Настоящее изобретение относится к соединениям, которые применяют в качестве ингибиторов активности Wee-1 киназы. Настоящее изобретение также относится к фармацевтическим композициям, включающим эти соединения, и к способам применения этих соединений для лечения рака и к способам лечения рака.
УРОВЕНЬ ТЕХНИКИ
Клетки ежедневно и непрерывно подвергаются проверке на иммунность к специфическому агенту, что приводит к многочисленным повреждениям, возникающим в ДНК. Повреждения, если их не устранять, могут вызывать мутации или гибель клеток, поэтому, существуют сложные сигнальные сети, которые для сохранения целостности ДНК обеспечивают обнаружение повреждений и их устранение.
Обнаружение повреждений ДНК инициирует ряд событий, которые играют ключевую роль в поддержании генома. Контрольные точки клеточного цикла предназначены для остановки клеточного цикла и позволяют устранить повреждения до того, как клеткам будет дана возможность продолжить процесс митоза.
Были идентифицированы две ключевых контрольных точки, одна в конце фазы G1, а вторая в фазе G2, которые, работая в тандеме, гарантируют, что все повреждения будут выявлены и устранены. В 50% случаев рака у человека, контрольная точка G1 не функционирует из-за мутаций в гене-супрессоре опухолевого роста р53. В то время как контрольная точка G2 мутируют редко, и часто обнаруживается, что она активирована в раковых клетках. Раковые клетки используют ее, для того чтобы обрести резистентность к воздействию различных методов лечения, в том числе к воздействию лекарственных средств, повреждающих ДНК, и лучевой терапии.
В качестве ключевых регуляторов контрольной точки G2 были идентифицированы три киназы, а именно Chk1, Chk2 и Wee-1. Исследование ингибиторов этих киназ в настоящее время находится в стадии клинических испытаний.
Wee-1 представляет собой тирозинкиназу с ядерной локализацией, которая негативно регулирует вступление клеток в фазу митоза в контрольной точке G2/M путем катализа реакции фосфорилирования киназного комплекса cdc2/циклин В. Фосфорилирование происходит на остатке тирозина 15 и приводит к инактивации комплекса cdc2/циклин B, что, в конечном счете, предотвращает митоз. Функция Wee-1 тесно связана с функцией Chk1 и Chk2 вследствие их фосфорилирования и инактивации cdc25 на серине 216, а также в связи с обнаружением активации Wee-1 под воздействием Chk 1 и Chk 2 (Ashwell et al., 2012, DNA Repair in Cancer Therapy, DOI: 10,1016/B978-0-12-384999-1,10010-1).
Wee-1 следует вслед за семейством Chk и является одним из важнейших компонентов контрольной точки сигнального каскада, так как она предотвращает вход клеток в фазу митоза в случае, если обнаружены повреждения (Do et al., Cell Cycle 2013 12 (19) 3159-3164.
Обычно применяемые противораковые соединения вызывают повреждение ДНК, и они включают антиметаболиты, средства на основе соединений платины, ингибиторы топоизомеразы и алкилирующие средства. Однако их эффективность ограничена вследствие их чрезмерной токсичности, резистентности опухолей и отсутствия селективности в отношении опухоли. Поэтому, было бы очень важным иметь соединения, которые работают в комбинации с этими лекарственными средствами, селективно предотвращая репарацию ДНК в опухолевых клетках. Так как ген-супрессор опухолевого роста р53 обычно мутирует в линиях опухолевых клеток, введение ингибитора киназы Wee-1, который прекращает действие контрольной точки G2, может приводить к повышенной чувствительности опухолевых клеток к воздействию средств, повреждающих ДНК. И по поводу такой возможности уже сообщалось. Как было обнаружено, подавление активности Wee-1 является достаточным для того, чтобы повысить чувствительность клеток HeLa к действию доксорубицина в результате отмены блокирования G2. В отличие от этого, в нормальном эпителии молочной железы из-за полностью компетентного белка р53, подавление функции Wee-1 практически не оказывало дополнительного действия по сравнению с действием только одного доксорубицина (Wang et al., 2004, Cancer Biology и Therapy, 3(3), 305-313).
Сообщалось, что клеточные линии, несущие мутации в гене-супрессоре опухолевого роста р53, проявляют повышенную чувствительность к средствам, повреждающим ДНК, при совместном введении с низкомолекулярными ингибиторами Wee-1. Сообщалось о синергетическом действии in vitro и in vivo при объединении низкомолекулярных ингибиторов с гемцитабином, 5-фторурацилом, карбоплатином, цисплатином (Hirai et al 2010, Cancer Biology & Therapy 9:7, 514-522), цитарабином (Tibes et al., 2012, Blood, 119(12), 2863-2872), ингибиторами Chk-1 (Carrasa et al., 2012 Cell Cycle 1:11(13):2507-2517), (Russell et al., 2013 Cancer Res. 15; 73 (2) 776-784) и ингибиторами Src (Cozzi et al., 2012, Cell Cycle 11(5), 1-11). Сообщалось об апоптическом действии взятого в одиночку лекарственного средства, независимо от статуса p53, в отношении линии клеток саркомы и образцов, взятых у пациентов с саркомой (Kreahling et al., 2012, Mol. Cancer Ther., 11(1), 174-182), и была продемонстрирована эффективность in vivo в отношении панели линий раковых клеток (Guertin et al., 2013 Mol Cancer Ther, 12 (2) 141-151).
Известно, что облучение увеличивает фосфорилирование остатков Tyr15 и Thr14 из cdc2, что приводит к радиорезистентному фенотипу. Ингибирование активности Wee-1 с помощью низкомолекулярных ингибиторов (Wang et al., 2004, Cancer Biology и Therapy 3(3), 305-313), (Caretti et al., 2013 Mol Cancer Ther. 12 (2) 141-150) приводит к снижению фосфорилирования и к радиосенсибилизации, причем эффект становится более выраженным в линиях р53 мутантных клеток.
Сообщалось, что в случае меланомы, сверхэкспрессия Wee-1 коррелирует с неблагоприятным клиническим исходом (Magnusson et al., 2012 PLoS One 7; (6)e38254), что указывает на то, что сверхэкспрессия Wee-1 может рассматриваться в качестве важного биомаркера и в качестве мишени при таргетной терапии.
Соединения, обладающие ингибирующим действием в отношении киназы, например, ингибирующим действием в отношении Wee-1 киназы, описаны в патентных документах WO 2007/126122, US 2010/0063024, EP 2,213,673, WO 2008/133866, US 2007/0254892, WO 2012/161812, WO 2013/126656, US 2013/0102590, WO 2013/059485 и WO 2013/013031.
В патентных документах WO 2010/067886, WO 2010/067888, US 2011/0135601, EP 2168966, WO 2005/090344, US 2009/0048277 и публикации Bioorg. Med. Chem. Lett., 2005, 15, 1931-1935 описаны различные соединения, обладающие ингибирующим действием в отношении киназы, такие как производные дигидропиримидопиримидина и пиридопиримидинона. В частности, сообщается, что соединения, описанные в патентном документе WO 2005/090344, проявляют активность в качестве ингибиторов протеинкиназы, в частности ингибиторов Src семейства тирозинкиназы. Сообщается, что соединения, описанные в публикации Bioorg. Med. Chem. Lett., 2005, 15, p1931-1935, проявляют в 10-100 раз более высокую ингибирующую активность в отношении с-Src, чем в отношении Wee-1, и изменение заместителей на 6-фенильном кольце заметно не изменяет это соотношение. Сообщается, что 5-алкилзамещенные аналоги обычно являются селективными в отношении Wee-1, но за счет связывающей активности.
В патентном документе WO 2013/013031 описан пиридазино[4,5-d]пиримидин-(6H)-он в качестве ингибитора Wee-1 киназы, который предлагают применять для ингибирования киназ, таких как Wee-1, и в способах лечения таких заболеваний, как рак. Соединения, описанные в патентном документе WO 2013/013031, имеют атом азота в "положении 3" кольца относительно карбонильной группы.
В патентном документе США 2013/0018045 описаны различные трициклические сульфонамидные соединения, которые применяют для ингибирования киназ, таких как Wee-1, и способы лечения заболеваний, таких как рак. Соединения, описанные в патентном документе США 2013/0018045, имеют сульфонамидную группу в "1-м положении" на кольце, и атомы в "3- и 4-положениях" образуют часть конденсированного арильного или гетероарильного кольца ("A").
Одной задачей настоящего изобретения является устранение, по меньшей мере, некоторых из недостатков предшествующего уровня техники или создание коммерчески применимой альтернативы этому.
Другой задачей настоящего изобретения является создание соединений, обладающих повышенной селективностью по отношению к Wee-1 киназе по сравнению с известными соединениями или композициями.
Следующей задачей настоящего изобретения является создание соединений, обладающих улучшенной стабильностью в микросомах человека, например, микросомах печени человека, по сравнению с известными соединениями или композициями.
Еще одной задачей настоящего изобретения является создание соединения, обладающего более сильным или аналогичным ингибирующим действием в отношении киназы по сравнению с известными соединениями или композициями.
Следующей задачей настоящего изобретения является создание соединений, обладающих более высокой эффективностью по сравнению с известными соединениями или композициями.
Еще одной задачей настоящего изобретения является создание соединения, характеризующегося более высокой эффективностью и улучшенной переносимостью при введении в комбинации с другими методами лечения по сравнению с известными соединениями или композициями.
Следующей задачей настоящего изобретения является создание соединения, характеризующегося улучшенной переносимостью по сравнению с известными соединениями или композициями.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
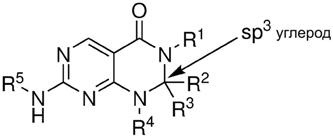
В первом аспекте, настоящее изобретение предлагает соединение формулы (I):

(I)
или его фармацевтически приемлемую соль или его N-оксидное производное, где:
R1 представляет собой необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу;
R2 и R3 независимо выбирают из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной алкильной группы, необязательно замещенной циклоалкильной группы, необязательно замещенной алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной арильной группы и необязательно замещенной гетероарильной группы; или R2, R3 и углеродный атом, к которому они оба присоединены, взятые вместе, образуют необязательно замещенную циклоалкильную группу или необязательно замещенную гетероциклильную группу;
R4 представляет собой атом водорода, необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу;
или R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную группу; и
R5 представляет собой необязательно замещенную алкильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу.
Каждый определенный в изобретении аспект или вариант осуществления может быть объединен с любым другим аспектом (аспектами) или вариантом осуществления (вариантами осуществления), если однозначно не указано иначе. В частности, любой отличительный признак, указанный в качестве предпочтительного или желательного, может быть объединен с любым другим отличительным признаком, указанным в качестве предпочтительного или желательного.
Во втором аспекте, настоящее изобретение предлагает фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль или его N-оксидное производное и, по меньшей мере, один фармацевтически приемлемый носитель.
В третьем аспекте, настоящее изобретение предлагает соединение формулы (I) или его фармацевтически приемлемую соль или его N-оксидное производное или фармацевтическую композицию, включающую соединение формулы (I), для применения в терапии.
В четвертом аспекте, настоящее изобретение предлагает соединение формулы (I) для применения в качестве лекарственного препарата.
В пятом аспекте, настоящее изобретение предлагает соединение формулы (I) для применения при лечении рака или для его предотвращения.
В шестом аспекте, настоящее изобретение предлагает применение соединения формулы (I) для производства лекарственного препарата для лечения или предотвращения рака.
В седьмом аспекте, настоящее изобретение предлагает способ лечения или предотвращения рака у человека или животного, включающий введение пациенту, если он в этом нуждается, эффективного количества соединения формулы (I) или фармацевтической композиции, включающей соединение формулы (I).
Другие предпочтительные варианты осуществления соединений согласно изобретению приведены в описании изобретения и, в частности, в примерах. Особенно предпочтительными являются те названные соединения, которые по результатам испытаний обладают более высокой активностью. Соединения, обладающие более высокой активностью, являются более предпочтительными по сравнению с соединениями, обладающими более низкой активностью.
Авторы настоящего изобретения неожиданно обнаружили, что соединения настоящего изобретения, которые содержат sp3 гибридизированный углеродный атом в карбонилсодержащем конденсированном кольце, проявляют более сильное или аналогичное ингибирующее действие в отношении киназы по сравнению с известными соединениями или композициями. В частности, соединения настоящего изобретения предпочтительно проявляют более сильное или аналогичное ингибирующее действие в отношении Wee-1 киназы по сравнению с известными соединениями или композициями. Это является достаточно неожиданным, так как сообщалось, что известные соединения, которые проявляют высокое ингибирующее действие в отношении Wee-1 киназы, включают плоскую кольцевую систему с одним или более sp2 гибридизированными углеродными атомами и одним или более атомами азота, в то время как соединения настоящего изобретения включают sp3 гибридизированный углеродный атом, в которых, по меньшей мере, один из заместителей на sp3 углероде выступает за плоскость кольца.
Авторы настоящего изобретения неожиданно обнаружили, что соединения настоящего изобретения могут проявлять более высокую или сопоставимую селективность в отношении Wee-1 киназы по сравнению с соединениями предшествующего уровня техники. Предпочтительно, в частности, чтобы соединения изобретения были селективными в отношении представителей Src семейства киназ, например, в отношении LCK (лимфоидно-специфической протеин-тирозин-киназы) и c-Src.
Авторы настоящего изобретения неожиданно обнаружили, что описанные в изобретении соединения могут иметь исключительные физико-химические свойства по сравнению с соединениями предшествующего уровня техники. Например, кинетическая растворимость, приведенная в примерах далее, является более высокой, чем в соответствующем примере, приведенном в патентном документе WO 2013/126656.
Соединения настоящего изобретения могут также характеризоваться исключительной метаболической стабильностью по сравнению с соединениями предшествующего уровня техники. Например, величина собственного клиренса из микросом человека, приведенная в примерах далее, является более низкой, чем для соответствующих соединений, приведенных в патентных документах WO 2013/126656 и WO 2013/059485.
Авторы настоящего изобретения также неожиданно обнаружили, что соединения настоящего изобретения могут также проявлять пониженное или сопоставимой ингибирование hERG (смотрите примеры), улучшенную или сопоставимую стабильность в микросомах печени человека и пониженную или сопоставимую токсичность в отношении сердечно-сосудистой системы токсичность по сравнению с ингибиторами киназы предшествующего уровня техники.
Как уже отмечалось выше, можно высказать предположение, но не подтверждая его какой-либо теорией, что соединения настоящего изобретения обычно характеризуются обсужденными выше положительными эффектами, которые обусловлены, по меньшей мере, частично, присутствием sp3 гибридизированного углеродного атома, показанного в формуле (I), то есть, углерода, к которому присоединены R2 и R3.
(I)
Дополнительные факторы, которые позволяют получить обсужденные выше положительные эффекты, включают структурную взаимосвязь между упомянутым выше sp3 гибридизированным углеродным атомом в "3-положении", карбонильной (C=O) группой, группой N-R1 во "2-положении" и группой N-R4 в "4-положении".
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если в изобретении не указано иначе, то научные и технические термины, используемые при описании настоящего изобретения, имеют значения, которые являются общепринятыми среди обычных специалистов в этой области. Значение термина и охватываемая термином область должны быть очевидными, однако, в случае любой латентной неясности, определения, приводимые в изобретении, имеют преимущество по отношению к любому словарному определению или определению из внешнего документа.
Если не указано иначе, то используемые в описании изобретения и прилагаемых пунктах формулы изобретения следующие далее термины имеют указанные значения.
Термин "алкильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает линейный или разветвленный насыщенный гидрокарбильный заместитель, обычно содержащий от 1 до 15 углеродных атомов, например, от 1 до 10, от 1 до 8, от 1 до 6, или от 1 до 4 углеродных атомов. "Cn алкильная" группа относится к алифатической группе, содержащей n углеродных атомов. Например, C1-C10 алкильная группа содержит 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 углеродных атомов. Присоединение к алкильной группе происходит через углеродный атом. Примеры таких заместителей включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил, третбутил, пентил (разветвленный или неразветвленный), гексил (разветвленный или неразветвленный), гептил (разветвленный или неразветвленный), октил (разветвленный или неразветвленный), нонил (разветвленный или неразветвленный) и децил (разветвленный или неразветвленный).
Термин "алкенильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает линейный или разветвленный гидрокарбильный заместитель, содержащий одну или более двойных связей и обычно от 2 до 15 углеродных атомов; например, от 2 до 10, от 2 до 8, от 2 до 6 или от 2 до 4 углеродных атомов. Примеры таких заместителей включают этенил (винил), 1-пропенил, 3-пропенил, 1,4-пентадиенил, 1,4-бутадиенил, 1-бутенил, 2-бутенил, 3-бутенил, пентенил и гексенил.
Термин "алкинильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает линейный или разветвленный гидрокарбильный заместитель, содержащий одну или более тройных связей и обычно от 2 до 15 углеродных атомов; например, от 2 до 10, от 2 до 8, от 2 до 6 или от 2 до 4 углеродных атомов. Примеры таких заместителей включают этинил, 1-пропинил, 3-пропинил, 1-бутинил, 3-бутинил и 4-бутинил.
Термин "карбоциклильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает насыщенный циклический (то есть "циклоалкильный"), частично насыщенный циклический (то есть "циклоалкенильный") или полностью ненасыщенный (то есть "арильный") углеводородный заместитель, содержащий от 3 до 14 углеродных кольцевых атомов ("кольцевые атомы" представляют собой связанные между собой атомы, образующие кольцо или кольца циклического заместителя). Карбоциклил может представлять собой только одно кольцо (моноциклический карбоциклил) или полициклическую кольцевую структуру.
Карбоциклил может представлять собой структуру только с одним кольцом, которое обычно содержит от 3 до 8 кольцевых атомов, более типично, от 3 до 7 кольцевых атомов, и еще более типично, от 5 до 6 кольцевых атомов. Примеры таких карбоциклилов только с одним кольцом включают циклопропил (циклопропанил), циклобутил (циклобутанил), циклопентил (циклопентанил), циклопентенил, циклопентадиенил, циклогексил (циклогексанил), циклогексенил, циклогексадиенил и фенил. В качестве варианта, карбоциклил может быть полициклическим (то есть, может содержать более чем одно кольцо). Примеры полициклических карбоциклилов включают мостиковые, конденсированные и спироциклические карбоциклилы. В спироциклическом карбоциклиле, один атом является общим для двух различных колец. Примером спироциклического карбоциклила является спиропентанил. В мостиковом карбоциклиле, кольца совместно используют, по меньшей мере, два общих несмежных атома. Примеры мостиковых карбоциклилов включают бицикло[2,2,1]гептанил, бицикло-[2,2,1]гепт-2-енил и адамантил. В карбоциклиле с конденсированной кольцевой системой, два или более колец могут быть конденсированными, в результате чего два кольца делят между собой одну общую связь. Примеры карбоциклилов с двумя или тремя конденсированными кольцами включают нафталенил, тетрагидронафталенил (тетралинил), инденил, инданил (дигидроинденил), антраценил, фенантренил и декалинил.
Термин "циклоалкильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает насыщенный циклический углеводородный заместитель, содержащий от 3 до 14 углеродных кольцевых атомов. Циклоалкил может представлять собой только одно углеродное кольцо, которое обычно содержит от 3 до 8 углеродных кольцевых атомов, и более типично, от 3 до 6 кольцевых атомов. Следует иметь в виду, что присоединение к циклоалкильной группу происходит через кольцевой атом циклоалкильной группы. Примеры циклоалкилов с одним кольцом включают циклопропил, циклобутил, циклопентил и циклогексил. В качестве варианта, циклоалкил может быть полициклическим или содержать более чем одно кольцо. Примеры полициклических циклоалкилов включают мостиковые, конденсированные и спироциклические карбоциклилы.
Термин "арильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает ароматический карбоциклил, содержащий от 6 до 14 углеродных кольцевых атомов, или от 3 до 8, от 3 до 6 или от 5 до 6 углеродных кольцевых атомов. Арил может быть моноциклическим или полициклическим (то есть, может содержать более чем одно кольцо). В случае полициклических ароматических колец, требуется, чтобы только одно кольцо в полициклической системе было ненасыщенным, а другое кольцо (кольца) может быть насыщенным, частично насыщенным или ненасыщенным. Присоединение к арильной группе происходит через углеродный атом, содержащейся в кольце. Примеры арильных групп включают фенил, нафтил, акридинил, инденил, инданил и тетрагидронафтил.
Термин "гетероциклильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает насыщенную (то есть "гетероциклоалкильную"), частично насыщенную (то есть "гетероциклоалкенильную") или полностью ненасыщенную (то есть "гетероарильную") кольцевую структуру, содержащую суммарно от 3 до 14 кольцевых атомов, где, по меньшей мере, один из кольцевых атомов является гетероатомом (то есть кислородом, азотом или серой), при этом остальные кольцевые атомы являются углеродными атомами. Гетероциклильная группа может, например, содержать один, два, три, четыре или пять гетероатомов. Присоединение к гетероциклильной группе может происходить или через углеродный атом и/или через один или более гетероатомов, которые содержатся в кольце. Гетероциклил может представлять собой однокольцевую (моноциклическую) или полициклическую кольцевую структуру.
Гетероциклильная группа может представлять собой единственное кольцо, которое обычно содержит от 3 до 7 кольцевых атомов, более типично, от 3 до 6 кольцевых атомов, и еще более типично, от 5 до 6 кольцевых атомов. Примеры однокольцевых гетероциклилов включают фуранил, дигидрофуранил, тетрагидрофуранил, тиофенил (тиофуранил), дигидротиофенил, тетрагидротиофенил, пирролил, пирролинил, пирролидинил, имидазолил, имидазолинил, имидазолидинил, пиразолил, пиразолинил, пиразолидинил, триазолил, тетразолил, оксазолил, оксазолидинил, изоксазолидинил, изоксазолил, тиазолил, изотиазолил, тиазолинил, изотиазолинил, тиазолидинил, изотиазолидинил, тиодиазолил, оксадиазолил (в том числе 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил (фуразанил) или 1,3,4-оксадиазолил), оксатриазолил (в том числе 1,2,3,4-оксатриазолил или 1,2,3,5-оксатриазолил), диоксазолил (в том числе 1,2,3-диоксазолил, 1,2,4-диоксазолил, 1,3,2-диоксазолил или 1,3,4-диоксазолил), оксатиазолил, оксатиолил, оксатиоланил, пиранил, дигидропиранил, тиопиранил, тетрагидротиопиранил, пиридинил (азинил), пиперидинил, диазинил (в том числе пиридазинил (1,2-диазинил), пиримидинил (1,3-диазинил) или пиразинил (1,4-диазинил)), пиперазинил, триазинил (в том числе 1,3,5-триазинил, 1,2,4-триазинил и 1,2,3-триазинил)), оксазинил (в том числе 1,2-оксазинил, 1,3-оксазинил или 1,4-оксазинил)), оксатиазинил (в том числе 1,2,3-оксатиазинил, 1,2,4-оксатиазинил, 1,2,5-оксатиазинил или 1,2,6-оксатиазинил)), оксадиазинил (в том числе 1,2,3-оксадиазинил, 1,2,4-оксадиазинил, 1,4,2-оксадиазинил или 1,3,5-оксадиазинил)), морфолинил, азепинил, оксепинил, тиепинил и диазепенил.
В качестве варианта, гетероциклильная группа может быть полициклической (то есть, может содержать более чем одно кольцо). Примеры полициклических гетероциклильных групп включают мостиковые, конденсированные и спироциклические гетероциклильные группы. В спироциклической гетероциклильной группе, один атом является общим для двух различных колец. В мостиковой гетероциклильной группе, кольца совместно используют, по меньшей мере, два общих несмежных атома. В конденсированной кольцевой гетероциклильной группе, два или более колец могут быть конденсированными, в результате чего два кольца делят между собой одну общую связь. Примеры конденсированных кольцевых гетероциклильных групп, содержащих два или три кольца, включают индолизинил, пиранопирролил, 4H-хинолизинил, пуринил, нафтиридинил, пиридопиридинил (в том числе пиридо[3,4-b]-пиридинил, пиридо[3,2-b]пиридинил, или пиридо[4,3-b]пиридинил), и птеридинил. Другие примеры конденсированных кольцевых гетероциклильных групп включают бензоконденсированные гетероциклильные группы, такие как индолил, изоиндолил (изобензазолил, псевдоизоиндолил), индоленинил (псевдоиндолил), изоиндазолил (бензпиразолил), бензазинил (в том числе хинолинил (1-бензазинил) или изохинолинил (2-бензазинил)), фталазинил, хиноксалинил, хиназолинил, бензoдиазинил (в том числе циннолинил (1,2-бензoдиазинил) или хиназолинил (1,3-бензoдиазинил)), бензoпиранил (в том числе хроманил или изохроманил), бензоксазинил (в том числе 1,3,2-бензоксазинил, 1,4,2-бензоксазинил, 2,3,1-бензоксазинил или 3,1,4-бензоксазинил) и бензизоксазинил (в том числе 1,2-бензизоксазинил или 1,4-бензизоксазинил).
Термин "гетероциклоалкильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает насыщенный гетероциклил.
Термин "гетероарильная группа" (сам по себе или в комбинации с другим термином (терминами)) обозначает ароматический гетероциклил, содержащий от 5 до 14 кольцевых атомов. Гетероарил может представлять собой одно кольцо или 2 или 3 конденсированных кольца. Примеры гетероарильных заместителей включают 6-членные кольцевые заместители, такие как пиридил, пиразил, пиримидинил, пиридазинил и 1,3,5-, 1,2,4- или 1,2,3-триазинил; 5-членные кольцевые заместители, такие как имидазил, фуранил, тиофенил, пиразолил, оксазолил, изоксазолил, тиазолил, 1,2,3-, 1,2,4-, 1,2,5- или 1,3,4-оксадиазолил и изотиазолил; 6/5-членные конденсированные кольцевые заместители, такие как бензoтиофуранил, бензизоксазолил, бензоксазолил, пуринил и антранилил; и 6/6-членные конденсированные кольцевые заместители, такие как бензoпиранил, хинолинил, изохинолинил, циннолинил, хиназолинил и бензоксазинил.
Термин "азотсодержащая гетероциклильная группа" относится к моноциклической или бициклической гетероциклильной группе, содержащей, по меньшей мере, один атом азота, в которой каждое кольцо включает от 3 до 7 кольцевых атомов и необязательно содержит, помимо атома азота, ноль или один или два или более одинаковых или различных гетероатомов, но, предпочтительно, ноль или один гетероатом, выбранный из группы, состоящей из атома кислорода, атома азота и атома серы; и гетероциклильная группа может быть насыщенной (то есть "гетероциклоалкильной"), частично насыщенной (то есть "гетероциклоалкенильной") или полностью ненасыщенной (то есть "гетероарильной"). Бициклическая гетероциклильная группа может иметь спироструктуру, в которой два кольца совместно используют один и тот же кольцевой атом, или может иметь бициклическую структуру, в которой кольца совместно используют два или более кольцевых атомов. Примеры азотсодержащей гетероциклильной группы включают, например, пирролильную группу, имидазолильную группу, пиразолильную группу, тиазолильную группу, изотиазолильную группу, оксазолильную группу, изоксазолильную группу, триазолильную группу, тетразолильную группу, оксадиазолильную группу, 1,2,3-тиадиазолильную группу, 1,2,4-тиадиазолильную группу, 1,3,4-тиадиазолильную группу, пиридильную группу, пиразинильную группу, пиримидинильную группу, пиридазинильную группу, 1,2,4-триазинильную группу, 1,3,5-триазинильную группу, индолильную группу, бензимидазолильную группу, бензоксазолильную группу, бензизоксазолильную группу, бензoтиазолильную группу, бензизотиазолильную группу, индазолильную группу, пуринильную группу, хинолильную группу, изохинолильную группу, фталазинильную группу, нафтиридинильную группу, хиноксалинильную группу, хиназолинильную группу, циннолинильную группу, птеридинильную группу, пиридо[3,2-b]пиридильную группу, азетидильную группу, пирролидинильную группу, дигидро-1,2,4-триазолильную группу, дигидро-1,2,4-оксадиазолильную группу, дигидро-1,3,4-оксадиазолильную группу, дигидро-1,2,4-тиадиазолильную группу, дигидро-1,2,3,5-оксатиадиазолильную группу, пиперидинильную группу, пиперазинильную группу, дигидропиридильную группу, морфолинильную группу, тиоморфолинильную группу, 2,6-диазаспиро[3,5]нонильную группу, 2,7-диазаспиро[3,5]нонильную группу, 2,7-диазаспиро[4,5]-децильную группу или 2,7-диазабицикло[3,3,0]октильную группу, 3,6-диазабицикло[3,3,0]октильную группу.
Азотсодержащая гетероциклильная группа может быть необязательно замещена ("замещенная азотсодержащая гетероциклильная группа") с помощью одного или более заместителей, которые могут быть одинаковыми или разными.
Термин "аминогруппа" относится к группе -NH2. Аминогруппа может быть необязательно замещена ("замещенная амино") с помощью одного или более заместителей, которые могут быть одинаковыми или разными. Заместителями для аминогруппы могут быть, но этим не ограничивая, алкильная, алкенильная, алканоильная, арильная и/или гетероциклильная группа.
Термин "амидогруппа" относится к группе -C(=O)-NR-. присоединение может быть осуществлено через углеродный атом и/или атом азота. Например, амидогруппа может быть присоединена в качестве заместителя только через углеродный атом, и в этом случае атом азота имеет две присоединенных группы R (-C(=O)-NR2). Амидогруппа может быть присоединена только через атом азота, и в этом случае углеродный атом имеет две присоединенных группы R (-NR-C(=O)R.
Термин "иминильная" группа относится к группе -C(=NR)-. Присоединение может быть осуществлено через углеродный атом.
Группа "=N-R" относится к замещающей группе азот-R, соединенной с другим атомом двойной связью. Например, иминильная группа (группа -C(=NR)-) представляет собой атом азота, соединенный двойной связью с углеродным атомом, при этом азота также соединен с группой R одинарной связью.
Термин "алкоксильная группа" относится к -O-алкильной группе. Алкоксильная группа может относиться к линейным, разветвленным или циклическим насыщенным или ненасыщенным оксо-углеводородным цепям, включающим, например, метоксил, этоксил, пропоксил, изопропоксил, бутоксил, третбутоксил и пентоксил. Алкоксильная группа может быть необязательно замещена ("замещенная алкокси") с помощью одной или более групп, замещающих алкокси.
Термин "гидроксил" относится к группе -OH.
Термин "алканоильная группа" (то есть ацильная группа) относится к группе органической кислоты, в которой -OH карбоксильной группы был заменен на другой заместитель. Поэтому, алканоильная группа может быть представлена формулой RC(=O)-, где R включает, но этим не ограничивая, алкильную, аралкильную или арильную группу, которая, в свою очередь, может быть необязательно замещена одним или более заместителями. Примеры алканоильных групп включают ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, валерильную группу, изовалерильную группу и пивалоильную группу.
Термин "сульфонильная группа" относится к группе сульфоновой кислоты, в которой -OH сульфонильной группы был заменен на другой заместитель. Например, заместителем может быть алкильная группа ("алкилсульфонильная группа"). Алкилсульфонильная группа может быть представлена формулой RS(O)2-, где R представляет собой алкильную группу, необязательно замещенную с помощью одного или более заместителей. Примеры алкилсульфонильных групп включают метилсульфонильную группу, этилсульфонильную группу, пропилсульфонильную группу, изопропилсульфонильную группу, бутилсульфонильную группу, вторбутилсульфонильную группу, изобутилсульфонильную группу, третбутилсульфонильную группу, пентилсульфонильную группу, изопентилсульфонильную группу, гексилсульфонильную группу и изогексилсульфонильную группу.
Термин "сульфинильная группа" относится к двухвалентной группе -S(=O).
Термин "сульфоксиминильная группа" относится к группе "-S(=O)(=NR)(R)-".
Термин "тиоморфолинсульфоксиминильная группа" относится к группе формулы (h):

(h).
Термин "оксогруппа" относится к группе (=O), то есть к замещающему атому кислорода, соединенному с другим атомом двойной связью. Например, карбонильная группа (-C(=O)- представляет собой углеродный атом, соединенный двойной связью с атомом кислорода, то есть оксогруппу, присоединенную к углеродному атому.
Термин "галогеновая группа" относится к группе, выбранной из хлора, фтора, брома и йода. Предпочтительно, чтобы галогеновую группу выбирали из хлора и фтора.
Алкил, алкенил, алкинил, амино, амидо, иминил, алкокси, карбоциклил (в том числе циклоалкил, циклоалкенил и арил), гетероциклил (в том числе гетероциклоалкил, гетероциклоалкенил и гетероарил), сульфонил, сульфинил, сульфоксиминил и азотсодержащая гетероциклильная группа могут быть необязательно замещены с помощью одного или более заместителей, которые могут быть одинаковыми или разными. Заместитель может быть присоединен через углеродный атом и/или гетероатом в алкиле, алкениле, алкиниле, амино, амидо, иминиле, алкокси, карбоциклиле (в том числе циклоалкиле, циклоалкениле и ариле), гетероциклиле (в том числе гетероциклоалкиле, гетероциклоалкениле и гетероариле), сульфониле, сульфиниле, сульфоксиминиле и азотсодержащей гетероциклильной группе. Термин "заместитель" (или "радикал") включает, но этим не ограничивая, алкил, замещенный алкил, аралкил, замещенный аралкил, алкенил, замещенный алкенил, алкинил, замещенный алкинил, галоген, циано, амино, амидо, алкиламино, ариламино, карбоциклил, циклоалкил, замещенный циклоалкил, циклоалкенил, замещенный циклоалкенил, арил, замещенный арил, нитро, тио, алканоил, гидроксил, арилоксил, алкоксил, алкилтио, арилтио, аралкилоксил, аралкилтио, карбоксил, алкоксикарбонил, оксо, алкилсульфонил и арилсульфонил.
Если группа, например алкильная группа, является "необязательно замещенной", то это означает, что группа имеет один или более присоединенных заместителей (то есть является замещенной) или не имеет никаких присоединенных заместителей (то есть является незамещенной).
Кроме того, следует отметить, что некоторые химические формулы, используемые в изобретении, определяют делокализованные системы. В химии этим определением характеризуют ароматичность, и это определение может указывать на присутствие, например, моно-, ди- или трициклической системы, которая содержит (4n+2) электронов, где n представляет собой целое число. Другими словами, эти системы могут проявлять ароматичность Хюккеля.
В любом аспекте, соединения настоящего изобретения могут иметь некоторые признаки стереохимии. Например, соединения могут иметь хиральные центры и/или плоскости и/или оси симметрии. В силу этого, соединения могут быть получены в виде индивидуальных стереоизомеров, индивидуальных диастереомеров, смесей стереоизомеров или в виде рацемических смесей. Стереоизомерами называют молекулы, которые имеют одинаковую молекулярную формулу и последовательность связанных атомов, но которые отличаются пространственными ориентациями их атомов и/или групп.
Кроме того, соединения настоящего изобретения может характеризоваться таутомеризмом. Предполагается, что каждая таутомерная форма входит в объем изобретения.
Кроме того, соединения настоящего изобретения могут быть получены в форме пролекарств. Пролекарства превращаются, обычно in vivo, из одной формы в активные формы лекарственных средств, описанных в изобретении. Например, пролекарство может быть образовано путем защиты группы -N-H, к которой присоединен R3, с помощью способной к гидролизу группы, которая дает в результате гидролиза -NH. В качестве варианта или дополнительно, любая group -NH в соединение может быть защищена в форме амида, способного к гидролизу в физиологических условиях.
Кроме того, следует иметь в виду, что описанные в изобретении химические элементы могут представлять собой обычный изотоп или изотоп, который не является обычным изотопом. Например, атом водорода может представлять собой 1H, 2H (дейтерий) или 3H (тритий).
Кроме того, соединения настоящего изобретения могут быть получены в форме их фармацевтически приемлемых солей или сокристаллов. Например, могут быть получены соединения, имеющие протонированные аминогруппы.
Термин "фармацевтически приемлемая соль" относится к ионным соединениям, образованным путем добавления кислоты к основанию. Термин относится к таким солям, которые считают подходящими для применения при контакте с пациентом, например, in vivo, и фармацевтически приемлемые соли обычно выбирают с учетом отсутствия у них токсичности и раздражающего действия.
Термин "сокристалл" относится к многокомпонентному молекулярному кристаллу, который может включать неионные взаимодействия.
Фармацевтически приемлемые соли и сокристаллы могут быть получены методом ионообменной хроматографии или путем реакции свободного основания или кислотной формы соединения с стехиометрическими количествами или с избытком требуемой солеобразующей неорганической или органической кислоты или основания в одном или более подходящих растворителях, или путем смешения соединения с другим фармацевтически приемлемым соединением, способным образовывать сокристалл.
Известные соли, которые обычно применяют для контакта с пациентом, включают соли, полученные из неорганических и/или органических кислот, в том числе гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, олеат, пальмитат, стеарат, лаурат, бензoат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат и тартрат. Они могут включать катионы щелочных и щелочноземельных металлов, таких как натрий, калий, кальций и магний, а также аммоний, тетраметиламмоний, тетраэтиламмоний. Можно привести ссылки на ряд литературных источников, в которых рассмотрены подходящие фармацевтически приемлемые соли, например, на руководство Handbook of pharmaceutical salts, изданное Международным союзом теоретической и прикладной химии (IUPAC).
Кроме того, соединения настоящего изобретения могут иногда существовать в форме цвиттер-ионов, которые рассматриваются в качестве части изобретения.
Авторы настоящего изобретения обнаружили, что соединения настоящего изобретения могут применяться при лечении медицинских состояний, связанных с нарушением клеточного роста, включающих, но этим не ограничивая, рак, в частности, онкологические заболевания, связанные с мутациями гена-супрессора опухолевого роста р53.
Например, онкологические заболевания включают рак сердца, рак легких, рак желудочно-кишечного тракта, рак урогенитального тракта, рак печени, рак костей, опухоль нервной системы, гинекологическая опухоль, гемобластоз, рак кожи и рак надпочечника.
Например, онкологические заболевания включают опухоль надпочечника, рак желчного протока, рак мочевого пузыря, рак крови, рак кости и соединительной ткани, рак мозга и центральной нервной системы, рак молочной железы, рак шейки матки, рак толстой кишки и прямой кишки (колоректальный рак), эндометриальный рак, рак пищевода, рак желчного пузыря, рак головы и шеи, лимфому Ходжкина, гипофарингеальный рак, рак почки, рак гортани, лейкозы, рак печени, рак легкого, лимфомы, медиастинальные опухоли, меланому (злокачественную меланому), мезотелиому, множественную миелому, рак носовой полости, назофарингеальный рак, нейроэндокринные опухоли, неходжкинскую лимфому, рак полости рта, рак пищевод, орофарингеальный рак, рак яичников, рак поджелудочной железы, рак носовой пазухи, рак паращитовидной железы, рак полового члена, опухоль гипофиза, рак предстательной железы, рак слюнной железы, саркому, рак кожи, рак позвоночника, рак желудка, рак яичек, рак щитовидной железы, рак уретры, рак матки, вагинальный рак и рак влагалища.
Соединения настоящего изобретения также используют при приготовлении лекарственного препарата, который применяют при лечении описанных выше заболеваний, в частности, рака.
Кроме того, настоящее изобретение относится к способу ингибирования активности Wee-1, который включает введение млекопитающему, если оно в этом нуждается, фармацевтически эффективного количества соединения настоящего изобретения.
Соединения этого изобретения могут быть введены млекопитающим, в том числе людям, или в чистом виде, или в комбинации с фармацевтически приемлемыми носителями, вспомогательными веществами или разбавителями в виде фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Соединения могут быть введены перорально или парентерально, в том числе внутривенно, внутримышечно, интраперитонеально, подкожно, ректально и местно.
В объем настоящего изобретения также входит применение соединения настоящего изобретения в комбинации со вторым или дополнительным лекарственным средством при лечении рака. Второе или дополнительное лекарственное средство может представлять собой лекарственное средство, применение которого при лечении рака является уже известным.
Настоящее изобретение также включает применение соединений изобретения в схеме лечения, включающей стадию лучевой терапии. Лучевая терапия может представлять собой общепринятый метод лечения с помощью рентгеновского облучения, γ-облучения, нейтронного облучения, облучения α-частицами или облучения электронным пучком. Совместное введение соединений, описанных в этом изобретении, может приводить к усилению действия лучевой терапии, и, поэтому, их можно классифицировать как средства для повышения чувствительности пациентов к лучевой терапии.
В частности, онкологические заболевания часто становятся резистентными к применяемому лечению. Развитие резистентности может быть отсрочено или предотвращено путем введения комбинации лекарственных средств, которая включает соединения настоящего изобретения, например, при онкологических заболеваниях, по поводу которых известно, что они резистентны к действию средств, повреждающих ДНК, или воздействию лучевой терапии.
Например, лекарственные средства, которые могут использоваться в комбинации с соединениями настоящего изобретения, могут быть нацелены на тот же самый или аналогичный каскад биологических реакций, на который нацелены соединения настоящего изобретения, или могут воздействовать на другой или независимый каскад реакций.
В зависимости от заболевания, которое подвергают лечению, с соединениями настоящего изобретения могут быть совместно введены самые разнообразные лекарственные средства. Второй активный ингредиент может включать, но этим не ограничивая, алкилирующие средства, в том числе циклофосфамид, ифосфамид, тиотепу, мелфалан, хлорэтилнитрозомочевину и бендамустин; производные платины, в том числе цисплатин, оксалиплатин, карбоплатин и сатраплатин; антимитотические средства, в том числе алкалоиды барвинка (винкристин, винорелбин и винбластин), таксаны (паклитаксел, доцетаксел), эпотилоны и ингибиторы митотических киназ, в том числе ингибиторы киназ Аврора и поло-киназ; ингибиторы топоизомеразы, в том числе антрациклины, эпидофиллотоксины, камптотецин и аналоги камптотецина; антиметаболиты, в том числе 5-фторурацил, капецитабин, цитарабин, гемцитабин, 6-меркаптопурин, 6-тиогуанин, флударабин, метотрексат и преметрексед; ингибиторы протеинкиназы, в том числе иматиниб, гефитиниб, сорафениб, сунитиниб, эрлотиниб, дазатиниб и лапатиниб; ингибиторы протеосом, в том числе бортезомиб; ингибиторы гистондеацетилазы, в том числе вальпроат и SAHA; антиангиогенные лекарственные средства, в том числе бевацизумаб; моноклональные антитела, в том числе трастузумаб, ритуксимаб, алемтузумаб, тозитумомаб, цетуксимаб, панитумумаб; конъюгаты моноклональных антител, в том числе гемтузумаб озогамицин, ибритутомаб тиуксетан; гормональные препараты, в том числе антиэстрогены (тамоксифен, ралоксифен, анастрозол, летрозол, эксеместан) антиандрогены (флутамид, бикалутамид) и аналоги или антагонисты лютеинизирующего гормона.
Применительно к комбинированной терапии, соединения настоящего изобретения могут быть введены раздельно, последовательно, одновременно, параллельно или могут быть хронологически разнесены по времени с одним или более стандартными терапевтическими средствами, такими как любое из упомянутых выше средств.
Настоящее изобретение предлагает, в предпочтительном варианте, соединение формулы (I):

(I)
или его фармацевтически приемлемую соль или его N-оксидное производное, где:
R1 представляет собой необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу;
R2 и R3 независимо выбирают из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной алкильной группы, необязательно замещенной циклоалкильной группы, необязательно замещенной алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной арильной группы и необязательно замещенной гетероарильной группы; или R2, R3 и углеродный атом, к которому они оба присоединены, взятые вместе, образуют необязательно замещенную циклоалкильную группу или необязательно замещенную гетероциклильную группу;
R4 представляет собой атом водорода, необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу;
или R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную группу; и
R5 представляет собой необязательно замещенную алкильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу.
Предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной алкильной группы, необязательно замещенной циклоалкильной группы, необязательно замещенной алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной арильной группы и необязательно замещенной гетероарильной группы; и
R4 представляет собой атом водорода, необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу;
или R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную группу.
В качестве варианта, предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной алкильной группы, необязательно замещенной циклоалкильной группы, необязательно замещенной алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной арильной группы и необязательно замещенной гетероарильной группы; и
R4 представляет собой атом водорода, необязательно замещенную алкильную группу, необязательно замещенную алкенильную группу, необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу.
Предпочтительно, чтобы R1 представлял собой необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу. Предпочтительно, чтобы R1 представлял собой замещенную арильную группу или замещенную гетероарильную группу. Более предпочтительно, чтобы R1 представлял собой замещенную арильную группу. Еще более предпочтительно, чтобы R1 представлял собой замещенную 6-членную арильную группу.
Предпочтительно, чтобы R1 представлял собой группу, представленную формулой (a):

(a)
где R1a и R1b каждый независимо выбирают из группы, состоящей из атома водорода, галогеновой группы, гидроксильной группы, цианогруппы, аминогруппы, C1-C6 алкильной группы, C1-C6 алкоксильной группы и C1-C6 алкокси-Cl-C6 алкильной группы. Предпочтительно, чтобы R1a и R1b каждый независимо выбирали из группы, состоящей из атома водорода и галогеновой группы. Более предпочтительно, чтобы R1a и R1b каждый независимо представлял собой галогеновую группу. Еще более предпочтительно, чтобы R1a и R1b каждый независимо выбирали из группы, состоящей из атома хлора и атома фтора.
Предпочтительно, чтобы R1 представлял собой группу, представленную формулой (b):
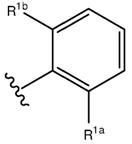
(b)
где R1a и R1b каждый независимо выбирают из группы, состоящей из атома водорода, галогеновой группы, гидроксильной группы, цианогруппы, аминогруппы, C1-C6 алкильной группы, C1-C6 алкоксильной группы и C1-C6 алкокси-Cl-C6 алкильной группы. Предпочтительно, чтобы R1a и R1b каждый независимо выбирали из группы, состоящей из атома водорода и галогеновой группы. Более предпочтительно, чтобы R1a и R1b каждый независимо представлял собой галогеновую группу. Еще более предпочтительно, чтобы R1a и R1b каждый независимо выбирали из группы, состоящей из атома хлора и атома фтора.
Предпочтительно, чтобы в формуле (a) и/или (b), R1a представлял собой атом водорода, галогеновую группу, цианогруппу, метильную группу или метоксильную группу; и R1b представлял собой галогеновую группу.
Предпочтительно, чтобы в формуле (a) и/или (b), R1a представлял собой атом водорода или галогеновую группу; и R1b представлял собой галогеновую группу.
Предпочтительно, чтобы в формуле (a) и/или (b), R1a выбирали из группы, состоящей из атома хлора и атома фтора; и R1b представлял собой атом хлора.
Предпочтительно, чтобы R1 представлял собой 2-хлорфенильную группу.
В качестве варианта, предпочтительно, чтобы R1 представлял собой 2,6-дихлорфенильную группу.
В качестве варианта, предпочтительно, чтобы R1 представлял собой 2-хлор-6-фторфенильную группу.
В качестве варианта, предпочтительно, чтобы R1 представлял собой необязательно замещенную алкильную группу или необязательно замещенную алкенильную группу. Более предпочтительно, чтобы R1 представлял собой необязательно замещенную C1-C6 алкильную группу или необязательно замещенную C1-C6 алкенильную группу. Еще более предпочтительно, чтобы R1 представлял собой необязательно замещенную C1-C3 алкильную группу или необязательно замещенную C1-C3 алкенильную группу.
Предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной алкильной группы, необязательно замещенной циклоалкильной группы, необязательно замещенной алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной арильной группы и необязательно замещенной гетероарильной группы. Более предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной C1-C6 или C1-C3 алкильной группы, необязательно замещенной C3-C6 циклоалкильной группы, необязательно замещенной C1-C6 или C1-C3 алкоксильной группы, необязательно замещенной аминогруппы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-7-членной гетероарильной группы. Еще более предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной метильной группы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-7-членной гетероарильной группы. Еще более предпочтительно, чтобы R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, метильной группы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-7-членной гетероарильной группы.
Предпочтительно, чтобы R2 и R3 каждый независимо представлял собой атом водорода.
В качестве варианта, предпочтительно, чтобы R2 и/или R3 представляли собой атом дейтерия.
В качестве варианта, предпочтительно, чтобы R2 выбирали из группы, состоящей из необязательно замещенной алкильной группы и необязательно замещенной C3-C6 циклоалкильной группы; и R3 выбирали из группы, состоящей из атома водорода, необязательно замещенной алкильной группы и необязательно замещенной C3-C6 циклоалкильной группы. Более предпочтительно, чтобы R2 представлял собой необязательно замещенную метильную группу; и R3 выбирали из группы, состоящей из атома водорода и метильной группы. Еще более предпочтительно, чтобы R2 представлял собой метильную группу; и R3 представлял собой атом водорода.
В качестве варианта, предпочтительно, чтобы R2 выбирали из группы, состоящей из необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-7- или 5-6-членной гетероарильной группы; и R3 представлял собой атом водорода. Более предпочтительно, чтобы R2 выбирали из группы, состоящей из необязательно замещенной пирролильной группы, необязательно замещенной пиразолильной группы и необязательно замещенной имидазолильной группы; и R3 представлял собой атом водорода.
В качестве варианта, предпочтительно, чтобы R2 выбирали из группы, состоящей из необязательно замещенной фенильной группы и необязательно замещенной пиридинильной группы; и R3 представлял собой атом водорода. Более предпочтительно, чтобы R2 выбирали из группы, состоящей из фенильной группы и пиридинильной группы; и R3 представлял собой атом водорода.
В качестве варианта, R1 выбирают из группы, состоящей из C2-C3 алкенильной группы и метильной группы, замещенной с помощью циклопропанильной группы; и R2 выбирают из группы, состоящей из необязательно замещенной фенильной группы и необязательно замещенной пиридинильной группы; и R3 представляет собой атом водорода.
Предпочтительно, чтобы R4 представлял собой атом водорода, необязательно замещенную C1-C6 алкильную группу, необязательно замещенную C1-C6 алкенильную группу, необязательно замещенную 6-членную арильную группу или необязательно замещенную пяти- семи-членную гетероарильную группу.
Предпочтительно, чтобы R4 представлял собой необязательно замещенную C1-C6 алкильную группу. Более предпочтительно, чтобы R4 представлял собой необязательно замещенную C1-C3 алкильную группу. Еще более предпочтительно, чтобы R4 представлял собой метильную группу.
В качестве варианта, предпочтительно, чтобы R4 представлял собой необязательно замещенную гетероарильную группу. Предпочтительно, чтобы R4 представлял собой необязательно замещенную 5-7- или 5-6-членную гетероарильную группу. Более предпочтительно, чтобы R4 выбирали из группы, состоящей из необязательно замещенной пиразолильной группы, необязательно замещенной имидазолильной группы, необязательно замещенной триазолильной группы, необязательно замещенной оксазолильной группы, необязательно замещенной тиазолильной группы и необязательно замещенной пиридинильной группы.
В качестве варианта, R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную группу. Предпочтительно, чтобы R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 4-7-членную, 5-7-членную или 5-6-членную гетероциклильную группу. Еще более предпочтительно, чтобы R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 5-членную гетероциклильную группу.
Предпочтительно, чтобы R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную гетероциклоалкильную группу. Более предпочтительно, чтобы R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 4-7-членную или 5-7-членную или 5-6-членную гетероциклоалкильную группу. Еще более предпочтительно, чтобы R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 5-членную гетероциклоалкильную группу.
Наиболее предпочтительно, чтобы гетероциклильная группа представляла собой пирролидинильную группу, то есть R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную пирролидинильную группу.
В качестве варианта, когда R4 и R2 или R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную/гетероциклоалкильную группу, R1 представляет собой необязательно замещенную алкильную группу или необязательно замещенную алкенильную группу. Предпочтительно, чтобы R1 представлял собой необязательно замещенную алкенильную группу, более предпочтительно, чтобы R1 представлял собой необязательно замещенную C2-C3 алкенильную группу, наиболее предпочтительно, чтобы R1 представлял собой  .
.
Гетероциклильная/гетероциклоалкильная группа может быть замещена с помощью одного или более заместителей, выбранных из группы, состоящей из гидроксильной группы, C1-C3 алкоксильной группы, необязательно замещенной аминогруппы, оксогруппы и необязательно замещенной C1-C3 алкильной группы. В одном варианте осуществления, гетероциклильная/гетероциклоалкильная группа замещена с помощью одного или более заместителей, выбранных из группы, состоящей из оксогруппы и необязательно замещенной C1-C3 алкильной группы.
Предпочтительно, чтобы необязательно замещенная гетероциклоалкильная группа представляла собой замещенную пирролидинильную группу.
Предпочтительно, чтобы R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную гетероциклильную/гетероциклоалкильную группу; и R3 выбирали из группы, состоящей из атома водорода и C1-C3 алкильной группы. Более предпочтительно, чтобы R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 4-7-членную или 5-7-членную или 5-6-членную гетероциклильную/гетероцикло-алкильную группу; и R3 выбирали из группы, состоящей из атома водорода и метильной группы. Еще более предпочтительно, чтобы R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали необязательно замещенную 5-членную гетероциклильную/гетероциклоалкильную группу; и R3 представлял собой атом водорода. Наиболее предпочтительно, чтобы необязательно замещенная гетероциклильная/гетероциклоалкильная группа представляла собой замещенную пирролидинильную группу, то есть R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образовывали замещенную пирролидинильную группу; и R3 представлял собой атом водорода.
Предпочтительно, чтобы, когда R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную гетероциклильную/гетероциклоалкильную группу, R1 представлял собой необязательно замещенную алкильную группу или необязательно замещенную алкенильную группу; и R3 представлял собой атом водорода.
Предпочтительно, чтобы R5 представлял собой необязательно замещенную C1-C6 или C1-C3 алкильную группу, необязательно замещенную 6-членную арильную группу или необязательно замещенную 5-7-членную гетероарильную группу.
Предпочтительно, чтобы R5 представлял собой необязательно замещенную арильную группу или необязательно замещенную гетероарильную группу. Более предпочтительно, чтобы R5 представлял собой необязательно замещенную 6-членную арильную группу или необязательно замещенную 5-7-членную гетероарильную группу. Еще более предпочтительно, чтобы R5 представлял собой необязательно замещенную 6-членную арильную группу или необязательно замещенную 6-членную гетероарильную группу. Еще более предпочтительно, чтобы R5 представлял собой замещенную фенильную группу или замещенную пиридинильную группу.
Предпочтительно, чтобы R5 представлял собой группу, представленную формулой (c):

(c)
где Z представляет собой атом азота или необязательно замещенную метиновую группу;
R5a и R5b независимо выбирают из группы, состоящей из атома водорода, галогеновой группы, необязательно замещенной C1-C6 алкильной группы, необязательно замещенной C1-C6 нитрильной группы, необязательно замещенной аминогруппы, необязательно замещенной C1-C6 алкоксильной группы, необязательно замещенной сульфанильной группы, необязательно замещенной сульфонильной группы, необязательно замещенной сульфоксиминильной группы и необязательно замещенной 4-7-членной азотсодержащей гетероциклильной группы;
где необязательно замещенная 4-7-членная азотсодержащая гетероциклильная группа необязательно замещена с помощью одного или более заместителей, выбранных из группы, состоящей из галогеновой группы, необязательно замещенной C1-C6 алкильной группы, оксогруппы, гидроксильной группы, необязательно замещенной аминогруппы и группы =N-R5g;
или, в формуле (c), R5a и R5b существуют на смежных кольцевых атомах, и R5a и R5b и кольцевые атомы, к которым они присоединены, могут образовывать, взятые вместе, 3-7-членную циклоалкильную группу или 3-7-членную гетероциклильную группу, где один или два из кольцевых атомов, из которых состоит трет-семи-членная гетероциклильная группа, необязательно независимо заменены на атом кислорода, атом азота, группу -N(R5c)-, сульфинильную группу, сульфонильную группу и сульфоксиминильную группу, где 3-7-членная циклоалкильная или 3-7-членная гетероциклильная группа могут быть замещены с помощью одного или более заместителей, выбранных из группы, состоящей из галогеновой группы и C1-C6 алкильной группы;
или R5a и R5b и кольцевые атомы, к которым они присоединены, могут образовывать, взятые вместе, спироциклическую группу или бициклическую группу, образованную из 5-7-членного алифатического кольца и любого другого 3-7-членного алифатического кольца, в котором одна или две или более метиленовых групп, из которых состоит спироциклическая группа или бициклическая группа, могут быть каждая независимо заменены на атом кислорода, aтом серы, сульфинильную группу, сульфонильную группу, сульфоксиминильную группу, оксогруппу или группу -N(R5d)-, и спироциклическая группа или бициклическая группа могут быть каждая независимо замещена с помощью заместителя, выбранного из группы, состоящей из галогеновой группы, гидроксильной группы или C1-C6 алкильной группы; где
R5c, R5d и R5g каждый независимо представляет собой атом водорода или C1-C6 алкильную группу, необязательно замещенную с помощью заместителя, выбранного из группы, состоящей из галогеновой группы, гидроксильной группы, цианогруппы, оксогруппы, C1-C6 алкильной группы, C1-C6 алкоксильной группы, an аминогруппы, замещенной аминогруппы и азотсодержащей гетероциклильной группы.
Более предпочтительно, чтобы R5 представлял собой группу, представленную формулой (d):

(d)
R5a и R5b независимо выбирают из группы, состоящей из атома водорода, галогеновой группы, необязательно замещенной C1-C6 алкильной группы, необязательно замещенной C1-C6 нитрильной группы, необязательно замещенный аминогруппы, необязательно замещенной C1-C6 алкоксильной группы, необязательно замещенной сульфанильной группы, необязательно замещенной сульфонильной группы, необязательно замещенной сульфоксиминильной группы и необязательно замещенной 4-7-членной азотсодержащей гетероциклильной группы;
где необязательно замещенная четерех-семи-членная азотсодержащая гетероциклильная группа необязательно замещена с помощью одного или более заместителей, выбранных из группы, состоящей из галогеновой группы, необязательно замещенной C1-C6 алкильной группы, оксогруппы, гидроксильной группы, группы =N-R5g и группы -Q-N(R5e)R5e’;
R5e, R5e’ и R5g каждый независимо представляет собой атом водорода или C1-C6 алкильную группу, или R5e и R5e’ и атом азота, к которому они присоединены, взятые вместе, могут образовывать необязательно замещенную 6-членную гетероциклильную группу; и
Q представляет собой одинарную связь или C1-C3 алкильную группу.
Предпочтительно, чтобы 4-7-членная азотсодержащая гетероциклильная группа представляла собой 4-7-членную азотсодержащую гетероциклоалкильную группу.
Более предпочтительно, чтобы 4-7-членную азотсодержащую гетероциклильную группу выбирали из группы, состоящей из азетидинильной группы, пирролидинильной группы, пиперидинильной группы, морфолинильной группы, тиоморфолинильной группы, тиоморфолин-S,S-диоксидной группы, тиоморфолин-S-оксо-S-иминилсульфоксиминильной группы и гомопиперазинильной группы, каждая из которых может быть необязательно замещенной.
Предпочтительно, чтобы R5a представлял собой C1-C3 алкоксильную группу, замещенную с помощью аминогруппы, или R5a представлял собой C1-C3 алкильную группу, замещенный замещенную с помощью необязательно замещенной 5-7-членной гетероциклильной группы, или R5a представлял собой 5-7-членную азотсодержащую гетероциклильную группу, необязательно замещенную с помощью одного или более заместителей, выбранных из группы, состоящей из C1-C3 алкильной группы и C1-C3 алкильной группы, замещенный с помощью одного или более заместителей, выбранных из группы, состоящей из гидроксильной группы, карбоксильной группы, оксогруппы и аминогруппы; и
R5b представляет собой атом водорода, галогеновую группу, C1-C3 нитрильную группу, C1-C3 алкоксильную группу или C1-C3 алкильную группу, замещенную с помощью заместителя, выбранного из группы, состоящей из аминогруппы и гидроксильной группы.
Предпочтительно, чтобы R5a представлял собой азотсодержащую гетероциклильную группу, необязательно замещенную с помощью метильной группы; и
R5b представляет собой атом водорода, метильную группу, метоксильную группу или метильную группу, замещенную с помощью гидроксильной группы.
В качестве варианта, предпочтительно, чтобы R5a и/или R5b независимо выбирали из группы, состоящей из необязательно замещенной сульфанильной группы, необязательно замещенной сульфонильной группы, необязательно замещенной сульфоксиминильной группы и необязательно замещенной тиоморфолин-сульфоксиминильной группы.
Более предпочтительно, чтобы R5a представлял собой необязательно замещенную сульфоксиминильную группу.
Предпочтительно, чтобы необязательно замещенная сульфоксиминильная группа представляла собой группу формулы (g):

(g)
где R5h выбирают из группы, состоящей из атома водорода, необязательно замещенной C1-C6 или C1-C3 алкильной группы, необязательно замещенной цианогруппы, необязательно замещенной ацильной группы, необязательно замещенной C1-C6 или C1-C3 алкоксикарбонильной группы и необязательно замещенной гетероциклильной группы;
R5i выбирают из группы, состоящей из необязательно замещенной C1-C6 или C1-C3 алкильной группы и необязательно замещенной гетероциклильной группы;
или R5h и R5i и кольцевые атомы, к которым они присоединены, могут образовывать, взятые вместе, 5-7-членную гетероциклильную группу, где 5-7-членная гетероциклильная группа может быть замещена с помощью одного или более заместителей, выбранных из группы, состоящей из гидроксильной группы, C1-C6 или C1-C3 алкильной группы, необязательно замещенной аминогруппы и оксогруппы.
Предпочтительно, чтобы R5h и/или R5i представлял собой независимо C1-C6 или C1-C3 алкильную группу, необязательно замещенную с помощью группы, выбранной из группы, состоящей из гидроксильной группы и необязательно замещенной аминогруппы.
Предпочтительно, чтобы R5h выбирали из группы, состоящей из атома водорода, необязательно замещенной C1-C6 или C1-C3 алкильной группы, необязательно замещенной цианогруппы, необязательно замещенной ацильной группы, необязательно замещенной C1-C6 или C1-C3 алкоксикарбонильной группы и необязательно замещенной гетероциклильной группы; и
R5i представляет собой метильную группу.
В качестве варианта, предпочтительно, чтобы R5i выбирали из группы, состоящей из необязательно замещенной C1-C6 или C1-C3 алкильной группы и необязательно замещенной гетероциклильной группы; и R5h представлял собой метильную группу.
В качестве варианта, предпочтительно, чтобы R5 представлял собой группу, представленную формулой (f):
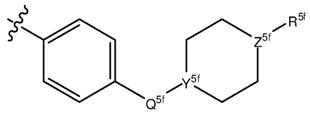
(f)
где R5f выбирают из группы, состоящей из атома водорода и необязательно замещенной C1-C6 алкильной группы; Q5f выбирают из группы, состоящей из одинарной связи и C1-C3 алкильной группы; Y5f выбирают из группы, состоящей из атома азота и группы C-H (то есть углеродного атома, соединенного с атомом водорода); и Z5f выбирают из группы, состоящей из атома азота и атома кислорода. Более предпочтительно, чтобы R5f выбирали из группы, состоящей из атома водорода и необязательно замещенной C1-C4 или C1-C3 алкильной группы; Q5f выбирали из группы, состоящей из одинарной связи и метиленовой группы (то есть группы -C(H)2-); Y5f выбирали из группы, состоящей из атома азота и группы C-H (то есть углеродного атома, соединенного с атомом водорода); и Z5f выбирали из группы, состоящей из атома азота и атома кислорода. Еще более предпочтительно, чтобы R5f выбирали из группы, состоящей из атома водорода, метильной группы и C1-C3 алкильной группы, необязательно замещенной с помощью группы, выбранной из группы, состоящей из гидроксильной группы и C1-C3 алкильной группы; Q5f выбирали из группы, состоящей из одинарной связи и метиленовой группы (то есть группы -C(H)2-); Y5f выбирали из группы, состоящей из атома азота и группы C-H (то есть углеродного атома, соединенного с атомом водорода); и Z5f выбирали из группы, состоящей из атома азота и атома кислорода. Наиболее предпочтительно, чтобы R5f выбирали из группы, состоящей из атома водорода, метильной группы и C1-C3 алкильной группы, необязательно замещенной с помощью группы, выбранной из группы, состоящей из гидроксильной группы и C1-C3 алкильной группы; Q5f выбирали из группы, состоящей из одинарной связи и метиленовой группы (то есть группы -C(H)2-); Y5f представлял собой атом азота; и Z5f выбирали из группы, состоящей из атома азота и атома кислорода.
В качестве варианта, предпочтительно, чтобы R5 представлял собой группу, представленную формулой (e):

(e)
где R5g выбирают из группы, состоящей из атома водорода и необязательно замещенной C1-C3 алкильной группы. Более предпочтительно, чтобы R5g выбирали из группы, состоящей из атома водорода и метильной группы.
Предпочтительно, в соединении формулы (I), чтобы R1 представлял собой группу, представленную формулой (a), определенной выше; и/или R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, необязательно замещенной метильной группы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-7-членной гетероарильной группы; и/или R4 представлял собой атом водорода, необязательно замещенную C1-C6 алкильную группу, необязательно замещенную C1-C6 алкенильную группу, необязательно замещенную 6-членную арильную группу или необязательно замещенную 5-7-членную гетероарильную группу; и/или R5 представлял собой группу, представленную формулой (c), определенной выше.
Более предпочтительно, чтобы R1 представлял собой группу, представленную формулой (a), определенной выше; и/или R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, метильной группы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-6-членной гетероарильной группы; и/или R4 представлял собой атом водорода, необязательно замещенную C1-C6 алкильную группу, необязательно замещенную C1-C6 алкенильную группу, необязательно замещенную 6-членную арильную группу или необязательно замещенную 5-7-членную гетероарильную группу; и/или R5 представлял собой группу, представленную формулой (d) или формулой (f), определенными выше.
Предпочтительно, чтобы R1 представлял собой группу, представленную формулой (b), определенной выше; и/или R2 и R3 независимо выбирали из группы, состоящей из атома водорода, атома дейтерия, метильной группы, необязательно замещенной 6-членной арильной группы и необязательно замещенной 5-6-членной гетероарильной группы; и/или R4 представлял собой атом водорода или метильную группу; и/или R5 представлял собой группу, представленную формулой (d) или формулой (f), определенными выше.
Предпочтительно, чтобы R1 представлял собой 2-хлорфенильную группу, 2,6-дихлорфенильную группу или 2-хлор-6-фторфенильную группу, и/или R2 выбирали из группы, состоящей из атома водорода, атома дейтерия, метильной группы и фенильной группы; и/или R3 выбирали из группы, состоящей из атома водорода и атома дейтерия; и/или R5 представлял собой группу, представленную формулой (d) или формулой (f), определенными выше.
В качестве варианта, R1 представляет собой необязательно замещенную C1-C3 алкильную группу или необязательно замещенную C1-C3 алкенильную группу; и/или R4 и R2 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют необязательно замещенную 4-7-членную или 5-7-членную или 5-6-членную гетероциклильную группу; и/или R3 представляет собой атом водорода; и/или R5 представляет собой группу, представленную формулой (f), определенной выше.
Предпочтительно, чтобы соединение формулы (I) выбирали из следующих соединений:
(1) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(2) 3-(2,6-дихлорфенил)-1-метил-7-((4-(2-(метиламино)-этокси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(3) 3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(4) 2-(4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)уксусной кислота гидрохлорид;
(5) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)метил)-4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(6) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(7) 3-(2,6-дихлорфенил)-7-((3-фтор-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(8) 3-(2,6-дихлорфенил)-1-(4-метоксибензил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(9) 3-(2,6-дихлорфенил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(10) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(11) 3-(2,6-дихлорфенил)-7-((3-циано-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(12) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил-метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(13) 7-((4-(4-(2-аминоацетил)пиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(14) 3-(2,6-дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-изохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(15) 3-(2,6-дихлорфенил)-2,2-дидейтеро-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(16) (R)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(17) (S)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(18) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(19) (R)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(20) (S)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(21) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(22) 3-(2,6-дихлорфенил)-1-метил-7-((2-метил-1,2,3,4-тетра-гидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(23) (рац)-3-(2,6-дихлорфенил)-1-метил-2-фенил-7-((1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(24) 3-(2-хлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(25) 3-(2-хлор-6-фторфенил)-1-метил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(26) 3-(2,6-дихлорфенил)-1,2-диметил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(27) 3-(2,6-дихлорфенил)-1-метил-7-((4-(морфолинометил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(28) 6-(2,6-дихлорфенил)-2-((4-(пиперазин-1-ил)фенил)-амино)-6a,7,8,9-тетрагидропиримидо[5,4-e]пирроло[1,2-a]-пиримидин-5(6H)-он;
(29) 3-(2,6-дихлорфенил)-1-метил-7-((4-((4-метилпиперазин-1-ил)метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он; и
(30) (рац)-3-(2,6-дихлорфенил)-1-метил-7-((4-(S-метил-сульфонимидоил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он.
Предлагается, предпочтительно, соединение формулы (I) или его фармацевтически приемлемая соль или его N-оксидное производное и, по меньшей мере, один фармацевтически приемлемый носитель.
Подходящие фармацевтически приемлемые носители хорошо известны специалисту в этой области, такие как, например, жиры, вода, физиологический раствор, спирт (например, этанол), глицерин, полиолы, водный раствор глюкозы, наполнитель, разрыхлитель, связующее вещество, скользящее вещество, увлажняющее вещество, стабилизатор, эмульгатор, диспергирующее вещество, консервант, подсластитель, окрашивающее вещество, корригирующее вкус вещество или ароматизатор, концентрирующее вещество, разбавитель, буферное вещество, растворитель или солюбилизирующее вещество, химический реагент, обеспечивающий длительное хранение лекарственного препарата, соль для изменения осмотического давления, вещество для нанесения покрытия или антиоксидант, сахариды, такие как лактоза или глюкоза; кукурузный, пшеничный или рисовый крахмал; жирные кислоты, такие как стеариновая кислота; неорганические соли, такие как алюминат метасиликата магния или безводный фосфат кальция; синтетические полимеры, такие как поливинилпирролидон или полиалкиленгликоль; спирты, такие как стеариловый спирт или бензиловый спирт; синтетические производные целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза, этилцеллюлоза или гидроксипропилметил-целлюлоза; и другие традиционно используемые добавки, такие как желатин, тальк, растительное масло и гуммиарабик.
Предлагается, предпочтительно, фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемая соль или его N-оксидное производное и, по меньшей мере, один фармацевтически приемлемый носитель.
Предлагается, предпочтительно, фармацевтическая композиция, включающая соединение формулы (I) и одно или более дополнительных фармацевтически активных средств.
Предлагается, предпочтительно, соединение формулы (I) или его фармацевтически приемлемая соль или его N-оксидное производное или фармацевтическая композиция, включающая соединение формулы (I), для применения в терапии.
Предлагается, предпочтительно, соединение формулы (I) для применения в качестве лекарственного препарата.
Предлагается, предпочтительно, соединение формулы (I) для применения при лечении или профилактики рака.
Предлагается, предпочтительно, соединение формулы (I) или его фармацевтически приемлемая соль или его N-оксидное производное или фармацевтическая композиция, включающая соединение формулы (I), для применения в качестве лекарственного препарата и/или для применения при лечении или профилактики рака.
Предлагается, предпочтительно, применение соединения формулы (I) для производства лекарственного препарата для лечения или профилактики рака.
Предлагается, предпочтительно, способ лечения или профилактики рака у человека или животного, включающий введение пациенту, если он в этом нуждается, эффективного количества соединения формулы (I) или фармацевтической композиции, включающей соединение формулы (I).
Предпочтительно, чтобы соединения настоящего изобретения имели величину IC50 в отношении Wee-1 киназы от приблизительно 1 нM до приблизительно 1000 нM, более предпочтительно, от приблизительно 1 нM до приблизительно 500 нM, или от приблизительно 1 нM до приблизительно 300 нM, или от приблизительно 1 нM до приблизительно 100 нM, или от приблизительно 1 нM до приблизительно 50 нM, или от приблизительно 1 нM до приблизительно 30 нM, или от приблизительно 1 нM до приблизительно 15 нM, или от приблизительно 1 нM до приблизительно 10 нM, наиболее предпочтительно, менее чем 10 нM. Метод определения величины IC50 для соединения в отношении Wee-1 киназы описан ниже (смотрите примеры).
При представлении элементов настоящего изобретения или его предпочтительного варианта осуществления (вариантов осуществления), предполагается, что форма единственного числа обозначает наличие одного или более элементов. Предполагается, что термины "состоящий", "включающий" и "имеющий" являются включающе-отличающими и означают, что помимо перечисленных элементов могут существовать дополнительные элементы.
Далее приводится подробное описание изобретения с помощью примеров и иллюстраций, но это не означает, что они ограничивают объем прилагаемых пунктов формулы изобретения. Для любого специалиста в этой области является очевидным, что может существовать множество различных вариантов приведенных в изобретении предпочтительных на настоящий момент вариантов осуществления, и все они входят в объем прилагаемых пунктов формулы изобретения и их эквивалентов.
Далее настоящее изобретение и иллюстрируется с помощью неограничивающих примеров.
ПРИМЕРЫ
Далее настоящее изобретение описывается с помощью различных примеров.
Соединения примеров 1-185 были синтезированы методами, описанными далее. Величины IC50 в отношении Wee-1 и другие значения определяли, как описано ниже, и они представлены в следующей таблице.
Таблица 1
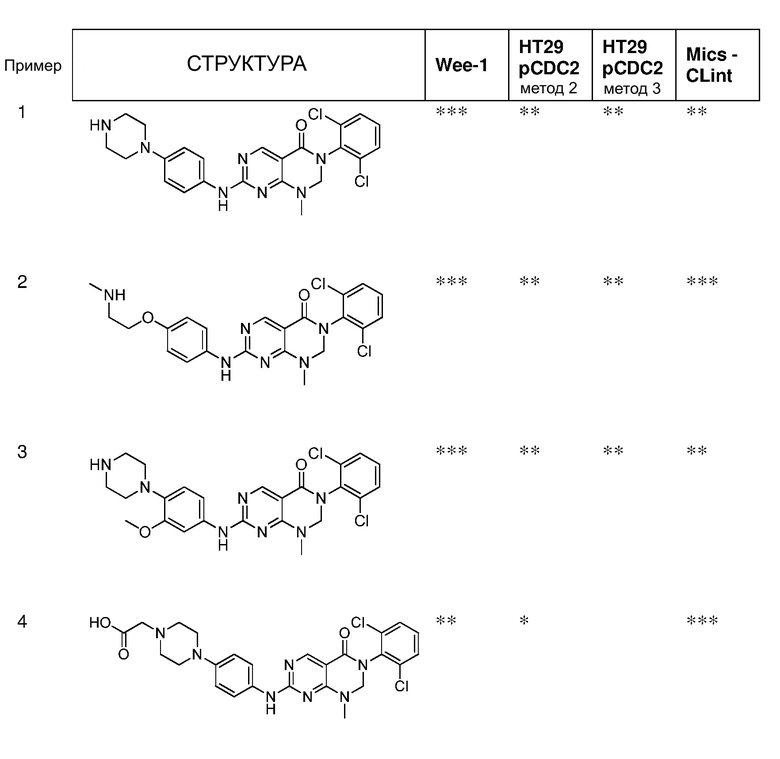



