Область техники, к которой относится изобретение
Настоящее изобретение относится к новому производному азолобензола и его кристаллу, которые полезны в качестве терапевтического средства или профилактического средства для заболеваний, связанных с ксантиноксидазой, таких как подагра, гиперурикемия, синдром лизиса опухоли, мочекаменная болезнь, гипертония, дислипидемия, диабет, сердечно-сосудистые заболевания, такие как артериосклероз или сердечная недостаточность, заболевания почек, такие как диабетическая нефропатия, респираторные заболеваний, такие как хронические обструктивные легочные заболевания, воспалительные заболевания кишечника или аутоиммунные заболевания, а также к способу получения такого кристалла.
Уровень техники
Ксантиноксидаза представляет собой фермент, катализирующий превращение гипоксантина в ксантин и далее в мочевую кислоту в процессе метаболизма нуклеиновых кислот.
С учетом действия ксантиноксидазы ингибитор ксантиноксидазы снижает уровень мочевой кислоты в крови, ингибируя ее синтез. То есть, ингибитор ксантиноксидазы эффективен в качестве терапевтического средства для лечения гиперурикемии и различных заболеваний, вызванных гиперурикемией. С другой стороны, подагрический артрит и подагрическое отложения солей, называемое подагрой, известны, как клиническое состояние, вызванное осаждением кристаллов урата после длительной гиперурикемии. Кроме того, гиперурикемия считается важным фактором заболеваний, связанных с образом жизни, ассоциированных с ожирением, гипертонией, дислипидемией и диабетом или метаболическим синдромом, а в последнее время в результате эпидемиологических исследований было выявлено, что гиперурикемия является фактором риска повреждения почек, мочекаменной болезни и сердечно-сосудистых заболеваний (The Guideline Revising Committee of Japanese Society of Gout and Nucleic Acid Metabolism, ed., Guideline for the management of hyperuricemia and gout, второе издание, Medical Review (2010)). Кроме того, ингибитор ксантиноксидазы, как ожидается, будет полезен при лечении заболеваний, связанных с активными формами кислорода из-за ингибирующей активности в отношении образования активных форм кислорода, например для лечения сердечно-сосудистых заболеваний посредством воздействия, улучшающего сосудистую функцию (Circulation, 2006; 114: 2508-2516).
В качестве терапевтического средства для лечения гиперурикемии клинически используют аллопуринол и фебуксостат, но аллопуринол, как сообщается, имеет побочные эффекты, такие как синдром Стивенса-Джонсона, токсический эпидермальный некролиз, расстройство печени и почечная дисфункция (Nippon Rinsho, 2003;. 61, Suppl 1: 197-201).
В качестве соединения, имеющего ингибирующую активность ксантиноксидазы, например, сообщалось о производном 2-фенилтиазола (см. ссылки PTL 1 -3).
С другой стороны, в документах PTL 4 и 5 сообщалось о производном дитиазол-карбоновой кислоты, имеющей бензольное кольцо в центре. Кроме того, в документах PTL 6 и 7 сообщалось о производном бифенил-тиазол-карбоновой кислоты.
Список процитированной литературы
Патентная литература]
PTL 1 Международная публикация № 92/09279
PTL 2 выложенный для всеобщего ознакомления патент Японии No. 2002-105067
PTL 3 Международная публикация № 96/31211
PTL 4 Международная публикация № 2011/139886
PTL 5 Международная публикация № 2011/101867
PTL 6 Международная публикация № 2010/018458
PTL 7 Международная публикация № 2010/128163
Сущность изобретения
Техническая задача
Задачей настоящего изобретения является создание нового соединения и его кристалла, полезных в качестве терапевтического или профилактического агента для лечения заболеваний, связанных с ксантиноксидазой, таких как подагра, гиперурикемия, синдром лизиса опухоли, мочекаменная болезнь, гипертония, дислипидемия, диабет, сердечно-сосудистые заболевания, такие как артериосклероз или сердечная недостаточность, заболевания почек, такие как диабетическая нефропатия, заболевания дыхательных путей, такие как хронические обструктивные заболевания легких, воспалительные заболевания кишечника или аутоиммунные заболевания. Кроме того, другой задачей изобретения является создание воспроизводимого способа получения кристалла, который является химически стабильным и подходит для активного фармацевтического ингредиента.
Решение поставленной задачи
В результате интенсивных исследований в соответствии с указанными задачами авторы настоящего изобретения обнаружили, что 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновая кислота (в дальнейшем также упоминаемая как соединение (I)) обеспечивает отличный эффект понижения мочевой кислоты при использовании в качестве ингибитора ксантиноксидазы, способна кристаллизоваться и существует, по меньшей мере, в виде трех типов кристаллических полиморфов. Кроме того, авторы настоящего изобретения обнаружили, что эти кристаллические полиморфные формы могут быть получены селективно. Кроме того, авторы настоящего изобретения обнаружили, что натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил) фенил]-1,3-тиазол-5-карбоксилат (далее также именуемый как соединение (II)) обеспечивает прекрасный эффект понижения мочевой кислоты при использовании в качестве ингибитора ксантиноксидазы и что соединение (II) может быть кристаллизовано.
Таким образом, настоящее изобретение относится к следующему.
[1] Кристалл 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты;
[2] Кристалл в соответствии с [1], имеющий кристаллическую форму А;
[3] Кристалл в соответствии с [2], который имеет характерные пики при углах дифракции 2θ = 8,6°, 10,2°, 13,3°, 14,4°, 18,5°, 19,9°, 21,8°, 25,1°, 25,6°, 26,6°, 27,1° и 29,5° в порошковой рентгеновской дифрактограмме;
[4] Кристалл в соответствии с [2], порошковая рентгеновская дифрактограмма которого имеет вид, представленный на фиг. 1;
[5] Кристалл в соответствии с [2], который имеет характерные пики при значениях химических сдвигов 116,3 ppm, 117,6 ppm, 120,0 ppm, 123,6 ppm, 125,9 ppm, 127,4 ppm, 143,7 ppm, 151,8 ppm, 161,1 ppm, 162,3 ppm и 165,5 ppm в его твердотельном спектре 13C ЯМР;
