Область Техники
Настоящее изобретение относится к области медицинской технологии, в частности, к новому соединению, обладающему активностью ингибитора циклинзависимой киназы 9 (CDK9) или его фармацевтически приемлемой соли, или стереоизомеру, изотопному производному, или пролекарству, и его применению в качестве пролекарства.
Уровень Техники
Циклинзависимые киназы (CDK) представляют собой группу серин/треониновых протеинкиназ, которые играют ключевую роль в регуляции клеточного цикла и транскрипции. На сегодняшний день у людей присутствует около 20 CDK и около 30 генов циклинов (Cao et al. 2014). Эти CDK могут активироваться циклинами и выполнять различные биологические функции. Среди них CDK1, CDK2, CDK3, CDK4 и CDK6 непосредственно участвуют в клеточном цикле. CDK5 не регулирует клеточный цикл, но играет ключевую роль в сложной миграции постмитотических нейронов. Тогда как CDK7 лишь косвенно действует как активатор этих CDK. Кроме того, CDK9, CDK7 и CDK8 участвуют в контроле транскрипции, опосредованной РНК-полимеразой II (RNAPII).
CDK9 была первоначально обнаружена в 1990-х годах и охарактеризована мотивом Pro-Ile-Thr-Ala-Leu-Arg-Glu и была идентифицирована как родственная CDC2 киназа, называемая PITALRE (Grana et al, 1994). При дальнейших исследованиях было обнаружено, что это должна быть протеинкиназа, зависящая от клеточного цикла, и существуют два изофермента, CDK9-42 (372 а.о., 42 кДа) и CDK9-55 (489 а.о., 55 кДа), которые связываются с четырьмя циклинами: циклин Т1, циклин Т2а, циклин T2b и циклин K (Fu et al., 1999). CDK9 не участвует в регуляции клеточного цикла, но играет решающую роль в регуляции транскрипции (de Falco & Giordano 1998). CDK9 и его регуляторная субъединица циклин Τ или циклин К (Fu et al. 1999) являются основными компонентами так называемого положительного фактора b элонгации транскрипции (Р-TEFb). CDK9 и циклин Τ образуют комплекс P-TEFb, который обеспечивает фосфорилирование С-концевого домена (CTD) РНК-полимеразы II (RNAPII) и активирует продуктивное удлинение транскриптов мРНК.
CDK9 представляет собой каталитическую субъединицу позитивных факторов элонгации транскрипции. Когда негативные факторы элонгации транскрипции (NELF, NELFs) участвуют в негативной регуляции клеточной транскрипции, транскрипция ингибируется, и P-TEFb рекрутируется в систему, в которой элонгация транскрипции ингибируется негативными факторами элонгации транскрипции, он катализирует фосфорилирование на С-конце RNAPII и одновременно катализирует фосфорилирование субъединицы SPT5 NELF и субъединицы RD NELF, что приводит к отделению негативных факторов элонгации транскрипции от транскрипционного комплекса, тем самым позволяя транскрипции продолжаться. Следовательно, путем ингибирования CDK9 для блокировки фосфорилирования P-TEFb в С-концевой области RNAPII транскрипция ингибируется, а уровни внутриклеточной мРНК и белков с короткими периодами полужизни быстро снижаются, что может привести к апоптозу опухолевых клеток.
Опухоли обычно вызываются потерей экспрессии ингибиторов циклинзависимых киназ (CDKI) или сверхэкспрессией циклинов, что делает клетки нерегулируемыми и чрезмерно пролиферирующими. Ингибиторы CDK9 предотвращают фосфорилирование С-конца RNAPII с помощью CDK9, еще больше препятствуют отходу NEFL, усиливают отрицательное ингибирование и в конечном итоге вызывают остановку транскрипции. CDK9 стал потенциальной белковой мишенью для разработки эффективной терапии рака, поэтому нацеливание на ингибирование CDK9 будет полезно для лечения заболеваний человека. Недавно фармацевтические компании начали исследования по использованию ингибиторов CDK9 для лечения рака, включая AZD4573, который в настоящее время проходит фазу I клинических испытаний компании AstraZeneca, для лечения рецидивов гематологических злокачественных новообразований, включая острый миелоидный лейкоз (AML) и неходжкинскую лимфому. Кроме того, компания Bayer также изучает применение ингибитора CDK9 BAY-1251152 при прогрессирующих солидных опухолях и гематомах, который в настоящее время проходит I фазу клинических испытаний.
Это показано по опубликованным данным: WO 2009047359 относится к производным полициклических амидов, которые можно использовать для лечения гиперпролиферативных заболеваний, WO 2014076091 относится к производным 5-фтор-N-(пиридин-2-ил)пиридин-2-амина, содержащим сульфоксиминовую группу (для лечения и/или предотвращения заболеваний, в частности, гиперпролиферативных заболеваний, вирусных инфекционных заболеваний и/или сердечно-сосудистых заболеваний), и способу их получения, в WO 2011116951 раскрыты производные замещенного триазина в качестве селективных ингибиторов CDK9, в WO 2011110612 раскрыт ингибитор протеинкиназы для противовоспалительного действия, обладающий эффектом селективного ингибирования CDK9. Таким образом, ингибирование CDK9 теоретически может обеспечить терапевтический эффект при вышеупомянутых заболеваниях.
Хотя в настоящее время раскрыты некоторые низкомолекулярные ингибиторы CDK9, все еще необходимо разработать новые соединения с более высокой эффективностью и меньшей токсичностью, а также более благоприятные для получения лекарственных средств. Путем непрерывных исследований было разработано соединение со структурой, показанной в общей формуле (I), и эксперименты in vitro и in vivo показывают, что соединение со структурой демонстрирует превосходные эффекты и функции, в частности, эффективная активность in vivo явно выше, чем у аналогичных молекул предшествующего уровня техники, и соединение с этой структурой имеет более высокую вероятность стать лекарственным средством.
Краткое Описание Изобретения
Технические проблемы
Настоящее изобретение направлено на решение одной из следующих технических проблем:
- предоставление низкомолекулярного соединения с новой структурой, то есть ингибитора CDK9 с превосходной ингибирующей активностью в отношении CDK9, селективностью и ингибирующей активностью в отношении опухолевых клеток;
- предоставление низкомолекулярного соединения, которое представляет собой ингибитор CDK9, обладающего лучшей противоопухолевой активностью in vivo по сравнению с известным соединением и сохраняющего относительно хорошую ингибирующую активность в отношении CDK9, селективность и ингибирующую активность в отношении опухолевых клеток;
- предоставление низкомолекулярного соединения, которое представляет собой ингибитор CDK9, которое имеет более высокую безопасность по сравнению с известным соединением и сохраняет относительно хорошую ингибирующую активность в отношении CDK9, селективность и ингибирующую активность в отношении опухолевых клеток;
- предоставление низкомолекулярного соединения, которое представляет собой ингибитор CDK9, обладающего лучшей противоопухолевой активностью in vivo и лучшей безопасностью по сравнению с известными соединениями; и
- предоставление низкомолекулярного соединения, которое представляет собой ингибитор CDK9, обладающего лучшей противоопухолевой активностью in vivo и лучшей безопасностью по сравнению с известными соединениями и сохраняющего относительно хорошую ингибирующую активность в отношении CDK9, селективность и ингибирующую активность в отношении опухолевых клеток.
Технические решения настоящего раскрытия
Настоящее изобретение направлено на предоставление соединения или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства, а также относится к применению соединения или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства в качестве ингибитора циклинзависимой киназы 9 (CDK9).
В частности, настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному, или пролекарству,
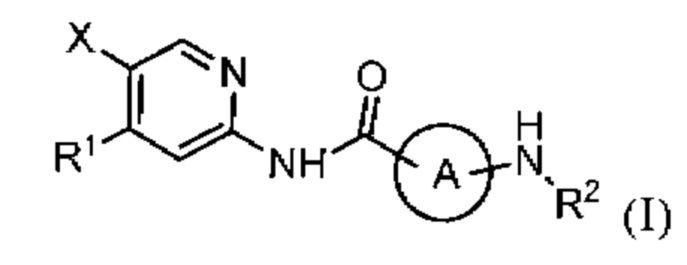
где,
X представляет собой Cl или F, предпочтительно F;
R1 представляет собой замещенный или незамещенный ар ил или замещенный или незамещенный гетероарил; термин «замещенный» в R1 относится к замещению 1, 2, 3, 4 или 5 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -NH2, -ОН, -SH, -CN, -NO2, -N3, -С≡СН, -СООН, -R3, -(CH2)wO(CH2)nR3, -(CH2)wNH(CH2)nR3, -(CH2)wNR3(CH2)nR4, -(CH2)wS(CH2)nR3, -(CH2)wC(O)(CH2)nR3, -(CH2)wCO(CH2)nR3, -(CH2)wOC(O)(CH2)nR3, -(CH2)wC(O)nH(CH2)nR3, -(CH2)wNHC(O)(CH2)nR3, -(CH2)wC(O)nR3(CH2)nR4, -(CH2)wNR3C(O)(CH2)nR4, -(CH2)wOS(O)2(CH2)nR3 и -(CH2)wS(O)2O(CH2)nR3; где w и n в каждом случае независимо выбраны из 0, 1, 2, 3 и 4; каждый из R3 и R4 независимо выбирается из замещенного или незамещенного арила, замещенного или незамещенного гетероарила, замещенного или незамещенного C1-6алкила, замещенного или незамещенного C1-6галогеналкила, замещенного или незамещенного С2-6алкенила, замещенного или незамещенного С2-6алкинила, замещенного или незамещенного C1-6алкилокси, замещенного или незамещенного С1-6галогеналкилокси, замещенного или незамещенного циклоалкила и замещенного или незамещенного гетероциклоалкила, или когда R3 и R4 оба присоединены к одному и тому же атому азота, R3, R4 и атом азота, к которому они присоединены, вместе образуют замещенный или незамещенный гетероциклоалкил; термин «замещенный» в R3 и R4 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -NH2, -ОН, -SH, -CN, -NO2, -N3, -С≡СН, -СООН, C1-6алкила, С1-6галогеналкила, C1-6алкилокси, C1-6галогеналкилокси и тому подобного;
Кольцо А представляет собой замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил; термин «замещенный» в кольце А относится к замещению 1, 2, 3, 4 или 5 группой(ами), каждая из которых независимо выбрана из -F, -С1, -Br, ОН, NH2, SH, CN, NO2, -N3, -C≡CH, COOH, R5, OR5, -NHR5, -NR5R6, -SR5, -NHCOR5, -CONHR5, -NHS(O)2R5, -S(O)2NHR5, -NR5S(O)2R6 и -S(O)2NR5R6, или одна, две или более группа(ы) -СН2- в структуре кольца А может быть необязательно замещена группой(ами) -С(О)-; где R5 и R6 независимо представляют собой C1-6алкил или С1-6галогеналкил;
R2 выбирается из Н, R7, -(CH2)xR7, -(CH2)xNH(CH2)yR7, -(CH2)xO(CH2)yR7, -(CH2)xNR7(CH2)yR8, -(СН2)хС(O)(СН2)уН, -(CH2)xC(O)(CH2)yR7, -(CH2)xS(O)2(CH2)yR7, -(CH2)xC(O)C(O)(CH2)yR7, -(CH2)xS(O)2NH2, -(CH2)xNHS(O)2H, -(CH2)xS(O)2NH(CH2)yR7, -(CH2)xNHS(O)2(CH2)yR7, -(CH2)xS(O)2NR7(CH2)yR8, -(CH2)xNR7S(O)2(CH2)yR8, -(CH2)xC(O)O(CH2)yR7, -(CH2)xOC(O)(CH2)yR7, -(CH2)xC(O)nH2, -(CH2)xNHC(O)H, -(CH2)xC(O)nH(CH2)yR7, -(CH2)xNHC(O)(CH2)yR7, -(CH2)xC(O)nR7(CH2)yR8 и -(CH2)xNR7C(O)(CH2)yR8; где одна, две или более группа(ы) -СН2- может быть необязательно замещена группой(ами) -С(О)-; х и у в каждом случае каждый независимо выбирается из 0, 1, 2, 3 и 4;
R7 и R8 независимо выбираются из следующих замещенных или незамещенных групп: R9, OR9, -R10-O-R9, -R10-NH-R9, -R10-C(O)-R9, -R10-NHC(O)-R9, -R10-C(O)nH-R9, -R10-S-R9, -R10-S(O)-R9, -R10-S-C(O)-R9, циклоалкил, гетероциклоалкил, арил, гетероарил, -R10-арил, R10-гетероарил, -О-R10-арил, -О-R10-гетероарил, R10-О-арил, -R10-O-гетероарил, - циклоалкиларил, -циклоалкилгетероарил, -гетероциклоалкиларил, гетероциклоалкилгетероарил, С2-6алкенил и С2-6алкинил, или когда R7 и R8 оба присоединены к одному и тому же атому азота, R7 и R8 и атом азота, к которому они присоединены, вместе образуют замещенный или незамещенный гетероциклоалкил; где R9 представляет собой С1-6алкил, R10 представляет собой С1-6алкилен, С2-6алкенилен или С2-6алкинилен; термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -ОН, -NH2, -SH, -CN, -NO2, -N3, -C≡CH, -COOH, C1-6алкила, C1-6алкилокси, C1-6галогеналкил, C1-6галогеналкилокси, -NHCN, -NHCONH2, NHC(O)CH3, N(CH3)2, N(C2H5)2, -SC(O)CH3, -ОС(O)-С1-6алкила и тому подобного;
Предпочтительно изотопное производное представляет собой производное в дейтерированной форме.
Определения «арила», «гетероарила», «циклоалкила» и «гетероциклоалкила» в настоящем описании приведены в части «Определения» следующим образом.
В предпочтительных вариантах выполнения изобретения арил предпочтительно содержит 6-10 атомов углерода, циклоалкил предпочтительно содержит 3-6 атомов углерода, гетероарил предпочтительно представляет собой 5-10-членный гетероарил, и гетероциклоалкил предпочтительно представляет собой 3-8-членный гетероциклоалкил; гетероарил или гетероциклоалкил предпочтительно содержат один, два или три гетероатома(ов), каждый из которых независимо выбран из N, О и S, а остальные представляют собой атом(ы) углерода.
Как описано выше, каждый из «w», «n», «x» и «у» в настоящем раскрытии может быть независимо выбран из 0, 1, 2, 3 и 4. Когда «w» и «n» или «х» и «у» одновременно существуют в группе, в частности, комбинации числовых значений «w» и «n» и «х» и «у» могут быть выбраны из (0,0), (0,1), (0,2), (0,3), (0,4), (1,0), (1,1), (1,2), (1,3), (1,4), (2,0), (2,1), (2,2), (2,3), (2,4), (3,0), (3,1), (3,2), (3,3), (3,4), (4,0), (4,1), (4,2), (4,3) и (4,4). Комбинация числовых значений применяется к каждой соответствующей группе в определениях R1 и R2. Например, -(CH2)wO(CH2)nR3 в определении R1 эквивалентно раскрытию таких групп, как -OR3, -OCH2R3, -O(CH2)2R3, -O(CH2)3R3, -O(CH2)4R3, -CH2OR3, -CH2OCH2R3, -CH2O(CH2)2R3, -CH2O(CH2)3R3, -CH2O(CH2)4R3, -(CH2)2OR3, -(CH2)2OCH2R3, -(CH2)2O(CH2)2R3, -(CH2)2(CH2)3R3, -(CH2)2O(CH2)4R3, -(CH2)3OR3, -(CH2)3OCH2R3, -(CH2)3O(CH2)3R3, -(CH2)3O(CH2)3R3, -(CH2)3O(CH2)4R3, -(CH2)4OR3, -(CH2)4OCH2R3, -(CH2)4O(CH2)4R3, -(CH2)4O(CH2)3R3, -(CH2)4O(CH2)4R3 и тому подобных. Подобным образом, другие соответствующие группы в R1 и/или R2 также раскрывают такие варианты выбора и не будут подробно описаны.
Предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где кольцо А выбирается из замещенного или незамещенного 4-6-членного циклоалкила, замещенного или незамещенного 4-6-членного гетероциклоалкила, замещенного или незамещенного 5-6-членного циклоалкила и замещенного или незамещенного 5-6-членного гетероциклоалкила; более предпочтительно кольцо А представляет собой замещенный или незамещенный 5-6-членный циклоалкил или замещенный или незамещенный 5-6-членный гетероциклоалкил; еще более предпочтительно кольцо А представляет собой замещенный или незамещенный 5-6-членный циклоалкил; в другом предпочтительном варианте выполнения изобретения кольцо А выбирается из циклогексанила, тетрагидропирролила, пиперидинила, пиперазинила и морфолинила, более предпочтительно кольцо А представляет собой циклогексанил или тетрагидропирролил.
Дополнительно предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, причем термин «замещенный» в кольце А означает замещение 1, 2, 3, 4 или 5 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, ОН, NH2, SH, CN, R5 и OR5, где R5 представляет собой C1-6алкил или C1-6алкилокси, R5 может дополнительно представлять собой С1-4алкил или С1-4алкилокси.
Более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где указанное соединение имеет структуру, представленную формулой (II):
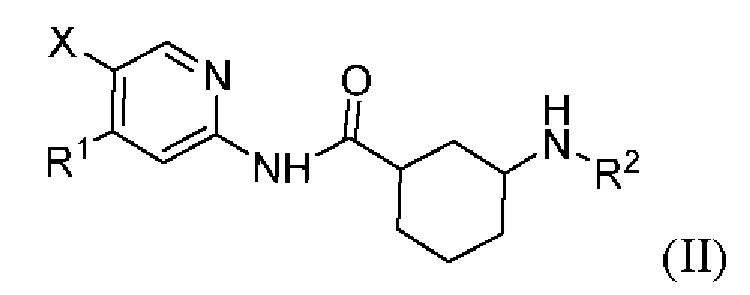
где R1, R2 и X показаны в формуле (I).
Более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где указанное соединение имеет структуру, представленную формулой (IIa):
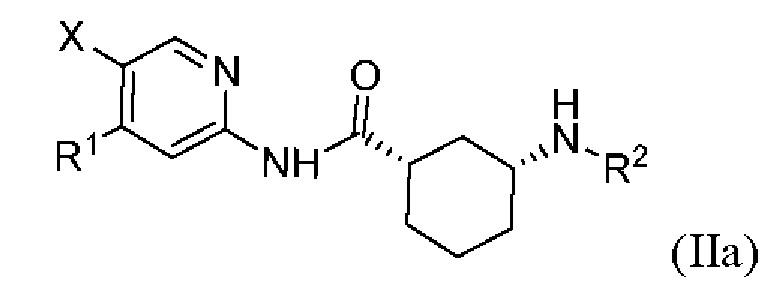
где R1, R2 и X показаны в формуле (I).
Более предпочтительно настоящее раскрытие относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где указанное соединение имеет структуру, представленную формулой (III):
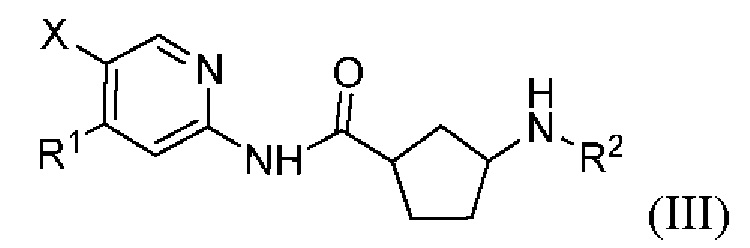
где R1, R2 и X показаны в формуле (I).
Более предпочтительно настоящее раскрытие относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где указанное соединение имеет структуру, представленную формулой (IIIa):
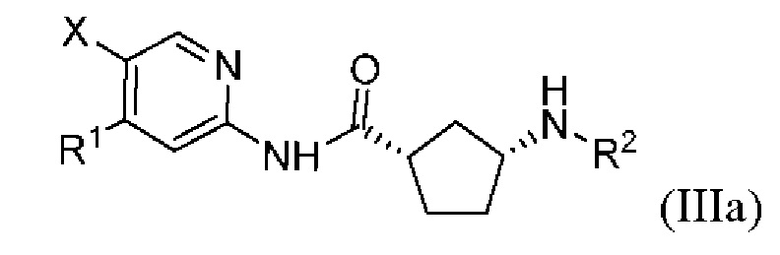
где R1, R2 и X показаны в формуле (I).
Предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где R1 представляет собой замещенный или незамещенный 6-10-членный арил, или замещенный или незамещенный 5-10-членный гетероарил; гетероарил содержит 1 или 2 гетероатома, каждый из которых независимо выбран из N и О; число заместителей равно 1, 2 или 3.
Более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, стереоизомеру, изотопному производному или пролекарству, где R1 выбирается из замещенного или незамещенного бензольного кольца, замещенного или незамещенного пиридинового кольца, замещенного или незамещенного индольного кольца, замещенного или незамещенного индазольного кольца, замещенного или незамещенного бензофуранового кольца и замещенного или незамещенного пирролопиридинового кольца; предпочтительно выбирается из замещенного или незамещенного бензольного кольца, замещенного или незамещенного пиридинового кольца, замещенного или незамещенного индольного кольца, замещенного или незамещенного бензофуранового кольца и замещенного или незамещенного пирролопиридинового кольца; более предпочтительно выбирается из замещенного бензольного кольца, замещенного пиридинового кольца, незамещенного индольного кольца, незамещенного бензофуранового кольца и незамещенного пирролопиридинового кольца; еще более предпочтительно замещенного бензольного кольца.
Более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где каждый из заместителей указанного R1 независимо выбирается из -F, -Cl, -ОН, -NH2, -R3, -(CH2)wO(CH2)nR3 и -(CH2)wOC(O)(CH2)nR3; w и n в каждом случае независимо выбираются из 0, 1 и 2, где R3 определен, как описано в формуле (I). Предпочтительно каждый из заместителей указанного R1 независимо выбирается из -F, -Cl, -OH, -R3 и -(CH2)wO(CH2)nR3; более предпочтительно каждый из заместителей указанного R1 независимо выбирается из -F, -ОН, -R3 и -(CH2)wO(CH2)nR3. Например, когда R1 представляет собой замещенное бензольное кольцо, заместитель выбирается из -F, -ОН и алкокси, предпочтительно он замещен одним или двумя атомами фтора и одним -ОН или алкокси.
Предпочтительно настоящее изобретениеи относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где каждый из R3 и R4 независимо выбираются из замещенного или незамещенного 6-членного арила, замещенного или незамещенного 5-6-членного арила, замещенного или незамещенного С1-3алкила, замещенного или незамещенного С1-3алкилокси, замещенного или незамещенного С3-6циклоалкила и замещенного или незамещенного С3-6гетероциклоалкила, или когда R3 и R4 оба присоединены к одному и тому же атому азота, R3, R4 и атом азота, к которому они присоединены, вместе образуют 3-7-членный замещенный или незамещенный гетероциклоалкил; гетероциклоалкил содержит один или два гетероатома(ов), независимо выбранных из N, О и S; термин «замещенный» в R3 и R4 относится к замещению 1, 2 или 3 заместителем(ями), каждый из которых независимо выбран из -F, -Cl, -Br, -ОН, -СН3, -С2Н5, -ОСН3 и -ОС2Н5; предпочтительно каждый из R3 и R4 независимо выбирается из замещенного или незамещенного С1-3алкила и замещенного или незамещенного C1-3алкилокси.
Более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где каждый из R3 и R4 независимо выбирается из следующих замещенных или незамещенных групп: бензольное кольцо, пиридиновое кольцо, метил, этил, пропил, изопропил, метилокси, этилокси, пропилокси, изопропилокси, циклопропил, циклобутил, циклопентил и циклогексил, термин «замещенный» в R3 и R4 означает замещение 1, 2 или 3 заместителем(ями), каждый из которых независимо выбран из -F, -Cl, -Br, -ОН, -СН3, -С2Н5, -ОСН3 и -ОС2Н5. предпочтительно каждый из R3 и R4 независимо выбирается из следующих замещенных или незамещенных групп: метил, этил, изопропил, метилокси, этилокси, изопропилокси, циклопропил и пиридиновое кольцо; более предпочтительно каждый из R3 и R4 независимо выбирается из следующих замещенных или незамещенных групп: метил, этил, изопропил, метилокси, этилокси и изопропилокси; еще более предпочтительно каждый из R3 и R4 независимо выбирается из следующих замещенных или незамещенных групп: метил и метилокси.
Предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где R2 выбирается из R7, -(CH2)xR7, -(CH2)xNH(CH2)yR7, -(CH2)xC(O)(CH2)yR7, -(CH2)xS(O)2(CH2)yR7, -(СН2)хС(O)С(O)(СН2)yR7, (CH2)xC(O)O(CH2)yR7, -(CH2)xC(O)nH(CH2)yR7, -(CH2)xC(O)nR7(CH2)yR8 и (CH2)xNR7C(O)(CH2)yR8, где R7 и R8 имеют значения, определенные в формуле (I). Более предпочтительно R2 выбирается из R7, -(CH2)xR7, -(CH2)xC(O)(CH2)yR7, (CH2)xS(O)2(CH2)yR7, -(CH2)xC(O)C(O)(CH2)yR7, -(CH2)xC(O)O(CH2)yR7 и -(CH2)xC(O)nH(CH2)yR7; еще более предпочтительно R2 выбирается из R7, -(CH2)xR7 и -(CH2)xC(O)(CH2)yR7.
Более предпочтительно настоящее раскрытие относится к соединению или его фармацевтически приемлемой соли, стереоизомеру, изотопному производному или пролекарству, где каждый из R7 и R8 независимо выбран из следующих замещенных или незамещенных групп: R9, OR9, -R10-O-R9, -R10-NH-R9, -R10-C(O)-R9, -R10-NHC(O)-R9, -R10-C(O)nH-R9, -R10-S-R9, -R10-S-C(O)-R9, С3-6циклоалкил, 3-6-членный гетероциклоалкил, С6-10арил, 5-10-членный гетероарил, -R10-C6-10арил, -R10-5-10-членный гетероарил, -O-R10-C6-10арил, -O-R10-5-10-членный гетероарил, -R10-O-C6-10арил, R10-О-5-10-членный гетероарил, С2-6алкен и С2-6алкин, или когда R7 и R8 оба присоединены к одному и тому же атому азота, R7 и R8 и атом азота, к которому они присоединены, вместе образуют замещенный или незамещенный 3-6-членный гетероциклоалкил; где R9 представляет собой C1-6алкил, R10 представляет собой C1-6алкилен, С2-6алкенилен или С2-6алкинилен; термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -ОН, -NH2, -SH, -CN, С1-3алкила, С1-3алкилокси, C1-3галогеналкила, С1-3галогеналкилокси, -NHCN, -NHCONH2, NHC(O)CH3, N(CH3)2, N(C2H5)2, -SC(O)CH3, -ОС(O)-С1-6алкила и тому подобного; предпочтительно R7 независимо выбран из следующих замещенных или незамещенных групп: R9, OR9, -R10-O-R9, -R10-NHC(O)-R9, С3-6циклоалкил, 3-6-членный гетероциклоалкил, С6-10арил, 5-10-членный гетероарил, С2-6алкен и С2-6алкин, или когда R7 и R8 оба присоединены к одному и тому же атому азота, R7 и R8 и атом азота, к которому они присоединены, вместе образуют замещенный или незамещенный 3-6-членный гетероциклоалкил; более предпочтительно R7 независимо выбирается из следующих замещенных или незамещенных групп: R9, OR9, -R10-O-R9, С3-6циклоалкил, 3-6-членный гетероциклоалкил, С6-10арил и 5-10-членный гетероарил; еще более предпочтительно R7 независимо выбирается из следующих замещенных или незамещенных групп: R9, OR9, -R10-O-R9, С3-6циклоалкил и 3-6-членный гетероциклоалкил; термин «замещенный» в R7 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -ОН, -NH2, -SH, -CN, C1-3алкила, C1-3алкилокси, C1-3галогеналкила, C1-3галогеналкилокси, -NHCN, -NHCONH2, NHC(O)CH3, N(CH3)2, N(C2H5)2, -SC(O)CH3, -ОС(O)-С1-6алкила и тому подобного; предпочтительно термин «замещенный» в R7 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -ОН, -NH2, -SH, -CN, С1-3алкила, C1-3алкилокси, C1-3галогеналкила, NHC(O)CH3, N(СН3)2, -ОС(O)-С1-6алкила и тому подобного; более предпочтительно термин «замещенный» в R7 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -ОН, -NH2, -CN, С1-3алкила, C1-3алкилокси и тому подобного.
Дополнительно предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, стереоизомеру, изотопному производному или пролекарству, где каждый из R7 и R8 независимо выбирается из следующих замещенных или незамещенных групп: метил, этил, пропил, изопропил, бутил, пентил, метилокси, этилокси, пропилокси, изопропилокси, -СН2ОСН3, -СН2ОСН2СН3, -СН2СН2ОСН3, -СН2СН2ОСН2СН3, циклопропил, циклобутил, цикло пентил, циклогексил, азиридинил, азетидинил, пиррол ид инил, пиперидил, оксиранил, оксетанил, оксациклопентил, оксациклогексил, фенил, пиридил, пиразолил, оксазолил, изоксазолил, тиенил, тиазолил, бензил, фенилэтил, винил, пропенил, этинил и пропинил. Предпочтительно R7 независимо выбирается из следующих замещенных или незамещенных групп: метил, этил, метилокси, этилокси, циклопропил, циклобутил, азетидинил, пиперидил, оксетанил, оксациклогексил, фенил, пиридил, пиразолил, изоксазолил, тиенил, тиазолил, бензил, винил, пропенил и этинил; более предпочтительно R7 независимо выбирается из следующих замещенных или незамещенных групп: метил, этил, метилокси, циклопропил, циклобутил, пиперидил, оксетанил, оксациклогексил, пиридил, пиразолил, изоксазолил, винил, пропенил и этинил; еще более предпочтительно R7 независимо выбирается из метила, этила, циклопропила, пиперидила, оксетанила, пиразолила, винила, пропенила и этинила; еще более предпочтительно R7 независимо выбирается из метила, этила и циклопропила. Термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -Cl, -Br, -ОН, -NH2, -SH, -CN, C1-3алкила, C1-3алкилокси, C1-3галогеналкила, C1-3галогеналкилокси, -NHCN, -NHCONH2, NHC(O)CH3, N(CH3)2, N(C2H5)2, -SC(O)CH3, -ОС(O)-С1-6алкила и тому подобного; предпочтительно термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -ОН, -SH, -CN, C1-3алкила, C1-3алкилокси, C1-3галогеналкилокси, NHC(O)CH3, N(СН3)2, N(C2H5)2, -ОС(O)-C1-6алкила и тому подобного; более предпочтительно термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -ОН, -NH2, -CN, C1-3галогеналкилокси и тому подобного; еще более предпочтительно термин «замещенный» в R7 и R8 относится к замещению 1, 2 или 3 группой(ами), каждая из которых независимо выбрана из -F, -ОН, -CN и тому подобного.
Еще более предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, стереоизомеру, изотопному производному или пролекарству, где R9 представляет собой C1-4алкил, R10 представляет собой С1-4алкилен.
