1,0*&
2,0*&
6,0*&
8,7 &
3,2&
5,3 &$
7 &$
4,7 &$
3,1*&$
5,6&
5,3 &$
7,5 &$
4,9 &$
3,0*&$
*- различия статистически значимы относительно интактной группы (p<0.05);
$ - статистически значимо относительно соответствующей группы «Контроль, леченный плацебо», при p<0.05;
& - статистически значимо относительно соответствующей группы «Контроль без плацебо», при p<0.05.
Из таблицы 1 видно, что Соединение 1 снижало прирост толщины правого (пораженного) уха мышей до уровня интактных животных.
Таким образом, Соединение 1 оказывает выраженный эффект в модели псориаза на ухе у мышей, не уступая глюкокортикостероидному препарату Дермовейт (клобетазолу), предназначенному для лечения псориаза.
Пример 12. Исследование активности Соединения 1 на модели имиквимод-индуцированного псориаза на спине у мышей
Псориаз моделировали у самок мышей линии balb/c путем нанесения крема Алдара (5% имиквимод) на предварительно выбритый участок кожи 3×4 см на спине ежедневно 1 раз в сутки в течение 15 дней. (J Immunol. 2009 May 1;182(9):5836-45) Интактным животным наносили вазелин: 120 мг на выбритый участок спины.
Оценку развития патологии проводили на 10, 12, 13 и 15 сутки перед очередным нанесением крема Алдара по показателю: толщина кожной складки на спине. Толщину кожной складки на спине измеряли микрометром Digimatic MK-25 (Mitutoyo, Japan).
Результаты исследования представлены в таблице 2.
Таблица 2 - Прирост толщины кожи спины в определенный день исследования к толщине кожи спины до начала исследования при изучении активности Соединения 1 на модели псориаза на спине, % (M±m, n=10)
Из таблицы 2 видно, что Соединение 1 снижало прирост толщины кожной складки пораженного участка кожи в 2 раза. Препарат сравнения Дермовейт показал токсическое действие, истончив кожу и вызвав тотальную гибель животных на 12 ые сутки исследования.
Таким образом, можно заключить, что на модели псориаза на спине у мышей Соединение 1 оказывает выраженное лечебное действие, снижая прирост толщины кожи пораженного участка. По профилю безопасности Соединение 1 превосходит Дермовейт.
Пример 13. Исследование активности Соединения 1 на модели болезни Крона у мышей
Исследование проводили на мышах-самцах линии balb/c. Голодавшим в течение 24 ч животным вводили 150 мкл/мышь раствора TNBS в 50% этаноле в ректальное отверстие мыши на глубину 4 см с помощью катетера 3.5 F. Далее мышей переворачивали вверх ногами и держали 60 секунд. Здоровому контролю (интактным) вводили 150 мкл 50% раствора этанола. Исследование длилось 7 дней. Развитие патологии оценивали по гибели животных.
Таблица 4 - Гибель животных при проведении исследования активности Соединения 1 на модели болезни Крона
Из данных, приведенных в таблице 4 видно, что введение Соединения 1 позволило более чем в 2 раза снизить гибель животных. По выраженности действия Соединение 1 превосходит стероидный препарат преднизолон, который снижал летальность на 20-40%.
Пример 14. Исследование активности Соединения 1 на модели индометацинового колита у мышей
Исследование выполняли на крысах-самцах линии Wistar, которым для индукции колита подкожно вводили раствор индометацина в дозе 9 мг/кг 2 дня подряд. Раствор для введения готовили следующим образом: сначала индометацин растворяли в 100% этаноле, затем разводили в 5% растворе NaHCO3. На 4-й день после эвтаназии в CO2-камере, у животных извлекали желудок и кишечник, затем иссекали слепую кишку, от подвздошной кишки и толстого кишечника отрезали по 10 см, а от слепой кишки - 5 см для оценки макроскопических повреждений (J Ethnopharmacol. 2004 Feb;90(2-3):195-204).
Таблица 5 - Макроскопическая оценка поражения слепой, подвздошной и толстой кишки при тестировании Соединения 1 на модели колита, индуцированного введением индометацина, баллы (M±m, n=10)
(Учтены только животные с патологией)
*- различия статистически значимы относительно интактной группы (p<0.05);
&- различия статистически значимы относительно контрольной группы (p<0.05).
