Настоящее изобретение относится к (5,6-дигидро)пиримидо[4,5-е]индолизинам, к содержащим эти соединения фармацевтическим композициям и к их применению в терапии. В частности, настоящее изобретение относится к применению (5,6-дигидро)пиримидо[4,5-е]индолизинов в лечении рака.
Настоящее изобретение относится к химическим соединениям, которые модулируют активность протеинкиназ, в частности, ингибируют активность протеинкиназы TTK (EC 2.7.12.1). TTK, которую часто называют Mps1, является компонентом митотической контрольной точки, механизма, который обеспечивает правильность сегрегации сестринских хроматид по двум дочерним клеткам во время деления клеток. Дефекты митотической контрольной точки или ингибирование митотической контрольной точки приводят к анеуплоидии. В ходе эмбрионального развития подавляющее большинство единичных ошибок сегрегации не являются переносимыми (Cohen, J., Science 296: 2164, 2002). Напротив, анеуплоидия является частым генетическим отклонением в солидных опухолях у человека (Lengauer, С. et al., Nature 396, 643; 1998) и показателем неблагоприятного прогноза при раке молочной железы, легкого, мозга (Carter, S.L., et al., Nat. Genet. 38: 1043; 2006; Tannous, B.A., et al., J. Nat. Cane. Inst. 105, 1322; 2013) и колоректальном раке (Walther, A. et al., Gut 57: 941; 2008; Sheffer, M. et al., Proc. Natl. Acad. Sci. USA 106: 7131, 2009). Экспрессия TTK увеличивается при раке молочной железы с хромосомной нестабильностью (Yuan, В. et al., Clin. Cancer Res. 12: 405; 2006) и, в частности, при трижды негативном раке молочной железы, наиболее агрессивном типе рака молочной железы (Maire, V. et al., PLoS ONE 8(5) e63712; 2013). Уменьшение экспрессии TTK путем РНК-интерференции приводило к значительной неправильной сегрегации хромосом и гибели клеток, но не влияло на жизнеспособность нормальных (нетрансформированных) клеток (Yuan, В. et al.; Maire, V. et al.; Janssen, A. et al., Proc. Natl. Acad. Sci. USA 106: 19108; 2009). Частичное ингибирование экспрессии TTK путем РНК-интерференции в различных опухолевых клеточных линиях вызывало небольшие неправильные сегрегации хромосом, но не летальность (Janssen, А. et al.). Однако эти клетки являлись более чувствительными к обработке небольшими дозами антимитотических агентов, таких как паклитаксел (Janssen, A. et al.) и винкристин (Tannous, В.А., et al.). Приведенные выше сведения являются биологической основой для рассмотрения ингибиторов TTK в качестве подхода для селективной противораковой терапии.
Ингибиторы киназы TTK являются полезными для лечения множества видов рака и их можно применять в качестве отдельных агентов или в комбинации с другими химиотерапевтическими агентами.
WO 2012/101032 A1 (Nerviano Medical Sciences SRL) относится к трициклическим пиррольным производным, которые модулируют активность протеинкиназ и поэтому полезны в лечении заболеваний, вызванных разрегулированной активностью протеинкиназ.
Компанией Bayer Schering Pharma A.G. раскрыты в качестве ингибиторов TTK триазолопиридины в WO 2011/063908 A1 и имидазопиразины в WO 2012/080229 A1 и WO 2011/013729 A1. После определения характеристик трех репрезентативных ингибиторов было выявлено ингибирование активности киназы TTK с IC50 от 1 до 10 нМ и ингибирование пролиферации раковых клеточных линий человека с IC50 в диапазоне от 160 нМ до более чем 10 мкМ ( , М. et al., Cell Death Different. 20: 1532; 2013). Было показано, что одно из соединений, Mps-BAY2b, ингибировало рост клеток HeLa-Matu опухоли шейки матки в мышиной модели ксенотрансплантата и увеличивало эффективность паклитаксела в этой модели (, М. et al.), что, таким образом, подтверждало химиотерапевтический сенсибилизирующий эффект ингибирования TTK, ранее продемонстрированный с помощью методик РНК-интерференции (Janssen, A. et al.).
, М. et al., Cell Death Different. 20: 1532; 2013). Было показано, что одно из соединений, Mps-BAY2b, ингибировало рост клеток HeLa-Matu опухоли шейки матки в мышиной модели ксенотрансплантата и увеличивало эффективность паклитаксела в этой модели (, М. et al.), что, таким образом, подтверждало химиотерапевтический сенсибилизирующий эффект ингибирования TTK, ранее продемонстрированный с помощью методик РНК-интерференции (Janssen, A. et al.).
Компанией Nerviano Medical Sciences S.R.L. в WO 2009/156315 A1 раскрыты пиразолохиназолины в качестве ингибиторов TTK. Соединение NMS-Р715, описанное в этом патенте, ингибировало активность киназы TTK с полумаксимальной ингибирующей концентрацией (IC50) от 8 до 182 нМ, в зависимости от того, была ли включена в тест стадия предварительного инкубирования или нет (Colombo, R., et al., Cancer Res. 70: 10255; 2010). NMS-P715 также ингибировало пролиферацию раковых клеточных линий различного опухолевого происхождения с IC50 1 мкМ и выше и ингибировало рост опухолей в мышиных моделях ксенотрансплантата А375 и А2780 (Colombo, R. et al.).
Компанией Myriad Pharmaceuticals, Inc. в WO 2010/111406 A2 раскрыты соединения, представляющие собой ингибиторы TTK. Репрезентативное соединение, MPI 4079605, ингибировало TTK с IC50 1,8 нМ (Tardif, K.D., et al., Mol. Cancer Res. 10: 2267; 2011). Обработка раковых клеточных линий в течение 72 часов выявила много клеточных линий с небольшой чувствительностью (Tardif, K.D., et al.). MPI 4079605 является структурно схожим с реверсином и MPS1-IN-1, двумя другими опубликованными ингибиторами TTK (Kwiatkowski, N., et al., Nat. Chem. Biol. 6: 359; 2010; Santiguida, S., et al., J. Cell. Biol. 190: 73; 2010).
Существует явная необходимость в ингибиторах TTK с мощной активностью в отношении ингибирования киназы и антипролиферативной активностью.
Авторы изобретения синтезировали ряд (5,6-дигидро)пиримидо[4,5-е]индолизинов и обнаружили, что эти соединения являются очень эффективными ингибиторами активности киназы TTK и пролиферации роста опухолевых клеток.
Согласно настоящему изобретению предложены (5,6-дигидро)пиримидо[4,5-е]индолизиновые производные.
Более конкретно, согласно настоящему изобретению предложены (5,6-дигидро)пиримидо[4,5-е]индолизиновые производные формулы I или их фамрмацевтически приемлемые соли.
Согласно настоящему изобретению предложены соединения, которые ингибируют активность TTK, их применение для лечения гиперпролиферативных расстройств, в частности видов рака, вызванных или ассоциированных с хромосомной нестабильностью или анеуплоидией, в виде отдельного агента или в комбинации с другими активными ингредиентами, а также фармацевтические композиции, содержащие такие соединения и фармацевтические носители.
Цель настоящего изобретения заключается в предоставлении (5,6-дигидро)пиримидо[4,5-е]индолизинов, фармацевтических композиций, содержащих эти соединения, и их применения в терапии. В частности, настоящее изобретение относится к применению (5,6-дигидро)пиримидо[4,5-е]индолизинов в лечении рака.
Более конкретно, согласно настоящему изобретению предложены (5,6-дигидро)пиримидо[4,5-е]индолизины формулы I или их фармацевтически приемлемые соли
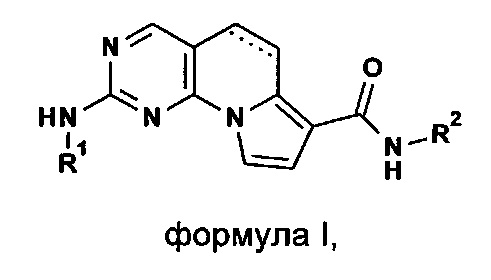
где
R1 и R2 независимо выбраны из группы, состоящей из:
а) (6-10C)арила,
б) (1-5C)гетероарила,
где обе группы возможно могут быть замещены.
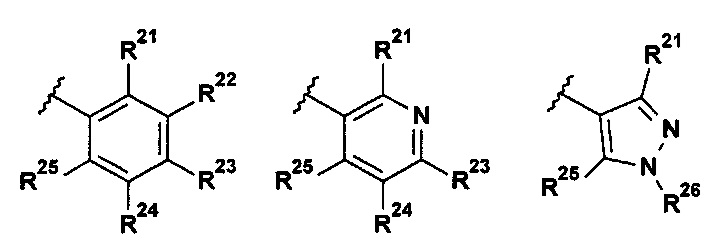
В одном воплощении R1 выбран из группы, состоящей из:
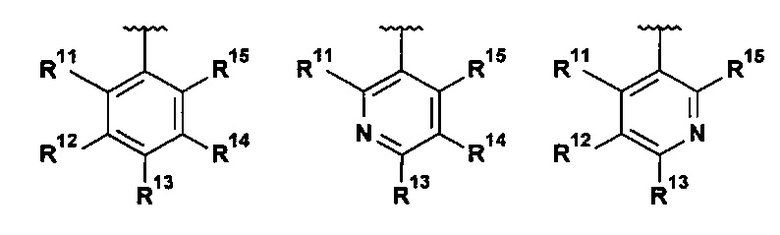
 ;
;
R11 представляет собой H, галоген, (1-2C)алкил, (2-3C)алкенил, (2-3C)алкинил, (1-2C)алкокси или OC2H3, где все алкильные и алкокси группы возможно замещены одним или более галогенами;
R12 представляет собой H, галоген, (1-2C)алкил или (1-2C)алкокси;
R13 представляет собой R131CH2, R132O, R133R134N, R135C(O), R136S, R136S(O), R136S(O)(NH), R137SO2, (2-7C)гетероциклоалкил или (1-5C)гетероарил, где каждый гетероциклоалкил или гетероарил возможно замещен (1-2C)алкилом, фтором, гидроксилом, оксо, (1-2C)алкокси, (1-6C)алкилкарбонилом, (1-6C)алкилсульфонилом, (1-5C)алкоксикарбонилом, (1-6C)алкиламинокарбонилом, (3-6C)циклоалкилкарбонилом, (2-7C)гетероциклоалкилкарбонилом или ди[(1-2C)алкил]амино, где каждый алкилкарбонил, алкилсульфонил, алкоксикарбонил, алкиламинокарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором, гидроксилом, циано, оксо или (1-2С)алкокси;
R131 представляет собой (1-6С)алкилкарбониламино, (3-6С)циклоалкилкарбониламино или (2-7С)гетероциклоалкилкарбониламино, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила или (1-2С)алкокси;
R132 представляет собой (1-6C)алкил, (3-6C)циклоалкил, (2-7С)гетероциклоалкил, (6-10C)арил или (1-5С)гетерогарил, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, галогена, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино или (2-7С)гетероциклоалкила;
R133 представляет собой (1-6C)алкил, (3-6C)циклоалкил, (2-7C)гетероциклоалкил, (1-6С)алкилкарбонил, (1-5С)алкоксикарбонил, (3-6С)циклоалкилкарбонил или (2-7С)гетероциклоалкилкарбонил, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, галогена, гидроксила или (1-2С)алкокси, ди[(1-2С)алкил]амино или (2-7С)гетероциклоалкила;
R134 представляет собой водород или (1-2C)алкил;
R135 представляет собой (2-7C)гетероциклоалкил, (1-6С)алкиламино, ди[(1-6С)алкил]амино, (2-7С)гетероциклоалкиламино или (3-6С)циклоалкиламино, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино, (2-7С)гетероциклоалкила, оксо, циано или амино;
R136 представляет собой (1-6C)алкил, (3-6C)циклоалкил, (2-7С)гетероциклоалкил, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила или (1-2С)алкокси;
R137 представляет собой (1-6C)алкил, (3-6C)циклоалкил, (2-7С)гетероциклоалкил, (1-6C)алкиламино, ди[(1-6С)алкил]амино, (2-7С)гетероциклоалкиламино или (3-6C)циклоалкиламино, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила или (1-2С)алкокси;
R14 представляет собой H, галоген, (1-2C)алкил или (1-2С)алкокси; и
R15 представляет собой H, галоген.
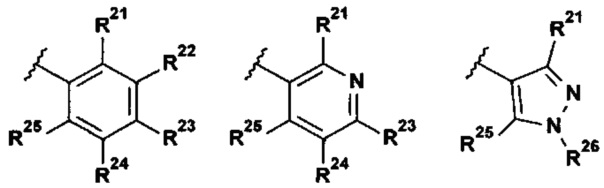
В указанной выше формуле I R2 выбран из группы, состоящей из:
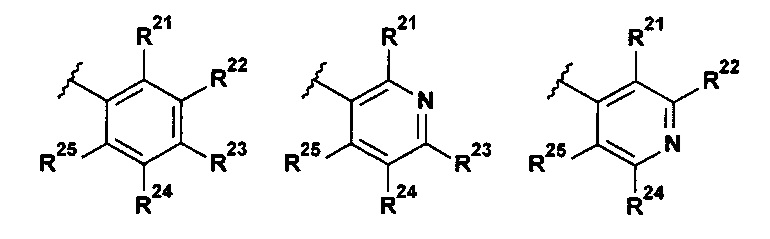
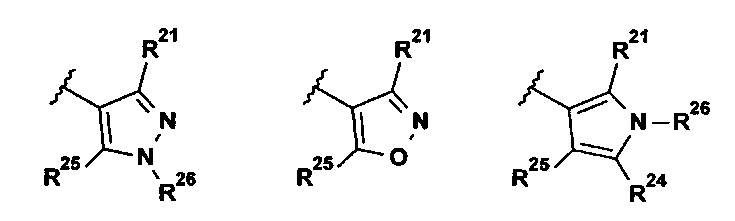 ;
;
R21 представляет собой Н, галоген, (1-3C)алкил, (1-2С)алкокси, гидрокси(1-2С)алкил, (3-4С)циклоалкил, (2-3C)алкенил или циано;
R22 представляет собой Н, галоген, (1-2C)алкил или (1-2С)алкокси;
R23 представляет собой H, галоген, (1-2C)алкил, (1-2C)алкокси, циано или гидрокси;
R24 представляет собой H, галоген, (1-2C)алкил или (1-2С)алкокси;
R25 представляет собой H, галоген, (1-3C)алкил, (1-2С)алкокси, гидрокси(1-2С)алкил, (3-4С)циклоалкил, (2-3C)алкенил или циано;
R26 представляет собой Н, (1-6C)алкил, (3-6C)циклоалкил, (2-5С)гетероциклоалкил, (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкил, где n представляет собой целое число 1, 2, 3 или 4, все алкильные, гетероциклоалкильные и (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкильные группы возможно замещены одной или более группами, выбранными из (1-2С)алкила, (1-2С)алкокси, гидроксила, оксо, амино, (3-6C)циклоалкила, ди[(1-2С)алкил]амино или (2-5C)гетероциклоалкила.
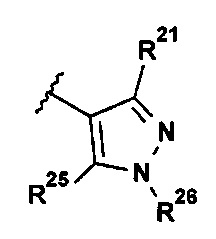
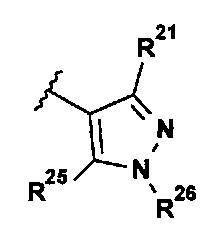
В представляющем интерес воплощении R2 выбран из группы, состоящей из:
 .
.
Кроме того, были обнаружены очень мощные ингибиторы TTK с превосходной селективностью относительно Polo-подобной киназы 1 (PLK1), где R2 представляет собой:
 .
.
Вследствие этой селективности относительно PLK1 соединения, соответствующие указанному определению R2, часто демонстрируют высокую селективность в отношении киназ Aurora A (AurA) и/или Aurora C (AurC).
В указанной выше формуле I только один из R21 и R25 в R2 может представлять собой H.
Термины, использованные в данной заявке, относятся к следующему:
Галоген означает фтор, хлор, бром или йод, где фтор, хлор или бром являются предпочтительными галогенами, при этом фтор или хлор являются более предпочтительными.
(1-2C)Алкил означает алкильную группу, имеющую от 1 до 2 атомов углерода, представляющую собой метил или этил. Метильная группа может быть обозначена как Me или CH3.
(1-3C)Алкил означает разветвленную или неразветвленную алкильную группу, имеющую 1-3 атома углерода, представляющую собой метил, этил, пропил или изопропил.
(1-4C)Алкил означает разветвленную или неразветвленную алкильную группу, имеющую 1-4 атома углерода, представляющую собой метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил или трет-бутил, где (1-3C)алкильные группы являются предпочтительными.
(1-5С)Алкил означает разветвленную или неразветвленную алкильную группу, имеющую 1-5 атомов углерода, например метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил и изопентил, где (1-4С)алкильные группы являются предпочтительными.
(1-6С)Алкил означает разветвленную или неразветвленную алкильную группу, имеющую 1-6 атомов углерода, например метил, этил, пропил, изопропил, бутил, трет-бутил, н-пентил и н-гексил. (1-5С)алкильные группы являются предпочтительными, (1-4С)алкил является более предпочтительным.
(1-2С)Алкокси означает группу алкокси, имеющую 1-2 атома углерода, где алкильная группировка имеет такое же значение, как определено ранее.
(2-4С)Алкокси означает группу алкокси, имеющую 2-4 атома углерода, например этокси, пропилокси, бутилокси, изопропилокси, изобутилокси и трет-бутилокси. Этилокси и пропилокси являются предпочтительными. Группы этилокси являются более предпочтительными.
(1-3C)Алкокси означает группу алкокси, имеющую 1-3 атома углерода, где алкильная группировка имеет такое же значение, как определено ранее. Группы (1-2С)алкокси являются предпочтительными.
(1-4С)Алкокси означает группу алкокси, имеющую 1-4 атома углерода, где алкильная группировка имеет такое же значение, как определено ранее. Группы (1-3C)алкокси являются предпочтительными, группы (1-2С)алкокси являются наиболее предпочтительными.
(1-5С)Алкокси означает группу алкокси, имеющую 1-5 атомов углерода, где алкильная группировка имеет такое же значение, как определено ранее. Группы (1-4С)алкокси являются предпочтительными, группы (1-3C)алкокси являются более предпочтительными.
(2-3C)Алкенил означает разветвленную или неразветвленную алкенильную группу, имеющую 2-3 атома углерода, например этенил или 2-пропенил.
(2-3C)Алкинил означает этинил или 2-пропинил.
(3-4С)Циклоалкил означает циклоалкильную группу, имеющую 3-4 атома углерода, представляющую собой циклопропил или циклобутил.
(3-6C)Циклоалкил означает циклоалкильную группу, имеющую 3-6 атомов углерода, представляющую собой циклопропил, циклобутил, циклопентил или циклогексил. Циклопропил и циклобутил являются предпочтительными.
(2-5С)Гетероциклоалкил означает гетероциклоалкильную группу, имеющую 2-5 атомов углерода, предпочтительно 3-5 атомов углерода; и один или два гетероатома, выбранных из N, O и/или S, которая может быть присоединена посредством гетероатома, если это возможно, или посредством атома углерода. Предпочтительными гетероатомами являются N или O. Предпочтительными являются оксетанил, азетидинил, пиперидинил, морфолинил, пирролидинил и пиперазинил. Наиболее предпочтительными (2-5C)гетероциклоалкилами являются оксетанил и азетидинил.
(2-7C)Гетероциклоалкил означает гетероциклоалкильную группу, имеющую 2-7 атомов углерода, предпочтительно 2-5 атомов углерода, и один или два гетероатома, выбранные из N, O и/или S. Предпочтительными гетероатомами являются N или O. Предпочтительными (2-7С)гетероциклоалкильными группами являются азетидинил, пирролидинил, пиперидинил, пиперазинил, гомопиперидинил, морфолинил или тиоморфолинил. Гетероциклоалкильная группа может быть присоединена посредством гетероатома, если это возможно.
(6-10C)Арил означает ароматическую углеводородную группу, имеющую 6-10 атомов углерода, например фенил, нафтил, тетрагидронафтил или инденил. Предпочтительная (6-10C)арильная группа представляет собой фенил.
(1-5С)Гетероарил означает замещенную или незамещенную ароматическую группу, имеющую 1-5 атомов углерода и 1-4 гетероатома, выбранных из N, О и/или S. (1-5С)Гетероарил возможно может быть замещен. Предпочтительные (1-5С)гетероарильные группы представляют собой изоксазолил, пиразолил, пиридил, пиримидил, пиразинил, более предпочтительные (1-5С)гетероарилы представляют собой пиразолил, изоксазолил, пиридинил и пиримидил.
(3-6C)Циклоалкиламино означает аминогруппу, монозамещеную циклоалкильной группой, содержащей 3-6 атомов углерода, имеющей такое же значение, как определено ранее.
(1-6С)Алкиламино означает аминогруппу, монозамещеную алкильной группой, содержащей 1-6 атомов углерода, имеющей такое же значение, как определено ранее. Предпочтительная (1-6C)алкиламиногруппа представляет собой метиламино.
Ди[(1-2C)алкил]амино означает аминогруппу, дизамещенную алкильной(ыми) группой(ами), где каждая независимо содержит 1-2 атома углерода и имеет такое же значение, как определено ранее. Предпочтительная ди[(1-2C)алкил]аминогруппа представляет собой диметиламино.
Ди[(1-6С)алкил]амино означает аминогруппу, дизамещенную алкильной(ыми) группой(ами), где каждая независимо содержит 1-6 атомов углерода и имеет такое же значение, как определено ранее. Предпочтительная ди[(1-6С)алкил]аминогруппа представляет собой N-метилпропан-1-амино.
(2-7С)Гетероциклоалкиламино означает аминогруппу, монозамещенную (2-7)гетероциклоалкильной группой, содержащей 2-7 атомов углерода, имеющей такое же значение, как определено ранее.
(1-6С)Алкиламинокарбонил означает карбонильную группу, замещенную аминогруппой. Указанная аминогруппа монозамещена алкильной группой, имеющей 1-6 атомов углерода и имеющей такое же значение, как определено ранее.
(2-7С)Гетероциклоалкилкарбонил означает карбонильную группу, замещенную (2-7С)гетероциклоалкильной группой, имеющей 2-7 атомов углерода и имеющей такое же значение, как определено ранее.
(1-5С)Алкоксикарбонил означает карбонильную группу, замещенную группой алкокси, алкильная группировка которой имеет 1-6 атомов углерода, как определено ранее.
(1-6С)Алкилсульфонил означает сульфонильную группу, замещенную (1-6С)алкильной группой, имеющей 1-6 атомов углерода и имеющей такое же значение, как определено ранее.
(1-6С)Алкилкарбонил означает карбонильную группу, замещенную (1-6С)алкильной группой, имеющей 1-6 атомов углерода и имеющей такое же значение, как определено ранее.
(3-6C)Циклоалкилкарбонил означает карбонильную группу, замещенную (3-6C)циклоалкильной группой, имеющей 3-6 атомов углерода и имеющей такое же значение, как определено ранее.
(1-6С)Алкиламинокарбонил означает карбонильную группу, замещенную аминогруппой. Указанная аминогруппа монозамещена алкильной группой, имеющей 1-6 атомов углерода и имеющей такое же значение, как определено ранее.
(1-6С)Алкилкарбониламино означает аминогруппу, замещенную карбонильной группой. Указанная карбонильная группа монозамещена алкильной группой, имеющей 1-6 атомов углерода и имеющей такое же значение, как определено ранее.
(3-6C)Циклоалкилкарбониламино означает аминогруппу, замещенную карбонильной группой. Указанная карбонильная группа монозамещена циклоалкильной группой, имеющей 3-6 атомов углерода и имеющей такое же значение, как определено ранее.
(2-7С)Гетероциклоалкилкарбониламино означает аминогруппу, замещенную карбонильной группой. Указанная карбонильная группа монозамещена (2-7С)гетероциклоалкильной группой, имеющей 2-7 атома углерода и имеющей такое же значение, как определено ранее.
Гидрокси(1-2С)алкил означает (1-2С)алкильную группу, имеющую 1-2 атома углерода с таким же значением, как определено ранее, замещенную гидроксильной группой.
