ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
На основании § 119(e) раздела 35 свода законов США данная заявка испрашивает приоритет американской предварительной заявки на патент № 61/785460, поданной 14 марта 2013, которая полностью включена в данный документ в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Настоящее изобретение в целом ориентировано на новые соединения, обладающие активностью как ингибиторы киназ ALK2 и/или JAK2, и использование вышеуказанных соединений для лечения различных онкологических заболеваний.
ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
Янус-киназы (JAK) представляют собой семейство киназ, из которых четыре имеются у млекопитающих (JAK1, JAK2, JAK3 и TYK2), которые являются неотъемлемой частью системы передачи сигналов от внеклеточных цитокинов, включая интерлeйкины, интерфероны, а также многочисленных гормонов (Aringer, M., et al., Life Sci, 1999. 64(24): p. 2173-86; Briscoe, J., et al., Phitos Trans R Soc Lond В Biol Sci, 1996. 351(1336): p. 167-71; Ihle, J. N., Semin Immunol, 1995. 7(4): p. 247-54; Ihle, J. N., Philos Trans R Soc Lond В Biol Sci, 1996. 351(1336): p. 159-66; Firmbach-Kraft, I., et al., Oncogene, 1990. 5(9): p. 1329-36; Harpur, A. G., et al., Oncogene, 1992. 7(7): p. 1347-53; Rane, S. G. and E. P. Reddy, Oncogene, 1994. 9(8): p. 2415-23; Wilks, A. F., Methods Enzymol, 1991. 200: p. 533-46). Эти нерецепторные тирозинкиназы связываются с различными цитокиновыми рецепторами и принимают участие в передаче сигнала от внеклеточной связки лиганд-рецептор в цитоплазму, фосфорилируя молекулы STAT (передатчик сигнала и активатор транскрипции), которые затем проникают в ядро и управляют транскрипцией различных целевых генов, вовлеченных в рост и пролиферацию (Briscoe, J., et al.; Ihle, J. N. (1995); Ihle, J. N. (1996); Rawlings, J. S., K. M. Rosier and D. A. Harrison, J Cell Sci, 2004. 117(Pt 8): p.1281-3.). Четыре изоформы JAK передают различные сигналы, связываясь исключительно с некоторыми цитокиновыми рецепторами и активизируя подмножество расположенных далее в системе передачи сигнала генов. Например, JAK2 связывается с цитокиновыми рецепторами соответствующими интерлeйкину-3 (Silvennoinen, O., et al., Proc Natl Acad Sci USA, 1993. 90(18): p. 8429-33), erythropoietin (Witthuhn, B. A., et al., Cell, 1993. 74(2): p. 227-36), granulocyte colony stimulating factor (Nicholson, S. E., et al., Proc Natl Acad Sci USA, 1994. 91(8): p. 2985-8), and growth hormone (Argetsinger, L. S., et al., Cell, 1993. 74(2): p. 237-44).
Семейство ферментов JAK стало группой мишеней для терапии различных гематологических и иммунологических расстройств. В настоящее время JAK2 исследуется как перспективная мишень для терапии новообразований, в особенности лейкемии и лимфомы (Benekli, M., et al., Blood, 2003. 101(8): p. 2940-54; Peeters, P., et al., Blood, 1997. 90(7): p. 2535-40; Reiter, A., et al., Cancer Res, 2005. 65(7): p. 2662-7; Takemoto, S., et al., Proc Natl Acad Sci USA, 1997. 94(25): p. 13897-902), а также солидных опухолей (Walz, C., et al., J Biol Chem, 2006. 281(26): p. 18177-83) и других миелопролиферативных заболеваний, таких как полицитемия (Baxter, E. J., et al., Lancet, 2005. 365(9464): p. 1054-61; James, C., et al., Nature, 2005. 434(7037): p. 1144-8; Levine, R. L., et al., Cancer Cell, 2005. 7(4): p. 387-97; Shannon, K. and R. A. Van Etten, Cancer Cell, 2005. 7(4): p. 291-3), в связи с его активацией расположенных далее в системе передачи сигнала эффекторных генов, вовлеченных в пролиферацию. Ввиду его связи с новообразованиями и миелопролиферативными заболеваниями, а также его дерегуляцией в этих случаях, низкомолекулярные ингибиторы JAK2 представляют значительный интерес для лечения злокачественных новообразований у человека.
Костные морфогенетические белки (BMP) являются плейотропными факторами роста, играющими существенные роли в обеспечении архитектоники тканей по всему объему различных органов в теле. Лиганды BMP взаимодействуют с рецепторами костного морфогенетического белка (BMPR), которые относятся к суперсемейству серин/треонин-киназных рецепторов трансформирующего ростового фактора-бета (TGF-b) (Ikushima, H. and K. Miyazono, Biology of Transforming Growth Factor-beta Signalin. Curr Pharm Biotechnol, 2011). Лиганды связываются с рецепторами II типа, которые затем присоединяют рецепторы I типа, образуя гетеромерный комплекс. В виде комплекса, рецептор II типа фосфорилирует рецептор I типа, что позволяет рецептору I типа стать активным и фосфорилировать дальнейшие сигнальные молекулы. Дальнейшие эффекты активации этих рецепторов в первую очередь осуществляются семейством белков SMAD. SMAD становятся фосфорилированными и передают сигнал от клеточной мембраны в ядро, где они действуют как транскрипционные факторы в отношении регулируемой экспрессии гена (Massague, J., J. Seoane, and D. Wotton, Smad transcription factors. Genes Dev, 2005. 19(23): p. 2783-810).
У людей с хроническими заболеваниями, такими как онкологическое заболевание и воспалительный процесс, сигнальный путь BMP постоянно активизирован, что приводит к анемии. Это состояние обычно называется анемией хронического заболевания (ACD) и является симптомом истощения, ассоциируемым с больными онкологическим заболеванием (Cullis, J.O., Diagnosis and management of anaemia of chronic disease: current status. Br J Haematol, 2011. 154(3): p. 289-300). Хроническая анемия у больных онкологическим заболеванием приводит к крайней слабости и патологической усталости, что приводит к низкому качеству жизни этих людей. У таких больных передача сигнала с участием BMP посредством двух рецепторов BMP I типа, ALK2 (также известного как ACVR1) и ALK3, вызывает печеночную экспрессию пептидного гормона, называемого гепсидин (Steinbicker, A.U., et al., Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood, 2011. 118(15): p. 4224-30). Гепсидин понижает уровень содержания железа в сыворотке крови, способствуя разрушению экспортера железа, ферропортина, что приводит к увеличению количества железа, хранящегося в макрофагах и других типах клеток, и делает железо недоступным для функционирования эритроцитов (RBC) и гемоглобина. Обогащение рациона пациента железом не способствует устранению ACD, потому что принятое внутрь железо просто хранится из-за активизированного сигнального пути BMP и высокого уровня гепсидина в сыворотке крови. В настоящее время развитие ACD при онкологическом заболевании сдерживают путем ограничения физической активности больного, а в самых серьезных случаях используют переливание крови. Ингибирование сигнального пути BMP у этих больных может обеспечить реальное изменение качества их жизни, и в конечном счете может положительно влиять на то как они реагируют на терапию, излучение или хирургическую операцию (Steinbicker, A.U., et al., Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood, 2011. 117(18): p. 4915-23; Coyne, D.W., Hepcidin: clinical utility as a diagnostic tool and therapeutic target. Kidney Int, 2011. 80(3): p. 240-4; Theurl, I., et al., Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood, 2011).
Помимо его функции при ACD, сигнальный путь BMP играет основные роли в росте и метастазировании раковых клеток, особенно при раке молочной железы, простаты и других видах злокачественных новообразований, которые часто метастазируют в кости (Ye, L., M.D. Mason, and W.G. Jiang, Bone morphogenetic protein and bone metastasis, implication and therapeutic potential. Front Biosci, 2011. 16: p. 865-97). BMP и BMPR в большей степени экспрессируются в метастатических клетках рака молочной железы по сравнению с менее склонными к метастазированию клетками, а также в клетках рака простаты, которые порождают остеосклеротические костные метастазы (Bobinac, D., et al., Expression of bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat Med J, 2005. 46(3): p. 389-96). Было также показано, что помимо обеспечения инвазивности и метастазирования раковых клеток, сигнальный путь BMP влияет на микросреду кости. Взаимосвязь между раковыми клетками и микросредой кости через посредство сигнального пути BMP способствует метастазированию раковых клеток в кости. Исследования показали, что ингибирование сигнального пути BMP значительно ослабляет степень клинической тяжести новообразований в костях и остеолитический синдром в доклинической модели костного метастазирования рака простаты. Эти результаты предполагают то, что ингибитор BMP можно применять для предотвращения метастазирования в кости в дополнение к его активности против анемии, вызванной хроническим заболеванием.
Кроме того ингибитор BMP обладает потенциалом для лечения множества признаков болезни, не относящихся к онкологическому заболеванию. ACD представляет собой изнуряющее состояние, которое поражает людей, страдающих от других болезней, включающих ревматоидный артрит, системную волчанку, хроническую болезнь почек и множество других воспалительных заболеваний. Кроме того, редкое детское генетическое заболевание, называемое fibrodysplasia ossificans progressiva (прогрессирующая оссифицирующая фибродисплазия) (FOP), как было показано, вызвано активацией мутаций в гене alk2 (Kaplan, F.S., et al., Investigations of activated ACVR1/ALK2, a bone morphogenetic protein type I receptor, that causes fibrodysplasia ossificans progressiva. Methods Enzymol, 2010. 484: p. 357-73). Мутация в ALK2 при данном заболевании является причиной того, что волокнистая соединительная ткань (мышцы, сухожилия, связки и т.д.) оссифицируется при повреждении. Другими словами, когда пациенты с таким заболеванием подвержены повреждениям мышц или соединительных тканей, восстановленная ткань трансформируется в кость, приводя к тому, что суставы необратимо фиксируются в определенном положении. К подростковому возрасту такие дети теряют большую часть работоспособности суставов. Исследования, осуществленные на животных моделях FOP, позволяют считать, что ингибирование ALK2 снижает количество «внезапных обострений», связанных с FOP, и предотвращает оссификацию восстановленной ткани у модели. Медицинские и коммерческие преимущества ингибитора BMP (т.е. ALK2) довольно обширны и распространяются на множество признаков, не относящихся к онкологическому заболеванию.
В то время как были достигнуты успехи в данной области, имеется потребность в разработке специфичных и селективных ингибиторов для лечения онкологического заболевания и других состояний, которые опосредованы и/или связаны с протеинкиназами ALK2 и/или JAK2 (включая JAK2 V617F). Настоящее изобретение отвечает этим потребностям и предлагает другие соответствующие преимущества.
КРАТКАЯ СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Коротко говоря, настоящее изобретение ориентировано на соединения, обладающие активностью как ингибиторы киназы ALK2 и/или JAK2, включая их стереоизомеры, таутомеры, фармацевтически приемлемые соли и пролекарства, а также на использование таких соединений для лечения различных онкологических заболеваний.
В одном из вариантов осуществления обеспечиваются соединения, имеющие следующую структуру (I):
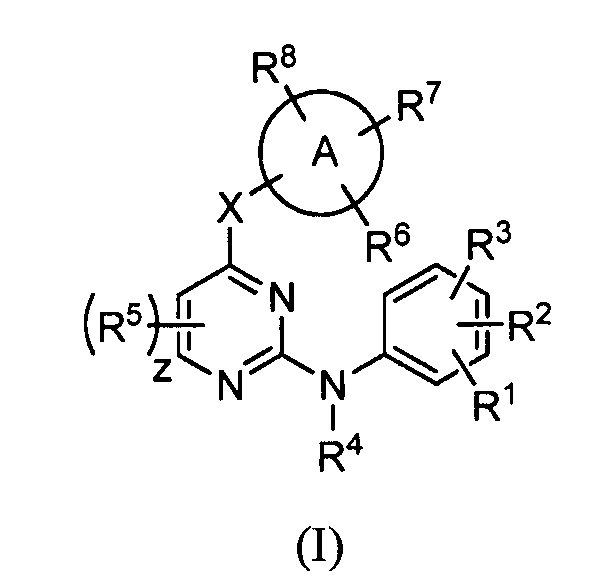
или их стереоизомеры, фармацевтически приемлемые соли, таутомеры или пролекарства, где X, A, z, R1, R2, R3, R4, R5, R6, R7 и R8 соответствуют указанным в данном документе.
В другом варианте осуществления обеспечивается фармацевтическая композиция, содержащая соединение, имеющее структуру (I), или его стереоизомер, фармацевтически приемлемую соль, таутомер или пролекарство, а также фармацевтически приемлемый носитель, разбавитель или наполнитель. В некоторых вариантах осуществления изобретение ориентировано на использование фармацевтической композиции для ингибирования киназ ALK2 и/или JAK2 у млекопитающего.
В другом варианте осуществления обеспечивается способ ингибирования киназы ALK2 и/или JAK2 у нуждающегося в этом млекопитающего, при этом способ включает введение в организм млекопитающего эффективной дозы соединения, имеющего структуру (I), или его стереоизомера, фармацевтически приемлемой соль, таутомера или пролекарства. В некоторых вариантах осуществления способ предназначен для лечения онкологического заболевания. В других вариантах осуществления способ предназначен для лечения анемии и/или состояний, сопровождающихся анемией.
Также обеспечивается использование соединения, имеющего структуру (I), для лечения связанных с киназой ALK2 и/или JAK2 состояний, таких как онкологическое заболевание. В других вариантах осуществления использование предназначено для лечения анемии и/или состояний, сопровождающихся анемией.
Эти и другие аспекты согласно изобретению станут очевидными при ссылке на последующее подробное описание.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигурах одинаковые ссылочные позиции обозначают схожие элементы. Размеры и относительные положения элементов на фигурах не обязательно вычерчены в масштабе, и некоторые из этих элементов произвольно увеличены и размещены для повышения удобства восприятия фигуры. Кроме того конкретные формы элементов в начерченном виде не предназначены для передачи какой-либо информации относительно фактической формы конкретных элементов и выбирались исключительно для простоты распознавания на фигурах.
На фигуре 1 представлены данные касательно экспрессии гепсидина.
Фигура 2 представляет собой гистограмму зависимости экспрессии гепсидина от концентрации соединения № 4 (левые столбики) и соединения № 12 (правые столбики).
На фигуре 3 показаны данные касательно экспрессии гепсидина в присутствии и отсутствии BMP-2.
Фигура 4 представляет собой гистограмму, показывающую экспрессию гепсидина у мыши для репрезентативных соединений и сравнительного соединения.
На фигуре 5 показана экспрессия гепсидина в живом организме у LPS(липополисахарид)- ндуцированной мышиной модели.
На фигуре 6 представлены данные по дозозависимому эффекту для репрезентативных соединений.
На фигурах 7A и 7B показаны содержания IL-5 в живом организме при различных дозах сравнительного соединения и репрезентативных соединений, соответственно.
На фигуре 8 представлены фармакокинетические данные.
Фигура 9 представляет собой график, показывающий зависимость уровней концентрации в плазме приводимого в качестве примера соединения от времени.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В нижеследующем описании изложены некоторые конкретные детали, чтобы обеспечить полное понимание различных вариантов осуществления изобретения. Однако специалисту в данной области будет понятно, что изобретение может быть осуществлено без этих деталей.
Если контекст не требует иного, по всему объему настоящего описания и формулы изобретения слово «содержать» и его вариации, такие как «содержит», и «содержащий» должны рассматриваться в открытом, инклюзивном смысле, т.е. как «включающий, но не ограничивающийся».
Ссылка в объеме данного описания на «один из вариантов осуществления» или «вариант осуществления» означает, что конкретная особенность, структура или характеристика, описанная с привязкой к варианту осуществления, включена, по меньшей мере, в один из вариантов осуществления настоящего изобретения. Таким образом, появления фраз «в одном варианте осуществления» или «в одном из вариантов осуществления» в различных местах по всему объему данного описания не обязательно всегда отсылают к одному и тому же варианту осуществления. Кроме того конкретные особенности, структуры или характеристики могут объединяться любым подходящим образом в одном или нескольких вариантах осуществления.
I. Определения
«Амино» обозначает радикал -NH2.
«Циано» или «нитрил» обозначают радикал -CN.
«Гидрокси» или «гидроксил» обозначают радикал -ОH.
«Имино» обозначает заместитель =NH.
«Нитро» обозначает радикал -NO2.
«Оксо» обозначает заместитель =0.
«Тиоксо» обозначает заместитель =S.
«Алкил» обозначает радикал в виде прямой или разветвленной углеводородной цепи, состоящей исключительно из атомов углерода и водорода, которая является насыщенной или ненасыщенной (т.е. содержит одну или более двойных (алкенильных) и/или тройных (алкинильных) связей), содержащей от одного до двенадцати атомов углерода (алкил C1-C12), предпочтительно от одного до восьми атомов углерода (алкил C1-C8) или от одного до шести атомов углерода (алкил C1-C6), и которая присоединена к остальной части молекулы посредством одинарной связи, например, метил, этил, н-пропил, 1-метилэтил (изопропил), н-бутил, н-пентил, 1,1-диметилэтил (трет-бутил), 3-метилгексил, 2-метилгексил, этенил, проп-1-енил, бут-1-енил, пент-1-енил, пента-1,4-диенил, этинил, пропинил, бутинил, пентинил, гексинил, и т.п. Алкил, содержащий одну или более двойных углерод-углеродных связей является алкенилом. Алкил, содержащий одну или более тройных углерод-углеродных связей является алкинилом. Если в описании определенно не указано иначе, алкильная группа необязательно может быть замещенной.
«Алкилен» или «алкиленовая цепь» обозначает прямую или разветвленную двухвалентную углеводородную цепь, соединяющую остальную часть молекулы с радикальную группой, состоящую исключительно из углерода и водорода, которая является насыщенной или ненасыщенной (т.е. содержит одну или более двойных и/или тройных связей) и содержит от одного до двенадцати атомов углерода, например, метилен, этилен, пропилен, н-бутилен, этенилен, пропенилен, н-бутенилен, пропинилен, н-бутинилен и т.п. Алкиленовая цепь присоединена к остальной части молекулы посредством одинарной или двойной связи, а также к радикальной группе посредством одинарной или двойной связи. Точки присоединения алкиленовой цепи к остальной части молекулы и к радикальной группе могут быть обеспечены посредством одного углерода или любых двух углеродов в пределах цепи. Если в описании определенно не указано иначе, алкиленовая цепь необязательно может быть замещенной.
«Алкокси» обозначает радикал, имеющий формулу -ORa, где Ra представляет собой алкильный радикал согласно вышеприведенному определению, содержащий от одного до двенадцати атомов углерода. Если в описании определенно не указано иначе, алкоксигруппа необязательно может быть замещенной.
«Алкоксиалкил» обозначает радикал, имеющий формулу -RbORa, где Ra является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода, а Rb алкиленовым радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, алкоксиалкильная группа необязательно может быть замещенной.
«Алкиламино» обозначает радикал, имеющий формулу -NHRa или -NRaRa, где каждый Ra независимо является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода. Если в описании определенно не указано иначе, алкиламиногруппа необязательно может быть замещенной.
«Алкиламиноалкил» обозначает радикал, имеющий формулу -RbNHRa или -NRaRa, где каждый Ra независимо является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода, а Rb является алкиленовым радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, алкиламиноалкильная группа необязательно может быть замещенной.
«Алкилсульфон» обозначает радикал, имеющий формулу -S(O)2Ra, где Ra является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода, а Rb является алкиленовым радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, алкилсульфоновая группа необязательно может быть замещенной.
«Гидроксиалкил» относится к алкильному радикалу согласно вышеприведенному определению, содержащему от одного до двенадцати атомов углерода, который замещен одной или более гидроксильными группами. Если в описании определенно не указано иначе, гидроксиалкильная группа необязательно может быть замещенной.
«Тиоалкил» обозначает радикал, имеющий формулу -SRa, где Ra является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода. Если в описании определенно не указано иначе, тиоалкильная группа необязательно может быть замещенной.
«Арил» обозначает радикал с кольцевой углеводородной системой содержащей водород, от 6 до 18 атомов углерода и, по меньшей мере, одно ароматическое кольцо. Применительно к данному изобретению, арильный радикал может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или соединенные мостиковой связью кольцевые системы. Арильные радикалы включают, в частности, арильные радикалы, являющиеся производными ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, флуорантена, флуорена, ассим-индацена, сим-индацена, индана, индена, нафталина, феналена, фенантрена, плейадена, пирена и трифенилена. Если в описании определенно не указано иначе, термин «арил» или префикс «ар-» (как, например, у «аралкила») предусматривает включение арильных радикалов, которые необязательно являются замещенными.
«Аралкил» обозначает радикал, имеющий формулу -Rb-Rc, где Rb является алкиленовой цепью согласно вышеприведенному определению, а Rc представляет собой один или более арильных радикалов согласно вышеприведенному определению, например, бензил, дифенилметил и т.п. Если в описании определенно не указано иначе, аралкильная группа необязательно может быть замещенной.
«Циклоалкил» или «карбоциклическое кольцо» обозначает стабильный неароматический моноциклический или полициклический углеводородный радикал, состоящий исключительно из атомов углерода и водорода, который может включать конденсированные или соединенные мостиковой связью кольцевые системы, содержащие от трех до пятнадцати атомов углерода, предпочтительно содержащие от трех до десяти атомов углерода, и которые являются насыщенными или ненасыщенными и присоединены к остальной части молекулы посредством одинарной связи. Моноциклические радикалы включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Полициклические радикалы включают, например, адамантил, норборнил, декалинил, 7,7-диметил-бицикло[2.2.1]гептанил и т.п. Если в описании определенно не указано иначе, циклоалкильная группа необязательно может быть замещенной.
«Циклоалкилалкил» обозначает радикал, имеющий формулу -RbRd где Rb является алкиленовой цепью согласно вышеприведенному определению, а Rd является циклоалкильным радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, циклоалкилалкильная группа необязательно может быть замещенной.
«Циклоалкокси» обозначает радикал, имеющий формулу -ORa, где Ra является циклоалкильным радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, циклоалкоксигруппа необязательно может быть замещенной.
«Циклоалкоксиалкил» обозначает радикал, имеющий формулу -RbORa, где Ra является циклоалкильным радикалом согласно вышеприведенному определению, а Rb является алкильным радикалом согласно вышеприведенному определению, содержащим от одного до двенадцати атомов углерода. Если в описании определенно не указано иначе, циклоалкоксиалкильная группа необязательно может быть замещенной.
«Конденсированной» именуется любая кольцевая структура, описанная в данном документе, которая конденсирована с имеющейся кольцевой структурой в соединениях согласно изобретению. Когда конденсированное кольцо является гетероциклильным кольцом или гетероарильным кольцом, любой атом углерода в имеющейся кольцевой структуре, который становится частью конденсированного гетероциклильного кольца или конденсированного гетероарильного кольца, можно заменить атомом азота.
«Гало» или «галоген» обозначает бром, хлор, фтор или йод.
«Галогеналкил» обозначает алкильный радикал согласно вышеприведенному определению, который замещен одним или более галогенрадикалами согласно вышеприведенному определению, например, трифторметил, дифторметил, трихлорметил, 2,2,2-трифторэтил, 1,2-дифторэтил, 3-бром-2-фторпропил, 1,2-дибромэтил и т.п. Если в описании определенно не указано иначе, галогеналкильная группа необязательно может быть замещенной.
«Гетероциклил» или «гетероциклическое кольцо» обозначает стабильный радикал, представляющий собой от 3 до 18-членное неароматическое кольцо, которое содержит от двух до двенадцати атомов углерода и от одного до шести гетероатомов, выбираемых из группы, состоящей из азота, кислорода и серы. Если в описании определенно не указано иначе, гетероциклильный радикал может являться моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или соединенные мостиковой связью кольцевые системы; при этом атомы азота, углерода или серы в гетероциклильном радикале необязательно могут быть окислены; атом азота необязательно может быть кватернизированным; и гетероциклильный радикал может быть частично или полностью насыщенным. Примеры таких гетероциклильных радикалов включают, в частности, диоксоланил, тиенил[1,3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4-пиперидонил, пирролидинил, пиразолидинил, хинуклидинил, тиазолидинил, тетрагидрофурил, тритианил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1-оксо-тиоморфолинил, и 1,1-диоксо-тиоморфолинил. Если в описании определенно не указано иначе, гетероциклильная группа необязательно может быть замещенной.
«N-гетероциклил» обозначает гетероциклильный радикал согласно вышеприведенному определению, содержащий, по меньшей мере, один атом азота, и в котором точка присоединения гетероциклильного радикала к остальной части молекулы обеспечивается посредством атома азота в гетероциклильном радикале. Если в описании определенно не указано иначе, N-гетероциклильная группа необязательно может быть замещенной.
«Гетероциклилалкил» обозначает радикал, имеющий формул -RbRe, где Rb является алкиленовой цепью согласно вышеприведенному определению, а Rе является гетероциклильным радикалом согласно вышеприведенному определению, при этом, если гетероциклил представляет собой азотсодержащий гетероциклил, гетероциклил может быть присоединен к алкильному радикалу посредством атома азота. Если в описании определенно не указано иначе, гетероциклилалкильная группа необязательно может быть замещенной.
«Гетероарил» обозначает радикал, являющийся от 5 до 14-членной кольцевой системой, содержащей атомы водорода, от одного до тринадцати атомов углерода, от одного до шести гетероатомов, выбираемых из группы, состоящей из азота, кислорода и серы, и, по меньшей мере, одно ароматическое кольцо. Применительно к данному изобретению гетероарильный радикал может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или соединенные мостиковой связью кольцевые системы; при этом атомы азота, углерода или серы в гетероарильном радикале необязательно могут быть окислены; атом азота необязательно может быть кватернизированным. Примеры включают, в частности, азепинил, акридинил, бензимидазолил, бензотиазолил, бензиндолил, бензодиоксолил, бензофуранил, бензооксазолил, бензотиазолил, бензотиадиазолил, бензо[b][l,4]диоксепинил, 1,4-бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензопиранил, бензопиранонил, бензофуранил, бензофуранонил, бензотиенил (бензотиофенил), бензотриазолил, бензо[4,6]имидазо[l,2-a]пиридинил, карбазолил, циннолинил, дибензофуранил, дибензотиофенил, фуранил, фуранонил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолил, индолизинил, изоксазолил, нафтиридинил, оксадиазолил, 2-оксазепинил, оксазолил, оксиранил, 1-оксидопиридинил, 1-оксидопиримидинил, 1-оксидопиразинил, 1-оксидопиридазинил, 1-фенил-1H-пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пирролил, пиразолил, пиридинил, пиразинил, пиримидинил, пиридазинил, хиназолинил, хиноксалинил, хинолинил, хинуклидинил, изохинолинил, тетрагидрохинолинил, тиазолил, тиадиазолил, триазолил, тетразолил, триазинил и тиофенил (т.е. тиенил). Если в описании определенно не указано иначе, гетероарильная группа необязательно может быть замещенной.
«N-гетероарил» обозначает гетероарильный радикал согласно вышеприведенному определению, содержащий, по меньшей мере, один азот, и где точка присоединения гетероарильного радикала к остальной части молекулы обеспечивается посредством атом азота в гетероарильном радикале. Если в описании определенно не указано иначе, N-гетероарильная группа необязательно может быть замещенной.
«Гетероарилалкил» обозначает радикал, имеющий формулу -RbRf, где Rb является алкиленовой цепью согласно вышеприведенному определению, а Rf является гетероарильным радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, гетероарилалкильная группа необязательно может быть замещенной.
«Цианоалкил» представляет собой алкил согласно вышеприведенному определению, который содержит одну или более замещающих групп -CN. Если в описании определенно не указано иначе, цианоалкильная группа необязательно может быть замещенной.
«Цианоциклоалкил» представляет собой циклоалкил согласно вышеприведенному определению, который содержит одну или более замещающих групп -CN. Если в описании определенно не указано иначе, цианоциклоалкильная группа необязательно может быть замещенной.
«Цианоциклоалкилалкил» обозначает радикал, имеющий формулу -RbRd, где Rb является алкиленовой цепью согласно вышеприведенному определению, а Rd является цианоциклоалкильным радикалом согласно вышеприведенному определению. Если в описании определенно не указано иначе, цианоциклоалкилалкильная группа необязательно может быть замещенной.
«Сложный эфир аминокислоты» обозначает аминокислоту, содержащую сложноэфирную группу вместо кислотной группы. Если в описании определенно не указано иначе, группа, представляющая собой сложный эфир аминокислоты, необязательно может быть замещенной.
Используемый в данном документе термин «замещенная» обозначает любую из вышеуказанных групп (т.е. алкильная, алкиленовая, алкокси, алкоксиалкильная, алкиламино, алкиламиноалкильная, алкилсульфо, гидроксиалкильная, тиоалкильная, арильная, аралкильная, циклоалкильная, циклоалкилалкильная, циклоалкокси, циклоалкоксиалкильная, галогеналкильная, гетероциклильная, N-гетероциклильная, гетероциклилалкильная, гетероарильная, N-гетероарильная, гетероарилалкильная, цианоалкильная, цианоциклоалкильная, цианоциклоалкилалкильная и/или сложный эфир аминокислоты) в которой, по меньшей мере, один атом водорода заменен химической связью с неводородными атомами, такими как, в том числе: атом галогена, такой как F, Cl, Br и I; атом кислорода в таких группах как гидроксильные группы, алкоксигруппы и сложноэфирные группы; атом серы в таких группах как тиольные группы, тиоалкильные группы, сульфогруппы, сульфонильные группы и сульфоксидные группы; атом азота в таких группах как аминогруппы, амидогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, алкилариламиногруппы, диариламиногруппы, N-оксидные группы, имидогруппы и енаминогруппы; атом кремния в таких группах как триалкилсилильные группы, диалкиларилсилильные группы, алкилдиарилсилильные группы и триарилсилильные группы; и другой гетероатом в различных других группах. «Замещенная» также обозначает любую из вышеуказанных групп, в которой один или более атомов водорода заменены химической связью более высокого порядка (например, двойной или тройной связи) с гетероатомом, таким как кислород в оксо, карбонильных, карбоксильных и сложноэфирных группах; и азот в таких группах как иминогруппы, оксимные группы, гидразонные группы и нитрильные группы. Например, «замещенная» включает любую из вышеуказанных групп, в которой один или более водородных атомов заменены на -NRgRh, -NRgC(=O)Rh, -NRgC(=O)NRgRh, -NRgC(=O)ORh, -NRgSO2Rh, -OC(=O)NRgRh, -ORg, -SRg, -SORg, -SO2Rg, -OSO2Rg, -SO2ORg, =NSO2Rg и -SO2NRgRh. «Замещенная» также означает любую из вышеуказанных групп, в которых один или более атомов водорода заменены на -C(=O)Rg, -C(=O)ORg, -C(=O)NRgRh, -CH2SO2Rg, -CH2SO2NRgRh. В вышеуказанных группах Rg и Rh являются одинаковыми или отличающимися и независимо представляют собой водород, алкил, алкокси, алкиламино, тиоалкил, арил, аралкил, циклоалкил, циклоалкилалкил, галогеналкил, гетероциклил, N-гетероциклил, гетероциклилалкил, гетероарил, N-гетероарил и/или гетероарилалкил. «Замещенная» также обозначает любую из вышеуказанных групп, в которых один или более атомов водорода заменены химической связью с амино, алкиламино, циано, гидрокси, имино, нитро, оксо, тиоксо, галогеном, алкильной, алкокси, алкиламино, тиоалкильной, арильной, аралкильной, циклоалкильной, циклоалкилалкильной, галогеналкильной, гетероциклильной, N-гетероциклильной, гетероциклилалкильной, гетероарильной, N-гетероарильной и/или гетероарилалкильной группой. Кроме того, каждый из вышеуказанных заместителей может также необязательно быть замещенным одним или более из вышеуказанных заместителей.
Термин «пролекарство» предназначен для обозначения соединения, которое может превращаться в физиологических условиях или путем сольволиза в биологически активное соединению согласно изобретению. Таким образом, термин «пролекарство» обозначает метаболического предшественника соединения согласно изобретению, который является фармацевтически приемлемым. Пролекарство может быть неактивным, когда вводится в организм нуждающегося в этом больного, но превращаться в активное соединение согласно изобретению в живом организме. Пролекарства, как правило, быстро трансформируются в живом организме с образованием исходного вещества согласно изобретению, например, путем гидролиза в крови. Пролекарственное соединение часто обеспечивает такие преимущества, как растворимость, тканевая совместимость или отсроченное высвобождение в организме млекопитающих (см., Bundgard, H., Design of Prodrugs (1985), pp. 7-9, 21-24 (Elsevier, Amsterdam)). Обсуждение пролекарств приводится в Higuchi, T., et al., A.C.S. Symposium Series, Vol. 14 и в Bioreversible Carriers in Drug Design, Ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Термин «пролекарство» также предусматривает включение любых ковалентно связанных носителей, которые высвобождают активное соединение согласно изобретению в живом организме, когда такое пролекарство вводится в организм больного млекопитающего. Пролекарства соединения согласно изобретению могут быть получены путем модификации функциональных групп, присутствующих в соединении согласно изобретению таким способом, при котором модификацирующие фрагменты отщепляются либо при стандартной обработке, либо в живом организме, с образованием исходного вещества согласно изобретению. Пролекарства включают соединения согласно изобретению, у которых гидрокси-, амино- или меркаптогруппа связана с любой группой, которая при введении пролекарства соединения согласно изобретению в организм больного млекопитающего отщепляется с образованием свободной гидрокси-, свободной амино- или свободной меркаптогруппы, соответственно. Примеры пролекарств включают, в частности, ацетатную, формиатную и бензоатную производные спиртовой или амидную производную амино- функциональных групп в соединениях согласно изобретению и т.п.
Изобретение, раскрытое в данном документе, также подразумевает включение всех фармацевтически приемлемых соединений, имеющих структуру (I), которые являются изотопно мечеными за счет замены одного или более атомов на атом, имеющий другую атомную массу или массовое число. Примеры изотопов, которые могут быть введены в раскрытые соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15О, 17О, 18О, 31P, 32P, 35S, 18F, 36Cl, 123I и 125I, соответственно. Эти меченые радиоактивным изотопом соединения могут быть подходящими, чтобы помогать определять или измерять эффективность соединений путем определения, например, места приложения или способа действия, или аффинности связывания с фармакологически важными местом приложения действия. Некоторые изотопно меченые соединения, имеющие структуру (I), например, соединения, которые содержат радиоактивный изотоп, подходят для изучения распределения в тканях лекарственного вещества и/или субстрата. Радиоактивные изотопы тритий, т.е. 3H, и углерод-14, т.е. 14C, особенно подходят для этой цели исходя из простоты их введения и легкодоступных средств обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить некоторые терапевтические преимущества, обусловленные большей устойчивостью к инактивации в процессе метаболизма, например, увеличением периода полуразложения в живом организме, или снижением требований к дозировке, и, следовательно, может быть предпочтительным при некоторых обстоятельствах.