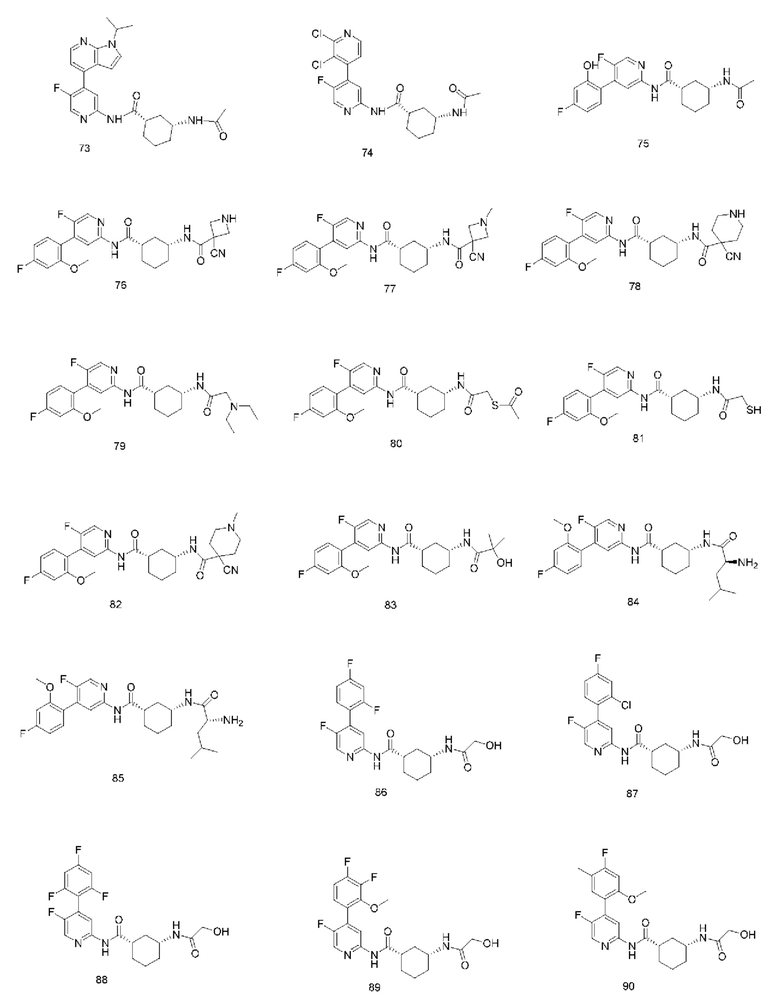
Предпочтительно настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, где соединения имеют следующие структуры:
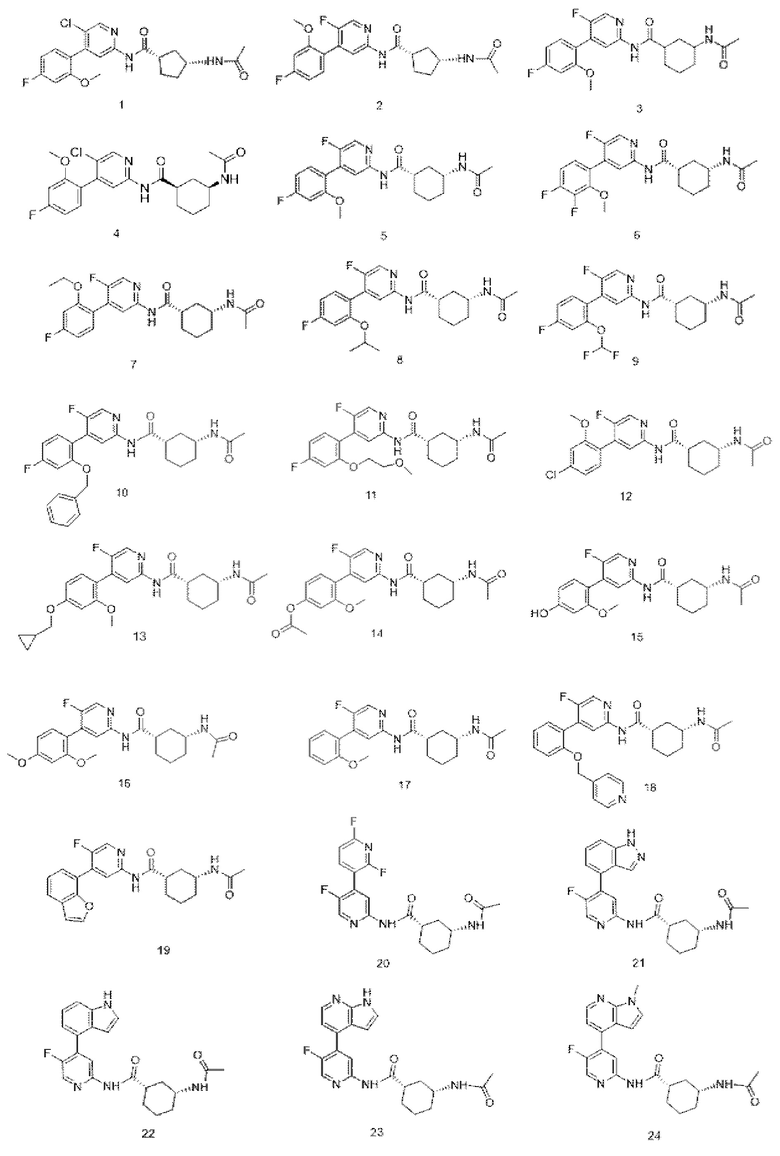
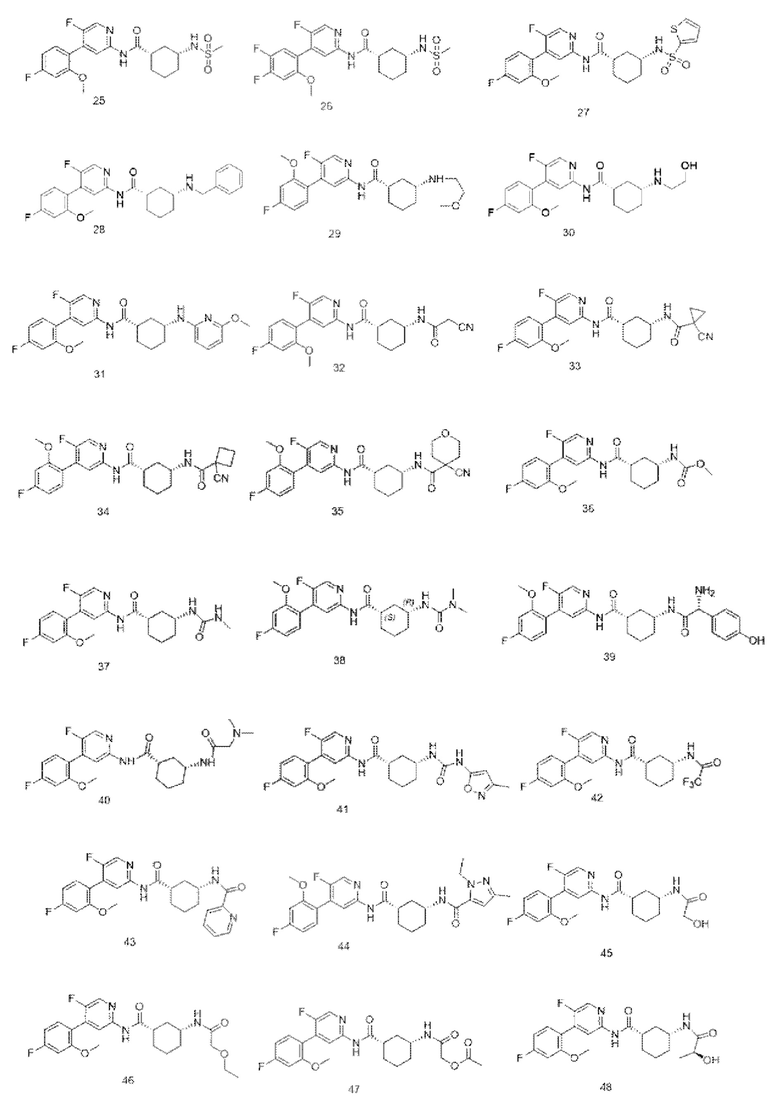
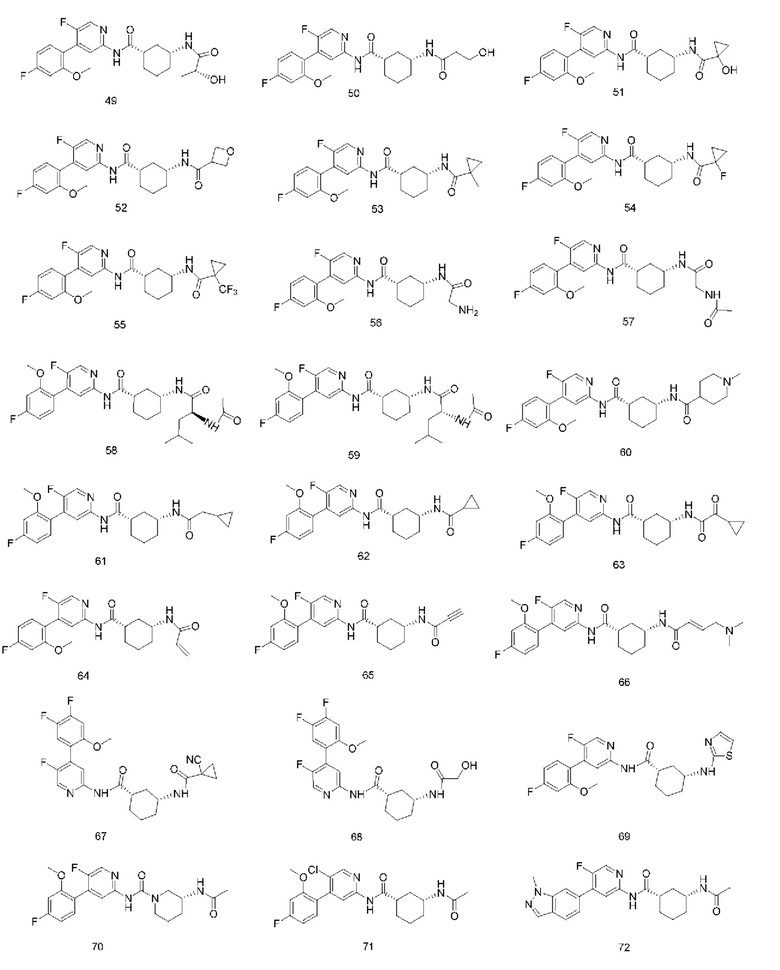
В другом аспекте настоящее изобретение также относится к фармацевтической композиции, содержащей соединение по настоящему изобретению или его фармацевтически приемлемую соль, или его стереоизомер, изотопное производное или пролекарство.
В другом аспекте настоящее изобретение также относится к применению соединения по настоящему изобретению или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства, или фармацевтической композиции по настоящему изобретению в производстве медикамента для предотвращения и/или лечения заболеваний, опосредованных активностью циклинзависимой киназы 9 (CDK9); предпочтительно заболевание представляет собой гиперпролиферативное заболевание или воспалительное заболевание; более предпочтительно гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; еще более предпочтительно гематологическая опухоль представляет собой лейкоз; еще более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
В другом аспекте настоящее изобретение также относится к соединению по настоящему изобретению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, или фармацевтической композиции по настоящему изобретению для применения в медикаменте.
Дополнительно в соответствии с применением в медикаменте, предусмотренным настоящим изобретением, медикамент применяется для лечения гиперпролиферативных заболеваний или воспалительных заболеваний.
Дополнительно в соответствии с применением в медикаменте, предусмотренным настоящим изобретением, гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; предпочтительно гематологическая опухоль представляет собой лейкоз; более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
В другом аспекте настоящее изобретение также относится к применению соединения по настоящему изобретению или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства, или фармацевтической композиции по настоящему изобретению для применения в предотвращении и/или лечении заболеваний, опосредованных активностью циклинзависимой киназы 9 (CDK9); предпочтительно заболевание представляет собой гиперпролиферативное заболевание или воспалительное заболевание; более предпочтительно гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; еще более предпочтительно гематологическая опухоль представляет собой лейкоз; еще более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
В другом аспекте настоящее изобретение также относится к соединению по настоящему изобретению или его фармацевтически приемлемой соли, или его стереоизомеру, изотопному производному или пролекарству, или фармацевтическаой композиции по настоящему изобретению для применения в лечении гиперпролиферативных заболеваний или воспалительных заболеваний, предпочтительно гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; более предпочтительно гематологическая опухоль представляет собой лейкоз; еще более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
В другом аспекте настоящее изобретение также относится к способу лечения заболеваний и/или состояний, включающему введение вышеупомянутого соединения или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства, или фармацевтической композиции, субъекту, нуждающемуся в этом, где указанные заболевания и/или состояния у субъекта представляют собой заболевания, опосредованные активностью циклинзависимой киназы 9 (CDK9); предпочтительно заболевание представляет собой гиперпролиферативное заболевание или воспалительное заболевание; более предпочтительно гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; еще более предпочтительно гематологическая опухоль представляет собой лейкоз; еще более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
В другом аспекте настоящее изобретение также относится к способу лечения заболеваний и/или состояний, включающему введение вышеупомянутого соединения или его фармацевтически приемлемой соли, или его стереоизомера, изотопного производного или пролекарства, или фармацевтической композиции, субъекту, нуждающемуся в этом, где указанное заболевание и/или состояние у субъекта представляет собой гиперпролиферативное заболевание или воспалительное заболевание; предпочтительно гиперпролиферативное заболевание представляет собой гематологическую опухоль или солидную опухоль; более предпочтительно гематологическая опухоль представляет собой лейкоз; еще более предпочтительно лейкоз представляет собой острый миелоидный лейкоз.
Соединение по настоящему изобретению может обладать оптической активностью. Соединение по настоящему изобретению может присутствовать в виде рацемата, оптического изомера или их смеси. Для того чтобы синтезировать оптический изомер соединения по настоящему изобретению, его можно получить из оптического изомера исходного материала или можно получить путем разделения рацемата соединения по настоящему изобретению.
Определение
Термин «алкил» относится к одновалентной насыщенной алифатической углеводородной группе с прямой или разветвленной цепью, содержащей 1-20 атомов углерода, предпочтительно содержащей 1-10 атомов углерода (т.е. С1-10алкил), более предпочтительно содержащей 1-8 атомов углерода (С1-8алкил), еще более предпочтительно содержащей 1-6 атомов углерода (т.е. C1-6алкил). Например, «С1-6алкил» относится к такой группе, которая представляет собой алкильную группу, и число атомов углерода в углеродной цепи составляет от 1 до 6 (в частности, 1, 2, 3, 4, 5 или 6). Примеры включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, неопентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, н-гептил, н-октил и тому подобное.
Термин «циклоалкил» относится к моноциклической насыщенной алифатической углеводородной группе, имеющей определенное число атомов углерода, предпочтительно содержащей 3-12 атомов углерода (т.е. С3-12циклоалкил), более предпочтительно содержащей 3-10 атомов углерода (С3-10циклоалкил), еще более предпочтительно 3-6 атомов углерода (С3-6циклоалкил), 4-6 атомов углерода (С4-6циклоалкил) и 5-6 атомов углерода (С5-6циклоалкил). Примеры включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, метилциклопропил, 2-этилциклопентил, диметилциклобутил и тому подобное.
Термин «алкилокси» относится к -О-алкильной группе, где алкил является таким, как определено выше, т.е. содержит 1-20 атомов углерода, предпочтительно 1-10 атомов углерода, более предпочтительно 1-8 атомов углерода, еще более предпочтительно 1-6 атомов углерода (в частности, 1, 2, 3, 4, 5 или 6). Репрезентативные примеры включают, но не ограничиваются ими, метилокси, этилокси, пропилокси, изопропилокси, бутилокси, 1-метилпропилокси, 2-метилпропилокси, трет-бутилокси, пентилокси, 1-метилбутилокси, 2-метилбутилокси, 3-метилбутилокси, 1,1-диметилпропилокси, 1,2-диметилпропилокси, 2,2-диметилпропилокси, 1-этилпропилокси и тому подобное.
Термин «галоген» или «гало» относится к F, Cl, Br и I. Термин «галогеналкил» относится к алкильной группе, как определено выше, в которой один, два, более или все атом(ы) водорода замещен(ы) атомом(ами) галогена. Репрезентативные примеры галогеналкилов включают CCl3, CF3, CHCl2, CH2Cl, CH2Br, CH2I, CH2CF3, CF2CF3 и тому подобное.
Термин «гетероциклическая группа» относится к насыщенному или частично ненасыщенному моноциклическому, бициклическому или полициклическому циклоуглеводородному заместителю, который представляет собой неароматическую структуру, содержащую 3-20 атомов в кольце (в которой 1, 2, 3 или более атом(ов) в кольце представляет собой/выбираются из N, О и S, а остальные атомы кольца представляют собой С), предпочтительно содержащую 3-12 атомов в кольце (С3-12 гетероциклическая группа), более предпочтительно содержащую 3-10 атомов в кольце (С3-10 гетероциклическая группа), или 3-8 атомов в кольце (С3-8 гетероциклическая группа), или 3-6 атомов в кольце (С3-6 гетероциклическая группа), или 4-6 атомов в кольце (С4-6 гетероциклическая группа), или 5-6 атомов в кольце (С5-6 гетероциклическая группа); и предпочтительно имеющую 1-4 гетероатома, более предпочтительно 1-3 (т.е. 1, 2 или 3) гетероатома. Примеры моноциклических гетероциклических групп включают пирролидинил, имидазолидинил, тетрагидрофуранил, дигидропирролил, пиперидинил, пиперазинил, пиранил и тому подобное. Полициклические гетероциклические группы включают спиро, конденсированные и мостиковые гетероциклические группы.
Термин «гетероциклоалкил» относится к насыщенной «гетероциклической группе», как определено выше, содержащей 3-20 атомов в кольце (где 1, 2, 3 или более атом(ов) в кольце представляет собой/выбираются из N, О и S, а остальные атомы кольца представляют собой С), предпочтительно содержащей 3-12 атомов в кольце (С3-12 гетероциклоалкил), более предпочтительно содержащей 3-10 атомов в кольце (С3-10 гетероциклоалкил) или 3-8 атомов в кольце (С3-8 гетероциклоалкил), или 3-7 атомов в кольце (С3-7 гетероциклоалкил), или 3-6 атомов в кольце (С3-6 гетероциклоалкил), или 4-6 атомов в кольце (С4-6 гетероциклоалкил), или 5-6 атомов в кольце (С5-6 гетероциклоалкил); и предпочтительно имеющей 1-4 гетероатома, более предпочтительно 1-3 (т.е. 1, 2 или 3) гетероатома. Примеры включают азиридинил, оксиранил, тииранил, азетидинил, оксетанил, тиетанил, пирролидинил, тетрагидрофуранил, оксациклогексанил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, диоксанил, дитиациклогексил, оксазолидинил, тиазолидинил, пиразолидинил, имидазолидинил и тому подобное.
Термин «арил» относится к моноциклическим, бициклическим и трициклическим ароматическим углеродным кольцевым системам, содержащим 6-16 атомов углерода, или 6-14 атомов углерода, или 6-12 атомов углерода, или 6-10 атомов углерода, предпочтительно 6-10 атомов углерода. Термин «арил» может использоваться взаимозаменяемо с термином «ароматическое кольцо». Примеры арильных групп могут включать, но не ограничиваются ими, фенил, нафталенил, антраценил, фенантренил, пиренил и тому подобное.
Термин «гетероарил» относится к ароматической моноциклической или полициклической кольцевой системе, содержащей 5-12-членную структуру или предпочтительно 5-10-членную структуру, 5-8-членную структуру и более предпочтительно 5-6-членную структуру, где 1, 2, 3 или более атом(ов) в кольце представляет(ют) собой гетероатом(ы), а остальные атомы представляют собой углерод, гетероатомы независимо выбираются из О, N и S, а количество гетероатомов предпочтительно равно 1, 2 или 3. Примеры гетероарильных групп включают, но не ограничиваются ими, фурил, тиенил, оксазолил, тиазолил, изоксазолил, оксадиазолил, тиадиазолил, пирролил, пиразолил, имидазолил, триазолил, тетразолил, пиридил, пиримидинил, пиразинил, пиридазинил, тиодиазолил, триазинил, фталазинил, хинолинил, изохинолинил, птеридил, пуринил, индолил, изоиндолил, индазолил, бензофуранил, бензотиенил, бензопиридил, бензо пиримидинил, бензопиразинил, бензимидазолил, бензо фталазинил, пирроло [2,3-b] пиридил, имидазо[1,2-а]пиридил, пиразоло [1,5-а]пиридил, пиразоло[1,5-а]пиримидинил, имидазо[1,2-b] пиридазинил, [1,2,4]триазоло [4,3-b] пиридазинил, [1,2,4]триазоло[1,5-а]пиримидил, [1,2,4]триазоло[1,5-а]пиридил и тому подобное.
Термин «фармацевтически приемлемая соль» относится к тем солям, которые, согласно здравому медицинскому мнению, подходят для использования в контакте с тканями млекопитающих, в частности, человека, без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного, и соизмеримы разумному соотношению польза/риск. Например, фармацевтически приемлемые соли аминов, карбоновых кислот и других типов соединений хорошо известны в данной области техники.
Термин «соль» включает соли, полученные из неорганических кислот, таких как соляная кислота, серная кислота, сернистая кислота, азотная кислота, фосфорная кислота, бромоводородная кислота и тому подобных, а также включает соли, полученные из органических кислот, таких как уксусная кислота, пропионовая кислота, янтарная кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, малеиновая кислота, фумаровая кислота, салициловая кислота и тому подобных. Когда соединение по настоящему изобретению является кислым, его соответствующую соль можно легко получить из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из таких неорганических оснований, включают соль алюминия, соль аммония, соль кальция, соль меди (ic и ous), соль трехвалентного железа, соль двухвалентного железа, соль лития, соль магния, соль цинка и тому подобные. Соли, полученные из органических нетоксичных оснований, включают соль первичного амина, соль вторичного амина, соль третичного амина и тому подобные.
Термин «стереоизомер» относится к изомерам, образованным различным расположением атомов в молекуле в пространстве, включая конфигурационные изомеры и конформационные изомеры, где конфигурационные изомеры, в свою очередь, включают геометрические изомеры (или цис-транс-изомеры) и оптические изомеры (включая энантиомеры и диастереомеры).
В настоящих соединениях могут существовать геометрические изомеры. Соединения по данному изобретению могут содержать двойные углерод-углеродные связи или двойные углерод-азотные связи в конфигурации Ε или Ζ, где термин «Е» представляет заместители более высокого порядка на противоположных сторонах двойной углерод-углеродной или углерод-азотной связи, и термин «Ζ» представляет заместители более высокого порядка на той же стороне двойной углерод-углеродной или углерод-азотной связи, как определено Правилами Приоритета Кана-Ингольда-Прелога. Соединения по этому изобретению также могут существовать в виде смеси изомеров «Е» и «Ζ». Заместители вокруг циклоалкила или гетероциклоалкила обозначены как имеющие цис- или транс-конфигурацию.
Оптические изомеры относятся к веществам с одинаковыми молекулярными структурами, сходными физическими и химическими свойствами, но с разным оптическими вращениями.
Соединения по этому изобретению могут содержать асимметрично замещенные атомы углерода в R- или S-конфигурации, в которой термины «R» и «S» определены в Рекомендациях IUPAC 1974 г. для Section Ε, Fundamental Stereochemistry, Pure Appl. Chem. (1976) 45, 13-10. Соединения, имеющие асимметрично замещенные атомы углерода с равным количеством R- и S-конфигураций, являются рацемическими по этим атомам углерода. Атомам с избытком одной конфигурации над другой присваивается конфигурация, присутствующая в большем количестве, предпочтительно с избытком около 85%-90%, более предпочтительно с избытком около 95%-99% и еще более предпочтительно с избытком более чем около 99%. Соответственно, данное изобретение включает рацемические смеси, относительные и абсолютные стереоизомеры и смеси относительных и абсолютных стереоизомеров.
Соединения по этому изобретению могут существовать в меченной изотопами или обогащенной изотопами форме, содержащей один или несколько атомов, имеющих атомную массу или массовое число, отличающиеся от атомной массы или массового числа, наиболее часто встречающихся в природе. Изотопы могут быть радиоактивными или нерадиоактивными изотопами. Изотопы атомов, таких как водород, углерод, фосфор, сера, фтор, хлор и йод, включают, но не ограничиваются ими, 2Н, 3Н, 13С, 14С, 15N, 18O, 32Р, 35S, 18F, 36Cl и 125I. Соединения, которые содержат другие изотопы этих и/или других атомов, входят в объем настоящего изобретения.
В другом варианте выполнения изобретения меченные изотопом соединения содержат изотопы дейтерия (2Н), трития (3Н) или 14С. Меченные изотопами соединения по настоящему изобретению могут быть получены обычными способами, хорошо известными специалистам в данной области техники. В этом аспекте соответствующая литература включает Lizondo, J et al., Drugs Fut, 21(11), 1116 (1996); Brickner, S J et al, J Med Chem, 39(3), 673 (1996); Mallesham, В et al., Org Lett, 5(7), 963 (2003).
Содержащие изотопы соединения использовались в фармацевтических исследованиях для изучения метаболического изменения соединений in vivo путем оценки механизма действия и метаболического пути исходного соединения, не меченного изотопом (Blake et al. J. Pharm. Sci. 64, 3, 367-391 (1975)). Такие метаболические исследования важны для разработки безопасных, эффективных терапевтических препаратов либо потому, что активное in vivo соединение вводили пациенту либо потому, что метаболиты, полученные из исходного соединения, оказываются токсичными или канцерогенными (Kushner et al., Can. J. Physiol. Pharmacol., 77, 79-88 (1999); Foster et al., Advances in Drug Research Vol.14, pp.2-36, Academic Press, London, 1985; Kato et al., J. Labelled Сотр. Radiopharmaceut, 36(10):927-932 (1995)).
Кроме того, для лечения заболеваний и состояний можно использовать нерадиоактивные изотопсодержащие препараты, такие как дейтерированные препараты, называемые «тяжелыми лекарственными средствами». Увеличение количества изотопа, присутствующего в соединении, выше его естественного содержания, называется обогащением. Примеры количества обогащения включают от около 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 21, 25, 29, 33, 37, 42, 46, 50, 54, 58, 63, 67, 71, 75, 79, 84, 88, 92, 96 до около 100 мол. %.
Замещение изотопами может присутствовать в любом возможном месте молекулярной структуры для получения изотопных производных. Например, замещение дейтерием (2Н) может присутствовать в любом возможном месте молекулы с образованием производных в дейтерированной форме.
Лекарственные средства, меченные стабильными изотопами, могут изменять свои физико-химические свойства, такие как pKa и растворимость в липидах. Если изотопное замещение влияет на области, участвующие во взаимодействиях лиганд-рецептор, то эти эффекты и изменения могут повлиять на фармакодинамический ответ молекулы лекарственного средства. Хотя некоторые физические свойства молекул, меченных стабильными изотопами, отличаются от свойств немеченых молекул, химические и биологические свойства одинаковы, с одним важным отличием: из-за увеличенной массы тяжелых изотопов участие тяжелых изотопов и любая связь с другим атомом сильнее, чем такая же связь между легким изотопом и этим атомом. Соответственно, связывание изотопа в месте метаболизма или ферментативного превращения потенциально может замедлить реакцию, что может изменить фармакокинетические свойства или эффекты по сравнению с неизотопными соединениями.
Пролекарства представляют собой разработанные производные активных лекарственных средств, которые улучшают определенные нежелательные физические или биологические свойства. Физические свойства обычно связаны с растворимостью (излишняя или недостаточная растворимость в липидах или воде) или стабильностью, в то время как проблемные биологические свойства включают слишком быстрый метаболизм или плохую биодоступность, которые сами по себе могут быть связаны с физико-химическими свойствами.
Пролекарства обычно получают путем: а) образования сложных эфиров, полу сложных эфиров, карбонатных сложных эфиров, нитратных сложных эфиров, амидов, гидроксамовых кислот, карбаматов, иминов, оснований Манниха, фосфатов, фосфатных сложных эфиров и енаминов активного лекарственного средства, b) функционализации лекарственного средства азо-, гликозидными, пептидными и простоэфирными функциональными группами, с) применения аминалей, полуаминалей, полимеров, солей, комплексов, фосфорамидов, ацеталей, полуацеталей и кетальных форм лекарственных средств. Например, см. Andrejus Korolkovas's, "Essentials of Medicinal Chemistry", John Wiley-Interscience Pulications, John Wiley and Sons, New York (1988), pp.97-118, которая полностью включена в настоящий документ посредством ссылки. Сложные эфиры могут быть получены из субстратов, содержащих либо гидроксильную группу, либо карбоксильную группу, общими способами, известными специалистам в данной области техники. Типичными реакциями этих соединений являются замещения одного из гетероатомов другим атомом. Аналогичным образом амиды могут быть получены из субстратов, содержащих либо аминогруппу, либо карбоксильную группу. Сложные эфиры также могут реагировать с аминами или аммиаком с образованием амидов. Другой способ получения амидов состоит в совместном нагревании карбоновой кислоты и амина.
Благоприятные эффекты настоящего изобретения заключаются в следующем:
В настоящем изобретении разработан ряд соединений с новыми структурами, что обеспечивает новое направление для лечения гиперпролиферативных заболеваний, особенно для лечения развития гематологических опухолей и солидных опухолей.
Результаты анализа активности киназы in vitro и анализа клеток показывают, что соединения по настоящему изобретению обладают хорошей in vitro ингибирующей активностью киназы в отношении CDK9, хорошей селективностью в отношении других субъединиц CDK; и относительно сильным ингибирующим действием на клетки MV4;11. Предпочтительные соединения по настоящему изобретению имеют IC50 ниже 300 нМ, предпочтительно ниже 200 нМ, более предпочтительно ниже 100 нМ и еще более предпочтительно ниже десятков нМ в отношении ингибирующей активности клеток MV4; 11 in vitro.
Анализы ингибирующей активности hERG in vitro показывают, что соединения по настоящему изобретению имеют более низкий риск кардиотоксичности.
Результаты анализа in vivo показывают, что по сравнению с контрольным соединением соединения по настоящему изобретению обладают лучшим in vivo противоопухолевым действием, меньшей токсичностью и более высокой вероятностью стать лекарственнным средством, что обеспечивает лучший выбор для лекарственных средств, ингибирующих целевую CDK9.
Разработка соединений по настоящему изобретению расширила выбор лекарственных средств для лечения рака. Кроме того, в настоящем изобретении исследуется конкретный способ синтеза, который является простым в процессе, удобным в работе и способствует крупномасштабному промышленному производству и применению.
Подробное описание изобретения
Настоящее изобретение дополнительно описано ниже в сочетании с конкретными примерами. Следует понимать, что эти примеры используются только для иллюстрации настоящего изобретения, а не для ограничения объема настоящего изобретения. Экспериментальные методы без конкретных условий в следующих примерах выполняются в соответствии с обычными условиями или в соответствии с условиями, предложенными производителем. Если не указано иное, все используемые в настоящем документе профессиональные и научные термины имеют те же значения, что и термины, известные специалистам в данной области техники. Кроме того, в способах по настоящему изобретению можно использовать любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем документе. Предпочтительные реализации и материалы, показанные в настоящем документе, представлены только в иллюстративных целях.
Пример 1:
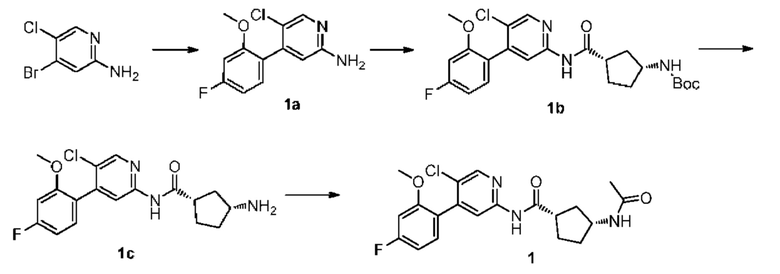
Синтез Промежуточного соединения 1а:
4-бром-5-хлорпиридин-2-амин (3,00 г, 14,50 ммоль) растворяли в диметиловом эфире этиленгликоля (50 мл) и воде (10 мл) с последующим добавлением 4-фтор-2-метоксифенилбороновой кислоты (2,50 г, 14,70 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (1,06 г, 1,45 ммоль) и карбоната калия (6,00 г, 44,10 ммоль). Реакционную смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и кипятили с обратным холодильником при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан:метанол=50:1-10:1) с получением 1а (3,11 г, выход 85%).
Синтез Промежуточного соединения 1b:
1a (0,89 г, 3,50 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклопентанкарбоновую кислоту (0,85 г, 3,50 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,60 г, 4,20 ммоль) и Ν,Ν-диизопропилэтиламин (0,91 г, 7,00 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением 1b (0,96 г, выход 59%).
Синтез Промежуточного соединения 1с:
1b (0,96 г, 2,07 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 млх2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 1 с (0,66 г, выход 88%).
Синтез конечного продукта 1:
1с (0,66 г, 1,80 ммоль) растворяли в дихлорметане (35 мл), затем добавляли уксусный ангидрид (0,92 г, 9,00 ммоль) итриэтиламин (0,91 г, 9,00 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 млх2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением конечного продукта 1 (0,48 г, выход 64%). MS m/z (ESI): 406,1 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 9.16 (s, 1 Η), 8.28-8.25 (m, 2Η), 7.55 (s, 1Η), 7.18 (d, J=7.2 Гц, 1H), 6.79-6.71 (m, 2Н), 4.44 (s, 1H), 3.80 (s, 3Н), 2.98 (t, J=4.8 Гц,1 Η), 2.20-2.17 (m, 3Н), 1.97 (s, 3Н), 1.88-1.82 (m, 3Н).
Пример 2:
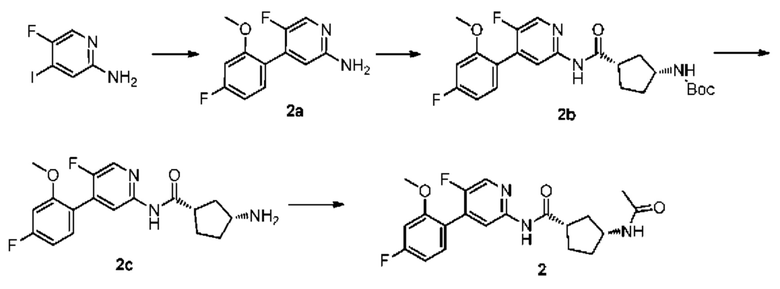
Синтез Промежуточного соединения 2а:
5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и воде (4 мл) и затем добавляли 4-фтор-2-метоксифенилбороновую кислоту (0,71 г, 4,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,31 г, 0,42 ммоль) и карбонат калия (1,70 г, 12,60 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и кипятили с обратным холодильником при 100°С в течение 4 часов. Исходный материал не был обнаружен с помощью ТСХ. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-10:1) с получением 2а (0,80 г, выход 81%).
Синтез Промежуточного соединения 2b:
2а (0,80 г, 3,40 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклопентанкарбоновую кислоту (0,83 г, 3,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,56 г, 4,10 ммоль) и Ν,Ν-диизопропилэтиламин (0,88 г, 6,80 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением 2b (0,82 г, выход 54%).
Синтез Промежуточного соединения 2 с:
2b (0,82 г, 1,83 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией
(дихлорметан:метанол=50:1-8:1) с получением 2 с (0,59 г, выход 93%).
Синтез конечного продукта 2:
2 с (0,59 г, 1,70 ммоль) растворяли в дихлорметане (35 мл), затем добавляли уксусный ангидрид (0,87 г, 8,50 ммоль) итриэтиламин (0,86 г, 8,50 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением конечного продукта 2 (0,42 г, выход 63%).
MS m/z (ESI): 390,2 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 9.20 (s, 1H), 8.32 (s, 1H), 8.12 (s, 1H), 7.30 (d, J=6.6 Гц, 1Η), 6.82-6.75 (m, 3H), 4.44 (s, 1H), 3.84 (s, 3H), 2.99 (q, J=3.6 Гц,1Н), 2.22-2.15 (m, 3H), 1.99 (s, 3H), 1.89-1.85 (m, 3H).
Пример 3:
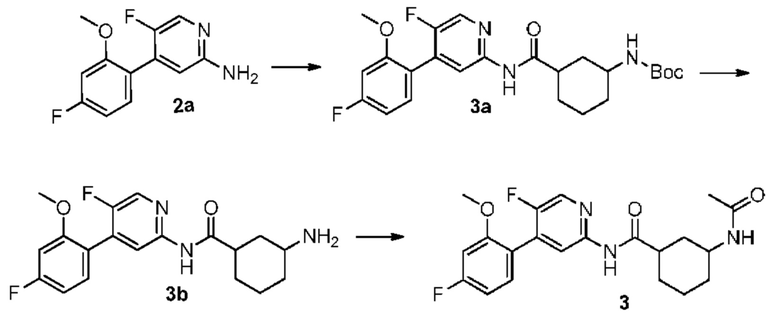
Синтез Промежуточного соединения За:
2а (0,80 г, 3,40 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли цис-3- [(трет-бутилоксикарбонил)амино] циклогексанкарбоновую кислоту (0,83 г, 3,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,56 г, 4,10 ммоль) и Ν,Ν-диизопропилэтиламин (0,88 г, 6,80 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением За (0,90 г, выход 57%).
Синтез Промежуточного соединения 3b:
За (0,90 г, 1,95 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом. Реакционную смесь перемешивали в течение ночи при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 3b (0,61 г, выход 87%).
Синтез конечного продукта 3:
3b (0,61 г, 1,69 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,86 г, 8,40 ммоль) итриэтиламин (0,85 г, 8,40 ммоль). Реакционную смесь перемешивали при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением конечного продукта 3 (0,38 г, выход 56%).
MS m/z (ESI): 404,2 [М+Н]+.
1H ЯМР (600 МГц, CDCl3) δ 8.88 (s, 1H), 8.29 (s, 1H), 8.10 (s, 1H), 7.28 (s, 1H), 6.77-6.72 (m, 3H), 3.82 (s, 3H), 2.52-2.49 (m, 1H), 2.24-2.22 (m, 1H), 2.00-1.95 (m, 4H), 1.98 (s, 3H), 1.48-1.38 (m, 3H), 1.18-1.13 (m, 1H).
Пример 4:
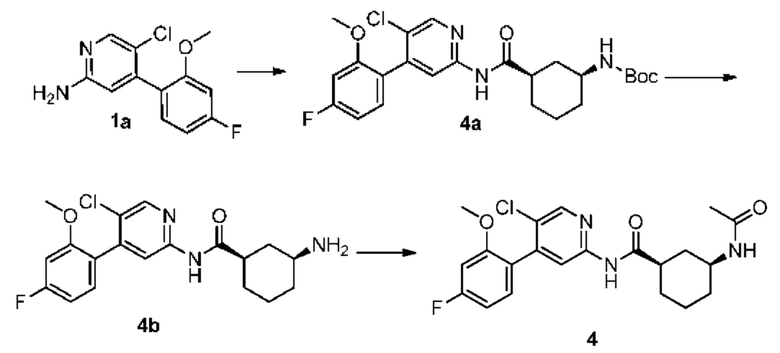
Синтез Промежуточного соединения 4а:
1а (0.40 г, 1.58 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1R,3S)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,38 г, 1,58 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,72 г, 1,90 ммоль) и Ν,Ν-диизопропилэтиламин (0,41 г, 3,16 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением 4а (0,45 г, выход 60%).
