ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в целом относится к кристаллическим формам (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. Настоящее изобретение в целом также относится к фармацевтической композиции, содержащей кристаллические формы, а также к способам получения таких кристаллических форм и способам применения таких кристаллических форм при лечении заболеваний и расстройств, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H. Такие заболевания и нарушения могут включать воспалительные и аутоиммунные нарушения, а также воспаление легких и дыхательных путей.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
(S)-3-Амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота впервые была описана как соль HCl в патентном документе WO2015/092740, поданном 18 декабря 2014 г., который включен посредством ссылки во всей своей полноте, и является ингибитором LTA4H, характеризующимся структурой формулы (I):

Формула (I)
Соединение формулы (I) применимо при лечении заболеваний и нарушений, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H. Такие заболевания и состояния включают воспалительные и аутоиммунные нарушения, а также воспаление легких и дыхательных путей.
Соответственно, соединение формулы (I) является применим при лечении следующих заболеваний или нарушений: острое или хроническое воспаление, анафилактические реакции, аллергические реакции, виды атопического дерматита, псориаз, синдром острой дыхательной недостаточности, опосредованное иммунокомплексами повреждение легких и хроническое обструктивное заболевание легких, воспалительные заболевания кишечника (включая язвенный колит, болезнь Крона и послеоперационную травму), язвы желудочно-кишечного тракта, виды нейтрофильного дерматоза (включая без ограничения гангренозную приодермию, синдром Свита, акне и нейтрофильную крапивницу), опосредованный иммунокомплексами гломерулонефрит, аутоиммунные заболевания (включая инсулинозависимый сахарный диабет, рассеянный склероз, ревматоидный артрит, остеоартроз и системную красную волчанку), виды васкулита (включая без ограничения виды кожного васкулита, болезнь Бехчета и болезнь Шенлейн-Геноха), нарушения сердечно-сосудистой системы (включая без ограничения гипертонию, атеросклероз, аневризму, критическую ишемию нижних конечностей, окклюзионную болезнь периферических артерий, легочную артериальную гипертензию и болезнь Рейно), сепсис, воспалительная и нейропатическая боль, в том числе артритная боль, пародонтоз, в том числе гингивит, воспаления среднего уха, мигрень, доброкачественная гиперплазия предстательной железы, синдром Шегрена-Ларссона и виды рака (включая без ограничения виды лейкоза и лимфомы, рак предстательной железы, рак молочной железы, рак легких, злокачественную меланому, рак почки, опухоли головы и шеи и колоректальный рак).
Соединение формулы (I) является особенно применимым при лечении острого или хронического воспаления, особенно аутовоспалительных нарушений, таких как «стерильные» нейтрофильные воспалительные нарушения, воспалительное заболевание кишечника (включая язвенный колит и болезнь Крона), виды нейтрофильного дерматоза (включая гангренозную приодермию и акне), виды васкулита, ревматоидный артрит, подагра и заболевания сердечно-сосудистой системы.
Твердая форма активного фармацевтического ингредиента (API) конкретного лекарственного средства зачастую является важным определяющим фактором в отношении простоты получения лекарственного средства, гигроскопичности, стабильности, растворимости, стабильности при хранении, простоты составления, скорости растворения в жидкостях желудочно-кишечного тракта и биологической доступности in vivo. Кристаллические формы возникают тогда, когда вещество одного и того же состава кристаллизуется в виде различных структурных решеток, что приводит к различным термодинамическим свойствам и показателям стабильности, характерным для конкретной кристаллической формы. Кристаллические формы также могут включать различные гидраты или сольваты того самого соединения. При принятии решения о том, какая форма является предпочтительной, сравнивают ряд свойств форм и выбирают предпочтительную форму, исходя из множества переменных физических свойств. Вполне возможно, что при некоторых обстоятельствах предпочтительной может быть одна форма, когда крайне важными считаются определенные аспекты, такие как простота получения, стабильность и т.д. В других ситуациях может быть предпочтительной другая форма с точки зрения более высокой скорости растворения и/или лучшей биологической доступности.
Следовательно, такая способность химического вещества кристаллизоваться в более чем одной кристаллической форме может оказывать огромное влияние на срок хранения, растворимость, свойства состава и свойства при обработке лекарственного средства. Кроме того, действие лекарственного средство может зависеть от полиморфизма молекулы лекарственного средства. Разные полиморфы могут характеризоваться разными скоростями всасывания в организме, что приводит к пониженной или повышенной биологической активности, чем необходимо. В экстремальных случаях нежелательный полиморф может даже проявлять токсичность. Появление в ходе производства неизвестной кристаллической формы может иметь значительные последствия.
Пока невозможно предсказать, будут ли конкретное соединение или соль соединения образовывать полиморфы, будут ли какие-либо такие полиморфы пригодными для коммерческого применения в терапевтической композиции, или какие полиморфы будут проявлять такие желательные свойства.
КРАТКОЕ ОПИСАНИЕ
Настоящее изобретение предусматривает кристаллические формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме (т.е. несолевой форме). В конкретном варианте осуществления свободная форма дополнительно включает воду (называемая в данном документе гидратом).
Следовательно, настоящее изобретение предусматривает кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме.
Настоящее изобретение дополнительно предусматривает кристаллическую форму гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
Варианты осуществления таких кристаллических форм включают те формы, которые обозначены в данном документе как форма B и форма HB. Названия, применяемые в данном документе для распознавания конкретной формы, например, "формы B" или "формы HB, не следует рассматривать как ограничивающие по отношению к любому другому веществу, обладающему подобными или идентичными физическими и химическими характеристиками, а скорее следует понимать, что такие обозначения являются просто идентификаторами, которые следует интерпретировать в соответствии с информацией о характеристиках, также представленной в данном документе.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 представлен иллюстративный спектр XRPD для свободной формы соединения формулы (I), обозначенной в данном документе как форма B, при этом по оси X отмечены градусы 2θ (2-тета), а по оси Y - относительная интенсивность.
На фигуре 2 представлены иллюстративные результаты DSC для свободной формы соединения формулы (I), обозначенной в данном документе как форма B.
На фигуре 3 представлены иллюстративные результаты TGA для свободной формы соединения формулы (I), обозначенной в данном документе как форма B.
На фигуре 4 представлен иллюстративный спектр XRPD для гидратной формы соединения формулы (I), обозначенной в данном документе как форма HB, при этом по оси X отмечены градусы 2θ (2-тета), и по оси Y - относительная интенсивность.
На фигуре 5 представлены иллюстративные результаты DSC для гидратной формы соединения формулы (I), обозначенной в данном документе как форма HB.
На фигуре 6 представлены иллюстративные результаты TGA для гидратной формы соединения формулы (I), обозначенной в данном документе как форма HB.
Более подробные перечни пиков XRPD для каждой из форм B и HB приведены ниже в таблицах 1 и 2 соответственно, в которых также приведены значения относительной интенсивности в %(I/I0 × 100). Следует понимать, что на спектрах порошковой рентгеновской дифракции или на порошковых рентгеновских дифрактограммах присутствует естественная изменчивость в отношении значений, измеренных в градусах 2θ (°2θ), в результате, например, погрешности прибора (включая различия между приборами). В связи с этим следует понимать, что в измерениях пиков XRPD присутствует изменчивость 2θ, составляющая до ±0,2°, и, тем не менее, такие значения пиков также будут считаться характерными для конкретной твердой формы кристаллических веществ, описанных в данном документе. Также следует понимать, что другие измеренные значения из экспериментов XRPD и экспериментов DSC/TGA, такие как относительная интенсивность и содержание воды, могут изменяться в результате, например, условий получения образца, и/или хранения, и/или окружающей среды, и измеренные значения также будут считаться характерными для конкретной твердой формы кристаллических веществ, описанных в данном документе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определение
Используемые в данном документе термины "приблизительно" и "по сути" указывают в отношении таких характеристик, как эндотермы, эндотермический пик, экзотермы, смещения базовой линии и т.д., на то, что их значения могут изменяться. В отношении положений пиков рентгеновской дифракции термин "приблизительно" или "по сути" означает то, что учитываются типичная изменчивость положения и интенсивности пика. Например, специалист в данной области поймет, что положения пика (2θ) будут демонстрировать некоторую изменчивость между устройствами, обычно вплоть до 0,2°. Иногда изменчивость может составлять более 0,2° в зависимости от различий в калибровке устройств. Кроме того, специалист в данной области поймет, что значения относительной интенсивности пика будут демонстрировать изменчивость между устройствами, а также изменчивость, обусловленную степенью кристалличности, предпочтительной ориентацией, полученной поверхностью образца и другими факторами, известными специалистам в данной области, и должны рассматриваться исключительно как качественные показатели. В случае DSC изменение наблюдаемых значений температуры будет зависеть от скорости изменения температуры, а также методики получения образца и конкретного используемого прибора. Таким образом, приведенные в данном документе значения эндотермы/точки плавления, относящиеся к термограммам DSC/TGA, могут изменяться на ±5°C (и также считаются характерными для конкретной кристаллической формы, описанной в данном документе). При использовании в контексте других характеристик, таких как, например, процент по весу (% по весу), температуры реакций, термин "приблизительно" указывает на отклонение в ±5%.
Термины "кристаллическая форма(-ы)", или "кристаллическая модификация(-и)", или "полиморфная форма(-ы)", или "полиморф(-ы)" будут применяться в данном документе взаимозаменяемо. Используемый в данном документе термин "полиморф" относится к кристаллическим формам, характеризующимся подобным химическим составом, но различными расположениями в пространстве молекул, атомов и/или ионов, образующих кристалл.
Используемый в данном документе термин "аморфный" относится к твердой форме молекулы, атома и/или ионов, которая не является кристаллической. Аморфное твердое вещество не демонстрирует определенную дифракционную рентгенограмму.
Используемое в данном документе выражение "по сути чистая фаза", в случае использования в отношении любой кристаллической формы соединения формулы (I), означает соединение, характеризующееся чистотой фазы, составляющей более чем приблизительно 90% по весу, в том числе более чем приблизительно 90, 91, 92, 93, 94, 95, 96, 97, 98 и приблизительно 99% по весу, а также в том числе равной приблизительно 100% по весу соединения формулы (I), в пересчете на вес соединения на безводной основе. Термин "чистая фаза" или "чистота фазы" в данном документе относится к однородности фазы в отношении конкретной твердой формы соединения формулы (I) и не подразумевает обязательно высокую степень химической чистоты при отсутствии прямого указания на это. Чистота фазы может быть определена в соответствии со способами, известными из уровня техники, например, с использованием XRPD, для выполнения количественного фазового анализа с использованием одного или нескольких подходов, известных из уровня техники, например, с помощью способа внешнего стандарта, прямых сопоставлений линейных (пиковых) характеристик, которые относятся к различным фазам в конкретных спектрах, или с помощью способа внутреннего стандарта. Однако количественное определение чистоты фазы с помощью XRPD может быть осложнено присутствием аморфного вещества. Соответственно, другие способы, которые можно применять для определения чистоты фазы, включают, например, ЯМР-спектроскопию твердого тела, рамановскую спектроскопию и/или инфракрасную спектроскопию. Специалист в данной области техники сможет легко разобраться в таких способах и в том, как применять такие дополнительные (или альтернативные) способы определения чистоты фазы.
Используемое в данном документе выражение "по сути химически чистый", в случае использования в отношении любой кристаллической формы соединения формулы (I), означает соединение, характеризующееся химической чистотой, составляющей более чем приблизительно 90% по весу, в том числе более чем приблизительно 90, 91, 92, 93, 94, 95, 96, 97, 98 и приблизительно 99% по весу, а также в том числе равной приблизительно 100% по весу соединения формулы (I), в пересчете на вес соли (на безводной основе). Остальное вещество в целом содержит другие соединения, такие как, например, другие стереоизомеры соединения формулы (I), примеси, образующиеся в результате реакции, исходные вещества, реагенты, побочные продукты и/или другие примеси, образующиеся при обработке, возникающие в результате получения, и/или выделения, и/или очищения конкретной кристаллической формы. Например, кристаллическая форма соединения формулы (I) может считаться по сути химически чистой, если было определено, что она характеризуется химической чистотой, составляющей более чем приблизительно 90% по весу, измеренной посредством стандартных и общепринятых способов, известных из уровня техники, где остальную часть, менее чем приблизительно 10% по весу, составляют другие вещества, такие как другие стереоизомеры соединения формулы (I), примеси, образующиеся в результате реакции, исходные вещества, реагенты, побочные продукты и/или примеси, образующиеся при обработке. Химическая чистота может быть определена в соответствии со способами, известными из уровня техники, например, с высокоэффективной жидкостной хроматографией (HPLC), LC-MS (жидкостной хроматографией с масс-спектрометрией), со спектроскопией ядерного магнитного резонанса (ЯМР) или с инфракрасной спектроскопией. Специалист в данной области техники сможет легко разобраться в таких способах и в том, как применять такие дополнительные (или альтернативные) способы определения химической чистоты.
