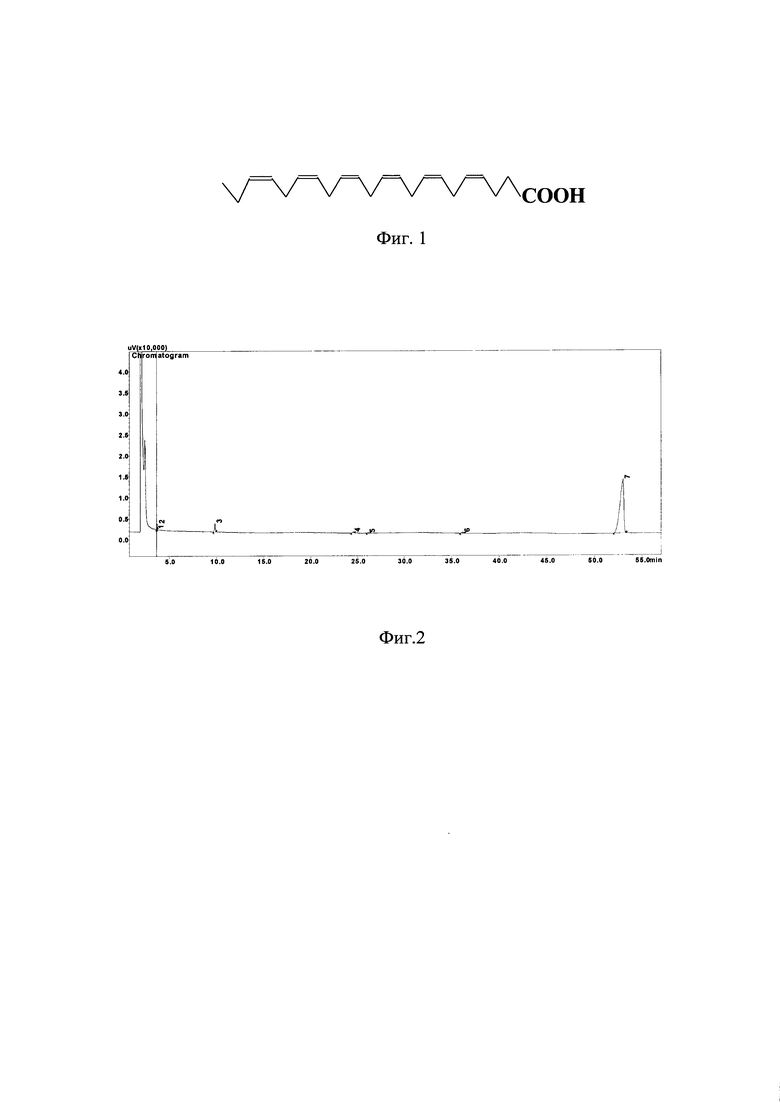
Изобретение относится к пищевой и фармацевтической промышленности и может быть использовано для получения композиций с высоким содержанием докозагексаеновой кислоты (ДГК), обладающей выраженным биологическим действием. Формула докозагексаеновой кислоты приведена на фиг.1.
Докозагексаеновая кислота, ДГК (Δ4,7,10,13,16,19-C22:6, 22:6ω3), наряду с арахидоновой и эйкозапентаеновой кислотами, наиболее востребованная медиками, диетологами и фармацевтической промышленностью, как жирная кислота обширного спектра действия. В организме человека она находится, преимущественно, в полярных липидах – в фосфолипидах и их плазмалогенных формах. Набольшие ее концентрации отмечены в нервной ткани, особенно в головном мозге. Данная кислота в организме практически не синтезируется и должна поступать с пищей. ДГК в составе мембранных липидов влияет на пластичность мембран, в липидных рафтах нервной ткани в составе плазмалогенов контролирует транспорт белков и холестерина, препятствуя образованию амилоидного белка β42, ответственного за развитие болезни Альцгеймера (Habeck C., Stern Y. Alzheimer’s Disease Neuroimaging Initiative. Multivariate data analysis for neuroimaging data: overview and application to Alzheimer's disease //Cell Biochem. Biophys., 2010. Vol.58, Р. 53-67). Помимо этого, ДГК является эффективным ингибитором циклооксигиназы, снижая, тем самым, уровень провоспалительных простагландинов 2-й серии, а также является субстратом при образовании защитных для нервных тканей соединений – нейропротектина D1, резолвинов и мирисинов (Kuda О. «Bioactive metabolites of docosahexaenoic acid» // Biochimie, 2017. Р. 1-9).
ДГК важна для формирующегося организма в утробе матери и, в дальнейшем, при его развитии (Carlson S.E. «Docosahexaenoic acid supplementation in pregnancy and lactation» // Am.J.Clin.Nutr., 2009. Vol. 89 (suppl.), P. 678-684S). При этом дефицит ДГК на данных этапах развития может привести к снижению когнитивных функций ребенка, вплоть до умственной отсталости. У взрослых и пожилых людей по причинам несоответствия питания, наследственным заболеваниям, сбоям в метаболизме липидов содержание ДГК в нервных тканях организма падает. Поэтому необходимо компенсировать это за счет дополнительного потребления ДГК.