Из Таблицы 6 видно, что Соединение 1 снижало степень поражения кишечника. Таким образом, можно заключить что Соединение 1 оказывает выраженный терапевтический эффект на модели индометацинового колита у мышей.
Пример 15. Исследование активности Соединения 1 на модели диареи, индуцированной касторовым маслом у мышей
Голодавшим в течение 12 часов мышам линии balb/c внутрижелудочно вводили касторовое масло. Затем животных рассаживали по индивидуальным клеткам с дном, застеленным белой бумагой, и засекали время до начала диареи. Время наблюдения - 4 часа (J Pharm Pharmacol. 2015 Feb;67(2):244-54).
Таблица 6 - Время наступления диареи при изучении активности Соединения 1 на модели диареи, вызванной касторовым маслом, мин (M±m, n=10)
(50 мг/кг)
(1 мг/кг)
(1 мг/кг)
Из таблицы 6 видно, что введение Соединения 1 увеличило время до наступления диареи в 2 раза. Это дает основание заключить, что Соединение 1 оказывает выраженное терапевтическое действие в моделях расстройств желудочно-кишечного тракта.
Пример 16. Исследование активности Соединения 1 на модели синдрома раздраженного кишечника
Влияние на моторику ЖКТ было изучено на модели синдрома раздраженного кишечника на нелинейных мышах-самцах массой 24 -30 г. Животным внутрижелудочно вводили раствор активированного угля (50 мг/мл, в объеме 10 мл/кг) и оценивали скорость (в минутах) продвижения активированного угля по кишечнику животных. Исследуемые соединения вводили однократно внутрижелудочно за 1 час до введения активированного угля. В качестве препаратов сравнения использовали Дротаверин (6.7 мг/кг), Бускопан (3 мг/кг) и Тримедат (33 мг/кг).
Таблица 7 - Результаты исследования влияния Соединения 1 на моторику желудочно-кишечного тракта in vivo
Из таблицы 7 видно, что введение Соединения 1 увеличило время эвакуации активированного угля в 2 раза. Это дает основание заключить, что Соединение 1 оказывает выраженное спазмолитическое действие.
Пример 17. Исследование стабильности Соединения 1 в плазме крови животных и человека
Соединение 1 в концентрации 1 мкМ инкубировали в течение 24 часов в плазме крови человека и разных видов животных (крысы, мыши, морские свинки, кролики, лошади, собаки, бык, карликовые свиньи) при 37˚С. Аликвоты отбирали во временных точках 0, 0,25, 1, 2, 4, 8 и 24 часа. В случае плазмы крови кроликов и обезьян Соединение 1 инкубировали в течение 4 часов, а аликвоты отбирали во временных точках 0, 0,25, 1, 4. После осаждения белков с помощью ацетонитрила, образцы анализировали методом ВЭЖХ-МС/МС на для определения концентрации Соединения 1. В качестве стабильного контроля использовали Верапамил.
Проведенные исследования показали, что Соединение 1 уже через 15 минут практически полностью гидролизуется в плазме крови мышей, крыс, кроликов и морских свинок. По остальным видам животных стабильность Соединения 1 увеличивается в ряду обезьяна - карликовая свинка - человек - собака. Результаты исследования представлены в таблице 8.
Таблица 8 - Стабильность Соединения 1 в плазме крови человека и разных видов животных
Таким образом, в ходе исследования была показана низкая стабильность Соединения 1 в плазме крови человека и разных видов животных. Это неожиданное свойство Соединения 1 позволяет данному соединению оказывать исключительно топическое действие. Таким образом, применение Соединения 1 будет безопасно, т.к. не будут возникать никакие системные побочные эффекты связанные с мультитаргентым действием данного лекарственного препарата.
Пример 18. Исследование фармакокинетики Соединения 1 в плазме крови животных после перорального введения
Для подтверждения низкой системной доступности Соединения 1 было проведено исследование фармакокинетики и биодоступности Соединения 1 после перорального введения крысам в дозе 3 мг/кг. Отбор образцов крови у животных проводили в заданных временных точках течение 24 часов после введения препарата. Содержание Соединения 1 в образцах плазмы анализировали методом ВЭЖХ-МС/МС, предел количественного определения составил 1 нг/мл.