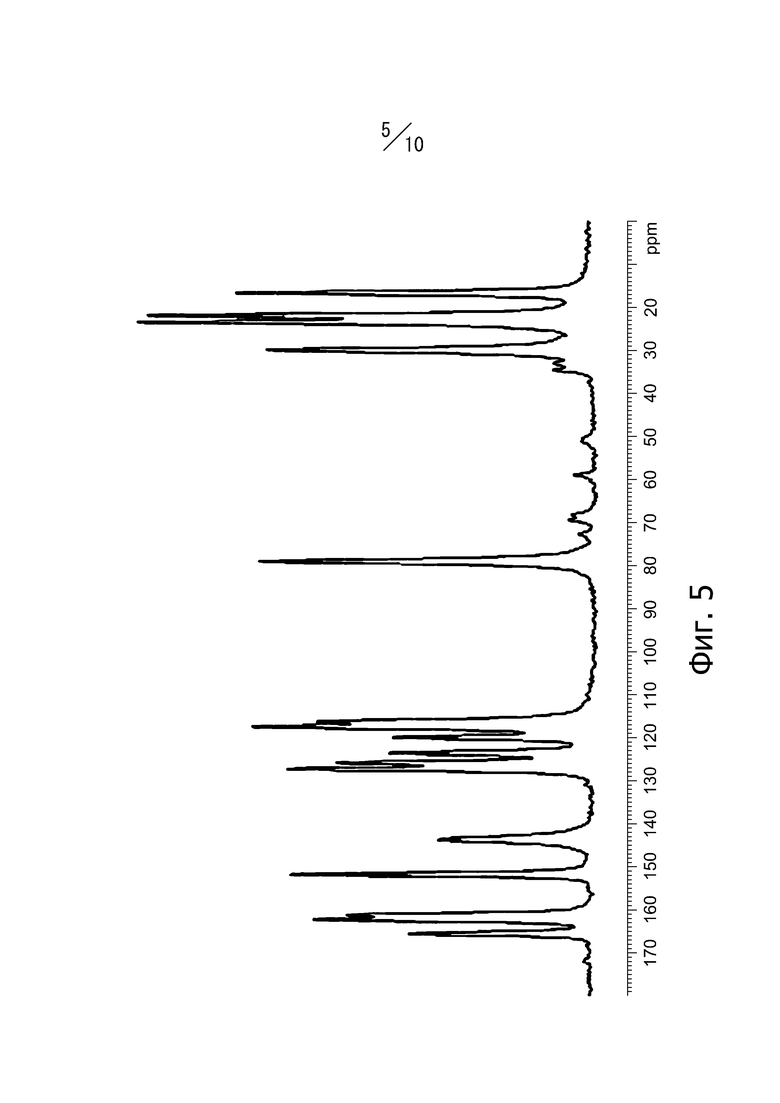
[6] Кристалл в соответствии с [2], твердотельный спектр 13C ЯМР которого имеет вид, показанный на фиг. 5;
[7] Кристалл в соответствии с [2], который имеет характерные пики при волновых числах 745 см-1, 822 см-1, 889 см-1, 975 см-1, 997 см-1, 1611 см-1 и 1705 см-1 в его ИК-спектре поглощения (способ KBr);
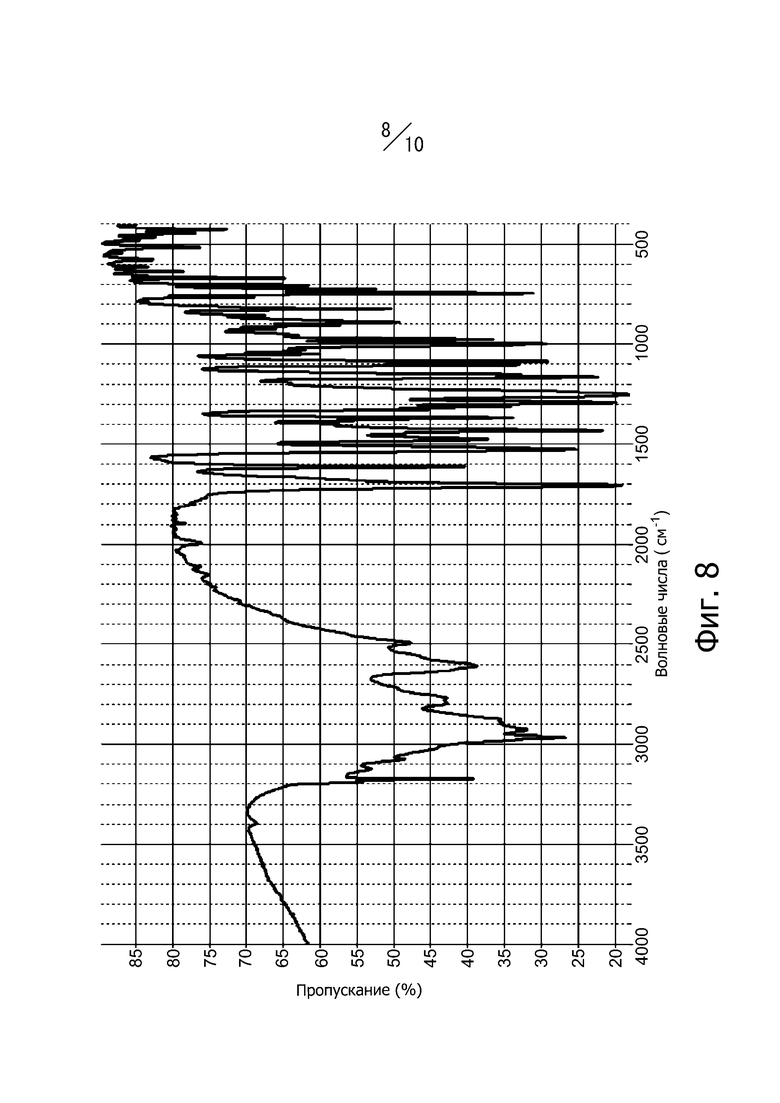
[8] Кристалл в соответствии с [2], инфракрасный спектр поглощения которого (метод KBr) имеет вид, показанный на фиг. 8;
[9] Кристалл в соответствии с [2], экзотермический пик которого в термогравиметрическом/дифференциальном термическом анализе располагается при 222°C;
[10] Кристалл в соответствии с [1], имеющий кристаллическую форму В;
[11] Кристалл в соответствии с [10], который имеет характерные пики при углах дифракции 2θ = 10,1°, 12,6°, 13,1°, 14,0°, 18,6°, 24,2°, 25,2°, 25,7°, 27,2° и 30,5° в его порошковой рентгеновской дифрактограмме;
[12] Кристалл в соответствии с [10], порошковая рентгеновская дифрактограмма которого имеет вид, показанный на фиг. 2;
[13] Кристалл в соответствии с [10], который имеет характерные пики при значениях химических сдвигов 115,4 ppm, 118,0 ppm, 119,8 ppm, 123,2 ppm, 126,4 ppm, 129,1 ppm, 142,7 ppm, 151,2 ppm, 160,9 ppm и 166,6 ppm в твердотельном спектре 13C ЯМР;
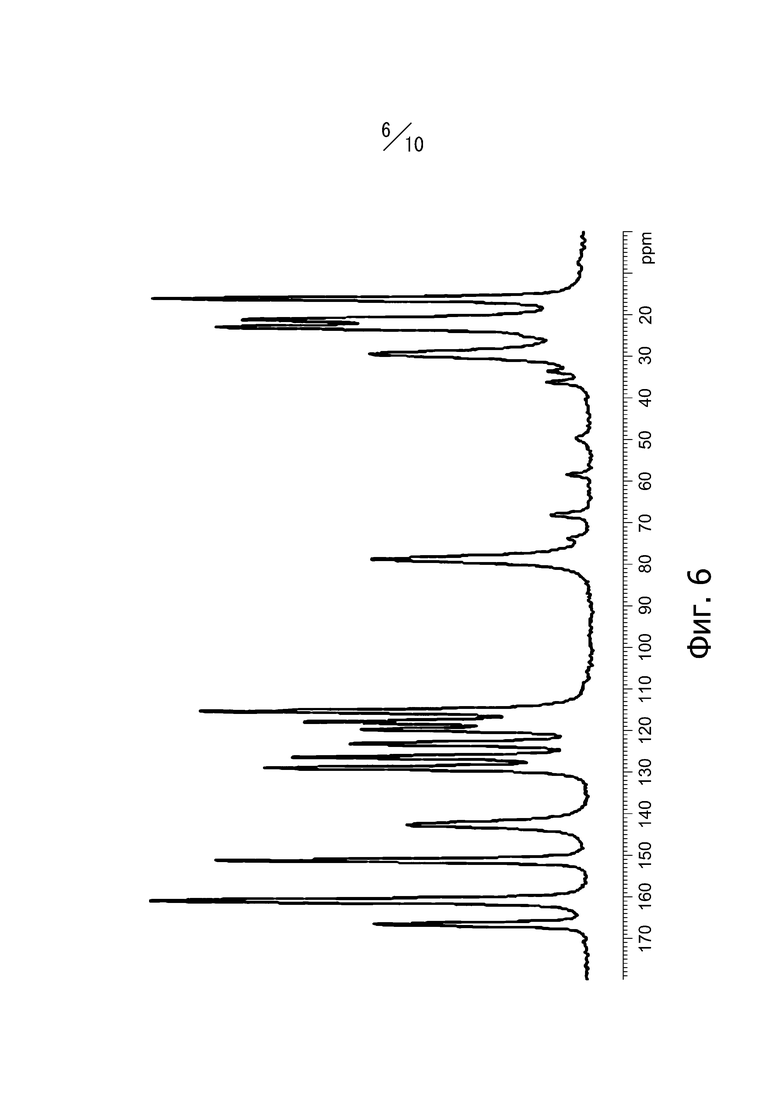
[14] Кристалл в соответствии с [10], твердотельный 13C-ЯМР-спектр которого имеет вид, показанный на фиг. 6;
[15] Кристалл в соответствии с [10], который имеет характерные пики при волновых числах 744 см-1, 810 см-1, 972 см-1, 997 см-1, 1005 см-1, 1611 см-1 и 1710 см-1 в ИК-спектре поглощения (способ KBr);
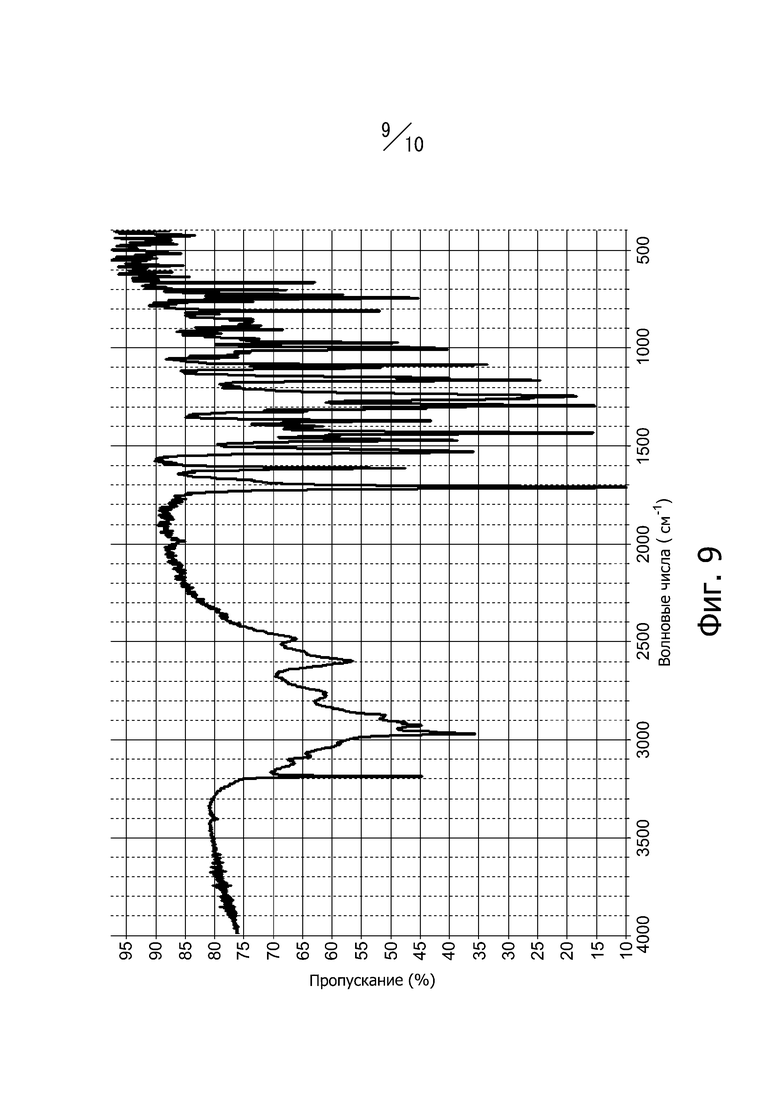
[16] Кристалл в соответствии с [10], ИК-спектр поглощения которого (способ KBr) имеет вид, показанный на фиг. 9;
[17] Кристалл в соответствии с [10], экзотермический пик которого в термогравиметрическом/дифференциальном термическом анализе располагается при 225°С, причем кристалл представляет собой безводный кристалл;
[18] Кристалл в соответствии с [1], имеющий кристаллическую форму С;
[19] Кристалл в соответствии с [18], который имеет характерные пики при углах дифракции 2θ = 7,2°, 12,5°, 13,0°, 14,7°, 19,2°, 20,0°, 21,4°, 21,7°, 24,7° и 26,0° в порошковой рентгеновской дифрактограмме;
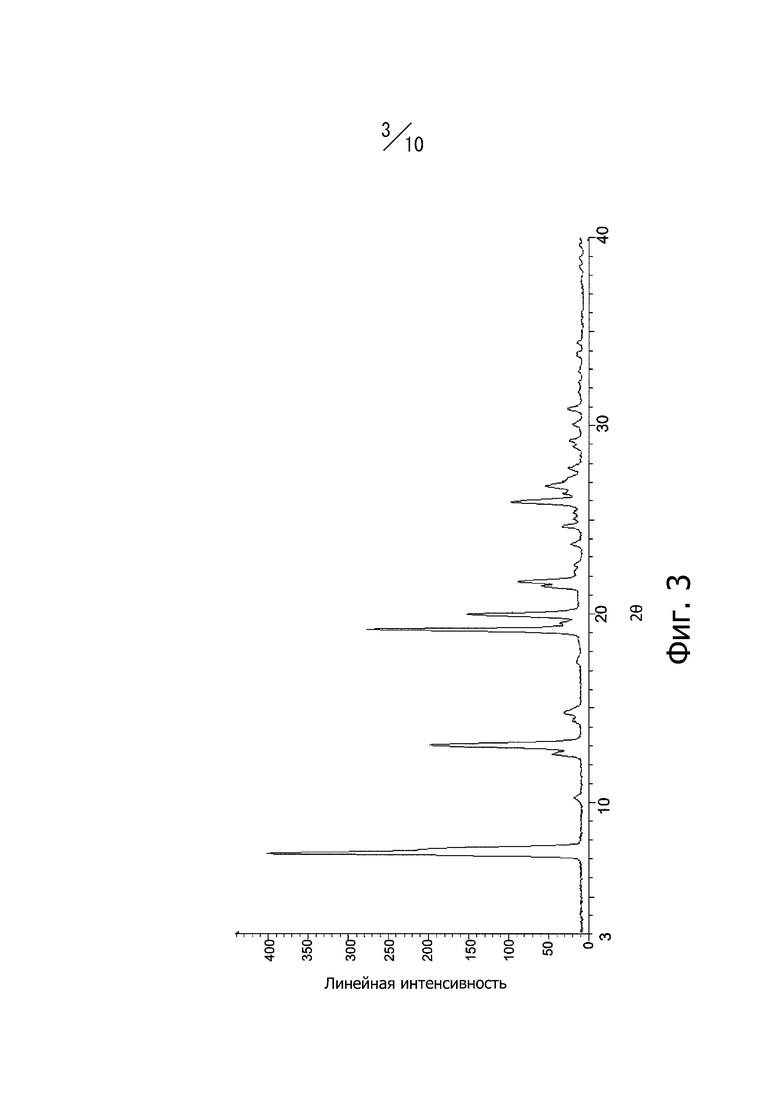
[20] Кристалл в соответствии с [18], порошковая рентгеновская дифрактограмма которого имеет вид, показанный на фиг. 3;
[21] Кристалл в соответствии с [18], который имеет характерные пики при значениях химических сдвигов 116,1 ppm, 119,6 ppm, 123,1 ppm, 126,1 ppm, 127,1 ppm, 130,0 ppm, 143,6 ppm, 150,3 ppm, 158,3 ppm, 160,7 ppm, 163,9 ppm, 165,5 ppm и 167,0 ppm в твердотельном спектре 13C ЯМР;
[22] Кристалл в соответствии с [18], твердотельный спектр 13C-ЯМР которого имеет вид, показанный на фиг. 7;
[23] Кристалл в соответствии с [18], который имеет характерные пики при волновых числах 745 см-1, 751 см-1, 809 см-1, 820 см-1, 971 см-1, 1006 см-1, 1613 см-1, 1682 см-1 и 1710 см-1 в ИК-спектре поглощения (способ KBr);
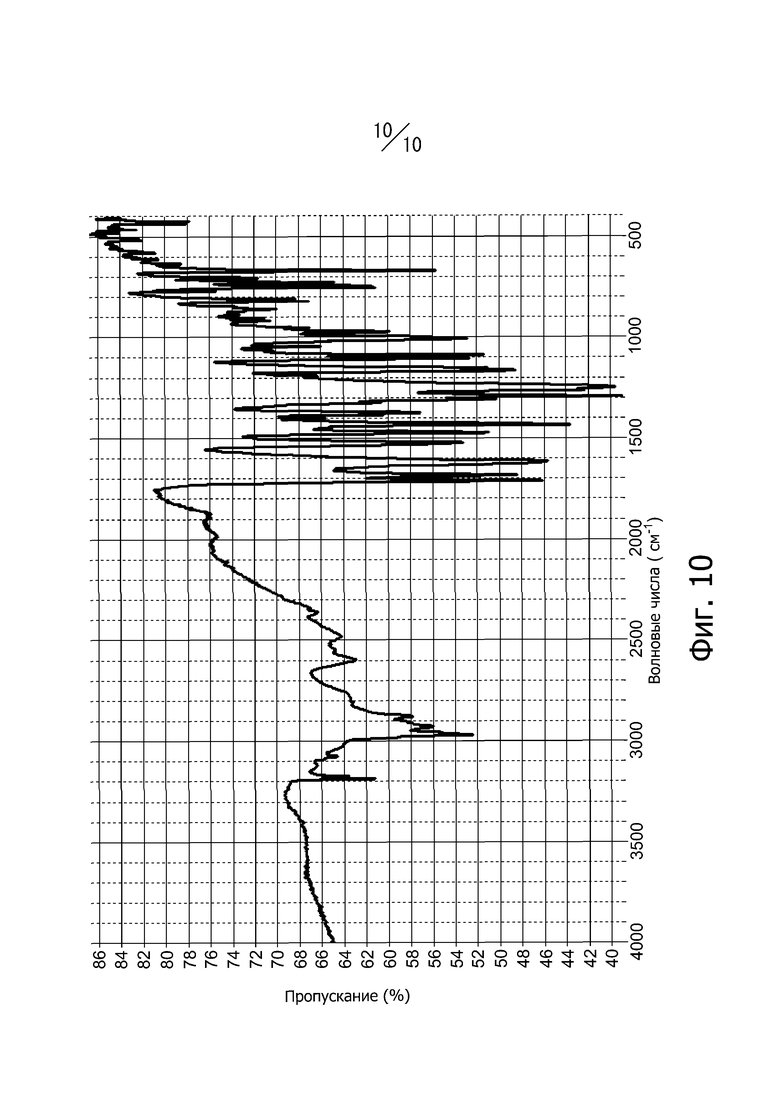
[24] Кристалл в соответствии с [18], ИК-спектр поглощения которого (способ KBr), имеет вид, показанный на фиг. 10;
[25] Кристалл в соответствии с [18], имеющий эндотермический пик при 88°С и экзотермический пик при 225°С в термогравиметрическом/дифференциальном термическом анализе;
[26] Натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилат;
[27] Кристалл соединения в соответствии с [26];
[28] Кристалл в соответствии с [27], который имеет характерные пики при углах дифракции 2θ = 7,2°, 10,9°, 13,3°, 15,9°, 18,2°, 20,8°, 22,1°, 25,2°, 26,1° и 29,1° в порошковой рентгеновской дифрактограмме;
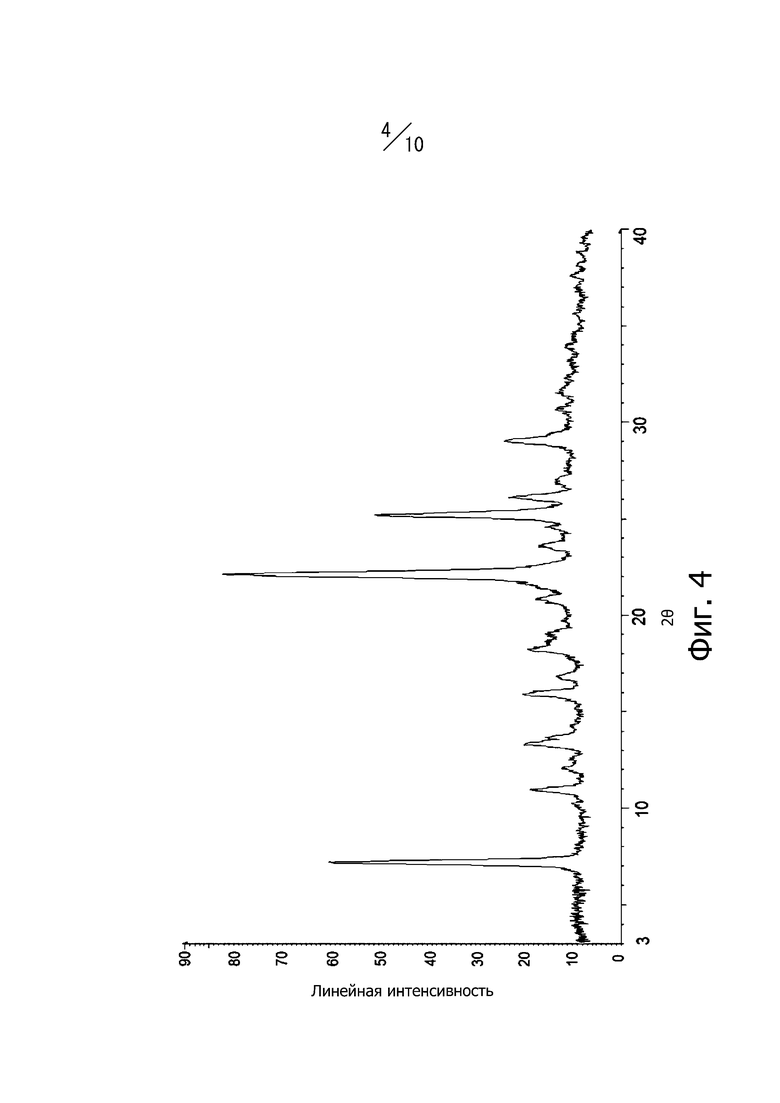
[29] Кристалл в соответствии с [27], порошковая рентгеновская дифрактограмма которого имеет вид, показанный на фиг. 4;
[30] Кристалл в соответствии с [27], экзотермический пик которого в термогравиметрическом/дифференциальном термическом анализе располагается при 281°C;
[31] Фармацевтическая композиция, содержащая соединение или его кристалл по любому из [1]-[30], и фармацевтически приемлемый носитель;
[32] Ингибитор ксантиноксидазы, содержащий соединение или его кристалл по любому из [1]-[30] в качестве активного ингредиента;
[33] Терапевтическое или профилактическое средство для одного или более заболеваний, выбранных из группы, состоящей из подагры, гиперурикемии, синдрома лизиса опухоли, мочекаменной болезни, гипертонии, дислипидемии, диабета, сердечно-сосудистых заболеваний, заболеваний почек, респираторных заболеваний, воспалительных заболеваний кишечника и аутоиммунных заболеваний, содержащее соединение или его кристалл по любому из [1]-[30] в качестве активного ингредиента;
[34] Способ получения кристаллической формы А из 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты, включающий следующие стадии:
суспендирование алкилового эфира 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты в растворителе и проведение гидролиза путем добавления водного раствора основания и
нейтрализации продукта реакции;
[35] Способ получения в соответствии с [34], дополнительно включающий стадию добавления воды к нейтрализованному продукту и его перемешивание;
[36] Способ получения кристаллической формы В 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты, включающий стадию суспендирования кристаллической формы А 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты в растворителе;
[37] Способ получения в соответствии с [36], дополнительно включающий стадию нагревания суспензии;
[38] Способ получения кристалла в соответствии с [36] или [37], в котором растворитель выбирают из группы, состоящей из простых эфиров, кетонов, сложных эфиров, спиртов, воды и смесей этих растворителей; и
[39] Способ получения кристаллической формы С 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты, включающий кристаллизацию из ее раствора в N,N-диметилформамиде.