Научные эксперименты с ДГК должны быть доступны для всех желающих принять в них участие исследовательских лабораторий. Именно поэтому, разработанные методики получения высокообогащенных концентратов ДГК, при отсутствии какого-либо дорогостоящего оборудования и возможностью их применения персоналом средней квалификации, будут востребованы, что даст импульс к интенсификации продвижения препаратов ДГК, как эффективных средств оздоровления организма.
На данный момент из научной и патентной литературы нам не известно о методах получения фармацевтических композиций с высоким содержанием ДГК, получение которых проходило бы с возможно минимальным количеством стадий технологического процесса при высокой чистоте целевого продукта и кардинальном упрощении стадии концентрирования с повышением ее эффективности в отношении концентрирования ДГК. Тем самым, обеспечивается доступность для научных лабораторий и фармпредприятий к быстрому и технологически простому получению высокообогащенных препаратов ДГК для биохимических, медицинских и диетологических исследований.
Известен способ получения эфиров полиненасыщенных жирных кислот из рыбных жиров и препаратов на их основе (EP Application №0 292 846 A2 « A process for the extraction of polyunsaturated fatty acid esters from fish oils and pharmaceutical and/or dietetic compositions containing said esters», 18.05.88, Int. Cl.4 C11C 3/04, A61K 31/23, A23D 5/00, A23L 1/30). Способ основан на разной летучести эфиров жирных кислот в вакууме при высокой температуре и осуществляется следующим образом. Рыбный жир для перевода жирных кислот из триглицеридов в этиловые эфиры обрабатывают 5%-ным раствором серной кислоты в этаноле, охлаждают, добавляют воды, экстрагируют этиловые эфиры жирных кислот гексаном, для удаления окисленных производных гексановый раствор подвергают хроматографической очистке, гексан упаривают, а сам продукт используют для молекулярной дистилляции при температуре 80-1250С и остаточном давлении 10-3 мм рт. ст. Исходный рыбный жир (как и получаемые из него этиловые эфиры жирных кислот) содержит 10,7% ДГК. После описанных процедур конечный продукт содержит 39,9% ДГК.
Способ имеет ряд недостатков:
1. Достаточно трудоемкая подготовка к осуществлению способа – кислотный этанолиз и хроматографическая очистка от окисленных примесей.
2. Высокая температура, до 1250С, вызывает такие явления как окисление полиненасыщенных жирных кислот, образование диеновых конъюгатов, миграцию двойных связей в цепочке жирной кислоты, полимеризацию продукта, а также деградацию кислоты на короткоцепочечные летучие продукты.
3. Использование конечного продукта, полученного по данному способу, допустимо лишь при очень тщательной хроматографической очистке, которую авторы, по какой-то причине, не указывают, поскольку привнесение в фармацевтическую композицию измененного по химической структуре продукта будет опасным для потребителей.
4. Способ не обеспечивает высокую концентрацию ДГК в конечном продукте (39,9%), наблюдаемое увеличение только в 3,7 раза. Полученная композиция, после очистки, действительно может быть использована в пищевых целях, но не в качестве препарата высокой эффективности.
Известен способ получения композиции, содержащей докозагексаеновую кислоту (UK Patent Application GB 2 098 065 «Antithrombotic compositions containing docosahexaenoic acid», 17.11.82, Int. Cl.3 A61K 31/20, A61K 31/16). Способ основан на комбинировании различных методов – низкотемпературной кристаллизации, молекулярной дистилляции и хроматографии. Метод низкотемпературной кристаллизации использует физические свойства индивидуальных жирных кислот - разные точки их плавления, как в свободном виде, так и в растворах органических растворителей при различных температурах, что обеспечивает первичное концентрирование полиненасыщенных жирных кислот. Полученный концентрат подвергают обогащению по 2-му методу - молекулярной перегонке в вакууме и в заключении используют 3-й метод - хроматографию. Способ осуществляют следующим образом (пример 1): из свежего мяса скумбрии (100 кг) тепловым способом выделяют жир (15 кг), его гидролизуют до получения свободных жирных кислот (9 кг), смесь охлаждают до -600С, получая 4 кг ненасыщенных жирных кислот, далее концентрат подвергают молекулярной перегонке при остаточном давлении 10-2 мм рт. ст. и дополнительно концентрируют колоночной хроматографией, получая 900 г концентрата ДГК с чистотой 82%.
К недостаткам способа следует отнести:
1. Проведение процесса при очень низкой температуре (-600С) является весьма затратным, ибо низкие температуры предполагают специализированные мощности, предназначенные для этого и, возможно, функционирование больших помещений при этой температуре, если речь идет о получении промышленных партий препарата.
2. Вторая стадия – молекулярная перегонка, использующая высокие температуры (до 1100С) способствует значительному окислению ДГК и общим потерям при ее производстве.
3. Способ изначально предполагает существенное образование окисленных и полимеризованных соединений, почему на конечном этапе используют адсорбционную хроматографию. Это достаточно дорогая процедура при получении продукта, учитывая, что сорбент (обычно силикагель) в дальнейшем не регенерируют.
4. Концентрирование ДГК, при столь длительных и затратных процедурах, составляет только 82% при выходе по ДГК – приблизительно 24,6% (авторы не указывают содержание ДГК в жире японской скумбрии, но основываясь на известных данных можно предположить, что это в пределах 30% (Bae J.H. Heavy Metal Contents and Chemical Compositions of Atlantic (Scomber scombrus), Blue (Scomber australasicus), and Chub (Scomber japonicus) Mackerel Muscles //Food Sci. Biotechnol., 2011. Vol.20, P. 709-714).