Замещение позитронно-активными изотопами, такими как 11C, 18F, 15О и 13N, может применяться для исследований с помощью позитронно-эмиссионной топографии (PET) для изучения степени занятости рецептора субстратом. Изотопно меченые соединения, имеющие структуру (I), как правило, можно получить обычными известными специалистам в данной области способами или способами, аналогичными описанным в разделах «препараты» и «примеры», в соответствии с нижеизложенным, путем использования подходящего изотопно меченого реактива вместо использованного ранее немаркированного реактива.
Раскрытое в данном документе изобретение также подразумевает включение продуктов, образующихся из раскрытых соединений в результате метаболизма в живом организме. Такие продукты могут образовываться в результате, например, окисления, восстановления, гидролиза, амидирования, этерификации и т.п. введенного в организм соединения, в первую очередь из-за ферментативных процессов. Соответственно, изобретение включает соединения, полученные в результате процесса, включающего введение соединения согласно данному изобретению в организм млекопитающего, в течение промежутка времени, достаточного для образования его метаболического продукта. Такие продукты, как правило, определяются путем введения в поддающейся обнаружению дозе меченого радиоактивным изотопом соединения согласно изобретению в организм животного, такого как крыса, мышь, морская свинка, обезьяна или в организм человека, предоставляя достаточное количество времени для протекания метаболизма, и выделения продуктов его превращения из мочи, крови или других биологических образцов.
Термины «стабильное соединение» и «стабильная структура» предназначены для обозначения соединения, которое является достаточно прочным, чтобы выдержать выделение из реакционной смеси с получением подходящей степени чистоты, а также составления на его основе эффективного терапевтического агента.
Термин «млекопитающее» включает людей, а также как домашних животных, таких как лабораторные животные и домашние питомцы (например, кошки, собаки, свинья, крупный рогатый скот, овцы, козы, лошади, кролики), так и не домашних животных, таких как дикие животные и т.п.
«Необязательный» или «необязательно» означает, что описанное впоследствии стечение обстоятельств может произойти или может не произойти, и что описание включает случаи, где указанное явление или обстоятельство возникает, а также случаи, в которых они не возникают. Например, «необязательно замещенный арил» означает, что арильный радикал может быть или может не быть замещенным, и что описание включает как замещенные арильные радикалы, так и арильные радикалы, не содержащие замещающего фрагмента.
Термин «фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество» включает без ограничения любые вспомогательное средство, носитель, вспомогательное вещество, скользящее вещество, подсластитель, разбавитель, консервант, краситель/красящее вещество, интенсификатор вкусоароматических свойств, поверхностно-активное вещество, увлажняющий компонент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, которые были одобрены управлением по контролю качества продуктов питания и лекарственных средств США, как являющиеся приемлемыми для использования на людях или домашних животных.
«Фармацевтически приемлемая соль» включает соли присоединения как кислоты, так и основания.
Термин «Фармацевтически приемлемая соль присоединения кислоты» обозначает те соли, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются неприемлемыми с биологической или иной точки зрения, и которые образуются с неорганическими кислотами такими как, в частности, соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п. и органическими кислотами, такими как, в том числе, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфокислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфокислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламиновая кислота, додецилсерная кислота, этан-1,2-дисульфокислота, этансульфокислота, 2-гидроксиэтансульфокислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксо-глутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфокислота, слизевая кислота, нафталин-1,5-дисульфокислота, нафталин-2-сульфокислота, l-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памоевая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфокислота, трифторуксусная кислота, ундециленовая кислота и т.п.
Термин «Фармацевтически приемлемая соль присоединения основания» обозначает те соли, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются неприемлемыми с биологической или иной точки зрения. Эти соли получают путем присоединения неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, в частности, натриевые, калиевые, литиевые, аммониевые, кальциевые, магниевые, железные, цинковые, медные, марганцевые, алюминиевые соли и т.п. Предпочтительными неорганическими солями являются аммониевые, натриевые, калиевые, кальциевые и магниевые соли. Соли, полученные из органических оснований, включают, в частности, соли первичных, вторичных, и третичных аминов, замещенных аминов включая замещенные амины природного происхождения, циклических аминов и основных ионообменных смол, таких как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и т.п. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто кристаллизация дает сольват соединения согласно изобретению. В данном контексте термин «сольват» обозначает агрегат, который содержит одну или более молекул соединения согласно изобретению с одной или более молекулами растворителя. Растворитель может являться водой, и в этом случае сольват может являться гидратом. В соответствии с другим вариантом растворитель может являться органическим растворителем. Таким образом, соединения согласно настоящему изобретению могут существовать в виде гидрата, включая моногидрат, дигидрат, полугидрат, полуторагидрат, тригидрат, тетрагидрат и т.п., а также в виде соответствующих сольватированных форм. Соединение согласно изобретению может представлять собой истинные сольваты, в то время как в других случаях соединение согласно изобретению может просто удерживать дополнительную воду или являться смесью, содержащей воду, а также некоторое количество дополнительного растворителя.
«Фармацевтическая композиция» обозначает рецептуру, состоящую из соединения согласно изобретению и общепринятой в данной области среды для доставки биологически активного соединения в организм млекопитающих, например, людей. Такая среда включает все соответствующие фармацевтически приемлемые носители, разбавители или вспомогательные вещества.
Термины «Эффективная доза» или «терапевтически эффективная доза» обозначают такую дозу соединения согласно изобретению, которое является достаточным для осуществления лечения, согласно нижеприведенному определено, онкологического заболевания у млекопитающего, предпочтительно человека, при введении в организм млекопитающего, предпочтительно человека. Количество соединения согласно изобретению, которое составляет «терапевтически эффективную дозу», будет меняться в зависимости от соединения, состояния и его серьезности, способа введения в организм и возраста подлежащего лечению млекопитающего, но может легко определяться обычным специалистом в данной области исходя из его собственного знания и данного раскрытия.
Термин «терапия» или «лечение» в данном контексте охватывает лечение интересующей болезни или состояния у млекопитающего, предпочтительно человека, имеющего интересующую болезнь или состояние, и включает:
(i) предотвращение возникновения болезни или состояния у млекопитающего, в частности, когда такое млекопитающее предрасположено к такому состоянию, но ему еще не поставлен диагноз, что оно его имеет;
(ii) подавление болезни или состояния, т.е. остановка его развития;
(iii) облегчение болезни или состояния, т.е. вызов обратного развития болезни или состояния; или
(iv) облегчение симптомов, вызванных болезнью или состоянием, т.е. облегчение боли без принятия мер в отношении основной болезни или состояния. В данном контексте термины «болезнь» и «состояние» могут использоваться как взаимозаменяемые или могут отличаться в том, что определенное расстройство или состояние может не иметь известный этиологический фактор (следовательно, этиология еще не была определена), и поэтому оно еще не признано болезнью, а только нежелательным состоянием или синдромом, для которого клиническими врачами был установлен более или менее точный набор симптомов.
Соединения согласно изобретению, или их фармацевтически приемлемые соли или таутомеры могут содержать один или более центров ассиметрии и таким образом могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены с точки зрения абсолютной стереохимии как (R)- или (S)-, или как (D)- или (L)- для аминокислот. Настоящее изобретение предусматривает включение всех таких возможных изомеров, а также их рацемических и оптически чистых форм. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)- изомеры можно получить с использованием хиральных синтонов или хиральных реактивов или выделить с использованием обычных способов, например, хроматографии и фракционной кристаллизации. Обычные способы для получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного), используя, например, хиральную высокоэффективную жидкостную хроматографию (HPLC). Когда соединения, описанные в данном документе, содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иначе, предполагается, что соединения включают как E, так и Z геометрические изомеры. Аналогичным образом также предполагается, что включаются все таутомерные формы.
Термин «стереоизомер» обозначает соединение, состоящее из тех же атомов, соединенных теми же связями, но имеющее отличающиеся трехмерные структуры, которые не являются взаимозаменяемыми. Настоящее изобретение рассматривает различные стереоизомеры и их смеси и включает термин «энантиомеры», который обозначает два стереоизомера, молекулы которых представляют собой несовпадающие при наложении зеркальные отражения друг друга.
Термин «таутомер» обозначает перенос протона с одного атома молекулы на другой атом той же самой молекулы, например, превращение кетона в енол путем переноса протона. Настоящее изобретение включает таутомеры любых указанных соединений.
«Химиотерапевтическое средство» - химическое вещество, которое уничтожает, останавливает или замедляет развитие раковых клеток.
II. Соединения
Как отмечалось выше, в одном из вариантов осуществления настоящего изобретения обеспечиваются соединения, обладающие активностью как ингибиторы киназы ALK2 и/или JAK2, при этом соединения имеют следующую структуру (I):
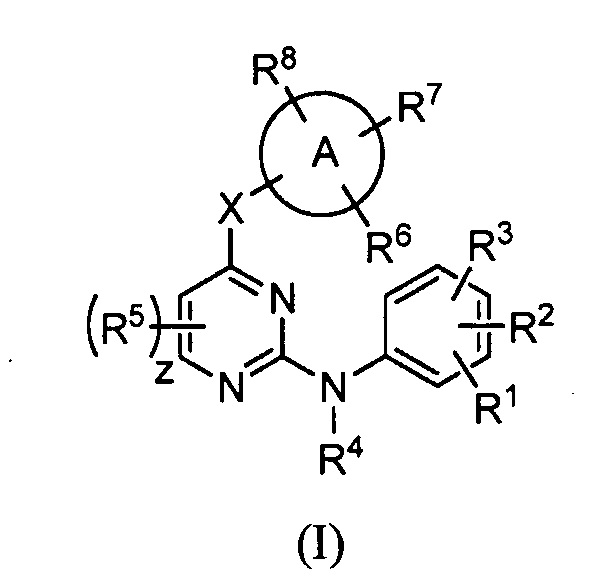
или их стереоизомеры, фармацевтически приемлемые соли, таутомеры или пролекарства, где:
A обозначает 6-членное ароматическое кольцо или 5- или 6-членное гетероарильное кольцо;
X является -NH-, -О-, -S(О)m-, -CH2-, -CHOH- или -C(=О)-;
R1 является H, галогеном, C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, C1-C6 галогеналкилом, C1-C6 алкоксигруппой, -S(О)m C1-C6 алкилом, C1-C6 гидроксиалкилом, -OCH2CH2R9, -(CH2)nNRaRb или -CONRaRb;
R2 является галогеном, C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, C1-C6 галогеналкилом, C1-C6 алкоксигруппой, -S(О)m C1-C6 алкилом, C1-C6 гидроксиалкилом, -OCH2CH2R9, -(CH2)nNRaRb или -CONRaRb;
R3 является галогеном, C1-C6 алкилом, C2-C6 алкенилом, C2-C6 алкинилом, C1-C6 галогеналкил, C1-C6 алкоксигруппой, -S(О)m C1-C6 алкилом, C1-C6 гидроксиалкилом, -OCH2CH2R9, -(CH2)nNRaRb, -CONRaRb или -NHCHRaRb;
R4 является H или C1-C6 алкилом;
R5 в каждом случае наличия независимо является H, галогеном, C1-C6 алкилом, C1-C6 алкоксигруппой, C3-C6 циклоалкоксигруппой, -CN, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом;
Каждый из R6 и R7 независимо является H, галогеном, гидроксилом, C1-C6 алкилом, C1-C6 алкоксигруппой, C3-C6 циклоалкоксигруппой, C1-C6 цианоалкилом, C3-C6 цианоциклоалкилом, C3-C6 цианоциклоалкилалкилом или -(CH2)nNRaRb;
R8 является H, галогеном, гидроксилом, C1-C6 алкилом, C1-C6 алкоксигруппой, C3-C6 циклоалкоксигруппой, C1-C6 цианоалкилом, C3-C6 цианоциклоалкилом, C3-C6 цианоциклоалкилалкилом, -(CH2)nNRaRb, арилом или гетероарилом;
R9 является -H, -F, -Cl, C1-C4 алкилом, C2-C3 алкенилом, C2-C3 алкинилом, C3-C4 циклоалкилом, -CH2OH, -OCH3, -OCH2CH3, -S(О)mCH3, -CH2CN, -CH2OCH3, -CH2S(О)mCH3, -CN, -CHCH3CN, -C(CH3)2CN или 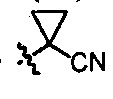 ;
;
Каждый из Ra и Rb независимо является -H, C1-C6 алкилом, C1-C6 гидроксиалкилом, или Ra и Rb образуют необязательно замещенное 5- или 6-членное насыщенное карбоциклическое или гетероциклическое кольцо совместно с атомом азота или углерода, к которому они присоединены;
m равняется 0, 1 или 2; и
n равняется 0, 1, 2 или 3.
В некоторых вариантах осуществления соединения (I), R5 не является H или ни один заместитель из R6 или R7 не является -CH2CN, когда X является NH, и один заместитель из R1, R2 или R3 является 4-метилпиперазин-1-илом, а другой заместитель из R1, R2 или R3 является -F.
В других вариантах осуществления соединения (I):
Либо R5 не является H, либо ни один заместитель из R6, R7 или R8 не является -CH2CN, когда X представляет собой NH, A является 6-членным ароматическим кольцом и один заместитель из R1, R2 или R3 является 4-метилпиперазин-1-илом, а другой заместитель из R1, R2 или R3 является -F или CF3; и
C1-C6 алкоксигруппа не содержит гетероциклильного заместителя.
Еще в нескольких вариантах осуществления z равняется 1, а R5 является H, галогеном, C1-C6 алкилом, C1-C6 алкоксигруппой, C3-C6 циклоалкоксигруппой или-CN.
В связи с этим понятно, что варианты осуществления, которые включают одно или несколько из вышеуказанных условий, не включают конкретные соединения, раскрытые в публикации PCT № WO 2008/106635.
В других вариантах осуществления соединения (I), R8 является гетероарилом, выбираемым из пиридила, пирролила и тиазолила.
В некоторых других вариантах осуществления вышеизложенного, соединение имеет следующую структуру (II):
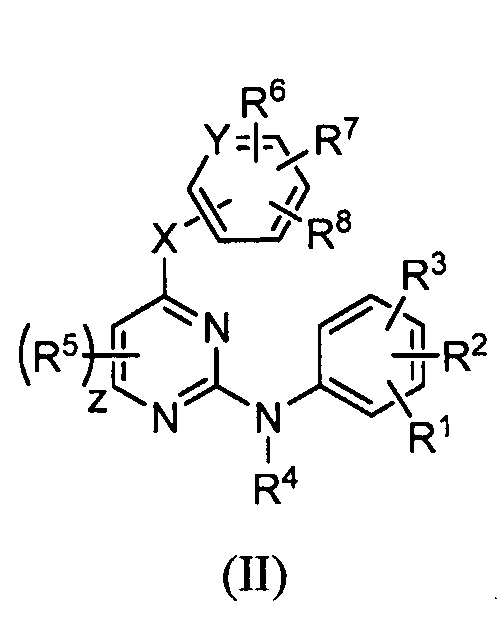 ,
,
в которой:
X является -NH-;
Y является Н или CH;
R1 является H или C1-C6 алкоксигруппой;
R2 является галогеном или C1-C6 алкоксигруппой;
R3 является C1-C6 алкоксигруппой или -NHCHRaRb;
R4 является H;
R5 в каждом случае наличия независимо является H, галогеном, C1-C6 алкилом, C1-C6 алкоксигруппой, -CN или C1-C6 цианоалкилом;
Каждый из R6 и R7 независимо является H, галогеном, C1-C6 алкилом, C1-C6 цианоалкилом, C3-C6 цианоциклоалкилом;
R8 является H или гетероарилом; и
z равняется 0, 1 или 2.
В некоторых вариантах осуществления соединения, имеющего структуру (II), R6 является H, C1-C6 алкилом, C1-C6 цианоалкилом, C3-C6 цианоциклоалкилом, а R7 является H, галогеном, C1-C6 алкилом, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом.
В некоторых других вариантах осуществления соединения, имеющего структуру (II), R5 является H, галогеном, C1-C6 алкилом, C1-C6 алкоксигруппой или -CN. В некоторых из этих вариантов осуществления z равняется 0.
В некоторых вариантах осуществления R8 является гетероарилом, выбираемым из пиридинила, пирролила и тиазолила.
В других вариантах осуществления вышеизложенного, X является -NH-. В нескольких вариантах осуществления Y является CH. Еще в некоторых вариантах осуществления Y является N.
Еще в других вариантах осуществления вышеуказанных соединений, имеющих структуру (I) или (II), R1 является H. В некоторых различных вариантах осуществления R1 является C1-C6 алкоксигруппой. В других вариантах осуществления R1 является метоксигруппой.
В некоторых других вариантах осуществления каждый заместитель из R1 и R2 является C1-C6 алкоксигруппой. Например, в некоторых вариантах осуществления каждый заместитель из R1 и R2 является метоксигруппой.
В некоторых других вариантах осуществления каждый заместитель из R1, R2 и R3 является C1-C6 алкоксигруппой. Например, в некоторых вариантах осуществления каждый заместитель из R1, R2 и R3 является метоксигруппой.
Еще в других вариантах осуществления любого из вышеуказанных соединений, имеющих структуру (I) или (II), R2 является галогеном. Например, в некоторых вариантах осуществления R2 является F или Cl. В других вариантах осуществления R2 является C1-C6 алкоксигруппой. Например, в некоторых вариантах осуществления R2 является метоксигруппой.
В нескольких вариантах осуществления вышеуказанных соединений, имеющих структуру (I) или (II), R3 является -NHCHRaRb, при этом Ra и Rb соединяются с образованием гетероциклического кольца. В некоторых вариантах осуществления гетероциклическое кольцо является замещенным или незамещенным пиперазинильным кольцом. Например, в некоторых вариантах осуществления замещенное пиперазинильное кольцо является N-замещенным пиперазинильным кольцом, а заместитель выбирают из C1-C6 алкила, C1-C6 карбоксиалкилкарбонила и C1-C6 гидроксиалкила. В некоторых вариантах осуществления исключаются соединения, в которых R3 является незамещенной пиперазин-1-ильной группой.
В нескольких вариантах осуществления R3 является -NHCHRaRb, при этом Ra и Rb соединяются с образованием гетероциклического кольца и один или более заместителей из R1 и R2 являются C1-C6 алкоксигруппой. Например, в некоторых вариантах осуществления R3 является пиперазинилом, а R1 является C1-C6 алкоксигруппой, например метоксигруппой. В некоторых вариантах осуществления пиперазинил является N-метилпиперазинилом. В дополнительных вариантах осуществления вышеизложенного, R2 является H.
Еще в других вариантах осуществления любого из вышеуказанных соединений, имеющих структуру (I) или (II), R3 является C1-C6 алкоксигруппой. Например, в некоторых вариантах осуществления R3 является метоксигруппой.
В других вариантах осуществления любого из вышеуказанных соединений, соединение имеет одну из следующих структур:
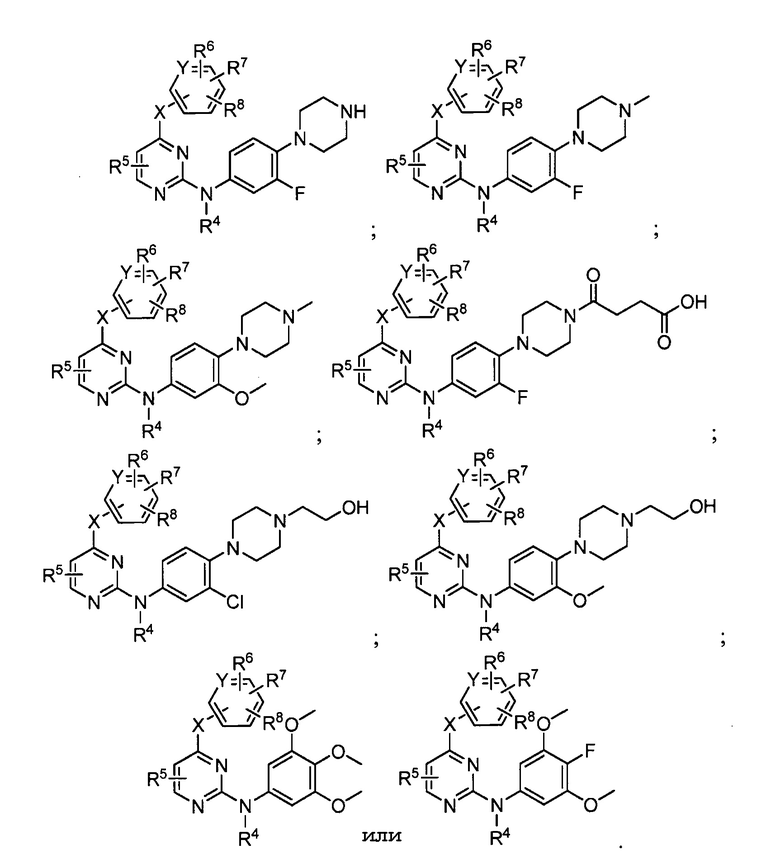
В некоторых из вышеуказанных вариантов осуществления R5 является H. В других вариантах осуществления R5 является метилом. В нескольких вариантах осуществления R5 является хлором или фтором. В еще нескольких вариантах осуществления R5 является цианогруппой. В других аспектах R5 является метоксигруппой.
Еще в других вариантах осуществления вышеуказанных соединений, имеющих структуру (I) или (II), по меньшей мере, один заместитель из R6 и R7 является H.
В нескольких вариантах осуществления любого из вышеуказанных соединений, имеющих структуру (I) или (II), по меньшей мере, один заместитель из R6 или R7 является фтором или хлором.
В других вариантах осуществления любого из вышеуказанных соединений, имеющих структуру (I) или (II), по меньшей мере, один заместитель из R6 или R7 является C1-C6 алкилом. Например, в некоторых вариантах осуществления C1-C6 алкил является метилом.
Еще в других нескольких вариантах осуществления вышеуказанных соединений, имеющих структуру (I) или (II), один заместитель из R6 или R7 является C1-C6 цианоалкилом. Например, в некоторых вариантах осуществления C1-C6 цианоалкил является -CH2CN. В некоторых из этих вариантов осуществления R3 является пиперазинилом. В дополнительных вариантах осуществления R2 является галогеном, таким как хлор или фтор, а R1 является H. Еще в других из этих вариантов осуществления R3 является пиперазинилом, R2 является C1-C6 алкоксигруппой, такой как метоксигруппа, а R1 является H.
Еще в других вариантах осуществления вышеуказанных соединений, имеющих структуру (I) или (II), R6 или R7 является C3-C6 цианоциклоалкилом. Например, в некоторых вариантах осуществления C3-C6 цианоциклоалкил является 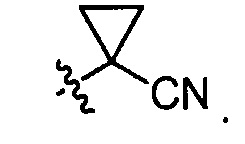
В некоторых других вариантах осуществления A является фенилом, R6 является C3-C6 цианоциклоалкилом, а R2 является C1-C6 алкоксигруппой. В дополнительных вариантах осуществления вышеизложенного R3 является пиперазинилом, а R1 является H.
В некоторых других вариантах осуществления A является фенилом, R6 является C3-C6 цианоциклоалкилом, а R2 является галогеном, таким как фтор или хлор. В дополнительных вариантах осуществления вышеизложенного R3 является пиперазинилом, а R1 является H.
В некоторых других вариантах осуществления A является фенилом, R6 является C3-C6 цианоциклоалкилом, а R2 является C1-C6 алкоксигруппой, такой как метоксигруппа. В дополнительных вариантах осуществления вышеизложенного каждый заместитель из R3 и R1 является C1-C6 алкоксигруппой, такой как метоксигруппа.
В некоторых вариантах осуществления R8 является H. В других вариантах осуществления R8 является гетероарилом. Например, в некоторых вариантах осуществления гетероарил является замещенным или незамещенным пиридинилом. В некоторых из этих вариантов осуществления A является гетероарилом, таким как пиридинил. Еще в других вариантах осуществления A является пиридинилом, R8 является пиридинилом и один или более, например, каждый заместитель из R1, R2 или R3 является C1-C6 алкоксигруппой, такой как метоксигруппа.
В различных вариантах осуществления вышеизложенного соединение имеет одну из следующих структур:
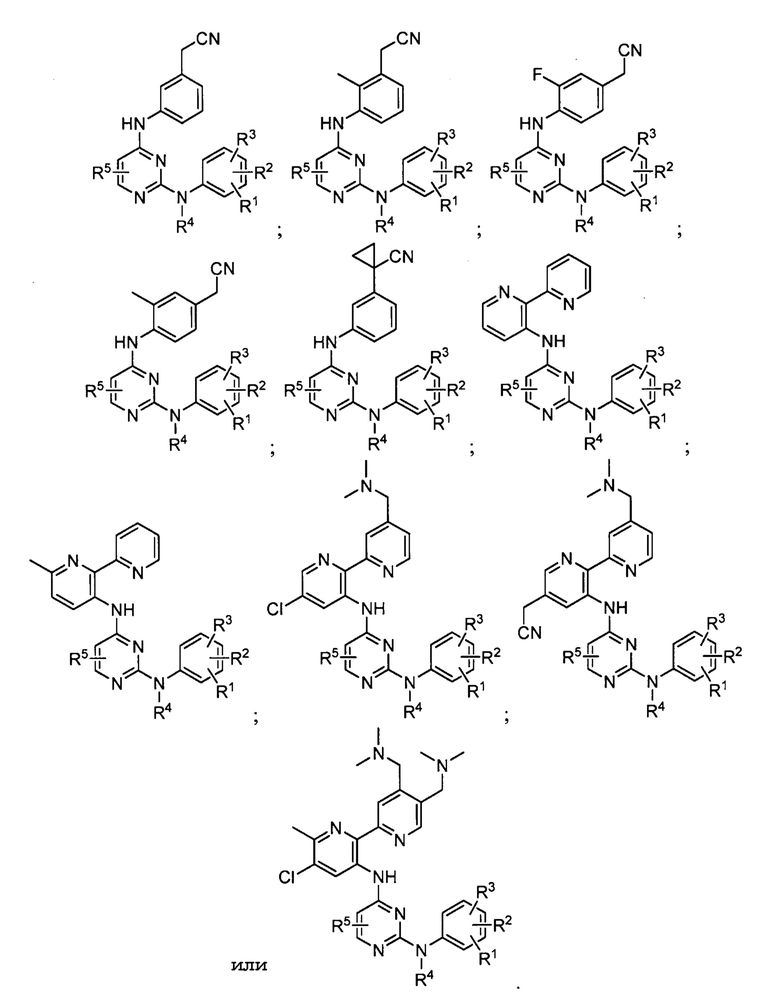
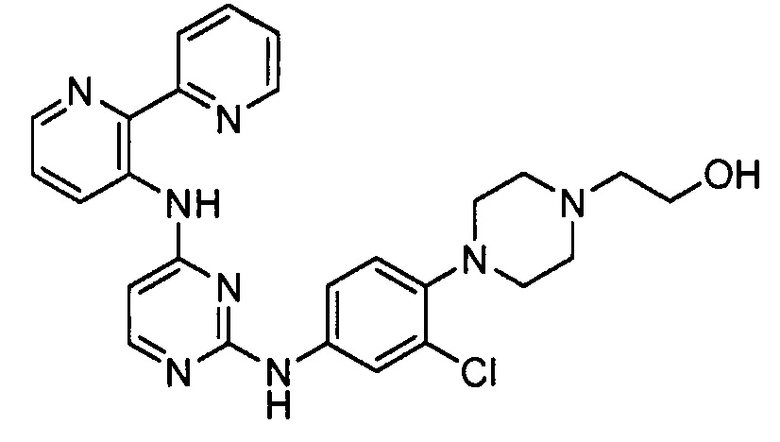
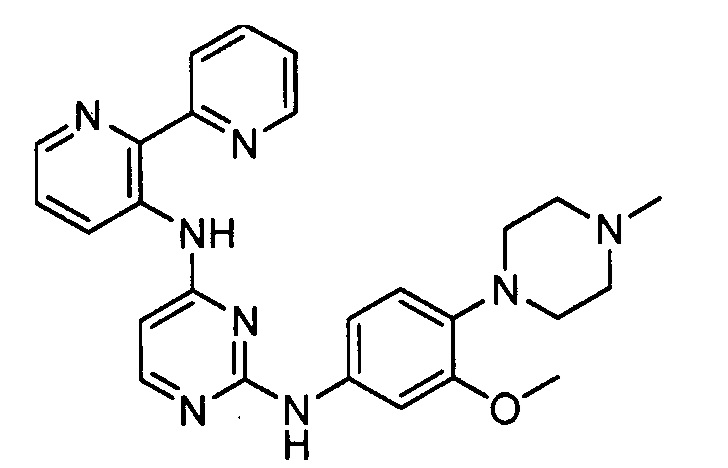
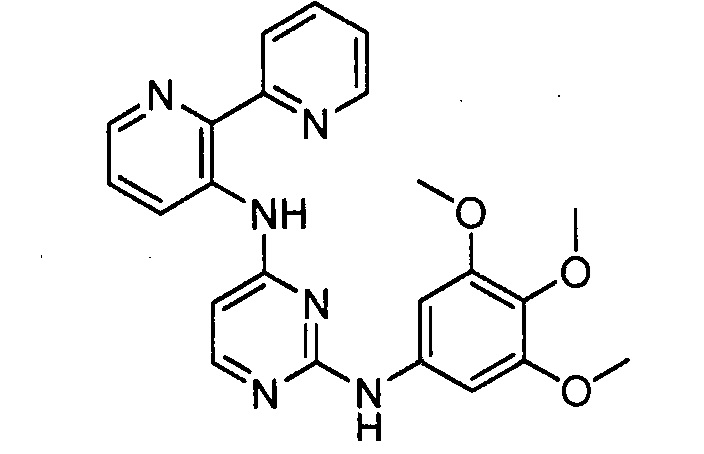
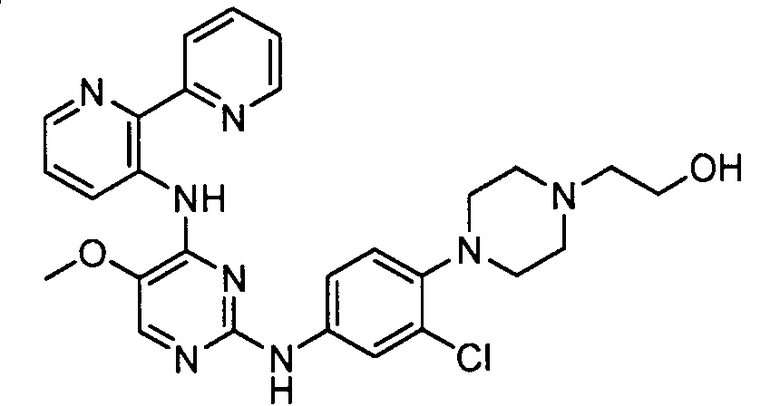
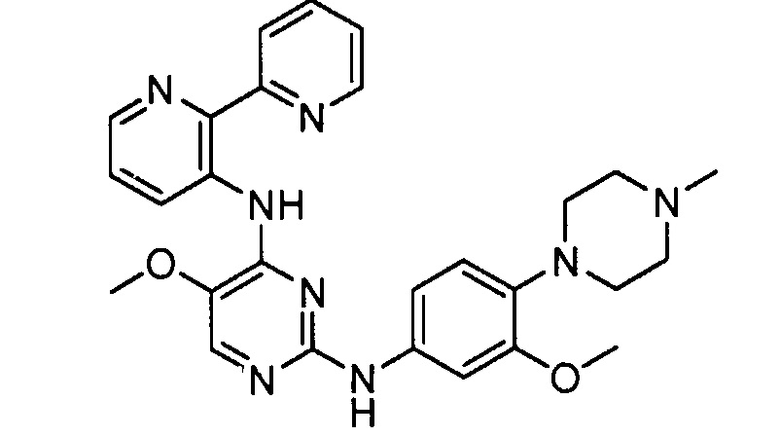
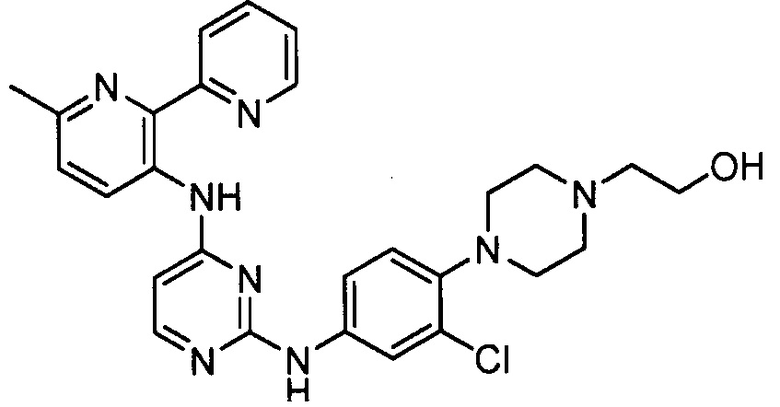
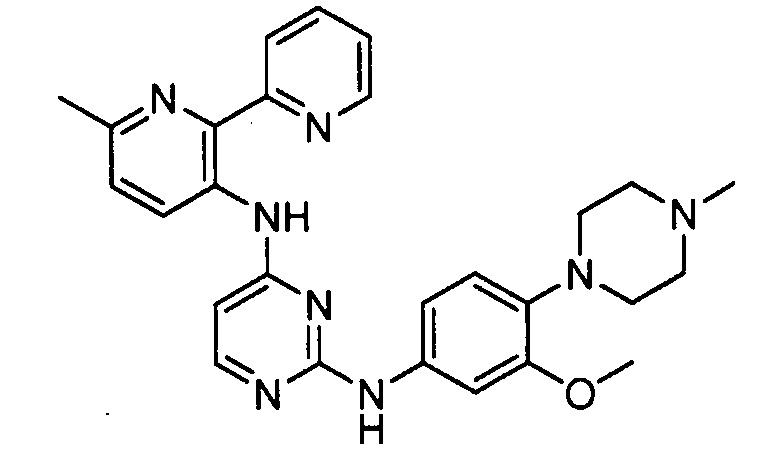
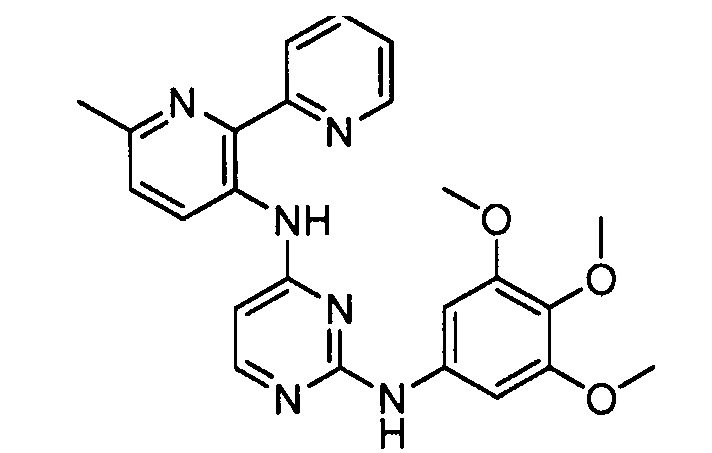
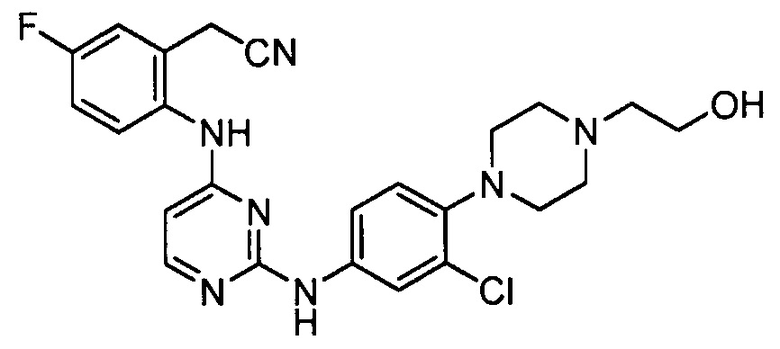
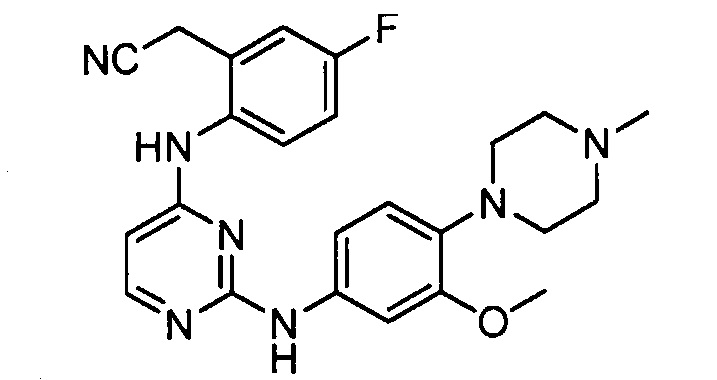
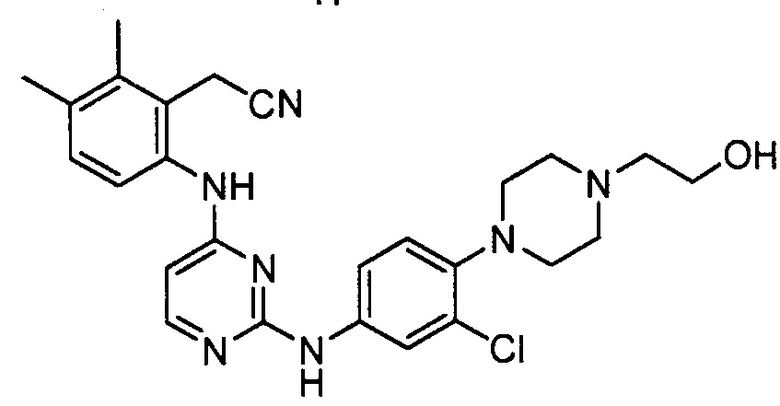
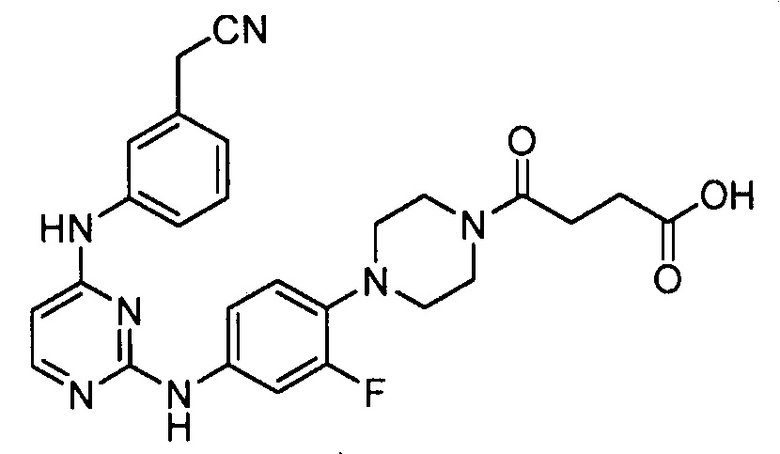
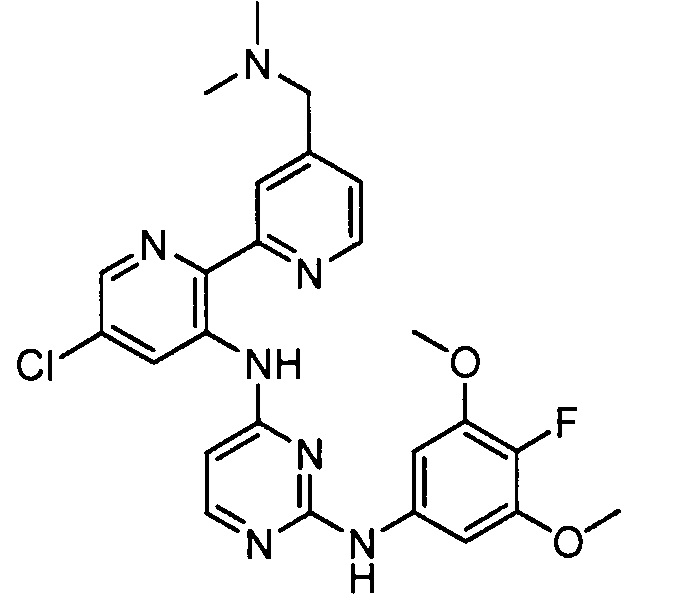
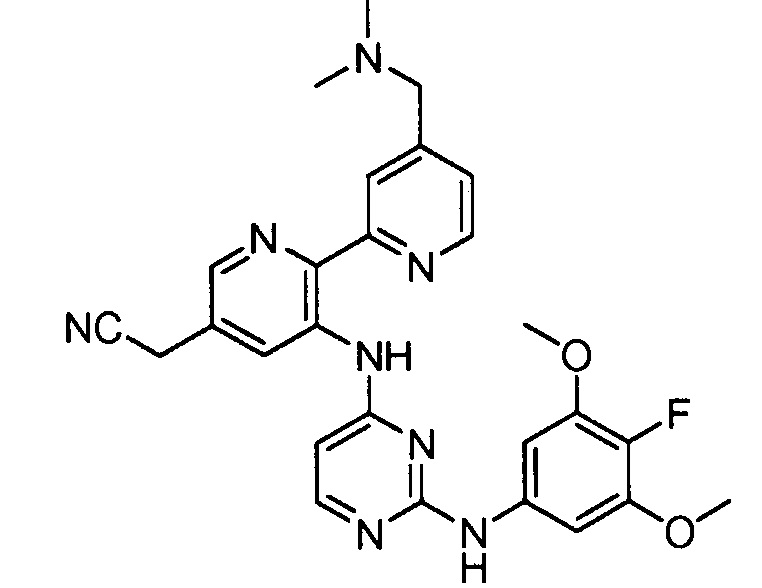
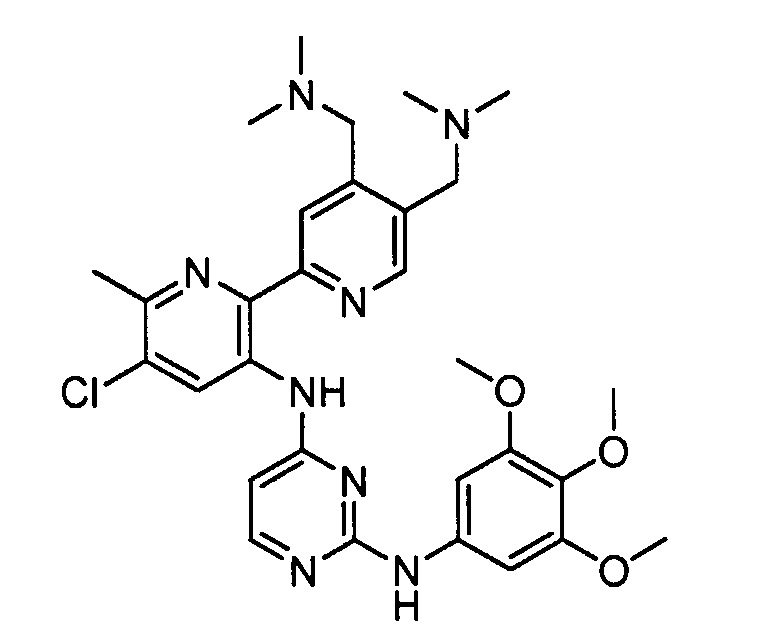
В некоторых других вариантах осуществления соединение выбирают из
соединений, представленных в таблице 1.
