Ссылка на родственные заявки
Согласно настоящей заявке испрашивается приоритет в соответствии с китайской заявкой на выдачу патента с № CN201710698086,3, поданной 15 августа 2017 года.
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к ингибитору FGFR и его применению в изготовлении медикамента для лечения связанного с FGFR заболевания. В частности, настоящее изобретение относится к соединению, представленному формулой (I), и к его фармацевтически приемлемой соли.
Предшествующий уровень техники настоящего изобретения
Рецептор фактора роста фибробластов (FGFR) представляет собой рецептор для передачи сигнала фактора роста фибробластов (FGF). Его семейство включает в себя четырех представителей (FGFR1, FGFR2, FGFR3, FGFR4) и представляет собой гликопротеин, состоящий из внеклеточного подобного иммуноглобулину (Ig) структурного домена, гидрофобной чрезмембранной области и внутриклеточной части, включающей в себя область тирозинкиназы. Фактор роста фибробластов (FGF) посредством этих рецепторов (FGFR) играет важную роль во многих процессах физиологической регуляции, таких как пролиферация клеток, дифференцировка клеток, миграция клеток и ангиогенез. Существует много доказательств того, что аномалии сигнальных путей FGF (высокая экспрессия, амплификация генов, мутация генов, хромосомная рекомбинация и т.п.) напрямую связаны со многими патологическими процессами, такими как пролиферация, миграция, инвазия и ангиогенез опухолевых клеток. Таким образом, FGFR стал важной терапевтической целью, вызывающей широкий круг интересов в области исследований и разработок.
В международной заявке на выдачу патента WO 2015008844 раскрывается серия соединений, обладающих ингибиторной активностью в отношении FGFR, в том числе эталонные соединения 1 и 2. В международных заявках на выдачу патентов WO2013124316, WO2013087647 и в заявке на выдачу патента США US20130158000 раскрывается серия соединений, обладающих ингибиторной активностью в отношении FGFR, включающих в себя бензотиофеновую структуру, применяемую в настоящем изобретении, и эталонное соединение 3.
Краткое описание настоящего изобретения
Настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли
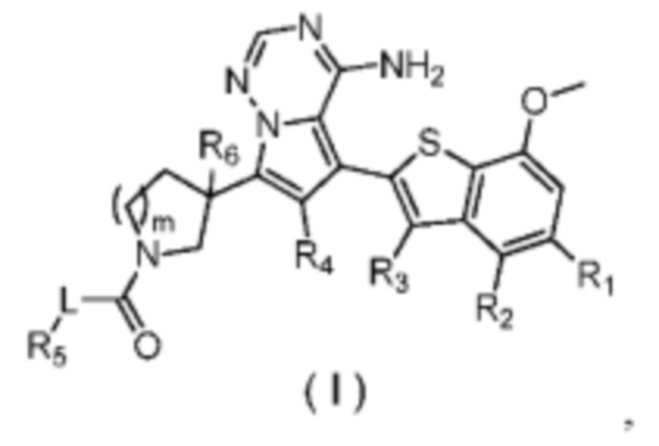
где
m представляет собой 1 или 2;
L выбран из простой связи, С2-4алкенила и С2-4алкинила;
R1 выбран из Н, галогена, ОН и NH2 или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1, 2 или 3 R группами;
R2 выбран из Н, F, Cl, Br, I, ОН и NH2;
R3 выбран из Н, галогена, ОН, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1, 2 или 3 R группами;
R4 выбран из Н, галогена, ОН, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1, 2 или 3 R группами;
R5 представляет собой Н или выбран из C1-3алкила, C1-3гетероалкила, C3-6циклоалкила и 4-6-членного гетероциклоалкила, которые необязательно замещены 1, 2 или 3 R группами;
R6 выбран из Н, галогена, ОН и NH2 или выбран из C1-3алкила, необязательно замещенного 1, 2 или 3 R группами;
R выбран из F, Cl, Br, I, ОН, NH2, CN, Me, CF3, N(CH3)2 и 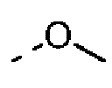 ;
;
гетероатом или гетерогруппа в C1-3гетероалкиле и 4-6-членном гетероциклоалкиле независимо и отдельно выбраны из-NH-, N, -О- и -S-;
в любом из вышеуказанных случаев число гетероатомов или число гетерогрупп независимо и отдельно выбраны из 1, 2 или 3.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R1 выбран из Н, галогена, ОН и NH2 или выбран из C1-3алкила и C1-3алкоксила, которые необязательно замещены 1, 2 или 3 R группами, и R группы определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R1 выбран из Н, F, Cl, Br, I, ОН, NH2, Me и .
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R3 выбран из Н, галогена, ОН, NH2 и CN или выбран из C1-3алкила, C1-3алкоксила и C1-3алкиламино, которые необязательно замещены 1, 2 или 3 R группами, и R группы определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R3 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, Me, CF3, 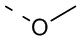 и
и 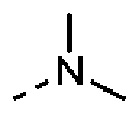 .
.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R4 выбран из Н, галогена, ОН, NH2 и CN или выбран из C1-3алкила, C1-3алкоксила и C1-3алкиламино, которые необязательно замещены 1, 2 или 3 R группами, и R группы определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R4 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, Me, CF3, 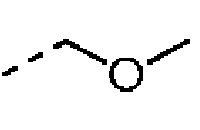 и .
и .
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R5 представляет собой Н или выбран из C1-3алкила, C1-3алкиламино и морфолинила, которые необязательно замещены 1, 2 или 3 R группами, и R группы определены в настоящем изобретении.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R5 выбран из Н, Me, Et, 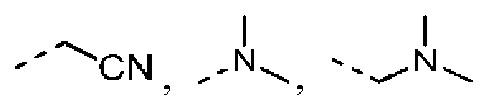 и
и 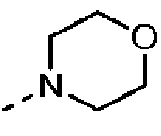 .
.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R6 выбран из Н, F, Cl, Br, I, ОН, NH2 и Me.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный L выбран из простой связи, 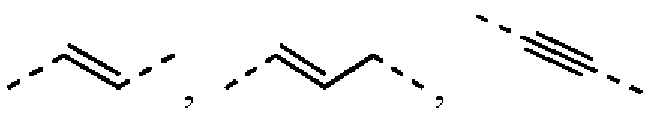 и
и  .
.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанная структурная единица 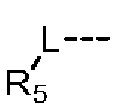 выбрана из
выбрана из  и
и  .
.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанная структурная единица 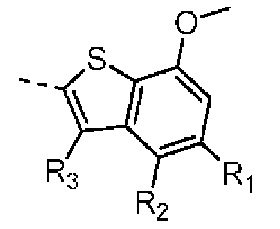 выбрана из:
выбрана из: 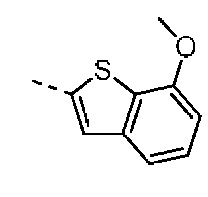 ,
, 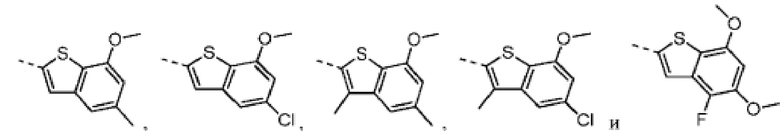 .
.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R1 выбран из Н, галогена, ОН и NH2, или выбран из С1-3алкила и C1-3алкоксила, которые необязательно замещены 1, 2 или 3 R группами, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R1 выбран из Н, F, Cl, Br, I, ОН, NH2, Me и , а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R3 выбран из Н, галогена, ОН, NH2 и CN или выбран из С1-3алкила, C1-3алкоксила и C1-3алкиламино, которые необязательно замещены 1, 2 или 3 R группами, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R3 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, Me, CF3,  и
и  , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R4 выбран из Н, галогена, ОН, NH2 и CN или выбран из C1-3алкила, C1-3алкоксила и C1-3алкиламино, которые необязательно замещены 1, 2 или 3 R группами, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R4 выбран из Н, F, Cl, Br, I, ОН, NH2, CN, Me, CF3, и 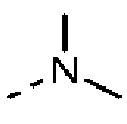 , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R5 представляет собой Н или выбран из C1-3алкила, C1-3алкиламино и морфолинила, которые необязательно замещены 1, 2 или 3 R группами, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R5 выбран из Н, Me, Et, 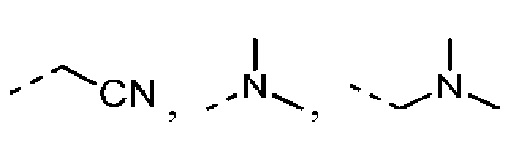 и
и 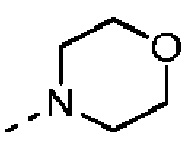 , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный R6 выбран из Н, F, Cl, Br, I, ОН, NH2 и Me, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанный L выбран из простой связи, 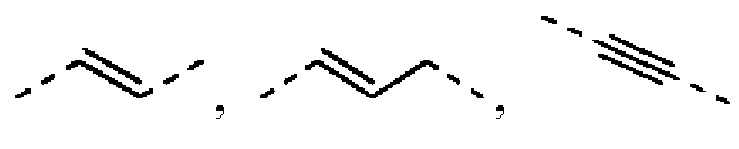 и
и  , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанная структурная единица 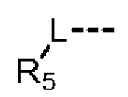 выбрана из
выбрана из  и
и  , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанная структурная единица 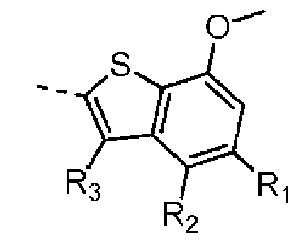 выбрана из
выбрана из 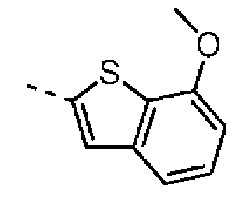 ,
,  и
и 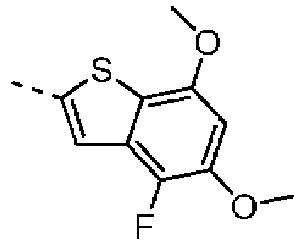 , а другие переменные определены выше.
, а другие переменные определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения оно относится к вышеуказанному соединению или его фармацевтически приемлемой соли, причем соединение выбрано из:
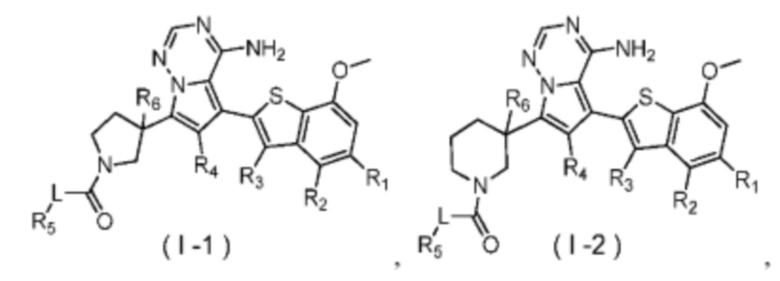
где
R1, R2, R3, R4, R5, R6 и L определены выше.
Согласно некоторым вариантам осуществления настоящего изобретения оно относится к вышеуказанному соединению или его фармацевтически приемлемой соли, причем соединение выбрано из:
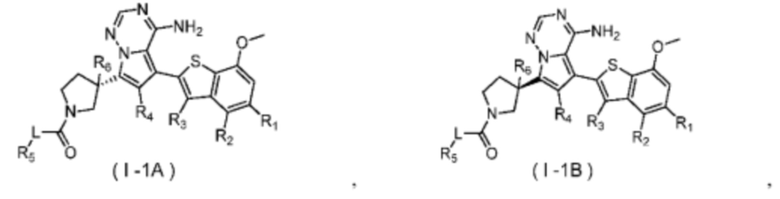
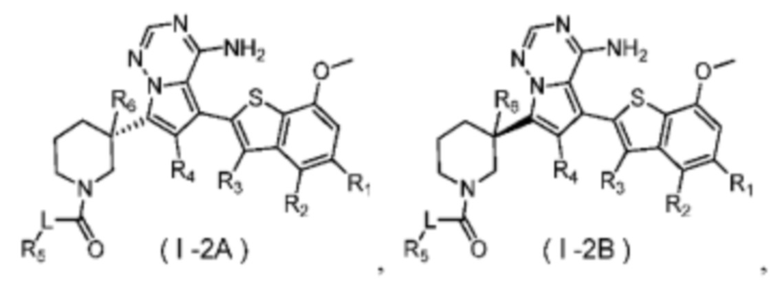
где
R1, R2, R3, R4, R5, R6 и L определены выше.
Настоящее изобретение также включает в себя некоторые варианты осуществления, которые получали из любой комбинации вышеуказанных переменных.
Настоящее изобретение дополнительно относится к соединению, представленному в любой из следующих формул, или его фармацевтически приемлемой соли:
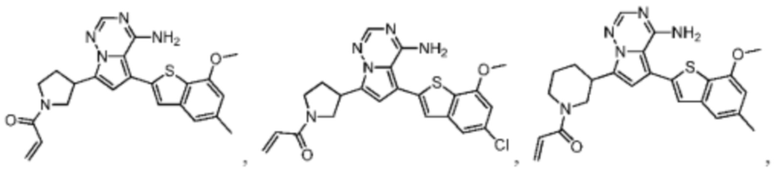
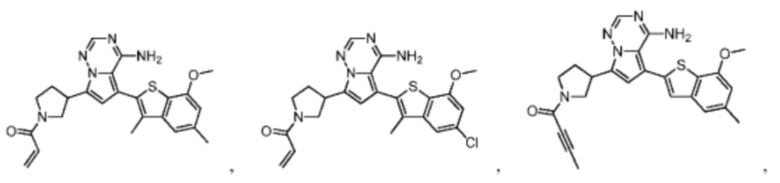
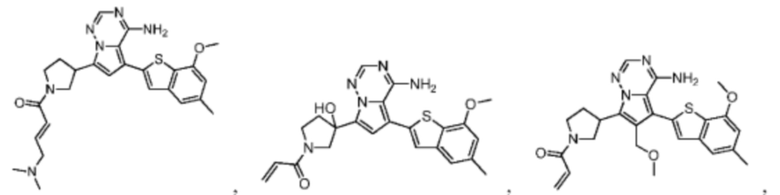
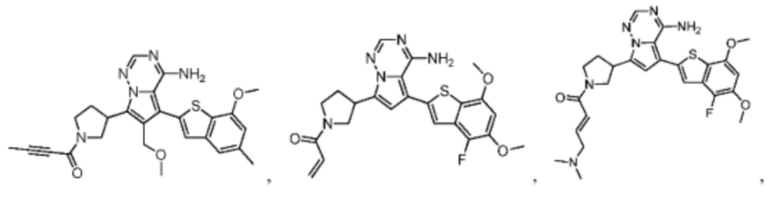
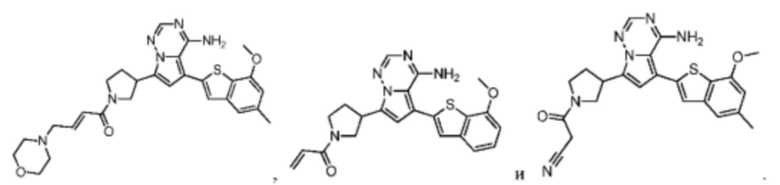
Согласно некоторым вариантам осуществления настоящего изобретения оно относится к вышеуказанному соединению или его фармацевтически приемлемой соли, причем соединение выбрано из:
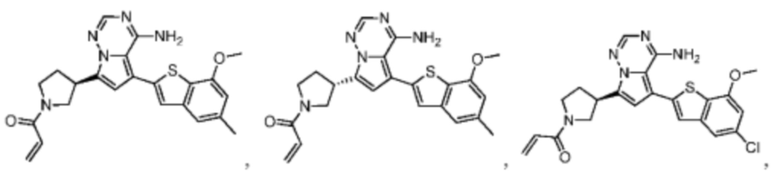
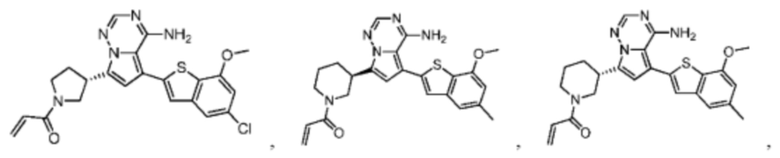
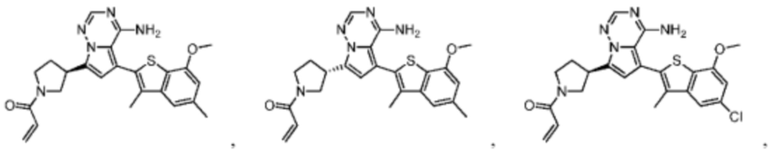
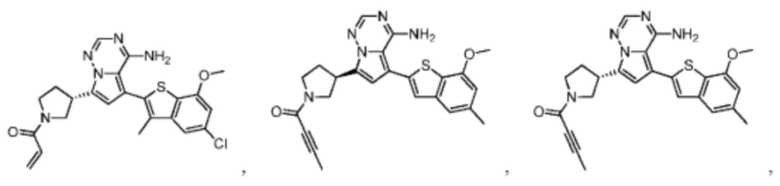
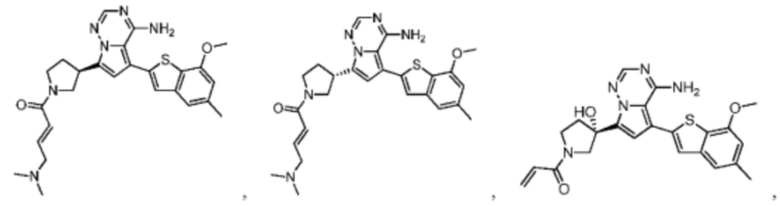
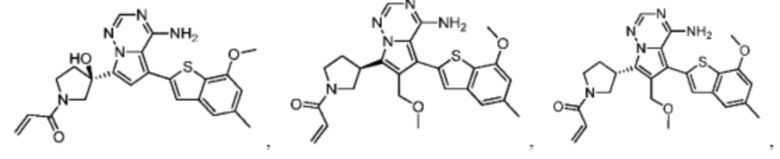
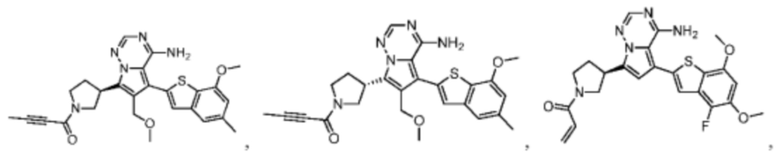
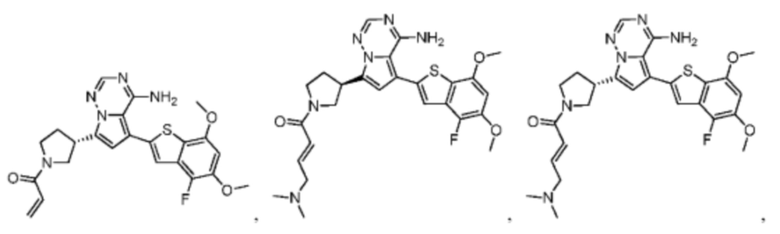
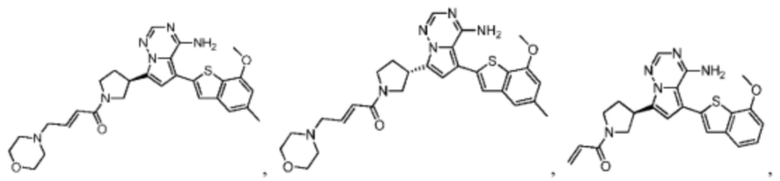
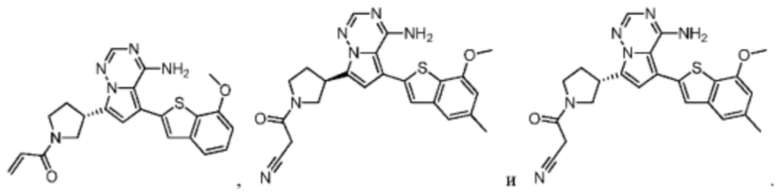
Настоящее изобретение дополнительно относится к фармацевтической композиции, которая содержит терапевтически эффективное количество вышеуказанного соединения или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя.
Настоящее изобретение дополнительно относится к применению вышеуказанного соединения или его фармацевтически приемлемой соли или вышеуказанной композиции при изготовлении лекарственного препарата для лечения связанного с FGFR заболевания.
Согласно некоторым вариантам осуществления настоящего изобретения вышеуказанное связанное с FGFR заболевание относится к солидной опухоли.
Технический эффект настоящего изобретения
Некоторые соединения по настоящему изобретению проявляют более высокие ингибиторные активности по отношению к FGFR дикого типа и мутантные.
Связанные определения
Если не отмечено иное, предусмотрено, что следующие термины и фразы, используемые в настоящем описании, характеризуются следующими значениями. Конкретный термин или фраза не должны рассматриваться как неопределенные или неясные без особого определения, а их следует понимать в их оригинальном значении. Если в настоящем описании встречается торговое наименование, предусмотрено, что оно относится к своему соответствующему коммерческому продукту или его активному ингредиенту. Используемый в настоящем описании термин «фармацевтически приемлемый» относится к таким соединениям, веществам, композициям и/или лекарственным формам, которые в пределах объема тщательной медицинской оценки являются подходящими для применения в контакте с тканями людей и животных без неспецифической токсичности, раздражения, аллергической реакции или других проблем или осложнений и в соответствии с подходящим соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, полученной из соединения, содержащего конкретный заместитель, который встречается в настоящем изобретении, и относительно не токсичной кислоты или основания. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, основно-аддитивная соль может быть получена приведением в контакт достаточного количества основания с нейтральной формой такого соединения в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые основно-аддитивные соли включают в себя соли натрия, калия, кальция, аммония, органического аммония или магния или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, кислотно-аддитивная соль может быть получена приведением в контакт достаточного количества кислоты с нейтральной формой такого соединения в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают в себя соли неорганических кислот, включая, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, гидрокарбонат, фосфорную кислоту, вторичный кислый фосфат, первичный кислый фосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфорную кислоту и т.п.; соли органических кислот, включая, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, пара-толуолсульфоновую кислоту, лимонную кислоту, виннокаменную кислоту, метансульфоновую кислоту и т.п.; соли аминокислот (такие как аргинин); и соли органических кислот, таких как глюкуроновая кислота (см. Berge et al., «Pharmaceutical Salts», Journal of Pharmaceutical Science 66: 1-19 (1977)). Определенные конкретные соединения по настоящему изобретению содержит как основные, так и кислотные функциональные группы, и, таким образом, могут быть превращены в их любые основные или кислотные аддитивные соли.
Предпочтительно, соль находится в контакте с основанием или кислотой традиционным способом, а затем исходное соединение выделяли, тем самым восстанавливая нейтральную форму соединения. Исходная форма соединения отличается от его различных солевых форм определенными физическими свойствами, такими как различная растворимость в полярном растворителе.
Используемая в настоящем описании «фармацевтически приемлемая соль» представляет собой производное соединения по настоящему изобретению, где исходное соединение модифицировано образованием соли с кислотой или с основанием. Примеры фармацевтически приемлемых солей включают в себя без ограничения неорганической или органической кислоты соли оснований, такие как амины, щелочного металла или органические соли кислот, таких как карбоновые кислоты, и т.п. Фармацевтически приемлемые соли включают в себя традиционные не токсичные соли или четвертичные аммонийные соли исходного соединения, например, соли, образованные из не токсичных неорганических или органических кислот. Традиционные не токсичные соли включают в себя без ограничения соли, полученные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодата, гидроксила, гидроксинафталина, изетионовой кислоты, молочной кислоты, лактозы, додецилсульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактуроновой кислоты, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, иминодиуксусной кислоты, янтарной кислоты, сульфамовой кислоты, сульфаниловой кислоты, серной кислоты, дубильной кислоты, виннокаменной кислоты и пара-толуолсульфоновой кислоты.
Фармацевтически приемлемая соль по настоящему изобретению может быть синтезирована из исходного соединения, содержащего кислотную группу или основную группу, традиционным химическим способом. Как правило, такие соли получали путем осуществления взаимодействия таких соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе или их смеси. Как правило, предпочтительной является неводная среда, такая как этиловый эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. В настоящем изобретении рассмотрены все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-пары энантиомеров, (R)- и (S)-энантиомеры, дистереоизомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и их другие смеси, такие как энантиомерно или диастереоизомерно обогащенные смеси, все из которых подпадали в пределы объема настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в таком заместителе, как алкильная группа. Все такие изомеры и их смеси включены в объем настоящего изобретения.
Если не отмечено иное, термины «энантиомеры» или «оптические изомеры» относятся к стереоизомерам в отношении зеркального отражения друг к другу.
Если не отмечено иное, термин «цис-транс-изомер» или «геометрический изомер» образуется путем невозможности двойных связей или простых связей образующих кольцо атомов углерода свободно вращаться.
Если не отмечено иное, термин «диастереоизомер» относится к стереоизомеру для которого каждая из молекул содержит два или более хиральных центров и молекулы находятся в отношении не зеркального отражения друг к другу.
Если не отмечено иное, «(D)» или «(+)» означает декстроротацию, «(L)» или «(-)» означает левостороннее вращение и «(DL)» или «(±)» означает рацемический.