Используемый в данном документе термин "затравка" может использоваться как существительное для описания одного или нескольких кристаллов кристаллического соединения формулы (I). Термин "затравка" также может использоваться как глагол для описания действия внесения указанного одного или несколько кристаллов кристаллического соединения формулы (I) в среду (включая, но без ограничения, например, раствор, смесь, суспензию или дисперсию), что приводит к образованию большего количества кристаллов кристаллического соединения формулы (I).
Термин "терапевтически эффективное количество" соединения по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта, например, снижение или ингибирование активности фермента или белка, или уменьшать интенсивность проявления симптомов, облегчать состояния, замедлять или задерживать прогрессирование заболевания или предупреждать заболевание и т. д. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при введении субъекту является эффективным для(1) по меньшей мере частичного облегчения, ингибирования, предупреждения и/или уменьшения интенсивности проявления состояния, или нарушения, или заболевания, (i) опосредованного LTA4H, или (ii) ассоциированного с активностью LTA4H , или (iii) характеризующегося активностью (нормальной или аномальной) LTA4H; или (2) снижения или ингибирования активности LTA4H; или (3) снижения или ингибирования экспрессии LTA4H. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при введении в клетку, или ткань, или неклеточный биологический материал, или среду является эффективным в отношении по меньшей мере частичного снижения или ингибирования активности LTA4H или частичного или полного снижения или ингибирования экспрессии LTA4H.
Используемый в данном документе термин "субъект" относится к животному. Предпочтительно животное является млекопитающим. Субъект относится, например, к приматам (например, людям), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т. п. В предпочтительном варианте осуществления субъектом является человек.
Используемые в данном документе термины в форме единственного числа и подобные термины, используемые в контексте настоящего изобретения (в частности, в контексте формулы изобретения), следует истолковывать как охватывающие как формы единственного числа, так и формы множественного числа, если в данном документе не указано иное или это явно не противоречит контексту.
Все способы, описанные в данном документе, можно осуществлять в любом подходящем порядке, если в данном документе не указано иное или это иным образом явно не противоречит контексту. Использование всех без исключения примеров или вводных слов перед примерами (например, "такой как"), предусмотренных в данном документе, предназначено исключительно для лучшего объяснения настоящего изобретения и не накладывает ограничения на объем настоящего изобретения, заявленного иным образом.
Используемые в данном документе термины "ингибировать", "ингибирование" или "ингибирующий" относятся к снижению или подавлению данного состояния, симптома, или нарушения, или заболевания, или значительному уменьшению исходной активности в отношении биологической активности или процесса.
Используемые в данном документе термины "лечить", "осуществление лечения" или "лечение" любого заболевания или нарушения относится в одном варианте осуществления к уменьшению интенсивности проявления заболевания или нарушения (т. е. замедлению, или остановке, или снижению развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечить", "осуществление лечения" или "лечение" относится к облегчению или уменьшению интенсивности проявления по меньшей мере одного физического параметра, в том числе параметров, которые могут быть неощутимы для пациента. В еще одном варианте осуществления "лечить", "осуществление лечения" или "лечение" относится к модулированию заболевания или нарушения либо физическим путем (например, путем стабилизации явного симптома), либо физиологическим путем (например, путем стабилизации физического параметра), или как тем, так и другим. В одном варианте осуществления "лечить" или "осуществление лечения" относится к задержке прогрессирования заболевания или нарушения.
Используемый в данном документе термин "предупреждать", "осуществление предупреждения" или "предупреждение" любого заболевания или нарушения относится к профилактическому лечению заболевания или нарушения или задержке возникновения заболевания или нарушения.
Как используется в данном документе, субъект "нуждается в" или является "нуждающимся в" лечении, если такое лечение принесет пользу такому субъекту с биологической, медицинской точки зрения или с точки зрения качества его жизни.
Термин "содержащий" охватывает "включающий", а также "состоящий"; например, композиция, содержащая X, может состоять исключительно из X или может включать дополнительный элемент, например, X и Y.
Используемый в данном документе термин "комбинация" относится либо к фиксированной комбинации в одной единичной лекарственной форме, либо комбинированному введению, где кристаллическую форму соединения формулы (I) и партнер по комбинации (т. е. иммунотерапевтическое средство) можно вводить независимо в одно и то же время или по отдельности с промежутками времени, особенно если такие промежутки времени позволяют партнерам по комбинации продемонстрировать суммарный, например синергетический, эффект. Отдельные компоненты могут быть упакованы в набор или предоставлены по отдельности. Один или оба компонента (например, порошки или жидкости) могут быть восстановлены или разбавлены до требуемой дозы перед введением.
Термины "совместное введение" или "комбинированное введение" или им подобные, используемые в данном документе, подразумеваются как охватывающие введение выбранного партнера по комбинации одному субъекту, нуждающемуся в этом (например, пациенту), и предполагают включение схем лечения, в которых средства не обязательно вводят одним и тем же путем введения или в одно и то же время.
Термины "фармацевтическая комбинация" и "комбинированный продукт" используются взаимозаменяемо и относятся либо к фиксированной комбинации в одной единичной лекарственной форме, либо нефиксированной комбинации или набору из частей для комбинированного введения, где два или более терапевтических средств можно вводить независимо в одно и то же время или по отдельности с интервалами времени, особенно если такие промежутки времени позволяют партнерам по комбинации продемонстрировать суммарный, например синергетический, эффект. Термин "фиксированная комбинация" означает, что как кристаллическую форму соединения формулы (I), так и партнер по комбинации (т.е. иммунотерапевтическое средство) вводят пациенту одновременно в форме единого объекта или дозы. Термин "нефиксированная комбинация" означает, что как кристаллическую форму соединения формулы (I), так и партнер по комбинации (т. е. иммунотерапевтическое средство) вводят пациенту в виде отдельных объектов одновременно, параллельно или последовательно без конкретных ограничений по времени, при этом такое введение обеспечивает терапевтически эффективные уровни двух соединений в организме пациента. Последнее также применяется в отношении "коктейльной терапии", например введения трех или более терапевтических средств. В предпочтительном варианте осуществления фармацевтическая комбинация представляет собой нефиксированную комбинацию.
Термин "комбинированная терапия" относится к введению двух или более терапевтических средств для лечения связанного с LTA4H заболевания, описанного в настоящем изобретении. Такое введение охватывает совместное введение данных терапевтических средств по сути одновременно, как, например, в одной капсуле, содержащей фиксированное соотношение активных ингредиентов. В качестве альтернативы такое введение охватывает совместное введение в нескольких или в отдельных контейнерах (например, таблетки, капсулы, порошки и жидкости) для каждого активного ингредиента. Порошки и/или жидкости могут быть восстановлены или разбавлены до требуемой дозы перед введением. Кроме того, такое введение также охватывает применение каждого типа терапевтического средства последовательно, при этом примерно в одно и то же время, либо в разное время. В любом случае режим лечения будет обеспечивать благоприятные эффекты комбинации лекарственных средств при лечении состояний или нарушений, описанных в данном документе.
Кристаллическая формы и варианты их применения
Настоящее изобретение относится к кристаллической форме свободной формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения формулы (I)), которая описана и охарактеризована в данном документе.
Настоящее изобретение также относится к кристаллической форме гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. Более конкретно, настоящее изобретение относится к кристаллической форме моногидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, которая описана и охарактеризована в данном документе.
В одном варианте осуществления в настоящем изобретении предусмотрена кристаллическая форма свободной формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форма B), характеризующаяся порошковой рентгеновской дифрактограммой (XRPD), содержащей характеристический пик, выраженный в °2θ, при значении 2θ, составляющем 24,1 ± 0,2°, измеренный при температуре, составляющей приблизительно 25°C. В другом варианте осуществления дифрактограмма XRPD дополнительно содержит один или несколько дополнительных характеристических пиков при значениях, выбранных из значения 2θ, составляющего 22,6 ± 0,2°, и значения 2θ, составляющего 26,3 ± 0,2°. В одном аспекте предыдущего варианта осуществления дифрактограмма XRPD дополнительно содержит один или несколько дополнительных характеристических пиков при значениях, выбранных из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, и значения 2θ, составляющего 28,5 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C. Соответственно, дифрактограмма XRPD для кристаллической формы свободной формы соединения формулы (I) может содержать один, два, три или четыре характеристических пика при значениях, выбранных из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, и значения 2θ, составляющего 28,5 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C. В другом варианте осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется дифрактограммой XRPD, которая может дополнительно содержать один или несколько дополнительных характеристических пиков при значениях, выбранных из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 15,2 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 20,0 ± 0,2°, значения 2θ, составляющего 20,3 ± 0,2°, значения 2θ, составляющего 21,0 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 24,4 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, значения 2θ, составляющего 28,5 ± 0,2°, и значения 2θ, составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C. Таким образом, дифрактограмма XRPD для кристаллической формы свободной формы соединения формулы (I) может содержать один или несколько (например, один, два, три, четыре, пять или шесть) характеристических пиков при значениях, выбранных из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 15,2 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 20,0 ± 0,2°, значения 2θ, составляющего 20,3 ± 0,2°, значения 2θ, составляющего 21,0 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 24,4 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, значения 2θ, составляющего 28,5 ± 0,2°, и значения 2θ, составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C. Дифрактограмма XRPD для кристаллической формы свободной формы соединения формулы (I) может содержать один или несколько (один, два, три, четыре, пять или шесть) характеристических пиков, выбранных из пиков, раскрытых в таблице 1 и измеренных при температуре, составляющей приблизительно 25°C.
В другом аспекте вышеуказанного варианта осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значений 2θ (CuKα λ=1,54184  ), выбранных из группы, состоящей из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 15,2 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 20,0 ± 0,2°, значения 2θ, составляющего 20,3 ± 0,2°, значения 2θ, составляющего 21,0 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 24,4 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, значения 2θ, составляющего 28,5 ± 0,2°, и значения 2θ, составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
), выбранных из группы, состоящей из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 15,2 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 20,0 ± 0,2°, значения 2θ, составляющего 20,3 ± 0,2°, значения 2θ, составляющего 21,0 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 24,4 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, значения 2θ, составляющего 28,5 ± 0,2°, и значения 2θ, составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
В другом аспекте вышеуказанного варианта осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ (CuKα λ=1,54184 ), выбранных из группы, состоящей из значения 2θ, составляющего 11,3 ± 0,2°, значения 2θ, составляющего 12,8 ± 0,2°, значения 2θ, составляющего 15,2 ± 0,2°, значения 2θ, составляющего 19,7 ± 0,2°, значения 2θ, составляющего 20,0 ± 0,2°, значения 2θ, составляющего 20,3 ± 0,2°, значения 2θ, составляющего 21,0 ± 0,2°, значения 2θ, составляющего 22,6 ± 0,2°, значения 2θ, составляющего 24,1 ± 0,2°, значения 2θ, составляющего 24,4 ± 0,2°, значения 2θ, составляющего 25,1 ± 0,2°, значения 2θ, составляющего 26,3 ± 0,2°, значения 2θ, составляющего 28,5 ± 0,2°, и значения 2θ, составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
В еще одном аспекте вышеуказанного варианта осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется дифрактограммой XRPD, которая является по сути такой, как показана на фигуре 1. Следует понимать, что содержание воды в форме B может находиться в диапазоне от приблизительно 0% до приблизительно 1%, и при этом она считается кристаллической формой, характеризующейся дифрактограммой XRPD, содержащей один, два, три, четыре, пять или шесть характеристических пиков, описанных выше или в таблице 1. Содержание воды, определенное с помощью способа титрования по Карлу Фишеру, для формы B составляет 0,1%.
Кристаллическая форма свободной формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты может быть охарактеризована с помощью термического анализа. В одном варианте осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется термическим профилем, измеренным с помощью дифференциальной сканирующей калориметрии (DSC) со скоростью нагревания 10°C/мин, содержащим один эндотермический пик, который начинается при приблизительно 197,4°C (что соответствует плавлению при разложении).
В другом варианте осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется термограммой DSC, которая является по сути такой, как показана на фигуре 2. Следует понимать, что для гидратированных форм могут быть получены различные термограммы (с точки зрения формы пика и профиля) в зависимости от параметров прибора, таким образом одно и то же вещество может характеризоваться термограммами, которые выглядят существенно отличающимися друг от друга, если данные получены на двух разных приборах.
В другом варианте осуществления кристаллическая форма свободной формы соединения формулы (I) характеризуется диаграммой термогравиметрического анализа (TGA), которая является по сути такой же, как показанная на представленной фиг. 3. Потеря веса при TGA составляет приблизительно 0,32% при 150°C.
В еще одном варианте осуществления кристаллическая форма B представляет собой по сути чистую фазу.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, при этом указанный способ включает стадии:
a) суспендирования формы HB (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в спирте или в смеси вода/спирт с образованием смеси в виде суспензии;
b) перемешивания и нагревания смеси в виде суспензии до температуры выше 50oC;
c) охлаждения раствора до комнатной температуры с образованием смеси в виде суспензии;
d) сбор кристаллической формы B из смеси в виде суспензии.
