Изобретение относится к пищевой и фармацевтической промышленности и может быть использовано для получения композиций с высоким содержанием арахидоновой кислоты (АК), обладающей выраженным биологическим действием.
На данный момент из научной и патентной литературы нам не известно о методах получения фармацевтических композиций с высоким содержанием АК, получение которых проходило бы при одновременно высоком выходе и высокой чистоте целевого продукта, при сохранении эффективности технологического процесса - кардинальном упрощении технологических стадий, сокращении времени проведения процесса, повышении его экологичности. Тем самым, достигается интенсификация процесса, что дает фармацевтическим предприятиям возможность получения новых препаратов (композиций) с высоким содержанием АК, оказывающих лечебно-профилактическое действие при нарушениях обмена липидов в организме.
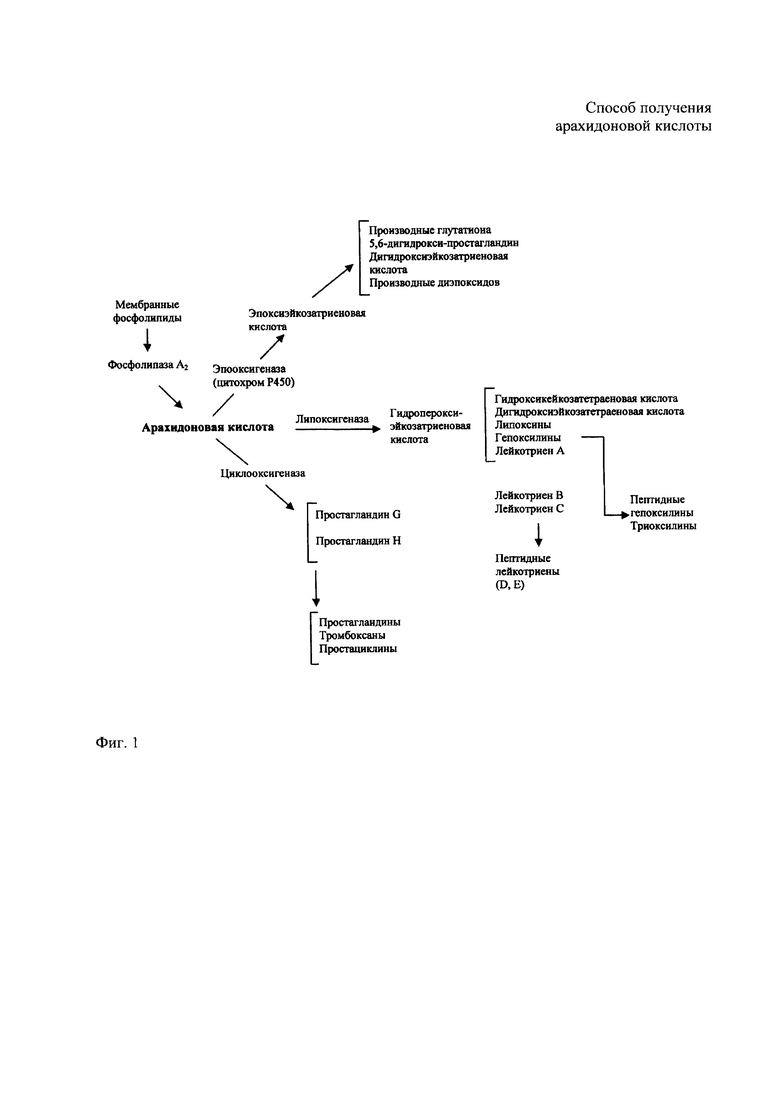
Арахидоновая кислота - биохимически важная эссенциальная кислота, предшественник различных эйкозаноидов и других активных медиаторов. В животных АК образуется в несколько стадий из линолевой кислоты путем элонгации и десатурации (Sprecher Н. «Metabolism of highly unsaturated n-3 and n-6 fatty acids» // BBA. - 2000. - Vol. 1486. - P. 219-231), либо поступает с пищей. В природе АК аккумулируется в фосфолипидах клеточных мембран, из которых высвобождается фосфолипазой А2 при действии воспалительных агентов (интерлейкин-1, -6; фактор некроза опухоли; форболовые эфиры и проч.) или других стимуляторов. Арахидоновая кислота является исходным материалом для биосинтеза ряда биологически активных окисленных производных (в особенности гидрокси-, гидроперокси- и эпоксисоединений), известных как эйкозаноиды. Их подразделяют на различные группы в каскаде арахидоновой кислоты, который представлен на Фиг. 1.
Среди известных «сигнальных» соединений арахидоновой кислоты следует упомянуть изопростаны (неферментативный путь окисления), 2 - арахидоноил-глицерин и этаноламид арахидоновой кислоты (анандамид) - эндогенный каннабиноид (Сергеева М.Г., Варфоломеева А.Т. «Каскад арахидоновой кислоты». М.: Народное образование, 2006. - 256 с).
Исследования арахидоновой кислоты, как потенциального субстрата для синтеза и биосинтеза биологически активных веществ, продолжаются из года в год неослабевающими темпами. Однако для большинства лабораторий остро стоит вопрос о приобретении АК для исследований, поскольку стоимость чистых препаратов АК чрезвычайно велика. Так, например, фирма «Sigma-Aldrich» предлагает АК (98,5%) по 1130 евро/г. Практически любое исследование, связанное с АК, начинается со значительных затрат. Такое положение сложилось, в основном, из-за того, что большинство экспериментаторов, даже имея в наличии необходимые приборы, не обладают опытом по получению АК.
Известно много лабораторных и промышленных методов получения АК, но их применение в том или ином процессе не является очевидным и должно быть обосновано использованием соответствующего сырья, возможностями самого метода и наличием определенного оборудования.
Ранее в мировой практике для получения АК использовали такие методы как - низкотемпературная кристаллизация жирных кислот (ЖК) из растворителей, молекулярная дистилляция метиловых эфиров жирных кислот (МЭЖК) в вакууме, комплексообразование ЖК с мочевиной, хроматографическое разделение ЖК на сорбентах, включая силикагель, импрегнированный солями серебра. Хорошей иллюстрацией этих методов являются ниже приведенные работы.
Известен способ выделения АК из липидов надпочечников крупного рогатого скота (Herb S.F., Riemenschneider R.W., Donaldson J. «Isolation of natural arachidonic acid as its methyl esters» // JAOCS. 1951. Vol. 28, N 2. P 55-58). Способ осуществляли следующим способом: выделенные липиды из надпочечников гидролизовали до свободных жирных кислот (8-9% АК), кристаллизовали из ацетона последовательно при 0°С, -18°С, -40°С, переводили ЖК в метиловые эфиры и далее фракционировали АК на силикагеле (silicic acid), по мнению авторов получен концентрат АК чистоты 99,15% (слишком оптимистичный результат для того времени и возможности действительного анализа).
Недостатки способа:
1) метод кристаллизации ЖК из ацетона обладает единственной возможностью - отделение из смеси ЖК насыщенных кислот; другие модификации с пониженными температурами не несут эффективного концентрирования полиненасыщенных ЖК;
2) фракционирование ЖК на силикагеле, как показывает практика, не производительный метод; принципиально метод осуществимый, но нагрузка на колонку малая, а расход органических растворителей высокий;
3) выход авторами не оговаривается, но судя по приведенным таблицам очень незначительный, а чистота АК вызывает большие сомнения.
Известен способ получения АК из липидов надпочечников млекопитающих (А.с. СССР №247461, МПК A61K 31/22, А61Р 3/00, С11С 1/02, опубл. 01.01.1969), при осуществлении которого липиды гидролизуют щелочью, подкисляют для выделения свободных жирных кислот, экстрагируют толуолом, растворитель упаривают, а липиды перерастворяют в ацетоне и осаждают из него насыщенную фракцию при минус 15°С, фильтруют, жирные кислоты этерифицируют этиловым спиртом (содержание АК 11,6%) и подвергают дистилляции при температуре 193-205°С и остаточном давлении 3 мм рт. ст.
Недостатки способа:
1) при экстракции гидролизованных липидов авторы используют толуол, плохо в дальнейшем подвергающийся отгонке (температура кипения 110°С), к тому же весьма токсичный;
2) кристаллизация свободных ЖК из ацетона малоэффективный процесс, а температура (-15°С) не такая низкая, чтобы можно было ощутимо поднять концентрацию АК;
3) метод молекулярной дистилляции вызывает разрушение и окисление АК, в особенности при столь высоких температурах и недостаточно полном вакууме;
4) АК получена с 25%_ной чистотой - малый показатель для каких-либо биохимических исследований, а выход 20-25% отражает неэффективность набора используемых методов.