Синтез Промежуточного соединения 4b:
4а (0,45 г, 0,94 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 4b (0,30 г, выход 85%).
Синтез конечного продукта 4:
4b (0,30 г, 0,80 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,24 г, 2,39 ммоль) итриэтиламин (0,24 г, 2,39 ммоль). Реакционную смесь перемешивали при комнатной температуре, и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-10:1) с получением конечного продукта 4 (0,17 г, выход 51%).
MS m/z (ESI): 420,14 [М+Н]+.
1Н ЯМР (600 МГц, CDCb) δ 10.69 (s, 1Н), 8.41 (s, 1H), 8.05 (s, 1H), 7.78-7.73 (m, 1H), 7.27-7.22 (m, 1H), 7.10-7.08 (m, 1H), 6.91-6.88 (m, 1H), 3.76 (s, 3H), 3.57-3.54 (m, 1H), 2.62-2.59 (m, 1 H), 1.86 (d, J=12.6 Гц, 1H), 1.76 (s, 6H), 1.31-1.23 (m, 3H), 1.07-1.05 (m, 1H).
Пример 5:
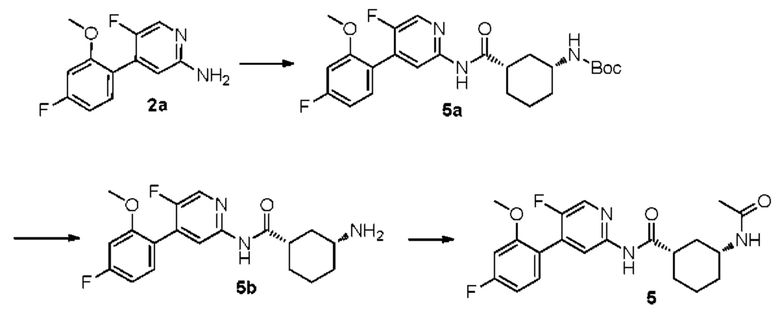
Синтез Промежуточного соединения 5а:
2а (0,80 г, 3,40 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,83 г, 3,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,56 г, 4,10 ммоль) и Ν,Ν-диизопропилэтиламин (0,88 г, 6,80 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением 5а (0,90 г, выход 58%).
Синтез Промежуточного соединения 5b:
5а (0,90 г, 1,95 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 5b (0,61 г, выход 87%).
Синтез конечного продукта 5:
5b (0,61 г, 1,69 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,86 г, 8,40 ммоль) итриэтиламин (0,85 г, 8,40 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением конечного продукта 5 (0,38 г, выход 56%).
MS m/z (ESI): 404,2 [М+Н]+.
1H ЯМР (600 МГц, CD3OD) δ 8.18 (s, 1H), 8.09 (s, 1H), 7.33-7.29 (m, 1Н), 6.94 (d, J=10.8 Гц, 1H), 6.84-6.81 (m, 1H), 3.83 (s, 3H), 3.76-3.72 (m, 1H), 2.60-2.57 (m, 1H), 2.06 (d, J=12.0 Гц, 1H), 1.96-1.90 (m, 3H), 1.93 (s, 3H), 1.51-1.39 (m, 3H), 1.24-1.21 (m, 1H).
Пример 6:
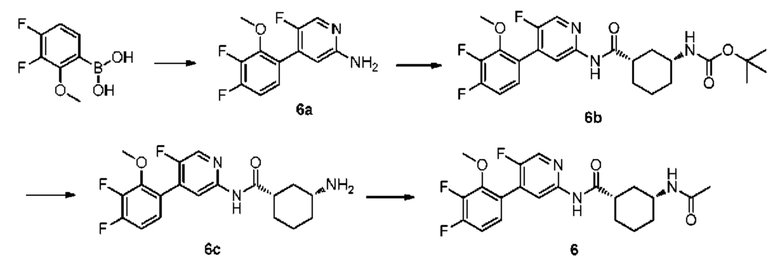
Синтез Промежуточного соединения 6а:
3,4-дифтор-2-метоксифенилбороновую кислоту (0,57 г, 3,03 ммоль) растворяли в диоксане (50 мл) и затем добавляли 5-фтор-4-йодпиридин-2-амин (0,60 г, 2,52 ммоль), тетракис(трифенилфосфин)палладий (150 мг, 0,13 ммоль) и тригидрат фосфата калия (1,00 г, 3,78 ммоль). Реакционную смесь нагревали вплоть до 100°С в атмосфере азота и оставляли реагировать в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-1:1) с получением 6а (0,40 г, выход 52%).
Синтез Промежуточного соединения 6b:
(1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (348 мг, 1,43 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли пиридин (572 мг, 7,24 ммоль) и тионилхлорид (300 мг, 2,52 ммоль). Полученную смесь оставляли реагировать при комнатной температуре в течение 4 часов, и затем непосредственно к указанному выше реакционному раствору добавляли 6а (400 мг, 1,57 ммоль). Реакционная смесь продолжала реагировать при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (30 мл). Полученную смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением 6b (250 мг, выход 33%).
Синтез Промежуточного соединения 6с:
6b (250 мг, 0,52 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли трифторуксусную кислоту (1 мл). Полученную смесь подвергали реакции при комнатной температуре в течение 1,5 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 6 с (130 мг, выход 65%).
Синтез конечного продукта 6:
6 с (130 мг, 0,34 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли уксусный ангидрид (45 мг, 0,44 ммоль) и триэтиламин (44 мг, 0,44 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 1,5 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-30:1) с получением конечного продукта 6 (120 мг, выход 84%).
MS m/z (ESI): 422,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.68 (s, 1H), 8.43 (s, 1H), 8.20 (s, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.33(t, 7=7.8 Гц, 1H), 7.21(t, 7=7.8 Гц, 1H), 3.95 (s, 3H), 3.59-3.54 (m, 1H), 2.63-2.59 (m, 1H), 1.88-1.84 (m, 1H), 1.80-1.72 (m, 3H), 1.78 (s, 3H), 1.31-1.25 (m, 3H), 1.10-1.04 (m, 1H).
Пример 7:
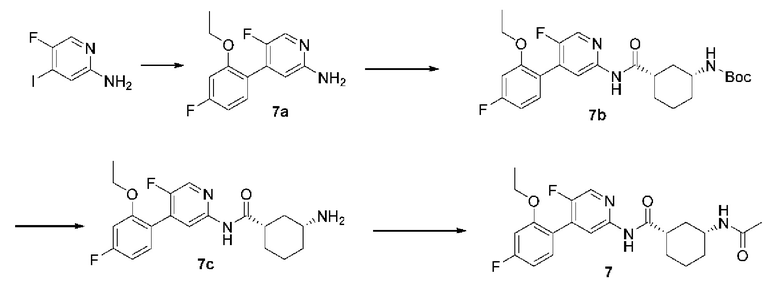
Синтез Промежуточного соединения 7а:
2-амино-5-фтор-4-йодпиридин (0,50 г, 2,10 ммоль) и 5-фтор-2-этоксифенилбороновую кислоту (0,46 г, 2,50 ммоль) растворяли в диметиловом эфире этиленгликоля (10 мл) и воде (2 мл) и затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (71 мг, 0,10 ммоль) и карбонат калия (0,87 г, 6,30 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь подвергали реакции при 100°С в течение 2 часов. До тех пор, пока с помощью ТСХ не было обнаружено исходного материала, смесь охлаждали и затем растворитель удаляли путем концентрирования. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением 7а (0,50 г, выход 95%).
Синтез Промежуточного соединения 7b:
Соединение (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,29 г, 1,20 ммоль) растворяли в дихлорметане (10 мл) и добавляли пиридин (395 мг, 5,00 ммоль) и тионилхлорид (202 мг, 1,70 ммоль) на бане со льдом. Реакционную смесь оставляли реагировать при комнатной температуре в течение 2 часов и концентрировали для удаления растворителя и избыточного тионилхлорида. Затем добавляли дихлорметан (10 мл) и соединение 7а (250 мг, 1,00 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-1:1) с получением 7b (100 мг, выход 21%).
Синтез Промежуточного соединения 7с:
7b (47 мг, 0,10 ммоль) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 1 часа до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали с получением 7с (50 мг, неочищенный продукт). Продукт не подвергали дальнейшей очистке и сразу использовали в реакции следующей стадии.
Синтез конечного продукта 7:
7с (37 мг, 0,10 ммоль) растворяли в дихлорметане (2 мл), добавляли триэтиламин (20 мг, 0,20 ммоль) и уксусный ангидрид (20 мг, 0,20 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 1 часа до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали. Полученный неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол=10:1) с получением конечного продукта 7 (30 мг, выход 72%).
MS:(m/z, ESI): 417,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.57 (s, 1H), 8.33 (s, 1H), 8.11 (d, J=5.4 Гц, 1H), 7.79(d, J=7.8 Гц, 1H), 7.35 (d, J=7.8 Гц, 1H), 7.09 (d,.7=11.4 Гц, 1H), 6.90 (d, J=7.8 Гц, 1H), 4.10-4.07 (m, 2H), 3.57-3.55 (m, 1H), 2.59-2.57 (m, 1H), 1.87-1.84 (m, 1H), 1.77-1.75 (m, 5H), 1.70-1.50 (m, 1H), 1.31-1.26 (m, 3H), 1.24-1.21 (m, 3H), 1.07-1.05 (m, 1H).
Пример 8:
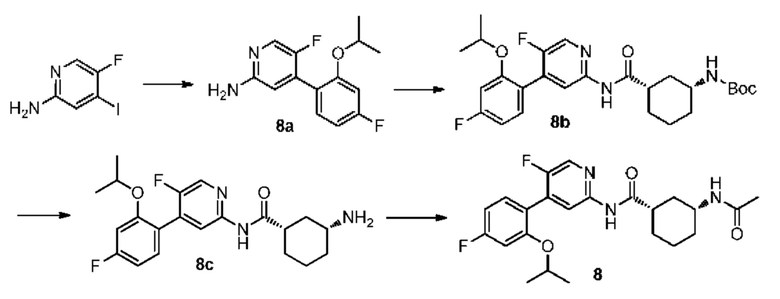
Синтез Промежуточного соединения 8а:
5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и воде (4 мл) и затем добавляли 4-фтор-2-изопропоксифенилбороновую кислоту (0,83 г, 4,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,31 г, 0,42 ммоль) и карбонат калия (1,74 г, 12,60 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали при кипячении с обратным холодильником при 100°С и подвергали реакции в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-10:1) с получением 8а (0,85 г, выход 77%).
Синтез Промежуточного соединения 8b:
8а (0,85 г, 3,20 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,78 г, 3,20 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,44 г, 3,80 ммоль) и Ν,Ν-диизопропилэтиламин (0,83 г, 6,40 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением 8b (0,90 г, выход 57%).
Синтез Промежуточного соединения 8с:
8b (0,90 г, 1,84 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 8 с (0,64 г, выход 89%).
Синтез конечного продукта 8:
8с (0,64 г, 1,64 ммоль) растворяли в дихлорметане (35 мл), затем добавляли уксусный ангидрид (0,84 г, 8,20 ммоль) итриэтиламин (0,83 г, 8,20 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением конечного продукта 8 (0,42 г, выход 59%).
MS:(m/z, ESI): 431,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.57 (s, 1H), 8.32 (s, 1Н), 8.12 (s, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.34 (d, J=7.2 Гц, 1H), 7.10 (s, 1H), 6.88 (d, 7=8.4 Гц, 1H), 4.72-4.69 (m, 1H), 3.56 (s, 1H), 2.62-2.58 (m, 1H), 1.78 (s, 6H), 1.31-1.23 (m, 4H), 1.20 (s, 6H), 1.09-1.04 (m, 1H).
Пример 9:
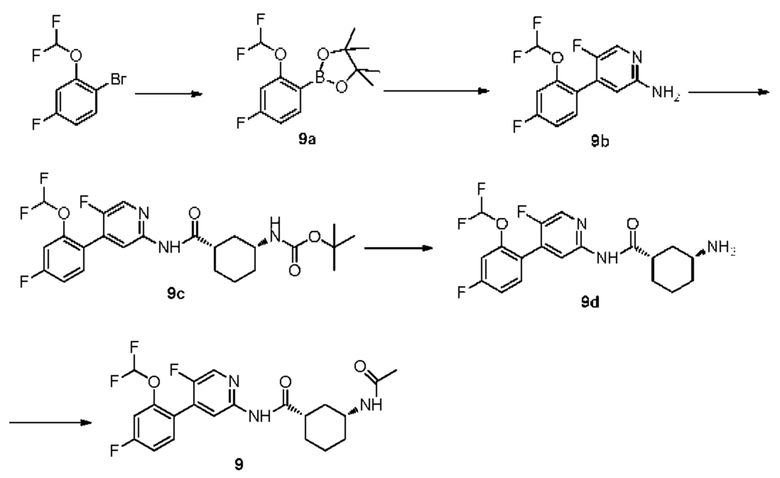
Синтез Промежуточного соединения 9а:
1-бром-2-дифторметокси-4-фторбензол (1,00 г, 4,15 ммоль), бис(пинаколато)дибор (1,26 г, 4,98 ммоль), ацетат калия (1,22 г, 12,45 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,24 г, 0,33 ммоль) и диметиловый эфир этиленгликоля (30 мл) добавляли в реактор. Реакционную смесь нагревали до 100°С в атмосфере азота и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь охлаждали до комнатной температуры. Смесь концентрировали при пониженном давлении и неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат=10:1) с получением 9а (0,50 г, выход 42%).
Синтез Промежуточного соединения 9b:
9а (0,50 г, 1,74 ммоль), 5-фтор-4-йодпиридин-2-амин (0,33 г, 1,39 ммоль), тетракис(трифенилфосфин)палладий (0,12 г, 0,10 ммоль), тригидрат трикалийфосфата (0,60 г, 2,26 ммоль) и диоксан (30 мл) добавляли в реактор. Реакционную смесь нагревали до 100°С в атмосфере азота и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Нагрев прекращали и реакционную смесь охлаждали до комнатной температуры. Смесь концентрировали при пониженном давлении и неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат=1:1) с получением 9b (0,39 г, выход 83%).
Синтез Промежуточного соединения 9с:
9b (0,39 г, 1,43 ммоль), (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,35 г, 1,43 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,65 г, 1,72 ммоль), Ν,Ν-диизопропилэтиламин (0,37 г, 2,86 ммоль) и Ν,Ν-диметилформамид (20 мл) добавляли в реактор. Реакционную смесь подвергали реакции при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (30 мл). Смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат=1:1) с получением 9 с (0,16 г, выход 23%).
Синтез Промежуточного соединения 9d:
9с (0,16 г, 0,33 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (4 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор промывали водой (20 мл×3) и водные фазы объединяли. Объединенную водную фазу доводили карбонатом натрия до рН 8-9 и экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 9d (0,10 г, выход 76%).
Синтез конечного продукта 9:
9d (0,10 г, 0,26 ммоль) растворяли в дихлорметане (20 мл) и добавляли уксусный ангидрид (0,05 г, 0,52 ммоль) и триэтиламин (0,05 г, 0,52 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол=25:1) с получением конечного продукта 9 (0,05 г, выход 44%).
MS m/z (ESI):440,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.67 (s, 1H), 8.41 (s, 1Н), 8.12 (d, J=5.4 Гц, 1H), 7.78 (d, J=7.2 Гц, 1H), 7.58-7.56 (m, 1H), 7.42 (s, 1H), 7.34-7.28 (m, 1H), 3.57-3.56 (m, 1H), 2.61-2.59 (m, 1H), 1.89-1.78 (m, 4H), 1.77 (s, 3H), 1.31-1.24 (m, 3H), 1.08-1.06 (m, 1H).
Пример 10:
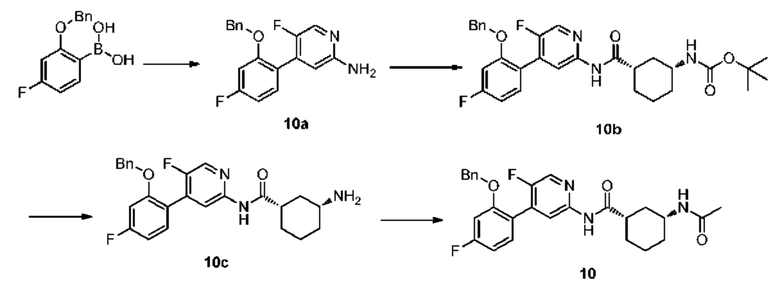
Синтез Промежуточного соединения 10а:
5-фтор-4-йодпиридин-2-амин (500 мг, 2,10 ммоль) растворяли в смешанном растворителе из ДМЭ (20 мл) и воды (4 мл) и затем добавляли 2-бензилокси-4-фторфенилбороновую кислоту (620 мг, 2,52 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия дихлорметана (90 мг, 0,11 ммоль) и карбонат калия (870 мг, 6,30 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и кипятили с обратным холодильником при 100°С и подвергали реакции в течение 4 часов до полной конверсии исходного материала, что контролировали с помощью ТСХ. Реакционный раствор очищали непосредственно колоночной хроматографией (петролейный эфир:этилацетат=5:1-2:1) с получением 10а (0,50 г, выход 76%).
Синтез Промежуточного соединения 10b:
10а (0,30 г, 0,96 ммоль) растворяли в Ν,Ν-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,25 г, 1,01 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,43 г, 1,15 ммоль) и Ν,Ν-диизопропилэтиламин (0,25 г, 1,92 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат=10:1-2:1) с получением 10b (0,30 г, выход 58%).
Синтез Промежуточного соединения 10с:
10b (0,30 г, 0,56 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-8:1) с получением 10 с (0,20 г, выход 82%).
Синтез конечного продукта 10:
10с (0,20 г, 0,46 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли уксусный ангидрид (94 мг, 0,92 ммоль) и триэтиламин (93 мг, 0,92 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 1,5 часов, и исходный материал полностью прореагировал, что контролировали с помощью ТСХ (этилацетат). К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=50:1-20:1) с получением конечного продукта 10 (150 мг, выход 68%).
MS m/z (ESI): 480,2 [М+Н]+.
1Н ЯМР(600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.32 (s, 1H), 8.14 (d, J=5.4 Гц, 1H), 7.76 (d, J=7.8 Гц, 1H), 7.37 (dd, J=7.8 Гц, J=7.2 Гц, 1H), 7.32-7.27 (m, 5H), 7.16 (d,.7=11.4 Гц, 1H), 6.92 (dd, J=8.4 Гц, J=8.4 Гц, 1H), 5.15 (s, 2H), 3.55-3.54 (m, 1H), 2.58-2.56 (m, 1H), 1.86-1.84 (m, 1H), 1.75-1.70 (m, 6H), 1.33-1.15 (m, 4H).
Пример 11:
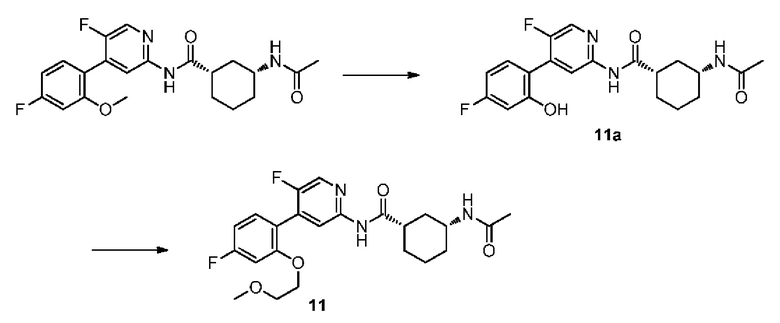
Синтез Промежуточного соединения 11а:
5 (0,10 г, 0,25 ммоль) растворяли в дихлорметане (20 мл) и добавляли трибромид бора (0,12 г, 0,50 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор доводили водным раствором бикарбоната натрия до рН около 6 и органическую фазу сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением Па (0,08 г, выход 82%).
Синтез конечного продукта 11:
11а (0,08 г, 0,21 ммоль) растворяли в N,N-диметилформамиде (10 мл) и добавляли 2-бромэтилметиловый простой эфир (0,03 г, 0,25 ммоль) и карбонат калия (0,06 г, 0,42 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 и с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (20 мл). Смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Органические фазы очищали колоночной хроматографией (дихлорметан:метанол = 25:1) с получением конечного продукта 11 (0,05 г, выход 53%).
MS m/z (ESI): 448,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1Н), 8.31 (s, 1Н), 8.11-8.10 (m, 1Н), 7.78-7.58 (m, 1Н), 7.36-7.33 (m, 1H), 7.12-7.01 (m, 1Н), 6.92-6.89 (m, 1Н), 4.15-4.13 (m, 2Н), 3.55-3.54 (m, 2Н), 3.53-3.52 (m, 1H), 3.15 (s, 3Н), 2.59-2.58 (m, 1H), 1.85 (s, 3Н), 1.83-1.47 (m, 4Н), 1.29-1.04 (m, 4Н).
Пример 12:
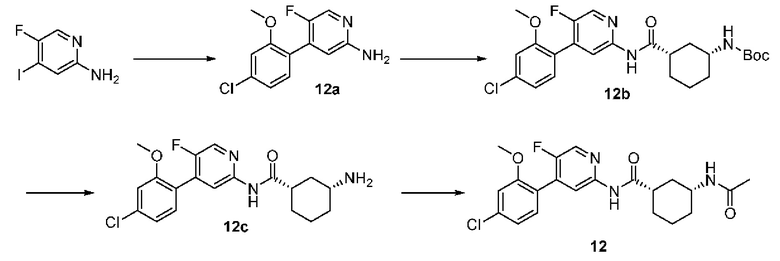
Синтез Промежуточного соединения 12а:
2-амино-5-фтор-4-йодпиридин (0,50 г, 2,10 ммоль) и 5-хлор-2-метоксифенилбороновую кислоту (0,47 г, 2,50 ммоль) растворяли в диметиловом эфире этиленгликоля (10 мл) и добавляли [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий (73 мг, 0,10 ммоль), карбонат калия (0,87 г, 6,3 ммоль) и воду (2 мл). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь подвергали реакции при 100°С в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь охлаждали и затем концентрировали для удаления растворителя. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением 12а (0,50 г, выход 95%).
Синтез Промежуточного соединения 12b:
(1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,29 г, 1,20 ммоль) растворяли в дихлорметане (10 мл) и добавляли пиридин (0,40 г, 5,00 ммоль) и тионилхлорид (0,20 г, 1,70 ммоль) на бане со льдом. Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов и концентрировали. Затем добавляли дихлорметан (10 мл) и соединение 12а (0,25 г, 1,00 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи. После того как с помощью ТСХ не было обнаружено исходного материала, реакционную смесь концентрировали. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-1:1) с получением 12b (0,10 г, выход 21%).
Синтез Промежуточного соединения 12с:
12b (48 мг, 0,10 ммоль) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (2 мл). Реакцию проводили при комнатной температуре в течение 1 часа до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали с получением 12 с (50 мг, неочищенный продукт). Продукт не подвергали дальнейшей очистке и сразу использовали в реакции следующей стадии.
Синтез конечного продукта 12:
12 с (50 мг, 0,10 ммоль) растворяли в дихлорметане (2 мл) и добавляли триэтиламин (20 мг, 0,20 ммоль) и уксусный ангидрид (20 мг, 0,20 ммоль). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь концентрировали. Полученный неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол = 10:1) с получением конечного продукта 12 (30 мг, выход 72%).
MS:(m/z, ESI): 420,1 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.60 (s, 1H), 8.34 (s, 1H), 8.08 (d, J=5.4 Гц, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.33 (d, J=8.4 Гц, 1H), 7.27 (s, 1H), 7.20-7.15 (m, 1H), 3.80 (s, 3Н), 3.65-3.55 (m, 1H), 2.68-2.62 (m, 1H), 1.90-1.85 (m, 1H), 1.76-1.55 (m, 6Н), 1.28-1.23 (m, 3Н), 1.15-1.10 (m, 1H).
Пример 13:
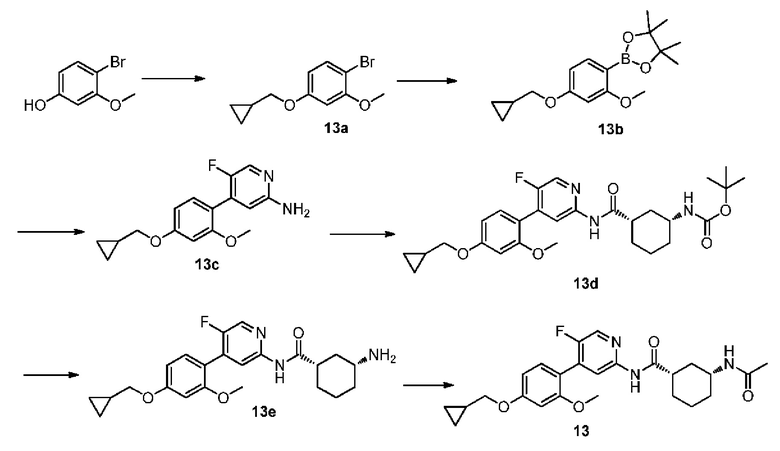
Синтез Промежуточного соединения 13а:
3-метокси-4-бромфенол (3,00 г, 14,80 ммоль) растворяли в ацетоне (50 мл) и затем добавляли бромметилциклопропан (2,20 г, 16,30 ммоль), йодид натрия (1,11 г, 7,40 ммоль) и карбонат цезия (9,64 г, 29,60 ммоль). Реакционную смесь перемешивали при кипячении с обратным холодильником и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь охлаждали до комнатной температуры. Ацетон удаляли при пониженном давлении. К остатку добавляли воду (50 мл). Смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 13а (3,70 г, выход 97%).
Синтез Промежуточного соединения 13b:
13а (3,20 г, 12,40 ммоль) растворяли в диметиловом эфире этиленгликоля (50 мл) и затем добавляли бис(пинаколато)дибор (3,79 г, 14,90 ммоль), ацетат калия (3,65 г, 37,2 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,88 г, 1,2 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и подвергали реакции в течение 8 часов и с помощью ТСХ не было обнаружено исходного материала. Нагрев прекращали и смесь охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат = 5:1) с получением 13b (2,50 г, выход 66%).
Синтез Промежуточного соединения 13с:
13b (2,19 г, 7,20 ммоль) растворяли в диоксане (50 мл) и затем добавляли 5-фтор-4-иодпиридин-2-амин (1,38 г, 5,80 ммоль), тетракис(трифенилфосфин)палладий (0,46 г, 0,40 ммоль) и тригидрат фосфата калия (2,50 г, 9,40 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Нагрев прекращали и реакционную смесь охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат = 1:1) с получением 13 с (1,90 г, выход 92%).
Синтез Промежуточного соединения 13d:
13с (0,60 г, 2,10 ммоль) растворяли в N,N-диметилформамиде (20 мл) и затем добавляли (1S,3R-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,51 г, 2,10 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,95 г, 2,50 ммоль) и N,N-диизопропилэтиламин (0,54 г, 4,20 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат = 1:1) с получением 13d (0,50 г, выход 46%).
Синтез Промежуточного соединения 13е:
13d (0,50 г, 0,97 ммоль) растворяли в дихлорметане (20 мл) и добавляли трифторуксусную кислоту (4 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор промывали водой (30 мл×3) и водные фазы объединяли. Объединенную водную фазу доводили карбонатом натрия до рН 8-9 и экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 13е (0,35 г, выход 88%).
Синтез конечного продукта 13:
13е (0,06 г, 0,14 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли уксусный ангидрид (0,03 г, 0,28 ммоль) итриэтиламин (0,03 г, 0,28 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (DCM:MeOH=25:l) с получением конечного продукта 13 (0,02 г, выход 31%).
MS m/z (ESI): 456,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.52 (s, 1H), 8.28 (s, 1H), 8.06 (d, J=4.8 Гц, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.18 (d, J=8.4 Гц, 1H), 6.71 (s, 1H), 6.63 (d, J=8.4 Гц, 1H), 3.89 (d, J=7.2 Гц, 2Н), 3.76 (s, 3Н), 3.57-3.55 (m, 1H), 2.61-2.60 (m, 1H), 1.88-1.86 (m, 1H), 1.77 (s, 3Н), 1.76-1.75 (m, 3Н), 1.31-1.23 (m, 4Н), 1.09-1.04 (m, 1H), 0.60-0.59 (m, 2Н), 0.35-0.34 (m, 2Н).
Пример 14:
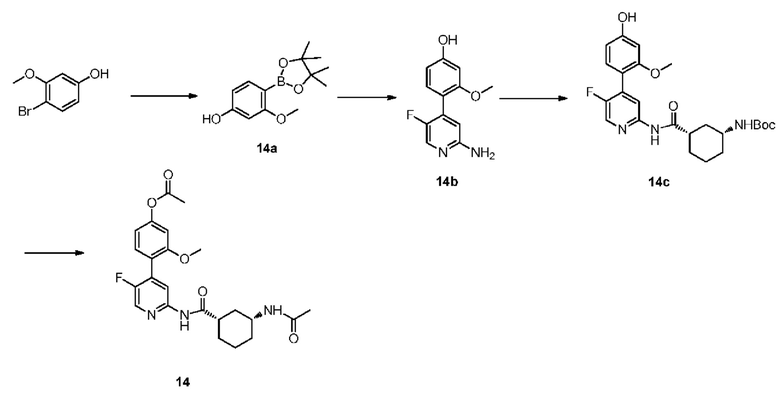
Синтез Промежуточного соединения 14а:
4-бром-3-метоксифенол (0,40 г, 2,00 ммоль) растворяли в диоксане (50 мл) и затем добавляли бис(пинаколато)дибор (0,60 г, 2,40 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,12 г, 0,16 ммоль) и ацетат калия (0,59 г, 6,00 ммоль). Реакционную смесь перемешивали и подвергали реакции в течение 5 часов при 100°С в атмосфере азота, и исходный материал полностью прореагировал, что контролировали с помощью ТСХ. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 4:1) с получением 14а (0,36 г, выход 72%).
Синтез Промежуточного соединения 14b:
14а (0,36 г, 1,44 ммоль) растворяли в диметиловом эфире диэтиленгликоля (50 мл) и воде (10 мл) и затем добавляли 5-фтор-4-йодпиридин-2-амин (0,29 г, 1,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,44 г, 0,60 ммоль) и карбонат калия (0,50 г, 3,60 ммоль). Реакционную смесь перемешивали при 80°С в течение 5 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:3) с получением 14b (0,25 г, выход 74%).
Синтез Промежуточного соединения 14с:
14b (0,15 г, 0,64 ммоль) растворяли в N,N-диметилформамиде (50 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,18 г, 0,72 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,58 г, 0,72 ммоль) и диизопропилэтиламин (0,20 мл, 1,20 ммоль). Реакционную смесь перемешивали и подвергали реакции при 25°С в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением 14 с (0,15 г, выход 51%).
Синтез конечного продукта 14:
14с (0,15 г, 0,32 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли трифторуксусную кислоту (2 мл). Смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К смеси добавляли насыщенный водный раствор карбоната натрия. Значение рН реакционной смеси доводили до 9-10. Реакционную смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли и концентрировали до объема около 50 мл. Добавляли триэтиламин (2 мл) и уксусный ангидрид (2 мл). Смесь подвергали реакции при комнатной температуре в течение 30 минут. Для промывки органической фазы добавляли водный раствор карбоната натрия. Водную фазу отделяли и затем экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией (этилацетат) с получением конечного продукта 14 (90 мг, выход 63%).
MS m/z (ESI): 444,2 [М+Н]+.
1H ЯМР (600 МГц, CD3OD) δ 8.16 (s, 1H), 8.10 (s, 1H), 7.29 (d, J=7.8 Гц, 1H), 6.87 (s, 1H), 6.78 (t, J=7.8 Гц, 1H), 3.79 (s, 3Н), 3.78-3.73 (m, 1H), 2.72 (t, J=12.0 Гц, 1H), 2.31-2.29 (m, 1H), 2.20-2.10 (m, 1H), 2.00-1.80 (m, 6Н), 1.51-1.38 (m, 3Н), 1.27-1.21 (m, 2Н).
Пример 15:
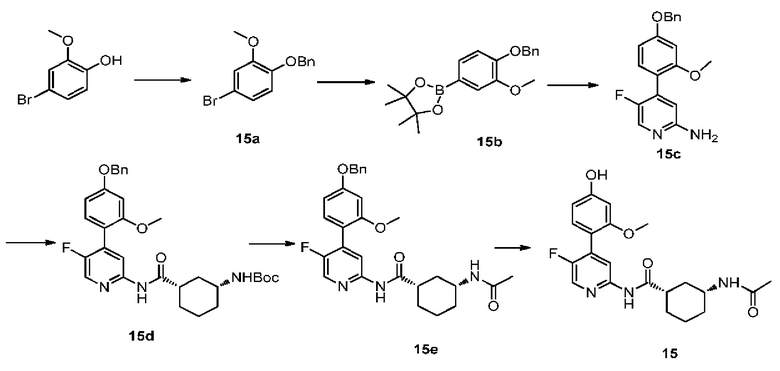
Синтез Промежуточного соединения 15а:
3-метокси-4-бромфенол (540 мг, 2,66 ммоль) растворяли в N,N-диметилформамиде (50 мл) и затем добавляли карбонат калия (735 мг, 5,32 ммоль) и бензилбромид (910 мг, 5,32 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 4:1) с получением 15а (662 мг, выход 85%).
Синтез Промежуточного соединения 15b:
Соединение 15а (662 мг, 2,27 ммоль), бис(пинаколато)дибор (1,15 г, 4,54 ммоль), ацетат калия (667 мг, 6,81 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (146 мг, 0,20 ммоль) растворяли в безводном 1,4-диоксане (100 мл). Реакционную смесь подвергали реакции под защитой газообразного азота при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 4:1) с получением 15b (510 мг, выход 65%).
Синтез Промежуточного соединения 15 с:
15b (510 мг, 1,50 ммоль) растворяли в диметиловом эфире диэтиленгликоля (150 мл). 5-фтор-4-йодпиридин-2-амин (536 мг, 2,25 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (110 мг, 0,15 ммоль) и карбонат калия (621 мг, 4,50 ммоль) добавляли при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 1:1) с получением 15 с (263 мг, выход 54%).
Синтез Промежуточного соединения 15d:
15с (263 мг, 0,81 ммоль) и (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (290 мг, 1,23 ммоль) растворяли в N,N-диметилформамиде (20 мл). Последовательно добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (470 мг, 1,23 ммоль) и диизопропилэтиламин (480 мг, 3,69 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением 15d (250 мг, выход 56%).
Синтез Промежуточного соединения 15е:
15d (250 мг, 0,45 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли трифторуксусную кислоту (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К смеси добавляли насыщенный водный раствор карбоната натрия. Значение рН реакционной смеси доводили до 9-10. Водную фазу отделяли и затем экстрагировали дихлорметаном (100 мл×3). Органические фазы объединяли и концентрировали до объема около 50 мл. Добавляли триэтиламин (2 мл) и уксусный ангидрид (2 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 30 минут. Для промывки органической фазы добавляли водный раствор карбоната натрия и смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (этилацетат) с получением 15е (176 мг, выход 79%).