Выгодные эффекты, обеспечиваемые изобретением
Настоящее изобретение относится к кристаллам производного азолобензола, которые полезны в качестве терапевтических или профилактических средств при заболеваниях, связанных с ксантиноксидазой, таких как подагра, гиперурикемия, синдром лизиса опухоли, мочекаменная болезнь, гипертония, дислипидемия, диабет, сердечно-сосудистые заболевания, такие как артериосклероз или сердечная недостаточность, заболевания почек, такие как диабетическая нефропатия, заболевания дыхательных путей, такие как хронические обструктивные заболевания легких, воспалительные заболевания кишечника или аутоиммунные заболевания, а также к способу их получения. Кристалл соединения (I) или соединение (II) или его кристалл могут быть использованы в качестве активного фармацевтического ингредиента для получения фармацевтического агента. Кроме того, способы по настоящему изобретению для получения кристалла соединения (I) или соединения (II) или его кристалла пригодны для промышленного производства.
Краткое описание чертежей
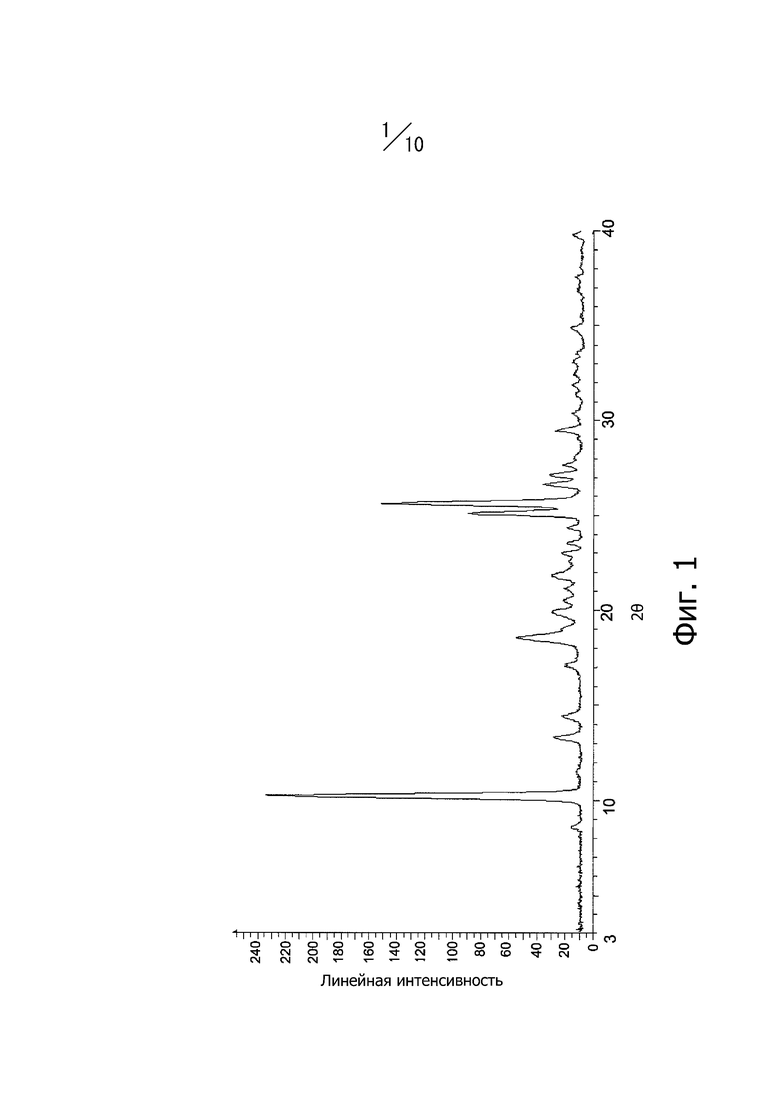
Фиг. 1 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы А соединения (I).
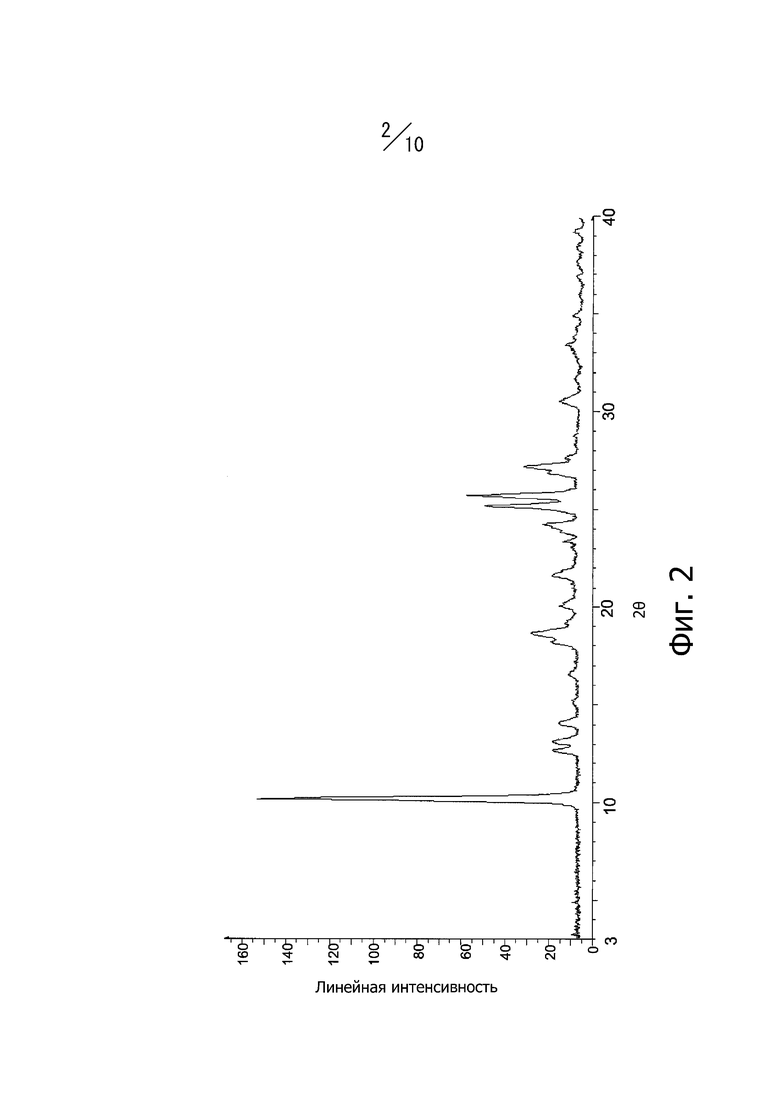
Фиг. 2 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы В соединения (I).
Фиг. 3 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы С соединения (I).
Фиг. 4 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы А соединения (II).
Фиг. 5 представляет собой твердотельный спектр 13C-ЯМР кристаллической формы А соединения (I).
Фиг. 6 представляет собой твердотельный спектр 13C-ЯМР кристаллической формы В соединения (I).
Фиг. 7 представляет собой твердотельный спектр 13C-ЯМР кристаллической формы С соединения (I).
Фиг. 8 представляет собой ИК-спектр поглощения (метод KBr) кристаллической формы А соединения (I).
Фиг. 9 представляет собой ИК-спектр поглощения (метод KBr) кристаллической формы В соединения (I).
Фиг. 10 представляет собой ИК-спектр поглощения (метод KBr) кристаллической формы С соединения (I).
Описание вариантов осуществления изобретения
Термин «ксантиноксидаза» используется как в широком смысле, согласно которому он означает фермент для катализа реакции окисления гипоксантина в ксантин и далее в мочевую кислоту, так и в узком смысле, согласно которому он означает ксантиноксидоредуктазу типа оксидазы, и является одним из ферментов, катализирующих ту же реакцию. В настоящем изобретении, если не указано иное, «ксантиноксидазой» собирательно называют фермент, который катализирует реакцию окисления гипоксантина в ксантин и далее в мочевую кислоту. Среди ксантиноксидоредуктаз, которые отвечают за эту реакцию, имеется два типа оксидоредуктаз: типа оксидазы и типа дегидрогеназы, и оба вида включены в ксантиноксидазы по настоящему изобретению. Если не указано иное, «ксантиноксидаза» в выражениях «активность ингибирования ксантиноксидазы», «ингибитор ксантиноксидазы» и т.п., имеет такое же значение, что и указанное выше.
Кристаллы по настоящему изобретению характеризуются порошковыми рентгеновскими дифрактограммами, твердотельными спектрами 13C-ЯМР, ИК-спектрами поглощения (способ KBr), и/или термогравиметрическим/дифференциальным термическим анализом (TG/DTA) и т.п. Порошковая рентгеновская дифрактограмма (XRD), твердотельный спектр 13C-ЯМР и ИК-спектр поглощения (метод KBr) этих кристаллов демонстрируют характерные паттерны, и каждый кристалл имеет специфические значения угла дифракции 2θ. Кроме того, каждый из этих кристаллов также проявляет свое собственное характерное термическое поведение в термогравиметрическом/дифференциальном термическом анализе (TG/DTA).
Кристаллическая форма А соединения (I) имеет характерные пики при углах дифракции 2θ = 8,6°, 10,2°, 13,3°, 14,4°, 18,5°, 19,9°, 21,8°, 25,1°, 25,6°, 26,6°, 27.1° и 29,5° в порошковой рентгеновской дифрактограмме. Кроме того, порошковая рентгеновская дифрактограмма кристаллической формы А соединения (I) имеет вид, показанный на фиг. 1.
Кристаллическая форма А соединения (I) имеет пики при значениях химических сдвигов 116,3 ppm, 117,6 ppm, 120,0 ppm, 123,6 ppm, 125,9 ppm, 127,4 ppm, 143,7 ppm, 151,8 ppm, 161,1 ppm, 162,3 ppm и 165,5 ppm в твердотельном спектре 13C-ЯМР. Кроме того, твердотельный спектр 13С-ЯМР кристаллической формы А соединения (I) имеет вид, показанный на фиг. 5.