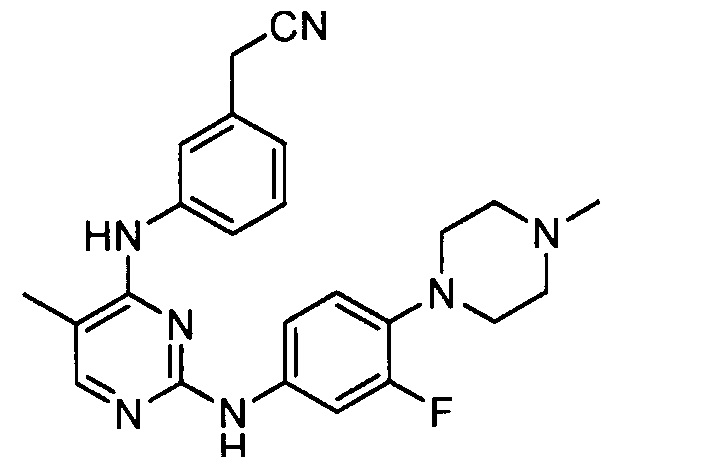
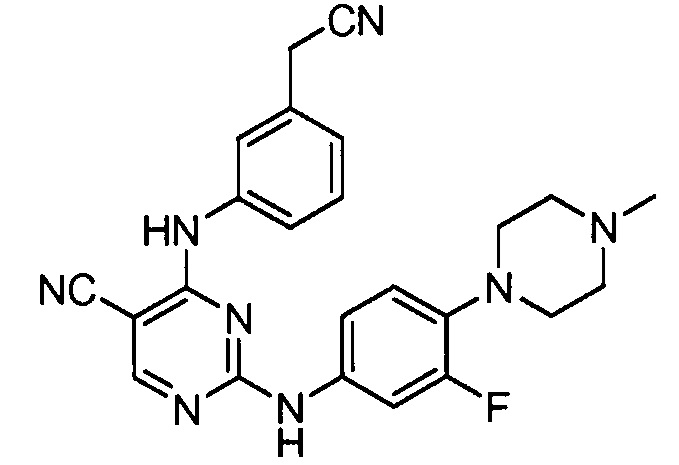
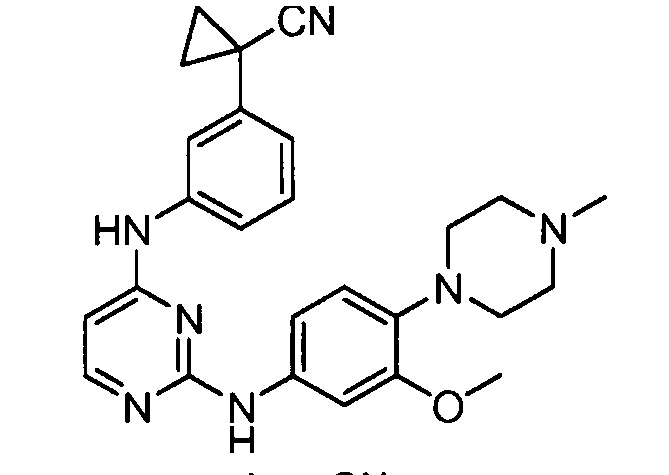
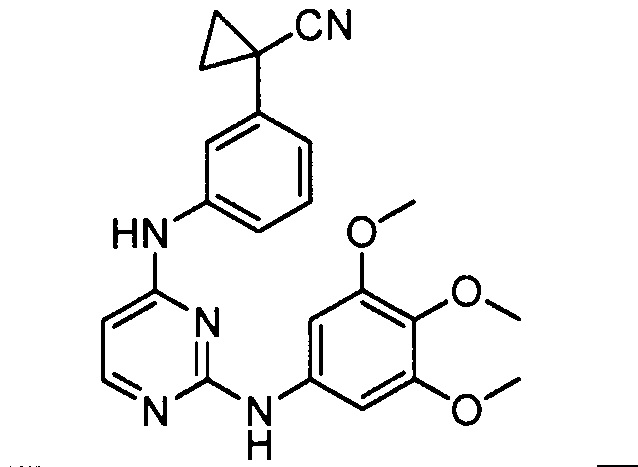
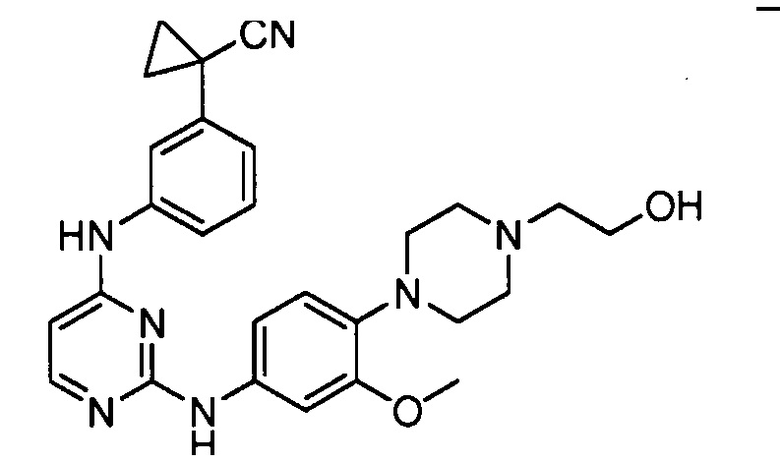
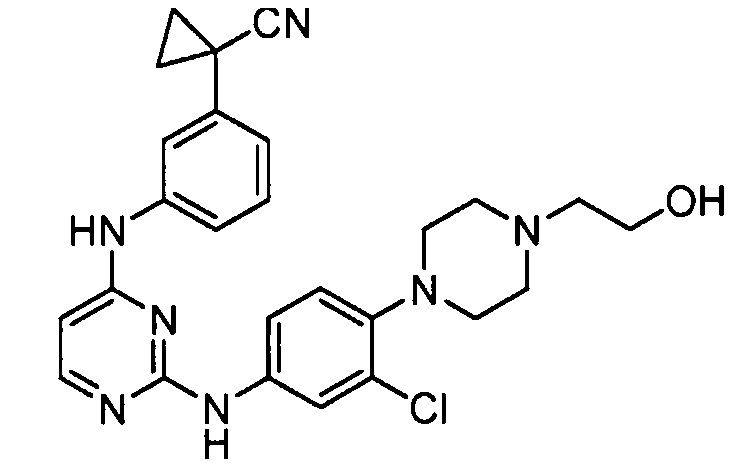
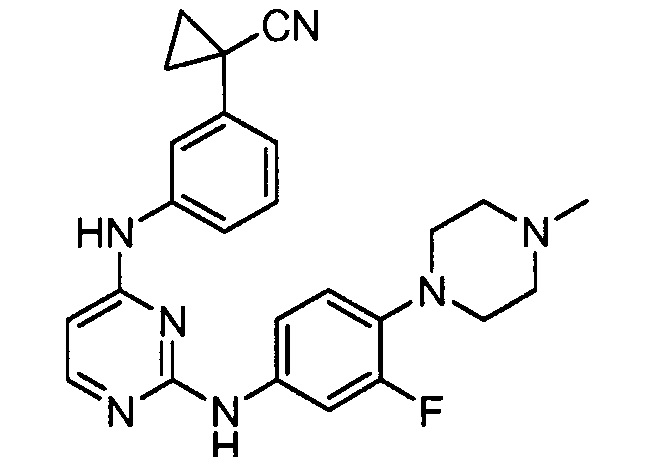
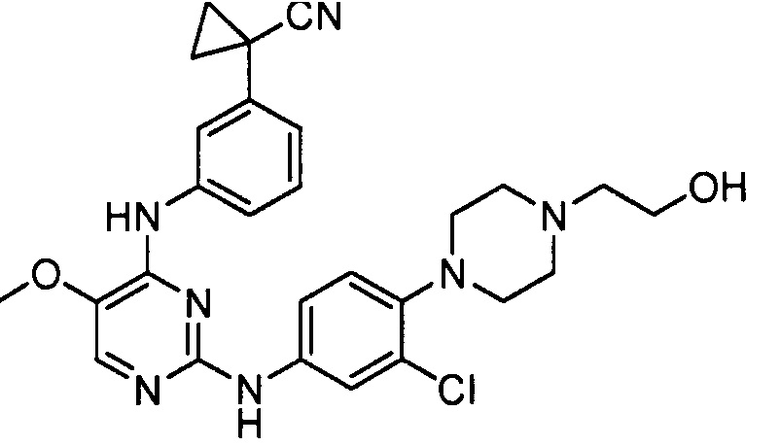
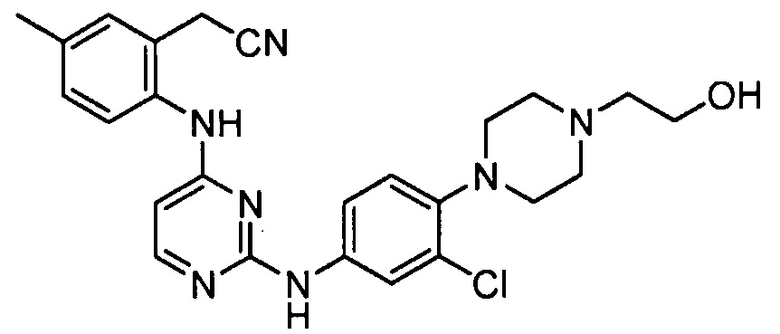
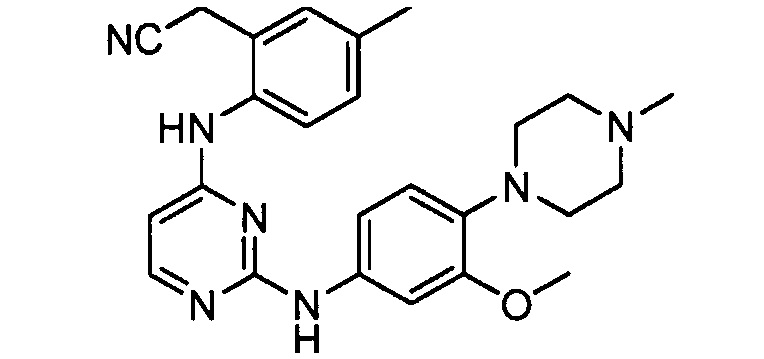
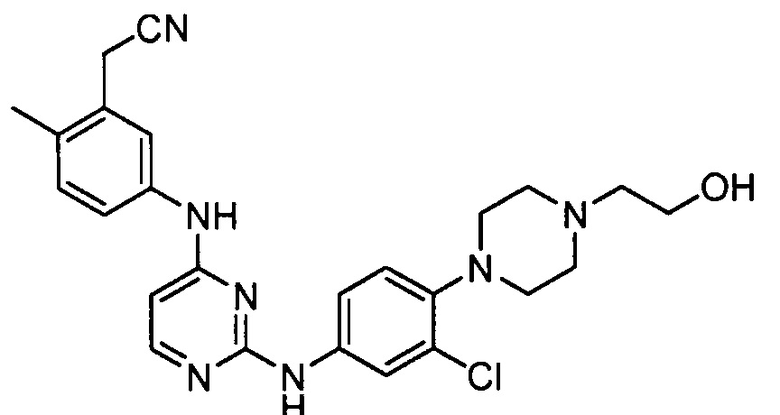
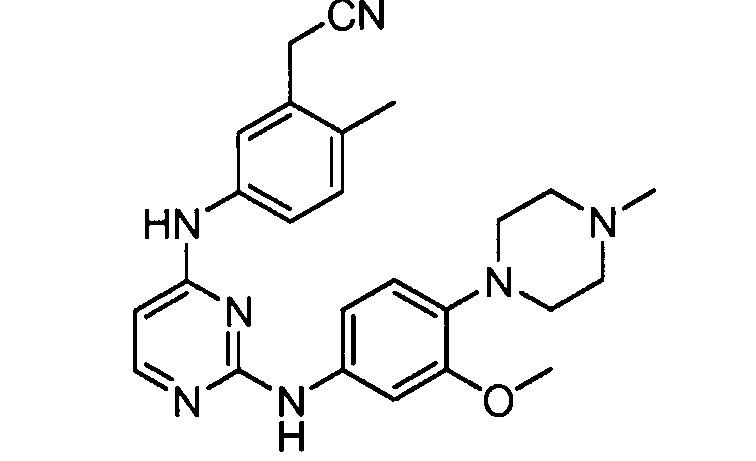
Подразумевается, что любой вариант осуществления соединений, имеющих структуру (I), в соответствии с изложенным выше, и любой из представленных в данном документе отдельных заместителей (например, R1-R9) в соединениях, имеющих структуры (I) и (II), в соответствии с изложенным выше, может независимо комбинироваться с другими вариантами осуществления и/или заместителями соединений, имеющих структуры (I) и (II), с получением вариантов осуществления изобретения, которые не были конкретно изложены выше. Кроме того, в том случае, когда перечень заместителей перечисляется для любой отдельной группы R в конкретном варианте осуществления и/или пункте формулы изобретения, подразумевается, что каждый отдельный заместитель может быть удален из особого варианта осуществления и/или пункта формулы изобретения, и что оставшийся перечень заместителей будет считаться находящимся в рамках объема изобретения. Подразумевается, что в настоящем описании комбинации заместителей и/или переменных характеристик для изображенных формул допустимы, только если такие вклады приводят к стабильным соединениям.
Соединения согласно настоящему изобретению могут быть получены в соответствии с любым количеством известных в данной области способов, включая те способы, которые конкретно описаны ниже в примерах. Следующая общая схема I реакции иллюстрирует способ получения соединений согласно данному изобретению, т.е. соединений, имеющих структуру (I), в которой R1-R8, A и X соответствуют определенным выше, а LG и LG’ независимо являются уходящими группами.
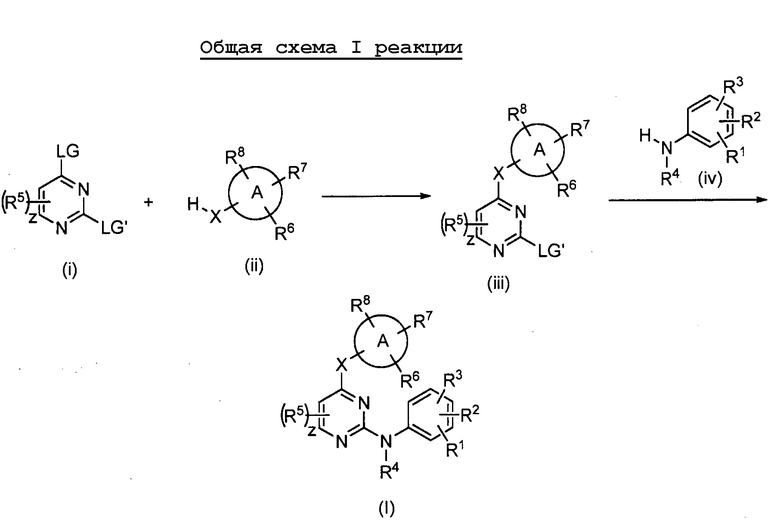 .
.
Что касается общей схемы I реакции, соединения, имеющие структуры (i), (ii) и (iv), могут быть получены согласно известным в данной области способам (например, согласно приведенным в примерах) или приобретены в коммерческих источниках. Реакция соединения (i) с соединением (ii) в подходящих условиях (например, в присутствии основания) приводит к получению соединений, имеющих структуру (iii). Кроме того реакция соединения (iii) с соединением (iv) в подходящих условиях (например, в присутствии основания) дает соединение, имеющее структуру (I).
Подразумевается, что специалист в данной области может быть способным получить эти соединения аналогичными способами или путем комбинирования других известных специалисту в данной области способов. Также подразумевается, что специалист в данной области согласно способу, аналогичному описанному ниже, потенциально может получить другие соединения, имеющие структуру (I), которые конкретно не проиллюстрированы ниже, при использовании подходящих исходных компонентов и изменении параметров синтеза по мере необходимости. Как правило, исходные компоненты могут быть получены из таких источников, как Sigma Aldrich, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI и Fluorochem USA и т.д., либо синтезированы в соответствии с известными специалистам в данной области источниками (см., например, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)) или получены согласно описанному в данном изобретении.
Специалистам в данной области понятно, что последовательность стадий, показанная на общей схеме I реакции (а также другие модификации), может быть осуществлена для получения соединения, имеющего структуру (I). Специалистам в данной области также понятно, что в способе, описанном в данном документе, функциональные группы промежуточных соединений могут нуждаться в защите подходящими защитными группами. Такие функциональные группы включают гидроксигруппу, аминогруппу, меркаптогруппу и карбоксильную кислотную группу. Подходящие защитные группы для гидроксигруппы включают триалкилсилил или диарилалкилсилил (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и т.п. Подходящие защитные группы для аминогруппы, амидиногруппы и гуанидиногруппы включают трет-бутоксикарбонил, бензилоксикарбонил и т.п. Подходящие защитные группы для меркаптогруппы включают -C(О)-R’’ (где R’’ является алкилом, арилом или арилалкилом), п-метоксибензил, тритил и т.п. Подходящие защитные группы для карбоксильной кислотной группы включают алкиловые, ариловые или арилалкиловые сложноэфирные группы. Защитные группы могут присоединяться или удаляться в соответствии со стандартными способами, которые известны специалисту в данной области, и в соответствии с описанным в данном документе. Использование защитных групп подробно описано в Green, T.W. and P.G.M. Wutz, Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley. Как понятно специалисту в данной области, защитная группа может также представлять собой полимерную смолу, такую как смола Ванга, смола Ринка или 2-хлортритилхлоридная смола.
Специалистам в данной области также понятно, что хотя такие защищенные производные соединений согласно данному изобретению могут не обладать фармакологической активностью как таковой, они могут вводиться в организм млекопитающего и затем претерпевать превращения в процессе обмена веществ в теле с образованием соединений согласно изобретению, которые являются фармакологически активными. Поэтому такие производные могут описываться как «пролекарства». Все пролекарства соединений согласно данному изобретению включены в объем изобретения.
Кроме того все соединения согласно изобретению, которые существуют в форме свободного основания или кислоты, могут подвергаться превращению в их фармацевтически приемлемые соли при обработке подходящими неорганическими или органическими основанием или кислотой согласно известным специалисту в данной области способам. Соли соединений согласно изобретению могут подвергаться превращению в соответствующие им формы свободного основания или кислоты с использованием стандартных способов.
III. Композиции и введение в организм
В других вариантах осуществления настоящее изобретение ориентировано на фармацевтическую композицию, содержащую соединение, имеющее структуру (I) или (II), или его стереоизомер, фармацевтически приемлемую соль, таутомер или пролекарство, а также фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
Для осуществления введения в организм, соединение согласно настоящему изобретению может вводиться в организм в виде необработанного химического вещества или может входить в рецептуру фармацевтических композиций. Фармацевтические композиции согласно настоящему изобретению содержат соединение, имеющее структуру (I), а также фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. Соединение, имеющее структуру (I), присутствует в композиции в таком количестве, которое является эффективным для лечения конкретного рассматриваемого заболевания или состояния, то есть, в количестве, достаточном для лечения различных онкологических заболеваний, и предпочтительно с допустимым токсическим воздействием на пациента. Активность соединений, имеющих структуру (I), в отношении JAK2 и/или ALK2 киназы может определяться специалистом в данной области, например, согласно описанному ниже в примерах. Подходящие концентрации и дозировки могут легко определяться специалистом в данной области.
Введение в организм соединений согласно изобретению или их фармацевтически приемлемых солей, в чистом виде или в составе подходящей фармацевтической композиции, может проводиться посредством любого из принятых способов введения в организм агентов для обеспечения аналогичных полезных эффектов. Фармацевтические композиции согласно изобретению могут быть получены путем комбинирования соединения согласно изобретению с подходящим фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом, и могут использоваться для создания препаратов в твердой, полутвердой, жидкой или газообразной формах, таких как таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, инъекции, лекарственные формы для ингаляции, гели, микросферы и аэрозоли. Обычные способы введения таких фармацевтических композиций включают, в том числе, пероральный, местное применение, трансдермальный, ингаляцию, парентеральный, сублингвальный, трансбуккальный, ректальный, вагинальный и интраназальный. В данном контексте термин парентеральный включает подкожные инъекции, внутривенную, внутримышечную, интрастернальную инъекцию или инфузионные способы. Фармацевтические композиции согласно изобретению составляются таким образом, чтобы обеспечить биодоступность содержащихся в них активных ингредиентов при введении композиции в организм пациента. Композиции, которые будут вводить больному или пациенту, принимают форму, состоящую из одной или более дозированных единиц, где например, таблетка может представлять собой одну дозированную единицу, а емкость, содержащая соединение согласно изобретению в форме аэрозоля, может содержать множество дозированных единиц. Актуальные способы получения таких лекарственных форм являются известными или будут очевидными для специалистов в данной области; например, см. Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). Композиция, которая подлежит введению в организм, в любом случае будет содержать терапевтически эффективную дозу соединения согласно изобретению или его фармацевтически приемлемой соли для лечения интересующей болезни или состояния в соответствии с указаниями данного изобретения.
Фармацевтическая композиция согласно изобретению может находиться в форме твердого вещества или жидкости. В одном аспекте носитель(и) имеет(ют) форму частиц, поэтому композиции находятся, например, в форме таблеток или порошка. Носитель(и) может(гут) быть жидкостью, при этом композиции являются, например, сиропом для перорального применения, инъекционной жидкостью или аэрозолем, который является подходящим, например, при ингаляционном введении в организм.
В случае предназначения для перорального приема, фармацевтическая композиция предпочтительно находится либо в твердой, либо в жидкой форме, где полутвердая, полужидкая, суспензионная и гелевая формы входят в число форм, которые в данном документе рассматриваются либо как твердое вещество, либо как жидкость.
В качестве твердой композиции для перорального приема, фармацевтическая композиция может использоваться для создания порошка, гранул, прессованных таблеток, пилюль, капсул, жевательной резинки, пластинок или тому подобных форм. Такая твердая композиция, как правило, будет содержать один или более инертных разбавителей или съедобных носителей. Кроме того, может присутствовать один или несколько из следующих компонентов: связующие вещества, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, трагант или желатин; вспомогательные вещества, такие как крахмал, лактоза или декстрины, вещества для улучшения распадаемости таблеток, такие как альгиновая кислота, альгинат натрия, Primogel, кукурузный крахмал и т.п.; смазывающие вещества, такие как стеарат магния или Sterotex; скользящие вещества, такие как коллоидный диоксид кремния; подсластители, такие как сахароза или сахарин; ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор; и красящее вещество.
Когда фармацевтическая композиция находится в форме капсулы, например, желатиновой капсулы, помимо материалов вышеуказанного типа она может содержать жидкий носитель, такой как полиэтиленгликоль или масло.
Фармацевтическая композиция может находиться в форме жидкости, например, настойки, сиропа, раствора, эмульсии или суспензии. В качестве двух примеров, жидкость может предназначаться для перорального приема или для доставки путем инъекции. В случае предназначения для перорального приема, помимо настоящих соединений предпочтительная композиция содержит один или более компонентов из подсластителя, консервантов, красителя/красящего вещества и интенсификатора вкусоароматических свойств. В композицию, предназначенную для введения путем инъекции, могут быть включены один или более компонентов из поверхностно-активного вещества, консерванта, увлажняющего компонента, диспергирующиего агента, суспендирующего агента, буфера, стабилизатора и изотонического агента.
Жидкие фармацевтические композиции согласно изобретению, независимо от того, являются они растворами, суспензиями или другим аналогичными формами, включают одно или более из следующих вспомогательных средств: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический раствор, раствор Рингера, изотонический хлорид натрия, нелетучие масла, такие как синтетические моно или диглицериды, которые могут служить в качестве растворителя или суспензионной среды, полиэтиленгликоли, глицерин, пропиленгликоль или другие растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетатные, цитратные или фосфатные и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Препараты для парентерального введения могут быть заключены в ампулах, шприцах одноразового применения или пузырьках для многократного применения, изготовленных из стекла или пластмассы. Физиологический раствор является предпочтительным вспомогательным средством. Вводимая фармацевтическая композиция предпочтительно является стерильной.
Жидкая фармацевтическая композиция согласно изобретению предназначенная либо для парентерального, либо для перорального введения, должна содержать такое количество соединения согласно изобретению, чтобы была получена подходящая дозировка.
Фармацевтическая композиция согласно изобретению может быть предназначена для местного применения, и в этом случае носитель может соответственно содержать раствор, эмульсионную, мазевую или гелевую основу. Основа, например, может содержать один или более из следующих компонентов: петролатум, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, а также эмульгаторы и стабилизаторы. В фармацевтической композиции для местного применения может присутствовать загуститель. В случае предназначения для трансдермального введения, композиция может включать трансдермальный пластырь или устройство для ионтофореза.
Фармацевтическая композиция согласно изобретению может предназначаться для ректального введения в форме, например, суппозитория, который будет таять в прямой кишке и высвобождать лекарственное вещество. Композиция для ректального введения может содержать масляную основу в качестве подходящего не вызывающего раздражения вспомогательного вещества. Такие основы включают, в том числе, ланолин, масло какао и полиэтиленгликоль.
Фармацевтическая композиция согласно изобретению может включать различные материалы, которые модифицируют физическую форму твердой или жидкой дозированной единицы. Например, композиция может включать материалы, которые образуют покрывающую оболочку вокруг активных компонентов. Материалы, которые образуют покрывающую оболочку, как правило, являются инертными и могут быть выбраны, например, из сахара, шеллака и других агентов для создания кишечнорастворимой оболочки. В соответствии с другим вариантом активные ингредиенты могут быть заключены в желатиновую капсулу.
Фармацевтическая композиция согласно изобретению в твердом или жидком виде может включать агент, который связывается с соединением согласно изобретению и таким образом способствует доставке соединения. Подходящие агенты, которые могут выступать в этой роли, включают моноклональное или поликлональное антитело, белок или липосома.
Фармацевтическая композиция согласно изобретению может состоять из дозированных единиц, которые могут вводиться в организм в виде аэрозоля. Термин аэрозоль используется для обозначения множества систем, начиная от систем коллоидной природы и заканчивая системами, состоящими из аэрозольных упаковок. Доставка может осуществляться с помощью сжиженного или сжатого газа или с помощью подходящей нагнетающей системы, которая дозирует активные компоненты. Для доставки активного(ых) компонента(ов), аэрозоли соединений согласно изобретению могут доставляться в виде отдельной фазы, двухфазной или трехфазной системы. Система доставки аэрозоля включает необходимые емкость, элементы приведения в действие, клапаны, малые емкости и т.п., которые могут вместе образовывать набор. Специалист в данной области может определить предпочтительные аэрозоли без проведения лишних экспериментов.