Синтез конечного продукта 15:
15е (175 мг, 0,36 ммоль) растворяли в метаноле (20 мл) и добавляли палладий/уголь (10 мг). Реакционную смесь подвергали реакции при комнатной температуре под защитой газообразного водорода в течение 10 часов до тех пор, пока с помощью LC-MS не было обнаружено исходного материала. Палладий/уголь отфильтровывали и реакционный раствор концентрировали при пониженном давлении с получением конечного продукта 15 (117 мг, выход 81%).
MS m/z (ESI): 402,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.49 (s, 1H), 10.09 (s, 1H), 8.25 (s, 1H), 8.05 (s, J=5.4 Гц, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.07 (d, J=8.4 Гц, 1H), 6.55 (d, J=2.4 Гц, 1H), 6.48 (s, 1H), 3.71 (s, 3Н), 3.55-3.53 (m, 1H), 2.61-2.57 (m, 1Н), 1.87 (d, J=11.4 Гц, 1H), 1.77 (s, 3Н), 1.77-1.75 (m, 3Н), 1.29-1.27 (m, 4Н).
Пример 16:
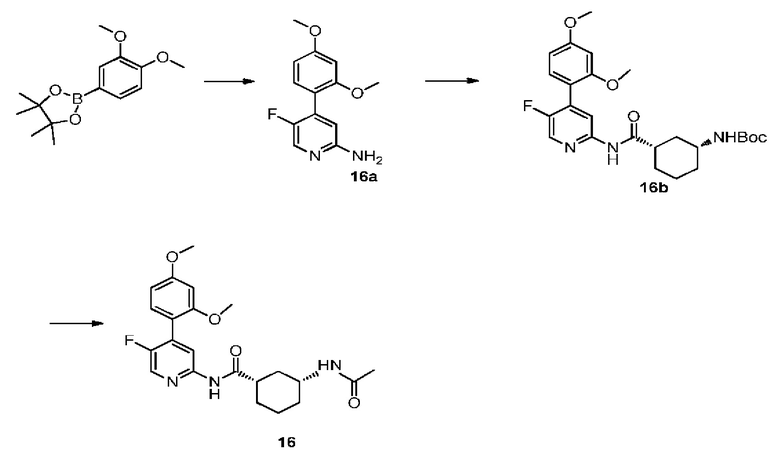
Синтез Промежуточного соединения 16а:
2-(3,4-диметоксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (150 мг, 0,57 ммоль) и 5-фтор-4-йодпиридин-2-амин (202 мг, 0,85 ммоль) растворяли в диметиловом эфире диэтиленгликоля (50 мл) и последовательно добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (37 мг, 0,05 ммоль), карбонат калия (117 мг, 0,85 ммоль) и воду (10 мл). Реакционную смесь подвергали реакции при 80°С под защитой газообразного азота в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-1:1) с получением 16а (124 мг, выход 88%).
Синтез Промежуточного соединения 16b:
Соединение 16а (36 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (10 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (71 мг, 0,29 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (ПО мг, 0,29 ммоль) и диизопропилэтиламин (57 мг, 0,44 ммоль). Реакционную смесь перемешивали и подвергали реакции при 25°С в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (15 мл). Смесь экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением 16b (35 мг, выход 49%).
Синтез конечного продукта 16:
16b (35 мг, 0,074 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К смеси добавляли насыщенный водный раствор карбоната натрия. Значение рН реакционной смеси доводили до 9-10. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли и концентрировали до объема около 20 мл. Добавляли триэтиламин (2 мл) и уксусный ангидрид (2 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 30 минут. Для промывки органической фазы добавляли водный раствор карбоната натрия. Водную фазу экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (этилацетат) с получением конечного продукта 16 (15 мг, выход 49%).
MS m/z (ESI): 416,2 [М+Н]+.
1H ЯМР (600 МГц, CD3OD) δ 8.12 (s, 1H), 8.08 (d, J=5.4 Гц, 1H), 7.19 (d, 7=8.4 Гц, 1H), 6.64 (s, 1H), 6.60 (s, 1H), 3.88 (s, 3Н), 3.79 (s, 3Н), 3.71 (t, J=4.8 Гц, 2Н), 2.71-2.57 (m, 1H), 2.24-2.22 (m, 1H), 1.91 (s, 3Н), 1.89 (s, 2Н), 1.49-1.19 (m, 6Н).
Пример 17:
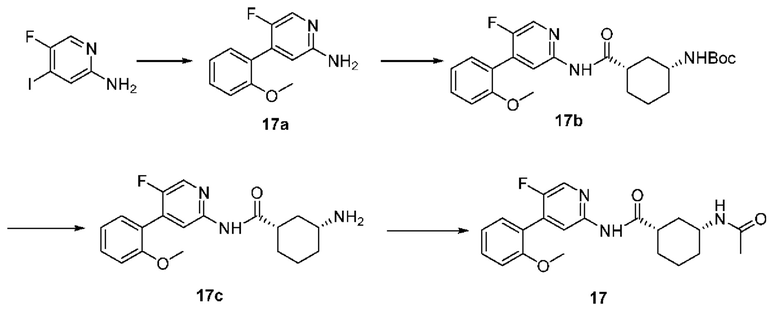
Синтез Промежуточного соединения 17а:
2-метоксифенилбороновую кислоту (0,42 г, 2,77 ммоль) растворяли в диметиловом эфире диэтиленгликоля (30 мл) и воде (6 мл) и затем добавляли 5-фтор-4-йодпиридин-2-амин (0,60 г, 2,52 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (95 мг, 0,13 ммоль) и карбонат калия (1,04 г, 7,56 ммоль). Реакционную смесь перемешивали и подвергали реакции при 100°С в атмосфере азота в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:3) с получением 17а (0,60 г, выход 99%).
Синтез Промежуточного соединения 17b:
17а (0,60 г, 2,77 ммоль) растворяли в N,N-диметилформамиде (50 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,79 г, 3,20 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,22 г, 3,20 ммоль) и диизопропилэтиламин (1,2 мл, 7,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:3) с получением 17b (0,60 г, выход 49%).
Синтез Промежуточного соединения 17с:
17b (0,60 г, 1,35 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли трифторуксусную кислоту (3 мл). Реакционную смесь перемешивали и подвергали реакции при 25°С до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли водный раствор карбоната натрия (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт 17 с может быть непосредственно использован в следующей стадии реакции.
Синтез конечного продукта 17:
17с растворяли в дихлорметане (50 мл) и затем добавляли триэтиламин (0,4 мл, 2,70 ммоль) и уксусный ангидрид (0,3 мл, 2,70 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, и исходный материал полностью прореагировал, что контролировали с помощью ТСХ. К смеси добавляли насыщенный водный раствор карбоната натрия. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением конечного продукта 17 (0,35 г, выход 68%).
MS m/z (ESI): 386,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.55 (s, 1H), 8.30 (s, 1Н), 8.07 (d, J=5.4 Гц, 1H), 7.76 (d, J=7.8 Гц, 1H), 7.46 (t, J=7.8 Гц, 1H), 7.27 (d, J=7.2 Гц, 1H), 7.15 (d, J=8.4 Гц, 1H), 7.06 (t, J=7.8 Гц, 1H), 3.75 (s, 3Н), 3.55-3.54 (m, 1H), 2.56-2.48 (m, 1H), 1.86-1.84 (m, 1H), 1.77-1.74 (m, 6Н), 1.31-1.02 (m, 4Н).
Пример 18:
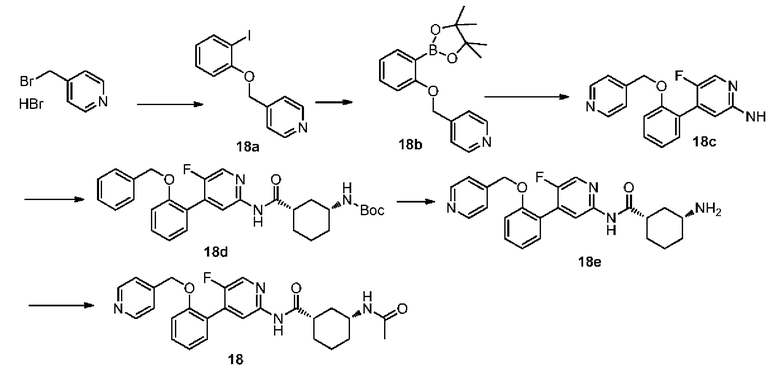
Синтез Промежуточного соединения 18а:
Гидробромид 4-(бромметил)пиридина (1,72 г, 6,81 ммоль) растворяли в N,N-диметилформамиде (20 мл) и затем добавляли 2-йодофенол (1,50 г, 6,81 ммоль), карбонат калия (2,83 г, 20,45 ммоль) и йодид натрия (1,02 г, 6,81 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (петролейный эфир:этилацетат = 2:1-петролейный эфир:этилацетат = 1:1) с получением 18а (1,90 г, выход 90%).
Синтез Промежуточного соединения 18b:
18а (1,00 г, 3,21 ммоль) растворяли в диметиловом эфире этиленгликоля (15 мл) и затем добавляли бис(пинаколато)дибор (980 мг, 3,85 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (234 мг, 0,32 ммоль) и ацетат калия (0,95 г, 9,64 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (петролейный эфир:этилацетат = 2:1-петролейный эфир:этилацетат = 1:1) с получением 18b (0,80 г, выход 80%).
Синтез Промежуточного соединения 18с:
5-фтор-4-йодпиридин-2-амин (802 мг, 3,37 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и затем добавляли 18b (700 мг, 2,25 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (168 мг, 0,23 ммоль) и карбонат калия (932 мг, 6,75 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1-дихлорметан:метанол = 10:1) с получением 18 с (220 мг, выход 33%).
Синтез Промежуточного соединения 18d:
18с (220 мг, 0,75 ммоль) растворяли в N,N-диметилформамиде (10 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (217 мг, 0,89 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (340 мг, 0,89 ммоль) и N,N-диметилэтиламин (192 мг, 1,49 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-дихлорметан:метанол = 20:1) с получением 18d (300 мг, выход 77%).
Синтез Промежуточного соединения 18е:
18d (0,30 г, 0,58 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-дихлорметан:метанол = 8:1) с получением 18е (0,16 г, выход 66%).
Синтез конечного продукта 18:
18е (160 мг, 0,38 ммоль) растворяли в дихлорметане (5 мл) и затем добавляли уксусный ангидрид (116 мг, 1,14 ммоль) и триэтиламин (115 мг, 1,14 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-петролейный эфир:этилацетат = 2:1) с получением конечного продукта 18 (60 мг, выход 34%).
MS m/z (ESI): 463,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.61 (s, 1H), 8.50 (d, J=5.4 Гц, 2Н), 8.36 (s, 1Н), 8.20 (d, J=5.4 Гц, 1H), 7.80 (d, J=7.8 Гц, 1H), 7.47-7.44 (m, 1H), 7.35 (d, J=7.2 Гц, 1H), 7.30 (d, J=5.4 Гц, 2Н), 7.18 (d, J=9.0 Гц, 1H), 7.15 (t, J=7.8 Гц, 1H), 5.22 (s, 2Н), 3.58-3.57 (m, 1H), 2.62-2.53 (m, 1H), 1.89-1.75 (m, 7Н), 1.30-1.04 (m, 4Н).
Пример 19:
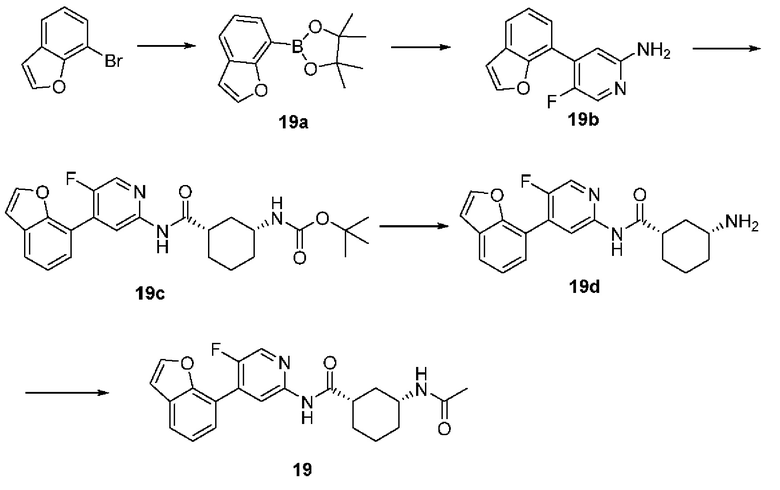
Синтез Промежуточного соединения 19а:
7-бромбензофуран (600 мг, 3,00 ммоль) растворяли в диметиловом эфире этиленгликоля (50 мл) и затем добавляли бис(пинаколато)дибор (928 мг, 3,70 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия дихлорметана (163 мг, 0,20 ммоль) и ацетат калия (892 мг, 9,10 ммоль). Реакционную смесь нагревали до 100°С под защитой газообразного азота и подвергали реакции в течение 4 часов. Исходный материал полностью прореагировал, что контролировалось с помощью ТСХ. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 100:1-50:1) с получением 19а (450 мг, выход 62%).
Синтез Промежуточного соединения 19b:
19а (450 мг, 1,84 ммоль) растворяли в диметиловом эфире этиленгликоля (50 мл) и воде (10 мл) и затем добавляли 5-фтор-4-йодпиридин-2-амин (350 мг, 1,47 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия дихлорметана (ПО мг, 0,15 ммоль) и карбонат калия (405 мг, 2,94 ммоль). Реакционную смесь нагревали до 100°С под защитой газообразного азота и подвергали реакции в течение 4 часов. Исходный материал полностью прореагировал, что контролировалось с помощью ТСХ. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением 19b (300 мг, выход 71%).
Синтез Промежуточного соединения 19 с:
(1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (292 мг, 1,20 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли пиридин (474 мг, 6,00 ммоль) и тионилхлорид (242 мг, 2,04 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов, и затем к указанному выше реакционному раствору добавляли 19b (300 мг, 1,32 ммоль). Реакцию продолжали проводить при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор разбавляли водой (30 мл) и экстрагировали. Органическую фазу собирали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением 19 с (200 мг, выход 39%).
Синтез Промежуточного соединения 19d:
19с (200 мг, 0,47 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли трифторуксусную кислоту (1 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 1,5 часов до полной конверсии исходного материала, что контролировалось с помощью ТСХ. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 19d (128 мг, выход 83%).
Синтез конечного продукта 19:
19d (128 мг, 0,39 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли уксусный ангидрид (48 мг, 0,47 ммоль) и триэтиламин (47 мг, 0,47 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 1,5 часов до полной конверсии исходного материала, что контролировалось с помощью ТСХ. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 19 (117 мг, выход 76%).
MS m/z (ESI): 396,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.68 (s, 1H), 8.48 (s, 1H), 8.41 (d, J=5.4 Гц, 1Н), 8.09 (s, 1H), 7.83-7.78 (m, 2Н), 7.46 (d, J=7.2 Гц, 1Н), 7.41 (dd, J=7.2 Гц, 7=7.2 Гц, 1H), 7.09 (s, 1H), 3.58-3.56 (m, 1H), 2.64-2.60 (m, 1H), 1.90-1.84 (m, 1H), 1.78-1.76 (m, 6Н), 1.31-1.25 (m, 3Н), 1.10-1.06 (m, 1H).
Пример 20:
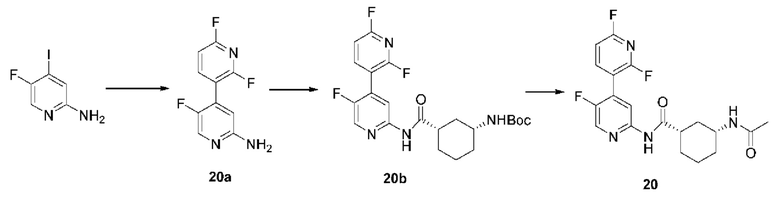
Синтез Промежуточного соединения 20а:
2,6-дифторпиридин-3-бороновую кислоту (550 мг, 3,14 ммоль) и 5-фтор-4-йодпиридин-2-амин (898 мг, 3,77 ммоль) растворяли в 1,4-диоксане (15 мл), и последовательно добавляли 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (219 мг, 0,30 ммоль) и карбонат калия (1,30 г, 9,42 ммоль), и затем добавляли воду (6 мл). Реакционную смесь перемешивали и подвергали реакции в течение 5 часов при 100°С под защитой газообразного азота до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:3) с получением 20а (700 мг, выход 99%).
Синтез Промежуточного соединения 20b:
20а (700 мг, 3,14 ммоль) растворяли в ацетонитриле (50 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (778 мг, 3,20 ммоль), гексафторфосфат тетраметилхлорформамидиния (898 мг, 3,20 ммоль) и N-метилимидазол (0,87 мл, 11,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Растворитель удаляли концентрированием и полученный неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:3) с получением 20b (1,40 г, выход 99%).
Синтез конечного продукта 20:
20b (1,40 г, 3,14 ммоль) растворяли в дихлорметане (50 мл) и добавляли трифторуксусную кислоту (5 мл) при комнатной температуре. Реакционную смесь подвергали реакции в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь промывали насыщенным водным раствором карбоната натрия до слабой щелочности и экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель концентрировали до 50 мл и затем добавляли триэтиламин (2 мл) и уксусный ангидрид (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь промывали безводным раствором карбоната натрия и затем экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией с получением конечного продукта 20 (750 мг, выход 61%).
MS m/z (ESI): 393,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.74 (s, 1H), 8.50 (s, 1H), 8.40 (dd, J=16.8 Гц, J=7.8 Гц, 1H), 8.26 (d, J=6.0 Гц, 1Н), 7.79 (d, J=7.8 Гц, 1H), 7.41 (dd, J=7.8 Гц, J=2.4 Гц, 1H), 3.60-3.55 (m, 1H), 2.64-2.60 (m, 1H), 1.91-1.88 (m, 1H), 1.78-1.76 (m, 6Н), 1.29-1.27 (m, 3Н), 1.09-1.06 (m, 1H).
Пример 21:
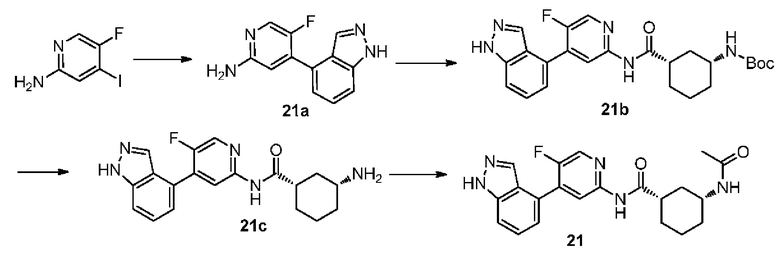
Синтез Промежуточного соединения 21а:
5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и воде (4 мл) и затем добавляли индазол-4-бороновую кислоту (0,68 г, 4,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,34 г, 0,42 ммоль) и карбонат калия (1,74 г, 12,60 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением 21а (0,77 г, выход 80%).
Синтез Промежуточного соединения 21b:
21а (0,75 г, 3,30 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,80 г, 3,30 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,56 г, 4,10 ммоль) и N,N-диизопропилэтиламин (0,85 г, 6,60 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением 21b (0,91 г, выход 60%).
Синтез Промежуточного соединения 21с:
21b (0,91 г, 2,00 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-8:1) с получением 21 с (0,61 г, выход 86%).
Синтез конечного продукта 21:
21с (0,61 г, 1,72 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,53 г, 5,20 ммоль) итриэтиламин (0,52 г, 5,20 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 21 (0,32 г, выход 47%).
MS m/z (ESI): 396,1 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 8.60 (s, 1H), 8.27 (s, 1H), 8.14 (s, 1H), 7.63 (d, 7=7.8 Гц, 1H), 7.51 (d, 7=7.8 Гц, 1H), 7.39 (d, 7=6.6 Гц, 1H), 7.15 (d, 7=7.2 Гц, 1H), 3.89-3.87 (m, 1Н), 2.54-2.50 (m, 1H), 2.29 (d, 7=12.0 Гц, 1H), 1.79-1.77 (m, 6H), 1.44-1.28 (m, 3H), 1.12-1.09 (m, 1H).
Пример 22:
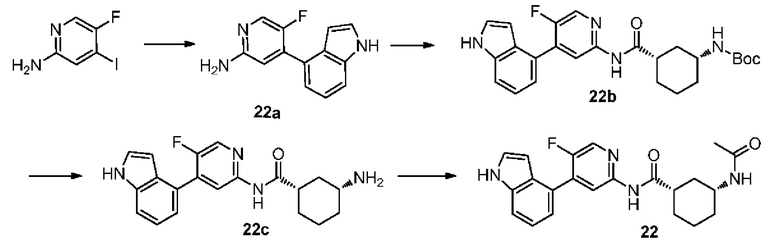
Синтез Промежуточного соединения 22а:
5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и воде (4 мл) и затем добавляли индол-4-бороновую кислоту (0,68 г, 4,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,34 г, 0,42 ммоль) и карбонат калия (1,74 г, 12,60 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением 22а (0,78 г, выход 82%).
Синтез Промежуточного соединения 22b:
22а (0,78 г, 3,40 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,83 г, 3,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,56 г, 4,10 ммоль) и N,N-диизопропилэтиламин (0,88 г, 6,80 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением 22b (0,92 г, выход 59%).
Синтез Промежуточного соединения 22 с:
22b (0,92 г, 2,03 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-8:1) с получением 22с (0,61 г, выход 85%).
Синтез конечного продукта 22:
22с (0,61 г, 1,73 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,53 г, 5,20 ммоль) итриэтиламин (0,52 г, 5,20 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 22 (0,35 г, выход 51%). MS m/z (ESI): 395,1 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 11.38 (s, 1Н), 10.59 (s, 1H), 8.42 (s, 1Н), 8.36 (d, J=6.0 Гц, 1H), 7.77 (d, J=7.8 Гц, 1Н), 7.54 (d, J=8.4 Гц, 1H), 7.46 (d, J=3.0 Гц, 1H), 7.23 (d, J=8.4 Гц, 1H), 7.15 (d, J=7.2 Гц, 1H), 6.41 (s, 1H), 3.60-3.54 (m, 1H), 2.64-2.60 (m, 1H), 1.89 (d, J=12.0 Гц, 1H), 1.77-1.76 (m, 6Н), 1.34-1.23 (m, 3Н), 1.10-1.08 (m, 1Н).
Пример 23:
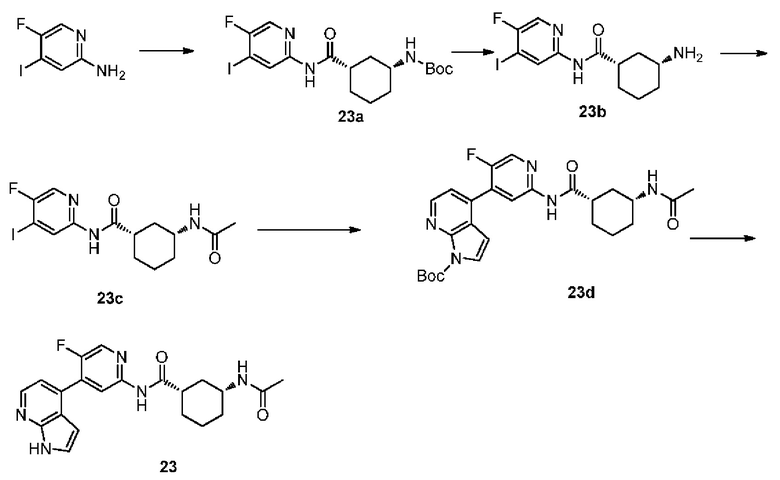
Синтез Промежуточного соединения 23а:
5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль) растворяли в N,N-диметилформамиде (20 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (1,32 г, 5,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (2,39 г, 6,29 ммоль) и М,М-диметилэтиламин (2,08 мл, 12,6 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением 23а (806 мг, выход 41%).
Синтез Промежуточного соединения 23b:
23а (806 мг, 1,74 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли трифторуксусную кислоту (5 мл) на бане со льдом и водой. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении с получением 23b (770 мг, неочищенный продукт).
Синтез Промежуточного соединения 23с:
23b (770 мг, неочищенный продукт) растворяли в дихлорметане (10 мл) и затем добавляли триэтиламин (883 мкл, 6,35 ммоль) и уксусный ангидрид (297 мкл, 3,18 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (этилацетат) с получением 23 с (490 мг, выход 69%).
Синтез Промежуточного соединения 23d:
23с (100 мг, 0,25 ммоль) растворяли в 1,4-диоксане (10 мл) и воде (5 мл) и затем добавляли трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин-1-карбоксилат (167 мг, 0,49 ммоль), [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий (15 мг, 0,02 ммоль) и карбонат калия (68 мг, 0,49 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением 23d (122 мг, выход 99%).
Синтез конечного продукта 23:
23d (122 мг, 0,24 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли трифторуксусную кислоту (5 мл) на бане со льдом и водой. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10. Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением конечного продукта 23 (57 мг, выход 59%).
MS m/z (ESI): 396,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 11.97 (s, 1Н), 10.70 (s, 1H), 8.42 (s, 1Н), 8.41 (s, 1H), 8.36 (d, J=4.8 Гц, 1H), 7.79 (d, J=7.8 Гц, 1H), 7.63-7.62 (m, 1H), 7.23 (d, J=4.2 Гц, 1H), 6.47 (s, 1H), 3.58-3.56 (m, 1H), 2.62-2.61 (m, 1H), 1.99 (d, 7=4.8 Гц, 1H), 1.91-1.89 (m, 6Н), 1.39-1.21 (m, 3Н), 1.20-1.09 (m, 1Н).
Пример 24:
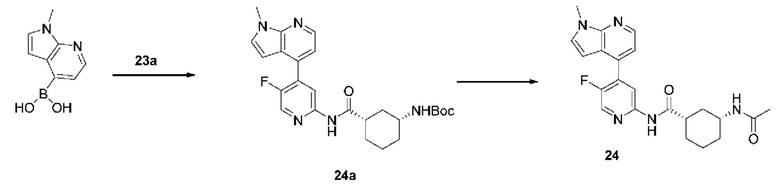
Синтез Промежуточного соединения 24а:
(1-метил-1Н-пирроло[2,3-b]пиридин-4-ил)бороновую кислоту (176 мг, 1,00 ммоль) и 23а (461 мг, 1,00 ммоль) растворяли в 1,4-диоксане (20 мл) и последовательно добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (73 мг, 0,10 ммоль) и карбонат калия (414 мг, 3,00 ммоль). Реакционную смесь перемешивали и подвергали реакции при 100°С под защитой газообразного азота в течение 4,5 часов, и исходный материал полностью прореагировал, что контролировали с помощью ТСХ. К реакционной смеси добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-петролейный эфир:этилацетат = 1:1) с получением 24а (400 мг, выход 86%).
Синтез конечного продукта 24:
24а (400 мг, 0,86 ммоль) растворяли в дихлорметане (25 мл) и добавляли трифторуксусную кислоту (3 мл) при комнатной температуре. Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь промывали насыщенным раствором карбоната натрия до слабой щелочности. Водную фазу экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Растворитель концентрировали до объема 15 мл. Добавляли триэтиламин (2 мл) и уксусный ангидрид (2 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 30 минут до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь промывали насыщенным раствором карбоната натрия и растворитель удаляли путем концентрирования. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол = 50:1-10:1) с получением конечного продукта 24 (169 мг, выход 48%).
MS m/z (ESI): 410,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.69 (s, 1H), 8.50 (s, 1H), 8.41 (s, 1H), 8.40 (s, 1H), 7.77 (d, J=7.8 Гц, 1H), 7.68 (d, J=3.6 Гц, 1H), 7.26 (d, J=4.8 Гц, 1H), 6.47 (d, J=2.4 Гц, 1Н), 3.89 (s, 3H), 3.58-3.56 (m, 1H), 2.64-2.60 (m, 1Н), 1.91-1.89 (m, 1H), 1.79-1.77 (m, 6H), 1.30-1.26 (m, 3Н), 1.09-1.07 (m, 1H).
Пример 25:
Синтез конечного продукта 25:
5b (0,61 г, 1,69 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли метилсульфонилхлорид (0,29 г, 2,54 ммоль) и триэтиламин (0,34 г, 3,38 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 25 (0,41 г, выход 55%).
MS m/z (ESI): 440,1 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.60 (s, 1H), 8.33 (s, 1Н), 8.08 (d, J=4.8 Гц, 1H), 7.34 (d, J=7.2 Гц, 1H), 7.10 (d, J=7.8 Гц, 2Н), 6.92 (d, J=8.4 Гц, 1H), 3.80 (s, 3Н), 3.13-3.11 (m, 1H), 2.92 (s, 3H), 2.62-2.57 (m, 1H), 2.03 (d,.7=12 Гц, 1H), 2.25 (d, 7=12 Гц, 1H), 1.77-1.73 (m, 2H), 1.36-1.11 (m, 4H).
Пример 26:
Синтез Промежуточного соединения 26а:
5-фтор-4-йодпиридин-2-амин (0,50 г, 2,10 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл) и воде (4 мл) и затем добавляли 4,5-дифтор-2-метоксифенилбороновую кислоту (0,39 г, 2,10 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,15 г, 0,21 ммоль) и карбонат калия (0,87 г, 6,3 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением 26а (0,40 г, выход 75%).
Синтез Промежуточного соединения 26b:
26а (0,30 г, 1,18 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,35 г, 1,42 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,54 г, 1,42 ммоль) и N,N-диметилэтиламин (0,30 г, 2,36 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением 26b (0,33 г, выход 58%).
Синтез Промежуточного соединения 26с:
26b (0,20 г, 0,42 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-8:1) с получением 26 с (0,11 г, выход 70%).
Синтез конечного продукта 26:
26с (0,11 г, 0,29 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли метилсульфонилхлорид (0,40 г, 0,35 ммоль) и триэтиламин (44 мг, 0,46 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 26 (40 мг, выход 30%).
MS m/z (ESI): 458,1 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 8.59 (s, 1H), 8.31 (s, 1Н), 8.14 (d, J=10.8 Гц, 1H), 7.17 (d, J=9.0 Гц, 1H), 6.85-6.82 (m, 1H), 5.46 (s, 1H), 3.88 (s, 1H), 3.83 (s, 3H), 2.82 (s, 1H), 2.48 (s, 1H), 2.27-2.25 (m, 1H), 2.05-1.94 (m, 5H), 1.45-1.16 (m, 4H).
Пример 27:
Синтез конечного продукта 27:
5b (0,61 г, 1,69 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли тиофенсульфонилхлорид (0,46 г, 2,53 ммоль) и триэтиламин (0,34 г, 3,38 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 27 (0,52 г, выход 61%).
MS m/z (ESI): 508,1 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.55 (s, 1H), 8.31 (s, 1H), 8.03 (s, 1H), 7.96 (d, J=7.2 Гц, 1H), 7.89 (s, 1H), 7.57 (s, 1H), 7.32 (d, J=7.2 Гц, 1Н), 7.15 (s, 1Н), 7.08 (d, J=11.4 Гц, 1H), 6.89 (d, J=8.4 Гц, 1H), 4.02 (s, 3Н), 3.04-3.03 (m, 1Н), 1.78-1.62 (m, 5Н), 1.17-1.08 (m, 4Н).
Пример 28:
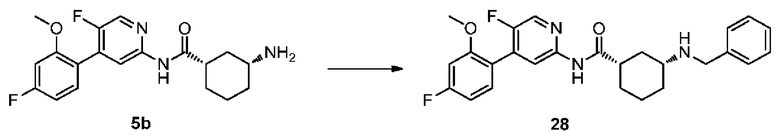
Синтез конечного продукта 28:
5b (79 мг, 0,22 ммоль) растворяли в ацетонитриле (5 мл) и добавляли карбонат натрия (47 мг, 0,44 ммоль) и бензилбромид (37 мг, 0,22 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Растворитель удаляли концентрированием. Неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол = 10:1) с получением конечного продукта 28 (30 мг, выход 30%).
MS:(m/z, ESI): 451,2 [М+Н]+.
1Н ЯМР (600 МГц, CD3OD) δ 8.16 (s, 1H), 8.09 (d, J=4.8 Гц, 1H), 7.37-7.32 (m, 4Н), 7.30-7.26 (m, 2Н), 6.93 (d, J=11.4 Гц, 1H), 6.80 (dd, J=8.4 Гц, 1H), 3.86 (s, 2Н), 3.81 (s, 3Н), 2.71-2.67 (m, 1H), 2.51-2.47 (m, 1H), 2.20-2.18 (m, 1H), 2.06-2.04 (m, 1H), 1.89-1.85 (m, 2Н), 1.49-1.39 (m, 3Н), 1.21-1.16 (m, 1H).
Пример 29:
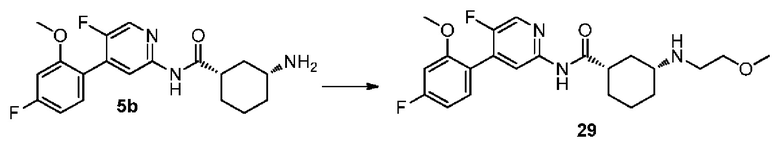
Синтез конечного продукта 29:
5b (0,61 г, 1,69 ммоль) растворяли в N,N-диметилформамиде (35 мл) и затем добавляли бромэтилметиловый эфир (0,26 г, 1,86 ммоль) и карбонат калия (0,47 г, 3,38 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 29 (0,42 г, выход 59%).
MS m/z (ESI): 420,2 [М+Н]+.
1H ЯМР (600 МГц, CDCl3) δ 8.25 (d, J=5.4 Гц, 1H), 8.12 (s, 1H), 7.25 (d, J=6.6 Гц, 1H), 6.75-6.70 (m, 2Н), 3.80 (s, 3Н), 3.57 (t, J=4.8 Гц, 2Н), 3.36 (s, 3Н), 2.90 (t, J=4.8 Гц, 2Н), 2.70 (s, 1H), 2.37 (t, J=5.4 Гц, 1H), 2.26 (d, J=12.0 Гц, 1H), 2.05-1.90 (m, 3Н), 1.52-1.22 (m, 4Н).
Пример 30:

Синтез конечного продукта 30:
5b (155 мг, 0,43 ммоль) растворяли в ацетонитриле (5 мл) и добавляли триэтиламин (86 мг, 0,86 ммоль), бромэтанол (54 мг, 0,43 ммоль) и йодид натрия (10 мг, 0,06 ммоль). Реакционную смесь подвергали реакции при 60°С в течение 12 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь охлаждали и затем концентрировали для удаления растворителя. Неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол:триэтиламин = 40:2:1) с получением конечного продукта 30 (30 мг, выход 17%).
MS m/z (ESI): 406,2 [М+Н]+.