(1-2С)Алкокси[(2-4С)алкокси]n(1-6С)алкил означает (1-6С)алкильную группу, имеющую 1-6 атомов углерода с таким же значением, как определено ранее, замещенную одной или более группами (2-4С)алкилокси, где n означает целое число 1, 2, 3 или 4, где группы алкокси линейно соединены друг с другом. Последняя группа (2-4С)алкилокси замещена группой (1-2С)алкилокси. В (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкильной группе предпочтительная группа (1-2С)алкокси представляет собой метокси, предпочтительная группа (2-4С)алкокси представляет собой этокси, и предпочтительный (1-6С)алкил представляет собой этил, n предпочтительно представляет собой 1, 2, 3, 4, где наиболее предпочтительно n представляет собой 1 или 2.
В указанных выше определениях с многофункциональными группами точка присоединения находится на последней группе.
Когда в определении заместителя указано, что "все из алкильных групп" указанного заместителя возможно замещены, это также учитывает алкильную группировку группы алкокси.
Термин "замещенный" означает, что один или более атомов водорода на указанном атоме/атомах заменен/заменены выборкой из указанной группы, при условии, что нормальная валентность указанного атома в существующих условиях не превышена, и что в результате замещения получается стабильное соединение. Комбинации заместителей и/или переменных допустимы только, если в результате таких комбинаций получаются стабильные соединения.
"Стабильное соединение" или "стабильная структура" определена как соединение или структура, которое(ая) является достаточно прочным(ой) для того, чтобы выдержать выделение из реакционной смеси до полезной степени чистоты и изготовление в виде эффективного терапевтического агента.
Термин "возможно замещенный" означает возможное замещение конкретными группами, радикалами или группировками.
В одном воплощении изобретение относится к соединению формулы I, где R13 представляет собой R132O, R135C(O), (2-7C)гетероциклоалкил или (1-5C)гетероарил, где каждый гетероциклоалкил или гетероарил возможно замещен (1-2С)алкилом, (1-6С)алкилкарбонилом, (1-6С)алкилсульфонилом, (1-5С)алкоксикарбонилом, (1-6С)алкиламинокарбонилом, (3-6С)циклоалкилкарбонилом или (2-7С)гетероциклоалкилкарбонилом, где каждый алкилкарбонил, алкилсульфонил, алкоксикарбонил, алкиламинокарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором, (1-2С)алкокси; R132 выбран из группы, состоящей из (1-6С)алкила, (3-6C)циклоалкила, (2-7С)гетероциклоалкила, (6-10C)арила или (1-5С)гетерогарила, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, галогена, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино или (2-7С)гетероциклоалкила, и R135 выбран из группы, состоящей из (2-7С)гетероциклоалкила, (1-6С)алкиламино, ди[(1-6С)алкил]амино, (2-7С)гетероциклоалкиламино или (3-6C)циклоалкиламино, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино, (2-7С)гетероциклоалкила, оксо, циано или амино.
В еще одном воплощении изобретение относится к соединению формулы I, где R13 представляет собой R132O, R135C(O); или R13 представляет собой пиперидинил, пиперазинил, морфолинил, пиразолил или изоксазолил (каждый из которых возможно замещен (1-2С)алкилом, (1-6С)алкилкарбонилом, (1-6С)алкилсульфонилом, (1-5С)алкоксикарбонилом, (1-6С)алкиламинокарбонилом, (3-6C)циклоалкилкарбонилом или (2-7С)гетероциклоалкилкарбонилом, где каждый алкилкарбонил, алкилсульфонил, алкоксикарбонил, алкиламинокарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором или (1-2С)алкокси. R132 выбран из группы, состоящей из (1-6C)алкила, пиперидинила, пирролидинила или азетидинила, каждый из которых возможно замещен одной или более группами, выбранными из (1-2C)алкила, (1-2С)алкокси или ди[(1-2С)алкил]амино, и R135 выбран из группы, состоящей из пиперидинила, тиоморфолинила, морфолинила, гомопиперазинила, (1-6С)алкиламино, (3-6С)циклоалкиламино или пиперидиниламино, азетидиниламино, тетрагидропираниламино или 3-оксабицикло[3.1.0]гексан-6-амино, каждый из которых возможно замещен одной или более группами, выбранными из (1-2С)алкила, фтора, гидроксила или (1-2С)алкокси, ди[(1-2С)алкил]амино, (2-7С)гетероциклоалкила, оксо, циано или амино.
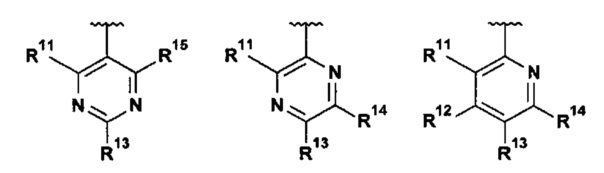
В другом воплощении настоящее изобретение относится к соединению формулы I, где R1 выбран из группы, состоящей из:
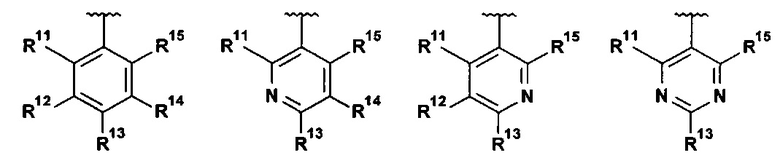 .
.
В еще одном воплощении настоящее изобретение относится к соединению формулы I, где каждый из R12 и R15 представляет собой Н, и R14 представляет собой Н, фтор, хлор или (1-2C)алкил.
Изобретение также относится к соединениям формулы I, где R11 представляет собой Н, (1-2C)алкил или (1-2C)алкокси, где все алкильные и алкокси группы возможно замещены одним или более фторами.
В еще одном воплощении изобретение относится к соединению формулы I, где R2 выбран из группы, состоящей из:
 .
.
В конкретном воплощении изобретение относится к соединению формулы I, в котором R2 представляет собой:
 .
.
В еще одном воплощении изобретение относится к соединению формулы I, где R23 представляет собой Н или (1-6C)алкил, и каждый из R22 и R24 представляет собой Н.
В другом воплощении изобретение относится к соединению формулы, где R21 выбран из группы, состоящей из H, галогена, (1-6C)алкила или циано.
В еще одном воплощении изобретение относится к соединению формулы I, где R23 представляет собой Н или (1-2C)алкил, и каждый из R22 и R24 представляет собой Н, и R21 и R25 независимо выбраны из группы, состоящей из галогена, (1-3C)алкила, метокси, гидроксиметила или циано.
В другом воплощении изобретение относится к соединению формулы I, где R26 представляет собой Н, (1-6C)алкил, оксетанил, азетидинил или (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкил, где n представляет собой целое число 1 или 2, все алкильные, оксетанильные и азетидинильные группы возможно замещены одной или более группами, выбранными из (1-2С)алкила, (1-2С)алкокси, гидроксила, ди[(1-2С)алкил]амино или оксетанила.
Изобретение также относится к тем соединениям, у которых все конкретные определения R1, R2, R11-15, R21-26 и R131-137 и все группы-заместители в различных аспектах изобретений, определенные выше, встречаются в любой комбинации в пределах определения соединения формулы I.
В другом аспекте изобретения соединения по изобретению обладают ингибирующей активностью в отношении TTK с IC50 10 мкМ или ниже. В другом аспекте изобретение относится к соединениям формулы I, которые обладают ингибирующей активностью в отношении TTK с IC50 менее чем 100 нМ. В еще одном аспекте изобретение относится к соединениям формулы I, которые обладают ингибирующей активностью в отношении TTK с IC50 менее чем 10 нМ.
Термин IC50 означает концентрацию тестируемого соединения, которая требуется для 50%-ного ингибирования его максимального эффекта in vitro.
Ингибирование активности киназы TTK может быть измерено с использованием анализа фосфорсодержащих химических продуктов на иммобилизованных металлах (IMAP). IMAP представляет собой гомогенный флуоресцентный поляризационный (FP) анализ, основанный на аффинном захвате фосфорилированных пептидных субстратов. В IMAP используют меченые флуоресцеином пептидные субстраты, которые при фосфорилировании протеинкиназой связываются с так называемыми наночастицами IMAP, которые дериватизированы комплексами трехвалентных металлов. Связывание вызывает изменение скорости молекулярного вращения пептида и приводит к увеличению значения FP, наблюдаемого для флуоресцеиновой метки, присоединенной к субстратному пептиду (Gaudet, Е.А. et al. J. Biomol. Screen 8: 164; 2003).
Биологическая активность ингибиторов TTK может быть измерена в пролиферативных анализах с опухолевыми клеточными линиями. Активность соединений в отношении опухолевых клеток также может быть определена в тестах на колониеобразование, а в контексте животной модели - у мышей с пересаженными человеческими или мышиными клеточными линиями или опухолевой тканью.
Соединения формулы I могут образовывать соли, которые также находятся в объеме настоящего изобретения. Ссылка в данной заявке на соединение формулы I, как понимают, включает ссылку на его соли, если не указано иное. Термин "соль(и)", используемый в данной заявке, означает кислотные соли, образованные с неорганическими и/или органическими кислотами, а также  соли, образованные с неорганическими и/или органическими основаниями. Кроме того, если соединение формулы I может содержать и основную группировку, такую как, но не ограничиваясь этим, пиридин или имидазол, и кислотную группировку, то могут быть образованы цвиттер-ионы ("внутренние соли"), и они включены в термин "соль(и)", используемый в данной заявке. Предпочтительными являются фармацевтически приемлемые (то есть, нетокчисные физиологически приемлемые) соли. Соли соединений формулы I могут быть образованы, например, путем взаимодействия соединения формулы I с кислотой или основанием в количестве, таком как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
соли, образованные с неорганическими и/или органическими основаниями. Кроме того, если соединение формулы I может содержать и основную группировку, такую как, но не ограничиваясь этим, пиридин или имидазол, и кислотную группировку, то могут быть образованы цвиттер-ионы ("внутренние соли"), и они включены в термин "соль(и)", используемый в данной заявке. Предпочтительными являются фармацевтически приемлемые (то есть, нетокчисные физиологически приемлемые) соли. Соли соединений формулы I могут быть образованы, например, путем взаимодействия соединения формулы I с кислотой или основанием в количестве, таком как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Иллюстративные соли присоединения кислоты включают ацетаты, аскорбаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, фумараты, гидрохлориды, гидробромиды, гидройодиды, лактаты, малеаты, метансульфонаты, нафталинсульфонаты, натраты, оксалаты, фосфаты, пропионаты, салицилаты, сукцинаты, сульфаты, тартраты, тиоцианаты, толуол сульфонаты (также известные как тозилаты) и тому подобное. Кроме того, кислоты, которые обычно считаются подходящими для образования фармацевтически полезных солей из  фармацевтических соединений, рассмотрены, например, в P. Stahl et al, Camille G. (eds.), Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, J. of Pharm. Sci. (1977) 66(1), 1-19; P. Gould, Int. J. Pharm. (1986) 33, 201-21 7; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; и в The Orange Book (Food & Drug Administration, Washington, D.C. на их вебсайте).
фармацевтических соединений, рассмотрены, например, в P. Stahl et al, Camille G. (eds.), Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, J. of Pharm. Sci. (1977) 66(1), 1-19; P. Gould, Int. J. Pharm. (1986) 33, 201-21 7; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; и в The Orange Book (Food & Drug Administration, Washington, D.C. на их вебсайте).
Иллюстративные основные соли включают аммониевые соли, соли щелочных металлов, например соли натрия, лития и калия, соли щелочноземельных металлов, например соли кальция и магния, соли с органическими основаниями (например с органическими аминами), например с дициклогексиламинами, трет-бутиламинами, и соли с аминокислотами, например с аргинином, лизином и тому подобным. Основные азотсодержащие группы могут быть кватернизированы агентами, такими как низшие алкилгалогениды (например, метил-, этил- и бутилхлориды, -бромиды и -йодиды), диалкилсульфаты (например, диметил-, диэтил- и дибутилсульфаты), длинноцепочечные галогениды (например, децил-, лаурил- и стеарилхлориды, -бромиды и -йодиды), аралкилгалогениды (например, бензил- и фенэтилбромиды) и другие.
Соединения формулы I могут обладать способностью к кристаллизации в более чем одной форме; это свойство известно как полиморфизм, и понимают, что такие полиморфные формы ("полиморфы") находятся в объеме формулы I. Обычно полиморфизм может происходить в результате ответа на изменения в температуре, или давлении, или в обоих этих параметрах, а также может возникать в результате различий в процессе кристаллизации. Полиморфы можно различить с помощью разных физических характеристик, известных в области техники, таких как картины дифракции рентгеновских лучей, растворимость и температура плавления.
Соединения формулы I могут содержать асимметрические или хиральные центры и, следовательно, существовать в разных стереоизомерных формах. Подразумевают, что все стереоизомерные формы соединений формулы I, а также их смеси, включая рацемические смеси, являются частью настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические изомеры и изомеры положения. Например, если соединение формулы I содержит двойную связь или конденсированное кольцо, то и цис-, и транс-формы, также как и смеси, содержатся в объеме этого изобретения.
Диастереомерные смеси можно разделить на их индивидуальные диастереомеры на основании их физико-химических различий способами, хорошо известными специалистам в области техники, такими как, например, хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь путем взаимодействия с подходящим оптически активным соединением (например, с хиральной добавкой, такой как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, путем гидролиза) индивидуальных диастереомеров в соответствующие чистые энантиомеры. Кроме того, некоторые соединения формулы I могут представлять собой атропоизомеры (например, замещенные биарилы), и они считаются частью этого изобретения. Энантиомеры также можно разделить путем использования хиральной колонки для высокоэффективной жидкостной хроматографии (HPLC).
Также возможно, что соединения формулы I могут существовать в различных таутомерных формах, и все такие формы содержатся в объеме этого изобретения. Также в изобретение включены, например, все кето-енольные и имин-енаминные формы соединений.
Все стереоизомеры (например, геометрические изомеры, оптические изомеры и тому подобные) заявленных соединений (включая стереоизомеры солей, сольватов, сложных эфиров и пролекарств соединений, а также солей, сольватов и сложных эфиров пролекарств), такие как те, которые могут существовать благодаря асимметрическим атомам углерода с различными заместителями, включая энантиомерные формы (которые могут существовать даже при отсутствии асимметрических атомов углерода), ротамерные формы, атропоизомеры и диастереомерные формы, рассматриваются в объеме этого изобретения. Хиральные центры по настоящему изобретению могут иметь S- или R-конфигурацию, как определено Рекомендациями IUPAC от 1974 года. Подразумевают, что использование терминов "соль", "сольват", "сложный эфир", "пролекарство" и тому подобных в равной степени применимо к соли, сольвату, сложному эфиру и пролекарству энантиомеров, стереоизомеров, ротамеров, таутомеров, изомеров положения, рацематов или пролекарств соединений по изобретению.
Соединения, имеющие формулу (I), или фармацевтически приемлемые соли могут образовывать гидраты или сольваты. Специалистам в области техники известно, что заряженные соединения образуют гидратированные соединения, когда лиофилизированы с водой, или образуют сольватированные соединения при концентрировании в растворе с подходящим органическим растворителем. Соединения по настоящему изобретению включают гидраты или сольваты перечисленных соединений.
В соединениях формулы I атомы могут демонстрировать их природный изотопный состав, или один или более атомов могут быть искусственно обогащены конкретным изотопом, имеющим тот же самый атомный номер и имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, которые в основном встречаются в природе. Подразумевают, что настоящее изобретение включает все подходящие изотопные вариации соединений формулы I. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H). Протий представляет собой преобладающий изотоп водорода, встречающийся в природе.
Замещение более тяжелыми изотопами, например дейтерием (то есть, 2H), может давать определенные терапевтические преимущества, вытекающие из большей метаболической стабильности (например, увеличенного времени полувыведения in vivo или сниженных требований по дозировке), и поэтому может являться предпочтительным в некоторых обстоятельствах. Меченые изотопом соединения формулы I обычно могут быть получены, следуя методикам, аналогичным описанным в Схемах и/или Примерах в данной заявке ниже, путем замены немеченого изотопом реагента на подходящий меченый изотопом реагент.
Соединения по настоящему изобретению можно применять в терапии.
В одном аспекте соединения по настоящему изобретению могут быть использованы для лечения TTK-опосредованных заболеваний или состояний. В частности, соединения формулы I или их соли и их фармацевтические композиции могут быть использованы для лечения заболеваний и состояний, вызванных или ассоциированных с чрезмерной экспрессией или чрезмерной активностью белка TTK и/или аномальной экспрессией, активностью или регуляцией любых регуляторов активности TTK или других регуляторов митотической контрольной точки, например MAD1, MAD2, BUB1, BUBR1, BUB3 и других (Kops, G.J.P.L., et al.; Nature Rev. Cancer 5: 773; 2005).
Таким образом, в одном аспекте изобретения соединения формулы I или их фармацевтически приемлемые соли могут быть использованы для лечения заболеваний или состояний, вызванных или ассоциированных с чрезмерной экспрессией или чрезмерной активностью белка TTK.
В другом аспекте соединения формулы I или их фармацевтически приемлемые соли могут быть использованы для лечения гиперпролиферативных расстройств.
Таким образом, изобретение относится к способу регулирования, модулирования или ингибирования TTK для предотвращения и/или лечения гиперпролиферативных расстройств.
В другом аспекте соединения по настоящему изобретению могут быть использованы для лечения заболеваний или состояний, вызванных аномальной пролиферацией клеток, и/или заболеваний, ассоциированных с хромосомной нестабильностью, хромосомными перестройками и/или анеуплоидией.
В еще одном аспекте соединения по настоящему изобретению могут быть использованы для лечения рака, в частности для лечения или профилактики заболеваний, вызванных или ассоциированных с неконтролируемым ростом клеток, пролиферацией клеток и/или выживанием клеток.
В другом аспекте соединения по настоящему изобретению могут быть использованы для лечения солидных опухолей, гематологических опухолей и/или их метастазов, например опухолей молочной железы и гинекологических опухолей, опухолей головы и шеи, опухолей мозга и метастазов в мозге, опухолей грудной клетки, включая немелкоклеточные и мелкоклеточные опухоли легкого, желудочно-кишечных опухолей, эндокринных опухолей, урологических опухолей, включая опухоли почки, мочевого пузыря и предстательной железы, кожной опухоли, и сарком, лейкемий и миелодиспластического синдрома, злокачественных лимфом и/или их метастазов.
Другой аспект изобретения заключается в применении соединения формулы I или его фармацевтически приемлемой соли для изготовления лекарственного средства, предназначенного для использования для лечения заболеваний и состояний, вызванных или ассоциированных с чрезмерной экспрессией или чрезмерной активностью белка TTK, и для лечения расстройств, в которых гиперпролиферативные клетки играют важную роль.
В настоящую заявку включены способы лечения и/или фармацевтические композиции, где по меньшей мере одно соединение формулы I или его фармацевтически приемлемую соль вводят в комбинации с по меньшей мере одним другим активным агентом. Другим активным агентом может быть химиотерапевтический агент, антитело или активный полипептид.
Другой аспект изобретения относится к соединению формулы I в комбинации с одним или более чем одним другим лекарственным средством.
В изобретении также предложена фармацевтическая композиция, содержащая соединение формулы I и его соли и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми с точки зрения совместимости с другими ингредиентами композиции и не являться вредными для ее реципиента.
Смешанный с такими фармацевтически приемлемыми дополнительными компонентами, как, например, описанные в типовом источнике Gennaro, A.R. et al., Remington: The Science and Practice of Pharmacy (20th Edition, Lippincott Williams & Wilkins, 2000; в частности Part 5: Pharmaceutical Manufacturing), активный агент может быть спрессован в твердые стандартные лекарственные формы, такие как пилюли, таблетки, или может быть изготовлен в виде капсул или суппозиториев. С помощью фармацевтически приемлемых жидкостей активный агент можно применять в виде жидкой композиции, например в виде инъекционной композиции, в форме раствора, суспензии, эмульсии или в виде спрея, например назального спрея.
Фармацевтические композиции по настоящему изобретению могут быть представлены в стандартных лекарственных формах, содержащих предварительно определенное количество активного ингредиента на стандартную дозу. Такая стандартная форма может содержать, например, от 5 мкг до 1 г, предпочтительно от 1 мг до 700 мг, более предпочтительно от 5 мг до 100 мг соединения формулы I, в зависимости от состояния, подлежащего лечению, пути введения и возраста, массы и состояния пациента. Таким образом, такие стандартные дозы можно вводить более одного раза в сутки. Предпочтительными композициями со стандартной дозировкой являются композиции, содержащие суточную дозу или субдозу (для введения более одного раза в сутки), как изложено в данной заявке выше, или ее подходящую часть, активного ингредиента. Более того, такие фармацевтические композиции могут быть получены любым из способов, хорошо известных в фармации.
Фармацевтические композиции по настоящему изобретению могут быть приспособлены для введения любым подходящим путем, например пероральным (включая трансбуккальный или подъязычный), ректальным, местным, ингаляционным, назальным, окулярным, подъязычным, подкожным, местным или парентеральным (включая внутривенный и внутримышечный) путем и тому подобным, все в стандартных лекарственных формах для введения. Такие композиции могут быть получены любым способом, известным в фармации, например путем объединения активного ингредиента с носителем(ями) или эксципиентом(ами). Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и тому подобное.
Соединение по настоящему изобретению также можно вводить в виде конъюгата белок-лекарственное средство. Соединение может быть ковалентно связано, возможно с помощью линкерной молекулы, с пептидом или белком, например связующим белком, например антителом.. Используя этот подход, в целевую ткань можно доставлять конъюгат. Способы получения таких конъюгатов хорошо известны специалистам в области техники.
Следует понимать, что когда соединение по настоящему изобретению вводят в комбинации с другими терапевтическими агентами, обычно вводимыми ингаляционным, внутривенным или интраназальным путем, то полученную фармацевтическую композицию можно вводить такими же путями.
Терапевтически эффективное количество соединения по настоящему изобретению будет зависеть от ряда факторов, включая, например, возраст и массу животного, точное состояние, требующее лечения, и его тяжесть, конкретное соединение, имеющее формулу I, природу препарата и путь введения, и в конечном итоге будет оставлено на усмотрение лечащего врача или ветеринара. Однако, эффективное количество соединения формулы I для лечения заболеваний или состояний, ассоциированных с неподходящей активностью TTK, обычно будет находиться в диапазоне от 5 мкг до 100 мг/кг массы тела реципиента (млекопитающего) в сутки и более обычно в диапазоне от 5 мкг до 10 мг/кг массы тела в сутки. Это количество может быть выдано в виде однократной дозы в сутки или, более обычно, в виде нескольких (например двух, трех, четырех, пяти или шести) субдоз в сутки, так чтобы общая суточная доза была такой же. Эффективное количество его соли или сольвата может быть определено как часть эффективного количества соединения формулы I как такового.
Обычно для парентерального введения требуются меньшие дозировки, чем для других способов введения, которые больше зависят от абсорбции. Однако дозировка для людей предпочтительно включает 0,0001-25 мг соединения формулы I или его фармацевтически приемлемых солей на кг массы тела. Целевая доза может быть представлена как одна доза или как несколько субдоз, вводимых через подходящие интервалы на протяжении суток, или, в случае реципиентов женского пола, в виде доз, предназначенных для введения через определенные суточные интервалы на протяжении менструального цикла. Дозировка, а также режим введения, может варьироваться между реципиентом женского пола и реципиентом мужского пола.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль в смеси с фармацевтически приемлемыми дополнительными веществами и возможно другими терапевтическими агентами. Дополнительные вещества должны быть "приемлемыми" с точки зрения совместимости с другими ингредиентами композиции и не являться вредными для их реципиентов.
Изобретение также включает фармацевтическую композицию, содержащую по меньшей мере одно соединение формулы I или его фармацевтически приемлемые соли в комбинации с по меньшей мере одним другим терапевтически активным агентом.
Для лечения рака соединение формулы I можно объединять с одним или более противораковыми агентами. Примеры таких агентов можно найти в Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001), Lippincott Williams & Wilkins Publishers. Специалист в области техники сможет распознать, какие комбинации агентов будут полезными на основе конкретных характеристик лекарственных средств и вовлеченного рака.
(5,6-Дигидро)пиримидо[4,5-е]индолизиновые производные по настоящему изобретению могут быть получены способами, хорошо известными в области органической химии. См., например, J. March, 'Advanced Organic Chemistry', 4th Edition, John Wiley and Sons. В ходе синтетических последовательностей может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы любых рассматриваемых молекул. Этого достигают с помощью традиционных защитных групп, например описанных в T.W. Greene and P.G.M. Wutts 'Protective Groups in Organic Synthesis', 3rd Edition, John Wiley and Sons, 1999. Защитные группы могут быть удалены на подходящей последующей стадии с использованием способов, хорошо известных в области техники.