В ходе проведенных исследований Соединение 1 не было обнаружено в плазме крови опытных животных.
Пример 19. Исследование влияния хронического перорального введения Соединения 1 на массу тела кроликов
Было изучено влияние 90 дневного, перорального введения Соединения 1 в дозах 1,5 мг/кг, 7.5 мг/кг и 15 мг/кг на массу тела самцов и самок кроликов породы Шиншилла. Общее состояние кроликов, внешний вид, подвижность были на протяжении всего эксперимента удовлетворительны и не различались в опытных и контрольных группах.
Животные, получавшие Соединение 1, несколько отставали в приросте массы тела по сравнению с параллельным контролем. У кроликов-самцов, получавших Соединение 1 в дозе 1,5 мг/кг, отставание прироста массы тела отмечено на 4-5 и 9-11 неделях введения; у самцов, получавших препарат в дозе 7.5 мг/кг - на 4-5 и 10-11 неделях; у самцов, получавших препарат в дозе 15 мг/кг на 2; 4-5; 8-11 неделях эксперимента. У самок кроликов, на фоне ежедневного внутрижелудочного введения Соединения 1 в дозе 1,5 мг/кг отставание прироста массы тела наблюдалось на 5-6 и 9-ой неделях; на фоне доз 7.5 мг/кг и 15 мг/кг - практически, в течение всего периода введения препарата. В восстановительном периоде отличий в динамике массы тела между контрольными и опытными животными не отмечено. Уменьшение прироста массы тела подопытных животных сопровождалось уменьшением потребления корма и воды.
Данные исследования показывать, что Соединение 1 может так же быть эффективным для контроля массы тела, и при ожирении.
Таким образом, в ходе проведенных исследований было показано, что Соединение 1 является ингибитором катепсина S, агонистом каннабиноидных рецепторов первого типа, антагонистом тахикининовых рецепторов первого и второго типа, антагонистом прокинетициновых рецепторов первого и второго типа, антагонистом брадикининового рецептора первого типа, антагонистом меланокортиновых рецепторов MC4R, антагонистом серотониновых рецепторов 5-HT2B и ингибитором сигнального пути NB-kB. Действие на данные терапевтические мишени позволяет Соединения 1 оказывать выраженный терапевтический эффект в моделях ожирения, псориаза, болезни Крона, колита, синдрома раздраженного кишечника и диареи. Крайне низкая стабильность Соединения 1 в плазме крови животных и человека позволяет исключить возможность возникновения побочных эффектов, которые могли бы возникнуть при системном применении подобного мультитаргетного средства.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРУЮЩИЙ РАСТВОР И СПОСОБ ПОДАВЛЕНИЯ БОЛИ, ВОСПАЛЕНИЯ ИЛИ СПАЗМА | 1995 |
|
RU2180852C2 |
| НОВЫЙ АНТАГОНИСТ ТАХИКИНИНОВЫХ РЕЦЕПТОРОВ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2697414C1 |
| ЛИГАНД С ШИРОКИМ СПЕКТРОМ ОДНОВРЕМЕННОЙ РЕЦЕПТОРНОЙ АКТИВНОСТИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2008 |
|
RU2374245C1 |
| РАСТВОРЫ И СПОСОБЫ ИНГИБИРОВАНИЯ БОЛИ, ВОСПАЛЕНИЯ И РАЗРУШЕНИЯ ХРЯЩА | 2000 |
|
RU2271825C2 |
| ПРИМЕНЕНИЕ КАННАБИНОИДОВ В КОМБИНАЦИИ С АНТИПСИХОТИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ | 2008 |
|
RU2503448C2 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ОСНОВЕ ВЕЩЕСТВА, ВЛИЯЮЩЕГО НА ЭНДОКАННАБИНОИДНУЮ СИСТЕМУ | 2013 |
|
RU2568896C2 |
| СПОСОБ ЛЕЧЕНИЯ ОЖИРЕНИЯ И СОПУТСТВУЮЩИХ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ И ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОЖИРЕНИЯ И СОПУТСТВУЮЩИХ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ | 2010 |
|
RU2552221C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЖЕЛУДОЧНО-КИШЕЧНЫХ ЗАБОЛЕВАНИЙ | 2000 |
|
RU2264215C2 |
| КОМБИНАЦИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2465915C2 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
Группа изобретений относится к медицине, и касается применения соединения, представленного формулой I или его аддукта, гидрата или сольвата, для предупреждения и/или лечения ожирения. Также предложено применение соединения для лечения псориаза, и заболеваний желудочно-кишечного тракта, выбранного из болезни крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты. Группа изобретений обеспечивает лечение указанных патологий, при этом низкая стабильность соединения в плазме крови у человека и разных видов животных позволяет оказывать исключительно топическое действие, следовательно, соединение безопасно, т.к. не вызывает системных побочных эффектов. 6 н.п. ф-лы, 7 табл., 19 пр.