1Н ЯМР (600 МГц, CD3OD) δ 8.18 (s, 1H), 8.08 (d, J=4.8 Гц, 1H), 7.28 (d, J=7.2 Гц, 1H), 6.92 (d, J=11.4 Гц, 1H), 6.79 (d, J=8.4, 1H), 3.82-3.80 (m, 5Н), 3.20-3.25 (m, 1H), 3.20-3.18 (m, 2H), 2.65-2.62 (m, 1H), 2.31-2.29 (m, 1H), 2.18-2.16 (m, 1H), 1.99-1.91 (m, 2H), 1.70-1.64 (m, 1H), 1.53-1.34 (m, 3H).
Пример 31:
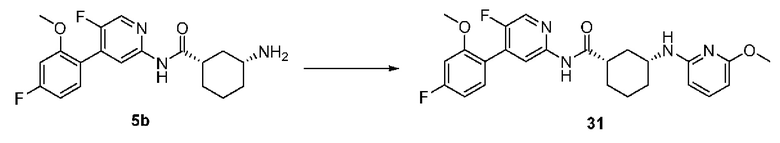
Синтез конечного продукта 31:
5b (79 мг, 0,22 ммоль) растворяли в диоксане (5 мл) и затем добавляли 2-бром-6-метоксипиридин (40 мг, 0,22 ммоль), трис(дибензилиденацетон)дипалладий (20 мг, 0,022 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (12,7 мг, 0,022 ммоль) и карбонат цезия (215 мг, 0,66 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь подвергали реакции при 120°С в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Смесь охлаждали и затем концентрировали для удаления растворителя. Неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол = 10:1) с получением конечного продукта 31 (30 мг, выход 29%).
MS:(m/z, ESI): 469,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.07 (d, J=5.4 Гц, 1H), 7.32 (d, J=7.2 Гц, 1H), 7.24 (d, J=7.8 Гц, 1H), 7.08 (d, J=11.4 Гц, 1H), 6.89 (d, J=8.4 Гц, 1H), 6.33 (d, J=7.2 Гц, 1H), 5.97 (d, J=7.8 Гц, 1Н), 5.80 (d, J=7.8 Гц, 1Н), 3.76 (s, 3Н), 3.72 (s, 3Н), 3.63-3.61 (m, 1H), 2.64-2.62 (m, 1Н), 2.06-2.04 (m, 1H), 1.97-1.95 (m, 1Н), 1.79-1.77 (m, 2Н), 1.36-1.30 (m, 3Н), 1.11-1.09 (m, 1H).
Пример 32:
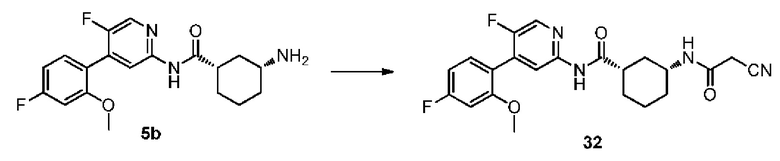
Синтез конечного продукта 32:
5b (61 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли цианоуксусную кислоту (16 мг, 0,18 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N,N,N',N'-тетраметилурония гексафторфосфат (69 мг, 0,18 ммоль) и N,N-диизопропилэтиламин (83 мкл, 0,50 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 32 (50 мг, выход 71%).
MS m/z (ESI): 429,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.31 (s, 1H), 8.20 (d, J=7.8 Гц, 1H), 8.06 (d, J=5.4 Гц, 1H), 7.32 (t, J=7.2 Гц, 1H), 7.08 (d, J=11.4 Гц, 1H), 6.90 (t, J=8.4 Гц, 1H), 3.77 (s, 3Н), 3.56 (m, 3Н), 2.61-2.57 (m, 1H), 1.89-1.76 (m, 4Н), 1.30-1.07 (m, 4Н).
Пример 33:
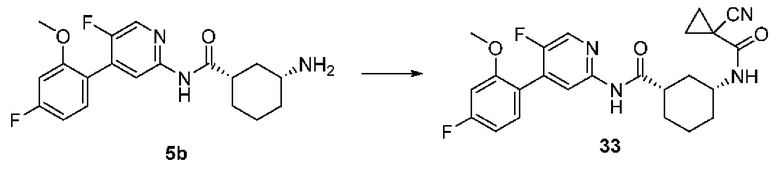
Синтез конечного продукта 33:
5b (0,16 г, 0,43 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли 1-циано-1-циклопропанкарбоновую кислоту (72 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и М,М-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан: метанол = 50:1-20:1) с получением конечного продукта 33 (96 мг, выход 49%).
MS m/z (ESI): 455,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.05 (d, J=4.8 Гц, 1H), 8.09 (d, J=7.8 Гц, 1Н), 7.32 (dd, J=7.8 Гц, J=7.2 Гц, 1Н), 7.09 (d, J=11.4 Гц, 1H), 6.90 (dd, J=8.4 Гц, J=7.8 Гц, 1Н), 3.77 (s, 3Н), 3.68-3.56 (m, 1Н), 3.59-3.55 (m, 1H), 1.81-1.67 (m, 4Н), 1.53-1.46 (m, 5Н)), 1.28-1.23 (m, 3Н).
Пример 34:
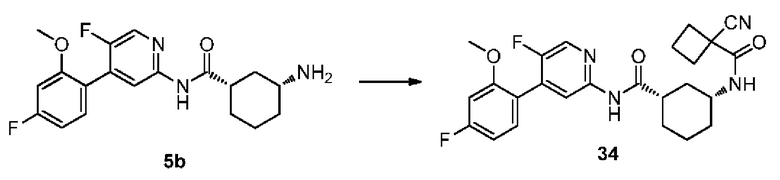
Синтез конечного продукта 34:
5b (0,16 г, 0,43 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли 1-цианоциклобутанкарбоновую кислоту (81 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и М,К-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 34 (119 мг, выход 59%).
MS m/z (ESI): 469,2 [М+Н]+.
1H ЯМР(600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.05 (d, J=4.8 Гц, 1Н), 8.09 (d, J=7.8 Гц, 1H), 7.34-7.31 (m, 1H), 7.09 (s, 1H), 6.91-6.88 (m, 1H), 3.77 (s, 3Н), 3.68-3.56 (m, 1H), 2.59-2.55 (m, 1H), 1.81-1.67 (m, 4Н), 1.53-1.46 (m, 5Н), 1.30-1.23 (m, 5Н).
Пример 35:
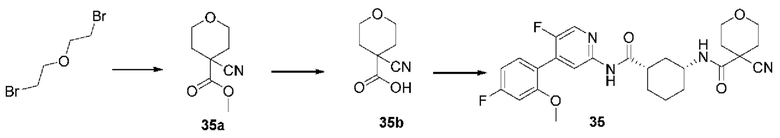
Синтез Промежуточного соединения 35а:
2,2'-дибромдиэтиловый эфир (12,85 г, 55,60 ммоль) растворяли в N,N-диметилформамиде (10 мл) и затем добавляли метилцианоацетат (5,00 г, 50,45 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (11,50 г, 75,68 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при 85°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 1:1) с получением 35а (5,00 г, выход 59%).
Синтез Промежуточного соединения 35b:
35а (2,37 г, 14,00 ммоль) растворяли в этаноле (36 мл) и воде (5 мл) и затем добавляли гидроксид натрия (2,24 г, 56,00 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь доводили водным раствором бикарбоната натрия до рН 8. Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 35b (1,50 г, выход 69%).
Синтез конечного продукта 35:
35b (123 мг, 0,79 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли (1S,3R)-3-амино-N-(5-фтор-4-(4-фтор-2-метоксифенил)пиридин-2-ил)циклогексанкарбоксамид (260 мг, 0,72 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (548 мг, 1,44 ммоль) и М,М-диизопропилэтиламин (357 мкл, 2,16 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 35 (214 мг, выход 60%).
MS m/z (ESI): 499,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.33 (s, 1H), 8.17 (d, J=7.8 Гц, 1H), 8.08 (d, J=6.0 Гц, 1H), 7.35-7.33 (m, 1H), 7.11-7.09 (m, 1H), 6.93-6.89 (m, 1H), 3.88-3.86 (m, 2Н), 3.78 (s, 3Н), 3.66-3.63 (m, 1H), 3.52-3.51 (m, 2Н), 2.63 (m, 1H), 1.97-1.96 (m, 4Н), 1.85-1.75 (m, 4Н), 1.38-1.21 (m, 4Н).
Пример 36:
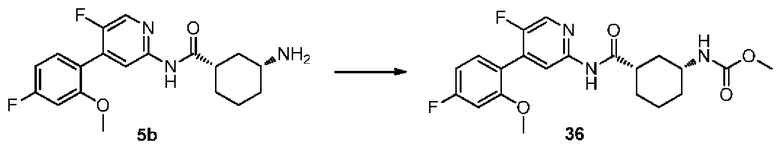
Синтез конечного продукта 36:
Трифосген (33 мг, 0,11 ммоль) растворяли в дихлорметане (2 мл) и затем по каплям добавляли раствор 5b (61 мг, 0,17 ммоль) и триэтиламин (22 мг, 0,22 ммоль), растворенный в дихлорметане (2 мл). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 2 часов, и затем добавляли раствор метанола (2 мл). Реакция продолжалась в течение 3 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 36 (30 мг, выход 42%).
MS m/z (ESI): 420,2[М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1Н), 8.06 (d, J=5.4 Гц, 1Н), 7.33 (d, J=7.8 Гц, 1H), 7.10-7.07 (m, 2Н), 6.90 (d, J=8.4 Гц, 1H), 3.77 (s, 3Н), 3.48 (s, 3Н), 3.32 (m, 1H), 2.56 (m, 1H), 1.87-1.71 (m, 4H), 1.31-1.06 (m, 4H).
Пример 37:
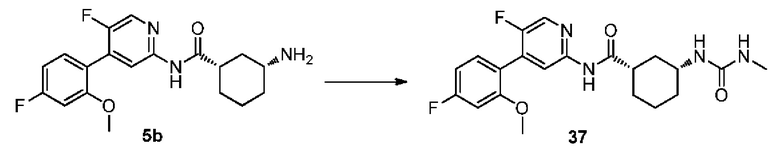
Синтез конечного продукта 37:
Трифосген (33 мг, 0,11 ммоль) растворяли в дихлорметане (2 мл) и затем по каплям добавляли раствор 5b (61 мг, 0,17 ммоль) и триэтиламин (22 мг, 0,22 ммоль), растворенный в дихлорметане. Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 2 часов, и затем добавляли раствор метиламина в этаноле (33%, 3 мл). Реакция продолжалась в течение 3 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 37 (15 мг, выход 21%).
MS m/z (ESI): 419,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.06 (d, J=5A Гц, 1Н), 7.33 (t, J=8.4 Гц, 1H), 7.10-7.07 (m, 1H), 6.91-6.88 (m, 1H), 5.78 (d, J=8.4 Гц, 1H), 5.56 (d, J=4.8 Гц, 1H), 3.76 (s, 3H), 3.36-3.34 (m, 1H), 2.65-2.55 (m, 1H), 2.51 (s, 3H), 1.88-1.73 (m, 4H), 1.28-0.82 (m, 4H).
Пример 38:
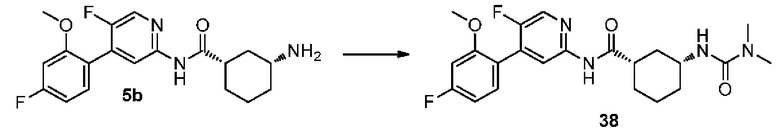
Синтез конечного продукта 38:
5b (0,61 г, 1,69 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли диметилкарбаминхлорид (0,27 г, 2,54 ммоль) и триэтиламин (0,34 г, 3,38 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 100:1-40:1) с получением конечного продукта 38 (0,38 г, выход 53%).
MS m/z (ESI): 433,1 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 8.28-8.25 (m, 2Н), 8.11 (s, 1H), 7.27-7.24 (m, 2H), 6.76-6.70 (m, 2H), 3.81 (s, 3Н), 3.76-3.73 (m, 1H), 2.88 (s, 6H), 2.47-2.43 (m, 1H), 2.30 (d, J=12.0 Гц, 1H), 2.02-1.90 (m, 3Н), 1.47-1.12 (m, 4H).
Пример 39:
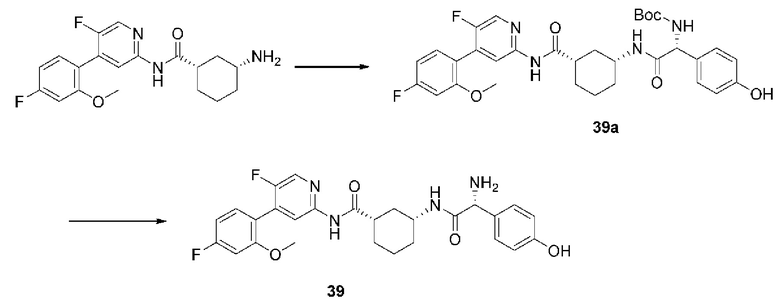
Синтез Промежуточного соединения 39а:
5b (0,12 г, 0,33 ммоль) растворяли в N,N-диметилформамиде (10 мл) и добавляли (S)-2-((трет-бутилоксикарбонил)амино)-2-(4-гидроксифенил)уксусную кислоту (0,10 г, 0,36 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,15 г, 0,40 ммоль), N,N-диизопропилэтиламин (0,09 г, 0,66 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (20 мл). Смесь экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Органическую фазу очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 10:1) с получением 39а (0,12 г, выход 60%).
Синтез конечного продукта 39:
39а (0,12 г, 0,20 ммоль) растворяли в дихлорметане (20 мл) и добавляли трифторуксусную кислоту (4 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор промывали водой (20 мл×3) и водные фазы объединяли. Объединенную водную фазу доводили карбонатом натрия до рН 8-9 и экстрагировали дихлорметаном (30 мл×3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Органическую фазу удаляли выпариванием при пониженном давлении с получением конечного продукта 39 (0,04 г, выход 39%).
MS m/z (ESI): 511,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 9.23 (s, 1H), 8.32 (s, 1H), 8.08 (d, J=5.4 Гц, 1H), 7.90-7.89 (m, 1H), 7.36-7.34 (m, 1Н), 7.17-7.15 (m, 2Н), 7.12-7.10 (m, 1Н), 6.93-6.91 (m, 1H), 6.68-6.66 (m, 2Н), 4.19 (s, 1H), 4.04 (d, J=7.2 Гц, 2Н), 3.80 (s, 3Н), 3.58-3.57 (m, 1H), 2.59-2.57 (m, 1H), 1.80-1.76 (m, 4Н), 1.33-1.25 (m, 4Н).
Пример 40:
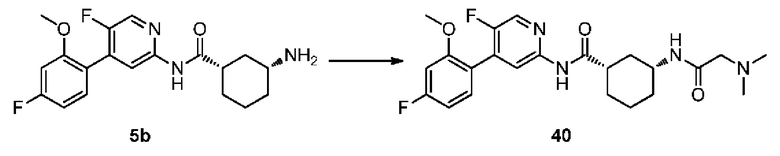
Синтез конечного продукта 40:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли N,N-диметилглицин (67 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 40 (100 мг, выход 41%).
MS m/z (ESI): 447,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1Н), 8.06 (d, J=4.2 Гц, 1H), 7.58 (d, J=8.4 Гц, 1H), 7.32 (dd, J=7.8 Гц, J=7.2 Гц, 1H), 7.08(d, J=10.8 Гц, 1H), 6.90 (dd, J=7.8 Гц, J=7.8 Гц, 1H), 3.76 (s, 3Н), 3.65-3.63 (m, 1H), 2.84-2.77 (m, 2Н), 2.66-2.60 (m, 1H), 2.16 (s, 6Н), 1.85-1.84 (m, 1H), 1.75-1.69 (m, 3Н), 1.41-1.35 (m, 1H), 1.32-1.23 (m, 3Н).
Пример 41:
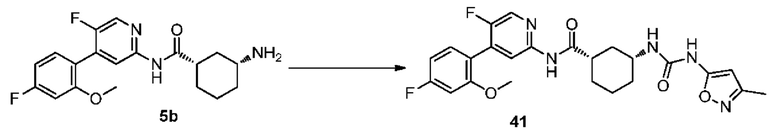
Синтез соединения 41:
Трифосген (0,027 г, 0,09 ммоль) растворяли в безводном тетрагидрофуране (20 мл) и добавляли раствор 3-метилизоксазол-5-амина (0,027 г, 0,28 ммоль) в тетрагидрофуране и триэтиламин (0,028 г, 0,28 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 6 часов. К реакционному раствору добавляли раствор 5b (0,10 г, 0,28 ммоль) в тетрагидрофуране и триэтиламин (0,028 г, 0,28 ммоль). Реакционную смесь нагревали до 70°С и подвергали реакции в течение 6 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь охлаждали до комнатной температуры. Реакционный раствор очищали колоночной хроматографией (петролейный эфир:этилацетат = 1:1) с получением конечного продукта 41 (45 мг, выход 33%).
MS m/z (ESI): 486,2 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 8.65 (s, 1Н), 8.29 (s, 1Н), 8.12 (s, 1H), 6.76-6.71 (m, 2Н), 6.23 (s, 2Н), 5.43 (s, 1H), 4.03-4.01 (m, 1H), 3.81 (s, 3Н), 2.54-2.53 (m, 1Н), 2.35 (s, 3Н), 2.36-2.24 (m, 2Н), 2.05-1.94 (m, 3Н), 1.56-1.50 (m, 3Н).
Пример 42:
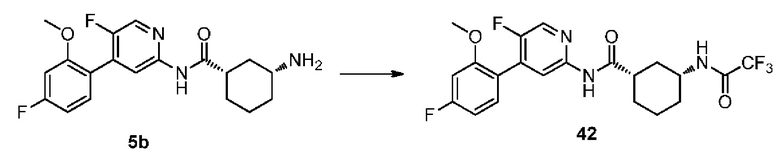
Синтез конечного продукта 42:
5b (100 мг, 0,28 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли трифторуксусный ангидрид (56 мг, 0,55 ммоль) и триэтиламин (56 мг, 0,55 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 1,5 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 42 (100 мг, выход 78%).
MS m/z (ESI): 458,1 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.59 (s, 1H), 9.33 (d, J=8.4 Гц, 1H), 8.31 (s, 1Н), 8.05 (d, J=6 Гц, 1H), 7.32 (dd, J=7.8 Гц, J=7.2 Гц, 1H), 7.08 (m, 1H), 6.89 (m, 1H), 3.76 (s, 3Н), 3.70-3.68 (m, 1H), 2.63-2.59 (m, 1H), 1.86-1.83 (m, 1H), 1.80-1.76 (m, 2Н), 1.51-1.44 (m, 1H), 1.33-1.24 (m, 4Н).
Пример 43:
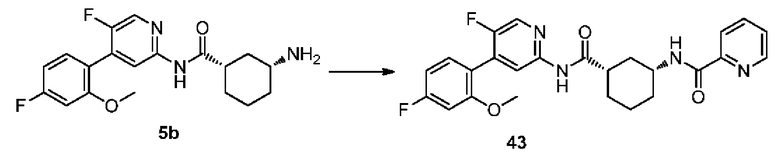
Синтез соединения 43:
5b (0,11 г, 0,30 ммоль) и 2-пиридинкарбоновую кислоту (0,04 г, 0,33 ммоль) растворяли в N,N-диметилформамиде (15 мл) и добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,14 г, 0,36 ммоль) и N,N-диизопропилэтиламин (0,08 г, 0,6 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (30 мл). Твердое вещество осаждали и фильтровали с получением конечного продукта 43 (0,07 г, 50%).
MS m/z (ESI): 467,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.63 (s, 1Н), 8.63-8.62 (m, 2H), 8.31 (s, 1H), 8.07 (d, J=5.4 Гц, 1H), 8.01 (d, J=7.8 Гц, 1H), 7.98-7.95 (m, 1H), 7.58-7.56 (m, 1H), 7.34-7.31 (m, 1H), 7.09-7.07 (m, 1H), 6.91-6.88 (m, 1H), 3.89-3.87 (m, 1H), 3.77 (s, 3H), 2.71-2.63 (m, 1H), 1.92-1.91 (m, 1H), 1.82-1.77 (m, 3H), 1.61-1.55 (m, 1H), 1.45-1.28 (m, 3H).
Пример 44:
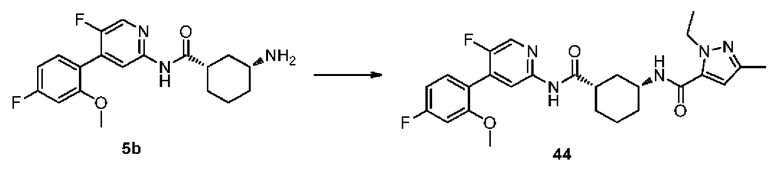
Синтез соединения конечного продукта 44:
5b (188 мг, 0,52 ммоль) и 1-этил-3-метил-1Н-пиразол-4-карбоновую кислоту (161 мг, 1,04 ммоль) растворяли в N,N-диметилформамиде (5 мл). Последовательно добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (395 мг, 1,04 ммоль) и диизопропилэтиламин (0,26 мл, 1,56 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением конечного продукта 44 (230 мг, выход 88%).
MS m/z (ESI): 498,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.63 (s, 1H), 8.34 (s, 1H), 8.22 (d, J=8.4 Гц, 1H), 8.09 (d, J=6.0 Гц, 1Н), 7.35 (m, 1Н), 7.11 (m, 1Н), 6.92 (m, 1Н), 6.59 (s, 1H), 4.38 (dd, J=7.2 Гц,7=3.0 Гц, 2Н), 3.79 (s, 4Н), 2.15 (s, 3Н), 2.00 (d,J=11.4ni, 1H), 1.84-1.80 (m, 4Н), 1.49-1.46 (m, 2Н), 5 1.32-1.30 (m, 5Н).
Пример 45:
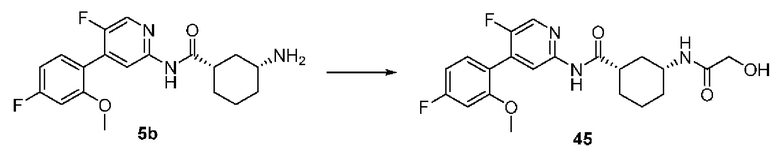
Синтез конечного продукта 45:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли гидроксиуксусную кислоту (16 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметлурония гексафторфосфат (80 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 45 (30 мг, выход 52%).
MS m/z (ESI): 420,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.06 (d, J=5.4 Гц, 1H), 7.56 (d, J=9.0 Гц, 1H), 7.34-7.31 (m, 1H), 7.09-7.07 (m, 1H), 6.91-6.88 (m, 1H), 5.38-5.36 (m, 1H), 3.77-3.75 (m, 5Н), 3.66-3.63 (m, 1H), 2.61-2.57 (m, 1H), 1.83-1.68 (m, 4Н), 1.34-1.26 (m, 4Н).
Пример 46:
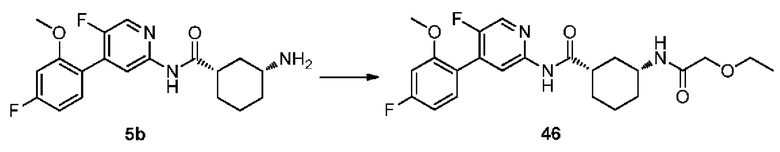
Синтез конечного продукта 46:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли этоксиуксусную кислоту (68 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 46 (96 мг, выход 39%).
MS m/z (ESI): 448,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.33 (s, 1Н), 8.07 (d, J=5.4 Гц, 1H), 7.59 (d, J=8.4 Гц, 1H), 7.35-7.33 (m, 1H), 7.11-7.09 (m, 1H), 6.93-6.90 (m, 1H), 3.78-3.76 (m, 5Н), 3.69-3.67 (m, 1Н), 3.47-3.44 (m, 2H), 2.63-2.61 (m, 1H), 1.84-1.69 (m, 4H), 1.47-1.43(m, 1H), 1.30-1.28(m, 6H).
Пример 47:
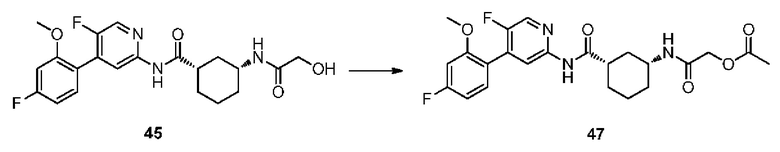
Синтез конечного продукта 47:
45 (0,20 г, 0,48 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли ацетилхлорид (45 мг, 0,57 ммоль) и триэтиламин (97 мг, 0,96 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 47 (100 мг, выход 45%).
MS m/z (ESI): 462,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.33 (s, 1H), 8.07 (d, J=5.4 Гц, 1H), 7.59 (d, J=8.4 Гц, 1H), 7.35-7.34 (m, 1H), 7.33-7.11 (m, 1H), 7.09-6.90 (m, 1H), 4.39 (s, 2Н), 3.78 (s, 3Н), 3.65-3.63 (m, 1H), 2.66-2.60 (m, 1H), 2.58 (s, 3Н), 1.85-1.84 (m, 1Н), 1.75-1.69 (m, 3Н), 1.39-1.24 (m, 3Н), 1.16-1.14 (m, 1H).
Пример 48:
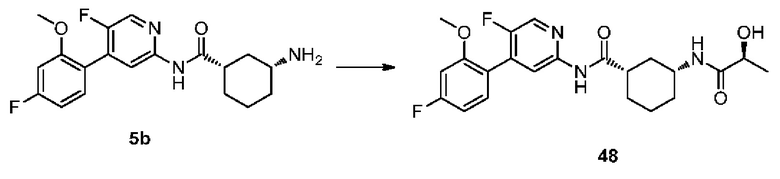
Синтез конечного продукта 48:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли L-молочную кислоту (58,5 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл×2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении.
Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 48 (96 мг, выход 40%).
MS m/z (ESI): 434,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.57 (s, 1H), 8.31 (s, 1H), 8.05 (d, J=6 Гц, 1H), 7.51 (d, J=8.4 Гц, 1H), 7.34-7.31 (m, 1H), 7.10-7.07 (m, 1H), 6.91-6.88 (m, 1H), 5.38 (d, J=5.4 Гц, 1H), 3.92-3.88 (m, 1H), 3.77 (s, 3Н), 3.62-3.59 (m, 1H), 2.60-2.56 (m, 1H), 1.83-1.81 (m, 1H), 1.77-1.72 (m, 2Н), 1.41-1.35 (m, 1H), 1.31-1.26 (m, 4Н), 1.23 (d, J=13.2 Гц, 3Н).
Пример 49:
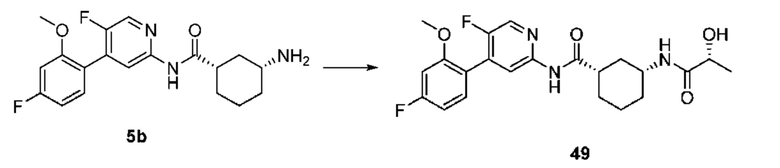
Синтез конечного продукта 49:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли D-молочную кислоту (58,5 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 49 (100 мг, выход 42%).
MS m/z (ESI): 434,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.57 (s, 1Н), 8.30 (s, 1H), 8.05 (d, J=4.8 Гц, 1H), 7.51 (d, J=7.8 Гц, 1H), 7.33(t, J=7.8 Гц, 1H), 7.08 (d, J=11.4 Гц, 1H), 6.89 (t, J=7.8 Гц, 1H), 5.38 (d, J=5.4 Гц, 1H), 3.92-3.88 (m, 1H), 3.77 (s, 3Н), 3.62-3.59 (m, 1H), 2.60-2.56 (m, 1H), 1.81 (d,J=12 Гц, 1H), 1.77-1.72 (m, 2Н), 1.67 (d, J=11.4 Гц, 1H), 1.41-1.35 (m, 1H), 1.31-1.25 (m, 4Н), 1.23(s, 3Н).
Пример 50:
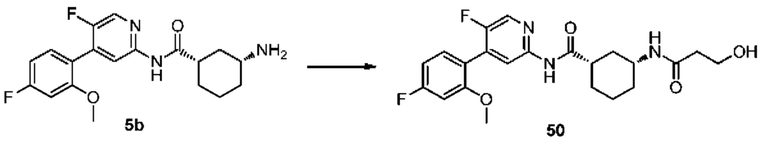
Синтез конечного продукта 50:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 3-гидроксилпропионовую кислоту (19 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=40:1-20:1) с получением конечного продукта 50 (32 мг, выход 53%).
MS m/z (ESI): 434,2[М+Н]+.
1Н ЯМР (600МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.31 (s, 1H), 8.06-8.05 (d, J=5.4 Гц, 1Н), 7.57-7.55 (d, J=9.0 Гц, 1Н), 7.34-7.31 (m, 1Н),7.09-7.07 (m, 1Н), 6.91-6.88 (m, 1H), 5.38-5.36 (t, J=5.4 Гц, 1Н), 3.77-3.75 (m, 5H), 3.66-3.63 (m, 1H), 2.61-2.56 (m, 3H), 1.83-1.68 (m, 4H), 1.34-1.26 (m, 4H).
Пример 51:
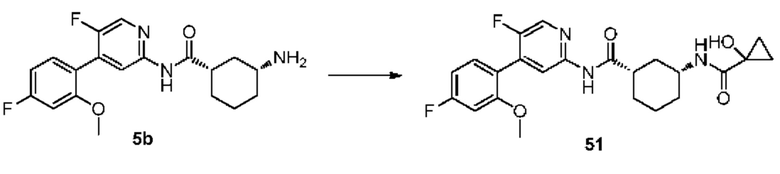
Синтез конечного продукта 51:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 1-гидроксициклопропанкарбоновую кислоту (21 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 51 (36 мг, выход 59%).
MS m/z (ESI): 446,2[М+Н]+.
1Н ЯМР (600МГц, ДМСО-d6) δ 10.61 (s, 1H), 8.34 (s, 1H), 8.09-8.08 (m, 1H), 7.73-7.71 (m, 1H), 7.37-7.34 (m, 1H),7.13-7.10 (m, 1H),6.95-6.91 (m, 1H), 3.80 (s, 3H), 2.61-2.55 (m, 2H), 1.85-1.72 (m, 4H), 1.50-1.47 (m, 1H), 1.32-1.30 (m, 3H), 1.00-0.98 (m, 2H), 0.81-0.80 (m, 2H).
Пример 52:
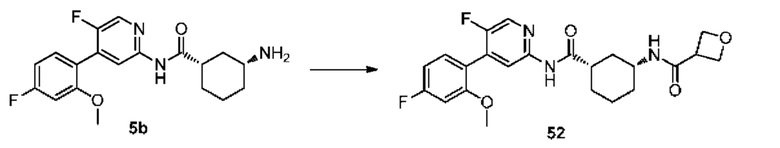
Синтез конечного продукта 52:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 3-оксетанкарбоновую кислоту (21 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=40:1-20:1) с получением конечного продукта 52 (36 мг, выход 59%).
MS m/z (ESI): 446,2[М+Н]+.
1Н ЯМР (600МГц, ДМСО-d6) δ 10.59 (s, 1H), 8.33 (s, 1H), 8.07-8.06 (m, 1H), 7.98-7.97 (m, 1H), 7.35-7.32 (m, 1H), 7.10-7.08 (m, 1H), 6.93-6.91 (m, 1H), 4.61-4.56 (m, 2Н), 3.78 (s, 3Н), 3.69-3.67 (m, 2Н), 3.42-3.40 (m, 1H), 2.69-2.55 (m, 2Н), 1.89-1.87 (m, 1H), 1.77-1.75 (m, 3Н), 1.29-1.25 (m, 3Н), 1.18-1.09 (m, 1H).
Пример 53:
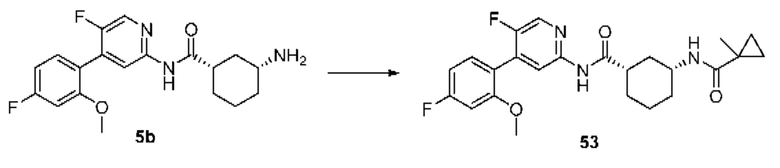
Синтез конечного продукта 53:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 1-метилциклопропан-1-карбоновую кислоту (21 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол=40:1-20:1) с получением конечного продукта 53 (40 мг, выход 65%).
MS m/z (ESI): 444,2[М+Н]+.
1H ЯМР (600МГц, ДМСО-d6) δ 10.57 (s, 1H), 8.32 (s, 1H), 8.08-8.06 (m, 1H), 7.34-7.32 (m, 1Н), 7.24-7.22 (m, 1H), 7.11-7.09 (m, 1H), 6.93-6.91 (m, 1Н), 3.78 (s, 3Н), 3.66-3.58 (m, 1H), 2.59-2.50 (m, 1H), 1.80-1.69 (m, 4Н), 1.47-1.44 (m, 1H), 1.30-1.22 (m, 6Н), 0.92-0.90 (m, 2Н), 0.46-0.44 (m, 2Н).
Пример 54:
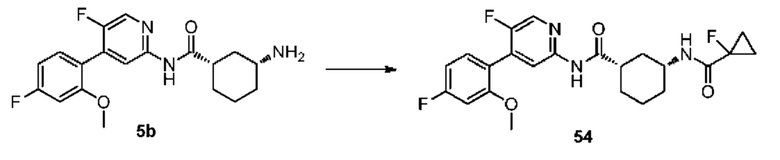
Синтез конечного продукта 54:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 1-фторциклопропанкарбоновую кислоту (22 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 54 (37 мг, выход 60%).
MS m/z (ESI): 448,2[М+Н]+.
1Н ЯМР (600МГц, CDCl3) δ 8.23-8.20 (m, 1H), 8.13 (s, 1H), 7.86 (s, 1H), 7.27-7.25 (m, 1H), 6.80-6.70 (m, 2H), 6.40-6.25 (m, 1H), 4.00-3.85 (m, 1H), 3.82 (s, 3H), 2.50-2.40 (m, 1H), 2.35-2.25 (m, 1H), 2.10-1.90 (m, 3H), 1.55-1.45 (m, 3H), 1.40-1.30 (m, 2H), 1.25-1.15 (m, 3H).
Пример 55:
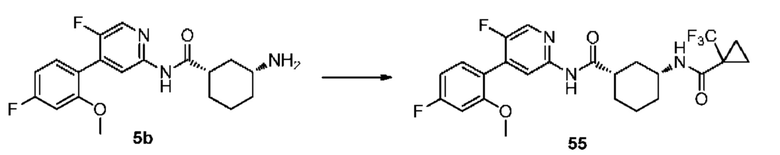
Синтез конечного продукта 55:
5b (50 мг, 0,138 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 1-трифторметилциклопропан-1-карбоновую кислоту (32 мг, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (79 мг, 0,21 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,42 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 55 (45 мг, выход 65%).
MS m/z (ESI): 498,2[М+Н]+.
1Н ЯМР (600МГц, CDCl3) δ 8.23-8.20 (m, 1Н), 8.13 (s, 1H), 7.86 (s, 1H), 7.27-7.25 (m, 1H), 6.79-6.69 (m, 2H), 6.05-5.95 (m, 1H), 4.00-3.86(m, 1H), 3.82 (s, 3H), 2.50-2.35 (m, 1H), 2.30-2.20 (m, 1H), 2.05-1.90 (m, 3H), 1.56-1.35 (m, 5H), 1.21-1.15 (m, 3H).