Продукты реакций возможно выделяют и очищают, если необходимо, с использованием традиционных методик, включающих без ограничения фильтрование, перегонку, кристаллизацию, хроматографию и тому подобное. Такие вещества возможно охарактеризовывают с использованием традиционных способов, в том числе с помощью физических констант и спектральных данных.
5,6-Дигидропиримидо[4,5-e]индолизиновые соединения формулы I, где R1-Rx имеют значения, определенные ранее, могут быть получены общим путем синтеза, изображенным на схеме I.
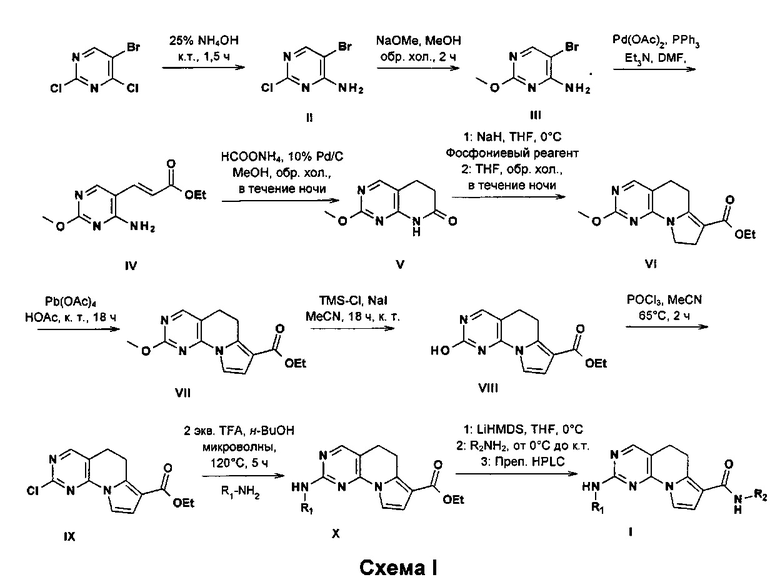
5-Бром-2-хлор-пиримидин-4-амин (II) может быть получен из имеющегося в продаже 5-бром-2,4-дихлор-пиримидина с использованием 25%-ного водного аммиака при температуре окружающей среды. Затем полученный продукт можно подвергнуть взаимодействию с метанолятом натрия в метаноле при повышенных температурах с получением 5-бром-2-метокси-пиримидин-4-амина (III). Затем из соединения III может быть получено соединение IV с использованием этилакрилата в присутствии подходящей палладиевой каталитической системы, например ацетата палладия(II), органического основания, подобного триэтиламину, или неорганического основания, подобного карбонату калия, карбонату цезия или фосфату калия, в подходящей системе растворителей, подобной комбинациям диоксана и воды или диметилформамида. Восстановление двойной связи и последующую циклизацию можно осуществлять путем гидрирования в присутствии подходящей каталитической системы и растворителя, например палладия на древесном угле в метаноле, с получением лактама V. Сложный этиловый эфир VI может быть получен из лактама V с использованием тетрафторбората [1-(этоксикарбонил)циклопропил]трис(фенил)фосфония в THF и подходящего основания, например гидрида натрия. Окисление VI можно осуществлять с использованием окислителей, например оксида марганца или ацетата свинца(IV), с получением производного VII. Соединение VII может быть превращено в производное IX после подходящего снятия защиты триметилсилилйодидом и последующего превращения с использованием оксихлорида фосфора(V) в условиях нагревания. Замещение этил-2-хлор-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксилата с помощью R1NH2 можно проводить в кислотных условиях с использованием трифторуксусной кислоты или концентрированного раствора хлористого водорода и подходящего растворителя, подобного н-бутанолу или изопропиловому спирту, при микроволновом излучении. Альтернативно, R1NH2 можно вводить в присутствии подходящей палладиевой каталитической системы, например ацетата палладия(II) или тетракис(трифенилфосфин)палладия(0), в присутствии неорганического основания, например карбоната калия, карбоната цезия или фосфата калия, в подходящей системе растворителей, подобной диоксану и воде, с получением производного X. Наконец, превращение производного X в соединения формулы I можно осуществлять или сначала путем омыления сложноэфирной функциональной группы в соеди
нении X и последующей конденсации с амидом с использованием способов, хорошо известных в области техники, или путем аминолиза сложноэфирной функциональной группы с использованием сильного основания, например бис(триметилсилил)амида лития.
Пиримидо[4,5-e]индолизиновые соединения формулы I, где R1-Rx имеют значения, определенные ранее, могут быть получены общим путем синтеза, изображенным на схеме II.
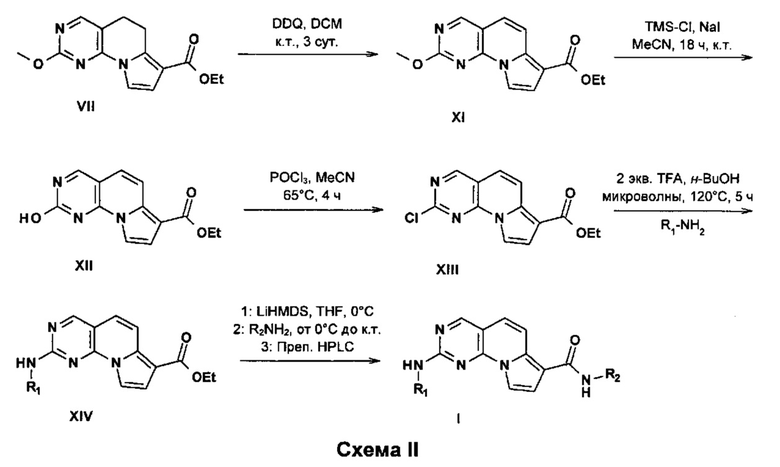
Окисление VII можно осуществлять с использованием окислителей, например оксида марганца, кислорода или DDQ, с получением производного XI. Соединение XI может быть превращено в хлорид XIII после подходящего снятия защиты триметилсилилйодидом и последующего превращения с использованием оксихлорида фосфора(V) в условиях нагревания. Замещение этил-2-хлор-пиримидо[4,5-e]индолизин-7-карбоксилата с помощью R1NH2 можно проводить в кислотных условиях с использованием трифторуксусной кислоты или концентрированного раствора хлористого водорода и подходящего растворителя, подобного н-бутанолу или изопропиловому спирту, при микроволновом излучении. Альтернативно, R1NH2 можно вводить в присутствии подходящей палладиевой каталитической системы, например ацетата палладия(II) или тетракис(трифенилфосфин)палладия(0) в присутствии неорганического основания, например карбоната калия, карбоната цезия или фосфата калия, в подходящей системе растворителей, подобной диоксану и воде, с получением производного XIV. Наконец, превращение производного XIV в соединения формулы I можно осуществлять или сначала путем омыления сложноэфирной функциональной группы соединения XIV и последующей конденсации с амидом с использованием способов, хорошо известных в области техники, или путем аминолиза сложноэфирной функциональной группы с использованием сильного основания, например бис(триметилсилил)амида лития.
Изобретение проиллюстрировано следующими примерами.
Примеры
Следующие примеры представляют собой иллюстративные воплощения изобретения, не ограничивающие никоим образом объем изобретения. Реагенты или имеются в продаже, или их получают в соответствии с методиками, описанными в литературе.
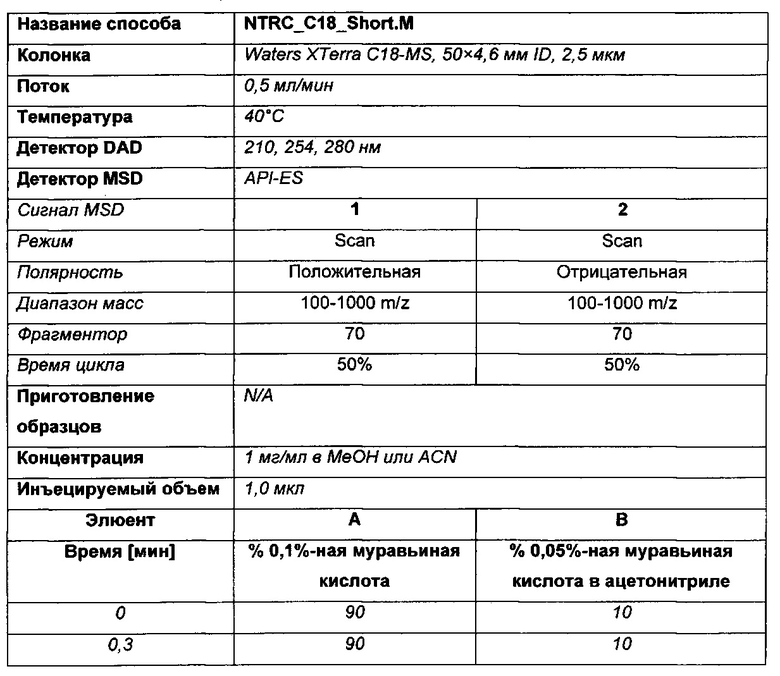
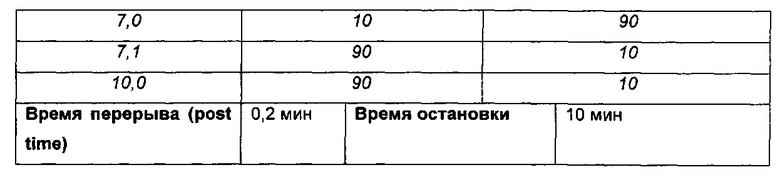
Способ LCMS (А)
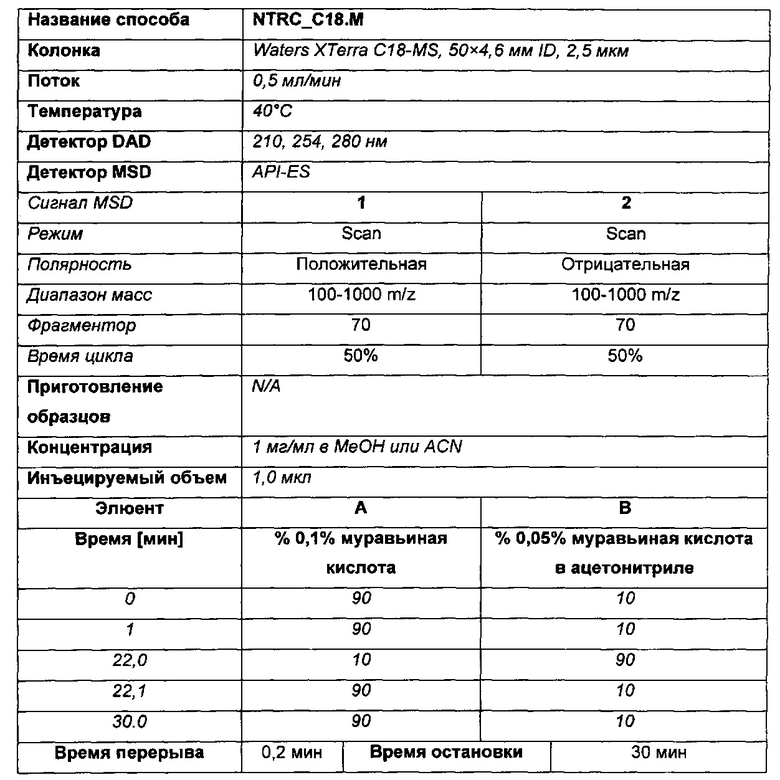
Способ LCMS (В)
Способ LCMS (С)

Способ Препаративной HPLC

Следующие сокращения используют во всем описании заявки по отношению к химической терминологии:
TFA Трифторуксусная кислота
HATU Гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония
DMF N,N-Диметилформамид
THF Тетрагидрофуран
MeOH Метанол
EtOAc Этилацетат
DCM Дихлорметан
Na2SO4 Сульфат натрия
TMS-Cl Хлортриметилсилан
DiPEA N,N-Диизопропилэтиламин
EtOH Этанол
10% Pd/C 10% палладия на древесном угле
HPLC Высокоэффективная жидкостная хроматография
LCMS Жидкостная хроматография с масс-спектрометрическим детектированием
NaOH Гидроксид натрия
HCl Хлористый водород
KOH Гидроксид калия
NaHCO3 Бикарбонат натрия
4-DMAP 4-Диметиламинопиридин
Boc Бутилоксикарбонил
Cbz Бензилоксикарбонил
HNO3 Азотная кислота
LiHMDS Бис(триметилсилил)амид лития
DDQ 2,3-Дихлор-5,6-дициано-л-бензохинон
DBU 1,8-Диазабицикло[5.4.0]ундец-7-ен
DEAD Диэтилазодикарбоксилат
Названия конечных продуктов в примерах созданы с использованием Accelrys Draw (версия 4.1).
Промежуточное соединение 1
Этил-2-хлор-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
а) 5-Бром-2-хлор-пиримидин-4-амин
К раствору 5-бром-2,4-дихлор-пиримидина (150 г; 658 ммоль) в THF (445 мл) добавляли гидроксид аммония (25%-ный в воде, 250 мл) и полученную реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Затем смесь упаривали до небольшого объема и распределяли между этилацетатом и водой. Органическую фазу отделяли и промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали с получением 137,3 г (количественный выход) 5-бром-2-хлор-пиримидин-4-амина.
б) 5-Бром-2-метокси-пиримидин-4-амин
К суспензии 5-бром-2-хлор-пиримидин-4-амина (137,3 г, 658 ммоль) в метаноле (1 л) порциями добавляли метоксид натрия (83,5 г; 1,54 моль). Реакционную смесь перемешивали в течение 2 ч при температуре флегмообразования. Реакционную смесь концентрировали до небольшого объема (примерно 400 мл) и вливали в насыщенный раствор хлорида аммония в воде (1,2 л). Эту смесь оставляли перемешиваться в течение 15 мин, после чего водный слой экстрагировали этилацетатом. Объединенные этилацетатные слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали с получением 5-бром-2-метоксипиримидин-4-амина (133,7 г, 99,4%).
в) Этил-(Е)-3-(4-амино-2-метокси-пиримидин-5-ил)проп-2-еноат
Ацетат палладия (II) (1,21 г, 5,5 ммоль) и трифенилфосфин (3,40 г, 13,0 ммоль) растворяли в безводном и не содержащем кислорода DMF (53 мл) и перемешивали в течение 5 мин при 30°C с получением оранжевой суспензии. К этой суспензии добавляли раствор 5-бром-2-метоксипиримидин-4-амина (44,1 г, 216 ммоль) в DMF (270 мл), триэтиламина (60,2 мл, 432 ммоль) и раствор этилакрилата (23,5 мл, 216 ммоль) в DMF (50 мл). Реакционную смесь перемешивали при 100°C в течение ночи в атмосфере азота. Реакционную смесь упаривали до небольшого объема. К смеси добавляли воду (300 мл) и рассол (300 мл) с последующим экстрагированием этилацетатом (300 мл, дважды). Объединенные органические слои промывали водой, рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (этилацетат : гептан = 2:1 об./об. %) с получением указанного в заголовке соединения (38,2 г, 77%).
г) 2-Метокси-6,8-дигидро-5Н-пиридо[2,3-d]пиримидин-7-он
К перемешиваемому раствору этил-(E)-3-(4-амино-2-метокси-пиримидин-5-ил)проп-2-еноата (12,52 г, 56,1 ммоль) в метаноле (250 мл) добавляли суспензию 10% Pd на древесном угле (1,19 г) в смеси метанол/этанол = 3/1 об./об. % (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин в атмосфере азота. Затем добавляли формиат аммония (35,3 г, 561 ммоль) и полученную реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения реакционной смеси добавляли новую порцию формиата аммония (20 г, 317 ммоль) и перемешивание продолжали в течение последующей ночи при температуре флегмообразования. Реакционную смесь фильтровали через Decalite® и остаток Pd-С/ Decalite® промывали смесью дихлорметан/метанол = 8/2 об./об. % и фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане и промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 9,4 г (94%) 2-метокси-6,8-дигидро-5Н-пиридо[2,3-d]пиримидин-7-она.
д) Этил-2-метокси-5,6,8,9-тетрагидропиримидо[4,5-e]индолизин-7-карбоксилат
2-Метокси-6,8-дигидро-5Н-пиридо[2,3-d]пиримидин-7-он (4,79 г, 26,8 ммоль) суспендировали в THF (200 мл) в трехгорлой колбе (500 мл), снабженной механической мешалкой, термометром и обратным холодильником. Смесь охлаждали до 0°C и двумя порциями добавляли гидрид натрия (60%-ная дисперсия в масле, 1,18 г, 29,4 ммоль). Смесь перемешивали при 0°C в течение 30 мин. Добавляли тетрафторборат (1-этоксикарбонилциклопропил)трифенилфосфония (13,6 г, 29,4 ммоль) и полученную суспензию нагревали до образования флегмы и выдерживали при температуре флегмообразования в течение 3 суток. Реакционную смесь охлаждали до комнатной температуры и вливали в смесь 1/1/1 рассол/вода/EtOAc (450 мл). Водный слой экстрагировали этилацетатом (2×). Объединенные органические слои промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 18,05 г оранжевого масла. Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
е) Этил-2-метокси-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
К перемешиваемому раствору этил-2-метокси-5,6,8,9-тетрагидропиримидо[4,5-e]индолизин-7-карбоксилата (18,05 г, 26,2 ммоль) в дихлорметане (100 мл) добавляли уксусную кислоту (3,15 г, 3 мл) и ацетат свинца(IV) (13,9 г, 31,4 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, затем фильтровали через PE фильтр для удаления солей Pb и остаток Pb промывали 2×30 мл DCM. Фильтрат концентрировали в вакууме и полученный остаток растворяли в этилацетате (300 мл). Добавляли раствор бикарбоната натрия (5%) до pH примерно 8,5. И органические, и водные слои фильтровали через Decalite® для удаления всех оставшихся солей. Затем водный слой экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали 5%-ным раствором бикарбоната натрия (100 мл), водой (100 мл), рассолом (50 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (гептан : этилацетат = от 1/0 до 1/1 об./об. %) с получением указанного в заголовке соединения (4,74 г, 66% за две стадии).
ж) Этил-2-гидрокси-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
Йодид натрия (7,83 г, 52,2 ммоль) добавляли к перемешиваемому раствору этил-2-метокси-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксилата (4,74 г, 17,3 ммоль) в ацетонитриле (150 мл). В реакционную смесь по каплям добавляли триметилсилилхлорид (5,64 г, 6,59 мл), растворенный в ацетонитриле (30 мл), и смесь перемешивали при комнатной температуре в течение ночи. Добавляли NaI (1 экв.) и по каплям добавляли дополнительное количество TMS-Cl (0,94 г, 1,1 ммоль) в ацетонитриле (6 мл) и реакционную смесь перемешивали в течение 3 суток при комнатной температуре. Смесь концентрировали и остаток суспендировали в 200 мл DCM/MeOH (4/1) и экстрагировали смесью насыщенного раствора тиосульфата натрия (200 мл) и воды (200 мл). Водный слой экстрагировали 3×150 мл DCM/MeOH (4/1). Объединенные органические слои сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением желтого твердого вещества. Остаток сушили при 40°C в вакууме в течение 18 ч с получением 3,89 г этил-2-гидрокси-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксилата (86%).
з) Этил-2-хлор-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксилат (Промежуточное соединение 1)
N,N-диметиланилин (182 мг, 191 мкл, 1,50 ммоль) добавляли к раствору этил-2-гидрокси-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата (3,89 г, 15,0 ммоль) в ацетонитриле (100 мл). К реакционной смеси по каплям добавляли раствор оксихлорида фосфора(V) (11,5 г, 7,00 мл, 75,0 ммоль) в ацетонитриле (15 мл). Желтую суспензию нагревали в течение 4 часов до 65°C, в ходе чего суспензия превратилась в прозрачный раствор. После охлаждения смесь медленно вливали в перемешиваемую смесь 25%-ного водн. аммиака (200 мл, 86,7 экв.) и ледяной воды (250 мл), поддерживая температуру ниже 10°C в течение 15-20 минут. После перемешивания в течение последующих 15 минут твердые вещества отфильтровывали. Твердые вещества растворяли в 200 мл EtOAc и промывали рассолом (20 мл). Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением не совсем белого твердого вещества. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (гептан/этилацетат = от 1/0 до 1/1 об./об. %) с получением указанного в заголовке соединения (3,05 г, 73%).
Промежуточное соединение A
4-Амино-N-(2-гидрокси-2-метил-пропил)-3-метокси-бензамид
а) N-(2-гидрокси-2-метил-пропил)-3-метокси-4-нитро-бензамид
Смесь 3-метокси-4-нитробензойной кислоты (1 г, 5,07 ммоль), HATU (2,31 г, 6,1 ммоль) и 1-амино-2-метил-пропан-2-ола (1,13 г, 12,7 ммоль) в дихлорметане (20 мл) перемешивали в бане со льдом-холодной водой и добавляли DiPEA (2,2 мл, 12,7 ммоль). Через 10 мин смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Смесь разбавляли этилацетатом (100 мл) и затем промывали 5%-ным раствором бикарбоната натрия (3×75 мл), водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме с получением 1,47 г неочищенного N-(2-гидрокси-2-метил-пропил)-3-метокси-4-нитро-бензамида (количеств.).
б) 4-Амино-N-(2-гидрокси-2-метил-пропил)-3-метокси-бензамид (Промежуточное соединение А)
Раствор N-(2-гидрокси-2-метил-пропил)-3-метокси-4-нитро-бензамида (1,46 г, 5,44 ммоль) в EtOH (70 мл) гидрировали с использованием реактора с непрерывным потоком H-Cube, 10% Pd/C, при 30°C, 1 бар, режим полного H2, 1 мл/мин. Полученный раствор концентрировали в вакууме и неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (DCM/MeOH/25% NH3 = от 10/0/0 до 9/1/0 до 9/0,9/0,1 об./об. %) с получением указанного в заголовке соединения (775 мг, 60%).
Промежуточное соединение B
2-Метил-4-(4-метилпиперазин-1-ил)анилин
а) 1-Метил-4-(3-метил-4-нитро-фенил)пиперазин
N-Метилпиперазин (752 мкл, 6,79 ммоль) добавляли к 4-фтор-2-метил-1-нитро-бензолу (527 мг, 3,39 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 ч. К реакционной смеси добавляли воду и осуществляли экстрагирование этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (DCM/MeOH = от 1/0 до 9/1 об./об. %) с получением указанного в заголовке соединения (786 мг, 98%).
б) 2-Метил-4-(4-метилпиперазин-1-ил)анилин (Промежуточное соединение В)
К перемешиваемому раствору 1-метил-4-(3-метил-4-нитро-фенил)пиперазина (393 мг, 1,67 ммоль) в этаноле (10 мл) добавляли суспензию 10% Pd на древесном угле (35 мг) в этаноле (6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин в атмосфере азота. Затем добавляли формиат аммония (1,05 г; 16,7 ммоль) и реакционную смесь нагревали до температуры флегмообразования в течение 15 мин. Реакционную смесь охлаждали, фильтровали через Decalite® и концентрировали в вакууме. Остаток растворяли в дихлорметане и промывали насыщенным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 268 мг (78%) 2-метил-4-(4-метилпиперазин-1-ил)анилина.
Промежуточное соединение 2
2-[2-Метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-e]индолизин-7-карбонилхлорид
а) Этил-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
К суспензии этил-2-хлор-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата (Промежуточное соединение 1, 381 мг, 1,37 ммоль) в н-бутаноле (11 мл) добавляли 2-метил-4-(4-метилпиперазин-1-ил)анилин (Промежуточное соединение B, 268 мг, 1,3 ммоль) и трифторуксусную кислоту (200 мкл; 2,6 ммоль). Реакционную смесь нагревали в течение 12 часов при 120°C при микроволновом излучении. Реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством препаративной HPLC. Фракции, содержащие продукт, собирали и упаривали с получением этил-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата (211 мг, 34%-ный выход).
б) 2-[2-Метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбонилхлорид (Промежуточное соединение 2)
К раствору этил-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата (211 мг, 0,47 ммоль) в 8 мл абсолютного этанола добавляли 2М раствор NaOH (591 мкл (2,5 экв.), 1,18 ммоль). Реакционную смесь нагревали при 65°C в течение ночи. Реакционную смесь упаривали досуха и сушили в глубоком вакууме. Полученный остаток растворяли в воде, перемешивали в течение ночи при комнатной температуре и лиофилизировали с получением неочищенного указанного в заголовке соединения.
