Изобретение относится к соединениям формулы (I)

в которой
Х представляет собой СН, CF, S или N,
Y представляет собой СН, S или N,
Z представляет собой С или N,
 образует, если Z=С, двойную связь совместно с простой связью,
образует, если Z=С, двойную связь совместно с простой связью,
отсутствует, если Z=N,
n равен 1 или 2, где
если n=1, X=S,
и если n=2, оба Х=СН, или X, связанный с пиримидиновым кольцом, представляет собой CF и X, несвязанный с пиримидиновым кольцом, представляет собой СН, или один Х представляет собой СН, а другой Х представляет собой N;
m равен 1 или 2,
где если m=1, Y=S,
и если m=2, оба Y=СН, или один Y представляет собой СН, а другой Y представляет собой N;
R1, R2, R3, R4, независимо друг от друга, представляют собой Н, Hal, CN, ОН, CONH2, CONH(LA) или LA;
R5 представляет собой Н, Hal, CN или С≡СН;
Сус представляет собой фенил, который может быть незамещен или моно- или дизамещен, независимо друг от друга, с помощью R6, или представляет собой Het1;
Het1 представляет собой моно- или бициклический, 5-10-членный гетероцикл, имеющий 1-3 атома N, О и/или S, или 1-4 атома N, который может быть незамещен или моно-, ди- или тризамещен, независимо друг от друга, с помощью R6, или может быть монозамещен с помощью Het2;
R6 представляет собой Hal, LA, оксо, CN, или NH2;
LA представляет собой неразветвленный или разветвленный алкил, имеющий 1-5 атомов С, который может быть насыщенным или частично ненасыщенным, в котором 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две СН2 группы могут быть заменены О, NH, NH2, N(CH3) или СО;
Het2 представляет собой 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атома N, О и/или S, который незамещен;
Hal представляет собой F, Cl, Br или I;
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Соединения формулы (I) можно использовать для ингибирования серин/треонин протеинкиназ и для сенсибилизации злокачественных клеток к противораковым средствам и/или ионизирующему излучению. Изобретение также относится к применению соединений формулы (I) для профилактики, терапии или контроля прогрессирования злокачественного новообразования, опухолей или метастаз, в комбинации с лучевой терапией и/или противораковым средством. Кроме того, изобретение относится к способу получения соединений формулы (I) путем взаимодействия соединений формул (IV) и (V) и необязательно превращения основания или кислоты соединений формулы (I) в его соль.
ДНК-зависимая протеинкиназа (ДНК-ПК) представляет собой серин/треонин протеинкиназу, которая активируется в комплексе с ДНК. Биохимические и генетические данные свидетельствуют о том, что ДНК-ПК состоит из (а) каталитической субъединицы, которая называется ДНК-ПКкс, и (б) двух регуляторных компонентов (Ku70 и Ku80). По функциональным показателям, ДНК-ПК является чрезвычайно важной составляющей, с одной стороны, репарации двухцепочечных разрывов ДНК (ДЦР), а с другой стороны, соматической или V(D)J рекомбинации. Дополнительно, ДНК-ПК и ее компоненты связаны с множеством других физиологических процессов, включая модуляцию структуры хроматина и поддержание теломер (Smith & Jackson (1999) Genes и Dev 13: 916; Goytisolo и др. (2001) Mol. Cell. Biol. 21: 3642; Williams и др. (2009) Cancer Res. 69: 2100).
Генетический материал человека в форме ДНК постоянно подвержен атакам активных форм кислорода (АФК), которые образуются главным образом в качестве побочных продуктов окислительного метаболизма. АФК способны вызывать повреждение ДНК в форме одноцепочечных разрывов. Двухцепочечные разрывы могут возникать, если предшествующие одноцепочечные разрывы происходят в непосредственной близости. Дополнительно, одно- и двухцепочечные разрывы могут возникать, если ДНК репликативная вилка наталкивается на поврежденные структуры оснований. Кроме того, экзогенные влияния, такие как ионизирующее излучение (например, гамма или корпускулярное излучение), и определенные противораковые лекарственные средства (например, блеомицин) способны вызывать двухцепочечные разрывы ДНК. ДЦР кроме того, могут встречаться в качестве промежуточных продуктов соматической рекомбинации, процесса, который является важным для образования функциональной иммунной системы всех позвоночных. Если двухцепочечные разрывы ДНК не репарированы или репарированы неправильно, то могут встречаться мутации и/или хромосомные аберрации, которые в результате могут привести к клеточной гибели. Для противодействия тяжелым повреждениям, возникающим вследствие двухцепочечных разрывов ДНК, эукариотические клетки разработали различные механизмы для их репарации. Высшие эукариоты используют главным образом так называемое негомологичное соединение концов, в котором ключевую роль выполняет ДНК-зависимая протеинкиназа. В биохимических исследованиях было показано, что ДНК-ПК наиболее эффективно активируется посредством случаев ДНК-ДЦР. Клеточные линии, у которых ДНК-ПК компоненты были мутированы и являются нефункциональными, оказываются чувствительными к действию радиации (Smith и Jackson, 1999).
Благодаря ее каталитическому домену, который находится на С-концевой каталитической субъединице (ДНК-ПКкс), которая состоит из приблизительно 500 аминокислот, ДНК-ПК относится к семейству фосфатидилинозитол-3-киназа-родственных киназ (PIKK), где ДНК-ПК представляет собой нелипидную киназу (Hartley и др. (1995) Cell 82: 849; Smith & Jackson (1999) Genes и Dev 13: 916;  (2009) EMBO J. 28: 3067).
(2009) EMBO J. 28: 3067).
Izzard и др. ((1999) Cancer Res. 59: 2581) было показано, что ингибитор PI3 киназы LY294002 ингибирует функцию ДНК-ПК в экспериментах in-vitro. IC50 значение (концентрация, при которой активность фермента ингибируется на 50%) составляет при относительной неэффективности 1,25 мкМ (5,0 мМ АТФ). Несмотря на наличие данных, свидетельствующих о том, что ингибитор LY294002 предоставляет возможность клеткам млекопитающих становиться чувствительными к излучению, то есть цитотоксичность ионизирующего излучения повышается, в принципе для предполагаемого применения при лучевой терапии, например, солидных раковых опухолей, было показано только слабое повышение чувствительности к ионизирующему излучению для LY294002 на клетках (Rosenzweig и др. (1999) Clin. Cancer Res. 3: 1149). KuDOS Pharmaceuticals Ltd. оптимизированы лидерную структуру LY294002 и представили различные ДНК-ПК ингибиторы. Интродукция дибензотиофенильной группы привела к ингибитору NU-7441, АФТ-конкурирующему соединению, имеющему IC50 значение 20,0 нМ (Hardcastle и др. (2005) J. Med. Chem. 48: 7829). KU-0060648 комбинирует ингибирующие свойства по отношению к ДНК-ПК с улучшенным профилем растворимости в водной среде, но киназы семейства изофермента PI3K, более того, потенциально ингибируются с помощью KU-0060648. Следовательно, существующая в течение продолжительного времени потребность в эффективном и селективном ДНК-ПК ингибиторе не удовлетворена до настоящего времени.
Изобретение основано на задаче преодоления недостатков, указанных в известном уровне техники, и разработки эффективных ингибиторов ДНК-ПК, которые является селективными по отношению к родственным киназам семейства PIKK и имеют низкий молекулярный вес и, в особенности, предоставляют возможность эффективного применения для лечения злокачественного новообразования в качестве радио- и хемосенсибилизаторов - с целью улучшения терапевтической эффективности с одновременным уменьшением побочных действий.
Задача изобретение решается в соответствии с независимыми пунктами формулы изобретения. Зависимые пункты содержат предпочтительные варианты осуществления. В соответствии с изобретением, обеспечиваются соединения формулы (I).
Неожиданно, было обнаружено, что соединения в соответствии с изобретением обеспечиваются с ингибирующими свойствами для серин/треонин протеинкиназ. Соединения формулы (I) создавались таким образом, чтобы происходило эффективное и селективное ингибирование ДНК-ПК. Таким образом, соединения в соответствии с изобретением открывают совершенно новые возможности по отношению к антиканцерогенному действию противораковых средств. Соединения формулы (I) выполняют терапевтическую роль в настоящем изобретении в качестве радио- и хемосенсибилизаторов посредством специфического ингибирования репарации двухцепочечных разрывов ДНК (негомологичное соединение концов) для лечения злокачественного новообразования.
До настоящего времени, из WO 1992/07844 было известно, что производные 2,4-диаминохиназолина являются усилителями химиотерапевтических средств для лечения злокачественного новообразования. Производные нацелены на множественную устойчивость опухолевых клеток вследствие сверхэкспрессии гена mdr1, генный продукт которого эффлюксного Р гликопротеинового насоса поддерживает внутриклеточную концентрацию активного соединения низкой. Не были известны ни физико-химические или фармакологические описанные данные, ни поставляемый на рынок препарат. Другие производные хиназолина в качестве ДНК-ПК ингибиторов описаны в WO 2011/113512.
Настоящее изобретение обеспечивает новую генерацию ДНК-ПК ингибиторов, которые не только способны специфически ингибировать, что проявляется, в особенности, в случае клеточных анализов. Дополнительно, они также отличаются отсутствием часто наблюдаемого, нежелательного ингибирования ионных каналов сердца, в особенности Kv1.11 hERG, блокада которых может приводить к опасным для жизни аритмиям.
Следовательно, соединения в соответствии с изобретением и их соли имеют ценные фармакологические свойства, при этом являются хорошо переносимыми. Для целей настоящего изобретения, соединения формулы (I) определяются таким образом, что под ними также понимают фармацевтически приемлемые производные, соли, сольваты, сольваты солей, предшественники соединений, таутомеры и оптически активные формы (такие как, например, стереоизомеры, диастереомеры, энантиомеры, рацематы). Под сольватами соединений понимают аддукцию молекул инертного растворителя на соединениях, которые образуются благодаря их взаимным силам притяжения. Сольваты представляют собой, например, моно- или дигидраты или алкоголяты. Под фармацевтически приемлемыми производными понимают, например, соли соединений в соответствии с изобретением и так называемые предшественники соединений. Под предшественниками понимают, например, соединения формулы (I), модифицированные с помощью алкильных или ацильных групп, Сахаров или олигопептидов, которые быстро расщепляются в организме с образованием эффективных соединений в соответствии с изобретением. Они также включают производные биоразлагаемых полимеров соединений в соответствии с изобретением, как описано, например, в Int. J. Pharm. 115, 61-67 (1995). Любое соединение, которое может быть превращено in vivo в биоактивное средство, то есть соединения формулы (I), представляет собой предшественник в контексте настоящего изобретения. Любое биологически активное соединение, которое возникает при метаболизации in-vivo соединения в соответствии с изобретением, представляет собой метаболит в контексте настоящего изобретения. Соединения формулы (I) могут иметь один или несколько хиральных центров, и, следовательно, встречаться в различных стереоизомерных формах. Формула (I) охватывает все эти формы.
Изобретение также относится к применению смесей соединений формулы (I), например, смесей двух диастереомеров, например, в соотношении 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 или 1:1000. Особенно предпочтительными в данной заявке являются смеси стереоизомерных соединений.
Выше и ниже, радикалы X, Y, R1, R2, R3, R4, R5, R6, LA, Cyc, Het1, Het2 и Hal, a также m и n, имеют значения, указанные для формулы (I), если специально не указано иначе. Если индивидуальные радикалы встречаются несколько раз в соединении или радикале, то радикалы принимают, независимо друг от друга, указанные значения, если специально не указано иначе. Термины, используемые в данной заявке для определения соединений, в целом основываются на правилах организации ИЮПАК для химических соединений, в частности, органических соединений. Термины для пояснения вышеуказанных соединений согласно изобретению, всегда имеют вышеуказанные значения, если специально не указано иначе в описании или пунктах формулы.
"LA" в контексте изобретения обозначает насыщенный или частично ненасыщенный углеводородный радикал, которые является неразветвленным (линейным) или разветвленным и имеет 1, 2, 3, 4 или 5 атомов С. Примерами LA являются метил, этил, пропил, изопропил, 1,1-, 1,2- или 2,2-диметилпропил, 1-этилпропил, бутил, изобутил, втор-бутил, трет-бутил. Тем не менее, углеводородный радикал также может быть замещен таким образом, что 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две CH2 группы могут быть заменены О, NH, N(СН3) или СО. Их примерами являются метокси, метилсульфанил, этокси, цианометокси, 2-пропионитрилокси, оксетан-3-илокси, N-метиламинокарбонил, карбоксамидо, 2-метоксиэтокси, 2,2,2-трифторэтокси, или 2-гидроксиэтокси.
"Het1" в контексте изобретения обозначает моно- или бициклический алифатический или ароматический углеводородный гетероцикл, имеющий 3, 4, 5, 6, 7, 8, 9 или 10 атомов С и 0, 1, 2 или 3 атома N, О и/или S, которые могут быть замещены. Примерами подходящих "Сус" являются фенил, пиридин, пиразин, пиридазин, пиразоло[1,5-а]пиримидинил, или имидазо[1,2-b]пиридазинил.
"Het2" в контексте изобретения обозначает а 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атомов N, О или S. Примерами Het2 являются оксетан, пирролидин или циклопропил.
В предпочтительном варианте осуществления настоящего изобретения, обеспечиваются производные арилхиназолина формулы (Ia)
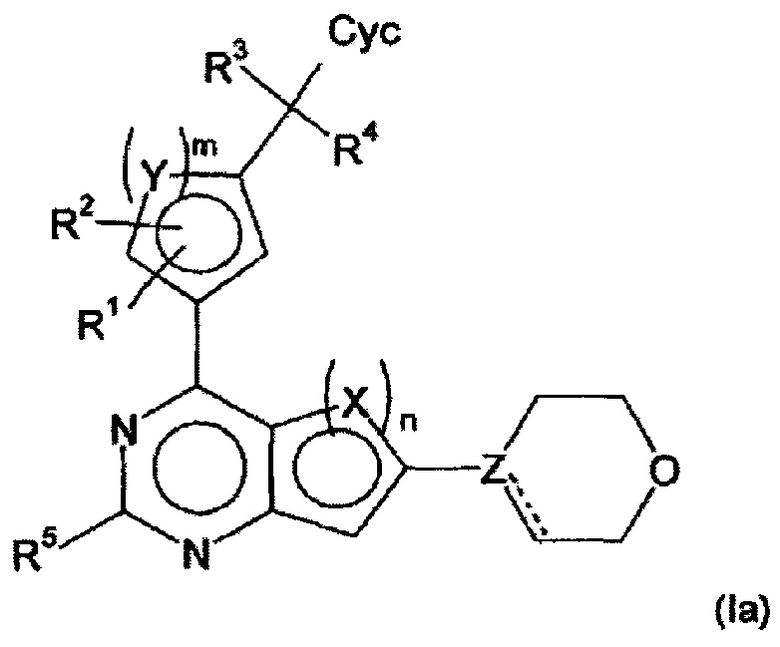
в которой
X, Y, независимо друг от друга, представляют собой СН, S или N,
Z представляет собой С или N,
 образует, если Z=С, двойную связь совместно с простой связью,
образует, если Z=С, двойную связь совместно с простой связью,
отсутствует, если Z=N,
n равен 1 или 2, где
если n=1, Х=S,
и если n=2, оба Х=СН, или X, связанный с пиримидиновым кольцом, представляет собой СН и X, несвязанный с пиримидиновым кольцом, представляет собой N;
m равен 1 или 2, где
если m=1, Y=S,
и если m=2, оба Y=СН, или один Y представляет собой СН, а другой Y представляет собой N;
R1, R2, R3, R4, независимо друг от друга, представляют собой Н, Hal, CN, ОН, CONH2 или LA;
R5 представляет собой Н, Hal, CN или С≡СН;
Сус представляет собой фенил, который может быть незамещен или моно- или дизамещен, независимо друг от друга, с помощью R6, или Het1;
Het1 представляет собой моно- или бициклический, 5-10-членный гетероцикл, имеющий 1-3 атома N, О и/или S, который может быть незамещен или моно- или дизамещен, независимо друг от друга, с помощью R6;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
LA представляет собой неразветвленный или разветвленный алкил, имеющий 1-5 атомов С, который может быть насыщенным или частично ненасыщенным, в котором 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две СН2 группы могут быть заменены О, NH, NH2, N(CH3) или СО;
Het2 представляет собой 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атома N, О и/или S, который незамещен;
Hal представляет собой F, Cl, Br или I;
Кроме того, предпочтительные производные арилхиназолина соответствуют формуле (Ib)
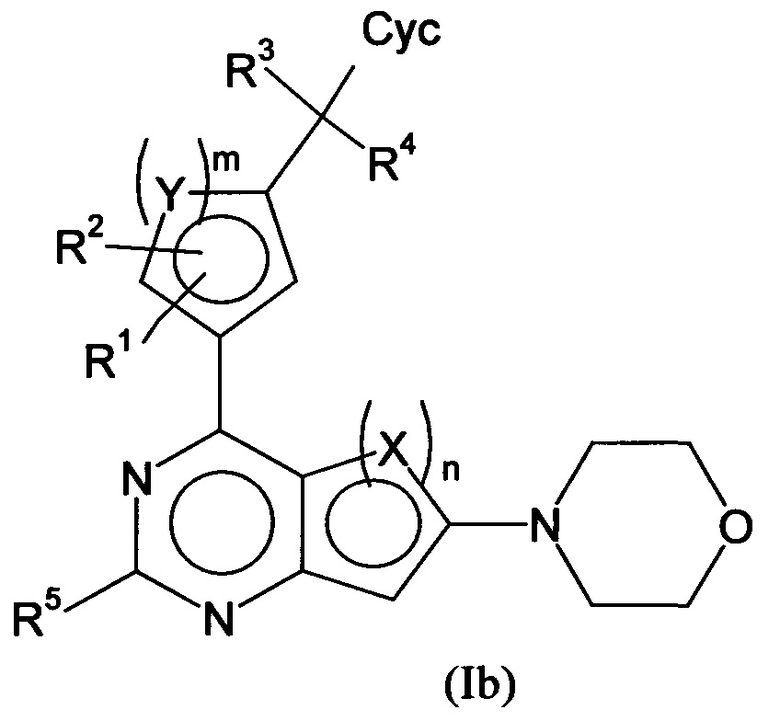
в которой все заместители имеют значения, указанные для формул (I) или (Ia), и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
В дальнейшем предпочтительном варианте осуществления настоящего изобретения, обеспечиваются производные арилхиназолина формулы (II)
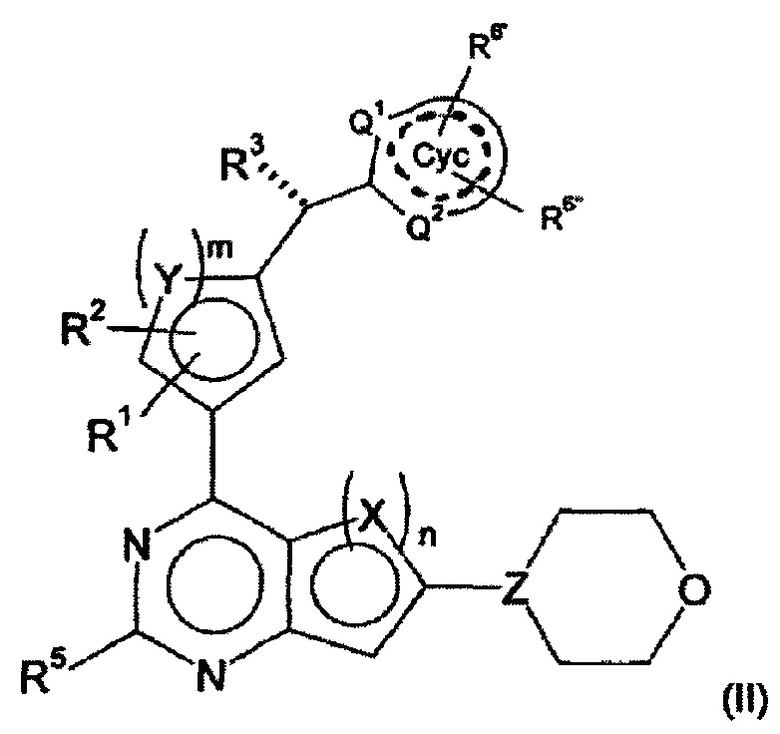
в которой
R3 представляет собой Hal, CN, ОН, CONH2, CONH(LA) или LA;
R6', R6'', независимо друг от друга, представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2, независимо друг от друга, представляют собой СН, N или NH и в каждом случае являются незамещенными;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
А именно, было обнаружено, что активность соединений в соответствии с изобретением является особенно высокой, если R3 имеет конфигурацию, изображенную в формуле (II), и Q не несет заместителей.
В дальнейшем предпочтительном варианте осуществления настоящего изобретения, обеспечиваются производные арилхиназолина формулы (III)

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CONH(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой Н, Hal, LA, оксо, CN, NH2 или Het2;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
А именно, было обнаружено, что активность соединений в соответствии с изобретением является особенно высокой, если R3 имеет конфигурацию, изображенную в формуле (III) и Сус замещен в орто-положении с помощью R6.
Чрезвычайно предпочтительными являются подформулы (IIa), (IIb), (IIIa) и (IIIb) формул (II) и (III):

в которой
R2, R3, независимо друг от друга, представляют собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6', R6'', независимо друг от друга, представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2, независимо друг от друга, представляют собой СН, N или NH и в каждом случае являются незамещенными;
Х1 представляет собой СН, CF или N;
Х2 представляет собой СН или N,
где Х1, Х2 одновременно не представляют собой N;
Y представляет собой СН или N;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях;

в которой
R2, R3, независимо друг от друга, представляют собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6', R6'' независимо друг от друга, представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2, независимо друг от друга, представляют собой СН, N или NH и в каждом случае являются незамещенными;
Y представляет собой СН или N,
обозначает присутствие или отсутствие двойных связей в Сус;
и все другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях;

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Х1 представляет собой СН, CF или N;
Х2 представляет собой СН или N,
где Х1, Х2 одновременно не представляют собой N;
Y представляет собой СН или N;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях;

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Y представляет собой СН или N,
обозначает присутствие или отсутствие двойных связей в Сус;
и все другие заместители имеют значения, указанные для формулы (I),
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Кроме того предпочтительные подгруппы соединений формулы (IIa) могут быть выражены следующими подформулами (IIa-А) - (IIa-O), которые соответствуют формуле (IIa), но в которых
в случае подформулы (IIa-А)
Х1 представляет собой СН,
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
в случае подформулы (IIa-В)
R1 представляет собой F,
R2 представляет собой F или Cl,
в случае подформулы (IIa-C)
Х1, Х2 представляет собой СН,
в случае подформулы (IIa-D)
Х1 представляет собой СН,
R5 представляет собой Н,
в случае подформулы (IIa-Е)
R3 представляет собой Н, ОН,
в случае подформулы (IIa-F)
Х1 представляет собой СН,
R3 представляет собой ОН,
в случае подформулы (IIa-G)
Х1 представляет собой СН,
Y представляет собой СН,
в случае подформулы (IIa-Н)
Х1 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIa-J)
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d}пиридазинил,
тиено[2,3-d}пиридазинил, тиено[2,3-d}пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещен, или может быть моно- или дизамещен с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIa-K)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
в случае подформулы (IIa-L)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
в случае подформулы (IIa-М)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Х1, Х2 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIa-N)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-а}пиридазинил, тиено[2,3-d}пиридазинил, тиено[2,3-d}пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещен, или может быть моно- или дизамещен с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIa-O)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
Сус представляет собой 5-метоксипиридазин-3-ил, имидазо[1,2-b]пиридазин-6-ил, 3-хлор-6-метоксипиразин-2-ил, 3-хлорпиразин-2-ил, пиридазин-4-ил, 3-метоксипиразин-2-ил, 6-метоксипиридазин-3-ил,
3-дифторметоксипиридин-2-ил, 3-метилпиразин-2-ил, тиено[2,3-а}пиримидин-4-ил, 1-метил-1Н-пиридин-2-он-6-ил, 1H-пиридазин-6-он-3-ил, фуро[2,3-d}пиридазин-7-ил, тиено[2,3-d}пиридазин-7-ил, 3,5-диметилпиразин-2-ил, фуро[2,3-а}пиримидин-4-ил, 3-метил-3Н-имидазо[4,5-с]пиридин-4-ил,
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Кроме того, предпочтительные подгруппы соединений формулы (IIIa) могут быть выражены следующими подформулами (IIIa-В)-(IIIa-O), которые соответствуют формуле (IIIa), но в которых
в случае подформулы (IIIa-В)
R1 представляет собой F,
в случае подформулы (IIIa-С)
X1, X2 представляет собой СН,
в случае подформулы (IIIa-D)
Х1 представляет собой СН,
R5 представляет собой Н,
в случае подформулы IIIa-(E)
R3 представляет собой Н, ОН,
в случае подформулы (IIIa-F)
Х1 представляет собой СН,
R3 представляет собой ОН,
в случае подформулы (IIIa-G)
Х1 представляет собой СН,
Y представляет собой СН,
в случае подформулы (IIIa-Н)
Х1 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIIa-J)
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d}пиридазинил, тиено[2,3-d}пиридазинил, тиено[2,3-d}пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещен, или может быть моно- или дизамещен с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIIa-K)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
в случае подформулы (IIIa-L)
R1 представляет собой F,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
в случае подформулы (IIIa-M)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Х1, Х2 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIIa-N)
R1 представляет собой F,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d}пиридазинил, тиено[2,3-d}пиридазинил, тиено[2,3-d}пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещен, или может быть моно- или дизамещен с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIIa-O)
R1 представляет собой F,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
Сус представляет собой 5-метоксипиридазин-3-ил, имидазо[1,2-b]пиридазин-6-ил, 3-хлор-6-метоксипиразин-2-ил, 3-хлорпиразин-2-ил, пиридазин-4-ил, 3-метоксипиразин-2-ил, 6-метоксипиридазин-3-ил, 3-дифторметоксипиридин-2-ил, 3-метилпиразин-2-ил, тиено[2,3-d}пиримидин-4-ил, 1-метил-1Н-пиридин-2-он-6-ил, 1Н-пиридазин-6-он-3-ил, фуро[2,3-d}пиридазин-7-ил, тиено[2,3-d}пиридазин-7-ил, 3,5-диметилпиразин-2-ил, фуро[2,3-d}пиримидин-4-ил, 3-метил-3Н-имидазо[4,5-с]пиридин-4-ил,
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Кроме того предпочтительные подгруппы соединений формулы (IIb) могут быть выражены следующими подформулами (IIb-Q) - (IIb-U), которые соответствуют формуле (IIb), но в которых
в случае подформулы (IIb-Q)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIb-R)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIb-S)
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIb-T)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIb-U)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин или 3-метилпиразин-2-ил,
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Кроме того предпочтительные подгруппы соединений формулы (IIIb) могут быть выражены следующими подформулами (IIIb-Q) - (IIIb-U), которые соответствуют формуле (IIIb), но в которых
в случае подформулы (IIIb-Q)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIIb-R)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIIb-S)
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIIb-T)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIIb-U)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин или 3-метилпиразин-2-ил,
и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях.
Чрезвычайно предпочтительными являются те соединения формулы (I) и их подформулы, и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях, которые собраны в таблицах 1-8.
Соединения формулы (I), а также исходные вещества для их получения, приготавливают с помощью методов, известных per se, которые описаны в литературе (например, в стандартных работах, таких как Houben-Weyl, Methoden der organischen Chemie [Методы органической химии], Georg-Thieme-Verlag, Stuttgart) и/или известны специалисту в данной области техники, и в реакционных условиях, которые известны и пригодны для указанных реакций. Также здесь можно осуществлять варианты, известные per se, но которые здесь подробно не описаны.
В зависимости от используемых условий, время реакции составляет от нескольких минут до 14 дней, температура реакции находится в диапазоне от -70°С до 150°С, обычно в диапазоне от -50°С до 100°С, особенно предпочтительно в диапазоне от -10°С до 70°С.
Реакцию осуществляют в инертном растворителе и, как правило, в присутствии вещества, связывающего кислоту, предпочтительно органического основания, такого как DIPEA, триэтиламин, диметиланилин, пиридин, хинолин, пиперидин или диэтаноламин. Добавление гидроксида, карбоната или бикарбоната щелочного или щелочно-земельного металла или другой соли слабой кислоты щелочных или щелочно-земельных металлов, предпочтительно калия, натрия, кальция или цезия, также может являться благоприятным. Подходящими основаниями являются оксиды металлов, такие как, например, оксид алюминия, гидроксиды щелочных металлов (включая гидроксид калия, гидроксид натрия и гидроксид лития), гидроксиды щелочно-земельных металлов (например, гидроксид бария и гидроксид кальция) и алкоголяты щелочных металлов (например, этоксид калия и пропоксид натрия).
Подходящими инертными растворителями являются, в частности, углеводороды, такие как циклогексан, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, четыреххлористый углерод, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как простой диэтиловый эфир, простой диизопропиловый эфир, трет-бутил метиловый эфир, тетрагидрофуран (ТГФ) или диоксан; простые гликолевые эфиры, такие как этиленгликоль монометиловый или моноэтиловый эфир, этиленгликоль диметиловый эфир (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметил сульфоксид (ДМСО); сероуглерод; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей. Особенно предпочтительными являются ДМФА, метанол, дихлорметан, ТГФ, уксусная кислота и ацетонитрил.
Получение и последующая обычная обработка реакционной смеси может по существу осуществляться в виде периодической реакции или в непрерывной реакционной процедуре. Непрерывная реакционная процедура включает, например, реакцию в непрерывно перемешиваемом котловом реакторе, перемешиваемом котловом каскаде, петлевом или поперечноточном реакторе, трубке Вентури или в микрореакторе. Реакционные смеси необязательно подвергают обычной обработке, при необходимости, путем фильтрации через твердые фазы, хроматографии, разделения между несмешиваемыми фазами (например, экстракция), адсорбции на твердые подложки, удаления растворителей и/или азеотпропирования смесей путем перегонки, селективной перегонки, сублимации, кристаллизации, совместной кристаллизации или путем нанофильтрации на мембранах.
Соединения формулы (I) предпочтительно можно получать путем взаимодействия соединений формулы (V) и (VI). Следовательно, настоящее изобретение также относится к способу получения соединений формулы (I), их подформул и/или их физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, включающему следующие стадии:
(а) взаимодействие соединения формулы (V)