Известен способ получения метиловых эфиров полиненасыщенных жирных кислот из рыбного жира (Adolf R.O. et al., "The Isolation of Omega-3 Polyunsaturated Fatty Acids and Methyl Esters of Fish Oils by Silver Resin" //JAOCS, 1985. Vol.62, P. 1592-1595). Реализация данного способа основана на способности полиненасыщенных соединений образовывать координационные связи с атомами серебра – чем больше двойных связей, тем сильнее устойчивость таких соединений. Нанося соли серебра на хроматографические сорбенты можно регулировать равновесие в системе насыщенных - ненасыщенных жирных кислот. Способ осуществляют следующим образом: рыбный жир (американская сельдь) переводят в метиловые эфиры жирных кислот, далее, используя хроматографическую колонку со смолой, содержащей ионы серебра, хроматографируют их в системе полярных растворителей, получая при элюции концентрат с содержанием 20,4% ДГК (исходное содержание ДГК – 11,1%).
Способ имеет следующие недостатки:
1. Несмотря на возможность получения высокообогащенных концентратов полиненасыщенных жирных кислот, способ не дает высокие выходы конечного продукта.
2. Использование в способе солей серебра чрезвычайно удорожает стоимость конечного продукта. Системы элюции вымывают импрегнированные соли серебра с сорбента и, в дальнейшем, регенерация растворов с солями серебра, также как и сорбента с их остатками в комплексе реализуемого процесса невозможна.
3. Производительность процесса очень мала. Используют значительные по объему колонки (340-750 мл) для разделения 0,5-10,0 г вещества. Требуются значительные количества растворителей, постоянная их регенерация.
Известен способ получения докозагексаеновой кислоты из жира печени трески (Medina А.R. et al. Concentration and Purification of Stearidonic, Eicosapentaenoic, and Docosahexaenoic Acids from Cod Liver Oil and the Marine microalga Isochrysis galbana //JAOCS, 1995. Vol.72, P. 575-583). Способ основан на разной подвижности жирных кислот в условиях хроматографии высокого давления на специализированных сорбентах. Способ осуществляют следующим образом: жир печени трески гидролизуют с выделением свободных жирных кислот (содержание ДГК – 11%), далее разделяют по составу жирных кислот на хроматографической колонке с обращенной фазой (С-18), элюируя колонку различными системами растворителей. Способ позволяет получать концентраты ДГК с выходом 70,5-100%, но с содержанием данной кислоты не более 72-85%.
Способ имеет многие недостатки:
1. Колоночная хроматография обеспечивает разделения нужных продуктов, но при этом известно, что соотношение разделяемая смесь-сорбент обычно составляет 1 : 10-100 г/г. Это очень большое использование сорбента для разделения малых количеств веществ. К тому же после разделения веществ, сорбент на колонке необходимо регенерировать и для этого требуются растворители в не меньшем объеме, чем для выделения целевого продукта.
2. Способ использует различные системы элюции при различном соотношении растворителя с водой. Это предполагает регенерацию растворителя и, исходя из приведенных объемов, она очень значительна.
3. Использование в качестве растворителя метанола, а именно в примерах с его участием в системах элюции концентрация ДГК самая высокая, вызывает опасения по дальнейшему использованию такого продукта в пищевой индустрии.
4. Реализация способа не обеспечивает получение препаратов с высоким содержанием ДГК, а учитывая затраты на их производство и малую производимость, не может претендовать на массовое использование.
5. Способ предполагает использование препаративного жидкостного хроматографа, прибора, недоступного для многих лабораторий.
Известен способ получения докозагексаеновой кислоты из жира печени трески (Wright S.W., Kuo E.Y., Corey E.J. An Effective Process for the Isolation of Docosahexaenoic Acid in Quantity from Cod Liver Oil //J. Org. Chem., 1987. Vol.52, P 4399-4401). Способ осуществляли следующим образом: жир печени трески (200 г) подвергали щелочному гидролизу, отделяли неомыляемые вещества, подкисляли для выделения свободных жирных кислот (180 г). Далее полиненасыщенные жирные кислоты концентрировали методом разделения литиевых солей жирных кислот в ацетоне (получили концентрат с содержанием 42% ДГК, 50 г). Затем проводили реакцию йод-лактонизации жирных кислот в тетрагидрофуране с участием йода (соотношение 1,2 моль йода на 1 моль ДГК) и калия йодистого в течение 48 часов, после чего йод-лактоны экстрагировали этилацетатом, упаривали, перерастворяли в гексане, отмывали непрореагировавшие жирные кислоты водно-метанольным раствором карбоната калия, гексановый раствор последовательно промывали водой и солевым раствором, упаривали с получением 24,11 г йод-лактона ДГК (выход в расчете на йод-лактон ДГК 81%). Далее полученный йод-лактон ДГК раскрывали с высвобождением исходной ДГК в растворе йодистого натрия и триметилхлорсилана (соотношение йод-лактон ДГК / триметилхлорсилан, TMS-Cl – 1 : 2, моль/моль) в ацетонитриле. Выделившийся свободный йод связывали раствором сульфита натрия и цитрата натрия. Жирные кислоты экстрагировали смесью гексана и дихлорметана, объединенные экстракты промывали водой, солевым раствором, высушивали и упаривали до получения 13,55 г ДГК с чистотой 100% (выход на стадии 78%).