Если не отмечено иное, абсолютная конфигурация стереоцентра выражена при помощи связи в виде жирной клинообразной линии 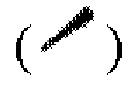 и при помощи связи в виде пунктирной клинообразной линии
и при помощи связи в виде пунктирной клинообразной линии 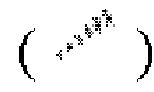 , относительная конфигурация стереоцентра выражена при помощи связи в виде жирной прямой линии
, относительная конфигурация стереоцентра выражена при помощи связи в виде жирной прямой линии 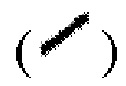 и при помощи связи в виде пунктирной прямой линии
и при помощи связи в виде пунктирной прямой линии 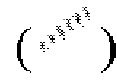 , связь в виде жирной клинообразной линии
, связь в виде жирной клинообразной линии 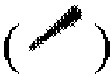 и/или связь в виде пунктирной клинообразной линии
и/или связь в виде пунктирной клинообразной линии 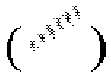 выражены при помощи волнистой линии
выражены при помощи волнистой линии 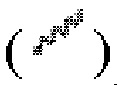 , или связь в виде жирной прямой линии
, или связь в виде жирной прямой линии 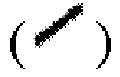 и/или связь в виде пунктирной прямой линии
и/или связь в виде пунктирной прямой линии  выражены при помощи волнистой линии
выражены при помощи волнистой линии  .
.
Соединения по настоящему изобретению могут существовать в конкретных формах. Если не отмечено иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре изомеры, содержащие разные функциональные группы, находятся в динамическом равновесии и могут быть быстро превращены друг в друга. Если таутомеры возможны (например, в растворе), может быть достигнуто химическое равновесие таутомеров. Например, протонные таутомеры (также известные как прототропные таутомеры) включают в себя взаимные превращения путем миграции протона, такие как кето-енольная изомеризация и имин-енаминовая изомеризация. Валентные таутомеры включают в себя рекомбинацию некоторых связывающих электронов для взаимного превращения. Среди прочих, конкретный пример кето-енольной таутомеризации представляет собой взаимное превращение между двумя таутомерами пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
Если не отмечено иное, термин «обогащенный в изомере», «изомерно обогащенный», «обогащенный в энантиомере» или «энантиомерно обогащенный» относится к содержимому изомера или энантиомера, что менее чем 100%, и содержимое изомера или энантиомера составляет более чем или равно 60%, или более чем или равно 70%, или более чем или равно 80%, или более чем или равно 90%, или более чем или равно 95%, или более чем или равно 96%, или более чем или равно 97%, или более чем или равно 98%, или более чем или равно 99%, или более чем или равно 99,5%, или более чем или равно 99,6%, или более чем или равно 99,7%, или более чем или равно 99,8%, или более чем или равно 99,9%.
Если не отмечено иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разнице между относительным процентным содержанием двух изомеров или энантиомеров. Например, если содержимое одного изомера или энантиомера составляет 90%, а содержимое другого изомера или энантиомера составляет 10%, изомерный или энантиомерный избыток (э. и. значение) составляет 80%.
Оптически активные (R)- и (S)-изомеры, а также D и L изомеры, могут быть получены при помощи хирального синтеза, или хиральных реагентов, или другими традиционными методиками. Если необходим энантиомер соединения по настоящему изобретению, он может быть получен асимметричным синтезом или дериватизацией при помощи хирального вспомогательного элемента, при этом полученную диастереомерную смесь разделяли, а чистый требуемый энантиомер обеспечивали при помощи отщепления группы. Альтернативно, если молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), диастереомерная соль образуется с соответствующей оптически активной кислотой или основанием, а затем диастереомерное расщепление проводили традиционным способом, хорошо известным из области техники, а затем чистый энантиомер восстанавливали и получали. Кроме того, разделение энантиомеров и диастереомеров обычно проводили с применением хроматографии, при которой использовали хиральную неподвижную фазу, и ее необязательно объединяли с химической дериватизацией (например, образование карбамата из амина). Соединения по настоящему изобретению могут содержать атомный изотоп в неестественной пропорции при одном или нескольких атомах, что составляют соединение. Например, соединения могут быть помечены радиоактивными изотопами, такими как тритий (3Н), йод-125 (125I) или С-14 (14С). Превращения всех изотопных композиций соединений по настоящему изобретению, радиоактивных или нет, включены в объем настоящего изобретения.
«Необязательный» или «необязательно» относится к событиям или условиям, описанным позже, которые могут, но не обязательно, происходить, и это описание включает в себя ситуации, в которых события или условия происходят, и ситуации, в которых события или условия не происходят.
Термин «замещенный» относится к замене любого одного или нескольких атомов водорода, которые могут включать в себя дейтерий и варианты водорода на конкретном атоме с заместителем, поскольку валентность конкретного атома является нормальной и замещенное соединение является стабильным. Если заместителем является кислород (=O), это означает, что два атома водорода являются замещенными. Кислородное замещение не происходит на ароматической группе. Термин «необязательно замещенный» означает, что он может быть или не быть замещенным, и, если не отмечено иное, вид и число заместителей могут быть производными на основе химической доступности.
Если любая переменная (такая как R) встречается более чем один раз в композиции или структуре соединения, ее определение в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R заместителями, группа может быть необязательно замещенной не более чем двумя R заместителями, и для каждого заместителя R содержит независимый вариант. Кроме того, комбинация заместителей и/или их вариантов разрешена, только если такая комбинация приводит к стабильному соединению.
Если число связывающей группы равно 0, такое как -(CRR)0-, это означает, что связывающая группа представляет собой простую связь.
Если одна из переменных выбрана из простой связи, две группы, соединенные с ней, прямо соединены. Например, если L представляет собой простую связь в A-L-Z, на самом деле структурой является A-Z.
Если заместитель свободный, это означает, что заместитель отсутствует. Например, если X в А-Х свободный, это означает, что на самом деле структурой является А. Если заместитель может быть присоединен к более чем одному атому на кольце, этот заместитель может быть связан с любым атомом на кольце. Например, структурная единица 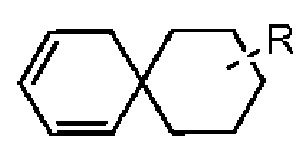 или
или 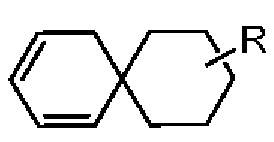 представляет, что замещение R заместителем может возникать на любом положении циклогексила или циклогексадиена. В случае отсутствия указания того, какой атом в изложенном заместителе будет присоединен к замещаемой группе, такой заместитель может быть присоединен через любой ее атом. Например, пиридильная группа в качестве замещающей группы может быть присоединена к замещаемой группе при помощи любого атома углерода на пиридиновом кольце. В случае отсутствия указания направления связывания изложенной связывающей группы, его направление связывания является произвольным. Например, в
представляет, что замещение R заместителем может возникать на любом положении циклогексила или циклогексадиена. В случае отсутствия указания того, какой атом в изложенном заместителе будет присоединен к замещаемой группе, такой заместитель может быть присоединен через любой ее атом. Например, пиридильная группа в качестве замещающей группы может быть присоединена к замещаемой группе при помощи любого атома углерода на пиридиновом кольце. В случае отсутствия указания направления связывания изложенной связывающей группы, его направление связывания является произвольным. Например, в 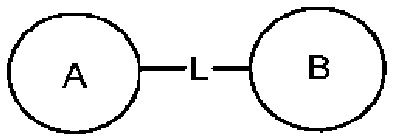 связывающая группа L представляет собой -M-W-; в это же время -M-W- может или связывать кольцо А и кольцо В в том же направлении, что и порядок чтения слева направо с образованием
связывающая группа L представляет собой -M-W-; в это же время -M-W- может или связывать кольцо А и кольцо В в том же направлении, что и порядок чтения слева направо с образованием 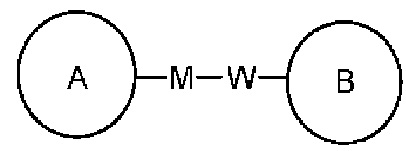 , или связывать кольцо А и кольцо В в направлении, противоположном порядку чтения слева направо с образованием
, или связывать кольцо А и кольцо В в направлении, противоположном порядку чтения слева направо с образованием 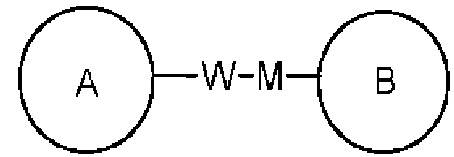 . Комбинация связывающей группы, заместителей и/или их вариантов разрешена, только если такая комбинация приводит к стабильному соединению.
. Комбинация связывающей группы, заместителей и/или их вариантов разрешена, только если такая комбинация приводит к стабильному соединению.
Если не отмечено иное, «кольцо» представляет собой замещенный или незамещенный циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил, арил или гетероарил. Так называемое кольцо включает в себя простое кольцо, соединенное кольцо, спирокольцо, конденсированное кольцо или кольцо с мостиковыми связями. Число атомов на кольце обычно определено как номер элемента колец. Например, «5-7-членное кольцо» означает 5-7 атомов, расположенных по кругу. Если не отмечено иное, кольцо необязательно содержит 1-3 гетероатома. Таким образом, «5-7-членное кольцо» включает в себя, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает в себя пиридил и пиперидил, но не включает в себя фенил. Термин «кольцо» также включает в себя кольцевую систему, содержащую по меньшей мере одно кольцо, каждое из «колец» независимо попадает под указанное определение.
Если не отмечено иное, термин «гетероцикл» или «гетероциклил» означает стабильное, содержащее гетероатом или гетерогруппу моноциклическое, бициклическое или трициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранные из N, О и S, где любой из вышеуказанных гетероциклов может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы азота и серы необязательно могут быть окислены (т.е., NO и S(O)p, где р представляет собой 1 или 2). Атом азота может быть замещенным или незамещенным (т.е., N или NR, где R представляет собой Н или другие заместители, которые были определены в настоящем описании). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, описанный в настоящем изобретении гетероцикл может подвергаться замещению в положении углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. Предпочтительным вариантом осуществления является тот, в котором общее число S и О атомов в гетероцикле превышает 1, такие гетероатомы не являются смежными друг с другом. Другим предпочтительным вариантом осуществления является тот, в котором общее число S и О атомов в гетероцикле не превышает 1. Используемый в настоящем описании термин «ароматическая гетероциклическая группа» или «гетероарил» означает стабильное 5-, 6-, 7-членное моноциклическое или бициклическое или 7-, 8-, 9- или 10-членное бициклическое гетероциклическое ароматическое кольцо, которое содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, которые независимо выбраны из N, О и S. Атом азота может быть замещенным или незамещенным (т.е., N или NR, если R представляет собой Н или другие заместители, которые были определены в настоящем описании). Гетероатомы азота и серы необязательно могут быть окислены (т.е., NO и S(O)p, где р представляет собой 1 или 2). Следует отметить, что общее число атомов S и О в ароматическом гетероцикле не превышает 1. Кольцо с мостиковыми связями также включено в определение гетероцикла. Если один или несколько атомов (т.е., С, О, N или S) соединены с двумя не смежными атомами углерода или азота, образуется кольцо с мостиковыми связями. Предпочтительное кольцо с мостиковыми связями включает в себя без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостик всегда превращает простое кольцо в трициклическое кольцо. В кольце с мостиковыми связями заместитель в кольце также может располагаться на мостике.
Пример гетероциклического соединения включает в себя без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, меркаптобензофуранил, меркаптобензофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензо фуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонин, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридинооксазол, пиридиноимидазол, пиридинотиазол, пиридинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил, 1Н-1,2,4-триазолил, 4Н-1,2,4-триазолил и ксантенил. Также включены соединение с конденсированным кольцом и соединение со спирокольцом.
Если не отмечено иное, термин «гидрокарбил» или его родовое понятие (например, алкил, алкенил, алкинил, арил и т.п.), сам по себе или в комбинации с другим заместителем, относится к углеводородному радикалу с неразветвленной, разветвленной цепью или циклическому углеводородному радикалу или их любой комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно- или полизамещенными, могут быть моно валентными (например, метил), дивалентными (например, метилен) или поливалентными (например, метинил), также могут включать в себя дивалентную или поливалентную группу, содержат конкретное число атомов углерода (например, С1-С12 означает 1-12 атомов углерода, С1-12 выбран из C1, С2, С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12; С3-12 выбран из С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12). Термин «гидрокарбил» включает в себя без ограничения алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает в себя неразветвленный и циклический гидрокарбил, особенно включая в себя без ограничения алкил, алкенил и алкинил. Ароматический гидрокарбил включает в себя без ограничения 6-12-членный ароматический гидрокарбил, такой как фенил, нафталенил и т.п. Согласно некоторым вариантам осуществления термин «гидрокарбил» относится к неразветвленной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать в себя дивалентную или поливалентную группу. Пример насыщенной гидрокарбильной группы включает в себя без ограничения метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, цикло пропил метил и гомолог или изомер н-пентила, н-гексила, н-гептила, н-октила и подобные группы. Ненасыщенный гидрокарбил содержит одну или более чем одну двойную или тройную связь, и их пример включает в себя без ограничения этенил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и более высокие гомологи и изомеры.
Если не отмечено иное, термин «гетерогидрокарбил» или его родовое понятие (например, гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.п.), сам по себе или в комбинации с другим термином, относится к стабильной неразветвленной, разветвленной или циклической углеводородной группе или любой их комбинации, которая состоит из конкретного числа атомов углерода и по меньшей мере одного гетероатома. Согласно некоторым вариантам осуществления, термин «гетероалкил», сам по себе или в комбинации с другим термином, относится к стабильной неразветвленной или разветвленной углеводородной группе или любой их комбинации, которая состоит из конкретного числа атомов углерода и по меньшей мере одного гетероатома. Согласно конкретному варианту осуществления гетероатом выбран из В, О, N и S, где атомы азота и серы необязательно окислены, а атом азота является необязательно кватернизирован. Гетероатом или гетерогруппа могут быть расположены в любом внутреннем положении гетерогидрокарбила, включая положение, где гидрокарбил присоединен к оставшейся части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или алкоксил, в которой О замене S) принадлежат к идиоматическому выражению и относятся к алкильной группе, соединенной с оставшейся частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Пример включает в себя без ограничения -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -СН=СН-O-СН3, -CH2-CH=N-OCH3 и -СН=СН-N(CH3)-СН3. Вплоть до двух гетероатомов могут быть последовательными, например, -CH2-NH-OCH3.
Если не отмечено иное, термин «циклогидрокарбил», «гетероциклогидрокарбил» или его родовое понятие (например, арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.п.) сам по себе или в комбинации с другим термином относится к циклизированному «гидрокарбилу» и «гетерогидрокарбилу», соответственно. Более того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила и гетероциклоалкила), гетероатом может занимать положение, в котором гетероцикл присоединен к оставшемуся положению молекулы. Пример циклогидрокарбила включает в себя без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.п. Неограничивающий пример гетероциклоалкила включает в себя 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран индол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если не отмечено иное, термин «алкил» относится к неразветвленному или разветвленному насыщенному гидрокарбилу, который может быть монозамещенным (например, -CH2F) или полизамещенным (например, -CF3), и может быть моновалентным (например, метил), дивалентным (например, метилен) или поливалентным (например, метенил). Пример алкила включает в себя метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т.п.
Если не отмечено иное, термин «алкенил» относится к алкильной группе, содержащей одну или более чем одну двойную связь углерод-углерод в любом положении цепи, которая может быть монозамещенной или полизамещенной и может быть моновалентной, дивалентной или поливалентной. Пример алкенила включает в себя этенил, пропенил, бутенил, пентенил, гексенил, 1,3-бутадиенил, 1,3-пентадиенил, 1,3-гексадиенил и т.п.
Если не отмечено иное, термин «алкинил» относится к алкильной группе, содержащей одну или более чем одну тройную связь углерод-углерод в любом положении цепи, которая может быть монозамещенной или полизамещенной и может быть моновалентной, дивалентной или поливалентной. Пример алкинила включает в себя этинил, пропинил, бутинил, пентинил и т.п.
Если не отмечено иное, циклоалкил включает в себя любой стабильный циклический или полициклический гидрокарбил, любой атом углерода которого является насыщенным, и который может быть монозамещенным или полизамещенным и может быть моновалентным, дивалентным или поливалентным. Пример циклоалкила включает в себя без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодекан и т.п.
Если не отмечено иное, термин «гало» или «галоген», сам по себе или как часть другого заместителя, относится к атому фтора, хлора, брома или йода. Более того, подразумевается, что термин «галогеналкил» включает в себя моногалогеналкил и полигало геналкил. Например, подразумевается, что термин «галоген(С1-С4)алкил» включает в себя без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п. Если не отмечено иное, пример галогеалкила включает в себя без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
«Алкокси» представляет собой любой алкил, определенный выше, с конкретным числом атомов углерода, присоединенный кислородным мостиком. Если не отмечено иное, C1-6 алкокси включает в себя C1, С2, С3, С4, С5 и С6 алкокси. Пример алкокси включает в себя без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
Соединение по настоящему изобретению может быть получено различными способами синтеза, хорошо известными специалистам настоящей области техники, включая следующие перечисленные варианты осуществления, варианты осуществления, образованные следующими перечисленными вариантами осуществления в комбинации с другими химическими способами синтеза, и эквивалентными режимами подстановки, хорошо известными специалистам настоящей области техники. Предпочтительный вариант осуществления включает в себя без ограничения примеры по настоящему изобретению.
Все используемые в настоящем изобретении растворители являются коммерчески доступными. В настоящем изобретении используют следующие аббревиатуры: водн. представляет собой воду; HATU представляет собой O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC представляет собой N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; мета-СРВА представляет собой 3-хлорпероксибензойную кислоту; экв. представляет собой эквивалент, эквивалентное количество; CDI представляет собой карбонилдиимидазол; DCM представляет собой метиленхлорид; РЕ представляет собой петролейный эфир; DIAD представляет собой диизопропилазодиформиат; DMF представляет собой N,N-диметилформамид; DMSO представляет собой диметилсульфоксид; EtOAc представляет собой этилацетат; EtOH представляет собой этанол; МеОН представляет собой метанол; CBz представляет собой бензилоксикарбонил, аминозащитную группу; ВОС представляет собой трет-бутоксилкарбонил, аминозащитную группу; НОАс представляет собой уксусную кислоту; NaCNBH3 представляет собой цианоборгидрид натрия; к.т. представляет собой комнатную температуру; O/N представляет собой всю ночь; THF представляет собой тетрагидрофуран; Boc2O представляет собой ди-трет-бутилдикарбонат; TFA представляет собой трифторуксусную кислоту; DIPEA представляет собой диизопропилэтиламин; SOCl2 представляет собой тионилхлорид; CS2 представляет собой бисульфид углерода; TsOH представляет собой паратолуолсульфоновую кислоту; NFSI представляет собой N-фтор-N-(бензолсульфонил)бензолсульфонамид; NCS представляет собой 1-хлорпирролидин-2,5-дион; н-Bu4NF представляет собой тетрабутиламмония фторид; iPrOH представляет собой 2-пропиловый спирт; т.пл. представляет собой точку плавления; LDA представляет собой диизопропиламид лития.
Соединения называли вручную или при помощи программного обеспечения ChemDraw®, и в коммерчески доступных соединениях использовали названия из каталогов их поставщиков.
Подробное описание предпочтительных вариантов осуществления
Настоящее изобретение конкретно будет описано ниже при помощи примеров, но оно не подразумевает никаких неприемлемых ограничений настоящего изобретения. Настоящее изобретение подробно было описано здесь и его конкретные варианты осуществления также раскрыты. Специалистам настоящей области техники будет очевидно, что различные изменения и улучшения могут быть сделаны по отношению к конкретным вариантам осуществления настоящего изобретения без отклонения от сущности и объема настоящего изобретения.
Иллюстративный пример 1: WXR1
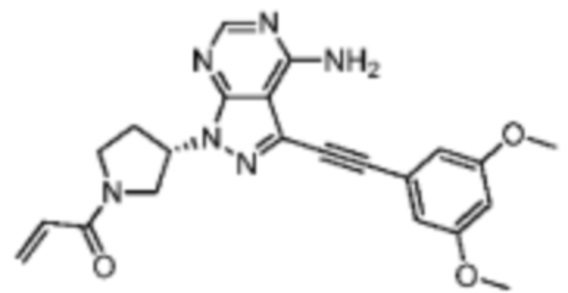
Соединение WXR1 синтезировали на основании пути, описанного в патентной заявке WO2015008844. 1Н ЯМР (400 МГц, DMSO-d6) δ=8.40 (d, J=3.0 Гц, 1 Н), 6.93 (d, J=2.5 Гц, 2 Н), 6.74-6.52 (m, 2 H), 6.20-6.16 (m, 1 Н), 5.74-5.69 (m, 1 Н), 5.45-5.61 (m, 1 Н), 4.12-3.90 (m, 2 Н), 3.90-3.79 (m, 8Н), 2.47-2.30 (m, 2Н). MS масса/заряд: 419.1 [М+Н]+.
Иллюстративный пример 2: WXR2
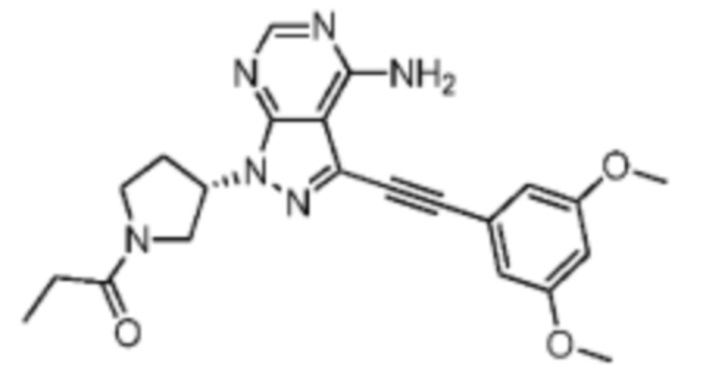
Соединение WXR2 синтезировали на основании пути, описанного в патентной заявке WO2015008844. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=8.28 (s, 1Н), 6.83 (br s, 2H), 6.60 (d, J=2.4 Гц, 1H), 5.65-5.44 (m, 1H), 4.12-3.98 (m, 1H), 3.97-3.88 (m, 2H), 3.83 (s, 6H), 3.82-3.74 (m, 1H), 3.74-3.63 (m, 1H), 2.63-2.53 (m, 1H), 2.51-2.35 (m, 3H), 1.22-1.12 (m, 3H). MS масса/заряд: 421.1 [M+H]+.
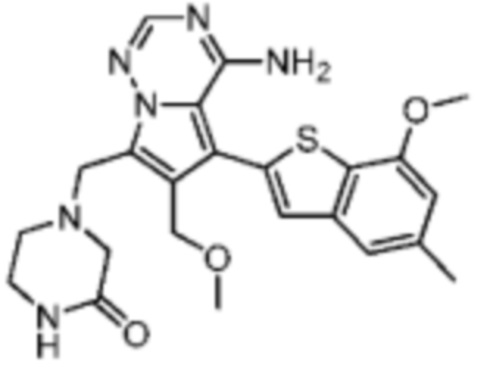
Иллюстративный пример 3: WXR3
Иллюстративный пример 4: Синтез соединения AZD4547
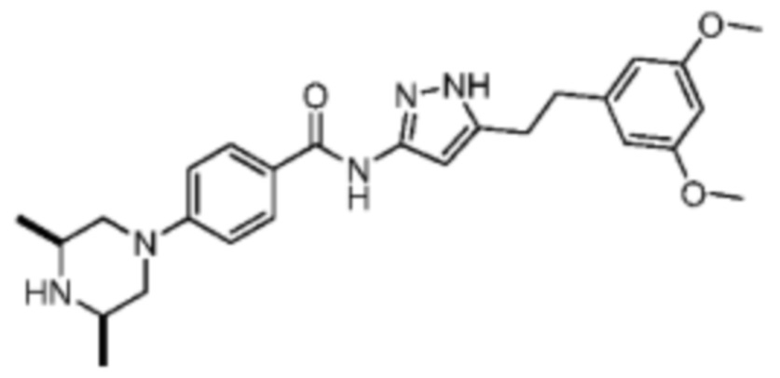
Соединение AZD4547 синтезировали на основании пути, описанного в патентной заявке WO2009153592. 1H ЯМР (400 МГц, дейтерированный метанол) δ: 7.93 (d, J=8.8 Гц, 2 Н), 7.13 (d, J=8.8 Гц, 2 Н), 6.37 (s, 2 Н), 6.33 (s, 1 Н), 6.31 (s, 1 Н), 4.12-4.09 (m, 2 Н), 3.74 (s, 6 H), 3.51-3.48 (m, 2 H), 2.99-2.93 (m, 4 H), 2.84 (t, J=12.4 Гц, 2 H), 1.42 (d, J=6.4 Гц, 6 H). MS масса/заряд: 464.4 [М+Н]+.
Иллюстративный пример 5: Синтез соединения BGJ398
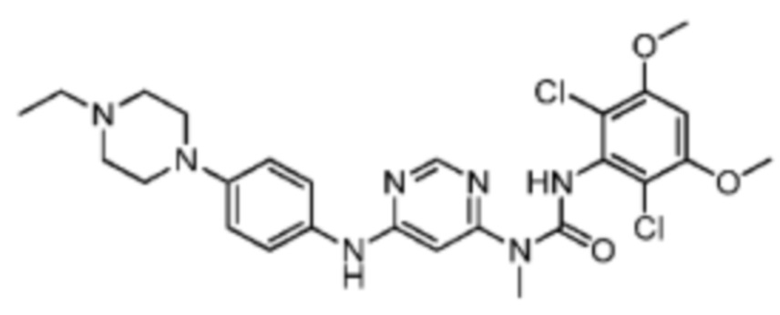
Соединение BGJ398 синтезировали на основании пути, описанного в патентной заявке WO2006000420. 1Н ЯМР (400 МГц, дейтерированный метанол) δ: 8.40 (s, 1 H), 7.46 (d, J=8.8 Гц, 2 H), 7.09 (d, J=8.8 Гц, 2 Н), 6.82 (s, 1 Н), 6.49 (s, 1 H), 3.96 (s, 6 H), 3.86 (d, J=12.0 Гц, 2 H), 3.69 (d, J=12.0 Гц, 2 Н), 3.43 (s, 3 H), 3.33-3.20 (m, 4 Н), 3.08 (t, J=12.4 Гц, 2 Н), 1.42 (t, J=7.2 Гц, 3 H). MS масса/заряд: 560.1 [М+Н]+.