Известен способ извлечения АК из липидов семенников с/х животных (А.с. СССР №641963, МПК A61K 31/557, A61K 31/20, A61K 31/202, А61Р 15/00, С07С 57/03, опубл. 15.01.1979) последовательным применением методов низкотемпературной кристаллизации (-20°С), молекулярной дистилляции (3 мм рт. ст., 220°-235°С) и колоночной хроматографии на силикагеле с AgNO3. Выход 96-99%-ной АК не превышал 9,2%.
Недостатками способа следует считать:
1) использование при концентрировании АК молекулярной дистилляции при высоких температурах, способствующих быстрому окислению и разрушению структуры арахидоновой кислоты;
2) применение хроматографии с AgNO3 - нагрузка на колонку невелика, 1:10, для столь значительного содержания на сорбенте соли серебра -50%, к тому же полноценное регенерирование для повторного использования такой «дорогой» колонки проблематично, а ее эффективность критически падает уже после первого использования;
3) малый выход конечного продукта (9,2%), практически 9/10 АК теряется в результате использования способа.
Известен способ выделения АК из липидов надпочечников (А.с. СССР № 643496, МПК С07С 57/02, С11В 13/00, опубл. 25.01.1979) фракционированием смеси ЖК два раза в петролейном эфире (-28°С 78°С) и два раза в ацетоне (-78°С), а далее с использованием колоночной хроматографии на AgNO3. Выход 96-99%-ной АК чуть более 20%.
Недостатками способа являются:
1) избыточное, на наш взгляд, применение неселективного метода - низко-температурной кристаллизации - четыре раза, а в итоге увеличение с 16-18% до 65-67%, а суммарная продолжительность этих операций составляет 40-45 часов, выход только 24%;
2) «серебряная» хроматография очень увеличивает затраты способа, ибо делает использование колонки одноразовым;
3) общий выход АК невысокий - около 20%.
Известен способ выделении АК из липидов поджелудочной железы (А.с. СССР №585152, МПК С07С 57/02, С11В 13/00, опубл. 25.12.1977), включающий сложный набор методов: вначале дробную низкотемпературную кристаллизацию ЖК (-65°С), затем комплексообразование ЖК с мочевиной, 2-х кратную молекулярную дистилляцию и окончательную рафинацию АК на колонке с AgNO3. Выход АК с чистотой 97-98% составил 27-28%.
Недостатки способа:
1) низкотемпературная кристаллизация ЖК - малопроизводительный и достаточно трудоемкий метод, так как требует манипуляций при очень низких температурах;
2) комплексообразование ЖК с мочевиной «компактный» и удобный в исполнении метод, но у него есть пределы эффективности - не более чем 2-3-х кратное увеличение полиненасыщенных жирных кислот (ПНЖК) в концентрате и большие потери ПНЖК при осуществлении метода (часть ценных кислот остается в комплексе с мочевиной);
3) молекулярная дистилляция ПНЖК чрезвычайно губительная для них процедура, так высокая температура (150°С) провоцирует миграцию двойных связей в углеродной цепи и деградацию ПНЖК;
4) хроматография на сорбентах с AgNO3 - малопроизводительная и не оправдывающая себя по себестоимости процедура;
5) в результате такого объемного набора методов низкий выход - не более 28%.
Известен способ получения АК из отходов производства инсулина (А.с. СССР №897766, МПК С07С 57/02, С07С 51/00, опубл. 15.01.1982) при использовании следующего набора методов: низкотемпературной кристаллизации ЖК из диэтилового эфира (-25° - 50°С) три раза, затем молекулярной перегонки ЖК при 0,05-1 мм рт. ст. и температуре 126° - 131°С, а далее - метода «серебряной» хроматографии. Выход 99,9%-ной АК, в расчете на ее исходное содержание в смеси, составляет 42,2%. Недостатки метода:
1) использование диэтилового эфира, как растворителя при фракционировании ЖК, не самое удачное решение, так как эффективность разделения в нем ЖК оставляет желать лучшего, поэтому в настоящее время диэтиловый эфир никто не использует в этих целях, к тому же он чрезвычайно пожароопасен и его применение в технологии не желательно;
2) молекулярная дистилляция при температуре до 130°С не способствует сохранению высоколабильной АК и, нужно отметить, что этот метод требует соответствующего аппаратурного оформления, не доступного для многих лабораторий;
3) метод «серебряной» хроматографии - самое малопроизводительное звено в способе, существенно ограничивающее объемы выпуска АК;
4) как результат всего вышесказанного, низкий выход - 42,2%. Известен способ концентрирования АК из жирных кислот поджелудочной железы (А.с. СССР, №908356, МПК A61K 31/20, опубл. 28.02.1982) использующий только одну стадию - комплексообразование ЖК с мочевиной при температуре до -20°С. Выход 40-60%-ной АК составлял 60-78%.
Недостатки способа:
1) как уже упоминалось ранее, комплексообразование ЖК с мочевиной не является методом получения индивидуальных ПНЖК, его эффективность ограничена 2-3-х кратным увеличением их содержания в концентрате, но не более, причем потери целевого вещества очень значительны;
2) концентрирование АК до 60% не обеспечивает потребности биохимических и медицинских исследований, так как для этого нужны высокочистые препараты; фактически способ обеспечивает только начальное обогащение смеси АК для дальнейшей очистки.
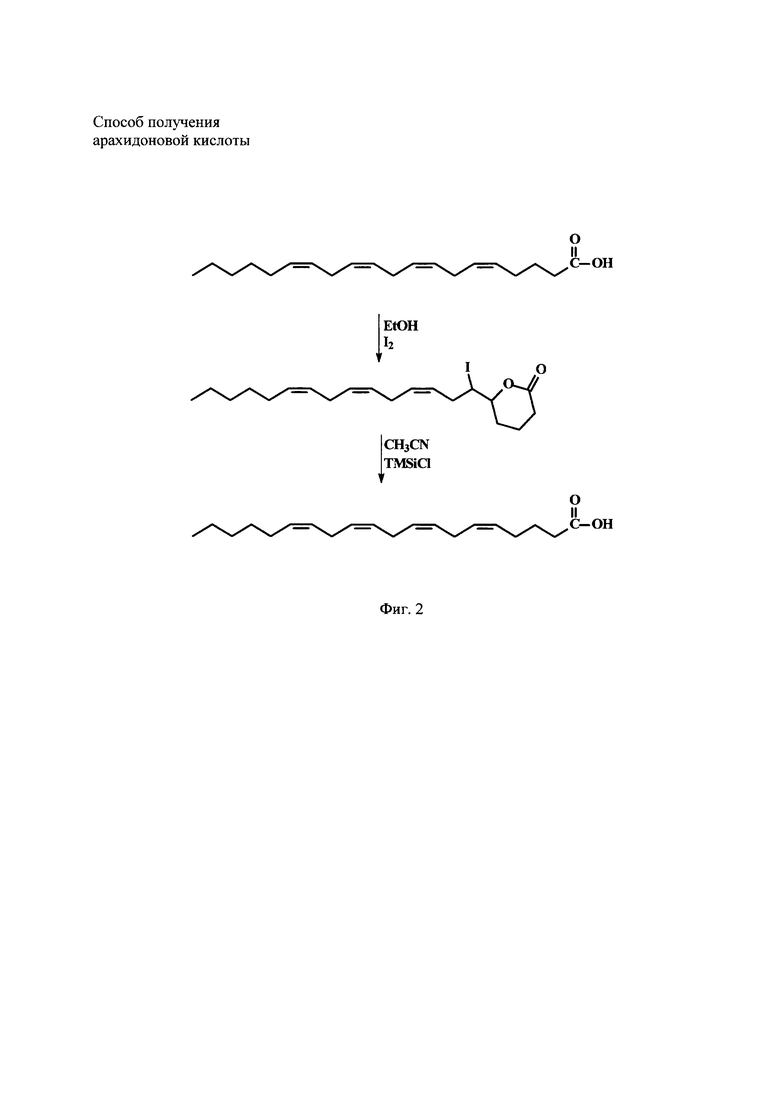
Среди других новых методов, появившихся в 1980-90 г. замечателен своей технологичностью и определенной простотой метод йод-лактонизации (ЙЛ) Δ 4 и Δ 5 ПНЖК. Сущность метода заключается в возможности образования такими кислотами как АК, ЭПК и ДГК (докозагексаеновая кислота, 22:6n-3) йод-лактонов с 5 и 6-ти членными циклами (дельта-йодлактоны для АК и ЭПК, гамма-йод-лактон для ДГК) в отличие от других кислот. На Фиг. 2 приведена химическая схема образования и раскрытия дельта-йод-лактона АК.
Йод-лактоны ЖК имеют другие физические и химические свойства, чем ЖК, поэтому их выделение из реакционной смеси и дальнейшее раскрытие до исходных ЖК не столь трудоемко.