Кристаллическая форма А соединения (I) имеет пики поглощения при волновых числах 745 см-1, 822 см-1, 889 см-1, 975 см-1, 997 см-1, 1611 см-1 и 1705 см-1 в инфракрасном спектре поглощения (способ KBr). Кроме того, инфракрасный спектр поглощения кристаллической формы А соединения (I) (способ KBr) имеет вид, показанный на фиг. 8.
Кристаллическая форма А соединения (I) имеет экзотермический пик при 222°С в термогравиметрическом/дифференциальном термическом анализе (TG/DTA). Такая кристаллическая форма А представляет собой безводный кристалл.
Кристаллическая форма В соединения (I) имеет характерные пики при углах дифракции 2θ = 10,1°, 12,6°, 13,1°, 14,0°, 18,6°, 24,2°, 25,2°, 25,7°, 27,2° и 30,5° в порошковой рентгеновской дифрактограмме. Кроме того, порошковая рентгеновская дифрактограмма кристаллической формы В соединения (I) имеет вид, показанный на фиг. 2.
Кристаллическая форма В соединения (I) имеет пики при значениях химических сдвигов 115,4 ppm, 118,0 ppm, 119,8 ppm, 123,2 ppm, 126,4 ppm, 129,1 ppm, 142,7 ppm, 151,2 ppm, 160,9 ppm и 166,6 ppm в твердотельном спектре 13C-ЯМР. Кроме того, твердотельный спектр 13С-ЯМР кристаллической формы В соединения (I) имеет вид, показанный на фиг. 6.
Кристаллическая форма В соединения (I) имеет пики поглощения при волновых числах 744 см-1, 810 см-1, 972 см-1, 997 см-1, 1005 см-1, 1611 см-1 и 1710 см-1 в инфракрасном спектре поглощения (способ KBr). Кроме того, инфракрасный спектр поглощения кристаллической формы В соединения (I) (метод KBr) имеет вид, показанный на фиг. 9.
Кристаллическая форма В соединения (I) имеет экзотермический пик при 225°С в термогравиметрическом/дифференциальном термическом анализе (TG/DTA). Такая кристаллическая форма В представляет собой безводный кристалл.
Кристаллическая форма С соединения (I) имеет характерные пики при углах дифракции 2θ = 7,2°, 12,5°, 13,0°, 14,7°, 19,2°, 20,0°, 21,4°, 21,7°, 24,7° и 26,0° в порошковой рентгеновской дифрактограмме. Кроме того, порошковая рентгеновская дифрактограмма кристаллической формы С соединения (I) имеет вид, показанный на фиг. 3.
Кристаллическая форма С соединения (I) имеет пики при значениях химических сдвигов 116,1 ppm, 119,6 ppm, 123,1 ppm, 126,1 ppm, 127,1 ppm, 130,0 ppm, 143,6 ppm, 150,3 ppm, 158,3 ppm, 160,7 ppm, 163,9 ppm, 165,5 ppm и 167,0 ppm в твердотельном спектре 13С-ЯМР. Кроме того, твердотельный спектр 13С-ЯМР кристаллической формы С соединения (I) имеет вид, показанный на фиг. 7.
Кристаллическая форма С соединения (I) имеет пики поглощения при волновых числах 745 см-1, 751 см-1, 809 см-1, 820 см-1, 971 см-1, 1006 см-1, 1613 см-1, 1682 см-1 и 1710 см-1 в ИК-спектре поглощения (способ KBr). Кроме того, инфракрасный спектр поглощения кристаллической формы С соединения (I) (метод KBr) имеет вид, показанный на фиг. 10.
Кристаллическая форма С соединения (I) имеет эндотермический пик при 88° С и экзотермический пик при 225°С в термогравиметрическом/дифференциальном термическом анализе (TG/DTA). Кристаллическая форма С, как полагают, образует сольват с диметилформамидом в соотношении 1:1.
Кристаллическая форма А соединения (II) имеет характерные пики при углах дифракции 2θ = 7.2°, 10,9°, 13,3°, 15,9°, 18,2°, 20,8°, 22,1°, 25,2°, 26,1° и 29,1° в порошковой рентгеновской дифрактограмме. Кроме того, порошковая рентгеновская дифрактограмма кристаллической формы А соединения (II) имеет вид, показанный на фиг. 4. В дополнение, кристаллическая форма А соединения (II) имеет экзотермический пик при 281°С в термогравиметрическом/дифференциальном термическом анализе (TG/DTA). Такая кристаллическая форма А представляет собой безводный кристалл.
Используемый в данном описании термин «характерные пики» означают главные пики и уникальные пики, которые наблюдаются в порошковой рентгеновской дифрактограмме, твердотельном спектре 13C ЯМР и ИК-спектре поглощения (метод KBr) каждого кристаллического полиморфа. Кристаллы, идентифицируемые углами дифракции по настоящему изобретению, также имеют пики, отличные от тех, которые наблюдаются в качестве характерных пиков, как описано выше.
Положение и относительная интенсивность дифракционного угла 2θ в порошковой рентгеновской дифрактограмме могут незначительно варьироваться в зависимости от условий измерения, и, следовательно, даже если 2θ немного отличается, природу кристаллической формы можно распознать путем детального сравнения с видом всего спектра. Кристаллы в пределах диапазона таких ошибок также включены в настоящее изобретение. Ошибки в 2θ могут быть, например, в диапазоне ±0,5° или ±0,2°. Другими словами, кристаллы, идентифицированные описанными выше дифракционными углами, включают также и те, которые имеют углы дифракции в пределах погрешности ± 0,5° или ± 0,2°.
Как правило, ошибки могут возникать также и в химических сдвигах в твердотельном спектре 13С ЯМР. Такие ошибки находятся в диапазоне, например, ±0,25 ppm, и, как правило, ±0,5 ppm Иными словами, кристаллы, идентифицированные вышеприведенными химическими сдвигами, включают также и те, которые имеют химические сдвиги в пределах погрешности ±0,25 ppm или ±0,5 ppm
Как правило, ошибки могут возникать также в пиках поглощения в инфракрасном спектре поглощения (способ KBr). Такие ошибки находятся в диапазоне, например, ±2 см-1, а обычно ±5 см-1. Иными словами, кристаллы, идентифицированные описанными выше волновыми числами, включают также и те, которые имеют волновые числа в пределах погрешности ±2 см-1 или ±5 см-1.
В термогравиметрическом/дифференциальном термическом анализе (TG/DTA) «экзотермический пик» и «эндотермический пик» определяются как температура в начальной точке пика, и означают начальную экзотермическую и эндотермическую температуру, определенную путем экстраполяции. «Экзотермический пик» и «эндотермический пик » в TG/DTA могут немного варьироваться в зависимости от условий измерения. Например, ошибка, как полагают, лежит в пределах ±5°С или ±2°C. Иными словами, кристаллы, идентифицированные описанными выше пиками, включают также и те, которые имеют пики в пределах диапазона погрешности ±5°С или ±2°С.
Кроме того, для порошковой рентгеновской дифрактограммы, спектра твердотельного 13C-ЯМР, ИК-спектра поглощения (способ KBr) и TG/DTA, различие между измеренными значениями для эталонного материала кристаллов, например каждого из кристаллов, полученных способом, описанным в настоящих примерах, и числовыми значениями, приведенным в настоящем документе, может быть принято в качестве ошибки измерения. То есть, кристаллы, которые имеют одинаковые углы дифракции, химические сдвиги, инфракрасные пики поглощения или экзотермические и эндотермические пики в пределах диапазона ошибок, определяемых такими способами, включены в кристаллы по настоящему изобретению.
Кристаллическую форма А соединения (I) получают способом, включающим следующие стадии:
суспендирование алкилового эфира соединения (I) в растворителе и проведение гидролиза путем добавления водного раствора основания; и
нейтрализации продукта реакции.
Кроме того, способ получения кристаллической формы А соединения (I) может дополнительно включать стадию добавления воды к нейтрализованному веществу и последующую стадию перемешивания реакционного раствора.
Растворитель, используемый для суспендирования алкилового эфира соединения (I), включает, например, ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан, 1,2-диэтоксиэтан и метил-трет-бутиловый эфир; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол, бутанол гексафтор-2-пропанола и трифторэтанол; N,N-диметилформамид (DMF); N,N-диметилацетамид (DMA); N-метилпирролидон; диметилсульфоксид (DMSO); ацетонитрил; ацетон; этилацетат; и воду, и смешанный растворитель из них. Предпочтительным растворителем является простой эфир или спирт, вода или смешанный растворитель из них.
Примеры алкилового эфира соединения (I) предпочтительно включают алкиловый эфир, имеющий от 1 до 6 атомов углерода и более предпочтительно этиловый эфир. Здесь алкиловый эфир относится к линейному или разветвленному алифатическому насыщенному углеводородному сложному эфиру. Конкретные примеры алкилового эфира, имеющего от 1 до 6 атомов углерода, включают метиловый эфир, этиловый эфир, изопропиловый эфир и трет-бутиловый эфир.
Реакция гидролиза алкилового эфира соединения (I) в соединение (I) протекает путем суспендирования алкилового эфира соединения (I) в растворителе, упомянутом выше (например, в 15-кратном количестве относительно количества алкилового эфира), с последующей реакцией суспензии с основанием в эквивалентном или слегка избыточном количестве по отношению к алкиловому эфиру. Примеры предпочтительных оснований включают гидроксид натрия, гидроксид калия и гидроксид лития. Реакция протекает в диапазоне от 0°С до 100°С и предпочтительно проводится в диапазоне от 20°С до 30°С.
После реакции гидролиза продукт реакции нейтрализуют с помощью реакции используемого основания с кислотой в эквивалентном или слегка избыточном количестве по отношению к используемому основанию. Пример предпочтительной кислоты включает соляную кислоту. Реакция нейтрализации протекает в диапазоне от 0°С до 100°С и предпочтительно в диапазоне от 0°С до 30°С.
Затем к нейтрализованному продукту реакции добавляют воду (например, в 5 кратном количестве относительно алкилового эфира) и полученную смесь перемешивают в течение одного часа, после чего осадок отфильтровывают и сушат с получением кристаллов. Хотя количество растворителя, количество добавляемой воды, условия перемешивания и количество времени до разделения с помощью фильтрации особо не ограничены, но поскольку эти условия могут оказывать влияние на выход кристаллов, химическую чистоту, распределение диаметров частиц и размер частиц, то эти условия предпочтительно комбинируют и устанавливают в соответствии с целью. Фильтрация может быть осуществлена с использованием обычного способа, например естественной фильтрации, фильтрации под давлением, вакуумной фильтрации или отделения центрифугированием. Сушка может быть осуществлена с использованием обычного способа, например, могут быть использованы естественная сушка, вакуумная сушка, сушка с нагреванием и вакуумная сушки с нагреванием.
Алкиловый эфир соединения (I) может быть синтезирован с помощью любого из способов, например, с помощью следующих способов.
Синтез соединения (А-2)
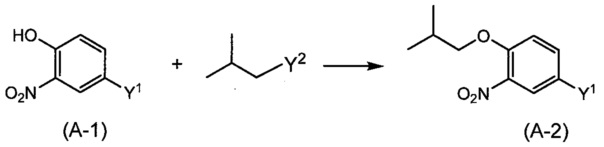
(где Y1 и Y2 представляют собой уходящую группу).
Примеры уходящей группы, представленной Y1 и Y2, включают атом галогена, метансульфонилоксигруппу, п-толуолсульфонилоксигруппу, трифторметансульфонилоксигруппу и т.п. Реакция является способом синтеза соединения (А-2) путем реакции алкилирующего реагента, имеющего уходящую группу, с фенольной гидроксильной группой в соединении (A-1) в присутствии основания. Примеры используемого основания включают неорганические соли, такие как гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид лития, карбонат натрия, карбонат калия, карбонат цезия; алкоксид металла, такой как этоксид натрия, метоксид натрия и трет-бутоксид калия; и органические амины, такой как триэтиламин, пиридин, 4-аминопиридин, N-этил-N,N-диизопропиламин (DIPEA) и 1,8-диазабицикло [5.4.0]-7-ундецен (DBU). Реакцию осуществляют путем взаимодействия соединения (A-1) с основанием в эквивалентном или слегка избыточном количестве по отношению к соединению (A-1), в растворителе, неактивном по отношению к реакции, в диапазоне от 0°С и 140°C, с последующим добавлением алкилирующего агента в эквивалентном или избыточном количестве по отношению к соединению (A-1), и проведением реакции обычно в течение от 0,5 до 16 часов. Реакцию предпочтительно проводят в атмосфере инертного газа, такого как азот. Здесь, растворитель включает, хотя и без особых ограничений, например, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан и 1,2-диэтоксиэтан; N,N-диметилформамид (DMF); N-метилпирролидон; диметилсульфоксид (DMSO); воду; и смешанный растворитель из них.
Синтез соединения (А-4)
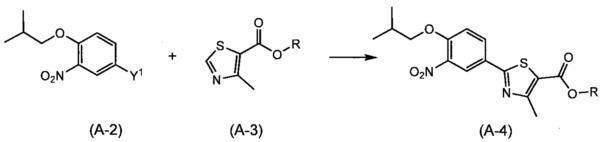
(Где R представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода).