Фармацевтические композиции согласно изобретению могут быть получены в соответствии с известными в области фармацевтики способами. Например, фармацевтическую композицию, предназначенную для введения путем инъекции, можно получить путем комбинирования соединения согласно изобретению со стерильной дистиллированной водой, чтобы образовался раствор. Чтобы облегчить образование однородного раствора или суспензии можно добавить поверхностно-активное вещество. Поверхностно-активные вещества представляют собой соединения, которые нековалентно взаимодействуют с соединением согласно изобретению таким образом, чтобы облегчить растворение или получение однородной суспензии соединения в водной системе доставки.
Соединения согласно изобретению или их фармацевтически приемлемые соли вводятся в организм в терапевтически эффективной дозе, которая будет меняться в зависимости от множества факторов, включающих активность конкретного используемого соединения; устойчивость к инактивации в процессе метаболизма и продолжительность действия соединения; возраст, масса тела, общее состояние здоровья, пол и режим питания пациента; способ и время введения в организм; скорость экскреции; сочетание лекарственных веществ; серьезность конкретного расстройства или состояния; и терапию, которую проходит больной.
Соединения согласно изобретению или их фармацевтически приемлемые производные также могут вводиться в организм одновременно с, до или после введения в организм одного или более других терапевтических средств. Такая комбинированная терапия включает введение в организм единственного дозированного лекарственного состава, который содержит соединение согласно изобретению и один или более дополнительных активных компонентов, а также введение в организм соединения согласно изобретению и каждого активного компонента в виде собственного отдельного дозированного лекарственного состава. Например, соединение согласно изобретению и другой активный компонент могут вводиться в организм пациента совместно в одной пероральной дозированной композиции, такой как таблетка или капсула, или каждый компонент вводится в виде отдельных пероральных дозированных составов. В том случае, когда используются отдельные дозированные составы, соединения согласно изобретению и один или более дополнительных активных компонентов могут вводиться в организм практически в одно и то же время, т.е. одновременно, или в отдельные моменты времени с некоторым интервалом, т.е. последовательно; Подразумевается, что комбинированная терапия включает все указанные режимы.
Для любого соединения, используемого в способах согласно изобретению, терапевтически эффективное количество или дозу можно первоначально оценить исходя из испытаний на клеточных культурах. Затем можно определить дозу для использования на животных моделях таким образом, чтобы достичь диапазона циркулирующей концентрации, который включает IC50, соответствующую установленной на клеточной культуре (то есть, концентрацию испытываемого соединения, которая обеспечивает полумаксимальное ингибирование активности протеинкиназы). Такая информация может затем использоваться для более точного определения подходящих доз у людей.
Токсичность и терапевтическая эффективность описанных в данном документе соединений могут быть определены с использованием стандартных фармацевтических методик на клеточных культурах или экспериментальных животных, например, при определении IC50 и LD50 (обе из которых обсуждаются в другой части данного документа) для исследуемого соединения. Данные, полученные в результате таких испытаний на клеточных культурах и исследований на животных, могут использоваться при установлении диапазона дозировок для использования на людях. Дозировка может меняться в зависимости от используемой лекарственной формы и используемого способа введения. Точная рецептура, способ введения и дозировка может выбираться отдельным врачом исходя из состояния пациента. (См., например, GOODMAN & GILMAN'S THE PHARMACOLOGICAL BASIS OL THERAPEUTICS, Ch. 3, 9th ed., Ed. by Hardman, J., and Limbard, L., McGraw-Hill, New York City, 1996, p.46.)
Размер дозировки и интервал дозирования можно подбирать индивидуально, чтобы обеспечивать концентрации в плазме крови молекул активного вещества, которые являются достаточными для поддержания эффектов модуляции киназы. Эти концентрации в плазме крови называются минимальными эффективными концентрациями (MEC). MEC будет меняться для каждого соединения, но ее можно оценить исходя из данных, полученных вне живого организма, например, концентрация, необходимая для достижения ингибирования киназы на 50-90%, может быть установлена путем проведения описанных в данном документе испытаний. Необходимые для достижения MEC дозировки будут зависеть от индивидуальных характеристик и способа введения в организм. Для определения концентраций в плазме крови могут использоваться HPLC анализ или биопробы.
Интервалы дозирования могут также быть определены с использованием значения MEC. Соединения следует вводить в организм согласно режиму, который позволяет поддерживать концентрации в плазме крови выше MEC в течение 10-90% времени, предпочтительно в интервале от 30 до 90% и наиболее предпочтительно в интервале от 50 до 90%.
В настоящее время терапевтически эффективное количество соединений согласно настоящему изобретению может иметь значение в интервале от приблизительно 2,5 мг/м2 до 1500 мг/м2 в день.
Дополнительные иллюстративные количества имеют значения в диапазоне от 0,2 до 1000 мг 4 раза в сутки, от 2 до 500 мг 4 раза в сутки и от 20 до 250 мг 4 раза в сутки.
В случаях местного применения или селективного накопления, эффективная местная концентрация лекарственного вещества не может быть связана с концентрацией в плазме крови, и для определения правильного размера дозировки и интервала дозирования могут использоваться другие известные в данной области методики.
Количество введенной в организм композиции будет, конечно, зависеть от проходящего лечение больного, серьезности болезни, способа введения в организм, мнения лечащего врача и т.д.
При необходимости композиции могут предоставляться в упаковке или дозирующем приспособлении, например, в одобренном FDA наборе, который может содержать одну или более лекарственных форм с однократной дозировкой, содержащих активный компонент. Упаковка может, например, содержать металлическую фольгу или полимерную пленку, как например блистерная упаковка. К упаковке или дозирующему приспособлению могут прилагаться инструкции по применению. К упаковке или дозатору может также прилагаться сопровождающее тару уведомление в форме, предписанной правительственным учреждением, регулирующим изготовление, использование или продажу лекарственных препаратов, в котором отражается одобрение учреждением данной формы композиций, или введения в организм человека или животного. Такое уведомление, например, может представлять собой маркировку, одобренную управлением по санитарному надзору за пищевыми продуктами и медикаментами США для отпускаемых по рецепту лекарств, или одобренный листок-вкладыш. Композиции, содержащие соединение согласно изобретению, объединенное с совместимым фармацевтическим носителем, также могут быть получены, помещены в подходящую тару и обеспечены маркировкой, указывающей на их использование для лечения отмеченного состояния. Подходящие указываемые на маркировке состояния могут включать лечение новообразования, ингибирование ангиогенеза, лечение фиброза, диабета и т.п.
IV. Способы лечения
В различных других вариантах осуществления изобретение ориентировано на способ ингибирования киназы ALK2 или киназы JAK2, или их комбинации у нуждающегося в этом млекопитающего, при этом способ включает введение в организм млекопитающего эффективной дозы любого из вышеуказанных соединений (т.е. соединений, имеющих структуру (I) или (II)) или фармацевтической композиции по пункту формулы изобретения, содержащей соединение.
В некоторых вариантах осуществления способ предназначен для ингибирования киназы ALK2. В других вариантах осуществления способ предназначен для ингибирования киназы JAK2.
Еще в нескольких вариантах осуществления ингибирование осуществляется для лечения онкологического заболевания. В нескольких вариантах осуществления ингибирование осуществляется для лечения анемии хронического заболевания, анемии хронического воспаления, анемии онкологического заболевания или прогрессирующей оссифицирующей фибродисплазии.
В другом варианте осуществления настоящее раскрытие ориентировано на способ лечения онкологического заболевания у нуждающегося в этом млекопитающего, при этом способ включает введение в организм млекопитающего эффективной дозы любого из вышеуказанных соединений (т.е. соединений, имеющих структуру (I) или (II)) или фармацевтической композиции по пункту формулы изобретения, содержащей соединение.
В некоторых вариантах осуществления вышеуказанного способа онкологическое заболевание представляет собой миелопролиферативное заболевание, лимфому или солидную опухоль. Например, в некоторых вариантах осуществления миелопролиферативное заболевание является миелофиброзом, полицитемией или эссенциальной тромбоцитемей.
В других вариантах осуществления солидная опухоль представляет собой опухоль молочной железы, простаты или поджелудочной железы.
Еще в нескольких вариантах осуществления онкологическое заболевание представляет собой рак простаты, яичников или головы и шеи.
Изобретение также обеспечивает лечение различных других онкологических заболеваний путем введения в организм соединений, имеющих структуру (I) или (II), согласно описанному ниже.
Преимущественно настоящие соединения обнаруживают полезность в способах лечения и облегчения симптомов онкологического заболевания. Соответственно, в некоторых вариантах осуществления способ предназначен для обеспечения поддерживающей терапии нуждающегося в этом больного онкологическим заболеванием (т.е. пациента, такого как обследуемый человек, у которого было диагностировано наличие онкологического заболевания), при этом способ включает введение в организм пациента эффективной дозы любого из вышеуказанных соединений (т.е. соединений, имеющих структуру (I) или (II)) или фармацевтической композиции по пункту формулы изобретения, содержащей соединение. Например, в некоторых вариантах осуществления способ предназначен для лечения анемии и патологической усталости, связанных с онкологическим заболеванием.
Как упомянуто выше, соединения и композиции согласно изобретению найдут полезность в широком диапазоне болезней и состояний, опосредованных протеинкиназами ALK2 и/или JAK2. Такие болезни могут включать в качестве примера, в том числе, онкологические заболевания, такие как гематологические злокачественные опухоли (например, острый миелоидный лейкоз (AML) и хронический миелоидный лейкоз (CML)), рак легких, NSCLC (немелкоклеточный рак легких), мелкоклеточный рак легких, рак костей, рак поджелудочной железы, рак кожи, возвышающаяся дерматофибросаркома, рак головы и шеи, кожная или внутриглазная меланома, рак матки, рак яичников, колоректальный рак, рак анального канала, рак желудка, рак толстой кишки, рак молочной железы, гинекологические новообразования (например, саркомы матки, карцинома маточных труб, карцинома оболочки матки, карцинома шейки матки, карцинома влагалища или карцинома вульвы), болезнь Ходжкина, гепатоцеллюлярный рак, рак пищевода, рак тонкой кишки, новообразования эндокринной системы (например, рак щитовидной железы, поджелудочной железы, паращитовидной железы или надпочечников), саркомы мягких тканей, рак уретры, рак полового члена, рак яичка, рак простаты (особенно гормонорезистентный), хроническая или острая лейкемия, солидные опухоли детского возраста, гиперэозинофилия, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника (например, почечно-клеточная карцинома, карцинома почечной лоханки), злокачественные новообразования у детей, новообразования центральной нервной системы (например, первичная лимфома CNS (ЦНС), новоорбазования позвоночника, медуллобластома, глиомы ствола мозга или аденомы гипофиза), пищевод Баррета (предраковый синдром), опухолевое поражение кожи, псориаз, фунгоидные гранулемы и доброкачественную гиперплазия предстательной железы, болезни, связанные с диабетом, такие как диабетическая ретинопатия, ретинальная ишемия и ретинальная неоваскуляризация, цирроз печени, ангиогенез, сердечно-сосудистое заболевание, такое как атеросклероз, иммунологическую болезнь, такую как аутоиммунная болезнь и почечное заболевание.
В некоторых вариантах осуществления соединения и композиции согласно изобретению могут использоваться в способах лечения онкологических заболеваний, таких как гематологические злокачественные новообразования. Например, в некоторых вариантах осуществления соединения и композиции согласно изобретению могут использоваться в способах лечения острой миелоидной лейкемии (AML). Другие способы включают лечение рака мочевого пузыря или лечение рака простаты.
Разработанные соединения (т.е. соединения, имеющие структуру (I)) могут использоваться в комбинации с одним или более другими химиотерапевтическими средствами. Дозировку разработанных соединений можно подобрать для любого межлекарственного взаимодействия. В одном из вариантов осуществления химиотерапевтическое средство выбирают из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, таких как CAMPTOSAR (иринотекан), модификаторов биологического отклика, антигормонов, антиангиогенных агентов, таких как ингибиторы MMP-2, MMP-9 и COX-2, антиандрогенов, координационных комплексов платины (цисплатин и т.д.), замещенных мочевин, таких как гидроксимочевина; производных метилгидразина, например, прокарбазина; адренокортикальных супрессивных средств, например, митотана, аминоглютетимида, гормонов и гормональных антагонистов, таких как, адренокортикостероиды (например, преднизон), прогестины (например, капроат гидроксипрогестерона), эстрогены (например, диэтилстилбэстрол), антиэстрогены, такие как тамоксифен, андрогены, например, пропионат тестостерона и ингибиторов ароматазы, таких как анастрозол и AROMASIN (эксеместан).
Примеры алкилирующих агентов, в сочетании с которыми может быть осуществлен вышеуказанный способ, включают, в том числе, фторурацил (5-FU) взятый отдельно или дополнительно в комбинации с лейковорином; другие аналоги пиримидина, такие как UFT, капецитабин, гемцитабин и цитарабин, алкилсульфонаты, например, бусульфан (используемый при лечении хронического миелолейкоза), импросульфан и пипосульфан; азиридины, например, бензодепа, карбоквон, метуредепа и уредепа; этиленимины и метилмеламины, например, алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилолмеламин; а также азотистый иприт, например, хлорамбуцил (используемый при лечении хронического лимфолейкоза, первичной макроглобулинемии и неходжкинской лимфомы), циклофосфамид (используемый при лечении болезни Ходжкина, множественной миеломы, нейробластомы, рака молочной железы, рака яичников, рака легких, опухоли Вилма и рабдомиосаркомы), эстрамустин, ифосфамид, новембрихин, преднимустин и урациловый иприт (используемый при лечении первичного тромбоцитоза, неходжкинской лимфомы, Болезни Ходжкина и рака яичников); и триазины, например, дакарбазин (используемый при лечении саркомы мягкой ткани)
Примеры антиметаболитных химиотерапевтических средств, в сочетании с которыми может быть осуществлен вышеуказанный способ, включают, в том числе, аналоги фолиевой кислоты, например, метотрексат (используемый при лечении острого лимфолейкоза, хориокарциномы, фунгоидного микоза, рака молочной железы, рака головы и шеи, а также остеогенной саркомы) и птероптерин; и аналоги пурина, такие как меркаптопурин и тиогуанин, которые находят использование при лечении острых гранулоцитарных, острых лимфоцитарных и хронических гранулоцитарных лейкозов.
Примеры химиотерапевтических средств на основе природных продуков, в сочетании с которыми может быть осуществлен вышеуказанный способ, включают, в том числе, алкалоиды барвинка, например, винбластин (используемый при лечении рака молочной железы и яичка), винкристин и виндезин; эпиподофиллотоксины, например, этопозид и тенипозид, оба из которых подходят при лечении рака яичка и саркомы Капоши; антибиотические химиотерапевтические средства, например, даунорубицин, доксорубицин, эпирубицин, митомицин (используется для лечения рака желудка, шейки матки, толстой кишки, молочной железы, мочевого пузыря и поджелудочной железы), дактиномицин, темозоломид, пликамицин, блеомицин (используется при лечении рака кожи, пищевода и мочеполовой системы); и ферментативные химиотерапевтические средства, такие как L-аспарагиназа.
Примеры подходящих ингибиторов COX-II включают Vioxx, CELEBREX (целеоксиб), валдекоксиб, паракоксиб, рофекоксиб и Cox 189.
Примеры подходящих ингибиторов матриксной металлопротеиназы описаны в WO 96/33172 (опубликованном 24 октября 1996), WO 96/27583 (опубликованном 7 марта 1996), европейской заявке на патент № 97304971.1 (поданной 8 июля 1997), европейской заявке на патент № 99308617.2 (поданной 29 октября 1999), WO 98/07697 (опубликованном 26 февраля 1998), WO 98/03516 (опубликованном 29 января 1998), WO 98/34918 (опубликованном 13 августа 1998), WO 98/34915 (опубликованном 13 августа 1998), WO 98/33768 (опубликованном 6 августа, 1998), WO 98/30566 (опубликованном 16 июля 1998), европейской патентной публикации 606046 (опубликованной 13 июля 1994), европейской патентной публикации 931788 (опубликованной 28 июля, 1999), WO 90/05719 (опубликованном 31 мая 1990), WO 99/52910 (опубликованном 21 октября 1999), WO 99/52889 (опубликованном 21 октября 1999), WO 99/29667 (опубликованном 17 июня 1999), международной заявке PCT № PCT/IB98/01113 (поданной 21 июля 1998), европейской заявке на патент № 99302232.1 (поданной 25 марта 1999), заявке на патент Великобритании номер 9912961.1 (поданной 3 июня 1999), патенте США № 5863949 (изданном 26 января 1999), патенте США № 5861510 (изданном 19 января 1999) и европейской патентной публикации 780386 (опубликованной 25 июня 1997), все из которых полностью включены в данный документ в качестве ссылки. Предпочтительными ингибиторами MMP-2 и MMP-9 являются те, которые имеют небольшую или вообще не имеют активности в отношении ингибирования MMP-1. Более предпочтительными являются те, которые селективно ингибируют MMP-2 и/или MMP-9 по сравнению с другими матриксными металлопротеиназами (т.е. MMP-1, MMP-3, MMP-4, MMP-5, MMP-6, MMP-7, MMP-8, MMP-10, MMP-11, MMP-12 и MMP-13).
Некоторые конкретные примеры ингибиторов MMP, подходящих для использования в настоящем изобретении, представляют собой AG-3340, RO 32-3555, RS 13-0830, а также соединения, выбираемые из: 3-[[4-(4-фторфенокси)-бензолсульфонил]-(1-гидроксикарбамоилциклопентил)-амино]-пропионовой кислоты; гидроксиамида 3-экзо-3-[4-(4-фторфенокси)-бензолсульфониламино]-8-окса-бицикло[3.2.l]октан-3-карбоновой кислоты; гидроксиамида (2R,3R) 1-[4-(2-хлор-4-фторбензилокси)-бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; гидроксиамида 4-[4-(4-фторфенокси)-бензолсульфониламино]-тетрагидропиран-4-карбоновой кислоты; 3-[[4-(4-фторфенокси)-бензолсульфонил]-(1-гидроксикарбамоилциклобутил)-амино]-пропионовой кислоты; гидроксиамида 4-[4-(4-хлорфенокси)-бензолсульфониламино]-тетрагидропиран-4-карбоновой кислоты; гидроксиамид (R) 3-[4-(4-хлорфенокси)-бензолсульфониламино]-тетрагидропиран-3-карбоновой кислоты; гидроксиамид (2R,3R) 1-[4-(4-фтор-2-метилбензилокси)-бензолсульфонил]-3-гидрокси-3-метил-пиперидин-2-карбоновой кислоты; 3-[[(4-(4-фторфенокси)-бензолсульфонил]-(1-гидроксикарбамоил-1-метилэтил)-амино]-пропионовой кислоты; 3-[[4-(4-фторфенокси)-бензолсульфонил]-(4-гидроксикарбамоил-тетрагидропиран-4-ил)-амино]-пропионовой кислоты; гидроксиамид 3-экзо-3-[4-(4-хлорфенокси)-бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты; гидроксиамид 3-эндо-3-[4-(4-фторфенокси)-бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты; и гидроксиамид (R) 3-[4-(4-фторфенокси)-бензолсульфониламино]-тетрагидрофуран-3-карбоновой кислоты; и фармацевтически приемлемых солей и сольватов этих соединений.
Другие агенты антиангиогенеза, другие ингибиторы COX-II и другие ингибиторы MMP также могут использоваться в настоящем изобретении.
Разработанное соединение также может использоваться с ингибиторами передачи сигнала, такими как агенты, которые могут ингибировать сигнальные ответы EGFR (рецептор эпидермального фактора роста), такие как антитела EGFR, антитела EGF и молекулы, которые являются ингибиторами EGFR; ингибиторами VEGF (фактор роста эндотелия сосудов); и ингибиторами рецептора erbB2, такими как органические молекулы или антитела, которые связываются рецептором erbB2, такие как E1ERCEPTIN (Genentech, Inc., Южный Сан-Франциско, Калифорния). Ингибиторы EGFR описаны, например, в WO 95/19970 (опубликованном 27 июля 1995), WO 98/14451 (опубликованном 9 апреля 1998), WO 98/02434 (опубликованном 22 января 1998), и патенте США № 5747498 (изданном 5 мая 1998), при этом такие вещества могут использоваться в настоящем изобретении в соответствии с описанным в данном документе.
Ингибирующие EGFR агенты включают, в частности, моноклональные антитела C225 и anti-EGFR 22Mab (ImClone Systems, Inc., Нью-Йорк, штат Нью-Йорк), соединения ZD-1839 (AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDX-447 (Medarex Inc., Аннандейл, Нью-Джерси), а также OFX-103 (Merck & Co., Уайтхаус-Стейшн, Нью-Джерси) и гибридный токсин EGF (Seragen Inc., Хопкинтон, Массачусетс).
Эти и другие ингибирующие EGFR агенты могут использоваться в настоящем изобретении. Также с разработанным соединением можно комбинировать ингибиторы VEGF, например SU-5416 и SU-6668 (Sugen Inc., Южный Сан-Франциско, Калифорния). Ингибиторы VEGF описаны, например, в WO 01/60814 A3 (опубликованном 23 августа 2001), WO 99/24440 (опубликованном 20 мая 1999), международной заявке PCT PCT/IB99/00797 (поданной 3 мая 1999), WO 95/21613 (опубликованном 17 августа 1995), WO 99/61422 (опубликованном 2 декабря 1999), патенте США № 5834504 (изданном 10 ноября 1998), WO 01/60814, WO 98/50356 (опубликованном 12 ноября 1998), патенте США № 5883113 (изданном 16 марта 1999), патенте США № 5886020 (изданном 23 марта 1999), патенте США № 5792783 (изданном 11 августа 1998), WO 99/10349 (опубликованном 4 марта 1999), WO 97/32856 (опубликованном 12 сентября 1997), WO 97/22596 (опубликованном 26 июня 1997), WO 98/54093 (опубликованном 3 декабря 1998), WO 98/02438 (опубликованном 22 января 1998), WO 99/16755 (опубликованном 8 апреля 1999) и WO 98/02437 (опубликованном 22 января 1998), все из которых полностью включены в данный документ в качестве ссылки. Другие примеры некоторых специфичных ингибиторов VEGF, подходящих для использования в настоящем изобретении, представляют собой IM862 (Cytran Inc., Киркланд, Вашингтон); моноклональное антитело anti-VEGF от Genentech, Inc.; и ангиозим, синтетический рибозим от Ribozyme (Боулдер, Колорадо) и Chiron (Эмеривилл, Калифорния). Эти и другие ингибиторы VEGF могут использоваться в настоящем изобретении в соответствии с описанным в данном документе. Ингибиторы рецептора pErbB2, такие как GW 282974 (Glaxo Wellcome plc), и моноклональные антитела AR-209 (Aronex Pharmaceuticals Inc., Вудлендс, Техас) и 2B-1 (Хирон) также можно комбинировать с разработанным соединением, например, те, которые указаны в WO 98/02434 (опубликованном 22 января 1998), WO 99/35146 (опубликованном 15 июля 1999), WO 99/35132 (опубликованном 15 июля 1999), WO 98/02437 (опубликованном 22 января 1998), WO 97/13760 (опубликованном 17 апреля 1997), WO 95/19970 (опубликованном 27 июля 1995), в патенте США № 5587458 (изданном 24 декабря 1996), и патенте США № 5877305 (изданном 2 марта 1999), все из которых таким образом полностью включены в данный документ в качестве ссылки. Ингибиторы рецептора ErbB2, подходящие для использования в настоящем изобретении, также описаны в патенте США № 6284764 (изданном 4 сентября 2001), который полностью включен в данный документ в качестве ссылки. Ингибирующие рецептор erbB2 соединения и вещества, описанные в вышеупомянутых заявках PCT, патентах США и предварительных заявках США, так же как и другие соединения и вещества, которые ингибируют рецептор erbB2, можно использовать совместно с разработанным соединением в соответствии с настоящим изобретением.
Разработанное соединение может также использоваться совместно с другими агентами, подходящими для лечения онкологического заболевания, включая, в том числе, агенты, способные усиливать противоопухолевые иммунные реакции, такие как антитела CTLA4 (антиген цитотоксического лимфоцита 4) и другие агенты, способные блокировать CTLA4; а также антипролиферативные агенты, такие как другие ингибиторы фарнезилпротеинтрансферазы, например ингибиторы фарнезилпротеинтрансферазы, описанные в литературе, процитированной в разделе «уровень техники» патента США № 6258824 B1.
Вышеописанный способ можно также осуществлять в комбинации с радиотерапией, в котором доза разработанного соединения в комбинации с радиотерапией является эффективной для лечения вышеуказанных заболеваний.
Способы применения радиотерапии известны в данной области, и эти способы могут использоваться при комбинированной терапии, описанной в данном документе. Способ введения в организм соединения согласно изобретению при такой комбинированной терапии может определяться согласно описанному в данном документе.
Следующие примеры приведены в целях пояснения, а не ограничения.
Пример 1
Синтез соединений
Получение 2-(4-(4-амино-2-метоксифенил)пиперазин-1-ил)этанола(анилин A)
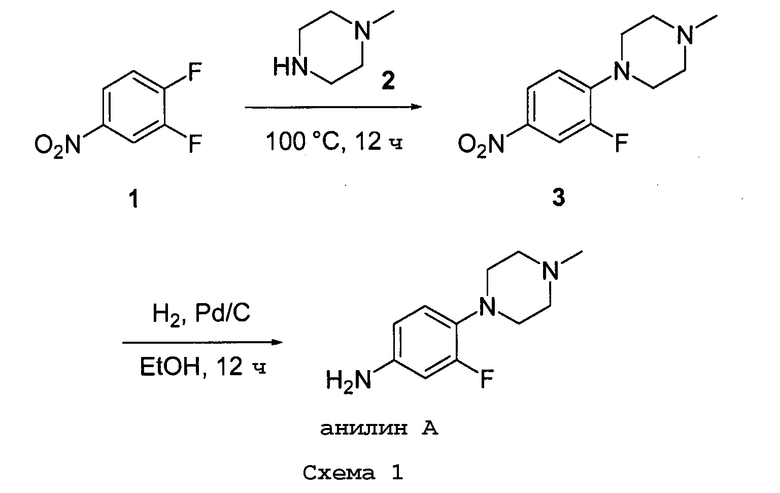
Смесь соединения 1 на схеме 1 (5,0 г, 31,4 ммоль) и соединения 2 на схеме 1 (70 мл, 628,0 ммоль) нагревали до 100°C в течение 12 ч в запаянной трубке. После того, как анализ TLC (ТСХ) показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (5×100 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения соединения 3 на схеме 1 (3,0 г, выход 62%) в виде желтого твердого вещества, которое использовали без дополнительно очистки. 1Н-ЯМР (400 МГц, CDCl3): δ 8,09–7,71 (м, 2H), 6,90 (т, J=8,8 Гц, 1H), 3,31 (дд, J=5,9, 3,9 Гц, 4H), 2,73-2,52 (м, 4H), 2,35 (с, 3H). Масс-спектр [ИЭС, MH+] = 240,15.
К соединению 3 на схеме 1 (1,0 г, 4,18 ммоль) в этаноле (10 мл) добавили Pd/C (10%, 200 мг) и перемешивали полученную смесь в атмосфере H2 (баллон давлением) в течение 12 ч. После того как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc (30 мл). Фильтрат высушили над Na2SO4, профильтровали и выпарили для получения анилина А (600 мг, выход 69%) в виде коричневого полутвердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,75 (дд, J=10,0, 8,3 Гц, 1H), 6,50-6,21 (м, 2H), 4,97 (с, 2H), 2,81 (т, J= 4,9 Гц, 4H), 2,41 (с, 4H), 2,19 (с, 3H). Масс-спектр [ИЭС, MH+] = 210,13.
Получение 3-метокси-4-(4-метилпиперазин-1-ил)анилина (анилин B):
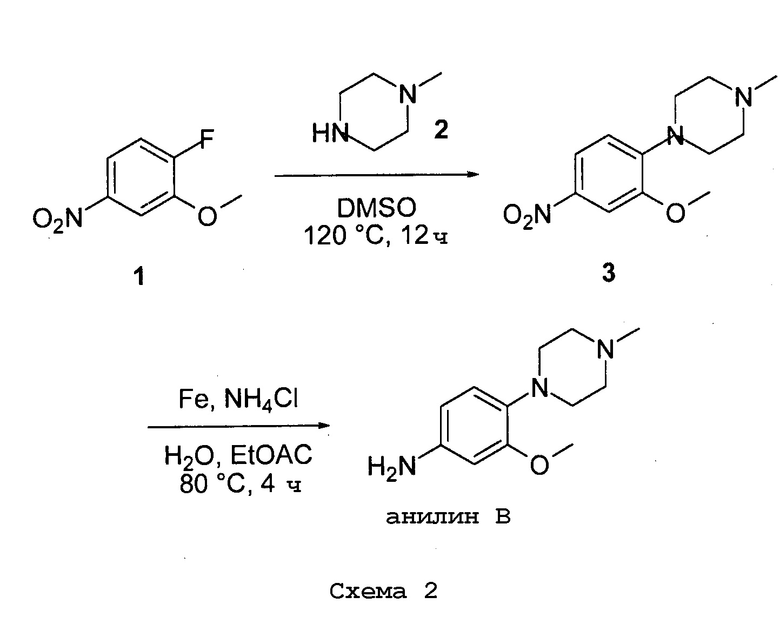
Смесь соединения 1 на схеме 2 (2,00 г, 11,68 ммоль) и соединения 2 на схеме 2 (1,17 г, 11,68 ммоль) в безводном ДМСО (ДМСО) (5 мл) нагрели до 120°C в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (5×50 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения соединения 3 на схеме 2 (2,00 г, выход 69%) в виде коричневого твердого вещества, которое использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3): δ 7,87 (дд, J=8,8, 2,6 Гц, 1H), 7,71 (д, J=2,5 Гц, 1H), 6,90 (д, J=8,8 Гц, 1H), 3,95 (с, 3H), 3,26 (д, J=4,9 Гц, 4H), 2,61 (т, J=4,9 Гц, 4H), 2,37 (с, 3H). Масс-спектр [ИЭС, MH+] = 252,13.
Смесь неочищенного соединения 3 на схеме 2 (1,00 г, 3,98 ммоль), Fe (0,89 г, 15,93 ммоль) и NH4Cl (2,70 г, 39,80 ммоль) в EtOAc/H2O (20 мл, 1/1) нагрели до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc (50 мл). Фильтрат выпарили при пониженном давлении для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 99/1, постепенно увеличивая до 80/20) для получения анилина B (0,70 г, выход 79%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,60 (дд, J=8,4, 2,0 Гц, 1H), 6,21 (д, J=2,3 Гц, 1H), 6,06 (м, 1H), 4,71 (с, 2H), 3,67 (д, J=2,0 Гц, 3H), 2,78 (с, 4H), 2,46-2,33 (м, 4H), 2,19 (д, J=2,1 Гц, 3H). Масс-спектр [ИЭС, MH+]=222,16.
Получение 2-(4-(4-амино-2-метоксифенил)пиперазин-1-ил)этанола (анилин C):
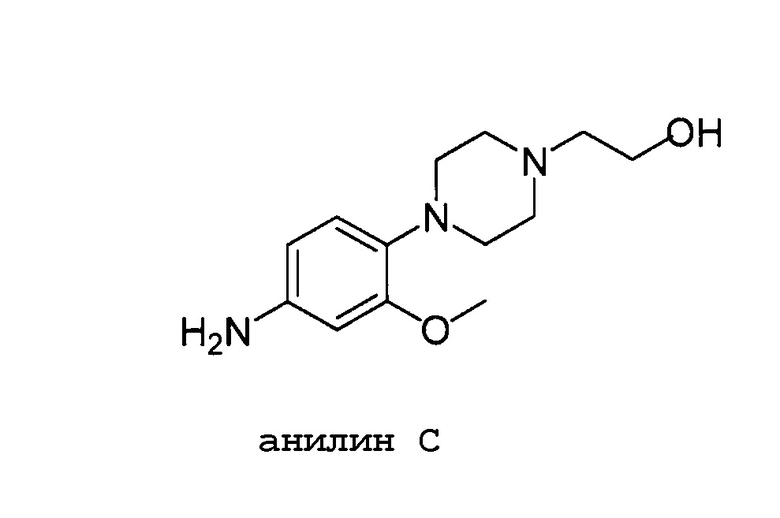
Указанное в заголовке соединение было синтезировано аналогично анилину B с использованием 2-(пиперазин-1-ил)этанола на первой стадии. Анилин C был получен в виде коричневого твердого вещества (5,0 г, выход 34% после 2 стадий). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,60 (д, J=8,3 Гц, 1H), 6,22 (д, J=2,4 Гц, 1H), 6,07 (дд, J=8,3, 2,4 Гц, 1H), 4,73 (с, 3H), 3,68 (с, 3H), 3,53 (д, J=27,0 Гц, 2H), 2,84 (с, 4H), 2,54 (с, 6H). Масс-спектр [ИЭС, MH+] = 252,17.
Получение 2-(4-(4-амино-2-метоксифенил)пиперазин-1-ил)этанола (анилин D):
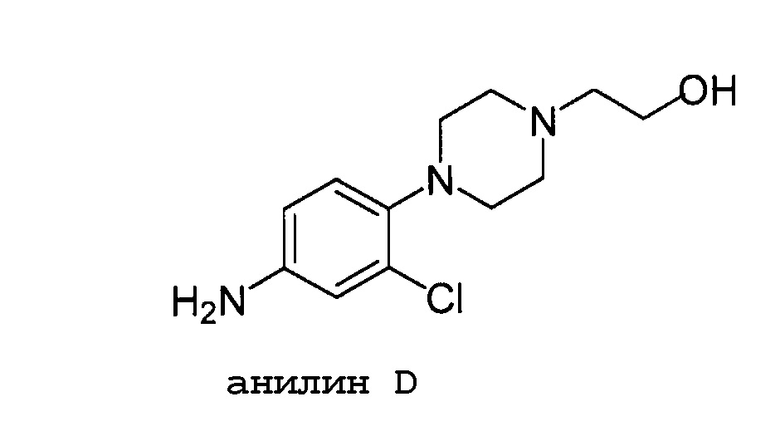
Указанное в заголовке соединение было синтезировано аналогично анилину B с использованием 2-хлор-1-фтор-4-нитробензола и 2-(пиперазин-1-ил)этанола на первой стадии. Анилин D был получен в виде коричневого твердого вещества (1,5 г, выход 51% после 2 стадий). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,91 (д, J= 8,6 Гц, 1H), 6,65 (д, J=2,5 Гц, 1H), 6,51 (дд, J=8,6, 2,5 ГЦ, 1H), 5,34 (с, 1H), 5,18 (с, 2H), 3,78 (д, J=5,0 Гц, 2H), 3,52 (с, 2H), 3,15 (д, J=46,2 Гц, 10H). Масс-спектр [ИЭС, MH+] = 256,12.
