Ссылка на родственные заявки
Согласно настоящей заявке испрашивается приоритет в соответствии с предварительной заявкой на выдачу патента США с серийным № 62/322017, поданной 13 апреля 2016 года, полное содержание которой включено в настоящий документ посредством ссылки.
Область техники, к которой относится настоящее изобретение
Настоящая заявка относится к агонистам активируемых пролифератором пероксисом рецепторов (PPAR), в частности, PPAR-дельта (PPARδ), и к способам их применения, таким как для лечения или предупреждения одного или нескольких связанных с PPARδ заболеваний.
Предшествующий уровень техники настоящего изобретения
Активируемый пролифератором пероксисом рецептор-дельта (PPARδ) является ядерным рецептором, который способен регулировать митохондриальный биосинтез. Как показано в PCT/2014/033088, включенной в настоящий документ посредством ссылки, модуляция активности PPARδ применима для лечения заболеваний, задержек развития и симптомов, связанных с дисфункцией митохондрий, таких как болезнь Альперса, MERRF – заболевание миоклонической эпилепсии с рваными мышечными волокнами, синдром Пирсона и подобных. Модуляция активности PPARδ эффективна при лечении других состояний, таких как мышечные заболевания, демиелинизирующие заболевания, сосудистые заболевания и метаболические заболевания. Действительно, PPARδ является важной биологической целью для соединений, используемых для лечения и предупреждения митохондриальных заболеваний, связанных с мышцами заболеваний и расстройств, а также других родственных состояний.
Следовательно, в данной области сохраняется потребность в новых соединениях, способных эффективно и безопасно активировать PPARδ in vitro и in vivo. Также существует потребность в активирующих PPARδ соединениях с улучшенными фармакокинетическими свойствами и улучшенной метаболической стабильностью. Настоящее изобретение отвечает этим и другим таким потребностям.
Краткое раскрытие настоящего изобретения
В настоящем документе представлены, inter alia, соединения и композиции, содержащие такие соединения, которые применимы для усиления активности PPARδ. В частности, в настоящем документе раскрываются способы модулирования активности PPARδ для лечения заболеваний, задержек развития и симптомов, связанных с дисфункцией митохондрий (см., например, пример 1). Например, раскрываемые соединения и композиции применимы в лечении митохондриальных заболеваний, таких как болезнь Альперса, CPEO - хроническая прогрессирующая внешняя офтальмоплегия, синдром Кирнса-Сейра (KSS), наследственная оптическая нейропатия Лебера (LHON), MELAS - митохондриальная миопатия, энцефаломиопатия, лактатацидоз и подобные инсульту эпизоды, MERRF - заболевание миоклонической эпилепсии с рваными мышечными волокнами, NARP - нейрогенная мышечная слабость, атаксия, пигментный ретинит и синдром Пирсона. В качестве альтернативы, раскрываемые соединения и композиции применимы в лечении других связанных с PPARδ заболеваний, таких как почечные заболевания, мышечные заболевания, демиелинизирующие заболевания, сосудистые заболевания и метаболические заболевания.
Более того, соединения, раскрываемые в настоящем документе, обладают определенными преимуществами над подобными соединениями, известными в уровне техники. В частности, соединения, раскрываемые в настоящем документе, лишь слегка ингибируют, если вообще ингибируют, активность hERG даже при высоких концентрациях. Дополнительные подробности, касающиеся активности раскрываемых в настоящем документе соединений против hERG представлены в примере 1a.
Согласно одному варианту осуществления, в настоящем документе представлено соединение формулы (I),
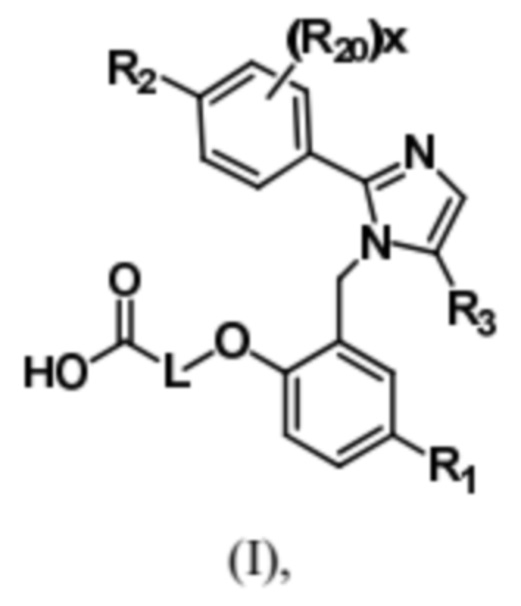
или его фармацевтически приемлемая соль, в которых
L представляет собой (CH2)5, который необязательно замещен одной метильной группой;
R1 представляет собой водород, галоген, C1–C4-алкил, C1–C4-галогеналкил, CN, C1–C4-алкокси, C1–C4-галогеналкокси или C3–C6-циклоалкил;
R2 представляет собой водород, галоген, CN, C1–C4-алкил, C1–C4-галогеналкил, C3–C6-циклоалкил, C1–C4-алкокси, C1–C4-галогеналкокси, S(C1–C4-алкил), SO2(C1–C4-алкил), 5- или 6-членный гетероцикл, арил, 5-членный гетероарил,  –R2A, O(CH2)mR2B, NH(C1–C4-алкил), N(C1–C4-алкил)2 или C(O)(C1–C4-алкил), при этом арил и гетероарил необязательно замещены галогеном, OH, CN, C1–C4-алкилом, формилом, ацетилом, ацетокси или карбокси, и при этом m представляет собой целое число, имеющее значение 1, 2 или 3;
–R2A, O(CH2)mR2B, NH(C1–C4-алкил), N(C1–C4-алкил)2 или C(O)(C1–C4-алкил), при этом арил и гетероарил необязательно замещены галогеном, OH, CN, C1–C4-алкилом, формилом, ацетилом, ацетокси или карбокси, и при этом m представляет собой целое число, имеющее значение 1, 2 или 3;
x представляет собой целое число, имеющее значение 0 или 1;
каждый R2A и R2B независимо представляет собой C1–C4-алкил, C1–C4-галогеналкил или C3–C6-циклоалкил;
R3 представляет собой С1-C4-галогеналкил, -NO2, -CN, галоген или C(O)O(C1–C4-алкил); и
каждый R20 независимо представляет собой галоген, C1–C4-алкил, CN или C1–C4-алкокси.
Также в настоящем документе раскрываются фармацевтические композиции раскрываемых соединений. Конкретные варианты осуществления предусматривают фармацевтически приемлемые носитель или наполнитель и одно или несколько раскрываемых соединений или их фармацевтически приемлемую соль. Фармацевтические композиции в соответствии с настоящим изобретением могут быть использованы в терапии, например, для лечения связанного с PPARδ заболевания или состояния у субъекта.
Другой вариант осуществления предусматривает лечение связанного с PPARδ заболевания или состояния у субъекта путем введения субъекту терапевтически эффективного количества одного или нескольких раскрываемых соединений, или их фармацевтически приемлемой соли, или фармацевтической композиции, содержащей соединение(ия).
Также в настоящем документе представлено применение одного или нескольких раскрываемых соединений, или их фармацевтически приемлемой соли, или фармацевтической композиции, содержащей одно или несколько раскрываемых соединений, для получения медицинского препарата для лечения связанного с PPARδ заболевания или состояния.
Согласно другому варианту осуществления в настоящем документе представлены раскрываемые соединения, или их фармацевтически приемлемая соль, или фармацевтическая композиция, содержащая одно или несколько раскрываемых соединений, для применения в лечении связанного с PPARδ заболевания или состояния.
Подробное раскрытие настоящего изобретения
Активируемый пролифератором пероксисом рецептор-дельта (PPAR-δ), также известный как активируемый пролифератором пероксисом рецептор-бета (PPAR-β) или как NR1C2 (подсемейство ядерных рецепторов 1, группа C, представитель 2), относится к белку ядерного рецептора, который функционирует как фактор транскрипции, регулирующий экспрессию генов. Лиганды PPARδ могут обеспечивать пролиферацию миобластов после повреждения, такого как повреждение скелетной мускулатуры. Последовательности PPARδ (OMIM 600409) являются общедоступными, например, из базы данных последовательностей GenBank® (например, под номерами доступа NP_001165289.1 (человек, белок), NP_035275 (мышь, белок), NM_001171818 (человек, нуклеиновая кислота) и NM_011145 (мышь, нуклеиновая кислота)).
В настоящем документе фраза «агонист PPARδ» относится к веществам, которые повышают активность PPARδ. Вещества могут быть тестированы по их агонистической активности в отношении PPARδ путем введения вещества в контакт с клетками, экспрессирующими PPARδ, выявления их связывания с PPARδ, а затем выявления сигналов, которые служат индикатором активации PPARδ. См., например, пример 1a.
Определения
Термин «алкил», используемый отдельно или как часть более крупного фрагмента, такого как «алкокси», «галогеналкил», «галогеналкокси», «циклоалкил» и подобные, означает насыщенный алифатический прямоцепочечный или разветвленный одновалентный углеводородный радикал. Если не указано иное, алкильная группа, как правило, имеет 1-4 атома углерода, т.e. C1–C4-алкил. Используемый в настоящем документе термин «C1–C4-алкильная» группа означает радикал, имеющий от 1 до 4 атомов углерода в линейном или разветвленном расположении, и включает в себя метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил.
Термин «алкокси» означает алкильный радикал, присоединяющийся через атом кислорода, представленный как –O-алкил. Например, термин «C1–C4-алкокси» включает в себя метокси, этокси, пропокси, изопропокси и бутокси.
Термины «галогеналкил» и «галогеналкокси» означают алкил или алкокси, который в зависимости от случая может быть замещен одним или несколькими атомами галогена. Например, термин «C1–C4-галогеналкил» включает в себя фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, бромметил, фторэтил, дифторэтил, дихлорэтил и хлорпропил, а «C1–C4-галогеналкокси» включает в себя фторметокси, дифторметокси, трифторметокси, хлорметокси, дихлорметокси, бромметокси, фторэтокси, дифторэтокси, дихлорэтокси и хлорпропокси.
Термин «галоген» означает фтор или фторо (F), хлор или хлоро (Cl), бром или бромо (Br), или йод или йодо (I).
Термин «арил» относится к карбоциклической ароматической группе. Примеры «арила» включают в себя фенил, нафтил, антраценил, 1,2-дигидронафтил, 1,2,3,4-тетрагидронафтил, флуоренил, инданил и инденил.
Термин «циклоалкил» означает 3–6 членный насыщенный алифатический циклический углеводородный радикал. Он может быть моноциклическим, бициклическим (например, мостиковым или слитым бициклическим кольцом) или трициклическим. Например, моноциклический C3-C6-циклоалкил означает радикал, имеющий от 3 до 6 атомов углерода, расположенных в моноциклическом кольце. Например, термин «C3–C6-циклоалкил» включает в себя без ограничения циклопропил, циклобутил, циклопентил и циклогексил.
Термин «5- или 6-членный гетероцикл» означает неароматический радикал, имеющий 5 или 6 кольцевых атомов (в том числе 1-3 кольцевых гетероатома), расположенных в моноциклическом кольце. Примеры «5- или 6-членного гетероцикла» включают в себя без ограничения морфолинил, тиоморфолинил, пирролидинонил, пирролидинил, пиперидинил, пиперазинил, гидантоинил, валеролактамил, дигидроимидазол, дигидрофуранил, дигидропиранил, дигидропиридинил, дигидропиримидинил, дигидротиенил, дигидротиофенил, дигидротиопиранил, тетрагидроимидазол, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил и тетрагидротиопиранил.
Термин «5-членный гетероарил» означает моноциклическую ароматическую кольцевую систему, имеющую пять кольцевых атомов, выбранных из углерода и по меньшей мере одного (как правило, 1-3, чаще 1 или 2) кольцевых гетероатомов (например, кислорода, азота или серы). Типичным примером является 5-членный гетероарил, содержащий 1 или 2 атома, независимо выбранных из атомов азота, атомов серы и атомов кислорода, такой как пирролил, тиенил, фурил, имидазолил, пиразолил, изотиазолил, изоксазолил и подобные.
Если группа описывается как являющаяся «замещенной», то отличный от водорода заместитель находится на месте водорода на углероде, сере или азоте группы. Таким образом, например, замещенный алкил представляет собой алкил, в котором по меньшей мере один отличный от водорода заместитель находится на месте водорода на алкиле. В качестве иллюстрации, монофторалкил представляет собой алкил, замещенный заместителем фтором, а дифторалкил представляет собой алкил, замещенный двумя заместителями фторами. Следует учитывать, что, если в группе имеется более чем одно замещение, то каждый отличный от водорода заместитель может быть идентичным или отличающимся (если не указано иное).
Соединения, имеющие один или несколько хиральных центров, могут существовать в различных стереоизомерных формах. Стереоизомеры представляют собой соединения, которые отличаются только своим пространственным расположением. Стереоизомеры включают в себя все диастереомерные, энантиомерные и эпимерные формы, а также рацематы и их смеси. Термин «геометрический изомер» относится к соединениям, имеющим по меньшей мере одну двойную связь, при этом двойная связь(связи) может существовать в формах цис (также называемой «syn» или «entgegen (E)») или транс (entgegen «anti» или «zusammen (Z)»), а также в их смесях. Если раскрываемое соединение называют или изображают с помощью структуры без указания стереохимии, то это означает, что название или структура охватывает один или несколько из возможных стереоизомеров или геометрических изомеров, или смесь охваченных стереоизомеров или геометрических изомеров.
Если геометрический изомер или стереоизомер указывают с помощью названия или структуры, то следует учитывать, что названный или изображенный изомер присутствует в большей степени, чем другой изомер, то есть, что геометрическая изомерная чистота названного или изображенного геометрического изомера превышает 50%, например, чистота составляет по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% по массе. Геометрическую изомерную чистоту определяют путем деления массы названного или изображенного геометрического изомера в смеси на суммарную массу всех геометрических изомеров в смеси.
Рацемическая смесь означает, что 50% составляет один энантиомер, и 50% составляет соответствующий энантиомер. Если соединение с одним хиральным центром называют или изображают с указанием стереохимии хирального центра, то следует учитывать, что название или структура охватывает как энантиомерно чистые, так и энантиомерно обогащенные или рацемические формы соединения. Если соединения с двумя или более хиральными центрами называют или изображают с указанием стереохимии хиральных центров, то следует учитывать, что название или структура охватывает все возможные диастереомерные формы (например, диастереомерно чистые, диастереомерно обогащенные и эквимолярные смеси одного или нескольких диастереомеров (например, рацемические смеси) соединения.
Энантиомерные и диастереомерные смеси могут быть разделены на составляющие их энантиомеры или стереоизомеры хорошо известными способами, такими как хиральная газовая хроматография, хиральная высокоэффективная жидкостная хроматография, кристаллизация соединения как хирального солевого комплекса или кристаллизация соединения в хиральном растворителе. Энантиомеры и диастереомеры также могут быть получены из диастереомерно- или энантиомерно чистых промежуточных соединений, реагентов и катализаторов хорошо известными способами асимметричного синтеза.
Если соединение обозначают названием или структурой, которые указывают на один энантиомер, если не указано иное, соединение является по меньшей мере на 60%, 70%, 80%, 90%, 99% или 99,9% оптически чистым (также называется «энантиомерно чистым»). Оптическая чистота представляет собой массу в смеси названного или обозначенного энантиомера, поделенную на суммарную массу в смеси обоих энантиомеров.
Если стереохимию раскрываемого соединения называют или изображают с помощью структуры, и названная или изображенная структура охватывает более чем один стереоизомер (например, как в диастереомерной паре), то следует учитывать, что предусматривается один из охваченных стереоизомеров или любая смесь охваченных стереоизомеров. Кроме того, следует учитывать, что стереоизомерная чистота названных или изображенных стереоизомеров составляет по меньшей мере 60%, 70%, 80%, 90%, 99% или 99,9% по массе. Стереоизомерную чистоту в данном случае определяют путем деления суммарной массы в смеси стереоизомеров, охватываемых названием или структурой, на суммарную массу в смеси всех стереоизомеров.
Принципы настоящего изобретения предусматривают фармацевтически приемлемые соли соединений, раскрываемых в настоящем документе. Раскрываемые соединения содержат оснóвные аминные группы и, поэтому, могут формировать фармацевтически приемлемые соли с фармацевтически приемлемой кислотой(ами). Подходящие фармацевтически приемлемые соли присоединения кислоты соединений, описываемых в настоящем документе, включают в себя соли неорганических кислот (таких как хлористоводородная кислота, бромистоводородная, фосфорная, азотная и серная кислоты) и органических кислот (таких как, например, уксусная кислота, бензолсульфоновая, бензойная, метансульфоновая и п-толуолсульфоновая кислоты). Раскрываемые соединения содержат карбоциклическую группу и, поэтому, могут формировать фармацевтически приемлемые соли с фармацевтически приемлемой кислотой(ами). Соединения в соответствии с принципами настоящего изобретения с кислотными группами, такие как карбоновые кислоты, могут формировать фармацевтически приемлемые соли с фармацевтически приемлемым основанием(ями). Подходящие фармацевтически приемлемые оснóвные соли включают в себя соли аммония, соли щелочных металлов (такие как соли натрия и калия) и соли щелочноземельных металлов (такие как соли магния и кальция).
Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к фармацевтическим солям, которые с медицинской точки зрения подходят для применения в контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения и аллергической реакции и соизмеримы с разумным соотношением пользы и риска. Фармацевтически приемлемые соли хорошо известны в уровне техники. Например, S. M. Berge, et al. описывают фармакологически приемлемые соли в J. Pharm. Sci., 1977, 66:1-19.
Нейтральные формы соединений в соответствии с настоящим изобретением повторно создают из их соответствующих солей путем введения в контакт соли с основанием или кислотой и выделения исходного соединения традиционным способом. Исходная форма соединения может отличаться от различных форм соли определенными физическими свойствами, такими как растворимость в полярных растворителях. Нейтральные формы соединений, раскрываемых в настоящем документе, также охватываются настоящим изобретением.
Используемые в настоящем документе термины «вводить», «процесс введения», «введение» и подобные относятся к способам, которые могут быть использованы для обеспечения доставки композиций в желаемый участок биологического действия. Такие способы включают в себя без ограничения внутрисуставной (в суставы), внутривенный, внутримышечный, внутриопухолевый, внутрикожный, внутриперитонеальный, подкожный, пероральный, местный, интратекальный, ингаляционный, чрескожный, ректальный и подобные. Методики введения, которые могут быть использованы со средствами и способами, описываемыми в настоящем документе, находятся, например, в Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; и Remington’s, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pa.
Используемые в настоящем документе термины «совместное введение», «введенный в комбинации с» и их грамматические эквиваленты предусматривают введение двух или более терапевтических средств одному субъекту и включают в себя режимы лечения, при которых средства вводят одинаковым или отличным путем введения или в одни и те же, или в разные моменты времени. Согласно некоторым вариантам осуществления одно или несколько соединений, описываемых в настоящем документе, будут вводить совместно с другими средствами. Данные термины охватывают введение двух или более средств субъекту так, что оба средства и/или их метаболиты присутствуют у субъекта в одно и то же время. Они включают в себя одновременное введение в отдельных композициях, введение в разные моменты времени в отдельных композициях и/или введение в композиции, в которой присутствуют оба средства. Таким образом, согласно некоторым вариантам осуществления соединения, описываемые в настоящем документе, и другое средство(а) вводят в одной композиции. Согласно некоторым вариантам осуществления соединения, описываемые в настоящем документе, и другое средство(а) смешивают в композицию.
Как правило, эффективное количество соединения, раскрываемого в настоящем документе, варьирует в зависимости от различных факторов, таких как данное лекарственное средство или соединение, фармацевтический состав, путь введения, тип заболевания или нарушения, персональные особенности субъекта или реципиента, подлежащего лечению, и подобные, но тем не менее оно может быть определено специалистом в данной области. Эффективные количество соединения в соответствии с принципами настоящего изобретения может быть легко определено специалистом в данной области рутинными способами, известными в уровне техники.
Термин «эффективное количество» или «терапевтически эффективное количество» означает количество, ведение которого субъекту дает полезные или желаемые результаты, в том числе клинические результаты, например, ингибирует, подавляет или уменьшает симптомы состояния, подлежащего лечению, у субъекта по сравнению с контролем. Например, терапевтически эффективное количество может быть обеспечено в единичной дозированной форме (например, от 1 мг до приблизительно 50 г в сутки, например, от 1 мг до приблизительно 5 грамм в сутки).
Конкретный способ введения и режим введения дозы будет выбран лечащим врачом с учетом конкретных особенностей случая (например, субъекта, заболевания, состояния сопутствующего заболевания, конкретного лечения и того, является ли лечение профилактическим). Лечение может предусматривать дозы один раз в сутки или несколько раз в сутки или менее чем один раз в сутки (например, один раз в неделю или один раз в месяц и т.д.) на протяжении периода от нескольких суток до месяцев или даже лет. Однако специалист в данной области сможет сразу же определить подходящие и/или эквивалентные дозы, рассматривая дозировки композиций, одобренных для лечения связанного с PPARδ заболевания, с использованием раскрываемых агонистов PPAR в качестве руководства.
Термин «субъект» означает млекопитающего, предпочтительно человека, но также может означать животное, нуждающееся в ветеринарном лечении, например, животных-компаньонов (например, собак, кошек и подобных), сельскохозяйственных животных (например, коров, овец, свиней, лошадей и подобных) и лабораторных животных (например, крыс, мышей, морских свинок и подобных).
Термины «фармацевтически приемлемый наполнитель» и «фармацевтически приемлемый носитель» относятся к веществам, которые обеспечивают составление, и/или введение активного средства, и/или абсорбирование у субъекта и могут быть включены в композиции в соответствии с настоящим раскрытием без причинения субъекту существенного неблагоприятного токсикологического эффекта. Неограничивающие примеры фармацевтически приемлемых носителей и наполнителей включают в себя воду, NaCl, нормальные солевые растворы, лактированный раствор Рингера, нормальную сахарозу, нормальную глюкозу, связующие, наполнители, разрыхлители, смазки, покрытия, подсластители, ароматизаторы, солевые растворы (такие как раствор Рингера), спирты, масла, желатины, углеводы, такие как лактоза, амилоза или крахмал, сложные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон, красители и подобные. Такие препараты могут быть стерилизованы и, при необходимости, смешаны со вспомогательными средствами, такими как смазки, консерванты, стабилизаторы, увлажняющие средства, эмульгаторы, соли для воздействия на осмотическое давление, буферы, красители и/или ароматические вещества и подобные, которые не оказывают вредного воздействия или влияния на активность соединений, представленных в настоящем документе. Специалисту в данной области будет понятно, что для применения с раскрываемыми соединениями подходят и другие фармацевтические носители и наполнители.
Соединения в соответствии с настоящим изобретением
Согласно вариантам осуществления в настоящем документе раскрывается соединение, характеризующееся общей формулой (I),
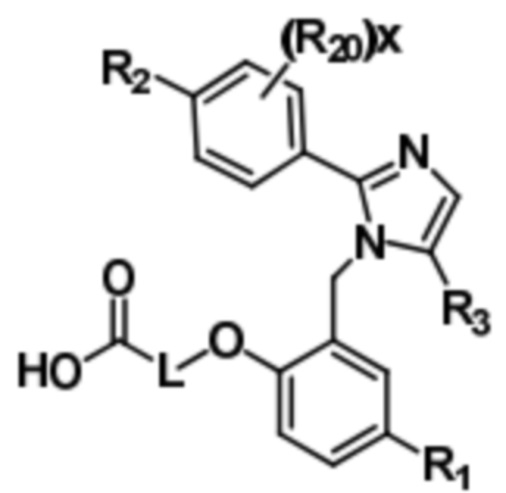
соединение формулы (I),
или его фармацевтически приемлемая соль, в которых
L представляет собой (CH2)5, который необязательно замещен одной метильной группой;
R1 представляет собой водород, галоген, C1–C4-алкил, C1–C4-галогеналкил, CN, C1–C4-алкокси, C1–C4-галогеналкокси или C3–C6-циклоалкил;
R2 представляет собой водород, галоген, CN, C1–C4-алкил, C1–C4-галогеналкил, C3–C6-циклоалкил, C1–C4-алкокси, C1–C4-галогеналкокси, S(C1–C4-алкил), SO2(C1–C4-алкил), 5- или 6-членный гетероцикл, арил, 5-членный гетероарил, –R2A, O(CH2)mR2B, NH(C1–C4-алкил), N(C1–C4-алкил)2 или C(O)(C1–C4-алкил), при этом арил и гетероарил необязательно замещены галогеном, OH, CN, C1–C4-алкилом, формилом, ацетилом, ацетокси или карбокси, и при этом m представляет собой целое число имеющее значение 1, 2 или 3;
x представляет собой целое число, имеющее значение 0 или 1;
каждый R2A и R2B независимо представляет собой C1–C4-алкил, C1–C4-галогеналкил или C3–C6-циклоалкил;
R3 представляет собой С1-C4-галогеналкил, -NO2, -CN, галоген или C(O)O(C1–C4-алкил); и
каждый R20 независимо представляет собой галоген, C1–C4-алкил, CN или C1–C4-алкокси.