Способ синтеза представляет собой способ синтеза соединения (А-4) путем сочетания соединений (А-2) и (А-3). Примеры уходящей группы, представленной Y1, включают атом галогена, метансульфонилокси группу, п-толуолсульфонилокси группу и трифторметансульфонилокси группу. Реакцию осуществляют путем взаимодействия соединения (А-2) и (А-3) с использованием эквивалентного или избыточного количества одного соединения относительно другого, в растворителе, неактивном по отношению к реакции, в присутствии основания и катализатора на основе переходного металла, путем добавления лиганда, карбоновой кислоты и моно- или двухосновной соли меди в случае необходимости, в интервале от комнатной температуры до температуры флегмы в целом в течение 0,5 часов до 2 дней. Реакцию предпочтительно проводят в атмосфере инертного газа, такого как азот. Здесь, растворитель включает, хотя и без особых ограничений, например, ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан и 1,2-диэтоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол и бутанол; N,N-диметилформамид (DMF); N-метилпирролидон; диметилсульфоксид (DMSO); воду; и смешанный растворитель из них. Примеры оснований включают гидрид лития, гидрид натрия, гидрид калия, гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, карбонат цезия, фторид калия, фторид цезия, трикалийфосфат, ацетат натрия и ацетат калия; соль металла алкоксида, имеющего от 1 до 6 атомов углерода (соль лития, соль натрия, соль калия и соль магния); соль металла алкильного аниона, имеющего от 1 до 6 атомов углерода (соль лития, соль натрия, соль калия и соль магния); тетра (алкил, содержащий от 1 до 4 атомов углерода), соль аммония (фторид, хлорид и бромид); диизопропилэтиламин; трибутиламин; N-метилморфолин; диазабициклоундецен; диазабициклооктан; и имидазол. Примеры катализатора на основе переходного металла включают медь, палладий, кобальт, железо, родий, рутений и иридий. Примеры лигандов включают три(трет-бутил)фосфин, три(циклогексил)фосфин, трет-бутилдициклогексилфосфин, ди(трет-бутил)циклогексилфосфин и ди(трет-бутил)метилфосфин. Примеры соли одновалентной или двухвалентной меди включают хлорид меди (I), бромид меди (I), йодид меди (I), ацетат меди (I), фторид меди (II), хлорид меди (II), бромид меди (II), йодид меди (II), ацетат меди (II), их гидрат и смесь. Примеры карбоновой кислоты включают муравьиную кислоту, уксусную кислоту, пропионовую кислоту, н-масляную кислоту, изомасляную кислоту, пентановую кислоту, изопентановую кислоту, пивалиновую кислоту и трифторуксусную кислоту.
Синтез соединения (А-5)
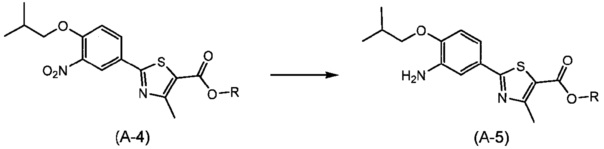
(Где R представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода).
Способ синтеза представляет собой способ синтеза соединения (А-5) путем восстановления нитрогруппы соединения (А-4). Реакцию осуществляют путем взаимодействия соединения (A-4) в атмосфере газообразного водорода, в растворителе, неактивном по отношению к реакции, в присутствии катализатора на основе переходного металла в интервале от комнатной температуры до температуры кипения обычно в течение от 0,5 часов до 2 дней. Здесь растворитель включает (хотя и не ограничивается этим), например, ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан и 1,2-диэтоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол и бутанол; N,N-диметилформамид (DMF); N-метилпирролидон; диметилсульфоксид (DMSO); этилацетат; и смешанный растворитель из них. Предпочтительные примеры катализатора на основе переходного металла включают палладий на угле, гидроксид палладия, палладиевую чернь, платину-на-угле, никель Рене и т.п.
Синтез соединения (А-6)
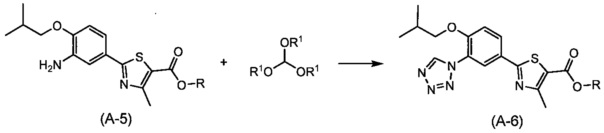
(где R и R1 независимо друг от друга представляют собой алкильную группу, имеющую от 1 до 6 атомов углерода).
Способ синтеза представляет собой способ синтеза тетразольного кольца путем взаимодействия соединения (A-5) с орто-формиатом и соединением азида. Реакцию осуществляют путем взаимодействия соединения (А-5), ортоформиата и соединения азида, с использованием эквивалентного или избыточного количества одного из соединений по отношению к другим в растворителе, неактивном по отношению к реакции, в присутствии кислоты в диапазоне от комнатной температуры до температуры кипения обычно в течение от 0,5 часов до 2 дней. Реакцию предпочтительно проводят в атмосфере инертного газа, такого как азот. Примеры орто-формиата включают триметилортоформиат и триэтилортоформиат. Кроме того, примеры соединения азида включают азид натрия и триметил силилазид. Примеры используемой кислоты включают органическую кислоту, такую как муравьиная кислота и уксусная кислота, неорганическую кислоту, такую как соляная кислота и серная кислота, и кислоту Льюиса, такую как трифлат индия, трифлат иттербия, трифлат цинка и трихлориндий. Растворитель, который можно использовать для этих реакций, включает (хотя и не ограничивается этим), например, бензол, толуол, дихлорметан, дихлорэтан, хлороформ, четыреххлористый углерод, диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, N,N-диметилформамид (DMF), N-метилпирролидон, диметилсульфоксид (DMSO), и смеси этих растворителей, и также в качестве растворителя может быть использована кислота, такая как уксусная кислота.
Промежуточные соединения реакции могут быть очищены с помощью обычного способа, такого как перекристаллизация, переосаждение и разнообразные хроматографические способы, при необходимости, в процессе синтеза.
Кристалл В соединения (I) может быть получен способом, включающим стадию суспендирования кристалла А соединения (I) в растворителе.
Кроме того, способ получения кристалла B соединения (I) может дополнительно включать стадию последующего нагрева реакционного раствора.
При получении кристалла В соединения (I) примеры растворителей для суспендирования кристалла А включают: ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан, 1,2-диэтоксиэтан и метил-трет-бутиловый эфир; кетоны, такие как ацетон и 2-бутанон, сложные эфиры, такие как этилацетат и изобутилацетат; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол, бутанол, гексафтор-2-пропанол и трифторэтанол; N,N-диметилформамид (DMF); N,N-диметилактамид (DMA); N-метилпирролидон; диметилсульфоксид (DMSO); ацетонитрил; ацетон; этилацетат; метилэтилкетон; воду; или смешанный растворитель из них. Предпочтительные примеры растворителя включают простые эфиры, кетоны, сложные эфиры, спирты, воду или смешанный растворитель из них.
Превращение кристалла А соединения (I) в кристалл В соединения (I) протекает путем суспендирования кристалла А соединения (I) в вышеуказанном растворителе (например, 5-60 кратном количестве относительно количества кристалла А соединения (I)), с последующим нагреванием реакционного раствора с обратным холодильником в течение 6 часов.
Затем, после перемешивания реакционного раствора при 25°С осадок отфильтровывают и сушат с получением кристаллов. Хотя количество растворителя, время нагрева, условия перемешивания и время до разделения фильтрованием никак особенное не ограничиваются, но поскольку эти условия могут оказывать влияние на выход кристаллов, химическую чистоту, диаметр частиц и распределение частиц по размерам, эти условия предпочтительно объединяются и устанавливаются в соответствии с поставленными целями. Фильтрация может быть осуществлена с использованием обычного способа, например естественной фильтрации, фильтрации под давлением, вакуумной фильтрации или разделения центрифугированием. Сушка может быть осуществлена с использованием обычного способа, например естественной сушки, вакуумной сушки, сушки с нагреванием и вакуумной сушки с нагреванием.
Кристалл С соединения (I) получают путем кристаллизации соединения (I) с использованием растворителя N,N-диметилформамида.
Реакционный раствор готовят путем добавления N,N-диметилформамида (например, 10-кратно превышающем количество соединения (I)) к соединению (I), и растворения смеси при нагревании и перемешивании при 80°С. Реакционный раствор охлаждают до температуры в интервале от 20°С до 30°С и перемешивают в течение двух часов. Осадок отфильтровывают и отфильтрованный продукт промывают этанолом (например, 10-кратным количеством относительно соединения (I)). Маточную жидкость оставляют отстаиваться в диапазоне от 20°С до 30°С в течение 7 дней, и осадок отфильтровывают и сушат с получением кристаллов. Хотя растворитель, условия перемешивания и время до разделения с помощью фильтрации никак особенно не ограничиваются, но поскольку эти условия могут оказывать влияние на выход кристаллов, химическою чистоту, диаметр частиц и распределение частиц по размерам, то эти условия предпочтительно объединяются и устанавливаются в соответствии с поставленными целями. Фильтрация может быть осуществлена с использованием обычного способа, например естественной фильтрации, фильтрации под давлением, вакуумной фильтрации или разделения центрифугированием. Сушка может быть осуществлена с использованием обычного способа, например естественной сушки, вакуумной сушки, сушки с нагреванием и вакуумной сушки с нагреванием.
Кристалл С соединения (I) может быть получен способом, включающим стадию суспендирования кристалла B соединения (I) в растворителе.
Смешанный раствор (например, 10-кратно превышающий количество соединения (I)) N,N-диметилформамида и этилацетата в соотношении 1:1 добавляют к кристаллу В соединения (I) с последующим перемешиванием при комнатной температуре в течение 14 дней. Полученный раствор фильтруют, и отфильтрованный продукт сушат при комнатной температуре с получением кристаллов. Хотя растворитель, условия перемешивания и время до разделения с помощью фильтрации никак особенно не ограничиваются, но, поскольку эти условия могут оказывать влияние на выход кристаллов, химическую чистоту, диаметр частиц и распределение частиц по размерам, эти условия предпочтительно объединяются и устанавливаются в соответствии с поставленными целями. Фильтрация может быть осуществлена с использованием обычного способа, например естественной фильтрации, фильтрации под давлением, вакуумной фильтрации или разделения центрифугированием. Сушка может быть осуществлена с использованием обычного способа, например естественной сушки, вакуумной сушки, сушки с нагреванием и вакуумной сушки с нагреванием.
Соединение (I) может быть получено, например, по следующему способу.
(где R2 представляет собой защитную группу карбоксильной группы).
Способ синтеза представляет собой способ синтеза соединения (I) удалением защитной группы R2 соединения (А-7) с помощью кислоты, основания или т.п. Термин «защитная группа карбоксильной группы» означает, например, общую защитную группу карбоксильной группы, которая описана в PROTECTIVE GROUPS in ORGANIC SYNTHESIS, THIRD EDITION, H John Wiley & Sons, Inc., и примеры защитных групп включают метильную группу, этильную группу, изопропильную группу, гептильную группу, трет-бутильную группу, метоксиметильную группу, метилтиометильную группу, метоксиэтоксиметильную группу, метоксиэтильную группу, бензильную группу и т.п. Соединение (А-7) может быть синтезировано с помощью способа, в котором R заменяется R2 в способе синтеза соединения (А-6). Реакцию осуществляют путем взаимодействия соединения (А-7) с эквивалентным или избыточным, относительно соединения, количеством кислоты или основания, в растворителе, неактивном по отношению к реакции в диапазоне от комнатной температуры до температуры кипения обычно в течение от 0,5 ч до 5 дней. Здесь растворитель включает, хотя и не ограничивается этим, например, ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан и 1,2-диэтоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол и бутанол; N,N-диметилформамид (DMF); N-метилпирролидон; диметилсульфоксид (DMSO); воду; или смешанный растворитель из них. Примеры кислоты включают неорганическую кислоту, такие как хлористый водород, бромистый водород, серная кислота, азотная кислота и фосфорная кислота, или раствор, полученный разбавлением этих кислот водой или органическим растворителем. Примеры оснований включают неорганические основания, такие как гидроксид натрия, гидроксид калия, гидроксид лития, карбонат натрия и карбонат калия; алкоксид металла, такой как этилат натрия и метилат натрия; или раствор, полученный путем разбавления этих оснований водой или органическим растворителем. При использовании основания для удаления защитной группы, соединение (I) получали добавлением кислоты к реакционному раствору и его нейтрализации. Вышеуказанные примеры кислот могут быть использованы для нейтрализации. Полученное соединение (I) может быть очищено обычным способом, таким как перекристаллизация, переосаждение и разнообразные хроматографические способы, если это необходимо.
Кристалл А соединения (II) может быть получен, например, следующим способом.
Соединение (II) может быть получено способом, включающим стадию суспендирования соединения (I) в растворителе и добавление гидроксида натрия. Кроме того, способ может дополнительно включать стадию перемешивания реакционного раствора. Растворитель для суспендирования соединения (I) включает, например, ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (THF), 1,4-диоксан, 1,2-диметоксиэтан и 1,2-диэтоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; спирты, такие как метанол, этанол, 2-пропанол и бутанол; N,N-диметилформамид (DMF); N-метилпирролидон; диметилсульфоксид (DMSO); воду; или смешанный растворитель из них. Предпочтительными растворителями являются простые эфиры или спирты, вода или смешанный растворитель из них. Реакция образования соли из соединения (I) в соединение (II) протекает путем суспендирования соединения (I) в растворителе, упомянутом выше (например, в количестве, 10 кратно превышающем количество карбоновой кислоты) с последующим взаимодействием соединения (I) с эквивалентным или слегка избыточным количеством едкого натра. Реакция протекает в диапазоне от 0°С и 100°С, но предпочтительно осуществляется в диапазоне от 0°С до 30°С.
Затем смесь перемешивают в течение одного часа, а затем осадок отфильтровывают и сушат с получением кристаллов. Хотя количество растворителя, количество добавляемой воды, условия перемешивания и количество времени до разделения с помощью фильтрации особо не ограничиваются, но поскольку эти условия могут оказывать влияние на выход кристаллов, химическую чистоту, распределение диаметров частиц и размер частиц, то эти условия предпочтительно комбинируют и устанавливают в соответствии с поставленными целями. Фильтрация может быть осуществлена с использованием обычного способа, например естественной фильтрации, фильтрации под давлением, вакуумной фильтрации или разделения центрифугированием. Сушка может быть осуществлена с использованием обычного способа, например естественной сушки, вакуумной сушки, нагрева сушки и нагрева вакуумной сушки.