Иллюстративный пример 6: Соединение JNJ493
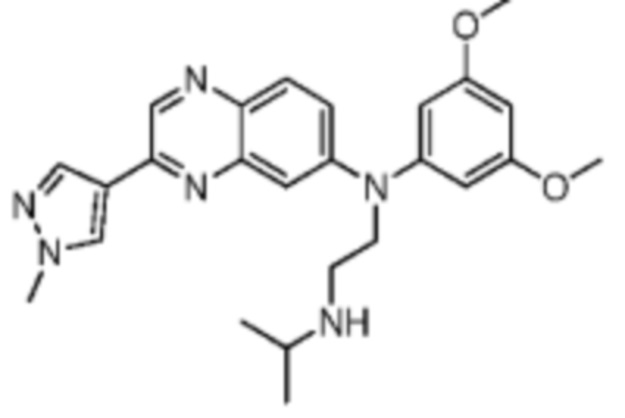
JNJ493 покупали у Shanghai Haoyuan Biotechnology Co., Ltd. (CAS: 1346242-81-6). 1H ЯМР (400 МГц, дейтерированный метанол) δ: 8.86 (s, 1H), 8.41 (s, 1H), 8.24 (s, 1H), 7.77 (d, J=9.2 Гц, 1H), 7.35 (dd, J=2.4, 9.3 Гц, 1H), 7.24 (d, J=2.4 Гц, 1H), 6.53-6.43 (m, 3H), 4.08-3.97 (m, 5H), 3.80 (s, 6H), 2.97 (t, J=7.2 Гц, 2H), 2.90-2.78 (m, 1H), 1.10 (d, J=6.4 Гц, 6H). MS масса/заряд: 447.2 [M+H]+.
Иллюстративный пример 7: Соединение WXR4
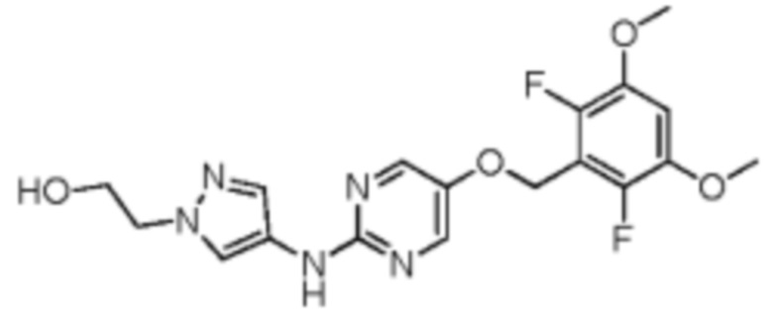
Соединение WXR4 синтезировали на основании пути, описанного в патентной заявке US20140142084. 1H ЯМР (400 МГц, дейтерированный метанол) δ: 8.41 (s, 2 Н), 8.13 (s, 1 Н), 7.86 (s, 1 Н), 6.98 (t, J=8.3 Гц, 1 Н), 5.25 (s, 2 Н), 4.34 (t, J=5.0 Гц, 2 Н), 3.95 (t, J=5.3 Гц, 2 Н), 3.91 (s, 6 Н). MS масса/заряд: 408.1 [М+Н]+.
Промежуточное соединение А1:
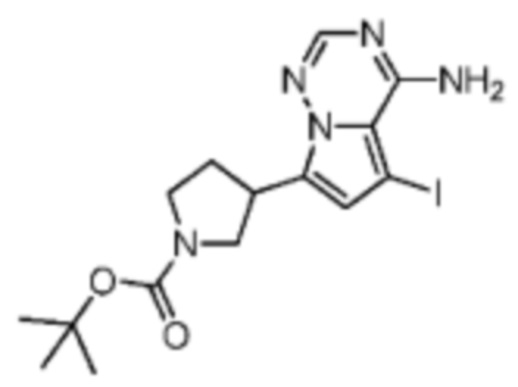
Путь синтеза:
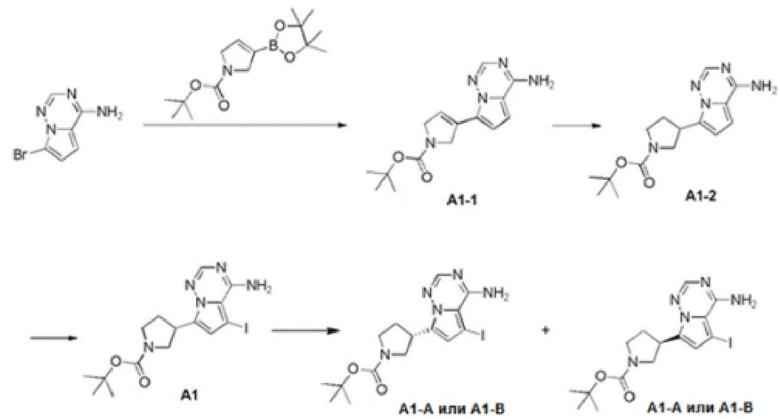
Стадия 1: Синтез соединения A1-1
При комнатной температуре 4-амино-7-бромпирроло[2,1-f][1,2,4]триазин (3,00 г, 14,1 ммоль, 1,00 экв.) сначала растворяли в смешанном растворе 1,4-д и океан а (40 мл) и воды (8 мл), а затем к смешанному раствору последовательно добавляли Н-Вос-2,5-дигадро-1Н-пиррол-1-пинакол6орат (4,36 г, 14,8 ммоль, 1,05 экв.), фосфат калия (8,97 г, 42,2 ммоль, 3,00 экв.) и 1,1'-бис(цифенилфосфино)ферроцен палладия хлорид (1,03 г, 1,41 ммоль, 0,10 экв.). При защите при помощи азота реакционный раствор нагревали до 80°С и перемешивали в течение 2 часов. После завершения реакции реакционный раствор охлаждали до 25°С и выливали в 20 мл воды. Образовывалось черное твердое вещество. Черное твердое вещество собирали фильтрацией, а затем растворяли в смешанном растворе дихлорметана/метанола (100 мл, 5/1) и снова фильтровали. Фильтрат сушили над безводным сульфатом натрия, а органический растворитель удаляли ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт переводили во взвесь при помощи этилацетата (30 мл) и фильтровали с получением соединения A1-1. LCMS (EST) масса/заряд: 302.1 [М+Н]+, 1H ЯМР (400 МГц, дейтерированный хлороформ) δ=8.05 (s, 1Н), 6.98-6.84 (m, 1Н), 6.72-6.54 (m, 2Н), 4.67-4.49 (m, 2Н), 4.44-4.30 (m, 2Н).
Стадия 2: Синтез соединения А1-2
При комнатной температуре гидр оксид палладия (615 мг, 438 ммоль) добавляли к раствору соединения А1-1 (1,20 г, 3,98 ммоль, 1,00 экв.) в метаноле (30 мл). После замены водородом 3 раза реакционный раствор нагревали до 50°С. Под 50 фунт/кв. дюйм водорода реакционный раствор перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали с удалением катализатора. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением соединения А1-2. 1H ЯМР (400 МГц, деитерированный метанол) δ: 7.80 (s, 1H), 6.86 (d, J=4.4 Гц, 1H), 6.53 (d, J=4.4 Гц, 1H), 3.96-3.79 (m, 2Н), 3.60-3.51 (m, 1H), 3.49-3.38 (m, 2Н), 2.39-2.36 (m, 1H), 2.19-2.13 (m, 1Н), 1.49 (d, J=3.6 Гц, 9Н).
Стадия 3: Синтез соединения А1
При комнатной температуре йодсукцинимид (26,7 г, 119 ммоль, 3,00 экв.) добавляли к смеси раствора соединения А1-2 (12,0 г, 39,6 ммоль, 1,00 экв.) в N,N-диметилформамиде (150 мл). Затем реакционный раствор перемешивали при комнатной температуре в течение 1 часа, реакционный раствор медленно добавляли к ледяной воде (200 мл) и образовывалось твердое вещество. Растворитель удаляли фильтрацией и фильтрационный кек сушили ротационным испарением при пониженном давлении с получением соединения А1. Соединение А1 хирально расщепляли (колонка: IC (250 мм×0 мм, 10 мкм); подвижная фаза: [0,1% аммиачная вода/этанол]; В%: 30%-30%) с получением соединения А1-А (время удержания 2,94 минуты) и соединения А1-В (время удержания 3,28 минуты).
Промежуточное соединение А2:
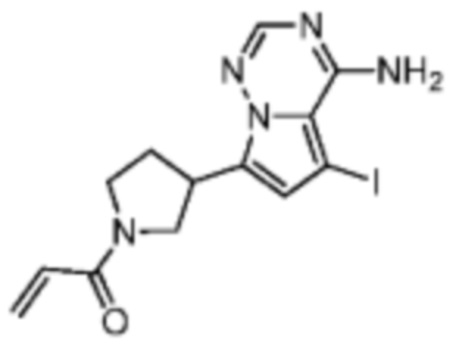
Путь синтеза:
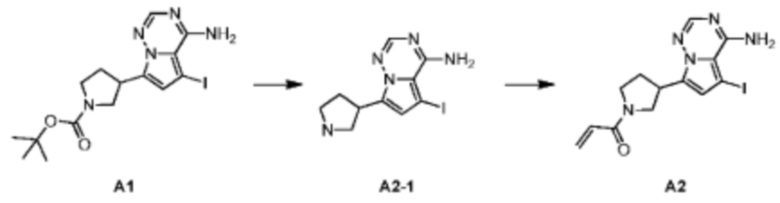
Стадия 1: Синтез соединения А2-1
При комнатной температуре хлористоводородную кислоту/этилацетат (4 М, 20,00 мл, 6,87 экв.) медленно добавляли к раствору соединения А1 (5,00 г, 11,65 ммоль, 1,00 экв.) в этилацетате (30 мл). Реакционный раствор перемешивали в течение двух часов, а затем фильтровали. Растворитель удаляли из фильтрационного кека ротационным испарением при пониженном давлении с получением соединения А2-1 в виде гидрохлорида. TCMS (ESI) масса/заряд: 329.9 [М+Н]+, 1Н ЯМР (400 МГц, дейтерированный метанол) δ=8.11 (s, 1Н), 7.20 (s, 1Н), 4.12 (m, 1H), 3.84 (m, 1Н), 3.67-3.54 (m, 1H), 3.51-3.37 (m, 2Н), 2.71-2.51 (m, 1H), 2.35-2.27 (m, 1H).
Стадия 2: Синтез соединения A2
При 0°С триэтиламин (3,60 г, 35,55 ммоль, 4,93 мл, 5,00 экв.) и акрилоил хлорид (707,88 мг, 7,82 ммоль, 1,10 экв.) последовательно добавляли к раствору соединения А2-1 (2,60 г, 7,11 ммоль, 1,00 экв., гидрохлорид) в дихлорметане (20,00 мл). После перемешивания в течение 1 часа реакционный раствор выливали в 50 мл воды. После разделения фаз водную фазу экстрагировали при помощи дихлорметана (20 мл×5). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли при пониженном давлении с получением соединения А2. LCMS (ESI) масса/заряд: 384.0 [М+Н]+, 406.0 [M+Na]+.
Промежуточное соединение A3:
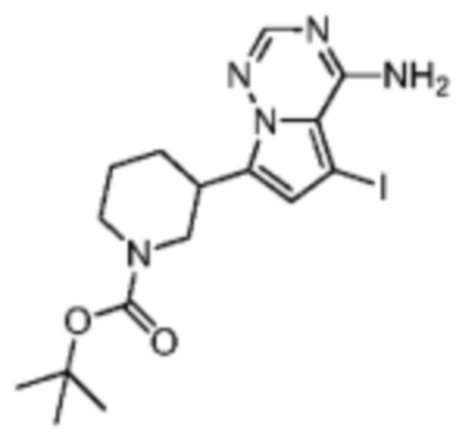
Его синтезировали на основании способа синтеза промежуточного соединения А1.
Промежуточное соединение В1:
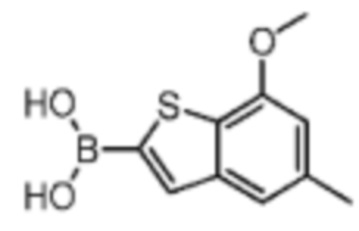
Путь синтеза:
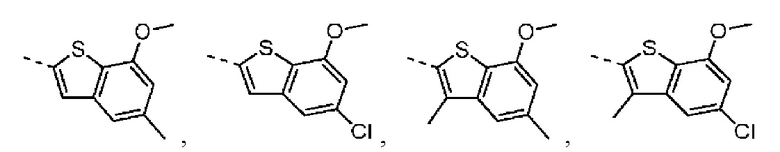
Раствор 7-метокси-5-метилбензотиофена (2,00 г, 11,22 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл) охлаждали до -70°С. Раствор бутиллития в н-гексане (2,5 М, 8,98 мл, 2,00 экв.) медленно по каплям добавляли к охлажденному раствору. После добавления по каплям перемешивание продолжали в течение 1 часа. Затем добавляли триизопропилбороновую кислоту (2,11 г, 11,22 ммоль, 1,00 экв.). После завершения добавления перемешивание продолжали в течение 1 часа. По каплям добавляли воду (10 мл) для гашения реакции. Погашенную реакционную смесь концентрировали с удалением тетрагидрофурана. Остаток сначала промывали петролейным эфиром (50 мл), а затем доводили при помощи разбавленной хлористоводородной кислоты значение рН до 5. Получали белое твердое вещество. После фильтрации фильтрационный кек промывали водой (50 мл), а затем сушили под вакуумом с получением промежуточного соединения В1. 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ=7.72 (s, 1H), 7.28 (s, 1H), 6.67 (s, 1Н), 4.01 (s, 3Н), 2.50 (s, 3Н).
Промежуточное соединение В2:
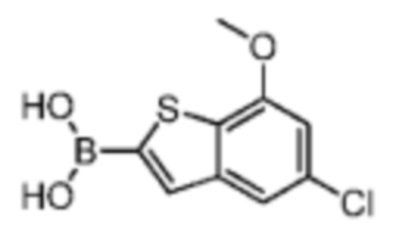
4-Хлор-2-метокситиофенол получали из исходного вещества 2-бром-5-хлоранизола (со ссылкой на J.O. Jilek et al., Collection of Czechoslovak Chemical Communications, Vol. 43, 1978, p. 1747-1759) и промежуточное соединение B2 синтезировали на основании способа синтеза соединения В1. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7.75 (s, 1H), 7.46 (s, 1H), 6.87 (s, 1H), 4.00 (s, 3Н).
Промежуточное соединение В3:
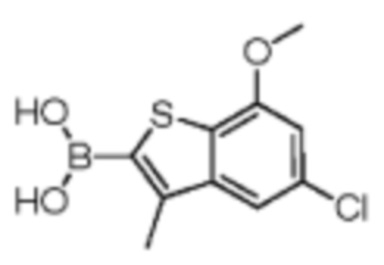
Путь синтеза:
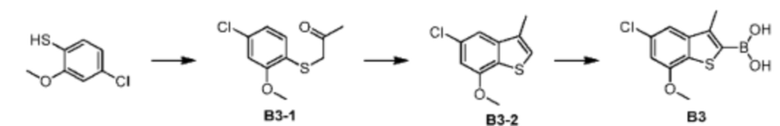
Стадия 1: Синтез соединения В3-1
При комнатной температуре карбонат цезия (149,24 г, 458,06 ммоль, 2,00 экв.) добавляли к раствору 4-хлор-2-метокситиофенола (40,00 г, 229,03 ммоль, 1,00 экв.) и 1-хлорацетона (31,78 г, 343,55 ммоль, 1,50 экв.) в N,N-диметилформамиде (500,00 мл). После перемешивания в течение 16 часов при защите азота реакционный раствор добавляли к 250 мл воды и смесь экстрагировали при помощи этилацетата (100 мл) 3 раза. Органические фазы объединяли, промывали насыщенным солевым раствором (250 мл) 3 раза, сушили над безводным сульфатом натрия и фильтровали. Органический растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир - петролейный эфир/этилацетат = 10/1) с получением соединения В3-1. 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ=7.25 (d, J=8.0 Гц, 1H), 7.16 (d, J=8.0 Гц, 1H), 6.84-6.80 (m, 1H), 3.80-3.76 (m, 2Н), 3.72 (s, 3Н), 1.35 (s, 3Н).
Стадия 2-3: Промежуточное соединение В3 синтезировали на основании способа синтеза соединения В1. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7.25 (d, J=1.6 Гц, 1Н), 7.16 (d, J=2.0 Гц, 1H), 3.87 (s, 2H), 2.41 (s, 3H).
Промежуточное соединение B4:
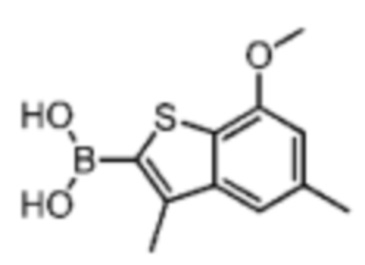
Его синтезировали на основании способов синтеза соединения В1 и соединения В3. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7.75 (s, 1H), 7.46 (s, 1H), 6.87 (s, 1H), 4.00 (s, 3Н).
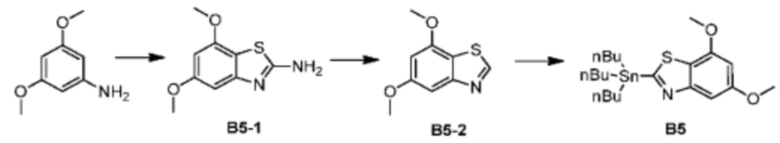
Промежуточное соединение В5:
Путь синтеза:
Стадия 1: Синтез соединения В5-1
При комнатной температуре 3,5-диметоксианилин (43,00 г, 280,72 ммоль, 1,00 экв.) и тиоцианат аммония (47,01 г, 617,58 ммоль, 2,20 экв.) сначала растворяли в ледяной уксусной кислоте (500 мл). Затем реакционный раствор охлаждали до 10°С в бане с ледяной водой. Жидкий бром (43,00 г, 280,72 ммоль, 1,00 экв.) медленно добавляли по каплям в течение 1 часа. Реакционную смесь перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционный раствор выливали в 1000 мл воды, нейтрализовали и доводили при помощи 2 М раствора NaOH значение рН до 9, экстрагировали при помощи дихлорметана (500 мл) 5 раз. Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир/этилацетат = 10/1 - этилацетат) с получением соединения В5-1. LCMS (ESI) масса/заряд: 210.8 [М+Н]+, 1Н ЯМР (400 МГц, дейтерированный метанол) δ=6.51 (d, J=2.0 Гц, 1H), 6.22 (d, J=2.0 Гц, 1H), 3.77 (s, 3Н), 3.70 (s, 3Н).
Стадия 2: Синтез соединения В5-2
При комнатной температуре соединение В5-1 (5 г, 23,78 ммоль, 1 экв.) добавляли к раствору диоксана (50 мл) и при комнатной температуре добавляли изоамилнитрит (4,18 г, 35,67 ммоль, 4,80 мл, 1,5 экв.). Реакционный раствор нагревали до 90°С и перемешивали с защитной азота в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и выливали в 100 мл воды. Смесь экстрагировали при помощи дихлорметана (20 мл) 5 раз. Органические фазы объединяли. Объединенные органические фазы сначала промывали безводным сульфатом натрия, а затем растворитель удаляли ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир - петролейный эфир/этилацетат = 10/1) с получением соединения В5-2. LCMS (ESI) масса/заряд: 195.9 [М+Н]+, 1Н ЯМР (400 МГц, дейтерированный метанол) δ=9.16 (s, 1Н), 7.18 (d, J=1.6 Гц, 1H), 6.66 (d, J=2.0 Гц, 1H), 3.98 (s, 3Н), 3.90 (s, 3Н).
Стадия 3: Синтез соединения В5
В трехгорлую колбу объемом 100 мл, оборудованную мешалкой и низкотемпературным термометром, соединение В5-1 (1 г, 5,12 ммоль, 1 экв.) и тетрагидрофуран (20 мл) добавляли при защите азота. Пока температуру системы понижали до -78°С, медленно по каплям добавляли раствор н-бутиллития в н-гексане (2,5 М, 2,46 мл, 1,2 экв.), и реакционную систему поддерживали при -78°С и перемешивали в течение 1 часа. Далее медленно по каплям добавляли хлорид трибутилолова (2,4 г, 7,37 ммоль, 1,98 мл, 1,44 экв.) при -78°С. После завершения добавления по каплям смесь нагревали до -10°С и продолжали реакцию в течение 1 часа. Далее тетрагидрофуран удаляли из реакционного раствора ротационным испарением. Добавляли 1,4-диоксан и растворяли. Нерастворимые вещества удаляли фильтрацией. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением промежуточного соединения В5.
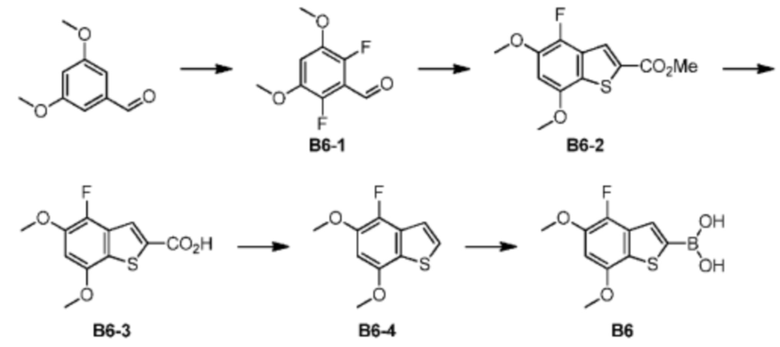
Промежуточное соединение В6
Путь синтеза:
Стадия 1: Синтез соединения В6-1
При 0°С к раствору 3,5-диметоксибензальдегида (125 г, 752,23 ммоль, 1 экв.) в ацетонитриле (3000 мл) добавляли к партии 1-хлорметил-4-фтор-1,4-диазабицикло[2.2.2]октана бис(тетрафторбората) (532,97 г, 1,50 моль, 2 экв.). После завершения добавления реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 48 часов. После завершения реакции твердое вещество удаляли из реакционного раствора фильтрацией. Большую часть растворителя удаляли из фильтрата ротационным испарением при пониженном давлении. Реакционный раствор разбавляли 1000 мл этилацетата и доводили при помощи насыщенного водного раствора бикарбоната натрия значение рН до 7-8. В заключение, фазы разделяли при помощи делительной воронки и водную фазу экстрагировали при помощи этилацетата (1800 мл) 3 раза. Органические фазы объединяли, один раз промывали 2000 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и в заключение фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (100-200 меш силикагеля, элюент: петролейный эфир/этилацетат = 1/0-3/1) с получением соединения В6-1.
Стадия 2: Синтез соединения В6-2
При 40°С реакционный раствор соединения В6-1 (10 г, 49,47 ммоль, 1 экв.), метилмеркаптоацетата (5,78 г, 54,41 ммоль, 4,94 мл, 1,1 экв.) и карбоната калия (6,84 г, 49,47 ммоль, 1 экв.) в N,N-диметилформамиде (100 мл) перемешивали в течение 20 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры. К реакционному раствору добавляли 400 мл воды. Смесь экстрагировали при помощи 200 мл этилацетата. Органическую фазу сушили над безводным сульфатом натрия. После фильтрации растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали на колонке (ISCO®; 200 г SepaFlash® быстрая колонка с силикагелем, подвижная фаза: 0-100% этилацетат/петролейный эфир приблизительно 100 мл/мин) с получением соединения В6-2.
Стадия 3: Синтез соединения В6-3
При 90°С смешанный раствор соединения В6-2 (5 г, 18,50 ммоль, 1 экв.) и моногидрата гидроксида лития (7,76 г, 185,00 ммоль, 10 экв.) в диоксане (50 мл) и воде (10 мл) перемешивали в течение 18 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, а затем органический растворитель удаляли ротационным испарением при пониженном давлении. 1 М разбавленную хлористоводородную кислоту использовали для доведения значения рН до 6 и полученную смесь экстрагировали 100 мл этилацетата 5 раз. Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением соединения В6-3. 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ=8.18 (s, 1H), 6.68 (d, J=6.0 Гц, 1H), 4.01 (s, 3Н), 3.99 (s, 3Н).
Стадия 4: Синтез соединения В6-4
При 200°С смесь соединения В6-3 (2,4 г, 9,37 ммоль, 1 экв.), закиси меди (2,68 г, 18,73 ммоль, 1,91 мл, 2 экв.) и хинолина (20 мл) перемешивали в течение 1 часа. После завершения реакции реакционную смесь охлаждали до комнатной температуры. 50 мл этилацетата добавляли к реакционному раствору, а затем 1 М разбавленную хлористоводородную кислоту использовали для доведения значения рН до 6. После фазового разделения при помощи делительной воронки органическую фазу сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением с получением неочищенного продукта. Неочищенный продукт очищали на колонке (ISCO®; 24g SepaFlash® быстрая колонка с силикагелем, подвижная фаза: 0-100% этилацетат/петролейный эфир приблизительно 35 мл/мин) с получением соединения В6-4. 1H ЯМР (400МГц, дейтерированный хлороформ) δ=7.47-7.41 (m, 1H), 7.41-7.35 (m, 1H), 6.57 (d, J=5.6 Гц, 1H), 3.99 (s, 3Н), 3.98 (s, 3Н).
Стадия 5: Синтез соединения В6
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения В6-4 в качестве исходного вещества. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7.80 (s, 1Н), 6.74 (d, J=6.0 Гц, 1H), 3.97 (s, 3Н), 3.95 (s. 3Н).
Промежуточное соединение В7
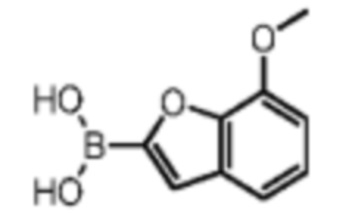
Его синтезировали на основании способа синтеза промежуточного соединения В1 с применением 7-метоксибензофурана в качестве исходного вещества. 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7.34 (s, 1H), 7.25-7.11 (m, 2Н), 6.91 (d, J=7.6 Гц, 1Н), 4.12-3.91 (m, 3Н).