в которой LG представляет собой общепринятую уходящую группу, такую как Hal,
с соединением формулы (IV)
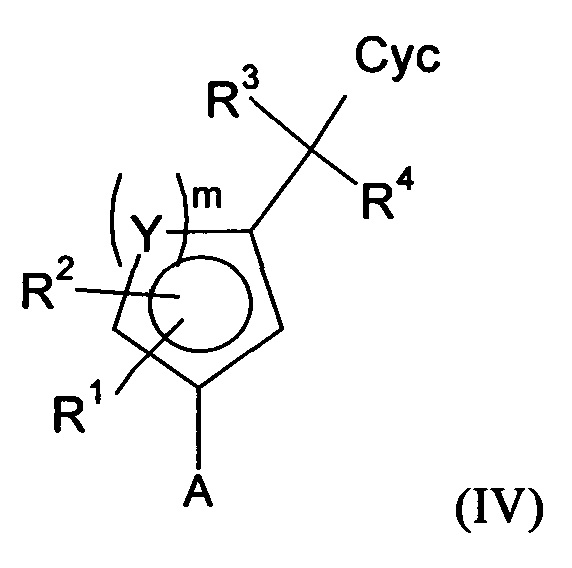
в которой А представляет собой бороновую кислоту или сложный эфир бороновой кислоты,
получая соединения формулы (I) и необязательно
(б) превращения основания или кислоты соединений формулы (I) в одну из их солей.
Исходные соединения, как правило, известны. Если они являются новыми, то они могут быть получены с помощью методов, известных per se. Соединения формулы (I), (Ia), (Ib), (II), (IIa), (IIb), (III), (IIIa), (IIIb), (IV) и (V) могут быть приготовлены с помощью известных методов. Если это является желательным, то исходные вещества могут образовываться in situ, таким образом, что они не выделяются из реакционной смеси, но вместо этого они незамедлительно в дальнейшем превращаются в соединения в соответствии с изобретением. Также возможно осуществлять реакцию постадийно.
Указанные соединения в соответствии с изобретением могут использоваться в их конечной несолевой форме. В другой стороны, настоящее изобретение также охватывает применение этих соединений в форме их фармацевтически приемлемых солей, которые могут иметь происхождение из различных органических и неорганических кислот и оснований с помощью процедур, известных в данной области техники. Фармацевтически приемлемые солевые формы соединений формулы (I) и ее подформул главным образом приготавливают с помощью общепринятых методов. Если соединения содержат карбоксильную группу, то одна из их приемлемых солей может быть образована путем взаимодействия соединения с подходящим основанием для получения соответствующей соли присоединения основания. Такими основаниями являются, например, гидроксиды щелочных металлов (например, гидроксид калия, гидроксид натрия и гидроксид лития), гидроксиды щелочно-земельных металлов (например, гидроксид бария и гидроксид кальция), алкоголяты щелочных металлов (например, этоксид калия и пропоксид натрия) и различные органические основания, такие как пиперидин, диэтаноламин и N-метилглутамин. Основание формулы (I) и ее подформул может быть превращено в ассоциированную соль присоединения кислоты, используя кислоты, например, путем реакции эквивалентных количество основания и кислоты в инертном растворителе, таком как, например, этанол, с последующим упариванием. Подходящие кислоты для этой реакции представляют собой, в частности, те, которые образуют физиологически приемлемые соли, такие как, например, галогеноводороды (например, хлористый водород, бромистый водород или йодистый водород), другие минеральные кислоты и их соответствующие соли (например, сульфат, нитрат или фосфат и другие), алкил- и моноарилсульфонаты (например, этансульфонат, толуолсульфонат и бензолсульфонат) и другие органические кислоты и их соответствующие соли (например, ацетат, трифторацетат, тартрат, малеат, сукцинат, цитрат, бензоат, салицилат, аскорбат и другие. Соли с физиологически неприемлемыми кислотами, например, пикраты, можно использовать для выделения и/или очистки соединений формулы (I).
Принимая во внимание указанное выше, понятно, что выражение "фармацевтически приемлемая соль" в контексте настоящей заявке обозначает активное соединение, которое содержит соединение формулы (I) в форме одной из его солей, в особенности, если эта солевая форма придает улучшенные фармакокинетические свойства активному соединению по сравнению со свободной формой активного соединения. Фармацевтически приемлемая солевая форма активного соединения также может обеспечивать это активное соединение впервые с желательным фармакокинетическим свойством и может даже оказывать положительное влияние на фармакодинамику этого активного соединения по отношению к его терапевтической эффективности в организме.
Соединения в соответствии с изобретением могут быть хиральными благодаря их молекулярной структуре и могут, следовательно, встречаться в различных энантиомерных формах. Таким образом, они могут быть представлены в рацемической или оптически активной форме. Поскольку фармацевтическая эффективность рацематов или стереоизомеров соединений формулы (I) может отличаться, то может являться желательным использовать энантиомеры. В этих случаях, конечный продукт, или даже промежуточный продукт, может быть разделен на энантиомерные соединения с помощью химических или физических средств, известных специалисту в данной области техники или уже применяться, как таковой, в синтезе.
В целом известно, что атомы могут иметь их атомные массы или массовые числа, которые отличаются от атомных масс или массовых чисел, обычно встречающихся в природе. Примерами изотопов, которые являются коммерчески доступными и которые могут быть инкорпорированы в соединение в соответствии с изобретением с помощью известных методов, являются изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например, 2Н, 3Н, 13С, 14С, 15N, 18О, 17О, 31Р, 32Р, 35S, 18F и 36CI. Инкорпорация более тяжелых изотопов, в частности дейтерия (2Н), в соединение в соответствии с изобретением, имеет терапевтические преимущества благодаря более высокой метаболической стабильности этого меченного изотопом соединения. Большая метаболическая стабильность приводит непосредственно к повышенному периоду полураспада in vivo, который предоставляет возможность более низкого дозирования.
Определения атомов Н, С, N, и т.д., как используется в соединениях в соответствии с изобретением, обычно также относится к более тяжелым изотопам этих атомов.
Особенно предпочтительным в соответствии с изобретением является применение D (дейтерий, 2Н) вместо водорода (1Н).
Было обнаружено, что соединения в соответствии с изобретением вызывают специфическое ингибирование серин/треонин протеинкиназ. Следовательно, изобретение также относится к применению соединений формулы (I) или ее подформул и/или их физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, для ингибирования серин/треонин протеинкиназ, предпочтительно PIK.K, в особенности предпочтительно ДНК-ПК. Особенно предпочтительным является ингибирование вышеуказанных серин/треонин протеинкиназ ex vivo или in vitro. Термин "ингибирование" относится к любому уменьшению активности, которое основано на действии специфического соединения в соответствии с изобретением в том смысле, что последнее способно взаимодействовать с целевой молекулой таким образом, что является возможным распознавание, связывание и блокирование. Соединения отличаются высокой аффинностью по меньшей мере к одной серин/треонин протеинкиназе, обеспечивая надежное связывание и предпочтительно полностью блокируя активность киназы. Соединения особенно предпочтительно являются моноспецифическими для гарантирования эксклюзивного и прямого распознавания выбранной киназы. Термин "распознавание" относится в настоящей заявке к любому типу взаимодействия между соединением и указанными целевыми молекулами, в частности, ковалентные или нековалентные связи, такие как, например, ковалентная связь, гидрофобные/гидрофильные взаимодействия, ван-дер-ваальсовские силы, ионные притяжения, водородные связи, лиганд/рецепторные взаимодействия, спаривание оснований нуклеотидов или взаимодействия между эпитопом и связывающим сайтом антитела.
Соединения в соответствии с изобретением проявляют благоприятную биологическую активность, которая может быть продемонстрирована в тестах, описанных в настоящей заявке, такие как, например, ферментативные анализы. Измерения активности киназы представляет собой технологию, хорошо известную квалифицированному специалисту в данной области техники. Обычные тест-системы для определения активности киназы, используя субстраты, например, гистон (Alessi и др. (1996) FEBS Lett. 399(3): 333) или основной миелиновый белок, описаны в литературе ( (1992) JBC 267: 14535). Доступны различные системы для анализов для идентификации ингибиторов киназ. В сцинтилляционном анализе сближения (Sorg и др. (2002) J Biomolecular Screening 7: 11) и анализе флэш-планшета, измеряют радиоактивное фосфорилирование белка или пептида в качестве субстрата, используя АТФ. В присутствии ингибирующего соединения, обнаруживают снижение радиоактивного сигнала, или сигнал отсутствует. Кроме того, метод резонансного переноса энергии гомогенной флуоресценции с временным разрешением (HTR-FRET) и технологии флуоресцентной поляризации (FP) пригодны в качестве методов определения (Sills и др. (2002) J Biomolecular Screening 191). В других нерадиоактивных методах ELISA используются специфические фосфо-антитела (фосфо-АТ). Фосфо-АТ связывают только фосфорилированный субстрат. Это связывание можно обнаружить с помощью хемилюминесценции, используя вторичное конъюгированное с пероксидазой антиовечье антитело.
(1992) JBC 267: 14535). Доступны различные системы для анализов для идентификации ингибиторов киназ. В сцинтилляционном анализе сближения (Sorg и др. (2002) J Biomolecular Screening 7: 11) и анализе флэш-планшета, измеряют радиоактивное фосфорилирование белка или пептида в качестве субстрата, используя АТФ. В присутствии ингибирующего соединения, обнаруживают снижение радиоактивного сигнала, или сигнал отсутствует. Кроме того, метод резонансного переноса энергии гомогенной флуоресценции с временным разрешением (HTR-FRET) и технологии флуоресцентной поляризации (FP) пригодны в качестве методов определения (Sills и др. (2002) J Biomolecular Screening 191). В других нерадиоактивных методах ELISA используются специфические фосфо-антитела (фосфо-АТ). Фосфо-АТ связывают только фосфорилированный субстрат. Это связывание можно обнаружить с помощью хемилюминесценции, используя вторичное конъюгированное с пероксидазой антиовечье антитело.
Вышеуказанное применение соединений можно осуществлять на моделях in-vitro или in-vivo. Чувствительность конкретной клетки к лечению с применением соединения в соответствии с изобретением можно определять путем тестирования in vitro. Типично, культуру клеток инкубируют с соединением в соответствии с изобретением при различных концентрациях в течение периода времени, которого достаточно для того, чтобы активные агенты индуцировали клеточную гибель или ингибировали клеточную пролиферацию, жизнеспособность клеток или миграцию, обычно в интервале от приблизительно одного часа до вплоть до 9 дней. Для тестирования in vitro, можно использовать культивированные клетки из образца биопсии. После этого определяют количество клеток, оставшихся после лечения. Применение in vitro осуществляют, в особенности, на образцах из видов млекопитающих, которые страдают от злокачественного новообразования, опухолей или метастаз. Хозяин или пациент может принадлежать к млекопитающим любых видов, например, виды приматов, в особенности люди, но также и грызуны (включая мышей, крыс и хомяков), кролики, лошади, коровы, собаки, кошки и др. Животные модели представляют интерес для экспериментальных исследований, обеспечивая модель лечения заболевания человека.
Тестирования множества специфических соединений предоставляет возможность выбора активного соединения, которое проявляет наибольшую пригодность для лечения пациента. Дозу in-vivo выбранного соединения благоприятно подгоняют к чувствительности киназы и/или тяжести заболевания пациента, принимая во внимания данные in-vitro, в результате чего заметно повышается терапевтическая эффективность. Доза изменяется в зависимости от специфического используемого соединения, специфического заболевания, состояния пациента и др. Терапевтической дозы типично достаточно в значительной степени для уменьшения популяции нежелательных клеток в целевой ткани, при этом поддерживается жизнеспособность пациента. Последующее раскрытие изобретения и вариантов его осуществления, относящиеся к применению соединений формулы (I) для приготовления лекарственного средства для профилактики, терапии и/или контроля прогрессирования, является действительным и может применяться без органический к применению соединений для ингибирования активности киназы, если это является подходящим.
Лечение обычно продолжается до тех пор, пока не произойдет значительное уменьшение, например, уменьшение по меньшей мере на приблизительно 50% клеточной нагрузки, и может продолжаться до тех пор, пока по существу не будет больше обнаруживаться нежелательных клеток в организме. В тестах этого типа, соединения в соответствии с изобретением проявляют и вызывают ингибирующий эффект, который обычно подтверждается значениями IC50 в подходящем диапазоне, предпочтительно в микромолярном диапазоне и более предпочтительно в наномолярном до пикомолярного диапазона. Киназа ингибируется, в особенности, до уровня 50%, если концентрация соединений составляет меньше, чем 1 мкМ, предпочтительно равна или меньше, чем 0,5 мкМ, особенно предпочтительно, меньше, чем 0,1 мкМ. Эту концентрацию называют IC50 значение.
Изобретение также относится к лекарственному средству, содержащему по меньшей мере одно соединение формулы (I) или ее подформул и/или его физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях. Изобретение также относится к фармацевтической композиции, содержащей, в качестве активного соединение, эффективное количество по меньшей мере одного соединения формулы (I) или ее подформул и/или его физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, совместно с фармацевтически допустимыми вспомогательными веществами.
"Медикамент", "лекарственное средство" и "фармацевтическая композиция" или "фармацевтический препарат" в настоящей заявке представляет собой любую композицию, которая может применяться для профилактики, терапии, контроля прогрессирования или последующего лечения пациентов, к которых, по меньшей мере временно, проявляется патогенная модификация общего состояния или состояния отдельных компонентов организма пациента, предпочтительно вследствие злокачественного новообразования, опухолей, или метастаз.
Для повышения защитного или терапевтического действия соединений в соответствии с изобретением, можно добавлять фармацевтически допустимые адъюванты. Для целей изобретения, любое вещество, которое способствует, усиливает или модифицирует эффект с соединениями в соответствии с изобретением, представляет собой "адъювант". Известными адъювантами являются, например, соединения алюминия, такие как, например, гидроксид алюминия или фосфат алюминия, сапонины, такие как, например, QS 21, мурамил дипептид или мурамил трипептид, белки, такие как, например, гамма-интерферон или TNF, MF 59, фосфатидилхолин, сквален или многоатомные спирты. Совместное применение яичного альбумина в полном адъюванте Фрейнда может, вероятно, повышать опосредованный клетками иммунитет и, следовательно, поддерживать действие по нейтрализации образующихся антител. Кроме того, ДНК, которая имеет иммуностимулирующие свойства, или которая кодирует белок с эффектом адъюванта, такой как, например, цитокин, может применяться параллельно или в конструкции.
Введение фармацевтической композиции в клетку или организм может осуществляться в соответствии с изобретением любым способом, который предоставляет возможность киназам осуществлять контакт с соединениями, присутствующими в композиции, вследствие чего индуцируется ответ. Фармацевтическую композицию согласно настоящему изобретению можно вводить перорально, трансдермально, трансмукозально, трансуретрально, вагинально, ректально, легочно, энтерально и/или парентерально. Выбранный тип введения зависит от показаний, вводимой дозы, параметров, специфических для индивидуума, и т.д. В частности, различные типы введения облегчают сайт-специфическую терапию, которая минимизирует побочные действия и уменьшает дозу активного соединения. Чрезвычайно предпочтительные инъекции представляют собой внутрикожные, подкожные, внутримышечные или внутривенные инъекции. Введение можно осуществлять, например, с помощью так называемых пистолетов для вакцинации или с помощью шприцов. Также представляется возможным приготавливать вещество в виде аэрозоля, который ингалируется в организм, предпочтительно организм человека.
Формы для введения фармацевтической композиции приготавливают в соответствии с желательным типом введения в подходящей дозировке и с помощью способа, известного per se, используя общепринятые твердые или жидкие носители и/или разбавители и вспомогательные вещества, которые обычно применяются. Следовательно, фармацевтически приемлемые наполнители, известные квалифицированному специалисту в данной области техники, могут в большинстве составлять часть фармацевтической композиции в соответствии с изобретением, где количество вещества-наполнителя, которое объединяют с активным соединением для приготовления однократной дозы, изменяется в зависимости от индивидуума, подвергаемого лечению, и типа введения. Эти фармацевтически допустимые вспомогательные вещества включают соли, буферы, заполнители, стабилизаторы, комплексообразующие средства, антиоксиданты, растворители, связующие, смазывающие вещества, вещества для нанесения оболочек на таблетки, ароматизаторы, красители, консерванты, корректирующие вещества и другие. Примерами наполнителей этого типа являются вода, растительные масла, бензиловые спирты, алкиленгликоль, полиэтиленгликоль, Kolliphor, глицерол триацетат, желатин, гидроксипропилметилцеллюлоза (НРМС), углеводы, такие как, например, лактоза или крахмал, стеарат магния, тальк и вазелин.
Фармацевтический препарат может быть представлен в форме таблетки, таблетки с пленочной оболочкой, драже, пастилки, капсулы, пилюли, порошка, гранул, сиропа, сока, каплей, раствора, дисперсии, суспензии, суппозитория, эмульсии, экструдата, импланта, крема, геля, мази, пасты, лосьона, сыворотки, масла, спрея, аэрозоля, адгезива, пластыря или бандажа. Формы для перорального введения, которые приготавливаются, предпочтительно представляют собой таблетки, таблетки с пленочным покрытием, драже, пастилки, капсулы, пилюли, порошки, гранулы, сиропы, соки, капли, растворы, дисперсии или суспензии - включая в виде депо-формы. Кроме того, следует рассматривать формы парентерального медикамента, такие как, например, суппозитории, суспензии, эмульсии, импланты или растворы, предпочтительно масляные или водные растворы. Для местного применения, активное соединение лекарственного средства приготавливают общепринятым способом по меньшей мере с одним фармацевтически приемлемым носителем, таким как, например, микрокристаллическая целлюлоза, и необязательно дополнительными вспомогательными веществами, такими как, например, увлажнители, для получения твердых препаратов, которые можно наносить на кожу, такие как, например, кремы, гели, мази, пасты, порошки или эмульсии, или для получения жидких препаратов, которые можно наносить на кожу, такие как, например, растворы, суспензии, лосьоны, сыворотки, масла, спреи или аэрозоли. Фармацевтическая композиция предпочтительно представлена в форме инъекционного раствора. Для приготовления инъекционного раствора, можно использовать водные среды, такие как, например, дистиллированная вода или физиологические солевые растворы, где последние включают соли присоединения кислот и оснований. Фармацевтическая композиция также может быть представлена в форме твердой композиции, например, в лиофилизированном состоянии, и затем может быть приготовлена перед использованием путем добавления растворителя, такого как, например, дистиллированная вода. Для квалифицированного специалиста в данной области техники известны основные принципы приготовления лиофилизатов.
Концентрация активного соединения в препарате может составлять от 0,1 до 100 процентов по весу. Является ключевым, что фармацевтическая композиция содержит, в качестве активного соединения, эффективное количество соединения совместно с фармацевтически допустимыми вспомогательными веществами. Термины "эффективное количество" или "эффективная доза" используются в данной заявке взаимозаменяемо и обозначают количество фармацевтически активного соединения, которое имеет профилактически или терапевтически релевантное действие на заболевание или патологическое изменение в клетке, ткани, органе или млекопитающем. "Профилактическое действие" предотвращает начало заболевания или даже инфицирование патогеном после попадания в организм индивидуальных типичных представителей таким образом, что его последующее распространение существенно уменьшается или они даже полностью деактивируются. "Профилактическое действие" также включает повышение нормальной физиологической функции. Профилактика целесообразна, в частности, если индивидуум имеет предрасположенности к началу вышеуказанных заболеваний, такие как, например, семейный анамнез, дефект гена или недавно перенесенное заболевание. "Терапевтически релевантное действие" освобождает частично или полностью от одного, больше или всех симптомов заболевания или приводит к частичной или полной реверсии одного, нескольких или всех физиологических или биохимических параметров, которые ассоциированы с или причинно вовлечены в заболевание или патологическое изменение нормального состояния. Контроль прогрессирования также является типом терапевтического лечения, если соединения вводят в определенные временные промежутки, например, для полной элиминации симптомов заболевания. Соответствующая доза или дозируемый диапазон для введения соединений в соответствии с изобретением является достаточно большим для достижения желательного профилактического или терапевтического эффекта индукции биологической или ответной реакции. В целом, доза будет изменяться в зависимости от возраста, конституции и пола пациента, и тяжесть заболевания также следует учитывать. Совершенно очевидно, что специфическая доза, частота и продолжительность введения, дополнительно, зависят от множества факторов, таких как, например, нацеливание и связывающая способность соединений, пищевые привычки индивидуума, подлежащего лечению, тип введения, скорость экскреции и комбинации с другими лекарственными средствами. Индивидуальная доза может корректироваться как по отношению к первичному заболеванию, так и по отношению к появлению любых осложнений. Точная доза может быть установлена квалифицированным специалистом в данной области техники, используя известные средства и методы. Это раскрытие изобретения является действительным и может применяться без ограничений к фармацевтической композиции, содержащей соединения формулы (I), если это является подходящим.
В варианте осуществления изобретения, соединения вводятся в дозе от 0,01 мг до 1 г на дозируемую единицу, предпочтительно в диапазоне от 1 до 700 мг, особенно предпочтительно от 5 до 200 мг. Суточная доза, в частности, находится в диапазоне от 0,02 до 100 мг/кг веса тела.
Для поддержания медицинского эффекта, фармацевтическая композиция также может содержать, в варианте осуществления изобретения, одно или несколько дополнительных активных соединений, где возможно одновременное или последовательное введение. Терапевтический эффект фармацевтической композиции в соответствии с изобретением может состоять, например, для определенных противораковых средств, имеющих лучшее действие путем ингибирования ДНК-ПК в качестве желательного побочного эффекта или в нескольких побочных действиях этих лекарственных средств, которые уменьшаются путем уменьшения дозы.
В предпочтительном варианте осуществления изобретения, фармацевтическая композиция в соответствии с изобретением комбинируется с противораковым средством. Как используется в настоящей заявке, термин "противораковое средство" относится к любому средству, которое вводится пациенту со злокачественным новообразованием, опухолями или метастазами для целей лечения злокачественного новообразования. Противораковые средства, которые являются предпочтительными в соответствии с изобретением, представляют собой те средства, которые повреждают ДНК опухолевых клеток и, следовательно, вовлечены в репликацию ДНК, транскрипцию ДНК или экспрессию генов. Следующие в особенности являются подходящими для этой цели:
- алкилирующие средства, такие как алтретамин, бендамустин, бусульфан, кармустин, хлорамбуцил, хлорметин, циклофосфамид, дакарбазин, ифосфамид, импросульфан тозилат, ломустин, мельфалан, митобронитол, митолактол, нимустин, ранимустин, темозоломид, тиотепа, треосульфан, мехлоретамин, карбоквон, апазиквон, фотемустин, глуфосфамид, палифосфамид, пипоброман, трофосфамид, урамустин;
- соединения платины, такие как карбоплатин, цисплатин, эптаплатин, мириплатин гидрат, оксалиплатин, лобаплатин, недаплатин, пикоплатин, страплатин;
- ингибиторы топоизомеразы, такие как этопозид, иринотекан, разоксан, собузоксан,
- ДНК-модифицирующие средства, такие как амрубицин, бисантрен, децитабин, митоксантрон, прокарбазин, трабектедин, клофарабин, амсакрин, бросталлицин, пиксантрон, ларомустин;
- противораковые антибиотики, такие как блеомицин, дактиномицин, доксорубицин, эпирубицин, идарубицин, левамизол, милтефозин, митомицин С, ромидепсин, стрептозоцин, валрубицин, зиностатин, зорубицин, даунорубицин, пликамицин, акларубицин, пепломицин, пирарубицин;
- вещества, излучающие альфа-частицы, такие как альфарадин (223Ra дихлорид, Xofgio), 211At, 213Bi, 225Ac, 227Th;
особенно предпочтительными являются блеомицин и альфарадин.
Изобретение также может практически реализовываться в виде набора, который содержит соединения в соответствии с изобретением. Набор состоит из отдельных упаковок (а) эффективного количества соединения формулы (I) и/или его физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, и (б) эффективного количества противоракового средства. Набор содержит подходящие контейнеры, такие как, например, коробки или картонные коробки, индивидуальные флаконы, пакеты или ампулы. Набор может содержать, например, отдельные ампулы, каждая из которых содержит эффективное количество соединения формулы (I) и/или его фармацевтически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, и эффективное количество противоракового средства в растворенной или лиофиллизированной форме. Набор согласно изобретению также может содержать изделие, которое содержит письменные инструкции или указывает пользователю на письменные инструкции, которые поясняют использование соединений согласно изобретению.