Тионилхлорид (682 мкл, 9,4 ммоль) добавляли к холодной (0°C) суспензии неочищенного 2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата натрия (242 мг, 0,47 ммоль теор.) в дихлорметане (10 мл). Полученную суспензию перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме и остаток совместно упаривали с толуолом (2×10 мл) с получением 2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбонилхлорида в виде желтого/коричневого порошка (354 мг, количеств. выход неочищенного вещества).
Промежуточное соединение C
2-Метокси-4-морфолино-анилин
Это соединение получали аналогичным способом, как описано в Промежуточном соединении B-a, начиная с морфолина и 4-фтор-2-метокси-1-нитро-бензола (1,3 г, 95%). Полученный таким образом 4-(3-метокси-4-нитро-фенил)морфолин (1,3 г, 5,46 ммоль) растворяли в THF (45 мл) и добавляли уксусную кислоту (5 мл). Смесь охлаждали до 0°C и небольшими порциями добавляли цинк (7,09 г, 109 ммоль) для поддержания температуры ниже 20°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После того, как анализ посредством TLC (тонкослойная хроматография) показал полное превращение исходного вещества, смесь фильтровали через Decalite® и остаток Zn-Decalite® промывали EtOAc (20 мл). Объединенные фильтраты промывали 1 н. раствором NaOH (25 мл), а затем водой (25 мл) и рассолом (25 мл). Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением 2-метокси-4-морфолино-анилина (1,05 г, 92%).
Промежуточное соединение D
2-Метил-6-морфолино-пиридин-3-амин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с морфолина и 6-хлор-3-нитро-2-пиколина, с получением указанного в заголовке соединения (156,9 мг, 82%).
Промежуточное соединение E
2-Хлор-4-(4-метилпиперазин-1-ил)анилин
а) трет-Бутил-N-(4-бром-2-хлор-фенил)-N-трет-бутоксикарбонил-карбамат
К раствору 4-бром-2-хлоранилина (3,0 г, 14,52 ммоль) и карбоната калия (6 г, 43,6 ммоль) в DMF (100 мл) добавляли ди-трет-бутилдикарбонат (1,8 г, 28,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, после чего смесь вливали в смесь вода/рассол и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали рассолом (3×50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (EtOAc/гептан = 1/9 об./об. %) с получением трет-бутил-(4-бром-2-хлор-фенил)-N-трет-бутоксикарбонил-карбамата (2,5 г, 42,3%) в виде желтого/оранжевого масла.
б) трет-Бутил-N-трет-бутоксикарбонил-N-[2-хлор-4-(4-метилпиперазин-1-ил)фенил]карбамат
Смесь трет-бутил-N-(4-бром-2-хлор-фенил)-N-трет-бутоксикарбонил-карбамата (500 мг, 1,23 ммоль), 1-метилпиперазина (166 мкл, 1,48 ммоль), ацетата палладия(II) (27,6 мг, 0,12 ммоль), (+/-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (119 мг, 0,20 ммоль) и карбоната цезия (1,2 г, 3,69 ммоль) в толуоле (20 мл) нагревали при 100°C в течение 16 часов в атмосфере азота. После охлаждения до температуры окружающей среды смесь концентрировали и остаток разбавляли дихлорметаном, промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (дихлорметан/метанол = от 100/0 до 95/5 об./об. %) с получением трет-бутил-N-трет-бутоксикарбонил-N-[2-хлор-4-метилпиперазин-1-ил)фенил]карбамата (480 мг, 91,6%).
в) 2-Хлор-4-(4-метилпиперазин-1-ил)анилин (Промежуточное соединение E)
трет-Бутил-N-трет-бутоксикарбонил-N-[2-хлор-4-метилпиперазин-1-ил)фенил]карбамат (260 мг; 0,61 ммоль) растворяли в DCM (4 мл). Добавляли TFA (4 мл) и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Смесь концентрировали и остаток растворяли в DCM (10 мл) и вливали в 5%-ный раствор бикарбоната натрия (10 мл). Водный слой экстрагировали DCM (2×10 мл). Объединенные органические слои фильтровали через РЕ фильтр и концентрировали в вакууме с получением коричневого масла (105 мг, 76%), которое использовали без дальнейшей очистки.
Промежуточное соединение F
2-Метокси-4-(4-метилпиперазин-1-ил)анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с N-метилпиперазина и 2-метокси-4-фторнитробензола, с получением указанного в заголовке соединения (1,38 г, 94%).
Промежуточное соединение G
2-Этокси-4-(4-метилпиперазин-1-ил)анилин
а) 2-Этокси-4-фтор-1-нитро-бензол
К холодной смеси (0°C) этанола (0,735 мл, 12,6 ммоль) в THF (15 мл) добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 553 мг, 13,83 ммоль). Реакционную смесь перемешивали при 0°C в течение 15 мин, после чего по каплям добавляли раствор 2,4-дифтор-1-нитробензола (1,38 мл, 12,6 ммоль) в THF (25 мл). После перемешивания в течение последующих 90 мин при комнатной температуре реакцию гасили водой, смесь экстрагировали EtOAc. Объединенные органические слои промывали водой и рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = от 100/0 до 85/15 об./об. %) с получением 2-этокси-4-фтор-1-нитро-бензола (2,15 г, 92%).
б) 1-(3-Этокси-4-нитро-фенил)-4-метил-пиперазин
N-Метилпиперазин (603 мкл, 5,44 ммоль) добавляли к 2-этокси-4-фтор-1-нитро-бензолу (500 мг, 2,7 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли воду и затем экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (DCM/MeOH = от 10/0 до 9/1 об./об. %) с получением указанного в заголовке соединения (646 мг, 90%).
в) 2-Этокси-4-(4-метилпиперазин-1-ил)анилин (Промежуточное соединение G)
К перемешиваемому раствору 1-(3-этокси-4-нитро-фенил)-4-метил-пиперазина (265 мг, 1,0 ммоль) в этаноле (5 мл) добавляли суспензию 10% Pd на древесном угле (22 мг) в этаноле (6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин в атмосфере азота. Добавляли формиат аммония (630 мг; 10,0 ммоль) и полученную реакционную смесь нагревали при температуре флегмообразования в течение 15 мин. Реакционную смесь охлаждали и фильтровали через Decalite®. Фильтрат концентрировали в вакууме и затем остаток растворяли в дихлорметане, промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 231 мг (98%) 2-этокси-4-(4-метилпиперазин-1-ил)анилина.
Промежуточное соединение H
трет-Бутил-4-(4-амино-3-метокси-фенил)пиперазин-1-карбоксилат
Это соединение получали аналогичным способом, как описано в Промежуточном соединении B, начиная с трет-бутил-пиперазин-1-карбоксилата и 2-метокси-4-фторнитробензола, с получением указанного в заголовке соединения (245 мг, 91%).
Промежуточное соединение I
Бензил-4-(4-амино-3-метил-фенил)пиперазин-1-карбоксилат
Это соединение получали аналогичным способом, как описано для Промежуточного соединения C, начиная с бензилпиперазин-1-карбоксилата и 4-фтор-2-метил-1-нитро-бензола, с получением указанного в заголовке соединения (327 мг, количеств.).
Промежуточное соединение J
2-(Дифторметокси)-4-(4-метилпиперазин-1-ил)анилин
К раствору 5-фтор-2-нитро-фенола (500 мг, 3,18 ммоль) в DMF (6 мл) добавляли 2-хлор-2,2-дифтор-ацетат натрия (970 мг, 6,36 ммоль) и динатрия карбонат (405 мг, 3,82 ммоль). Реакционную смесь перемешивали при 100°C в течение 3,5 часов и затем при комнатной температуре в течение 3 суток. Добавляли 4М раствор HCl до получения прозрачного раствора и смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои промывали 1М раствором NaOH, рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = от 10/0 до 8/2 об./об. %) с получением 2-(дифторметокси)-4-фтор-1-нитро-бензола (493 мг, 75%).
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с N-метилпиперазина и 2-(дифторметокси)-4-фтор-1-нитро-бензола, с получением 180 мг (80%).
Промежуточное соединение K
4-Метил-6-морфолино-пиридин-3-амин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с морфолина и 2-хлор-5-нитро-4-пиколина, с получением указанного в заголовке соединения (122,9 мг, 81,5%).
Промежуточное соединение L
4-Амино-3-метокси-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения A, начиная с 4-амино-1-метилпиперидина и 3-метокси-4-нитробензйной кислоты, с получением указанного в заголовке соединения (980 мг, 82%).
Промежуточное соединение M
4-Амино-3-метил-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения A-a, начиная с 4-амино-1-метилпиперидина и 4-амино-3-метилбензойной кислоты, с получением указанного в заголовке соединения (700 мг, 70%).
Промежуточное соединение N
4-Амино-3-хлор-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения A-a, начиная с 4-амино-1-метилпиперидина и 4-амино-3-хлорбензойной кислоты, с получением указанного в заголовке соединения (1,64 г, количеств.).
Промежуточное соединение O
4-Амино-3-фтор-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения А-а, начиная с 4-амино-1-метилпиперидина и 4-амино-3-фторбензойной кислоты, с получением указанного в заголовке соединения (170 мг, 16%).
Промежуточное соединение P
4-Амино-3-этокси-N-(1-метил-4-пиперидил)бензамид
а) 3-Этокси-4-нитро-бензойная кислота
3-фтор-4-нитробензойную кислоту (1,50 г, 8,11 ммоль) и гидроксид калия (1,05 г, 18,6 ммоль) перемешивали в этаноле (25 мл) при комнатной температуре. Полученную суспензию медленно нагревали до температуры флегмообразования (10 минут), в ходе чего реакционная смесь превратилась в темно-красный раствор. После нагревания в течение 10 минут при температуре флегмообразования и при интенсивном перемешивании толстым слоем осаждалось твердое вещество. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (10 мл). Далее добавляли 2М раствор HCl (9,3 мл) до pH меньше 2. Полученный осадок интенсивно перемешивали, фильтровали и остаток промывали водой (2×10 мл). Остаток сушили при 40°C под вакуумом с получением 1,53 г 3-этокси-4-нитро-бензойной кислоты (89%).
б) 4-Амино-3-этокси-N-(1-метил-4-пиперидил)бензамид (Промежуточное соединение Р)
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения А, начиная с 4-амино-1-метилпиперидина и 3-этокси-4-нитробензойной кислоты, с получением 190 мг 4-амино-3-этокси-N-(1-метил-4-пиперидил)бензамида (70%).
Промежуточное соединение Q
4-Амино-3-(дифторметокси)-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения А-а, начиная с 4-амино-1-метилпиперидина и 4-амино-3-(дифторметокси)бензоата, с получением указанного в заголовке соединения (130 мг, 29,5%).
Промежуточное соединение R
2-Метокси-4-(1,3,5-триметилпиразол-4-ил)анилин
а) трет-Бутил-N-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)фенил]карбамат
Смесь трет-бутил-N-(4-бром-2-метокси-фенил)карбамата (150 мг, 0,5 ммоль), 1,3,5-триметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразола (118 мг, 0,5 ммоль), тетракис(три-фенилфосфин)палладия(0) (58 мг, 0,05 ммоль) и карбоната калия (207 мг, 1,5 ммоль) в диоксане (4 мл) нагревали при 100°C при микроволновом излучении в течение 20 минут в герметичной пробирке. После охлаждения до температуры окружающей среды смесь концентрировали и остаток разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = от 100/0 до 25/75 об./об. %) с получением трет-бутил-N-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)фенил]карбамата (126,8 мг, 77%).
б) 2-Метокси-4-(1,3,5-триметилпиразол-4-ил)анилин (Промежуточное соединение R)
трет-Бутил-N-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)фенил]карбамат (127 мг, 0,38 ммоль) растворяли в DCM (2 мл). Добавляли TFA (3 мл) и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Смесь концентрировали в вакууме с получением коричневого масла (313 мг), которое использовали без дальнейшей очистки.
Промежуточное соединение S
2-Метокси-4-[(1-метил-4-пиперидил)окси]анилин
а) 4-(3-Метокси-4-нитро-фенокси)-1-метил-пиперидин
К раствору 4-фтор-2-метокси-1-нитро-бензола (750 мг, 4,38 ммоль) в толуоле (10 мл) добавляли 10 мл 25%-ного раствора KOH, 4-гидрокси-N-метилпиперидин (1009 мг, 8,76 ммоль) и бромид тетра-н-бутиламмония (282 мг, 0,876 ммоль). Смесь нагревали при 60°C в течение ночи. Затем реакционную смесь разбавляли этилацетатом и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (дихлорметан/метанол = от 99/1 до 9/1 об./об. %) с получением указанного в заголовке соединения (650 мг, 55,7%).
б) 2-Метокси-4-[(1-метил-4-пиперидил)окси]анилин (Промежуточное соединение S)
10% Pd/C (20 мг) добавляли в виде суспензии в этаноле к раствору 4-(3-метокси-4-нитро-фенокси)-1-метил-пиперидина (200 мг, 0,75 ммоль) в этаноле (5 мл). Полученную смесь перемешивали в течение 15 мин при комнатной температуре. Добавляли формиат аммония (473 мг, 7,5 ммоль) и реакционную смесь перемешивали в течение 1 часа при температуре флегмообразования в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры и фильтровали через Decalite®. Фильтрат концентрировали в вакууме, после чего добавляли дихлорметан и органическую фазу промывали 5%-ным раствором NaHCO3. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 2-метокси-4-[(1-метил-4-пиперидил)окси]анилина (169,5 мг, 95,6%).
Промежуточное соединение T
2-Метил-4-[(1-метил-4-пиперидил)окси]анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения S, начиная с 1-метилпиперидин-4-ола и 4-фтор-2-метил-1-нитро-бензола, с получением указанного в заголовке соединения (151,7 мг, 81%).
Промежуточное соединение U
4-(2-Диметиламиноэтилокси)-2-метокси-анилин
а) 2-(3-метокси-4-нитро-фенокси)-N,N-диметил-этанамин
К холодному (0°C) раствору N,N-диметилэтаноламина (651 мкл, 6,43 ммоль) в THF (5 мл) порциями добавляли NaH (60%-ная дисперсия в минеральном масле, 385 мг). Суспензию перемешивали в течение последующих 30 мин, затем по каплям добавляли 4-фтор-2-метокси-1-нитробензол (1 г, 5,4 ммоль) в 5 мл сухого THF. Раствор нагревали с обратным холодильником в течение ночи. Растворитель удаляли в вакууме и остаток распределяли между этилацетатом и водой (каждого по 50 мл). Органический слой собирали и затем водный слой экстрагировали этилацетатом. Объединенные органические слои промывали 1М раствором HCl, водой, 5%-ным раствором бикарбоната натрия и рассолом. Органический слой сушили над сульфатом натрия и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (гептан/этилацетат = от 9/1 до 1/1 об./об. %, качестве элюента) с получением указанного в заголовке соединения (720 мг, 51,3%).
б) 4-(2-Диметиламиноэтилокси)-2-метокси-анилин (Промежуточное соединение U)
Это соединение получали аналогичным способом, как описано для Промежуточного соединения Sb, с получением указанного в заголовке соединения (144,8 мг, 68,9%).
Промежуточное соединение V
4-Амино-N-(1-метил-4-пиперидил)бензамид
Это соединение получали аналогичным способом, как описано для Промежуточного соединения А-а, начиная с 4-амино-1-метилпиперидина и 4-амино-бензойной кислоты, с получением указанного в заголовке соединения (170 мг, 16%).
Промежуточное соединение W
2-Метил-4-морфолино-анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения C, начиная с морфолина и 5-фтор-2-нитротолуола, с получением указанного в заголовке соединения (8,77 г, 92%).
Промежуточное соединение X
2-Изопропокси-4-(4-метилпиперазин-1-ил)анилин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения G, начиная с 1-метилпиперазина и 4-фтор-2-изопропокси-1-нитро-бензола, с получением 2-изопропокси-4-(4-метилпиперазин-1-ил)анилина (337 мг, 50%).
Промежуточное соединение Y
2-Метокси-4-(1-метилпирролидин-3-ил)окси-анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения U, начиная с 1-метилпирролидин-3-ола и 4-фтор-2-метокси-1-нитро-бензола, с получением указанного в заголовке соединения (199 мг, 84%).
Промежуточное соединение Z
Бензил-3-(4-амино-3-метил-фенокси)азетидин-1-карбоксилат
а) трет-Бутил-3-(3-метил-4-нитро-фенокси)азетидин-1-карбоксилат
К холодному (0°C) раствору N-Boc-3-гидрокси-азетидина (1,84 г, 10,6 ммоль) в THF (10 мл) порциями добавляли NaH (60%-ная дисперсия в минеральном масле, 382 мг). Суспензию перемешивали в течение последующих 30 мин, затем по каплям добавляли 2-нитро-5-фтортолуол (1,5 г, 9,79 ммоль) в 10 мл сухого THF. Раствор перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме и остаток распределяли между этилацетатом и водой (каждого по 50 мл). Органический слой собирали и затем водный слой экстрагировали этилацетатом. Объединенные органические слои промывали 1М раствором HCl, водой, 5%-ным раствором бикарбоната натрия и рассолом. Органический слой сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = от 9/1 до 1/1 об./об. %) с получением 2,24 г (75,1%) указанного в заголовке соединения.
б) Бензил-3-(3-метил-4-нитро-фенокси)азетидин-1-карбоксилат
К раствору трет-Бутил-3-(3-метил-4-нитро-фенокси)азетидин-1-карбоксилата (2,24 г, 7,3 ммоль) в DCM (20 мл) добавляли 4 н. раствор HCl в диоксане (9,1 мл) и реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Смесь концентрировали в вакууме и остаток дважды совместно упаривали с этанолом с получением соединения со снятой защитой (1,7 г, 95%), которое использовали без дальнейшей очистки. К перемешиваемой суспензии неочищенного гидрохлорида 3-(3-метил-4-нитро-фенокси)азетидина (1,7 г, 7,97 ммоль) в DCM (25 мл) добавляли триэтиламин (2,8 мл, 20 ммоль). К реакционной смеси добавляли N-(бензилоксикарбонилокси)-сукцинимид (2,19 г; 8,77 ммоль) и смесь перемешивали в течение 30 мин при комнатной температуре. К реакционной смеси добавляли 5%-ный раствор бикарбоната натрия. Органическую фазу собирали и водную фазу экстрагировали дихлорметаном. Объединенные органические слои сушили (Na2SO4), фильтровали и растворитель упаривали. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = от 100/0 до 50/50 об./об. %) с получением 2,17 г (80%) бензил-3-(3-метил-4-нитро-фенокси)азетидин-1-карбоксилата.
в) Бензил-3-(4-амино-3-метил-фенокси)азетидин-1-карбоксилат (Промежуточное соединение Z)
Это соединение получали аналогичным способом, как описано для Промежуточного соединения С, начиная с бензил-3-(3-метил-4-нитро-фенокси)азетидин-1-карбоксилата, с получением указанного в заголовке соединения (145,3 мг, 39%).
Промежуточное соединение ZA
4-(3,5-Диметилизоксазол-4-ил)-2-метокси-анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения R, начиная с трет-бутил-N-(4-бром-2-метокси-фенил)карбамата и 3,5-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоксазола, с получением указанного в заголовке соединения (96 мг).
Промежуточное соединение ZB
Бензил-3-(4-амино-3-метокси-Фенокси)азетидин-1-карбоксилат
Это соединение получали аналогичным способом, как описано для Промежуточного соединения Z, начиная с 4-фтор-2-метокси-1-нитро-бензола и N-Вос-3-гидрокси-азетидина, с получением указанного в заголовке соединения (215,1 мг, 55%).
Промежуточное соединение ZC
4-(4-Метилпиперазин-1-ил)-2-(тридейтериометокси)анилин
а) 4-Фтор-1-нитро-2-(тридейтериометокси)бензол
К раствору 5-фтор-2-нитрофенола (1,5 г, 9,55 ммоль) в ацетоне (20 мл) добавляли К2CO3 (2,31 г, 16,7 ммоль) при комнатной температуре. К полученной суспензии добавляли дейтерированный йодметан (0,71 мл, 11,46 ммоль) и реакционную смесь перемешивали при температуре флегмообразования в течение ночи. После концентрирования реакционной смеси остаток распределяли между водой и этилацетатом. Этилацетатный слой промывали водой, рассолом, сушили над сульфатом натрия и упаривали в вакууме с получением 4-фтор-1-нитро-2-(тридейтериометокси)бензола (1,67 г, 100%).
б) 1-Метил-4-[4-нитро-3-(тридейтериометокси)фенил]пиперазин
4-Фтор-1-нитро-2-(тридейтериометокси)бензол (1,67 г, 9,55 ммоль) и N-метилпиперазин (2,12 мл, 19,1 ммоль) объединяли в THF (1 мл) и перемешивали в течение 18 ч при комнатной температуре. Желтый осадок, образовавшийся после добавления воды, фильтровали, промывали водой и сушили в вакууме с получением 1-метил-4-[4-нитро-3-(тридейтериометокси)фенил]пиперазина (1,67 г, 80,7%).
в) 4-(4-Метилпиперазин-1-ил)-2-(тридейтериометокси)анилин (Промежуточное соединение ZC)
Это соединение получали аналогичным способом, как описано для Промежуточного соединения В, стадии б), начиная с 1-метил-4-[4-нитро-3-(тридейтериометокси)фенил]пиперазина, с получением указанного в заголовке соединения (177,8 мг).
Промежуточное соединение ZD
2-Метил-6-(4-метилпиперазин-1-ил)пиридин-3-амин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с N-метилпиперазина и 6-хлор-3-нитро-2-пиколина, с получением указанного в заголовке соединения (188,9 мг, 92%).
Промежуточное соединение ZE
Бензил-4-(4-амино-3-метокси-фенил)пиперазин-1-карбоксилат
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B-a, начиная с бензилпиперазин-1-карбоксилата и 2-метокси-4-фторнитробензола. После восстановления с использованием методики, описанной для Промежуточного соединения C, получали указанное в заголовке соединение (1,2 г, 95%).
Промежуточное соединение ZF
4-Метокси-2-(4-метилпиперазин-1-ил)пиримидин-5-амин
а) 2-Хлор-4-метокси-5-нитро-пиримидин
К раствору 2,4-дихлор-5-нитро-пиримидина (1,00 г; 5,2 ммоль) в метаноле (30 мл) по каплям добавляли раствор метоксида натрия (278 мг, 5,2 ммоль) в метаноле (5 мл) при -10°C. Реакционную смесь перемешивали в течение 10 минут при -10°C. Добавляли уксусную кислоту (5 мл) и смесь оставляли нагреваться до комнатной температуры. После упаривания смеси остаток распределяли между 5%-ным раствором NaHCO3 и этилацетатом. Этилацетатный слой промывали водой, рассолом, сушили над сульфатом натрия и упаривали в вакууме. Остаток очищали посредством колоночной хроматографии (гептан/этилацетат = 4/1 об./об. %) с получением 281,9 мг (29%) 2-хлор-4-метокси-5-нитро-пиримидина.
б) 4-Метокси-2-(4-метилпиперазин-1-ил)пиримидин-5-амин (Промежуточное соединение ZF)
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, стадии б), начиная с 2-хлор-4-метокси-5-нитро-пиримидина и N-метилпиперазина, с получением указанного в заголовке соединения (212,8 мг, 89%).
Промежуточное соединение ZG
2-Метокси-5-метил-4-(4-метилпиперазин-1-ил)анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с N-метилпиперазина и 5-фтор-4-метил-2-нитроанизола, с получением указанного в заголовке соединения (94,4 мг, количеств.).
Промежуточное соединение ZH
Бензил-4-(4-амино-3-метокси-фенил)-2-метил-пиперазин-1-карбоксилат
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B-a, начиная с бензил-2-метилпиперазин-1-карбоксилата и 2-метокси-4-фторнитробензола. После восстановления с использованием методики, описанной для Промежуточного соединения C, получали указанное в заголовке соединение (295,4 мг, 97%).
Промежуточное соединение ZI
N4-[3-(диметиламино)пропил]-2-метокси-N4-метил-бензол-1,4-диамин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, начиная с N,N',N'-триметилпропан-1,3-диамина и 4-фтор-2-метокси-1-нитро-бензола, с получением указанного в заголовке соединения (148,8 мг, 94%).
Промежуточное соединение ZJ
2-Метокси-4-(2-метоксиэтокси)анилин
Это соединение получали аналогичным способом, как описано для Промежуточного соединения S, начиная с 2-метоксиэтанола и 4-фтор-2-метокси-1-нитро-бензола, с получением указанного в заголовке соединения (113,4 мг, количеств.).