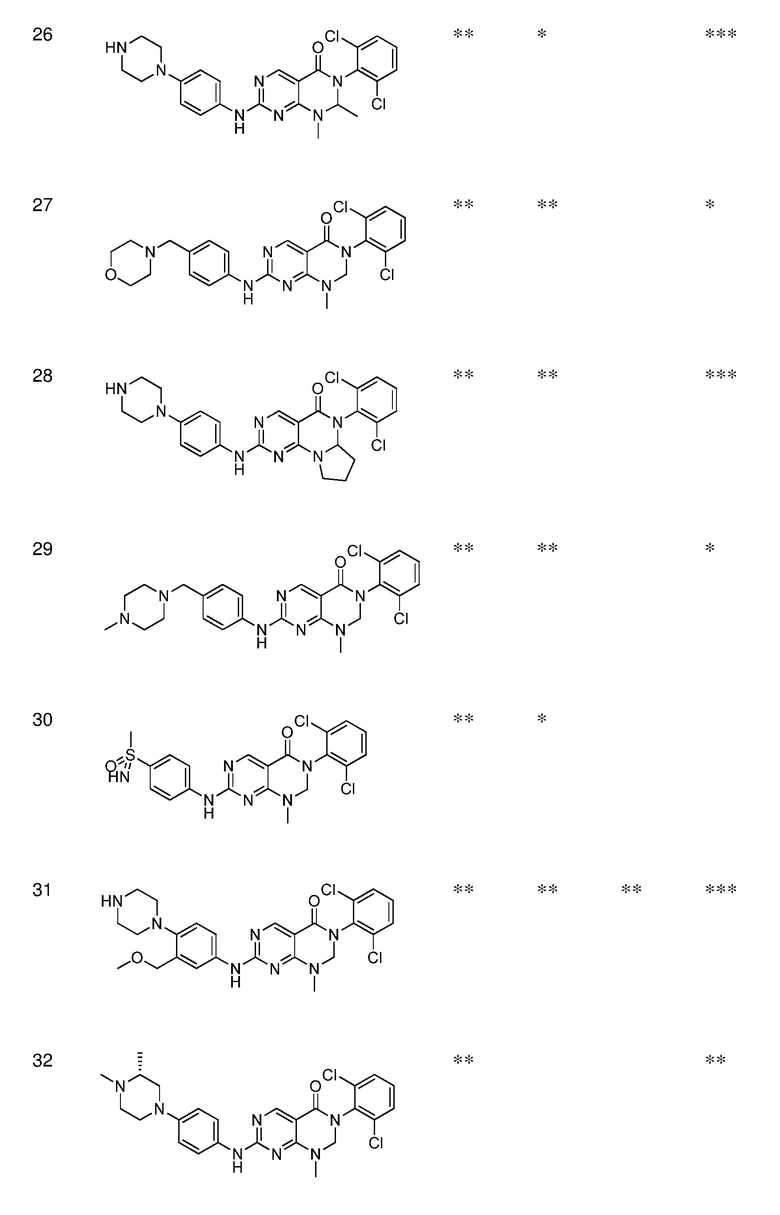
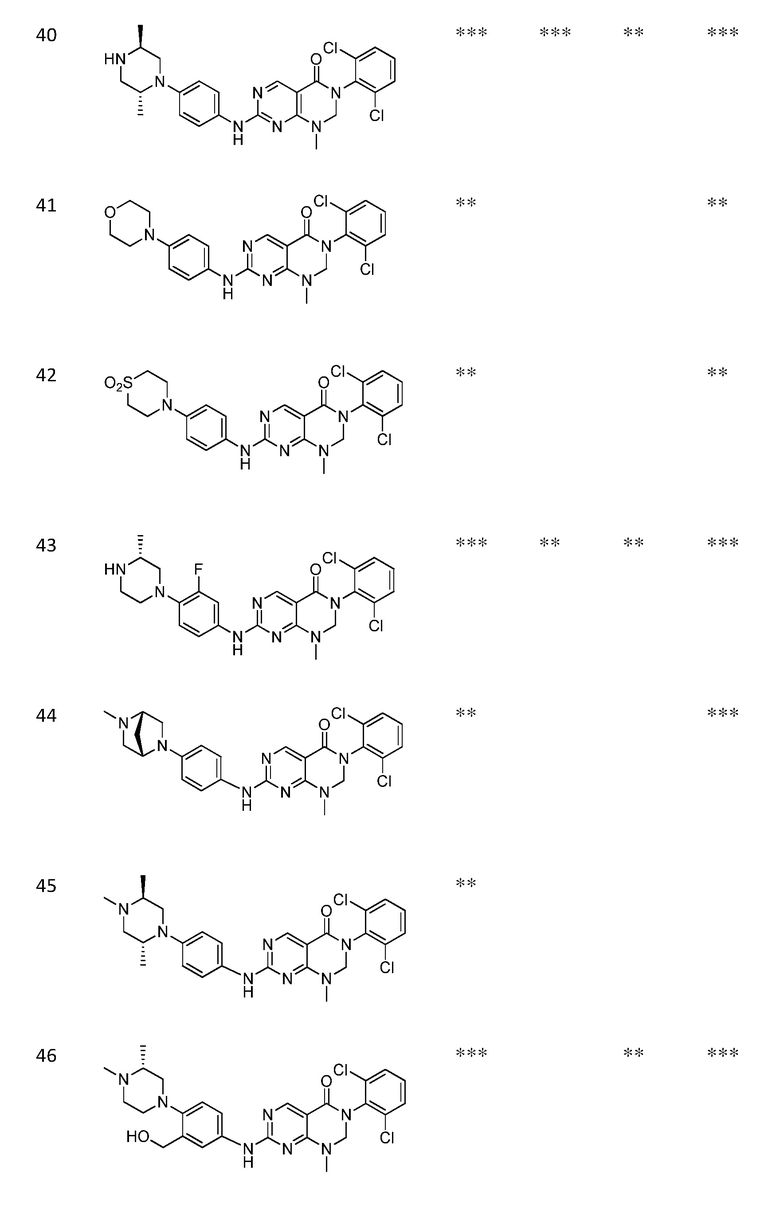
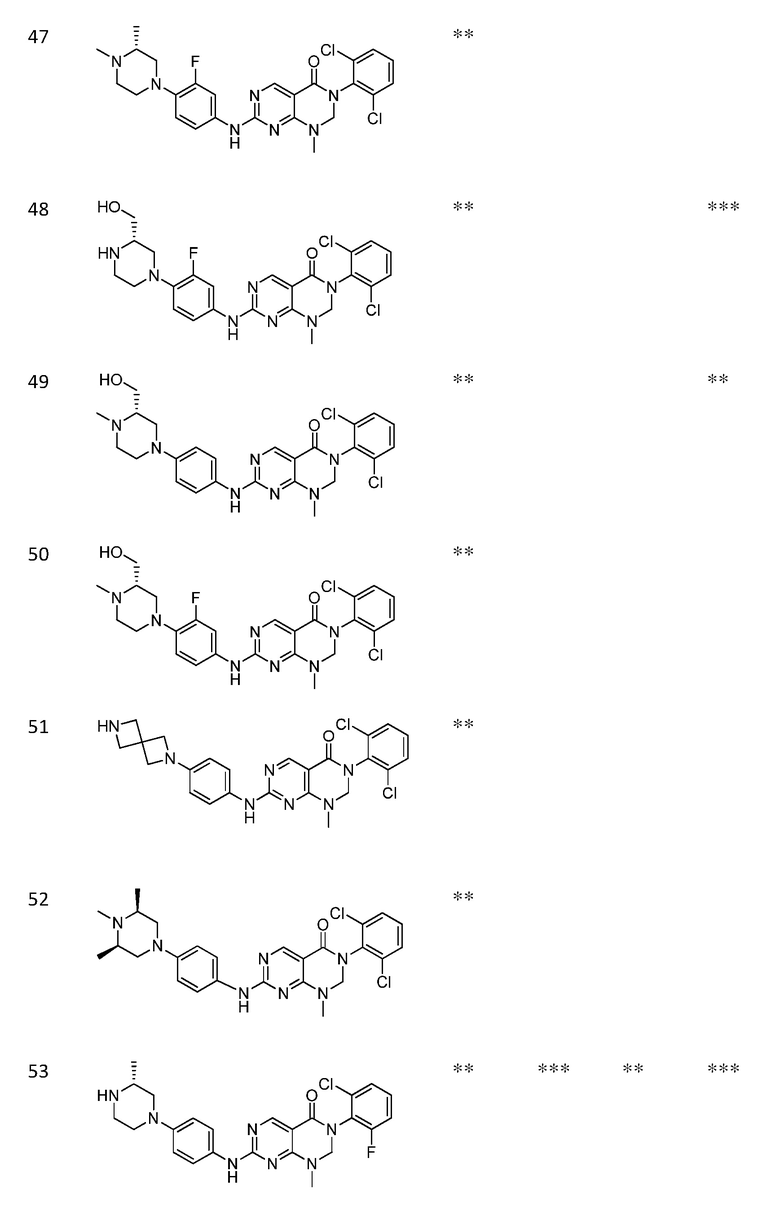
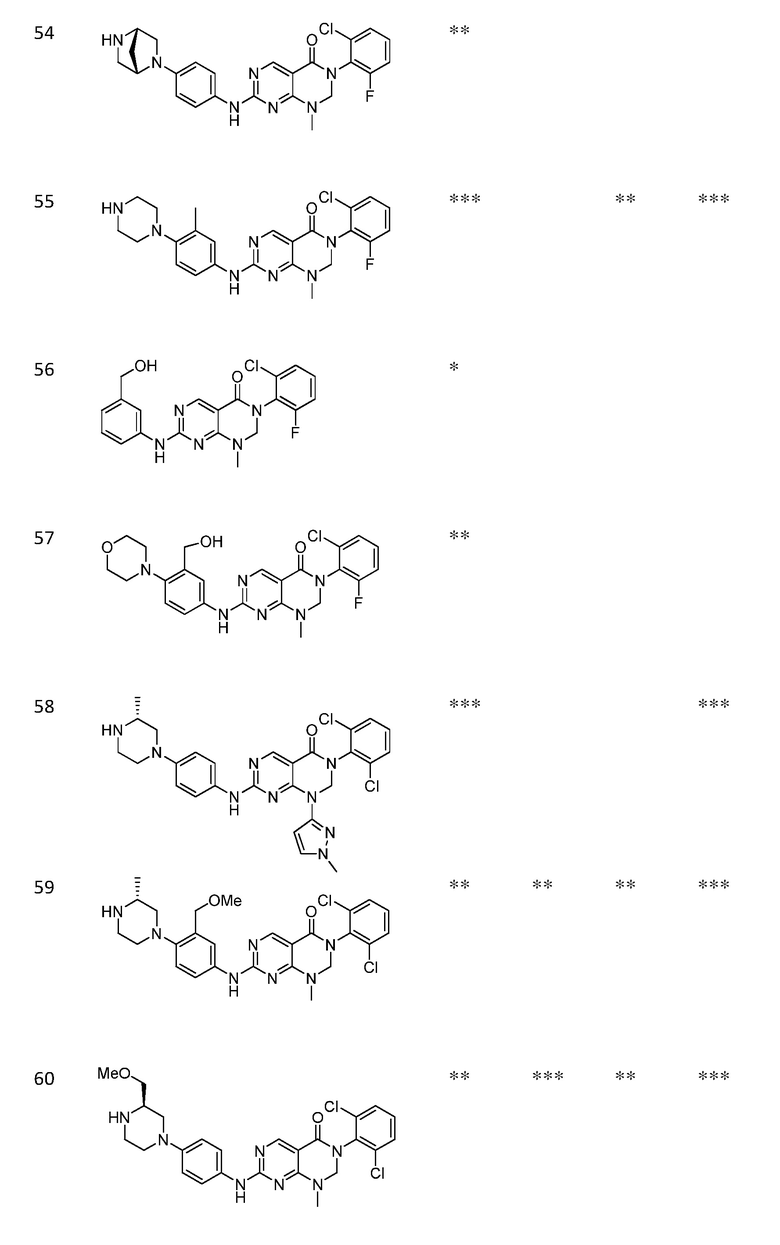
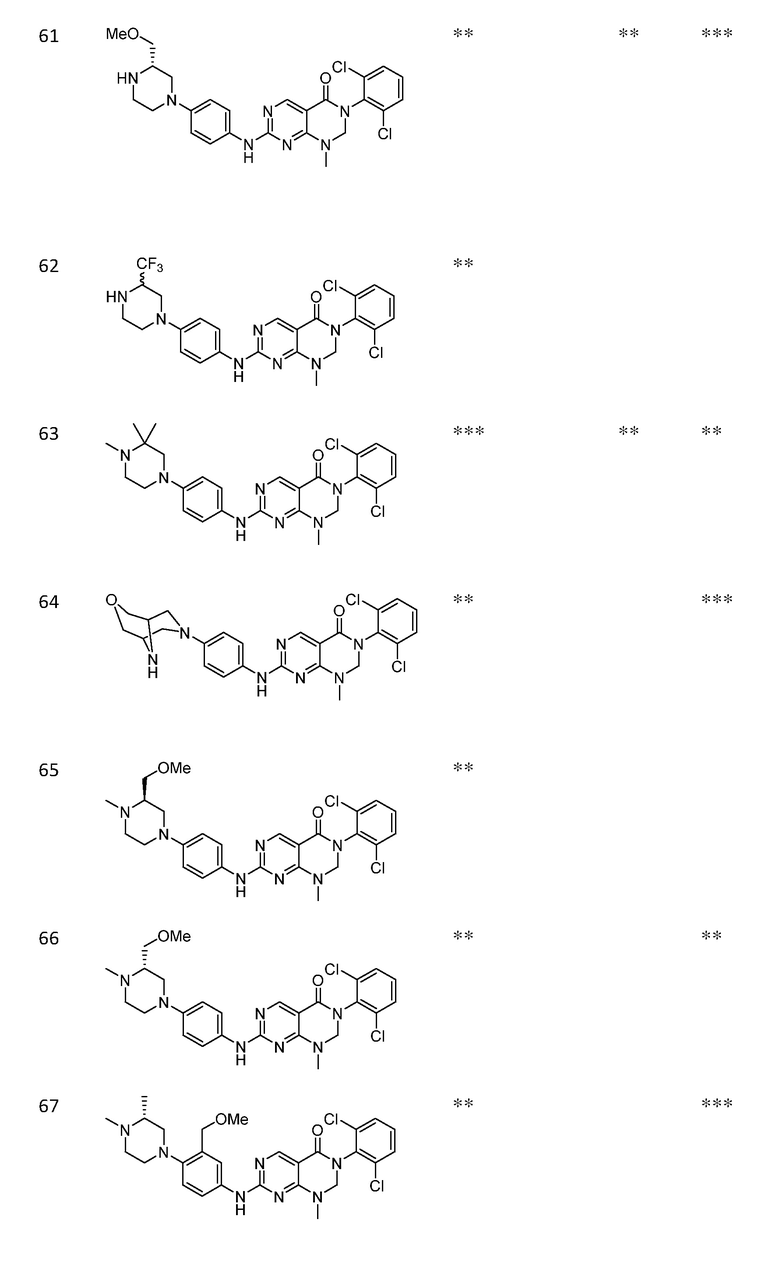


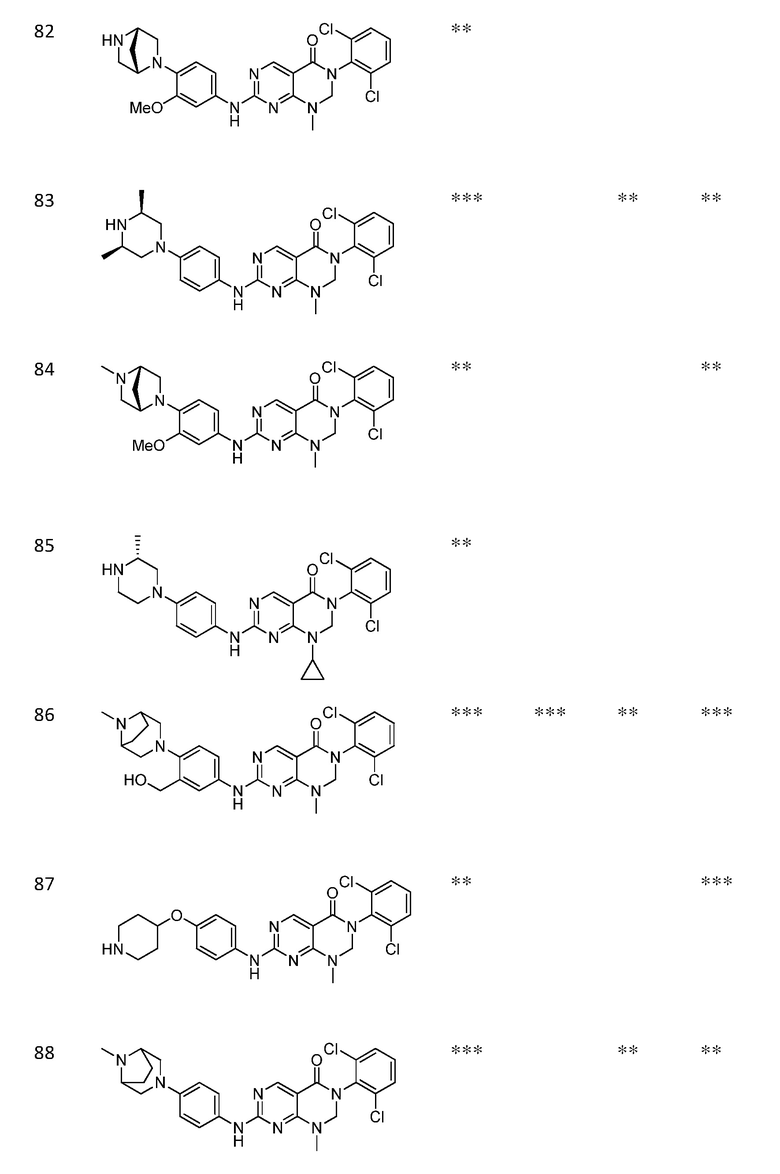

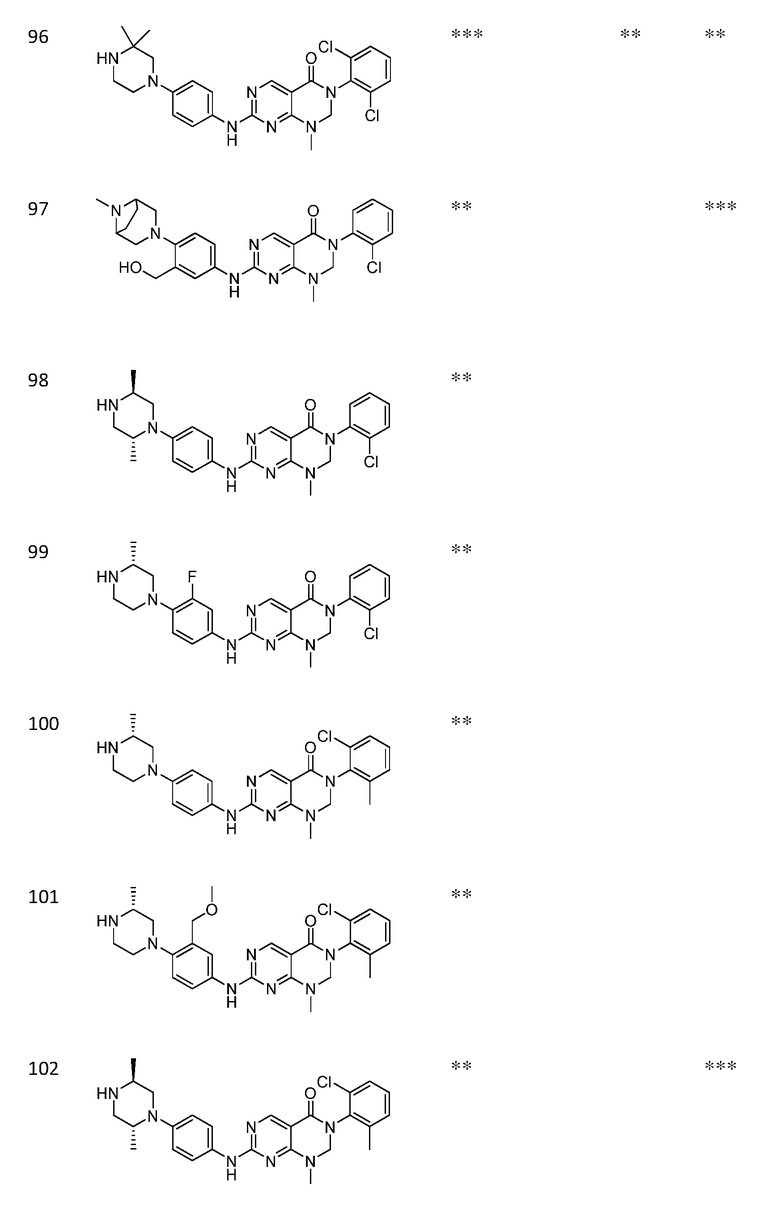

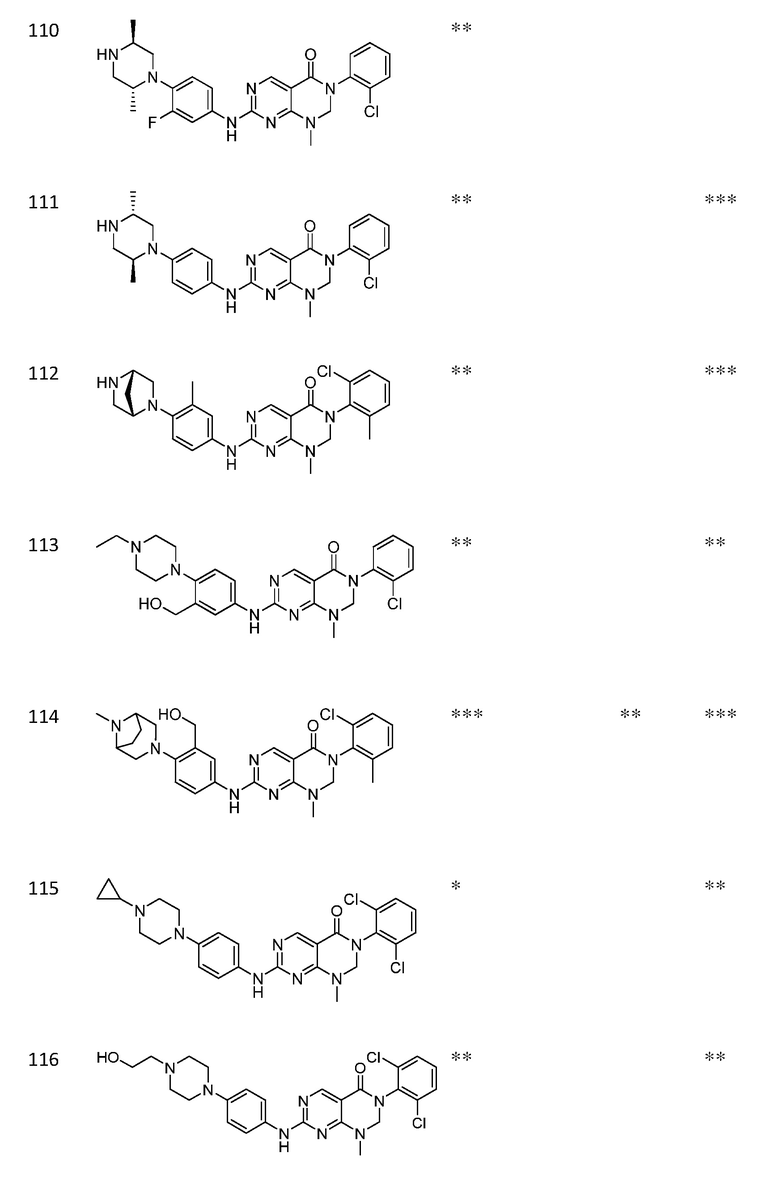
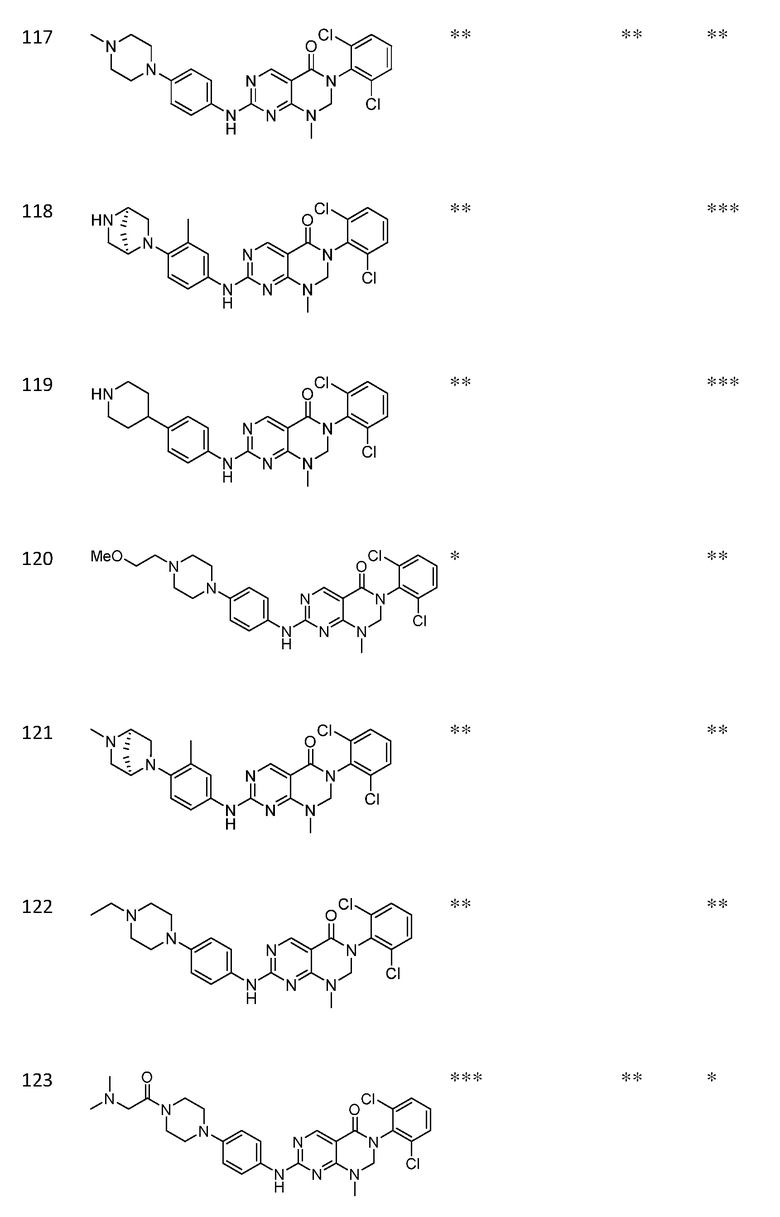
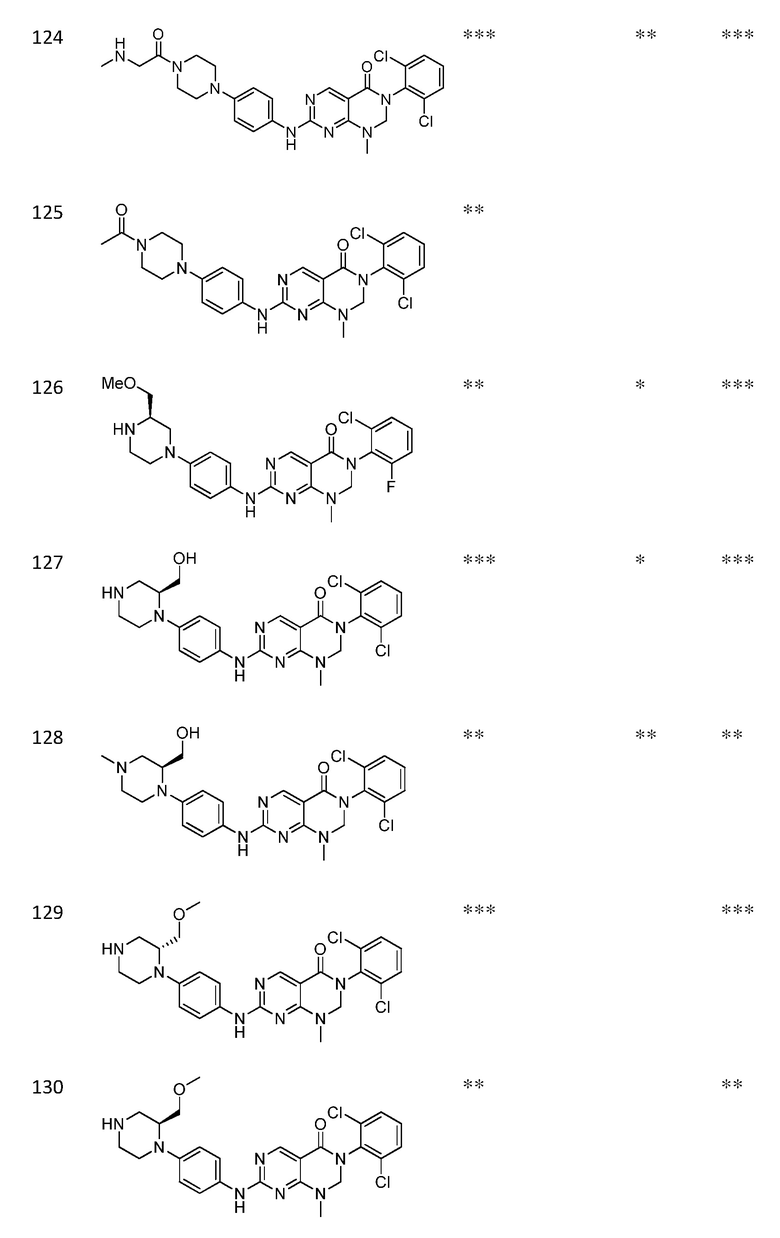


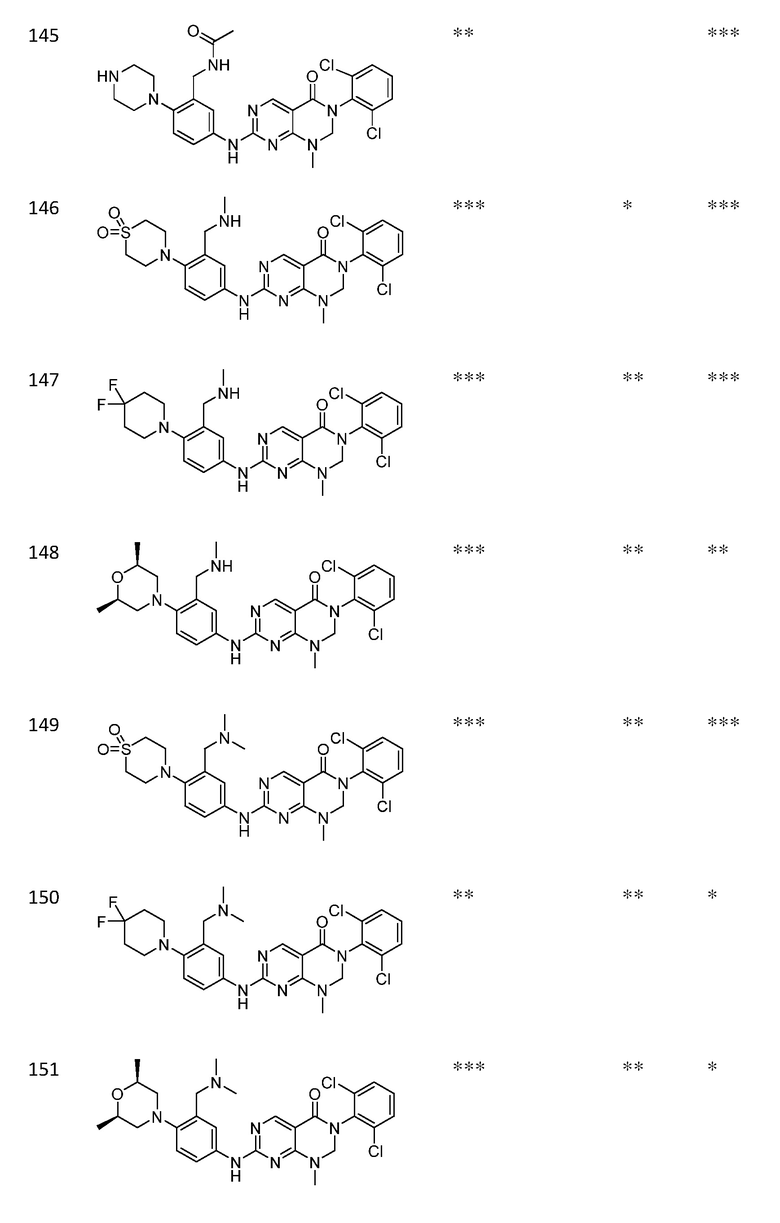

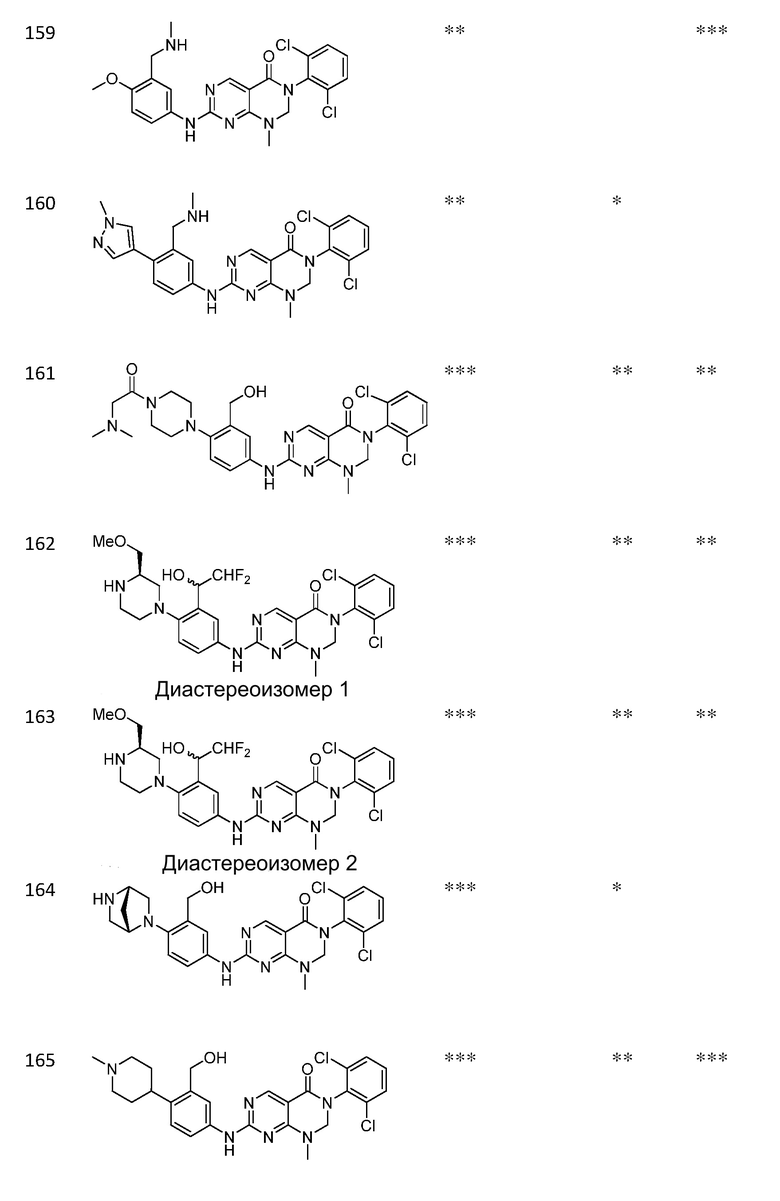
Для представленных в таблице 1 примеров, величины активности в отношении Wee-1, HT29 pCDC2 и HLM систематизированы следующим образом:
ЭКCПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Условные сокращенные обозначения
aq: водный; Boc: третбутоксикарбонил; c-hex: циклогексан; cv: объемы колонки; dba: дибензилиденацетон; DCM: дихлорметан; DIPEA: диизопропилэтиламин; DMF: N,N-диметилформамид; DMSO: диметилсульфоксид; dppf: 1,1′-бис(дифенилфосфино)ферроцен; EtOAc: этилацетат; ESI: ионизация методом электрораспыления; h: час; HATU: 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]-пиридиния 3-оксида гексафторфосфат; ВЭЖХ: высокоэффективная жидкостная хроматография; LC: жидкостная хроматография; LCMS: жидкостная хроматомасс-спектрометрия; M: молярный; m/z: отношение массы к заряду; mCPBA: 3-хлорпербензойная кислота; MeOH: метанол; мин: минуты; MS: масс-спектрометрия; NBS: N-бромсукцинимид; ЯМР: ядерный магнитный резонанс; RT: время удерживания; RT: комнатная температура; нас.: насыщенный; SM: исходный материал; TFA: трифторуксусная кислота; THF: тетрагидрофуран.
Общие условия проведения эксперимента
Растворители и реагенты
Обычные органические растворители, которые использовали в реакциях (например THF, DMF, DCM и метанол), приобретали безводными у фирмы Sigma-Aldrich® в герметичных бутылях Sure/SealTM и хранили соответствующим образом в атмосфере азота. Воду деионизировали, используя систему для получения воды Elga PURELAB Option-Q. Все другие используемые растворители (то есть для методик выделения и очистки) имели обычно квалификацию "для ВЭЖХ" и их использовали в том виде, в котором они поставлялись различными фирмами-производителями. Если не указано иначе, то все используемые исходные материалы приобретались у фирм-производителей и использовались в том виде, в котором их поставляли.
Микроволновый синтез
Если не указано иначе, то эксперименты с применением микроволнового излучения проводили, используя систему CEM Discover™/Explorer24™ под управлением компьютерной программы Synergy 1,5. В других случаях использовали систему Biotage Initiator™ Eight. Обе установки характеризовались хорошей воспроизводимостью и контролированием температуры в диапазоне 60-250°C и давления до 2 МПа.
Флэш-хроматография
Очистку соединений методом флэш-хроматографии проводили с помощью системы Biotage Isolera Four. Если не указано иначе, то использовали картриджи Biotage KP-Sil SNAP или Grace Resolv (10-340 г) вместе с указанной системой растворителей и соответствующим градиентом растворителя в зависимости от полярности соединения. В случае более полярных соединений и соединений с основными свойствами использовали картриджи Biotage KP-NH SNAP (11-28 г).
ЯМР спектроскопия
1H ЯМР спектры регистрировали при температуре окружающей среды на спектрометре Bruker Avance (500 МГц) или Bruker Avance (400 МГц) или Bruker Avance (300 МГц). Значения всех химических сдвигов (δ) выражали в ppm (в частях на миллион). Сигналы остаточного растворителя использовали в качестве внутреннего стандарта, и характеристические пики растворителя корректировали в соответствии со справочными данными, приведенными в публикации J. Org. Chem., 1997, 62, p 7512-7515; в других случаях, растворители для ЯМР содержали тетраметилсилан, который использовали в качестве внутреннего стандарта.
Высокоэффективная жидкостная хроматография
Эксперименты по жидкостной хроматомасс-спектрометрии (LCMS) для определения времен удерживания (RT) и присоединенной массы ионов проводили, используя один из следующих методов.
Метод A: Система состоит из одноквадрупольного масс-спектрометра Agilent Technologies 6140, соединенного с жидкостной хроматографической системой Agilent Technologies 1290 Infinity с УФ-детектором на диодной матрице и автоматическим дозатором. Спектрометр состоит из многорежимного источника ионизации (ионизация электрораспылением и химическая ионизации при атмосферном давлении), работающего в режиме положительных и отрицательных ионов. Эксперименты по жидкостной хроматомасс-спектрометрии (LCMS) проводили для каждого представленного образца, используя следующие условия: колонка для жидкостной хроматографии: Zorbax Eclipse Plus C18 RRHD 1,8 микрон 50×2,1 мм при температуре 40°C. Подвижные фазы: A) 0,1% (по объему) муравьиной кислоты в воде; B) 0,1% (по объему) муравьиной кислоты в ацетонитриле.
(мин)
(мл/мин)
Метод B: Система состоит из установки для жидкостной хроматографии с масс-спектрометрией Varian Prostar 1200, в которой используется колонка Luna Phenomenex 50×4,6 мм ID 3 мкм C18(2). УФ-детектирование проводят при двух длинах волн (214 и 254 нм) с масс-спектральным анализом в режиме положительной ионизации. Подвижные фазы: A) 0,05% (по объему) муравьиной кислоты в воде; B) 0,05% (по объему) муравьиной кислоты в ацетонитриле.
градиентного элюирования
(мин)
(мл/мин)
Метод C: Система состоит из одноквадрупольного масс-спектрометра Agilent Technologies 6130, соединенного с жидкостной хроматографической системой Agilent Technologies 1290 Infinity с УФ-детектором на диодной матрице и автоматическим дозатором. Спектрометр состоит из источника ионизации электрораспылением, работающего в режиме положительных и отрицательных ионов. Эксперименты по жидкостной хроматомасс-спектрометрии (LCMS) проводили для каждого представленного образца, используя следующие условия: колонка для жидкостной хроматографии: Agilent Eclipse Plus C18 RRHD 1,8 micron 50×2,1 мм при температуре 40°C. Подвижные фазы: A) 0,1% (по объему) муравьиной кислоты в воде; B) 0,1% (по объему) муравьиной кислоты в ацетонитриле.
градиентного
элюирования
(мин)
(мл/мин)
Препаративная высокоэффективная жидкостная хроматография
Система состоит из одноквадрупольного масс-спектрометра Agilent Technologies 6120, соединенного с препаративной жидкостной хроматографической системой Agilent Technologies 1200 с многоволновым детектором и автоматическим дозатором. В масс-спектрометре используется многорежимный источник ионизации (ионизация электрораспылением и химическая ионизации при атмосферном давлении), работающий в режиме положительных и отрицательных ионов. Сбор фракций контролировался по массе (многорежимный источник положительных и отрицательных ионов). Эксперименты по очистке, если не указано иначе, проводили при основных условиях при соответствующем градиенте растворителя, который обычно определяли по времени удерживания, обнаруженного методом A высокоэффективной жидкостной хроматографии. В случаях, когда щелочные условия не давали положительного результата, использовали кислотные условия.
Щелочные условия: Колонка для жидкостной хроматографии: колонка Waters XBridge™ Prep C18 5 мкм OBDTM 19×50 мм при комнатной температуре или колонка Water XBridgeTM Prep C18 5 мкм OBDTM 30×100 мм при комнатной температуре. Подвижная фаза: A) 0,1% (по объему) гидроксида аммония в воде; B) 0,1% (по объему) гидроксида аммония в смеси 95:5 ацетонитрил/вода. Суммарное время эксперимента составляло приблизительно 10 или 20 минут, и пример осуществления метода приводится ниже:
градиентного
элюирования
(мин)
(мл/мин)
Кислотные условия: Колонка для жидкостной хроматографии: колонка Waters XBridge™ Prep C18 5 мкм OBDTM 19×50 мм при комнатной температуре или колонка Water XBridgeTM Prep C18 5 мкм OBDTM 30×100 мм при комнатной температуре. Подвижная фаза: A) 0,1% (по объему) муравьиной кислоты в воде; B) 0,1% (по объему) муравьиной кислоты в смеси 95:5 ацетонитрил/вода. Суммарное время эксперимента составляло приблизительно 10 или 20 минут, и пример осуществления метода приводится ниже:
градиентного
элюирования
(мин)
(мл/мин)
Чистые фракции объединяли и концентрировали, используя испаритель Genevac EZ-2 Elite, если не указано иначе.
Номенклатура
Если не указано иначе, то номенклатуру структур определяли с помощью функции "Convert Structure to Name" ("Преврати структуру в химическое название") компьютерной программы ChemBioDraw Ultra 12,0,2 (CambridgeSoft/PerkinElmer).
Масса ионов
Для соединений, содержащих атомы хлора, регистрируемыми ионами являются ионы, содержащие только изотоп 35Cl.
Пример 1: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: N-(2,6-Дихлорфенил)-4-(метиламино)-2-(метилтио)-пиримидин-5-карбоксамид:
4-(Метиламино)-2-(метилтио)пиримидин-5-карбоновую кислоту [Bioorg. Med. Chem. 2005, 13 (16), 4936] (0,5 г, 2,51 ммоль) суспендировали в хлорбензоле (10 мл) и добавляли 2,6-дихлоранилин (0,407 г, 2,51 ммоль). Добавляли трихлорид фосфора (0,220 мл, 2,51 ммоль), и смесь перемешивали при 120°C в течение 16 часов. Реакцию прерывали 2 М водным раствором Na2CO3, затем экстрагировали этилацетатом (x 2). Объединенные органические экстракты промывали солевым раствором, затем сушили (MgSO4), фильтровали и концентрировали (отгоняли в виде азеотропа с толуолом). Остаток растирали с диэтиловым эфиром, и названное соединение собирали фильтрацией с получением названного соединения в виде слегка желтого твердого вещества (180 мг, 21%). Маточный раствор концентрировали с получением дополнительно слегка менее чистого образца названного соединения (300 мг; суммарный выход 56%). ¹H ЯМР (500 МГц, DMSO-d6): δ 10,27 (с, 1H), 8,72 (уш.с, 1H), 8,61 (уш.с, 1H), 7,60 (м, 2H), 7,45 (м, 1H), 2,96 (с, 3H), пик SMe, по-видимому, перекрывался растворителем. LCMS (метод A): RT=1,24 мин, m/z=343 [M+H]+.
Стадия 2: 3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамид (500 мг, 1,46 ммоль) суспендировали в ацетонитриле (10 мл) и добавляли карбонат цезия (1,90 г, 5,83 ммоль). Добавляли дибромметан (0,304 мл, 4,37 ммоль), и реакционную смесь перемешивали при 80°C в течение 16 часов. Смесь разбавляли водой, затем экстрагировали этилацетатом (x 3). Объединенные органические экстракты промывали солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-70% EtOAc/циклогексан 20 колоночных объемов) с получением названного соединения в виде кристаллического белого твердого вещества (190 мг, 37%). ¹H ЯМР (500 МГц, CDCl3): δ 8,72 (с, 1H), 7,44 (д, 2H), 7,30 (т, 1H), 4,91 (с, 2H), 3,20 (с, 3H), 2,58 (с, 3H). LCMS (метод A): RT=1,29 мин, m/z=355 [M+H]+.
Стадия 3: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (30 мг, 0,084 ммоль) суспендировали в толуоле (2 мл) и mCPBA (34,4 мг, 0,110 ммоль) добавляли в виде суспензии в толуоле (2 мл) при комнатной температуре. Через 15 минут добавляли DIPEA (0,044 мл, 0,253 ммоль), затем третбутил 4-(4-аминофенил)пиперазин-1-карбоксилат [Activate Scientific] (23,4 мг, 0,084 ммоль). Полученную смесь нагревали при 80°C в течение 1 часа, затем концентрировали под вакуумом и отгоняли в виде азеотропа с этилацетатом. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-90% EtOAc/циклогексан 20 колоночных объемов) с получением полутвердого материала. Этот материал суспендировали в диэтиловом эфире, и желтое твердое вещество собирали фильтрацией. Твердое вещество растворяли в DCM (1 мл) и добавляли TFA (1 мл). Смесь перемешивали в течение 30 минут, затем концентрировали. Остаток растворяли в метаноле и загружали в картридж 2 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH. Щелочную фракцию концентрировали с получением воскообразного твердого вещества. Это вещество суспендировали в диэтиловом эфире, и твердое вещество собирали фильтрацией с получением названного соединения в виде белого твердого вещества (7 мг, 17%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,70 (с, 1H), 8,42 (с, 1H), 7,63 (м, 4H), 7,48 (м, 1H), 6,90 (д, 2H), 4,95 (с, 2H), 3,10 (с, 3H), 2,97 (м, 4H), 2,81 (м, 4H). LCMS (метод A): RT=0,71 мин, m/z=484 [M+H]+.
Пример 2: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(2-(метил-амино)этокси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (82 мг, 0,231 ммоль) подвергали взаимодействию с третбутил (2-(4-аминофенокси)-этил)(метил)карбаматом (61,5 мг, 0,231 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (27 мг, 24%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,71 (с, 1H), 8,45 (с, 1H), 7,69 (м, 4H), 7,47 (м, 1H), 6,89 (д, 2H), 4,88 (с, 2H), 3,97 (м, 2H), 3,10 (с, 3H), 2,81 (т, 2H), 2,32 (с, 3H). LCMS (метод A): RT=0,77 мин, m/z=473 [M+H]+.
Пример 3: 3-(2,6-Дихлорфенил)-7-((3-метокси-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,141 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-метокси-фенил)пиперазин-1-карбоксилатом (43 мг, 0,14 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (34 мг, 47%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,75 (с, 1H), 8,48 (с, 1H), 7,65 (м, 3H), 7,47 (м, 1H), 7,21 (д, 1H), 6,85 (д, 1H), 4,97 (с, 2H), 3,69 (с, 3H), 3,13 (с, 3H), 2,92 (м, 8H). LCMS (метод A): RT=0,75 мин, m/z=514 [M+H]+.
Пример 4: 2-(4-(4-((6-(2,6-Дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-пиперазин-1-ил)уксусной кислоты гидрохлорид