В еще одном варианте осуществления способ включает суспендирование формы HB (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в метаноле, или в смеси вода/метанол, или в смеси вода/1-пропанол.
В еще одном варианте осуществления способ включает суспендирование формы HB (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в метаноле.
В еще одном варианте осуществления способ включает суспендирование формы HB (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в смеси вода/1-пропанол, где соотношение вода/1-пропанол составляет 30/70% по весу.
В еще одном варианте осуществления способ включает суспендирование формы HB (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в смеси вода/метанол, где соотношение вода/метанол составляет от 1:2 до 1:9 объем к объему (об./об.). Предпочтительно соотношение вода/метанол выбрано из 1:2, 1:4 и 1:9 (об./об.). Более предпочтительно соотношение вода/метанол составляет 1:2 об./об.
В еще одном варианте осуществления температура, применяемая на стадии b) способа, предпочтительно выше 60oC, наиболее предпочтительно выше 70°C. В дополнительном аспекте данного варианта осуществления перемешивание на стадии b) длится больше 24 ч, предпочтительно больше 30 ч.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, где на стадии d) сбор кристаллической формы B из суспензии проводят путем фильтрации.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, где на стадии d) сбор кристаллической формы B из суспензии проводят путем перегонки.
В настоящем изобретении дополнительно предусмотрена кристаллическая форма гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форма HB), характеризующаяся порошковой рентгеновской дифрактограммой (XRPD), содержащей характеристический пик, выраженный в °2θ, при значении 2θ, составляющем 24,7 ± 0,2°, измеренный при температуре, составляющей приблизительно 25°C. В другом варианте осуществления дифрактограмма XRPD дополнительно содержит один или несколько дополнительных характеристических пиков при значениях, выбранных из значения 2θ, составляющего 22,1 ± 0,2°, значения 2θ, составляющего 23,8 ± 0,2°, и значения 2θ, составляющего 28,7 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
Альтернативно, дифрактограмма XRPD для кристаллической формы указанного гидрата соединения формулы (I) может содержать один, два, три или четыре характеристических пика при значениях, выбранных из значения 2θ, составляющего 22,1 ± 0,2°, значения 2θ, составляющего 23,8 ± 0,2°, значения 2θ, составляющего 24,7 ± 0,2°, и значения 2θ, составляющего 28,7 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
В другом варианте осуществления кристаллическая форма указанного гидрата соединения формулы (I) характеризуется дифрактограммой XRPD, которая может дополнительно включать один или несколько дополнительных характеристических пиков при значениях, выбранных из значения 2θ, составляющего 13,4 ± 0,2°, значения 2θ, составляющего 20,8 ± 0,2°, значения 2θ, составляющего 26,1 ± 0,2°, и значения 2θ, составляющего 33,8 ± 0,2°. Таким образом, дифрактограмма XRPD кристаллической формы указанного гидрата соединения формулы (I) может содержать один, два, три, четыре, пять или шесть характеристических пиков при значениях, выбранных из значения 2θ, составляющего 13,4 ± 0,2°, значения 2θ, составляющего 20,8 ± 0,2°, значения 2θ, составляющего 22,1 ± 0,2°, значения 2θ, составляющего 23,8 ± 0,2°, значения 2θ, составляющего 24,7 ± 0,2°, значения 2θ, составляющего 26,1 ± 0,2°, значения 2θ, составляющего 28,7 ± 0,2°, и значения 2θ, составляющего 33,8 ± 0,2°. Дифрактограмма XRPD кристаллической формы свободной формы соединения формулы (I) может содержать один или несколько (например, один, два, три, четыре, пять или шесть) характеристических пиков, выбранных из пиков, раскрытых в таблице 2 и измеренных при температуре, составляющей приблизительно 25°C.
В другом варианте осуществления указанная гидратная форма характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значений 2θ (CuKα λ=1,54184 ), выбранных из группы, состоящей из 13,4 ± 0,2°, 20,8 ± 0,2°, 22,1 ± 0,2°, 23,5 ± 0,2°, 23,5 ± 0,2°, 23,8 ± 0,2°, значения 2θ, составляющего 24,7 ± 0,2°, значения 2θ, составляющего 26,1 ± 0,2°, значения 2θ, составляющего 26,9 ± 0,2°, значения 2θ, составляющего 28,7 ± 0,2°, значения 2θ, составляющего 30,4 ± 0,2°, значения 2θ, составляющего 31,2 ± 0,2°, значения 2θ, составляющего 33,8 ± 0,2°, и значения 2θ, составляющего 38,7 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C.
В другом варианте осуществления указанная гидратная форма характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ (CuKα λ=1,54184 ), выбранных из группы, состоящей из 13,4 ± 0,2°, 20,8 ± 0,2°, 22,1 ± 0,2°, 23,5 ± 0,2°, 23,5 ± 0,2°, 23,8 ± 0,2°, значения 2θ, составляющего 24,7 ± 0,2°, значения 2θ, составляющего 26,1 ± 0,2°, значения 2θ, составляющего 26,9 ± 0,2°, значения 2θ, составляющего 28,7 ± 0,2°, значения 2θ, составляющего 30,4 ± 0,2°, значения 2θ, составляющего 31,2 ± 0,2°, значения 2θ, составляющего 33,8 ± 0,2°, и значения 2θ, составляющего 38,7 ± 0,2°, измеренных при температуре приблизительно 25°C.
В еще одном варианте осуществления кристаллическая форма гидрата соединения формулы (I) характеризуется дифрактограммой XRPD, которая является по сути такой, как показана на фигуре 4. Следует понимать, что содержание воды в форме HB может находиться в диапазоне от приблизительно 2% до приблизительно 6%, и при этом она считается гидратом, характеризующимся дифрактограммой XRPD, содержащей один, два, три, четыре, пять или шесть характеристических пиков, описанных выше. Содержание воды, определенное с помощью способа титрования по Карлу Фишеру, для формы HB составляет 5,1%.
Кристаллическая форма гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты может быть охарактеризована с помощью термического анализа. В одном варианте осуществления кристаллическая форма гидрата соединения формулы (I) характеризуется дифференциальным термогравиметрическим профилем, содержащим эндотермический пик, начинающийся при приблизительно 95°C (что соответствует дегидратации), и эндотермический пик, начинающийся при приблизительно 198,5°C (что соответствует плавлению при разложении).
В другом варианте осуществления кристаллическая форма гидрата соединения формулы (I) характеризуется термограммой DSC, которая является по сути такой, как показана на фигуре 5. Следует понимать, что для гидратированных форм могут быть получены различные термограммы (с точки зрения формы пика и профиля) в зависимости от параметров прибора, таким образом одно и то же вещество может характеризоваться термограммами, которые выглядят существенно отличающимися друг от друга, если данные получены на двух разных приборах.
В другом варианте осуществления кристаллическая форма гидрата соединения формулы (I) характеризуется диаграммой термогравиметрического анализа (TGA), которая является по сути такой же, как показанная на представленной фиг. 6. Потеря веса при TGA составляет приблизительно 4,46% при 150°C.
В еще одном варианте осуществления кристаллическая форма HB, описанная выше, представляет собой моногидратную форму.
В еще одном варианте осуществления кристаллическая форма HB представляет собой по сути чистую фазу.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, при этом указанный способ включает стадии:
a) растворения гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в воде или в смеси растворителей THF/вода с образованием раствора;
b) регулирования pH раствора с помощью водного раствора NaHCO3 или водного раствора NaOH до значения pH, составляющего от 3,3 до 7,5, при перемешивании с получением в результате суспензии;
c) сбора кристаллической формы HB из суспензии.
В конкретном аспекте вышеуказанного варианта осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где на стадии a) гидрохлоридную соль (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты растворяют в воде, и при этом коэффициент разбавления составляет по меньшей мере 30 объемов на вес (т.е. 30 объемов воды на вес соединения, например, 30 л воды на 1 кг соединения).
В другом аспекте вышеуказанного варианта осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где смесь растворителей на стадии a) представляет собой смесь THF/вода, и при этом коэффициент разбавления составляет по меньшей мере 15 объемов на вес (т.е. по меньшей мере 15 объемов смеси THF/вода на вес соединения; например, 15 л смеси THF/вода на 1 кг соединения). В конкретном аспекте данного варианта осуществления соотношение THF/вода составляет приблизительно 10:90 (вес/вес: весовое соотношение).
В другом аспекте вышеуказанного варианта осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где стадию a) проводят при комнатной температуре или, если это необходимо, при температуре, составляющей не более 35°C, пока не будет получен прозрачный раствор.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где на стадии b) pH регулируют до значения pH от 3,3 до 7,5 с помощью водного раствора NaHCO3, предпочтительно 5-10% вес/вес водного раствора NaHCO3. Предпочтительно pH доводят до приблизительно 4 или приблизительно 5.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где на стадии b) pH регулируют до значения pH от 3,3 до 7,5 с помощью водного раствора NaOH, предпочтительно 32% вес/вес водного раствора NaOH. Предпочтительно pH доводят до приблизительно 4 или приблизительно 5.
В еще одном варианте осуществления настоящее изобретение относится к способам получения кристаллической формы HB, где на стадии b) pH регулируют до значения pH от 3,3 до 7,5 при перемешивании, и при этом перемешивание продолжают не более 20 ч.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где на стадии c) сбор формы HB из суспензии проводят путем фильтрации.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, при этом указанный способ включает стадии:
a) растворения гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в воде или в смеси растворителей THF/вода с образованием раствора;
b) регулирования pH раствора с помощью водного раствора NaHCO3 или водного раствора NaOH до значения pH, составляющего от 3,3 до 7,5 при перемешивании с получением в результате суспензии;
c) сбора кристаллической формы HB из суспензии;
и дополнительно включает стадии d)-i) для получения гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
d) добавления толуола к (S)-3-((трет-бутоксикарбонил)амино)-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоте;
e) добавления водного раствора HCl с образованием реакционной смеси;
f) перемешивания и нагревания реакционной смеси до температуры, составляющей от приблизительно 50oC до приблизительно 65oC, необязательно под давлением;
g) охлаждения реакционной смеси до приблизительно 35°C,
h) отделения водного слоя и охлаждения полученного органического слоя до температуры, составляющей от 18°C до 25°C, с получением в результате суспензии,
i) сбора гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты из суспензии.
В одном варианте осуществления настоящее изобретение относится к способу получения формы HB, описанной выше, где на стадии d) толуол добавляют в таком количестве, чтобы соотношение (S)-3-((трет-бутоксикарбонил)амино)-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота/толуол составляло от 1:5 до 1:10 (вес к весу).
В другом варианте осуществления настоящее изобретение относится к способу получения формы HB, как описано выше, где на стадии e) водный раствор HCl представляет собой 30-40% водный раствор HCl, предпочтительно приблизительно 30%, и где количество HCl составляет приблизительно 15 эквивалентов гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В другом варианте осуществления настоящее изобретение относится к способу получения формы HB, описанной выше, где на стадии f) поводят нагревание реакционной смеси при температуре от приблизительно 60°C до приблизительно 65°C и перемешивание в течение 10-15 ч.
В еще одном варианте осуществления настоящее изобретение относится к способу получения формы HB, описанной выше, где стадию f) проводят под давлением, т.е. устанавливают сбрасывание давления реактора для сведения к минимуму количества HCl, улетучивающейся из реакционной смеси. Предпочтительно, сбрасывание давления устанавливают на уровне приблизительно 2000 мбар.
В еще одном варианте осуществления настоящее изобретение относится к способу получения кристаллической формы HB, где на стадии i) сбор гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты из суспензии проводят путем фильтрации или центрифугирования.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы B или формы HB или их комбинации) и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. В конкретном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму B и один или несколько фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму B в по сути чистой фазе. В еще одном варианте осуществления настоящее изобретение относится к фармацевтическому составу, содержащему кристаллическую форму B и дополнительно содержащему по меньшей мере одну другую твердую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. В одном аспекте данного варианта осуществления другая твердая форма представляет собой кристаллическую форму HB. В еще одном варианте осуществления другая твердая форма представляет собой аморфную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В конкретном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму HB и один или несколько фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму HB в по сути фазово-чистой форме. В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму HB и дополнительно содержащей по меньшей мере одну другую твердую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. В одном аспекте данного варианта осуществления другая твердая форма представляет собой кристаллическую форму B. В еще одном варианте осуществления другая твердая форма представляет собой аморфную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В других вариантах осуществления настоящее изобретение относится к комбинациям, в частности фармацевтическим комбинациям, содержащим терапевтически эффективное количество кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы B, формы HB или их комбинации) и одно или несколько терапевтических средств.