При необходимости промежуточные соединения реакции могут быть очищены с помощью обычного способа, такого как перекристаллизация, переосаждение и разнообразные хроматографические способы, в процессе синтеза.
Хотя кристаллы по настоящему изобретению могут быть идентифицированы с помощью характерных порошковой рентгеновской дифрактограммы, твердотельного спектра 13C-ЯМР, ИК-спектра поглощения (метод KBr) или термогравиметрического/дифференциального термического анализа (TG/DTA), если присутствует другая форма кристалла, количества ее не упоминается. Когда получают только специфическую форму кристалла, включение других кристаллических форм может по меньшей мере иметь место в такой степени, в которой оно не может детектироваться указанными способами измерения. Кроме того, когда конкретная форма кристалла используется в качестве активного фармацевтического ингредиента для фармацевтического агента, это вовсе не означает, что включение других форм кристаллов является неприемлемым.
Каждый кристалл по настоящему изобретению может быть использован в качестве фармацевтически активного ингредиента. Кроме того, могут быть использованы не только одна форма кристаллов, но также смеси двух или более форм кристаллов.
В настоящем изобретении кристаллы соединения (I) и соединения (II) обеспечивают преимущества по сравнению с некристаллическими соединениями в том, что касается обрабатываемости в процессе производства, воспроизводимости, стабильности и устойчивости при хранении.
Фармацевтическая композиция может быть получена путем использования кристалла соединения (I) или соединения (II) или его кристалла по настоящему изобретению и фармацевтически приемлемого носителя.
Состав, содержащий кристалл соединения (I) или соединение (II) или его кристаллы, согласно настоящему изобретению получают с использованием добавок, обычно используемых при получении таких составов. Примеры добавок для твердого состава включают наполнитель, такой как лактоза, сахароза, глюкоза, кукурузный крахмал, белый картофельный крахмал, кристаллическая целлюлоза, легкая безводная кремниевая кислота, синтетический силикат алюминия, алюмометасиликат магния и гидрофосфат кальция; связующее вещество, такое как кристаллическая целлюлоза, карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, натрий карбоксиметилцеллюлоза и поливинилпирролидон; разрыхлитель, такой как крахмал, натрий карбоксиметилцеллюлоза, кальций карбоксиметилцеллюлоза, кроскармеллоза натрия и натрий карбоксиметилкрахмал; смазывающее вещество, такое как тальк и стеариновая кислота; покрывающий агент, такой как гидроксиметилпропилцеллюлоза, гидроксипропилметилцеллюлоза фталат и этилцеллюлоза; и окрашивающий агент, добавки для полутвердого состава включают субстрат, такой как белый вазелин, а добавки для жидкого состава включают растворитель, таком как этанол; солюбилизирующий агент, такой как этанол; консервант, такой как пара-гидроксибензоат; агент, регулирующий тоничность, такой как глюкоза; буферное вещество, такие как лимонная кислота; антиоксидант, такой как L-аскорбиновая кислота; хелатирующий агент, такой как EDTA; и суспендирующий агент и эмульгатор, такой как полисорбат 80.
Кристаллы данного соединения (I) или соединение (II) или его кристаллы в настоящем изобретении могут быть использованы в любых лекарственных формах, таких как твердый состав, полутвердый состав и жидкий состав, и могут быть использованы при получении формы для любого типа введения, такой как пероральный состав и состав для парентерального введения (например, в виде состава для инъекции, чрескожного состава, офтальмологического состава, состава-суппозитория, трансназального состава и состава для ингаляции).
Фармацевтическая композиция, содержащая кристалл соединения (I) или соединение (II) или его кристаллы по настоящему изобретению в качестве активного ингредиента, может быть использована в качестве ингибитора ксантиноксидазы или терапевтического агента или профилактического агента против заболеваний, связанных с ксантиноксидазой, таких как подагра, гиперурикемия, синдром лизиса опухоли, мочекаменная болезнь, гипертония, дислипидемия, диабет, сердечно-сосудистые заболевания, таких как атеросклероз и сердечная недостаточность, заболевания почек, такие как диабетическая нефропатия, респираторные заболевания, такие как хроническое обструктивное заболевание легких, воспалительное заболевание кишечника заболеваний или аутоиммунные заболевания. Здесь термин «профилактический» означает снижение частоты возникновения или предотвращение возникновения заболеваний у человека, который не страдает от указанных болезней, или у которого еще не развилось заболевание, а термин «терапевтический» означает лечение, подавление или устранение заболеваний или симптомов у индивидуума, который уже затронут этими заболеваниями или у которого развились эти заболевания.
Примеры
Способ измерения
Порошковую рентгеновскую дифракцию кристаллов по настоящему изобретению измеряли в следующих условиях.
Устройство: D8 DISCOVER с GADDS CS производства Bruker AXS
Источник излучения: Cu Kα, Длина волны: 1,541838 (10-10 м), 40 кВ-40 мА, плоский графитовый монохроматор падающего луча, диаметр коллиматора 300 мкМ, двумерный детектор PSPC, сканирование от 3 до 40°
Способ измерения
Твердотельный спектр 13C-ЯМР кристаллов по настоящему изобретению измеряли в следующих условиях.
Устройство: DSX300WB производства Bruker
Измеряемое ядро: 13C
Частота повторения импульсов: 5 секунд
Импульсный режим: измерение CP/MAS
Способ измерения
Инфракрасное поглощение (метод KBr) кристаллов по настоящему изобретению измеряли в следующих условиях способом с таблеткой бромида калия метода инфракрасной абсорбции, описанного в общих методах тестирования Японской фармакопеи.
Устройство: AVATAR 320 производства ThermoFischer Scientific
Диапазон измерений: от 4000 до 400 см-1
Разрешение: 4 см-1
Номер интеграции: 64
Термогравиметрический/дифференциальный термический анализ (TG/DTA) кристаллов по настоящему изобретению проводили в следующих условиях.
Устройство: TG8120 производства Rigaku
Скорость подъёма температуры: 10°С/мин, Атмосфера: азот, кювета для образцов: алюминий, эталон: оксид алюминия, отбор образцов: 1,0 сек, Диапазон рабочих температур: от 25 до 300°C
Что касается соединений, для которых измеряли спектр 1Н-ЯМР (400 МГц, DMSO-d6 или CDCl3), то показаны химический сдвиг (δ:ppm) и постоянная взаимодействия (J: Гц).
Устройство: JMTC-400/54/SS производства JEOL
Аббревиатуры представляют следующее:
с = синглет, д = дублет, т = триплет, кв = квартет, шир.с. = широкий синглет, м = мультиплет
Справочный пример 1
Синтез этил 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилата
(1) К смеси, полученной путем суспендированием 20,0 г 4-бром-2-нитрофенола и 19,0 г карбоната калия в 200 мл N,N-диметилформамида, добавляли 20,1 г изобутил бромида и полученную смесь нагревали при 110°С в течение 22 часов в атмосфере азота. После добавления воды к раствору реакционной смеси осуществляли экстракцию с использованием этилацетата. Органический слой промывали солевым раствором, с последующей сушкой и концентрированием при пониженном давлении, с получением 24,8 г 4-бром-1-(2-метилпропокси)-2-нитробензола.
(2) Смесь, полученную добавлением 2,10 г бикарбоната калия, 43,5 мг хлорида палладия (II) и 205 мг бромида меди (I) 2,74 г 4-бром-1-(2-метилпропокси)-2-нитробензола, суспендировали в 35 мл толуола. Затем раствор реакционной смеси, полученный добавлением 2,05 г этил-4-метил-1,3-тиазол-5-карбоновой кислоты, 92 мкл изомасляной кислоты и 230 мкл ди-трет-бутилциклогексилфосфина к суспензии, нагревали при 120°С в течение 14 часов в атмосфере азота. Раствор реакционной смеси фильтровали через целит для удаления нерастворимых веществ, добавляли к фильтрату воду и проводили экстракцию с использованием этилацетата. Органический слой промывали насыщенным солевым раствором и затем подвергали сушке и концентрированию при пониженном давлении с последующим очищением с помощью обычного способа с получением 3,16 г этил-4-метил-2-[3-нитро-4-(2-метилпропокси)фенил]-1,3-тиазол-5-карбоксилата.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,07 (6H, д, J = 6,8 Гц), 1,39 (3H, т, J = 6,8 Гц), 2,14-2,22 (1H, м), 2,77 (3H, с), 3,92 (2H, д, J = 6,0 Гц), 4,36 (2H, д, J = 6,8 Гц), 7,12 (1H, д, J = 8,8 Гц), 8,09 (1H, дд, J = 2,0 Гц, 8,8 Гц), 8,43 (1H, д, J = 2,0 Гц)
(3) Раствор реакционной смеси получали суспендированием 3,16 г этил-4-метил-2-[3-нитро-4-(2-метилпропокси)фенил]-1,3-тиазол-5-карбоксилата в 30 мл этанола и добавлением 200 мг палладий/уголь (10% по массе) к суспензии, и реакционную смесь перемешивали при температуре 50° с в течение 14 часов в атмосфере водорода. Раствор реакционной смеси фильтровали и фильтрат концентрировали при пониженном давлении с получением 2,12 г этил-2-[3-амино-4-(2-метилпропокси)фенил]-4-метил-1,3-тиазол-5-карбоксилата.
(4) Раствор реакционной смеси, полученный суспендированием 2,12 г этил 2-[3-амино-4-(2-метилпрокси)фенил]-4-метил-1,3-тиазол-5-карбоксилата в 20 мл уксусной кислоты, и добавлением 780 мг азида натрия и 1,78 г триэтилортоформиата, нагревали при 70°С в течение 2 часов в атмосфере азота. После охлаждения реакционной смеси до комнатной температуры, добавляли 20 мл воды, с последующим очищением с помощью обычного способа с получением 2,25 г этил-4-метил-2-[4-(2-метилпропокси)-3-(1H-1, 2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилата.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,02 (6H, д, J = 6,8 Гц), 1,40 (3H, т, J = 6,8 Гц), 2.10-2.17 (1H, м), 2,78 (3H, с), 3,95 (2H, д, J = 6,8 Гц), 4,36 (2H, д, J = 6,8 Гц), 7,18 (1H, д, J = 8,8 Гц), 8,07 (1H, дд, J = 2,4 Гц, 8,8 Гц), 8,46 (1H, д, J = 2,8 Гц), 9,21 (1H, с)
Пример 1
Получение кристаллической формы А 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты: соединение (I)
Раствор реакционной смеси, полученный растворением 883 г этил-4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилата в 13,2 л смешанного раствора тетрагидрофуран/метанол = 1/1, и добавлением в смесь 2,28 л водного раствора 2 М гидроксида натрия, перемешивали в диапазоне от 20°С до 30°C в течение 3 часов. При перемешивании в диапазоне от 20°С до 30°С 2,28 л 2 М соляной кислоты медленно добавляли к раствору реакционной смеси и дополнительно медленно добавляли 4,4 л воды. Раствор реакционной смеси перемешивали в диапазоне от 20°С до 30°С в течение одного часа и кристаллы получали фильтрацией. Полученные кристаллы промывали 4,4 л смешанного раствора метанол/вода = 1/1 и 4,4 л воды. Кристаллы сушили под вакуумом при 50°С с получением 811 г кристаллов 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты. Дифракционный рентгеновский анализ полученных кристаллов показан на фиг. 1. Пики наблюдали при углах дифракции 2θ = 8,6°, 10,2°, 13,3°, 14,4°, 18,5°, 19,9°, 21,8°, 25,1°, 25,6°, 26,6°, 27,1° и 29,5°. Твердотельный спектр 13C ЯМР полученных кристаллов показан на фиг. 5. Пики наблюдали при химических сдвигах 116,3 ppm, 117,6 ppm, 120,0 ppm, 123,6 ppm, 125,9 ppm, 127,4 ppm, 143,7 ppm, 151,8 ppm, 161.1 ppm, 162,3 ppm и 165,5 ppm. Инфракрасный спектр (способ KBr) полученных кристаллов показан на фиг. 8. Пики наблюдали при волновых числах 745 см-1, 822 см-1, 889 см-1, 975 см-1, 997 см-1, 1611 см-1 и 1705 см-1. Кроме того, наблюдали экзотермический пик в термогравиметрическом/дифференциальном термическом анализе (TG/DTA) при 222° С.
1Н-ЯМР (400 МГц, DMSO-d 6) δ: 0,85 (6Н, д, J = 6,8 Гц), 1,93-2,00 (1H, м), 2,66 (3H, с), 3,96 (2H, д, J = 6,0 Гц), 7,48 (1H, д, J = 8,8 Гц), 8,18 (1H, дд, J = 2,4 Гц, 8,8 Гц), 8,27 (1H, д, J = 2,4 Гц), 9,79 (1H, с), 13,41 ( 1H, с)
Пример 2
Получение кристаллической формы В 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты: соединение (I)
Суспензию, полученную суспендированием 3,0 г кристалла А соединения (I), в 21,0 мл этилацетата, подвергали нагреванию с обратным холодильником в течение 6 часов. Суспензию охлаждали до 25°С и перемешивали при 25°С в течение 30 минут. Полученные кристаллы отфильтровывали и промывали 15 мл этилацетата. Кристаллы сушили под вакуумом при 45°С с получением 2,9 г кристаллов 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил-1,3-тиазол-5-карбоновой кислоты. Дифракционный рентгеновский анализ полученных кристаллов показан на фиг. 2. Пики наблюдали при углах дифракции 2θ = 10,1°, 12,6°, 13,1°, 14,0°, 18,6°, 24,2°, 25,2°, 25,7°, 27,2° и 30,5°. Твердотельный спектр 13C ЯМР полученных кристаллов показан на фиг. 6. Пики наблюдали при химических сдвигах = 115,4 ppm, 118,0 ppm, 119,8 ppm, 123,2 ppm, 126,4 ppm, 129,1 ppm, 142,7 ppm, 151,2 ppm, 160,9 ppm и 166,6 ppm. Инфракрасный спектр поглощения (способ KBr) полученных кристаллов показан на фиг. 9. Пики наблюдали при волновых числах = 744 см-1, 810 см-1, 972 см-1, 997 см-1, 1005 см-1, 1611 см-1 и 1710 см-1. Кроме того, наблюдали экзотермический пик в термогравиметрическом/дифференциальном термическом анализе (TG/DTA) при 225° С.