Стадия 1: третбутил 2-(4-(4-нитрофенил)пиперазин-1-ил)-ацетат:
1-(4-Нитрофенил)пиперазин (3 г, 14,5 ммоль) растворяли в ацетонитриле (25 мл) и добавляли карбонат цезия (6,43 г, 19,7 ммоль), затем третбутил 2-бромацетат (1,95 мл, 13,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали под вакуумом. Остаток растворяли в дихлорметане и промывали последовательно водным раствором NaHCO3, водой и солевым раствором. Органический компонент сушили, используя картридж для разделения фаз, затем концентрировали с получением названного соединения (3,5 г, 85%), которое использовали без дополнительной очистки. LCMS (метод A): RT=0,90 мин, m/z=322 [M+H]+.
Стадия 2: третбутил 2-(4-(4-аминофенил)пиперазин-1-ил)-ацетат:
Третбутил 2-(4-(4-нитрофенил)пиперазин-1-ил)ацетат (1,5 г, 4,67 ммоль) растворяли в этаноле (50 мл) и добавляли 10% палладий на угле (0,497 г, 0,467 ммоль) в атмосфере азота. Добавляли формиат аммония (2,94 г, 46,7 ммоль), и смесь нагревали до 40°C. Добавляли дополнительную порцию формиата аммония (2,94 г, 46,7 ммоль), и полученную смесь перемешивали при 40°C в течение 16 часов. Смесь фильтровали через целит® и концентрировали под вакуумом. Остаток растворяли в водном растворе бикарбоната натрия и экстрагировали дихлорметаном (x 3). Объединенные органические экстракты сушили над Na2SO4, затем фильтровали и концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 50 g Si; элюировали 1:1 CH2Cl2:EtOAc) с получением названного соединения (1,2 г, 88%), которое использовали без дополнительной очистки.
Стадия 3: 2-(4-(4-((6-(2,6-Дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-пиперазин-1-ил)уксусной кислоты гидрохлорид:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,141 ммоль) суспендировали в толуоле (1 мл) и добавляли mCPBA (57,4 мг, 0,183 ммоль) в виде суспензии в толуоле (1 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли DIPEA (0,074 мл, 0,422 ммоль), затем третбутил 2-(4-(4-аминофенил)пиперазин-1-ил)ацетат (41 мг, 0,141 ммоль). Полученную смесь нагревали при 80°C в течение 16 часов, затем концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 10 g Si; элюировали 0-90% EtOAc/циклогексан 20 колоночных объемов) с получением желтого масла. Это масло растирали с диэтиловым эфиром с получением желтовато-белого твердого вещества, которое суспендировали в 1 мл 4 M HCl в диоксане и нагревали при 70°C в течение 16 часов. Смесь концентрировали под вакуумом, и к этому моменту метод ЯМР показывал, что удаление защитной группы прошло полностью. Остаток растворяли в 2 мл 2 M HCl в воде и нагревали при 60°C в течение 3 часов, затем концентрировали под вакуумом и отгоняли в виде азеотропа с толуолом (x 3). Оставшееся стеклообразное вещество растворяли в смеси ацетонитрил/вода и лиофилизировали с получением названного соединения в виде желтого твердого вещества (24 мг, 32%). ¹H ЯМР (500 МГц, DMSO-d6): δ 10,40 (с, 1H), 10,05 (с, 1H), 8,46 (с, 1H), 7,77 (м, 4H), 7,50 (т, 1H), 7,00 (д, 2H), 5,01 (с, 2H), 4,22 (с, 2H), 3,85 (м, 8H), 3,12 (с, 3H). LCMS (метод A): RT=0,74 мин, m/z=542 [M+H]+.
Пример 5: 3-(2,6-Дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
Стадия 1: 2-Морфолино-5-нитробензальдегид:
К раствору 2-хлор-5-нитробензальдегида (3,00 г, 16,2 ммоль) в DMF (20 мл) добавляли DIPEA (4,24 мл, 24,3 ммоль) и морфолин (1,55 г, 17,8 ммоль). Реакционную смесь перемешивали при 90°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли воду. Смесь экстрагировали с помощью EtOAc. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, и растворители удаляли под вакуумом с получением названного соединения (неочищенное 3,2 г, 85%), которое использовали на следующей стадии без дополнительной очистки. LCMS (метод A): RT=0,99 мин, m/z=237 [M+H]+.
Стадия 2: N-Метил-1-(2-морфолино-5-нитрофенил)метанамин:
К суспензии бикарбоната натрия (0,356 г, 4,23 ммоль) в MeOH (6 мл) добавляли 2-морфолино-5-нитробензальдегид (0,5 г, 2,12 ммоль) и метиламин (2 M в MeOH) (1,27 мл, 2,54 ммоль). Реакционную смесь перемешивали при 70°C. Через 4 часа, реакционную смесь охлаждали до 0°C и добавляли боргидрид натрия (0,096 г, 2,54 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли несколько капель воды, и растворители удаляли под вакуумом. Полученный остаток распределяли между DCM и солевым раствором, фазы разделяли, экстрагировали дополнительным количеством DCM, сушили над Na2SO4, фильтровали, и растворители удаляли под вакуумом с получением названного соединения (неочищенное, 450 мг, 85%), которое использовали на следующей стадии без дополнительной очистки. LCMS (метод A): RT=0,47 мин, m/z=221 [M+H-30(CH3NH)]+.
Стадия 3: третбутил метил(2-морфолино-5-нитробензил)-карбамат:
К раствору N-метил-1-(2-морфолино-5-нитрофенил)метанамина (450 мг, 1,79 ммоль) в THF (5 мл) добавляли триэтиламин (0,50 мл, 3,58 ммоль) и Boc2O (0,46 мл, 1,97 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли воду, и реакционную смесь экстрагировали, используя EtOAc. Объединенную органическую фазу промывали водой и солевым раствором, сушили над Na2SO4, фильтровали, и растворители удаляли под вакуумом с получением названного соединения (неочищенное, 622 мг, 99%), которое использовали на следующей стадии без дополнительной очистки. LCMS (метод A): RT=1,39 мин, m/z=296 [M+H-56(tBu)]+.
Стадия 4: третбутил 5-амино-2-морфолинобензил(метил)-карбамат:
К раствору третбутил метил(2-морфолино-5-нитробензил)-карбамата (640 мг, 1,82 ммоль) в этаноле (4 мл) добавляли 10% палладий на угле (194 мг, 0,182 ммоль) и формиат аммония (230 мг, 3,64 ммоль). Реакционную смесь перемешивали при 60°C в течение 3 часов. Смесь фильтровали через целит®, и фильтрат концентрировали под вакуумом. Полученный остаток распределяли между EtOAc и насыщенным водным раствором бикарбоната натрия, органическую фазу промывали солевым раствором, сушили над Na2SO4, фильтровали, и растворители удаляли под вакуумом с получением названного соединения (неочищенное, 580 мг, 99%), которое использовали на следующей стадии без дополнительной очистки. LCMS (метод A): RT=0,83 мин, m/z=322 [M+H]+.
Стадия 5: 3-(2,6-Дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,141 ммоль) подвергали взаимодействию с третбутил 5-амино-2-морфолинобензил-(метил)карбаматом (45,2 мг, 0,141 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (6 мг, 8%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,49 (с, 1H), 8,45 (с, 1H), 7,85 (с, 1H), 7,65 (м, 2H), 7,57 (д, 1H), 7,47 (т, 1H), 7,05 (д, 1H), 4,96 (с, 2H), 3,97 (м, 4H), 3,69 (с, 2H), 3,12 (с, 3H), 2,82 (м, 4H), 2,31 (с, 3H). LCMS (метод A): RT=0,85 мин, m/z=528 [M+H]+.
Пример 6: 3-(2,6-Дихлорфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,141 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-метилфенил)пиперазин-1-карбоксилатом (41 мг, 0,141 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (14 мг, 20%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,70 (с, 1H), 8,46 (с, 1H), 7,68 (м, 2H), 7,56 (м, 2H), 7,47 (м, 2H), 6,97 (д, 2H), 4,96 (с, 2H), 3,08 (с, 3H), 2,81 (м, 4H), 2,72 (м, 4H), 2,23 (с, 3H). LCMS (метод A): RT=0,78 мин, m/z=499 [M+H]+.
Пример 7: 3-(2,6-Дихлорфенил)-7-((3-фтор-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
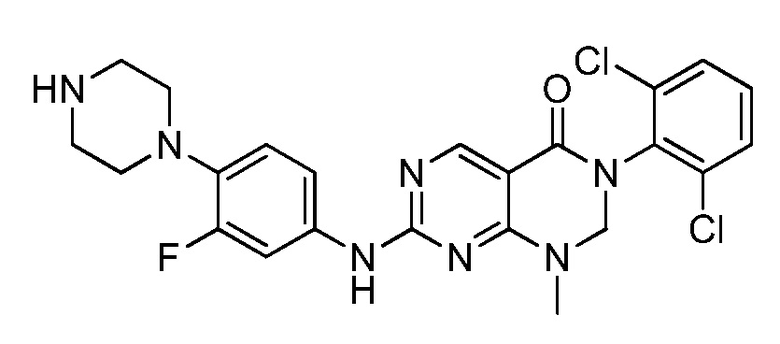
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,141 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-фторфенил)-пиперазин-1-карбоксилатом (42 мг, 0,141 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (22 мг, 31%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,87 (с, 1H), 8,48 (с, 1H), 7,74 (м, 3H), 7,45 (м, 2H), 6,96 (т, 1H), 4,96 (с, 2H), 3,11 (с, 3H), 2,85 (м, 8H). LCMS (метод A): RT=0,76 мин, m/z=503 [M+H]+.
Пример 8: 3-(2,6-Дихлорфенил)-1-(4-метоксибензил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
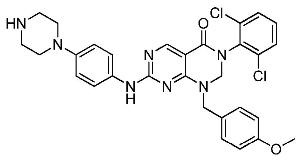
Стадия 1: 4-((4-Метоксибензил)амино)-2-(метилтио)пиримидин-5-карбоновая кислота:
Этил 4-хлор-2-(метилтио)пиримидин-5-карбоксилат (3 г, 12,9 ммоль) растворяли в DMF (10 мл) и охлаждали до 0°C. Медленно добавляли раствор триэтиламина (2 мл, 14,4 ммоль) и (4-метоксифенил)метанамина (1,85 мл, 14,2 ммоль) в DMF (10 мл). Смесь перемешивали в течение 1 часа, затем выливали в лед/10% раствор лимонной кислоты. Смесь экстрагировали этилацетатом (x 2). Объединенные органические слои промывали раствором лимонной кислоты, затем солевым раствором, затем сушили над MgSO4, фильтровали и концентрировали под вакуумом. Остаток отгоняли в виде азеотропа с циклогексан/DCM с получением сиропа, из которого кристаллизовалось 5,20 г белого твердого вещества при стоянии. Это вещество растворяли в этаноле (25 мл) и 2 M NaOH (25 мл), и раствор перемешивали в течение ночи. Смесь концентрировали под вакуумом, затем подкисляли (2 M HCl). Осажденное твердое вещество собирали фильтрацией, затем сушили под вакуумом с получением названного соединения в виде белого твердого вещества (4,1 г, количественно). ¹H ЯМР (500 МГц, DMSO-d6): δ 13,30 (уш.с, 1H), 8,85 (т, 1H), 8,53 (с, 1H), 7,29 (т, 2H), 6,90 (т, 2H), 4,63 (д, 2H), 3,74 (с, 3H), 2,45 (с, 3H). LCMS (метод A): RT=1,05 мин, m/z=306 [M+H]+.
Стадия 2: N-(2,6-Дихлорфенил)-4-((4-метоксибензил)амино)-2-(метилтио)пиримидин-5-карбоксамид:
4-((4-Метоксибензил)амино)-2-(метилтио)пиримидин-5-карбоновую кислоту (4,1 г, 13,4 ммоль) суспендировали в хлорбензоле (30 мл). Добавляли 2,6-дихлоранилин (2,18 г, 13,4 ммоль), затем трихлорид фосфора (1,18 мл, 13,4 ммоль). Смесь перемешивали при 120°C в течение 60 часов. Реакцию прерывали с помощью Na2CO3 и добавляли этилацетат. Нерастворимое твердое вещество собирали фильтрацией. Двухфазный маточный раствор экстрагировали с помощью DCM (x 3). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали до сиропа, который превращался в твердое вещество при стоянии в течение ночи. Это вещество объединяли с твердым веществом, собранным фильтрацией. Объединенный материал суспендировали в диэтиловом эфире и подвергали воздействию ультразвука, затем твердое вещество собирали фильтрацией с получением названного соединения (3,19 г, 53%) в виде желтовато-белого твердого вещества, которое использовали без дополнительной очистки. ¹H ЯМР (500 МГц, DMSO-d6): δ 9,05 (с, 1H), 8,77 (с, 1H), 7,58 (д, 2H), 7,41 (м, 2H), 7,27 (д, 2H), 6,88 (д, 2H), 4,60 (д, 2H), 3,72 (с, 3H). LCMS (метод A): RT=1,49 мин, m/z=449 [M+H]+.
Стадия 3: 3-(2,6-Дихлорфенил)-1-(4-метоксибензил)-7-(метил-тио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-((4-метоксибензил)амино)-2-(метил-тио)пиримидин-5-карбоксамид (760 мг, 1,69 ммоль) суспендировали в ацетонитриле (20 мл) и добавляли карбонат цезия (2,76 г, 8,46 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли дийодметан (0,341 мл, 4,23 ммоль), и смесь перемешивали в течение 1 часа при 70°C. Добавляли дополнительную порцию дийодметана (0,341 мл, 4,23 ммоль) и смесь перемешивали еще 1 час при 80°C. Смесь разбавляли водой и экстрагировали дихлорметаном (x 2). Объединенные органические слои промывали солевым раствором, затем сушили (MgSO4), фильтровали и концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 25 g; элюировали 0-80% EtOAc/циклогексан) с получением названного соединения в виде липкого стеклообразного вещества (125 мг, 16%), которое непосредственно использовали без дополнительной очистки. LCMS (метод A): RT=1,48 мин, m/z=461 [M+H]+.
Стадия 4: 3-(2,6-Дихлорфенил)-1-(4-метоксибензил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-(4-метоксибензил)-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (123 мг, 0,267 ммоль) суспендировали в толуоле (1 мл) и добавляли mCPBA (109 мг, 0,347 ммоль) в виде суспензии в толуоле (1 мл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли DIPEA (0,140 мл, 0,800 ммоль), затем третбутил 4-(4-аминофенил)-пиперазин-1-карбоксилат (73,9 мг, 0,267 ммоль). Смесь нагревали при 80°C в течение 4 часов, затем охлаждали и концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-80% EtOAc/циклогексан 20 колоночных объемов) с получением желтого стеклообразного вещества. Это вещество растворяли в DCM (1 мл) и TFA (1 мл) и перемешивали в течение 1 часа при комнатной температуре, затем концентрировали под вакуумом. Остаток растворяли в метаноле и загружали в картридж 2 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH, и растворитель концентрировали под вакуумом с получением желтого стеклообразного вещества. Это вещество растирали этилацетатом с получением названного соединения в виде оранжевого твердого вещества (43 мг, 27%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,70 (с, 1H), 8,49 (с, 1H), 7,62 (д, 2H), 7,54 (д, 2H), 7,45 (м, 1H), 7,35 (д, 2H), 6,87 (м, 4H), 4,90 (с, 2H), 4,77 (с, 2H), 3,71 (с, 3H), 2,96 (м, 4H), 2,80 (м, 4H). LCMS (метод A): RT=0,97 мин, m/z=590 [M+H]+.
Пример 9: 3-(2,6-Дихлорфенил)-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
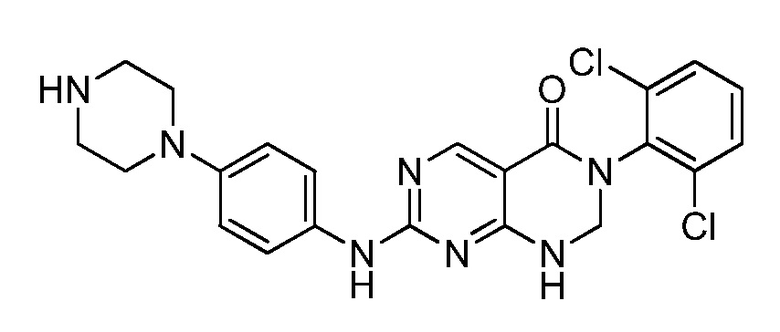
3-(2,6-Дихлорфенил)-1-(4-метоксибензил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (35 мг, 0,059 ммоль) растворяли в TFA (2 мл, 26,0 ммоль) в пробирке для проведения реакций при микроволновом излучении. Пробирку герметизировали и нагревали при 150°C в течение 20 минут при микроволновом излучении. Смесь концентрировали под вакуумом. Остаток растворяли в метаноле и загружали в картридж 5 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH и концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera KP-NH 11 g; элюировали 0-100% EtOAc/циклогексан; затем 0-25% MeOH/EtOAc 25 колоночных объемов) с получением стеклообразного твердого вещества. Это вещество растирали с диэтиловым эфиром с получением названного соединения в виде желтовато-белого твердого вещества (6 мг, 21%). ¹H ЯМР (500 МГц, DMSO-d6): δ 9,52 (с, 1H), 8,46 (м, 1H), 8,25 (м, 1H), 7,57 (м, 4H), 7,46 (т, 1H), 6,87 (д, 1H), 4,89 (с, 2H), 2,98 (м, 4H), 2,83 (м, 4H). LCMS (метод A): RT=0,62 мин, m/z=470 [M+H]+.
Пример 10: 3-(2,6-Дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
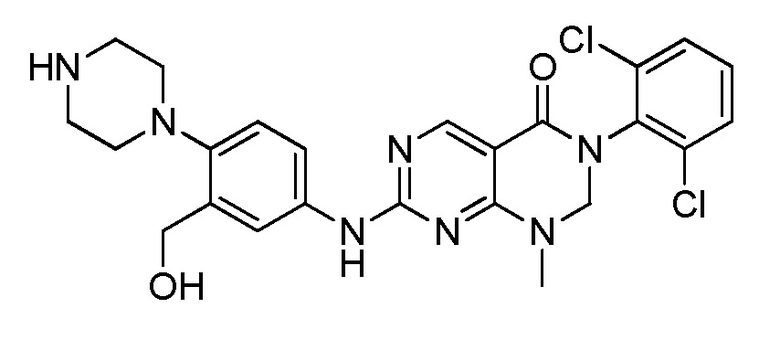
Стадия 1: третбутил 4-(4-амино-2-(гидроксиметил)фенил)-пиперазин-1-карбоксилат:
Формиат аммония (1,47 г, 23,2 ммоль) осторожно добавляли [примечание: происходит разогрев] к перемешиваемому раствору третбутил 4-(2-(гидроксиметил)-4-нитрофенил)пиперазин-1-карбоксилата (1,57 г, 4,65 ммоль) [J. Med. Chem., 2011, 54(13), 4638-4658] и 10% палладия на угле (0,247 г, 0,232 ммоль) в этаноле (15 мл) в круглодонную колбу объемом 100 мл при комнатной температуре в атмосфере азота. Через 16 часов, реакционную смесь фильтровали через целит®, и растворители удаляли под вакуумом. Полученный остаток распределяли между насыщенным водным раствором бикарбоната натрия (30 мл) и DCM (30 мл), фазы разделяли, и экстрагировали, используя дополнительное количество DCM (2×15 мл). Объединенную органическую фазу сушили (фазовый сепаратор), и растворители удаляли под вакуумом с получением названного соединения (1,40 г, 98%) в виде бледно-коричневой пены. LCMS (метод A): RT=0,72 мин, m/z=308 [M+H]+.
Стадия 2: 3-(2,6-Дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (58 мг, 0,163 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-(гидрокси-метил)фенил)пиперазин-1-карбоксилатом (55,2 мг, 0,180 ммоль), следуя методике примера 1, с получением названного продукта (10 мг, 12%) в виде бежевого твердого вещества. ¹H ЯМР (400 МГц, DMSO-d6): δ 9,80 (с, 1H), 8,46 (с, 1H), 7,99 (с, 1H), 7,65 (д, 2H), 7,57 (д, 1H), 7,48 (т, 1H),7,01 (д, 1H), 5,07 (т, 1H), 4,98 (с, 2H), 4,66 (д, 2H), 3,14 (с, 3H), 2,78 (м, 4H), 2,70 (м, 4H). LCMS (метод B): RT=2,20 мин, m/z=514 [M+H]+.
Пример 11: 3-(2,6-Дихлорфенил)-7-((3-циано-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
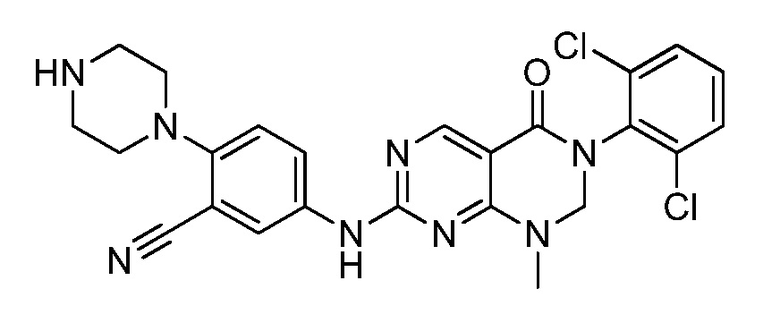
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (75 мг, 0,211 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-цианофенил)-пиперазин-1-карбоксилатом (70,2 мг, 0,232 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (15 мг, 14%). ¹H ЯМР (400 МГц, DMSO-d6): δ 10,05 (с, 1H), 8,51 (с, 1H), 8,15 (с, 1H), 7,94 (д, 1H), 7,68 (д, 2H), 7,52 (т, 1H), 7,16 (д, 1H), 5,01 (с, 2H), 3,13 (с, 3H), 2,99 (м, 4H), 2,85 (м, 4H). LCMS (метод B): RT=3,35 мин, m/z=509 [M+H]+.
Пример 12: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-илметил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
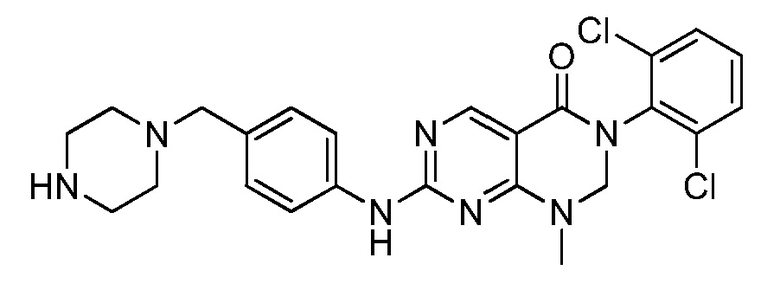
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (75 мг, 0,211 ммоль) подвергали взаимодействию с третбутил 4-(4-аминобензил)-пиперазин-1-карбоксилатом (67,7 мг, 0,232 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (15 мг, 14%). ¹H ЯМР (400 МГц, CDCl3): δ 8,73 (с, 1H), 7,61 (д, 2H), 7,44 (д, 2H), 7,31 (м, 4H), 4,90 (с, 2H), 3,48 (с, 2H), 3,20 (с, 3H), 2,90 (м, 4H), 2,44 (м, 4H). LCMS (метод B): RT=3,38 мин, m/z=498 [M+H]+.
Пример 13: 7-((4-(4-(2-Аминоацетил)пиперазин-1-ил)фенил)-амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (85 мг, 0,175 ммоль) суспендировали в THF (1 мл) и пиридине (0,1 мл, 1,24 ммоль). Добавляли N-Boc-глицин (32,3 мг, 0,184 ммоль), затем HATU (73,4 мг, 0,193 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-100% EtOAc/циклогексан затем 0-15% MeOH/EtOAc) с получением желтого стеклообразного вещества. Это вещество растворяли в DCM (1 мл) и TFA (1 мл) и перемешивали при комнатной температуре в течение 30 минут, затем концентрировали под вакуумом. Остаток растворяли в MeOH и загружали в картридж 5 g SCX-2. Колонку промывали метанолом? затем элюировали c помощью 2 M NH3/MeOH, и растворитель концентрировали под вакуумом с получением желтого стеклообразного вещества. Это вещество растирали с этилацетатом, и твердое вещество собирали фильтрацией и сушили под вакуумом с получением названного соединения в виде бежевого твердого вещества (54 мг, 57%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,72 (с, 1H), 8,45 (с, 1H), 7,65 (м, 4H), 7,49 (т, 1H), 6,93 (д, 2H), 4,95 (с, 2H), 3,62 (м, 3H), 3,10 (м, 8H), несколько пиков перекрыты уширенным сигналом воды. LCMS (метод A): RT=0,70 мин, m/z=541 [M+H]+.
Пример 14: 3-(2,6-Дихлорфенил)-1-метил-7-((1,2,3,4-тетра-гидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (80 мг, 0,225 ммоль) подвергали взаимодействию с третбутил 7-амино-3,4-дигидро-изохинолин-2(1H)-карбоксилатом (61,5 мг, 0,248 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (8 мг, 8%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,76 (с, 1H), 8,46 (с, 1H), 7,64 (д, 2H), 7,48 (м, 4H), 6,97 (м, 1H), 4,98 (д, 2H), 3,83 (m 2H), 3,11 (с, 3H), 2,95 (м, 2H), 2,65 (м, 2H). LCMS (метод A): RT=0,76 мин, m/z=455 [M+H]+.
Пример 15: 3-(2,6-Дихлорфенил)-2,2-дидейтеро-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 3-(2,6-Дихлорфенил)-2,2-дидейтеро-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамид (500 мг, 1,46 ммоль) суспендировали в ацетонитриле (10 мл) и добавляли карбонат цезия (1,90 г, 5,83 ммоль). Добавляли дибромметан-d2 (0,307 мл, 4,37 ммоль), и смесь перемешивали при 80°C в течение 16 часов, затем концентрировали под вакуумом. Остаток разбавляли водой, затем экстрагировали этилацетатом (x 3). Объединенные органические слои промывали солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-90% EtOAc/циклогексан 20 колоночных объемов) с получением 3-(2,6-дихлорфенил)-2,2-дидейтеро-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она в виде кристаллического белого твердого вещества (85 мг, 16%). Это вещество использовали непосредственно без дополнительной очистки.
Стадия 2: 3-(2,6-Дихлорфенил)-2,2-дидейтеро-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-2,2-дидейтеро-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (80 мг, 0,224 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)пиперазин-1-карбоксилатом (68,3 мг, 0,246 ммоль), следуя методике примера 1, с получением названного соединения в виде бледно-коричневого твердого вещества (51 мг, 47%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,68 (с, 1H), 8,44 (с, 1H), 7,65 (м, 4H), 7,49 (т, 1H), 6,88 (д, 2H), 3,11 (с, 3H), 2,98 (м, 4H), 2,85 (м, 4H). LCMS (метод A): RT=0,72 мин, m/z=486 [M+H]+.
Пример 16: (R)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилат:
Смесь (R)-третбутил 2-метилпиперазин-1-карбоксилата (1,42 г, 7,09 ммоль), 1-фтор-4-нитробензола (1 г, 7,09 ммоль) и карбоната калия (1,47 г, 10,6 ммоль) в DMF (10 мл) нагревали при 60°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (120 мл) и перемешивали в течение 1 часа. Полученное твердое вещество собирали фильтрацией и промывали водой (3х40 мл), затем сушили под вакуумом с получением названного соединения (1,9 г, 81%). LCMS (метод A): RT=1,45 мин, m/z=322 [M+H]+.
Стадия 2: (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилат:
Смесь (R)-третбутил 2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилата (1,9 г, 5,91 ммоль) и палладия на угле (0,4 г, 0,376 ммоль) в EtOH (50 мл) нагревали до 50°C. Добавляли одной порцией формиат аммония (2,2 г, 34,9 ммоль). Полученную реакционную смесь перемешивали при 50°C в течение 2 часов, затем охлаждали и фильтровали через целит®. Фильтрат концентрировали, и остаток распределяли между этилацетатом (80 мл) и раствором бикарбоната натрия (70 мл). Органическую фазу отделяли и концентрировали с получением названного соединения (1,7 г, 98%). LCMS (метод A): RT=0,77 мин, m/z=236 [M-56]+.
Стадия 3: (R)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (107 мг, 0,301 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (88 мг, 0,301 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (50 мг, 33%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,71 (с, 1H), 8,44 (с, 1H), 7,65 (м, 4H), 7,49 (т, 1H), 6,91 (д, 2H), 4,95 (с, 2H), 3,46 (м, 2H), 3,11 (с, 3H), 2,94 (м, 1H), 2,78 (м, 2H), 2,15 (т, 1H), 1,02 (д, 3H), один пик, по-видимому, перекрывался растворителем. LCMS (метод A): RT=0,76 мин, m/z=498 [M+H]+.
Пример 17: (S)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
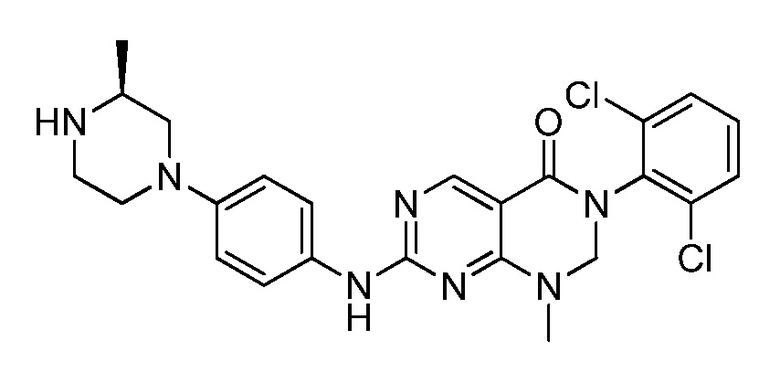
Стадия 1: (S)-третбутил 2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилат:
Смесь (S)-третбутил 2-метилпиперазин-1-карбоксилата (1,42 г, 7,09 ммоль), 1-фтор-4-нитробензола (1 г, 7,09 ммоль) и карбоната калия (1,47 г, 10,6 ммоль) в DMF (6 мл) нагревали при 50°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (50 мл). Полученный осадок собирали фильтрацией и промывали водой с получением названного соединения (1,83 г, 80%). LCMS (метод A): RT=1,45 мин, m/z=266 [M+H-tBu]+.
Стадия 2: (S)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилат:
Смесь (S)-третбутил 2-метил-4-(4-нитрофенил)пиперазин-1-карбоксилата (1,82 г, 5,66 ммоль) и палладия на угле (0,301 г, 0,283 ммоль) в этаноле (30 мл) подогревали до 50°C. Добавляли порциями формиат аммония (1,43 г, 22,7 ммоль), и смесь нагревали при 50°C в течение 1 часа, затем охлаждали. Смесь фильтровали через целит®, и фильтрат концентрировали под вакуумом с получением названного соединения (1,46 г, 88%). LCMS (метод A): RT=0,77 мин, m/z=236 [M+H-tBu]+.
Стадия 3: (S)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (107 мг, 0,301 ммоль) подвергали взаимодействию с (S)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (88 мг, 0,301 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (32 мг, 21%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,71 (с, 1H), 8,45 (с, 1H), 7,65 (м, 4H), 7,49 (т, 1H), 6,90 (д, 2H), 4,95 (с, 2H), 3,46 (м, 2H), 3,11 (с, 3H), 2,94 (м, 1H), 2,78 (м, 2H), 2,15 (т, 1H), 1,02 (д, 3H), один пик, по-видимому, перекрывался растворителем. LCMS (метод A): RT=0,75 мин, m/z=498 [M+H]+.
Пример 18: 3-(2,6-Дихлорфенил)-1-метил-7-((3-метил-4-(пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (80 мг, 0,225 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-метилфенил)-пиперидин-1-карбоксилатом (78 мг, 0,270 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (14 мг, 13%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,77 (с, 1H), 8,47 (с, 1H), 7,55 (м, 4H), 7,13 (м, 1H), 4,97 (с, 2H), 3,11 (с, 3H), 3,02 (м, 2H), 2,57 (м, 5H), 2,30 (с, 3H), 1,62 (м, 2H), 1,51 (м, 2H). LCMS (метод A): RT=0,82 мин, m/z=497 [M+H]+.
Пример 19: (R)-3-(2,6-Дихлорфенил)-7-((4-(3-(гидрокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (70 мг, 0,197 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-(гидроксиметил)пиперазин-1-карбоксилатом [полученным из (R)-третбутил 2-(гидроксиметил)пиперазин-1-карбоксилата и 1-фтор-4-нитробензола аналогично примеру 17, стадиям 1 и 2] (60 мг, 0,195 ммоль), следуя методике примера 1, с получением названного соединения в виде оранжевого твердого вещества (23 мг, 20%). ¹H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,53 (д, 2H), 7,46 (д, 2H), 7,30 (м, 2H), 6,94 (м, 2H), 4,88 (с, 2H), 3,72 (дд, 1H), 3,64 (дд, 1H), 3,48 (д, 2H), 3,20 (м, 1H), 3,18 (с, 3H), 3,08 (м, 2H), 2,78 (т, 1H), 2,60 (т, 1H), 1,27 (м, 1H). LCMS (метод A): RT=0,71 мин, m/z=514 [M+H]+.
Пример 20: (S)-3-(2,6-Дихлорфенил)-7-((4-(3-(гидрокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
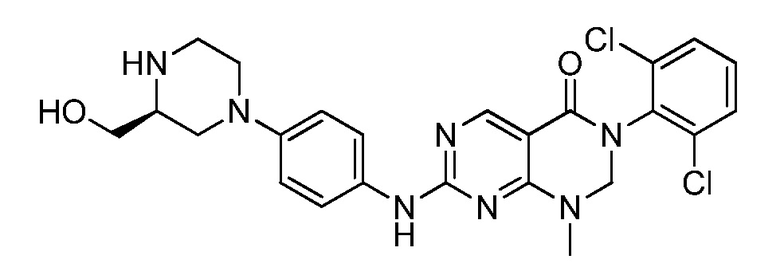
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (80 мг, 0,225 ммоль) подвергали взаимодействию с (S)-третбутил 4-(4-аминофенил)-2-(гидроксиметил)пиперазин-1-карбоксилатом [полученным из (S)-третбутил 2-(гидроксиметил)пиперазин-1-карбоксилата и 1-фтор-4-нитробензола аналогично примеру 17, стадиям 1 и 2] (83 мг, 0,270 ммоль), следуя методике примера 1, с получением названного соединения в виде желтого твердого вещества (19 мг, 16%). ¹H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,53 (д, 2H), 7,46 (д, 2H), 7,30 (м, 2H), 6,94 (м, 2H), 4,88 (с, 2H), 3,72 (дд, 1H), 3,64 (дд, 1H), 3,48 (д, 2H), 3,20 (м, 1H), 3,18 (с, 3H), 3,08 (м, 2H), 2,78 (т, 1H), 2,60 (т, 1H), 1,27 (м, 1H). LCMS (метод A): RT=0,71 мин, m/z=514 [M+H]+.
Пример 21: 3-(2,6-Дихлорфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 2-(4-Изопропилпиперазин-1-ил)-5-нитро-бензальдегид:
Смесь 2-хлор-5-нитробензальдегида (2 г, 10,8 ммоль), 1-изопропилпиперазина (2,07 г, 16,2 ммоль) и карбоната калия (2,68 г, 19,4 ммоль) в DMF (10 мл) нагревали при 90°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Полученный осадок собирали фильтрацией, затем промывали на фильтре водой и сушили в вакуумном сушильном шкафу с получением названного соединения (2,8 г, 96%). LCMS (метод A): RT=0,49 мин, m/z=278 [M+H]+.
Стадия 2: (2-(4-Изопропилпиперазин-1-ил)-5-нитрофенил)-метанол:
Раствор 2-(4-изопропилпиперазин-1-ил)-5-нитробензальдегида (2,8 г, 10,1 ммоль) в THF (25 мл) охлаждали до 0°C. Добавляли боргидрид натрия (0,382 г, 10,1 ммоль), и полученную суспензию перемешивали при 0°C в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали с помощью DCM (x 3). Объединенные органические экстракты промывали водой, сушили, фильтровали и концентрировали. Остаток подвергали хроматографии (Isolera KP-Sil 50g, элюировали смесью 0-100% этилацетат/циклогексан) с получением названного соединения (1,5 г, 53%). LCMS (метод A): RT=0,50 мин, m/z=280 [M+H]+.
Стадия 3: (5-Амино-2-(4-изопропилпиперазин-1-ил)фенил)-метанол:
Палладий на угле (0,419 г, 0,39 ммоль) добавляли к раствору (2-(4-изопропилпиперазин-1-ил)-5-нитрофенил)метанола (1,1 г, 3,94 ммоль) в этаноле (35 мл). Полученный раствор нагревали до 50°C. Добавляли формиат аммония (0,745 г, 11,8 ммоль), и смесь перемешивали при 50°C в течение 30 минут. Смесь охлаждали до комнатной температуры и фильтровали через целит®. Фильтрат концентрировали под вакуумом, и остаток распределяли между DCM и водным раствором бикарбоната натрия. Органическую фазу отделяли и концентрировали под вакуумом с получением названного соединения (0,65 г, 66%). LCMS (метод A): RT=0,13 мин, m/z=250 [M+H]+.
Стадия 4: 3-(2,6-Дихлорфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (80 мг, 0,225 ммоль) подвергали взаимодействию с (5-амино-2-(4-изопропилпиперазин-1-ил)фенил)метанолом (67,4 мг, 0,270 ммоль), следуя методике примера 1, с получением названного соединения в виде желтовато-белого твердого вещества (11 мг, 8%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,82 (с, 1H), 8,47 (с, 1H), 8,01 (с, 1H), 7,66 (д, 2H), 7,55 (м, 1H), 7,50 (т, 1H), 7,01 (д, 1H), 5,08 (м, 1H), 4,97 (с, 2H), 4,55 (с, 2H), 3,12 (с, 3H), 2,79 (м, 4H), 2,70 (м, 1H), 2,58 (м, 4H), 1,03 (д, 6H). LCMS (метод A): RT=0,76 мин, m/z=556 [M+H]+.
Пример 22: 3-(2,6-Дихлорфенил)-1-метил-7-((2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
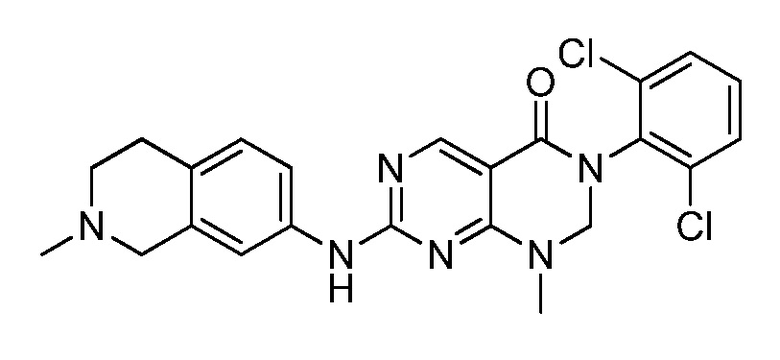
3-(2,6-Дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-изохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (51 мг, 0,112 ммоль) [пример 14] растворяли в 1,2-дихлорэтане (1 мл) и добавляли формалин (0,017 мл, 0,224 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, затем охлаждали до 0°C. Добавляли триацетоксиборгидрид натрия (28,5 мг, 0,134 ммоль), и смесь подогревали до комнатной температуры при перемешивании в течение 1 часа. Добавляли дополнительные порции формалина (0,1 мл) и триацетоксиборгидрида натрия (28,5 мг, 0,134 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов. Добавляли 1,4-диоксан вместе с дополнительными порциями формалина (0,1 мл) и триацетоксиборгидрида натрия (28,5 мг, 0,134 ммоль). Еще через 1 час, добавляли дополнительные порции формалина (0,1 мл) и триацетоксиборгидрида натрия (28,5 мг, 0,134 ммоль). Еще через 1 час перемешивания, реакцию прерывали путем добавления 1 N HCl (1 мл), и реакционную смесь перемешивали в течение 20 минут. Смесь загружали в картридж 5 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH, и щелочные фракции концентрировали под вакуумом с получением желтого стеклообразного вещества. Это вещество растирали с диэтиловым эфиром с получением названного соединения в виде желтовато-белого твердого вещества (20 мг, 38%). ¹H ЯМР (400 МГц, CDCl3): δ 8,74 (с, 1H), 7,40 (м, 4H), 7,30 (м, 2H), 7,10-7,00 (м, 1H), 4,90 (с, 2H), 3,58 (с, 2H), 3,17 (с, 3H), 2,91 (м, 2H), 2,70 (м, 2H), 2,48 (с, 3H). LCMS (метод A): RT=0,75 мин, m/z=469 [M+H]+.
Пример 23: (рац)-3-(2,6-Дихлорфенил)-1-метил-2-фенил-7-((1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (рац)-3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2-фенил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамид (200 мг, 0,58 ммоль) растворяли в ацетонитриле (10 мл) и добавляли бензальдегид (0,25 мл, 2,47 ммоль), затем гидрат п-толуолсульфоновой кислоты (25 мг, 0,15 ммоль). Пробирку герметизировали, и смесь подвергали микроволновому нагреву при 220°C в течение 16 часов. Смесь концентрировали под вакуумом и подвергали хроматографии (картридж Isolera Si 10 g, элюировали 0-60% EtOAc/циклогексан), затем опять подвергали хроматографии (картридж 11 g KP-NH, элюировали 0-70% DCM/циклогексан) с получением названного соединения в виде желтой пены (59 мг, 23%). ¹H ЯМР (400 МГц, CDCl3): δ 8,76 (с, 1H), 7,45 (м, 1H), 7,38 (м, 1H), 7,29 (м, 4H), 7,10 (д, 2H), 5,85 (с, 1H), 3,08 (с, 3H), 2,61 (с, 3H). LCMS (метод A): RT=1,50 мин, m/z=431 [M+H]+.
Стадия 2: (рац)-3-(2,6-Дихлорфенил)-1-метил-2-фенил-7-((1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
(рац)-3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2-фенил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (57 мг, 0,132 ммоль) растворяли в толуоле (2 мл) и добавляли mCPBA (49,8 мг, 0,159 ммоль). Смесь перемешивали при комнатной температуре в течение 20 минут, затем добавляли DIPEA (0,069 мл, 0,396 ммоль), затем третбутил 7-амино-3,4-дигидроизохинолинe-2(1H)-карбоксилат (32,8 мг, 0,132 ммоль). Смесь нагревали при 80°C в течение 64 часов, затем концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 10 g GraceResolv; элюировали 0-70% EtOAc/циклогексан 20 колоночных объемов) с получением коричневого стеклообразного вещества. Это вещество растворяли в DCM (1 мл) и TFA (1 мл) и перемешивали в течение 30 минут, затем концентрировали под вакуумом. Остаток растворяли в метаноле и загружали в картридж 2 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH, и растворитель удаляли под вакуумом с получением стеклообразного вещества. Остаток подвергали хроматографии (Isolera KP-NH 11 г, элюировали 0-100% EtOAc/циклогексан, затем 0-10% MeOH/EtOAc), затем растирали с диэтиловым эфиром с получением названного соединения в виде белого твердого вещества (11 мг, 16%). ¹H ЯМР (400 МГц, CDCl3): δ 8,77 (с, 1H), 7,37 (м, 6H), 7,15 (м, 6H), 5,83 (с, 1H), 4,02 (м, 2H), 3,15 (м, 2H), 3,07 (с, 3H), 2,79 (м, 2H). LCMS (метод A): RT=0,90 мин, m/z=531 [M+H]+.
Пример 24: 3-(2-Хлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: N-(2-Хлорфенил)-4-(метиламино)-2-(метилтио)-пиримидин-5-карбоксамид:
4-(Метиламино)-2-(метилтио)пиримидин-5-карбоновую кислоту (400 мг, 2,01 ммоль) суспендировали в хлорбензоле (10 мл) и добавляли трихлорид фосфора (0,184 мл, 2,11 ммоль), затем 2-хлоранилин (282 мг, 2,21 ммоль). Смесь перемешивали при 120°C в течение 64 часов, затем концентрировали под вакуумом. Остаток растворяли в водном растворе Na2CO3 и экстрагировали этилацетатом (x 2). Органические фазы сушили (Na2SO4), фильтровали и концентрировали под вакуумом с получением желтовато-белого твердого вещества. Это вещество растирали с диэтиловым эфиром с получением названного соединения в виде белого твердого вещества (460 мг, 74%). ¹H ЯМР (400 МГц, CDCl3): δ 8,53 (с, 1H), 8,42 (с, 1H), 8,32 (с, 1H), 8,21 (с, 1H), 7,42 (д, 1H), 7,31 (т, 1H), 7,10 (т, 1H), 3,10 (д, 3H), 2,57 (с, 3H). LCMS (метод A): RT=0,58/0,65 мин (раздвоенный пик), m/z=450 [M+H]+.
Стадия 2: 3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2-Хлорфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамид (250 мг, 0,810 ммоль) растворяли в ацетонитриле (10 мл) и добавляли карбонат цезия (1055 мг, 3,24 ммоль), затем дибромметан (0,170 мл, 2,429 ммоль). Смесь нагревали при 80°C в течение 1 часа. Добавляли дополнительно 0,06 мл дибромметана, и смесь нагревали при 90°C в течение 16 часов. Добавляли дополнительно 0,12 мл дибромметана, и смесь нагревали при 125°C при микроволновом излучении в течение 1 часа. Смесь концентрировали под вакуумом, и остаток подвергали хроматографии (Isolera 25 g; элюировали 0-100% EtOAc/циклогексан 20 колоночных объемов) с получением полутвердого материала, который использовали без дополнительной очистки (92 мг, 35%).
Стадия 3: 3-(2-Хлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (90 мг, 0,281 ммоль) растворяли в толуоле (4 мл) и добавляли mCPBA (106 мг, 0,337 ммоль). Смесь перемешивали в течение 15 минут, затем добавляли третбутил 4-(4-аминофенил)пиперазин-1-карбоксилат (78 мг, 0,281 ммоль), затем DIPEA (0,064 мл, 0,365 ммоль), и смесь перемешивали при 80°C в течение 16 часов. Смесь концентрировали под вакуумом затем подвергали хроматографии (Isolera 10 g GraceResolv, элюировали 0-100% EtOAc/циклогексан 20 колоночных объемов) и содержащие соединение фракции концентрировали под вакуумом с получением желтого стеклообразного вещества. Это вещество растворяли в DCM (1 мл) и TFA (1 мл) и перемешивали в течение 1 часа, затем концентрировали под вакуумом. Остаток растворяли в метаноле и загружали в картридж 2 g SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH, и растворитель концентрировали под вакуумом с получением желтого стеклообразного вещества. Это вещество подвергали хроматографии (Isolera 11 g KP-NH, элюировали 0-100% EtOAc/циклогексан, затем 0-15% MeOH/EtOAc 20 колоночных объемов) с получением названного соединения в виде бледно-желтого твердого вещества (11 мг, 8%). ¹H ЯМР (400 МГц, CDCl3): δ 8,69 (с, 1H), 7,52 (м, 3H), 7,35 (м, 3H), 7,22 (с, 1H), 6,94 (д, 2H), 4,90 (уш.м, 2H), 3,15 (м, 7H), 3,06 (м, 4H). LCMS (метод A): RT=0,65 мин, m/z=450 [M+H]+.
Пример 25: 3-(2-Хлор-6-фторфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: N-(2-Хлор-6-фторфенил)-4-(метиламино)-2-(метил-тио)пиримидин-5-карбоксамид:
Получали из 4-(метиламино)-2-(метилтио)пиримидин-5-карбоновой кислоты и 2-хлор-6-фторанилина, следуя методике, описанной в примере 1, стадии 1. ¹H ЯМР (400 МГц, DMSO-d6): δ 10,16 (с, 1H), 8,72 (с, 1H), 8,61 (уш.д, 1H), 7,41 (м, 3H), 2,95 (д, 3H), SMe сигнал, по-видимому, перекрывался растворителем. LCMS (метод A): RT=1,17 мин, m/z=327 [M+H]+.
Стадия 2: 3-(2-Хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Получали из N-(2-хлор-6-фторфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамида, следуя методике, описанной в примере 1, стадии 2. Получали неочищенное вещество и использовали его без очистки. LCMS (метод A): RT=1,19 мин, m/z=339 [M+H]+.
Стадия 3: 3-(2-Хлор-6-фторфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Получали из 3-(2-хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она, следуя методике, описанной в примере 1, стадии 3. 1H ЯМР (400 МГц, DMSO-d6): δ 9,78 (с, 1H), 8,53 (с, 1H), 7,69 (д, 2H), 7,57 (м, 2H), 7,48 (м, 1H), 6,97 (д, 2H), 5,07 (д, 1H), 5,02 (д, 1H), 3,17 (с, 3H), 3,07 (м, 4H), 2,91 (м, 4H). LCMS (метод A): RT=0,68 мин, m/z=468 [M+H]+.
Пример 26: 3-(2,6-Дихлорфенил)-1,2-диметил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
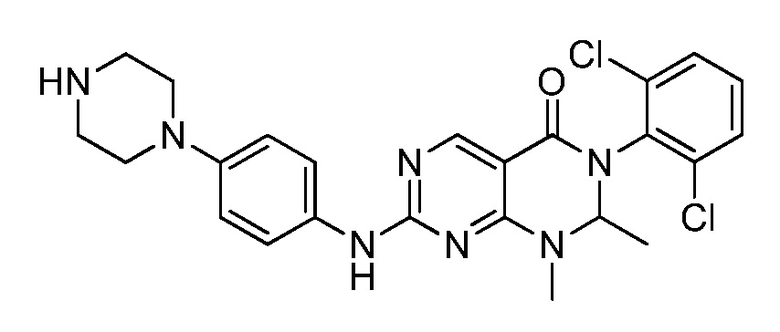
Стадия 1: 3-(2,6-Дихлорфенил)-1,2-диметил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(метиламино)-2-(метилтио)пиримидин-5-карбоксамид (200 мг) суспендировали в ацетальдегиде (1,5 мл). Смесь нагревали при микроволновом излучении при 150°C в течение 7 часов. Добавляли воду, и смесь экстрагировали этилацетатом (x 2). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток подвергали хроматографии (Isolera 10 g Si, элюировали 0-60% EtOAc/циклогексан) с получением названного соединения в виде желтого сиропа (65 мг, 29%). 1H ЯМР (400 МГц, DMSO-d6): δ 8,72 (с, 1H), 7,47 (т, 2H), 7,29 (д, 1H), 4,97 (кв, 1H), 3,22 (с, 3H), 2,58 (с, 3H), 1,56 (д, 3H). LCMS (метод A): RT=1,30 мин, m/z=369 [M+H]+.
Стадия 2: 3-(2,6-Дихлорфенил)-1,2-диметил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Получали из 3-(2,6-дихлорфенил)-1,2-диметил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она, следуя методике, описанной в примере 1, стадии 3. 1H ЯМР (400 МГц, DMSO-d6): δ 9,63 (с, 1H), 8,44 (с, 1H), 7,67 (м, 4H), 7,48 (т, 1H), 6,89 (д, 1H), 5,15 (кв, 1H), 3,14 (с, 3H), 2,98 (м, 4H), 2,83 (м, 4H), 1,43 (д, 3H). LCMS (метод A): RT=0,75 мин, m/z=498 [M+H]+.
Пример 27: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(морфолино-метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,17 ммоль) подвергали взаимодействию с 4-(морфолинометил)анилином (36 мг, 0,19 ммоль), следуя методике примера 1, с получением названного соединения в виде белого твердого вещества (37 мг, 44%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,89 (с, 1H), 8,49 (с, 1H), 7,74 (д, 2H), 7,65 (д, 2H), 7,49 (м, 1H), 7,24 (д, 2H), 4,98 (с, 2H), 3,57 (м, 4H), 3,41 (с, 2H), 3,12 (с, 3H), 2,35 (м, 4H). LCMS (метод A): RT=0,78 мин, m/z=499 [M+H]+.
Пример 28: 6-(2,6-Дихлорфенил)-2-((4-(пиперазин-1-ил)-фенил)амино)-6a,7,8,9-тетрагидропиримидо[5,4-e]пирроло[1,2-a]-пиримидин-5(6H)-он

Стадия 1: 2-Хлор-1-(2,6-дихлорфенил)-4-((4-гидроксибутил)-амино)пиримидин-5-карбоксамид:
К раствору 2,4-дихлор-N-(2,6-дихлорфенил)пиримидин-5-карбоксамида (350 мг, 1,04 ммоль) (полученного, как описано в патентном документе US 2005/0209221) и DIPEA (0,200 мл, 1,14 ммоль) в THF (5 мл) при 0°C добавляли 4-аминобутан-1-ол (0,096 мл, 1,04 ммоль) в THF (1 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 30 минут, концентрировали под вакуумом, и остаток распределяли между водой и DCM. Слои затем разделяли, используя фазовый сепаратор, и органический слой концентрировали под вакуумом с получением названного соединения (0,4 г, 1,03 ммоль, 99%) в виде белого твердого вещества. LCMS (метод A): RT=1,08 мин, m/z=389 [M+H]+.
Стадия 2: третбутил 4-(4-((5-((2,6-дихлорфенил)карбамоил)-4-((4-гидроксибутил)амино)пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилат:
Раствор 2-хлор-N-(2,6-дихлорфенил)-4-((4-гидроксибутил)-амино)пиримидин-5-карбоксамида (401 мг, 1,03 ммоль), третбутил 4-(4-аминофенил)пиперазин-1-карбоксилата (400 мг, 1,44 ммоль) и DIPEA (0,36 мл, 2,06 ммоль) в безводном DMF (7 мл) перемешивали при 90°C в течение 60 часов. Реакционную смесь разбавляли с помощью EtOAc и воды, и слои разделяли, водный слой затем экстрагировали еще раз с помощью EtOAc, и органические слои объединяли, промывали водой 4 раза, сушили (MgSO4), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (SiO2, 30-70% этилацетат в циклогексане) с получением названного соединения (480 мг, 74%) в виде коричневого твердого вещества. LCMS (метод A): RT=1,17 мин, m/z=630 [M+H]+.
Стадия 3: третбутил 4-(4-((5-((2,6-дихлорфенил)карбамоил)-4-((4-оксобутил)амино)пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилат:
К раствору третбутил 4-(4-((5-((2,6-дихлорфенил)карбамоил)-4-((4-гидроксибутил)амино)пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилата (480 мг, 0,761 ммоль) в THF (10 мл) добавляли 1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензиодоксол-3-(1H)-он [реактив Десса-Мартина] (517 мг, 1,218 ммоль). Гетерогенную смесь затем перемешивали при 50°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали, фильтрат концентрировали под вакуумом и очищали флэш-хроматографией (SiO2, 30-65% этилацетат в циклогексане) с получением названного соединения (110 мг, 23%) в виде коричневой смолы. LCMS (метод A): RT=1,27 мин, m/z=628 [M+H]+.
Стадия 4: третбутил 4-(4-((6-(2,6-дихлорфенил)-5-оксо-5,6,6a,7,8,9-гексагидропиримидо[5,4-e]пирроло[1,2-a]пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилат:
Третбутил 4-(4-((5-((2,6-дихлорфенил)карбамоил)-4-((4-оксо-бутил)амино)пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилат (55 мг, 0,088 ммоль) и моногидрат п-толуолсульфоновой кислоты (1,7 мг, 8,75 мкмоль) в толуоле (1 мл) нагревали при 150°C в течение 30 минут при микроволновом излучении. Растворители удаляли под вакуумом, и остаток очищали флэш-хроматографией (SiO2, 20-60% этилацетат в циклогексане) с получением названного соединения (35 мг, 66%) LCMS (метод A): RT=1,46 мин, m/z=610 [M+H]+.
Стадия 5: 6-(2,6-Дихлорфенил)-2-((4-(пиперазин-1-ил)фенил)-амино)-6a,7,8,9-тетрагидропиримидо[5,4-e]пирроло[1,2-a]-пиримидин-5(6H)-он:
К раствору третбутил 4-(4-((6-(2,6-дихлорфенил)-5-оксо-5,6,6a,7,8,9-гексагидропиримидо[5,4-e]пирроло[1,2-a]пиримидин-2-ил)амино)фенил)пиперазин-1-карбоксилата (70 мг, 0,115 ммоль) в DCM добавляли TFA (8,8 мкл, 0,115 ммоль) при комнатной температуре, реакционную смесь затем перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь затем концентрировали под вакуумом, и остаток растирали с этилацетатом с получением коричневого твердого вещества. Твердое вещество собирали и растворяли в MeOH и загружали в картридж Biotage SCX-2, картридж промывали с помощью MeOH, и продукт элюировали, используя 2 M NH3 в MeOH. Фракции, содержащие продукт, концентрировали под вакуумом, и продукт сушили (Genevac EZ-2) с получением названного соединения (26 мг, 44%) в виде коричневого твердого вещества. ¹H ЯМР (400 МГц, DMSO-d6): δ 9,72 (с, 1H), 8,43 (с, 1H), 7,66-7,63 (м, 4H), 7,49 (т, 1H), 6,90 (д, 2H), 5,63-5,60 (м, 1H), 4,77-3,64 (м, 2H), 3,08-3,05 (м, 4H), 2,95-2,93 (м, 4H), 2,10-1,91 (м, 2H), 1,86-1,71 (м, 2H). LCMS (метод A): RT=0,79 мин, m/z=510 [M+H]+.
Пример 29: 3-(2,6-Дихлорфенил)-1-метил-7-((4-((4-метил-пиперазин-1-ил)метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-илметил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,241 ммоль) кипятили с обратным холодильником в смеси муравьиной кислоты (2 мл) и формальдегида (37 масс.% в воде, 2,5 мл) в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и загружали в картридж SCX-2. Колонку промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH, и растворитель концентрировали под вакуумом. Полученный остаток очищали препаративной ВЭЖХ с получением названного соединения (25 мг, 20%) в виде белого твердого вещества. ¹H ЯМР (400 МГц, DMSO-d6): δ 9,88 (с, 1H), 8,48 (с, 1H), 7,73 (д, 2H), 7,65 (д, 2H), 7,48 (м, 1H), 7,22 (д, 2H), 4,98 (с, 2H), 3,39 (с, 2H), 3,11 (с, 3H), 2,33 (уш.с, 8H), 2,14 (м, 3H). LCMS (метод A): RT=0,74 мин, m/z=512 (weak) [M+H]+.
Пример 30: (рац)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(S-метилсульфонимидоил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 3-(2,6-Дихлорфенил)-1-метил-7-((4-(метилтио)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Получали из 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она, следуя методике, описанной в примере 1, стадии 3. Получали неочищенное вещество и использовали без очистки. LCMS (метод A): RT=1,34 мин, m/z=446 [M+H]+.
Стадия 2: (рац)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(метил-сульфинил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-((4-(метилтио)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (148 мг, 0,332 ммоль) растворяли в ацетонитриле (5 мл) и добавляли хлорид железа(III) (1,6 мг, 0,001 ммоль), затем йодную кислоту (79 мг, 0,348 ммоль). Полученную смесь перемешивали в течение 16 часов и концентрировали под вакуумом. Остаток растворяли в водном растворе тиосульфата натрия и экстрагировали с помощью DCM (x 2). Объединенные органические экстракты промывали солевым раствором затем сушили (Na2SO4), фильтровали и концентрировали с получением названного соединения в виде коричневой пены (150 мг), которое использовали неочищенным. LCMS (метод A): RT=0,91 мин, m/z=462 [M+H]+.
Стадия 3: (рац)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(S-метил-сульфонимидоил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(рац)-3-(2,6-Дихлорфенил)-1-метил-7-((4-(метилсульфинил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,216 ммоль) растворяли в DCM (3 мл) и добавляли димер ацетата родия(II) (5,0 мг, 1,13 мкмоль), затем оксид магния (52 мг, 1,30 ммоль) и 2,2,2-трифторацетамид (74 мг, 0,65 ммоль). Затем добавляли диацетат йодбензола (157 мг, 0,49 ммоль), и полученную смесь перемешивали при 40°C в течение 16 часов. Добавляли еще одну порцию димера ацетата родия(II) (5,0 мг, 1,13 мкмоль), затем 2,2,2-трифторацетамид (37 мг, 0,32 ммоль) и диацетат йодбензола (78 мг, 0,24 ммоль). Смесь перемешивали при 40°C еще 16 часов, затем фильтровали через целит® и концентрировали. Остаток подвергали хроматографии (Isolera 10 g; элюировали 0-100% EtOAc/циклогексан, затем 0-20% MeOH/EtOAc) с получением белого твердого вещества. Это вещество растворяли в метаноле (2 мл) и добавляли карбонат калия (30 мг, 0,217 ммоль), и смесь перемешивали при комнатной температуре в течение 16 часов, затем концентрировали под вакуумом. Остаток подвергали хроматографии (Isolera 12 г GraceResolv; элюировали 0-100% EtOAc/циклогексан, затем 0-25% MeOH/EtOAc) с получением названного соединения в виде серого твердого вещества (5,3 мг, 5%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,31 (с, 1H), 8,55 (с, 1H), 8,00 (д, 2H), 7,87 (д, 2H), 7,68 (д, 2H), 7,65 (т, 1H), 5,03 (с, 2H), 4,05 (с, 1H), 3,16 (с, 3H), 3,04 (с, 3H). LCMS (метод A): RT=0,91 мин, m/z=477 [M+H]+.
Пример 31: 3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 4-(2-формил-4-нитрофенил)пиперазин-1-карбоксилат:
Суспензию 2-фтор-5-нитробензальдегида (3 г, 17,74 ммоль), третбутил пиперазин-1-карбоксилата (3,30 г, 17,74 ммоль) и карбоната калия (3,68 г, 26,6 ммоль) в безводном DMF (15 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (5,89 г, 99%). 1H ЯМР (300 МГц, CDCl3): δ 10,11 (с, 1H), 8,65 (д, 1H), 8,33 (дд, 1H), 7,09 (д, 1H), 3,68 (т, 4H), 3,26 (т, 4H), 1,49 (с, 9H). LCMS (метод C) RT=1,60 мин, m/z=336 [M+H]+.
Стадия 2: третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-пиперазин-1-карбоксилат:
Раствор третбутил 4-(2-формил-4-нитрофенил)пиперазин-1-карбоксилата (5,89 г, 17,56 ммоль) в безводном THF (32,5 мл) охлаждали до 0°C, затем порциями добавляли боргидрид натрия (0,664 г, 17,56 ммоль). Реакционную смесь перемешивали при 0°C в течение 90 минут, затем гасили водой (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (5,42 г, 91%). 1H ЯМР (300 МГц, CDCl3): δ 8,32 (д, 1H), 8,13 (дд, 1H), 7,10 (д, 1H), 4,80 (д, 2H), 3,61 (т, 4H), 3,05 (т, 1H), 2,99 (т, 4H), 1,48 (с, 9H). LCMS (метод C): RT=1,48 мин, m/z=338 [M+H]+.
Стадия 3: третбутил 4-(2-(метоксиметил)-4-нитрофенил)-пиперазин-1-карбоксилат:
Раствор третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-пиперазин-1-карбоксилата (2,00 г, 5,93 ммоль) в безводном THF (19,7 мл) охлаждали до 0°C, затем добавляли гидрид натрия (60% в минеральном масле, 0,26 г, 6,52 ммоль). После перемешивания при 0°C в атмосфере азота в течение 10 минут, добавляли метилйодид (0,45 мл, 7,11 ммоль). Полученную смесь подогревали до комнатной температуры и перемешивали в течение 60 минут. Добавляли дополнительное количество метилйодида (0,13 мл, 2,08 ммоль), и смесь перемешивали в течение еще 60 минут. Реакционную смесь разбавляли раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и подвергали хроматографии (картридж с диоксидом кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 50:50) с получением названного соединения в виде желтого твердого вещества (1,48 г, 71%). 1H ЯМР (300 МГц, CDCl3): δ 8,35 (д, 1H), 8,13 (дд, 1H), 7,04 (д, 1H), 4,48 (с, 2H), 3,60 (т, 4H), 3,46 (с, 3H), 3,00 (т, 4H), 1,49 (с, 9H). LCMS (метод C) RT=1,79 мин, m/z=352 [M+H]+.
Стадия 4: третбутил 4-(4-амино-2-(метоксиметил)фенил)-пиперазин-1-карбоксилат:
Раствор третбутил 4-(2-(метоксиметил)-4-нитрофенил)-пиперазин-1-карбоксилата (1,48 г, 4,21 ммоль) в метаноле (100 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (1,33 г, 98%). 1H ЯМР (300 МГц, CDCl3): δ 6,91 (д, 1H), 6,78 (д, 1H), 6,59 (дд, 1H), 4,49 (с, 2H), 3,47-3,61 (м, 6H), 3,42 (с, 3H), 2,77 (т, 4H), 1,48 (с, 9H). LCMS (метод C): RT=0,94 мин, m/z=322 [M+H]+.
Стадия 5: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(метоксиметил)фенил)пиперазин-1-карбоксилат:
Раствор mCPBA 50% чистоты (291 мг, 0,844 ммоль) в дихлорметане (1,50 мл) пропускали через фазовый сепаратор и промывали дополнительным количеством дихлорметана (0,750 мл). Этот раствор добавляли к перемешиваемой суспензии 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-она (250 мг, 0,704 ммоль) в безводном толуоле (7,5 мл), и смесь перемешивали в течение 30 минут. Добавляли основание Хунига (0,369 мл, 2,111 ммоль), затем третбутил 4-(4-амино-2-(метоксиметил)фенил)пиперазин-1-карбоксилат (215 мг, 0,669 ммоль), и смесь нагревали до 75°C в течение 40 часов. После охлаждения, непосредственно реакционную смесь очищали флэш-хроматографией (5-65% этилацетат в циклогексане) с получением названного соединения (360 мг, 81%). LCMS (метод C): RT=1,81 мин, m/z=628 [M+H]+.
Стадия 6: 3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
К перемешиваемому раствору третбутил 4-(4-((6-(2,6-дихлор-фенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]-пиримидин-2-ил)амино)-2-(метоксиметил)фенил)пиперазин-1-карбоксилата в безводном дихлорметан (6 мл) добавляли трифторуксусную кислоту (1,5 мл, 19,5 ммоль), и раствор перемешивали в течение 2 часов. Картридж с диоксидом кремния SCX-2 (10 г) предварительно обрабатывали смесью 20 об.% метанола в дихлорметане (100 мл). Реакционную смесь загружали в колонку SCX-2, используя дихлорметан (3×2 мл) для споласкивания колбы. Через 5 минут, колонку промывали смесью 20 об.% метанола в дихлорметане (100 мл), смесью затем 20% об.% (7 M аммиак в метаноле) в дихлорметане (50 мл). Содержащую аммиак фракцию концентрировали под вакуумом. Остаток растворяли в метаноле (3 мл) и воде (12 мл) и лиофилизировали с получением названного соединения (260 мг, 86%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,55 (с, 1H), 7,84-8,01 (м, 1H), 7,53-7,64 (м, 3H), 7,44 (дд, 1H), 7,14 (д, 1H), 5,00 (с, 2H), 4,59 (с, 2H), 3,45 (с, 3H), 3,21 (с, 3H), 3,00 (т, 4H), 2,90 (т, 4H). LCMS (метод C): RT=0,77 мин, m/z=528 [M+H]+.
Пример 32: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
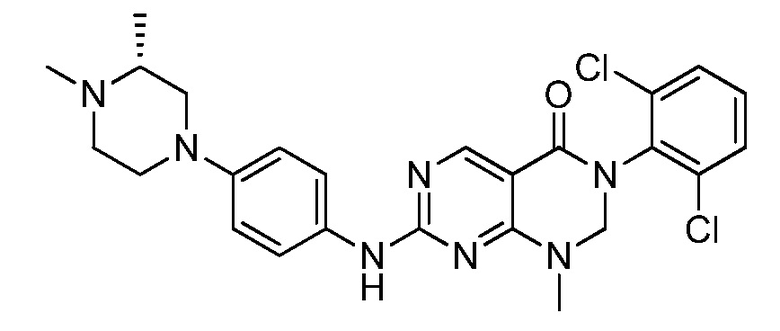
Стадия 1: (R)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,42 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (123 мг, 0,422 ммоль), следуя методике примера 31, с получением названного соединения (120 мг, 41%), которое использовали без дополнительной очистки.
Стадия 2: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
(R)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,12 ммоль) подвергали взаимодействию с формальдегидом (0,018 мл, 0,24 ммоль), следуя методике примера 35, с получением названного соединения (56 мг, 91%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,72 (с, 1H), 8,43 (с, 1H), 7,66 (м, 3H), 7,46 (м, 1H), 6,91 (д, 2H), 4,95 (с, 2H), 3,45 (м, 2H), 3,34 (с, 1H), 3,09 (с, 3H), 2,83 (м, 1H), 2,67 (м, 1H), 2,30 (м, 1H), 1,05 (д, 3H). LCMS (метод C): RT=0,72 мин, m/z=512 [M+H]+.
Пример 33: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,117 ммоль) подвергали взаимодействию с формальдегидом (0,018 мл), следуя методике примера 35, с получением названного соединения (59 мг, 96%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,82 (с, 1H), 8,45 (с, 1H), 8,12 (с, 1H), 7,99 (с, 1H), 7,65 (д, 2H), 7,48 (м, 1H), 7,01 (д, 1H), 5,09 (т, 1H), 4,97 (с, 2H), 4,54 (д, 2H), 3,17 (с, 3H), 2,80 (м, 4H), 2,4 (м, 4H), 2,25 (с, 3H). LCMS (метод C): RT=0,69 мин, m/z=528 [M+H]+.
Пример 34: (R)-3-(2,6-дихлорфенил)-7-((4-(3-(гидрокси-метил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

(R)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,117 ммоль) подвергали взаимодействию с формальдегидом (0,017 мл), следуя методике примера 35, с получением названного соединения (60 мг, 97%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,72 (с, 1H), 8,44 (с, 1H), 7,7-7,6 (м, 4H), 7,48 (м, 1H), 6,91 (д, 2H), 4,95 (с, 2H), 3,65 (м, 2H), 3,43 (м, 1H), 3,17 (д, 1H), 3,09 (с, 3H), 2,80 (м, 1H), 2,65 (м, 1H),2,45 (м, 1H), 2,31 (м, 1H), 2,25 (с, 3H), 2,13 (м, 1H). LCMS (метод C): RT=0,69 мин, m/z=528 [M+H]+.
Пример 35: 3-(2,6-дихлорфенил)-1-метил-7-((4-(5-метилгекса-гидропирроло[3,4-c]пиррол-2(1H)-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 5-(4-нитрофенил)гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат:
Суспензию 1-фтор-4-нитробензола (1,662 г, 11,78 ммоль), третбутил гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилата (2,5 г, 11,78 ммоль) и карбоната калия (2,441 г, 17,66 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение ночи. Добавляли дополнительное количество DMF (5 мл), и смесь нагревали при 50°C в течение еще 24 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой и воду отсасывали с получением названного соединения в виде желтого твердого вещества (3,21 г, 82%). 1H ЯМР (300 МГц, CDCl3): δ 8,13 (дт, 2H), 6,47 (дт, 2H), 3,59-3,74 (м, 4H), 3,21-3,43 (м, 4H), 2,98-3,14 (м, 2H), 1,45 (с, 9H). LCMS (метод C): RT=1,66 мин, m/z=334 [M+H]+.
Стадия 2: третбутил 5-(4-аминофенил)гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат:
Раствор третбутил 5-(4-нитрофенил)гексагидропирроло[3,4-c]-пиррол-2(1H)-карбоксилата (2,3 г, 6,90 ммоль) в метаноле (500 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1,5 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде серого твердого вещества (1,35 г, 65%). 1H ЯМР (300 МГц, CDCl3): δ 6,67 (д, 2H), 6,45 (д, 2H), 3,63 (дд, 2H), 2,87-3,55 (м, 10H), 1,45 (с, 9H). LCMS (метод C): RT=0,76 мин, m/z=304 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-метил-7-((4-(5-метилгекса-гидропирроло[3,4-c]пиррол-2(1H)-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
Раствор третбутил 5-(4-аминофенил гексагидропирроло[3,4-c]-пиррол-2(1H)-карбоксилата (50 мг, 0,098 ммоль), формальдегида (37% в воде, 0,015 мл, 0,196 ммоль, 2 экв.) и триацетоксиборгидрида натрия (0,062 г, 0,294 ммоль, 3 экв.) перемешивали при комнатной температуре в течение 1 часа. Раствор затем загружали в колонку SCX-2. Проводили элюирование смесью DCM/MeOH (20/80, 20 мл), затем 7 N раствором аммиака в метаноле. Аммиачную фракцию концентрировали под вакуумом. Остаток разбавляли метанолом и водой и лиофилизировали с получением названного соединения (25 мг, 49%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,66 (уш.с, 1H), 8,43 (с, 1H), 7,64 (м, 2H), 7,56-7,45 (м, 3H), 6,62 (д, 2H), 4,94 (с, 2H), 3,34 (м, 3H), 3,08 (с, 3H), 3,02 (м, 2H), 2,85 (м, 2H), 2,56 (м, 1H), 2,37 (м, 2H). LCMS (метод C): RT=0,72 мин, m/z=524 [M+H]+.
Пример 36: 3-(2,6-дихлорфенил)-7-((4-(гексагидропирроло- [3,4-c]пиррол-2(1H)-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
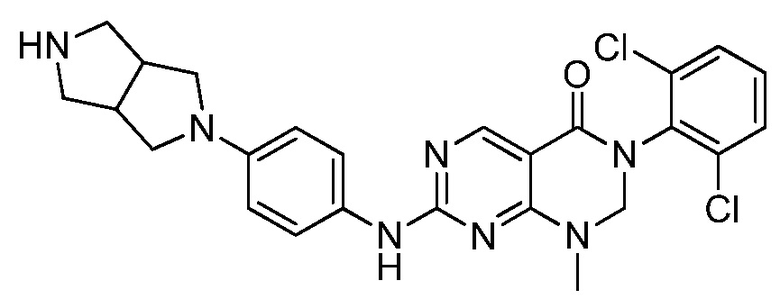
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,422 ммоль) подвергали взаимодействию с третбутил 5-(4-аминофенил)гекса-гидропирроло[3,4-c]пиррол-2(1H)-карбоксилатом (128 мг, 0,422 ммоль), следуя методике примера 31, с получением названного соединения (120 мг, 56%).¹H ЯМР (400 МГц, DMSO-d6): δ 9,66 (с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,56-7,45 (м, 3H), 6,62 (д, 2H), 4,94 (с, 2H), 3,40-3,32 (м, 3H), 3,08 (с, 3H), 3,10-2,95 (м, 3H), 2,85 (м, 2H), 2,66 (м, 2H). LCMS (метод C): RT=0,72 мин, m/z=510 [M+H]+.
Пример 37: 7-((4-(1-аминоциклобутил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
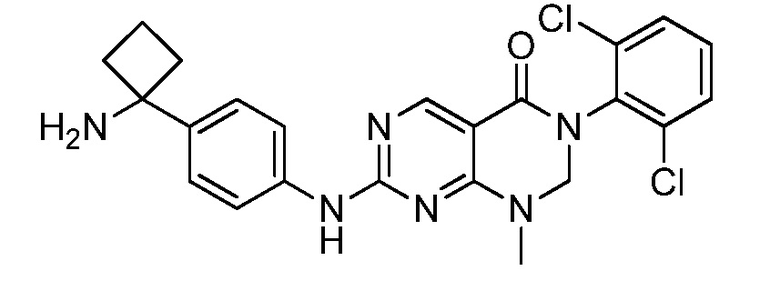
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,422 ммоль) подвергали взаимодействию с третбутил (1-(4-аминофенил)цикло-бутил)карбаматом (111 мг, 0,422 ммоль), следуя методике примера 31, с получением названного соединения (45 мг, 83%).¹H ЯМР (400 МГц, DMSO-d6): δ 9,89 (с, 1H), 8,49 (с, 1H), 7,73 (д, 2H), 7,65 (д, 2H), 7,48 (т, 1H), 7,40 (д, 2H), 4,98 (с, 2H), 3,12 (с, 3H), 2,38 (м, 2H), 2,07 (м, 2H), 1,95 (м, 1H), 1,60 (м, 1H). LCMS (метод A): RT=0,81 мин, m/z=469 [M+H]+
Пример 38: (R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(2-формил-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Суспензию 2-фтор-5-нитробензальдегида (2,53 г, 14,98 ммоль), (R)-третбутил 2-метилпиперазин-1-карбоксилата (3 г, 14,98 ммоль) и карбоната калия (3,11 г, 22,47 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Смесь затем экстрагировали этилацетатом (100 мл) и промывали 50:50 вода:солевой раствор. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (4,72 г, 90%). 1H ЯМР (300 МГц, CDCl3): δ 10,15 (с, 1H), 8,63 (д, 1H), 8,31 (дд, 1H), 7,08 (д, 1H), 4,41 (уш.с, 1H), 3,97 (дт, 1H), 3,20-3,53 (м, 4H), 3,08 (тд, 1H), 1,48 (д, 9H), 1,31 (д, 3H). LCMS (метод C): RT=1,68 мин, m/z=350 [M+H]+.
Стадия 2: (R)-третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-формил-4-нитрофенил)-2-метил-пиперазин-1-карбоксилат (4,72 г, 13,51 ммоль) в безводном THF (25,02 мл) охлаждали до 0°C, затем порциями добавляли боргидрид натрия (0,511 г, 13,51 ммоль). Реакционную смесь перемешивали при 0°C в течение 90 минут, затем гасили водой (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (4,39 г, 92%). 1H ЯМР (300 МГц, CDCl3): δ 8,34 (д, 1H), 8,12 (дд, 1H), 7,09 (д, 1H), 4,81 (уш.с, 2H), 4,37 (уш.с, 1H), 3,99 (дт, 1H), 3,27 (тд, 1H), 3,12 (ддд, 1H), 3,05 (дт, 1H), 2,88-3,01 (м, 2H), 2,80 (тд, 1H), 1,48 (с, 9H), 1,36 (д, 3H). LCMS (метод C): RT=1,56 мин, m/z=352 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-амино-2-(гидроксиметил)фенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат (2,4 г, 6,83 ммоль) в метаноле (100 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (2,05 г, 93%). 1H ЯМР (300 МГц, CDCl3): δ 6,99 (д, 1H), 6,57 (дд, 1H), 6,53 (д, 1H), 4,96 (уш.с, 1H), 4,69 (с, 2H), 4,26-4,39 (м, 1H), 3,94 ((уш.д, 1H), 3,62 (уш.с, 2H), 3,21 (тд, 1H), 2,87-2,97 (м, 2H), 2,65-2,82 (м, 2H), 1,48 (с, 9H), 1,33 (д, 3H). LCMS (метод C): RT=0,81 мин, m/z=322 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(гидроксиметил)фенил)-2-метилпиперазин-1-карбоксилатом (109 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения (57,5 мг, 76%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,55 (с, 1H), 7,90 (уш.с, 1H), 7,54-7,61 (м, 3H), 7,44 (дд, 1H), 7,15 (д, 1H), 5,00 (с, 2H), 4,77 (с, 2H), 3,21 (с, 3H), 2,94-3,06 (м, 5H), 2,77 (тд, 1H), 2,46 (т, 1H), 1,13 (д, 3H). LCMS (метод C): RT=0,71 мин, m/z=528 [M+H]+.
Пример 39: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 5-(4-нитрофенил)-2,5-диазабицикло-[2,2,1]гептан-2-карбоксилат:
К перемешиваемой суспензии 1-фтор-4-нитробензола (1,294 г, 9,17 ммоль) и карбоната калия (5,07 г, 36,7 ммоль) в безводном DMF (10 мл) добавляли третбутил 2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (2 г, 10,09 ммоль), и смесь нагревали при 80°C в течение 40 часов. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (50 мл). Водный слой отделяли и экстрагировали этилацетатом (3×50 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток подвергали хроматографии (градиент 0-50% этилацетат в циклогексане) с получением названного соединения (2,11 г, 72,0%). 1H ЯМР (400 МГц, CDCl3): δ 8,12 (д, 2H), 6,49 (д, 2H), 4,53-4,71 (м, 2H), 3,59 (д, 1H), 3,23-3,51 (м, 3H), 2,00 (м, 2H), 1,44 (д, 9H). LCMS (метод C): RT=1,57 мин, m/z=320 [M+H]+.
Стадия 2: третбутил 5-(4-аминофенил)-2,5-диазабицикло-[2,2,1]гептан-2-карбоксилат:
Раствор третбутил 5-(4-нитрофенил)-2,5-диазабицикло-[2,2,1]гептан-2-карбоксилата (1,20 г, 3,76 ммоль) в тетрагидрофуране (50 мл) пропускали два раза через проточный реактор H-Cube, оснащенный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 20°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (1,03 г, 95%). LCMS (метод C): RT=0,68 мин, m/z=290 [M+H]+.
Стадия 3: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-аминофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (98 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения (84,0 мг, 100%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,50 (с, 1H), 7,57 (д, 2H), 7,49 (уш.с, 2H), 7,43 (дд, 1H), 6,64 (д, 2H), 4,97 (с, 2H), 4,39 (с, 1H), 3,78 (с, 1H), 3,63 (дд, 1H), 3,16 (с, 3H), 3,06 (т, 2H), 2,97 (дд, 1H), 1,99 (д, 1H), 1,82 (д, 1H). LCMS (метод C): RT=0,72 мин, m/z=496 [M+H]+.
Пример 40: 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (2S,5R)-третбутил 2,5-диметил-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
К перемешиваемой суспензии 1-фтор-4-нитробензола (0,60 г, 4,24 ммоль) и карбоната калия (2,34 г, 16,97 ммоль) в безводном DMF (5 мл) добавляли (2S,5R)-третбутил 2,5-диметилпиперазин-1-карбоксилат (1 г, 4,67 ммоль), и смесь нагревали при 90°C в течение 120 часов. После охлаждения, смесь распределяли между солевой раствор/вода (1:1, 50 мл) и этилацетатом (25 мл). Водный слой отделяли и экстрагировали этилацетатом (3×25 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×12,5 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетат в циклогексане) с получением названного соединения (1,03 г, 72,4%). 1H ЯМР (400 МГц, CDCl3): δ 8,12 (д, 2H), 6,77 (д, 2H), 4,24-4,58 (м, 1H), 4,03-4,16 (м, 1H), 3,73-3,93 (м, 1H), 3,31-3,49 (м, 3H), 1,49 (с, 9H), 1,24 (д, 3H), 1,18 (д, 3H). LCMS (метод C): RT=1,80 мин, m/z=336 [M+H]+.
Стадия 2: (2S,5R)-третбутил 4-(4-аминофенил)-2,5-диметил-пиперазин-1-карбоксилат:
Раствор (2S,5R)-третбутил 2,5-диметил-4-(4-нитрофенил)-пиперазин-1-карбоксилата (1,03 г, 3,07 ммоль) в метаноле (100 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (900 мг, 96%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,62 (д, 2H), 6,49 (д, 2H), 4,51 (с, 2H), 4,21-4,29 (м, 1H), 3,67-3,75 (м, 1H), 3,63 (д, 1H), 3,33 (дд, 1H), 3,04 (дд, 1H), 2,79 (д, 1H), 1,41 (с, 9H), 1,17 (д, 3H), 0,78 (д, 3H). LCMS (метод C): RT=0,82 мин, m/z=306 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (200 мг, 0,563 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (163 мг, 0,535 ммоль), следуя методике примера 31, с получением названного соединения (151 мг, 92%). 1H ЯМР (300 МГц, MeOH-d4): δ 8,56 (с, 1H), 7,69 (д, 2H), 7,57 (д, 2H), 7,43 (дд, 1H), 7,18 (д, 2H), 5,00(с, 2H), 3,19 (с, 3H), 2,92-3,11 (м, 4H), 2,64 (дд, 1H), 2,50 (т, 1H), 1,09 (д, 3H), 0,90 (д, 3H). LCMS (метод C): RT=0,79 мин, m/z=512 [M+H]+.
Пример 41: 3-(2,6-дихлорфенил)-1-метил-7-((4-морфолино-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
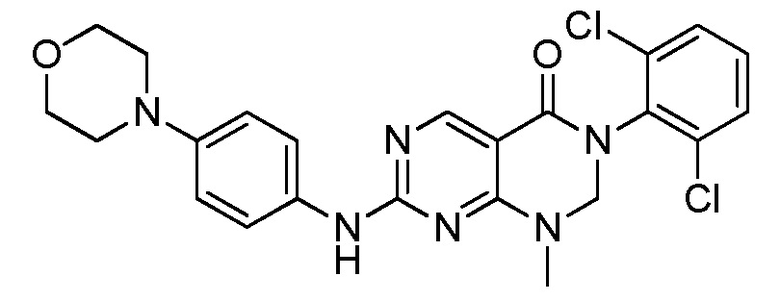
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с 4-морфолиноанилином (60 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (67 мг, 41%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,74 (с, 1H), 8,45 (с, 1H), 7,64 (д, 4H), 7,50 (т, 1H), 6,91 (д, 2H),4,97 (с, 2H), 3,74 (м, 4H), 3,09 (с, 3H), 3,04 (м, 4H). LCMS (метод C): RT=1,17 мин, m/z=485 [M+H]+.
Пример 42: 3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидотио-морфолино)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
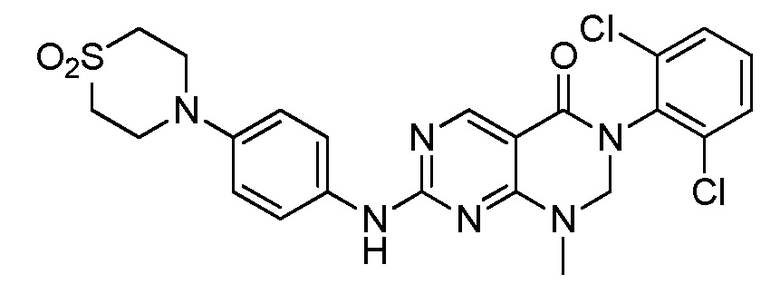
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с трет-5-бутил 4-(4-амино-2-метокси-фенил)пиперазин-1-карбоксилатом (76 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения (91 мг, 53%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,78 (с, 1H), 8,45 (с, 1H), 7,7-7,6 (м, 4H), 7,48 (м, 1H), 7,01 (д, 2H), 4,96 (с, 2H), 3,72 (м, 4H), 3,13 (м, 4H), 3,03 (с, 3H). LCMS (метод C): RT=1,09 мин, m/z=533 [M+H]+.
Пример 43: (R)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
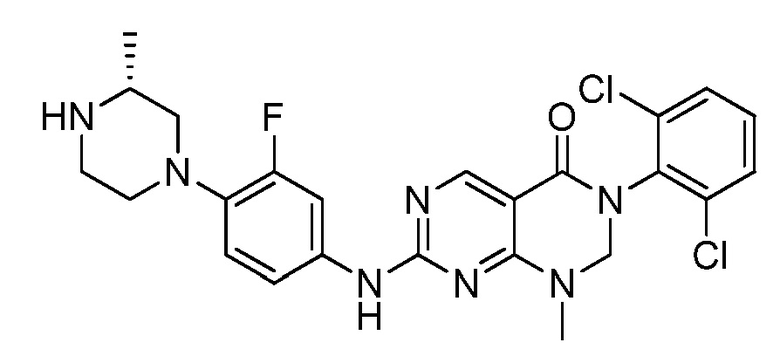
Стадия 1: (R)-третбутил 4-(2-фтор-4-нитрофенил)-2-метил-пиперазин-1-карбоксилат:
К перемешиваемой суспензии 1,2-дифтор-4-нитробензола (1,444 г, 9,08 ммоль) и карбоната калия (5,02 г, 36,3 ммоль) в безводном DMF (10 мл) добавляли (R)-третбутил 2-метилпиперазин-1-карбоксилат (2 г, 9,99 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетат (50 мл). Водный слой отделяли и экстрагировали этилацетатом (3×50 мл). Объединенные этилацетатные фракции промывали солевым раствором/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток подвергали хроматографии (градиент 0-30% этилацетат в циклогексане) с получением названного соединения (2,56 г, 83%). 1H ЯМР (400 МГц, CDCl3): δ 7,98 (дд, 1H), 7,90 (дд, 1H), 6,89 (т, 1H), 4,30-4,40 (м, 1H), 3,97 (д, 1H), 3,44-3,56 (м, 2H), 3,28 (тд, 1H), 3,07 (дд, 1H), 2,93 (тд, 1H), 1,49 (с, 9H), 1,31 (д, 3H). LCMS (метод C): RT=1,87 мин, m/z=284 [M-бутен]+.
Стадия 2: (R)-третбутил 4-(4-амино-2-фторфенил)-2-метил-пиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-фтор-4-нитрофенил)-2-метил-пиперазин-1-карбоксилата (1,3 г, 3,83 ммоль) в тетрагидрофуране (40 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (1,17 г, 99%). 1H ЯМР (300 МГц, CDCl3): δ 6,74 (дд, 1H), 6,32 (дд, 1H), 6,29 (дд, 1H), 5,01 (уш.с, 2H), 4,07-4,20 (м, 1H), 3,75 (д, 1H), 3,08 (тд, 1H), 2,98 (д, 1H), 2,91 (д, 1H), 2,62 (дд, 1H), 2,55 (дд, 1H), 1,41 (с, 9H), 1,23 (д, 3H). LCMS (метод C): RT=1,32 мин, m/z=310 [M+H]+.
Стадия 3: (R)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (250 мг, 0,704 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-фтор-фенил)-2-метилпиперазин-1-карбоксилатом (207 мг, 0,669 ммоль), следуя методике примера 31, с получением названного соединения (227 мг, 88%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,56 (с, 1H), 7,70 (дд, 1H), 7,58 (д, 2H), 7,44 (дд, 1H), 7,36 (д, 1H), 7,03 (т, 1H), 5,01 (с, 2H), 3,28 (д, 2H), 3,20 (с, 3H), 2,98-3,11 (м, 3H), 2,75 (тд, 1H), 2,42 (т, 1H), 1,14 (д, 3H). LCMS (метод C): RT=0,83 мин, m/z=516 [M+H]+.
Пример 44: 3-(2,6-дихлорфенил)-1-метил-7-((4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
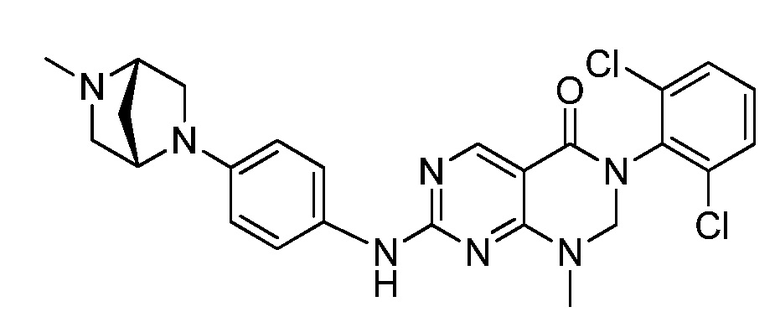
Стадия 1: 3-(2,6-дихлорфенил)-1-метил-7-((4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
7-((4-(2,5-Диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (25 мг, 0,050 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (25,0 мг, 97%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,50 (с, 1H), 7,57 (д, 2H), 7,46-7,53 (м, 2H), 7,43 (дд, 1H), 6,64 (д, 2H), 4,97 (с, 2H), 4,32 (с, 1H), 3,54 (с, 1H), 3,46 (дд, 1H), 3,31 (д, 1H), 3,16 (с, 3H), 2,77-2,88 (м, 2H), 2,39 (с, 3H), 1,97 (квAB, 2H). LCMS (метод C): RT=0,72 мин, m/z=510 [M+H]+.
Пример 45: 3-(2,6-дихлорфенил)-1-метил-7-((4-((2R,5S)-2,4,5-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: 3-(2,6-дихлорфенил)-1-метил-7-((4-((2R,5S)-2,4,5-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (26 мг, 0,051 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (26,5 мг, 99%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,56 (с, 1H), 7,70 (д, 2H), 7,58 (д, 2H), 7,44 (дд, 1H), 7,18 (д, 2H), 5,00 (с, 2H), 3,20 (с, 3H), 3,13-3,19 (м, 1H), 3,01 (дд, 1H), 2,93 (дд, 1H), 2,66 (дд, 1H), 2,37-2,43 (м, 1H), 2,36 (с, 3H), 2,19 (дд, 1H), 1,11 (д, 3H), 0,92 (д, 3H). LCMS (метод C): RT=0,84 мин, m/z=526 [M+H]+.
Пример 46: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил-пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
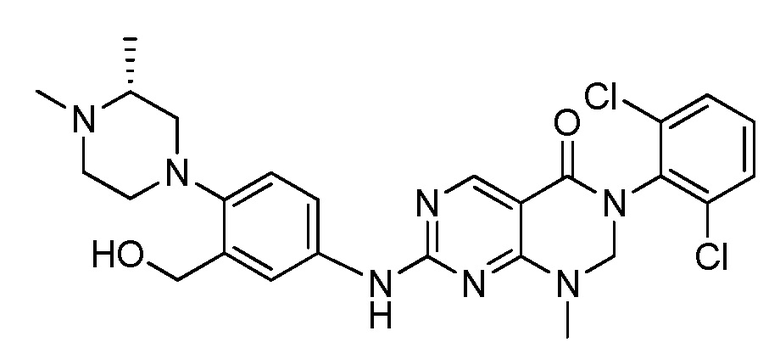
Стадия 1: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил-пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(R)-3-(2,6-Дихлорфенил)-7-((3-(гидроксиметил)-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он (26 мг, 0,051 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (25,3 мг, 99%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,55 (с, 1H), 7,90 (с, 1H), 7,57-7,62 (м, 1H), 7,58 (д, 2H), 7,44 (дд, 1H), 7,15 (д, 1H), 5,00 (с, 2H), 4,76 (с, 2H), 3,21 (с, 3H), 2,88-3,02 (м, 4H), 2,61 (дд, 1H), 2,52 (тд, 1H), 2,35-2,45 (м, 1H), 2,38 (с, 3H), 1,15 (д, 3H). LCMS (метод C): RT=0,73 мин, m/z=542 [M+H]+.
Пример 47: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
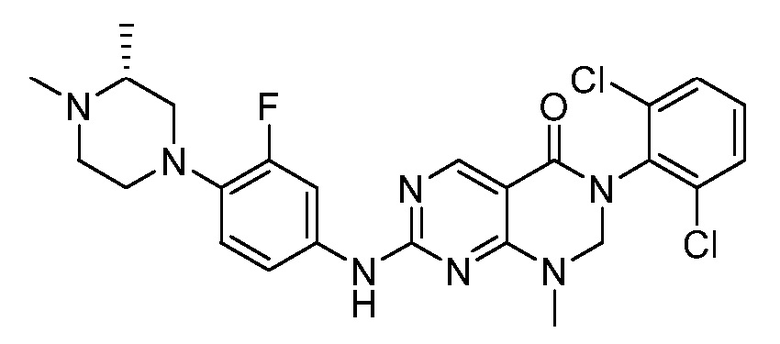
(R)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (23 мг, 0,045 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (20 мг, 85%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,91 (уш.с, 1H), 8,48 (с, 1H), 7,69 (дд, 1H), 7,65 (д, 2H), 7,43-7,52 (м, 2H), 6,98 (т, 1H), 4,98 (с, 2H), 3,07-3,20 (м, 5H), 2,73-2,82 (м, 2H), 2,42 (т, 1H), 2,29 (тд, 1H), 2,14-2,24 (м, 4H), 1,01 (д, 3H). LCMS (метод C): RT=0,87 мин, m/z=530 [M+H]+.
Пример 48: (S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидроксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
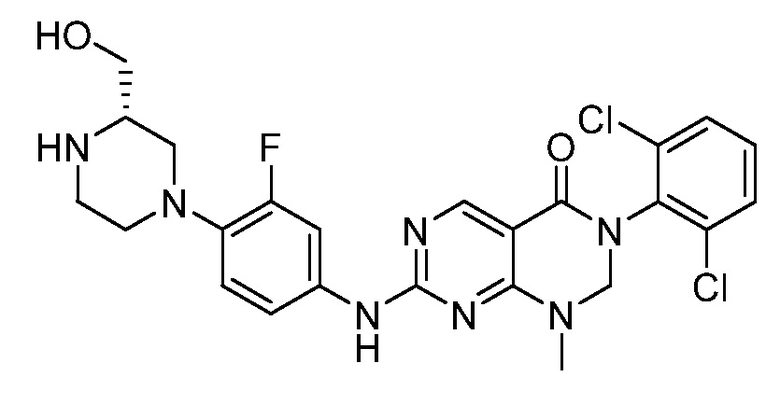
Стадия 1: (S)-третбутил 4-(2-фтор-4-нитрофенил)-2-(гидроксиметил)пиперазин-1-карбоксилат:
К перемешиваемой суспензии 1,2-дифтор-4-нитробензола (0,464 мл, 4,20 ммоль) и карбоната калия (2,324 г, 16,81 ммоль) в безводном DMF (5 мл) добавляли (S)-третбутил 2-(гидрокси-метил)пиперазин-1-карбоксилат (1 г, 4,62 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (50 мл). Водный слой отделяли и экстрагировали этилацетатом (3×50 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 20-100% этилацетат в циклогексане) с получением названного соединения (1,14 г, 76%). 1H ЯМР (300 МГц, CDCl3): δ 8,00 (ддд, 1H), 7,92 (дд, 1H), 6,94 (т, 1H), 4,22-4,35 (м, 1H), 4,03 (д, 1H), 3,90 (д, 2H), 3,74 (д, 1H), 3,50 (д, 1H), 3,30 (т, 1H), 3,05 (дд, 1H), 2,97 (тд, 1H), 1,97 (уш.с, 1H), 1,49 (с, 9H). LCMS (метод C): RT=1,48 мин, m/z=356 [M+H]+.
Стадия 2: (S)-третбутил 4-(4-амино-2-фторфенил)-2-(гидроксиметил)пиперазин-1-карбоксилат:
Раствор (S)-третбутил 4-(2-фтор-4-нитрофенил)-2-(гидрокси-метил)пиперазин-1-карбоксилата (1,14 г, 3,21 ммоль) в метаноле (100 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2] двумя порциями. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (1,04 г, 100%). 1H ЯМР (300 МГц, DMSO-d6): δ 6,77 (т, 1H), 6,27-6,36 (м, 2H), 5,01 (уш.с, 2H), 4,72 (т, 1H), 3,90-4,02 (м, 1H), 3,67-3,93 (м, 2H), 3,42-3,54 (м, 1H), 3,17 (д, 1H), 2,92-3,09 (м, 2H), 2,57 (дд, 1H), 2,44-2,54 (м, 1H), 1,41 (с, 9H). LCMS (метод C): RT=0,89 мин, m/z=326 [M+H]+.
Стадия 3: (S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-фторфенил)-2-(гидроксиметил)пиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с (S)-третбутил 4-(4-амино-2-фторфенил)-2-(гидроксиметил)пиперазин-1-карбоксилатом (110 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения (108 мг, 51%).
Стадия 4: (S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидроксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
(S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-фторфенил)-2-(гидроксиметил)пиперазин-1-карбоксилат (108 мг, 0,171 ммоль) подвергали реакции по удалению защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (41 мг, 45%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,91 (уш.с, 1H), 8,48 (с, 1H), 7,69 (дд, 1H), 7,65 (д, 2H), 7,42-7,52 (м, 2H), 6,97 (т, 1H), 4,98 (с, 2H), 4,60 (т, 1H), 3,30-3,38 (м, 1H), 3,22 (д, 1H), 3,07-3,16 (м, 4H), 2,94 (д, 1H), 2,76-2,88 (м, 2H), 2,61 (тд, 1H), 2,26-2,33 (м, 1H), 2,18 (уш.с, 1H). LCMS (метод C): RT=0,79 мин, m/z=532 [M+H]+.
Пример 49: (S)-3-(2,6-дихлорфенил)-7-((4-(3-(гидрокси-метил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