Согласно 1му варианту осуществления соединение характеризуется структурными формулами (Ia) или (Ib),
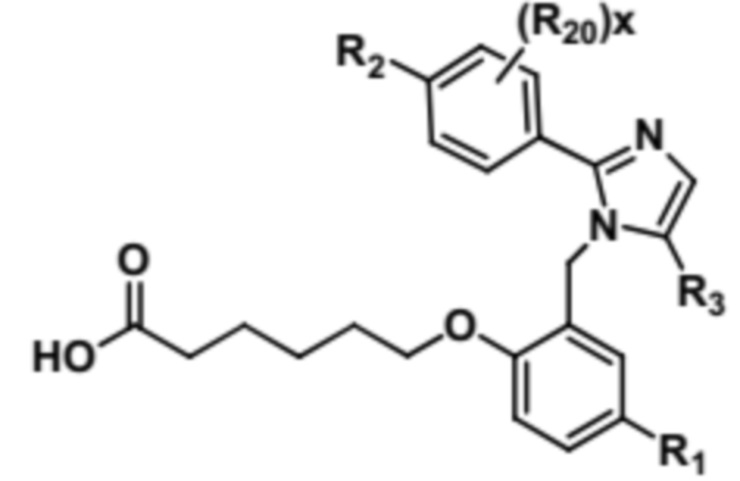
(Ia) или
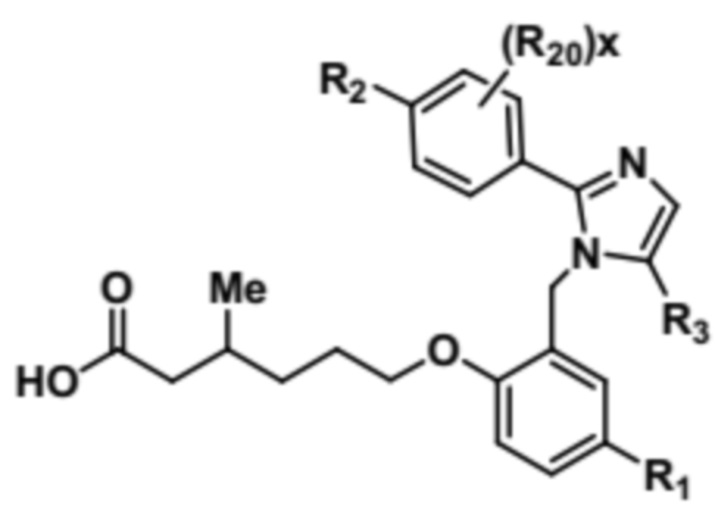
(Ib);
или его фармацевтически приемлемой солью, при этом переменные определяются также, как в формуле (I).
Согласно 2му варианту осуществления соединение характеризуется структурной формулой (Ibb),
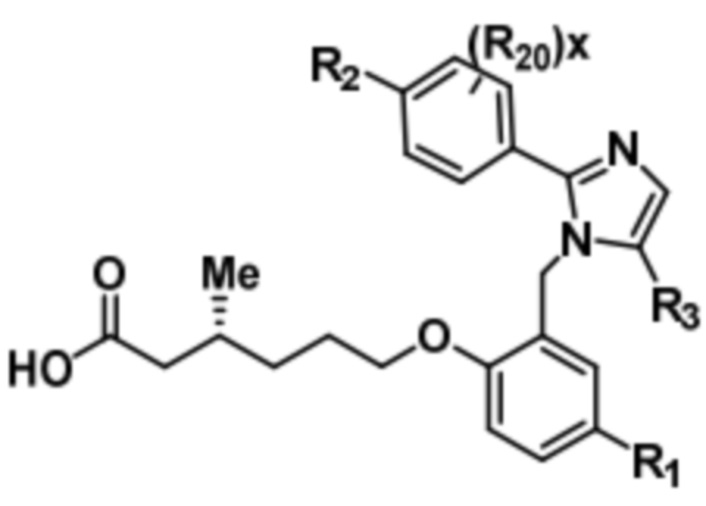
(Ibb);
или его фармацевтически приемлемой солью, при этом переменные определяются также, как в формуле (I).
Согласно 3му варианту осуществления соединение характеризуется структурными формулами (I), (Ia), (Ib) или (Ibb), при этом R3 представляет собой галогенметил, CN или галоген; а остальные переменные определяются также, как в формуле (I) или как в 1ом варианте осуществления.
Согласно 4му варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R3 представляет собой CF3, Cl или CN; а остальные переменные определяются также, как в формуле (I) или как в 1ом варианте осуществления.
Согласно 5му варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R2 представляет собой водород, галоген, CN, C1–C4-алкил, C1-C4 алкокси, C1–C4-галогеналкил, C1–C4-галогеналкокси, S(C1–C4-алкил) или фуранил, при этом фуранил необязательно может быть замещен C1–C4-алкилом; и x равняется 0 или 1; а остальные переменные определяются также, как в формуле (I) или как в 1ом, 2ом, 3ем или 4ом вариантах осуществления.
Согласно 6ому варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R2 представляет собой H, галоген, CN, CH3, галогенметил, галогенметокси, метокси или фуранил, при этом фуранил необязательно может быть замещен CH3; и R20 представляет собой метил или галоген; а остальные переменные определяются также, как в 5ом варианте осуществления.
Согласно 7ому варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R2 представляет собой H, F, Cl, CN, CF3, OCF3 или фуранил; и x равняется 0; а остальные переменные определяются также, как в 6ом варианте осуществления.
Согласно 8ому варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R1 представляет собой водород; а остальные переменные определяются также, как в формуле (I) или как в 1ом, 2ом, 3ем, 4ом, 5ом, 6ом или 7ом варианте осуществления.
Согласно 9ому варианту осуществления соединение характеризуется структурой формул (I), (Ia), (Ib) или (Ibb), при этом R1 представляет собой водород или фтор; а остальные переменные определяются также, как в формуле (I) или как в 1ом, 2ом, 3ем, 4ом, 5ом, 6ом, 7ом или 8ом варианте осуществления.
Согласно некоторым вариантам осуществления настоящее изобретение относится к любому из соединений, показанных в разделе примеров настоящей заявки; при этом фармацевтически приемлемые соли, а также нейтральные формы этих соединений также включены в настоящее изобретение. В частности, настоящее изобретение относится к любому из соединений, показанных в примерах 2A–2I; при этом фармацевтически приемлемые соли, а также нейтральные формы этих соединений также включены в настоящее изобретение. Согласно предпочтительным вариантам осуществления настоящее изобретение относится к любому из соединений 2a – 2i; при этом фармацевтически приемлемые соли, а также нейтральные формы этих соединений также включены в настоящее изобретение.
Способы получения соединений в соответствии с настоящим изобретением
Раскрываются способы получения соединений формул (I), (Ia) и (Ib). Как правило, соединение формулы (I) может быть получено путем осуществления реагирования соединения формулы (II),
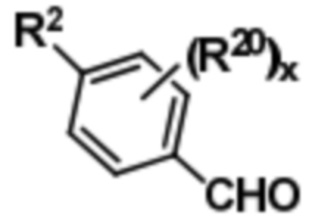
(II),
с этан-1,2-диамином с получением соединения формулы (III),
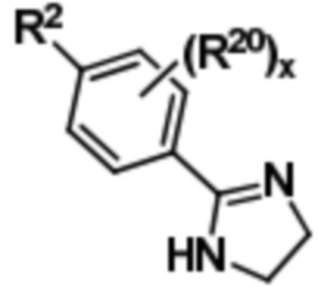
(III).
Соединение формулы (III) может быть подвергнуто окислительным условиям с получением соединения формулы (IV),
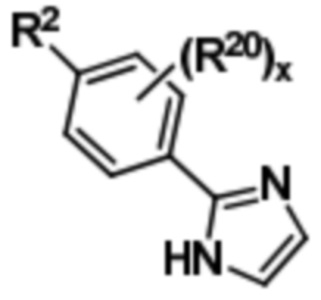
(IV).
Затем соединение формулы (IV) может быть подвергнуто реагированию с 2-метокси-5-(R1)-бензилбромидом с получением соединения формулы (V),
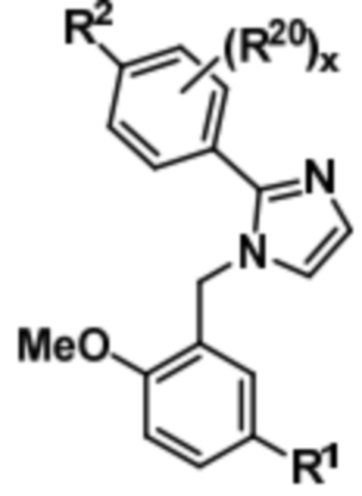
(V).
Соединение формулы (V) может быть подвергнуто реагированию с N-йодсукцинимидом (NIS) с получением соединения формулы (VI),
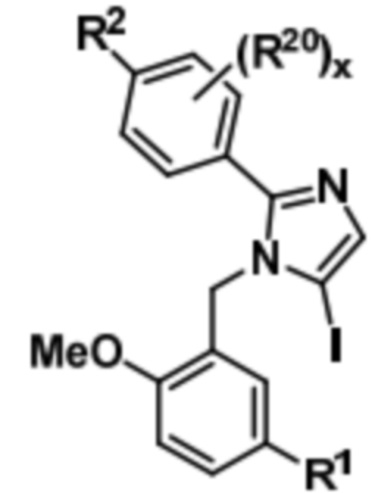
(VI).
Затем соединение формулы (VI) может быть подвергнуто реагированию с R3-Xa, при этом Xa представляет собой уходящую группу, с получением соединения формулы (VII),
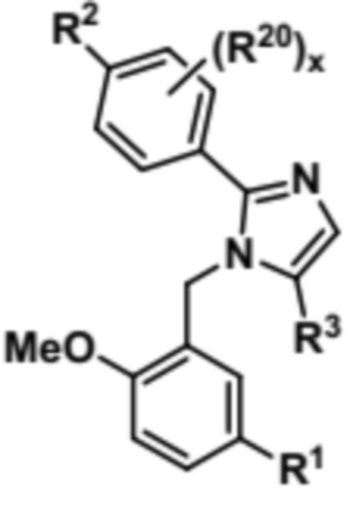
(VII).
Затем соединение формулы (VII) может быть подвергнуто условиям O-деметилирования с образованием соединения формулы (VIII),
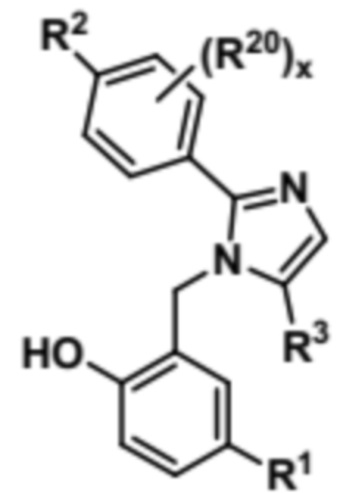
(VIII).
Затем соединение формулы (VIII) может быть подвергнуто реагированию с соединением формулы EtOCO-L-Br (IX) с получением соединения формулы (X),
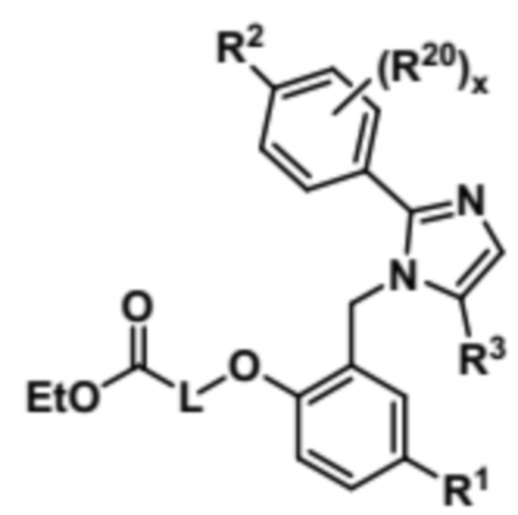
(X).
Наконец, соединение формулы (X) может быть подвергнуто условиям гидролиза с образованием соединения формулы (I).
Подобным образом, соединение формулы (Ia) может быть получено путем осуществления реагирования соединения формулы (VIII) с этил-6-бром-3-гексаноатом с получением соединения формулы (Xa),
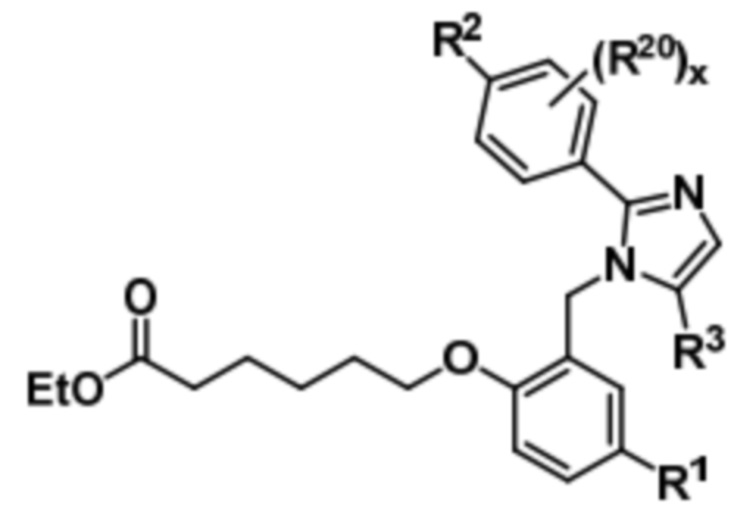
(Xa).
Последующий гидролиз соединения формулы (Xa) дает соединение формулы (Ia).
Подобным образом, соединение формулы (Ib) может быть получено путем осуществления реагирования соединения формулы (VIII) с этил-6-бром-3-метилгексаноатом с получением соединения формулы (Xb),
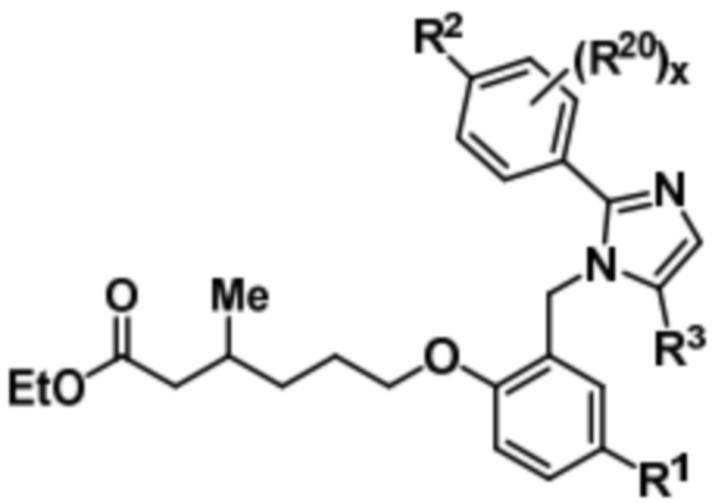
(Xb).
Последующий гидролиз соединения формулы (Xb) дает соединение формулы (Ib).
Подобным образом, соединение формулы (Ibb) может быть получено путем осуществления реагирования соединения формулы (VIII) с этил-(R)-6-бром-3-метилгексаноатом с получением соединения формулы (Xbb),
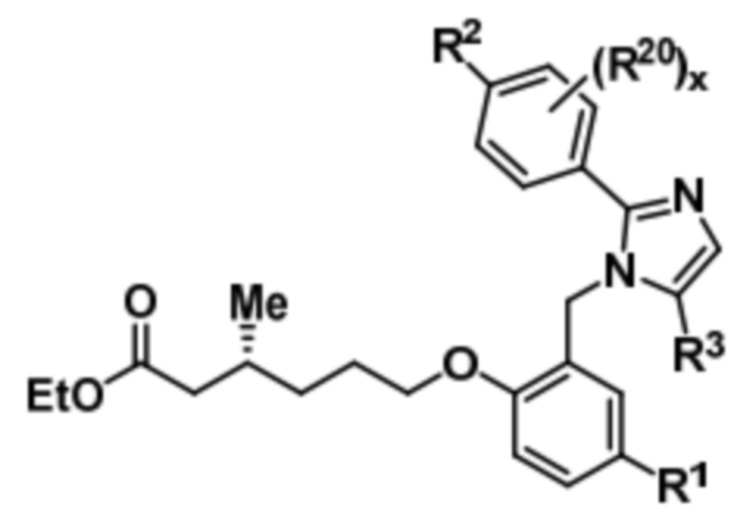
(Xbb).
Последующий гидролиз соединения формулы (Xbb) дает соединение формулы (Ibb).
Подробные протоколы синтеза для получения типичных соединений формул (I), (Ia), (Ib) и (Ibb) представлены в примерах 2A–2I.
Способы лечения
Раскрываются способы лечения связанного с PPARδ заболевания или состояния у субъекта. Способы могут предусматривать введение субъекту терапевтически эффективного количества одного или нескольких соединений или композиций, представленных в настоящем документе.
Согласно одному варианту осуществления связанным с PPARδ заболеванием является митохондриальное заболевание. Примеры митохондриальных заболеваний включают в себя без ограничения болезнь Альперса, CPEO - хроническую прогрессирующую внешнюю офтальмоплегию, синдром Кирнса-Сейра (KSS), наследственную оптическую нейропатию Лебера (LHON), MELAS - митохондриальную миопатию, энцефаломиопатию, лактатацидоз и подобные инсульту эпизоды, MERRF - заболевание миоклонической эпилепсии с рваными мышечными волокнами, NARP - нейрогенную мышечную слабость, атаксию, пигментный ретинит и синдром Пирсона.
Согласно другим вариантам осуществления связанным с PPARδ заболеванием является сосудистое заболевание (такое как сердечно-сосудистое заболевание или любое заболевание, при котором будет полезным усиление васкуляризации в тканях, демонстрирующих ухудшенный или недостаточный кровоток). Согласно другим вариантам осуществления связанным с PPARδ заболеванием является мышечное заболевание, такое как мышечная дистрофия. Примеры мышечной дистрофии включают в себя без ограничения мышечную дистрофию Дюшенна, мышечную дистрофию Беккера, тазово-плечевую мышечную дистрофию, врожденную мышечную дистрофию, плече-лопаточно-лицевую мышечную дистрофию, миотоническую мышечную дистрофию, окулофарингеальную мышечную дистрофию, дистальную мышечную дистрофию и мышечную дистрофию Эмери-Дрейфуса.
Согласно некоторым вариантам осуществления связанным с PPARδ заболеванием или состоянием является демиелинизирующее заболевание, такое как рассеянный склероз, болезнь Шарко-Мари-Тута, болезнь Пелицеуса-Мерцбахера, энцефаломиелит, нейромиелит зрительного нерва, адренолейкодистрофия или синдром Гийена-Барре.
Согласно другим вариантам осуществления связанным с PPARδ заболеванием является метаболическое заболевание. Примеры метаболических заболеваний включают в себя без ограничения ожирение, гипертриглицеридемию, гиперлипидемию, гипо-альфа-липопротеинемию, гиперхолестеринемию, дислипидемию, синдром X и сахарный диабет II типа.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является нарушение мышечной структуры. Примеры нарушений мышечной структуры включают в себя без ограничения миопатию Бетлема, болезнь центрального стержня, врожденную диспропорцию волокнистого типа, дистальную мышечную дистрофию (MD), MD Дюшенна и Беккера, MD Эмери-Дрейфуса, плече-лопаточно-лицевую MD, миопатию гиалиновых телец, тазово-плечевую MD, мышечные нарушения, связанные с натриевым каналами, миотоническую хондродистрофию, миотоническую дистрофию, миотубулярную миопатию, заболевание с образованием немалиновых телец, окулофарингеальную MD и недержание мочи при напряжении.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является нарушение нейрональной активации. Примеры нарушений нейрональной активации включают в себя без ограничения амиотрофический латеральный склероз, болезнь Шарко-Мари-Тута, синдром Гийена-Барре, синдром Ламберта-Итона, рассеянный склероз, миастению гравис, повреждение нерва, периферическую нейропатию, спинальную мышечную атрофию, поздний паралич локтевого нерва и токсическое нервно-мышечное нарушение.
Согласно другим вариантам осуществления связанным с PPARδ заболеванием является связанное с мышечным утомлением нарушение. Примеры связанных с мышечным утомлением нарушений включают в себя без ограничения синдром хронической усталости, сахарный диабет (I или II типа), болезнь накопления гликогена, фибромиалгию, атаксию Фридрейха, перемежающуюся хромоту, миопатию, обусловленную накоплением липидов, MELAS, мукополисахаридоз, болезнь Помпе и тиреотоксическую миопатию.
Согласно некоторым вариантам осуществления связанным с PPARδ заболеванием является связанное с мышечной массой нарушение. Примеры связанных с мышечной массой нарушений включают в себя без ограничения кахексию, дегенерацию хряща, церебральный паралич, синдром сдавливания, миопатию критических состояний, миозит с включенными тельцами, мышечную атрофию (дисфункциональную), саркопению, стероидную миопатию и системную красную волчанку.
Согласно другим вариантам осуществления связанным с PPARδ заболеванием является связанное с бета-окислением заболевание. Примеры связанных с бета-окислением заболеваний включают в себя без ограничения системный дефицит транспортера карнитина, дефицит карнитинпальмитоилтрансферазы (CPT) II, дефицит длинноцепочечной ацил-КоА-дегидрогеназы (LCHAD или VLCAD), дефицит трифункционального фермента, дефицит среднецепочечной ацил-КоА-дегидрогеназы (MCAD), дефицит короткоцепочечной ацил-КоА-дегидрогеназы (SCAD) и рибофлавин-чувствительные нарушения β-окисления (RR-MADD).
Согласно некоторым вариантам осуществления связанным с PPARδ заболеванием является сосудистое заболевание. Примеры сосудистых заболеваний включают в себя без ограничения недостаточность периферических сосудов, заболевание периферических сосудов, перемежающуюся хромоту, заболевание периферических сосудов (PVD), заболевание периферических артерий (PAD), окклюзионное заболевание периферических артерий (PAOD) и периферическую облитерирующую артериопатию.
Согласно другим вариантам осуществления связанным с PPARδ заболеванием является глазное сосудистое заболевание. Примеры глазных сосудистых заболеваний включают в себя без ограничения возрастную макулярную дегенерацию (AMD), болезнь Штаргардта, гипертензивную ретинопатию, диабетическую ретинопатию, ретинопатию, макулярную дегенерацию, кровоизлияние в сетчатку и глаукому.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является заболевание мышечного аппарата глаза. Примеры заболеваний мышечного аппарата глаза включают в себя без ограничения страбизм (косоглазие/блуждающий взгляд/дивергентный страбизм), прогрессивную внешнюю офтальмоплегию, изотропию, экзотропию, нарушение рефракции и аккомодации, гиперметропию, миопию, астигматизм, анизометропию, пресбиопию, нарушение аккомодации или внутреннюю офтальмоплегию.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является метаболическое заболевание. Примеры метаболических нарушений включают в себя без ограничения гиперлипидемию, дислипидемию, гиперхолестеринемию, гипертриглицеридемию, гипохолестеринемию за счет HDL, гиперхолестеринемию за счет LDL и/или холестеринемию не за счет HLD, гиперпротеинемию за счет VLDL, дислипопротеинемию, гипопротеинемию аполипротеина A-I, атеросклероз, заболевание артериального склероза, заболевание сердечно-сосудистой системы, сосудистое заболевание головного мозга, заболевание периферического кровообращения, метаболический синдром, синдром X, ожирение, сахарный диабет (I или II типа), гипергликемию, инсулиновую резистентность, нарушенную толерантность к глюкозе, гиперинсулинизм, диабетические осложнения, сердечную недостаточность, инфаркт миокарда, кардиомиопатию, гипертензию, неалкогольную жировую болезнь печени (NAFLD), неалкогольный стеатогепатит (NASH), тромб, болезнь Альцгеймера, нейродегенеративное заболевание, демиелинизирующее заболевание, рассеянный склероз, лейкодистрофию надпочечника, дерматит, псориаз, акне, старение кожи, трихоз, воспаление, артрит, астму, синдром повышенной чувствительности кишечника, язвенный колит, болезнь Крона и панкреатит.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является злокачественная опухоль. Примеры злокачественной опухоли включают в себя без ограничения злокачественные опухоли ободочной кишки, толстого кишечника, кожи, молочной железы, предстательной железы, яичника и/или легкого.
Согласно следующим вариантам осуществления связанным с PPARδ заболеванием является почечное заболевание. Примеры почечных заболеваний включают в себя без ограничения гломерулонефрит, гломерулосклероз, нефротический синдром, гипертонический нефросклероз, острый нефрит, рецидивную гематурию, персистирующую гематурию, хронический нефрит, быстро прогрессирующий нефрит, острое поражение почек (также известное как острая почечная недостаточность), хроническую почечную недостаточность, диабетическую нефропатию или синдром Барттера. В PCT/US2014/033088, включенной в настоящий документ посредством ссылки, показано, что генетическая и фармакологическая активация PPARδ способствует мышечной регенерация на мышиной модели острого термического поражения. Следовательно, также представлено применение PPARδ в качестве терапевтической цели для усиления регенеративной эффективности в отношении скелетной мускулатуры.
Фармацевтические композиции и их введение
Дополнительные терапевтические средства
Раскрываются фармацевтические композиции, которые включают в себя одно или несколько представленных в настоящем документе соединений (например, 1, 2, 3, 4 или 5 таких соединений) и, как правило, по меньшей мере одно дополнительное вещество, такое как наполнитель, известное терапевтическое средство, отличное от средств в соответствии с настоящим раскрытием, и их комбинации. Согласно некоторым вариантам осуществления раскрываемые агонисты PPAR могут быть использованы в комбинации с другими средствами, которые, как известно, обладают благоприятной активностью с раскрываемыми агонистами PPAR. Например, раскрываемые соединения могут быть введены отдельно или в комбинации с одним или несколькими другими агонистами PPAR, такими как тиазолидиндион, в том числе розиглитазон, пиоглитазон, троглитазон и их комбинации, или сульфонилмочевинное средство или его фармацевтически приемлемая соль, такие как толбутамид, толазамид, глипизид, карбутамид, глисоксепид, глизентид, глиборнурид, глибенкламид, гликвидон глимепирид, гликлазид и фармацевтически приемлемые соли этих соединений, или мураглитазар, фарглитазар, навеглитазар, нетоглитазон, ривоглитазон, K-111, GW-677954, (-)-галофенат, кислота, арахидоновая кислота, клофбрат, гемфиброзил, фенофибрат, ципрофибрат, безафибрат, ловастатин, правастатин, симвастатин, мевастатин, флувастатин, индометацин, фенопрофен, ибупрофен и фармацевтически приемлемые соли этих соединений.