Промежуточное соединение Aa
1-(2-Метоксиэтил)-3,5-диметил-пиразол-4-амин
а) 1-(2-Метоксиэтил)-3,5-диметил-4-нитро-пиразол
К раствору 3,5-диметил-4-нитро-1H-пиразола (2,5 г, 17,7 ммоль) и карбоната цезия (6,06 г, 18,6 ммоль) в DMF (50 мл) добавляли 2-бромэтилметиловый эфир (2,59 г, 1,75 мл, 18,6 ммоль). Смесь нагревали при 100°C в течение 3,5 ч. После охлаждения до комнатной температуры смесь вливали в воду и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (EtOAc/гептаны = 1/4 об./об. %) с получением 1-(2-метоксиэтил)-3,5-диметил-4-нитро-пиразола (2,66 г, 75,4%) в виде белого кристаллического твердого вещества.
б) 1-(2-Метоксиэтил)-3,5-диметил-пиразол-4-амин
1-(2-Метоксиэтил)-3,5-диметил-4-нитро-пиразол (245 мг, 1,22 ммоль) растворяли в метаноле (25 мл). Полученный раствор гидрировали с использованием реактора гидрирования с непрерывным потоком H-Cube, 10% Pd/C, при 30°C, 8-10 бар, 1 мл/мин, режим полного H2. Полученный раствор концентрировали в вакууме с получением 208 мг (количественный выход) указанного в заголовке соединения в виде светло-коричневого масла.
Промежуточное соединение Ab
3,5-Диметил-1H-пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Aa-b, начиная с 3,5-диметил-4-нитро-1H-пиразола, с получением 110 мг 3,5-диметил-1Н-пиразол-4-амина (количеств.).
Промежуточное соединение Ac
3,5-Диэтил-1H-пиразол-4-амин
а) 3,5-Диэтил-1H-пиразол
К раствору 3,5-гептандиона (2 г, 15,6 ммоль) и гидрата гидразина (0,77 г, 15,8 ммоль) в воде (10 мл) добавляли уксусную кислоту (1 капля) и реакционную смесь нагревали до температуры флегмообразования в течение 1 ч. Затем реакционную смесь охлаждали и концентрировали при пониженном давлении с получением 1,8 г указанного в заголовке соединения. Это соединение использовали непосредственно на следующей стадии без очистки.
б) 3,5-Диэтил-4-нитро-1H-пиразол
К холодной (0°C) смеси 3,5-диэтил-1Н-пиразола (1,8 г, 14,5 ммоль) и концентрированной серной кислоты (1,5 мл) медленно добавляли при интенсивном перемешивании дымящую HNO3 (4,35 мл). Реакционную смесь перемешивали в течение ночи при 60°C. Затем смесь охлаждали до комнатной температуры, потом осторожно добавляли к ледяному насыщенному раствору бикарбоната натрия и перемешивали в течение 15 мин. Затем смесь три раза экстрагировали EtOAc и объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и упаривали в вакууме с получением 2,52 г 3,5-диэтил-4-нитро-1Н-пиразола.
в) 3,5-Диэтил-1H-пиразол-4-амин (Промежуточное соединение Ac)
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Aa-b, начиная с 3,5-диэтил-4-нитро-1Н-пиразола, с получением 3,5-диэтил-1Н-пиразол-4-амина (174 мг, 71%).
Промежуточное соединение Ad
5-Хлор-1,3-диметил-пиразол-4-амин
а) 5-Хлор-1,3-диметил-4-нитро-пиразол
К холодной (0°C) смеси 5-хлор-1,3-диметил-пиразола (1 г, 7,66 ммоль) и концентрированной серной кислоты (750 мкл) медленно добавляли при интенсивном перемешивании дымящую HNO3 (2,1 мл). Реакционную смесь перемешивали в течение ночи при 60°C. Затем смесь охлаждали до комнатной температуры, потом осторожно добавляли к ледяному насыщенному раствору бикарбоната натрия и перемешивали в течение 15 мин. Затем смесь три раза экстрагировали EtOAc и объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и упаривали в вакууме с получением 5-хлор-1,3-диметил-4-нитро-пиразола (1,1 г, 82%).
б) 5-Хлор-1,3-диметил-пиразол-4-амин (Промежуточное соединение Ad)
К перемешиваемому раствору 5-хлор-1,3-диметил-4-нитро-пиразола (146 мг, 1 ммоль) в THF (5 мл) добавляли уксусную кислоту (1,1 мл, 16 ммоль). Смесь охлаждали до 0°C и небольшими порциями добавляли цинк (1,31 г, 20 ммоль), поддерживая температуру ниже 20°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После того, как анализ посредством TLC показал полное превращение исходного вещества, смесь фильтровали через Decalite® и остаток Zn-Decalite® промывали DCM/MeOH 9:1. Объединенные фильтраты промывали 5%-ным раствором NaHCO3, затем промывали водой и рассолом. Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (78 мг, 53%).
Промежуточное соединение Ae
3,5-диэтил-1-(2-метоксиэтил)пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Aa, начиная с 2-бромэтилметилового эфира и 3,5-диэтил-4-нитро-1Н-пиразола, с получением 3,5-диэтил-1Н-пиразол-4-амина (143 мг, 36%).
Промежуточное соединение Af
1-(4-Амино-3,5-диметил-пиразол-1-ил)-2-метил-пропан-2-ол
а) 1-(3,5-Диметил-4-нитро-пиразол-1-ил)-2-метил-пропан-2-ол
К раствору 3,5-диметил-4-нитро-1Н-пиразола (706 мг, 5 ммоль) и DBU (1,49 мл, 10 ммоль) в ацетонитриле (10 мл) добавляли изобутиленоксид (669 мкл, 7,5 ммоль). Смесь нагревали при 65°C в течение 72 ч. После охлаждения до комнатной температуры смесь вливали в воду и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали 1М раствором HCl, водой и рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (гептан/EtOAc = от 100/0 до 60/40 об./об. %) с получением 1-(3,5-диметил-4-нитро-пиразол-1-ил)-2-метил-пропан-2-ола (799 мг, 75%).
б) 1-(4-Амино-3,5-диметил-пиразол-1-ил)-2-метил-пропан-2-ол (Промежуточное соединение Af)
Это соединение получали аналогичным способом, как описано для Промежуточного соединения B, стадии б), начиная с 1-(3,5-диметил-4-нитро-пиразол-1-ил)-2-метил-пропан-2-ола, с получением указанного в заголовке соединения (229 мг, 39%).
Промежуточное соединение Ag
3-Этил-5-метил-изоксазол-4-амин
а) трет-Бутил-N-(3-этил-5-метил-изоксазол-4-ил)карбамат
Дифенилфосфорилазид (2,51 мл, 11,6 ммоль) добавляли к раствору 3-этил-5-метил-изоксазол-4-карбоновой кислоты (1,5 г, 9,67 ммоль), триэтиламина (2,7 мл, 19,3 ммоль), и трет-бутилового спирта (0,92 мл, 9,67 ммоль) в толуоле (50 мл) и перемешивали в течение 4 ч при 100°C. Растворитель удаляли путем выпаривания и остаток переносили в EtOAc (50 мл). Органический слой промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии на силикагеле в смеси гептан/этилацетат = от 10/0 до 7/3 об./об. % в качестве элюента. Фракции, содержащие указанное в заголовке соединение, объединяли и упаривали с получением указанного в заголовке соединения (1,67 г, 76,3%).
б) 3-Этил-5-метил-изоксазол-4-амин (Промежуточное соединение Ag)
трет-Бутил-N-(3-этил-5-метил-изоксазол-4-ил)карбамат (500 мг, 2,2 ммоль) растворяли в смеси TFA/Дихлорметан = 1/1 об./об. % (5 мл) и перемешивали при комнатной температуре в течение 1 ч. Полученную смесь упаривали и остаток растворяли в метаноле и затем фильтровали через колонку SCX-2. После промывки колонки метанолом целевой продукт элюировали 0,7 н. раствором аммиак/метанол. Полученный элюат концентрировали в вакууме с получением указанного в заголовке соединения (302,7 мг, 92%).
Промежуточное соединение Ah
1-[2-[2-(2-Метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-амин
а) 1-[2-[2-(2-Метоксиэтокси)этокси]этил]-3,5-диметил-4-нитро-пиразол
К холодному (0°C) раствору 3,5-диметил-4-нитро-1H-пиразола (250 мг, 1,77 ммоль), монометилового эфира триэтиленгликоля (482 мкл, 3,01 ммоль) и трифенилфосфина (789 мг, 3,01 ммоль) в THF (10 мл) по каплям добавляли раствор 40%-ного DEAD в толуоле (1,31 мл, 3,01 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 3 ч. Добавляли этилацетат и промывали 10%-ным раствором NaCl. Органический слой сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (DCM/MeOH = от 99/1 до 95/5 об./об. %) с получением 1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-4-нитро-пиразола (1,7 г, неочищенный), который непосредственно использовали на следующей стадии.
б) 1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-амин (Промежуточное соединение Ah)
1-[2-[2-(2-Метоксиэтокси)этокси]этил]-3,5-диметил-4-нитро-пиразол (1,5 г, 1,77 ммоль теор.) растворяли в THF (15 мл) и добавляли уксусную кислоту (1,6 мл). Смесь охлаждали до 0°C и небольшими порциями добавляли цинк (2,3 г, 35,4 ммоль), поддерживая температуру ниже 20°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После того, как анализ посредством TLC показал полное превращение исходного соединения, смесь фильтровали через Decalite® и остаток Zn-Decalite® промывали этилацетатом. Объединенные фильтраты промывали 1 н. раствором NaOH, затем водой и рассолом. Органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в метаноле и затем фильтровали через колонку SCX-2. После промывки колонки метанолом целевой продукт элюировали 0,7 н. раствором аммиак/метанол с получением указанного в заголовке соединения (340,1 мг, 74,7%).
Промежуточное соединение Ai
трет-Бутил-3-(4-амино-3,5-диметил-пиразол-1-ил)азетидин-1-карбоксилат
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Ah, начиная с 3,5-диметил-4-нитро-1Н-пиразола и 1-Вос-3-гидроксиазетидина, с получением 261,1 мг трет-бутил-3-(4-амино-3,5-диметил-пиразол-1-ил)азетидин-1-карбоксилата (количеств.).
Промежуточное соединение Aj
3,5-Диметил-1-(оксетан-2-илметил)пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Ah, начиная с 3,5-диметил-4-нитро-1Н-пиразола и 2-гидроксиметилоксетана, с получением 116 мг 3,5-диметил-1-(оксетан-2-илметил)пиразол-4-амина (36,2%.).
Промежуточное соединение Ak
3,5-Диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Aa, начиная с 3,5-диэтил-4-нитро-1Н-пиразола (Промежуточное соединение Ас-b) и 1-бром-2-(2-метоксиэтокси)-этана, с получением 290 мг 3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-амина (72,2%.).
Промежуточное соединение Al
3,5-Диэтил-1-(оксетан-3-ил)пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Aa, начиная с 3,5-диэтил-4-нитро-1Н-пиразола (Промежуточное соединение Ас-b) и оксетан-3-илового эфира толуол-4-сульфоновой кислоты, с получением 165 мг 3,5-диэтил-1-(оксетан-3-ил)пиразол-4-амина (47,7%.).
Промежуточное соединение Am
1-(2-Диметиламиноэтил)-3,5-диметил-пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения Ah, начиная с 3,5-диметил-4-нитро-1H-пиразола и N,N-диметилэтаноламина, с получением 380,4 мг 1-(2-диметиламиноэтил)-3,5-диметил-пиразол-4-амина (количеств.).
Пример 1
N-(2,6-диметилфенил)-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
а) Этил-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
Этил-2-хлор-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат (Промежуточное соединение 1, 200 мг, 0,72 ммоль), 4-амино-N-(2-гидрокси-2-метил-пропил)-3-метокси-бензамид (Промежуточное соединение A, 172 мг, 0,72 ммоль) и карбонат цезия (937 мг; 2,89 ммоль) суспендировали в диоксане (20 мл). Азот барботировали через смесь при температуре 30°C в течение 5 минут с последующим добавлением 9,9-бис-диметил-4,5-бис(дифенилфосфино)ксантена (41,7 мг, 72 мкмоль) и трис(дибензилиденацетон)дипалладия(0) (33,0 мг, 36 мкмоль). Реакционную смесь перемешивали при 80°C в течение 20 часов в потоке газообразного азота. К реакционной смеси добавляли смесь этилацетат/вода/рассол = 1/1/1 об./об. % (50 мл) и перемешивание продолжали в течение 15 мин. После фильтрования через Decalite® водный слой отделяли и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои последовательно промывали водой (40 мл), рассолом (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (DCM/MeOH = от 10/0 до 9/1 об./об. %) с получением указанного в заголовке соединения (190 мг, 55,3%).
б) N-(2,6-диметилфенил)-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
LiHDMS (1Mb смеси THF/этилбензол, 0,61 мл, 0,61 ммоль) добавляли к холодному (0°C) раствору 2,6-диметиланилина (38,5 мкл, 0,31 ммоль) в THF (1 мл). После перемешивания в течение 15 минут при 0°C к реакционной смеси по каплям добавляли этил-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат (50 мг, 0,10 ммоль) в THF (3 мл) и перемешивание продолжали при 0°C в течение 90 мин при 0°C. По каплям при комнатной температуре добавляли дополнительное количество LiHMDS (100 мкл) и перемешивание продолжали в течение 2 часов при комнатной температуре. Реакционную смесь гасили 20 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной HPLC. Фракции, содержащие продукт, собирали и лиофилизировали с получением N-(2,6-диметилфенил)-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (10 мг, 18%). Данные: LCMS (В) Rt: 14,432 мин; m/z 555,3 (М+Н)+.
Пример 2
N-(2-хлор-6-метил-фенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
К суспензии 2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбонилхлорида (Промежуточное соединение 2, 52 мг, 0,07 ммоль теор.) в ацетонитриле (2 мл) добавляли 2-хлор-6-метиланилин (15 мкл, 0,12 ммоль) и каталитическое количество 4-DMAP. Реакционную смесь перемешивали при 50°C в течение ночи. После выпаривания растворителя неочищенный продукт очищали посредством препаративной HPLC. Фракции, содержащие продукт, собирали и концентрировали в вакууме. Остаток распределяли между дихлорметаном и 5%-ным раствором NaHCO3. Органическую фазу отделяли через РЕ фильтр и упаривали с получением 28 мг указанного в заголовке соединения (выход 74%). Данные: LCMS (В) Rt: 9,852 мин; m/z 542,2/544,2 (М+Н)+ (хлоридный образец).
Пример 3
N-(2-бром-6-метил-фенил)-2-(2-метокси-4-морфолино-анилино)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения C в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,8 мг, 17%). Данные: LCMS (A) Rt: 8,031 мин; m/z 587,1/589,1 (М+Н)+ (бромидный образец).
Пример 4
N-(2-этил-6-метил-фенил)-2-[4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием имеющегося в продаже 4-(4-метилпиперазино)анилина в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18 мг, 58%). Данные: LCMS (В) Rt: 10,456 мин; m/z 522,3 (М+Н)+.
Пример 5
N-(2-хлор-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения D в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18 мг, 58%). Данные: LCMS (В) Rt: 10,991 мин; m/z 530,2/532,2 (М+Н)+ (хлоридный образец).
Пример 6
N-(2-хлор-6-метил-фенил)-2-[2-хлор-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения E в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 11%). Данные: LCMS (В) Rt: 10,886 мин; m/z 562,2 (М+Н)+.
Пример 7
N-(2,6-диметилфенил)-2-[2-фтор-4-[(1-метил-4-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения O в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 20%). Данные: LCMS (В) Rt: 10,469 мин; m/z 568,3 (М+Н)+.
Пример 8
2-[2-Хлор-4-[(1-метил-4-пиперидил)карбамоил]анилино]-N-(2,6-диэтилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения N в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,7 мг, 10%). Данные: LCMS (В) Rt: 10,222 мин; m/z 612,2/614,3 (М+Н)+.
Пример 9
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
2-[2-Метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоновую кислоту получали аналогичным способом, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения В.
2-[2-Метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоновую кислоту (26 мг, 0,06 ммоль) растворяли в N,N-диметилформамиде (1,5 мл). Затем добавляли HATU (25 мг, 0,066 ммоль) и триэтиламин (25 мкл, 0,178 ммоль) и смесь перемешивали в течение 10 минут при комнатной температуре. Добавляли 3,5-диэтил-1H-пиразол-4-амин (7 мг, 0,07 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Смесь вливали в смесь этилацетат/вода/рассол 1/1/1 и перемешивали в течение 15 мин. Органический слой отделяли, промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14,0 мг, 43%). Данные: LCMS (В) Rt: 7,530 мин; m/z 540,3 (М+Н)+.
Пример 10
N-(2-метокси-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-метокси-6-метил-анилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (16,1 мг, 29,1%). Данные: LCMS (В) Rt: 9,925 мин; m/z 554,3 (М+Н)+.
Пример 11
N-(2,6-диизопропилфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диизопропиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (15,9 мг, 35,7%). Данные: LCMS (В) Rt: 12,686 мин; m/z 594,4 (М+Н)+.
Пример 12
N-(3,5-диметилизоксазол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 3,5-диметилизоксазол-4-амином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (24,4 мг, 46,1%). Данные: LCMS (В) Rt: 8,408 мин; m/z 529,3 (М+Н)+.
Пример 13
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения T в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ae в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14 мг, 27%). Данные: LCMS (В) Rt: 9,531 мин; m/z 613,3 (М+Н)+.
Пример 14
2-[2-Этокси-4-[(1-метил-4-пиперидил)карбамоил]анилино]-N-(2-этил-6-метил-фенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения P в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,3 мг, 14%). Данные: LCMS (В) Rt: 12,313 мин; m/z 608,3 (М+Н)+.
Пример 15
N-(3,5-диметил-1H-пиразол-4-ил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ab в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14,1 мг, 37%). Данные: LCMS (В) Rt: 7,902 мин; m/z 543,2 (М+Н)+.
Пример 16
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Примера 9, с использованием Промежуточного соединения H в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Aa аналогичным образом, как описано для Примера 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (5,2 мг, 28%). Данные: LCMS (В) Rt: 8,140 мин; m/z 572,3 (М+Н)+.
Пример 17
N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного эфира карбоновой кислоты с использованием той же последовательности реакций, как описано для Примера 9, с использованием Промежуточного соединения Н в качестве исходного вещества. Затем сложный эфир карбоновой кислоты подвергали взаимодействию с 2,6-диметиланилином аналогичным образом, как описано для Примера 1. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (16 мг, 52%). Данные: LCMS (В) Rt: 10,268 мин; m/z 524,3 (М+Н)+.
Пример 18
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Примера 9, с использованием Промежуточного соединения Н в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ac аналогичным образом, как описано для Примера 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (7 мг, 32%). Данные: LCMS (В) Rt: 8,065 мин; m/z 542,3 (М+Н)+.
Пример 19
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Примера 9 и с использованием Промежуточного соединения H в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ae аналогичным образом, как описано для Примера 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 32%). Данные: LCMS (В) Rt: 9,146 мин; m/z 600,3 (М+Н)+.
Пример 20
N-(2,6-диэтилфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного эфира с использованием той же последовательности реакций, как описано для Примера 1, с использованием Промежуточного соединения F в качестве исходного вещества. Затем сложный эфир подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 1. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,5 мг, 23,2%). Данные: LCMS (С) Rt: 12,686 мин; m/z 566,4 (М+Н)+.
Пример 21
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ac в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (22 мг, 36%). Данные: LCMS (В) Rt: 8,319 мин; m/z 557,3 (М+Н)+.
Пример 22
2-[2-Метокси-4-(4-метилпиперазин-1-ил)анилино]-N-(2,4,6-триметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,4,6-триметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (16 мг, 26%). Данные: LCMS (В) Rt: 11,240 мин; m/z 552,3 (М+Н)+.
Пример 23
N-[2-(гидроксиметил)-6-метил-фенил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с (2-амино-3-метил-фенил)метанолом в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6 мг, 10%). Данные: LCMS (В) Rt: 8,876 мин; m/z 554,3 (М+Н)+.
Пример 24
N-(2-фтор-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-фтор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (21,4 мг, 39,6%). Данные: LCMS (В) Rt: 9,878 мин; m/z 542,3 (М+Н)+.
Пример 25
N-(2-хлор-4-метил-3-пиридил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 3-амино-2-хлор-4-метилпиридином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,9 мг, 28,4%). Данные: LCMS (В) Rt: 8,790 мин; m/z 559,2/561,2 (М+Н)+ (хлоридный образец).
Пример 26
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (25,0 мг, 42,7%). Данные: LCMS (В) Rt: 8,128 мин; m/z 586,3 (М+Н)+.
Пример 27
N-(3,5-диметил-1Н-пиразол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ab в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14,0 мг, 26,5%). Данные: LCMS (В) Rt: 7,146 мин; m/z 528,3 (М+Н)+.
Пример 28
N-(5-хлор-1,3-диметил-пиразол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ad в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18 мг, 29%). Данные: LCMS (В) Rt: 8,563 мин; m/z 562,2/564,2 (М+Н)+ (хлоридный образец).
Пример 29
N-(2-этилфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-этиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14 мг, 26%). Данные: LCMS (В) Rt: 10,708 мин; m/z 538,3 (М+Н)+.
Пример 30
N-(2,6-дифторфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-дифторанилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (20,5 мг, 37,5%). Данные: LCMS (В) Rt: 9,607 мин; m/z 546,2 (М+Н)+.
Пример 31
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-этокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения G в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ас в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (10 мг, 37%). Данные: LCMS (В) Rt: 8,939 мин; m/z 570,3 (М+Н)+.
Пример 32
2-[2-Этокси-4-(4-метилпиперазин-1-ил)анилино]-N-(2-этил-6-метил-фенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения G в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5 мг, 9%). Данные: LCMS (В) Rt: 11,636 мин; m/z 566,3 (М+Н)+.
Пример 33
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения J в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ас аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (9,6 мг, 23%). Данные: LCMS (В) Rt: 8,864 мин; m/z 592,3 (М+Н)+.
Пример 34
N-(2,6-диметилфенил)-2-[4-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения K в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,2 мг, 17,4%). Данные: LCMS (В) Rt: 10,575 мин; m/z 510,3 (М+Н)+.
Пример 35
N-(2-хлор-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения L в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (9,8 мг, 21%). Данные: LCMS (В) Rt: 11,000 мин; m/z 600,2/602,2 (М+Н)+ (хлоридный образец).
Пример 36
N-(2,6-диэтилфенил)-2-[2-метил-4-[(1-метил-4-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения М в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (2,5 мг, 11%). Данные: LCMS (В) Rt: 11,332 мин; m/z 592,3 (М+Н)+.
Пример 37
2-[2-Дифторметокси)-4-[(1-метил-4-пиперидил)карбамоил]анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения Q в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 12%). Данные: LCMS (В) Rt: 11,282 мин; m/z 616,3 (М+Н)+.
Пример 38
N-(2,6-диэтилфенил)-2-[2-метокси-4-(1-метилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
а) 2-(4-Бром-2-метокси-анилино)-N-(2,6-диэтилфенил)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием имеющегося в продаже 4-бром-2-метоксианилина в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (50 мг).
б) N-(2,6-диэтилфенил)-2-[2-метокси-4-(1-метилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Смесь 2-(4-бром-2-метокси-анилино)-N-(2,6-диэтилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (22,6 мг, 0,041 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (17,2 мг, 0,083 ммоль), хлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия(II), комплекса с дихлорметаном (3,4 мг), и карбоната калия (28,3 мг, 0,205 ммоль) в смеси диоксан/вода (1,5 мл/0,3 мл) нагревали в микроволновой печи при 140°C в течение 60 минут в гермитчной пробирке. После охлаждения до температуры окружающей среды смесь концентрировали в вакууме. Полученный остаток разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (15,2 мг, 38%). Данные: LCMS (В) Rt: 17,769 мин; m/z 548,3 (М+Н)+.
Пример 39
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,8 мг, 37,7%). Данные: LCMS (В) Rt: 11,129 мин; m/z 553,3 (М+Н)+.
Пример 40
N-(2-этил-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8,7 мг, 21,9%). Данные: LCMS (В) Rt: 11,778 мин; m/z 567,3 (М+Н)+.
Пример 41
N-(2-хлор-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,4 мг, 28,4%). Данные: LCMS (В) Rt: 11,395 мин; m/z 573,2/575,2 (М+Н)+ (хлоридный образец).