Общий выход:
1) при расчете от 180 г жирных кислот при среднем содержании ДГК в жире печени трески 12,5% - 58,9% (авторы не указывают исходную концентрацию ДГК, но по литературным данным в жире печени атлантической трески Gadus morhua содержится 10,5-14,3% ДГК, Gruger E.H. «Fatty acid composition». In: Fish oils/Ed. Stansby M.E. Westport, Connecticut: The AVI Publ. Comp., Inc., 1967. P. 15).
2) при расчете от 50 г концентрата полиненасыщенных жирных кислот – 63,2%.
Способ имеет следующие недостатки:
1. Растворители, используемые при осуществлении способа – тетрагидрофуран и метанол чрезвычайно ядовиты и не должны быть использованы при получении препаратов, назначаемых человеку. Полученные таким способом препараты следует отнести к веществам для биохимических исследований.
2. Способ осуществляют в 4-е стадии, что значительно усложняет процесс и увеличивает время его осуществления почти до 3-х суток (66 часов и более).
3. Предварительное концентрирование, использующее метод осаждения литиевых солей насыщенных кислот из ацетона, сугубо лабораторный метод, так как объемы растворителя в 20 раз превышают объем разделяемой массы. Масштабирование такого метода для производства необоснованно и не эффективно.
4. Реакция йод-лактонизации проведена с низким выходом (81%), то есть часть ДГК из полученного на предыдущей стадии концентрата просто теряется. Это увеличивает затраты на выпуск данного препарата.
5. Выход на стадии раскрытия йод-лактона ДГК также низок – 78%. Можно заключить, что условия проведения реакции не оптимальны. Это зависит и от соотношения компонентов в среде и от времени проведения реакции и от других немаловажных деталей.
6. Авторы указывают, что продукт, выделенный таким образом, чистый (100%). Однако 4-х стадийное получение ДГК малоэффективно, в виду не высоких выходов на каждой из стадий, общий выход очень низкий – 58,9%, хотя достоинство любого процесса – максимальное использование исходного сырья, минимизация потерь продукта, труда и времени. Данный способ для возможного применения в промышленном производстве должен быть критически воспринят и существенным образом доработан.
Наиболее близким к заявляемому способу является способ получения концентрата этиловых эфиров эйкозапентаеновой (ЭПК) и докозагексаеновой кислот (Авторское свидетельство SU №1581737 А1 «Способ получения концентрата этиловых эфиров эйкозапентаеновой и докозагексаеновой кислот», 30.07.90, МПК С11С 1/00, С11В 7/00). Способ осуществляют следующим образом. Рыбный жир подвергают щелочному гидролизу для получения свободных жирных кислот, после чего смесь нейтрализуют 15% серной кислотой до рН 1,5, добавляют воду и отбирают свободные жирные кислоты, которые растворяют в растворе мочевины в этаноле в соотношении 0,5:3:12 (жирные кислоты-мочевина-этанол), выдерживают смесь 1-3 часа при комнатной температуре, затем 8-12 часов при температуре от (+5) до (-40)0С, осадок отфильтровывают, а из раствора спирта выделяют концентрат ненасыщенных жирных кислот. Для этого фильтрат упаривают до 1/6 объема, добавляют 2%-ый раствор серной кислоты до рН 1,5, затем экстрагируют выделившиеся эфиры жирных кислот хлороформом. Экстракты упаривают и подвергают адсорбционной очистке. Так, из 4 г свободных жирных кислот (пример 2), полученных гидролизом рыбного жира, с содержанием ДГК 5,3% по способу получено 330 мг концентрата полиненасыщенных жирных кислот с содержанием ДГК – 37,0%. Выход по ДГК составляет 57,8%.
Способ имеет следующие недостатки:
1. технология использует концентрированные растворы серной кислоты (15%-ные), которые самым негативным образом отражаются на сохранности полиненасыщенных жирных кислот – происходит их окисление, осмоление, а оставшиеся жирные кислоты могут изменить исходную структуру из-за миграции двойных связей. Концентрат ДГК в виде свободных жирных кислот, полученный по данному способу, представляет собой темно-коричневое масло с выраженным запахом окисления;
2. концентрирование ЭПК и ДГК в продукте происходит с равным увеличением, поэтому такие концентраты следует называть концентратами омега-3 жирных кислот, но никак не концентратом докозагексаеновой кислоты. Концентраты, полученные по данному методу, не могут быть использованы в биохимических и медицинских целях для выявления индивидуальных фармакологических свойств ДГК;
3. способ не обеспечивает оптимальных условий (соотношение компонентов, температура, время) для получения концентратов разного состава, в первую очередь, ДГК;
4. в конечном итоге, концентрирование ДГК в продукте не превышает 37.0%.
Техническая проблема, решаемая изобретением - разработка простого и экономичного способа получения целевого продукта, с высоким содержанием ДГК при одновременном снижении затрат производства.