Получение 2-(3-((5-хлор-2-((3-фтор-4-(4-метилпиперазин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)ацетонитрила (Соединение 1)
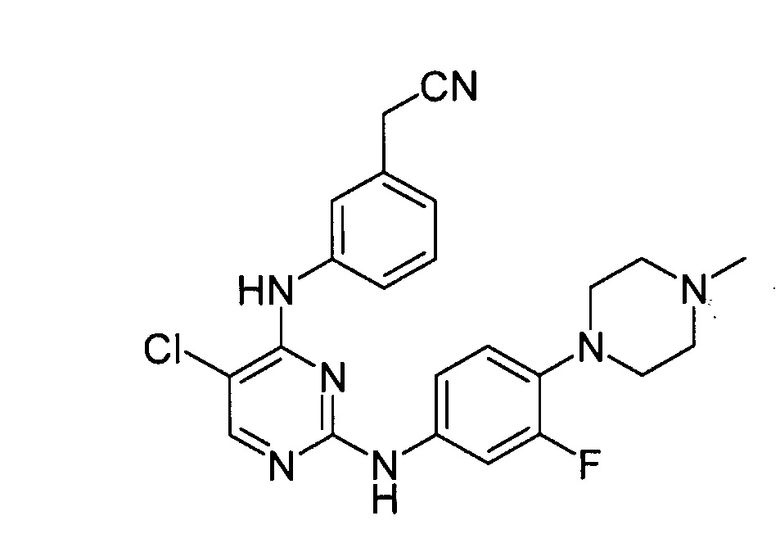
Указанное в заголовке соединение было синтезировано согласно отображенной на схеме 3 методике.
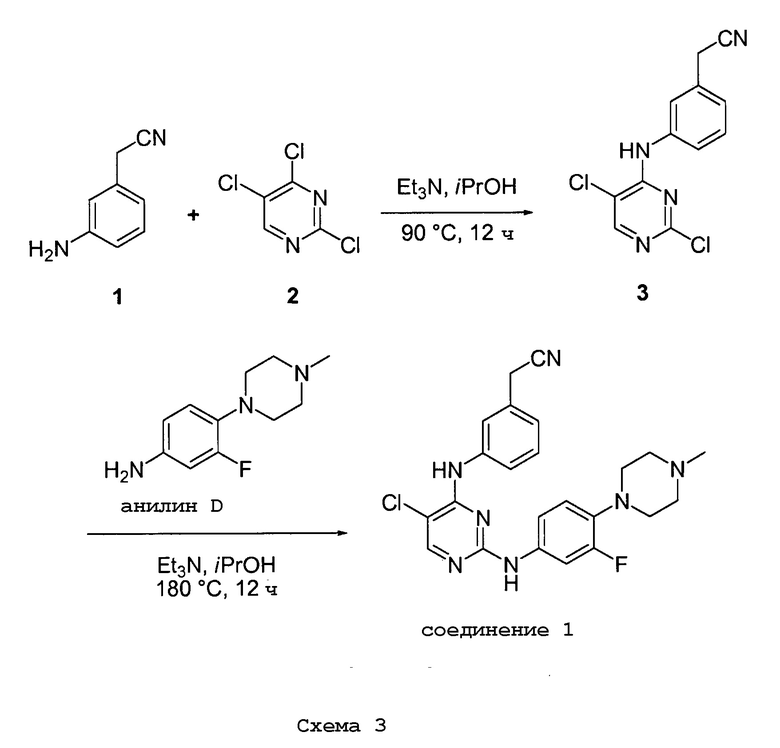
К смеси соединения 1 на схеме 3 (100 мг, 0,55 ммоль) и соединения 2 на схеме 3 (76 мг, 0,57 ммоль) в i-PrOH (4 мл) добавили Et3N (0,16 мл) и полученную смесь нагревали до 90°C в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, растворитель выпарили, а остаток очистили с помощью флэш-хроматографии на силикагеле для получения соединения 3 на схеме 3 (91 мг, выход 59%).
К смеси соединения 3 на схеме 3 (50 мг, 0,179 ммоль) и анилина А (49 мг, 0,234 ммоль) в i-PrOH (5 мл) добавили Et3N (0,5 мл) и полученную смесь нагревали до 90°C в течение 12 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, растворитель выпарили, а остаток очистили с помощью флэш-хроматографии на силикагеле, для получения соединения 1 (63 мг, выход 77%). Mасс-спектр [ИЭС, (M-CH3+H)+]=438,16.
Получение 2-(3-((2-((3-фтор-4-(4-метилпиперазин-1-ил)фенил)амино)-5-метилпиримидин-4-ил)амино)фенил)ацетонитрила (Соединение 2)
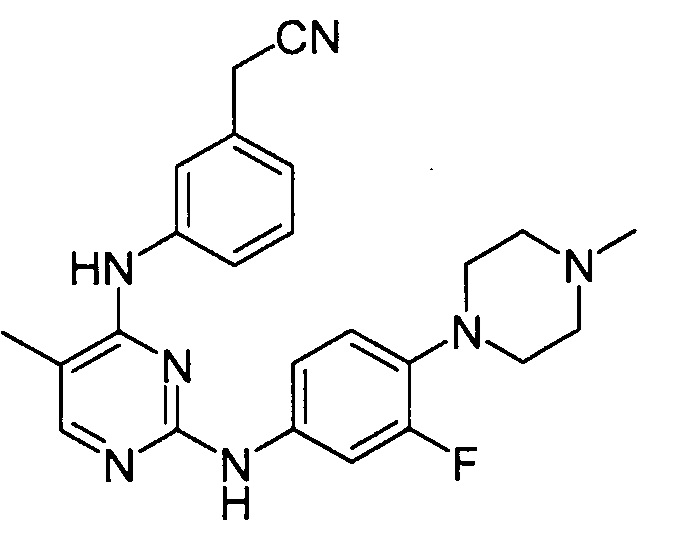
Указанное в заголовке соединение было синтезировано аналогично соединению 1 с использованием 2,4-дихлор-5-метилпиримидина на первой стадии сочетания. Соединение 2 было получено в виде белого твердого вещества (18 мг, выход 5% после 2 стадий). Mасс-спектр [ИЭС, (M-CH3+H)+]=418,29.
Получение 4-((3-(цианометил)фенил)амино)-2-((3-фтор-4-(4-метилпиперазин-1-ил)фенил)амино)пиримидин-5-карбонитрила (Соединение 3)
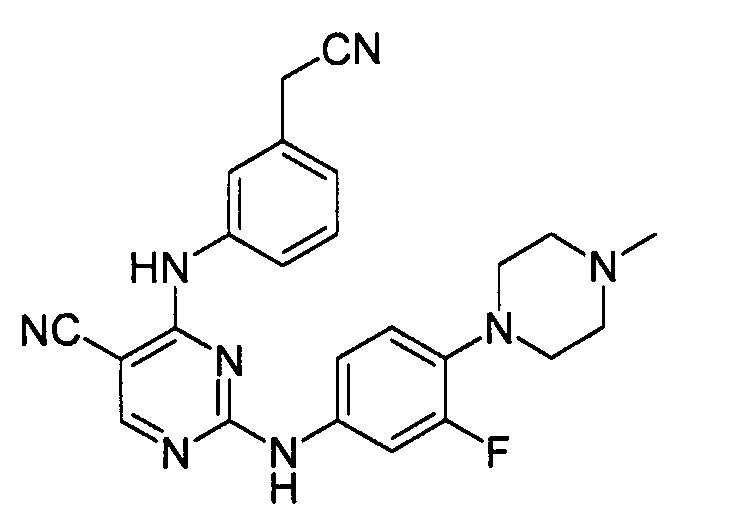
Указанное в заголовке соединение было синтезировано аналогично соединению 1 с использованием 2,4-дихлорпиримидин-5-карбонитрила на первой стадии сочетания и при снижении температуры для данной стадии до комнатной температуры. Соединение 3 было получено в виде белого твердого вещества (47 мг, выход 39% после 2 стадий). Mасс-спектр [ИЭС, (M-CH3+H)+]=429,21.
Получение 1-(3-(2-(3-метокси-4-(4-метилпиперазин-1-ил)фениламино)пиримидин-4-иламино)фенил)циклопропанкарбонитрила (Соединение 4)
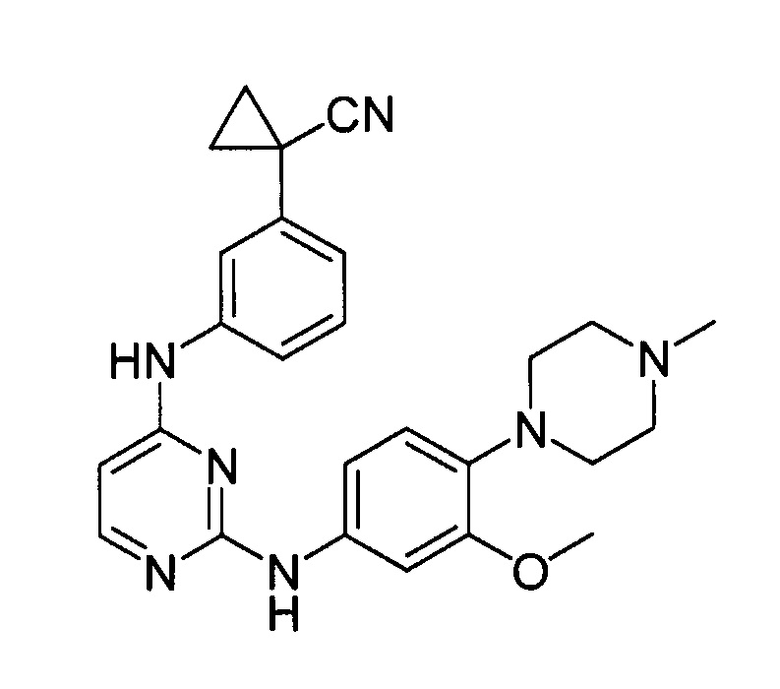
Указанное в заголовке соединение было синтезировано согласно отображенной на схеме 4 методике. Получение 1-(3-аминофенил)циклопропанкарбонитрила (соединения 1 на схеме 4) описано ниже на схеме 5.
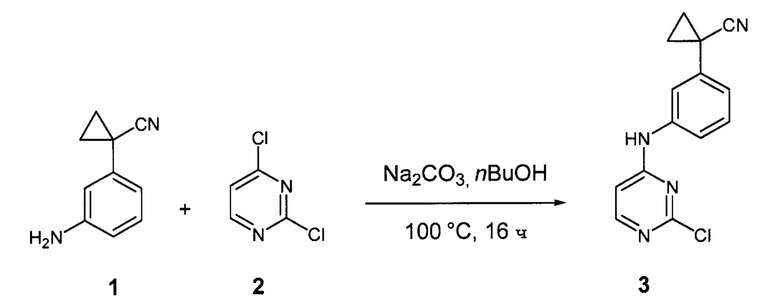
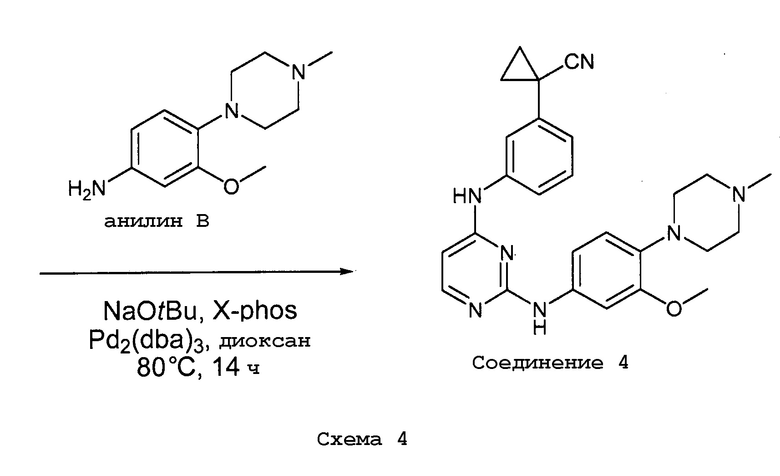
Смесь соединения 1 на схеме 4 (400 мг, 2,53 ммоль), соединения 2 на схеме 4 (450 мг, 3,03 ммоль) и Na2C03 (536 мг, 5,06 ммоль) в n-BuOH (5 мл) нагревали до 100°C в течение 16 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который растерли со смесью Et2O и н-пентана (20 мл, 1/4) для получения соединения 3 на схеме 4 (300 мг, выход 44%) в виде не совсем белого твердого вещества. . 1Н-ЯМР (400 МГц, ДМСО-d6): δ 10,15 (с, 1H), 8,19 (м, 1H), 7,78–7,47 (м, 2H) 7,49–7,23 (м, 1H), 7,14–6,92 (м, 1H), 6,77 (м, 1H), 1,79 (д, J=5,0 Гц, 2H), 1,51 (т, J=3,9 Гц, 2H). Масс-спектр [ИЭС, MH+] = 271,05.
Соединение 3 на схеме 4 (150 мг, 0,55 ммоль) и анилин B (200 мг, 0,83 ммоль) растворили в безводном диоксане (10 мл). К этой смеси в атмосфере азота добавили t-BuONa (160 мг, 1,66 ммоль), X-PHOS (64 мг, 0,11 ммоль) и Pd2(dba)3 (50 мг, 0,05 ммоль, 5% мол.). Реакционную смесь перемешивали при 80°C в течение 14 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, смесь погасили добавлением воды (10 мл) и подвергли экстракции этилацетатом (30 мл). Органический слой промыли рассолом (10 мл) и высушили над безводным Na2SO4, затем профильтровали, выпарили для получения остатка, который очистили с помощью препаративной HPLC для получения соединения 4 (70 мг, выход 28%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,07 (д, J=5,7 Гц, 1H), 7,43 (с, 1H), 7,40-7,27 (м, 2H), 7,16 (д, J=2,4 ГЦ, 1H), 7,06 (м, 1H), 6,98 (м, 1H), 6,94–6,86 (м, 2H), 6,58 (с, 1H), 6,13 (д, J=5,8 Гц, 1H), 3,80 (с, 3H), 3,07 (с, 4H), 2,63 (с, 4H), 2,36 (с, 3H), 1,72 (д, J=5,0 Гц, 2H), 1,38 (д, J=5,1 ГЦ, 2H). Масс-спектр [ИЭС, MH+] = 456,20.
1-(3-Аминофенил)циклопропанкарбонитрил (соединение 1 на схеме 4) был синтезирован как показано на схеме 5.
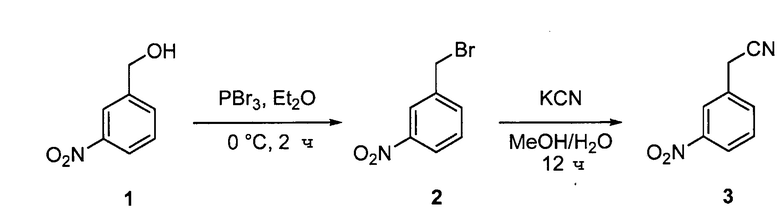
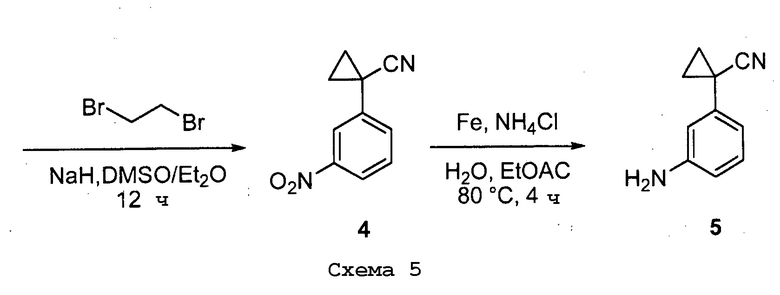
К холодному (0°C) раствору соединения 1 на схеме 5 (55,0 г, 359,4 ммоль) в Et2O (500 мл) добавили по каплям PBr3 (38,9 г, 270,7 ммоль) и реакционную смесь перемешивали 2 ч при 0°C. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, смесь разбавили водой (50 мл) и подвергли экстракции Et2O (2×200 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения соединения 2 на схеме 5 (55,0 г, выход 71%) в виде не совсем белого твердого вещества, которое использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3): δ 8,27 (т, J=2,0 Гц, 1H), 8,17 (дд, J=8,6, 2,0 Гц, 1H), 7,74 (м, 1H), 7,55 (т, J=8,0 Гц, 1H), 4,54 (с, 2H). Масс-спектр [ИЭС, MH+] = 215,96.
К раствору соединения 2 на схеме 5 (55,0 г, 254,6 ммоль) в смеси MeOH/вода (250 мл, 4/1) добавили KCN (21,5 г, 331,0 ммоль) и реакционную смесь перемешивали при комнатной температуре 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (50 мл) и подвергли экстракции EtOAc (200 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 40/60) для получения соединения 3 на схеме 5 (38,0 г, выход 91%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,31-8,12 (м, 2H), 7,78-7,69 (м, 1H), 7,68–7,51 (м, 1H), 3,91 (с, 2H). Масс-спектр [ИЭС, MH+] = 163,05.
К раствору NaH (5,41 г, 271,4 ммоль) в диметилсульфоксиде (20 мл) добавили по каплям смесь соединения 3 на схеме 5 (20,00 г, 123,0 ммоль) и 1,2-дибромэтана (23,08 г, 123,0 ммоль) в ДМСО/Et2O (60 мл, 1/2) и полученную смесь перемешивали при комнатной температуре в течение 1 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь погасили добавлением i-PrOH (20 мл), а затем воды (20 мл) и подвергли экстракции EtOAc (30 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 80/20) для получения соединения 4 на схеме 5 (13,0 г, выход 56%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,22–8,13 (м, 1H), 8,05 (т, J=2,2 Гц, 1H), 7,75 (д, J=7,9 Гц, 1H), 7,57 (т, J=8,0 Гц, 1H), 1,88 (д, J=2,7 Гц, 2H), 1,54-1,50 (м, 2H). Масс-спектр [ИЭС, MH+] = 189,04.
Смесь соединения 4 на схеме 5 (10,0 г, 52,6 ммоль), Zn пыли (13,7 г, 210,5 ммоль) и NH4Cl (28,1 г, 526,3 ммоль) в EtOAc/H2O (60 мл, 1/1) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc (200 мл). Фильтрат выпарили для получения остатка, который очистили с использованием флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 70/30) для получения соединения 5 на схеме 5 (6,0 г, выход 71%) в виде коричневой жидкости. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,99 (т, J=7,8 Гц, 1H), 6,57 (т, J=2,1 Гц, 1H), 6,47 (дд, J=8,0, 2,1 Гц, 1H), 6,39-6,34 (м, 1H), 5,21 (с, 2H), 1,66 (м, 2H), 1,36 (кв., J=4,7 ГЦ, 2H). Масс-спектр [ИЭС, MH+] = 159,09
Получение 1-(3-(2-(3,4,5-триметоксифениламино)пиримидин-4-иламино)фенил)циклопропанкарбонитрил (Соединение 5)
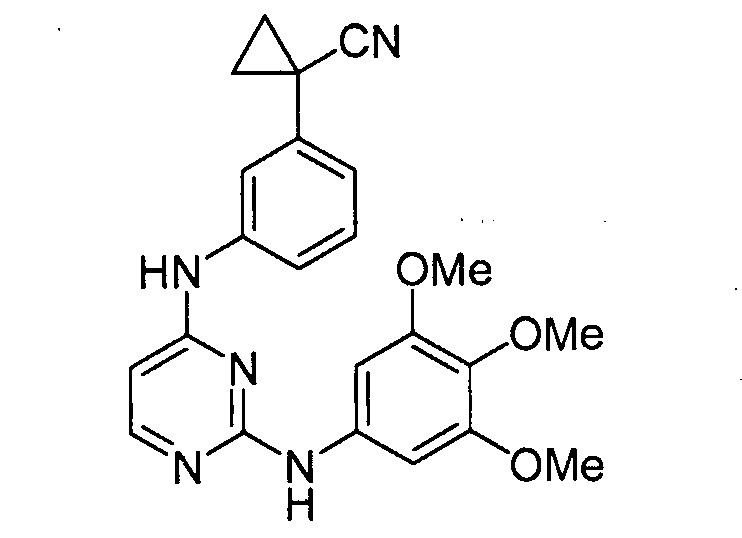
Указанное в заголовке соединение синтезировали аналогично соединению 4 c использованием 3,4,5-триметоксианилина на последней стадии сочетания.
Соединение 3 на схеме 4 (120 мг, 0,440 ммоль) и 3,4,5- триметоксианилин (122 мг, 0,660 ммоль) растворили в безводном диоксане (10 мл). К этой смеси под атмосферой азота добавили t-BuONa (126 мг, 1,320 ммоль), X-PHOS (50 мг, 0,088 ммоль) и Pd2(dba)3 (40 мг, 0,044 ммоль, 10% мол.). Реакционную смесь перемешивали при 80°C 14 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, смесь погасили добавлением воды (10 мл) и подвергли экстракции этилацетатом (30 мл). Органический слой промыли рассолом (10 мл) и высушили над безводным Na2SO4, затем профильтровали и выпарили для получения остатка, который очистили с помощью препаративной HPLC для получения соединения 5 (120 мг, выход 66%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,09 (д, J=5,7 Гц, 1H), 7,50 (с, 1H), 7,44–7,28 (м, 2H), 7,03–6,88 (м, 2H), 6,85 (с, 2H), 6,56 (с, 1H), 6,15 (д, J=5,7 Гц, 1H), 3,82 (д, J=7,9 Гц, 9H), 1,74 (д, J=5,1 Гц, 2H), 1,39 (д, J=5,0 Гц, 2H). Масс-спектр [ИЭС, MH+] – 418,15.
Получение 1-(3-(2-(4-(4-(2-гидроксиэтил)пиперазин-1-ил)-3-метоксифениламино)пиримидин-4-иламино)фенил)циклопропанкарбонитрила (Соединение 6)
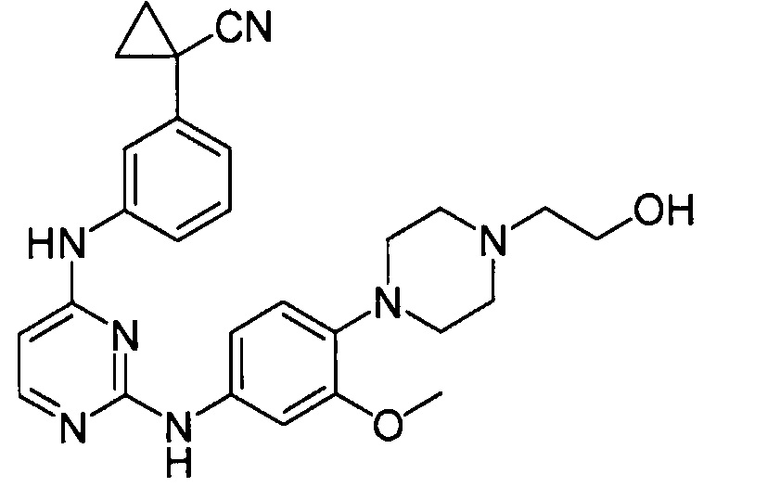
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 6.
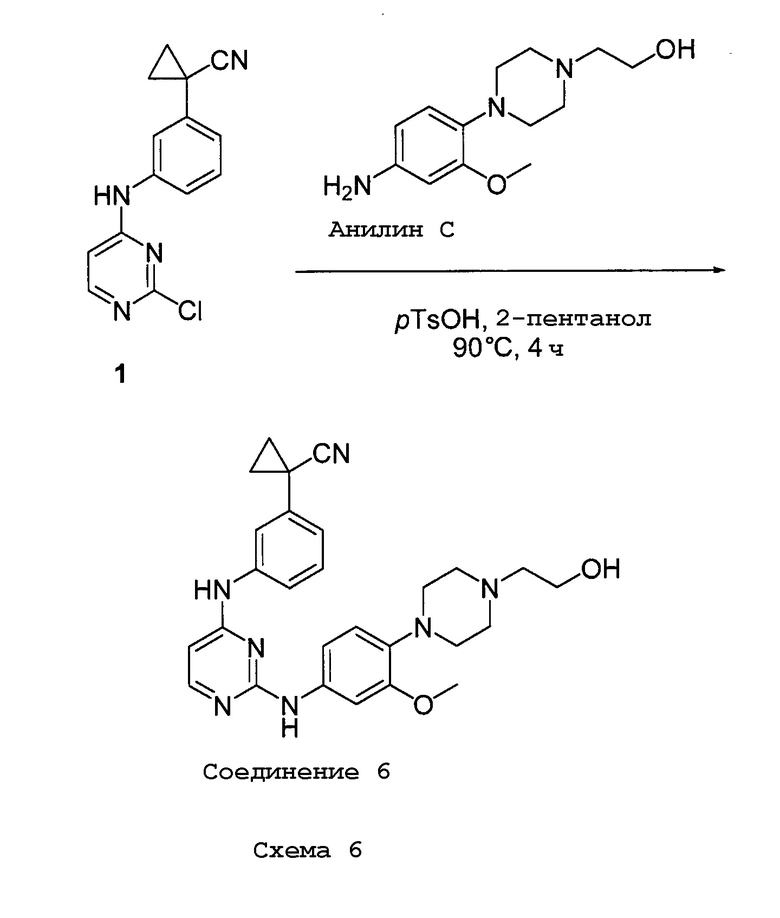
Смесь соединения 1 на схеме 6 (2,0 г, 7,38 ммоль), анилина C (2,2 г, 8,56 ммоль) и pTsOH (1,2 г, 7,38 ммоль) в 2-пентаноле (40 мл) нагревали до 90°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×50 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 6 (1,3 г, выход 36%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,94 (с, 1H), 8,00 (д, J=5,6 Гц, 1H), 7,90 (д, J=8,1 Гц, 1H), 7,40 (т, J=2,1 Гц, 1H), 7,32-7,19 (м, 3H), 6,91 (д, J=7,7 Гц, 1H), 6,78 (д, J=8,4 Гц, 1H), 6,18 (д, J=5,7 Гц, 1H), 4,40 (с, 1H), 3,65 (с, 3H), 3,53 (с, 2H), 2,91 (с, 4H), 2,46 (с, 4H), 1,70 (д, J=4,8 Гц, 2H), 1,42 (д, J=4,7 Гц, 2H). Масс-спектр [ИЭС, MH+] = 486,25.
Получение 1-(3-((2-((3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)циклопропанкарбонитрила (Соединение 7)
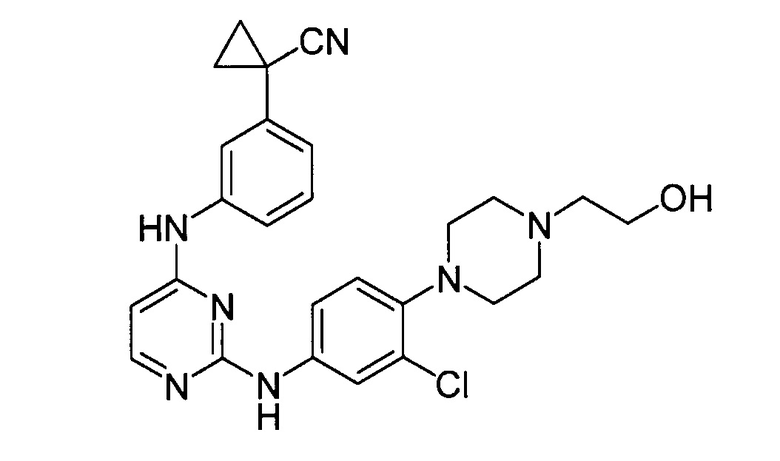
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 7.
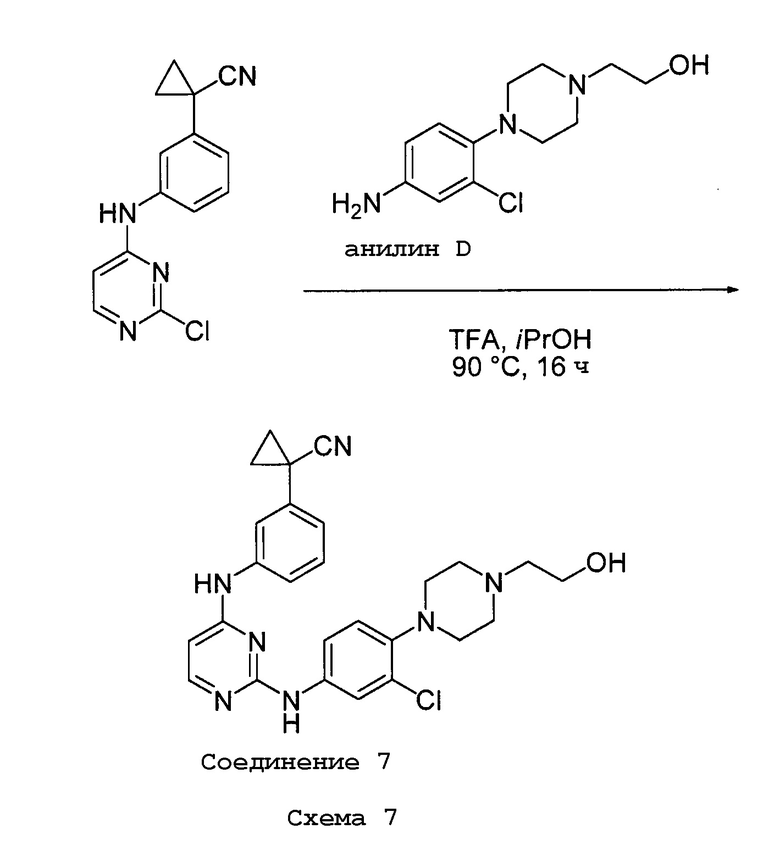
Смесь соединения 1 на схеме 7 (50,0 мг, 0,185 ммоль), анилина D (47,3 мг, 0,185 ммоль) и TFA (трифторуксусная кислота) (0,5 мл) в i-PrOH (10 мл) нагревали до 90°C в течение 16 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, растворитель удалили при пониженном давлении, а остаток очистили с помощью препаративной HPLC для получения соединения 7 (7,6 мг, выход 22%). 1Н-ЯМР (400 МГц, CD3OD): δ 8,31 (с, 2H), 7,95 (д, J=6,0 Гц, 1H), 7,81 (д, J=2,4 Гц, 1H), 7,66 (д, J=8,4 Гц, 1H), 7,49 (с, 1H), 7,46 (дд, J=8,8, 2,4 Гц, 1H), 7,35 (т, 1H), 7,12 (д, J=8,8 Гц, 1H), 7,05 (д, J=7,6 Гц, 1H), 6,24 (д, J=6,0 Гц, 1H), 3,92 (т, 2H), 3,45 (с, 4H), 3,28 (м, 6H), 1,67-1,64 (м, 2H), 1,42-1,38 (м, 2H). Масс-спектр [ИЭС, MH+] = 490,2.
Получение 1-(3-((2-((3-фтор-4-(4-метилпиперазин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)циклопропанкарбонитрила (Соединение 8)
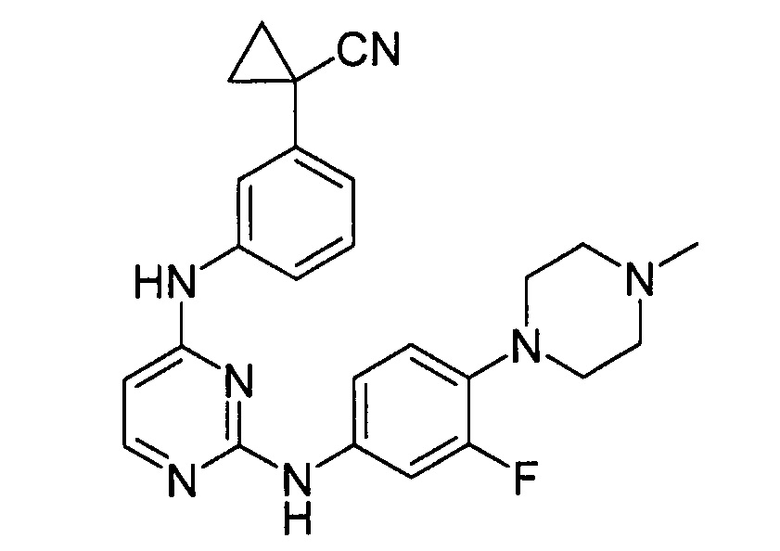
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 8.
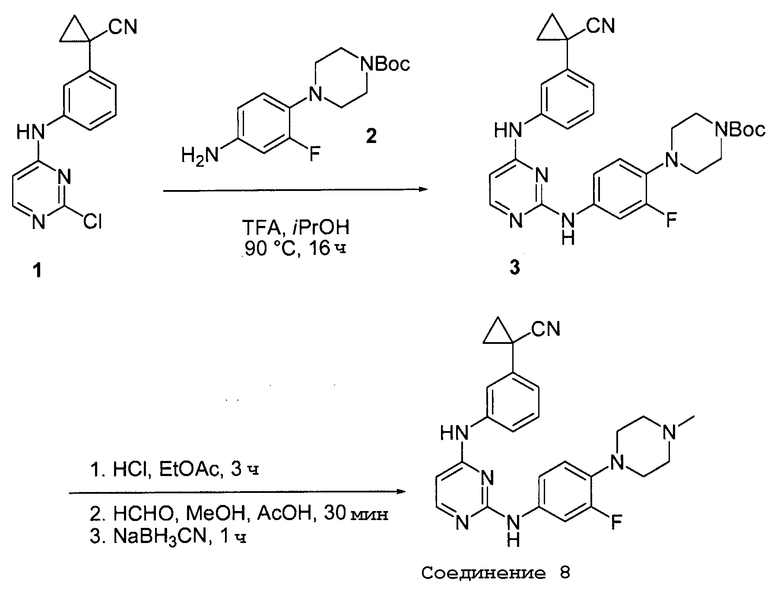
Схема 8
Смесь соединения 1 на схеме 8 (50,0 мг, 0,185 ммоль), соединения 2 на схеме 8 (54,6 мг, 0,185 ммоль) и TFA (0,5 мл) в i-PrOH (10 мл) нагревали до 90°C в течение 16 ч. После того, как анализ LCMS (ЖХМС) показал, что реакция завершилась, растворитель удалили при пониженном давлении для получения соединения 3 на схеме 8 (100 мг), которое использовали без дополнительной очистки. Масс-спектр [ИЭС, MH+]=530,0.