Пример 42
N-(2-бром-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,8 мг, 27,3%). Данные: LCMS (В) Rt: 11,527 мин; m/z 617,2/619,2 (М+Н)+ (бромидный образец).
Пример 43
N-(2,6-диэтилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,6 мг, 33,6%). Данные: LCMS (В) Rt: 12,578 мин; m/z 581,3 (М+Н)+.
Пример 44
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (16,9 мг, 40,1%). Данные: LCMS (В) Rt: 8,940 мин; m/z 601,3 (М+Н)+.
Пример 45
N-(2,6-диметилфенил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения E в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14,2 мг, 40%). Данные: LCMS (В) Rt: 10,765 мин; m/z 537,3 (М+Н)+.
Пример 46
N-(2-хлор-6-метил-фенил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения T в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (10,5 мг, 26%). Данные: LCMS (В) Rt: 10,077 мин; m/z 557,2/559,2 (М+Н)+ (хлоридный образец).
Пример 47
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения T в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ac в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (7,6 мг, 19%). Данные: LCMS (В) Rt: 8,307 мин; m/z 555,3 (М+Н)+.
Пример 48
2-[4-(2-Диметиламиноэтилокси)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (16,7 мг, 52,9%). Данные: LCMS (В) Rt: 10,556 мин; m/z 527,3 (М+Н)+.
Пример 49
N-(2-хлор-6-метил-фенил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (15,6 мг, 52,9%). Данные: LCMS (В) Rt: 10,566 мин; m/z 547,2/549,2 (М+Н)+ (хлоридный образец).
Пример 50
2-[4-(2-Диметиламиноэтилокси)-2-метокси-анилино]-N-(2-этил-6-метил-фенил)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,2 мг, 53,1%). Данные: LCMS (В) Rt: 11,075 мин; m/z 541,3 (М+Н)+.
Пример 51
N-(2-бром-6-метил-фенил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (9,38 мг, 26,4%). Данные: LCMS (В) Rt: 10,760 мин; m/z 590,2/592,2 (М+Н)+ (бромидный образец).
Пример 52
N-(2,6-диэтилфенил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,54 мг, 16,6%). Данные: LCMS (В) Rt: 11,832 мин; m/z 555,3 (М+Н)+.
Пример 53
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ас в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,4 мг, 41,0%). Данные: LCMS (В) Rt: 8,189 мин; m/z 545,3 (М+Н)+.
Пример 54
2-[4-(2-Диметиламиноэтилокси)-2-метокси-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения U в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18,3 мг, 53,2%). Данные: LCMS (В) Rt: 8,234 мин; m/z 575,3 (М+Н)+.
Пример 55
N-(2,6-диэтилфенил)-2-[4-[(1-метил-4-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 1-а и Промежуточного соединения 2, с использованием Промежуточного соединения V в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (3,7 мг, 17%). Данные: LCMS (В) Rt: 11,254 мин; m/z 578,3 (М+Н)+.
Пример 56
N-(2,6-диметилфенил)-2-(2-метил-4-морфолино-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного этилового эфира, с использованием той же последовательности реакций, как описано для Примера 2а, с использованием Промежуточного соединения W. Затем сложный эфир подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 1b. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,5 мг, 22%). Данные: LCMS (В) Rt: 13,950 мин; m/z 509,3 (М+Н)+.
Пример 57
N-(2-этил-6-метил-фенил)-2-[2-изоргорокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения X в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 6-этил-о-толуидином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13 мг, 28%). Данные: LCMS (В) Rt: 12,031 мин; m/z 580,3 (М+Н)+.
Пример 58
N-(2,6-диметилфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (20 мг, 64%). Данные: LCMS (В) Rt: 9,706 мин; m/z 508,3 (М+Н)+.
Пример 59
N-(2,6-диэтилфенил)-2-[2-метокси-4-(1-метилпирролидин-3-ил)окси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения Y в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8,1 мг, 25%). Данные: LCMS (В) Rt: 12,158 мин; m/z 567,3 (М+Н)+.
Пример 60
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-3-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
а) 4-[(7-Этоксикарбонил-5,6-дигидропиримидо[4,5-e]индолизин-2-ил)амино]-3-метокси-бензойная кислота
Это соединение получали способом, аналогичным описанному для Промежуточного соединения 1а, с использованием 4-амино-3-метоксибензойной кислоты в качестве исходного вещества. Выход: 160 мг (43,5%).
б) Этил-2-[2-метокси-4-[(1-метил-3-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилат
К перемешиваемой суспензии 4-[(7-этоксикарбонил-5,6-дигидропиримидо[4,5-e]индолизин-2-ил)амино]-3-метокси-бензойной кислоты (80 мг, 0,20 ммоль) и дигидрохлорида 3-амино-1-метилпиперидина (36,6 мг, 0,20 ммоль) в DMF (2 мл) добавляли DiPEA (139 мкл, 0,84 ммоль). Полученный раствор охлаждали до 0°C и добавляли HATU (83,6 мг, 0,22 ммоль). Охлаждение удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь по каплям добавляли к интенсивно перемешиваемой смеси EtOAc/вода/рассол 1/1/1 (30 мл). Затем водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические слои промывали водой (20 мл), рассолом (20 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (70 мг, 70%).
в) N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-3-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
После омыления неочищенного этил-2-[2-метокси-4-[(1-метил-3-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата и последующего взаимодействия с тионилхлоридом, как описано для Промежуточного соединения 2b, получали соответствующий хлорангидрид. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методикой, описанной в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8,4 мг, 19%). Данные: LCMS (В) Rt: 11,406 мин; m/z 580,3 (М+Н)+.
Пример 61
N-(2,6-дихлорфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-дихлоранилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (9 мг, 20,7%). Данные: LCMS (В) Rt: 10,404 мин; m/z 578,2 (М+Н)+ (хлоридный образец).
Промежуточное соединение 3
Этил-2-хлорпиримидо[4,5-e]индолизин-7-карбоксилат
а) Этил-2-метоксипиримидо[4,5-e]индолизин-7-карбоксилат
DDQ (1,53 г, 6,76 ммоль) добавляли к перемешиваемому раствору этил-2-метокси-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксилата (1,54 г, 5,63 ммоль) в DCM (50 мл). Реакционную смесь перемешивали в течение 3 суток при комнатной температуре. Добавляли дополнительное количество 200 мг DDQ и реакционную смесь перемешивали в течение еще 7 суток при комнатной температуре. Смесь фильтровали и концентрировали в вакууме до небольшого объема. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (гептан/этилацетат = от 1/0 до 1/1 об./об. %) с получением указанного в заголовке соединения (750 мг, 50%).
б) Этил-2-гидроксипиримидо[4,5-e]индолизин-7-карбоксилат
Йодид натрия (1,24 г, 8,29 ммоль) добавляли к перемешиваемому раствору этил-2-метокси-пиримидо[4,5-e]индолизин-7-карбоксилата (750 мг, 2,76 ммоль) в ацетонитриле (19 мл). К реакционной смеси по каплям добавляли раствор триметилсилилхлорида (896 мг, 1,05 мл) в ацетонитриле (3 мл). Смесь перемешивали при комнатной температуре в течение ночи. По каплям добавляли дополнительное количество йодида натрия (3,33 г), TMS-Cl (2,4 г, 2,8 мл) в ацетонитриле (6 мл) и реакционную смесь перемешивали в течение 3 суток при комнатной температуре. Смесь концентрировали при пониженном давлении. Остаток суспендировали в 200 мл DCM/MeOH (4/1) и экстрагировали смесью насыщенного раствора тиосульфата натрия (50 мл) и воды (100 мл). Водный слой экстрагировали DCM/MeOH (4/1, 2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением твердого вещества. Твердое вещество растирали в кипящем этилацетате (50 мл). После охлаждения твердое вещество перемешивали 1 час при комнатной температуре и фильтровали. Остаток сушили при 40°C в вакууме с получением 1,0 г неочищенного этил-2-гидрокси-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксилата (количеств, выход).
в) Этил-2-хлорпиримидо[4,5-e]индолизин-7-карбоксилат (Промежуточное соединение 3)
N,N-Диметиланилин (47 мг, 50 мкл, 1,50 ммоль) добавляли к раствору этил-2-гидроксипиримидо[4,5-е]индолизин-7-карбоксилата (1,0 г, 3,89 ммоль) в ацетонитриле (30 мл). К реакционной смеси по каплям добавляли раствор оксихлорида фосфора(V) (2,99 г, 1,81 мл, 19,5 ммоль) в ацетонитриле (4 мл). Коричневую/красную суспензию нагревали до 65°C в течение 4 часов. После охлаждения смесь медленно вливали в перемешиваемую смесь 25%-ного водн. аммиака (50 мл) и ледяной воды (100 мл), поддерживая температуру ниже 10°C. После перемешивания в течение последующих 15 минут смесь экстрагировали этилацетатом. Затем объединенные органические слои промывали водой (50 мл), 0,2 н. HCl (50 мл), рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали посредством колоночной хроматографии на диоксиде кремния (гептан/этилацетат = от 1/0 до 1/1 об./об. %) с получением 200 мг указанного в заголовке соединения.
Пример 62
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, начиная с Промежуточного соединения 3 и Промежуточного соединения S в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (30 мг, 45%). Данные: LCMS (В) Rt: 12,491 мин; m/z 551,3 (М+Н)+.
Пример 63
N-(2,6-диметилфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения B в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17 мг, 47%). Данные: LCMS (В) Rt: 9,602 мин; m/z 522,3 (М+Н)+.
Пример 64
N-(2-этил-6-метил-сЬенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения B в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-этил-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14 мг, 65%). Данные: LCMS (В) Rt: 10,303 мин; m/z 536,3 (М+Н)+.
Пример 65
N-(2-бром-6-метил-фенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения B в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (30 мг, 73%). Данные: LCMS (В) Rt: 9,914 мин; m/z 586,2/588,2 (М+Н)+ (бромидный образец).
Пример 66
N-(2,6-дихлорфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения B в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-дихлоранилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (10 мг, 36%). Данные: LCMS (В) Rt: 9,770 мин; m/z 562,2 (М+Н)+ (хлоридный образец).
Пример 67
N-(2,6-диэтилфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения B в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13 мг, 34%). Данные: LCMS (В) Rt: 10,954 мин; m/z 550,3 (М+Н)+.
Пример 68
N-(2,6-диметилфенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения D в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (9,6 мг, 27%). Данные: LCMS (В) Rt: 10,844 мин; m/z 510,2 (М+Н)+.
Пример 69
N-(2-этил-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения D в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-этил-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,2 мг, 30,4%). Данные: LCMS (В) Rt: 11,599 мин; m/z 524,3 (М+Н)+.
Пример 70
N-(2-бром-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения D в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (12 мг, 30%). Данные: LCMS (В) Rt: 11,151 мин; m/z 574,2/576,2 (М+Н)+ (бромидный образец).
Пример 71
N-(2,6-диэтилфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, начиная с Промежуточного соединения 3 и Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (0,7 мг). Данные: LCMS (A) Rt: 6,446 мин; m/z 564,3 (М+Н)+.
Пример 72
N-(2-этил-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-этил-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,8 мг, 32,2%). Данные: LCMS (В) Rt: 10,836 мин; m/z 552,3 (М+Н)+.
Пример 73
N-(2-бром-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18,1 мг, 30%). Данные: LCMS (В) Rt: 10,427 мин; m/z 602,2/604,2 (М+Н)+ (бромидный образец).
Пример 74
N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного этилового эфира с использованием той же последовательности реакций, как описано для Примера 1, начиная с Промежуточного соединения 3 и Промежуточного соединения Н в качестве исходных веществ. Затем сложный этиловый эфир подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 1. После снятия Boc-защитной группы с использованием смеси 50% TFA/дихлорметан осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (1,4 мг, 5%). Данные: LCMS (В) Rt: 10,283 мин; m/z 524,3 (М+Н)+.
Пример 75
N-(2-хлор-6-метил-фенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения H в качестве исходных веществ. Затем сложный этиловый эфир подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 1. После снятия Boc-защитной группы с использованием смеси 50% TFA/дихлорметан осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (0,9 мг, 4%). Данные: LCMS (A) Rt: 5,794 мин; m/z 544,2/546,2 (М+Н)+ (хлоридный образец).
Пример 76
N-(2,6-диэтилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного этилового эфира с использованием той же последовательности реакций, как описано для Примера 1, начиная с Промежуточного соединения 1 и Промежуточного соединения Н в качестве исходных веществ. Затем сложный этиловый эфир подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 1. После снятия Boc-защитной группы с использованием смеси 50% TFA/дихлорметан осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (20,7 мг, 72%). Данные: LCMS (A) Rt: 6,213 мин; m/z 552,3 (М+Н)+.
Пример 77
N-(2,6-диэтилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение выделяли в качестве побочного продукта в ходе очистки Примера 76 посредством препаративной HPLC с получением указанного в заголовке соединения (2 мг, 7%). Данные: LCMS (A) Rt: 6,512 мин; m/z 550,3 (М+Н)+.
Пример 78
N-(2-хлор-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (24 мг, 81%). Данные: LCMS (В) Rt: 9,831 мин; m/z 528,2/530,2 (М+Н)+ (хлоридный образец).
Пример 79
N-(2,6-дихлорфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-дихлоранилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (27 мг, 96%). Данные: LCMS (В) Rt: 9,853 мин; m/z 548,2 (М+Н)+ (хлоридный образец).
Пример 80
N-(2,6-диэтилфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (18 мг, 40%). Данные: LCMS (В) Rt: 10,916 мин; m/z 536,3 (М+Н)+.
Пример 81
N-(2-бром-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-бром-6-метиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (16 мг, 51%). Данные: LCMS (В) Rt: 9,922 мин; m/z 572,2/574,2 (М+Н)+ (бромидный образец).
Пример 82
N-(2-этил-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения I в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-этил-6-метиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (22 мг, 77%). Данные: LCMS (В) Rt: 10,361 мин; m/z 522,3 (М+Н)+.
Пример 83
N-(2,6-диэтилфенил)-2-(2-метил-4-морфолино-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему сложного этилового эфира с использованием той же последовательности реакций, как описано для Промежуточного соединения 2а, с использованием Промежуточного соединения W. Затем сложный эфир подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 1b. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,2 мг, 13%). Данные: LCMS (В) Rt: 15,695 мин; m/z 537,3 (М+Н)+.
Пример 84
N-(2,6-диметилфенил)-2-[2-метокси-4-(1-метилпирролидин-3-ил)окси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения Y в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 12%). Данные: LCMS (В) Rt: 10,832 мин; m/z 539,2 (М+Н)+.
Пример 85
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[4-(1-метил-4-пиперидил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием имеющегося в продаже 4-(1-метил-пиперидин-4-ил)-анилина в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (18,3 мг, 44%). Данные: LCMS (В) Rt: 8,405 мин; m/z 555,3 (М+Н)+.
Пример 86
N-(2-хлор-6-метил-фенил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения J в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-хлор-6-метиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,2 мг, 41%). Данные: LCMS (В) Rt: 11,569 мин; m/z 594,2/596,2 (М+Н)+ (хлоридный образец).
Пример 87
N-(2,6-диэтилфенил)-2-[2-этил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 38-а, с использованием 4-бром-2-этиланилина в качестве исходного вещества. Затем бромид подвергали взаимодействию с N-метилпиперазином в соответствии с методиками, описанными в Примере 38-b. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,5 мг, 15%). Данные: LCMS (В) Rt: 12,136 мин; m/z 564,4 (М+Н)+.
Пример 88
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[4-[3-(диметиламино)пропил-метил-карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Примера 60, с использованием N,N,N'-триметил-1,3-пропандиамина в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ae в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,8 мг, 20%). Данные: LCMS (В) Rt: 10,220 мин; m/z 658,4 (М+Н)+.
Пример 89
2-[4-(Азетидин-3-илокси)-2-метил-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения Z в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (6,6 мг, 17%). Данные: LCMS (В) Rt: 7,975 мин; m/z 543,3 (М+Н)+.
Пример 90
2-[4-(3,5-Диметилизоксазол-4-ил)-2-метокси-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZA в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (16,2 мг, 46%). Данные: LCMS (В) Rt: 14,477 мин; m/z 583,3 (М+Н)+.
Пример 91
2-[4-(Азетидин-3-илокси)-2-метокси-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZB в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (12,8 мг, 25%). Данные: LCMS (В) Rt: 8,441 мин; m/z 559,3 (М+Н)+.
Пример 92
2-[4-(Азетидин-3-илокси)-2-метокси-анилино]-N-(3,5-диэтил-1Н-пиразол-4-ил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZB в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ac в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (8,9 мг, 19%). Данные: LCMS (В) Rt: 8,324 мин; m/z 529,3 (М+Н)+.
Пример 93
2-[4-(Азетидин-3-илокси)-2-метокси-анилино]-N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZB в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ae в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (14,9 мг, 34%). Данные: LCMS (В) Rt: 9,618 мин; m/z 587,4 (М+Н)+.
Пример 94
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(1.3,5-триметилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения R в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8,7 мг, 21%). Данные: LCMS (В) Rt: 12,704 мин; m/z 596,3 (М+Н)+.
Пример 95
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, начиная с Промежуточного соединения 3 и Промежуточного соединения S в качестве исходных веществ. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (23 мг, 32%). Данные: LCMS (В) Rt: 9,796 мин; m/z 599,3 (M+H)+.
Пример 96
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, начиная с Промежуточного соединения 3 и Промежуточного соединения ZE в качестве исходных веществ. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (14 мг, 61%). Данные: LCMS (В) Rt: 9,058 мин; m/z 570,3 (М+Н)+.
Пример 97
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, начиная с Промежуточного соединения 3 и Промежуточного соединения ZE в качестве исходных веществ. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ас в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (10,6 мг, 39%). Данные: LCMS (В) Rt: 8,908 мин; m/z 540,3 (М+Н)+.
Пример 98
N-[1-(2-гидрокси-2-метил-пропил)-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZE в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Af в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (11,4 мг, 28%). Данные: LCMS (В) Rt: 7,882 мин; m/z 586,3 (М+Н)+.
Пример 99
N-(2,6-диэтилфенил)-2-[4-(4-метилпиперазин-1-ил)-2-(тридейтериометокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZC в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (14,6 мг, 23%). Данные: LCMS (В) Rt: 12,878 мин; m/z 569,4 (М+Н)+.
Пример 100
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[4-(4-метилпиперазин-1-ил)-2-(тридейтериометокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZC в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ae в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,2 мг, 25%). Данные: LCMS (В) Rt: 9,443 мин; m/z 617,4 (М+Н)+.
Пример 101
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[[2-метил-6-(4-метилпиперазин-1-ил)-3-пиридил]амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZD в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,8 мг, 27%). Данные: LCMS (В) Rt: 7,187 мин; m/z 571,4 (М+Н)+.
Пример 102
2-[5-Хлор-2-метокси-4-(4-метилпиперазин-1-ил)анилино]-N-(2,6-диэтилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение выделяли в качестве побочного продукта из соединения, описанного в Примере 20, с использованием препаративной HPLC с получением указанного в заголовке соединения (9,1 мг). Данные: LCMS (В) Rt: 14,555 мин; m/z 601,3/603,3 (М+Н)+ (хлоридный образец).
Пример 103
N-(2,6-диметилфенил)-2-[2-метокси-4-(тетрагидропиран-4-илкарбамоил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
а) 2-(4-Бром-2-метокси-анилино)-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием имеющегося в продаже 4-бром-2-метоксианилина в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2, с получением указанного в заголовке соединения (1,35 г, 84%).
б) 2-(4-ииано-2-метокси-анилино)-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
К раствору 2-(4-бром-2-метокси-анилино)-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (1,35 г, 2,6 ммоль) и цианида цинка (321 мг, 2,73 ммоль) в DMF (4 мл) добавляли тетракис(трифенилфосфино)палладий(0) (300 мг, 0,26 ммоль). Реакционную смесь нагревали в течение 30 минут при 170°C при микроволновом излучении. После охлаждения до температуры окружающей среды смесь концентрировали и остаток разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (1,05 г, 87%).
в) 4-[[7-[(2,6-Диметилфенил)карбамоил]-5,6-дигидропиримидоИ,5-е]индолизин-2-ил]амино]-3-метокси-бензойная кислота
К перемешиваемой суспензии 2-(4-циано-2-метокси-анилино)-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (750 мг, 1,61 ммоль) в MeOH (25 мл) добавляли раствор гидроксида калия (453 мг, 8,07 ммоль) в воде (12,5 мл). Реакционную смесь нагревали в течение 2 часов при 120°C при микроволновом излучении. После упаривания метанольной фракции полученный водный слой подкисляли путем добавления 2 н. раствора HCl до pH примерно 2. После экстрагирования дихлорметаном объединенные органические слои фильтровали через РЕ фильтр с получением 330 мг указанного в заголовке соединения (выход: 42%).
г) N-(2,6-диметилфенил)-2-[2-метокси-4-(тетрагидропиран-4-илкарбамоил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида 4-аминотетрагидропирана с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5 мг, 18%). Данные: LCMS (В) Rt: 14,407 мин; m/z 567,3 (М+Н)+.
Пример 104
2-[4-(Азетидин-3-илкарбамоил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и 3-амино-1-N-Вос-азетидина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (6,8 мг, 20%). Данные: LCMS (В) Rt: 11,597 мин; m/z 538,3 (М+Н)+.
Пример 105
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 17) и метоксиуксусной кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (10 мг, 49%). Данные: LCMS (В) Rt: 12,973 мин; m/z 596,3 (М+Н)+.
Пример 106
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[[4-метокси-2-(4-метилпиперазин-1-ил)пиримидин-5-ил]амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZF в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,9 мг, 20%). Данные: LCMS (В) Rt: 7,658 мин; m/z 588,4 (М+Н)+.
Пример 107
N-(2,6-диэтилфенил)-2-[[4-метокси-2-(4-метилпиперазин-1-ил)пиримидин-5-ил]амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZF в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (3,1 мг, 11%). Данные: LCMS (В) Rt: 12,119 мин; m/z 568,4 (М+Н)+.
Пример 108
N-(3-этил-5-метил-изоксазол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ag аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,3 мг, 45%). Данные: LCMS (В) Rt: 9,102 мин; m/z 543,3 (М+Н)+.
Пример 109
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ae аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (21,5 мг, 76%). Данные: LCMS (В) Rt: 9,108 мин; m/z 614,4 (М+Н)+.
Пример 110
N-(2,6-диметилфенил)-2-[4-[4-(3-фторциклобутанкарбонил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 17) и 3-фторцикло-бутанкарбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (26 мг, 99%). Данные: LCMS (В) Rt: 15,532 мин; m/z 624,3 (М+Н)+.
Пример 111
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(3-метилоксетан-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 17) и 3-метил-оксетан-3-карбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4 мг, 17%). Данные: LCMS (В) Rt: 13,824 мин; m/z 622,3 (М+Н)+.
Пример 112
2-[4-[1-(Циклопропанкарбонил)пиперазин-1-ил]-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 17) и циклопропанкарбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11 мг, 49%). Данные: LCMS (В) Rt: 14,744 мин; m/z 592,3 (М+Н)+.
Пример 113
2-[4-(4-БутилсульФонилпиперазин-1-ил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
К перемешиваемому раствору N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (Пример 17, 20 мг, 0,038 ммоль) в DCM (2 мл) добавляли триэтиламин (18 мкл, 0,13 ммоль) и бутан-1-сульфонилхлорид (5 мкл, 0,042 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали и остаток очищали посредством препаративной HPLC с получением указанного в заголовке соединения (3 мг, 13%). Данные: LCMS (В) Rt: 17,533 мин; m/z 644,3 (М+Н)+.
Пример 114
N-(2,6-диметилфенил)-2-[4-[4-(этилкарбамоил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
К перемешиваемому раствору N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (Пример 17, 20 мг, 0,038 ммоль) в DCM (2 мл) добавляли изоцианатоэтан (4 мкл, 0,042 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали и остаток очищали посредством препаративной HPLC с получением указанного в заголовке соединения (24 мг, 99%). Данные: LCMS (В) Rt: 13,414 мин; m/z 595,3 (М+Н)+.
Пример 115
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-5-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZG в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aa в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (19,2 мг, 29%). Данные: LCMS (В) Rt: 9,296 мин; m/z 600,4 (М+Н)+.