Поставленная техническая проблема решается тем, что в известном способе получения докозагексаеновой кислоты, включающим гидролиз рыбного жира, нейтрализацию солей жирных кислот минеральной кислотой, выделение свободных жирных кислот, двухэтапную кристаллизацию свободных жирных кислот в этиловом спирте в присутствии мочевины, фильтрацию, добавление к фильтрату воды, выделение докозагексаеновой кислоты из фильтрата органическим растворителем, упаривание с получением целевого продукта, согласно изобретению, перед проведением двухэтапной кристаллизации проводят экстракцию свободных жирных кислот гексаном; кристаллизацию ведут при следующем соотношении компонентов свободные жирные кислот – мочевина – этанол 1 : 3 : 18, кг/кг/л, на первом этапе кристаллизацию ведут 3-5 часов при температуре 18-220С, а кристаллизацию на втором этапе - 12-24 часа при температуре минус 20-250С; в качестве минеральной кислоты для нейтрализации солей жирных кислот после гидролиза применяют соляную кислоту; а в качестве органического растворителя для выделения докозагексаеновой кислоты из фильтрата используют гексан. Упаривание фильтрата с целью получения докозагексаеновой кислоты ведут под вакуумом в среде инертного газа.
Щелочной гидролиз морского жира ведут раствором гидроксидов калия или натрия. Как показали наши предыдущие исследования, для проведения технологического процесса это не имеет значения, так как гидролиз проходит быстро и полностью, но это значимо, если в наличие только один из реагентов.
Важным фактором, выступающим за сохранность ДГК, является применение в технологии малоагрессивных веществ. Отсутствие серной кислоты при производстве ДГК можно рассматривать как правильный прием работы с высоко неустойчивыми соединениями. Серная кислота, самая доступная на рынке по цене, но, в какой бы концентрации она не присутствовала, она может повредить значительную часть ПНЖК. Фактически, в свободных жирных кислотах после стадии гидролиза присутствует определенное количество окисленных смолоподобных соединений. Поэтому применение в процессе гидролиза соляной кислоты, не обладающей разрушительными свойствами на липиды, положительно отразится на качестве продукта.
Для полного выделения свободных жирных кислот из нейтрализованной смеси на стадии гидролиза и после кристаллизации с мочевиной используют гексан. Данная процедура обеспечивает деликатное выделение лабильных веществ из раствора. К тому же продукт, а это жировые субстанции, полностью переходит в растворитель, что снижает производственные потери, позволяет легко отделять этот объем от водной части и сохраняет продукт из-за снижения его прямого контакта с кислородом воздуха.
При кристаллизации с мочевиной используют соотношение компонентов свободные жирные кислоты – мочевина – этанол 1 : 3 : 18, кг/кг/л. Данное соотношение очень критичный показатель. В данном случае мы имеем баланс компонентов, нарушение которого приводит к невозможности получения целевого продукта - концентрата ДГК.
Именно данное соотношение приводит к оптимальному образованию кристаллов жирных кислот с мочевиной. Оно обеспечивает, даже при низких температурах, достаточность количества мочевины для ускоряющегося процесса образования кристаллов. Если взять бόльшее количество мочевины, то процесс протекает неконтролируемо, в образующиеся кристаллы захватываются значительные количества полиненасыщенных жирных кислот, в том числе и ДГК. Обогащение продукта ДГК уже не происходит, мочевина забирает из раствора все кислоты без всякой селективности. Происходит неоправданная потеря вещества, так как осадок – это отход, и, в итоге низкий выход без всякого увеличения концентрации ДГК.
При уменьшении содержания мочевины в растворе, наблюдается обратный эффект – мочевины недостаточно для образования кристаллов с другими кислотами. Процесс, в результате, усложняется. Необходимо использовать низкие температуры, чтобы остатки мочевины выпадали из раствора, образуя кристаллы с кислотами, что крайне неэкономично.
Другим важным показателем, характеризующим систему кристаллизации, является соотношение количеств мочевины и растворителя (в данном случае этанола). Поскольку при температурах 0-50С, как показывает прототип, высокообогащенный концентрат ДГК получить нельзя, так как другие менее ненасыщенные жирные кислоты еще слабо образуют кристаллы с мочевиной, то, предпринятое нами техническое решение продемонстрировало, что, как оказывается, при правильно выбранных соотношения компонентов, этот процесс можно сдвинуть в нужном направлении, применяя более низкие температуры при кристаллизации. Однако при этом есть особенность: растворы с высокой концентрацией мочевины при низких температурах кристаллизации провоцируют слишком быстрое образование кристаллов. В результате падает селективность разделения полиненасыщенных жирных кислот, что отражается на качестве продукта.
Предлагаемое нами соотношение мочевина – этанол 3 : 18 полностью обеспечивает контролируемое протекание кристаллизации с постепенным образованием осадка. Если долю растворителя уменьшать, то мы сталкиваемся с проблемами, указанными выше, если, наоборот, увеличивать, то это приводит к снижению концентрации ДГК, снижению выхода целевого продукта, увеличению технологических объемов и связанного с этим аппаратурного оформления.
При первой кристаллизации используют температуру +18-220С. Снижение температуры менее 180С приводит к неравномерности образования первых кристаллов, а повышение свыше 220С не обеспечивает окончание этого процесса. И в том, и в другом случае, это отражается на целевом продукте – снижается концентрация ДГК в целевом продукте.