К раствору соединения 3 на схеме 8 (100,0 мг, 0,19 ммоль) в EtOAc (5 мл) добавили HCl в EtOAc (10%, 5 мл) и смесь перемешивали при комнатной температуре в течение 3 ч. После того, как анализ LCMS показал, что реакция завершилась, растворитель выпарили, остаток растворили в MeOH (5 мл) и добавили HCHO (5,7 мг, 0,19 ммоль) и AcOH (0,05 мл). Смесь перемешивали при комнатной температуре 30 мин по истечение которых добавили NaBH3CN (15,7 мг, 0,25 ммоль) и продолжили перемешивание при комнатной температуре в течение 1 ч. После того, как анализ LCMS показал, что реакция завершилась, растворитель выпарили для получения остатка, который очистили с помощью препаративной HPLC для получения соединения 8 (7,9 мг, выход 18%).1Н-ЯМР (400 МГц, CD3OD): δ м.д.: 8,30 (с, 2H), 7,95 (д, J=6,0 Гц, 1H), 7,63–7,57 (м, 2H), 7,52 (с, 1H), 7,34 (т, 1H), 7,24 (д, J=8,0 Гц, 1H), 7,05-6,98 (м, 2H), 6,23 (д, J=6,0 Гц, 1H), 3,30 (м, 8H), 2,87 (с, 3H), 1,68-1,64 (м, 2H), 1,43-1,39 (м, 2H). Масс-спектр [ИЭС, MH+] = 444,2
Получение 1-(3-(2-(3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)-5-метоксипиримидин-4-иламино)фенил)циклопропанкарбонитрила (Соединение 9)
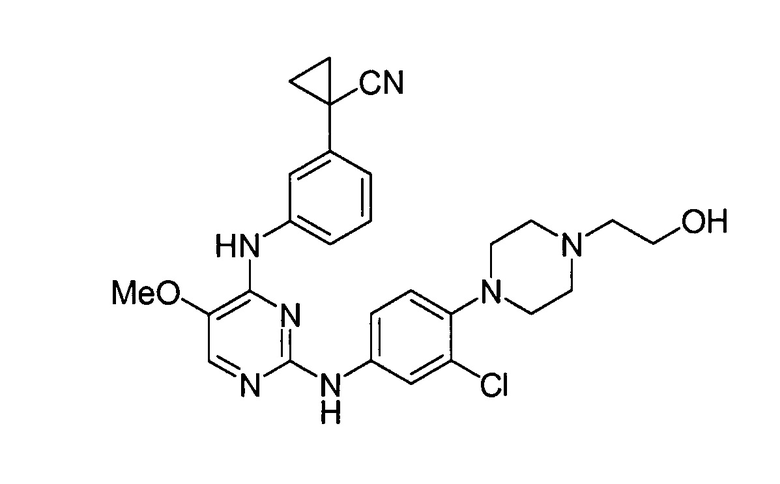
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 9.
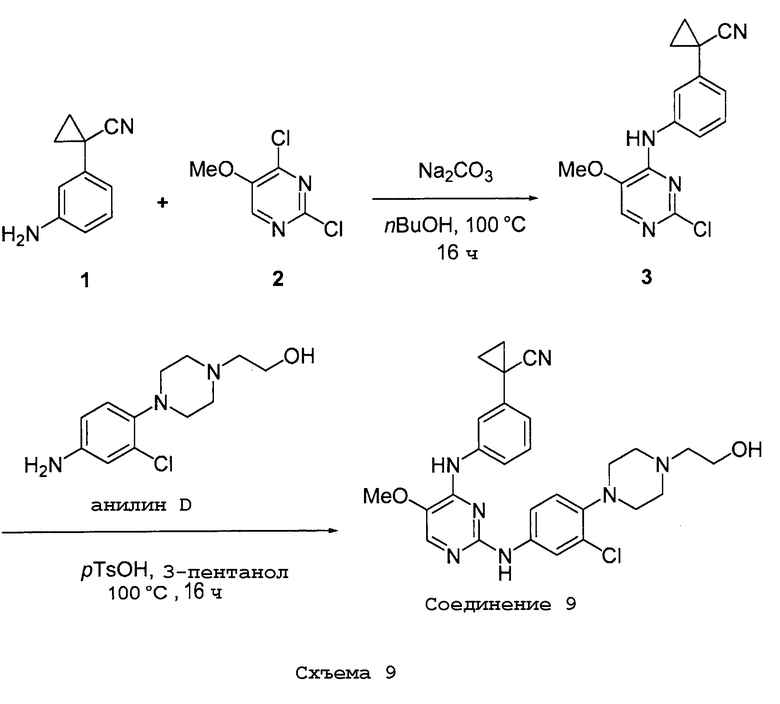
Смесь соединения 1 на схеме 9 (400 мг, 2,53 ммоль), соединения 2 на схеме 9 (678 мг, 3,79 ммоль) и Na2CO3 (804 мг, 7,59 ммоль) в n-BuOH (10 мл) нагревали до 100°C в течение 16 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 3 на схеме 9 (180 мг, выход 27%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ 8,96 (д, J=8,6 Гц, 1H), 8,72–8,63 (м, 1H), 8,51 (д, J=8,2 Гц, 1H), 7,96 (м, 2H), 7,42 (м, 1H), 7,28 (д, J=2,3 Гц, 1H), 7,18 (д, J=8,5 Гц, 1H), 7,08 (м, 1H), 6,95 (д, J=8,6 Гц, 1H), 6,28 (д, J=5,9 Гц, 1H), 3,79 (с, 3H), 3,15 (с, 4H), 2,93 (с, 4H), 2,56 (д, J=8,0 Гц, 6H). Масс-спектр [ИЭС, MH+] = 301,05.
Смесь соединения 3 на схеме 9 (180 мг, 0,59 ммоль), анилина D (183 мг, 0,72 ммоль) и pTsOH (100 мг, 0,59 ммоль) в 3-пентаноле (10 мл) нагревали до 100°C в течение 16 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 9 (100 мг, выход 32%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,96 (с, 1H), 8,85 (с, 1H), 8,02 (д, J=8,0 Гц, 1H), 7,88 (д, J=2,4 Гц, 2H), 7,54 (т, J=2,0 Гц, 1H), 7,48 (м, 1H), 7,34 (т, J=7,9 Гц, 1H), 7,05-6,97 (м, 2H), 4,41 (т, J=5,3 Гц, 1H), 3,86 (с, 3H), 3,52 (д, J=6,0 Гц, 2H), 2,89 (т, J=4,6 Гц, 4H), 2,56 (с, 4H), 2,44 (т, J=6,2 Гц, 2H), 1,72 (д, J=4,9 Гц, 2H), 1,58-1,30 (м, 2H). Масс-спектр [ИЭС, MH+] = 520,12.
Получение 2-(4-(4-(4-(2,2’-бипиридин-3-иламино)пиримидин-2-иламино)-2-хлорфенил)пиперазин-1-ил)этанола (Соединение 10)
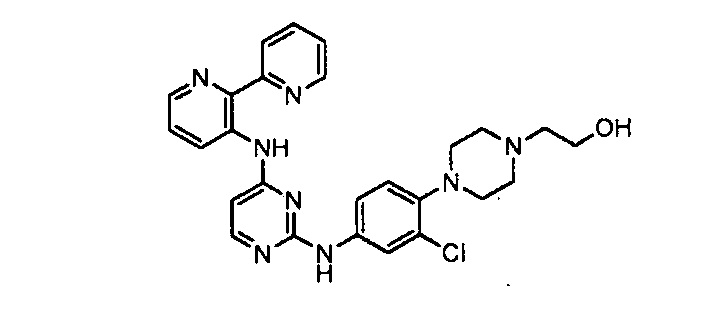
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 10. Получение 2,2’-бипиридин-3-амина (соединение 1 на схеме 10) описано ниже на схеме 11.
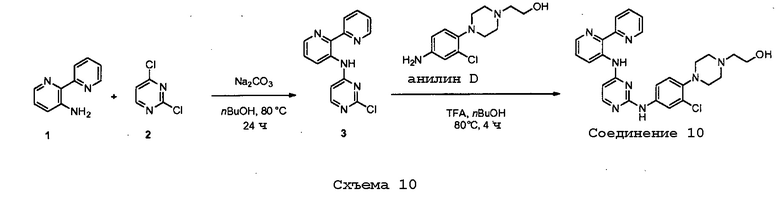
Смесь соединения 1 на схеме 10 (400 мг, 2,33 ммоль), соединения 2 на схеме 10 (410 мг, 2,80 ммоль) и Na2CO3 (500 мг, 4,68 ммоль) в n-BuOH (5 мл) нагревали до 80°C в течение 24 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 80/20) для получения соединения 3 на схеме 10 (320 мг, выход 48%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 13,16 (с, 1H), 8,86 (дд, J=8,4, 1,6 ГЦ, 1H), 8,82-8,76 (м, 1H), 8,53 (д, J=8,2 Гц, 1H), 8,48-8,40 (м, 1H), 8,29 (д, J=5,8 ГЦ, 1H), 8,07 (м, 1H), 7,65-7,49 (м, 2H), 7,13 (д, J=5,9 Гц, 1H). Масс-спектр [ИЭС, MH+] = 284,07.
Смесь соединения 3 на схеме 10 (150 мг, 0,53 ммоль), анилина D (148 мг, 0,58 ммоль) и TFA (2 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH = 100/0, постепенно увеличивая до 90/10) для получения соединения 10 (50 мг, выход 19%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 12,88 (с, 1H), 9,39 (с, 1H), 9,14 (с, 1H), 8,88–8,73 (м, 1H), 8,58 (д, J=8,2 Гц, 1H), 8,37 (дд, J=4,4, 1,5 Гц, 1H), 8,15 (д, J=5,6 Гц, 1H), 8,09-8,00 (м, 1H), 7,95 (д, 7=2,3 Гц, 1H), 7,61-7,38 (м, 3H), 7,11 (д, J=8,7 Гц, 1H), 6,50 (д, J=5,7 Гц, 1H), 5,76 (с, 1H), 4,43 (т, J=5,3 Гц, 1H), 3,53 (д, J=6,1 Гц, 2H), 2,93 (с, 4H), 2,58 (с, 4H), 2,47 (д, J=9,4 Гц, 2H). Масс-спектр [ИЭС, MH+] = 503,10.
2,2’-Бипиридин-3-амин (соединение 1 на схеме 10) синтезировали как показано на схеме 11.
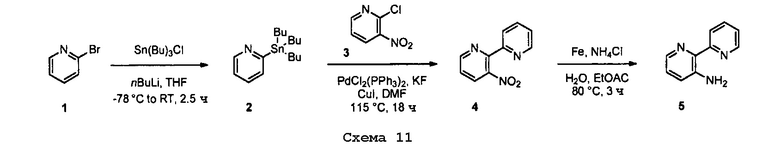
К холодному (-78°C) раствору соединения 1 на схеме 11 (2,00 г, 12,7 ммоль) в безводном THF (ТГФ) (20 мл) добавили nBuLi (1,6 М в гексане, 7,9 мл, 12,7 ммоль). После 30 минут перемешивания добавили по каплям хлорид трибутилолова (4,14 г, 12,7 ммоль) и полученный раствор перемешивали 1 ч при -78°C, по истечении которого ему дали нагреться до комнатной температуры и перемешивали еще 1 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь погасили водным NH4Cl (20 мл) и подвергли экстракции Et2O (50 мл), после чего высушили над Na2SO4 и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 80/20) для получения соединения 2 на схеме 11 (3,20 г, выход 68%) в виде коричневой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ 8,73 (д, J=4,7 Гц, 1H), 7,49 (м, 1H), 7,40 (д, J=7,6 Гц, 1H), 7,16-7,04 (м, 1H), 1,56 (т, J=7,7 Гц, 6H), 1,33 (д, J=7,3 Гц, 6H), 1,18–1,08 (м, 6H), 0,88 (т, J=7,3 ГЦ, 9H). Масс-спектр [ИЭС, MH+] = 370,12.
К перемешиваемому дегазированному раствору соединения 2 на схеме 11 (1,56 г, 4,23 ммоль), соединения 3 на схеме 11 (600 мг, 3,84 ммоль) и CuI (7,3 мг, 0,038 ммоль) в безводном DMF (ДМФА) (20 мл) добавили PdCl2(PPh3)3 (27 мг, 0,04 ммоль) и полученную смесь нагревали до 115°C в течение 18 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь охладили до комнатной температуры, погасили 1 Н водным KF (6 мл) и перемешивали в течение 30 мин. Твердую фазу удалили путем фильтрации через подушку из целита и промыли с Et2O (20 мл). Фильтрат выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 50/50) для получения соединения 4 на схеме 11 (400 мг, выход 52%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,84 (м, 1H), 8,70–8,52 (м, 1H), 8,08 (м, 2H), 7,89 (м, 1H), 7,49 (м, 1H), 7,37 (м, 1H). Масс-спектр [ИЭС, MH+] = 202,06.
Соединение 4 на схеме 11 (400 мг, 1,99 ммоль), Fe (445 мг, 7,96 ммоль) и NH4Cl (1,06 г, 19,90 ммоль) было растворили в смеси EtOAc/вода (20 мл, 1/1) и перемешивали при 80°C 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь охладили до комнатной температуры и профильтровали через подушку из целита. Твердые вещества промыли EtOAc (20 мл) и фильтрат выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 0/100) для получения соединения 5 на схеме 11 (260 мг, выход 77%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,65–8,58 (м, 1H), 8,44 (д, J=8,2 Гц, 1H), 7,91 (д, J=3,9 Гц, 2H), 7,40–7,28 (м, 1H), 7,23 (с, 2H), 7,15 (м, 2H). Масс-спектр [ИЭС, MH+] = 172,08.
Получение N4-(2,2’-бипиридин-3-ил)-N2-(3-метокси-4-(4-метилпиперазин-1-ил)фенил)пиримидин-2,4-диамина (Соединение 11)
Указанное в заголовке соединение синтезировали аналогично соединению 10 с использованием 3-метокси-4-(4-метилпиперазин-1-ил)анилина на последней стадии сочетания.
Смесь соединения 3 на схеме 10 (150 мг, 0,543 ммоль), анилина B (160 мг, 0,630 ммоль) и TFA (1,5 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 11 (60 мг, выход 24%) в виде не достаточно белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6):δ 12,87 (с, 1H), 9,24 (д, J=7,5 Гц, 1H), 9,18 (с, 1H), 8,79 (д, J=4,4 Гц, 1H), 8,58 (д, J=8,3 Гц, 1H), 8,36 (дд, J=4,4, 1,6 Гц, 1H), 8,12 (д, J=5,7 Гц, 1H), 8,04–8,08 (м, 1H), 7,55 (м, 1H), 7,40–7,30 (м, 1H), 7,27 (д, J=8,8 Гц, 1H), 6,84 (д, J=8,3 Гц, 1H), 6,45 (д, J=5,7 Гц, 1H), 3,72 (с, 3H), 2,98 (м, 4H), 2,67-2,55 (м, 4H), 2,36 (с, 3H). Масс-спектр [ИЭС, MH+] = 470,25.
Получение N4-(2,2’-бипиридин-3-ил)-N2-(3,4,5-триметоксифенил)пиримидин-2,4-диамина (Соединение 12)
Указанное в заголовке соединение синтезировали аналогично соединению 10 с использованием 3,4,5-триметоксианилина на последней стадии сочетания.
Смесь соединения 3 на схеме 10 (150 мг, 0,53 ммоль), 3,4,5-триметоксианилина (116 мг, 0,630 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 12 (50 мг, выход 22%) в виде не достаточно белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 12,91 (с, 1H), 9,25 (д, J=12,3 Гц, 2H), 8,79 (дд, J=4,9, 1,8 Гц, 1H), 8,58 (д, J=8,1 Гц, 1H), 8,36 (дд, J=4,4, 1,6 Гц, 1H), 8,15 (д, J=5,7 Гц, 1H), 8,06 (м, 1H), 7,55 (м, 1H), 7,40 (дд, J=8,6, 4,4 Гц, 1H), 7,13 (с, 2H), 6,49 (д, J=5,7 Гц, 1H), 3,72 (с, 6H), 3,63 (с, 3H). Масс-спектр [ИЭС, MH+] = 430,18.
Получение 2-(4-(4-(4-(2,2’-бипиридин-3-иламино)-5-метоксипиримидин-2-иламино)-2-хлорфенил)пиперазин-1-ил)этанола (Соединение 13)
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 12.
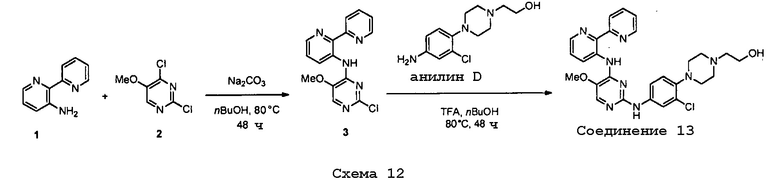
Смесь соединения 1 на схеме 12 (400 мг, 2,33 ммоль), соединения 2 на схеме 12 (502 мг, 2,80 ммоль), и Na2CO3 (493 мг, 4,66 ммоль) в n-BuOH (5 мл) нагревали до 80°C в течение 48 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 80/20) для получения соединения 3 на схеме 12 (200 мг, выход 27%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 13,95 (с, 1H), 9,20 (д, J=8,4 Гц, 1H), 8,79 (д, J=4,7 Гц, 1H), 8,60 (д, J=8,1 Гц, 1H), 8,43 (д, J=4,4 Гц, 1H), 8,08 (д, J=6,8 Гц, 2H), 7,57 (дд, J=8,0, 4,6 Гц, 2H), 4,10 (с, 3H). Масс-спектр [ИЭС, MH+] = 314,08.
Смесь соединения 3 на схеме 12 (200 мг, 0,63 ммоль), анилина D (178 мг, 0,70 ммоль) и TFA (2 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 48 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 13 (100 мг, выход 30%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 13,48 (с, 1H), 9,48 (м, 1H), 9,17 (с, 1H), 8,95–8,65 (м, 1H), 8,70-8,48 (м, 1H), 8,38 (м, 1H), 8,08 (м, 1H), 7,99 (д, J=2,5 Гц, 1H), 7,97 (с, 1H), 7,55 (м, 1H), 7,48 (м, 2H), 7,10 (д, J=8,8 Гц, 1H), 4,43 (т, J=5,3 Гц, 1H), 4,00 (с, 3H), 3,53 (д, J=6,0 Гц, 2H), 2,92 (т, J=4,5 Гц, 4H), 2,56 (д, J=15,6 Гц, 4H), 2,45 (т, J=6,3 ГЦ, 2H). Масс-спектр [ИЭС, MH+] = 533,21.
Получение N4-([2,2’-бипиридин]-3-ил)-5-метокси-N2-(3-метокси-4-(4-метилпиперазин-1-ил)фенил)пиримидин-2,4-диамина (Соединение 14)
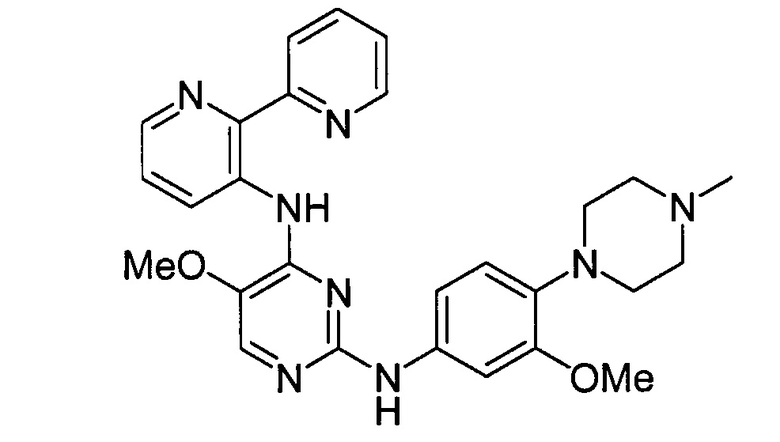
Указанное в заголовке соединение синтезировали аналогично соединению 13 с использованием анилина B на последней стадии сочетания.
Смесь соединения 3 на схеме 12 (130 мг, 0,41 ммоль), анилина B (125 мг, 0,49 ммоль) и TFA (1,5 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 14 (40 мг, выход 20%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 13,45 (с, 1H), 9,61-9,47 (м, 1H), 8,92 (с, 1H), 8,82–8,71 (м, 1H), 8,60 (д, J=8,2 Гц, 1H), 8,37 (м, 1H), 8,07 (м, 1H), 7,93 (с, 1H), 7,55 (м, 1H), 7,43 (м, 1H), 7,34 (д, J=2,4 Гц, 1H), 7,26 (м, 1H), 6,81 (д, J=8,5 Гц, 1H), 3,99 (с, 3H), 3,74 (с, 3H), 2,92 (с, 4H), 2,46 (с, 4H), 2,22 (с, 3H). Масс-спектр [ИЭС, MH+] = 499,13.
Получение 2-(4-(2-хлор-4-(4-(6-метил-2,2’-бипиридин-3-иламино)пиримидин-2-иламино)фенил)пиперазин-1-ил)этанола (Соединение 15)
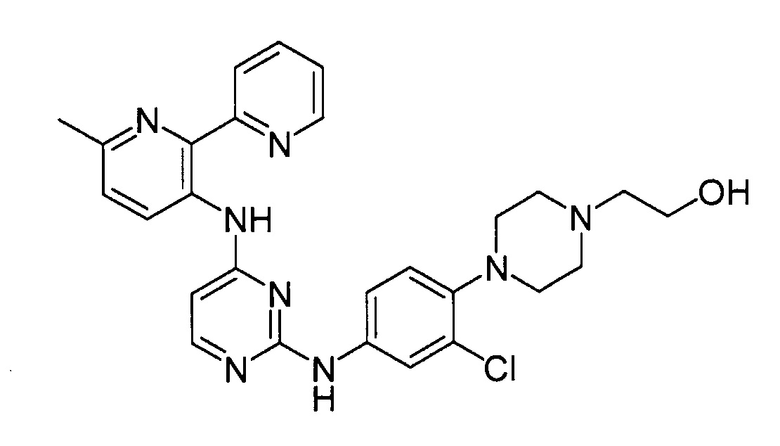
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 13. Получение 6-метил-2,2’бипиридин-3-амина (соединение 1 на схеме 13) описано ниже.
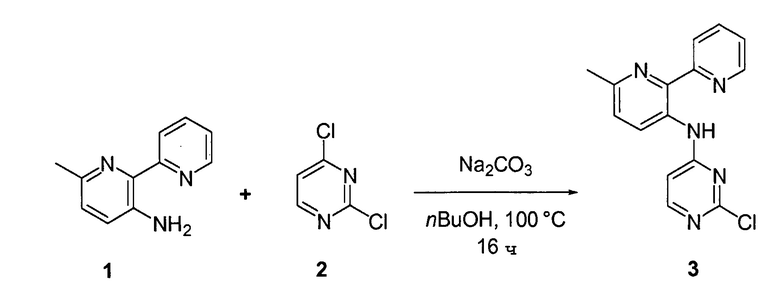
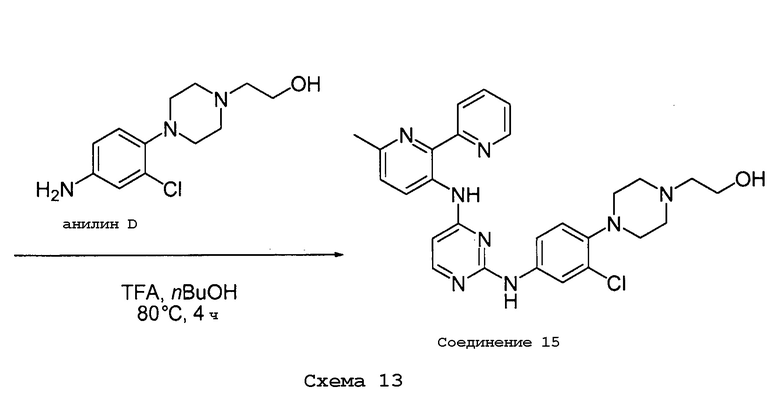
Смесь соединения 1 на схеме 13 (400 мг, 2,18 ммоль), соединения 2 на схеме 13 (488 мг, 3,27 ммоль) и Na2CO3 (462 мг, 4,36 ммоль) в n-BuOH (10 мл) нагревали до 100°C в течение 16 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения, соединения 3 на схеме 13 (230 мг, выход 35%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 12,88 (с, 1H), 8,75 (м, 1H), 8,69 (д, J=8,5 Гц, 1H), 8,49 (м, 1H), 8,24 (д, J=5,8 ГЦ, 1H), 8,05 (м, 1H), 7,52 (м, 1H), 7,40 (д, J=8,6 Гц, 1H), 7,07 (д, J=5,9 ГЦ, 1H), 2,54 (с, 3H). Масс-спектр [ИЭС, MH+] = 298,08.
Смесь соединения 3 на схеме 13 (200 мг, 0,67 ммоль), анилина D (188 мг, 0,74 ммоль) и TFA (2 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 15 (50 мг, выход 15%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 12,62 (с, 1H), 9,35 (с, 1H), 8,97 (д, J=8,4 Гц, 1H), 8,77 (д, J=4,7 Гц, 1H), 8,55 (д, J=8,2 ГЦ, 1H), 8,11 (д, J=5,7 Гц, 1H), 8,04 (т, J=7,7 Гц, 1H), 7,96 (д, J=2,6 Гц, 1H), 7,51 (м, 2H), 7,32 (д, J=8,6 Гц, 1H), 7,10 (д,J=8,7 Гц, 1H), 6,45 (д, J=5,8 Гц, 1H), 4,44 (т, J=5,4 Гц, 1H), 3,54 (д, J=6,0 Гц, 2H), 2,93 (с, 4H), 2,56 (д, J=16,1 Гц, 7H), 2,46 (т, J=9,0 Гц, 2H). Масс-спектр [ИЭС, MH+] = 517,08.
6-Метил-2,2'-бипиридин-3-амин (соединение 1 на схеме 13) синтезировали аналогично соединению 5 на схеме 11 с использованием 2-хлор-6-метил-3-нитропиридина на второй стадии и выделили в виде коричневого твердого вещества (700 мг, выход 32% после 2 стадий, начиная от соединения 2 на схеме 11). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,76–8,66 (м, 1H), 8,65-8,55 (м, 1H), 8,44 (д, J=8,2 Гц, 1H), 8,40 (д, J=8,1 Гц, 1H), 7,99–7,92 (м, 1H), 7,92-7,86 (м, 1H), 7,47 (м , 1H), 7,35-7,27 (м, 1H), 7,11 (д, J=8,3 Гц, 1H), 7,05 (с, 2H), 7,01 (д, J=8,4 Гц, 1H), 2,38 (с, 3H). Масс-спектр [ИЭС, MH+] = 186,09.
Получение N2-(3-метокси-4-(4-метилпиперазин-1-ил)фенил)-N4-(6-метил-2,2’-бипиридин-3-ил)пиримидин-2,4-диамина (Соединение 16)
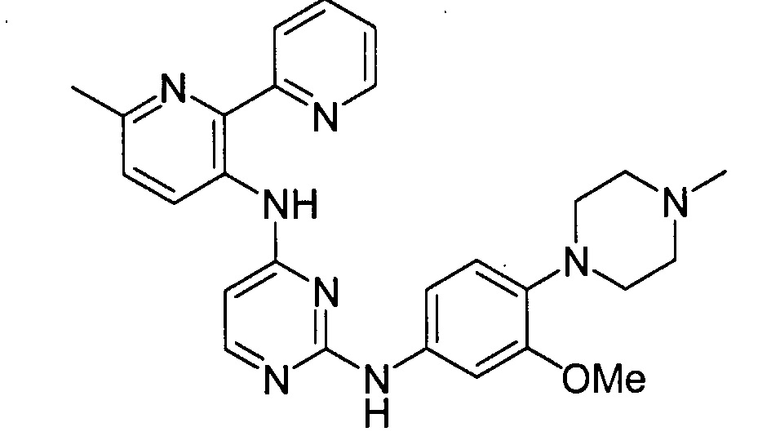
Указанное в заголовке соединение синтезировали аналогично соединению 15 с использованием анилина B на последней стадии сочетания.
Смесь соединения 3 на схеме 13 (90 мг, 0,30 ммоль), анилина B (114 мг, 0,45 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 16 (35 мг, выход 24%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ 8,96 (д, J=8,6 Гц, 1H), 8,72–8,63 (м, 1H), 8,51 (д, J=8,2 Гц, 1H), 7,96 (м, 2H), 7,42 5 (м, 1H), 7,28 (д, J=2,3 Гц, 1H), 7,18 (д, J=8,5 Гц, 1H), 7,08 (м, 1H), 6,95 (д, J=8,6 Гц, 1H), 6,28 (д, J=5,9 Гц, 1H), 3,79 (с, 3H), 3,15 (с, 4H), 2,93 (с, 4H), 2,56 (д, J=8,0 Гц, 6H). Масс-спектр [ИЭС, MH+] = 482,01.
Получение N4-(6-метил-2,2’-бипиридин-3-ил)-N2-(3,4,5-триметоксифенил)пиримидин-2,4-диамина (Соединение 17)
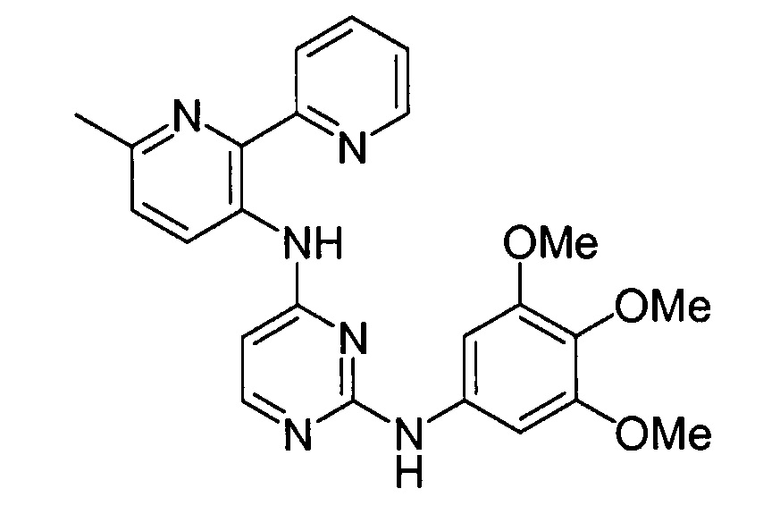
Указанное в заголовке соединение синтезировали аналогично соединению 15 с использованием 3,4,5-триметоксианилина на последней стадии сочетания.
Смесь соединения 3 на схеме 13 (90 мг, 0,30 ммоль), 3,4,5-триметоксианилина (55 мг, 0,30 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 6 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 17 (24 мг, выход 19%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 12,67 (с, 1H), 9,19 (с, 1H), 9,07 (д, J=8,4 Гц, 1H), 8,81–8,68 (м, 1H), 8,56 (д, J=8,1 Гц, 1H), 8,12 (д, J=5,7 Гц, 1H), 8,07–7,98 (м, 1H), 7,58–7,44 (м, 1H), 7,26 (д, J=8,6 Гц, 1H), 7,12 (с, 2H), 6,44 (д, J=5,6 Гц, 1H), 3,71 (с, 6H), 3,63 (с, 3H), 2,54 (с, 3H). Масс-спектр [ИЭС, MH+] = 445,12.
Получение 2-(2-(2-(3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)пиримидин-4-иламино)-5-метилфенил)ацетонитрила (Соединение 18)
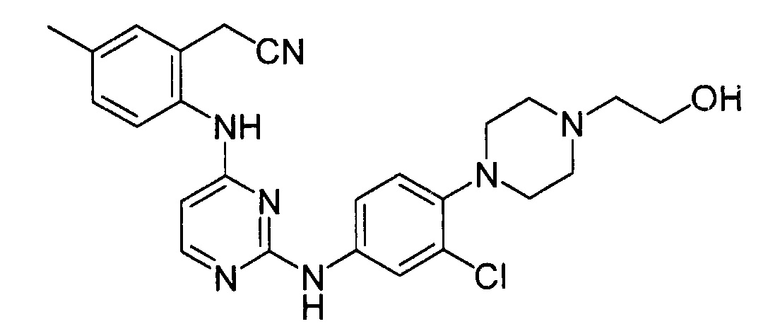
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 14. Получение 2-(2-амино-5-метилфенил)ацетонитрила (соединение 1 на схеме 14) описано ниже на схеме 15.
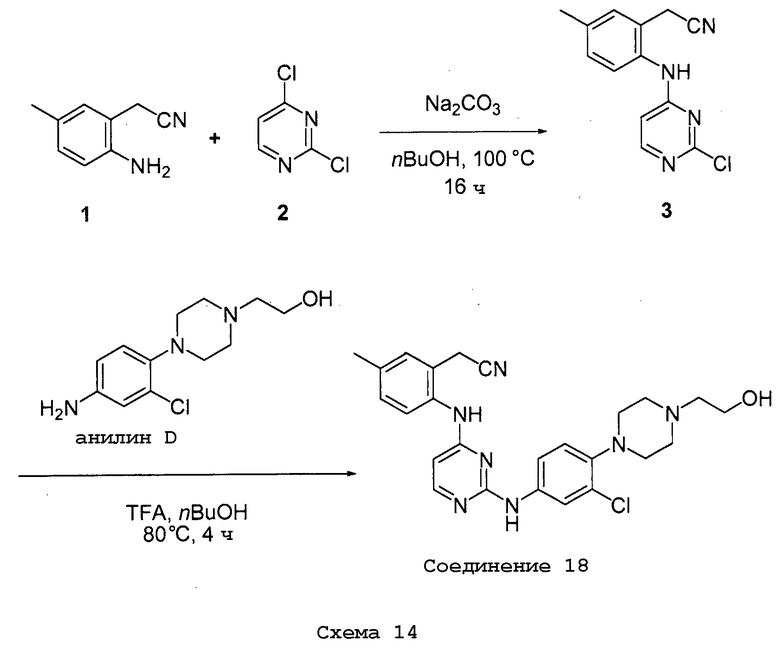
Смесь соединения 1 на схеме 14 (420 мг, 2,87 ммоль), соединения 2 на схеме 14 (640 мг, 4,31 ммоль) и Na2CO3 (608 мг, 5,74 ммоль) в n-BuOH (10 мл) нагревали до 100°C в течение 48 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 3 на схеме 14 (200 мг, выход 27%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 9,62 (с, 1H), 8,10 (д, J=5,9 Гц, 1H), 7,47-7,00 (м, 3H), 6,53 (с, 1H), 3,90 (с, 2H), 2,34 (с, 3H). Масс-спектр [ИЭС, MH+] = 259,07
Смесь соединения 3 на схеме 14 (180 мг, 0,69 ммоль), анилина D (195 мг, 0,76 ммоль) и TFA (2 мл) в n-BuOH (8 мл) нагревали до 80°C в течение 6 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью препаративной HPLC для получения соединения 18 (50 мг, выход 15%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 9,07 (с, 1H), 8,89 (с, 1H), 7,97 (д, J=5,5 Гц, 1H), 7,79 (д, J=2,7 Гц, 1H), 7,50-7,36 (м, 1H), 7,30 (д, J=8,4 Гц, 2H), 7,20 (м, 1H), 6,94 (д, J=8,7 Гц, 1H), 6,10 (д, J=5,8 Гц, 1H), 4,44 (с, 1H), 3,90 (с, 2H), 3,52 (д, J=5,8 Гц, 2H), 2,87 (с, 3H), 2,50 (д, J=1,7 Гц, 6H), 2,35 (с, 3H). Масс-спектр [ИЭС, MH+] = 478,10.