Известен способ получения АК (Corey E.J., Wright S.W. «А simple process for the purification of arachidonic acid» // Tetrahedron letters. - 1984. - Vol. 25, №26. - P. 2729-2730) из смеси ЖК, где АК была единственной полиненасыщенной ЖК (50% АК). Способ был осуществлен следующим образом: реакцию образования йод-лактонов АК (ЙЛ-АК) проводили в тетрагидрофуране, после чего ЙЛ-АК отделяли экстракцией, раскрывали в ацетонитриле, добавляя триметилйодсилан, TMSi-I, и экстрагировали из смеси 97%-ную АК.
Недостатки способа:
1) сложно найти смесь природных липидов, где бы среди ЖК единственной полиненасыщенной кислотой была АК, так как в смесях всегда присутствуют ЭПК и ДГК, также образующие лактоны, что вносит существенные коррективы в осуществление способа;
2) авторы не указывают общий выход при осуществлении способа, но известно, что первая стадия - образование ЙЛ-АК - самая проблематичная и требует наибольших знаний при осуществлении, даже незначительные недоработки на этой стадии приводят к существенным потерям в АК.
Известен способ получения АК, описанный в Патенте РФ №1631067, МПК С11С 1/02, опубл. 28.02.1991. Способ осуществляли следующим образом: липиды печени крысы (АК - 17%, ДГК - 7%) омыляли действием раствора щелочи, затем подкисляли кислотой для выделения свободных жирных кислот, далее переводили их в калиевые соли и обрабатывали раствором йода (с добавлением KI) в расчете на ДГК при соотношении ДГК /I2 = 1 : 1,1 моль / моль, перемешивали при 25°С 5 час, образовавшийся йод-лактон ДГК экстрагировали гексаном, а к водному слою добавляли йодистый калий и йод (количество йода рассчитывали на АК, содержащуюся в исходном сырье, больше стехиометрического в 5 раз, соотношение йодистого калия и йода равно KI / I2 = 0,9 моль / моль) и перемешивали при 10°С 2 часа, непрореагировавший йод нейтрализовали водным раствором тиосульфата натрия (Na2S2O3), экстрагировали ЙЛ-АК смесью гексан-этилацетат, экстракт упаривали, снова перерастворяли смесь в гексане, отмывали свободные ЖК от йод-лактона АК водно-спиртовыми растворами бикарбоната или карбоната калия, упаривали гексан и раскрывали йод-лактон АК в безводном ацетонитриле силирующим агентом (триметилйодсилан, TMSi-I) в соотношении йод-лактон АК / силирующий агент = 1 : 0,86, моль /моль, в течение 3-х минут, нейтрализовали выделившийся йод раствором тиосульфата натрия и экстрагировали АК из смеси хлороформом. Чистота выделенной АК - 98,5%, общий выход - 50,0%.
1) Использование в смеси для йод-лактонизации калия йодида (KI) - известно, что его добавление к раствору йода замедляет реакцию йод-лактонизации и позволяет более «плавно» проводить процесс. Поэтому йод даже добавляют в большом избытке на содержание АК до 5 моль / моль, а время реакции ограничивают 2-мя часами, при этом температура процесса должна быть пониженной (10°С) из-за возможности протекания побочных реакций, снижающих чистоту АК. Данная стадия процесса при упомянутых условиях имеет большие недостатки - неоправданно высокое использование реактивов (йода, калия йодистого), а также необходимость специального контроля за температурным режимом;
2) Авторы, в качестве силирующего агента, необходимого для раскрытия йод-лактона АК, используют только триметилйодсилан (TMSi-I), при этом наиболее доступный и недорогой триметилхлорсилан (в несколько раз более дешевый на рынке, чем TMSi-I) для раскрытия йод-лактона АК не применяют, что удорожает продукт и сужает рамки технологического процесса.
Известен способ получения АК методом высокоэффективной жидкостной хроматографии, ВЭЖХ (Yuan С., Wang X., Yu Z. «Separation and preparative purification of arachidonic acid from microbial lipids by urea inclusion reaction and HPLC» // Preparative Biochemistry and Biotechnology. 2007. Vol. 37, N 2.
P. 149-159). Способ осуществляли следующим образом: микробиальные липиды с содержанием АК 38,3% гидролизовали раствором натриевой щелочи, подкисляли соляной кислотой до выделения свободных ЖК, которые подвергали комплексообразованию с мочевиной в соотношении ЖК: мочевина: метанол - 1:2:8, по массе при -10°С (АК - 77,3%, выход на стадии 68,0%), далее полученный концентрат АК переводили в метиловые эфиры ЖК и получали чистую АК (99,5%), используя ВЭЖХ на препаративной колонке С-18 (300 мм × 30 мм i.d., диаметр частиц сорбента 15 мкм), элюирующая система 95:5, метанол-вода, об./об., детектор - рефрактометр.
Общий выход в процессе получения АК не указан.
Общее время осуществления способа 15 час.
Недостатки способа:
1) использование метода комплексообразования ЖК с мочевиной резко снижает конечный выход целевого продукта - АК;
2) применение в комплексообразовании ЖК с мочевиной метанола - чрезвычайно ядовитого соединения - не позволяет использовать данную методику в медицинской и пищевой промышленности;
3) известно, что производительность ВЭЖХ сильно зависит от размера частиц сорбента, поэтому использование для выделения АК частиц размером 15 мкм, возможно, приемлемо для снижения давления в системе, но одновременно накладывает малую производительность по целевому веществу (скорость подвижной фазы всего 5 мл/мин), такой способ реализуем, как показывает данный пример, но будет обеспечивать очень малые выходы по АК;
4) авторы используют в элюирующей системе метанол, хотя задача большинства исследователей, предлагающих свои способы получения активных веществ, максимально снизить применение токсичных растворителей; способ интересен, так как позволяет выделять АК, но не может быть тиражирован из-за требований, предъявляемых к чистым производствам.
Наиболее близким к заявляемому нами способу по количеству существенных признаков является способ получения арахидоновой кислоты, описанный Gaiday et al. «Separation of Natural Polyunsaturated Fatty Acids by Means of Iodolactonization», JAOCS, v. 68, no 4, 1991, p. 231). Способ осуществляли следующим образом: липиды печени крысы гидролизовали, гидролизат подкисляли, и выделяли свободные жирные кислоты низкотемпературной кристаллизацией, далее полученный концентрат полиненасыщенных жирных кислот переводили в калиевые соли и обрабатывали смесью йода и калия йодида в реакции йод-лактонизации, по окончании реакции йод-лактоны АК экстрагировали органическим растворителем и далее раскрывали TMS-I до исходной АК, нейтрализовали выделившийся йод тиосульфатом натрия и экстрагировали жирные кислоты органическим растворителем.
Недостатки способа:
1. в статье авторы не приводят кинетику образования йод-лактона АК, в то время, как для других кислот (ЭПК и ДГК) она приведена даже в табличной форме, только потому, что процесс образования йод-лактона АК, предложенный авторами не совершенен и должен быть подвергнут всестороннему критическому изучению;
2. процесс получения АК очень длительный - более 2 суток (55+ часов);
3. на протяжении всего процесса используют разные температурные режимы (-25°С, +10°С, +15°С), что создает определенные сложности в аппаратурном оформлении при масштабировании;
4. при осуществлении процесса выделения АК были получены следующие результаты:
- выход 40,5% (исходя из того, что было взято 222 мг АА (1 г с содержанием 22,2% АК), а получено 92 мг 97,8% АК или на 100% АК - 90 мг);
- чистота АК - 97,8%;
полученные данные в полной мере отражают незавершенность проведенного исследования, так как большая часть АК теряется, а чистота продукта не соответствует требованиям, ныне предъявляемым к использованию в биохимии.
Задача, решаемая изобретением, - разработка простого и экономичного способа получения арахидоновой кислоты, с высоким качеством и выходом продукта при одновременном снижении затрат производства и времени технологического цикла.
Заявляемый технический результат достигается тем, что в известном способе получения арахидоновой кислоты из сырья животного происхождения, включающим щелочной гидролиз липидов, выделенных из сырья, с последующим подкислением гидролизата, проведение реакции йод-лактонизации, экстракцию йод-лактонов из реакционной смеси органическим растворителем, раскрытие йод-лактонов силирующим агентом, нейтрализацию выделившегося йода тиосульфатом натрия и экстракцию жирных кислот органическим растворителем, согласно изобретению, перед реакцией йод-лактонизации гидролизат липидов обрабатывают литиевой щелочью в ацетоне и отделяют насыщенные кислоты в виде осадка; йод-лактонизацию ведут путем перевода литиевых солей ненасыщенных жирных кислот в натриевые соли, которые обрабатывают раствором йода для образования йод-лактонов жирных кислот при соотношении суммы (арахидоновой, эйкозапентаеновой и докозагексаеновой) кислот:йод, равном 1,0:(1,0-1,2) моль/моль, при температуре смеси 18-22°С, при продолжительности добавления раствора йода к реакционной смеси 30-60 минут, с последующей выдержкой растворов в течение 60 мин.; после экстракции йод-лактонов из реакционной смеси органическим растворителем, дополнительно осуществляют разделение дельта- и гамма-йод-лактонов хроматографией на силикагеле, используя в качестве элюэнта бензол; а раскрытие дельта-йод-лактонов арахидоновой и эйкозапентаеновой кислот проводят силирующим агентом при соотношении йод-лактоны арахидоновой + эйкозапентаеновой кислот / силирующий агент = 1,0:(1,0-1,2) моль / моль, температуре смеси 18-22°С, продолжительности реакции 120-180 минут; после экстракции жирных кислот органическим растворителем их этерифицируют этиловым спиртом и выделяют арахидоновую кислоту в виде этилового эфира методом высокоэффективной жидкостной хроматографии (ВЭЖХ) на обращенной фазе (С-18) с использованием элюирующей системы этанол-вода 8,0:(1,0-2,5) об./об.