Пример 56:
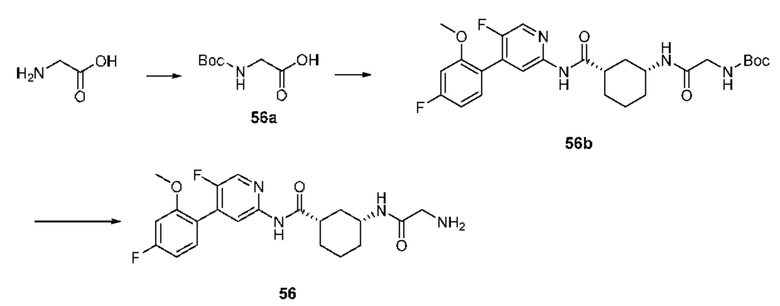
Синтез Промежуточного соединения 56а:
Глицин (500 мг, 6,70 ммоль) растворяли в диоксане (10 мл) и воде (10 мл) и добавляли триэтиламин (1,40 г, 14,40 ммоль) и ди-трет-бутилдикарбонат (1,74 г, 8,00 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи. Исходный материал полностью прореагировал, что контролировалось с помощью ТСХ. Смесь концентрировали. Добавляли водный раствор карбоната натрия (100 мл). Смесь промывали этилацетатом (100 × 3). Водную фазу доводили разбавленной соляной кислотой до рН 3 и экстрагировали этилацетатом (100 × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением 56а (1,00 г, выход 85%).
Синтез Промежуточного соединения 56b:
56а (46 мг, 0,26 ммоль) растворяли в N,N-диметилформамиде (3 мл) и добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (125 мг, 0,33 ммоль) и диизопропилэтиламин (57 мг, 0,44 ммоль). Реакционную смесь перемешивали в течение 10 минут и затем добавляли 5b (78 мг, 0,22 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение ночи. Исходный материал полностью прореагировал, что контролировалось с помощью ТСХ. К реакционной смеси добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали. Неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол = 10:1) с получением 56b (80 мг, выход 70%).
Синтез конечного продукта 56:
56b (80 мг, 0,22 ммоль) растворяли в дихлорметане (4 мл) и добавляли трифторуксусную кислоту (1 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов до полной конверсии исходного материала, что контролировалось с помощью ТСХ. Реакционную смесь концентрировали.
Неочищенный продукт очищали тонкослойной хроматографией (дихлорметан:метанол = 10:1) с получением конечного продукта 56 (40 мг, выход 43%).
MS m/z(ESI): 419,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.59 (s, 1H), 8.31 (s, 1Н), 8.05 (d, J=6.0 Гц, 1H), 7.94 (br, 2Н), 7.32-7.30 (m, 1H), 7.09-7.07 (m, 1Н), 6.91-6.88 (m, 1Н), 3.76 (s, 3Н), 3.64-3.59 (m, 1H), 3.15(d,J=4.8 Гц, 2Н), 2.62-2.59 (m, 1H), 1.89-1.87 (m, 1H), 1.79-1.77 (m, 3Н), 1.34-1.28 (m, 3Н), 1.18-1.10 (m, 1Н).
Пример 57:
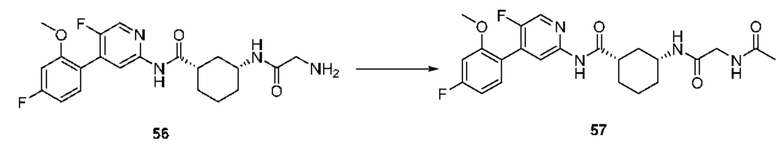
Синтез конечного продукта 57:
Соединение 56 (205 мг, 0,49 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (149 мг, 1,47 ммоль) и триэтиламин (148 мг, 1,47 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением конечного продукта 57 (138 мг, выход 61%).
MS m/z (ESI): 461,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.60 (s, 1H), 8.33 (s, 1H), 8.07 (d, J=6.0 Гц, 1H), 7.95-7.92 (m, 2Н), 7.35-7.33 (m, 1Н), 7.11 (d, J=1.8 Гц, 1Н), 6.93-6.90 (m, 1H), 4.09 (s, 2Н), 3.79 (s, 3Н), 3.58-3.56 (m, 1H), 2.59-2.50 (m, 1H), 1.86-1.73 (m, 4Н), 1.83 (s, 3Н), 1.39-1.14 (m, 4Н).
Пример 58:
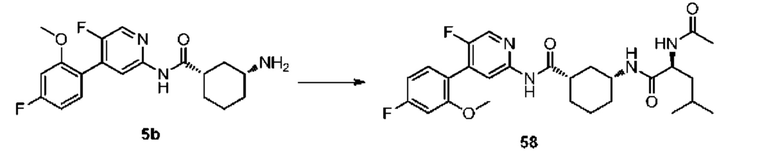
Синтез конечного продукта 58:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли N-ацетил-L-лейцин (112 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 58 (190 мг, выход 67%).
MS m/z (ESI): 517,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.60(s, 1H), 8.33 (s, 1H), 8.07 (d, J=6.0 Гц, 1H), 7.94 (dd, J=10.2, Гц, 7=8.4 Гц, 2Н), 7.35(dd, J=7.8 Гц, J=7.2 Гц, 1H), 7.11-7.10 (m, 1Н), 6.93-6.90 (m, 1H), 4.24-4.23 (m, 1H), 3.79(s, 3Н), 3.58-3.56 (m, 1H), 2.59-2.50 (m, 1H), 1.83-1.71 (m, 7Н), 1.53-1.51 (m, 1H), 1.38-1.36 (m, 2Н), 1.31-1.28 (m, 6Н), 0.91 (m, 6Н).
Пример 59:
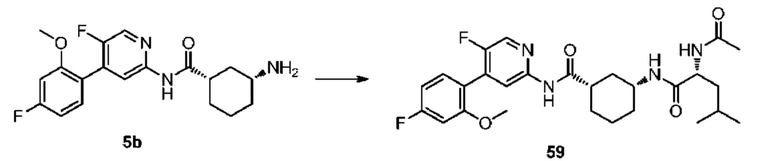
Синтез конечного продукта 59:
5b (200 мг, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли N-ацетил-D-лейцин (112 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 59 (181 мг, выход 64%).
MS m/z (ESI): 517,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.59 (s, 1H), 8.33 (s, 1Н), 8.07 (d, J=6.0 Гц, 1H), 7.93 (dd, J=10.2, Гц, J=8.4 Гц, 2Н), 7.34 (dd, J=7.8 Гц, J=7.2 Гц, 1Н), 7.11-7.10 (m, 1H), 6.92-6.89 (m, 1H), 4.24-4.23 (m, 1H), 3.80 (s, 3Н), 3.57-3.55 (m, 1H), 2.59-2.50 (m, 1H), 1.83-1.71 (m, 7Н), 1.53-1.51 (m, 1H), 1.38-1.36 (m, 2Н), 1.30-1.27 (m, 6Н), 0.91 (m, 6Н).
Пример 60:
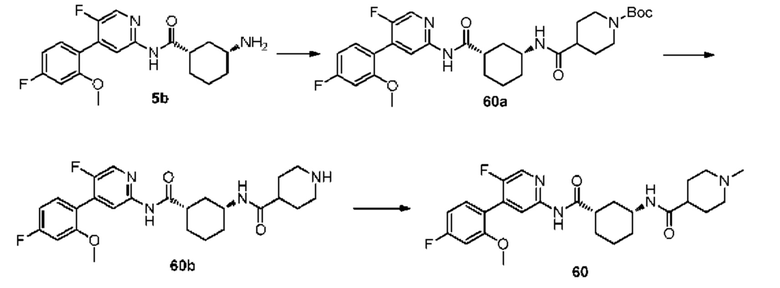
Синтез Промежуточного соединения 60а:
5b (500 мг, 1,385 ммоль) растворяли в N,N-диметилформамиде (15 мл) и затем добавляли l-Boc-4-пиперидинкарбоновую кислоту (380 мг, 1,66 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (787 мг, 2,07 ммоль) и N,N-диизопропилэтиламин (687 мкл, 4,15 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь перемешивали в течение 30 минут, фильтровали и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении с получением 60а (623 мг, выход 79%).
Синтез Промежуточного соединения 60b:
60а (623 мг, 1,09 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении с получением 60b (500 мг, выход 97%).
Синтез конечного продукта 60:
60b (300 мг, 0,63 ммоль) растворяли в метаноле (10 мл) и затем добавляли водный раствор формальдегида (1 мл), триацетоксиборгидрид натрия (269 мг, 1,27 ммоль) и одну каплю уксусной кислоты. Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением конечного продукта 60 (214 мг, выход 70%).
MS m/z (ESI): 487,2[М+Н]+.
1Н ЯМР (600МГц, ДМСО-d6) δ 10.59 (s, 1H), 8.34 (s, 1H), 8.08 (d, J=4.8 Гц, 1H), 7.68 (d, J=7.8 Гц, 1H), 7.35 (d, J=7.8 Гц, 1H), 7.11 (d, J=11.4 Гц, 1H), 6.93 (d,J=8.4Hi, 1H), 3.79 (s, 3Н), 3.58-3.56 (m, 1H), 2.76-2.74 (m, 2Н), 2.62-2.58 (m, 1H), 2.51 (s, 3Н), 1.99-1.96 (m, 1H), 1.86-1.84 (m, 1H), 1.81-1.72 (m, 5Н), 1.57-1.53 (m, 4Н), 1.31-1.24 (m, 3Н), 1.18-1.08 (m, 1H).
Пример 61:
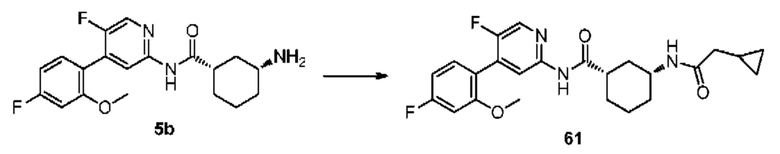
Синтез конечного продукта 61:
5b (0,07 г, 0,19 ммоль) растворяли в N,N-диметилформамиде (10 мл) и добавляли циклопропилуксусную кислоту (0,02 г, 0,21 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,09 г, 0,23 ммоль) и N,N-диизопропилэтиламин (0,05 г, 0,38 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (20 мл). Смесь экстрагировали этилацетатом (15 × 3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Смесь концентрировали при пониженном давлении и неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 20:1) с получением конечного продукта 61 (0,04 г, выход 47%).
MS m/z (ESI): 444,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 5 8.33 (s, 1Н), 8.07 (d, J=5.4 Гц, 1H), 7.63 (d, J=8.4 Гц, 1H), 7.35-7.33 (m, 1H), 7.11-7.01 (m, 1H), 6.93-6.90 (m, 1H), 3.79 (s, 3Н), 3.60-3.57 (m, 1H), 2.60-2.61 (m, 1H), 1.94-1.93 (m, 2H), 1.88-1.75 (m, 4H), 1.32-1.23 (m, 4H), 1.10-1.08 (m, 1H), 0.54-0.45 (m, 2H), 0.19-0.15 (m, 2H).
Пример 62:
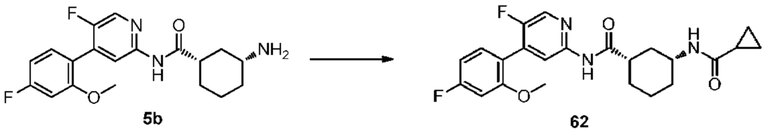
Синтез конечного продукта 62:
5b (0,07 г, 0,19 ммоль) растворяли в дихлорметане (20 мл) и добавляли циклопропилкарбоновую кислоту (0,02 г, 0,21 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (0,07 г, 0,38 ммоль) и 4-диметиламинопиридин (2,3 мг, 0,019 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор последовательно промывали водой и водным раствором бикарбоната натрия и сушили над безводным сульфатом натрия. Органическую фазу очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1) с получением конечного продукта 62 (0,04 г, выход 48%).
MS m/z (ESI): 430,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.32 (s, 1H), 8.08 (d, J=5.4 Гц, 1H), 7.99 (d, J=8.4 Гц, 1Н), 7.36-7.33 (m, 1Н), 7.11-7.09 (m, 1Н), 6.93-6.90 (m, 1Н), 3.79 (s, 3Н), 3.60-3.58 (m, 1H), 2.59-2.50 (m, 1H), 1.89-1.77 (m, 4Н), 1.50-1.47 (m, 1H), 1.34-1.26 (m, 3Н), 1.10-1.08 (m, 1H), 0.64-0.60 (m, 4Н).
Пример 63:
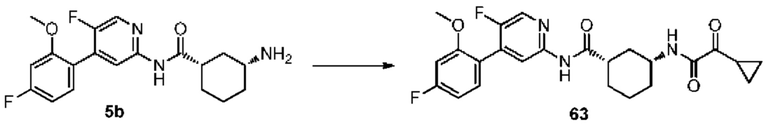
Синтез конечного продукта 63:
5b (0,10 г, 0,28 ммоль) растворяли в N,N-диметилформамиде (25 мл) и затем добавляли 2-циклопропил-2-карбонилуксусную кислоту (35 мг, 0,31 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (118 мг, 0,31 ммоль) и N,N-диизопропилэтиламин (0,07 г, 0,55 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 100:1-40:1) с получением конечного продукта 63 (54 мг, выход 43%).
MS m/z (ESI): 458,1 [М+Н]+.
1H ЯМР (600 МГц, CDCl3) δ 8.55 (s, 1H), 8.32 (d, J=6.0 Гц, 1H), 8.11 (s, 1Н), 7.28-7.25 (m, 1H), 6.94 (d, J=8.4 Гц, 1H), 6.77-6.71 (m, 2H), 3.86-3.82 (m, 1H), 3.81 (s, 3H), 3.10-3.08 (m, 1H), 2.48-2.46 (m, 1H), 2.25 (d, J=12.0 Гц, 1H), 2.04-1.95 (m, 3H), 1.53-1.47 (m, 3H), 1.26-1.24 (m, 1H), 1.18-1.15 (m, 4H).
Пример 64:
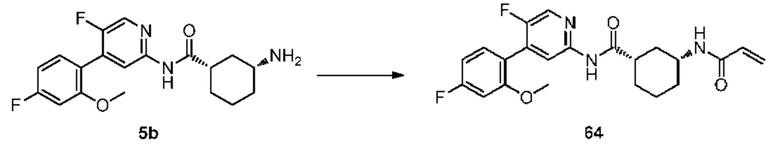
Синтез конечного продукта 64:
5b (0,16 г, 0,44 ммоль) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (0,05 г, 0,53 ммоль). На бане со льдом добавляли акрилоилхлорид (0,05 г, 0,53 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (30 мл). Смесь экстрагировали дихлорметаном (20 × 3). Органические фазы объединяли и сушили над безводным сульфатом натрия. Смесь концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1) с получением конечного продукта 64 (0,10 г, выход 55%).
MS m/z (ESI): 416,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.59 (s, 1H), 8.31 (s, 1H), 8.06 (d, J=6.0 Гц, 1H), 8.02 (d, J=7.8 Гц, 1H), 7.34-7.31(m, 1H), 7.10-7.07 (m, 1H), 6.91-6.89 (m, 1H), 6.18-6.14 (m, 1H), 6.06-6.03 (m, 1H), 5.55-5.53 (m, 1H), 3.77 (s, 3Н), 3.65-3.63 (m, 1H), 2.61-2.60 (m, 1H), 1.90-1.77 (m, 4Н), 1.34-1.08 (m, 4Н).
Пример 65:
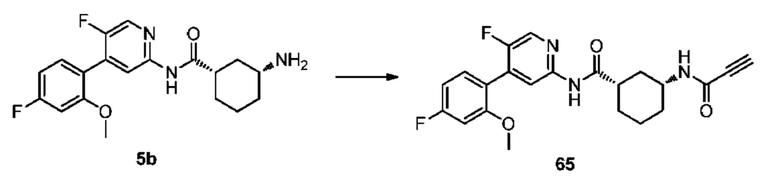
Синтез конечного продукта 65:
5b (180 мг, 0,50 ммоль) и пропиоловую кислоту (70 мг, 1,00 ммоль) растворяли в N,N-диметилформамиде (20 мл). Последовательно добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (380 мг, 1,00 ммоль) и N,N-диизопропилэтиламин (0,25 мл, 1,50 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 15 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением конечного продукта 65 (200 мг, выход 97%).
MS m/z (ESI): 414,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.60 (s, 1H), 8.73 (d, J=7.8 Гц, 1H), 8.33 (s, 1H), 8.08 (d, J=5.4 Гц, 1H), 7.35 (dd, J=8.4 Гц, J=7.8 Гц, 1H), 7.11 (dd, J=11.4 Гц, J=2.4 Гц, 1H), 6.92-6.90 (m, 1H), 4.12 (s, 1H), 3.79 (s, 3Н), 3.64-3.62 (m, 1H), 2.61-2.57 (m, 1H), 1.85-1.74 (m, 4Н), 1.35-1.23 (m, 4Н).
Пример 66:
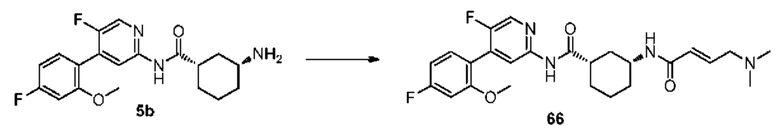
Синтез конечного продукта 66:
5b (0,25 г, 0,69 ммоль) растворяли в N,N-диметилформамиде (20 мл) и добавляли гидрохлорид транс-4-диметиламинокротоновой кислоты (0,13 г, 0,76 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,32 г, 0,83 ммоль) и N,N-диизопропилэтиламин (0,27 г, 2,07 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (30 мл). Смесь экстрагировали этилацетатом (20 × 5). Органические фазы объединяли и сушили над безводным сульфатом натрия. Органическую фазу очищали непосредственно колоночной хроматографией (дихлорметан:метанол=10:1) с получением конечного продукта 66 (0,18 г, выход 55%).
MS m/z (ESI): 472,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.61 (s, 1H), 8.33 (s, 1H), 8.08 (d, J=6.0 Гц, 1H), 8.01 (d, J=6.0 Гц, 1H), 7.35-7.33 (m, 1H), 7.11-7.09 (m, 1H), 6.93-6.90 (m, 1H), 6.56-6.51 (m, 1H), 6.04 (d, J=15.0 Гц, 1H), 3.78 (s, 3H), 3.66 (m, 1H), 3.14 (s, 2H), 2.64-2.60 (m, 1H), 2.25 (s, 6H), 1.91-1.90 (m, 1H), 1.84-1.78 (m, 3H), 1.35-1.13 (m, 4H).
Пример 67:
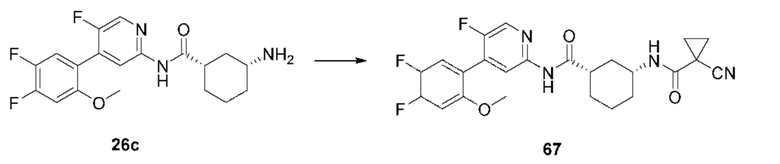
Синтез конечного продукта 67:
26 с (99 мг, 0,26 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли 1-циано-1-циклопропанкарбоновую кислоту (44 мг, 0,40 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (152 мг, 0,40 ммоль) и N,N-диизопропилэтиламин (131 мкл, 0,79 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 67 (50 мг, выход 41%).
MS m/z (ESI): 473,2 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 8.32-8.31 (m, 1H), 8.13 (s, 1H), 7.18-7.15 (m, 1H), 6.84-6.81 (m, 1H), 6.38-6.37 (m, 1H), 3.90-3.85 (m, 1H), 3.79 (s, 3H), 2.49-2.47 (m, 1H), 2.27-2.25 (m, 1H), 2.01-1.94 (m, 3H), 1.70-1.62 (m, 2H), 1.60-48 (m, 5H), 1.32-1.23 (m, 1H).
Пример 68:
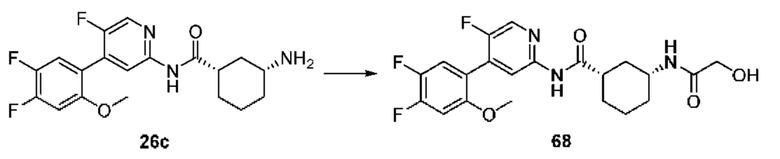
Синтез конечного продукта 68:
26 с (99 мг, 0,26 ммоль) растворяли в N,N-диметилформамиде (5 мл) и затем добавляли гидроксилуксусную кислоту (20 мг, 0,26 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (152 мг, 0,40 ммоль) и N,N-диизопропилэтиламин (131 мкл, 0,79 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 68 (50 мг, выход 44%).
MS m/z (ESI): 438,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.60 (s, 1H), 8.35 (s, 1Н), 8.08 (d, J=6.0 Гц, 1H), 7.58-7.56 (m, 1H), 7.53-7.51 (m, 1H), 7.37-7.34 (m, 1H), 5.38 (t, J=6.0 Гц, 1H), 3.82 (s, 5Н), 3.74-3.63 (m,1Н), 2.69-2.61 (m, 1H), 1.85-1.70 (m, 4Н), 1.33-1.22 (m, 4Н).
Пример 69:
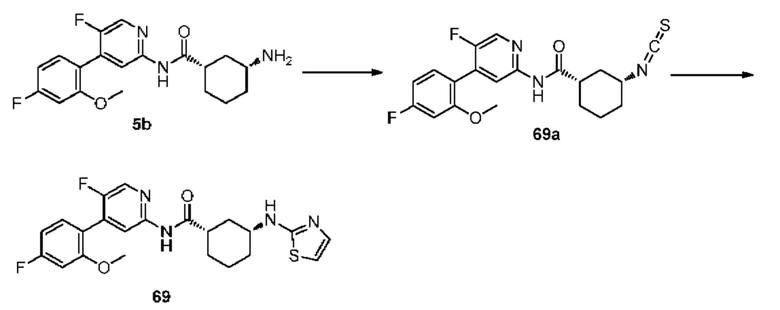
Синтез Промежуточного соединения 69а:
5b (0,07 г, 0,19 ммоль) растворяли в абсолютном этиловом спирте (20 мл) и добавляли триэтиламин (0,02 г, 0,19 ммоль) и бисульфид углерода (0,02 г, 0,26 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 30 минут. К реакционному раствору добавляли ди-трет-бутилдикарбонат (0,04 г, 0,19 ммоль) и 4-диметиламинопиридин (2,3 мг, 0,019 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Этанол удаляли при пониженном давлении. Остаток очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1) с получением 69а (0,03 г, выход 39%).
Синтез конечного продукта 69:
69а (0,03 г, 0,07 ммоль) растворяли в абсолютном этиловом спирте (10 мл) и добавляли раствор амина/этанола (2,0 М, 0,004 г, 0,28 ммоль). Реакционную смесь нагревали до 90°С и подвергали реакции в течение 4 часов в запаянной пробирке до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Растворитель удаляли при пониженном давлении. Остаток растворяли в абсолютном этиловом спирте (10 мл). Добавляли водный раствор дихлорацетальдегида (40%, 0,005 г, 0,07 ммоль). Реакционную смесь нагревали до 90°С и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Абсолютный этиловый спирт удаляли при пониженном давлении. Остаток очищали непосредственно колоночной хроматографией (дихлорметан:метанол=25:1) с получением конечного продукта 69 (0,02 г, выход 64%).
MS m/z (ESI): 445,1 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.67 (s, 1Н), 8.33 (s, 1Н), 8.33-8.08 (m, 1H), 7.50 (d, J=7.8 Гц, 1H), 7.35-7.33 (m, 1H), 7.11 (d, J=2.4 Гц, 1H), 7.09 (d, J=2.4 Гц, 1H), 6.99-6.90 (m, 1H), 6.58 (d, J=4.2 Гц, 1H), 3.79 (s, 3Н), 3.52-3.50 (m, 1H), 2.63 (m, 1H), 2.14-2.12 (m, 1H), 2.02-2.00 (m, 1H), 1.82-1.78 (m, 2Н), 1.36-1.30 (m, 3Н), 1.14-1.12 (m, 1H).
Пример 70:
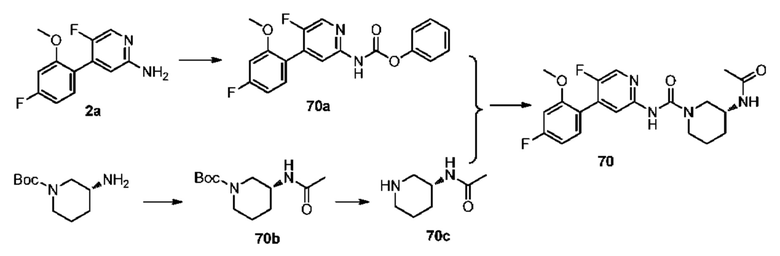
Синтез Промежуточного соединения 70а:
2а (160 мг, 0,68 ммоль) растворяли в тетрагидрофуране (10 мл), затем добавляли фенилхлорформиат (191 мг, 1,22 ммоль) и карбонат калия (187 мг, 1,35 ммоль). Реакционную смесь перемешивали в течение ночи до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (петролейный эфир:этилацетат = 10:1-2:1) с получением 70а (200 мг, выход 83%).
Синтез Промежуточного соединения 70b:
(R)-1-трет-бутилоксикарбонил-3-аминопиперидин (1,00 г, 5,00 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли триэтиламин (1,52 г, 15,0 ммоль) и уксусный ангидрид (701 мкл, 7,50 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 70b (1,17 г, выход 97%).
Синтез Промежуточного соединения 70 с:
70b (1,17 г, 4,83 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (10 мл) на бане со льдом и водой. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли при пониженном давлении с получением 70 с (1,50 г), который непосредственно использовали на следующей стадии.
Синтез конечного продукта 70:
70а (235 мг, 0,66 ммоль) растворяли в тетрагидрофуране (10 мл) и затем добавляли 70 с (339 мг, 1,32 ммоль) и триэтиламин (458 мкл, 3,30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1-20:1) с получением конечного продукта 70 (80 мг, выход 30%).
MS m/z (ESI): 405,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 9.22 (s, 1H), 8.25 (s, 1H), 7.84 (d, J=7.2 Гц, 1Н), 7.75 (d, J=5.4 Гц, 1H), 7.33 (t, J=7.8 Гц, 1H), 7.10 (d, J=11.4 Гц, 1H), 6.92 (t, J=7.8 Гц, 1Н), 3.93 (d, J=12.6 Гц, 1H), 3.83 (d, J=12.6 Гц, 1H), 3.80 (s, 3Н), 3.64-3.59 (m, 1H), 2.98-2.93 (m, 1H), 2.79-2.76 (m, 1H), 1.82-1.79 (m, 4Н), 1.71-1.69 (m, 1H), 1.45-1.36 (m, 2Н).
Пример 71:
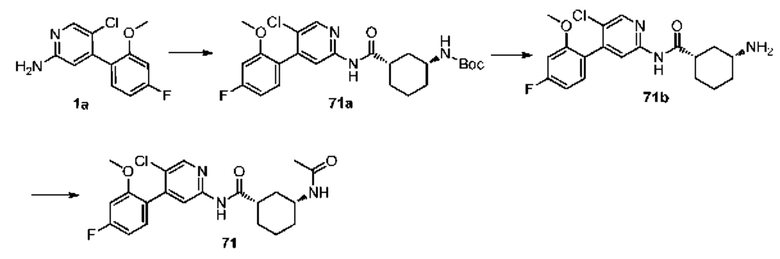
Синтез Промежуточного соединения 71а:
1а (0,40 г, 1,58 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,38 г, 1,58 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (0,72 г, 1,90 ммоль) и N,N-диизопропилэтиламин (0,41 г, 3,16 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением 71а (0,45 г, выход 60%).
Синтез Промежуточного соединения 71b:
71а (0,45 г, 0,94 ммоль) растворяли в дихлорметане (30 мл) и затем добавляли трифторуксусную кислоту (2 мл) на бане со льдом и водой. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Затем смесь доводили насыщенным водным раствором бикарбоната натрия до рН 9-10 и экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-8:1) с получением 71b (0,31 г, выход 87%).
Синтез конечного продукта 71:
71b (0,31 г, 0,82 ммоль) растворяли в дихлорметане (35 мл) и затем добавляли уксусный ангидрид (0,25 г, 2,47 ммоль) итриэтиламин (0,25 г, 2,47 ммоль). Реакционную смесь перемешивали при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением конечного продукта 71 (0,18 г, выход 52%).
MS m/z (ESI): 420,1 [М+Н]+.
1Н ЯМР (600 МГц, CDCl3) δ 10.69 (s, 1H), 8.41 (s, 1H), 8.05 (s, 1H), 7.78 (d, J=7.8 Гц, 1H), 7.25 (d, J=7.2 Гц, 1H), 7.09 (d, J=2.4 Гц, 1H), 6.91-6.88 (m, 1H), 3.76 (s, 3Н), 3.57-3.54 (m, 1H), 2.62-2.59 (m, 1H), 1.86-1.84 (m, 1H), 1.76-1.74 (m, 6H), 1.31-1.23 (m, 3H), 1.07-1.05 (m, 1H).
Пример 72:
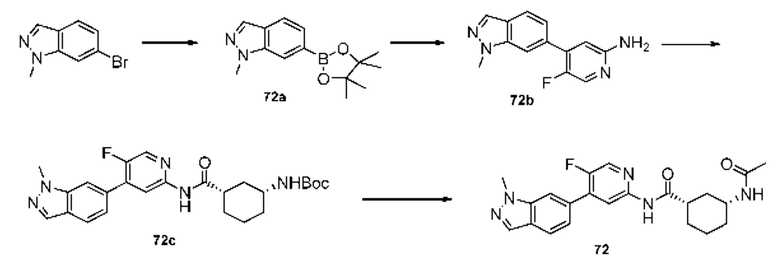
Синтез Промежуточного соединения 72а:
6-бром-1-метил-1Н-индазол (211 мг, 1,00 ммоль), бис(пинаколато)дибор (508 мг, 2,00 ммоль), ацетат калия (294 мг, 3,00 ммоль) и [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий (73 мг, 0,10 ммоль) растворяли в 1,4-диоксане (20 мл). Реакционную смесь подвергали реакции под защитой газообразного азота при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала, нагревание прекращали. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 4:1) с получением 72а (240 мг, выход 93%).
Синтез Промежуточного соединения 72b:
72а (258 мг, 1,00 ммоль) растворяли в диметиловом эфире диэтиленгликоля (40 мл) и при комнатной температуре добавляли 5-фтор-4-йодпиридин-2-амин (286 мг, 1,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (73 мг, 0,10 ммоль), карбонат калия (414 мг, 3,00 ммоль) и воду (10 мл) Реакционную смесь перемешивали при 80°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала, реакцию останавливали. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (100 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир-петролейный эфир:этилацетат = 1:1) с получением 72b (175 мг, выход 72%).
Синтез Промежуточного соединения 72 с:
72b (175 мг, 0,72 ммоль) и (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (350 мг, 1,44 ммоль) растворяли в N,N-диметилформамиде (20 мл). Последовательно добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (548 мг, 1,44 ммоль) и диизопропилэтиламин (0,36 мл, 2,16 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь экстрагировали этилацетатом (100 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир:этилацетат = 4:1-2:1) с получением 72 с (200 мг, выход 56%).
Синтез конечного продукта 72:
72 с (200 мг, 0,40 ммоль) растворяли в дихлорметане (50 мл) и затем добавляли трифторуксусную кислоту (5,00 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционной смеси добавляли насыщенный водный раствор карбоната натрия. Значение рН реакционной смеси доводили до слабощелочного. Водную фазу отделяли и экстрагировали дихлорметаном (100 × 3). Органические фазы объединяли и концентрировали до объема около 50 мл. Добавляли триэтиламин (2,00 мл) и уксусный ангидрид (2,00 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 30 минут. Для промывки органической фазы добавляли водный раствор карбоната натрия. Водную фазу отделяли и затем экстрагировали дихлорметаном (20 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (этилацетат) с получением конечного продукта 72 (135 мг, выход 83%).
MS m/z (ESI): 410,2 [М+Н]+.
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.64 (s, 1Н), 8.44 (s, 1H), 8.36 (s, 1H), 8.14 (s, 1H), 7.96 (s, 1Н), 7.90 (s, 1H), 7.79 (d, J=7.8 Гц, 1Н), 7.33 (d, J=7.8 Гц, 1H), 4.12 (s, 3Н), 3.59-3.58 (т.1H), 2.99-2.95 (m, 1H), 1.91 (s, 3Н), 1.82-1.78 (m, 4Н), 1.14-1.12 (m, 4Н).
Пример 73:
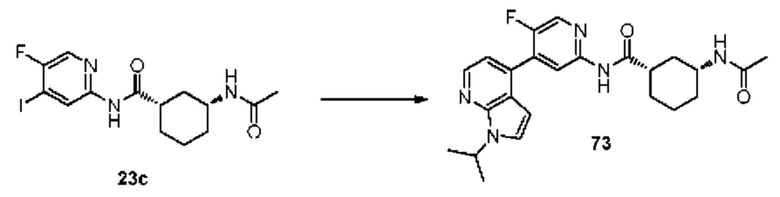
Синтез конечного продукта 73:
23 с (101 мг, 0,25 ммоль) растворяли в 1,4-диоксане (10 мл) и воде (5 мл) и затем добавляли 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин (106 мг, 0,37 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (15 мг, 0,02 ммоль) и калий карбонат (68 мг, 0,49 ммоль). Смесь дегазировали и заполняли N2 (3 раза). Реакционную смесь перемешивали и подвергали реакции при кипячении с обратным холодильником при 100°С в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1-10:1) с получением конечного продукта 73 (60 мг, выход 55%).
MS m/z (ESI): 438,2 [М+Н]+
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.70 (s, 1H), 8.51 (s, 1H), 8.41-8.38 (m, 2Н), 7.83 (d, J=3.6 Гц, 1Н), 7.79 (d, J=7.8 Гц, 1H), 7.25-7.24 (m, 1Н), 6.50-6.49 (m, 1H), 5.17-5.15 (m, 1H), 3.58-3.56 (m, 1H), 2.64-2.60 (m, 1Н), 1.90-1.88 (m, 1Н), 1.77-1.76 (m, 6Н), 1.51 (s, 3Н), 1.49 (s, 3Н), 1.40-1.25 (m, 3Н), 1.15-1.05 (m, 1Н).