В соответствии с изобретением, соединения формулы (I) или ее подформул и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях, пригодны для профилактики, терапии и/или контроля прогрессирования заболеваний, которые вызываются, поддерживаются и/или распространяются посредством активности серин/ треонин протеинкиназ. Следовательно, настоящее изобретение также относится к применению соединений формулы (I) или ее подформул и/или их физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, для приготовления лекарственного средства для профилактики, терапии и/или контроля прогрессирования заболеваний, которые вызываются, поддерживаются и/или распространяются посредством активности серин/треонин протеинкиназ. В соответствии с изобретением, соединения формулы (I) или ее подформул и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях, пригодны для применения для профилактики, терапии и/или контроля прогрессирования заболеваний, которые вызываются, поддерживаются и/или распространяются посредством активности серин/протеинкиназ. Для идентификации соответствующего пути передачи сигналов и для определения взаимодействий между различными путями передачи сигналов, были разработаны подходящие модели или модельные системы, например, модели на основе культуры клеток (Khwaja и др. (1997) ЕМВО 16: 2783) и модели трансгенных животных (White и др. (2001) Oncogene 20: 7064). Для определения определенных стадий в каскаде передачи сигналов, взаимодействующие соединения можно использовать для модуляции сигнала (Stephens и др. (2000) Biochemical J 351: 95). Дополнительно, соединения в соответствии с изобретением также можно использовать в качестве реагентов для тестирования зависимых от киназ путей передачи сигналов на животных моделях и/или моделях культур клеток или на клинических заболеваний, указанных в данной заявке. Как обсуждается в данной заявке, эти пути передачи сигналов являются релевантными для различных заболеваний. Таким образом, соединения в соответствии с изобретением пригодны для профилактики, терапии и/или контроля прогрессирования заболеваний, которые зависят от пути передачи сигналов с участием серин/треонин протеинкиназ.
В соответствии с изобретением, соединения формулы (I) или ее подформул и/или их физиологически приемлемые соли, таутомеры и/или стереоизомеры, включая их смеси во всех соотношениях, пригодны для применения для профилактики, терапии и/или контроля прогрессирования злокачественного новообразования, опухолей и/или метастаз.
Опухоль выбирают, в частности, из группы злокачественных заболеваний мочевого пузыря, желудка, почек, головы, шеи, пищевода, шейки матки, щитовидной железы, кишечника, печени, головного мозга, предстательной железы, мочеполового тракта, лимфатической системы, гортани, легких, кожи, крови, костей и иммунной системы, и/или злокачественное новообразование выбирают из группы моноцитарного лейкоза, не-мелкоклеточного рака легкого, мелкоклеточного рака легкого, рака поджелудочной железы, глиобластомы, колоректальной карциномы, карциномы молочной железы, острого миелолейкоза, хронического миелолейкоза, острого лимфолейкоза, хронического лимфолейкоза, ходжкинской лимфомы и не-ходжкинской лимфомы.
Дальнейший вариант осуществления настоящего изобретения относится к соединениям в соответствии с изобретением в комбинации с лучевой терапией и/или с по меньшей мере одним дополнительным активным соединением, предпочтительно в комбинации с лучевой терапией и/или противораковым средством. Промышленные способы облучения, которые используются клинически, предпочтительно включают фотонное облучение (классическое, электромагнитное рентгеновское /гамма излучение), протонное облучение, облучение пучком тяжелых ионов (ионизированный углерод) и нейронное облучение, не ограничиваясь только ими. Дополнительно, близкофокусная лучевая терапия используется клинически в качестве подходящего источника излучения (например, излучатели альфа-частиц) в форме поверхностного применения и внутриполостного и интерстициального введения. Эти радиотерапии и другие подходящие облучающие терапии в контексте изобретения известны специалисту в данной области техники, такие как, например, из Herrmann и др. (2006) Klinische Strahlenbiologie [Клиническая лучевая биология], Elsevier Munich, 4-ое изд., 67-68; Bhide & Nutting (2010) BMC Medicine 8: 25; Choi & Hung (2010) Current Urology Reports 11(3): 172. В качестве наиболее частого применения, фотонное облучение корректируется технически с помощью метода IMRT (радиотерапия с модулированием пучка) и с помощью методов визуализации (трехмерная конформационная лучевая терапия) в запланированном облучении и осуществления для наиболее точной возможной фокусировки. Соединения в соответствии с изобретением обеспечивают синергетические эффекты с существующими в настоящее время химиотерапиями и облучениями при злокачественных новообразованиях и/или восстанавливают эффективность существующих в настоящее время химиотерапии и облучений при злокачественных новообразованиях.
Еще дальнейший вариант осуществления изобретения относится к применению по меньшей мере одного соединения формулы (I) и/или его физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, для сенсибилизации злокачественных клеток к противораковому средству и/или ионизирующему излучению, при условии, что не происходит сенсибилизации in vivo в организме человека или животного. Сенсибилизация предпочтительно происходит ех vivo или in vitro путем введения соединений в клетки, культуры клеток, ткани или органы, которые содержат серин/треонин протеинкиназы. Применение ex-vivo используется, в частности, в случае животных клеток, имеющих происхождение из организма животного, который поражен заболеванием, выбранным из группы злокачественного новообразования, опухолей или метастаз. Клетки, обработанные ех vivo, могут либо продолжать поддерживаться в культуре для последующих исследований или переноситься в животное, которое может представлять собой животное-хозяин или другое животные, ex-vivo сенсибилизация в соответствии с изобретением является особенно благоприятной для тестирования специфического действия соединений, таким образом, что in-vivo доза может предварительно корректироваться в соответствии с оценкой этих данных ex-vivo. В результате этого, существенно повышается терапевтический эффект. Альтернативно, изобретение также предназначено для применения в in vivo и относится к по меньшей мере одному соединению формулы (I) и/или его физиологически приемлемым солям, таутомерам и/или стереоизомерам, включая их смеси во всех соотношениях, для применения для сенсибилизации злокачественных клеток к противораковому средству и/или ионизирующему излучению.
Изобретение также относится к способу профилактики, терапии и/или контроля прогрессирования злокачественного новообразования, опухолей или метастаз, в котором эффективное количество по меньшей мере одного соединения в соответствии с изобретением и/или его физиологически приемлемых солей, таутомеров и/или стереоизомеров, включая их смеси во всех соотношениях, вводят субъекту, подвергаемому лечению. Предпочтительными субъектами в контексте изобретения являются люди или животные, особенно предпочтительно люди. Для квалифицированного специалиста в данной области техники известно в данном случае, как он может вводить соединения в соответствии с изобретением, который также, несомненно, может использовать в виде фармацевтической композиции в соответствии с изобретением, в различных дозах в организм, в особенности в организм человека. Эффективное количество и тип введения может быть определен специалистом в данной области техники с помощью традиционных экспериментов. Раскрытое выше описание изобретения и варианты его осуществления являются действительными и могут применяться без ограничений к способу лечения, если это является подходящим.
Все указанные и дополнительные составляющие или компоненты известны квалифицированному специалисту в данной области техники и могут быть испытаны в специфическом варианте осуществления для идей в соответствии с изобретением в общепринятых экспериментах. Все документы, процитированные в описании, таким образом полностью включены в раскрытие настоящего изобретения в качестве ссылки.
В качестве части изобретения, представленного в данной заявке, впервые обеспечиваются новые соединения арилхиназолина формулы (I). Соединения в соответствии с изобретением контролируют серин/протеинкиназы, в особенности ДНК-ПК, аффинно и/или селективно. Соединения согласно формуле (I) и их производные отличаются высокой специфичностью и стабильностью, низкой стоимостью получения и простой обработкой. Эти свойства составляют основание для воспроизводимого способа действия, и надежного и безопасного взаимодействия с соответствующими целевыми структурами. Изобретение также включает применение данных производных арилхиназолина для ингибирования, регуляции и/или модуляции сигнального каскада серин/треонин протеинкиназ, в особенности ДНК-ПК, и, следовательно, предоставляют новые наборы для исследования и/или диагностики.
Лекарственные средства и фармацевтические композиции, которые содержат указанные соединения, и применения указанных соединений для лечения нарушений, промотируемых киназами, составляют, дополнительно, чрезвычайно перспективный подход для широкого спектра терапий, предоставляя возможность прямого и непосредственного облегчения симптомов у людей и животных. Это является особенно благоприятным для эффективной борьбы с тяжелыми заболеваниями, такими как злокачественное новообразование, либо в виде монотерапии или в комбинации с другими противоопухолевыми терапиями. Ключевое участие ДНК-ПК в процессах репарации ДНК и данные о том, что ДНК-ПК ингибиторы предоставляют возможность клеткам млекопитающих становиться более чувствительными к излучению, обеспечивают возможность использования ДНК-ПК-специфических ингибиторов в качестве части лечения, например, солидных злокачественных опухолей путем лучевой терапии и/или химиотерапии, направленных на ДНК-ДЦР.
Соединения формулы (I), их соли, изомеры, таутомеры, энантиомеры, диастереомеры, рацематы, производные, пролекарства и/или метаболиты являются эффективными не только в случае указанных клинических картин заболеваний, но, вероятно, при диагностике и терапии всех заболеваний, связанных с сигнальным каскадом ДНК-ПК, в особенности, по отношению к ингибированию клеточной пролиферации и миграции. Дополнительно, ингибиторы в соответствии с изобретением можно использовать для лечения ретровирусных заболеваний путем супрессии интеграции ретровирусов (R. Daniel (1999) Science 284: 644). В завершение, ингибиторы в соответствии с изобретением могут применяться в качестве иммуномодуляторов и модуляторов поддержания теломер. Низкомолекулярные ингибиторы используются индивидуально и/или в комбинации с другими терапевтическими средствами, такими как, например, хирургические вмешательства, иммунотерапия, лучевая терапия и/или химиотерапия. Последняя относится к нацеливающей терапии с любым желательным NME (то есть NCE и/или NBE) в качестве монотерапии и/или целевой /нецелевой комбинированной терапии.
Благодаря их неожиданно сильному и/или селективному ингибированию ферментов, которые регулируют клеточные процессы посредством репарации дцДНК, соединения согласно изобретению можно вводить в благоприятной низкой дозе, в то время как они обеспечивают сходную или даже превосходящую биологическую эффективность по сравнению с менее эффективными или менее селективными ингибиторами, известными из уровня техники. Уменьшенная доза также сопровождается уменьшенными или отсутствием медицинских побочных действий. Дополнительно, чрезвычайно высокая селективность ингибирования соединениями в соответствии с изобретением также сопровождается уменьшением нежелательных побочных действий, которые не зависят от дозы. В особенности, соединения в соответствии с изобретением не имеют физиологически релевантных ингибирований или блокад Kv 11.1 hERG калиевого ионного канала.
Также является понятным, что настоящее изобретение не ограничивается специфическими соединениями, фармацевтическими композициями, применениями и способами, как описано в настоящей заявке, поскольку такие аспекты могут изменяться. Кроме того, является очевидным, что терминология, используемая в настоящей заявке, служит исключительно для целей описания предпочтительных вариантов осуществления изобретения и не предназначена для ограничения объема защиты изобретения. Как используется в настоящей заявке в описании, включая приложенные пункты формулы, словоформы в единственном числе включают эквиваленты во множественном числе. Например, ссылка на "соединение" включает единичное соединение или множество соединений, которые могут быть, в свою очередь, идентичными или различными, или ссылка на "способ" включает эквивалентные стадии и способы, которые известны специалисту в данной области техники.
Изобретение более подробно поясняется ниже со ссылкой на неограничивающие примеры специфических вариантов осуществления изобретения. Примеры должны, в частности, интерпретироваться как неограничивающие комбинациями характерных признаков, проиллюстрированных специфически, но вместо этого проиллюстрированные характерные признаки могут, в свою очередь, свободно комбинироваться до тех пор, пока достигается цель изобретения.
Примеры
Обзор демонстрационных примеров представлен в Таблицах 1-7.
Следующие диапазоны применяются к биологическим данным, воспроизведенным в данной заявке:
ДНК-ПК (ферментативный):
пДНК-ПК (клеточный):
Kv11.1 hERG:
Анализ
ЯМР (1H) осуществляли при следующих параметрах.
Приборы: Broker Avance DRX 500, Broker Avance 400, Broker DPX 300
Эталон: TMS
TD (временной интервал = количество измерительных точек или вес младшего разряда): 65536
Растворитель ДМСО-d6
NS (число сканирований = частота сканирований): 32
SF (частота спектрометра = частота передачи): 400 или 500 МГц
ТЕ (температура): 303 K, 363 K или 393 K
Константы связи (J) указаны в Герцах (Гц)
ВЭЖХ: высокоэффективная жидкостная хроматография с УФ детектором
ЖХ-МС: высокоэффективная жидкостная хроматография с УФ и МС детектором
SFC: сверхкритическая жидкостная хроматография с УФ детектором
Идентификация промежуточных соединений при синтезе и конечных продуктов синтеза с помощью ЖХ-МС:
ЖХ-МС метод А:
Колонка: Chromolith SpeedROD RP-18e 50-4,6 мм, скорость потока: 2,4 мл/минут, длина волны: 220 нм, элюент А: вода + 0,05 об. % муравьиной кислоты, элюент В: ацетонитрил + 0,4 об. % муравьиной кислоты, градиент: 4 об. % - 100 об. % элюента В в течение 2,8 минут, затем 100% элюента В в течение периода времени 0,5 минуты.
ЖХ-МС метод В:
Колонка: Chromolith SpeedROD RP-18e 50-4,6 мм, скорость потока: 2,4 мл/минут, длина волны: 220 нм, элюент А: вода + 0,1 об. % трифторуксусной кислоты, элюент В: ацетонитрил + 0,1 об. % трифторуксусной кислоты, градиент: 4 об. % - 100 об. % элюента В в течение 2,8 минут, затем 100 об. % элюента В течение периода времени 0,5 минуты.
Разделение стереоизомерных смесей с помощью ВЭЖХ и SFC:
ВЭЖХ: сначала, осуществляли скрининг колонок для каждой стереоизомерной смеси, со следующими колонками: Chiralpak AD-H, Chiralpak AS-H, Chiralpak IA, Chiralpak IB, Chiralpak IC, Chiralcel OD-H, Chiralcel OJ-H, Lux Целлюлоза-2, Lux-Амилоза-2, все колонки: 250-4,6 мм. Наиболее подходящую колонку использовали для дальнейших измерений (например, определение энантиомерного соотношения). Скорость потока: 0,8 мл/минут, длина волны: переменная, адаптировали в соответствии с максимумом экстинкции и используемым элюентами. Элюент: следующие растворители или смеси растворителей использовали для элюентов: н-гептан, н-гексан, этанол, метанол, 2-пропанол, ацетонитрил, этилацетат, дихлорметан; следующие можно использовать в качестве дополнительного элюента: 0,01-0,5 об. % муравьиной кислоты, 0,01-0,5 об. % диэтиламина; использовали градиенты или условия изократических измерений, в соответствии с указаниями.
SFC: сначала, осуществляли скрининг колонок для каждой стереоизомерной смеси, со следующими колонками: Chiralpak AD-H, Chiralpak AS-H, Chiralpak IA, Chiralpak IB, Chiralpak IC, Chiralcel OD-H, Chiralcel OJ-H, Lux Целлюлоза-2, Lus-Амилоза-2, все колонки: 250-4,6 мм. Наиболее подходящую колонку использовали для дальнейших измерений (например, определение энантиомерного соотношения). Скорость потока: 5 мл/минут, длина волны: переменная, адаптировали в соответствии с максимумом экстинкции и используемым элюентами. Элюент: диоксид углерода в жидком состоянии (>70 бар), со-элюент: следующие растворители или смеси растворителей использовали для со-элюентов: этанол, метанол, изопропанол, ацетонитрил, этилацетат, дихлорметан. Следующие можно использовать в качестве дополнительного элюента: 0,01-0,5 об. % муравьиной кислоты, 0,01-0,5 об. % диэтиламина. Использовали градиенты или условия изократических измерений, в соответствии с указаниями.
Биологическое тестирование
А) ДНК-ПК АНАЛИЗ (БИОХИМИЧЕСКИЙ)
Киназный анализ осуществляли в покрытых стрептавидином микротитровальных флэшпланшетах на 348-лунки. Для этого, 1,5 мкг комплекса ДНК-ПК/белок и 100 нг биотилированного субстрата, такого как, например, PESQEAFADLWKK-биотин-NH2 ("биотин-ДНК-ПК пептид"), инкубировали в течение 90 минут при комнатной температуре в общем объеме 36,5 мкл (34,25 мМ HEPES/KOH; 7,85 мМ Трис HCl; 68,5 мМ KCl; 5 мкМ АТФ; 6,85 мМ MgCl2; 0,5 мМ EDTA; 0,14 мМ EGTA; 0,69 мМ DTT; рН 7,4) с 500 нг ДНК из тимуса теленка, 0,1 мкКи 33Р-АТФ и 1,8% ДМСО на лунку с и без тестируемого соединения. Реакцию останавливали, используя 50 мкл/лунку 200 мМ EDTA. После инкубирования дополнительно в течение 30 минут при комнатной температуре, жидкость удаляли. Каждую лунку промывали три раза с помощью 100 мкл 0,9% солевого раствор. Неспецифическую реакцию (холостое значение) определяли, используя 10 мкМ природного ингибитора киназы. Измерения радиоактивности осуществляли, используя TopCount. IC50 значения рассчитывали в RS1.
Литература: Kashishian и др. (2003) Molecular Cancer Therapeutics 1257.
Б) ДНК-ПК ФОСФОРИЛИРОВАНИЕ НА СЕРИНЕ 2056 (КЛЕТОЧНЫЙ)
НСТ116 клетки культивировали при 37°С и 10% CO2 в MEM альфа-среде с 10% фетальной бычьей сыворотки и 2 мМ глутамина. Клетки отсоединяли от основания культуральных сосудов с помощью трипсина /EDTA, центрифугировали в центрифужных пробирках, ресуспендировали в свежей среде, и определяли плотность клеток. 100000 клеток высевали в 1 мл культуральной среды на лунку планшета для культивирования клеток на 24 лунки и культивировали в течение ночи. На следующий день, к клеткам добавляли 10 мкМ блеомицина (ДНК интеркалятор и индуктор двухцепочечных разрывов ДНК) и тестируемые вещества в свежей культуральной среде, и их культивировали дополнительно в течение шести часов. После этого осуществляли лизис клеток, и клеточные лизаты добавляли к блокирующему ELISA планшету на 96 лунок, покрытому ДНК-ПК-специфическими антителами (Sigma-Aldrich WH0005591M2: общая ДНК-ПК; Abeam abl8192 или Epitomics EM09912: фосфо-серин 2056 ДНК-ПК) и инкубировали при 4°С в течение ночи. После этого планшеты ELISA на 96 лунок обрабатывали детекторным антителом (Abcam ab79444: общая ДНК-ПК) и конъюгатом стрептавидин-HRP. Развитие ферментативной реакции осуществляли с помощью хемилюминесцентного реагента, хемилюминесценцию измеряли с помощью Mithras LB940. Сигналы с фосфо-ДНК-ПК-специфическим антителом стандартизировали с сигналом с антителом к общему белку ДНК-ПКс. Определение IC50 значений или процентных значений осуществляли путем сравнения с эталоном с уровнем сигнала обработанной блеомицином контрольной группы с носителем (100% контроля). ДМСО контроль использовали в качестве холостого.
В) Активность ионного канала Kv11.1 (hERG) (анализ фиксации потенциала)
Метод для обнаружения и характеристики тестируемых веществ, которые интерферируют с Kv11.1 (hERG) каналом: Kv11.1 (hERG, ген специфических калиевых каналов сердца человека) представляет собой калиевый канал, которые играет центральную роль для реполяризации клеток в желудочковых кардиомиоцитах.
Измерения фиксации потенциала осуществляли при комнатной температуре в конфигурации целой клетки на эмбриональных почечных клетках человека (HEK293), в которые трансфектировали подходящим образом ген hERG. Конфигурации целой клетки осуществляли, используя автоматизированное устройство фиксации потенциала (PatchlinerTM, Nanion Technologies, Munich). Оно представляло собой систему на основании стеклянного чипа, для которого возможно одновременное измерение целых клеток на вплоть до 8 клетках. Стеклянный чип представляет собой отверстие определенного размера, к которому клетку переносят на Gigaseal путем применения сниженного давления и переводят в конфигурацию целой клетки. Буфер, клеточную суспензию и тестируемые вещества добавляют через микроканалы чипа, используя пипетку, покрытую тефлоном.
Клетки фиксировали к исходному потенциалу -80 мВ. Для измерения опосредованного веществом ингибирования Kv11.1 канала применяли следующий протокол напряжения с интервалами 10 секунд: 51 мс / -80 мВ, 500 мс / +40 мВ, 500 мс / -40 мВ, 200 мс / -80 мВ. Ток утечки вычитали с помощью Р4 метода. Клетки ресупендировали в экстраклеточном буфере (ЕС) и применяли на чипе. После сбора клеток, уплотнение улучшали путем добавления буфера, улучшающего уплотнение. Как только достигали конфигурации целой клетки, буфер, улучшающий уплотнение, отмывали и заменяли на экстраклеточный буфер. Измерения начинали на ЕС в течение 1,5 минуты. Затем применяли ДМСО (контрольный наполнитель, 0,1% ДМСО), и записывали контрольный ток в течение 3 минут. После этого добавляли тестируемое вещество дважды в одной и той же концентрации, и калиевый ток записывали в течение 3,5 минут в каждом случае.
Если измеренный результат для тестируемого вещества при начальной концентрации 10 мкМ был меньше, чем (-)50% эффекта (пороговое значение) (например, (-)60% эффекта), то тестируемое вещество, для определения взаимосвязи доза/действие, добавляли кумулятивно в возрастающей концентрации, где каждую концентрацию измеряли в течение 5 минут. Используемым эталонным веществом был блокатор ионного канала Kv11.1 (hERG) хинидин. Эффекты тестируемых веществ и хинидина стандартизировали с ассоциированным контрольным носителем. Эффект на активность канала Kv11.1 (hERG) оценивали на основании калиевого тока при -40 мВ. Для расчетов, ток оценивали для соответствующей конечной записи. Ингибирование канала Kv11.1 (hERG), индуцированное тестируемым веществом, стандартизировали с контрольным носителем (0,1% ДМСО).
Во время измерения, отбирали аликвоту тестируемого вещества для определения концентрации. Пробу сразу измеряли с помощью ВЭЖХ, и конечную концентрацию определяли согласно калибровочной кривой. Если измеренный результат для тестируемого вещества при начальной концентрации 10 мкМ больше, чем или равен (-)50% эффекта (пороговое значение) (например, (-)30% эффекта, то есть 30% ингибирование при 10 мкМ), то Ki рассчитывали в соответствии со следующей формулой: Ki=1,0Е-5 × (100 + % эффекта) / (-% эффекта), [М].
Измеренный результат (-)30% эффекта при концентрации тестируемого вещества 10 мкМ обеспечивает Ki 23 мкМ.
Г) Анализ связывания ионного канала Kv11.1
Kv11.1 (hERG = ген специфических калиевых каналов сердца человека) представляет собой сердечный K+ канал, которые должен, если это возможно, не взаимодействовать с тестируемыми веществами. Это взаимодействие количественно определяют с помощью Predictor™ hERG Fluorescence Polarizations (FP) Assays от Life Technologies. Согласно принципу этого анализа, мембраны клеток кардиомиоцитов, имеющих определенную пропорцию Kv11.1 каналов, выделяли. Меченный красителем Kv11.1 связывающий партнер обеспечивает высокий сигнал флуоресцентной поляризации путем взаимодействия с Kv11.1. В случае вытеснения красителя, происходит уменьшение сигнала флуоресцентной поляризации.
Этот анализ осуществляли автоматически следующим образом: 15 нл тестируемых веществ (наивысшая концентрация: 10 мМ, 10 концентрации: факторы разведения 1:3,16, ДМСО) переносили в пустые микротитровальные планшеты, имеющие 384 лунок, с помощью акустической пипетки. После этого добавляли 3 мкл выделенных мембран. Мембраны и тестируемые вещества инкубировали при 22°С в течение 15 минут (+/-5 минут). На следующей стадии, добавляли меченный красителем связывающий партнер, затем инкубировали при 22°С. После инкубирования в течение двух часов, осуществляли измерения флуоресцентной поляризации на Envision multimode Reader. Измеренные необработанные данные стандартизировали с помощью Genedata Assay Analyzer. Значения IC50 и % эффекта рассчитывали на Genedata Condoseo.
Химический синтез
Выше и ниже, все температуры указаны в °С.
Распределение стереохимической конфигурации энантиомерных примеров 27, 72, 82, 83, 135, 136, 185, 234, 251, 455 и 456 подтверждали с помощью рентгеноструктурных анализов. Для примеров 234 и 251, идентификацию осуществляли путем кристаллизации и рентгеноструктурного анализа предшественника.
Другие соединения, обозначенные как хиральные в таблицах (звездочка на ассиметричном атоме С) получали с помощью хроматографии на хиральной твердой фазе. Энантиомер, который элюируется первым в соответствующих условиях, называли "Эна1", энантиомер, который элюируется вторым, называли "Эна2".
ПРИМЕРЫ 1 и 2:
(3,5-Дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (1)
(4-Хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (2)