Промежуточное соединение В8
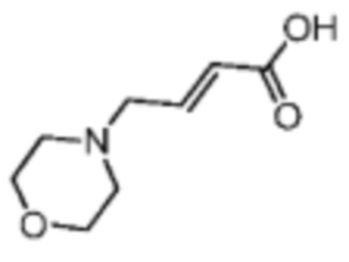
Путь синтеза:
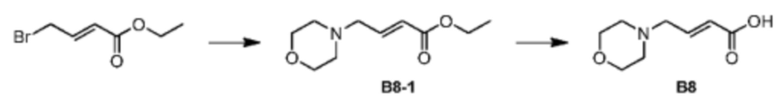
Стадия 1: Синтез соединения В8-1
При комнатной температуре тетрагидрофуран (100 мл) и этил-4-бромкротонат (10,0 г, 51,80 ммоль, 7,14 мл, 1,00 экв.) добавляли в предварительно высушенную колбу объемом 250 мл и смесь перемешивали при 25°С. Добавляли K2CO3 (14,32 г, 103,61 ммоль, 2,00 экв.) и морфолин (4,74 г, 54,39 ммоль, 4,79 мл, 1,05 экв.) при 25°С и смесь перемешивали при 25°С в течение 12 часов. После завершения реакции реакционный раствор медленно выливали в воду (50 мл). Смесь экстрагировали этилацетатом (50 мл) 3 раза. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали на колонке (петролейный эфир/этилацетат = 10:1-3:1) с получением соединения В8-1. 1Н ЯМР (400 МГц, дейтерированный хлороформ) 5=7.27-6.88 (m, 1H), 6.00-5.95 (m, 1H), 4.15 (q, J=7.2 Гц, 2Н), 3.75-3.59 (m, 4Н), 3.12-3.10 (m, 2Н), 2.51-2.31 (m, 4Н), 1.35-1.09 (m, 3Н).
Стадия 2: Синтез соединения В8
Готовили прозрачную трехгорлую колбу объемом 100 мл. Соединение В8-1 (1 г, 5,02 ммоль, 1 экв.) растворяли в метаноле (20 мл) и воде (10 мл) при 25°С, а затем начинали перемешивание смеси. Реакционный раствор охлаждали до 0°С. К вышеуказанному реакционному раствору добавляли NaOH (602,27 мг, 15,06 ммоль, 3 экв.). Реакционную систему нагревали до 25°С. После перемешивания в течение 1 часа реакционный раствор концентрировали ротационным испарением при пониженном давлении. Осаждалось твердое вещество. Твердое вещество пропитывали дихлорметаном/метанолом (10/1). Смесь фильтровали. Фильтрат концентрировали с получением соединения В8. 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.04-6.85 (m, 1Н), 6.43-6.23 (m, 1Н), 4.02-4.00 (m, 4Н), 3.94-3.81 (m, 2Н), 3.57-3.36 (m, 2Н), 3.27-3.17 (m, 2Н).
Промежуточное соединение В9
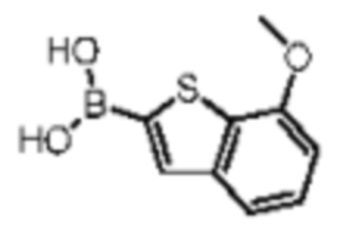
Его синтезировали на основании способа синтеза промежуточного соединения В1 с применением 7-метоксибензстиофена в качестве исходного вещества. 1H ЯМР (400МГц, DMSO-d6) δ = 7.95-7.79 (m, 1Н), 7.61-7.39 (m, 1H), 7.37-7.24 (m, 1H), 6.99-6.83 (m, 1H), 3.96-3.87 (m, 3H).
Примеры 1 и 2: Синтез соединения WX001 (WX001A и WX001B)
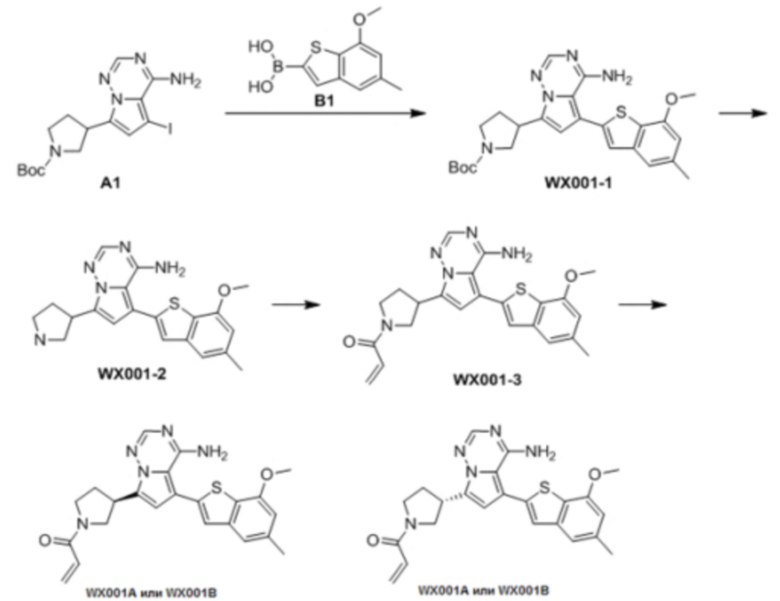
Стадия 1: Синтез соединения WX001-1
При комнатной температуре соединение В1 (777,25 мг, 3,50 ммоль, 2,50 экв.), карбонат натрия (296,77 мг, 2,80 ммоль, 2,00 экв.) и тетра(трифенмпфосфин)палладий (161,78 мг, 140,00 мкмоль, 0,10 экв.) последовательно добавляли к смешанному раствору соединения A1 (600,00 мг, 1,40 ммоль, 1,00 экв.) в этиленгликольдиметиловом эфире (9 мл)/этаноле (3 мл)/воде (0,5 мл). После замены азотом 3 раза смесь нагревали до 90°С, перемешивали в течение 5 часов, охлаждали до комнатной температуры и выливали в 30 мл воды. Полученную смесь экстрагировали при помощи дихлорметана (10 мл) 5 раз. Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир/этилацетат = 10/1 - 1/3) с получением соединения WX001-1. LCMS (ESI) масса/заряд: 480.2 [М+Н]+, 502.2 [M+Na]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.91 (s, 1Н), 7.27 (s, 2Н), 6.77 (s, 1Н), 6.70 (s, 1Н), 4.00 (s, 3Н), 3.96-3.90 (m, 2Н), 3.64-3.50 (m, 3Н), 2.49 (s, 3Н), 2.44-2.36 (m, 2Н), 1.50 (s, 9Н).
Стадия 2: Синтез соединения WX001-2
При комнатной температуре раствор хлористоводородной кислоты и этилацетата (4 М, 2,00 мл, 9,51 экв.) медленно по каплям добавляли к раствору соединения WX001-1 (350,00 мг, 729,79 мкмоль, 1,00 экв.) в этилацетате (2 мл). Смесь перемешивали в течение 1 часа и фильтровали с получением твердого вещества. Твердое вещество сушили при пониженном давлении с получением гидрохлорида соединения WX001-2. LCMS (ESI) масса/заряд: 380.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 8.17 (s, 1Н), 7.46 (s, 1Н), 7.33 (s, 1Н), 7.12-7.06 (m, 1Н), 6.84 (s, 1Н), 4.12-4.06 (m, 1Н), 4.02 (s, 3Н), 3.92-3.82 (m, 2Н), 3.67-3.58 (m, 2Н), 2.66-2.60 (m, 1Н), 2.51 (s, 3Н), 2.39-2.32 (m, 1Н).
Стадия 3: Синтез соединения WX001 (WX001A и WX001B)
При 0°С диизопропилэтиламин (258,56 мг, 2,00 ммоль, 349,41 мкл, 4,00 экв.) и раствор акрилоилхлорида в дихлорметане (0,25 М, 1,80 мл, 0,90 экв.) добавляли к раствору хлористоводородной соли соединения WX001-2 (200,00 мг, 500,16 мкмоль, 1,00 экв.) в дихлорметане (4,00 мл). Смесь перемешивали в течение 5 минут. Реакционный раствор выливали в 2 мл воды. После разделения фаз водную фазу экстрагировали при помощи дихлорметана (1 мл) 3 раза. Органические фазы объединяли. Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали тонкослойной препаративной пластины (дихлорметан/метанол = 10/1) с получением соединения WX001. Соединение WX001 хирально расщепляли (колонка: AS (250 мм × 30 мм, 5 мкм); подвижная фаза: [0,1% аммиачная вода/этанол]; В%: 40%-40%) с получением соединения WX001A (время удержания: 6,16 минут) и соединения WX001B (время удержания: 6,98 минуты). Время удержания измеряли при помощи следующей аналитической колонки: колонка: Chiralpak AS-3 150×4,6 мм I.D., 3 мкм, подвижная фаза: А: диоксид углерода В: метанол (0,05% диэтиламин), 40% В, скорость потока: 2,5 мл/мин, температура колонки: 35°С. Соединение WX001A, LCMS (ESI) масса/заряд: 434.2 [М+Н]+, 456.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.75 (d, J=2.8 Гц, 1Н), 7.12-7.04 (m, 2Н), 6.61 (s, 1Н), 6.56-6.40 (m, 2Н), 6.20-6.15 (m, 1Н), 5.65-5.60 (m, 1Н), 4.11-3.94 (m, 1Н), 3.85 (s, 3Н), 3.81-3.38 (m, 4Н), 2.48-2.26 (m, 4Н), 2.22-1.93 (m, 1Н).
Соединение WX001B, LCMS (ESI) масса/заряд: 434.2 [М+Н]+, 456.1[M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.75 (d, J=2.8 Гц, 1Н), 7.08 (s, 2Н), 6.61 (s, 1Н), 6.54 (d, J=6.4 Гц, 1Н), 6.41-6.51 (m, 1Н), 6.20-6.16 (m, 1Н), 5.66-5.42 (m, 1Н), 4.09-3.96 (m, 1Н), 3.85 (s, 3Н), 3.80-3.38 (m, 4Н), 2.44-2.25 (m, 4Н), 2.21-1.99 (m, 1Н).
Пример 3: Синтез соединения WX001C 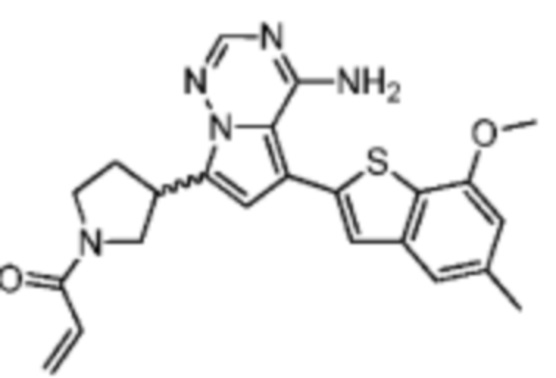
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-В и промежуточного соединения В1 в качестве исходных веществ. Его объединяли с соединением WX001A и определяли при помощи SFC (способ анализа SFC соединения WX001, время удержания: 6,14 минут) в виде соединения WX001A.
Пример 4: Синтез соединения WX002A
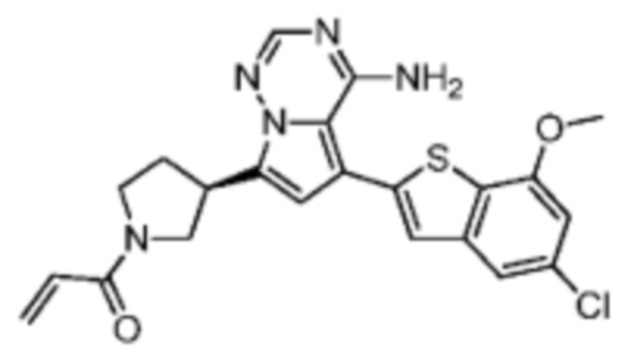
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-В и промежуточного соединения В2 в качестве исходных веществ. LCMS (ESI) масса/заряд: 454.1 [М+Н]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.81 (d, J=2.0 Гц, 1Н), 7.36 (d, J=1.6 Гц, 1Н), 7.21 (s, 1Н), 6.82 (s, 1Н), 6.64 (d, J=9.2 Гц, 1Н), 6.58-6.50 (m, 1Н), 6.22-6.17 (m, 1Н), 5.68-5.63 (т 1Н), 4.20-3.93 (m, 2Н), 3.91 (s, 3Н), 3.77-3.44 (m, 3Н), 2.52-2.31 (m, 1Н), 2.27-2.05 (m, 1Н).
Пример 5: Синтез соединения WX002B
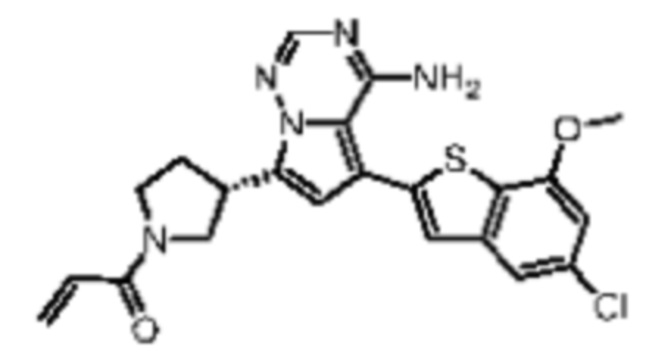
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-А и промежуточного соединения В2 в качестве исходных веществ. LCMS (ESI) масса/заряд: 454.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.81 (d, J=2.4 Гц, 1Н), 7.36 (d, J=1.6 Гц, 1Н), 7.20 (s, 1Н), 6.81 (s, 1Н), 6.63 (d, J=8.8 Гц, 1Н), 6.58-6.50 (m, 1Н), 6.22-6.17(m, 1Н), 5.74-5.56 (m, 1Н), 4.18-3.92 (m, 2Н), 3.90 (s, 3Н), 3.81-3.59 (m, 2Н), 3.57-3.42 (m, 1Н), 2.50-2.30 (m, 1Н), 2.28-2.02 (m, 1Н).
Пример 6: Синтез соединения WX003
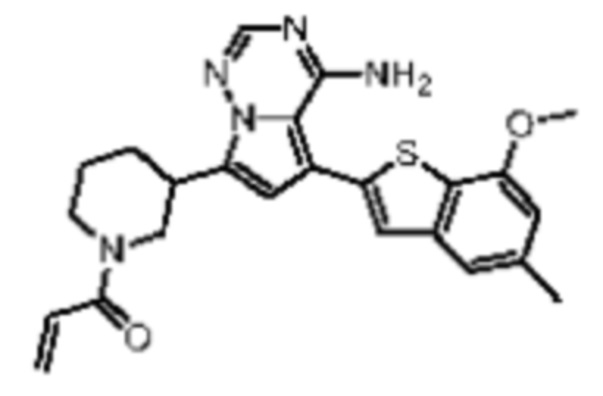
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения A3 и промежуточного соединения В1 в качестве исходных веществ. LCMS (ESI) масса/заряд: 448.1 [М+Н]+, 470.2 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.83-7.76 (m, 1Н), 7.15 (s, 2Н), 6.75-6.68 (m, 1Н), 6.66 (s, 1Н), 6.61 (s, 1Н), 6.14-6.06 (m, 1Н), 5.68-5.58 (m, 1Н), 4.42-4.27 (m, 1Н), 3.88 (s, 3Н), 3.40-3.27 (m, 2Н), 3.08-2.83 (m, 2Н), 2.37 (s, 3Н), 2.13-2.02 (m, 1Н), 1.86-1.72 (m, 2Н), 1.63-1.54 (m, 1Н).
Примеры 7 и 8: Синтез соединения WX004 (WX004A, WX004B)
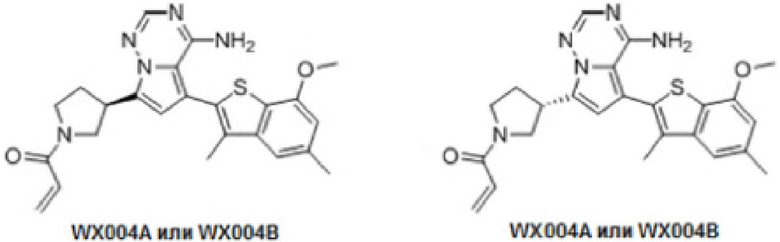
Его синтезировали на основании способа на стадии 1, пример 1, с применением промежуточного соединения А2 и промежуточного соединения В4 в качестве исходных веществ. После синтеза продукт хирально расщепляли (колонка: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: [0.1% аммиачная вода/метанол]; В%: 40%-40%) с получением соединений WX004A (время удержания: 5,58 минут) и WX004B (время удержания: 6,14 минут). Время удержания измеряли на следующей аналитической колонке: колонка: Chiralpak AS-3 150×4,6 мм I.D., 3 мкм, подвижная фаза: А: диоксид углерода В: метанол (0,05% диэтиламин), 40% В, скорость потока: 2,5 ил/мин, температура колонки: 35°С.
Соединение WX004A, LCMS (ESI) масса/заряд: 448.2 [М+Н]+, 470.2 [M+Na]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.90 (d, J=2.0 Гц, 1Н), 7.21 (s, 1Н), 6.81 (s, 1Н), 6.73-6.57 (m, 2Н), 6.34-6.33 (m, 1Н), 5.80-5.75 (m, 1Н), 4.32-4.12 (m, 1Н), 4.11-3.95 (m, 4Н), 3.93-3.72 (m, 2Н), 3.70-3.64 (m, 1Н), 2.64-2.42 (m, 4Н), 2.39-2.16 (m, 4Н).
Соединение WX004B, LCMS (ESI) масса/заряд: 448.2 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.78 (d, J=2.0 Гц, 1Н), 7.09 (s, 1Н), 6.69 (s, 1Н), 6.63-6.44 (m, 2Н), 6.22-6.17 (m, 1Н), 5.73-5.54 (m, 1Н), 4.24-4.01 (m, 1Н), 4.00-3.89 (m, 1Н), 3.87 (s, 3Н), 3.80-3.60 (m, 2Н), 3.57-3.44 (m, 1Н), 2.52-2.32 (m, 4Н), 2.30-2.10 (m, 4Н).
Пример 9: Синтез соединения WX005
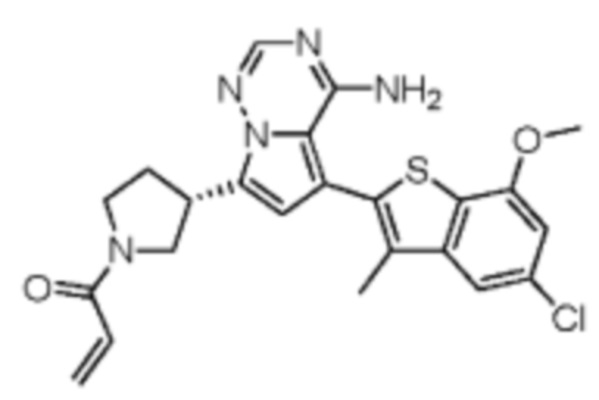
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-А и промежуточного соединения В3 в качестве исходных веществ. LCMS (ESI) масса/заряд: 490.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.80 (d, J=2.0 Гц, 1Н), 7.32 (d, J=1.2 Гц, 1Н), 6.87 (s, 1Н), 6.67-6.49 (m, 2Н), 6.25-6.03 (m, 1Н), 5.70-5.59 (m, 1Н), 4.24-4.00 (m, 1Н), 3.99-3.85 (m, 3Н), 3.83-3.60 (m, 2Н), 3.58-3.41 (m, 1Н), 3.40-3.26 (m, 1Н), 2.50-2.34 (m, 1Н), 2.32-2.19 (m, 1Н), 2.17 (s, 3Н).
Пример 10: Синтез соединения WX006A
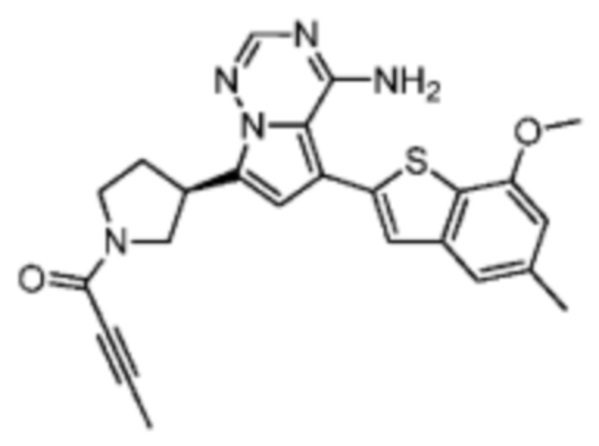
Путь синтеза:
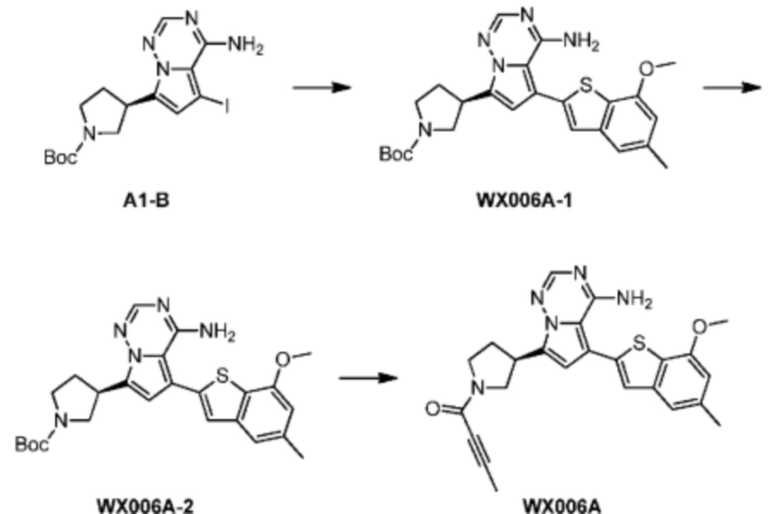
Стадия 1-2: Синтез соединения WX006A-2
Его синтезировали на основании способа синтеза на стадии 1 и стадии 2 примера 1 с применением промежуточного соединения А1-В и промежуточного соединения В1 в качестве исходных веществ.
Стадия 3: Синтез соединения WX006A
При 0°С O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфофат (68,56 мг, 180,31 мкмоль, 1,50 экв.) добавляли к раствору 2-бутиновой кислоты (10,11 мг, 120,21 мкмоль, 1,00 экв.) в дихлорметане (2,00 мл). Смесь перемешивали в течение 30 минут. При 0°С соединение WX006A-2 (50,00 мг, 120,21 мкмоль, 1,00 экв., HCl) и триэтиламин (36,49 мг, 360,63 мкмоль, 49,99 мкл, 3,00 экв.) добавляли к реакционному раствору и полученную смесь медленно нагревали до 20°С и перемешивали в течение 16 часов. После завершения реакции реакционный раствор разбавляли 10 мл дихлорметана, промывали 15 мл воды 3 раза, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт выделяли при помощи тонкослойной препаративной пластины (петролейный эфир/этилацетат = 1/1) с получением соединения WX006A. LCMS (ESI) масса/заряд: 446.1 [М+Н]+, 468.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.78 (d, J=7.2 Гц, 1Н), 7.13 (s, 2Н), 6.64 (s, 1Н), 6.59 (d, J=6.8 Гц, 1Н), 3.98-3.74(m, 5Н), 3.69-3.55 (m, 2Н), 3.47-3.34 (m, 1Н), 2.46-2.31 (m, 4Н), 2.22-2.07 (m, 1Н), 1.93 (d, J=9.2 Гц, 3Н).
Пример 11: Синтез соединения WX006B
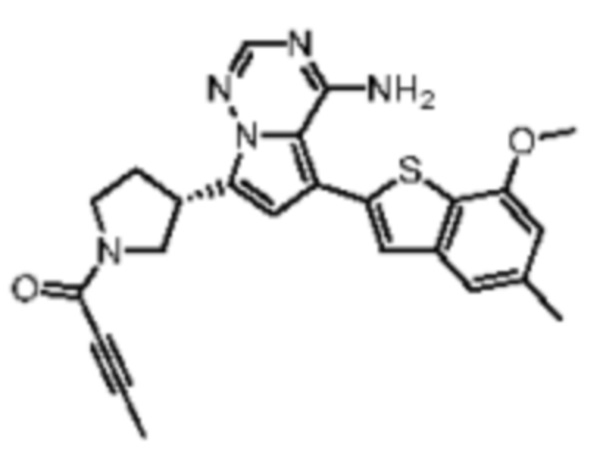
Его синтезировали на основании способов синтеза примеров 1 и 9 с применением промежуточного соединения А1-А и промежуточного соединения В1 в качестве исходных веществ. LCMS (ESI) масса/заряд: 446.1 [М+Н]+, 468.0 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.78 (d, J=7.2 Гц, 1Н), 7.13 (s, 2Н), 6.64 (s, 1Н), 6.59 (d, J=6.4 Гц, 1Н), 4.00-3.73 (m, 5Н), 3.70-3.51 (m, 2Н), 3.45-3.34 (m, 1Н), 2.36 (s, 4Н), 2.23-2.06 (m, 1Н), 1.93 (d, J=9.2 Гц, 3Н).