Согласно одному варианту осуществления раскрываемые соединения могут быть введены в комбинации с дексамфетамином, амфетамином, мазиндолом или фентермином, а также могут быть введены в комбинации с медицинскими препаратами, обладающими противовоспалительным эффектом.
Кроме того, при использовании для лечения метаболического состояния фармацевтические композиции, представленные в настоящем документе, могут быть введены в качестве комбинированной терапии с одним или несколькими фармакологически активными веществами, обладающими благоприятными эффектами в отношении метаболических расстройств или нарушений. Например, раскрываемые фармацевтические композиции могут быть введены в комбинации с агонистами RXR для лечения метаболических и сердечно-сосудистых заболеваний, медицинскими препаратами, которые снижают содержание глюкозы в крови; противодиабетическими средствами, такими как инсулины и инсулиновые производные, в том числе лантус, апидра и другие инсулины быстрого действия, и модуляторами рецептора GLP-1; активными ингредиентами для лечения дислипидемий; противоатеросклеротическими медицинскими препаратами; средствами против ожирения; противовоспалительными активными ингредиентами; активными ингредиентами для лечения злокачественных опухолей; противотромботическими активными ингредиентами; активными ингредиентами для лечения высокого кровяного давления; активными ингредиентами для лечения сердечной недостаточности и их комбинациями.
Примеры
Пример 1a
Скрининг активности PPARδ
Клеточная культура и трансфекция. Клетки CV-1 выращивали в DMEM + 10% очищенного активированным углем FCS. Клетки высевали в 384-луночные планшеты за сутки до трансфекции с получением конфлюентности 50-80% при трансфекции. Трансфицировали всего 0,8 г ДНК, содержащей 0,64 микрограмма pCMX-PPARDelta LBD, 0,1 микрограмма pCMX.beta.Gal, 0,08 микрограмма репортера pGLMH2004 и 0,02 микрограмма pCMX пустого вектора, на лунку с использованием реагента для трансфекции FuGene в соответствии с инструкциями изготовителя (Roche). В клетках обеспечивали экспрессию белка в течение 48 часов после добавления соединения.
Плазмиды. Человеческий PPARδ использовали для ПЦР-амплификации PPARδ LBD. Амплифицировали кДНК лиганд-связывающего домена (LBD) изоформы PPARδ (от аминокислоты 128 PPARδ до C-конца) и сливали с ДНК-связывающим доменом (DBD) фактора транскрипции дрожжей GAL4 путем субклонирования фрагментов в рамку в векторе pCMX GAL (Sadowski et al. (1992), Gene 118, 137) с образованием плазмид pCMX-PPARDelta LBD. Осуществление слияний подтверждали путем секвенирования. Люциферазный репортер pCMXMH2004 содержит множество копий элемента отклика ДНК GAL4 под минимальным эукариотическим промотором (Hollenberg and Evans, 1988). Создавали pCMXβGal.
Соединения. Все соединения растворяли в DMSO и разбавляли 1:1000 при добавлении в клетки. Соединения тестировали в четырех повторностях при концентрациях, варьирующих от 0,001 до 100 мкM. Клетки обрабатывали соединением в течение 24 часов с последующим люциферазным анализом. Каждое соединение тестировали по меньшей мере в двух отдельных экспериментах.
Люциферазный анализ. Среду, содержащую тестируемое соединение, отсасывали и вымывали с помощью PBS. Затем в каждую лунку добавляли 50 мкл PBS, содержащего 1 мM Mg++ и Ca++. Люциферазный анализ выполняли с использованием набора LucLite в соответствии с инструкциями изготовителя (Packard Instruments). Испускание света количесвенно определяли путем подсчета на устройстве для считывания планшетов Perkin Elmer Envision. Для измерения 3-галактозидазной активности 25 мкл супернатанта из каждого лизата трансфекции переносили в новый 384-луночный микропланшет. Анализы с использованием бета-галактозидазы выполняли в планшетах с микролунками с использованием набора от Promega и считывали на устройстве для считывания планшетов Perkin Elmer Envision. Данные по бета-галактозидазе использовали для нормализации (эффективность трансфекции, клеточный рост и т.д.) данных по люциферазе.
Статистические способы. Активность соединения вычисляли как кратность индукции по сравнению с необработанным образцом. Для каждого соединения обеспечивали эффективность (максимальную активность) как относительную активность по сравнению с GW501516 - агонистом PPARδ. EC50 представляет собой концентрацию, обеспечивающую 50% максимальной наблюдаемой активности. Значения EC50 вычисляли с помощью нелинейной регрессии с использованием GraphPad PRISM (GraphPad Software, San Diego, Calif.).
Скрининг ингибирования hERG
Клеточная культура и сбор клеток. Клетки культивировали в среде DMEM/F-12, дополненной 10% эмбриональной телячьей сыворотки и 400 мкг/мл генетицина. Клетки выращивали в 75-см2 колбах для культуры ткани, поддерживали при 37ºC с 5% CO2 и пересевали каждые 3 суток с использованием 0,05/0,02% трипсина/EDTA (конфлюентность < 80%). Для электрофизиологического исследования клетки высевали в 25-см2 колбы для культуры ткани. Через трое суток клетки собирали с использованием трипсина/EDTA (0,025/0,01%) в качестве отсоединяющего средства и повторно суспендировали в наружном растворе для измерений.
Состав тестируемого препарата. Предварительно отвешенное количество тестируемого препарата растворяли в DMSO для культуры ткани с получением первичного маточного раствора. Концентрация первичного маточного раствора была 1000-кратной по отношению к предполагаемой наибольшей тестируемой концентрации. Затем готовили рабочие маточные растворы подходящих концентраций путем разбавления первичного маточного раствора с помощью DMSO. Маточные растворы аликвотировали в пробирки Эппендорфа и хранили при -20ºC замороженными (< 0ºC) до применения. В день эксперимента аликвоты размораживали и использовали для приготовления аналитического раствора (тестируемого раствора).
Тестируемые растворы готовили свежими путем добавления 4 мкл маточного раствора в 3996 мкл наружного раствора для измерений так, что конечная концентрация DMSO в аналитическом растворе составляла 0,1% объем/объем. При добавлении раствор тщательно проверяли против света невооруженным глазом на предмет каких-либо признаков осаждения.
Электрофизиологические процедуры
Тип фиксации потенциала: полуавтоматический.
Изготовитель: Nanion technologies, GmBH Germany.
Электрофизиологические чипы: покрытые стеклом чипы NPC-1.
Процентное ингибирование тока hERG. Установившийся ток после применения 0,1% DMSO рассматривали как исходный уровень (контрольный ток). Установившийся ток, полученный в конце добавления каждой тестируемой концентрации, использовали для вычисления % ингибирования тока hERG при каждой концентрации. Корректировали любое снижение из-за добавления среды-носителя для вычисления % ингибирования тока hERG. Каждая клетка действовала в качестве своего собственного контроля. Учитывали средний ток последних 3-5 выборок приемлемого качества для вычисления % ингибирования. Выборки с артефактами и шумом не учитывали при вычислении.
Таблица 1. Данные трансактивации PPARδ и ингибирования hERG
Пример 1b
Фармакокинетический скрининг
В данном примере определяли PK профиль некоторых агонистов PPARδ, раскрываемых в настоящем документе, у самцов мышей CD1 или у крыс Wistar способами, раскрытыми в Boxenbaum H. (1980), Interspecies variation in liver weight, hepatic blood flow and antipyrine intrinsic clearance in extrapolation of Benzodiazepines and phenytoin. J. Pharmacokinet Biopharm 8: 165-176. Подобные способы могут быть использованы для анализа других соединений, представленных в настоящем документе.
Все соединения вводили отдельно мышам CD1 при 1 мг/кг i.v. или при 3 мг/кг p.o. Соединения вводили отдельно самцам крыс Wistar при 1 мг/кг i.v. или при 3 мг/кг p.o. В следующей таблице вид M относится к мыши, а R относится к крысе. NA означает отсутствие данных.
Пример 2
Синтетическое получение вариантов осуществления соединения
Сокращения
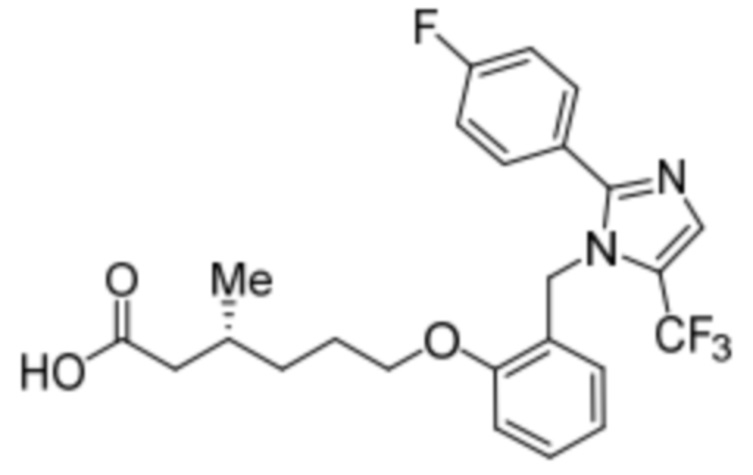
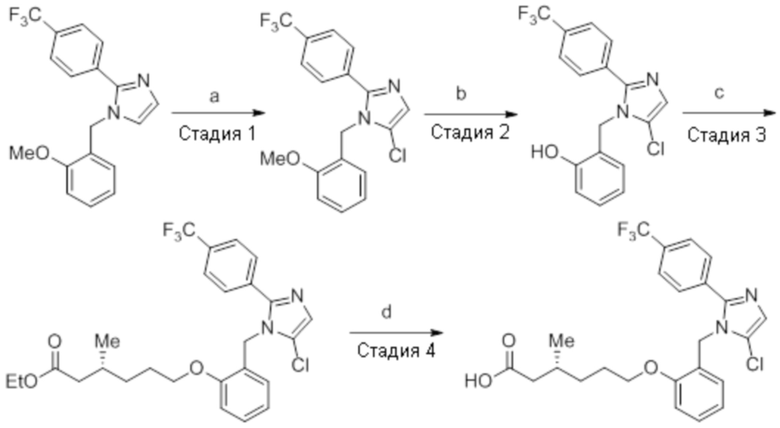
Пример 2A: Синтез Соединения 2a
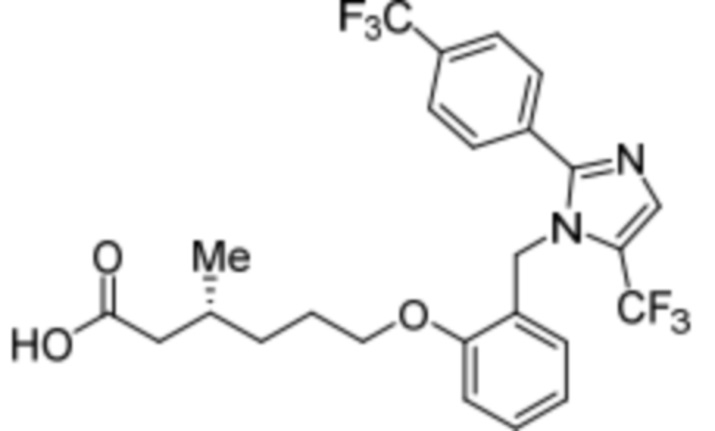
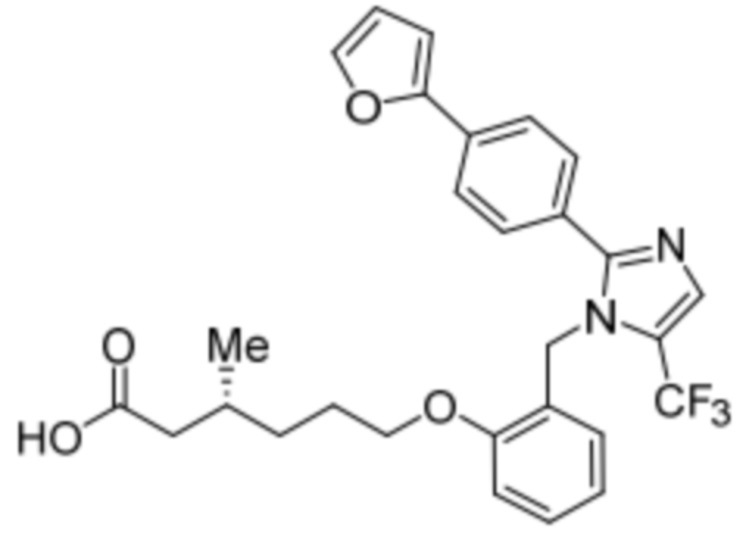
Синтез (3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоты
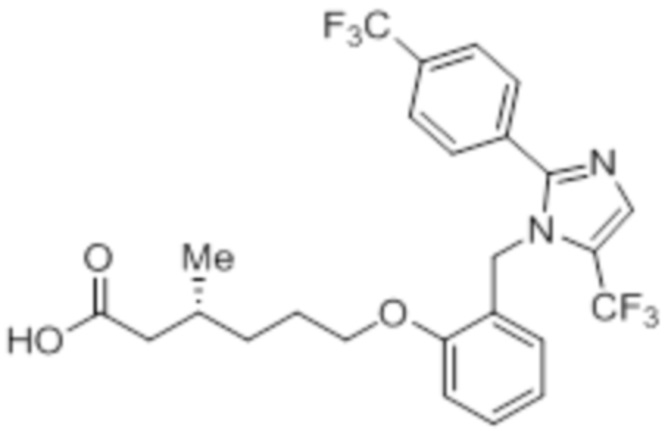
Схема 1:
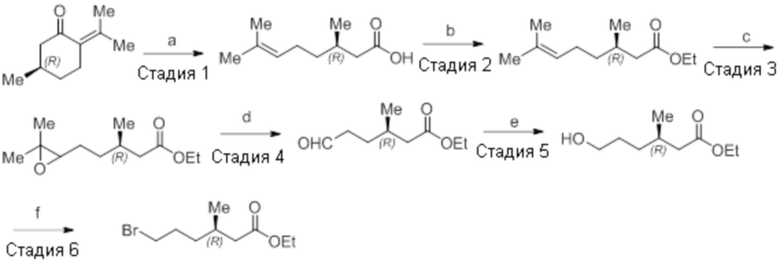
Реагенты и условия: a) i) HCl (газ), -30°C – к.т., 12 ч; ii) 4н NaOH, к.т., 12 ч; b) этилбромид, K2CO3, DMF, к.т., 2 ч; c) m-CPBA, Et2O, -30°C – 0°C, 20 ч; d) NaIO4, 1,4-диоксан, к.т., 12 ч; e) NaBH4, MeOH, к.т., 3 ч; f) PBr3, DCM, 0°C – к.т., 3 ч
Схема 2:
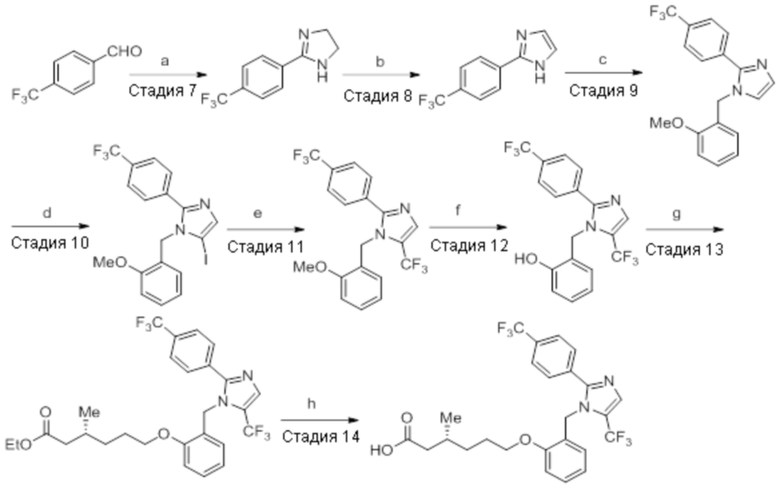
Реагенты и условия: a) этан-1,2-диамин, I2, K2CO3, tBuOH, 85°C, 5 ч; b) (диацетоксийод)бензол, K2CO3, DMSO, 12 ч; c) 2-метоксибензилбромид, NaH (60% дисперсия), DMF 0°C – к.т., 4 ч; d) NIS, MeCN, 70°C, 12 ч; e) TMSCF3, Ag2CO3, 1,10-фенантролин, KF, CuI, 100°C, 12 ч; f) BBr3, DCM, к.т., 3 ч; г) этил-(3R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 12 ч; h) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез (3R)-3,7-диметилокт-6-еновой кислоты:
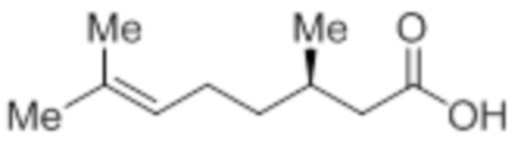
В трехгорлой круглодонной колбе емкостью 5 л, (R)-пулегон (150,0 г, 986,84 ммоль) продували газообразным HCl в течение 3 ч при -30°C. Реакционную смесь переносили в герметизируемый реакционный сосуд, и оставляли смесь отстаиваться при к.т. в течение 12 ч. Смесь обрабатывали раствором NaOH (4н, 3 л), и перемешивали полученную смесь при к.т. дополнительно в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой (1 л) и промывали диэтиловым эфиром (3×1 л). Водный слой подкисляли (pH 4) разбавленным HCl после чего экстрагировали диэтиловым эфиром (3×1 л). Объединенный органический слой сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (125 г, 74,8%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,01 (с, 1H), 5,07 (т, J=6,9 Гц, 1H), 2,22 (дд, J=15,0, 6,0 Гц, 1H), 2,03-1,78 (м, 4H), 1,64 (с, 3H), 1,56 (с, 3H), 1,36-1,17 (м, 2H), 0,88 (д, J=6,6 Гц, 3H).
Стадия 2: Синтез этил-(3R)-3,7-диметилокт-6-еноата:
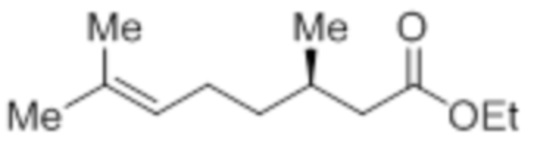
В круглодонной колбе емкостью 5 л, суспензию (3R)-3,7-диметилокт-6-еновой кислоты (100,0 г, 587,41 ммоль) и K2CO3 (243,59 г, 1762,23 ммоль) в DMF (1 л) при к.т. обрабатывали этилбромидом (95,94 г, 881,12 ммоль). Реакционную смесь перемешивали при к.т. в течение 2 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой (1 л) и экстрагировали диэтиловым эфиром (3×1 л). Объединенные органические экстракты сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения(101,1 г, 86,7%).
1H-ЯМР (300 МГц, CDCl3): δ 5,08 (т, J=6,9 Гц, 1H), 4,12 (кв, J=7,2 Гц, 2H), 2,29 (дд, J=14,7, 6,0 Гц, 1H), 2,12-2,05 (м, 1H), 1,99-1,94 (м, 3H), 1,66 (с, 3H), 1,58 (с, 3H), 1,39-1,16 (м, 2H), 1,24 (т, J=6,9 Гц, 3H), 0,93 (д, J=6,6 Гц, 3H).
Стадия 3: Синтез этил-(3R)-5-(3,3-диметилоксиран-2-ил)-3-метилпентаноата:
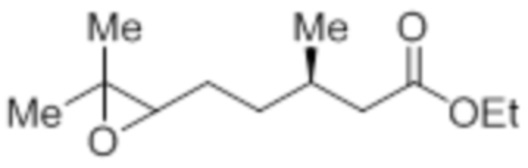
В круглодонной колбе емкостью 5 л, к раствору этил-(3R)-3,7-диметилокт-6-еноата (100,0 г, 504,51 ммоль) в диэтиловом эфире (1 л) по каплям при -30°C добавляли 65% раствор m-CPBA (267,51 г, 1,01 моль) в диэтиловом эфире (1 л). После завершения добавления, смесь нагревали до 0°C и перемешивали при той же температуре в течение 6 ч, после чего оставляли ее отстаиваться в течение ночи (~14 ч) при 0 – 3°C. После завершения реакции (согласно TLC), реакционную смесь разбавляли диэтиловым эфиром (1 л) и промывали н NaOH (2×1 л), а затем водой (1 л). Органический слой промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (99,5 г, 92,0%).
1H-ЯМР (300 МГц, CDCl3): δ 4,12 (кв, J=7,2 Гц, 2H), 2,69 (т, J=5,4 Гц, 1H), 2,30 (дд, J=8,7, 1,5 Гц 1H), 2,17-2,09 (м, 1H), 2,04-1,97 (м, 1H), 1,55-1,42 (м, 4H), 1,30 (с, 3H), 1,27 (с, 3H), 1,25 (т, J=7,2 Гц, 3H), 0,95 (д, J=6,6 Гц, 3H).
Стадия 4: Синтез этил-(3R)-3-метил-6-оксогексаноата:
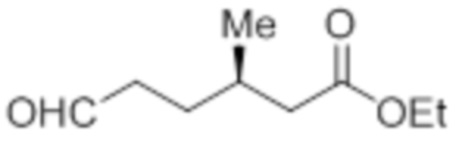
В круглодонной колбе емкостью 5 л, раствор этил-(3R)-5-(3,3-диметилоксиран-2-ил)-3-метилпентаноата (99,0 г, 462,07 ммоль) в 1,4-диоксане (1 л) при к.т. обрабатывали раствором NaIO4 (296,49 г, 1,386 моль) в воде (1 л). Реакционную смесь перемешивали при той же температуре в течение 12 ч. После завершения реакции (согласно TLC), неорганические соли фильтровали через слой Celite®, и экстрагировали фильтрат EtOAc (3×1 л). Объединенный органический экстракт промывали водой, солевым раствором и сушили над безводным Na2SO4. Раствор концентрировали в условиях пониженного давления с получением указанного в заголовке соединения (79,56 г, 99,3%).
1H-ЯМР (300 МГц, CDCl3): δ 9,79 (с, 1H), 4,11 (кв, J=7,2 Гц, 2H), 2,48-2,43 (м, 2H), 2,27 (дд, J=15, 6,6 Гц, 1H), 2,17-2,10 (м, 1H), 2,02-1,96 (м, 1H), 1,72-1,66 (м, 1H), 1,54-1,50 (м, 1H), 1,25 (т, J=7,2 Гц, 3H), 0,96 (д, J=6,6 Гц, 3H).
Стадия 5: Синтез этил-(3R)-6-гидрокси-3-метилгексаноата:
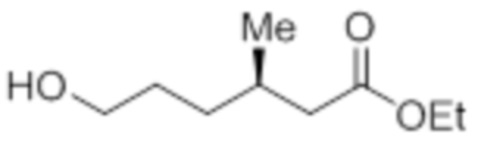
В круглодонной колбе емкостью 1 л, раствор этил-(3R)-3-метил-6-оксогексаноата (79,0 г, 458,76 ммоль) в метаноле (400 мл) при к.т. обрабатывали NaBH4 (27,75 г, 734,02 ммоль). Реакционную смесь перемешивали при к.т. в течение 2 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой (500 мл) и экстрагировали EtOAc (3×500 мл). Объединенный органический экстракт сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения(70,0 г).
1H-ЯМР (300 МГц, CDCl3): δ 4,12 (кв, J=7,2 Гц, 2H), 3,64 (т, J=6,3 Гц, 2H), 2,30 (дд, J=14,7, 6,6 Гц, 1H), 2,17-2,09 (м, 1H), 2,02-1,96 (м, 1H), 1,67-1,56 (м, 5H), 1,26 (т, J=7,2 Гц, 3H), 0,95 (д, J=6,6 Гц, 3H).
Стадия 6: Синтез этил-(3R)-6-бром-3-метилгексаноата:
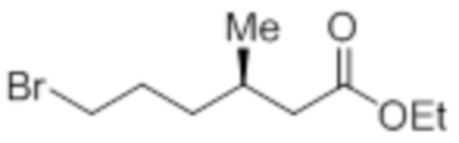
В круглодонной колбе емкостью 1 л, раствор этил-(3R)-6-гидрокси-3-метилгексаноата (65,0 г, 373,56 ммоль) в DCM (650 мл) при к.т. обрабатывали PBr3 (101,0 г, 373,56 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой (500 мл) и экстрагировали диэтиловым эфиром (3×500 мл). Органический экстракт отделяли и сушили над безводным Na2SO4. Растворитель выпаривали в условиях пониженного давления. Полученную жидкость (57,12 г) использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 7: Синтез 2-(4-(трифторметил)фенил)-4,5-дигидро-1H-имидазола:
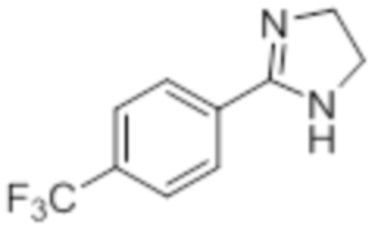
В круглодонной колбе емкостью 250 мл, перемешанный раствор 4-(трифторметил)бензальдегида (5,0 г, 27,17 ммоль) и этан-1,2-диамина (1,80 г, 29,89 ммоль) в tBuOH (80 мл) при к.т. обрабатывали йодом (8,60 г, 33,96 ммоль) и K2CO3 (11,30 г, 81,51 ммоль). Реакционную смесь нагревали в течение 3 ч при 85°C в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора Na2S2O3 и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением целевого продукта в виде желтого твердого вещества, которое переносили на следующую стадию без какой-либо очистки (5,1 г, 83,1%).
1H-ЯМР (300 МГц, DMSO-d6): δ 8,02 (д, J=8,1 Гц, 2H), 7,81 (д, J=8,1 Гц, 2H), 3,64 (с, 4H). 19F-ЯМР (300 МГц, DMSO-d6) : δ -66,22. LCMS (ESI+, m/z): 215,2 (M+H)+. HPLC (210 нм): 90,59%.
Стадия 8: Синтез 2-(4-(трифторметил)фенил)-1H-имидазола:
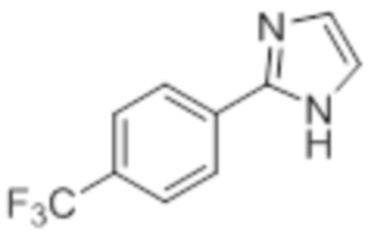
В круглодонной колбе емкостью 250 мл, перемешанный раствор 2-(4-(трифторметил)фенил)-4,5-дигидро-1H-имидазола (5,0 г, 23,36 ммоль) в DMSO (80 мл) при к.т. в атмосфере азота обрабатывали K2CO3 (3,55 г, 25,7 ммоль) и (диацетоксийод)бензолом (8,30 г, 25,7 ммоль). Реакционную смесь перемешивали в течение 12 ч при к.т. в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь разбавляли ледяной водой и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4.и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование 40% EtOAc в гексанах) с получением указанного в заголовке соединения в виде желтого твердого вещества (2,70 г, 54,7%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,81 (ушир. с, 1H), 8,14 (д, J=8,8 Гц, 2H), 7,81 (д, J=8,8 Гц, 2H), 7,23 (с, 2H). 19F-ЯМР (400 МГц, DMSO-d6) : δ -60,98. LCMS (ESI+, m/z): 213,0 (M+H)+.
Стадия 9: Синтез 1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола:
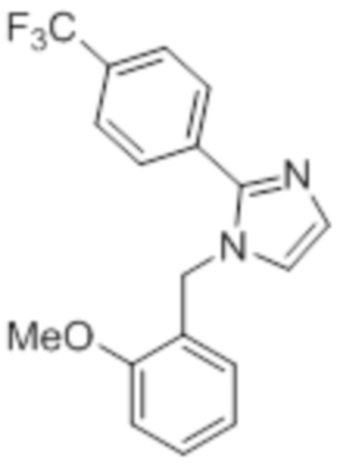
В круглодонной колбе емкостью 250 мл, перемешанный раствор 2-(4-(трифторметил)фенил)-1H-имидазола (6,5 г, 30,66 ммоль) в DMF (70 мл) при 0°C обрабатывали NaH (60% дисперсия, 1,41 г, 36,79 ммоль) и перемешивали в течение 30 мин при той же температуре в атмосфере азота. Спустя 30 мин, смесь обрабатывали 2-метоксибензилбромидом (7,40 г, 36,79 ммоль), и перемешивали реакционную смесь в течение 4 ч при к.т. в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора NH4Cl и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование 20% EtOAc в гексанах) с получением указанного в заголовке соединения в виде бесцветного твердого вещества (8 г, 82,5%).
1H-ЯМР (300 МГц, DMSO-d6): δ 7,80 (ушир. с, 4H), 7,30-7,26 (м, 2H), 7,10 (с, 1H), 7,01 (д, J=8,1 Гц, 1H), 6,89 (т, J=6,9 Гц, 1H) 6,75 (дд, J=7,5, 1,8 Гц, 1H), 5,29 (с, 2H), 3,68 (с, 3H). 19F-ЯМР (300 МГц, DMSO-d6) : δ -61,10. LCMS (ESI+, m/z): 333,2 (M+H)+.
Стадия 10: Синтез 5-йод-1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола:
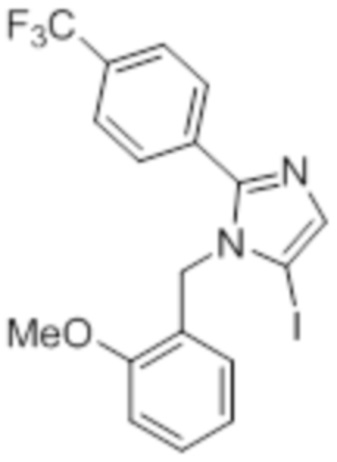
В круглодонной колбе емкостью 250 мл, перемешанный раствор 1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола (5 г, 15,06 ммоль) в ацетонитриле (50 мл) при к.т. в атмосфере азота обрабатывали NIS (4,0 г, 18,07 ммоль). Реакционную смесь нагревали при 70°C в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора Na2S2O3 и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование 8% EtOAc в гексанах) с получением указанного в заголовке соединения в виде бесцветного твердого вещества (2,5 г, 36,3%).
LCMS (ESI+, m/z): 459,0 (M+H)+.
Стадия 11: Синтез 1-(2-метоксибензил)-5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазола:
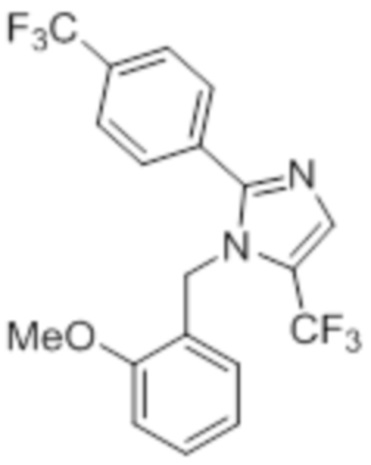
В герметизируемом реакционном сосуде емкостью 100 мл, перемешанный раствор 5-йод-1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола (0,5 г, 1,09 ммоль) в DMF (15 мл) продували аргоном при к.т. К описанной выше реакционной смеси в атмосфере аргона последовательно добавляли Ag2CO3 (0,6 г, 2,18 ммоль), KF (0,189 г, 3,27 ммоль), 1,10-фенантролин (0,196 г, 1,09 ммоль) и CuI (0,207 г,1,09 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали TMSCF3 (0,464 г, 3,27 ммоль) в атмосфере азота. Реакционную смесь нагревали в течение 12 ч при 100°C в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь разбавляли этилацетатом (30 мл), фильтровали через слой Celite® и промывали этилацетатом (20 мл×2). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 3-5% EtOAc в гексанах) с получением указанного в заголовке соединения в виде прозрачного масла (0,26 г, 59,6%).
1H-ЯМР (300 МГц, DMSO-d6): δ 7,65-7,62 (м, 5H), 7,32-7,29 (м, 1H), 6,90 (т, J=7,2 Гц 2H), 6,57 (д, J=7,2 Гц, 1H), 5,31 (с, 2H), 3,82 (с, 3H). LCMS (ESI+, m/z): 401,0 (M+H)+.
Стадия 12: Синтез 2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенола:
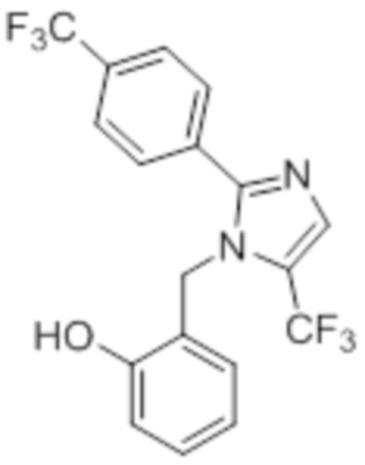
В круглодонной колбе емкостью 50 мл, раствор 1-(2-метоксибензил)-5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазола (0,5 г, 1,25 ммоль) в DCM (5 мл) при 0°C по каплям обрабатывали BBr3 (1 M в DCM, 1 мл). Реакционную смесь перемешивали при к.т. в течение 3 ч. После завершения реакции (согласно TLC), реакционную смесь подщелачивали добавлением насыщенного раствора NaHCO3 и экстрагировали EtOAc (20 мл×3). Объединенный органический экстракт сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения. Выход: 0,32 г (66,4%).
1H-ЯМР (300 МГц, DMSO-d6): δ 9,91 (с, 1H), 7,87-7,83 (м, 5H), 7,08 (т, J=9,0 Гц, 1H), 6,81-6,68 (м, 2H), 6,32 (д, J=7,5 Гц, 1H), 5,31 (с, 2H). 19F-ЯМР (300 МГц, DMSO-d6) : δ -57,93, -61,33. LCMS (ESI+, m/z): 387,0 (M+H)+.
Стадия 13: Синтез этил-(3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)гексаноата:
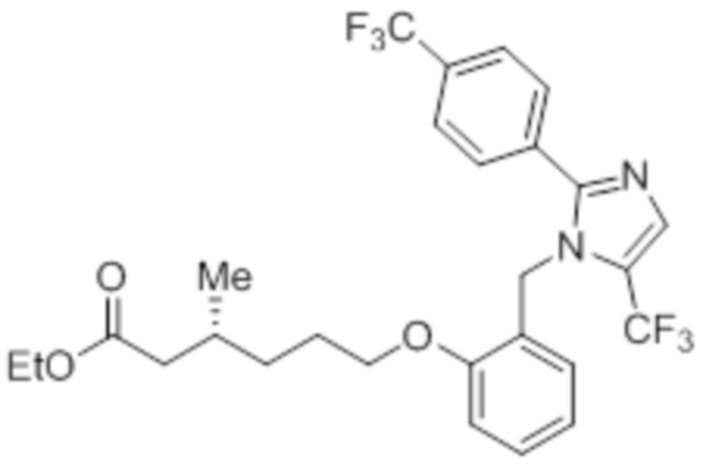
В круглодонной колбе емкостью 50 мл, перемешанный раствор 2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенола (0,3 г, 0,775 ммоль) в DMF (5 мл) при к.т. в атмосфере азота обрабатывали K2CO3 (0,642 г, 0,465 ммоль) и этил-(3R)-6-бром-3-метилгексаноатом (0,548 г, 2,36 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением ледяной воды и экстрагировали этилацетатом (25 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 15-30% EtOAc в гексанах) с получением указанного в заголовке соединения. Выход: 0,285 г (67,8%).
LCMS (ESI+, m/z): 543,0 (M+H)+.
Стадия 14: Синтез (3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоты:
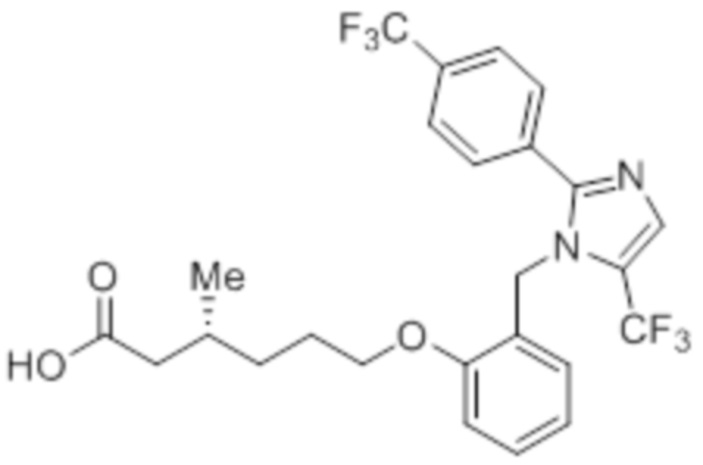
В круглодонной колбе емкостью 100 мл, перемешанный раствор этил-(3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-гексаноата (0,38 г, 0,701 ммоль) в THF (5 мл), этаноле (5 мл) и воде (5 мл) при к.т. обрабатывали моногидратом гидроксида лития (0,147 г, 3,50 ммоль). Реакционную смесь перемешивали при к.т. в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь концентрировали в условиях пониженного давления. Полученный остаток промывали EtOAc, разбавляли холодной водой и подкисляли (pH ~5) добавлением 1н HCl. Полученное твердое вещество очищали методом обращенно-фазовой препаративной HPLC [Zorbax C18 (21,2 мм×150 мм, 5 мкм); поток: 20 мл/мин; подвижная фаза: A/B =0,1% TFA в воде/MeCN; T/%B = 0/40, 2/50, 7/80] с получением указанного в заголовке соединения. Выход: 0,185 г (51,1%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,0 (с, 1H), 7,82-7,76 (м, 5H), 7,22 (т, J=7,2 Гц, 1H), 6,97 (д, J=8,1Гц, 1H), 6,83 (т, J=7,5 Гц,1H), 6,44 (д, J=7,5 Гц, 1H ), 5,34 (с, 2H), 3,94 (т, J=6,0 Гц, 2H),2,24-2,17 (м, 1H), 2,02-1,95 (м, 1H),1,90-1,80 (м,1H), 1,68-1,61 (м, 2H), 1,40-1,30 (м, 1H),1,30-1,15 (м, 1H), 0,87 (д, J=6,6 Гц, 3H). 19F-ЯМР (300 МГц, DMSO-d6): δ -57,86, -61,38. LCMS (ESI+, m/z): 515,1 (M+H)+. HPLC (210 нм): 99,77%.
Пример 2B: Синтез Соединения 2b
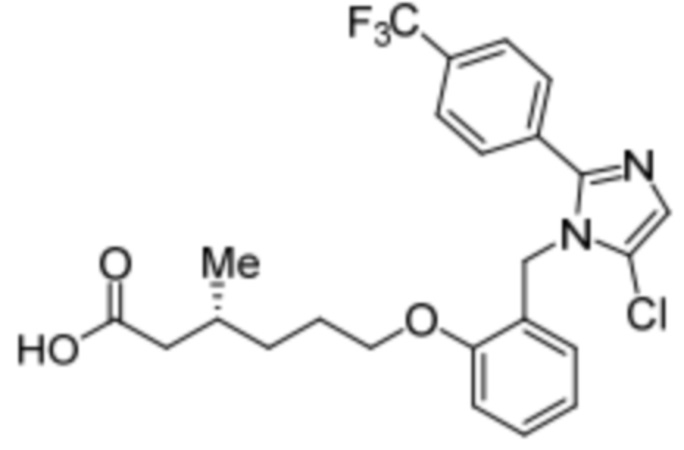
Синтез (R)-6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)-фенокси)-3-метилгексановой кислоты
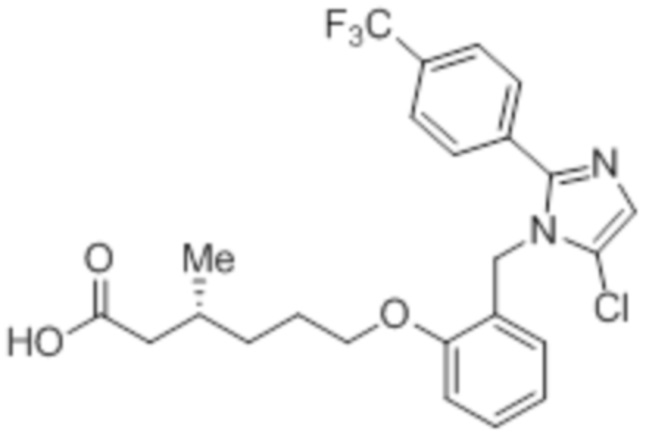
Схема:
Реагенты и условия: a) NCS, DMF, 45°C, 3 ч; b) BBr3, DCM, -78°C–к.т., 2 ч; c) этил-(R)-6-гидрокси-3-метилгексаноат, PPh3, DIAD, PhMe, 65°C, 12 ч; d) LiOH·H2O, THF, EtOH, H2O, к.т., 16 ч.
Стадия 1: Синтез 5-хлор-1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола:
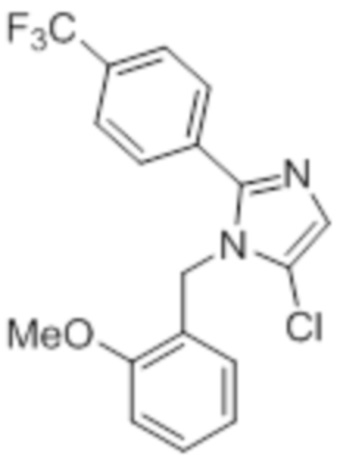
В круглодонной колбе емкостью 250 мл, перемешанный раствор 1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола (9 г, 27,1 ммоль), который был получен посредством способов, описанных в Примере 2A, в DMF (90 мл) при к.т. обрабатывали NCS (4,32 г, 32,0 ммоль). Реакционную смесь нагревали при 45°C в течение 3 ч. После завершения реакции, реакционную смесь гасили добавлением воды со льдом и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 5% EtOAc в гексанах) с получением указанного в заголовке соединения в виде белого твердого вещества (4,0 г, 40,4%).
1H-ЯМР (400 МГц, CDCl3): δ 7,60 (с, 4H), 7,33-7,29 (м, 1H), 7,20 (с, 1H), 6,94-6,90 (м, 2H), 6,70-6,65 (дд, J=8,0, 2,0 Гц, 1H), 5,23 (с, 2H), 3,84 (с, 3H). LCMS (ESI+, m/z): 367,3 (M+H)+.
Стадия 2: Синтез 2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)-метил)фенола:
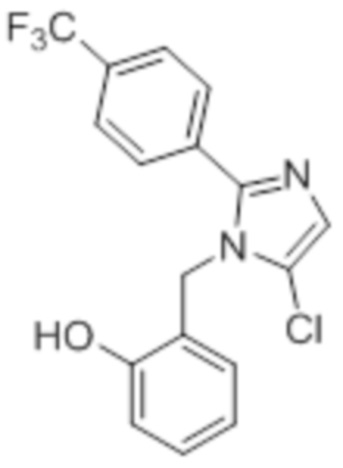
В круглодонной колбе емкостью 500 мл, раствор 5-хлор-1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола (6,0 г, 16,0 ммоль) в дихлорметане (60 мл) при -78°C по каплям обрабатывали BBr3 (6,0 мл ). Реакционную смесь постепенно нагревали до к.т. и перемешивали при к.т. в течение 2 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением воды со льдом и подщелачивали добавлением водного раствора NaHCO3. Твердое вещество фильтровали, промывали EtOAc и сушили в условиях пониженного давления с получением указанного в заголовке соединения (5,5 г, 96,5%).
1H-ЯМР (400 МГц, DMSO-d6): δ 9,92 (с, 1H), 7,81 (д, J=8,4 Гц, 2H), 7,74 (д, J=8,0 Гц, 2H), 7,31 (с, 1H), 7,12 (т, J=8,0 Гц, 1H), 6,83 (д, J=7,2 Гц, 1H), 6,72 (т, J=8,0 Гц, 1H), 6,38 (д, J=7,2 Гц, 1H), 5,26 (с, 2H). LCMS (ESI+, m/z): 353,2 (M+H)+.
Стадия 3: Синтез этил-(R)-6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
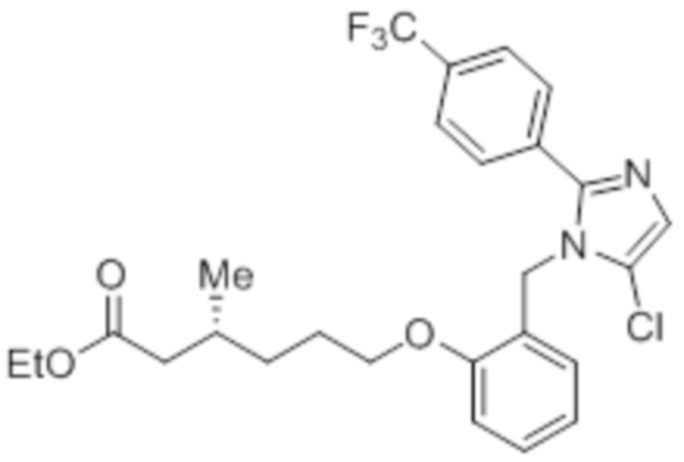
В круглодонной колбе емкостью 250 мл, перемешанный раствор 2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенола (5,5 г, 15,0 ммоль) в толуоле (60 мл) при к.т. в атмосфере азота обрабатывали DIAD (4,7 г, 23,0 ммоль) и PPh3 (6,1 г, 23,0 ммоль). Реакционную смесь перемешивали при к.т. в течение 15 мин и обрабатывали в атмосфере азота этил-(3R)-6-гидрокси-3-метилгексаноатом (3,2 г, 18,0 ммоль), который был получен посредством способов, описанных в Примере 2A. Затем, полученную реакционную смесь нагревали до 65°C в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением ледяной воды и экстрагировали н-гексаном (100 мл×2). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 5-10% EtOAc в гексанах) с получением указанного в заголовке соединения (6,5 г, 81,9%).
LCMS (ESI+, m/z): 509,3 (M+H)+.
Стадия 4: Синтез (R)-6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
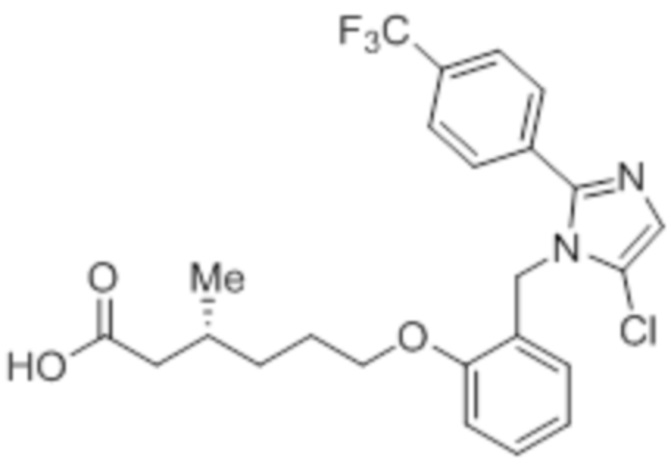
В круглодонной колбе емкостью 500 мл, перемешанный раствор этил-(R) -6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (8,0 г, 15,0 ммоль) в THF (100 мл) и воде (100 мл) при к.т. обрабатывали моногидратом гидроксида лития (8,0 г, 191,0 ммоль). Реакционную смесь перемешивали при к.т. в течение 16 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой и промывали диэтиловым эфиром. Водный слой нейтрализовали добавлением 1н HCl, и фильтровали полученное твердое вещество. Твердое вещество перекристаллизовывали в этаноле и промывали н-гексаном с получением чистого соединения (3,5 г, 46,7%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,03 (с, 1H), 7,78 (д, J=8,4 Гц, 2H), 7,70 (д, J=8,4 Гц, 2H), 7,30 (с, 1H), 7,23 (т, J=8,0 Гц, 1H), 7,02 (д, J=8,0 Гц, 1H), 6,85 (т, J=7,6 Гц, 1H), 6,48 (дд, J=7,6, 1,6 Гц, 1H), 5,26 (с, 2H), 3,98 (т, J=6,4 Гц, 2H), 2,22-2,17 (м, 1H), 2,01-1,95 (м, 1H), 1,85-1,78 (м, 1H), 1,69-1,63 (м, 2H), 1,37-1,33 (м, 1H), 1,24-1,22 (м, 1H), 0,85 (д, J=6,4 Гц, 3H). 19F-ЯМР (400 МГц, DMSO-d6) : δ -61,27. LCMS (ESI+, m/z): 481,3 (M+H)+. HPLC: 98,39% (210 нм).
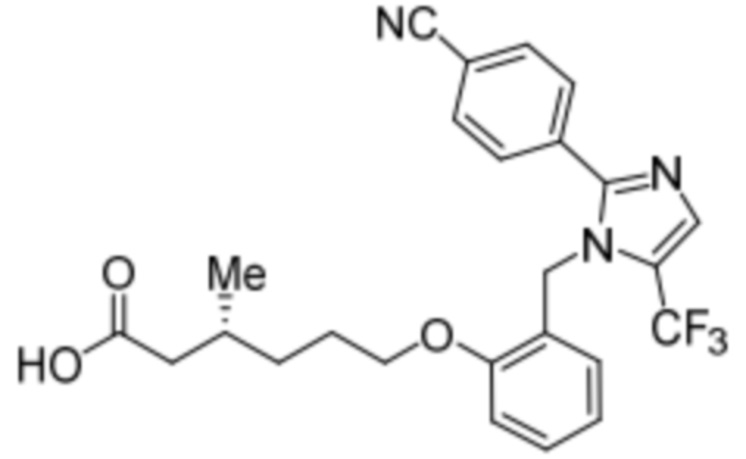
Пример 2C:Синтез Соединения 2c
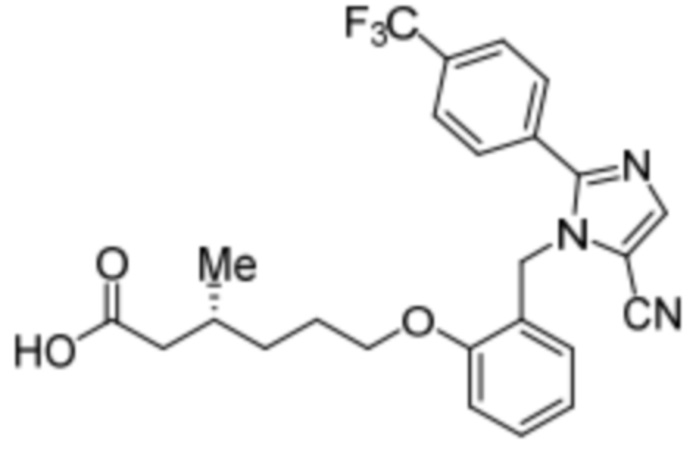
Синтез (3R)-6-(2-((5-циано-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты
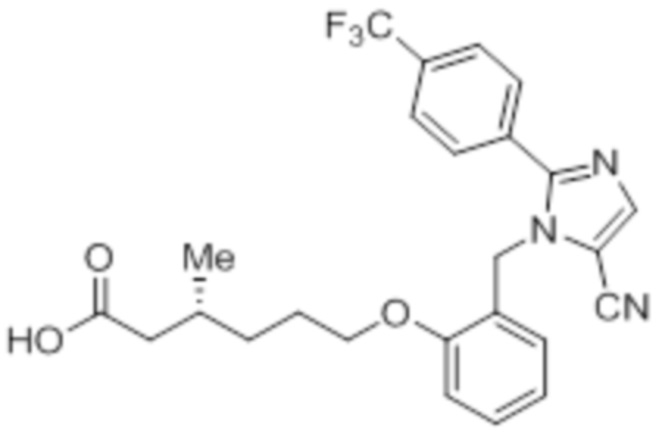
Схема:
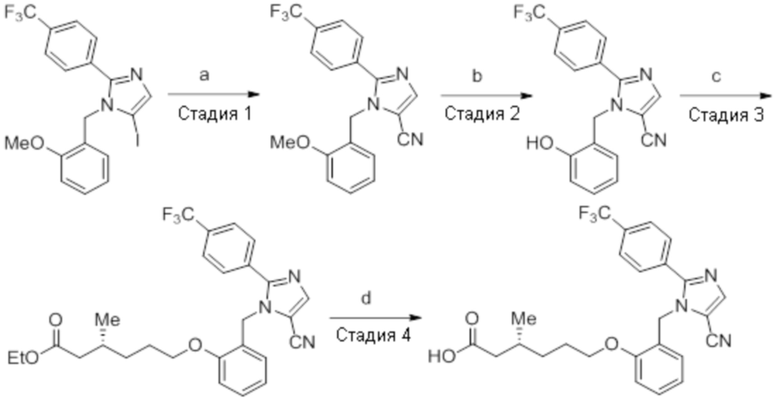
Реагенты и условия: a) CuCN, Pd(PPh3)4, микроволновое излучение, 150°C, 2 ч; b) BBr3, DCM, к.т., 36 ч; c) этил-(3R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 24 ч; d) LiOH·H2O, THF, EtOH, H2O, 0°C – 10°C, 36 ч.
Стадия 1: Синтез 1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазол-5-карбонитрила:
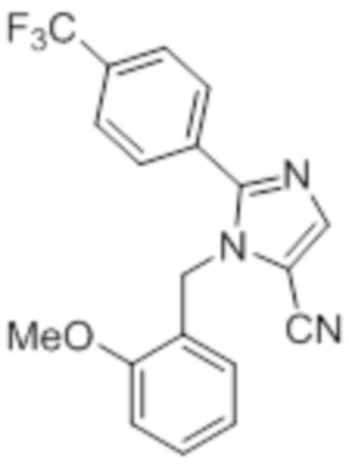
Во флаконе для микроволновой обработки емкостью 20 мл, перемешанный раствор 5-йод-1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазола (2 г, 4,36 ммоль) в DMF (10 мл) при к.т. продували аргоном. К описанной выше смеси в атмосфере аргона последовательно добавляли CuCN (0,97 г, 10,917 ммоль) и Pd(PPh3)4 (0,2 г, 0,174 ммоль). Реакционную смесь нагревали при 150°C в микроволновом реакторе в течение 2 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли этилацетатом (30 мл) и водой (20 мл), фильтровали через слой Celite® и промывали этилацетатом (20 мл×2). Объединенный фильтрат промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование 10% EtOAc в гексанах) с получением указанного в заголовке соединения в виде белого твердого вещества (0,7 г, 45,2%).
1H-ЯМР (300 МГц, CDCl3): δ 7,83 (с, 1H), 7,68 (д, J=1,2 Гц, 4H), 7,32 (м, 1H), 6,93-6,88 (м, 2H), 6,73-6,71 (д, J=7,5 Гц, 1H), 5,36 (с, 2H), 3,77 (с, 3H). LCMS (ESI+, m/z): 357,9 (M+H)+.
Стадия 2: Синтез 1-(2-гидроксибензил)-2-(4-(трифторметил)фенил)-1H-имидазол-5-карбонитрила:
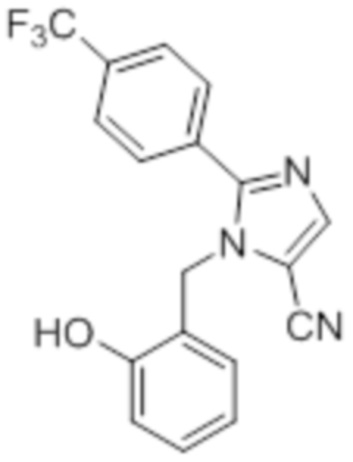
В круглодонной колбе емкостью 50 мл, перемешанный раствор 1-(2-метоксибензил)-2-(4-(трифторметил)фенил)-1H-имидазол-5-карбонитрила (0,7 г, 1,96 ммоль) в DCM (5 мл) при 0°C в атмосфере азота обрабатывали раствором BBr3 (1 M в DCM, 4,9 г, 19,60 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 12 ч и снова обрабатывали при 0°C в атмосфере азота раствором BBr3 в DCM (4,9 г, 19,60 ммоль). Реакционную смесь перемешивали при к.т. в атмосфере азота дополнительно в течение 24 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением ледяного раствора NaHCO3 и экстрагировали DCM (50 мл×2). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 10-15% MeOH в CHCl3) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (0,31 г, 44,8%).
1H-ЯМР (300 МГц, DMSO-d6): δ 9,90 (ушир. с, 1H), 8,10 (с, 1H), 8,0-7,81 (м, 4H), 7,15-7,10 (т, J=7,5 Гц, 1H), 6,82-6,62 (м, 3H), 5,35 (с, 2H). LCMS (ESI+, m/z): 344,2 (M+H)+.
Стадия 3: Синтез этил-(3R)-6-(2-((5-циано-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
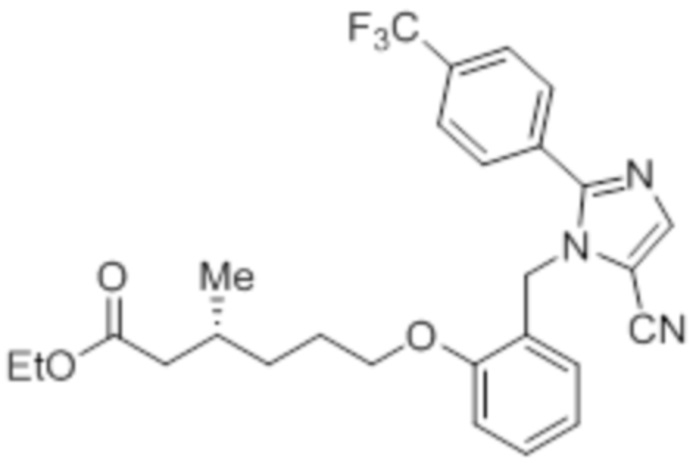
Указанное в заголовке соединение синтезировали из 1-(2-гидроксибензил)-2-(4-(трифторметил)фенил)-1H-имидазол-5-карбонитрила (0,15 г, 0,437 ммоль) и этил-(3R)-6-бром-3-метилгексаноата (0,31 г, 1,311 ммоль), следуя экспериментальной методике, описанной на стадии 13 Примера 2A. Выход: 0,11 г (47,6%).
1H-ЯМР (300 МГц, CDCl3): δ 7,84 (с, 1H), 7,67 (м, 4H), 7,30-7,27 (м, 1H), 6,92-6,87 (т, J=8,1 Гц, 2H), 6,76 (д, J=6,6 Гц, 1H), 5,36 (с, 2H), 4,13 (кв, J=6,6 Гц, 2H), 4,01 (т, J=6,9 Гц, 2H), 2,26-2,24 (м, 1H), 2,16-2,08 (м, 2H), 1,77-1,76 (м, 2H), 1,41 (м, 1H), 1,22-1,18 (м, 4H), 0,95 (д, J=6,6 Гц, 3H). LCMS (ESI+, m/z): 500,1 (M+H)+.
Стадия 4: Синтез (3R)-6-(2-((5-циано-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
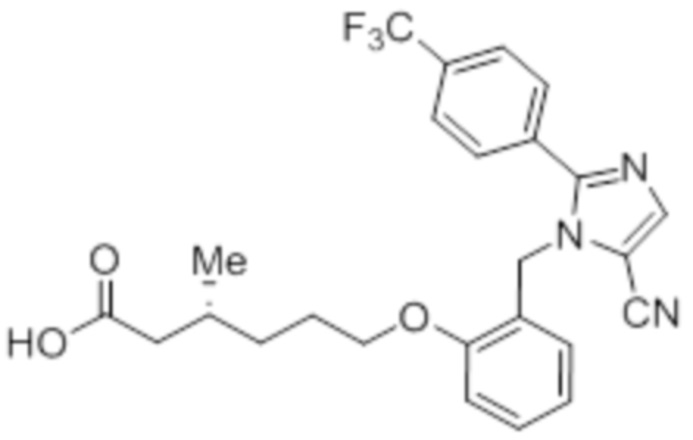
Указанное в заголовке соединение синтезировали из этил-(3R)-6-(2-((5-циано-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (0,075 г, 0,150 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A, и проводили очистку методом обращенно-фазовой препаративной HPLC [Kinetex EVO C18: 21,2 мм×150 мм); поток: 15 мл/мин; подвижная фаза: A/B = вода/MeCN; T/%B = 0/45, 2/55, 12/75]. Выход: 0,032 г (45,1%).
1H-ЯМР (400 МГц, CD3OD): δ 7,92 (с, 1H), 7,80-7,73 (м, 4H), 7,31-7,27 (м, 1H), 6,97 (д, J=8,0 Гц, 1H), 6,86-6,80 (м, 2H), 5,24 (с, 2H), 3,94 (т, J=6,4 Гц, 2H), 2,29-2,14 (м, 1H), 1,98-1,92 (м, 1H), 1,83-1,78 (м, 1H), 1,67-1,6 (м, 2H), 1,37-1,331 (м, 1H), 1,22-1,18 (м, 1H), 0,97 (д, J=6,4 Гц, 3H). 19F-ЯМР (400 МГц, CD3OD): δ - 61,38. LCMS (ESI+, m/z): 472,3 (M+H)+. HPLC: 95,05% (210 нм).
Пример 2D: Синтез Соединения 2d
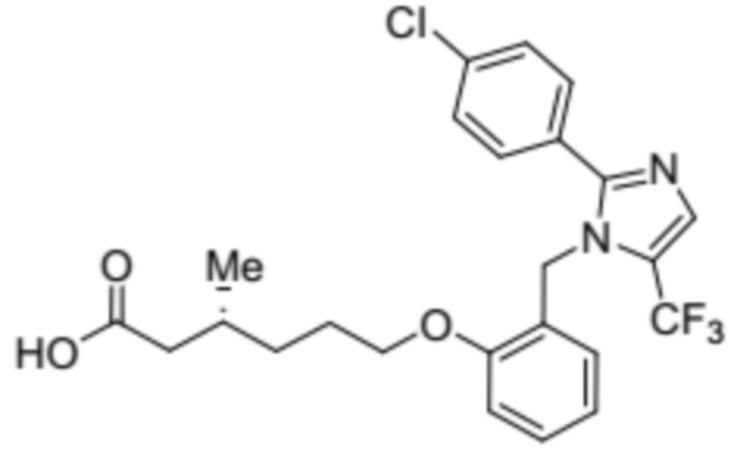
Синтез (3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)-метил)фенокси)-3-метилгексановой кислоты
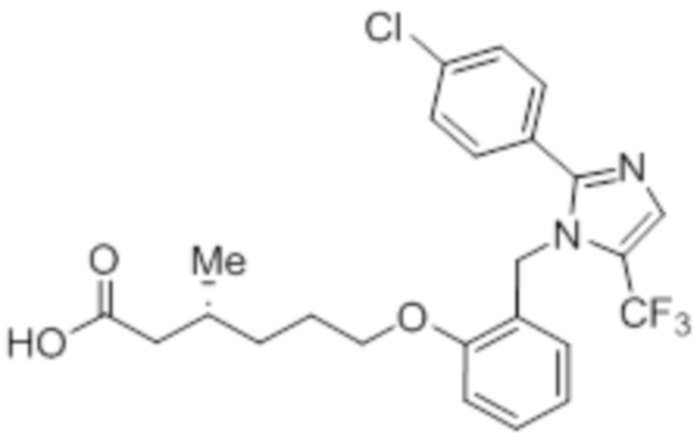
Схема:
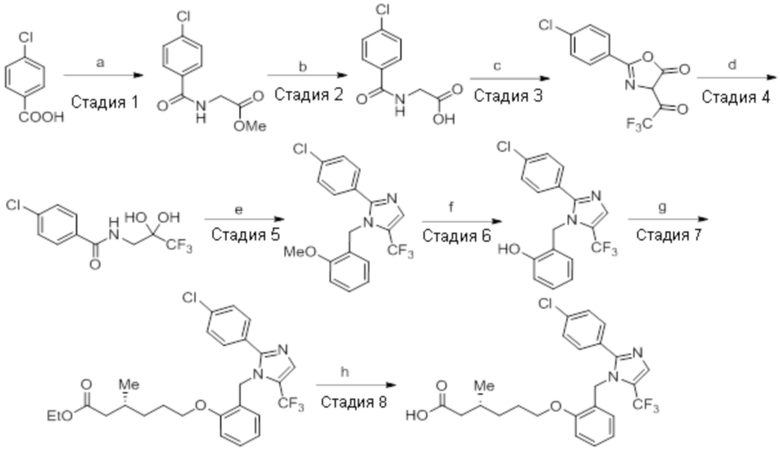
Реагенты и условия: a) метилглицината гидрохлорид, EDCI·HCl, HOBt, Et3N, DMF, 12 ч; b) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч; c) 2,2,2-трифторуксусный ангидрид, ацетон, 0°C, 12 ч; d) 1,4-диоксан, H2O, 3 ч; e) 2-метоксибензиламин, AcOH, толуол, 120°C, 12 ч; f) BBr3, DCM, -78°C – к.т., 3 ч; г) этил-(3R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 12 ч; h) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез метил-2-(4-хлорбензамидо)ацетата:
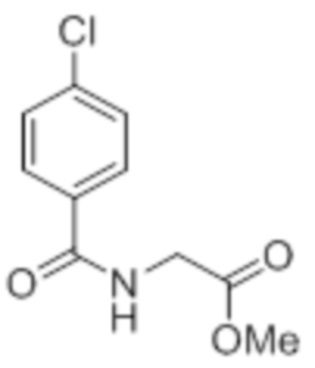
В круглодонной колбе емкостью 1000 мл, перемешанный раствор 4-хлорбензойной кислоты (25,0 г, 160 ммоль) и метилглицината гидрохлорида (30,12 г, 240 ммоль) в DMF (250 мл) при к.т. в атмосфере азота последовательно обрабатывали EDCI·HCl (61,28 г, 320 ммоль), HOBt (43,23 г, 320 ммоль) и Et3N (111 мл, 800 ммоль). Реакционную смесь перемешивали в течение 12 ч при к.т. в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь разбавляли ледяной водой и экстрагировали этилацетатом (500 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 15-30% EtOAc в гексанах) с получением указанного в заголовке соединения (20,5 г, 56,4%).
1H-ЯМР (300 МГц, CDCl3): δ 7,75 (д, J=8,4 Гц, 2H), 7,42 (д, J=8,4 Гц, 2H),6,66 (ушир. с, 1H), 4,24 (д, J=4,8 Гц, 2H),3,80 (с, 3H). LCMS (ESI+, m/z): 227,9, 229,9 (M+H)+.
Стадия 2: Синтез (4-хлорбензоил)глицина:
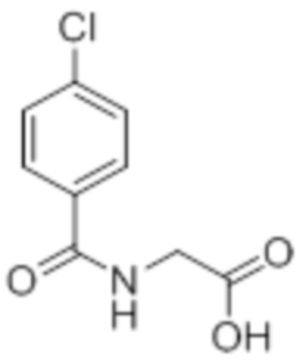
В круглодонной колбе емкостью 500 мл, перемешанный раствор метил-2-(4-хлорбензамидо)ацетата (20 г, 88,1 ммоль) в THF (100 мл), метаноле (100 мл) и воде (100 мл) при к.т. обрабатывали моногидратом гидроксида лития (18,5 г, 441 ммоль). Реакционную смесь перемешивали при к.т. в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь концентрировали в условиях пониженного давления. Полученный остаток промывали EtOAc, разбавляли холодной водой и подкисляли (pH~5) добавлением 1н HCl. Твердое вещество фильтровали и сушили в условиях пониженного давления с получением указанного в заголовке соединения (14,21 г, 75,6%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,7 (ушир. с, 1H), 8,93 (т, J=5,7 Гц, 1H), 7,88-7,84 (м, 2H), 7,54 (д, J=8,7 Гц, 2H), 3,90 (д, J=6,3 Гц, 2H). LCMS (ESI+, m/z): 214,0, 216,0 (M+H)+.
Стадия 3: Синтез 2-(4-хлорфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она:
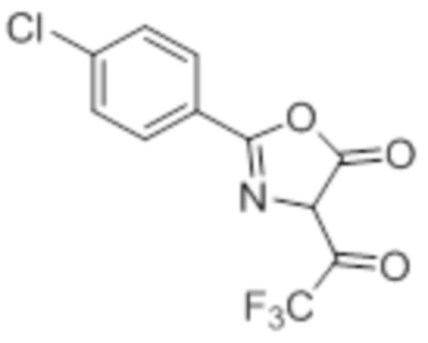
В круглодонной колбе емкостью 250 мл, перемешанный раствор (4-хлорбензоил)-глицина (10 г, 46,9 ммоль) в ацетоне (100 мл) при 0°C в атмосфере аргона обрабатывали 2,2,2-трифторуксусным ангидридом (29,8 г, 140 ммоль). Реакционную смесь перемешивали при к.т. в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь концентрировали в условиях пониженного давления. Полученный остаток разбавляли холодной водой, и фильтровали выпавшее в осадок твердое вещество. Твердое вещество промывали водой (100 мл) и сушили в условиях пониженного давления с получением целевого продукта в виде коричневого твердого вещества, которое переносили на следующую стадию без какой-либо очистки (9,92 г).
1H-ЯМР (300 МГц, DMSO-d6): δ 7,91 (д, J=8,7 Гц, 1H), 7,22(д, J=8,4 Гц, 1H), 7,60 (д, J=8,7 Гц, 1H), 7,47 (д, J=8,7 Гц, 1H).
Стадия 4: Синтез 4-хлор-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида:
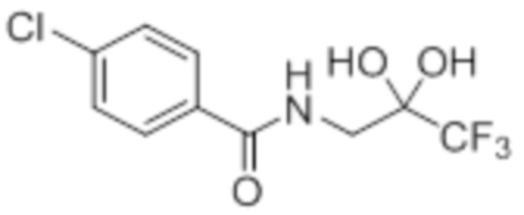
В круглодонной колбе емкостью 250 мл, перемешанный раствор 2-(4-хлорфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она (9,9 г, 34,1 ммоль) в 1,4-диоксане (100 мл) и воде (100 мл) нагревали при 100°C в атмосфере аргона в течение 3 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли ледяной водой и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением целевого продукта в виде коричневого твердого вещества, которое переносили на следующую стадию без какой-либо очистки (8,82 г).
1H-ЯМР (300 МГц, DMSO-d6): δ 8,61 (т, J=6,0 Гц, 1H) 7,88 (д, J=8,7 Гц, 2H), 7,56(д, J=8,7 Гц, 2H), 7,22 (ушир. с, 2H), 3,60 (д, J=6,0 Гц, 2H)
Стадия 5: Синтез 2-(4-хлорфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола:
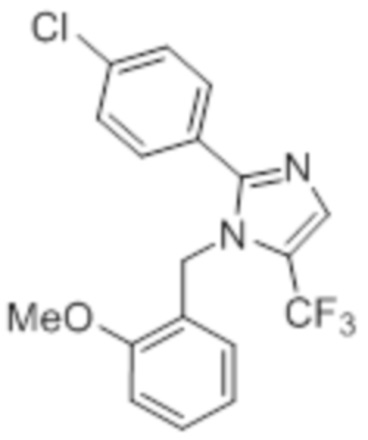
В герметизируемом реакционном сосуде емкостью 100 мл, перемешанный раствор 4-хлор-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида (2 г, 7,06 ммоль) в толуоле (20 мл) при к.т. обрабатывали 2-метоксибензиламином (1,46 г, 10,70 ммоль) и уксусной кислотой (0,6 мл). Реакционную смесь нагревали при 120°C в атмосфере аргона в течение 18 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора NaHCO3 и экстрагировали этилацетатом (25 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование 5% EtOAc в гексанах) с получением указанного в заголовке соединения в виде прозрачного масла (0,186 г, 7,2%).
LCMS (ESI+, m/z): 367,0, 369,0 (M+H)+.
Стадия 6: Синтез 2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)-метил)фенола:
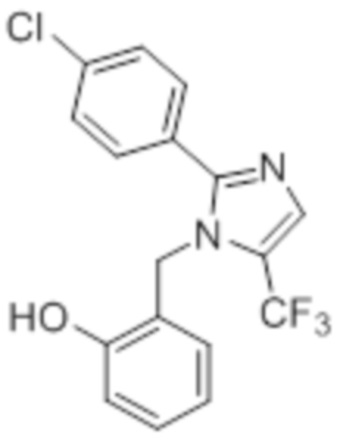
Указанное в заголовке соединение синтезировали из 2-(4-хлорфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола (0,4 г, 1,09 ммоль), следуя экспериментальной методике, описанной на стадии 12 Примера 2A. Выход: 0,15 г.
LCMS (ESI+, m/z): 352,9, 354,9 (M+H)+.
Стадия 7: Синтез этил-(3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
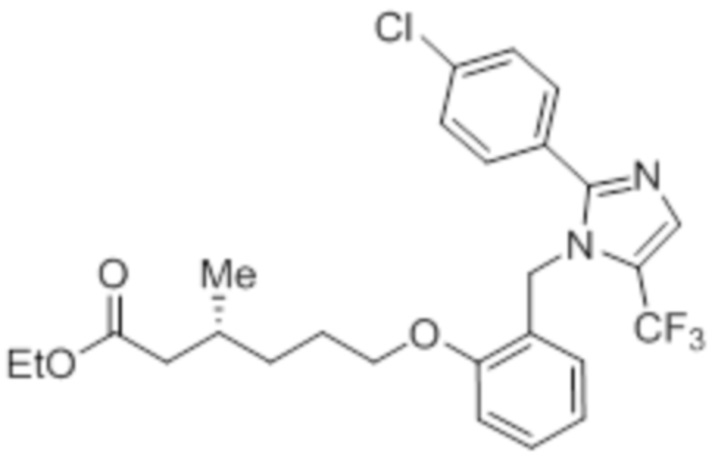
Указанное в заголовке соединение синтезировали из 2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола (0,15 г, 0,426 ммоль) и этил-(3R)-6-бром-3-метилгексаноата (0,29 г, 1,29 ммоль), следуя экспериментальной методике, описанной на стадии 13 Примера 2A. Выход: 0,141 г (65,3%).
LCMS (ESI+, m/z): 508,9, 510,9 (M+H)+.
Стадия 8: Синтез (3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
Указанное в заголовке соединение синтезировали из этил-(3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (0,140 г, 0,275 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A. Выход: 0,032 г (24,2%).
1H-ЯМР (400 МГц, CD3OD): δ 7,67 (д, J=1,2 Гц, 1H), 7,47-7,41 (м, 4H), 7,26 (т, J=8,4 Гц, 1H), 6,95 (д, J=7,6 Гц, 1H), 6,86 (т, J=7,6 Гц, 1H), 6,53 (д, J=6,4 Гц, 1H ), 5,36 (с, 2H), 3,99 (т, J=6,0 Гц, 2H), 2,34-2,29 (м, 1H), 2,15-2,10 (м, 1H), 2,00-1,82 (м, 1H), 1,80-1,73 (м, 2H),1,54-1,45 (м, 1H),1,36-1,29 (м, 1H), 0,95 (д, J=6,8 Гц, 3H). 19F-ЯМР (400 МГц, CD3OD): δ -60,58. LCMS (ESI+, m/z): 481,0, 483,0 (M+H)+. HPLC (210 нм): 96,02%
Пример 2E: Синтез Соединения 2e
Синтез (3R)-6-(2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)-метил)фенокси)-3-метилгексановой кислоты
Схема:
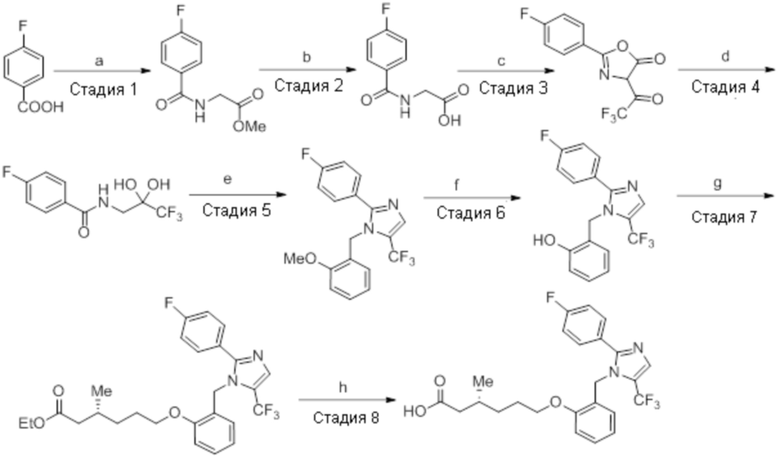
Реагенты и условия: a) метилглицината гидрохлорид, EDCI·HCl, HOBt, Et3N, DMF, к.т., 12 ч; b) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч; c) 2,2,2-трифторуксусный ангидрид, ацетон, 0°C, к.т., 4 ч; d) 1,4-диоксан, H2O, 100°C, 3 ч; e) 2-метоксибензиламин, AcOH, толуол, 120°C, 12 ч; f) BBr3, DCM -78°C – к.т.; г) этил-(3R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 12 ч; h) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез метил-(4-фторбензамидо)ацетата:
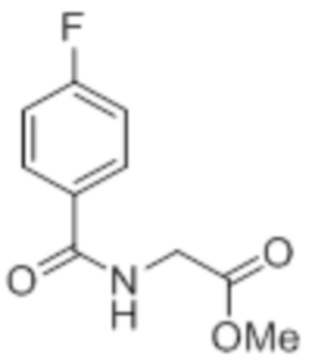
Указанное в заголовке соединение синтезировали из 4-фторбензойной кислоты (20,0 г, 142,74 ммоль) и метилглицината гидрохлорида (26,87 г, 214,11 ммоль), следуя экспериментальной методике, описанной на стадии 1 Примера 2D. Выход: 24,65 г (81,7%).
1H-ЯМР (400 МГц, DMSO-d6): δ 9,0 (т, J=6,4 Гц, 1H), 7,96-7,92 (м, 2H), 7,32 (т, J=8,8 Гц, 2H), 4,01 (д, J=6,0 Гц, 2H), 3,65 (с, 3H). LCMS (ESI+, m/z): 212,0 (M+H)+.
Стадия 2: Синтез (4-фторбензоил)глицина:
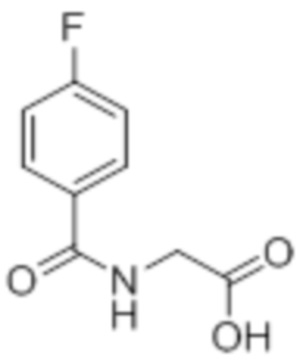
Указанное в заголовке соединение синтезировали из метил-(4-фторбензамидо)-ацетата (12,5 г, 59,1 ммоль), следуя экспериментальной методике, описанной на стадии 2 Примера 2D. Выход: 7,15 г (61,3%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,7 (ушир. с, 1H), 8,88 (т, J=6,0 Гц, 1H), 7,96-7,91 (м, 2H), 7,35-7,29 (м, 2H), 3,92 (т, J=6,0 Гц, 2H). 19F-ЯМР (400 МГц, DMSO-d6): δ -109,18
Стадия 3: Синтез 2-(4-фторфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она:
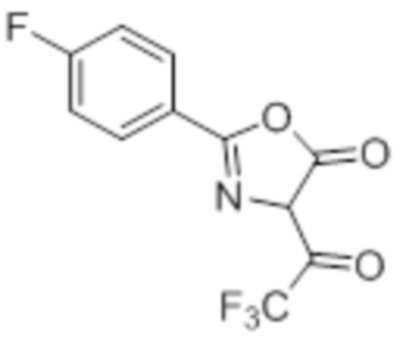
Указанное в заголовке соединение синтезировали из (4-фторбензоил)глицина (11,2 г, 56,8 ммоль), следуя экспериментальной методике, описанной на стадии 3 Примера 2D. Выход: 11,7 г (74,8%).
LCMS (ESI+, m/z): 276,1 (M+H)+.
Стадия 4: Синтез 4-фтор-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида:
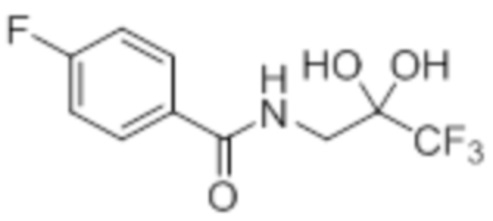
Указанное в заголовке соединение синтезировали из 2-(4-фторфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она (11,7 г, 42,5 ммоль), следуя экспериментальной методике, описанной на стадии 4 Примера 2D. Выход: 9,75 г (85,8%).
LCMS (ESI+, m/z): 268,0 (M+H)+.
Стадия 5: Синтез 2-(4-фторфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола:
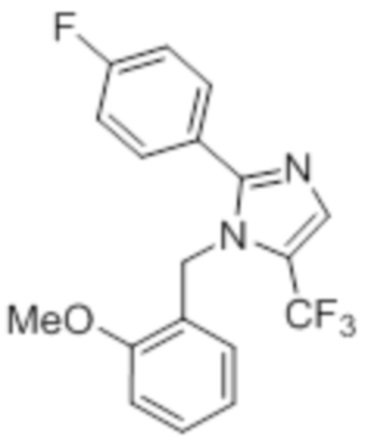
Указанное в заголовке соединение синтезировали из 4-фтор-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида (1,0 г, 3,74 ммоль) и 2-метоксибензиламина (0,769 г, 5,61 ммоль), следуя экспериментальной методике, описанной на стадии 5 Примера 2D. Выход: 0,12 г (9,2%).
1H-ЯМР (400 МГц, CDCl3): δ 7,62 (ушир. с, 1H), 7,47-7,44 (м, 2H), 7,29-7,26 (м, 1H), 7,06-7,02 (м, 2H), 6,89 (т, J=8,0 Гц, 2H), 6,56 (д, J=7,6 Гц, 1H), 5,27 (с, 2H), 3,81 (с, 3H). 19F-ЯМР (400 МГц, CDCl3): δ -110,50, -59,24. LCMS (ESI+, m/z): 350,9 (M+H)+.
Стадия 6: Синтез 2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола:
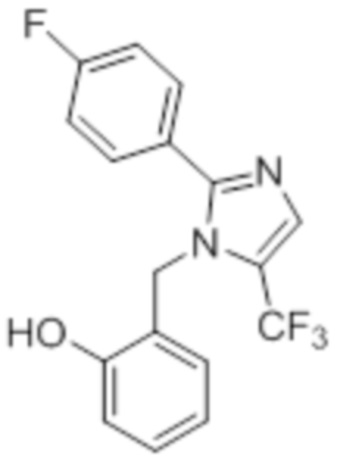
Указанное в заголовке соединение синтезировали из 2-(4-фторфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола (0,12 г, 0,34 ммоль), следуя экспериментальной методике, описанной на стадии 12 Примера 2A. Выход: 0,10 г (неочищ.).
1H-ЯМР (400 МГц, DMSO-d6): δ 9,85 (ушир. с, 1H), 8,06 (ушир. с, 1H), 7,64 (дд, J=5,2 Гц, 2H), 7,35 (т, J=8,8 Гц, 2H), 7,08 (т, J=7,6 Гц, 1H), 6,80-6,68 (м, 2H), 6,45 (т, J=8,0 Гц, 1H), 5,27 (с, 2H). 19F-ЯМР (400 МГц, DMSO-d6): δ -109,0, -58,20. LCMS (ESI+, m/z): 337,2 (M+H)+.
Стадия 7: Синтез этил-(3R)-6-(2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
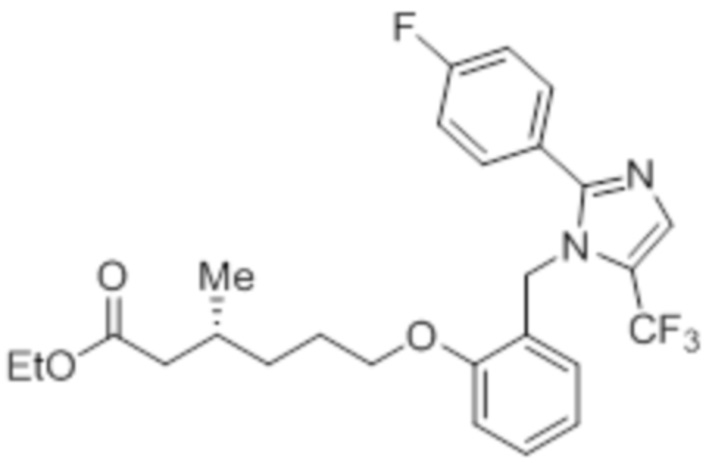
Указанное в заголовке соединение синтезировали из 2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола (0,1 г, 0,29 ммоль) и этил-(3R)-6-бром-3-метилгексаноата (0,14 г, 0,59 ммоль), следуя экспериментальной методике, описанной на стадии 13 Примера 2A. Выход: 0,05 г (34,2%).
LCMS (ESI+, m/z): 492,9 (M+H)+.
Стадия 8: Синтез (3R)-6-(2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
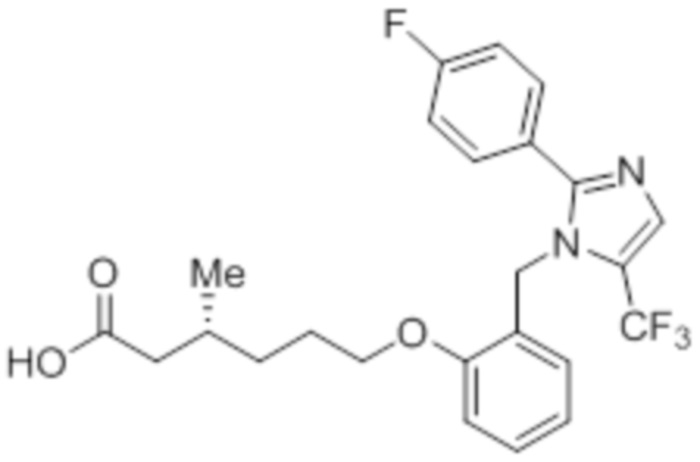
Указанное в заголовке соединение синтезировали из этил-(3R)-6-(2-((2-(4-фторфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (0,62 г, 1,25 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A. Выход: 0,018 г (38,3%).
1H-ЯМР (400 МГц, CD3OD): δ 7,64 (ушир. с, 1H), 7,48-7,45 (м, 2H), 7,21 (т, J=7,2 Гц, 1H), 7,14-7,09 (м, 2H), 6,91 (д, J=8,0 Гц, 1H), 6,82 (т, J=7,6 Гц, 1H), 6,49 (д, J=7,2 Гц, 1H), 5,31 (с, 2H), 3,95 (т, J=6,0 Гц, 2H), 2,28-2,23 (м, 1H), 2,13-2,06 (м, 1H), 1,94-1,93 (м, 1H), 1,75-1,71 (м, 2H), 1,44 (м, 1H), 1,31-1,26 (м, 1H), 0,94 (д, J=6,8 Гц, 3H). 19F-ЯМР (400 МГц, CD3OD): δ -112,00, -60,62. LCMS (ESI+, m/z): 465,3 (M+H)+. HPLC: 93,72% (210 нм).
Пример 2F: Синтез Соединения 2f
Синтез (R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)-метил)фенокси)-3-метилгексановой кислоты
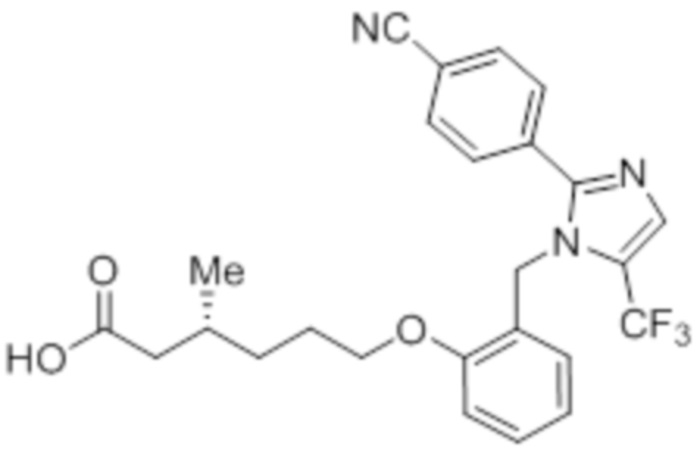
Схема:
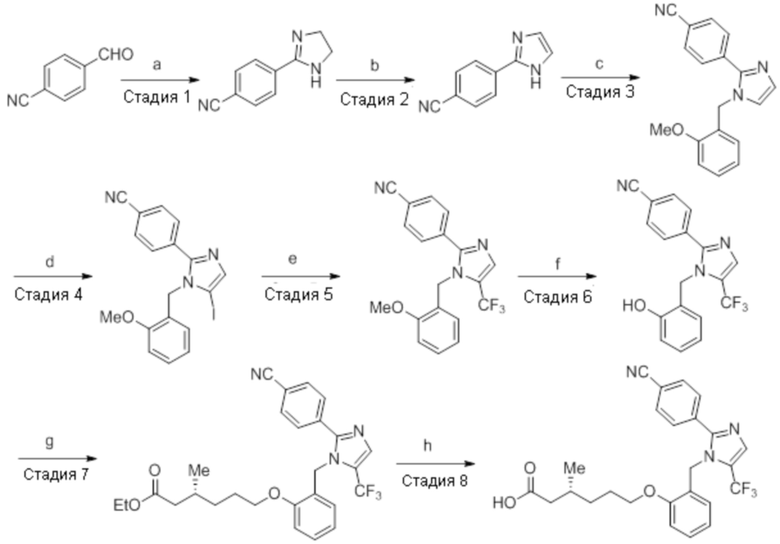
Реагенты и условия: a) этан-1,2-диамин, I2, K2CO3, tBuOH, 85°C, 5 ч; b) (диацетоксийод)бензол, K2CO3, DMSO, 12 ч; c) 2-метоксибензилбромид, NaH, DMF, 0°C – к.т., 4 ч; d) NIS, DMF, 80°C, 12 ч; e) TMSCF3, Ag2CO3, 1,10-фенантролин, KF, CuI, 100°C, 12 ч; f) BBr3, DCM, -78°C – к.т., 3 ч; г) этил-(R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 16 ч; h) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез 4-(4,5-дигидро-1H-имидазол-2-ил)бензонитрила:
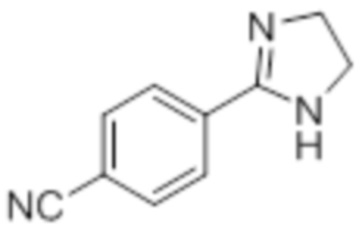
В круглодонной колбе емкостью 1000 мл, перемешанный раствор 4-формилбензонитрила (25,0 г, 190,65. ммоль) и этан-1,2-диамина (12,60 г, 209,7 ммоль) в tBuOH (250 мл) перемешивали в течение 30 мин при к.т. в атмосфере азота. Затем, добавляли йод (58,11 г, 228,78 ммоль) и K2CO3 (79,04 г, 571,95 ммоль), и нагревали реакционную смесь в течение 5 ч при 85°C в атмосфере азота. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора Na2S2O3 и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4.и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения в виде желтого твердого вещества, которое переносили на следующую стадию без какой-либо очистки (24,0 г, 73,5%).
LCMS (ESI+, m/z): 172,2 (M+H)+.
Стадия 2: Синтез 4-(1H-имидазол-2-ил)бензонитрила:
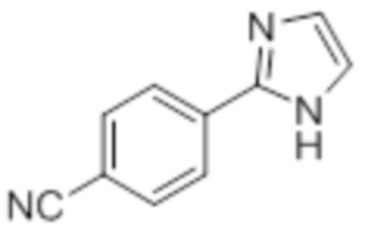
В круглодонной колбе емкостью 1000 мл, перемешанный раствор 4-(4,5-дигидро-1H-имидазол-2-ил)бензонитрила (24,0 г, 140,18 ммоль) в DMSO (400 мл) при к.т. в атмосфере азота обрабатывали K2CO3 (23,24 г, 168,21 ммоль) и (диацетоксийод)бензолом (54,18 г, 168,21 ммоль ). Реакционную смесь перемешивали при к.т. в атмосфере азота в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли ледяной водой и экстрагировали этилацетатом (200 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 40% EtOAc в гексанах) с получением указанного в заголовке соединения в виде желтого твердого вещества (18,5 г, 78,0%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,81 (ушир. с, 1H), 8,09 (д, J=8,8 Гц, 2H), 7,90 (д, J=8,4 Гц, 2H), 7,36 (bs, 1H), 7,12 (ушир. с, 1H). LCMS (ESI+, m/z): 170,3 (M+H)+.
Стадия 3: Синтез 4-(1-(2-метоксибензил)-1H-имидазол-2-ил)бензонитрила:
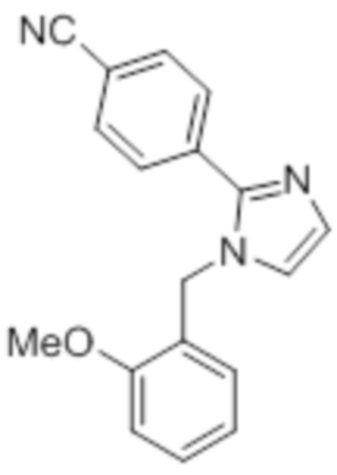
В круглодонной колбе емкостью 500 мл, перемешанный раствор 4-(1H-имидазол-2-ил)бензонитрила (10 г, 59,10 ммоль) в DMF (100 мл) при 0°C в атмосфере азота обрабатывали NaH (60%, 4,72 г, 118,20 ммоль). Реакционную смесь перемешивали в атмосфере азота в течение 30 мин при той же температуре. Смесь обрабатывали 2-метоксибензилбромидом (15,48 г, 76,83 ммоль), и перемешивали полученную реакционную смесь при к.т. в атмосфере азота в течение 4 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора NH4Cl и экстрагировали этилацетатом (300 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 20% EtOAc в гексанах) с получением указанного в заголовке соединения в виде белого твердого вещества (9,1 г, 53,2%).
1H-ЯМР (400 МГц, CDCl3): δ 7,72-7,64 (м, 4H), 7,32 (т, J=7,6 Гц,1H), 7,22 (с, 1H), 7,02 (с, 1H), 6,94- 6,90 (м, 2H), 6,81 (д, J=6,8 Гц, 1H), 5,22 (с, 2H), 3,81 (с, 3H). LCMS (ESI+, m/z): 290,3 (M+H)+.
Стадия 4: Синтез 4-(5-йод-1-(2-метоксибензил)-1H-имидазол-2-ил)-бензонитрила:
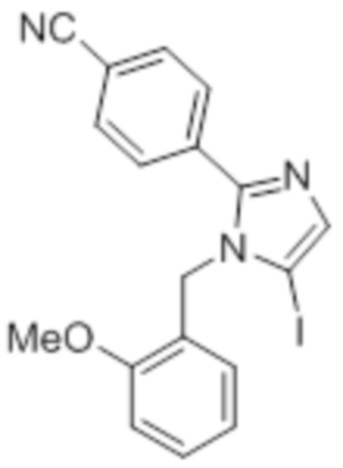
В круглодонной колбе емкостью 250 мл, перемешанный раствор 4-(1-(2-метоксибензил)-1H-имидазол-2-ил)бензонитрила (5 г, 17,30 ммоль) в DMF (60 мл) при к.т. обрабатывали NIS (3,89 г, 17,30 ммоль). Реакционную смесь нагревали при 80°C в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением насыщенного раствора Na2S2O3 и экстрагировали этилацетатом (100 мл×3). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 7-8% EtOAc в гексанах) с получением указанного в заголовке соединения в виде белого твердого вещества (1,41 г, 19,8%).
1H-ЯМР (300 МГц, CDCl3): δ 7,64-7,56 (м, 4H), 7,38 (с, 1H), 7,32 (т, J=8,1 Гц, 1H), 6,95- 6,89 (м, 2H), 6,56 (д, J=7,5 Гц, 1H), 5,22 (с, 2H), 3,87 (с, 3H). LCMS (ESI+, m/z): 415,6 (M+H)+.
Стадия 5: Синтез 4-(1-(2-метоксибензил)-5-(трифторметил)-1H-имидазол-2-ил)бензонитрила:
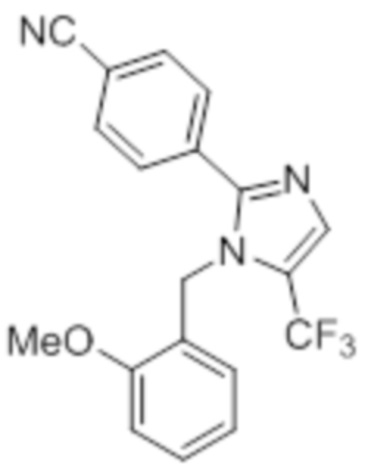
В герметизируемом реакционном сосуде емкостью 100 мл, перемешанный раствор 4-(5-йод-1-(2-метоксибензил)-1H-имидазол-2-ил)бензонитрила (2,5 г, 6,02 ммоль) в DMF (15 мл) дегазировали при к.т. путем продувки аргоном в течение 5 мин. К описанной выше смеси в атмосфере азота последовательно добавляли Ag2CO3 (3,31 г, 12,04 ммоль), KF (1,047 г, 18,06 ммоль), 1,10-фенантролин (1,08 г, 6,02 ммоль), CuI (1,143 г, 6,02 ммоль). Полученную смесь охлаждали до 0°C и по каплям обрабатывали TMSCF3 (2,56 г, 18,06 ммоль) в атмосфере азота. Реакционную смесь нагревали при 100°C в течение 12 ч в атмосфере азота и обрабатывали дополнительным количеством TMSCF3 (1,28 г, 9,03 ммоль) для гарантированного завершения реакции (согласно TLC). Реакционную смесь охлаждали до к.т. и разбавляли этилацетатом (30 мл), после чего фильтровали через слой Celite®. Твердый остаток промывали этилацетатом (20 мл×2). Объединенный фильтрат промывали водой, солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 3-5% EtOAc в гексанах) с получением указанного в заголовке соединения в виде прозрачного масла (1,73 г, 74,4%).
1H-ЯМР (400 МГц, CDCl3): δ 7,67-7,60 (м, 5H), 7,32 (т, J=8,4 Гц, 1H), 6,96- 6,89 (м, 2H), 6,57 (д, J=7,2 Гц, 1H), 5,33 (с, 2H), 3,85 (с, 3H). 19F-ЯМР (400 МГц, CDCl3): δ -59,25. LCMS (ESI+, m/z): 358,8 (M+H)+.
Стадия 6: Синтез 4-(1-(2-гидроксибензил)-5-(трифторметил)-1H-имидазол-2-ил)бензонитрила:
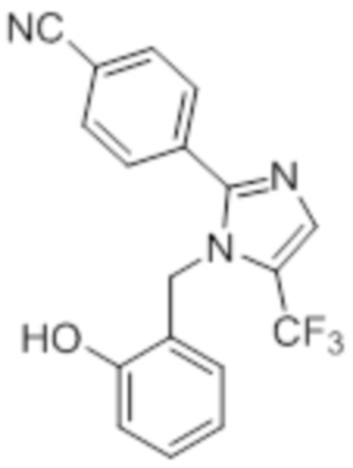
В круглодонной колбе емкостью 1000 мл, раствор 4-(1-(2-метоксибензил)-5-(трифторметил)-1H-имидазол-2-ил)бензонитрила (1,5 г, 4,20 ммоль) в дихлорметане (15 мл) при -78°C в атмосфере азота по каплям обрабатывали BBr3 (1,5 мл). Реакционную смесь перемешивали при к.т. в течение 3 ч. После завершения реакции (согласно TLC), реакционную смесь подщелачивали добавлением водного NaHCO3 и экстрагировали DCM (30 мл×2). Объединенный органический экстракт разделяли, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток промывали н-гексаном (3×10 мл) и сушили в условиях пониженного давления с получением указанного в заголовке соединения (1,13 г).
LCMS (ESI+, m/z): 344,4 (M+H)+.
Стадия 7: Синтез этил-(R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
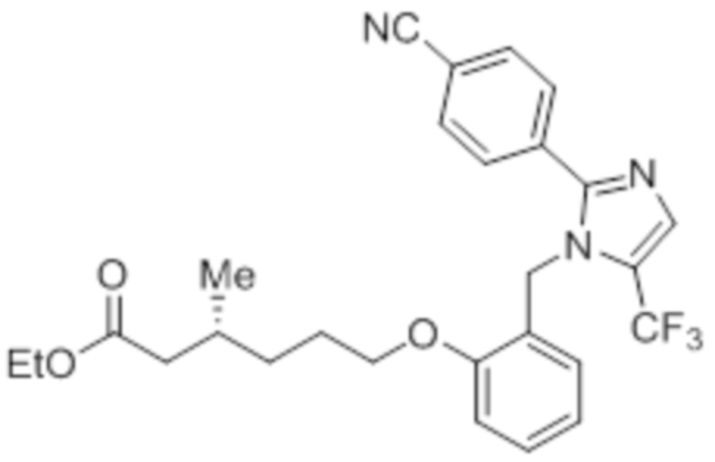
В круглодонной колбе емкостью 250 мл, перемешанный раствор 4-(1-(2-гидроксибензил)-5-(трифторметил)-1H-имидазол-2-ил)бензонитрила (1,0 г, 2,91 ммоль) в DMF (5 мл) при к.т. в атмосфере азота обрабатывали K2CO3 (1,2 г, 8,73 ммоль) и этил-(R)-6-бром-3-метилгексаноатом (1,37 г, 5,83 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 16 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли холодной водой (50 мл), после чего экстрагировали этилацетатом (50 мл). Органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом препаративной HPLC [колонка: GEMINI NX C18, 21,2 мм×150 мм); поток: 20 мл/мин; подвижная фаза: A/B = 0,05% TFA в воде/MeCN-MeOH (2:1); T/%B = 0/40, 2/50, 5/80] с получением указанного в заголовке соединения.
1H-ЯМР (300 МГц, CDCl3): δ 7,65-7,63 (м, 5H), 7,30-7,26 (м, 1H), 6,90- 6,85 (м, 2H), 6,54 (д, J=7,8 Гц, 1H), 5,31 (с, 2H), 4,12 (кв, J=6,9 Гц, 2H), 3,98 (т, J=6,3 Гц, 2H), 2,32-2,25 (м, 1H), 2,18-2,10 (с, 1H), 2,09-1,86 (м, 1H), 1,77-1,65 (м, 2H), 1,51-1,42 (м, 1H), 1,35-1,26 (м, 1H), 1,24 (т, J=6,9 Гц, 3H), 0,96 (д, J=6,3 Гц, 3H). 19F-ЯМР (300 МГц, CDCl3): δ -59,27. LCMS (ESI+, m/z): 500,5 (M+H)+.
Стадия 8: Синтез (R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
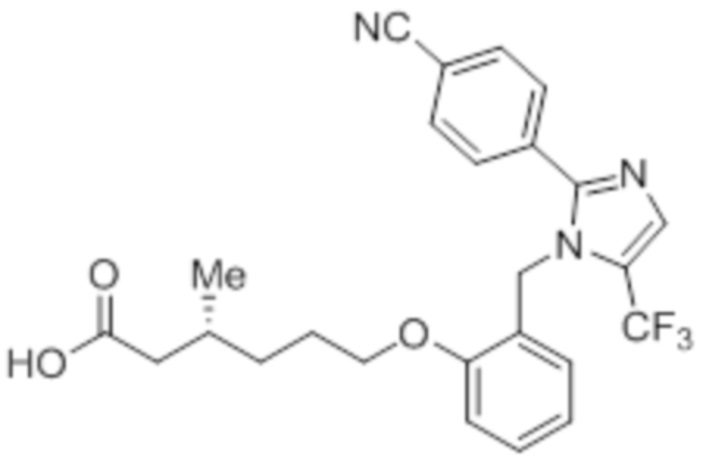
В круглодонной колбе емкостью 500 мл, перемешанный раствор этил-(R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (140 мг, 0,280 ммоль) в THF (5 мл) и воде (5 мл) при к.т. обрабатывали моногидратом гидроксида лития (14,0 г, 0,336 ммоль). Реакционную смесь перемешивали при к.т. в течение 12 ч. После завершения реакции (согласно TLC), реакционную смесь разбавляли водой и промывали диэтиловым эфиром. Водный слой подкисляли добавлением 1н HCl и экстрагировали этилацетатом (50 мл). Органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом препаративной HPLC (Kinetex C18, 21,2 мм×150 мм; поток: 18,0 мл/мин; подвижная фаза: A/B = вода:MeCN, T/%B = 0/10, 2/20/ 8/80) с получением указанного в заголовке соединения (81 мг, 61,8%).
1H-ЯМР (400 МГц, CD3OD): δ 7,75 (д, J=8,4 Гц, 2H), 7,71 (с, 1H), 7,66 (д, J=8,4 Гц, 2H), 7,23 (т, J=7,2 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,82 (т, J=8,4 Гц, 1H), 6,52 (д, J=7,2 Гц, 1H), 5,38 (с, 2H), 3,96 (т, J=6,0 Гц, 2H), 2,31-2,26 (м, 1H), 2,13-2,07 (м, 1H), 2,03-1,91 (м, 1H), 1,79-1,71 (м, 2H), 1,47-1,44 (м, 1H), 1,34-1,28 (м, 1H), 0,96 (д, J=6,8 Гц, 3H). 1H-ЯМР (400 МГц, CD3OD): δ - 60,58. LCMS (ESI+, m/z): 472,4 (M+H)+. HPLC: 92,02% (210 нм).
Пример 2G: Синтез Соединения 2g
Синтез (3R)-3-метил-6-(2-((2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоты
Схема:
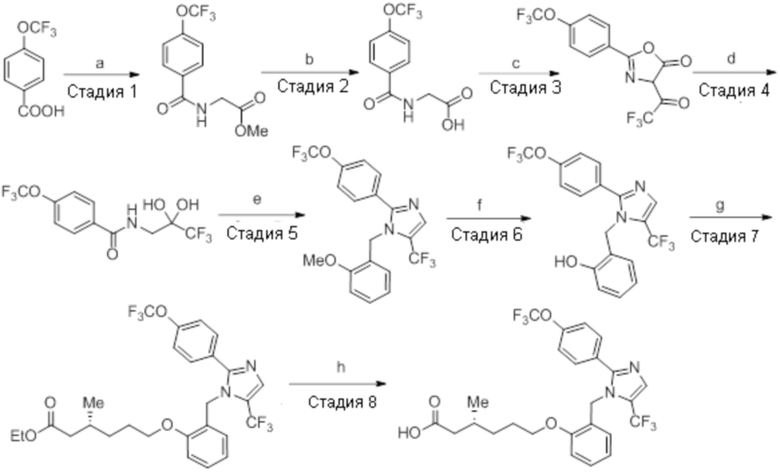
Реагенты и условия: a) метилглицината гидрохлорид, EDCI·HCl, HOBt, Et3N, DMF, к.т., 12 ч; b) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч; c) 2,2,2-трифторуксусный ангидрид, ацетон, 0°C – к.т., 12 ч; d) 1,4-диоксан-H2O, 100°C, 3 ч; e) 2-метоксибензиламин, AcOH, толуол, 120°C, 12 ч; f) BBr3, DCM, 0°C – к.т., 3 ч; г) этил-(3R)-6-бром-3-метилгексаноат, K2CO3, DMF, к.т., 12 ч; h) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез метил-(4-(трифторметокси)бензамидо)ацетата:
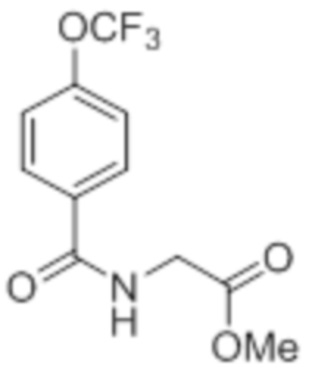
Указанное в заголовке соединение синтезировали из 4-(трифторметокси)бензойной кислоты (10,0 г, 48,53ммоль) и метилглицината гидрохлорида (9,13 г, 72,80 ммоль), следуя экспериментальной методике, описанной на стадии 1 Примера 2D. Выход: 9,8 г (72,8%).
1H-ЯМР (300 МГц, CDCl3): δ 7,86 (д, J=6,9 Гц, 2H), 7,28 (д, J=6,9 Гц, 2H), 6,72 (ушир. с, 1H), 4,25 (д, J=5,4 Гц, 2H), 3,81 (с, 3H). 19F-ЯМР (300 МГц, CDCl3): δ -57,73. LCMS (ESI+, m/z): 277,6 (M+H)+.
Стадия 2: Синтез (4-(трифторметокси)бензоил)глицина:
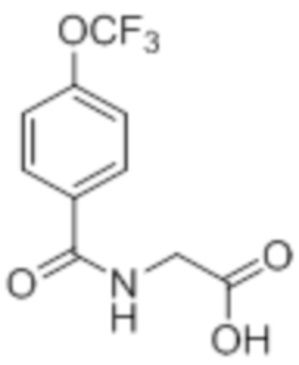
Указанное в заголовке соединение синтезировали из метил-(4-(трифторметокси)-бензамидо)ацетата (9,8 г, 35,35 ммоль), следуя экспериментальной методике, описанной на стадии 2 Примера 2D. Выход: 7,5 г (80,6%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,73 (ушир. с, 1H), 8,97 (т, J=5,4 Гц, 1H), 7,98 (д, J=8,7 Гц, 2H), 7,47 (д, J=8,1 Гц, 2H), 3,91 (д, J=6,0 Гц, 2H). 19F-ЯМР (300 МГц, DMSO-d6): δ -56,69. LCMS (ESI+, m/z): 263,6 (M+H)+.
Стадия 3: Синтез 4-(2,2,2-трифторацетил)-2-(4-(трифторметокси)фенил)-оксазол-5(4H)-она:
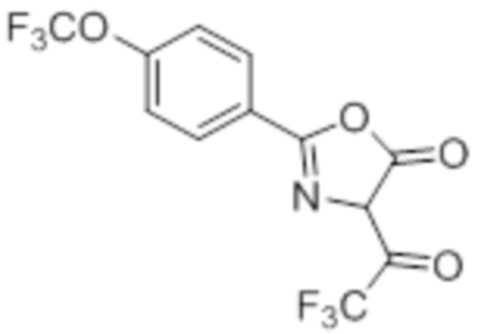
Указанное в заголовке соединение синтезировали из (4-(трифторметокси)бензоил)-глицина (1,0 г, 3,80 ммоль), следуя экспериментальной методике, описанной на стадии 3 Примера 2D. Выход: 1,1 г.
1H-ЯМР (400 МГц, DMSO-d6): δ 7,99 (д, J=8,8 Гц, 1H), 7,81 (д, J=8,8 Гц, 1H), 7,50 (д, J=8,4 Гц, 1H), 7,40 (д, J=8,4 Гц, 1H).
Стадия 4: Синтез N-(3,3,3-трифтор-2,2-дигидроксипропил)-4-(трифторметокси)бензамида:
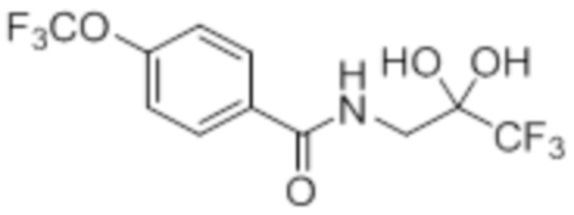
Указанное в заголовке соединение синтезировали из 4-(2,2,2-трифторацетил)-2-(4-(трифторметокси)фенил)оксазол-5(4H)-она (1,0 г, 4,14 ммоль), следуя экспериментальной методике, описанной на стадии 4 Примера 2D. Выход: 0,92 г.
LCMS (ESI+, m/z): 333,9 (M+H)+.
Стадия 5: Синтез 1-(2-метоксибензил)-2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазола:
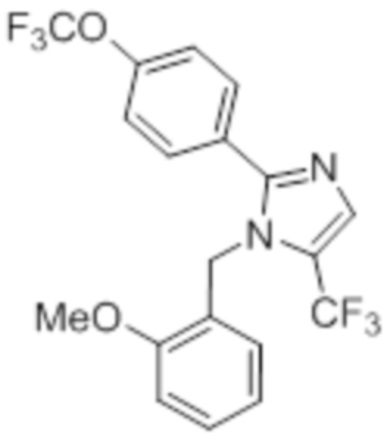
Указанное в заголовке соединение синтезировали из N-(3,3,3-трифтор-2,2-дигидроксипропил)-4-(трифторметокси)бензамида (0,9 г, 2,83 ммоль) и 2-метоксибензиламина (2,33 мл, 17,03 ммоль), следуя экспериментальной методике, описанной на стадии 5 Примера 2D. Выход: 0,14 г (13,4%).
1H-ЯМР (300 МГц, CDCl3): δ 7,64 (ушир. с, 1H), 7,55-7,52 (м, 2H), 7,21 (д, J=8,1 Гц, 2H), 6,90 (д, J=8,1 Гц, 2H), 6,59 (д, J=6,9 Гц, 2H), 5,31 (с, 2H), 3,84 (с, 3H). LCMS (ESI+, m/z): 416,9 (M+H)+.
Стадия 6: Синтез 2-((2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола:
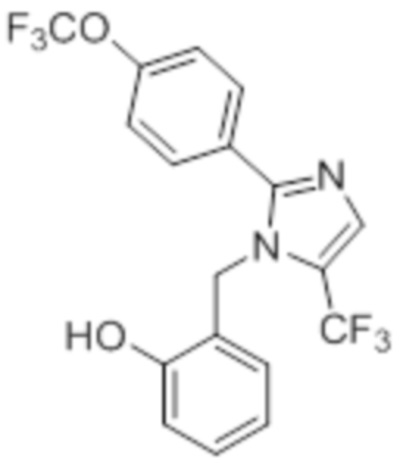
Указанное в заголовке соединение синтезировали из 1-(2-метоксибензил)-2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазола (0,14 г, 0,33 ммоль), следуя экспериментальной методике, описанной на стадии 12 Примера 2A. Выход: 0,115 г (неочищ.).
1H-ЯМР (400 МГц, DMSO-d6): δ 9,80 (ушир. с, 1H), 7,84 (с, 1H), 7,68 (д, J=8,8 Гц, 2H), 7,43 (д, J=8,0 Гц, 2H), 7,06-7,02 (м, 1H), 6,79-6,64 (м, 1H), 6,59 (т, J=6,9 Гц, 1H), 6,32 (д, J=7,6 Гц, 1H ), 5,24 (с, 2H).
Стадия 7: Синтез этил-(3R)-3-метил-6-(2-((2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноата:
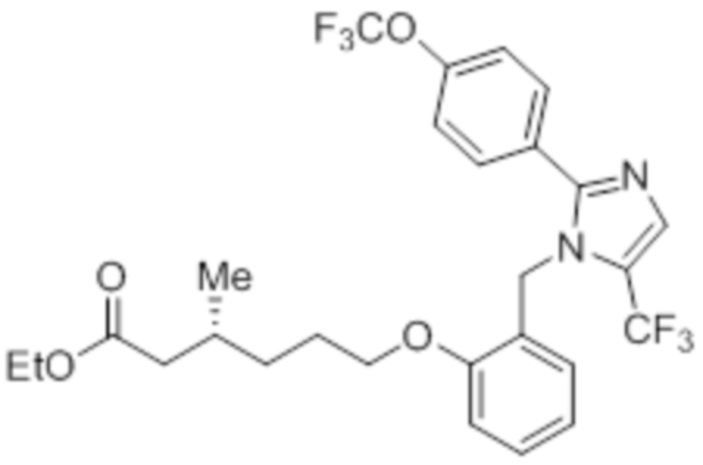
Указанное в заголовке соединение синтезировали из 2-((2-(4-(трифторметокси)-фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола (0,110 г, 0,27 ммоль) и этил-(3R)-6-бром-3-метилгексаноата (0,185 г, 0,81 ммоль), следуя экспериментальной методике, описанной на стадии 13 Примера 2A. Выход: 0,075 г.
LCMS (ESI+, m/z): 558,8 (M+H)+.
Стадия 8: Синтез (3R)-3-метил-6-(2-((2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоты:
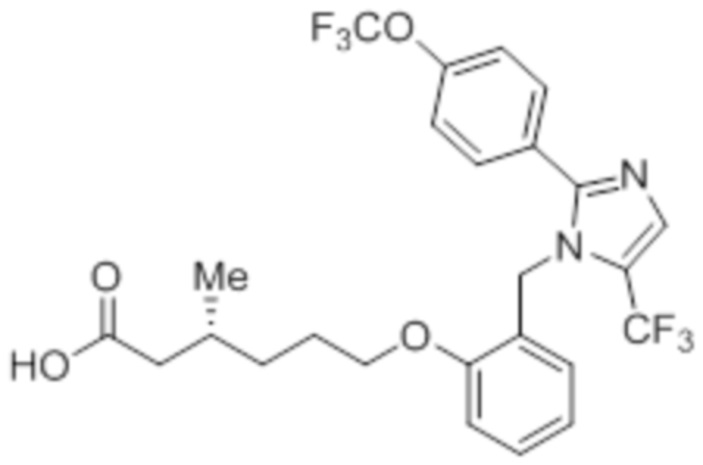
Указанное в заголовке соединение синтезировали из этил-(3R)-3-метил-6-(2-((2-(4-(трифторметокси)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноата (0,07 г, 0,12 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A. Соединение очищали методом обращенно-фазовой препаративной HPLC [EVO C18 (21,2 мм×150 мм, 5 мкм; поток: 15 мл/мин; подвижная фаза: A/B = 0,1% TFA в воде/MeCN; время/% B в подвижной фазе = 0/40, 2/45, 10/70]. Выход: 0,021 г (31,8%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,10 (ушир. с, 1H), 7,77 (с, 1H), 7,68-7,66 (м, 2H), 7,42 (д, J=8,0 Гц, 2H), 7,21 (т, J=6,8 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 6,82 (т, J=7,2 Гц, 1H), 6,42 (д, J=7,2 Гц, 1H), 5,31 (с, 2H), 3,93 (т, J=6,4 Гц, 2H), 2,34-2,29 (м, 1H), 2,01-1,95 (м, 1H), 1,86-1,84 (м, 1H), 1,66-1,63 (м, 2H), 1,48-1,46 (м, 1H), 1,30-1,23 (м, 1H), 0,87 (д, J=6,4 Гц, 3H). 19F-ЯМР (400 МГц, CDCl3): δ -57,80, -59,25. LCMS (ESI+, m/z): 531,0 (M+H)+. HPLC: 95,19% (210 нм).
Пример 2H: Синтез Соединения 2h
Синтез 6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)-метил)фенокси)гексановой кислоты
Схема:
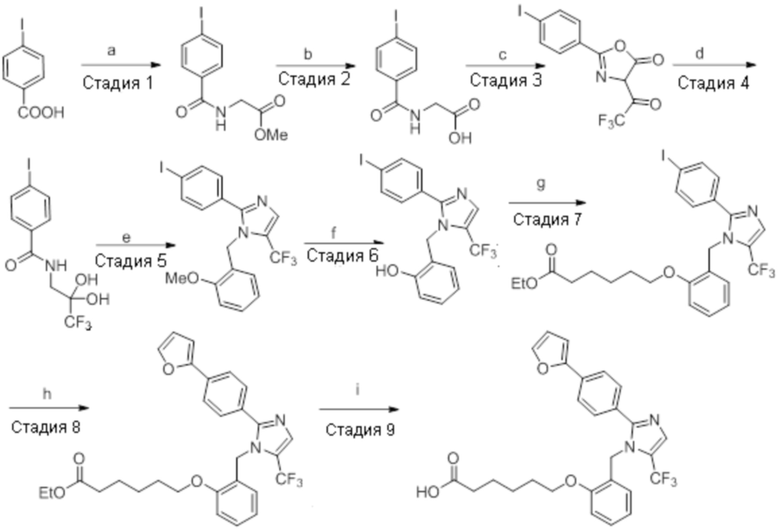
Реагенты и условия: a) метилглицината гидрохлорид, EDCI·HCl, HOBt, Et3N, DMF, 12 ч; b) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч; c) 2,2,2-трифторуксусный ангидрид, ацетон, 0°C - к.т., 12 ч; d) 1,4-диоксан, H2O, 100°C, 3 ч; e) 2-метоксибензиламин, AcOH, толуол, 120°C, 12 ч; f) BBr3, DCM, -78°C; г) этил-6-бромгексаноат, K2CO3, DMF, к.т., 12 ч; h) фуран-2-бороновая кислота, Pd(PPh3)4, Na2CO3, DME, EtOH, H2O, 90°C, 12 ч; i) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез метил-(4-йодбензамидо)ацетата:
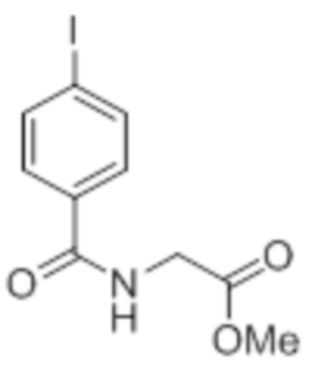
Указанное в заголовке соединение синтезировали из 4-йодбензойной кислоты (20,0 г, 142,74 ммоль) и метилглицината гидрохлорида (26,87 г, 214,11 ммоль), следуя экспериментальной методике, описанной на стадии 1 Примера 2D. Выход: (21,1 г, 81,6%)
LCMS (ESI+, m/z): 319,9 (M+H)+.
Стадия 2: Синтез (4-йодбензоил)глицина:
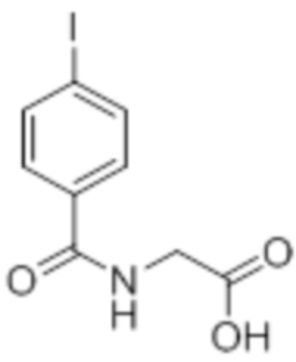
Указанное в заголовке соединение синтезировали из метил-(4-йодбензамидо)-ацетата (21 г, 65,83 ммоль), следуя экспериментальной методике, описанной на стадии 2 Примера 2D. Выход: (17,1 г, 84,7%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,63 (ушир. с, 1H), 8,93 (т, J=5,7 Гц, 1H), 7,89 (д, J=8,1 Гц, 2H), 7,65 (д, J=8,1 Гц, 2H), 3,91 (д, J=5,7 Гц, 2H). LCMS (ESI+, m/z): 306,0 (M+H)+.
Стадия 3: Синтез 2-(4-йодфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она:
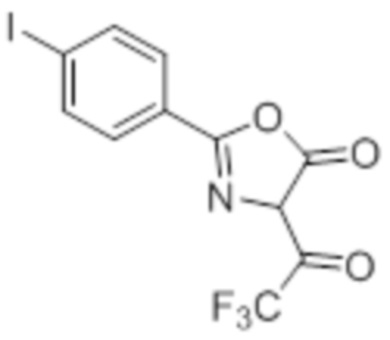
Указанное в заголовке соединение синтезировали из (4-йодбензоил)глицина (17 г, 55,73 ммоль) и 2,2,2-трифторуксусного ангидрида (23,5 мл, 167,21 ммоль), следуя экспериментальной методике, описанной на стадии 3 Примера 2D. Выход: (11,7 г, 66,5%)
1H-ЯМР (400 МГц, DMSO-d6): δ 7,87 (д, J=8,0 Гц, 1H), 7,77 (д, J=8,4 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), 7,49 (д, J=8,0 Гц, 1H). LCMS (ESI-, m/z): 382,1 (M-H)+.
Стадия 4: Синтез 4-йод-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида:
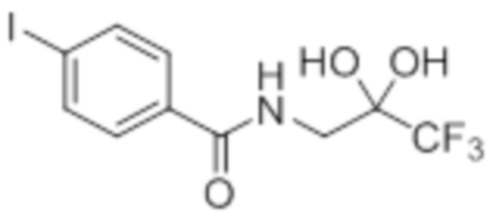
Указанное в заголовке соединение синтезировали из 2-(4-йодфенил)-4-(2,2,2-трифторацетил)оксазол-5(4H)-она (14 г, 36,55 ммоль), следуя экспериментальной методике, описанной на стадии 4 Примера 2D. Выход: 9,75 г (59,1%)
LCMS (ESI+, m/z): 376,1 (M+H)+.
Стадия 5: Синтез 2-(4-йодфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола:
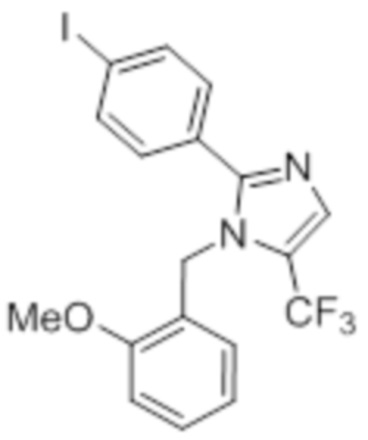
Указанное в заголовке соединение синтезировали из 4-йод-N-(3,3,3-трифтор-2,2-дигидроксипропил)бензамида (3 г, 8,0 ммоль) и 2-метоксибензиламина (1,7 г, 12,0 ммоль), следуя экспериментальной методике, описанной на стадии 5 Примера 2D. Выход: 0,53 г (14,5%)
1H-ЯМР (300 МГц, CDCl3): δ 7,69 (д, J=6,6 Гц, 2H), 7,06 (д, J=1,2 Гц, 1H), 7,30-7,27 (м, 1H), 7,2 (д, J=6,6 Гц, 2H), 6,90-6,85 (м, 2H), 6,53 (д, J=6,6 Гц, 1H), 5,27 (с, 2H), 3,81 (с, 3H). LCMS (ESI+, m/z): 459,1 (M+H)+.
Стадия 6: Синтез 2-((2-(4-йодфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола:
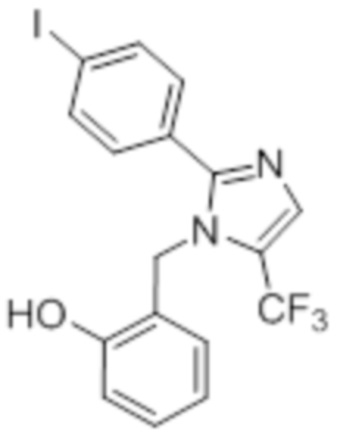
Указанное в заголовке соединение синтезировали из 2-(4-йодфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола (0,5 г, 1,09 ммоль), следуя экспериментальной методике, описанной на стадии 12 Примера 2A. Выход: 0,31 г (61,9%)
LCMS (ESI+, m/z): 444,8 (M+H)+.
Стадия 7: Синтез этил-6-(2-((2-(4-йодфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноата:
Указанное в заголовке соединение синтезировали из 2-((2-(4-йодфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола (0,15 г, 0,337 ммоль) и этил-6-бромгексаноата (0,115 г, 0,506 ммоль), следуя экспериментальной методике, описанной на стадии 13 Примера 2A. Выход: 0,21 г.
LCMS (ESI+, m/z): 586,9 (M+H)+.
Стадия 8: Синтез этил-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноата:
В герметизируемом реакционном сосуде емкостью 100 мл, этил-6-(2-((2-(4-йодфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноат (0,2 г, 0,341 ммоль) и фуран-2-бороновую кислоту (0,075 г, 0,682 ммоль) при к.т. в атмосфере азота растворяли в дегазированной смеси DME (10 мл), EtOH (10 мл) и воды (10 мл). К описанному выше раствору в атмосфере азота добавляли Pd(PPh3)4 (0,040 г, 0,0341 ммоль) и Na2CO3 (0,110 г, 1,023 ммоль). Полученную смесь дегазировали путем продувки аргоном в течение 15 мин, и нагревали реакционную смесь до 90°C до завершения реакции (согласно TLC). Реакционную смесь охлаждали до к.т., разбавляли холодной водой и экстрагировали этилацетатом (3×30 мл). Объединенный экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления с получением указанного в заголовке соединения. Выход: 0,11 г (55,5%).
LCMS (ESI+, m/z): 527,3 (M+H)+.
Стадия 9: Синтез 6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоты:
Указанное в заголовке соединение синтезировали из этил-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)гексаноата (0,14 г, 0,265 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A. Выход: 0,03 г (23,1%).
1H-ЯМР (400 МГц, DMSO-d6, 80°C): δ 12,1 (ушир. с, 1H), 7,76 (ушир. с, 2H), 7,72 (д, J=8,4 Гц, 2H), 7,59 (д, J=8,4 Гц, 2H), 7,21 (т, J=7,6 Гц, 1H), 7,03 (д, J=3,2 Гц, 1H), 6,97 (д, J=8,0 Гц, 1H), 6,83 (т, J=8,0 Гц, 1H), 6,60-6,59 (м, 1H), 6,40 (д, J=7,2 Гц, 1H), 5,32 (с, 2H), 3,95 (т, J=6,0 Гц, 2H), 2,2-2,1 (м, 2H), 1,66-1,63 (м, 2H), 1,52-1,46 (м, 2H), 1,36-1,34 (м, 2H). 19F-ЯМР (400 МГц, DMSO-d6): δ -57,85. LCMS (ESI+, m/z): 499,3 (M+H)+. HPLC: 98,97% (210 нм).
Пример 2I: Синтез Соединения 2i
Синтез (3R)-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты
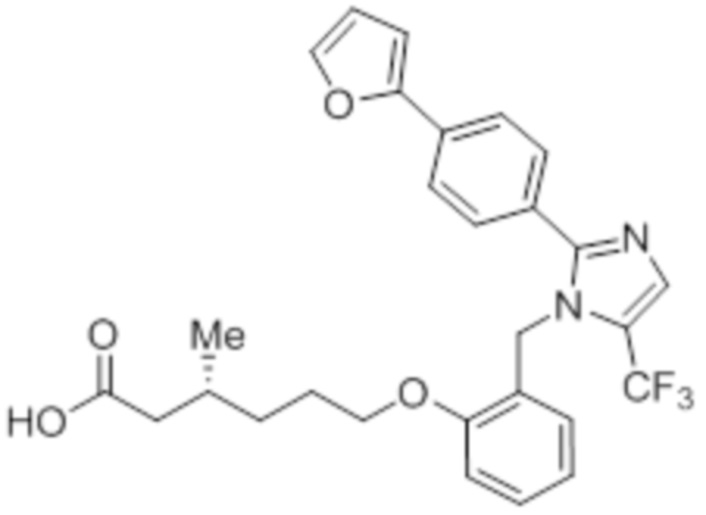
Схема:
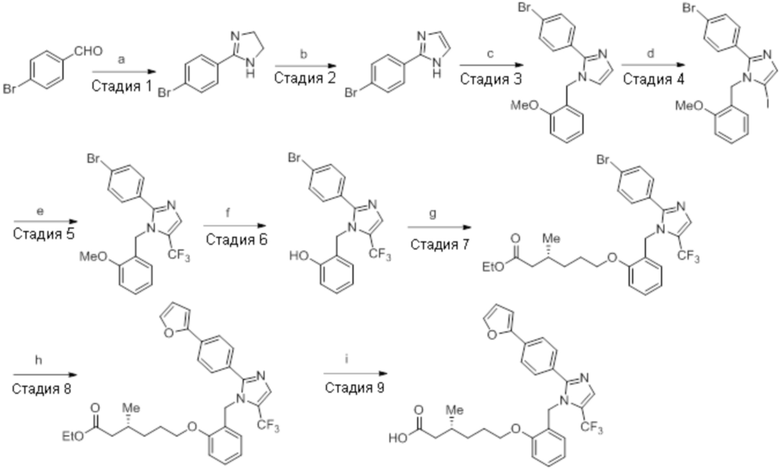
Реагенты и условия: a) этан-1,2-диамин, I2, K2CO3, трет-BuOH, 85°C, 5 ч; b) (диацетоксийод)бензол, K2CO3, DMSO, 12 ч; c) 2-метоксибензилбромид, NaH (60% дисперсия), DMF 0°C – к.т., 4 ч; d) NIS, DMF, 70°C, 12 ч; e) TMSCF3, Ag2CO3, 1,10-фенантролин, KF, CuI, 100°C, 12 ч; f) BBr3, DCM, 0°C – к.т., 3 ч; г) этил-(3R)-6-бром-3-метилгексаноат, tBuOK, DMF, к.т., 12 ч; h) фуран-2-бороновая кислота, Pd(PPh3)4, Na2CO3, DME, EtOH, H2O, 90°C, 12 ч; i) LiOH·H2O, THF, EtOH, H2O, к.т., 12 ч.
Стадия 1: Синтез 2-(4-бромфенил)-4,5-дигидро-1H-имидазола:
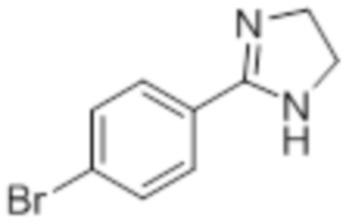
Указанное в заголовке соединение синтезировали из 4-бромбензальдегида (25,0 г, 131,52 ммоль), следуя экспериментальной методике, описанной на стадии 7 Примера 2A. Выход: 22,3 г (77,13%).
1H-ЯМР (400 МГц, DMSO-d6): δ 7,73 (д, J=8,0 Гц, 2H), 7,62 (д, J=8,4 Гц, 2H), 3,59 (с, 4H), 3,48 (ушир. с, 1H). LCMS (ESI+, m/z): 225,0, 227,0 (M+H)+.
Стадия 2: Синтез 2-(4-бромфенил)-1H-имидазола:
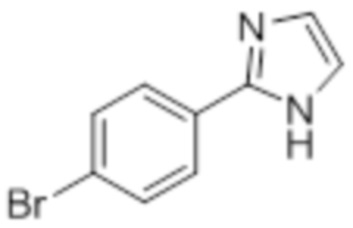
Указанное в заголовке соединение синтезировали из 2-(4-бромфенил)-4,5-дигидро-1H-имидазола (10,0 г, 44,44 ммоль), следуя экспериментальной методике, описанной на стадии 8 Примера 2A. Выход: 4,5 г (45,4%).
1H-ЯМР (300 МГц, DMSO-d6): δ 12,6 (с, 1H), 7,85 (дд, J=9,0, 1,8 Гц, 2H), 7,62 (дд, J=6,6, 1,8 Гц, 2H), 7,13 (ушир. с, 2H). LCMS (ESI+, m/z): 223,1, 225,1 (M+H)+.
Стадия 3: Синтез 2-(4-бромфенил)-1-(2-метоксибензил)-1H-имидазола:
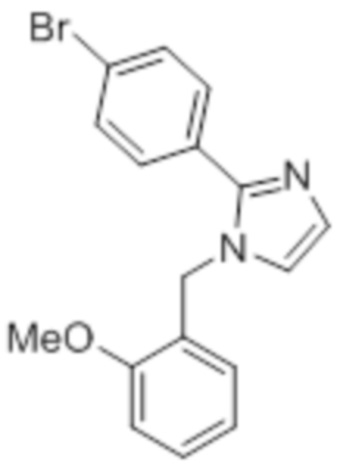
Указанное в заголовке соединение синтезировали из 2-(4-бромфенил)-1H-имидазола (4,5 г, 20,17 ммоль), следуя экспериментальной методике, описанной на стадии 9 Примера 2A. Выход: 3,1 г (82,5%).
LCMS (ESI+, m/z): 342,9, 344,9 (M+H)+.
Стадия 4: Синтез 2-(4-бромфенил)-5-йод-1-(2-метоксибензил)-1H-имидазола:
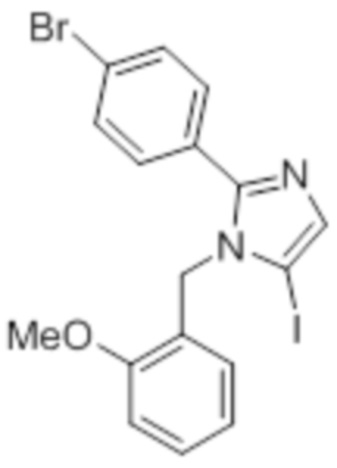
Указанное в заголовке соединение синтезировали из 2-(4-бромфенил)-1-(2-метоксибензил)-1H-имидазола (3 г, 8,746 ммоль), следуя экспериментальной методике, описанной на стадии 10 Примера 2A. Выход: 2,1 г (51,2%).
1H-ЯМР (300 МГц, CDCl3): δ 7,47 (д, J=8,7 Гц, 2H), 7,33 (д, J=5,2 Гц, 2H), 7,31 (с, 1H), 6,92 (д, J=7,5 Гц, 2H), 6,53 (д, J=8,1 Гц, 2H), 5,18 (с, 2H), 3,86 (с, 3H). LCMS (ESI+, m/z): 468,9, 470,9 (M+H)+.
Стадия 5: Синтез 2-(4-бромфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола:
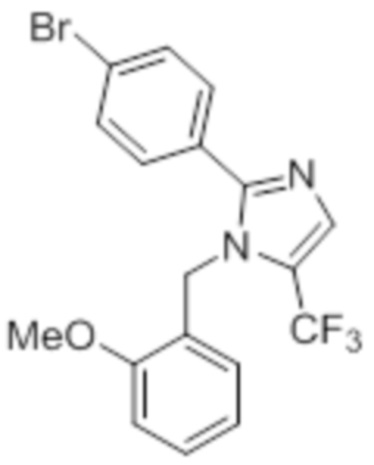
Указанное в заголовке соединение синтезировали из 2-(4-бромфенил)-5-йод-1-(2-метоксибензил)-1H-имидазола (0,5 г, 1,06 ммоль), следуя экспериментальной методике, описанной на стадии 11 Примера 2A. Выход: 0,22 г (50,2%).
1H-ЯМР (400 МГц, CDCl3): δ 7,61 (с, 1H), 7,49 (д, J=6,3 Гц, 2H), 7,35 (д, J=6,3 Гц, 2H), 7,30-7,26 (м, 1H), 6,91-6,75 (м, 2H), 6,55 (д, J=5,7 Гц, 1H), 5,28 (с, 2H), 3,82 (с, 3H). 19F-ЯМР (400 МГц, CDCl3) : δ -63,21. LCMS (ESI+, m/z): 411,1, 413,1(M+H)+.
Стадия 6: Синтез 2-((2-(4-бромфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола:
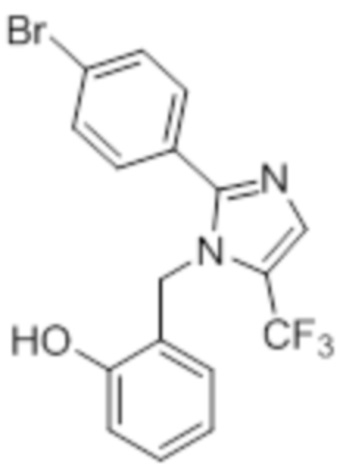
Указанное в заголовке соединение синтезировали из 2-(4-бромфенил)-1-(2-метоксибензил)-5-(трифторметил)-1H-имидазола (0,5 г, 1,216 ммоль), следуя экспериментальной методике, описанной на стадии 12 Примера 2A. Выход: 0,252 г, (52,08%).
1H-ЯМР (400 МГц, DMSO-d6): δ 9,91 (с, 1H), 7,86 (с, 1H), 7,66 (д, J=8,4 Гц, 2H), 7,52 (д, J=8,4 Гц, 2H), 7,07 (т, J=7,2 Гц, 1H), 6,79 (д, J=8,0 Гц, 1H), 6,69 (т, J=7,6 Гц, 1H), 6,32 (д, J=7,2 Гц, 1H), 5,26 (с, 2H). 19F-ЯМР (400 МГц, DMSO-d6) : δ -58,01.
Стадия 7: Синтез этил-(3R)-6-(2-((2-(4-бромфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
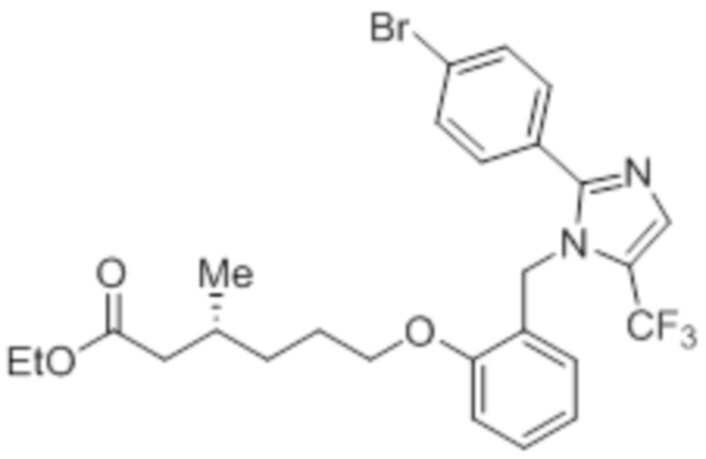
В круглодонной колбе емкостью 100 мл, перемешанный раствор 2-((2-(4-бромфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенола (0,5 г, 1,259 ммоль) в DMF (20 мл) при к.т. в атмосфере азота обрабатывали KOtBu (0,435 г, 3,7 ммоль) и этил-(3R)-6-бром-3-метилгексаноатом (0,6 г, 2,518 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 2 ч. После завершения реакции (согласно TLC), реакционную смесь гасили добавлением ледяной воды и экстрагировали этилацетатом (3×25 мл). Объединенный органический экстракт промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюирование градиентом 15-30% EtOAc в гексанах) с получением указанного в заголовке соединения. Выход: 0,35 г (50,28%).
LCMS (ESI+, m/z): 553,3, 555,3 (M+H)+.
Стадия 8: Синтез этил-(3R)-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата:
Указанное в заголовке соединение синтезировали из этил-(3R)-6-(2-((2-(4-бромфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (0,2 г, 0,361 ммоль) и фуран-2-бороновой кислоты (81 мг, 0,723 ммоль), следуя экспериментальной методике, описанной на стадии 8 Примера 2H. Выход: 0,1 г (51,28%).
19F-ЯМР (400 МГц, CDCl3) : δ -59,18. LCMS (ESI+, m/z): 541,3 (M+H)+.
Стадия 9: Синтез (3R)-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоты:
Указанное в заголовке соединение синтезировали из этил-(3R)-6-(2-((2-(4-(фуран-2-ил)фенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексаноата (0,25 г, 0,46 ммоль), следуя экспериментальной методике, описанной на стадии 14 Примера 2A. Выход: 0,135 г (56,96%).
1H-ЯМР (400 МГц, DMSO-d6): δ 12,01 (с, 1H), 7,79 (с, 2H), 7,74 (д, J=8,4 Гц, 2H), 7,61 (д, J=8,8 Гц, 2H), 7,25 (т, J=8,0 Гц, 1H), 7,05 (д, J=3,2 Гц, 1H), 7,01 (д, J=8,0 Гц, 1H), 6,86 (т, J=8,0 Гц, 1H), 6,62 (с, 1H), 6,42 (д, J=6,8 Гц,1H), 5,34 (с, 2H), 3,97 (т, J=6,0 Гц, 2H), 2,24-2,19 (м, 1H), 2,02-1,96 (м, 1H), 1,90-1,80 (м,1H), 1,68-1,61 (м, 2H), 1,40-1,30 (м, 1H), 1,30-1,15 (м, 1H), 0,87 (д, J=6,4 Гц, 3H). 19F-ЯМР (400 МГц, DMSO-d6): δ -57,83. LCMS (ESI+, m/z): 512,6 (M+H)+. HPLC (210 нм): 94,62%.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ И СОЛЕВЫЕ ФОРМЫ СОЕДИНЕНИЙ АГОНИСТОВ PPAR | 2017 |
|
RU2759724C2 |
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2525116C2 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ОСТРОГО ПОВРЕЖДЕНИЯ ПОЧЕК | 2017 |
|
RU2753607C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его фармацевтически приемлемой соли, где L представляет собой (CH2)5, который необязательно замещен одной метильной группой; R1 представляет собой водород; R2 представляет собой галоген, CN, C1–C4-галогеналкил, C1–C4-галогеналкокси или фуранил; R3 представляет собой С1-C4-галогеналкил, -CN или галоген. Также изобретение относится к (3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоте, (R)-6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоте, (R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановой кислоте, (3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)гексановой кислоте или к их фармацевтически приемлемым солям, фармацевтической композиции на основе соединения формулы (I) или конкретных указанных выше соединений, способу лечения связанного с PPARδ заболевания или состояния у субъекта, предусматривающему введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или конкретных указанных выше соединений. Технический результат: получены новые гетероциклические соединения, полезные для лечения связанных с PPARδ заболеваний. 7 н. и 8 з.п. ф-лы, 1 табл., 11 пр.
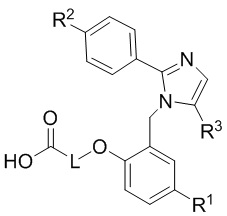 (I)
(I)
1. Соединение формулы (I),
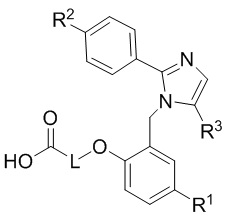 (I)
(I)
или его фармацевтически приемлемая соль, где
L представляет собой (CH2)5, который необязательно замещен одной метильной группой;
R1 представляет собой водород;
R2 представляет собой галоген, CN, C1–C4-галогеналкил, C1–C4-галогеналкокси или фуранил;
R3 представляет собой С1-C4-галогеналкил, -CN или галоген.
2. Соединение по п. 1, имеющее структуру любой из формул (Ia) или (Ib),
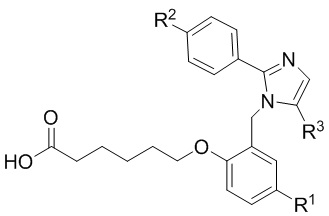
(Ia) или
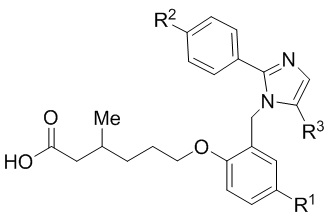
(Ib);
или его фармацевтически приемлемая соль.
3. Соединение по п. 2, имеющее структуру формулы (Ibb),
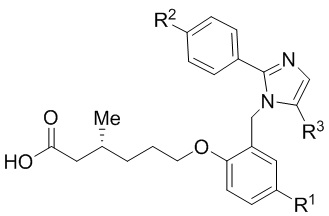 (Ibb);
(Ibb);
или его фармацевтически приемлемая соль.
4. Соединение по любому из пп. 1–3, в котором R3 представляет собой галогенметил, CN или галоген.
5. Соединение по любому из пп. 1–3, в котором R3 представляет собой CF3, Cl или CN.
6. Соединение по любому из пп. 1–5, в котором R2 представляет собой галоген, CN, галогенметил, галогенметокси, или фуранил.
7. Соединение по п. 6, в котором R2 представляет собой F, Cl, CN, CF3, OCF3 или фуранил.
8. (3R)-6-(2-((2-(4-хлорфенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановая кислота или ее фармацевтически приемлемая соль.
9. (R)-6-(2-((5-хлор-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановая кислота или ее фармацевтически приемлемая соль.
10. (R)-6-(2-((2-(4-цианофенил)-5-(трифторметил)-1H-имидазол-1-ил)метил)фенокси)-3-метилгексановая кислота или ее фармацевтически приемлемая соль.
11. (3R)-3-метил-6-(2-((5-(трифторметил)-2-(4-(трифторметил)фенил)-1H-имидазол-1-ил)метил)фенокси)гексановая кислота или ее фармацевтически приемлемая соль.
12. Фармацевтическая композиция для лечения связанного с PPARδ заболевания или состояния у субъекта, содержащая фармацевтически приемлемый носитель или наполнитель и терапевтически эффективное количество соединения по любому из пп. 1–11 или его фармацевтически приемлемую соль.
13. Способ лечения связанного с PPARδ заболевания или состояния у субъекта, предусматривающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества одного или более из соединений по любому из пп. 1–11, или его фармацевтически приемлемой соли, или фармацевтической композиции по п. 12.
14. Способ по п. 13, причем связанным с PPARδ заболеванием является нарушение мышечной структуры, нарушение нейрональной активации, связанное с мышечным утомлением нарушение, связанное с мышечной массой нарушение, митохондриальное заболевание, связанное с бета-окислением заболевание, метаболическое заболевание, злокачественная опухоль, сосудистое заболевание, глазное сосудистое заболевание, заболевание мышечного аппарата глаза или почечное заболевание.
15. Способ по п. 14, причем:
нарушение мышечной структуры выбрано из миопатии Бетлема, болезни центрального стержня, врожденной диспропорции волокнистого типа, дистальной мышечной дистрофии (MD), MD Дюшенна и Беккера, MD Эмери-Дрейфуса, плече-лопаточно-лицевой MD, миопатии гиалиновых телец, тазово-плечевой MD, мышечных нарушений, связанных с натриевыми каналами, миотонической хондродистрофии, миотонической дистрофии, миотубулярной миопатии, заболевания с образованием немалиновых телец, окулофарингеальной MD или недержания мочи при напряжении;
нарушение нейрональной активации выбрано из амиотрофического латерального склероза, болезни Шарко-Мари-Тута, синдрома Гийена-Барре, синдрома Ламберта-Итона, рассеянного склероза, миастении гравис, повреждения нерва, периферической нейропатии, спинальной мышечной атрофии, позднего паралича локтевого нерва или токсического нервно-мышечного нарушения;
связанное с мышечным утомлением нарушение выбрано из синдрома хронической усталости, сахарного диабета (I или II типа), болезни накопления гликогена, фибромиалгии, атаксии Фридрейха, перемежающейся хромоты, миопатии, обусловленной накоплением липидов, MELAS, мукополисахаридоза, болезни Помпе или тиреотоксической миопатии;
связанное с мышечной массой нарушение представляет собой кахексию, дегенерацию хряща, церебральный паралич, синдром сдавливания, миопатию критических состояний, миозит с включенными тельцами, мышечную атрофию (дисфункциональную), саркопению, стероидную миопатию или системную красную волчанку;
митохондриальное заболевание выбрано из болезни Альперса, CPEO - хронической прогрессирующей внешней офтальмоплегии, синдрома Кирнса-Сейра (KSS), наследственной оптической нейропатии Лебера (LHON), MELAS - митохондриальной миопатии, энцефаломиопатии, лактатацидоза и подобных инсульту эпизодов, MERRF - заболевания миоклонической эпилепсии с рваными мышечными волокнами, NARP - нейрогенной мышечной слабости, атаксии, пигментного ретинита или синдрома Пирсона;
связанное с бета-окислением заболевание выбрано из системного дефицита транспортера карнитина, дефицита карнитинпальмитоилтрансферазы (CPT) II, дефицита длинноцепочечной ацил-КоА-дегидрогеназы (LCHAD или VLCAD), дефицита трифункционального фермента, дефицита среднецепочечной ацил-КоА-дегидрогеназы (MCAD), дефицита короткоцепочечной ацил-КоА-дегидрогеназы (SCAD) или рибофлавин-чувствительных нарушений β-окисления (RR-MADD);
метаболическое заболевание выбрано из гиперлипидемии, дислипидемии, гиперхолестеринемии, гипертриглицеридемии, гипохолестеринемии за счет HDL, гиперхолестеринемии за счет LDL и/или холестеринемии не за счет HLD, гиперпротеинемии за счет VLDL, дислипопротеинемии, гипопротеинемии аполипопротеина A-I, атеросклероза, заболевания артериального склероза, заболевания сердечно-сосудистой системы, сосудистого заболевания головного мозга, заболевания периферического кровообращения, метаболического синдрома, синдрома X, ожирения, сахарного диабета (I или II типа), гипергликемии, инсулиновой резистентности, нарушенной толерантности к глюкозе, гиперинсулинизма, диабетического осложнения, сердечной недостаточности, инфаркта миокарда, кардиомиопатии, гипертензии, неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), тромба, болезни Альцгеймера, нейродегенеративного заболевания, демиелинизирующего заболевания, рассеянного склероза, лейкодистрофии надпочечника, дерматита, псориаза, акне, старения кожи, трихоза, воспаления, артрита, астмы, синдрома повышенной чувствительности кишечника, язвенного колита, болезни Крона или панкреатита;
злокачественная опухоль представляет собой злокачественную опухоль ободочной кишки, толстого кишечника, кожи, молочной железы, предстательной железы, яичника или легкого;
сосудистое заболевание выбрано из недостаточности периферических сосудов, заболевания периферических сосудов, перемежающейся хромоты, заболевания периферических сосудов (PVD), заболевания периферических артерий (PAD), окклюзионного заболевания периферических артерий (PAOD) или периферической облитерирующей артериопатии;
глазное сосудистое заболевание выбрано из возрастной макулярной дегенерации (AMD), болезни Штаргардта, гипертензивной ретинопатии, диабетической ретинопатии, ретинопатии, макулярной дегенерации, кровоизлияния в сетчатку или глаукомы;
заболевание мышечного аппарата глаза выбрано из страбизма, прогрессивной внешней офтальмоплегии, изотропии, экзотропии, нарушения рефракции и аккомодации, гиперметропии, миопии, астигматизма, анизометропии, пресбиопии, нарушения аккомодации или внутренней офтальмоплегии; и
почечное заболевание выбрано из гломерулонефрита, гломерулосклероза, нефротического синдрома, гипертонического нефросклероза, острого нефрита, рецидивной гематурии, персистирующей гематурии, хронического нефрита, быстро прогрессирующего нефрита, острого поражения почек, хронической почечной недостаточности, диабетической нефропатии или синдрома Барттера.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Телефонная трансляция | 1926 |
|
SU5532A1 |