(3-Бром-4-фторфенил)ацетонитрил (4,00 г, 18,32 ммоль), бис(пинаколато)дибор (5,22 г, 20,15 ммоль), ацетат калия (55,86 ммоль) и бис(трифенилфосфин)палладий(II) хлорид (15,2% Pd) (393,53 мг, 0,55 ммоль) растворяли в безкислородном 1,4-диоксане (40 мл, макс. 0,005% воды) в атмосфере аргона. После этого реакционную смесь нагревали при температуре 130°С в течение 90 минут. После завершения реакционного превращения, смесь фильтровали через диатомовую землю. Фильтрат разводили дихлорметаном (200 мл) и водой (50 мл) и экстрагировали. Органическую фазу высушивали над сульфатом натрия, после этого фильтровали и упаривали насухо в вакууме, получая [4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]ацетонитрил в виде масла (7,59 г, чистота 81%, МС: 262,2 [М+Н+]), который подвергали дальнейшей реакции без дополнительной обработки.

4-Фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]ацетонитрил (7,60 г, 23,53 ммоль), 1,4-диоксан (85,6 мл, макс. 0,005% воды), 4-хлор-7-морфолин-4-илхиназолин (5,00 г, 20,02 ммоль), бис(трициклогексилфосфин)палладий(II) дихлорид (597,24 мг, 0,80 ммоль) и раствор карбоната натрия (2,0 М, 30 мл, 60,07 ммоль) изначально вносили в трехгорлую колбу. Полученную суспензию нагревали при температуре 140°С в атмосфере азота при перемешивании в течение 2,5 ч. После завершения реакции, смесь охлаждали до комнатной температуры и фильтровали через диатомовую землю. Фильтрат разводили этилацетатом (250 мл) и водой (100 мл) и экстрагировали. Водную фазу промывали дважды этилацетатом (75 мл в каждом случае). Объединенные органические фазы высушивали над сульфатом натрия, после этого фильтровали и упаривали насухо в вакууме. Для дальнейшей обработки, остаток суспендировали в трет-бутил метиловом эфире, отфильтровывали и промывали дважды с дополнительным количеством трет-бутил метилового эфира (30 мл в каждом случае). Осадок после фильтрации высушивали в течение ночи в вакууме, получая [4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрил (4,91 г, 13,49 ммоль, МС: 349,1 [М+Н+], 67% выход) в виде желтого твердого вещества.
[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрил (400 мг, 1,12 ммоль), 4-хлор-3,5-дифторпиридин (189,9 мг, 1,23 ммоль) растворяли в безкислородном, дегазированном тетрагидрофуране (8 мл, макс. 0,0075% воды) в атмосфере безводного аргона. После этого к реакционной смеси добавляли трет-бутилат калия (263,9 мг, 2,35 ммоль), при этом образовывался темно-красный раствор, который перемешивали при комнатной температуре дополнительно в течение 30 минут. После завершения реакции, смесь разводили насыщенным раствором хлорида аммония (20 мл) и водой (50 мл). После этого водную фазу дважды экстрагировали с помощью дихлорметана (60 мл в каждом случае). Органическую фазу высушивали над сульфатом натрия, отфильтровывали и упаривали насухо в вакууме на роторном испарителе. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан / 0-4 об. % этанола), получая смесь (420 мг, приблизительно 5:3) масел (3,5-дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрил (263 мг, 0,57 ммоль, МС: 462,1 [М+Н+], 50% выход) и (4-хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрил (157 мг, 0,33 ммоль, МС: 478,1/480,1 [М+Н+], 29% выход).

Смесь (420 мг, приблизительно 5:3) (3,5-дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрила и (4-хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрила растворяли в ацетонитриле (12,7 мл). Трет-бутилат калия (80,90 мг, 0,72 ммоль) добавляли к реакционному раствору при перемешивании, при этом образовывался темно-красный раствор. После перемешивания в течение 15 минут, смесь охлаждали до 0°С, и затем по каплям добавляли 30% раствор перекиси водорода (276 мкл, 2,70 ммоль). Охлаждающую баню удаляли после перемешивания в течение 5 минут при 0°. Реакционный раствор перемешивали при комнатной температуре дополнительно в течение 1 ч. После завершения реакционного превращения, добавляли 10% раствор тиосульфата натрия (5 мл), и смесь разводили водой (25 мл). Водный раствор дважды экстрагировали с помощью дихлорметана (50 мл в каждом случае). Объединенные органические фазы высушивали над сульфатом натрия, отфильтровывали и упаривали насухо в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан / 0-4 об. % этанола), получая смесь (310 мг, приблизительно 3:1) масел (3,5-дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанон (233 мг, 0,50 ммоль, МС: 451,1 [М+Н+]) и (4-хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанон (77 мг, 0,16 ммоль, МС: 467,1/469,1 [М+Н+]).

Смесь (310 мг, приблизительно 3:1) (3,5-дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанона и (4-хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанона растворяли в метаноле (15 мл). После этого добавляли борогидрид натрия (30,4 мг, 0,80 ммоль) (выделение газа). Реакционный раствор перемешивали при комнатной температуре в течение 45 минут. После завершения реакции, смесь разводили насыщенным раствором хлорида аммония (5 мл) и водой (15 мл). После этого водную фазу экстрагировали три раза с помощью дихлорметана (20 мл в каждом случае). Объединенные органические фазы высушивали над сульфатом натрия, отфильтровывали и упаривали насухо в вакууме на роторном испарителе. Остаток растворяли в диметилсульфоксиде (4,8 мл) и очищали с помощью хроматографии путем препаративной ВЭЖХ (градиент: вода/1-50 об. % ацетонитрила в течение 21 минут, скорость потока 50 мл/минут). Фракции, содержащие продукт, объединяли, разводили насыщенным раствором гидрокарбоната натрия (5 мл в каждом случае) и дважды экстрагировали с помощью дихлорметана (40 мл в каждом случае). Органические фазы упаривали в вакууме, после этого остатки ресуспендировали в 1,4-диоксане (5 мл) и воде (30 мл) и лиофилизировали, получая чистый (3,5-дифторпиридин-4-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (ПРИМЕР 1, 42,50 мг, 0,09 ммоль, МС: 453,1 [М+Н+]) и (4-хлор-5-фторпиридин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (ПРИМЕР 2, 28,40 мг, 0,06 ммоль, МС: 469,1/471,1 [М+Н+]) в виде твердых веществ.
ПРИМЕР 37
(3-Хлорпиразин-2-ил)-[4-фтор-3-(6-морфолин-4-илтиено[3,2-d}пиримидин-4-ил)фенил]метанол (37)

Гидрид натрия (60% суспензия в парафиновом масле, 1,41 г, 35,0 ммоль) суспендировали в безводном тетрагидрофуране (25 мл) в атмосфере аргона в стеклянном сосуде. Затем медленно добавляли по каплям 4-метоксифенилметанол (4,21 г, 30,0 ммоль), растворенный в безводном тетрагидрофуране (5 мл), при перемешивании, и смесь перемешивали при комнатной температуре в течение 1 ч. После этого медленно добавляли суспензию 4-хлортиено[3,2-d}пиримидина (4,00 г, 23,4 ммоль) в безводном тетрагидрофуране (20 мл), и смесь перемешивали дополнительно в течение часа. После завершения реакционного превращения, осторожно добавляли метанол (15 мл), после этого смесь упаривали в вакууме и разводили смесью воды (100 мл) и этилацетата (150 мл). Водную фазу экстрагировали три раза этилацетатом (100 мл в каждом случае), высушивали над сульфатом натрия, отфильтровывали с отсасыванием, и фильтрат упаривали в вакууме. Остаток, не содержащий растворителя, ресуспендировали в этаноле (40 мл) и осторожно перемешивали приблизительно при 5°С в течение 16 часов. Кристаллы, которые осаждались в течение ночи, отфильтровывали с отсасыванием, промывали небольшим количеством холодного этанола и высушивали при комнатной температуре, получая 4-(4-метоксибензилокси)тиено[3,2-d}пиримидин (4,15 г, 15,24 ммоль, МС: 273,0 [М+Н+]), 65% выход) в виде кристаллического твердого вещества.

4-(4-Метоксибензилокси)тиено[3,2-d}пиримидин (2,60 г, 9,55 ммоль) растворяли в безводном тетрагидрофуране (35 мл) и охлаждали до (-)55°С. Свежеприготовленный раствор диизопропиламида лития (21 ммоль, приготовленный из диизопропиламина [2,13 г, 21 ммоль] и н-BuLi [15% раствор из н-гексана, 13,13 мл, 21 ммоль в безводном тетрагидрофуране [35 мл] при [-]10°С) добавляли по каплям при (-)55°С в течение 10 минут. Полученную суспензию перемешивали дополнительно. После этого добавляли 1,2-дибромэтан (10,76 г, 6,0 ммоль). Через дополнительные 10 минут, смесь нагревали до (-)20°С и перемешивали в течение 1 ч. При завершении реакции, реакционный раствор добавляли к 50% водному раствору гидрокарбоната натрия/ тиосульфата натрия (объемное соотношение 1:1, 120 мл). Водную фазу экстрагировали три раза с помощью этилацетата. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, после этого высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе. Остаток очищали с помощью колоночной флэш-хроматографии (градиент циклогексан / 0-18 об. % этилацетата, CombiFlash Rf 200, колонка 80 г диоксида кремния, скорость потока = 50 мл/минут, λ=220 нм), получая 6-бром-4-(4-метоксибензилокси)тиено[3,2-d}пиримидин (1,39 г, 3,95 ммоль, МС: 351,0/353,0 [М+Н+]), 41% выход) в виде твердого вещества.
6-Бром-4-(4-метоксибензилокси)тиено[3,2-d}пиримидин (1,38 г, 3,93 ммоль), морфолин (1,03 г, 11,79 ммоль), трет-бутилат натрия (1,13 г, 11,79 ммоль), (S)-(-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (S-BINAP, 122,3 мг, 0,196 ммоль) и трис(дибензилиденацетон)дипалладий (179,9 мг, 0,196 ммоль) растворяли в толуоле (20 мл) в атмосфере азота в микроволновом сосуде. Реакционный раствор нагревали при 95°С в течение 4 ч. После этого смесь разводили водой (60 мл) и дихлорметаном (60 мл). Водную фазу экстрагировали три раза с помощью дихлорметана. Объединенные органические фазы высушивали над сульфатом натрия и упаривали на роторном испарителе. Остаток очищали с помощью флэш-хроматографии (градиент дихлорметан / 5-25 об. % [дихлорметан/этанол 9:1], CombiFlash Rf 200, колонка 80 г диоксида кремния, λ=220 нм). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая 4-(4-метоксибензилокси)-6-морфолин-4-илтиено[3,2-d}пиримидин (756,0 мг, 2,12 ммоль, МС: 358,2 [М+Н+]) 54% выход) в виде твердого вещества.

4-(4-Метоксибензилокси)-6-морфолин-4-илтиено[3,2-d}пиримидин (923 мг, 2,58 ммоль) растворяли в тетрагидрофуране (5 мл) и метаноле (5 мл). Добавляли порциями Pd/C (5%, 1,9 г) (в начале реакции, через дополнительные 7 ч. и 24 ч.), и смесь гидрировали при максимальном давлении водорода 5 бар (Н2, чистота 3,0, 57,9 г) в течение 36 ч. Полученный раствор фильтровали через диатомовую землю и упаривали на роторном испарителе. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан/10-20 об. % [метанол/аммиак 10:1], CombiFlash Rf 200, колонка 24 г диоксида кремния, λ=220 нм). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая 6-морфолин-4-ил-3Н-тиено[3,2-d}пиримидин-4-он (361,0 мг, 1,521 ммоль, МС: 238,0 [М+Н+], 59% выход) в виде твердого вещества.
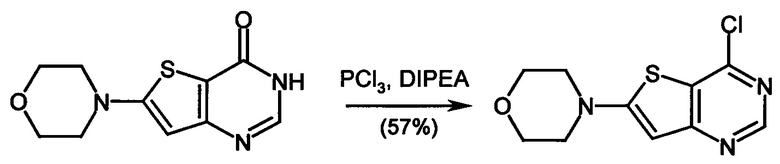
6-Морфолин-4-ил-3Н-тиено[3,2-d}пиримидин-4-он (206 мг, 0,87 ммоль) суспендировали в фосфорилхлориде (1,67 г, 10,89 ммоль). После этого к суспензии добавляли N-этилдиизопропиламин (56,1 мг, 0,43 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Для осуществления обычной обработки, добавляли смесь насыщенного раствора гидрокарбоната натрия (30 мл) и дихлорметана (20 мл). Полученный раствор экстрагировали три раза с помощью дихлорметана. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали насухо в вакууме, получая 4-хлор-6-морфолин-4-илтиено[3,2-d}пиримидин (127 мг, 0,497 ммоль, МС: 256,0/258,0 [М+Н+], 57% выход) в виде твердого вещества.

Используя в качестве исходных соединений 4-хлор-6-морфолин-4-илтиено[3,2-d}пиримидин, (3-хлорпиразин-2-ил)-[4-фтор-3-(6-морфолин-4-илтиено[3,2-d}пиримидин-4-ил)фенил]метанол (ПРИМЕР 37) получали аналогично последовательности синтеза, описанной для Примеров 1 и 2.
Соединения, которые получены в соответствии с Примерам 1, 2 и 37, можно найти в таблице 1 ниже.


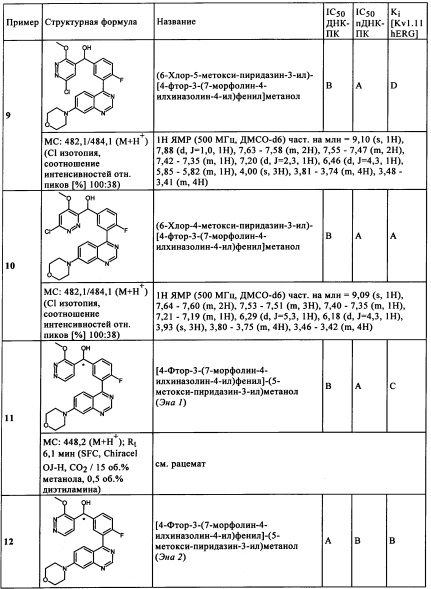
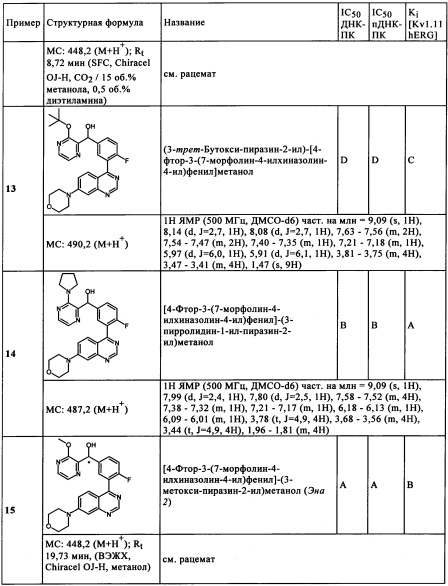
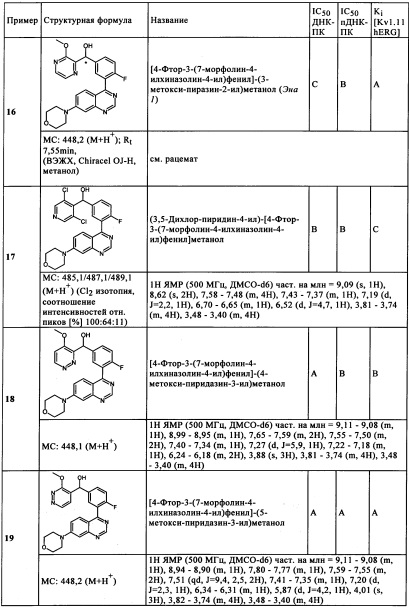


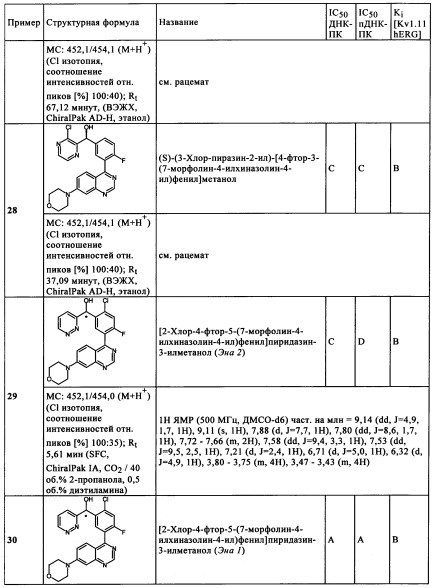














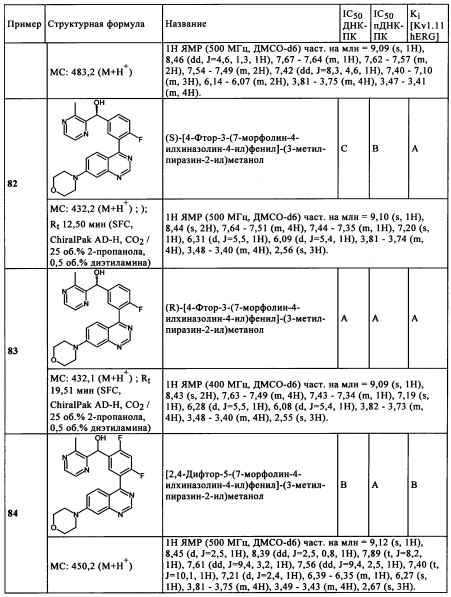


ПРИМЕРЫ 92 и 93:
3-[[2-Хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]гидроксиметил]-1Н-пиридазин-6-он (92)
6-{[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-2-этил-2Н-пиридазин-3-он (93)
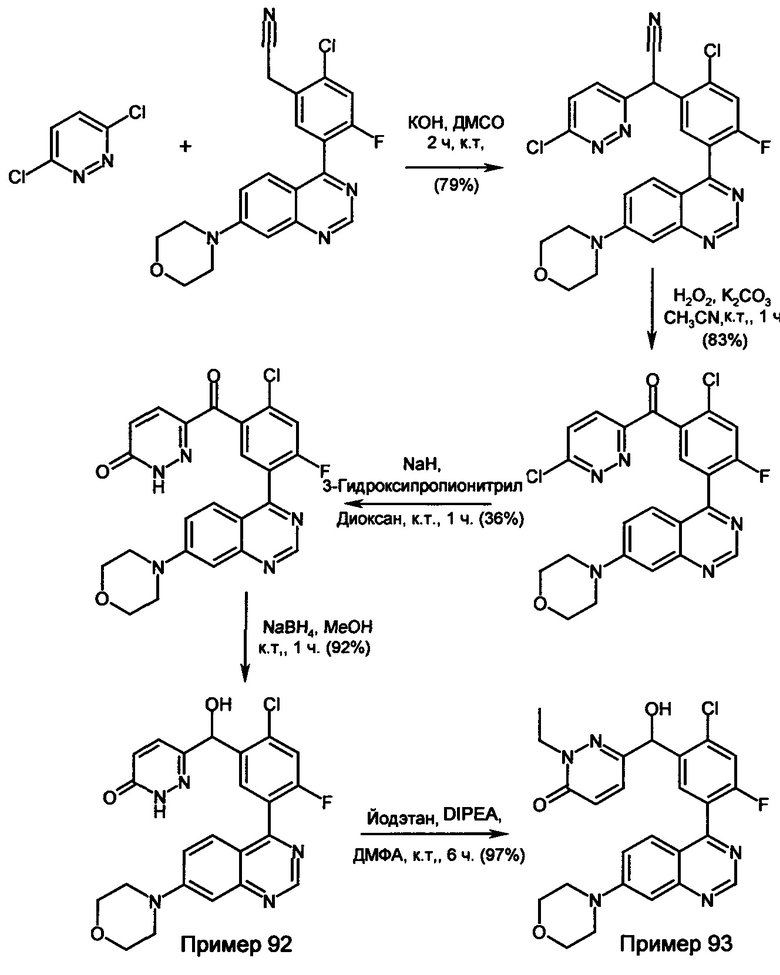
[2-Хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]-(6-хлорпиридазин-3-ил)метанон, используя в качестве исходных соединений 2,6-дихлорпиридазин и 2-[2-хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]ацетонитрил, и 3-[[2-хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]гидроксиметил]-1Н-пиридазин-6-он (ПРИМЕР 92) получали аналогично способам синтеза, описанным для ПРИМЕРОВ 1 и 2.
Получение 3-[2-хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)бензоил]-1Н-пиридазин-6-она из [2-хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]-(6-хлорпиридазин-3-ил)метанона:
[2-Хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]-(6-хлорпиридазин-3-ил)метанон (2,0 г, 4,13 ммоль) растворяли в 1,4-диоксане (80 мл, макс. 0,005% воды) в атмосфере аргона. После этого добавляли 3-гидроксипропионитрил (570 мкл мл, 8,27 ммоль) и гидрид натрия (60% дисперсия в парафиновом масле) (215 мг; 5,37 ммоль) (выделение газа). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После завершения реакции, смесь осторожно разводили водой (100 мл) и нейтрализовали, используя соляную кислоту (1,0 М). После этого водную фазу дважды экстрагировали этилацетатом (200 мл в каждом случае). Объединенные органические фазы промывали насыщенным раствором хлорида натрия, после этого высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе. Остаток очищали с помощью колоночной флэш-хроматографии (дихлорметан /0-10 об. % этанола, CombiFlash Rf 200), получая 3-[2-хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)бензоил]-1Н-пиридазин-6-он (695 мг, 1,47 ммоль, МС: 466,1/468,1 [М+Н+]), 36% выход) в виде твердого вещества.
Получение 6-{[2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-2-этил-2Н-пиридазин-3-она (ПРИМЕР 93) из 3-[[2-хлор-4-фтор-5 -(7-морфолинилхиназолин-4-ил)фенил]гидроксиметил]-1Н-пиридазин-6-она (ПРИМЕР 92):
3-[[2-Хлор-4-фтор-5-(7-морфолинилхиназолин-4-ил)фенил]гидроксиметил]-1Н-пиридазин-6-он (150 мг; 0,316 ммоль) растворяли в N,N-диметилформамиде (5,0 мл). После этого добавляли йодэтан (52 мкл, 0,632 ммоль) и карбонат калия (132 мг, 0,947 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. После завершения реакции, смесь декантировали на воду (100 мл). После этого водную фазу дважды экстрагировали этилацетатом (100 мл в каждом случае). Объединенные органические фазы промывали водой (40 мл), после этого высушивали над сульфатом натрия, фильтровали и упаривали насухо в вакууме. Остаток суспендировали в ацетоне и отфильтровывали с отсасыванием. Осадок после фильтрации высушивали при комнатной температуре в высоком вакууме, получая 6-{[2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-2-этил-2Н-пиридазин-3-он (ПРИМЕР 93, 157 мг, 0,31 ммоль, МС: 496,1/498,1 [М+Н+], 97% выход) в виде твердого вещества.
Соединения, которые получены в соответствии с ПРИМЕРОМ 93, можно найти в таблице 2 ниже.



ПРИМЕР 100:
[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-[6-(оксетан-3-илокси)пиридазин-3-ил]метанол (100)

[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-хлорпиридазин-3-ил)метанон (700 мг, 1,30 ммоль) и оксетан-3-ол (112 мг, 1,43 ммоль) изначально вносили растворенные в 1,4-диоксане (25 мл, макс. 0,005% воды) в атмосфере аргона. После этого добавляли гидрид натрия (60% дисперсия в парафиновом масле, 62 мг, 1,56 ммоль) (выделение газа). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения реакции, смесь осторожно разводили водой (80 мл) и нейтрализовали, используя соляную кислоту (1,0 М). После этого водную фазу дважды экстрагировали этилацетатом (80 мл в каждом случае). Объединенные органические фазы промывали водой (20 мл), после этого высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе. Остаток очищали с помощью колоночной флэш-хроматографии (дихлорметан / 0-10 об. % этанола, CombiFlash Rf 200), получая [2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-[6-(оксетан-3-илокси)пиридазин-3-ил]метанон (264 мг, 0,506 ммоль, 522,2 [М+Н+]), 39% выход) в виде твердого вещества.
[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-[6-(оксетан-3-илокси)пиридазин-3-ил]метанол (ПРИМЕР 100) получали аналогично способу синтеза, описанному для ПРИМЕРОВ 1 и 2, используя в качестве исходных соединений [2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-[6-(оксетан-3-илокси)пиридазин-3-ил]метанон.
Соединения, которые получены в соответствии с ПРИМЕРОМ 100, можно найти в таблице 3 ниже.