Пример 74:
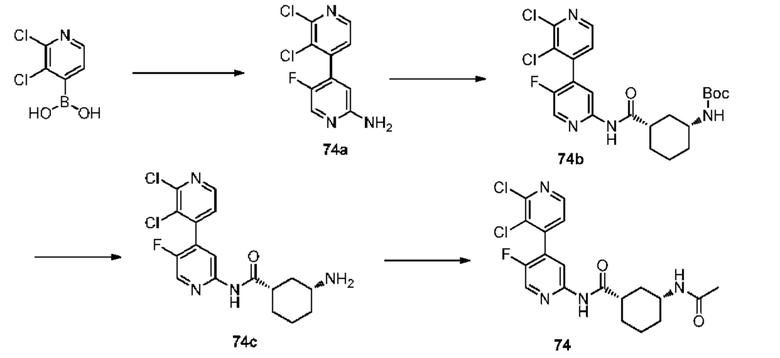
Синтез Промежуточного соединения 74а:
2,3-дихлорпиридин-4-бороновую кислоту (1,05 г, 5,50 ммоль) растворяли в диоксане (30 мл) и воде (5 мл) и затем добавляли 5-фтор-4-йодпиридин-2-амин (1,00 г, 4,20 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия и дихлорметана (0,34 г, 0,42 ммоль) и карбонат калия (1,74 г, 12,6 ммоль). Реакционную смесь нагревали до 100°С в атмосфере азота и подвергали реакции в течение 8 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционную смесь охлаждали до комнатной температуры. Реакционный раствор очищали непосредственно колоночной хроматографией (н-гексан:этилацетат = 1:1) с получением промежуточного соединения 74а (0,40 г, выход 37%).
Синтез Промежуточного соединения 74b:
74а (0,13 г, 0,50 ммоль) растворяли в ацетонитриле (10 мл) и затем добавляли (1S,3R)-3-[(трет-бутилоксикарбонил)амино]циклогексанкарбоновую кислоту (0,16 г, 0,65 ммоль), N,N,N',N'-тетраметилхлорформамидиния гексафторфосфат (0,17 г, 0,60 ммоль) и N-метилимидазол (0,14 г, 1,75 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 12 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (н-гексан:этилацетат = 2:1) с получением 74b (0,12 г, выход 50%).
Синтез Промежуточного соединения 74 с:
74b (0,12 г, 0,25 ммоль) растворяли в дихлорметане (20 мл) и добавляли трифторуксусную кислоту (4 мл). Реакционную смесь подвергали реакции при комнатной температуре в течение 4 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор промывали водой (30 × 3) и водные фазы объединяли. Объединенную водную фазу доводили карбонатом натрия до рН 8-9 и экстрагировали дихлорметаном (30 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 74 с (0,08 г, выход 84%).
Синтез конечного продукта 74:
74 с (0,08 г, 0,21 ммоль) растворяли в дихлорметане (20 мл), затем добавляли уксусный ангидрид (0,04 г, 0,42 ммоль) итриэтиламин (0,04 г, 0,42 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор очищали непосредственно колоночной хроматографией (дихлорметан:метанол = 50:1) с получением конечного продукта 74 (0,02 г, выход 22%).
MS m/z (ESI): 426,2 [М+Н]+.
1H ЯМР (600 МГц, ДМСО-d6) δ 10.81 (s, 1H), 8.55-8.54(m, 2Н), 8.18 (d, J=5.4 Гц, 1H), 7.80 (d, J=7.8 Гц, 1H), 7.6 (d, J=5.4 Гц, 1H), 3.60-3.54 (m, 1H), 2.64-2.60 (m, 1H), 1.90-1.88 (m, 1H), 1.77-1.76 (m, 6Н), 1.40-1.20 (m, 3Н), 1.19-1.05 (m, 1H).
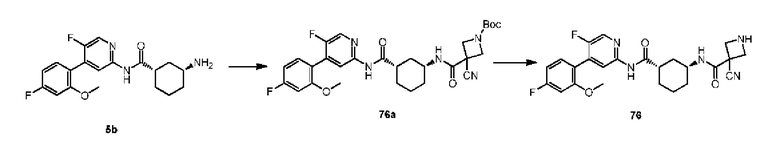
Синтез Промежуточного соединения 76а:
5b (500 мг, 1,385 ммоль) растворяли в N,N-диметилформамиде (15 мл) и затем добавляли 1-[(трет-бутокси)карбонил]-3-цианоазетидин-3-карбоновую кислоту (375 мг, 1,66 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (787 мг, 2,07 ммоль) и N,N-диизопропилэтиламин (687 мкл, 4,15 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Реакционную смесь перемешивали в течение 30 минут, фильтровали и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении с получением 76а (497 мг, выход 63%).
Синтез конечного продукта 76:
76а (497 мг, 0,87 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1) с получением конечного продукта 76 (396 мг, выход 97%).
MS m/z (ESI): 470,2 [М+Н]+.
1Н ЯМР (600 МГц, CD3OD) δ 8.38 (s, 1H), 7.75 (d, J=5.4 Гц, 1H), 7.50-7.42 (m, 1Н), 7.04 (d, J=10.8 Гц, 1H), 6.96-6.86 (m, 1H), 4.60-4.32 (m, 4Н), 3.93-3.76 (m, 4Н), 2.70 (m, 1H), 2.23 (m, 1H), 2.00 (m, 3Н), 1.71-1.46 (m, 3Н), 1.43-1.27 (m, 1H).
Пример 77:
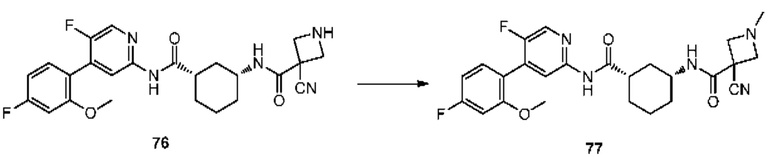
Синтез конечного продукта 77:
76 (296 мг, 0,63 ммоль) растворяли в метаноле (10 мл) и затем добавляли водный раствор формальдегида (1 мл), триацетоксиборогидрид натрия (249 мг, 1,27 ммоль) и одну каплю уксусной кислоты. Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением конечного продукта 77 (186 мг, выход 61%).
MS m/z (ESI): 484,2[М+Н]+
1Н ЯМР(600 МГц, ДМСО-d6) δ 10.56 (s, 1H), 8.32 (s, 1H), 8.05 (d, J=4.8 Гц, 1H), 8.10 (d, J=7.8 Гц, 1H), 7.34-7.31 (m, 1H), 7.09 (s, 1H), 6.91-6.88 (m, 1H), 3.79 (s, 3Н), 3.68-3.56 (m, 1H), 2.98-2.73 (m, 4Н), 2.59-2.55 (m, 1H), 2.42 (s, 3Н), 1.81-1.67 (m, 4Н), 1.30-1.09 (m, 4Н).
Пример 78:
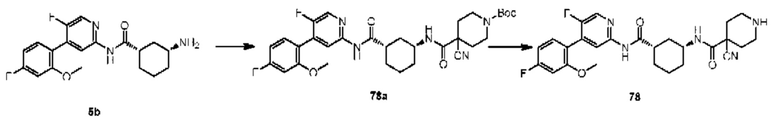
Синтез Промежуточного соединения 78а:
5b (500 мг, 1,385 ммоль) растворяли в N,N-диметилформамиде (15 мл) и затем добавляли 1-(трет-бутоксикарбонил)-4-цианопиперидин-4-карбоновую кислоту (422 мг, 1,66 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (787 мг, 2,07 ммоль) и N,N-диизопропилэтиламин (687 мкл, 4,15 ммоль). Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь перемешивали в течение 30 минут, фильтровали и сушили над безводным сульфатом натрия с получением 78а (504 мг, выход 61%).
Синтез конечного продукта 78:
78а (504 мг, 0,84 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 40:1) с получением конечного продукта 78 (384 мг, выход 92%).
MS m/z (ESI): 498,7 [М+Н]+.
1H ЯМР (600 МГц, CD3OD) δ 8.32 (s, 1H), 8.07 (d, J=8.4 Гц, 1H), 7.35-7.33 (m, 1Н), 7.11-7.10 (m, 1H), 7.08-6.91 (m, 1H), 3.78 (s, 3Н), 3.78-3.66 (m, 1H), 3.30-3.28 (m, 2Н), 2.99-2.97 (m, 2Н), 2.62-2.52 (m, 1H), 2.30-2.22 (m, 4Н), 1.79-1.77 (m, 4Н), 1.46-1.43 (m, 1H), 1.29-1.26 (m, 3Н).
Пример 79:
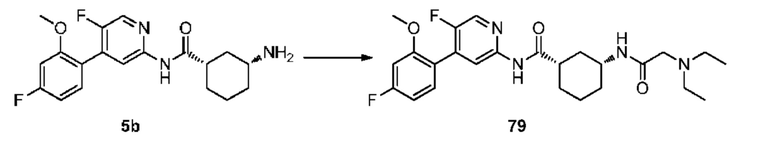
Синтез конечного продукта 79:
5b (0,16 г, 0,43 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли N,N-диэтилглицина гидрохлорид (109 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэтиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией
(дихлорметан:метанол=50:1-20:1) с получением конечного продукта 79 (116 мг, выход 57%).
MS m/z (ESI): 475,3 [М+Н]+
1Н ЯМР (600 МГц, CDCl3) δ 8.23 (d, J=7.8 Гц, 1H). 8.13 (s, 1H), 8.07 (s,lH), 7.40 (m, 1H), 7.277.22 (m, 1H), 6.87-6.60 (m, 2H), 3.94-3.84 (m, 1H), 3.81 (s, 3H), 3.01 (s, 2H), 2.54 (m, 4H), 2.45 (m, 1H), 2.22 (m, 1H), 1.96 (m, 3H), 1.56-1.36 (m, 3H), 1.26-1.12 (m, 1H), 1.01 (m, 6H).
Пример 80:
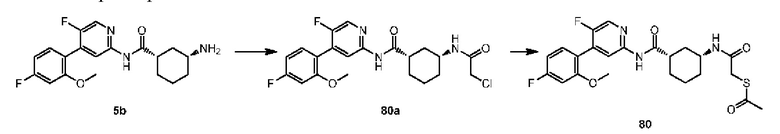
Синтез Промежуточного соединения 80а:
5b (0,30 г, 0,83 ммоль) растворяли в дихлорметане (10 мл) и затем добавляли триэтиламин (420 мг, 4,15 ммоль) и хлорацетилхлорид (186 мг, 1,66 ммоль) при 0°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (20 мл). Смесь экстрагировали этилацетатом (10 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат = 1:0-0:1) с получением 80а (200 мг, выход 55%).
Синтез конечного продукта 80:
80а (200 мг, 0,46 ммоль) растворяли в N,N-диметилформамиде (10 мл) и затем добавляли тиоацетат калия (78,25 мг, 0,69 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. Реакционный раствор добавляли к водному раствору HCl (1 М, 10 мл) и затем добавляли воду (10 мл) и дихлорметан (15 мл). Смесь экстрагировали дихлорметаном (10 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат=1:0-0:1) с получением конечного продукта 80 (200 мг, выход 91%).
MS m/z (ESI): 478,2 [М+Н]+
1H ЯМР (600 МГц, ДМСО-d6) δ 10.58 (s, 1H), 8.33 (s, 1Н), 8.07 (d, J=5.4 Гц, 1H), 7.59 (d, J=8.4 Гц, 1H), 7.35-7.34 (m, 1H), 7.33-7.11 (m, 1H), 7.09-6.90 (m, 1H), 4.39 (s, 2Н), 3.78 (s, 3Н), 3.65-3.63 (m, 1H), 2.66-2.60 (m, 1H), 2.58 (s, 3Н), 1.85-1.69 (m, 4Н), 1.24-1.14 (m, 4Н).
Пример 81:
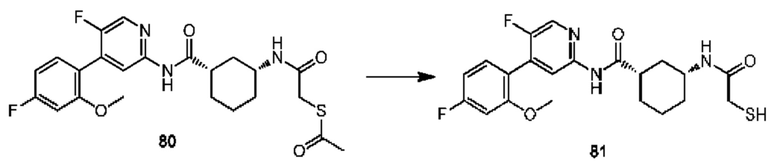
Синтез конечного продукта 81:
80 (220 мг, 0,46 ммоль) растворяли в метаноле (5 мл) и затем добавляли карбонат калия (317 мг, 2,30 ммоль). Реакционную смесь подвергали реакции при комнатной температуре в течение 20 минут, а затем нагревали вплоть до 55°С и подвергали реакции в течение 40 минут до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (20 мл). Смесь экстрагировали этилацетатом (10 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (н-гексан:этилацетат=1:0-0:1) с получением конечного продукта 81 (41 мг, выход 20%).
MS m/z (ESI): 436,2 [М+Н]+
1H ЯМР (600МГц, ДМСО-d6) δ 10.55 (s, 1H), 8.31 (s, 1H), 8.07 (d, J=5.4 Гц, 1Н), 7.56 (d, J=9.0 Гц, 1H), 7.35-7.32 (m, 1H), 7.09-7.07 (m, 1H), 6.91-6.88 (m, 1H), 3.89-3.84 (m, 4Н), 3.66-3.63 (m, 1H), 3.24-3.22 (m, 2Н), 2.61-2.57 (m, 1H), 1.83-1.68 (m, 4Н), 1.34-1.26 (m, 4Н).
Пример 82:
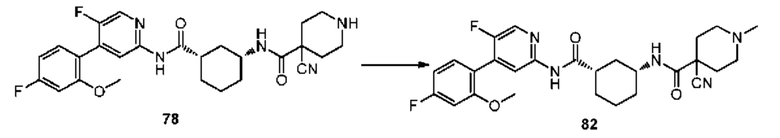
Синтез конечного продукта 82:
78 (300 мг, 0,60 ммоль) растворяли в метаноле (10 мл) и затем добавляли водный раствор формальдегида (1 мл), триацетоксиборгидрид натрия (235 мг, 1,20 ммоль) и одну каплю уксусной кислоты. Реакционную смесь перемешивали и подвергали реакции при комнатной температуре в течение 10 часов до тех пор, пока с помощью ТСХ не было обнаружено исходного материала. К реакционному раствору добавляли воду (50 мл). Смесь доводили бикарбонатом натрия до рН 8 и экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 × 2) и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением конечного продукта 82 (205 мг, выход 67%).
MS m/z (ESI): 512,2[М+Н]+
1Н ЯМР (600 МГц, ДМСО-d6) δ 10.28 (s, 1H), 8.28 (s, 1H), 8.22-8.09 (m, 1H), 8.05 (d, J=8.4 Гц, 1H), 7.32-7.30 (m, 1H), 7.09-7.02 (m, 1H), 6.91-6.87 (m, 1H), 3.80 (s, 3Н), 3.75-3.66 (m, 1Н), 3.57-3.52 (m, 1H), 3.16-2.96 (m, 2Н), 2.81-2.59 (m, 4Н), 2.50-2.48 (m, 2Н), 2.43-2.31 (m, 3Н), 2.00-1.74 (m, 4Н), 1.52-1.48 (m, 1H), 1.41-1.22 (m, 3Н).
Пример 83:
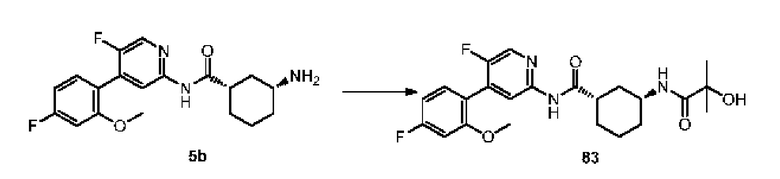
Синтез конечного продукта 83:
5b (0,20 г, 0,55 ммоль) растворяли в N,N-диметилформамиде (30 мл) и затем добавляли 2-гидр о кс и из о масляную кислоту (68 мг, 0,65 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (178 мг, 0,47 ммоль) и N,N-диизопропилэгиламин (111 мг, 0,86 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре до тех пор, пока с помощью ТСХ не было обнаружено исходного материала К реакционному раствору добавляли воду (100 мл). Смесь экстрагировали этилацетатом (50 × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 2) и сушили над безводным сульфатом натрия. Затем растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (дихлорметан:метанол = 50:1-20:1) с получением конечного продукта 83 (81 мг, выход 33%).
MS m/z (ESI): 448,2 [М+Н]+
1Н ЯМР (600 МГц, CDCl3) δ 8.31 (s, 1H), 8.24 (d, j=8.4 Гц, 1H), 8.12 (s, 1Н), 7.26-7.24 (m, 1H), 6.76-6.73 (m, 2H), 6.63 (d, J=12.6 Гц, 1H), 3.90-3.82 (m, 1Н), 3.81 (s, 3Н), 2.65 (s, 1H), 2.56-2.42 (m, 1H), 2.26-2.23 (m, 1H), 2.00-1.89 (m, 3Н), 1.61-1.49 (m, 2H), 1.45 (d, J=4.8 Гц, 6H), 1.30-1.18 (m, 1H).
Примеры 75 и 84-90:
В соответствии со способами синтеза Примеров 1-72 были выбраны соответствующие исходные материалы для синтеза соединений 75 и 84-90, которые имели следующие структуры:
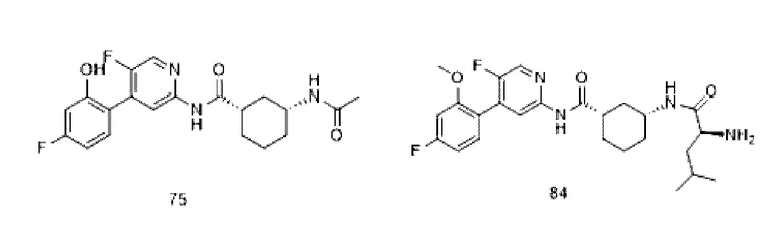
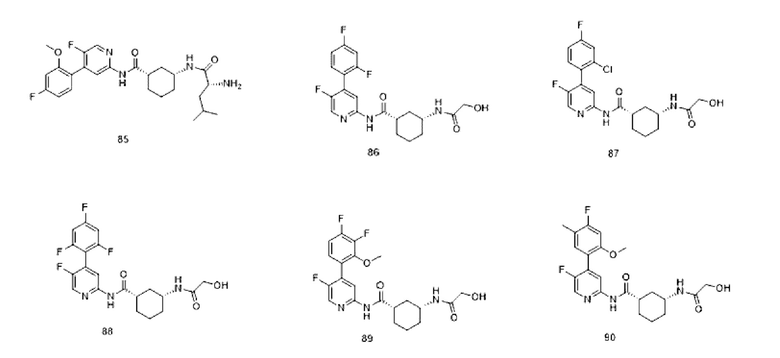
Пример Анализа 1: Анализ ингибирующего действия соединений по настоящему изобретению на CDK9, CDK1, CDK2, CDK4, CDK5, CDK6 и CDK7
1. Объект анализа
Тестировали ингибирующее действие соединений на киназы CDK 1/2/4/5/6/7/9 и путем подгонки и расчета получили эффективные значения IC50.
2. Семейство тестируемых CDK
CDK1/CD/K2/CDK4/CDK5/CDK6/CDK7/CDK9
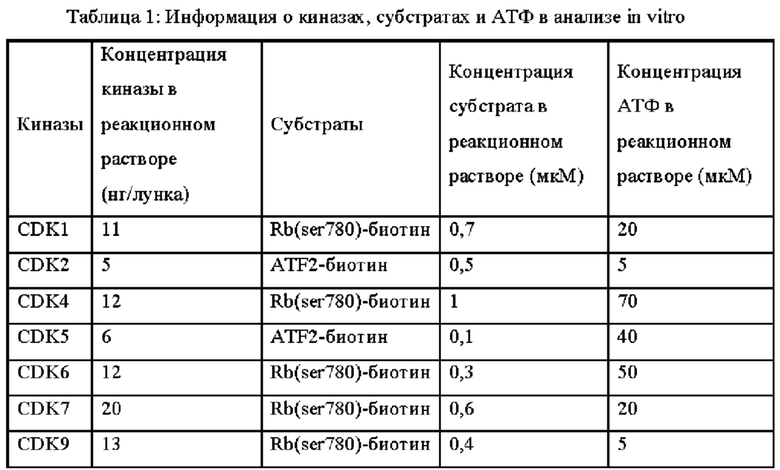
3. Процедур а тестирования
3.1 Разбавление соединений
Соединения разбавляли ДМСО до 11 концентраций путем 3-кратного разведения, при этом самая высокая концентрация референсного соединения стауроспорина составляла 1 мкМ, а самая высокая концентрация тестируемых соединений составляла 10 мкМ.
3.2 Киназная реакция
Соединение, растворенное в ДМСО (50 нл), переносили на планшет для киназной реакции с использованием акустической технологии (Echo). 5 мкл раствора для разведения киназы CDK добавляли в планшет для киназной реакции и инкубировали при комнатной температуре в течение 10 минут после центрифугирования. В планшет добавляли 5 мкл раствора премикса субстрата и конечные концентрации субстрата и АТФ в каждой лунке показаны в Таблице 1. После центрифугирования реакцию проводили при 30°С в течение 120 минут.
3.3 Остановка реакции и обнаружение сигнала
В каждую лунку добавляли по 10 мкл стоп-раствора, и после центрифугирования планшет инкубировали при комнатной температуре в течение 120 минут, и затем инкубировали при 4°С в течение ночи. Значения сигналов считывали на приборе Envision с использованием программы HTRF и проводили анализ данных. Значения IC50 (ингибирующая концентрация при 50% максимального эффекта) выражали в нМ. Результаты показаны в Таблице 2.
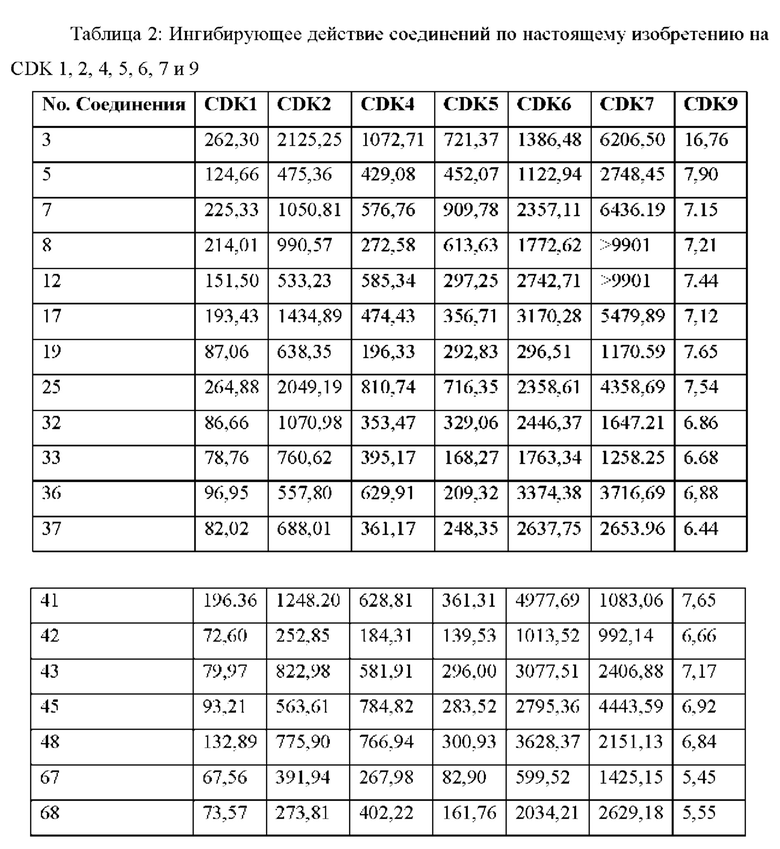
Приведенный выше анализ показал, что соединения, оцененные в настоящем раскрытии, проявляют селективность в отношении CDK9 и обладают сильным ингибирующим действием в отношении CDK9.
Пример Анализа 2: Ингибирующее действие ингибитора CDK9 in vitro на пролиферацию клеток MV4; 11
1. Объект анализа
Исследовали ингибирующее действие in vitro синтезированных соединений на пролиферацию клеток.
2. Принцип анализа
Торговое название МТТ представляло собой тиазоловый синий, который является тетразолиевой солью красителя, способного принимать атомы водорода. Сукцинатдегидрогеназа в митохондриях живых клеток могла восстанавливать экзогенный МТТ до нерастворимых сине-фиолетовых кристаллов и откладываться в клетках, но мертвые клетки не обладали этой функцией. Диметилсульфоксид может растворять сине-фиолетовые комплексы в клетках, а его показатель светопоглощения можно измерить при длине волны 490-550 нм с помощью иммуноферментного детектора, который может косвенно отражать количество клеток. В определенном диапазоне числа клеток количество образовавшихся кристаллов МТТ было пропорционально числу клеток. Исследуемое лекарственное средство последовательно разводили до различных концентраций и добавляли в 96-луночный планшет. Величину OD определяли после воздействия лекарственного средства в течение определенного времени. Размер величины OD мог отражать количество живых клеток, и его значение IC50 рассчитывали с помощью SPSS 19.0.
3. Аппарат для анализа
СО2-Инкубатор Модели 371: Thermo
Инвертированный Флуоресцентный Микроскоп Модели IX70-142: Olympus
Шкаф биологической безопасности HFsafe-1500 Type: Shanghai Lishen Scientific Instrument Co., Ltd.
Флэш-ридер для Микропланшетов Varloskan: Thermo Company
Прецизионные электронные весы: Mettler AL204 Type
4. Материал для анализа:
4.1 Клетки и культуральная среда

4.2 Материал для анализа
4.3 Подготовка реагентов
Рабочий раствор МТТ 5 мг/мл: 0,5 г МТТ растворяли в 100 мл PBS, стерилизовали фильтрованием через микропористый фильтр 0,22 мкм и хранили при 4°С в холодильнике (использование в течение двух недель) или при -20°С для длительного хранения.
5. Метод анализа
5.1 Посев
Суспензия клеток: Клетки ресуспендировали центрифугированием и подсчитывали. Суспензию клеток определенной плотности готовили с полной культуральной средой, равномерно продували, инокулировали на 96-луночный планшет по 100 мкл на лунку и затем культивировали в CO2-инкубаторе.
5.2 Состав лекарственного средства
Соответствующее количество лекарственного средства добавляли к рассчитанному количеству ДМСО и растворяли. Полученную смесь распределяли и хранили при -20°С. Концентрация лекарственного средства составляла 10 мМ.
5.3 Добавление лекарственного средства
Исходный раствор соединения с концентрацией 10 мМ разводили в различных концентрациях (8 концентраций) растворов соединения в ДМСО (3-кратное разведение, 20-кратная конечная концентрация). Каждую концентрацию растворов (10 мкл) соединения в ДМСО соответственно разбавляли средой для культивирования клеток (90 мкл) для приготовления рабочих растворов (2хконечная концентрация). Рабочие растворы каждой концентрации (100 мкл) соединения добавляли в 96-луночный планшет, инокулированный клетками (1х конечная концентрация, самая высокая конечная концентрация составляет 1000 нМ). Полученный планшет продолжали культивировать в СО2-инкубаторе.
6. Тестирование
Извлекали 96-луночный планшет и наблюдали за плотностью клеток под микроскопом. Исследование проводили методом МТТ.
Метод МТТ: в каждую лунку добавляли по 20 мкл МТТ; планшет культивировали около 4 часов в инкубаторе; жидкость из лунке удаляли; в каждую лунку добавляли по 150 мкл ДМСО; планшет помещали в шейкер и встряхивали в течение 5-10 минут, а исследование проводили с помощью устройства для считывания микропланшетов при длине волны 550 нм.
7. Анализ данных: значение IC50 лекарственного средства рассчитывали с помощью статистического программного обеспечения SPSS 19.0.
Результат применения соединений на ингибирование пролиферации клеток показан в Таблице 3:
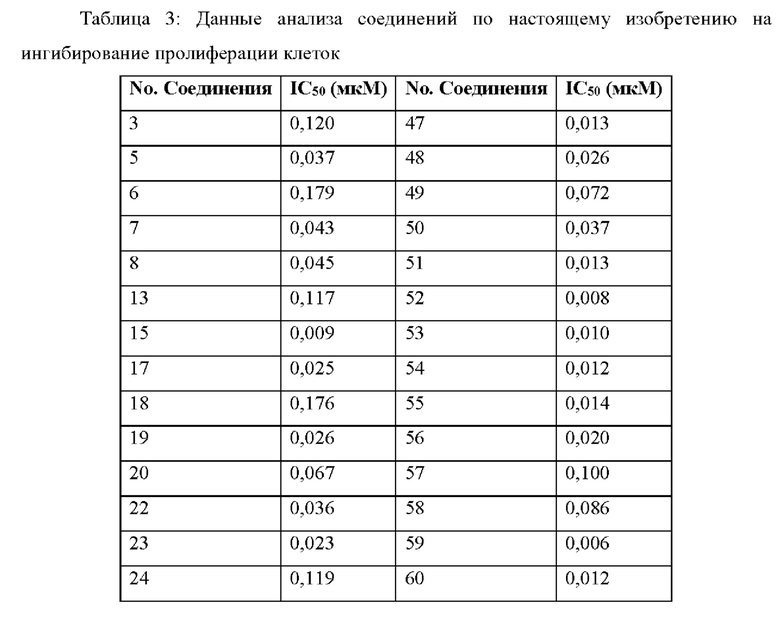
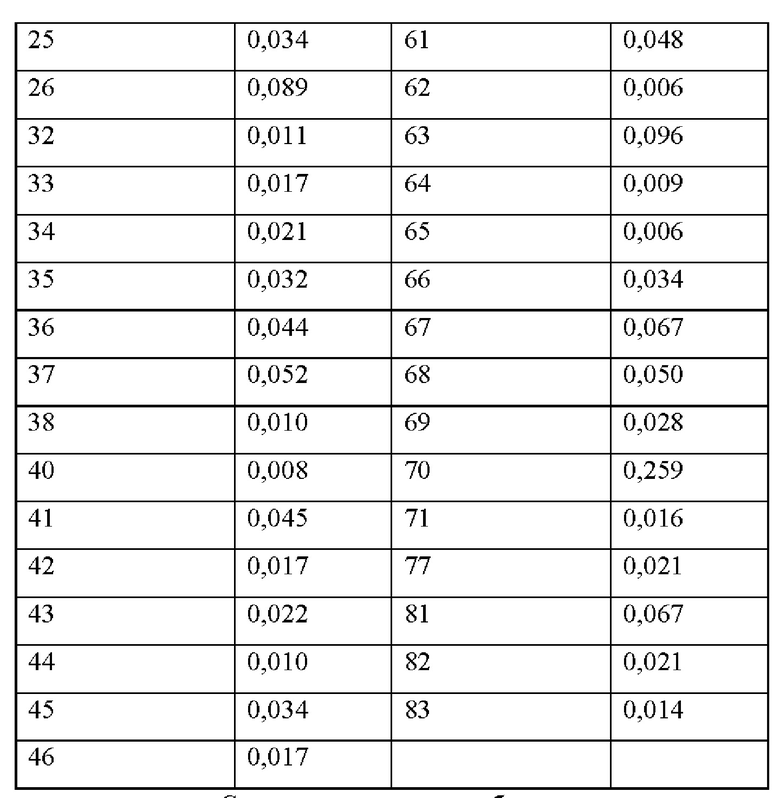
Заключение анализа: Согласно данным в таблице, соединения по настоящему изобретению обладают относительно сильным ингибирующим действием на CDK9, и ингибирующая активность клеток in vitro была существенно ниже 300 нМ, и оптимальное значение может достигать нескольких нМ.
Пример Анализа 3: Исследование ингибирующей активности hERG in vitro
1. Объект анализа:
Быстрый компонент выпрямляющего тока (IKr) в основном опосредуется ионным каналом hERG и участвует в реполяризации кардиомиоцитов человека. Ингибирование IKr было ведущей причиной клинического синдрома удлинения интервала QT, даже острой сердечной аритмии и даже внезапной смерти. С использованием технологии патч-кламп для цельных клеток блокирующий эффект соединения на канал hERG был обнаружен на клеточной линии СНО-К1, стабильно экспрессирующей канал hERG, и была определена концентрация половинного ингибирования IC50 соединения. Она использовалась как часть комплексной оценки безопасности для сердца и первоначально оценивалась в ходе скрининга безопасности in vitro на кардиотоксичность.
2. Метод анализа:
Этот анализ включал следующие аспекты:
- Регистрировали ток hERG на клеточной линии СНО-K1, стабильно экспрессирующей канал hERG, с использованием ручной технологии патч-кламп;
- Степень ингибирования при каждой концентрации рассчитывали в соответствии с хвостовым током hERG;
- Измерение проводили при 5 концентрациях каждого соединения для расчета значения IC50;
- Тестировали по 3 клетки при каждой концентрации;
- Был предоставлен один положительный контрольный препарат.
Ток hERG регистрировали с использованием технологии патч-кламп для целых клеток. Клеточную суспензию добавляли в чашку, и чашку помещали на предметный столик прямого микроскопа. После адгезии клеток осуществляли перфузию внеклеточной жидкостью со скоростью 1-2 мл/мин. Стеклянный микроэлектрод вытягивали с помощью съемника микроэлектрода посредством двухэтапного процесса, а сопротивление разрыву пипетки составляло 2-5 МОм. После того как была установлена регистрация целых клеток, зажимной потенциал удерживался на уровне -80 мВ. Клетки были деполяризованы до +60 мВ путем применения стимула напряжения, и затем реполяризованы до -50 мВ, чтобы вызвать хвостовой ток hERG. Все записи проводились после того, как ток стабилизировался. Внеклеточное перфузионное введение начинали с наименьшей концентрации, выдерживая при каждой концентрации по 5-10 мин до стабилизации тока, и затем проводили со следующей концентрацией. Концентрацию половинного ингибирования (IC50) соединения получали путем наилучшего соответствия Логистическому уравнению.
Амитриптилин был одним из наиболее широко используемых препаратов для блокирования тока hERG. Поэтому он использовался в качестве положительного контрольного лекарственного средства в исследовании.
3. Результат показан в Таблице 4:
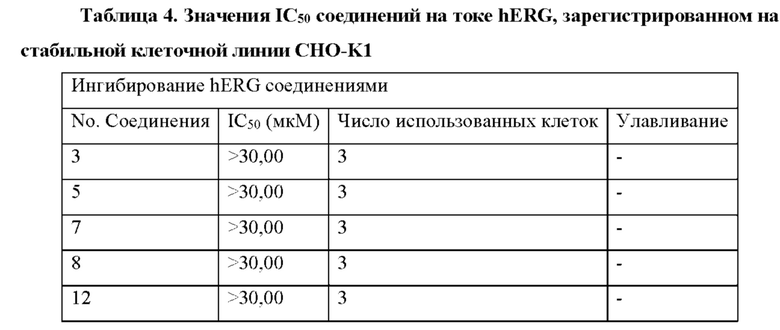
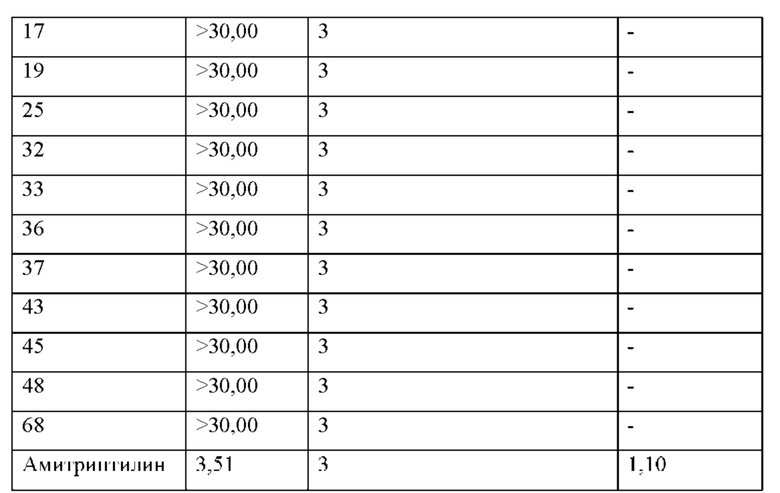
IC50 положительного контрольного лекарственного средства Амитриптилин в отношении ингибирования тока hERG согласуется с предыдущими результатами партии анализов, а также с результатами, опубликованными в литературе, что указывает на то, что результаты этого анализа заслуживают доверия. Результаты вышеуказанного анализа показали, что исследуемые соединения не могли достичь половинного ингибирования тока hERG при самой высокой тестируемой концентрации, поэтому IC50 не могла быть определена, что указывает на то, что соединения по настоящему изобретению не оказывали очевидного ингибирующего действия на канал hERG в пределах диапазона исследованных концентраций в этом анализе. В определенной степени это может отражать то, что соединения по настоящему изобретению обладают более низкой кардиотоксичностью или не обладают ею, и имеют положительное значение для оценки безопасности лекарственных средств.