Пример 116
N-(2,6-диэтилфенил)-2-[2-метокси-4-(3-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZH в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (12,2 мг, 17%). Данные: LCMS (В) Rt: 12,915 мин; m/z 566,4 (М+Н)+.
Пример 117
N-(2,6-диметилфенил)-2-[2-метокси-4-[[(3S,4R)-3-метокси-4-пиперидил]карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и (3S,4R)-4-амино-1-Вос-3-метокси-пиперидина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (2,7 мг, 7%). Данные: LCMS (В) Rt: 11,799 мин; m/z 596,4 (М+Н)+.
Пример 118
N-(2,6-димeтилфeнил)-2-[4-[[(3S,4R)-3-фтop-4-пипepидил]кapбaмoил]-2-мeтoкcи-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и трет-бутил-(3S,4R)-4-амино-3-фтор-пиперидин-1-карбоксилата с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (2,2 мг, 5%). Данные: LCMS (В) Rt: 11,671 мин; m/z 584,3 (М+Н)+.
Пример 119
2-[4-[(4-цис-Аминоциклогексил)карбамоил]-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида 1-N-Boc-цис-1,4-циклогексилдиамина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (4,6 мг, 15%). Данные: LCMS (В) Rt: 11,581 мин; m/z 580,3 (М+Н)+.
Пример 120
N-(2,6-диметилфенил)-2-[4-(изопропилкарбамоил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида изопропиламина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (2,9 мг, 8%). Данные: LCMS (В) Rt: 15,663 мин; m/z 525,3 (М+Н)+.
Пример 121
N-(2,6-диметилфенил)-2-[2-метокси-4-[[(1R,5S)-3-оксабицикло[3.1.0]гексан-6-ил]карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида (1R,5S)-3-оксабицикло[3.1.0]гексан-6-амина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,5 мг, 15%). Данные: LCMS (В) Rt: 14,186 мин; m/z 565,2 (М+Н)+.
Пример 122
N-(2,6-диметилфенил)-2-[2-метокси-4-(морфолин-4-карбонил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и морфолина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8,7 мг, 15%). Данные: LCMS (В) Rt: 14,262 мин; m/z 553,3 (М+Н)+.
Пример 123
N-(2,6-диметилфенил)-2-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и тиоморфолин-1,1-диоксида с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (10,5 мг, 14%). Данные: LCMS (В) Rt: 13,936 мин; m/z 601,2 (М+Н)+.
Пример 124
N-(2,6-диметилфенил)-2-[4-(4-этил-1,4-диазепан-1-карбонил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и дигидрохлорида 1-этил-1,4-диазепана с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,4 мг, 7%). Данные: LCMS (В) Rt: 11,510 мин; m/z 594,4 (М+Н)+.
Пример 125
2-[4-[(3,3-Дифторциклобутил)карбамоил]-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида 3,3-дифторциклобутанамина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,1 мг, 7%). Данные: LCMS (В) Rt: 16,415 мин; m/z 573,3 (М+Н)+.
Пример 126
2-[4-(Циклопропилкарбамоил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и циклопропиламина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,7 мг, 10%). Данные: LCMS (В) Rt: 14,833 мин; m/z 523,3 (М+Н)+.
Пример 127
N-(2,6-диметилфенил)-2-[2-метокси-4-(2-метоксиэтилкарбамоил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и 2-метоксиэтиламина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,9 мг, 8%). Данные: LCMS (В) Rt: 14,283 мин; m/z 541,3 (М+Н)+.
Пример 128
N-(2,6-диметилфенил)-2-[2-метокси-4-[2-(1-пиперидил)этилкарбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и 2-(1-пиперидил)этанамина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (4,2 мг, 5%). Данные: LCMS (В) Rt: 12,340 мин; m/z 594,3 (М+Н)+.
Пример 129
2-[4-(4-ииано-4-метил-пиперидин-1-карбонил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему карбоновой кислоты (Пример 103-с) и гидрохлорида 4-метилпиперидин-4-карбонитрила с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку проводили посредством флэш-хроматографии на силикагеле (дихлорметан/метанол = от 99/1 до 9/1 об./об. %) с получением указанного в заголовке соединения (3,9 мг, 5%). Данные: LCMS (В) Rt: 15,853 мин; m/z 590,3 (М+Н)+.
Пример 130
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(3-метилазетидин-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 17) и 1-трет-бутоксикарбонил-3-метил-азетидин-3-карбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (10 мг, 39%). Данные: LCMS (В) Rt: 11,186 мин; m/z 621,4 (М+Н)+.
Пример 131
2-[4-(4-Ацетилпиперазин-1-ил)-2-метокси-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 16) и уксусной кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17 мг, 45%). Данные: LCMS (В) Rt: 10,096 мин; m/z 614,3 (М+Н)+.
Пример 132
2-[4-[4-(3-Фторциклобутанкарбонил)пиперазин-1-ил]-2-метокси-анилино]-N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 16) и 3-фторциклобутанкарбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (8 мг, 20%). Данные: LCMS (В) Rt: 12,238 мин; m/z 672,3 (М+Н)+.
Пример 133
N-(2-циано-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2-амино-3-метилбензонитрилом в соответствии с методиками, описанными в Примере 2. Очистку проводили флэш-хроматографией на силикагеле (дихлорметан/метанол = от 99/1 до 9/1 об./об. %) с получением указанного в заголовке соединения (8,3 мг, 10%). Данные: LCMS (В) Rt: 10,090 мин; m/z 549,3 (М+Н)+.
Пример 134
N-(2,6-диэтилфенил)-2-[4-[3-(диметиламино)пропил-метил-амино]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZI в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,6 мг, 14%). Данные: LCMS (В) Rt: 12,601 мин; m/z 582,4 (М+Н)+.
Пример 135
2-[4-[3-Диметиламино)пропил-метил-амино]-2-метокси-анилино]-N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZI в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ah в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (5,7 мг, 11%). Данные: LCMS (В) Rt: 8,660 мин; m/z 690,4 (М+Н)+.
Пример 136
N-(2,6-диэтилфенил)-2-[2-метокси-4-(2-метоксиэтокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZJ в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диэтиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (13,4 мг, 30%). Данные: LCMS (В) Rt: 17,314 мин; m/z 542,3 (М+Н)+.
Пример 137
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(2-метоксиэтокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZJ в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ah в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (6,7 мг, 13%). Данные: LCMS (В) Rt: 12,186 мин; m/z 650,3 (М+Н)+.
Пример 138
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ah в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,6 мг, 28,6%). Данные: LCMS (В) Rt: 6,985 мин; m/z 674,3 (М+Н)+.
Пример 139
N-[1-(азетидин-3-ил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ai в соответствии с методиками, описанными в Примере 2. После снятия Boc-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (6,9 мг, 19,7%). Данные: LCMS (В) Rt: 4,784 мин; m/z 583,3 (М+Н)+.
Пример 140
N-[3,5-диметил-1-(оксетан-2-илметил)пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Aj в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством флэш-хроматографии на силикагеле (дихлорметан/метанол = от 99/1 до 9/1 об./об. %) с получением указанного в заголовке соединения (22,1 мг, 61,6%). Данные: LCMS (В) Rt: 6,495 мин; m/z 598,3 (М+Н)+.
Пример 141
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Ak в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (22 мг, 54,4%). Данные: LCMS (В) Rt: 7,845 мин; m/z 658,3 (М+Н)+.
Пример 142
N-[3,5-диэтил-1-(оксетан-3-ил)пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Al в соответствии с методиками, описанными в Примере 2. Очистку проводили посредством флэш-хроматографии на силикагеле (дихлорметан/метанол = от 99/1 до 9/1 об./об. %) с получением указанного в заголовке соединения (23,8 мг, 64,8%). Данные: LCMS (В) Rt: 7,412 мин; m/z 612,3 (М+Н)+.
Пример 143
N-[1-(2-диметиламиноэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с Промежуточным соединением Am в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11,5 мг, 32,0%). Данные: LCMS (В) Rt: 4,944 мин; m/z 599,3 (М+Н)+.
Пример 144
N-(2,6-диметил-фенил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему хлорангидрида с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения J в качестве исходного вещества. Затем хлорангидрид подвергали взаимодействию с 2,6-диметиланилином в соответствии с методиками, описанными в Примере 2. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (11 мг, 27%). Данные: LCMS (В) Rt: 11,461 мин; m/z 574,3 (М+Н)+.
Пример 145
N-(2,6-диметилфенил)-2-[4-[4-(изопропилкарбамоил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
К перемешиваемому раствору N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида (Пример 17, 37 мг, 0,063 ммоль) в DCM (2 мл) добавляли 2-изоцианатопропан (8 мкл, 0,07 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали и остаток очищали посредством препаративной HPLC с получением указанного в заголовке соединения (6 мг, 16%). Данные: LCMS (В) Rt: 14,299 мин; m/z 609,4 (М+Н)+.
Пример 146
Биохимический киназный анализ
Для определения ингибирующей активности соединений в отношении активности фермента TTK использовали анализ IMAP® (Molecular Devices). Соединения серийно разводили в диметилсульфоксиде (DMSO), а затем в 4% DMSO в реакционном буфере IMAP, который состоит из 10 мМ Трис-HCl, pH 7,5, 10 мМ MgCl2) 0,01% Tween-20, 0,1% NaN3 и 1 мМ свежеприготовленного дитиотреитола (DTT). Раствор соединения смешивали с равным объемом полноразмерного фермента TTK (Life Technologies, кат. № PV 3792) в реакционном буфере IMAP. После предварительного инкубирования в течение 1 часа в темноте при комнатной температуре, добавляли меченый флуоресцеином полученный из МВР субстратный пептид (Molecular Devices, кат. № RP 7123) с последующим добавлением ATP для начала реакции. Конечная концентрация фермента составляла 3,9 нМ, конечная концентрация субстрата составляла 50 нМ, и конечная концентрация АТР составляла 5 мкМ. Реакцию оставляли протекать в течение 2 часов при комнатной температуре в темноте. Реакцию останавливали путем гашения прогрессирующим связывающим раствором IMAP в соответствии с протоколом производителя (Molecular Devices). Поляризацию флуоресцеина измеряли на многорежимном ридере Envision (Perkin Elmer, Waltham, MA, U.S.A.). Значения IC50 рассчитывали с использованием программного обеспечения XLfit™5 (ID Business Solutions, Ltd., Surrey, U.K.). Было обнаружено, что значения IC50 всех приведенных соединений составляли менее чем 100 нМ.
Пример 147
Анализ пролиферации клеток
Линию раковых клеток MOLT-4 приобретали в American Type Culture Collection (АТСС, Manassas, VA, U.S.A.) и выращивали в среде RPMI 1640 (LifeTechnologies, Bleiswijk, The Netherlands) с добавлением 10% телячьей сыворотки. Соединения серийно разводили 3,16-кратными шагами в 100% DMSO с последующим дополнительным разведением в водном буфере. Клетки в среде высевали в количестве 45 мкл на лунку в лунки 384-луночного планшета и инкубировали в течение 24 часов в увлажненной атмосфере 5% CO2 при 37°C. Добавляли 5 мкл раствора соединения и планшеты инкубировали в течение дополнительных 72 часов, после чего в каждую лунку добавляли 25 мкл раствора ATPIite 1Step™ (PerkinElmer, Groningen, The Netherlands). Люминесценцию регистрировали на многорежимном ридере Envision. Клеточный сигнал в начале инкубирования регистрировали отдельно для того, чтобы проводить различие между ростом популяции клеток и гибелью клеток. Кроме того, максимальный рост определяли путем инкубирования дубликата без соединения в присутствии 0,4% DMSO. Процент роста использовали в качестве сигнала основной оси Y. Значения IC50 интерполировали с помощью нелинейной регрессии с использованием IDBS XLfit™5 с использованием 4-параметрической логистической кривой с получением максимального сигнала, минимального сигнала, параметра выпуклости (hill-parameter) и IC50.
Было обнаружено, что значения IC50 всех приведенных соединений составляли менее чем 300 нМ. Соединения примеров 3, 6, 7, 10, 11, 14-16, 20, 23-29, 31, 32, 34, 36, 38, 44, 52, 53, 77, 88, 90-92, 98, 102, 104, 128, 131, 132, 136, 140 и 142 показали значение IC50 от ≥50 нМ до меньше 150 нМ, а Соединения примеров 1, 2, 4, 5, 9, 13, 17-19, 21, 22, 33, 35, 37, 39-43,45-51, 56, 58-61, 62-76, 78-84, 86, 93-97, 99, 100, 103, 105, 107, 109-114, 116-124, 126, 127, 129, 130, 133-135, 137, 138, 141, 144 и 145 показали IC50 меньше 50 нМ.
Промежуточное соединение An
1-[2-(2-этоксиэтокси)этил]-3,5-диметил-пиразол-4-амин
а) 2-(2-этоксиэтокси)этил-4-метилбензолсульфонат
К раствору ди(этиленгликоль)этилового эфира (4,92 мл, 36,2 ммоль) в 15 мл THF, охлажденному до 0°C, добавляли гидроксид натрия (2,46 г, 61,5 ммоль), растворенный в 15 мл воды, при интенсивном перемешивании. К этой смеси по каплям добавляли раствор тозилхлорида (8,28 г, 43,4 ммоль) в 15 мл THF в течение периода времени 10 мин при 0°C. Затем охлаждение удаляли и реакционную смесь перемешивали в течение 1 ч под азотом. После того, как анализ посредством TLC показал полное превращение исходного вещества, смесь дважды экстрагировали диэтиловым эфиром (2×50 мл) и органический слой промывали 1 н. раствором NaOH (25 мл) и водой (25 мл). Органический слой сушили (Na2SO4), фильтровали и растворитель удаляли при пониженном давлении с получением неочищенного 2-(2-этоксиэтокси)этил-4-метилбензолсульфоната в виде бесцветной жидкости, выход 10 г (95,8%).
б) 1-[2-(2-этоксиэтокси)этил]-3,5-диметил-4-нитро-пиразол
К раствору 3,5-диметил-4-нитро-1H-пиразола (1 г, 7,08 ммоль) и карбоната цезия (2,31 г, 7,08 ммоль) в DMF (10 мл) добавляли 2-(2-этоксиэтокси)этил-4-метилбензолсульфонат (2,04 г, 7,08 ммоль). Смесь нагревали при 100°C в течение 1 ч. Смесь охлаждали до комнатной температуры и вливали в смесь вода/рассол и экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 1,69 г указанного в заголовке соединения (92,8%).
в) 1-[2-(2-этоксиэтокси)этил]-3,5-диметил-пиразол-4-амин
К перемешиваемому раствору 1-[2-(2-этоксиэтокси)этил]-3,5-диметил-4-нитро-пиразола (1,69 г, 6,57 ммоль) в метаноле (25 мл) добавляли суспензию 10% Pd на древесном угле (200 мг) в этаноле (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин в атмосфере азота. Затем добавляли формиат аммония (4,14 г, 65,7 ммоль) и реакционную смесь нагревали до температуры флегмообразования в течение 15 мин. Реакционную смесь охлаждали, фильтровали через Decalite® и концентрировали в вакууме. Остаток растворяли в метаноле и затем фильтровали через колонку SCX-2. После промывки колонки метанолом целевой продукт элюировали 0,7 н. раствором аммиак/метанол. Полученный элюат концентрировали в вакууме с получением указанного в заголовке соединения (520 мг, 34,8%).
Промежуточное соединение Ao
1-[2-(2-Метоксиэтокси)этил]-3,5-диметил-пиразол-4-амин
Указанное в заготовке соединение получали аналогичным способом, как описано для Промежуточного соединения An, начиная с метилового эфира диэтиленгликоля и 3,5-диметил-4-нитро-1H-пиразола, с получением 500 мг 1-[2-(2-метоксиэтокси)этил]-3,5-диметил-пиразол-4-амина (34,8%).
Промежуточное соединение Ap
1-[2-(2-Этоксиэтокси)этил]-3,5-диэтил-пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения An, начиная с 3,5-дизтил-4-нитро-1H-пиразола (Промежуточное соединение Ас-b) и ди(этиленгликоль)этилового эфира, с получением 550 мг 1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-амина (79,8%).
Промежуточное соединение Aq
3,5-Диэтил-1-[2-[2-(2-метоксиэтокси)этокси]этил]пиразол-4-амин
Указанное в заголовке соединение получали аналогичным способом, как описано для Промежуточного соединения An, начиная с 3,5-диэтил-4-нитро-1H-пиразола (Промежуточное соединение Ас-b) и монометилового эфира триэтиленгликоля, с получением 660 мг 3,5-диэтил-1-[2-[2-(2-метоксиэтокси)этокси]этил]пиразол-4-амина (41,7%).
Пример 148
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением An аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (32,3 мг, 53,9%). Данные: LCMS (В) Rt: 7,432 мин; m/z 644,6 (М+Н)+.
Пример 149
N-[1-[2-(2-метоксиэтокси)этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ao аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (31,5 мг, 53,7%). Данные: LCMS (В) Rt: 6,806 мин; m/z 630,7 (М+Н)+.
Пример 150
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения F в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ap аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (34,2 мг, 54,8%). Данные: LCMS (В) Rt: 8,430 мин; m/z 672,7 (М+Н)+.
Пример 151
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZE в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ap аналогичным образом, как описано в Примере 9. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (147,4 мг, 93,4%). Данные: LCMS (A) Rt: 4,376 мин; m/z 658,7 (М+Н)+.
Пример 152
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 151) и метоксиуксусной кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,0 мг, 61,4%). Данные: LCMS (В) Rt: 10,554 мин; m/z 730,7 (М+Н)+.
Пример 153
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 151) и Вос-N-этил-глицина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Вос-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (15,0 мг, 53,1%). Данные: LCMS (В) Rt: 8,619 мин; m/z 743,8 (М+Н)+.
Пример 154
N-[3,5-диэтил-1-[2-[2-(2-метоксиэтокси)этокси]этил]пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина, полученного аналогичным способом, как описано для Примера 151, с использованием Промежуточного соединения 2, Промежуточного соединения ZE и Промежуточного соединения Aq, и метоксиуксусной кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,8 мг, 57,1%). Данные: LCMS (В) Rt: 9,908 мин; m/z 760,8 (М+Н)+.
Пример 155
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-[4-(1-метилазетидин-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-e]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 16) и 1-метил-3-азетидинкарбоновой кислоты с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (0,5 мг, 1%). Данные: LCMS (A) Rt: 3,892 мин; m/z 669,7 (М+Н)+.
Пример 156
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZE в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ah аналогичным образом, как описано для Примера 9. После снятия Cbz-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (189,4 мг, 95,7%). Данные: LCMS (A) Rt: 3,811 мин; m/z 660,7 (М+Н)+.
Пример 157
2-[4-[4-[2-(Этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил 1-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующего ему амина (Пример 156) и Вос-N-этил-глицина с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Вос-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (18,9 мг, 61,8%). Данные: LCMS (В) Rt: 7,255 мин; m/z 745,8 (М+Н)+.
Пример 158
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZE в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ak аналогичным образом, как описано для Примера 9. После снятия Cbz-защитной группы вводили Вос-N-этил-глицин с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. После снятия Вос-защитной группы осуществляли очистку посредством препаративной HPLC с получением указанного в заголовке соединения (15,8 мг, 54,2%). Данные: LCMS (В) Rt: 8,002 мин; m/z 729,8 (М+Н)+.
Пример 159
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения ZE в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ak аналогичным образом, как описано для Примера 9. После снятия Cbz-защитной группы вводили метоксиуксусную кислоту с использованием стандартных методик сочетания с помощью HATU, как описано в Примере 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (17,3 мг, 60,4%). Данные: LCMS (В) Rt: 9,848 мин; m/z 716,7 (М+Н)+.
Пример 160
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е1индолизин-7-карбоксамид
Это соединение получали из соответствующей ему кислоты с использованием той же последовательности реакций, как описано для Промежуточного соединения 2, с использованием Промежуточного соединения R в качестве исходного вещества. Затем карбоновую кислоту подвергали взаимодействию с Промежуточным соединением Ah аналогичным образом, как описано для Примера 9. Очистку осуществляли посредством препаративной HPLC с получением указанного в заголовке соединения (19,5 мг, 42,6%). Данные: LCMS (В) Rt: 10,946 мин; m/z 684,7 (М+Н)+.
Соединения примеров 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 и 160 показали значение IC50 меньше 50 нМ в биохимическом анализе TTK.
Соединения примеров 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 и 160 показали значение IC50 меньше 50 нМ в клеточном анализе TTK.
Пример 161
Биохимический анализ киназы Aurora A
Для определения ингибирующей активности соединений в отношении Aurora A использовали анализ LANCE® Ultra TR-FRET (Perkin Elmer). Соединения серийно разводили в диметилсульфоксиде (DMSO), а затем в 4% DMSO в киназном буфере LANCE®, которой состоит из 50 мМ Hepes, pH 7,5, 10 мМ MgCl2, 1 мМ EGTA, 0,01% Tween-20 и 2 мМ дитиотреитола (DTT). 2,5 мкл раствора соединения смешивали с равным объемом полноразмерного фермента Aurora A (Carna Biosciences, кат. №05-101) в киназном буфере LANCE®. После предварительного инкубирования в течение 1 часа в темноте при комнатной температуре, добавляли ULight™-меченый PLK (Ser137) субстратный пептид (Perkin Elmer, кат. № TRF-0110) и АТР для начала реакции. Конечная концентрация фермента составляла 2,5 нМ; конечная концентрация субстрата составляла 25 нМ; конечная концентрация АТР составляла 2 мкМ; конечная концентрация DMSO в каждой лунке составляла 1%. Через 2 часа реакцию останавливали путем добавления 5 мкл 100 мМ EDTA. После 5 минут инкубирования при комнатной температуре добавляли 5 мкл меченого европием субстратного антитела анти-фосфо-PLK (Ser137) (Perkin Elmer, кат. № TRF-0203), и инкубирование продолжали в течение 1 ч в темноте при комнатной температуре. Флуоресценцию с временным разрешением измеряли на многозначном ридере Envision (Perkin Elmer, Waltham, MA, USA). Значения IC50 рассчитывали с использованием программного обеспечения XLfit™5 (ID Business Solutions, Ltd., Surrey, U.K.). Соединения примеров 33, 138, 141, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 и 160 показали значение IC50 ≥100 нМ.
Пример 162
Биохимический анализ киназы Aurora C
Для определения ингибирующей активности соединений в отношении Aurora C использовали анализ LANCE® Ultra TR-FRET (Perkin Elmer). Соединения серийно разводили в диметилсульфоксиде (DMSO), а затем в 4% DMSO в киназном буфере LANCE®, которой состоит из 50 мМ Hepes, pH 7,5, 10 мМ MgCl2, 1 мМ EGTA, 0,01% Tween-20 и 2 мМ дитиотреитола (DTT). 2,5 мкл раствора соединения смешивали с равным объемом полноразмерного фермента Aurora C (Cama, кат. №05-103) в киназном буфере LANCE®. После предварительного инкубирования в течение 1 часа в темноте при комнатной температуре, добавляли ULight™-меченый PLK (Ser137) субстратный пептид (Perkin Elmer, кат. № TRF-0110) и АТР для начала реакции. Конечная концентрация фермента составляла 10 нМ; конечная концентрация субстрата составляла 25 нМ; конечная концентрация АТР составляла 5 мкМ; конечная концентрация DMSO в каждой лунке составляла 1%. Через 3 часа реакцию останавливали путем добавления 5 мкл 100 мМ EDTA. После 5 минут инкубирования при комнатной температуре добавляли 5 мкл меченого европием субстратного антитела анти-фосфо-PLK (Ser137) (Perkin Elmer, кат. № TRF-0203), и инкубирование продолжали в течение 1 ч в темноте при комнатной температуре. Флуоресценцию с временным разрешением измеряли на многозначном ридере Envision (Perkin Elmer, Waltham, MA, USA). Значения IC50 рассчитывали с использованием программного обеспечения XLfit™5 (ID Business Solutions, Ltd., Surrey, U.K.).
Соединения примеров 33, 138, 141, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 и 160 показали значение IC50 ≥250 нМ.