Время первой кристаллизации, после смешения компонентов, составляет 3-5 часов. Для того чтобы образовались первые кристаллы, которые будут служить центрами, инициирующими процесс образования кристаллов в дальнейшем, необходимо дать выдержку смеси не менее 3-х часов. Если время сократить, то в смеси при кристаллизации при 18-220С начнется хаотичный процесс образования кристаллов, не способствующий желаемому разделению жирных кислот, а полученный концентрат будет менее обогащенным ДГК. Если время кристаллизации будет более 5-ти часов, то никакого влияния на качество конечного продукта это уже не имеет. 5 час - время достаточное для полной кристаллизации ДГК, дальнейшее увеличение времени нецелесообразно.
При второй кристаллизации используют температуру минус 20-250С. Понижение температуры, менее минус 250С, приводит к снижению селективности процесса кристаллизации – в кристаллы, наряду с другими жирными кислотами, включаются значительные количества ДГК. Это отражается на выходе продукта – он снижается и на составе – падает концентрация ДГК. Увеличение температуры, свыше минус 200С, не обеспечивает необходимой концентрации ДГК в продукте.
Время второй кристаллизации составляет 12-24 часа. Уменьшение времени, менее 12-ти часов, приводит к снижению выхода целевого продукта, а его увеличение, свыше 24-х часов, на выходе и концентрации ДГК не отражается.
Для получения чистой фракции ДКГ целевой продукт подвергают очистке методом высокоэффективной жидкостной хроматографии.
Способ осуществляют следующим образом.
Гидролиз морского жира
Морской (рыбный) жир загружали в стеклянную емкость с подогревом и мешалкой и добавляли раствор NaOH или КОН в 70%-м этанола. Смесь перемешивали 1 час при температуре 650С. Далее реакционную смесь охлаждали до комнатной температуры и подкисляли 10%-ной соляной кислотой до рН=2. Свободные жирные кислоты экстрагировали гексаном, 3 х 150 мл. Объединенный гексановый экстракт промывали дистиллированной водой, 2 х 200 мл. Упаривали под вакуумом.
Кристаллизация жирных кислот с мочевиной
Мочевину добавляли к 96%-ному этанолу (из расчета 3 : 18, кг/л) и нагревали до 500С при перемешивании. После полного растворения мочевины добавляли свободные жирные кислоты. Кристаллизацию проводили в два этапа: первый - при температуре 18-220С в течение 3-5 ч, второй при температуре минус 20-250С в течение 12-24 часов. Образовавшиеся кристаллы отделяли от супернатанта фильтрованием при температуре кристаллизации. К фильтрату добавляли три объема воды и экстрагировали свободные жирные кислоты гексаном. Объединенные гексановые экстракты дважды промывали дистиллированной водой. Для обезвоживания экстракты пропускали через слой сульфата натрия и упаривали под вакуумом в токе азота.
Высокоэффективная жидкостная хроматография
При необходимости и, в большей степени при наличии соответствующего оборудования, ДГК можно получить в очень концентрированном виде, используя метод препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) известным методом (Патент РФ № 2 620 164 «Способ получения арахидоновой кислоты из морской красной водоросли рода Gracilaria», 23.05.2017, МКП А61К 31/20, А61К 36/04).
Концентрат докозагексаеновой кислоты для разделения ВЭЖХ предварительно переводили в этиловые эфиры жирных кислот (ЭЭЖК). К свободным жирным кислотам добавляли 5%-ный раствор хлористого ацетила в безводном этаноле. Реакционную смесь перемешивали при температуре 400С в течение 1 ч. Контроль за прохождением реакции осуществляли тонкослойной хроматографией, как описано выше. После завершения реакции к смеси добавляли дистиллированную воду и экстрагировали ЭЭЖК гексаном. Гексан промывали водой, обезвоживали пропуская через слой безводного сульфата натрия, упаривали под вакуумом в токе азота.
ВЭЖХ была выполнена на хроматографе LC-8A (Shimadzu, Япония) с детекторами UV/VIS SPD-20A (210 нм) и RID-10A. Препаративное разделение проводили на колонке Discovery HS С-18, 10 µм 25 см Ч 50 мм (Supelco, США). Элюирование осуществляли в изократическом режиме системой растворителей этанол-вода (80:20, об./об.). Скорость элюирования составила 50 мл/мин. Элюаты этиловых эфиров ДГК объединяли и упаривали под вакуумом в токе азота.
Изобретение иллюстрируется следующими материалами: на фиг.1. приведена структурная формула докозагексаеновой кислоты, на фиг.2. – хроматограмма ВЭЖХ целевого продукта, в табл. представлен состав главных жирных кислот исходных жиров и концентратов ДГК, полученных кристаллизацией с мочевиной, полученных по примерам 1 и 2.
Способ осуществляют следующим образом.
Реактивы и общие аналитические процедуры для всех примеров
Все используемые реактивы и растворители были марки «хч»; гидроксид натрия, гидроксид калия, этанол, гексан и петролейный эфир, 400-600С («Реагент», Москва), мочевина (карбамид) (АО «ЛенРеактив»).
Тонкослойную хроматографию (ТСХ) липидов для контроля за процессами осуществляли на пластинках с закрепленным слоем силикагеля («Sorbfil», Россия) при использовании элюирующей системы 80:20:1, гексан-диэтиловый эфир-уксусная кислота, об./об./об., для определения состава нейтральных липидов.