(S)-3-(2,6-Дихлорфенил)-7-((4-(3-(гидроксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (27 мг, 0,052 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде бледно-желтого твердого вещества (27 мг, 97%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,69 (уш.с, 1H), 8,44 (с, 1H), 7,57-7,67 (уш.м, 4H), 7,48 (дд, 1H), 6,89 (д, 2H), 4,95 (с, 2H), 4,56 (т, 1H), 3,66 (ддд, 1H), 3,59 (уш.д, 1H), 3,44 (уш.д, 1H), 3,33-3,39 (м, 1H), 3,09 (с, 3H), 2,79 (дт, 1H), 2,65-2,73 (м, 1H), 2,41-2,48 (м, 1H), 2,21-2,33 (м, 4H), 2,07-2,16 (м, 1H). LCMS (метод C): RT=0,70 мин, m/z=528 [M+H]+.
Пример 50: (S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидроксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

(S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он (19 мг, 0,036 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (15 мг, 77%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,91 (уш.с, 1H), 8,48 (с, 1H), 7,70 (дд, 1H), 7,65 (д, 2H), 7,48 (дд, 2H), 6,98 (т, 1H), 4,98 (с, 2H), 4,53 (т, 1H), 3,61-3,69 (м, 1H), 3,26-3,38 (м, 2H), 3,07-3,17 (м, 4H), 2,71-2,81 (м, 2H), 2,44-2,51 (м, 1H), 2,28-2,36 (м, 1H), 2,25 (с, 3H), 2,13-2,21 (м, 1H). LCMS (метод C): RT=0,81 мин, m/z=546 [M+H]+.
Пример 51: 7-((4-(2,6-диазаспиро[3,3]гептан-2-ил)фенил)-амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 6-(4-нитрофенил)-2,6-диазаспиро-[3,3]гептан-2-карбоксилат:
Суспензию 1-фтор-4-нитробензола (356 мг, 2,52 ммоль), третбутил 2,6-диазаспиро[3,3]гептан-2-карбоксилата (500 мг, 2,52 ммоль) и карбонат калия (523 мг, 3,78 ммоль) в безводном DMF (3 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали и очищали на хроматографической системе Biotage (картридж диоксид кремния 25g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 20:80) с получением названного соединения в виде желтого твердого вещества (400 мг, 50%). 1H ЯМР (300 МГц, CDCl3): δ 8,10 (дт, 2H), 6,31 (дт, 2H), 4,15 (с, 4H), 4,13 (с, 4H), 1,45 (с, 9H). LCMS (метод C): RT=1,64 мин, m/z=320 [M+H]+.
Стадия 2: третбутил 6-(4-аминофенил)-2,6-диазаспиро-[3,3]гептан-2-карбоксилат:
Раствор третбутил 6-(4-нитрофенил)-2,6-диазаспиро-[3,3]-гептан-2-карбоксилата (400 мг, 1,253 ммоль) в метаноле (100 мл) [примечание: низкая растворимость, поэтому требуется высокое разбавление] гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде бледно-коричневого твердого вещества (315 мг, 87%). 1H ЯМР (400 МГц, CDCl3): δ 6,63 (дт, 2H), 6,35 (дт, 2H), 4,06 (с, 4H), 3,87 (с, 4H), 3,34 (уш.с, 2H), 1,44 (с, 9H). LCMS (метод C): RT=0,76 мин, m/z=290 [M+H]+.
Стадия 3: 7-((4-(2,6-диазаспиро[3,3]гептан-2-ил)фенил)-амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,338 ммоль) подвергали взаимодействию с третбутил 6-(4-аминофенил)-2,6-диазаспиро[3,3]гептан-2-карбоксилатом (98 мг, 0,338 ммоль), следуя методике примера 31, с получением названного соединения (71,6 мг, 81%). 1H ЯМР: (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,57 (д, 2H), 7,47-7,55 (м, 2H), 7,43 (дд, 1H), 6,53 (д, 2H), 4,97 (с, 2H), 3,94 (с, 4H), 3,82 (с, 4H), 3,15 (с, 3H). LCMS (метод C): RT=0,68 мин, m/z=496 [M+H]+.
Пример 52: 3-(2,6-дихлорфенил)-1-метил-7-((4-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-7-((4-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (57 мг, 0,111 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (33,6 мг, 57,4%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,53 (с, 1H), 7,53-7,65 (м, 4H), 7,44 (дд, 1H), 6,99 (д, 2H), 4,98 (с, 2H), 3,52 (д, 2H), 3,17 (с, 3H), 2,42-2,57 (м, 4H), 2,97 (с, 3H), 1,22 (д, 6H). LCMS (метод C): RT=0,79 мин, m/z=526 [M+H]+.
Пример 53:(R)-3-(2-хлор-6-фторфенил)-1-метил-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2-Хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,295 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (86 мг, 0,295 ммоль), следуя методике примера 31, с получением названного соединения (45 мг, 33%). ¹H ЯМР (400 МГц, CDCl3): δ 9,73 (с, 1H), 8,45 (с, 1H), 7,60 (д, 2H), 7,51 (м, 2H), 7,42-7,37 (м, 1H), 6,88 (д, 1H), 4,98 (д, 1H), 4,93 (д, 1H), 3,48 (м, 1H), 3,08 (с, 3H), 2,93 (м, 1H), 2,75 (м, 2H), 2,13 (м, 2H), 1,01 (д, 3H). LCMS (метод C): RT=0,62 мин, m/z=470 [M+H]+.
Пример 54: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-3-(2-хлор-6-фторфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

3-(2-Хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (85 мг, 0,295 ммоль) подвергали взаимодействию с трет-5-бутил 4-(4-амино-2-метоксифенил)пиперазин-1-карбоксилатом (100 мг, 0,295 ммоль), следуя методике примера 31, с получением названного соединения (36 мг, 27%). ¹H ЯМР (400 МГц, CDCl3): δ 8,69 (с, 1H), 7,44 (д, 2H), 7,31 (м, 2H), 7,13 (м, 1H), 6,55 (д, 2H), 4,91 (д, 1H), 4,80 (д, 1H), 4,29 (м, 1H), 3,80 (м, 1H), 3,66 (м, 1H), 3,14 (с, 3H), 3,05 (м, 1H), 2,95 (м, 1H), 2,00 (д, 1H), 1,84 (д, 1H). LCMS (метод C): RT=0,67 мин, m/z=480 [M+H]+.
Пример 55: 3-(2-хлор-6-фторфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
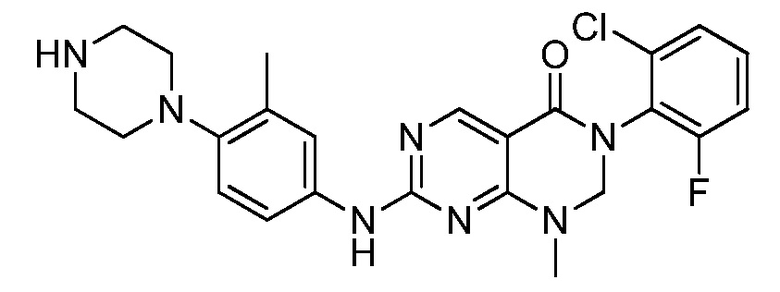
Стадия 1: третбутил 4-(2-метил-4-нитрофенил)пиперазин-1-карбоксилат:
Суспензию 1-фтор-2-метил-4-нитробензола (2,00 г, 12,9 ммоль), третбутил пиперазин-1-карбоксилата (2,40 г, 12,9 ммоль) и карбоната калия (2,67 г, 19,3 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (200 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, и воду отсасывали с получением названного соединения в виде желтого твердого вещества (1,51 г, 36%). LCMS (метод A): RT=1,52 мин, m/z=222 [M-Boc+H]+.
Стадия 2: третбутил 4-(4-амино-2-метилфенил)пиперазин-1-карбоксилат:
К раствору третбутил 4-(2-метил-4-нитрофенил)пиперазин-1-карбоксилата (0,85 г, 2,64 ммоль) в этаноле (10 мл) добавляли 10% палладий на угле (0,141 г, 0,132 ммоль). Реакционную смесь нагревали до 50 °C. При перемешивании, добавляли порциями формиат аммония (0,667 г, 10,58 ммоль), затем нагревали при 50°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры в течение ночи и фильтровали через целит®. Фильтраты концентрировали, и остаток распределяли между DCM и раствором бикарбоната натрия. Органическую фазу отделяли, сушили (фазовый сепаратор) и концентрировали с получением названного соединения в виде желтовато-белого твердого вещества (632 мг, 82%). LCMS (метод A): RT=0,88 мин, m/z=236 [M-tBu+H]+.
Стадия 3: 3-(2-хлор-6-фторфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2-Хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он) (100 мг, 0,295 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-метил-фенил)пиперазин-1-карбоксилатом (86 мг, 0,295 ммоль), следуя методике примера 31, с получением названного соединения (56 мг, 39%). ¹H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,48 (м, 1H), 7,43 (м, 1H), 7,33 (м, 2H), 7,15 (м, 1H), 6,92 (1, 2H), 4,93 (д, 1H), 4,83 (д, 1H), 3,18 (с, 3H), 3,06 (м, 4H), 2,90 (м, 4H), 2,33 (с, 3H). LCMS (метод C): RT=0,75 мин, m/z=482 [M+H]+.
Пример 56: 3-(2-хлор-6-фторфенил)-7-((3-(гидроксиметил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2-хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,295 ммоль) подвергали взаимодействию с (3-аминофенил)метанолом (36 мг, 0,295 ммоль), следуя методике примера 31, с получением названного соединения (35 мг, 29%). ¹H ЯМР (400 МГц, CDCl3): δ 8,65 (с, 1H), 7,81 (м, 1H), 7,67 (с,1H), 7,49 (м, 1H), 7,28 (м, 1H), 7,10 (м, 1H), 6,98 (м, 1H), 4,93 (д, 1H), 4,86 (д, 1H), 4,78 (д, 1H), 4,53 (с, 3H). LCMS (метод C): RT=0,93 мин, m/z=414 [M+H]+.
Пример 57: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2-хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг,0,295 ммоль) подвергали взаимодействию с (5-амино-2-морфолинофенил)метанолом (61 мг, 0,295 ммоль), следуя методике примера 31, с получением названного соединения (57 мг, 39%). ¹H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,48 (м, 1H), 7,43 (м,1H), 7,33 (м, 2H), 7,15 (м, 1H), 6,92 (1, 2H), 4,95 (д, 1H), 4,83 (д, 1H), 4,78 (с, 2H), 3,18 (с, 3H), 3,72 (м, 4H), 3,04 (м, 4H). LCMS (метод C): RT=0,93 мин, m/z=499 [M+H]+.
Пример 58: (R)-3-(2,6-дихлорфенил)-1-(1-метил-1H-пиразол-3-ил)-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 4-хлор-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамид:
Смесь 4-хлор-2-(метилтио)пиримидин-5-карбонилхлорида (35 г, 157 ммоль) и Amberlyst A21 (6 г, 149 ммоль) в этилацетате (250 мл) нагревали до 40°C, затем добавляли по каплям раствор 2,6-дихлоранилина (24,21 г, 149 ммоль) в этилацетате (250 мл). Реакционную смесь нагревали при 40°C в атмосфере азота в течение ночи. Полученную суспензию охлаждали до комнатной температуры и фильтровали. Выделенные твердые вещества растворяли в горячем тетрагидрофуране (100 мл) и фильтровали, повторяя этот процесс с нерастворенным твердым веществом еще два раза до тех пор, пока большая часть твердого вещества не растворялась. Объединенные фильтраты концентрировали досуха при пониженном давлении с получением бледно-желтого твердого вещества. Это вещество суспендировали в дихлорметане (100 мл) с получением названного соединения в виде белого твердого вещества (22,1 г, 42%). 1H ЯМР (300 МГц, DMSO-d6): δ 10,71 (с, 1H), 8,82 (с, 1H), 7,62 (д, 2H), 7,43 (т, 1H), 2,60 (с, 3H). LCMS (метод C): RT=1,46 мин, m/z=348 [M+H]+.
Стадия 2: N-(2,6-дихлорфенил)-4-((1-метил-1H-пиразол-3-ил)-амино)-2-(метилтио)пиримидин-5-карбоксамид:
К перемешиваемому раствору 4-хлор-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамида (218 мг, 0,625 ммоль) и основание Хунига (0,328 мл, 1,876 ммоль) в безводном тетрагидрофуране (4 мл) добавляли 1-метил-1H-пиразол-3-амин (0,052 мл, 0,657 ммоль), и смесь нагревали до 50°C в течение 16 часов. Растворитель удаляли под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-5% метанол в дихлорметане) с получением названного соединения (117 мг, 45,6%). LCMS (метод C): RT=1,45 мин, m/z=409 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-(1-метил-1H-пиразол-3-ил)-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
В пробирку для проведения реакций при микроволновом излучении объемом 10 мл добавляли N-(2,6-дихлорфенил)-4-((1-метил-1H-пиразол-3-ил)амино)-2-(метилтио)пиримидин-5-карбоксамид (112 мг, 0,274 ммоль) в безводном ацетонитриле (3 мл), затем карбонат цезия (535 мг, 1,642 ммоль) и дибромметан (0,058 мл, 0,821 ммоль). Пробирку герметизировали, и смесь нагревали до 90°C при перемешивании в течение 40 часов. Растворитель удаляли под вакуумом. Остаток распределяли между водой (10 мл) и дихлорметаном (20 мл). Водную фазу отделяли и экстрагировали дихлорметаном (2×10 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор) и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетат в циклогексане) с получением названного соединения (54,1 мг, 46,9%). LCMS (метод C): RT=1,53 мин, m/z=421 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-1-(1-метил-1H-пиразол-3-ил)-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидро-пиримидо [4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-(1-метил-1H-пиразол-3-ил)-7-(метил-тио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (50 мг,0,119 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-фенил)-2-метилпиперазин-1-карбоксилатом (38 мг, 0,131 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (25 мг,37%). ¹H ЯМР (400 МГц, метанол-d4): δ 8,69 (с, 1H), 7,60-7,35 (м, 5H), 6,93 (д, 2H), 6,61 (уш.с, 1H), 5,49 (с, 2H), 3,82 (с, 3H), 3,50 (м, 2H), 3,15-2,9 (м, 3H), 2,67 (дт, 1H), 2,35 (м, 1H), 1,17 (д, 3H). LCMS (метод C): RT=0,88 мин, m/z=564 [M+H]+.
Пример 59: (R)-3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(2-(метоксиметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилата (1,00 г, 2,85 ммоль) в безводном THF (9,49 мл) охлаждали до 0°C,затем добавляли гидрид натрия (60% в минеральном масле, 0,125 г, 3,13 ммоль). После перемешивания при 0°C в атмосфере азота в течение 10 минут, добавляли метилйодид (0,214 мл, 3,41 ммоль). Полученную смесь подогревали до комнатной температуры и перемешивали в течение 60 минут. Добавляли дополнительное количество метилйодида (0,071 мл, 1,138 ммоль), и смесь перемешивали в течение еще 60 минут. Реакционную смесь разбавляли раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 50:50) с получением названного соединения в виде желтого масла, которое превращалось в твердое вещество при стоянии (870 мг, 84%). 1H ЯМР (300 МГц, CDCl3): δ 8,34 (д, 1H), 8,13 (дд, 1H), 7,04 (д, 1H), 4,56 (д, 1H), 4,50 (д, 1H), 4,36 (уш.с, 1H), 3,98 (дт, 1H), 3,47 (с, 3H), 3,29 (тд, 1H), 3,18 (ддд, 1H), 3,10 (дт, 1H), 2,97 (дд, 1H), 2,80 (тд, 1H), 1,49 (с, 9H), 1,37 (д, 3H). LCMS (метод C): RT=1,87 мин, m/z=366 [M+H]+.
Стадия 2: (R)-третбутил 4-(4-амино-2-(метоксиметил)фенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(метоксиметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилата (870 мг, 2,381 ммоль) в метаноле (40 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин) за три прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого масла (730 мг, 91%). 1H ЯМР (400 МГц, CDCl3): δ 6,90 (д, 1H), 6,78 (д, 1H), 6,59 (дд, 1H), 4,55 (д, 1H), 4,51 (д, 1H), 4,28 (уш.с, 1H), 3,88 (д, 1H), 3,42 (с, 3H), 3,22 (тд, 1H), 2,80-2,90 (м, 2H), 2,76 (дт, 1H), 2,66 (тд, 1H), 1,48 (с, 9H), 1,36 (д, 3H). LCMS (метод C): RT=1,05 мин, m/z=336 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(метоксиметил)фенил)-2-метилпиперазин-1-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(метокси-метил)фенил)-2-метилпиперазин-1-карбоксилатом (95 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (98 мг, 54%). 1H ЯМР (300 МГц, CDCl3) δ 8,72 (с, 1H), 7,73-7,93 (уш.м, 2H), 7,53 (дд, 1H), 7,43 (д, 2H), 7,27 (дд, 1H), 7,04 (д, 1H), 4,88 (с, 2H), 4,61 (д, 1H), 4,56 (д, 1H), 4,24-4,38 (м, 1H), 3,92 (уш.д, 1H), 3,44 (с, 3H), 3,25 (тд, 1H), 3,19 (с, 3H), 2,96 (уш.д, 1H), 2,82-2,89 (м, 2H), 2,71 (тд, 1H), 1,48 (с, 9H), 1,37 (д, 3H). LCMS (метод C): RT=1,88 мин, m/z=642 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
(R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(метоксиметил)фенил)-2-метилпиперазин-1-карбоксилат (98 мг, 0,153 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (70 мг, 85%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,53 (с, 1H), 7,90 (уш.с, 1H), 7,53-7,60 (м, 3H), 7,42 (дд, 1H), 7,11 (д, 1H), 4,98 (с, 2H), 4,56 (с, 2H), 3,43 (с, 3H), 3,19 (с, 3H), 2,92-3,05 (м, 5H), 2,69-2,78 (м, 1H), 2,42 (т, 1H), 1,11 (д, 3H). LCMS (метод C): RT=0,82 мин, m/z=542 [M+H]+.
Пример 60: (R)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 2-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Суспензию 1-фтор-4-нитробензола (2,86 г, 20,30 ммоль), (R)-третбутил 2-(гидроксиметил)пиперазин-1-карбоксилата (4,39 г, 20,30 ммоль) и карбоната калия (4,21 г, 30,4 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Маслянистый продукт экстрагировали этилацетатом (50 мл) и промывали смесью 50:50 вода:солевой раствор (3×50 мл). Органическую фазу затем сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток суспендировали в смеси циклогексан (20 мл) и диэтиловый эфир (2 мл) с получением названного соединения в виде желтого твердого вещества (4,78 г, 70%). 1H ЯМР (300 МГц, CDCl3): δ 8,11 (дт, 2H), 6,78 (дт, 2H), 4,25 (уш.с, 1H), 3,87-4,03 (м, 2H), 3,62-3,80 (м, 3H), 3,26-3,42 (м, 2H), 3,18 (ддд, 1H), 1,49 (с, 9H). LCMS (метод C): RT=1,39 мин, m/z=338 [M+H]+.
Стадия 2: (R)-третбутил 2-(метоксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Раствор (R)-третбутил 2-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилата (500 мг, 1,482 ммоль) в безводном THF (4,94 мл) охлаждали до 0°C, затем добавляли гидрид натрия (60% в минеральном масле, 65,2 мг, 1,630 ммоль). После перемешивания при 0°C в атмосфере азота в течение 10 минут, добавляли метилйодид (111 мкл, 1,778 ммоль). Полученную смесь подогревали до комнатной температуры и перемешивали в течение 60 минут. Реакционную смесь разбавляли раствором хлорида аммония (10 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 25g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 20:80) с получением названного соединения в виде желтого масла (200 мг, 38% выход). 1H ЯМР (300 МГц, CDCl3): δ 8,12 (д, 2H), 6,79 (д, 2H), 4,29 (уш.с, 1H), 4,04 (уш.д, 1H), 3,95 (уш.д, 1H), 3,73 (уш.д, 1H), 3,36-3,43 (м, 2H), 3,33 (с, 3H), 3,08-3,30 (м, 3H), 1,49 (с, 9H). LCMS (метод C): RT=1,69 мин, m/z=352 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-аминофенил)-2-(метоксиметил)-пиперазин-1-карбоксилат:
Раствор (R)-третбутил 2-(метоксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилата (200 мг, 0,569 ммоль) в метаноле (30 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде коричневого масла (160 мг, 87% выход). 1H ЯМР (300 МГц, CDCl3): δ 6,79 (дт, 2H), 6,65 (дт, 2H), 4,28 (уш.с, 1H), 3,97 (уш.д, 1H), 3,78 (т, 1H), 3,37-3,55 (м, 4H), 3,39 (с, 3H), 3,27 (уш.д, 1H), 3,11 (тд, 1H), 2,56-2,73 (м, 2H), 1,48 (с, 9H). LCMS (метод C): RT=0,74 мин, m/z=322 [M+H]+.
Стадия 4: (R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2-(метоксиметил)пиперазин-1-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-(метоксиметил)пиперазин-1-карбоксилатом (91 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (113 мг, 64%). 1H ЯМР (300 МГц, CDCl3): δ 8,70 (с, 1H), 7,51 (д, 2H), 7,44 (д, 2H), 7,28 (дд, 1H), 6,92 (д, 2H), 4,87 (с, 2H), 4,31 (уш.с, 1H), 4,00 (уш.д, 1H), 3,65-3,80 (м, 2H), 3,40-3,50 (м, 2H), 3,40 (д, 3H), 3,07-3,20 (м, 4H), 2,80 (дд, 1H), 2,74 (тд, 1H), 1,49 (с, 9H). LCMS (метод C): RT=1,67 мин, m/z=628 [M+H]+.
Стадия 5: (R)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-2-(метоксиметил)пиперазин-1-карбоксилат (113 мг, 0,180 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (83 мг, 87%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,52-7,61 (м, 4H), 7,42 (дд, 1H), 6,98 (д, 2H), 4,96 (с, 2H), 3,39-3,57 (м, 4H), 3,39 (с, 3H), 3,15 (с, 3H), 3,04-3,14 (м, 2H), 2,96 (тд, 1H), 2,72 (тд, 1H), 2,48 (т, 1H). LCMS (метод C): RT=0,75 мин, m/z=528 [M+H]+.
Пример 61: (S)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
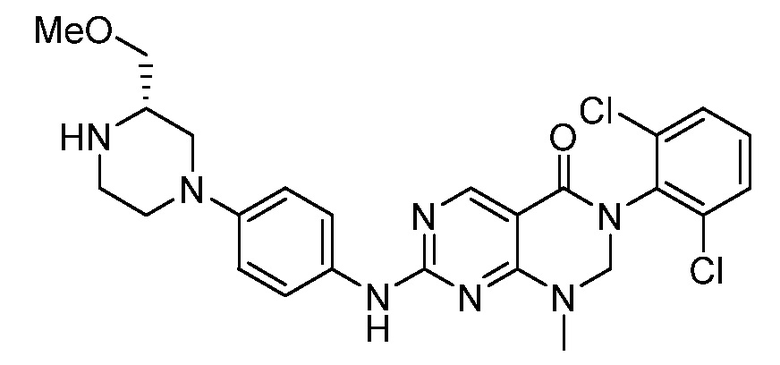
Стадия 1: (S)-третбутил 2-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Суспензию 1-фтор-4-нитробензола (1,305 г, 9,25 ммоль), (S)-третбутил 2-(гидроксиметил)пиперазин-1-карбоксилата (2,00 г, 9,25 ммоль) и карбоната калия (1,917 г, 13,87 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой (50 мл), воду отсасывали и лиофилизировали в течение ночи с получением названного соединения в виде оранжевого твердого вещества (2,13 г, 68%). 1H ЯМР (300 МГц, CDCl3): δ 8,11 (дт, 2H), 6,78 (дт, 2H), 4,25 (уш.с, 1H), 3,85-4,04 (м, 2H), 3,61-3,80 (м, 3H), 3,25-3,43 (м, 2H), 3,18 (ддд, 1H), 1,49 (с, 9H). LCMS (метод C): RT=1,39 мин, m/z=338 [M+H]+.
Стадия 2: (S)-третбутил 2-(метоксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Раствор (S)-третбутил 2-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат (1,00 г, 2,96 ммоль) в безводном THF (9,88 мл) охлаждали до 0°C затем добавляли гидрид натрия (60% в минеральном масле, 0,130 г, 3,26 ммоль). После перемешивания при 0°C в атмосфере азота в течение 10 минут, добавляли метилйодид (0,222 мл, 3,56 ммоль). Полученную смесь подогревали до комнатной температуры и перемешивали в течение 60 минут. Реакционную смесь разбавляли раствором хлорида аммония (20 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 20:80) с получением названного соединения в виде желтого масла (282 мг, 27% выход). 1H ЯМР (300 МГц, CDCl3): δ 8,13 (дт, 2H), 6,79 (дт, 2H), 4,29 (уш.с, 1H), 4,05 (дт, 1H), 3,95 (уш.д, 1H), 3,74 (уш.д, 1H), 3,37-3,43 (м, 2H), 3,33 (с, 3H), 3,08-3,28 (м, 3H), 1,49 (с, 9H). LCMS (метод C): RT=1,69 мин, m/z=352 [M+H]+.
Стадия 3: (S)-третбутил 4-(4-аминофенил)-2-(метоксиметил)-пиперазин-1-карбоксилат:
Раствор (S)-третбутил 2-(метоксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилата (282 мг, 0,803 ммоль) в метаноле (30 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (236 мг, 91% выход). 1H ЯМР (300 МГц, CDCl3) δ 6,78 (дт, 2H), 6,65 (дт, 2H), 4,28 (уш.с, 1H), 3,98 (уш.д, 1H), 3,78 (т, 1H), 3,43-3,54 (м, 2H), 3,39 (с, 3H), 3,27 (уш.д, 1H), 3,11 (тд, 1H), 2,56-2,74 (м, 2H), 1,48 (с, 9H). LCMS (метод C): RT=0,78 мин, m/z=322 [M+H]+.
Стадия 4: (S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2-(метоксиметил)пиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (200 мг, 0,563 ммоль) подвергали взаимодействию с (S)-третбутил 4-(4-аминофенил)-2-(метоксиметил)пиперазин-1-карбоксилатом (181 мг, 0,563 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (190 мг, 54%). 1H ЯМР (300 МГц, CDCl3): δ 8,70 (с, 1H), 7,51 (д, 2H), 7,44 (д, 2H), 7,41 (уш.с, 1H), 7,28 (дд, 1H), 6,91 (д, 2H), 4,87 (с, 2H), 4,32 (уш.с, 1H), 4,00 (уш.д, 1H), 3,64-3,80 (м, 2H), 3,39-3,52 (м, 2H), 3,39 (с, 3H), 3,06-3,21 (м, 4H), 2,67-2,85 (м, 2H), 1,49 (с, 9H). LCMS (метод C): RT=1,66 мин, m/z=628 [M+H]+.
Стадия 5: (S)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-2-(метоксиметил)пиперазин-1-карбоксилат (190 мг, 0,302 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (135 мг, 85%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,51-7,62 (м, 4H), 7,42 (дд, 1H), 6,98 (д, 2H), 4,96 (с, 2H), 3,39-3,57 (м, 4H), 3,39 (с, 3H), 3,15 (с, 3H), 3,04-3,14 (м, 2H), 2,96 (тд, 1H), 2,72 (тд, 1H), 2,48 (т, 1H). LCMS (метод C): RT=0,76 мин, m/z=528 [M+H]+.
Пример 62: 3-(2,6-дихлорфенил)-1-метил-7-((4-(3-(трифтор-метил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 1-(4-нитрофенил)-3-(трифторметил)пиперазин:
Суспензию 1-фтор-4-нитробензола (0,915 г, 6,49 ммоль), 2-(трифторметил)пиперазина (1,00 г, 6,49 ммоль) и карбоната калия (1,345 г, 9,73 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, и воду отсасывали с получением названного соединения в виде желтого твердого вещества (1,47 г, 82%). 1H ЯМР (300 МГц, CDCl3): δ 8,15 (дт, 2H), 6,87 (дт, 2H), 3,91 (уш.д, 1H), 3,74 (ддд, 1H), 3,39-3,54 (м, 1H), 3,25 (дд, 1H), 2,93-3,16 (м, 3H), 2,00 (уш.с, 1H). LCMS (метод C): RT=1,12 мин, m/z=276 [M+H]+.
Стадия 2: третбутил 4-(4-нитрофенил)-2-(трифторметил)пиперазин-1-карбоксилат:
К раствору 1-(4-нитрофенил)-3-(трифторметил)пиперазина (1,20 г, 4,36 ммоль) в безводном дихлорметане (14,53 мл) добавляли дитретбутилдикарбонат (2,025 мл, 8,72 ммоль), DIPEA (1,904 мл, 10,90 ммоль) и DMAP (0,053 г, 0,436 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 3 дней. Добавляли дополнительное количество дитретбутил- дикарбоната (2,025 мл, 8,72 ммоль), DIPEA (1,904 мл, 10,90 ммоль) и DMAP (0,053 г, 0,436 ммоль), и смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли дополнительное количество дитретбутилдикарбоната (2,025 мл, 8,72 ммоль), DIPEA (1,904 мл, 10,90 ммоль) и DMAP (0,053 г, 0,436 ммоль), и смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли дитретбутилдикарбоната (2,025 мл, 8,72 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали насыщенным раствором бикарбоната натрия (15 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Полученный остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 60:40) с получением названного соединения в виде желтого твердого вещества (1,49 г, 91%). 1H ЯМР (300 МГц, CDCl3): δ 8,14 (дт, 2H), 6,82 (дт, 2H), 4,54-4,99 (уш.м, 1H), 3,97-4,27 (уш.м, 2H), 3,79 (д, 1H), 3,23-3,48 (уш.м, 2H), 3,10 (тд, 1H), 1,49 (с, 9H). LCMS (метод C): RT=1,78 мин, m/z=376 [M+H]+.
Стадия 3: третбутил 4-(4-аминофенил)-2-(трифторметил)-пиперазин-1-карбоксилат:
Раствор третбутил 4-(4-нитрофенил)-2-(трифторметил)-пиперазин-1-карбоксилата (1,49 г, 3,97 ммоль) в метаноле (50 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (1,34 г, 98%). 1H ЯМР (400 МГц, CDCl3): δ 6,80 (дт, 2H), 6,65 (дт, 2H), 4,49-4,88 (уш.м, 1H), 4,10 (уш.с, 1H), 3,65 (д, 1H), 3,23-3,59 (уш.м, 4H), 2,85 (ддд, 1H), 2,66 (тд, 1H), 1,49 (с, 9H). LCMS (метод C): RT=0,99 мин, m/z=346 [M+H]+.
Стадия 4: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2-(трифторметил)пиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)-2-(трифторметил)пиперазин-1-карбоксилатом (98 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (126 мг, 68%). 1H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,54 (д, 2H), 7,44 (д, 2H), 7,28 (дд, 1H), 7,24 (уш.с, 1H), 6,93 (д, 2H), 4,88 (с, 2H), 4,65 (уш.с, 1H), 4,13 (уш.с, 1H), 3,82 (д, 1H), 3,46 (д, 1H), 3,35 (уш.с, 1H), 3,17 (с, 3H), 2,97 (ддд, 1H), 2,76 (тд, 1H), 1,50 (с, 9H). LCMS (метод C): RT=1,80 мин, m/z=652 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-1-метил-7-((4-(3-(трифтор-метил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-2-(трифторметил)пиперазин-1-карбоксилат (126 мг, 0,193 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (92 мг, 86%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,60 (уш.д, 2H), 7,56 (д, 2H), 7,42 (дд, 1H), 7,00 (д, 2H), 4,97 (с, 2H), 3,66 (уш.д, 1H), 3,43-3,61 (м, 2H), 3,10-3,19 (м, 4H), 2,98 (тд, 1H), 2,70-2,81 (м, 2H). LCMS (метод C): RT=1,06 мин, m/z=552 [M+H]+.
Пример 63: 3-(2,6-дихлорфенил)-1-метил-7-((4-(3,3,4-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
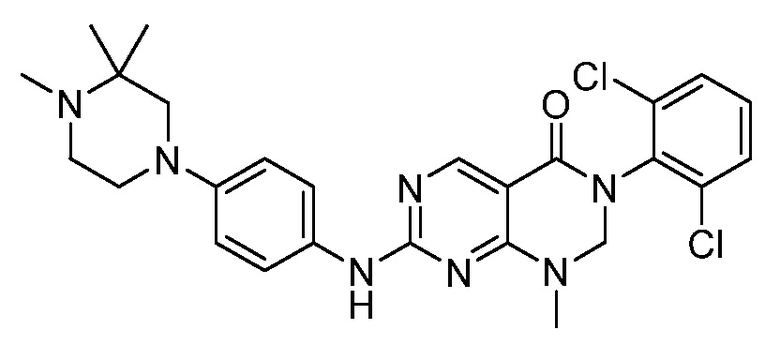
Стадия 1: 1,2,2-триметил-4-(4-нитрофенил)пиперазин:
К перемешиваемой суспензии 1-фтор-4-нитробензола (0,668 г, 4,74 ммоль) и карбоната калия (3,27 г, 23,68 ммоль) в безводном DMF (5 мл) добавляли 1,2,2-триметилпиперазин⋅2HCl (1 г, 4,97 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (50 мл). Водный слой отделяли и экстрагировали этилацетатом (3×50 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали досуха при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (градиент 50-100% этилацетат в циклогексане, затем 0-20% метанол в этилацетате) с получением названного соединения (1,15 г, 97%). 1H ЯМР (400 МГц, CDCl3): δ 8,10 (д, 2H), 6,78 (д, 2H), 3,46 (т, 2H), 3,18 (с, 2H), 2,67 (т, 2H), 2,29 (с, 3H), 1,08 (с, 6H). LCMS (метод C) RT=0,48 мин, m/z=250 [M+H]+.
Стадия 2: 4-(3,3,4-триметилпиперазин-1-ил)анилин:
Раствор 1,2,2-триметил-4-(4-нитрофенил)пиперазина (1,15 г, 4,61 ммоль) в метаноле (30 мл) и тетрагидрофуране (5,00 мл) пропускали два раза через проточный реактор H-Cube, оснащенный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения (1,02 г, 101%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,64 (д, 2H), 6,48 (д, 2H), 4,54 (уш.с, 2H), 2,85 (т, 2H), 2,60 (с, 2H), 2,52 (т, 2H), 2,13 (с, 3H), 1,01 (с, 6H). LCMS (метод C) RT=0,21 мин, m/z=220 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-метил-7-((4-(3,3,4-три-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с 4-(3,3,4-триметилпиперазин-1-ил)-анилином (62 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (19 мг, 13%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,50 (с, 1H), 7,50-7,60 (м, 4H), 7,42 (дд, 1H), 6,93 (д, 2H), 4,96 (с, 2H), 3,12-3,19 (м, 5H), 2,91 (с, 2H), 2,73 (т, 2H), 2,29 (с, 3H), 1,16 (с, 6H). LCMS (метод C): RT=0,80 мин, m/z=526 [M+H]+.
Пример 64: 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 7-(4-нитрофенил)-3-окса-7,9-диаза-бицикло[3,3,1]нонан-9-карбоксилат:
К перемешиваемой суспензии 1-фтор-4-нитробензола (124 мг, 0,876 ммоль) и карбоната калия (484 мг, 3,50 ммоль) в безводном DMF (2 мл) добавляли третбутил 3-окса-7,9-диазабицикло-[3,3,1]нонан-9-карбоксилат (200 мг, 0,876 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (20 мл) и этилацетатом (10 мл). Водную фазу отделяли и экстрагировали этилацетатом (3×10 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×5 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали досуха при пониженном давлении. Полученный остаток подвергали хроматографии (градиент 0-50% этилацетат в циклогексане) с получением названного соединения (237 мг, 77%). 1H ЯМР (400 МГц, CDCl3): δ 8,15 (д, 2H), 6,78 (д, 2H), 4,24-4,31 (м, 1H), 4,11-4,18 (м, 1H), 3,95 (т, 2H), 3,84 (т, 4H), 3,37 (т, 2H), 1,49 (с, 9H). LCMS (метод C): RT=1,54 мин, m/z=350 [M-бутен]+.
Стадия 2: третбутил 7-(4-аминофенил)-3-окса-7,9-диаза-бицикло[3,3,1]нонан-9-карбоксилат:
Раствор третбутил 7-(4-нитрофенил)-3-окса-7,9-диаза-бицикло[3,3,1]нонан-9-карбоксилата (230 мг, 0,658 ммоль) в метаноле (30 мл) и тетрагидрофуране (5 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения (208 мг, 99%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,62 (д, 2H), 6,50 (д, 2H), 4,67 (уш.с, 2H), 3,96 (д, 2H), 3,85 (д, 2H), 3,64 (д, 2H), 3,50 (т, 2H), 2,76 (д, 2H), 1,42 (с, 9H). LCMS (метод C) RT=0,67 мин, m/z=320 [M+H]+.
Стадия 3: третбутил 7-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 7-(4-аминофенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилатом (90 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (108 мг, 61%). 1H ЯМР (300 МГц, CDCl3): δ 8,69 (с, 1H), 7,50 (д, 2H), 7,44 (д, 2H), 7,28 (дд, 1H), 7,16 (уш.с, 1H), 6,87 (д, 2H), 4,87 (с, 2H), 4,22 (уш.с, 1H), 4,09 (уш.с, 1H), 4,00 (д, 1H), 3,96 (д, 1H), 3,87 (ддд, 2H), 3,71 (д, 2H), 3,09-3,21 (м, 5H), 1,49 (с, 9H). LCMS (метод C): RT=1,46 мин, m/z=626 [M+H]+.
Стадия 4: 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
Третбутил 7-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат (108 мг, 0,172 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (60 мг, 66%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,50 (с, 1H), 7,49-7,62 (м, 4H), 7,41 (дд, 1H), 6,93 (д, 2H), 4,96 (с, 2H), 3,91-4,00 (м, 4H), 3,79 (д, 2H), 3,09-3,18 (м, 5H), 3,03 (с, 2H). LCMS (метод C): RT=0,66 мин, m/z=526 [M+H]+.
Пример 65: (R)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

(R)-3-(2,6-Дихлорфенил)-7-((4-(3-(метоксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (46 мг, 0,087 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде бледно-желтого твердого вещества (28 мг, 59%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,52-7,62 (м, 4H), 7,42 (дд, 1H), 6,97 (д, 2H), 4,96 (с, 2H), 3,54-3,62 (м, 2H), 3,44-3,54 (м, 2H), 3,38 (с, 3H), 3,16 (с, 3H), 2,93 (дт, 1H), 2,84 (тд, 1H), 2,67 (дд, 1H), 2,41-2,55 (м, 2H), 2,39 (с, 3H). LCMS (метод C): RT=0,78 мин, m/z=542 [M+H]+.
Пример 66: (S)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
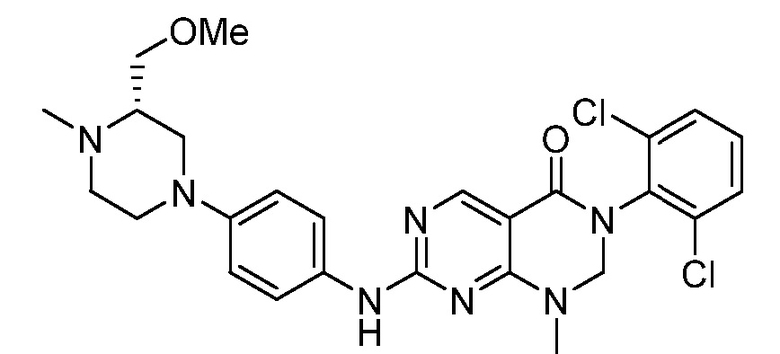
(S)-3-(2,6-Дихлорфенил)-7-((4-(3-(метоксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (41 мг, 0,078 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде бледно-желтого твердого вещества (28 мг, 67%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,50-7,62 (м, 4H), 7,42 (дд, 1H), 6,97 (д, 2H), 4,96 (с, 2H), 3,54-3,62 (м, 2H), 3,44-3,54 (м, 2H), 3,37 (с, 3H), 3,15 (с, 3H), 2,92 (дт, 1H), 2,84 (тд, 1H), 2,67 (дд, 1H), 2,40-2,55 (м, 2H), 2,38 (с, 3H). LCMS (метод C): RT=0,78 мин, m/z=542 [M+H]+.
Пример 67: (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил-пиперазин-1-ил)-3-(метоксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

(R)-3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он (40 мг, 0,074 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (33 мг, 80%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,53 (с, 1H), 7,90 (уш.с, 1H), 7,52-7,60 (м, 3H), 7,42 (дд, 1H), 7,12 (д, 1H), 4,98 (с, 2H), 4,55 (с, 2H), 3,42 (с, 3H), 3,19 (с, 3H), 2,86-3,04 (м, 4H), 2,48-2,62 (м, 2H), 2,33-2,47 (м, 4H), 1,13 (д, 3H). LCMS (метод C): RT=0,83 мин, m/z=556 [M+H]+.
Пример 68: 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-метил-3-(трифторметил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-((4-(3-(трифторметил)-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (57 мг, 0,103 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде бледно-желтого твердого вещества (17 мг, 29%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,47 (с, 1H), 7,49-7,62 (м, 4H), 7,43 (дд, 1H), 6,99 (д, 2H), 5,01 (с, 2H), 3,58 (д, 1H), 3,33-3,42 (м, 1H), 3,19 (с, 3H), 3,08-3,17 (м, 1H), 2,97-3,09 (м, 3H), 2,61-2,71 (м, 1H), 2,53 (д, 3H). LCMS (метод C): RT=1,46 мин, m/z=566 [M+H]+.
Пример 69: 3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
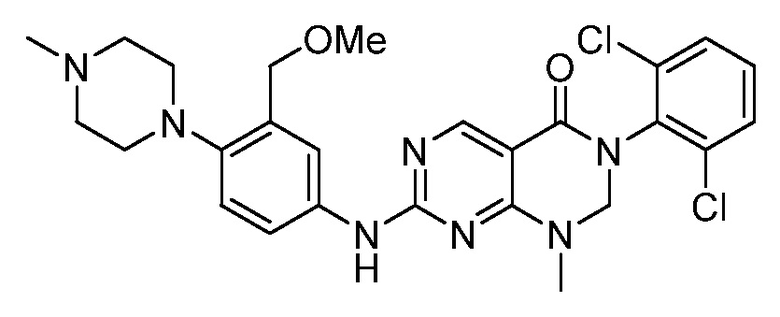
3-(2,6-Дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (55 мг, 0,104 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (29 мг, 51%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,81 (уш.с, 1H), 8,46 (с, 1H), 7,95 (уш.с, 1H), 7,64 (д, 2H), 7,60 (дд, 1H), 7,48 (дд, 1H), 7,06 (д, 1H), 4,97 (с, 2H), 4,45 (с, 2H), 3,34 (с, 3H), 3,12 (с, 3H), 2,84 (т, 4H), 2,54 (уш.с, 4H), 2,29 (с, 3H). LCMS (метод C): RT=0,81 мин, m/z=542 [M+H]+.
Пример 70: 3-(2,6-дихлорфенил)-1-метил-7-((6,7,8,9-тетра-гидро-5H-5,8-эпиминобензo[7]аннулен-3-ил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
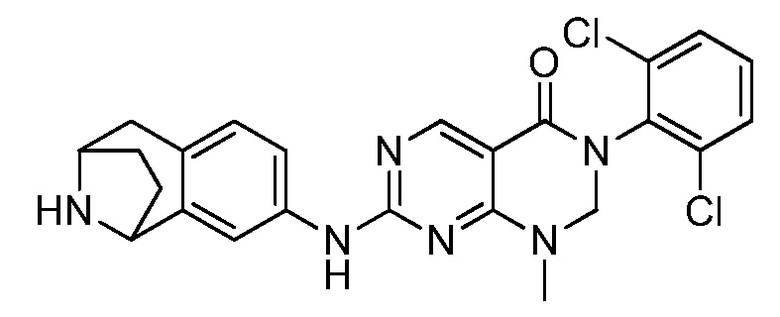
Стадия 1: третбутил 3-амино-6,7,8,9-тетрагидро-5H-5,8-эпиминобензo[7]аннулен-10-карбоксилат:
Раствор 6,7,8,9-тетрагидро-5H-5,8-эпиминобензo[7]аннулен-3-амина (WO 2008051547, 0,11 г, 0,631 ммоль), дитретбутил- дикарбоната (0,138 г, 0,631 ммоль) и триэтиламина (0,128 г, 1,26 ммоль) перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Остаток очищали на хроматографической системе Biotage с получением названного соединения (0,137 г, 79%). LCMS (метод A): RT=1,01 мин, m/z=275 [M+H]+
Стадия 2: 3-(2,6-дихлорфенил)-1-метил-7-((6,7,8,9-тетра-гидро-5H-5,8-эпиминобензo[7]аннулен-3-ил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (177 мг,0,498 ммоль) подвергали взаимодействию с третбутил 3-амино-6,7,8,9-тетра-гидро-5H-5,8-эпиминобензo[7]аннулен-10-карбоксилатом (130 мг, 0,474 ммоль), следуя методике примера 31, с получением названного соединения (58 мг, 25%). ¹H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,77 (уш.с, 1H), 7,44 (д, 2H), 7,36 (м, 2H), 7,28 (м, 1H), 7,02 (м, 1H), 4,90 (с, 2H), 4,20 (м, 1H), 3,91 (м, 1H), 3,18 (с, 3H), 3,10 (м, 1H), 2,55 (м, 1H), 2,11 (м, 2H), 1,92 (м, 1H), 1,62 (м, 1H). LCMS (метод C): RT=0,79 мин, m/z=481 [M+H]+.
Пример 71: 3-(2,6-дихлорфенил)-1-метил-7-((4-(9-метил-3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
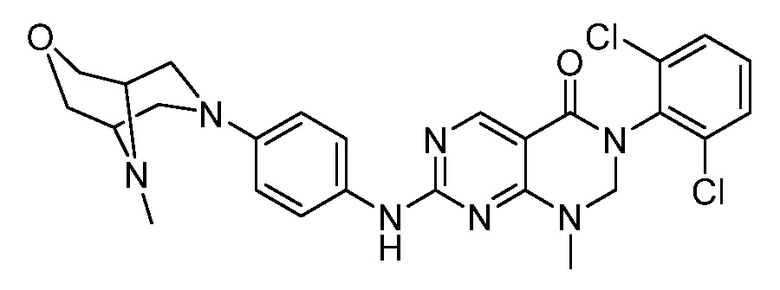
7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)фенил)-амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он (30 мг, 0,057 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде желтого твердого вещества (13 мг, 42%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,64 (уш.с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,59 (уш.с, 2H), 7,47 (т, 1H), 6,80 (д, 2H), 4,94 (с, 2H), 3,84 (д, 2H), 3,74 (д, 2H), 3,41 (д, 2H), 3,20 (дд, 2H), 3,08 (с, 3H), 2,76 (уш.с, 2H), 2,46 (с, 3H). LCMS (метод C): RT=0,69 мин, m/z=540 [M+H]+.
Пример 72: 3-(2,6-дихлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
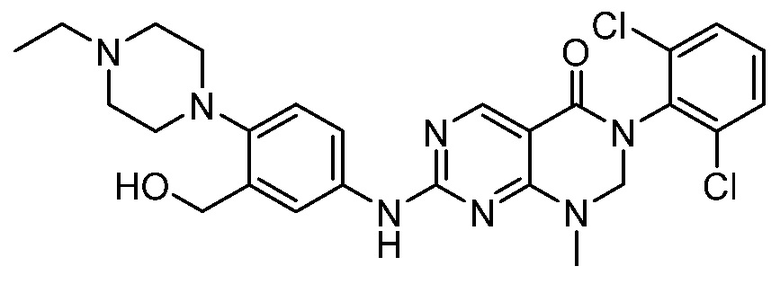
Стадия 1: 2-(4-этилпиперазин-1-ил)-5-нитробензальдегид:
Суспензию 2-фтор-5-нитробензальдегида (3 г, 17,74 ммоль), 1-этилпиперазина (2,026 г, 17,74 ммоль) и карбоната калия (3,68 г, 26,6 ммоль) в безводном DMF (15 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали, и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (4,38 г, 94%). 1H ЯМР (300 МГц, CDCl3): δ 10,10 (с, 1H), 8,62 (д, 1H), 8,30 (дд, 1H), 7,09 (д, 1H), 3,35 (т, 4H), 2,68 (т, 4H), 2,52 (кв, 2H), 1,14 (т, 3H). LCMS (метод C): RT=0,38 мин, m/z=264 [M+H]+.
Стадия 2: (2-(4-этилпиперазин-1-ил)-5-нитрофенил)метанол:
Раствор 2-(4-этилпиперазин-1-ил)-5-нитробензальдегида (4,38 г, 16,64 ммоль) в безводном THF (30,8 мл) охлаждали до 0°C, затем порциями добавляли боргидрид натрия (0,629 г, 16,64 ммоль). Реакционную смесь перемешивали при 0°C в течение 90 минут. Реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, дихлорметан:метанол, градиентное элюирование от 100:0 до 85:15) с получением названного соединения в виде желтого твердого вещества (1,70 г, 39%). 1H ЯМР (300 МГц, CDCl3): δ 8,26 (д, 1H), 8,12 (дд, 1H), 7,14 (д, 1H), 4,79 (с, 2H), 3,61 (уш.с, 1H), 3,08 (т, 4H), 2,64 (уш.с, 4H), 2,50 (кв, 2H), 1,12 (т, 3H). LCMS (метод C): RT=0,37 мин, m/z=266 [M+H]+.
Стадия 3: (5-амино-2-(4-этилпиперазин-1-ил)фенил)метанол:
Раствор (2-(4-этилпиперазин-1-ил)-5-нитрофенил)метанола (1,70 г, 6,41 ммоль) в метаноле (100 мл) гидрировали в проточном реакторе H-Cube (масштабируемый картридж 10% Pd/C, заполненный H2, 40°C, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (1,48 г, 98%). 1H ЯМР (300 МГц, DMSO-d6): δ 6,80 (д, 1H), 6,66 (д, 1H), 6,39 (дд, 1H), 4,96 (т, 1H), 4,76 (уш.с, 2H), 4,45 (д, 2H), 2,70 (т, 4H), 2,44 (уш.с, 4H), 2,35 (кв, 2H), 1,01 (т, 3H). LCMS (метод C): RT=0,21 мин, m/z=236 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (5-амино-2-(4-этилпиперазин-1-ил)фенил)метанолом (67 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (24 мг, 16%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,81 (уш.с, 1H), 8,46 (с, 1H), 7,99 (уш.с, 1H), 7,64 (д, 2H), 7,56 (дд, 1H), 7,48 (дд, 1H), 7,01 (д, 1H), 5,06 (т, 1H), 4,96 (с, 2H), 4,54 (д, 2H), 3,11 (с, 3H), 2,81 (т, 4H), 2,50 (т, 4H), 2,38 (кв, 2H), 1,02 (т, 3H). LCMS (метод C): RT=0,71 мин, m/z=542 [M+H]+.
Пример 73: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Раствор mCPBA 50% чистоты (117 мг, 0,338 ммоль) в дихлорметане (0,400 мл) пропускали через фазовый сепаратор и промывали дополнительно дихлорметаном (0,400 мл). Этот раствор добавляли к перемешиваемой суспензии 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она (100 мг, 0,281 ммоль) в безводном толуоле (2 мл), и смесь перемешивали в течение 30 минут. Добавляли основание Хунига (0,147 мл, 0,844 ммоль), затем (5-амино-2-морфолинофенил)метанол (58,6 мг, 0,281 ммоль), и смесь нагревали до 75°C в течение 30 минут. Добавляли DMF (0,5 мл), и нагревание продолжали в течение 16 часов. После охлаждения, непосредственно реакционную смесь очищали флэш-хроматографией (0-100%, этилацетат в циклогексане, колонка KP-NH). Остаток растворяли в метаноле (1 мл) и воде (8 мл), затем лиофилизировали. Остаток перемешивали в диэтиловом эфире (2 мл) в течение 20 минут. Диэтиловый эфир удаляли декантированием, затем твердое вещество сушили под вакуумом при 50°C. Твердое вещество растворяли в метаноле (0,5 мл) и воде (1,5 мл), затем лиофилизировали с получением названного соединения (40,8 мг, 28,1%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,46 (с, 1H), 8,00 (уш.с, 1H), 7,65 (д, 2H), 7,58 (дд, 1H), 7,48 (дд, 1H), 7,03 (д, 1H), 5,06 (т, 1H), 4,96 (с, 2H), 4,56 (д, 2H), 3,71 (т, 4H), 3,12 (с, 3H), 2,80 (т, 4H). LCMS (метод C): RT=1,08 мин, m/z=515 [M+H]+.
Пример 74: 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)фенил) амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 3-(4-нитрофенил)-3,8-диазабицикло- [3,2,1]октан-8-карбоксилат:
Суспензию 1-фтор-4-нитробензола (0,266 г, 1,884 ммоль), третбутил 3,8-диазабицикло[3,2,1]октан-8-карбоксилата (0,4 г, 1,884 ммоль) и карбоната калия (0,39 г, 2,83 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между водой (50 мл) и этилацетатом (50 мл). Этилацетат отделяли и промывали водой (3×20 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (0-100% этилацетат в циклогексане) с получением названного соединения (0,62 г, 100%). LCMS (метод A): RT=1,61 мин, m/z=334 [M+H]+.
Стадия 2: третбутил 3-(4-аминофенил)-3,8-диазабицикло-[3,2,1]октан-8-карбоксилат:
Раствор третбутил 2,2-диметил-4-(4-нитрофенил)пиперазин-1-карбоксилата (250 мг, 0,75 ммоль) в метаноле (10 мл) пропускали через проточный реактор H-Cube, снабженный картриджем с 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2)]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (230 мг, 100%). LCMS (метод C): RT=0,84 мин, m/z=304 [M+H]+.
Стадия 3: 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)фенил) амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (250 мг, 0,704 ммоль) подвергали взаимодействию с третбутил 3-(4-аминофенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилатом (203 мг,0,67 ммоль), следуя методике примера 31, с получением названного соединения (168 мг, 41%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,65 (с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,56 (м, 2H), 7,47 (м, 1H), 6,76 (д, 2H), 4,91 (с, 2H), 3,48 (м, 2H), 3,31 (м, 1H), 3,08 (с, 3H), 2,70 (м, 2H), 2,33 (м, 1H), 1,69 (м, 4H). LCMS (метод C): RT=0,77 мин, m/z=510 [M+H]+.
Пример 75: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
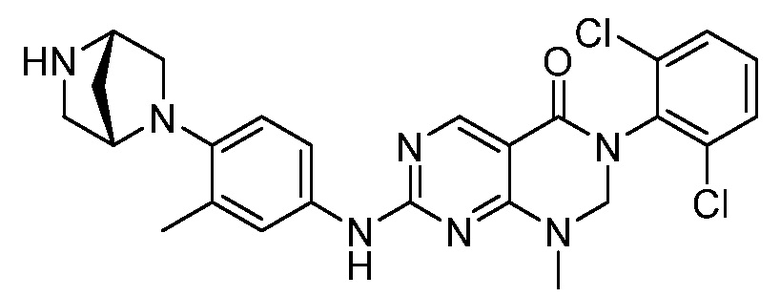
Стадия 1: (1S,4S)-третбутил 5-(2-метил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию 2-фтор-5-нитротолуола (1,565 г, 10,09 ммоль), (1S,4S)-третбутил 2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (2,00 г, 10,09 ммоль) и карбоната калия (2,091 г, 15,13 ммоль) в безводном DMF (8 мл) нагревали до 50°C в атмосфере азота в течение 2 дней, затем при 100°C в течение 3 дней. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 0:100) с получением названного соединения в виде желтого твердого вещества (1,87 г, 56%). 1H ЯМР (300 МГц, CDCl3): δ 7,92-8,02 (м, 2H), 6,60 (дд, 1H), 4,56 (д, 1H), 4,44 (с, 1H), 3,83 (д, 1H), 3,62 (дд, 1H), 3,47 (т, 1H), 3,32 (дд, 1H), 2,36 (с, 3H), 1,86-2,06 (м, 2H), 1,38-1,49 (м, 9H). LCMS (метод C): RT=1,69 мин, m/z=334 [M+H]+.
Стадия 2: (1S,4S)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Раствор (1S,4S)-третбутил 5-(2-метил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (1,87 г, 5,61 ммоль) в THF (140 мл) гидрировали в проточном реакторе H-Cube (масштабируемый картридж 10% Pd/C, заполненный H2, 40°C, 1 мл/мин) за три прохода. Реакционную смесь концентрировали досуха при пониженном давлении, суспендировали в диэтиловом эфире (25 мл), фильтровали, и воду отсасывали с получением названного соединения в виде желтовато-коричневого твердого вещества (1,21 г, 71%). 1H ЯМР (400 МГц, CDCl3): δ 6,71 (дд, 1H), 6,55 (с, 1H), 6,48 (д, 1H), 4,46 (д, 1H), 3,91 (с, 1H), 3,54 (дд, 1H), 3,19-3,47 (м, 5H), 2,21 (с, 3H), 1,96 (т, 1H), 1,82 (т, 1H), 1,42-1,52 (м, 9H). LCMS (метод C): RT=0,81 мин, m/z=304 [M+H]+.
Стадия 3: (1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (86 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (109 мг, 63%). 1H ЯМР (300 МГц, CDCl3): δ 8,70 (с, 1H), 7,73 (уш.с, 1H), 7,43 (д, 2H), 7,32-7,42 (м, 2H), 7,27 (дд, 1H), 6,79 (дд, 1H), 4,86 (с, 2H), 4,50 (д, 1H), 4,10 (уш.с, 1H), 3,61 (дд, 1H), 3,19-3,50 (м, 3H), 3,16 (с, 3H), 2,28 (с, 3H), 1,75-2,01 (м, 2H), 1,41-1,50 (м, 9H). LCMS (метод C): RT=1,65 мин, m/z=610 [M+H]+.
Стадия 4: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
(1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (109 мг, 0,179 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (76 мг, 83%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,63 (уш.с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,39-7,53 (уш.м, 3H), 6,75 (д, 1H), 4,95 (с, 2H), 3,98 (с, 1H), 3,54 (с, 1H), 3,41 (дд, 1H), 3,09 (с, 3H), 3,04 (д, 1H), 2,95 (д, 1H), 2,84 (дд, 1H), 2,18 (с, 3H), 1,75 (д, 1H), 1,58 (д, 1H). LCMS (метод C): RT=0,78 мин, m/z=510 [M+H]+.
Пример 76: 3-(2,6-дихлорфенил)-1-метил-7-((2'-метил-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-изохинолин]-7'-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 3-(2,6-дихлорфенил)-1-метил-7-((2'-метил-2',3'-дигидро-1'H-spiro[циклопропанe-1,4'-изохинолин]-7'-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с дигидрохлоридной солью 2'-метил-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-изохинолин]-7'-амина [WO 2009/151997] (74 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (8 мг, 6%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,79 (уш.с, 1H), 8,47 (с, 1H), 7,64 (д, 2H), 2,43-7,56 (уш.м, 3H), 6,65 (д, 1H), 4,97 (с, 2H), 3,55 (с, 2H), 3,10 (с, 3H), 2,43 (с, 2H), 2,30 (с, 3H), 0,85 (дт, 4H). LCMS (метод C): RT=0,84 мин, m/z=495 [M+H]+.
Пример 77: 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (41 мг, 0,080 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде бледно-желтого твердого вещества (30 мг, 71%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,50 (с, 1H), 7,56 (д, 2H), 7,35-7,48 (м, 3H), 6,86 (д, 1H), 4,97 (с, 2H), 4,03 (с, 1H), 3,52 (с, 1H), 3,40 (д, 1H), 3,28 (дд, 1H), 3,17 (с, 3H), 3,01 (д, 1H), 2,85 (дд, 1H), 2,45 (с, 3H), 2,29 (с, 3H), 1,98 (д, 1H), 1,91 (д, 1H). LCMS (метод C): RT=0,80 мин, m/z=524 [M+H]+.
Пример 78: (R)-3-(2,6-дихлорфенил)-1-d3-метил-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
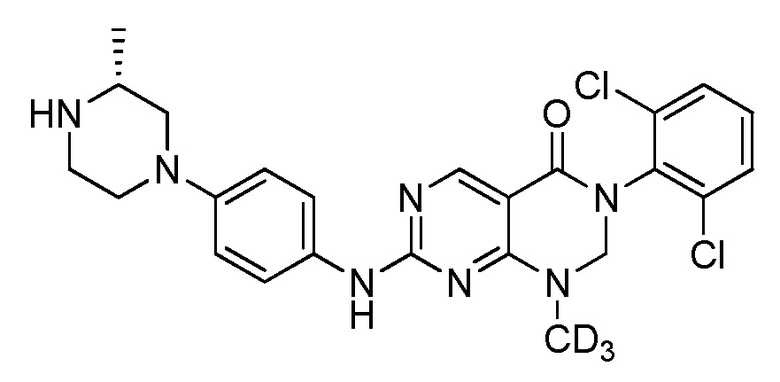
Стадия 1: N-(2,6-дихлорфенил)-4-(d3-метиламино)-2-(метил-тио)пиримидин-5-карбоксамид:
4-хлор-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамид (3,1 г, 8,89 ммоль) суспендировали в безводном метаноле (30 мл). Добавляли метил-d3-амина гидрохлорид (0,627 г, 8,89 ммоль) и основание Хунига (4,66 мл, 26,7 ммоль). Смесь разделяли на две пробирки для проведения реакций при микроволновом излучении, которые герметизировали и нагревали до 50°C в течение 1 часа. объединенную смесь распределяли между водой (25 мл) и дихлорметаном (50 мл). Водный слой отделяли и экстрагировали дихлорметаном (2×50 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор) и концентрировали под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (3,31 г, 108%). 1H ЯМР (400 МГц, CDCl3): δ 8,49 (с, 1H), 8,45 (уш.с, 1H), 7,50 (уш.с, 1H), 7,42 (д, 2H), 7,22 (дд, 1H), 2,56 (с, 3H). LCMS (метод C): RT=1,43 мин, m/z=346 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-1-d3-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(d3-метиламино)-2-(метилтио)пиримидин-5-карбоксамид (3,31 г, 8,89 ммоль) суспендировали в безводном ацетонитриле (70 мл) в сосуде Шленка. Добавляли карбонат цезия (11,59 г, 35,6 ммоль), затем дийодметан (1,434 мл, 17,78 ммоль). Пробирку герметизировали, и смесь нагревали до 80°C при перемешивании в течение 40 часов. Добавляли дополнительное количество дийодметана (1,434 мл, 17,78 ммоль), и нагревание продолжали в течение 48 часов. Растворитель удаляли под вакуумом. Остаток распределяли между водой (50 мл) и дихлорметаном (100 мл). Водный слой отделяли и экстрагировали дихлорметаном (2×25 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор) и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетат в циклогексане). Полученный остаток дополнительно очищали хроматографией на силикагеле (изократический 25% этилацетат в циклогексане) с получением названного соединения (779 мг, 24,5%). 1H ЯМР (400 МГц, CDCl3): δ 8,72 (с, 1H), 7,45 (д, 2H), 7,30 (дд, 1H), 4,91 (с, 2H), 2,57 (с, 3H). LCMS (метод C): RT=1,44 мин, m/z [M+H]+.
Стадия 3: (R)-3-(2,6-дихлорфенил)-1-d3-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,279 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (81 мг, 0,279 ммоль), следуя методике примера 31, с получением названного соединения (84,4 мг, 96%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,60 (уш.д, 2H), 7,48 (т, 1H), 6,88 (д, 2H), 4,95 (с, 2H), 3,44 (т, 2H), 2,93 (д, 1H), 2,71-2,84 (м, 2H), 2,23 (уш.с, 1H), 2,14 (т, 1H), 1,01 (д, 3H). LCMS (метод C): RT=0,78 мин, m/z=501 [M+H]+.
Пример 79: 3-(2,6-дихлорфенил)-7-((4-((2R,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (2R,5R)-третбутил 2,5-диметил-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Суспензию 1-фтор-4-нитробензола (329 мг, 2,333 ммоль), (2R,5R)-третбутил 2,5-диметилпиперазин-1-карбоксилата (500 мг, 2,333 ммоль) и карбоната калия (484 мг, 3,50 ммоль) в безводном DMF (2 мл) нагревали до 50°C в атмосфере азота в течение 7 дней. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Смесь экстрагировали этилацетатом (3×20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали с помощью двух соединенных последовательно хроматографических колонок Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 50:50) с получением названного соединения в виде желтого твердого вещества (200 мг, 26%). 1H ЯМР (400 МГц, CDCl3): δ 8,11 (д, 2H), 6,66 (д, 2H), 3,93-4,21 (м, 3H), 3,79 (дд, 1H), 3,08 (дд, 1H), 2,90 (дд, 1H), 1,41 (с, 9H), 1,20 (дд, 6H). LCMS (метод C): RT=1,74 мин, m/z=336 [M+H]+.
Стадия 2: (2R,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилат:
Раствор (2R,5R)-третбутил 2,5-диметил-4-(4-нитрофенил)пиперазин-1-карбоксилата (0,20 г, 0,596 ммоль) в метаноле (20 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 50°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде серого твердого вещества (150 мг, 82%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,79 (д, 2H), 6,54 (д, 2H), 5,49 (уш.с, 2H), 4,08 (уш.с, 1H), 3,74 (дд, 1H), 2,92 (дд, 1H), 2,75-2,87 (м, 1H), 2,63-2,75 (м, 2H), 1,40 (с, 9H), 1,22 (д, 3H), 0,79 (д, 3H). LCMS (метод C): RT=0,84 мин, m/z=306 [M+H]+.
Стадия 3: (2R,5R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)фенил)-2,5-диметилпиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (2R,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (86 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (105 мг, 61%). 1H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,64 (уш.с, 1H), 7,53 (д, 2H), 7,43 (д, 2H), 7,28 (дд, 1H), 6,98 (д, 2H), 4,87 (с, 2H), 4,20 (уш.с, 1H), 3,93 (д, 1H), 3,08-3,23 (м, 5H), 2,89 (дд, 1H), 2,83 (дд, 1H), 1,46 (с, 9H), 1,30 (д, 3H), 0,97 (д, 3H). LCMS (метод C): RT=1,76 мин, m/z=612 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((4-((2R,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]-пиримидин-4(1H)-он:
(2R,5R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2,5-диметилпиперазин-1-карбоксилат (105 мг, 0,171 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (50 мг, 57%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,65 (уш.с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,58 (уш.с, 2H), 7,47 (дд, 1H), 6,82 (д, 2H), 4,94 (с, 2H), 3,84-3,94 (м, 1H), 3,14 (дд, 1H), 3,08 (с, 3H), 2,97 (дд, 1H), 2,79 (д, 1H), 2,67-2,77 (м, 1H), 2,40 (т, 1H), 1,06 (д, 3H), 0,96 (д, 3H). LCMS (метод C): RT=0,77 мин, m/z=512 [M+H]+.
Пример 80: 3-(2,6-дихлорфенил)-7-((4-((2S,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (2R,5S)-третбутил 2,5-диметил-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
Суспензию 1-фтор-4-нитробензола (329 мг, 2,333 ммоль), (2R,5S)-третбутил 2,5-диметилпиперазин-1-карбоксилата (500 мг, 2,333 ммоль) и карбоната калия (484 мг, 3,50 ммоль) в безводном ацетонитриле (3 мл) нагревали до 100°C в атмосфере азота в течение 11 дней. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 60:40) с получением названного соединения в виде желтого твердого вещества (530 мг, 68%). 1H ЯМР (400 МГц, CDCl3): δ 8,12 (д, 2H), 6,77 (д, 2H), 4,42 (уш.д, 1H), 4,00-4,19 (м, 1H), 3,84 (дд, 1H), 3,29-3,50 (м, 3H), 1,24 (д, 3H), 1,49 (с, 9H), 1,19 (д, 3H). LCMS (метод C): RT=1,80 мин, m/z=336 [M+H]+.
Стадия 2: (2R,5S)-третбутил 4-(4-аминофенил)-2,5-диметил-пиперазин-1-карбоксилат:
Раствор (2R,5S)-третбутил 2,5-диметил-4-(4-нитрофенил)-пиперазин-1-карбоксилата (0,53 г, 1,580 ммоль) в метаноле (50 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 50°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде серого твердого вещества (480 мг, 99%). 1H ЯМР (400 МГц, CDCl3): δ 6,72 (дт, 2H), 6,65 (дт, 2H), 4,39 (уш.с, 1H), 3,70-3,82 (м, 2H), 3,42 (дд, 1H), 3,39 (уш.с, 2H), 3,22 (дд, 1H), 2,85 (дд, 1H), 1,48 (с, 9H), 1,26 (д, 3H), 0,93 (д, 3H). LCMS (метод C): RT=0,90 мин, m/z=306 [M+H]+.
Стадия 3: (2R,5S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)фенил)-2,5-диметилпиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (2R,5S)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (86 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (133 мг, 77%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 8,13 (уш.с, 1H), 7,50 (д, 2H), 7,42 (д, 2H), 7,26 (дд, 1H), 6,84 (д, 2H), 4,86 (с, 2H), 4,41 (уш.с, 1H), 3,92 (уш.с, 1H), 3,79 (д, 1H), 3,42 (дд, 1H), 3,27 (дд, 1H), 3,15 (с, 3H), 3,05 (д, 1H), 1,48 (с, 9H), 1,27 (д, 3H), 1,02 (д, 3H). LCMS (метод C): RT=1,78 мин, m/z=612 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((4-((2S,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(2R,5S)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2,5-диметилпиперазин-1-карбоксилат (133 мг, 0,217 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (79 мг, 71%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,80 (уш.с, 1H), 8,47 (с, 1H), 7,68 (д, 2H), 7,64 (д, 2H), 7,48 (дд, 1H), 7,03 (д, 2H), 4,96 (с, 2H), 3,10 (с, 3H), 2,80-3,00 (м, 4H), 2,45 (дд, 1H), 2,33 (дд, 1H), 0,95 (д, 3H), 0,80 (д, 3H). LCMS (метод C): RT=0,80 мин, m/z=512 [M+H]+.
Пример 81: 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
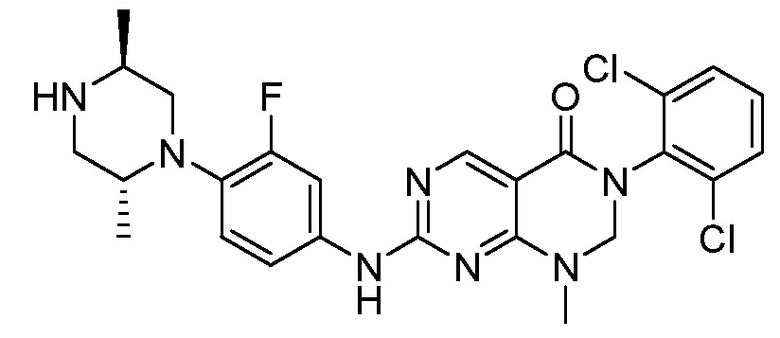
Стадия 1: (2S,5R)-третбутил 4-(2-фтор-4-нитрофенил)-2,5-диметилпиперазин-1-карбоксилат:
Суспензию 1,2-дифтор-4-нитробензола (371 мг, 2,333 ммоль), (2S,5R)-третбутил 2,5-диметилпиперазин-1-карбоксилата (500 мг, 2,333 ммоль) и карбоната калия (484 мг, 3,50 ммоль) в безводном ацетонитриле (3 мл) нагревали до 100°C в атмосфере азота в течение 8 дней. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 60:40) с получением названного соединения в виде желтого масла (800 мг, 97%). 1H ЯМР (400 МГц, CDCl3): δ 7,97 (дд, 1H), 7,90 (дд, 1H), 6,84 (т, 1H), 4,42 (уш.с, 1H), 4,00 (уш.с, 1H), 3,76 (д, 1H), 3,54 (дд, 1H), 3,46 (д, 1H), 3,16 (д, 1H), 1,49 (с, 9H), 1,28 (д, 3H), 1,15 (д, 3H). LCMS (метод C): RT=1,92 мин, m/z=376 [M+Na]+.
Стадия 2: (2S,5R)-третбутил 4-(4-амино-2-фторфенил)-2,5-диметилпиперазин-1-карбоксилат:
Раствор (2S,5R)-третбутил 4-(2-фтор-4-нитрофенил)-2,5-диметилпиперазин-1-карбоксилата (800 мг, 2,264 ммоль) в метаноле (80 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 50°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде серого твердого вещества (623 мг, 85%). 1H ЯМР (300 МГц, CDCl3) δ 6,61-6,71 (м, 1H), 6,34-6,45 (м, 2H), 4,37 (квин, 1H), 3,69 (д, 1H), 3,34-3,61 (м, 5H), 2,58 (дд, 1H), 1,47 (с, 9H), 1,29 (д, 3H), 0,91 (д, 3H). LCMS (метод C): RT=1,37 мин, m/z=324 [M+H]+.
Стадия 3: (2S,5R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-фторфенил)-2,5-диметилпиперазин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-амино-2-фторфенил)-2,5-диметилпиперазин-1-карбоксилатом (91 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (86 мг, 48%). 1H ЯМР (400 МГц, CDCl3) δ 8,72 (с, 1H), 7,93 (уш.с, 1H), 7,65 (д, 1H), 7,43 (д, 2H), 7,28 (дд, 1H), 7,09 (дд, 1H), 6,80 (т, 1H), 4,89 (с, 2H), 4,40 (уш.с, 1H), 3,65-3,79 (м, 2H), 3,47 (ддд, 2H), 3,18 (с, 3H), 2,73 (д, 1H), 1,48 (с, 9H), 1,30 (д, 3H), 0,98 (д, 3H). LCMS (метод C): RT=1,98 мин, m/z=630 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
(2S,5R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-фторфенил)-2,5-диметилпиперазин-1-карбоксилат (105 мг, 0,167 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (31 мг, 35%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,01 (уш.с, 1H), 8,50 (с, 1H), 7,75 (дд, 1H), 7,65 (д, 2H), 7,43-7,53 (м, 2H), 7,18 (т, 1H), 4,99 (с, 2H), 3,12 (с, 3H), 2,80-2,99 (м, 4H), 2,46 (дд, 1H), 2,36 (т, 1H), 0,94 (д, 3H), 0,78 (д, 3H). LCMS (метод C): RT=0,90 мин, m/z=530 [M+H]+.
Пример 82: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
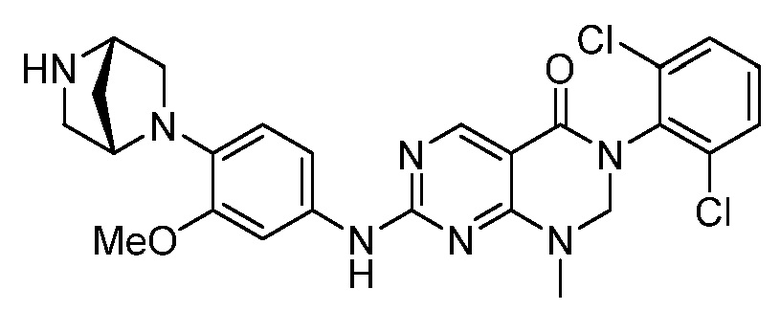
Стадия 1: (1S,4S)-третбутил 5-(2-метокси-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию 1-фтор-2-метокси-4-нитробензола (2,59 г, 15,13 ммоль), (1S,4S)-третбутил 2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (3,00 г, 15,13 ммоль) и карбоната калия (3,14 г, 22,7 ммоль) в безводном DMF (40 мл) нагревали до 50°C в атмосфере азота в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (120 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией и сушили под вакуумом с получением названного соединения в виде желтого твердого вещества (4,49 г, 85%). LCMS (метод C): RT=1,44 мин, m/z=294 [M-tBu+H]+.
Стадия 2: (1S,4S)-третбутил 5-(4-амино-2-метоксифенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию третбутил 4-(2-метокси-4-нитрофенил)пиперазин-1-карбоксилата (3,15 г, 9,34 ммоль), порошок железа (3,13 г, 56,0 ммоль) и хлорид аммония (4,50 г, 84 ммоль) в смеси метанола (38,6 мл) и воды (8,12 мл) кипятили с обратным холодильником в атмосфере азота в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит®. Фильтраты разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия (x3). объединенные органические фазы сушили (Na2SO4) и концентрировали досуха при пониженном давлении. Остаток разбавляли этилацетатом, и твердое вещество собирали фильтрацией с получением названного соединения в виде розового твердого вещества (2,78 г, 68%). LCMS (метод C): RT=0,71 мин, m/z=320 [M+H]+.
Стадия 3: (1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-метоксифенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-метоксифенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (90 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (88 мг, 50%). 1H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,75 (уш.с, 1H), 7,43 (д, 2H), 7,23-7,35 (м, 2H), 7,00 (дд, 1H), 6,61 (д, 1H), 4,87 (с, 2H), 4,48 (д, 1H), 4,40 (с, 1H), 3,83 (с, 3H), 3,71 (дд, 1H), 3,61 (дд, 1H), 3,30 (т, 1H), 3,26 (дд, 1H), 3,17 (с, 3H), 1,96 (д, 1H), 1,84 (дд, 1H), 1,43 (д, 9H). LCMS (метод C): RT=1,53 мин, m/z=626 [M+H]+.
Стадия 4: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метоксифенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (88 мг, 0,140 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (36 мг, 49%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,67 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,57 (уш.с, 1H), 7,48 (дд, 1H), 7,11 (дд, 1H), 6,55 (д, 1H), 4,96 (с, 2H), 4,20 (с, 1H), 3,73 (с, 3H), 3,59 (дд, 1H), 3,48 (с, 1H), 3,11 (с, 3H), 2,99 (д, 1H), 2,88 (д, 1H), 2,81 (дд, 1H), 1,70 (д, 1H), 1,55 (д, 1H). LCMS (метод C): RT=0,77 мин, m/z=526 [M+H]+.
Пример 83: 3-(2,6-дихлорфенил)-7-((4-((3R,5S)-3,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (3S,5R)-3,5-диметил-1-(4-нитрофенил)пиперазин:
К перемешиваемой суспензии 1-фтор-4-нитробензола (562 мг, 3,98 ммоль) и карбоната калия (688 мг, 4,98 ммоль) в безводном DMSO (2 мл) добавляли (2S,6R)-2,6-диметилпиперазин (500 мг, 4,38 ммоль), и смесь нагревали при 100°C в течение 16 часов. После охлаждения, смесь суспендировали в воде (40 мл) и фильтровали под вакуумом с получением названного соединения (889 мг, 95%). 1H ЯМР (500 МГц, DMSO-d6): δ 8,03 (д, 2H), 7,03 (д, 2H), 3,88 (дд, 2H), 2,75-2,79 (м, 2H), 2,37-2,42 (м, 2H), 2,28 (уш.с, 1H), 1,03 (д, 6H). LCMS (метод C): RT=0,51 мин, m/z=236 [M+H]+.
Стадия 2: 4-((3S,5R)-3,5-диметилпиперазин-1-ил)анилин:
Перемешиваемый раствор (3S,5R)-3,5-диметил-1-(4-нитро-фенил)пиперазина (888,8 мг, 3,78 ммоль) в этаноле (15 мл) нагревали до 50°C. Добавляли Pd/C (201 мг, 0,189 ммоль), затем порциями добавляли формиат аммония (1191 мг, 18,89 ммоль), и суспензию перемешивали в течение 10 минут. Суспензию фильтровали, промывая свежим этанолом (6 мл). Этанол удаляли под вакуумом. Остаток распределяли между дихлорметаном (15 мл) и водой (10 мл). Водную фазу отделяли и экстрагировали дихлорметаном (2×5 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор) и концентрировали под вакуумом с получением названного соединения (111 мг, 14%). 1H ЯМР (500 МГц, DMSO-d6): δ 6,66 (д, 2H), 6,47 (д, 2H), 4,49 (уш.с, 2H), 3,18 (дд, 2H), 2,81-2,84 (м, 2H), 1,82-2,10 (м, 1H), 1,96 (т, 2H), 0,98 (д, 6H). LCMS (метод C): RT=0,12 мин, m/z=206 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-((3R,5S)-3,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с 4-((3R,5S)-3,5-диметилпиперазин-1-ил)анилином (58 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (44 мг, 30%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,60 (уш.д, 2H), 7,47 (дд, 1H), 6,88 (д, 2H), 4,95 (с, 2H), 3,45 (дд, 2H), 3,08 (с, 3H), 2,77-2,90 (м, 2H), 2,00-2,13 (м, 3H), 1,01 (д, 6H). LCMS (метод C): RT=0,76 мин, m/z=512 [M+H]+.
Пример 84: 3-(2,6-дихлорфенил)-7-((3-метокси-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (21 мг, 0,040 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде желтого твердого вещества (9 мг, 42%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,58 (уш.с, 1H), 7,48 (дд, 1H), 7,12 (дд, 1H), 6,55 (д, 1H), 4,96 (с, 2H), 4,17 (с, 1H), 3,74 (с, 3H), 3,41 (дд, 1H), 3,28 (с, 1H), 3,05-3,17 (м, 4H), 2,71 (дд, 1H), 2,63 (д, 1H), 2,24 (с, 3H), 1,76 (д, 1H), 1,66 (д, 1H). LCMS (метод C): RT=0,78 мин, m/z=540 [M+H]+.
Пример 85: (R)-1-циклопропил-3-(2,6-дихлорфенил)-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
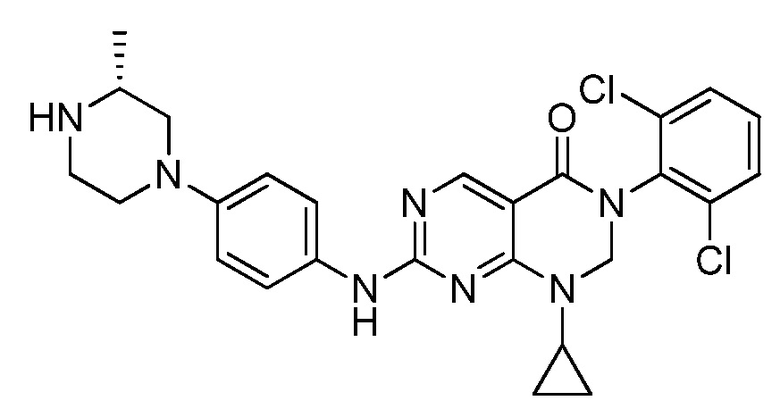
Стадия 1: 4-(циклопропиламино)-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамид:
К суспензии 4-хлор-N-(2,6-дихлорфенил)-2-(метилтио)-пиримидин-5-карбоксамида (2 г, 5,74 ммоль) в тетрагидрофуране (20 мл) при 0°C добавляли основание Хунига (2,004 мл, 11,47 ммоль), затем циклопропанамин (0,437 мл, 6,31 ммоль). Реакционную смесь перемешивали при этой температуре в течение 15 минут и при комнатной температуре в течение 30 минут. Реакционную смесь разбавляли водой и этилацетатом, и слои разделяли, водный слой затем экстрагировали два раза дополнительным количеством этилацетата, и объединенные органические экстракты промывали солевым раствором, сушили (безводный сульфат магния), фильтровали и концентрировали под вакуумом с получением названного соединения (2,2 г) в виде белого твердого вещества. LCMS (метод C) RT=1,65 мин, m/z=369 [M+H]+.
Стадия 2: 1-циклопропил-3-(2,6-дихлорфенил)-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
4-(циклопропиламино)-N-(2,6-дихлорфенил)-2-(метилтио)-пиримидин-5-карбоксамид (2,2 г, 5,96 ммоль) суспендировали в безводном ацетонитриле (25 мл) в сосуде Шленка. Добавляли карбонат цезия (11,65 г, 35,7 ммоль), затем дибромметан (1,254 мл, 17,87 ммоль). Пробирку герметизировали, и смесь нагревали до 80°C при перемешивании в течение 40 часов. Растворитель удаляли под вакуумом. Остаток распределяли между водой (50 мл) и дихлорметаном (100 мл). Водную фазу отделяли и экстрагировали дихлорметаном (2×25 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор) и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-40% этилацетат в циклогексане) с получением названного соединения (1,22 г, 53,7%). LCMS (метод C): RT=1,63 мин, m/z=381 [M+H]+.
Стадия 3: (R)-1-циклопропил-3-(2,6-дихлорфенил)-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
1-циклопропил-3-(2,6-дихлорфенил)-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,262 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метилпиперазин-1-карбоксилатом (76 мг, 0,262 ммоль), следуя методике примера 31, с получением названного соединения (47,2 мг, 47,5%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,56 (с, 1H), 7,64-7,90 (м, 2H), 7,57 (д, 2H), 7,43 (дд, 1H), 7,00 (д, 2H), 4,99 (с, 2H), 3,52 (т, 2H), 3,09 (дт, 1H), 2,91-.3,04 (м, 2H), 2,63-2,77 (м, 2H), 2,34 (т, 1H), 1,17 (д, 3H), 1,02 (кв, 2H), 0,83-0,89 (м, 2H). LCMS (метод C): RT=0,88 мин, m/z=524 [M+H]+.
Пример 86: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 3-(2-формил-4-нитрофенил)-3,8-диаза-бицикло[3,2,1]октан-8-карбоксилат:
Суспензию 2-фтор-5-нитробензальдегида (0,358 г, 2,120 ммоль), третбутил 3,8-диазабицикло[3,2,1]октан-8-карбоксилата (0,45 г, 2,120 ммоль) и карбоната калия (0,439 г, 3,18 ммоль) в безводном DMF (2 мл) нагревали до 50°C в атмосфере азота в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между водой (50 мл) и этилацетатом (50 мл). Этилацетат отделяли и промывали водой (3×20 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (0-100% этилацетат в циклогексане) с получением названного соединения (360 мг, 0,996 ммоль). LCMS (метод C): RT=1,69 мин, m/z=384 [M+Na]+.
Стадия 2: третбутил 3-(2-(гидроксиметил)-4-нитрофенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат:
К перемешиваемому раствору третбутил 3-(2-формил-4-нитро-фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилата (360 мг, 0,996 ммоль) в безводном тетрагидрофуране (4 мл) при 0°C добавляли боргидрид натрия (41,5 мг, 1,096 ммоль). Перемешивание продолжали в течение 3 часов. Смесь концентрировали под вакуумом. Остаток распределяли между водой (20 мл) и дихлорметаном (10 мл). Водную фазу отделяли и экстрагировали дихлорметаном (3×5 мл). Объединенные дихлорметановые фракции сушили (фазовый сепаратор), фильтровали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 0-50% этилацетат в циклогексане) с получением названного соединения (343 мг, 95%). 1H ЯМР (400 МГц, CDCl3): δ 8,34 (д, 1H), 8,12 (дд, 1H), 7,13 (д, 1H), 4,82 (д, 2H), 4,20-4,47 (м, 2H), 2,90-3,22 (м, 4H), 2,60 (т, 1H), 1,98-2,04 (м, 4H), 1,49 (с, 9H). LCMS (метод C): RT=1,56 мин, m/z=364 [M+H]+.
Стадия 3: третбутил 3-(4-амино-2-(гидроксиметил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат:
Раствор третбутил 3-(2-(гидроксиметил)-4-нитрофенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилата (335 мг, 0,922 ммоль) в метаноле (10 мл) и тетрагидрофуране (2,000 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2)]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (295,6 мг, 96%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,79 (д, 1H), 6,67 (д, 1H), 6,38 (дд, 1H), 4,86 (т, 1H), 4,81 (уш.с, 2H), 4,04-4,13 (м, 2H), 4,53 (д, 2H), 2,75 (д, 2H), 2,59 (д, 2H), 1,93 (д, 2H), 1,72-1,84 (м, 2H), 1,43 (с, 9H). LCMS (метод C): RT=0,84 мин, m/z=334 [M+H]+.
Стадия 4: 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,281 ммоль) подвергали взаимодействию с третбутил 3-(4-амино-2-(гидрокси-метил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилатом (94 мг, 0,281 ммоль), следуя методике примера 31, с получением названного соединения (81,3 мг, 88%). 1H ЯМР (400 МГц, CDCl3): δ 8,72 (с, 1H), 7,56-7,60 (м, 1H), 7,52 (дд, 1H), 7,44 (д, 2H), 7,28 (дд, 1H), 7,25 (д, 1H), 4,89 (с, 2H), 4,80 (с, 2H), 3,55-3,60 (м, 2H), 3,19 (с, 3H), 3,02 (д, 2H), 2,86 (дд, 2H), 1,99-2,08 (м, 2H), 1,83-1,89 (м, 2H). LCMS (метод C) RT=0,73 мин, m/z=540 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)-3-(гидрокси-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (77 мг, 0,142 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (47,1 мг, 59,6%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,80 (уш.с, 1H), 8,46 (с, 1H), 8,00 (уш.с, 1H), 7,64 (д, 2H), 7,53 (дд, 1H), 7,48 (дд, 1H), 7,04 (д, 1H), 5,05 (уш.с, 1H), 4,96 (с, 2H), 4,60 (с, 2H), 3,12 (с, 3H), 3,04-3,10 (м, 2H), 2,88 (д, 2H), 2,62 (д, 2H), 2,20 (с, 3H), 1,88-1,98 (м, 2H), 1,80-1,98 (м, 2H). LCMS (метод C): RT=0,72 мин, m/z=554 [M+H]+.
Пример 87: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-илокси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
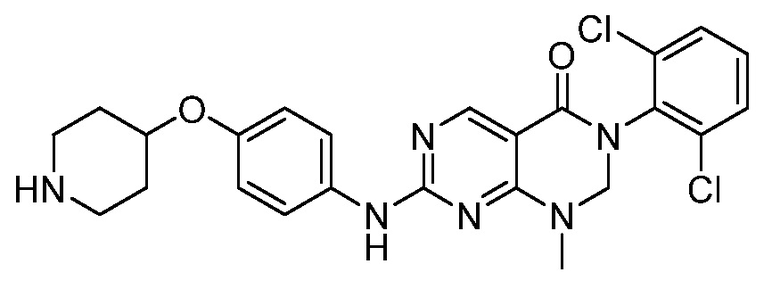
Стадия 1: третбутил 4-(4-нитрофенокси)пиперидин-1-карбоксилат
К суспензии третбутил 4-гидроксипиперидин-1-карбоксилата (3,00 г, 14,91 ммоль) и 1-фтор-4-нитробензола (3,89 г, 27,6 ммоль) в водном растворе гидроксида калия (25 масс.% раствор) (21,84 мл, 97 ммоль) добавляли тетрабутиламмония бромид (0,625 г, 1,938 ммоль), и полученную смесь перемешивали при 35°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, и осажденное твердое вещество выделяли фильтрацией, промывали водой (2×20 мл), и воду отсасывали. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 100:0 до 80:20) с получением названного соединения в виде желтовато-белого твердого вещества (611 мг, 13%). 1H ЯМР (300 МГц, CDCl3): δ 8,20 (дт, 2H), 6,95 (дт, 2H), (септ, 1H), 3,70 (ддд, 2H), 3,38 (ддд, 2H), 1,90-2,04 (м, 2H), 1,72-1,86 (м, 2H), 1,47 (с, 9H). LCMS (метод C): RT=1,78 мин, m/z=345 [M+Na]+.
Стадия 2: третбутил 4-(4-аминофенокси)пиперидин-1-карбоксилат
Раствор третбутил 4-(4-нитрофенокси)пиперидин-1-карбоксилата (611 мг, 1,895 ммоль) в метаноле (60 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 50°C, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде бледно-розового твердого вещества (517 мг, 93%). 1H ЯМР (300 МГц, CDCl3): δ 6,76 (дт, 2H), 6,63 (дт, 2H), 4,26 (септ, 1H), 3,71 (ддд, 2H), 3,45 (уш.с, 2H), 3,26 (ддд, 2H), 1,80-1,95 (м, 2H), 1,61-1,78 (м, 2H), 1,46 (с, 9H). LCMS (метод C): RT=0,85 мин, m/z=237 [M-tBu+H]+.
Стадия 3: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенокси)пиперидин-1-карбоксилат
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (200 мг, 0,563 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенокси)-пиперидин-1-карбоксилатом (165 мг, 0,563 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (190 мг, 56%). 1H ЯМР (300 МГц, CDCl3): δ 8,71 (с, 1H), 7,52 (д, 2H), 7,44 (д, 2H), 7,38 (уш.с, 1H), 7,28 (дд, 1H), 6,91 (д, 2H), 4,88 (с, 2H), 4,43 (септ, 1H), 3,71 (ддд, 2H), 3,32 (ддд, 2H), 3,16 (с, 3H), 1,83-1,99 (м, 2H), 1,65-1,83 (м, 2H), 1,47 (с, 9H). LCMS (метод C): RT=1,75 мин, m/z=599 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-илокси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенокси)-пиперидин-1-карбоксилат (150 мг, 0,250 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (123 мг, 98%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,74 (уш.с, 1H), 8,45 (с, 1H), 7,64 (д, 4H), 7,48 (дд, 1H), 6,90 (д, 2H), 4,95 (с, 2H), 4,31 (тт, 1H), 3,09 (с, 3H), 2,94 (дт, 2H), 2,55 (ддд, 2H), 1,84-1,95 (м, 2H), 1,36-1,49 (м, 2H). LCMS (метод C): RT=0,76 мин, m/z=499 [M+H]+.
Пример 88: 3-(2,6-дихлорфенил)-1-метил-7-((4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (67 мг, 0,131 ммоль) подвергали взаимодействию с формальдегидом (0,018 мл), следуя методике примера 35, с получением названного соединения (65 мг, 94%). ¹H ЯМР (400 МГц, CDCl3): δ 8,69 (с, 1H), 7,50-7,40 (м, 3H), 7,30 (д, 2H), 6,80 (д, 2H), 4,85 (с, 2H), 3,33 (м, 2H), 3,25 (м, 2H), 3,15 (с, 3H), 3,02 (м, 2H), 2,35 (с, 3H), 2,02 (м, 2H), 1,78 (м, 2H). LCMS (метод C): RT=0,72 мин, m/z=524 [M+H]+.
Пример 89: 3-(2-хлор-6-фторфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