Примеры 12 и 13: Синтез соединения WX007 (WX007A, WX007B)
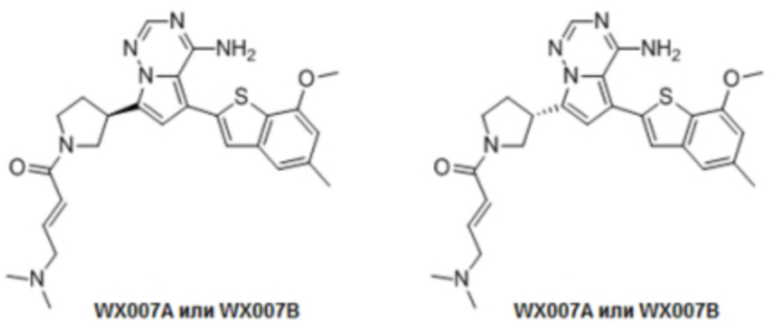
Его синтезировали на основании способа на стадии 3, пример 9, с применением промежуточного соединения WX001-2 и олеиновой кислоты в качестве исходных веществ. После синтеза продукт хирально расщепляли (колонка: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% аммиачная вода/этанол]; В%: 45%-45%) с получением соединений WX007A (время удержания: 1,70 минут) и WX007B (время удержания: 2,02 минут). Время удержания измеряли на следующей аналитической колонке: колонка: Chiralpak AS-H 150×4,6 мм ID., 5 мкм, подвижная фаза: 40% этанол (0,05% диэтиламин) в диоксиде углерода, скорость потока: 3 мл/мин, температура колонки: 40°С.
Соединение WX007A, LCMS (ESI) масса/заряд: 491.2 [М+Н]+, 513.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.76 (d, J=3.6 Гц, 1Н), 7.09 (s, 2Н), 6.79-6.67 (m, 1Н), 6.62 (s, 1Н), 6.55 (d, J=8.8 Гц, 1Н), 6.37-6.32 (m, 1Н), 4.12-3.92 (m, 1Н), 3.86 (s, 3Н), 3.81-3.72 (m, 1Н), 3.70-3.39 (m, 3Н), 3.07-2.99 (m, 2Н), 2.39-2.25 (m, 4Н), 2.16 (s, 3Н), 2.15 (s, 3Н), 2.12-1.99 (m, 1Н).
Соединение WX007B, LCMS (ESI) масса/заряд: 491.2 [М+Н]+, 513.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.76 (d, J=3.2 Гц, 1Н), 7.10 (s, 2Н), 6.79-6.68 (m, 1Н), 6.63 (s, 1Н), 6.56 (d, J=9.2 Гц, 1Н), 6.38-6.33 (m, 1Н), 4.15-3.94 (m, 1Н), 3.86 (s, 3Н), 3.83-3.71 (m, 1Н), 3.69-3.40 (m, 3Н), 3.06-3.03 (m, 2Н), 2.40-2.32 (m, 4Н), 2.17 (s, 3Н), 2.15 (s, 3Н), 2.12-1.97 (m, 1Н).
Пример 14: Синтез соединения WX008
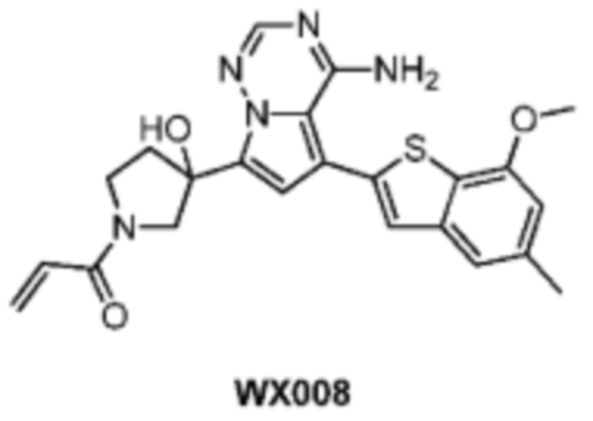
Путь синтеза:
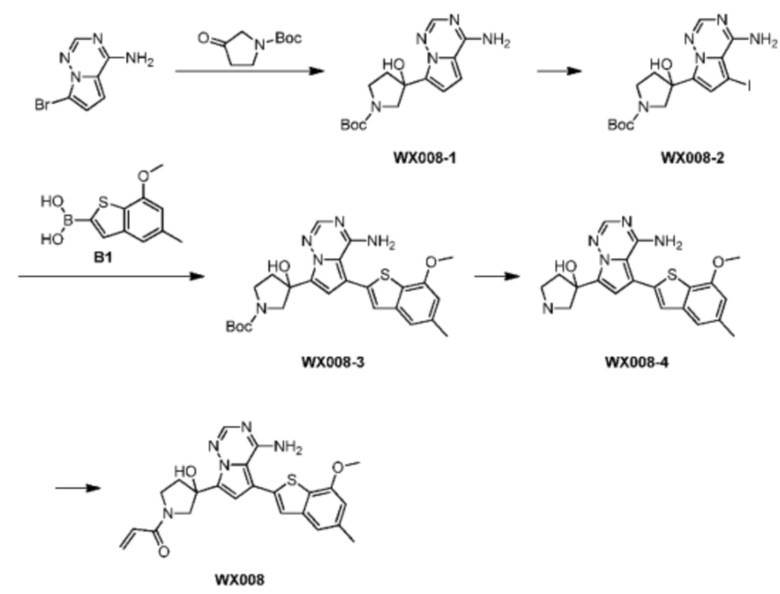
Стадия 1: Синтез соединения WX008-1
При -60°С при защите при помощи азота метиллитий (1,6 М, 616,10 мкл, 1,05 экв.) добавляли по каплям к раствору 4-амино-7-бромпиррол[2,1-f][1,2,4]триазина (0,2 г, 938,81 мкмоль, 1 экв.) в растворе тетрагидрофурана (15 мл) в пределах 15 минут. Через 30 минут после реакции медленно по каплям добавляли н-бутиллитий (2,5 М, 413,08 мкл, 1,1 экв.) к реакционному раствору. Реакционный раствор перемешивали в течение 1 часа от -60°С до 40°С, а затем к реакционному раствору добавляли N-ВОС-3-пирролидон (347,77 мг, 1,88 ммоль, 2 экв.). Реакционный раствор медленно нагревали до 20°С и перемешивали в течение 16 часов. Реакционный раствор охлаждали до 0°С и 1 мл воды добавляли к реакционному раствору для гашения реакции. Реакционный раствор разбавляли 5 мл воды и экстрагировали этилацетатом (5 мл) 3 раза. Органические фазы промывали 10 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и фильтровали. Фильтрат ротационно выпаривали при пониженном давлении с получением соединения WX008-1 в виде неочищенного продукта. Неочищенный продукт использовали сразу в следующей реакции. LCMS (ESI) масса/заряд: 319,9 [М+Н]+.
Стадия 2: Синтез соединения WX008-2
Его синтезировали на основании способа синтеза соединения А1 с применением промежуточного соединения WX008-1 в качестве исходного вещества. LCMS (ESI) масса/заряд: 446.0 [М+Н]+.
Стадия 3-5: Синтез соединения WX008
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения WX008-2 в качестве исходного вещества. LCMS (ESI) масса/заряд: 432.1 [М+Н]+, 450.1 [M+Na]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.81 (d, J=3.6 Гц, 1Н), 7.15 (d, J=2.4 Гц, 2Н), 6.77 (d, J=3.6 Гц, 1Н), 6.65 (s, 1Н), 6.63-6.44 (m, 1Н), 6.24-6.19 (m, 1Н), 5.72-5.57 (m, 1Н), 4.17-3.93 (m, 2Н), 3.88 (s, 3Н), 3.85-3.56 (m, 2Н), 2.86-2.61 (m, 1Н), 2.37 (s, 3Н), 2.34-2.20 (m, 1Н).
Примеры 15 и 16: Синтез соединений WX009A и WX009B
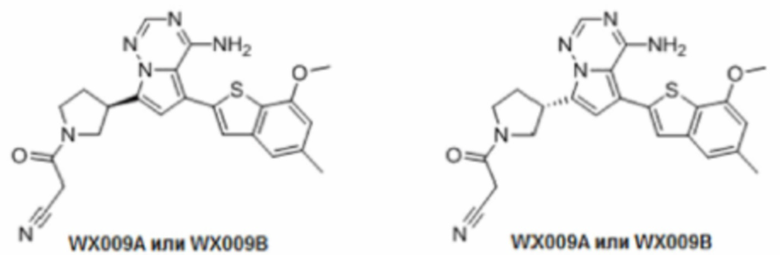
Соединение WX009 синтезировали на основании способов синтеза примеров 1 и 9 с применением промежуточного соединения А1, промежуточного соединения В1 и цианоуксусной кислоты в качестве исходных веществ. Разделением при помощи SFC (колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% аммиачная вода/изопропанол]; В%: 55%-55%) получали соединение WX009A (время удержания: 5,08 минут) и соединение WX009B (время удержания: 7,89 минут). Время удержания измеряли на следующей аналитической колонке: колонка: Chiralpak AD-3 50×4,6 мм I.D., 3 мкм, подвижная фаза: 40% изопропанол (0,05% эилендиамин) в диоксиде углерода, скорость потока: 4 мл/мин, температура колонки: 40°С.
Соединение WX009A: LCMS (ESI) масса/заряд: 447.2[М+Н]+, 469.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.79 (d, J=1.2 Гц, 1Н), 7.16 (d, J=1.6 Гц, 2Н), 6.75-6.54 (m, 2Н), 4.05-3.93 (m, 2Н), 3.88 (s, 3Н), 3.70-3.59 (m, 1Н), 3.56-3.39 (m, 2Н), 2.48-2.33 (m, 4Н), 2.25-2.01 (m, 1Н).
Соединение WX009B: LCMS (ESI) масса/заряд: 447.2[М+Н]+, 469.4 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.79 (d, J=1.6 Гц, 1Н), 7.16 (d, J=1.6 Гц, 2Н), 6.74-6.51 (m, 2Н), 4.10-3.91 (m, 2Н), 3.88 (s, 3Н), 3.71-3.59 (m, 1Н), 3.58-3.40 (m, 2Н), 2.50-2.27 (m, 4Н), 2.26-1.99 (m, 1Н).
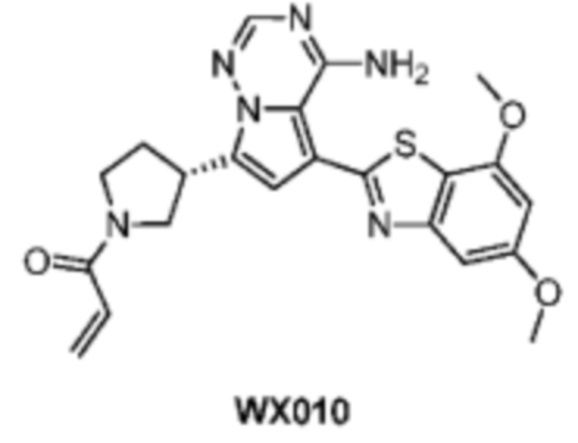
Пример 17: Синтез соединения WX010
Путь синтеза:
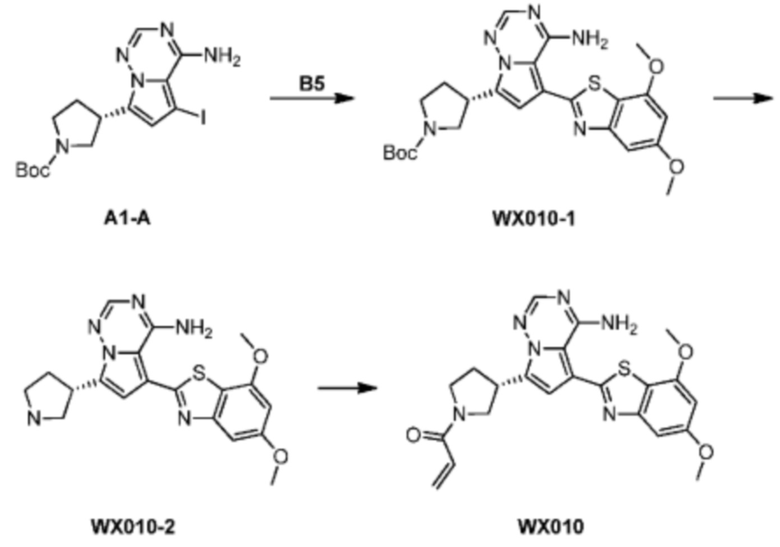
Стадия 1: Синтез соединения WX010-1
В трехгорлую колбу объемом 100 мл, оснащенную мешалкой под защитой водорода, последовательно добавляли соединение А1 (1,10 г, 2,56 ммоль, 1 экв.), йодид меди (97,53 мг, 512,00 мкмоль, 0,2 экв.), бис(трифенилфосфин)палладия дихлорид (359,44 мг, 512,00 мкмоль, 0,2 экв.), триэтиламин (1,04 г, 10,24 ммоль, 1,43 мл, 4 экв.) и 1,4-диоксан (5 мл), а затем добавляли свежеприготовленное соединение В 5 (2,48 г, 5,12 ммоль, 2 экв.). После замены азотом 3 раза реакционный раствор помещали в масляную баню при 100°C и проводили реакцию в течение 12 часов. После завершения реакции нерастворимый материал удаляли фильтрацией и фильтрат ротационно выпаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали колоночной хроматографии (петролейный эфир/этилацетат = 3/1 - этилацетат) с получением продукта WX010-1. LCMS (ESI) масса/заряд: 467,1 [М+Н]+.
Стадии 2 и 3: Синтез соединения WX010
Соединение WX010 синтезировали на основании способа на стадиях 2 и 3, пример 1, с применением промежуточного соединения WX010-1 в качестве исходного вещества. LCMS (ESI) масса/заряд: 473.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.86 (s, 1Н), 7.09-6.93 (m, 2Н), 6.70-6.64 (m, 1Н), 6.55 (s, 1Н), 6.37-6.31 (m, 1Н), 5.82-5.79 (m, 1Н), 4.34-4.09 (m, 1Н), 3.97 (s, 3Н), 3.89-3.83 (m, 5Н), 3.82-3.69 (m, 1Н), 3.68-3.55 (m, 1Н), 2.62-2.41 (m, 1Н), 2.38-2.12 (m, 1Н).
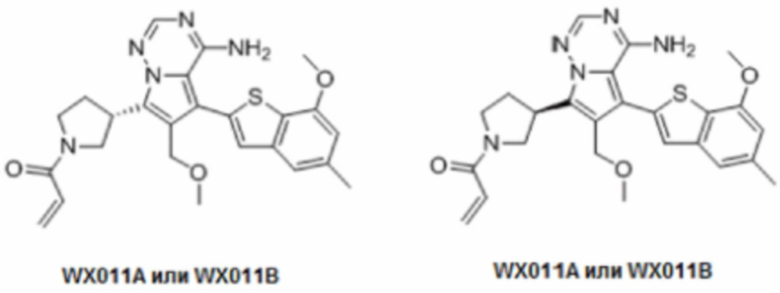
Примеры 18 и 19: Синтез соединений WX011 (WX011A и WX011B)
Путь синтеза:
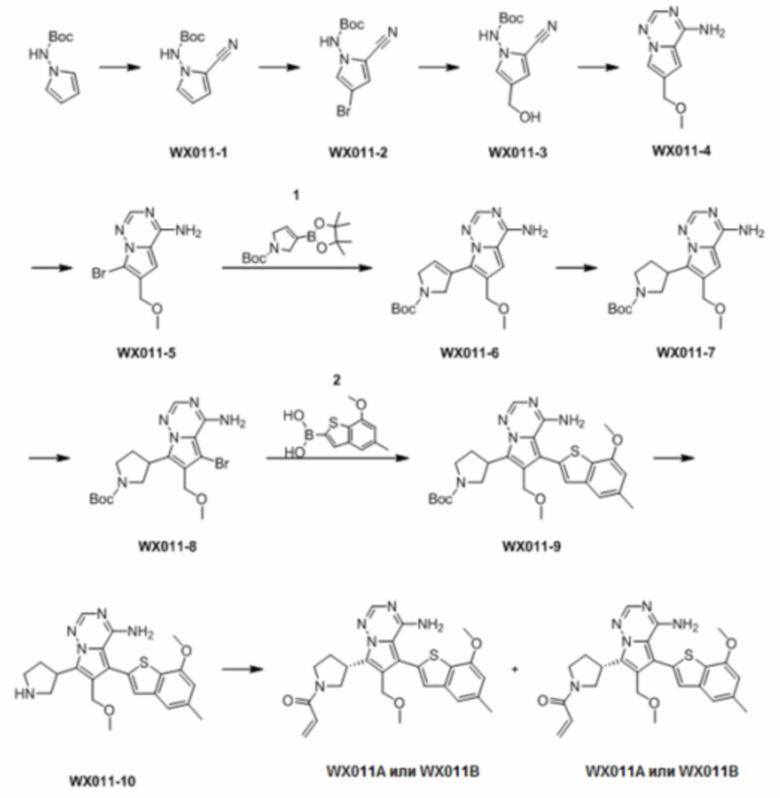
Стадия 1: Синтез соединения WX011-1
Раствор трет-бутил(1Н-пиррол-1-ил)карбамата (25,00 г, 137,20 ммоль, 1,00 экв.) в ацетонитриле (200,00 мл) охлаждали до 0°С, и медленно по каплям при помощи шприца добавляли хлорсульфонилизоцианат (20,39 г, 144,06 ммоль, 12,51 мл, 1,05 экв.) к реакционному раствору Осадок образовывался после перемешивания в течение 30 минут. После продолжения перемешивания в течение 45 минут при 0°С по каплям добавляли N,N,-диметилформамид (14,84 г, 203,05 ммоль, 15,62 мл, 2,50 экв.) при помощи шприца к реакционному раствору и осадок в реакционном растворе исчезал. После продолжения перемешивания при такой температуре в течение 45 минут реакционный раствор медленно нагревали до 25°С и реакцию завершали. Реакционный раствор медленно выливали в 200 мл ледяной воды. Смесь экстрагировали при помощи 200 мл этилацетата. Органическую фазу сначала сушили над безводным сульфатом магния, а затем фильтровали на воронке с песчаным стержнем, заполненной си лика гелем. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением соединения WX011-1.
Стадия 2: Синтез соединения WX011-2
При -30°С дибромгидантоин (10,69 г, 37,40 ммоль, 0,50 экв.) добавляли к партии раствора соединения WX011-1 (15,50 г, 74,80 ммоль, 1,00 экв.) в ацетонитриле (150 мл). Реакционный раствор медленно нагревали до 25°С и перемешивали в течение 2 часов. После завершения реакции реакционный раствор добавляли к 100 мл воды и водную фазу экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли. Объединенную органическую фазу один раз промывали 100 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали путем ротационного вращения при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир/этилацетат = 9/1 - 1/1) с получением соединения WX011-2. 1Н ЯМР (400МГц, CDCl3) δ = 7.23 (brs, 1Н), 6.85 (d, J=2.0 Гц, 1H), 6.71 (d, J=2.0 Гц, 1H), 1.44 (s, 9H).
Стадия 3: Синтез соединения WX011-3
В атмосфере азота раствор соединения WX011-2 (5,30 г, 18,52 ммоль, 1,0 экв.) в тетрагидрофуране (100 мл) охлаждали до -60°С. Метилмагния бромид (3 моль/л раствора тетрагидрофурана, 6,80 мл, 20,38 ммоль, 1,1 экв.) медленно добавляли по каплям к реакционному раствору и полученную смесь перемешивали в течение 30 минут. Затем по каплям добавляли н-бутиллитий (2,0 моль/л раствора н-гексана, 14,80 мл, 37,05 ммоль, 2,0 экв.) к реакционному раствору. Реакционный раствор поддерживали при внутренней температуре от -40°С до -60°С и перемешивали в течение 1 часа. Параформальдегид (1,67 г, 18,52 ммоль, 1,0 экв.) добавляли к реакционному раствору. Затем полученную смесь нагревали до комнатной температуры и перемешивали всю ночь. После завершения реакции реакционный раствор медленно выливали в 100 мл насыщенного солевого раствора. Полученную смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли. Объединенную органическую фазу промывали один раз 100 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и фильтровали. Затем фильтрат выпаривали при помощи роторного вращения при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали на колонке (ISCO®; 220 г SepaFlash® испарительная колонка с силикагелем, подвижная фаза 0-50% этилацетат/петролейный эфир приблизительно 100 мл/мин) с получением соединения WX011-3. 1Н ЯМР (400МГц, DMSO-d6): δ = 10.77 (brs, 1Н), 7.09 (d, J=1.6 Гц, 1Н), 6.86 (d, J=1.6 Гц, 1Н), 4.97 (t, J=5.6 Гц, 1Н), 4.28 (d, J=5.6 Гц, 2Н), 1.45 (s, 9Н).
Стадия 4: Синтез соединения WX011-4
При комнатной температуре раствор хлорида водорода/диоксана (12 мл) добавляли к соединению WX011-3 (4,70 г, 19,81 ммоль, 1 экв.). Смесь перемешивали в течение 5 часов, а затем к реакционному раствору добавляли метанол (60 мл). Полученную смесь перемешивали всю ночь. В заключение к реакционному раствору добавляли фосфат калия (42,05 г, 198,10 ммоль, 10 экв.) и формамидина ацетат (10,31 г, 99,05 ммоль, 5 экв.), а затем полученную смесь нагревали до 65°С и перемешивали в течение 20 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали и очищали на колонке (ISCO®; 220 г SepaFlash® испарительная колонка с силикагелем, подвижная фаза 0-3% метанол/дихлорметан приблизительно 100 мл/мин) с получением соединения WX011-4. LCMS (ESI) масса/заряд: 178.9 [М+Н]+, 1Н ЯМР (400МГц, DMSO-d6) δ = 7.78 (s, 1Н), 7.69 (brs, 2H), 7.58 (d, J=1.2 Гц, 1H), 6.82 (d, J=1.6 Гц, 1H), 4.42 (s, 2H), 3.26 (s, 3H).
Стадия 5: Синтез соединения WX011-5
При -30°C дибромгидантоин (1,85 г, 6,46 ммоль, 0,50 экв.) добавляли к партии раствора соединения WX011-4 (2,30 г, 12,91 ммоль, 1,00 экв.) в тетрагидрофуране (20 мл). Реакционный раствор перемешивали при 15°С в течение 16 часов. Затем реакционный раствор концентрировали и очищали на колонке (ISCO®; 40 г SepaFlash® испарительная колонка с силикагелем, подвижная фаза 0-10% дихлорметан/метанол приблизительно 60 мл/мин) с получением соединения WX011-5 в виде белого твердого вещества. LCMS (ESI) масса/заряд: 256.8 [М+Н]+, 1Н ЯМР (400МГц, DMSO-d6) δ = 7.92 (s, 1Н), 7.88 (brs, 1Н), 7.04 (s, 1Н), 4.42 (s, 2H), 3.27 (s, 3Н).
Стадия 6: Синтез соединения WX011-6
При комнатной температуре соединение WX011-5 (6,40 г, 24,89 ммоль, 1,00 экв.) растворяли в смешанном растворе 1,4-диоксана (100 мл) и воды (20 мл). Затем к смешанному раствору последовательно добавляли N-Вос-2,5-дигидро-1Н-пиррол-1-пинаколборат (7,35 г, 24,89 ммоль, 1,00 экв.), фосфат калия (15,85 г, 74,68 ммоль, 3,00 экв.) и 1,1'-бис(дифенилфосфин)ферроцен палладия хлорид (1,82 г, 2,49 ммоль, 0,10 экв.). При защите при помощи азота реакционный раствор нагревали до 80°С и перемешивали в течение 16 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и медленно выливали в 100 мл воды. Смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали при ротационном вращении при пониженном давлении с получением соединения WX011-6. LCMS (ESI) масса/заряд: 346.0 [М+Н]+.
Стадия 7: Синтез соединения WX011-7
При комнатной температуре гидроксид палладия (65,05 мг, 463,24 мкмоль, 0,1 экв.) добавляли к раствору соединения WX011-6 (1,60 г, 4,63 ммоль, 1,00 экв.) в метаноле (30 мл). После замены водородом 3 раза реакционный раствор нагревали до 50°С и перемешивали в течение 16 часов при 50 фунт/кв. дюйм водорода. Реакционный раствор охлаждали до комнатной температуры и фильтровали с удалением катализатора. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением соединения WX011-7. LCMS (ESI) масса/заряд: 348.1 [М+Н]+.
Стадия 8: Синтез соединения WX011-8
При комнатной температуре бромсукцинимид (563,55 мг, 3,17 ммоль, 1,10 экв.) добавляли к партии раствора соединения WX011-7 (1,00 г, 2,88 ммоль, 1,00 экв.) в тетрагидрофуране (20 мл). Затем реакционный раствор перемешивали при 20°С в течение 1 часа, реакционный раствор добавляли к этилацетату (50 мл). Полученную смесь последовательно промывали один раз 30 мл воды и 30 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и выпаривали при ротационном вращении при пониженном давлении с получением соединения WX011-8. LCMS (ESI) масса/заряд: 425.9 [М+Н]+.
Стадия 9: Синтез соединения WX011-9
При комнатной температуре соединение WX011-8 (1,25 г, 5,63 ммоль, 1,50 экв.), фторид цезия (2,85 г, 18,77 ммоль, 5,00 экв.) и хлор(2-дициклогексилфосфино-2,4,6-триизопропил-1,1-бифенил)[2-(2-амино-1,1-бифенил)]палладий(II) (295,3 мг, 375,32 мкмоль, 0,10 экв.) последовательно добавляли к смешанному раствору соединения WX001-9 (1,60 мг, 3,75 ммоль, 1,00 экв.) в тетрагидрофуране (20 мл)/воде (2 мл). После замены азотом 3 раза смесь нагревали до 60°С, перемешивали в течение 16 часов, охлаждали до комнатной температуры и выливали в 30 мл воды. Полученную смесь экстрагировали при помощи дихлорметана (10 мл) 3 раза и органические фазы объединяли. Объединенную органическую фазу промывали один раз 10 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением соединения WX011-9. LCMS (ESI) масса/заряд: 524,1 [М+Н]+.
Стадия 10: Синтез соединения WX011-10
При комнатной температуре раствор хлористоводородной кислоты в этилацетате (4 М, 20,00 мл) добавляли к соединению WX011-9 (1,60 г, 3,06 ммоль, 1,00 экв.). Смесь перемешивали в течение 1 часа и фильтровали с получением твердого вещества. Твердое вещество сушили при пониженном давлении с получением хлористоводородной соли соединения WX011-10. LCMS (ESI) масса/заряд: 424,1 [M+H]+.