2-(2-Амино-5-метилфенил)ацетонитрила (соединение 1 на схеме 14) синтезировали как показано на схеме 15.
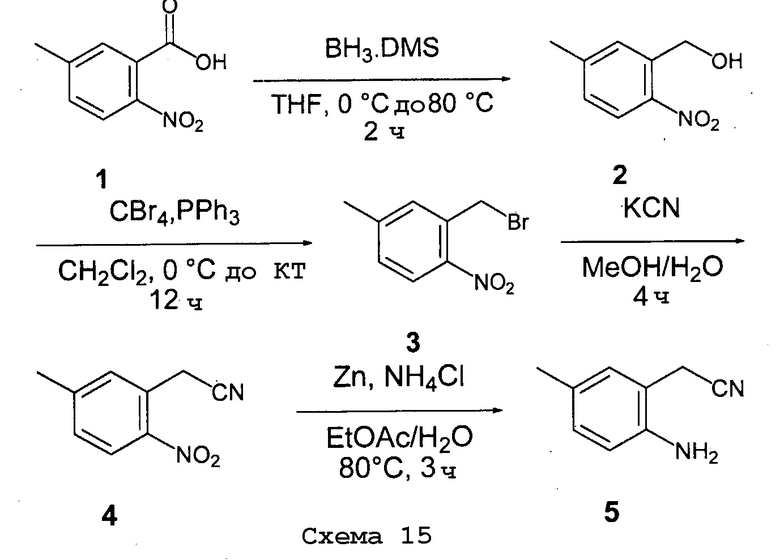
К холодному (0°C) перемешиваемому раствору соединения 1 на схеме 15 (5,0 г, 27,6 ммоль) в безводном THF (20 мл) добавили по каплям BH3.DMS (1 M в THF, 110 мл, 110,0 ммоль) и реакционную смесь перемешивали при 80°C 2 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили ледяной водой (30 мл) и подвергли экстракции EtOAc (2×50 мл). Органический слой промыли рассолом (20 мл), высушили над Na2SO4 и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 30/70) для получения соединения 2 на схеме 15 (3,5 г, выход 76%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,97 (д, J=8,3 Гц, 1H), 7,65 (д, J=2,3 Гц, 1H), 7,37–7,14 (м, 1H), 5,53 (с, 1H), 4,81 (с, 2H), 2,43 (с, 3H). Масс-спектр [ИЭС, MH+] = 168,06.
К холодному (0°C) перемешиваемому раствору соединения 2 на схеме 15 (1,0 г, 5,98 ммоль) в безводном CH2Cl2 (10 мл) добавили PPh3 (2,5 г, 9,76 ммоль) и CBr4 (3,2 г, 9,76 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь выпарили при пониженном давлении для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 3 на схеме 15 (900 мг, выход 69%) в виде коричневой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ 7,99 (д, J=8,3 Гц, 1H), 7,35 (д, J=1,9 Гц, 1H), 7,28 (д, J=1,9 Гц, 1H), 4,83 (с, 2H), 2,45 (с, 3H). Масс-спектр [ИЭС, MH+] = 229.98.
К раствору соединения 3 на схеме 15 (900 мг, 3,9 ммоль) в смеси MeOH/вода (8 мл, 3/1) добавили KCN (330 мг, 5,1 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (30 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 40/60) для получения соединения 4 на схеме 15 (350 мг, выход 51%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,13 (д, J=8,4 Гц, 1H), 7,62–7,46 (м, 1H), 7,41–7,30 (м, 1H), 4,21 (с, 2H), 2,50 (с, 3H). Масс-спектр [ИЭС, MH+] = 177,06.
Смесь соединения 4 на схеме 15 (350 мг, 1,98 ммоль), Zn пыли (297 мг, 4,54 ммоль) и NH4Cl (607 г, 11,36 ммоль) в EtOAc/H2O (10 мл, 1/1) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc. Фильтрат выпарили при пониженном давлении для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 5 на схеме 15 (220 мг, выход 76%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,91 (д, J=2,2 Гц, 1H), 6,84 (дд, J=8,1, 2,1 Гц, 1H), 6,59 (д, J=8,0 Гц, 1H), 4,93 (с, 2H), 3,73 (с, 2H), 2,15 (с, 3H). Масс-спектр [ИЭС, MH+] = 147,09.
Получение 2-(2-(2-(3-метокси-4-(4-метилпиперазин-1-ил)фениламино)пиримидин-4-иламино)-5-метилфенил)ацетонитрила (Соединение 19)
Указанное в заголовке соединение синтезировали аналогично соединению 18 с использованием анилина B на последней стадии сочетания.
Смесь соединения 3 на схеме 14 (170 мг, 0,65 ммоль), анилина B (125 мг, 0,78 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 19 (25 мг, выход 9%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,00 (д, J=5,8 Гц, 1H), 7,34 (с, 1H), 7,19 (дд, J=8,2, 1,8 Гц, 3H), 6,98 (дд, J=8,4, 2,4 Гц, 1H), 6,92–6,79 (м, 2H), 6,28 (с, 1H), 5,80 (д, J=5,7 Гц, 1H), 3,78 (с, 3H), 3,68 (с, 2H), 3,11 (с, 4H), 2,72 (с, 4H), 2,43 (с, 3H), 2,41 (с, 3H). Масс-спектр [ИЭС, MH+] = 444,12.
Получение 2-(2-(2-(3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)пиримидин-4-иламино)-5-метилфенил)ацетонитрила (Соединение 20)
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 16. Получение 2-(5-амино-2-метилфенил)ацетонитрила (соединение 1 на схеме 16) описано ниже.
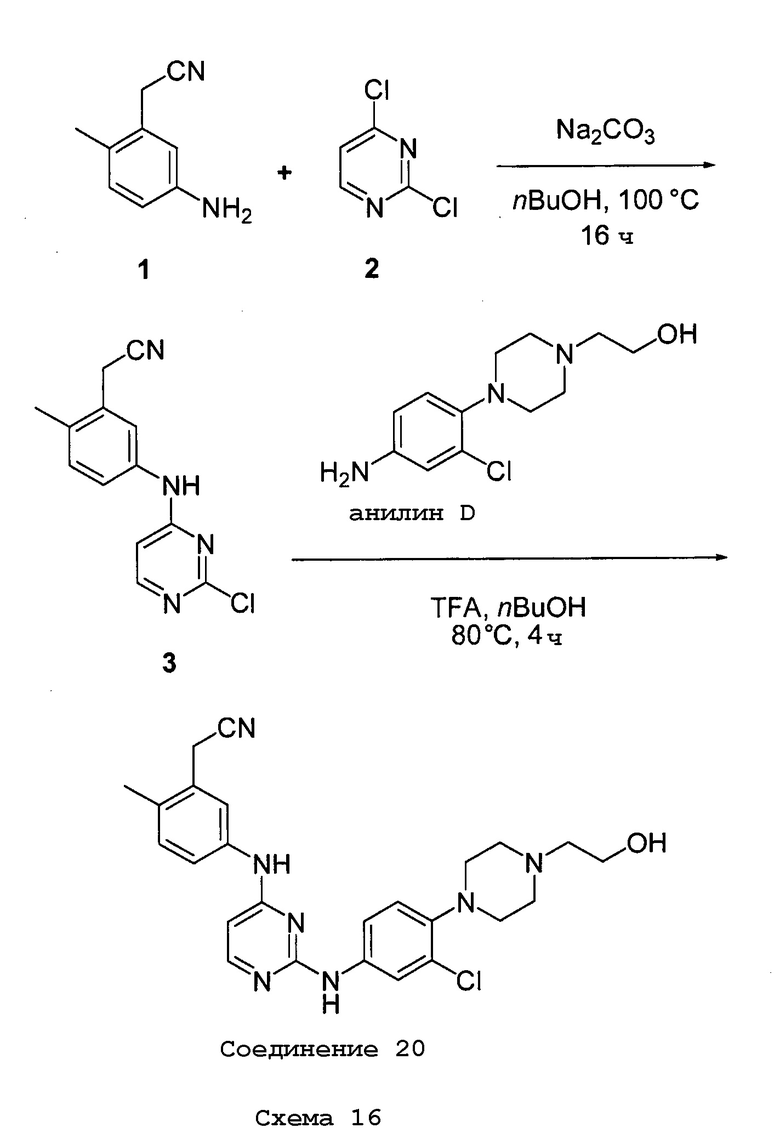
Смесь соединения 1 на схеме 16 (700 мг, 4,79 ммоль), соединения 2 на схеме 16 (851 мг, 5,75 ммоль) и Na2CO3 (952 мг, 8,98 ммоль) в n-BuOH (5 мл) нагревали до 80°C в течение 48 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 70/30) для получения соединения 3 на схеме 16 (350 мг, выход 47%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 10,06 (с, 1H), 8,15 (д, J=5,9 Гц, 1H), 7,60 (с, 1H), 7,52 (д, J=8,4 Гц, 1H), 7,24 (д, J=8,2 Гц, 1H), 6,74 (д, J=5,9 Гц, 1H), 4,01 (с, 2H), 2,25 (с, 3H). Масс-спектр [ИЭС, MH+] = 259,07.
Смесь соединения 3 на схеме 16 (200 мг, 0,77 ммоль), анилина D (237 мг, 0,93 ммоль) и TFA (2 мл) в n-BuOH (10 мл) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 20 (66 мг, выход 18%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 9,42 (с, 1H), 9,16 (с, 1H), 8,01 (д, J=5,7 Гц, 1H), 7,90 (д, 7=2,6 Гц, 1H), 7,72 (д, J=7,5 Гц, 1H), 7,59–7,51 (м, 2H), 7,18 (д, J=8,3 Гц, 1H), 7,07 (д, J=8,7 Гц, 1H), 6,21 (д, J=5,7 Гц, 1H), 4,43 (т, J=5,3 Гц, 1H), 3,97 (с, 2H), 3,53 (д, J=6,0 Гц, 2H), 2,95–2,87 (м, 4H), 2,57 (с, 4H), 2,45 (т, J=6,3 Гц, 2H), 2,26 (с, 3H). Масс-спектр [ИЭС, MH+] = 478,12.
2-(5-Амино-2-метилфенил)ацетонитрил (соединение 1 на схеме 16) синтезировали аналогично соединению 5 на схеме 15 с использованием 2-метил-5-нитробензойной кислоты на первой стадии и выделили в виде коричневого твердого вещества (700 мг, выход 25% после четырех стадий). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,91 (д, J=2,2 Гц, 1H), 6,80 (с, 1H), 6,59 (д, J=8,0 Гц, 1H), 4,93 (с, 2H), 3,80 (с, 2H), 2,25 (с, 3H). Масс-спектр [ИЭС, MH+] = 147,05.
Получение 2-(5-(2-(3-метокси-4-(4-метилпиперазин-1-ил)фениламино)пиримидин-4-иламино)-2-метилфенил)ацетонитрила (Соединение 21)
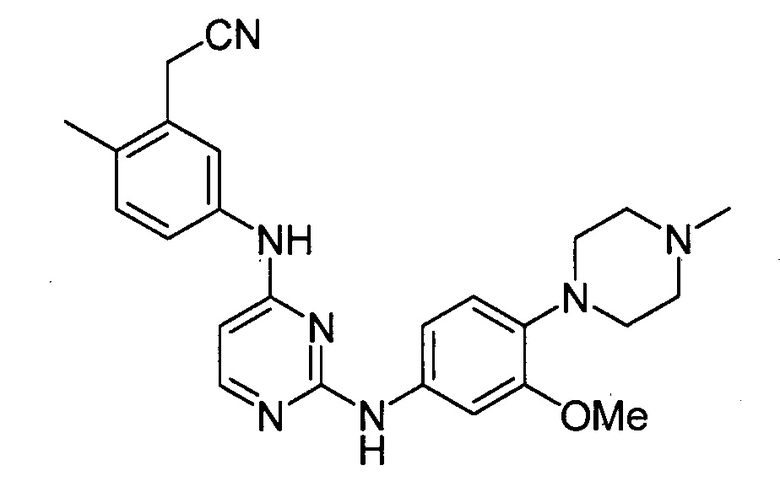
Указанное в заголовке соединение синтезировалось аналогично соединению 20 с использованием анилина B на последней стадии сочетания.
Смесь соединения 3 на схеме 15 (150 мг, 0,58 ммоль), анилина B (160 мг, 0,63 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 21 (20 мг, выход 8%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,05 (д, J=5,7 Гц, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,21-7,15 (м, 2H), 7,08 (дд, J=8,5, 2,4 Гц, 1H), 6,92 (д, J=8,5 Гц, 1H), 6,84 (с, 1H), 6,49 (с, 1H), 6,10 (д, J=5,8 Гц, 1H), 3,83 (с, 3H), 3,65 (с, 2H), 3,09 (с, 4H), 2,66 (с, 4H), 2,38 (с, 3H), 2,32 (с, 3H). Масс-спектр [ИЭС, MH+] = 444,12.
Получение 2-(2-(2-(3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)пиримидин-4-иламино)-5-фторфенил)ацетонитрила (Соединение 22)
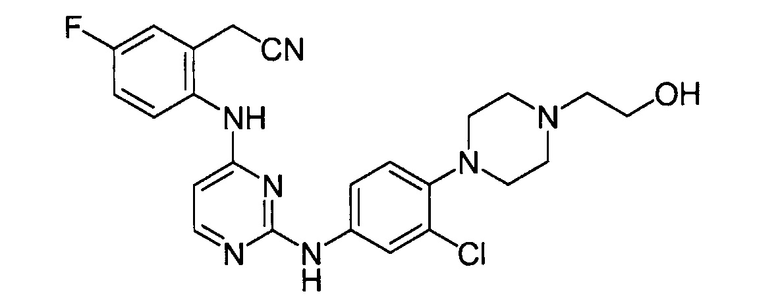
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 17.
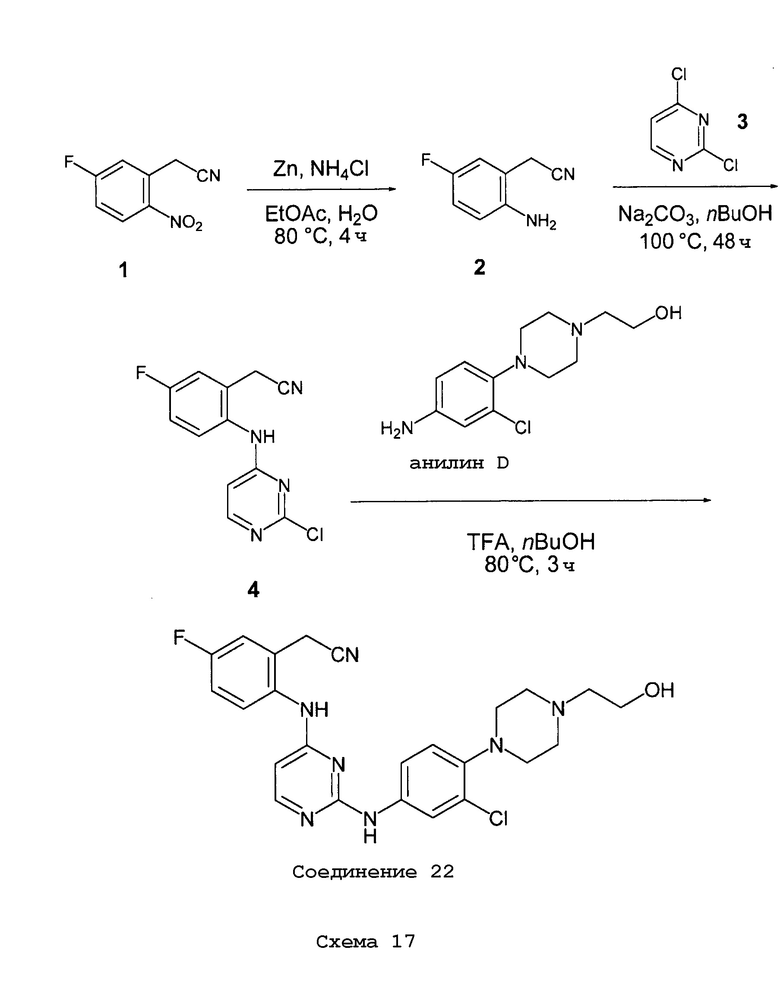
Смесь соединения 1 на схеме 17 (0,80 г, 4,45 ммоль), Zn пыли (1,12 г, 17,77 ммоль) и NH4Cl (2,40 г, 44,5 ммоль) в EtOAc/H2O (20 мл, 1/1) нагревали до 80°C в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc. Фильтрат выпарили при пониженном давлении для получения остатка, который очистили с помощью хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 2 на схеме 17 (0,50 г, выход 72%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,00–6,86 (м, 2H), 6,68 (м Гц, 1H), 5,05 (с, 2H), 3,79 (с, 2H). Масс-спектр [ИЭС, MH+]= 151,02.
Смесь соединения 2 на схеме 17 (500 мг, 3,34 ммоль), соединения 3 на схеме 17 (740 мг, 5,00 ммоль), и Na2CO3 (708 мг, 6,68 ммоль) в n-BuOH (5 мл) нагревали до 100°C в течение 48 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 50/50) для получения соединения 4 на схеме 17 (280 мг, выход 32%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 20 8,16 (д, J=5,8 Гц, 1H), 7,33 (м, 2H), 7,20–7,12 (м, 1H), 6,81–6,72 (м, 1H), 6,20 (д, J=5,9 Гц, 1H), 3,71 (с, 2H). Масс-спектр [ИЭС, MH+] = 263,04.
Смесь соединения 4 на схеме 17 (200 мг, 0,76 ммоль), анилина D (213 мг, 0,85 ммоль) и TFA (2 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 22 (45 мг, выход 12%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 9,11 (с, 1H), 8,96 (с, 1H), 8,00 (д, J=5,7 Гц, 1H), 7,76 (с, 1H), 7,46 (м, 1H), 7,43–7,29 (м, 2H), 7,25 (м, 1H), 6,95 (д, J=8,8 Гц, 1H), 6,13 (д, J=5,7 Гц, 1H), 4,41 (д, J-6,2 Гц, 1H), 3,96 (с, 2H), 3,52 (д, J=6,0 Гц, 2H), 2,86 (с, 4H), 2,56 (с, 4H), 2,44 (д, J=5,9 Гц, 2H). Масс-спектр [ИЭС, MH+] = 482,03.
Получение 2-(5-фтор-2-(2-(3-метокси-4-(4-метилпиперазин-1-ил)фениламино)пиримидин-4-иламино)фенил)ацетонитрила (Соединение 23)
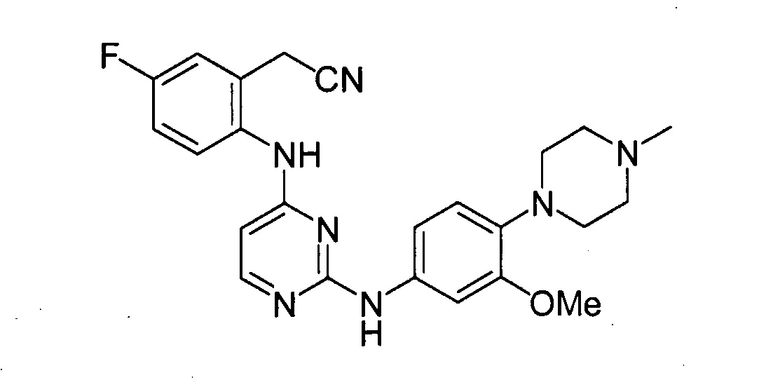
Указанное в заголовке соединение синтезировалось аналогично соединению 22 с использованием анилина B на последней стадии сочетания.
Смесь соединения 4 на схеме 17 (200 мг, 0,76 ммоль), анилина B (229 мг, 0,91 ммоль) и TFA (2 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 23 (60 мг, выход 17%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,03 (д, J=5,7 Гц, 1H), 7,34–7,28 (м, 2H), 7,12 (д, J=2,9 Гц, 2H), 6,98 (дд, J=8,5, 2,4 Гц, 1H), 6,90–6,81 (м, 2H), 6,21 (с, 1H), 5,81 (д, J=5,7 Гц, 1H), 3,79 (с, 3H), 3,71 (с, 2H), 3,09 (с, 4H), 2,69 (с, 4H), 2,40 (с, 3H). МАСС-СПЕКТР [ИЭС, MH+] = 448,02.
Получение 2-(6-(2-(3-хлор-4-(4-(2-гидроксиэтил)пиперазин-1-ил)фениламино)пиримидин-4-иламино)-2,3-диметилфенил)ацетонитрила (Соединение 24)
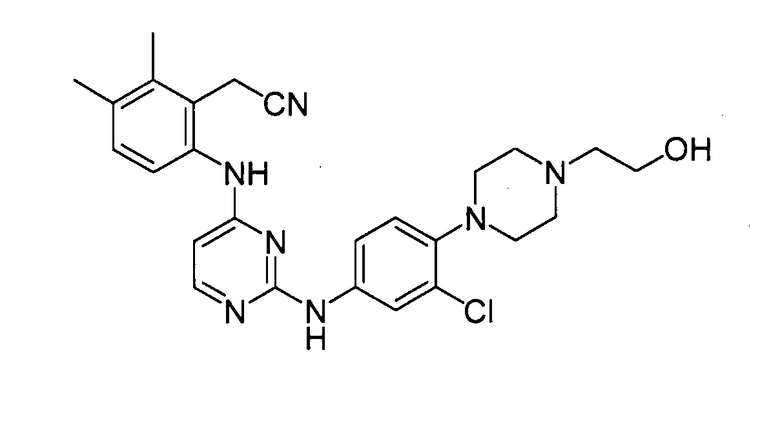
Указанное в заголовке соединение синтезировали согласно методике, изображенной на схеме 18. Получение 2-(6-амино-2,3-диметилфенил)ацетонитрила (соединение 1 на схеме 18) описано ниже на схеме 19.
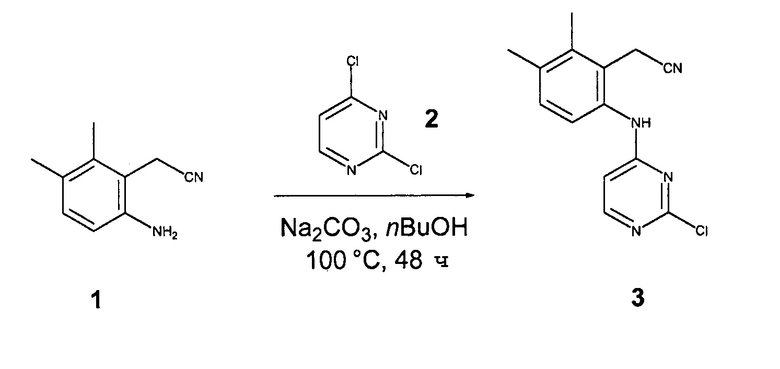
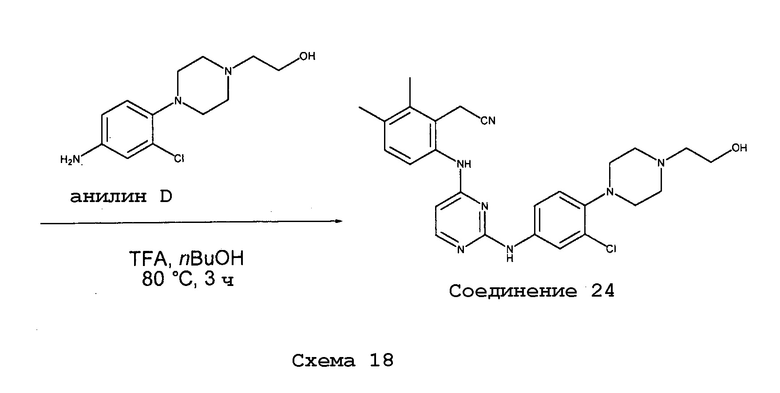
Смесь соединения 1 на схеме 18 (500 мг, 3,12 ммоль), соединения 2 на схеме 18 (925 мг, 6,25 ммоль) и Na2CO3 (662 мг, 6,25 ммоль) в n-BuOH (10 мл) нагревали до 100°C в течение 16 ч в запаянной трубке. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 50/50) для получения соединения 3 на схеме 18 (200 мг, выход 24%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 8,07 (д, J=5,9 Гц, 1H), 7,56 (с, 1H), 7,16 (с, 2H), 6,48 (д, J=5,9 Гц, 1H), 4,76 (с, 3H), 2,36 (с, 3H), 2,32 (с, 3H). МАСС-СПЕКТР [ИЭС, MH+] = 273,01.
Соединение 3 на схеме 18 (150 мг, 0,55 ммоль), анилин D (168 мг, 0,66 ммоль) и TFA (1 мл) в n-BuOH (5 мл) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл), нейтрализовали водным NaHCO3 (pH 8) и подвергли экстракции EtOAc (2×20 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на нейтральном оксиде алюминия (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 24 (25 мг, выход 9%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 7,99 (д, J=5,7 Гц, 1H), 7,68 (д, J=2,6 Гц, 1H), 7,26–7,24 (м, 1H), 7,18 (д, J=7,9 Гц, 1H), 7,09 (с, 1H), 6,96 (д, J=8,80 Гц, 1 H), 6,76 (с, 1H), 6,05 (д, J=5,7 Гц, 1H), 4,76 (с, 2H), 3,66 (т, J=5,3 Гц, 2H), 3,03 (м, 4H), 2,71 (м, 4H), 2,64 (т, J=5,3 Гц, 2H), 2,35 (с, 3H), 2,32 (с, 3H). MS [ИЭС, MH+] = 492,10.
2-(6-Амино-2,3-диметилфенил)ацетонитрил (соединение 1 на схеме 18) синтезировали как показано на схеме 19.
К раствору соединения 1 на схеме 19 (10,00 г, 60,24 ммоль) в AcOH (25 мл) и 6 Н HC1 (30 мл) добавили по каплям NaNO2 (4,98 г, 72,28 ммоль) в воде (30 мл), а затем NaHCO3 (30,00 г, 35,71 ммоль) и толуол (25 мл) и полученныую смесь перемешивали при 0°C 30 мин. Этот раствор затем добавили к перемешиваемой смеси KCN (31,30 г, 481,90 ммоль) и CuCN (11,79 г, 132,50 ммоль) в EtOAc (50 мл) и воде (70 мл) при 0°C и реакционную смесь медленно нагрели до комнатной температуры в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили EtOAc (60 мл). Органический слой отделили и высушили над Na2SO4, после чего профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 2 на схеме 19 (6,60 г, выход 62%) в виде желтого твердого вещества. 1HNMR(400 МГц, ДМСО-d6): δ 8,15 (д, J=8,4 Гц, 1H), 7,76 (д, J=8,4 Гц, 1H), 2,54 (с, 3H), 2,43 (с, 3H). МАСС-СПЕКТР [ИЭС, MH+] = 177,06.
Раствор соединения 2 на схеме 19 (6,60 г, 37,5 ммоль) в воде (60 мл), AcOH (60 мл) и H2SO4 (60 мл) перемешивали при 160°C в течение 6 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили ледяной водой (30 мл) и подвергли экстракции EtOAc (2×100 мл). Органический слой промыли рассолом (20 мл) высушили над (Na2SO4) и выпарили для получения остатка, который растворили в 25% водном H2SO4 и нагрели до 160°C. К полученному раствору, которому дали охладиться до комнатной температуры в течение 4 ч, добавили по каплям NaNO2 (3,98 г, 56,7 ммоль) в воде (30 мл). После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили ледяной водой (30 мл) и подвергли экстракции EtOAc (2×100 мл). Органический слой промыли рассолом (20 мл), высушили над (Na2SO4) и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента CH2Cl2/MeOH 100/0, постепенно увеличивая до 90/10) для получения соединения 3 на схеме 19 (3,00 г, выход 55%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 13,67 (с, 1H), 7,92 (д, J=8,4 Гц, 1H), 7,47 (д, J=8,4 Гц, 1H), 2,36 (с, 3H), 2,24 (с, 3H). МАСС-СПЕКТР [ИЭС, (M-H)-] = 194,07.
К холодному (0°C) перемешиваемому раствору соединения 3 на схеме 19 (3,0 г, 15,38 ммоль) в безводном THF (30 мл) добавили по каплям BH3.DMS (2 M в THF, 30,7 мл, 61,53 ммоль) и реакционную смесь перемешивали при 80°C в течение 2 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили талой водой (30 мл) и подвергли экстракции EtOAc (2×50 мл). Органический слой промыли рассолом (20 мл), затем высушили над (Na2SO4) и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 30/70) для получения соединения 4 на схеме 19 (2,5 г, выход 89%) в виде не совсем белого твердого вещества. 1H NMR (400 МГц, ДМСО-d6): δ 7,52 (д, J=8,2 Гц, 1H), 7,30 (д, J=8,2 Гц, 1H), 5,18 (т, J=5,4 Гц, 1H), 4,61 (д, J=3,3 Гц, 2H), 2,33 (с, 3H), 2,31 (с, 3H). МАСС-СПЕКТР [ИЭС, MH+] = 182,05.
К холодному (0°C) перемешиваемому раствору соединения 4 на схеме 19 (2,5 г, 13,81 ммоль) в безводном CH2Cl2 (25 мл) добавили PPh3 (7,2 г, 27,62 ммоль) и CBr4 (9,2 г, 27,62 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь выпарили при пониженном давлении для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 5 на схеме 19 (2,3 г, выход 67%) в виде коричневой жидкости. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,72 (д, J=8,3 Гц, 1H), 7,41 (д, J=8,3 Гц, 1H), 4,79 (с, 2H), 2,36 (с, 3H), 2,35 (с, 3H). МАСС-СПЕКТР [ИЭС, MH+] = 243,96.
К раствору соединения 5 на схеме 19 (2,30 г, 9,23 ммоль) в MeOH (20 мл) и воде (6 мл) добавили NaCN (0,59 г, 12,00 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь разбавили водой (20 мл) и подвергли экстракции EtOAc (30 мл). Органический слой высушили над Na2SO4, профильтровали и выпарили для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 40/60) для получения соединения 6 на схеме 19 (1,10 г, выход 64%) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ 7,79 (д, J=8,4 Гц, 1H), 7,32 (д, J=8,4 Гц, 1H), 3,96 (с, 2H), 2,43 (д, J=1,9 Гц, 6H). Масс-спектр [ИЭС, MH+] = 191,08.
Смесь соединения 6 на схеме 19 (1,10 г, 5,78 ммоль), Zn пыли (1,51 г, 23,15 ммоль) и NH4Cl (3,09 г, 57,8 ммоль) в EtOAc/H2О (20 мл, 1/1) нагревали до 80°C в течение 3 ч. После того, как анализ TLC показал, что исходное вещество полностью прореагировало, реакционную смесь пропустили через подушку из целита и промыли твердую фазу EtOAc. Фильтрат выпарили при пониженном давлении для получения остатка, который очистили с помощью флэш-хроматографии на силикагеле (с использованием в качестве элюента смеси петролейный эфир/EtOAc 100/0, постепенно увеличивая до 60/40) для получения соединения 7 на схеме 19 (0,60 мг, выход 65%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ 6,82 (д, J=8,1 Гц, 1H), 6,47 (д, J=8,0 Гц, 1H), 4,96 (с, 2H), 3,76 (с, 2H), 2,14 (с, 3H), 2,10 (с, 3H). Масс-спектр [ИЭС, MH+] = 161,10.
Другие соединения, имеющие структуру (I) (или структуру (II)), получают согласно способу, аналогичному описанному выше.
Пример 2
Испытание соединений
Биохимические испытания для измерения ингибирующих действий соединений были осуществлены службой поиска и разработки лекарственных веществ при Life Technologies (Мадисон, Висконсин). Испытания с киназой JAK2 были выполнены с использованием технологии Z'-LYTE®, в то время как ингибирование ALK2 было испытано с применением анализа связывания LanthaScreen®. Результаты показаны выше в таблице 1.
Была испытана экспрессия гепсидина в клетках HEPG2, обработанных приведенными в качестве примера соединениями согласно изобретению. Результаты показаны на фигурах 1-3. Ссылка: В PNAS об. 103no27 10289-10293 показано, что BMP2 при 1OO нг/мкл индуцирует синтез гепсидина в клетках HepG2 сильнее, чем IL-6, BMP4 и BMP9. BMP2 и BMP4 связываются с ALK2, ALK3 и ALK6. Для определения способности соединений влиять на исходный уровень экспрессии гепсидина, клетки HEPG2 (гепатоцеллюлярная карцинома) обрабатывали соединением в течение 6 часов и затем оценивали экспрессию гепсидина с помощью полимеразной цепной реакции с обратной транскрипцией (RT-PCR) в реальном времени (относительно уровней β-актина). Экспрессия гепсидина снижалась дозозависимым образом. Экспрессия гепсидина была ингибирована на 90% при концентрации соединения 12 равной 3 мкмоль/л. Второй подход позволил оценить способность соединений ингибировать экспрессию гепсидина, индуцированную BMP2. BMP2 индуцирует экспрессию гепсидина путем связывания с ALK2 и ее активации. Клетки HEPG2 обработали соединением, а затем BMP2 при 100 нг/мкл. Добавление BMP2 вызвало повышение концентрации гепсидина более чем в 20 раз. Для сравнения обработанные соединением 7 клетки показали 50% снижение индукции при обработке 0,3 мкммоль/л. Как показано на фигуре 3, способность блокировать сигнальный путь BMP2 является дозозависимой.
Экспрессию гепсидина у мышей, обработанных соединениями 4 и 12 сравнили с экспрессией гепсидина у мышей, обработанных соединением A. В данном эксперименте мышей лечили перорально разовой дозой испытываемого соединения. После шести часов у подвергнутых эвтаназии животных удаляли печень и экстрагировали RNA (РНК). Содержание mRNA (матричной РНК) гепсидина определяли с помощью RT-PCR в реальном времени согласно описанному выше. Как видно на фигуре 4, соединения 4 и 12 ингибируют экспрессию гепсидина в большей степени, чем соединение А, при испытанной дозе (250 мг/кг).
 .
.
По аналогии с вышеуказанным исследованием, соединения 4 и 12 оценили на LPS-индуцированной мышиной модели. LPS широко используется при исследованиях на животных, чтобы добиться опосредованного цитокинами иммунного ответа и сопутствующей анемии. В данном эксперименте соединение 4 или 12 вводили в организм перорально в виде разовой дозы, составляющей 250 мг/кг, после чего осуществляли внутрибрюшное введение 1 мг/кг LPS. После шести часов анализировали экспрессию гепсидина с помощью RT-PCR согласно описанному выше. Результаты представлены на фигуре 5.