В конкретном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму B и одно или несколько терапевтических средств. В еще одном аспекте настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму B в по сути чистой фазе и одно или несколько терапевтических средств. В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму B и дополнительно содержащей по меньшей мере одну другую твердую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. В одном аспекте данного варианта осуществления другая твердая форма представляет собой кристаллическую форму HB. В еще одном варианте осуществления другая твердая форма представляет собой аморфную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В конкретном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму HB и одно или несколько терапевтических средств. В еще одном аспекте настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму HB в по сути фазово-чистой форме и одно или несколько терапевтических средств. В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллическую форму HB и дополнительно содержащей по меньшей мере одну другую твердую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. В одном аспекте данного варианта осуществления другая твердая форма представляет собой кристаллическую форму B. В еще одном варианте осуществления другая твердая форма представляет собой аморфную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В другом варианте осуществления в настоящем изобретении предусматриваются фармацевтические комбинации, описанные выше, где терапевтическое средство является независимо выбранным из группы, состоящей из
иммуносупрессивных или иммуномодулирующих средств или других противовоспалительных средств. Более конкретно терапевтическое средство выбрано из группы, состоящей из ингибитора COX, антагониста цистеинил-лейкотриеновых рецепторов (включая монтелукаст, пранлукаст, зафирлукаст), ингибитора лейкотриен-С4-синтазы (LTC4S), статина, сульфасалазина, месалазина, ингибитора кальциневрина, например, циклоспорина A или FK 506; ингибитора mTOR, например рапамицина, 40-О-(2-гидроксиэтил)рапамицина, биолимус-7 или биолимус-9; аскомицина, обладающего иммуносупрессивными свойствами, например, ABT-281, ASM981; кортикостероидов; циклофосфамида; азатиоприна; метотрексата; лефлуномида; мизорибина; микофеноловой кислоты или соли; микофенолата мофетила; ингибитора IL-1-бета.
В одном варианте осуществления настоящее изобретение относится к способу лечения заболеваний или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, у субъекта, нуждающегося в этом, при этом способ включает: введение субъекту, нуждающемуся в этом, терапевтически эффективного количества кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы B, формы HB или их комбинации) отдельно или в комбинации с одним или несколькими терапевтическими средствами.
В другом варианте осуществления настоящее изобретение относится к способу лечения заболевания или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, у субъекта, нуждающегося в этом, включающему введение указанному субъекту фармацевтической композиции, описанной выше, отдельно или в комбинации с одним или несколькими терапевтическими средствами.
В другом варианте осуществления настоящее изобретение относится к способу лечения заболевания или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, у субъекта, нуждающегося в этом, включающему введение указанному субъекту фармацевтической комбинации, описанной выше.
В одном варианте осуществления настоящее изобретение относится к применению кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы B, формы HB или их комбинации) отдельно или в комбинации с одним или несколькими терапевтическими средствами для лечения заболевания или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H.
В еще одном варианте осуществления настоящее изобретение относится к кристаллической форме (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форме B, форме HB или их комбинации) для применения в лечении заболевания или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H.
В еще одном варианте осуществления настоящее изобретение относится к комбинации кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы B, формы HB или их комбинации) и одного или нескольких терапевтических средств для применения в лечении заболевания или нарушения, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H.
В одном варианте осуществления настоящее изобретение относится к способу лечения, применению, соединению для применения или комбинации для применения, как описано выше, где заболевание или нарушение, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, выбрано из воспалительных и аутоиммунных нарушений, а также воспаления легких и дыхательных путей. Более конкретно, заболевание или нарушение, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, выбрано из острого или хронического воспаления, анафилактических реакций, аллергических реакций, видов атопического дерматита, псориаза, синдрома острой дыхательной недостаточности, опосредованного иммунокомплексами повреждения легких и хронического обструктивного заболевания легких, воспалительных заболеваний кишечника (включая язвенный колит, болезнь Крона и послеоперационную травму), видов язвы желудочно-кишечного тракта, видов нейтрофильного дерматоза (включая без ограничения гангренозную приодермию, синдром Свита, акне и нейтрофильную крапивницу), опосредованного иммунокомплексами гломерулонефрита, аутоиммунных заболеваний (включая инсулинозависимый сахарный диабет, рассеянный склероз, ревматоидный артрит, остеоартроз и системную красную волчанку), видов васкулита (включая без ограничения виды кожного васкулита, болезнь Бехчета и болезнь Шенлейн-Геноха), нарушений сердечно-сосудистой системы (включая без ограничения гипертонию, атеросклероз, аневризму, критическую ишемию нижних конечностей, окклюзионную болезнь периферических артерий, легочную артериальную гипертензию и болезнь Рейно), сепсиса, воспалительной и нейропатической боли, в том числе артритной боли, пародонтоза, в том числе гингивита, видов воспаления среднего уха, мигрени, доброкачественной гиперплазии предстательной железы, синдрома Шегрена-Ларссона и видов рака (включая без ограничения виды лейкоза и лимфомы, рак предстательной железы, рак молочной железы, рак легких, злокачествленную меланому, рак почки, опухоли головы и шеи и колоректальный рак). В предпочтительном варианте осуществления заболевание или нарушение, интенсивность проявления которых обычно уменьшается за счет ингибирования LTA4H, выбрано из воспалительных заболеваний кишечника (включая язвенный колит, болезнь Крона и послеоперационную травму) и видов нейтрофильного дерматоза (включая без ограничения гангренозную приодермию, синдром Свита, акне и нейтрофильную крапивницу).
В одном варианте осуществления настоящее изобретение относится к способу, применению или комбинации для применения в соответствии с вышеуказанным вариантом осуществления, где терапевтическое средство вводится сообща в одной композиции или вводится разрозненно в двух или более различных формах композиций.
В одном варианте осуществления настоящее изобретение относится к способу, применению или комбинации для применения, как описано выше, где терапевтическое средство вводится одновременно с кристаллической формой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, до или после нее.
Свойства кристаллических форм по настоящему изобретению
Было установлено, что кристаллические формы, описанные в данном документе, обладают преимущественными свойствами. Критериями отбора являются токсикологические факторы, кристалличность, точка плавления, гигроскопичность, стабильность в нерасфасованном виде, совместимость со вспомогательными веществами, pH водного раствора, растворимость в воде и водных средах, морфология, поведение при обработке и полиморфное поведение. Свободная форма B и гидратная форма HB продемонстрировали превосходные варианты поведения, особенно по сравнению с HCl-солью, которая была ранее известна и описана в WO2015/092740. Было установлено, что HCl-соль является веществом, вызывающим коррозию, причем оказывающим сильное коррозионное действие на пробный образец стали марки 1.2767 и умеренное коррозионное действие на пробный образец стали марки 1.2803, поэтому она была признана непригодной для дальнейшей разработки.
Кристаллическая форма B представляет собой безводную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, которая является высококристаллической, демонстрируя столбчатый габитус. Она является слабогигроскопичной с максимальным водопоглощением, составляющим менее 0,7% при 25°C и относительной влажности 90% согласно DVS.
Кристаллическая форма HB представляет собой гидратированную форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. Данные TGA и DVS указывают на то, что она является моногидратом. Кристаллическая форма HB не гигроскопична при относительной влажности 10-90%, но теряет свою воду при значении относительной влажности ниже 10%. Постепенная регидрация HB происходит при относительной влажности 10% и относительной влажности выше 60%.
Стабильность в растворителе
Проводили тесты на установление равновесия с растворителем для изучения относительной стабильности формы HB в растворителе. 50 мг формы HB уравновешивали с помощью 1 мл растворителя в течение 24 ч (или 7 дней, или 28 дней) на водяной бане при 25°C или в течение 7 дней при 50°C. Растворы фильтровали и высушивали в течение 10 минут на воздухе. Полученное твердое вещество анализировали с помощью XRPD.
Изучали различные растворители: ацетон, ацетонитрил, дихлорметан, диоксан, этанол, гептан, метанол, THF, смеси ацетон/вода в соотношении от 10/90 до 90/10, смеси ацетонитрил/вода в соотношении от 10/90 до 90/10, смеси диоксан/вода в соотношении от 10/90 до 90/10 и смесь этанол/вода в соотношении от 10/90 до 90/10, тетрагидрофуран/вода (50/50) и 1-пропанол/вода (70/30).
Полиморф HB не продемонстрировал изменений в ряде растворителей при 25°C в течение 7 дней или 28 дней, но превращается в аморфный материал или гелеобразный материал в ацетоне, диоксане, этаноле, метаноле и THF. Однако при 50°C в течение 7 дней форма HB становится аморфной во многих растворителях. В метаноле форма HB превращается в форму B. Преобразование формы HB в форму B в целом происходит при низком содержании воды (таблица 3).
Таблица 3. Стабильность формы HB в различных растворителях при 50°C
Проводили сравнительные эксперименты со взвесью при разных значениях активности воды при 25°C и 50°C. Также готовили сравнительные взвеси физической смеси (1:1) модификаций HB и B при разных значениях активности воды.
Таблица 4. Сравнительные эксперименты со взвесью при разных значениях активности воды при 25 градусах Цельсия
(97:3)
При 25°C смесь превратилась в чистую форму B при значениях активности воды aw ≤0,5. При значениях активности воды, указанных выше, смесь оставалась без изменений. При 50°C можно было наблюдать подобную тенденцию, однако, максимальная активность воды для преобразования в форму B повышалась до aw ≤0,7.
Таблица 4B. Сравнительные эксперименты со взвесью при разных значениях активности воды при 50 градусах
(97:3)
(92:8)
(88:12)
(83:17)
(97:3)
(89:11)
(77:23)
(57:43)
(22:78)
Было установлено, что модификация HB преобразовывается в безводную модификацию B посредством помещения взвеси в горячий или кипящий метанол (т.е. температура составляет от приблизительно 50°C до приблизительно 65°C). Модификация B также может быть получена посредством помещения взвеси при температуре 80°C в водные смеси растворителей, при этом также из модификации HB. Модификация B не преобразовывается в другую форму и поэтому демонстрирует лучшую стабильность.
Стабильность твердой формы в нерасфасованном виде и со смесями вспомогательных веществ
Изучали химическую и физическую стабильность формы B и формы HB, подвергая отдельные формы действию высокой влажности при значениях температуры в диапазоне от 25 до 80°C. Обе формы подвергали различным условиям испытания, описанным ниже.
Условие испытания 1: 1 неделя в герметичном контейнере при 80°C, 50°C, 80°C/75% относительной влажности (RH) или 50°C/75% RH.
Условие испытания 2: 1 неделя в открытом контейнере при 80°C.
Условие испытания 3: 1 неделя в герметичном контейнере при 50°C.
Условие испытания 4: 1 неделя в открытом контейнере при 50°C/75% RH.
Условие испытания 5: 1 неделя в открытом контейнере при комнатной температуре/92% RH.
Продукты распада анализировали с помощью HPLC, а образец анализировали с помощью XPRD для выявления каких-либо изменений твердой формы.
Способ HPLC
Прибор Water Acquity UPLC
Колонка: Water Acquity UPLC BEH Shield RP18
Размер частицы (мкм): 1,7
Размеры (мм): 2,1×100
Температура (°C): 30
Расход (мл/мин.): 0,5
Объем вводимой пробы (мкл): 1
Растворитель: ацетонитрил/вода (50:50)
Концентрация (мкг/мл): 500
Длина волны детектора (нм): 248
Подвижная фаза A: 0,1% TFA в смеси ацетонитрил/вода (5:95)
Подвижная фаза B: 0,1% TFA в смеси ацетонитрил/вода (95:5)
Время анализа (мин.): 4 (22)
Градиент % B Мин.
0 Исходное значение
45 15
100 19
100 20
При описанных выше условиях испытания форма B продемонстрировала хорошую стабильность и абсолютное отсутствие превращения. Модификация HB была стабильной при всех условиях, кроме 80°C без воздействия влажности.
В дополнение к этому было установлено, что форма B является стабильной после 1 недели при 40°C/90% RH, причем образования гидратов не обнаружено. Следовательно, форму B идентифицировали как предпочтительную форму, поскольку она не гидратируется при воздействий высокой относительной влажности (RH).
Физическая стабильность
Поведение при прессовании. Оценивали также физическую стабильность кристаллической формы HB. 300 мф кристаллической формы HB подвергали прессованию в течение 5 минут при давлении 10 тонн с помощью гидравлического пресса (диаметр таблеток 13 мм). Свойства образца затем определяли с помощью XRPD для выявления любого изменения в твердой фазе.
С помощью XRPD не наблюдали никакого изменения кристаллической формы для кристаллической формы HB. Следовательно, было показано, что кристаллическая форма HB обладают хорошими свойствами в отношении физической стабильности.
Поведение при измельчении. Оценивали также физическую стабильность кристаллической формы для формы HB. 50 мг кристаллической формы B или HB вручную измельчали в ступке в течение 3 минут. Согласно XRPD измельчение кристаллической формы HB не приводило к какому-либо изменению.
Поведение при эксперименте с моделированием грануляции. Физическую стабильность кристаллических форм HB также оценивали в экспериментах с моделированием грануляции. В таких экспериментах к кристаллической форме HB добавляли по каплям растворитель для грануляции до тех пор, пока твердое вещество не было достаточно увлажнено. Смесь затем перемешивали вихревым способом между каждым добавлением при 25°C. Смесь затем высушивали под вакуумом. Кристалличность материала (после измельчения) повторно оценивали с помощью XRPD и/или DSC. Растворителем для грануляции были вода или этанол. При грануляции с применением этанола или воды в качестве растворителя для грануляции XRPD для полиморфа HB свидетельствовала об отсутствии изменения формы.