Справочный пример 2
Получение 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты: соединение (I)
Раствор реакционной смеси, полученный растворением 4,00 г этил-4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты в 60 мл смешанного раствора тетрагидрофурана/метанола = 1/1, и добавлением 10,3 мл водного раствора 2 М гидроксида натрия в смесь, перемешивали при комнатной температуре в течение 3 часов. После добавления 10,3 мл 2 М соляной кислоты к раствору реакционной смеси при перемешивании, добавляли 60 мл воды и полученную смесь очищали с использованием обычного способа, чтобы получить 3,50 г 4-метил-2-[4-(2- метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновую кислоту.
1Н-ЯМР (400 МГц, DMSO d6) : δ: 0,85 (6Н, д, J = 6,8 Гц), 1,93-2,00 (1H, м), 2,66 (3H, с), 3,96 (2H, д, J = 6,0 Гц ), 7,48 (1H, д, J = 8,8 Гц), 8,18 (1H, дд, J = 2,4 Гц, 8,8 Гц), 8,27 (1H, д, J = 2,4 Гц), 9,79 (1H, с), 13,41 (1H, с)
Пример 3
Получение кристаллической формы С 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты: соединение (I)
Смесь, полученную добавлением 90 мл N,N-диметилформамида к 8,5 г 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил-1,3-тиазол-5-карбоновой кислоты, растворяли нагреванием и перемешиванием при 80°С. После охлаждения раствора до комнатной температуры его перемешивали в течение 2-х часов при комнатной температуре, а затем фильтровали. Отфильтрованный продукт промывали 90 мл этанола. Маточный раствор оставляли стоять при комнатной температуре в течение 7 дней, и полученные кристаллы промывали 30 мл этанола. Кристаллы сушили под вакуумом при 40°С с получением 1,0 г кристаллов соединения (I). Дифракционный рентгеновский анализ полученных кристаллов показан на фиг. 3. Пики наблюдали при углах дифракции 2θ = 7,2°, 12,5°, 13,0°, 14,7°, 19,2°, 20,0°, 21,4°, 21,7°, 24,7° и 26,0°. Твердотельный спектр 13C ЯМР полученных кристаллов показана на фиг. 7. Пики наблюдали при химических сдвигах = 116,1 ppm, 119,6 ppm, 123,1 ppm, 126,1 ppm, 127,1 ppm, 130,0 ppm, 143,6 ppm, 150,3 ppm, 158,3 ppm, 160,7 ppm, 163,9 ppm, 165,5 ppm и 167,0 ppm. Инфракрасный спектр (способ KBr) полученных кристаллов показан на фиг. 10. Пики наблюдали при волновых числах = 745 см-1, 751 см-1, 809 см-1, 820 см-1, 971 см-1, 1006 см-1, 1613 cм-1, 1682 см-1 и 1710 см-1. Кроме того, наблюдали эндотермический пик при 88°С и экзотермический пик при 225°С в термогравиметрическом/дифференциальном термическом анализе (TG/DTA).
Пример 4
Получение кристаллической формы А натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол 5-карбоксилата: соединение (II)
К раствору, полученному растворением 400 мг гидроксида натрия в 36 мл этанола, добавляли 3,59 г соединения (I), и полученную смесь перемешивали в интервале от 20°С до 30°С в течение одного часа. После этого кристаллы отфильтровывали. Полученные кристаллы промывали 10 мл этанола. Кристаллы сушили под вакуумом при 50°С с получением 3,53 г соединения (II). Дифракционный рентгеновский анализ полученных кристаллов показан на фиг. 4. Пики наблюдали при углах дифракции от 2θ = 7,2°, 10,9°, 13,3°, 15,9°, 18,2°, 20,8°, 22,1°, 25,2°, 26,1 и 29,1°. Кроме того, наблюдали экзотермический пик в термогравиметрическом/дифференциальном термическом анализе (TG/DTA) при 281°С.
1Н-ЯМР (400Mz, DMSO-d 6) δ: 0,85 (6Н, д, J = 6,8 Гц), 1.92-1.99 (1H, м), 2,59 (3H, с), 3,92 (2H, д, J = 6,4 Гц), 7,41 (1H, д, J = 8,8 Гц), 8,02 (1H, дд, J = 1,6 Гц, 8,8 Гц), 8,12 (1H, д, J = 2,0 Гц), 9,78 (1H, с)
Пример 5
Гипоурикемический эффект (Нормальные Крысы)
Гипоурикемический эффект был подтвержден для соединения (II). Исследуемое соединение суспендировали в 0,5% растворе метилцеллюлозы, вводили 8-9-недельным самцам крыс Sprague-Dawley (Japan Charles River Co.) при пероральном введении через желудочный зонд с помощью иглы для кормления. После отбора крови из хвостовой вены через 2 часа после введения отделяли плазму. Уровень мочевой кислоты в пробе крови измеряли с использованием способа с уриказой с использованием абсорбционного спектрометра, а также набора для определения мочевой кислоты (L type Wako UA F: Wako Pure Chemical Industries, Ltd.). Процент гипоурикемического эффекта определяли следующим выражением:
Процент гипоурикемического эффекта (%) = (уровень мочевой кислоты контрольного животного-уровень мочевой кислоты у животного, которому вводили тестируемое соединение) × 100/уровень мочевой кислоты контрольного животного.
Кристалл А соединения (II) показал гипоурикемический эффект в 50% или более в дозах 10 мг/кг и 1 мг/кг.
На основании приведенных выше результатов было показано, что соединение (II), обладает мощным гипоурикемическим эффектом.
Пример 6
Пролонгированный гипоурикемический эффект (Нормальные Крысы)
Кристаллическую форму А соединения (II) вводили самцам-крысам Sprague-Dawley так же, как и в примере 5.
После отбора крови из хвостовой вены через 24 часа после введения отделяли плазму. Уровень мочевой кислоты в пробе крови измеряли с помощью способа с уриказой с использованием абсорбционного спектрометра, а также набора для определения мочевой кислоты (L type Wako UA F: Wako Pure Chemical Industries, Ltd.). Процент гипоурикемического эффекта определяли следующим выражением:
Процент гипоурикемического эффекта (%) = (уровень мочевой кислоты контрольного животного-уровень мочевой кислоты у животного, которому вводили тестируемое соединение) × 100/уровень мочевой кислоты контрольного животного.
Кристаллическая форма А соединения (II) показала гипоурикемический эффект 50% или более в течение 24 часов после введения в дозе 10 мг/кг и 40% или более в течение 24 часов после введения в дозе 3 мг/кг.
На основании приведенных выше результатов было показано, что кристаллическая форма А соединения (II) обладает пролонгированным гипоурикемическим эффектом в течение длительного периода времени.
Пример 7
Растворимость в JP2-растворе
Была подтверждена растворимость соединения (II) в растворе JP2 (рН 6,8). Раствор готовили умеренным измельчением кристаллов А соединения (II) в ступке с добавлением приблизительно 4 мг измельченных кристаллов к 20 мл JP2 раствора при перемешивании при комнатной температуре. Оптическую плотность (At) каждого раствора при длине волны 280 нм последовательно измеряли сразу же после добавления кристаллов с использованием способа измерения оптической плотности в ультрфиолете-видимой области и растворимость в каждый момент времени измерения определяли следующим выражением с использованием оптической плотности (As) стандартного раствора при 280 нм, определенного заранее в качестве маркера. Измерение длилось до 120 минут.
Растворимость = At/As × Cs (где Cs представляет собой концентрацию стандартного раствора)
Условия измерения
Измерительное устройство: μ DISS Profiler производства Pion Inc.
Скорость перемешивания: 700 оборотов в минуту
Длина волны измерения в УФ: 280 нм
Интервал измерения: С начала измерения по 1 минуту: 3 секунды
От 1 до 8 минут: 20 секунд
От 8 до 120 минут: 2 минуты
Общее количество кристалла А соединения (II) растворяли в течение 30 секунд после добавления и растворимость кристалла А соединения (II) составляла 187 мкг/мл. Кроме того, растворимость в 120 минут после добавления составляла 189 мкг/мл, осаждение не наблюдалось и растворенное состояние сохранялось.
На основании приведенных выше результатов было показано, что соединение (II) обладает отличной растворимостью.
Справочный пример 3
Измерение активности ингибирования ксантиноксидазы
(1) Получение тестируемого соединения
После растворения соединения (I) в DMSO (производства Sigma Corporation) для получения концентрации в 20 мМ, полученный раствор использовали доведением концентрации до искомого значения для целей во время применения.
(2) Способ измерения
Оценку активности ингибирования ксантиноксидазы соединения (I) проводили способом, описанным в данном источнике (Method Enzymatic Analysis, 1, 521-522, 1974), с частичной модификацией. Эту оценку проводили измерением активности ксантиноксидоредуктазы типа оксидазы. В частности, раствор ксантина (изготовитель Sigma Co.) готовили в концентрации 10 мМ с использованием 20 мМ раствора гидроксида натрия и затем смешивали с 100 мМ фосфатного буфера с доведением до 30 мкМ. 75 мкл раствора добавляли в каждую лунку 96-луночного планшета. Исследуемое соединение разбавляли DMSO в 100 раз до конечной концентрации и добавляли в каждую лунку по 1,5 мкл на лунку. После перемешивания планшета измеряли оптическую плотность при 290 нм микропланшетным ридером SPECTRA MAX Plus 384 (производства Molecular Devices, LLC). Затем ксантиноксидоредуктазу типа оксидазы (из пахты, производства компании Calbiochem Novabiochem Corp.) готовили в концентрации 30,6 мЕд/мл, с помощью 100 мМ фосфатного буферного раствора и добавляли в каждую лунку по 73,5 мкл на лунку. Сразу же после смешивания проводили измерение изменения оптической плотности при 290 нм в течение 5 минут. Ферментативную активность раствора DMSO без тестируемого соединения использовали в качестве контроля 100%, и рассчитывали степень ингибирования испытуемых соединений. 50% ингибирующую концентрацию тестируемых соединений на активность ксантиноксидоредуктазы типа оксидазы рассчитывали путем подгонки кривой доза-ответ.
Соединение (I) продемонстрировало активность ингибирования ксантиноксидазы 1,0 нмоль ≤ IC 50 <5,0 нм.
Справочный пример 4
Гипоурикемический эффект (Нормальные Крысы)
Гипоурикемический эффект был подтвержден для соединения (I) так же, как в Примере 5. Соединение (I) показало гипоурикемический эффект 50% или более в дозах 10 мг/кг и 1 мг/кг.
На основании приведенных выше результатов было показано, что соединение (I) обладает мощным гипоурикемическим эффектом.
Справочный Пример 5
Пролонгированный гипоурикемический эффект (Нормальные Крысы)
Пролонгированный гипоурикемический эффект был подтвержден для соединения (I) так же, как в Примере 6.
Соединение (I) показало гипоурикемический эффект 50% или более в течение 24 часов после введения в дозе 10 мг/кг и 40% или более в течение 24 часов после введения в дозе 3 мг/кг.
На основании приведенных выше результатов было показано, что соединение (I) обладает пролонгированным гипоурикемическим эффектом в течение длительного периода времени.
Справочный пример 6
Гипоурикемический эффект (гиперурикемические собаки породы бигль)
Гипоурикемический эффект был подтвержден для соединений (I) на собаках породы бигль с индуцируемой оксоновой кислотой гиперурикемией. Исследуемое соединение суспендировали в 0,5% растворе метилцеллюлозы, вводили собакам породы бигль (Kitayama labes) путем перорального введения через зонд. Калий оксонат (50 мг/кг) вводили подкожно до и через 4 часа после введения соединения. После отбора крови из латеральной подкожной вены через 8 часов после введения отделяли плазму. Уровень мочевой кислоты в плазме образца определяли способом LC-MS/MS и процент гипоурикемического эффекта определяли следующим выражением:
Процент гипоурикемического эффекта (%) = (уровень мочевой кислоты контрольного животного-уровень мочевой кислоты у животного, которому вводили тестируемое соединение) × 100/уровень мочевой кислоты контрольного животного.
Соединение (I) показало гипоурикемический эффект в дозе 10 мг/кг через 8 часов после введения.
На основании приведенных выше результатов было показано, что соединение (I) обладает мощным гипоурикемическим эффектом у собак породы бигль.
Справочный пример 7
Пролонгированный эффект ингибирования ксантиноксидазы в ткани и плазме.
Для «ксантиноксидазы» в настоящем изобретении в отношении данного примера различают катализирующие окислительную реакцию активности, вызванные только ксантиноксидоредуктазой типа оксидазы и вызванные обоими оксидоредуктазами типа оксидазы и типа дегидрогеназы. Первая из них является «XO-активностью», а вторая является «XOR-активностью». «XO-активность в ткани», «ХО-активность в плазме», «ингибирование XOR-активности в ткани», «ингибирование XOR-активности в ткани», «ингибирование XOR-активности в ткани» и т.п., «XO-активность» и «XOR-активность» имеют те же значения, что и приведенные выше. Ткань включает печень, почки, жировую ткань, кишечник и сосуды. Кроме того, процент ингибирования ХО-активности и ингибирование XO-активности в том же образце понимаются как схожие величины, согласно приведенным ниже результатам.