Пример 163
Биохимический анализ Polo-подобной киназы 1
Для определения ингибирующей активности соединений в отношении Polo-подобной киназы 1 (PLK1) использовали анализ LANCE® Ultra TR-FRET (Perkin Elmer). Соединения серийно разводили в диметилсульфоксиде (DMSO), а затем в 4% DMSO в киназном буфере LANCE®, которой состоит из 50 мМ Hepes, pH 7,5, 10 мМ MgCl2, 1 мМ EGTA, 0,01% Tween-20 и 2 мМ дитиотреитола (DTT). 2,5 мкл раствора соединения смешивали с равным объемом полноразмерного фермента PLK1 (Carna, кат. №05-157) в киназном буфере LANCE®. После предварительного инкубирования в течение 1 часа в темноте при комнатной температуре, добавляли ULight™-меченый p70S6K (Thr389) субстратный пептид (Perkin Elmer, кат. № TRF-0126) и ATP для начала реакции. Конечная концентрация фермента составляла 7,5 нМ; конечная концентрация субстрата составляла 50 нМ; конечная концентрация АТР составляла 5 мкМ; конечная концентрация DMSO в каждой лунке составляла 1%. Через 4 часа реакцию останавливали путем добавления 5 мкл 100 мМ EDTA. После 5 минут инкубирования при комнатной температуре добавляли 5 мкл меченого европием субстратного антитела анти-фосфо-p70S6K (Thr389) (Perkin Elmer, кат. № TRF-0203), и инкубирование продолжали в течение 1 ч в темноте при комнатной температуре. Флуоресценцию с временным разрешением измеряли на многозначном ридере Envision (Perkin Elmer, Waltham, MA, USA). Значения IC50 рассчитывали с использованием программного обеспечения XLfit™5 (ID Business Solutions, Ltd., Surrey, U.K.).
Соединения примеров 33, 138, 141, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159 и 160 показали значение IC50 ≥250 нМ.
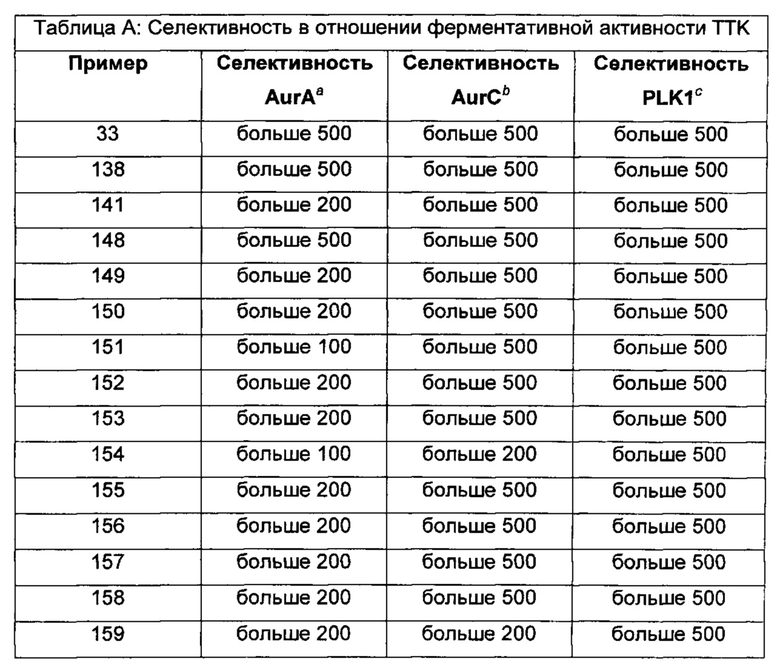

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
| ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2691105C1 |
| ЛЕЧЕНИЕ МИОДИСТРОФИИ ДЮШЕНА | 2007 |
|
RU2462458C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| АНАЛОГИ СОЕДИНЕНИЙ 4Н-ПИРАЗОЛО[1,5-А]БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2015 |
|
RU2672722C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
Изобретение относится к соединению формулы I или к его фармацевтически приемлемой соли, где R1 выбран из группы, состоящей из:
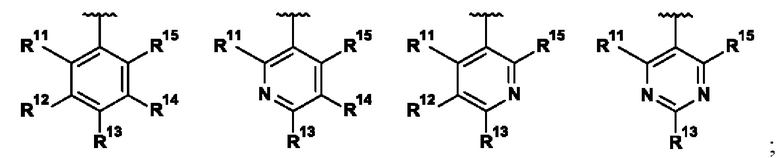
R11 представляет собой Н, галоген, (1-2С)алкил, (1-2С)алкокси или ОС2Н3, где все алкильные и алкокси группы возможно замещены одним или двумя галогенами; R12 представляет собой Н; R13 представляет собой R132O, R133R134N, R135C(O), (2-7С)гетероциклоалкил, выбранный из пиперидинила, пиперазинила, морфолинила или азетидинила, или (1-5С)гетероарил, выбранный из пиразолила или изоксазолила, где каждый гетероциклоалкил или гетероарил возможно замещен 1-3 заместителями (1-2С)алкил, (1-2С)алкокси, (1-6С)алкилкарбонил, (1-6С)алкилсульфонил, (1-5С)алкоксикарбонил, (1-6С)алкиламинокарбонил, (3-6С)циклоалкилкарбонил или (2-7С)гетероциклоалкилкарбонил, в которых указанный гетероциклоалкил выбран из азетидинила или оксетанила, где каждый алкилкарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором или (1-2С)алкокси; R132 представляет собой (1-6С)алкил или (2-7С)гетероциклоалкил, выбранный из пиперидинила, пирролидинила, азетидинила или тетрагидропиранила, каждый из которых возможно замещен одной группой, выбранной из (1-2С)алкила, (1-2С)алкокси или ди[(1-2С)алкил]амино; R133 представляет собой (1-6С)алкил, возможно замещенный одной группой, выбранной из ди[(1-2С)алкил]амино; R134 представляет собой водород или (1-2С)алкил; R135 представляет собой (2-7С)гетероциклоалкил, выбранный из морфолина, тиазинана, диазепана или пиперидина; (1-6С)алкиламино; ди[(1-6С)алкил]амино; (2-7С)гетероциклоалкиламино, в котором указанный гетероциклоалкил выбран из пиперидина, тетрагидропирана, азетидина и оксабициклогексана; или (3-6С)циклоалкиламино, каждый из которых возможно замещен одной или двумя группами, выбранными из (1-2С)алкила, фтора, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино, пиперидинила, оксо, циано или амино; R14 представляет собой Н, галоген или (1-2С)алкил; R15 представляет собой Н; R2 выбран из группы, состоящей из:
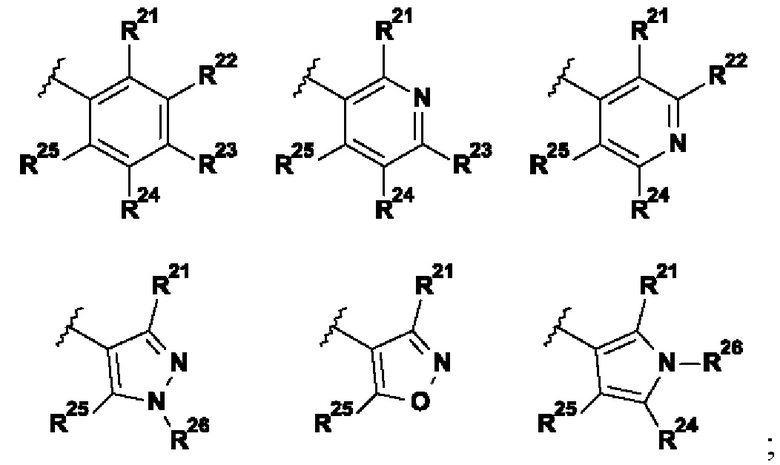
R21 представляет собой Н, галоген, (1-3С)алкил, (1-2С)алкокси, гидрокси(1-2С)алкил или циано; R22 представляет собой Н; R23 представляет собой Н, галоген или (1-2С)алкил; R24 представляет собой Н; R25 представляет собой Н, галоген или (1-3С)алкил; R26 представляет собой Н, (1-6С)алкил, азетидинил, оксетанил, (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкил, где n представляет собой целое число 1 или 2, все алкильные группы возможно замещены одной группой, выбранной из (1-2С)алкокси, гидроксила, ди[(1-2С)алкил]амино или оксетанила; при условии, что только один из R21 или R25 в R2 может представлять собой Н. Изобретение также относится к фармацевтической композиции в качестве ингибитора ТТК на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине в лечении рака. 2 н. и 24 з.п. ф-лы, 5 табл., 163 пр.
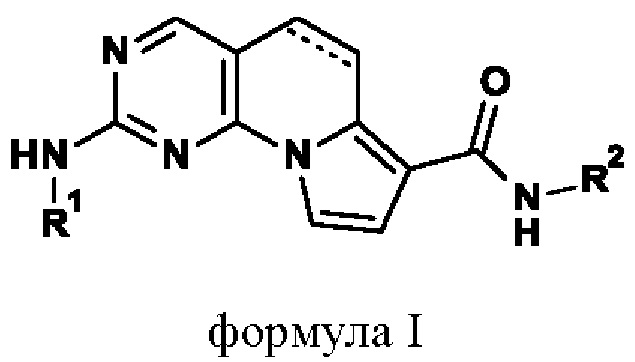
1. Соединение формулы I:
или его фармацевтически приемлемая соль, где
R1 выбран из группы, состоящей из:
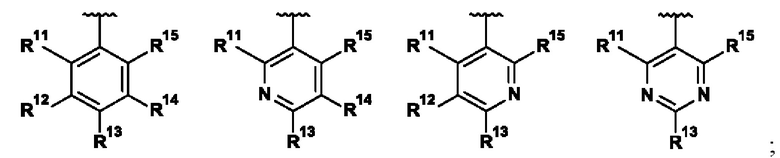
R11 представляет собой Н, галоген, (1-2С)алкил, (1-2С)алкокси или ОС2Н3, где все алкильные и алкокси группы возможно замещены одним или двумя галогенами;
R12 представляет собой Н;
R13 представляет собой R132O, R133R134N, R135C(O), (2-7С)гетероциклоалкил, выбранный из пиперидинила, пиперазинила, морфолинила или азетидинила, или (1-5С)гетероарил, выбранный из пиразолила или изоксазолила, где каждый гетероциклоалкил или гетероарил возможно замещен 1-3 заместителями (1-2С)алкил, (1-2С)алкокси, (1-6С)алкилкарбонил, (1-6С)алкилсульфонил, (1-5С)алкоксикарбонил, (1-6С)алкиламинокарбонил, (3-6С)циклоалкилкарбонил или (2-7С)гетероциклоалкилкарбонил, в котором указанный гетероциклоалкил выбран из азетидинила или оксетанила, где каждый алкилкарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором или (1-2С)алкокси;
R132 представляет собой (1-6С)алкил или (2-7С)гетероциклоалкил, выбранный из пиперидинила, пирролидинила, азетидинила или тетрагидропиранила, каждый из которых возможно замещен одной группой, выбранной из (1-2С)алкила, (1-2С)алкокси или ди[(1-2С)алкил]амино;
R133 представляет собой (1-6С)алкил, возможно замещенный одной группой, выбранной из ди[(1-2С)алкил]амино;
R134 представляет собой водород или (1-2С)алкил;
R135 представляет собой (2-7С)гетероциклоалкил, выбранный из морфолина, тиазинана, диазепана или пиперидина; (1-6С)алкиламино; ди[(1-6С)алкил]амино; (2-7С)гетероциклоалкиламино, в котором указанный гетероциклоалкил выбран из пиперидина, тетрагидропирана, азетидина и оксабициклогексана; или (3-6С)циклоалкиламино, каждый из которых возможно замещен одной или двумя группами, выбранными из (1-2С)алкила, фтора, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино, пиперидинила, оксо, циано или амино;
R14 представляет собой Н, галоген или (1-2С)алкил;
R15 представляет собой Н;
R2 выбран из группы, состоящей из:
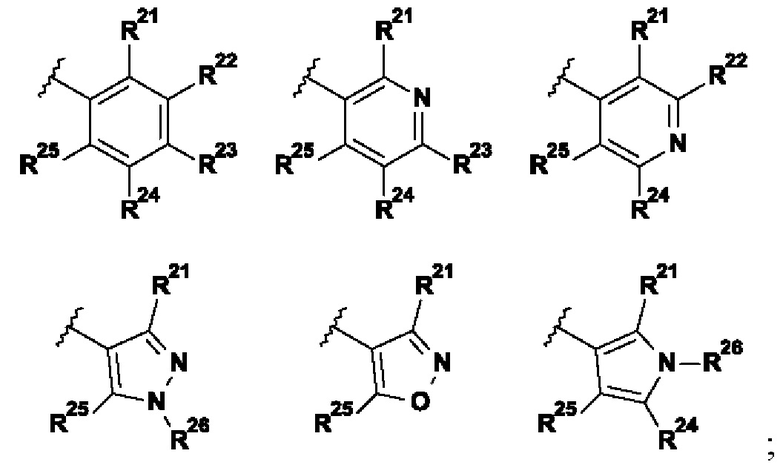
R21 представляет собой Н, галоген, (1-3С)алкил, (1-2С)алкокси, гидрокси(1-2С)алкил или циано;
R22 представляет собой Н;
R23 представляет собой Н, галоген или (1-2С)алкил;
R24 представляет собой Н;
R25 представляет собой Н, галоген или (1-3С)алкил;
R26 представляет собой Н, (1-6С)алкил, азетидинил, оксетанил, (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкил, где n представляет собой целое число 1 или 2, все алкильные группы возможно замещены одной группой, выбранной из (1-2С)алкокси, гидроксила, ди[(1-2С)алкил]амино или оксетанила;
при условии, что только один из R21 или R25 в R2 может представлять собой Н.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где
R13 представляет собой R132O, R135C(O), (2-7С)гетероциклоалкил, выбранный из пиперидинила, пиперазинила, морфолинила или азетидинила, или (1-5С)гетероарил, выбранный из пиразолила или изоксазолила, где каждый гетероциклоалкил или гетероарил возможно замещен 1-3 заместителями (1-2С)алкил, (1-3С)алкилкарбонил, (1-2С)алкокси, (1-6С)алкилсульфонил, (1-5С)алкоксикарбонил, (1-6С)алкиламинокарбонил, (3-бС)циклоалкилкарбонил или (2-7С)гетероциклоалкилкарбонил, в котором указанный гетероциклоалкил выбран из азетидинила или оксетанила, где каждый алкилкарбонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором или (1-2С)алкокси;
R132 выбран из группы, состоящей из (1-6С)алкила, (2-7С)гетероциклоалкила, выбранного из пиперидинила, пирролидинила, азетидинила или тетрагидропиранила, каждый из которых возможно замещен одной группой, выбранной из (1-2С)алкила, (1-2С)алкокси или ди[(1-2С)алкил]амино;
R135 выбран из группы, состоящей из (2-7С)гетероциклоалкила, выбранного из морфолина, тиазинана, диазепана или пиперидина; (1-6С)алкиламино; (2-7С)гетероциклоалкиламино, в котором указанный гетероциклоалкил выбран из пиперидина, тетрагидропирана, азетидина, оксабициклогексана или морфолина; или (3-6С)циклоалкиламино, каждый из которых возможно замещен одной или двумя группами, выбранными из (1-2С)алкила, фтора, гидроксила, (1-2С)алкокси, ди[(1-2С)алкил]амино, пиперидинила, оксо, циано или амино.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где
R13 представляет собой R132O, R135C(О); или R13 представляет собой пиперидинил, пиперазинил, морфолинил, пиразолил или изоксазолил (каждый из которых возможно замещен 1-3 заместителями (1-2С)алкил, (1-6С)алкилкарбонил, (1-6С)алкилсульфонил, (1-5С)алкоксикарбонил, (1-6С)алкиламинокарбонил, (3-6С)циклоалкилкарбонил или (2-7С)гетероциклоалкилкарбонил, в котором указанный гетероциклоалкил выбран из азетидинила или оксетанила, где каждый алкилкарбонил, алкилсульфонил, циклоалкилкарбонил или гетероциклоалкилкарбонил возможно замещен (1-2С)алкилом, фтором или (1-2С)алкокси;
R132 выбран из группы, состоящей из (1-6С)алкила, пиперидинила, пирролидинила или азетидинила, каждый из которых возможно замещен одной группой, выбранной из (1-2С)алкила, (1-2С)алкокси или ди[(1-2С)алкил]амино;
R135 выбран из группы, состоящей из морфолина, тиазинана, диазепана, пиперидинила, (1-6С)алкиламино, (3-6С)циклоалкиламино или пиперидиниламино, азетидиниламино, тетрагидропираниламино, 3-оксабицикло[3.1.0]гексан-6-амино или морфолиниламино, каждый из которых возможно замещен одной или двумя группами, выбранными из (1-2С)алкила, фтора, гидроксила или (1-2С)алкокси, ди[(1-2С)алкил]амино, пиперидинила, оксо, циано или амино.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где
каждый из R12 и R15 представляет собой Н, и R14 представляет собой Н, фтор, хлор или (1-2С)алкил.
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль, где
R11 представляет собой Н, (1-2С)алкил или (1-2С)алкокси, где все алкильные и алкокси группы возможно замещены одним или двумя фторами.
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из:
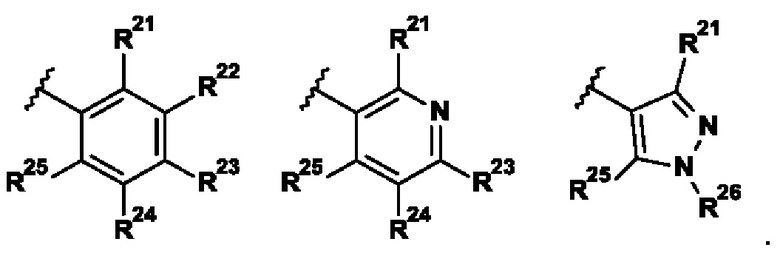
7. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, где R2 представляет собой:
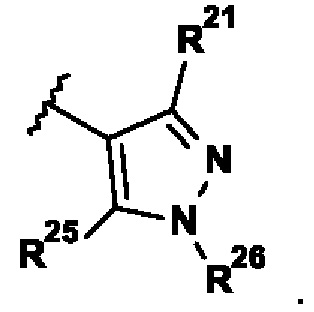
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, где
R23 представляет собой Н или (1-2С)алкил, и каждый из R22 и R24 представляет собой Н, и R21 и R25 независимо выбраны из группы, состоящей из галогена, (1-3С)алкила, метокси, гидроксиметила или циано.
9. Соединение по любому из пп. 1-8 или его фармацевтически приемлемая соль, где
R26 представляет собой Н, (1-6С)алкил, оксетанил, азетидинил или (1-2С)алкокси[(2-4С)алкокси]n(1-6С)алкил, где n означает целое число 1 или 2, где все алкильные, оксетанильные и азетидинильные группы возможно замещены одной группой, выбранной из (1-2С)алкокси, гидроксила, ди[(1-2С)алкил]амино или оксетанила.
10. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение выбрано из группы, состоящей из:
N-(2,6-диметилфенил)-2-[4-[(2-гидрокси-2-метил-пропил)карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-[4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1H-пиразол-4-ил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-N-(2,4,6-триметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[2-(дифторметокси)-4-[(1-метил-4-пиперидил)карбамоил]анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-метил-4-[(1-метил-4-пиперидил)окси]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-N-(2-этил-6-метил-фенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-[4-(2-диметиламиноэтилокси)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-(2-метил-4-морфолино-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[2-метокси-4-(1-метилпирролидин-3-ил)окси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-3-пиперидил)карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-дихлорфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]пиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-дихлорфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[2-метил-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-[(2-метил-6-морфолино-3-пиридил)амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]пиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-дихлорфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-бром-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-этил-6-метил-фенил)-2-(2-метил-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-(2-метил-4-морфолино-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-(1-метилпирролидин-3-ил)окси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-хлор-6-метил-фенил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(азетидин-3-илокси)-2-метокси-анилино]-N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-[(1-метил-4-пиперидил)окси]анилино]пиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамида;
N-(3,5-диэтил-1Н-пиразол-4-ил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[4-(4-метилпиперазин-1-ил)-2-(тридейтериометокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[4-(4-метилпиперазин-1-ил)-2-(тридейтериометокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-(тетрагидропиран-4-илкарбамоил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[[4-метокси-2-(4-метилпиперазин-1-ил)пиримидин-5-ил]амино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-(2-метоксиэтил)пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-[4-(3-фторциклобутанкарбонил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(3-метилоксетан-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-[4-(циклопропанкарбонил)пиперазин-1-ил]-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(4-бутилсульфонилпиперазин-1-ил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-[4-(этилкарбамоил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[2-метокси-4-(3-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[[(3S,4R)-3-метокси-4-пиперидил]карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-[[(3S,4R)-3-фтор-4-пиперидил]карбамоил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-[(4-цис-аминоциклогексил)карбамоил]-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-(изопропилкарбамоил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[[(1R,5S)-3-оксабицикло[3.1.0]гексан-6-ил]карбамоил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-(морфолин-4-карбонил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-(1,1-диоксо-1,4-тиазинан-4-карбонил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-(4-этил-1,4-диазепан-1-карбонил)-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(циклопропилкарбамоил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-(2-метоксиэтилкарбамоил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-(4-циано-4-метил-пиперидин-1-карбонил)-2-метокси-анилино]-N-(2,6-диметилфенил)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(3-метилазетидин-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2-циано-6-метил-фенил)-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диэтилфенил)-2-[4-[3-(диметиламино)пропил-метил-амино]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-[3-(диметиламино)пропил-метил-амино]-2-метокси-анилино]-N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(2-метоксиэтокси)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметил-фенил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-(2,6-диметилфенил)-2-[4-[4-(изопропилкарбамоил)пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-метоксиэтокси)этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-[2-[2-(2-метоксиэтокси)этокси]этил]пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-(2-метоксиэтил)-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-[4-(1-метилазетидин-3-карбонил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида;
N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамида.
11. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
12. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-(3,5-диэтил-1Н-пиразол-4-ил)-2-[2-(дифторметокси)-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
13. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-(2,6-диметилфенил)-2-(2-метокси-4-пиперазин-1-ил-анилино)пиримидо[4,5-е]индолизин-7-карбоксамид.
14. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-(2,6-диметилфенил)-2-[2-метокси-4-[4-(2-метоксиацетил)пиперазин-1-ил]анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
15. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
16. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-[3,5-диэтил-1-[2-(2-метоксиэтокси)этил]пиразол-4-ил]-2-[2-метокси-4-(4-метилпиперазин-1-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
17. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-[1-[2-(2-этоксиэтокси)этил]-3,5-диэтил-пиразол-4-ил]-2-[4-[4-[2-(этиламино)ацетил]пиперазин-1-ил]-2-метокси-анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
18. Соединение по п. 1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой N-[1-[2-[2-(2-метоксиэтокси)этокси]этил]-3,5-диметил-пиразол-4-ил]-2-[2-метокси-4-(1,3,5-триметилпиразол-4-ил)анилино]-5,6-дигидропиримидо[4,5-е]индолизин-7-карбоксамид.
19. Соединение по любому из пп. 1-18 или его фармацевтически приемлемая соль для применения в терапии для модулирования активности протеинкиназ.
20. Соединение по п. 19 или его фармацевтически приемлемая соль для применения в терапии в качестве ингибитора ТТК.
21. Соединение по любому из пп. 1-18 или его фармацевтически приемлемая соль для лечения ТТК-опосредованных заболеваний или состояний.
22. Соединение по любому из пп. 1-18 или его фармацевтически приемлемая соль для изготовления лекарственного средства для лечения ТТК-опосредованных заболеваний или состояний.
23. Соединение по п. 21 или 22 или его фармацевтически приемлемая соль, где ТТК-опосредованные заболевания или состояния представляют собой рак, солидные опухоли, гематологические опухоли и/или их метастазы.
24. Соединение по п. 23 или его фармацевтически приемлемая соль, где солидные опухоли, гематологические опухоли и/или их метастазы выбраны из опухолей молочной железы и гинекологических опухолей, опухолей головы и шеи, опухолей мозга и метастазов в мозге, опухолей грудной клетки, включая немелкоклеточные и мелкоклеточные опухоли легкого, желудочно-кишечных опухолей, эндокринных опухолей, урологических опухолей, включая опухоли почки, мочевого пузыря и предстательной железы, кожной опухоли, и сарком, лейкемий и миелодиспластического синдрома, злокачественных лимфом и/или их метастазов.
25. Фармацевтическая композиция для модулирования активности протеинкиназ, содержащая соединение формулы I по любому из пп. 1-18 или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых эксципиентов.
26. Фармацевтическая композиция по п. 25 для применения в качестве ингибитора ТТК.
| WO 2012101032 A1, 02.08.2012 | |||
| WO 2009156315 A1, 30.12.2009 | |||
| Приспособление к телеграфному аппарату Бодо для печатания строками | 1926 |
|
SU7983A1 |