Растворимость
Форма B характеризуется растворимостью, составляющей 0,05 мг/мл при значениях pH от 4 до 7.
Форма B демонстрирует растворимость в водных буферах в биорелевантных средах, составляющую 0,07 мг/мл в FaSSIF-V2 (pH=6,6) и 0,18 мг/мл в FeSSIF-V2 (pH=5,9).
Форма HB характеризуется растворимостью в водных буферах в биорелевантных средах, составляющей 0,75 мг/мл в искусственном желудочном соке (SGF при pH=2), 0,09 мг/мл в FaSSIF (pH=6,5) и 0,009 мг/мл в FaSSIF (pH=5,8).
В заключение, кристаллическая форма B продемонстрировала химическую и физическую стабильность как в растворенном, так и в твердом состояниях. Кристаллическая форма HB является стабильной в большом диапазоне значений влажности и является высококристаллической, но превращается в форму B в метаноле или в водном растворителе при высоких температурах.
Фармацевтическая композиция, комбинация, дозировка и введение
В некоторых вариантах осуществления кристаллические формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, описанные в данном документе, могут использоваться отдельно или они могут быть составлены в фармацевтическую композицию, которая также содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество и зачастую содержит по меньшей мере два или более фармацевтически приемлемых вспомогательных веществ. Некоторые подходящие вспомогательные вещества раскрыты в данном документе. Могут применяться другие вспомогательные вещества, которые известны из уровня техники, без отступления от сути и объема данной заявки.
В некоторых вариантах осуществления в настоящем изобретении применяют фармацевтическую композицию, содержащую соединение по настоящему изобретению и фармацевтически приемлемое вспомогательное вещество.
Используемый в данном документе термин "фармацевтически приемлемые вспомогательные вещества" включает все без исключения растворители, носители, разбавители, дисперсионные среды, средства для нанесения покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства, антиоксиданты), средства для обеспечения изотоничности, замедляющие абсорбцию средства, соли, стабилизаторы лекарственных средств, связующие вещества, добавки, объемообразующие средства, разрыхляющие средства, смазывающие вещества, подслащивающие средства, ароматизирующие средства, красители и т.п. и их комбинации, которые будут известны специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). Следует понимать, что если традиционное вспомогательное вещество является несовместимым с активным ингредиентом, то данной заявкой предусмотрено применение любого традиционного вспомогательного вещества в любых терапевтических или фармацевтических композициях.
Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение, ректальное введение и т.д. Кроме того, фармацевтические композиции по настоящему изобретению могут быть изготовлены в твердой форме (в том числе без ограничения в виде капсул, таблеток, пилюль, гранул, порошков или суппозиториев) или в жидкой форме (в том числе без ограничения в виде растворов, суспензий или эмульсий). Фармацевтические композиции могут подвергаться традиционным операциям фармацевтической отрасли, таким как стерилизация, и/или они могут содержать традиционные инертные разбавители, смазывающие средства, носители или буферные средства, а также вспомогательные средства, такие как растворители, консерванты, стабилизаторы, смачивающие средства, эмульгаторы, объемообразующие средства и т.д.
Как правило, фармацевтические композиции представляют собой таблетки или капсулы, содержащие активный ингредиент вместе с по меньшей мере одним вспомогательным веществом, таким как:
a) разбавители, например, лактоза, декстроза, сахароза, маннит, сорбит, целлюлоза и/или глицин;
b) смазывающие вещества, например, диоксид кремния, тальк, стеариновая кислота, ее магниевая или кальциевая соль и/или полиэтиленгликоль; в случае таблеток также
c) связующие вещества, например, алюмосиликат магния, крахмальная паста, желатин, трагакант, метилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон, при необходимости;
d) носители, такие как водная среда-носитель, содержащая совместно растворяющееся вещество, такое как каптизол, PEG, глицерин, циклодекстрин или т.п.;
e) разрыхлители, например, виды крахмала, агар, альгиновая кислота или ее натриевая соль или шипучие смеси; и/или
f) абсорбенты, красящие вещества, ароматизаторы и подсластители.
Таблетки могут быть покрыты либо пленочной оболочкой, либо энтеросолюбильной оболочкой в соответствии со способами, известными из уровня техники.
Предпочтительно соединение или композицию готовят для перорального введения, например, в виде таблетки или, например, капсулы, и необязательно упаковывают в формате нескольких доз, что подходит для хранения и/или дозирования однократных доз фармацевтического продукта. Примеры подходящей упаковки включают без ограничения виды герметично закрываемой фольги, контейнеры с однократной дозой (например, флаконы), блистерные упаковки и контурные безъячейковые упаковки.
Таблетки могут содержать активный ингредиент в смеси с нетоксичными, фармацевтически приемлемыми вспомогательными веществами, которые являются подходящими для изготовления таблеток.
Такие вспомогательные вещества представляют собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактозу, фосфат кальция или фосфат натрия; средства для грануляции и разрыхляющие средства, например, кукурузный крахмал или альгиновую кислоту; связующие средства, например крахмал, желатин или аравийскую камедь; и смазывающие средства, например, стеарат магния, стеариновую кислоту или тальк. Таблетки являются непокрытыми или покрытыми посредством известных методик для замедления распада и всасывания в желудочно-кишечном тракте и тем самым обеспечивают устойчивое действие в течение более длительного периода. Например, можно использовать вещество для замедленного действия, такое как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
В настоящем изобретении дополнительно предусмотрены безводные фармацевтические композиции и лекарственные формы, содержащие соединения по настоящему изобретению в качестве активных ингредиентов, поскольку вода может способствовать разрушению некоторых соединений.
Безводные фармацевтические композиции и лекарственные формы по настоящему изобретению можно получать с применением безводных ингредиентов или ингредиентов с низким содержанием влаги и условий с низким содержанием влаги или низкой влажностью. Безводную фармацевтическую композицию можно получать и хранить таким образом, чтобы сохранялась ее безводная природа. Соответственно, безводные композиции предпочтительно упаковывают с применением материалов, которые, как известно, предотвращают воздействие воды, так чтобы их можно было включать в подходящие рецептурные наборы. Примеры подходящей упаковки включают без ограничения виды герметично закрываемой фольги, пластиковые материалы, контейнеры с однократной дозой (например, флаконы), блистерные упаковки и контурные безъячейковые упаковки.
В настоящем изобретении дополнительно предусмотрены фармацевтические композиции и лекарственные формы, которые содержат одно или несколько средств, которые снижают скорость, с которой соединение по настоящему изобретению в качестве активного ингредиента будет распадаться. Такие средства, которые упоминаются в данном документе как "стабилизаторы", включают без ограничения антиоксиданты, такие как аскорбиновая кислота, буферы для поддержания pH или солевые буферы и т.д.
Фармацевтическая композиция или комбинация по настоящему изобретению может быть представлена в однократной дозировке, составляющей приблизительно 1-1000 мг активного(-ых) ингредиента(-ов) для субъекта весом приблизительно 50-70 кг, или приблизительно 1-500 мг, или приблизительно 1-250 мг, или приблизительно 1-150 мг, или приблизительно 0,5-100 мг, или приблизительно 10-50 мг активных ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, массы тела, возраста и индивидуального состояния, нарушения или заболевания, лечение которых осуществляют, или их тяжести. Лечащий врач, клиницист или ветеринар средней квалификации может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или ингибирования прогрессирования нарушения или заболевания.
Вышеупомянутые параметры дозировки являются очевидными в тестах in vitro и in vivo с применением преимущественно млекопитающих, например, мышей, крыс, собак, обезьян, или выделенных органов, тканей и их препаратов. Соединения по настоящему изобретению можно применять in vitro в виде растворов, например, предпочтительно водных растворов, и in vivo либо энтерально, либо парентерально, преимущественно внутривенно, например, в виде суспензии или водного раствора. Дозировка in vitro может находиться в диапазоне значений концентрации от приблизительно 10-3 моль/л до 10-9 моль/л. В зависимости от пути введения терапевтически эффективное количество in vivo может находиться в диапазоне приблизительно 0,1-500 мг/кг или приблизительно 1-100 мг/кг.
В других вариантах осуществления предусмотрена фармацевтическая композиция, которая содержит по меньшей мере одно соединение в соответствии с вариантами осуществления выше по тексту и по меньшей мере один носитель.
Кристаллические формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, описанные в данном документе, также являются применимыми в качестве активных фармацевтических ингредиентов (API), а также материалов для получения составов, которые включают одно или несколько фармацевтически приемлемых вспомогательных веществ и являются подходящими для введения субъектам-людям.
Соответственно, в одном варианте осуществления настоящего изобретения кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форма B или форма HB) предусмотрена в по сути фазово-чистой форме. Данная кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форма B или форма HB) в по сути фазово-чистой форме может применяться для получения фармацевтических композиций, которые могут дополнительно содержать одно или несколько фармацевтически приемлемых вспомогательных веществ. В некоторых вариантах осуществления кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты может не сохранять свою кристалличность в фармацевтической композиции. Например, в некоторых вариантах осуществления кристаллическая форма B или HB может применяться в способе получения фармацевтической композиции, который, например, включает распылительное высушивание или влажную грануляцию; таким образом возможно, что в полученной фармацевтической композиции будет выявлено небольшое количество кристаллической формы B или HB или она не будет выявлена вовсе.
Терапевтические наборы
В одном варианте осуществления в настоящем изобретении предусмотрен набор, содержащий две или более отдельных фармацевтических композиций, по меньшей мере одна из которых содержит кристаллическую форму соединения формулы (I) (форму B или форму HB). В одном варианте осуществления набор содержит средства для раздельного содержания указанных композиций, такие как контейнер, секционная бутылка или секционный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковки таблеток, капсул и т. п.
Набор по настоящему изобретению можно применять для введения различных лекарственных форм, например, для перорального и парентерального применения, для введения отдельных композиций с различными интервалами между введениями доз или для титрования отдельных композиций относительно друг друга. В целях содействия соблюдению режима лечения набор по настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии по настоящему изобретению кристаллическая форма соединения формулы (I) (т. е. форма B или форма HB) и другое терапевтическое средство могут быть изготовлены и/или составлены одним и тем же или разными производителями. Более того, кристаллическая форма соединения формулы (I) и другое терапевтическое средство могут быть объединены в средство комбинированной терапии: (i) до того, как комбинированный продукт попадает к врачам (например, в случае набора, содержащего кристаллическую форму соединения формулы (I) и другое терапевтическое средство); (ii) самими врачами (или под руководством врача) незадолго до введения; (iii) самими пациентами, например, во время последовательного введения кристаллической формы соединения формулы (I) и другого терапевтического средства.
Соответственно, в настоящем изобретении предусмотрено применение кристаллической формы, описанной в данном документе (т. е. формы B или формы HB), для лечения заболеваний, интенсивность проявления которых уменьшается за счет ингибирования LTA4H (например, воспалительных, аутоиммунных и респираторных заболеваний), где лекарственный препарат получен для введения с другим терапевтическим средством. В настоящем изобретении также предусмотрено применение терапевтического средства для лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные и респираторные заболевания), где лекарственный препарат вводят с кристаллической формой соединения формулы (I).
В настоящем изобретении также предусмотрена кристаллическая форма соединения формулы (I) (т.е. форма B или форма HB) для применения в способе лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные и респираторные заболевания), где кристаллическая форма соединения формулы (I) получена для введения с другим терапевтическим средством. В настоящем изобретении также предусмотрено другое иммунотерапевтическое средство для применения в способе лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные или респираторные заболевания), где другое терапевтическое средство получено для введения с кристаллической формой соединения формулы (I). В настоящем изобретении также предусмотрена кристаллическая форма соединения формулы (I) для применения в способе лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные и респираторные заболевания), где кристаллическая форма соединения формулы (I) вводится с другим терапевтическим средством. В настоящем изобретении также предусмотрено другое терапевтическое средство для применения в способе лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например воспалительные, аутоиммунные или респираторные заболевания), где другое терапевтическое средство вводится с кристаллической формой соединения формулы (I).
В настоящем изобретении также предусмотрено применение кристаллической формы соединения формулы (I) для лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные и респираторные заболевания), где пациент ранее (например, в течение 24 часов) подвергался лечению с помощью другого терапевтического средства. В настоящем изобретении также предусмотрено применение другого терапевтического средства для лечения заболевания, интенсивность проявления которого уменьшается за счет ингибирования LTA4H (например, воспалительные, аутоиммунные и респираторные заболевания), где пациент ранее (например, в течение 24 часов) подвергался лечению с помощью кристаллической формы соединения формулы (I).
Комбинация
Дополнительные терапевтические средства, применяемые в комбинации с кристаллической формой по настоящему изобретению, включают без ограничения противовоспалительные средства, иммуномодулирующие средства, иммуносупрессивные средства или химиотерапевтическое средство.