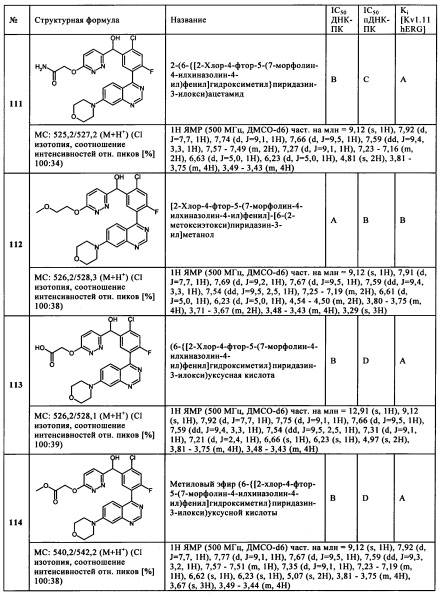


ПРИМЕРЫ 120, 121 и 122:
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-метоксифенил)метанол (ПРИМЕР 120)
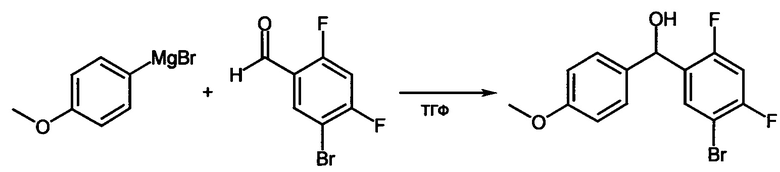
5-Бром-2,4-дифторбензальдегид (280 мг, 1,27 ммоль) в безводном тетрагидрофуране (10 мл) изначально вносили в трехгорлую колбу с встроенным термометром, входным отверстием для защитного газа, диафрагмой и магнитной мешалкой, которую высушивали путем нагревания. Медленно по каплям добавляли бромид 4-метоксифенилмагния (1 М в ТГФ, 1,39 мл, 1,39 ммоль) при 5°С, и реакционный раствор перемешивали при комнатной температуре в течение 18 ч. После этого к реакционному раствору добавляли воду (20 мл). Фазы разделяли, и водную фазу дважды экстрагировали этилацетатом (20 мл). Объединенные органические фазы промывали водой, высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе, получая (5-бром-2,4-дифторфенил)-(4-метоксифенил)метанол (530 мг, 1,61 ммоль, МС: 353 [М+Н+]) в виде маслянистого неочищенного продукта, который использовали без дополнительной очистки на следующей стадии синтеза.
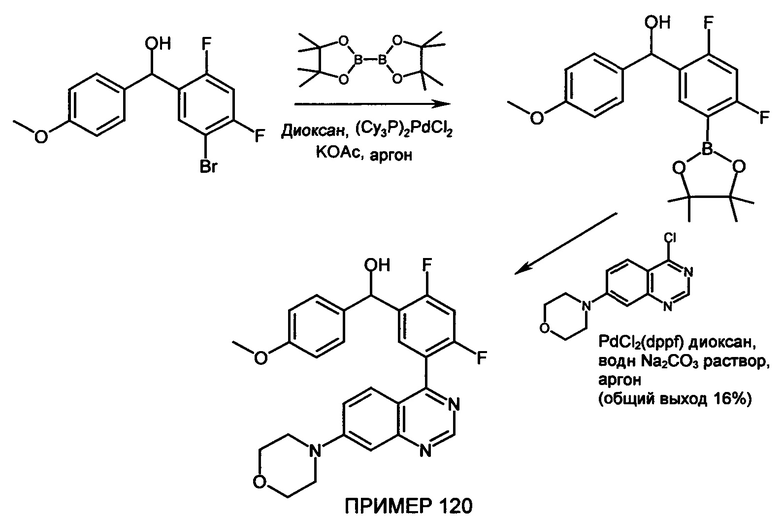
Используя в качестве исходных соединений (5-бром-2,4-дифторфенил)-(4-метоксифенил)метанол, [2,4-дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-метоксифенил)метанол (ПРИМЕР 120) получали аналогично способам синтеза, описанным для ПРИМЕРОВ 1 и 2.
(6-Дифторметоксипиридазин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (ПРИМЕР 121)
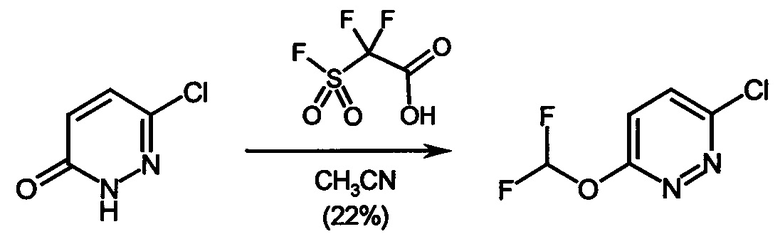
6-Хлор-2Н-пиридазин-3-он (944 мг, 7,23 ммоль) и дифтор(фторсульфонил)уксусную кислоту (1,42 г, 7,96 ммоль) растворяли в ацетонитриле (19 мл) в сосуде с магнитной мешалкой и перемешивали при комнатной температуре в течение 40 ч. После этого реакционный раствор разводили этилацетатом (150 мл) и промывали последовательно водой, насыщенным раствором гидрокарбоната натрия и снова водой. Органическую фазу высушивали, используя сульфат натрия, фильтровали и упаривали насухо на роторном испарителе. Остаток ресуспендировали в циклогексане, повторно фильтровали, и растворитель удаляли на роторном испарителе. Полученный остаток очищали с помощью колоночной флэш-хроматографии (градиент циклогексан/0-50 об. % этилацетата, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая 3-хлор-6-(дифторметокси)пиридазин (285 мг, 1,58 ммоль, МС: 181,0/183,1 [М+Н+]), 22% выход) в виде бесцветной жидкости.

Порошкообразный гидроксид калия (603 мг, 10,75 ммоль) суспендировали в безводном N,N-диметилформамиде (2 мл) в стеклянном сосуде с магнитной мешалкой и перемешивали при комнатной температуре в течение 30 минут. После этого по каплям добавляли (3-бром-4-фторфенил)ацетонитрил (1,0 г, 4,67 ммоль), растворенный в N,N'-диметилформамиде (1,3 мл). Реакционную смесь перемешивали при комнатной температуре дополнительно в течение 30 минут. После этого к реакционной смеси порциями добавляли (5-бром-2,4-дифторфенил)-(4-метоксифенил)метанол (506 мг, 2,80 ммоль) и перемешивали при 50°С в течение 2 ч. в бескислородной атмосфере защитного газа аргона. Реакционную смесь добавляли к смеси воды (50 мл) и насыщенного раствора хлорида натрия (35 мл) и дважды экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе. Остаток очищали с помощью колоночной хроматографии с обращенной фазой (градиент вода/ацетонитрил с 0,1 об. % муравьиной кислоты, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая (3-бром-4-фторфенил)-(6-дифторметоксипиридазин-3-ил)ацетонитрил (146 мг, 0,41 ммоль, МС: 358,0/360,0 [М+Н+], 14% выход) в виде жидкости. 2-(3-Бром-4-фторфенил)-2-(6-хлорпиридазин-3-ил)ацетонитрил образовывался в качестве побочного продукта.
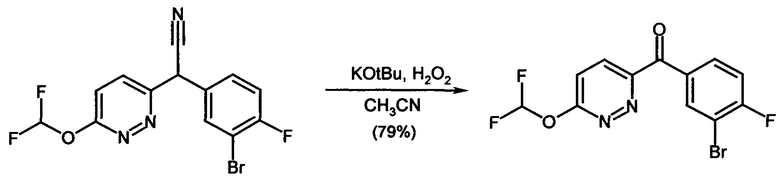
(3-Бром-4-фторфенил)-(6-дифторметоксипиридазин-3-ил)ацетонитрил (146 мг, 0,41 ммоль) растворяли в безводном ацетонитриле (4 мл). После этого добавляли трет-бутилат калия (43,6 мг, 0,388 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 25 минут. После этого реакционный раствор охлаждали до 0°С на ледяной бане, добавляли по каплям перекись водорода (30% в воде, 92 мкл, 0,90 ммоль), и реакционную смесь перемешивали сначала при 0°С дополнительно в течение 25 минут и затем при комнатной температуре в течение 1 ч. Для осуществления обычной обработки, реакционную смесь добавляли к воде (40 мл) и дважды экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе, получая (3-бром-4-фторфенил)-(6-дифторметоксипиридазин-3-ил)метанон (113 мг, 0,32 ммоль, МС: 346,9/349,0 [М+Н+], 79% выход) в виде твердого вещества.

(3-Бром-4-фторфенил)-(6-дифторметоксипиридазин-3-ил)метанон (126 мг, 0,36 ммоль) растворяли в метаноле (4 мл). После этого добавляли порциями борогидрид натрия (60,4 мг, 1,60 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции, смесь разводили насыщенным раствором хлорида аммония (5 мл) и после этого дважды экстрагировали этилацетатом (30 мл). Объединенные органические фазы промывали водой, высушивали над сульфатом натрия, фильтровали и упаривали насухо на роторном испарителе, получая (3-бром-4-фторфенил)-(6-дифторметоксипиридазин-3-ил)метанол (127 мг, МС: 349/351[М+Н+]) в виде неочищенного продукта в форме твердого вещества, которое использовали без дополнительной очистки для дальнейших стадий синтеза.
(6-Дифторметоксипиридазин-3-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол (ПРИМЕР 121) получали аналогично с помощью способа синтеза, описанного для [2,4-дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-метоксифенил)метанола (ПРИМЕР 120).
1-[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-1-(6-метоксипиридазин-3-ил)проп-2-ин-1-ол (ПРИМЕР 122)
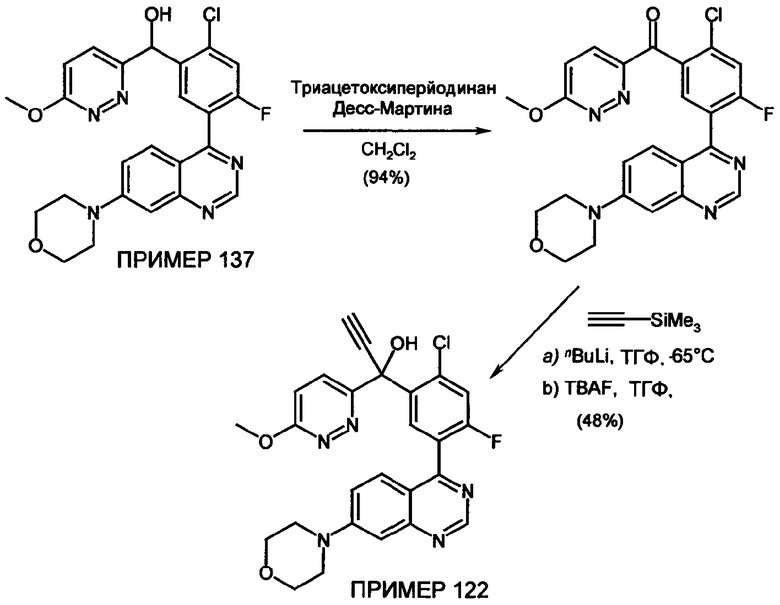
([2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанол (ПРИМЕР 137, 898 мг, 1,75 ммоль) растворяли в диохлорметане (15 мл). После этого добавляли триацетоксиперйодинан Десс-Мартина (15% в дихлорметане, 7,23 мл, 3,50 ммоль). Реакционную суспензию перемешивали при комнатной температуре в течение 1 ч. Для осуществления обычной обработки, добавляли воду (60 мл) и 10%, водный раствор тиосульфата натрия. Водную фазу дважды экстрагировали этилацетатом (80 мл в каждом случае). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), высушивали над сульфатом натрия, фильтровали, и фильтрат упаривали насухо в вакууме, получая 2,1 г неочищенного продукта в форме масла. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан / 0-25 об. % дихлорметана/этанола 9:1, CombiFlash Rf 200), получая [2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанон (792 мг, 1,65 ммоль, МС: 480,1/482,1 [М+Н+], 94% выход) в виде пены.
Триметилсилилацетилен (179 мкл, 125 мг, 1,25 ммоль), растворенный в безводном тетрагидрофуране (3 мл), изначально вносили в стеклянный сосуд с магнитной мешалкой и встроенным термометром в атмосфере аргона. Реакционный раствор охлаждали до (-)20°С, и медленно по каплям добавляли н-бутиллитий (1,6 М в н-гексане, 781 мкл, 1,25 ммоль). Реакционную смесь перемешивали при (-)20°С дополнительно в течение 30 минут. После этого реакционный раствор охлаждали до (-)70°С, и после этого по каплям добавляли [2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанон (200 мг, 0,417 ммоль), растворенный в безводном тетрагидрофуране (6 мл). Температуру реакционной смеси повышали до (-)40°С в течение периода 1 ч. После этого добавляли воду (40 мл), и фазы разделяли. Органическую фазу дважды экстрагировали с помощью дихлорметана. Объединенные органические фазы высушивали, используя сульфат натрия, фильтровали и упаривали насухо в вакууме. Остаток растворяли в безводном тетрагидрофуране (4 мл), и добавляли тригидрат фторида тетра-н-бутиламмония (109 мг, 0,42 ммоль). После этого смесь перемешивали при комнатной температуре в течение 18 ч. После этого летучие компоненты реакции удаляли на роторном испарителе. Остаток предварительно очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан / 0-34 об. % дихлорметана/этанола 1:1, CombiFlash Rf 200). Фракции, содержащие продукт, объединяли, и растворители удаляли в вакууме на роторном испарителе. Остаток в заключение очищали с помощью препаративной хроматографии с обращенной фазой (Chromolith RP-18e 21,2×100 мм, скорость потока: 50 мл/минут, длина волны: 220 нм). Летучие компоненты растворителя подходящих фракций удаляли с помощью вакуумной центрифуги (Genevac HT-12), и продукт лиофилизировали из ацетонитрила/воды (1:3 объемных частей), получая 1-[2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-1-(6-метоксипиридазин-3-ил)проп-2-ин-1-ол (ПРИМЕР 122, 102 мг, 0,20 ммоль, МС: 506,1/508,1 [М+Н+], 48% выход) в виде твердого вещества.
Соединения, которые получены в соответствии с ПРИМЕРАМИ 120, 121 и 122, можно найти в таблице 4 ниже.

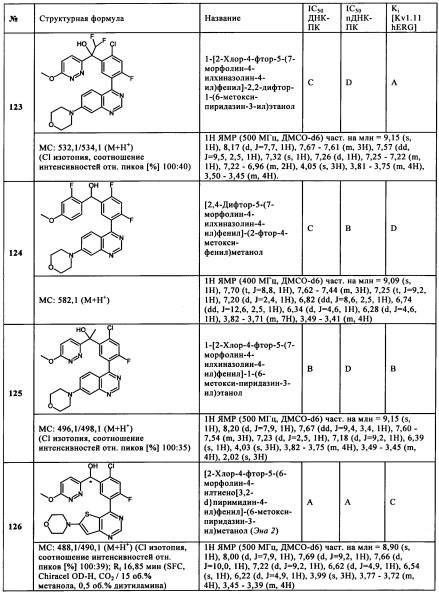
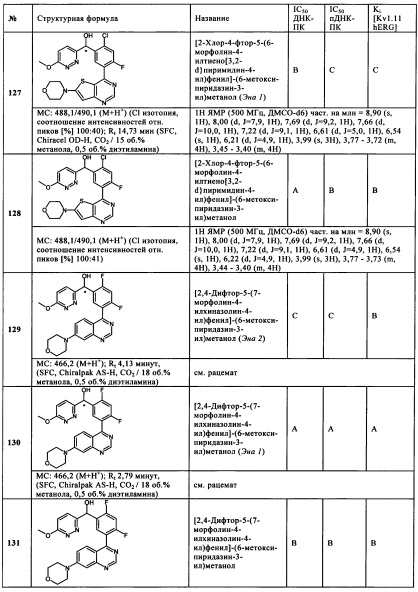


ПРИМЕР 138
1-[5-(7-Морфолин-4-илхиназолин-4-ил)пиридин-3-ил]-1-тиазол-2-илэтанол (ПРИМЕР 138)

Тиазол (143 мкл, 2,0 ммоль) в безводном тетрагидрофуране (10 мл) изначально вносили в трехгорлую колбу, которую высушивали путем нагревания. Реакционный раствор охлаждали до (-)78°С с помощью ацетона/бани с сухим льдом. Добавляли по каплям н-бутиллитий (15% раствор в н-гексане, 1,63 мл, 2,6 ммоль) в течение периода 10 минут при постоянной температуре. Реакционную смесь перемешивали дополнительно в течение 10 минут. После этого суспензию нагревали до (-)30°С и повторно охлаждали до (-)55°С, и добавляли по каплям 1-(5-бромпиридин-3-ил)этанон (380 мг, 1,90 ммоль), растворенный в безводном тетрагидрофуране (6 мл), при (-)40°С. Температуре реакции предоставляли возможность повышаться до (-)10°С в течение 1,5 ч. После завершения реакции (ВЭЖХ контроль), добавляли насыщенный раствор хлорида аммония, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь добавляли к двухфазному раствору воды (60 мл) и этилацетата (80 мл) и экстрагировали три раза с помощью этилацетата. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия, фильтровали и упаривали на роторном испарителе. Маслянистый неочищенный продукт очищали с помощью колоночной флэш-хроматографии (растворитель: дихлорметан/2,0 об. % метанола, затем дихлорметан/3,0 об. % метанола + 1,0 об. % аммиака, количество флэш-силикагеля 30 г). Фракции, содержащие продукт, объединяли, и растворители удаляли в вакууме на роторном испарителе, получая 1-(5-бромпиридин-3-ил)-1-тиазол-2-илэтанол (479 мг, 1,68 ммоль, МС: 285,0/287,0 [М+Н+], 84% выход) в виде масла.

1-(5-Бромпиридин-3-ил)-1-тиазол-2-илэтанол (162 мг, 0,55 ммоль), бис(пинаколато)дибор (140 мг, 0,55 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (Dppf, 7,1 мг, 0,013 ммоль), 1,1'-бис(дифенилфосфино)ферроценпалладий(II) дихлорид [Pd(dppf)Cl2, 10,4 мг, 0,013 ммоль] и ацетат калия (167 мг, 1,7 ммоль) суспендировали в безводном, бескислородном 1,4-диоксане в стеклянном сосуде с магнитной мешалкой. Стеклянный сосуд запечатывали, используя мембрану. Реакционный раствор перемешивали и нагревали при 115°С в течение 2,5 ч. За осуществлением реакции наблюдали с помощью ВЭЖХ. К реакционному раствору добавляли 4-хлор-7-морфолин-4-илхиназолин (106 мг, 0,43 ммоль), бис(трициклогексилфосфин)палладий(II) дихлорид (9,4 мг, 0,013 ммоль) и 2,0 М раствор карбоната натрия (531 мкл). После этого реакционную смесь перемешивали при температуре 125°С в течение 1,5 ч. Смесь декантировали в воду /дихлорметан (1:1 объемных частей, 40 мл), и полученный раствор экстрагировали три раза с помощью дихлорметана. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали на роторном испарителе. Остаток очищали с помощью флэш-хроматографии [градиент: дихлорметан / 20-58 об. % смеси растворителей дихлорметан/9:1 (объемных частей), CombiFlash Rf 200]. Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая 1-[5-(7-морфолин-4-илхиназолин-4-ил)пиридин-3-ил]-1-тиазол-2-илэтанол (ПРИМЕР 138, 95 мг, 0,23 ммоль, МС: 420,2 [М+Н+], 53% выход) в виде масла.
ПРИМЕР 139
{3-[7-(3,6-Дигидро-2Н-пиран-4-ил)хиназолин-4-ил]-4-фторфенил}тиазол-2-илметанол (139)
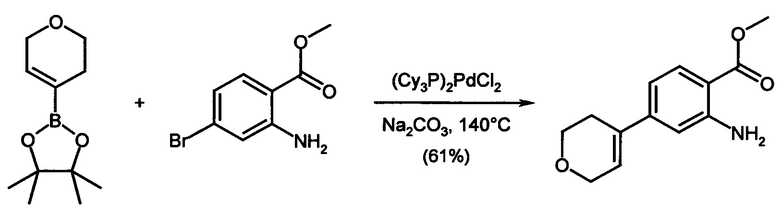
4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидро-2Н-пиран (575 мг, 2,74 ммоль), метил 2-амино-4-бромбензоат (600 мг, 2,61 ммоль), бис(трициклогексилфосфин)палладий(II) дихлорид (57,8 мг, 0,078 ммоль) и бескислородный 2,0 М раствор карбоната натрия (3,26 мл, 6,52 ммоль) в дегазированном, бескислородном 1,4-диоксане (12 мл) изначально вносили в микроволновой стеклянный сосуд с магнитной мешалкой. Смесь веществ нагревали при 135°С в течение периода 55 минут в Personal Chemistry Microwave Synthesiser при 100 Вт. После этого реакционный раствор декантировали в смесь воды (40 мл) и смесь этилацетата (30 мл). Полученный раствор экстрагировали три раза с помощью этилацетата. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия, фильтровали и упаривали в вакууме на роторном испарителе. Остаток очищали с помощью флэш-хроматографии [градиент: дихлорметан / 0-10 об. % смеси растворителей дихлорметан/метанол 10:1 (объемных частей), CombiFlash Rf 200]. Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая метил 2-амино-4-(3,6-дигидро-2Н-пиран-4-ил)бензоат (371,1 мг, 1,59 ммоль, МС: 234,2 [М+Н+], 61% выход) в виде твердого вещества
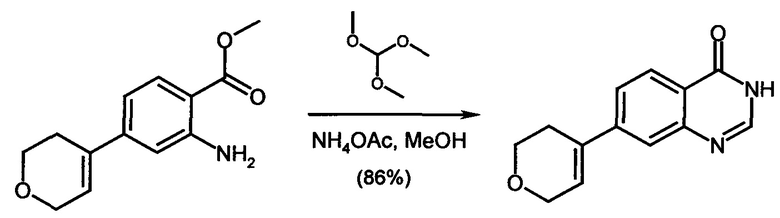
Метил 2-амино-4-(3,6-дигидро-2Н-пиран-4-ил)бензоат (620 мг, 2,66 ммоль), триметил ортоформиат (564,1 мг, 5,32 ммоль) и ацетат аммония (410 мг, 5,32 ммоль), растворенный в метаноле (20 мл), изначально вносили в стеклянный сосуд с магнитной мешалкой. Смесь веществ перемешивали в течение ночи при 80°С. После этого добавляли воду (10 мл), и твердое вещество, которое осаждались, отфильтровывали с отсасыванием, промывали небольшим количеством воды и после этого высушивали в вакууме, получая 7-(3,6-дигидро-2Н-пиран-4-ил)-3Н-хиназолин-4-он (520 мг, 2,28 ммоль, МС: 229,1 [М+Н+], 86% выход) в виде твердого вещества.
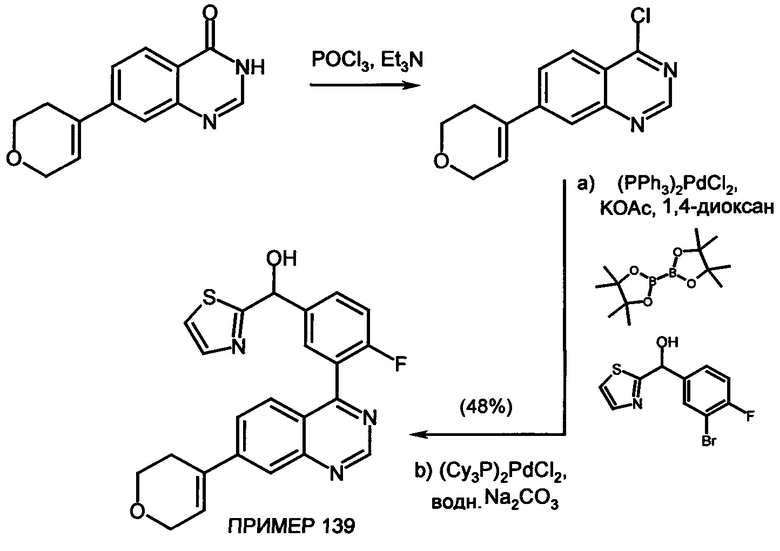
{3-[7-(3,6-Дигидро-2Н-пиран-4-ил)хиназолин-4-ил]-4-фторфенил}тиазол-2-илметанол (ПРИМЕР 139) получали аналогично способам синтеза для получения 1-[5-(7-морфолин-4-илхиназолин-4-ил)пиридин-3-ил]-1-тиазол-2-илэтанола (ПРИМЕР 138).
ПРИМЕР 140
[4-Фтор-3-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]тиазол-2-илметанол (140)
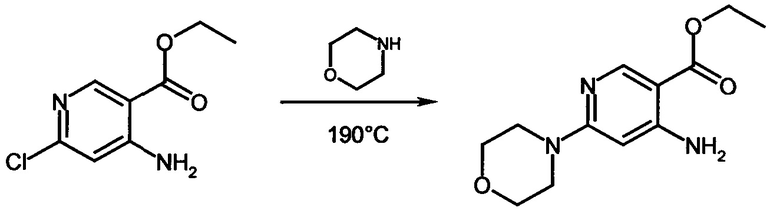
Этил 4-амино-6-хлорникотинат (8,38 г, 39,7 ммоль) растворяли в морфолине (40 мл). Смесь веществ нагревали при 120°С в течение 4 ч. После завершения реакции, охлажденный реакционный раствор декантировали в воду (400 мл). Вводную суспензию перемешивали в течение 10 минут, и после этого осадок отфильтровывали. Осадок после фильтрации промывали небольшим количеством воды и высушивали в течение ночи при 60°С в вакууме, получая чистый этил 4-амино-6-морфолин-4-илникотинат (8,55 г, 34,03 ммоль, МС: 252,2 [М+Н+], 85% выход) в виде бесцветного твердого вещества.

Аналогично процессу синтеза, описанного для примера 139, с помощью этил 4-амино-6-морфолин-4-илникотината (3,54 г, 14,1 ммоль) получали 7-морфолин-4-ил-3Н-пиридо[4,3-d}пиримидин-4-он (2,29 г, 9,86 ммоль, МС: 233,1 [М+Н+] 70% выход) в виде твердого вещества.