3-(2-хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,443 ммоль) подвергали взаимодействию с (5-амино-2-(4-изопропилпиперазин-1-ил)фенил)метанолом (105 мг, 0,422 ммоль), следуя методике примера 31, с получением названного соединения (109 мг, 48%). ¹H ЯМР (400 МГц, CDCl3): δ 8,72 (с, 1H), 7,51 (д, 2H), 7,40-7,3 (м, 3H), 7,23 (д, 1H), 7,15 (м, 1H), 4,94 (д, 1H), 4,85 (д, 1H), 4,81 (с, 2H), 3,19 (с, 3H), 3,03 (м, 4H), 2,73 (м, 4H), 2,78 (м, 1H), 1,11 (д, 3H). LCMS (метод C): RT=0,709 мин, m/z=540 [M+H]+.
Пример 90: 3-(2,6-дихлорфенил)-1-метил-7-((4-((1-метил-пиперидин-4-ил)окси)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-илокси)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (58 мг, 0,116 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (54 мг, 91%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,74 (уш.с, 1H), 8,45 (с, 1H), 7,64 (д, 4H), 7,48 (дд, 1H), 6,91 (д, 2H), 4,95 (с, 2H), 4,28 (септ, 1H), 3,09 (с, 3H), 2,55-2,65 (м, 2H), 2,09-2,20 (м, 5H), 1,85-1,96 (м, 2H), 1,54-1,68 (м, 2H). LCMS (метод C): RT=0,78 мин, m/z=513 [M+H]+.
Пример 91: 3-(2,6-дихлорфенил)-1-метил-7-((3-((1-метил-пиперидин-4-ил)амино)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 4-((3-нитрофенил)амино)пиперидин-1-карбоксилат:
К перемешиваемому раствору 3-нитроанилина (2 г, 14,48 ммоль) в безводном дихлорметане (40 мл) добавляли третбутил 4-оксопиперидин-1-карбоксилат (5,77 г, 29,0 ммоль), затем триацетоксиборгидрид натрия (9,21 г, 43,4 ммоль). Смесь перемешивали при комнатной температуре в течение 40 часов. Смесь интенсивно перемешивали с водой (40 мл), затем разделяли фазы и сушили (фазовый сепаратор). Водную фазу промывали дополнительным количеством дихлорметана (2×15 мл), который отделяли и сушили (фазовый сепаратор). Объединенные дихлорметановые фракции концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 10-15% этилацетат в циклогексане) с получением названного соединения (3,64 г, 78%). 1H ЯМР (400 МГц, CDCl3): δ 7,51 (дд, 1H), 7,38 (т, 1H), 7,27 (т, 1H), 6,84 (дд, 1H), 3,97-4,17 (м, 2H), 3,90 (д, 1H), 3,44-3,53 (м, 1H), 2,95 (т, 2H), 2,05(д, 2H), 1,47 (с, 9H), 1,31-1,41 (м, 2H). LCMS (метод C): RT=1,74 мин, m/z=266 [M-бутен]+.
Стадия 2: третбутил 4-((3-аминофенил)амино)пиперидин-1-карбоксилат:
Раствор третбутил 4-((3-нитрофенил)амино)пиперидин-1-карбоксилата (810 мг, 2,52 ммоль) в метаноле (50 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2)]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (735 мг, 100%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,70 (т, 1H), 5,84 (с, 1H), 5,81 (т, 2H), 5,05 (д, 1H), 4,68 (с, 2H), 3,86 (д, 2H), 3,54-3,60 (м, 1H), 3,37-3,47 (м, 1H), 3,22-3,31 (м, 1H), 1,85 (д, 2H), 1,41 (с, 9H), 1,21 (д, 2H). LCMS (метод C): RT=0,82 мин, m/z=292 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-метил-7-((3-(пиперидин-4-иламино)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,281 ммоль) подвергали взаимодействию с третбутил 4-((3-аминофенил)-амино)пиперидин-1-карбоксилатом (82 мг, 0,281 ммоль), следуя методике примера 31, затем очищали препаративной ВЭЖХ с получением названного соединения (41,9 мг, 54,4%). LCMS (метод C): RT=0,79 мин, m/z=498 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-1-метил-7-((3-((1-метил-пиперидин-4-ил)амино)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-((3-(пиперидин-4-иламино)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (41,9 мг, 0,084 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (33,2 мг, 77%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,55 (с, 1H) 7,58 (д, 2H), 7,44 (дд, 1H), 7,05-7,13 (м, 2H), 6,99 (д, 1H), 6,43 (дд, 1H), 5,00 (с, 2H), 3,21 (с, 3H), 2,92 (д, 2H), 2,33 (с, 3H), 2,24 (т, 2H), 2,07 (д, 2H), 1,54 (кв, 2H). LCMS (метод C): RT=0,80 мин, m/z=512 [M+H]+.
Пример 92: 3-(2-хлор-6-метилфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: N-(2-хлор-6-метилфенил)-4-(метиламино)-2-(метил-тио)пиримидин-5-карбоксамид:
К раствору 4-хлор-2-(метилтио)пиримидин-5-карбонилхлорида (24,81 г, 111 ммоль) в этилацетате (250 мл), добавляли Amberlyst A21 (7 г, 106 ммоль), и реакционную смесь нагревали до 40°C. Затем добавляли раствор 2-хлор-6-метиланилина (15 г, 106 ммоль) в этилацетате (250 мл), и реакционную смесь перемешивали при 40°C в течение ночи. Образовывался белый осадок. Его фильтровали, затем растворяли в осушенном THF (1 L) и нагревали при 60°C в течение 15 минут, затем опять фильтровали. Фильтрат концентрировали под вакуумом. Остаток суспендировали в DCM (200 мл), затем фильтровали и сушили. Суспендировали его в THF (1 л) и добавляли метиламин (2 M в THF, 50 мл) при 0°C. После перемешивания в течение 1 часа при комнатной температуре, раствор фильтровали. Фильтрат концентрировали под вакуумом. Полученное масло растирали в ацетонитриле до образования белого осадка, который собирали и сушили при температуре окружающей среды с получением названного соединения (3,5 г, 11%). LCMS (метод A): RT=1,43 мин, m/z=323 [M+H]+
Стадия 2: 3-(2-хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
Суспензию N-(2-хлор-6-метилфенил)-4-(метиламино)-2-(метил-тио)пиримидин-5-карбоксамида (3,5 г, 10,8 ммоль), дибромметана (2,3 мл, 32,5 ммоль) и карбоната цезия (21 г, 65,1 ммоль) в ацетонитриле (120 мл) нагревали при 80°C в герметизированной пробирке в течение 72 часов. После охлаждения, реакционную смесь разбавляли водой и экстрагировали три раза этилацетатом. Объединенные органические экстракты промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали под вакуумом. Остаток очищали на хроматографической системе Biotage с получением названного соединения (0,87 г, 24%). LCMS (метод A): RT=1,48 мин, m/z=335 [M+H]+
Стадия 3: 3-(2-хлор-6-метилфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,358 ммоль) подвергали взаимодействию с трет-5-бутил 4-(4-амино-2-метокси-фенил)пиперазин-1-карбоксилатом (95 мг, 0,341 ммоль), следуя методике примера 31, с получением названного соединения (57 мг, 36%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,65 (с, 1H), 8,43 (с, 1H), 7,62 (м, 2H), 7,43 (м, 1H), 7,23 (м, 2H), 6,90 (д, 2H), 4,92 (д, 1H), 4,87 (д, 1H), 3,09 (с, 3H), 3,03 (м, 4H), 2,90 (м, 4H), 2,25 (с, 3H). LCMS (метод C): RT=0,65 мин, m/z=464 [M+H]+.
Пример 93: 3-(2-хлор-6-фторфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2-Хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,295 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (86 мг, 0,281 ммоль), следуя методике примера 31, с получением названного соединения (42 мг, 36%). ¹H ЯМР (400 МГц, CDCl3): δ 8,72 (с, 1H), 7,57 (д, 2H), 7,32 (м, 2H), 7,13 (м, 3H), 4,93 (д, 1H), 4,85 (д, 1H), 3,18 (с, 3H), 3,09 (м, 3H), 2,94 (м, 1H), 2,71 (м,1H), 2,47 (м, 1H), 1,07 (д, 3H), 0,91 (д, 3H). LCMS (метод C): RT=0,74 мин, m/z=496 [M+H]+.
Пример 94: (R)-3-(2-хлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (R)-3-(2-хлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (200 мг, 0,623 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-метил-пиперазин-1-карбоксилатом (154 мг, 0,530 ммоль), следуя методике примера 31, с получением названного соединения (160 мг, 93%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,52 (с, 1H), 7,54-7,66 (м, 3H), 7,39-7,53 (м, 3H), 7,00 (д, 2H), 4,95-5,06 (м, 2H), 3,53 (т, 2H), 3,17 (с, 3H), 3,06-3,14 (м, 1H), 2,93-3,05 (м, 2H), 2,69 (тд, 1H), 2,36 (дд, 1H), 1,17 (д, 3H). LCMS (метод C): RT=0,63 мин, m/z=464 [M+H]+.
Пример 95: 3-(2,6-дихлорфенил)-1-метил-7-((4-(1-метил-пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
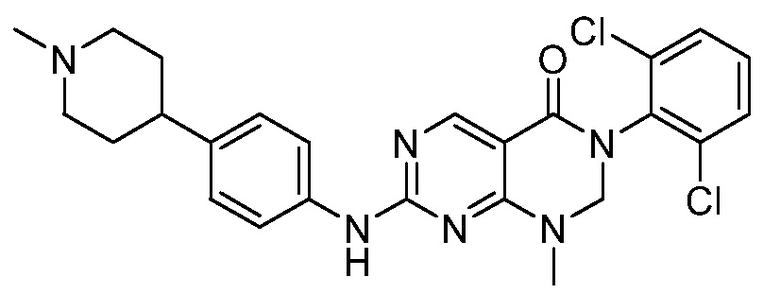
3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперидин-4-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (54 мг, 0,112 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде желтовато-белого твердого вещества (10 мг, 18%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,47 (с, 1H), 7,71 (д, 2H), 7,65 (д, 2H), 7,48 (дд, 1H), 7,18 (д, 2H), 4,97 (с, 2H), 3,11 (с, 3H), 3,02 (д, 2H), 2,43-2,54 (м, 1H), 2,11-2,41 (м, 5H), 1,63-1,83 (м, 4H). LCMS (метод C): RT=0,80 мин, m/z=497 [M+H]+.
Пример 96: 3-(2,6-дихлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
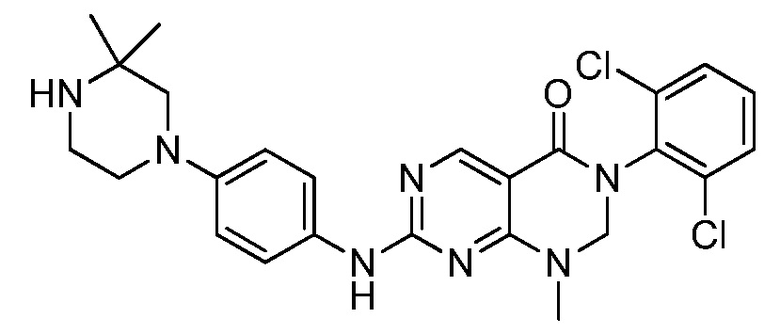
Стадия 1: третбутил 2,2-диметил-4-(4-нитрофенил)пиперазин-1-карбоксилат:
Суспензию 1-фтор-4-нитробензола (0,468 г, 3,32 ммоль), третбутил 2,2-диметилпиперазин-1-карбоксилата гидрохлорида (1,0 г, 3,48 ммоль) и карбоната калия (1,83 г, 13,26 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между водой (50 мл) и этилацетатом (50 мл). Этилацетат отделяли и промывали водой (3×20 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (0-100% этилацетат в циклогексане) с получением названного соединения (1,15 г, 100%). LCMS (метод C): RT=1,80 мин, m/z=336 [M+H]+.
Стадия 2: третбутил 4-(4-аминофенил)-2,2-диметилпиперазин-1-карбоксилат:
Раствор третбутил 2,2-диметил-4-(4-нитрофенил) пиперазин-1-карбоксилата (150 мг, 0,447 ммоль) в метаноле (10 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (137 мг, 100%). LCMS (метод C): RT=0,94 мин, m/z=306 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (180 мг,0,507 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)-2,2-диметилпиперазин-1-карбоксилатом (147 мг, 0,483 ммоль), следуя методике примера 31, с получением названного соединения (87 мг, 34%). ¹H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,71 (уш.с, 1H), 7,50 (д, 2H), 7,44 (д,2H), 7,29 (м, 1H), 6,90 (д,2H), 4,87 (с, 2H), 3,16 (с, 3H), 3,34 (с, 1H), 3,07 (м, 4H), 2,87 (м, 2H), 2,87 (м, 2H), 1,27 (с, 6H). LCMS (метод C): RT=0,759 мин, m/z=510 [M+H]+.
Пример 97: 3-(2-хлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
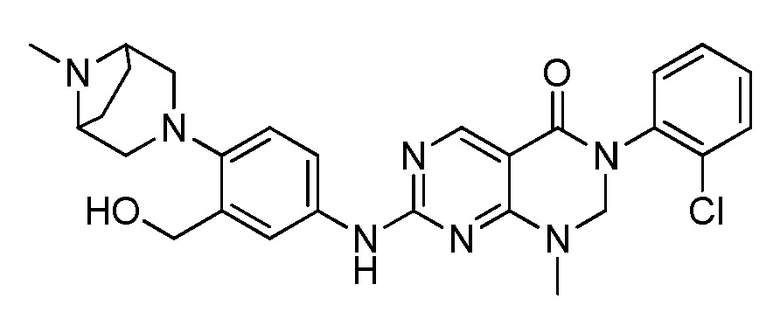
Стадия 1: 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)-3-(гидроксиметил)фенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (100 мг, 0,312 ммоль) подвергали взаимодействию с третбутил 3-(4-амино-2-(гидроксиметил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилатом (88 мг, 0,265 ммоль), следуя методике примера 31, с получением названного соединения (76,4 мг, 83%). LCMS (метод C): RT=0,63 мин, m/z=506 [M+H]+.
Стадия 2: 3-(2-хлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)-3-(гидрокси-метил)фенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (76,4 мг, 0,151 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (71,1 мг, 91%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,76 (с, 1H), 8,44 (с, 1H), 7,89-8,09 (м, 1H), 7,62 (дд, 1H), 7,49-7,57 (м, 2H), 7,38-7,48 (м, 2H), 7,04 (д, 1H), 5,04 (т, 1H), 4,83-5,13 (м, 2H), 4,60 (д, 2H), 3,11 (с, 3H), 3,04-3,10 (м, 2H), 2,89 (д, 2H), 2,62 (дд, 2H), 2,20 (с, 3H), 1,88-2,00 (м, 2H), 1,77-1,88 (м, 2H). LCMS (метод C): RT=0,66 мин, m/z=520 [M+H]+.
Пример 98: 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (2S,5R)-третбутил 4-(4-((6-(2-хлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2,5-диметилпиперазин-1-карбоксилат:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (76 мг, 0,249 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (74 мг, 51%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,29-7,54 (м, 7H), 6,84 (д, 2H), 4,88 (уш.д, 2H), 4,42 (уш.с, 1H), 3,93 (уш.с, 1H), 3,80 (д, 1H), 3,43 (дд, 1H), 3,28 (дд, 1H), 3,16 (с, 3H), 3,05 (дд, 1H), 1,49 (с, 9H), 1,27 (д, 3H), 1,02 (д, 3H). LCMS (метод C): RT=1,63 мин, m/z=578 [M+H]+.
Стадия 2: 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(2S,5R)-третбутил 4-(4-((6-(2-хлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-2,5-диметилпиперазин-1-карбоксилат (74 мг, 0,128 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (61 мг, 100%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,77 (уш.с, 1H), 8,45 (с, 1H), 7,68 (д, 2H), 7,62 (дд, 1H), 7,52 (дд, 1H), 7,39-7,48 (м, 2H), 7,03 (д, 2H), 5,07 (д, 1H), 4,90 (д, 1H), 3,10 (с, 3H), 2,79-2,98 (м, 4H), 2,45 (дд, 1H), 2,32 (дд, 1H), 2,19 (уш.с, 1H), 0,94 (д, 3H), 0,80 (д, 3H). LCMS (метод C): RT=0,72 мин, m/z=478 [M+H]+.
Пример 99: (R)-3-(2-хлорфенил)-7-((3-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(4-((6-(2-хлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-фторфенил)-2-метилпиперазин-1-карбоксилат:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-фторфенил)-2-метилпиперазин-1-карбоксилатом (77 мг, 0,249 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (64 мг, 44%). 1H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,68 (д, 1H), 7,65 (уш.с, 1H), 7,52 (дд, 1H), 7,30-7,43 (м, 3H), 7,10 (дд, 1H), 6,88 (т, 1H), 4,91 (уш.д, 2H), 4,31 (уш.с, 1H), 3,94 (д, 1H), 3,15-3,33 (м, 6H), 2,83 (дд, 1H), 2,73 (тд, 1H), 1,49 (с, 9H), 1,36 (д, 3H). LCMS (метод C): RT=1,83 мин, m/z=582 [M+H]+.
Стадия 2: (R)-3-(2-хлорфенил)-7-((3-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(R)-третбутил 4-(4-((6-(2-хлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-фтор-фенил)-2-метилпиперазин-1-карбоксилат (64 мг, 0,110 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (41 мг, 77%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,88 (уш.с, 1H), 8,47 (с, 1H), 7,69 (дд, 1H), 7,62 (дд, 1H), 7,52 (дд, 1H), 7,39-7,49 (м, 3H), 6,97 (т, 1H), 5,09 (д, 1H), 4,91 (д, 1H), 3,06-3,16 (м, 5H), 2,78-2,95 (м, 3H), 2,57 (тд, 1H), 2,24 (т, 1H), 0,99 (д, 3H). LCMS (метод C): RT=0,77 мин, m/z=482 [M+H]+.
Пример 100: (R)-3-(2-хлор-6-метилфенил)-1-метил-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,358 ммоль) подвергали взаимодействию с (R)-4-(3-метилпиперазин-1-ил)анилином (99 мг, 0,341 ммоль), следуя методике примера 31, с получением названного соединения (39 мг, 24%). ¹H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,51 (д, 1H), 7,35 (м, 1H), 7,22 (м, 2H), 6,94 (д, 2H), 5,01 (д, 1H), 4,65 (д, 1H), 3,50 (м, 2H), 3,14 (с, 3H), 3,12-2,95 (м, 3H), 2,70 (м, 1H), 2,37 (м, 1H), 2,32 (с, 3H), 1,13 (д, 3H). LCMS (метод C): RT=0,67 мин, m/z=478 [M+H]+.
Пример 101: (R)-3-(2-хлор-6-метилфенил)-7-((3-(метокси-метил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
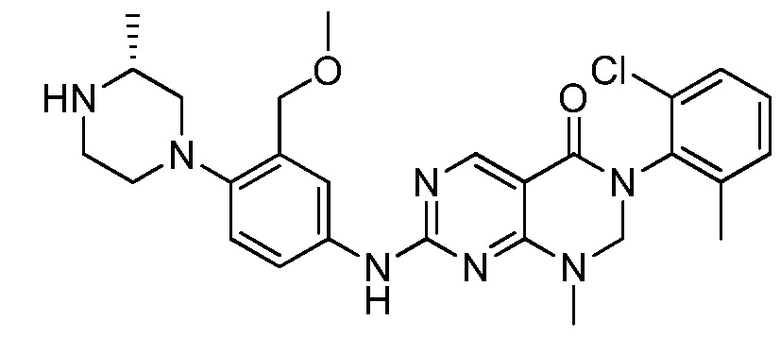
3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (120 мг, 0,358 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(метокси-метил)фенил)-2-метилпиперазин-1-карбоксилатом (114 мг, 0,341 ммоль), следуя методике примера 31, с получением названного соединения (30 мг, 17%). ¹H ЯМР (400 МГц, CDCl3): δ 8,71 (с, 1H), 7,78 (с, 1H), 7,53 (м, 1H), 7,35 (м, 1H), 7,23 (м, 2H), 7,08 (д, 1H), 5,03 (д, 1H), 4,66 (д, 1H), 4,56 (с, 2H), 3,44 (с, 3H), 3,19 (с, 3H), 3,02 (м, 3H), 2,92 (м, 2H), 2,71 (м, 1H), 2,45 (м, 1H), 2,23 (с, 3H). LCMS (метод C): RT=0,77 мин, m/z=522 [M+H]+.
Пример 102: 3-(2-хлор-6-метилфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (2S,5R)-третбутил 4-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)фенил)-2,5-диметилпиперазин-1-карбоксилат:
3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,180 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (55 мг, 0,180 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (85 мг, 80%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,54 (уш.с, 1H), 7,50 (д, 2H), 7,30-7,37 (м, 1H), 7,18-7,23 (м, 2H), 6,84 (д, 2H), 5,01 (д, 1H), 4,64 (д, 1H), 4,42 (уш.с, 1H), 3,93 (уш.с, 1H), 3,80 (д, 1H), 3,43 (дд, 1H), 3,27 (дд, 1H), 3,14 (с, 3H), 3,05 (дд, 1H), 2,32 (с, 3H), 1,49 (с, 9H), 1,27 (д, 3H), 1,02 (д, 3H). LCMS (метод C): RT=1,73 мин, m/z=592 [M+H]+.
Стадия 2: 3-(2-хлор-6-метилфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
(2S,5R)-третбутил 4-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)-2,5-диметилпиперазин-1-карбоксилат (85 мг, 0,144 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (60 мг, 85%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,76 (уш.с, 1H), 8,45 (с, 1H), 7,68 (д, 2H), 7,41-7,48 (м, 1H), 7,30-7,37 (м, 2H), 7,03 (д, 2H), 4,92 (дд, 2H), 3,10 (с, 3H), 2,80-2,98 (м, 4H), 2,45 (дд, 1H), 2,33 (дд, 1H), 2,26 (с, 3H), 0,95 (д, 3H), 0,80 (д, 3H). LCMS (метод C): RT=0,78 мин, m/z=492 [M+H]+.
Пример 103: 3-(2-хлорфенил)-7-((4-((3S,5R)-3,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
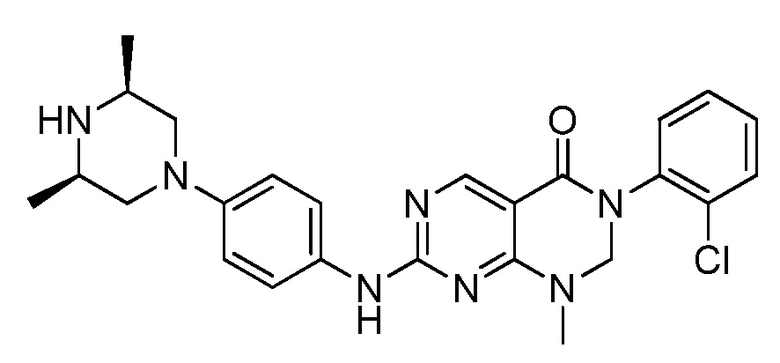
Стадия 1: 3-(2-хлорфенил)-7-((4-((3S,5R)-3,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Раствор mCPBA 50% чистоты (103 мг, 0,299 ммоль) в дихлорметане (0,400 мл) пропускали через фазовый сепаратор и промывали дополнительным количеством дихлорметана (0,200 мл). Этот раствор добавляли к перемешиваемой суспензии 3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-она (80 мг, 0,249 ммоль) в безводном толуоле (2 мл), и смесь перемешивали в течение 30 минут. Добавляли основание Хунига (0,131 мл, 0,748 ммоль), затем 4-((3S,5R)-3,5-диметилпиперазин-1-ил)анилин (43,5 мг, 0,212 ммоль), и смесь нагревали до 80°C в течение 16 часов. После охлаждения, непосредственно реакционную смесь очищали флэш-хроматографией (0-100%, этилацетат в циклогексане, KP-NH). Остаток растворяли в метаноле (1 мл) и воде (10 мл) и лиофилизировали с получением названного соединения (29,6 мг, 24,83%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,64 (уш.с, 1H), 8,43 (с, 1H), 7,55-7,67 (м, 3H), 7,51 (дд, 1H), 7,44 (дтд, 2H), 6,88 (д, 2H), 4,80-5,15 (м, 2H), 3,45 (д, 2H), 3,08 (с, 3H), 2,78-2,90 (м, 2H), 2,12 (уш.с, 1H), 2,06 (т, 2H), 1,01 (д, 6H). LCMS (метод C): RT=0,67 мин, m/z=478 [M+H]+.
Пример 104: 7-((4-((1S,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (60 мг, 0,187 ммоль) подвергали взаимодействию с (1R,4S)-третбутил 5-(4-аминофенил)-2,5-диаза-бицикло[2,2,1]гептан-2-карбоксилатом (46,0 мг, 0,159 ммоль), следуя методике примера 31, с получением названного соединения (42,0 мг, 88%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,49 (с, 1H), 7,57-7,62 (м, 1H), 7,46-7,57 (м, 3H), 7,40-7,46 (м, 2H), 6,63 (д, 2H), 4,92-5,03 (м, 2H), 4,39 (с, 1H), 3,79 (с, 1H), 3,63 (дд, 1H), 3,15 (с, 3H), 3,05 (т, 2H), 2,98 (дд, 1H), 1,99 (д, 1H), 1,81 (д, 1H). LCMS (метод C): RT=0,60 мин, m/z=462 [M+H]+.
Пример 105: 7-((4-(4,7-диазаспиро[2,5]октан-7-ил)фенил)-амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 7-(4-нитрофенил)-4,7-диазаспиро-[2,5]октан-4-карбоксилат:
Суспензию 1-фтор-4-нитробензола (0,166 г, 1,177 ммоль), третбутил 4,7-диазаспиро[2,5]октан-7-карбоксилата (0,25 г, 1,178 ммоль) и карбоната калия (0,642 г, 4,64 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, затем распределяли между водой (50 мл) и этилацетатом (50 мл). Этилацетат отделяли и промывали водой (3×20 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (0-100% этилацетат в циклогексане) с получением названного соединения (0,334 г, 85%). LCMS (метод C): RT=1,78 мин, m/z=334 [M+H]+.
Стадия 2: третбутил 7-(4-аминофенил)-4,7-диазаспиро[2,5]-октан-4-карбоксилат:
Раствор третбутил 7-(4-нитрофенил)-4,7-диазаспиро[2,5]-октан-4-карбоксилата (149 мг, 0,447 ммоль) в метаноле (10 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (136 мг, 100%). LCMS (метод C): RT=0,82 мин, m/z=304 [M+H]+.
Стадия 3: 7-((4-(4,7-диазаспиро[2,5]октан-7-ил)фенил)-амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с третбутил 7-(4-аминофенил)-4,7-диазаспиро-[2,5]октан-4-карбоксилатом (64,3 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (25,7 мг, 93%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,52 (с, 1H), 7,53-7,66 (м, 3H), 7,38-7,52 (м, 3H), 6,98 (д, 2H), 4,93-5,06 (м, 2H), 3,11-3,21 (м, 5H), 3,05-3,11 (м, 2H), 2,97-3,02 (м, 2H), 0,63-0,75 (м, 4H). LCMS (метод C): RT=0,67 мин, m/z=476 [M+H]+.
Пример 106: 3-(2-хлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 3-(2-хлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)-2,2-диметил-пиперазин-1-карбоксилатом (64,7 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (71,2 мг, 90%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,52 (с, 1H), 7,52-7,65 (м, 3H), 7,39-7,52 (м, 3H), 6,95 (д, 2H), 4,93-5,04 (м, 2H), 3,16 (с, 3H), 3,00-3,09 (м, 4H), 2,89 (с, 2H), 1,26 (с, 6H). LCMS (метод C): RT=0,70 мин, m/z=478 [M+H]+.
Пример 107: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-метоксифенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (67,7 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (68,5 мг, 86%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,49 (с, 1H), 7,54-7,61 (м, 1H), 7,35-7,50 (м, 4H), 7,11 (д, 1H), 6,70 (д, 1H), 4,90-5,03 (м, 2H), 4,33 (с, 1H), 3,82 (с, 3H), 3,63-3,70 (м, 2H), 3,12-3,22 (м, 4H), 3,05 (с, 1H), 2,91 (дд, 1H), 1,91 (д, 1H), 1,71 (д, 1H). LCMS (метод C): RT=0,69 мин, m/z=492 [M+H]+.
Пример 108: 3-(2-хлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 3-(2-хлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-(метоксиметил)фенил)-пиперазин-1-карбоксилатом (68,1 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (46,6 мг, 96%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,54 (с, 1H), 7,92 (уш.с, 1H), 7,55-7,63 (м, 2H), 7,40-7,52 (м, 3H), 7,14 (д, 1H), 4,93-5,07 (м, 2H), 4,59 (с, 2H), 3,45 (с, 3H), 3,20 (с, 3H), 2,97-3,03 (м, 4H), 2,87-2,93 (м, 4H). LCMS (метод C): RT=0,70 мин, m/z=494 [M+H]+.
Пример 109: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (64,3 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (58,8 мг, 83%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,57-7,63 (м, 1H), 7,36-7,54 (м, 5H), 6,90 (д, 1H), 4,92-5,05 (м, 2H), 4,10 (с, 1H), 3,74 (с, 1H), 3,45 (дд, 1H), 3,23 (д, 1H), 3,13-3,20 (м, 4H), 2,98 (дд, 1H), 2,96 (с, 3H), 1,98 (д, 1H), 1,76 (д, 1H). LCMS (метод C): RT=0,68 мин, m/z=476 [M+H]+.
Пример 110: 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
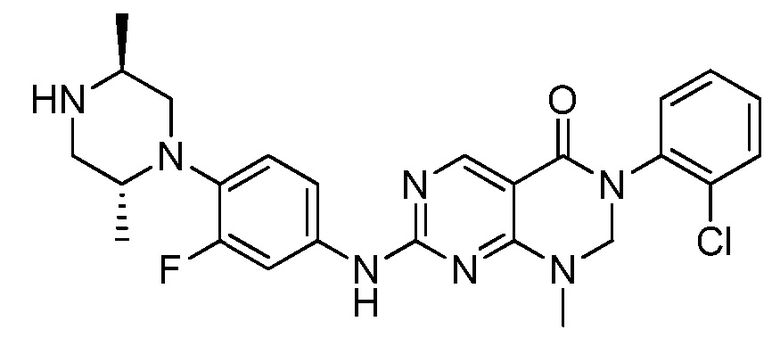
Стадия 1: 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (2S,5R)-третбутил 4-(4-амино-2-фторфенил)-2,5-диметилпиперазин-1-карбоксилатом (68,6 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (49,7 мг, 92%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,57 (с, 1H), 7,77 (дд, 1H), 7,57-7,64 (м, 1H), 7,41-7,53 (м, 3H), 7,38 (дд, 1H), 7,21 (т, 1H), 4,95-5,10 (м, 2H), 3,21 (с, 3H), 3,06-3,14 (м, 1H), 2,96-3,06 (м, 3H), 2,63 (дд, 1H), 2,58 (т, 1H), 1,08 (д, 3H), 0,89 (д, 3H). LCMS (метод C): RT=0,81мин, m/z=496 [M+H]+.
Пример 111: 3-(2-хлорфенил)-7-((4-((2S,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
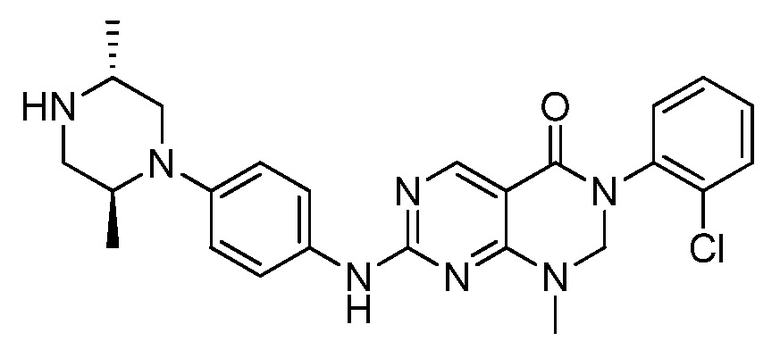
Стадия 1: 3-(2-хлорфенил)-7-((4-((2S,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2-Хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (80 мг, 0,249 ммоль) подвергали взаимодействию с (2R,5S)-третбутил 4-(4-аминофенил)-2,5-диметилпиперазин-1-карбоксилатом (64,7 мг, 0,212 ммоль), следуя методике примера 31, с получением названного соединения (66,1 мг, 100%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,55 (с, 1H), 7,70 (д, 2H), 7,57-7,64 (м, 1H), 7,40-7,53 (м, 3H), 7,18 (д, 2H), 4,95-5,07 (м, 2H), 3,19 (с, 3H), 2,93-3,09 (м, 4H), 2,64 (дд, 1H), 2,51 (дд, 1H), 1,10 (д, 3H), 0,90 (д, 3H). LCMS (метод C): RT=0,71 мин, m/z=478 [M+H]+.
Пример 112: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлор-6-метилфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
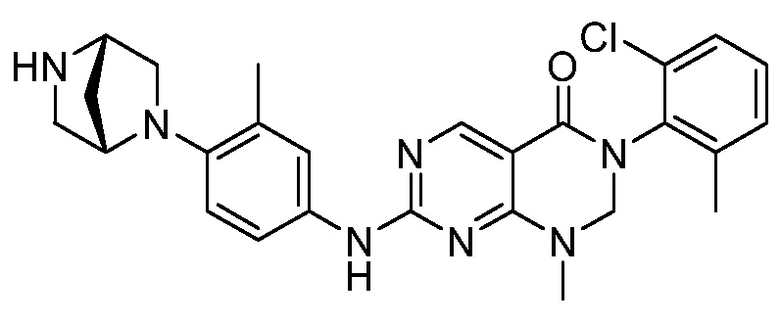
Стадия 1: (1S,4S)-третбутил 5-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,180 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (55 мг, 0,180 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (65 мг, 61%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,30-7,55 (м, 4H), 7,18-7,24 (м, 2H), 6,80 (т, 1H), 5,01 (д, 1H), 4,65 (д, 1H), 4,50 (д, 1H), 4,11 (уш.с, 1H), 3,62 (дд, 1H), 3,45 (т, 1H), 3,20-3,40 (м, 2H), 3,15 (с, 3H), 2,32 (с, 3H), 2,28 (с, 3H), 1,98 (д, 1H), 1,86 (т, 1H), 1,42-1,50 (м, 9H). LCMS (метод C): RT=1,62 мин, m/z=590 [M+H]+.
Стадия 2: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлор-6-метилфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(1S,4S)-третбутил 5-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (65 мг, 0,110 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (36 мг, 67%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,57 (уш.с, 1H), 8,42 (с, 1H), 7,39-7,58 (м, 3H), 7,29-7,37 (м, 2H), 6,76 (д, 1H), 4,90 (дд, 2H), 3,99 (с, 1H), 3,56 (с, 1H), 3,41 (дд, 1H), 3,08 (с, 3H), 3,05 (д, 1H), 2,96 (д, 1H), 2,85 (дд, 1H), 2,25 (д, 3H), 2,18 (с, 3H), 1,75 (д, 1H), 1,59 (д, 1H). LCMS (метод C): RT=0,75 мин, m/z=490 [M+H]+.
Пример 113: 3-(2-хлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
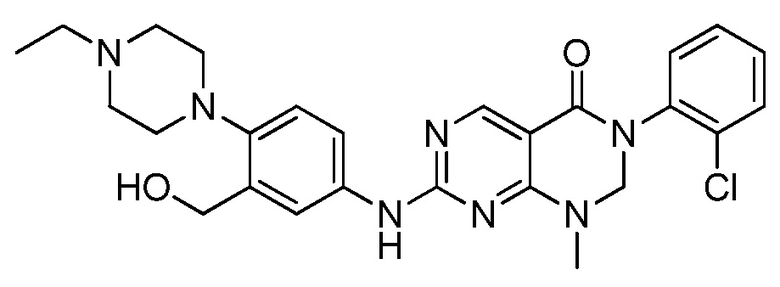
Стадия 1: 3-(2-хлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Раствор mCPBA 50% чистоты (103 мг, 0,299 ммоль) в дихлорметане (0,400 мл) пропускали через фазовый сепаратор и промывали дополнительным количеством дихлорметана (0,200 мл). Этот раствор добавляли к перемешиваемой суспензии 3-(2-хлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-она (80 мг, 0,249 ммоль) в безводном толуоле (2 мл), и смесь перемешивали в течение 30 минут. Добавляли основание Хунига (0,131 мл, 0,748 ммоль), затем (5-амино-2-(4-этилпиперазин-1-ил)фенил)метанол (49,9 мг, 0,212 ммоль), и смесь нагревали до 80°C в течение 16 часов. После охлаждения, непосредственно реакционную смесь очищали флэш-хроматографией (0-100%, этилацетат в циклогексане, KP-NH). Остаток суспендировали в дихлорметане (4 мл). Твердое вещество собирали фильтрацией и промывали свежим дихлорметаном (3 мл). Остаток суспендировали в метаноле (1 мл) и воде (10 мл) и лиофилизировали с получением названного соединения (58,3 мг, 46,0%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,77 (с, 1H), 8,45 (с, 1H), 7,99 (уш.с, 1H), 7,62 (дд, 1H), 7,56 (дд, 1H), 7,52 (дд, 1H), 7,38-7,48 (м, 2H), 7,01 (д, 1H), 4,85-5,12 (м, 3H), 4,54 (д, 2H), 3,12 (с, 3H), 2,76-2,88 (м, 4H), 2,40 (кв, 2H), 1,03 (т, 3H). 1H ЯМР (300 МГц, MeOH-d4): δ 8,54 (с, 1H), 7,90 (с, 1H), 7,54-7,68 (м, 2H), 7,39-7,53 (м, 3H), 7,18 (д, 1H), 4,97-5,07 (м, 2H), 4,76 (с, 2H), 3,21 (с, 3H), 2,95-3,06 (м, 4H), 4,64-2,82 (м, 4H), 2,59 (кв, 2H), 1,19 (т, 3H). LCMS (метод C): RT=0,61 мин, m/z=508 [M+H]+.
Пример 114: 3-(2-хлор-6-метилфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 3-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат:
3-(2-Хлор-6-метилфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (60 мг, 0,180 ммоль) подвергали взаимодействию с третбутил 3-(4-амино-2-(гидрокси-метил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилатом (60 мг, 0,180 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (42 мг, 38%). 1H ЯМР (400 МГц, CDCl3): δ 8,73 (с, 1H), 7,69 (уш.с, 1H), 7,61 (д, 1H), 7,56 (дд, 1H), 7,31-7,38 (м, 1H), 7,16-7,24 (м, 3H), 5,03 (д, 1H), 4,81 (д, 2H), 4,67 (д, 1H), 4,19-4,42 (м, 3H), 3,17 (с, 3H), 2,94-3,14 (м, 2H), 2,82 (д, 2H), 2,32 (с, 3H), 2,00 (уш.с, 4H), 1,50 (с, 9H). LCMS (метод C): RT=1,54 мин, m/z=620 [M+H]+.
Стадия 2: 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)-3-(гидроксиметил)фенил)амино)-3-(2-хлор-6-метилфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 3-(4-((6-(2-хлор-6-метилфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)-3,8-диазабицикло[3,2,1]октан-8-карбоксилат (42 мг, 0,068 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (32 мг, 91%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,76 (уш.с, 1H), 8,44 (с, 1H), 8,00 (уш.с, 1H), 7,54 (дд, 1H), 7,41-7,48 (м, 1H), 7,30-7,37 (м, 2H), 7,03 (д, 1H), 5,05 (т, 1H), 4,91 (дд, 2H), 4,61 (д, 2H), 3,41 (уш.с, 2H), 3,11 (с, 3H), 2,82 (д, 2H), 2,67 (дд, 2H), 2,26 (с, 3H), 1,91 (дд, 2H), 1,67 (дд, 2H). LCMS (метод C): RT=0,71 мин, m/z=520 [M+H]+.
Стадия 3: 3-(2-хлор-6-метилфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
7-((4-(3,8-Диазабицикло[3,2,1]октан-3-ил)-3-(гидрокси-метил)фенил)амино)-3-(2-хлор-6-метилфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (32 мг, 0,062 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (15 мг, 46%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,69 (уш.с, 1H), 8,37 (с, 1H), 7,93 (уш.с, 1H), 7,46 (дд, 1H), 7,35-7,40 (м, 1H), 7,24-7,30 (м, 2H), 6,98 (д, 1H), 4,98 (т, 1H), 4,87 (д, 1H), 4,82 (д, 1H), 4,54 (д, 2H), 3,04 (с, 3H), 3,01 (уш.с, 2H), 2,82 (д, 2H), 2,55 (дд, 2H), 2,19 (с, 3H), 2,13 (с, 3H), 1,71-1,92 (м, 4H). LCMS (метод C): RT=0,71 мин, m/z=534 [M+H]+.
Пример 115: 7-((4-(4-циклопропилпиперазин-1-ил)фенил)-амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 1-циклопропил-4-(4-нитрофенил)пиперазин:
Суспензию 1-фтор-4-нитробензол (1,096 г, 7,77 ммоль), 1-циклопропилпиперазина (1,00 г, 7,92 ммоль) и карбоната калия (1,61 г, 11,65 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры и выливали в смесь льда с водой (50 мл). Осажденное твердое вещество выделяли фильтрацией, промывали водой, и воду отсасывали, с получением названного соединения в виде желтого твердого вещества (1,11 г, 58%). 1H ЯМР (500 МГц, CDCl3): δ 8,12 (д, 2H), 7,01 (д, 2H), 3,47 (т, 4H), 2,80 (т, 4H), 1,70-1,80 (м, 1H), 0,45-0,60 (м, 4H). LCMS (метод C): RT=0,51 мин, m/z=248 [M+H]+.
Стадия 2: 4-(4-циклопропилпиперазин-1-ил)анилин:
Суспензию 1-циклопропил-4-(4-нитрофенил)пиперазина (1,11 г, 4,49 ммоль), порошка железа (1,504 г, 26,9 ммоль) и хлорида аммония (2,161 г, 40,4 ммоль) в смеси метанола (18,54 мл) и воды (3,90 мл) кипятили с обратным холодильником в атмосфере азота в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит® и концентрировали досуха при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали насыщенным раствором бикарбоната натрия (3×70 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде коричневого масла, которое превращалось в твердое вещество при стоянии (910 мг, 93%). 1H ЯМР (500 МГц, CDCl3): δ 6,81 (д, 2H), 6,64 (д, 2H), 3,41 (уш.с, 2H), 3,02 (т, 4H), 2,77 (т, 4H), 1,64-1,69 (м, 1H), 0,40-0,51 (м, 4H). LCMS (метод C): RT=0,13 мин, m/z=218 [M+H]+.
Стадия 3: 7-((4-(4-циклопропилпиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с 4-(4-циклопропилпиперазин-1-ил)-анилином (62 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (60 мг, 41%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,70 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,60 (уш.д, 2H), 7,48 (т, 1H), 6,89 (д, 2H), 4,95 (с, 2H), 3,08 (с, 3H), 3,04 (т, 4H), 2,67 (т, 4H), 1,65 (септ, 1H), 0,39-0,48 (м, 2H), 0,29-0,39 (м, 2H). LCMS (метод C): RT=0,79 мин, m/z=524 [M+H]+.
Пример 116: 3-(2,6-дихлорфенил)-7-((4-(4-(2-гидроксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 2-(4-(4-нитрофенил)пиперазин-1-ил)этанол:
К перемешиваемой суспензии 1-фтор-4-нитробензола (2 г, 14,17 ммоль) и карбоната калия (3,92 г, 28,3 ммоль) в безводном диметилсульфоксиде (10 мл) добавляли 2-(пиперазин-1-ил)этанол (2,089 мл, 17,01 ммоль), и смесь нагревали при 80°C в течение 16 часов. После охлаждения, смесь распределяли между водой (100 мл) и этилацетатом (30 мл). Водный слой отделяли и экстрагировали этилацетатом (2×30 мл). Объединенные органические фракции концентрировали под вакуумом. Остаток растирали в воде (100 мл). Твердое вещество собирали фильтрацией под вакуумом и сушили в течение 16 часов под вакуумом и продували азотом с получением названного соединения (3,45 г, 97%). 1H ЯМР (400 МГц, CDCl3): δ 8,11 (д, 2H), 6,82 (д, 2H), 3,68 (т, 2H), 3,44 (т, 4H), 2,67 (т, 4H), 2,62 (т, 2H), 2,55 (уш.с, 1H). LCMS (метод C): RT=0,45 мин, m/z=252 [M+H]+.
Стадия 2: 2-(4-(4-аминофенил)пиперазин-1-ил)этанол:
Перемешиваемый раствор 2-(4-(4-нитрофенил)пиперазин-1-ил)-этанола (1,2 г, 4,78 ммоль) в этаноле (20 мл) нагревали до 50°C. Добавляли 10% палладий на угле (0,254 г, 0,239 ммоль), затем порциями добавляли формиат аммония (1,506 г, 23,88 ммоль), и суспензию перемешивали в течение 1 часа. Суспензию фильтровали через целит®, промывая свежим этанолом (20 мл). Этанол удаляли под вакуумом с получением названного соединения (1,10 г, 104%). 1H ЯМР (400 МГц, CDCl3): δ 6,81 (д, 2H), 6,66 (д, 2H), 3,69 (т, 2H), 3,09 (т, 4H), 3,02 (уш.с, 3H), 2,74 (т, 4H), 2,66 (т, 2H). LCMS (метод C): RT=0,13 мин, m/z=222 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-(4-(2-гидроксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с 2-(4-(4-аминофенил)пиперазин-1-ил)-этанолом (63 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (67 мг, 45%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,70 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,61 (уш.д, 2H), 7,48 (дд, 1H), 6,90 (д, 2H), 4,95 (с, 2H), 4,42 (т, 1H), 3,53 (кв, 2H), 3,02-3,14 (м, 7H), 2,55 (т, 4H), 2,43 (т, 2H). LCMS (метод C): RT=0,71 мин, m/z=528 [M+H]+.
Пример 117: 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,422 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)пиперазин-1-карбоксилатом (100 мг, 0,359 ммоль), следуя методике примера 31, с получением названного соединения (143,5 мг, 86%). LCMS (метод C): RT=0,71 мин, m/z=484 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (44 мг, 0,091 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения (41,2 мг, 91%). 1H ЯМР (400 МГц, MeOH-d4): δ 9,70 (уш.с, 1H), 8,44 (с, 1H), 7,53-7,72 (м, 3H), 7,47 (1H), 6,90 (д, 2H), 4,95 (с, 1H), 3,01-3,14 (м, 7H), 2,41-2,48 (м, 4H), 2,23 (м, 3H). LCMS (метод C): RT=0,72 мин, m/z=498 [M+H]+.
Пример 118: 7-((4-((1R,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
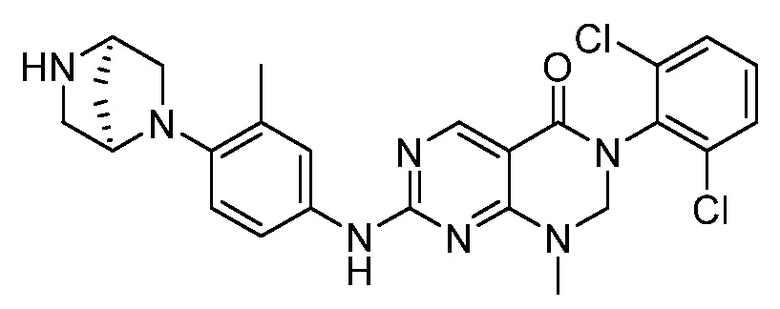
Стадия 1: (1R,4R)-третбутил 5-(2-метил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию 2-фтор-5-нитротолуола (391 мг, 2,52 ммоль), (1R,4R)-третбутил 2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (500 мг, 2,52 ммоль) и карбоната калия (523 мг, 3,78 ммоль) в безводном DMF (2 мл) нагревали до 100°C в атмосфере азота в течение 5 дней. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 30:70) с получением названного соединения в виде желтого твердого вещества (503 мг, 60%). 1H ЯМР (300 МГц, CDCl3): δ 7,97 (уш.с, 2H), 6,60 (д, 1H), 4,56 (д, 1H), 4,44 (с, 1H), 3,83 (д, 1H), 3,62 (дд, 1H), 3,47 (т, 1H), 3,32 (дд, 1H), 2,36 (с, 3H), 1,86-2,06 (м, 2H), 1,34-1,57 (м, 9H). LCMS (метод C): RT=1,68 мин, m/z=334 [M+H]+.
Стадия 2: (1R,4R)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Раствор (1R,4R)-третбутил 5-(2-метил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (503 мг, 1,509 ммоль) в метаноле (75 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, комнатная температура, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 0:100) с получением названного соединения в виде коричневого твердого вещества (319 мг, 70%). 1H ЯМР (300 МГц, CDCl3): δ 6,71 (дд, 1H), 6,55 (с, 1H), 6,48 (д, 1H), 4,46 (д, 1H), 3,91 (с, 1H), 3,13-3,67 (м, 6H), 2,20 (с, 3H), 1,96 (д, 1H), 1,82 (дд, 1H), 1,46 (д, 9H). LCMS (метод C): RT=0,78 мин, m/z=304 [M+H]+.
Стадия 3: (1R,4R)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (1R,4R)-третбутил 5-(4-амино-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (86 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (95 мг, 55%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,44 (д, 2H), 7,32-7,42 (м, 2H), 7,28 (дд, 1H), 7,11 (уш.с, 1H), 6,80 (т, 1H), 4,87 (с, 2H), 4,50 (д, 1H), 4,11 (с, 1H), 3,62 (дд, 1H), 3,46 (т, 1H), 3,20-3,41 (м, 2H), 3,17 (с, 3H), 2,29 (с, 3H), 1,98 (д, 1H), 1,87 (т, 1H), 1,40-1,51 (м, 9H). LCMS (метод C): RT=1,68 мин, m/z=610 [M+H]+.
Стадия 4: 7-((4-((1R,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
(1R,4R)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (95 мг, 0,156 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (16 мг, 20%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,62 (уш.с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,37-7,55 (м, 3H), 6,75 (д, 1H), 4,95 (с, 2H), 3,98 (с, 1H), 3,52 (с, 1H), 3,41 (дд, 1H), 3,09 (с, 3H), 3,04 (д, 1H), 2,94 (д, 1H), 2,83 (дд, 1H), 2,18 (с, 3H), 1,74 (д, 1H), 1,57 (д, 1H). LCMS (метод C): RT=0,80 мин, m/z=510 [M+H]+.
Пример 119: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
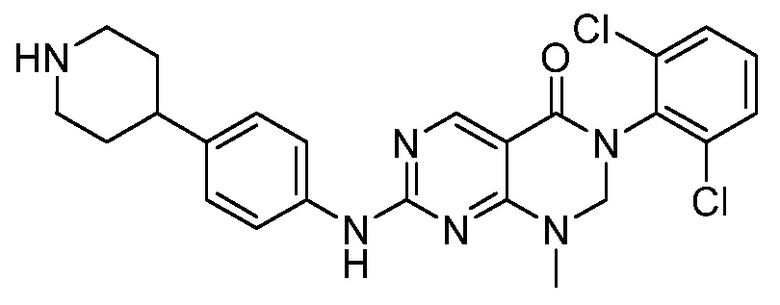
Стадия 1: третбутил 4-(4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
Суспензию 1-бром-4-нитробензола (0,205 г, 1,012 ммоль), третбутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (0,36 г, 1,164 ммоль) и карбоната натрия (0,322 г, 3,04 ммоль) в смеси 1,4-диоксана (3,5 мл) и воды (0,700 мл) дегазировали с помощью N2 в течение 5 минут. Затем к смеси добавляли аддукт PdCl2(dppf)-дихлорметан (0,083 г, 0,101 ммоль), и реакционную смесь дегазировали с помощью N2 в течение 5 минут, затем нагревали в микроволновом реакторе при 100°C в течение 10 минут. Нагревание повторяли еще пять раз. После охлаждения, смесь распределяли между насыщенным раствором гидрокарбоната натрия (25 мл) и дихлорметаном (25 мл). Слои разделяли, и водную фазу экстрагировали дихлорметаном (3×25 мл). Объединенные органические фазы сушили, фильтровали, концентрировали досуха при пониженном давлении и очищали хроматографией на силикагеле (градиент 0-15% этилацетат в циклогексане) с получением названного соединения (303 мг, 98%). LCMS (метод C): RT=1,53 мин, m/z=304 [M]-.
Стадия 2: третбутил 4-(4-аминофенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
К перемешиваемому раствору третбутил 4-(4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (303,1 мг, 0,996 ммоль) в этаноле (3,5 мл) добавляли хлорид олова(II) (944 мг, 4,98 ммоль), и смесь нагревали при 55°C в течение 3 часов. Смесь концентрировали досуха при пониженном давлении, и остаток распределяли между 4 M раствором гидроксида натрия (50 мл) и этилацетатом (15 мл). Водную фазу отделяли и экстрагировали этилацетатом (3×15 мл). Объединенные этилацетатные фракции сушили (над безводным сульфатом натрия), фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения (310 мг, 100%). 1H ЯМР (400 МГц, DMSO- d6): δ 7,11 (д, 2H), 6,52 (д, 2H), 5,88 (уш.с, 1H), 5,08 (уш.с, 2H), 3,91-3,96 (м, 2H), 3,49 (т, 2H), 2,34-2,39 (м, 2H), 1,42 (с, 9H). LCMS (метод C): RT=1,07 мин, m/z=275 [M+H]+.
Стадия 3: третбутил 4-(4-аминофенил)пиперидин-1-карбоксилат:
Перемешиваемую суспензию третбутил 4-(4-аминофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (79 мг, 0,253 ммоль) и 10% Pd/C (33,7 мг, 0,032 ммоль) в этаноле (2 мл) герметизировали, затем колбу вакуумировали и заполняли азотом. Эту операцию повторяли еще два раза, затем колбу вакуумировали и заполняли водородом (из баллона). Суспензию перемешивали в течение 5 часов. Оставшийся водород удаляли под вакуумом. Смесь фильтровали через целит®, промывая этанолом (5 мл), и фильтрат концентрировали досуха при пониженном давлении. Остаток очищали флэш-хроматографией (0-10% метанол в дихлорметане) с получением названного соединения (51,3 мг, 73,3%). LCMS (метод C): RT=0,92 мин, m/z=221 [M-Бутен]+.
Стадия 4: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)пиперидин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (200 мг, 0,563 ммоль) подвергали взаимодействию с третбутил 4-(4-аминофенил)пиперидин-1-карбоксилатом (156 мг, 0,563 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (182 мг, 55%). 1H ЯМР (300 МГц, CDCl3): δ 8,72 (с, 1H), 7,58 (уш.д, 3H), 7,44 (д, 2H), 7,28 (дд, 1H), 7,18 (д, 2H), 4,88 (с, 2H), 4,24 (уш.с, 2H), 3,18 (с, 3H), 2,80 (уш.т, 2H), 2,63 (тт, 1H), 1,83 (уш.д, 2H), 1,54-1,71 (м, 2H), 1,49 (с, 9H). LCMS (метод C): RT=1,82 мин, m/z=583 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-пиперидин-1-карбоксилат (182 мг, 0,312 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (24 мг, 16%). 1H ЯМР (400 МГц, MeOH-d4) δ 8,54 (с, 1H), 7,66 (д, 2H), 7,58 (д, 2H), 7,44 (дд, 1H), 7,22 (д, 2H), 4,99 (с, 2H), 3,12-3,22 (м, 5H), 2,75 (тд, 2H), 2,67 (тт, 1H), 1,84 (д, 2H), 1,68 (квт, 2H). LCMS (метод C): RT=0,80 мин, m/z=483 [M+H]+.
Пример 120: 3-(2,6-дихлорфенил)-7-((4-(4-(2-метоксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: 1-(2-метоксиэтил)-4-(4-нитрофенил)пиперазин:
Суспензию 1-фтор-4-нитробензола (1,00 г, 7,09 ммоль), 1-(2-метоксиэтил)пиперазина (1,02 г, 7,09 ммоль) и карбоната калия (1,47 г, 10,63 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали водой (5×20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (1,10 г, 59%). LCMS (метод C): RT=0,51 мин, m/z=266 [M+H]+.
Стадия 2: 4-(4-(2-метоксиэтил)пиперазин-1-ил)анилин:
Раствор 1-(2-метоксиэтил)-4-(4-нитрофенил)пиперазина (1 г, 3,77 ммоль) в этаноле (20 мл) нагревали до 50 °C. При перемешивании, добавляли порциями формиат аммония (1,188 г, 18,85 ммоль). Реакционную смесь нагревали при 50°C в течение еще 30 минут. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит® и концентрировали досуха при пониженном давлении с получением названного соединения в виде коричневого масла (650 мг, 73%). LCMS (метод C): RT=0,13 мин, m/z=236 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-(4-(2-метоксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с 4-(4-(2-метоксиэтил)пиперазин-1-ил)-анилином (67 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (53 мг, 35%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,70 (уш.с, 1H), 8,45 (с, 1H), 7,65 (д, 2H), 7,62 (уш.д, 2H), 7,48 (дд, 1H), 6,91 (д, 2H), 4,96 (с, 2H), 3,47 (т, 2H), 3,25 (с, 3H), 3,02-3,15 (м, 7H), 2,51-2,62 (м, 6H). LCMS (метод C): RT=0,78 мин, m/z=542 [M+H]+.
Пример 121: 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-((1R,4R)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
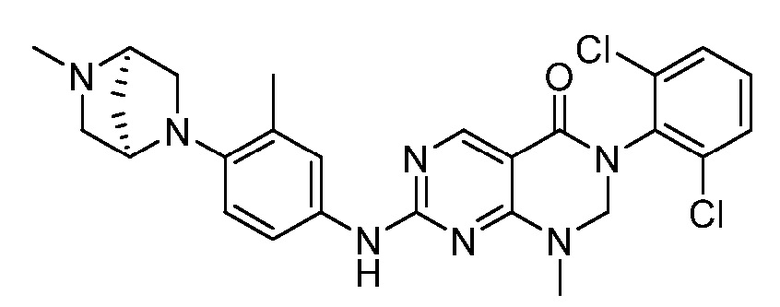
7-((4-((1R,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метил-фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он (32 мг, 0,063 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде желтого твердого вещества (17 мг, 52%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,63 (уш.с, 1H), 8,43 (с, 1H), 7,64 (д, 2H), 7,35-7,57 (уш.м, 3H), 6,73 (д, 1H), 4,95 (с, 2H), 3,92 (с, 1H), 3,32 (с, 1H), 3,20 (с, 2H), 3,09 (с, 3H), 2,75 (дд, 1H), 2,68 (д, 1H), 2,27 (с, 3H), 2,19 (с, 3H), 1,78 (д, 1H), 1,69 (д, 1H). LCMS (метод C): RT=0,80 мин, m/z=524 [M+H]+.
Пример 122: 3-(2,6-дихлорфенил)-7-((4-(4-этилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
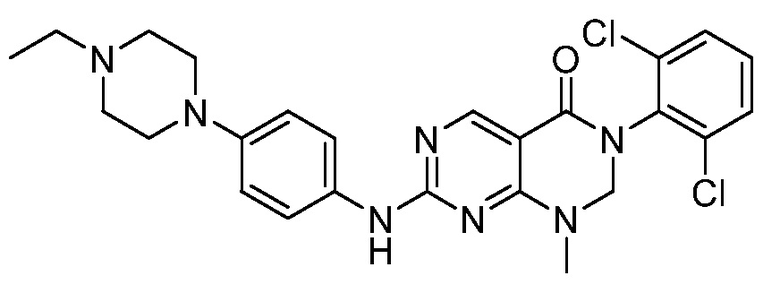
К суспензии 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она (60 мг, 0,124 ммоль) и карбоната калия (51,4 мг, 0,372 ммоль) в безводном DMF (2 мл) добавляли йодэтан (0,012 мл, 0,149 ммоль), и смесь нагревали при 90°C в течение 21 часа. Реакционную смесь охлаждали до комнатной температуры и распределяли между солевой раствор/вода (20 мл) и этилацетатом (5 мл). Водную фазу отделяли и экстрагировали этилацетатом (3×5 мл). Объединенные органические фазы промывали смесью 1:1 солевой раствор:вода (4×5 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали хроматографией на силикагеле (градиент 50-100% этилацетат в циклогексане, KP-NH) и лиофилизировали в течение ночи с получением названного соединения в виде бледно-желтого твердого вещества (28 мг, 44%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,70 (уш.с, 1H), 8,44 (с, 1H), 7,64 (д, 2H), 7,61 (уш.д, 2H), 7,47 (дд, 1H), 6,90 (д, 2H), 4,95 (с, 2H), 3,01-3,15 (м, 7H), 2,45-2,50 (м, 4H), 2,36 (кв, 2H), 1,03 (т, 3H). LCMS (метод C): RT=0,75 мин, m/z=512 [M+H]+.
Пример 123: 3-(2,6-дихлорфенил)-7-((4-(4-(2-(диметиламино)-ацетил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