Например, соединения по настоящему изобретению можно применять в комбинации с ингибитором COX, антагонистом цистеинил-лейкотриеновых рецепторов (включая монтелукаст, пранлукаст, зафирлукаст), ингибитором лейкотриен-С4-синтазы (LTC4S), статином, сульфасалазином, месалазином, ингибитором кальциневрина, например, циклоспорином A или FK 506; ингибитором mTOR, например рапамицином, 40-О-(2-гидроксиэтил)рапамицином, биолимус-7 или биолимус-9; аскомицином, обладающим иммуносупрессивными свойствами, например, ABT-281, ASM981; кортикостероидами; циклофосфамидом; азатиоприном; метотрексатом; лефлуномидом; мизорибином; микофеноловой кислотой или солью; микофенолата мофетилом; ингибитором IL-1-бета.
Получение кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты
Кристаллические формы можно получать различными способами, включая, например, кристаллизацию или перекристаллизацию из подходящего растворителя, сублимацию, выращивание из расплава, преобразование в твердом состоянии из другой фазы, кристаллизацию из сверхкритической жидкости и струйное распыление. Методики кристаллизации или перекристаллизации кристаллических форм из смеси растворителей включают, например, выпаривание растворителя, понижение температуры смеси растворителей, затравку кристаллами перенасыщенной смеси растворителей молекулы и/или соли, сублимационную сушку смеси растворителей и добавление антирастворителей (противорастворителей) к смеси растворителей. Иллюстративные способы получения кристаллических форм, описанных в данном документе, подробно изложены ниже.
Кристаллы лекарственных средств, в том числе полиморфы, способы получения и определение характеристик кристаллов лекарственных средств рассмотрены в Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, Уэст-Лафейетт, Индиана (1999).
В случае методик кристаллизации, в которых используются растворители, выбор растворителя или растворителей обычно зависит от одного или нескольких факторов, таких как растворимость соединения, методика кристаллизации и давление паров растворителя. Можно использовать комбинации растворителей, например, соединение может быть растворено в первом растворителе с получением раствора, за чем следует добавление антирастворителя для уменьшения растворимости соединения в растворе и обеспечения образования кристаллов. Антирастворитель представляет собой растворитель, в котором соединение характеризуется низкой растворимостью.
В одном способе получения кристаллов соединение растворяют и/или перемешивают в подходящем растворителе с получением взвеси, которую можно нагревать для обеспечения растворения. Используемый в данном документе термин "взвесь" означает насыщенный раствор соединения, который также может содержать дополнительное количество соединения для получения гетерогенной смеси соединения и растворителя при заданной температуре. Он также может называться суспензией.
К любой кристаллизационной смеси можно добавлять затравочные кристаллы для ускорения кристаллизации. Введение затравки можно применять для регулирования роста конкретного полиморфа или для регулирования распределения частиц кристаллического продукта по размеру. Соответственно, расчет необходимого количества затравочных кристаллов зависит от размера доступного затравочного кристалла и необходимого размера средней частицы продукта, как описано, например, в "Programmed Cooling of Batch Crystallizers," J.W. Mullin and J. Nyvlt, Chemical Engineering Science, 1971,26, 369-377. Как правило, затравочные кристаллы малого размера необходимы для эффективного регулирования роста кристаллов в партии. Затравочный кристалл малого размера может быть получен путем просеивания, размалывания или тонкого измельчения крупных кристаллов, или путем микрокристаллизации растворов. Необходимо проявлять осторожность, чтобы размалывание или тонкое измельчение кристаллов не приводило к какому-либо изменению формы кристалличности необходимой формы кристалла (т.е. к превращению в аморфную форму или в другой полиморф).
Охлажденную кристаллизационную смесь можно фильтровать под вакуумом, и выделенные твердые вещества можно промывать с помощью подходящего растворителя, такого как холодный растворитель для перекристаллизации, и высушивать при продувании азотом с получением необходимой кристаллической формы. Выделенные твердые вещества можно анализировать с помощью подходящей спектроскопической или аналитической методики, такой как ядерный магнитный резонанс твердого тела, дифференциальная сканирующая калориметрия, порошковая рентгеновская дифрактометрия или т.п., для обеспечения образования предпочтительной кристаллической формы продукта. Как правило, полученную в результате кристаллическую форму получают в количестве, соответствующем практическому выходу, составляющему более чем приблизительно 70 вес. %, предпочтительно практическому выходу, составляющему более чем 90 вес. %, в пересчете на вес соединения, изначально используемого в процедуре кристаллизации. При необходимости продукт может быть совместно размолот или пропущен через сетчатое сито для удаления комков продукта.
В качестве альтернативы кристаллические формы можно получать непосредственно из реакционной среды конечной стадии способа получения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты. Этого можно достичь, например, посредством использования на конечной стадии способа растворителя или смеси растворителей, из которых может быть выкристаллизована (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота. Кроме того, кристаллические формы можно получать с помощью методик перегонки или добавления растворителя.
В дополнение к способам, кратко рассмотренным ниже, следует понимать, что для определения характеристик любого из веществ, описанных в данном документе, можно применять различные аналитические способы.
Следующие неограничивающие примеры иллюстрируют настоящее изобретение.
ПРИМЕРЫ
Сокращения:
IT: внутренняя температура
Tj: температура рубашки реактора
Tr: температура реакции
THF: тетрагидрофуран
Об./мин.: обороты в минуту
Пример 1. Получение кристаллической формы HB
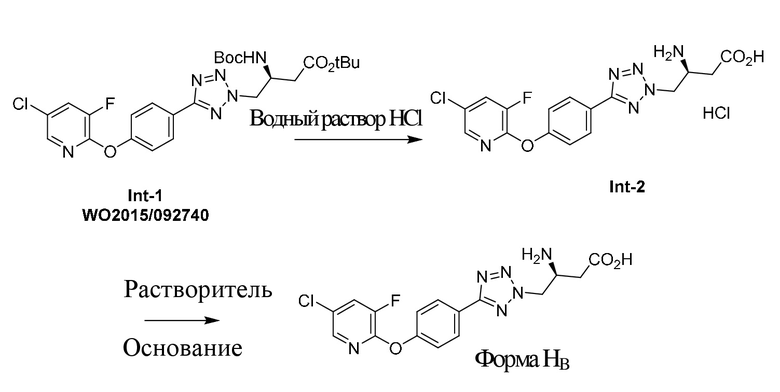
В реактор, футерованный стеклом, емкостью 2500 л добавляли Int-1 (55 кг) и толуол (810 кг) и смесь перемешивали (55 об/мин, якорная мешалка). Добавляли 31% HCl (232 кг). Сбрасывание давления реактора устанавливали на уровне 2000 мбар. Внутреннюю температуру регулировали на уровне 65°C и выполняли перемешивание (60 об/мин) при внутренней температуре (IT), составляющей от 65 до 66oC, в течение 11 ч 29 мин. Затем внутреннюю температуру регулировали на уровне 35oC. Затем 82 кг водного раствора отделяли при пониженном давлении с помощью устройства по типу ловушки Дина-Старка (ITмакс.=44°C), при этом внутреннюю температуру регулировали до уровня ~20. Систему перемешивали при 19~24°C в течение 80 мин. Твердое вещество собирали путем центрифугирования. Влажный осадок промывали толуолом (50 кг с добавлением 20 мл антистатика) и дихлорметаном (50 кг). Твердое вещество высушивали в титановой полочной сушилке при 195 мбар с помощью раствора бисульфита натрия JT Baker при установленной температуре, равной 60°C, в течение 8 ч 26 мин.
Получали гидрохлоридную соль (Int-2).
В реактор, футерованный стеклом, емкостью 50 л добавляли 5 кг Int-2, и также в реактор добавляли 59 кг смеси растворителей THF/вода (вес/вес=7,2/64). Смесь перемешивали при 32~34°C в течение 2 ч (при этом наблюдали некоторое количество нерастворимых частиц). Раствор фильтровали через рукавный фильтр и
после этого через фильтр частиц с размером пор 0,2 мкм.
Фильтрат переносили в отдельный сосуд через другой фильтр частиц с размером пор 0,2 мкм. В отдельный сосуд добавляли раствор NaHCO3 (0,989 кг) в воде (15 кг). Во время добавления осаждалось белое твердое вещество. После завершения добавления твердое вещество стабильно суспендировалось в жидкости и не оседало заметно при прекращении перемешивания. После перемешивания при 300 об/мин в течение 67 мин отбирали образец из системы и проводили испытание с помощью индикаторной бумаги для определения pH, при этом получали результат, равный 4. После перемешивания при
300 об/мин в течение 20 часов систему фильтровали. Твердое вещество на фильтре промывали с помощью 50 кг очищенной воды с получением формы HB.
В качестве альтернативы пример 29, описанный в WO2015/092740 (28 г, 35 ммоль), и смесь растворителей, содержащую 360 г воды и 40 г THF, смешивали вместе и перемешивали в течение 20 минут. Смесь фильтровали и фильтрат регулировали до pH=5 с помощью водного раствора NaHCO3. Перемешивание продолжали в течение 18 ч, после чего смесь фильтровали с получением полиморфа HB во влажном осадке весом 25,6 г, который использовали для получения полиморфа формы B (см. пример 2) без дополнительной очистки.
В качестве альтернативы, пример 29, описанный в WO2015/092740 (15,0 г, 65 ммоль, HCl-соль), добавляли к 450 мл воды и перемешивали в течение 1 часа. Раствор регулировали до pH~4 с помощью водного раствора NaHCO3. Смесь перемешивали при комнатной температуре в течение 17 часов, перед тем как фильтровали. Осадок на фильтре промывали водой и высушивали in vacuo с получением 14,7 г формы HB, характеристики которой определяли с помощью XRPD, DSC и TGA (фигуры 4-6 соответственно).
Пример 2. Получение гидрата кристаллической формы B

Первый способ: взвешивали 505 мг формы HB в стеклянном флаконе объемом 20 мл и добавляли 6 мл
метанола. Взвесь нагревали до 50°C и перемешивали в течение 4 дней с применением магнитной мешалки. Суспензию
охлаждали до комнатной температуры и фильтровали. Извлеченное твердое вещество высушивали при 40°C в течение 2,5 ч. под вакуумом. Белое твердое вещество анализировали с помощью XRPD, DSC и TGA (фигуры 1-3 соответственно).
Второй способ: перемешивание формы HB в смеси вода/MeOH (2:8, 1:9, 1:2 об./об.) при температуре выше 60°C также приведет к получению формы B. Например, 20,7 г формы HB (вес в сухом состоянии) добавляли в предварительно перемешанную смесь растворителей, содержащую 810 мл MeOH и 90 мл H2O. Полученную смесь нагревали до 68°C в течение 36 ч, после чего охлаждали до 25°C. Смесь перемешивали еще в течение 2 ч и фильтровали. Осадок на фильтре высушивали in vacuo с получением формы B (15,8 г, выход 76%) в виде белого порошка.
Масса/заряд=393,1 [M+H]+, Rt=2,47 мин (условия UPLC-MS), Rt=14,85 мин (условия HPLC), 1H ЯМР (400 МГц, MeOD-d4) δ=8,24 (d, 2H), 7,98 (d, 1H), 7,91 (dd, 1H), 7,36 (d, 2H), 5,17 (d, 2H), 4,25-4,34 (m, 1H), 2,96 (dd, 1H), 2,80 (dd, 1H) ppm, 19F ЯМР (376 МГц, MeOD-d4) δ = -135,7 (d, 1F) ppm
Для определения характеристик формы B применяли следующие условия UPLC-MS и HPLC. Условия UPLC-MS: прибор: Waters Acquity UPLC; колонка: Waters Aquity BEH C18 (50 мм, внутренний диаметр 2,1 мм, размер частиц: 1,7 мкм); температура колонки: 30°C, подвижная фаза (градиент): 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле (об./об. = от 90/10 до 15/85). Расход 0,5 мл/мин. Длина волны детектора: 210 нм. Условия HPLC: прибор: Agilent 1200 HPLC, колонка: Waters Xbridge C18 (150 мм, внутренний диаметр 3,0 мм, размер частиц 3,5 мкм); температура колонки 30°C; подвижная фаза (градиент): 0,1% фосфорной кислоты в воде и ацетонитрил (об./об. = от 90/10 до 15/85). Расход 0,7 мл/мин. Длина волны детектора: 210 нм.
В третьем способе исходный материал в виде формы HB (1 часть по весу) загружали в реактор 1 как суспензию в смеси 1-пропанол/вода в соотношении 70/30% по весу (41,3 части по весу) при комнатной температуре. Суспензию нагревали до температуры рубашки (Tj), равной 90°C (температура реакции (Tr) > 80°C), для достижения полного растворения. Раствор фильтровали через предварительно прогретое оборудование для фильтрации (температура рубашки фильтра составляла 85-90°C) при давлении, составляющем 500 мбар, и переносили в реактор 2. Фильтрацию с получением прозрачного раствора проводили с применением гидрофильного фильтра Millipore с размером пор 0,45 мкм и целлюлозных фильтров Seitz K250P. Оба фильтры предварительно увлажняли смесью 1-пропанол/вода в соотношении 70/30. Транспортные линии и фильтры промывали с помощью смеси 1-пропанол/вода в соотношении 70/30% по весу (4,5 части по весу) и соединяли снова с исходным веществом в виде суспензии/раствора в реакторе 2 (в собирающем реакторе могло наблюдаться самопроизвольное образование твердого вещества из-за перепада температуры).