7-Морфолин-4-ил-3Н-пиридо[4,3-d}пиримидин-4-он (600 мг, 2,58 ммоль) суспендировали в 1,4-диоксане (10 мл). К реакционной смеси добавляли фосфорил хлорид (POCl3, 546 мкл, 5,9 ммоль) и основание Хьюнига (N-этилдиизопропиламин, 220 мкл, 1,29 ммоль). После этого смесь перемешивали при температуре 100°С в течение 3 ч. После завершения реакции, реакционный раствор декантировали в полу-насыщенный раствор гидрокарбоната натрия (80 мл). Водную фазу экстрагировали три раза с помощью дихлорметана (40 мл в каждом случае). Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали в вакууме на роторном испарителе, получая 4-хлор-7-морфолин-4-илпиридо[4,3-d}пиримидин (627 мг, 2,50 ммоль, МС: 251,0/253,0 [М+Н+], 96% выход) в виде твердого вещества.
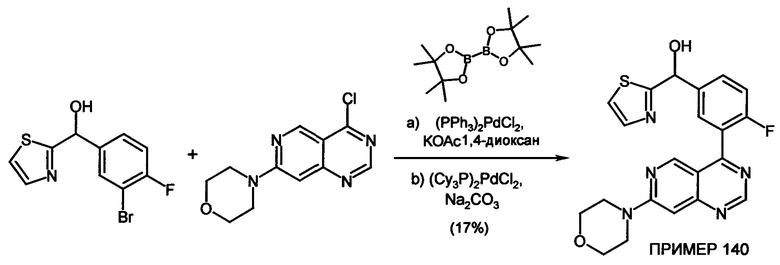
[4-Фтор-3-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 140) получали аналогично способам синтеза для получения 1-[5-(7-морфолин-4-илхиназолин-4-ил)пиридин-3-ил]-1-тиазол-2-илэтанола (ПРИМЕР 138).
ПРИМЕР 141
[2-Хлор-4-фтор-5-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]-(4-гидроксиметилтиазол-2-ил)метанол (141)

4-(трет-Бутилдиметилсиланилоксиметил)тиазол (10,15 г, 43,5 ммоль) растворенный в безводном тетрагидрофуране (78 мл) изначально вносили в двугорлую колбу с магнитной мешалкой, встроенным термометром и мембраной в атмосфере аргона. Реакционный раствор охлаждали до (-)75°С с помощью ацетона/бани с сухим льдом. Затем медленно по каплям к реакционному раствору добавляли н-бутиллитий (15% раствор в н-гексане, 29,3 мл, 46,6 ммоль) при постоянной температуре. Реакционный раствор перемешивали при (-)75°С дополнительно в течение 30 минут, после этого нагревали до 0°С. Затем повторно охлаждали до (-)50°С. Раствор, предварительно охлажденный до (-)50°С, 5-бром-2-хлор-4-фтор-N-метокси-N-метилбензамида (5,58 г, 12,4 ммоль), растворенного в безводном тетрагидрофуране (21 мл), медленно добавляли по каплям к реакционному раствору при (-)50°С в течение периода 1,5 ч. Реакционный раствор перемешивали при (-)50°С дополнительно в течение 30 минут. После завершения реакции, к реакционному раствору добавляли воду (20 мл). После этого реакционному раствору предоставляли возможность нагреться до комнатной температуры при перемешивании. Реакционный раствор разводили этилацетатом (400 мл) и насыщенным раствором хлорида натрия (100 мл). Фазы разделяли, и водную фазу экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и упаривали в вакууме на роторном испарителе. Остаток очищали с помощью флэш-хроматографии (градиент: циклогексан/-7 об. % этилацетата, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и органические растворители удаляли на роторном испарителе, получая (5-бром-2-хлор-4-фторфенил)-[4-(трет-бутилдиметилсиланилоксиметил)тиазол-2-ил]метанон (4,94 г, 10,39 ммоль, МС: основной пик 466 [М+Н+], 84% выход) в виде масла.
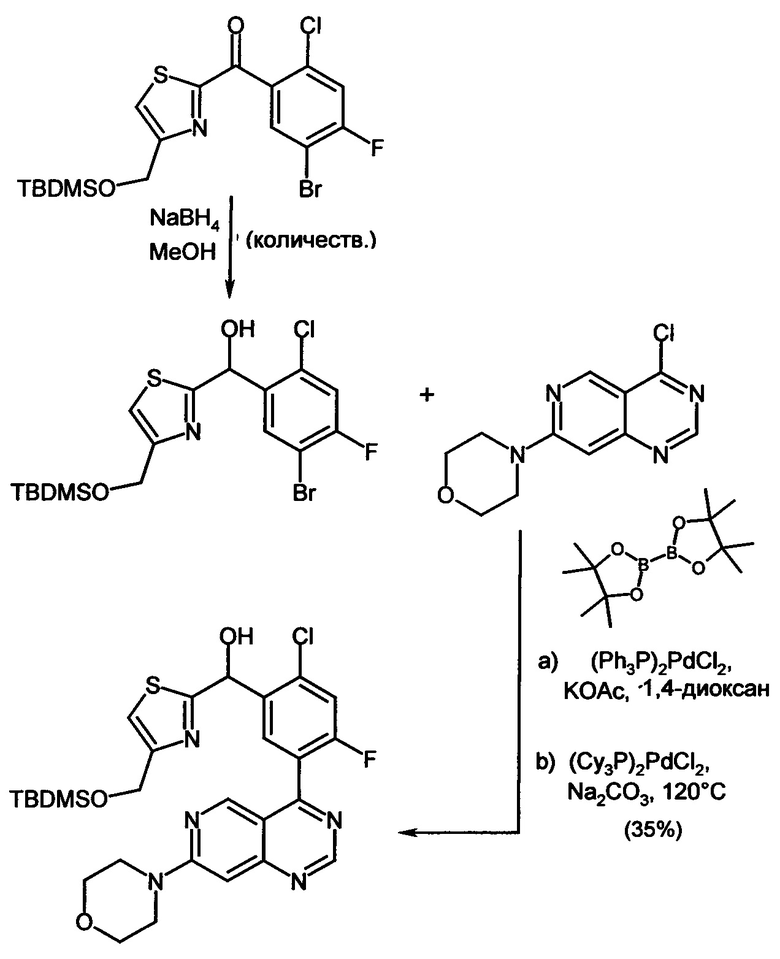
[4-[[трет-Бутил(диметил)силил]оксиметил]тиазол-2-ил]-[2-хлор-4-фтор-5-(7-морфолинопиридо[4,3-d}пиримидин-4-ил)фенил]метанол получали аналогично способам синтеза для ПРИМЕРОВ 1 и2и 138 из (5-бром-2-хлор-4-фторфенил)-[4-(трет-бутилдиметилсиланилоксиметил)тиазол-2-ил]метанонаи 4-хлор-7-морфолин-4-илпиридо[4,3-d}пиримидина.

[4-(трет-Бутилдиметилсиланилоксиметил)тиазол-2-ил]-[2-хлор-4-фтор-5-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]метанол (333 мг, 0,55 ммоль) растворяли в 1,4-диоксане (7 мл). Добавляли 4,0 М HCl, растворенную в 1,4-диоксане (1,38 мл, 5,53 ммоль), и после этого реакционный раствор перемешивали при комнатной температуре в течение 30 минут. После завершения реакции, реакционный раствор фильтровали, и растворители удаляли на роторном испарителе. Остаток очищали с помощью флэш-хроматографии (градиент: дихлорметан/0-15 об. % этанола, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и растворители удаляли в вакууме. Остаток ресуспендировали в дихлорметане, экстрагировали насыщенным раствором гидрокарбоната натрия, высушивали над сульфатом натрия, фильтровали, и фильтрат упаривали насухо, где [2-хлор-4-фтор-5-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]-(4-гидроксиметилтиазол-2-ил)метанол (ПРИМЕР 141, 229 мг, 0,47 ммоль, МС: 488,0/490,0 [М+Н+], 85% выход) в виде твердого вещества.
ПРИМЕРЫ 142 и 143
2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-гидроксиметилтиазол-2-ил)метанол (142)
[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-метиламинометилтиазол-2-ил)метанол (143)
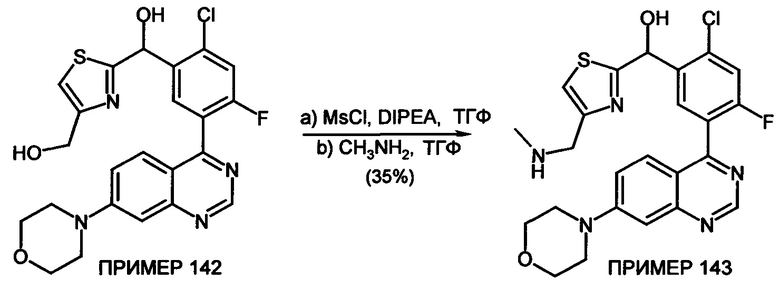
[2-Хлор-4-фтор-5-(7-морфолин-4-илпиридо[4,3-d}пиримидин-4-ил)фенил]-(4-гидроксиметилтиазол-2-ил)метанол (46,6 мг, 96 мкмоль) растворяли в безводном тетрагидрофуране (3,1 мл) в атмосфере аргона. Добавляли N-этилдиизопропиламин (98 мкл, 57,4 мкмоль) и метансульфонил хлорид (14,8 мкл, 191 мкмоль). Реакционный раствор перемешивали при комнатной температуре в течение 30 минут. После этого добавляли метиламин (40% раствор в воде, 183 мкл, 1,91 моль), и смесь перемешивали при комнатной температуре дополнительно в течение 2 ч. После завершения реакции, к реакционному раствору добавляли этилацетат (15 мл) и насыщенный раствор хлорида натрия (10 мл). Фазы разделяли, и водную фазу экстрагировали этилацетатом. Объединенные органические фазы высушивали над сульфатом натрия и фильтровали. Фильтрат упаривали на роторном испарителе, и остаток очищали с помощью препаративной ВЭЖХ с обращенной фазой (градиент вода + 0,1% трифторуксусной кислоты/ацетонитрил + 0,1% трифторуксусной кислоты, Sunfire Prep С-18 150-21 мм, скорость потока: 50 мл/минут, λ=220 нм). Подходящие фракции продукта объединяли, и растворители удаляли в вакууме на роторном испарителе, и остаток лиофилизировали из диоксана/воды, где [2-хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(4-метиламинометилтиазол-2-ил)метанол (ПРИМЕР 143, 14,8 мг, 0,030 ммоль, МС: 500, 1/502,0 [М+Н+], 31% выход) в виде твердого вещества.
ПРИМЕРЫ 144, 145 и 146

Рацемический [4-фтор-3-(6-морфолин-4-илтиено[3,2-d}пиримидин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 144, 35 мг, 0,082 ммоль) разделяли с помощью хроматографии на его энантиомеры на хиральной неподвижной фазе, используя препаративную SFC: после скрининга аналитических колонок для идентификации наиболее подходящей хиральной фазы, имеющей наивысшую селективность, выбирали фазу Lux амилоза-2 от Phenomex. Условия SFC: прибор: SFC Berger milligram; колонка: Lux амилоза-2, 250×4,6 мм; элюент: углекислый газ + 20 об. % метанола + 0,5 об. % диэтиламина, скорость потока: 5 мл/минут, длина волны: 220 нм. Препаративное разделение на энантиомеры осуществляли в тех же самых условиях в SFC Berger minigram Stacked Injection Mode. Подходящие фракции собирали, и растворители удаляли в вакууме на роторном испарителе, получая энантиомерно чистые [4-фтор-3-(6-морфолин-4-илтиено[3,2-d}пиримидин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 145, Rt=7,85 минут 12 мг, 0,028 ммоль, >99% ее Эна 1) и [4-фтор-3-(6-морфолин-4-илтиено[3,2-d}пиримидин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 146, Rt=8,82 минут, 12,0 мг, 0,028 ммоль, 92% ее, Эна 2) в виде твердых веществ.
Соединения, которые получали аналогично ПРИМЕРАМ 138-146, можно найти в таблице 5 ниже.


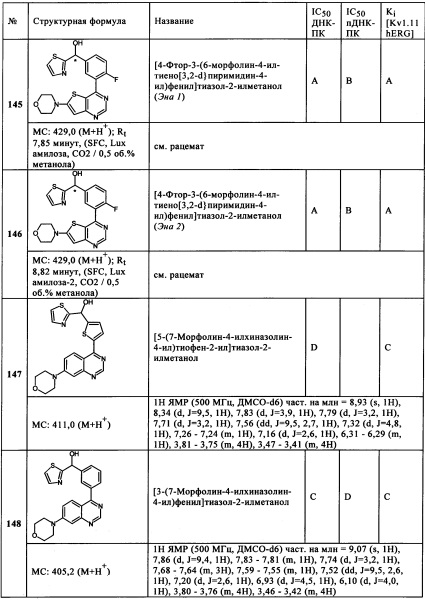


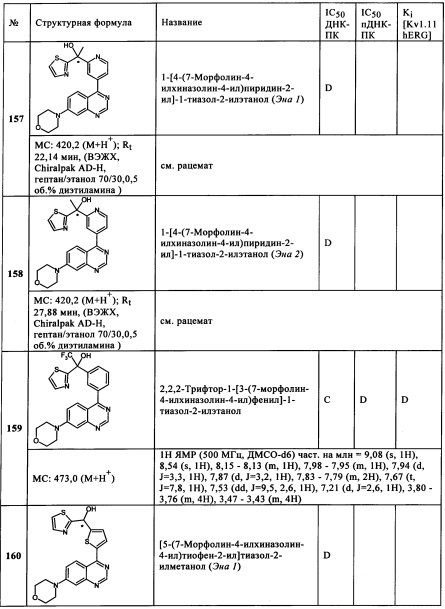

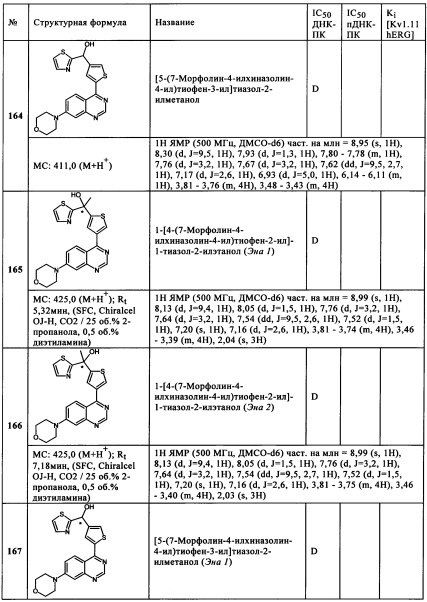
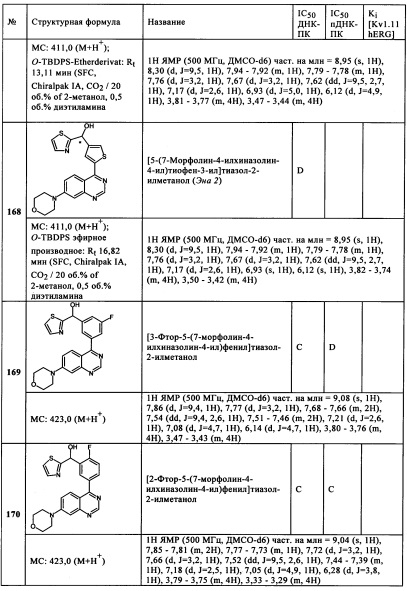

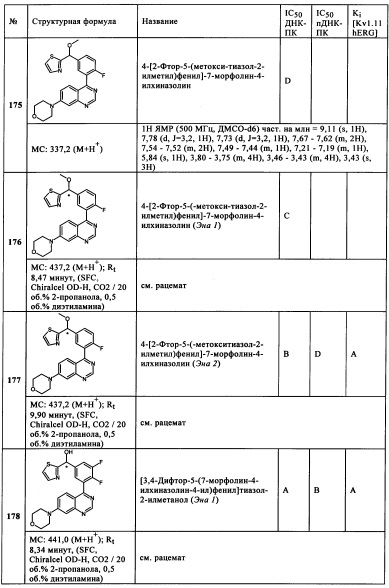



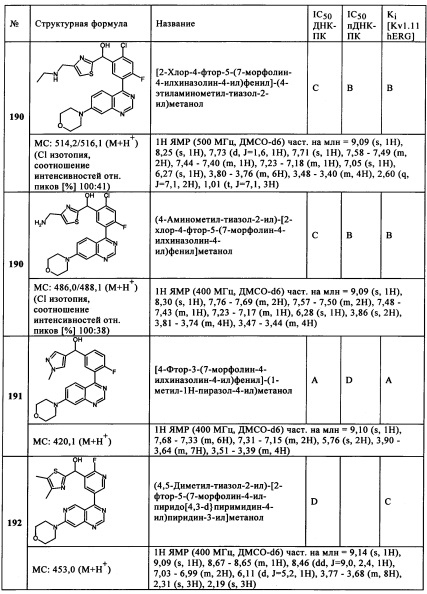




ПРИМЕР 207
[4-Фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 207)

Метил 4-фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)бензоат получали аналогично способам синтеза, описанным для ПРИМЕРОВ 1 и 2, используя в качестве исходных соединений 4-хлор-7-морфолин-4-илхиназолин и метил 4-фтор-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат.

[4-Фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)фенил]тиазол-2-илметанон получали аналогично способам синтеза, описанным для ПРИМЕРА 138, используя в качестве исходных соединений тиазол и метил 4-фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)бензоат.

[4-Фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)фенил]тиазол-2-илметанол (ПРИМЕР 207) получали аналогично способам синтеза, описанным для ПРИМЕРОВ 1 и 2, используя в качестве исходных соединений [4-фтор-2-метил-5-(7-морфолинилхиназолин-4-ил)фенил]тиазол-2-илметанон.
ПРИМЕР 208
[3-(2-Этинил-7-морфолинилхиназолин-4-ил)-4-фторфенил]тиазол-2-илметанол (ПРИМЕР 208)
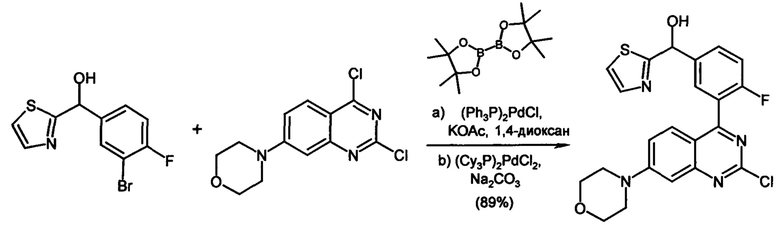
[3-(2-Хлор-7-морфолинилхиназолин-4-ил)-4-фторфенил]тиазол-2-илметанол получали аналогично способам синтеза, описанным для ПРИМЕРА 138, используя в качестве исходных соединений (3-бром-4-фторфенил)тиазол-2-илметанол и 4-(2,4-дихлорхиназолин-7-ил)морфолин.

[3-(2-Хлор-7-морфолинилхиназолин-4-ил)-4-фторфенил]тиазол-2-илметанол (102 мг, 0,225 ммоль) растворяли в бескислородном N,N-диметилформамиде (4 мл) в атмосфере аргона. После этого добавляли CuI (17 мг, 90 мкмоль), (Ph3P)2PdCl2 (63 мг, 90 мкмоль), 2-дифенилфосфанилпиридин (95 мг, 0,359 ммоль), DIPEA (765 мкл, 4,49 ммоль) и триэтилэтинилсилан (275 мкл, 1,48 ммоль). Затем реакционную смесь нагревали при температуре 140°С в течение 1 ч. Для осуществления обычной обработки, добавляли этилацетат (50 мл), воду (10 мл) и насыщенный раствор хлорида натрия (15 мл). Водную фазу отделяли и экстрагировали этилацетатом (20 мл). Объединенные органические фазы высушивали над сульфатом натрия, фильтровали, и фильтрат упаривали насухо в вакууме. Остаток растворяли в диметилсульфоксиде (2 мл), очищали с помощью хроматографии с обращенной фазой (растворитель: ацетонитрил/вода/0,1 об. % НСООН, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая [4-фтор-3-[7-морфолинил-2-(2-триэтилсилилэтинил)хиназолин-4-ил]фенил]тиазол-2-илметанол (64 мг, 0,114 ммоль, МС: 561,2 [М+Н+], 50% выход) в виде воскообразного твердого вещества.
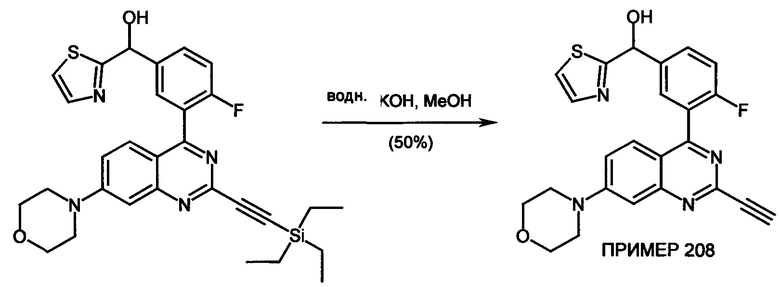
[4-фтор-3-[7-морфолинил-2-(2-триэтилсилилэтинил)хиназолин-4-ил]фенил]тиазол-2-илметанол (552 мг, 0,985 ммоль) растворяли в метаноле (102 мл). После этого добавляли раствор гидроксида калия (1,0 М, 15 мл, 15 ммоль), и смесь перемешивали при комнатной температуре в течение 90 минут. После завершения реакции, смесь осторожно нейтрализовали, используя соляную кислоту (1,0 М, 15 мл, 15 ммоль). После этого добавляли этилацетат (500 мл), вода (100 мл) и насыщенный раствор хлорида натрия (150 мл). Фазы разделяли, и водную фазу экстрагировали этилацетатом (100 мл). Объединенные органические фазы высушивали над сульфатом натрия, фильтровали, и фильтрат упаривали насухо в вакууме. Остаток, растворенный в диметилсульфоксиде (8 мл), очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан/0-5 об. % этанола, CombiFlash Rf 200). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая [3-(2-этинил-7-морфолинилхиназолин-4-ил)-4-фторфенил]тиазол-2-илметанол (ПРИМЕР 208, 221 мг, 0,495 ммоль, МС: 447,1 [М+Н+], 50% выход) в виде твердого вещества.
Соединения, которые получали аналогично ПРИМЕРАМ 207 и 208, можно найти в таблице 6 ниже.

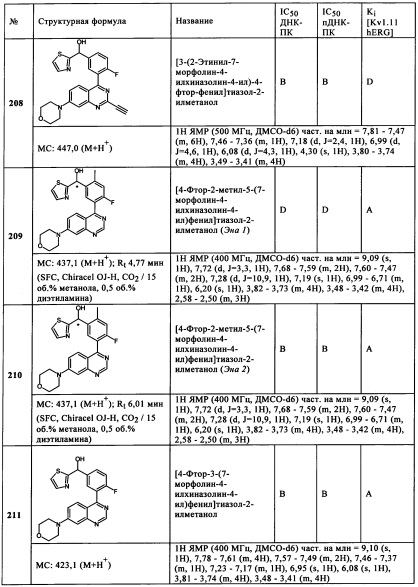


ПРИМЕР 217
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]пиридин-3-илметанол (217)

1,5-Дибром-2,4-дифторбензол (500 мг, 1,78 ммоль) растворяли в безводном простом диэтиловом эфире (10 мл) в атмосфере аргона. Реакционный раствор охлаждали до (-)65°С с помощью ацетона/бани с сухим льдом. Добавляли по каплям н-бутиллитий (15% раствор в н-гексане, 1,23 мл, 1,96 ммоль) в течение 15 минут при постоянной температуре (-)65°С, и реакционный раствор перемешивали при (-)65°С дополнительно в течение 30 минут. После этого по каплям добавляли предварительно приготовленный раствор никотинальдегида (201 мкл, 2,14 ммоль) в безводном простом диэтиловом эфире (5 мл) в течение периода 15 минут при (-)65°С, и реакционную смесь перемешивали дополнительно в течение 10 минут и затем медленно нагревали до 0°С в течение одного часа. После завершения реакции, к реакционному раствору добавляли насыщенный раствор хлорида аммония (5 мл) и водой (30 мл). Водную фазу экстрагировали три раза трет-бутил метиловым эфиром. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия, фильтровали и упаривали на роторном испарителе. Неочищенный продукт (масло) очищали путем препаративной колоночной хроматографии с обращенной фазой (растворитель градиент вода/ацетонитрил/0,1 об. % трифторуксусной кислоты [5,5 минут], CombiFlash Rf 200). Подходящие фракции продукта объединяли и упаривали в вакууме. Водный остаток нейтрализовали, используя насыщенный раствор гидрокарбоната натрия, и экстрагировали три раза с помощью этилацетата. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия, фильтровали и упаривали насухо в вакууме на роторном испарителе, получая (5-бром-2,4-дифторфенил)-(3-пиридил)метанол (215 мг, 0,717 ммоль, МС: 300,0/302,0 [М+Н+], 40% выход) в виде бесцветного масла.