К раствору 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она (40 мг, 0,083 ммоль), 2-(диметиламино)уксусной кислоты (8,52 мг, 0,083 ммоль) и N-метилморфолина (0,036 мл, 0,330 ммоль) в безводном DMF (1 мл) добавляли HBTU (37,6 мг, 0,099 ммоль), и полученную смесь перемешивали в течение ночи. Реакционную смесь разбавляли водой (8 мл) и экстрагировали этилацетатом (3×4 мл). Объединенные органические фазы промывали смесью 50:50 вода:солевой раствор (3×8 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж KP-NH 11g, циклогексан:этилацетат:метанол, градиентное элюирование от 90:10:0 до 0:100:0 до 0:80:20), суспендировали в диэтиловом эфире и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (30 мг, 64%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,73 (уш.с, 1H), 8,45 (с, 1H), 7,54-7,74 (м, 4H), 7,48 (т, 1H), 6,94 (д, 2H), 4,95 (с, 2H), 3,68 (т, 2H), 3,59 (т, 2H), 2,97-3,17 (м, 9H), 2,19 (с, 6H). LCMS (метод C): RT=0,75 мин, m/z=569 [M+H]+.
Пример 124: 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-(2-(метил-амино)ацетил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил (2-(4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-фенил)пиперазин-1-ил)-2-оксоэтил)(метил)карбамат:
3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (40 мг, 0,083 ммоль) подвергали взаимодействию с 2-((третбутокси-карбонил)(метил)амино)уксусной кислотой (15,62 мг, 0,083 ммоль), следуя методике примера 123, с получением названного соединения в виде желтого твердого вещества (35 мг, 65%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (с, 1H), 7,58 (уш.с, 1H), 7,54 (уш.д, 2H), 7,43 (д, 2H), 7,28 (дд, 1H), 6,93 (уш.д, 2H), 4,87 (с, 2H), 4,06 (уш.д, 2H), 3,74-3,83 (м, 2H), 3,54-3,65 (м, 2H), 3,16 (с, 3H), 3,13 (т, 4H), 2,94 (с, 3H), 1,41-1,50 (м, 9H). LCMS (метод C): RT=1,31 мин, m/z=655 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-(2-(метил-амино)ацетил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
Третбутил (2-(4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)-пиперазин-1-ил)-2-оксоэтил)(метил)карбамат (35 мг, 0,053 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (25 мг, 84%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,73 (уш.с, 1H), 8,45 (с, 1H), 7,54-7,77 (м, 4H), 7,48 (т, 1H), 6,94 (д, 2H), 4,95 (с, 2H), 3,60 (уш.с, 2H), 3,55 (уш.с, 2H), 3,34 (с, 2H), 2,97-3,17 (м, 7H), 2,28 (с, 3H), 1,95 (уш.с, 1H). LCMS (метод C): RT=0,73 мин, m/z=555 [M+H]+.
Пример 125: 7-((4-(4-ацетилпиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Суспензию 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-она (43 мг, 0,089 ммоль) и триэтиламина (0,062 мл, 0,444 ммоль) в безводном дихлорметан (1 мл) охлаждали до 0°C. Добавляли уксусный ангидрид (0,017 мл, 0,178 ммоль), и полученный раствор подогревали до комнатной температуры и перемешивали в течение 3 часов. Смесь промывали насыщенным раствором бикарбоната натрия (3×1 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток подвергали хроматографии (картридж KP-NH 11g, циклогексан:этилацетат:метанол, градиентное элюирование от 90:10:0 до 0:100:0 до 0:80:20), суспендировали в диэтиловом эфире и лиофилизировали в течение ночи с получением названного соединения в виде белого твердого вещества (29 мг, 62%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,73 (уш.с, 1H), 8,45 (с, 1H), 7,53-7,77 (м, 4H), 7,48 (т, 1H), 6,94 (д, 2H), 4,95 (с, 2H), 3,57 (уш.с, 4H), 2,95-3,16 (м, 7H), 2,04 (с, 3H). LCMS (метод C): RT=1,02 мин, m/z=526 [M+H]+.
Пример 126: (R)-3-(2-хлор-6-фторфенил)-7-((4-(3-(метокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2-хлор-6-фторфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (67 мг, 0,197 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-аминофенил)-2-(метоксиметил)пиперазин-1-карбоксилатом (60 мг, 0,186 ммоль), следуя методике примера 31, с получением названного соединения (37 мг,39%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,73 (с, 1H), 8,45 (с, 1H), 7,60 (д, 2H), 7,51 (м, 2H), 7,42-7,37 (м, 1H), 6,88 (д, 1H), 4,98 (д, 1H), 4,93 (д, 1H), 3,54 (м, 1H), 3,42 (м, 1H), 3,38 (с, 3H), 3,14 (с, 3H), 3,05 (м, 2H), 2,93 (м, 1H), 2,75 (м, 2H), 2,42 (м, 2H). LCMS (метод C): RT=0,73 мин, m/z=482 [M+H]+.
Пример 127: (S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидрокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (S)-третбутил 3-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
1-фтор-4-нитробензол (1,460 г, 10,4 ммоль) и карбонат калия (4,29 г, 31,0 ммоль) суспендировали в безводном DMF (10 мл). Добавляли (S)-третбутил 3-(гидроксиметил)пиперазин-1-карбоксилат (2,35 г, 10,9 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (25 мл). Водный слой отделяли и дополнительно экстрагировали этилацетатом (3×25 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 25-100% этилацетат в циклогексане) с получением названного соединения (0,99 г, 28,4%). 1H ЯМР (400 МГц, CDCl3): δ 8,12 (д, 2H), 6,83 (д, 2H), 4,20-4,46 (м, 1H), 4,02-4,14 (м, 2H), 3,48-3,76 (м, 3H), 3,08-3,30 (м, 3H), 2,86 (уш.с, 1H), 1,50 (с, 9H). LCMS (метод C): RT=1,38 мин, m/z=338 [M+H]+.
Стадия 2: (S)-третбутил 4-(4-аминофенил)-3-(гидроксиметил)-пиперазин-1-карбоксилат:
(S)-третбутил 3-(гидроксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат (450 мг. 1,33 ммоль) подвергали непрерывному гидрированию (проточный реактор ThalesNano H-cube apparatus, картридж 10% Pd/C, комнатная температура, режим заполненный H2, растворитель MeOH). Неочищенный продукт подвергали хроматографии (картридж 10g Si, элюировали 0-20% MeOH/DCM) с получением названного соединения в виде стеклообразного вещества (95 мг, 23%). LCMS (метод C): RT=0,62 мин, m/z=308 [M+H]+.
Стадия 3: (S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(S)-третбутил 4-(4-аминофенил)-3-(гидроксиметил)пиперазин-1-карбоксилат (88 мг, 0,42 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (150 мг, 0,42 ммоль), как описано в примере 1, с получением названного соединения в виде желтого твердого вещества (6 мг, 3%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,44 (с, 1H), 7,60 (м, 4H), 7,48 (т, 1H), 6,85 (д, 2H), 4,95 (с, 2H), 3,71 (т, 1H), 3,55 (м, 1H), 3,25 (дд, 1H), 3,13 (м, 5H), 2,94 (м, 1H), 2,81 (м, 2H), 2,71 (м, 1H). LCMS (метод C): RT=0,72 мин, m/z=514 [M+H]+.
Пример 128: (S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидрокси-метил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
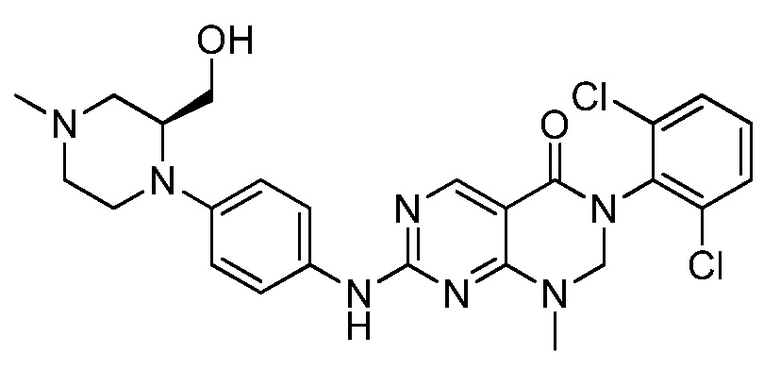
(S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидроксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (52 мг, 0,101 ммоль) растворяли в метаноле (1 мл) и добавляли водный раствор формальдегида (0,015 мл, 0,20 ммоль), затем триацетоксиборгидрид натрия (107 мг, 0,51 ммоль). Смесь перемешивали в течение 3 часов, затем загружали в картридж 2 g SCX. Картридж промывали метанолом, затем элюировали с помощью 2 M NH3/MeOH с получением стеклообразного твердого вещества. Это вещество суспендировали в эфире, и полученное твердое вещество собирали фильтрацией с получением названного соединения в виде желтого твердого вещества (29 мг, 54%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,67 (уш.с, 1H), 8,44 (с, 1H), 7,60 (м, 4H), 7,48 (т, 1H), 6,86 (д, 2H), 4,95 (с, 2H), 4,59 (уш.с, 1H), 3,66 (с, 2H), 3,27 (м, 2H), 3,09 (с, 3H), 2,96 (м, 2H), 2,75 (м, 1H), 2,22 (с, 3H), 2,10 (м, 2H). LCMS (метод C): RT=0,74 мин, m/z=528 [M+H]+.
Пример 129: (R)-3-(2,6-дихлорфенил)-7-((4-(2-(метокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 3-(гидроксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
1-Фтор-4-нитробензол (1,056 г, 7,49 ммоль) и карбонат калия (3,10 г, 22,5 ммоль) суспендировали в безводном DMF (10 мл). Добавляли (R)-третбутил 3-(гидроксиметил)пиперазин-1-карбоксилат (1,70 г, 7,86 ммоль), и смесь нагревали при 90°C в течение 21 часа. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (25 мл). Водный слой отделяли и дополнительно экстрагировали этилацетатом (3×25 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле (градиент 20-100% этилацетат в циклогексане) с получением названного соединения (0,69 г, 27,3%). LCMS (метод C): RT=1,38 мин, m/z=338 [M+H]+.
Стадия 2: (R)-третбутил 3-(метоксиметил)-4-(4-нитрофенил)-пиперазин-1-карбоксилат:
(R)-третбутил 3-(гидроксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат (430 мг, 1,28 ммоль) растворяли в THF (10 мл) и охлаждали до 0°C. Добавляли гидрид натрия (61,3 мг, 1,53 ммоль), и смесь перемешивали в течение 5 минут. Добавляли йодметан (0,105 мл, 1,67 ммоль), и смесь перемешивали в течение ночи. Смесь затем концентрировали под вакуумом и подвергали хроматографии (картридж Isolera 25 g Si; элюировали 0-50% EtOAc/циклогексан) с получением названного соединения в виде желтого масла (281 мг, 63%). LCMS (метод C): RT=1,67 мин, m/z=352 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-аминофенил)-3-(метоксиметил)-пиперазин-1-карбоксилат:
(R)-третбутил 3-(метоксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат (270 мг, 0,77 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, заполненный H2, растворитель MeOH). Смесь концентрировали с получением неочищенного названного соединения в виде желтого стеклообразного твердого вещества (280 мг, 113%), которое использовали без дополнительной очистки. LCMS (метод C): RT=0,83 мин, m/z=322 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((4-(2-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(R)-третбутил 4-(4-аминофенил)-3-(метоксиметил)пиперазин-1-карбоксилат (90 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения в виде желтого твердого вещества (45 мг, 30%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,69 (уш.с, 1H), 8,45 (с, 1H), 7,60 (м, 4H), 7,48 (м, 1H), 6,86 (д, 2H), 4,96 (с, 2H), 3,72 (м, 2H), 3,19 (с, 3H), 3,13 (м, 5H), 2,98 (м, 2H), 2,83 (м, 2H), 2,70 (м, 1H). LCMS (метод C): RT 0,81 мин, m/z=528 [M+H]+.
Пример 130: (S)-3-(2,6-дихлорфенил)-7-((4-(2-(метокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (S)-третбутил 3-(метоксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат:
(S)-третбутил 3-(гидроксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат (470 мг, 1,39 ммоль) растворяли в THF (10 мл) и охлаждали до 0 °C. Добавляли гидрид натрия (61,3 мг, 1,53 ммоль), и смесь перемешивали в течение 5 минут. Добавляли йодметан (0,105 мл, 1,67 ммоль), и смесь перемешивали в течение ночи. Смесь затем концентрировали под вакуумом и подвергали хроматографии (картридж 25 g Si; элюировали 0-50% EtOAc/циклогексан) с получением названного соединения в виде желтого масла (380 мг, 78%). LCMS (метод C): RT=1,67 мин, m/z=352 [M+H]+.
Стадия 2: (S)-третбутил 4-(4-аминофенил)-3-(метоксиметил)-пиперазин-1-карбоксилат:
(S)-третбутил 3-(метоксиметил)-4-(4-нитрофенил)пиперазин-1-карбоксилат (450 мг, 1,28 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, режим заполненный H2, растворитель MeOH). Неочищенный продукт подвергали хроматографии (картридж Isolera 10 g Si; элюировали 0-20% MeOH/DCM) с получением названного соединения в виде стеклообразного твердого вещества (90 мг, 22%). LCMS (метод C): RT=0,82 мин, m/z=322 [M+H]+.
Стадия 3: (S)-3-(2,6-дихлорфенил)-7-((4-(2-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
(S)-третбутил 4-(4-аминофенил)-3-(метоксиметил)пиперазин-1-карбоксилат (62 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения в виде желтого твердого вещества (12 мг, 6%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,45 (с, 1H), 7,60 (м, 4H), 7,48 (м, 1H), 6,86 (д, 2H), 4,96 (с, 2H), 3,72 (м, 2H), 3,19 (с, 3H), 3,13 (м, 5H), 2,98 (м, 2H), 2,83 (м, 2H), 2,70 (м, 1H). LCMS (метод C): RT=0,81 мин, m/z=528 [M+H]+.
Пример 131: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
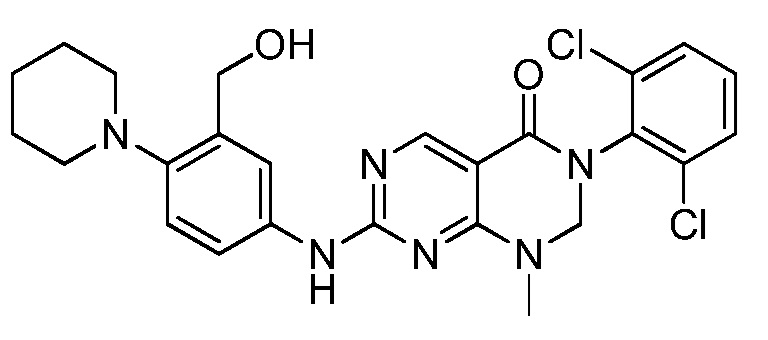
Стадия 1: (5-амино-2-(пиперидин-1-ил)фенил)метанол:
2-фтор-5-нитробензальдегид (1 г, 5,91 ммоль) растворяли в DMF (10 мл) и добавляли пиперидин (0,644 мл, 6,50 ммоль), затем DIPEA (2,272 мл, 13,0 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и полученный желтый осадок собирали фильтрацией. Этот осадок растворяли в этаноле (10,0 мл) и добавляли боргидрид натрия (0,336 г, 8,87 ммоль). Полученный раствор перемешивали в течение 30 минут, затем концентрировали под вакуумом. Остаток распределяли между DCM и насыщенным водным раствором NH4Cl. Органический слой отделяли, промывали солевым раствором, затем концентрировали под вакуумом. Остаток растворяли в метаноле и фильтровали через хлопковую вату, затем подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения в виде оранжевого масла (410 мг, 34%), которое использовали без дополнительной очистки. LCMS (метод C): RT=0,25 мин, m/z=207 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (300 мг, 0,084 ммоль) суспендировали в толуоле (5 мл) и добавляли mCPBA (291 мг, 0,93 ммоль) в виде раствора в DCM (3 мл) при комнатной температуре. Через 15 минут, добавляли DIPEA (0,44 мл, 2,53 ммоль), затем (5-амино-2-(пиперидин-1-ил)фенил)метанол (174 мг, 0,84 ммоль). Полученную смесь нагревали при 70°C в течение 16 часов, затем концентрировали под вакуумом и отгоняли в виде азеотропа с этилацетатом. Остаток подвергали хроматографии (картридж 10 g Si; элюировали 0-100% EtOAc/циклогексан, затем 0-5% MeOH/EtOAc) с получением стеклообразного твердого вещества. Это вещество суспендировали в диэтиловом эфире, и твердое вещество собирали фильтрацией с получением названного соединения в виде желтовато-белого твердого вещества (271 мг, 63%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,69 (уш.с, 1H), 8,46 (с, 1H), 7,66 (уш.с, 1H), 7,65 (д, 2H), 7,55 (дд, 1H), 7,48 (т, 1H), 7,00 (д, 1H), 5,04 (уш.с, 1H), 4,97 (с, 2H), 4,55 (с, 2H), 3,12 (с, 3H), 2,76 (м, 4H), 1,64 (м, 4H), 1,51 (м, 2H). LCMS (метод C): RT=0,97 мин, m/z=513 [M+H]+.
Пример 132: (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметокси)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 1-бром-2-(дифторметокси)-4-нитробензол:
Суспензию 2-бром-5-нитрофенола (1,00 г, 4,59 ммоль), этил хлордифторацетата (0,581 мл, 4,59 ммоль) и карбоната калия (0,634 г, 4,59 ммоль) в безводном DMF (10,35 мл) нагревали до 70°C в атмосфере азота в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали этилацетатом (3×25 мл). Объединенные органические фазы промывали смесью 50:50 вода:солевой раствор (3×30 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан: этилацетат, градиентное элюирование от 100:0 до 80:20) с получением названного соединения в виде бесцветного масла (920 мг, 75%). 1H ЯМР (400 МГц, DMSO-d6): δ 8,10 (д, 1H), 8,00 (дд, 1H), 7,83 (д, 1H), 6,66 (т, 1H).
Стадия 2: (R)-третбутил 4-(2-(дифторметокси)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Суспензию 1-бром-2-(дифторметокси)-4-нитробензола (400 мг, 1,492 ммоль), (R)-третбутил 2-метилпиперазин-1-карбоксилата (299 мг, 1,492 ммоль), тетрабутиламмония бромида (48,1 мг, 0,149 ммоль) и карбоната калия (309 мг, 2,239 ммоль) в безводном DMSO (2 мл) нагревали до 80°C в атмосфере азота в течение 24 часов, затем до 120°C в течение еще 24 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали смесью 50:50 вода:солевой раствор (3×10 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 60:40) с получением названного соединения в виде оранжевого масла, которое превращалось в твердое вещество при стоянии (320 мг, 55%). 1H ЯМР (300 МГц, CDCl3): δ 8,08 (дд, 1H), 7,99 (д, 1H), 6,98 (д, 1H), 6,53 (т, 1H), 4,37 (уш.с, 1H), 3,98 (уш.д, 1H), 3,51 (ддд, 1H), 3,44 (дт, 1H), 3,28 (тд, 1H), 2,99 (дд, 1H), 2,85 (тд, 1H), 1,48 (с, 9H), 1,31 (д, 3H). LCMS (метод C): RT=1,87 мин, m/z=410 [M+Na]+.
Стадия 3: (R)-третбутил 4-(4-амино-2-(дифторметокси)фенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(дифторметокси)-4-нитрофенил)-2-метилпиперазин-1-карбоксилата (320 мг, 0,826 ммоль) в метаноле (50 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 40 МПа, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде бледно-желтого твердого вещества (262 мг, 89%). 1H ЯМР (400 МГц, CDCl3): δ 6,75-6,83 (м, 1H), 6,37-3,60 (м, 3H), 4,30 (уш.с, 1H), 3,89 (д, 1H), 3,58 (с, 2H), 3,23 (тд, 1H), 3,13 (дд, 1H), 2,98 (дт, 1H), 2,79 (дд, 1H), 2,58 (тд, 1H), 1,47 (с, 9H), 1,30 (д, 3H). LCMS (метод C): RT=1,54 мин, m/z=358 [M+H]+.
Стадия 4: (R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(дифторметокси)фенил)-2-метилпиперазин-1-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(дифторметокси)фенил)-2-метилпиперазин-1-карбоксилатом (101 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (88 мг, 47%). 1H ЯМР (300 МГц, CDCl3): δ 8,70 (с, 1H), 8,13 (уш.с, 1H), 7,89 (уш.с, 1H), 7,44 (дд, 2H), 7,29 (дд, 1H), 7,16 (уш.д, 1H), 6,94 (д, 1H), 6,59 (т, 1H), 4,90 (с, 2H), 4,33 (уш.с, 1H), 3,93 (д, 1H), 3,08-3,33 (м, 6H), 2,83 (дд, 1H), 2,65 (тд, 1H), 1,48 (с, 9H), 1,32 (д, 3H). LCMS (метод C): RT=1,96 мин, m/z=664 [M+H]+.
Стадия 5: (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметокси)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
(R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(дифторметокси)фенил)-2-метилпиперазин-1-карбоксилат (88 мг, 0,132 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (27 мг, 36%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,95 (уш.с, 1H), 8,48 (с, 1H), 7,82 (уш.с, 1H), 7,65 (д, 2H), 7,43-7,53 (м, 2H), 7,05 (д, 1H), 7,02 (т, 1H), 4,98 (с, 2H), 3,11 (с, 3H), 3,07 (д, 2H), 2,75-2,94 (м, 3H), 2,43-2,58 (м, 1H), 2,23 (т, 1H), 0,98 (д, 3H). LCMS (метод C): RT=0,93 мин, m/z=564 [M+H]+.
Пример 133: (S)-3-(2,6-дихлорфенил)-7-((4-(гексагидро-пиразино[2,1-c][1,4]оксазин-8(1H)-ил)-3-(гидроксиметил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
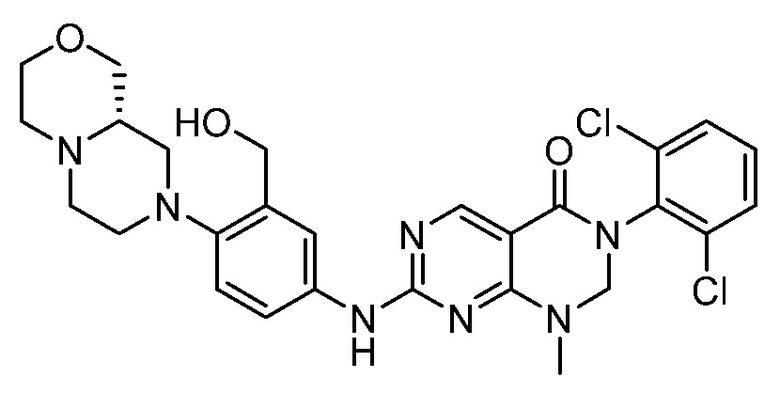
Стадия 1: (S)-2-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)-5-нитробензальдегид:
Суспензию 2-фтор-5-нитробензальдегида (400 мг, 2,365 ммоль), (S)-октагидропиразино[2,1-c][1,4]оксазина (336 мг, 2,365 ммоль) [полученного, как описано для R-энантиомера в публикации J. Med. Chem., 2012, 55(12), 5887-5900, но используя противоположный энантиомер исходного материала] и карбоната калия (490 мг, 3,55 ммоль) в безводном DMF (2,5 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (25 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток подвергали хроматографии (картридж диоксид кремния 50g, циклогексан:этилацетат:метанол, градиентное элюирование от 90:10:0 до 0:100:0 до 0:80:20) с получением названного соединения в виде оранжевого твердого вещества (506 мг, 73%). 1H ЯМР (300 МГц, CDCl3): δ 10,09 (с, 1H), 8,62 (д, 1H), 8,31 (дд, 1H), 7,08 (д, 1H), 3,90 (дд, 1H), 3,65-3,79 (м, 2H), 3,25-3,47 (м, 3H), 3,20 (дт, 1H), 2,79-2,93 (м, 2H), 2,74 (дт, 1H), 2,43-2,68 (м, 3H). LCMS (метод C): RT=0,48 мин, m/z=292 [M+H]+.
Стадия 2: (S)-(2-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)-5-нитрофенил)метанол:
Раствор (S)-2-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)-5-нитробензальдегида (506 мг, 1,737 ммоль) в безводном THF (3217 мкл) охлаждали до 0°C, затем добавляли порциями боргидрид натрия (65,7 мг, 1,737 ммоль). Смесь перемешивали при 0°C в течение 90 минут, затем гасили водой (20 мл) и экстрагировали дихлорметаном (3×15 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (390 мг, 77%). 1H ЯМР (300 МГц, CDCl3): δ 8,29 (д, 1H), 8,14 (дд, 1H), 7,12 (д, 1H), 4,72-4,86 (м, 2H), 3,90 (дд, 1H), 3,65-3,78 (м, 2H), 3,31 (т, 1H), 3,01-3,23 (м, 3H), 2,98 (дт, 1H), 2,89 (дт, 1H), 2,72 (дт, 1H), 2,42-2,67 (м, 4H). LCMS (метод C): RT=0,26 мин, m/z=294 [M+H]+.
Стадия 3: (S)-(5-амино-2-(гексагидропиразино[2,1-c][1,4]-оксазин-8(1H)-ил)фенил)метанол:
Раствор (S)-(2-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)-5-нитрофенил)метанола (390 мг, 1,330 ммоль) в метаноле (40 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 30 МПа H2, комнатная температура, 1 мл/мин) за два прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-коричневого твердого вещества (296 мг, 85%). 1H ЯМР (300 МГц, CDCl3): δ 7,03 (д, 1H), 6,57 (дд, 1H), 6,46 (д, 1H), 4,73 (д, 1H), 4,67 (д, 1H), 3,88 (дд, 1H), 3,71 (тд, 2H), 3,61 (уш.с, 2H), 3,29 (дд, 1H), 2,92-3,08 (м, 2H), 2,86 (дт, 1H), 2,78 (дт, 1H), 2,70 (дт, 1H), 2,37-2,61 (м, 4H). LCMS (метод C): RT=0,21 мин, m/z=264 [M+H]+.
Стадия 4: (S)-3-(2,6-дихлорфенил)-7-((4-(гексагидро-пиразино[2,1-c][1,4]оксазин-8(1H)-ил)-3-(гидроксиметил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с (S)-(5-амино-2-(гексагидро-пиразино[2,1-c][1,4]оксазин-8(1H)-ил)фенил)метанолом (75 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (35 мг, 22%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,81 (уш.с, 1H), 8,46 (с, 1H), 7,99 (уш.с, 1H), 7,64 (д, 2H), 7,56 (дд, 1H), 7,48 (т, 1H), 7,00 (д, 1H), 5,06 (т, 1H), 4,96 (с, 2H), 4,48-4,61 (м, 2H), 3,76 (д, 1H), 3,64 (дд, 1H), 3,52 (т, 1H), 3,07-3,18 (м, 4H), 2,90 (д, 1H), 2,70-2,85 (м, 3H), 2,65 (д, 1H), 2,19-2,42 (м, 4H). LCMS (метод C): RT=0,72 мин, m/z=570 [M+H]+.
Пример 134: 3-(2,6-дихлорфенил)-7-((3-(2,2-дифтор-1-гидроксиэтил)-4-((R)-3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
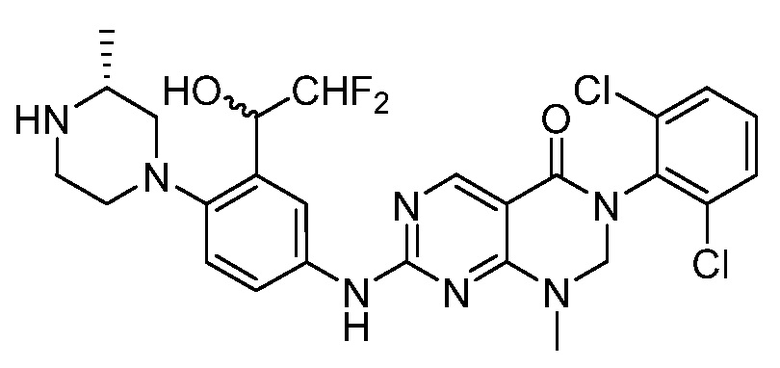
Стадия 1: (2R)-третбутил 4-(2-(2,2-дифтор-1-гидроксиэтил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат
В атмосфере N2, добавляли CsF (10 мг, 0,07 ммоль) к раствору (R)-третбутил 4-(2-формил-4-нитрофенил)-2-метилпиперазин-1-карбоксилата (1 г, 2,86 ммоль) и (дифторметил)триметилсилана (0,81 мл, 5,72 ммоль) в 10 мл DMF, затем смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли раствор TBAF (5,7 мл, 1 M в THF), и всю смесь перемешивали в течение еще 1 часа. После экстракции в системе Et2O и H2O, органическую фазу промывали солевым раствором, и затем сушили над безводным MgSO4. Затем раствор фильтровали, и растворитель испаряли под вакуумом, остаток подвергали хроматографии с получением названного соединения (0,9 г, 78%).
Стадия 2: (2R)-третбутил 4-(4-амино-2-(2,2-дифтор-1-гидроксиэтил)фенил)-2-метилпиперазин-1-карбоксилат
(2R)-третбутил 4-(2-(2,2-дифтор-1-гидроксиэтил)-4-нитро-фенил)-2-метилпиперазин-1-карбоксилат (0,3 г, 0,747 ммоль) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполнен H2, 25°C, 1 мл/мин). Раствор концентрировали под вакуумом с получением названного соединения (0,27 г, 97%).
Стадия 3: 3-(2,6-дихлорфенил)-7-((3-(2,2-дифтор-1-гидрокси-этил)-4-((R)-3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (191 мг, 0,538 ммоль) подвергали взаимодействию с (2R)-третбутил 4-(4-амино-2-(2,2-дифтор-1-гидроксиэтил)фенил)-2-метилпиперазин-1-карбоксилатом (183 мг, 0,512 ммоль), следуя методике примера 31, с получением названного соединения (154 мг, 52%). LCMS (метод C): RT=0,78 мин, m/z=578 [M+H]+.
Пример 135:(R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-(метоксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(2-формил-4-нитрофенил)-2-(метоксиметил)пиперазин-1-карбоксилат
2-фтор-5-нитробензальдегид (1 г, 5,91 ммоль) подвергали взаимодействию с (R)-третбутил 2-(метоксиметил)пиперазин-1-карбоксилатом (1 г, 4,34 ммоль), следуя методике примера 21, с получением названного соединения (1,30 г, 79%).
Стадия 2: (R)-третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-2-(метоксиметил)пиперазин-1-карбоксилат
(R)-третбутил 4-(2-формил-4-нитрофенил)-2-(метоксиметил) пиперазин-1-карбоксилат (0,27 г, 0,712 ммоль) подвергали взаимодействию с боргидридом натрия (0,032 г, 0,854 ммоль), следуя методике примера 21, с получением названного соединения (0,24 г, 88%).
Стадия 3: (R)-третбутил 4-(4-амино-2-(гидроксиметил)фенил)-2-(метоксиметил)пиперазин-1-карбоксилат:
(R)-третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-2-(метокси-метил)пиперазин-1-карбоксилат (0,24 г, 0,63 ммоль) гидрировали в проточном реакторе H-cube (картридж 10% Pd/C, заполнен H2, 25°C, 1 мл/мин). Раствор концентрировали под вакуумом с получением названного соединения (0,21 г, 95%).
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-(метоксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (244 мг, 0,687 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(гидроксиметил)фенил)-2-(метоксиметил)пиперазин-1-карбоксилатом (230 мг, ммоль), следуя методике примера 31, с получением названного соединения (190 мг, 52%).¹H ЯМР (400 МГц, MeOD): δ 8,52 (с, 1H), 7,88 (с, 1H), 7,54 (м, 3H), 7,41 (м, 1H), 7,12 (д, 2H), 4,98 (с, 2H), 4,74 (с, 2H), 3,80 (м, 1H), 3,28 (с, 3H), 3,19 (с, 3H), 3,05 (м, 1H), 2,95 (м, 1H), 2,00 (д, 1H), 1,84 (д, 1H). LCMS (метод C): RT=0,70 мин, m/z=558 [M+H]+.
Пример 136: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метил-амино)метил)-4-(пиперидин-1-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
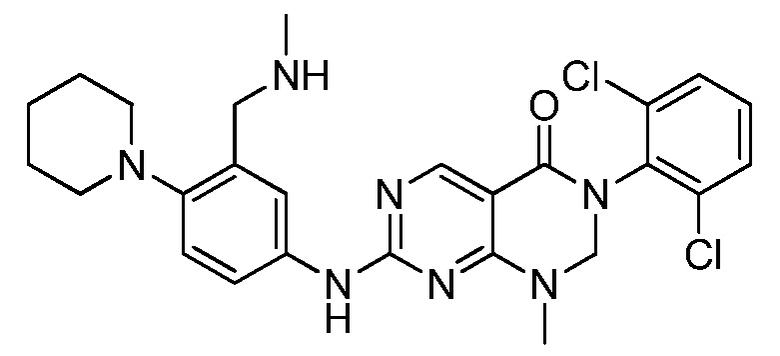
Стадия 1: 5-нитро-2-(пиперидин-1-ил)бензальдегид:
2-фтор-5-нитробензальдегид (1 г, 5,91 ммоль) растворяли в DMF (10 мл) и добавляли пиперидин (0,64 мл, 6,50 ммоль), затем DIPEA (2,27 мл, 13,0 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и полученный осадок собирали фильтрацией и сушили под вакуумом с получением названного соединения (1,3 г, 94%) в виде желтого твердого вещества, которое использовали без дополнительной очистки. LCMS (метод C): RT=1,59 мин, m/z=235 [M+H]+.
Стадия 2: N-метил-1-(5-нитро-2-(пиперидин-1-ил)фенил)-метанамин:
5-нитро-2-(пиперидин-1-ил)бензальдегид (250 мг, 1,067 ммоль) растворяли в метаноле (1 мл) и добавляли бикарбонат натрия (108 мг, 1,281 ммоль), затем метиламин (2 M в THF, 0,640 мл, 1,281 ммоль). Смесь перемешивали в течение 2 часов при 70°C, затем охлаждали до 0 °C. Добавляли боргидрид натрия (48,5 мг, 1,281 ммоль), и смесь перемешивали в течение ночи (подогревали до комнатной температуры). Добавляли каплю воду, и смесь концентрировали под вакуумом. Остаток растворяли в метаноле и подкисляли с помощью HCl. Полученный раствор загружали в картридж 5 g SCX и промывали с помощью MeOH, затем элюировали, используя 2 M NH3/MeOH, с получением названного соединения в виде стеклообразного твердого вещества (245 мг, 92%). LCMS (метод C): RT=0,70 мин, m/z=250 [M+H]+.
Стадия 3: третбутил метил(5-нитро-2-(пиперидин-1-ил)-бензил)карбамат:
Раствор дитретбутилдикарбоната (236 мг, 1,081 ммоль) в DCM (5 мл) добавляли к N-метил-1-(5-нитро-2-(пиперидин-1-ил)фенил)-метанамину (245 мг, 0,983 ммоль). Добавляли триэтиламин (0,274 мл, 1,965 ммоль), и смесь перемешивали при комнатной температуре в течение 90 минут. Смесь разбавляли водой, и органический слой собирали, используя картридж для разделения фаз, и концентрировали с получением названного соединения (344 мг, 99%) в виде стеклообразного твердого вещества. LCMS (метод C): RT=2,05 мин, m/z=350 [M+H]+.
Стадия 4: третбутил 5-амино-2-(пиперидин-1-ил)бензил-(метил)карбамат:
Третбутил метил(5-нитро-2-(пиперидин-1-ил)бензил)карбамат (344 мг, 0,98 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (306 мг, 96%) в виде желтого сиропа, которое использовали без дополнительной очистки. LCMS (метод C): RT=1,03 мин, m/z=320 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиперидин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-амино-2-(пиперидин-1-ил)бензил(метил)карбамат (71,9 мг, 0,22 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-оном (80 мг, 0,22 ммоль), как описано в примере 1, с получением названного соединения (70 мг, 58%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,76 (уш.с, 1H), 8,46 (с, 1H), 7,64 (уш.с, 1H), 7,52 (д, 2H), 7,49 (м, 2H), 7,03 (д, 1H), 4,97 (с, 2H), 3,67 (м, 2H), 3,15 (с, 3H), 2,78 (м, 4H), 2,33 (с, 3H), 1,64 (м, 4H), 1,52 (м, 2H). LCMS (метод C): RT=1,13 мин, m/z=526 [M+H]+.
Пример 137: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метил-амино)метил)-4-(пирролидин-1-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (5-нитро-2-(пирролидин-1-ил)фенил)метанол:
2-фтор-5-нитробензальдегид (1 г, 5,91 ммоль) растворяли в DMF (10 мл) и добавляли пирролидин (0,54 мл, 6,50 ммоль), затем DIPEA (2,27 мл, 13,0 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и полученный осадок собирали фильтрацией и сушили под вакуумом с получением названного соединения (1,30 г, 99%) в виде желтого твердого вещества которое использовали без дополнительной очистки. LCMS (метод C): RT=1,34 мин, m/z=221 [M+H]+.
Стадия 2: N-метил-1-(5-нитро-2-(пирролидин-1-ил)фенил)-метанамин:
(5-нитро-2-(пирролидин-1-ил)фенил)метанол (235 мг, 1,07 ммоль) растворяли в метаноле (1 мл) и добавляли бикарбонат натрия (108 мг, 1,281 ммоль), затем метиламин (2M в THF, 0,640 мл, 1,281 ммоль). Смесь перемешивали в течение 2 часов при 70°C, затем охлаждали до 0 °C. Добавляли боргидрид натрия (48,5 мг, 1,28 ммоль), и смесь перемешивали в течение ночи (подогревали до комнатной температуры). Добавляли каплю воды, и смесь концентрировали под вакуумом. Остаток растворяли в метаноле и подкисляли с помощью HCl. Полученный раствор загружали в картридж 5 g SCX и промывали, используя MeOH, затем элюировали с помощью 2 M NH3/MeOH с получением названного соединения в виде стеклообразного твердого вещества (240 мг, 96%). LCMS (метод C): RT=0,61 мин, m/z=236 [M+H]+.
Стадия 3: третбутил метил(5-нитро-2-(пирролидин-1-ил)бензил)карбамат:
Раствор дитретбутилдикарбоната (236 мг, 1,08 ммоль) в DCM (5 мл) добавляли к N-метил-1-(5-нитро-2-(пирролидин-1-ил)фенил)-метанамину (235 мг, 1,00 ммоль). Добавляли триэтиламин (0,274 мл, 1,965 ммоль), и смесь перемешивали при комнатной температуре в течение 90 минут. Смесь разбавляли водой, и органический слой собирали, используя картридж для разделения фаз, и концентрировали с получением названного соединения (333 мг, 99%) в виде стеклообразного твердого вещества. LCMS (метод C): RT=1,84 мин, m/z=336 [M+H]+.
Стадия 4: третбутил 5-амино-2-(пирролидин-1-ил)бензил-(метил)карбамат:
Третбутил метил(5-нитро-2-(пирролидин-1-ил)бензил)карбамат (333 мг, 0,99 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (312 мг, 102%) в виде желтого сиропа, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,91 мин, m/z=306 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пирролидин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-амино-2-(пирролидин-1-ил)бензил(метил)карбамат (69 мг, 0,22 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-оном (80 мг, 0,22 ммоль), как описано в примере 1, с получением названного соединения (51 мг, 50%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,70 (уш.с, 1H), 8,45 (с, 1H), 7,92 (уш.с, 1H), 7,65 (м, 2H), 7,48 (т, 2H), 6,91 (д, 1H), 4,96 (с, 2H), 3,65 (с, 2H), 3,12 (с, 3H), 3,06 (м, 4H), 2,32 (с, 3H), 1,86 (м, 4H). LCMS (метод C): RT=0,96 мин, m/z=512 [M+H]+.
Пример 138: 7-((4-(4-ацетилпиперазин-1-ил)-3-(пирролидин-1-илметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 1-(4-(4-нитро-2-(пирролидин-1-илметил)фенил)-пиперазин-1-ил)этанон:
2-(4-Ацетилпиперазин-1-ил)-5-нитробензальдегид (296 мг, 1,07 ммоль) растворяли в метаноле (1 мл) и добавляли бикарбонат натрия (108 мг, 1,28 ммоль), затем пирролидин (0,11 мл, 1,28 ммоль). Смесь перемешивали в течение 2 часов при 70°C, затем охлаждали до 0°C. Добавляли боргидрид натрия (48,5 мг, 1,28 ммоль), и смесь перемешивали в течение ночи (подогревали до комнатной температуры). Добавляли каплю воды, и смесь концентрировали под вакуумом. Остаток растворяли в метаноле и подкисляли с помощью HCl. Полученный раствор загружали в картридж 5 g SCX и промывали, используя MeOH, затем элюировали с помощью 2 M NH3/MeOH с получением названного соединения в виде стеклообразного твердого вещества (212 мг, 60%). LCMS (метод C): RT=0,25 мин, m/z=333 [M+H]+.
Стадия 2: 1-(4-(4-амино-2-(пирролидин-1-илметил)фенил)-пиперазин-1-ил)этанон:
1-(4-(4-нитро-2-(пирролидин-1-илметил)фенил)пиперазин-1-ил)этанон (205 мг, 0,61 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 Мпа, растворитель метанол) с получением названного соединения (196 мг, 106%) в виде желтого сиропа, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,24 мин, m/z=303 [M+H]+.
Стадия 3: 7-((4-(4-ацетилпиперазин-1-ил)-3-(пирролидин-1-илметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
1-(4-(4-Амино-2-(пирролидин-1-илметил)фенил)пиперазин-1-ил)этанон (68 мг, 0,22 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (80 мг, 0,22 ммоль), как описано в примере 131, с получением названного соединения (4 мг, 1%) в виде желтого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,77 (уш.с, 1H), 8,47 (с, 1H), 7,98 (уш.с, 1H), 7,66 (д, 2H), 7,50 (м, 2H), 7,03 (д, 1H), 4,98 (с, 2H), 3,65 (с, 2H), 3,57 (м, 4H), 3,14 (с, 3H), 2,80 (м, 4H), 2,04 (с, 3H), 1,70 (м, 4H); один сигнал, по-видимому относящийся к 4H, обычно перекрывался растворителем при 2,50. LCMS (метод C): RT=0,90 мин, m/z=609 [M+H]+.
Пример 139: (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(2-(дифторметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-формил-4-нитрофенил)-2-метил-пиперазин-1-карбоксилат (0,5 г, 1,43 ммоль) и DAST (0,6 г, 3,72 ммоль) в DCM (20 мл) перемешивали при комнатной температуре в течение ночи. Затем его разбавляли насыщенным раствором бикарбоната натрия (5 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали на хроматографической системе Biotage с получением названного соединения (0,43 г, 81%). LCMS (метод A) RT=0. 1,65 мин, m/z=372 [M+H]+.
Стадия 2: (R)-третбутил 4-(4-амино-2-(дифторметил)фенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(дифторметил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилат (0,3 г, 0,8 ммоль) в метаноле (30 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2)]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (270 мг, 97%). LCMS (метод A): RT=1,65 мин, m/z=342 [M+H]+.
Стадия 3: (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (183 мг,0,516 ммоль) подвергали взаимодействию с трет-5-бутил 4-(4-амино-2-метоксифенил)пиперазин-1-карбоксилатом (160 мг, 0,47 ммоль), следуя методике примера 31, с получением названного соединения (121 мг, 47%). ¹H ЯМР (400 МГц, CDCl3): δ 8,72 (с, 1H), 8,27 (с, 1H), 7,83 (с, 1H), 7,50 (уш.с, 1H), 7,44 (д, 2H), 7,30 (м, 1H), 7,22 (с, 1H), 7,09 (т, 1H), 5,01 (д, 1H), 4,91 (с, 2H), 3,21 (с, 3H), 3,09 (м, 3H), 2,93 (м, 2H), 2,77 (м, 1H), 2,48 (м, 1H), 1,11 (д, 3H). LCMS (метод C): RT=0,93 мин, m/z=548 [M+H]+.
Пример 140: (R)-3-(2,6-дихлорфенил)-7-((3-(2-гидроксипропан -2-ил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (R)-третбутил 4-(2-(этоксикарбонил)-4-нитро-фенил)-2-метилпиперазин-1-карбоксилат:
Суспензию этил 2-фтор-5-нитробензoата (2,13 г, 10 ммоль), (R)-третбутил 2-метилпиперазин-1-карбоксилата (2 г, 10 ммоль) и карбоната калия (2,1 г, 15 ммоль) в безводном DMF (20 мл) нагревали до 50°C в атмосфере азота в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между водой (100 мл) и этилацетатом (100 мл). Этилацетат отделяли и промывали водой (3×20 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Остаток очищали флэш-хроматографией (0-100% этилацетат в циклогексане) с получением названного соединения (2,8 г, 71%). LCMS (метод A): RT=1,83 мин, m/z=394 [M+H]+.
Стадия 2: (R)-третбутил 4-(4-амино-2-(этоксикарбонил)-фенил)-2-метилпиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(2-(этоксикарбонил)-4-нитрофенил)-2-метилпиперазин-1-карбоксилата (500 мг, 1,27 ммоль) в метаноле (30 мл) пропускали через проточный реактор H-Cube, снабженный картриджем 10% Pd-C, при следующих условиях [расход 1,0 мл/мин, 40°C, режим заполненный H2)]. Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (425 мг, 92%). LCMS (метод A) RT=1,2 мин, m/z=364 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-амино-2-(2-гидроксипропан-2-ил)фенил)-2-метилпиперазин-1-карбоксилат:
К раствору (R)-третбутил 4-(4-амино-2-(этоксикарбонил)- фенил)-2-метилпиперазин-1-карбоксилата (0,8 г, 2,2 ммоль) в осушенном THF (30 мл) при 0°C в атмосфере азота добавляли по каплям раствор метилмагния бромид в THF (1,4M, 7,26 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи, затем реакционную смесь гасили насыщенным раствором хлорида аммония. Полученный раствор разбавляли этилацетатом (100 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали на хроматографической системе Biotage с получением названного соединения (0,16 г, 20,8%). LCMS (метод A): RT=0,86 мин, m/z=350 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((3-(2-гидроксипропан-2-ил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (178 мг,0,501 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-2-(2-гидроксипропан-2-ил)фенил)-2-метилпиперазин-1-карбоксилатом (160 мг,4,77 ммоль), следуя методике примера 31, с получением названного соединения (30 мг, 9,8%). ¹H ЯМР (400 МГц, CDCl3): δ 9,18 (с, 1H), 8,72 (с, 1H), 7,60 (с, 1H), 7,52 (дд, 1H), 7,47 (м, 1H), 7,44 (м, 1H), 7,32 (м, 2H), 5,30 (с, 1H), 4,90 (с, 2H), 3,21 (с, 3H), 3,2-3,05 (м, 3H), 2,99 (м, 3H), 2,67 (м, 1H), 1,61 (с, 3H), 1,60 (с, 3H), 1,15 (д, 3H). LCMS (метод C): RT=0,75 мин, m/z=556 [M+H]+.
Пример 141: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)-3-(((2,2,2-трифторэтил)амино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 4-(2-(хлорметил)-4-нитрофенил)-пиперазин-1-карбоксилат:
Третбутил 4-(2-(гидроксиметил)-4-нитрофенил)пиперазин-1-карбоксилат (900 мг, 2,67 ммоль) растворяли в DMF (10 мл). Добавляли триэтиламин (1,859 мл, 13,3 ммоль), затем метансульфонилхлорид (0,624 мл, 8,00 ммоль), и смесь перемешивали в течение ночи при 60 °C. Растворитель удаляли под вакуумом. Остаток разбавляли водой и экстрагировали этилацетатом (x2). Объединенные органические экстракты сушили над MgSO4 и концентрировали. Остаток подвергали хроматографии (25 g Si; элюировали 0-100% EtOAc/циклогексан) с получением названного соединения (410 мг, 43%) в виде желтого сиропа, который кристаллизовался (желтые розетки) при стоянии. LCMS (метод C): RT=1,86 мин, m/z=356 [M+H]+.
Стадия 2: третбутил 4-(4-нитро-2-(((2,2,2-трифторэтил)-амино)метил)фенил)пиперазин-1-карбоксилат:
Третбутил 4-(2-(хлорметил)-4-нитрофенил)пиперазин-1-карбоксилат (200 мг, 0,562 ммоль) суспендировали в смеси 2,2,2-трифторэтиламина (0,5 мл, 6,37 ммоль), DIPEA (0,196 мл, 1,124 ммоль) и DMF (0,5 мл). Смесь нагревали при 50°C в течение ночи, затем концентрировали под вакуумом (отгоняли в виде азеотропа с толуолом x3). Остаток растворяли в дихлорметане, и раствор промывали насыщенным водным раствором карбоната натрия, затем концентрировали с получением названного соединения (180 мг, 77%) в виде желтого масла, которое использовали без дополнительной очистки. LCMS (метод C): RT=1,82 мин, m/z=418 [M+H]+.
Стадия 3: третбутил 4-(4-амино-2-(((2,2,2-трифторэтил)-амино)метил)фенил)пиперазин-1-карбоксилат:
Третбутил 4-(4-нитро-2-(((2,2,2-трифторэтил)амино)метил)-фенил)пиперазин-1-карбоксилат (180 мг, 0,43 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (125 мг, 75%) в виде коричневого стеклообразного твердого вещества, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,92 мин, m/z=389 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)-3-(((2,2,2-трифторэтил)амино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-амино-2-(((2,2,2-трифторэтил)амино)метил)-фенил)пиперазин-1-карбоксилат (109 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения (14 мг, 8%) в виде желтовато-белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,46 (с, 1H), 7,97 (уш.с, 1H), 7,65 (д, 2H), 7,49 (м, 2H), 7,05 (д, 1H), 4,97 (с, 2H), 3,81 (д, 2H), 3,26 (м, 2H), 3,11 (с, 3H), 2,76 (м, 8H). LCMS (метод C): RT=0,84 мин, m/z=595 [M+H]+.
Пример 142: (R)-3-(2,6-дихлорфенил)-7-((2-фтор-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
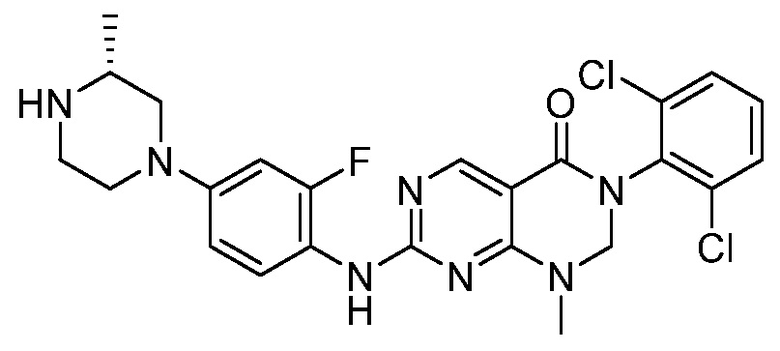
Стадия 1: (R)-третбутил 4-(3-фтор-4-нитрофенил)-2-метилпиперазин-1-карбоксилат:
Суспензию 2,4-дифтор-1-нитробензола (0,827 мл, 7,54 ммоль), (R)-третбутил 2-метилпиперазин-1-карбоксилата (1,511 г, 7,54 ммоль) и карбоната калия (1,564 г, 11,31 ммоль) в безводном DMSO (6 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали этилацетатом (3×25 мл). Объединенные органические фазы промывали смесью 50:50 вода:солевой раствор (3×40 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 40:60) с получением названного соединения в виде желтого масла (911 мг, 36%). 1H ЯМР (300 МГц, CDCl3) δ 8,03 (т, 1H), 6,40-6,56 (м, 2H), 4,26-4,40 (м, 1H), 3,93 (дт, 1H), 3,70 (дтд, 1H), 3,57 (ддд, 1H), 3,28-3,41 (м, 2H), 3,16 (ддд, 1H), 1,48 (с, 9H), 1,21 (д, 3H). LCMS (метод C): RT=1,71 мин, m/z=340 [M+H]+.
Стадия 2: (R)-третбутил 4-(4-амино-3-фторфенил)-2-метил-пиперазин-1-карбоксилат:
Раствор (R)-третбутил 4-(3-фтор-4-нитрофенил)-2-метил-пиперазин-1-карбоксилата (911 мг, 2,68 ммоль) в метаноле (60 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 30 МПа H2, комнатная температура, 1 мл/мин) за три прохода. Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде голубого масла (690 мг, 83%). 1H ЯМР (300 МГц, CDCl3) δ 6,72 (дд, 1H), 6,62 (дд, 1H), 6,54 (ддд, 1H), 4,24-4,38 (м, 1H), 3,87-3,97 (м, 1H), 3,46 (уш.с, 2H), 3,09-3,34 (м, 3H), 2,79 (дд, 1H), 2,61 (тд, 1H), 1,48 (с, 9H), 1,30 (д, 3H). LCMS (метод C): RT=1,34 мин, m/z=310 [M+H]+.
Стадия 3: (R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-3-фторфенил)-2-метилпиперазин-1-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (200 мг, 0,566 ммоль) подвергали взаимодействию с (R)-третбутил 4-(4-амино-3-фторфенил)-2-метилпиперазин-1-карбоксилатом (175 мг, 0,566 ммоль), следуя методике примера 31, с получением названного соединения в виде бледно-коричневого твердого вещества (104 мг, 30%). 1H ЯМР (300 МГц, CDCl3): δ 8,71 (с, 1H), 8,12 (т, 1H), 7,44 (дд, 2H), 7,28 (дд, 1H), 7,24 (уш.с, 1H), 6,61-6,72 (м, 2H), 4,88 (с, 2H), 4,34 (уш.с, 1H), 3,95 (тд, 1H), 3,46 (уш.д, 1H), 3,18-3,36 (м, 2H), 3,16 (с, 3H), 2,93 (дд, 1H), 2,74 (тд, 1H), 1,48 (с, 9H), 1,29 (д, 3H). LCMS (метод C): RT=1,80 мин, m/z=616 [M+H]+.
Стадия 4: (R)-3-(2,6-дихлорфенил)-7-((2-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
((R)-третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-3-фтор-фенил)-2-метилпиперазин-1-карбоксилат (104 мг, 0,169 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (55 мг, 63%). 1H ЯМР (300 МГц, DMSO-d6): δ 9,18 (с, 1H), 8,38 (с, 1H), 7,64 (д, 2H), 7,47 (т, 1H), 7,35 (уш.с, 1H), 6,66-6,86 (м, 2H), 4,91 (с, 2H), 3,52 (т, 2H), 2,86-3,06 (м, 4H), 2,76 (т, 2H), 2,53-2,61 (м, 1H), 2,18 (т, 1H), 1,02 (д, 3H). LCMS (метод C): RT=0,81 мин, m/z=516 [M+H]+.
Пример 143: 7-((4-(4-ацетилпиперазин-1-ил)-3-((метиламино)-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
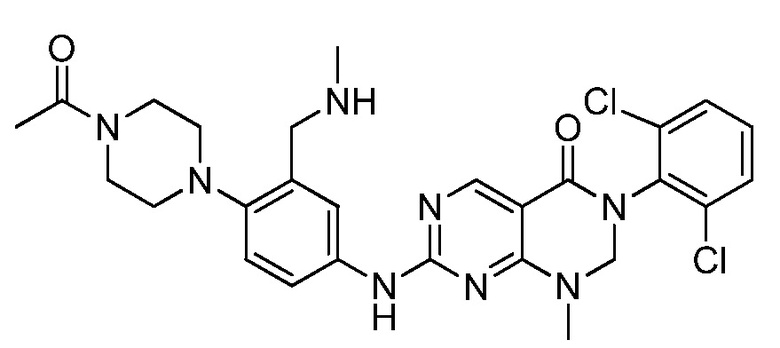
Стадия 1: 2-(4-ацетилпиперазин-1-ил)-5-нитробензальдегид:
2-Фтор-5-нитробензальдегид (1,00 г, 5,91 ммоль) растворяли в DMF (10 мл) и добавляли 1-(пиперазин-1-ил)этанон (0,83 г, 6,50 ммоль), затем DIPEA (2,27 мл, 13,0 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и смесь экстрагировали этилацетатом (x2). Объединенные органические слои промывали солевым раствором и концентрировали под вакуумом. Остаток сушили путем отгонки азеотропа с толуолом с получением названного соединения (1,20 г, 73%) в виде желтого сиропа, который частично кристаллизовался при стоянии. LCMS (метод C): RT=0,95 мин, m/z=278 [M+H]+.
Стадия 2: 1-(4-(2-((метиламино)метил)-4-нитрофенил)-пиперазин-1-ил)этанон:
2-(4-ацетилпиперазин-1-ил)-5-нитробензальдегид (296 мг, 1,07 ммоль) растворяли в метаноле (1 мл) и добавляли бикарбонат натрия (108 мг, 1,281 ммоль), затем метиламин (2M в THF, 0,640 мл, 1,281 ммоль). Смесь перемешивали в течение 2 часов при 70°C, затем охлаждали до 0°C. Добавляли боргидрид натрия (48,5 мг, 1,28 ммоль), и смесь перемешивали в течение ночи (подогревали до комнатной температуры). Добавляли каплю воды, и смесь концентрировали под вакуумом. Остаток растворяли в метаноле и подкисляли с помощью HCl. Полученный раствор загружали в картридж 5 g SCX и промывали, используя MeOH, затем элюировали с помощью 2 M NH3/MeOH с получением названного соединения в виде стеклообразного твердого вещества (256 мг, 82%). LCMS (метод C): RT=0,26 мин, m/z=293 [M+H]+.
Стадия 3: третбутил 2-(4-ацетилпиперазин-1-ил)-5-нитро-бензил(метил)карбамат:
Раствор дитретбутилдикарбоната (236 мг, 1,08 ммоль) в DCM (5 мл) добавляли к 1-(4-(2-((метиламино)метил)-4-нитрофенил)-пиперазин-1-ил)этанону (250 мг, 0,85 ммоль). Добавляли триэтиламин (0,274 мл, 1,965 ммоль), и смесь перемешивали при комнатной температуре в течение 90 минут. Смесь разбавляли водой, и органический слой собирали, используя картридж для разделения фаз, и концентрировали с получением названного соединения (334 мг, 92%) в виде стеклообразного твердого вещества. LCMS (метод C): RT=1,41 мин, m/z=393 [M+H]+.
Стадия 4: третбутил 2-(4-ацетилпиперазин-1-ил)-5-амино-бензил(метил)карбамат
Третбутил 2-(4-ацетилпиперазин-1-ил)-5-нитробензил(метил)-карбамат (332 мг, 0,85 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (300 мг, 98%) в виде желтого сиропа, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,77 мин, m/z=363 [M+H]+.
Стадия 5: 7-((4-(4-ацетилпиперазин-1-ил)-3-((метиламино)-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 2-(4-ацетилпиперазин-1-ил)-5-аминобензил(метил)-карбамат (82 мг, 0,22 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-оном (80 мг, 0,22 ммоль), как описано в примере 1, с получением названного соединения (32 мг, 28%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,87 (уш.с, 1H), 8,47 (с, 1H), 7,93 (уш.с, 1H), 7,67 (м, 4H), 7,08 (д, 1H), 4,97 (с, 2H), 3,79 (с, 2H), 3,57 (м, 4H), 3,12 (с, 3H), 2,78 (м, 4H), 2,27 (с, 3H), 2,04 (с, 3H). LCMS (метод C): RT=0,83 мин, m/z=569 [M+H]+.
Пример 144: 7-((4-(4-ацетилпиперазин-1-ил)-3-((диметил-амино)метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 1-(4-(2-((диметиламино)метил)-4-нитрофенил)-пиперазин-1-ил)этанон:
2-(4-ацетилпиперазин-1-ил)-5-нитробензальдегид (296 мг, 1,07 ммоль) растворяли в метаноле (1 мл) и добавляли бикарбонат натрия (108 мг, 1,281 ммоль), затем диметиламин (2M в THF, 0,640 мл, 1,281 ммоль). Смесь перемешивали в течение 2 часов при 70°C, затем охлаждали до 0 °C. Добавляли боргидрид натрия (48,5 мг, 1,28 ммоль), и смесь перемешивали в течение ночи (подогревали до комнатной температуры). Добавляли каплю воды, и смесь концентрировали под вакуумом. Остаток растворяли в метаноле и подкисляли с помощью HCl. Полученный раствор загружали в картридж 5 g SCX и промывали, используя MeOH, затем элюировали с помощью 2 M NH3/MeOH с получением названного соединения в виде стеклообразного твердого вещества (198 мг, 61%). LCMS (метод C): RT=0,26 мин, m/z=307 [M+H]+.
Стадия 2: 1-(4-(4-амино-2-((диметиламино)метил)фенил)-пиперазин-1-ил)этанон:
1-(4-(2-((диметиламино)метил)-4-нитрофенил)пиперазин-1-ил)-этанон (190 мг, 0,62 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (185 мг, 94%) в виде желтого сиропа, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,23 мин, m/z=277 [M+H]+.
Стадия 3: 7-((4-(4-ацетилпиперазин-1-ил)-3-((диметиламино)-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
1-(4-(4-Амино-2-((диметиламино)метил)фенил)пиперазин-1-ил)-этанон (62 мг, 0,22 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-оном (80 мг, 0,22 ммоль), как описано в примере 131, с получением названного соединения (33 мг, 25%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,85 (уш.с, 1H), 8,4 (с, 1H), 8,03 (уш.с, 1H), 7,64 (м, 2H), 7,48 (м, 2H), 7,04 (д, 1H), 4,97 (с, 2H), 3,56 (м, 4H), 3,45 (с, 2H), 3,13 (с, 3H), 2,82 (м, 2H), 2,78 (м, 2H), 2,18 (с, 6H), 2,04 (с, 3H). LCMS (метод C): RT=0,83 мин, m/z=583 [M+H]+.
Пример 145: N-(5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиперазин-1-ил)бензил)ацетамид