Чтобы определить активность в живом организме соединений 4 и 12 при величине дозы ниже 250 мг/кг, мышей лечили разовой дозой испытываемого соединения при 75, 150 или 300 мг/кг. В этом случае тоже измеряли уровни гепсидина в печени согласно описанному выше. На фигуре 6 показаны результаты исследований влияния величины дозы на мышиных моделях для соединений 4 и 12.
Помимо понижения уровня гепсидина эти соединения также продемонстрировали многообещающую активность при модуляции уровня цитокина в живом организме. В этом случае снова использовали LPS, чтобы индуцировать цитокиновый ответ, при этом испытываемые соединения исследовались для определения, можно ли отменить или предотвратить индуцированные цитокиновые ответы путем лечения соединением 4 или 12. В это исследование было включено несколько цитокинов, и на фигуре 7 сравнивается модуляция цитокина IL-5 (в качестве примера) соединениями 4 и 12, а также соединением A. Для соединения № 4 был также представлен фармакокинетический профиль при использовании как внутривенного так и перорального введения в организм. Результаты сведены в Таблице 2. Данные демонстрируют, что бионакопление может быть увеличено при использовании солевых форм соединения № 4. Фармакокинетические свойства соединения 4 определялись на женских особях крыс. На фигуре 8 показаны концентрации в плазме крови в заданные моменты времени как для крыс, которым ввели дозу внутривенно, так и для крыс, которым ввели дозу перорально, (на графике показано среднее значение для 3 особей), а также фармакокинетические параметры для соединения, принятого перорально. Данные показывают, что уровни концентрации в плазме крови соединения 4 остаются высокими даже после 24 часов.
Фармакокинетические свойства соединения 12 были количественно определены на женских особях крыс. На фигуре 9 показаны концентрации в плазме крови в заданные моменты времени как для крыс, которым ввели дозу внутривенно, так и для крыс, которым ввели дозу перорально, (на графике показано среднее значение для 3 особей), а также фармакокинетические параметры для соединения, принятого перорально. Концентрация соединения для соединения 12 в плазме оставалась высокой даже после 24 ч при пероральном приеме. Пероральная биодоступность соединения 12 является исключительно хорошей, около 95%.
Различные описанные выше варианты осуществления можно объединять для обеспечения дополнительных вариантов осуществления. Все патенты США, публикации заявок на патент США, заявки на патент США, патенты других стран, заявки на патент других стран и непатентные публикации, упомянутые в данном описании и/или перечисленные в инструкции по применению, полностью включены в данный документ в качестве ссылки. Аспекты вариантов осуществления при необходимости могут быть модифицированы, чтобы использовать сведения из различных патентов, заявок и публикаций для обеспечения дополнительных вариантов осуществления.
Эти и другие изменения в вариантах осуществления могут быть сделаны в свете вышеизложенного описания. В целом в последующей формуле изобретения используемые термины не должны рассматриваться как ограничивающие формулу изобретения конкретными вариантами осуществления, раскрытыми в описании и формуле изобретения, но должны рассматриваться как включающие все возможные варианты осуществления наряду с полным объемом эквивалентов, на которые распространяется право такой формулы изобретения. Соответственно, формула изобретения не ограничивается раскрытием.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2016 |
|
RU2718915C2 |
| БОРСОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE4 | 2019 |
|
RU2793936C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2019 |
|
RU2801140C2 |
| НОВЫЕ СОЕДИНЕНИЯ ЗАМЕЩЕННОГО N-(3-ФТОРПРОПИЛ)ПИРРОЛИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2742278C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| СОЕДИНЕНИЯ ИМИДАЗОТРИАЗИНОНА | 2011 |
|
RU2603140C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2808165C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2665571C2 |
| АГОНИСТЫ PPAR, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2746602C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743360C2 |
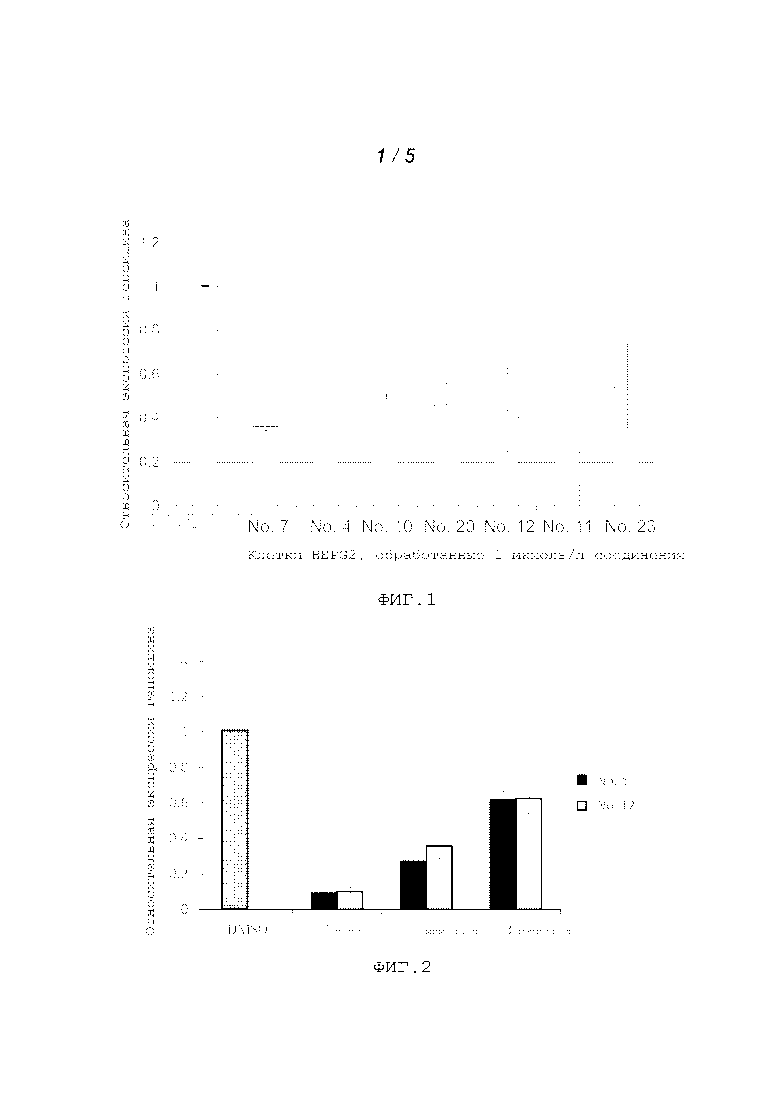


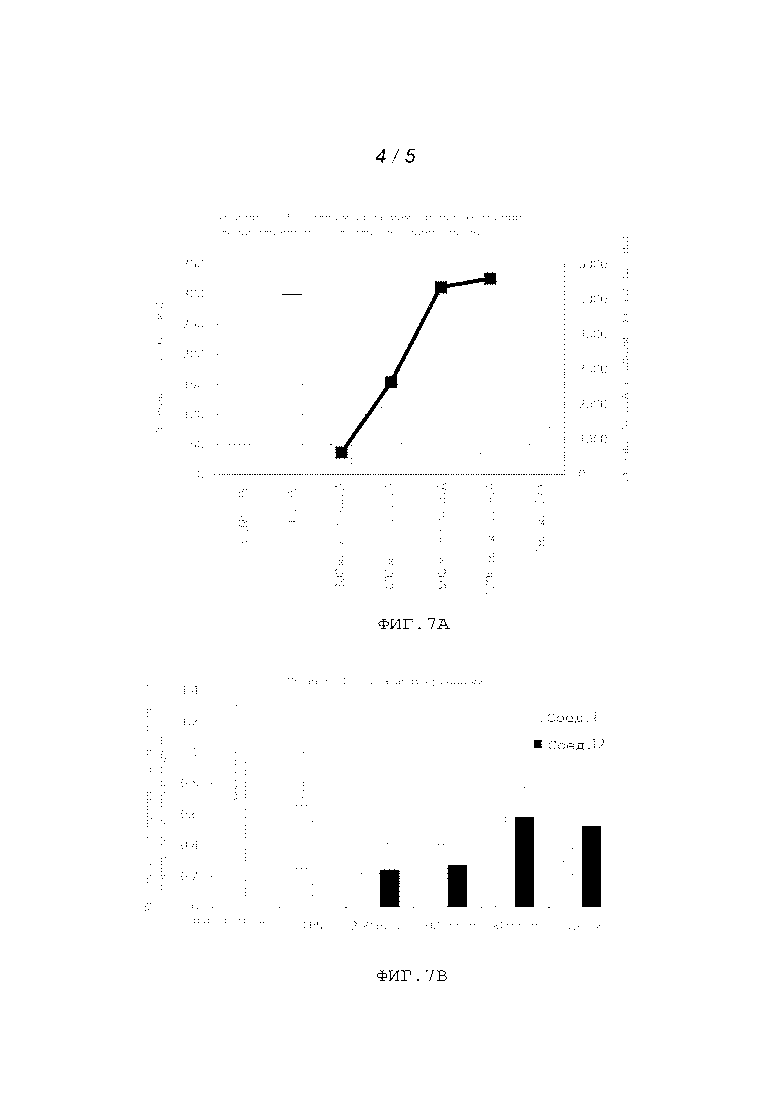
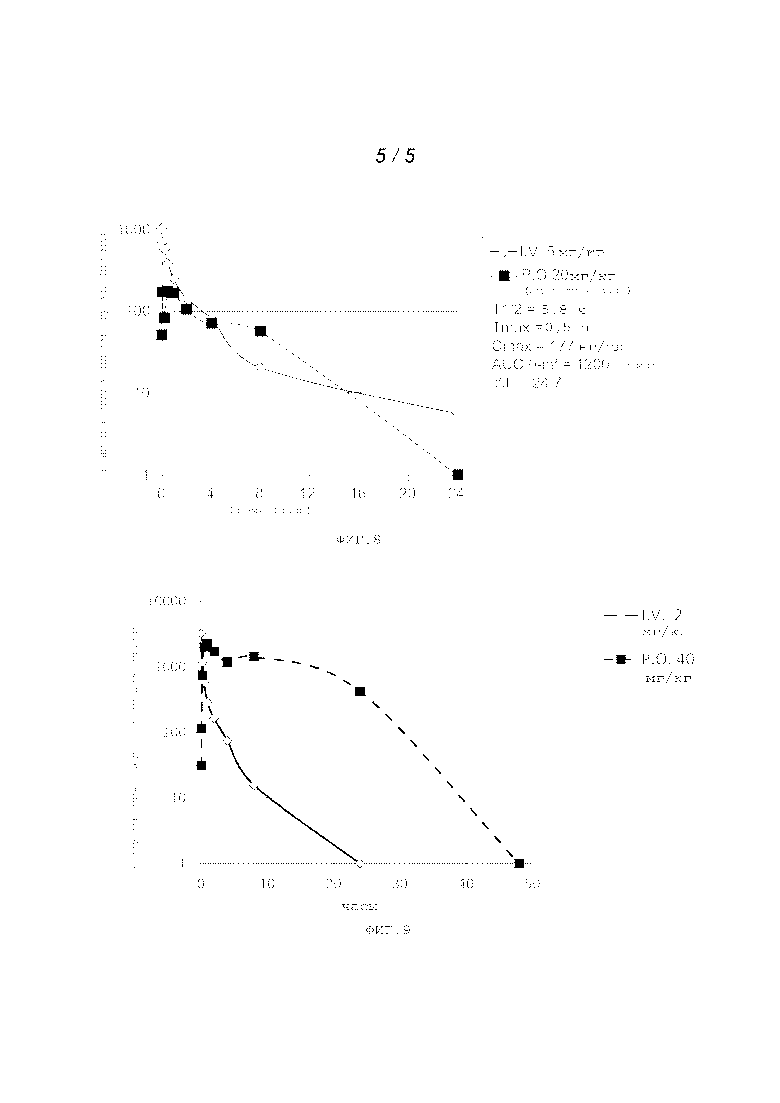
Изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли. Соединения обладают свойствами ингибиторов киназы ALK2 и/или киназы JAK2. При этом соединения проявляют селективную активность в отношении ALK2 относительно JAK2. Соединения могут найти применение для лечения анемии хронического заболевания, анемии хронического воспаления, анемии онкологического заболевания или прогрессирующей оссифицирующей фибродисплазии, а также для лечения миелопролиферативного расстройства, лимфомы, солидной опухоли, глиомы, фибродисплазии. Ингибирование активности ALK2 позволяет снизить количество внезапных обострений детского генетического заболевания, называемого прогрессирующей оссифицирующей фибродисплазии (FOR). В формуле (I)
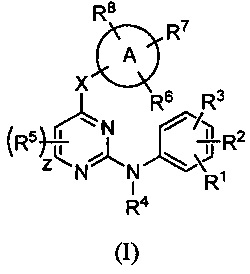
A обозначает 6-членное гетероарильное кольцо, содержащее атом азота; X является -NH-; R1 является H, галогеном или C1-C6 алкоксигруппой; R2 является галогеном или C1-C6 алкоксигруппой; R3 является галогеном, C1-C6 алкоксигруппой или -(CH2)nNRaRb; R4 является H; R5 является H; каждый из R6 и R7 независимо является H, галогеном, гидроксилом, C1-C6 алкилом, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом; R8 является 6-членным гетероарилом, содержащим атом азота, необязательно замещенным алкиламиноалкилом; каждый из Ra и Rb независимо является -H, C1-C6 алкилом, C1-C6 гидроксиалкилом, или Ra и Rb совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членное насыщенное карбоциклическое или гетероциклическое кольцо, необязательно замещенное одним или более заместителем, выбранным из C1-C6алкила, C1-C6карбоксиалкилкарбонила и C1-C6 гидроксиалкила; n равняется 0 или 1; и z равняется 0 или 1. Изобретение также относится к способу получения соединений формулы I. Способ осуществляют по следующей схеме
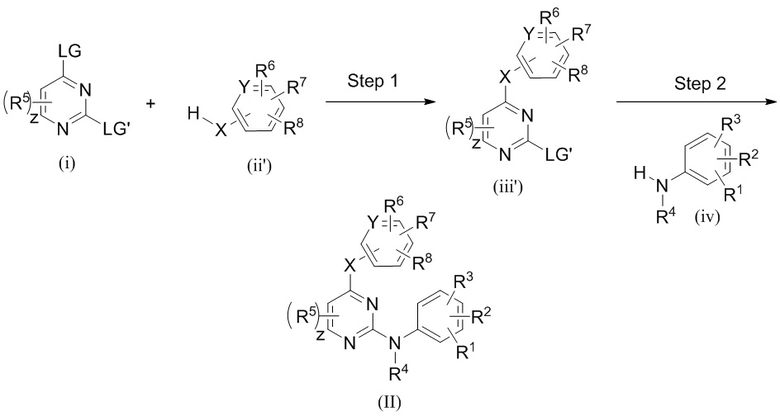
Значения радикалов соответствуют значениям, указанным выше. LG и LG' представляет уходящую группу –галоген. 11 н. и 33 з.п. ф-лы, 9 ил., 2 табл., 2 пр.
1. Соединение, имеющее следующую структуру (I):
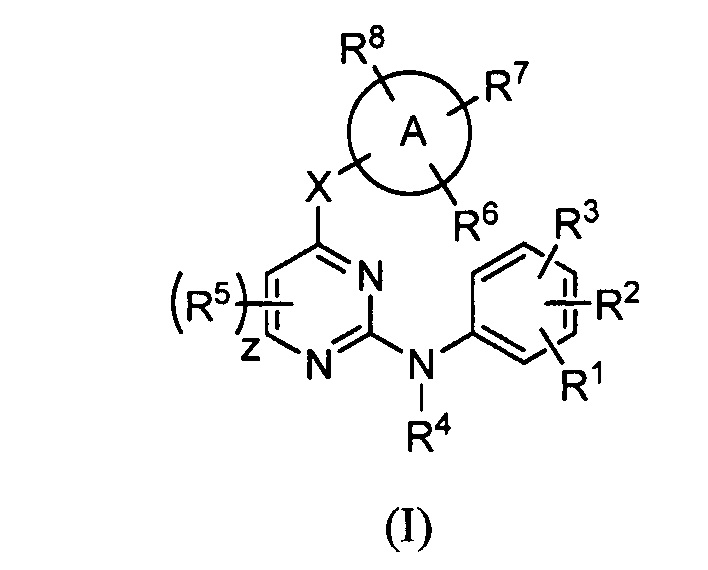 .
.
или его фармацевтически приемлемая соль, где:
A обозначает 6-членное гетероарильное кольцо, содержащее атом азота;
X является -NH-;
R1 является H, галогеном или C1-C6 алкоксигруппой;
R2 является галогеном или C1-C6 алкоксигруппой;
R3 является галогеном, C1-C6 алкоксигруппой или -(CH2)nNRaRb;
R4 является H;
R5 является H;
каждый из R6 и R7 независимо является H, галогеном, гидроксилом, C1-C6 алкилом, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом;
R8 является 6-членным гетероарилом, содержащим атом азота, необязательно замещенным алкиламиноалкилом;
каждый из Ra и Rb независимо является -H, C1-C6 алкилом, C1-C6 гидроксиалкилом, или Ra и Rb совместно с атомом азота, к которому они присоединены, образуют 5- или 6-членное насыщенное карбоциклическое или гетероциклическое кольцо, необязательно замещенное одним или более заместителем, выбранным из C1-C6алкила, C1-C6карбоксиалкилкарбонила и C1-C6 гидроксиалкила;
n равняется 0 или 1; и
z равняется 0 или 1.
2. Соединение по п. 1, в котором соединение имеет следующую структуру (II)
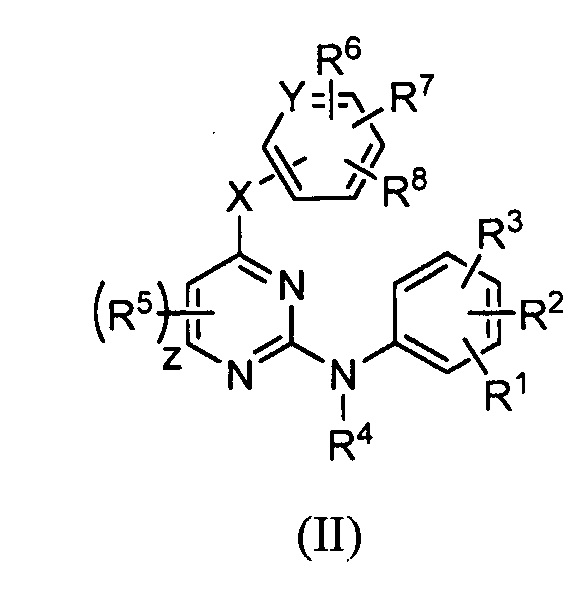 ,
,
или его фармацевтически приемлемая соль,
где:
X является -NH-;
Y является N;
R1 является H или C1-C6 алкоксигруппой;
R2 является галогеном или C1-C6 алкоксигруппой;
R3 является C1-C6 алкоксигруппой или -(CH2)nNRaRb;
R4 является H;
R5 является H;
R6 является H, C1-C6 алкилом, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом;
R7 является H, C1-C6 алкилом, C1-C6 цианоалкилом или C3-C6 цианоциклоалкилом;
R8 является 6-членным гетероарилом, содержащим атом азота, необязательно замещенным алкиламиноалкилом; и
n равняется 0 или 1
z равняется 0, или 1.
3. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R1 является H.
4. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R1 является C1-C6 алкоксигруппой.
5. Соединение или его фармацевтически приемлемая соль по п. 4, в котором R1 является метоксигруппой.
6. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R2 является галогеном.
7. Соединение или его фармацевтически приемлемая соль по п. 6, в котором R2 является F или Cl.
8. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R2 является C1-C6 алкоксигруппой.
9. Соединение или его фармацевтически приемлемая соль по п. 8, в котором R2 является метоксигруппой.
10. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R3 является -NRaRb, при этом Ra и Rb соединяются с образованием необязательно замещенного гетероциклического кольца.
11. Соединение или его фармацевтически приемлемая соль по п. 10, в котором гетероциклическое кольцо является замещенным или незамещенным пиперазинильным кольцом.
12. Соединение или его фармацевтически приемлемая соль по п. 11, в котором замещенное пиперазинильное кольцо является N-замещенным пиперазинильным кольцом.
13. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R3 является C1-C6 алкоксигруппой.
14. Соединение или его фармацевтически приемлемая соль по п. 13, в котором R3 является метоксигруппой.
15. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где соединение имеет одну из следующих структур:
16. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором по меньшей мере один заместитель из R6 и R7 является H.
17. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором R7 является фтором или хлором.
18. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором по меньшей мере один заместитель из R6 и R7 является C1-C6 алкилом.
19. Соединение или его фармацевтически приемлемая соль по п. 18, в котором C1-C6 алкил является метилом.
20. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором один заместитель из R6 или R7 является C1-C6 цианоалкилом.
21. Соединение или его фармацевтически приемлемая соль по п. 20, в котором C1-C6 цианоалкил является -CH2CN.
22. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, в котором один заместитель из R6 и R7 является C3-C6 цианоциклоалкилом.
23. Соединение или его фармацевтически приемлемая соль по п. 22, в котором C3-C6 цианоциклоалкил является 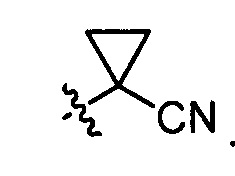 .
.
24. Соединение или его фармацевтически приемлемая соль по п. 1, в котором R8 является замещенным или незамещенным пиридинилом.
25. Соединение по п. 1 или 2, где соединение имеет одну из следующих структур:
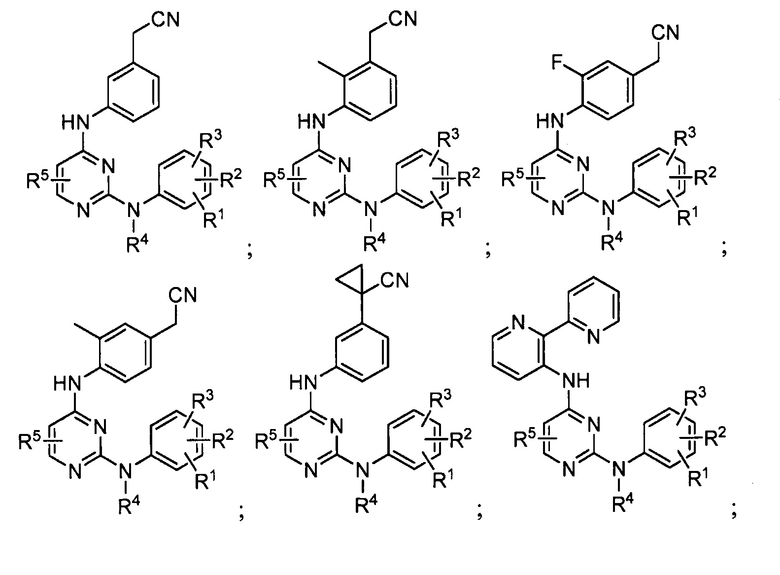
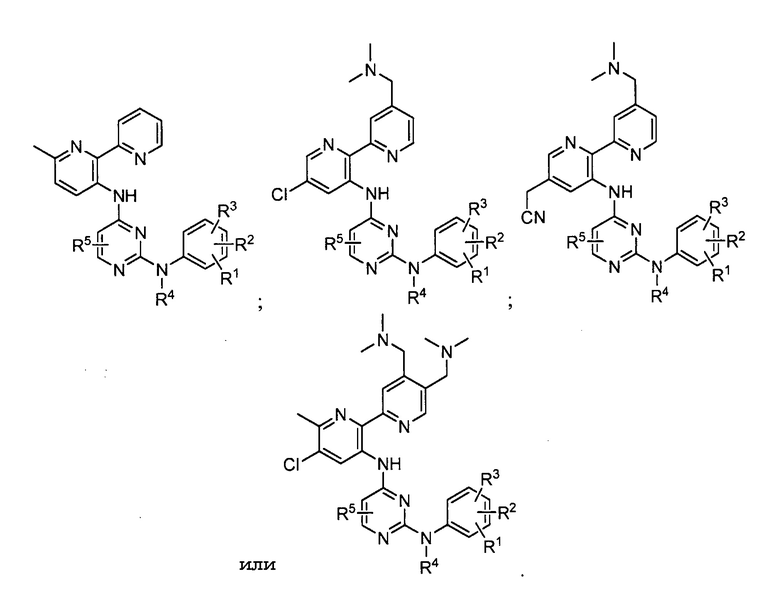
или его фармацевтически приемлемая соль.
26. Соединение по п. 1, где соединение выбирают из соединений, имеющих следующие структуры:
 ;
;  ;
;
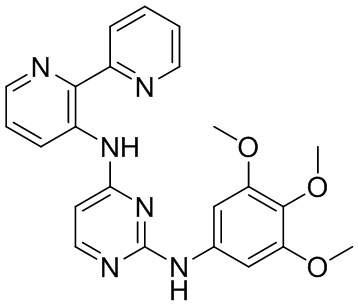 ;
; 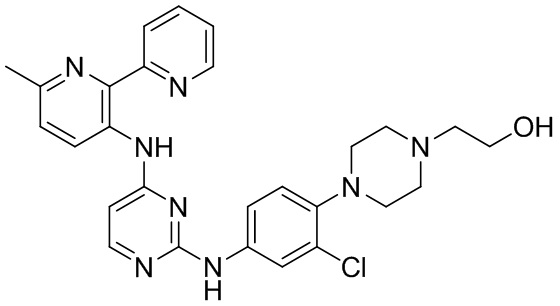 ;
;
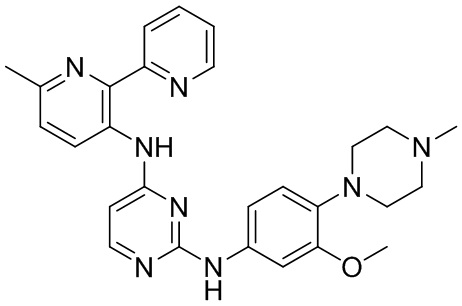 или
или 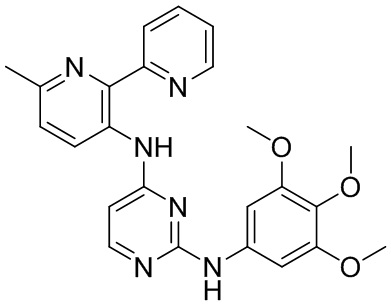 ,
,
или его фармацевтически приемлемая соль.
27. Соединение, имеющее следующую структуру
или его фармацевтически приемлемая соль.
28. Фармацевтическая композиция, обладающая свойствами ингибитора киназы ALK2, содержащая в эффективном количестве соединение по любому из пп. 1, 2, 26 или 27 или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
29. Способ ингибирования киназы ALK2 у нуждающегося в этом млекопитающего, где способ включает введение в организм млекопитающего эффективной дозы соединения по любому из пп. 1, 2, 26 или 27 или его фармацевтически приемлемой соли.
30. Способ по п. 29, в котором ингибирование осуществляется для лечения онкологического заболевания.
31. Способ по п. 29, в котором ингибирование осуществляется для лечения анемии хронического заболевания, анемии хронического воспаления, анемии онкологического заболевания или прогрессирующей оссифицирующей фибродисплазии.
32. Способ лечения миелопролиферативного расстройства, лимфомы, солидной опухоли, глиомы, анемии хронического заболевания, анемии хронического воспаления, анемии онкологического заболевания или прогрессирующей оссифицирующей фибродисплазии, опосредованных активностью киназы ALK2, у нуждающегося в этом млекопитающего, где способ включает введение в организм млекопитающего эффективной дозы соединения по любому из пп. 1, 2, 26 или 27 или его фармацевтически приемлемой соли.
33. Способ по п. 32, в котором миелопролиферативное заболевание является миелофиброзом, полицитемией или эссенциальной тромбоцитемией.
34. Способ по п. 32, в котором солидная опухоль является опухолью молочной железы, простаты или поджелудочной железы.
35. Способ по п. 32, в котором солидная опухоль является злокачественной солидной опухолью простаты, яичников или головы и шеи.
36. Применение соединения по любому из пп. 1, 2, 26 или 27 или его фармацевтически приемлемой соли в эффективной дозе в качестве средства для лечения и облегчения симптомов онкологического заболевания у нуждающегося в этом пациента, где симптомы опосредованы активностью киназы ALK2.
37. Применение по п. 36, где симптомы представляют собой анемию или патологическую усталость, связанную с онкологическим заболеванием.
38. Способ получения соединения по п. 2 или его фармацевтически приемлемой соли
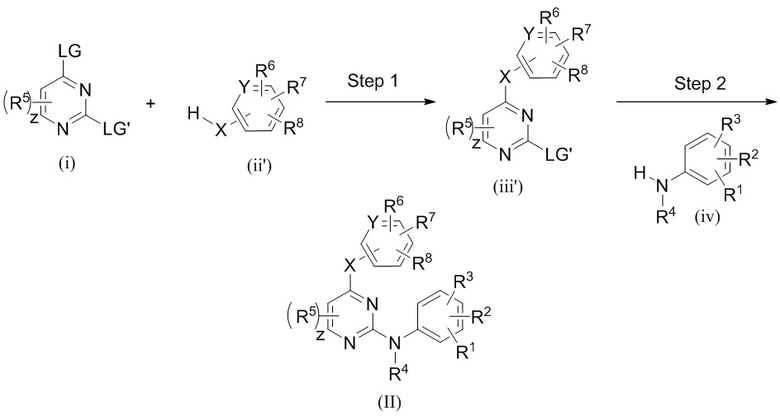 ,
,
где LG и LG' каждый независимо представляет собой уходящую группу, выбранную из галогена, и все остальные заместители имеют значения, определенные в п. 1,
где способ включает следующие стадии:
Стадию 1 (Step 1): взаимодействие Соединения (i) с Соединением (ii') с образованием Соединения (iii'); и
Стадию 2 (Step 2): взаимодействие Соединения (iii') с Соединением (iv) с образованием Соединения (II).
39. Способ по п. 38, где соединение, имеющее структуру (II), выбирают из следующих соединений:
; и
,
или их фармацевтически приемлемых солей.
40. Способ получения соединения, имеющего следующую структуру:
,
или его фармацевтически приемлемой соли, где способ включает следующую Стадию 1 (Step 1):
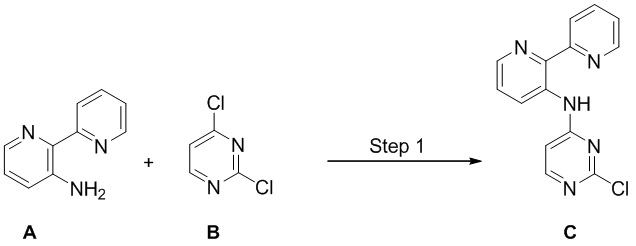 ,
,
и Стадию 2 (Step 2):
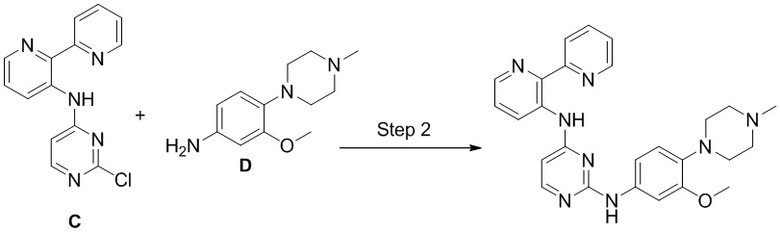 .
.
41. Способ по п. 40, где Стадия 1 включает comprises нагревание A и B в присутствии Na2CO3, и Стадия 2 включает нагревание C и D в присутствии трифторуксусной кислоты.
42. Способ получения соединения, имеющего следующую структуру:
,
или его фармацевтически приемлемой соли, где способ включает следующую Стадию 1(Step 1):
,
и Стадию 2(Step 2):
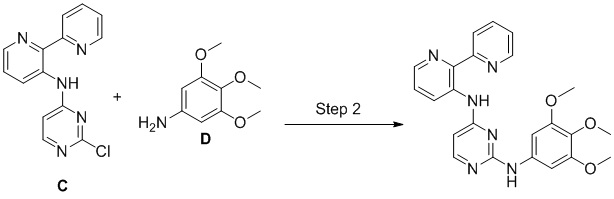 .
.
43. Способ получения соединения, имеющего следующую структуру:
,
или его фармацевтически приемлемой соли, где способ включает следующую Стадию 1 (Step 1):
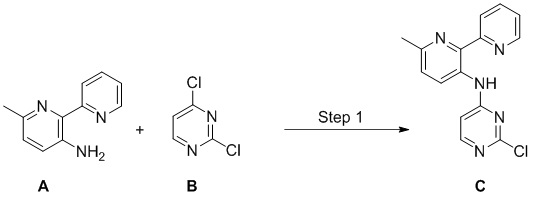 ,
,
и Стадию 2 (Step 2):
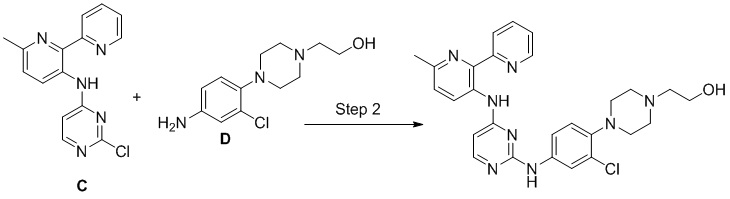 .
.
44. Способ получения соединения, имеющего следующую структуру:
,
или его фармацевтически приемлемой соли, где способ включает следующую Стадию 1 (Step 1):
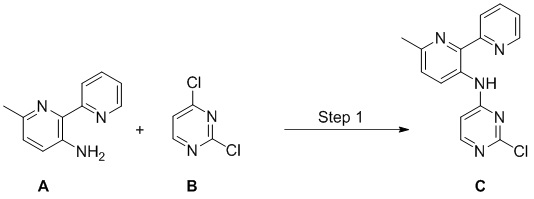 ,
,
и Стадию 2 (Step 2):
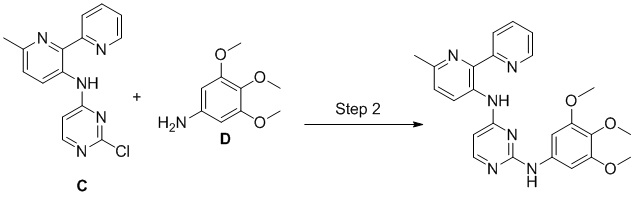
| WO2008106635 A1, 04.09.2008 | |||
| WO 200106816, 23.08.2001 | |||
| RU 2343148 C2, 10.01.2009 | |||
| RU 2010151355 A1, 10.08.2012 & RU 2536584, 27.12.2014 | |||
| RU 2008152195 А1, 20.07.2010. |