Перегонка
Cуспензию (в реакторе 2) снова нагревали до приблизительно Tj=85°C (Tr > 80°C) для достижения полного растворения. После достижения полного растворения температуру реактора регулировали на уровне 70-75°C и добавляли затравочный кристалл (форма B) с уменьшенным размером затравочного кристалла (0,01 части по весу), суспендированный в 1-пропаноле (0,1 части по весу). Температуру рубашки повышали для начала перегонки (приблизительно 80-85°C) и создавали вакуум при 500 мбар. Процесс перегонки начинался при следующих условиях: внутреннюю температуру реактора регулировали на уровне 70-75°C и создавали вакуум при 450-500 мбар. В ходе процесса перегонки добавляли новую порцию растворителя 1-пропанола (70 частей по весу), чтобы заменить отогнанное вещество (перегонка при постоянном объеме).
В конце процесса перегонки сбрасывали вакуум и содержимое реактора охлаждали до 20°. Отбирали образец суспензии для IPC-контроля (остаточное количество воды составляло ниже 2 вес. %).
Фильтрация и промывание
Твердое вещество фильтровали и промывали с помощью 1-пропанола (2×10 частей по весу). Твердое вещество охарактеризовали продолговатыми частицами иглоподобной формы.
Твердое вещество высушивали при 45-50°C под вакуумом (остаточное давление составляло 10-20 мбар) при перемешивании в течение 24 часов.
Порошковая рентгеновская дифракция
Порошковые рентгеновские дифрактограммы (XRPD) получали с применением Bruker Discovery D8 в геометрии отражения. Порошки анализировали с применением держателя для образца с Si-подложкой и нулевым фоном. Излучение представляло собой Cu Kα (λ=1,5418 ). Дифрактограммы измеряли при значениях 2-тета, составляющих от 2° до 40°.
Таблица 1. Данные порошковой рентгеновской дифракции для кристаллической гидратной формы B
Таблица 2. Данные порошковой рентгеновской дифракции для кристаллической формы HB
DSC
Дифференциальную сканирующую калориметрию выполняли для каждой кристаллической формы с применением прибора от TA Instruments (DSC 2500). При каждом анализе 2-4 мг образца помещали в алюминиевый тигель T-zero, который закрывали крышкой с отверстием для иглы. Скорость нагревания составляла 10°C в минуту в диапазоне значений температуры от 30 до 300°C. Значения температуры приведены в градусах Цельсия (°C), и значения энтальпии приведены в Джоулях на грамм (Дж/г). На графиках эндотермические пики показаны направленными вниз. Эндотермический пик плавления (точку плавления) устанавливали по экстраполированной температуре начала плавления. Точность измеренной температуры образца по данному способу составляет приблизительно ±1°C, и теплота плавления может быть измерена с относительной погрешностью, составляющей приблизительно ±5%.
Иллюстративные профили DSC, полученные с применением кристаллических форм B и HB, показаны на фигурах 2 и 4 соответственно.
Форма B: эндотерма плавления: Tначала=197,4°C (плавление при разложении)
Форма HB: эндотерма плавления: Tначала=95°C (дегидратация) и Tначала=198,5°C (плавление при разложении).
Термогравиметрический анализ (TGA)
Кривые TGA получали с применением прибора от TA Instruments Q5000. 5-15 мг образца помещали в алюминиевый тигель и герметично закрывали. Непосредственно перед анализом автоматизированный пробоотборник пробивал отверстие в герметично закрытом тигле. Измерения для кривой TGA проводили в диапазоне значений температуры 30-300°C с шагом 10°C/мин. Рассчитывали LoD (потеря массы при высушивании) при температуре от 40°C до 150°C. Строили график зависимости потери массы от измеренной температуры образца. Значения температуры приведены в градусах Цельсия (°C), а потеря веса - в %.
Иллюстративные профили TGA, полученные с применением кристаллических форм B и HB, показаны на фигурах 3 и 6 соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ LTA4H ДЛЯ ЛЕЧЕНИЯ ГНОЙНОГО ГИДРАДЕНИТА | 2020 |
|
RU2821362C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СВОБОДНОГО ОСНОВАНИЯ | 2014 |
|
RU2675851C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНИЛА И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2839891C1 |
| СОЛИ И ТВЕРДЫЕ ФОРМЫ МОНОБАКТАМНОГО АНТИБИОТИКА | 2016 |
|
RU2754180C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2814498C2 |
| СОЛЬ ПРИСОЕДИНЕНИЯ КИСЛОТЫ СОЕДИНЕНИЯ, ИНГИБИРУЮЩЕГО Trk | 2015 |
|
RU2708236C2 |
| КРИСТАЛЛЫ СОЛИ | 2013 |
|
RU2652116C2 |
| КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОГО КАСКАДА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2569299C2 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2692766C1 |
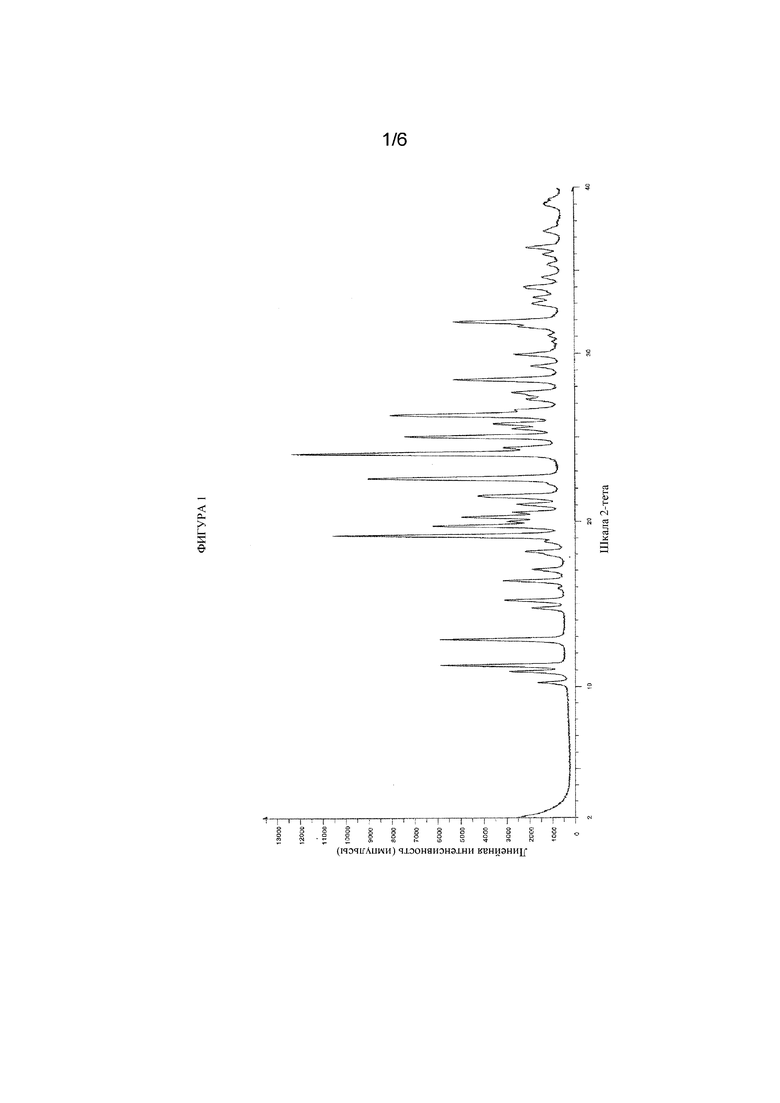
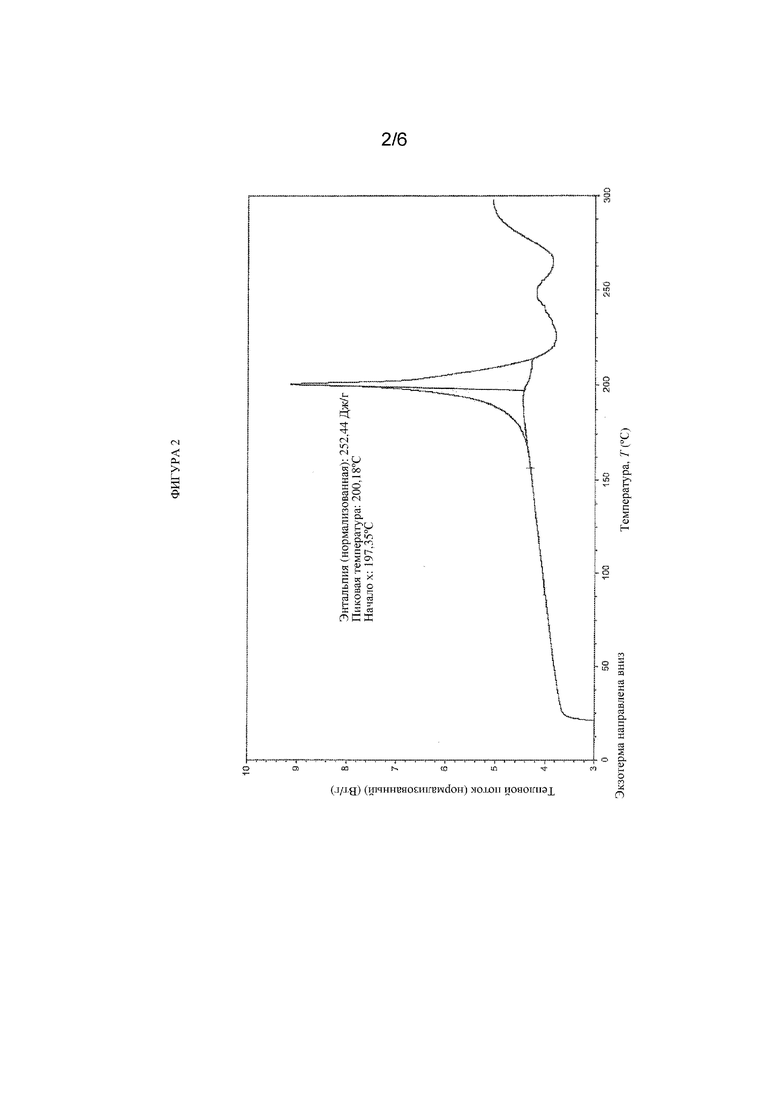
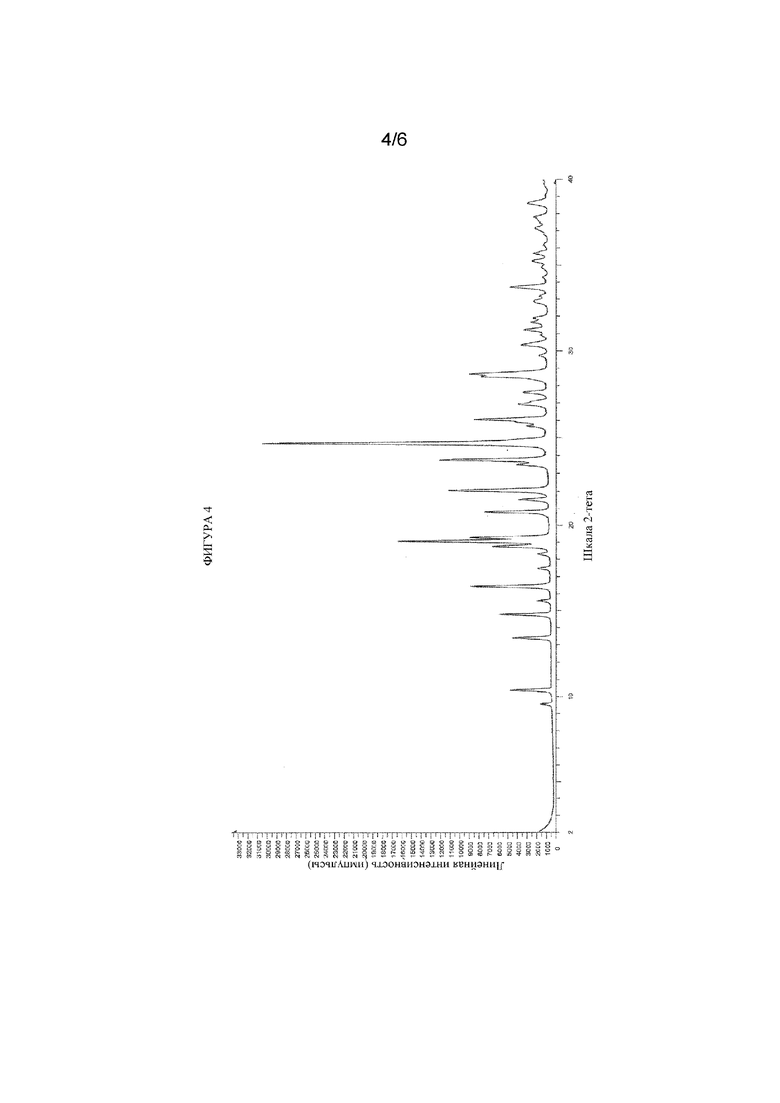

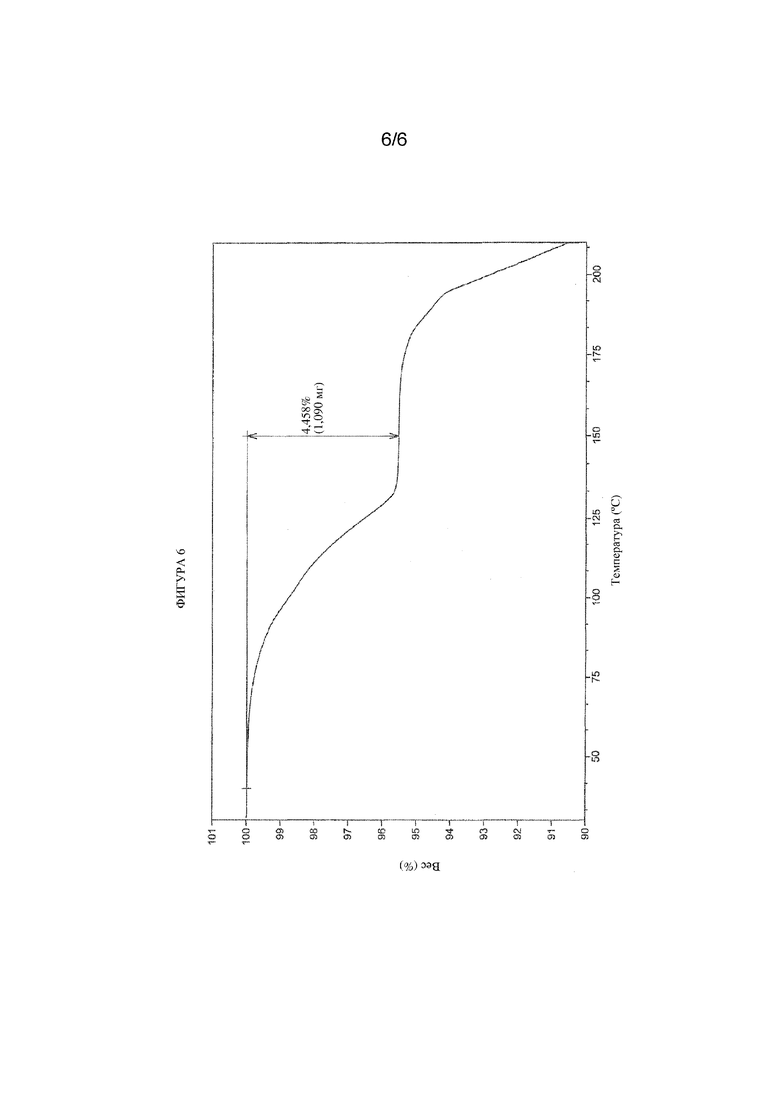
Изобретение относится к способу получения кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме, которая характеризуется порошковой рентгенограммой, включающей четыре или более значений 2θ, выбранных из группы, состоящей из 11,3±0,2°2θ, 12,8±0,2°2θ, 15,2±0,2°2θ, 19,7±0,2°2θ, 20,0±0,2°2θ, 20,3±0,2°2θ, 21,0±0,2°2θ, 22,6±0,2°2θ, 24,1±0,2°2θ, 24,4±0,2°2θ, 25,1±0,2°2θ, 26,3±0,2°2θ, 28,5±0,2°2θ и 30,0±0,2°2θ, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418  . Способ осуществляют путем (а) суспендирования в метаноле или суспендирование в смеси вода/метанол, где указанная смесь имеет активность воды меньше или равную 0,7, или супендирование в смеси 1-пропанол/вода в соотношении 70/30% по весу кристаллической формы гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы HB), которая характеризуется порошковой рентгенограммой, включающей четыре или более значений 2θ, выбранных из группы, состоящей из 13,4±0,2°2θ, 20,8±0,2°2θ, 22,1±0,2°2θ, 23,5±0,2°2θ, 23,5±0,2°2θ, 23,8±0,2°2θ, 24,7±0,2°2θ, 26,1±0,2°2θ, 26,9±0,2°2θ, 28,7±0,2°2θ, 30,4±0,2°2θ, 31,2±0,2°2θ, 33,8±0,2°2θ и 38,7±0,2°2θ, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 , с образованием смеси в виде суспензии. Затем (b) нагревают и перемешивают смесь в виде суспензии до температуры от приблизительно 50°C до приблизительно 65°С в случае суспендирования в метаноле, или до температуры от приблизительно 50°С до приблизительно 68°С в случае суспендирования в смеси метанол/вода, или до температуры выше 80°С до приблизительно 90°С для достижения полного растворения в случае суспендирования в смеси 1-пропанол/вода. После (c) охлаждают раствор до комнатной температуры с образованием смеси в виде суспензии; и (d) собирают кристаллическую форму B из суспензии, при этом термин «приблизительно» указывает на отклонение ±5%. Технический результат - получения высококристаллической и слабогигроскопичной кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме. 18 з.п. ф-лы, 6 ил., 5 табл., 2 пр.