Заявленный способ до выделения чистого целевого продукта методом ВЭЖХ, в отличие от прототипа, включает 3-й этапа: 1)
осаждение в виде Li-солей насыщенной фракции; 2) проведение процесса йод-лактонизации в новых технологических условиях; 3) использование хроматографического разделение гамма- и дельта-йод-лактонов на силикагеле.
Осаждение в виде литиевых солей позволяет удалить насыщенные жирные кислоты из гидролизата, что, в конечном результате, способствует получению целевого продукта с высокой степенью чистоты. Перевод жирных кислот в Li-соли осуществляют известным методом, в частности, из ацетона по Хилдичу (Hildich Т.Р. «The Chemical constitution of natural fats». London: Chapman&Hall Ltd., 1949. - P. 471).
Проведение йод-лактонизации в предлагаемых условиях обеспечивает не только выделение из гидролизата ненасыщенных жирных кислот смеси арахидоновой, эйкозапентаеновой и докозагексаеновой кислот, поскольку только они образуют йод-лактоны, но и отделение от этой смеси докозагексаеновой кислоты, которая образует гамма-йод-лактон. Докозагексаеновую кислоту отделяют используя метод хроматографического разделения гамма- и дельта-йод-лактонов на силикагеле.
Соотношение компонентов реакционной смеси при образовании йод-лактонов (сумма арахидоновой, эйкозапентаеновой и докозагексаеновой кислот) / I2 = 1 / 1,2 моль/моль) обеспечивает высокий выход йод-лактонов ЖК. Однако при снижении количества взятого в реакцию йода выход йод-лактонов падает пропорционально его недостаче, но при избытке - более 1,2 моль 1 г на 1 моль ЖК, образующих йод-лактоны, это приводит, с одной стороны, к ненужным тратам на реактивы, а с другой, к невозвратной потере АК из-за побочных реакций. Применение такого подхода позволило достичь нового технического результата, обеспечивающего высокий выход АК при сокращении времени реакции и экономии химических реактивов.
Температурный режим реакции йод-лактонизации, 18°С-22°С, гарантирует высокий выход йод-лактонов ЖК. При снижении температуры реакции менее 18°С процесс образования замедляется и осуществление способа значительно увеличивается. При повышении температуры более 22°С протекают химические процессы ухудшения целевого продукта.
Продолжительность процесса йод-лактонизации очень важна, так как определяет необходимую меру воздействия йода на образующие ЖК йод-лактоны. Моделированием процесса было определено, что наилучшие условия для данного способа находятся в пределах 90-120 минут. Менее 90 минут, как оказалось, йод-лактонизация не проходит полностью, а свыше 120 минут никакого увеличения выхода более не происходит.
Соотношение компонентов реакционной среды при раскрытии йод-лактонов до свободных ЖК - (сумма йод-лактонов АК, ЭПК и ДГК) : силирующий агент = 1:1,2 моль/моль), в данных соотношениях гарантирует высокий выход ЖК. При снижении молярного соотношения силирующего агента менее 1 моль, раскрытие йод-лактонов ЖК резко падает, а при увеличении более 1,2 моль на 1 моль йод-лактонов ЖК, наблюдается протекание побочных реакций, вследствие чего, снижается чистота целевого продукта.
В качестве силирующего агента могут быть использованы: триметилйодсилан (TMS-I) или триметилхлорсилан (TMS-Cl). Проведение стадии раскрытия йод-лактонов жирных кислот силирующим агентом, TMS-Cl, в среде ацетонитрила, содержащего йодид натрия способствует снижению производственных затрат на получение целевого продукта (АК), в виду невысокой стоимости данных реактивов.
Температурный режим раскрытия йод-лактонов ЖК оптимален при 18-22°С. Проведение процесса менее нижнего предела (18°С) не препятствует самому процессу реакции, но делает этот процесс более долгим, а превышение температуры более 22°С не только не способствует ускорению процесса, но активирует деградацию ЖК и, в первую очередь, АК. В конечном результате, это приводит не только к увеличению длительности всего процесса получения арахидоновой кислоты, но и отрицательно отражается на ее выходе.
В качестве органического растворителя в способе получения АК при экстракции жирных кислот и йод-лактонов могут быть использованы, в частности, петролейный эфир, гексан, гептан.
Для упрощения способа и уменьшение производственных затрат на получения АК целесообразно образовавшиеся в процессе йод-лактонизации йод-лактон ЭПК, ДГК и АК экстрагировать из реакционной смеси пентаном или гексаном. Эти растворители в меньшей степени растворяются в реакционной смеси, не образуют эмульсий и имеют высокую экстракционную способность к йод-лактонам. Преимуществом использования данных растворителей является также то, что после экстракции их объединенные растворы не нужно упаривать и заново перерастворять йод-лактон ЖК в гексане для того, чтобы удалить из раствора непрореагировавшие свободные жирные кислоты. Объединенные экстракционные растворы сразу обрабатывают для удаления свободных жирных кислот.
Соблюдение заявляемых режимных параметров на всех этапах технологического процесса обеспечивает высокий выход и качество целевого продукта - АК.
Использование метода хроматографии на силикагеле позволяет отделить гамма-йод-лактоны ДГК, от дельта-йод-лактонов ЭПК и АК. Элюирование чистым бензолом в дальнейшем дает возможность многократного использования регенерированного растворителя, что делает весь процесс более рентабельным.
Окончательную очистку АК в виде этилового эфира методом ВЭЖХ проводят на обращенной фазе (С-18) с использованием водно-этанольного элюента, этанол-вода, 8,0:(1,0-2,5) об./об.
Использование водно-этанольного элюента позволяет отказаться от применения в процессе вредных веществ в отличие от прототипа, загрязняющих как целевой продукт, но и делает процесс получения АК безвредным и экологически безопасным. Предложенная новая элюирующая система позволяет выделять чистую АК в виде этилового эфира, при этом система не токсична (этанол-вода 8,0:(1,0-2,5) об./об.), хорошо регенерируется и обеспечивает получение качественного целевого препарата.
Заявленные параметры системы (этанол-вода 8,0:(1,0-2,5) об./об.) позволяют в широком диапазоне применять полярный компонент (Н2О) для увеличения общей полярности системы, причем в применяемом при ВЭЖХ изократическом режиме выделения АК не требуется изменений (градиента) в содержании элюирующей системы - от начала до конца процесса параметры сохраняются неизменными.
Данная элюирующая система обладает большими преимуществами перед использованием систем на основе метанола, так как позволяет без каких-либо ограничений применять данный метод на фармацевтических и, главное, на пищевых производствах, что открывает большие перспективы получения продукта (АК) без ядовитого метилового спирта, экологичным методом.
Доступность такой элюирующей системы не вызывает сомнений, а себестоимость ее меньше, чем традиционной с метиловым спиртом.
Наиболее предпочтительной системой для элюции является система этанол-вода при соотношении 8,0:(1,8-2,2) об./об., поскольку именно при этом соотношении компонентов наряду с получением арахидоновой кислоты с высокими степенью чистоты и выходом дополнительно сокращается время выделения АК методом ВЭЖХ.
Соблюдение заявляемых режимных параметров на всех этапах технологического процесса обеспечивает достижение заявленного технического результата - получения АК с высоким качеством и выходом простым и экономичным способа при одновременном снижении затрат производства и времени технологического цикла.
Заявленный способ иллюстрируется следующими фиг.: на фиг. 1 представлен каскад арахидоновой кислоты; на фиг. 2 - схема реакции образования и раскрытия йод-лактона арахидоновой кислоты.
Способ осуществляют следующим образом.
Экстракция
Сырье животного происхождения измельчали на гомогенизаторе и экстрагировали липиды смесью этанол-хлороформ 1:1 об./об., в соотношении печень - экстракционная смесь 1:3 кг/л. Далее экстракт фильтровали через плотную ткань, осадок отжимали. Отделенный осадок повторно экстрагировали таким же объемом хлороформа. Оба фильтрата объединяли, приупаривали, добавляли равный объем воды и отбирали хлороформный слой. Водно-спиртовую часть повторно экстрагировали хлороформом. Экстракты объединяли и упаривали.