Метиловые эфиры жирных кислот липидов для ГЖХ-анализа получали согласно методике (Carreau J.P., Dubacq J.P. «Adaptation of a macro-scale method to the micro-scale for fatty acid methyl transesterification of biological lipid extracts» // J. Chromatogr. – 1978. – Vol. 151, № 3. - 384–390). Анализ метиловых эфиров жирных кислот проводили газовой хроматографией на хроматографе Shimadzu GC-17A («Shimadzu», Kyoto, Japan) с пламенно-ионизационным детектором и капиллярной колонкой 30 m × 0.25 mm i.d. «Supelcowax 10» (Bellefonte, USA), условия анализа: температура колонки 1900С, инжектора и детектора 2400C, газ-носитель гелий. Идентификацию пиков метиловых эфиров жирных кислот проводили по временам удерживания индивидуальных эфиров жирных кислот и с использованием стандартных смесей ПНЖК («Supelco», Bellefonte, USA) и с использованием значений эквивалентной длины цепи (Christie W.W. «Lipid Analysis: Isolation, separation, identification and structural analysis of lipids». - The oily Press, Bridgwater (UK), 2003. – 416 р.).
Пример 1
Гидролиз морского жира (жир дальневосточного лосося, Берингово море)
100 г морского (рыбного) жира загружали в 2 л стеклянную емкость с подогревом и мешалкой и добавляли раствор 14,5 г NaOH в 500 мл 70%-ного этанола. Смесь перемешивали 1 час при температуре 650С. Контроль за окончанием реакции гидролиза жира проводили тонкослойной хроматографией продуктов реакции на пластинках со слоем силикагеля («Sorbfil», Россия) в системе 80:20:1, гексан-диэтиловый эфир-уксусная кислота (объемные %). Далее реакционную смесь охлаждали до комнатной температуры и подкисляли 10%-ной соляной кислотой до рН=2. Свободные жирные кислоты экстрагировали гексаном, 3 х 150 мл. Объединенный гексановый экстракт промывали дистиллированной водой, 2 х 200 мл. Упаривали под вакуумом.
Получено 91,5 г свободных жирных кислот.
Кристаллизация жирных кислот с мочевиной
Мочевину в количестве 274,5 г добавляли к 1647 мл 96%-ного этанола (из расчета 3 : 18, кг/л) и нагревали до 500С при перемешивании. После полного растворения мочевины добавляли свободные жирные кислоты. Кристаллизацию проводили в два этапа: первый - при температуре 180С в течение 5 ч, второй при температуре минус 250С в течение 12 часов. Образовавшиеся кристаллы были отделены от супернатанта фильтрованием при температуре кристаллизации. К фильтрату добавляли три объема воды и экстрагировали свободные жирные кислоты гексаном, 3 х 150 мл. Объединенные гексановые экстракты промывали дистиллированной водой. Для обезвоживания пропускали через слой сульфата натрия и упаривали под вакуумом в токе азота.
Состав жирных кислот липидов экстракта определяли газо-жидкостной хроматографией (Таблица).
Получено: 4,77 г концентрата с содержанием 53,18% ДГК.
Выход: 35,0% в расчете на ДГК.
Для выделения из концентрата чистой докозагексаеновой кислоты использовали следующий метод.
Очистка докозагексаеновой кислоты препаративной высокоэффективной жидкостной хроматографией
Концентрат докозагексаеновой кислоты, 4,77 г, для разделения высокоэффективной жидкостной хроматографией (ВЭЖХ) предварительно переводили в этиловые эфиры жирных кислот (ЭЭЖК). К свободным жирным кислотам добавляли 50 мл 5%-ного раствора хлористого ацетила в безводном этаноле. Реакционную смесь перемешивали при температуре 400С в течение 1 ч. Контроль за прохождением реакции осуществляли тонкослойной хроматографией, как описано выше. После завершения реакции к смеси добавляли дистиллированную воду, 50 мл, и экстрагировали ЭЭЖК гексаном, 3 х 30мл. Гексан промывали водой, 2 х 20мл, обезвоживали пропуская через слой безводного сульфата натрия, упаривали под вакуумом.
ВЭЖХ была выполнена на хроматографе LC-8A (Shimadzu, Япония) с детекторами UV/VIS SPD-20A (210 нм) и RID-10A. Препаративное разделение проводили на колонке Discovery HS С-18, 10 µм 25 см Ч 50 мм (Supelco, США). Элюирование осуществляли в изократическом режиме системой растворителей этанол-вода (80:20, об./об.). Скорость элюирования составила 50 мл/мин. Элюаты этиловых эфиров ДГК объединяли и упаривали под вакуумом в токе азота. Состав жирных кислот препарата ДГК в виде этилового эфира приведен в Таблице 1 а на и Фиг. 2 приведена хроматограмма анализа.
Получено: 2,40 г концентрата с содержанием 94,11% ДГК в виде этилового эфира.
Выход на стадии ВЭЖХ: 89,1%.
Общий выход: 31,1% в расчете на ДГК
Пример 2.
Все операции в данном примере проводили также как в примере 1, за исключением того, что:
- в качестве морского жира использовали жир из печени сельдевой акулы (Охотское море);
- гидролиз жира проводили калиевой щелочью, КОН;
- первую кристаллизацию проводили при температуре 220С и продолжительностью 3 часов;
- вторая кристаллизация проведена при температуре минус 200С и продолжительностью 24 часа;
- в качестве инертного газа при упаривании применяли аргон.