[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]пиридин-3-илметанол (ПРИМЕР 217) получали аналогично способам синтеза для получения 1-[5-(7-морфолин-4-илхиназолин-4-ил)пиридин-3-ил]-1-тиазол-2-илэтанола (ПРИМЕР 138), используя в качестве исходных соединений (5-бром-2,4-дифторфенил)-(3-пиридил)метанол.
Соединения, которые получали аналогично ПРИМЕРУ 217, можно найти в таблице 7 ниже.
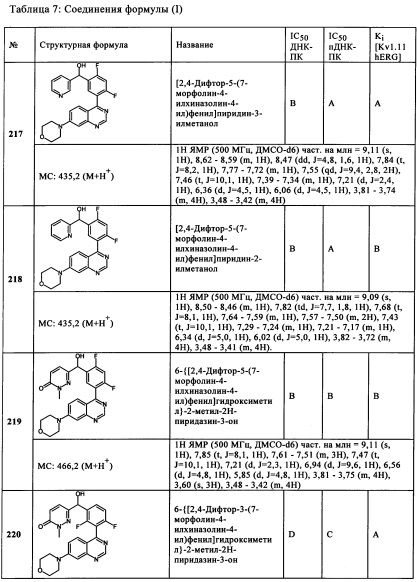

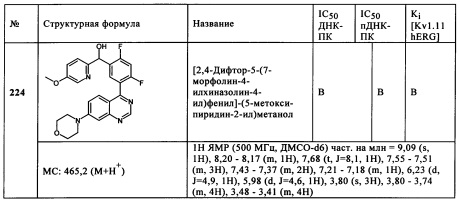
ПРИМЕР 225:
[2-Хлор-5-(5,6-дидейтерио-7-морфолинилхиназолин-4-ил)-4-фторфенил]-(6-метоксипиридазин-3-ил)метанол (225)

При взаимодействии 5,6,8-тридейтерио-7-морфолинил-3Н-хиназолин-4-она с оксихлоридом фосфора получали 4-хлор-5,6-дидейтерио-7-N-морфолинилхиназолин.
ПРИМЕР 237:
[4-Фтор-3-(7-морфолин-4-илпиридо[3,2-d}пиримидин-4-ил)фенил]-(3-метилпиразин-2-ил)метанол (237)

ПРИМЕР 258:
[2-Хлор-4-фтор-5-[7-(2,2,3,3,5,5,6,6-октадейтериоморфолин-4-ил)хиназолин-4-ил]фенил]-(3-метоксипиразин-2-ил)метанол (258)

ПРИМЕРЫ 268 и 278:
[4-Фтор-3-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метилпиразин-2-ил)метанол (268), [2-хлор-4-фтор-5-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанол (278)

ПРИМЕР 319:
2-[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-2-(3-метоксипиразин-2-ил)ацетамид (319)
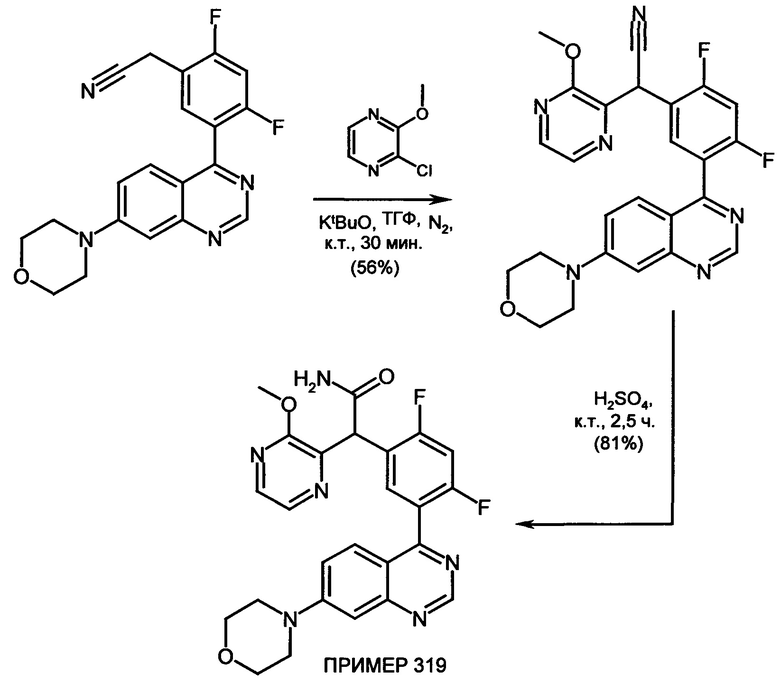
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]ацетонитрил (300 мг, 0,82 ммоль) и 2-хлор-3-метоксипиразин (297 мг, 1,97 ммоль) растворяли в тетрагидрофуране. После этого азот пропускали в раствор в течение периода 10 минут. После этого к реакционному раствору добавляли трет-бутилат калия (193 мг, 1,72 ммоль), и смесь перемешивали при комнатной температуре в атмосфере аргона в течение периода 30 минут. После завершения реакции, реакционную смесь нейтрализовали, используя насыщенный раствор NH4Cl, разводили дистиллированной водой (30 мл) и экстрагировали три раза с помощью дихлорметана (30 мл в каждом случае). Органическую фазу высушивали над NaSO4, отфильтровывали с отсасыванием и упаривали насухо в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан/0-5 об. % этанола, CombiFlash Rf 200, колонка 40 г диоксида кремния, λ=220 нм). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая [2,4-дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метоксипиразин-2-ил)ацетонитрил (218 мг, 0,46 ммоль; МС: 475,2 [М+Н+], 56% выход) в виде твердого вещества.
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метоксипиразин-2-ил)ацетонитрил (218 мг, 0,46 ммоль) изначально вносили в реакционную колбу и после этого растворяли с H2SO4 (95-98%, 3,53 мл, 64 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 2,5 ч. После завершения реакции, к реакционному раствору добавляли лед (80 г). После этого смесь осторожно нейтрализовали, используя раствор NaOH (32%, 10,6 мл). Полученную суспензию разводили дистиллированной водой (50 мл) и экстрагировали три раза с помощью дихлорметана (100 мл в каждом случае). Органическую фазу высушивали над NaSO4, отфильтровывали с отсасыванием и упаривали насухо в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (градиент: дихлорметан/0-12 об. % этанола, CombiFlash Rf 200, колонка 40 г диоксида кремния, λ=220 нм). Подходящие фракции продукта объединяли, и растворители удаляли на роторном испарителе, получая 2-[2,4-дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-2-(3-метоксипиразин-2-ил)ацетамид (182 мг, 0,37 ммоль, МС: 493,4 [М+Н+], 81% выход) в виде твердого вещества.
Соединения, которые получали в соответствии с ПРИМЕРАМИ 225, 237, 258, 268, 278 и 319, и аналогично последовательностям синтеза из ПРИМЕРОВ 1, 2, 37, 137, 121, 217, можно найти в таблице 8 ниже:


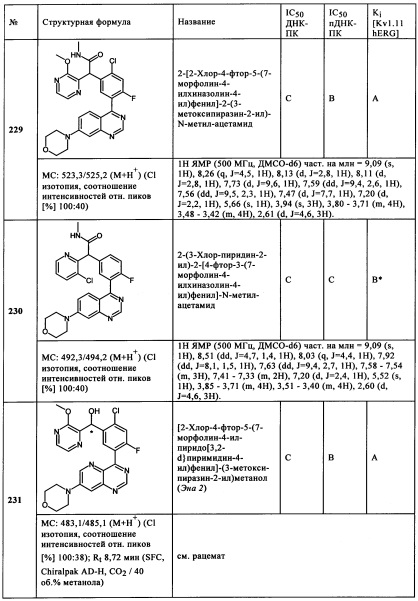

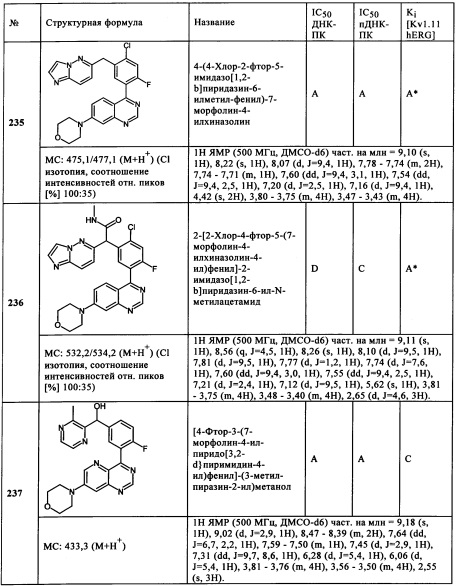
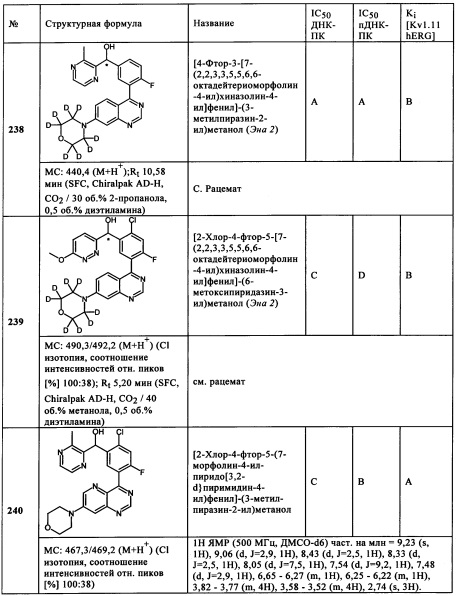







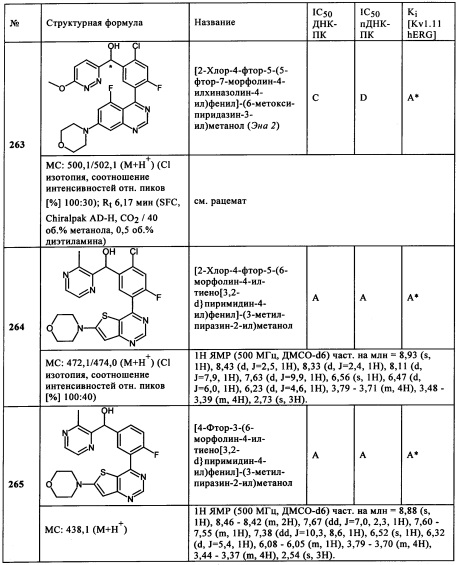


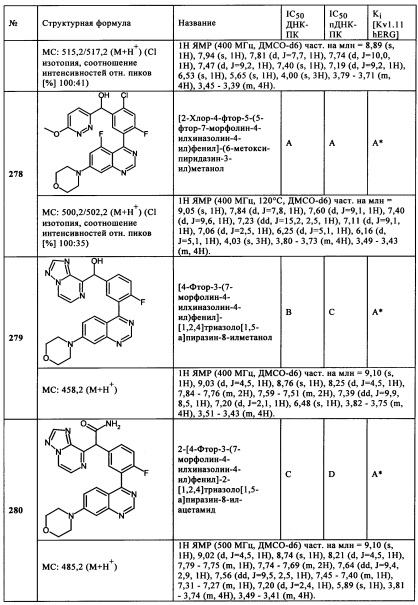
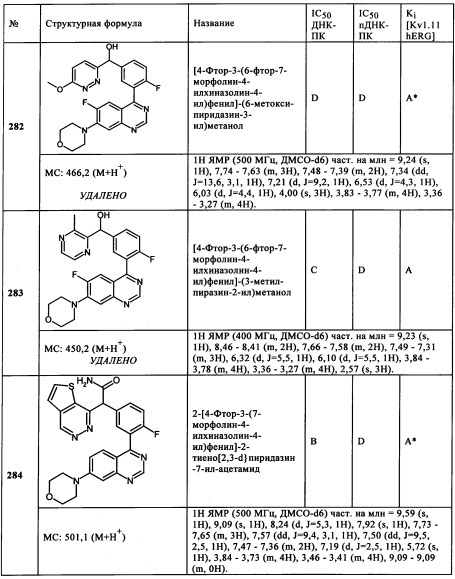




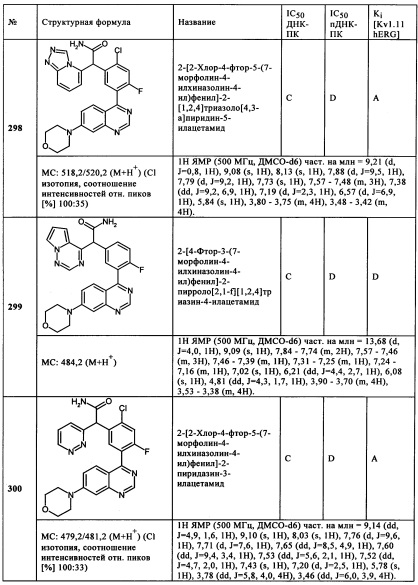

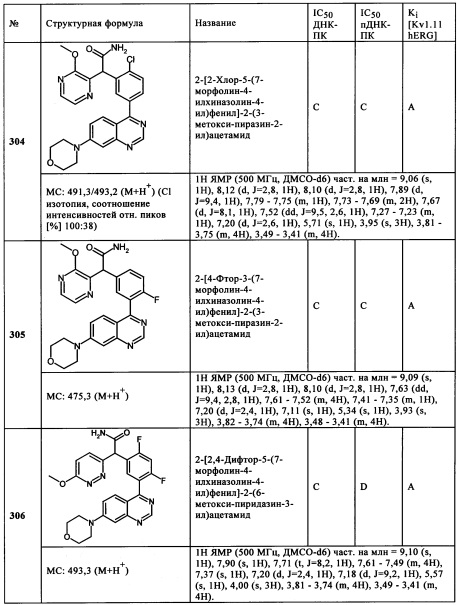









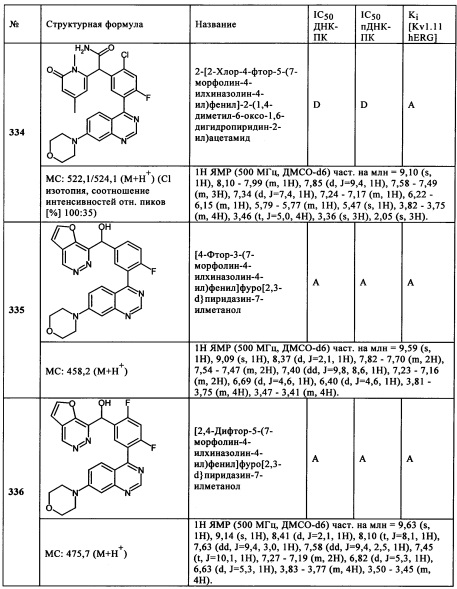
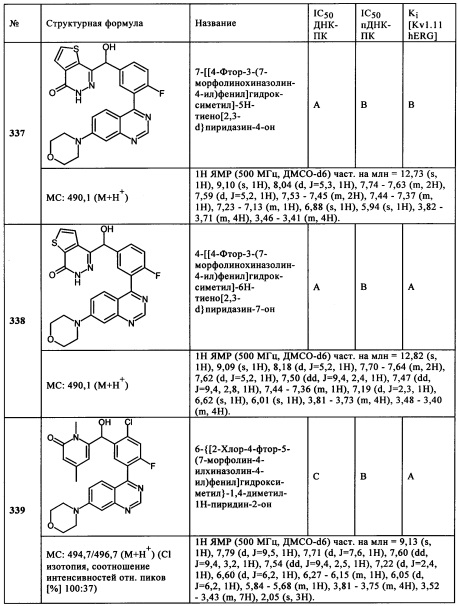

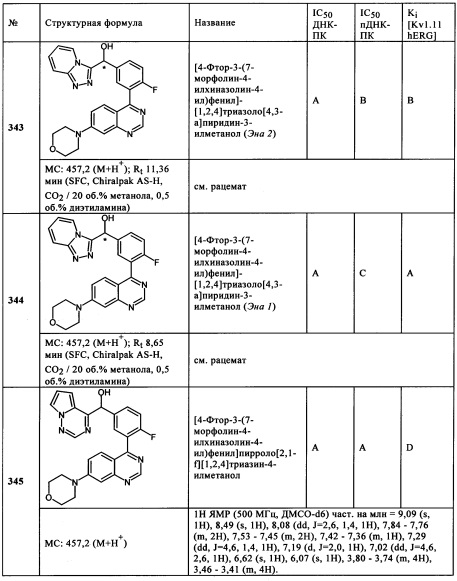


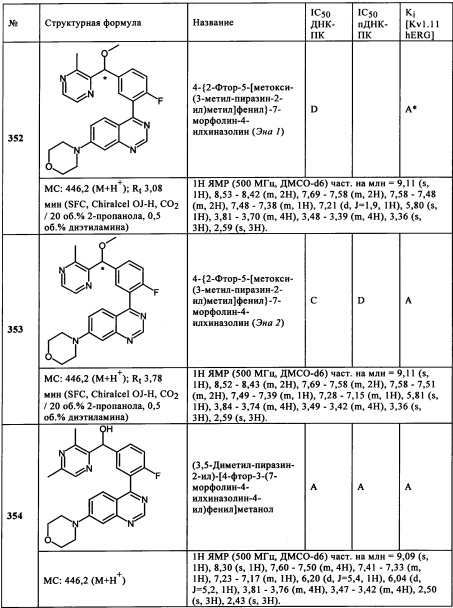
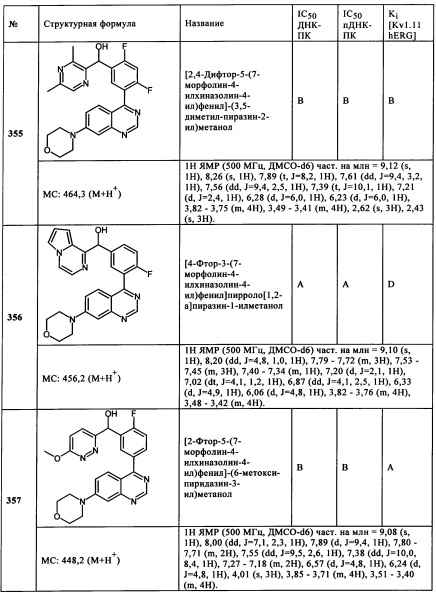
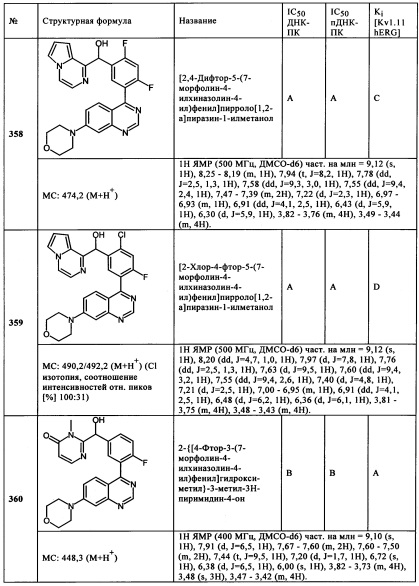
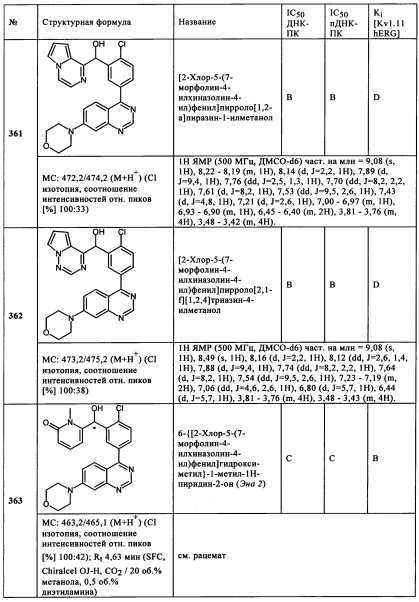



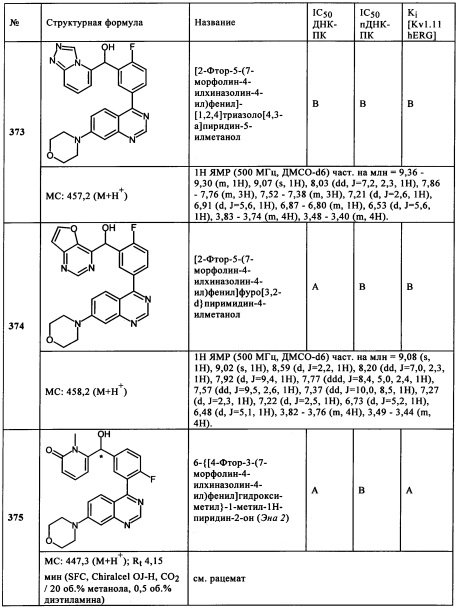






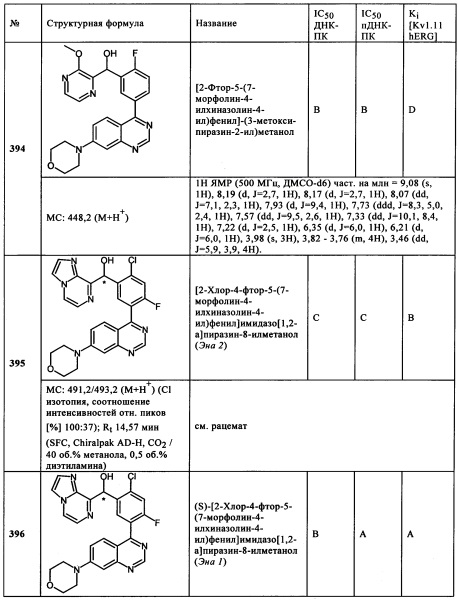
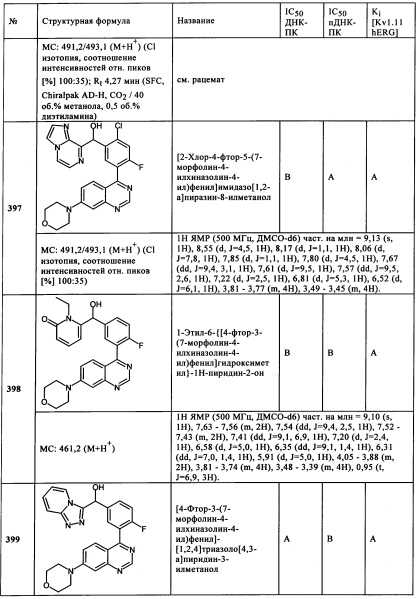


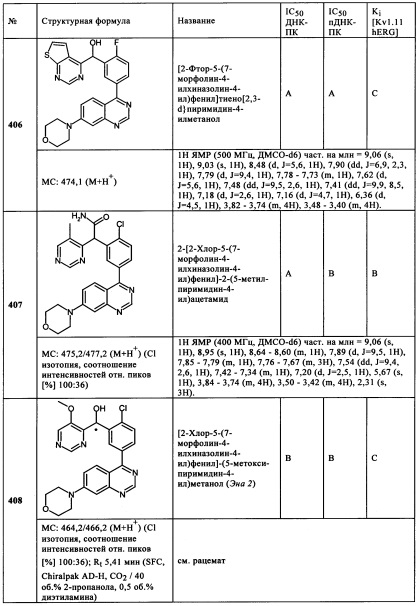



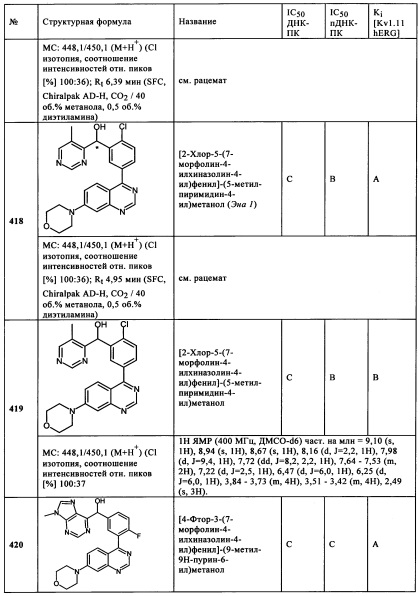


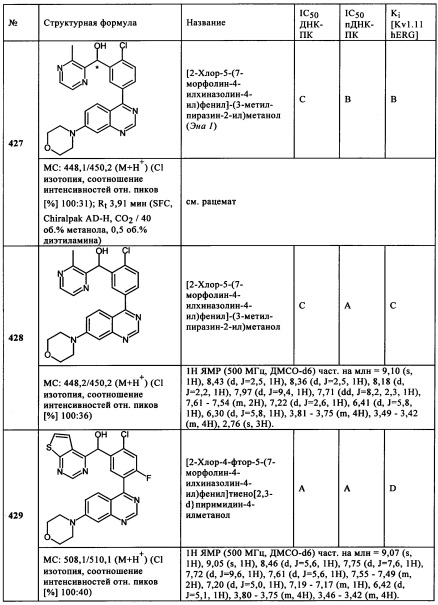



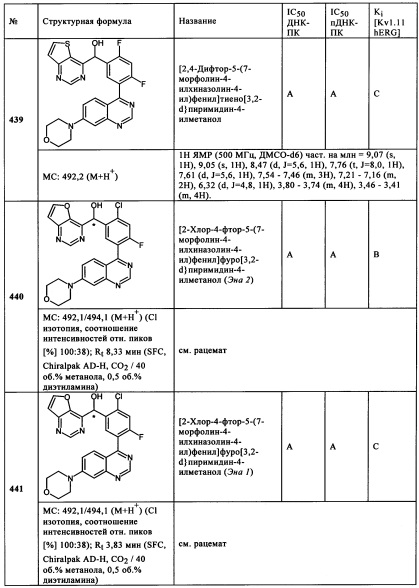


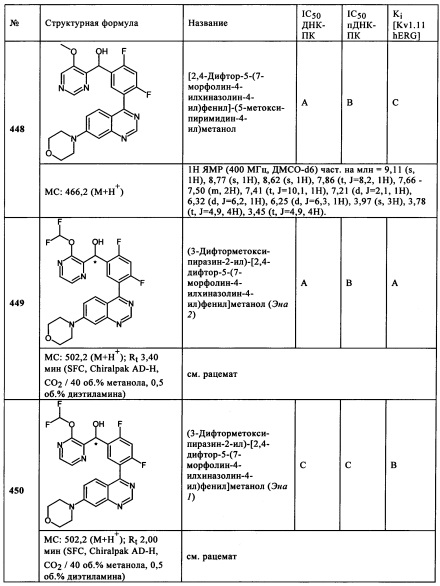







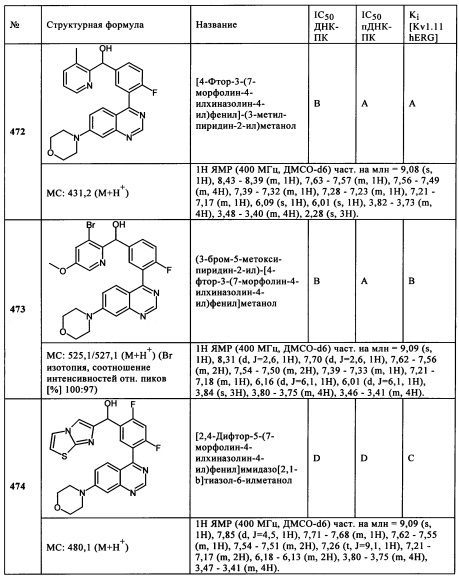


| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОЛОНИЛХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ АТМ | 2016 |
|
RU2743343C2 |
| 4-(ИМИДАЗО[1,2-а]ПИРИДИН-3-ИЛ)-ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2822388C2 |
| ИНДАЗОЛИЛИЗОКСАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ТАКИХ КАК ЗЛОКАЧЕСТВЕННОЕ НОВООБРАЗОВАНИЕ | 2020 |
|
RU2836662C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2003 |
|
RU2296757C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ МОДУЛИРОВАНИЯ КИНАЗЫ И ПОКАЗАНИЯ К ИХ ПРИМЕНЕНИЮ | 2013 |
|
RU2666146C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА И ИХ ПРИМЕНЕНИЕ ПРИ НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЯХ | 2011 |
|
RU2581504C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 2018 |
|
RU2809144C2 |
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]- И ТИЕНО[3,2-В]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ TBK1 И IKKε | 2013 |
|
RU2622034C2 |
| ПРОИЗВОДНЫЕ 4-АМИНО-6-ФЕНИЛПИРРОЛО[2,3] ПИРИМИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ ДЕЙСТВИЯ ТИРОЗИНКИНАЗЫ, ИХ ПРИМЕНЕНИЕ И СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2002 |
|
RU2318826C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА | 2000 |
|
RU2256659C2 |
Изобретение относится к новому соединению формулы (I), его физиологически приемлемой соли и/или стереоизомеру, обладающим свойствами ДНК-зависимой протеинкиназы (ДНК-ПК). Соединения могут найти применение при приготовления лекарственного средства для сенсибилизации злокачественных клеток к противораковым средствам и/или ионизирующему излучению. В формуле (I)