Стадия 11: Синтез соединения WX011
При 0°С акрилоил хлорид (216,44 мг, 2,39 ммоль, 1,00 экв.) добавляли к раствору триэтиламина (2,42 г, 23,91 ммоль, 10,00 экв.) и гидрохлорида соединения WX011-10 (1,10 г, 2,39 ммоль, 1,00 экв.) в дихлорметане (10 мл). Смесь перемешивали в течение 60 минут и реакционный раствор выливали в 25 мл дихлорметана. Органические фазы дважды промывали водой (25 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли из фильтрата ротационным испарением при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали при помощи тонкослойной препаративной пластины (этилацетат) с получением соединения WX011. Соединение WX011 хирально расщепляли (колонка: AD (250 мм × 30 мм, 5 мкм); подвижная фаза: [0,1% аммиачная вода/этанол]; В%: 45%-45%) с получением соединения WX011A (время удержания: 0,58 минуты) и соединения WX011B (время удержания: 0,74 минуты). Время удержания измеряли на следующей аналитической колонке: колонка: Chiralpak AD-3 50×4,6 мм I.D., 3 мкм, подвижная фаза: 40% изопропанол (0,05% этилендиамин) в диоксиде углерода, скорость потока: 4 мл/мин, температура колонки: 40°С.
Соединение WX011A, LCMS (ESI) масса/заряд: 478.1 [М+Н]+, 500.1 [M+Na]+, 1H ЯМР (400МГц, CDCl3) δ: 7.85 (d, J=8.4 Гц, 1Н), 7.20 (s, 1Н), 7.16 (s, 1Н), 6.62 (s, 1Н), 6.51-6.30 (m, 2H), 5.68-5.60 (m, 1Н), 5.47 (2H, brs), 4.34 (s, 2H), 4.18-3.78 (m, 7H), 3.65-3.48 (m, 1Н), 3.22 (d, J=10.0 Гц, 3Н), 2.85-2.65 (m, 1Н), 2.44 (s, 3Н).
Соединение WX011B, LCMS (ESI) масса/заряд: 478.1 [М+Н]+, 500.0 [M+Na]+, 1Н ЯМР (400МГц, CDCl3) δ: 7.84 (d, J=8.8 Гц, 1Н), 7.20 (s, 1Н), 7.16 (s, 1Н), 6.62 (s, 1Н), 6.51-6.30 (m, 2H), 5.68-5.60 (m, 1Н), 5.42 (2H, brs), 4.34 (s, 2H), 4.15-3.72 (m, 7H), 3.62-3.48 (m, 1Н), 3.22 (d, J=10.4 Гц, 3Н), 2.90-2.65 (m, 1Н), 2.44 (s, 3Н).
Пример 20: Синтез соединения WX012
Путь синтеза:
Его синтезировали на основании способа синтеза примера 9 с применением промежуточного соединения WX011-10 в качестве промежуточного соединения. LCMS (ESI) масса/заряд: 490.0 [М+Н]+, 512.0 [M+Na]+, 1H ЯМР (400МГц, CDCl3) δ = 7.90-7.80 (m, 1Н), 7.22-7.18 (m, 1Н), 7.16 (s, 1H), 6.65-6.60 (m, 1H), 6.57 (brs, 2H), 4.38-4.30 (m, 2H), 4.20-3.90 (m, 6H), 3.89-3.70 (m, 1H), 3.68-3.38 (m, 1H), 3.28-3.18 (m, 3H), 2.80-2.65 (m, 1H), 2.44 (s, 3H), 2.18-2.08 (m, 1H), 1.98-1.85 (m, 3H).
Пример 21: Синтез соединения WX013
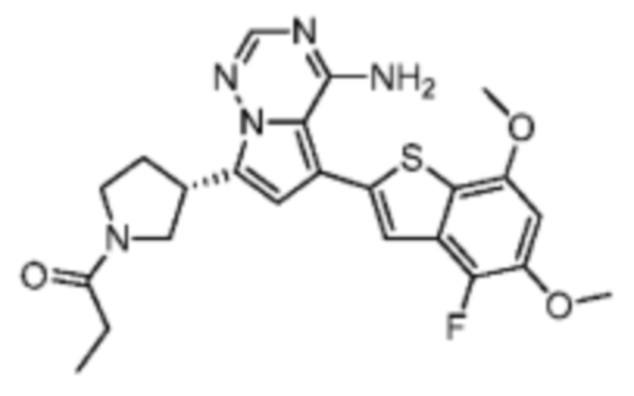
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-А и промежуточного соединения В6 в качестве исходных веществ. LCMS (ESI) масса/заряд: 470.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.79 (d, J=3.2 Гц, 1Н), 7.20 (s, 1Н), 6.72-6.54 (m, 2Н), 4.09-3.91 (m, 1Н), 3.88 (s, 3Н), 3.87 (s, 3Н), 3.85-3.72 (m, 1Н), 3.67-3.37 (m, 4Н), 2.32-2.25 (m, 2Н), 1.05-1.00 (m, 3Н).
Пример 22: Синтез соединения WX014
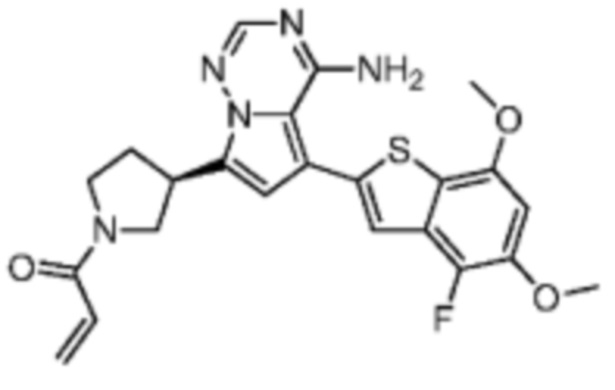
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-В и промежуточного соединения В6 в качестве исходных веществ. LCMS (ESI) масса/заряд: 468.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.90 (br s, 1Н), 7.31 (br d, J=10.0 Гц, 1Н), 6.83-6.69 (m, 2Н), 6.69-6.55 (m, 1Н), 6.39-6.21 (m, 1H), 5.83-5.70 (m, 1H), 4.69 (br d, J=2.0 Гц, 1H), 4.28-4.04 (m, 1H), 3.99 (d, J=3.6 Гц, 3Н), 3.97 (d, J=2.4 Гц, 4H), 3.89-3.70 (m, 2H), 3.64-3.54 (m, 1H), 2.63-2.42 (m, 1H), 2.33-2.13 (m, 2H).
Пример 23: Синтез соединения WX015
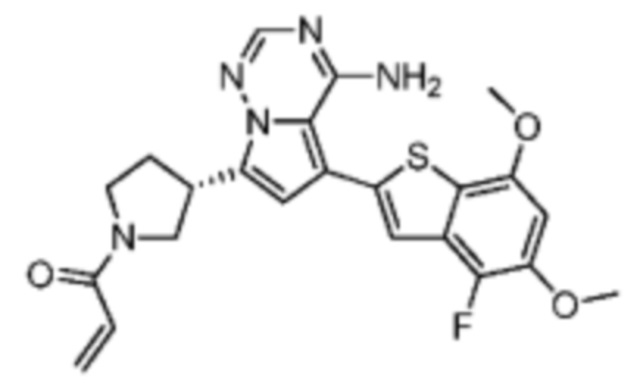
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-А и промежуточного соединения В6 в качестве исходных веществ. LCMS (ESI) масса/заряд: 468.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.79 (d, J=1.6 Гц, 1Н), 7.18 (d, J=1.6 Гц, 1Н), 6.65-6.60 (m, 2Н), 6.56-6.47 (m, 1Н), 6.28-6.03 (m, 1Н), 5.74-5.57 (m, 1Н), 4.17-3.97 (m, 1Н), 3.88 (s, 3Н), 3.86 (s, 3Н), 3.77-3.59 (m, 2Н), 3.50-3.38 (m, 1Н), 23.07-2.96 (m, 1Н), 2.47-2.31 (m, 1Н), 2.20-2.01 (m, 1Н).
Пример 24: Синтез соединения WX016
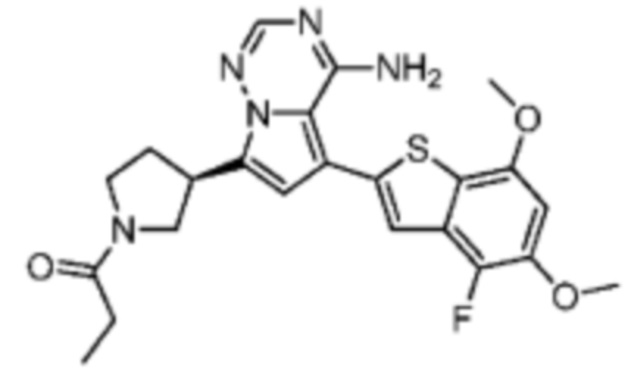
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-В и промежуточного соединения В6 в качестве исходных веществ. LCMS (ESI) масса/заряд: 470.1 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.79 (d, J=3.2 Гц, 1Н), 7.20 (s, 1Н), 6.71-6.49 (m, 2Н), 4.06-3.78 (m, 8Н), 3.68-3.58 (m, 1Н), 3.57-3.34 (m, 3Н), 3.15-3.05 (m, 1Н), 2.24-1.98 (m, 2Н), 1.07-1.00(m, 3Н).
Пример 25: Синтез соединения WX017
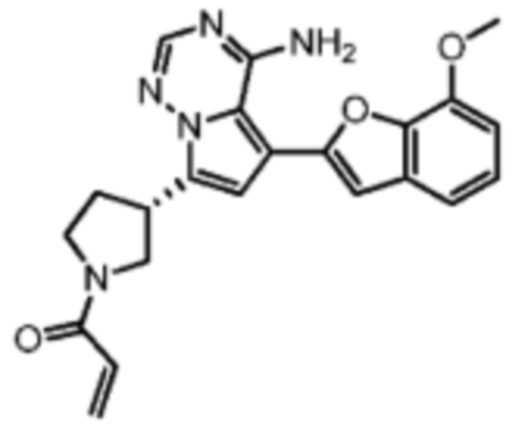
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-А и промежуточного соединения В7 в качестве исходных веществ. LCMS (ESI) масса/заряд: 404.2 [М+Н]+, 426.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.74 (d, J=2.4 Гц, 1Н), 7.12-7.02 (m, 2Н), 6.97-6.88 (m, 2Н), 6.79 (d, J=7.2 Гц, 1Н), 6.59-6.50 (m, 1Н), 6.24-6.10 (m, 1Н), 5.70-5.64 (m, 1Н), 4.28-3.94 (m, 1Н), 3.90 (s, 3Н), 3.84-3.45 (m, 3Н), 3.20-3.18 (m, 1Н), 2.49-2.31 (m, 1Н), 2.24-2.01 (m, 1Н).
Пример 26: Синтез соединения WX018
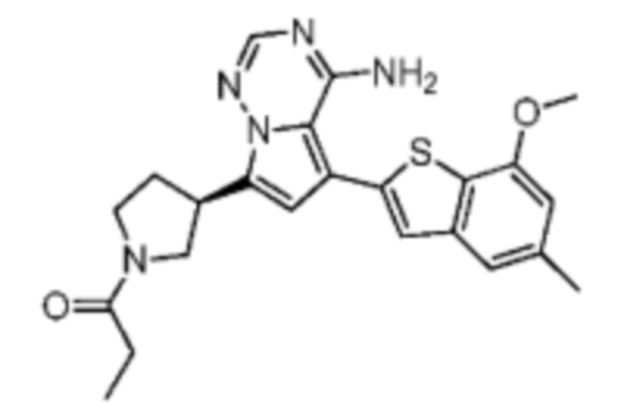
Его синтезировали на основании способа синтеза соединения WX001 с применением промежуточного соединения А1-В и промежуточного соединения В1 в качестве исходных веществ. LCMS (ESI) масса/заряд: 436.1 [М+Н]+, 458.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.76 (d, J=4.4 Гц, 1Н), 7.10 (s, 2Н), 6.67-6.61 (m, 1Н), 6.56 (d, J=13.2 Гц, 1Н), 3.97-3.89 (m, 1Н), 3.86 (s, 3Н), 3.65-3.55 (m, 1Н), 3.53-3.30 (m, 2Н), 3.20-3.18 (m, 1Н), 2.35 (s, 3Н), 2.33-2.16 (m, 3Н), 2.15-1.98 (m, 1Н), 1.07-0.97(m, 3Н).
Пример 27: Синтез соединения WX019
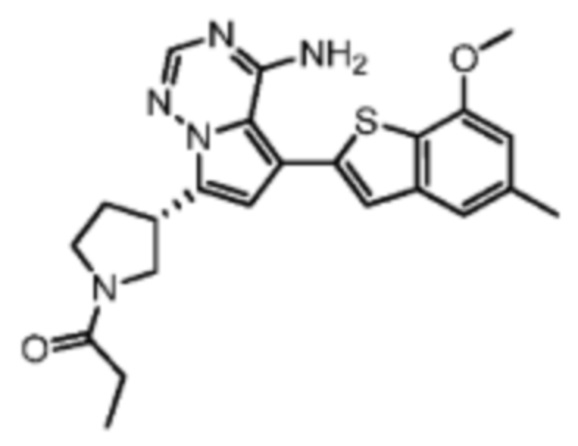
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-А и промежуточного соединения В1 в качестве исходных веществ. LCMS (ESI) масса/заряд: 436.1 [М+Н]+, 458.1 [M+Na]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.78 (d, J=4.4 Гц, 1Н), 7.14-7.10 (m, 2Н), 6.64 (s, 1Н), 6.58 (d, J=13.2 Гц, 1Н), 4.03-3.91 (m, 1Н), 3.87 (s, 3Н), 3.66-3.57 (m, 1Н), 3.55-3.32 (m, 2Н), 3.21-3.18 (m, 1Н), 2.45-2.32 (m, 4Н), 2.28 (q, J=7.2 Гц, 2Н), 2.19-2.01 (m, 1Н), 1.05-1.01 (m, 3Н).
Пример 28: Синтез соединения WX020
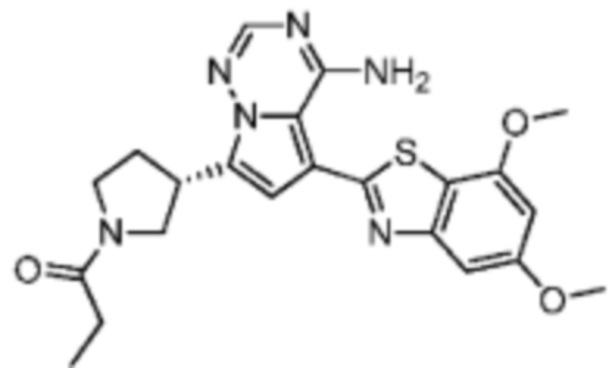
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения WX010-2 в качестве исходного вещества. LCMS (ESI) масса/заряд: 453.1 [М+Н]+.
Пример 29: Синтез соединения WX021
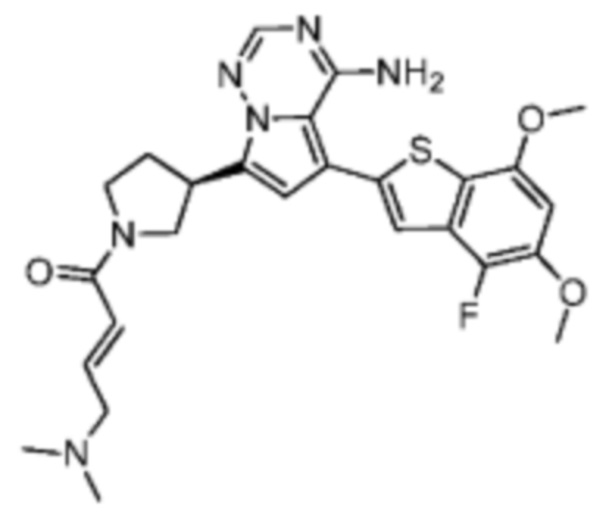
Его синтезировали на основании способов синтеза примера 1 и примера 9 с применением промежуточного соединения А1-В, промежуточного соединения В6 и масляной кислоты в качестве исходных веществ. LCMS (ESI) масса/заряд: 525.1 [М+Н]+, 547.0 [M+Na]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 7.91 (d, J=2.4 Гц, 1Н), 7.30 (d, J=1.2 Гц, 1Н), 6.93-6.80 (m, 1Н), 6.79-6.65 (m, 2Н), 6.53-6.47 (m, 1Н), 4.28-4.06 (m, 1Н), 4.06-3.91 (m, 6Н), 3.90-3.70 (m, 2Н), 3.69-3.52 (m, 1Н), 3.32-3.28 (m, 1Н), 3.27-3.09 (m, 2Н), 2.59-2.40 (m, 1Н), 2.38-2.28 (m, 6Н), 2.28-2.16 (m, 1Н).
Пример 30: Синтез соединения WX022
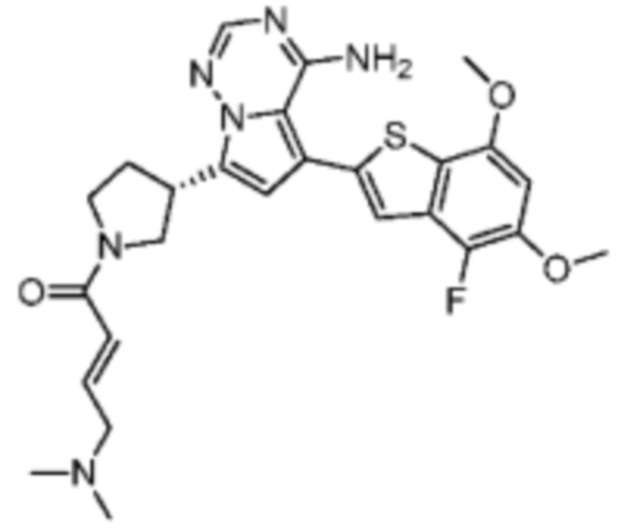
Его синтезировали на основании способов синтеза примера 1 и примера 9 с применением промежуточного соединения А1-А, промежуточного соединения В6 и масляной кислоты в качестве исходных веществ. LCMS (ESI) масса/заряд: 525.2 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный метанол) δ = 7.78 (d, J=2.4 Гц, 1Н), 7.17 (s, 1Н), 6.80-6.66 (m, 1Н), 6.65-6.53 (m, 2H), 6.50-6.39 (m, 1H), 4.18-3.94 (m, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.83-3.58 (m, 3H), 3.56-3.42 (m, 1H), 3.10 (q, J=7.2 Гц, 2H), 2.48-2.36 (m, 1H), 2.34 (s, 3H), 2.33 (s, 3H), 2.25-2.03 (m, 1H).
Пример 31: Синтез соединения WX023A
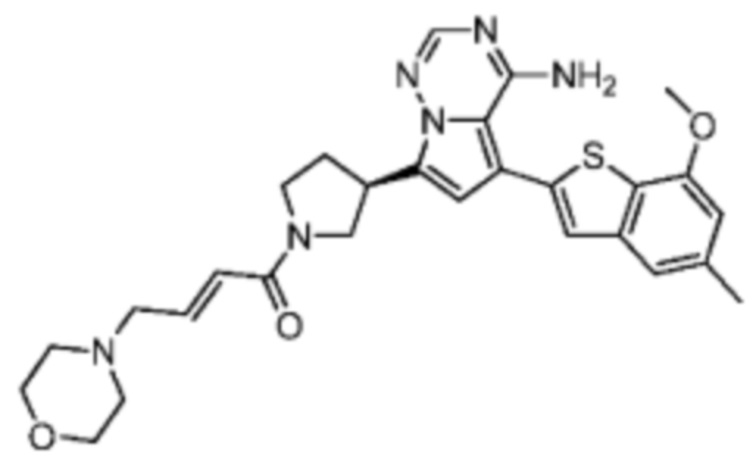
Его синтезировали на основании способов синтеза примера 1 и примера 9 с применением промежуточного соединения А1-В, промежуточного соединения В6 и промежуточного соединения В8 в качестве исходных веществ. LCMS (ESI) масса/заряд: 533.5[М+Н]+, 1H ЯМР (400МГц, дейтерированный хлороформ) δ = 7.97 (d, J=5.2 Гц, 1Н), 7.25-7.17 (m, 2Н), 7.04-6.82 (m, 1Н), 6.73-6.55 (m, 2Н), 6.37-6.31 (m, 1Н), 5.81 (br s, 2Н), 4.34-4.11 (m, 1Н), 4.10-3.94 (m, 4Н), 3.91-3.86 (m, 1Н), 3.81-3.59 (m, 6Н), 3.17 (t, J=7.2 Гц, 2Н), 2.60-2.38 (m, 8Н), 2.30-2.14 (m, 1Н).
Пример 32: Синтез соединения WX023B
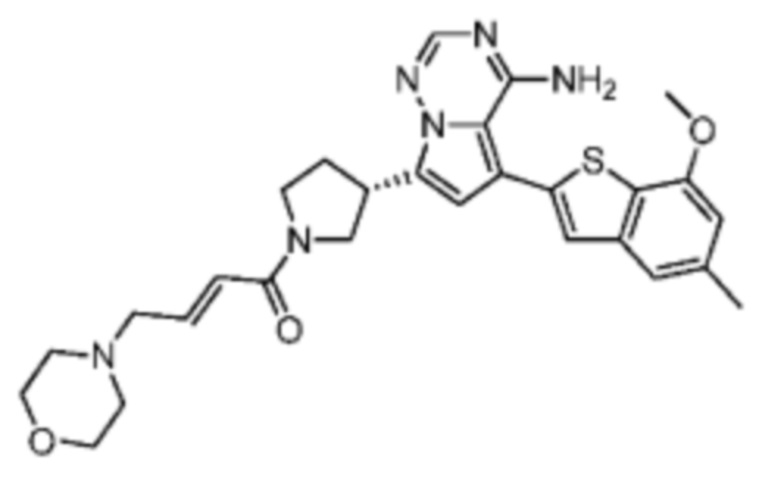
Его синтезировали на основании способов синтеза примера 1 и примера 9 с применением промежуточного соединения А1-А, промежуточного соединения В1 и промежуточного соединения В8 в качестве исходных веществ. LCMS (ESI) масса/заряд: 533.2 [М+Н]+, 1H ЯМР (400МГц, дейтерированный хлороформ) δ = 7.96 (d, J=5.6 Гц, 1Н), 7.25-7.17 (m, 2Н), 7.00-6.84 (m, 1Н), 6.73-6.60 (m, 2Н), 6.42-6.28 (m, 1Н), 5.87 (br s, 2Н), 4.25-4.10 (m, 1Н), 4.09-3.95 (m, 4Н), 3.90-3.78 (m, 1Н), 3.77-3.59 (m, 6Н), 3.26-3.12 (m, 2Н), 2.62-2.39 (m, 8Н), 2.31-2.15 (m, 1Н).
Пример 33: Синтез соединения WX024
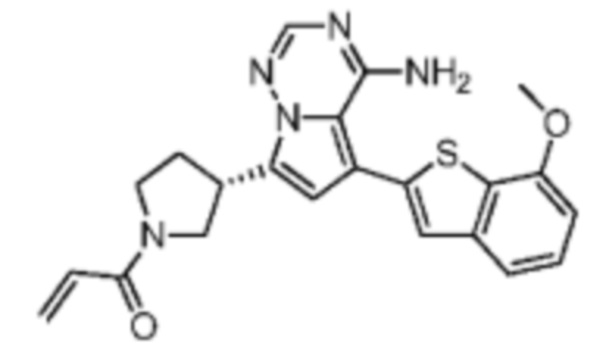
Его синтезировали на основании способа синтеза примера 1 с применением промежуточного соединения А1-А и промежуточного соединения В9 в качестве исходных веществ. LCMS (ESI) масса/заряд: 420.1[М+Н]+, 1H ЯМР (400МГц, дейтерированный метанол) δ = 8.12 (d, J=1.6 Гц, 1Н), 7.56-7.45 (m, 2Н), 7.43-7.31 (m, 1Н), 7.04-6.87 (m, 2Н), 6.72-6.54 (m, 1Н), 6.35-6.21 (m, 1Н), 5.80-5.68 (m, 1Н), 4.25-4.02 (m, 2Н), 4.00 (s, 3Н), 3.95-3.71 (m, 2Н), 3.68-3.56 (m, 1Н), 2.64-2.43 (m, 1Н), 2.41-2.17 (m, 1Н).
Пример 34: Синтез соединения WX025
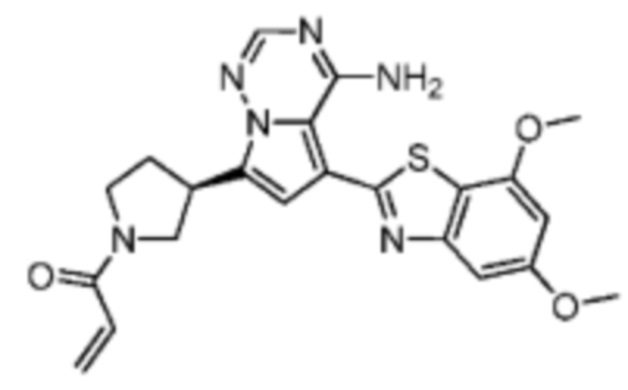
Его синтезировали на основании способов синтеза примеров 15 и 16, а также примера 1, с применением промежуточного соединения А1-В и промежуточного соединения В5 в качестве исходных веществ. LCMS (ESI) масса/заряд: 451.0 [М+Н]+, 1Н ЯМР (400МГц, дейтерированный хлороформ) δ = 7.94 (d, J=3.2 Гц, 1Н), 7.01 (s, 1Н), 6.90 (s, 1Н), 6.60-6.38 (m, 3Н), 5.76-5.69 (m, 1Н), 4.31-4.13 (m, 1Н), 4.11-3.86 (m, 7Н), 3.80-3.54 (m, 3Н), 2.70-2.38 (m, 1Н), 2.31-2.22 (m, 1Н).