Гидролиз липидов
Липиды гидролизовали 7%-ным раствором КОН в 50%-ом этаноле в соотношении липиды - раствор, 1 : 4, кг/л, при температуре 50°С в течение 60 мин. Смесь охлаждали и нейтрализовали щелочь концентрированной соляной кислотой до рН 3. Далее разбавляли в 2-а раза водой и экстрагировали жирные кислоты и др. липиды петролейным эфиром, упаривали.
Осаждение ЖК в виде Li-солей
Насыщенные жирные кислоты удаляли процедурой осаждения в виде литиевых солей из ацетона по Хилдичу (Hildich Т.P. «The Chemical constitution of natural fats». London: Chapman&Hall Ltd., 1949. P. 471).
Гидролизованные липиды растворяли в 4-х кратном объеме чистого ацетона. Добавляли 2 мл 2%-ного раствора фенолфталеина в ацетоне и при нагреве (40°С) оттитровывали свободные жирные кислоты насыщенным водным раствором LiOH (≈ 4N) до появления характерной малиновой окраски. Фиксировали объем использованной литиевой щелочи. Далее, к смеси добавляли ацетон до содержания в ней воды 5% по объему. Смесь охлаждали до комнатной температуры, выдерживали 1 час, осадок отфильтровывали на бумажном фильтре, ацетоновый фильтрат подкисляли соляной кислотой до рН 3, упаривали.
Йод-лактонизация
Реакцию йод-лактонизации ЖК проводили в расчете на все жирные кислоты, образующие лактоны.
Концентрат жирных кислот и др. липидов растворяли в 5-ти кратном объеме 95% этанола и для перевода свободных жирных кислот в Na-соли добавляли по стехиометрии водный раствор бикарбоната натрия.
Далее по стехиометрии рассчитывали необходимое количество йода растворяли в 95%-ном этиловом спирте до концентрации раствора 5% и добавляли из делительной воронки порциями к перемешиваемой смеси солей ЖК в соотношении (АК+ЭПК+ДГК) / I2 = 1,0 / 1-1,2 моль/моль, при комнатной температуре в течение 30 минут, после добавления выдерживали еще 90 минут.
Непрореагировавший йод нейтрализовали до бесцветной окраски насыщенным водным раствором тиосульфата натрия, разбавляли смесь 2-м объемом воды и экстрагировали йод-лактоны ЖК три раза неполярным органическим растворителем. Объединенные экстракты упаривали в два раза и экстрагировали не образовавшие йод-лактоны ЖК 2%-ным NaCO3 в 40%-ном этаноле. Слой органического растворителя промывали водой, подкисляли концентрированной соляной кислотой до рН 3, фильтровали через сульфат натрия для обезвоживания, упаривали.
Хроматографическое разделение йод-лактонов
Силикагель, 100-160 мкм, суспендировали в бензоле, наносили на хроматографическую колонку, загружали суммарные йод-лактоны ЖК (соотношение йод-лактоны ЖК : силикагель = 1:10 г/г) и элюировали бензолом. Контроль осуществляли ТСХ.
Фракции с чистым δ-йод-лактоном АК объединяли и упаривали.
Раскрытие йод-лактона АК
Раскрытие йод-лактона АК проводили в 3%-ном растворе безводного NaI в ацетонитриле. К смеси при перемешивании добавляли TMSi-Cl в соотношении йод-лактоны ЖК - TMSi-Cl = 1,0:1-1,2 моль / моль, по каплям, при общей продолжительности реакции 120-180 минут, после чего выделившийся йод нейтрализовали насыщенным водным раствором тиосульфата натрия, смесь разбавляли 4-мя объемами воды и экстрагировали липиды неполярным растворителем.
Свободные ЖК экстрагировали из неполярного растворителя 2%-ным NaCO3 в 40%-ном этаноле, промывали щелочной экстракт неполярным растворителем, добавляли равный объем воды, подкисляли концентрированной соляной кислотой до рН 3 и экстрагировали свободные ЖК неполярным растворителем, экстракт высушивали, фильтруя через сульфат натрия, упаривали.
Получение этиловых эфиров ЖК
Жирные кислоты вносили в 1%-ный раствор серной кислоты в абсолютированном этаноле при соотношении ЖК : этанол 1:10 об./об. Смесь нагревали (50°С) при перемешивании в течение 1,5 часов, далее разбавляли 3-мя объемами воды и экстрагировали этиловые эфиры ЖК неполярным растворителем.
Упаренный экстракт очищали на колонке с силикагелем, элюент - неполярный растворитель.
Выделение АК в виде этилового эфира методом ВЭЖХ
Разделение смеси этиловых эфиров ЖК осуществляли на колонке «Supelco Discovery HS С-18», 250 мм × 50 мм i.d. (размер частиц 10 мкм) на жидкостном хроматографе «Shimadzu-LC8A» с UV-детектором SPD-20A. Элюирование осуществляли предварительно дегазированной и отфильтрованной системой растворителей этанол-вода 8:(1,0-2,5) об./об., при скорости 50 мл/мин.
Способ иллюстрируется следующими фигурами: фиг. 1, на которой представлен каскад арахидоновой кислоты, фиг. 2. - представлена схема реакции образования и раскрытия йод-лактона арахидоновой кислоты, подтвержденная примерами конкретного осуществления способа.
Пример 1
В эксперименте была использована печень крупного рогатого скота. До использования весь материал хранили при температуре минус 20°С.
Все используемые реактивы и растворители были марки «хч»; силикагель производства «Chemapol» (Чехия), безводный NaI («Sigma-Aldrich», США), триметилхлорсилан, TMSi-Cl («Acros Organics», Бельгия), гидроксид лития, этанол и петролейный эфир, 40°-60°С («Реагент», Москва).
Тонкослойную хроматографию (ТСХ) липидов для контроля за процессами осуществляли на пластинках с закрепленным слоем силикагеля («Sorbfil», Россия) с элюирующими системами: 1) 80:20:1, гексан-диэтиловый эфир-уксусная кислота, об./об./об., для определения состава нейтральных липидов, 2) 2-х кратное элюирование бензолом для идентификации йод-лактонов ЖК.
Метиловые жирных кислот липидов для ГЖХ-анализа получали согласно методике (Carreau J.P., Dubacq J.P. «Adaptation of a macro-scale method to the micro-scale for fatty acid methyl transesterification of biological lipid extracts» // J. Chromatogr. - 1978. - Vol. 151, №3. - 384-390). Анализ метиловых эфиров жирных кислот проводили газовой хроматографией на хроматографе Shimadzu GC-17А («Shimadzu», Kyoto, Japan) с пламенно-ионизационным детектором и капиллярной колонкой 30 m × 0.25 mm i.d. «Supelcowax 10» (Bellefonte, USA), условия анализа: температура колонки 190°С, инжектора и детектора 240°С, газ-носитель гелий. Идентификацию пиков метиловых (этиловых) эфиров жирных кислот проводили по временам удерживания индивидуальных эфиров жирных кислот и с использованием стандартных смесей ПНЖК («Supelco», Bellefonte, USA) и с использованием значений эквивалентной длины цепи (Christie W.W. «Lipid Analysis: Isolation, separation, identification and structural analysis of lipids». - The oily Press, Bridgwater (UK), 2003. - 416 p.).
Для экстракции липидов замороженную говяжью печень (500 г) измельчали. на гомогенизаторе и экстрагировали липиды смесью этанол-хлороформ 1:1 об./об. 1,5 л. Далее экстракт фильтровали через плотную ткань, осадок отжимали. Отделенный осадок повторно экстрагировали 1,5 л хлороформа. Оба фильтрата объединяли, приупаривали до 2 л, добавляли равный объем воды и отбирали хлороформный слой. Водно-спиртовую часть повторно экстрагировали хлороформом, 500 мл. Экстракты объединяли и упаривали. Получено 52,5 г липидов.
Для гидролиза липидов с предыдущей стадии использовали 210 мл 7%-ного раствора КОН в 50%-ом этаноле при температуре 50°С в течение 60 минут. Смесь охлаждали и нейтрализовали щелочь концентрированной соляной кислотой до рН 3. Далее разбавляли водой, 500 мл, и экстрагировали жирные кислоты и другие липиды петролейным эфиром, 2 × 200 мл, упаривали. Получено 45,6 г ЖК.
Насыщенные жирные кислоты удаляли из смеси ЖК процедурой осаждения в виде литиевых солей из ацетона следующим образом: гидролизованные липиды растворяли в 180 мл ацетона, добавляли 2 мл 2%-ного раствора фенолфталеина в ацетоне и при нагреве (40°С) оттитровывали свободные жирные кислоты насыщенным водным раствором LiOH (≈ 4N), 36 мл, до появления характерной малиновой окраски. Далее, к смеси добавляли еще 500 мл ацетона. Смесь охлаждали до - комнатной температуры, выдерживали 1 час, осадок отфильтровывали на бумажном фильтре, ацетоновый фильтрат подкисляли соляной кислотой до рН 3, упаривали. Получено 25,4 г ЖК.