1. Применение соединения, представленного формулой
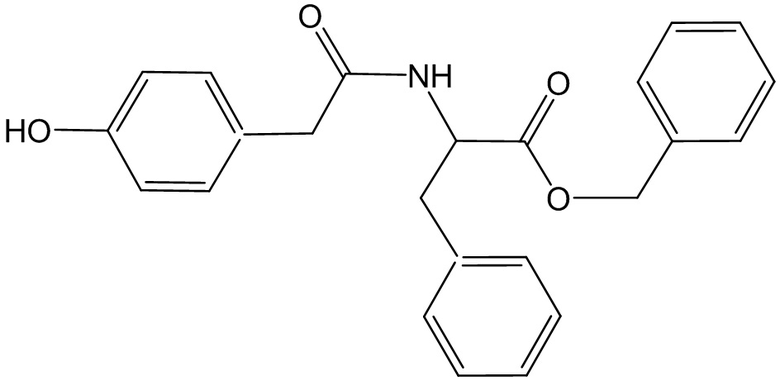
или его аддукта, гидрата или сольвата,
для предупреждения и/или лечения ожирения.
2. Применение соединения, представленного формулой
или его аддукта, гидрата или сольвата,
для предупреждения и/или лечения псориаза.
3. Применение соединения, представленного формулой
или его аддукта, гидрата или сольвата,
для предупреждения и/или лечения заболевания желудочно-кишечного тракта, выбранного из болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты.
4. Способ предупреждения и/или лечения ожирения у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения или его аддукта, гидрата или сольвата, определенного в п.1, указанному субъекту.
5. Способ предупреждения и/или лечения псориаза у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения или его аддукта, гидрата или сольвата, определенного в п.1, указанному субъекту.
6. Способ предупреждения и/или лечения заболевания желудочно-кишечного тракта, выбранного из болезни Крона, колита, синдрома раздраженного кишечника, диареи, тошноты и рвоты, у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения или его аддукта, гидрата или сольвата, определенного в п.1, указанному субъекту.
| ПОЛИПЕПТИД ДЛЯ УВЕЛИЧЕНИЯ АКТИВНОСТИ РЕЦЕПТОРА ГУАНИЛАТЦИКЛАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНОГО РАССТРОЙСТВА У ПАЦИЕНТА, СПОСОБ ПОВЫШЕНИЯ АКТИВНОСТИ РЕЦЕПТОРА ГУАНИЛАТЦИКЛАЗЫ У ПАЦИЕНТА, СПОСОБ ЛЕЧЕНИЯ ВИСЦЕРАЛЬНОЙ БОЛИ, СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДА (ВАРИАНТЫ), ИЗОЛИРОВАННАЯ МОЛЕКУЛА НУКЛЕИНОВОЙ КИСЛОТЫ, БАКТЕРИАЛЬНЫЙ ВЕКТОР ЭКСПРЕССИИ И ИЗОЛИРОВАННАЯ БАКТЕРИАЛЬНАЯ КЛЕТКА | 2005 |
|
RU2433133C2 |
| WO 2006101422 A1, 28.09.2006 | |||
| ПРУЖИНЯЩАЯ ЛЕРКА | 1930 |
|
SU20921A1 |
| DONNA M | |||
| SMALL et al | |||
| The Emerging Relevance of the Cysteine Protease Cathepsin S in Disease// Clinical Reviews in Bone and Mineral Metabolism | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |

