Область изобретения
Настоящее изобретение относится к новым способам получения гетероциклических соединений 1,3-бензодиоксола и его промежуточных соединений. Соединения пригодны для использования в качестве ингибиторов PDE4.
Уровень техники
В WO2011/160632 раскрыты гетероциклические соединения бензодиоксола и бензодиоксепина, пригодные для использования в качестве ингибиторов PDE4, а также подходящие способы их получения.
В WO2008/104175 раскрыты гетероциклические соединения бензодиоксола и бензодиоксепина, пригодные для использования в качестве ингибиторов PDE4, а также подходящие способы их получения.
В WO2008/077404 раскрыты замещенные ацетофеноны, пригодные для использования в качестве ингибиторов PDE4, а также подходящие способы их получения.
В PCT/EP2015/063942, самая ранняя дата приоритета 23 июня 2014 г., раскрыты способы получения гетероциклических соединений 1,3-бензодиоксола.
Zafrani et al.Tetrahedron 65, 2009, pp 5278-5283, описали способ дифторметилирования фенолов и тиофенолов.
Sperry et al. Org. Process Res. Dev. 15, 2011, pp 721-725, тоже описали дифторметилирование фенолов.
Frey et al. Tetrahedron 2003, 59, pp. 6363-6373 тоже описали диметилирование и дифторметилирование фенолов
Zhang et al. J. Org. Chem. 2006, 71, 9845-9848 тоже описали дифторметилирование фенолов.
Zheng et al. Chem. Commun.2007, 5149-5151 тоже описали дифторметилирование фенолов.
При разработке новых потенциальных лекарственных препаратов, весьма желательно иметь доступ к альтернативным способам приготовления потенциальных лекарственных препаратов, поскольку расширение некоторых эффективных способов маломасштабного синтеза до производства промышленных количеств продукта может представлять проблему. Кроме того, маломасштабный синтез может включать реагенты и растворители, которые нецелесообразно использовать на промышленном уровне.
Таким образом, целью настоящего изобретения является предложение альтернативных способов получения гетероциклических соединений 1,3-бензодиоксола того типа, который раскрыт в WO 2011/160632 и PCT/EP2015/063942, в случаях, когда такие альтернативные способы создают преимущества в отношении одной или более характеристик, таких как количество стадий реакции, чистота, выход, легкость очистки, экономичность процесса, доступность исходных материалов и реагентов, безопасность, предсказуемость и т. д.
Неожиданно обнаружилось, что на стадии (2a) по настоящему изобретению, выходы ≥80%, которые намного превосходят выход 32%, полученный при удалении защиты фенольной группы в способе, описанном в WO 2011/160632. Кроме того, по способу, описанному в WO 2011/160632, используется хроматографическая очистка продукта, тогда как способ по настоящему изобретению дает возможность очищать продукт простыми однократными операциями, легко масштабируемыми в промышленную установку.
Далее, неожиданно обнаружилось, что на стадии (3) по настоящему изобретению, дифторметиловая группа теперь может быть введена с выходом ≥76% с или без выделения промежуточной соли. Это превосходит выход 52%, получаемый способом, описанным в WO 2011/160632. Кроме того, по способу, описанному в WO 2011/160632, используется хроматографическая очистка продукта, тогда как способ по настоящему изобретению дает возможность очищать продукт очень простыми однократными операциями, легко масштабируемыми в промышленную установку.
Улучшенный выход и простота масштабирования процесса по настоящему изобретению по сравнению со способом, описанным в WO 2011/160632, оказались неожиданными.
Сущность изобретения
Авторы изобретения обнаружили, что альтернативный способ, раскрытый в данном документе, обеспечивает преимущества перед известными способами, обусловленные меньшим количеством стадий, улучшенным общим химическим и объемным выходом и сопутствующим снижением производственных затрат.
Таким образом, в настоящем изобретении предлагается способ получения соединений 1,3-бензодиоксола, например, соединения формулы (I).
Кроме того, в объем изобретения входят промежуточные соединения, применяемые в указанном выше способе для получения соединений формулы (I).
Подробное описание изобретения
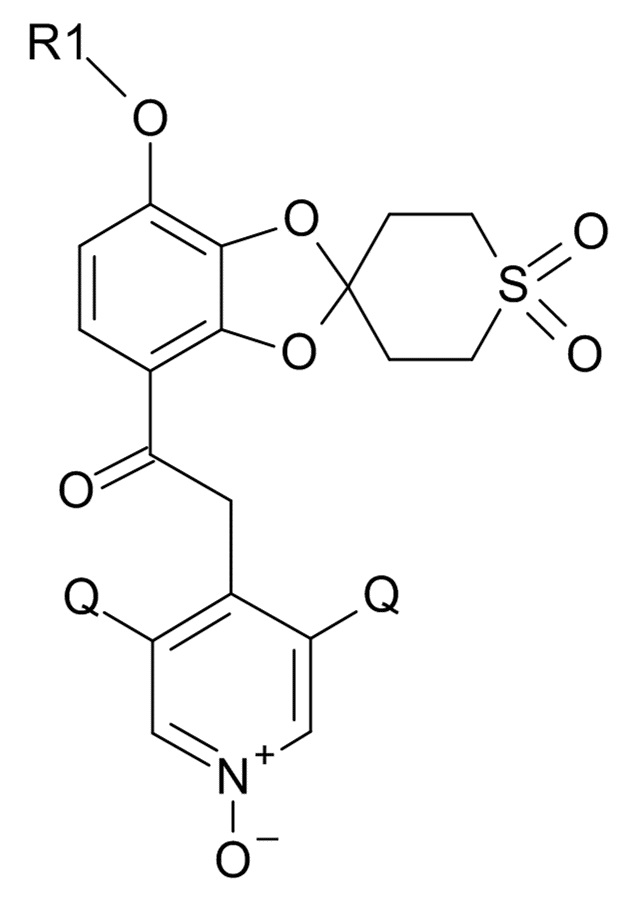
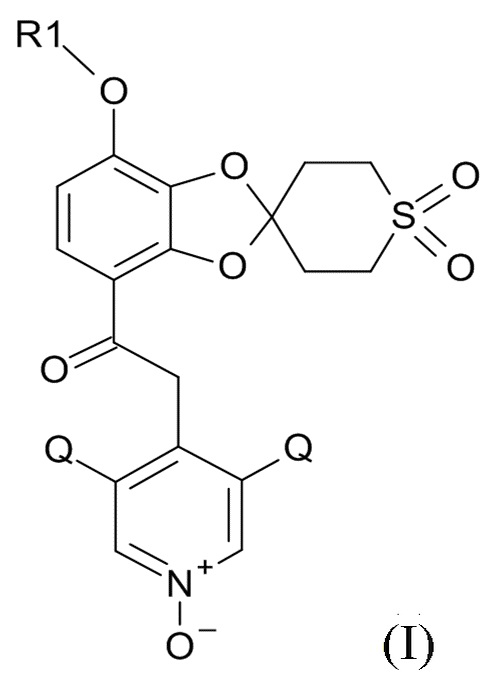
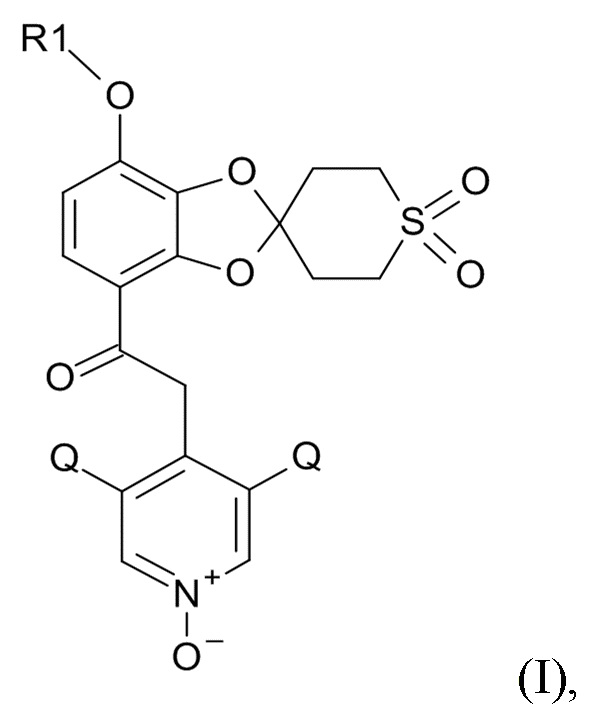
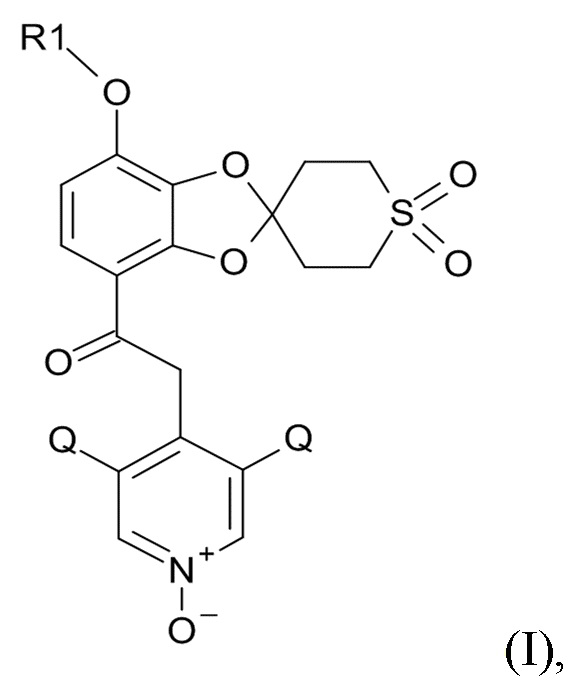
В первом аспекте, настоящее изобретение относится к способу получения соединения формулы (I)
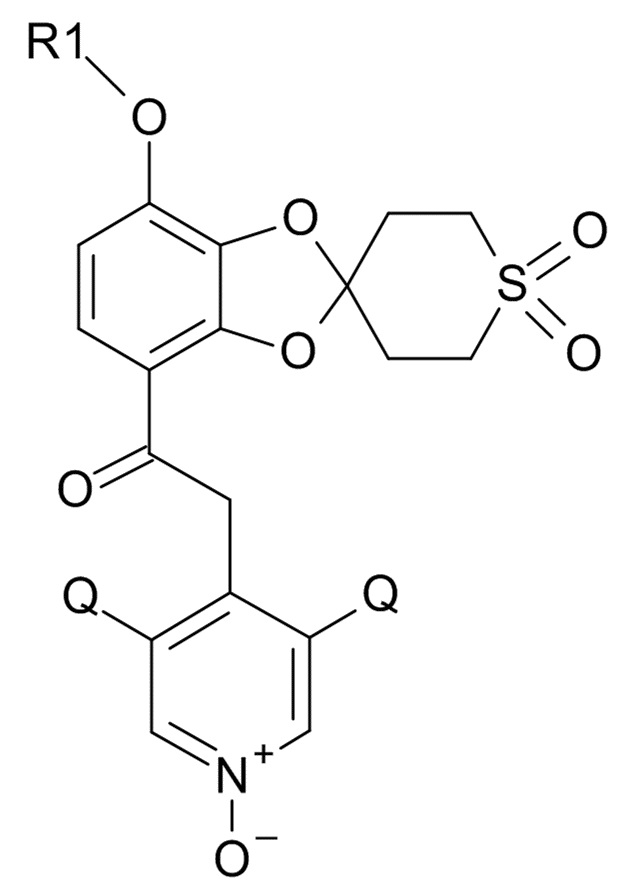 (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора.
В соединении формулы (I), R1 обычно обозначает CHF2, Q обычно выбрано из хлора, брома и фтора, предпочтительно, хлора, где Q, предпочтительно, являются одинаковыми. В одном варианте реализации изобретения, оба Q представляют собой хлор.
Определения
Термин ʺC1-6-алкилʺ используется для обозначения насыщенной, линейной или разветвленной углеводородной цепи, содержащей от одного до шести атомов углерода, включая метил, этил, пропил, изопропил, бутил, изобутил, вторичный бутил, третичный бутил, пентил, изопентил, неопентил, третичный пентил, гексил и изогексил. В некоторых вариантах реализации изобретения, ʺC1-6-алкилʺ представляет собой C1-4-алкильную группу, например, метил, этил, пропил, изопропил, бутил, изобутил, вторичный бутил и третичный бутил. Соответственно, ʺC1-3-алкилʺ включает метил, этил, пропил и изопропил.
Термин ʺгалогенʺ используется для обозначения одного из элементов, фтора, хлора, брома и иода. В одном варианте реализации изобретения, термин ʺгалогенʺ обозначает фтор или хлор. В другом варианте реализации изобретения, термин ʺгалогенʺ обозначает хлор.
Термин ʺарилʺ используется для обозначения карбоциклической ароматической кольцевой системы, полученной из ароматического углеводорода посредством удаления атома водорода. Кроме того, арил включает би-, три- и полициклические кольцевые системы. Примеры предпочтительных ариловых соединений включают фенил, нафтил, инденил, инданил, флуоренил и бифенил. Предпочтительный "арил" представляет собой фенил, нафтил или инданил, особенно фенил, в случаях, когда не указано иное.
Термин ʺарилалкилʺ используется для обозначения арильного радикала, как определено выше, ковалентно связанного с алкильной группой, например, бензила.
Способы получения
Обнаружилось, что способ обеспечивает преимущества перед известными способами за счет использования дешевых исходных материалов, простоты способа производства и повышения выходов в реакциях.
Стадия (1)
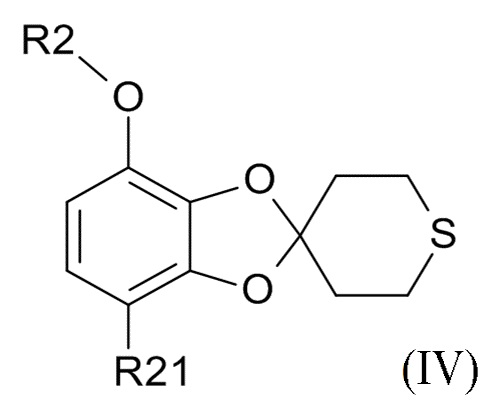
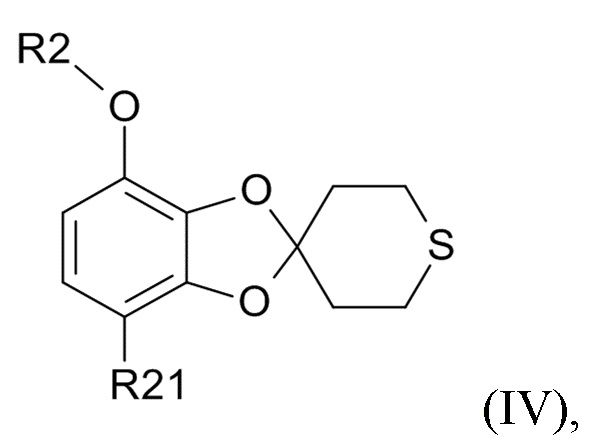
Способ получения соединения формулы (I) включает образование соединения формулы (IV), которое получают посредством
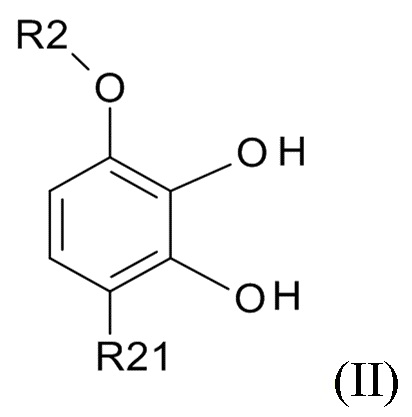
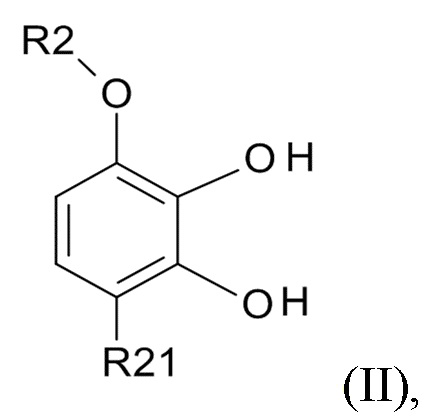
проведения реакции соединения формулы (II)
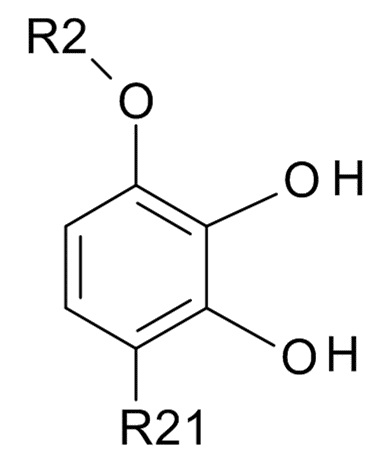 (II)
(II)
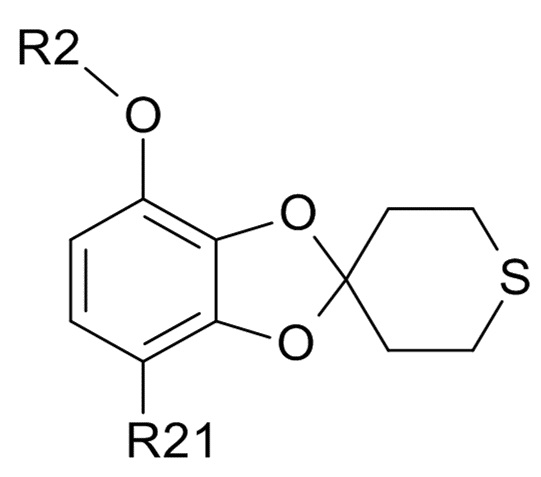
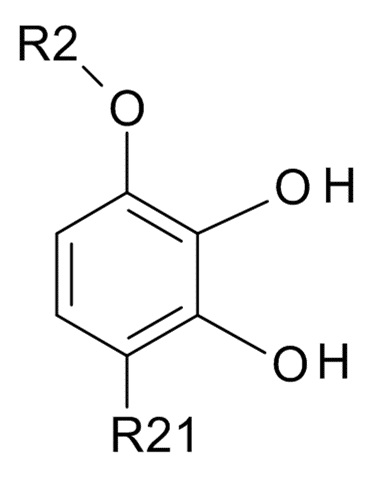
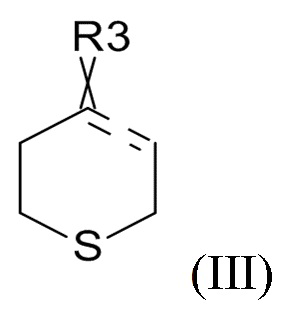
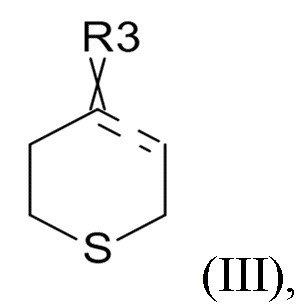
где R2 выбрано из водорода, C1-6-алкила и арилалкила, R21 выбрано из водорода и C(O)R22 и R22 выбрано из водорода и C1-6-алкила; с соединением формулы (III)
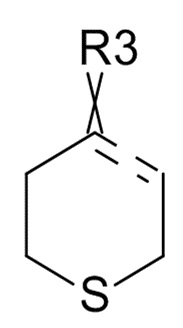 (III)
(III)
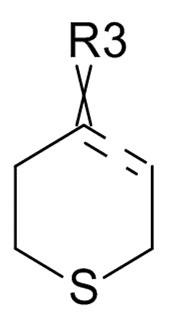
где ʺ ʺ обозначает одинарную связь, двойную связь или две одинарных связи и, в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺ
ʺ обозначает одинарную связь, двойную связь или две одинарных связи и, в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺ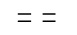 ʺ обозначает одинарную связь, и, в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь; R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарных связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
ʺ обозначает одинарную связь, и, в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь; R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарных связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
 (IV)
(IV)
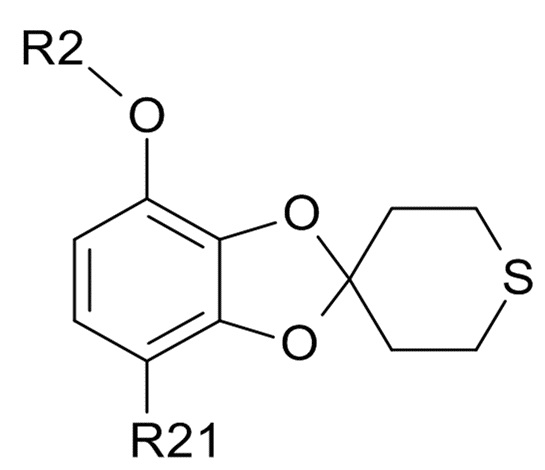
где R2 и R21 соответствуют приведенным выше определениям.
Кислотный катализатор обычно находится в виде силикатного минерала. Силикатные минералы обычно выбирают из монтмориллонита K10, монтмориллонита K30, монтмориллонита KSF, цеолита HSZ-341NHA, цеолита HSZ-331NHA, цеолита HSZ-350HUA и цеолита HSZ-360HUA. В одном варианте реализации изобретения, силикатный минерал выбрано из монтмориллонита K10 и цеолита HSZ-360HUA. В другом варианте реализации изобретения, силикатный минерал представляет собой монтмориллонит K10.
Соединения формулы (III) обычно выбирают из
 (IIIa),
(IIIa), 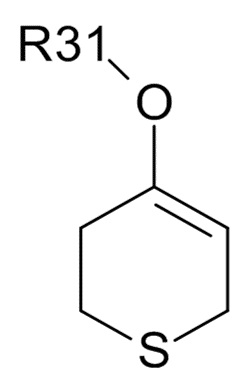 (IIIb), и
(IIIb), и 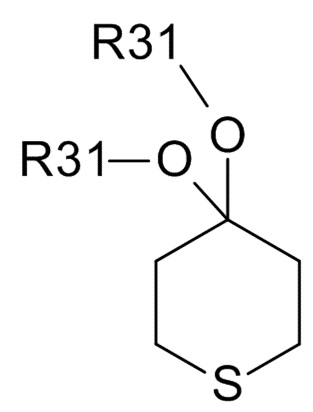 (IIIc)
(IIIc)
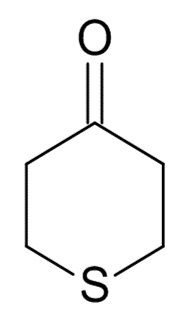
где R31 представляет собой C1-6-алкил. В одном варианте реализации изобретения, соединение формулы (III) выбрано из соединений формулы (IIIa) и формулы (IIIb), где R31 представляет собой метил. В другом варианте реализации изобретения, соединение формулы (III) представляет собой тетрагидротиопиран-4-он.
Соотношение между силикатным минералом и соединением формулы (II) может оказывать влияние на конверсию и время фильтрации. Следовательно, обычно предпочтительно, чтобы количество минерала составляло от 25%-мас./мас. до 500%-мас./мас. относительно массы соединения формулы (II). В частности, количество минерала должно составлять от 25%-мас./мас. до 75%-мас./мас., предпочтительно, должно находиться в диапазоне от 45%-мас./мас. до 55%-мас./мас.
Обычно реакцию проводят в толуоле, бензоле, 2-метил-ТГФ (2-метил-тетрагидрофуране), EtOAc (этилацетате), ксиленах, гептане, октане, хлорбензоле и дихлорбензоле. В одном варианте реализации изобретения, растворитель представляет собой толуол.
Обычно реакцию проводят при температуре выше 80°C, для ускорения реакции. Следовательно, обычно предпочтительно, чтобы температура находилась в диапазоне 80-200°C, такой как 100-160°C, особенно такой как 105-115°C. В одном варианте реализации изобретения, реакцию проводят при конденсации реакционной смеси. Обычно реакции дают протекать в течение 4-96 час, например, 24-84 час, особенно, 48-84 час.
Полученное соединение формулы (IV) можно извлекать традиционными средствами, известными специалистам в данной области техники, например, водной обработкой с последующей экстракцией, дальнейшим осаждением и фильтрацией.
В одном варианте реализации изобретения, используется соединение формулы (II), в котором R2 выбрано из водорода или метила и R21 выбрано из водорода, COCH3 или COOH. В другом варианте реализации изобретения, соединение формулы (II) представляет собой 1-(2,3-дигидрокси-4-метоксифенил)этанон.
В одном варианте реализации изобретения, соединение формулы (III) представляет собой тетрагидротиопиран-4-он.
В одном варианте реализации изобретения, соединение формулы (IV) представляет собой, где R2 обозначает водород, метил, этил, пропил, изопропил, изобутил, вторичный бутил, третичный бутил или бензил, и R21 выбрано из водорода, COCH3 или COOH. В другом варианте реализации изобретения, соединение формулы (IV) представляет собой, где R2 обозначает метил и R21 обозначает COCH3.
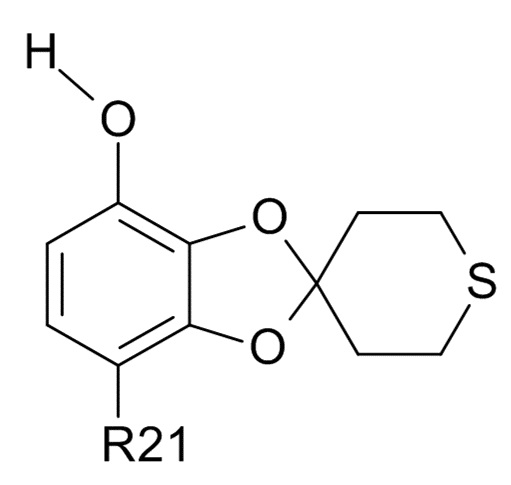
Стадия (2a)
Соединение формулы (IV)
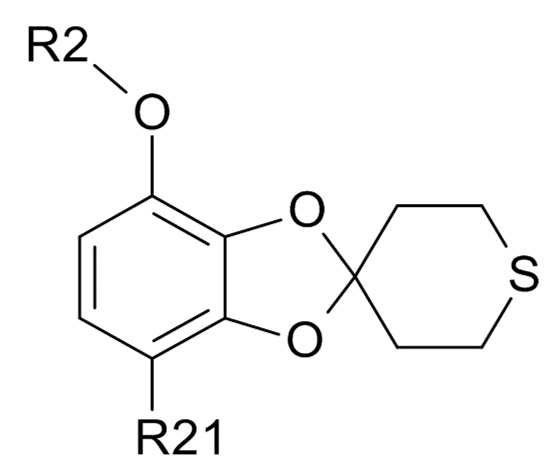 (IV)
(IV)
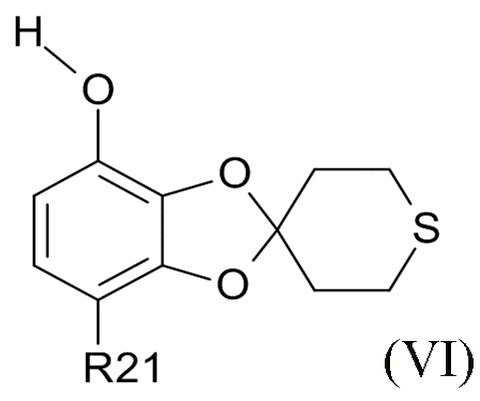
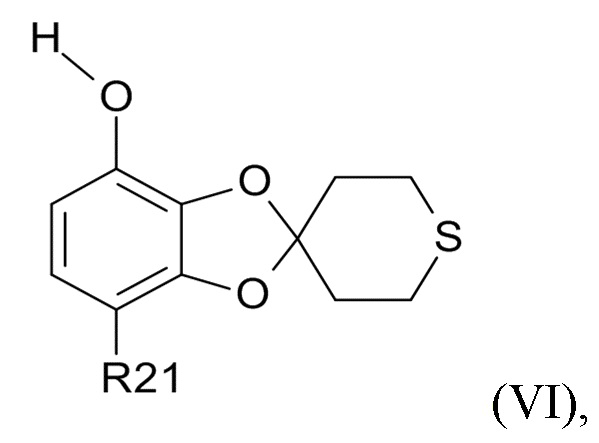
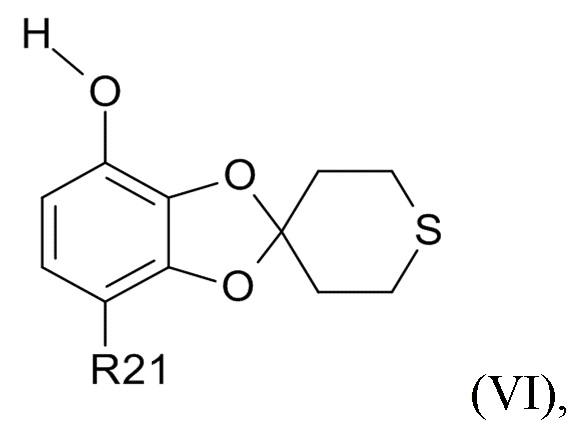
где R2 и R21 соответствуют приведенным выше определениям, преобразуют в соединение формулы (VI)
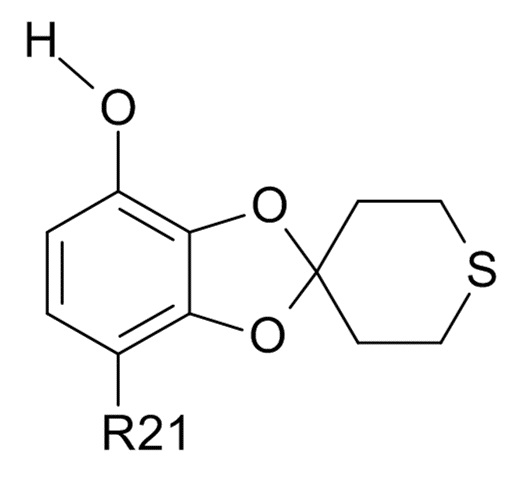 (VI)
(VI)
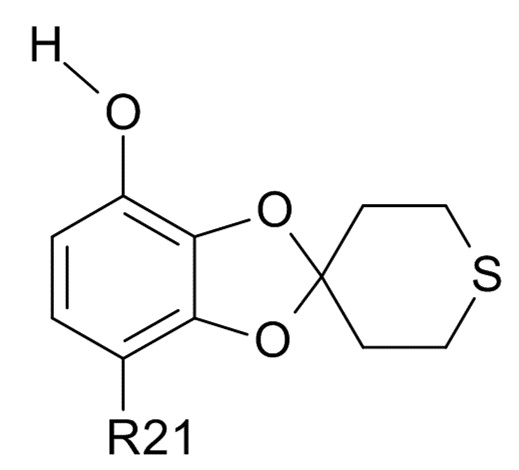
где R21 определено выше, удалением защиты фенольной группы.
Это можно осуществить путем проведения реакции соединения формулы (IV) с ароматическим или алифатическим тиолом в комбинации с основанием.
Ароматический тиол может представлять собой, например, но не ограничиваясь этим, бензолтиол, 4-метилбензолтиол, 3,5-диметилбензолтиол, 2,5-диметилбензолтиол, 4-изопропилбензолтиол или 5-трет-бутил-2-метил-бензолтиол. В одном варианте реализации изобретения, ароматический тиол представляет собой 5-трет-бутил-2-метил-бензолтиол.
Алифатический тиол может представлять собой, например, но не ограничиваясь этим, 1-додекантиол, 1-тетрадекантиол, 1-гексадекантиол или трет-додекантиол. В одном варианте реализации изобретения, алифатический тиол представляет собой 1-додекантиол.
Удаление защиты фенольной группы на стадии (2a) можно произвести с использованием различных растворителей, например, выбранных из ДМФА (N,N-диметилформамид), NMP (N-метилпирролидон), ДМСО (диметилсульфоксид), метанола или этанола и их смесей. В одном варианте реализации изобретения, растворитель представляет собой ДМФА. В другом варианте реализации изобретения, растворитель представляет собой смесь ДМФА и метанола.
Удаление защиты фенольной группы производится в присутствии основания, например, выбранного из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, TEA (триэтиламина),трет-бутоксида калия, трет-BuOLi (трет-бутоксида лития), метоксида натрия, этоксида натрия и DIPEA (N,N-диизопропилэтиламина). В одном варианте реализации изобретения, основание представляет собой K2CO3. В другом варианте реализации изобретения, основание представляет собой метоксид натрия.
Обычно реакцию проводят при температуре из диапазона 50-120 °C, такой как из диапазона 70-100 °C. Обычно реакции дают протекать в течение 2-36 час, например, 3-24 час. Обычно реакции дают протекать до достижения конверсии ≥98%.
Полученное соединение формулы (VI) можно извлекать традиционными средствами, известными специалистам в данной области техники, например, водной обработкой с последующей экстракцией, дальнейшим осаждением и фильтрацией.
В одном варианте реализации изобретения, соединение формулы (VI) представляет собой, где R21 обозначает C(O)R22, а R22 выбрано из водорода и C1-6-алкила. В другом варианте реализации изобретения, соединение формулы (VI) представляет собой 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
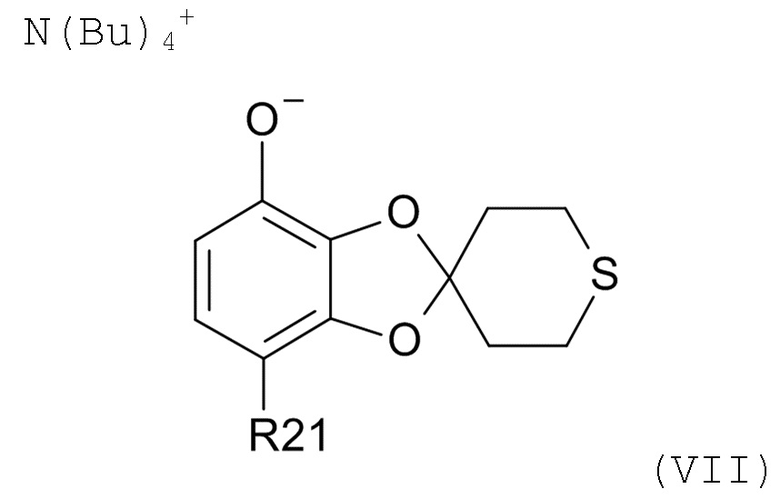
Стадия (2b)
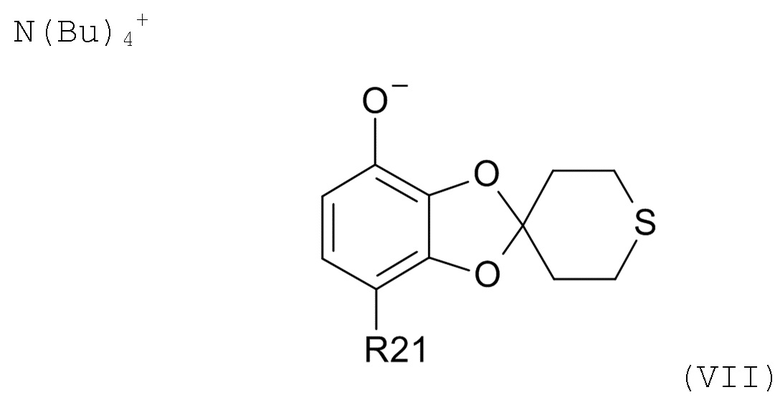
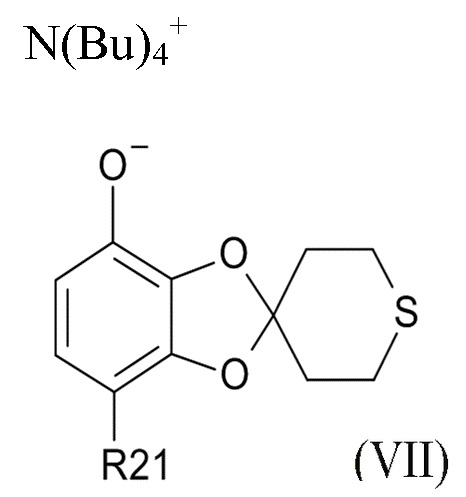
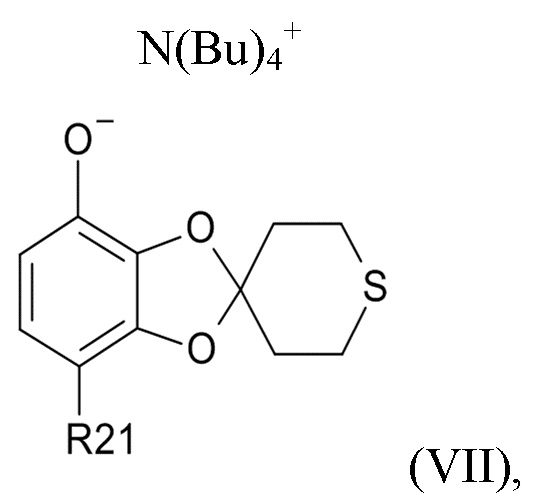
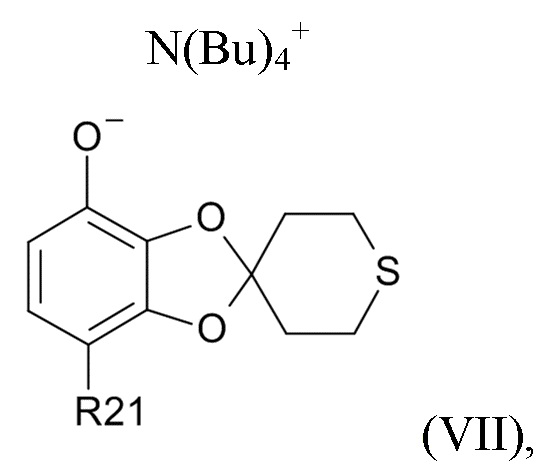
На стадии (2b) проводят реакцию соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения формулы (VII)
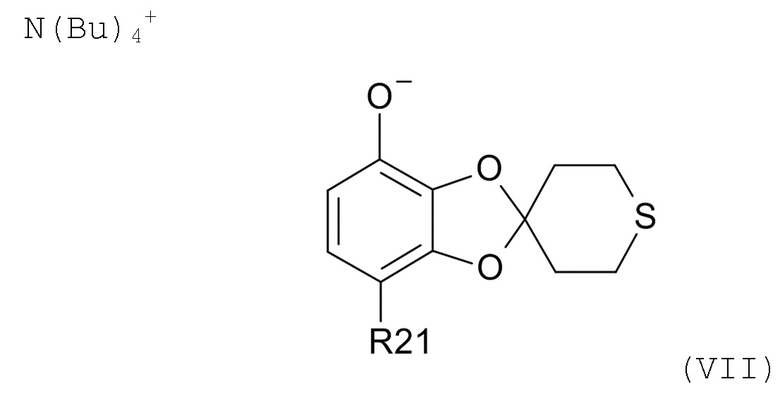
где R21соответствуют приведенным выше определениям.
Смесь обычно нагревают до температуры из диапазона 20-80°C, такой как 55-65°C, пока не растворятся все компоненты.
Получившийся раствор обычно промывают раствором хлорида натрия в воде путем перемешивания при температуре из диапазона 20-80°C, такой как 55-65°C, в течение > 20 мин. Последующее добавление смеси воды и хлорида натрия с последующим охлаждением смеси от ≥35°C до 0-20°C, например, до 5°C, в течение периода 1-24 час, такого как 1-4 час, обуславливает осаждение соли TBA (тетрабутиламмоний). Соль TBA выделяют, например, фильтрацией, и высушивают.
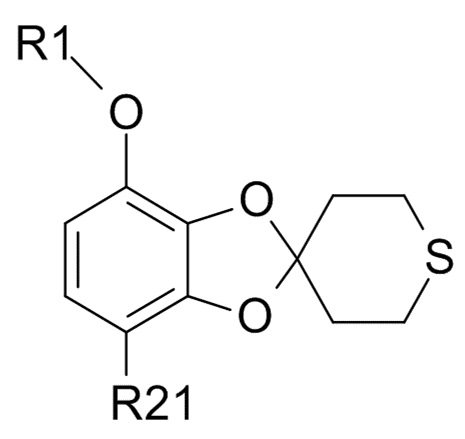
Стадия (3)
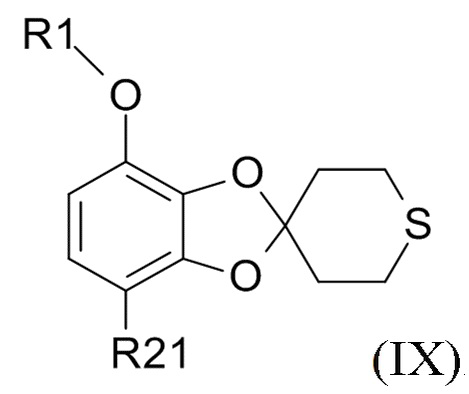
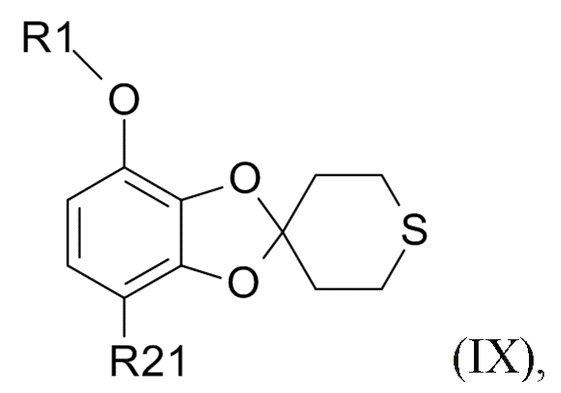
Соединение формулы (IX)
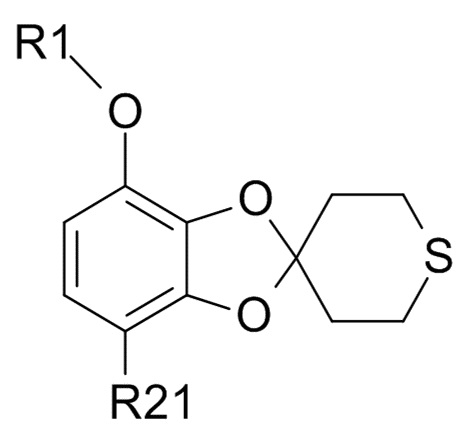 (IX)
(IX)
где R1 и R21 соответствуют приведенным выше определениям, может быть получено алкилированием полученного соединения формулы (VII)
где R21 соответствуют приведенным выше определениям, путем проведения реакции с гидрохлорфторуглеродным реагентом,
R1-Cl (VIII)
где R1 соответствует приведенным выше определениям.
Алкилирование может быть проведено с использованием одного из различных возможных реагентов, таких как различные гидрохлорфторуглеродные газы. В одном варианте реализации изобретения, реакцию алкилирования проводят с использованием хлордифторметана в апротонном полярном растворителе, например, выбранном из ДМФА (N,N-диметилформамид), NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил) и ТГФ (тетрагидрофуран) и их смесей. В одном предпочтительном варианте реализации изобретения, апротонный растворитель выбран из ДМФА и NMP. В конкретном варианте реализации изобретения, реакцию проводят с использованием хлордифторметана в ДМФА.
Обычно реакцию проводят при температуре из диапазона 40-120°C, такой как из диапазона 50-70°C. Обычно реакции дают протекать до тех пор, пока в реакционной смеси не останется ≤4% фенола.
Полученное соединение формулы (IX) можно извлекать традиционными средствами, известными специалистам в данной области техники, например, водной обработкой с последующим осаждением и фильтрацией.
В одном варианте реализации изобретения, соединение формулы (IX) представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Альтернативная стадия (2b+3)
Альтернативно, соединение формулы (IX),
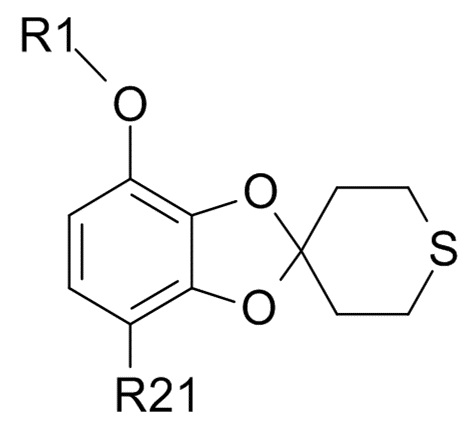 (IX)
(IX)
где R1 и R21 соответствуют приведенным выше определениям, может быть получено из соединения формулы (VI),
 (VI)
(VI)
где R21 определено выше, без формирования промежуточной соли формулы (VII), путем использования источника дифторкарбена в полярном растворителе в присутствии основания.
Источник дифторкарбена выбран из, например, но не ограничиваясь этим, хлордифторацетата натрия, диэтилбромдифторметилфосфоната, хлордифторметилфенилсульфона и 2-хлор-2,2-дифторацетофенона. Специалисты в данной области техники легко могут выбрать другие подходящие, аналогичные указанным, источники дифторкарбена. В одном варианте реализации изобретения, источником дифторкарбена является хлордифторацетат натрия. В другом варианте реализации изобретения, источником дифторкарбена является диэтил бромдифторметилфосфонат.
Реакцию проводят в растворителе, выбранном из, например, NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), EtOAc (этилацетат), MeCN (ацетонитрил), ТГФ (тетрагидрофуран), этанола, метанола, воды и их смесей. В одном варианте реализации изобретения, растворитель представляет собой смесь воды и ДМФА. В другом варианте реализации изобретения, растворитель представляет собой смесь воды и ацетонитрила.
Реакцию проводят в присутствии основания, выбранного из, например, K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, TEA (триэтиламин), трет-BuOLi (трет-бутоксид лития), метоксида натрия, этоксида натрия, DIPEA (N,N-диизопропилэтиламин), KOH, NaOH, LiOH. В одном варианте реализации изобретения, основание представляет собой K2CO3. В другом варианте реализации изобретения, основание представляет собой NaOH.
Обычно реакцию проводят при температуре из диапазона 0-120°C, такой как 6-115°C. В одном варианте реализации изобретения, реакцию проводят при 6-20°C с использованием диэтилбромдифторметилфосфоната в качестве источника дифторкарбена. В другом варианте реализации изобретения, реакцию проводят при температуре от температуры окружающей среды до 111°C, с использованием хлордифторацетата натрия в качестве источника дифторкарбена.
В одном варианте реализации изобретения, соединение формулы (IX) представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Полученное соединение формулы (IX), где R1 и R21 соответствуют приведенным выше определениям, можно извлекать традиционными средствами, известными специалистам в данной области техники, например, водной обработкой с последующим осаждением и фильтрацией.
Стадия (4)
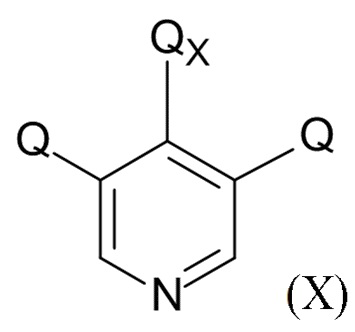
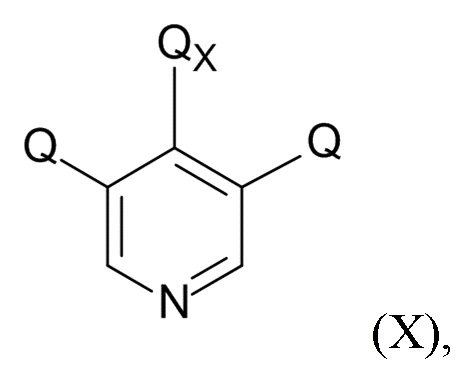
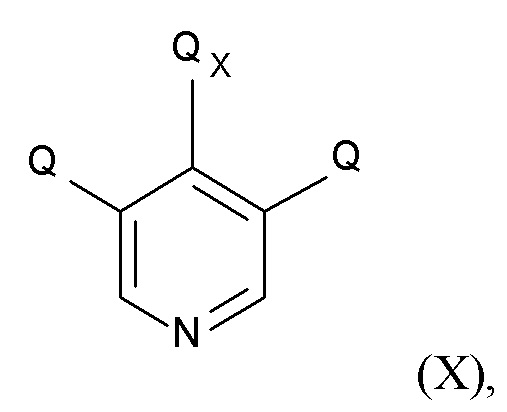
На стадии (4), проводят реакцию соединения формулы (IX) с пиридиновым соединением формулы (X)
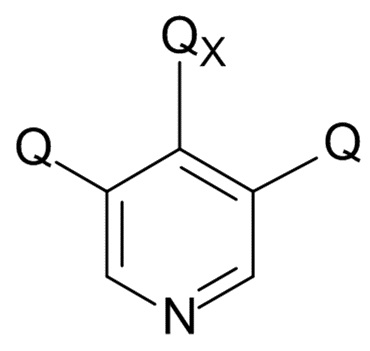 (X)
(X)
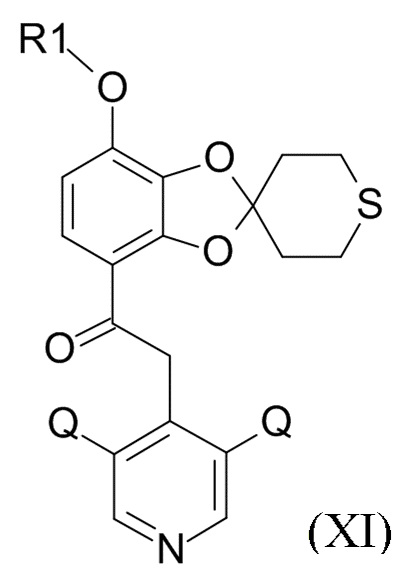
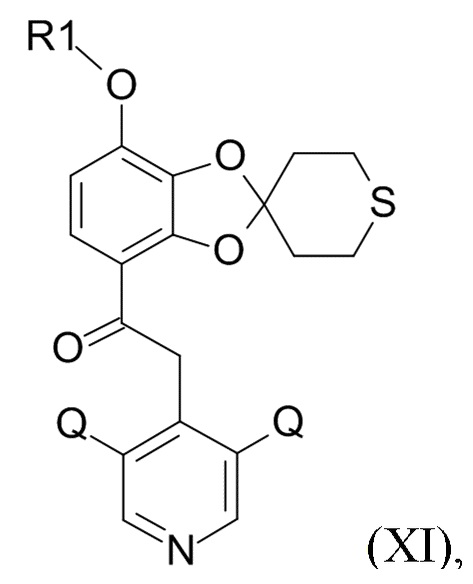
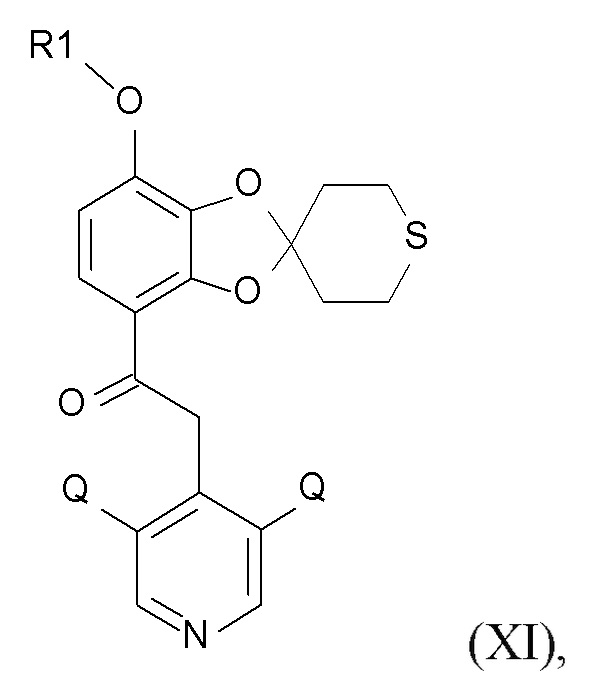
где Q соответствует приведенному выше определению и QX выбрано из хлора, брома, фтора и иода, для образования соединения формулы (XI)
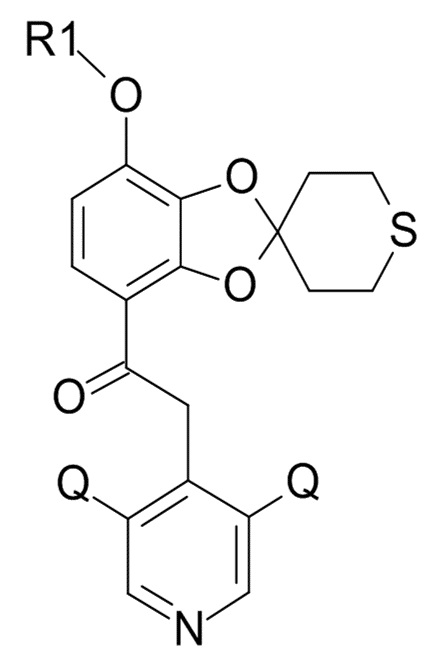 (XI)
(XI)
где R1 и Q соответствуют приведенным выше определениям.
Реакцию сочетания пиридина на стадии (4) обычно проводят в полярном растворителе, например, выбранном из ДМФА (N,N-диметилформамид), NMP (N-метилпирролидон), DMI (1,3-диметил-2-имидазолидинон), ДМСО (диметилсульфоксид), MeCN (ацетонитрил) и ТГФ (тетрагидрофуран) и их смесей, в присутствии основания, например, выбранного из трет-BuOK (трет-бутоксид калия), трет-BuOLi (трет-бутоксид лития), трет-BuONa (трет-бутоксид натрия), метоксида натрия или калия, этоксида натрия или калия, K2CO3, Na2CO3, KHCO3, NaHCO3, Et3N (триэтиламин) и DIPEA (N,N-диизопропилэтиламин). В одном варианте реализации изобретения, растворитель представляет собой ДМФА и основание представляет собой трет-BuOK.
Обычно используется два или более эквивалента основания относительно соединения формулы (IX), так, чтобы молярное отношение (основание)/(формула IX) составляло от 5:1 до 2:1, например, от 3:1 до 2:1, особенно от 2,4:1 до 2,7:1.
Реакцию на этапе (4) обычно проводят при температуре 0-40°C, такой как 5-25°C.
В одном варианте реализации изобретения, соединение формулы (X) представляет собой 3,4,5-трихлорпиридин.
В одном варианте реализации изобретения, соединение формулы (XI) представляет собой 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
В ходе реакции, образуется в значительных количествах примесь формулы (XII).
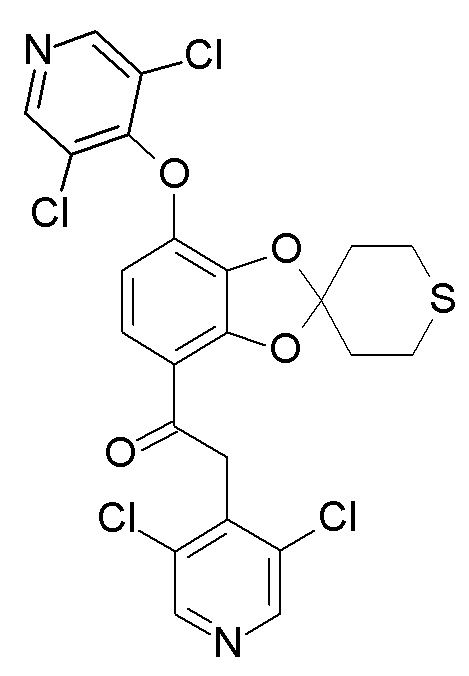 (XII)
(XII)
Очистку продукта от этой примеси осуществляют путем кристаллизации продукта из растворителя, выбранного из, например, диметилформамида (ДМФА), этанола, метанола, этилацетата, гексана, гептана и их смесей. В одном варианте реализации изобретения, растворитель представляет собой смесь этилацетата и этанола.
Стадия (5)
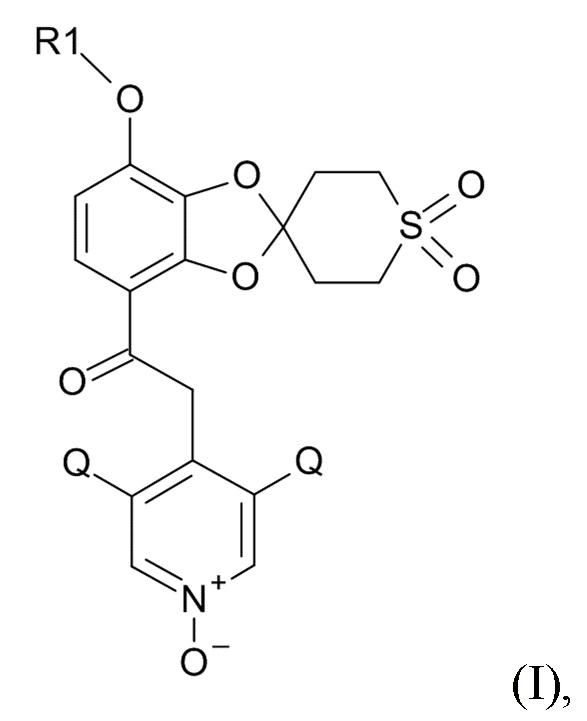
Окисление полученного соединения формулы (XI) проводят для образования соединения формулы (I)
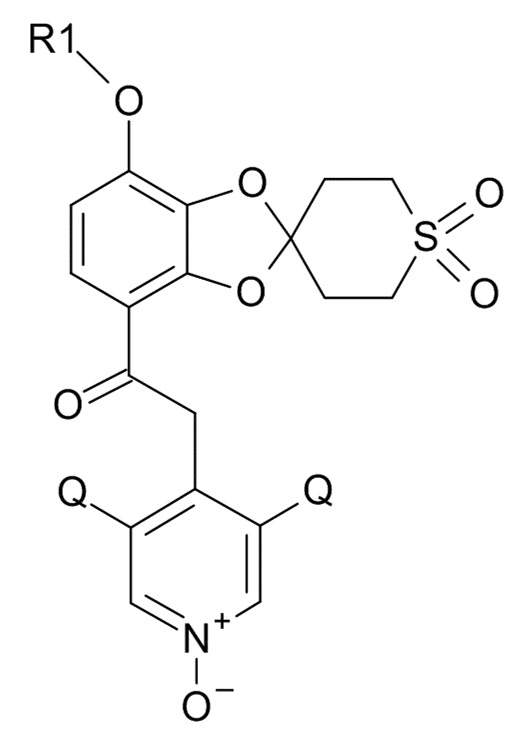 (I)
(I)
где R1 и Q соответствуют приведенным выше определениям, путем проведения реакции указанного соединения формулы (XI) с окислительным реагентом.
Обычно окислительный реагент выбирают из PAA (надуксусная кислота) в AcOH (уксусная кислота) и H2O2 (водн.) в муравьиной кислоте или уксусной кислоте. В одном предпочтительном варианте реализации изобретения, окислительным реагентом является PAA в AcOH. В одном варианте реализации изобретения, количество использованной PAA относительно (XI) (молярное отношение) составляет обычно от 3 до 6, например, от 3,8 до 4,2. Обычно окислительный реагент медленно добавляют в течение периода 1-8 час, такого как 3-5 час, поддерживая температуру в диапазоне 15-100°C, такую как в диапазоне 15-50°C, особенно в диапазоне 15-40°C.
Обычно реакцию проводят при температуре в диапазоне 30-70 °C, таком как 40-60°C, особенно 48-52°C, и перемешивают в течение 3-48 час, например, 16-24 час.
Очистка соединения формулы (I)
Полученный неочищенный продукт формулы (I) можно предпочтительно очищать кристаллизацией, осаждением, хроматографически или т. п.
В одном варианте реализации изобретения, полученный неочищенный продукт формулы (I) кристаллизуют из смеси воды и EtOH (этанол), и выделяют фильтрацией с последующей сушкой.
Промежуточные соединения
В другом аспекте, настоящее изобретение относится к промежуточным соединениям, которые используются при получении соединения формулы (I), где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора.
В одном варианте реализации, изобретение относится к промежуточным соединениям формулы (VI)
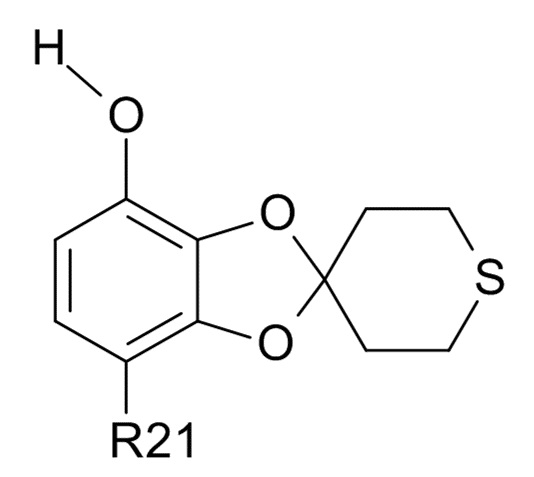 (VI)
(VI)
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила. В другом варианте реализации изобретения, промежуточное соединение формулы (VI) представляет собой 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
В другом варианте реализации, изобретение относится к промежуточному соединению формулы (VII)
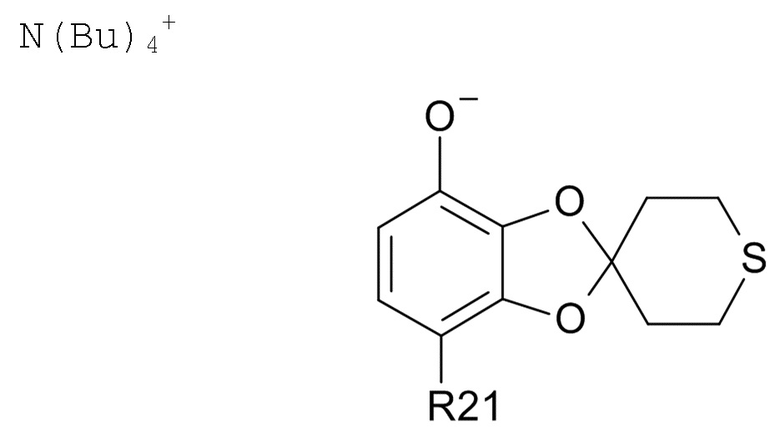
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила. В другом варианте реализации изобретения, промежуточное соединение формулы (VII) представляет собой тетрабутиламмоний 7-ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат.
В другом варианте реализации, изобретение относится к промежуточному соединению формулы (IX)
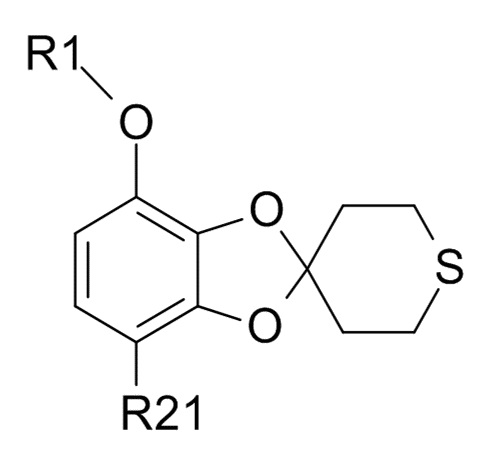 (IX)
(IX)
где R1 выбрано из CHF2 и CF3, а R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила.
В другом варианте реализации изобретения, R1 представляет собой CHF2, а R21 представляет собой C(O)R22 и R22 выбрано из водорода и C1-6-алкила. В другом варианте реализации изобретения, промежуточное соединение формулы (IX) представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методики и реагенты
Все использованные химические вещества и реагенты имеются в продаже.
Спектры 1H ядерного магнитного резонанса (ЯМР) были сняты при заданном магнитном поле и значения химического сдвига (δ, в м.д..) приведены в специфическом растворителе относительно тетраметилсилана (δ=0,00).
ВЭЖХ: Колонка: Aeris Peptide 3.6 мкм XB-C18, 100×4,6 мм, элюент был градиентом A: 10% MeCN; 90% H2O; 0,1% ТФК и B: 90% MeCN; 10% H2O; 0,1% ТФК, температура колонки: 35°C, УФ-детектирование при 220 нм, скорость потока: 1,5 мл/мин. Использовались следующие градиенты элюентов:
Шаги градиента 2a, 2b,3, и 5
Шаг градиента 4
ПРИМЕР 1
Стадия (1): Приготовление 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
В реактор загрузили 1-(2,3-дигидрокси-4-метокси-фенил)этанон (60,0 кг, 329 моль), тетрагидротиопиран-4-он (37,2 кг, 320 моль), монтмориллонит K 10 (30,0 кг) и толуол (720,0 л). Смесь перемешивали при нагревании до появления конденсата, применяя температуру в рубашке 140-150°C в течение 84 час. Смесь охладили до 86-90°C и профильтровали через слой фильтрующего вещества. Реактор прополоснули горячим (86-90°C) толуолом (120 л), и затем горячий толуол использовали для промывки слоя фильтрующего вещества. Прополаскивание реактора и последующую промывку слоя фильтрующего вещества горячим толуолом (120 л) повторили дважды, и один раз промыли горячим (70°C) этилацетатом (60 л). Все фильтраты толуола и этилацетата объединили и охладили до 2-6°C в течение приблизительно 6 час. Смесь перемешивали при 2-6°C в течение приблизительно получаса.
Непрореагировавший исходный материал собрали фильтрацией и высушили под вакуумом при 43-47°C. Выход составил 32,0 кг.
Фильтрат от выделения непрореагировавшего исходного материала охладили до 10-16°C при перемешивании, и добавили смесь гидроксида натрия (26,40 кг) и воды (162,0 л) при 10-16°C. Затем реакционную смесь перемешивали приблизительно в течение получаса при 10-16°C, после этого перемешивание прекратили и дали возможность фазам осаждаться. Более низкую водную фазу удалили, затем добавили смесь гидроксида натрия (26,40 кг) и воды (162,0 л) при перемешивании при 10-16°C. Смесь перемешивали приблизительно в течение одного часа, после этого перемешивание прекратили и дали возможность фазам осаждаться. Более низкую водную фазу удалили и органическую фазу перенесли в контейнер. Реактор прополоскали толуолом и затем перенесли органическую фазу обратно в реактор через картриджный фильтр.
Произвели максимально возможное концентрирование раствора под вакуумом, поддерживая температурк ≤70°C. Добавили этанол (90,0 л), нагрели смесь до 47-53°C, и перемешивали при этой температуре в течение 10-15 минут. Затем произвели максимально возможное концентрирование смеси под вакуумом при температуре ≤55°C. Добавили в реактор этанол (120,0 л), смесь нагрели до появления конденсата при перемешивании и добавили воду (90,0 л) при подогреве, поддерживая смесь при конденсации. Смесь охладили до 2-8°C приблизительно за 10 час, и перемешивали при этой температуре в течение приблизительно получаса.
Продукт выделили фильтрацией, промыли смесью этанола (30,0 л) и воды (22,8 л) и высушили под вакуумом при 43-47°C. Выход составил 21,80 кг (24%, но 51%, если сделать поправку на извлеченный исходный материал). 1H NMR (600 MHz, ДМСО-d6) δ 7.30 (d, J=9.0 Hz, 1H), 6.75 (d, J=9.0 Hz, 1H), 3.88 (s, 3H), 2.91-2.84 (m, 2H), 2.84-2.77 (m, 2H), 2.49 (s, 3H), 2.30-2.22 (m, 2H), 2.22-2.12 (m, 2H).
При необходимости, стадию (1) повторяли для получения нужного количества 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона.
Стадия (2a) Приготовление 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона
В реактор загрузили 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (26,0 кг, 92,7 моль), карбонат калия (14 кг, 101 моль), диметилформамид (104 л) и 5-трет-бутил-2-метил-бензолтиол (26,8 кг, 149 моль). Смесь нагревали при перемешивании до 85-92°C, пока не была достигнута конверсия ≥98% по результатам ВЭЖХ. Затем смесь охладили до 25°C, добавили воду (104 л) и гидроксид натрия (28% в воде, 21,4 кг), и перемешивали в течение ≥10 мин. В случаях, когда pH смеси был ниже 12, добавляли больше гидроксида натрия (28% в воде). Затем добавили толуол (65 л), и продолжали перемешивание в течение ≥15 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Фазы разделили и удалили органическую фазу. Две нижних водных фазы смешали с толуолом (65 л) и перемешивали смесь в течение ≥15 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Фазы разделили и удалили органическую фазу. Две водные фазы вернули в реактор и добавили соляную кислоту (18% в воде, 67,6 кг), добавляли медленно, при перемешивании, чтобы контролировать выделение газа. Полученную смесь перемешивали в течение ≥10 мин. Для достижения pH ≤6 добавили больше соляной кислоты (18% в воде, 10,2 кг).
Температуру смеси довели до 35-45°C и поддерживали на этом уровне в течение последующих экстракций. Добавили этилацетат (156 л) и перемешивали смесь в течение ≥30 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Произвели разделение фаз. Водную фазу перемешивали с этилацетатом (78 л) в течение ≥30 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Водные фазы удалили. Две этилацетатные фазы объединили объединили в реакторе и перемешивали с водой (78 л) в течение ≥15 мин. Перемешивание прекратили и дали возможность фазам разделиться. Водную фазу удалили.
Произвели максимально возможное концентрирование органических фаз при температуре рубашки 50-60°C и с применением вакуума. Затем добавили гептан (39 л) и полученную смесь охладили до ≤5°C со скоростью ≤10°C/час, и выдерживали при этой температуре в течение ≥3 час. Титульное соединение выделили фильтрацией, промыли холодной (≤5°C) смесью этилацетата (10 л) и гептана (10 л), и высушили под вакуумом при 40-50°C. Выход составил 19,75 кг (80%). 1H NMR (600 MHz, ДМСО-d6) δ 10.51 (s, 1H), 7.18 (d, J=9.0 Hz, 1H), 6.50 (d, J=9.0 Hz, 1H), 2.93-2.85 (m, 2H), 2.84-2.78 (m, 2H), 2.46 (s, 3H), 2.31-2.23 (m, 2H), 2.20-2.11 (m, 2H).
Стадия (2b): Тетрабутиламмоний 7-ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат
В подходящий реактор загрузили 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (19,75 кг, 74,16 моль), затем добавили гидроксид тетрабутиламмония (40% раствор в воде, 53,0 кг, 81,7 моль). Температуру рубашки установили на уровне 60°C, и перемешивали смесь до полного растворения. Добавили насыщенный раствор хлорида натрия в воде (59,2 кг) и продолжали перемешивание в течение ≥20 мин при температуре рубашки 60°C. Перемешивание прекратили, создав возможность резделения фаз. Нижнюю водную фазу удалили. Смесь в реакторе опять перемешали при температуре рубашки 60°C. Добавили насыщенный раствор хлорида натрия в воде (29,6 кг), а затем воду (25 л). Смесь перемешивали в течение ≥15 мин при температуре смеси ≥35°C. Смесь охладили до 0-5°C со скоростью приблизительно 20°C/час, в смесь ввели затравку при 40°C и еще раз при 35°C. Смесь перемешивали при 0-5°C в течение ≥2 час, а затем титульное соединение выделили фильтрацией и высушили под вакуумом при 40-50°C. Выход составил 32,9 кг (87%). 1H NMR (600 MHz, ДМСО-d6) δ 6.94 (d, J=9.1 Hz, 1H), 5.74 (d, J=9.1 Hz, 1H), 3.23-3.07 (m, 8H), 2.87-2.72 (m, 4H), 2.25 (s, 3H), 2.16-2.07 (m, 2H), 2.06-1.96 (m, 2H), 1.62-1.51 (m, 8H), 1.30 (h, J=7.4 Hz, 8H), 0.93 (t, J=7.4 Hz, 12H).
Стадия (3): 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
В реактор поместили тетрабутиламмоний 7-ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат (32,93 кг, 64,85 моль) и диметилформамид (198 л). Смесь перемешивали до полного растворения. Через погружную трубу реактора добавили к раствору хлордифторметан (39,5 кг, 457 моль). Реакционную смесь нагрели до 50-55°C и перемешивали до тех пор, пока, по результатам ВЭЖХ, не оставалось ≤4% исходного материала. Реакционную смесь охладили до 20-25°C и перенесли в контейнер через фильтр. Реактор и твердую фазу на фильтре промыли диметилформамидом (10 л), который также добавили в контейнер.
Воду (198 л) и гидроксид натрия (28% в воде, 11.0 кг) загрузили в реактор и нагрели до 45-55°C. Реакционную смесь в контейнере медленно добавили в реактор при перемешивании, поддерживая температуру на уровне 45-55°C. Затем смесь охладили до 5-10°C и перемешивали при этой температуре в течение ≥2 час. Продукт выделили фильтрацией, промыли водой (82 л) и высушили под вакуумом при 45-55°C в токе азота. Выход составил 19,08 кг (94%). 1H NMR (600 MHz, ДМСО-d6) δ 7.34 (t, J=73.1 Hz, 1H), 7.32 (d, J=9.1 Hz, 1H), 6.86 (d, J=9.1 Hz, 1H), 2.92-2.80 (m, 4H), 2.54 (s, 3H), 2.34-2.27 (m, 2H), 2.27-2.19 (m, 2H).
Стадия (4) 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
В подходящий реактор загрузили диметилформамид (96 л) с последующим добавлением трет-бутоксида калия (17,60 кг, 156,8 моль). Перенос трет-бутоксида калия обеспечили промывкой диметилформамидом (3 л), и перемешивали смесь до растворения трет-бутоксида калия. Раствор перенесли из реактора в контейнер, реактор промыли диметилформамидом (6 л), который также был перенесен в контейнер.
В реактор загрузили 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (19,08 кг, 60,32 моль), 3,4,5-трихлорпиридин (14,30 кг, 78,38 моль) и диметилформамид (96 л). Смесь перемешали и охладили до 10-15°C, затем медленно добавили раствор трет-бутоксида калия в диметилформамиде, поддерживая температуру реакционной смеси на уровне 5-25°C. Перенос раствора трет-бутоксида калия обеспечили промывкой диметилформамидом (6 л). Смесь нагрели до 20-25°C и перемешивали до тех пор, пока степень конверсии, по данным ВЭЖХ, не составила ≥98%. Содержание примеси формулы (XII) в реакционной смеси: 12%
К реакционной смеси медленно добавили воду (96 л), поддерживая температуру в диапазоне между 20-30°C. За этим последовало добавление хлорида натрия в воде (115,2 кг) и этилацетата (134 л). Смесь перемешивали в течение 20-60 мин, затем перемешивание прекратили и дали возможность фазам осаждаться. Произвели разделение фаз, и водную фазувернули в реактор. Добавили этилацетат (96 л), и перемешивали смесь в течение 20-60 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Произвели разделение фаз. Органические фазы объединили в реакторе и перемешивали с водой (48 л) и насыщенным хлоридом натрия в воде (57,8 кг) в течение ≥20 мин. Перемешивание прекратили и дали возможность фазам осаждаться. Нижнюю водную фазу удалили и добавили воду (48 л) и насыщенный хлорид натрия (57,6 кг). Смесь перемешивали в течение 20-60 мин, затем перемешивание прекратили и дали возможность фазам осаждаться. Нижнюю водную фазу удалили и добавили воду (84 л) и гидроксид натрия (28% в воде, 14,0 кг). Смесь перемешивали в течение 20-60 мин, затем перемешивание прекратили и дали возможность фазам осаждаться. Нижнюю водную фазу удалили.
Произвели концентрирование органической фазы в реакторе с использованием вакуума и нагрева при температуре рубашки 50-65°C до остаточного объема приблизительно 40 л. В реактор загрузили этанол (57 л) и нагревали смесь до появления конденсата, пока не был получен прозрачный раствор. Смесь охладили до 5°C приблизительно за ≥5 час, и перемешивали при этой температуре в течение приблизительно ≥3 hours. Продукт выделили фильтрацией, перенос обеспечили промывкой этанолом (10 л). Продукт промыли холодным (≤5°) этанолом (48 л) и высушили под вакуумом при 45-55°C. Выход составил 15,57 кг (56%). 1H NMR (600 MHz, Chloroform-d) δ 8.52 (s, 2H), 7.46 (d, J=8.9 Hz, 1H), 6.80 (d, J=8.9 Hz, 1H), 6.73 (t, J=73.3 Hz, 1H), 4.59 (s, 2H), 3.01-2.85 (m, 4H), 2.47-2.30 (m, 4H). ВЭЖХ: Чистота: 97,8%, содержание примеси формулы (XII): 1,0%.
Стадия (5): 2-(3,5-дихлор-1-оксидо-пиридин-1-ий-4-ил)-1-[7-(дифторметокси)-1',1'-диоксо-спиро[1,3-бензодиоксол-2,4'-тиан]-4-ил]этанон
В реактор загрузили 2-(3,5-дихлор-4-пиридил)-1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон (15,6 кг, 33,7 моль) и ледяную уксусную кислоту (78,0 кг), и охладили смесь до 13-20°C. Медленно добавили надуксусную кислоту (36-40% в уксусной кислоте, 6,52 кг, 32,6 моль), поддерживая температуру ниже 40°C. Смесь нагрели до 40-50°C и перемешивали в течение 10-25 мин. Смесь охладили до 13-20°C и медленно добавили вторую порцию надуксусной кислоты (36-40% в уксусной кислоте, 6,51 кг, 32,5 моль), поддерживая температуру ниже 40°C. Смесь нагрели до 40-50°C и перемешивали в течение 10-25 мин. Смесь охладили до 20-30°C и медленно добавили третью порцию надуксусной кислоты (36-40% в уксусной кислоте, 14,3 кг, 71,5 моль). Смесь нагрели до 48-55°C и перемешивали до тех пор, пока конверсия не составила ≥98,5%. Смесь охладили до 20-25°C и медленно добавили смесь метабисульфита натрия (7,21 кг, 37,9 моль) и воду (46 л)поддерживая температуру ниже 35°C.
Добавили 2-пропанол (78 л), нагрели смесь до 60-65°C и отфильтровали в горячем состоянии. Реактор очистили, и отфильтрованную реакционную смесь вернули в реактор. Смесь нагрели до 60-65°C и медленно добавили воду (234 л), поддерживая температуру выше 55°C. Смесь перемешивали в течение 30-60 мин при 60-65°C, медленно охладили до 5°C за 12 час и перемешивали при 0-10°C в течение ≥2 час. Неочищенный продукт выделили фильтрацией, промыли водой (27 л), и высушивали под вакуумом в течение приблизительно двух часов.
Твердую фазу вернули в реактор и нагревали с этанолом (390 л) до появления конденсата. Затем смесь охладили до 68-72°C и ввели затравку. Смесь охладили до 5°C за 13 час, и перемешивали при 0-10°C в течение ≥2 час. Продукт выделили фильтрацией, промыли холодной (0-10°C) смесью воды (4 л) и этанола (39 л) и высушили под вакуумом при 45-55°C в токе азота. Выход составил 14,6 кг (85%). 1H NMR (600 MHz, хлороформ-d) δ 8.23 (s, 2H), 7.52 (d, J=9.1 Hz, 1H), 6.90 (d, J=9.1 Hz, 1H), 6.71 (t, J=72.3 Hz, 1H), 4.49 (s, 2H), 3.47- 3.38 (m, 2H), 3.33-3.24 (m, 2H), 2.83-2.75 (m, 2H), 2.75-2.68 (m 2H). ВЭЖХ: чистота 98,6%.
ПРИМЕР 2
1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон
Метоксид натрия в метаноле (30%, 64.2 мл, 0,34 моль) добавили к раствору 1-(7-метоксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанона (50,0 г, 0,178 моль) в диметилформамиде (250 мл) при 25-30°C. Затем добавили 1-додекан-тиол (64,88 мл, 0,271 моль) при 25-30°C и смесь нагревали до 95-100°C в течение трех часов. Реакционную смесь охладили до 25-30°C и добавили гидроксид натрия (28% в воде, 50 мл) и воду (250 мл). Полученную смесь перемешивали в течение получаса, а затем смесь трижды экстрагировали толуолом (250 мл). Водный раствор подкислили соляной кислотой (6M) до приблизительно pH 6 и экстрагировали этилацетатом (250 мл) четыре раза. Экстракты этилацетата объединили, четыре раза промыли рассолом (250 мл) и сконцентрировали до приблизительно 50 мл с использованием ротационного испарителя. Добавили гептан (300 мл) перемешивали смесь в течение одного часа при температуре окружающей среды. Продукт выделили фильтрацией, промыли гептаном (100 мл) и высушили. Выход составил 44,3 г (93%). Результаты ЯМР соответствовали данным ЯМР продукта, полученного на стадии (2a) в Примере 1.
ПРИМЕР 3
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Смесь 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (221,6 г, 0,8322 моль), карбоната калия (161,3 г, 1,167 моль), хлордифторацетата натрия (292,0 г, 1,915 моль), диметилформамида (1,50 л) и воды (500 мл) перемешали в 5-литровой реакционной колбе и медленно нагрели до 106-111°C, при этом, начиная приблизительно от 78°C, наблюдалось выделение газа. Реакционную смесь перемешивали при 106-111°C до прекращения выделения газа, приблизительно в течение двух часов. Смесь охладили на ледяной бане и медленно добавили воду (1,00 л) при 30-32°C. Полученную суспензию далее охладили до 6°C при перемешивании. Неочищенный продукт выделили фильтрацией и промыли водой.
Влажный неочищенный продукт перемешивали с этилацетатом (1,66 л) и гидроксидом натрия (1 M, 560 мл) в течение приблизительно 20 мин, а затем разделили фазы в делительной воронке. Нижнюю водную фазу удалили, а органическую фазу дважды промыли водой (два раза по 560 мл). Органическую фазу сконцентрировали с использованием ротационного испарителя (под вакуумом при 60°C на водяной бане) до приблизительно 450 мл. Добавили этилацетат (1,56 л) и опять произвели концентрирование смеси с использованием ротационного испарителя, как и ранее, до приблизительно 450 мл. Добавили этилацетат (1,44 л), и непрозрачный раствор отфильтровали, перенесли и промыли свежей порцией этилацетата (100 мл). Объединенный фильтраты профильтровали через пробку из активированного угля (6,0 г), перенесли и промыли этилацетатом (200 мл). Произвели концентрирование объединенных фильтратов с использованием ротационного испарителя до приблизительно 450 мл. Полученный горячий раствор (приблизительно 60°C) перемешивали при температуре окружающей среды, в то же время медленно добавляя гептан (2,00 л) приблизительно в течение получаса. Суспензию перемешивали при температуре окружающей среды в течение 14 час.
Смесь перемешивали на ледяной бане в течение приблизительно 2,5 час, в это время температура смеси составляла 4°C. Продукт выделили фильтрацией, промыли охлажденной до температуры льда смесью гептана и этилацетата (10:1, 200мл) и высушили под вакуумом при 50°C в токе азота. Выход составил 201 г (76%). Результаты ЯМР соответствовали данным ЯМР продукта, полученного на стадии 3 в Примере 1.
ПРИМЕР 4
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Гидроксид натрия (6,16 g, 154 ммоль) растворили в воде (40 мл), и раствор перемешивали с охлаждением на водяной бане. Добавили 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (2,00 г, 7,51 ммоль) и ацетонитрил (20 мл) и продолжили перемешивание при охлаждении. Добавили одной порцией диэтил-бромдифторметилфосфонат (2,67 мл, 15,0 ммоль) при 6°C и продолжали перемешивание при охлаждении приблизительно 20 мин. Убрали охлаждающую баню и перемешивали смесь в течение приблизительно 21 час при температуре окружающей среды.
Произвели разделение фаз с использованием делительной воронки и экстрагировали водную фазу этилацетатом (20 мл). Объединенные органические фазы промыли водой (20 мл), а затем рассолом (20 мл). Произвели концентрирование органических фаз до высушивания с использованием ротационного испарителя, как в Примере 3. Добавили к остатку этилацетат (20 мл) и произвели концентрирование смеси до высушивания, опять с использованием ротационного испарителя.
Остаток растворили в этилацетате (30 мл) и отфильтровали, перенесли и промыли этилацетатом (20 мл). Произвели концентрирование объединенных фильтратов до высушивания, опять с использованием ротационного испарителя, как и ранее, что привело к получению титульного соединения в виде желтоватой твердой фазы. Выход составил 2,14 г (90%). Результаты ЯМР соответствовали данным ЯМР продукта, полученного на стадии 3 в Примере 1.
ПРИМЕР 5
1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон
Гидроксид натрия (301 г, 7,52 моль) перемешали с водой (2,0 л) и полученный раствор охладили на ледяной бане. Добавили 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон (100,1 г, 0,3757 моль) и ацетонитрил (1,0 л). Медленно добавили в реакционную смесь диэтилбромдифторфосфонат (150,5 г, 0,5637 моль) за приблизительно 40 мин при температуре 15-20°C. Перемешивание продолжали еще в течение приблизительно двух часов при 15-20°C. Произвели разделение фаз.
К органической фазе медленно добавили воду (920 мл) при перемешивании и полученную суспензию перемешивали при температуре окружающей среды в течение приблизительно 18 час. Продукт выделили фильтрацией, промыли смесью 1:1 ацетонитрила и воды (120 мл) и высушили под вакуумом при 50°C в токе воздуха. Выход составил 108 г (91%). Результаты ЯМР соответствовали данным ЯМР продукта, полученного на стадии 3 в Примере 1.
КЛАУЗУЛА
С точки зрения данного описания, авторы изобретения, в частности, предложили:
Пункт 0. Способ получения соединения формулы (I)
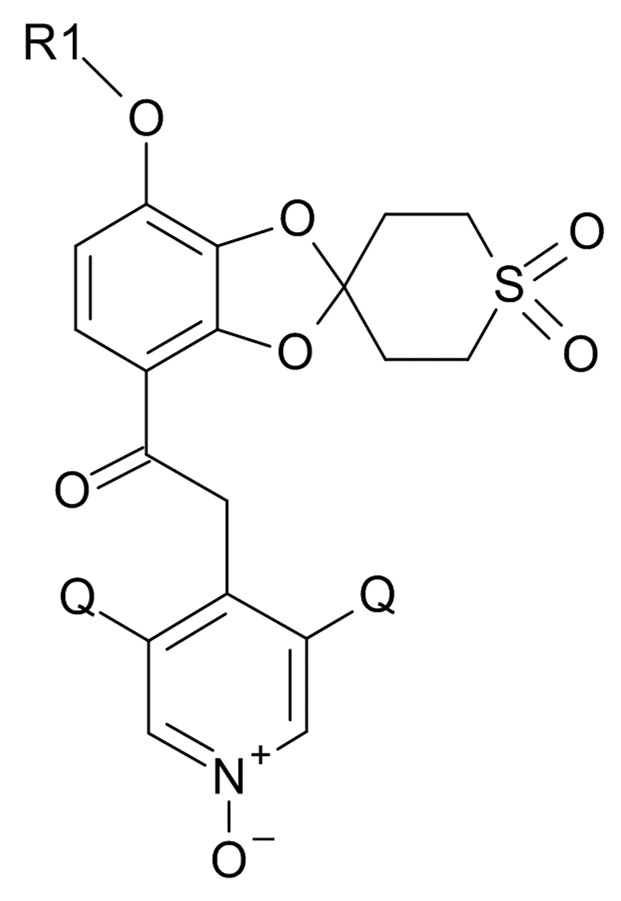 (I)
(I)
где R1 выбрано из CHF2 и CF3, Q выбрано из хлора, брома и фтора,
включающий одну или более из следующих стадий:
(1) проведение реакции соединения формулы (II)
 (II)
(II)
где R2 выбрано из водорода, C1-6-алкила и арилалкила, R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила; с соединением формулы (III)
 (III)
(III)
где ʺʺ обозначает одинарную связь, двойную связь или две одинарных связи и в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺʺ обозначает одинарную связь, и, в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь, и R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарных связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
 (IV)
(IV)
где R2 и R21 соответствуют приведенным выше определениям;
(2a) проведение реакции полученного соединения формулы (IV) с ароматическим или алифатическим тиолом, для образования соединения формулы (VI)
 (VI)
(VI)
где R21 соответствует приведенным выше определениям;
(2b) проведение реакции соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения соединения формулы (VII)
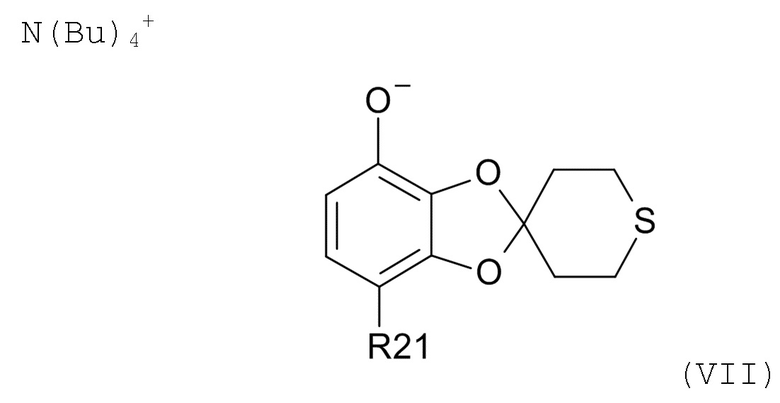
где R21 соответствуют приведенным выше определениям;
(3) алкилирование полученного соединения формулы (VII) гидрохлорфторуглеродным реагентом,
R1-Cl (VIII)
где R1 соответствуют приведенным выше определениям, до образования соединения формулы (IX)
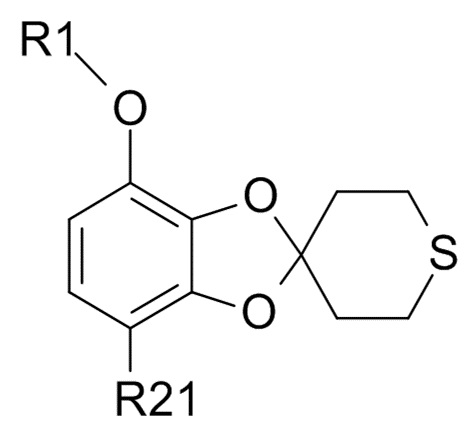 (IX)
(IX)
где R1 и R21 соответствуют приведенным выше определениям;
(4) проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X)
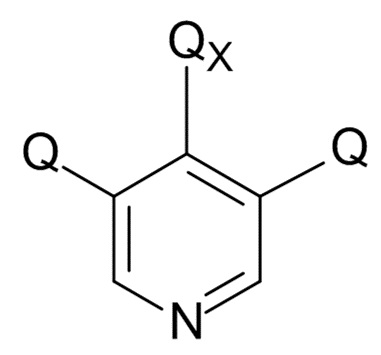 (X)
(X)
где Q соответствует приведенному выше определению и QX выбрано из хлора, брома, фтора и иода, для образования соединения формулы (XI)
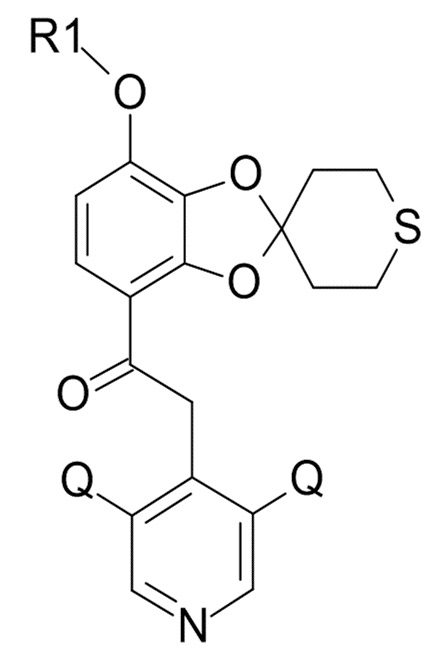 (XI)
(XI)
где R1 и Q соответствуют приведенным выше определениям; и
(5) окисление полученного соединения формулы (XI) для образования соединения формулы (I)
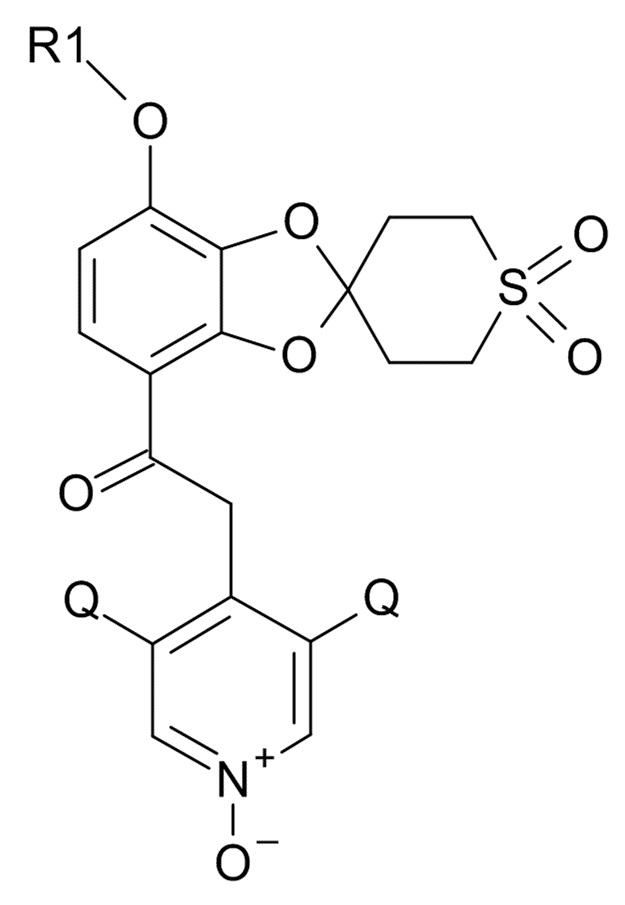 (I)
(I)
где R1 и Q соответствуют приведенным выше определениям.
Пункт 1. Способ получения соединения формулы (I)
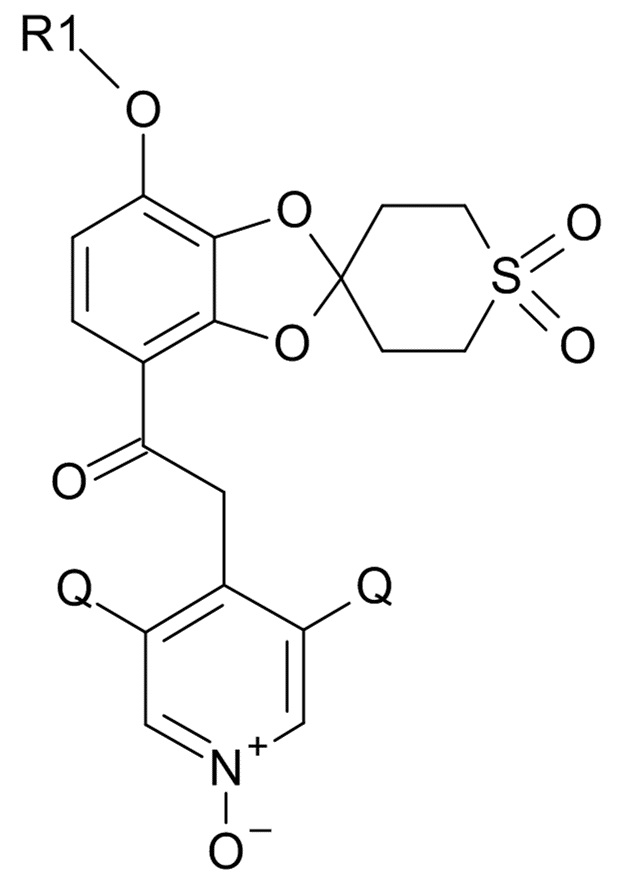 (I)
(I)
где R1 выбрано из CHF2 и CF3, Q выбрано из хлора, брома и фтора,
включающий каждую из следующих стадий:
(1) проведение реакции соединения формулы (II)
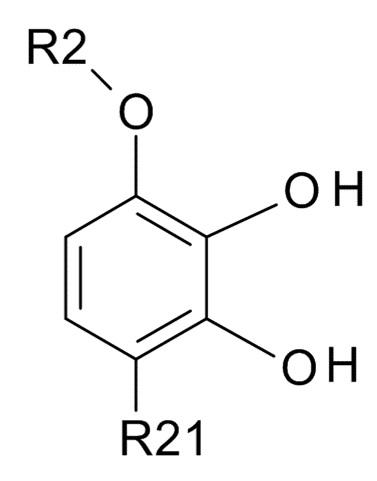 (II)
(II)
где R2 выбрано из водорода, C1-6-алкила и арилалкила, R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила; с соединением формулы (III)
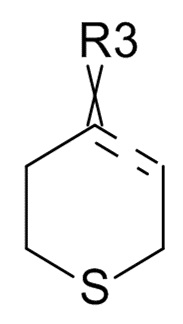 (III)
(III)
где ʺʺ обозначает одинарную связь, двойную связь или две одинарных связи и в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺʺ обозначает одинарную связь, и, в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь, и R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарных связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
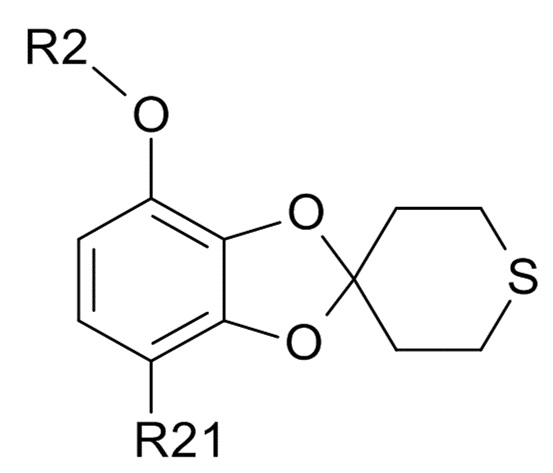 (IV)
(IV)
где R2 и R21 соответствуют приведенным выше определениям;
(2a) проведение реакции полученного соединения формулы (IV) с ароматическим или алифатическим тиолом, для образования соединения формулы (VI)
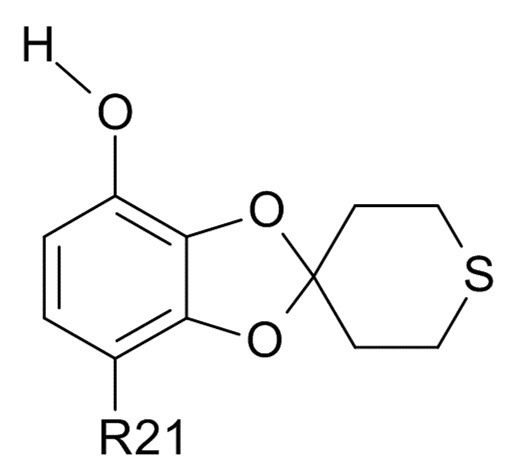 (VI)
(VI)
где R21 соответствует приведенным выше определениям;
(2b) проведение реакции соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения соединения формулы (VII) 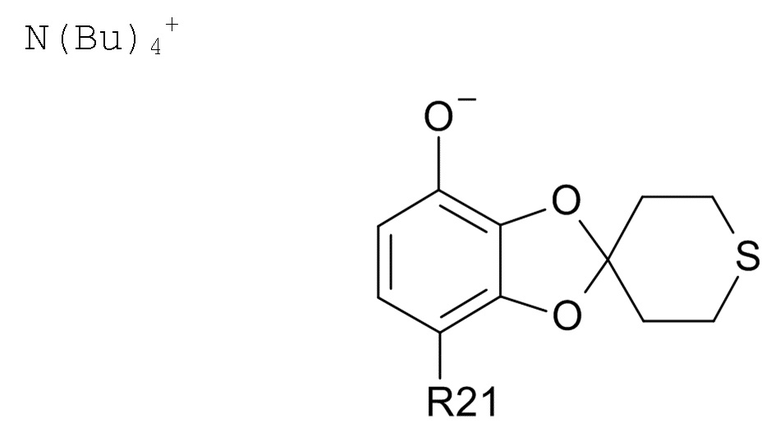 (VII)
(VII)
где R21 соответствуют приведенным выше определениям;
(3) алкилирование полученного соединения формулы (VII) гидрохлорфторуглеродным реагентом,
R1-Cl (VIII)
где R1 соответствуют приведенным выше определениям, до образования соединения формулы (IX)
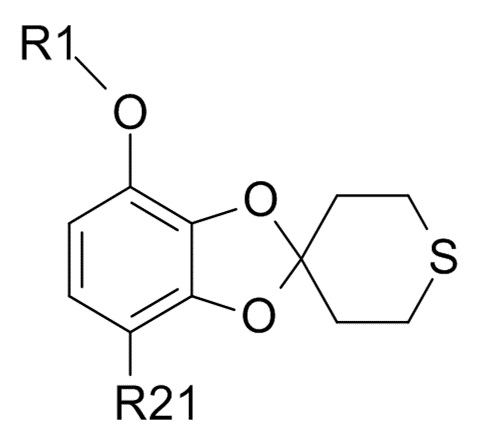 (IX)
(IX)
где R1 и R21 соответствуют приведенным выше определениям;
(4) проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X)
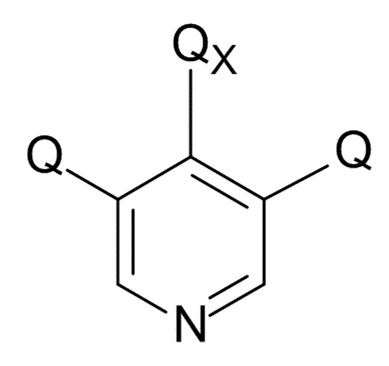 (X)
(X)
где Q соответствует приведенному выше определению и QX выбрано из хлора, брома, фтора и иода, для образования соединения формулы (XI)
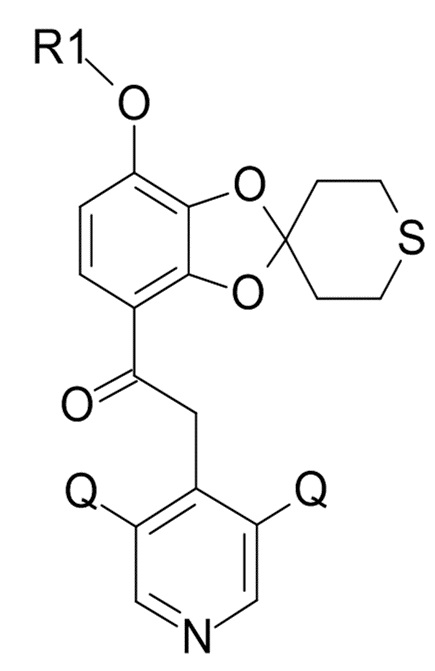 (XI)
(XI)
где R1 и Q соответствуют приведенным выше определениям; и
(5) окисление полученного соединения формулы (XI) для образования соединения формулы (I)
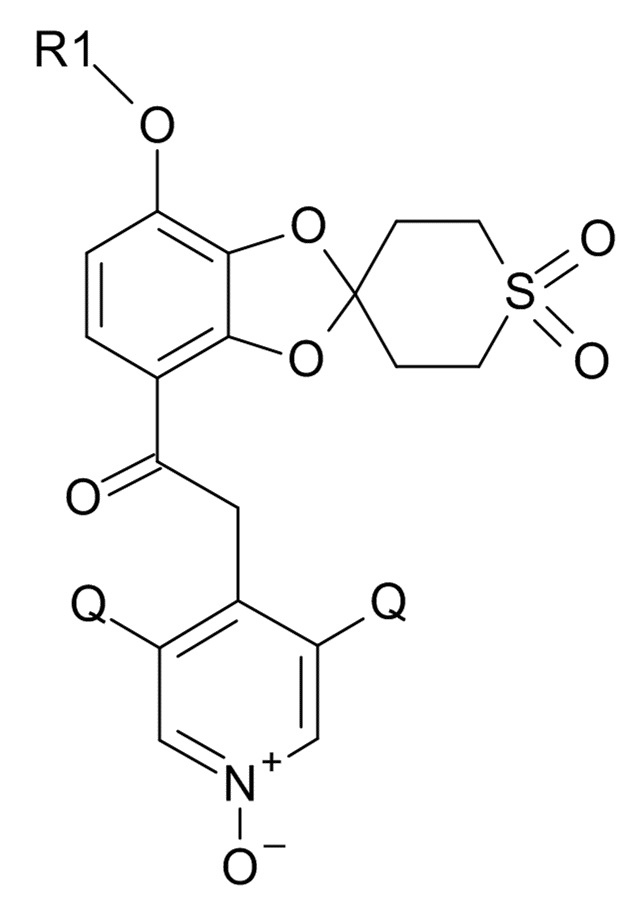 (I)
(I)
где R1 и Q соответствуют приведенным выше определениям.
Пункт 2. Способ по п. 1, отличающийся тем, что удаление защиты на стадии (2a) производят в растворителе, например, выбранном из NMP, ДМСО, ДМФА, метанола, этанола и их смесей, в присутствии основания, например, выбранного из K2CO3, Na2CO3, KHCO3, NaHCO3, CsCO3, TEA и DIPEA, трет-бутоксида калия, трет-бутоксида лития, метоксида натрия, этоксида натрия.
Пункт 3. Способ по п. 2, отличающийся тем, что растворитель представляет собой ДМФА и основание представляет собой K2CO3.
Пункт 4. Способ по п. 2, отличающийся тем, что растворитель представляет собой смесь ДМФА и метанола, а основание представляет собой метоксид натрия.
Пункт 5. Способ по любому из предшествующих пп., отличающийся тем, что реакцию на стадии (3) проводят с использованиемгидрохлорфторуглеродного соединения R1-Cl в присутствии полярного растворителя, например, выбранного из ДМФА, NMP, DMI, ДМСО, EtOAc и ТГФ.
Пункт 6. Способ по п. 5 отличающийся тем, что реакцию проводят с использованием хлордифторметана в ДМФА.
Пункт 7. Способ по любому из предшествующих пп., отличающийся тем, что на стадии (4) связывание проводят в полярном растворителе, например, выбранном из NMP, ДМФА, DMI, ДМСО, MeCN и ТГФ и их смесей, в присутствии основания, например, выбранного из трет-BuOK, трет-BuOLi, трет-BuONa, метоксида натрия или калия, этоксида натрия или калия, K2CO3, Na2CO3, KHCO3, NaHCO3, Et3N и DIPEA.
Пункт 8. Способ по п. 7 отличающийся тем, что полярный растворитель представляет собой ДМФА и основание представляет собой трет-BuOK.
Пункт 9. Способ по любому из предшествующих пп., отличающийся тем, что R1 представляет собой CHF2.
Пункт 10. Способ по любому из предшествующих пп., отличающийся тем, что все Q и Qx представляют собой хлор.
Пункт 11. Способ по любому из предшествующих пп., отличающийся тем, что примесь формулы (XII)
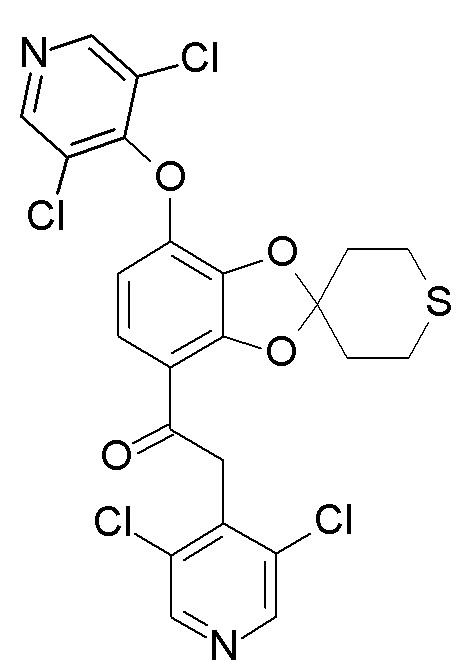 (XII)
(XII)
удаляют из продукта, полученного на стадии (4), кристаллизацией указанного продукта из растворителя, выбранного из, например, диметилформамида (ДМФА), этанола, метанола, этилацетата, гексана, гептана и их смесей.
Пункт 12. Способ по п. 11, отличающийся тем, что растворитель представляет собой смесь этилацетата и этанола.
Пункт 13. Промежуточное соединение формулы(VI)
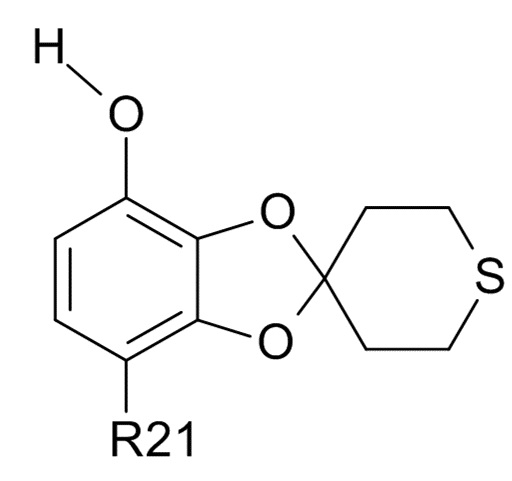 (VI)
(VI)
где R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила.
Пункт 14. Промежуточное соединение по п. 13, которое представляет собой 1-(7-гидроксиспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил)этанон.
Пункт 15. Промежуточное соединение формулы(VII)
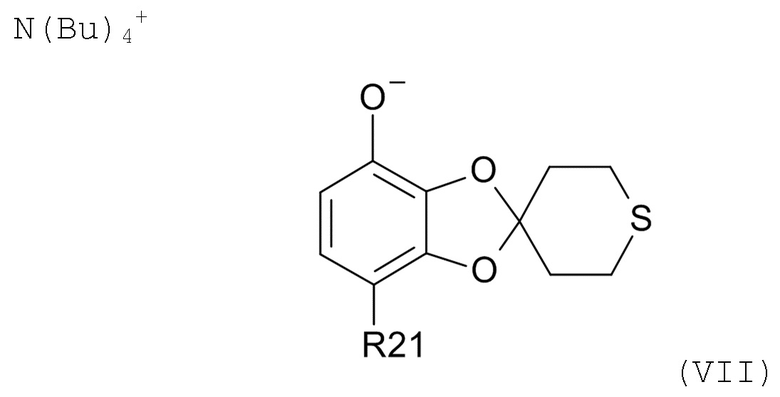
где R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила.
Пункт 16. Промежуточное соединение по п. 15, которое представляет собой тетрабутиламмоний 7-ацетилспиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-олат.
Пункт 17. Промежуточное соединение формулы (IX)
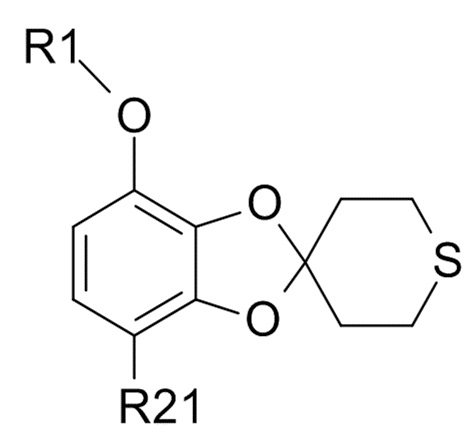 (IX)
(IX)
где R1 выбрано из CHF2 и CF3, а R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила.
Пункт 18. Промежуточное соединение по п. 17, которое представляет собой 1-[7-(дифторметокси)спиро[1,3-бензодиоксол-2,4'-тетрагидротиопиран]-4-ил]этанон.
Пункт 19. Способ получения соединения формулы (VI)
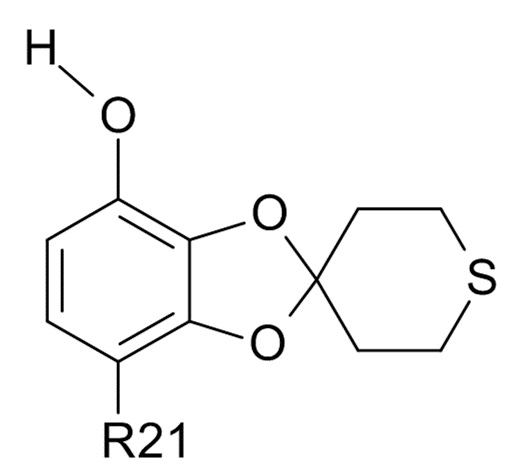 (VI)
(VI)
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила, включающий проведение реакции соединения формулы (IV) с алифатическим или ароматическим тиолом.
Пункт 20. Способ по п. 19, отличающийся тем, что тиол представляет собой 1-додекан-тиол.
Пункт 21. Способ по п. 19, отличающийся тем, что тиол представляет собой 5-трет-бутил-2-метил-бензолтиол
Пункт 22. Способ получения соединения формулы(VII)
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила, включающий стадии (1), (2a) и (2b), которые определены в п. 1.
Пункт 23. Способ получения соединения формулы(VII)
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила, включающий стадии (2a) и (2b), которые определены в п. 1.
Пункт 24. Способ получения соединения формулы(VII)
где R21 выбрано из водорода и C(O)R22, а R22 выбрано из водорода и C1-6-алкила, включающий стадию (2b), которая определена в п. 1.
Пункт 25. Способ получения соединения формулы(IX)
 (IX)
(IX)
где R1 выбрано из CHF2 и CF3, а R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила; включающий стадии (2b) и (3), которые определены в п. 1.
Пункт 26. Способ получения соединения формулы(IX)
(IX)
где R1 выбрано из CHF2 и CF3, а R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила; включающий стадию (3), которая определена в п. 1.
Пункт 27. Способ получения соединения формулы(IX)
(IX)
где R1 выбрано из CHF2 и CF3, а R21 выбрано из водорода и C(O)R22, и R22 выбрано из водорода и C1-6-алкила; включающий проведение реакции соединения формулы (VI) с источником дифторкарбена в полярном растворителе в присутствии основания.
Пункт 28. Способ по п. 27, отличающийся тем, что источник дифторкарбена представляет собой хлордифторацетат натрия.
Пункт 29. Способ по п. 27, отличающийся тем, что источник дифторкарбена представляет собой диэтилбромдифторметилфосфонат.
Пункт 30. Способ по п. 28, отличающийся тем, что полярный растворитель представляет собой смесь воды и ДМФА.
Пункт 31. Способ по п. 29, отличающийся тем, что полярный растворитель представляет собой смесь воды и ацетонитрила.
Пункт 32. Способ по п. 30, отличающийся тем, что основание представляет собой K2CO3.
Пункт 33. Способ по п. 31, отличающийся тем, что основание представляет собой NaOH.
Пункт 34. Способ получения соединения формулы (I)
 (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное способом по п. 1.
Пункт 35. Способ получения соединения формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий (2a), (2b), (3) и (4), как определено в п. 1, и последующее окисление полученного соединения.
Пункт 36. Способ получения соединения формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий (2b), (3) и (4), как определено в п. 1, и последующее окисление полученного соединения.
Пункт 37. Способ получения соединения формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий (3) и (4), как определено в п. 1, и последующее окисление полученного соединения.
Пункт 38. Способ получения соединения формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий: (2a), (2b+3) и (4), и последующее окисление полученного соединения.
Пункт 39. Соединение формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное способом по п. 1.
Пункт 40. Соединение формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное путем реализации стадий (2a), (2b), (3) и (4), как определено в п. 1, с последующим окислением полученного соединения.
Пункт 41. Соединение формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное путем реализации стадий (2b), (3) и (4), как определено в п. 1, с последующим окислением полученного соединения.
Пункт 42. Соединение формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное путем реализации стадий (3) и (4), как определено в п. 1, с последующим окислением полученного соединения.
Пункт 43. Соединение формулы (I)
(I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, полученное путем реализации стадий (2a), (2b+3) и (4), с последующим окислением полученного соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ 1, 3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2015 |
|
RU2697251C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1,3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2018 |
|
RU2814175C2 |
| КОНДЕНСИРОВАННЫЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2015 |
|
RU2706007C2 |
| ФУНГИЦИДНЫЕ КОМПОЗИЦИИ | 2012 |
|
RU2592554C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ СИЛИЛИРОВАННЫЕ ПОЛИМЕРЫ | 2014 |
|
RU2643990C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2680100C9 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2016 |
|
RU2727194C2 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2012 |
|
RU2621695C2 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2007 |
|
RU2478620C2 |
| АМИНО-ИМИННЫЙ МЕТАЛЛОКОМПЛЕКС И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2827476C1 |
Изобретение относится к способу получения соединения формулы (I), в которой R1 выбран из CHF2 и CF3, а Q выбран из хлора, брома и фтора, включающему каждую из следующих стадий: (1) проведение реакции соединения формулы (II), в которой R2 выбран из C1-6-алкила и арилалкила, R21 представляет собой C(O)CH3; с соединением формулы (III), в которой  обозначает одинарную связь, двойную связь или две одинарные связи, и в случаях, когда обозначает двойную связь или две одинарные связи,
обозначает одинарную связь, двойную связь или две одинарные связи, и в случаях, когда обозначает двойную связь или две одинарные связи, 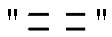 обозначает одинарную связь, а в случаях, когда обозначает одинарную связь, обозначает двойную связь; R3 представляет собой кислород в случаях, когда обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда обозначает одинарную связь или две одинарные связи; в присутствии кислотного катализатора для образования соединения формулы (IV); (2a) проведение реакции полученного соединения формулы (IV) с ароматическим или алифатическим тиолом в растворителе в присутствии основания для образования соединения формулы (VI); (2b) проведение реакции соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения формулы (VII); (3) алкилирование полученного соединения формулы (VII) гидрохлорфторуглеродным реагентом R1-Cl (VIII) в присутствии полярного растворителя, где R1 соответствует приведенным выше определениям, до образования соединения формулы (IX); (4) проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X), в котором QX выбрано из хлора, брома, фтора и иода, в присутствии полярного растворителя в присутствии основания для образования соединения формулы (XI); и (5) окисление полученного соединения формулы (XI) для образования соединения формулы (I). Изобретение также относится к промежуточным соединениям формулы (VI), формулы (VII), формулы (IX). Технический результат – разработан новый способ получения соединения формулы (I) с повышенной чистотой и выходом, которые могут найти применение в медицине для использования в качестве ингибиторов PDE4. 6 н. и 2 з.п. ф-лы, 5 пр.
обозначает одинарную связь, а в случаях, когда обозначает одинарную связь, обозначает двойную связь; R3 представляет собой кислород в случаях, когда обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда обозначает одинарную связь или две одинарные связи; в присутствии кислотного катализатора для образования соединения формулы (IV); (2a) проведение реакции полученного соединения формулы (IV) с ароматическим или алифатическим тиолом в растворителе в присутствии основания для образования соединения формулы (VI); (2b) проведение реакции соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения формулы (VII); (3) алкилирование полученного соединения формулы (VII) гидрохлорфторуглеродным реагентом R1-Cl (VIII) в присутствии полярного растворителя, где R1 соответствует приведенным выше определениям, до образования соединения формулы (IX); (4) проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X), в котором QX выбрано из хлора, брома, фтора и иода, в присутствии полярного растворителя в присутствии основания для образования соединения формулы (XI); и (5) окисление полученного соединения формулы (XI) для образования соединения формулы (I). Изобретение также относится к промежуточным соединениям формулы (VI), формулы (VII), формулы (IX). Технический результат – разработан новый способ получения соединения формулы (I) с повышенной чистотой и выходом, которые могут найти применение в медицине для использования в качестве ингибиторов PDE4. 6 н. и 2 з.п. ф-лы, 5 пр.
1. Способ получения соединения формулы (I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора,
включающий каждую из следующих стадий:
(1) проведение реакции соединения формулы (II)
где R2 выбрано из C1-6-алкила и арилалкила, R21 представляет собой C(O)CH3; с соединением формулы (III)
где ʺ ʺ обозначает одинарную связь, двойную связь или две одинарные связи, и в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺ
ʺ обозначает одинарную связь, двойную связь или две одинарные связи, и в случаях, когда ʺʺ обозначает двойную связь или две одинарных связи, ʺ ʺ обозначает одинарную связь, а в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь; R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарные связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
ʺ обозначает одинарную связь, а в случаях, когда ʺʺ обозначает одинарную связь, ʺʺ обозначает двойную связь; R3 представляет собой кислород в случаях, когда ʺʺ обозначает двойную связь, и R3 представляет собой O-C1-6-алкил в случаях, когда ʺʺ обозначает одинарную связь или две одинарные связи; в присутствии кислотного катализатора для образования соединения формулы (IV)
где R2 и R21 соответствуют приведенным выше определениям;
(2a) проведение реакции полученного соединения формулы (IV) с ароматическим или алифатическим тиолом в растворителе в присутствии основания для образования соединения формулы (VI)
где R21 соответствует приведенным выше определениям;
(2b) проведение реакции соединения формулы (VI) с водным N(Bu)4+OH- для образования соединения формулы (VII)
где R21 соответствует приведенным выше определениям;
(3) алкилирование полученного соединения формулы (VII) гидрохлорфторуглеродным реагентом
R1-Cl (VIII)
в присутствии полярного растворителя, где R1 соответствует приведенным выше определениям, до образования соединения формулы (IX)
где R1 и R21 соответствуют приведенным выше определениям;
(4) проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X) в присутствии полярного растворителя в присутствии основания
где Q соответствует приведенному выше определению, а QX выбрано из хлора, брома, фтора и иода, для образования соединения формулы (XI)
где R1 и Q соответствуют приведенным выше определениям; и
(5) окисление полученного соединения формулы (XI) для образования соединения формулы (I)
где R1 и Q соответствуют приведенным выше определениям.
2. Способ по п.1, отличающийся тем, что R1 представляет собой CHF2.
3. Способ по п.1 или 2, отличающийся тем, что все Q и Qx представляют собой хлор.
4. Промежуточное соединение формулы (VI)
где R21 представляет собой C(O)R22, а R22 представляет собой C1-6-алкил.
5. Промежуточное соединение формулы (VII)
где R21 представляет собой C(O)R22, а R22 представляет собой C1-6-алкил.
6. Промежуточное соединение формулы (IX)
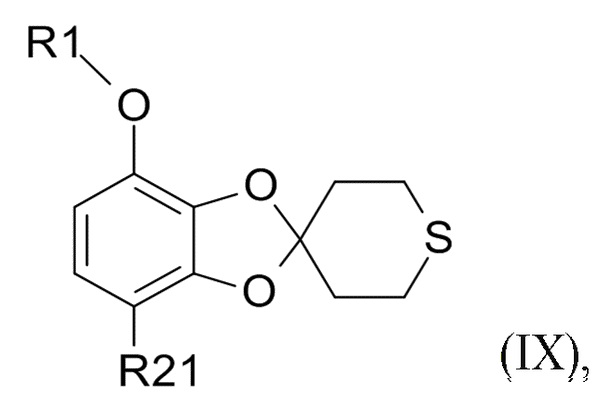
где R1 выбрано из CHF2 и CF3, а R21 представляет собой C(O)R22 и R22 представляет собой C1-6-алкил.
7. Способ получения соединения формулы (I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий (2a), (2b), (3) и (4), как определено в п. 1, и последующее окисление полученного соединения.
8. Способ получения соединения формулы (I)
где R1 выбрано из CHF2 и CF3, а Q выбрано из хлора, брома и фтора, включающий каждую из стадий: (2a), (2b+3) и (4), и последующее окисление полученного соединения, где
стадия (2a) содержит проведение реакции соединения формулы (IV)
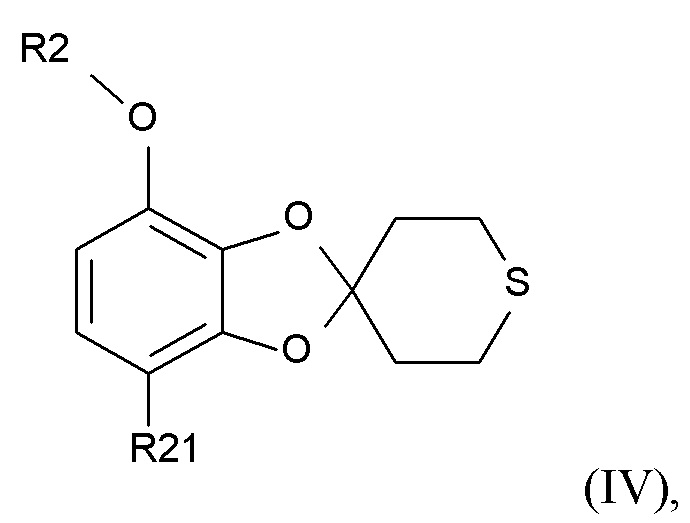
где R2 выбрано из C1-6-алкила и арилалкила, и R21 представляет собой C(O)CH3, с ароматическим или алифатическим тиолом для образования соединения формулы (VI)
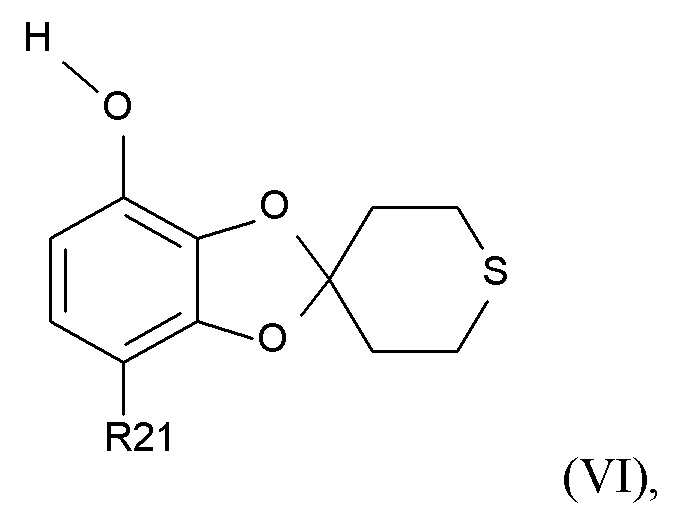
где R21 представляет собой C(O)CH3;
стадия (2b+3) содержит получение соединения формулы (IX)
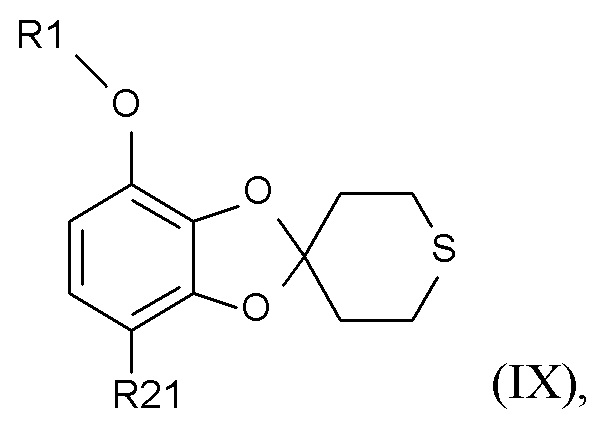
где R1 и R21 соответствуют приведенным выше определениям, из соединения формулы (VI)
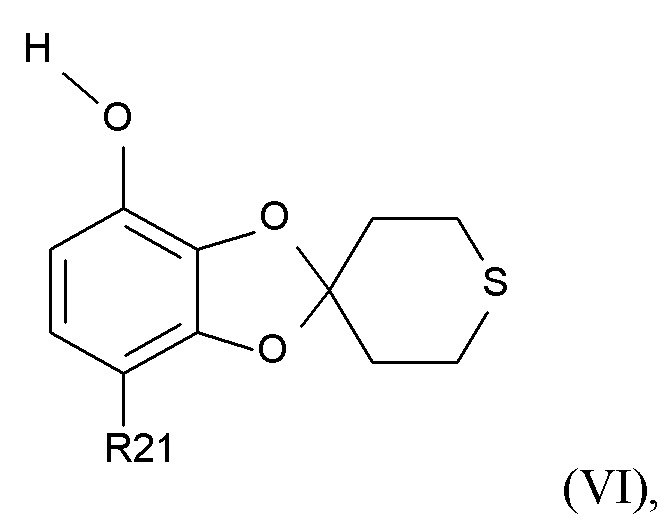
где R21 соответствует приведенным выше определениям, без образования промежуточной соли формулы (VII), путем использования дифторкарбена в полярном растворителе в присутствии основания; и
стадия (4) содержит проведение реакции соединения формулы (IX) с пиридиновым соединением формулы (X)
где Q соответствует приведенному выше определению и QX выбрано из хлора, брома, фтора и иода, для образования соединения формулы (XI)
где R1 и Q соответствуют приведенным выше определениям.
| WO 2011160632 A1, 29.12.2011 | |||
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| US 8980905 B2, 17.03.2015. | |||