Стадия 1: третбутил 4-(2-(аминометил)-4-нитрофенил)-пиперазин-1-карбоксилат:
Третбутил 4-(2-(хлорметил)-4-нитрофенил)пиперазин-1-карбоксилат (200 мг, 0,56 ммоль) суспендировали в смеси метанола (2 мл) и гидроксида аммония (2 мл). Смесь перемешивали в течение ночи, затем концентрировали под вакуумом. Остаток подвергали хроматографии (картридж 10 g Si; элюировали 0-100% этилацетат/циклогексан, затем 0-10% метанол/этилацетат) с получением названного соединения (95 мг, 50%) в виде стеклообразного твердого вещества, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,90 мин, m/z=337 [M+H]+.
Стадия 2: третбутил 4-(2-(ацетамидометил)-4-аминофенил)-пиперазин-1-карбоксилат:
Третбутил 4-(2-(аминометил)-4-нитрофенил)пиперазин-1-карбоксилат (90 мг, 0,27 ммоль) растворяли в тетрагидрофуране (2 мл). Добавляли триэтиламин (0,09 мл, 0,67 ммоль), затем уксусный ангидрид (0,038 мл, 0,40 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и концентрировали. Остаток подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (74 мг, 79%) в виде желтого масла, которое непосредственно использовали без дополнительной очистки. LCMS (метод C): RT=0,79 мин, m/z=349 [M+H]+.
Стадия 3: N-(5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиперазин-1-ил)бензил)ацетамид:
Третбутил 4-(2-(ацетамидометил)-4-аминофенил)пиперазин-1-карбоксилат (98 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения (36 мг, 23%) в виде желтовато-белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,84 (уш.с, 1H), 8,45 (с, 1H), 8,20 (т, 1H), 7,65 (м, 4H), 7,46 (м, 1H), 7,05 (д, 1H), 4,97 (с, 2H), 4,33 (с, 2H), 3,09 (с, 3H), 2,84 (м, 4H), 2,73 (м, 4H), 1,89 (с, 3H). LCMS (метод C): RT=0,72 мин, m/z=556 [M+H]+.
Пример 146: 3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидо-тиоморфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
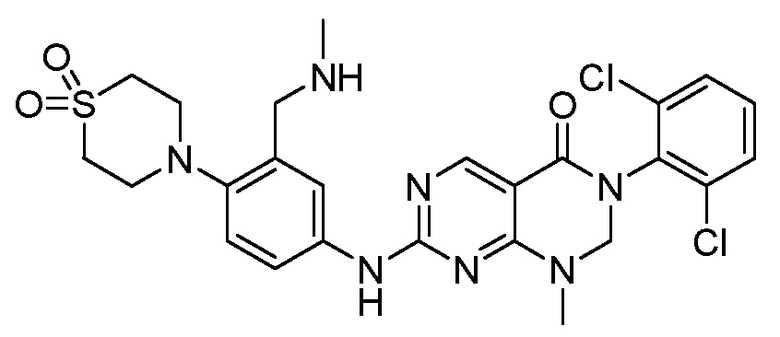
Стадия 1: 2-(1,1-диоксидотиоморфолино)-5-нитробензальдегид:
Суспензию 2-фтор-5-нитробензальдегида (2 г, 11,83 ммоль), тиоморфолина 1,1-диоксида (1,599 г, 11,83 ммоль) и карбоната калия (2,452 г, 17,74 ммоль) в безводном DMF (10 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, суспендировали в 1M растворе HCl (50 мл), выделяли фильтрацией, воду отсасывали, и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (2,68 г, 80%). 1H ЯМР (300 МГц, DMSO-d6): δ 10,11 (с, 1H), 8,54 (д, 1H), 8,34 (дд, 1H), 7,43 (д, 1H), 3,69-3,80 (м, 4H), 3,36-3,45 (м, 4H). LCMS (метод C): RT=0,95 мин, m/z=285 [M+H]+.
Стадия 2: 4-(2-((метиламино)метил)-4-нитрофенил)тио-морфолина 1,1-диоксид:
К суспензии 2-(1,1-диоксидотиоморфолино)-5-нитро-бензальдегида (800 мг, 2,81 ммоль) и бикарбоната натрия (473 мг, 5,63 ммоль) в метаноле (6 мл) добавляли метиламин (2M в метаноле, 1,688 мл, 3,38 ммоль), и реакционную смесь нагревали до 70°C в течение 2 часов. Смесь затем охлаждали до 0°C, затем добавляли боргидрид натрия (128 мг, 3,38 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли дополнительное количество метиламина (2M в метаноле, 0,492 мл, 0,985 ммоль) и боргидрида натрия (38,8 мг, 1,688 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили несколькими каплями воды, затем концентрировали досуха при пониженном давлении. Остаток растворяли в дихлорметане (20 мл), промывали солевым раствором (20 мл) и пропускали через фазовый сепаратор. Органическую фазу концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (500 мг, 59%). 1H ЯМР (300 МГц, CDCl3): δ 8,30 (д, 1H), 8,12 (дд, 1H), 7,15 (д, 1H), 3,79 (с, 2H), 3,60-3,69 (м, 4H), 3,20-3,29 (м, 4H), 2,50 (с, 3H). LCMS (метод C): RT=0,36 мин, m/z=300 [M+H]+.
Стадия 3: третбутил 2-(1,1-диоксидотиоморфолино)-5-нитро-бензил(метил)карбамат:
К раствору 4-(2-((метиламино)метил)-4-нитрофенил)тио-морфолина 1,1-диоксида (500 мг, 1,670 ммоль) и триэтиламина (0,512 мл, 3,67 ммоль) в безводном THF (5 мл) добавляли дитретбутилдикарбонат (0,427 мл, 1,837 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли солевым раствором (20 мл) и экстрагировали этилацетатом (3×15 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и подвергали хроматографии (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 0:100) с получением названного соединения в виде желтого масла (350 мг, 53%). 1H ЯМР (400 МГц, CDCl3): δ 8,16 (дд, 1H), 8,04 (уш.с, 1H), 7,23 (д, 1H), 4,53 (уш.с, 2H), 3,49 (дд, 4H), 3,26 (дд, 4H), 2,88 (с, 3H), 1,52 (уш.с, 9H). LCMS (метод C): RT=1,43 мин, m/z=400 [M+H]+.
Стадия 4: третбутил 5-амино-2-(1,1-диоксидотио-морфолино)бензил(метил)карбамат:
Раствор третбутил 2-(1,1-диоксидотиоморфолино)-5-нитро-бензил(метил)карбамата (350 мг, 0,876 ммоль) в метаноле (40 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 20 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (260 мг, 80%). 1H ЯМР (300 МГц, CDCl3): δ 6,99 (д, 1H), 6,57 (дд, 1H), 6,47 (уш.с, 1H), 4,47 (уш.с, 2H), 3,62 (уш.с, 2H), 3,24-3,36 (м, 4H), 3,11-3,24 (м, 4H), 2,70-2,90 (уш.м, 3H), 1,39-1,56 (уш.м, 9H). LCMS (метод C): RT=0,80 мин, m/z=370 [M+H]+.
Стадия 5: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(1,1-диоксидотиоморфолино)бензил(метил)карбамат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,423 ммоль) подвергали взаимодействию с третбутил 5-амино-2-(1,1-диоксидо-тиоморфолино)бензил(метил)карбаматом (156 мг, 0,423 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (170 мг, 59%). 1H ЯМР (300 МГц, CDCl3): δ 8,68 (с, 1H), 8,10 (уш.с, 1H), 7,58 (дд, 1H), 7,37-7,47 (м, 3H), 7,27 (дд, 1H), 7,16 (д, 1H), 4,88 (с, 2H), 4,54 (уш.с, 2H), 3,30-3,43 (м, 4H), 3,15-3,25 (м, 4H), 3,15 (с, 3H), 2,70-2,90 (уш.м, 3H), 1,47 (уш.с, 9H). LCMS (метод C): RT=1,46 мин, m/z=676 [M+H]+.
Стадия 6: 3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидотио-морфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(1,1-диоксидо-тиоморфолино)бензил(метил)карбамат (170 мг, 0,251 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (121 мг, 84%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,46 (с, 1H), 7,92 (уш.с, 1H), 7,65 (д, 2H), 7,56 (дд, 1H), 7,48 (т, 1H), 7,13 (д, 1H), 4,97 (с, 2H), 3,69 (с, 2H), 3,29-3,33 (м, 4H), 3,21-3,29 (м, 4H), 3,12 (с, 3H), 2,33 (с, 3H). LCMS (метод C): RT=0,83 мин, m/z=576 [M+H]+.
Пример 147: 3-(2,6-дихлорфенил)-7-((4-(4,4-дифторпиперидин-1-ил)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: 2-(4,4-дифторпиперидин-1-ил)-5-нитробензальдегид:
Суспензию 2-фтор-5-нитробензальдегида (1 г, 5,91 ммоль), 4,4-дифторпиперидина гидрохлорида (0,932 г, 5,91 ммоль) и карбоната калия (1,635 г, 11,83 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (40 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали, и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (1,52 г, 95%). 1H ЯМР (300 МГц, CDCl3): δ 10,09 (с, 1H), 8,65 (д, 1H), 8,33 (дд, 1H), 7,13 (д, 1H), 3,41 (дд, 4H), 2,17-2,34 (м, 4H). LCMS (метод C): RT=1,47 мин, m/z=271 [M+H]+.
Стадия 2: 1-(2-(4,4-дифторпиперидин-1-ил)-5-нитрофенил)-N-метилметанамин:
К суспензии 2-(4,4-дифторпиперидин-1-ил)-5-нитро-бензальдегида (700 мг, 2,59 ммоль) и бикарбоната натрия (435 мг, 5,18 ммоль) в метаноле (5 мл) добавляли метиламин (2M в метаноле, 1,554 мл, 3,11 ммоль), и реакционную смесь нагревали до 70°C в течение 2 часов. Смесь затем охлаждали до 0°C, затем добавляли боргидрид натрия (118 мг, 3,11 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили несколькими каплями воды, затем концентрировали досуха при пониженном давлении. Остаток растворяли в дихлорметане (20 мл), промывали солевым раствором (20 мл) и пропускали через фазовый сепаратор. Органическую фазу концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого масла, которое превращалось в твердое вещество при стоянии (611 мг, 83%). 1H ЯМР (300 МГц, CDCl3): δ 8,32 (д, 1H), 8,09 (дд, 1H), 7,09 (д, 1H), 3,78 (с, 2H), 3,18 (дд, 4H), 2,49 (с, 3H), 2,17 (тт, 4H). LCMS (метод C): RT=0,74 мин, m/z=286 [M+H]+.
Стадия 3: третбутил 2-(4,4-дифторпиперидин-1-ил)-5-нитро-бензил(метил)карбамат:
К раствору 1-(2-(4,4-дифторпиперидин-1-ил)-5-нитрофенил)-N-метилметанамина (611 мг, 2,142 ммоль) и триэтиламина (0,657 мл, 4,71 ммоль) в безводном THF (6 мл) добавляли дитретбутил-дикарбонат (0,547 мл, 2,356 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли солевым раствором (20 мл) и экстрагировали этилацетатом (3×15 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 70:30) с получением названного соединения в виде желтого масла (850 мг, количественный выход). 1H ЯМР (400 МГц, CDCl3): δ 8,11 (дд, 1H), 7,99 (уш.с, 1H), 7,14 (д, 1H), 4,39-4,60 (уш.м, 2H), 3,08 (т, 4H), 2,75-2,94 (уш.м, 3H), 2,08-2,26 (м, 4H), 1,38-1,59 (м, 9H). LCMS (метод C): RT=1,85 мин, m/z=386 [M+H]+.
Стадия 4: третбутил 5-амино-2-(4,4-дифторпиперидин-1-ил)-бензил(метил)карбамат:
Раствор третбутил 2-(4,4-дифторпиперидин-1-ил)-5-нитро-бензил(метил)карбамата (850 мг, 2,205 ммоль) в метаноле (60 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 20 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (670 мг, 85%). 1H ЯМР (300 МГц, CDCl3): δ 6,96 (д, 1H), 6,56 (дд, 1H), 6,49 (уш.д, 1H), 4,48 (д, 2H), 3,55 (с, 2H), 2,88 (т, 4H), 2,80 (д, 3H), 1,98-2,18 (м, 4H), 1,36-1,56 (м, 9H). LCMS (метод C): RT=1,26 мин, m/z=356 [M+H]+.
Стадия 5: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(4,4-дифторпиперидин-1-ил)бензил(метил)карбамат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 5-амино-2-(4,4-дифтор-пиперидин-1-ил)бензил(метил)карбаматом (101 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (120 мг, 64%). 1H ЯМР (300 МГц, CDCl3) δ 8,70 (с, 1H), 7,77 (уш.с, 1H), 7,55 (уш.д, 1H), 7,43 (дд, 2H), 7,33-7,43 (уш.м, 1H), 7,28 (дд, 1H), 7,12 (д, 1H), 4,87 (с, 2H), 4,54 (уш.д, 2H), 3,17 (с, 3H), 2,90-3,00 (м, 4H), 2,71-2,90 (уш.м, 3H), 2,00-2,21 (м, 4H), 1,35-1,57 (уш.м, 9H). LCMS (метод C): RT=1,91 мин, m/z=662 [M+H]+.
Стадия 6: 7-((4-((1R,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(4,4-дифтор-пиперидин-1-ил)бензил(метил)карбамат (120 мг, 0,181 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (94 мг, 92%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,80 (уш.с, 1H), 8,46 (с, 1H), 7,93 (уш.с, 1H), 7,65 (д, 2H), 7,54 (дд, 1H), 7,48 (т, 1H), 7,08 (д, 1H), 4,97 (с, 2H), 3,68 (с, 2H), 3,12 (с, 3H), 2,92-3,02 (м, 4H), 2,33 (с, 3H), 2,02-2,18 (м, 4H). LCMS (метод C): RT=1,05 мин, m/z=562 [M+H]+.
Пример 148: 3-(2,6-дихлорфенил)-7-((4-((2S,6R)-2,6-диметил-морфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
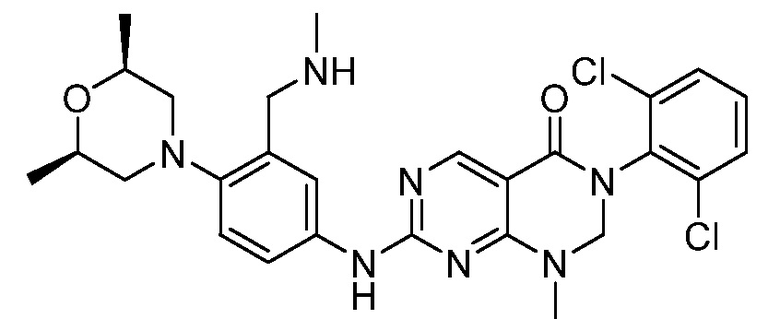
Стадия 1: 2-((2S,6R)-2,6-диметилморфолино)-5-нитро-бензальдегид:
Суспензию 2-фтор-5-нитробензальдегида (2 г, 11,83 ммоль), цис-2,6-диметилморфолина (1,362 г, 11,83 ммоль) и карбоната калия (2,452 г, 17,74 ммоль) в безводном DMF (20 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (70 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали, и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (2,94 г, 94%). 1H ЯМР (300 МГц, CDCl3): δ 10,07 (с, 1H), 8,63 (д, 1H), 8,31 (дд, 1H), 7,06 (д, 1H), 3,85-3,99 (м, 2H), 3,30 (д, 2H), 2,82 (дд, 2H), 1,25 (д, 6H). LCMS (метод C): RT=1,43 мин, m/z=265 [M+H]+.
Стадия 2: 1-(2-((2S,6R)-2,6-диметилморфолино)-5-нитро-фенил)-N-метилметанамин:
К суспензии 2-((2S,6R)-2,6-диметилморфолино)-5-нитро-бензальдегида (800 мг, 3,03 ммоль) и бикарбоната натрия (509 мг, 6,05 ммоль) в метаноле (6 мл) добавляли метиламин (2M в метаноле, 1,816 мл, 3,63 ммоль), и реакционную смесь нагревали до 70°C в течение 2 часов. Смесь затем охлаждали до 0°C, затем добавляли боргидрид натрия (137 мг, 3,63 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили несколькими каплями воды, затем концентрировали досуха при пониженном давлении. Остаток растворяли в дихлорметане (20 мл), промывали солевым раствором (20 мл) и пропускали через фазовый сепаратор. Органическую фазу концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого масла (880 мг, количественный выход). 1H ЯМР (300 МГц, CDCl3): δ 8,29 (д, 1H), 8,08 (дд, 1H), 7,02 (д, 1H), 3,79-3,93 (м, 2H), 3,77 (с, 2H), 3,17 (д, 2H), 2,53 (дд, 2H), 2,46 (с, 3H), 1,23 (д, 6H). LCMS (метод C): RT=0,62 мин, m/z=280 [M+H]+.
Стадия 3: третбутил 2-((2S,6R)-2,6-диметилморфолино)-5-нитробензил(метил)карбамат:
К раствору 1-(2-((2S,6R)-2,6-диметилморфолино)-5-нитро-фенил)-N-метилметанамина (880 мг, 3,15 ммоль) и триэтиламина (0,966 мл, 6,93 ммоль) в безводном THF (9 мл) добавляли дитретбутилдикарбонат (0,805 мл, 3,47 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли солевым раствором (40 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 70:30) с получением названного соединения в виде желтого масла (1,06 г, 89%). 1H ЯМР (400 МГц, CDCl3): δ 8,11 (дд, 1H), 7,99 (уш.с, 1H), 7,07 (д, 1H), 4,39-4,58 (м, 2H), 3,85 (уш.с, 2H), 2,72-3,03 (м, 5H), 2,54 (т, 2H), 1,41-1,59 (м, 9H), 1,23 (д, 6H). LCMS (метод C): RT=1,87 мин, m/z=380 [M+H]+.
Стадия 4: третбутил 5-амино-2-((2S,6R)-2,6-диметил-морфолино)бензил(метил)карбамат:
Раствор третбутил 2-((2S,6R)-2,6-диметилморфолино)-5-нитро-бензил(метил)карбамата (1,06 г, 2,79 ммоль) в метаноле (60 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 20 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (840 мг, 86%). 1H ЯМР (300 МГц, CDCl3): δ 6,93 (д, 1H), 6,57 (дд, 1H), 6,50 (уш.д, 1H), 4,40-4,58 (м, 2H), 3,71-3,87 (м, 2H), 3,54 (уш.с, 2H), 2,61-2,88 (м, 5H), 2,41 (т, 2H), 1,37-1,55 (м, 9H), 1,18 (д, 6H). LCMS (метод C): RT=1,08 мин, m/z=350 [M+H]+.
Стадия 5: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-((2S,6R)-2,6-диметилморфолино)бензил(метил)карбамат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 5-амино-2-((2S,6R)-2,6-диметилморфолино)бензил(метил)карбаматом (99 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (109 мг, 59%). 1H ЯМР (300 МГц, CDCl3): δ 8,69 (с, 1H), 7,78 (уш.с, 1H), 7,54 (уш.д, 1H), 7,43 (дд, 2H), 7,32-7,42 (уш.м, 1H), 7,28 (дд, 1H), 7,08 (уш.д, 1H), 4,86 (с, 2H), 4,55 (уш.д, 2H), 3,75-3,90 (м, 2H), 3,16 (с, 3H), 2,67-2,91 (м, 5H), 2,46 (т, 2H), 1,35-1,56 (м, 9H), 1,20 (д, 6H). LCMS (метод C): RT=1,88 мин, m/z=656 [M+H]+.
Стадия 6: 3-(2,6-дихлорфенил)-7-((4-((2S,6R)-2,6-диметил-морфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-((2S,6R)-2,6-диметилморфолино)бензил(метил)карбамат (109 мг, 0,166 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (64 мг, 69%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,78 (уш.с, 1H), 8,46 (с, 1H), 7,90 (уш.с, 1H), 7,65 (д, 2H), 7,55 (дд, 1H), 7,48 (т, 1H), 7,02 (д, 1H), 4,96 (с, 2H), 3,72 (септ, 2H), 3,66 (с, 2H), 3,12 (с, 3H), 2,94 (д, 2H), 2,24-2,38 (м, 5H), 1,10 (д, 6H). LCMS (метод C): RT=0,97 мин, m/z=556 [M+H]+.
Пример 149: 3-(2,6-дихлорфенил)-7-((3-((диметиламино)-метил)-4-(1,1-диоксидотиоморфолино)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
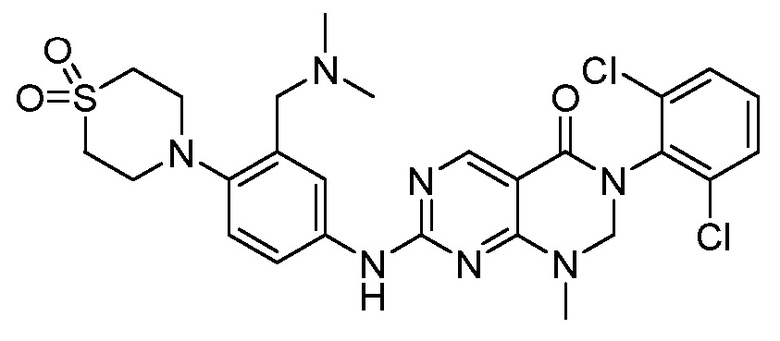
3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидотиоморфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (66 мг, 0,114 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (58 мг, 86%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,85 (уш.с, 1H), 8,47 (с, 1H), 7,99 (уш.с, 1H), 7,65 (д, 2H), 7,55 (д, 1H), 7,48 (дд, 1H), 7,15 (д, 1H), 4,97 (с, 2H), 3,44 (с, 2H), 3,32-3,39 (м, 4H), 3,21-3,30 (м, 4H), 3,13 (с, 3H), 2,20 (с, 6H). LCMS (метод C): RT=0,85 мин, m/z=590 [M+H]+.
Пример 150: 3-(2,6-дихлорфенил)-7-((4-(4,4-дифторпиперидин-1-ил)-3-((диметиламино)метил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-дихлорфенил)-7-((4-(4,4-дифторпиперидин-1-ил)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (45 мг, 0,80 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (32 мг, 69%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,82 (уш.с, 1H), 8,46 (с, 1H), 8,03 (уш.с, 1H), 7,64 (д, 2H), 7,53 (д, 1H), 7,48 (т, 1H), 7,09 (д, 1H), 4,97 (с, 2H), 3,42 (с, 2H), 3,13 (с, 3H), 2,92-3,04 (м, 4H), 2,20 (с, 6H), 2,01-2,17 (м, 4H). LCMS (метод C): RT=1,07 мин, m/z=576 [M+H]+.
Пример 151: 3-(2,6-дихлорфенил)-7-((3-((диметиламино)-метил)-4-((2S,6R)-2,6-диметилморфолино)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-дихлорфенил)-7-((4-((2S,6R)-2,6-диметилморфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (35 мг, 0,63 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (12 мг, 33%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,80 (уш.с, 1H), 8,45 (с, 1H), 8,01 (уш.с, 1H), 7,65 (д, 2H), 7,54 (дд, 1H), 7,48 (дд, 1H), 7,02 (д, 1H), 4,96 (с, 2H), 3,74 (септ, 2H), 3,41 (с, 2H), 3,13 (с, 3H), 3,02 (д, 2H), 2,32 (т, 2H), 2,19 (с, 6H), 1,10 (д, 6H). LCMS (метод C): RT=0,99 мин, m/z=570 [M+H]+.
Пример 152: 3-(2,6-дихлорфенил)-7-((4-фтор-3-((метиламино)-метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
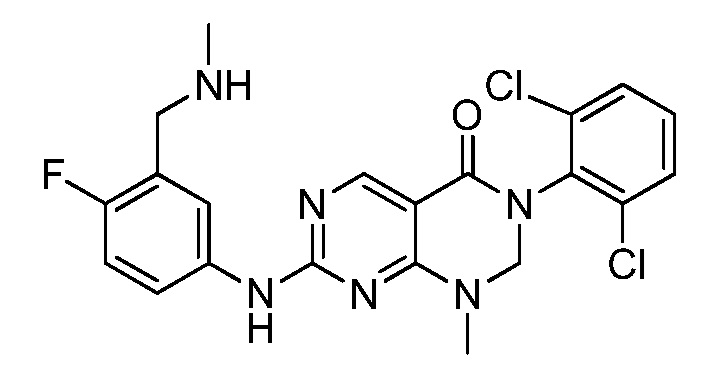
Стадия 1: третбутил 2-фтор-5-нитробензил(метил)карбамат:
2-Фтор-5-нитробензальдегид (1 г, 5,91 ммоль) растворяли в метиламине (2 M в метаноле, 3,25 мл, 6,50 ммоль) и этаноле (7 мл), и смесь перемешивали в течение 1 часа. Добавляли боргидрид натрия (0,447 г, 11,83 ммоль), и смесь перемешивали в течение 1 часа, затем концентрировали под вакуумом. Остаток растворяли в этилацетате и промывали водой и солевым раствором, затем концентрировали до желтого масла. Это масло растворяли в дихлорметане (15 мл) и добавляли DIPEA (1,343 мл, 7,69 ммоль), затем дитретбутилдикарбонат (1,42 г, 6,50 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Остаток подвергали хроматографии (картридж 25 g Si; элюировали 0-50% EtOAc/циклогексан) с получением названного соединения (0,90 г, 54%) в виде желтого масла. ¹H ЯМР (300 МГц, CDCl3): δ 8,17 (м, 2H), 7,24 (м, 1H), 4,50 (м, 2H), 2,93 (м, 3H), 1,49 (с, 9H). LCMS (метод C): RT=1,66 мин, m/z=185 [M+H-Boc]+.
Стадия 2: третбутил 5-амино-2-фторбензил(метил)карбамат:
Третбутил 2-фтор-5-нитробензил(метил)карбамат (180 мг, 0,63 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол). Неочищенный продукт подвергали хроматографии (картридж 10 g Si; элюировали 0-100% EtOAc/циклогексан) с получением названного соединения (105 мг, 65%) в виде бесцветного масла. LCMS (метод C): RT=1,05 мин, m/z=199 [M+H-tBu]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((4-фтор-3-((метиламино)-метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-амино-2-фторбензил(метил)карбамат (72 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения (45 мг, 34%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,48 (с, 1H), 7,98 (уш.с, 1H), 7,64 (д, 2H), 7,47 (м, 2H), 7,09 (т, 1H), 4,98 (с, 2H), 3,65 (с, 2H), 3,12 (с, 3H), 2,28 (с, 3H), 2,03 (уш.с, 1H). LCMS (метод C): RT=0,84 мин, m/z=461 [M+H]+.
Пример 153: 3-(2,6-дихлорфенил)-7-((4-(диметиламино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 5-амино-2-(диметиламино)бензил(метил)-карбамат:
Третбутил 2-фтор-5-нитробензил(метил)карбамат (190 мг, 0,668 ммоль) растворяли в 2 M растворе диметиламина в THF (3 мл, 6,00 ммоль). Смесь нагревали при 140°C в течение 1 часа при микроволновом излучении. Раствор концентрировали под вакуумом, остаток распределяли между водой и DCM, и органический слой отделяли, используя картридж для разделения фаз, затем концентрировали под вакуумом. Остаток подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения в виде бесцветного масла (110 мг, 59%), которое использовали без дополнительной очистки. LCMS (метод C): RT 0,76 мин, m/z 280 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-7-((4-(диметиламино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-амино-2-(диметиламино)бензил(метил)карбамат (79 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения (20 мг, 7%) в виде желтовато-белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,79 (уш.с, 1H), 8,46 (с, 1H), 7,64 (уш.с, 1H), 7,53 (м, 2H), 7,48 (м, 2H), 7,05 (д, 1H), 4,97 (с, 2H), 3,72 (с, 2H), 3,12 (с, 3H), 2,60 (с, 6H), 2,34 (с, 3H). LCMS (метод C): RT=0,92 мин, m/z=486 [M+H]+.
Пример 154: 3-(2,6-дихлорфенил)-7-((3-((диметиламино)-метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
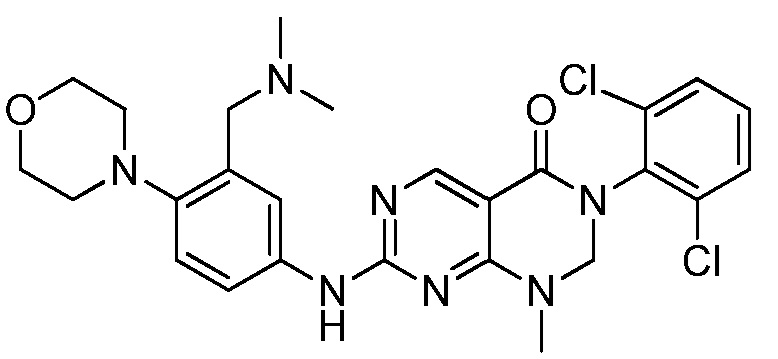
Стадия 1: 3-((диметиламино)метил)-4-морфолиноанилин:
2-Морфолино-5-нитробензальдегид (0,5 г, 2,117 ммоль) суспендировали в метаноле (6 мл). Добавляли бикарбонат натрия (0,356 г, 4,23 ммоль), затем 2 M раствор диметиламина в метаноле (1,270 мл, 2,54 ммоль). Смесь нагревали при 80°C в течение 3 часов, затем охлаждали до комнатной температуры. Добавляли боргидрид натрия (0,096 г, 2,54 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию прерывали путем добавления нескольких капель воды, и раствор концентрировали под вакуумом. Остаток распределяли между водой и DCM, и органический слой отделяли, используя картридж для разделения фаз, и концентрировали под вакуумом. Остаток подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения в виде белого твердого вещества (160 мг, 32%), которое использовали без дополнительной очистки. LCMS (метод C): RT=0,24 мин, m/z=236 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-7-((3-((диметиламино)метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-((Диметиламино)метил)-4-морфолиноанилин (66 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 131, с получением названного соединения (75 мг, 49%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,84 (уш.с, 1H), 8,46 (с, 1H), 8,01 (уш.с, 1H), 7,64 (м, 2H), 7,50 (м, 2H), 7,06 (д, 1H), 4,97 (с, 2H), 3,72 (м, 4H), 3,43 (с, 2H), 3,13 (с, 3H), 2,86 (м, 4H), 2,19 (с, 6H). LCMS (метод C): RT=0,88 мин, m/z=542 [M+H]+.
Пример 155: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метил-амино)метил)-4-(пиридин-4-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

Стадия 1: 2-бром-5-нитробензальдегид:
Смесь концентрированной серной кислоты (17,01 мл) и дымящейся азотной кислоты (2,250 мл) охлаждали до 5°C, затем добавляли по каплям 2-бромбензальдегид (3,15 мл, 27,0 ммоль) в течение 30 минут. Смесь затем подогревали до комнатной температуры и перемешивали в течение 60 минут. Реакционную смесь выливали в ледяную воду (200 мл), и осажденное твердое вещество выделяли фильтрацией, промывали водой, и воду отсасывали с получением бледно-желтого твердого вещества. Это вещество перекристаллизовывали из смеси 50:50 циклогексан:этилацетат (30 мл) с получением названного соединения в виде желтовато-белого твердого вещества (3,39 г, 55%). 1H ЯМР (300 МГц, CDCl3): δ 10,39 (с, 1H), 8,72 (д, 1H), 8,29 (дд, 1H), 7,89 (д, 1H).
Стадия 2: 1-(2-бром-5-нитрофенил)-N-метилметанамин:
К суспензии 2-бром-5-нитробензальдегида (2,00 г, 8,70 ммоль) в метаноле (22,53 мл) добавляли метиламин (2M в метаноле, 13,04 мл, 26,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 60 минут. После охлаждения до 0°C, добавляли боргидрид натрия (0,658 г, 17,39 ммоль), и смесь подогревали до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь гасили путем добавления нескольких капель воды и концентрировали досуха при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали водой (2×30 мл) и солевым раствором (30 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде оранжевого твердого вещества (1,84 г, 86%). 1H ЯМР (400 МГц, CDCl3): δ 8,31 (д, 1H), 7,98 (дд, 1H), 7,72 (д, 1H), 3,90 (с, 2H), 2,50 (с, 3H). LCMS (метод C): RT=0,33 мин, m/z=245 [M+H]+.
Стадия 3: третбутил 2-бром-5-нитробензил(метил)карбамат:
К раствору 1-(2-бром-5-нитрофенил)-N-метилметанамина (1,84 г, 7,51 ммоль) в безводном THF (18 мл) добавляли триэтиламин (2,302 мл, 16,52 ммоль) и дитретбутилдикарбонат (1,917 мл, 8,26 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали насыщенным раствором бикарбоната натрия (50 мл) и солевым раствором (50 мл). Органическую фазу сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и суспендировали в циклогексане с получением названного соединения в виде желтого твердого вещества. Фильтрат концентрировали и подвергали хроматографии (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 100:0 до 80:20) с получением дополнительного количества названного соединения. Суммарный выход (2,34 г, 90%). 1H ЯМР (300 МГц, CDCl3): δ 7,92-8,11 (м, 2H), 7,74 (д, 1H), 4,47-4,63 (м, 2H), 2,96 (с, 3H), 1,33-1,61 (м, 9H). LCMS (метод C): RT=1,81 мин, m/z=289 [M-tBu+H]+.
Стадия 4: третбутил метил(5-нитро-2-(пиридин-4-ил)бензил)-карбамат:
Суспензию третбутил 2-бром-5-нитробензил(метил)карбамата (200 мг, 0,579 ммоль), пиридин-4-илбороновой кислоты (107 мг, 0,869 ммоль) и 2M раствора карбоната натрия (869 мкл, 1,738 ммоль) в 1,4-диоксане (1745 мкл) дегазировали путем барботирования азота в течение 10 минут, затем добавляли аддукт PdCl2(dppf)-дихлорметан (47,3 мг, 0,058 ммоль). Реакционную смесь нагревали до 120°C при микроволновом излучении в течение 15 минут, разбавляли водой (10 мл) и экстрагировали этилацетатом (3×6 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж GraceResolv диоксид кремния 24g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 0:100) с получением названного соединения в виде желтого масла (161 мг, 81%). 1H ЯМР (400 МГц, CDCl3): δ 8,73 (с, 2H), 8,09-8,28 (м, 2H), 7,39 (д, 1H), 7,23 (с, 2H), 4,32-4,51 (уш.м, 2H), 2,76 (с, 3H), 1,35-1,55 (уш.м, 9H). LCMS (метод C): RT=1,25 мин, m/z=344 [M+H]+.
Стадия 5: третбутил 5-амино-2-(пиридин-4-ил)бензил(метил)-карбамат:
Раствор третбутил метил(5-нитро-2-(пиридин-4-ил)бензил)-карбамата (241 мг, 0,702 ммоль) в метаноле (25 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 10 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого масла (206 мг, 94%). 1H ЯМР (300 МГц, CDCl3): δ 8,60 (д, 2H), 7,19 (д, 2H), 7,03 (д, 1H), 6,65 (дд, 1H), 6,60 (уш.с, 1H), 4,38 (уш.д, 2H), 3,81 (уш.с, 2H), 2,67 (уш.д, 3H), 1,44 (с, 9H). LCMS (метод C): RT=0,70 мин, m/z=314 [M+H]+.
Стадия 6: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиридин-4-ил)бензил(метил)карбамат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 5-амино-2-(пиридин-4-ил)бензил(метил)карбаматом (89 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (14 мг, 8%). 1H ЯМР (300 МГц, CDCl3): δ 8,74 (с, 1H), 8,64 (уш.с, 2H), 7,73 (уш.с, 1H), 7,52 (уш.с, 2H), 7,45 (д, 2H), 7,29 (дд, 1H), 7,15-7,27 (м, 3H), 4,90 (с, 2H), 4,31-4,56 (м, 2H), 3,20 (с, 3H), 2,51-2,84 (м, 3H), 1,27-1,52 (м, 9H). LCMS (метод C): RT=1,22 мин, m/z=620 [M+H]+.
Стадия 7: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиридин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиридин-4-ил)бензил(метил)карбамат (14 мг, 0,023 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (5 мг, 43%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,05 (с, 1H), 8,60 (дд, 2H), 8,51 (с, 1H), 8,19 (уш.с, 1H), 7,72 (д, 1H), 7,65 (д, 2H), 7,45-7,52 (м, 3H), 7,24 (д, 1H), 5,00 (с, 2H), 3,57 (с, 2H), 3,17 (с, 3H), 2,26 (с, 3H). LCMS (метод C): RT=0,71 мин, m/z=520 [M+H]+.
Пример 156: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метил-амино)метил)-4-(пиридин-3-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
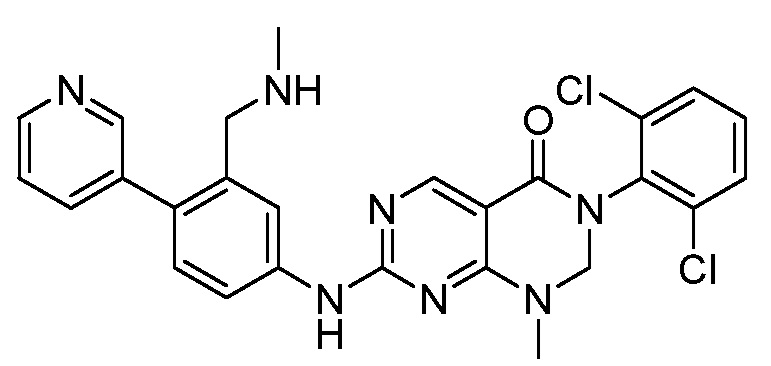
Стадия 1: третбутил метил(5-нитро-2-(пиридин-3-ил)бензил)-карбамат:
Третбутил 2-бром-5-нитробензил(метил)карбамат (300 мг, 0,869 ммоль) подвергали взаимодействию с пиридин-3-илбороновой кислотой (160 мг, 1,30 ммоль), следуя методике примера 155, с получением названного соединения в виде желтого твердого вещества (255 мг, 85%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (д, 1H), 8,57 (д, 1H), 8,10-8,28 (м, 2H), 7,57-7,71 (м, 1H), 7,34-7,49 (м, 2H), 4,33-4,51 (уш.м, 2H), 2,76 (с, 3H), 1,31-1,57 (уш.м, 9H). LCMS (метод C): RT=1,30 мин, m/z=344 [M+H]+.
Стадия 2: третбутил 5-амино-2-(пиридин-3-ил)бензил(метил)-карбамат:
Третбутил метил(5-нитро-2-(пиридин-3-ил)бензил)карбамат (255 мг, 0,743 ммоль) гидрировали, следуя методике примера 155, с получением названного соединения в виде желтого масла (243 мг, количественный выход). LCMS (метод C): RT=0,72 мин, m/z=314 [M+H]+.
Стадия 3: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиридин-3-ил)бензил(метил)карбамат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,283 ммоль) подвергали взаимодействию с третбутил 5-амино-2-(пиридин-3-ил)-бензил(метил)карбаматом (89 мг, 0,283 ммоль), следуя методике примера 31, с получением названного соединения в виде коричневого твердого вещества (14 мг, 8%). LCMS (метод C): RT=1,36 мин, m/z=620 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиридин-3-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиридин-3-ил)-бензил(метил)карбамат (14 мг, 0,23 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде розового твердого вещества (7 мг, 60%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,03 (с, 1H), 8,62 (д, 1H), 8,55 (дд, 1H), 8,51 (с, 1H), 8,18 (уш.с, 1H), 7,87 (дт, 1H), 7,70 (дд, 1H), 7,66 (д, 2H), 7,49 (дд, 1H), 7,45 (дд, 1H), 7,22 (д, 1H), 5,00 (с, 2H), 3,54 (с, 2H), 3,17 (с, 3H), 2,24 (с, 3H). LCMS (метод C): RT=0,78 мин, m/z=520 [M+H]+.
Пример 157: 7-((4-бром-3-((метиламино)метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
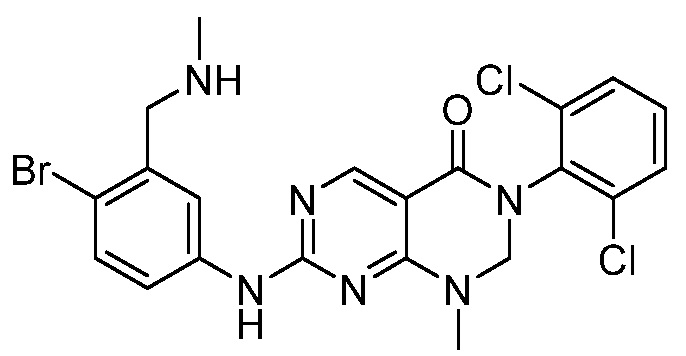
Стадия 1: третбутил 5-амино-2-бромбензил(метил)карбамат:
К раствору третбутил 2-бром-5-нитробензил(метил)карбамата (0,72 г, 2,086 ммоль) в метаноле (11,99 мл) и тетрагидрофуране (2,397 мл) добавляли цинковую пыль (1,364 г, 20,86 ммоль), затем добавляли порциями хлорид аммония (2,231 г, 41,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 60 минут, затем нагревали до 40°C в течение 4 часов. Реакционную смесь концентрировали досуха при пониженном давлении и распределяли между насыщенным раствором карбоната натрия (30 мл) и этилацетатом (30 мл). Смесь интенсивно перемешивали в течение 10 минут, фильтровали через целит® и промывали этилацетатом (2×30 мл). Объединенные органические фазы промывали насыщенным раствором карбоната натрия (2×20 мл), водой (2×20 мл) и солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде бесцветного масла (544 мг, 83%). 1H ЯМР (300 МГц, CDCl3): δ 7,27 (д, 1H), 6,40-6,57 (м, 2H), 4,34-4,52 (м, 2H), 3,37 (уш.с, 2H), 2,76-2,94 (м, 3H), 1,36-1,54 (м, 9H). LCMS (метод C): RT=1,55 мин, m/z=259 [M-tBu+H]+.
Стадия 2: третбутил 2-бром-5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-бензил(метил)карбамат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (300 мг, 0,846 ммоль) подвергали взаимодействию с третбутил 5-амино-2-бромбензил-(метил)карбаматом (267 мг, 0,846 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (223 мг, 42%). 1H ЯМР (300 МГц, CDCl3): δ 8,71 (с, 1H), 7,32-7,61 (м, 6H), 7,29 (дд, 1H), 4,89 (с, 2H), 4,45-4,60 (уш.м, 2H), 3,18 (с, 3H), 2,80-2,96 (уш.м, 3H), 1,31-1,57 (уш.м, 9H). LCMS (метод C): RT=1,98 мин, m/z=621 [M+H]+.
Стадия 3: 7-((4-бром-3-((метиламино)метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 2-бром-5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)бензил-(метил)карбамат (25 мг, 0,040 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (15 мг, 72%). 1H ЯМР (400 МГц, DMSO-d6): δ 10,01 (с, 1H), 8,49 (с, 1H), 8,15 (уш.с, 1H), 7,65 (д, 2H), 7,43-7,58 (м, 3H), 4,99 (с, 2H), 3,67 (с, 2H), 3,15 (с, 3H), 2,33 (с, 3H). LCMS (метод C): RT=0,91 мин, m/z=521 [M+H]+.
Пример 158: 3-(2,6-дихлорфенил)-7-((4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
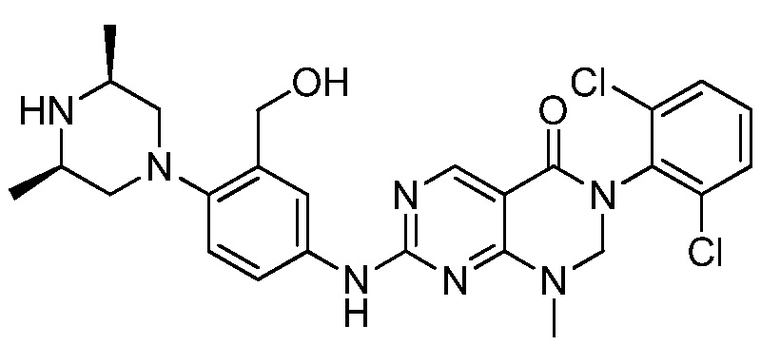
Стадия 1: 2-((3R,5S)-3,5-диметилпиперазин-1-ил)-5-нитро-бензальдегид:
К перемешиваемой суспензии 2-фтор-5-нитробензальдегида (2,69 г, 15,9 ммоль) и карбоната калия (8,80 г, 63,7 ммоль) в безводном DMF (10 мл) добавляли (2S,6R)-2,6-диметилпиперазин (2 г, 17,51 ммоль), и смесь нагревали при 90°C в течение 16 часов. После охлаждения, смесь распределяли между солевой раствор/вода (100 мл) и этилацетатом (25 мл). Водный слой отделяли и экстрагировали этилацетатом (3×25 мл). Объединенные этилацетатные фракции промывали смесью солевой раствор/вода (1:1, 4×25 мл), сушили (над безводным сульфатом натрия), фильтровали и концентрировали под вакуумом. Полученный остаток подвергали хроматографии (градиент 20-100% этилацетат в циклогексане) с получением названного соединения (2,77 г, 66,1%). 1H ЯМР (400 МГц, CDCl3): δ 10,07 (с, 1H), 8,62 (д, 1H), 8,28 (дд, 1H), 7,06 (д, 1H), 3,35 (д, 2H), 3,14-3,17 (м, 2H), 2,72 (дд, 2H), 1,13 (д, 6H). LCMS (метод C): RT=0,43 мин, m/z=264 [M+H]+.
Стадия 2: (2-((3R,5S)-3,5-диметилпиперазин-1-ил)-5-нитро-фенил)метанол:
К перемешиваемому раствору 2-((3R,5S)-3,5-диметилпиперазин-1-ил)-5-нитробензальдегида (2,77 г, 10,52 ммоль) в безводном тетрагидрофуран (35 мл) при 0°C добавляли порциями боргидрид натрия (0,438 г, 11,57 ммоль). Перемешивание продолжали в течение 1 часа. Смесь концентрировали под вакуумом. Остаток суспендировали в дихлорметане (40 мл) и перемешивали в течение 1 часа. Суспензию фильтровали, и влажный осадок на фильтре промывали свежим дихлорметаном (2×5 мл). Объединенные фильтраты концентрировали под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (2,79 г, 100%). 1H ЯМР (400 МГц, CDCl3): δ 8,25 (д, 1H), 8,12 (дд, 1H), 7,13 (д, 1H), 4,79 (с, 2H), 3,46 (уш.с, 1H), 3,09 (д, 4H), 2,44 (т, 2H), 1,41 (уш.с, 1H), 1,12 (д, 6H). LCMS (метод C) RT=0,38 мин, m/z=266 [M+H]+.
Стадия 3: (5-амино-2-((3R,5S)-3,5-диметилпиперазин-1-ил)-фенил)метанол:
(2-((3R,5S)-3,5-Диметилпиперазин-1-ил)-5-нитрофенил)метанол (1,40 г, 5,28 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, заполненный H2, растворитель метанол 100 мл). Растворитель удаляли под вакуумом с получением названного соединения, которое использовали без дополнительной очистки (1,16 г, 93%). 1H ЯМР (400 МГц, DMSO-d6): δ 6,75 (д, 1H), 6,64 (д, 1H), 6,38 (дд, 1H), 4,97 (уш.с, 1H), 4,71 (с, 2H), 4,45 (с, 2H), 2,76-2,88 (м, 2H), 2,66 (д, 2H), 2,09 (т, 2H), 1,91 (т, 1H), 0,93 (д, 6H). LCMS (метод C): RT=0,22 мин, m/z=236 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((4-((3S,5R)-3,5-диметил-пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(5-Амино-2-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)метанол (66 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 131, с получением названного соединения в виде белого твердого вещества (9 мг, 6%). ¹H ЯМР (300 МГц, DMSO-d6): δ 9,83 (с, 1H), 8,46 (с, 1H), 7,99 (уш.с, 1H), 7,65 (д, 2H), 7,54 (дд, 1H), 7,48 (дд, 1H), 6,96 (д, 1H), 5,07 (т, 1H), 4,96 (с, 2H), 4,54 (д, 2H), 3,11 (с, 3H), 2,83 (м, 4H), 2,16 (т, 2H), 0,97 (д, 6H). LCMS (метод C): RT=0,78 мин, m/z=542 [M+H]+.
Пример 159: 3-(2,6-дихлорфенил)-7-((4-метокси-3-((метил-амино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил 5-амино-2-метоксибензил(метил)карбамат:
Третбутил 2-фтор-5-нитробензил(метил)карбамат (185 мг, 0,65 ммоль) растворяли в DMF (4 мл) и добавляли раствор 28% метоксида натрия в метаноле (0,16 мл, 0,78 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и полученный осадок собирали фильтрацией с получением твердого вещества. Это вещество подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 50 МПа, растворитель метанол) с получением названного соединения (95 мг, 55%) в виде бесцветного масла, которое использовали без дополнительной очистки. LCMS (метод C): RT=0,82 мин, m/z=267 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-7-((4-метокси-3-((метил-амино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 5-амино-2-метоксибензил(метил)карбамат (75 мг, 0,28 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,28 ммоль), как описано в примере 1, с получением названного соединения (59 мг, 22%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,76 (уш.с, 1H), 8,45 (с, 1H), 7,78 (уш.с, 1H), 7,64 (д, 2H), 7,49 (м, 2H), 6,91 (д, 1H), 4,96 (с, 2H), 3,76 (с, 3H), 3,60 (с, 2H), 3,10 (с, 3H), 2,28 (с, 3H). LCMS (метод C): RT=0,78 мин, m/z=473 [M+H]+.
Пример 160: 3-(2,6-дихлорфенил)-1-метил-7-((4-(1-метил-1H-пиразол-4-ил)-3-((метиламино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(1-метил-1H-пиразол-4-ил)бензил(метил)карбамат:
Суспензию третбутил 2-бром-5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-бензил(метил)карбамата (40 мг, 0,064 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (20,06 мг, 0,096 ммоль) и 2M раствора карбоната натрия (193 мкл, 0,386 ммоль) в 1,4-диоксане (400 мкл) дегазировали путем барботирования азота в течение 10 минут. Добавляли аддукт PdCl2(dppf)-дихлорметан (5,25 мг, 6,43 мкмоль), и реакционную смесь нагревали до 120°C при микроволновом излучении в течение 10 минут, затем до 150°C в течение 15 минут, затем до 180°C в течение 10 минут. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3×6 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток подвергали хроматографии (картридж KP-NH 11g, циклогексан:этилацетат, градиентное элюирование от 90:10 до 0:100) с получением названного соединения в виде желтовато-белого твердого вещества (17 мг, 42%). 1H ЯМР (400 МГц, CDCl3): δ 8,73 (с, 1H), 7,65 (уш.с, 1H), 7,53 (с, 1H), 7,36-7,48 (м, 4H), 7,25-7,35 (м, 3H), 4,89 (с, 2H), 4,55 (уш.с, 2H), 3,96 (с, 3H), 3,20 (с, 3H), 3,64-3,87 (уш.м, 3H), 1,33-1,56 (уш.м, 9H). LCMS (метод C): RT=1,50 мин, m/z=623 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-1-метил-7-((4-(1-метил-1H-пиразол-4-ил)-3-((метиламино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(1-метил-1H-пиразол-4-ил)бензил(метил)карбамат (17 мг, 0,27 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (7 мг, 49%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,90 (с, 1H), 8,49 (с, 1H), 8,03 (уш.с, 1H), 7,90 (с, 1H), 7,55-7,73 (м, 4H), 7,48 (т, 1H), 7,29 (д, 1H), 4,98 (с, 2H), 3,88 (с, 3H), 3,64 (с, 2H), 3,15 (с, 3H), 2,34 (с, 3H). LCMS (метод C): RT=0,85 мин, m/z=523 [M+H]+.
Пример 161: 3-(2,6-дихлорфенил)-7-((4-(4-(2-(диметиламино)-ацетил)пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
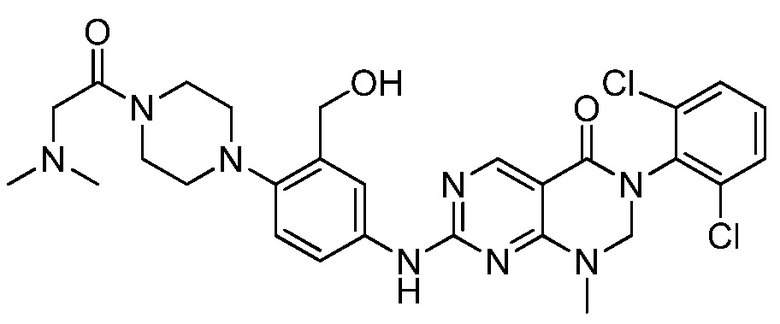
3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (47 мг, 0,091 ммоль) растворяли в THF (1 мл) и добавляли DIPEA (0,040 мл, 0,23 ммоль). Добавляли 2-хлорацетил- хлорид (7,99 мкл, 0,101 ммоль), и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли 2 M раствор диметиламина в THF (4,63 мкл, 0,091 ммоль), и смесь нагревали при 150°C в течение 30 минут при микроволновом излучении. Смесь концентрировали под вакуумом, и остаток подвергали хроматографии (картридж 11 g KPNH; элюировали 10-100% EtOAc/циклогексан, затем 0-10% MeOH/EtOAc) с получением бесцветного стеклообразного вещества. Это вещество растирали с эфиром с получением названного соединения (12 мг, 22%) в виде бесцветного стеклообразного вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,87 (уш.с, 1H), 8,46 (с, 1H), 8,02 (уш.с, 1H), 7,6 (д, 2H), 7,50 (м, 2H), 7,00 (д, 1H), 4,97 (с, 2), 3,59 (м, 4H), 3,39 (с, 2H), 3,12 (с, 3H), 2,77 (м, 4H), 2,35 (с, 6H). LCMS (метод C): RT=0,76 мин, m/z=599 [M+H]+.
Примеры 162-163: 3-(2,6-дихлорфенил)-7-((3-((R)-2,2-дифтор-1-гидроксиэтил)-4-((R)-3-(метоксиметил)пиперазин-1-ил)фенил)-амино)-1-метил -2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

В атмосфере N2, CsF (16 мг, 0,103 ммоль) добавляли к раствору (R)-третбутил 4-(2-формил-4-нитрофенил)-2-(метокси-метил)пиперазин-1-карбоксилата (300 мг, 0,50 ммоль) и (дифторметил)триметилсилана (196 мг, 1,581 ммоль) в 5 мл DMF, затем смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли раствор TBAF (2,5 мл, 1 M в THF), и всю смесь перемешивали в течение еще 1 часа. После экстракции в системе этилацетат и вода, органическую фазу промывали солевым раствором и затем сушили над безводным MgSO4. Раствор фильтровали, и растворитель испаряли под вакуумом. Анализ неочищенного остатка методом LCMS показывал присутствие двух диастереомеров, которые разделяли на хроматографической системе Biotage. Каждый диастереомер затем отдельно гидрировали, используя проточный реактор H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин). Каждый полученный диастереоизомерный анилин (1 экв) затем подвергали реакции сочетания с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (1,1 экв), следуя методике примера 31, с получением каждого индивидуального диастереоизомера названного продукта.
Диастереоизомер 1 (27 мг): ¹H ЯМР (400 МГц, DMSO-d6): δ 9,92 (с, 1H), 8,47 (с,1H), 8,11 (уш.с, 1H), 7,65 (д, 2H), 7,59(м,1H),7,48 (дд, 1H), 7,21 (д, 1H), 6,12 (д, 1H), 6,03 (дт,1H), 5,23 (м, 1H), 4,98 (с,2H),3,31(м,2H), 3,26(с, 3H), 2,95 (м, 2H), 3,12 (с, 3H), 2,80 (м, 3H), 2,61 (м, 2H). LCMS (метод C): RT=0,87 мин, m/z=608 [M+H]+
Диастереоизомер 2 (60 мг): ¹H ЯМР (400 МГц, DMSO-d6): δ 9,92 (с, 1H), 8,47 (с, 1H),8,12 (уш.с, 1H), 7,65 (д, 2H), 7,58 (м, 1H),7,48 (дд, 1H), 7,21 (д, 1H), 6,12 (д, 1H), 6,02 (дт,1H), 5,25 (м, 1H), 4,89 (с, 2H),3,28(м, 2H), 3,25 (с, 3H), 3,12 (с, 3H), 3,00-2,60 (м, 7H), 2,37 (м, 2H). LCMS (метод C): RT=0,77 мин, m/z=608 [M+H]+.
Пример 164: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: (1S,4S)-третбутил 5-(2-формил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию 2-фтор-5-нитробензальдегида (1,00 г, 5,91 ммоль), (1S,4S)-третбутил 2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (1,172 г, 5,91 ммоль) и карбоната калия (2,452 г, 17,74 ммоль) в безводном DMF (5 мл) нагревали до 50°C в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и перемешивали при комнатной температуре в течение 15 минут. Осажденное твердое вещество выделяли фильтрацией, промывали водой, воду отсасывали, и лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества (1,82 г, 89%). 1H ЯМР (300 МГц, CDCl3): δ 9,91 (с, 1H), 8,62 (д, 1H), 8,22 (дд, 1H), 6,78 (дд, 1H), 4,50-4,76 (м, 2H), 3,96 (дд, 1H), 3,46-3,66 (м, 2H), 2,91 (дд, 1H), 1,95-2,19 (м, 2H), 1,42 (с, 9H). LCMS (метод C): RT=1,47 мин, m/z=348 [M+H]+.
Стадия 2: (1S,4S)-третбутил 5-(2-(гидроксиметил)-4-нитро-фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Суспензию (1S,4S)-третбутил 5-(2-формил-4-нитрофенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (1,82 г, 5,24 ммоль) в безводном THF (9,70 мл) охлаждали до 0°C, затем порциями добавляли боргидрид натрия (0,198 г, 5,24 ммоль). Реакционную смесь перемешивали при 0°C в течение 90 минут. Реакционную смесь гасили водой (30 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде желтого твердого вещества (1,80 г, 98%). 1H ЯМР (300 МГц, CDCl3): δ 8,18 (дд, 1H), 7,98-8,08 (м, 1H), 6,59 (д, 1H), 4,79 (с, 1H), 4,49-4,73 (м, 3H), 3,81 (д, 1H), 3,36-3,64 (м, 3H), 2,21 (кв, 1H), 2,00 (уш.с, 2H), 1,37-1,53 (м, 9H). LCMS (метод C): RT=1,34 мин, m/z=350 [M+H]+.
Стадия 3: (1S,4S)-третбутил 5-(4-амино-2-(гидроксиметил)-фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
Раствор (1S,4S)-третбутил 5-(2-(гидроксиметил)-4-нитро-фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (1,80 г, 5,15 ммоль) в метаноле (100 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 40 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде коричневого твердого вещества (1,49 г, 91%). 1H ЯМР (300 МГц, CDCl3): δ 6,86 (д, 1H), 6,51-6,60 (м, 2H), 4,83 (д, 1H), 4,53 (д, 1H), 4,45 (д, 1H), 3,86 (с, 1H), 3,30-3,79 (м, 4H), 3,12-3,29 (м, 2H), 2,02 (д, 1H), 1,91 (т, 1H), 1,65 (уш.с, 1H), 1,40-1,55 (м, 9H). LCMS (метод C): RT=0,68 мин, m/z=320 [M+H]+.
Стадия 4: (1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)-амино)-2-(гидроксиметил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,423 ммоль) подвергали взаимодействию с (1S,4S)-третбутил 5-(4-амино-2-(гидроксиметил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилатом (135 мг, 0,423 ммоль), следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (190 мг, 72%). 1H ЯМР (300 МГц, CDCl3): δ 8,71 (с, 1H), 7,59 (уш.с, 1H), 7,48 (дт, 1H), 7,44 (дд, 2H), 7,35 (уш.с, 1H), 7,29 (дд, 1H), 6,95 (д, 1H), 4,88 (с, 2H), 4,84 (д, 1H), 4,45-4,67 (м, 2H), 4,06-4,13 (м, 1H), 3,85 (уш.д, 1H), 3,26-3,63 (м, 4H), 3,18 (с, 3H), 1,86-2,05 (м, 2H), 1,40-1,53 (м, 9H). LCMS (метод C): RT=1,37 мин, m/z=626 [M+H]+.
Стадия 5: 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
(1S,4S)-третбутил 5-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (190 мг, 0,303 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтого твердого вещества (49 мг, 31%). 1H ЯМР (400 МГц, MeOH-d4): δ 8,51 (с, 1H), 7,83 (уш.с, 1H), 7,56 (д, 2H), 7,49 (дд, 1H), 7,42 (дд, 1H), 6,97 (д, 1H), 4,97 (с, 2H), 4,70 (д, 1H), 4,61 (д, 1H), 4,15 (с, 1H), 3,88 (с, 1H), 3,42 (дд, 1H), 3,22-3,30 (м, 3H), 3,19 (с, 3H), 3,03 (дд, 1H), 2,05 (д, 1H), 1,80 (д, 1H). LCMS (метод C): RT=0,66 мин, m/z=526 [M+H]+.
Пример 165: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1-метилпиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он

3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (31 мг, 0,060 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (18 мг, 57%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,47 (с, 1H), 7,92 (уш.с, 1H), 7,65 (д, 2H), 7,57 (дд, 1H), 7,48 (т, 1H), 7,18 (д, 1H), 5,07 (т, 1H), 4,97 (с, 2H), 4,53 (д, 2H), 3,12 (с, 3H), 2,85 (д, 2H), 2,57-2,70 (м, 1H), 2,18 (с, 3H), 1,95 (тд, 2H), 1,51-1,73 (м, 4H). LCMS (метод C): RT=0,70 мин, m/z=527 [M+H]+.
Пример 166: 3-(2,6-дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он
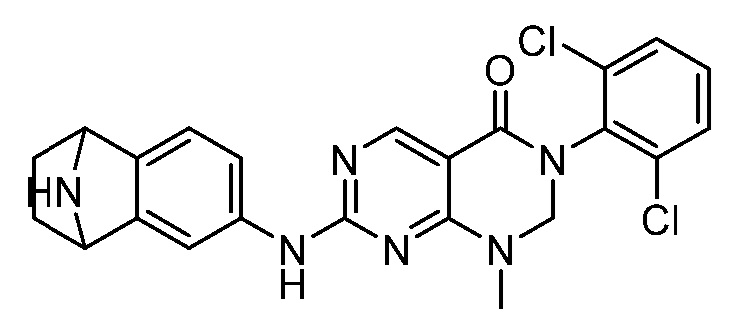
Стадия 1: третбутил 6-амино-1,2,3,4-тетрагидро-1,4-эпимино-нафталинe-9-карбоксилат:
Раствор третбутил 6-нитро-1,4-дигидро-1,4-эпиминонафталин-9-карбоксилата [Bioorg. Med. Chem., 2011, 19(8), 2726-2741] (500 мг, 1,734 ммоль) в метаноле (50 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 40 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде белого твердого вещества (440 мг, 97%). 1H ЯМР (300 МГц, CDCl3): δ 8,06 (д, 1H), 7,97 (дд, 1H), 7,37 (д, 1H), 7,01 (уш.д, 2H), 5,58 (уш.с, 2H), 1,37 (с, 9H). LCMS (метод C): RT=1,62 мин, m/z=233 [M-tBu+H]+.
Стадия 2: третбутил 6-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-1,2,3,4-тетрагидро-1,4-эпиминонафталинe-9-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,423 ммоль) подвергали взаимодействию с третбутил 6-амино-1,2,3,4-тетрагидро-1,4-эпиминонафталин-9-карбоксилатом (110 мг, 0,423 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (143 мг, 60%). 1H ЯМР (300 МГц, CDCl3): δ 8,72 (с, 1H), 7,62 (уш.с, 1H), 7,44 (дд, 2H), 7,34 (д, 2H), 7,29 (дд, 1H), 7,19 (д, 1H), 5,10 (уш.с, 2H), 4,89 (с, 2H), 3,18 (с, 3H), 2,12 (д, 2H), 1,40 (с, 9H), 1,32 (д, 2H). LCMS (метод C): RT=1,76 мин, m/z=567 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-метил-7-((1,2,3,4-тетра-гидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 6-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-1,2,3,4-тетра-гидро-1,4-эпиминонафталин-9-карбоксилат (143 мг, 0,252 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде бледно-желтого твердого вещества (75 мг, 64%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,79 (уш.с, 1H), 8,46 (с, 1H), 7,61-7,67 (м, 3H), 7,48 (дд, 1H), 7,42 (д, 1H), 7,12 (д, 1H), 4,96 (с, 2H), 4,41 (дд, 2H), 3,09 (с, 3H), 1,85 (дд, 2H), 1,10 (дд, 2H). LCMS (метод C): RT=0,75 мин, m/z=467 [M+H]+.
Пример 167: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
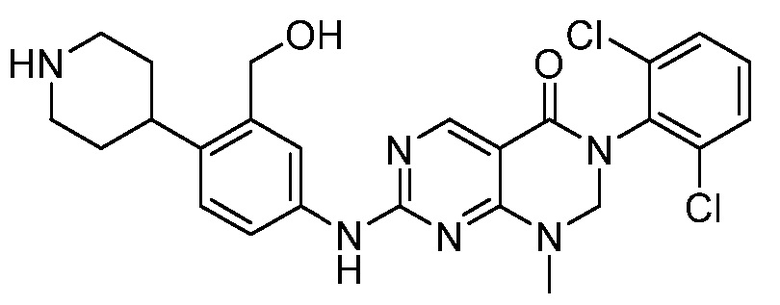
Стадия 1: третбутил 4-(4-амино-2-(гидроксиметил)фенил)-пиперидин-1-карбоксилат:
Раствор третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата в метаноле (15 мл) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, 40 МПа H2, комнатная температура, 1 мл/мин; затем картридж Pd(OH)2/C, 40 МПа H2, комнатная температура, 1 мл/мин; затем картридж Pd(OH)2/C, 60 МПа H2, комнатная температура, 1 мл/мин). Реакционную смесь концентрировали досуха при пониженном давлении с получением названного соединения в виде желтовато-белого твердого вещества (120 мг, 94%). 1H ЯМР (300 МГц, CDCl3): δ 7,01 (д, 1H), 6,70 (д, 1H), 6,62 (дд, 1H), 4,62 (с, 2H), 4,19 (уш.с, 2H), 3,17 (уш.с, 2H), 2,64-2,90 (м, 3H), 1,50-1,73 (м, 4H), 1,47 (с, 9H). LCMS (метод C): RT=0,78 мин, m/z=314 [M-tBu-H2O+H]+.
Стадия 2: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)пиперидин-1-карбоксилат:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (150 мг, 0,423 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-(гидрокси-метил)фенил)пиперидин-1-карбоксилатом (130 мг, 0,423 ммоль), следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (83 мг, 32%). 1H ЯМР (300 МГц, CDCl3): δ 8,73 (с, 1H), 8,38 (уш.с, 1H), 7,58-7,69 (м, 2H), 7,43 (дд, 2H), 7,28 (дд, 1H), 7,20 (д, 1H), 4,88 (с, 2H), 4,74 (с, 2H), 4,23 (уш.с, 2H), 3,18 (с, 3H), 3,63-2,97 (м, 4H), 1,52-1,84 (м, 4H), 1,48 (с, 9H). LCMS (метод C): RT=1,54 мин, m/z=613 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)пиперидин-1-карбоксилат (83 мг, 0,135 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде желтовато-белого твердого вещества (56 мг, 81%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,84 (уш.с, 1H), 8,47 (с, 1H), 7,93 (уш.с, 1H), 7,65 (д, 2H), 7,58 (дд, 1H), 7,48 (дд, 1H), 7,16 (д, 1H), 5,08 (уш.с, 1H), 4,97 (с, 2H), 4,54 (с, 2H), 3,13 (с, 3H), 3,05 (д, 2H), 2,80 (тт, 1H), 2,63 (тд, 2H), 1,62 (д, 2H), 1,53 (квд, 2H). LCMS (метод C): RT=0,85 мин, m/z=513 [M+H]+.
Пример 168: 3-(2,6-дихлорфенил)-1-(3-((метиламино)метил)-фенил)-7-((4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
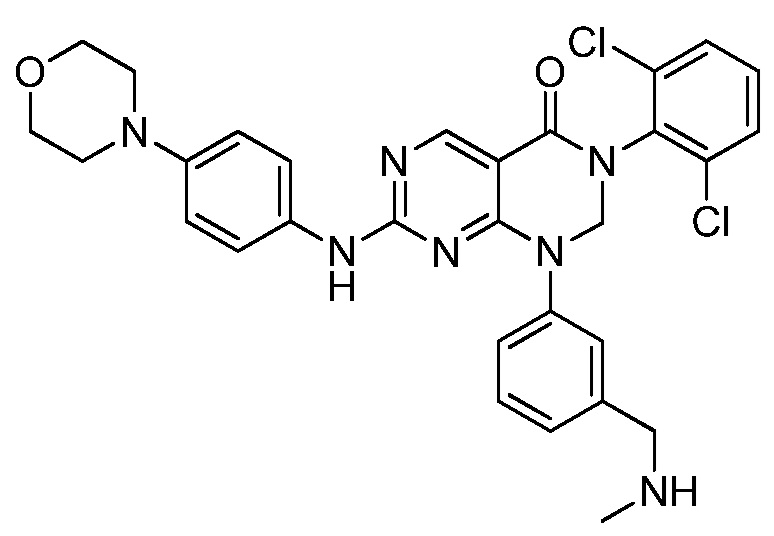
Третбутил 3-(3-(2,6-дихлорфенил)-7-(метилтио)-4-оксо-3,4-дигидропиримидо[4,5-d]пиримидин-1(2H)-ил)бензил(метил)карбамат (60 мг, 0,107 ммоль) подвергали взаимодействию с 4-морфолино-анилином (19 мг, 0,107 ммоль), как описано в примере 1, с получением названного соединения (27 мг, 47%) в виде желтовато-белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6): δ 9,78 (уш.с, 1H), 8,63 (с, 1H), 7,62 (д, 2H), 7,40 (7 H, m), 6,70 (м, 2H), 5,37 (с, 2H), 3,70 (м, 6H), 2,98 (м, 4H). LCMS (метод C): RT=0,81 мин, m/z=590 [M+H]+.
Пример 169: 3-(2,6-дихлорфенил)-1-этил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: N-(2,6-дихлорфенил)-4-(этиламино)-2-(метилтио)-пиримидин-5-карбоксамид:
4-Хлор-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамид (2 г, 5,74 ммоль) суспендировали в THF (15 мл) и добавляли DIPEA (2,00 мл, 11,5 ммоль), затем 2 M раствор этиламина в THF (3,16 мл, 6,31 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали под вакуумом, затем разбавляли водой и экстрагировали этилацетатом (x3). Объединенные органические слои промывали солевым раствором, сушили (MgSO4) и концентрировали под вакуумом с получением названного соединения (2,20 г, 107%) в виде белого твердого вещества, которое использовали без дополнительной очистки. LCMS (метод C): RT=1,59 мин, m/z=357 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-1-этил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-4-(этиламино)-2-(метилтио)пиримидин-5-карбоксамид (1,50 г, 4,20 ммоль) суспендировали в ацетонитриле (15 мл). Добавляли карбонат цезия (6,84 г, 21,0 ммоль), затем дибромметан (1,17 мл, 16,8 ммоль). Смесь кипятили с обратным холодильником в течение ночи. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном (x2). Объединенные органические слои промывали солевым раствором, затем сушили (MgSO4) и концентрировали. Остаток подвергали хроматографии (картридж 24 g Si; элюировали 0-100% EtOAc/циклогексан) с получением названного соединения (460 мг, 30%) в виде бесцветного стеклообразного вещества, которое кристаллизовалось при стоянии. 1H ЯМР (300 МГц, CDCl3): δ 8,72 (с, 1H), 7,46 (д, 2H), 7,31 (м, 1H), 4,94 (с, 2H), 3,75 (кв, 2H), 2,57 (с, 3H), 1,26 (т, 3H). LCMS (метод C): RT=1,57 мин, m/z=369 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-этил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-аминофенил)пиперазин-1-карбоксилат (75 мг, 0,27 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-этил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (100 мг, 0,27 ммоль), как описано в примере 1, с получением названного соединения (60 мг, 45%) в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6): δ 9,70 (с, 1H), 8,45 (с, 1H), 7,63 (м, 4H), 7,48 (м, 1H), 6,88 (д, 2H), 4,99 (с, 2H), 3,65 (кв, 2H), 2,99 (м, 4H), 2,84 (м, 4H), 1,20 (т, 3H). LCMS (метод C): RT=0,80 мин, m/z=498 [M+H]+.
Пример 170: 3-(2,6-дихлорфенил)-7-((4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-этил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

(5-Амино-2-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)метанол (64 мг, 0,27 ммоль) подвергали взаимодействию с 3-(2,6-дихлор-фенил)-1-этил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,27 ммоль), как описано в примере 131, с получением названного соединения (60 мг, 45%) в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6): δ 9,81 (с, 1H), 8,46 (с, 1H), 7,99 (с, 1H), 7,65 (м, 2H), 7,50 (м, 2H), 6,96 (д, 1H), 5,05 (т, 1H), 5,00 (с, 2H), 4,54 (д, 2H), 3,68 (кв, 2H), 2,85 (м, 4H), 2,16 (т, 2H), 1,20 (т, 3H), 0,97 (д, 6H). LCMS (метод C): RT=0,84 мин, m/z=556 [M+H]+.
Пример 171: 3-(2,6-дихлорфенил)-1-фенил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
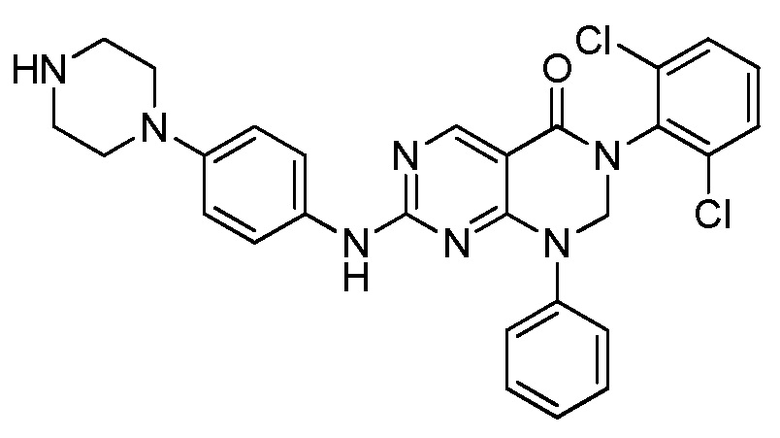
Стадия 1: N-(2,6-дихлорфенил)-2-(метилтио)-4-(фениламино)-пиримидин-5-карбоксамид:
4-Хлор-N-(2,6-дихлорфенил)-2-(метилтио)пиримидин-5-карбоксамид (2 г, 5,74 ммоль) суспендировали в THF (15 мл). Добавляли DIPEA (2,00 мл, 11,47 ммоль), затем анилин (0,58 мл, 6,31 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали под вакуумом, затем разбавляли водой и DCM. Органический слой концентрировали с получением твердого вещества, которое промывали этилацетатом с получением названного соединения (2,20 г, 95%) в виде желтовато-белого твердого вещества, которое использовали без дополнительной очистки. LCMS (метод C): RT=1,84 мин, m/z=405 [M+H]+.
Стадия 2: 3-(2,6-дихлорфенил)-7-(метилтио)-1-фенил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
N-(2,6-Дихлорфенил)-2-(метилтио)-4-(фениламино)пиримидин-5-карбоксамид (1 г, 2,467 ммоль) растворяли в ацетонитриле (10 мл) и добавляли дибромметан (0,69 мл, 9,87 ммоль), затем карбонат цезия (4,02 г, 12,34 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 64 часов. Смесь разбавляли водой и экстрагировали дихлорметаном (x3). Объединенные органические слои промывали солевым раствором, затем сушили (MgSO4) и концентрировали под вакуумом. Остаток подвергали хроматографии (картридж 24 g Si; элюировали 0-50% EtOAc/циклогексан) с получением названного соединения (293 мг, 29%) в виде желтовато-белой пены. LCMS (метод C): RT=1,72 мин, m/z=417 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-фенил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-аминофенил)пиперазин-1-карбоксилат (67 мг, 0,24 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-7-(метилтио)-1-фенил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (100 мг, 0,24 ммоль), как описано в примере 1, с получением названного соединения (60 мг, 45%) в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6): δ 9,83 (уш.с, 1), 8,62 (с, 1H), 7,62 (м, 2H), 7,40 (м, 8H), 6,63 (м, 2H), 5,37 (с, 2H), 2,91 (м, 4H), 2,81 (м, 4H). LCMS (метод C): RT=0,95 мин, m/z=546 [M+H]+.
Пример 172: 3-(2,6-дихлорфенил)-7-((3-(2-(диметиламино)-1-гидроксиэтил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
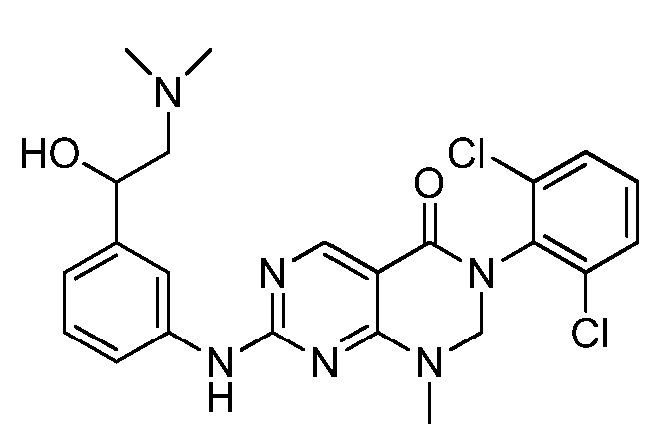
Стадия 1: 2-(3-нитрофенил)оксиран:
5 г 2-бром-1-(3-нитро-фенил)этанона растворяли в 100 мл этанола, обрабатывали с помощью 0,755 г боргидрида натрия и перемешивали в течение 1 часа при комнатной температуре. Добавляли 1,15 г гидроксида калия, и смесь перемешивали в течение еще 15 часов при комнатной температуре. Смесь разбавляли 500 мл этилацетата, и полученный раствор промывали два раза 300 мл полунасыщенного раствора хлорида аммония и один раз 100 мл воды. Органическую фазу сушили над сульфатом натрия и концентрировали под вакуумом. Остаток подвергали хроматографии с получением названного соединения (1,50 г, 44%). 1H ЯМР (300 МГц, CDCl3): δ 2,79 (дд, 1H); 3,19 (дд, 1H); 3,93 (дд, 1H); 7,50 (т, 1H); 7,60 (д, 1H); 8,08-8,16 (м, 2H) ppm.
Стадия 2: третбутил (2-гидрокси-2-(3-нитрофенил)этил) (метил)карбамат:
2-(3-Нитрофенил)оксиран (1,5 г,9,08 ммоль) и метиламин (2N в THF, 20 мл, 40 ммоль) перемешивали при комнатной температуре в течение ночи. Раствор затем концентрировали под вакуумом. Остаток растворяли в THF. Затем добавляли дитретбутил-дикарбонат (1,87 г, 8,56 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Остаток очищали на хроматографической системе Biotage с получением названного соединения (0,57 г, 23,6%).
Стадия 3: третбутил (2-(3-аминофенил)-2-гидроксиэтил) (метил)карбамат:
Третбутил (2-гидрокси-2-(3-нитрофенил)этил)(метил)карбамат (0,3 г, 1,012 ммоль) гидрировали в проточном реакторе H-Cube, следуя методике примера 1, с получением названного соединения (0,17 г, 63%), которое непосредственно использовали на следующей стадии.
Стадия 4: 3-(2,6-дихлорфенил)-7-((3-(2-(диметиламино)-1-гидроксиэтил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (249 мг, 0,702 ммоль) подвергали взаимодействию с третбутил (2-(3-аминофенил)-2-гидроксиэтил)(метил)карбаматом (170 мг, 0,638 ммоль), следуя методике примера 1, с получением названного соединения (130 мг, 43%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,92 (с, 1H), 8,49 (с, 1H), 7,97 (с, 1H), 7,67 (д, 2H), 7,55-7,45 (м, 2H), 7,24 (т, 1H), 6,98 (д, 1H), 5,01 (д, 1H), 4,99 (с, 2H), 3,15 (с, 3H), 2,41 (дд, 1H), 2,31 (дд, 1H). LCMS (метод C): RT=0,72 мин, m/z=487 [M+H]+.
Пример 173: 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 7-(2-формил-4-нитрофенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат:
2-Фтор-5-нитробензальдегид (37 мг, 0,219 ммоль) подвергали взаимодействию с третбутил 3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилатом, следуя методике примера 21, с получением названного соединения (85 мг, 93%), которое непосредственно использовали на следующей стадии.
Стадия 2: третбутил 7-(2-(гидроксиметил)-4-нитрофенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат:
Третбутил 7-(2-формил-4-нитрофенил)-3-окса-7,9-диаза-бицикло[3,3,1]нонан-9-карбоксилат (85 мг, 0,225 ммоль) подвергали взаимодействию с боргидридом натрия (10,23 мг, 0,27 ммоль), следуя методике примера 21, с получением названного соединения (78 мг, 91%), которое непосредственно использовали на следующей стадии.
Стадия 3: третбутил 7-(4-амино-2-(гидроксиметил)фенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат:
Третбутил 7-(2-(гидроксиметил)-4-нитрофенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилат (78 мг, 0,206 ммоль) гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин). Раствор концентрировали под вакуумом с получением названного соединения (66,8 мг, 93%), которое непосредственно использовали на следующей стадии.
Стадия 4: 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
3-(2,6-Дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (84 мг, 0,236 ммоль) подвергали взаимодействию с третбутил 7-(4-амино-2-(гидроксиметил)фенил)-3-окса-7,9-диазабицикло[3,3,1]нонан-9-карбоксилатом (75 мг, 0,215 ммоль), следуя методике примера 31, с получением названного соединения (26 мг, 22%). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,80 (с, 1H), 8,46 (с, 1H), 7,86 (уш.с, 1H), 7,64 (д, 1H), 7,59 (дд, 1H), 7,48 (т, 1H), 7,05 (д, 1H), 5,22 (т, 1H), 4,96 (с, 2H), 4,63 (д, 2H), 3,87 (м, 4H), 3,11 (с, 3H), 3,10 (м, 4H), 2,85 (м, 2H). LCMS (метод C): RT=0,71 мин, m/z=556 [M+H]+.
Пример 174: 3-(2,6-дихлорфенил)-7-((3-((этиламино)метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он