Эффект ингибирования XO-активности в ткани, XOR-активности в ткани и XO-активности в плазме был подтвержден для соединений (I). Исследуемое соединение суспендировали в 0,5% растворе метилцеллюлозы, вводили 7-9-недельным самцам крыс Sprague-Dawley (Japan Charles River Co.) пероральным образом через желудочный зонд с помощью иглы для кормления. Кровь собирали из брюшной вены и ткани собирали через 24 или 27 часов после введения. Образец плазмы получали центрифугированием.
ХО-активность в ткани, XOR-активность в ткани и ХО-активность в плазме измеряли с помощью анализа в котором используется реакция, в которой птерин окисляется каждым типом ксантиноксидоредуктазы с получением флуоресцентного изоксантоптерина. Вкратце, замороженные ткани гомогенизировали с калий-фосфатным буфером, рН 7,4, содержащим 1 мМ этилендиаминтетрауксусной кислоты (EDTA) и ингибиторы протеаз для получения концентрации ткани, указанной ниже (печень: 25 мг/мл, почка: 25 мг/мл, кишечник: 5 мг/мл, жировая ткань: 5 мг/мл, сосуды: 30 мг/мл). Затем гомогенаты центрифугировали при 12000 оборотов в минуту в течение 15 мин при 4°С. При измерении XO-активности надосадочную жидкость ткани и плазмы соответственно совместно инкубировали с раствором 50 мкМ птерина при 37°С. При измерении XOR-активности надосадочную жидкость тканевого гомогената совместно инкубировали с раствором 50 мкМ птерина и 50 мкМ метиленового синего при 37°С. В качестве контроля ксантиноксидоредуктазу типа оксидазы (из пахты, производства фирмы Calbiochem Novabiochem Corp.) также инкубировали с раствором птерина аналогичным образом. ХО-активность и XOR-активность образцов определяли по интенсивности флуоресценции, которую нормализовали по значению интенсивности контроля и концентрации белка.
Процент ингибирования ХО-активности и ингибирования XOR-активности определяли следующим выражением:
Процент ХО- или XOR-активности ингибирования (%) = (ХО- или XOR-активность контрольного животного - ХО- или XOR-активность животного, которому вводили тестируемое соединение) × 100/ ХО- или XOR-активность контрольного животного.
Активность в ткани и плазме и ХО-активность в плазме через 27 часов после введения показаны в таблице ниже.
Таблица 1. XO-ингибирующая активность в ткани и плазме (при препарировании около 27 часов после введения), % ингибирования (по сравнению с носителем)
Соединение (I) ингибирует на 80% или более XO-активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в печени.
Соединение (I) ингибирует на 70% или более XO-активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в почках.
Соединение (I) ингибирует на 40% или более XO активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в плазме.
Соединение (I) ингибирует на 80% или более XO-активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в печени.
Соединение (I) ингибирует на 60% или более XO-активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в почке.
Соединение (I) ингибирует на 25% или более XO-активность в течение 27 ч после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в плазме.
Кроме того, XO и XOR ингибирующая активность в ткани в течение 24 часов после введения показаны в таблице ниже.
Таблица 2. ХО- и XOR-ингибирующая активность в ткани (при препарировании около 24 ч после введения), % ингибирования (относительно носителя)
Соединение (I) ингибирует на 80% или более XOR-активность и XO-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в печени.
Соединение (I) ингибирует на 70% или более XOR-активность и XO-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в почке.
Соединение (I) ингибирует на 80% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в кишечнике.
Соединение (I) ингибирует на 60% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в жировых тканях.
Соединение (I) ингибирует на 40% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 10 мг/кг в сосудах.
Соединение (I) ингибирует на 80% или более XOR-активность и XO-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в печени.
Соединение (I) ингибирует на 60% или более XOR-активность и XO-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в почках.
Соединение (I) ингибирует на 60% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в кишечнике.
Соединение (I) ингибирует на 30% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в жировых тканях.
Соединение (I) ингибирует на 25% или более XOR-активность через 24 часа после введения лекарственного средства по сравнению с контрольным животным в дозе 1 мг/кг в сосудах.
Из приведенных выше результатов видно, что соединения по настоящему изобретению имеют пролонгированное ингибирующее действие на XO-активность или XOR-активность.
Промышленная применимость
Кристалл соединения (I) и соль и кристалл соединения (II) по настоящему изобретению использовали в качестве фармацевтического агента. Кроме того, эти соединения могут быть использованы в качестве активного фармацевтического ингредиента для получения фармацевтического агента.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА АЗОЛБЕНЗОЛЬНОГО ПРОИЗВОДНОГО | 2015 |
|
RU2675854C2 |
| АЗОЛОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОЛА | 2014 |
|
RU2641891C2 |
| АМИНОВАЯ СОЛЬ И ЕЕ КРИСТАЛЛЫ | 2013 |
|
RU2658823C2 |
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО 1,2-ДИГИДРОПИРИДИНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2323930C1 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| ПОЛИМОРФНАЯ ФОРМА 4-{ [4-({ [4-(2,2,2-ТРИФТОРЭТОКСИ)-1,2-БЕНЗИЗОКСАЗОЛ-3-ИЛ]ОКСИ} МЕТИЛ)ПИПЕРИДИН-1-ИЛ]МЕТИЛ} -ТЕТРАГИДРО-2Н-ПИРАН-4-КАРБОНОВОЙ КИСЛОТЫ | 2012 |
|
RU2616978C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| МЕХАНОАКТИВИРОВАННЫЕ АМОРФНЫЕ И АМОРФНО-КРИСТАЛЛИЧЕСКИЕ КАЛЬЦИЕВЫЕ СОЛИ ГЛЮКОНОВОЙ КИСЛОТЫ, КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2007 |
|
RU2373185C2 |

Изобретение относится к кристаллическим формам 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты и натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилата, предназначенным для использования в качестве терапевтического или профилактического средства для заболеваний, связанных с ксантиноксидазой. Кристаллические формы по изобретению характеризуются по меньшей мере одним из признаков (i)-(vii): (i) характерные пики при углах дифракции 2θ (±0,5°) в порошковой рентгеновской дифрактограмме; (ii) порошковая рентгеновская дифрактограмма, имеющая вид, показанный на фигурах; (iii) характерные пики при значениях химических сдвигов (±0,5 ppm) в твердотельном спектре 13C-ЯМР; (iv) твердотельный спектр 13C-ЯМР, имеющий вид, показанный на фигурах; (v) характерные пики при волновых числах (±5 см-1) в инфракрасном спектре поглощения (способ KBr); (vi) ИК-спектр поглощения (способ KBr), имеющий вид, показанный на фигурах; или (vii) экзотермический пик в термогравиметрическом/дифференциальном термическом анализе. Также изобретение относится к способам получения кристаллических форм 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты. 11 н. и 1 з.п. ф-лы, 2 табл., 15 пр.
1. Кристалл 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты (кристаллическая форма А), характеризующийся по меньшей мере одним из признаков (i)-(vii):
(i) в его порошковой рентгеновской дифрактограмме присутствуют характерные пики при углах дифракции 2θ (±0,5°) = 8,6°, 10,2°, 13,3°, 14,4°, 18,5°, 19,9°, 21,8°, 25,1°, 25,6°, 26,6°, 27,1° и 29,5°;
(ii) его порошковая рентгеновская дифрактограмма имеет вид, показанный на фиг. 1;
(iii) в его твердотельном спектре 13C-ЯМР присутствуют характерные пики при значениях химических сдвигов (±0,5 ppm) 116,3 ppm, 117,6 ppm, 120,0 ppm, 123,6 ppm, 125,9 ppm, 127,4 ppm, 143,7 ppm, 151,8 ppm, 161,1 ppm, 162,3 ppm и 165,5 ppm;
(iv) его твердотельный спектр 13C-ЯМР имеет вид, показанный на фиг. 5;
(v) в его инфракрасном спектре поглощения (способ KBr) присутствуют характерные пики при волновых числах (±5 см-1) 745 см-1, 822 см-1, 889 см-1, 975 см-1, 997 см-1, 1611 см-1 и 1705 см-1;
(vi) его ИК-спектр поглощения (способ KBr), имеет вид, показанный на фиг. 8; или
(vii) он имеет экзотермический пик в термогравиметрическом/дифференциальном термическом анализе при 222±2°С.
2. Кристалл 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты (кристаллическая форма В), характеризующийся по меньшей мере одним из признаков (i)-(vii):
(i) в его порошковой рентгеновской дифрактограмме присутствуют характерные пики при углах дифракции 2θ (±0,5°) = 10,1°, 12,6°, 13,1°, 14,0°, 18,6°, 24,2°, 25,2°, 25,7°, 27,2° и 30,5°;
(ii) его порошковая рентгеновская дифрактограмма имеет вид, показанный на фиг. 2;
(iii) в его твердотельном спектре 13С ЯМР присутствуют характерные пики при значениях химических сдвигов (±0,5 ppm) 115,4 ppm, 118,0 ppm, 119,8 ppm, 123,2 ppm, 126,4 ppm, 129,1 ppm, 142,7 ppm, 151,2 ppm, 160,9 ppm и 166,6 ppm;
(iv) его твердотельный спектр 13C-ЯМР имеет вид, показанный на фиг. 6;
(v) в его инфракрасном спектре поглощения (способ KBr) присутствуют характерные пики при волновых числах (±5 см-1) 744 см-1, 810 см-1, 972 см-1, 997 см-1, 1005 см-1, 1611 см-1 и 1710 см-1;
(vi) его инфракрасный спектр поглощения (способ KBr) имеет вид, показанный на фиг. 9; или
(vii) он имеет экзотермический пик в термогравиметрическом/дифференциальном термическом анализе при 225±2°С и представляет собой безводный кристалл.
3. Кристалл 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты (кристаллическая форма С), характеризующийся по меньшей мере одним из признаков (i)-(vii):
(i) в его порошковой рентгеновской дифрактограмме присутствуют характерные пики при углах дифракции 2θ (±0,5°) = 7,2°, 12,5°, 13,0°, 14,7°, 19,2°, 20,0°, 21,4°, 21,7°, 24,7° и 26,0;
(ii) его порошковая рентгеновская дифрактограмма имеет вид, показанный на фиг. 3;
(iii) в его твердотельном спектре 13С ЯМР присутствуют характерные пики при значениях химических сдвигов (±0,5 ppm) 116,1 ppm, 119,6 ppm, 123,1 ppm, 126,1 ppm, 127,1 ppm, 130,0 ppm, 143,6 ppm, 150,3 ppm, 158,3 ppm, 160,7 ppm, 163,9 ppm, 165,5 ppm и 167.0 ppm;
(iv) его твердотельный спектр 13C-ЯМР имеет вид, показанный на фиг. 7;
(v) в его ИК-спектре поглощения (способ KBr) присутствуют характерные пики при волновых числах (±5 см-1) 745 см-1, 751 см-1, 809 см-1, 820 см-1, 971 см-1, 1006 см-1, 1613 см-1, 1682 см-1 и 1710 см-1;
(vi) его ИК-спектр поглощения (способ KBr) имеет вид, показанный на фиг. 10; или
(vii) он имеет эндотермический пик при 88±2°С и экзотермический пик при 225±2°С в термогравиметрическом/дифференциальном термическом анализе.
4. Натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилат.
5. Кристалл натрий 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоксилата, характеризующийся по меньшей мере одним из признаков (i)-(iii):
(i) в его порошковой рентгеновской дифрактограмме присутствуют характерные пики при углах дифракции 2θ (±0,5°) = 7,2°, 10,9°, 13,3°, 15,9°, 18,2°, 20,8°, 22,1°, 25,2°, 26,1° и 29,1°;
(ii) его порошковая рентгеновская дифрактограмма имеет вид, показанный на фиг. 4; или
(iii) он имеет экзотермический пик в термогравиметрическом/дифференциальном термическом анализе при 281±2°С.
6. Фармацевтическая композиция для использования в качестве терапевтического или профилактического средства для заболеваний, связанных с ксантиноксидазой, содержащая соединение по п. 4 или кристалл по любому из пп. 1-3, 5 в качестве активного ингредиента и фармацевтически приемлемый носитель.
7. Ингибитор ксантиноксидазы, содержащий соединение по п. 4 или кристалл по любому из пп. 1-3, 5 в качестве активного ингредиента.
8. Терапевтический или профилактический агент для одного или более заболеваний, выбранных из группы, состоящей из подагры, гиперурикемии, синдрома лизиса опухоли, мочекаменной болезни, гипертонии, дислипидемии, диабета, сердечно-сосудистых заболеваний, заболеваний почек, заболеваний дыхательных путей, воспалительных заболеваний кишечника и аутоиммунных заболеваний, включающий соединение по п. 4 или кристалл по любому из пп. 1-3, 5 в качестве активного ингредиента.
9. Способ получения кристаллической формы А 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты по п. 1, включающий следующие стадии:
суспендирование алкилового эфира 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты в смешанном растворе тетрагидрофурана и метанола в диапазоне между 20°C и 30°C, и осуществление гидролиза путем добавления водного раствора основания; и
нейтрализацию продукта реакции.
10. Способ по п. 9, дополнительно включающий стадию добавления воды к нейтрализованному продукту и его перемешивание.
11. Способ получения кристаллической формы В 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты по п. 2, включающий стадию суспендирования и нагревания кристаллической формы А 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты в этилацетате или этаноле в условиях нагревания с обратным холодильником.
12. Способ получения кристаллической формы С 4-метил-2-[4-(2-метилпропокси)-3-(1H-1,2,3,4-тетразол-1-ил)фенил]-1,3-тиазол-5-карбоновой кислоты по п. 3, включающий кристаллизацию из ее раствора в N,N-диметилформамиде.
| АЗОЛОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОЛА | 2014 |
|
RU2641891C2 |
| Магнитная головка | 1974 |
|
SU513379A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