Данные ЯМР и МС для каждого примера

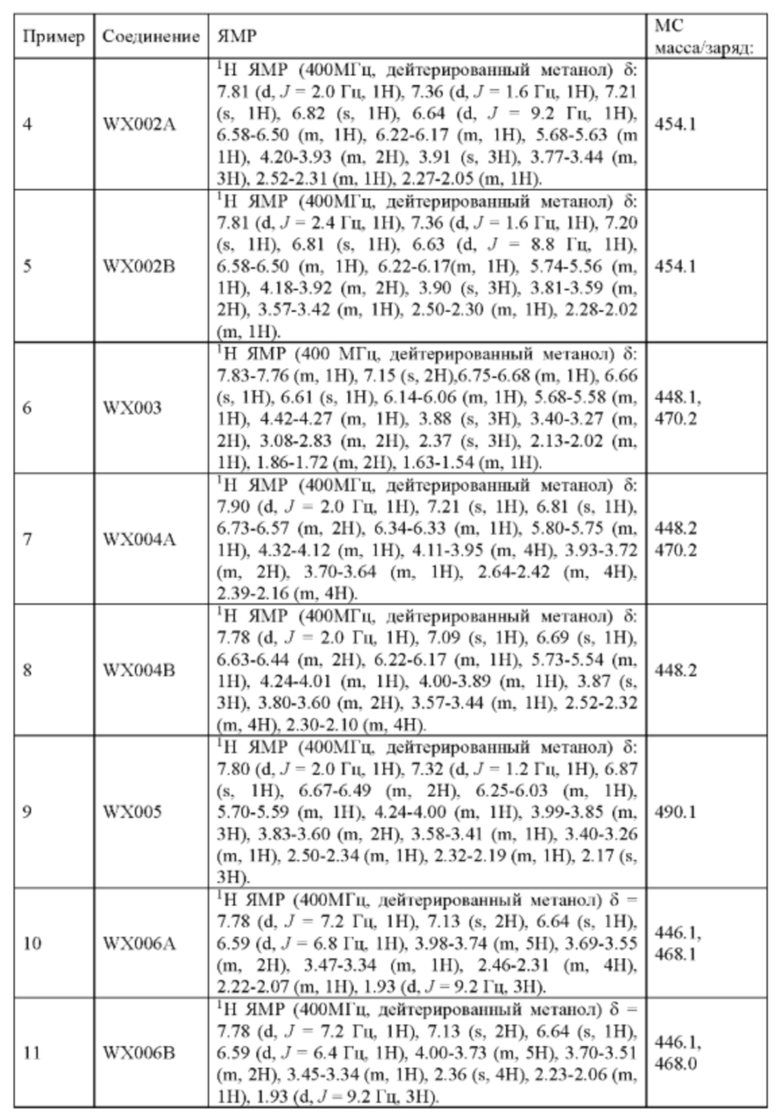
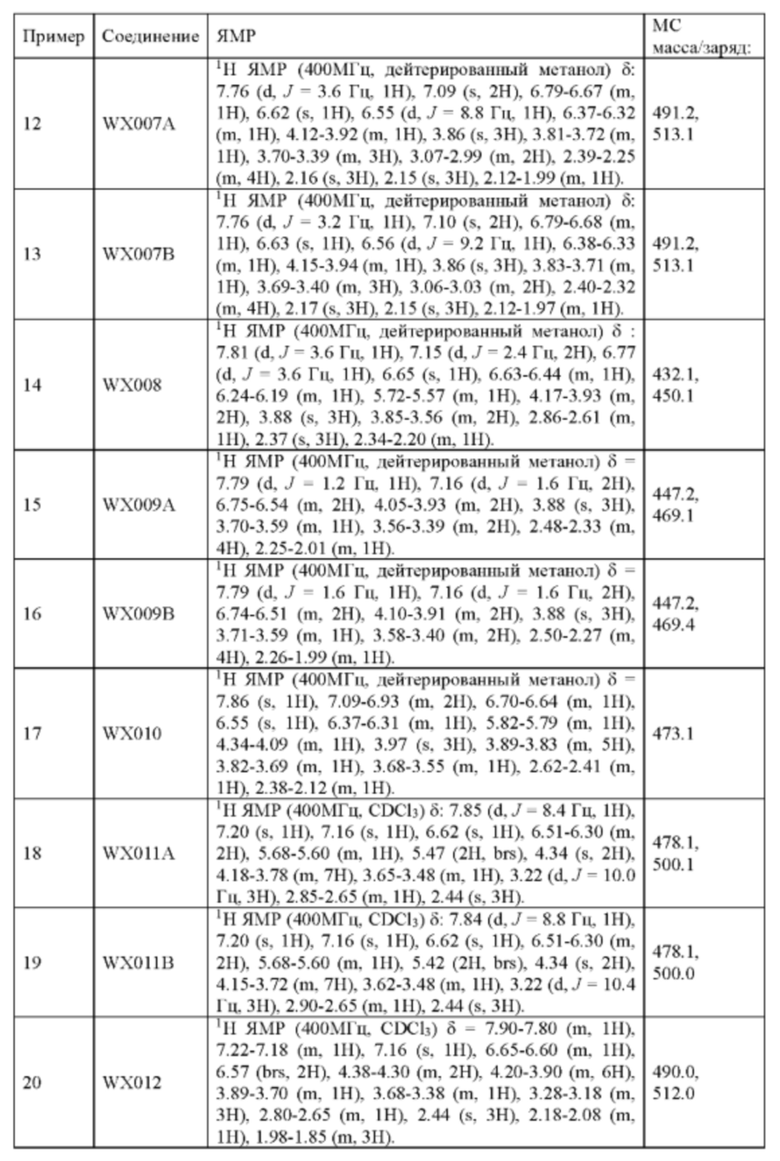
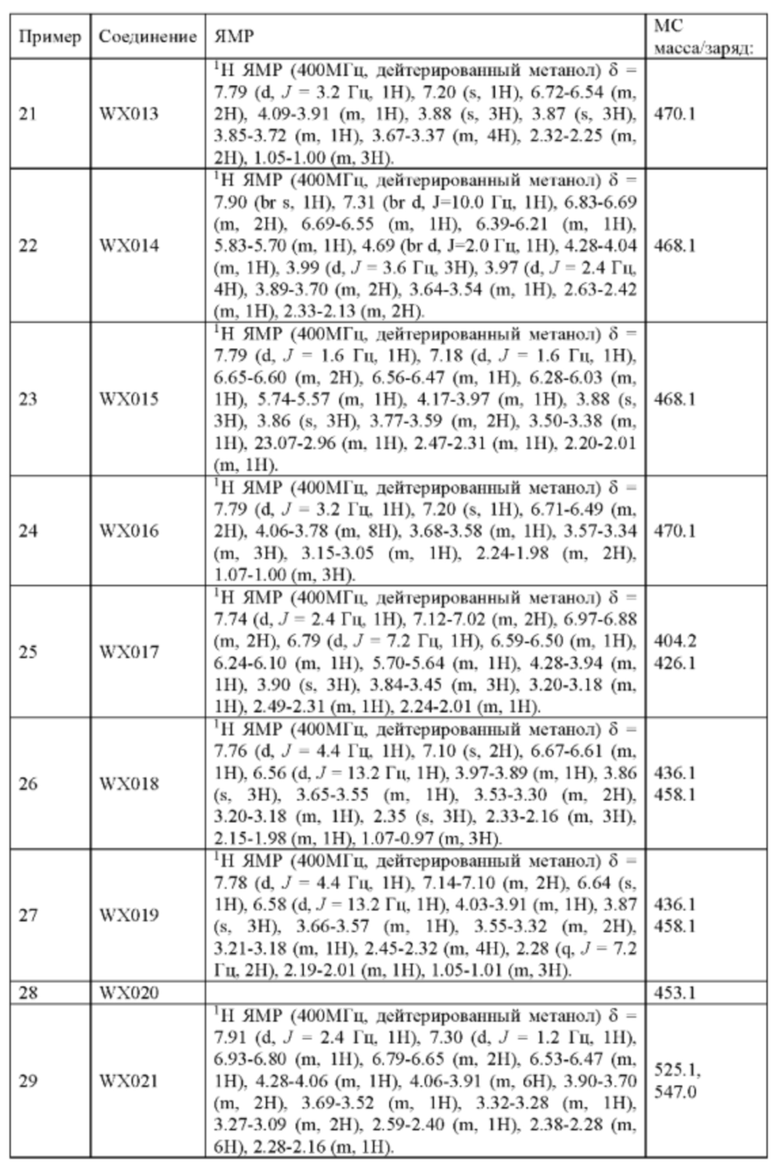
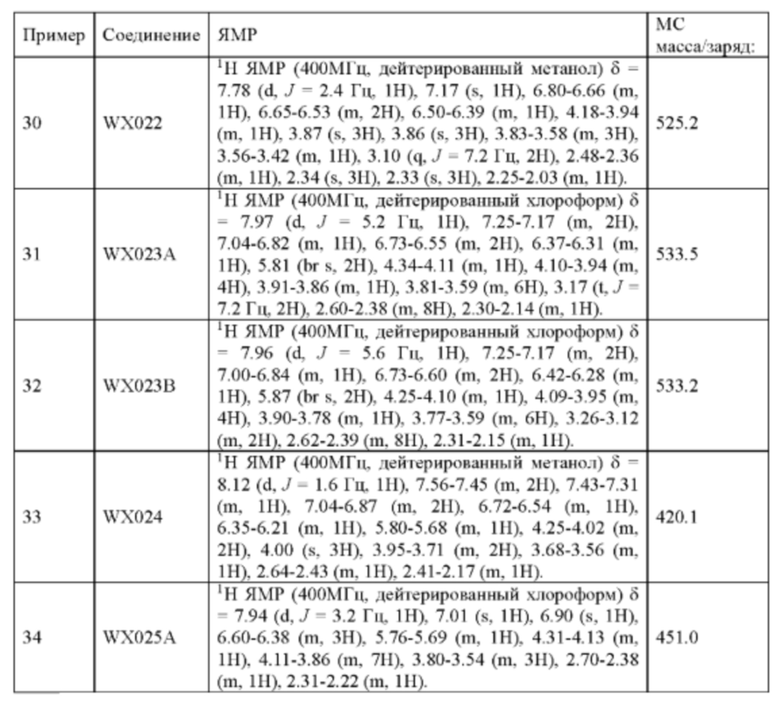
Анализ 1. Оценивание ингибиторной активности киназы дикого типа in vitro Применяли тест активности меченной изотопом 33Р киназы (Reaction Biology Corp) для определения значения IC50 в целях оценивания ингибиторной способности тестируемого соединения в отношении человеческого FGFR1 и FGFR4.
Буферные условия. 20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO.
Стадия анализа. Тестируемое соединение растворяли в DMSO при комнатной температуре с получением 10 мМ раствора для применения. Субстрат растворяли в свежеприготовленном буфере, в него добавляли тестируемую киназу и полученный в результате реакционный раствор смешивали до однородности. Путем применения акустического метода (Echo 550) добавляли раствор DMSO, в котором растворяли тестируемое соединение, в упомянутый выше однородно смешанный реакционный раствор. Концентрации соединений в реакционных растворах составляли 10 мкМ, 3,33 мкМ, 1,11 мкМ, 0,370 мкМ, 0,123 мкМ, 41,2 нМ, 13,7 нМ, 4,57 нМ, 1,52 нМ и 0,508 нМ, соответственно, или составляли 10 мкМ, 2,50 мкМ, 0,62 мкМ, 0,156 мкМ, 39,1 нМ, 9,8 нМ, 2,4 нМ, 0,61 нМ, 0,15 нМ и 0,038 нМ, соответственно. Через 15 минут после инкубации добавляли 33Р-АТФ (активность 0,01 мкСi/мкл, соответствующая концентрация показана в таблице 1) для начала реакции. Информация о номере категории поставщика, номере партии и концентрации в реакционном растворе FGFR1, FGFR4 и их субстратах приведена в таблице 1. После осуществления реакции при комнатной температуре в течении 120 минут реакционный раствор наносили пятнами на ионообменную фильтровальную бумагу Р81 (Whatman №3698-915). После многократной промывки фильтровальной бумаги 0,75% раствором фосфорной кислоты измеряли радиоактивность фосфорилированного субстрата, оставшегося на фильтровальной бумаге. Данные по активности киназы выражали путем сравнения киназной активности тестируемого соединения и киназной активности пустой группы (содержащей только DMSO). Значения IC50 получали путем вычерчивания кривой с использованием программного обеспечения Prism4 (GraphPad). Экспериментальные результаты приведены в таблице 2.
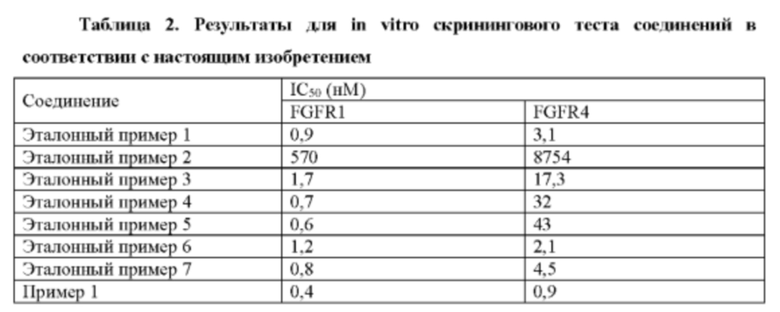
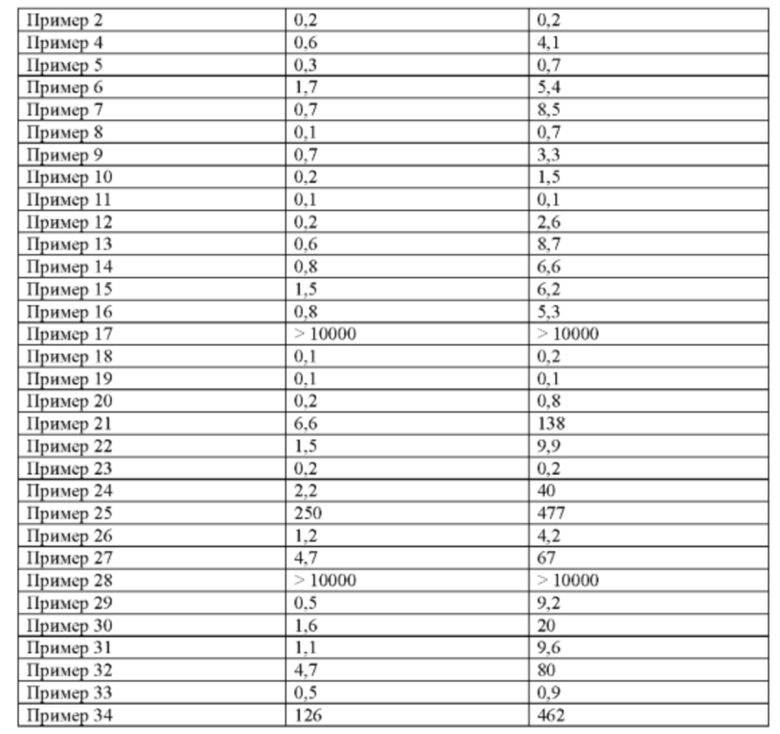
Заключение. Соединения в соответствии с настоящим изобретением демонстрировали хорошую ингибиторную активность в отношении киназ дикого типа.
Анализ 2. Оценивание ингибиторной активности мутантной киназы in vitro
Применяли тест активности меченной изотопом 33Р киназы (Reaction Biology Corp) для определения значения IC50 в целях оценивания ингибиторной способности тестируемого соединения в отношении мутантного штамма FGFR.
Буферные условия. 20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO.
Стадия анализа. Тестируемое соединение растворяли в DMSO при комнатной температуре с получением 10 мМ раствора для применения. Субстрат растворяли в свежеприготовленном буфере, в него добавляли тестируемую киназу и полученный в результате реакционный раствор смешивали до однородности. Путем применения акустического метода (Echo 550) добавляли раствор DMSO, в котором растворяли тестируемое соединение, в упомянутый выше однородно смешанный реакционный раствор. Концентрации соединений в реакционных растворах составляли 10 мкМ, 3,33 мкМ, 1,11 мкМ, 0,370 мкМ, 0,123 мкМ, 41,2 нМ, 13,7 нМ, 4,57 нМ, 1,52 нМ и 0,508 нМ, соответственно, или составляли 10 мкМ, 2,50 мкМ, 0,62 мкМ, 0,156 мкМ, 39,1 нМ, 9,8 нМ, 2,4 нМ, 0,61 нМ, 0,15 нМ и 0,038 нМ, соответственно. Через 15 минут после инкубации добавляли 33Р-АТФ (активность 0,01 мкCi/мкл, соответствующая концентрация показана в таблице 3) для начала реакции. Информация о номере категории поставщика, номере партии и концентрации в реакционном растворе FGFR1, FGFR4 и их субстратах приведена в таблице 3. После осуществления реакции при комнатной температуре в течении 120 минут реакционный раствор наносили пятнами на ионообменную фильтровальную бумагу Р81 (Whatman №3698-915). После многократной промывки фильтровальной бумаги 0,75% раствором фосфорной кислоты измеряли радиоактивность фосфорилированного субстрата, оставшегося на фильтровальной бумаге. Данные по активности киназы выражали путем сравнения киназной активности тестируемого соединения и киназной активности пустой группы (содержащей только DMSO). Значения IC50 получали путем вычерчивания кривой с использованием программного обеспечения Prism4 (GraphPad). Экспериментальные результаты приведены в таблице 4.
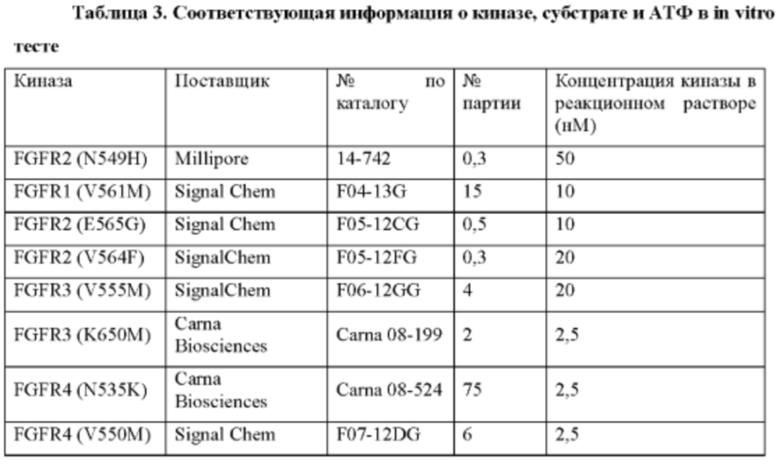


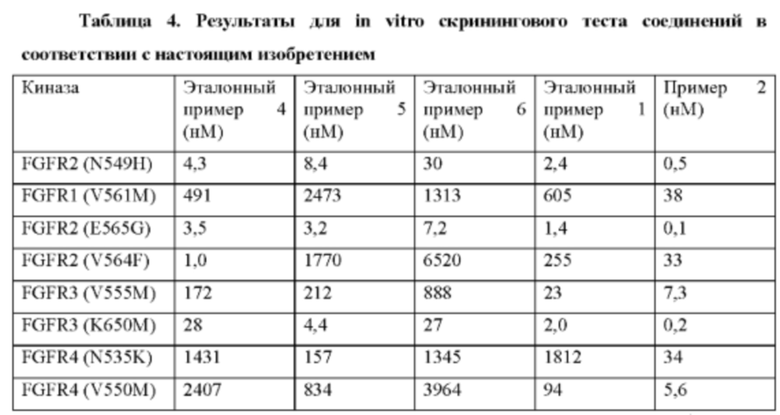
Заключение. Некоторые соединения в соответствии с настоящим изобретением демонстрировали хорошую ингибиторную активность в отношении как FGFR дикого типа, так и мутантного FGFR.
Анализ 3. Оценивание фармакокинетических показателей соединений Цель эксперимента. Тестирование фармакокинетических показателей соединения у мышей in vivo.
Экспериментальные материалы
Мыши Balb/c (самки).
Экспериментальные процедуры
Тестировали профили фармакокинетических показателей у грызунов после внутривенной инъекции и перорального введения соединения с использованием стандартного протокола. В экспериментах получали кандидатное соединение в виде прозрачного раствора, который вводили мышам в виде однократной внутривенной инъекции и перорального введения. Среда для внутривенной инъекции представляла собой 10% DMSO/10% солютола/80% воды, а среда для перорального введения представляла собой 0,5% карбоксиметилцеллюлозы натрия + 0,2% Tween. Собирали образцы цельной крови в пределах 24 часов. Все образцы крови добавляли в корректно маркированные пластиковые центрифужные пробирки, в которые заранее добавляли 0,5 М K2-EDTA антикоагулянта. После сбора образцов крови образцы крови центрифугировали при 4°С и 3000 g в течение 10 минут. Супернатантную плазму собирали, незамедлительно помещали в сухой лед и держали при температуре -20°С или ниже. Применяли аналитический метод LC-MS/MS для количественного анализа концентрации в крови и вычисляли фармакокинетические параметры, такие как пиковая концентрация, пиковое время, скорость выведения, период полужизни, площадь под кривой и биодоступость.
Результаты эксперимента
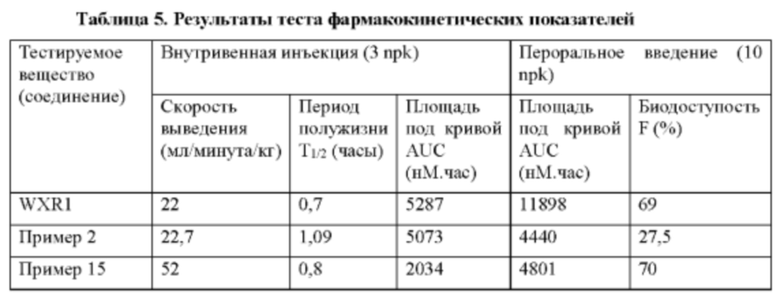
Заключение. Фармакокинетический индекс соединений в соответствии с настоящим изобретением был хорошим у мышей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| АНАЛОГИ СОЕДИНЕНИЙ 4Н-ПИРАЗОЛО[1,5-А]БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2015 |
|
RU2672722C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ИЗОТИАЗОЛО[5,4-d]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРА IRAK4 | 2019 |
|
RU2801942C2 |
| Соединение ингибитора FGFR в твердой форме и способ его получения | 2020 |
|
RU2810067C2 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2016 |
|
RU2766543C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
Изобретение относится к гетероароматическим соединениям. Предложено соединение формулы (I) или его фармацевтически приемлемая соль, где L выбран из С2-4алкенила, C2-4алкинила и R5, который представляет собой H или выбран из C1-3алкила, C1-3гетероалкила, C3-6циклоалкила и 4-6-членного гетероциклоалкила, которые необязательно замещены 1, 2 или 3 R группами; или R5 и L вместе образуют структурную единицу  , которая представляет собой
, которая представляет собой  ; R1 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила и C1-3гетероалкила; R2 выбран из H, F, Cl, Br, I, OH и NH2; R3 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила; R4 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1R группой; R6 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила; гетероатом или гетерогруппа независимо и отдельно выбраны из -NH-, N, -O- и -S- в количестве 1, 2 или 3. Также раскрываются конкретные соединения и применения соединений. Технический результат – получение соединений, подходящих для использования в качестве FGFR ингибитора. 4 н. и 15 з.п. ф-лы, 6 табл., 44 пр.
; R1 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила и C1-3гетероалкила; R2 выбран из H, F, Cl, Br, I, OH и NH2; R3 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила; R4 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1R группой; R6 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила; гетероатом или гетерогруппа независимо и отдельно выбраны из -NH-, N, -O- и -S- в количестве 1, 2 или 3. Также раскрываются конкретные соединения и применения соединений. Технический результат – получение соединений, подходящих для использования в качестве FGFR ингибитора. 4 н. и 15 з.п. ф-лы, 6 табл., 44 пр.
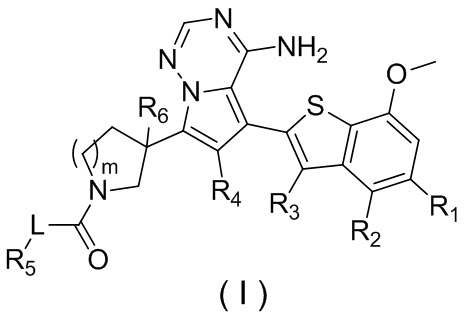
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль
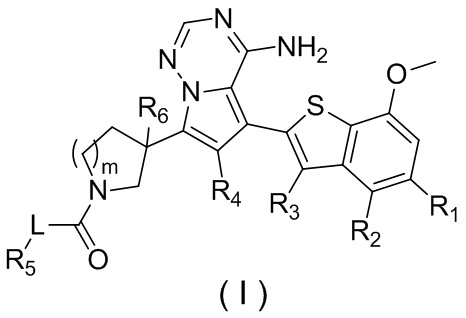 ,
,
где
m представляет собой 1 или 2;
L выбран из C2-4алкенила, C2-4алкинила и R5, который представляет собой H или выбран из C1-3алкила, C1-3гетероалкила, C3-6циклоалкила и 4-6-членного гетероциклоалкила, который необязательно замещен 1R группой; или R5 и L вместе образуют структурную единицу 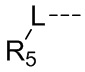 , которая представляет собой
, которая представляет собой  ;
;
R1 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила и C1-3гетероалкила;
R2 выбран из H, F, Cl, Br, I, OH и NH2;
R3 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила;
R4 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила и C1-3гетероалкила, которые необязательно замещены 1R группой;
R6 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила;
R выбран из F, Cl, Br, I, OH, NH2, CN, Me, CF3, N(CH3)2 и  ;
;
гетероатом или гетерогруппа в C1-3гетероалкиле и 4-6-членном гетероциклоалкиле независимо и отдельно выбраны из -NH-, N, -O- и-S-;
в любом из вышеуказанных случаев число гетероатомов или число гетерогрупп независимо и отдельно выбраны из 1, 2 или 3.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 выбран из H, галогена, OH и NH2 или выбран из C1-3алкила и C1-3алкоксила.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R1 выбран из H, F, Cl, Br, I, Me и.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R3 выбран из H и C1-3алкила.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, где R3 выбран из H и Me.
6. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R4 выбран из H, галогена, OH, NH2 и CN или выбран из C1-3алкила, C1-3алкоксила и C1-3алкиламино, которые необязательно замещены 1R группой.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где R4 выбран из H и  .
.
8. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R5 представляет собой H или выбран из C1-3алкила, C1-3алкиламино и морфолинила, которые необязательно замещены 1R группой.
9. Соединение по п. 8 или его фармацевтически приемлемая соль, где R5 выбран из H, Me, Et,  ,
,  и
и  .
.
10. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R6 выбран из H и OH.
11. Соединение по п. 1 или его фармацевтически приемлемая соль, где L выбран из  ,
,  ,
,  и
и  .
.
12. Соединение по п. 9 или 11 или его фармацевтически приемлемая соль, где структурная единица выбрана из  ,
,  ,
,  ,
,  и .
и .
13. Соединение по любому из пп. 1, 3 или 5 или его фармацевтически приемлемая соль, где структурная единица 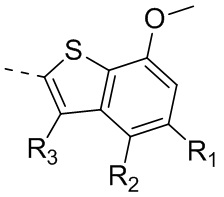 выбрана из
выбрана из 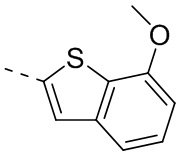 ,
, 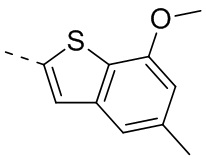 ,
, 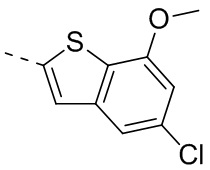 ,
, 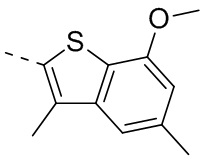 ,
, 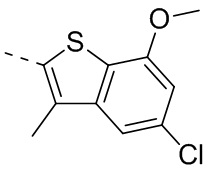 и
и 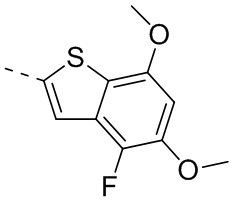 .
.
14. Соединение по любому из пп. 1-13 или его фармацевтически приемлемая соль, которые выбраны из:
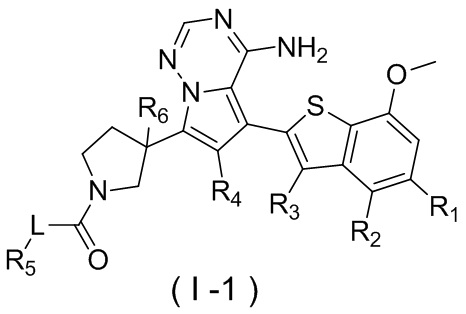 ,
, 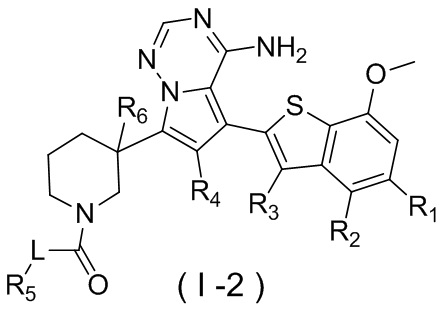 ,
,
где R1, R2, R3, R4, R5, R6 и L определены в любом из пп. 1-12.
15. Соединение по п. 14 или его фармацевтически приемлемая соль, которые выбраны из:
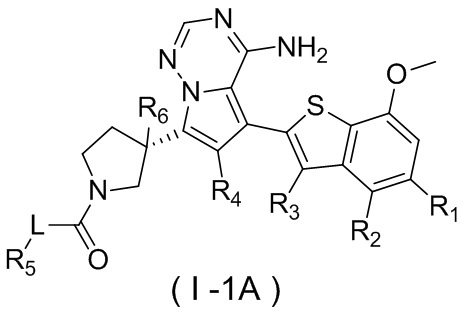 ,
, 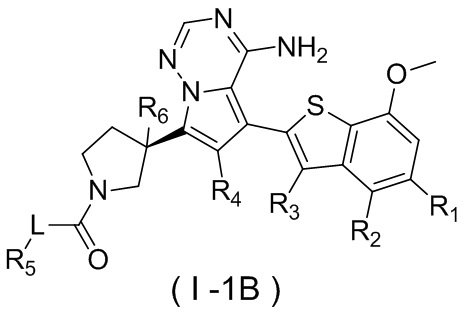 ,
,
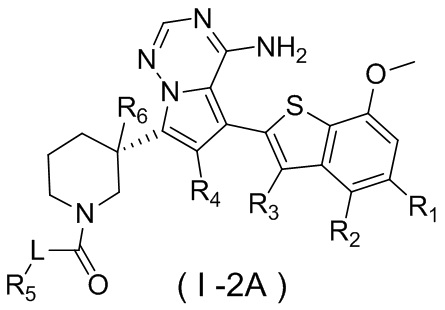 ,
, 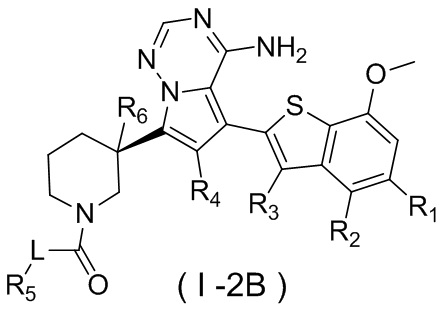 ,
,
где
R1, R2, R3, R4, R5, R6 и L определены в любом из пп. 1-12.
16. Соединение, представленное в любой из следующих формул:
 ,
,  ,
,  ,
,
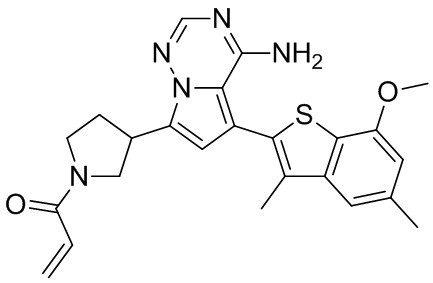 ,
, 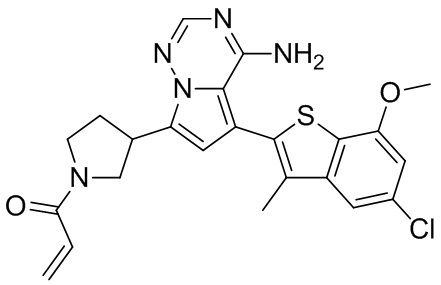 ,
, 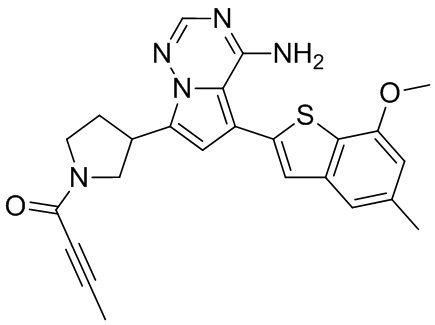 ,
,
 ,
, 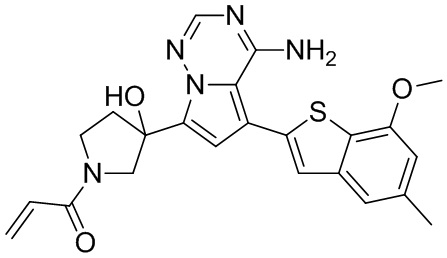 ,
, 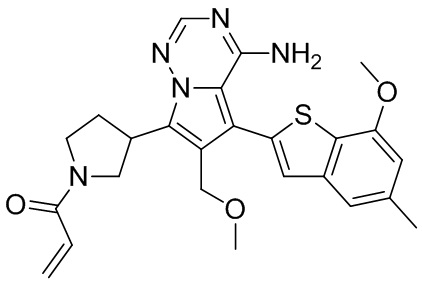 ,
,
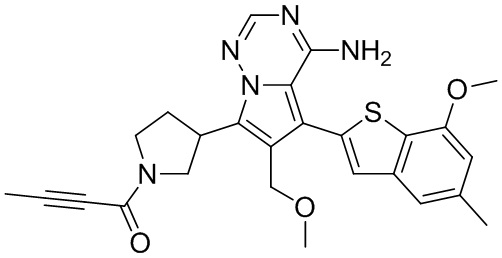 ,
, 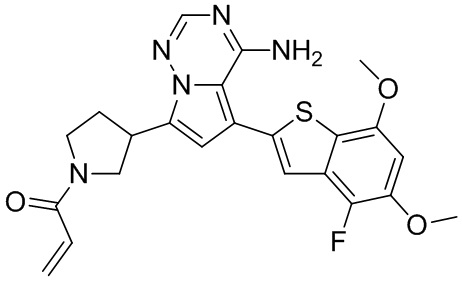 ,
, 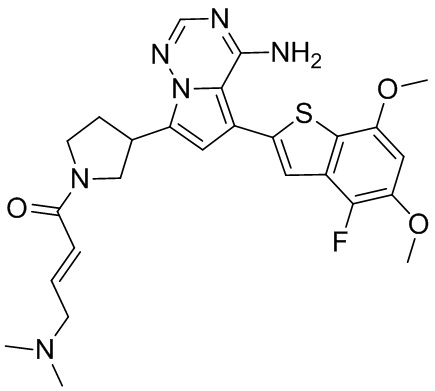 ,
,
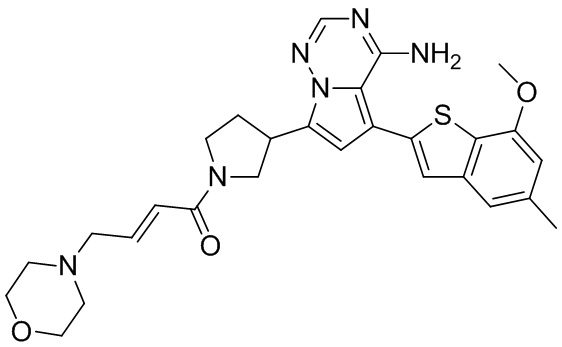 ,
, 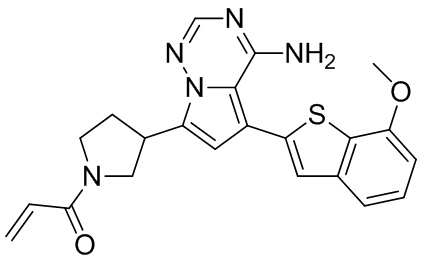 ,
, 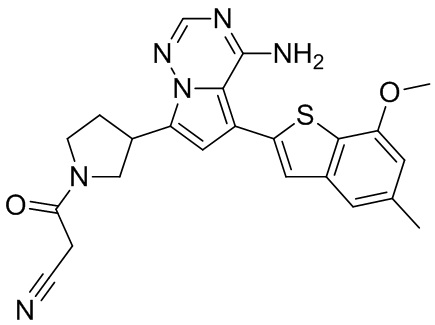 ,
,
или его фармацевтически приемлемая соль.
17. Соединение по п. 16, которое выбрано из:
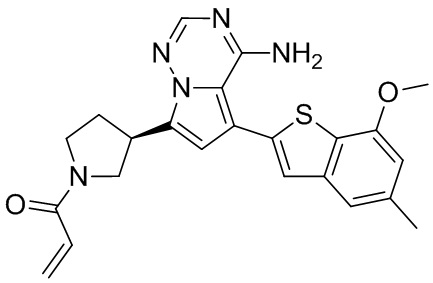 ,
, 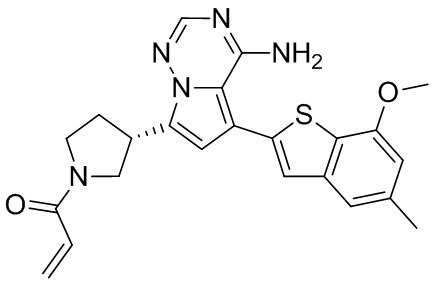 ,
, 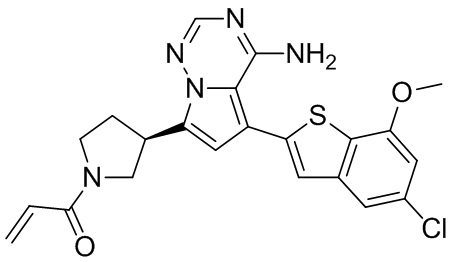 ,
,
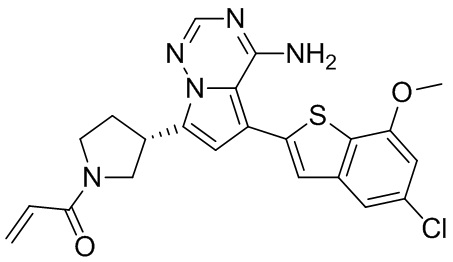 ,
, 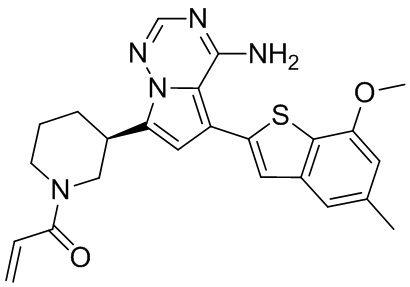 ,
, 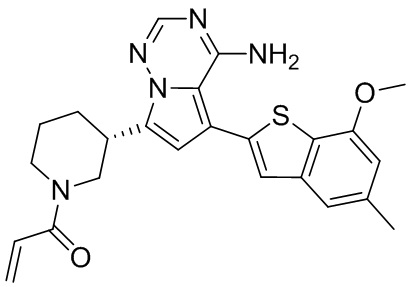 ,
,
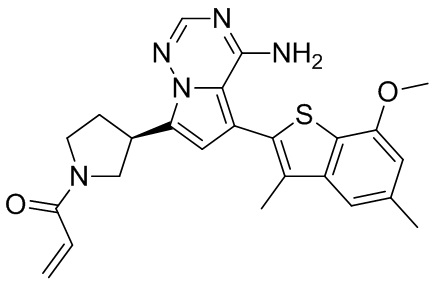 ,
, 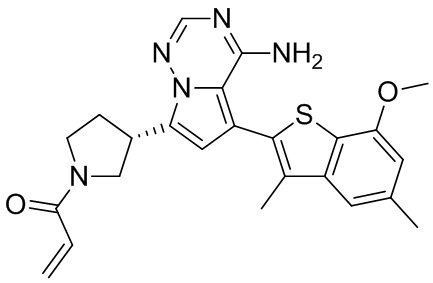 ,
, 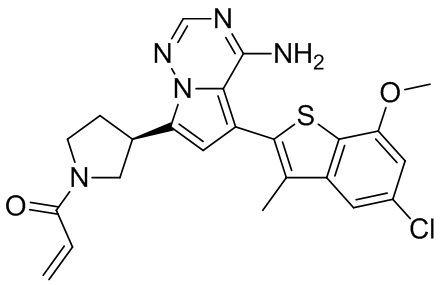 ,
,
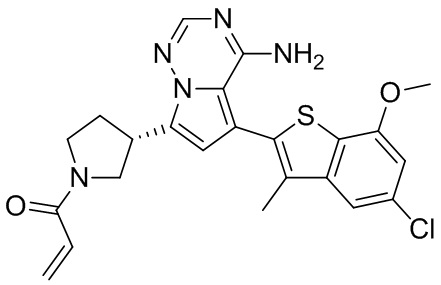 ,
, 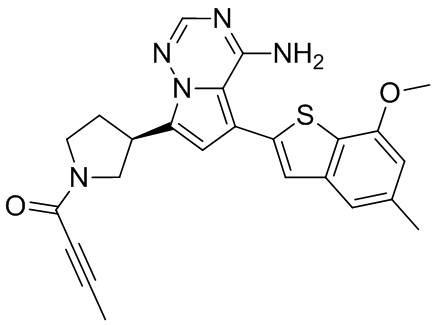 ,
, 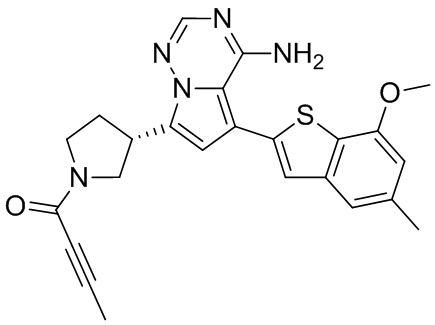 ,
,
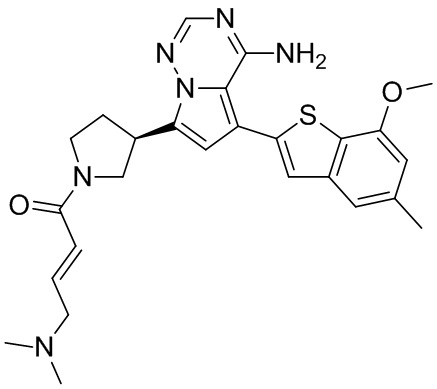 ,
, 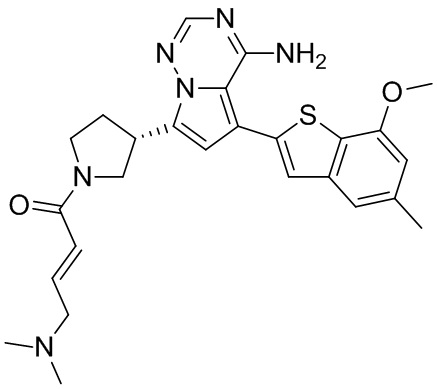 ,
, 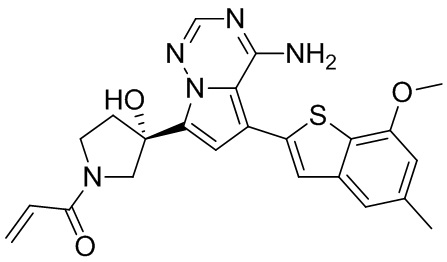
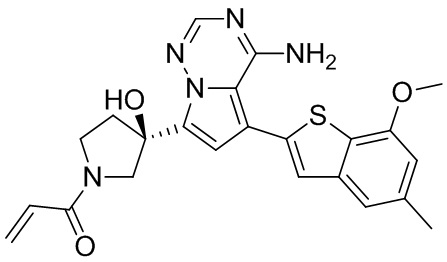 ,
, 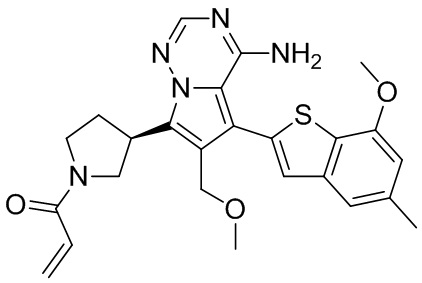 ,
, 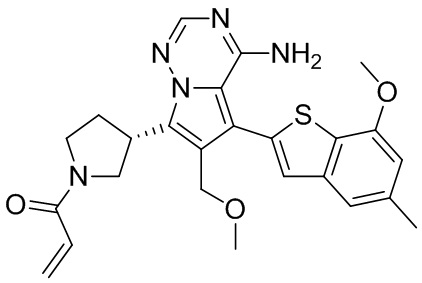 ,
,
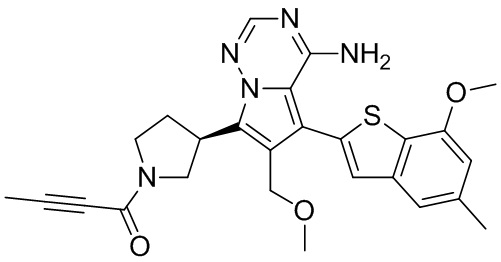 ,
, 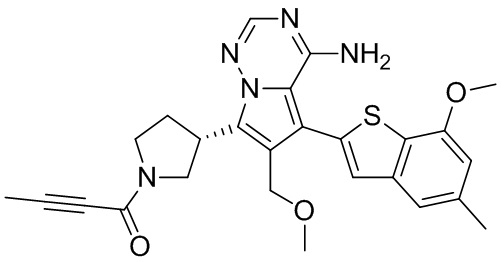 ,
, 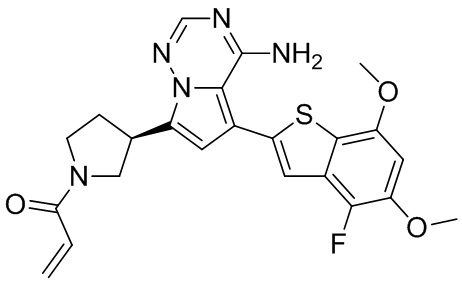 ,
,
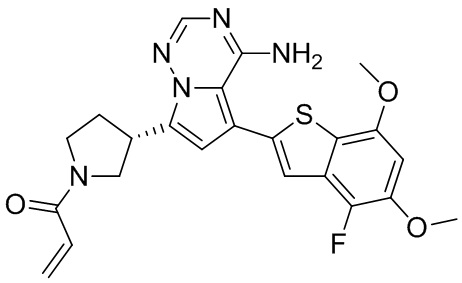 ,
, 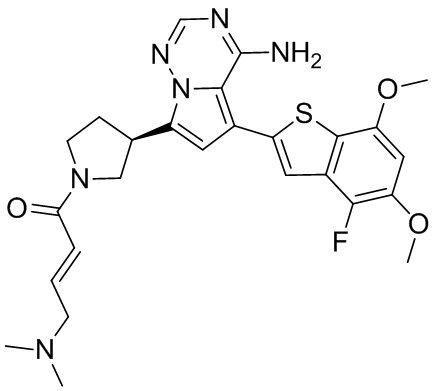 ,
, 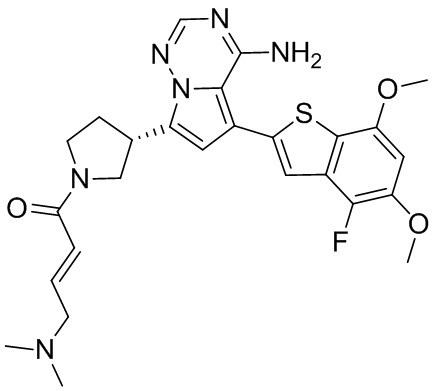 ,
,
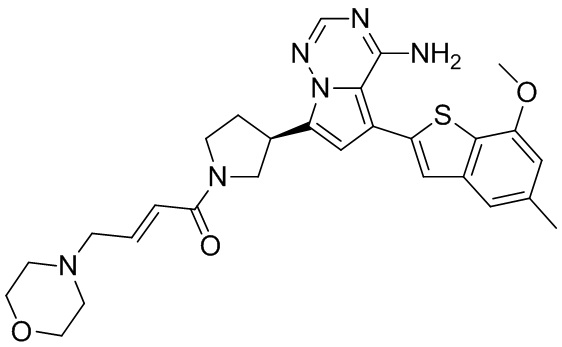 ,
, 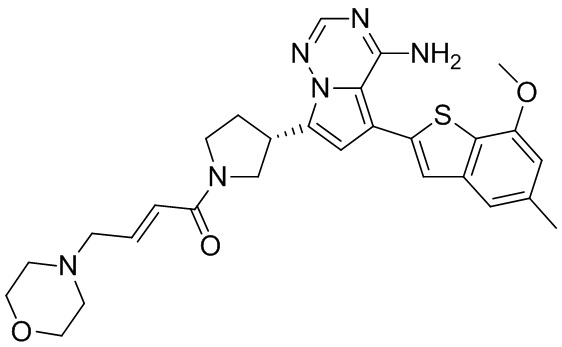 ,
, 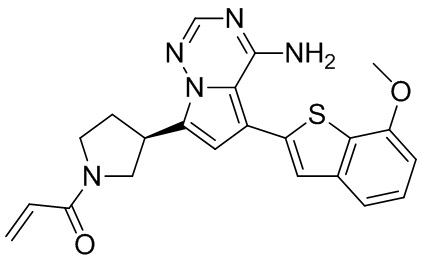 ,
,
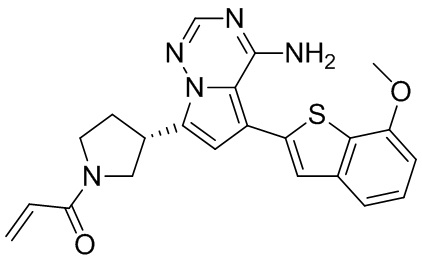 ,
, 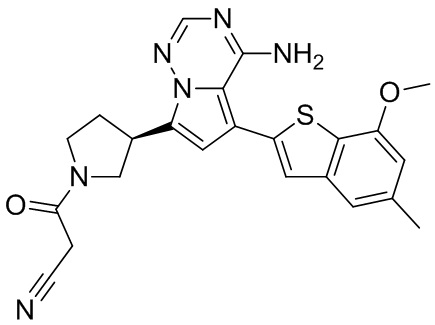 и
и 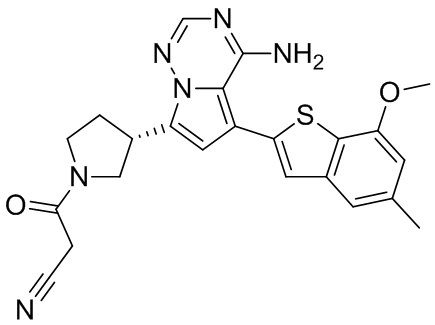 ,
,
или его фармацевтически приемлемая соль.
18. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли при изготовлении FGFR ингибитора.
19. Применение соединения по любому из пп. 1-17 или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для лечения солидной опухоли.
| WO 2013087578 A1, 20.06.2013 | |||
| WO 2013124316 A1, 29.08.2013 | |||
| WO 2007056170 A2, 18.05.2007 | |||
| ПИРРОЛО[2,1-f][1,2,4]ТРИАЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2589053C1 |
| Устройство для сцепления продольного бруса (развода) повозочной рамы со шкворнем | 1929 |
|
SU15463A1 |