. Способ осуществляют путем (а) суспендирования в метаноле или суспендирование в смеси вода/метанол, где указанная смесь имеет активность воды меньше или равную 0,7, или супендирование в смеси 1-пропанол/вода в соотношении 70/30% по весу кристаллической формы гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы HB), которая характеризуется порошковой рентгенограммой, включающей четыре или более значений 2θ, выбранных из группы, состоящей из 13,4±0,2°2θ, 20,8±0,2°2θ, 22,1±0,2°2θ, 23,5±0,2°2θ, 23,5±0,2°2θ, 23,8±0,2°2θ, 24,7±0,2°2θ, 26,1±0,2°2θ, 26,9±0,2°2θ, 28,7±0,2°2θ, 30,4±0,2°2θ, 31,2±0,2°2θ, 33,8±0,2°2θ и 38,7±0,2°2θ, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 , с образованием смеси в виде суспензии. Затем (b) нагревают и перемешивают смесь в виде суспензии до температуры от приблизительно 50°C до приблизительно 65°С в случае суспендирования в метаноле, или до температуры от приблизительно 50°С до приблизительно 68°С в случае суспендирования в смеси метанол/вода, или до температуры выше 80°С до приблизительно 90°С для достижения полного растворения в случае суспендирования в смеси 1-пропанол/вода. После (c) охлаждают раствор до комнатной температуры с образованием смеси в виде суспензии; и (d) собирают кристаллическую форму B из суспензии, при этом термин «приблизительно» указывает на отклонение ±5%. Технический результат - получения высококристаллической и слабогигроскопичной кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме. 18 з.п. ф-лы, 6 ил., 5 табл., 2 пр.
1. Способ получения кристаллической формы B соединения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в свободной форме, которая характеризуется порошковой рентгенограммой, включающей четыре или более значений 2θ, выбранных из группы, состоящей из 11,3±0,2°2θ, 12,8±0,2°2θ, 15,2±0,2°2θ, 19,7±0,2°2θ, 20,0±0,2°2θ, 20,3±0,2°2θ, 21,0±0,2°2θ, 22,6±0,2°2θ, 24,1±0,2°2θ, 24,4±0,2°2θ, 25,1±0,2°2θ, 26,3±0,2°2θ, 28,5±0,2°2θ и 30,0±0,2°2θ, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418  , при этом указанный способ включает стадии:
, при этом указанный способ включает стадии:
а) суспендирования в метаноле или суспендирование в смеси вода/метанол, где указанная смесь имеет активность воды меньше или равную 0,7, или супендирование в смеси 1-пропанол/вода в соотношении 70/30% по весу кристаллической формы гидрата (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (формы HB), которая характеризуется порошковой рентгенограммой, включающей четыре или более значений 2θ, выбранных из группы, состоящей из 13,4±0,2°2θ, 20,8±0,2°2θ, 22,1±0,2°2θ, 23,5±0,2°2θ, 23,5±0,2°2θ, 23,8±0,2°2θ, 24,7±0,2°2θ, 26,1±0,2°2θ, 26,9±0,2°2θ, 28,7±0,2°2θ, 30,4±0,2°2θ, 31,2±0,2°2θ, 33,8±0,2°2θ и 38,7±0,2°2θ, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 , с образованием смеси в виде суспензии;
b) нагревания и перемешивания смеси в виде суспензии до температуры от приблизительно 50°C до приблизительно 65°С в случае суспендирования в метаноле, или до температуры от приблизительно 50°С до приблизительно 68°С в случае суспендирования в смеси метанол/вода, или до температуры выше 80°С до приблизительно 90°С для достижения полного растворения в случае суспендирования в смеси 1-пропанол/вода;
c) охлаждения раствора до комнатной температуры с образованием смеси в виде суспензии;
d) сбора кристаллической формы B из суспензии,
где термин «приблизительно» указывает на отклонение ±5%.
2. Способ по п.1, в котором форму НВ суспендируют в смеси вода/метанол, где соотношение вода/метанол составляет от 1:2 до 1:9 по объему (об./об.).
3. Способ по п.2, в котором соотношение вода/метанол выбрано из 1:2; 1:4 и 1:9 по объему (об./об.).
4. Способ по п.2, в котором соотношение вода/метанол составляет 1:2 по объему (об./об.).
5. Способ по любому из пп.1-4, в котором на стадии d) собирают кристаллическую форму В из суспензии путем фильтрации.
6. Способ по любому из пп.1-4, в котором на стадии d) сбор кристаллической формы В из суспензии проводят путем перегонки.
7. Способ по любому из пп.1-6, дополнительно включающий стадии:
e) растворения гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в воде или в смеси растворителей THF/вода с образованием раствора;
f) регулирования pH раствора с помощью водного раствора NaHCO3 или водного раствора NaOH до значения pH, составляющего от 3,3 до 7,5, при перемешивании с получением в результате суспензии;
g) сбора кристаллической формы HB из суспензии.
8. Способ по п.7, в котором соотношение ТГФ/вода составляет приблизительно 10:90 по весу, термин "приблизительно" указывает на отклонение в ±5%.
9. Способ по п.7 или 8, в котором стадию e) проводят при температуре до 35°С до получения прозрачного раствора.
10. Способ по любому из пп.7-9, в котором на стадии f) рН доводят до значения рН от 3,3 до 7,5 с использованием водного раствора NaOH.
11. Способ по п.10, в котором на стадии f) перемешивание продолжают до 20 часов.
12. Способ по любому из пп.7-11, в котором на стадии g) сбор кристаллической формы HB осуществляют путем фильтрации.
13. Способ по любому из пп.7-12, дополнительно включающий стадии h)-m) для получения гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, где осуществляют:
h) добавление толуола к (S)-3-((трет-бутоксикарбонил)амино)-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоте;
i) добавление водного раствора HCl с образованием реакционной смеси;
j) перемешивание и нагревание реакционной смеси до температуры, составляющей от приблизительно 50°C до приблизительно 65°C;
k) охлаждение реакционной смеси до приблизительно 35°C;
l) отделение водного слоя и охлаждение полученного органического слоя до температуры, составляющей от 18°C до 25°C, с получением в результате суспензии;
m) сбор гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты из суспензии, где термин "приблизительно" указывает на отклонение в ±5%.
14. Способ по п.13, в котором на стадии h) добавляют толуол в таком количестве, чтобы соотношение (S)-3-((трет-бутоксикарбонил)амино)-4-(5-(4-((5-хлор)-3-фторпиридин-2-ил)окси)фенил)-2Н-тетразол-2-ил)бутановая кислота/толуол составляло от 1:5 до 1:10 (вес/вес).
15. Способ по п.13 или 14, в котором на стадии i) водный раствор HCl представляет собой 30-40% водный раствор HCl, а количество HCl составляет приблизительно 15 эквивалентов гидрохлоридной соли (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты и где термин "приблизительно" указывает на отклонение ±5%.
16. Способ по любому из пп.13-15, в котором на стадии j) реакционную смесь нагревают при температуре от приблизительно 60°С до приблизительно 65°С и перемешивают в течение 10-15 часов и где термин "приблизительно" указывает на отклонение ±5%.
17. Способ по п.16, в котором стадию j) проводят под давлением.
18. Способ по любому из пп.13-17, в котором на стадии m) сбор гидрохлоридной соли(S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2Н-тетразол-2-ил)бутановой кислоты из суспензии проводят фильтрованием или центрифугированием.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| MINO R.CAIRA, Crystalline Polymorphism of Organic Compounds, 1998, p.163-208 | |||
| L.Morissette et al.: "High-through put crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids", ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, pp.275-300 | |||
| Митькина Л.И., Ковалева Е.Л., Прокопов И.А | |||