Стадия 1: третбутил этил(2-морфолино-5-нитробензил)-карбамат:
2-Морфолино-5-нитробензальдегид (0,5 г, 2,12 ммоль) суспендировали в метаноле (6 мл). Добавляли бикарбонат натрия (0,36 г, 4,23 ммоль), затем 2 M раствор этиламина в THF (1,27 мл, 2,54 ммоль). Реакционную смесь нагревали при 80°C в течение 3 часов. Смесь охлаждали до комнатной температуры. Добавляли боргидрид натрия (0,096 г, 2,54 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию прерывали путем добавления воды и концентрировали под вакуумом. Остаток разбавляли водой и экстрагировали дихлорметаном. Слои разделяли, используя картридж для разделения фаз, и органический слой концентрировали. Остаток растворяли в дихлорметане (5 мл) и добавляли DIPEA (0,481 мл, 2,75 ммоль), затем BOC-ангидрид (0,54 мл, 2,33 ммоль). Реакционную смесь перемешивали в течение ночи. Смесь концентрировали под вакуумом. Остаток подвергали хроматографии (картридж 10 g Si; элюировали 0-70% EtOAc/циклогексан) с получением названного соединения (680 мг, 88%) в виде желтого масла. 1H ЯМР (300 МГц, CDCl3): δ 8,11 (м, 2H), 7,10 (д, 1H), 4,53 (м, 2H), 3,88 (м, 4H), 3,19 (м, 2H), 2,99 (м, 4H), 1,54 (м, 9H), 1,10 (м, 3H). LCMS (метод C): RT=1,73 мин, m/z=366 [M+H]+.
Стадия 2: третбутил 5-амино-2-морфолинобензил(этил)-карбамат:
Третбутил этил(2-морфолино-5-нитробензил)карбамат (200 мг, 0,55 ммоль) подвергали непрерывному гидрированию (проточный реактор H-cube, картридж 10% Pd/C, комнатная температура, 60 МПа, растворитель метанол). Неочищенный продукт подвергали хроматографии (картридж 12 g Si; элюировали 0-80% EtOAc/циклогексан) с получением названного соединения в виде белого твердого вещества (85 мг, 46%). LCMS (метод C): RT=1,00 мин, m/z=336 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-7-((3-((этиламино)метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он:
Третбутил 5-амино-2-морфолинобензил(этил)карбамат (85 мг, 0,25 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (90 мг, 0,25 ммоль), как описано в примере 1, с получением названного соединения (35 мг, 26%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,45 (с, 1H), 7,97 (уш.с, 1H), 7,65 (м, 2H), 7,53 (дд, 1H), 7,47 (м, 1H), 7,08 (д, 1H), 4,97 (с, 2H), 3,74 (м, 6H), 3,17 (м, 1H), 3,13 (с, 3H), 2,84 (м, 4H), 2,62 (кв, 2H), 1,07 (т, 3H). LCMS (метод C): RT=0,91 мин, m/z=542 [M+H]+.
Пример 175: 3-(2,6-дихлорфенил)-1-метил-7-((9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (40 мг, 0,086 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (18 мг, 44%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,83 (уш.с, 1H), 8,48 (с, 1H), 7,68 (с, 1H), 7,65 (д, 2H), 7,44-7,56 (м, 2H), 7,18 (д, 1H), 4,96 (с, 2H), 4,01 (д, 2H), 3,10 (с, 3H), 1,86-2,03 (м, 5H), 1,01-1,11 (м, 2H). LCMS (метод C): RT=0,81 мин, m/z=481 [M+H]+.
Пример 176: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

7-((4-((1S,4S)-2,5-Диазабицикло[2,2,1]гептан-2-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (34 мг, 0,065 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде желтого твердого вещества (12 мг, 34%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,68 (уш.с, 1H), 8,43 (с, 1H), 7,94 (уш.с, 1H), 7,64 (д, 2H), 7,44-7,52 (м, 2H), 6,75 (д, 1H), 5,03 (т, 1H), 4,95 (с, 2H), 4,50 (дд, 1H), 4,39 (дд, 1H), 3,93 (с, 1H), 3,23 (д, 1H), 3,28-3,35 (м, 1H), 3,07-3,15 (м, 4H), 2,74 (дд, 1H), 2,64 (д, 1H), 2,28 (с, 3H), 1,77 (д, 1H), 1,69 (д, 1H), пик при 3,28-3,35 ppm частично перекрывался растворителем. LCMS (метод C): RT=0,70 мин, m/z=540 [M+H]+.
Пример 177: 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
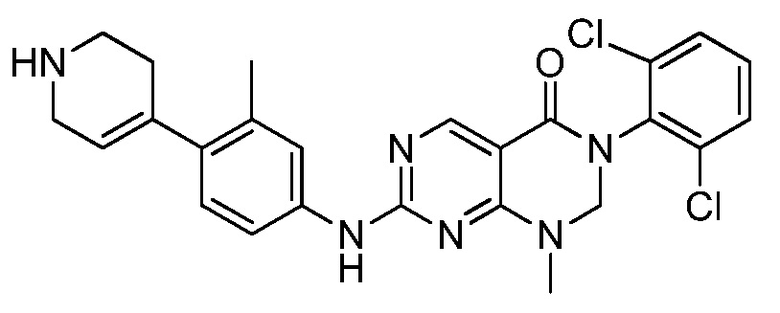
Стадия 1: третбутил 4-(2-метил-4-нитрофенил)-5,6-дигидро-пиридин-1(2H)-карбоксилат:
1-Йод-2-метил-4-нитробензол (0,61 г, 2,32 ммоль) и третбутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (0,932 г, 3,01 ммоль) растворяли в 1,4-диоксане (7 мл) и добавляли 2M раствор карбоната натрия (3,48 мл, 6,96 ммоль). Смесь дегазировали и помещали в атмосферу азота. Добавляли аддукт PdCl2(dppf) и дихлорметана (0,189 г, 0,232 ммоль), и смесь нагревали до 100°C при микроволновом излучении в течение 20 минут. Смесь разбавляли водой и экстрагировали этилацетатом (x3). Объединенные органические экстракты промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали под вакуумом. Остаток подвергали хроматографии (картридж 24 g Si, элюировали 0-50% EtOAc/циклогексан) с получением названного соединения в виде коричневого масла (798 мг, 2,51 ммоль, 90% выход). LCMS (метод C): RT=1,89 мин, m/z=263 [M+H-tBu]+.
Стадия 2: третбутил 4-(4-амино-2-метилфенил)-5,6-дигидро-пиридин-1(2H)-карбоксилат:
Третбутил 4-(2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат(390 мг, 1,23 ммоль) растворяли в этаноле (7 мл). Добавляли хлорид олова(II) (1,16 г, 6,12 ммоль), и смесь нагревали при 65°C в течение 3 часов. Раствор концентрировали под вакуумом. Остаток растворяли в 4 M водном растворе гидроксида натрия и экстрагировали этилацетатом (x3). Объединенные органические экстракты промывали солевым раствором, затем сушили (MgSO4) и концентрировали с получением названного соединения (246 мг, 70) в виде коричневого сиропа, которое непосредственно использовали без дополнительной очистки. LCMS (метод C): RT=1,24 мин, m/z=289 [M+H]+.
Стадия 3: 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-амино-2-метилфенил)-5,6-дигидропиридин-1(2H)-карбоксилат (179 мг, 0,62 ммоль) подвергали взаимодействию с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-оном (200 мг, 0,56 ммоль), как описано в примере 1, с получением названного соединения (41 мг, 15%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,85 (с, 1H), 8,48 (с, 1H), 7,64 (м, 3H), 7,50 (м, 2H), 6,99 (д, 1H), 5,54 (с, 1H), 4,98 (с, 2H), 3,34 (м, по-видимому, 2H частично закрыты пиком воды), 3,12 (с, 3H), 2,87 (т, 3H), 2,24 (с, 3H), 2,14 (м, 2H). LCMS (метод C): RT=0,87 мин, m/z=495 [M+H]+.
Пример 178: 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он

3-(2,6-Дихлорфенил)-1-метил-7-((3-метил-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он (18 мг, 0,036 ммоль) растворяли в метаноле (1 мл). Добавляли концентрированный водный раствор формальдегида (5,41 мкл, 0,073 ммоль), затем триацетоксиборгидрид натрия (38,5 мг, 0,18 ммоль). Смесь перемешивали в течение 16 часов. Добавляли дополнительную порцию водного раствора формальдегида (5,41 мкл, 0,073 ммоль), затем добавляли триацетоксиборгидрид натрия (38,5 мг, 0,18 ммоль). Смесь перемешивали в течение 30 минут. Реакцию прерывали путем добавления нескольких капель концентрированной HCl. Смесь загружали в картридж 2 g SCX, который промывали метанолом, затем элюировали с помощью 2 M NH3 в метаноле с получением белого твердого вещества. Это вещество суспендировали в диэтиловом эфире, и твердое вещество собирали фильтрацией с получением названного соединения (8 мг, 43%) в виде белого твердого вещества. ¹H ЯМР (300 МГц, DMSO-d6): δ 9,86 (с, 1H), 8,48 (с, 1H), 7,62 (м, 3H), 7,55 (д, 1H), 7,48 (м, 1H), 7,00 (д, 1H), 5,50 (с, 1H), 4,98 (с, 2H), 3,12 (с, 3H), 2,97 (м, 2H), 2,73 (м, 2H), 2,28 (м, 5H). LCMS (метод C): RT=0,90 мин, m/z=509 [M+H]+.
Пример 179: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
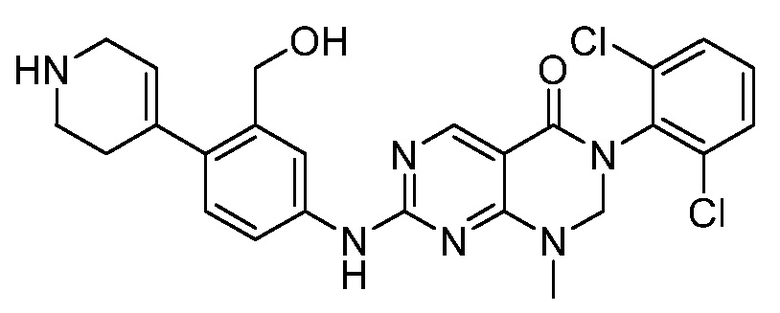
Стадия 1: (2-бром-5-нитрофенил)метанол:
Раствор 2-бром-5-нитробензoйной кислоты (2,460 г, 10,00 ммоль) в безводном THF (50 мл) охлаждали до 0°C, затем добавляли по каплям тетрагидрофурановый комплекс борана (1M в THF, 25 мл, 25,00 ммоль). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь охлаждали до 0°C и гасили путем добавления солевого раствора (50 мл). Слои разделяли, и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж диоксид кремния 100g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 60:40) с получением названного соединения в виде белого твердого вещества (1,80 г, 78%). 1H ЯМР (300 МГц, CDCl3): δ 8,43 (д, 1H), 8,02 (дд, 1H), 7,72 (д, 1H), 4,83 (д, 2H), 2,16 (т, 1H).
Стадия 2: третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
Суспензию (2-бром-5-нитрофенил)метанола (500 мг, 2,155 ммоль), третбутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (999 мг, 3,23 ммоль) и 2M раствора карбоната натрия (3232 мкл, 6,46 ммоль) в 1,4-диоксане (6491 мкл) дегазировали путем барботирования азота в течение 10 минут, затем добавляли аддукт PdCl2(dppf)-дихлорметан (88 мг, 0,108 ммоль). Реакционную смесь нагревали до 120°C при микроволновом излучении в течение 20 минут. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (3×12 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали на хроматографической системе Biotage (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 50:50) с получением названного соединения в виде желтого масла (743 мг, количественный выход). 1H ЯМР (300 МГц, CDCl3) δ 8,41 (д, 1H), 8,11 (дд, 1H), 7,28 (д, 1H), 5,68 (уш.с, 1H), 4,75 (д, 2H), 4,06 (кв, 2H), 3,64 (т, 2H), 2,37 (уш.с, 2H), 1,50 (с, 9H). LCMS (метод C): RT=1,52 мин, m/z=235 [M-Boc+H]+.
Стадия 3: третбутил 4-(4-амино-2-(гидроксиметил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
К раствору третбутил 4-(2-(гидроксиметил)-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (370 мг, 1,107 ммоль) в этаноле (7377 мкл) добавляли хлорид олова(II) (1049 мг, 5,53 ммоль), и полученную смесь нагревали до 65°C в течение 2,5 часов. Реакционную смесь концентрировали досуха при пониженном давлении, разбавляли 4M раствором NaOH (15 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали досуха при пониженном давлении и очищали на хроматографической системе Biotage (картридж GraceResolv диоксид кремния 12g, циклогексан:этилацетат:метанол, градиентное элюирование от 90:10:0 до 0:100:0 до 0:90:10) с получением названного соединения в виде белого твердого вещества (45 мг, 13%). 1H ЯМР (300 МГц, CDCl3): δ 6,92 (д, 1H), 6,79 (д, 1H), 6,58 (дд, 1H), 5,54 (уш.с, 1H), 4,57 (с, 2H), 3,99 (кв, 2H), 3,69 (уш.с, 2H), 3,58 (т, 2H), 2,32 (уш.с, 2H), 1,49 (с, 9H). LCMS (метод C): RT=0,92 мин, m/z=305 [M+H]+.
Стадия 4: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (53 мг, 0,148 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-(гидроксиметил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилатом (45 мг, 0,148 ммоль), следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (58 мг, 64%). 1H ЯМР (300 МГц, CDCl3): δ 8,75 (с, 1H), 7,84 (д, 1H), 7,80 (уш.с, 1H), 7,57 (дд, 1H), 7,44 (д, 2H), 7,29 (дд, 1H), 7,11 (д, 1H), 5,61 (уш.с, 1H), 4,90 (с, 2H), 4,68 (д, 2H), 4,03 (д, 2H), 3,62 (т, 2H), 3,21 (с, 3H), 2,37 (уш.с, 2H), 1,50 (с, 9H). LCMS (метод C): RT=1,58 мин, m/z=611 [M+H]+.
Стадия 5: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(гидроксиметил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат (58 мг, 0,095 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (39 мг, 80%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,88 (уш.с, 1H), 8,48 (с, 1H), 8,07 (уш.с, 1H), 7,65 (д, 2H), 7,55 (дд, 1H), 7,48 (дд, 1H), 7,01 (д, 1H), 5,56 (уш.с, 1H), 5,04 (т, 1H), 4,97 (с, 2H), 4,48 (д, 2H), 3,13 (с, 3H), 2,89 (т, 2H), 2,50-2,53 (м, 2H), 2,15 (уш.с, 2H), протоны при 2,50-2,53 частично перекрыты растворителем. LCMS (метод C): RT=0,74 мин, m/z=511 [M+H]+.
Пример 180: 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он
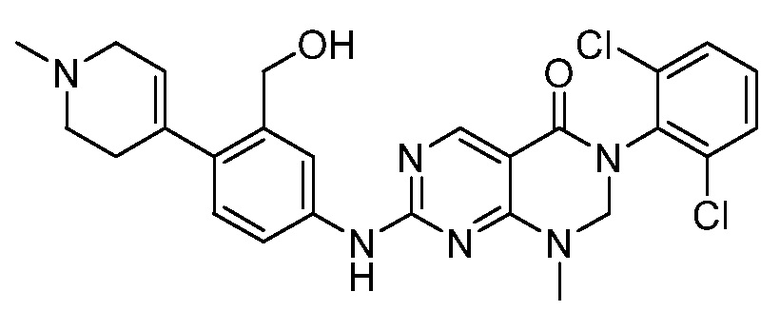
3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1,2,3,6-тетра-гидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (27 мг, 0,91 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (27 мг, 91%). 1H ЯМР (400 МГц, DMSO-d6): δ 9,89 (уш.с, 1H), 8,48 (с, 1H), 8,07 (уш.с, 1H), 7,65 (д, 2H), 7,55 (д, 1H), 7,48 (т, 1H), 7,02 (д, 1H), 5,52 (уш.с, 1H), 5,05 (т, 1H), 4,97 (с, 2H), 4,46 (д, 2H), 3,13 (с, 3H), 2,96 (с, 2H), 2,50-2,57 (м, 2H), 2,22-2,35 (м, 5H), протоны при 2,50-2,57 частично перекрыты растворителем. LCMS (метод C): RT=0,75 мин, m/z=525 [M+H]+.
Пример 181: 7-((4-(1-циклобутилпиперидин-4-ил)-3-(гидрокси-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он
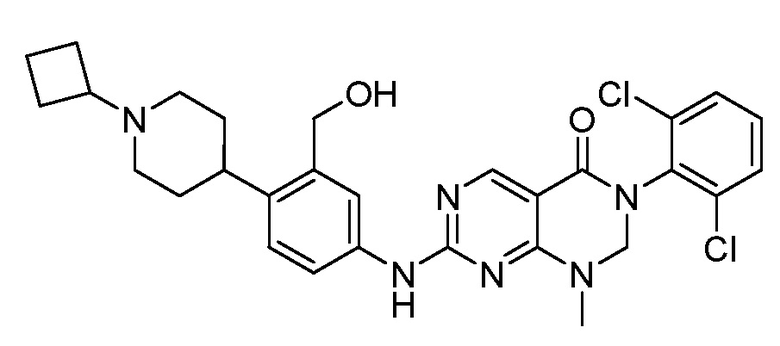
Суспензию 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-она (25 мг, 0,049 ммоль), циклобутанона (10,92 мкл, 0,146 ммоль) и уксусной кислоты (8,36 мкл, 0,146 ммоль) в 1,2-дихлорэтане (1 мл) перемешивали при комнатной температуре в течение 15 минут, затем добавляли триацетоксиборгидрид натрия (31,0 мг, 0,146 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Добавляли дополнительное количество циклобутанона (10,92 мкл, 0,146 ммоль) и триацетоксиборгидрида натрия (31,0 мг, 0,146 ммоль), и смесь нагревали при 60°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли дополнительное количество циклобутанона (10,92 мкл, 0,146 ммоль), триацетоксиборгидрида натрия (31,0 мг, 0,146 ммоль) и уксусной кислоты (8,36 мкл, 0,146 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь загружали в предварительно промытый картридж 5g SCX-2, давали возможность произойти связыванию в течение 10 минут, промывали 80:20 дихлорметан: метанол, затем продукт элюировали смесью 80:20 дихлорметан:7M аммиак в метаноле. Полученный раствор концентрировали досуха, очищали хроматографией (картридж GraceResolv диоксид кремния 12g, дихлорметан:2M аммиак в метаноле, градиентное элюирование от 100:0 до 80:20) и лиофилизировали в течение ночи с получением названного соединения в виде белого твердого вещества (10 мг, 36%). 1H ЯМР (300 МГц, DMSO-d6): δ 9,87 (уш.с, 1H), 8,47 (с, 1H), 7,95 (уш.с, 1H), 7,65 (д, 2H), 7,56 (уш.д, 1H), 7,48 (дд, 1H), 7,18 (д, 1H), 5,10 (т, 1H), 4,97 (с, 2H), 4,53 (д, 2H), 3,12 (с, 3H), 2,57-3,05 (уш.м, 3H), 1,48-2,13 (уш.м, 13H). LCMS (метод C): RT=0,80 мин, m/z=567 [M+H]+.
Пример 182: 3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

Стадия 1: третбутил 4-(2-метокси-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат:
Суспензию 1-йод-2-метокси-4-нитробензола (600 мг, 2,150 ммоль), третбутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (997 мг, 3,23 ммоль) и 2M раствора карбоната натрия (3225 мкл, 6,45 ммоль) в 1,4-диоксане (6477 мкл) дегазировали путем барботирования азота в течение 10 минут, затем добавляли аддукт PdCl2(dppf)-дихлорметан (88 мг, 0,108 ммоль). Реакционную смесь нагревали до 120°C при микроволновом излучении в течение 20 минут. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (3×12 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении. Остаток очищали хроматографией (картридж диоксид кремния 50g, циклогексан:этилацетат, градиентное элюирование от 95:5 до 70:30) с получением названного соединения в виде желтого масла, которое превращалось в твердое вещество при стоянии (661 мг, 92%). 1H ЯМР (300 МГц, CDCl3): δ 7,82 (дд, 1H), 7,71 (д, 1H), 7,28 (д, 1H), 5,87 (уш.с, 1H), 4,07 (кв, 2H), 3,91 (с, 3H), 3,60 (т, 2H), 2,48 (уш.с, 2H), 1,49 (с, 9H). LCMS (метод C): RT=1,85 мин, m/z=279 [M-tBu+H]+.
Стадия 2: третбутил 4-(4-амино-2-метоксифенил)пиперидин-1-карбоксилат:
К суспензии третбутил 4-(2-метокси-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (400 мг, 1,196 ммоль) и 10% палладий на угле (127 мг, 0,120 ммоль) в этаноле (5 мл) добавляли формиат аммония (377 мг, 5,98 ммоль), и полученную смесь нагревали до 60°C в атмосфере азота в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит® и концентрировали досуха при пониженном давлении. Остаток повторно растворяли в дихлорметане (10 мл) и промывали насыщенным раствором бикарбоната натрия (10 мл). Водную фазу промывали дихлорметаном (2×10 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали досуха при пониженном давлении с получением названного соединения в виде розового твердого вещества (270 мг, 74%). 1H ЯМР (300 МГц, CDCl3): δ 6,90 (д, 1H), 6,20-6,30 (м, 2H), 4,20 (уш.с, 2H), 3,77 (с, 3H), 3,60 (уш.с, 2H), 2,94 (тт, 1H), 2,79 (уш.т, 2H), 1,74 (уш.д, 2H), 1,51 (тд, 2H), 1,47 (с, 9H). LCMS (метод C): RT=1,22 мин, m/z=251 [M-tBu+H]+.
Стадия 3: третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метоксифенил)пиперидин-1-карбоксилат:
3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он (210 мг, 0,590 ммоль) подвергали взаимодействию с третбутил 4-(4-амино-2-метокси-фенил)пиперидин-1-карбоксилатом (181 мг, 0,590 ммоль), следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (210 мг, 58%). 1H ЯМР (300 МГц, CDCl3): δ 8,73 (с, 1H), 7,59 (уш.с, 1H), 7,39-7,47 (м, 3H), 7,29 (дд, 1H), 7,02-7,12 (м, 2H), 4,89 (с, 2H), 4,22 (уш.с, 2H), 3,85 (с, 3H), 3,20 (с, 3H), 3,04 (тт, 1H), 2,82 (уш.т, 2H), 1,79 (уш.д, 2H), 1,59 (тд, 2H), 1,48 (с, 9H). LCMS (метод C): RT=1,89 мин, m/z=613 [M+H]+.
Стадия 4: 3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он:
Третбутил 4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-метоксифенил)пиперидин-1-карбоксилат (210 мг, 0,342 ммоль) подвергали реакции удаления защитной группы, следуя методике примера 31, с получением названного соединения в виде белого твердого вещества (168 мг, 96%). 1H ЯМР (300 МГц, DMSO-d6): δ 9,87 (уш.с, 1H), 8,48 (с, 1H), 7,68 (уш.с, 1H), 7,65 (д, 2H), 7,48 (дд, 1H), 7,20 (уш.д, 1H), 7,06 (д, 1H), 4,98 (с, 2H), 3,78 (с, 3H), 3,14 (с, 3H), 2,99 (д, 2H), 2,88 (тт, 1H), 2,49-2,62 (м, 2H), 1,60 (д, 2H), 1,45 (квд, 2H). LCMS (метод C): RT=0,85 мин, m/z=513 [M+H]+.
Пример 183: 3-(2,6-дихлорфенил)-7-((3-метокси-4-(1-метил-пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он
3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперидин-4-ил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он (50 мг, 0,97 ммоль) подвергали метилированию, следуя методике примера 35, с получением названного соединения в виде белого твердого вещества (42 мг, 82%). 1H ЯМР (300 МГц, DMSO-d6): δ 9,88 (уш.с, 1H), 8,48 (с, 1H), 7,69 (уш.с, 1H), 7,65 (д, 2H), 7,48 (дд, 1H), 7,21 (уш.д, 1H), 7,08 (д, 1H), 4,99 (с, 2H), 3,78 (с, 3H), 3,13 (с, 3H), 2,84 (д, 2H), 2,67-2,79 (м, 1H), 2,17 (с, 3H), 1,84-2,00 (м, 2H), 1,52-1,72 (м, 4H). LCMS (метод C): RT=0,86 мин, m/z=527 [M+H]+.
Примеры 184-185: 3-(2,6-дихлорфенил)-7-((4-((R)-3-(метокси-метил)пиперазин-1-ил)-3-((R)-2,2,2-трифтор-1-гидроксиэтил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он

(R)-третбутил 2-(метоксиметил)-4-(4-нитро-2-((R)-2,2,2-трифтор-1-гидроксиэтил)фенил)пиперазин-1-карбоксилат и (R)-трет-бутил 2-(метоксиметил)-4-(4-нитро-2-((S)-2,2,2-трифтор-1-гидроксиэтил)фенил)пиперазин-1-карбоксилат:
Тетрабутиламмония фторид (2 капли) добавляли к раствору 4-(R)-третбутил 4-(2-формил-4-нитрофенил)-2-(метоксиметил) пиперазин-1-карбоксилата (220 мг, 0,58 ммоль) и триметил(трифторметил)силана (165 мг, 1,160 ммоль) в 5 мл THF, затем смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли раствор TBAF (1,7 мл, 1 M в THF), и всю смесь перемешивали в течение еще 1 часа. После экстракции в системе этилацетат и вода, органическую фазу промывали солевым раствором и затем сушили над безводным MgSO4. Раствор фильтровали, и растворитель испаряли под вакуумом. Анализ методом LCMS (метод C) неочищенного остатка показывал присутствие двух диастереоизомеров (1,69 и 1,74 мин, m/z=420), которые разделяли методом хроматографии. Каждый диастереоизомер затем отдельно гидрировали в проточном реакторе H-Cube (картридж 10% Pd/C, заполненный H2, 25°C, 1 мл/мин). Каждый полученный диастереоизомерный анилин (1 экв) затем подвергали реакции сочетания с 3-(2,6-дихлорфенил)-1-метил-7-(метилтио)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-оном (1,1 экв), следуя методике примера 31, с получением двух диастереоизомеров названного продукта.
Диастереоизомер 1 (28 мг). ¹H ЯМР (400 МГц, DMSO-d6): δ 9,98 (с, 1H), 8,48 (с,1H),8,26 (уш.с, 1H), 7,65 (д, 2H), 7,61 (уш.с, 1H), 7,48 (дд, 1H), 7,27 (д, 1H), 6,75 (д, 1H), 5,69 (м, 1H), 4,98 (с, 2H),3,29 (м, 2H), 3,25 (с, 3H), 3,12 (с, 3H), 2,97 (м, 2H), 2,80 (м, 3H), 2,61 (м, 2H). LCMS (метод C): RT=0,87 мин, m/z=626 [M+H]+.
Диастереоизомер 2 (18 мг).¹H ЯМР (400 МГц, DMSO-d6): δ 9,98 (с, 1H), 8,48 (с, 1H),8,26 (уш.с, 1H), 7,65 (д, 2H), 7,61 (уш.с,1H),7,48 (дд, 1H), 7,27 (д, 1H), 6,75 (д, 1H), 5,67 (м, 1H), 4,98 (с, 2H),3,32 (м, 2H), 3,25(с, 3H), 3,12 (с, 3H), 3,00 (м, 2H), 2,9-2,55 (м, 6H), 2,61 (м, 2H). LCMS (метод C): RT=0,91 мин, m/z=626 [M+H]+.
Сравнение насыщенных соединений с предшествующим уровнем техники:
Соединения изобретения могут превосходить по своим физико-химическим свойствам соединения предшествующего уровня техники. Например, кинетическая растворимость соединений примеров, приведенных ниже, превосходит кинетическую растворимость соответствующих соединений примеров, приведенных в патентном документе WO2013126656.

Соединения настоящего изобретения могут также иметь более высокую метаболическую стабильность по сравнению с соединениями предшествующего уровня техники. Например, собственный клиренс из микросом человека соединений примеров, приведенных ниже, существенно меньше, чем для соответствующих соединений, раскрытых в патентных документах WO 2013126656 и WO 2013059485.

Кроме того, соединения настоящего изобретения могут иметь более низкую нецелевую активность и, поэтому, более высокий терапевтический индекс in vivo. Например, in vitro ингибирование hERG связано с сердечно-сосудистой токсичностью in vivo. Соединения настоящего изобретения обычно имеют низкую активность в отношении hERG, например, соединения примеров 1, 6, 10, 12, 16, 20 все имеют величину hERG IC50>10 мкM. Кроме того, соединения настоящего изобретения могут иметь более низкую активность в отношении hERG, чем соединения предшествующего уровня техники, например, соединение примера 5 настоящего изобретения характеризуется более низкой величиной ингибирования hERG, чем соответствующие аналоги из патентных документов WO2013126656, WO2013013031 и WO2013059485
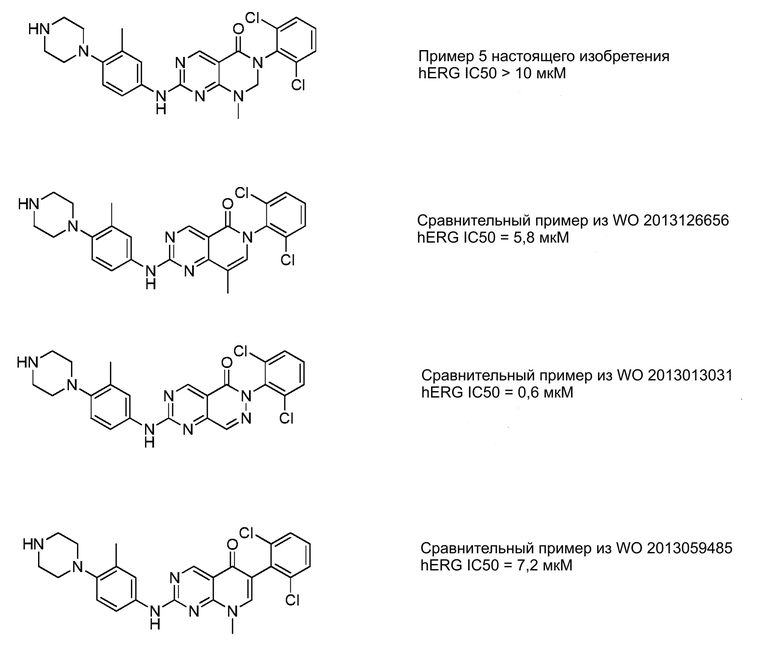
Метод 1: Измерение активности Wee-1 киназы
Для измерения активности Wee-1, приобретали производимый фирмой Sigma Aldrich пептид Poly(Lys Tyr(4:1)) гидробромид и использовали его в качестве субстрата. Активированную Wee-1 киназе приобретали у фирмы Nitrogen (PV3817), а набор для люминесцентного анализа ADP-Glo приобретали у фирмы Promega.
Все реакции проводили в объемах 60 мкл в реакционном буфере, содержащем 40 мM Трис-HCl и 20 мM хлорида магния, дополненном 0,1 мг/мл альбумином бычьей сыворотки и 2 мM DTT. Соединения последовательно разбавляли в буфере, и 5 мкл каждой концентрации соединения раскапывали пипеткой в белый 384-луночный планшет (Sigma Aldrich M6186). В каждую лунку добавляли 5 мкл Wee-1 фермента, и планшет центрифугировали в течение 1 минуты с целью смешения фермента и ингибитора.
Планшет инкубировали при комнатной температуре в течение 30 минут, затем добавляли 2,0 мкг/мл субстрата и 30 мкM АТФ в аликвоте 5 мкл. Планшет центрифугировали в течение одной минуты и инкубировали в течение 1 часа при комнатной температуре.
В каждую лунку добавляли 15 мкл останавливающего реагента ADP-Glo для прерывания реакции и уничтожения непрореагировавшего АТФ. Планшет инкубировали в течение еще 40 минут в темноте при комнатной температуре.
В каждую лунку добавляли 30 мкл реагента ADP-Glo для обнаружения киназы, превращающего АДФ в АТФ, катализирующего образование люциферина из люциферазы. Планшет встряхивали в течение 1 минуты и инкубировали в темноте в течение еще одного часа.
Люминесценцию каждой лунки определяли с помощью планшет-ридера Biotek Synergy4 HD, и для каждого испытуемого ингибитора вычисляли процент ингибирования активности киназы. В каждый планшет добавляли положительный (только киназа) и отрицательный (отсутствует киназа) контроль для выявления специфического взаимодействия киназы и ингибитора. Величину IC50 концентрации для каждого ингибитора рассчитывали из графической зависимости процента ингибирования киназы от концентрации ингибитора и кривой, полученной в результате аппроксимации моделью нелинейной регрессии.
Метод 2: Определение воздействия соединений на фосфорилирование cdc2 в Tyr15
Линии клеток колоректального рака HT-29 и HCT-116 получали из Американской коллекции типовых культур (ATCC) и в соответствии с предписанием сохраняли в среде Мак-Коя (Invitrogen), дополненной 10% эмбриональной бычьей сывороткой.
Клетки трипсинизировали из камеры для выращивания и подсчитывали, 100 мкл суспензии клеток, содержащих 6000 клеток, раскапывали в черные 96-луночные планшеты Co-star и инкубировали в течение ночи для прилипания клеток к поверхности при температуре 37°C и 5% CO2. Испытуемые соединения приготавливали в DMSO и разбавляли в среде, дополненной эмбриональной бычьей сывороткой. Инкубационную среду удаляли путем отсасывания, и в каждую лунку добавляли среду, дополненную разбавленным лекарственным средством.
Планшет помещали в инкубатор еще на восемь часов при 37°C и 5% CO2. После инкубации, среду, дополненную лекарственным средством, отсасывали из каждой лунки, и клетки промывали один раз в ледяном забуференном фосфатом физиологическом растворе (PBS). В каждую лунку 96-луночного планшета добавляли 100 мкл буфера для лизиса клеток (Cell Signalling Technologies #9803), содержащего 20 мM Трис, 150 мM NaCl, 1 мM EDTA, 1 мM EGTA, 1% Triton-X100, 2,5 мM пирофосфата натрия, 1 мM глицерофосфата, 1 мM Na3VO4 и 1 мкг/мл лейпептина, и инкубировали при 4°C в течение 30 минут. Образцы в планшете быстро замораживали при -80°C до момента использования. Непосредственно перед продолжением проведения анализа, планшет с образцами размораживали и центрифугировали при 4°C в течение 10 минут, и надосадочную жидкость переносили во вспомогательные пробирки или на 96-луночный планшет.
Клетки в надосадочной жидкости смешивали в соотношении 1:1 с буфером для разбавления образцов и встряхивали в течение одной минуты. 100 мкл разбавленного образца раскапывали в планшеты с нанесенным покрытием, содержащим кроличье поликлональное антитело для фосфо-cdc2 (Tyr15) (Cell Signalling Technologies PathScan kit #7176). Планшет герметизировали и инкубировали в течение ночи при 4 °C.
Удаляли с планшета крышку и отсасывали содержимое лунок, затем 3×5 минут промывали с помощью 200 мкл разбавленного промывного буфера. Между каждой промывкой, планшет плотно прижимали к промокательной бумаге для полного удаления раствора. В каждую лунку добавляли 100 мкл идентифицирующего антитела из набора, и планшет повторно герметизировали и инкубировали при 37°C в течение 1 часа. После инкубации, планшет промывали и обрабатывали таким же образом, как было описано выше.
В каждую испытуемую лунку добавляли 100 мкл связанного с пероксидазой хрена вторичного антитела, планшет герметизировали и инкубировали в течение тридцати минут при 37°C. После инкубации, планшет промывали, как было описано выше, затем добавляли 100 мкл 3,3’,5,5’-тетраметилбензидина (реагент TMB). Планшет герметизировали и инкубировали при комнатной температуре в течение 30 минут.
В каждую лунку добавляли 100 мкл останавливающего раствора, и днище планшета протирали безворсовой тканью перед спектрофотометрическим определением. Величину поглощения для каждой лунки считывали при 450 нм в течение 30 минут после добавления останавливающего раствора.
Рассчитывали процент фосфо-cdc2 относительно DMSO контроля и строили графическую зависимость от концентрации ингибитора, используя компьютерную программу GraphPad Prism. Данные аппроксимировали, используя нелинейный регрессионный анализ, и получали значения IC50.
Метод 3: Определение воздействия соединений на cdc2 фосфорилирование в Tyr15 с использованием набора для твердофазного иммуноферментного клеточного анализа ELISA (R&D Systems, KCB001)
Линии клеток колоректального рака HT-29 получали из Американской коллекции типовых культур (ATCC) и сохраняли в среде Мак-Коя 5A (Gibco®, Life technologies), дополненной 10% эмбриональной бычьей сывороткой (FCS).
Клетки трипсинизировали и подсчитывали, 100 мкл, содержащих 8000 клеток, высевали в каждой лунке черного 96-луночного микропланшета с прозрачным дном и инкубировали в течение ночи для прилипания клеток к поверхности при температуре 37°C и 5% CO2 в инкубаторе для культивирования клеток.
Испытуемые соединения приготавливали в DMSO и разбавляли в среде для культивирования клеток, дополненной FCS. Среду для культивирования удаляли из клеток, и среду, содержащую разбавления соединений, добавляли в каждую лунку. Планшет опять помещали в инкубатор еще на восемь часов при 37°C и 5% CO2.
После инкубации, клетки фиксировали путем замены среды, дополненной соединениями, на 100 мкл 4% формальдегида в 1x забуференном фосфатом физиологическом растворе (PBS) в течение 20 минут при комнатной температуре. Раствор формальдегида удаляли, и клетки промывали три раза с помощью PBS. Твердофазный иммуноферментный клеточный анализ ELISA (R&D Systems) проводили сразу после фиксации клеток, или планшет герметизировали и фиксированные клетки хранили в PBS при 4°C до момента использования (не более чем две недели).
Для продолжения анализа, PBS удаляли, и клетки промывали 3 раза в течение 5 минут с помощью 200 мкл 1x промывочного буфера. Каждую стадию промывки проводили при осторожном встряхивании. После промывки, добавляли 100 мкл 0,6% H2O2 в 1x промывном буфере (буфер для гашения), планшет герметизировали и инкубировали в течение 20 минут при комнатной температуре. После инкубации, клетки промывали 3 раза в течение 5 минут с помощью 200 мкл 1x промывного буфера при осторожном встряхивании. В каждую лунку добавляли 100 мкл блокирующего буфера, планшет опять герметизировали и инкубировали при комнатной температуре в течение 1 часа.
После блокирования, клетки промывали, как было описано выше. Приготавливали смесь первичных антител путем смешения двух первичных антител, кроличьего моноклонального антитела для фосфо-cdc2 (Tyr15) (Cell Signaling, #4539) и мышиного моноклонального антитела для суммы cdc2 p34 (Santa Cruz Biotechnology, sc-54), разбавляли 1:25 и 1:50, соответственно, в блокирующем буфере. В каждую лунку добавляли 60 мкл смеси первичных антител, планшет герметизировали и инкубировали в течение ночи при 4 °C.
Первичные тела удаляли, и клетки промывали, как было описано выше. В каждую испытуемую лунку добавляли 100 мкл смеси вторичных антител, содержавшей пероксидазу хрена (HRP), конъюгированную с козьим анти-кроличьим IgG, и щелочную фосфотазу (AP), конъюгированную с козьим анти-мышиным IgG, разбавленные 1:100 в блокирующем буфере. Планшет опять герметизировали и инкубировали в течение 2 часов при комнатной температуре.
Вторичные антитела удаляли, и клетки промывали 2 раза промывочным буфером, затем промывали 2 раза с помощью PBS. Каждую стадию промывки проводили в течение 5 минут при осторожном встряхивании. PBS удаляли, и в каждую лунку добавляли 75 мкл флуорогенного субстрата для HRP (субстрата F1). Планшет герметизировали и инкубировали в течение 1 часа при комнатной температуре в темноте. В каждую лунку непосредственно добавляли 75 мкл флуорогенного субстрата для AP (субстрата F2), планшет снова герметизировали и инкубировали в течение еще 40 минут при комнатной температуре, защищали от действия прямого солнечного света.
После инкубации, считывали флуоресценцию каждой лунки при 600 нм (сигнал субстрата 1, фосфорилированного белка) и затем при 450 нм (сигнал субстрата 2, общего белка).
Рассчитывали процент фосфо-cdc2 относительно DMSO контроля, нормализовали относительно общего cdc2 и строили графическую зависимость от концентрации ингибитора, используя компьютерную программу GraphPad Prism. Данные аппроксимировали, используя нелинейный регрессионный анализ, и получали значения IC50.
Метод 4: Определение CLint оценок, используя микросомы человеческой печени
Испытуемые соединения (конечная концентрация=1 мкM; конечная концентрация DMSO=0,1%) инкубировали в 0,1 M фосфатном буфере pH 7,4 с микросомами человеческой печени (0,5 мг белок/мл) при 37°C. Реакции инициировали путем добавления НАДФH в 0,1 M фосфатном буфере pH 7,4 (конечная концентрация 1 мM). Аликвоты объемом 40 мкл отбирали через 2, 5, 10, 15, 20, 30, 40 и 50 минут. Реакции прерывали с помощью 80 мкл ледяного метанола. Затем образцы замораживали в течение ночи, затем центрифугировали при 3500 об/мин в течение 20 минут при 4°C. Отбирали надосадочные жидкости и переносили их в аналитические планшеты и анализировали методом жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS).
Метод LC/MS/MS
Все образцы анализировали на хроматографе Waters Acquity I-Class, соединенном с масс-спектрометром Waters Xevo TQD. Применяли колонку Waters BEH C18 2,1x50 мм 1,7 мкм, и в качестве подвижных фаз использовали воду и метанол, содержащий 0,1% муравьиной кислоты в качестве модификатора. Анализ осуществляли путем мониторинга множественных реакций, и условия оптимизировали для каждого испытуемого соединения.
Анализ данных
Из графической зависимости натурального логарифма площади пика от времени определяли градиент линии. Затем рассчитывали, период полувыведения и собственный клиренс, используя следующие уравнения.
Элиминированная константа скорости (k)=(- градиент)
Период полувыведения (t1/2)(мин)=
Собственный клиренс (CLint)(мкл/мин/миллион клеток)=
где V=объем инкубации (мкл)/число клеток
Метод 5: Определение кинетической растворимости (в PBS буфере)
Испытуемые соединения (5 мкл; 10 мM DMSO исходный раствор) добавляли к 245 мкл PBS буфера pH 7,4 (Dulbecco A) в фильтрующем планшете Millipore MultiScreen® Solubility и смешивали при 300 об/мин при комнатной температуре на планшетном шейкере в течение 90 минут. В то же время были получены 5-точечные калибровочные кривые для каждого соединения в смеси ацетонитрил/PBS буфер (наивысшая концентрация 200 мкM). После фильтрации и компенсации матричного эффекта, калибровочные и исследовательские планшеты анализировали на планшет-ридере Bioteck Synergy 4 (240-400 нм). Вычисляли конечную концентрацию испытуемого соединения в фильтрате, используя наклон калибровочной кривой. Все 20 измерений проводили по три раза.
Метод 6: Определение hERG активности
Соединения испытывали на ингибирование гена специфических калиевых каналов сердца человека (hERG), используя систему для фиксация электрофизиологического потенциала IonWorks фирмы Essen BioScience. Для соединения строили 8-точечные кривые концентрация-воздействие на ток hERG как процент от сигнала до введения соединения, используя 3-кратные последовательные разбавления от 11 мкM, и результаты представлены в виде IC50 в мкM.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРИДО[4,3-d]ПИРИМИДИН-5-ОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ WEE-1 | 2014 |
|
RU2656342C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
| ЛЕЧЕНИЕ МИОДИСТРОФИИ ДЮШЕНА | 2007 |
|
RU2462458C2 |
| (5,6-дигидро)пиримидо[4,5-е]индолизины | 2015 |
|
RU2692479C2 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С PI3 КИНАЗАМИ, СОДЕРЖАЩАЯ ДАННОЕ ДЕЙСТВУЮЩЕЕ ВЕЩЕСТВО | 2016 |
|
RU2719367C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ФЕНИЛКАРБАМАТЫ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА ГИДРОЛАЗЫ АМИДОВ ЖИРНЫХ КИСЛОТ (FAAH) И МОДУЛЯТОРОВ D3 ДОПАМИНОВОГО РЕЦЕПТОРА (D3DR) | 2014 |
|
RU2693457C2 |
Изобретение относится к новому соединению формулы (I) и его фармацевтически приемлемой соли. Соединения обладают свойствами ингибиторов активности Wee-1 киназы и могут найти применение для производства лекарственного препарата для лечения или профилактики рака, опосредованного активностью Мее1 киназы. В формуле (I)
 (I)
(I)
R1 представляет собой фенильную группу, замещенную одним или несколькими атомами галогена и необязательно метильной группой; R2 и R3 независимо выбирают из группы, состоящей из атома водорода, атома дейтерия, метильной группы и фенильной группы; R4 представляет собой атом водорода, C1-C6-алкильную группу, которая необязательно дейтеризована или замещена метоксифенильной группой, фенильной группой, необязательно замещенной метильной группой, С1-C6-алкиламино-С1-C6-алкильной группой, циклопропильной группой или метилпиразолильной группой; или R2 представляет собой водород и R4 и R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют 5-членную гетероциклоалкильную группу; и R5 представляет собой фенильную группу, замещенную одной или несколькими группами, состоящими из С1-C6-алкильной группы, необязательно замещенной одним или несколькими атомами галогена и гидроксильной группой, C1-C6-алкоксигруппы, необязательно замещенной одним или несколькими атомами галогена, амино, С1-C6-алкиламино, гидрокси-С1-C6-алкильной группы, C1-C4-алкиламино-C1-C6-алкильной группы, необязательно замещенной одной или несколькими гидроксильными группами или карбонильной группой, аминосульфонильной группы, алкилиминосульфоксидной группы, атома галогена, цианогруппы, пиридинильной группы, пиразолильной группы, замещенной метильной группой, циклобутильной группы, замещенной аминогруппой, 5-6-членной гетероциклоалкильной группы, где 5-6-членная гетероциклоалкильная группа содержит в цикле 1-2 атома азота, или атом азота и атом кислорода, или атом азота и атом серы и его диоксид и где 5-6-членная гетероциклоалкильная группа необязательно замещена одной или несколькими С1-C4-алкильными группами, необязательно замещенными одним или несколькими атомами галогена, гидрокси-С1-C4-алкильной группы, C1-C4-алкокси-C1-C4-алкильной группы, атома галогена, гидроксикарбонил-C1-C4-алкильной группы, C3-C4-циклоалкильной группы, карбонильной группы, замещенной С1-C4-алкилом, С0-C4-алкиламино-С1-C4-алкильной группой; или R5 представляет собой фенильную группу, замещенную 7-8-членной моно- или бициклической гетероциклоалкильной группой, содержащей один или два атома азота и необязательно атом кислорода в цикле, где 7-8-членная моно- или бициклическая гетероциклоалкильная группа необязательно замещена C1-C4-алкильной группой; или R5 представляет собой фенильную группу, слитую с 6-8-членной моно- или бициклической гетероциклоалкильной группой, содержащей атом азота и необязательно атом кислорода в цикле, которая необязательно замещена C1-C4-алкильной группой. 4 н. и 22 з.п. ф-лы, 1 табл., 191 пр.
1. Соединение формулы (I)
 (I)
(I)
или его фармацевтически приемлемая соль,
где R1 представляет собой фенильную группу, замещенную одним или несколькими атомами галогена и необязательно метильной группой;
R2 и R3 независимо выбирают из группы, состоящей из атома водорода, атома дейтерия, метильной группы и фенильной группы;
R4 представляет собой атом водорода, C1-C6-алкильную группу, которая необязательно дейтеризована или замещена метоксифенильной группой, фенильной группой, необязательно замещенной метильной группой, С1-C6-алкиламино-С1-C6-алкильной группой, циклопропильной группой или метилпиразолильной группой;
или R2 представляет собой водород и R4 и R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют 5-членную гетероциклоалкильную группу; и
R5 представляет собой фенильную группу, замещенную одной или несколькими группами, состоящими из С1-C6-алкильной группы, необязательно замещенной одним или несколькими атомами галогена и гидроксильной группой, C1-C6-алкоксигруппы, необязательно замещенной одним или несколькими атомами галогена, амино, С1-C6-алкиламино, гидрокси-С1-C6-алкильной группы, C1-C4-алкиламино-C1-C6-алкильной группы, необязательно замещенной одной или несколькими гидроксильными группами или карбонильной группой, аминосульфонильной группы, алкилиминосульфоксидной группы, атома галогена, цианогруппы, пиридинильной группы, пиразолильной группы, замещенной метильной группой, циклобутильной группы, замещенной аминогруппой, 5-6-членной гетероциклоалкильной группы, где 5-6-членная гетероциклоалкильная группа содержит в цикле 1-2 атома азота, или атом азота и атом кислорода, или атом азота и атом серы и его диоксид и где 5-6-членная гетероциклоалкильная группа необязательно замещена одной или несколькими С1-C4-алкильными группами, необязательно замещенными одним или несколькими атомами галогена, гидрокси-С1-C4-алкильной группы, C1-C4-алкокси-C1-C4-алкильной группы, атома галогена, гидроксикарбонил-C1-C4-алкильной группы, C3-C4-циклоалкильной группы, карбонильной группы, замещенной С1-C4-алкилом, С0-C4-алкиламино-С1-C4-алкильной группой;
или R5 представляет собой фенильную группу, замещенную 7-8-членной моно- или бициклической гетероциклоалкильной группой, содержащей один или два атома азота и необязательно атом кислорода в цикле, где 7-8-членная моно- или бициклическая гетероциклоалкильная группа необязательно замещена C1-C4-алкильной группой;
или R5 представляет собой фенильную группу, слитую с 6-8-членной моно- или бициклической гетероциклоалкильной группой, содержащей атом азота и необязательно атом кислорода в цикле, которая необязательно замещена C1-C4-алкильной группой.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой группу, представленную формулой (a)
 (a)
(a)
где R1a и R1b каждый независимо выбирают из группы, состоящей из атома водорода, галогеновой группы и метильной группы, при условии, что по меньшей мере одна группа представляет собой галоген.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R1 представляет собой группу, представленную формулой (b)
 (b)
(b)
где R1a и R1b каждый независимо выбирают из группы, состоящей из атома водорода, галогеновой группы, метильной группы, при условии, что по меньшей мере одна группа представляет собой галоген.
4. Соединение по п. 3 или его фармацевтически приемлемая соль, где
R1a представляет собой атом водорода, галогеновую группу или метильную группу; и
R1b представляет собой галогеновую группу.
5. Соединение по любому одному из пп. 1, 2, 3 или 4 или его фармацевтически приемлемая соль, где R1 представляет собой 2-хлорфенильную группу.
6. Соединение по любому одному из пп. 1, 2, 3 или 4 или его фармацевтически приемлемая соль, где R1 представляет собой 2,6-дихлорфенильную группу.
7. Соединение по любому одному из пп. 1, 2, 3 или 4 или его фармацевтически приемлемая соль, где R1 представляет собой 2-хлор-6-фторфенильную группу.
8. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6 или 7 или его фармацевтически приемлемая соль, где R2 и R3 каждый независимо представляет собой атом водорода.
9. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6 или 7 или его фармацевтически приемлемая соль, где R2 и/или R3 представляет собой атом дейтерия.
10. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6 или 7 или его фармацевтически приемлемая соль, где R2 представляет собой метильную группу и R3 представляет собой атом водорода.
11. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6 или 7 или его фармацевтически приемлемая соль, где R2 представляет собой фенильную группу и R3 представляет собой атом водорода.
12. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 или его фармацевтически приемлемая соль, где R4 представляет собой C1-C6-алкильную группу.
13. Соединение по п. 12 или его фармацевтически приемлемая соль, где R4 представляет собой метильную группу.
14. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6 или 7 или его фармацевтически приемлемая соль, где R2 представляет собой атом водорода, R4 и R3 и кольцевые атомы, к которым они присоединены, взятые вместе, образуют 5-членную гетероциклоалкильную группу.
15. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14, или его фармацевтически приемлемая соль, где R5 представляет собой группу, представленную формулой (d)
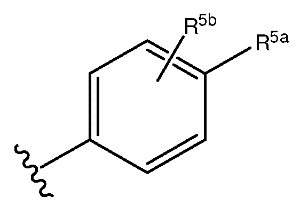 (d)
(d)
R5a выбирают из группы, состоящей из пиридинильной группы, пиразолильной группы, замещенной метильной группой, циклобутильной группы, замещенной аминогруппой и 5-6-членной гетероциклоалкильной группой, где 5-6-членная гетероциклоалкильная группа содержит в цикле 1-2 атома азота, или атом азота и атом кислорода, или атом азота и атом серы и его диоксид и где 5-6-членная гетероциклоалкильная группа необязательно замещена одной или несколькими C1-C4-алкильными группами, необязательно замещенными одним или несколькими атомами галогена, гидрокси-С1-C4-алкильной группы, C1-C4-алкокси-C1-C4-алкильной группы, атома галогена, гидроксикарбонил-C1-C4-алкильной группы, C3-C4-циклоалкильной группы и C1-C4-карбонильной группы, необязательно замещенной С1-C4-алкиламиногруппой;
R5b выбирают из группы, состоящей из атома водорода, атома галогена, C1-C6-алкильной группы, необязательно замещенной одним или несколькими атомами галогена и гидроксильной группой, C1-C6-алкоксигруппы, необязательно замещенной одним или несколькими атомами галогена, гидрокси-C1-C6-алкильной группы, C1-C4-алкиламино-C1-C6-алкильной группы, необязательно замещенной одной или несколькими гидроксильными группами или карбонильными группами, аминосульфонильной группы, цианогруппы.
16. Соединение по п. 15 или его фармацевтически приемлемая соль, где 5-6-членную гетероциклоалкильную группу выбирают из группы, состоящей из пиперидинильной группы, морфолинильной группы и пиперазинильной группы.
17. Соединение по любому одному из пп. 15 или 16 или его фармацевтически приемлемая соль, где
R5a представляет собой 5-6-членную гетероциклоалкильную группу, необязательно замещенную с помощью одного или более заместителей, выбранных из группы, состоящей из C1-C3-алкильной группы, гидрокси-C1-C3-алкильной группы и C1-C3-алкокси-C1-C3-алкильной группы; и
R5b представляет собой атом водорода, атом галогена, C1-C3-алкоксигруппу, гидрокси-C1-C3-алкильную группу или C1-C3-алкильную группу.
18. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 или его фармацевтически приемлемая соль, где
R5 представляет собой группу, представленную формулой (e)
 (e)
(e)
где R5g выбирают из группы, состоящей из атома водорода и C1-C3-алкильной группы.
19. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение выбирают из следующих соединений:
(1) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(2) 3-(2,6-дихлорфенил)-1-метил-7-((4-(2-(метиламино)-этокси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(3) 3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(4) 2-(4-(4-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)уксусной кислоты гидрохлорид;
(5) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)метил)-4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(6) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(7) 3-(2,6-дихлорфенил)-7-((3-фтор-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(8) 3-(2,6-дихлорфенил)-1-(4-метоксибензил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(9) 3-(2,6-дихлорфенил)-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(10) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(11) 3-(2,6-дихлорфенил)-7-((3-циано-4-(пиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(12) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил-метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(13) 7-((4-(4-(2-аминоацетил)пиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(14) 3-(2,6-дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-изохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(15) 3-(2,6-дихлорфенил)-2,2-дидейтеро-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(16) (R)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(17) (S)-3-(2,6-дихлорфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(18) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(пиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(19) (R)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(20) (S)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(21) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(22) 3-(2,6-дихлорфенил)-1-метил-7-((2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(23) (рац)-3-(2,6-дихлорфенил)-1-метил-2-фенил-7-((1,2,3,4-тетрагидроизохинолин-7-ил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(24) 3-(2-хлорфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(25) 3-(2-хлор-6-фторфенил)-1-метил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(26) 3-(2,6-дихлорфенил)-1,2-диметил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(27) 3-(2,6-дихлорфенил)-1-метил-7-((4-(морфолинометил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(28) 6-(2,6-дихлорфенил)-2-((4-(пиперазин-1-ил)фенил)-амино)-6a,7,8,9-тетрагидропиримидо[5,4-e]пирроло[1,2-a]-пиримидин-5(6H)-он;
(29) 3-(2,6-дихлорфенил)-1-метил-7-((4-((4-метилпиперазин-1-ил)метил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он; и
(30) (рац)-3-(2,6-дихлорфенил)-1-метил-7-((4-(S-метил-сульфонимидоил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он.
20. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение выбирают из следующих соединений:
(31) 3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(32) (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметил пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(33) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(4-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(34) (R)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(35) 3-(2,6-дихлорфенил)-1-метил-7-((4-(5-метилгексагидро-пирроло[3,4-c]пиррол-2(1H)-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(36) 3-(2,6-дихлорфенил)-7-((4-(гексагидропирроло[3,4-c]-пиррол-2(1H)-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(37) 7-((4-(1-аминоциклобутил)фенил)амино)-3-(2,6-дихлор-фенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(38) (R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(39) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(40) 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(41) 3-(2,6-дихлорфенил)-1-метил-7-((4-морфолинофенил)-амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(42) 3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидотиоморфолино) фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(43) (R)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(44) 3-(2,6-дихлорфенил)-1-метил-7-((4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(45) 3-(2,6-дихлорфенил)-1-метил-7-((4-((2R,5S)-2,4,5-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(46) (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(47) (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметилпиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(48) (S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидрокси-метил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(49) (S)-3-(2,6-дихлорфенил)-7-((4-(3-(гидроксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(50) (S)-3-(2,6-дихлорфенил)-7-((3-фтор-4-(3-(гидроксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(51) 7-((4-(2,6-диазаспиро[3,3]гептан-2-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(52) 3-(2,6-дихлорфенил)-1-метил-7-((4-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(53) (R)-3-(2-хлор-6-фторфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(54) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-фенил)амино)-3-(2-хлор-6-фторфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(55) 3-(2-хлор-6-фторфенил)-1-метил-7-((3-метил-4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(56) 3-(2-хлор-6-фторфенил)-7-((3-(гидроксиметил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(57) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-морфолино-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(58) (R)-3-(2,6-дихлорфенил)-1-(1-метил-1H-пиразол-3-ил)-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(59) (R)-3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(60) (R)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(61) (S)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(62) 3-(2,6-дихлорфенил)-1-метил-7-((4-(3-(трифторметил)-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(63) 3-(2,6-дихлорфенил)-1-метил-7-((4-(3,3,4-триметил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(64) 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)-фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(65) (R)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(66) (S)-3-(2,6-дихлорфенил)-7-((4-(3-(метоксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(67) (R)-3-(2,6-дихлорфенил)-7-((4-(3,4-диметилпиперазин-1-ил)-3-(метоксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(68) 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-метил-3-(трифторметил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(69) 3-(2,6-дихлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(70) 3-(2,6-дихлорфенил)-1-метил-7-((6,7,8,9-тетрагидро-5H-5,8-эпиминобензо[7]аннулен-3-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(71) 3-(2,6-дихлорфенил)-1-метил-7-((4-(9-метил-3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(72) 3-(2,6-дихлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(73) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-морфолино-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(74) 7-((4-(3,8-диазабицикло[3,2,1]октан-3-ил)фенил) амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(75) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(76) 3-(2,6-дихлорфенил)-1-метил-7-((2'-метил-2',3'-дигидро-1'H-спиро[циклопропан-1,4'-изохинолин]-7'-ил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(77) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(78) (R)-3-(2,6-дихлорфенил)-1-d3-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(79) 3-(2,6-дихлорфенил)-7-((4-((2R,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(80) 3-(2,6-дихлорфенил)-7-((4-((2S,5R)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(81) 3-(2,6-дихлорфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(82) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(83) 3-(2,6-дихлорфенил)-7-((4-((3R,5S)-3,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(84) 3-(2,6-дихлорфенил)-7-((3-метокси-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(85) (R)-1-циклопропил-3-(2,6-дихлорфенил)-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(86) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(87) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-ил-окси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(88) 3-(2,6-дихлорфенил)-1-метил-7-((4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(89) 3-(2-хлор-6-фторфенил)-7-((3-(гидроксиметил)-4-(4-изопропилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(90) 3-(2,6-дихлорфенил)-1-метил-7-((4-((1-метилпиперидин-4-ил)окси)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(91) 3-(2,6-дихлорфенил)-1-метил-7-((3-((1-метилпиперидин-4-ил)амино)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(92) 3-(2-хлор-6-метилфенил)-1-метил-7-((4-(пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(93) 3-(2-хлор-6-фторфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(94) (R)-3-(2-хлорфенил)-1-метил-7-((4-(3-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(95) 3-(2,6-дихлорфенил)-1-метил-7-((4-(1-метилпиперидин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(96) 3-(2,6-дихлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(97) 3-(2-хлорфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(98) 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(99) (R)-3-(2-хлорфенил)-7-((3-фтор-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(100) (R)-3-(2-хлор-6-метилфенил)-1-метил-7-((4-(3-метил-пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(101) (R)-3-(2-хлор-6-метилфенил)-7-((3-(метоксиметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(102) 3-(2-хлор-6-метилфенил)-7-((4-((2R,5S)-2,5-диметил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(103) 3-(2-хлорфенил)-7-((4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(104) 7-((4-((1S,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-фенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(105) 7-((4-(4,7-диазаспиро[2,5]октан-7-ил)фенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(106) 3-(2-хлорфенил)-7-((4-(3,3-диметилпиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(107) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метоксифенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(108) 3-(2-хлорфенил)-7-((3-(метоксиметил)-4-(пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(109) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(110) 3-(2-хлорфенил)-7-((4-((2R,5S)-2,5-диметилпиперазин-1-ил)-3-фторфенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(111) 3-(2-хлорфенил)-7-((4-((2S,5R)-2,5-диметилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(112) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2-хлор-6-метилфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(113) 3-(2-хлорфенил)-7-((4-(4-этилпиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(114) 3-(2-хлор-6-метилфенил)-7-((3-(гидроксиметил)-4-(8-метил-3,8-диазабицикло[3,2,1]октан-3-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(115) 7-((4-(4-циклопропилпиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(116) 3-(2,6-дихлорфенил)-7-((4-(4-(2-гидроксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(117) 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-метилпиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(118) 7-((4-((1R,4R)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-метилфенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(119) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперидин-4-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(120) 3-(2,6-дихлорфенил)-7-((4-(4-(2-метоксиэтил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(121) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-((1R,4R)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(122) 3-(2,6-дихлорфенил)-7-((4-(4-этилпиперазин-1-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(123) 3-(2,6-дихлорфенил)-7-((4-(4-(2-(диметиламино)-ацетил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(124) 3-(2,6-дихлорфенил)-1-метил-7-((4-(4-(2-(метиламино)-ацетил)пиперазин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(125) 7-((4-(4-ацетилпиперазин-1-ил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(126) (R)-3-(2-хлор-6-фторфенил)-7-((4-(3-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(127) (S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидроксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(128) (S)-3-(2,6-дихлорфенил)-7-((4-(2-(гидроксиметил)-4-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(129) (R)-3-(2,6-дихлорфенил)-7-((4-(2-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(130) (S)-3-(2,6-дихлорфенил)-7-((4-(2-(метоксиметил)-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(131) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(132) (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметокси)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(133) (S)-3-(2,6-дихлорфенил)-7-((4-(гексагидропиразино-[2,1-c][1,4]оксазин-8(1H)-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(134) 3-(2,6-дихлорфенил)-7-((3-(2,2-дифтор-1-гидрокси-этил)-4-((R)-3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(135)(R)-3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(3-(метоксиметил)пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(136) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиперидин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(137) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пирролидин-1-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(138) 7-((4-(4-ацетилпиперазин-1-ил)-3-(пирролидин-1-ил-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(139) (R)-3-(2,6-дихлорфенил)-7-((3-(дифторметил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(140) (R)-3-(2,6-дихлорфенил)-7-((3-(2-гидроксипропан-2-ил)-4-(3-метилпиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо [4,5-d]пиримидин-4(1H)-он;
(141) 3-(2,6-дихлорфенил)-1-метил-7-((4-(пиперазин-1-ил)-3-(((2,2,2-трифторэтил)амино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(142) (R)-3-(2,6-дихлорфенил)-7-((2-фтор-4-(3-метил-пиперазин-1-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(143) 7-((4-(4-ацетилпиперазин-1-ил)-3-((метиламино)метил)-фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(144) 7-((4-(4-ацетилпиперазин-1-ил)-3-((диметиламино)-метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(145) N-(5-((6-(2,6-дихлорфенил)-8-метил-5-оксо-5,6,7,8-тетрагидропиримидо[4,5-d]пиримидин-2-ил)амино)-2-(пиперазин-1-ил)бензил)ацетамид;
(146) 3-(2,6-дихлорфенил)-7-((4-(1,1-диоксидотиоморфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(147) 3-(2,6-дихлорфенил)-7-((4-(4,4-дифторпиперидин-1-ил)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(148) 3-(2,6-дихлорфенил)-7-((4-((2S,6R)-2,6-диметил-морфолино)-3-((метиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(149) 3-(2,6-дихлорфенил)-7-((3-((диметиламино)метил)-4-(1,1-диоксидотиоморфолино)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(150) 3-(2,6-дихлорфенил)-7-((4-(4,4-дифторпиперидин-1-ил)-3-((диметиламино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(151) 3-(2,6-дихлорфенил)-7-((3-((диметиламино)метил)-4-((2S,6R)-2,6-диметилморфолино)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(152) 3-(2,6-дихлорфенил)-7-((4-фтор-3-((метиламино)метил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(153) 3-(2,6-дихлорфенил)-7-((4-(диметиламино)-3-((метил-амино)метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(154) 3-(2,6-дихлорфенил)-7-((3-((диметиламино)метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(155) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиридин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(156) 3-(2,6-дихлорфенил)-1-метил-7-((3-((метиламино)-метил)-4-(пиридин-3-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(157) 7-((4-бром-3-((метиламино)метил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(158) 3-(2,6-дихлорфенил)-7-((4-((3S,5R)-3,5-диметил-пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(159) 3-(2,6-дихлорфенил)-7-((4-метокси-3-((метиламино)-метил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(160) 3-(2,6-дихлорфенил)-1-метил-7-((4-(1-метил-1H-пиразол-4-ил)-3-((метиламино)метил)фенил)амино)-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(161) 3-(2,6-дихлорфенил)-7-((4-(4-(2-(диметиламино)-ацетил)пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(162, 163, диастереомеры) 3-(2,6-дихлорфенил)-7-((3-((R)-2,2-дифтор-1-гидроксиэтил)-4-((R)-3-(метоксиметил)пиперазин-1-ил)фенил)-амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(164) 7-((4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(165) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1-метил-пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(166) 3-(2,6-дихлорфенил)-1-метил-7-((1,2,3,4-тетрагидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(167) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(168) 3-(2,6-дихлорфенил)-1-(3-((метиламино)метил)фенил)-7-((4-морфолинофенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(169) 3-(2,6-дихлорфенил)-1-этил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(170) 3-(2,6-дихлорфенил)-7-((4-((3S,5R)-3,5-диметил-пиперазин-1-ил)-3-(гидроксиметил)фенил)амино)-1-этил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(171) 3-(2,6-дихлорфенил)-1-фенил-7-((4-(пиперазин-1-ил)-фенил)амино)-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(172) 3-(2,6-дихлорфенил)-7-((3-(2-(диметиламино)-1-гидроксиэтил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(173) 7-((4-(3-окса-7,9-диазабицикло[3,3,1]нонан-7-ил)-3-(гидроксиметил)фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(174) 3-(2,6-дихлорфенил)-7-((3-((этиламино)метил)-4-морфолинофенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(175) 3-(2,6-дихлорфенил)-1-метил-7-((9-метил-1,2,3,4-тетрагидро-1,4-эпиминонафталин-6-ил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(176) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(177) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он;
(178) 3-(2,6-дихлорфенил)-1-метил-7-((3-метил-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(179) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(180) 3-(2,6-дихлорфенил)-7-((3-(гидроксиметил)-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фенил)амино)-1-метил-2,3-дигидро-пиримидо[4,5-d]пиримидин-4(1H)-он;
(181) 7-((4-(1-циклобутилпиперидин-4-ил)-3-(гидроксиметил)-фенил)амино)-3-(2,6-дихлорфенил)-1-метил-2,3-дигидропиримидо-[4,5-d]пиримидин-4(1H)-он;
(182) 3-(2,6-дихлорфенил)-7-((3-метокси-4-(пиперидин-4-ил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он;
(183) 3-(2,6-дихлорфенил)-7-((3-метокси-4-(1-метил-пиперидин-4-ил)фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]-пиримидин-4(1H)-он; и
(184, 185, диастереомеры) 3-(2,6-дихлорфенил)-7-((4-((R)-3-(метоксиметил)пиперазин-1-ил)-3-((R)-2,2,2-трифтор-1-гидроксиэтил)-фенил)амино)-1-метил-2,3-дигидропиримидо[4,5-d]пиримидин-4(1H)-он.
21. Фармацевтическая композиция, обладающая свойствами ингибитора Мее-1 киназы, включающая эффективное количество соединения по любому из пп. 1-20 или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель.
22. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 или его фармацевтически приемлемая соль для применения в терапии лечения и профилактики рака, опосредованного активностью Мее1 киназы.
23. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 для применения в качестве лекарственного препарата для применения в терапии лечения и профилактики рака, опосредованного активностью Мее1 киназы.
24. Соединение по любому одному из пп. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 для применения при лечении или профилактики рака, опосредованного активностью Мее1 киназы.
25. Применение соединения по любому из пп. 1-20 для производства лекарственного препарата для лечения или профилактики рака, опосредованного активностью Мее1 киназы.
26. Способ лечения или профилактики рака, опосредованного активностью Мее1 киназы, включающий введение пациенту, если он в этом нуждается, эффективного количества соединения по любому одному из пп. 1-20 или фармацевтической композиции по п. 21.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| СОСУД ДЛЯ БЫСТРО ИСПАРЯЮЩИХСЯ КЛЕЙКИХ ВЕЩЕСТВ | 1930 |
|
SU23796A1 |
| Xu,Tianfeng et al., Pyrimido[4,5-d]pyrimidin-4(1H)-one Derivatives as Selective Inhibitors of EGFR Threonine 790 to Methionine 790 (T790M) Mutants | |||
| Angewandte Chemie, International Edition, 2013, 52(32), 8387-8390 (English) (реферат, найдено DATABASE CASREACT (Online), Retrieved from STN, AN 159:339869, соединения с RN 1449423-84-0P, RN 1449423-85-1P, RN 1449423-86-2P, RN 1449423-87-3P, RN 1449423-88-4P, RN 1449423-89-5P, RN 1449423-90-8P, RN 1449423-91-9P | |||
| Martin, Mathew W | |||
| et al., Structure-Based Design of Novel 2-Amino-6-phenyl-pyrimido[5',4':5,6]pyrimido[1,2-a]benzimidazol-5(6H)-ones as Potent and Orally Active Inhibitors of Lymphocyte Specific Kinase (Lck): Synthesis, SAR, and In Vivo Anti-Inflammatory Activity | |||
| Journal of Medicinal Chemistry, 2008, 51(6), 1637-1648 (English) | |||
| Liew, Chin Y | |||
| et al., SVM Model for Virtual Screening of Lck Inhibitors | |||
| Journal of Chemical Information and Modeling, (English), 2009, 49 (4), 877-885 (реферат, найдено DATABASE CASREACT (Online), Retrieved from STN, AN 150:413026, соединения с RN 847950-09-8, RN 847950-16-7, RN 847950-17-8, RN 847950-18-9, RN 847950-19-0, RN 847950-25-8, RN 847950-26-9, RN 847950-27-0, RN 847950-28-1, RN 847950-36-1, RN 847950-38-3, RN 847950-42-9, RN 847950-71-4, RN 847950-79-2, RN 847950-80-5, RN 847950-81-6, RN 847950-84-9, RN 847950-85-0, RN 1140480-47-2. | |||