Получено: 18,97 г концентрата с содержанием 73,01% ДГК.
Выход: 35,9% в расчете на ДГК.
Состав главных жирных кислот исходных жиров и концентратов ДГК, полученных кристаллизацией с мочевиной, полученных по примерам 1 и 2 представлен в таблице.
Как видно из представленных результатов, осуществление способа в заявленных режимах обеспечивает получение высокообогащенного ДКГ препарата практически в одну стадию. Способ прост в исполнении, не использует агрессивные реактивы и может быть реализован в практических целях в любой лаборатории.
Таблица
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДОКОЗАГЕКСАЕНОВОЙ КИСЛОТЫ | 2013 |
|
RU2537252C1 |
| Способ получения арахидоновой кислоты | 2016 |
|
RU2627273C1 |
| Способ получения концентрата этиловых эфиров эйкозапентаеновой и докозагексаеновой кислот | 1988 |
|
SU1581737A1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЦЕНТРАТА ЭТИЛОВЫХ ЭФИРОВ ПОЛИНЕНАСЫЩЕННЫХ ВЫСШИХ ЖИРНЫХ КИСЛОТ | 2000 |
|
RU2209235C2 |
| Способ получения витаминизированного концентрата этиловых эфиров полиненасыщенных высших жирных кислот из рыбного жира | 2015 |
|
RU2614587C1 |
| Способ получения арахидоновой кислоты из морской красной водоросли рода Gracilaria | 2016 |
|
RU2620164C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОНЦЕНТРАТА ЭТИЛОВЫХ ЭФИРОВ ПОЛИНЕНАСЫЩЕННЫХ ВЫСШИХ ЖИРНЫХ КИСЛОТ | 1994 |
|
RU2078130C1 |
| Способ получения концентрата ненасыщенных алкил-глицериновых эфиров из морских липидов | 2017 |
|
RU2649014C1 |
| Способ получения концентрата ненасыщенных алкил-глицериновых эфиров из морских гидробионтов | 2017 |
|
RU2642294C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ НЕНАСЫЩЕННЫЕ СОЕДИНЕНИЯ | 2004 |
|
RU2360952C2 |
Настоящее изобретение относится к способу получения докозагексаеновой кислоты, обладающей выраженным биологическим действием. Способ включает щелочной гидролиз рыбного жира, нейтрализацию солей жирных кислот минеральной кислотой, выделение свободных жирных кислот, двухэтапную кристаллизацию свободных жирных кислот в этиловом спирте в присутствии мочевины, фильтрацию, добавление к фильтрату воды, выделение докозагексаеновой кислоты из фильтрата органическим растворителем и упаривание с получением целевого продукта. При этом перед проведением двухэтапной кристаллизации проводят экстракцию свободных жирных кислот гексаном, кристаллизацию ведут при следующем соотношении компонентов: свободные жирные кислоты : мочевина : этанол - 1:3:18, кг/кг/л, на первом этапе кристаллизацию ведут 3-5 часов при температуре 18-22°С, а кристаллизацию на втором этапе - 12-24 часа при температуре минус 20-25°С, в качестве минеральной кислоты при нейтрализации солей жирных кислот после гидролиза применяют соляную кислоту, а в качестве органического растворителя для выделения докозагексаеновой кислоты из фильтрата используют гексан. 1 з.п. ф-лы, 2 ил., 1 табл., 2 пр.
1. Способ получения докозагексаеновой кислоты, включающий щелочной гидролиз рыбного жира, нейтрализацию солей жирных кислот минеральной кислотой, выделение свободных жирных кислот, двухэтапную кристаллизацию свободных жирных кислот в этиловом спирте в присутствии мочевины, фильтрацию, добавление к фильтрату воды, выделение докозагексаеновой кислоты из фильтрата органическим растворителем, упаривание с получением целевого продукта, отличающийся тем, что перед проведением двухэтапной кристаллизации проводят экстракцию свободных жирных кислот гексаном; кристаллизацию ведут при следующем соотношении компонентов: свободные жирные кислоты : мочевина : этанол - 1:3:18, кг/кг/л, на первом этапе кристаллизацию ведут 3-5 часов при температуре 18-22°С, а кристаллизацию на втором этапе - 12-24 часа при температуре минус 20-25°С; в качестве минеральной кислоты при нейтрализации солей жирных кислот после гидролиза применяют соляную кислоту; а в качестве органического растворителя для выделения докозагексаеновой кислоты из фильтрата используют гексан.
2. Способ по п. 1, отличающийся тем, что целевой продукт подвергают очистке методом высокоэффективной жидкостной хроматографии.
| Способ получения концентрата этиловых эфиров эйкозапентаеновой и докозагексаеновой кислот | 1988 |
|
SU1581737A1 |
| WO 1989011521 A1, 30.11.1989 | |||
| Wright, S.W | |||
| et al | |||
| An effective process for the isolation of Docosahexaenoic acid in quantity from cod liver oil | |||
| J.Org.Chem., 1987, 52(19), 4399-4401 | |||
| Е.В | |||
| Ермоленко и др | |||
| Комплексная переработка липидов печени командорского кальмара BERRYTEUTHIS MAGISTER | |||
| Известия ТИНРО, 2014, том 176, 288-294. | |||