Реакцию йод-лактонизации ЖК проводили в расчете на все жирные кислоты, образующие лактоны.
Концентрат жирных кислот и др. липидов, 25,4 г, растворяли в 125 мл 95% этанола и для перевода свободных жирных кислот в Na-соли добавляли по стехиометрии водный раствор бикарбоната натрия (7 г в 150 мл).
Далее по стехиометрии рассчитывали необходимое количество йода ((АК+ЭПК+ДГК) / I2 = 1,0 / 1,0 моль/моль), 4,1 г йода растворяли в 8 0 мл 95%-ного этилового спирта и добавляли из делительной воронки порциями к перемешиваемой смеси солей ЖК при температуре 18°С в течение 30 минут, после добавления выдерживали еще 60 минут.
Непрореагировавший йод нейтрализовали до бесцветной окраски насыщенным водным раствором тиосульфата натрия (20 г в 100 мл воды), разбавляли смесь 400 мл воды и экстрагировали йод-лактоны ЖК 3-й раза петролейным эфиром по 150 мл. Объединенные экстракты приупаривали в 2 раза и экстрагировали не образовавшие йод-лактоны ЖК 2%-ным NaCO3 в 40%-ном этаноле, три раз по 100 мл. Петролейный слой промывали 200 мл воды, подкисляли концентрированной соляной кислотой до рН 3, фильтровали через сульфат натрия для обезвоживания, упаривали. Получено 6,96 г йод-лактонов ЖК.
Хроматографическое разделение йод-лактонов ЖК проводили на силикагеле, 100-160 мкм, суспендированном в бензоле: наносили силикагель на хроматографическую стеклянную колонку (50 см × 4 см), загружали суммарные йод-лактоны ЖК, 6,9 г, (соотношение йод-лактоны ЖК - силикагель, 1 : 10, г / г) и элюировали бензолом. Контроль осуществляли ТСХ. Фракции с чистым δ-йод-лактонами. АК и ЭПК объединяли и упаривали. Получено 5,54 г δ-йод-лактонов ЖК.
Раскрытие δ-йод-лактонов ЖК, 5,54 г, проводили в растворе безводного NaI - 6 г соли в 200 мл ацетонитрила. К смеси при перемешивании добавляли TMSi-Cl, взятый по стехиометрии в соотношении йод-лактоны ЖК - TMSi-Cl = 1,0 : 1,0, моль / моль, по каплям в течение 60 минут, затем выдерживали еще 60 минут, после чего выделившийся йод нейтрализовали насыщенным водным раствором тиосульфата натрия (10 г в 100 мл воды), смесь разбавляли 1,2 л воды и экстрагировали липиды петролейным эфиром, 3 × 150 мл.
Свободные ЖК экстрагировали из петролейного эфира 2%-ным NaCO3 в 40%-ном этаноле, 3 × 100 мл, промывали щелочной экстракт 100 мл петролейного эфира, добавляли 300 мл воды, подкисляли концентрированной соляной кислотой до рН 3 и экстрагировали свободные ЖК петролейным эфиром, 3 × 100 мл, экстракт высушивали, фильтруя через сульфат натрия, упаривали. Получено 3,93 г ЖК.
Жирные кислоты, 3,93 г, переводили в этиловые эфиры ЖК внесением их в 1%-ный раствор серной кислоты в абсолютированном этаноле, 40 мл. Смесь нагревали (50°С) при перемешивании в течение 1,5 часов, далее разбавляли 220 мл воды и экстрагировали этиловые эфиры ЖК петролейным эфиром, 3 × 100 мл.
Упаренный экстракт очищали на колонке с силикагелем (10 г силикагеля суспендированного в бензоле), элюент - петролейный эфир, упаривали. Получено 3,85 г этиловых эфиров ЖК.
Разделение смеси ЭЭ-ЖК, 3,80 г, осуществляли методом ВЭЖХ на колонке «Supelco Discovery HS С-18», 250 мм × 50 мм i.d. (размер частиц 10 мкм). Элюирование осуществляли предварительно - дегазированной и отфильтрованной системой растворителей этанол-вода 8,0:1,0 об./об. при скорости 50 мл/мин. Количество смеси ЖК в виде этиловых эфиров, наносимых на колонку, составило 1 мл (0,95 г), расход элюирующей системы на одно разделение составлял в среднем 2,7 л. Процедура была повторена 4-е раза.
После разделения, фракции с чистой АК в виде этилового эфира упаривали.
Получена АК в виде этилового эфира, 3,43 г, с чистотой 99,67%. Общий выход, в расчете на содержание АК в исходном сырье, 50,9%.
Общее время осуществления способа 12 часов.
Пример 2
Сырье, реактивы, растворители и аналитические методы, используемые в Примере 2, были как в Примере 1.
Для экстракции липидов замороженную говяжью печень (500 г) измельчали на гомогенизаторе и экстрагировали липиды смесью этанол-хлороформ 1,0:1,2 об./об. 1,5 л. Далее экстракт фильтровали через плотную ткань, осадок отжимали. Отделенный осадок повторно экстрагировали 1,5 л хлороформа. Оба фильтрата объединяли, приупаривали до 2 л, добавляли равный объем воды и отбирали хлороформный слой. Водно-спиртовую часть повторно экстрагировали хлороформом, 500 мл. Экстракты объединяли и упаривали. Получено 52,1 г липидов.
Для гидролиза липидов с предыдущей стадии использовали 210 мл 7%-ный раствор КОН в 50%-ом этаноле при температуре 50°С в течение 60 мин. Смесь охлаждали и нейтрализовали щелочь концентрированной соляной кислотой до рН=3. Далее разбавляли водой, 500 мл, и экстрагировали жирные кислоты и другие липиды петролейным эфиром, 2 × 200 мл, упаривали. Получено 4 5,3 г ЖК.
Насыщенные жирные кислоты удаляли из смеси ЖК процедурой осаждения в виде литиевых солей из ацетона следующим образом: гидролизованные липиды растворяли в 180 мл ацетона, добавляли 2 мл 2%-ного раствора фенолфталеина в ацетоне и при нагреве (40°С) оттитровывали свободные жирные кислоты насыщенным водным раствором LiOH (≈ 4N), 36 мл, до появления характерной малиновой окраски. Далее, к смеси добавляли еще 500 мл ацетона. Смесь охлаждали до комнатной температуры, выдерживали 1 час, осадок отфильтровывали на бумажном фильтре, ацетоновый фильтрат подкисляли соляной кислотой до рН 3, упаривали. Получено 25,2 г ЖК.
Реакцию йод-лактонизации ЖК проводили в расчете на все жирные кислоты, образующие лактоны.
Концентрат жирных кислот и др. липидов, 25,2 г, растворяли в 125 мл 95% этанола и для перевода свободных жирных кислот в Na-соли добавляли водный раствор бикарбоната натрия (7 г в 150 мл).
Далее рассчитывали необходимое количество йода
(АК+ЭПК+ДГК) / I2 = 1,0 / 1,2, моль/моль), 4,9 г йода растворяли в 100 мл 95%-ного этилового спирта и добавляли из делительной воронки порциями к перемешиваемой смеси солей ЖК при 22°С в течение 60 минут, после добавления выдерживали еще 60 минут.
Непрореагировавший йод нейтрализовали до бесцветной окраски насыщенным водным раствором тиосульфата натрия (20 г в 100 мл воды), разбавляли смесь 400 мл воды и экстрагировали йод-лактоны ЖК 3-й раза петролейным эфиром по 150 мл. Объединенные экстракты приупаривали в 2 раза и экстрагировали не образовавшие йод-лактоны ЖК 2%-ным NaCO3 в 40%-ном этаноле, 3 × 100 мл. Петролейный слой - промывали водой, 200 мл, подкисляли концентрированной соляной кислотой до рН=3, фильтровали через сульфат натрия для обезвоживания, упаривали. Получено 6,89 г йод-лактонов ЖК.
Хроматографическое разделение йод-лактонов проводили на силикагеле, 100-160 мкм, 69 г, суспендированном в бензоле: наносили силикагель на хроматографическую стеклянную колонку (50 см × 4 см), загружали суммарные йод-лактоны ЖК, 6,89 г, и элюировали бензолом. Контроль осуществляли ТСХ. Фракции с чистым δ-йод-лактонами АК и ЭПК объединяли и упаривали. Получено 5,50 г δ-йод-лактонов ЖК.