Пример Анализа 4: Исследование противоопухолевой активности лекарственных средств in vivo - фармакодинамика соединений по настоящему изобретению на модели подкожного ксенотрансплантата опухоли MV4; 11-клеточного острого миелоцитарного лейкоза человека
Культура клеток: культуральная среда IMDM, содержащая 10% фетальной бычьей сыворотки (FBS), 37°С, 5% CO2.
Мыши NOD-SCID, самки, 6-8 недель (18-22 г), каждой мыши подкожно инокулировали 0,1 мл (1×108) клеток MV4; 11 в правую часть спины каждой мыши. Когда средний объем опухоли достигал 150 мм3, начинали введение лекарственного средства. Дозировка и способ введения указаны в таблице ниже. Объем опухоли измеряли два раза в неделю, и объем рассчитывали в кубических миллиметрах. Когда средний объем опухоли в группе, получавшей носитель, достигал более 800 мм3, введение прекращали для сравнения разницы в среднем объеме опухоли между группами, получавшими соединение, и группой, получавшей носитель. Противоопухолевую эффективность соединений оценивали по TGI (%). TGI (%) отражал скорость ингибирования роста опухоли.
Расчет TGI(%): TGI(%) = [1-(средний объем опухоли в группе в конце введения соединения - средний объем опухоли в группе в начале введения соединенийя) / (средний объем опухоли в группе в конце введения носителя - средний объем опухоли в группе в начале введения носителя)] × 100%.
Результаты показаны в Таблицах 5-8.

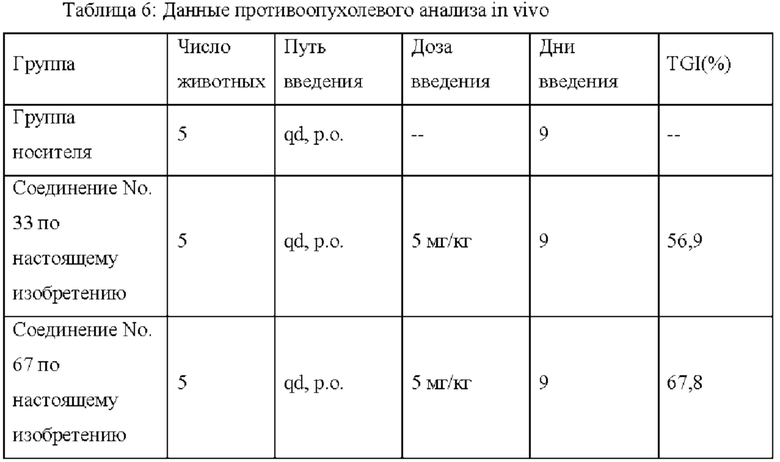
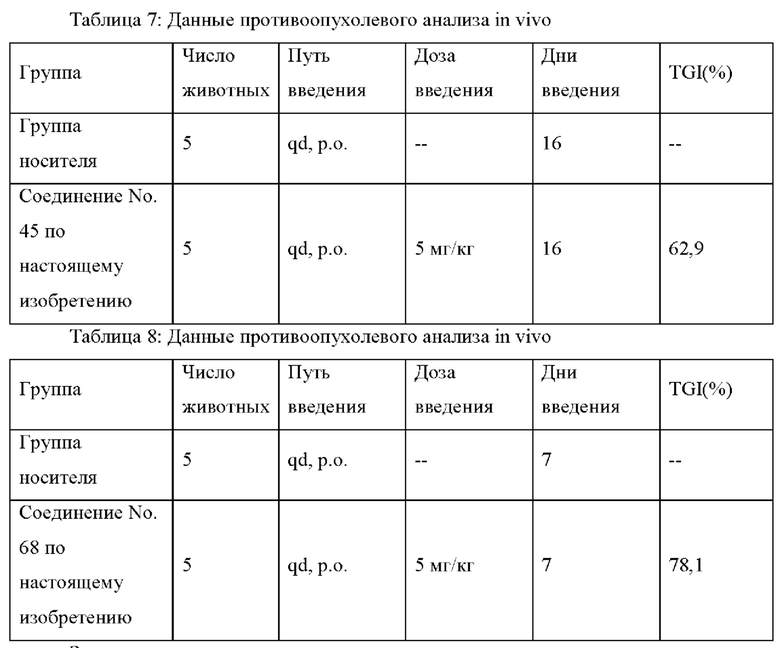
Заключение анализа:
Соединения по настоящему изобретению показали хорошую эффективность на модели in vivo подкожного ксенотрансплантата опухоли MV4; 11-клеточного острого миелоцитарного лейкоза человека и имели значительный противоопухолевый эффект.
Пример Анализа 5: Исследование противоопухолевой активности лекарственных средств in vivo - фармакодинамика соединений по настоящему изобретению на модели подкожного ксенотрансплантата опухоли HL-60-клеточного острого промиелоцитарного лейкоза человека
Культура клеток: культуральная среда IMDM, содержащая 20% фетальной бычьей сыворотки (FBS), 37°С, 5% CO2;
Мыши Nu/Nu, самки, 6-8 недель (18-22 г), каждой мыши подкожно инокулировали 0,1 мл клеточной суспензии HL-60 (содержащей около 1×107 клеток) в подмышечную область правой передней конечности. Когда средний объем опухоли достигал 150 мм3, начинали введение лекарственного средства. Дозировка и способ введения указаны в таблице ниже. Объем опухоли измеряли 2-3 раза в неделю и объем рассчитывали в кубических миллиметрах. Когда средний объем опухоли в группе, получавшей носитель, достигал более 800 мм3, введение прекращали для сравнения разницы в среднем объеме опухоли между группой, получавшей соединение, и группой, получавшей носитель. Противоопухолевую эффективность соединений оценивали по TGI (%). TGI (%) отражал скорость ингибирования роста опухоли.
Расчет TGI(%): TGI(%) = [1-(средний объем опухоли в группе в конце введения соединения - средний объем опухоли в группе в начале введения соединения) / (средний объем опухоли в контрольной группе в конце введения носителя - средний объем опухоли в контрольной группе в начале введения носителя)] × 100%.
Результаты показаны в Таблице 9.
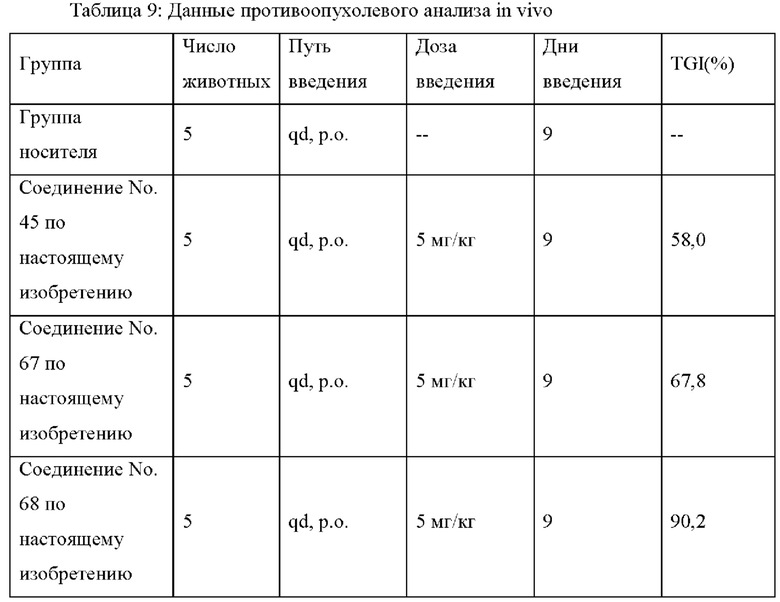
Заключение анализа:
Соединения по настоящему изобретению показали хорошую эффективность на модели in vivo подкожного ксенотрансплантата опухоли HL-60-клеточного острого промиелоцитарного лейкоза человека. Через девять дней после начала введения соединения по настоящему изобретению показали значительный противоопухолевый эффект.
Несмотря на то, что вышеприведенное раскрытие было описано достаточно подробно посредством описания и примеров с целью ясного понимания, из идей раскрытия будет очевидно, что специалисты в данной области техники, как правило, могут вносить определенные изменения и модификации в прилагаемую формулу изобретения без отклонения от сущности или объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| ИНГИБИТОР ATR И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2799047C2 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| НОВЫЙ ТИП ПРОИЗВОДНОГО ЦИТИДИНА И ЕГО ПРИМЕНЕНИЕ | 2015 |
|
RU2684402C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
Группа изобретений относится к органической химии и включает индивидуальные соединения, представленные в формуле изобретения, их фармацевтически приемлемые соли, фармацевтическую композицию на их основе и применение. Технический результат - производные пиридина, обладающие селективной ингибирующей активностью в отношении циклинзависимой киназы 9 (CDK9). 3 н. и 5 з.п. ф-лы, 9 табл., 95 пр.
1. Соединение или его фармацевтически приемлемая соль, или его стереоизомер, где соединения имеют следующие структуры:
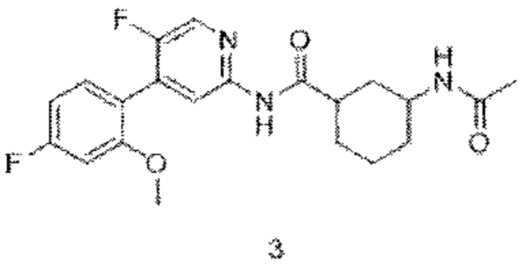
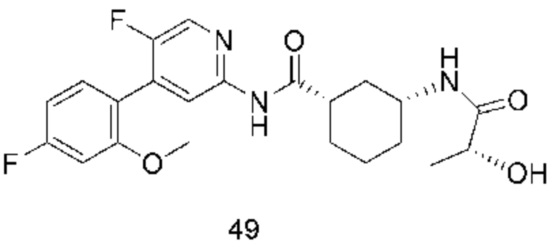
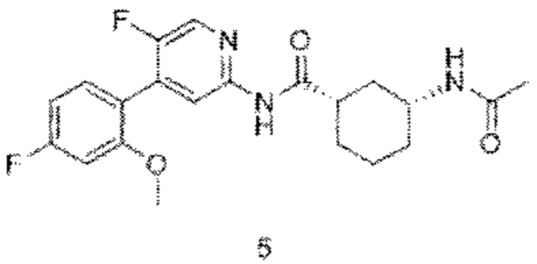
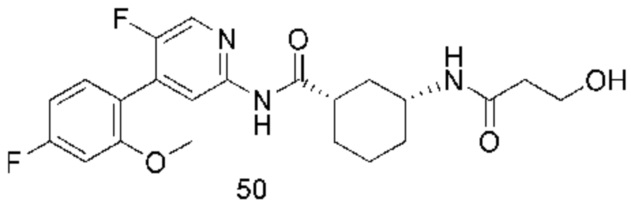
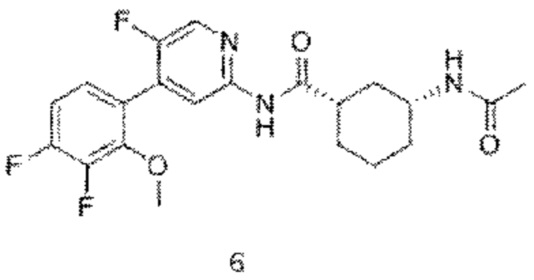
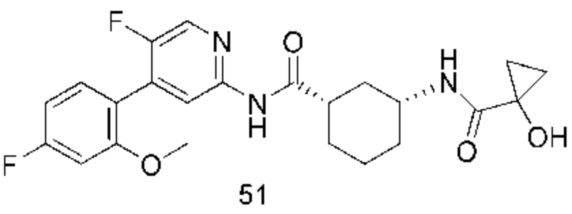
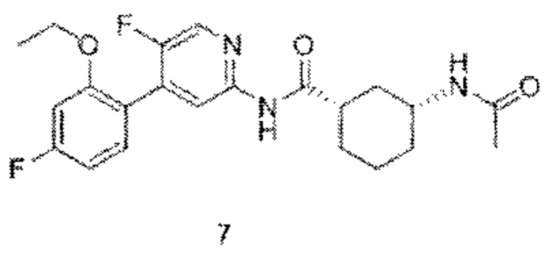
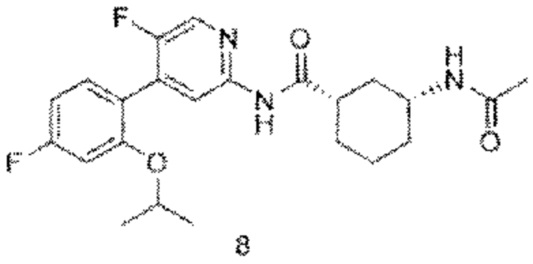
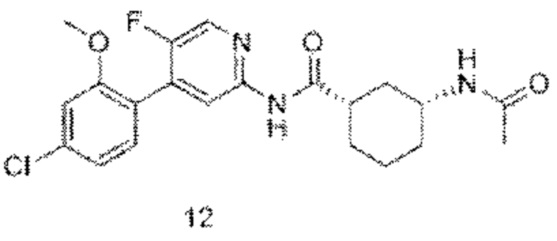
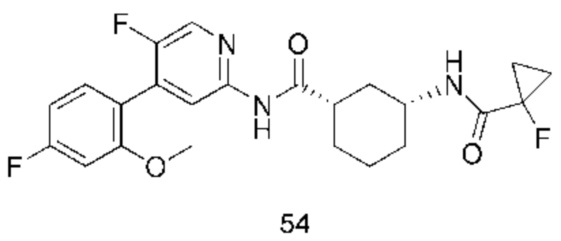
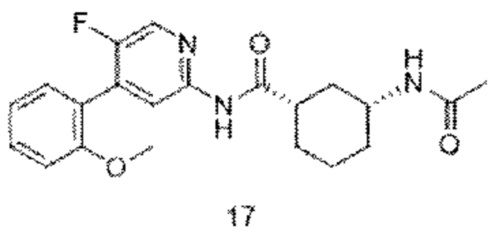
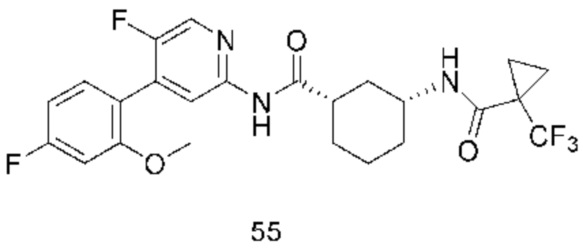
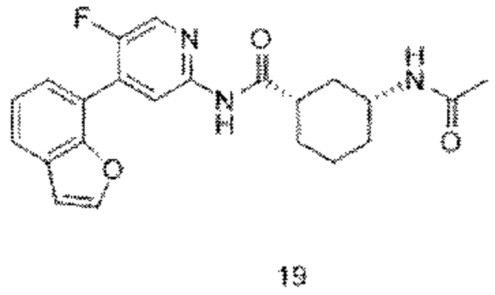
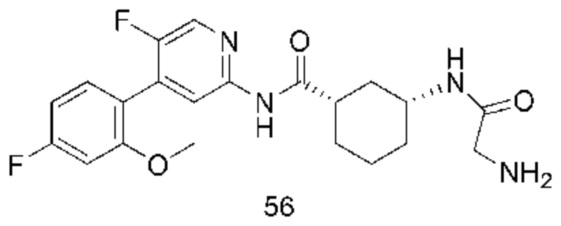
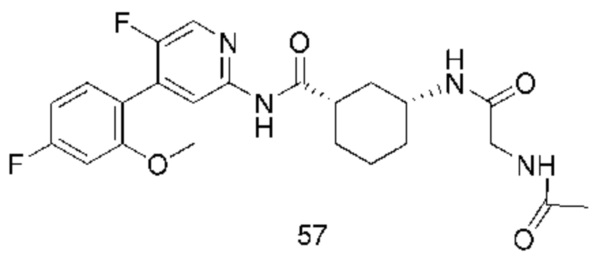
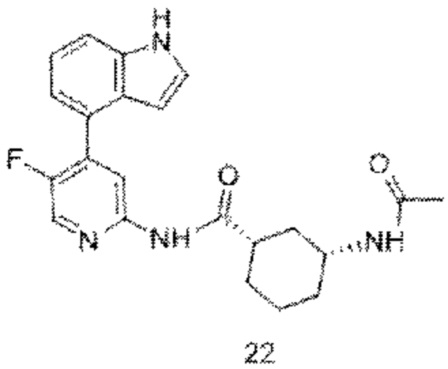
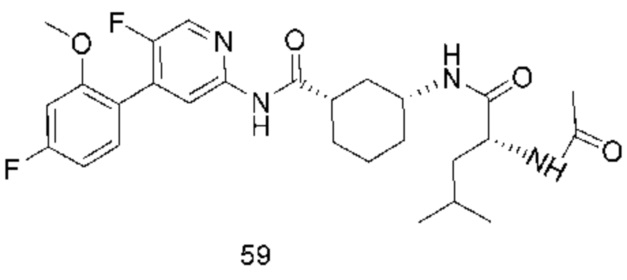
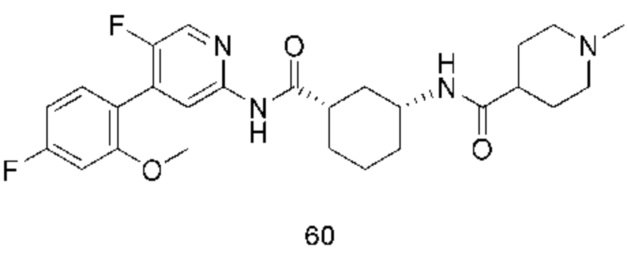
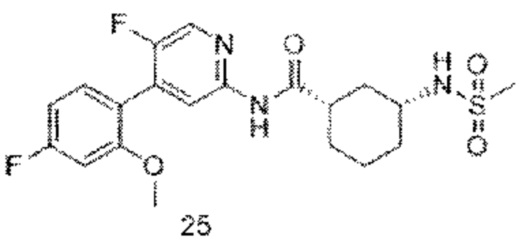
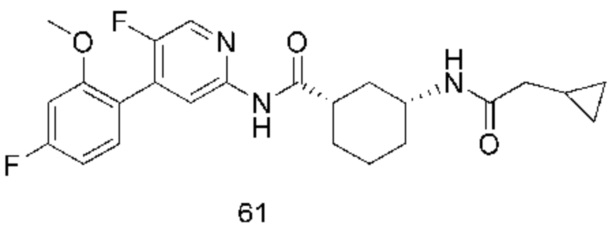
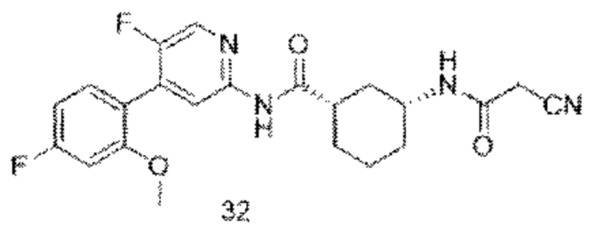
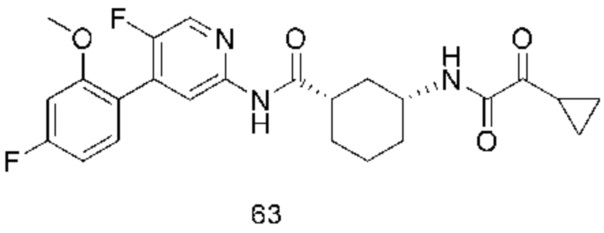
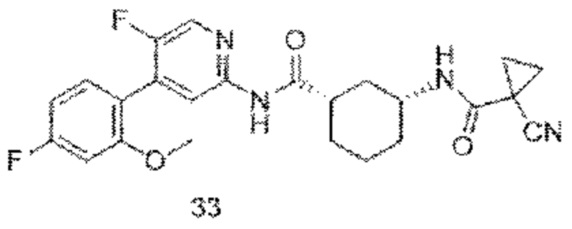
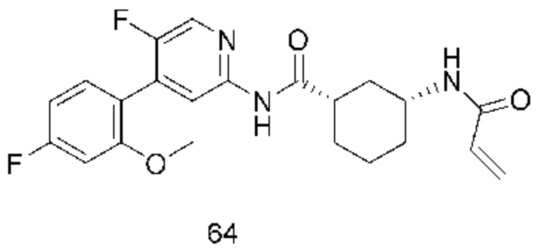
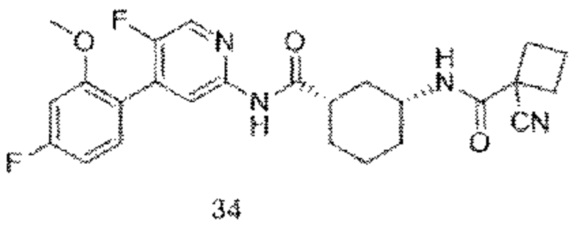
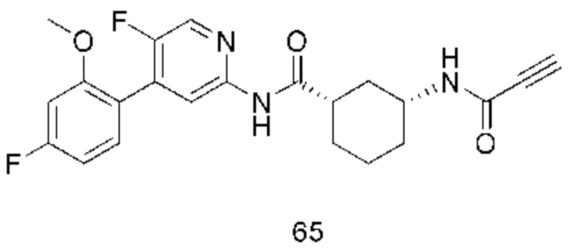
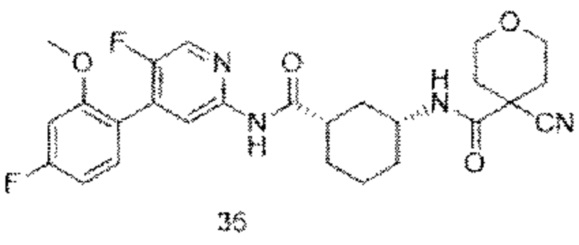
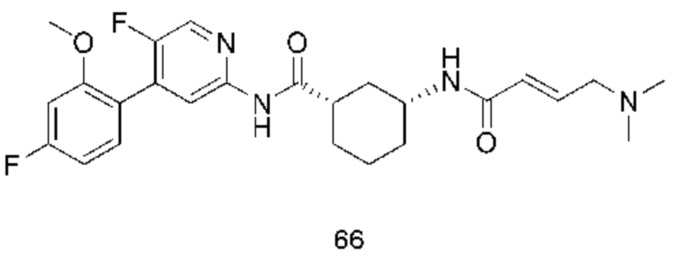
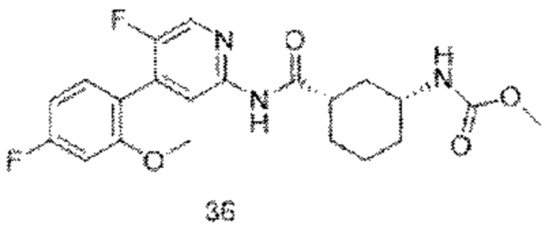
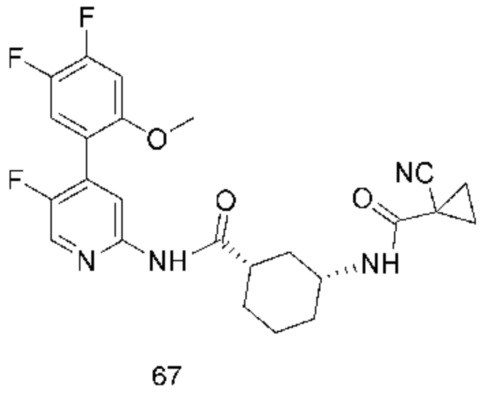
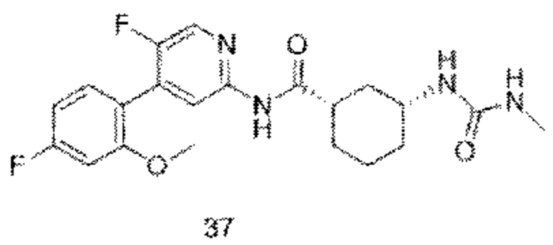
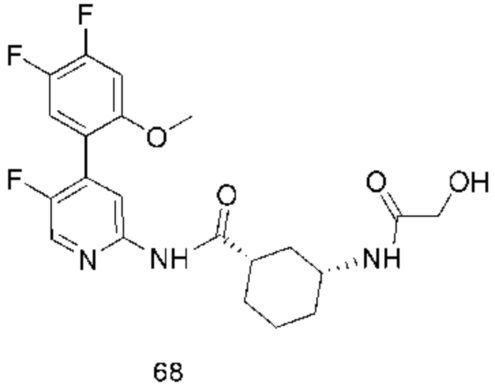
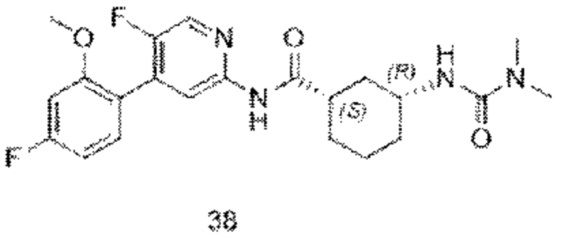
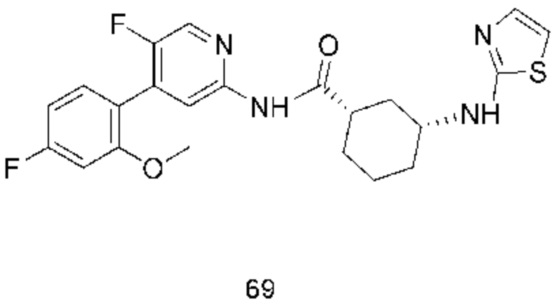
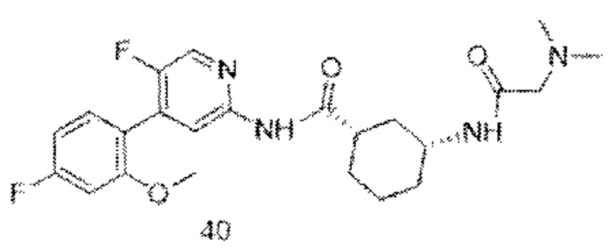
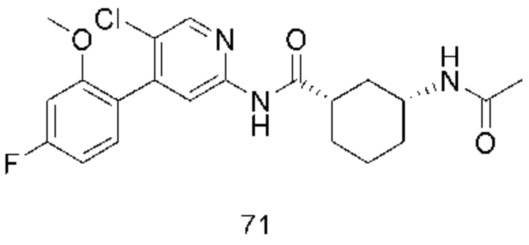
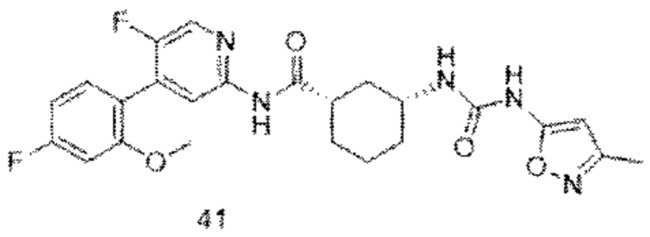
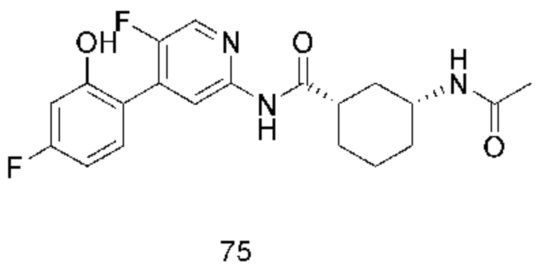
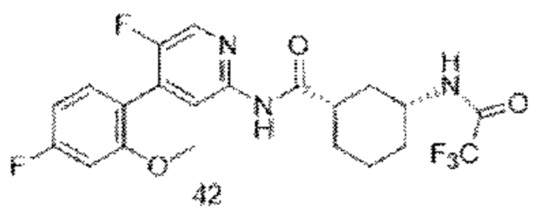
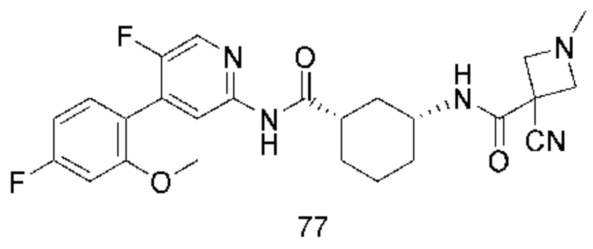
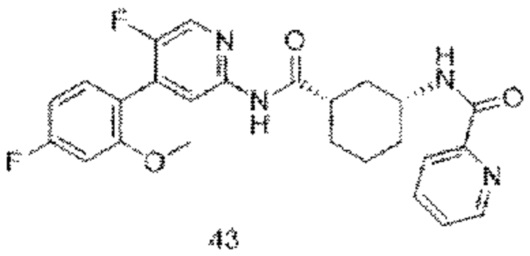
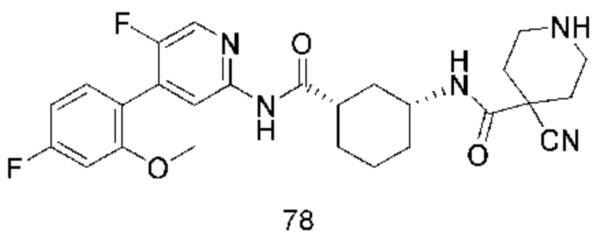
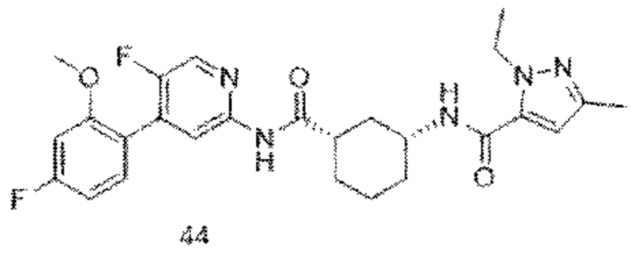
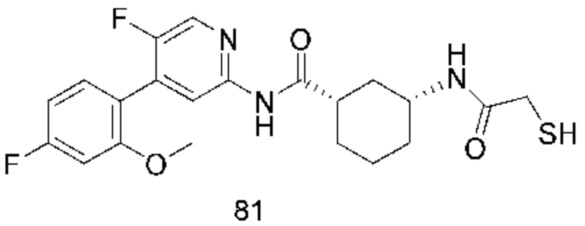
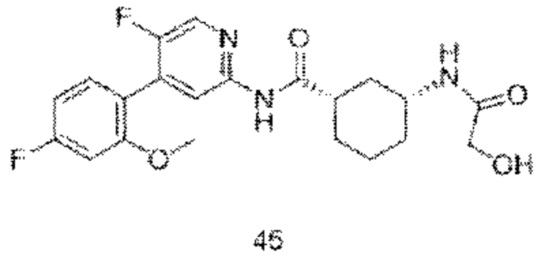
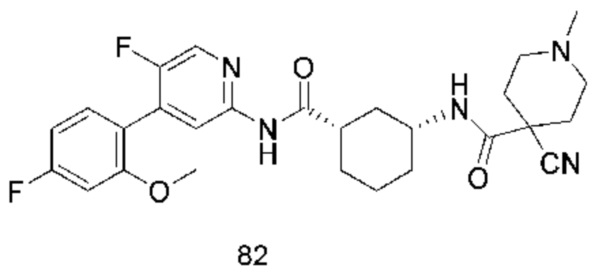
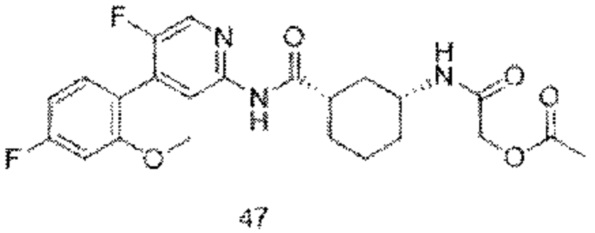
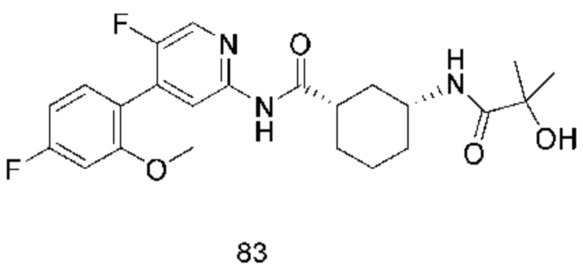
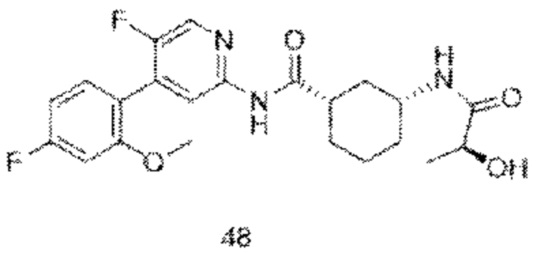
 .
.
2. Соединение по п.1 или его фармацевтически приемлемая соль, или его стереоизомер, где соединения имеют следующие структуры:
, , ,
, , ,
, , ,
, , ,
, , ,, , , и
.
3. Соединение по п.1 или его фармацевтически приемлемая соль, или его стереоизомер, где соединения имеют следующие структуры:
.
4. Фармацевтическая композиция с ингибирующей активностью в отношении циклинзависимой киназы 9, содержащая терапевтически эффективное количество соединения по любому из пп.1-3 или его фармацевтически приемлемую соль, или его стереоизомер.
5. Применение соединения по любому из пп.1-3 или его фармацевтически приемлемой соли, или его стереоизомера, или фармацевтической композиции по п.4 для предотвращения и/или лечения заболеваний, опосредованных активностью циклинзависимой киназы 9.
6. Применение соединения по п. 5, где заболевание представляет собой гематологическую опухоль или солидную опухоль.
7. Применение соединения по п. 6, где гематологическая опухоль представляет собой лейкоз.
8. Применение соединения по п. 7, где лейкоз представляет собой острый миелоидный лейкоз и острый промиелоцитарный лейкоз.
| WO 2011026911 A1, 10.03.2011 | |||
| WO 2011026904 A1, 10.03.2011 | |||
| WO 2011026917 A1, 10.03.2011 | |||
| WO 2011110612 A1, 15.09.2011 | |||
| EA 201890094 A1, 31.07.2018 | |||
| Albert T.K | |||
| et al | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| British Journal of Pharmacology, 2014, v.171, pp.55-68. | |||