X представляет собой СН, CF, S или N, Y представляет собой СН, S или N, Z представляет собой С или N,  если Z=С, то образует двойную связь совместно с простой связью, или отсутствует, если Z=N, n равен 1 или 2, где если n=1, то X=S, и если n=2, то оба X=СН, или X, связанный с пиримидиновым кольцом, представляет собой CF, а X, несвязанный с пиримидиновым кольцом, представляет собой СН, или один X представляет собой СН, а другой X представляет собой N; m равен 1 или 2, где если m=1, то Y=S, и если m=2, то оба Y=СН, или один Y представляет собой СН, а другой Y представляет собой N; R1, R2, R3, R4 независимо друг от друга представляют собой Н, Hal, CN, ОН, CONH2, CONH(LA) или LA; R5 представляет собой Н, Hal, CN или С≡СН; Сус представляет собой фенил, который может быть незамещенным или моно- или дизамещенным независимо друг от друга с помощью R6, или представляет собой Het1; Het1 представляет собой моно- или бициклический 5-10-членный гетероцикл, имеющий 1-3 атома N, О и/или S, или 1-4 атома N, который может быть незамещенным или моно-, ди- или тризамещенным независимо друг от друга с помощью R6, или может быть монозамещенным с помощью Het2; R6 представляет собой Hal, LA, оксо, CN или NH2; LA представляет собой неразветвленный или разветвленный алкил, имеющий 1-5 атомов С, который может быть насыщенным или частично ненасыщенным, в котором 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две СН2 группы могут быть заменены на О, NH, N(CH3) или СО; Het2 представляет собой 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атомов N, О и/или S, который является незамещенным; Hal представляет собой F, Cl, Br или I; где значение Н охватывает 1Н и дейтерий. Способ получения соединения формулы (I) и/или его физиологически приемлемой соли и/или стереоизомера включает следующие стадии:
если Z=С, то образует двойную связь совместно с простой связью, или отсутствует, если Z=N, n равен 1 или 2, где если n=1, то X=S, и если n=2, то оба X=СН, или X, связанный с пиримидиновым кольцом, представляет собой CF, а X, несвязанный с пиримидиновым кольцом, представляет собой СН, или один X представляет собой СН, а другой X представляет собой N; m равен 1 или 2, где если m=1, то Y=S, и если m=2, то оба Y=СН, или один Y представляет собой СН, а другой Y представляет собой N; R1, R2, R3, R4 независимо друг от друга представляют собой Н, Hal, CN, ОН, CONH2, CONH(LA) или LA; R5 представляет собой Н, Hal, CN или С≡СН; Сус представляет собой фенил, который может быть незамещенным или моно- или дизамещенным независимо друг от друга с помощью R6, или представляет собой Het1; Het1 представляет собой моно- или бициклический 5-10-членный гетероцикл, имеющий 1-3 атома N, О и/или S, или 1-4 атома N, который может быть незамещенным или моно-, ди- или тризамещенным независимо друг от друга с помощью R6, или может быть монозамещенным с помощью Het2; R6 представляет собой Hal, LA, оксо, CN или NH2; LA представляет собой неразветвленный или разветвленный алкил, имеющий 1-5 атомов С, который может быть насыщенным или частично ненасыщенным, в котором 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две СН2 группы могут быть заменены на О, NH, N(CH3) или СО; Het2 представляет собой 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атомов N, О и/или S, который является незамещенным; Hal представляет собой F, Cl, Br или I; где значение Н охватывает 1Н и дейтерий. Способ получения соединения формулы (I) и/или его физиологически приемлемой соли и/или стереоизомера включает следующие стадии:
(а) взаимодействие соединения формулы (V)


в которой LG представляет собой общепринятую уходящую группу, такую как Hal, и R5, X, n и Z имеют значения, указанные в п. 1, с соединением формулы (IV), в которой А представляет собой бороновую кислоту или сложный эфир бороновой кислоты, и R1, R2, R3, R4, Сус, Y и m имеют значения, указанные в п. 1, с получением соединения формулы (I), и необязательное (б) превращение основания или кислоты соединения формулы (I) в одну из его солей. 7 н. и 11 з.п. ф-лы, 8 табл., 469 пр.
1. Соединение формулы (I)
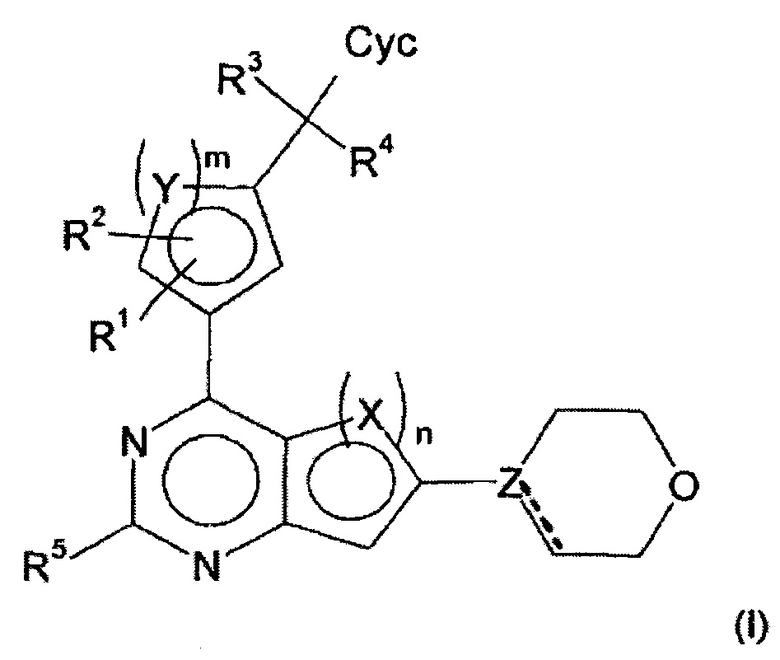
в которой
X представляет собой СН, CF, S или N,
Y представляет собой СН, S или N,
Z представляет собой С или N,
 если Z=С, то образует двойную связь совместно с простой связью,
если Z=С, то образует двойную связь совместно с простой связью,
или отсутствует, если Z=N,
n равен 1 или 2,
где если n=1, то X=S,
и если n=2, то оба X=СН, или X, связанный с пиримидиновым кольцом, представляет собой CF, а X, несвязанный с пиримидиновым кольцом, представляет собой СН, или один X представляет собой СН, а другой X представляет собой N;
m равен 1 или 2,
где если m=1, то Y=S,
и если m=2, то оба Y=СН, или один Y представляет собой СН, а другой Y представляет собой N;
R1, R2, R3, R4 независимо друг от друга представляют собой Н, Hal, CN, ОН, CONH2, CONH(LA) или LA;
R5 представляет собой Н, Hal, CN или С≡СН;
Сус представляет собой фенил, который может быть незамещенным или моно- или дизамещенным независимо друг от друга с помощью R6, или представляет собой Het1;
Het1 представляет собой моно- или бициклический 5-10-членный гетероцикл, имеющий 1-3 атома N, О и/или S, или 1-4 атома N, который может быть незамещенным или моно-, ди- или тризамещенным независимо друг от друга с помощью R6, или может быть монозамещенным с помощью Het2;
R6 представляет собой Hal, LA, оксо, CN или NH2;
LA представляет собой неразветвленный или разветвленный алкил, имеющий 1-5 атомов С, который может быть насыщенным или частично ненасыщенным, в котором 1-3 атома Н могут быть заменены Hal, и/или один атом Н может быть заменен CN или Het2, и/или одна или две СН2 группы могут быть заменены на О, NH, N(CH3) или СО;
Het2 представляет собой 3-5-членный алифатический гомо- или гетероцикл, имеющий 0, 1, 2 или 3 атомов N, О и/или S, который является незамещенным;
Hal представляет собой F, Cl, Br или I;
где значение Н охватывает 1Н и дейтерий,
и/или его физиологически приемлемая соль и/или стереоизомер.
2. Соединение по п. 1, которое соответствует формуле (Ib),
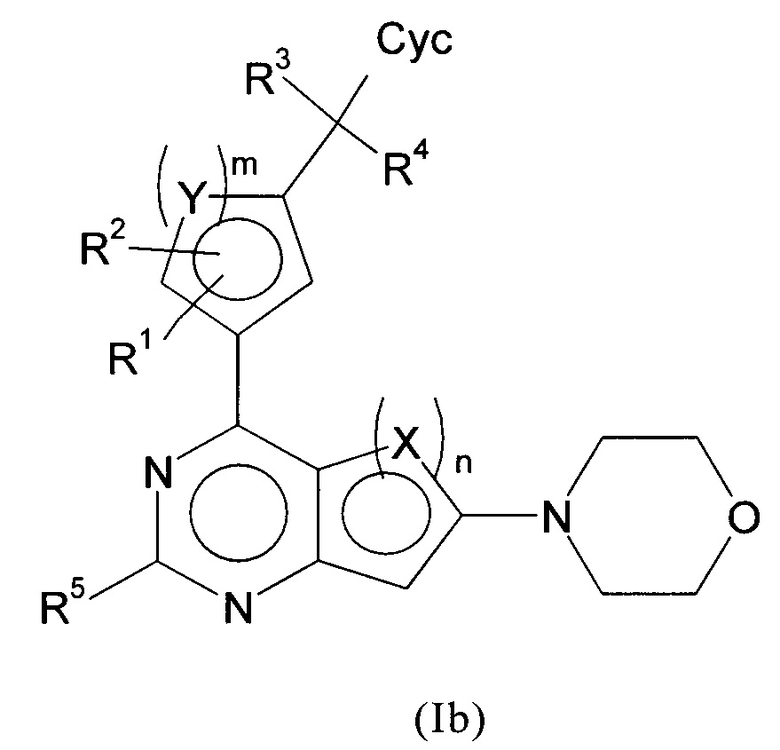
в которой все заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
3. Соединение по п. 1 или 2, которое соответствует формуле (II)

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CONH(LA) или LA;
R6', R6'' независимо друг от друга представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2 независимо друг от друга представляют собой СН, N или NH и в каждом случае являются незамещенными;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
4. Соединение по п. 1 или 2, которое соответствует формуле (III)

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CONH(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой H, Hal, LA, оксо, CN, NH2 или Het2;
 обозначает присутствие или отсутствие двойных связей в Сус;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
5. Соединение по п. 3, которое соответствует формуле (IIa)

в которой
R2, R3 независимо друг от друга представляют собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6', R6'' независимо друг от друга представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2 независимо друг от друга представляют собой СН, N или NH, и в каждом случае являются незамещенными;
X1 представляет собой СН, CF или N;
X2 представляет собой СН или N,
где X1, X2 одновременно не представляют собой N;
Y представляет собой СН или N;
 обозначает присутствие или отсутствие двойных связей в Сус;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
6. Соединение по п. 3, которое соответствует формуле (IIb)
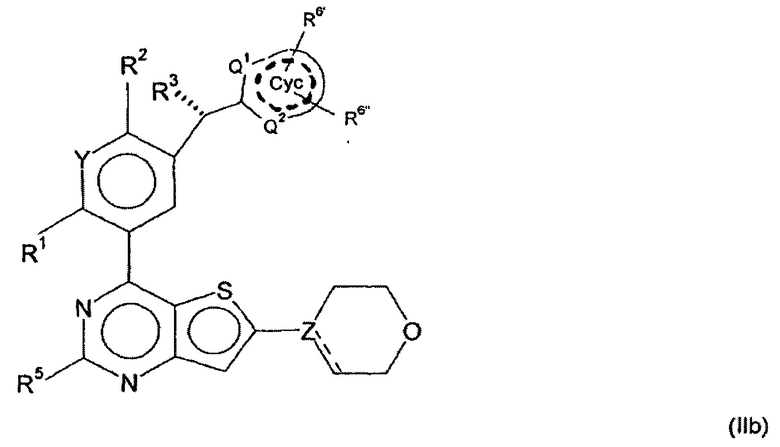
в которой
R2, R3 независимо друг от друга представляют собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6', R6'' независимо друг от друга представляют собой Н, Hal, LA, оксо, CN, NH2 или Het2;
Q1, Q2 независимо друг от друга представляют собой СН, N или NH, и в каждом случае являются незамещенными;
Y представляет собой СН или N,
 обозначает присутствие или отсутствие двойных связей в Сус;
обозначает присутствие или отсутствие двойных связей в Сус;
и все другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
7. Соединение по п. 4, которое соответствует формуле (IIIa)

в которой
R3 представляет собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой H, Hal, LA, оксо, CN, NH2 или Het2;
X1 представляет собой СН, CF или N;
X2 представляет собой СН или N,
где X1, X2 одновременно не представляют собой N;
Y представляет собой СН или N;
 обозначает присутствие или отсутствие двойных связей в Сус;
обозначает присутствие или отсутствие двойных связей в Сус;
и другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
8. Соединение по п. 4, которое соответствует формуле (IIIb)
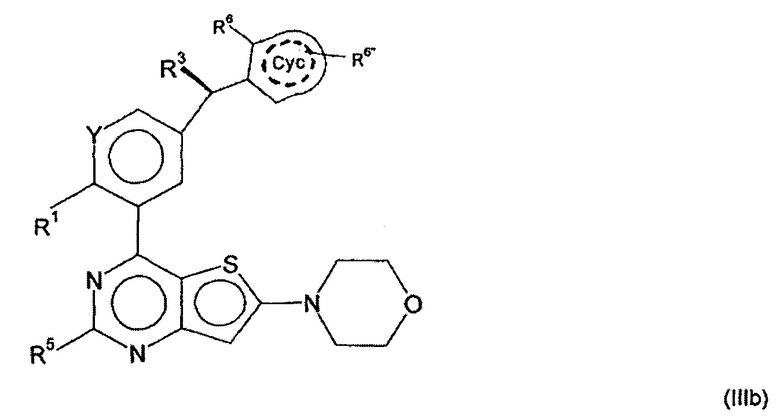
в которой
R3 представляет собой Hal, CN, ОН, CONH2, CON(LA) или LA;
R6 представляет собой Hal, LA, оксо, CN, NH2 или Het2;
R6'' представляет собой H, Hal, LA, оксо, CN, NH2 или Het2;
Y представляет собой СН или N,
 обозначает присутствие или отсутствие двойных связей в Сус;
обозначает присутствие или отсутствие двойных связей в Сус;
и все другие заместители имеют значения, указанные для формулы (I),
и/или его физиологически приемлемая соль и/или стереоизомер.
9. Соединение по п. 5, в котором радикалы, не обозначенные более подробно, имеют значения, указанные для формулы (IIa), но в которых
в случае подформулы (IIa-А)
X1 представляет собой СН,
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
в случае подформулы (IIa-В)
R1 представляет собой F,
R2 представляет собой F или Cl,
в случае подформулы (IIa-С)
X1, X2 представляет собой СН,
в случае подформулы (IIa-D)
X1 представляет собой СН,
R5 представляет собой Н,
в случае подформулы (IIa-Е)
R3 представляет собой ОН,
в случае подформулы (IIa-F)
X1 представляет собой СН,
R3 представляет собой ОН,
в случае подформулы (IIa-G)
X1 представляет собой СН,
Y представляет собой СН,
в случае подформулы (IIa-Н)
X1 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIa-J)
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]-пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d]-пиридазинил, тиено[2,3-d]пиридазинил, тиено[2,3-d]пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещенным или моно- или дизамещенным с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIa-K)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
в случае подформулы (IIa-L)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
в случае подформулы (IIa-М)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIa-N)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]-пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d]-пиридазинил, тиено[2,3-d]пиридазинил, тиено[2,3-d]пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещенным или моно- или дизамещенным с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIa-О)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой 5-метоксипиридазин-3-ил, имидазо[1,2-b]-пиридазин-6-ил, 3-хлор-6-метоксипиразин-2-ил, 3-хлорпиразин-2-ил, пиридазин-4-ил, 3-метоксипиразин-2-ил, 6-метоксипиридазин-3-ил, 3-дифторметокси-пиридин-2-ил, 3-метилпиразин-2-ил, тиено[2,3-d]пиримидин-4-ил, 1-метил-1Н-пиридин-2-он-6-ил, 1Н-пиридазин-6-он-3-ил, фуро[2,3-d]пиридазин-7-ил, тиено[2,3-d]пиридазин-7-ил, 3,5-диметилпиразин-2-ил, фуро[2,3-d]пиримидин-4-ил, 3-метил-3Н-имидазо[4,5-с]пиридин-4-ил,
и/или его физиологически приемлемая соль и/или стереоизомер.
10. Соединение по п. 7, в котором радикалы, не обозначенные более подробно, имеют значения, указанные для формулы (IIIa), но в которых
в случае подформулы (IIIa-В)
R1 представляет собой F,
в случае подформулы (IIIa-С)
X1, X2 представляет собой СН,
в случае подформулы (IIIa-D)
X1 представляет собой СН,
R5 представляет собой Н,
в случае подформулы IIIa-(Е)
R3 представляет собой ОН,
в случае подформулы (IIIa-F)
X1 представляет собой СН,
R3 представляет собой ОН,
в случае подформулы (IIIa-G)
X1 представляет собой СН,
Y представляет собой СН,
в случае подформулы (IIIa-Н)
X1 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIIa-J)
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]-пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d]пиридазинил, тиено[2,3-d]пиридазинил, тиено[2,3-d]пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещенным или моно- или дизамещенным с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIIa-K)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
в случае подформулы (IIIa-L)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
в случае подформулы (IIIa-М)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
X1, X2 представляет собой СН,
Сус представляет собой пиридин, пиразин или пиридазин, или пиразоло[1,5-а]пиримидинил или имидазо[1,2-b]пиридазинил,
в случае подформулы (IIIa-N)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин, пиразоло[1,5-а]-пиримидинил, имидазо[1,2-b]пиридазинил, фуро[2,3-с]пиридинил, фуро[2,3-d]-пиридазинил,
тиено[2,3-d]пиридазинил, тиено[2,3-d]пиримидинил или имидазо[4,5-с]пиридинил, каждый из которых может быть незамещенным или моно- или дизамещенным с помощью метокси, метила, оксо, Cl или CHF2O,
в случае подформулы (IIIa-О)
R1 представляет собой F,
R3 представляет собой Н или ОН,
R5 представляет собой Н,
Сус представляет собой 5-метоксипиридазин-3-ил, имидазо[1,2-b]-пиридазин-6-ил, 3-хлор-6-метоксипиразин-2-ил, 3-хлорпиразин-2-ил, пиридазин-4-ил, 3-метоксипиразин-2-ил, 6-метоксипиридазин-3-ил, 3-дифторметокси-пиридин-2-ил, 3-метилпиразин-2-ил, тиено[2,3-d]пиримидин-4-ил, 1-метил-1Н-пиридин-2-он-6-ил, 1Н-пиридазин-6-он-3-ил, фуро[2,3-d]пиридазин-7-ил, тиено[2,3-d]пиридазин-7-ил, 3,5-диметилпиразин-2-ил, фуро[2,3-d]пиримидин-4-ил, 3-метил-3Н-имидазо[4,5-с]пиридин-4-ил,
и/или его физиологически приемлемая соль и/или стереоизомер.
11. Соединение по п. 6, в котором радикалы, не обозначенные более подробно, имеют значения, указанные для формул (IIb), но в которых
в случае подформулы (IIb-Q)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIb-R)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIb-S)
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIb-Т)
R1 представляет собой F или Cl,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIb-U)
R1 представляет собой F,
R2 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин или 3-метилпиразин-2-ил,
и/или его физиологически приемлемая соль и/или стереоизомер.
12. Соединение по п. 8, в котором радикалы, не обозначенные более подробно, имеют значения, указанные для формул (IIIb), но в которых
в случае подформулы (IIIb-Q)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIIb-R)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
Y представляет собой СН,
в случае подформулы (IIIb-S)
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIIb-Т)
R1 представляет собой F или Cl,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин или пиридазин,
в случае подформулы (IIIb-U)
R1 представляет собой F,
R3 представляет собой ОН,
R5 представляет собой Н,
Сус представляет собой пиридин, пиразин, пиридазин или 3-метилпиразин-2-ил,
и/или его физиологически приемлемая соль и/или стереоизомер.
13. Соединение, выбранное из группы, включающей:
[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(5-метокси-пиридазин-3-ил)метанол,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]-(5-метоксипиридазин-3-ил)метанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]имидазо[1,2-b]-пиридазин-6-илметанол,
(3-Хлор-6-метоксипиразин-2-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол,
(R)-(3-Хлорпиразин-2-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)-фенил]метанол,
[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]пиридазин-4-илметанол,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метоксипиразин-2-ил)метанол,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанол,
(3-Дифторметоксипиридин-2-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]метанол,
(R)-[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метилпиразин-2-ил)метанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метилпиразин-2-ил)метанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]тиено[2,3-d]-пиримидин-4-илметанол,
6-{[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-1-метил-1Н-пиридин-2-он,
3-[[2-Хлор-4-фтор-5-(7-морфолинохиназолин-4-ил)фенил]гидроксиметил]-1Н-пиридазин-6-он,
(S)-[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метокси-пиридазин-3-ил)метанол,
(R)-[4-Фтор-3-(7-морфолин-4-илпиридо[3,2-d]пиримидин-4-ил)фенил]-(3-метил-пиразин-2-ил)метанол,
4-(4-Хлор-2-фтор-5-имидазо[1,2-b]пиридазин-6-илметилфенил)-7-морфолин-4-илхиназолин,
[4-Фтор-3-(6-морфолин-4-илтиено[3,2-d]пиримидин-4-ил)фенил]-(3-метил-пиразин-2-ил)метанол,
(R)-[4-Фтор-3-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метил-пиразин-2-ил)метанол,
[2-Хлор-4-фтор-5-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метоксипиразин-2-ил)метанол,
[4-Фтор-3-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метокси-пиразин-2-ил)метанол,
[4-Фтор-3-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метокси-пиридазин-3-ил)метанол,
[4-Фтор-3-[7-(2,2,3,3,5,5,6,6-октадейтериоморфолин-4-ил)хиназолин-4-ил]фенил]-(3-метилпиразин-2-ил)метанол,
[2-Хлор-4-фтор-5-[7-(2,2,3,3,5,5,6,6-октадейтериоморфолин-4-ил)-хиназолин-4-ил]фенил]-(6-метоксипиридазин-3-ил)метанол,
[2-Хлор-4-фтор-5-(6-морфолин-4-илтиено[3,2-d]пиримидин-4-ил)фенил]-(3-метилпиразин-2-ил)метанол,
[4-Фтор-3-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метил-пиразин-2-ил)метанол,
[2-Хлор-4-фтор-5-(5-фтор-7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанол,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]фуро[2,3-d]пиридазин-7-илметанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]фуро[2,3-d]-пиридазин-7-илметанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]тиено[2,3-d]-пиридазин-7-илметанол,
(3,5-Диметилпиразин-2-ил)-[4-фтор-3-(7-морфолин-4-илхиназолин-4-ил)-фенил]метанол,
6-{[2-Хлор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-1-метил-1Н-пиридин-2-он,
6-{[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-2Н-пиридазин-3-он,
6-{[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]гидроксиметил}-1-метил-1Н-пиридин-2-он,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]фуро[2,3-d]пиримидин-4-илметанол,
[4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]фуро[3,2-d]пиримидин-4-илметанол,
[2,4-Дифтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метоксипиразин-2-ил)метанол,
4-Фтор-3-(7-морфолин-4-илхиназолин-4-ил)фенил]-(3-метил-3Н-имидазо-[4,5-с]пиридин-4-ил)метанол,
и/или его физиологически приемлемая соль и/или стереоизомер.
14. Соединение, которое представляет собой (S)-[2-Хлор-4-фтор-5-(7-морфолин-4-илхиназолин-4-ил)фенил]-(6-метоксипиридазин-3-ил)метанол или его физиологически приемлемую соль.
15. Способ получения соединения формулы (I) по п. 1 и/или его физиологически приемлемой соли и/или стереоизомера, включающий следующие стадии:
(а) взаимодействие соединения формулы (V)
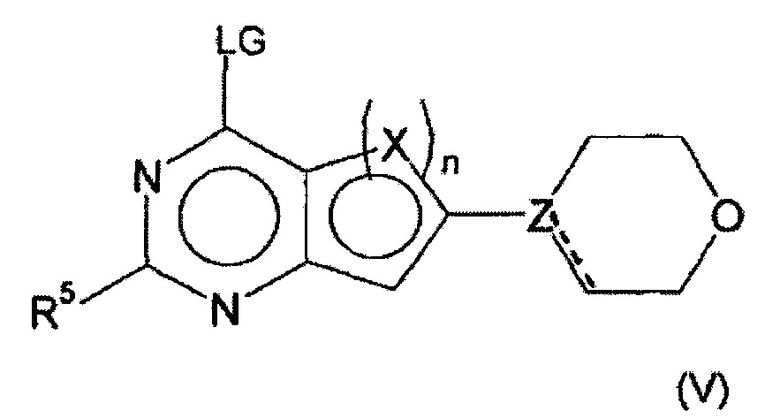
в которой LG представляет собой общепринятую уходящую группу, такую как Hal, и R5, X, n и Z имеют значения, указанные в п. 1,
с соединением формулы (IV)

в которой А представляет собой бороновую кислоту или сложный эфир бороновой кислоты, и R1, R2, R3, R4, Сус, Y и m имеют значения, указанные в п. 1,
с получением соединения формулы (I), и необязательно
(б) превращение основания или кислоты соединения формулы (I) в одну из его солей.
16. Применение соединения по любому из пп. 1-14 или его физиологически приемлемой соли и/или стереоизомера для приготовления лекарственного средства для сенсибилизации злокачественных клеток к противораковым средствам и/или ионизирующему излучению.
17. Применение соединения по любому из пп. 1-14 или его физиологически приемлемой соли и/или стереоизомера для приготовления лекарственного средства для профилактики и/или терапии злокачественного новообразования, опухолей или метастаз в комбинации с лучевой терапией и/или с по меньшей мере одним противораковым средством.
18. Лекарственное средство, обладающее свойствами ингибитора ДНК-зависимой протеинкиназы (ДНК-ПК), содержащее эффективное количество соединения по любому из пп. 1-14 или его физиологически приемлемую соль и/или стереоизомер.
| WO 2011113512 A1, 22.09.2011 & EA 201201289 A1, 30.08.2013 | |||
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2422449C2 |
| RU 2006137476 А, 20.06.2008. | |||