Раскрытие δ-йод-лактонов ЖК, 5,50 г, проводили в растворе безводного NaI - 6 г соли в 200 мл ацетонитрила. К смеси при перемешивании добавляли TMSi-Cl, взятый в соотношении йод-лактоны ЖК - TMSi-Cl = 1,0 : 1,2 моль/моль, по каплям в течение 120 минут, затем выдерживали 60 минут, после чего выделившийся йод нейтрализовали насыщенным водным раствором тиосульфата натрия (10 г в 100 мл воды), смесь разбавляли 1,2 л воды и экстрагировали липиды петролейным эфиром, 3 × 150 мл.
Свободные ЖК экстрагировали из петролейного эфира 2%-ным NaCO3 в 40%-ном этаноле, 3 × 100 мл, промывали щелочной экстракт петролейным эфиром, 100 мл, добавляли 300 мл воды, подкисляли концентрированной соляной кислотой до рН=3 и экстрагировали свободные ЖК петролейным эфиром, 3 × 100 мл, экстракт высушивали, фильтруя через сульфат натрия, упаривали. Получено 3,87 г ЖК.
Жирные кислоты, 3,87 г, переводили в этиловые эфиры ЖК внесением их в 1%-ный раствор серной кислоты в абсолютированном этаноле, 40 мл. Смесь нагревали (50°С) при перемешивании в течение 1,5 часов, далее разбавляли 220 мл воды и экстрагировали этиловые эфиры ЖК петролейным эфиром, 3 × 100 мл.
Упаренный экстракт очищали на колонке с силикагелем (10 г силикагеля суспендированного в бензоле), элюент - петролейный эфир, упаривали. Получено 3,76 г этиловых эфиров ЖК.
Разделение смеси ЭЭ-ЖК, 3,76 г, осуществляли методом ВЭЖХ на колонке «Supelco Discovery HS С-18», 250 мм × 50 мм i.d. (размер частиц 10 мкм). Элюирование осуществляли предварительно дегазированной и отфильтрованной системой растворителей этанол-вода 8,0:2,2 об./об. при скорости 50 мл/мин. Количество смеси ЖК в виде этиловых эфиров, наносимых на колонку, составило 0,94 г, расход элюирующей системы на одно разделение составлял в среднем 2,7 л. Процедура была повторена 4-е раза.
После разделения, фракции с чистой АК в виде этилового эфира упаривали.
Получена АК в виде этилового эфира, 3,38 г, с чистотой 99,18%. Общий выход, в расчете на содержание АК в исходном сырье, 50,1%.
Общее время осуществления способа 12,1 часов.
В таблице приведено поэтапное концентрирование АК из Примеров 1 и 2, а именно состав концентратов АК из сырья животного происхождения (Примеры 1 и 2)
Цифры по получению АК, приведенные в таблице, однозначно показывают, что способ обеспечивает получение АК в виде этилового эфира - с чистотой 99,18-99,67% и общим выходом в расчете на j содержание АК в исходном сырье - 50,1-56,2%. Осуществление данного способа решает проблемы, существующие на данный момент при получении АК в лабораторных и заводских производствах. Наиболее важным показателем выступает чистота продукта - свыше 99%, что позволяет АК использовать АК для очень тонких ферментативных синтезов, где недопустимо присутствие других кислот из-за побочных продуктов реакции, что при других способах осуществления требует дополнительных затрат по очистке полученных соединений. Суммарный выход по АК в способе - более 50%, это больше, чем в других аналогах при получении высокообогащенной АК.
Пример 3
Исходным сырьем в данном опыте служила печень крысы (лабораторные животные) - 500 г.
Содержание арахидоновой кислоты от общей суммы ЖК липидов печени - 14,52%, эйкозапентаеновой - 1,33%, докозагексаеновой - 3, 81%.
Растворители, реактивы и аналитические процедуры, как в Примере 1.
Экстракцию липидов, гидролиз, осаждение насыщенных жирных кислот в виде Li-солей, реакцию йод-лактонизации ЖК, хроматографическое разделение гамма- и дельта-йод-лактонов ЖК, раскрытие дельта-йод-лактонов ЖК до свободных жирных кислот, из этерификацию до получения этиловых эфиров ЖК и дальнейшее разделение методом ВЭЖХ проводили, как в Примере 1, за исключением того, что в качестве неполярного растворителя при экстракции йод-лактонов жирных кислот и ЖК после раскрытия йод-лактонов использовали петролейный эфир, а элюирование осуществляли системой растворителей этанол-вода 8,0:1,0 об./об.
Получена АК в виде этилового эфира, 4,56 г, с чистотой 99,43%.
Общий выход, в расчете на содержание АК в исходном сырье, 55,4%.
Общее время осуществления способа 13,0 часов.
Пример 4
Исходным сырьем в данном опыте служила печень мыши (лабораторные животные) - 50 г.
Содержание арахидоновой кислоты от общей суммы ЖК липидов печени - 8,03%, эйкозапентаеновой - 1,62%, докозагексаеновой 14,13%.
Растворители, реактивы и аналитические процедуры, как в Примере 2.
Экстракцию липидов, гидролиз, осаждение насыщенных жирных кислот в виде Li-солей, реакцию йод-лактонизации ЖК, хроматографическое разделение гамма- и дельта-йод-лактонов ЖК, раскрытие дельта-йод-лактонов ЖК до свободных жирных кислот, из этерификацию до получения этиловых эфиров ЖК и дальнейшее разделение методом ВЭЖХ проводили, как в Примере 1, за исключением того, что в качестве неполярного растворителя при экстракции йод-лактонов жирных кислот и ЖК после раскрытия йод-лактонов использовали пентан, раскрытие дельта-йод-лактонов ЖК проводили триметилйодсиланом в соотношении дельта-йод-лактоны ЖК:триметилйодсилан = 1:1 моль / моль, а элюирование осуществляли системой растворителей этанол-вода 8,0:2,0 об./об.
Получена АК в виде этилового эфира, 0,44 г, с чистотой 99,55%.
Общий выход, в расчете на содержание АК в исходном сырье, - 51,6%.
Общее время осуществления способа 11,0 часов.
Пример 5
Исходным сырьем в данном опыте служила продажная печень северного оленя - 500 г.
Содержание арахидоновой кислоты от общей суммы ЖК липидов печени - 10,66%, эйкозапентаеновой - 0,76%, докозагексаеновой 1,27%.
Растворители, реактивы и аналитические процедуры, как в Примере 1.
Экстракцию липидов, гидролиз, осаждение насыщенных жирных кислот в виде Li-солей, реакцию йод-лактонизации ЖК, хроматографическое разделение гамма- и дельта-йод-лактонов ЖК, раскрытие дельта-йод-лактонов ЖК до свободных жирных кислот, из этерификацию до получения этиловых эфиров ЖК и дальнейшее разделение методом ВЭЖХ проводили, как в Примере 1, за исключением того, что в качестве неполярного растворителя при экстракции йод-лактонов жирных кислот и ЖК после раскрытия йод-лактонов использовали гексан, а элюирование осуществляли системой растворителей этанол-вода 8,0:2,5 об./об.
Получена АК в виде этилового эфира, 4,41 г, с чистотой 99,60%.
Общий выход, в расчете на содержание АК в исходном сырье, 56,2%.
Общее время осуществления способа 13,8 часов.
Таким образом, заявителем предложен несложный и воспроизводимый способ выделения чистой АК из тканей животного происхождения, доступный для реализации в лабораториях и промышленных производствах, использующих АК в биохимических, медицинских и диетологических исследованиях.
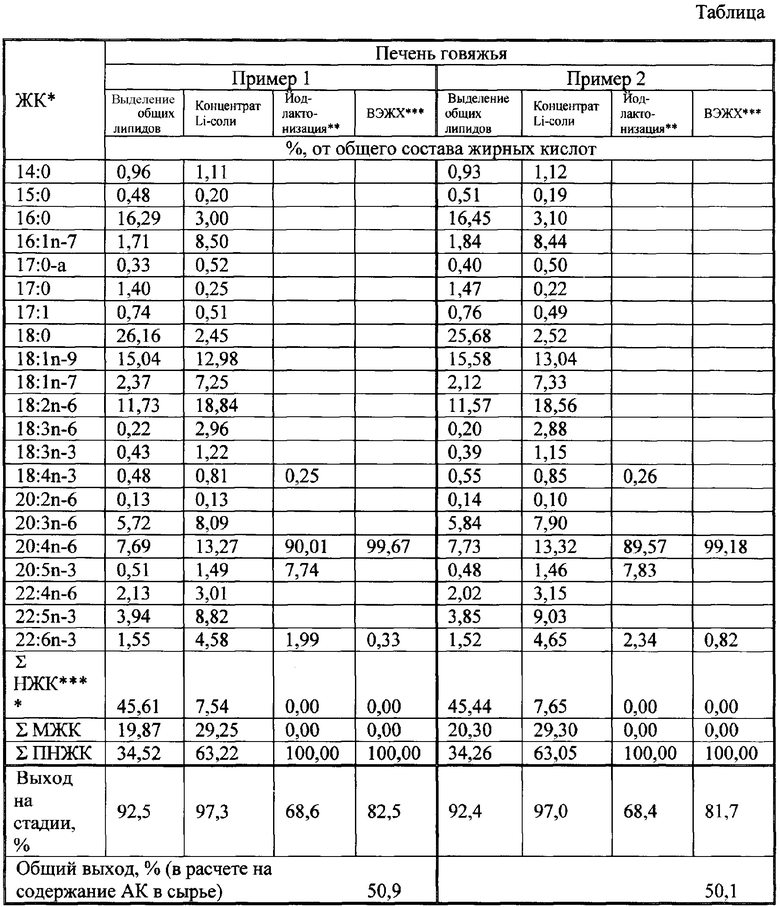
* - первая цифра обозначает количество углеродных атомов в цепи жирной кислоты, вторая - количество ненасыщенных связей, n-3 или др. - расположение первой двойной связи от метальной группы;
** - выход приведен на 3-й процедуре - йод-лактонизацию, хроматографическое разделение лактонов и их раскрытие;
*** - выход приведен на 2-е процедуры - получение этиловых эфиров ЖК и их разделение ВЭЖХ.
**** - Σ НЖК - сумма насыщенных жирных кислот, Σ МЖК - сумма мононенасыщенных жирных кислот, Σ ПНЖК - сумма полиненасыщенных жирных кислот.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДОКОЗАГЕКСАЕНОВОЙ КИСЛОТЫ | 2013 |
|
RU2537252C1 |
| Способ получения арахидоновой кислоты из морской красной водоросли рода Gracilaria | 2016 |
|
RU2620164C1 |
| Способ получения докозагексаеновой кислоты | 2019 |
|
RU2698720C1 |
| Способ получения докозагексаеновой, эйкозапентаеновой и арахидоновой кислот или их смеси | 1988 |
|
SU1631067A1 |
| Способ получения арахидоновой кислоты | 1977 |
|
SU897766A1 |
| СМЕСЬ ОМЕГА-3 ПОЛИНЕНАСЫЩЕННЫХ ЖИРНЫХ КИСЛОТ | 1995 |
|
RU2127115C1 |
| Способ получения концентрата ненасыщенных алкил-глицериновых эфиров из морских гидробионтов | 2017 |
|
RU2642294C1 |
| СПОСОБ ИДЕНТИФИКАЦИИ СРЕДЫ ОБИТАНИЯ ОСЕТРОВЫХ РЫБ С ПОМОЩЬЮ БИОХИМИЧЕСКИХ МАРКЕРОВ | 2021 |
|
RU2779424C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИПИДОВ (ВАРИАНТЫ) И ЛИПИДЫ, ПОЛУЧЕННЫЕ ЭТИМ СПОСОБОМ | 2001 |
|
RU2336307C2 |
| КОМПОЗИЦИЯ ДЛЯ ИММУНИЗАЦИИ РАСТЕНИЙ ОТ РАЗЛИЧНЫХ ФИТОПАТОГЕНОВ | 1994 |
|
RU2075933C1 |
Изобретение относится к пищевой и фармацевтической промышленности и может быть использовано для получения композиций с высоким содержанием арахидоновой кислоты (АК), обладающей выраженным биологическим действием. Заявленный способ получения арахидоновой кислоты из сырья животного происхождения включает щелочной гидролиз липидов, выделенных из сырья, с последующим подкислением гидролизата, проведение реакции йод-лактонизации, экстракцию йод-лактонов из реакционной смеси органическим растворителем, раскрытие йод-лактонов силирующим агентом, нейтрализацию выделившегося йода тиосульфатом натрия, экстракцию жирных кислот органическим растворителем, где перед реакцией йод-лактонизации гидролизат липидов обрабатывают литиевой щелочью в ацетоне и отделяют насыщенные кислоты в виде осадка, йод-лактонизацию ведут путем перевода литиевых солей ненасыщенных жирных кислот в натриевые соли, которые обрабатывают раствором йода для образования йод-лактонов жирных кислот при соотношении суммы (арахидоновой, эйкозапентаеновой и докозагексаеновой) кислот:йод, равном 1,0 : 1,0-1,2 моль/моль, при температуре смеси 18-22°С, при продолжительности добавления раствора йода к реакционной смеси 30-60 минут с последующей выдержкой растворов в течение 60 мин; после экстракции йод-лактонов из реакционной смеси органическим растворителем дополнительно осуществляют разделение дельта- и гамма-йод-лактонов хроматографией на силикагеле, используя в качестве элюэнта бензол; а раскрытие дельта-йод-лактонов арахидоновой и эйкозапентаеновой кислот проводят силирующим агентом при соотношении йод-лактоны арахидоновой + эйкозапентаеновой кислот / силирующий агент = 1,0 : 1,0-1,2, моль / моль, температуре смеси 18-22°С, продолжительности реакции 120-180 минут; после экстракции жирных кислот органическим растворителем их этерифицируют этиловым спиртом и выделяют арахидоновую кислоту в виде этилового эфира методом высокоэффективной жидкостной хроматографии на обращенной фазе (С-18) с использованием элюирующей системы этанол-вода 8,0 : 1,0-2,5 об./об. В изобретении предложен несложный и воспроизводимый способ выделения чистой АК из тканей животного происхождения, доступный для реализации в лабораториях и промышленных производствах, использующих АК в биохимических, медицинских и диетологических исследованиях. 3 з.п. ф-лы, 2 ил., 1 табл., 5 пр.
1. Способ получения арахидоновой кислоты из сырья животного происхождения, включающий щелочной гидролиз липидов, выделенных из сырья, с последующим подкислением гидролизата, проведение реакции йод-лактонизации, экстракцию йод-лактонов из реакционной смеси органическим растворителем, раскрытие йод-лактонов силирующим агентом, нейтрализацию выделившегося йода тиосульфатом натрия, экстракцию жирных кислот органическим растворителем, отличающийся тем, что перед реакцией йод-лактонизации гидролизат липидов обрабатывают литиевой щелочью в ацетоне и отделяют насыщенные кислоты в виде осадка, йод-лактонизацию ведут путем перевода литиевых солей ненасыщенных жирных кислот в натриевые соли, которые обрабатывают раствором йода для образования йод-лактонов жирных кислот при соотношении суммы (арахидоновой, эйкозапентаеновой и докозагексаеновой) кислот:йод, равном 1,0:(1,0-1,2) моль/моль, при температуре смеси 18-22°С, при продолжительности добавления раствора йода к реакционной смеси 30-60 минут с последующей выдержкой растворов в течение 60 мин; после экстракции йод-лактонов из реакционной смеси органическим растворителем дополнительно осуществляют разделение дельта- и гамма-йод-лактонов хроматографией на силикагеле, используя в качестве элюэнта бензол; а раскрытие дельта-йод-лактонов арахидоновой и эйкозапентаеновой кислот проводят силирующим агентом при соотношении йод-лактоны арахидоновой + эйкозапентаеновой кислот / силирующий агент = 1,0:(1,0-1,2) моль/моль, температуре смеси 18-22°С, продолжительности реакции 120-180 минут; после экстракции жирных кислот органическим растворителем их этерифицируют этиловым спиртом и выделяют арахидоновую кислоту в виде этилового эфира методом высокоэффективной жидкостной хроматографии на обращенной фазе (С-18) с использованием элюирующей системы этанол-вода 8,0:(1,0-2,5) об./об.
2. Способ по п. 1, отличающийся тем, что в качестве силирующего агента для раскрытия йод-лактонов арахидоновой и эйкозапентаеновой кислот используют трихлорсилан или триметилйодсилан.
3. Способ по п. 1 или 2, отличающийся тем, что раскрытие йод-лактонов арахидоновой и эйкозапентаеновой кислот трихлорсиланом ведут в присутствии йодида натрия в растворе ацетонитрила.
4. Способ по п. 1, отличающийся тем, что при осуществлении высокоэффективной жидкостной хроматографии на обращенной фазе (С-18) преимущественно используют элюирующую систему этанол-вода 8,0:(1,8-2,2) об./об.
| N.V | |||
| Gaiday et al | |||
| "Separation of Natural Polyunsaturated Fatty Acids by Means of Iodolactonization" JAOCS, v | |||
| Способ получения смеси хлоргидратов опийных алкалоидов (пантопона) из опийных вытяжек с любым содержанием морфия | 1921 |
|
SU68A1 |
| Канальная печь-сушильня | 1920 |
|
SU230A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОКОЗАГЕКСАЕНОВОЙ КИСЛОТЫ | 2013 |
|
RU2537252C1 |
| Способ получения докозагексаеновой, эйкозапентаеновой и арахидоновой кислот или их смеси | 1988 |
|
SU1631067A1 |
| N.A | |||
| Latyshev et al | |||
| "Concentration and purification of polyunsaturated fatty acids from squid liver processing wastes" Eur | |||
| J | |||
| Lipid Sci | |||
| Technol., 2014, 116, 1608-1613. | |||

